/






























































































































































































































































































Текст
А. СЕРРЕЙ
СПРАВОЧНИК
ПО ОРГАНИЧЕСКИМ
РЕАКЦИЯМ
ИМЕННЫЕ РЕАКЦИИ
В ОРГАНИЧЕСКОЙ ХИМИИ
ПЕРЕВОД С АНГЛИЙСКОГО
ПОД РЕДАКЦИЕЙ И С ДОПОЛНЕНИЯМИ
докт. хнм наук Я. С. ДУЛ6ФС0ЯЛ
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
ХИМИЧЕСКОЙ ЛИТЕРАТУРЫ
МОСКВА . 1 962
Содержание
СОДЕРЖАНИЕ
От редактора 8
Предисловие автора 9
Арбузов
Перегруппировка Арбузова 11
Арндт—Эйстерт
Синтез Арндтз—Эйстерта 13
Байер —Виллигер
Окисление по Банеру—Виллигеру 16
Барбье—Виланд
Расщепление по Барбье—Виланду 19
Барт
Реакция Барта 22
Бекман
Перегруппировка Бекмана 24
Берч
Восстановление по Берну 27
Бешан
Восстановление по Бешану 31
Бишлер—Напиральский
Реакция Бишлера—Напиральского 33
Блан
Хлорметилированне по Блану 37
Браун
Реакция Брауна 40
Расщеплетж аминов по Брауну 42
Буво—Блан
Восстановление л о Буво—Блану .... . . 44
Синтез альдегидов по Буво 46
Бухерер
Реакция Бухерер а 48
Синтез гидантоннов по Бухереру 51
Вагнер—Меервейн
Перегруппировка Ваги ера—Меервейна 53
баллах
Реакция В аллаха . . . . . , 57
Вильгеродт
Реакция Вильгеродтз 59
Внльямсон
Синтез Вильямсона , . . 62
Содержание 4
&ИТТИГ
Реакция Виттига 64
Воль—Циглер
Реакция Воля—Циглера 66
Вюрц
Реакция Вюрца 69
Габриэль
Синтез Габриэля 72
Ганч
Синтез Ганча 74
Гаттерман
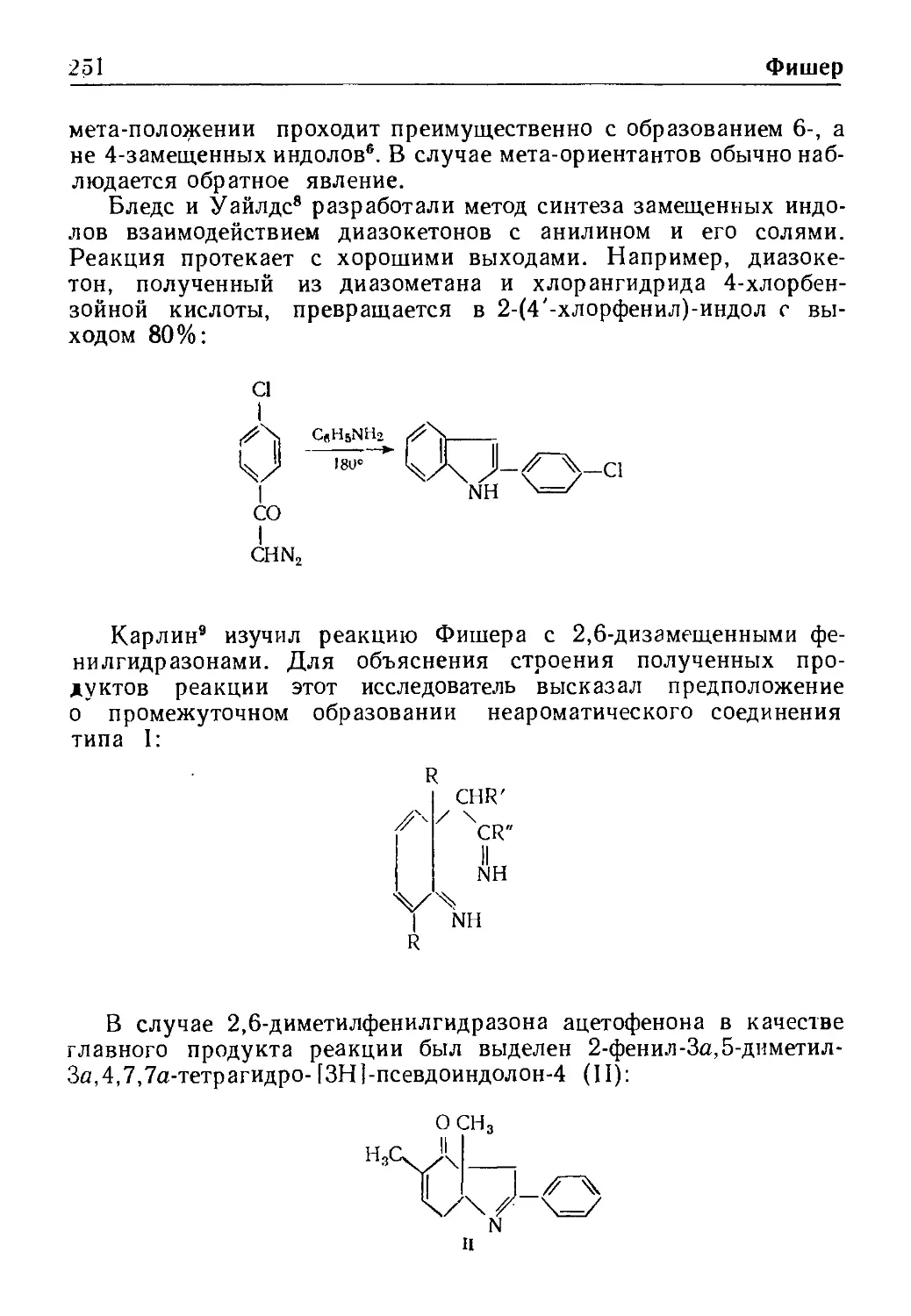
Реакция Гаттермана 77
Синтез альдегидов по Гаттерману 78
Реакция Гаттермана—Коха 81
Гелль—Фольгард—Зелинский 83
Реакция Гелля—Фольгарда—Зелинского 84
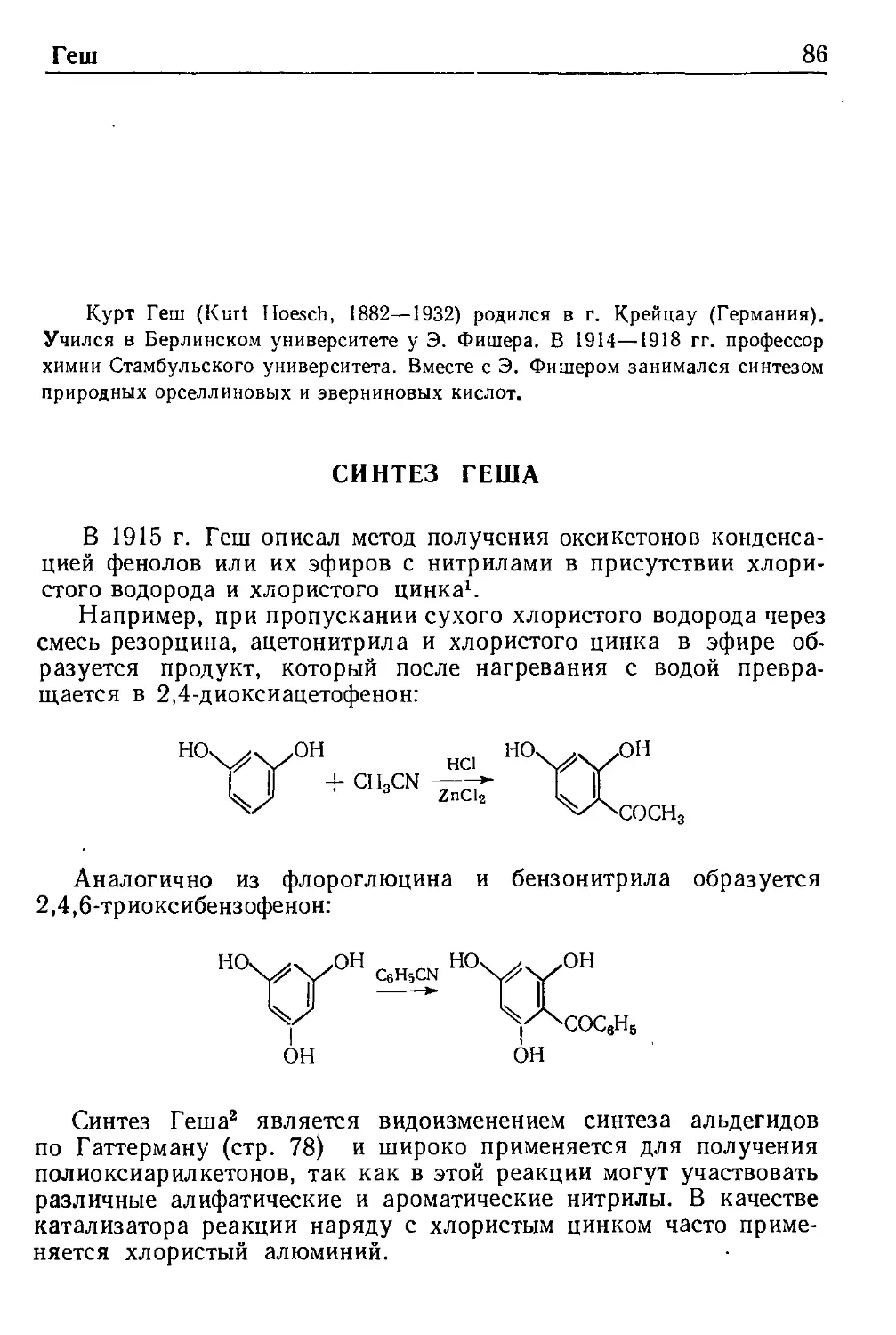
Геш
Синтез Геша 86
Гомберг—Бахман—Хей
Реакция Гомберга—Бахмана—Хея 89
Гоулд—Джекобе
Реакция Гоулда—Джекобса 91
Гофман
Перегруппировка Гофмана 93
Расщепление по Гофману 96
Гребе—Ульман
Синтез Гребе—Ульмана 99
Гриньяр
Реакция Гриньяра 101
Дарзан
Синтез глицидных эфиров по Дарзану 103
Синтез непредельных кетонов по Дарзану 106
Дафф
Реакция Даффа 109
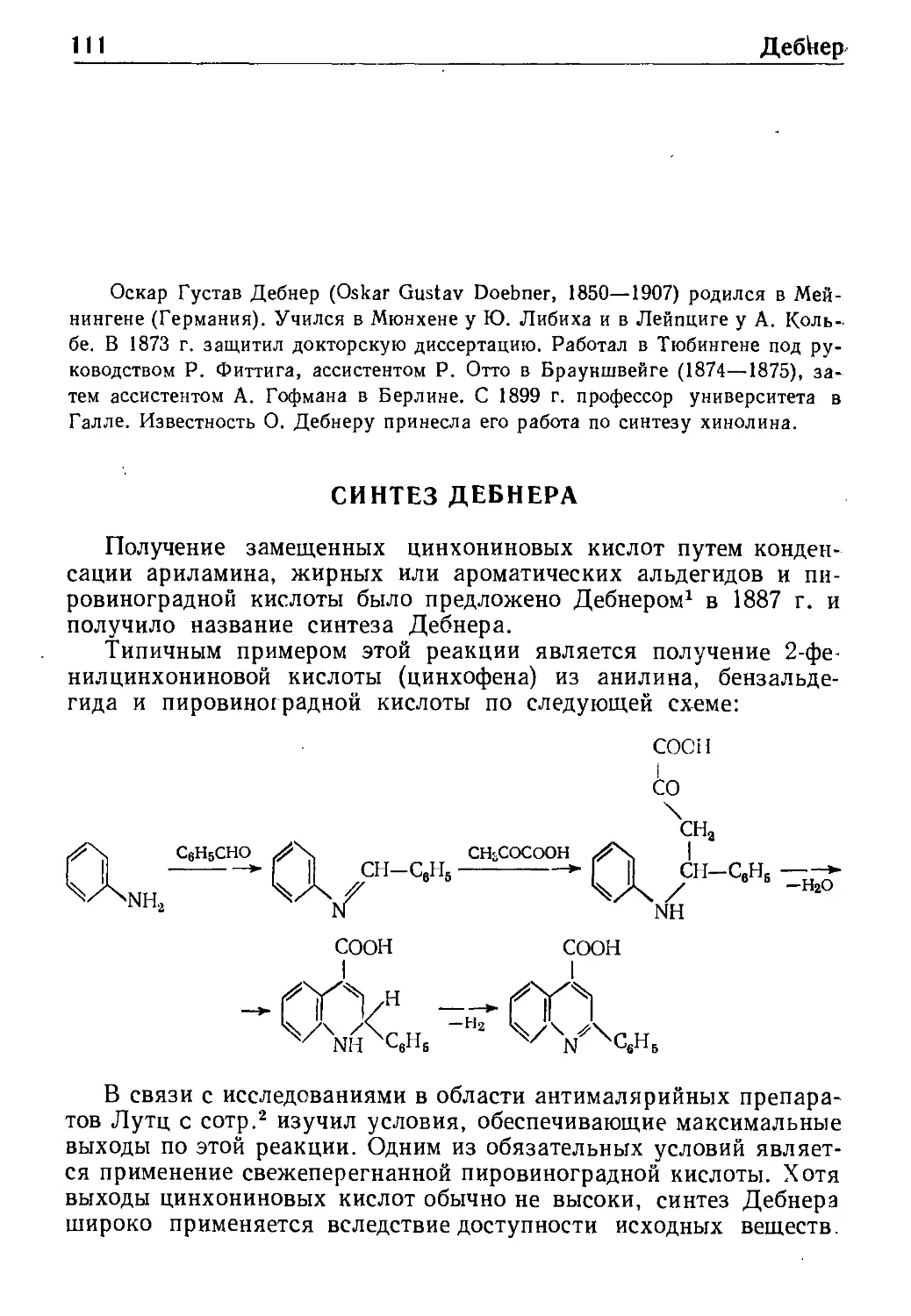
Дебнер
Синтез Дебнера * 111
Синтез Дебнера—Миллера 113
Делёпин
Реакция Делёпина , 114
Дикман
Реакция Дикмана 116
Дильс—Аль дер
Реакция Дильса—Аль дер а 119
Дэкин
Реакция Дэкина 122
Реакция Дэкина—Веста 124
Зандмейер
Реакция Зандмейера 126
Иванов
Реакция Иванова . . , . , . , . „ . . . . . 128
5 Содержание
Иоцич
Реакция Иоцича 130
Канниццаро
Реакция Канниццаро 132
Кижнер
Реакция Кижнера 135
Клайзен
Конденсация Клайзен а 139
Перегруппировка Клайзен а 142
Конденсация КлаЙзена—Шмидта 145
Клемменсен
Восстановление по Клеыменсену 147
Кневенагель
Реакция Кневенагеля 150
Кнорр
Синтез Кнорр а 153
Кольбе
Реакция Кольбе—Шмитта 155
Электрохимическая реакция Кольбе . ! 158
Комб
Синтез хинолина по Комбу 160
Коновалов
Реакция Коновалова 162
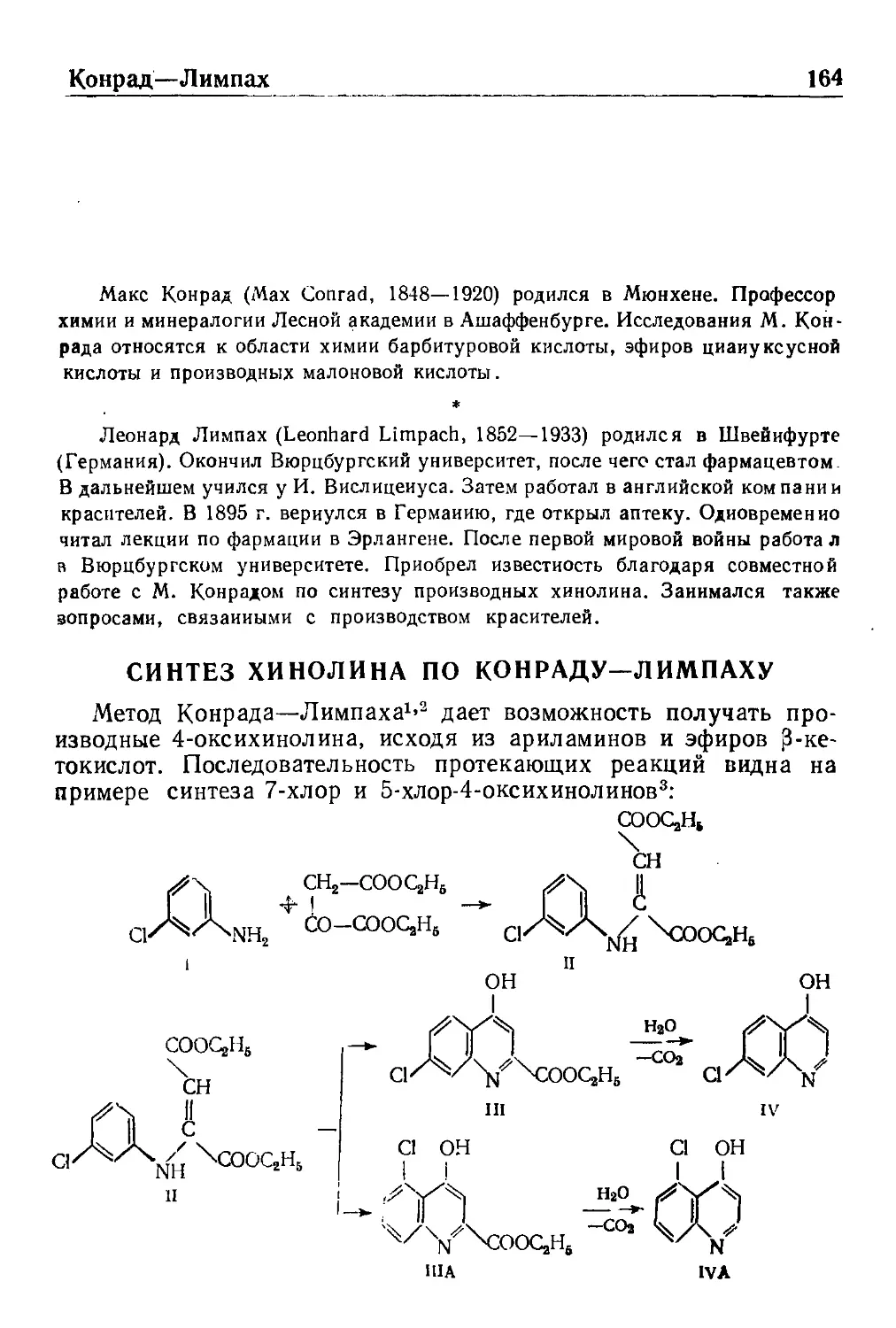
Конрад—Лимпах
Синтез хинолнна по Конраду—Лимпаху 164
Курциус
Реакция Куринуса 166
Кучеров
Реакция Кучерова 169
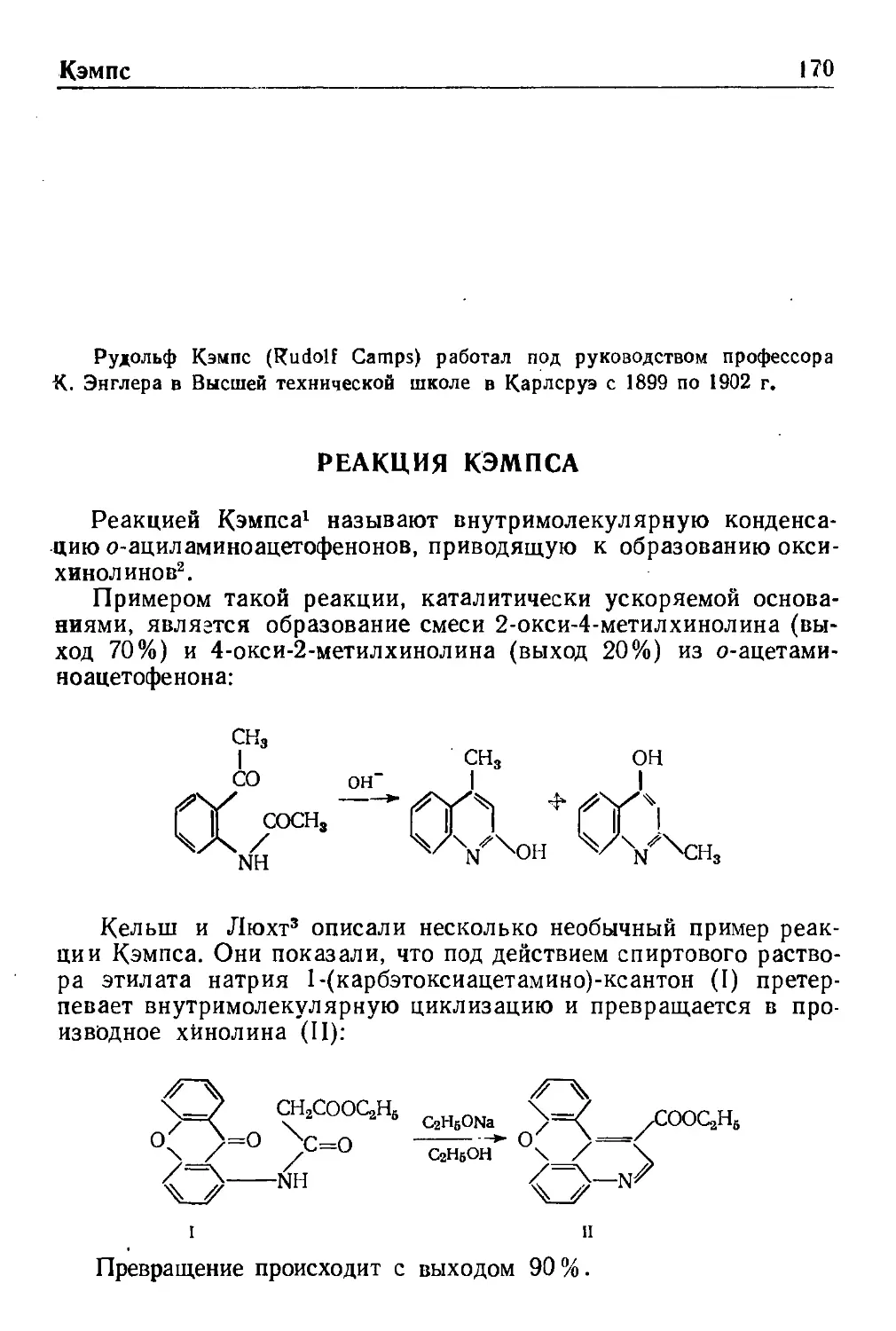
Кэмпс
Реакция Кэмпса 170
Лейкарт
Реакция Лейкарта 172
Лоссен
Реакция Лоссена 175
Мак-Фадиен—Стивене
Восстановление по Мак-Фадиену—Стивенсу 178
Манних
Реакция Манннха 180
Меервейн
Конденсация Меервейна . . 184
Восстановление по Меервейну—Понндорфу—Верлею 186
Михаэль
Реакция Михаэля ' 188
Наметкин
Перегруппировка Наметкина 190
Несмеянов
Реакция Несмеянова 192
Неф
Реакция Нефа 195
Содержание 6
Оппенауер
Окисление по Оппенауеру 197
Пассерини
Реакция Пассерини . . . ^ 199
Перкин
Реакция Перкнна . , . . . . . 201
Пехман
Конденсация Пехмана 204
Пиктэ—Шпенглер
Реакция Пнктэ—Шпенглера . . 207
Померанц—Фрич
Реакция Померанца—Фрича 208
Принс
Реакция Прннса .' . _. 210
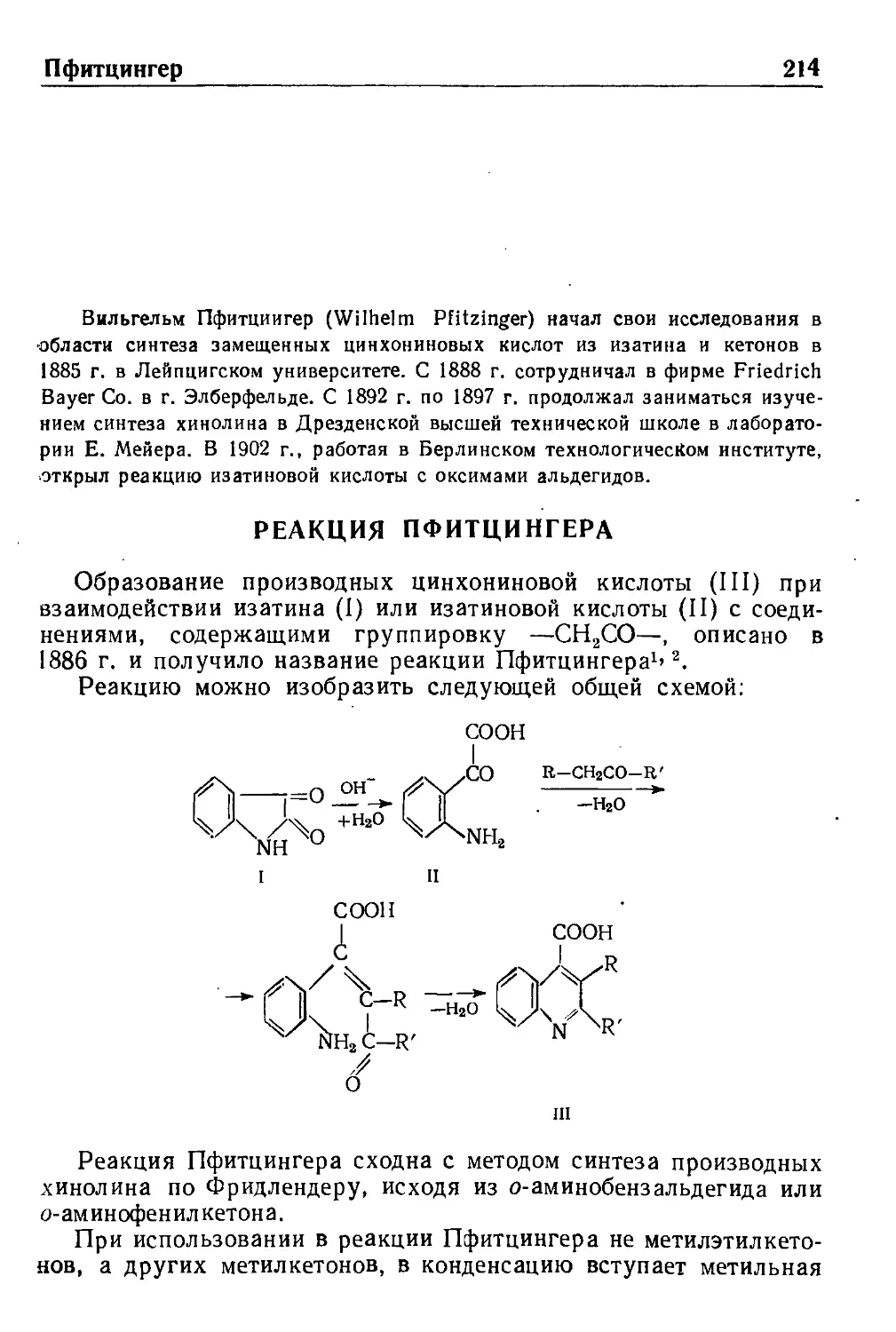
Пфитцингер
Реакция Пфитцингера 214
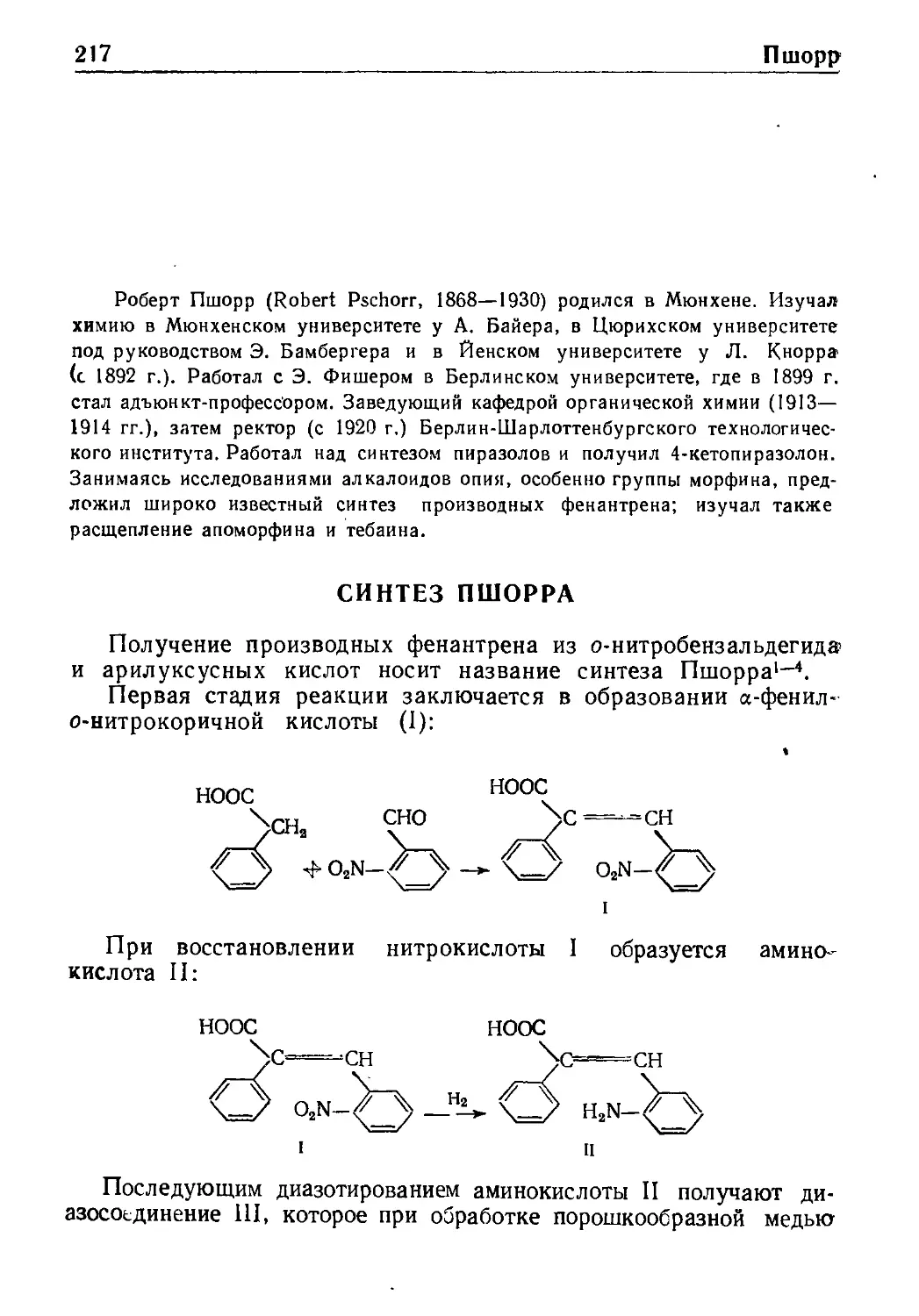
Пшорр
Синтез Пшорра . . , * . . . 217
Реймео—Тиман
Реакция Реймера—Тимана .... 219
Рейсерт
Реакция Ренсерта 222
Реформатский
Реакция Реформатского . . 224
Родионов
Реакция Родионова , - - . - 227
Розенмуид
Восстановление по Розенмунду 229
Скрауп
Реакция Скраупа . ,
Соммле
Реакция Соммле * .
Стефен
Реакция Стефена 233
Сгивенс
Перегруппировка Стивенса , 239
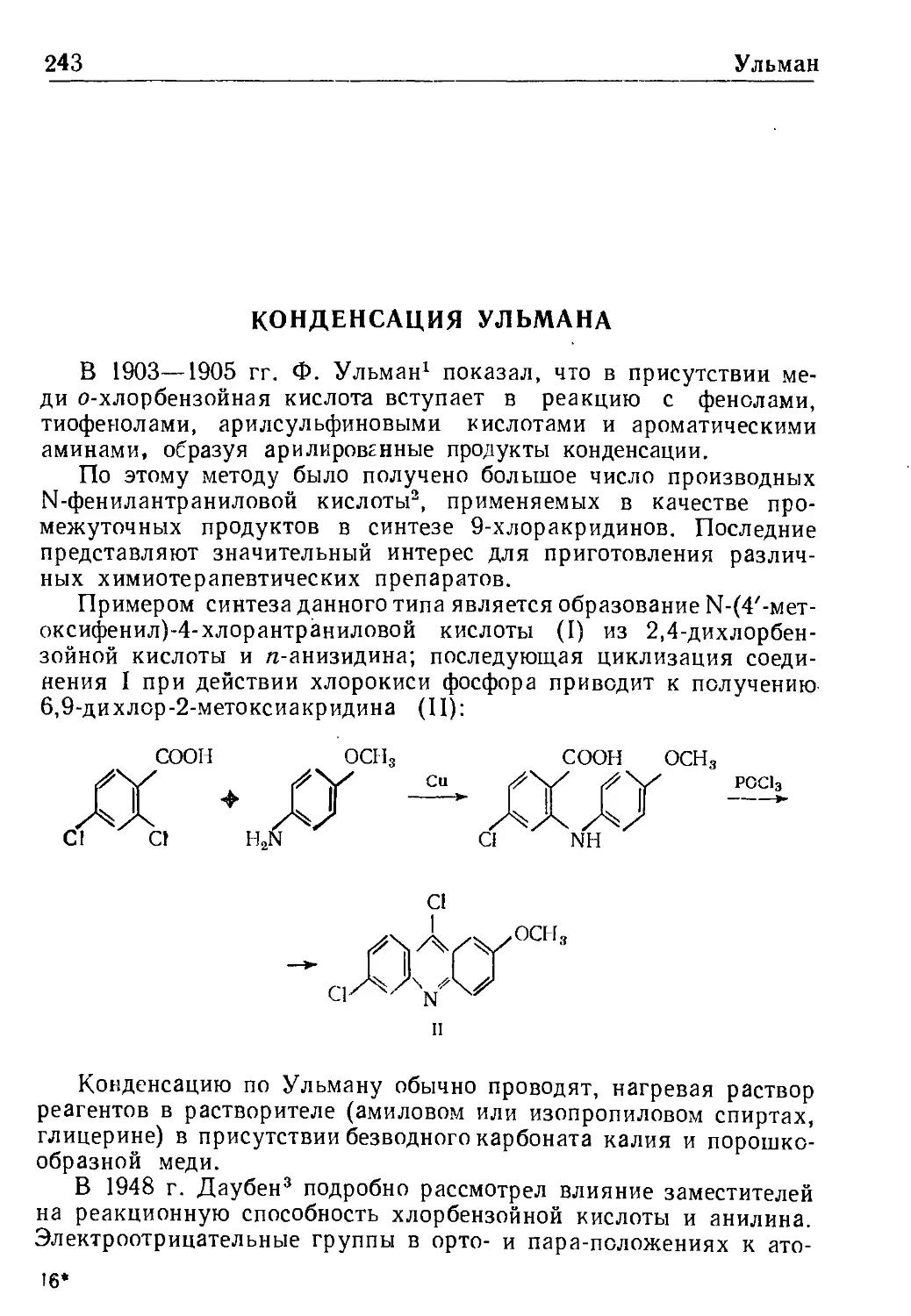
Ульман
Реакция Ульмана . * ' 241
Конденсация Ульмана 243
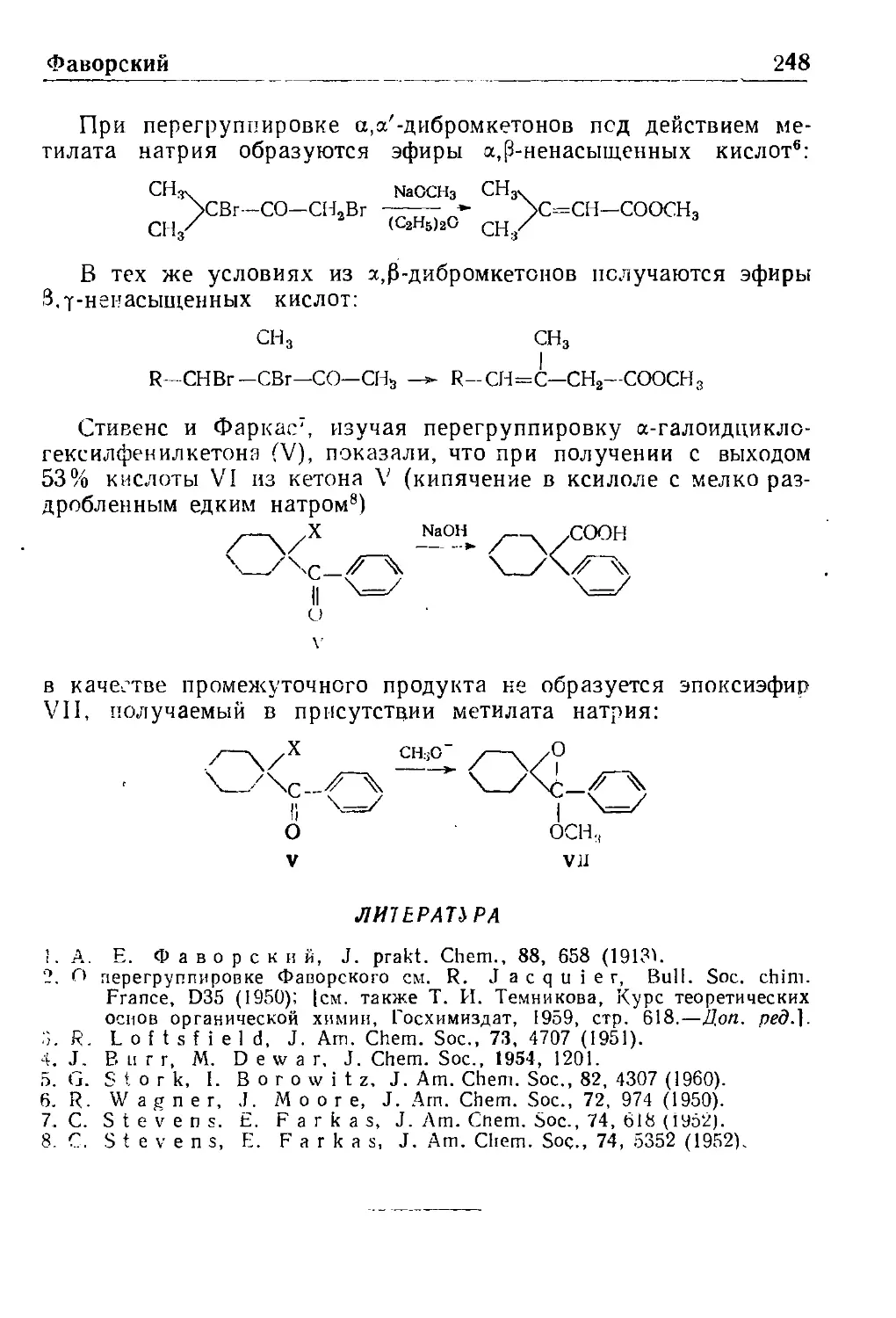
"Фаворский
Перегруппировка Фаворского ...,.,.. . . 24о
Фншер
Синтез индола по Фишеру . , , . .. .,
Фридель—Крафтс
Реакция Фриделя—Крафтса ..... ....
Фридлеыдер
Синтез Фридлендера *
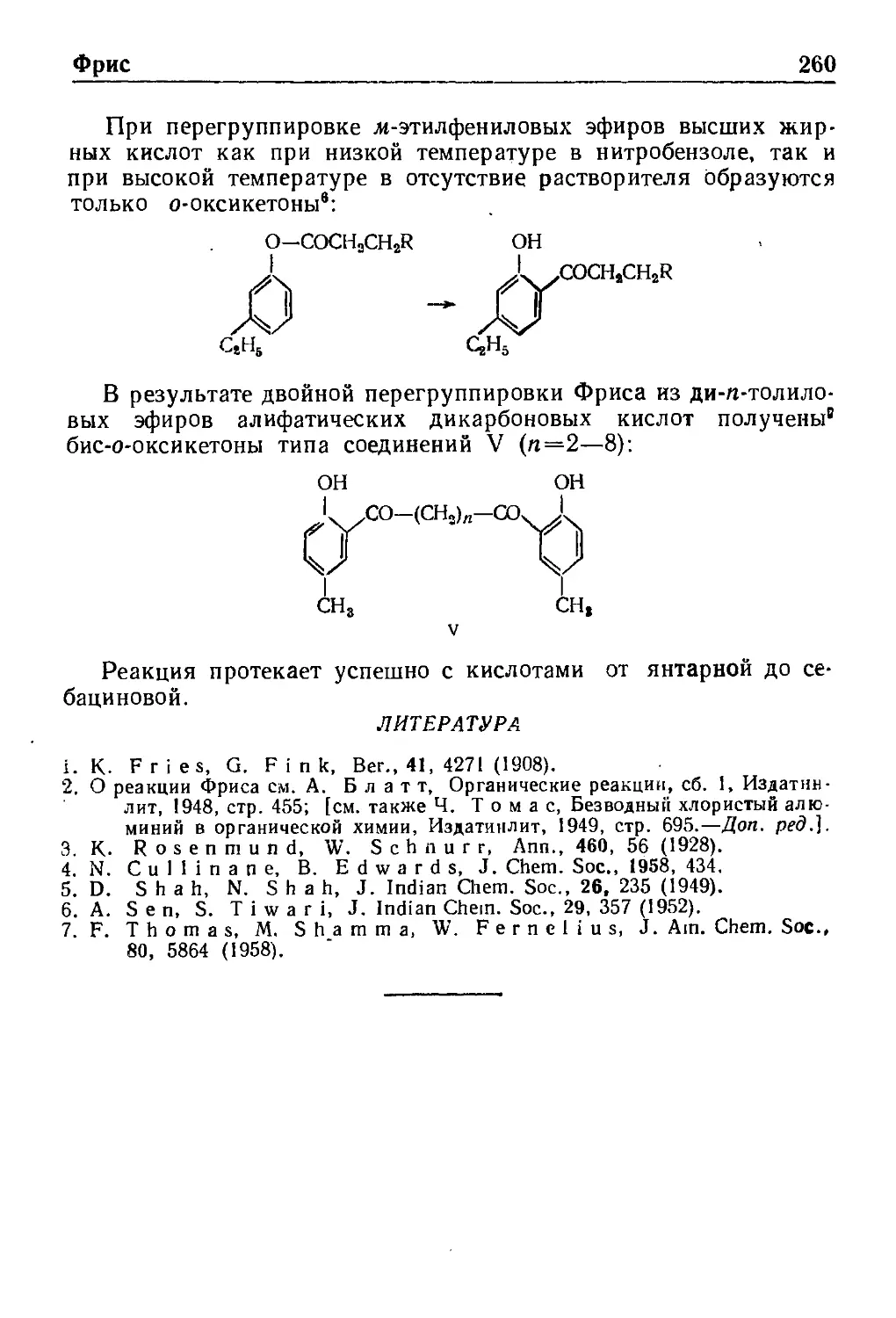
Реакция Фрнса
7 Содержание
Хунсдиккер
Реакция Хунсдиккера 261
Чичибабин
Реакция Чнчибабина 264
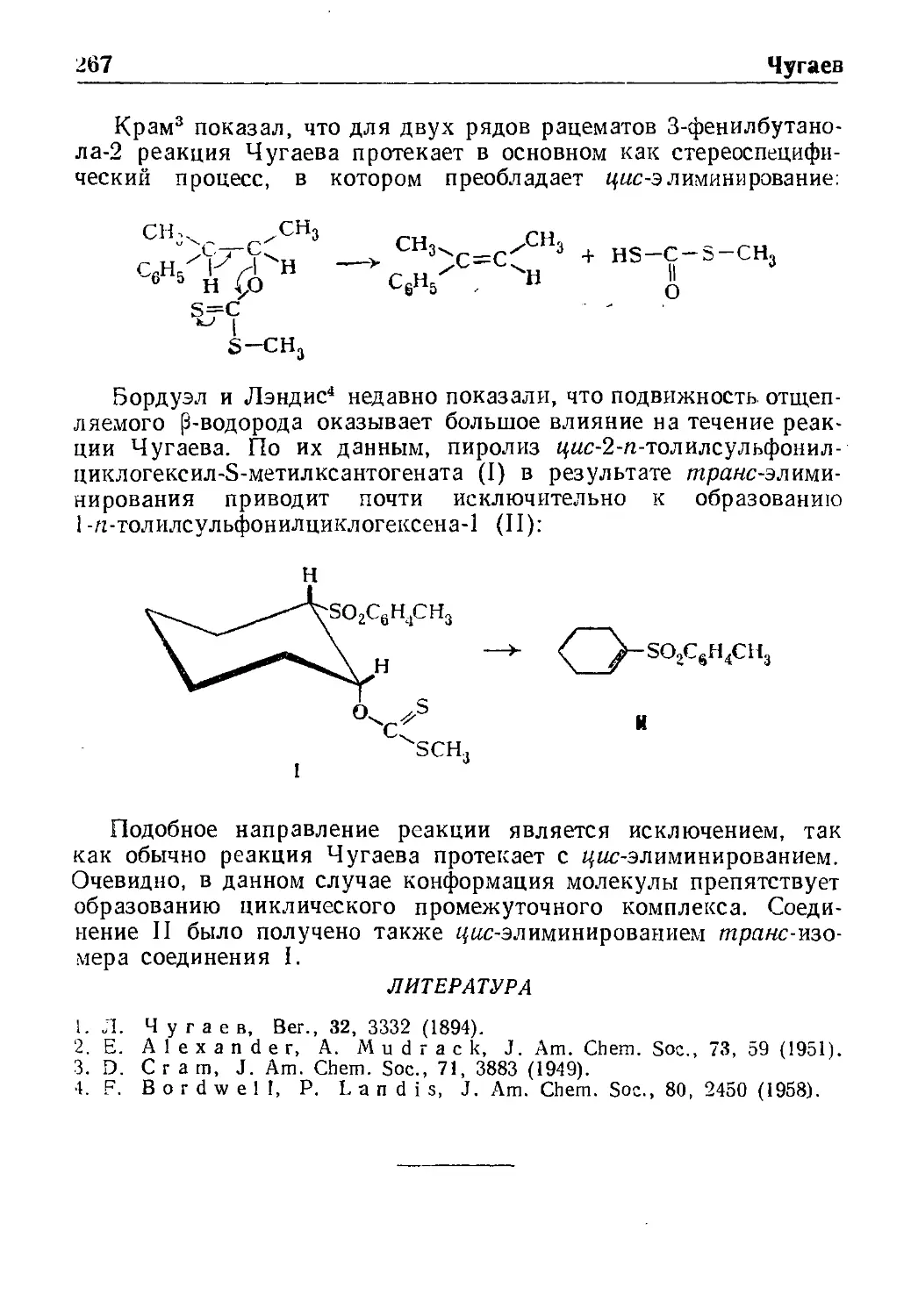
Чугаев
Реакция Чугаева 266
Шиман
Реакция Шнмана * 268
Шмидт
Реакция Шмидта . . . 260
Штоббе
Конденсация Штоббе 273
Шторк
Реакция Шторка . . , 276
Элбс
Реакция Элбса 279
Окисление персульфатами по ЭлЗсу ... 281
Эмде
Расщепление по Эмде 283
Эрленмейер—Плехль
Синтез азлактонов по Эрленыейеру—Плехлю ...... 285
Якобсен
Реакция Якобсена 288
Предметный указатель 290
От редактора 8
ОТ РЕДАКТОРА
Справочник А. Серрея «Именные реакции в органической
химии», вышедший первым изданием в 1954 г., сразу привлек к себе
внимание химиков-органиков. В дальнейшем в литературе
появилось большое число новых обзоров и статей, посвященных
«именным реакциям». Это побудило автора несколько расширить и
переработать книгу, после чего она была опубликована вторым
изданием в 1961 г.
Справочник А. Серрея содержит описание более 100 реакций,
широко известных в мировой литературе под именами авторов,
их открывших или наиболее обстоятельно изучивших. Выбор
материала для подобного справочника весьма затруднителен и
отчасти произволен, так как в литературе по имени автора часто
называют не только новые методы или реакции, но и определенные
приемы работы, реактивы, аналитические методы, цветные реакции
и т. д. В данном справочнике изложены только именные реакции
синтетической органической химии. При этом каждая реакция
описана достаточно обстоятельно, снабжена научно-биографической
справкой об ее авторах, а также краткой библиографией основных,
главным образом обзорных работ по данной теме. В других
книгах этого типа (см., например, J. E. Powan, T. S. Wheeler,
Name index of organic reactions, 1960; H. Krauch, W. Kunz,
Namenreaktionen der organischen Chemie, 1961, и др.) отбор
материала менее удачен и приводимые сведения не дают достаточно
полного представления ни о реакциях, ни о химиках, их
разработавших.
К сожалению, как в первом, так и во втором изданиях почти
полностью отсутствуют реакции, носящие имена отечественных
исследователей. Поэтому при подготовке к печати перевода
книги редактор счел необходимым дополнить ее описанием реакций
Иоцича, Коновалова, Кучерова, Несмеянова, Родионова, Чичи-
бабина, перегруппировок Арбузова, Вагнера—Меервеина,
Наметкина. Кроме того, были переработаны биографии Н. Д.
Зелинского, С. Н. Реформатского, А. Е. Фаворского, Л. А. Чугае-
ва и заново написан раздел, посвященный реакции Кижнера.
В ряде случаев приведенная автором библиография была
дополнена обзорными статьями, опубликованными в последнее время.
Я. С.
9 Предисловие автора
ПРЕДИСЛОВИЕ АВТОРА
В литературе по органической химии издавна существует
традиция называть многие реакции по имени химика, открывшего
или изучившего их. Эти «именные реакции» составляют
значительную часть наиболее важных реакций органической химии.
С момента открытия каждая такая реакция в большей или
меньшей степени применяется в органическом синтезе, претерпевая
зачастую различные изменения. Разработка новых процессов,
появление новых растворителей и конденсирующих агентов, а
также изменение условий проведения реакции способствуют ее
развитию и совершенствованию. Кроме того, исследование
механизма реакций приводит к лучшему пониманию и более
широкому использованию многих из них.
Нам казалось целесообразным собрать по возможности все
«именные реакции» в их современном состоянии и представить
в легко доступной для химиков форме справочника. Наша задача
заключалась в описании каждой реакции, сферы ее применения,
достоинств и недостатков, а также в изложении новейших
достижений в области изучения ее механизма.
Правильность подбора реакций, вошедших в книгу, мох:ет
вызвать споры, так как два химика никогда полностью не
согласятся, каким реакциям следует отдать предпочтение. Автор при
выборе материала основывался на том, какой интерес для
большинства химиков представляет та или иная реакция, в какой
мере она освещена в литературе, какой вклад внес ее автор в
развитие органической химии.
Вследствие быстрого роста органической химии легко потерять
из виду тех, кто являются пионерами в этой области. Доктор
Р. Э. Оспер* по этому поводу сказал: «Большинство химиков
употребляет имена просто как удобное обозначение направлений,
законов, реакций, типов оборудования. Обычно никто не думает
о человеке, имя которого он так часто произносит. Долгом
историка химии является превратить имя в человека».
Хотя автор и не претендует на звание историка химии, ему
кажется необходимым наряду с описанием реакций привести
R. Е. О е s p e г, J. Chem. Educ, 19, 444 (1942).
Предисловие автора 10
некоторые сведения о самих химиках, их образовании, работе
и сотрудниках, совместно с которыми они работали. Автор
надеется, что эти сведения, являющиеся частью истории химии,
представят определенный интерес для читателя.
В данной книге не дается подробный и полный перечень
литературы по каждой рассматриваемой реакции. Не было также
стремления конкурировать с книгой «Органические реакции»,
а при составлении библиографии не соблюдался строго принцип
приоритета. При подборе литературы автор больше старался
изложить материал, представляющий общий интерес, привести
соответствующие примеры и указать основные работы по данной
теме. Многие источники, на которые ссылается автор,
представляют собой общие обзоры.
В книге приводится предметный указатель, который поможет
читателю отыскать необходимую ему в данный момент реакцию.
Основными источниками биографических сведений были:
книга Поггендорфа «Литературно-биографический справочник»,
«Chemische Berichte», «Bulletin de la Societe chimique de France»,
«Journal of Chemical Education», «Journal of the Chemical Society
of London», некоторые другие журналы и личные сообщения.
Л.
11 Арбузов
Александр Ерминингельдовнч Арбузов родился в 1877 г. близ Казани.
В 1900 г. окончил Казанский университет, ученик А. М. Зайцева. В 1905 г,
защитил магистерскую, а в 1914 г.—докторскую диссертацию. С 1911 по 1930 г.
заведовал кафедрой органической химии Казанского университета, а в 1930 г.
стал профессором Казанского химико-технологического института. С 1945 г.
является председателем Казанского филиала АН СССР. В 1932 г. был избран
членом-корреспондентом, а в 1942 г. действительным членом АН СССР.
В 1957 г. ему присвоено звание Героя Социалистического Труда. Выдающиеся
по своему теоретическому и практическому значению работы А. Е. Арбузова
в основном посвящены исследованию фосфорорганическнх соединений.
ПЕРЕГРУППИРОВКА АРБУЗОВА
Изомеризация полных эфиров фосфористой кислоты (при
нагревании их с первичными г алощал килами) в эфиры моноалкил-
фосфиновых кислот называется перегруппировкой Арбузова*.
Примером такой перегруппировки может служить получение
диметилового эфира метилфосфиновой кислоты^ нагреванием три-
метилфосфита с бромистым метилом при 100°. По-видимому,
промежуточно образуется производное пятивалентного фосфора (I),
которое распадается с отщеплением галоидного ал кил а:
ОСИ* ОСИ, ОСНд
Р—ОСНп + CH^Br -^ ">Р—ОСН, -^ СНд—Р=О Ч- CHgBr
0CH3
Правильность этого предположения подтверждается, в
частности, тем, что из триэтилфосфита и бромистого метила
получается диэтиловыи эфир метилфосфиновой кислоты^ (выход 87%).
* Биография А. Е. Арбузова н изложение перегруппировки написаны
редактором.
Арбузов' [2
В дальнейшем было показано, что к подобной перегруппировке
способны и другие соединения, содержащие группу р
например* (QHJgP—CCHg, и группу /Р—SR, например
QHP(SQH)
В качестве галоидных алкилов для перегруппировки
Арбузова могут применяться первичные моно- и полигалоидуглеводоро-
ды^, эфиры галоидзамещенных кислот^, галоидзвмещенные
простые эфиры^* ^ и галоидкетоны^.
При помощи перегруппировки Арбузова были получены
также эфиры фосфиновых кислот с гетероциклическими радикалами,
например диэтиловый эфир 8-акридилфссфиновой кислоты*:
Р-ОСН,
При взаимодействии с галоидпроизводными, содержащими
галоид, связанный с неуглеродным атомом, триалкилфосфиты
также подвергаются перегруппировке. Так, триэти л фосфит при
нагревании с триэтилбромсиланом дает диэтиловый эфир фос-
фонтриэтилсилана^:
1
—Si—QH5 -» QH5—Si
Аналогично могут быть получены какодилфосфиновые эфиры^:
R
AsBr -^
1. О перегруппировке Арбузова см. Б. А. Арбузов, Реакции и методы
исследования органических соединений, кн. 3, Госхимиздат, 1954, стр. 7.
2. Г. X. Кама и, Труды Казанского химико-технологического института,
вып. 10, 1946, стр. 29.
3. А. Е. Арбузов, К. В. Н и к о н о р о в, ЖОХ, 18, 2008 (1948).
4. А. Е. Арбузов, Г. X. Камай, ЖРХО, 61,2039(1929).
5. А. Е. Арбузов, А. А. Дунин, ЖРХО, 46, 295 (1914);
А. Е. Арбузо в, ЖРХО, 69, 240 (1927).
6. В. С. Абрамов, Н. Ф. Т р я п и ц и и а, ЖОХ, 19, 929 (1949).
7. А. И. Разумов, А. Е. Арбузов, ЖОХ, 4, 838 (1934).
8. Kosolapoff, J. Am. Chem. Soc, 69, 1002 (1947).
9. Б. А. Арбузов, А. Н. Пудовик, ДАНСССР, 59, 1493(1948).
10. Г. X. Камай, О. Н. Б е л о р о с с о в я, Изв. АН СССР, ОХН,
193 (1947).
13 Арндт—Эйстерт
Фриц Арндт (Fritz Arndt) родился в 1885 г. в Гамбурге. Учился в Женеве
и Берлине, затем у Я. Говитца во Фрейбурге, где в 1908 г. получил степень
доктора. Работал ассистентом в ГреЙфсвальде, Фрейбурге н Киле. Во время первой
мировой войны был профессором Стамбульского университета, а с 1920 г.
профессор химии в Бреслау. В 1933 г. читал лекции в Оксфорде, после чего
вернулся в Стамбульский университет. Работал в различных областях
органической химии. Особенно интересовался синтезом диазометана и его реакциями с
альдегидами, кетонами и хлорангидридами кислот. Статьи Арндта по теории
резонанса разобраны в литературе^.
Берндт Эйстерт (Bernd Eistert) родился в 1902 г. в Олау (Германия). Учился
под руководством Ф. Арндта и Г. Бильтца в университете г. Бреслау, где в
1927 г. получил степень доктора философии. Работал ассистентом Ф. Арндта,
а затем П. Пфейффера в Бонне. В дальнейшем поступил на службу в Б
аденскую содо-анилиновую компанию в Людвигсхафене, с которой сотрудничает и
по настоящее время. С 1942 г. работал на химическом факультете Гейдельберг-
ского университета, а позднее в Высшей технической школе в Дармштадте
где в 1950 г. был назначен адъюнкт-профессором. Занимался исследованиями
диазосоединений, а также изучал зависимость цвета органических веществ от
их строения. Автор известных книг «Tautomerie und Mesomerie» и «Chemismus
und Konstitution». В 1951 г. Стокгольмское химическое общество присудило
ему медаль Шееле.
СИНТЕЗ АРНДТА—ЭЙСТЕРТА
Синтез Арндта—Эйстерта заключается в получении
ближайшего высшего гомолога карбоновой кислоты через ее хлорангид-
рид и диазометилкетон-' *.
Диазометилкетон образуется при взаимодействии хлорангид-
рида с диазометаном; последующее разложение диазокетона
влажной окисью серебра приводит к получению ближайшего высшего
гомолога исходной кислоты:
SOCl, CHaNa А*дО; Н%О
RCOOH >- RCOC1 >- RCOCHN » RCH,COOH
Арндт—Эйстерт
Последняя стадия синтеза, состоящая в перегруппировке
диазокетонов в карбоновые кислоты (или их производные), имеет
самостоятельное значение и называется лерег/и/ллироекоД Доль-
gk%*. Методика проведения этой перегруппировки была
усовершенствована Ньюменом и Билом*, которые показали, что при
применении раствора бензоата серебра в триэтиламине реакция
протекает в гомогенной среде. Весьма вероятно, что диазокетоны
сначала превращаются в кетены, которые далее реагируют с
водой или другими веществами, присутствующими в реакционной
смеси, с образованием карбоновых кислот или их производных;
RCOCHN, -» RCH=C=O
RCH=C=<
» RCHgCOOH
NH3
RCH,CONH*
R'OH
^ RCH,COOR'
Реакция Арндта—Эйстерта применяется для синтеза высших
гомологов алифатических, алициклических, ароматических и
гетероциклических карбоновых кислот. Диазокетоны получают из
хлорангидридов карбоновых кислот, постепенно добавляя их к
избытку диазометана в эфирном или бензольном растворе:
RCOC1 -f CHgNg -^- RCOCHN, ф HCI
Хлористый водород, образующийся в процессе реакции,
связывается избытком диазометана:
НО ф CH.N, -^ СНдС! ф Ng
В отсутствие избытка диазометана хлористый водород
реагирует с полученным диазокетоном, давая соответствующий хл ор-
метилкетон:
HCI + RCOCHN, -»- RCOCHaCl ^ N,
Ньюмен и Бил* показали, что прекрасные выходы
ароматических диазокетонов достигаются при взаимодействии
эквивалентных количеств хлорангидридов ароматических кислот,
диазометана и триэтиламина. Синтез арилхлорметилкетонов из
диазометана и ароилхлоридов часто называют paz/cywew //и/?е%ш/ла%«д\
По методу Арндта—Эйстерта ближайшие высшие гомологи
многих ненасыщенных кислот были получены в присутствии
раствора бензоата серебра в триэтил амине в качестве катализатора*.
Довольно хорошие выходы получены для кислот, содержащих
ненасыщенную связь в ином положении, нежели д, р
15 Арндт—Эйстерт
Стереохимия перегруппировки Вольфа была исследована
недавно Уибергом и Хаттоном*. Установлено, что
перегруппировка 6/пор-алкилдиазометилкетонов проходит в значительной
мере без изменения начальной конфигурации.
Применение высших алифатических диазосоединений (диазо-
углеводородов) в реакции Арндта—Эйстерта было описано Уайлд-
сом и Мидером^. Примером такой реакции является
взаимодействие л-хлорбензоилхлорида с диазоэтаном:
СН,
—GOC14- CM,—CHN, -^ C1—<f^S—CO—CN
Полученный замещенный диазокетон перегруппировывается
при нагревании в анилине:
СН,
а—^[А—^н—со—
Нагреванием в диметиланилине в присутствии бензилового спир
та был получен соответствующий сложный эфир:
а—^\—сн-соосн*с,н
*с,н,
Обычная реакция с применением окиси серебра в этом случае
не идет.
I.E. Campaign e, J. Chem. Educ, 36, 336 (1959).
2. F. A rn d t, В. Е ist ert, Ber., 68, 200 (1935).
3. О реакции Арндта—Эйстерта см. В. Б а х м а н, В. Струве,
Органические реакции, сб. 1, Издатинлит, 1948, стр. 53; [см. также К. И н-,
гольд, Механизм реакций и строение органических соединений,
Издатинлит, 1959, стр. 400.—Дол. реё.].
4. L. Wolf f, Ann., 394, 25 (1912).
5. At. Newman, P. В e a 1, J. Am. Chem. Soc, 72, 5163 (1950).
6. M. Newman, P. В e a 1, J. Am. Chem. Soc, 71, 1506 (1949).
7. D. С 1 i b b e n s, M. N i er e n s t e i n, J. Chem. Soc, 107, 1491 (1915):
M. Nierenstein, D. Wang, J. W а г r, J. Am. Chem. Soc...
46, 2551 (1924).
8. J. W о t i z, S. В u с o, J. Org. Chem., 20, 210 (1955).
9. K. W i b erg, T. Hu t t on, J. Am. Chem. Soc, 78, 1640 (1956).
10. A. WJ 1 d s, A. M ea de r, J. Org. Chem., 13, 763 (1948).
Байер —Виллигер 10
Адольф Байер (Adolf Ваеуег, 1835—1917) родился в Берлине. Учился в
Берлине и Гейдельберге у Р. Бунзена. В 1858 г. получил степень доктора наук
в Берлине, где он преподавал на кафедре, возглавляемой А. Гофманом.
С 1872 г. профессор и заведующий химическими лабораториями в Страсбурге;
с 1875 г. профессор химии Мюнхенского университета. Педагогическую
деятельность он не оставлял до восьмидесяти лет. Кафедру А. Байера занял
его известный ученик Р. Вильштеттер, получивший в 1915 г. Нобелевскую
премию за исследования по химии хлорофилла. Научные интересы А. Байера
были очень разнообразны. Он выполнил блестящую работу по установлению
строения н синтезу индиго. Занимался изучением фталеинов, мочевой кислоты,
пуринов, терпенов. Им' была предложена одна из формул строения бензола.
В 1905 г. А. Байеру была присуждена Нобелевская премия за работы в
области химии органических красителей н гндроароматическнх соединений.
ОКИСЛЕНИЕ ПО БАЙЕРУ—ВИЛЛИГЕРУ
Окисление альдегидов и кетонов перекисью водорода или над-
кислотами до сложных эфиров или их производных известно под
названием окисления по Байеру—Виллигеру^ \ Эта реакция
может быть изображена схемой:
О О
II Н*О% или II
R—C-R' — -» R—C—O—R'
надкислота
Первая работа Байера и Виллигера в этой области относится
к 1899 г. Она посвящена окислению алициклических кетонов мо-
нонадсерной кислотой до лактонов. Одним из окисленных ими
кетонов был ментон:
В 1948 г. Криге^ предположил, что реакция
Байера—Виллигера протекает по ионному механизму. Первая стадия этого
Байер—Виллигер
процесса, по-видимому, заключается в присоединении перекиси
к карбонильной группе. Радикал R вместе со своей парой
электронов мигрирует к атому кислорода с отщеплением иона R"O~
от перекисной связи; последующее отщепление протона приводит
к образованию эфира:
R
\с=О
-OR'
OR
H+
Перегруппировка не сопровождается изменением
конфигурации.
Установлено, что в случае диарилкетонов мигрирует та группа,
которая обладает большей способностью отдавать электроны*.
Например, при окислении д-метоксибензофенона надбензойной
кислотой получается монометиловый эфир гидрохинона и
бензойная кислота, т. е. в данном случае мигрирует л-метоксифениль-
ная группа:
О
НО—<fS-
В зависимости от растворимости реагентов в процессе
окисления по Байеру—Виллигеру применяются различные
растворители. Лучшим растворителем при окислении надбензойной
кислотой, вероятно, является хлороформ. В качестве окислителей
используют: надбензойную, мононадфталевую, мононадсерную и
над уксусную кислоты. Реакция обычно протекает в мягких
условиях и с удовлетворительными выходами.
Найдено, что трифторнадуксусная кислота в присутствии ди-
натрийфосфата служит прекрасным средством для окисления ке-
тонов до эфиров*. С ее помощью из метилциклопропилкетона был
получен циклопропиловый эфир уксусной кислоты с выходом
53%;
О О
II CF3CO2OH |\ ||
-С-СН, ^ )—О—О-СНз
Показано также, что в процессе окисления метилалкилкето-
нов всегда мигрирует только алкильная группа.
А. Серрей
Байер —Виллигер
Алициклические кетоны от циклобутанона до циклбгептаде-
канона при окислении надкислотами образуют соответствующие
лактоны. Рели окисление проводится в присутствии этилового
спирта, то получаются этиловые эфиры w-оксикислот (л=1 —14):
СН,
С
2—С=0
RCO#OH
з)п С=О —
сн.
О
Уд
сн
RCOgOH
Этим методом из циклобутанона был синтезирован бутиролак-
тон* с выходом 70%.
Алифатические и ароматические альдегиды при
взаимодействии с надкислотами обычно превращаются в кислоты. Окисление
ароматических альдегидов, содержащих в о- или л-положении
метоксильную или гидроксильную группу, перекисью водорода
в щелочной среде протекает с замещением альдегидной группы
гидроксильной. Данный метод, являющийся модификацией
окисления по Байеру—Виллигеру, известен под названием
(стр. 122),
1. А. В а е у е г, V i 1 1 i g е г, Вег., 32, 3625 (1899).
2. О реакции Байера—Виллигера см. Ч. X а с с е л, Органические реакции,
сб. 9, Издатинлит, 1959, стр. 82.
3. R. Criegee, Ann., 560, 127 (1948).
4. W. D о е г i n g, E. D orf m a n, J. Am. Chem. Soc, 75, 5595 (1953).
5. W. E m m о n s, С Lucas, J. Am. Chem. Soc, 77, 2287 (1955).
6. S. Fr I ess, P. Frankenburg, J. Am. Chem. Soc., 74, 2679 (1952).
19 Барбье—Виланд
Филипп Антуан Барбье (Phillippe Antoine Barbier, 1848—1922) родился в
г. Люзи (Франция). Ученик П. Бертло. В 1876 г. защитил докторскую
диссертацию, с 1880 г. профессор общей химии в Безансоне, а с 1884 по 1919 г.—
в Лионе. Работал в различных областях органической химии. [Синтезировал
основные составные части животных жиров, нагревая глицерин с различными
жирными кислотами; предложил общий метод восстановления органических
соединений действием йодистого водорода.—Дол. /W.] Занимаясь химией
терпенов, получил диметилгептенол из метилгептенона и йодистого метила в
присутствии магния. Именно Ф. Барбье порекомендовал своему ученику
В. Гриньяру применить магний в некоторых синтезах. Результаты этих работ,
как известно, вошли в историю.
Генрих Виланд (Heinrich Wieland, 1877—1957) родился в г. Пфорцгенмс
(Германия). Учился в Берлине и Штуттгарте, затем у Я. Тиле в Мюнхене.
В 1901 г. защитил докторскую диссертацию, с 1909 г. профессор химии в Мюнхен -
с ком университете, затем в Мюнхенской высшей школе и во Фрейбургском
университете. Г. Виланд—один из создателей химии органических нитросоеди-
нений. Значительное число его работ посвящено изучению строения нитросо-
единений. За выдающиеся труды в области желчных кислот, органических
радикалов и нитросоединений в 1927 г. ему была присуждена Нобелевская
премия. Исследование Г. Виландом механизма биологического окисления
является ценным вкладом в биохимию.
РАСЩЕПЛЕНИЕ ПО БАРБЬЕ—ВИЛАНДУ
Метод превращения карбоновых кислот (I) в их ближайшие
низшие гомологи (V) через третичные жирноароматические
спирты (Ш) и производные дифенилэтилена (IV) получил название
расщепления по Барбье—Виланду*.
Эфиры карбоновых кислот (II), реагируя с фенилмагнийброми-
дом, превращаются в третичные жирноароматические спирты (III),
которые при нагревании с уксусным ангидридом отщепляют воду
и образуют производные несимметричного дифенилэтилена (IV),
Барбье—Виланд 20
легко окисляющиеся хромовым ангидридом (в уксусной
кислоте) до карбоновых кислот (V):
R-CH,-COOH -* R
О
ОН
_ _,, I —Н%0
-->- R-CH,_C-C,H, >" R-CH=
R-COOH f (C,H,),CO
v
Примером расщепления по Барбье—Виланду является полу
чение ywc-2-фенилциклогексанкарбоновой кислоты' (VII) из
(2-фенилциклогексил)-уксусной кислоты (VI):
СН,ОООН уОООН
VII
Метод Барбье—Виланда оказался особенно ценным при
последовательном ступенчатом сокращении боковой,цепи стероидов;
при этом были использованы многочисленные модификации
основного процесса*. Мишер* предложил весьма интересное
видоизменение метода, что позволило одновременно удалять три атома
углерода:
)СН-СН,-СН=С(СА),
N-бромсукцинимид
СН*. —нвг
\СН—СНВг—СН=<
С=СН—CH=(g@),
R R
Недавно было показано*, что при окислении третичного
спирта VIII хромовой кислотой в водно-уксуснокислой среде
ожидаемая кислота X образуется лишь в незначительном количестве,
Барбье—Виланд
а главным продуктом реакции является кетон—4-фенил-4-метил-
пентанон-2 (IX):
с,н,
СгОз
СН,—С-СН.-С—ОН
СН@ СН,
VIII
CHg—С-СНд-СОСН;
сн,
IX
(1Н&
СН,—С-СООН ф (CHj,CO
СН,
X
1. Р. В а г b ier, R. Loc q u in, С. г., 156, 1443 (1913); H. W iel a n d,
0. Schli cht in g, R. J a cobi, Z. physiol. Chem., 161, 80 (1926)
2. С G u tsc h e, J. Am. Chem. Soc., 70, 4150 (1948).
3. C. S h о р р е e, Ann. Rept. Prog. Chem., 44, 184 (1947).
4. С. М е у s t г e, H. F г e y, A. W e 11 s t ei n, K. M i esc her, Helv.
chlm. acta, 27, 1815 (1944).
5. W. Baker, R. Curtis, J. M с О m i e, L. О I i v e, V. Rogers,
J. Chem. Soc, 1958, 1007.
Барт 22
Генрих Барт (Heinrich Bart) синтезировал мышьяковоорганические
препараты для борьбы с инфекционными заболеваниями, вызываемыми
простейшими микроорганизмами. С 1909 г. он начал разрабатывать методы
получения ароматических арсоновых кислот. Большую часть результатов этих
исследований Г. Барт опубликовал в 1922 г., работая в биохимическом институте
при Гейдельбергском университете.
РЕАКЦИЯ БАРТА
Реакцией Барта^ называется получение ароматических
арсоновых кислот взаимодействием ароматических солей диазония с
арсенитом натрия в присутствии таких катализаторов, как медь
или ее соли. Примером этой реакции может служить получение
фениларсоновой кислоты из хлористого фенилдиазония:
Си
С,НвМ,С1 ф Na*AsO, —-*" С,Н*АзОзМа, -f Nad ф N,
Реакцию проводят, добавляя раствор соли диазония к
щелочному раствору арсенита натрия, содержащему катализатор.
Реакционную смесь оставляют стоять или нагревают до
прекращения выделения азота, затем продукт реакции осаждают подкис-
лением.
Известны многочисленные модификации реакции Барта*.
Реакция может протекать в нейтральных растворах без катализатора
или в буферных растворах в присутствии катализатора.
Оптимальные условия меняются в зависимости от характера и
положения заместителей в ароматическом ядре. Удачной
модификацией* реакции Барта оказалось разложение фторборатов
диазония действием арсенита натрия. Фторбораты диазония*,
полученные диазотированием ароматических аминов в присутствии HBF*
(см. реакцию Шимана, стр. 268), добавляют к водному раствору
арсенита натрия, содержащему хлористую медь (Си,С1%). По
этому методу л-нитрофениларсоновая кислота* была получена из
л-нитрофенилдиазонийборфторида (выход 79%):
Барт
1. Н. Bart, герм. пат. 250264; 254092; 264924, 1910 г.; 268172, 1912 г.
2. О реакции Барта см. С. Гамильтон, Д. Морган, Органические
реакции, сб. 2, Издатинлит, 1950, стр. 449.
3. A. Ruddy, E. S t а г к е у, W. Н а г t u n g, J. Am. Chem. Soc, 64,
828 (1942).
4. A. P у д д и, Е. С т а р к и, Синтезы органических препаратов, т. 4,
Издатинлит, 1953, стр. 378.
Бекман 24
Эрнст Отто Бекман (Ernst Otto Beckman, 1853—:.1923) родился в Золингене
(Германия). Окончил Лейпциге кий университет, ученик А. Кольбе. В 1878 г.
защитил докторскую диссертацию. Затем работал ассистентом у И. Висли-
ценуса в том же университете. Э. Бекман был выдающимся педагогом. Он
занимал кафедру химии в Эрлангенскоы и Лейпцигском университетах, с 1912 г.
профессор Берлинского университета. Под руководством И. Вислиценуса
изучал пространственное строение оксимов ментона и при этом открыл реакцию,
соторую В. Мейер впоследствии назвал перегруппировкой Бекмана.
Э. Бекман разработал методы определения молекулярных весов органиче-
ских веществ по точкам их замерзания и кипения; изобрел термометр (носящий
его имя), позволяющий точно определять температуры вблизи этих точек.
Его методика применяется и в настоящее время.
ПЕРЕГРУППИРОВКА БЕКМАНА
Перегруппировкой Бекмана называют превращение кетокси-
мов в замещенные амиды кислот, протекающее под влиянием пяти-
хлористого фосфора, бензолсульфохлорида или серной кислоты*.
Реакция была впервые описана в 1886 г. Бекманом*, который
показал, что оксим бензофенона, энергично взаимодействуя с пя-
тихлористым фосфором, перегруппировывается в бензанилид:
PCI;
(C,H*),C=N—ОН » <%-С—NH-QH,
В дальнейшем было установлено, что перегруппировка может
проходить при действии большого числа различных реагентов,
в том числе (кроме перечисленных выше) ацетилхлорида, хлор-
окиси фосфора, хлораля и хлористого водорода. Простые и
сложные эфиры оксимов также способны к перегруппировке этого типа.
Перегруппировка Бекмана заключается в обмене гидроксиль-
ной группы оксима на радикал, находящийся в дк/гш-положении
к ней. Например, в случае несимметричных кетоксимов реакция
может быть представлена следующей схемой:
R—C-R' НО-С—R' R'—С—NHR
N-OH N-R О
25
Бекман
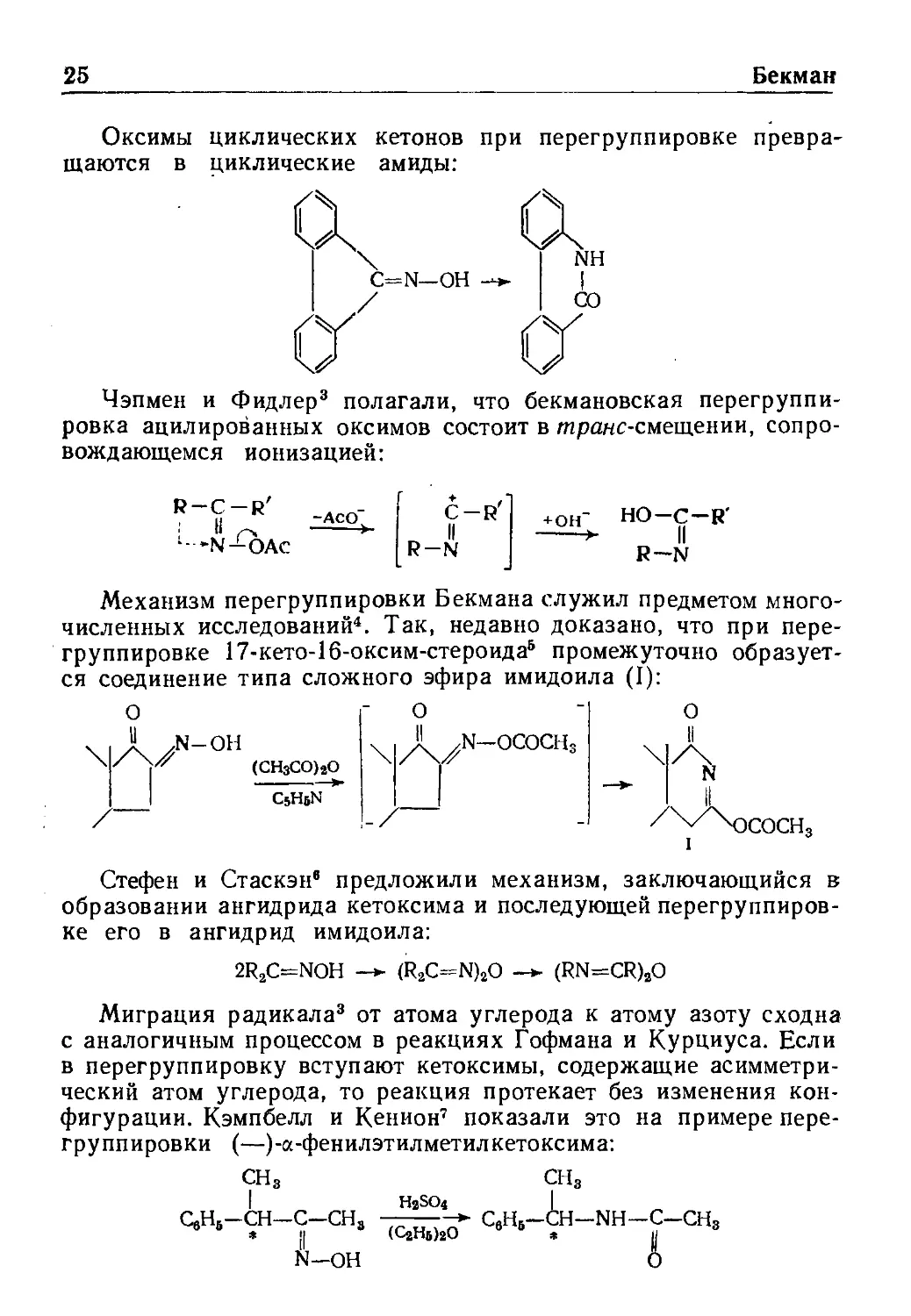
Оксимы циклических кетонов при перегруппировке
превращаются в циклические амиды:
Чэпмен и Фидлер* полагали, что бекмановская
перегруппировка ацилированных оксимов состоит в транс-смещении,
сопровождающемся ионизацией:
R-C-R'
-АСО"
R-N
+ОН" HO-C-R'
R-N
Механизм перегруппировки Бекмана служил предметом
многочисленных исследований*. Так, недавно доказано, что при
перегруппировке 17-кето-16-оксим-стероида* промежуточно
образуется соединение типа сложного эфира имидоила (I):
О
N-OH
(СНзСО)аО
—».
C5H5N
О
N—OCOCH,
ососн.
Стефен и Стаскэн* предложили механизм, заключающийся в
образовании ангидрида кетоксима и последующей
перегруппировке его в ангидрид имидоила:
2R,C=NOH
(RN=CR),0
Миграция радикала^ от атома углерода к атому азоту сходна
с аналогичным процессом в реакциях Гофмана и Курциуса. Если
в перегруппировку вступают кетоксимы, содержащие
асимметрический атом углерода, то реакция протекает без изменения
конфигурации. Кэмпбелл и Кенион? показали это на примере
перегруппировки (—)-а-фенилэтилметил кетоксима:
сн, сн,
N-OH
C
О
Бекман 26
Хорнинг недавно установил, что полифосфорная кислота
является прекрасным реагентом для перегруппировки кетоксимов^*^
и альдоксимов*. Так, оксим бензофенона под действием
полифосфорной кислоты количественно перегруппировывается в бензани-
лид, а %-гептальдоксим превращается в %-гептамид с выходом 92%.
1. О перегруппировке Бекмана см. А. В I a t t, Chem. Revs., 12, 215 (1933);
[И. Л. Кнунянц, Б. П. Фабричный, Усп. хим., 18, 633
(1949); Реакции и методы исследования органических соединений, сб. 3,
Госхимиздат, 1954, стр. 137.—Дол. рео\]
2. Е. Beckmann, Вег., 19, 988 (1886).
3. Данные о миграции различных групп см. A. Chapman, F. F i d 1 е г,
J. Chem. Soc, 1936, 448.
4. О механизме перегруппировки Бекмана см. В. Jones, Chem. Revs., 35,
335 (1944); Е. Alexander, Principles of Ionic Organic Reactions,
New York, 1950, стр. 72. [К. И н г о л ь д, Механизм реакций и
строение органических соединений, Издатинлит, 1959, стр. 400.—До/г. /W.]
5. R. Heard, M. Ryan, H. Bolker, J. Org. Chem., 24, 172 (1959).
6. Н. Stephen, В. . S t a s k u n, J. Chem. Soc, 1956, 980.
7. A. Campbell, J. К e n у о n, J. Chem. Soc, 1946, 25.
8. F. Horning, V. Stromberg, J. Am. Chem. Soc, 74, 2680 (1952).
9. F. Horning, V. Stromberg, J. Am. Chem. Soc, 74, 5151 (1952).
27 Берч
А. Дж. Берч (A. J. Birch) родился в 1915 г. в Сиднее (Австралия). Учился
8 университете в Сиднее, а затем в Оксфордском университете, где под
руководством Р. Робинсона в 1938 г. защитил докторскую диссертацию. В 1949 г.
получил от Английского королевского общества стипендию Смитсона и приехал
е Кембридж, где работал у А. Тодда. В 1952—1957 гг. профессор органической
химии в Сиднее, а с 1958 г.—профессор Манчестерского университета. В 1954 г.
он был избран членом Академии наук Австралии, а в 1958 г. стал членом
Английского королевского общества. Основными направлениями его исследований
являются: восстановление с помощью растворов металлов в жидком аммиаке,
синтезы аналогов стероидов, биосинтез и установление строения природных
соединений.
ВОССТАНОВЛЕНИЕ ПО БЕРНУ
Восстановление ароматических соединений в жидком аммиаке
при действии щелочного металла и спирта с образованием на
первой стадии циклогексадиенов-1,4 обычно называют
восстановлением по Берчу'-з.
Значение этой реакции, впервые открытой Вустером*, а
позднее исследованной и развитой Берчем, заключается в широте ее
применения и простоте эксперимента, а также в том, что
получаемые соединения очень сложно синтезировать другими методами.
Восстановлению по Верчу были подвергнуты органические
соединения различных классов*, включая метоксипроизводные ряда
бензола, бензойные кислоты, нафтойные кислоты, нафтол ы,
гетероциклические соединения, амидины, имидазолы и
стероиды.
Эту реакцию обычно проводят, прибавляя металлический
натрий к реакционной смеси, состоящей из восстанавливаемого
соединения, жидкого аммиака и абсолютного спирта. Для
повышения растворимости исходного вещества часто добавляют эфир,
тетрагидрофуран или 1,2-диметоксиэтан; иногда, в случае
необходимости, спирт приливают последним.
Примером реакции может служить восстановление 2-метокси-
5,6,7,8-тетрагидронафталина (I) до эфира енола II, который гид-
ролизуют в мягких условиях и получают кетон III с
несопряженными связями; при действии кислот или оснований кетон III пе-
Берч 28
регруппировывается в кетон IV с сопряженными двойными
связями—4,4<%,5,6,7,8-гексагидро-(ЗН)-нафталенон-2Ч
,ОСНо
Выход продукта IV, считая на исходное соединение I,
составляет 82%. .
Позднее была предложена модификация реакции Берча, за*
ключающаяся в замене натрия литием*. В присутствии лития
исключительно легко протекают процессы восстановления, которые
с трудом осуществляются при действии других восстановителей.
Например, Уайлдс и Нелсон^ в обычных условиях реакции Вер-
ча не смогли восстановить 4-циклогексиланизол (V). При
использовании же лития и добавлении спирта лишь в последнюю очередь
они получили в кристаллическом виде дигидросоединение VI с
выходом 88%:
Показано, что восстановление по Верчу нафтойной-2 кислоты,
а также ее 1- и 3-метоксипроизводных зависит от условий реакции
и соотношения реагентов и приводит в основном к образованию
1,2,3,4-тетрагидронафтойной-2 кислоты или 1,2,3,4,5,8-гексагидро-
нафтойной-2 кислоты?. Восстановление 2-метоксинафтойной-1
кислоты проходит без отщепления метоксильной группы; при
этом получается 2-метокси-1,4,5,8-тетрагидронафтойная-1 кислота^:
Проведение реакции Берча в жестких условиях описано
Джонсоном с сотр.* в работе по восстановлению ароматического
кольца производного додекагидрохризена. В данном случае
потребовалось относительно высокое соотношение спирта, причем
добавление лития регулировалось таким образом, чтобы происходило
отделение окрашенного в бронзовый цвет слоя, образующегося
при более высоких концентрациях металла в аммиаке.
Теоретические соображения о восстановлении по Верчу уже
ранее высказывались в литературе'. Недавно было опубликовано
еще несколько исследований, касающихся механизма этой реак-
Берч
^-ю. Вероятный механизм, предложенный Крапчо и Бот-
нер-Байем*, может быть изображен схемой, где М—щелочной
металл, S—растворитель (жидкий аммиак), (s) обозначает
сольватацию, a ROH—спирт:
1. М + S
н н
3. М (J)...
ROM(f)
н н
4.
н н
н н
5. М (J)...
ROH
ROMW
Авторы полагают, что стадией, определяющей скорость
реакции Берча, является взаимодействие пары сольватированных
ионов со спиртом, приводящее к образованию алкоголята
металла и промежуточного сольватированного свободного фадикала (I).
Последний реагирует затем аналогично бензолу, превращаясь в
дигидропроизводное (II).
Куэне и Ламберт*** изучили влияние заместителей,
оказывающих стабилизирующее и дестабилизирующее действие на
отрицательные ионы и на радикалы, возникающие при восстановлении
по Берчу ароматических кислот и амидов. Авторы показали,
что положение двойных связей в восстановленных соединениях
зависит от способности карбаниона воспринимать протон в месте
наибольшей плотности заряда и от стабильности продуктов реакции.
* Крупная точка в середине ядра и знак минус около него означают, что
речь идет о иин-раднкале с равномерным распределением заряда по всему ядру.
Мелкие точки в ядре радикала I символизируют частичную локализацию заряда.
—Ярил!. W
Берч30
1. А. В i г с h, J. Chem. Soc, 194-4, 430; 1946, 593.
2. А. В i r с h, Quart. Revs. (London), 4, 69 (1950).
3. А. В i г с h, H. S m i t h, Quart. Revs. (London), 12, 17 (1958).
4. C. W о о s t e г, пат. США 2182242, 1939 г.; С. W о о s t е г, К. G о d-
f г е у, J. Am. Chem. Soc, 59, 596 (1937).
5. А. К г а р с h о, А. В о t h n е г-В у, J. Am. Chem. Soc, 81, 3658 (1959).
6. A. W i 1 d s, N. N e I s о n, J. Am. Chem. Soc, 75, 5360 (1953).
7. E. E 1 i e 1, Т. Н о о v e r, J. Org. Chem., 24, 938 (1959).
8. W. J о h n s о n, В. В а п n i s t e r, R. P a p p o, J. Am. Chem. Soc,
78, 6331 (1956),
9. А. В i г с h, D. N a s i p u г i, Tetrahedron, 6, 148 (1959).
1С). M. К u e h n e, B. L a m b e r t, J. Am. Chem. Soc, 81, 4278 (1959).
31 Вешан
Антуан Вешан (Antoine J. Bechamp, 1816—1908) родился в г. Вассэне
(Франция). В 1853 г. ему была присуждена степень доктора наук, а в 1856 г.—
степень доктора медицины. С 1857 г. он—профессор химии и фармации
университета в Монпелье, а затем в университетах Нанси и Лилля. А. Бешану
принадлежит большое число различных работ. Он исследовал действие
ферментов, ферментацию вин, изучал многие другие проблемы биохимии.
Открытая им в 1854 г. реакция восстановления нитробензола железом в слабокислой
среде сыграла важную роль в развитии анилинокрасочной промышленности*.
Эта реакция была использована Перкиным для производства красителя мовеи-
на. В 1863 г. А. Бешан, нагревая анилин с арсенитом анилина, получил арса-
ниловую кислоту, строение которой и механизм реакции были установлены
позже П. Эрлихом и Бертгеймом. Образование ариларсоновых кислот при
сплавлении мышьяковой кислоты с ариламином или фенолом получило название
реакции Вешана*.
ВОССТАНОВЛЕНИЕ ПО БЕШАНУ
Химическое восстановление нитросоединений до
соответствующих аминов при действии металлического железа или солей
двухвалентного железа и разбавленных кислот известно под
названием восстановления по Бешану^. Вследствие простоты и
экономичности этот метод широко применяется в промышленности^.
С его помощью может быть восстановлено большое число
различных ароматических нитросоединений.
В лабораторных условиях восстановление обычно проводят,
постепенно добавляя нитросоединение в кипящую (с обратным
холодильником), хорошо перемешиваемую взвесь железных
опилок в разбавленном водой спирте, содержащем небольшое
количество уксусной кислоты. Скорость прибавления
нитросоединения определяется тепловым эффектом экзотермической реакции.
Соотношение вода—спирт подбирается в соответствии с
растворимостью нитросоединения. Кипячение с обратным холодильником
* Впервые анилин был получен синтетически восстановлением нитробензола
сульфидом аммония Н. Н. Зиминым (1842).
Н. Н. Зинин (1812—1880) окончил Казанский университет в 1833 г.
Профессор Медико-хирургической академии в Петербурге; с 1868 г. президент Рус
ского химического общества. /7pww. d
Бешан ^ ^^^^^^^^ 32
продолжают до тех пор, пока на внутренней поверхности
реакционного сссуда не образуется железное зеркало. По окончании
восстановления реакционную смесь обрабатывают содой и
фильтруют. Продукт реакции обычно выделяют, отгоняя спирт из
реакционной смеси.
Описанным методом можно, например, восстановить 7-хлор-
4-амино-З-нитрохинолин до 7-хлор-3,4-Диаминохинолина*
(выход 75%):
NH, NH,
СНзСООН
Примером восстановления с помощью сульфата железа может
служить превращение л-нитросалициловой кислоты в л-амино-
салициловую кислоту*.
[Полное восстановление нитросоединений может протекать
без добавления свободной кислоты даже в начальной стадии
процесса (при протравливании железа) с заменой кислоты
электролитами. Это доказывает, что процесс восстановления проходит
не в кислой, а в нейтральной среде. Наиболее активными
электролитами* являются FeCl* и NH*C1.
Механизм восстановления нитросоединений чугунными
стружками обстоятельно исследован В. О. Лукашевичем^ Подробнее
о восстановлении нитросоединений железом см. в литературеЛ—
Дол. ра).!
ЛЯГЕРЛГУРЛ
1. С. Гамильтон, Д. Морган, Органические реакции, сб. 2, Издатин-
лит, 1950, стр. 461.
2. A. Bechamp, Ann. chim. phys., [3], 42, 186 (1854).
3. J. Werner, Ind. Eng. Chem., 43, 1917 (1951).
4. A. Surrey, R. Cutler, J. Am. Chcm. Soc, 73, 2413 (1951).
5. J. McGhie, C. Morton, B. Reynolds, J. Spense, J. Soc.
Chem. Ind., (London), 68, 328 (1949).
6. A. Wohl, Ber., 27, 1432, 1815 (1894); R. Lyons, L. Smith, Ber.,
60, 173 (1927); В. О. Лукашевич, М. А. Ворошилова,
ПОХ, 4, 253 (1937); авт. свид. 51050; пат. США 1948330, Z., 1934, I, 365.—
Доа. ped.
7. В. О. Лукашевич, Усп. хим., 17, 692 (1948).—Дол. реЗ.
Д. Н. Н. Ворожцов, Основы синтеза промежуточных продуктов и
красителей, Госхимиздат, 1955, стр. 234.—До%. ^
Бишлер—Напиральский
Август Бишлер (Augustus Bischler, 1865—1957) изучал химию в
Цюрихском политехникуме под руководством В. Мей ер а и А. Ганча. В 1897—1910 гг.
научный руководитель фирмы «Циба» в Базеле (Швейцария), затем директор
химической фабрики в г. Монте. Продолжал научную деятельность в
различных лабораториях до 1955 г. В процессе исследования алкалоидов открыл
реакцию синтеза изохинолина.
Б. Напиральский—ученик А. Бишлера.
РЕАКЦИЯ БИШЛЕРА—НАПИРАЛЬСКОГО
При обработке N-ацилпроизводных фенилэтиламина (р-фе-
нилэтиламидов кислот) такими дегидратирующими агентами,
как хлорокись фосфора, пятиокись фосфора или полифосфорная
кислота, происходит циклодегидратация и образуются
производные 3,4-дигидроизохинолина:
сн,
СН, -
NH
с=о
I
R
N
R
Эта реакция, открытая Бишлером и Напиральским* в 1893 г.,
является ценным методом получения большого числа различных
соединений ряда изохинолина, особенно алкалоидов-**. В
реакции Бишлера—Напирал ьского могут с успехом применяться
ароматические, жирноароматические и алифатические амиды,
которые нагревают с дегидратирующим агентом* в среде инертных
растворителей (хлороформ, бензол, толуол или нитробензол).
Выбор растворителя определяется температурой, необходимой
для проведения реакции; обычно синтез проводят при
температуре кипения реакционной смеси. Полученный 3,4-дигидроизохино-
лин выделяют перегонкой с водяным паром или экстракцией.
Механизм реакции Бишлера—Напиральского заключается,
вероятно, в электрофильной атаке атома углерода, находящегося
3 А.
Бишлер—Напиральский
34
в орто-положении к аминоэтильной цепи, карбонильным атомом
углерода. Это подтверждается активирующим влиянием ал кок -
сильной группы, если в замещенном фенилэтиламине она
находится в мета-положении:
сн,
н
н*со
сн
- Дегидрирование полученных 3,4-дигидросоединений приводит
к производным изохинолина. Применяя Р-окси-(3-фенил этил амиды,
можно прямо получить соединения ряда изохинолина. Эта
модификация реакции, обычно называемая с%я/мгзол< У7ик/лэ—Гджш^,
протекает через стадию образования стириламидов,
дегидратирующихся затем в производные изохинолина:
ОН
сн
^ сн,
NH
I
R
сн
сн
NH
с=о
R
Недавно реакция Бишлера—Напиральского была
использована для синтеза замещенного бензофенонз, возможного
промежуточного продукта в синтезе пикроподофилина*. Замещенный
3,4-дигидроизохинолин (I), полученный по обычному методу
Бишлера—Напирал ьского, затем обрабатывали диметилсульфа-
том и избытком едкого натра (исчерпывающее метилирование по
Гофману); при этом был получен с прекрасным выходом замещен-
35
Бишлер—Н апиральский
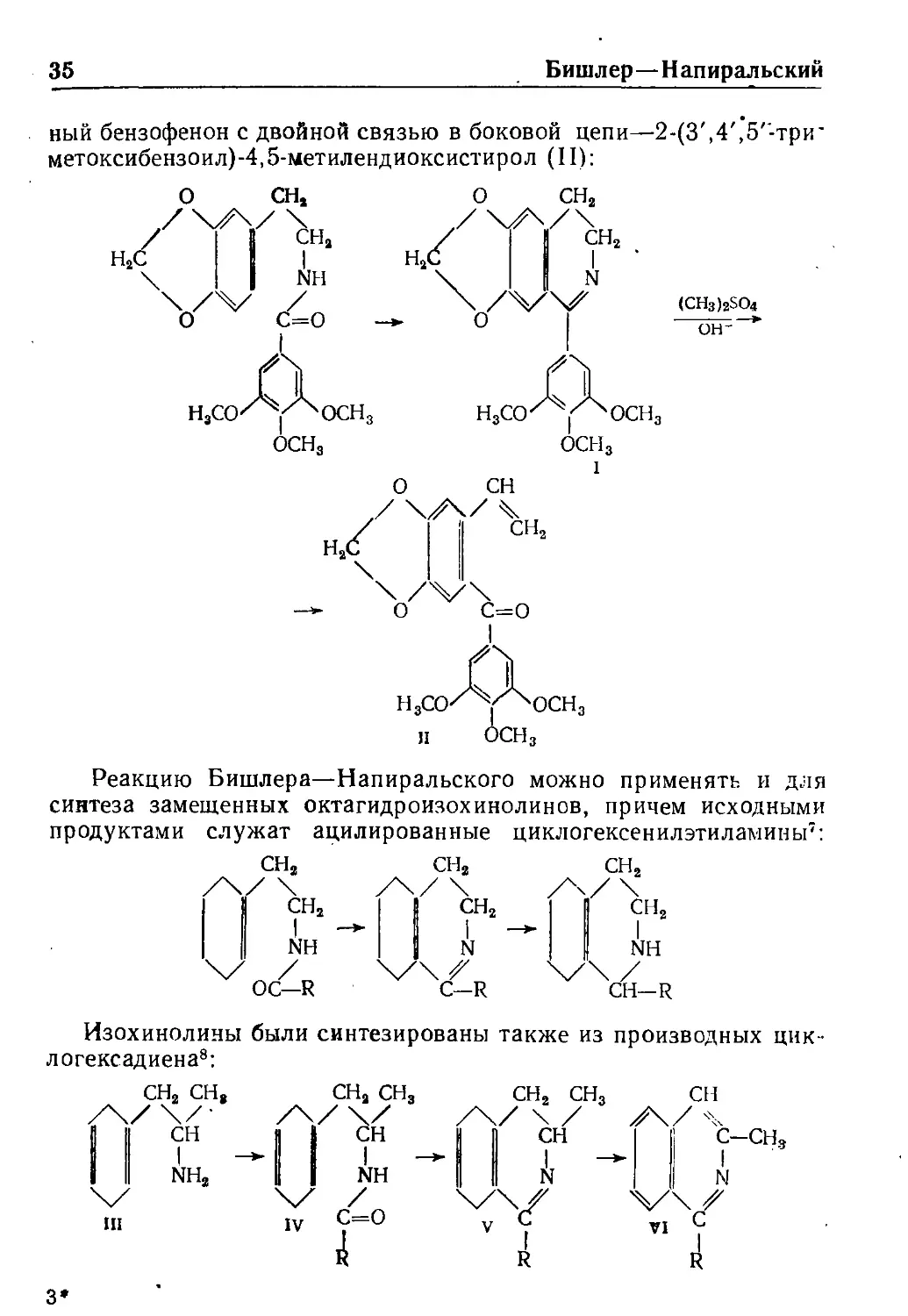
ный бензофенон с двойной связью в боковой цепи—2-(3',4',5'-три
метоксибензоил)-4,5-метилендиоксистирол (П):
Н,ОК у ^ОСНз
оси.
осн.
о с=о
Реакцию Бишлера—Напиральского можно применять и для
синтеза замещенных октагидроизохинолинов, причем исходными
продуктами служат ацилированные циклогексенилэтиламины":
сн,
СН,
С
СН,
СН, | СН, Ц СН^
NH | N I ^Н
ОС—R С—R СН—R
Изохинолины были синтезированы также из производных цнк-
логексадиена*:
СН* СН,
СН, СН,
NH,
m
IV
NH
СН, СИ,
СН
СН
С
С—СН@
N
R
Бишлер—Напиральский 36
Дигидросоединение III, полученное восстановлением по методу
Берча, обрабатывали 3,4-диметоксифенилацетилхлоридом и
образовавшийся ациламин IV циклизовали при помощи хлорокиси
фосфора в бензоле. Соединение V для превращения в производное
изохинолина VI было дегидрировано в две стадии: а) под дей-
"ствием Pd/C в ксилоле и б) в присутствии Pd/C и этилового эфира
комичной кислоты.
Реакция Бишлера—Напиральского была использована также
для синтеза производных пиридина. Так, циклизацией N-ацилиро-
ванного 5-фенилпентен-4-ил-1-амина были получены 2-замещенные
3-бензилиден-3,6-дигидропиридина*:
R/V
j. A. Bischler, В. Napiralski, Ber., 26, 1903 (1893).
2. О реакции Бишлера—Напиральского см. В. Уэли, Т. Г о в и н д а-
ч а р и, Органические реакции, сб. 6, Издатинлит, 1953, стр. 98.
3. См. также В. Г е н с л е р, Гетероциклические соединения, сб. 4,
Издатинлит, 1955, стр. 264.
4. О применении в реакции Бишлера—Напиральского смеси пятиокнси
фосфора с пиридином и песком см. N. I t о h, S. Sugasawa, Tetrahedron,
%, 45 (1957).
5. A. Pictet, A. Gams, Ber., 42, 2943 (1909).
6. W. G e n s I e r, C. S a m о u r, J. Am. Chem, Soc, 73, 5555 (1951).
7.0. Schnider, J. Hellerbach, Helv. chim. acla, 34, 2218
(1951).
8. R. Tachikawa, Tetrahedron, 7, 118 (1959).
9. T. Fuyisawa, S. Sugasawa, Tetrahedron, 7, 135 (1959).
37 Блан
Густав Луи Блан (Gustave Louis Blanc, 1872—1927) родился в Париже.
Учился в школе физики и промышленной химии. Работал ассистентом на
факультете наук Сорбонны, где в 1899 г. защитил докторскую диссертацию.
С 1906 г. возглавлял технические лаборатории управления военного интекдан
ства в Париже. Занимался химией терпенов, а также химией алифатических
и гидроароматических соединений. Исследуя некоторые производные камфоры,
получил взаимодействием сложного эфира с натрием и спиртом
соответствующий спирт и обнаружил, что аналогичным способом восстанавливаются эфиры
алифатических кислот. Совместно с Л. Буво разработал обший метод
восстановления сложных зфиров натрием и спиртом. Открыл общий способ
хлорметилирования ароматических углеводородов, который был использован для
приготовления бензилового спирта и толуола. Синтезировал бензальдегид
окислением бензилового спирта бихроматом. ^
ХЛОРМЕТИЛИРОВАНИЕ ПО БЛАНУ
Процесс хлорметилирования ароматических соединений путем
обработки их формальдегидом и хлористым водородом в
присутствии хлористого пинка часто называют реакцией Блана^ %
Примером хлорметилирования по Блану является
образование хлористого бензила из бензола (выход 79%):
Реакцию обычно проводят при 60°, пропуская сухой хлористый
водород через смесь бензола, параформальдегида и хлористого
цинка до прекращения абсорбции газа.
Другой иллюстрацией хлорметилирования может служить
получение из нафталина 1-хлорметилнафталина^ (выход 77%):,
СН*С1
I
CHoO; HC1
*.
Для проведения реакции хлорметилирования в качестве
катализаторов применяют также серную или фосфорную кислоту и
Блан 38
хлористый алюминий. Наиболее важной побочной реакцией,
сопровождающей хлорметилирование, является образование
производных диарилметана. Однако этот процесс и другие побочные
реакции можно подавить при помощи хлористого мышьяка или
трехокиси мышьяка*. Этим методом t-хлорметилнафталин
получается с выходом до 90% (считая на нафталин, вступивший в
реакцию).
Хлорметилирование метилового эфира а-фуранкарбоновой
кислоты (I) приводит к получению метилового эфира а'-хлорме-
тил-сс фуранкарбоновой кислоты* (II) с выходом 83%:
_ CHgO; HC1
" *
Н
Примером введения двух хлорметильных групп является
синтез 3,5-ди-(хлорметил)-2-оксиацетофенона (IIП п,и кипячении
с обратным холодильником смеси 2-оксиацетсфенона (IV) и
формальдегида в присутствии хлористого водорода*. При 25—30°
образуется только монохлорметилированное производное—5-хлор-
метил-2-оксиацетофенон (V):
иен,
III IV
На основании исследования скорости реакции хлорметилиро-
вания мезитилена по методу Блана был предложен следующий
механизм этого процесса?:
CH.O-MI+ ^ CHgOH
АгН 4- СН.ОН -^ АгСН^ОН ф Н+
ArCHgOH ф НО ^: АгСНдС! ф Н@О
Стадией, определяющей скорость реакции, является электро-
фильная атака группой СН%ОН. Ход реакции зависит от
характера заместителей в ароматическом кольце; электронодонорные
заместители облегчают реакцию, а электроноакцепторные
оказывают обратное действие.
39 Блан
Реакция хлорметилирования является довольно общей и ее
препаративное значение весьма велико, особенно если учесть
возможность легкого превращения группы —СН*С1 в другие
группы, например в группы —CHg, —CH%CN, —СНО, —CH^NHg,
—СН^ОН и т. д.
ЛЯГЕРЛГУРЛ
1. G. Blanc, Bull. Soc. chim. Paris, 33, 313 (1923).
2. P. Ф ь ю з о н, К. Ma к-К и в е р, Органические реакции, сб. 1, Издат-
инлит, 1948, стр. 84.
3. О. Г р у м м и т, А. Бак, Синтезы органических препаратов, сб. 3,
Издатинлит, 1952, стр. 481.
4. F. С о с к е г i I 1 е, пат. США 2541408, 1951 г.; С. А., 45, 6662 (1951).
5. R. An drisano, Ann. chim. (Rome), 40, 30 (1950); С. А., 45, 7563 (1951).
6. R. T г a v e, Gazz, chim. ital., 80, 502 (1950); C. A., 45, 7047 (1951).
7. Y. Ogata, M. Okane, J.Am. Chem. Soc, 78, 5423(1956).
Браун 40
Юлиус Браун (Julius Braun, 1875—1940) родился в Варшаве, учился в
Мюнхене и Геттингене. В 1909—1918 гг. адъюнкт-профессор в Бреслау, с 1921 г.
профессор химии во Франкфуртском университете. Ю. Браун опубликовал
большое число трудов в различных областях органической химии. Многие его
работы посвящены изучению азотсодержащих гетероциклических соединений.
РЕАКЦИЯ БРАУНА
Образование галоидпроизводного и бензонитрила при
нагревании N-замещенного бензамида с пятибромистым или пяти-
хлористым фосфором называется реакцией Брауна:
С,Н,—СО—NH—R + РВгд -^ QH^-CN Ч- RBr + POBr^+HBr
Реакцию обычно проводят, нагревая смесь замещенного
бензамида и пятигалоидного фосфора; конечные продукты выделяют
фракционированной перегонкой реакционной смеси.
Реакция Брауна применима также для превращения
алифатических аминов в соответствующие галоидпроизводные, так как
при этом (в противоположность дезаминированию азотистой
кислотой) не происходит перегруппировки в радикале R.
Превращение аминов в галоидпроизводные идет по схеме:
С*НэСОС1 Р%5
R—NH. -» R—NH—СО—СдН, » RX + QH,--CN
Из циклических аминов образуются соответствующие дигалоид-
производные. Например, 1,5-дибромпентан может быть получен
из бензоилпиперидинаЧ
/X CgHrCOCl /X
I I ^ I I
NH N
со—
Леонард и Номменсен^ в работе, посвященной исследованию
механизма реакции Брауна, показали, что алкильные группы,
связанные с атомом углерода, находящимся в %-положении по от-
41 Браун
ношению к атому азота, затрудняют течение реакции. Поэтому
из М-бензг*ил-2,2,6,6-тетраметнлпиридина (I) не образуются ди-
галоидзамещенные диметилгептана. Однако при
взаимодействии М-бензоил-2,5-ди-(трифенилметил)-2,5-дигидропиррола (II) с
пятибромистым фосфором получается с выходом 15%
соединение III—1,4-Ди-(трифенилметил)-1,2,3,4-тетрабромбутан, или что
то же—2,3,4,5-тетрабром-1,1,1,6,6,6-гексафенилгексан*:
i и
Нв)зС—СНВг—СНВг—СНВг—
HI
1. J. В г a u n, Бег., 37, 3210 (1904).
2. N. Leonard, E. N о m m e n s e n, J. Am. Chem. Soc., 71, 2808 (194
3. J. С о n a n t, В. Chow, J. Am. Chem. Soc., 55, 3475 (1933).
Браун
42
РАСЩЕПЛЕНИЕ АМИНОВ ПО БРАУНУ
Разрыв связи С—N в третичных аминах при взаимодействии
их с бромистым цианом известен под названием расщепления
аминов по Брауну^. При этом бром присоединяется к атому
углерода, а CN-группа—к атому азота. Вероятно, реакция идет через
стадию образования промежуточного комплекса I,
распадающегося затем на двухзамещенный цианамид и бромистый алкил:
R
R—N -f BrCN
R
R
R
i
R
Br
—CN+ RBr
Для несимметричных третичных аминов состав конечных
продуктов реакции определяется природой радикалов исходного
амина.
Эльдерфильд^'* изучил основные направления размыкания
кольца в несимметрично замещенных гетероциклических
системах. При взаимодействии 1-(м-бутил)-2-метилпирролидина* с
бромистым цианом в бензоле образуются два продукта—первичный
(II) и вторичный (III) галогениды, причем преимущественно
получается первичный бромид (II):
сн,—сн
/
СНз
СН,—СН—N
СН,—СНдВг
II
(выход 70%)
СН,—СНВг
in
(выход 26%)
Если же синтез проводится с 1-фенил-2-метилпирролидином*,
то соотношение первичного (IV) и вторичного (V) бромидов об-
43 Браун
ратное—основным продуктом реакции расщепления аминов
является вторичный бромид (V):
СН„ СН„ СН,
СН.-СН СН,—СН—М<Г СН,-СНВг
СНд—СНа СН,—СНдВг СН.^—CHg—N *
IV v
(выход 20%) (выход 80%)
Присутствие фенильной группы, понижающей основность
амина, определяет главное направление раскрытия кольца
пиррол идина*.
Другой метод расщепления третичных аминов до карбинол
аминов путем спонтанного разложения четвертичных солей ацилокси-
аммония получил наименование
1. J. Braun, Ber., 33, 1438 (1900).
2. R. Е 1 d е г f i e I d, H. H agem an, J. Org. Chem., 14, 605 (1949).
3. R. El derfiel d, M. Green, J. Org. Chem., 17, 431 (1952).
4. О реакции Брауна см. X. Хейгеман, Органические реакции, сб. 7,
Издатинлит, 1956, стр. 260.
5. Е. Wenkert, Experienta, 10, 346 (1954).
Буво—Блан 44
Лун Буво* (Louis Bouveault, 1864—1909) родился в г. Невере (Франция).
Всю жизнь занимался педагогической и научной работой. В 1890 г. защитил
докторскую диссертацию. Работал на медицинских факультетах
университетов Лиона, Лилля, Нанси и Парижа. С 1907 г. президент Французского
химического общества. Опубликовал очень много работ. Основные труды Буво
относятся к химии терпенов.
ВОССТАНОВЛЕНИЕ ПО БУВО—БЛАНУ
Восстановление сложных эфиров в соответствующие спирты
при действии металлического натрия в этиловом спирте было
впервые предложено Буво и Бланом в 1903 г.^.
Спиртовый раствор сложного эфира кипятят с обратным
холодильником в присутствии избытка натрия:
R—CO—OR' —--» R—СНд—ОНф R'
Для повышения температуры реакции в некоторых случаях
применяют бутиловый спирт.
В 1947 г. Хенсли* разработал улучшенную методику
восстановления сложных эфиров при помощи металлического натрия. В
соответствии с предложенным им механизмом реакции*, для
восстановления необходимы теоретические (эквимолекулярные)
количества металлического натрия и восстанавливающего спирта.
Смесь сложного эфира и спирта быстро добавляют при
перемешивании к расплавленному металлическому натрию в отсутствие
растворителя или в среде таких инертных растворителей, как
толуол или ксилол.
Дарзан* предложил новый общий метод получения спиртов
восстановлением кислот, сложных эфиров, кетонов или
альдегидов. Восстановителем служит не натрий, как при восстановлении
по Буво—Блану, а гидрид натрия.
Очень удобно для восстановления сложных эфиров до
соответствующих спиртов применять в качестве восстановителя ли-
тийалюминийгидрид^.
* О Блане см. стр. 37.
45 Буво— Блан
Каталитической гидрогенизации сложных эфиров до спиртов
над медно-хромовым или скелетноникелевым катализатором
посвящен обзор Адкинса*.
1. О реакции Буво—Блана см. V. Н а п s 1 е у, Ind. Eng. Cliem., 39, 55 (1947)'
2. L. Bouveault, G. Bl an с, С. г., 136. 1676 (1903); 137, 60, 328 (1903);
Bull. Soc. chim. Paris, 31, 666, 1203 (1904).
3. См. также L. P a I f г а у, Р. A ngl aret, С. г., 223, 860 (1946).
4. G. D а г z e n s, С. г., 224, 570 (1947).
5. О восстановлении литийалюминийгидридом см. В. Браун, Органические
реакции, сб. 6, Издатинлит, 1953, стр. 409; [Б. Марбур, М.
Михайлович, Алюмогидрид лития и его применение в органической химии,
Издатинлит, 1957 г.—До/z. /#&].
6. Г. А д к и н с, Органические реакции, сб. 8, Издатинлит, 1956, стр. 7.
Буво 46
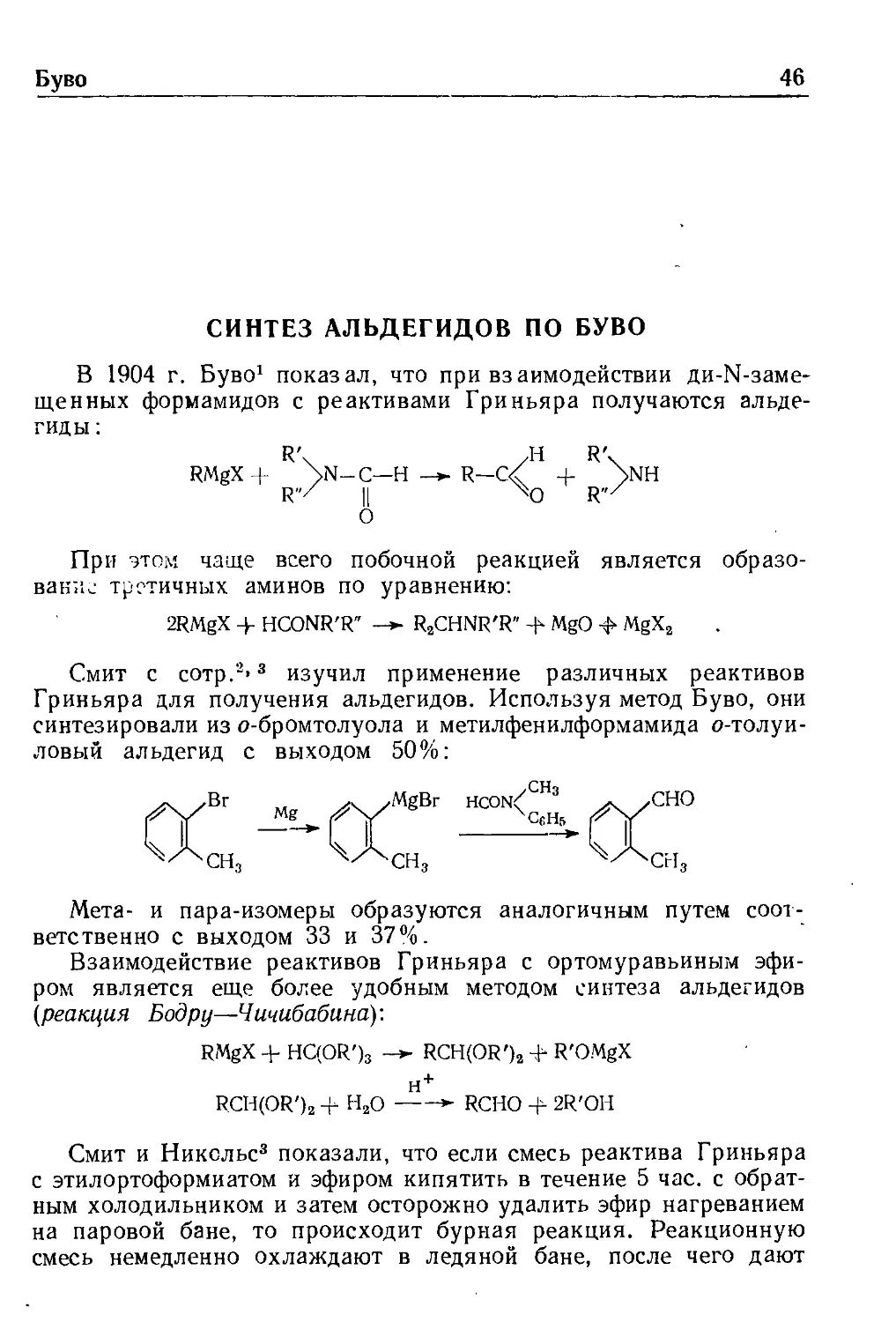
СИНТЕЗ АЛЬДЕГИДОВ ПО БУВО
В 1904 г. Буво* показал, что при взаимодействии ди-М-заме-
щенных формамидов с реактивами Гриньяра получаются
альдегиды:
RMgX + \м_С—H-^R—С/ +
О
При этом чаще всего побочной реакцией является
образовании третичных аминов по уравнению:
2RMgX 4- HCONR'R" -» R.CHNR'R' -h MgO ф MgX,
Смит с сотр.2' ^ изучил применение различных реактивов
Гриньяра для получения альдегидов. Используя метод Буво, они
синтезировали из о-бромтолуола и метилфенилформамида о-толуи-
ловый альдегид с выходом 50%:
сн,
Мета- и пара-изомеры образуются аналогичным путем
соответственно с выходом 33 и 37%.
Взаимодействие реактивов Гриньяра с ортомуравьиным
эфиром является еще более удобным методом синтеза альдегидов
RMgX + HQOR'); -^ RCH(OR')* ф R'OMgX
.+ H,0 ^ RCHO -h 2R'OH
Смит и Никольс^ показали, что если смесь реактива Гриньяра
с этил ортоформи атом и эфиром кипятить в течение 5 час. с
обратным холодильником и затем осторожно удалить эфир нагреванием
на паровой бане, то происходит бурная реакция. Реакционную
смесь немедленно охлаждают в ледяной бане, после чего дают
47 Буво
реакции закончиться. Этим методом авторы получили из о- и
л-бромтолуолов о- и я-толуиловые альдегиды соответственно с
выходом 74 и 73%, т. е. со значительно большими выходами, чем
по методу Буво.
Другим примером использования этилортоформиата является
получение капронового альдегида из амилмагнийбромида* (выход
45%):
QH^CHO
H^
1. L. Bouveault, Bull. Soc. chim. Paris, 31, 1306, 1322 (1904).
2. L. S m i t h, M. В а у 1 i s s, J. Org. Chem., 6, 437 (1941).
3 L Smith, J. Nichols, J. Org. Chem., 6, 489 (1941).
Б а х м а н, Синтезы органических препаратов, сб. 2, Издатинлит, 1949>
стр 295
2.
3. L.
4. Д.
стр. 295.
Бухерер 48
Ганс Теодор Бухерер (Hans Theodor Bucherer, 1869—1949) родился в
г. Эренфельде (Германия). Учился в Мюнхене и Карлсруе, а также у И. Висли-
ценуса в Лейпциге. В 1893 г. Г. Бухереру была присуждена степень доктора
наук. С 1894 по 1900 г. он работал на одном из предприятий Б аденской содо-
анилиновой компании в Людвигсхафене; затем доцент Мюнхенской высшей
технической школы, а через несколько лет—директор химического завода
в Берлине. С 1926 г. профессор химической технологии в Мюнхенской высшей
технической школе. Основные работы Г. Бухерера посвящены изучению
ароматических диазосоединении и их применению в производстве красителей.
РЕАКЦИЯ БУХЕРЕРА
Замещение аминогруппы на гидроксил с помощью водных
растворов сульфитов или бисульфитов обычно известно как
реакция Бухерера*. Хотя эта реакция была описана ранее (Лепти,
1903), ее общий характер и обратимость впервые показаны* By-
херером в 1904 г. Обратная реакция (замена гидроксильной
группы на аминогруппу) протекает при нагревании с водным
аммиаком и сульфитом аммония:
Примером реакции с участием а-замещенных производных
нафталина является получение 1-нафтол-4-сульфокислоты из наф-
тионовой кислоты:
ОН
Реакция Бухерера широко применяется в ряду производных
нафталина и имеет большое значение в синтезе промежуточных
продуктов и красителей. В ряду производных бензола, кроме
получения резорцина и флороглюцина, реакция обычно не при-
49
Бухерер
меняется. С ее помощью из 6- и 8-оксихинолинов получают
соответствующие аминохинолины*.
Как полагают в настоящее время, реакция Бухерера протекает
через стадию образования бисульфитного соединения (I), которое,
вероятно, получается в результате присоединения молекулы
бисульфита к нафтолу в кето-форме:
ОН
н н
НзО
SO3NH4
Продукт присоединения (1), реагируя затем с аммиаком,
превращается последовательно в аминопроизводное (II), имин (III)
и, наконец, в (З-нафтиламин (IV):
н н
н н
Коудрей и Хиншельвуд* подтвердили образование
промежуточных продуктов такого типа. Одновременно они показали, что
стадия образования аддукта определяет скорость всей реакции.
Дано также объяснение образования динафтиламинов как
побочных продуктов реакции превращения нафтиламинов в
нафтол ы.
Доказано*, что продукт присоединения бисульфита к 4-заме-
щенному а-нафтолу является 4-оксотетрагидронафталин-2-суль-
фокислотой;
4 А. Серрей
Бухерер50
1. О реакции Бухерера см. Н. Дрейк, Органические реакции, сб. 1, Иэдат-
инлит, 1949, стр. 133.
2. Н. Bucherer, J. prakt. Chem., 69, 49 (1904).
3. Н. Н. Ворожцов, И. М. Коган, Бег., 65, 142 (1932).
4. W. Cowdrey, С. Hinshelwood, J. Chem. Soc, 1946, 1036;
W. Cowdrey, J. Chem. Soc, 1946, 1044, 1046.
5. A. R e i с h e, H. Seeb ot h, Angew. Chem., 70, 312 (1958).
51
Бухерер
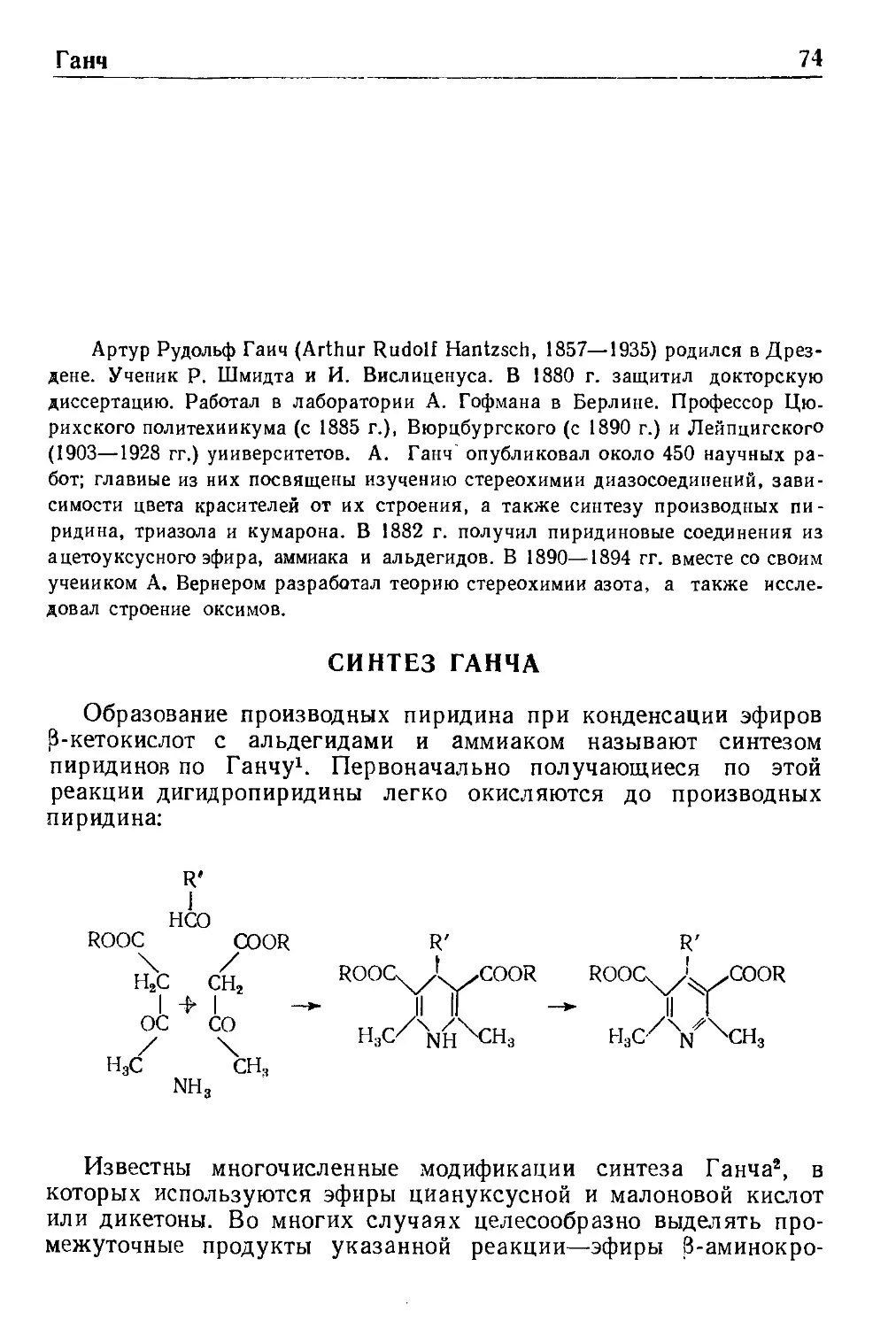
СИНТЕЗ ГИДАНТОИНОВ ПО БУХЕРЕРУ
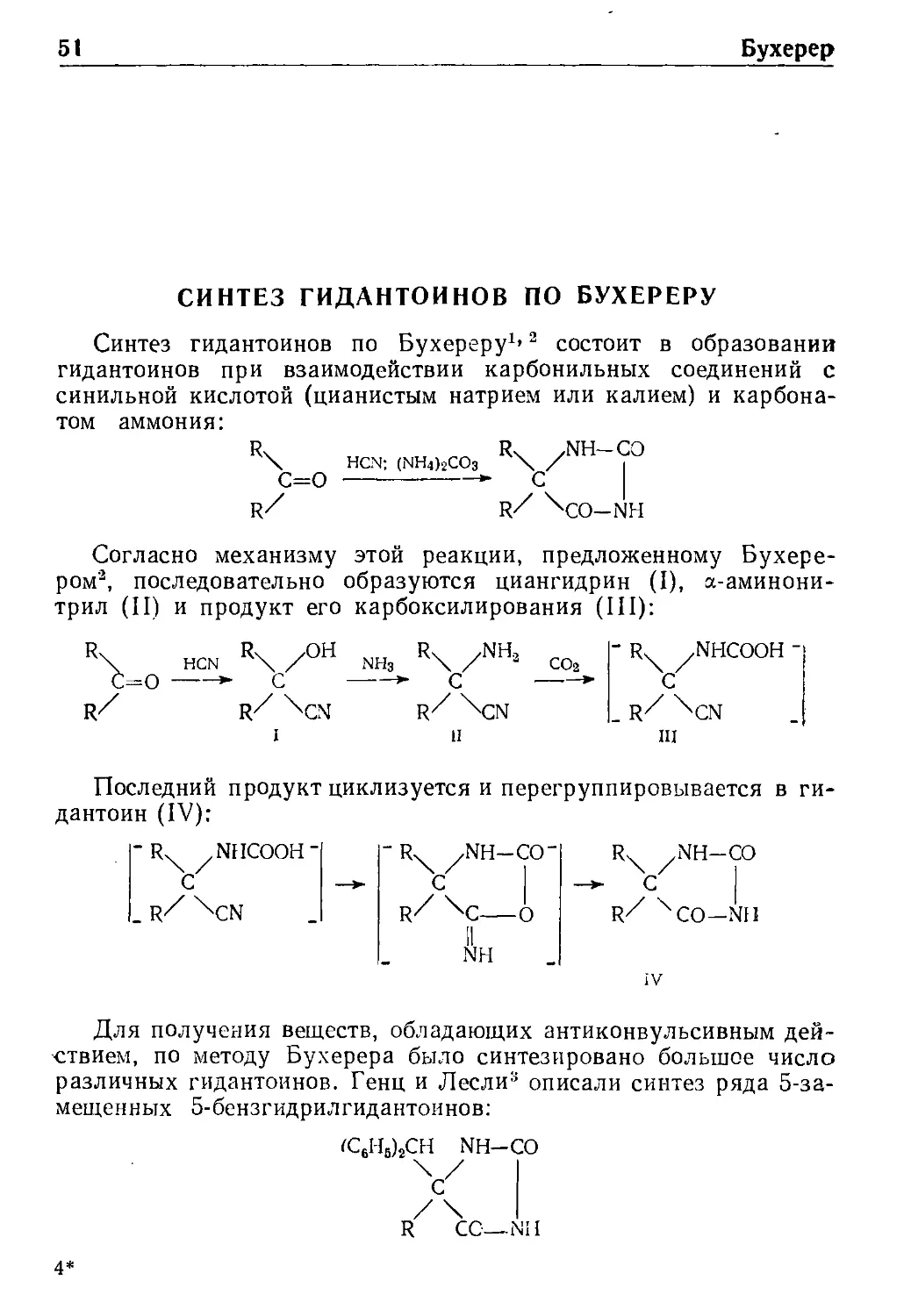
Синтез гидантоинов по Бухереру*' ^ состоит в образовании
гидантоинов при взаимодействии карбонильных соединений с
синильной кислотой (цианистым натрием или калием) и
карбонатом аммония:
Согласно механизму этой реакции, предложенному Бухере
ром\ последовательно образуются циангидрин (I), %-аминони
трил (II) и продукт его карбоксилирования (III):
с=о
HCN
R/
NH,
С
II
-R, yNHCOOH-
с
III
Последний продукт циклизуется и перегруппировывается в ги
дантоин (IV);
-R
_R
С
/\
R/\C
NH-CO"
-o
NH
R
V
NH-CO
Для получения веществ, обладающих антиконвульсивным
действием, по методу Бухерера было синтезировано большое число
различных гидантоинов. Генц и Лесли% описали синтез ряда 5-за-
мещенных 5-бензгидрилгидантоинов:
NH-CO
R CO—NH
Бухерер 52
Бухерер пытался распространить данный метод на получение
дитиогидантоинов, однако осуществить это ему не удалось.
Только в 1947 г. Каррингтон* показал, что 5,5-дизамещенные 2,4-ди-
тиогидантоины могут быть синтезированы кипячением смеси ке-
тона, сероуглерода и цианистого аммония в водном растворе
метилового спирта: ^
'
R/ ' R/ \]S—NH
В результате обработки '2,4-Дитиогидантоинов аммиаком или
первичными аминами получаются соответствующие 4-иминопро-
изводные, которые при кислотном гидролизе превращаются в
5,5-дизамещенные 2-тиогидантоины*:
с
R^/NH-CS
—NHfCHzCHsOH
—NH R/ ^C NH
. y
NCHgCHgOH
NH—CS
—NH
Из пиридинальдегидов получаются 5-(a-, P- и %-пиридил)-
гидантоины^. В случае 2-пиридииальдегида был выделен пр
межуточно образующийся оксазол.
r*'
1. Н. Bucherer, W. Steiner, J. prakt. Chem., [2], 140, 291 (1934).
2. H. Bucherer, V. Lieb, J. prakt. Chem., [2], 141, 5 (1934).
3. H. Henze, W. Leslie, J. Org. Chem., 16, 901 (1950); см. также
H. Henze, W. Craig, J. Org. Chem., 10, 2 (1945).
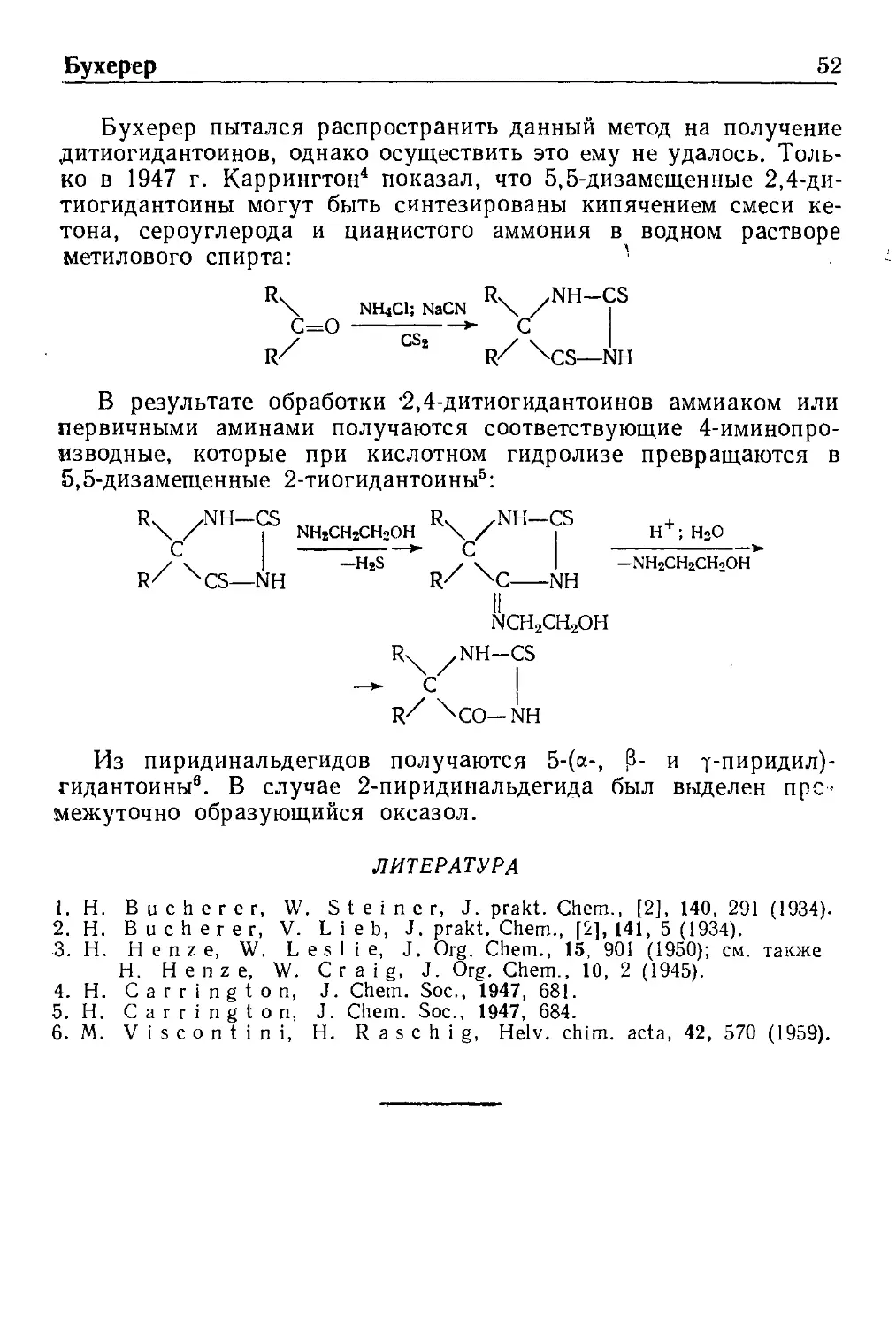
4. H. С a rr i ng t on, J. Chem. Soc., 1947, 681.
5. H. Carrington, J. Chem. Soc., 1947, 684.
6. M. V i s con t I n i, H. R a s с h ig, Helv. chim. act a, 42, 570 (1959).
53 Вагнер—Меервейн
Егор Егорович Вагнер (1849—1903) родился в Казани. В 1874 г. окончил
Казанский университет, ученик А. М. Зайцева. В 1875 г. работал с А. М.
Бутлеровым, с 1876 г.—с Н. А. Меншуткиным. С 1882 г. профессор
Ново-Александрийского сельскохозяйственного института. В 1885 г. защитил магистерскую
диссертацию. С 1886 г.—профессор Варшавского университета,
затем—Варшавского политехнического института. В 1888 г. защитил докторскую
диссертацию. Е. Е. Вагнер много сделал в области исследования реакции окисления
непредельных соединений и открыл общий способ окисления этиленовых
связей перманганатом, получивший в науке наименование «окисление по
Вагнеру», Его работы по изучению строения терпенов являются классическими; в
1895—1896 гг. он установил, в частности, строение основного компонента
скипидара—д-пинена.
ПЕРЕГРУППИРОВКА ВАГНЕРА—МЕЕРВЕЙНА**
- Изомеризация терпеновых углеводородов в процессе реакций
отщепления, присоединения или нуклеофильного замещения,
сопровождаемая перераспределением углерод-углеродных связей
в цикле, носит наименование перегруппировки Вагнера-Меервей-
на; ее называют также камфеновой перегруппировкой первого
рода*.
Указанная перегруппировка аналогична пинаколиновой и ре-
тропинаколиновой перегруппировкам, на что впервые обратил
внимание Вагнер\ Перегруппировка Вагнера—Меервейна имеет
особенно большое значение для понимания взаимных
превращений различных представителей ряда бицикл ических
терпенов.
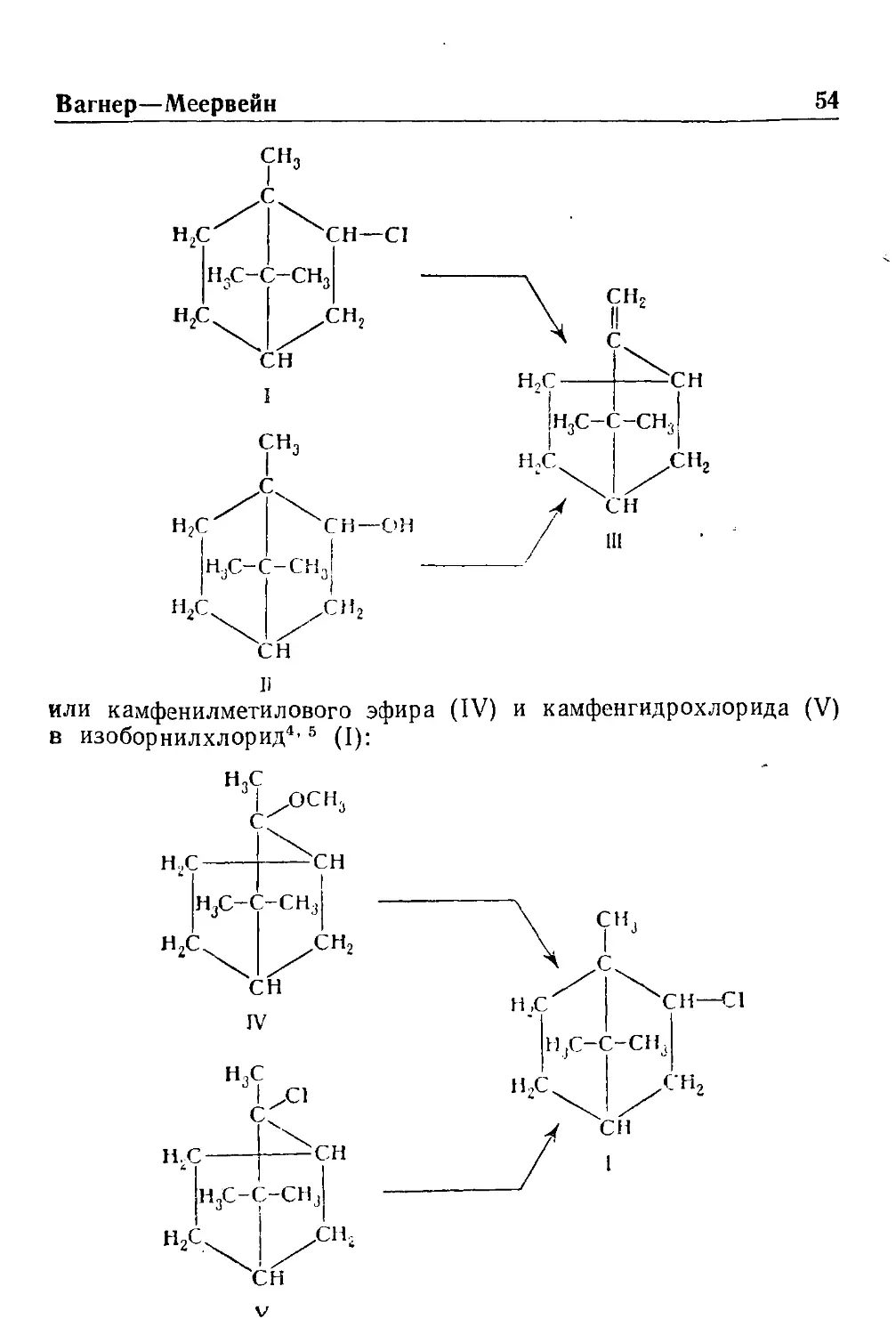
Примерами этой перегруппировки могут служить
превращения изоборнилхлорида (П и борнеола (II) в камфен^' * (III):
* Биография Вагнера и изложение перегруппировки
Вагнера—Меервейна написаны редактором.
** Биографические сведения о Г. Л. Меервейне см. при изложении
конденсации Меервейна, стр 184.
Вагнер—Меервеин
54
сн.
СН—CI
.сн.
сн
г
сн—он
н.с.
сн
III
сн
li
или камфенилметилового эфира (IV) и камфенгидрохлорида (V)
в изоборнилхлорид*' *
с
НС
дэсн.
сн
н^с-с-сн
н,с
сн
IV
с
НС
сн
сн
V
см
н,с
СН—€1
сн
55
Вагнер—Меервейн
Меервейн" считает, что сначала происходит диссоциация на
ионы и вследствие неустойчивости органического катиона
последний претерпевает перегруппировку. Таким образом, на примере
приведенной выше перегруппировки камфенгидрохлорида (V) в
изоборнилхорид (I) механизм реакции может быть изображен
схемой:
с
н,с
сн
Н.С-С-СН,
н,с
сн
V
сн
СН-С1
сн,
г
сн
нг-с-сн.
СИ,
с
сн
С1
.сн,
сн
Меервейн расширил представления о подобных
перегруппировках за пределы ряда бициклических терпенов, показав, например,
что 2,2-диметилциклогексанол (VI) при дегидратации в кислой
среде превращается в смесь изопропилиденциклопентана (VII)
и 1,2-диметилциклогексена? (VIII):
н„с
СН—ОН
(%
НдС СН;
н„с—с
н%с сн.
с
С—СИ,
Н,С
VI
VII
сн,
via
Вагнер—Меервейн 56
1. К. Ингольд, Механизм реакций и строение органических соединений,
Издатинлит, 1959, стр. 390; Т. И. Темников а, Курс теоретических
основ органической химии, Госхимиздат, 1959, стр. 521.
Е В ЖРХО, 31,680(1899).
В. Б р и кн ер, Вег., 32, 2302 (1899).
К. van E m s t e r, Вег., 53, 1815 (1920).
L. Gerard, Ann., 435, 174 (1923); Н. Meerwein,
, A. S ег i n I, J. V6 г s t e г, Ann., 453, 16 (1927).
К. van Emster, Вег., 55, 2500 (1922).
W. U nkel, Ann., 376, 152 (1910); Н. Meerwein,
(1913); 405, 129 (1914); 417, 255 (1918).
2.
3.
4.
5.
6.
7.
Е.
Е.
Н.
Н.
н.
н.
Е.
Е.
М
м
0.
м
м
е
е
е
е
Ann
В
В
е
е
Н
е
е
а г
а г
г w
г w
a m
г w
г w
396.
мер,
н е р
е i n,
е i n,
m e 1
е i n,
е i n,
, 200
57 Баллах:
Отто Баллах (Otto Wallach, 1847—1931) родился в Кенигсберге
(Германия). Изучал химию в Геттингенском университете у Ф. Велера и в Берлине
у А. Гофмана. В 1869 г. защитил докторскую диссертацию. С 1870 г. ассистент
А. Кекуле в Боннском университете, затем преподаватель фармацевтической
химии. В 1889—1915 гг. директор Химического института в Геттингене.
С 1879 г. начал заниматься химией терпенов. Систематическое изучение и
классификация терпенов, проведенные О. В аллахом, внесли ясность в эту
запутанную область химии. Его книга «Терпены и камфора» явилась основой для
последующих работ по химии терпенов. О. Баллах изучал также азо- и диазосо-
ед и нения. В 1910 г. ему была присуждена Нобелевская премия.
РЕАКЦИЯ ВАЛЛАХА
Восстановительное аминирование альдегидов и кетонов
первичными или вторичными аминами и муравьиной кислотой носит
название реакции Валлаха*. Эта реакция в значительной степени
аналогична реакции Лейкарта* и называется иногда реакцией
Лейкарта—В аллаха. Ее можно выразить следующим общим
уравнением:
С=О -h HN + НСООН -» СН- N ф Н%0 + СО
Недавно Штапль и Вагнер^ опубликовали работу,
посвященную реакции Валлаха. Авторы показали, что формильные
производные аминов обычно не вступают в эту реакцию и при их
образовании течение процесса затрудняется. Поэтому избыток
муравьиной кислоты нежелателен. Можно предположить, что
первичный продукт конденсации получается при взаимодействии
карбонильных соединений и аминов. Этот продукт конденсации
(алканоламин, алкилидендиамин или основание Шиффа)
восстанавливается затем муравьиной кислотой. Например, при
Изложение реакции Лейкарта см. стр. 172.
Баллах 58
действии муравьиной кислоты на 1, Г-бензилидендипиперидин
получается бензил пиперидин с количественным выходом:
\ нсоон
СН-С.Н, >-
Бенневилль и Макартней* исследовали поведение
алифатических альдегидов в реакции В аллаха. Им удалось получить ал-
килированные амины с хорошим выходом как при
непосредственном взаимодействии аминов, альдегидов и муравьиной
кислоты, так и при восстановлении муравьиной кислотой енаминов,
полученных из альдегидов и аминов.
Примером последней реакции является конденсация масляного
альдегида с морфолином. Енамин, образующийся издиимина,
можно выделить перегонкой и восстановить действием муравьиной
кислоты:
5—50» Г
2HNR, —^ C,H,
нсоон
-» CzH,—CH=CHNR, Q3ga
На основании этой работы можно предположить, что енамины
являются промежуточными продуктами в реакции Валлаха.
1. О. W а I I а с h, Ann., 343, 54 (1905).
2. Е. Staple, E. Wagner, J. Org. Chem., 14, 559 (1949).
3. P. В e n n e v i 1 1 e, J. Macartney, J. Am. Chem. Soc, 72, 3073
(1950).
59 Вильгеродт
Конрад Вильгеродт (Conrad Willgerodt, 1841—1930) родился в г. Харлин-
героде (Германия). В 1863 г. окончил учительский колледж и в течение
некоторого времени вел преподавательскую работу. Затем поступил в Врауншвейг-
ский технологический институт, где изучал естественные науки и зоологию.
В 1869 г. начал работать химиком в фирме по производству красителей в г. Эл-
берфельде. В дальнейшем учился во Оренбургском университете, где под
руководством Клауса защитил докторскую диссертацию, которая была посвящена
изучению ализарина и оксиантрахинона. С 1881 г. адъюнкт-профессор, с 1895 г.
профессор того же университета. Его преподавательская деятельность
продолжалась 37 лет. Работы Вильгеродта посвящены главным образом химии
ароматических соединений. Занимаясь галоидированием ароматических
углеводородов в присутствии металлов, он в 1886 г. синтезировал дихлориодбензол—
первое органическое вещество, содержащее трехвалентный иод, и из него
получил иодозобензол. Опубликовал книгу по химии трехвалентного иода.
РЕАКЦИЯ ВИЛЬГЕРОДТА
Превращение кетонов в амиды с тем же числом атомов углерода
действием водных растворов полисульфида аммония при
нагревании обычно называют реакцией ВильгеродтаЧ В конечном итоге
реакция сводится к восстановлению карбонильной группы и
окислению концевой метильной группы.
Примером реакции может служить образование амида фенил-
уксусной кислоты из ацетофенона:
CH,CONH,
Наряду с основным продуктом (амидом) всегда получается
некоторое количество аммонийной соли фенилуксусной кислоты.
В реакцию могут вступать высшие жирноароматические кето-
ны, олефины и ацетилены, а также кетоны и альдегиды жирного
ряда-:
АгСН=СНа -* g
АгС=СН -* ArCH.,CONH,
Вильгеродт 60
Равнозначность ArCOCHgCHg и ArCH=CHCHg в реакции
Вильгеродта иллюстрируется на примере получения 6-анизил-
пропионовой кислоты* из анетола (4-пропенил анизола):
сн=сн—сн, ,. .сн,—сн.—соон
Киндлер* установил, что вместо водного раствора
полисульфида аммония можно использовать серу и сухие амины; при этом
в качестве главного продукта реакции получаются тиоамиды.
Например, при взаимодействии ацетофенона с морфолином и
серой образуется морфолинтиоамид^:
C°™'.NC,H,0 /x/"'-|T" "
*v
s L U S
Механизм реакции Вильгеродта был предметом .длительной
дискуссии. В настоящее время считают, что все видоизменения
этой реакции имеют единый механизм, который заключается в
предварительном образовании промежуточного продукта,
содержащего в боковой цепи ненасыщенную углерод-углеродную связь»
перемещающуюся к концевой цетильной группе®:
АгСОСНдСНз ^± АгСН=СНСНз ^ АгСНдСН=СН,
Кинг и Мак-Миллан? предположили, что механизм
перемещения двойной связи состоит в последовательном присоединении
и отщеплении сероводорода, в результате чего образуется
первичный тиол, содержащий меркаптогруппу у концевого атома
углерода. Эти же авторы* изучили также механизм превращения
меркаптанов в производные карбоновых кислот:
RCOCHg -» RCH3CH2SH -» RCH,C
Механизм реакции Вильгеродта исследован^ на примере
ацетофенона, имеющего в карбонильной группе радиоактивный
атом С**. Согласно полученным данным, основная реакция, как
и предполагалось ранее, не сопровождается перегруппировкой
углеродного скелета. Превращением продукта реакции в бензил-
амин по Гофману установлено, что радиоактивность сохраняется:
^ ,т ,.,. ^ (MWaSn NaClO
С,Н,—С"О-С
61 Вильгеродт
Реакция Вильгеродта была распространена и на
гетероциклические соединения. Так, из а-пиколина и хинальдина
взаимодействием (\серой в присутствии морфолина получены тиоморфоли-
ды, гидролиз которых приводит к соответствующим карбоновым
кислотам^:

Описано также образование 3-пиразол ил уксусных кислот из
соответствующих 3-ацетильных производных^.
1. С. W i I I g e г о d t, Ber., 20, 2467 (1887).
2. О реакции Вильгеродта см. М. К а р м а к, М. Шпильман,
Органические реакции, сб. 3, Издатинлит, 1951, стр. 88.
3. J. Schm i t t, M. Su quet, Bull. Soc. chim. France, 775 (1956).
4. К. К i n d 1 e r, Arch. d. Pharm., 265, 389 (1927).
5. E. Sch wenk, E. В I о с h, J. Am. Chem. Soc, 64, 3051 (1942).
6. M. С a r m а с k, D. D e Tar, J. Am. Chem. Soc, 68, 2029 (1946).
7. J. King, F. McMillan, J. Ami Chem. Soc, 68, 632(1946).
8. F. McMillan, J. King, J. Am. Chem. Soc, 70, 4143 (1948).
9. W. Daub en, J. Reid, P. Yankwich, M. Calvin, J. Am.
Chem. Soc, 72, 121 (1950).
10. E. Brain, I. Finar, J. Chem. Soc, 1957, 2356.
Вильямсон 62
Александр Вильям Вильямсон (Alexander William Williamson, 1824—
1904) родился в Лондоне. Изучал химию в Гейдельбергском университете у
Л. Гмелина (1840 г.), затем в Гиссене у Ю. Либиха (1844 г.). С 1849 г. профессор
Университетского колледжа в Лондоне. Провел большие исследования в
области теории этерификации, экспериментально доказал правильность
эмпирических формул спиртов и эфиров, предложенных Ш. Жераром (1816—1856)
и О. Лораном (1807—1853), впервые получил ортоэфирьт.
СИНТЕЗ ВИЛЬЯМСОНА
Общий метод получения эфироп взаимодействием алкоголятов
с галоидными алкилами обычно называют синтезом эфиров по
Вильямсону!.
Примером этого синтеза может служить получение этилпропи-
лового эфира из пропилата натрия и йодистого этила*:
CsH,ONa + C2H5J -» QH5OC3H, ф NaJ
Другим примером является синтез 4-хлорбутилметилового
эфира нагреванием в течение 30 час. смеси 1,4-дихлорбутана и
метилата натрия в метаноле*:
С1(СН,).С1 ф NaOCHg -* Q^^^OCHg
При соответствующем подборе реагентов этим путем можно
получать как симметричные, так и смешанные эфиры.
Использование первичных галоидных алкилов и алкоголятов первичных
спиртов обычно не вызывает никаких затруднений в ходе
реакции; для получения же эфиров с вторичными или третичными
радикалами необходимо применять алкоголяты калия.
Использование вторичных и третичных галоидных алкилов обычно не дает
* Образование ряда йодистых алкилов при взаимодействии
соответствующих хлоридов или бромидов с йодистым натрием в ацетоне или метилэтилкетоне
получило название ред/суди ФыякелбштеДядЗ:
СН3СОС2Н5
RC1 + NaJ » RJ Чг NaCl
63 Вильямсон
хороших результатов, так как при взаимодействии с алкоголятами
они превращаются в олефины.
В синтезе Вильямсона применяются также фенолы. В качестве
примера можно привести образование известного стимулятора
роста растений—2,4-Дихлорфеноксиуксусной кислоты:
ONa у^ .ОСНдСООН
Иногда О-алкилирование галоидными алкилами проводят в
ацетоне в присутствии карбоната калия. Например, реакция
о-нитрофенола с %-бутилбромидом в этих условиях приводит к
получению я-бутил-о-нитрофенилового эфира* (выхоц, 80 %):
N0,
Ниман с сотрЛ описал новый метод О-метилирования гидро-
ксилсодержащих соединений. Спиртовые гидрсксилы по этому
методу метилируются диазометаном в присутствии
незначительных количеств фторборной кислоты. Использование первичных
спиртов и вторичных спиртов, не создающих пространственных
затруднений, позволяет получать прекрасные выходы метиловых
эфиров. Так, выход a-холестанилметилового эфира,
синтезированного по этому методу из a-холестанола, достигает 98%. Новая
реакция особенно применима для гидроксилсодержащих
соединений, чувствительных к основаниям. По-видимому, на алкил
непосредственно замещается водород гидроксильной группы,
причем первоначальная конфигурация исходного спирта сохраняется.
1. A. Williamson, J. Chem. Soc, 4, 229 (1852).
2. R. E I d erf i el d, В. P i 11, I. W e m p en, J. Am. Chem. Soc, 72,
1334 (1950).
3. H. F i n k e 1 s t e i n, Ber., 43, 1528 (1910).
4. 4. Алле н, Д. Гейтс, Синтезы органических препаратов, сб. 3, Из-
датинлит, 1952, стр. 134.
5. М. N е е m а п, М. С a s i е г о, J. Roberts, W. Johnson,
Tetrahedron, 6, 36 (1959).
Виттиг 64
Георг Виттиг (Georg Wittig) родился в 1897 г. в Берлине. Защитил
докторскую диссертацию у К. Ауерса в г. Марбурге. С 1932 г. преподавал в
Технологическом институте в Брауншвейге, а через пять лет перешел в университет
во Фрейбурге. В 1949 г. стал профессором и деканом химического факультета
Тюбингенского университета. С 1956 г. по настоящее время занимает такой же
пост в Гейдельберге. Основными областями его исследований являются
стереохимия, а также радикальные и анионотропные реакции. Он награжден медалью
А. Байера. В 1957 г. Сорбонна присудила ему почетную степень доктора наук.
РЕАКЦИЯ ВИТТИГА
Взаимодействие трифенилфосфиналкилиденов с карбонилсо-
держащими соединениями с образованием олефинов (путем
замещения карбонильного кислорода метиленовой группой) называется
реакцией Вит/гига**-.
Эта реакция может быть иллюстрирована следующим
примером:
+ (С,Н,),СО -^ (С,Н,),С=СН, + (С,Н*)зРО
Трифенилфосфин реагирует с бромистым метилом, давая
бромистый трифенилметилфосфоний; действуя на эту соль фенилли-
тием, получают алкилиденфосфин, который при взаимодействии
с бензофеноном превращается в 1,1-дифенилэтилен и окись три-
фенилфосфина. Последнюю можно восстановить для регенерации
трифенилфосфина.
В реакции Виттига используются различные основания: ли-
тийорганические соединения, амид натрия, алкоголят натрия и
карбонат натрия. Недавно предложенная модификация этой
реакции заключается в применении алкоголята лития*, что
упрощает реакцию и дает хорошие выходы олефинов.
Взаимодействие фосфониевых солей с основаниями проводят
в эфире, тетрагидрофуране, спирте и воде, а затем к
полученному раствору алкилиденфосфина прибавляют карбонилсодержащее
65 Виттиг
соединение. Иногда приходится добавлять реагенты в обратном
порядке, особенно в тех случаях, когда альдегиды или кетоны
содержат сложноэфирную группу.
Механизм реакции Виттига изображается следующей схемой*:
"Оg
В реакцию с алкилиденфосфинами вступают соединения
разных классов: алифатические и ароматические альдегиды,
алифатические, алициклические и ароматические кетоны, формамиды,
изоцианаты, кетены и нитрозосоединения. Во всех случаях
образуются соответствующие олефины.
< Недавно предложен новый метод синтеза альдегидов**", первой
стадией которого является получение ониевой соли с помощью
хлорметилового эфира:
—С=СН—
-с=сн—осн, —^ -сн-сно
При взаимодействии флуоренона с избытком триметилфосфин-
w-бутилидена образуется производное циклопропана^.
Реакция Виттига была использована в синтезах многих
природных соединений, в том числе предшественника холестерина—
сквалена, а также ликопина и сложных эфиров биксина. Область
применения этой реакции довольно обширна.
1. G. Wittig, U. Schollkopf, Ber., 87, 1318 (1954).
2. О реакции Виттига см. J. L e v i s a I I e s, Bull. Soc. chim. France, 1021
(1958); U. Schollkopf, Angew. Chem., 71, 260 (1959); S. T г i p-
pett, Advances in Organic Chemistry, v. 1, New York, I960, 83
[Л. А. Яновская, У сп. хим., № 7, 813 (1961)—Дол. /Ы.].
3. Т. Campbell, R. McDonald, J. Org. Chem., 24, 1246 (1959).
4. G. Wittig, Angew. Chem., 68, 505 (1956).
5. S. L e v i n e, J. Am. Chem. Soc., 80, 6150 (1958).
6. G. Wittig, E. Knauss, Angew. Chem., 71, 127(1959).
7. R. Mechoulam, F. Sondheimer, J. Am. Chem. Soc, 80, 4386
(1958).
5 А. Серрей
Воль—Циглер 66
Альфред Воль (Alfred Wohl, 1863—1939) родился в г. Грауденце (Германия).
Учился в Гейдельбергском университете, затем в Берлине, где в 1886 г. получил
степень доктора философии. В дальнейшем доцент Берлинского университета,
с 1904 г. профессор химии Высшей технической школыЪДанциге.^Наибольшую
известность приобрел благодаря разработке метода деградации альдоз.
Карл Циглер (Karl Ziegler) родился в 1898 г. в г. Хельса (Германия). Окон -
чил Марбургский университет; ученик К. Ауэрса. В 1920 г. ему была присуждена
степень доктора философии. Работал сначала ассистентом, затем доцентом в
Марбурге. Профессор Франкфуртского университета, потом заведующий ка-
федрой органической химии Гейдельбергского университета. В 1936 г. был
приглашен читать курс лекций в Чикагском университете. Вернувшись в
Германию, возглавил химический факультет университета в Галле. С 1943 г. ди -
ректор научно-исследовательского института в Мюльгейме. В течение
нескольких лет был президентом немецкого химического общества. В 1959 г.
избран почетным членом Общества химии и промышленности. Синтезировал кэн-
таридин, а также мускон и другие полиметиленовые кетоны. Применил N-бром-
сукцинимид в качестве селективного бромирующего агента. Известен своими
исследованиями в области металлоорганических соединений и свободных
радикалов.
РЕАКЦИЯ ВОЛЯ—ЦИГЛЕРА
Очень удобный метод избирательного бронированна
различных типов органических соединений при помощи N-бромамидов
и особенно N-бромимида янтарной кислоты (N-бромсукцинимида)
был предложен Волем^ и подробно разработан Циглером*. Этот
метод получил название реакции Воля—Циглера*. Бромирова-
ние олефинов идет в %-положение и не затрагивает двойной связи.
Примером такой реакции может служить бромирование
метилового эфира кротоновой кислоты N-бромсукцинимидом,
приводящее к получению метилового эфира у-бромкротонсвой кислоты
(выход 86%): .
СН.—СО СН*—СО
СНзСН=СНСООСН,+ NBr -^ СН.СН=СНСООСНз4-
СН,—СО 1 СН,-СО
\
NH
67 Воль— Циглер
Реакцию обычно проводят в кипящем четыреххлористом угле-
роде, нагревая смесь до полного растворения N-бромсукцинимида.
Другой иллюстрацией бромирования в у-положение является
образование ацетата 7-бромхолестерина (II) из ацетата
холестерина* (I):
СНяООО
При отщеплении бромистого водорода от соединения II
получается ацетат 7-дегидрохолестерина—промежуточный продукт в
синтезе витамина Оз.
Реакция Воля—Циглера была использована также для
расщепления боковой цепи желчных кислот*. Показано, что
эффективным ускорителем этой реакции является интенсивное
облучение.
Бромирование в ядро* при помощи N-бромсукцинимида
успешно протекает в присутствии хлоридов ряда металлов (Aids,
ZnCl* и FeCIg). Таким путем можно, например, получить л-бром-
толуол из толуола:
Шмид и Каррер^ расширили область применения реакции
Воля—Циглера. Они показали, что перекись бензоила служит
прекрасным катализатором бромирования N-бромсукцинимидом.
Применяя этот катализатор, можно бромировать соединения с
третичным атомом углерода, сопряженные диены и боковую цепь
ароматических углеводородов. Так, при бромировании толуола
образуется бромистый бензил с выходом 64%:
(С*Н*СО)аОв
При бромировании (3-фенилпропионовой кислоты с помощью
N-бромсукцинимида получается |3-бром-|3-фенилпропионовая кис-
* Модификацию Мишера реакции Барбье—Виланда см. стр. 20.
Воль—Циглер _^_^____ 68
лота'' (выход 58%), что указывает на образование
промежуточного радикала CgH^CHCHgCOOH:
-* С@Н,СНВгСНдСООН
Поскольку обе метиленовые группы ^-(Ьенилпропионовой
кислоты реакционноспособны, можно сделать вывод, что фенильная
группа оказывает более сильное стабилизирующее влияние на
промежуточный свободный радикал, чем карбоксильная группа.
Всестороннее исследование реакции бромирования циклогек-
сена в ^-положение действием N-бромсукцинимида в четыреххло-
ристом углероде опубликовано Даубеном и Мак-Коем®. В обзоре
рассмотрено влияние на течение этой реакции внешних факторов,
примесей, добавок и продолжительности процесса.
I. A. Woh I, Вег., 52, 51 (1919).
2. К. Z i eg I е г, A. Spaeth, E. Schaaf, W. Schumanm.
Е. Winkelmann, Ann.. 551, 80 (1942).
3. О реакции Воля—Циглера см. С. D j e r a s s i, Chem. Revs., 43, 271 (1948);
[И. В. Мачинская, В. А. Бархаш, Реакции и методы иссле-
дования органических соединений, кн. 9, Госхнмиздат, стр. 287;
L. Home г, Е. Winkelmann, Angew. Chem., 71, 349 (1959).—
Доя. pgd.].
4. A. Bide, Н. Н enh es t, E. Jones, R. P e e v e г s, P. Wik
k i n s о n, J. Chem. Soc, 1783 (1948).
5. H. Sch m I d, Helv. chim. acta, 29. 1144 (1946).
6. H. Sch m i d, P. К а г г e r, Helv. chim. acta, 29, 573 (1946).
7. R. Huang, P. Williams, J. Chem. Soc, 1958, 2637.
8. H. D au ben, L. McCoy, J. Am. Chem. Soc., 81. 4863 (1959).
69 Вюрц
Шарль Адольф Вюрц (Charles Adolphe Wurtz, 1817—1884) родился в
Страсбурге (Франция). В 1843 г. получил степень доктора медицины. Затем работал
под руководством Ю. Либиха в Гиссенском университете. С 1845 г. ассистент
Ж. Дюма (1800—1884) в Парижской медицинской академии, с 1854 г. работает
на медицинском факультете Сорбонны, с 1874 г. профессор органической химии.
Учениками Ш. Вюрца были Я. Вант-Гофф, Д. Крафтс, Р. Фиттиг и Ш. Фридель.
Совместно с Ж. Дюма, Л. Пастером и другими учеными А. Вюрц был одним из
организаторов Французского химического общества и его печатного органа.
В содружестве с Ж. Дюма, своим учителем и другом, внес большой вклад в
развитие атомно-молекулярного учения и в теорию замещения в органической
химии. Ему принадлежит также открытие метиламина (1849 г.) и этилен-
гликоля (1856 г.).
РЕАКЦИЯ ВЮРЦА
Реакцией Вюрца! обычно называют образование высших
предельных углеводородов при взаимодействии низших галоидных
алкилов с металлическим натрием.
Эта реакция является общим способом получения
углеводородов и дает хорошие результаты при использовании первичных
галоидных алкилов:
2RX + 2Na -» R—R -f- 2NaX
Например, две молекулы я-бутилиодида, реагируя с натрием,
образуют молекулу я-октана. Третичные галоидпроизводные в
процессе реакции превращаются в непредельные углеводороды
(олефины).
При применении галоидпроизводных с различными
углеводородными радикалами образуется трудно разделимая смесь
продуктов. Недавно было показано', что из я-бутилбромида и %-гек-
силбромида получается смесь углеводородов, содержащая 27,5%
я-октана, 43,5% N-декана и 29,0% м-додекана:
Na
CH*(CH,)gCH, ф СН,(СН,),СНз
Вюрц 70
Реакцию Вюрца можно рассматривать как свободнорадикаль-
ный процесс и как процесс, идущий через образование
промежуточных металлоорганических соединений^»*. Было установлено*,
что низшие первичные галоидные алкилы в мягких условиях
реагируют с натрием, превращаясь в натрийалкилы, которые
затем взаимодействуют с избытком галоидного алкила, образуя
углеводороды:
RX -f 2№ -» RNa + NaX
RNa ф XR -^ R—R -f NaX
При низких температурах реакцию можно остановить на
первой стадии—стадии образования металлоорганического
соединения, и присутствие его в реакционной смеси доказать карбокси-
лированием в соответствующую кислоту:
со, н+
RNa » RCOONa ^ RCOOH
Летзингерб показал, что при взаимодействии бензилнатрия с
оптически активным 2-бромбутаном образуется оптически
активный 2-бензилбутан:
CHoNa
Вг—С-Н
Выло также установлено, что реакция Вюрца между (—)-2-
хлороктаном и металлическим натрием сопровождается
инверсией конфигурации углеродного атома, связанного с хлором?.
Продуктом реакции является (—)-7,8-диметилтетрадекан:
I* Na I* I*
2C1—C—H —-*-H—С C—H
Эти данные рассматриваются как доказательство протекания
реакции нуклеофильного замещения, заключающейся в
образовании промежуточного натрийалкила. Инверсия и рацемизация
в ходе реакции Вюрца зависят, по-видимому, от природы натрий-
органического соединения и галоидного алкила.
71 Вюрц
Реакция ароматических галоидпроизводных и металлического
натрия, аналогичная реакции Вюрца, получила наименование
pewcqww Фшм/тшгд*. Выходы соответствующих диарилов обычно
очень низкие. *
Если в реакцию с металлическим натрием вступает смесь
жирных и ароматических галоидпроизводных, такой процесс
носит название редкуш* Дюруд—Фи/плшгд. Эта реакция является
весьма удачным методом получения жирноароматических
углеводородов. Примером может служить синтез я-бутилбензола из
я-бутилбромида и бромбензола^:
N. /Х
1. С. W u г t z, Ann., 96, 364 (1855).
2. Н. Davis, W. Gilkerson, H. Hernandez, J. Chem. Educ,
26, 606 (1949).
3. См. Е. Alexander, Principles of Ionic Organic Reactions, New York,
1950, 203.
4. О свободнорадикальном механизме реакции Вюрца, см. М. Tuot, Bull.
Soc. chim. France, 291 (1948).
5. A. Morton, I. Hechenbleikn e.r, J. Am. Chem. Soc, 58, 1697
(1936).
6. R. L e t s i n g er, J. Am. Chem. Soc, 70, 406 (1948).
, 7. E. L eG of f, S. U I г i ch, D. D e n n e y, J. Am. Chem. Soc, 80
622 (1958).
8. P. Рид, Л. Ф о с т е р, А. Рассел, В. С и м р и л, Синтезы
органических препаратов, сб. 3, Иэдатинлит, 1952, стр. 126.
* Рудольф Фиттиг (Rudolf Fittig, 1835—1910) родился в Гамбурге. Окончил
Геттингенский университет, ученик Ф. Велера, затем его ассистент. В 1858 г.
получил степень доктора философии. Преподавал в Тюбингене ком
университете, с 1876 г. профессор Страсбургского университета. Р. Фиттиг был очень
разносторонним химиком-органиком. Он открыл дифенил, фенантрен, кумарон,
диацетил и пинаконовую реакцию. Синтезировал мезитилен и сс-нафтол и по*
казал, что лактоны обычно образуются из j-оксикислот.
Габриэль
Зигмунд Габриэль (Siegmund Gabriel, 1851—1924) родился в Берлине
Учился у А. Гофмана в Берлине и у Р. Бунзена в Гейдельберге, где защитил
докторскую диссертацию. Преподавал неорганическую химию в Берлинском
университете. Впоследствии вице-президент и один из директоров Немецкого
химического общества. Работал в различных областях органической химии, в
частности занимался синтезом азотсодержащих циклических соединений.
Изучая соединения пуринового ряда, получил этиленимин* и оксазолидон из
[3-бромэтиламина и изохинолины из фталилглициновых эфиров*. Синтезировал
алифатические и ароматические кетоны из фталилглицинхлорида, а также хина-
золин из о-нитробензиламина.
СИНТЕЗ ГАБРИЭЛЯ
Образование первичных аминов взаимодействием
органических галоидпроизводных с фталимидом калия и последующим
гидролизом полученных N-замещенных фталимида обычно
называется синтезом Габриэля.
Одним из первых соединений, полученных Габриэлем^* поэтому
методу, был бензил амин. При нагревании фталимида калия с
хлористым бензилом при 170—180° образуется N-бензилфтал-
имид:
СО СО
NR + ClCHa-QH, -^ Г II N-CH.-СдНд ф КС1
СО СО
Гидролиз N-бензилфталимида дымящей соляной кислотой при
200° приводит к получению бензил амина и фталевс кислоты:
CH,-C,H, f HgO —^ Г ||
Метод Габриэля используют при синтезе различных аминосо-
единений, в том числе алифатических моно- и диаминов,
аминокислот и т. д. Метод является специфическим для получени
первичных аминов.
73
Габриэль
В 1926 г. Инг и Манске* предложили модификацию метода
Габриэля, значительно облегчающую расщепление
промежуточных N-замещенных фталимида. По этому варианту N-замещенным
фталимид обрабатывают гидразингидратом, получая С-замс-
щенный фталилгидразид (I), который легко разлагается
разбавленной соляной кислотой с образованием первичного амина и
гидразида фталевой кислоты:
NHR
CO
СО
NR-f NH,—NH,.H,0
NM
HC%
RNH.4-
NH
Фталилгидразид (I) получают при кипячении спиртового
раствора N-замещенного фталимида с эквивалентным количеством
гидразингидрата. Затем спирт отгоняют и остаток нагревают на
паровой бане с 10%-ной соляной кислотой. Гидразид фталевой
кислоты отфильтровывают, а в растворе получают хлоргидрат
первичного амина. Выходы первичных аминов по этому методу
обычно очень высокие.
Позднее было показано, что диметилформамид^ является
превосходным растворителем для конденсации фталимида калия
с органическими галоидпроизводными. Для таких реакционно-
способных галоидпроизводных, как, например, фенацилбромид,
реакция в диметилформамиде сопровождается выделением тепла
и заканчивается в течение 10 мин. Выход фталимидацетофенона
достигает 92%. Для малоактивных галоидпроизводных
конденсацию необходимо проводить при повышенной температуре.
S.
S.
S.
Н
5. J.
Gabriel, Вег.. 21, 1049 (1S88).
G a b г i e I, J. С о I m а п, Вег., 33, 980, 2630 (1900). /.
Gabriel, Ber., 20, 224 (1887).
Ing, R. Manske, J. Chem. Soc, 1926, 2348.
S h e e h a n, W. В о 1 h о f e r, J. Am. Chem. Soc, 72, 2786 (1950)
Ганч 74
Артур Рудольф Ганч (Arthur Rudolf Hantzsch, 1857—1935) родился в
Дрездене. Ученик Р. Шмидта и И. Вислиценуса. В 1880 г. защитил докторскую
диссертацию. Работал в лаборатории А. Гофмана в Берлине. Профессор
Цюрихского политехникума (с 1885 г.), Вюрцбургского (с 1890 г.) и Лейпцигского
(1903—1928 гг.) университетов. А. Ганч опубликовал около 450 научных
работ; главные из них посвящены изучению стереохимии диазосоединений,
зависимости цвета красителей от их строения, а также синтезу производных
пиридина, триазола и кумарона. В 1882 г. получил пиридиновые соединения из
ацетоуксусного эфира, аммиака и альдегидов. В 1890—1894 гг. вместе со своим
учеником А. Вернером разработал теорию стереохимии азота, а также
исследовал строение оксимов.
СИНТЕЗ ГАНЧА
Образование производных пиридина при конденсации эфиров
Р-кетокислот с альдегидами и аммиаком называют синтезом
пиридинов по Ганчу\ Первоначально получающиеся по этой
реакции дигидропиридины легко окисляются до производных
пиридина:
R'
нсо
ROOC OOOR R' R'
^ / ROOC. l/COOR ROOC\J. ^OOOR
NH,
Известны многочисленные модификации синтеза Ганча\ в
которых используются эфиры циануксусной и малоновой кислот
или дикетоны. Во многих случаях целесообразно выделять
промежуточные продукты указанной реакции—эфиры 8-аминокро-
75 Ганч
тоновой кислоты, так как их также можно применять для синтеза
в качестве исходных веществ:
R'
нсо
На
COOR
СН
II
с
N СН,
ROOC
ОС
Вследствие большого числа возможных видоизменений синтез
Ганча был успешно использован для получения многочисленных
производных пиридина. Например, при конденсации этоксиацетил-
ацетона с амидом циануксусной кислоты в присутствии
пиперидина был получен 3-циано-4-этоксиметил-2-окси-6-метилпири-
'—промежуточный продукт в синтезе витамина В*:
сн,ос,н,
I
СО CN СН,ОС,Н,
НдС СИ, /^/^
I Ф t -^ II I Ф 2НаО
ОС ,СО у
н,с нл
.
Другим примером служит получение 4-замещенного этилового
эфира 5-циано-2,6-Диметил-1,4-дигидроникотиновой кислоты при
реакции ароматического альдегида с нитрилом р-аминокротоно-
вой кислоты и этиловым эфиром |3-аминокротоновой кислоты^:
Аг
НСО
QH&OOC CN Аг
V^
НдС NH, H,N СН,
Недавно механизм образования производных пиридина по
Ганчу нашел подтверждение в работах Берсона и Брауна*. Взаимо-
Ганч 76
действием этилового эфира р-аминокротоновой кислоты и арили-
денацетилацетона они получили несколько несимметричных 4-арил-
1,4-дигидропиридинов:
CHAr COOQHg Ar
Y
H,N CH
1. А. Н ant zsch. Ann., 215, 1 (1882).
2. О синтезе Ганча н его видоизменениях см. Г. М о ш е р, Гетероциклические
соединения, сб. 1, Издатинлит, 1953, стр. 356.
3. S. Harris, К. Fo I k ers, J. Am. Chem. Soc., 61, 1245 (1939).
4. V. A. P e t г о w, J. Chem. Soc., 1946, 884.
5. J. Berson, E. Brown, J. Am. Chem. Soc, 77, 444 (1955).
77 Гаттерман
Людвиг Гаттерман (Ludwig Gattermann, 1860—1921) родился в г. Госларе
(Германия). Изучал химию в Гейдельбергском и Геттингенском университетах,
а также в Берлинской промышленной академии у К. Либермана (1882 г.).
Работал в Геттингене под руководством Т. Зандмейера и в Геидельберге у В. МеЙе-
ра. С 1900 г. профессор ФреЙбургского университета. В 1888 г. получил чистый
Трех хлористый азот. Кроме исследований по модификации реакции Зандмейера
и в области антрахиноновых красителей, разработал несколько методов синтеза
ароматических альдегидов. Автор популярного учебного руководства
«Практические работы по органической химии» (1894 г.).
РЕАКЦИЯ ГАТТЕРМАНА
По реакции Гаттермана* происходит замещение диазогруппы
на галоид или цианогруппу при каталитическом разложении
солей диазония в присутствии металлической меди.
Примером этой реакции может служить образование
хлорбензола при добавлении медного порошка к раствору хлористого
фенилдиазония в соляной кислоте;
НС1
Другим примером является получение о-бромтолуола* из
о-толуидина (выход 47%):
NaNOg ^\/ ^ Си \/
Реакция Гаттермана не получила широкого применения в
органическом синтезе. Выходы галоидпроизводных по этой
реакции обычно ниже выходов, получаемых по реакции Зандмейера
(см. стр. 126). Кроме того, тонко раздробленная
порошкообразная медь может значительно затруднить очистку конечных
продуктов.
1. L. Gattermann, Бег., 23, 1218 (1890).
2. Л. Бигелоу, Синтезы органических препаратов, сб. 1, Издатинлит,
1949, стр. 136.
Гаттерман 78
СИНТЕЗ АЛЬДЕГИДОВ ПО ГАТТЕРМАНУ
Получение ароматических оксиальдегидов (или
соответствующих им эфиров) при конденсации фенолов или их эфиров с
цианистым водородом в присутствии хлористого водорода и хлористого
цинка (или хлористого алюминия) обычно называют синтезом
альдегидов по Гаттерману^* ^.
Примером синтеза является образование /г-анисового
альдегида из анизола:
Эфирный раствор анизола и безводного цианистого
водорода насыщают хлористым водородом и добавляют хлористый
алюминий. Полученный хлоргидрат ЯЛьдимина (который можно
выделить) легко гидролизуется при нагревании в водной
среде. Выходы оксиальдегидов по методу Гаттермана обычно
хорошие.
Адаме и Левин* предложили модификацию синтеза
Гаттермана, заменив свободный цианистый водород более безопасным,
удобным и доступным реагентом—цианидом цинка; достигнутые
при этом выходы оксиальдегидов обычно близки к выходам,
полученным по классическому методу.
Другой модификацией синтеза Гаттермана является
использование хлористого алюминия, растворенного в эфире*. Этим
методом из метилового эфира (3-резорциловой кислоты (I) был
получен с выходом 70% метиловый эфир 2,4-диокси-З-формилбен-
зойной кислоты (П):
сно
79 Гаттерман
По обычной методике синтеза Гаттермана эту реакцию
провести не удавалось.
Гидролиз метилового эфира 2,4-Диокси-3-формил бензойной
кислоты (II) и последующее декарбоксилирование образовавшейся
кислоты III дают возможность синтезировать %-резорциловый
альдегид, или 2,6-диоксибензальдегид (IV), трудно получаемый
другими методами:
СНО СНО
-СО*
и m iv
Недавно эта модификация реакции была использована для
синтеза* метилового эфира 2,4-диокси-3-формил-6-метилбензой-
ной кислоты (V):
СНО
Для непосредственного введения альдегидной группы в
ароматические соединения предложен видоизмененный способ,
заключающийся в применении хлорокиси фосфора и N-метилформ-
амида или N-диметилформамида. Этот способ, названный редкцыгй
ДылбсиаЗерд*, распространяется только на «активированные»
соединения.
Спосрб Вильсмейера с успехом используется для прямого
формилирования некоторых ароматических углеводородов
(антрацен, пирен), а также гетероциклических соединений, включая
тиофен, пиррол и индол.
Джатц? получил по способу Вильсмейера несколько
ненасыщенных альдегидов из винилогов N-метилформамида. Например,
при взаимодействии N-метиланилинопропеналя (VI) с хлорокисью
фосфора и диметиланилином образуется N-диметиламинокорич-
ный альдегид (VIII).
Промежуточно получающийся анил VII был выделен в виде
кристаллического перхлората.
Гаттерман
80
Реакция получения N-диметиламинокоричного альдегида по
способу Вильсмейера может быть изображена следующей схемой:
N—CH=CH-CHO
снУ
С.Н,
CH/
N—CH=CH—CH—
/
с
N=CH
сн/
он'
VII
C,H,-NH—СМ
,H,
VIII
В процессе изучения механизма реакции Вильсмейера многие
амиды подвергались обработке хлорокисью фосфора, фосгеном и
тионилхлоридом*. Оказалось, что продукты реакции—аддукты
обладают исключительной устойчивостью. Им приписывают
строение амидо- или имидохлоридов:
О реакции Гаттемана см. У. Т р ю с. Органические реакции, сб. 9, Издат-
инлит, 1959, стр. 45; [И. В. Мачинская, Реакции и методы
исследования органических соединений, кн. 7, Госхимиэдат, 1958, стр. 307.—
До д. ре^ ].
31, 1149 (1898); L. Gatterm
2. L. Gattermann, Бег.,
W. В е г с h е 1 m a n п,
М. К б b n е г, Ber., 32
3. R. Adams, I. L e v i n e,
n о 1 d, J. Sprung, J
4. R. Shah, M. L a i w a I
5. W. W h a 1 1 e y, J. Chem. Soc, 1949, 3278.
6. A. V i 1 s m e i e г, А. Н а а с к, Ber., 60, 119 (1927).
7. C. J u t z, Chem. Ber., 91, 850 (1958).
8. H. Bosshard, R. Могу, M. S с h m i d, H. Z о 1 H n g e r,
chim. act a, 42, 1659 (1959).
(
Ber., 31, 1765 (1898); L. Gatterm
278(1899).
J. Am. Chem. Soc, 45. 2373 (1923); R
Am. Chem. Soc, 60, 1699 (1938).
I a, J. Chem. Soc, 1938, 1828.
ann,
ann,
A r-
Helv.
81 Гаттерман —Кох
РЕАКЦИЯ ГАТТЕРМАНА—КОХА
Реакцией Гаттермана—Коха*» ^ называют получение
ароматических альдегидов непосредственным формилированием при
помощи окиси углерода и хлористого водорода в присутствии
соответствующего катализатора. Эта реакция представляет собой
частный случай более общей реакции Фриделя—Крафтса (см.
стр. 253).
Примером реакции является синтез л-толуилового альдегида
из толуола* (выход 51%):
сно
Для проведения реакции суспензию однохлористой меди и
хлористого алюминия в толуоле насыщают смесью газообразных
окиси углерода и хлористого водорода.
Если реакция протекает при атмосферном давлении, то в
качестве активатора обычно используют однохлористую медь, роль
которой, вероятно, заключается в образовании с окисью углерода
комплекса, облегчающего получение формил хлорида; последний,
как считают, и является активным реагентом в синтезе
Гаттермана—Коха.
Изучив реакцию образования бензальдегида из бензола и
окиси углерода, Дилк и Эли* высказали предположение, что в
реакции участвует ион НСО+:
НО + СО + А1Вгз з±: С^[ +
который взаимодействует затем с ароматическим углеводородом:
В реакцию Гаттермана—Коха мо&ет вступать большая часть
ароматических и жирноароматических углеводородов. Присутст-
6 А. Серрей
Гаттерман—Кох 82
вне в ароматическом ядре сильных электроноакцепторных
заместителей затрудняет проведение реакции. Если реакция
проходит в автоклаве при повышенном давлении, применение
активатора не обязательно. Протекание побочных реакций (типа
миграции или отщепления алкильных групп) обусловливается
наличием катализатора.
1. L. Gattermann, J. Koch, Ber., 30, 1622 (1897).
2. О реакции Гаттермана—Коха см. Н. К р а у н з, Органические реакции,
сб. 5, Издатинлит, 1951, стр. 275; [И. В. Мачинская, Реакции и
методы исследования органических соединений, кн. 7, Госхимиздат,
1958, стр. 277; Ч. Томас, Безводный хлористый алюминий в
органической химии, Издатинлит, 1949, стр. 595.—Дол. реб.].
3. Г. К о л е м а н, Д. К р э г, Синтезы органических препаратов, сб. 2,
Издатинлит, 1949, стр. 464.
4. М. D i 1 к е, D. Е 1 е у, J. Chem. Soc, 1949, 2601, 2613.
83 Гелль—Фол ыард—Зелинский
Карл Магнус Гелль (Carl Magnus Hell, 1849—1926) родился в Штутгарте
(Германия). Учился в Штутгарте у Г. Фелинга и в Мюнхене у Э. Эрленмейера.
Работал ассистентом Г. Фелинга. С 1883 г. профессор Высшей технической
школы в Штутгарте. В 1889 г. синтезировал наиболее высокомолекулярный из
известных в то время предельных углеводородов—парафин CggM^. Приобрел
наибольшую известность как автор работ по бромированию кислот в присут*
ствии фосфора.
*
Якоб Фольгард (Jacob Volhard, 1834—1909) родился в Дармштадте
(Германия). Учился в Гиссене у Ю. Либиха и Билля. В 1855 г. защитил докторскую
диссертацию. Ассистент Ю. Либиха в Мюнхене (с 1857 г.) и А. Гофмана в
Лондоне (1860—1861 гг.). Работал в лабораториях Р. Бунзена в Гейдельберге и
А. Кольбе в Марбурге. Сначала адъюнкт-профессор, затем заведующий кафедрой
неорганической и аналитической химии Мюнхенского университета.
Синтезировал capкозин (1862дГ.), гуанидин и креатин. Изучая бромирование
органических кислот вместе с Н. Эрдманном, улучшил метод К. Гелля, синтезировал
<%-бромкарбоновые"кислоты, получил тиофен и производные янтарной кислоты;
занимался также%синтезом эфиров дифенилуксусной кислоты.
Николай Дмитриевич Зелинский* (1861—1953) родился в Тирасполе.
В 1884 г. окончил университет в Одессе, затем в течение трех лет работал в
Лейпциге и Геттингене в лаборатории В. Мейера. В 1889 г. защитил
магистерскую, а в 1891 г.—докторскую диссертации. С 1893 г. профессор кафедры
аналитической и органической химии в Московском университете, где работал до
конца жизни. С 1926 г. член-корреспондент, а с 1929 г. действительный член
Академии наук СССР. За выдающиеся научные заслуги ему в 1926 г. было
присвоено звание заслуженного деятеля науки, а в 1945 г.—звание Героя
Социалистического труда. Н. Д. Зелинский' опубликовал свыше 500 научных трудов в
области органической химии и физико-химических методов исследования. Ему
принадлежат классические работы в области органического катализа/' Важное
значение имеют его исследования в области химии нефти, разработка методов
получения из нефти ценных углеводородов, служащих исходными
материалами для синтеза красителей, искусственного каучука, пластмасс и т. д. Осо-
Биография Н. Д. Зелинского написана редактором.
Гелль—Фольгард—Зелинский 84
бенно значительными для химии нефти, моторных топлив и синтетического
каучука являются исследования в области нафтенов, каталитической
гидрогенизации и дегидрогенолиза, циклизации и размыкания циклов. Выдающееся
значение имеют также работы по химии белков и пептидов. Он первый предложил
использовать активированный уголь для защиты от отравляющих веществ.
Н. Д. Зелинский создал самую крупную научную школу химиков, к которой
принадлежат такие известные ученые, как Л. А. Чугаев, Н. А. Шилов,
С С. Наметкин, А. Н. Несмеянов, Б. А. Казанский, А. А. Баландин и другие.
РЕАКЦИЯ ГЕЛЛЯ—ФОЛЬГАРДА-ЗЕЛИНСКОГО
Реакцией Гелля—Фольгарда—Зелинского** ^ называют
получение %-хлор(а-бром)замещенных карбоновых кислот при
действии хлора или брома на кислоты в присутствии небольших
количеств треххлористого (соответственно трехбромистого) фосфора.
Галоидированию, вероятно, предшествует образование галоид-
ангидридов, с которыми галоиды реагируют значительно быстрее,
чем с незамещенными кислотами. Стадии реакции можно выразить
следующей схемой:
3RCH*—СООН + РХ, -* 3RCH,—СОХ 4- Н,РО,
RCH,—СОХ + X, -» RCHX—COX -f HX
RCHX—СОХ + RCH*—СООН -* RCHX—СООН + RCH,—СОХ
Первоначальная реакция Гелля—Фольгарда (с применением
брома и фосфора), по-видимому, также протекает через стадию
образования бромангидрида.
Примером бромирования кислот с использованием
треххлористого фосфора является реакция получения %-бромизокапроно-
вой кислоты^ (выход 66%):
Вг,
(СНз),СН—СН,—СНд-СООН » (СН^СН^СН,,—СНВг—СООН
РС1
Попытки бромирования |3-бромпропионовой кислоты в %-по-
ложение по методу Гелля—Фольгарда—Зелинского не увенчались
успехом и привели к образованию только бромангидрида (3-бром-
пропионовой кислоты*
й
Очень хороший метод получения моно-а-бромпроизводных
дикарбоновых кислот предложили Швенк и Папа*. Этот метод
заключается в обработке полуэфиров дикарбоновых кислот тио-
нилхлоридом и затем бромом. Действием спирта образующийся
85 Гелль—Фольгард—Зелинский
хлорангидрид д-бромзамещенного неполного эфира дикарбоно-
вой кислоты превращают в полный эфир, который очищают
перегонкой:
ROOC-(CH^n—CH,—COOH ^ ROOC—(CH^-CH,-COC1
R'OH
-^ ROOC-(CH%)n—СНВг—COCI —-» ROOC—(СН^д—СНВг—COOR'
Диэтиловый эфир %-бромадипиновой кислоты был получен по
этому методу с выходом 90%. Данный метод вполне применим и
для синтеза а-галоидзамещенных монокарбоновых кислот. Так,
этиловые эфиры ос-бромциклогексилуксусной и %-бромфенилуксус-
ной кислот были получены таким путем с практически
количественными выходами.
1.С. Hell, Вег., 14, 891 (1881); J. V о 1 h а г d, Ann., 242, 141 (1887);
Н. Д. Зелинский, Вег., 20, 2026 (1887).
2. Н. Watson, Chem. Rev., 7, 180 (1930).
3. К. М а р в е л, Синтезы органических препаратов, сб. 3, Издатинлит, 1952,
стр. 275.
4. Е. Charlesworth, H. Anderson, Can. J. Res., 288, 1 (1950).
5. E. Schwenk, D. Papa, J. Am. Chem. Soc, 70, 3626 (1948).
Геш 86
Курт Геш (Kurt Hoesch, 1882—1932) родился в г. Крейцау (Германия).
Учился в Берлинском университете у Э. Фишера. В 1914—1918 гг. профессор
химии Стамбульского университета. Вместе с Э. Фишером занимался синтезом
природных орселлиновых и эверниновых кислот.
СИНТЕЗ ГЕША
В 1915 г. Геш описал метод получения оксикетонов
конденсацией фенолов или их эфиров с нитрилами в присутствии
хлористого водорода и хлористого цинка*.
Например, при пропускании сухого хлористого водорода через
смесь резорцина, ацетонитрила и хлористого цинка в эфире
образуется продукт, который после нагревания с водой
превращается в 2,4-ДИОКсиацетофенон:
^\зэсн,
Аналогично из флороглюцина и бензонитрила образуется
2,4,6-триоксибензофенон:
ОН
Синтез Геша^ является видоизменением синтеза альдегидов
по Гаттерману (стр. 78) и широко применяется для получения
полиоксиарилкетонов, так как в этой реакции могут участвовать
различные алифатические и ароматические нитрилы. В качестве
катализатора реакции наряду с хлористым цинком часто
применяется хлористый алюминий.
87 , ^ Теш
В процессе синтеза первоначально образуются хлоргидраты
кетиминов, которые могут быть выделены. При гидролизе кет-,
имины превращаются в кетоны:
он ^, ноуу он
NH-HC1 О
Недавно Уэлли^ изучил реакцию Геша на примере нитрилов
трифтор- и трихлоруксусной кислот. Он показал, что резорцин
в эфирных растворах при —5° и насыщении хлористым водородом
присоединяет трифторацетонитрил. При гидролизе водой
образовавшийся твердый продукт превращается в ю-трифтор-2,4-Диокси-
ацетофенон:
НО,
Трихлорацетонитрил не реагирует с резорцином, орсином,
пирогаллолом и флороглюцином, а монометиловый эфир флоро-
глюцина образует о)-трихлор-2,4-Диокси-6-метоксиацетофенон. При
нагревании этого кетона с водным раствором бикарбоната натрия
получаются 2,4-диокси-6-метоксибензойная кислота и хлороформ:
,ОН
CCI,™ _,_ , „
coca,
осн, ' ^
сна.
Трихлорацетонитрил в условиях реакции Геша
взаимодействует со многими ароматическими соединениями, образуя
хлоргидраты кетиминов, которые при кислотном гидролизе превращаются
в кетоны. При щелочном гидролизе с хорошим выходом
образуются соответствующие нитрилы (/ж%с%%я Л/бенд—Фыше/%%*):
са, не; Аг—с—са,
CN ^^'" NH-HC1
он"
—*- Агсм Ф сна,
Геш88
1. К. Hoesch, Ber., 48, 1122 (1915).
2. О синтезе Геша см. П. С п е р р и, А. Дюбуа, Органические реакции,
сб. 5, Издатинлит, 1951, стр. 284.
3. W. W h а 1 1 е у, J. Chem. Soc, 1951, 665.
4. J. Н о u b e n, W. Fischer, J. prakt. Chem., 123, 313 (1929); см. также
D. Mo wry, Chem. Revs., 42, 221 (1948).
89 Гомберг—Бахман—Хей
Моисей Гомберг (Moses Gomberg, 1866—1947) родился в Елизаветграде
(ныне Кировоград). В 1885 г. семья Гомберга переселилась в США. Окончил
Мичиганский университет, где в 1894 г. ему была присуждена степень доктора
философии. Работал в Мюнхене у А. Байера и в Гейдельберге с Б. Мейером
Профессор, затем заведующий кафедрой химии (с 1927 г.) Мичиганского
университета. С 1931 г. президент Американского химического общества.
Основоположник химии свободных радикалов. Исследования полиарилметильных
соединений, начатые им еще в Гейдельберге, привели к открытию свободного
радикала трифенилметила (1900 г.). За свои выдающиеся труды М. Гомберг.
удостоен многих наград.
*
Вернер Эмануэль Бахман (Werner Emanuel Bachmann, 1901—1951)
родился в Детройте (США). В 1926 г. получил степень доктора в Мичиганском
университете. Затем он занимался исследовательской работой в Цюрихе, будучи
стипендиатом Рокфеллеровского фонда, и далее в Лондоне и Мюнхене, где получал
стипендию Гоггенхейма. В дальнейшем вернулся в Мичиганский университет
и стал профессором химии, сменив М. Гомберга. Занимался исследованием сво*
бодных радикалов и изучением молекулярных перегруппировок, а также
синтезом половых гормонов и взрывчатых веществ.
Дональд Холройд Хей (Donald Holroyde Hey) родился в 1904 г. в г. Суонси
(Англия). Получил степень магистра в Уэльсском университете, степень доктора
философии в Лондонском университете (где учился у Л. Кинкеля) и степень
доктора наук в Манчестерском университете. Читал лекции в Манчестерском
и Лондонском университетах. С 1941 по 1945 г. был директором Британского
научно-исследовательского института Шеринга. Затем вернулся в Лондонский
университет, профессором которого является в настоящее время.
РЕАКЦИЯ ГОМБЕРГА—БАХМАНА—ХЕЯ
Образование диарилов при обработке растворов солей диазо-
ния едким натром или ацетатом натрия в присутствии жидких
ароматических углеводородов обычно именуют реакцией
Гомберга. Эта реакция известна также под названием реакции
Гомберга—Бахмана—Хея*' ^.
Гомберг— Бахман—Хей ^^^ 90
Примером этой реакции может служить получение 4-нитроди-
фенила из хлористого л-нитрофенилдиазония и бензола^:
Лучшие выходы диарилов часто достигаются при замене едкого
натра ацетатом натрия. Применение стабилизированной соли
диазония*, полученной взаимодействием хлористого /г-нитрофе-
нилдиазония с нафталин-1,5-дисульфокислотой, способствует
повышению выхода 4-нитродифенила до 70%. Нитрозоацетильные
производные ароматических аминов также могут быть
использованы для синтеза несимметричных диарилов*.
Интересна дискуссия Хея и Уотерса" (1948 г.) с Ходжсоном?
(1949 г.) по поводу ионного и свободно-радикального
механизма реакции Гомберга. Ходжсон считает, что активным
реагентом является ион диазонии.
В недавно опубликованном обзоре Д. де Тара*, посвященном
синтезу Пшорра (стр. 217) и сходным с ним реакциям циклизации
диазониевых соединений, приведено сопоставление их с синтезом
Гомберга.
1. М. G о m b е г g, W. Bachmann, J. Am. Chem. Soc, 46, 2339 (1924)
2. J. Elks, J. H a wort h, D. Hey, J. Chem. Soc., 1940, 1284.
3. О реакции Гомберга—Бахмана—Хея см. В. Бахман, Р. Гофман,
Органические реакции, сб. 2, Издатинлит, 1950, стр. 244.
4. Н. Hodgson, Е. М а г s d е п, J. Chem. Soc, I940, 208.
5. W. Grieve, D. Hey, J. Chem. Soc, 1934, 1797; H. France,
I. Heilbron, D. Hey, J. Chem. Soc, 1940, 369.
6. D. Hey, W. Waters, J. Soc Dyers Colour., 64, 359 (1948).
7. H. Hodgson, J. Soc Dyers Colour., 65, 347 (1949).
8. Д. де Тар, Органические реакции, сб. 9, Издатинлит, 1959, стр. 529.
91 Гоулд—Джекобе
Р. Гордон Гоулд (R. Gordon Gould) родился в 1909 г. в Чикаго. Учился
в Гарвардском университете. Преподавал в Гарвардском и Айовском
университетах, затем в Рокфеллеровском институте медицинских исследований.
С 1947 г. доцент биохимической лаборатории Иллинойского медицинского
института.
Вальтер А. Джекобе (Walter A. Jacobs) родился в 1883 г. в Нью-Йорке.
Учился в Колумбийском и Берлинском университетах. Работал в
Рокфеллеровском институте медицинских исследований, с 1923 г. профессор этого
института. Основные работы Джекобса посвящены исследованию природных
соединений.
РЕАКЦИЯ ГОУЛДА—ДЖЕКОБСА
Конденсация ароматических аминов с эфирами этоксиметилен-
малоновой кислоты, приводящая к образованию замещенных
4-оксихинолинов, получила наименование реакции
Гоулда—ДжеПрайс и Роберте* получали этим методом промежуточные
продукты для синтеза химиотерапевтических препаратов, в частности
антималярийных.
Синтез 4-оксихинолина по Гоулду—Джекобсу состоит из
следующих стадий:
1/ Конденсация анилина с этиловым эфиром этоксиметилен-
малоновой кислоты, приводящая к образованию этилового эфира
а-карбэтокси-0-анилиноакриловой кислоты (1):
ооос,н,
С-СООС,Н*
+ С.Н.ОН
Гоулд^Джекобс 92
2. Циклизация эфира (I) при нагревании в даутерме А
(смесь дифенила и дифенилоксида) в З-карбэтокси-4-оксихинолин (II):
ОС COOQH,
—с*н&он
3. Гидролиз эфира (II) и последующее декарбоксилирование
кислоты (III) при нагревании приводит к образованию 4-окси-
хинолина (IV):
COOQH& ^_ ^ А ^СООН
—со.
II III
По этому удобному методу^ получаются обычно с хорошими
выходами многочисленные производные 4-оксихинолина с
различными заместителями в бензольном ядре.
Из мета-замещенных анилина по реакции Гоулда—Джекобса
образуются главным образом 7-замещенные4-оксихинолины.
Примером может служить синтез 7-хлор-4-оксихинолина* (выход 6096)
из jw-хлоранилина. Указанный выход значительно выше, чем
выход, получаемый по реакции Конрада—Лимпаха (стр. 164),
так как в последнем случае наряду с 7-хлор-4-оксихинолином
образуется большое количество 5-хлор-4-оксихинолина.
1. R. Gould, W. Jacobs, J. Am. Chem. Soc, 61, 2890 (1939).
2. С Price, R. Roberts, J. Am. Chem. Soc, 68, 1204 (1946).
3. R. R e i t s m a, Chem. Revs., 43, 43 (1948); см. также Р. Эльдерфильд,
Гетероциклические соединения, сб. 4, Издатинлит, 1955, стр. 29.
4. Ч. Прайс, Р. Роберте, Синтезы органических препаратов, сб. 4,
Издатинлит, 1953, стр. 239.
A3 Гофман
Август Вильгельм Гофман (August Wilhelm Hofmann, 1818—1892)
родился в г. Гиссене (Германия). Окончил Гиссенский университет, ученик Ю. Ли-
биха. В 1841 г. защитил докторскую диссертацию. В 1843—1844 гг. ассистент
Ю. Либиха, в 1845 г. приват-доцент Боннского университета. Затем профессор
и директор вновь основанного Королевского химического колледжа в Лондоне
(1845—1864 гг.). Среди его студентов были В. Перкин и П. Грисс (открывший
впоследствии реакцию диазотирования первичных аминов). С 1865 г.
профессор Берлинского университета. Был основателем Немецкого химического
общества (1868 г.) и до конца жизни его президентом. Его исследования в области
химии анилина и бензола положили начало производству синтетических краен
телей из каменноугольной смолы. Такие работы, как получение и разделение
жирных аминов, изучение алкалоидов, исследование расщепления четвертичных
аммониевых оснований, являются лишь частью его большого вклада в
органическую химию. Выдающуюся роль сыграл он и как педагог. В 1875 г. А. Гофман
был награжден медалью Копли.
ПЕРЕГРУППИРОВКА ГОФМАНА
Перегруппировкой Гофмана называют реакцию образования
первичных аминов из амидов кислот, связанную с
элиминированием карбонильной группы. Эта реакция, впервые описанная
Гофманом^ g 1881 г., заключается во взаимодействии амидов кислот
с водными растворами гипогалогенитов и может быть
представлена следующим уравнением:
RCONH, + X, + 2NaOH -* RNH* + 2NaX + CO, + Н,0
ИЛИ
RCONH, + NaXO -* RNH, + NaX + CO,
Первичными продуктами реакции являются изоцианаты
(RNCO); если синтез проводится в спиртовой среде, то сначала
образуются уретаны, которые легко гидролизуются до аминов.
В реакцию вступают амиды алифатических, ароматических, жир-
ноароматических и гетероциклических кислот (R—ал кил, арил,
аралкил и гетероциклический радикал). Из гипогалогенитов
обычно применяют гипохлориты или гипобромиты (X—С1 или Вг).
Перегруппировка Гофмана, как правило, протекает гладко и
с хорошими выходами*.
Гофман
94
Механизм реакции Гофмана был предметом многочисленных
исследований*. Установлено, что перегруппировка протекает
в несколько стадий: образование N-галоидаминов (а), переход их
в неустойчивую соль (6), распад соли (с) с образованием
промежуточного гипотетического продукта I и его
перегруппировка (d) в изоцианат II. ^
RCONH
RCONHX
Naox
ОН"
О
RCONHX
[RCONX]
[RCON:]
1
+ X
——^ R—N=C=O
Считают* что промежуточный продукт I представляет собой
тг-комплекс:
N=C=O
Гидролиз изоцианата II приводит к образованию первичных
аминов:
он"
R—N=C=O ^ RNHg (e>
Процесс распада (с) с образованием продукта I, по-видимому,
является стадией, определяющей скорость всей реакции. В связи
с этим было показано^, что в ряду замещенных бензамидов
реакция идет тем медленнее, чем более электроположителен атом
углерода карбонильной группы. Установлено также, что миграция
группы R никогда не сопровождается стереохимической
инверсией. Доказательством сохранения исходной конфигурации
группы R является постоянство оптической активности при
перегруппировке амидов, оптически активные центры которых
непосредственно связаны с карбонильной группой.
В результате исследования гофмановской перегруппировки
перфторамидов найдено, что в зависимости от условий процесса
образуются два различных соединения*. Так, пиролиз
промежуточной безводной соли [QFyCONBr]" Na+ приводит к ожидаемому
изоцианату QFyNCO, а в водном растворе NaOH отщепляется
ион изоцианата и получается гептафторпропилбромид QFyBr.
Показано, что при перегруппировке по Гофману амидов
высших жирных кислот добавка диоксана дает возможность умень-
95 Гофман
шить число побочных процессов?. Повышение выходов аминов в
присутствии диоксана наблюдалось при перегруппировке амидов
различных жирных кислот (от капроновой до лауриновой).
1. A. Hofmann, Ber., 14, 2725 (1881).
2. О реакции Гофмана см. Э. У э л л и с, Д. Л э н, Органические реакции,
сб. 3, Издатинлит, 1951, стр. 255.
3. О механизме реакции Гофмана см. К. И н г о л ь д, Механизм реакций и
строение органических соединений, Издатинлит, 1959, стр. 401.—
р
4. См. М. D e w a r, Electronic Theory of Organic Chemistry, London, 1949
p. 222.
5. С Mauser, W. Renfrew, J. Am. Chem. Soc., 59, 121 (1937).
6. D. Barr, R. Haszeldine, J. Chem. Soc, 1957, 30.
7. E. Magnien, R. В a 1 1 z 1 y, J. Org. Chem., 23, 2029 (1958).
Гофман 96
РАСЩЕПЛЕНИЕ ПО ГОФМАНУ
Разложение четвертичных аммониевых оснований на олефины
и третичные амины обычно называют расщеплением по Гофману
или реакцией исчерпывающего метилирования*.
Типичным примером этой реакции является получение
этилена и триметиламина из этил амина:
CHgJ Ag#O Г
QH.-NH, » МСН)]
3 + H.0
Соли четвертичных аммониевых оснований обычно разлагают
влажной окисью серебра. Реакция неприменима к производным
пиридина, хинолина, изохинолина и гидрированным хинолинам.
Если атом азота связан с различными заместителями, то при
разложении четвертичных аммониевых оснований олефин
образуется из заместителя с меньшим числом атомов углерода (л/хмило
/отжала). Например, из гидроокиси диметилэтилпропиламмония
получается этилен, а не пропилен:
сн,
R—CH,-CH,—N+—CH,—CM,
CHg
+ H,0
ОН
Это объясняется наличием гиперконъюгации^. Индукционный
эффект радикала R увеличивает электроотрицательность соседнего
атома углерода и затрудняет отщепление Р-водородного атома в
форме протона. Обычно считают, что реакция Гофмана
заключается в бимолекулярном ионном m/xzwc-отщеплении*. Механизм
гофмановского расщепления по Дерингу иМейслиху* представляет
собой одностадийный процесс с одним переходным состоянием
(реакционным комплексом).
Четвертичные аммонийные соли, содержащие фенэтильные
группы, могут расщепляться при нагревании с водными раствора-
97
Гофман
ми едкого натра, как было показано* на примере метилиоди-
да М-(3'-метоксибензил)-М-метил-3,4-метилендиокси-5-метоксифен-
этил амина:
СН,
ОСН,
Показано, что единственным установленным продуктом
расщепления четвертичных аммониевых оснований, полученных из
обоих %%с- и /лрдмс-2-фенилциклогексил аминов*, является 1-фе-
нилциклогексен:
Образование 1-фенилциклогексена из трдяс-изомера объясняет
ся прототропным смещением.
Термическое разложение окисей М,М-диметилалкиламинов ис
следовано Копом\ Полученные при этом соединения он сравни
вал с конечными продуктами гофмановского расщепления. Реак
ция термического разложения протекает с отщеплением цыс-р-во
дородного атома и аминоокисной группировки, приводя к образо
ванию олефина и М,М-диметилгидроксиламина по внутримолеку
лярному циклическому механизму:
В ряду алициклических соединений (из окисей циклононил- и
циклодецилдиметиламина), как и по реакции Гофмана,
получаются /лраяс-циклоолефины. Из окиси циклооктилдиметиламина
образуется %ыс-циклооктен, а из гидроокиси соответствующего
четвертичного основания—смесь у we- и /тгрдмс-изомеров. Из
алициклических производных с меньшим числом атомов в кольце
как при термическом разложении, так и при гофмановском
расщеплении получаются %%с-олефины.
А. Ссорей
Гофман 98
Одной из важнейших областей применения реакции гофманов-
ского расщепления является исследование структуры алкалоидов
и других сложных азотсодержащих гетероциклических систем*.
При последовательном проведении исчерпывающего
метилирования в конечном итоге образуется не содержащий азота продукт,
позволяющий установить строение исходного алкалоида.
1. A. Hofmann, Вег., 14, 659 (1881).
2. J. Baker, Hyperconjugation, London, 1952, p. 5.
3. К. J ewers, J. Me К en n a, J.Chem.Soc, 1958, 2209.
4. W. Doer ing, H. Meislich, J. Am. Chem. Soc, 74, 2099 (1952).
5. A. Surrey, J. Am. Chem. Soc, 70, 2887 (1948).
6. R. Arnold, P. Richardson, J. Am. Chem. Soc., 76, 3649 (1954).
7. A. Cope, D. McLean, N. Nelson, J. Am. Chem. Soc, 77, 1628
(1955).
8. A. Battersby, H. Oppenshaw, J.Chem.Soc, 1949,3207,559
99 Гребе—Ульман
Карл Гребе (Carl Graebe, 1841—1927) родился во Франкфурте. Работал у
Р. Бунзена в Гейдельбергском университете, под руководством А. Кольбе в
Марбурге, на фабрике красителей (короткое время), в лаборатории Э. Эрлен-
мейера, ассистентом А. Байера. Профессор химии Лейпцигского (1869 г.),
Кенигсбергского (с 1870 г.) и Женевского (1878—1906 гг.) университетов.
Вместе с К. Либерманом синтезировал природный краситель ализарин и установил
его строение, показав, что при восстановлении ализарина образуется антрацен.
Первый обнаружил в каменноугольной смоле акридин и карбазол. Получил
фталевую кислоту из нафталина и установил его бицикл ическую структуру.
За выдающиеся исследования был награжден медалями Перкина, Лавуазье
и Бертолле.
СИНТЕЗ ГРЕБЕ—УЛЬМАНА
Гребе и Ульман* установили, что при диазотировании о-ами-
нодифенил амина (I) получается 1-фенилбензотриазол (II), при
нагревании которого выделяется азот и образуется с хорошим
выходом карбазол*:
Применение синтеза Гребе—Ульмана ограничено вследствие
трудности получения производных о-аминодифенил амина и ин-
гибирующего влияния электроноакцепторных заместителей в
ароматических ядрах.
Недавно Кокер, Плент и Тернера изучили влияние различных
заместителей на образование карбазолов по реакции
Гребе—Ульмана. Они получили 3-хлор- и 1-хлор-6-нитрокарбазолы, однако
осуществить превращение некоторых других бензотриазолов им
не удалось. Был синтезирован также ряд
6-цианкарбазолов.Например, З-хлор-6-цианкарбазол получен путем нескольких по-
Об Ульмане см. стр. 241.
Гребе—Ульман
100
следовательных реакций, исходя из 4-хлор-З-нитробензонитрила
и /г-хлоранилина:
IK
HCI
NC
N
^ NaNO% \d^X/|| f^\/ 380*
HCI
"Yr-fY°+N.
Более удобным методом получения карбазолов является
/ягз Бг;ршб*, первая стадия которого заключается во
взаимодействии фенилгидразина с циклогексаноном*:
NH-NH
Тетрагидрокарбазолы I, образующиеся по этой реакции,
можно дегидрировать различными способами: перекисью свинца,
хлористым палладием, палладиевой чернью с коричной кислотой,
хлоранилом*.
1. С. G г а е b e, F. U I I ma n n, Ann., 291, 16 (1896).
2. Обзоры по химии карбазола см. N. Campbell, В. Barclay, Chem.
Revs., 43, 53 (1948); В. Фрейденберг, Гетероциклические
соединения, сб. 3, Издатинлит, 1954, стр. 231; Д. д е Тар, Органические
реакции, сб. 9, Издатинлит, 1959, стр. 529.
3. G. С оке г, S. Plant, P. Turner, J. Chem. Soc, 1951, 110.
4. W. Borsche, Ann., 359, 49 (1908).
* В. Борше (W. Borsche, p. 1877). Ученик О. Валлаха. В 1898 г. получил
степень доктора философии в Геттингенском университете.
Гриньяр
Виктор Гриньяр (Victor Grignard, 1871—1935) родился в Шербурге
(Франция). Работал преподавателем, а затем профессором университетов в Лионе,
Безансоне и Нанси. По совету своего учителя П. Барбье использовал магний
в качестве конденсирующего агента и нашел, что галоидные ал килы в
присутствии абсолютного эфира легко вступают в реакцию с магнием. В 1901 г.
закончил диссертацию, посвященную смешанным магнийорганическим соединениям
и их применению в синтезе спиртов, кислот и углеводородов. Значение этой
работы общеизвестно. Совместно с П. Барбье проводил исследования в области
химии терпенов, алюминийорганических соединений, по каталитическому
гидрированию под уменьшенным давлением. Занимался разработкой
номенклатуры органических соединений. Лауреат Нобелевской премии (1912).
РЕАКЦИЯ ГРИНЬЯРА
При взаимодействии галоидпроизводных с магнием в среде
эфира образуются эфирные растворы смешанных магнийгалоидор-
ганических соединений (реактивы Гриньяра). Реакции,
проводимые с помощью этих реактивов, объединяются под общим
названием реакции Гриньяра. Реактивы Гриньяра легко реагируют с
большинством веществ, содержащих функциональные груп-
Впервые подобную реакцию применил для синтеза
третичных спиртов Барбье®, который получал смешанное магнийгалоид-
органическое соединение непосредственно в присутствии второго
компонента реакции:
R^ R. yOMgJ
С=О + Mg + СНдЛ -» С
R^ /OMgJ R. /OH
С фН.0 -» с 4- MgJOH
R'/ \ / \
Гриньяр Ю2
В отличие от этого реакция Гриньяра протекает в две стадии:
1. Образование реактива Гриньяра;
Mg -» RMgX
2. Взаимодействие реактива Гриньяра со вторым компонентом
реакции.
За последнее время область применения реакции Гриньярд
расширилась благодаря использованию в качестве растворителей
тетрагидрофурана и эфиров ди- или триэтиленгликоля.
Повышенную активность органических галоидпроизводных в этих
растворителях приписывают способности ^-электронов кислородного
атома эфиров к образованию координационной связи с магнием.
Реактивы Гриньяра легко получаются из видилгалогенидов и
магния в присутствии тетрагидрофурана или эфиров ди- и
триэтиленгликоля. Взаимодействие винилмагнийбромида с
ангидридами кислот в растворе тетрагидрофурана при температуре от
—60 до —70° приводит к образованию винилкетонов* (выход
60—80%):
2CH,=CHMgBr + 2(RC0),0 -» 2CH,=CHCOR + MgBr, + Mg(OCOR),
Хауторн* описал превращение галоидных арилов в фенолы.
Для этого реактив Гриньяра обрабатывают триметилборатом, а
затем реакционную массу окисляют перекисью водорода.
1. Н. R h e i п b о I t, J. Chem. Educ, 27, 476 (1950); Bull. Soc. chim. France
897 (1950).
2. О реакции Гриньяра см. M. Knarasch, О. R e i n m u t h, Grignard
Reactions of Non-Metallic Substances, London, 1954.
3. P. В arb i er, С. г., 128, ПО (1899).
4. G. Martin, С. г., 245, 1933 (1957).
5. N. Hawthorne, J. Org. Chem., 22, 1001 (1957).
ЮЗ Дарзан
Георг Август Дарзан (Georges Auguste Darzens, 1867—1954) родился в
Москве. В 1895 г. получил степень доктора физических наук в Парижском
политехническом училище. В дальнейшем профессор химии в этом училище.
Получил известность благодаря работам по синтезу глицидных эфиров, произ-
вод ных тетралина и ненасыщенных кетонов. Позднее занимался
исследованием вальденовского обращения.
СИНТЕЗ ГЛИЦИДНЫХ ЭФИРОВ ПО ДАРЗАНУ
Конденсация альдегидов или кетонов с эфирами а-галоидо-
замещенных карбоновых кислот в присутствии основных
конденсирующих агентов приводит к получению глицидных эфиров и
называется реакцией Дарзана*»*.
Эта реакция может быть представлена следующим общим
уравнением:
С=О ф СН—COOQH, -i-
C-COOQH,
В качестве карбонильных компонентов конденсации
используются алифатические или ароматические альдегиды и кетоны;
сложноэфирными компонентами служат эфиры а-хлор- или а-бром-
карбоновых кислот; конденсирующими агентами обычно являются
амид натрия, металлический натрий или алкоголят натрия.
Реакцию проводят в атмосфере инертного газа, добавляя
конденсирующий агент в охлаждаемую смесь карбонильного соединения и
эфира а-галоидкислоты.
После некоторой выдержки реакционную смесь
обрабатывают разбавленной кислотой, экстрагируют полученный продукт
и очищают его перегонкой в вакууме.
Дарзан 104
Механизм конденсации Дарзана обычно изображается
следующей схемой*:
асн,—cooQH, + в -^ асн—cooQHg ч-вн+
c=oacH—ооосн. ф -$- с-
с—сн—сосед, f a
Глицидные эфиры могут использоваться для, синтеза многих
других соединений. Гидролиз глицидных эфиров приводит к
образованию глицидной кислоты, при декарбоксилировании
которой получаются альдегиды (R"=H) или кетоны (R"=Alk, Аг):
\ Л" н+ R\ Л" г «\ Л"
сс сс снс
сн-с
Например, из этилового эфира (3-метил-(3-фенилглицидной кис
лоты образуется а-фенилпропионовый альдегид^ с выходом 70%
:_/
сн—
Исходя из аддукта I, полученного конденсацией по Дильсу—
Альдеру /z-хинона с бутадиеном, по реакции Дарзана был
синтезирован^ кетоальдегид III:
О О—СН—COOR СНО
Промежуточно получающийся глицидный эфир II был
использован для полного синтеза иохимбина.
Показано, что алкоголят /м/?е/п-амилового спирта является
эффективным катализатором конденсации этилового эфира бен-
зоилпропионовой кислоты с этилхлорацетатом*. По-видимому,
это первый случай применения кетоэфира в реакции Дарзана.
1015 Дарзан
1. G. D a rz en s, С. г., 139, 1214 (1904).
2. О реакции Дарзана см. М. Ньюмен, Б. Мейджерлейн,
Органические реакции, сб. 5, Издатинлит, 1951, стр. 319.
3. О механизме конденсации Дарзана см. М. В а 1 1 е s t e г, Chem. Rev.,
55, 283 (1955); см. также М. В а 1 1 е s t e r, D. Perez-Bianco,
J. Org. Chem., 23, 652 (1958).
4. К. Аллеи, Д. Аллан, Синтезы органических препаратов, сб. 3,
Издатинлит, 1952, стр. 446.
5. Е. Т a m е 1 е п, J. Am. Chem. Soc, 80, 5006 (1958).
6. Е. Bergman n, S. Yaroslavsky, H. Weile r-F e i 1 с h e n-
f e I d, J. Am. Chem. Soc, 81, 2775 (1959).
Дарзан 106
СИНТЕЗ НЕПРЕДЕЛЬНЫХ КЕТОНОВ
ПО ДАРЗАНУ*
В 1910 г. Дарзан* разработал общий метод синтеза
ненасыщенных кетонов, заключающийся в обработке этиленовых
соединений хлористым ацетилом в присутствии хлористого
алюминия.
Примером этого метода является синтез тетрагидроацетофено-
на (I) из циклогексена:
/\ХЮШ» ^ ^СОСНд
CHgCOCl ^ [ | ->
В охлажденный до 0° раствор циклогексена и ацетилхлорида
в сероуглероде добавляют хлористый алюминий и
образовавшийся хлоркетон выделяют, выливая реакционную смесь в ледяную
воду. Обрабатывая хлоркетон третичными основаниями
(например, диэтиланилином), отщепляют хлористый водород и получают
тетрагидроацетофенон (I).
Реакция применима к алифатическим и циклическим олефи-
нам, а в качестве катализаторов можно использовать также
SnCl*, FeCl, и SbCb.
НеницескуЗ показал, что если растворителем является цикло-
гексан, то образуются предельные кетоны. Например, при
добавлении хлористого алюминия к смеси циклогексена и
ацетилхлорида в циклогексане (при —10°) и нагревании реакционной сме-
* Реакция ацетилирования этиленовых углеводородов хлористым
ацетилом или уксусным ангидридом в присутствии хлористого цинка была описана
И. Л. Кондаковым^ еще в 1892 г. и поэтому правильнее было бы называть ее
решсцыбД АомОдкодд или ЛГомбдкобд—Ддрлояд.
Иван Лаврентьевич Кондаков (1857—1931) родился в Вилюйске Якутской
губернии (ныне Якутская АССР). В 1884 г. окончил Петербургский
университет, ученик А. М. Бутлерова. В 1888—1895 гг. преподавал общую и
физиологическую химию в Варшавском университете, с 1895 г. профессор Юрьевского
(ныне Тартуского) университета. В 1894 г. защитил магистерскую
диссертацию. Основные работы И. Л. Кондакова посвящены изучению реакций
непредельных соединений.—/7рил*. ё
107 ^^^^^^^^^^^^^ Дарзан
си до 70° выделяется хлористый водород и получается метилцикло-
гексилкетон (II):
,сосн,
сн,соа -^|L [у + на
И
Вероятно, в этом случае непредельный кетон гидрируется за
счет растворителя.
Циклогептен в условиях реакции Неницеску* дает смесь двух
кетонов:
Неницеску^ установил также, что при добавлении циклогек
сена к смеси ацетилхлорида и хлористого алюминия в сероугле
роде при —15° и последующем введении бензола образуется 1-фе
нил-4-ацетилциклогексан^ (III):
СНзСОС! —-* { }-( )-СОСН
ш
По-видимому, это объясняется миграцией атома хлора из орто-
в пара-положение хлоркетона, с дальнейшим алкилированием по
Фриделю—Крафтсу:
III
Аналогично из пропилена был получен 2-фенилпентанон-4 (IV)
СНзСОС!
сн.—сн=сн. -
—СН—СН^—
IV
Дарзан ИЖ
Этот же кетон (IV) получается из непредельного кетона V и
бензола в присутствии хлористого алюминия:
CHg—CH=CH—СОСНз ——)- CHg—CH—CHs-COCHg
V IV
Описан^ синтез ацилированных олефинов взаимодействием кар-
боновых кислот, растворенных в трифторуксусном ангидриде, с
олефинами. Образующийся смешанный ангидрид присоединяется
при комнатной температуре по месту двойной связи. Продукт
реакции спонтанно разлагается с образованием ацилированного
олефина и трифторуксусной кислоты:
RCO—О—СОСРд 4- R'CH=CHR" -» RCO—CHR'—CHR"—O-COCF3 -^-
-#. RCO—CR'=CHR* ф CF3COOH
Тетрагидроацетофенон был получен^ перегруппировкой 1-эти-
нилциклогексанола по
он _ _ „. хюсн
1. О реакции Кондакова см. В. Н. Белов, Т. А. Рудольф и,
Реакции и методы исследования органических соединений, кн. 7, Госхимиз-
дат, 1958, стр. 255—Дол. рео\
2. G. D arzens, G. г., 150, 707 (1910).
3. С. Nenitzescu, E. Cioranescu, Бег., 69, 1820 (1936).
4. S. Friess, R. Pinson, J.Am. Chem. Soc, 73, 3512 (1951)
5. С. Nenitzescu, I. G a vat, Ann., 519, 260 (1935).
6. См. также W. Johnson, R. Offenhauer, J. Am. Chem. Soc,
67, 1045 (1945).
7. A. Henne, J. Tedder, J. Chem. Soc, 1953, 3628.
8. Д. Саундерс, Синтезы органических препаратов, сб. 4, Издатинлит,
1953, стр. 600.
109 Дафф
Джеймс Купер Дафф (James Cooper Duff) родился в 1888 г. в Глазго
(Шотландия). За самостоятельные исследования в области органической химии
получил степень магистра в Манчестерском университете. В 1923 г. защитил
докторскую диссертацию. В настоящее время профессор кафедры химии Вир-
мингамского технологического колледжа. Занимается исследованиями
комплексов металлов с аминами и изучением реакций фенолов с гексаметилентетр-
амином.
РЕАКЦИЯ ДАФФА
Образование производных о-оксибензальдегида из фенолов и
гексаметилентетрамина (уротропина) называется реакцией ДаффаЧ
он ^ ,он
сно
Реакция Даффа является общим методом получения
ароматических оксиальдегидов, позволяющим при меньшей затрате
времени получать более чистые продукты, чем по методу Реймера—
Тимана* (стр. 219).
Если проводить реакцию при 150—160° в безводном глицерине
в присутствии глицерин-борной кислоты и затем обрабатывать
смесь разбавленной серной кислотой, то промежуточные
продукты выделить не удается. Если же фенол, гексаметилентетрамин и
борную кислоту нагревать в 2-этоксиэтаноле, то могут быть
выделены промежуточно образующиеся вторичные амины*:
Нагревание полученных вторичных аминов I с гексаметилен-
тетрамином в уксусной кислоте приводит к их частичному
Дафф ПО
дегидрированию с образованием шиффовых оснований II, гидро-
лизующихся затем до д-оксиальдегидов III:
и
1. J. Duff, E. В i 1 1 s, J. Chem. Soc, 1932, 1987; J. Duff, J. Chem.
Soc, 547 (1941).
2. L. Ferguson, Chem. Revs., 38, 230(1946).
3. J. Duff, V. Fur ness, J. Chem. Soc, 1951. 1512.
Дебкер
Оскар Густав Дебнер (Oskar Gustav Doebner, 1850—1907) родился в Мей-
нингене (Германия). Учился в Мюнхене у Ю. Либиха и в Лейпциге у А. Коль-
бе. В 1873 г. защитил докторскую диссертацию. Работал в Тюбингене под
руководством Р. Фиттига, ассистентом Р. Отто в Брауншвейге (1874—1875),
затем ассистентом А. Гофмана в Берлине. С 1899 г. профессор университета в
Галле. Известность О. Дебнеру принесла его работа по синтезу хинолина.
СИНТЕЗ ДЕБНЕРА
Получение замещенных цинхониновых кислот путем
конденсации ариламина, жирных или ароматических альдегидов и
пировинограднои кислоты было предложено Дебнером* в 1887 г. и
получило название синтеза Дебнер а.
Типичным примером этой реакции является получение 2-фе-
нилцинхониновой кислоты (цинхофена) из анилина, бензальде-
гида и пировинограднои кислоты по следующей схеме:
СОСИ
СНьСОСООН
СН-С,Нд >
сн,
—На
В связи с исследованиями в области антималярийных
препаратов Лутц с сотр.* изучил условия, обеспечивающие максимальные
выходы по этой реакции. Одним из обязательных условий
является применение свежеперегнаннои пировинограднои кислоты. Хотя
выходы цинхониновых кислот обычно не высоки, синтез Дебнера
широко применяется вследствие доступности исходных веществ.
Дебнер 112
Образование производных дигидрохинолина в соответствии с
приведенной выше схемой аналогично синтезам промежуточных
продуктов по реакциям Скраупа (стр. 231) к Дебнер а—Миллера
(стр. ИЗ). Диспропорционирование, или, что более вероятно,
окисление промежуточными шиффовыми основаниями (бензил-
иденанилином), приводит к превращению дигидропроизводных
в цинхониновую кислоту.
ЛЯГЕРЛТУРЛ
l.O. D о е b n e г, Ann., 242, 265 (1887).
2. R. L u t z et al., J. Am. Chem. Soc, 68, 1813 (1946); см. также Р. Э л L-
д е р*ф и л ь д, Гетероциклические соединения, сб. 4, Издатинлит, 1955,
стр. 20.
113 Дебнер—Миллер
СИНТЕЗ ДЕБНЕРА—МИЛЛЕРА
Синтез Дебнера—Миллера^ дает возможность получать
производные хинолина при нагревании ароматических аминов с
альдегидами (смесью альдегидов) в концентрированной соляной или
серией кислотах.
Вероятно, первой стадией реакции является кротоновая
конденсация двух молекул альдегида с образованием а,
^-непредельного альдегида, взаимодействующего далее с ароматическим
амином. Дальнейшее течение реакции аналогично синтезу Скраупа
(стр. 231) и приводит к производным дигидрохинолина,
окисляющимся затем до соответствующих хинальдинов^:
I/
Небольшое изменение условий проведения реакции,
заключающееся в применении растворимого в воде окислителя (м-нитро-
бензолсульфокислоты) и 60—70%-ной серной кислоты, может
заметно увеличить выход хинальдина^. Спайви и Керд* изучили
условия образования 5- и 7-хлорхинальдинов из ж-хлоранилина
и пар альдегид а по этому видоизмененному методу и нашли, что
понижение концентрации кислоты способствует увеличению
количества 7-хлсрхинальдина.
1.0. D о е b п е г, W. Miller, Вег., 16, 2464 (1883).
2. См. Р. Эльдерфильд, Гетероциклические соединения, сб. 4, Издатин
лит, 1955, стр. 9.
3. Г. п. 567273; W. U t е г m о h 1 е п, J. Org. Chem., 8, 544 (1943)
4. A. S p i v е у, F. Curd, \ Chem. Soc, 1949, 2656.
8 А Ссррей
Делении I14
Марсель Делепнн (Marcel Delepine) родился в 1871 г. в г. Сен-Мартен-ле-
Гайар (Франция). Изучал медицину, неорганическую и органическую химию,
был ассистентом П. Бертло в Коллеж де Франс, затем профессором химии этого
колледжа. Президент Французского химического общества, почетный член
нескольких иностранных химических обществ. Многочисленные исследования
М. Делепина посвящены гидрированию в присутствии никелевого катализатора,
получению первичных аминов из бензилгалогенидов через четвертичные гекса-
метилентетраминовые соли, изучению различных сернистых соединений и
реакциям в ряду терпенов.
РЕАКЦИЯ ДЕЛЕПИНА
Реакцией Делепина называется метод получения первичных
аминов из бензил- или алкилгалоидпроизводных кислотным
гидролизом их четвертичных солей г гексаметилентетрамином\ При
этом наряду с галоидными солями первичных аминов образуются
также формальдегид и соли аммения:
ЗНС1 + 6НдО -^ RCH.NHgX + 6CH.0
Если гидролиз протекает в с шртовом растворе, то одним из
продуктов реакции является ацеталь формальдегида.
Галат и Элион- разработали упрощенную методику
проведения реакции Делепина, являющуюся прекрасным общим способом
получения первичных аминов. Авторы используют в качестве
растворителя спирт и в случае применения хлор- или бромпро-
изводных добавляют эквивалентное количество йодистого натрия.
Образовавшиеся четвертичные соли выделяют из реакционной
смеси и непосредственно гидролизуют газообразным хлористым
водородом.
Для проведения реакции с некоторыми дигалоидпроизводны-
ми было предложено^ использовать метиловый спирт, при этом
на аминогруппу замещается только один атом галоида. Таким
способом из 1,3-Дибромпропана был получен 3-бромпропиламин.
Греймор и Дэвис* разработали другую модификацию реакции
Делепина, заключающуюся в том, что четвертичные соли гекса-
115 Делепин
метилентетр амина выделяют, а затем гидролизуют при нагревании
с избытком концентрированной соляной кислоты.
Образовавшийся формальдегид отгоняют с водяным паром. По этому методу
из |3-бромпропионовой кислоты был синтезирован (3-аланин*.
1. М. D е 1 ё р i n e, Bull. Soc. chim. Paris, 13, 355 (1895); 17, 290 (1897); С. г.,
120, 501 (1895); 124, 292 (1897).
2. A. Ga I a t, G. E I ion, J. Am. Chem. Soc, 61, 3585 (1939).
3. L. Amundsen, A. P u I i t о, Доклад на 118 собрании Американского
химического общества, Чикаго, 1950.
4. J. Graymore, D. D a v i es, J. Chem. Soc., 1945, 293.
5. N. W e n d e r, J. Am. Chem. Soc, 71, 375 (1959).
Дикман 116
Вальтер Дикман (Walter Dieckmann, 1669—1925) родился в Гамбурге.
Изучал химию в Гейдельберге, в Техническом институте в Шарлоттенбурге
и в Мюнхене у Е. Бамбергера. В 1892 г. получил степень доктора философии за
исследование по химии тетрагидроизохинолинов. Был ассистентом А. Байера.
С 1898 г. преподавал в Мюнхенском университете, работал в Баварской
академии наук. Скончался во время работы в химической лаборатории. Открытая
им реакция циклизации, приводящая к образованию циклических р-кетоэфиров,
находит применение во многих областях органической химии. Опубликовал
также много работ по реакциям конденсации ацетоуксусного эфира и по
вопросам десмотропии.
РЕАКЦИЯ ДИКМАНА
Реакция Дикмана* заключается во внутримолекулярной
конденсации сложных эфиров двухосновных кислот в
присутствии основных катализаторов; при этом получаются (3-кетокис-
лоты.
Типичным примером реакции Дикмана является синтез 2-карб-
этоксициклопентанона из диэтилового эфир а адипиновой
кислоты:
сн* со
/\ /\
Н,С СООС,Нд NaOCA НдС CH-COOQHg
Н,С СООС,Нд Ндр CHg
\ /
сн.
Было показано^, что гидрид натрия служит очень хорошим
конденсирующим агентом для этой сложноэфнрной конденсации*.
Получаемые эфиры Ц-кетокарбоновых кислот легки гидролизуют-
ся и дскарбоксилируются с образованием циклических кетонов.
Реакция циклизации по Дикману представляет собой
частный случай конденсации Клайзена (см. стр. 139) и протекает
по сходному механизму. В результате изучения этого механизма
методом меченых атомов выяснилось, что стадией, определяют ей
* Выбор конденсирующего агента является строго специфическим и для
каждого случая требует экспериментальной проверки.—У7р%л*.
117
скорость реакции, является образование углерод-углеродной
связи при взаимодействии карбонильной группы с отрицательно
заряженным углеродным атомом второй д-метиленовой группы*
*:
сь
Na
Чакраварти* исследовал влияние алкильных групп (в исход-
ных эфирах дикарбоновых кислот) на процесс циклизации по
Дикману. В тех случаях, когда в результате циклизации
возможно образование двух изомеров, пространственные затруднения,
вызываемые алкильными группами, могут привести к понижению
активности одной из метиленовых групп и получается
преимущественно один из изомеров. Так, например, из этилового эфира
Р,р-диметиладипиновой кислоты образуется только 2-карбэтокси-
4,4-диметилциклопентанон*:
СН,—CHg-СООСдН
CHg/ ^CH,-COOQHg
дНд
СН
сн
с
сн,—со
По методу Дикмана, кроме эфиров алициклических
кислот, могут быть получены также эфиры (3-кетокарбоновых
кислот гетероциклического ряда. Примером синтеза азотсодержащей
гетероциклической системы* является образование 1-метил-4-карб-
этоксипиперидсна-3 (Н) из диэфира I:
—СН,—СН%—
з—N
N
CH,—СН,
\сн„-со
II
По методу Дикмана синтезированы также некоторые
производные тетрагидротисфена*. Исходя и? а,^-дикарбметоксиметил-
этилсульфида (III), были получены два изомерных циклических
Р-кетоэфира IV и V, причем, если реакцию проводить при 80°,
основным продуктом является [3-кетоэфир IV, а при более низкой
* Вопрос о направлении циклизации эфиров несимметричных
дикарбонокислот до настоящего времени полностью не выяснен.—/7р%,%. б
Дикман 118
температуре (в эфирном растворе) преобладает изомерный 3-кето-
2-карбметокситетрагидротиофен (V):
соосн,
I
Н^С COOCHg ^ HgCOOC-i =0 , | j=O
Н.С СН, ~" W W
\/ s
Ш IV
1. W. Dieckmann, Вег., 27, 102, 963 (1894); Вег., 33, 2670 (1900).
2. D. В а п е г j e e, P. Shafer, J. Am. Chem. Soc, 72, 1931 (1950);
W. Johnson, A. Jones, W. Schneider, J. Am. Chem.
Soc, 72, 2395 (1950).
3. W. Carrick, A. Fry, J. Am. Chem. Soc, 77, 4381 (1955).
4. R. С h а к г a v а г t i, J. Chem. So:., 1947, 1028.
5. E. Prill, S. M с E 1 v a i n, J. Am. Chem. Soc, 55, 1233 (1933).
6. Д. Вольф, К. Фолькерс, Органические реакции, сб. 6, Издатинлит,
1953, стр. 385.
% 9 Дильс—Альцер
Отто Дильс (Otto Diels, 1876—1954) родился в Гамбурге. В 1899 г. получил
докторскую степень в Берлине, где работал ассистентом Э. Фишера. В 1914 г.
стал адъюнкт-профессором. С 1916 г. декан химического отделения Кильского
университета. Большое внимание уделял химии стероидов. Показал, что при
дегидрировании стер и нов образуется углеводород С^Н^. Занимался также
синтезом и изучением строения кантаридина. В 1906 г. открыл недокись
углерода СзОз. В 1927 г. вместе со своим студентом К. Аль дером приступил к работе
над диеновым синтезом. За большие достижения в области химии сложных
органических соединений в 1950 г. ему была присуждена Нобелевская премия.
*
Курт Альдер (Kurt Alder, 1902—1958) родился в г. Кенигсхютте
(Германия). Учился в Берлине, затем у О. Дильса в Кильском университете, где в
1926 г. получил степень доктора философии. Преподавал там же, а с 1934 г.
профессор этого университета. В дальнейшем в течение нескольких лет был
уководителем исследовательского отдела на заводе красителей А. Байера.
В 1940 г. возглавил химический институт в Кельне. С 1951 г. декан химического
факультета Марбургского университета. Особую известность приобрел, работая
с О. Дильсом над диеновым синтезом. За этот труд в 1950 г. удостоен Нобелев-
с кой премии. Занимался также проблемами_стереохимии, аутоокисления и по
лимеризации органических соединений.
РЕАКЦИЯ ДИЛЬСА—АЛЬДЕРА
В 1928 г. Дильс и Альдер! установили, что бутадиен энергично
взаимодействует с малеиновым ангидридом, причем с
количественным выходом образуется шестичленный цикл—получается
ангидрид цис-1,2,3.6-тетрагидрофталевой кислоты:
HCf
1
НС
сн,
сн.
ч-
. со
/\
НС
II
НС
со
О -
/
4
Конденсация заключается в 1,4-присоединении этиленовой
системы к диену с образованием в продукте присоединения
(аддукте) двойной связи в положении 2,3.
Дильс—Альдер
120
Эта реакция конъюгированных диенов с соединениями,
содержащими этиленовые или ацетиленовые связи, обычно
активированные группами \(Х), —СООН, —CN или—N0%, является
весьма общей и имеет широкое применение*. В ней могут
использоваться многочисленные соединения с активированными двойными
и тройными связями (так называемые диенофилы) и
разнообразные диены.
Ацетиленовые диенофилы, такие, как ацетилендикарбоновая
кислота, реагируют с бутадиеном, образуя 3,6-дигидрофталевую
кислоту:
СН, СООН
СООН
НС
с
COOI
СН,
СООН
Реакция Дильса—Альдера позволила объяснить также
различные процессы полимеризации непредельных соединений,
поскольку диены могут вести себя и как диенофилы.
Во многих случаях реакция протекает уже при смешении
эквимолекулярных количеств диена и диенофила при комнатной
температуре. Для снижения скорости взаимодействия реакцию
можно проводить в инертных растворителях типа бензола или
эфира. Для мало активных диенофилов условия реакции могут
быть более жесткими. Выходы аддуктов по реакции Дильса—
Альдера обычно хорошие.
При проведении реакции в среде нитробензола возможно
дегидрирование гидроароматических аддуктов с образованием
ароматических соединений. В этих случаях реакция является
необратимой. Например, 1,4-дифенилбутадиен, реагируя с 1,4-нафтохи-
ноном в среде горячего нитробензола, дает 1Л-дифенилантрахи-
с выходом 70%:
с*н
гн,с, о
н.с
Н.С,
По реакции Дильса—Альдера можно получить ряд
«необычных» Костиковых структур. Одним из примеров такого синтеза
121
Дильс—Альдер
является присоединение 2,3-дигидротиофен-1-диоксида к цикло-
^:
пентадиену^:
V
140—150*
СНа
\/
О О
Взаимодействием 2-нитропропена и 2-нитробутеиа-2 с цикло-
пентадиеном получены третичные нитросоединения^, например:
Виттигом^ описано взаимодействие по Дильсу—Альдеру о-фтор-
бромбензола с фураном в присутствии амальгамы лития:
Li
С4Н4О
о.
Предполагается, что диенофилом служит промежуточно
образующийся дегидробензол.
Вудворд и Катц? недавно предложили несимметричный
механизм реакции Дильса—Альдера. Они считают, что реакция
протекает в две стадии, т. е. каждая из связей, соединяющих диен с
диенофилом, образуется самостоятельно. Высказано также
предположение*, что реакция имеет одностадийный механизм.
1. О. Di els, К. Alder, Ann., 460, 98(1928).
2. О реакции Дильса—Альдера см. М. Клетцел ь, Органические
реакции, сб. 4, Иэдатинлит, 1951, стр. 7; Г. Холмс, там же, стр. 86;
Л. Б у т ц, Р. Р и т и н а, Органические реакции, сб. 5, Издатинлит,
1951, стр. 93; [о стереохимии реакции Дильса—Альдера см. J. Martin,
R. Hill, Chem. Rev., 61, № 6, 537 (1961).—Дом. реЗ.]
3. Е. Bergman n, L. Haskelberg, F. Bergman n, J. Org.
Chem., 7, 303 (1942).
4. K. Alder, H. R i ckert, E. Wi n d emu t h, Ber., 71, 2451 (1938).
5 W. N о 1 a n d, R. В a m b u г у, J. Am. Chem. Soc, 77, 6386 (1957).
6 G. W i t t i g, Angew. Chem., 69, 245 (1957).
Л R. Woodward, Т. К a t z, Tetrahedron, 5, 70 (1959).
8. M. D e w a r, Tetrahedron Letters No., 4, 16 (1959); см. также R. Wood
wand, Т. К a t z, Tetrahedron Letters No., 5, 19(1959).
Дэкин 122
Генри Драйздол Дэкин (Henry Drysdale Dakin, 1880—1952) родился в
Лондоне. Изучал химию у Ю. Коэна в университете в Лидсе, где в 1907 г.
получил степень доктора философии. Исследовательскую работу начал в Листе-
ровском институт профилактической медицины. Во время первой мировой
войны уехал во Францию, где открыл возможность применения раствора гипохло-
рита (раствор Дэкина) для обработки ран. Продолжал исследования в Нью-
Йорке, работая консультантом в институте терапевтических исследований
Мерка. Один из крупнейших ученых-биохимиков. Открыл многие ферменты. Его
исследования действия ферментов привели к развитию понятия ферментно-
субстратного соединения. Известен также работами по метаболизму. Он
показал, что аминокислоты дезаминируются и декарбоксилируются перекисью
водорода, получил экспериментальное доказательство р-окисления жирных
кислот, предположенного Кнупом. В 1928 г. совместно с Р. Вестом (врач-
клиницист) синтезировал а-ациламинокетоны действием уксусного ангидрида
на пиридин и «-аминокислоты. За свои труды удостоен многих наград, в
частности Королевское общество присудило ему медаль Дэви. Ему присвоены
почетные степени Иельским, Лидским и Гейдельбергским университетами.
РЕАКЦИЯ ДЭКИНА
При обработке щелочного раствора о- или л-оксибензальде-
гида (или соответствующею оксикетона; разбавленной перекисью
водорода образуются многоатомные фенолы*. Таким путем,
например, л-оксибензальдегид превращается в гидрохинон:
СНО
i
НдО% %^\
НСООН
Этот метод окисления оксикарбонильных соединений
получил название реакции Дэкина, которая широко применяется
для получения многоатомных фенолов из встречающихся в
природе ароматических оксиальдегидов.
123 Дэкин
Окисление о-ванилина разбавленной перекисью водорода (в
атмосфере инертного газа) приводит к 1-монометиловому эфиру
пирогаллола* (выход 80%):
Примером реакции Дэкина с использованием кетона^ является
образование 3,4-Диоксифенилацетонитрила (II) из 5-цианометил-
2-оксиацетофенона (I):
Окислением 2-окси-3,4-ДИметилацетофенона перекисью
водорода в водном растворе гидроокиси тетраметиламмония
получен^ 3,4-диметилпирокатехин (III) с выходом 25%:
ОН
-к
СИ,
Ш
В водном растворе едкого кали выход фенола III всего 2,5%,
а в тритоне В (гидроокись бензилтриметиламмония) выход
достигает 18%.
1. Н. D a k i n, Am. Chem. J., 42, 477 (1909).
2. А. С е р р е и, Синтезы органических препаратов, сб. 4, Издатинлит, 1953,
стр. 340.
3. R. Т г a v e, Gaz., 80, 502 (1950); С. А., 45, 7047 (1951).
4. W. Baker, Н. В о n d у, J. Gump, D. Miles, J. Chem. Soc,
1953, 1615.
Дэкин—Вест 124
РЕАКЦИЯ ДЭКИНА—ВЕСТА
Реакция а-аминокислот с уксусным ангидридом в присутствии
пиридина, приводящая к образованию а-ацетаминоалкилметил-
кетонов, была предложена Дэкином и Вестом^ в 1928 г.
Примером реакции может служить получение 1-фенил-2-ацет-
аминобутанона-3 из фенилаланина:
СдНд—СНд—СН-СООН
| ^ (СНзСО)дО -^
СдНд—СНд-СН—СО—СНд
-» I ф COg 4- Н,0
NH—GO—CH,
В качестве основания в этой реакции применяется безводный
уксуснокислый натрий*.
Было показано*, что другие ангидриды (пропионовый,
масляный или бензойный) также могут с успехом использоваться в
реакции Дэкина—Веста.
Например, пропионовый ангидрид с фенилаланином дает
1-фенил-2-пропиониламинопентанон-3:
с,н*—сн,—сн—соон
| ф (С
СдНв—СНд-СН—СО—СНа-
I
NH—СО—СН,—д
Вероятно, реакция Дэкина—Веста протекает через стадии
образования и ацилирования оксазолонов^-з, аналогично описанной
125 Дэкин—Вест
ниже реакции Эрленмейера—Плехля (стр. 285):
R—СН—СООН (B'cohO R—CH—COOH R_CH—CO (R'CO)%o
NH, NH—COR' -НдО х О
COR' COR'
R—С CO R-C-COOH R—CH—COR'
NO N OH -^* NH—COR'
R' R'
1. H. Dakin, R. West, J.Biol.Chem., 78, 91, 745, 757(1928).
2. G. Cleland, С Niemann, J. Am. Chem. Soc, 71, 841 (1949)
3. J. Comfort h, D. Elliot, Science, 112, 534 (1950).
Зандмейер 126
Траугот Зандмейер (Traugott Sandmeyer, 1854—1922) родился в Веттин-
гене (Швейцария). Ассистент В. Мейера в Цюрихе (с 1882 г.) и Геттингене-
(с 1885 г.), затем ассистент А. Ганча. В 1888 г. поступил в фирму J. R. Geigy,
в которой работал 31 год. В 1891 г. ГеЙдельбергский университет присудил
ему степень почетного доктора философии. Известен своими многочисленными
исследованиями в области синтеза красителей, особенно трифенилметановых;
некоторые из разработанных им методов применяются и в настоящее время.
Открыл и изучил реакцию, носящую его имя. Предложил новый способ
получения изатина с количественным выходом при взаимодействии амина с хлоралем
и гидроксиламином!. Хотя обычно считают, что тиофен открыт В. Мейером,
первый, кто высказал предположение, что в техническом бензоле присутствует
какое-то вещество, дающее изатиновую реакцию с концентрированной серной
кислотой, был его ассистент Т. Зандмейер.
РЕАКЦИЯ ЗАНДМЕЙЕРА
В 1884 г. Зандмейер* установил, что ароматические соли ди-
азония легко разлагаются при нагревании их с однохлористой
мрдью в присутствии соляной кислоты и превращаются при этом
в ароматические хлорпроизводные:
C.H.NH,
Реакцию Зандмейера* обычно проводят, добавляя холодный
раствор соли диазония к солянокислому ре створ у однохлористой
меди (1 моль CugClg на 1 моль амина) с последующим нагреванием.
С бромистыми или цианистыми солями одновалентной меди
образуются соответственно бромистые или цианистые производные:
Cu^Brg
Реакция может быть использована для замещения диаззгруппы
на —SCN, —NOg, —SH. —N,, а также на —AsOgH/ и —F**.
* См. реакцию Барта, стр. 22.
'* См. реакцию Шимана, стр. 268.
127 ' Зандмейер
Ходжсон и Норрис* сообщили о получении хлор- и бромфено-
лов по модифицированной реакции Зандмейера. Нитрозогруппа
в |л-нитрозофеноле замещается на галоид при взаимодействии с
солью гидроксиламина в присутствии солей одновалентной меди
и соответствующей галоидоводородной кислоты:
ОН ОН
NHgOHHCl
; НС1
Активные нитрозосоединения легко выделяют азот, для
разрушения других ароматических нитрпзопроизводных требуется
нагревание.
Коудрей и Дэвис* изучили кинетику реакции Ззндмейера на
примере замещения диазогруппы галоидом и предложили новый
механизм реакции. Авторы считают, что скорость образования
хлорпроизводного прямо пропорциональна концентрации иона
диазония и растворенной однохлористон меди, т. е. реакция имеет
первый порядок. Ион OiClf является катализатором, который
реагирует с катионом ArN^, причем между атомом меди и
концевым атомом азота, вероятно, возникает координационная связь:
-^ [ArN%—
Этот комплекс легко разлагается с образованием конечного
продукта реакции ArCl:
g—CuCIJ -» 2АгС1
При такой интерпретации реакции Зандмейера становится
понятным, почему электроноакцепторные заместители ь
ароматическом ядре диазониевой соли повышают скорость реакции и
способствуют побочному образованию азосоединений*.
1. Герм. пат. 113848, 113981, 1899 г.
2. Т. Sandmeyer, Вег., 17, 1633 (1884); 23, 1880 (1890).
3. О реакции Зандмейера см. Н. Hodgson, Chem. Revs., 40, 251 (1947);
см. также W. С о w d г е у, D. D a v i e s, Quart. Rev. (London), 6,
358 (1952).
4. H. Hodgson, W. N о г r i s, J. Chem. Soc, 1949, S181.
5. W. С о w d г e y, D. D a v i es, J. Chem. Soc., 1949, 48.
6. Подробнее о механизме диазотирования первичных аминов см. Е. Н u g-
hes, С. I ngol d, J. R i d d, J. Chem. Soc, 1958, 58.
Иванов 128
Димитр Иванов (Dimiter Ivanov) родился в 1894 г. в Софии. В 192U г. окон*
чил Софийский университет. Затем учился в университете в Нанси (Франция),
где под руководством профессора Г. Вавона выполнил докторскую диссертацию,
которую защитил в 1923 г. Свою педагогическую деятельность начал в 1926 г.
в Софийском университете; с 1937 г. заведует там кафедрой органической химии.
Основными областями его исследований являются химия металлоорганических
соединений и соединений фосфора и особенно химия эфирных масел.
Член-корреспондент Болгарской академии наук, возглавляет отделение органической
химии. В 1932 г. получил звание лауреата Французской академии наук (1п-
stitut de France).
РЕАКЦИЯ ИВАНОВА
Взаимодействие магнийгалоидпроизводного соли фенилуксусной
кислоты (реактив Иванова) с карбонилсодержащими соединениями
известно под названием реакции ИвановэЧ
,—СОО№+(СНз),СН—MgQ -» QH^-CH—MgO + CgHg
COONa
C,H,—CH-MgCl ф СО -» СдНд—CN С
COONa . ^' GOOH OH^'
При карбоксилировании реактива Иванова получена фенилмало-
новая кислота^ с выходом 66%, а при взаимодействии реактива
Иванова с формальдегидом образуется троповая кислота* с
выходом 83%.
Иванов с сотр., а также Блике и другие исследователи
расширили область применения этой реакции. Нэйдено, что вместо соли
фенилуксусной кислоты для получения реактивов Иванова можно
применять соли ^-ненасыщенных алифатических кислот*. Магнин-
галоидпроизводное* и литиевое производное* фенилацетонитрила
действуют аналогично реактивам Иванова.
Недавно было установлено*, что при реакции натриевой соли
бензилсульфокислоты с изопропилмагнийхлоридом также
образуется реактив Иванова, который, конденсируясь с бензофеноноч,
Иванов
превращается в Р-окси-а,р,р-трифенилэтансульфоновую кислоту
(выход G4%):
QHb-CH—MgCl + С,Нв-СО-С,Н, -4. НО—С СН-С.Н,
! I !
SO^Na CgHg SO3H
Блике с сотр. использовали реакцию Иванова при получении
аминоспиртов, которые могут обладать физиологической
активностью. Они показали, что конденсация по методу Иванова
третичных амидов фенилуксусной кислоты с карбонилсодержащими
соединениями протекает с образованием р-оксиамидов.
Последние при восстановлении литийалюминийгидридом превращаются
в %-окситретичные амины^. Аминоспирты были получены также
взаимодействием реактивов Иванова с аминозамещенными кетонами^.
Лучшим способом приготовления реактива Иванова является
добавление натриевой соли фенилуксусной кислоты к изопро-
пилмагнийхлориду в эфире^ ^. Обработку реактивом Иванова
альдегидов и кетонов проводят так же, как и реактивом Гриньяра.
Циммерман и Трекслер^ рассматривают реактив Иванова как
полный енолят. Они установили, что с бензальдегидом этот
реактив образует преимущественно /npgo-изомер н некоторое
количество эри/лро-изомера (отношение трео- и аршл/ю-изомеров 76:24):
Ci-Ю ^ CH—CH
G OH COOH
CIMgO OMgCI
1. D. I v a n о v, A. S p a ss о v, Bull. Soc. chim. France, [4], 49, 19, 375
(1931).
2. F. В I i с k e, H. R a f f e 1 s о n, В. В a r n a, J. Am. Chem. Soc, 74,
253 (1952).
3. D. I v a n о v, G. P s h e n с h n i i, С. г., 197, 1230 (1933).
4. D. I v a no v, I. P ao u no v, С. г., 197, 923 (1933).
5. D. I v a no v, G, V a s i 1 e v, С. г. acad. bulgare Sci., 10, 53 (1957).
6. D. I v a n о v, N. M a r e k о v, Croat. Chem. Acta, 29, 347 (1959).
7. G. Dean, Dissertation Abstr., 19, 449 (1958).
8. F. В 1 i с k e, H. Z i п п es, J. Am. Chem. Soc, 77, 5168 (1955).
9. D. Ivanov, Ball. Soc chim. France, [5], 4, 682 (1937).
10. H. Zimmerman, M. T r a x 1 er, J. Am. Chem. Soc, 79, 1920 (1957).
9 A
Иоцич* 130
Живоин Ильич Иоцич (1870—1914), серб по национальности, родился в
г. Парачине (Югославия). В 1898 г. экстерном окончил Петербургский универ-
ситет. Ученик А. Е. Фаворского, работал у него лаборантом. Продолжал
исследования своего учителя в области изомеризации непредельных
углеводородов. Получил несимметричные галоидзамещенные углеводороды, которые
легко полимернзуются и окисляются свободным кислородом. Разработал методы
синтеза производных ацетилена и открыл некоторые новые ацетиленовые
производные.
РЕАКЦИЯ ИОЦИЧА
Образование производных ацетилена при взаимодействии маг-
нийгалоидацетилена (реактива Иоцича) с различными активными
органическими соединениями известно под названием реакции Ио-
цичаЧ
Реактив Иоцича получается действием ацетилена или моноза-
мещенного ацетилена на магнийгалоидалкил:
RCs=CH -ф- QH^MgBr -» RC=CMgBr ф СдН@
Ацетилен может реагировать и с двумя молекулами магнийгало-
идалкила, образуя димагнийгалоидацетилен. Изучением
кинетики взаимодействия ацетилена с магнийгалоидалкилами
установлено, что реакция протекает по схеме^:
СН=СН + RMgBr -^ CH=CMgBr ф RH
2CH=CMgBr -$-
Путем взаимодействия реактива Иоцича с галоидалкилами,
алкилсульфатами или алкилсульфокислотами получают
гомологи ацетилена. Например, я-бутилбромид с мономагнийбром-
ацетиленом дает гексин-1 (выход 60%)^:
CH=CMgBr + BrQH, -^ CH=CQH@ + MgBr,
С альдегидами, кетонами, сложными эфирами и эпоксисоеди-
нениями реакция Иоцича протекает аналогично реакции Гринья-
Биография Иоцича и описание его реакции составлены редактором.
131 ^^ ^^ Иоцич
ра (см. стр. 101), причем образуются соответствующие
производные ацетилена. С галоидами реактив Иоцича дает галоидацетиле-
ны\ а с д и цианом или с хлорцианом—-нитрилы ацетиленовых
кислот*.
1. О реакции Иоцича см. Л. Д. Бергельсон, Реакции и методы
исследования органических соединений, кн. 4, Госхимиздат, 1956, стр. 7.
2. Н. Kleinfeller, H. Lochmann, Бег., 71, 2608 (1938).
3. V. G г i g n а г d, L. Lapayre, ,Tsheou-Faki, С. г., 187, 517
(1928).
4. Ж. И. Иоцич, ЖРХО, 35, 1274 (1903).
5. V. G r i g n а г d, Н. Р е г г i с h о n, Ann. chim., (10), 5, 5 (1926);
V. Grignard, С. Coifrtot, Bull. Soc. chim. France, 1915, 226.
Канниццаро 132
Ста кисла о Канниццаро* (Stanislao Cannizzaro, 1826—1910) родился в
Палермо. Образование получил в университете—в Палермо и Пизе. С 1845 г.
работал препаратором в лаборатории профессора Р. Пириа. В 1847 г. принял
участие в национально-освободительном движении на о. Сицилия. После
подавления сицилийского восстания в 1849 г. эмигрировал во Францию, где
работал в химической лаборатории в Лиспе, а позднее у М. Шевреля в Париже.
В 1851 г. вернулся в Италию и возглавил кафедру физики, химии и механики
и Технологическом институте в Алессандрии (Пьемонт). С 1855 г. профессор
кафедры химии в Генуэзском университете, а с 1861 г.—в Палермо. С 1871 г.
н до конца жизни—декан химического факультета в Риме. В 1871 г. был избран
к Итальянский сенат, л затем стал его вице-президентом. В 1851 г. совместно
с Ф. Клоэзом открыл цианамид, получив его действием аммиака на хлористый
циан. В 1853 г. получил из бензойного альдегида бензойную кислоту и бен-
зиловый спирт (реакция Канниццаро). В 1891 г. ему была присуждена
медаль Копли за выдающееся исследование по установлению
различия в понятиях атомного и молекулярного весов. Эта работа явилась
большим вкладом в атомно-молекулярнуютеорию и вошла в историю химии.
РЕАКЦИЯ КАННИЦЦАРО
Реакцией Канниццаро^ называют
окислительно-восстановительное диспропорционирование** двух молекул ароматического
альдегида в присутствии гидроокиси натрия (калия), приводящее
к образованию соответствующих спиртов и кислот.
Примером этой реакции является превращение бензальдегида
в бензиловый спирт и бензойную кислоту:
Образование бензойной кислоты из бензальдегида при
обработке его водной щелочью впервые наблюдали еще Велер и Либих*,
* Биография Канниццаро дополнена редактором.
** Аналогичное диспропорционирование алифатических альдегидов в спирт
и кислоту впоследствии было открыто учеником А. М. Бутлерова и Д. И.
Менделеева—академиком В. Е. Тищенко (1861—1941) и называется реакцией
Тищенко.—/7/жл*. реё.
133
Канниццаро
однако только через много лет Канниццаро^ показал, что при
этом получается также и бензиловый спирт.
Широкое применение находит так называемая «перекрестная»
реакция Канниццаро, в которой исходный альдегид
восстанавливается до соответствующих спиртов под действием фсрмальде-
гида, окисляющегося в муравьиную кислоту:
NaOH
—СН^ОН -h HCOCNa
Этот вариант реакции Канниццаро используется для
восстановления большого числа различных альдегидов.
Перекрестная реакция Канниццаро оказалась наиболее
подходящим методом для синтеза е-нафтилкарбинола и 5-ыстилфурил-
карбкнола из соответствующих альдегидов*.
Реакцию Канниццаро мсжно совместить с реакцией Манвиха^
для получения 2,2-диметил-3-(4^-морфслин1-лрспгнс;;г 1 и
аналогичных ему аминоспиртов: ^'
R,NH.HC1 ф
R-
—CHg—C—СНО
CH,
CH3OH; КОН
R-
;—сн%—с—CH.o}i
Механизм реакции Канниццаро изучен Пфайлем*. Он
исследовал влияние растворителей, солей и гидроокисей различных
металлов на константу скорости реакции и показал, что слабые
основания более эффективны при диспрспсрционирсванни, чем
гидроокись натрия (калия). По его данным, лучшим катялизато-
ром является гидроокись кальция.
Пфайль предполагает, что в процессе реакции образуется
реакционный комплекс I, состоящий из двух молекул альдегида и
одной молекулы гидроокиси металла. Атом ведерода одной из
альдегидных групп мигрирует как анион Н" к углеродному атому
соседней альдегидной группы; при этом получается комплекс II,
после чего положительно заряженный атсм углерода, присоединяя
гидроксил, образует карбоксильную группу кислоты:
Г R 1
Me... ОН
L н R
1
J
Me- .-
Н—С—
LH R
R—CH,OMe
п
Канниццаро
Представление о присоединении аниона водорода (гидрид-иона)
к карбонильной группе используется также Гамметом?» * в
предложенном им механизме этой реакции.
i О реакции Канниццаро см. Т. Г е й с м а н. Органические реакции, сб. 2,
Издатинлит, 1950, стр. 106.
2. P. Wohler, J. Liebig, Ann., 3, 249 (1832).
3. S. Cannizzaro, Ann., 88, 129 (1853).
4. R. T i war i, N. Srivastava, Rec. trav. chim., 75, 254 (1956).
5. L. Cheney, J. Am. Chem. Soc., 73, 685 (1951).
5. E, Pf ei I, Ber., 84, 229 (1951).
7. L. Hammett, Physical Organic Chemistry, New York, 1940 p. 350.
см. также Е. Alexander, J. Am. Chem. Soc, 70, 2592 (1948).
6. О механизме реакции Канниццаро см. К. И н г о л ь д, Механизм реакций
н строение органических соединений, Издатинлит, 1959, стр. 563,—
Дол. d
135 Кижнер'
Николай Матвеевич Кижнер (1867—1935) родился в Москве. В 1890т. окон-
чнл Московский университет. Ученик В. Ф. Лугинина и В. В. Марковнякова,
работал в университете экстраординарным лаборантом. В 1895 г. защитил
магистерскую, а в 1900 г.—докторскую диссертацию. В 1901 г. занял кафедру
органической химии в Томском технологическом институте. В 1914—1917 гг.
преподавал в Народном университете Шанявского в Москве, а с 1918 г.
стал научным руководителем Научно-исследовательского института Анилтре-
ста (впоследствии НИОПиК). В 1929 г. избран
членом-корреспондентом АН СССР, а а 1934 г. почетным академиком. Особенно
известны исследования Н. М. Кижнера у области изомеризации циклов, а также ис
следования аминов и гидразинов, в частности носящая его имя реакция
каталитического разложения алкилиденгидразинов, позволяющая легко переходить
от карбонильных соединений к углеводородам. Разрабатывая методы получения
различных органических красителей, содействовал созданию и развитию
отечественной анилино-красочной промышленности. Труды Н. М. Кижнера были
отмечены в 1893 г. малой, а в 1914 г.—большой премией им. А. М. Бутлерова.
РЕАКЦИЯ КИЖНЕРА
Образование углеводородов при нагревании гидразонов
карбонильных соединений—алкилиденгидразинов в
присутствии катализаторов (едкое кали или едкое кали tмелко
раздробленная платина) носит название реакции КижнераЧ
Примером этой реакции может служить получение метилциклегексана
из 3-метилциклогексанона*:
CO C=N—N11, СН,
H,C СН, Н,С СНд Н,С СН
Н#С СН-СНз Н,С СН—СН^ Н.,С СН—СНд
G Н, СН, СН,
Реакция Кижнера, открытая в 1910 г., оказалась применимой
для получения не только предельных углеводородов, но и непре-
* Биография Кнжнера заноэо написана редактором, а описание реакции
переработано.
Кижнер 136
дельных, в случае если двойная связь в исходных карбонильных
соединениях не сопряжена с СО-группой^:
В дальнейшем указанная реакция была распространена также
на получение ароматических соединений (крезол из ванилина*,
этилбензол из ацетофенона*** и др.) и гетерсцкклов (сильван из
фурфурола*' в, 2-)1зтилпиррол из 2-пнррола.гьлстидаН и т. д.
Иначе протекает реакция Кижнера с непредельными
соединениями, содержащими конъюгированну^о с СО-группсй двойную
связь. Эти соединения, как правило, при взаимодействии с
гидразином дают нэ гидразоны, а пнразолиновые основания, которые
также претерпевают каталитическое разложение, превращаясь
в углеводороды, щспзводиые циклопропана. Tax, из окиси мезк-
ткла был пелучез; !,1,2-тр1шетнлцик;-г:ппопап®:
(^ С—N11 С
f
FLC
С=О С- N Ъ[
СН^ СИ., CIL
а из камферфорсна—2,6,6-триметнлбицикло-|0,1,3]-гексан^:
С С N (f
(!: GO -» ^зСус С -» НС СИ + N
I I
Ii*C CH—CHg Н%С СН—CHg HgC CH—CH3
Через полтора года после опубликования работы Н. М. Кижне-
ра появилась в печати статья Вольфа*, также посвященная ката-
* Иоганн Людвиг Вольф (Johann Ludwig Wolff, 1857—1919) родился в Ней-
штадте-на-Хаардте (Германия). Учился у Р. Фнттнга в Страсбургском
университете. В 1882 г. ему была присуждена степень доктора. В 1891 г. занял
должность профессора в Йенском университете на кафедре аналитической .химии,
возглавляемой Л- Кнорром. 1{роме изучения перегруппировки дназокетонов
(перегруппировка Вольфа) и восстановления гндразонов карбонильных
соединений, Вольф работал в области синтеза лактоноз, пнразинов, индолов из
р-бромлевулнновой кислоты и аминов, а также производных тетроновой
кислоты. Им был открыт первый В-лактон—о-оксикапролактон.
137 Кижнер
литическому разложению гидразонов*, однако не содержащая
ссылки на работу Кижнера. В последовавшей полемике Вольф
признал приоритет Кижнера. Несмотря на это, в иностранной
литературе до настоящего времени указанная реакция часто
называется «реакцией Кнжнера—Вольфа» и даже «реакцией
Вольфа—Кижнера».
Практическое выполнение реакции по Вольфу несколько иное,
чем по Кижнеру. В то время как по Кижнер у обычно гидра-
зон постепенно прибавляется к нагретому катализатору и
продукт реакции сразу же отгоняется (т. е. реакция не лимитируется
размерами аппаратуры), по Елльфу реакция проводится
исключительно в запаянных трубка\ и катализатором служит гтилзт
натрия.
Позднее были разработаны различные модификации реакции
Кижнера. В частности, была показана возможность
использования гликолем и высококипяших спиртов п качестве среды для
проведения реакции^, а также применения метилата натрия в три-
этиленгликоле^. Хуанг-Минлои^' предложил проводить реакцию
в среде ди- и триэтиленгллколя о присутствии едкого кали или
едкого натра. Этим методом л-феноксибензоилпропноиовзя
кислота (I) была превращена в %-феноксифенилмасляную кислоту
(II) с выходом 95%:
О—(^_\—СОСИ:
При восстановлении кетокислоты I по Клеммснсену (стр. 147)
кислота II получается только с выходом 54%. Преимущество
метода Кижнера перед методом Клемменсенг особенно наглядно
видно в случае восстановления карбонильных соединений,
неустойчивых в кислой среде или дающих, как, например, %-аминокето-
ны, при восстановлении по Клемменсену аномальные продукты.
При изучении кинетики реакции Кижнера, для нее был
предложен^ следующий ионный механизм. Сначала образуется
анион III, который при перемещении атома водорода и
одновременном выделении молекулы азота превращается з карбанион IV:
ш
+
IV
+ [ВН]+ -» R*CH,+ В
Кижнер 138
Вайсбергер и Грентам^ считают, что восстановление флуоре-
нона методом Кижнера протекает по радикальному механизму.
Для получения с хорошим выходом флуорена требуется
шестикратный избыток гидразина.
1. О реакции Кижнер а см. В. М. Р о д и о н о в, Н. Г. Ярцева,
Реакции и методы исследования органических соединений, кн. 1, Госхим-
издат, 1951, стр. 7; D. Т о d d, Organic Reactions, v. 4, New York, 1948,
p. 378.
2. H. M. Кнжнер, ЖРХО, 43, 582, 586 (1911).
3. H. M. Кижнер, ЖРХО, 45, 979 (1913).
4. J. Wolff, Ann., 394, 86 (1912).
5. G. Lock, K. S t а с h, Ber., 77, 293 (1944); A. Schmidt,
G. Hopp, V. Schoeller, Ber., 72, 1893(1939).
6. H. M. Кижнер, ЖРХО, 43. 1563 (1911); H. Д. Зелинский,
H. И. Ш у й к и н, ДАН СССР, № 2, 60 (1933).
7. L. Кпогг, К. Hess, Вег.,44,2758(1911);45, 2631 (1912).
8. Н. М. Кижнер, ЖРХО, 44, 165 (1912); 45, 1770 (1913).
9. Н. М. Кижнер, ЖРХО, 44, 849 (1912).
10. М. D. S о f f е г, М. В. S о f f ег, K.S k е г к, J. Am. Chem. Soc., 67, 1435
(1945).
И. С. Her r, F. Whitmor, R. Schiessler, J. Am. Chem. Soc,
67, 2061 (1945).
12. Huang-Min Ion, J. Am. Chem. Soc, 68, 2487 (1946).
13. H. S z m a n t, H. Harnsberger, T. Butler, W. В а г i e,
J. Am. Chem. Soc, 74, 2724 (1952).
14. J. Weisburger, P. G г a n t h a m. J. Org. Chem., 21, 1160 (1956).
139 Клайзен
РаЙнер Людвиг Клайзен Rainer Ludwig Claisen, 1851—1930) родился и
Кельне. Учился у А. Кекуле а Боннском университете н некоторое время
работал в лаборатории Ф. Велера в Геттннгене. Затем вернулся в Бонн, где
получил докторскую степень и стал ассистентом А. Кекуле. В дальнейшем около
четырех лет работал в Манчестерском колледже Оуенса, а с 1886 г. в Мюнхене
под руководством А. Байера. Профессор химии Высшего технического
училища в Аахене (с 1890 г.) и университетов в Киле (с 1897 г.) и в Берлине (с 1904 г.),
где сотрудничал с Э. Фишером. Работы Клайзена относятся к самым различным
областям органической химии. Из наиболее известных его трудов можно
назвать ацилнрование карбонильных соединений, перегруппировку аллиловых
ефиров фенолов в соответствующие аллилзамещенные фенолы, получение ко -
рнчной кислоты, синтез производных пиразола и изоксазола, приготовление
экснчетиленовых производных ацетоуксусного и малонового эфнров.
КОНДЕНСАЦИЯ КЛАЙЗЕНА
Конденсация сложных эфиров с соединениями, содержащими
активную метиленовую группу (сложные эфиры, альдегиды, ке-
тоны, нитрилы), протекающая в присутствии основных
катализаторов, получила наименование конденсации Клайзена. Эту
реакцию называют также ацилированием по Клайзену, поскольку ее
можно рассматривать как ацилнрование соединений, имеющих
активную метиленовую группу.
Классическим примером^ реакции является образование
ацетоуксусного эфира при конденсации двух молекул этил ацетата
мод действием этилата натрия:
2СН,—С-ОС.Н, » СНд—С—СНа—
II II
О О
Наиболее вероятен следующий механизм конденсации:
1. Образование аниона сложного эфира при отщеплении
прогона от активированной метильной группы:
ф В ^ СН,-С-ОС,Н, ф ВН+
II II
о о
Клайзен
2. Взаимодействие аниона с карбонильной группой второго
компонента реакции (в простейшем случае—с карбонилом второй
молекулы сложного эфира):
3. Отщепление этилат-иона от образовавшегося продукта,
приводящее к образованию эфира Р-кетокислоты (в простейшем
случае—аиетоуксусного эфира):
Полученный эфир 3-кетокислоты (если он содержит %-водо-
родный атсм) может далее взаимодействовать с одним молем
алкоголята и в свою очередь переходить в енолят-ион:
С}1.—С-СНо—COGQIlB + QHGO- -^ CHg—C=CH—C
о
о-
Клайзеновская конденсация нитрила фенилуксусной кислоты
с этилацетатом или с этилоксалатом под действием этилата
натрия приводит соответственно к образованию сс-фенилацетил-
ацетонитрила (1)^ и этилового эфира фенилцианпировиноградной
кислоты (II)®:
+ CH,—COOQHs
CH,ONa
СН-ШСН
NC
/
I
(выход 73%)
COOQH,
CH
COOQH
Hg
CH-C—COOQHg + СдЪ
II
II
(выход 75%)
В большинстве случаев при конденсации Клайзена в качестве
катализаторов используются металлический натрий, алкоголят
натрия или амид натрия. Позднее Суомер и Хайзер* исследовали
возможность применения гидрида натрия для проведения реак-
141 Клайзен
ции ацилирования и карбэтоксилирования по Клайзену. Авторы
показали, что по эффективности этот катализатор не уступает
другим конденсирующим агентам, а во многих случаях даже
превосходит их. При ацилировании кетонов сложными эфирами
применение гидрида натрия дает такие же хорошие результаты, как
использование амида натрия; выход продуктов
конденсации значительно выше, чем в присутствии металлического натрия
или этилата натрия. Лауроилацетон (из ацетона и этиллаурата)
был получен авторами с выходом 83%:
NaH
СП,—С-ШзфСиНю-СООСА » CIL—С—aiz-COCi;H=x
6 6
При самоконденсации сложных эфиров гидрид натрия
считается лучшим из всех конденсирующих агентов.
Другим синтезом, часто также называемым реакцией Клайзе-
на, является модификация реакции Перкина (стр. 201),
применяемая для получения коричных ф-арил акриловых) кислот
в присутствии металлического натрия как катализатора*.
Иллюстрацией этой реакции служит образование этилового
эфира коричной (3-фенилакриловой) кислоты из бензальдегида
и этил ацетата при действии металлического натрия и следов
спирта:
Na
дН^
1. О конденсации Клайзена см. Ч. X а й з е р, Б. X а д с о н, Органические
реакции, сб. 1, Издатинлит, 1948, стр. 345; [К. И н г о л ь д, Механизм
реакций и строение органических соединений, Издатинлит, 1959,
стр. 632.—Дол. ped.].
2. P. Julian, J. Oliver, R. К i m b a 1 1, A. Pike, G.
Jefferson, Синтезы органических препаратов, сб. 2, Издатинлит, 1949, стр. 503.
3. Р. Адаме, Г. Кальвери, Синтезы органических препаратов, сб. 2,
Издатинлит, 1949, стр. 600.
4. F. S w a m е г, С. Mauser, J. Am. Chem. Soc, 72, 1352 (1950).
5. Д. Джонсон, Органические реакции, сб. 1, Издатинлит, 1948, стр. 295.
Клайзен 142
ПЕРЕГРУППИРОВКА КЛАЙЗЕНА
Миграция алл ильного (или замещенного алл ильного)
радикала в алл иловых эфирах фенолов или енолов, приводящая к
образованию С-алл ильных производных, получила наименование
перегруппировки Клайзена*.
Примерами такой перегруппировки, протекающей при
нагревании алл иловых эфиров, могут быть следующие реакции:
—GOOQH, СНд—С—СН—4
-^ * II I
CH=CH, О СИ,—СН=СНа
II
ОН
^Ан_
IV
Клайзен* получил С-алл илацетоуксусный эфир (II) перегои
кой этилового эфира О-аллилацетоуксусной кислоты (I) под
атмосферным давлением при действии хлористого алюминия. При
перегруппировке алл иловых эфиров фенолов присутствие
катализаторов не является обязательным.
В образовании новой С—С-связи участвует %-углеродный атом
алл ильной группы. Так, в системе V у-углерод присоединяется
к атому углерода ароматического ядра (Уд), а двойная связь
перемещается при этом из р у-положения в аф-лоложение (Vb):
ИЗ Клайзе*
Перемещение двойной связи в аллильном радикале было
показано* на примере перегруппировки фенил-у-фенил алл илового
эфира (III) в о-(з-фенилаллил)-фенол (IV), а также при
перегруппировке л-крез ил алл илового эфира^, содержащего в у-положенин.
аллильного радикала меченый атом С**:
Сохранение оптической активности в процессе
перегруппировки Клайзена наблюдалось в случае оптически активного 3,%-ди-
метилаллилофенилового эфира*:
Обычно алл ильная группа мигрирует в орто-положение к ок-
сигруппе. Если же оба орто-положения заняты заместителями,
перегруппировка идет в пара-положение. При ультрафиолетовом
облучении раствора аллилфенилового эфира в изопропиловом
спирте перегруппировка также протекает с образованием пара-
производных*
В некоторых случаях перегруппировка Клайзена может
сопровождаться вытеснением о-заместителя; легче всего вытесняется
из орто-положения карбоксильная группа (R=—СН^СС)
Перегруппировка Клайзена оказалась особенно применимой
в синтезах некоторых встречающихся в природе эфирных масел.
При нагревании продуктов перегруппировки с сильными
щелочами двойная связь в аллильном радикале может перемещаться
и вступать в сопряжение с ароматическим ядром.
Клайзен 144
Фенил ал л иловый эфир реагирует при —80° с хлористым бором,
давая соединение, которое после гидролиза превращается
исключительно в о-аллилфенол\
Изучение механизма пара-перегруппировки Клаизена
показало, что промежуточными продуктами этой реакции являются
аллилциклогексадиеноны*. Взаимодействием бромистого аллила
с натриевой солью 2,6-диметилфенола в бензоле Кертин и Кроу-
фордо получили 6-аллил-2,6-диметнлциклогексадиен-2,4-он.
Последний перегруппировывается при температуре выше 70° с
образованием смеси 2,6-диметилфенилаллилового эфира и 4-аллил-
2,6-диметилфенола:
0 ОСИ,—CbI=CtI
1 СН,-СН=СН, Нд I
1. О перегруппировке Клаизена см. Д, Т а р б ел л, Органические
реакции, сб. 2, Издатинлит, 1950, стр. 7; |_К. И н г о л ь д, Механизм
реакций и строение органических соединений, Издатинлит, 1959, стр. 480.—
Доя. ped.].
2. L. Claisen, Ber., 45, 3157(1912).
3. L. Claisen, E. Tietze, Ber., 58, 275 (1925).
4. Н. S с h m i d, K. S с h m i d, Helv. chim. acta, 35, 1879 (1952).
5. E. Alexander, R. К 1 u i b e r, J. Am. Chem. Soc, 73, 4304 (1951).
6. M. Kharasch, G. Stamp a, W. Nudenberg, Science, П6,
309 (1952).
7. W. G e г г а г d, M. L a p p e г t, H. Silver, Proc. Chem. Soc, 19
(1957).
8. D. Cur t i n, H. Johnson, J. Am. Chem. Soc, 78, 2611 (1956).
9. D. Curt in, R. Crawford, J. Am. Chem. Soc, 79, 3156(1957).
145 Клайзен—Шмидт
КОНДЕНСАЦИЯ КЛАЙЗЕНА—ШМИДТА
Еще одной реакцией, связанной с именем Клайзена, является
так называемая конденсация Клайзена—Шмидта.
Реакция состоит во взаимодействии ароматических альдегидов
с алифатическими альдегидами или кетонами; в результате
конденсации в присутствии водных щелочей образуются
а,|3-ненасыщенные альдегиды или кетоны. По этому методу из бензальдегида и
ацетофенона получается бензилиденацетофенон* (I) с выходом
85%%
С,Н,—СНО + СН,—CO—CgH, -» СеН,-СН=СН—СО--СдНв
—HgO
I
Продукт I называют также халконом; его производные были
широко изучены для установления связи их строения с
антибактериальной активностью. Антибиотическое действие халконов
обусловлено их ненасыщенностью, так как гидрирование двойной
связи приводит к полной потере активности.
Недавно для терапевтических испытаний были предложены*
также некоторые производные кумарона. Например, 2-(4'-бром-
бензоил)-5-бромкумарон (II) был синтезирован из 5-бромсалици-
лового альдегида и 4-бромфенацилбромида в спиртовом растворе
едкого кали:
СвНвОН
кон
Джонсон^ использовал конденсацию Клайзена—Шмидта как
одну из стадий метода введения ангулярной метильной группы.
Вначале для защиты метнленовой группы декалона-1 получают
Ю А. Серрей
Клайзен—Шмидт 146
его бензальпроизводное, затем проводят метилирование и,
наконец, удаляют СдНдСН-группу:
О Н*С О НдС О
^3
СА-сж /VV="-CA ^A/H-Vi.
^;
Образование Р-нитростиролов при взаимодействии нитроал-
канов с ароматическими альдегидами в присутствии щелочей
также можно отнести к конденсации Клайзена—Шмидта.
Нитростиролы применяются как промежуточные продукты в
синтезе феяилэтиламинов, многие из которых обладают
фармакологической активностью и в то же время могут служить
полупродуктами в синтезе производных изохинолина.
Примером получения замещенного фенилэтиламина является
синтез |3-(3-метокси-4-бензилоксифенил)-этиламина* (iy) через
замещенный нитростирол (III), легко восстанавливаемый
Н@СО\ ,\.СНО Н,СО, ^ ,СН=СН—N0,
СН=СН-Ж)з,.„„ HgCO^ ^^ ^CH^-CH.-NH,
HI IV
Недавно Дэви и Тайви\ исследуя оптимальные условия
образования халконов, показали, что едкий натр и метилат натрия
являются более эффективными конденсирующими средствами, чем
хлористый водород, хлорокись фосфора и трехфтористый бор.
Они изучили также реакцию присоединения цианистого водорода
к халконам и исследовали продукты гидролиза полученных ад-
дуктов.
ЛЯГЕРЛГУРЛ
I.E. Колер, X. Ч а д в е л л, Синтезы органических препаратов, сб. 1»
Издатинлнт, 1949, стр. 77.
2. Е. Schraufstatter, S. D e u t s с h, Z. Naturforsch., 4b,
276 (1949); С. А., 44, 3568 (1950).
3. W. Johnson, J. Am. Chem. Soc, 65, 1317 (1943).
4. J. F i n k e 1 s t e i n, J. Am. Chem. Soc, 73, 550 (1951).
5. W. Davey, D. Tivey, J. Chem. Soc, 1958, 1230.
147 Клемменсен
Эрих Христиан Клемменсен (Erik Christian Clemmensen, 1876—1941)
родился в г. Оденсе (Дания). Учился в Королевском политехническом институте
в Копенгагене, где получил звание магистра наук. В 1900 г. переехал в США
где до 1914 г. работал химиком в фирме Parke, Davis and Company и
одновременно изучал восстановление карбонильных соединений амальгамированным
цинком. После публикации этой работы Копенгагенский университет присудил
ему степень доктора философии. Один из основателей фирмы Commonwealth
Chemical Corporation, образованной в 1914 г. В 1923 г. эта фирма слилась с
компанией Mathieson Alkali Works, предприятия которой в 1929 г. были
куплены фирмой Monsanto Chemical Company. После этого Э. Клемменсен начал
работать в исследовательском отделе фирмы Monsanto, возглавляемом Л. Ки-
р ид сом. Здесь он занимался синтезом ал кил- и арилфосфатов и тиофосфатов.
В 1933 г. основал фирму Clemmensen Chemical Corporation в г. Ньюарке н до
конца жизни был главой этой фирмы.
ВОССТАНОВЛЕНИЕ ПО КЛЕММЕНСЕНУ
Превращение карбонильной группы в метиленовую при
действии амальгамированного цинка и соляной кислоты получило
название восстановления по Клемменсену^ %:
Метод дает возможность восстанавливать самые различные
карбонильные соединения и оказался особенно удобным для
синтеза углеводородов из жирноароматических кетонов,
образующихся по реакции Фриделя—Крафтса. Восстановление
обычно проводят при кипячении раствора карбонильного соединения
в подходящем растворителе с амальгамированным цинком и
избытком соляной кислоты. В качестве растворителей применяются
как смешивающиеся, так и не смешивающиеся с соляной кислотой
вещества*.
10*
Клемменсен - 148
Примером реакции может служить получение м-амилбензола
из фенил-м-бутилкетонав (выход 88%):
,СО—QH,
Выходы продуктов восстановления по этому методу обычно
удовлетворительные. Галоиды и двойные связи, конъюгированные
с карбонильной группой, также восстанавливаются в условиях
реакции Клемменсена. Главной побочной реакцией является
бимолекулярное восстановление.
При изучении механизма реакции Клемменсена продукты
восстановления ацетофенона были исследованы методом газовой
хроматографии*. Полученные данные показывают, что процесс
восстановления ацетофенона до этилбензола протекает через
стадию образования а-метилбензилового спирта и
1-хлорэтилбензола:
CJH,—СО-СНд -» QH,-CH—СНд г^ CgHs—CH—CHg -^ С@Н,—СНд—СН,
он а
Установлено, что в условиях реакции восстановления по
Клемменсену а-метилбензиловый спирт также превращается в
этил бензол. Другим примером восстановления спирта действием
амальгамированного цинка в соляной кислоте служит получение
октанона-4 из бутироина*:
С,Н,-СО—СН—С3Н, ——--^ CgH,-CO—CHg-
I HCI
он
Интересной реакцией®, одна из стадий которой состоит в
восстановлении по Клемменсену, является расширение цикла 1-ме-
тил-2-пропионилпирролидина (I), приводящее к образованию
2-этил-1-метилпиперидина (П):
п
со—сн.^
сн,
II
Дэй и Линстед? описали молекулярную перегруппировку,
протекающую в процессе восстановления 1,1 -диметилциклогексан-
149 Клемменсен
диона-3,5; результатом перегруппировки является сокращение
шестичленного цикла до пятичленного:
О
Обстоятельное исследование перегруппировки а-аминокето
нов в процессе их восстановления по Клемменсену было прове
дено Леонардом*.
I.E. Clemmensen, Бег., 46, 1837 (1913); 47, 51 (1914).
2. О реакции Клемкенсена см. Э. Мартин, Органические реакции, сб. I,
Издатинлнт, 1948, стр. 194.
3. Н. F a h i m, A. Mustafa, J. Chem. Soc., 1949, 519.
4. М. Р о u t s m a, E. W о I t h i u s, J. Org. Chem., 24, 875 (1959); см.
также D. Staschewski, Angew. Chem., 71, 726 (1959).
5. W. S m i t h, J. Am. Chem. Soc., 73, 1883 (1951).
6. G. Cl emo, J. Vipon d, Chem. Ind. (London), 856 (1949); G. С 1 с mo,
R. R a p e r, H. V i p on d, J. Chem. Soc, 1949, 2095.
7. A. D ey, R. L i ns t ea d, J. Chem. Soc., 1935, 1063.
8. N. Leonard, E. N i с h о ] a i d es, J. Am. Chem. Soc., 73, 5210 (1951).
Кневенагель 150
Эмиль Кневенагель (Emil Knoevenagel, 1865—1921) родился в Ганновере
-(Германия). Учился в Ганноверском техническом институте, а с 1886 г. в Гет-
тингенском университете у В. Мейера и Л. Гаттермана. В 1889 г. получил
степень доктора философии и начал работать в Гейдельбергском университете.
С 1896 г. доцент, а с 1900 г. профессор кафедры органической химии этого
университета. Много работал в области стереоизомерии. Обстоятельно
исследовал реакцию альдегидов с ацетоуксусным эфиром в присутствии первичных
аминов. Изучал соединения пиридинового ряда и показал, что производные
пиридина могут быть получены нагреванием 1,5-дикетонов с гидроксиламином.
Занимался также проблемами неорганической и физической химии.
РЕАКЦИЯ КНЕВЕНАГЕЛЯ
Конденсацию альдегидов или кетонов в присутствии
оснований с соединениями, содержащими активные метиленовые группы,
обычно называют реакцией Кневенагеля^.
Эта реакция является модификацией известной реакции Пер-
кина (стр. 201) и имеет сходный с ней механизм. Основания
(первичные или вторичные амины) отщепляют протон от активной
метиленовой группы, а образовавшийся карбанион реагирует с
карбонильной группой альдегида или кетона. Следующая схема
дает общее представление о течении реакции:
н
н сг он
н,о
он
В реакции Кневенагеля могут использоваться самые
различные соединения, содержащие активную метиленовую группу, а
также ароматические и алифатические альдегиды и кетоны.
Согласно Копу*, лучшими катализаторами конденсации алифати-
151 Кневенагель
чёских и циклических кетонов с эфирами циануксусной кислоты
являются соли аминов и аммонийные соли органических кислот.
Очень эффективен в качестве катализатора также ацетамид в
уксуснокислом растворе. Непрерывное удаление воды,
выделяющейся в процессе реакции, приводит к получению с хорошим выходом
эфиров алкилиденциануксусной кислоты. Реакция протекает по
следующему уравнению:
, + ,—COOR' » R,C=C—COOR'
CN CN
Ароматические кетоны реагируют с этиловым эфиром циан-
уксусной кислоты в присутствии уксуснокислого аммония и
уксусной кислоты как конденсирующих агентов^. При
конденсации альдегидов с этиловым эфиром циануксусной кислоты
применение пиперидина в спиртовой среде оказалось более успешным,
чем использование ацетата пиперидина или ацетамида в среде
уксусной кислоты*.
Сравнительно недавно было показано^, что уксуснокислый
аммоний, добавляемый небольшими порциями, является очень
эффективным катализатором конденсации диарилкетонов и
некоторых малоактивных (пространственно-затрудненных) кетонов
с этиловым эфиром циануксусной кислоты.
При конденсации бензофенона по этому методу выход
продукта конденсации достигает 84%, т. е. значительно выше, чем
получали ранее.
Позднее было описано успешное проведение реакций
альдегидов или кетонов с малоновым, циануксусным и ацетоуксусным
эфирами** в присутствии эквимолекулярных количеств безводного
KF. Так, в кипящем спиртовом растворе этиловый эфир бутили-
денциануксусной кислоты получен с выходом 40,9%. При
конденсации параформальдегида с диэтиловым эфиром малоновой
кислоты в эфире (4,5 часа при 20—25°) в присутствии KF
получается этиловый эфир метиленмалоновой кислоты с выходом
61%:
к?
СН,0
Побочно образуется также этиловый эфир метилендималоно-
вой кислоты (выход 20 %).
Видоизменение реакции Кневенагеля, известное под названием
с%я/жзд Дублера?, заключается в конденсации ароматических
альдегидов с малоновой кислотой в присутствии пиридина и
пиперидина; при этом получаются производные коричной кислоты.
Кневенагель 152
1. Е. Knoevenagel, Бег., 29, 172 (1896); Бег., 31, 730 (1898).
2. А. Соре, J. Am. Chem. Soc., 59, 2327 (1937).
3. А. Соре, С. Hofmann, С. Wyckoff, E. Hardenbergh,
J. Am. Chem. Soc, 63, 3452 (1941).
4. A. Cope, С Hofmann, J. Am. Chem. Soc, 63, 3456 (1941).
5. E. С г a g о e, С R ob b, J. S p r a g u e, J. Org. Chem., 15, 381 (1950).
6. H. В ab a, H. Midorikawa, S. Aoy a m a, C. A., 53, 15960 (1959);
J. Scl. Res. Inst. (Tokyo), 52, 99 (1958).
7. O. Doebner, Ber., 33, 2140 (1900).
153 Кнорр-
Людвиг Кнорр (Ludwig Knorr, 1859—1921) родился в Мюнхене. Учился
у Я. Фольгарда, Э. Фишера и Р. Бунзена, затем работал ассистентом Э. Фишера
в Мюнхене, Эрлангене и Вюрцбурге. С 1889 г. профессор химии Йенского
университета. Занимался исследованием гетероциклических азотистых
соединений и разработал процессы синтеза производных пиррола, пиразола и хино-
лина. Впервые получил важное лекарственное вещество—антипирин,
являющееся производным пиразолона. В 1889 г. синтезировал морфолин. Изучал
строение кодеина, морфина, тебаина и других алкалоидов. Известен также
работами в области кето-енольной таутомерии.
СИНТЕЗ КНОРРА
Кнорре предложил в 1884 г. универсальный,* практически
удобный метод получения производных пиррола взаимодействием
а-аминокетонов с кетонами, содержащими реакционноспособную
метиленовую группу:
R'—C HgC-R" R'—С С—R"
R-C С—R~ R—С С—R"
н
В качестве исходных продуктов реакции удобно пользоваться
изонитрозокетонами, при восстановлении которых цинком в
ледяной уксусной кислоте образуются аминокетоны, причем
выделение последних не обязательно. Особенно высокие выходы
получаются, если R и R"—ацильные или карбалкоксильные
группы. Если R" = H, то кетон не вступает в реакцию Кнорра. В этом
случае основными продуктами реакции являются производные
пиразина, образующиеся при конденсации двух молекул аминоке-
тона:
R'—C^ C-R R'-C \-R
I + ! II I
R—С C-R' R—С С—R'
Кнорр 154
Кнорр показал также, что производные пиррола образуются
при взаимодействии у-дикетонов с аммиаком или с первичными
аминами^. Примером этой реакции является взаимодействие ди-
этилового эфира диацетилянтарной кислоты с аммиаком, в
результате чего образуется 3,4-ДИкарбэтокси-2,5-диметилпиррол:
!NH
Если реакцию проводить в эфирном растворе при О °С, то можно
выделить^ промежуточный продукт I:
C=C(
X)H
Синтез Кнорра^ был использован для получения производных
пиррола, необходимых для синтеза лорфиринов. В связи с этим
Мак-Дональд* сообщил о проведенном им синтезе пиррол ов,
родственных урапорфиринам. В качестве исходных веществ для
синтеза были применены диэтиловый эфир ацетондикарбоновой
кислоты и бензиловый эфир ацетоуксусной кислоты.
Недавно появились сообщения о применении гидросульфита
^SgOJ в качестве восстановителя в синтезе пирролов по Кнор-
. Изонитрозосоедмнения восстанавливаются гидросульфитом
натрия в водном растворе при комнатной температуре. Показано
также, что этиловый эфир фенилазоуксусной кислоты можно
восстановить цинком в ледяной уксусной кислоте^.
1. L. Кпогг, Вег., 17, 1635 (1884); Ann., 236, 290 (1886).
2. L. Knorr, p. Rabe, Вег., 33, 3801(1900).
3. Обзор по методам получения пиррола см. А. Кор вин, Гетероциклические,
соединения, сб. 1, Издатинлит, 1954, стр. 219.
4. S. М а с D о п a I d, Chem. Ind., 759 (1951).
5. А. Т г е i b s, R. Schmidt, R. Zinsmeister, Chem. Вег., 90
79(1957).
155 Кольбе—Шмитт
Адольф Вильгельм Герман Кольбе (Adolph Wilhelm Hermann Kolbe, 1818—
1884) родился в г. Эллихаузен (Германия). С 1838 по 1842 г. учился в Геттин-
генском университете у Ф. В ел ера. Затем работал в Марбурге в лаборатории
Р. Бунзена. В 1851 — 1864 гг. профессор Марбургского университета, а с 1865 г.
профессор Лейпцигского университета. А. Кольбе внес большой вклад в
развитие органической химии. Совместно с Ф. Вел ером изучал реакцию хлора с
сероуглеродом, что привело в дальнейшем к открытию трихлоруксусной
кислоты, которая была превращена в уксусную кислоту при помощи
амальгамы калия (1842). Это было второе искусственно синтезированное органическое
вещество, являющееся продуктом жизнедеятельности организмов (в 1828 г.
Ф. В ел ер синтезировал мочевину). Он определил строение глицина, аланина
и молочной кислоты, получил нитрометан из хлоруксусной кислоты и подробно
исследовал электролиз жирных кислот. Синтезировал салициловую кислоту
из фенолята натрия и двуокиси углерода и занимался вопросом ее
использования в качестве антисептического средства.
РЕАКЦИЯ КОЛЬБЕ—ШМИТТА
Синтез фенолкарбоновых кислот при нагревании фенолятов
щелочных металлов с двуокисью углерода известен под названием
реакции Кольбе—ШмиттаЧ
В I860 г. Кольбе'* разработал метод получения салициловой
кислоты нагреванием фенолята натрия с двуокисью углерода
под давлением при 180—200°:
В 1885 г. Шмитт^ улучшил метод Кольбе, предложив
проводить реакцию при менее высокой температуре (125°) в течение
более длительного времени. В этих условиях весь фенолят натрия
превращается в салициловую кислоту. Введение карбоксильной
группы в орто-положение к фенольному гидроксилу обычно
протекает с хорошим выходом. В некоторых случаях образуются
л-оксикислоты*.
Кольбе—Шмитт 156
Реакцию Кольбе—Шмитта можно проводить в разных
условиях, применяя различные производные фенолов. Так, например,
Р-резорциловая кислота получается с выходом 60% при быстром
пропускании двуокиси углерода через кипящий водный раствор
калиевой соли резорцина**:
ОН
I
СО*; КНСОд %^Х
у\он
соон
л-Аминосалициловая кислота образуется из л-аминофенола*
при нагревании с твердой углекислотой и раствором бикарбоната
калия под давлением (выход 80%):
СООН
Джоунс? предложил новый метод синтеза фенолкарбоновых
кислот, основанный на применении натриевой соли этилугольной
(NaOCOOQHg) или метил угольной кислоты. К спиртовому
раствору этилата натрия добавляют твердую углекислоту; выпадает
осадок натриевой соли этилугольной кислоты. При нагревании
ее с фенолом отгоняется этиловый спирт и образуется соль
соответствующей фенолкарбоновой кислоты (применение давления не
обязательно):
^\/ОН ^ Х)Н
Г |) + NaOCOCC,H, -»
Реакция Кольбе—Шмитта была модифицирована Марассе*.
Модифицированный способ предусматривает получение калиевой
соли ароматической оксикислоты нагреванием смеси фенола с
избытком безводного карбоната калия при повышенной
температуре (175°) в атмосфере двуокиси углерода под давлением 90—
135 д/лл*. Вместо карбоната калия можно применять также
карбонаты рубидия и цезия. Выходы достигаются хорошие, а во многих
случаях они даже лучше, чем в обычных условиях синтеза
Кольбе—Шмитта. Описанная модификация этой реакции была
проверена почти на ста примерах*.
157
Кольбе—Шмитт
[Механизм реакции Кольбе—Шмитта исследован многими
авторами. А. Н. Несмеянов и Н. Ф. Луценко^ полагают, что с СО,
реагирует неионизированный фенолят металла, причем карбоксил
вступает в ядро по месту повышенной электронной плотности.
Этой же точки зрения придерживаются английские исследователи^,
экспериментально показавшие, что первоначально из фенолята
металла и СО% образуется комплекс, который переходит затем в
мононатриевую соль салициловой кислоты:
ОН
ОХа
-Дол
1. О реакции Кольбе—Шмитта см. A. L i n d s e у, Н. J es k e у, Chem.
Revs., 57, 583 (1957).
2. Н. Kolbe, Ann., 113, 125 (1860).
3. R. S с h m i t t, J. prakt. Chem., 31, 397 (1885).
4. S. Hunt, J. Jones, A. L i n d s e y, D. К i I I o h, H. Turner,
J. Chem. Soc, 1958, 3152.
5. M. Ниренштейн, Д. Клнббенс, Синтезы органических
препаратов, сб. 2, Издатинлит, 1949, стр. 430.
6. Н. Erlenmeyer, В. Р г i j s, E. S о г k i n, E. S u t e г, Helv.
chim. acta, 31, 988 (1948).
7. J. Jones, Chem. Ind. (London), 889 (1957).
8. S. M а г a s s e, Герм. пат. 73279, 1893 г.; P. Friedlander, Fort-
schritte der Theerfarbenfabhkation, 3, 821 (1890—1894), Berlin, 1896.
9. О. В a i n e, G. A d a m s о n, J. Barton, J. Fitch, D. S w a-
y a m p a t e, H. J e s k e y, J. Org. Chem., 19, 510 (1954).
10. A. H. Несмеянов, Н. р. Л у ц е н к о, ДАН СССР, 59, 707(1948).—
11. J. Н а 1 е s, J. J о n e s, A. L i n d s 1 е у, J. Chem. Soc, 1954, 3145;
РЖхнм, 1955, 3666.
Кольбе 158
ЭЛЕКТРОХИМИЧЕСКАЯ РЕАКЦИЯ КОЛЬБЕ
Общий и достаточно простой метод получения углеводородов
путем электролиза щелочных солей органических кислот был
предложен Кольбе^ в 1849 г.
Реакция обычно проводится в водном или метанольном
растворе* и может быть изображена следующим уравнением:
2RCOCT -* R—R + 2СОз + 2е
где R—алкильная группа, обычно содержащая шесть или более
атомов углерода.
Реакция была широко изучена и является в настоящее время
ценным методом синтеза многих органических соединений. В
реакцию могут вступать также моноэфиры дикарбоновых кислот^;
при использовании смеси монокарбоновых кислот и моноэфиров
дикарбоновых кислот образуются как симметричные, так и
несимметричные продукты*. Например, при электролизе смеси
уксусной кислоты и монометилового эфира адипиновой кислоты
получается гексановая кислота (образующаяся после гидролиза
соответствующего эфира)
СНдООС—(СНд)4—СООН + СНд—СООН -м»
-» СНдООС-(СНз)*—СНз --» НООС—(СНг)*—СН,
одновременно с соответствующими симметричными продуктами.
При увеличении доли монокарбоновой кислоты относительно
моноэфира дикарбоновой кислоты увеличивается выход
несимметричного продукта.
Аналогичные результаты были получены со смесями кетокис-
лот\ Например, продуктами электролиза смеси кетокислот
д—СО—(СНа),—СООН f СНз—СО—(CH*)g—СООН
1 II
являются дикетоны:
-СО—(CHg),—СО—СНд СНз—CO-(CHJia—СО—
III IV
i*-CO—СН,
V
159 Кольбе
Описано применение в электрохимической реакции Кольбе
монобензиловых эфиров двухосновных кислот*;
соосн. соон соосн, соосн.
СООН OOOCHgCgHg COOCH, ggg
VI
Несимметричное соединение VI может быть каталитически
дебензилировано с помощью палладия, нанесенного на карбонат
стронция. При удалении бензильной группы образуется также
незначительное количество пробковой кислоты.
Линдсей и Петерсон? синтезировали много различных
соединений (олефинов, диенов и эфиров ненасыщенных дикарбоновых
кислот) путем электролиза карбоновых кислот и производных
дикарбоновых кислот в присутствии таких диенов, как бута-
диен-1,3.
1. Н. Kolbe, Ann., 69, 257 (1849).
2. О реакции Кольбе см. В. W e e d о n, Quart. Rev. (London), 6, 380 (1952);
см. также В. Wee don, Advances in Organic Chemistry, v. 1, New
York, 1960, p. 1.
3. A. Cr urn-Brown, G. Walker, Ann., 274, 71(1893).
4. W. Greaves, R. Linstead, B. Shephard, S. Thomas,
B. W e e d о n, J. Chem. Soc, 1950, 3326; R. Linstead, J. L u n t,
B. W e e d о n, J. Chem. Soc, 1950, 3331, 3333.
5. M. S t ol 1, Helv. chim. act a, 34, 1817 (1951).
6. L. D о 1 e j s, L. N о v о t n y, Coll. Czech. Chem. Comm., 19, 716 (1954),
7. R. L i n d s e у, М. Peterson, J. Am. Chem. Soc, 81, 2073 (1959).
Комб 160
Альфонс-Эдмонд Комб (Alphonse-Edmond Combes, 1858—1896) родился
в г. Сент-Ипполит-де-Фор (Франция). Учился у Ш. Вюрца в Париже, где
впоследствии читал лекции. В 1893 г. стал президентом Французского
химического общества. Широко исследовал реакции с хлористым алюминием. В
частности, действием хлористого алюминия на ацетилхлорид получил ацетилаце-
тон, изучил его свойства и показал, что взаимодействие анилина с ацетил-
ацетоном протекает с выделением воды и продукт реакции в присутствии серной
кислоты превращается в диметилхинолин. Вместе с Ш. Фриделем исследовал
действие фенилгидразина на камфорный ангидрид и синтезировал пиразолы.
СИНТЕЗ ХИНОЛИНА ПО КОМБУ
Конденсация ариламинов с 1,3-дикетонами с последующей
циклизацией полученного продукта в присутствии
концентрированной серной кислоты приводит к образованию производных
хинолина. Эта реакция является общим методом получения
замещенных хинолинов и называется синтезом Комба** *.
Примером реакции служит образование 2,4-диметилхинолина
из анилина и ацетил ацетона:
СИ,
со—сн,
Согласно Робертсу и Тернеру®, циклизация облегчается при
наличии сильных заместителей первого рода (электронодонорных),
находящихся в метаположении к аминогруппе. Те же
заместители в пара-положении затрудняют циклизацию. Нитроанилины в
условиях синтеза Комба не реагируют. Из мета-замещенных
анилинов преимущественно получаются 7-замещенные хинолины.
Например, при взаимодействии ж-хлоранилина с ацетил ацетоном
образуется 7-хлор-2,4-Диметилхинолин^. По методу Скраупа
(стр. 231) в данном случае получаются одновременно 5- и 7-хлор-
хинолины.
161
Комб
Джонсон и Метьюз* при изучении продуктов конденсации
^-нафтиламина с ацетил ацетоном по Комбу показали, что анил I
может быть получен с выходом 83%;
СО—СН
Циклизация анила I при добавлении концентрированной
серной кислоты или лучше безводного фтористого водорода приводит
к получению линеарного 2,4-диметилбенз- [gl-хинолина (II):
/СН;
и
Ангулярный изомер—2,4-диметилбенз-[/]-хинолин (III)
образуется с небольшим выходом при циклизации анила I в
присутствии концентрированной серной кислоты при 100°:
"^ СН, СН,
.сн,
—н,о
1. A. Combes, Bull. Soc. chim. Paris, 49, 89 (1888); С. г., 106, 142, 1536
(1888).
2. См. Р. Эльдерфильд, Гетероциклические соединения, сб. 4, Издат-
инлит, 1955, стр. 28.
3. Е. Roberts, E. Turner, J. Chem. Soc, 1927, 1832.
4. W. Johnson, F. Mat hews, J. Am. Chem. Soc, 66, 210 (1944).
И А. Сер pen
Коновалов* 162
Михаил Иванович Коновалов (1858—1906) родился в с. Будихино
Ярославской губернии. В 1884 г. окончил Московский университет; ученик В. В. Мар-
ковникова. В 1893 г. защитил докторскую диссертацию, с 1896 г. профессор
Московского сельскохозяйственного института, а с 1899 г.—профессор
Киевского политехнического института. М. И. Коновалов много труда посвятил
изучению природы кавказской нефти; исследовал действие разбавленной
азотной кислоты на углеводороды, открыв при этом реакцию (ныне носящую его
имя), применяемую для определения строения углеводородов; разработал
метод разделения нитросоединений и их очистки в виде солей.
РЕАКЦИЯ КОНОВАЛОВА
Реакция Коновалова* заключается в нитровании парафинов
и циклопарафинов разбавленной азотной кислотой. Примером
реакции может служить получение 2-нитрогексана^ с выходом 63%
нитрованием м-гексана при 140° азотной кислотой плотностью 1,075.
Часто по этой реакции образуется смесь нитропродуктов.
Например, при нитровании 2,5-диметилгексана кипячением с
азотной кислотой плотностью 1,38—1,40 получается смесь 1-нитро-,
2-нитро-, 3-нитро- и 2,5-динитро-2,5-диметилгексанов*' *.
Реакция Коновалова широко применяется также для
нитрования алифатической цепи жирноароматических соединений.
Так, при нитровании этил бензола кипячением с азотной кислотой
плотностью 1,075 образуется 1-нитро-1-фенилэтан* с выходом 33,7%:
аналогично из ж-ксилола получается ж-толилнитрометан (выход
26—62%)» и т. д.
Большое значение реакция Коновалова имеет для
определения строения парафиновых и циклопарафиновых соединений.
С этой целью исследуемое вещество нитруют, продукт нитрования
превращают в соответствующий альдегид или кетон, который
затем идентифицируют обычным способом. В. В. Марковников
показал, что при нитровании циклопарафинов разбавленной
азотной кислотой наряду с нитропроизводными получаются дикарбо-
новые кислоты с тем же числом атомов углерода, что и в цикле*.
* Биография М. И. Коновалова и изложение реакции составлены
редактором.
163 Коновалов
Дальнейшим развитием реакции Коновалова является
нитрование в газовой фазе. Так, при нагревании 2-метилпропана с
азотной кислотой в трубках при 150° с хорошим выходом
образуется 2-нитро-2-метилпропан?.
1. О реакции Коновалова см. А. С. Забродина, К. С. Забродина,
Реакции и методы исследования органических соединений, кн. 7. Гос-
химиздат, 1968, стр. 133.
2. М. И. Коновалов, ЖРХО, 25, 474 (1893).
3. М. И. КоновАлов, ЖРХО, 31, 57(1899).
4. М. И. Коновалов, ЖРХО, 38, 109 (1906).
5. М. И. Коновалов, ЖРХО, 31, 254 (1889); 37, 530 (1905).
6. В. В. Марковников, ЖРХО, 30, 151 (1898).
7. Н. Mass, E. Hodge, В. Vanderbilt, пат. США 1967667; С. А
28, 5830 (1934).
II
Конрад—Лимпах
164
Макс Конрад (Max Conrad, 1848—1920) родился в Мюнхене. Профессор
химии и минералогии Лесной академии в Ашаффенбурге. Исследования М.
Конрада относятся к области химии барбитуровой кислоты, эфиров циану ксусной
кислоты и производных малоновой кислоты.
Леонард Лимпах (Leonhard Limpach, 1852—1933) родился в Швейнфурте
(Германия). Окончил Вюрцбургский университет, после чего стал фармацевтом
В дальнейшем учился у И. Вислиценуса. Затем работал в английской компании
красителей. В 1895 г. вернулся в Германию, где открыл аптеку. Одновременно
читал лекции по фармации в Эрлангене. После первой мировой войны работал
в Вюрцбургским университете. Приобрел известность благодаря совместной
работе с М. Конрадом по синтезу производных хинолина. Занимался также
вопросами, связанными с производством красителей.
СИНТЕЗ ХИНОЛИНА ПО КОНРАДУ—ЛИМПАХУ
Метод Конрада—Лимпаха^ дает возможность получать
производные 4-оксихинолина, исходя из ариламинов и эфиров |3-ке-
токислот. Последовательность протекающих реакций видна на
примере синтеза 7-хлор и 5-хлор-4-оксихинолинов*:
COOQH*
СН,%
OO-GOOQH,
III
а он
а он
II
MIA
165 Конрад—Лимпах
При конденсации диэтилового эфира щавелевоуксусной
кислоты с ж-хлоранилином (I) промежуточно образуется анил II,
циклизующийся при нагревании в даутерме (смесь дифенила и
дифенилоксида) до 245°. При этом получаются этиловые эфиры
7-хлор- и 5-хлор-4-оксихинолин-2-карбоновой кислоты
(соответственно III и!НА). Увеличение количества растворителя в стадии
циклизации, по-видимому, не сказывается на общем выходе и
соотношении образующихся изомерных эфиров хлороксихинолин-
карбоновых кислот*. Изомеры разделяют дробной
кристаллизацией и гидролизуют, а образовавшиеся кислоты декарбоксилиру-
ют при нагревании в даутерме до 210°. Полученные
производные 4-оксихинолина (IV и IVA) используются в качестве
промежуточных продуктов в синтезе антималярийных препаратов.
При взаимодействии ариламинов с эфирами р-кетокарбоновых
кислот возможно образование как анилов, так и анилидов. В
последнем случае при циклизации образуются производные 2-окси-
хинолина* (синтез оксихинолина по Кнорру*):
сн,
со
\
сн,
1
со -*-
NH
сн,
1
Г |Г 1
V\^\QH
Хайзер и Рейнольде^ изучили факторы, определяющие
направление реакции в сторону образования анилов и анилидов.
Авторы показали, что обе реакции обратимы и анилы могут быть
превращены в анилиды при нагревании до 130—140° с водой,
содержащей следы кислоты.
1. М. С о п г a d, L. L i m p а с h, Бег., 20, 948 (1887).
2. R. R e i t s m a, Chem. Revs., 43, 43 (1948); см. также Р. Э л ь д е р -
ф и л ь *, Гетероциклические соединения, сб. 4, Издатинлит, 1955,
стр. 24.
3. A. Surrey, H. Hammer, J. Am. Chem. Soc., 68, 113 (1946).
4. A. S p I v e у, F. Curd, J. Chem. Soc, 1949, 2656.
5. L. К n о г г, Ann., 236, 69 (1886); 245, 357 (1888).
6. C. Hauser, G. Reynolds, J. Am. Chem. Soc, 70, 2402 (1948).
О Кнорре см. стр. 153.
Курциус 166
Теодор Курциус (Theodor Curtius, 1852—1928) родился в Дуйсбурге (Гер
мания). Изучал химию под руководством Р. Бунзена в Гейдельбер ге и у А. Коль.
5е в Лейпциге, где получил степень доктора философии. Профессор химии
Кильского и Гейдельбергского (с 1901 г.) университетов. Открыл диазоуксус-
ный эфир, гидразин, азотистоводородную кислоту и проводил исследования в
области химии этих новых соединений. При этом он синтезировал триазолы
гетразолы, азины, гидразоны и азиды кислот.
РЕАКЦИЯ КУРЦИУСА
Образование первичных аминов из азидов карбоновых кислот,
протекающее по общей схеме
R-COOH -» R—CON, -». R—N=C=O -^ R—NH,
было впервые описано Курциусомв 1890 г. и известно под
названием реакции или перегруппировки Курциуса^* ^. Наряду с
реакциями Гофмана и Шмидта^ реакция Курциуса является еще одним
методом замены карбоксильной группы на аминогруппу.
Азиды карбэновых кислот мокно получать либо из гидразидов
взаимодействием с азотистой кислотой, либо при обработке хлор-
ангидридов кислот азидом натрия;
ag MONO
R—OOOQH,; » R—CO—NH—NH% » R—CON,
R-COOH -^ R—COC1 *- R—CON,
Первичным продуктом разложения азидов карбоновых кислот
являются изоцианаты, которые можно выделить, если проводить
реакцию в инертном растворителе. В других условиях
проведения реакции могут получиться уретаны, замещенные мочевины
или амиды.
В реакции Курциуса были использованы разнообразные кар-
боновые кислоты. Дикарбоновые кислоты легко превращаются в
Курциус
соответствующие диамины, моногидразиды дикарбоновых кислот—
в аминокислоты. Из азидов а-оксикислот образуются кетоны или
альдегиды
R' R'
1 I
R^C—CON, -#. R-C=O + HNCO
R—CH—CONg -^ R-CHO ф НМСО
OH
Азиды* полученные из р-оксикислот, превращаются в урета-
ны, далее изомеризующиеся с образованием оксазолидонов I:
R—CH-CH, R—СН—СН, R-CH—
НО CON, НО N О NH
О О
I
Эта новая реакция была недавно применена для синтеза
некоторых фенилэтаноламинов^. При N-метилировании оксазолидо-
на I с последующим раскрытием цикла (при действии соляной
кислоты) и декарбоксилированием получается замещенный
этанол амин II (К=оксифенил):
R_CH—CHg R—CH—CHg R_CH—СН,
О NH О N—СН, ОН N—СН,
С С СООН
о о
R—СН—СН,,
-^ I I
ОН NH—СН„
и
Механизм перегруппировки Курциуса обычно связывают с
образованием промежуточного радикала (III), аналогичного
радикалам, образующимся при перегруппировках Гофмана и
Лоссена*:
R-C-N—N=N: -^ R-C—МфМ,
Курциус 168
Образование этого радикала, вероятно, сопровождается
одновременной миграцией радикала R от углерода к азоту*:
R—С—N --<- O=zC=N-_R
II
причем конфигурация группы R сохраняется и в образовавшемся
изоцианате она имеет то же пространственное строение, что и в
исходной кислоте.
Описано* влияние о-, ж- и я-заместителей на скорость
перегруппировки бензазидов по реакции Курциуса. Показано, что о-за-
мещенные реагируют значительно быстрее, чем соответствующие
л*- и м-производные. Результаты перегруппировки сильно зависят
от пространственных факторов.
1. Т. Си г t i u s, Вег., 23, 3023 (1890); J. prakt. Chem., 60, 275 (1894).
2. О реакции Курциуса см. П. Смит, Органические реакции, сб. 3, Издатин-
лит, 1951, стр. 322; сравнение реакций Курциуса, Гофмана и Шмидта
см. там же, стр. 344.
3. Е. Bergman n, М. Sulzbacher, J. Org. Chem., 16, 84 (1951).
4. К. И н г о л ь д, Механизм реакций и строение органических соединений.
Издатинлит, 1959, стр. 403.—Дол. /ж).
5. С. Н a u s e r, S. К a n t о г, J. Am. Chem. Soc, 72, 4284 (1950).
6. Y. Yukawa, Y. T s u n o, J. Am. Chem. Soc, 80, 6346 (1958).
169 Кучеров*
Михаил Григорьевич Кучеров (1860—1911) окончил в 1871 г.
Петербургский земледельческий институт (позже переименованный в Лесной институт)*
В 1903 г. стал профессором этого института. Основные работы М. Г. Кучерова
посвящены изучению непредельных углеводородов. Открытая им в 1881 г.
реакция гидратации ацетилена и его производных (реакция Кучерова) широко
применяется в промышленности и имеет особенно большое значение для про.
изводетва синтетического каучука.
РЕАКЦИЯ КУЧЕРОВА
Реакцией Кучерова^ называется присоединение воды к
веществам, содержащим тройную связь, каталитически ускоряемое
солями ртути и приводящее к получению карбонилсодержащих
соединений.
При гидратации ацетилена получается ацетальдегид^:
СН=Ш + Н,0 —-»- СН,=СНОН -» CHgCHO
В некоторых условиях, например, при высокой температуре
над окисью цинка, ацетилен гидратируется с образованием
ацетона®:
2СН=СН + ЗН,О -» CHgCOCH, + СО, 4 2Н,
При гидратации монозамещенных ацетиленов образуются ме
тилкетоны^, например, из монофенил ацетилен а получается ацето-
фенон:
Изучение кинетики реакции Кучерова показало, что вначале
образуется %-комплекс ацетилена с солями ртути^.
1. О реакции Кучерова см. А. Д. Петров, Усп. хим., 21, 250 (1952).
2. М. Г. Кучеров, ЖРХО, 13, 533, 542 (1880); А. Н. Несмеяй о„в.
Р. X. Фрейдлина, ДАНСССР, 24, 59(1940). "
3. BIOS, 1053, стр. 35.
4. М. Г. Кучеров, Вег., 14, 1532, 1540 (1882); ЖРХО, 16, 575 (1883);
24, 330 (1892).
5. И. В. Смирнов-Замков, Е. А. Шилов, ДАН СССР, 73, 72S
(1950).
* Биография М. Г. Кучерова и изложение реакции составлены
редактором.
Кэмпс 170
Рудольф Кэмпс (Rudolf Camps) работал под руководством профессора
К. Энглера в Высшей технической школе в Карлсруэ с 1899 по 1902 г.
РЕАКЦИЯ КЭМПСА
Реакцией Кэмпса^ называют внутримолекулярную
конденсацию о-ациламиноацетофенонов, приводящую к образованию окси
хинолинов^.
Примером такой реакции, каталитически ускоряемой
основаниями, является образование смеси 2-окси-4-метилхинолина
(выход 70%) и 4-окси-2-метилхинолина (выход 20%) из о-ацетами-
ноацетофенона:
он
Кельш и Люхт^ описали несколько необычный пример
реакции Кэмпса. Они показали, что под действием спиртового
раствора этилата натрия 1-(карбэтоксиацетамино)-ксантон (I)
претерпевает внутримолекулярную циклизацию и превращается в про-
извьдное хйнолина (II):
I II
Превращение происходит с выходом 90%.
171 Кэмпс
Недавно был описан* новый тип реакции"Кэмпса, при которой
из лактама III образуется 2,3-циклопентано-4-оксихинолин (IV):
ш
В этой же работе описано применение инфракрасной и
ультрафиолетовой спектроскопии для идентификации 2- и 4-оксихино
линов.
LR. Camps, Вег., 32, 3228(1899).
2. Р. Эльдерфильд, Гетероциклические соединения, сб. 4, Издатинлит,
1955, стр. 45.—Дом. рео\
3. С. Koelsch, F. Lucht, J. Am. Chem. Soc., 71, 3556 (1949).
4. B. W i t k о p, J. Patrick, M. R о s e n b I u m, J. Am. Chem. Soc,
73, 2641 (1951).
Лейкарт 172
Карл Луи Рудольф Александр Лейкарт (Carl Louis Rudolf Alexander Leu-
ckart, 1854—1889) родился в Гиссене (Германия). Изучал в Гейдельберге
физику у Г. Кирхгофа и химию под руководством Р. Бунзена. Работал у А. Коль-
бе в Лейпцигском университете, где в 1879 г. защитил докторскую диссертацию,
затем в течение трех лет сотрудничал в Мюнхенском университете с А.
Байером. Впоследствии доцент Геттингенского университета. Занимался
исследованиями мочевины и ее производных, синтезом тиофенолов и их эфиров иэ
диазониевых соединений н ксантогенатов* (тиофеноловая реакция Лейкарта).
а также восстановительным алкилированием тиофенолов.
РЕАКЦИЯ ЛЕЙКАРТА
Превращение альдегидов и кетонов в соответствующие амины
при нагревании с формамидом, формиатом аммония или со смесью
формамида и муравьиной кислоты известно под названием
реакции Лейкарта*' *:
RR'CO + 2HOOONH* -*» RR'CHNHCHO -f NH, ф GO, ф 2Н,0
Н*о
RR'CHNHCHO » RR'CHNH, ф НСООН
По Кросли и Муру* применение смеси формамида и
муравьиной кислоты дает лучшие результаты. Наиболее пригодны для
реакции Лейкарта ароматические альдегиды и высококипящие
кетоны. При использовании замещенных формамидов, а также
муравьинокислых солей первичных или вторичных аминов по
этому методу могут быть получены вторичные и третичные амины.
Реакция Лейкарта была применена для синтеза ряда %-арил-
пропил аминов, необходимых для фармакодинамических и химио-
терапевтических исследований*. Примером этих синтезов
является образование 1,3,3-трифенилпропиламина из |3,|3-дифенилпро
пиофенона:
((^,);ШСН,СОС,Н, -» (С,Н,),СНСН,СНС,Н,
NH,
При взаимодействии кетонов с диалкилформамидами необхо
димо применять в качестве катализатора хлористый магний*.
I7& Лейкарт
Например, циклогексанон и N-формилпиперидин в присутствии
хлористого магния дают N-циклогексилпиперидин (I); без
катализатора реакция не идет. Тот же третичный амин I был получен
Смитом и Макдональдсы? при кипячении смеси циклогексанона с
муравьинокислым пиперидином (выход 61%):
нсоо- ж ) -»( \-N
Эти авторы считали, что промежуточными продуктами
реакции являются муравьинокислые эфиры карбинол аминов (П)
С
и
которые путем 1,3-миграции гидрид-иона могут
перегруппировываться в третичные амины.
Механизм реакции Лейкарта был изучен Поллардом и Юнгом®.
На основании исследования кинетики первой стадии реакции и
спектрофотометрического анализа промежуточных продуктов ими
была предложена следующая схема реакции:
С=О _. —/, ^С^ .п ^ ^^Z.,\ ^<^ нсоо
н
В первой стадии реакции происходит присоединение формамида
к карбонильной группе. При повышении температуры от
продукта присоединения отщепляется вода и образуется промежуточный
продукт, имеющий конъюгированную систему двойных связей.
Вода гидролизует некоторое количество формамида, причем
образующийся муравьинокислый аммоний может служить
восстановителем. Перемещение гидрид-иона Н" вызывает отщепление
двуокиси углерода; затем ион аммония отдает протон полученному
Лейкарт
промежуточному соединению, что приводит к образованию фор-
милзамещенного первичного амина. Этот механизм не объясняет
образование вторичных и третичных аминов по реакции Лейкарта^.
Реакцией, близкой к реакции Лейкарта, является редкудя
сЫбеилера—Клдркд^'1\ которая применяется специально для
получения N-метилированных аминов путем взаимодействия
первичных или вторичных аминов с формальдегидом и муравьиной
кислотой:
RNH, -» RN(CH,),
R,NH -*- RgNCHg
Выходы третичных аминов, синтезируемых по этому методу,
обычно удовлетворительные.
Реакцию проводят при нагревании смеси амина (1 моль), 35—
40%-ного формальдегида (2,2 моля) и муравьиной кислоты (5 мо
лей) на паровой бане до полного прекращения выделения
двуокиси углерода. Продолжительное нагревание не оказывает отрнца
тельного влияния на ход процесса. Третичные амины могут быть
выделены из реакционной смеси при добавлении избытка основа
ний с последующей .экстракцией органическими растворителями.
1. R. Leuck ar t, J. prakt. Chem., 41, 179 (1890).
2. [О реакции Лейкарта см. Б. М. Богословский, Реакции и методы
исследования органических соединений, кн. 3, Госхимиздат, 1954,
стр. 253.—Дол. pal], см. также М. Moore, Organic Reactions, v. V.
New York, 1949, p. 301.
3. R. Leuck art, Ber., 18, 2341 (1885).
4. F. Cross ley, M. Moore, J.Org. Chem., 9, 529(1944).
5. J. Burckhalter, S. Johnson, J. Am. Chem. Soc, 73, 4830 (1951).
6. J. В u п п e t t, J. Marks, J. Am. Chem. Soc, 71, 1587 (1949).
7. P. Smith, A. Macdonald, J. Am. Chem. Soc, 72, 1037 (1950).
8. C. Pollard, D. Young, J. Org. Chem., 16, 661 (1950).
9 О механизме реакции Лейкарта см. V. Franz en, Chem. Ztg.,
80, 779 (1956).
10. W. Eschweiler, Ber., 38, 880 (1905).
11. H. Clarke, H. G i 1 1 e s p i e, S. Weisshaus, J. Am. Chem.
Soc., 55, 4571 (1933).
175 Лоссен
Вильгельм Клеменс Лоссен (Wilhelm Clemens Lossen, 1838—1906) родился
в г. Крейцнахе (Германия). В 1862 г. получил степень доктора философии в
Геттингенском университете. Работал доцентом в этом университете, затем
ассистентом в Карлсруэ, Галле и Гейдельберге, где позднее стал профессором
химии. С 1877 г. профессор Кенигсбергского университета. Его научные
интересы концентрировались в области синтеза и изучения свойств гидроксил-
аминов и их производных.
РЕАКЦИЯ ЛОССЕНА
Реакция Лоссена состоит в разложении гидроксамовых кислот
или их производных с образованием изоцианатов** *:
/о
R—C\ -» R—N=C=O -f» HgO
NH-OH
Общим методом получения моногидроксамовых кислот
является взаимодействие сложных эфиров карбоновых кислот с гидрок-
силамином в присутствии алкоголята натрия:
^%О МаОСжЩ J3 /ОН
R—Cf ф NH.-OH * R—Cf ^ R-%
X)R ^NH—ОН Ж—ОН
Для синтеза бензгидроксамовой кислоты используют едкое
кали^. Перегруппировку можно проводить путем нагревания с
добавкой дегидратирующих агентов типа тионилхлорида,
уксусного ангидрида или пятиокиси фосфора или без этих агентов.
Применение полифосфорной кислоты при перегруппировке
Лоссена описано Снайдером с сотр.*. Ароматические кислоты
при нагревании (150—170°) с гидроксиламином и полифосфорной
кислотой в течение, нескольких минут превращаются в амины.
Последние получены также при нагревании гидроксамовой
кислоты с полифосфорной кислотой.
Классическим примером использования перегруппировки
Лоссена является одна из стадий превращения камфоры в эпи-
камфору*:
Лоссен
176
Метиловый эфир борниленкарбоновой-3 кислоты (получаемый
из камфоры) действием гидроксиламина переводят в
соответствующую гидроксамовую кислоту. Образование изоцианата и
превращение его в непредельный циклический амин и далее в кетон
может быть выражено следующей схемой:
соосн,
CONHOH
NCO
NH.
При перегруппировке моногидроксамовых кислот—производ
ных дикарбоновых кислот могут получаться полимерные амиды*
Например, при нагревании в толуоле соли 5-карбоксивалеро-(бен
зоил)-гидроксамата образуется полиамид:
QHs-СО- О—N—СО—(СН^-СООН -» [—NH—(
Na
о)^—СО
Если перегруппировку проводить в водной среде, то конечным
продуктом является производное мочевины:
CO[NH—(CH,),—COOHj,
Алифатические дигидроксамовые кислоты путем нагревания
в бензоле (толуоле) в присутствии тионилхлорида или фосгена
превращаются в диизоцианаты?. Примером является
образование октаметилендиизоцианата из себациндигидроксамовой
кислоты:
/CO—NH—OH
XX)—NH-OH
OC=N—(CH,),-N=CO
Перегруппировка Лоссена® близка к перегруппировкам
Гофмана и Курциуса и является еще одним методом синтеза аминов
из карбоновых кислот. Сохранение конфигурации мигрирующего
177 Лоссен
радикала было показано при получении (—)-а-фенилэтиламнна из
бензоил-(фенилметилацетил)-гидроксамата* (R=CgH)
CHg—CH—COOH
R
- CHg—CH-CO—NH—ОН
R
CHg-CH-
-C-N/
|| X)—COR
и
-» сн
RCOC1
-^СНз
^-СН—СО-С1
R
СН„-
сн
R
-СН C=N
R ОН
N-CO -
к
HCI
» СНд—СН—NH
R
Бауэр и Майарка^ установили, что лоссеновская перегруппи
ровка некоторых N-феннлсульфонилоксимидов ^ис-циклогексан
дикарбоновой-1,2 кислоты стереоспецифична и приводит к обра
зованию ^wc-2-аминоциклогексанкарбоновой кислоты.
LW. Loss en, Ami., 16%, 347 (1872).
2. Н. Yale, Chem. Revs., 33, 209 (1943). -
3. С. X а у з e p, 8. P e н ф р о у, Синтезы органических препаратов, сб. 2,
Издатинлит, 1949, стр. 87.
4. Н. S n у d е г, С. Е I s t о n, D. 1\ е 1 I о m, J. Am. Chem. Soc, 75,
2014 (1953).
5. J. В г e d t, W. P e г k i n, J. prakt. Chem., 89, 209 (1914).
6. С Hurd, С Buess, L. Bauer, J.Org. Chem., 17, 865(1952).
7. J. D i с k e y, J. S t r a 1 e y, T. S t a v i n, пат. США 2394597, 1946 г.;
С. А., 40, 2848 (1946).
8. К. И н г о л ь д, Механизм реакций и строение органических
соединений, Издатинлит, 1959, стр. 402.—До/г. /W.
9. A. Campbell, J. К е п у о n, J. Chem. Soc, 1946, 25.
10. L. В a u e r, 6. M i a r k a. J. Org. Chem., 24, 1293 (1959).
12 А. Серрей
Мак-Фадиен—Стивене* 178
Дж. С. Мак-Фадиен (J. S. McFadyen) родился в 1908 г. в г. Торонто
(Канада). Учился у Т. С. Стивенса в университете в Глазго, где в 1936 г. получил
степень доктора философии. В течение пятнадцати лет работал в отделе
красителей фирмы Imperial Chemical Industries, затем вернулся в Канаду.
Работает в химическом отделе фирмы Canadian Industries Limited в г. Монреале.
ВОССТАНОВЛЕНИЕ ПО МАК ФАДИЕНУ—СТИВЕНСУ
В 1936 г. Мак-Фадиен и Стивене^ предложили новый метод
превращения ароматических карбоновых кислот в
соответствующие альдегиды.
В качестве примера этой реакции можно привести образование
бенз альдегид а из этилового эфира бензойной кислоты. Бензолсуль-
фонгидразид бензойной кислоты, полученный взаимодействием
соответствующего гидразида с бензолсульфонилхлоридом,
растворяют в этиленгликоле, нагревают раствор до 150—165° и
добавляют безводный карбонат натрия или калия. При этом
выделяется азот:
g CgH&SOsCl
С,Н*СООС*Нв * CgHgCONH—NHg "
NagCO"
-* CgHbCONH—NHSOaCgHs — ^ CgH^CHO + СдН„8О,Ма -f- N,
Примерно чергз минуту в реакционную массу быстро
приливают горячую воду и образовавшийся бензальдегид экстрагируют
подходящим органическим растворителем или отгоняют с
водяным паром. Выходы ароматических альдегидов обычно хорошие*.
Реакцию можно применять также для получения альдегидов
гетероциклического ряда. Например, восстановлением по Мак-
Фадиену—Стивенсу были синтезированы 2-пиридин и 3-пиридин
альдегиды", 3-, 5-, 6- и 8-хинолинальдегиды*. Как правило,
выходы гетероциклических альдегидов (13—45%) несколько ниже
выходов ароматических альдегидов.
* О Стивен се см. стр. 239.
179 Мак-Фадиен—Стивене
Роберте* недавно показал, что бензолсульфонгидразид цикло-
пропанкарбоновой кислоты восстанавливается по Мак-Фадиену—
Стивенсу до формилциклопропана с выходом 16%:
сн. сн.
'а
\
.CHCONH-NHSO,C.H, — у
СН. СН
сн-сно
1. J. McFadyen, Т. Stevens, J. Chem. Soc, 1936, 584.
2. О реакции Мак-Фадиена—Стнвенса, см. Э. Мозеттиг, Органические
реакции, сб. 8, Издатинлит, 1956, стр. 302.—До/г. ped.
3. С. N i е m a n n, R. Lewis, J. Hays, J. Am. Chem. Soc, 64, 16%8
(1942); C. N i e m a n n, J. Hays, J. Am. Chem. Soc, 65, 482 (1945).
4. A. Cook, J. Heilbron, L. Steger, J. Chem. Soc, 1943, 413.
5. J. Roberts, J. Am. Chem. Soc, 73, 2959 (1951).
12
Манних 180
Карл Ульрих Франц Манних (Carl Ulrich Franz Mannich, 1877—1947)
родился в Бреслау (Германия). Учился в Марбургском, Берлинском и Базель-
с ком университетах. В 1903 г. получил степень доктора. Преподавал в Геттин-
генском п Франкфуртском университетах. С 1927 г. профессор и директор
фармацевтического института в Берлине. Занимался главным образом синтезом
органических веществ, применяемых в фармакологии. Он выделил в
кристаллическом виде сердечные глюкознды из наперстянки и строфанта.
Начиная с 1917 г., в течение почти 30 лет, изучал реакцию, носящую его имя, и
возможности ее практического применения. Установил, что при взаимодействии
метиламина с формальдегидом и ацетоном образуется производное пиперидина
и получил из него метилареколин. Синтезировал большое число аминокетонов
н аминоспиртов, использовал аминоспирты для приготовления эфиров л-ами-
нобензойной кислоты, исследовал возможность применения этих эфиров в
качестве местных анестезирующих средств.
РЕАКЦИЯ МАННИХА
Реакцией Манниха называют конденсацию соединений,
содержащих один или несколько подвижных атомов водорода, с
формальдегидом и аммиаком или аминами (первичными или
вторичными); при этом атомы водорода замещаются на аминометильную
группу.
Примером реакции может служить синтез хлоргидрата 4(-ди-
этиламино)-бутанона-2 из ацетона, формальдегида и солянокислого
диэтиламина:
д—СО—СНз f CHgO ф (С,Нб)*МН.НС1 -*
СМ,—СО—Ш,—СН,—
Поскольку в молекуле ацетона имеется несколько подвижных
атомов водорода, их также можно заменить на диэтиламиноме-
тильные группы.
Отдельные примеры конденсации подобного типа были
известны и до Манниха!, до он первый показал общий характер этой
реакции и ее значение в органическом синтезе.
В реакции Манниха успешно используется большое число
различных соединений*, включая кетоны, альдегиды, сложные эфи-
181 Манних
ры и фенолы. Аминосоставляющие реакции также могут быть
самыми разнообразными.
Иллюстрацией применения ароматических оксисоединений для
получения оснований Манниха является конденсация 5-хлор-
8-оксихинолина^ с диэтиламином и параформальдегидом; при
этом образуется активный химиотерапевтический (амебоцидный)
препарат—5-хлор-7-диэтиламинометил-8-оксихинолин:
Присутствие следов железа оказывает большое влияние на
ход реакции Манниха, если в ней участвуют соединения с
концевой ацетиленовой группой*:
—С==СН f CH*O f N
В присутствии хлористого железа выходы продуктов реакции
достигали 80%, в то время как без него реакция практически не
протекала.
Основание Манниха—4-(диэтиламино)-бутанон-2 при перегонке
с водяным паром превращается в метилвинилкетон. Однако, по-
видимому, вследствие полимеризации выходы кетона обычно
низкие. Робинсон* показал, что для синтеза циклических кетонов
может применяться подметилат 4-(диэтиламино)-бутанона-2 как
источник метил винил кетона:
СН„
Недавно для этой же цели был предложен еще более удачный
реагент—четвертичная соль пиперидинового основания
Манниха*:
—СО—СН%—СНд—
В основном четвертичные соли ведут себя как алкилирующие
агенты: отщепляются третичные амины и образуется углерод-
углеродная связь. Можно считать, что вииилкетоны вступают
Манних 182
при этом в конденсацию по Михаэлю. Реакция этого типа была
использована для синтеза триптофана?, причем алкилирование
ацетаминомалонового эфира проводили подметил атом грамина:
„ CHfCOOQH,),
NH-CO-CHg
Показано, что иодметилаты и окиси оснований Манниха,
полученных из фенолов, легко реагируют в щелочной среде с
различными нуклеофильными реагентами^. Так, например, при
взаимодействии с ионом метоксила происходит замена четвертичного
атома азота на метоксигруппу.
Применение основания Манниха как алкилирующего агента
подробно описано Д одеоном и Солменом^. Авторы
показали, что продукт конденсации 6-(диметиламино)-4-кетокапроно-
вой кислоты (основание Манниха, образованное левулиновой
кислотой) с этиловым эфиром малоновой кислоты при
последующем гидролизе и декарбоксилировании превращается в у-кетосу-
бериновую кислоту:
(з)з
—(СН*)з—СООН ———*"(СНз);М—(СНа)%—СО—(CHJ^
CHO
Н*О
-CO-(СН^.-СООН —-
-^ НООС—(СНд)з—СО—(СН%)д—СООН
Механизм реакции Манниха не установлен; возможно, что
промежуточным продуктом реакции является аминометанол,
образующийся из амина и формальдегида^^Ч
+ CHgO
Предполагают также, что в качестве промежуточных
продуктов получаются метилен-бис-амины^, которые в условиях
реакции способны конденсироваться с соединениями, содержащими
активную группу СН,, в основания Манниха.
l.C. Mannlch, W. К rose he, Arch. Pharm., 250, 647(1912).
2. О реакции Манниха см. Ф. Блик, Органические реакции, сб. 1, Издат-
инлит, 1948, стр. 399; [В. Rei chert, Die Mannich-Reaktion,
Berlin, 1959.—Дод. ped.].
3. J. В u г с k h a 1 t er, S. Johnson, J. Am. Chem. Soc, 73, 4837 (1951).
4. И. Н. Назаров, Е. А. Мистряков, Изв. АН СССР, ОХН,
1958, стр. 335.
183 Манних
5. Е. Feu, F. М с Q u i I 1 i n, R. R о b i n s о n, J. Chem. Soc, 1937, 53.
6. A. Wilds, R. Werth, J.Org. Chem., 17, 1149(1952).
7. H. S п у d e r, C. Smith, J. Am. Chem. Soc, 66, 350 (1944); N. A 1-
bertson, S. Archer, С Suter, J. Am. Chem. Soc, 66, 500
(1944).
8. P. Gardner, H. Rafsanjani, L. Rand, J. Am. Chem. Soc,
81, 3364 (1959).
9. R. D о d s о n, P. S о 1 1 m a n n, J. Am. Chem. Soc, 73, 4197 (1951).
10. E. Alexander, E. Underbill, J. Am. Chem. Soc, 71, 4014
(1949).
11. Данные об этом типе промежуточных продуктов см. Е. Hope, R.
Robinson,, J. Chem. Soc, 99, 2114 (1911); о реакции катарнина с ацето-
феноном см. Е. Hope, R. Robinson, J. Chem. Soc, 103,361
(1913).
12. S. Lieberman, E. Wagner, J.Org. Chem., 14, 1001(1949).
Меервейн 184
Ганс Л. Меервейн (Hans L. Meerwein) родился в 1879 г. в Гамбурге.
Учился в Висбадене, Бонне и Шарлоттенбурге. В 1903 г. Боннский университет
присудил ему докторскую степень. С 1914 г. доцент этого университета, с
1922 г. профессор и директор Кенигсбергского химического институте, затем
ректор Марбургского университета. Работы Г. Меервейна являются
значительным вкладом в органическую химию. Среди них можно отметить реакцию
Меервейна, восстановление альдегидов и кетонов алкоголятом алюминия,
изучение пи на кол и новой перегруппировки, исследование реакций с диазо-
метаном.
КОНДЕНСАЦИЯ МЕЕРВЕЙНА
Взаимодействие хлористых (бромистых) солей арилдиазония
с д, |3-ненасыщенными карбонильными соединениями, в
результате которого ароматическое ядро вступает в а- или (3-положение
карбонильного соединения, носит название конденсации Меер-
вейна!, %
Меервейн использовал такие карбонильные соединения, при
участии которых в данной реакции получались д-арилпроизвод-
ные. Например, при конденсации коричной кислоты с хлористым
л-нитрофенилдиазонием образуется я-нитростильбен, так как в
этом случае реакция сопровождается декарбоксилированием:
_ . НС1
соон'
В 1943 г. Кельт* показал, что арильный радикал может
вступать и в 8-положение. Например, акрилонитрил и матилакрилат
превращаются в производные коричной кислоты.
Первоначальным продуктом реакции хлористого арилдиазония с акрилонитри-
лом является а-хлор-|3-арилпропионитрил. Обработка этого
промежуточного продукта диэтиланилином или едким кали приводит
к образованию Ц-арилакрилонитрила. При действии водного ам-
185 Меервейн
миака образуется а-аминопропионитрил-. Восстановление а-хлор-
В-арилпропионитрила цинком (в уксусной кислоте) приводит к
Р-арилпропионитрилу:
кон
» ArCH-CH—CN
ArN,Cl -f CH,=CH—CM >- ArCH,—CH—CN
AiCH,—CH-CN
Zn
ArCH,—CH,—CN
снзсоон ^
Реакция Меервейна, по-видимому, может протекать как по
радикальному"'*, так и по ионному механизму***. Недавно Ди-
керман* исследовал механизм реакции Меервейна, проводя ее в
растворе ацетона. Катализатором реакции, по-видимому, служит
хлористая медь, образующаяся из хлорной меди и ацетона.
Данные о кинетике этого процесса интерпретировались автором с
точки зрения радикального механизма.
1. Н. М е е г w e i п, Е. В и с h n е г, К. Е m s t е г, J. prakt. Chem.,
152, 237 (1939).
2. Подробно о конденсации Меервейна см. Е. Alii I I e r, Angew. Chem., 61,
179 (1949).
3. С. К о е 1 s с h, J. Am. Chem. Soc, 66, 57 (1943).
4. С. К о е 1 s с h, V. В о е k e 1 h e i d e, J. Am. Chem. Soc., 66, 412 (1944).
5. F. В е г g m а л n, D. S с h a p i r o, J. Org. Chem., 12, 57 (1947); см.
также W. С о w d г е у, D. D a v i e s, Quart. Revs. (London), 6, 365»
377 (1952).
6. S. Dickerman, K. Weiss, A. Ingberman, J. Am. Chem,
Soc, 80, 1904 (1958).
Меервейн—Понндорф—Верлей 186
ВОССТАНОВЛЕНИЕ ПО МЕЕРВЕЙНУ^ПОННДОРФУ—
ВЕРЛЕЮ
Избирательное восстановление карбонильных соединений до
спиртов в присутствии алкоголятов алюминия обычно называют
восстановлением по Меервейну—Понндорфу—Верлею^ ^.
Это превращение, которое является обратимым*, можно
представить следующим общим уравнением:
RR'CO + RgCHOH ^ RR'CHOH + R2CO
По данному методу алифатические и ароматические альдегиды
и кетоны восстанавливаются в соответствующие спирты с
удовлетворительными выходами. Реакцию обычно проводят путем
кипячения раствора карбонильного соединения в изопропиловом
спирте в присутствии изопропилата алюминия. Для того, чтобы
реакция закончилась, необходима непрерывная отгонка ацетона,
образующегося в соответствии с приведенным выше уравнением
(R"=CHg). Альдегиды обычно восстанавливаются быстрее кето-
нов. Другие группы (N0%, сложноэфирные, двойные углерод-
углеродные связи, активные галоиды) не восстанавливаются изо-
пропилатом алюминия*.
Для улучшения выхода спирта при восстановлении альдегидов
и реакционноспособных кетонов карбонильные соединения
рекомендуется вводить в реакционную смесь небольшими порциями*.
Недавно установлено^, что удаление ацетона в большинстве
случаев не является обязательным и высокие выходы продуктов
восстановления достигаются уже при непродолжительном
нагревании (с обратным холодильником) раствора карбонильного
соединения в изопропиловом спирте, содержащем изопропилат
алюминия. По этой видоизмененной методике из бензофенона был
получен бензгидрол с выходом 98%:
QH.-C-QH, -+. С^Нв
О ОН
Обратная реакция называется окислением по Олпенауеру (см. стр. 197).
187
Меервейн—Понндорф—Верлей
Механизм восстановления по Меервейну—Понндорфу—Вер-
лею, предложенный Джекманом и Милзом^, в некотором
отношении аналогичен механизму реакции Канниццаро (см. стр. 132).
Карбонильная группа образует с атомом металла изопропилата
алюминия комплекс I, строение которого можно изобразить одной
из шестичленных циклических структур II, показанных в
квадратных скобках.
R
R'
—О—А1
или
О О
| |
О О
/\
О
II
Переход гидрид-иона* облегчается близостью атомов
углерода в промежуточном циклическом комплексе II; при
разложении комплекса II кислотами получается продукт
восстановления RR'CHOH. В присутствии изопропилового спирта
происходит обменная реакция с комплексом II, приводящая к
регенерации изопропилата алюминия. Восстановление, однако, может
протекать и без изопропилового спирта.
ЛЯ7ЕРЛ7УРЛ
1. Н. Meerwein, R. Schmidt, Ann., 444, 221 (1925); W. Ponn-
d о г f, Angew. Chem., 39, 138 (1926); A. V e г 1 e y, Bull. Soc. chim.
Paris, 37, 537, 871 (1925).
2. О восстановлении по Меервейну—Понндорфу—В ер л ею см. А. У а й л д с,
Органические реакции, сб. 2, Издатинлит, 1950, стр. 194.
3. Недавнюю дискуссию по реакции Меервейна—Понндорфа—Верлея см.
W. Johnson, G. Skrimshire, Chem. Ind. (London) 380, (1951).
4. L. J ac kman, J. Mills, Nature, 164, 789 (1949).
W. Truett, W. Moulton, J. Am. Chem. Soc, 73, 5913(1951).
N. Deno, H. Peterson, G. Saines, Chem. Revs., 60, 7(1960).
5.
6.
Михаэль 188
Артур Михаэль (Arthur Michael, 1853—1942) родился в г. Буффало (США).
Учился в Гейдельбергском университете, где позднее стал ассистентом Р. Бун-
эена. Затем работал в Берлине под руководством А. Гофмана, в Париже у
Ш. Вюрца и в России с Д. И. Менделеевым. Вернувшись в США, занимался
педагогической и исследовательской работой в колледже Тафта. С 1909 г.
заведующий химической лабораторией во вновь открывшемся университете
Кларка, в дальнейшем профессор химии Гарвардского университета.
Значительную часть исследований выполнил в собственной лаборатории в г. Ньютоне
(США). Работал в разнообразных областях химии. Хорошо известны его
труды по химии соединений, содержащих активированные метиленовые группы. В
последние годы жизни занимался проблемами физической химии.
РЕАКЦИЯ МИХАЭЛЯ
Реакцией Михаэля* называют конденсацию соединений,
содержащих активную метиленовую (или метиновую) группу, с л,
^-непредельными соединениями (двойная связь которых
активирована электроотрицательными заместителями) в присутствии таких
основных катализаторов, как этилат натрия или пиперидин.
Примером этой реакции может служить конденсация ацетоук-
сусного эфира с нитрилом акриловой кислоты:
СНз—СО—CHg—COOQHb^-CHg^CHCN —» СНд—СО—СН(
С этил атом натрия реакцию можно проводить при комнатной
температуре, оставляя раствор реагентов на два (или более) дня.
Для предотвращения обратной реакции используется
минимальное количество катализатора. Если катализатором является
пиперидин, обычно требуется более высокая температура реакции*.
Реакцию Михаэля изображают следующей схемой:
L _ ^О /\ ^ RL.
сс:Г + снюнС5=м —» %с—см.—
к" "R а"
189 Михаэль
При удалении протона от активированной метиленовой группы
получается анион, который реагирует с а,|3-ненасыщенным
соединением с образованием нового аниона. Последний может отнять
протон от сопряженного основания (ВН) или от активированной
метиленовой группы.
К многочисленным соединениям, содержащим активную
метил еновую группу и применяемым в реакции Михаэля, относятся
эфиры малоновой, ацетоуксусной и циануксусной кислот, а также
бензилцианид; к соединениям с активной двойной связью
(акцепторам) относятся а,р-непредельные альдегиды, кетоны, сложные
эфиры и нитрилы.
Внутримолекулярная реакция Михаэля описана для
некоторых орто-замещенных эфиров коричной кислоты^ (Z=O, S,
/СН-СН—СООСНд у\
г \—г-
—СН,—СООСНз
Конденсация калиевой соли динитрометана с двумя
молекулами акрилонитрила* может служить иллюстрацией
использования нитросоединений в реакции Михаэля:
OgN—СН=МОдК Ч
Метод присоединения по Михаэлю нашел удачное применение
в реакции Шторка (см. стр. 276).
1. A. Michael, J. prakt. Chem., 35, 349 (1887).
2. О реакции Михаэля см. Е. Bergman п, D. G i n s b u г g, R. Р а р-
р о, Organic Reaction, v. X, New York, 1959, p. 179; [К. И н гольд,
Механизм реакций и строение органических соединений, Издатинлит,
1959, стр. 553.—Дол. /Ы.].
3. С. К о е 1 s с h, С. Stephens, J. Am. Chem. Soc, 72, 2209 (1950).
4. L. H e rz og, С Gold, R. G e с k 1 e r, J. Am. Chem. Soc, 73, 749
(1951).
Наметкин* 190
Сергей Семенович Наметкин (1876—1950) родился в Казани. В 1902 г.
окончил Московский университет и остался там работать. В 1912 г. защитил
магистерскую, а в 1917 г.—докторскую диссертацию. С 1912 г. профессор кафедры
органической химии Московских высших женских курсов, реорганизованных
в 1918 г. во 2-й Московский государственный университет, а в 1930 г.—в
Московский институт тонкой химической технологии. С 1938 г. профессор Московского
университета. Одновременно в 1926—1934 гг. работал в Государственном
исследовательском нефтяном институте, а в 1934—1948 гг.—в Институте
горючих ископаемых АН СССР, являясь с 1939 г. его директором. С 1948 г. директор
Института нефти АН СССР. В 1932 г. был избран членом-корреспондентом, а
в 1939 г. действительным членом АН СССР. С. С. Наметкин долгие годы
занимался исследованиями в области терпенов. Большая часть его трудов посвящена
химии и технологии нефти (каталитическая ароматизация нефтяных фракций,
синтез хлорпроизводных и спиртов на основе углеводородов нефти, окисление
парафинов в спирты и альдегиды, получение моющих средств и др.). Работал
также в области синтеза душистых веществ, металлоорганических соединений
и стимуляторов роста растений.
ПЕРЕГРУППИРОВКА НАМЕТКИНА
Перегруппировка Наметкина^ или камфеновая
перегруппировка второго рода, состоит в перемещении одной из гел-метиль-
ных групп производных камфена из положения 2 в положение 3
в присутствии катализатора (серная кислота); при этом
происходит рацемизация.
Примером подобной реакции, сопровождаемой
перегруппировкой Вагнера—Меервейна, или камфлювой перегруппировкой пер-
* Биография С. С. Наметкина и изложение его реакции составлены
редактором.
191
Наметкин
вого рода (см. стр. 53 ), является превращение %-метилкамфе
на (I) в 4-метилизоборнеол-' * (II):
сн
I
(^/
\ /СН<
перегруппировке
Наметкина
_>
,он
гн<
перегру п пировк а
Вагнера - Меервейна
сн,
f
хш.
с-сн,
или
но—с—н
СН;
7!
н
зс:
н
1. Т. И. Темников а, Курс теоретических основ органической химии,
Госхимиздат, 1959, стр. 536.
2. С. С. Наметкин, Л. Я. Б р ю с о в а, ЖРХО, 60, 265 Г(1928)"
J. prakt. Chem., 124, 144 (1930); 135, 155 (1932).
3. С. С. Наметкин, А. И. Шаврыгин, ЖОХ, 4, 847 (1934У 6
1314 (1936). \ ;, ,
Несмеянов*
Александр Николаевич Несмеянов родился в 1899 г. в Москве. В 1922 г.
окончил Московский университет. Ученик Н. Д. Зелинского. С 1935 г.
профессор Московского университета, в 1948—1951 гг. ректор университета.
Одновременно с 1934 г. заведующий лабораторией металлоорганических соединении
Института органической химии АН СССР, с 1939 г. директор этого института -
С 1954 г. директор организованного по его инициативе Института элементоор-
ганнческих соединений АН СССР. В 1939 г. был избран
членом-корреспондентом, а в 1943 г. действительным членом АН СССР. С 1951 по 1961 г. являлся
президентом АН СССР. Основной областью исследований А. Н. Несмеянов:;
является химия элементоорганических соединений. Ряд его трудов
посвящен дальнейшей разработке теории химического строения (вопросы
стереохимии и таутомерии органических соединений, изучение механизма электро-
фильного замещения у насыщенного углеродного атома и др.).
РЕАКЦИЯ НЕСМЕЯНОВА
Способ получения металлоорганических соединений
разложением двойных солей галоидных производных арилдиазония и га-
логенидов металлов в присутствии в качестве восстановителя
мелко раздробленного металла известен под названием реакции
(«диазометода») Несмеянова*.
Эта реакция открыта и наиболее детально изучена на примере
ртутноорганических соединений-* \ Так, при разложении
двойной соли хлористого фенилдиазония и хлорной ртути в
присутствии порошкообразной меди с выходом до 86,5% получается
хлористая фенилртуть-' *' в* G;
2Cu -» CgH&HgCl ф N
,
При применении большого количества восстановителя в при
сутствии аммиака образуются полностью замещенные ртутноорга
* Биография А. Н. Несмеянова и изложение реакции составлены
редактором.
193 Несмеянов
нические соединения. В этих условиях из двойной соли
хлористого фенилдиазония и хлорной ртути получается дифенилртуть?
с выходом 65%:
+ 6Cu4- 6NH, -»- (CgH&)%Hg + 2N% + Hg
В дальнейшем реакция Несмеянова была использована для
синтеза металлоорганических соединений других металлов:
олова®, свинца^, сурьмы^, мышьяка^ и т. д.
Модификацией реакции Несмеянова является разложение
двойных солей фторида арилдиазония с трехфтористым бором
в присутствии ртути^, свинца^, таллия^ и олова^, например:
С оловом, таллием и свинцом по этому способу получены
лучшие результаты, чем по основной реакции Несмеянова.
Другой модификацией реакции Несмеянова является
образование ртутноорганических**, сурьмяноорганических^ и мышья-
ковоорганических^ соединений при разложении солей арилазо-
карбоновых кислот хлебной ртутью, треххлористой сурьмой
или треххлористым мышьяком, например:
C,H,N=N-COOK + HgCl, -^ QH^Hga + N% + COg + КС1
Реакция Несмеянова применима также для получения
алифатических металлоорганических соединений.
Реакция Несмеянова протекает по гемолитическому
механизму. Уотерсом было доказано образование свободных арильных
радикалов при прямом воздействии раствора хлористого диазония
на металл^.
1. О реакции Несмеянова см. Л. Г. Макарова, Реакции и методы
исследования органических соединений, кн. 3, Гослитиздат, 1954, стр. 73.
О. А. Реутов, Tetrahedron, 1, 67 (1957).
2. А. Н. Несмеянов, ЖРХО, 61, 1393 (1929); Вег., 62, 1010 (1929).
3. А. Н. Несмеянов, Z. anorg. allg. Chem., 178, 300 (1929).
4. А. Н. Несмеянов, Н. Н. Новикова, Изв. АН СССР, ОХН,
1942, 372.
б. Англ. пат. 638665; С А., 44, 9478 (1950).
6. А. Н. Несмеянов, Ученые записки МГУ, сер. хим., 3, 291 (1934).
7. А. Н. Несмеянов, .Э. И. К а н, ЖРХО, 61, 1407 (1929); Вег.,
62, 1018 (1929).
8. А. Н. Несмеянов, К. А. Кочешков, В. А. Климова,
ЖОХ, 6, 167 (1936); Вег., 68, 1877 (1935).
9. К. А. Кочешков, А. Н. Несмеянов, Н. К. Г и п п, ЖОХ,
6, 172 (1936).
13 А. Се. рей
Несмеянов 194
10. К. А. Кочешков, А. Н. Несмеянов, Изв. АН СССР, ОХН,
1944, 416.
П. А. Н. Несмеянов, Усп. хим.. 14, 261 (1945).
12. D u n k e г, S t а г к е у, Jenkins, J. Am. Chem. Soc., 58, 2308 (1936);
Dunker, Star key, J. Am. Chem. Soc, 61, 3005 (1939).
13. A. H. Несмеянов, К. А. Кочешков, M. М. Надь, Изв.
АН СССР, ОХН, 1945, 522.
14. А. Н. Несмеянов, Л. Г. Макарова, ДАН СССР, 87, 417
(1952).
15. А. Н. Несмеянов, Л. Г. Макарова, ДАН СССР, 87, 421
(1952).
16. А. Н. Несмеянов, О. А. Реутов, Изв. АН СССР, ОХН, 1948,
316.
17. О. А. Реутов, О. А. П т и ц и н а, Изв. АН СССР, ОХН, 1952, 93.
18. О. А. Реутов, Ю. Г. Б у н д е л ь, Изв. АН СССР, ОХН, 1952,
1041.
19. Waters, J. Chem. Soc, 1937, 113; 1937, 2007; 1937, 2014; 1938, 1077;
1939, 864; Ma kin, Waters, J. Chem. Soc, 1938, 843.
195 Неф
Джон Ульрих Неф (John Ulric Nef, 1862—1915) родился в г. Херизау
(Швейцария). Шестнадцати лет уехал в США, где в 1880 г. поступил в
Гарвардский университет. Затем учился в Мюнхене у А. Байера. В 1886 г. получил
степень доктора, вернулся в США и стал преподавать химию сначала в
университете Парду, а через два года в университете Кларка. С 1892 г. профессор
Чикагского университета, в котором работал до конца жизни. Еще будучи студентом
Мюнхенского университета, проявил интерес к вопросам таутомерии. В
дальнейшем исследовал таутомерные соединения, особенно кето-енольного типа*
и нитропарафиновые соли. В противоположность В. Мейеру утверждал, что
металл в этих солях связан не с углеродом, а с кислородом. С целью
получения метилена изучал химию двухвалентного углерода.
РЕАКЦИЯ НЕФА
Реакция Нефа** * заключается в образовании альдегидов или
кетонов при взаимодействии натриевых солей первичных или
вторичных нитропарафннов (ду%-форма) с избытком холодных
минеральных кислот:
2RCH=NONa + 2Н+ -» 2RCHO -f 2Na+ ф N,0 ф Н^Э
4
R, R,
2 )C=NONa + 2Н+ -* 2 )С=:О ф 2Na+ + N,0 4» Н,О
R'/ I R'/
О
Примером реакции Нефа является превращение 2-нитропро-
пана в ацетон* (выход 85%)
2 /C=N. + 2H+ -^ 2 /C=0 + 2Na+ nk N,0 + Ii,0
СНд/ ^ONa CH/
а также получение некоторых 5-фенилпиклогексен-1-онов-4 из
соответствующих 4-нитро-5-фенилциклогексенов^:
^ /^>
Неф
196
Интересное исследование механизма реакции Нефа
опубликовали в 1952 г. Темелен и Тиде*, показавшие, что механизм этой
реакции аналогичен механизму гидролиза оксимов и может быть
представлен следующей схемой:
о
о
\
н
—^
R,
R
:с=
нм:
"ОН
Предложенный механизм был подтвержден работами Лейча*,
который исследовал реакцию Нефа при помощи дейтерирован-
ного нитроэтана CH3CD2NO2.
Описано образование фуранов по методу Нефа, исходя из
аддуктов ацетоуксусного эфира и а-нитрсолсфинсв?:
R-C
N б
/ \
НО О"
сн-соос,н* -
с-сн,
—н*о
is—CH-
R-C
-СН—СООС,Нв
С—СНз
н
С,Н,-С-
C-COOQH&
R—С С-СН,
v
О реакции Нефа см. W. N"o4 and, Chem. Revs., 55, 137 (1955).
J Nf A 280 263 (1894)
ф
Nef, Ann., 280, 263 (1894).
Johnson, E. Degering, J.Org. Chem.. 8, 10(1943).
Wildman, R. Wildman, J.Org. Chem., 17, 581(1952).
hd J A Ch S 2615 (I
2. J.
3. К.
5. Е. v a n fa me I en, R. T h i ed e, J. Am. Chem. Soc.i 74, 2615 (IO."2).
6. L. L e i t с h, Can. J. Chem., 33, 400 (1955).
7. F. Bob erg, G. Schu I tze, Chem. Ber., 90, 1215 (1957).
197 Оппенауер
Руперт В. Оппенауер (Rupert V. Oppenauer) родился в 1910 г. в Бургштале
(Италия). Учился в Цюрихской высшей технической школе у Л. Ружички и
Т. Рейхштейна. В 1934 г. получил степень доктора философии. Работал
ассистентом в Цюрихском университете, затем в Амстердамском университете у Ла-
кера и в Йенском университете под руководством Е. Мюллера. С 1946 г.
преподавал в Инсбрукском университете, с if.48 г. работал в фирме Hofmann—
La Roche в Базеле. В настоящее время работает в министерстве
здравоохранения Аргентины в Буэнос-Айресе. Большинство его работ посвящено синтезу
стероидов.
ОКИСЛЕНИЕ ПО ОППЕНАУЕРУ
Окисление вторичных спиртов до кетонов при обработке
спиртов смесью ацетона и /пре/п-бутилата (или изопропилата)
алюминия называют реакцией окисления по Оппенауеру^.Эта реакция
является обратной реакции восстановления по Меервейну—
Понндорфу—Верлею (см. стр. 186) и может быть представлена
следующим общим уравнением:
}4 }+ >
R'/ СН/ R'/ СН/
R СН%,
>снон
3
Другие кетоны (циклогексанон или метилэтилкетон) также
могут применяться вместо ацетона. Хотя некоторые первичные
спирты окисляются по методу Оппенауера, реакция имеет большее
значение как метод окисления вторичных спиртов, особенно в ряду
стероидов. Окисление является избирательным, и другие группы,
а также двойные углерод-углеродные связи не затрагиваются.
При большом избытке ацетона равновесие сдвигается вправо.
В результате обменной реакции с алкоголятом алюминия
образуются алюминиевые производные окисляемых оксисоединений.
Реакцию можно проводить в таких растворителях, как бензол или
толуол, при температуре их кипения. Присутствие аминогруппы
в молекуле спирта осложняет реакцию окисления*, так как, по-
видимому, образуется координационная связь между алюминием
и азотом.
Оппенауер
198
Весьма важным случаем окисления по Оппенауеру в ряду
стероидов* является превращение З-окси-А'-соединений в
соответствующие З-кето-А^-соединения. Иллюстрацией такого
окисления с миграцией двойной связи из |3,у-положения в а,Р-положение
служит превращение холестерина в холестенон* (выход 70—80%):
R
'\
x^
Найдено^, что хромовый ангидрид в пиридине является очень
аффективным окислителем стероидных спиртов. Этот реагент
(редк/шм Сар#л/лд) избирательно окисляет гидроксильные группы
в карбоксильные, не затрагивая двойные связи и чувствительные
к кислотам группы.
1. R. Oppenauer, Rec. trav. chim., 56, 137 (1937).
2. О реакции Оппенауера см. К. Дьерасси, Синтезы органических
препаратов, сб. 6, Издатинлит, 1953, стр. 235.
3. R. L u t z, R. Way I a nd, J. Am. Chem. Soc, 73, 1639 (1951).
4. W. Johnson, G. Skrimshire, Chem. Ind. (London), 380 (1951).
5. P. Оппенауер, Синтезы органических препаратов, сб. 3, Издатинлит,
1952, стр. 486.
6. G. Poos, G. А г t h, R. Beyler, L. Sarett, J. Am. Chem. Soc.,
75, 422 (1953).
199
Пассерини
Марио Пассерини (Mario Passerini) родился в 1891 г, в г. Скандиччи (Ита_
лия). Учился во Флорентийском университете, где ему была присуждена
степень доктора химии и фармации. Сначала ассистент, затем доцент института
химии и фармации во Флоренции. С 1930 г. преподавал в Сиенском
университете. В настоящее время работает во Флоре нции в области выделения
природных соединений из растительного сырья.
РЕАКЦИЯ ПАССЕРИНИ
Взаимодействие ароматических изонитрилов с карбонилсодер-
жащими соединениями и карбоновыми кислотами, в результате
которого образуются %-ацилоксианилиды, получило название
реакции ПассериниЧ
СО + ArNC KR'COOH -
yCONHAr
'"^OCOR"
Щелочной гидролиз образовавшегося продукта приводит к
соответствующим а-оксианилидам. Недавно было показано*, что
протекания побочных реакций можно избежать, если смешивать
эквимолекулярные количества реагентов при 0—25° и проводить
синтез при —20°. Это позволяет предположить*, что реакция
Пассерини протекает через стадию образования циклического
промежуточного реакционного комплекса I:
R
АгМСф "ХЗЭ
R'-
ArN=C
R'COOH
ArN
,R RCOOH
=C-C< ^
ArN=C
О"
R-
I \l)'
H—О О
v
(I)
»R'
о-
HO OCOR"
Пассерини 200
Если в реакции используется оптически активная карбоновая
кислота, то один из диастереоизомерных продуктов получается
в значительном избытке, что указывает на возможность
применения этой реакции для асимметрического синтеза^.
ЛЯГЕРЛГУРЛ
1. М. Passerini, Gazz. chim. ital., 51, 126 (1921); С. А., 16, 555 (1922).
2. R. Baker, D. S t a n о n i s, J. Am. Chem. Soc., 73, 699 (1951).
3. R. В aker, L. L inn, J. Am. Chem. Soc, 70, 3721 (1948).
201 Перкин
Вильям Генри Перкин (William Henry Perkin, 1838—1907) родился в
Лондоне. Учился у А. Гофмана в Лондонском королевском химическом колледже .
В 1856 г., работая в домашней лаборатории над синтезом хинина, получил при
окислении анилина первый синтетический органический краситель—м/эвеин.
Вскоре он открыл фабрику для производства этого красителя, а позднее по.
лучил другие красители, в том числе ализарин. В 1874 г. прекратил
коммерческую деятельность и посвятил себя исключительно исследовательской работе.
Открыл реакцию, названную его именем, синтезировал кумарин и коричную
кислоту. Изучал зависимость между химическим строением и физическими
свойствами веществ, в частности вращение плоскости поляризации света в
магнитном поле. За выдающиеся заслуги в области науки и промышленности удостоен
многих наград. Американская секция общества химической промышленности
учредила медаль Перкина, ежегодно присуждаемую за лучшие достижения в
американской химической промышленности.
РЕАКЦИЯ ПЕРКИНА
Реакция Перкина^ заключается в получении (3-замещенных
производных акриловой кислоты конденсацией ароматических
альдегидов с ангидридами карбоновых кислот в присутствии
оснований.
В качестве оснований обычно используются натриевые или
калиевые соли тех карбоновых кислот, ангидриды которых
участвуют в реакции. Примером реакции Перкина является синтез
коричной кислоты из бензальдегида и уксусного ангидрида в
присутствии ацетата калия:
СНзСООК
CgH,—СНО + (СНзС0),0 *-
СдН,—СН=СН—СООН + СНз—СООН
Смесь реагентов нагревают в течение 5 час. с обратным
холодильником при 175—180° и выливают плав в воду. Затем непро-
реагировавший бензальдегид отгоняют с водяным паром и после
подкисления выделяют продукт реакции. Вследствие жестких
условий процесса выходы обычно не очень хорошие. Однако при
Перкин 202
введении электроноакцепторных заместителей в ядро
ароматического альдегида выход продукта конденсации улучшается*.
Перкин считал, что альдегиды конденсируются с ангидридами,
а ацетат натрия является только основным катализатором*.
Кроме щелочных ацетатов, в реакции Перкина могут
использоваться и другие катализаторы основного характера. Примером
использования таких конденсирующих агентов служит
применение триэтиламина при конденсации /г-оксибензальдегида с фенокси-
уксусной кислотой*:
соон
но— ^^
Другим аналогичным примером является синтез кумарина из
салицилового альдегида? (выход 34%):
-К(СН,СО),О
^/V\)
В качестве побочного продукта этой реакции образуется
некоторое количество о-ацетоксикоричной кислоты.
Стереохимия взаимодействия бензальдегида с фенилуксусной
кислотой в условиях реакции Перкина обсуждалась
Циммерманом и Арамяном*. Основным продуктом этой конденсации
является а-фенилкоричная кислота, содержащая фенильные группы в
ffuc-положении. Механизм реакции можно изобразить следующей
схемой:
—CHO ф СдНд—СН,—СООН
CHg—COO/ XXX)—CO—g
QHg, /CgHg CgHg, /CgHg
/CH—C. -^ /C=(\
-<ХХХ \COO—CO—CH, H/ X:OO—OO-CH;
203 Перкин
Кроуфорд и Литли изучали реакцию Перкина с
пространственно незатрудненными алифатическими альдегидами в мягких
условиях.
1. W. *Р е г k i n, J. Chem. Soc, 21, 53, 181 (1869); J. Chem. Soc, 31, 388
(1877).
2. Д. Джонсон, Органические реакции, сб. I, Издатинлит, 1948, стр. 267.
3. О реакции Перкина см. P. Leak e, Chem. Revs., 56, 27 (1956).
4. G. Lock, E. Bauer, Ber., 72, 1064 (1939).
5. О механизме реакции Перкина см. D. Breslow, С. Hauser, J. Am.
Chem. Soc., 61, 786, 793 (1939); V. F г а п z e n, Chem. Ztg., 80, 166
(1956).
6. D. Papa, E. S с h w e n k, пат. США 2503296, 1950 г.; С. А., 44, 6886
(1950).
7. R. Buckles, J. Chem. Educ, 27, 210 (1950).
8. H. Zimmerman, L. A h г a m j i a n, J. Am. Chem. Soc, 8%, 2086
(1959).
9. M. Crawford, W. Little, J. Chem. Soc, 1959, 722.
Пехман 204
Ганс Пехман (Hans Pechmann, 1850—1902) родился в Нюрнберге. Учился
в Мюнхене и Гейдельберге, затем у Лимприхта в ГреАфсвальдском
университете, где в 1875 г. защитил докторскую диссертацию на тему: жСульфо кислоты
/1-тол у и дин а*. С 1875 по 1877 г. работал с Э. Франкландом в Лондонском
университете, а в дальнейшем с А. Байером в Мюнхенском университете, где стал
доцентом кафедры аналитической химии. С 1895 по 1902 г. преподавал в Тю-
бингенском университете. Синтез кумаринов является лишь одной из его
многочисленных работ. Он первый получил целый ряд важных органических
соединений, в том числе а цетонди кар боковую кислоту, ди ацетил и другие 1,2-ди-
кетоны, а также метилглиоксаль и диазометан. Обнаружил, что при бензоили-
ровании диазобензола образуется нитрозобензанилид.
КОНДЕНСАЦИЯ ПЕХМАНА
Образование производных кумарина при взаимодействии
фенолов с эфирами Р-кетокислот в присутствии таких
конденсирующих агентов, как серная кислота, хлористый алюминий или пяти-
окись фосфора, называется конденсацией Пехмана'-з.
Примером этой реакции может служить получение 4-метил-
кумарина* из фенола и ацетоуксусного эфира (выход 40—55%):
,—со—сн,-соосл
2^5
Другим примером является синтез метилового эфира 5-окси-
4,7-диметилкумаринкарбоновой-6 кислоты (II) из метилового
эфира орселлиновой кислоты (I) и ацетоуксусного эфира* (в
охлажденную смесь реагентов добавлялась концентрированная серная
кислота):
он он сн
|1
II
205 Пехман
В реакцию Пехмана могут вступать разнообразные фенолы
и эфиры р-кетокислот. При этом активность фенолов зависит
от присутствия в ядре тех или иных заместителей; галогены и
сильные электроноакцепторные заместители обычно затрудняют
конденсацию.
Показано, что прекрасным конденсирующим средством в
реакции Пехмана является полифосфорная кислота*' ?. При
взаимодействии разных фенолов с этиловым эфиром ацетоуксусной
кислоты в присутствии полифосфорной кислоты продукты
реакции получаются в более чистом виде и с большими выходами,
чем с серной кислотой.
В качестве конденсирующих средств в реакции Пехмана
применялись также катионообменные смолы® (например, амберлит
JR-120). В данном случае при проведении реакции в я-гексане
скорость процесса резко увеличивалась, а выход кумарина
повышался.
Фенолы, с трудом вступающие в реакцию или совсем не
реагирующие под действием серной кислоты, конденсируются с
образованием хромонов в присутствии пятиокиси фосфора (рткцшг Cw-
лкмшсд*). Выходы хромонов обычно низкие**» ^.
Наличие в молекуле ацетоуксусного эфира заместителей в а-по-
ложении облегчает течение реакции Симониса. Примером такой
реакции может служить синтез 8-хлор-2-метил-3-пропилхромона-4
при конденсации о-хлорфенола и этилового эфира а-пропилацето-
уксусной кислоты^:
О
—CO—CH—COOQH,
1. Н. Pechmann, С. D u i s b e rg, Ber., 16, 2119 (1883).
2. S. Sethna, N. Shah, Chem. Revs., 36, 10(1945).
3. О реакции Пехмана см. С. С е т н а, Р. П х а д к е, Органические
реакции, сб. 7, Издатинлит, 1956, стр. 7; см. также Гетероциклические
соединения, под ред. Р. Эльдерфильда, сб. 2, Издатинлит, 1954, стр. 139;
обзор по природным кумаринам см. F. Dean, Progress in the
Chemistry of Organic Natural Products, v. 9, Vienna, 1952, p; 225.
* Г. Симонис (Н. Sim on is) родился в 1874 г. в Дюссельдорфе (Германия).
В 1897 г. получил степень доктора философии во Фрейбургском
университете. С 1919 г. профессор органической химии в Берлинском техническом
училище.
Пехман 206
4. Э. Вудрафф, Синтезы органических препаратов, сб. 3, Издатинлит,
1952, стр. 305.
5. P. Sara i у a, R. Shah, Ргос. Indian Acad. Sci., 31, 213 (1950); С. А,
46, 5013 (1952).
6. J. К о о, Chem. Ind. (London), 445 (1955).
7. R. К а р i I, S. J о s h i, J. Indian Chem. Soc, 36, 596 (1959).
8. S. Israels! am, E. John, Chem. Ind. (London), 1262 (1958).
9. E. Petschek, H.Simon is, Ber., 46, 2014(1913).
10. S. Sethna, N. Shah, Chem. Revs., ?6, 14 (1945).
11. D. С h a k г a v а г t i, J. Indian Chem. Soc., 9, 25 (1932).
207 Пиктэ—Шпенглер
Аме Пиктэ (Ame Pictet, 1857—1937) родился в Женеве. Учился в Женеве,
Дрездене, Бонне и Париже. Работая под руководством К. Грэбе в Женевском
университете, защитил докторскую диссертацию. В дальнейшем преподавал
в этом университете и в 1899 г. стал профессором биологической и
фармацевтической химии. Изучая химию растительных алкалоидов, синтезировал
никотин, лауданозин и папаверин. В последние годы жизни занимался химией
углеводов.
РЕАКЦИЯ ПИКТЭ—ШПЕНГЛЕРА
Конденсацию Р-арилэтиламинов с карбонильными
соединениями, приводящую к образованию производных тетрагидроизохино-
лина, обычно называют реакцией Пиктэ—Шпенглера^%
Из используемых в реакции альдегидов лучшие результаты
дает формальдегид. Шиффпвы основания, образующиеся в первой
стадии реакции, могут быть в некоторых случаях выделены или
непосредственно циклизованы при нагревании с водной соляной
кислотой:
NH
CH,0 H,C
Активация орто-положения метокси- или оксигруппами
сходна с описанной активацией для реакции Бишлера—Напиральско-
го (см. стр. 33).
Во многих случаях реакция Ликтэ—Шпенглера проводится в
условиях (кислотность, температура, концентрация реагентов),
близких к природным. Возможно, что по этому типу протекают
реакции образования изохинолиновых алкалоидов в растениях.
1. A. Pictet, Т. S р е п % 1 е г, Вег., 44, 2030(1911).
2. О реакции Пиктэ—Шпенглера см. В. М. У э л и, Т. Р. Г о в и н д а-
ч а р и, Органические реакции, сб. 6, Издатинлит, 1953, стр. 177; см*
также статью В. Генслера в книге «Гетероциклические соединения»,
сб. 4, Издзтинлит, 1955, стр. 269.
Померанц—Фрич
208
Цезарь Померанц (Cesar Pomeranz, 1860—1926) получил степень доктора
философии в Венском университете, где позднее стал адъюнкт-профессором.
Затем работал в Черновицах.
Пауль Фрич (Paul Fritsch, 1859—1913) родился в г. Оэлсе (Германия).
Учился в Берлине и Мюнхене, где в 1884 г. ему была присуждена степень
доктора. Работал ассистентом в университетах Мюнхена, Бреслау и Ростока,
Профессор химии Марбургского университета. Занимался изучением реакций
конденсации хлорала и синтезом замещенных ароматических кислот,
ароматических аминов и производных трифенилметана.
РЕАКЦИЯ ПОМЕРАНЦА—ФРИЧА
Циклизация бензальаминоацеталей под действием серной
кислоты, приводящая к образованию производных изохинолина,
носит название реакции Померанца—Фрича\
В качестве конденсирующего агента в этой реакции
используется серная кислста или смеси серной кислоты с другими
кислотными реагентами* (хлористый водород, хлорокись фосфора,
уксусная кислота):
н
СН,
N
N
Бензальаминоацетали получаются из соответствующих
ароматических альдегидов и аминоацеталей. Шлиттлер и Мюллер*
видоизменили эту реакцию, применив бензиламин и полуацеталь
глиоксаля:
СН(ОС*Н,),
сн .
сно
N
/
сн
/
сн,
209
Померанц—Фрич
Наилучшие выходы изохинолинов по реакции Померанца—
Фрича достигаются приприменешиж-окси-, ж-алкокси- и jw-гало-
ид-бензальаминоацеталей; о- и л-замещенные бензальаминоацета-
ли не вступают в реакцию или дают низкие выходы
соответствующих производных изохинолина.
Папаверальдин, который в дальнейшем может быть
восстановлен до папаверина, синтезирован недавно с 8%-ным выходом из
неочищенного монооснования Шиффа—М-(%-вератроилвератрил-
иден)-аминоацеталя (I). Циклизация проводилась при комнатной
температуре под действием 72%-ной серной кислоты*:
HqCO
CH,-CH(OC,H*),
N
N
СО
Н.СО
НдСО
н,со
со
Вино* описал синтез производных тетрагидроизохинолина из
бензилами ноацеталей в присутствии трехфтористого бора:
R
NH
\/
R
ЛЯГЕРЛПРЛ
C. Pomeranz, Monatsh., 14, 116 (1893); P. Fr i tsch, Ber., 26, 419
(1893).
О реакции Померанца—Фрича см. В. Г е н с л е р, Органические реакции,
сб. 6, Издатинлит, 1953, стр. 218; В. Г е н с л е р, Гетероциклические
соединения, сб. 4, Издатинлит, 1955, стр. 279.
Е. Sch I i t t 1 ег, J. МО I I er, Helv. chim. acta, 31, 914 (1948).
D. G u t h г i e, A. Frank, C. P u г v es. Can. J. Chem., 33, 729 (1955).
[]
5. N. V i not, Ann. chim. (Paris), [13], 3, 461 (1958).
14 А. Сегрсй
Принс 210
Гейндрик Джекобус npHHc(HendrikJacobusPrins, 1889—1958) родился
в г. Заандаме (Голландия). В 1911 г. окончил Дельфтский технологический
институт, получив звание химика-технолога, а через год под руководством Дж. Бе*
зекена выполнил и защитил докторскую диссертацию. Работал химиком
сначала в компании эфирных масел, а с 1924 г. в компании термохимических фабрик
Голландии, где впоследствии занимал пост директора-председателя. Г. Принс
имел небольшую личную лабораторию, в которой в свободное время занимался
экспериментальными исследованиями. Его интересы в органической химии
были сосредоточены главным образом в области полихлоридов.
РЕАКЦИЯ ПРИНСА
Конденсация альдегидов с олефинами в присутствии
кислотных катализаторов обычно называется реакцией ПринсаЧ
Эта реакция с формальдегидом, по-видимому, носит общий
характер, но направление реакции зависит в большой мере от
природы катализатора и условий проведения процесса*. Чаще
всего применяются следующие три варианта реакции Принса:
конденсация в присутствии кислотного катализатора;
конденсация под действием галоидных металлов; термическая конденсация.
Конденсация в присутствии кислотного
катализатора. Если катализатором служит минеральная
кислота (обычно серная), а в качестве растворителя используют
воду, то основными продуктами реакции являются jw-диоксаны и
1,3-гликоли:
CHgv 25%-ная1^ЕбГ СНух у ^ -^*СНз\ /СН,—СН,—ОН
При удачном выборе условий конденсации один из продуктов
реакции может оказаться преобладающим. Например, из стирола*
синтезирован 4-фенил-ж-диоксан с выходом 71—88%. При
конденсации альдегидов с первичными олефинами при строгом
соблюдении температурного режима (выше 70°), времени реакции и
применении катализатора определенной концентрации в качестве
главного продукта могут быть получены 1,3-гликоли*. Если
растворителем служит уксусная кислота, то одним из конечных
продуктов будет диацетат соответствующего 1,3-гликоля.
211 Принс
3 Имеются данные*, что реакция Принса с /прдмс-коричной
кислотой в растворе смеси уксуснсй и серной кислот при 80*
приводит к образованию* 4-фенил-я-диоксанкарбоновой-5 кислоты с
выходом 25%. Амид указанной кислоты после реакции Гофмана
и размыкания кольца был превращен действием флороглюцина и
соляной кислоты в производное 1,3-гликоля. Осуществить
реакцию Принса с w-нитростиролом не удалось.
Конденсация под действием галоидных
металлов. Некоторые третичные олефины, содержащие 4—
8 атомов углерода, конденсируются с формальдегидом в безводной
среде, образуя ненасыщенные спирты^:
СН,
SnCl* I
,С= СН, + СН,0 » СН,=С-СН,—
Катализаторами этой реакции служат хлорное олово,
хлористый цинк и четыреххлористый кремний. Используя в качестве
катализатора хлористый алюминий или хлорное железо, можно
получить At-диоксаны. Первичные олефины так же как и
высшие альдегиды реагируют с образованием алкил-л*-диоксаноа.
Термическая конденсация. При нагревании
третичных олефинов с формальдегидом в отсутствие катализатора
получаются ненасыщенные спирты?. Так, например, метиленцикло-
гексан конденсируется с формальдегидом в запаянной трубке в
течение 4 час. при 200°, образуя 2-(циклогексенил-Н)-этанол:
СН.-СН.ОН
4- СН.0 -»
Оптимальная температура этой реакции, по-видимому, составляет
180—200°. При конденсации в присутствии ледяной уксусной
кислоты получается ацетат ненасыщенного спирта.
Соображения о механизме реакции Принса были высказаны
недавно Янгом*. Он полагает, что поляризованный комплекс
формальдегида с катализатором присоединяется к олефину согласно
правилу Марковникова:
НСНО 4» А ^ Н,С—О—А (А = SnCI, или кислота)
СН,
СН/ Ж . СН/ Y
О—А
И*
Принс
212
Промежуточное соединение может затем реагировать или как
шестичленный цикл, превращаясь в ненасыщенный спирт:
^
сн
или взаимодействовать с растворителем или формальдегидом,
образуя гликоль, сложный эфир или л-диоксан:
СН,
Растворитель
СН
СН
О—А
СН&О
СН,
сн.
_он он
сн„
СН
с сн%
о о
Арнольд и Доудэлл? предположили, что при термической кон
денсации по Принсу реакция также протекает между поляризо
ванной молекулой формальдегида и олефином:
СН;
О"
Стереохимия превращений, происходящих в процессе
конденсации по Принсу, изучена многими исследователями^-** .
Показано, что конденсация циклогексена с формальдегидом в
присутствии кислотного катализатора заключается в /мрдмс-д и
аксиальном присоединении по двойной связи. В качестве основных
продуктов этой реакции были выделены /n/xzwc-2-оксиметилцикло-
гексанол-1 и /мрд«с-4,5-тетраметилен-1,3-диоксан.
Четвертым вариантом реакции Принса является конденсация
альдегидов с олефинами под действием перекисей, при которой
213 Принс
образуются кетоны. Наилучшие выходы кетонов получевы при
взаимодействии альдегидов с большим молекулярным весом и
высших олефинов с концевой двойной связью^.
1. Н. Prins, Chem.. Weekbl., 14, 627 (1917).
2. О реакции Принса см. Е. А г u n d a I e, L. Mikeska, Chem. Revs.,
5%, 505 (1952).
3. Р. Ш р а Й н е р, П. Р ю б и, Органические препараты, сб. 5, Издатин-
лит, 1954, стр. 76.
4. L. Mikeska, Е. Arundale, пат. США 2449001, 1948 г.; С. А.,
43, 673 (1949).
5. F. Brugman, J. Areus, Rec. trav. chim., 74, 209 (1955).
6. E. A r u n d a 1 e, L. Mikeska. Chem. Revs., 51, 532 (1952).
7. R. Arnold, J. Dowd al 1, J. Am. Chem. Soc, 70, 2590 (1948).
8. N. Yang, D. Yang, C. Ross, J. Am. Chem. Soc., 81, 133 (1959).
9. С Fodor, O. Kovacs, I. Tomoskozi, J. Szilagyi, Bull.
Soc. chim. France, 357 (1957).
10. E. Sm issm an, R. Mode, J. Am. Chem. Soc, 79, 3447 (1957).
11. А. В I о m q u i s t, J. W о I i u s ky, J. Am. Chem. Soc., 79, 6025 (1957).
12. E. А г u n d a I e, L. Mikeska, Chem. Revs., 51, 544 (1952).
Пфитцингер 214
Вильгельм Пфитцингер (Wilhelm Pfitzinger) начал свои исследования в
области синтеза замещенных цинхониновых кислот из изатина и кетонов в
1885 г. в Лейпцигском университете. С 1888 г. сотрудничал в фирме Friedrich
Bayer Со. в г. Элберфельде. С 1892 г. по 1897 г. продолжал заниматься
изучением синтеза хинолина в Дрезденской высшей технической школе в
лаборатории Е. Мейера. В 1902 г., работая в Берлинском технологическом институте,
открыл реакцию изатиновой кислоты с оксимами альдегидов.
РЕАКЦИЯ ПФИТЦИНГЕРА
Образование производных цинхониновой кислоты (III) при
взаимодействии изатина (I) или изатиновой кислоты (II) с
соединениями, содержащими группировку —СН,СО—, описано в
1886 г. и получило название реакции Пфитцингера^^.
Реакцию можно изобразить следующей общей схемой:
соон
В-СН*СО—R'
ш
Реакция Пфитцингера сходна с методом синтеза производных
хинолина по Фридлендеру, исходя из о-аминобензальдегида или
о-аминофенилкетона.
При использовании в реакции Пфитцингера не метилэтилкето-
нов, а других метилкетонов, в конденсацию вступает метильная
215
Пфитцингер
группа кетона*. В соответствии с этим в качестве основного
продукта реакции метил-#-пропилкетона с изатином получается 2-м-про-
пилцинхониновая кислота:
соон
CO-QH,
Из продуктов взаимодействия с высшими несимметричными
%-алкилкетонами были выделены и идентифицированы также
изомерные 2,3-дизамещенные производные цинхониновой кислоты*.
Недавно пол учено* несколько производных этих кислот,
замещенных высшими алкильными группами в положении 2.
Установлено, что во многих случаях при пиролизе
производных цинхониновой кислоты наряду с ожидаемым 2-алкилхино-
лином образуются также соответствующие 4-оксипроизводные^:
соон
Реакция некоторых пропоксиметилкетонов с изатином
приводит к 2-алкил-З-пропоксицинхониновым кислотам (IV) (здесь
R =CgH, или высший алкильный радикал)^:
СООН
CO-R
Мэллер и Штобо? недавно сообщили о роли пространственных
затруднений в реакции Пфитцингера^. Было показано, что три-
метилдезоксибензоин (V)
СН,
v CH,
не вступает в реакцию конденсации с изатином.
Пфитцингер 216
1. W. Pf i tzi nger, J. prakt. Chem., 33, 100 (1886); 38, 582 (1888).
2. R. M a n s к e, Chem. Revs., 30, 126 (1942); P. Эльдерфильд, в
книге «Гетероциклические соединения», сб. 4, Издатинлит, 1955, стр. 36.
3. N. Buu-Hoi, R. Royer, J.Chem. Soc., 1948, 106.
4. H. H e n z e, D. Carroll, J. Am. Chem. Soc, 76, 4580 (1954).
5. N. В u u-H о i, P. J а с q u i g n о n, Buil. Soc. chim. France, 1567 (1958).
6. H. H e n z e, J. Melton, E. F о г m a n, J. Am. Chem. Soc, 70, 2622
(1948).
7. G. Mueller, R. Stobaugh, J. Am. Chem. Soc, 72, 1598 (1950).
8. N. В u u-H of, R. Royer, N. X u о n g, P. J а с q u i g n о n,
J. Org. Chem., 18, 1209 (1953).
217 Пшорр
Роберт Пшорр (Robert Pschorr, 1868—1930) родился в Мюнхене. Изучал
химию в Мюнхенском университете у А. Байера, в Цюрихском университете
под руководством Э. Бамбергера и в Йенском университете у Л. Кнорра
(с 1892 г.). Работал с Э. Фишером в Берлинском университете, где в 1899 г.
стал адъюнкт-профессором. Заведующий кафедрой органической химии (1913—
1914 гг.), затем ректор (с 1920 г,) Берлин-Шарлоттенбургского
технологического института. Работал над синтезом пиразолов и получил 4-кетопиразолон.
Занимаясь исследованиями алкалоидов опия, особенно группы морфина,
предложил широко известный синтез производных фенантрена; изучал также
расщепление апоморфина и тебаина.
СИНТЕЗ ПШОРРА
Получение производных фенантрена из о-нитробензальдегида
и арилуксусных кислот носит название синтеза Пшорра^-^.
Первая стадия реакции заключается в образовании а-фенил-
о-нитрокоричной кислоты (I):
сно
При восстановлении нитрокислоты I образуется
аминокислота II:
*сн \с-==сн
На
Последующим диазотированием аминокислоты II получают ди-
азосоьдинение 111, которое при обработке порошкообразной медью
Пшорр
218
щиклизуется в 9-фенантренкарбоновую кислоту (IV) с выделением
азота и хлористого водорода:
НООС
ноос
IV
Синтез Пшорра имеет широкое применение. Используя
замещенные о-нитробензальдегиды и фенилуксусные кислоты, по
этому методу можно получить ряд производных фенантрена. Из наф-
тилуксусных кислот образуются производные бензфенантрена.
Аналогичной циклизацией получают замещенные карбазолы,
флуорены и флуореноны*. Так, например, из 4-бром-2'-аминобен-
зофенона синтезирован 3-бромфлуоренон^:
О О
Синтез Пшорра с успехом применялся для установления строе
ния природных веществ, особенно алкалоидов опия?.
Хей и Осбонд* показали возможность удовлетворительного
использования различных модификаций синтеза Пшорра для
получения гетероциклических систем, в последней стадии
циклизации которых образуется пиридиновое ядро. Авторы нашли также,
что присутствие в %-фенильном ядре таких дезактивирующих
групп, как —N0%, —CN или —СООН, не препятствует ходу
реакции. Это подтверждает предположение о свободнорадикаль-
ном механизме синтеза Пшорра.
L
2.
3.
4.
5.
6.
7.
P.
д.
R
К
H
H
Л
8. D,
Leake, Chem. Revs., 56, 27 (1956).
Д е-Т а р, Органические реакции, сб. 9, Издатинлит, 1959, стр. 529.
Р s с h о г г, Вег., 29, 496 (1896).
Saunders, Reactions of the Diazo-Compounds, London, 1949, p. 254.
Hodgson, J. Chem. Soc, 1948, 348; K. Saunders, Reactions
of the Diazo-Compounds, London, 1949, p. 2581
Miller, G. Bachmann, J. Am. Chem. Soc, 57, 2443 (1935).
Ф и з е р, М. Ф и з е р, Химия природных соединений фенантренового
ряда, Госхимиздат, 1953.
Не у, J. Osbond, G. Chem. Soc, 1949, 3164, 3172.
219 Реймер—Тиман
Карл Людвиг Реймер (Karl Ludwig Reimer, 1845—1883) родился в Лейп-
щиге. Учился в Геттингенском и Грейфсвальдском университетах. Работал
в Гейдельберге и Берлине, где в 1871 г. получил степень доктора философии.
Преподавал в Лесной академии, затем работал химиком в фирме Kahlbaum Co.
В 1876 г. стал компаньоном фирмы Vanillin Co. В 1875 г. синтезировал из
фенола и хлороформа салициловый альдегид, что дало возможность получать
его в больших количествах.
Иоганн Карл Фердинанд Тиман (Johann Karl Ferdinand Tiemann, 1848—
1899) родился в г. Рюбеланд (Германия). Учился у А. Гофмана в Берлинском
университете, где получил степень доктора; с 1882 г. профессор химии этого
университета. Известен своими исследованиями в области нитрилов и терпенов.
Осуществленный им синтез ванилина, в котором он применил реакцию Реймера
для получения о-оксиальдегидов, сыграл большую роль в развитии
парфюмерной промышленности. В 1893 г. совместно с П. Крюгером выделил из корневища
ириса ирон—вещество с запахом фиалки, а в 1894 г. конденсацией цитраля с
ацетоном получил псевдоионок и превратил его в ионон, также имеющий
запах фиалки.
РЕАКЦИЯ РЕЙМЕРА—ТИМАНА
Получение ароматических о-оксиальдегидов путем введения
формильной группы в фенолы при нагревании их с хлороформом
в присутствии щелочи носит название реакции Реймера—Ти-
мана^ з.
Примером этой реакции может служить получение
салицилового альдегида из фенола и хлороформа*:
,ОН
+ СНС1, + ЗКОН -» I I 4* ЩС1 ф 2Н„О
Обычным продуктом реакции Реймера—Тимана является
о-формильное производное фенола, но наряду с ним образуется
также некоторое количество л-изомера. Это имеет особенно
важное значение, когда орто-положение в молекуле фенола занято.
Если в приведенном выше уравнении хлороформ заменить четы-
реххлористым углеродом и реакционную смесь нагревать при
Реймер—Тиман
220
100° в запаянной трубке, то образуется смесь салициловой и л-ок-
сибензойной кислот.
Другим примером реакции Реймера—Тимана является
превращение Р-нафтола в 2-окси-1-нафтальдегид* (выход 38—4б%);
СНО
ОСУ
ОН
Область применения реакции Реймера—Тимана довольно
ограничена. Электроноакцепторные заместители в ароматическом ядре
затрудняют течение реакции, и выходы ожидаемых продуктов
обычно невелики. В ряде случаев образуются другие продукты,
позволяющие установить возможный механизм реакции.
Например, при взаимодействии л-крезола с хлороформом были
выделены два продукта*: 6-окси-З-метилбензойный альдегид (I) и 4-ди-
хлорметил-4-метилциклогексадиен-2,5-он (П):
ОН О
СНО
сна,
Образование кетона II указывает на то, что дихлорметильные
производные являются, по-видимому, промежуточными
продуктами реакции Реймера—Тимана.
Вудворд' показал, что при помощи так называемой аномальной
реакции Реймера—Тимана ог-Р-тетралол может быть превращен
в /прояс-10-метилдекалон-2:
сна.
Этим же способом через соответствующий промежуточный дие-
нон получен? /л/%гяс-9-метилдекалон-1:
сна*
221 Реймер—-Тиман
Показано, что по реакции Реймера—Тимана из мезитола
наряду с небольшим количеством мезитилортоформиата образуются
два изомерных диенонаЧ
Обзор по реакции Реймера—Тимана опубликован недавно
Вайнбергом*.
1. О реакции Реймера—Тимана см: Н. W у п b e г g, Chem. Revs., 60, 169
(1960).
:2. К. R e i m е г, Бег., 9, 423 (1876); К. R e i m е г, F. T i e m a n п, Бег.,
9, 824 (1876).
3. К. Reimer, F. Tiemann, Ber., 9, 1285(1876).
4. А. Рассел, Л. Л о к х а р т, Синтезы органических препаратов, сб. 3,
Издатинлит, 1952, стр. 365.
5. К. Auwers, F. Winternitz, Ber., 35, 465(1902).
6. R. Woodward, J. Am. Chem. Soc, 62, 1208 (1940).
7. H. Wynberg, W. Johnson, J.Org. Chem., 24, 1424(1959).
Рейсерт 222
Карл Арнольд Рейсерт (Karl Arnold Reissert) родился в 1860 г. в г. По*
вайен (Германия). Учился в Гейдельберге и Кенигсберге, затем в Берлинском
университете, где в 1884 г. ему была присуждена степень доктора. С 1888 г.
доцент кафедры химии того же университета. В Берлине работал совместно с
И. Тиманом. В 1902 г. начал преподавать в Марбургском университете, с 1922 г_
адъюнкт-профессор этого университета.
РЕАКЦИЯ РЕЙСЕРТА
Реакция Рейсерта* заключается во взаимодействии хинолина
с цианистым калием (или синильной кислотой) и хлорангидри-
дом карбоновой кислоты и последующем гидролизе полученных
1-ацил-2-циано-1,2-дигидрохинолинов (I) кислотами с
образованием альдегидов и хинальдиновой кислоты (Ii):
Rcoa + KCN + QQ^Qg
COR
I
+ RCHO
Это общий метод получения жирных и ароматических альдеги-
;*, который может использоваться также
рых цианхинолинов из N-окисей хинолина^:
дов*, который может использоваться также для синтеза некото-
223
Рейсерт
Мак-Эвен и Хацлет* исследовали образование альдегидов из
соединений. Рейсерта (I) в присутствии кислотных катализаторов,
н предложили следующий механизм этой реакции:
CN
Первая стадия состоит в присоединении протона к кислороду
амидной группы. В результате отщепления протона из
положения 2 ядра хинолина и присоединения протона к карбонильному
углероду образуется промежуточный продукт III, который и
распадается на альдегид и нитрил хинальдиновой кислоты.
Присутствие в соединении Рейсерта двойной связи в положении 3,4,
по-видимому, необходимо для образования ароматического
альдегид а\
Дэвис* синтезировал недавно хинальдиновую кислоту с
количественным выходом из 1-бензоил-2-циано-1,2-дигидрохинолина
(соединение Рейсерта; R=QHJ путем его гидролиза бромистово-
дородной кислотой в уксуснокислой среде. Бензальдегид также
получен с хорошим выходом.
1. A, Reissert, Вег., 38, 1603, 3415 (1905).
2. О реакции Рейсерта см. W. McEwen, R. Cobb, Chem. Revs., 55, 511
(1955); Э. Мозеттиг, Органические реакции, сб. 8, Издатинлит,
1956, стр. 288.
3. М. Н е n z е, Вег., 69, 1566 (1936); Е. Ochiai, I. Nakayama_
С. А., 45, 8529 (1951).
4. W. McEwen, R. H azl et t, J. Am. Chem. Soc, 71, 1949 (1949).
5. R. Collins. Т. Н е п s h a 1 1, J. Chem. Soc, 1956, 1881.
6. J. Davis, J. Org. Chem., 24, 1691 (1959).
Реформатский 224
Сергей Николаевич Реформатский* (I860—1934) родился в с.
Борисоглебском Костромской губернии. В 1882 г. окончил Казанский университет;
ученик А. М. Зайцева. В 1889—1890 гг. работал под руководством В.Мейерав
Геттингенском и Гейдельбергском университетах и в Лейпцигском
университете у В. Оствальда. С 1891 г. профессор Киевского университета. В 1928 г.
избран членом-корреспондентом АН СССР. С. Н. Реформатский разработал
метод синтеза р-оксикислот с применением цинкорганических соединений
(реакция Реформатского). Особое значение реакция Реформатского имеет при
исследовании и синтезе сложных природных соединений (например, витаминов).
С. Н. Реформатский занимался также изучением замещенных глутаровых
кислот, многоатомных спиртов и проблемами стереоизомерии. Автор известного
учебника «Начальный курс органической химии».
РЕАКЦИЯ РЕФОРМАТСКОГО
Метод получения эфиров Р-оксикарбоновых или
соответствующих им непредельных кислот взаимодействием эфиров а-галоид-
кислот с альдегидами или кетонами в присутствии металлического
цинка известен под названием реакции Реформатского^-.
Эта широко распространенная реакция обычно протекает при
добавлении к цинку раствора смеси реагентов в инертных
растворителях (эфир, бензол или толуол); реагенты вводят в реакцию
такими порциями, чтобы поддерживать умеренное кипение
реакционной смеси. Металлоорганический комплекс, образующийся в
результате реакции, гидролизуют разбавленной кислотой.
Примером реакции Реформатского является синтез этилового
эфира а-метил-[3-фенил-|3-оксимасляной кислоты из этилового
эфира %-бромпропионовой кислоты и ацетофенона:
+Zn
СН*—СН—СООС,Н, » СНз—СН—СООС*Н,
QH.CH,
Вг
QH.CH,
СН,—С—CH-COOQH, -» СН,,—С — СН—COOQH,
О—ZnBr ОН
Биография С. Н. Реформатского переработана редактором.
225 Реформатский
Эфиры оксикарбоновых кислот при нагревании с
дегидратирующими агентами (уксусный ангидрид, сухой хлористый*
водород и многие другие) превращаются в эфиры соответствующих
непредельных кислот. Дегидратация обычно приводит к
образованию эфиров а,р-непредельных кислот. Однако в некоторых
случаях получаются как &,р-, так и (3,%-непредельные
производные. Соотношение &,(3- и р,у-изомеров в смеси зависит от строения
исходных эфиров Р-оксикислот и от природы применяемых
дегидратирующих агентов.
Большей частью в реакции Реформатского применяют только
эфиры а-бромкарбоновых кислот и цинк (иногда цинк заменяют
магнием). Миллер и Норд® показали, что в присутствии
соответствующих промоторов в реакцию могут вступать также а-хлор-
ацетаты и эфиры ,8-бромкарбоновых кислот. Из числа изученных
галогенидов металлов наиболее эффективным промотором
оказалась сулема (HgCl%). Авторы показали, что (3-бромэтиловый
эфир уксусной кислоты может вступать в реакцию^ с альдегидами
и кетонами, образуя р,?-ненасыщенные спирты:
^ он"
C=O + BrCH,—CH,—OCOCHg
C=CH—CHg—OCOCH
\c=CH—CH.OH
Хассей и Ньюмен* изучили продукты побочных реакций,
протекающих при синтезе Реформатского, и объяснили причины
повышения выходов основных продуктов реакции при
использовании избытка цинка и эфира а-бромкарбоновой кислоты.
Образование двух диастереоизомеров показано на примере
синтеза о-этил-р-фенилакриловой кислоты из этилового эфира
ц-броммасляной кислоты и бензальдегида**:
СН,-СНз СН,—СН,
! I
С,Нб—СНО ф СН—COOR -^ QHg—СН=С—СООН
Вг
[Недавно было установлено, что вместо а-галоидэфиров в
реакции Реформатского можно применять а-галоиднитрилы*.
Изменив порядок проведения реакции (сначала обрабатывали
этиловый эфир бромуксусной кислоты цинком, а затем вводили
карбонильную компоненту), удалось осуществить реакцию
Реформатского с нитроароматическими альдегидами?.—Доя. ]
15 А Сергей
Реформатский 226
1. S. Reformatsky, Вег., 20, 1210 (1887); J. prakt. Chem., 54, 469 (1896).
2. О реакции Реформатского см. Р. Ш р а й н е р, Органические реакции,
сб. 1, Издатинлит, 1948, стр. 9.
3. R. Mi I I ег, F. No rd, J. Org. Chem., 16, 728 (1951).
4. A. H u s s e y, M. Newman, J. Am. Chem. See, 70, 3024 (1948).
5. L. С а п о п i с a, F. P e 1 i z z о n i, Gazz. chim. ital., 84, 553 (1954)
C. A., 50, 879 (1956).
6. H. С. Вульфсон, Л. Х. Виноград, ДАН СССР, 106, 669 (1954);
ЖОХ, 29,245,1147, 2690, 2692 (1959).—Дол. рео\
7. Л. X. Виноград, Н. С. Вульфсон, ДАН СССР, 123, 97 (1958).—
Дол . \
227
Родионов*
Владимир Михайлович Родионов (1878—1954) родился в Москве. В 1901 г.
окончил Дрезденский политехнический институт, а в 1906 г. Московское высшее
техническое училище, после чего работал инженером на химических
предприятиях. С 1920 г. профессор Московского химико-технологического института и
2-го Московского государственного университета, где он создал кафедру химии
алкалоидов. С 1935 по 1944 г. заведовал кафедрой химической технологии
красителей Московского текстильного института, с 1938 г.—кафедрой органической
химии 2-го Московского медицинского института, а с 1943 г.—кафедрой
органической химии Московского химико-технологического института им. Д. И.
Менделеева. В 1939 г. был избран членом-корреспондентом, а в 1943 г.
действительным членом АН СССР. С 1950 г. президент ВХО им. Д. И. Менделеева. Один
из организаторов советской анилино-красочной и фармацевтической
промышленности и производства душистых веществ. Круг его научных интересов был
крайне широк и разносторонен. Особенно известны его работы в области химии
р-аминокислот, один из методов синтеза которых носит его имя.
РЕАКЦИЯ РОДИОНОВА
Синтез ^-аминокислот и их N-замещенных производных
конденсацией альдегидов с малоновой или алкилмалоновой кислотой
в присутствии спиртового раствора аммиака или аминов известен
под названием реакции Родионова.
Эта реакция была открыта в 1926 г. при изучении конденсации
пипероналя с малоновой кислотой в присутствии спиртового
раствора аммиака*:
HgC
,сно
СН,(СООН),
Н,С
СН—СН.-СООН
NH.
сн=сн-соон
II
* Биография В. М. Родионова и описание его реакции составлены
редактором.
15'
Родионов' 228
При этом с выходом 40% был выделен хлоргидрат |3-пиперонил-
(3-аминопролионовой кислоты (I) и незначительное количество
пиперонилакриловой кислоты (II).
В дальнейшем реакция была распространена* на другие
ароматические*'\ алифатические^, алициклические* и
гетероциклические^ альдегиды.
Для объяснения механизма реакции предложена схема*» *:
ОН
RCHO f NH3 ->- RCH=NH или RCH(
RCH=NH + СН,(СООН),
ОН
RCH-CH —-*- RCH—СИ,
COOH NH, COOH
Джонсон? видоизменил реакцию Родионова, заменив спирто-
вый раствор аммиака ацетатом аммония. Позднее В. М. Родионов
и Н. А. Кравченко применили смесь ацетата аммония и ледяной
уксусной кислоты*. В ряде случаев при этом получаются р-амино
кислоты с лучшими выходами. Так, по основной реакции
Родионова р-(2-нафтил)-|3-аланин образуется с выходом 16,5%, по
модификации Джонсона—с выходом 65%, а по модификации
Родионова—Кравченко—с выходом 69%.
Реакция Родионова применяется также для получения эфиров
(^-аминокислот^.
1. В. М. Родионов, Е. Ф. Малеви некая, Вег., 59,2952(1926).
2. В. М. Родионов, А. М. Федорова, Вег., 60, 804 (1927); Arch.
Pharm., 266, 116 (1928).
3. В. М. Родионов, В. К. Зворыкина, Изв. АН СССР, ОХН,
1943, 216; В. М. Родионов, Н. Г. Ярцева, Изв. АН СССР,
ОХН, 1952, 103.
4. В. М. Родионов, Т. С. Киселева, Изв. АН СССР, ОХН, 1952,
278; В. М. Родионов, Л. В. Антик, Изв. АН СССР, ОХН.
1953, 253.
5. В. М. Родионов, Т. К, В е с е л о в с к а я, ЖОХ, 20, 2203 (1950);
В. П. Мамаев, Н. Н. Суворов, Е. М. Рохлин, ДАН
СССР, 101, 269 (1955).
6. В. М. Родионов, Е. А. Постовская, J. Am. Chem. Soc, 51,
841 (1929).
7. Т. Johnson, J. Evans, J. Am. Chem. Soc, 52, 4993 (1930); Т.
Johnson, See. trav. chim., 48, 872 (1929).
8. В. М. Родионов, Б. И. Кур та ев, Изв. АН СССР, ОХН, 1952,
113.
9. В. М. Родионов, Н. Н. Безингер, Изв. АН СССР, ОХН, 1952,
696.
229 Розенмунд
Карл Вильгельм Розенмунд (Karl Wilhelm Rosenmund) родился в 1884 г.
в Берлине. Учился в Берлинском университете у О. Дильса. В 1906 г. получил
степень доктора, затем стал профессором химии. С 1925 г. профессор и
директор Кильского фармацевтического института. Приобрел широкую известность
благодаря исследованиям в области ароматических оксиаминокетонов,
разработке метода восстановления хлорангидридов карбоновых кислот и синтезу
фениларсоновых кислот взаимодействием галоидпронзводных бензола с арсе-
нитом калия (реакция Розенмунда).
ВОССТАНОВЛЕНИЕ ПО РОЗЕНМУНДУ
Эта реакция заключается в восстановлении хлорангидридов
карбоновых кислот до альдегидов в присутствии палладиевого
катализатора^» %.
Примером реакции является получение р-нафтальдегида из
хлорангидрида (3-нафтойной кислоты^:
1 Т —»
1
\/
1 Т
\^
/СНО
фна
Восстановление обычно протекает при пропускании водорода
через перемешиваемый кипящий раствор хлорангидрида в
инертном растворителе (толуол или ксилол). В большинстве случаев в
качестве катализатора используется 5%-ный палладий на
сульфате бария. Хлористый водород, выделяющийся в процессе
восстановления, может быть поглощен щелочью и оттитрован. Это
позволяет контролировать ход реакции и закончить ее, как
только прекратится выделение хлористого водорода.
Восстановление по Розенмунду, по-видимому, является
наиболее удобным методом превращения хлорангидридов жирных и
ароматических карбоновых кислот в соответствующие альдегиды/
В присутствии сернистых соединений, растворенных в хинолине
(катализаторный яд), другие функциональные группы, такие, как
N0, или CI, не оказывают влияния на реакцию восстановлении
хлорангидрнда и сами не восстанавливаются.
Розенмунд 230
Метод Розенмунда может успешно применяться* для
получения альдегндокислот из соответствующих монохлорангидридов
дикарбоновых кислот при использовании в качестве катализатора
палладия на угле:
Pd/C
ROOC(CH*),,COa ^
1. К. Rosenmund, Вег., 51, 585 (1918).
2. О восстановлении по Розенмунду см. Э. М о з е т т и г, Р. М о з н н г о,
Органические реакции, сб. 4, Издатинлит, 1951, стр. 337.
3. Э. Гершберг, Д. К е й з о н, Синтезы органических препаратов,
сб. 3, Издатинлит, 1952, стр. 328.
4. О восстановлении хлорангндридов в альдегиды с помощью литнйтрн-тре/л-
бутоксиалюминийгидрида см. Н. Brown, R. М с F а г 1 i n, J. Am.
Chem. Soc, 80, 5372 (1958).
5. F. P e n n i n g i о n, W. С e I m e r, M. McLamore, V. В о g e г t,
I. Solomons, J. Am. Chem. Soc, 75, 109 (1953).
231
Скрауп
Зденко Ганс Скрауп (Zdenko Hans Skraup, 1850—1910) родился в Праге.
Работал в Венском университете ассистентом у Рохледера, а затем у Либена.
Вместе с Рохледером изучал хи ноли новый алкалоид—цинхонин. Благодаря
работам 3. Скраупа в данной области удалось установить строение этого
важного алкалоида. В 1877 г. Прюдом описал краситель ализарин синий,
получаемый при нагревании нитроалиэарина с глицерином и серной кислотой, а в
1880 г. К. Гребе установил его строение и показал, что, реагируя с глицерином,
он образует пиридиновое кольцо. На основании этих работ 3. Скрауп
предположил и доказал, что при взаимодействии нитробензола с глицерином и
серной кислотой получается хинолин; он обнаружил, что при добавлении к смеси
реагентов анилина реакция протекает быстрее.
РЕАКЦИЯ СКРАУПА
Образование хинолина и его производных при нагревании
первичных ароматических аминов с соответствующими нитросоеди-
нениями, глицерином и концентрированной серной кислотой было
открыто Скраупом в 1880 г. и получило название реакции или
синтеза Скрауп а^.
Нитросоединения, действующие как окислители, в
большинстве случаев можно заменить мышьяковой кислотой. Есть
указания-* з, что применение в качестве окислителя м-нитробензолсуль-
фокислоты даег очень хорошие результаты. Для смягчения часто
бурного характера реакции добавляют борную кислоту.
Синтез Скраупа, вероятно, протекает через стадию
образования акролеина из глицерина*;
и
II
сн
сн
II
сно
' сн,
сн
^\н
^ сн,
LHg NH NH
СН
—^
1
\н
сн
о
Скрауп 232
Доказательство образования и строения 1,2-дигидрохинолииа
как промежуточного продукта реакции, идущей в соответствии с
указанной схемой, приведено Джонсоном и Белом*.
При взаимодействии анилина с акролеином не было
достигнуто хорошего выхода хинолина. Однако Иэль*,
модифицировавший реакцию Скраупа (синтез проводился при 100 °С в 85%-ной
фосфорной кислоте) получил с акролеином неплохие выходы
замещенных хинолинов. В этой модификации синтеза Скраупа
могут также применяться вещества, способные к образованию
акролеина.
Описано взаимодействие некоторых ароматических аминов с
глицерином, удовлетворительно протекающее в разбавленной
серной кислоте*' *. При использовании 60—70%-ной серной
кислоты реакция Скраупа проходит гладко и обычно достигаются
прекрасные выходы. Бредфорд, Эллиот и Роуз* показали, что
некоторые мета-з вмещенные анилины могут превращаться по методу
Скраупа в 5- и 7-замещенные хинолины, причем выход 5- и 7-про-
изводных зависит от природы мета-заместителя. Сильные о- и
n-ориентанты (электронодонорные заместители) приводят к
получению только 7-замещенных хинолина; слабые орто-пара-ориен-
танты дают смесь 5- и 7-замещенных с преобладанием
последних; в случае м-ориентантов образуется смесь, в которой
преобладают 5-звмещенные производные хинолина?.
1. Z. S k r a u p, Monatsh., 1, 316 (1880); 2, 139 (1881).
2. Англ. пат. 394416, 19%3 г.
3. L. Bradford, Т. Elliott, F. R о we, J. Chem. Soc, 1947, 437.
4. Дискуссию по реакции Скраупа см. F. Bergstrom, Chem. Revt.. 35,
1^2 (1944).
5. W. Johnson, В. В и el I, J. Am. Chem. Soc, 74, 4517 (1952).
6. H. Yale, J. Bernstein, J. Am. Chem. Soc, 70, 254 (1948).
7. О реакции Скраупа см. Р. М а н с к е, М. К у л к а, Органические
реакции, сб. 7, Издатинлит, 1956, стр. 100.
233 Соммле
Марсель Соммле (Marcel Sommelet, 1877—1952) родился в г. Ланж
(Франция). В 1902 г. окончил Парижский университет, получив диплом фармацевта»
затем работал под руководством Бехаля. В 1906 г. ему была присуждена
степень доктора. После первой мировой войны преподавал на фармацевтическом
факультете Парижского университета. С 1934 г. заведующий кафедрой
органической химии этого университета. Получил алифатические альдегиды из
замещенных оксиэтилэтиловых эфиров, а также хлористые бензилы, которые
используются для приготовления бензиловых спиртов и бензальдегидов (реакция
Соммле). Применил хлорметилэтиловый эфир для проведения реакции Гринья-
ра. Синтезировал л-изопропилбензальдегид и показал, что он идентичен
природному куминовому альдегиду. Открыл, что род действием серной кислоты
гидроокись бензгидрилтриметиламмония превращается в о-бензилбензилди-
метиламин (перегруппировка Соммле). В 1919 г. получил совместно с Фурно и
Майаром премию Джекера.
РЕАКЦИЯ СОММЛЕ
В 1913 г. Соммле показал, что четвертичные соли, получаемые
из галоидного бензила и гексаметилентетрамина (уротропина),
разлагаются при нагревании в присутствии воды с образованием
соответствующего ароматического альдегида. Реакция служит
общим методом синтеза ароматических альдегидов и обычно дает
удовлетворительные выходы. Ее можно представить следующей
схемой:
,i*4 H*O
Аг—СН,Х » [C,Hi,N,CH,Ar]+X- —» Аг—СНО
Из продуктов реакции выделены: аммиак, метил-, диметил-
и триметил амины, а также некоторое количество бензил амина.
Соммле установил, что выделять четвертичную соль не
обязательно. При простом нагревании хлористого бензила с гексаметиле'н-
тетрамином в водно-спиртовом растворе с хорошим выходом
получается бензальдегид.
Реакция Соммле была изучена Анжиалем с сотрЛ-*. Авторы
показали, что электроноакцепторные заместители в ароматическом
ядре снижают скорость образования и выход альдегидов. Лучшим
растворителем для проведения реакции они считают 50%-нуи>
Соммле 234
уксусную кислоту. Работами этих же авторов установлено^, что
при реакции Соммле сначала происходит гидролиз четвертичной
соли 1 с образованием арилметиламина II, который подвергается
дегидрогенизации до имина III, при гидролизе превращающегося
в альдегид IV:
—2Н
Аг—СН,—NH, »
и
Аг—CH=NH *- Аг—СНО
m iv
Возможно, что продукт гидролиза гексаметилентетрамина—
метиленимин (CH*=NH)—является акцептором водорода в
процессе превращения арилметиламина в имин и сам превращается
при этом в метиламин. Описанный механизм подтверждается
термодинамическими данными, полученными Шопи^. Дегидрирующее
действие гексаметилентетрамина было показано Даффом? на
примере синтеза щиффовых оснований из ди-(о-оксибензил)-аминов:
Образование ароматических диальдегидов по реакции Соммле
протекает удовлетворительно только для ж- или л-ди-(хлор-
метил)-производных при условии, что в ароматическом ядре
имеется хотя бы одно свободное орто-положение*' *. Из о-бис-(хлорме-
тил)-производных после гидролиза гексаметилентетраминовой
соли получаются азотсодержащие основания**.
Хасс и Бендер» недавно описали общий метод синтеза
замещенных бензальдегидов, довольно сходный с методом Соммле.
При обработке бензилгалогенидов спиртовым раствором
натриевой соли 2-нитропропана образуются ароматические альдегиды.
Например, из о-ксилилбромида получен о-кумиловый альдегид^
с выходом 70%:
,С№ ^ уСН,
По способу Хасс а и Бендера терефталевый альдегид
образуется с 70%-ным выходом при взаимодействии а,а'-дибром-л-ксмлола
с 2-нитропропаном и спиртовым раствором едкого кали**.
235 Соммле
Другим методом синтеза альдегидов из бензилгалогенидов
является рго%%%я Хренке*-. Она состоит в превращении соли пири-
диния в N-окись действием л-нитрозодиметиланилина. Кислотный
гидролиз N-окиси приводит к образованию альдегида и /г-диметил-
аминофенилгидроксил амина:
Аг—СН,Х ф QHgN -^ [Аг—СН.2—
—#. Аг—CH=N—<^\—М(СНз)д -^-^ Аг—СНО + (CHg)gN—<^\—NHOH
Эта реакция находит широкое применение. С ее помощью
могут быть получены в мягких условиях и с хорошими выходами
многие ароматические, непредельные и а-кетоальдегиды.
Гетероциклические соединения, содержащие активные метиль-
ные группы, реагируют с пиридином и иодом с образованием иоди-
дов пиридиния, которые могут быть превращены в альдегиды по
способу Кренке**. Из 2-метилбензтиазола этим методом получен
2-формилбензтиазол с выходом 84%.
1. М. S о m m e I e t, С. г., 157, 852 (1913).
2. S. Angyal, R. R ass а с k, Nature, 161, 723(1948).
3. S. Angyal, R. R a s s а с к, J. Chem. Soc, 1949, 2700.
4. S. Angyal, P. Morris, R. R a s s а с k, J. W a t e r e r,
J. Chem. Soc, 1949, 2704; о реакции Соммле см. С. Анджиал,
Органические реакции, сб. 8, Издатинлит, 1956, стр. 263.
5. V. Franz en, Ann., 600, 109 (1956).
6. С. S h о р е е, Nature, 162, 619 (1948).
7. J. Duff, V. F u r n e s s, J. Chem. Soc, 1951, 1512; см. также реакцию
Даффа, стр. 109.
8. J. Wood, С. Tung, M. Perry, R. Gibson, J. Am. Chem.
Soc, 72, 2992 (1950).
9. H. H a ss, M. Bender, J. Am. Chem. Soc, 71, 1767 (1949).
10. H. Mass, M. В e n d e г, Синтезы органических препаратов, сб. 4,
Издатинлит, 1953, стр. 486.
11. A. Surrey, J. Mayer, J. Med. Pharm. Chem. (в печати).
12. F. К г о h n k e, E. В 6 r n e r, Ber., 69, 2006 (1936).
13. W. Reid, H. Bender, Chem. Ber., 89, 1893(1956).
Стефен 23*
Генри Стефен (Henry Stephen) учился в университете Викторин в
Манчестере, где в 1920 г. ему была присуждена степень доктора наук. Работал в этом
университете сначала преподавателем, а с 1921 г. старшим лектором. С 1926 г.
и по настоящее время профессор химии и химической технологии Витветерс-
рандского университета в Иоганнесбурге (Южно-Африканский Союз).
Совместно с X. Вейцманом изучал реакции конденсации с применением эфиров цианук-
сусной малоновой и ацетоуксусной кислот.
РЕАКЦИЯ СТЕФЕНА
Реакция Стефен а заключается в превращении жирных или
ароматических нитрилов в альдегиды в присутствии хлористого
водорода и хлористого олова в абсолютном эфире*.
Для проведения реакции, иллюстрируемой приведенной ниже
схемой, обычно добавляют нитрил к насыщенному хлористым
водородом раствору безводного хлористого олова в абсолютном
эфире:
SnCl, HCl HgO
RCN » [RC==NH]zSnCl, -* [RCH=NH,),SnClg » RCHO
В ряде случаев комплекс иминхлорида с хлорным оловом
можно выделить и гидролизовать до альдегида при нагревании
с водой.
Примерами реакции Стефен а могут служить превращения
^-нафтонитрила в Р-нафтальдегид^ (выход 95%) и 4-метоксифенил-
ацетонитрила в 4-метоксифенилацетальдегид^ (выход 90%); %-нафт-
альдегид по этому методу не получается.
237 Стефен
Либер* недавно показал, что в случае нитрила лауриновой
кислоты альдиминкый комплекс может быть осажден из эфирного
раствора при стоянии на холоду. При изменении обычного
порядка проведения реакции Стефена, т. е. при добавлении хлористого
олова к лауронитрилу, растворенному в эфире, насыщенном
хлористым водородом, можно крличественно выделить растворимый
в эфире жидкий металлсодержащий комплекс. После кипячения
с водой комплекс превращается в мономерный лауриновый
альдегид.
Тернер* установил, что основным фактором, способствующим
доведению до конца реакции Стефена, является осаждение
комплекса альдимина с хлорным оловом. Он показал, что для
незамещенных алифатических нитрилов скорость реакции мало
зависит от их строения, в то время как присутствие электронодонор-
ных групп в замещенных бензонитрилах значительно ускоряет
реакцию. Добавляя хлористый ацетил и воду к дигидрату
хлористого олова в эфире, Тернер получил гомогенную реакционную
смесь, состоящую из безводной соли, уксусной кислоты и
хлористого водорода.
Позднее Стефен* видоизменил свою реакцию, заменив
абсолютный эфир другими растворителями—этилформиатом или этил-
ацетатом. Безводное хлористое олово и нитрилы хорошо
растворяются в обоих этих растворителях и не выпадают в осадок
после насыщения раствора хлористым водородом. Комплекс
альдимина очищают, выливают в воду и альдегид отгоняют с водяным
паром.
Браун с сотр.? описали превращение алифатических и
ароматических нитрилов в соответствующие альдегиды взаимодействием
г литийтриэтоксиалюминийгидридом; при этом достигнуты очень
хорошие выходы.
Для синтеза альдегидов^ было использовано также
восстановление нитрилов на скелетном никелевом катализаторе в
присутствии семикарбазида. По этому методу из бензилцианида получен
семикарбазон фенилацетальдегида с выходом 70%.
Восстановление по Стефену является модификацией редкуш/
Зиляо*—М/оллфд**, заключающейся в превращении анилидов
в альдегиды** ^. Обработка анилида пятихлористым фосфором
приводит к образованию имидохлорида, восстанавливаемого далее
хлористым оловом в эфире; соль шиффового основания, вероят-
* А. Зонп (A. Sonn) родился в 1882 г. В 1907 г. Берлинский университет
присудил ему степень доктора философии. В 1920 г. стал профессором химии
Кенигсбергского университета.—Дол. /W.
** Е. Мюллер (Е. Muller) родился в 1881 г. Окончил Гейдельбергский
университет, где в 1904 г. получил степень доктора философии.—До/%. d
Стефен 23»
но, образующаяся при восстановлении, разрушается путем
нагревания с разбавленной кислотой (или водой), превращаясь в
альдегид:
PCI, Snd, Н%О
R—СО—NH—Аг —-*- R—C=N—Аг *- R—CH=N—Аг —г» R—СНО
I н+
а
По этому способу о-толуиланилид был превращен в о-толу-
иловый альдегид^^ с выходом 7U%.
1. Н. Stephen, J. Chem. Soc, 127, 1874 (1925).
2. Д. Уильям с, Синтезы органических препаратов, сб. 3, Издатинлнт^
1952, стр. 331.
3. W. Evans, N. Walker, J. Chem. Soc, 1947, 1571.
4. E. Л ибер, J. Am. Chem. Soc, 71, 2862 (1949).
5. L. Turner, J. Chem. Soc, 1956, 1686.
6. T. Stephen, H. Stephen, J. Chem. Soc, 1956, 4695.
7. H. Brown, C. Shoaf, C. Garg, Tetrahedron Letters, №3, 9 (1959).
8. H. P 1 i e n i n g e r, G. Werst, Angew. Chem., 67, 156 (1955).
9. A. Sonn, E. M u I I e r, Ben, 52, 1927 (1919).
10. Э. М о з е т т и г, Органические реакции, сб. 8, Издатинлит, 1956, стр. 288.
11. Д. Уильяме, Ч. Витте н, Д. Криницкий, Синтезы
органических препаратов, сб. 4, Издатинлит, 1953, стр. 484.
239
Стивене
Томас С. Стивене (Thomas S. Stevens) родился в 1900 г. в г. Ренфру
(Шотландия). Учился в университете в Глазго, затем у В. Г. Леркина в Оксфордском
университете, где защитил докторскую диссертацию. В настоящее время
преподает органическую химию в Шеффилдском университете. В основном
занимается изучением алкалоидов и механизмов реакций, особенно молекулярных
перегруппировок.
ПЕРЕГРУППИРОВКА СТИВЕНСА
В 1928 г. Стивене^ показал, что фенацилбензилдиметиламмо-
нийбромид (I) в водных растворах под влиянием амальгамы
натрия или при нагревании с разбавленным раствором едкого натра
перегруппировывается* (с перемещением бензильной группы) в
производное ацетофенона (II):
C,H,COCH,-N
ОН"
С.Н.СОСН—N
СН.С.Н,
,с,
II
Электроотрицательные заместители в бензольном ядре фен-
ацильной группы затрудняют перемещение бензил ьной группы от
атома азота к соседнему атому углерода, в то время как те же
заместители в ядре бензильной группы ускоряют
перегруппировку^' \
Бревстер и Клайн* исследовали стереохимию перегруппировки
и подтвердили представления Стивенса о том, что в данном
случае происходит миграция бензильной группы. В этих работах
показано, что перегруппировка /-фенацил-а-фенилэтилдиметил-
аммонийбромида (III) в аминокетон IV сопровождается
незначительной рацемизацией:
ВГ
СН;
—нвг
з—СН-СдН,
*
III
С.Н.СОСН—N
СН,
СНч—СН—
С,Н,СОСН—
,Н,
-СН—С,Н,
*
ГУ
Стивене 240
Г. Т. Stevens, E. Cr eight on, A. Gordon, M. MacNicoi,
J. Chem. Soc, 1928, 3193.
2. О механизме реакции Стивенса см. К. И н г о л ъ д, Механизм реакций и
строение органических соединений, Издатинлит, 1959, стр. 422.—Дол.
p
3. J. Dunn, Т. Stevens, J.Chem. Soc, 1932, 1926.
4. См. также С. Н a u s е г, S. К a n t о г, J. Am. Chem. Soc, 73, 1437
(1951).
5. J. В г e w s t e г, М. Kline, J. Am. Chem. Soc., 74, 5179 (1952).
241 Ульман
Фриц Ульман (Fritz Ullmann, 1875—1939) родился в г. Фюрте (Германия).
Учился у К. Гребе в Женеве, в 1895—1905 гг. работал там же. С 1905 по 1925 г.
преподаватель Высшей технической школы в Берлине. В 1925—1939 гг. доцент
Женевского университета. Работал в области синтеза производных дифенила и
акридина. Синтезировал ряд диаминоакридинов для Л. Эрлиха, который
подвергал их химиотерапевтическому исследованию. Совместно с К. Гребе
получил карбазол. Впервые применил диметилсульфат в качеств метилирующего
средства. Осуществил конденсациюфталевого ангидрида с фенолами в
присутствии хлористого алюминия. Его «Энциклопедия технической химии»,
выходящая уже третьим изданием, получила широкую известность.
РЕАКЦИЯ УЛЬМАНА
Действием на ароматические моногалоидлроизводные
порошкообразной меди при повышенной температуре получаются дпарилы.
Эта реакция, известная в органической химии под названием
реакции Ульмана^-, применяется для синтеза симметричных и
несимметричных ди-, три- и полиарилов.
Примером этой реакции является синтез бифенила из иодбен-
зо л а (выход 82%):
В реакции Ульмана успешно используются иодпроизводные,
а также активные хлор- и бромпроизводные ароматического ряда.
Электроотрицательные заместители в орто- или пара-положении
к атому галоида благоприятствуют течению реакции. Обычно
синтез проводят, постепенно добавляя избыток медного
катализатора к ароматическому галоидпроизводному при
соответствующей температуре.
В некоторых случаях для предотвращения бурного течения
реакции применяют растворители. Корн блюм и Кендел^
недавно показали, что применение в реакции Ульмана диметилформами-
да в качестве растворителя дает возможность повысить выходы
конечных продуктов.
16 А. СерррП
Ульман 242
Например, 2,2'-динитродифенил был получен из о-хлорнитро-
бензола с выходом 80% (ранее выход его не превышал 52—61%):
Практически количественный выход этого же соединения был
достигнут^ путем добавления к о-иоднитробензолу при 190°
четырех эквивалентов свежеосажденной меди. Медный катализатор
приготовлялся по методике, описанной в литературе*.
Доказательство свободнорадикальниго механизма реакции
Ульмана приведено Нерстеном\ Он исследовал реакцию дегало-
генирования /%-иоднитробензола в присутствии пирокатехина
и резорцина в качестве антиоксидантов. С пирокатехином выход
нитробензола оказался выше, чем с резорцином. Так как
пирокатехин является лучшим антиоксидантом, чем резорцин, то
результаты этого исследования указывают на гемолитический
механизм реакции Ульмана:
1. F. UII mann, Ber., 29, 1878 (1896); Ann., 332, 38 (1904).
2. О реакции Ульмана см. P. F a n t a, Chem. Revs., 38, 139 (1946).
3. N. К о г п Ы u m, D. К en d а 1, J. Am. Chem. Sec, 74, 5782 (1952).
4. P. Gore, G. Hughes, J. Chem. Soc, 1959, 1615.
5. H. Nursten, J. Chem. Soc, 1955, 3081.
243
Ульман
КОНДЕНСАЦИЯ УЛЬМАНА
В 1903—1905 гг. Ф. Ульман* показал, что в присутствии
меди о-хлорбензойная кислота вступает в реакцию с фенолами,
тиофенолами, арилсульфиновыми кислотами и ароматическими
аминами, образуя арилировгнные продукты конденсации.
По этому методу было получено большое число производных
N-фенилантраниловой кислоты*, применяемых в качестве
промежуточных продуктов в синтезе 9-хлоракридинов. Последние
представляют значительный интерес для приготовления
различных химиотерапевтических препаратов.
Примером синтеза данного типа является образование М-(4'-мет-
оксифенил)-4-хлорантраниловой кислоты (I) из 2,4-дихлорбен-
зойной кислоты и л-анизидина; последующая циклизация
соединения I при действии хлорокиси фосфора приводит к получению
6,9-дихлор-2-метоксиакридина (II):
СООН
осн.
H,N
Си
осн,
РОС1з
NH
осн.
Конденсацию по Ульману обычно проводят, нагревая раствор
реагентов в растворителе (амиловом или изопропиловом спиртах,
глицерине) в присутствии безводного карбоната калия и
порошкообразной меди.
В 1948 г. Даубен* подробно рассмотрел влияние заместителей
на реакционную способность хлорбензойной кислоты и анилина.
Электроотрицательные группы в орто- и пара-положениях к ато-
16*
Ульман
244
му хлора повышают электроположительность атома углерода,
связанного с хлором; при этом активность хлора повышается.
Реакционная способность атома азота зависит от основности амина;
электроотрицательные заместители в ароматическом ядре амина,
снижая его основность, затрудняют течение реакции.
Механизм реакции Ульмана, вероятно, состоит в нуклеофиль-
ной атаке орто-углеродного атома связанного с хлором, атомом
азота ароматического амина:
С-ОН
-хх
CI
соон
Си
NH
Другим методом синтеза замещенных N-антраниловых кислот
служит лерегр^д/шр)бка Уалженд*, которая осуществляется
путем нагревания и последующего гидролиза имидоэфиров.
Например, если имидоэфир I, полученный при взаимодействии М-(4-мет-
оксифенил)-имидохлорида бензойной кислоты (II) с натриевой
солью метилового эфира 2-хлор-6-оксибензойной кислоты (III),
нагревать в течение 70 мин. при 210° и затем продукт реакции
омылить, то образуется М-(4'-метилоксифенил)-6-хлорантраниловая
кислота* (IV):
NaO
II
ш
Такая схема перегруппировки имидоэфира I аналогична
схеме механизма конденсации Ульмана, приведенной выше.
Электроположительные группы в орто- или пара-положении к атому
азота в кольце А имидоэфира I и электроотрицательные заместители
245 Ульман
в тех же положениях к атому кислорода в кольце Б облегчают
перегруппировку^.
Механизм перегруппировки Чэпмена был изучен Вайбергом
и Роуляндом?. Полученные ими данные подтверждают
возможность образования промежуточного четырехчленного
циклического комплекса (см. формулу I) при нуклеофильном замещении в
ароматическом ядре.
1. F. U И m апп, Вег., 36, 2382 (1903); 37, 2001 (1904); F. U I I m a n п,
Н. Kipper, Ber., 38, 2120 (1905).
2. См., например, A. G о 1 d b erg, W. Kelly, J. Chem. Soc, 1946, 102.
3. W. D a u be n, J. Am. Chem. Soc , 70, 2420 (1948).
4. A. Chapman, J. Chem. Soc., 127, 1992 (1925).
5. W. D a u b e n, R. Hodgson, J. Am. Chem. Soc, 72, 3479 (1950).
6. G. Singh, S. Singh, A. Singh, W. Singh, J. Indian Chem.
Soc., 28, 459 (1951).
7. K. W i b с rg, B. Rowland, J. Am. Chem. Soc, 77, 2205 (1955).
Фаворский 246
Алексей Евграфович Фаворский* (1860—1945) родился в с. Павлове Ниже
городской губернии. В 1882 г. окончил Петербургский университет; ученик
А. М. Бутлерова. Работал в университете лаборантом. В 1891 г. защитил
магистерскую, а в 1895 г.—докторскую диссертации. С 1896 г. профессор
Петербургского университета. После Октябрьской революции работал в Ленинградском
университете и Ленинградском технологическом институте. В 1934—
1938 гг. первый директор института органической химии АН СССР. В 1921 г.
был избран членом-корреспондентом, а в 1929 г. действительным членом
АН СССР. Среди его учеников—С. В. Лебедев, Б. В. Вызов, И. Н. Назаров,
А. Е. Порай-Кошиц и другие. А. Е. Фаворский является одним из
основоположников химии непредельных соединений, в частности химии ацетилена. Большое
значение имеют его работы по исследованию взаимодействия ацетилена и его
монозамещенных с кетонами, приведшие к открытию нового способа получения
третичных ацетиленовых спиртов. Он открыл и изучил явления изомеризации
и взаимных превращений ацетиленовых и алленовых углеводородов,
разработал метод получения простых виниловых эфиров при действии спиртов на
ацетилен в присутствии порошка едкого кали. Виниловые эфиры и полимеры на
их основе нашли широкое применение в разнообразных отраслях
промышленности и в медицине. За выдающиеся научные заслуги А. Е. Фаворскому в
1945 г. присвоено звание Героя Социалистического Труда.
ПЕРЕГРУППИРОВКА ФАВОРСКОГО
Перегруппировкой Фаворского^* - называется превращение
%-галоидкетонов а карбоновые кислоты или их уфиры.
Перегруппировка протекает под влиянием основных катализаторов и яь-
ляется общей реакцией %-моно-, а,&'- и сс,8 дигалоидкетонов.
Примером реакции может служить образование циклопенган-
карбоновой кислоты из 2-хлорциклогексанона под действием ал-
коголята калия:
соон
и
* Биография А. Е. Фаворского написана редактором.
247
Фаворский
Лофтсфилд* применил меченый 2-хлорциклогексанон-1-(2-С**]
и по данным изотопного распределения в продукте реакции и в
непрореагировавшем кетоне показал, что в процессе
перегруппировки Фаворского преимущественно образуется промежуточное
соединение, содержащее трехчленное кольцо циклопропана*:
JCOOR
Найдено, что самые лучшие выходы при этой
перегруппировке получаются в присутствии бензилата натрия*.
Шторк и Борович* показали также, что перегруппировка цис-
и /лрдяс-изомеров 1-хлор-1-ацетил-2-метилциклогексана в 1,2-диме-
тилциклогексанкарбоновую кислоту сопровождается инверсией у
углеродного атома, связанного с хлором:
=о
сн,
СООН
В ряду "алифатических галоидкетонов перегруппировка
Фаворского в зависимости от условий реакции приводит к различным
конечным продуктам. Например, а-хлоркетон (I) превращается
в присутствии этилата натрия в сложный эфир (II), при действии
метилата натрия в метиловом спирте—в оксиапеталь (III), при
обработке суспензией метилата натрия в эфире—в эфир (IV):
сн,
%—С—
СМ,
I
со—с—
NaOCHg
СНуОН
II
СН;
сн,—с—
он
ш
сн,—с—соосн,
I
СИ,
IV
Фаворский
248
При перегруппировке о,з'-дибромкетонов под действием
метил ата натрия образуются эфиры д,р-ненасыщенных кислот*:
СН,
%\ МаОСНз
CBr—СО-СН,Вг
с=сн—соосн,
В тех же условиях из %,|3-дибромкетонов получаются эфиры
8,%-ненасыщенных кислот:
R—СНВг —CBr—CO—
СН,
R-CH=6—СН,--СООСНз
Стивене и Фаркас", изучая перегруппировку а-галоидцикло-
гексилфенилкетоня (V), показали, что при получении с выходом
53% кислоты VI из кетона V (кипячение в ксилоле с мелко
раздробленным едким натром^)
X NaOH ^ ^ /СООН
в качестве промежуточного продукта не образуется эпоксиэфир
VII, получаемый в присутствии метилата натрия:
J.
G
R
С.
С
А. Е. Фаворский, J. prakt. Chem., 88, 658 (19I3V
П перегруппировке Фаворского см. R. J а с q u i е г, Bull. Soc chim.
France, D35 (1950); [см. также Т. PI. Темникова, Курс теоретических
основ органической химии, Госхимиздат, 1959, стр. 618.—Дол.
L о f t s f i e I d, J. Am. Chem. Soc, 73, 4707 (1951).
Burr, M. Dewar, J. Chem. Soc, 1954, 1201.
Stork, L В о г о w i t z, J. Am. Chem. Soc, 82, 4307 (1960).
J. Moore, J. Am. Chem. Soc, 72, 974 (1950).
E. F a r k a s, J. Am. Cnem. Soc, 74, 616 (1У52).
E. F а г k a s, J. Am. Chem. Soc, 74, 5352 (1952).
Wagner,
Stevens.
Stevens,
249
Фишер
Эмиль Фишер (Emil Fischer, I852—19I9) родился в г. Эйскирхен (Герма
ния). Высшее образование получил в университетах Бонна н Страсбурга.
Ученик А. Байера, с 1875 г. его ассистент. Профессор университетов Мюнхена
(с 1879 г.), Эрлангена (с 1882 г.), Вюрцбурга (с 1885 г.) и Берлина (с 1892 г.).
Среди разнообразных работ Э. Фишера наиболее известны исследования
Сахаров, аминокислот, полнпептидон, протеинов, производных пурина, депсидов и
дубильных веществ. Совместно со своим братом О. Фишером открыл фенил гидр -
азин (1875 г.), синтезировал мочевую кислоту (1897 г.), а также известное сно
творное средство—веронал (1903 г.). За выдающиеся работы по изучению
строения и синтезу Сахаров и пуринов в 1902 г. ему была присуждена
Нобелевская премия.
СИНТЕЗ ИНДОЛА ПО ФИШЕРУ
Реакцией Фишера или синтезом индола по Фишеру* обычно
называют метод получения производных индола из фенилгидр-
азонов альдегидов или жетонов.
Реакция протекает при нагревании арилгидразонов в
присутствии таких катализаторов, как хлористый цинк, разбавленная
серная кислота, ледяная уксусная кислота или спиртовый раствор
хлористого водорода. Эта реакция, имеющая общий характер*,
заключается во внутримолекулярной конденсации с отщеплением
аммиака:
R
Н,С К'
\ /
С leu
N
-R
NH
Аллен и Вильсон^, используя метод меченых атомов, показали,
что атом №*, занимающий ^-положение в молекуле фенил гидр-
азона, входит в ядро образовавшегося индола; если же атом №*
находится в 9-положении, то меченым оказывается отщепляющийся
аммиак. Последующие работы* подтвердили эти данные.
Фишер
250
Таким образом, общепринятый в настоящее время механизм
реакции Фишера можно представить следующей схемой:
R
I
H;C R'
\/
R
HC R'
NH
R
-NH3
R
С R'
^C
NH&
NH,
It
R
CH R'
NH
NH
Одним из последних примеров использования реакции Фишера
является получение 3-индолилуксусной кислоты из фенилгидра-
зона моноальдегида янтарной кислоты*:
соон
н,с
сн
N
сн,—соон
Конденсация хлоргидрата М-бензил-К-я-метоксифенилгидра-
зина с 5-фталимидопентаноном-2 в ледяной уксусной кислоте
протекает гладко с образованием ожидаемого индола* (1-бензил-
2-метил-5-метокситриптамина):
со
NH, Ч-
N СН;
i
сн-сн
сн,—сн.
СО
N ^СНд
I
Показано, что циклизация некоторых фенилгидразонов с
электронодонорными (орто-, пара-ориентантами) заместителями в
251
Фишер
мета-положении проходит преимущественно с образованием 6-, а
не 4-замещенных индолов*. В случае мета-ориентантов обычно
наблюдается обратное явление.
Бледс и Уайлдс* разработали метод синтеза замещенных
индолов взаимодействием диазокетонов с анилином и его солями.
Реакция протекает с хорошими выходами. Например, диазоке-
тон, полученный из диазометана и хлор ангидрида 4-хлорбен-
зойной кислоты, превращается в 2-(4'-хлорфенил)-индол с
выходом 80%:
изучил реакцию Фишера с 2,6-дизамещенными
фенил гидр азонами. Для объяснения строения полученных
продуктов реакции этот исследователь высказал предположение
о промежуточном образовании неароматического соединения
типа I:
R
CHR'
CR"
NH
NH
R
В случае 2,6-диметилфенилгидразона ацетофенона в качестве
главного продукта реакции был выделен 2-фенил-Зд,5-днметил-
Зд,4,7,7д-тетрагидро-[ЗН]-псевдоиндолон-4 (II):
Фишер 252
I.E. Fischer, F. Jourdan, Ber., 16, 2241 (1883).
2. О реакции Фишера см. R. Order, H. L i n d w a II, Chem. Revs., 30,
78 (1942); [см. также Н. И. Су в о ров, В. П. Мамаев,
В. М. Родионов, Реакции и методы исследования органических
соединений, кн. 9, Госхимиздат, 1959, стр. 7.—Доп. /W.].
3. С. Allen, С. Wilson, J. Am. Chem. Soc, 65, 611 (1943).
4. К. С I u s i u s, H. W e i sser, Helv. chim. acta, 35, 400 (1952).
5. S. Fox, M. Bullock, J. Am. Chem. Soc., 73, 2754, 2756 (1951).
6. D. Ockenden, K. S с hof i el d, J. Chem. Soc, 1957, 3175.
7. M. Sletzinger, W. G a i n e s, W. R u у I e, Chem. Ind. (London),
1215 (1957).
8. C. Blades, A. Wilds, J. Org. Chem., 21, 1013 (1956).
9. R. С a r I i n, D. Carlson, J. Am. Chem. Soc., 81, 4673 (1959).
253 Фридель—Крафтс
Шарль Фридель (Charles Friedel, 1832—1899) родился в Страсбурге
(Франция). В 1852 г. окончил Страсбургский университет. Изучал химию в Сорбонне
у Ш. Вюрца и минералогию в горной академии, где затем работал куратором.
В 1869 г. получил степень доктора. С 1876 г. профессор минералогии, а с 1884 г.
профессор органической химии Парижского университета. Получил молочную
кислоту из бромпропионовой кислоты (1861) н изопропнлоный спирт
восстановлением ацетона (1862), осуществил первый синтез глицерина (1873). Совместно
с Д. Крафтсом и А. Ладенбургом работал в области органических соединений
кремния и показал их сходство с соединениями углерода, получил впервые те-
траэтилкремний. В содружестве с Д. Крафтсом изучал применение хлористого
алюминия для синтеза алкил- и ацилпроизводных ароматических соединений.
В области минералогии представляют интерес его работы по искусственному
приготовлению минералов (кварца, тридимнта, рутила, топаза и др.). За свои
труды удостоен многих наград, в том числе медали Дэви. Был членом многих
зарубежных научных обществ. Один из основателей Французского химического
общества, четырежды избирался его президентом.
*
Джемс Мейсон Крафтс (James Mason Crafts, 1839—1917) родился в
Бостоне. Получив степень бакалавра, в течение года изучал инженерное дело в
Кембридже, а с 1859 г.—минералогию во Оренбурге. В дальнейшем
совершенствовался в Гейдельберге под руководством Р. Бунзена, а с 1861 г. в
Париже у Ш. Вюрца. С 1866 г. декан химического факультета Корнэллского
университета. В 1870—1874 гг. заведующий кафедрой общей химии
Массачусетсе ко го технологического института, япоследстиии ректор того же
института. С 1874 по 1891 г. работал совместно с Ш. Фриделем в Парижской горной
академии, где они открыли важную реакцию, названную впоследствии их
именем. Внес также большой вклад в термометрию.
РЕАКЦИЯ ФРИДЕЛЯ—КРАФТСА
Реакция Фрид ел я—Кряфтса заключается в алкилировании*
или ацилировании^ ароматических соединений действием
галоидных алкилов или галоидангидридов карбоновых кислот в
присутствии хлористого алюминия'*.
Следующие реакции являются типичными примерами алкили-
рования и ацилирования:
Aid,
Фридель—Крафтс 254
Указанная реакция была открыта* в 1877 г. и в дальнейшем
широко и подробно изучена; при этом было установлено, что
алкилирование и ацилирование по Фрид ел ю—Крафтсу возможно
для самых различных ароматических соединений—бензола, его
гомологов и производных, нафталина и его производных, ряда
гетероциклов и т. д. Показаня также возможность применения
различных алкилирующих агентов (галоидных алкилов, олсфл-
нов, спиртов, сложных эфиров) и катализаторов (AlClg, AlBr^,
H^SO^ BF,, HF, HgPO^, PA, ZnClg, SnClJ.
В зависимости от соотношения реагентов в ароматическое
ядро можно ввести одну или несколько алкильных групп. Во мнс-
гих случаях образуется смесь продуктов разной степени алкили-
рования. Например, при взаимодействии бензола с избытком
хлористого этила в присутствии хлористого алюминия получается
смесь тетра-, пента- и гексаэтилбензолов*:
(выход 6,5%) (выход 6,5%)
'5
(выход 38%) (выход 15%)
В противоположность алкилированию, при ацилировании по
Фриделю—Крафтсу (кислотами, галоидангидридами и
ангидридами кислот) возможно введение только одной ацильной группы.
Подобно другим дезактивирующим группам (—N0%, —СООН)
ацильная группа (—COR) оказывает ингибирующее действие на
процесс ацилирования по Фриделю—Крафтсу. Типичными
примерами реакции ацилирования являются образование ацетофено-
на из бензола и ацетилхлорида (см. стр. 253) и бензоилпропио-
новой кислоты из бензола и янтарного ангидрида:
СО—СН,—СН*—СООН
255
Фриде ль— Крафтс
Механизм реакции Фриделя—Крафтса, по всей вероятности,
включает стадию образования алкильных или ацильных карбо-
ниевых ионов:
RCH.C1 4
которые в дальнейшем реагируют с ароматическими ядрами:
R-CO-H
Н
н
COR
н
н
AIC1"
на
А1СГ
COR
НС1 +
Недавно Браун и Пирсел* высказали предположение о том, что
комплексы, образующиеся из ароматического углеводорода, га-
логенида алюминия и галоидоводорода, играют важную роль в
реакции Фриделя—Крафтса, создавая полярную среду,
облегчающую растворение галогенида алюминия и образование
промежуточных продуктов в ионной форме.
Установлено, что полифосфорная кислота является
эффективным катализатором для реакции Фриделя—Крафтса и особенно
применима в случаях, требующих более мягких условий
проведения процесса. Эфиры фенолов полифосфорной кислотой не
расщепляются*.
1. Обзор процессов алкилирования по реакции Фриделя—Крафтса см.
Ч. Прайс, Органические реакции, сб. 3, Издатинлит, 1951, стр. 7.
2. Обзор процессов ацилирования по реакции Фриделя—Крафтса см. Э. Б е р-
л и н е р, Органические реакции, сб. 5, Издатинлит, 1951, стр. 195;
см. также У. Джонсон, Органические реакции, сб. 2, Издатинлит,
1950, стр. 128.
3. Ч. Томас, Безводный хлористый алюминий в органической химии,
Издатинлит, 1949.
4. С. F r i e d e I, J. С г а f t s, С. г., 84, 1292, 1450 (1877).
5. S m i t h, G u ss, J. Am. Chem. Sec, 62, 2625 (1940).
6. H. Brown, H. P e s rs а 1 I, J. Am. Chem. Soc, 74, 191 (1952).
7. P. G a r d n e r, J. Am. Chem. Soc, 76, 4550 (1954); см. также F. U h I i g,
H. S n у d e r, Advances in Organic Chemistry, v. 1, New York, I960,
p. 35.
Фридлендер 256
Пауль Фридлендер (Paul Friedlander, 1857—1923) родился в Кенигсберге.
Учился в Кенигсбергском университете под руководством К. Гребе, а затем
в Мюнхене у А. Байера. С 1883 г. доцент Мюнхенского университета, с 1884 г.
директор небольшой фабрики красителей в Оффенбахе. В 1888—1895 гг. препо
давал химию красителей в Высшей технической школе в Карлсруэ, в 1895—
1911 гг. руководитель отдела химии промышленного музея в Вене. С 1911 г.
профессор Высшей технической школы в Дармштадте. В содружестве с А.
Байером занимался изучением строения и синтезом индиго. Выполнил
фундаментальное исследование производных изатина и получил индиго из хлоризатина.
Показал, что фенантрснхинон действием щелочи превращается в днфениленгли-
колевую кислоту и вместе с А. Басйром впервые описал индоксил.
Синтезировал тионафтен и тиоиндиго (1906), получил диброминдиго и доказал его
тождество с пурпуром древних (1909), занимался также исследованием флавонов и
фталеинов. Автор известного сборника немецких патентов по промежуточным
продуктами красителям «Fortschritte der Theerfarbenfabrikation und verwandter
Industriezweige», издававшегося с 1888 г.
СИНТЕЗ ФРИДЛЕНДЕРА
Конденсация о-аминпбензальдегида с соединениями,
содержащими группировку—СНдСО—, приводящая к образованию
производных хинолина, носит название синтеза Фридлендера^.
Эту реакцию обычно проводят при кипячении спиртовых
растворов реагентов в присутствии разбавленной щелочи. Реакция
протекает по схеме:
Здесь R—атом водорода, алкильный или арильный радикал,
МО.угруппа и т. д.; R'—алкильный или арильный радикал или
СООН-группа*.
Область применения реакции Фридлендера несколько
ограничивается сложностью получения замещенных о-аминобензаль-
дегидов.
Модифицированная методика реакции Фридлендера была ис-
пользованаБоршем и Ридом** в синтезе 6,7-диметоксихинолина (III).
257
Фридлендер
Шиффово основание I, полученное из 2-амино-4,5-диметокси
бензальдегида и /г-толуидина
н.
NH.
реагируя с пировиноградной кислотой, дает продукт конденсации—
6,7-диметоксихинальдиновую кислоту (II), декарбоксилирование
которой приводит к 6,7-диметоксихинолину (III):
Н.СО
Н.СО
CH=N—С«НаСНз-л СН
NH,
СО -CH3C6H4NH2
:оон
"
Найдено, что анионообменные смолы с сильноосновными
свойствами (например, Амберлит 1RA-400, Довекс 2) являются
эффективными катализаторами реакции Фридлендера*. Показано также,
что конденсация о-аминобензальдегида с диметилацеталем формил-
ацетонаподдействием катализатора приводит к образованию бен-
зилиденового производного IV, которое после обработки кислотой
превращается в 3-ацетилхинолин (V):
СНз—СН(ОСНз)г
СО—СН,
со—сн.
v
1. P. Friedlander, Ber., 35, 2572 (1882).
2. Р. Эльдерфильд, Гетероциклические соединения, сб. 4, Издатин-
лит. 1955, стр. 35.
3. W. В о rs с h e, W. R led, Ann., 554, 269 (1943).
4. S. Y a m a d a, I. Ch I b at a, Pharm. Bull. (Tokyo), 3, 21 (1955).
17 A. Ctp
Фрис 258
Карл Фрис (Karl Fries) родился в 1875 г. в г. Кидрих-на-Рейне (Германия).
Учился в Дармштадте, затем у Т. Цинке в Марбурге. В 1899 г. получил степень
доктора философии. Преподавал в Марбурге ком университете, где в 1912 г.
стал доцентом. С 1918 г. директор химического института при Брауншвейгской
высшей технической школе. Одной из важнейших его работ является
исследование бициклическнх соединений (бензтиазолы, бензоксазолы, тионафтены,
индазолы) н их сравнение с нафталином.
РЕАКЦИЯ ФРИСА
Образование ароматических оксикетонов при перегруппировке
сложных эфиров фенола в присутствии хлористого алюминия
называют реакцией (перегруппировкой или «сдвигом»)
В общем виде реакция иллюстрируется схемой
COR ROC^
(R—алифатический или ароматически^ радикал).
Миграция ацильной группы (—COR) в орто- или
пара-положение в значительной степени зависит от условий проведения
реакции и ст строения исходного сложного эфира. Во многих
случаях получается смесь обоих изомеров. Как правило, можно
считать, что при низких температурах образуются преимущественно
я-изомеры; под воздействием высокой температуры реакции
образуются в основном о-изомеры. Например, перегруппировка
259 Фрис
м-крезилацетата* при 25° дает до 80% л-изомера, а при 165°
получается 95% о-изомера:
ОСОСНд
I
Это правило подтверждается данными наблюдений за
изомеризацией я-кетофенолов в о-кетофенолы при нагревании я-изоме-
ров с хлористым алюминием*.
При использовании в качестве растворителя нитробензола
перегруппировка Фриса обычно протекает при более низкой
температуре, чем в отсутствие растворителя.
Влияние различных условий реакции на течение
перегруппировки Фриса было подробно изучено на примере изомерных ацет-
оксибензойных кислот*. Оказалось, что при добавлении о-изомера ]
к безводному хлористому алюминию в нитробензоле происходит
энергичная реакция. Для разрушения комплекса с хлористым
алюминием через 1 час после начала реакции в реакционную
смесь добавляют концентрированную соляную кислоту и лед,
затем отгоняют с водяным паром нитробензол и выделяют
образовавшуюся с 83%-ным выходом 5-ацетил-2-оксибензойную
кислоту (II):
Н,СОСууСООН
VkoH
II
Если проводить реакцию в отсутствие нитробензола при
температуре 120—125°, кислота (II) получается с выходом около 40%.
При проведении перегруппировки /z-ацетоксибензойной
кислоты (III) при 150—155° или 180—185° образуется с выходом
57% З-ацетил-4-оксибензойная кислота (IV):
у
COCHg
IV
В нитробензоле эта реакция не идет и о-кетопроизводное IV
не получается. ж-Ацетобензойная кислота ни при каких из
испытанных условий не дает продуктов миграции ацетильной группы
17*
Фрис 260
При перегруппировке л-этилфениловых эфиров высших
жирных кислот как при низкой температуре в нитробензоле, так и
при высокой температуре в отсутствие растворителя образуются
только о-оксикетоны":
O-COCHsCHsR
с,н,
В результате двойной перегруппировки Фриса из ди-л-толило-
вых эфиров алифатических дикарбоновых кислот получены*
бис-о-оксикетоны типа соединений V (п=2—8):
ОН
Реакция протекает успешно с кислотами от янтарной до се-
бациновой.
1. К. Fries, G. Fink, Ber., 41, 4271 (1908).
2. О реакции Фриса см. А. Б л а т т, Органические реакции, сб. 1, Издатнв-
лит, 1948, стр. 455; [см. также Ч. Томас, Безводный хлористый
алюминий в органической химии, Издатинлит, 1949, стр. 695.—Дол. рей.].
3. К. Rosenmund, W. Sch nurr, Ann., 460, 56 (1928).
4. N. С u 1 I i п а о e, B. Edwards, J. Chem. Soc., 1958, 434.
5. D. Shah, N. Shah, J. Indian Chem. Soc, 26, 235 (1949).
6. A. Sen, S. Ti wari, J. Indian Chem. Soc., 29, 357 (1952).
7. F. Thomas, M, S h a m m a, W. Per n e I i u s, J. Am. Chem. Soc,
80, 5864 (1958).
1261 Хунсдиккер
Хейнц Хунсдиккер (Heinz Hunsdiecker) родился в 1904 г. в Кельне. Учился
в Кельнском университете, где под руководством профессора Виндгена
выполнил и защитил докторскую диссертацию (1930 г.). Работал химиком в фирме
Dr. Vogt Co., директором и совладельцем которой является и в настоящее
время. Занимается исследованиями главным образом в области органического
синтеза. В связи с многочисленными работами по приготовлению различных
многочленных циклических кетонов и лактонов, необходимых для
парфюмерной промышленности, открыл способ получения галоидных органических
соединений, который носит его имя.
РЕАКЦИЯ ХУНСДИККЕРА
Синтез органических галогенидов взаимодействием сухих
серебряных солей карбоновых кислот с эквимолекулярным
количеством галоида* называют реакцией Хунсдиккера^* *.
Эта реакция может быть изображена схемой:
RCOOAg^X* -» RX+COs + AgX
Экспериментальные данные подтверждают справедливость сво-
боднорадикального механизма разложения промежуточно
образующегося ацилгипогалогенита:
RCOOX _* RX + GO,
Серебряные соли карбоновых кислот обычно получают из
соответствующих кислот и азотнокислого серебра; для
высокомолекулярных кислот применяют окись серебра. Высушенную
серебряную соль суспендируют в соответствующем растворителе,
например в четыреххлористом углероде, после чего добавляют
галоид. Чаще всего применяют бром, хотя в тех случаях, когда
использовали хлор, были достигнуты удовлетворительные выходы
хлорпроизводных. Шмид* показал, что при получении 1,4-Диброы-
бутана реагенты лучше добавлять в обратном порядке, т. е.
прибавлять серебряную соль адипиновой кислоты к раствору брома
а четыреххлористом углероде.
* Впервые способ получения бромпроизводных жирных кислот броыиро-
анием серебряных солей был разработан А. П. Бородиным^.—/7ргш.
Хунсдиккер __________ 262
В реакции Хунсдиккера применяют и другие растворители,
например бензол, нитробензол и хлороформ. Установлено, что
синтез высших галоидалкилов лучше проводить в кипящем четы-
реххлористом углероде, в то время как низкая температура
(ниже —20°) является, по-видимому, оптимальной для синтеза
циклобутилбромида*:
COOAg
Вг
В данном случае следует добавлять реагенты в обратном
порядке.
Реакция Хунсдиккера неприменима к большинству
ненасыщенных кислот. Она была с успехом использована для
получения а,ш-дигалоидных органических соединений из
соответствующих а,ю-дикарбоновых кислот. Лучшие выходы были достигнуты
с высшими гомологами. Показано, что из серебряных
солей моноэфиров указанных кислот образуются ю-галоидэфиры.
Примером может служить превращение серебряной соли
монометилового зфира адипиновой кислоты в эфир 5-бромвалериановой
кислоты* (выход 52%):
Вг_
CH_OOC(CH_)sCH_COOAg —->
Серебряные соли а-замещенных карбоновых кислот являются
исходными продуктами для синтеза различных органических
соединений. Например, из а-галоидных производных кислот можно
получать 1,1-дигалоидные соединения, а из а-окси- и и-амино-
кнслот—альдегиды:
R—CHX—GOOAg -& Xg -» R—CHX, -f CO, -^ AgX
R—CHOH—COOAg фХ, -^ R—CHO f CO_ f AgX ф НХ
H_o
R—CHNH_—COOAg ф Xg * R—СНО ф CO_ ^ AgX + NH^X
Недавно описано применение реакции Хунсдиккерп в ряду
гетероциклических соединений? на примере синтеза 4-бром-
1-фенилпиразола:
- COOAg |^ г-Вг
4 Вг, -*" N j| ф AgBr 4 СО_
N
263 Хунсдиккер
Осуществить реакцию Хунсдиккера с серебряной солью
изомерной 3-карбоновой кислоты не удалось.
Взаимодействие двух молей серебряной соли карбоновой
кислоты с одним молем иода приводит к образованию сложного
эфира*» в (реакция
2ROOOAg+J, -»
По этому методу из серебряной соли циклобутанкарбоновой
кислоты действием иода при 90—100° была получена
идентифицированная Робертсом и Симмонсом* смесь циклобутилового, цикло-
пропилметилового и 3-бутенилового эфиров
циклобутанкарбоновой кислоты.
Серебряные соли карбоновых кислот реагируют с галоидом в
присутствии олефинов или ацетиленов (раз/о^л Предо*' ^),
образуя следующие соединения:
RCOOAg + %,-*. R'CH=CHR* -^ R'CH—CHXR" + AgX
UCOR
RCOOAg + Xg ^ R^C=CH -* R'C=CX 4- RCOOH ф AgX
2RCOOAg ф X. ^r R'CH=CHR* -^ R'CH CHR" -f 2AgX
OCOR OGOR
Взаимодействием серебряной соли бензойной кислоты с иодом
в присутствии гексадецена-1 с последующим омылением
образовавшегося дибензойного эфира гликоля был получен** гексадехан-
диол-1,2.
1. A. Borodin, Ann., П9, 121 (1861).—До/%. p
2. С. Hunsdiecker, H. Hunsdiecker, E. V о g t, пат. США
2176181, 1939 г.;Н. Hunsdiecker, С. Hunsdiecker, Ber.,
78, 291 (1942).
3. О реакции Хундсдиккера и близких к ней превращениях см. Ч. В и л ь-
с о н, Органические реакции, сб. 9, Издатинлит, 1959, стр. 445.
4. Н. Schm id, Helv. chim. acta, 27, 127 (1944).
5. J. С as on, R. Way, J. Org Chem., 14, 32 (1949).
6. К. Аллеи, К. Вильсон, Синтезы органических препаратов, сб. 4,
Издатинлит, 1953, стр. 312.
7. Е. Brain, L. Finar, J.Chem.Soc., 2435(1958).
8. A. S i m о п i n i, Monatsb., 13, 320 (1892); Monatsh., 14, 81 (1893).
9. J. Roberts, H. Sim mo n s, J. Am. Chem. Soc, 73, 5487 (1951).
10. C. Prevost, С. г., 196, 1129 (1933).
И. С. Niemann, С. Wagner, J. Org. Chem., 7, 227 (1942).
Чичибабин* 264
Александр Евгеньевич Чичибабин (1871—1945) родился в местечке Кузе
мино Полтавской губернии. В 1892 г. окончил Московский университет; ученик
В. В. Марковникова. С 1899 г. ассистент кафедры химии Московского
сельскохозяйственного института, возглавляемой И. А. Каблуковым. С 1901 г.
приват-доцент Московского университета. В 1903 г. защитил магистерскую
диссертацию, с 1908 г. профессор органической химии Московского высшего
технического училища. В 1926 г. избран членом-корреспондентом, а в 1928 г. дебет
вительным членом АН СССР. В 1930 г. уехал за границу, где и умер в 1945 г
Основные работы А. Е. Чичибабина посвящены изучению химии
пиридина и других азотсодержащих гетероциклов, а также установлению строения
и синтезу ряда алкалоидов. Известны также исследования в области свободных
ароматических радикалов, метод получения альдегидов через магнийоруаниче-
ские соединения и др. Своими работами А. Е. Чичибабин способствовал
созданию и развитию отечественной химико-фармацевтической промышленности.
А. Е. Чичибабин является автором широко известного курса «Основные начала
органической химии», выдержавшего много изданий. В 1925 г. А. Е. Чичиба
бину была присуждена большая премия им. А. М. Бутлерова, а в 1926 г.—
премия им. В. И. Ленина.
РЕАКЦИЯ ЧИЧИБАБИНА
Реакцией Чичибабина называется взаимодействие
азотсодержащих гетероциклов с амидом натрия, приводящее к образование
а-аминопроизводных*. Эта реакция, открытая в 1914 г. на примере
получения а-аминопиридина*, протекает по схеме:
... . NaOH
V V^NHNa V^NH,
Синтез %-аминопроизводных всегда сопровождается
образованием небольших количеств %-аминопроизводных, (%,%'-диамино-
производных, дипиридилов и диниридиламинов.
При взаимодействии пиридина с избытком амида натрия основ
ным продуктом реакции является 2,6-диаминопиридин*. Если оба
Биография Чичибабина и описание реакции составлены редактором,
265 Чичибабин
%-положения пиридинового кольца заняты, то аминогруппа
вступает в т-положение. Тэк, из 2,6-диметилпиридина получается
4-ами но-2,6-диметилпиридинЗ» ^,
Предполагается, что первая стадия реакции Чичибабина
заключается в присоединении амида металла по связи —CH=N—.
полученный продукт присоединения [—CH(NH%)—NMe—] превра
щается в металлическое производное амина [—C(NHMe)=N—i
либо путем внутримолекулярной перегруппировки, либо при
разложении на амин и гидрид металла, которые затем реаги
руют между собой*:
\ <^ \.
Этот механизм реакции позволяет объяснить побочное образо
ванне у-аминопроизводных (1,4-присоединение) и отсутствие 8-ами
нопроизводных. При реакции с хинолином удалось доказать
образование нестойкого продукта присоединения**.
Как правило, препаративное значение реакции Чичибабина
ограничивается получением производных пиридина, хинолинв
и изохинолина. так как для других азотистых гетероциклов вы
ходы аминопроизводных незначительны.
L О реакции Чичибабина см. Л е ф ф л е р «Органические реакции», сб. ;
Издатинлит, 1948, стр. 115.
2. А. Е. Чичибабин, О. А. Зейде, РЖХО, 46, 1216(1914).
3. А. Е. Ч и ч и б а б и ы, РЖХО, 47, 835 (1915).
4. А. Е. Чичибабин, М. С. В и донов а, РЖХО, 53, 238(1921)
5. К. Z I eg I ег, Н. Z eisel, Ber., 63, 1848 (1930); А. В. Кирсанов
И. Иващенко. Bull. Soc. chim. France, (5) 2, 2109 (1935).
А. В. Кирсанов, И. Полякова, Bulk Soc. chim. France
(5) 3, 1600 (1936).
5. F. В е г g s t г о m, J. Org. Chem., 2, 411 (1937).
Чугаев 266
Лев Александрович Чугаев* (1873—1922) родился в Москве. В 1895 г-
окончил Московский университет. В 1904—1908 гг. профессор Московского
высшего технического училища. С 1909 г. заведующий Менделеевской кафедрой
Петербургского университета и одновременно профессор кафедры
неорганической химии Петербургского технологического института. Основатель и
директор (с 1918 г.) Государственного института по изучению платины и других
благородных металлов АН СССР. Работы Л. А. Чугаева по химии терпенов
привели к открытию нового метода превращения спиртов в олефины (ксанто-
геновый метод Чугаева). Изучая физико-химические свойства органических
соединений, он установил (1908 г.) зависимость оптической активности
соединений от их положения в гомологическом ряду (правило Чугаева) и открыл
(1911 г.) новый тип аномальной вращательной дисперсии. Большой вклад внес
Л. А. Чугаев в химию комплексных соединений. Он показал, что комплексные
циклические соединения значительно устойчивее соответствующих
ациклических соединений, открыл чувствительную реакцию на никель с диметнлглиок
симом (реактив Чугаева).
РЕАКЦИЯ ЧУГАЕВА
Превращение спиртов в олефины при термическом разложении
метилксантогенатов, полученных из этих спиртов, называется
реакцией ЧугаеваЧ
R CS,; NaOH R\ CH3J
—СНз—ОН -* )СН—СНз—О—С—SNa »
R R'/ ||
S
-*- \сН—СН.—О—С—S—СН, —-» \с=СН, ф HS—С—S—СНз
R'/ II R'/ II
Механизм реакции Чугаева был изучен Александером и
Мадрасом*. Отсутствие каких-либо перегруппировок в процессе
дегидратации объясняется промежуточным образованием циклического
реакционного комплекса.
* Биография Л. А. Чугаева переработана редактором.
267 Чугаев
показал, что для двух рядов рацематов 3-фенилбутано-
ла-2 реакция Чугаева протекает в основном как стереоспецифи-
ческий процесс, в котором преобладает /jwc-элиминирование:
Бордуэл и Лэндис* недавно показали, что подвижность
отщепляемого р-водорода оказывает большое влияние на течение
реакции Чугаева. По их данным, пиролиз ywc-2-л-толилсульфонил-
циклогексил-8-метилксантогената (I) в результате /лрдяс-элими-
нирования приводит почти исключительно к образованию
1-л-толилсульфонилциклогексена-1 (П):
Подобное направление реакции является исключением, так
как обычно реакция Чугаева протекает с %ис-элиминированием.
Очевидно, в данном случае конформация молекулы препятствует
образованию циклического промежуточного комплекса.
Соединение II было получено также цис-элиминированием /лрдмс-изо-
мера соединения I.
1. Л. Ч у г а е в, Вег., 32, 3332 (1894).
2. Е. Alexander, A. M u d г а с k, J. Am. Chem. Soc, 73, 59 (1951).
3. D. Cram, J. Am. Chem. Soc, 71, 3883 (1949).
4. F. В or d wel I, P. L a n d i s, J. Am. Chem. Soc, 80, 2450 (1958).
Шиман 268
Гюнтер Роберт Артур Шиман (Giinther Robert Arthur Schiemann) родился
a 1899 г. в Бреслау (Германия). Учился во Фрейбургском университете, затем
в Высшей технической школе в Бреслау, где в 1925 г. защитил докторскую
диссертацию и был назначен ассистентом кафедры органической химии. С 1929 Р
доцент Высшей технической школы в Ганновере, с 1950 г. заведующий кафед
рой технической химии Стамбульского университета. Основные работы поев я
сцены синтезу и изучению свойств фторароматических соединений,
РЕАКЦИЯ ШИМАНА
Термическое разложение борфторидов ароматических диазо-
соединений на ароматические фторпроизводные, азот и трехфто-
ристый бор называют реакцией [Лимана*.
Эта реакция является наиболее удобным методом получения
фторпроизводных из диазосоединений^:
NrnNO, HBF$ f*
Ароматические амины диазотируют обычным способом, затем
раствор соли диазония обрабатывают борфтористоводородной
кислотой (или фторборатом натрия), осаждая фторборат диазония.
По другому методу диазотирование сразу проводят в присутствии
борфтористоводородной кислоты. Нерастворимый фторборатдиазо*
ния отфильтровывают, промывают и сушат. В приведенном выше
примере сухую соль нагревают в колбе прибора для перегонки,
снабженного ловушкой для трехфтористого бора. Фторборат
фенилдиазония разлагается и фторбензол отгоняют при 75—87°.
Описаны различные методики разложения солей диазония.
По этим прописям обычно достигаются удовлетворительные
результаты^. Часто используются такие разбавители сухого фторбо-
рата диазония, как хлористый или бромистый калий, хлористый
аммоний, сульфат бария, фтористый натрий и песок.
Оптимальные условия реакции устанавливаются в зависимости от природы
и положения заместителей в ароматическом ядре соли диазония
1.0. В a I z, G. Schiemann, Вег., 60,1186 (1927).
2. О реакции Шимана см. А. Р оэ, Органические реакции, сб. 5, Издатинлнт.
1951, стр. 155.
3. Н. Schwechten, Ber., 65, 1605 (1932).
269 Шмидт
Карл Фридрих Шмидт (Karl Friedrich Schmidt) родился в 1887 г. Совмест
но с Т. Курциусоы изучал в Гейдельбергском университете воздействие суль
фурилазида |5Оз(Мз)2] на л-ксилол. Продолжил эту работу в 1922 г. в Швед
с ком университете г. Турку (Финляндия). В 1923 г. на собрании Немецкого
химического общества в Йене сделал доклад о действии азотистоводородной кис*
лоты на альдегиды и кетоны. После этого вернулся в Гейдельберг и позднее
стал профессором химии Гейдельбергского университета. Особенно много
работал над использованием азотистоводородной кислоты для синтеза
различных органических соединении.
РЕАКЦИИ ШМИДТА
Образование аминов из карбоновых кислот при обработке ид
азотистоводородной кислотой в присутствии концентрированной
серной кислоты носит название реакции Шмидта^.
В реакцию Шмидта наряду с карбоновыми кислотами могут
вступать и другие карбонильные соединениям Главными
продуктами взаимодействия альдегидов и кетонов с азотистоводородной
кислотой являются нитрилы и амиды. Протекающие реакции
можно выразить общими схемами:
RCOOHf
RCH0 4-
HN, "-^
HN, -^
RNHg
RCN
RCONHR
Реакция бензальдегида с азотистоводородной кислотой
приводит к образованию смеси бензонитрила и форманилида:
Соотношение выходов этих двух продуктов зависит от
количества взятой серной кислоты®: с увеличением количества серной
кислоты возрастает выход форманилида.
Нестойкие вещества или соединения, сульфирующиеся
концентрированной серной кислотой, не могут быть использованы
в реакции Шмидта*. В условиях этой реакции в малоновой кислоте
Шмидт 270
только одна карбоксильная группа вступает во взаимодействие
с HNg, поэтому конечным продуктом реакции является а-амино-
кислота. Дикарбоновые кислоты, в которых карбоксильные
группы разделены более чем одной метиленовой группой,
превращаются в соответствующие диамины*. По этому методу можно получить
также некоторые диаминокислоты* (лизин, орнитин).
Механизм реакции Шмидта был изучен Ньюменом и Гильден-
хорном?, которые на примере 2,4,6-триметилбензойной кислоты
показали, что при действии серной кислоты образуется оксокар-
бониевый ион R—С=О [I].
Согласно предложенному механизму, этот ион реагирует с
азотистоводородной кислотой, давая промежуточный продукт II;
последний теряет азот и перегруппировывается в ион III, причем
группа R со своей парой неподеденных электронов мигрирует от
атома углерода к атому азота:
R—C=O-t. :NH—A=
I II HI
При взаимодействии иона III с водой получается амин:
[OCNHR]+4H,O -^ КМНдфСОз
ш
Миграция группы R происходит с сохранением ее
конфигурации. Это же правило действительно для перегруппировок
Гофмана и Курциуса*, включающих аналогичную стадию
миграции группы R.
Конлей* успешно применил в реакции Шмидта с различными
кетонами полифосфорную кислоту, которая служит одновременно
растворителем и водоотнимающим средством. По этому способу
из циклопентанона получен пиперидон с выходом 83%:
О полифосфорная
Спиро-[4,4]-нонанон-1 действием азотистоводородной кислоты
в присутствии полифосфорной кислоты при 50° превращается в
271
Шмидт
Д8, в-гидринденон-4; при более низкой температуре и сокращении
времени реакции был получен ненасыщенный нитрил*:
50°; 8,5 час.
X
\) —
полифосфорная
кислота
25"
)=СН—
Смит и Хорвиц** изучили реакцию Шмидта для
несимметричных кетонов. Они нашли, что геометрические соотношения,
наблюдаемые при перегруппировке Шмидта, аналогичны
соотношениям, полученным для перегруппировки Бекмана^.
Конфигурация промежуточного иона III показывает, что в данном случае
мигрирует группа R:
О
II
R—С—R'
ОН
|
R—С—R'
ОН
I
R—С—R'
н.о
R—C-R'
N—A
ш
N=N-42—R'
||
N—R
=С—R'
|
NHR
Недавно установлено, что при шмидтовской перегруппировке
циклических кетонов, содержащих заместители основного
характера, преимущественно мигрирует группа, связанная с такими
заместителями^. Авторы объясняют это тем, что присоединившая
протон основная группа находится в дм/пи-положении по отноше-
нию к положительно заряженной группе —N=N.
Электростатический эффект падает по мере удаления атома азота основной
группы от реакционного центра.
1. К. Schmidt, Z. angew. Chem., 36, 511 (1923).
2. О реакции Шмидта см. Г. Воль ф, Органические реакции, сб. 3, Издат-
инлит, 1951, стр. 293.
3. W. McEwen, W. Conrad, С. VanderWerf, J. Am. Chem.
Soc, 74, 1168 (1952).
Шмидт 272
L Сравнение реакций Гофмана, Курциуса и Шмидта см. П. Смит,
Органические реакции, сб. 3, Издатннлит, 1951, стр. 344.
5. D. Hall, S. Mahbood, E. Turner, J. Chem. Soc., I960, 1842.
6. S. R о t h с h i I d, M. Fields, J. Org. Chem., 16, 1080(1951).
7. M. Newman^ H. Gildenhorn, J. Am. Chem. Soc., 70, 317 (1948)
8. R, Con I ey, J. Org. Chem., 23, 1330 (1958).
9. R. Con ley, B. N о w a k, Chem. Ind. (London), 1161 (1951).
10. P. Smith/j. Horwitz, J. Am. Chem. Soc., 72, 3718 (1950).
11. G. Badger, R. Howard, A. Simons, J. Chem. Soc, 2849(1952).
!2. H. S с h w e d, A. Hunger, K. Hoffmann, Helv. chim. acta,
33, 607 (1950%
273 Штоббе
Ганс Штоббе (Hans Stobbe, 1860—1938) родился в г. Тигенхофе (Германия).
Учился в Гейдельбергском, Мюнхенском и Страсбургском университетах,
затем у И. Вислиценуса в Лейпцигском университете. Здесь он защитил
докторскую диссертацию (1889 г.) и в 1892 г. стал профессором химии. Редактор
книги И. Поггендорфа кЛитературно-биографический справочник». Работа Г.
Штоббе по получению тераконовой кислоты из ацетона и диэтилового эфира
янтарной кислоты положила начало исследованию взаимодействия
карбонильных соединений с эфирами янтарной кислоты (конденсация Штоббе). Занимаясь
проблемами фотохимии, синтезировал около 60 фульгидов—ангидридов диал-
килиденянтарных кислот. Изучал также полимеризацию стирола под действием
света и тепла. Его труды в этой области имели большое значение для химии
полимеров.
КОНДЕНСАЦИЯ ШТОЬБЕ
Конденсация альдегидов и кетонов с эфирами янтарной
кислоты в присутствии основных катализаторов, приводящая к
образованию алкилиденянтарных кислот, называется реакцией или
конденсацией Штоббе^.
В качестве примера реакции можно привести получение
тераконовой кислоты из ацетона и диэтилового эфира янтарной кис-
(катализатор—этилат натрия):
CH,—COOQH, СзНвО№;Н+ СНз\ /СН.СООН
сн/ сн,—соосн. снУ \соон
Как правило, реакционную смесь оставляют на холоду на
несколько дней или недель. При добавлении воды с последующим
подкислением образуется моноэфир, который гидролиз уют до
дикарбоновой кислоты при помощи гидроокиси бария или едкого
натра.
Если исходный кетон имеет атом водорода в п-положении,
то продукт реакции может содержать примесь таутомерной алке-
18 А. Серрей
Штоббе 274
нилянтарной кислоты (I), которая в отдельных случаях является
единственным конечным продуктом конденсации:
R—CH, R-CH CH.COOH
"СО 4
\ СН.-СО(Х^Н, \ У
/ \
R СООН
Конденсация Штоббе осложняется побочной
реакцией—восстановлением некоторых кетонов до соответствующих спиртов.
Эту реакцию можно предотвратить, применяя диметиловый эфир
янтарной кислоты в присутствии mpe/м-бутилатов щелочных
металлов в /лрет-бутиловом спирте. В данных условиях выходы
получаемых продуктов выше, а время реакции меньше, чем при
использовании этилата натрия.
Конденсация бензофенона с диэтиловым эфиром янтарной
кислоты под действием /мре/м-бутилата калия приводит к
образованию Р-карбэтокси-%,т-дифенилвинилуксусной кислоты* (II)
с выходом 94% (время реакции 30 мин.):
СН,СООС,Н, m-c,H,OK ^^\ /
| Сс1
CH.O0OH
/С=ОФ | — ^ /
н
При конденсации диэтилового эфира фенилянтарной кислоты
с циклогептаноном в присутствии /пре/м-бутилата калия
образуется моноэфир—Р-карбэтокси-[3-(циклогептен-1-ил)-а-фенил пропио-
новая кислота*, т. е. двойная связь перемещается в цикл. Реакция
Штоббе была успешно применена для синтеза этиловых эфиров
р-ароилпропионовых кислот (АгСОСН,СН,СООС,Нд) с применением
гидрида натрия в качестве катализатора*.
Конденсация Штоббе имеет весьма широкое применение.
Компонентами реакции могут служить различные карбонильные
соединения, включая жирные и ароматические альдегиды и кетоны.
По методу Штоббе получается большое число веществ, в том числе
лактоны, нафтолы, индоны, тетралоны и фульгиДы (ангидриды
диалкилиденянтарных кислот).
Механизм конденсации Штоббе, вероятно, заключается в
образовании промежуточных продуктов-^производных эфиров пара-
коновой кислоты (Ш). Это объясняется особенностью поведения
эфиров янтарной кислоты в условиях реакции. Первичным про-
275 Штоббе
дуктом конденсации всегда являются моноэфиры (IV), которые
неоднократно выделялись с высокими выходами:
COOK' COOR'
R,CO+!CH-CH,-COOR' =<=±: РЛ-СН-СН^-С^
COOR' COOR'
Rtf
О
IH
COOR' COOR'
1. О конденсации Штоббе см. У. Джонсон, Г. Д о б, Оргечидескле резк
ции, сб. 6, Издатинлит, 1953, стр. 7,
В. Н. S t о b b е, Вег.,. 26, 2312 (1895).
8. У. Джонсон, У. Шнайдер, Синтезы органических препаратор
сб. 4, Издатинлит, 1953, стр. 279.
4. A. I s I a m, M. Z e m a i t у, J. Am. Chem. Soc., 80. 5896 (1958).
5. E. Bergman n, S. Yaroslavsky, H. Weiler-FeilchefeU
J. Am. Chem. Soc, 81, 2775 (1959).
Шторк 276
Жильберт Дж. Шторк (Gilbert J. Stork) родился в 1921 г. в Брюсселе.
В 1942 г. получил степень бакалавра в университете во Флориде. Под
руководством профессора Мак-Ильвейна выполнил докторскую диссертацию, которую
защитил в 1945 г. в Висконсинском университете. Преподавал в Гарвардском
университете. С 1953 г. и по настоящее время профессор Колумбийского
университета. Его труды посвящены главным образом исследованиям в области
механизма реакций и синтеза природных соединений. В 1957 г. удостоен премии
Американского химического общества за работу по теоретической химии. Пом о
щник редактора Journal of Organic Chemistry.
РЕАКЦИЯ ШТОРКА
Реакция С-алкилирования или ацилирования карбонилсодер-
жащих соединений с промежуточным образованием енамнна
известна под названием реакции Шторка^-%
Эта реакция обычно проводится следующим образом. При
взаимодействии альдегида или кетона с пирролидином в присутствии
катализатора (л-толуолсульфокислоты) получают енамин,
который ацилированием или алкилированием и последующим гидро
лизом превращают в соответствующее замещенное карбонилсо-
держащее соединение. Следует отметить, что реакция Шторка
нашла наибольшее применение при С-ацилировании и в про
цессах присоединения по Михаэлю (см. стр. 186).
В качестве примера реакции Шторка можно привести
имеющий важное значение синтез этилового эфира (2-оксоциклогек-
сил)-уксусной кислоты (II) из циклогексанона и этилового эфира
бромуксусной кислоты** *'* с промежуточным образованием
енамнна I:
Енамин I можно также алкилировать йодистым метилом,
причем получается 2-метилциклогексанон* с выходом 70%. Более
277
Шторк
ранними исследованиями было установлено, что реакция
образования енаминов* достаточно универсальна и легко обратима.
Поэтому ее использовали для введения защитной группировки, как,
например, в случае 3-кетостероидов.
Енамины применялись при получении тетрагидроциннолинов*,
в синтезе 4,/-протолихестериновой кислоты^ и при исследованиях в
области синтеза колхицина^ Недавно енамины были
использованы для введения атома фтора в а-положение молекулы стерои-
дов» (выход фторпроизводных 72%):
кто.
При изучении реакции Шторка с алициклическими кетонами
было показано, что промежуточно образующиеся енамины легко
алкилируются хлорцианом, акрилонитрилом или метилакрила-
том*. Например, енамин, полученный с 75%-ным выходом из цик-
лопентанона и пирролидина, реагирует с хлорцианом, давая 2-ци-
анциклопентанон^ (выход 46%).
Очень интересной модификацией реакции Шторка является
синтез А^^-окталона-2 (HI) при взаимодействии метилвинилке-
тона с енамином, полученным из циклогексанона^:
сн,=сн-со—сн.
Нуклеофильный характер промежуточного енамина становится
очевидным, если учесть эффект сопряжения:
С-Алкилирование с помощью хлорциана можно рассматривать
как нуклеофильную атаку соединением IV этого реагента с
образованием иммониевой соли V. Последняя, теряя протон, превра-
Шторк
278
щается в новый енамин VI, гидролиз которого приводит к циан
кетону VII:
CICN
-HCI
E.G.
8. M.
6 E
?, T
IV
V
ЛЯГЕРЛГУРЛ
Stork, R. Terrell, J. Szmuskovicz, J. Am. Chem. Soc
76, 2029 (1954).
Stork, H. Landsman, J. Am. Chem. Soc, 78, 5128 (1956).
Kuehne, J. Am. Chem. Soc, 81, 5400 (1959).
Baumgarten, P. Creger, С Villars, J.Am. Chem. Soc.
80, 6609 (1958).
Mannich, H. Davidsen, Ber., 698,2106 (1936); F. Hey!
M. Herr, J.Am. Chem. Soc, 75, 1918 (1953); M. Herr, F. Heyf
J. Am. Chem. Soc, 74, 3627 (1952).
T a m e 1 i n, S. В а с h, J. Am. Chem. Soc, 80, 3079 (1958).
Crabb, K. Schofield, J. Chem. Soc, 1958, 4276.
Gab bard, E. Jensen, J. Org. Chem., 23, 1406(1958).
279 Элбс
Карл Элбс (Karl Elbs, 1858—1933) родился в г. Альт-Брейзах (Германия).
8 1880 г. получил степень доктора философии во Фрейбургском университете,
с 1887 г. профессор этого университета. Преподавал физическую химию в Гис-
сенском университете, затем профессор экспериментальной химии того же
университета. Его книга, посвященная синтетическим методам получения
соединений углерода (1891 г.), явилась прототипом руководств Лассара—Кона и
Губена—Вейля. В 1902 г. написал книгу об электрохимических способах
получения различных соединений. Его исследования по электрохимическому
восстановлению и окислению органических веществ, особенно ароматических ни-
тросоединений, имеют большое научное и практическое значение. Разработал
методы получения надсерной кислоты и ее солей, которые использовал в
качестве окислителей. Установил, что смесь надсернокислого натрия и иода
является прекрасной средой для иодирования органических соединений.
РЕАКЦИЯ ЭЛБСА
Реакцией Элбса* называют процесс пиролитической
циклизации диарилкетонов, содержащих метильные или метиленовые
группы в орто-положении к карбонилу. Реакция приводит к
образованию полициклических ароматических систем.
Примером реакции может служить получение 2-метилантраце-
на из 2,5-диметилбензофенона^:
с
Реакция обычно проводится при нагревании замещенных ке-
тонов до температуры начала пиролиза^ (около 400°) и
продолжается до полного удаления воды. Во многих случаях побочные
процессы пиролиза затрудняют выделение и очистку конечных
продуктов. Хотя выходы целевых продуктов по реакции Элбса
обычно невысоки, этот метод все же часто применяется для синтеза
полициклических ароматических углеводородов вследствие
трудности их получения другими путями. Многие углеводороды этого
типа обладают канцерогенной активностью.
Элбс 280
Механизм реакции Элбса окончательно не установлен.
Возможно, что промежуточным продуктом реакции является дигидро-
антранол*' ^ (I):
Н ОН
\У 8 9 1
Недавно Херд и Азорлоза", используя соединения, меченные
дейтерием, показали, что, по-видимому, атом водорода,
находящийся в положении 9 молекулы антрацена, в исходном кетоне был
связан с ароматическим ядром.
1. О реакции Элбса см. Л. Ф и з е р. Органические реакции, сб. 1, Издатин-
лит, 1948, стр. 163.
2. К. Е 1 b s, E. L arsen, Бег., 17, 2847 (1884); К. Е I b s, J. prakt.
Chem., 41, 1 (1890).
3. L. Fieser, A. Seligman, J. Am. Chem. Soc., 58, 2482 (1936).
4. L. Fieser, E. Dietz, Ber., 62, 1827 (1929).
5. J. Cook, J. Chem. Soc, 1931, 487.
6. С H u r d, J. A z о г 1 os a, J. Am. Chem. Soc, 73, 37 (1951).
28] Элбс
ОКИСЛЕНИЕ ПЕРСУЛЬФАТАМИ ПО ЭЛБСУ
Процесс окисления одноатомных фенолов в двухатомные
при помощи персульфата калия в щелочной среде носит название
реакции персульфатного окисления или окисления по Элбсу*.
Реакция проводится при добавлении водного раствора
персульфата калия к охлажденному раствору фенола в разбавленной
щелочи; продукт реакции выделяют последующим подкислением
раствора. При окислении вторая гидроксильная группа обычно
вступает в пара-положение; если пара-положение занято,
получается орто-производное^.
г\%_г
Окисление по Элбсу* было применено также к замещенным
фенолам, нафтолам, кумаринам, флзвонам и азотсодержащим
гетероциклическим соединениям. Реакция протекает часто с
низкими выходами, но конечные продукты обычно получаются в
чистом виде. Примером реакции в ряду кумарина* может служить
образование 5,7-диметокси-6-оксикумарина (П) из 5,7-диметокси-
кумарина (I):
оси,
Недавно эта реакция была использована для окисления 2- и
3-оксипиридинов\ В обоих случаях после гидролиза сернокислых
эфиров в качестве главного продукта реакции был выделен 2,5-Ди-
оксипиридин:
А
V
Элбс282
1. К. El b s, 'J. prakt. Chem., 48, 179 (1893).
2. W. Baker, N. Brown, J. Chem. Soc., 1948, 2303.
3. О реакции Элбса см. S. S e t h n a, Chem. Revs., 49, 91 (1951).
4. V. D a I v i, R. Des a i, S. S e t h n a, J. Indian Chem. Soc, 28, 366
(1951).
5. E. В e h г m a n, B. P i 11, J. Am. Chem. Soc., 80, 3717 (1958).
283 Эмде
Герман Эмде (Hermann Emde, 1880—1935) родился в г. Опладен (Германия).
Изучал химию, фармацию и пищевую химию в Брауншвенгском
технологическом институте, а затем в Марбургском университете, где ему была
присуждена степень доктора. Затем преподавал в Брауншвейге, а в дальнейшем работал
под руководством Ф. Тиле в Страсбурге. В 1928 г. доцент Базельского универ.
ситета, в 1931 г. директор химико-фармацевтического института в Кенигсберге.
Многочисленные работы посвящены изучению природных соединений и
расщеплению четвертичных аммониевых солей.
РАСЩЕПЛЕНИЕ ПО ЭМДЕ
При обработке амальгамой натрия водных или спиртовых рас-
гворов галоидных солей четвертичных аммониевых оснований
происходит разрыв азот-углеродной связи. Этот метод
расщепления солей аммониевых оснований получил название расщепления
по Эмде! и был широко использован для установления строения
ряда алкалоидов и других азотсодержащих соединений^.
Эмде показал, что при обработке М,М-диметилтетрагидрохи-
аолиния (I) амальгамой натрия образуется о-(м-пропил)-М, N-диме-
тиланилин (II):
и
\
сн,
II
При гофмановском расщеплении четвертичной соли I не
происходит раскрытия кольца; в этих условиях отщепляется лишь
метиловый спирт и образуется N-метилтетрагидрохинолин:
он"
Эмде _^_______^_ 984
Галогениды N, N-диметилтетрагидроизохинолиния при рас
щеплении по Гофману и по Эмде образуют один и тот же (о-ви
нилбензил)-диметиламин (III):
Дальнейшее расщепление по Эмде полученной из соединения
III четвертичной аммониевой соли (IV) приводит к образованию
о-метилстирола (V):
ш iv v
Описано^ видоизменение метода расщепления по Эмде,
заключающееся в применении сплава никеля с алюминием вместе
амальгамы натрия. По этому методу восстановительное
расщепление четвертичных солей аммония быстро протекает в мягких
условиях и с хорошим выходом.
1. Н. Е m d e, Ann., 391, 88(1912).
2. Н. Em de, Н. К u 1 1, Arch. Pharm., 272, 469 (1934).
3. S. Sugasawa, S. Us h i о d a, Tetrahedron, 5, 48 (1959).
285 Эрленмейер— Плехль
Эмиль Эрленмейер мл. (Emil Erlenmeyer, Jr., 1864—1921) родился в
а. Гейдельберге (Германия). Его отец Эмиль Эрленмейер ст. (1825—1909) был
профессором химии Гейдельбергского университета. Эмиль Эрленмейер мл.
изучал естественные науки в Гейдельберге, а химию в Воине у А. Кекуле.
В дальнейшем совершенствовался в Марбурге, Дармштадте и в Геттннгене, где
в 1888 г. получил степень доктора. С 1891 г. преподаватель Боннского
университета, а с 1896 г. профессор химии в Страсбурге. В 1901 г. организовал
частную учебную лабораторию, в которой под его руководством работало много
студентов старших курсов. В 1907 г. его пригласили в Государственный
биологический институт в Далеме, где он получил возможность заняться
научной деятельностью, в частности исследованиями в области производных
коричной кислоты и вопросами физиологии растений.
СИНТЕЗ АЗЛАКТОНОВ ПО ЭРЛЕНМЕЙЕРУ—ПЛЕХЛЮ
Конденсация альдегидов с аудированными (обычно бензоил-
яли ацетил-) производными глицина в присутствии уксусного
ангидрида и уксуснокислого натрия, приводящая к получению
жсазолонов (азлактонов), называется синтезом азлактонов по
Эрленмейеру—Плехлю*-з.
Примером этой реакции может служить образование 4-бензили-
ден-2-метилоксазолона-5 из бензальдегида и ацетилглицина*:
С.Н,—СНО + СН.-СООН -» С,Н,—СН=С C=Oj
NH NO
СО С
сн, сн,
Эта реакция лучше изучена для ароматических альдегидов и
может рассматриваться как частный случай реакции Перкина
(см. стр. 201). Низшие алифатические альдегиды реагируют с
2-фенилоксазолоном-5 с образованием соответствующих
ненасыщенных азлактонов^.
Эрленмейер—Плехль 28G
Беннет и Ниман* недавно показали, что в ряде случаев
основная реакция конденсации сопровождается переацилированием.
т. е. обменом ацильного остатка ацилглицина (бензоила на ацетил).
Примером конденсации, сопровождающейся переацилированием,
является взаимодействие /г-фторбензальдегида с гиппуровой
кислотой в присутствии уксусного ангидрида и уксуснокислого
натрия. В результате реакции образуется смесь двух оксазолонов—
2-фенил- и 2-метил-4-(/г-фторбензилиден)-оксазолона-5:
сн,—соон
СО
4—CH=C С=0 o-FCgH^—СН=С С==О
->- N О ^ N О
с,н„ сн,
Азлактоны могут служить промежуточными продуктами^для
синтеза различных соединений, включая с-амино- и а-кетокислоты.
Гидролиз азлактойов приводит к а-ациламиноакриловым кислотам,
восстановлением которых получают а-аминокислоты:
R—CH=C—COOH R—CH,—СН—СООН
N О NH-COR' NH—COR'
R—CHg—СН—СООН
NH.
этому методу был получен фенил алан ин? (R=C,Hj с
выходом 86%.
Взаимодействием азлактонов с аминами и аминокислотами
образуются замещенные амиды и дипептиды. ^
а-Кетокислоты образуются при гидролизе азлактонов в
присутствии сильных минеральных кислот или щелочей. Окислением
287 Эрленмейер—Плехль
соответствующей а-кетокислоты, полученной этим путем, была
синтезирована 3,4-Диметоксифенилуксусная кислота*:
R—СН=С С=О ОН" Н:О„
| | » R—CHg—СО—СООН *"R—CHg^<
N О
R'
OCHg
Из азлактонов через а-кетокислоты и соответствующие оксимы
можно получать арилацетонитрилы*;
R—СН=С-—С=О
| | -* R—СН%—СО—СООН -^
N О
С
&
—» R—СНд—С—GOOH -» R—CHgCN
N—ОН
). J. PlochI, Бег., 17, 1616 (1884); Е. Erlenmeyer, Ann., 275, 1
(1893).
2. О методах получения азлактонов см. X. Картер, Органические реак
ции, сб. 3, Издатинлит, 1951, стр. 190; [СИ. Л у р ь е, Е. С. Ч а м а н.
Реакции и методы исследования органических соединений, кн. 9, Гос
химизат, 1959 г., стр. 155.—До/%. ped].
3. Обзор по химии 5-оксазолонов см. Е. В а 1 t a z z i, Quart. Rev. (London)
9, 150 (1955).
4. P. X e p б с т, Д. Ш е м и н, Синтезы органических препаратов, сб. 2,
Издатинлит, 1949, стр. 72.
5. М. Crawford, W. Little, J. Chem. Soc, 1959, 729.
6. E. Bennett, С Niemann, J. Am. Chem. Soc., 72, 1803 (1950).
7. P. X e p б с т, Д. Ш е м и н, Синтезы органических препаратов, сб. 2.
Издатинлит, 1949, стр. 498.
8. X. С н а и д е р, Д. Бек, В. А и д и, Синтезы органических
препаратов, сб. 2, Издатинлит, 1949, стр. 164.
9. J. Buck, R. Baltzly, W. Ide, J. Am. Chem. Soc, 60, 1789(1938),
Якобсен 288
Оскар Георг Фридрих Якобсен (Oskar Georg Friedrich Jacobsen, 1840—»
^889) родился в Аренбурге (Германия). Учился в Кольском университете,
где в 1868 г. получил степень доктора философии и работал ассистентом.
С 1873 г. профессор химии и фармации, позднее заведующий кафедрой химии,
а затем ректор Ростокского университета.
РЕАКЦИЯ ЯКОБСЕНА
Миграция алкильнои группы или атома галоида при действии
на полиалкилбензолсульфокислоты или галоидполиалкилбензол-
сульфокислоты концентрированной серной кислоты известна под
названием реакции Якобсена^ ^.
Примером этой реакции служит получение 2,3,4,5-тетраметил-
бензолсульфокислоты (выход 50%) из изомерной 2,3,5,6-тетра-
метилбензолсульфокислоты:
SO,H
,сн,;
Побочными продуктами данной реакции являются 2,4,5-триметил-
бензолсульфокислота, гексаметилбензол и двуокись серы.
Натриевая соль сульфокислоты I может быть гидролизована до
соответствующего углеводорода —1,2,3,4-тетраметилбензола (пренитола)*.
В качестве примера миграции атома галоида можно привести
образование 2,4-дихлор-1,3-ДИметилбензола (выход 12%) из 4,6-
дихлор-1,3-диметил бензола:
289 Якобсен
Реакция Якобсена может протекать как с внутримолекулярной,
так и с межмолекулярной миграцией алкильной группы (или
соответственно атома галоида). Во всех случаях образуются также и
вицинальные изомеры; механизм реакции пока не установлен.
1. О. Jacobsen, Вег., 19, 1209 (1866).
2. О реакции Якобсена см. Л. Смит, Органические реакции, сб. 1, Издат-
инлит, 1948, стр. 492.
3. S. Birch, R. Dean, F. F i d 1 е г, R. L о w г у, J. Am. Chem. Soc,
71, 1362 (1949).
19 A Ceppea
Предметный указатель
290
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
В указателе приведены названия конечных соединений, синтезир? емых по реакциям,
которые рассматриваются и книге; исходные и промежуточные продукты, как правило, в указатель
не вынесены.
Азиды кислот в реакции Курциуса
166
Азлактоны
превращения 286, 287
синтез по Эрленмейеру—Плехлю
285 ел.
Акридилфосфиновые эфиры 12
Акриловая кислота, производные
синтез по Перкину 201, 202
— по Реформатскому 225
— по Родионову 227
(З-Аланин 115
Алкилиденциануксусные эфиры 151
Алкилиденянтарные кислоты 273 ел.
Алкилирование
по Вильямсону 62, 63
по Манниху 180 ел.
по Фриделю—Крафтсу 107, 253 ел.
по Шторку 276 ел.
Алкилфосфиновые эфиры 11, 12
С-Аллилацетоуксусный эфир 142
6-Аллил-2,6-диметилциклогексадиен-
2,4-он 144
о-Аллилфенолы 143, 144
Альдегидокислоты 230
Альдегиды
восстановление по Дарзану 44
— по Меервейну—Понндорфу—
Верлею 186
диспропорционирование по Канниц-
царо 132, 133
окисление по Байеру—Виллигеру
16, 18
синтез по Б одр у—Чичибабину 46,
47
— по Буво 46, 47
— по Виттигу 65
— по Гаттерману 78 ел.
— по Гаттерману—Коху 81, 82
— по Дарзану 104
— по Зончу—Мюллеру 237, 238
Альдегиды
синтез по Клайзену—Шмидту 145
— по Кренке 235
— по Курциусу 167
— по Мак-Фадиену—Стивенсу 178,
179
— по Нефу 195
— по Рейсерту 222, 223
— по Розенмунду 229, 230
— по Соммле 233 ел.
— по Стефену 236 ел.
— по Хунсдиккеру 262
Альдимины в реакции Гаттермана 78
Амиды кислот, синтез
по Шмидту 269 ел.
по Эрленмейеру—Плехлю 286
Амилбензол 148
Аминирование
альдегидов 57, 58
кетонов 57
оксисоединений 48, 49
Аминокислоты, синтез
по Габриэлю 72
по Курциусу 166, 167
по Родионову 227. 228
по Шмидту 269, 270
по Эрленмейеру—Плехлю 286
Аминопиридины 264, 265
л-Аминосалициловая кислота, синтез
по Бешану 32
по Кольбе—Шмитту 156
Аминоспирты, синтез
по Иванову 129
по Канниццаро 133
Аминохинолины 49
Амины
гидролиз по Бухереру 48 ел.
расщепление по Брауну 42, 43
— по Полоновскому 43
синтез по Габриэлю 72, 73
— по Гофману 93 ел.
291
Предметный указатель
Амины
синтез по Делепину 114, 115
— по Иванову 129
— по Курциусу 166 ел.
— по Лейкарту 172 ел.
— по Лоссену 175 ел.
— по Соммле 233 ел.
— по Шмидту 269 ел.
З-Анизилпропионовая кислота 60
л-Анисовый альдегид 78
Лрб//зобд перегруппировка 11, 12
Арилакрилонигрнлы 184
Арилацетонитрилы 287
Арилпропнонитрилы 184, 185
Арилхлорметилкетоны 14
Лрм^гд—Эдсгерга синтез 13 ел.
(З-Ароилпропионовые кислоты 274
Арсоновые кислоты 22, 23
Асимметрический синтез 200
Ацетальде: ид 169
а-Ацетаминоалкилыетилкетоны 124,
125
Ацетоксибензойные кислоты 259
Ацетон, синтез
по Кучерову 169
по Нефу 195
Ацетофенон, синтез
по Кучерову 169
по Фриделю—Крафтсу 253, 254
Ацилгипогалогениты в реакции Хунс-
диккера 261
Ацилирование
по Клайзену 116, 139 ел.
по Фриделю—Крафтсу 253 ел.
по Шторку 276 ел.
по Эрленмейеру—Плехлю 285 ел.
о-Ацилоксианилиды 199
д—Диллыгерд реакция 16 ел.
—Дилдя^д реакция 19 ел.
5дргд реакция 22, 23
Беклюнд перегруппировка 24 ел., 271
Бензальаминоацетали в реакции
Померанца—Фрича 208, 209
Бензальдегид, синтез
по Гаттерману—Коху 81
по Мак-Фадиену—Стивенсу 178
по РеЙсерту 223
по Соммле 233, 234
Бензанилид 24, 26
Бензгидроксамовая кислота 175
Бензгидрол 186
Бензиламин, синтез
по Габриэлю 72
по Гофману 60
по Соммле 233
2-Бензилбутан 70
Бензилиденацетофенон 145
Бензиловый спирт 132, 133
Бензилпиперидин 57, 58
Бензилхлорид 37
Бензоилпропионовая кислота 254
Бензойная кислота 132
БенЗонитрил, синтез
по Брауну 40
по Шмидту 269
Бензфенантрен, производные 218
беруд реакция 27 ел., 36
Дешднд реакция 31, 32
Биксин 65
Бифенил 241
Д
р//длырдлбекого реакция
33 ел., 207
Длднд реакция 37 ел.
5o^p/—Уычкбдбимд реакция 46. 47
9%яд реакция 261
синтез 100
р^
расщепление аминов 42, 43
реакция 40, 41
5-оромвалерианоная кислота, эфир
262
З-Бромпропиламин 114
Бромтолуолы, синтез
по Волю—Циглеру 67
по Гаттерману 77
4-Бром-1-фенилгтнразол 262
5^60—блднд реакция 44, 45
Zx/go синтез альдегидов 46, 47
Бутилбензол 71
Бутил-о-нитрофениловый эфир 63
реакция 48 ел.
синтез гидантоинов 51, 52
р перегруппировка
53 ел., 190, 191
Ддллдхд реакция 57, 58
Д реакция 59 ел.
реакция 79, 80
синтез 62, 63
Винилкетоны 102
Дкттигд реакция 64, 65
Вольф
перегруппировка 14, 15
реакция 136, 137
Доля—Циглерд реакция 66 ел.
Восстановление
альдегидов 44, 59, 186
анилидов 237, 238
ацетиленов 59
карбоновых кислот 44
Предметный указатель
292
Восстановление
кетонов 59, 60, 186, 187
нитрилов 236, 237
нитросоединений 31, 32
олефинов 59
по Верчу 27 ел., 36
по Бешану 31, 32
по Буво—Блану 44, 45
по Вильгеродту 59 ел.
по Зонну—Мюллеру 237, 238
по Клемменсену 137, 147 ел.
по Мак-Фа диену—Стивен су 178,
179
по Меервейну—Понндорфу—Вер-
лею 186, 187, 197
по Розенмунду 229, 230
по Стефену 236 ел.
сложных эфиров 44, 45
спиртов 148
а реакция 69 ел.
реакция 71
синтез 72, 73
Галоидалкилы, синтез
по Бородину 261
по Брауну 40 ел.
по Хунсдиккеру 261 ел.
Галоидкислоты
полиалкилбензолсульфокислоты 288
синтез по Волю—Циглеру 66, 67
— по Геллю—Фольга
рдту—Зелинскому 84, 85
Галоидпроизводные, синтез
по Брауну 40 ел.
по Гаттерману 77
по Зандмейеру 126, 127
по Хунсдиккеру 261 ел.
по Шиману 268
Галоидэфиры 262
Г синтез 74 ел.
р
реакция 77
синтез альдегидов 78 ел.
/"дггерлюмд—Колгд реакция 81, 82
4, 4а, 5, 6, 7, 8-Гексагидро-(ЗН)-наф-
таленон-2 28
Гексадекандиол-1,2 263
Гексановая кислота 158
Гексин-1 130
Гелля—Фольгдр^а—Зеленского
реакция 84, 85
Гептамид 26
Гейш синтез 86 ел.
Гидантоины 51, 52
Гидридный сдвиг 134, 173, 187
А'.*-Гидринденон-4 270, 271
Гидроксамовые кислоты 175
Гндропиз
азлактонов 286
аминов 48 ел.
гексаметилентетрамина 114
глицндных эфиров 104
дитиогидантоинов 52
изоцианатов 94
имидоэфиров 244
кетиминов 87
окснмов 196
тиоморфолидов 61
уретанов 93
фталнмидов 72, 73
халконов 146
Гидрохинон
метиловый эфир 17
синтез по Дэкину 122
Гиперконъюгация 96
1,3-Гликоли 210 ел.
Глицидные кислоты и эфиры 103 ел.
—Хея реакция 89.
90
реакция 91, 92
перегруппировка 60, 93 ел., 166,
167, 176, 270
правило 96
расщепление аммониевых
оснований 96 ел., 284
Гребе—Ульлшнд синтез 99, 100
реактивы 46, 47
реакция 101, 102, 130, 131
бенд—Фишера реакция 87
Ддрздмд синтезы
глицидных эфиров 103 ел.
непредельных кетонов 106 ел.
реакция 109, 110
реакция 151
синтез 111, 112
Дебнерд—Миллера синтез 112, 113
Делемина реакция 114, 115
«Дназометод» Несмеянова 192, 193
Диалкилиианамиды 42, 43
Диаминокислоты 270
Диамнны, синтез
по Габриэлю 72
по Курчиусу 166, 167
по Шмидту 270
Диарилы, синтез
по Гомбергу—Бахману—Хею 89, 90
по Ульману 241, 242
1,4-Дибромбутан 261
293
Предметный указатель
1,5-Дибромпентан 40
Дигидропирндины в синтезе Ганча
74 ел.
3,6-Дигидрофталевая кислота 120
Дигидрохинолины, синтез
по Дебнеру 112
по Дебнеру—Миллеру 112, 113
по Рейсерту 222, 223
по Скраупу 232
Диеновые синтезы 119 ел.
Диизоцианаты 176
Дикарбоновые кислоты
бронирование 84, 85
восстановление 230
синтез по Коновалову 162
Дикетоны 158
Дшсяама реакция 116 ел.
Дкльсд—Лль^ерд реакция 104, 119 ел.
N-Диметиламинокоричный альдегид
80
л-Диметиламинофенилгидроксиламин
235
2,4-Диметилбензхинолины 161
М,М-Диметилгидроксиламин 97
3,4-Диметилпирокатехин 123
(—) -7,8-Диметилтетрадекан 70
1,2-Диметилциклогексен 55
3,4-Диметоксифенилуксусная кислота
287
2,2^-Динитродифенил 242
л(-Диоксаны 210 ел.
2,4-Диокси-6-метоксибензойная
кислота 87
2,5-Диоксипиридин 281
3,4-Диоксифенилацетонитрил 123
2,4-Диокси-З-формилбензойные
кислоты 78, 79
Дипептиды 286
Дипиридиламины 264
Дипиридилы 264
Диспропорционирование
альдегидов 132, 133
дигидрохинолинов 112
Дитиогидантоины 52
1,4-Дифенилантрахинон 120
Дифенилэтилены, синтез
по Барбье—Виланду 19, 20
по Виттигу 64
2,4-Дихлор-1,3-диметилбензол 288
4-Дихлорметил-4-метилциклогекса-
диен-2,5-он 220
3,5-Ди-(хлорметил)-2-оксиацетофе-
нон 38
6,9-Дихлор-2-метоксиакридин 243
2,4-Дихлорфеноксиуксусная кислота
63
4-Диэтиламинобутанон-2 180
Дэкимд—Веста реакция 124, 125
реакция 18, 122, 123
Замещение
нуклеофильное 38, 53, 70, 237, 244,
245, 250
радикальное 70, 90, 138, 185, 193,
218, 242, 261
электрофильное 38, 82, 99, 127, 201,
205, 220, 233
Здн&иеДерд реакция 77, 126, 127
Зоммд—Мюллера реакция 237, 238
#бднодд реакция 128, 129
Изоборнилхлорид 54, 55
Изомеризация
оксикетонов 259
терпеновых углеводородов 53 ел.
эфиров фосфористой кислоты 11, 12
Изопропнлиденциклопентан 55
Изохинолины, синтез
по Бншлеру—Налиральскому 33 ел.
по Пиктэ—Гамсу 34
по Померанцу—Фричу 208, 209
по Чнчибабину 265
Изоцианаты 175 ел.
Имидоэфиры в реакции Ульмана 244
З-Индолилуксусная кислота 250
Индолы 249 ел.
Индоны 274
Иохнмбин 104
Яо^чд реакция 130, 131
Какодилфосфиновые эфиры 12
Камфен 53, 54
Камфеновые перегруппировки 53 ел..
190, 191
Канниццаро
диспропорционирование альдегидов
132 ел., 187
«перекрестная» реакция 133
Капроновый альдегид 47
Карбазолы, синтез
по Борше 100
по Гребе—Ульману 99, 100
по Пшорру 218
Карбиноламины
в реакции ЛеАкарта 173
синтез по Лолоновскому 43
Карбоновые кислоты
бромирование 84, 85
восстановление 44, 178, 179, 229
синтез по Арндту—Эйстерту 13 ел
— по Канниццаро 132
— по Реформатскому 224, 225
— по Фаворскому 246 ел.
Предметный указатель
294
Кетимины в реакции Геша 87
Кетоальдегиды 235
Кетокарбоновые кислоты
синтез по Дикману 116, 117
— по Эрленмейеру—Плехлю 286
электролиз 158
Кетоны
восстановление 44, 147, 148, 186
синтез по Гешу 87
— по Дарзану 106 ел.
— по Клайзену—Шмидту 145, 146
— по Курциусу 167
— по Неницеску 106, 107
— по Нефу 195
— по Принсу 212, 213
%-Кетосубермновая кислота 182
;д реакция 135 ел.
конденсация 116, 139 ел.
перегруппировка 142 ел.
реакция 141
ш^т конденсация 145,
146
реакция 137, 147 ел.
я реакция 150 ел.
синтезы
оксихинолина 165
пирролов 153, 154
Колхицин 277
—Шл<иттд реакция 155 ел.
^ электрохимическая реакция
158, 159
синтез хинолина 160, 161
реакция 106
Конденсация
гидразонов 249 ел.
Клайзена 116, 139 ел.
Клайзена—Шмидта 145, 146
кротоновая 113
Меервейна 184, 185
Пеамана 204 ел.
Принса 210 ел.
сложноэфирная 116 ел.
Ульмана 243 ел.
Штоббе 273 ел.
Комоадлоед реакция 162, 163
/(омрд^д—Лнл(лдхд синтез хинолина
92, 164, 165
Коричная кислота
производные 141, 184, 217
синтез по Перкнну 201
/(pewce реакция 235
Кумарины, синтез
по Перкину 202
по Пехману 204 ел.
по Элбсу 281
Кумарон, производные 145
о-Кумиловый альдегид 234
%р%и#са реакция 25, 166 ел., 176,270
Л^цероед реакция 169
Кэлмсд реакция 170, 171
Лактоны, синтез
по Байеру—Виллигеру 16, 18
по Штоббе 274
ЛауриновыЙ альдегид 237
Лауроилацетон 141
ЛеДкдргд—Вдллдлгд реакция 57, 58
ЛеДкдргд реакция 57, 172 ел.
Ликопин 65
Лоссенд реакция 167, 175 ел.
стереоспецифичность 177
д<Э%е«—Сгыбемсд реакция 178,
179
реакция 133, 180 ел.
Мдркоеды/сое
правило 211
реакция 162
конденсация 184, 185
д—ZZ/усгерд перегруппировка
108
реДкд—/Уонн^одфя—Верл^л
реакция 186, 187, 197
Меркаптаны в реакции Вильгеродта
60
Металлоорганические соединения
синтез по Вюрцу 70
— по Несмеянову 192, 193
2-Метилантрацен 279
а-Метилбензиловый спирт в реакции
Клемменсена 148
грднс-Метилдекалоны 220
Метнленмалоновые эфиры 151
4-А1етилизоборнеол 190, 191
о-Метилстирол 284
N-Метилтетрагидрохинолин 283
5-Метилфурилкарбннол 133
Метнлциклогексан 135
1,2-Метилциклогексановая кислота
247
2-Метилциклогексанон 276
Метилциклогексилкетон 106, 107
4-Метоксифенилацетальдегид 236
Мшгдэля реакция 181, 182, 188, 189,
276
Мишррд реакция 20, 67
Мышьяковоорганические соединения
193
перегруппировка 190, 191
Натрийорганические соединения 70
295
Предметный указатель
р-Нафтальдегид, синтез
по Розенмунду 229
по Стефену 236
р-Нафтиламин 49
<%-Нафтилкарбичол 133
Нафтойные кислоты 28
I -Нафтол-4-сульфокислота 48
Нафтолы 274
реакция 106, 107
реакция 192, 193
реакция 195, 196
р реакция 14
Нитрилы, синтез
по Губену—Фишеру 87
по Зандмейеру 126
по Шмидту 269, 271
2-Нитрогексан 162
Нитро-2,5-днметилгексаны 162
2-Нитро-2-метмлпропан 163
м-Нитростильбен 184
1-Нитро-1-фенилэтан 162
Нуклеофильное замещение 38, 53. 70,
237, 244, 245, 250
присоединение 277, 278
Окисление
персульфатами по Элбсу 281, 282
по Байеру—Виллигеру 16 ел.
по Вильгеродту 59
по Дэкину 18, 122, 123
по Оппенауеру 186, 197. 198
Оксазолидоны в реакции Курциуся
167
Оксазолоны 124, 285 ел.
Оксиалвдегиды, синтез
по Гаттерману 78 ел.
по Даффу 109, ПО
по Реймеру—Тиману 219 ел.
В-Оксия^пды в реакции Иванова
129
Оксикарбоновые кислоты, синтез
по Кольбе—Шмитту 155 ел.
по Реймеру—Тиману 219, 220
по Реформатскому 224 ел.
Оксикетоны; синтез
по Гешу 86 ел.
по Фрису 258 ел.
/рдмс-2-Оксиметилцнклогексанол-1
212
р-Окси-а,р,р-трифенилэтансульфоно-
вая кислота 129
Оксихинолины, синтез
по Гоулду—Джекобсу 91, 92
по Конраду—Лимпаху 164, 165
по Кэмпсу 170, 171
ло Манниху 181
4-Оксотетрагидронафталин-2-сулъфо-
кислота 49
(2-Оксоииклогексил) -уксусная
кислота, эфир 276
А*.9-Окталон-2 277
Октанон-4 148
Олефины, синтез
по Вильямсону 63
по Виттигу 64, 65
по Вюрцу 69
по Гофману 96 ел.
по Кольбе 159
по Чугаеву 266, 267
Оловоорпанические соединения 193
Од реакция 186, 197, 198
Палаверальдин в реакции Фрича 209
Папаверин 209
Лдссерыш; реакция 199, 200
Перегруппировка(и)
Арбузова II, 12
Бекмана 24 ел., 271
Вагнера—Меервейна 53 ел., 190,
191
Вольфа 14, 15
Гофмана 60, 93 ел., 166, 167, 176
270
камфеновые 53 ел., 190, 191
Клайзена 142 ел.
Курциуса 25, 166 ел., 176, 270
Лоссена 167, 175 ел.
Наметкина 190, 191
МсЛера—Шуетрра 108
пинаколиновая 53
ретропинаколннопая 53
Стивенса 239, 240
Фаворского 246 ел.
Фриса 258 ел.
Чэпмена 244, 245
«Перекрестная» реакция Канннццаро
133
Перкина реакция 141, 150, 201 ел.,
285
/7ехл*а«о конденсация 204 ел.
Пикроподофилин 34
/7%ктэ—Голсо синтез 34
Яыкгэ—Шлемглера реакция 207
Пинаколиновая перегруппировка ЛЗ
Пиперидины, синтез
по Клемменсену 148
по Лейкарту 173
Пиперидон 270
Пиперонилакрилопая кислота 228
р-Пиперонил-р-аминопропионовая
кислота 228
Пиразины 153
Предметный указатель
296
З-Пиразолилуксусные кислоты 61
Пиридины, синтез
по Бншлеру—Напиральскому 36
по Ганчу 74 ел.
по Кренке 235
по Мак-Фаднену—Стивенсу 178
по Чичибабину 264, 265
Пирогаллол, эфир 123
Пирролы, синтез
по Кижнеру 136
по Кнорру 153, 154
Полиалкилбензолсульфокислоты 288
Полиамиды 176
Полиарилы 241
/7 реакция 43
—Фричд реакция 208, 209
Порфирины 154
Правило
Гофмана 96
Марковникова 211
/7pg@o реакция 263
Предельные углеводороды
нитрование 162, 163
синтез по Вюрцу 69, 70
— по Кижнеру 135 ел.
— по Клемменсену 147, 148
— по Кольбе 158, 159
Пренитол 288
/7ршгд реакция 210 ел.
л-Пропил-М-диметиланилин 283
Пропилцинхониновая кислота 215
^,/-Протолихестериновая кислота 277
реакция 214 ел.
а синтез 90, 217, 218
Радикальное замещение 70, 90, 138,
185, 193, 218, 242, 261
Рацемизация, в реакциях
Вюрца 70
Наметкина 190, 191
Стивенса 239
Расщепление
по Барбье—Виланду 19 ел.
по Брауну 42, 43
по Гофману 96 ел., 284
по Мишеру 20, 67
по Эмде 283, 284
Реактив(ы)
Гриньяра 46, 47, 101, 102, 129
Иванова 128, 129
Иоцича 130, 131
Саретта 198
Реакция(и)
Бодру—Чичибабина 46, 47
Бородина 261
Вильсмейера 79, 80
Реакция(и)
Вольфа 136, 137
восстановления 27 ел., 36, 44, 45,
59 ел., 137, 147 ел., 178, 179,
186, 187, 197, 229, 230, 236 ел.
Вюрца—Фиттига 71
Губена—Фишера 87
Зонна—Мюллера 237, 238
ионные 16, 17, 90, 137, 185
исчерпывающего метилирования 34,
96 ел.
Кондакова 106
* конденсации 113, 116 ел., 139 ел.,
145, 146, 184, 185, 204 ел.
210 ел., 243 ел., 249 ел., 273
Кренке 235
Лейкарта—В аллаха 57, 58
Марковннкова 162
Неницеску 106, 107
Ниренштейна 14
нуклеофильного замещения 38, 53,
70, 237, 244, 245, 250
присоединения 277, 278
окисления 16 ел., 59, 122, 123, 186,
197, 198, 281, 282
Полоновского 43
Прево 263
радикального замещения 70, 90,
138, 185, 193, 218, 242, 261
расщепления 19 ел., 42, 43, 67,
96 ел., 283, 284
Симонини 263
Симониса 205
Тишенко 132
Финкельштейна 62
Фиттига 71
хлорметилирования 37 ел.
циклизации 90, 116 ел., 160, 161,
207 ел., 217, 218
электроф ильного замещения и
присоединения 33, 34, 38, 82, 99,
127, 201, 205, 220, 233
Эшвейлера—Кларка 174
Р-Резорциловая кислота 156
у-Резорциловый альдегид 79
Рр7%лю«а реакция 109,
219 ел.
реакция 222, 223
Ретропинаколиновая
перегруппировка 53
реакция 224 ел.
—Крое цепко реакция 228
реакция 227, 228
д реакция 229
Ртутноорганические соединения 192,
193
297
Предметный указатель
Салициловая кислота, синтез
по Кольбе—Шмитту 155
по Реймеру—Тиману 219, 220
Салициловый альдегид 219
Со/?егтд реактив 198
Свинцовоорганические соединения 193
«Сдвиг» Фриса 258 ел.
Сильван 136
реакция 263
реакция 205
Синтез (ы)
азлактонов по Эрленмейеру—Плех-
лю 124, 125, 285 ел.
альдегидов
по Бодру—Чичибабину 46, 47
по Буво 46, 47
по Гаттерману 78 ел.
Арндта—Эйстертя 13 ел.
асимметрический 200
Борше 100
Вильямсона 62, 63
Габриэля 72, 73
Ганча 74 ел.
Геша 86 ел.
гидантоннов по Бухереру 51, 52
Гребе—Ульмана 99, 100
Дарзана
глицидных эфиров 103 ел.
непредельных кетонов 106 ел.
Дебнера Ш, 112
Дебнера—Миллера 112, 113
диеновые 119 ел.
индола по Фишеру 249 ел.
Кнорра
оксихинолина 165
пирролов 153, 154
Пиктэ—Гамса 34
Пшорра 90, 217, 218
Фридлендера 214, 256, 257
хинолина
по Комбу 160, 161
по Конраду—Лимпаху 92, 164,
165
Сквален 65
Скраулд реакция 112, 113, 160, 231
Сложные эфиры
восстановление по Буво—Блану 44,
45
конденсация 116 ел., 139 ел.
синч ез по Байеру—Виллигеру
16 ел.
Сол*лме реакция 233 ел.
Спирты
восстановление по Клемменсену
148
синтез по Буво—Блану 44, 45
Спирты
синтез по Канниццаро 132, 133
— по Меервейну—Лонндорфу—
Верлею 186, 187
— по Принсу 211, 212
реакция 236 ел.
перегруппировка 239, 240
Стириламиды в реакции
Пиктэ—Гамса 34
Сурьмяноорганические соединения 193
Терефталевый альдегид 234
Тетрагндроацетофенон 106, 108
Тетрагидроизохинолины 207
Тетрагидротиофены 117, 118
Тетрагидроциннолины 277
Тетралоны 274
1,2,3,4-Тетраметилбензол 288
7рдмс-4,5-Тетраметилен-1,3-диоксан
212
Тиоамиды 60
Тиоморфолиды 61
7uw<ewco реакция 132
а-Толилнитрометан 162
1-я-Толилсульфонилциклогексен-1 267
Толуиловые альдегиды, синтез
по Бодру—Чичибабину 46, 47
по Буво 46, 47
по Гаттерману—Коху 81
2,6,6-Триметилбицикло-[0,1,3]-гексан
136
1,1,2-Триметилциклопропэн 136
2-(3,4,5-Триметоксибензоил)-4,5-ме-
тилендиоксистирол 34, 35
Триптофан 182
1,3,3-Трифенилпропиламин 172
Трифторуксусная кислота 108
Троповая кислота 128
Углеводороды, синтез
по Вюрцу 69 ел
по Вюрцу—Фиттигу 71
по Кижнеру 135 ел.
по Клемменсену 147, 148
по Кольбе 158, 159
по Фиттигу 71
по Элбсу 279, 280
конденсация 243 ел.
реакция 241, 242
Уретаны в реакции Курциуса
166, 167
перегруппировка 246 ел.
Фенантрены 217, 218
(—)-а-Фенетиламин 176, 177
Предметный указатель
298
Фенилаланин 286
Фенилаллиловые эфиры 144
Фенилантраниловые кислоты, синтез
по Ульману 243, 244
по Чэпмену 244, 245
1-Фенил-4-ацетилциклогексан 107
а-Фенилацетоацетонитрил 140
1-Фенилбензотриазол в синтезе Гре-
бе—Ульмана 99
Фенилмалоновая кислота 128
4-Фенил-4-метилпентанон-2 20, 21
2-Фенилпентанон-4 107
1-Фенил-2-пропиониламинопента-
нон-3 124
а-Фенилпропионовый альдегид 104
Фенилуксусная кислота, амид 59
Фенилцианпировиноградная кислота,
эфир 140
уыс-2-Феннлциклогексанкарбонозая
кислота 2П
1-Фенилциклогексен 97
5-ФенмлциклогексенЛ -оны-4 195
Фенилэтаноламнны 167
Фенилэтиламины 146
м-Феноксифенилмасляная кислота 137
Фенолы, синтез
по Бухереру 48
по Дэкину 122, 123
по Зандменеру 127
реакция 62
реакция 71
р синтез индола 249 ел.
Флуореноны 218
Флуорены 218
Форманилид 269
2-Формилбензтиазол 235
Формулирование 79 ел., 219 ел.
Формилциклопропан 179
Фосфора ароматические соединения
11, 12
л^—/(рофтсд реакция 81, 107,
147, 253 ел.
р синтез 214, 256, 257
р реакция^ 258 ел.
Фталевая кислота 72
Фталнлгидразид в реакции Габриэля
73
Фталимиды 72, 73
Фторбензол 268
Фульгнды 274
Фураны 196
Халконы 145, 146
Хинальдины, синтез
по Дебнеру—Миллеру 113
по Рейсерту 222, 223
Хинолин(ы), синтез
но Комбу 160, 161
по Конраду—Лнмпаху 164, 165
по Скраупу 231, 232
по Фридлендеру 214, 256, 257
по Чичибабину 265
9-Хлоракридины 243
Хлорбензол 77
4-Хлорбутилметиловый эфир 62
7-Хлор-3,4-диаминохинолин 31
Хлорметилирование 37 ел.
1-Хлорэтилбензол в реакции Клем-
менсена 148
а-Холестанилметиловый эфир 63
Холестерин, производные
синтез по Волю—Циглеру 67
— по Оппенауеру 198
Хромоны 205
реакция 261 ел.
Цианциклопентанон 277
Циклизация
по Дикману 116 ел.
по Комбу 160, 161
по Лмктэ—Шпенглеру 207
по Померанцу—Фричу 208, 209
по Пшорру 90. 217, 218
Циклобутанкарбоновая кислота, эфи-
ры 263
Цнклобутилбромид 262
Цнклогекса диены в реакции Берча
27 ел.
Циклооктены 97
Циклопропаны, синтез
по Виттигу 65
по Кижнеру 136
Цинхониновые кислоты, синтез
по Дебнеру 111, 112
по Пфитцингеру 214 ел.
перегруппировка 244, 245
реакция 264, 265
реакция 266, 267
реакция 268
Шиффовы основания, в реакциях
Валлаха 57, 58
Даффа 109, 110
Дебнера 112
Зонна—Мюллера 237, 238
Пиктэ—Шпенглера 207
Соммле 234
Фридлендера 257
ШлшДт-д реакция 166, 269 ел.
Д/гоббе конденсация 273 ел.
Шгорка реакция 189, 276 ел.
299
Предметный указатель
окисление персульфатами 281, 282
реакция 279, 280
Электролиз кислот 158, 159
Электрофильное замещение 38, 82,
99, 127, 202, 205, 220, 233
присоединение 33, 34
Элиминирование, в реакциях
Гофмана 93 ел.
Чугаева 267
реакция 283, 284
Эпикамфора 175, 176
ЭрлемжбДбрд—/Улгхля синтез азлак-
тонов 124, 285 ел.
Этилбензол 136, 148
Этиллропиловый эфир 62
Эфиры
синтез по Вильямсону 62, 63
сложные 16 ел.
г—/(лдгрлсд реакция 174
реакция 288, 289
Сер ре(3
Справочник по органическим реакциям.
М., Госхимиздат, 1962.
ЭКЮ с. 547
Редактор М. Я.
Техн. редактор В. Ф.
Подписано к печати 21/V1II 1962 г. Бумага 60X90i/ig =
9,5 б. л.—19 п. л.
Учетно-изд. л. 15,6. Тираж 25000 экз.
Цена 1 руб. Зак. 3297.
Типография Госхимиздата. Москва, 88. Угрешская
NAME REACTIONS IN ORGANIC
CHEMISTRY
2nd Edition revised and enlarged
Academic press
New York and London* 1961
Перевод с английского
В справочнике содержатся сведения о перегруппировках и
основных синтетических реакциях современной органической химии.
Для каждой реакции приводятся ее механизм и области
применения, а также характеристики исходных продуктов и условия
проведения процесса. Описанию каждой реакции или перегруппировки
предшествует краткая научно-биографическая справка о химиках,
открывших реакцию или разработавших условия ее проведения.
Справочник предназначен для студентов, аспирантов и
преподавателей вузов и техникумов и будет полезен также научным
работникам и инженерам, работающим в области органической
химии.