/

























































































































































































































































































































































































Автор: Манк М.
Теги: биологические и этологические процессы: механизмы, взаимодействие с окружающей средой общая биология биология зоология млекопитающие фауна развитие млекопитающих
ISBN: 5-03-001333-4
Год: 1990




Текст
иология развития
млекопитающих
Методы
ИЗДАТЕЛЬСТВО «МИР»
MAMMALIAN DEVELOPMENT
A PRACTICAL APPROACH
Edited by M. Monk
MRC Mammalian Development Unit,
4 Stephenson Way, London NW1 2HE, UK
IRL Press
Oxford Washington DC
Биология развития
млекопитающих
Методы
Под редакцией
М. МАНК
Перевод с английского
канд. биол. наук Д. Г. ПОЛТЕВОЙ
под редакцией
д-ра мед. наук В. С. БАРАНОВА
Москва «Мир» 1990
ББК 28.0
Б63
УДК 57.02
Биология развития млекопитающих. Методы: Пер. с
Б63 англ./Под ред. М. Манк. — М.: Мир, 1990. — 406 с., ил.
ISBN 5-03-001333-4
В методическом руководстве, созданном авторитетными авторами из Велико-
британии и США, освещена вся совокупность подходов, применяемых при изуче-
нии ранних эмбрионов млекопитающих (в том числе и человека). Приведены ме-
тодики культивирования эмбрионов, анализа хромосом, маркерных ферментов,
клонирования кДНК. получения трансгенных животных, оплодотворения ооцитов
и культивирования предимплантацнонных эмбрионов человека.
Книга относится к хорошо зарекомендовавшей себя серии «Методы», изда-
ваемой «ИРЛ Пресс».
Для эмбриологов, молекулярных биологов, студентов-биологов старших
курсов.
Б 1902000000—343
Ь 041(01)—90
101-90
ББК 28.0
Редакция литературы по биологии
ISBN 5-03-001333-4
ISBN 1-85221-030-3
(русск.)
(англ.)
© 1987 IRL Press Limited
© перевод на русский язык, Полта-
ва Д. Г., 1990’
ПРЕДИСЛОВИЕ РЕДАКТОРА ПЕРЕВОДА
Биология развития млекопитающих — одно из магистральных
направлений современной науки. Вобрав в себя концептуальные
и методические достижения общей эмбриологии и генетики, био-
логия развития млекопитающих все более активно участвует в
решении многих фундаментальных проблем, в том числе в ис-
следовании функций генома на ранних стадиях онтогенеза, ана-
лизе контролирующих механизмов нормального и патологичес-
кого развития и многих других. Значительны и ее практические
успехи. Так, уже решена проблема глубокого замораживания и
длительной криоконсервации гамет и ранних зародышей, раз-
работаны научные основы получения трансгенных животных с
заданными наследственными свойствами, быстро совершенству-
ются методы ранней дородовой диагностики наследственных бо-
лезней, значительно повысилась эффективность лечения различ-
ных форм бесплодия. Все это — итог стремительного прогресса
эмбриологии млекопитающих, ее резко возросших методических
возможностей. Именно эта отрасль биологии сегодня может слу-
жить прекрасным примером известного положения о том, что
прогресс любой науки это, прежде всего, прогресс ее методов.
Одно из первых руководств по экспериментальной эмбриологии
млекопитающих появилось еще в 1971 г. [«Methods in Mamma-
lian Embryology», J. Daniel (ed.)]. Расширенное и существенно
дополненное описание методов экспериментальной эмбриологии
лабораторных и сельскохозяйственных животных [«Methods in
Mammalian Reproduction», J. Daniel (ed.)] опубликовано в
1978 г. В 1986 г. по материалам цикла лекций и практических
занятий школы по экспериментальной эмбриологии млекопитаю-
щих, состоявшейся в Колд-Спринг Харборе (США) было издано
практическое руководство по экспериментальной эмбриологии
лабораторной мыши (В. Hogan, F. Costantini, Е. Lacy «Manipu-
lating the Mouse Embryo»). Основное внимание в нем было об-
ращено на методы молекулярной биологии, адаптированные для
решения экспериментально-эмбриологических задач. К сожале-
нию, ни одно из вышеприведенных методических руководств не
было переведено на русский язык и ввиду резко ограниченного
числа библиотечных экземпляров, они практически остались не-
6 Предисловие редактора перевода
известными советским читателям и специалистам. Между тем,
отечественных аналогов таких книг до сих пор нет. Отдельные
методы экспериментальной эмбриологии млекопитающих описа-
ны в соответствующих главах коллективной монографии «Мето-
ды биологии развития», М. Наука, 1974. С цитогенетическими
приемами исследования митотических и мейотических хромосом
зародышей млекопитающих можно ознакомиться в книге
А. П. Дыбана и В. С. Баранова «Цитогенетика развития млеко-
питающих», М. Наука, 1978. Многие из приведенных в ней мето-
дов, однако, существенно изменены и не отражают реальный
уровень методических возможностей современной эксперимен-
тальной эмбриологии.
Предлагаемый вниманию читателей сборник —это первое
фундаментальное методическое руководство по биологии разви-
тия млекопитающих на русском языке. Его редактор — д-р Мэ-
рилин Манк — сотрудник Отдела эмбриологии млекопитающих
Лондонского Университета, хорошо известна исследованиями по
инактивации Х-хромосомы и биохимии доимплантационных за-
родышей.
В каждой главе в очень сжатой и лаконичной форме содер-
жится описание принципа метода (или методов), перечислены
необходимые реактивы, инструменты, оборудование (с обяза-
тельным указанием фирм-изготовителей), последовательность
манипуляций, возможные ошибки и пути их устранения.
Настоящая книга является наиболее полным и современным
методическим руководством по экспериментальной и молекуляр-
ной эмбриологии млекопитающих. Уверен, что среди специали-
стов она будет так же популярна, как и руководство по молеку-
лярной биологии под редакцией Ф. Маниатиса и соавт. (Мир,
М. 1985). Изложенный в книге материал представляет интерес
не только для биологов, занимающихся развитием млекопитаю-
щих, ио и для медицинских генетиков, иммунологов, биохимиков
и биотехнологов, так или иначе связанных с изучением онтогене-
за. Ценную информацию почерпнут для себя специалисты по
эмбриологии человека, по генетике и разведению сельскохозяй-
ственных животных. Наконец, книга должна привлечь внимание
преподавателей и студентов кафедр эмбриологии университетов
и медицинских вузов и способствовать росту популярности этого
перспективного направления.
В. С. Баранов
ПРЕДИСЛОВИЕ
Молекулярная эмбриология млекопитающих сформировалась
как новое научное направление благодаря применению обшир-
ного набора методов микроскопической техники к изучению раз-
вития млекопитающих, главным образом — мыши. Разработка
методов оплодотворения in vitro и имплантации эмбрионов Сде-
лала возможным использование этих методик в эмбриологии че-
ловека с целью культивирования предимплантационных зароды-
шей, эмбриональной биопсии, определения потенций развиваю-
щегося плода и ранней диагностики (а возможно и лечения)
наследственных заболеваний. Современные представления о мор-
фологических изменениях, сопровождающих нормальный рост и
развитие постимплантационного плода, создают основу для-йри-
менения новой техники манипулирования специфическими кле-
точными линиями, участвующими в органогенезе. Появилась
возможность изучать регуляцию экспрессии специфических ге-
нов в процессе развития.
В этой книге описаны способы выделения и культивирования
эмбрионов, приводится арсенал клеточных и субклеточных мето-
дик, используемых для изучения развития млекопитающих —
оплодотворения in vitro, маркирования и движения индивиду-
альных клеток, переноса генов, трансплантации ядер, анализа
мембран, внутриклеточной организации, хромосом, специфичес-
ких последовательностей ДНК, белков и ферментов. Большинст-
во этих процедур разработано для мыши, но может быть при-
менено к млекопитающим вообще и к их клеткам.
Я в высшей степени благодарна всем авторам этой книги за
сотрудничество и поддержку, а также работникам издательства
IRL-Press за их терпение, квалифицированную помощь и советы,
щедро предоставляемые в течение всего времени работы над
книгой.
Мэрилин Манк
СПИСОК СОКРАЩЕНИЙ
АБК авидин-биотин-пероксидазиый комплекс
АД адреналин
АДБ агглютинин Dolichos biflorus
АФРТ аденинфосфорибозилтрансфераза
АЗП агглютинин зародышей пшеницы
АХ ацетилхолин
БУДР бромуридиндезоксирибозид
БСА бычий сывороточный альбумин
ВАБ веронал-ацетатный буфер
ВКМ внутренняя клеточная масса
Г6ФД глюкозо-6-фосфат—дегидрогеназа
ГСЖК гонадотропин сыворотки жеребой кобылы
ГФРТ гипоксантинфосфорибозилтрансфераза
ДАБ диаминобензидин
ДМСО диметилсульфоксид
ДНФ динитрофенол
ДСН додецилсульфат натрия
ДЭПК диэтилпирокарбонат
ИЭФ изоэлектрическое фокусирование
КонА конканавалин А
ЛГ лютеинизирующий гормон
МНС минимальная необходимая среда
НА норадреналин
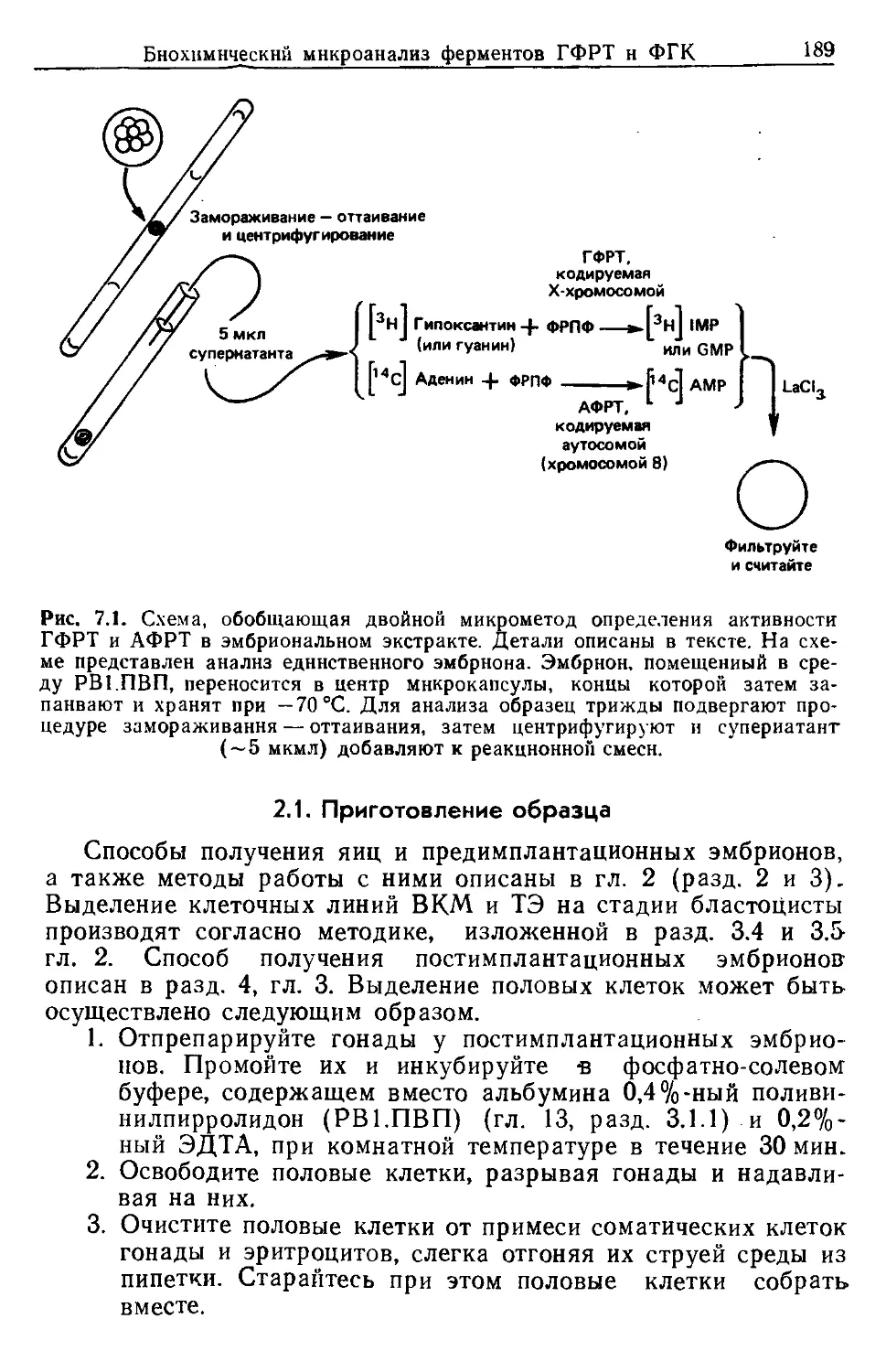
НАХР никотинацетилхолиновый рецептор
ОДАБ окисленный 3,3-аминобензидин
ПАП пероксидаза-антипероксидаза
ПВП поливинилпирролидон
ПГФТ перенос гамет в фаллопиевы трубы
ПЛП параформальдегид-лизин-периодат натрия
ПХ пероксидаза хрена
ПЭМ просвечивающая электронная микроскопия
РИА радиоиммунологический анализ
СК синаптонемпый комплекс
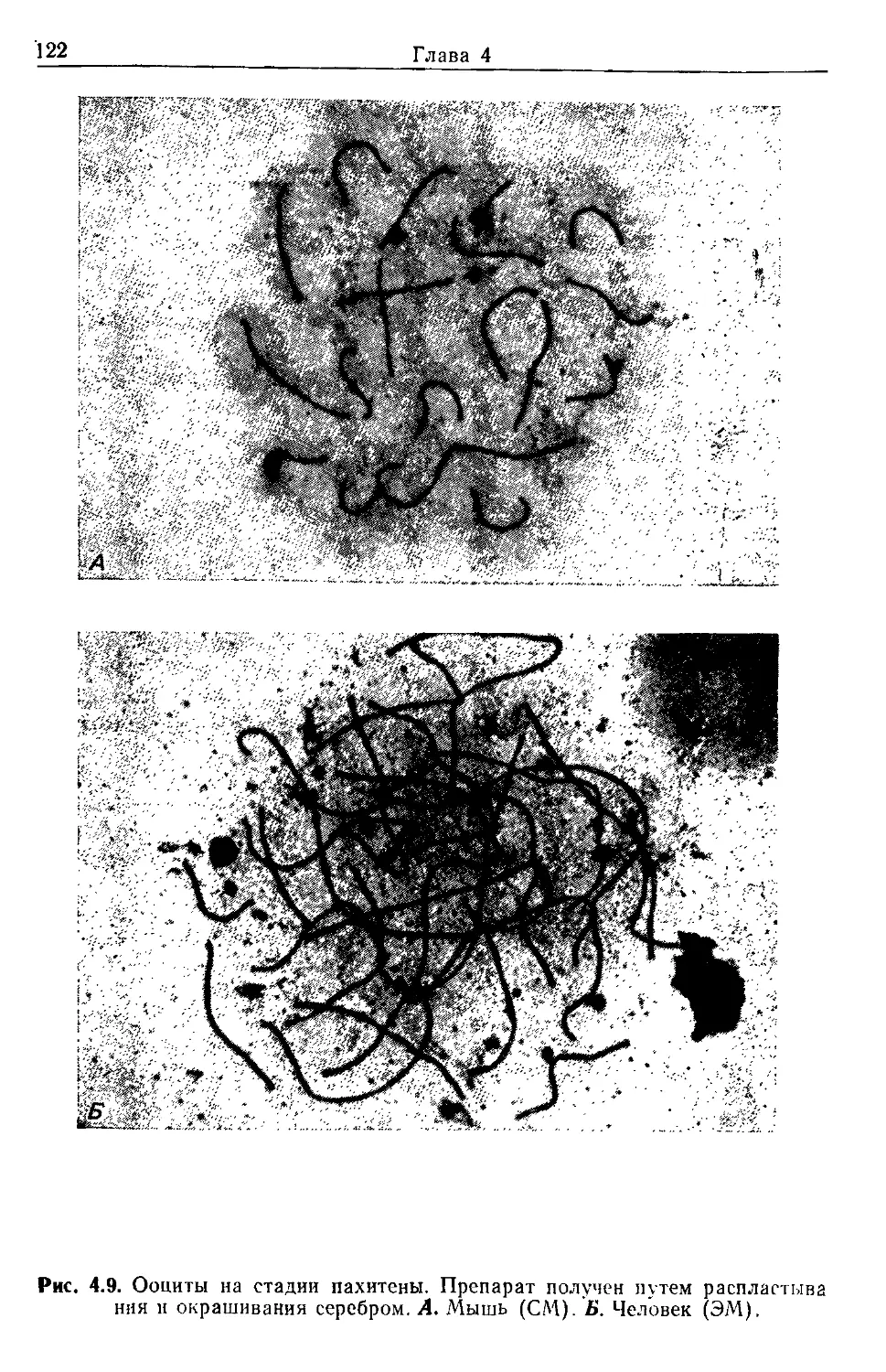
СМ световая микроскопия
СЭМ сканирующая электронная микроскопия
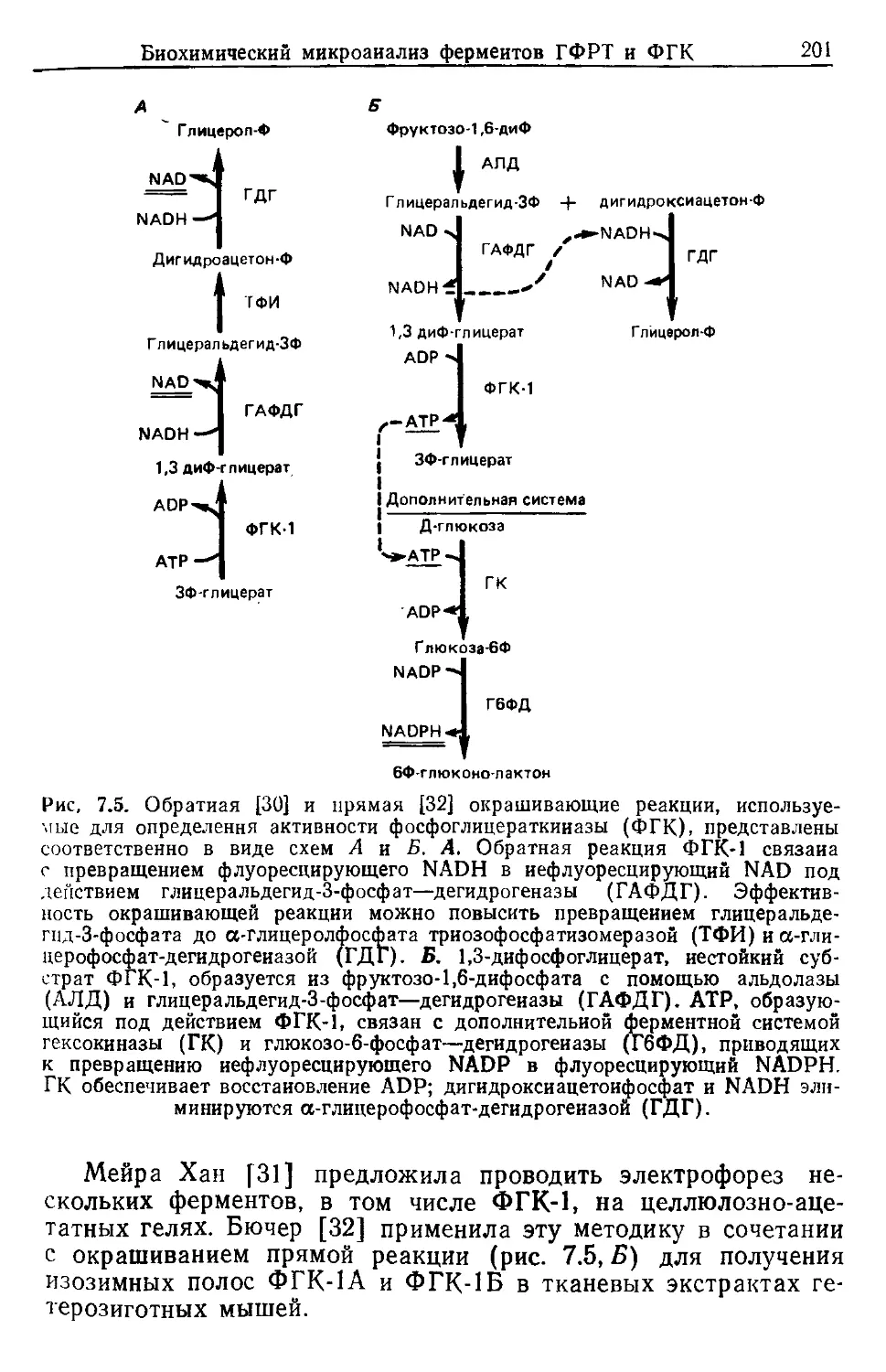
ТМБ тетраметилбензидин
Список сокращений
9
ТРИТЦ тех ТХУ тэ ФГА ФГЛ ФГК ФИТЦ ФМСФ ФРПФ цхд чХГ ЭДТА эк эм этс ЯОР CPMV EBS GABA HBSS HEPES изотиоцианат тетраметилродамииа тонкослойная хроматография трихлоруксусная кислота трофэктодерма фитогемагглютинин гидрохлорид фенилгидразина фосфоглицераткиназа изотиоцианат флуоресцеина фенилметилсульфонилфлуорид пирофосфат фосфорибозила цитохалазин Д человеческий гонадотропин этиленди амиитетраацетат эмбриональная карцинома электронная микроскопия эмбриональная телячья сыворотка область ядрышкового организатора вирус мозаики вигны сбалансированный солевой раствор Эрла у-аминобутириевая кислота сбалансированный солевой раствор Хэнкса Х[-2-гидроксиэтилпиперазин-М'-2-этансульфоновая кис- лота (от англ. N-2-hydroxyethylpiperazine-N'-2-ethane- sulfonic acid)
MED PBS SSC среда Игла, модифицированная Дульбекко солевой раствор, забуференный фосфатом физиологический раствор с цитратом
ВВЕДЕНИЕ
Развитие млекопитающих было предметом наблюдений и ги-
потез еще со времен Аристотеля, однако первые эксперименты в
этой области датируются концом XIX века. В 1890 г. Уолтеру
Хипу удалось перенести два дробящихся яйца кролика ангор-
ской породы в яйцевод бельгийского «зайца» (порода кроликов,
отличающихся маленькими размерами тела). Его интересовал
вопрос, мать или плод определяют длительность беременности и
влияет ли чужая среда на процесс развития зародыша. Оба
крольчонка родились живыми. Хипу понадобилось еще семь лет
упорного труда, чтобы осуществить следующий удачный опыт.
Его терпение и настойчивость должны служить примером для
современных исследователей.
Несмотря на столь давний интерес к этой области науки, мы
гораздо больше знаем о развитии морских ежей, нематод и дро-
зофил, даже рыб, лягушек и птиц, чем о развитии млекопитаю-
щих. В связи с недоступностью эмбрионов, находящихся в матке,
для прижизненных наблюдений, развитие млекопитающих как
научная дисциплина возникло скорее из биологин размножения
и генетики, чем из классической эмбриологии. Возможно именно
поэтому термин «эмбрион» в биологии развития млекопитающих
несет оттенок некой двусмысленности. Действительно, двуклеточ-
ная стадия или бластоциста по сути не являются эмбрионами,
эмбрион формируется лишь при гаструляции и затем развивает-
ся в плод. Наши успехи в понимании механизмов развития мле-
копитающих ограничены, главным образом, проэмбриональными
стадиями от оплодотворения до имплантации, которые сравни-
тельно легко наблюдать в культуре. Среди лучше изученных си-
стем следует выделить внезародышевые оболочки, а не сам за-
родыш.
Наиболее ценный вклад в изучение развития млекопитающих
внесла молекулярная биология. Методы рекомбинантных ДНК
обусловили настоящий переворот в этой области биологии раз-
вития. Его свидетельство — появление данной книги. Более ран-
ние руководства (К- A. Rafferty «Methods in Experimental Em-
bryology of the Mouse», 1970; «Methods of Mammalian Reproduc-
tion», 1978; «Methods of Mammalian Embryology», 1971, ed. by
Введение
11
J. С. Daniel) подробно описывают многие экспериментальные
методы, которыми пользовались в эпоху «До Клонирования», они
сохраняют ценность и сейчас. Единственное современное лабо-
раторное руководство, вышедшее в свет уже «После Клониро-
вания»— это превосходная книга Б. Хоган, Ф. Костантини и
Е. Лейси (В. Hogan, F. Costantini, Е. Lacy «Manipulating the
Mouse Embryo», 1986). Ее содержание до некоторой степени пе-
рекрывается с содержанием нашего руководства, однако в зна-
чительной мере эти книги являются взаимодополняющими.
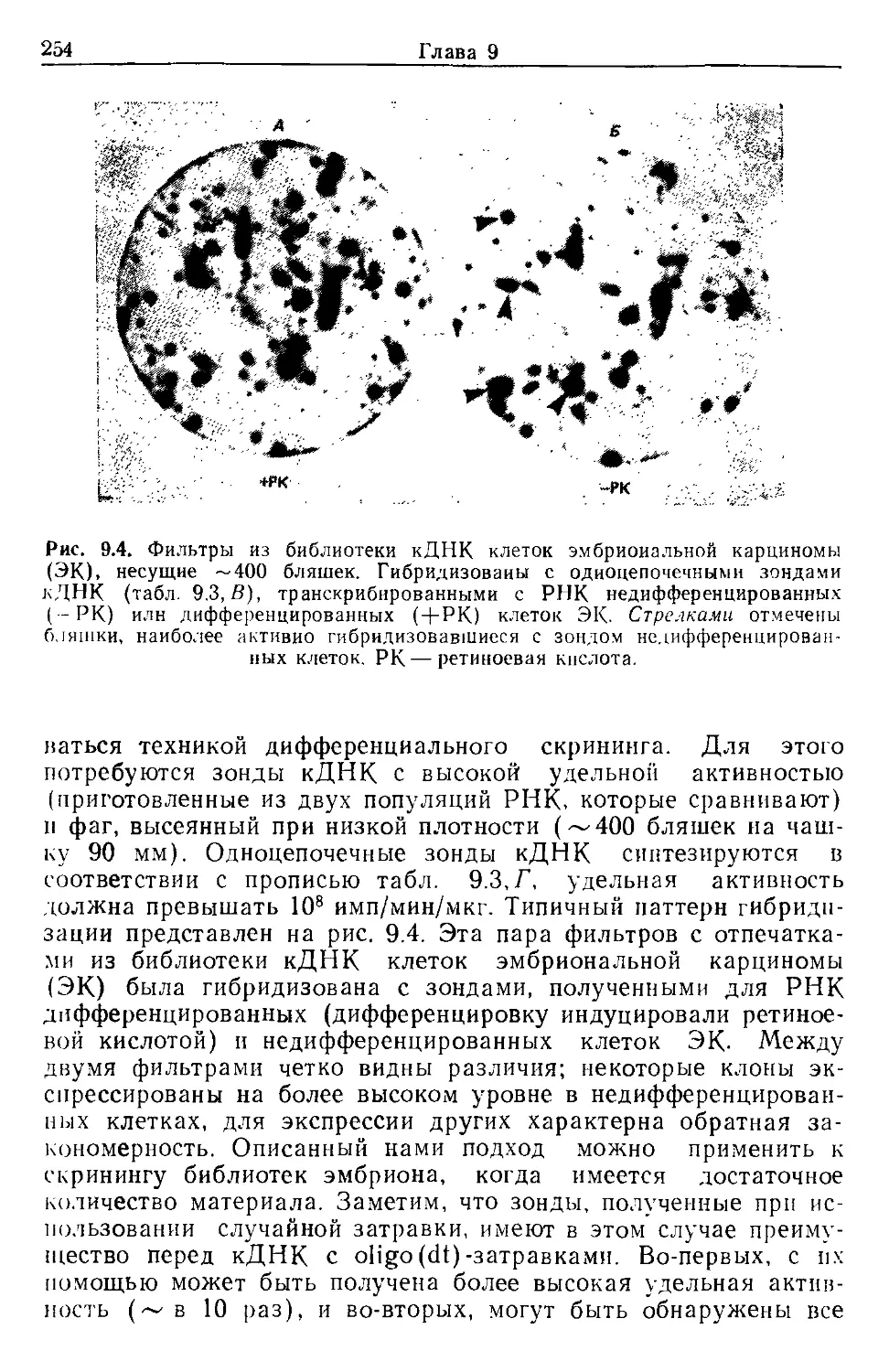
В настоящем сборнике представлены современные методы
получения экспериментального материала и анализа результа-
тов; описываются приемы работы с зародышами мышей предим-
плантационных и постимплантационных стадий развития, спосо-
бы получения, оплодотворения и культивирования человеческих
яйцеклеток. Отдельные разделы книги посвящены методам био-
химического и цитогенетического анализа эмбрионов, созданию
библиотек кДНК и гибридизации нуклеиновых кислот in situ.
Эти, а также другие, описанные в книге методические приемы,
позволяют анализировать молекулярные основы развития мле-
копитающих.
В настоящее время накоплено много сведений о репродукции
мыши, крысы, кролика, морской свинки, овцы и человека, но ге-
нетика мыши занимает особое положение. И именно это направ-
ление наиболее успешно развивается в Англии. Начиная с 50-х
годов здесь сосредоточены крупные силы. Прежде всего следует
упомянуть генетика Рональда Фишера (Кембридж). Хотя он
знаменит своими работами по статистике, велик его вклад и в
изучение генетики мыши. В Эдинбурге это направление возгла-
вил ученик Фишера Дуглас Фолконер, в Харуэлле — Мэри Лай-
он. С 1938 г. по приглашению Джона Холдейна в Лондонском
университетском колледже генетикой мыши стал заниматься
Грюнеберг. Не удивительно, что эти исследовательские центры,
хорошо оснащенные и высокопродуктивные,— обеспечили рас-
цвет указанного научного направления в Англии.
Мышь доминировала не только в генетике млекопитающих:
в 1960-е годы были разработаны системы культивирования in
vitro зародышей предимплантационных стадий этого вида. Но
если руководство Хоган посвящено исключительно мыши, наша
книга содержит материал и о человеке.
Нет сомнения в том, что проблема здоровья людей имеет вы-
сочайшую социальную значимость. Из истории науки известно,
что именно такие проблемы при появлении новых прогрессивных
методов исследования начинают бурно развиваться. Можно по-
лагать, что прогресс в технике оплодотворения in vitro позволит
нам добиться больших успехов в борьбе с бесплодием и хромо-
сомными нарушениями. Достижения биологии развития дают
12 Введение
возможность применить методы генной инженерии и в селекции
сельскохозяйственных животных.
И еще одна немаловажная проблема. Независимо от того, ка-
кую проблему развития млекопитающих решают ученые, они не
могут не восхищаться красотой своего объекта. Поразительная
симметрия 8-клеточных зародышей перед компактизацией бла-
стомеров; растущая мышиная бластоциста; беременная матка с
искусно имплантированными эмбрионами; маленький плод, в ко-
тором сквозь тончайшую прозрачную амниотическую оболочку
можно различить бьющееся сердце и массивную печень. Как все
это совершенно и загадочно! Я уверена, что наша книга помо-
жет современному поколению научных работников ответить на
многие вопросы. Конечно, еще больше вопросов при этом воз-
никнет,— но таков уж путь науки.
Анна МакЛарен
ГЛАВА 1
РАЗВЕДЕНИЕ МЫШЕИ
Колин Хедерингтон'
1. Введение
Мышь — наиболее распространенное лабораторное млекопи-
тающее. Благодаря малым размерам и высокой плодовитости
ее содержание сравнительно дешево. За последние 50 лет в
лабораториях выведены сотнн различных линий: инбредных,
близкородственных, рекомбинантных, аутбредных и мутантных.
Генетические и фенотипические характеристики многих изннх
хорошо изучены [1, 2].
2. Разведение
2.1. Источник мышей и общая оценка их здоровья
Для успешной научной работы требуются здоровые, генети-
чески определенные животные. В зависимости от обстоятельств
исследователь может приобретать их в питомниках или выво-
дить собственными руками. В том случае, когда потребность в
мышах периодическая и в эксперименте необходимы крупные
партии животных одного возраста, целесообразнее и дешевле
покупать их. Если же мыши используются регулярно, выгоднее
их разводить. Важно, чтобы с самого начала п постоянно жи-
вотные были здоровы, для этого следует строго контролиро-
вать условия их содержания.
Если при разведении мышей не принимаются специальные
меры, направленные против распространения инфекций, живот-
ных называют «обычными». Мыши, выведенные в условиях,
препятствующих инфекциям, известны как «барьерные». В по-
следнем случае в качестве синонима используется также тер-
мин «специфически патогенно-свободные» (СПС) животные.
В характеристике здоровья этих мышей указываются отсутст-
вующие патогены. Среди них, как правило, экто- и эндопара-1
зиты, такие, как клещи, кишечные черви, простейшие, некото-
рые бактерии, микоплазмы н вирусы. Гнотобиотические жнвот- *
! С. М. Hetherington. Biological Services, National Institute for Medical
Research. The Ridgeway, Mill Hill, London NW7 1AA, UK.
14
Глава 1
ные разводятся в специальных условиях, их микробиологичес-
кая флора известна. Аксенические мыши свободны от каких бы
то ни было организмов. Выбранная форма содержания живот-
ных зависит от характера имеющегося оборудования и природы
экспериментов. Если планируется длительное использование
одних и тех же мышей или предполагается, что они будут ос-
нователями колонии, барьерная система в той или иной фор-
ме — обязательна. Если никаких мер против инфекций не при-
менять, здоровье животных будет неизбежно ухудшаться, что
приведет к снижению их плодовитости или гибели. Минималь-
ная барьерная система предполагает, что люди, входящие в
виварий, снимают свою лабораторную одежду и обувь, тща-
тельно моют руки, прежде чем взять мышь, пользуются хирур-
гическими перчатками. Более строгий барьер включает требо-
вания принять душ и полностью сменить одежду.
Каковы бы ни были меры предосторожности, важно, чтобы
сотрудники, непосредственно не занятые уходом за животными,
не входили к ним вообще. Те, в чьи обязанности входит разве-
дение животных, по возможности не должны контактировать с
потенциально инфицированными мышами. Чем больше людей
имеет доступ к размножающейся колонии, тем выше риск вне-
сения инфекции.
2.2. Клетки
Наиболее удобны клетки двух размеров: с площадью дна
200 см2 или 1200 см2 и высотой 12 см (внутренние размеры).
Первые предполагают размещение мышей парами или по три
особи, в клетках второго типа может находиться до 25 мышей,
в зависимости от их размера. Для изготовления клеток приме-
няют прозрачный поликарбонат, матовый полипропилен или ме-
талл. Основания могут быть плотными или решетчатыми. Ко-
нечно, трудно судить о том, какого типа клетки предпочитают
мыши, исследователь руководствуется в их выборе собственным
вкусом. Как правило, используются пропиленовые клетки с не-
крашеными стальными крышками и плотными основаниями, по
при необходимости регулярного автоклавирования удобнее
клетки из металла. Для содержания диких мышей разработаны
специальные клетки. Их конструкция позволяет не брать мы-
шей руками при чистке. Клетки размещают на укрепленных
полках (чтобы было удобнее мыть пол) или специальных ме-
таллических стеллажах с колесиками.
2.3. Подстилка
В качестве подстилки обычно применяют древесные струж-
ки или опилки. Дерево не должно содержать инсектицидов
или гербицидов. Древесная пыль может вызывать аллергию у
Разведение мышей
15
обслуживающего персонала, поэтому рекомендуется пользо-
ваться свободными от пыли опилками и при работе с сухой
подстилкой надевать маски. Для подстилки подходят и другие
адсорбирующие материалы, важно только, чтобы в клетках с
размножающимися мышами они не способствовали обезвожи-
ванию приплода.
2.4. Питание
Важнейший фактор успешного разведения и содержания
здоровых и плодовитых мышей — полноценное питание. Разра-
ботаны разнообразные рационы. Одни из них используются для
размножающихся животных, другие — для всей линии. Корм,
приготовленный в виде гранул разной формы и размера или
порошка, лучше усваивается животными и занимает меньше
места в кормушке. Пища, предназначенная для автоклавиро-
вания, содержит дополнительные питательные вещества, компен-
сирующие их неизбежную деградацию при автоклавировании.
Некоторые типы корма снабжены специальными оболочками,
предупреждающими слипание гранул во время автоклавирова-
ния. При этом физические изменения гранул зависят как от их
свойств, так и от режима работы автоклава. Следует убедиться
в том, что после стерилизации пища осталась приемлемой в
отношении своих питательных и физических свойств. Чтобы
I ранулы при автоклавировании не склеивались, рекомендуется
их рассыпать на подносах. Можно приобретать корма, стерили-
зованные облучением; в этом случае следует убедиться в том,
что упаковка, обеспечивающая стерильность, цела.
2.5. Вода
Обычно используются автоматические поилки. Каким бы ни
было их устройство, следует убедиться в том, что нет утечки.
Бесконтрольно вытекающая из бутылок вода может привести
к гибели мышей, особенно молодняка.
Водопроводную воду перед употреблением необходимо про-
верить; это особенно важно при разведении барьерных живот-
ных. Для улучшения микробиологического состояния воды при-
меняют подкисление, хлорирование, ультрафиолетовое облуче-
ние или фильтрацию. Вода для гнотобиотпческих изоляторов
должна быть стерильной.
2.6. Среда обитания
Для удаления запахов и аллергенов помещение нужно вен-
тилировать. Температура должна поддерживаться в пределах
21 ±2 °C, относительная влажность 55±10%. Требуется полное
16
Глава 1
Рис. 1.1. Как правильно держать
мышь.
кондиционирование воздуха.
Важно, чтобы ofj в помещении
циркулировал, однако живот-
ные не должны находиться на
сквозняке. Число проветрива-
нии в течение часа зависит от
плотности стада и эффектив-
ности обмена воздуха в поме-
щении. Для комнаты, в кото-
рой содержатся 150—175 мы-
шей/м2 рекомендуется шест-
надцать проветриваний в час
(при условии регулярной чист-
ки клеток). Следует заметить,
что микроклимат внутри клет-
ки может значительно отли-
чаться от микроклимата по-
мещения, в котором она нахо-
дится, более высокой темпера-
турой, влажностью и кон-
центрацией аммония.
Чтобы получать эмбрионы определенного возраста (т. е. на-
ходящиеся на определенной стадии развития), необходим по-
стоянный световой режим. Поэтому в помещении, где содер-
жатся мыши, нет окон. Обычно пользуются световым циклом
12 ч темноты/12 ч света (см. также разд. 5.2).
2.7. Дезинфекция помещений
В ходе длительного эксперимента здоровье мышей неиз-
бежно ухудшается. При работе с неразмножающимися животны-
ми его можно сохранить, если здоровых особей перемещать в
чистую дезинфицированную комнату, так, чтобы к концу экс-
периментов «грязная» комната постепенно освободилась. Если
используются чистая и «грязная» комнаты, между ними должен
существовать барьер. Как только помещение освободится, оно
должно быть тщательно вычищено и дезинфицировано. Способ
дезинфекции зависит от возможности изолировать эту комна-
ту и ее вентиляционную систему от остальных помещений.
В случае эффективной изоляции наилучшим средством служит
формальдегид. Последний может быть получен несколькими
способами. Один из них состоит в нагревании раствора форма-
лина при помощи электрического нагревательного прибора или
смешивания двух частей формалина с одной частью перманга-
ната калия; в результате формальдегид будет улетучиваться.
Эффективность этой процедуры увеличивается при относитель-
ной влажности воздуха 75% или более. При выделении фор-
Разведение мышей
17
мальдегида комната должна быть изолирована на 24 часа. Дез-
инфекцию помещения следует производить с предосторожностя-
ми, рекомендуется надевать респиратор. Количество формалина
берут из расчета 10 мл на каждый кубометр помещения. Если
формальдегид получают нагреванием, следует пользоваться
10%-ным раствором формалина, чтобы одновременно повыша-
лась и влажность.
В том случае, когда комната не может быть изолирована,
в качестве дезинфицирующего средства эффективен препарат
Тегодор (Tegodor), в состав которого входит формальдегид,
глутаральдегид и еще некоторые вещества; этот препарат рас-
пыляют в помещении (рекомендуется делать это в респирато
ре).
3. Обращение с мышью
3.1. Подход
1. Возьмите мышь за основание хвоста большим и указа-
тельным пальцем и перенесите ее на решетку крышки
(рис. 1.1, Л) или на поверхность, в которую она может
вцепиться (например, рукав лабораторного халата). Есте-
ственная реакция животного—вырваться от удерживаю-
щего, поэтому, продолжая осторожно оттягивать мышь
назад за хвост, следует прижать ее к решетке большим и
согнутым указательным пальцами другой руки.
2. Приноровившись, плотно захватите большим и указатель-
ным пальцами кожу шеи за ушками.
3. Удерживайте хвост безымянным пальцем, мизинцем и
ладонью (рис. 1.1, Б). Если держать мышь только за за-
гривок или недостаточно плотно захватить ее кожу за
ушами, она может укусить. Чтобы захватить или удер-
жать мышь, часто пользуются большим анатомическим
пинцетом илн карнцангом.
Такие процедуры, как прищипывание ушей, подкожные и
внутрибрюшинные инъекции, пальпирование, делают, держа
мышь в положении, показанном на рис. 1.1.
Трудно иметь дело с дикими мышами, поскольку они пры-
гают. В этом случае, прежде чем открыть клетку, рекоменду-
ется поместить ее в глубокий бокс. При перемещении мышей из
клетки в клетку лучше заманить их в банку, чем брать руками.
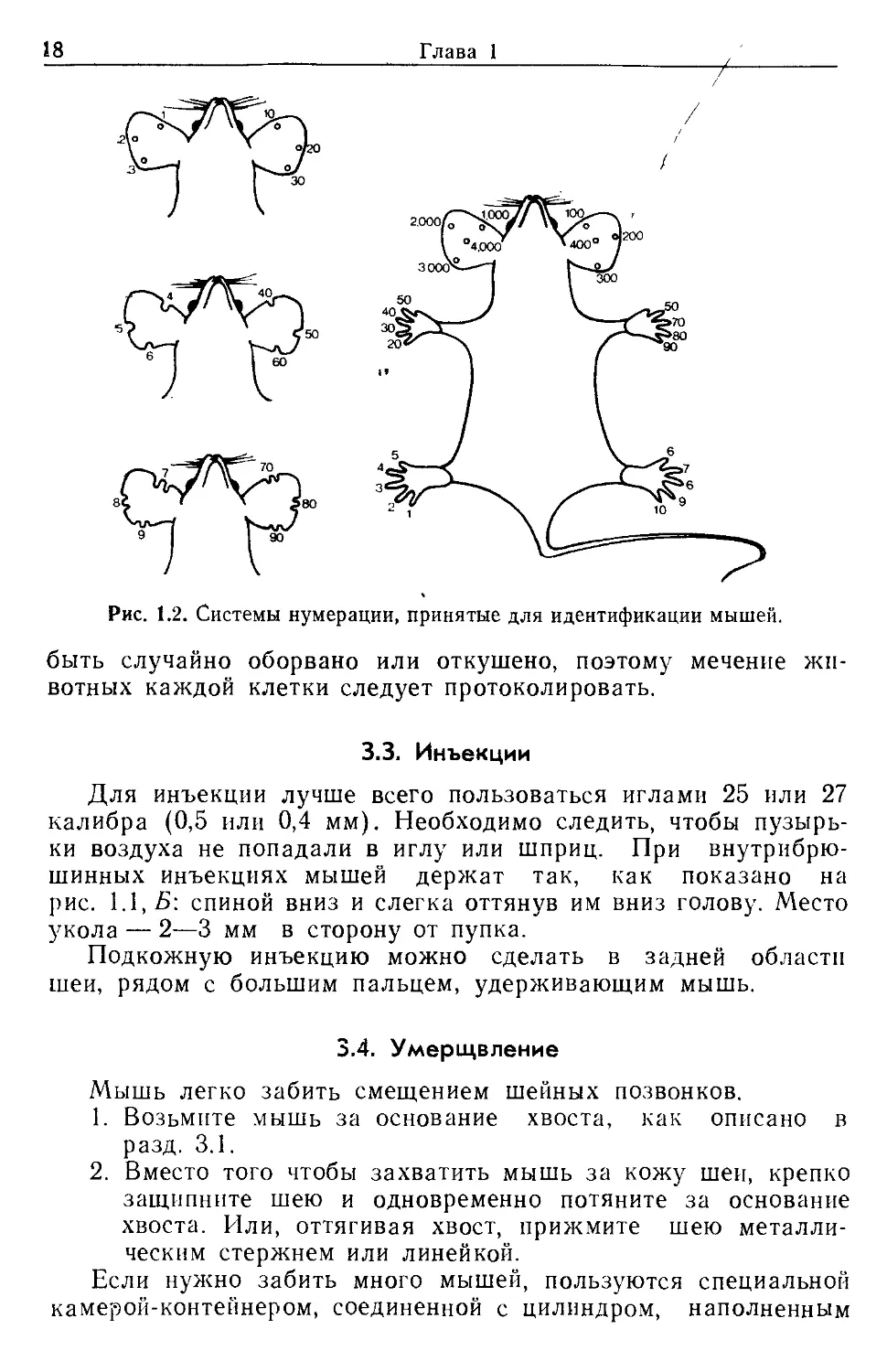
3.2. Идентификация
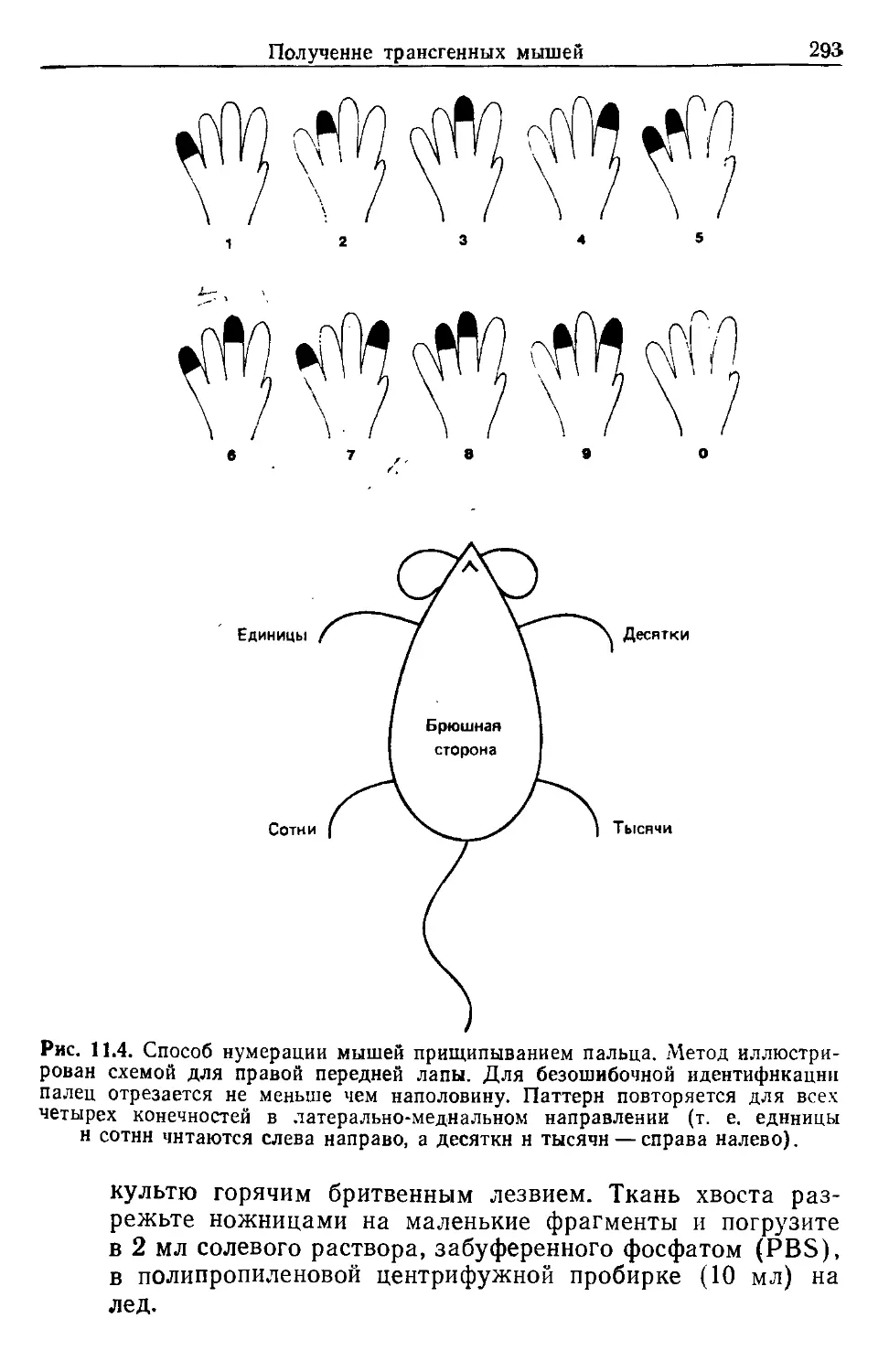
Существует два способа мечения мышей: прищипывание
ушей и прищипывание пальцев. Комбинируя их, можно полу-
чить до 9999 меток (рис. 1.2). Важно помнить, что ухо может
2-171
18
Глава 1
Рис. 1.2. Системы нумерации, принятые для идентификации мышей.
быть случайно оборвано или откушено, поэтому мечение жи-
вотных каждой клетки следует протоколировать.
3.3. Инъекции
Для инъекции лучше всего пользоваться иглами 25 или 27
калибра (0,5 или 0,4 мм). Необходимо следить, чтобы пузырь-
ки воздуха не попадали в иглу или шприц. При внутрибрю-
шинных инъекциях мышей держат так, как показано на
рис. 1.1,5: спиной вниз и слегка оттянув им вниз голову. Место
укола — 2—3 мм в сторону от пупка.
Подкожную инъекцию можно сделать в задней области
шеи, рядом с большим пальцем, удерживающим мышь.
3.4. Умерщвление
Мышь легко забить смещением шейных позвонков.
1. Возьмите мышь за основание хвоста, как описано в
разд. 3.1.
2. Вместо того чтобы захватить мышь за кожу шеи, крепко
защипните шею и одновременно потяните за основание
хвоста. Или, оттягивая хвост, прижмите шею металли-
ческим стержнем или линейкой.
Если нужно забить много мышей, пользуются специальной
камерой-контейнером, соединенной с цилиндром, наполненным
Разведение мышей
19
двуокисью углерода. Животных помещают в контейнер и заме-
няют воздух двуокисью углерода. Следует заметить, что взрос-
лые мыши погибают быстрее, чем молодняк в возрасте 1—8 су-
ток (см. также разд. 8).
4. Разведение
Мыши начинают размножаться в возрасте 6—8 недель; дли-
тельность репродуктивного периода у разных линий варьирует
и составляет приблизительно 200 дней. Беременность длится
20 дней, роды часто сопровождаются послеродовым эструсом.
Лактация, совпадающая с беременностью, может затормозить
имплантацию (т. е. эмбрионы остаются жизнеспособными, но
блокируются на стадии бластоцисты). Размножающихся мышей
обычно содержат парами или по три (один самец и две самки)
или в виде гаремов (один самец и больше двух самок). Це реко-
мендуется сажать в клетку более одного самца — это приво-
дит к дракам. Оптимальна одна пара на клетку. Другие про-
порции самцов и самок дают больший приплод на клетку, но
при этом увеличивается смертность молодняка. Мыши переста-
ют питаться материнским молоком примерно в трехнедельном
возрасте и должны быть удалены из клетки перед новыми ро-
дами. Чтобы предотвратить драки между молодыми самцами,
по прекращении вскармливания молоком их держат в одной
клетке. Взрослых самцов из разных клеток не смешивают.
Размножение следует протоколировать и непродуктивных
самцов и самок, приносящих потомство реже чем через 40дней,
выбраковывать.
Выбор методики разведения зависит от требуемого количе-
ства животных и от их принадлежности к инбредной или аут-
бредной линии. Линия инбредных мышей должна поддержи-
ваться спариванием сибсов (братХсестра), в то время как при
размножении мышей аутбредной линии инбридинг следует све-
сти к минимуму [1, 4].
Диких мышей можно разводить в обычных клетках, но при
этом нельзя не забывать об их способности прыгать.
5. Эксперименты
5.1. Самцы-производители
Для оценки самцов-производителей ставят контрольное
спаривание, кроме того, регистрируют все последующие спари-
вания, отмечая не только сам факт спаривания, но и. его ре-
зультаты.
2*
20
Глава 1
5.2. Получение мышей
с известным сроком беременности /
Эстральный цикл мышей обычно длится 4 дня, самки спо-
собны к оплодотворению только на стадии течки (эструса).
Если оплодотворенные самки требуются ежедневно, доста-
точно иметь в распоряжении серию клеток с самцом и тремя
самками в каждой, исходя из предположения, что одна самка
из четырех в данный день окажется в эструсе. Соотношение
один самец к трем самкам на клетку обычно дает наилучшие
результаты для большинства линий.
Число оплодотворенных самок к определенному дню можно
увеличить, воспользовавшись эффектом Уиттена [5]. Если груп-
пу самок поместить с самцом, их эстральный цикл синхронизи-
руется, и большинство животных будут оплодотворены на тре-
тью ночь. Для этого самец и самки содержатся в одной клетке,
разделенной перегородкой, в течение двух дней, а на третий день
им дают возможность спариваться [6J. Существует и другая
возможность иметь одновременно много оплодотворенных са-
мок. Для этого необходимо отобрать для спаривания самок,
находящихся в состоянии эструса. У таких животных влагали-
ще раскрыто, ткань его розовая, увлажненная, заметно исчер-
ченная.
Факт спаривания у мышей констатируют по наличию во
влагалище слизистого сгустка, образующегося в результате ко-
агуляции эякулята, т. е. вагинальной, или копулятивной, проб-
ки. Обычно пробка хорошо видна, но иногда для ее обнаруже-
ния пользуются зондом. Пробка может сохраняться во влага-
лище до 18 часов, однако чем скорее проверить животное, тем
меньше вероятности ее потери. Спаривание мышей обычно про-
исходит в середине темного времени суток. Время спаривания
и возраст эмбрионов в разных лабораториях определяют по-
разному: день обнаружения вагинальной пробки может быть
отмечен как «день 0», «день 1/2» и «день 1 беременности».
Время спаривания может быть изменено и приурочено к ра-
бочему дню затемнением вивария в дневное время и искусст-
венным освещением его ночью. Такое обращение цикла дает
возможность иметь нужные стадии эмбрионального развития в
удобное время.
Если животные получены из лаборатории с иным циклом
смены темноты и света или предыдущий цикл неизвестен, им
понадобится несколько дней адаптации к новым условиям.
5.3. Ложная беременность и вазэктомия
Псевдобеременных животных получают так же, как описа-
но в разд. 5.2, но в результате спаривания с вазэктомирован-
Разведение мышей
21
ным самцом. Наиболее удобный для операции возраст самцов —
около 6 недель, пока они не начали жиреть. Выбранные самцы
должны относиться к линии с хорошими репродуктивными спо-
собностями. Вазэктомия заключается в вырезании короткого
фрагмента семявыносящего протока. Операцию делают под
анестезией посредством единственного поперечного брюшного
разреза. Семявыносящий проток можно найти, не извлекая
семенников, — для этого достаточно оттянуть жировую подуш-
ку. Накладывать швы не обязательно. Края разреза соединя-
ют с помощью зажимов, которые нужно удалить через 12 дней.
Зашивать стенку тела нет необходимости.
В стерильности вазэктомированных самцов нужно убедиться
посредством контрольного спаривания.
5.4. Суперовуляция
В тех случаях, когда для работы требуется значительное
количество предимплантационных эмбрионов, овуляцию стиму-
лируют гонадотропинами ГСЖК (гонадотропин сыворотки же-
ребой кобылы) и чХГ (человеческий хориогонический гонадо-
тропин), вводя их самкам перед спариванием. Успешная индук-
ция суперовуляции зависит от ряда факторов — возраста, веса,
линии животных, времени инъекции гонадотропинов.
Наиболее подходящий возраст для суперовуляции 3—5 не-
дель, причем оптимум для данного генотипа находится в пре-
делах 4—6 дней. У некоторых линий лучший показатель воз-
раста — вес, а если экспериментатор имеет дело с коммерчес-
кими мышами, то вес служит единственным критерием воз-
раста.
Сроки введения гонадотропинов взаимно связаны и зависят
от светового цикла вивария. Они оказывают влияние на число
овулировавших яиц и синхронность их развития. Оптимальный
интервал между введением гормонов составляет 40—46 ч. Ову-
ляция обычно происходит через 10—13 ч после инъекции чХГ.
Для того чтобы добиться наилучшей синхронизации, нужно,
чтобы чХГ был введен до высвобождения эндогенного лютеини-
зирующего гормона (ЛГ). Считают, что эндогенный Л Г выделя-
ется через 15—20 ч от середины второго темного периода (по-
луночи), следующего за введением ГСЖК. Таким образом, рас-
писание инъекций для животных, содержащихся в помещении
со световым периодом от 8.00 до 20.00 ч, должно быть следую-
щим: ГСЖК вводят между 15.00 и 16.00, а чХГ спустя 46 ч —
между 13.00 и 14.00, т. е. за 3—4 ч до выделения эндогенного
Л Г.
Обычная доза ГСЖК и чХГ составляет 2—5 ME, но разные
линии и гибриды могут реагировать на нее по-разному.
22
Глава 1
После инъекции самку подсаживают в клетку к безусловно
плодовитому самцу и на следующее утро проверяют наличие
копулятивной пробки.
5,5. Анестетики
Несмотря на то, что мышей приходится анестезировать ча-
ще, чем других лабораторных животных, это процедура неред-
ко для них кончается смертью. Причиной гибели мышей может
быть не только сверхвысокая доза анестетика, но и гипотермия.
Установлено также, что реакция на анестетики сильно зависит
от линии, возраста, пола, веса и упитанности мыши.
Наиболее часто в качестве анестетиков применяют пенто-
барбитон, трибромоэтанол (Avertin) и фентанилфлюанизон
(Hypnorm, Grown Chemical Со. Limited) с мидазоламом (Hyp-
novel, Roche) [7, 8].
Пентобарбитон можно приготовить разведением порошка в
теплом нормальном солевом растворе в концентрации 5 мг/мл
или развести имеющийся в продаже раствор (60 мг/мл) 1 : 10.
Поскольку эффективная доза определяется многими факторами,
ее следует предварительно подобрать на контрольной группе
мышей той же линии, того же возраста, веса и пола. Для мы-
ши весом 25 г необходимая доза составляет 0,7—1,0 мг.
Трибромоэтанол готовят следующим-образом.
1. Для получения 100%-ного маточного раствора смешайте
10 г трибромоэтилового спирта с 10 мл третичного амило-
вого спирта.
2. Чтобы приготовить рабочий раствор, маточный раствор
следует развести стерильной водой или физиологическим
раствором до 2,5%.
3. Маточный и рабочие растворы храните в темноте при
4 °C.
Свойства препарата и чувствительность к нему подопытных
животных могут значительно варьировать, поэтому и в случае
трибромэтанола требуется контрольная группа животных. Нуж-
ная доза для мыши весом 25 г составляет около 3,0 мг. Есть
данные, свидетельствующие о том, что трибромоэтанол раздра-
жает внутренние органы мыши, повреждает кишечник и может
привести к гибели; сила этого побочного эффекта зависит от
линии мышей.
Фентанилфлуанизон в сочетании с мидазоламом обеспечива-
ет наилучшую хирургическую анестезию. Имеющиеся в прода-
же препараты следует развести в 2 раза стерильной водой (для
инъекций) и затем смешать их равные объемы. Полученная
смесь содержит 0,5 мг/мл мидазолама, 2,5 мг/мл флуанизона и
0,079 мг/мл фентанил-цитрата. Смесь стабильна при комнат^
Разведение мышей
23
ной температуре по крайней мере в течение 8 недель. Для мы-
ши весом 25 г требуется внутрибрюшинная инъекция 0,1—
0,2 мл смеси.
Эфир раздражает дыхательный тракт мыши и вызывает из-
быточную секрецию слизи. Нужный уровень анестезии эфиром
трудно контролировать, кроме того, это вещество взрывоопасно.
Поэтому эфир для анестезии не рекомендуется. Хлороформом
не следует пользоваться ни при каких обстоятельствах. Он вре-
ден не только для печени, следовые концентрации хлороформа
могут серьезно сказаться на репродуктивной способности сам-
цов.
Важно помнить, что анестезированных мышей нужно дер-
жать на теплой подушке под лампой или лучше в инкубаторе,
пока действие анестетика не прекратится.
5.6. Получение потомства путем гистерэктомии
Применение кесарева сечения способствует сохранению здо-
ровья колонии мышей. Этой же цели служит использование
приемных матерей.
Для того чтобы операция по передаче мышат прошла удач-
но, необходима синхронизация рождения собственных мышат
принимающей матери с завершением беременности самки ли-
нии, детеныши которой должны быть «усыновлены». Животных
для кесарева сечения отбирают посредством пальпации. Син-
хронизации можно добиться, рассчитывая время спаривания,
но, поскольку время родов в норме варьирует, всегда рекомен-
дуется пальпация.
Беременных самок пальпируют в положении, показанном на
рис. 1.1, Б. Большим и указательным пальцами свободной руки
осторожно нащупывают матку. На 18—19 сут беременности от-
дельные зародыши прощупываются как маленькие шарики. Не-
посредственно перед родами они принимают продольное поло-
жение и слегка перемещаются в просвете матки. Именно в это
время и нужно делать кесарево сечение. Больные животные или
животные, приступившие к родам, для операции не пригодны.
Приемных матерей и усыновляемый ими приплод не отсажива-
ют до тех пор, пока экспериментатор не будет уверен, что опе-
рация прошла благополучно. Сама процедура осуществляется
на открытом столике или в стерильной камере. Перед операци-
ей готовят стерильный контейнер с завинчивающейся крышкой,
содержащий дезинфицирующий раствор (смесь 10°/о-ногр фор-
малина и 10%-ного жидкого мыла в стерильной воде при36°С).
Кроме того, требуется еще стакан объемом 500 мл с 250 мл
такой же смеси. Брюшко беременной мыши нужно выбрить.
24
Глава 1
1. Забейте мышь смещением шейных позвонков, немедленно
погрузите ее в стакан с дезинфицирующей жидкостью,
промакните и перенесите на стерильную поверхность.
2. Захватив пальцами кожу брюшка по средней линии и од-
новременно оттягивая ее к голове и конечностям, приго-
товьте таким образом место разреза.
3. Сделайте продольный разрез стенки тела и обнажите
матку.
4. Удерживая влагалище пинцетом, сделайте каудальный
разрез. Постепенно поднимая матку, отрежьте мезенте-
рий. Старайтесь не проколоть матку и ни к чему ею не
прикасайтесь, чтобы не нарушить стерильность.
5. Перенесите матку в контейнер со стерильным раствором.
Приемная мать должна находиться поблизости. Всю про-
цедуру следует производить недалеко от входа в изоля-
тор.
6. Извлеките матку, слегка промокните.
7. Поместите матку на стерильную поверхность. Разрежьте
ее вдоль антимезометриальной поверхности и при помощи
второй пары ножниц и пинцета извлеките плоды из заро-
дышевых оболочек. Пупочный сосуд во избежание силь-
ного кровотечения нужно сжать и отвести, а не перере-
зать.
8. Промокните мышат салфеткой, осторожно тормошите их
салфеткой или хирургическим тампоном, пока они не по-
розовеют и не начнут ровно дышать. Дыхание можно
стимулировать, осторожно защемив хвост, если мышонок
пискнет, то, по всей вероятности, он будет жить.
Вся процедура — от умерщвления матери до извлечения по-
следнего мышонка — должна занять не более 4—4,5 минут.
Можно подсаживать мышат приемной матери, родившей собст-
венных детенышей на 2—3 дня раньше, но результат окажется
менее удачным.
В качестве контроля можно оставить некоторых мышат из ес-
тественного помета.
5.7. Усыновление
Усыновление требуется как в случаях гистерэктомии, так и
в тех случаях, когда естественная мать не способна вскормить
свой помет по состоянию здоровья или из-за генетических де-
фектов. Мышей подсаживают приемной матери при рождении
или в любое время до конца вскармливания. Собственный по-
мет приемной матери должен быть примерно того же возраста.
В качестве приемной матери выбирают мышь, принадлежащую
к линии, отличающейся особой заботой о потомстве, а также
Разведение мышей
25
иным цветом шерсти. В этом случае несколько мышат собст-
венного помета оставляют. Если в распоряжении исследовате-
ля есть несколько приемных матерей, к каждой из них подса-
живают по одному-двум приемышам, — это снижает риск поте-
ри потомства от каннибализма или неприятия. Когда выбрать
приемную мать с другим цветом шерсти не представляется
возможным, усыновленных мышат можно пометить отщипыва-
нием пальца. Заметим, однако, что новорожденных прищипы-
вать не рекомендуется, чтобы не стимулировать каннибализм.
Следует убедиться, что роды у приемной матери закончились,
а после вскармливания — проверить генетическую аутентичность
приемных мышат.
6. Транспортировка
Мышей можно перевозить по железной дороге, в автомоби-
ле, по воздуху. Для этого их помещают в специальные венти-
лируемые контейнеры из полипропилена или картона.
Животные должны быть обеспечены пищей и влагой на все
время пути. Источником воды служит увлажненная пища, ко-
торую помещают в тонкие пластиковые пакеты [9], или 1%-ная
желатина, содержащаяся в специальном контейнере. Для ко-
ротких перевозок бывает достаточно кусочка картофеля или
яблока, однако при транспортировке мышей за границу такая
пища не годится, поскольку существуют ограничения на ввоз
растительных материалов.
Контейнеры должны быть снабжены этикетками с указани-
ем адреса отправителя и получателя, описанием содержимого
(вид, линия, возраст) и регистрационного номера. Кроме того,
в этикетке должно быть указано, что запас пищи и влаги в
контейнере достаточен и открывать его в пути не нужно; это
особенно важно в тех случаях, когда к состоянию здоровья
транспортируемых животных предъявляются особые требова-
ния. Контейнер с мышами обязательно должен иметь надпись
«Внимание — живые животные» («Urgent — Live Animals»).
Получатель животных должен быть снабжен необходимыми
документами, куда может входить справка о состоянии здоро-
вья и пригодности животных для транспортировки, а также
разрешение на перевозку. В некоторых странах требуется сер-
тификат, подписанный инспектором министерства сельского хо-
зяйства, тогда как в других — достаточно справки от ветерина-
ра или главы учреждения, высылающего животных. При меж-
дународных пересылках все документы надо подготовить зара-
нее; они должны сопровождать животных в пути.
26
Глава 1
7. Ввоз
Если животные поступают в виварий со стороны, их реко-
мендуется поместить в изолятор на 28 дней — время, достаточ-
ное для контроля. Это особенно важно в тех случаях, когда
источник получения мышей — новый или состояние здоровья
исходной колонии неизвестно.
Если животные импортируются в Великобританию, требует-
ся справка-разрешение Министерства сельского хозяйства.
Мышей следует держать в карантине 6 месяцев, однако экспе-
рименты можно начать и раньше. Животных, родившихся в пе-
риод карантина разрешается брать спустя 3 недели после окон-
чания их вскармливания матерью.
8. Правила работы с лабораторными животными
Прежде чем начать экспериментальную работу, следует
убедиться в том, что ее методы не противоречат национальным
или международным правилам использования животных в на-
учных целях. В Великобритании, например, в 1986 г. принято
специальное постановление, в котором оговариваются требова-
ния к оборудованию вивария и определяются технические ус-
ловия экспериментов. Постановление предписывает применять
совершенные способы анестезии.
Литература
1. Fest'ing М. F. W. (1979). Inbred Strains in Biomedical Research. Macmillan
Press.
2. Festing M. F. W. (1987). International Index of Laboratory Animals. 5th
edition, Laboratory Animals Ltd, PO Box 101, Newbury, Berkshire, UK.
3. Small J. D. (1983). In: The Mouse in Biomedical Research. Foster H. L.,
Small J. D. and Fox J. G. (eds.), Academic Press, Vol. 3, p. 90.
4. The UFAW Handbook on the Care and Management of Laboratory Animals
(1967). Third edition, Livingstone, Edinburgh.
5. Whitten W. K. (1956). J. Endocrinol., 14, 160.
6. Ross.M, (1962). J. Anim. Technicians Assoc., 13, 1.
7. Green C. J. (1979). Animal Anaesthesia, Laboratory Animals Ltd, London.
8. Flecknell P. A., Mitchell M. (1984). Lab. Anim., 18, 143.
9. Peters A. G., Bywater P. M. (1983). Anim. Technol., 34, 71.
ГЛАВА 2
ЭКСПЕРИМЕНТЫ
С ПРЕДИМПЛАНТАЦИОННЫМИ ЭМБРИОНАМИ
МЫШИ
Хестер Пратт1
1. Введение
Культивирование предимплантационных эмбрионов in vitro
широко применяется в двух областях биологии развития — при
изучении эмбриона как автономно дифференцирующейся сис-
темы [1, 2] и для введения генов при получении трансгенных
мышей (гл. 11). В этой главе описаны способы выделения
яйцеклеток и предимплантационных эмбрионов, а также мето-
ды работы с эмбрионами разных стадий развития.
Но прежде следует сделать три замечания.
1. Мышь становится все более популярной моделью для
изучения предимплантационного эмбриогенеза млекопи-
тающих. Мышь — это единственный вид, за исключением
кролика, у которого эмбрионы можно культивировать in
vitro в течение всего предимплантационного периода.
Однако следует помнить, что выводы, полученные в ходе
таких исследований, вряд ли можно экстраполировать на
всех млекопитающих.
2. Эмбрионы, культивируемые в искусственной среде in
vitro, обычно развиваются более медленно, чем in
vivo, следовательно, для оптимального роста и развития
зародышей в культуре требуются некие специфические
факторы, содержащиеся в репродуктивном тракте.
3. Культивирование эмбрионов предполагает необходимость
тщательных и регулярных наблюдений. Только при этих
условиях можно овладеть данной методикой, научиться
отличать нормальное развитие от аномального и опреде-
лять нужную стадию развития эмбриона.
2. Извлечение эмбрионов
Извлечение и культивирование эмбрионов не обязательно
проводить в стерильных условиях, нужно лишь обеспечить чис-
тоту и отсутствие пыли. Поскольку культуры поддерживаются
не более 4—5 суток, в качестве мер, предохраняющих от бак-
териального заражения, вполне достаточно того, чтобы все ма-
1 Н. Р, М. Pratt. Embryo and Gamete Research Group, Department of Ana-
tomy, University of Cambridge, Downing Street, Cambridge CB2, 3DY, UK.
28
Глава 2
нипуляции проводились в стерильной среде при помощи вытя-
нутой в пламени стеклянной пипетки и под микроскопом, защи-
щенным от пыли специальным чехлом. Подробности о реакти-
вах, средах и оборудовании см. в разд. 5.
Яйцеклетки и эмбрионы (особенно на одно- и двухклеточной
стадиях развития) чрезвычайно чувствительны к внешним воз-
действиям. Между тем при культивировании часто оказывается
необходимым извлекать эмбрионы из инкубатора для наблю-
дений. Наиболее очевидное следствие возникающих при этом
температурных сдвигов — удлинение периодов G2 и М клеточ-
ного цикла [3J. Один из способов избежать неприятных по-
следствий — использовать при микроскопировании нагреватель-
ный столик, отрегулированный на 36±1°С и среду, нагретую
до 37 °C.
Общепринятой оптимальной культуральной среды нет; сос-
тав сред, применяемых в разных лабораториях, варьирует. Обя-
зательными компонентами во всех случаях служат содовый
буфер, лактат, пируват и бычий сывороточный альбумин.
Различия касаются концентраций этих и иных компонентов и
конечной осмолярности среды [4, 5].
Процедура извлечения эмбрионов в зависимости от стадии
развития отличается последовательностью операций. В общих
чертах процесс заключается в сборе зародышей в теплую
(37 °C) забуференную HEPES среду, содержащую БСА (М2 +
+ БСА) при помощи вытянутой в пламени пастеровской пипет-
ки, соединенной с мундштуком. Остаток клеток кумулюса и
слизи удаляют посредством нескольких переносов эмбрионов по
ряду капель теплой (37°C) культуральной среды М2 + БСА и
последующего споласкивания в культуральной среде Ml6 +БСА.
В заключение эмбрионы помещают в капли М16 + БСА
(~50 мкл), находящиеся под легким парафиновым маслом в
пластиковых чашках Петри, и инкубируют при 37 °C в атмо-
сфере 5%-ного СОг- В интенсивно работающих лабораториях
число таких чашек очень велико. В этом случае удобно уста-
навливать чашки на узкие подносы Регрех, увеличивающие глу-
бину инкубатора. Если необходим частый доступ к эмбрионам,
удобно иметь второй инкубатор для кратковременного культи-
вирования, что дает возможность долговременным культурам
находиться в стабильных условиях.
2.1. Оплодотворенные и неоплодотворенные яйцеклетки
2.1.1. Спаривание in vivo
Спаривание после естественной или экспериментально ин-
дуцированной овуляции (суперовуляции) описано в разд. 1.5
(гл. 1). По нашим данным, самцы-производители фунЕЦиони-
Эксперименты с предимплантацнонными эмбрионами мыши 29
руют наиболее эффективно в случае регулярных спариваний,
но не чаще, чем через ночь. Суперовуляция — удобный прием
для получения большого числа яйцеклеток и эмбрионов от мо-
лодых (3—4 недели) мышей [6]. Для эффективной синхрони-
зации оплодотворения и развития применяют гормональную
стимуляцию. Соответствующая схема инъекций представлена
в разд. 5.4 (гл. 1). Отклонения от нее обеспечивают асинхрон-
ность развивающейся популяции (если в эксперименте требуют-
ся эмбрионы разного возраста). Важно быть уверенным в том,
что из суперовулировавших яиц развиваются нормальные эмб-
рионы. Чтобы убедиться в этом, при сравнении таких эмбрионов
с контролем (т. е. с эмбрионами, развившимися из яиц, овули-
ровавших естественным образом) следует использовать надеж-
ные критерии.
Для получения оплодотворенных или неоплодотворенных
яйцеклеток проделайте следующие процедуры.
I. Забейте самку смещением шейных позвонков, отпрепари-
руйте яйцеводы и поместите их в теплый (37 °C) физио-
логический раствор или среду М2 + БСА (разд. 5). Все
манипуляции с яйцеводами производите в пластиковых
чашках Петри или в стеклянных камерах на нагреватель-
ном столике бинокулярного микроскопа. Если овуляция
произошла в течение предшествующих 10 ч (приблизи-
тельно), массы клеток кумулюса, окружающие яйцо, бу-
дут хорошо видны через тонкие стенки ампулы.
2. Отделите кумулюсные массы в культуральной среде М2 +
+ БСА с помощью тонкого глазного пинцета.
3. Удалите как можно больше среды (но не понижайте ме-
ниск настолько, чтобы он сдавил яйца) и замените ее
приблизительно 1 мл теплого раствора ( + 37°С) гиалу-
ронидазы (табл. 2.4). В течение следующих 2—4 мин
клетки кумулюса постепенно диссоциируют и освободят
яйцо. Этот процесс можно ускорить осторожным пипети-
рованием скопления яиц широкой стеклянной пипеткой.
Охлаждение и гиалуронидаза могут активировать неоп-
лодотворенные яйца, поэтому растворы должны быть теп-
лыми, а воздействие гиалуронидазой сокращено до мини-
мума.
4. После того, как яйца осядут на дно чашки, удалите как
можно больший объем раствора гиалуронидазы н замени-
те его теплой средой М2+БСА, соберите яйца пипеткой
и отмойте их от клеток кумулюса, проведя несколько раз
через среду М2 + БСА.
5. В тех случаях, когда нужно собрать тысячи яиц (напри-
мер, для выделения РНК, см. гл. 9), более эффективным
способом удаления клеток кумулюса служит нейлоновая
30
Глава 2
сетка (размер ячеек около 20—30 мкм), вставленная в
основание цилиндра пластикового шприца на 5—10 мл.
Смочите сетку средой, пропустите среду с яйцами через
это сито и соберите в чашку Петри прошедший через
ячейки раствор с клетками кумулюса. В результате этой
процедуры яйца окажутся почти свободными от клеток
кумулюса. Для удаления их остатков следует воспользо-
ваться пипеткой.
6. Если в течение 10 ч после инъекции чХГ яйца не будут
извлечены из яйцеводов, эндогенные энзимы высвободят
массы кумулюсных клеток. В этом случае разумно при-
бегнуть к получению яиц методом вымывания, описанным
в разд. 2.2 для дробящихся стадий, с использованием при
необходимости гиалуронидазы.
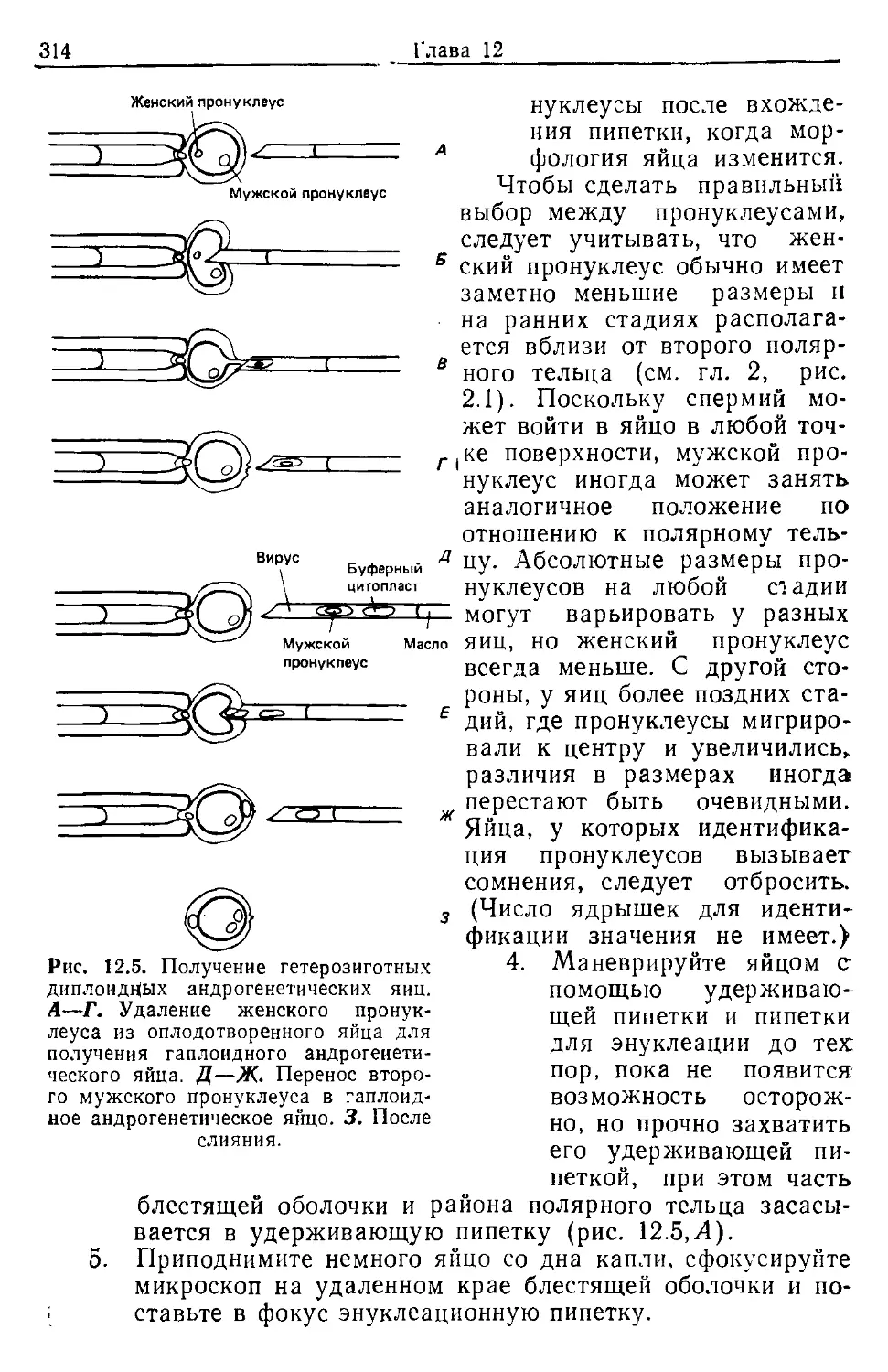
Оплодотворенные яйца отличаются от неоплодотворенных
присутствием хорошо заметного второго полярного тельца
(рис. 2.1,5) (первое полярное тельце у многих линий мышей
часто дегенерирует). Для определения стадий развития опло-
дотворенных яиц используют такие признаки, как наличие и
положение мужского и женского пронуклеусов (рис. 2.1). По-
следовательность событий во время первого клеточного цикла
(см. табл. 2.1) запускается оплодотворением и не зависит от
времени: овуляции [ 8J. Рекомендуется избегать оплодотворения
яиц, овулировавших более 12 ч назад, .так как с возрастом яиц
увеличивается вероятность их партеногенетической активации
(гл. 12).
2.1.2. Оплодотворение in vitro
Описанный способ получения яиц и эмбрионов удобен в
двух отношениях. Во-первых, он обеспечивает большую син-
хронность популяции, чем спаривание in vivo, так как момент
оплодотворения [8] можно, точно зарегистрировать. Во-вторых,
вся процедура перестает быть связанной с циклом смены дня и
ночи, и исследователь так выбирает время осеменения, чтобы ин-
тересующая его стадия развития пришлась на рабочий день.
Детали процедуры см. в гл. 13, разд. 6.2. Мы пользуемся сход-
ной методикой, поэтому на ней не останавливаемся.
Данные по первым пяти клеточным циклам получены для
•единичных клеток или пар клеток, синхронизированных в от-
ношении предыдущего деления. Данные по более поздним
циклам получены на интактных эмбрионах и поэтому более
вариабельны. Циклы 1 и 2 описаны для гибридов C57BL/CBA,
а более поздние циклы — для аутбредных мышей MFI. Все вре-
менные характеристики основаны на наблюдениях над группой
эмбрионов.
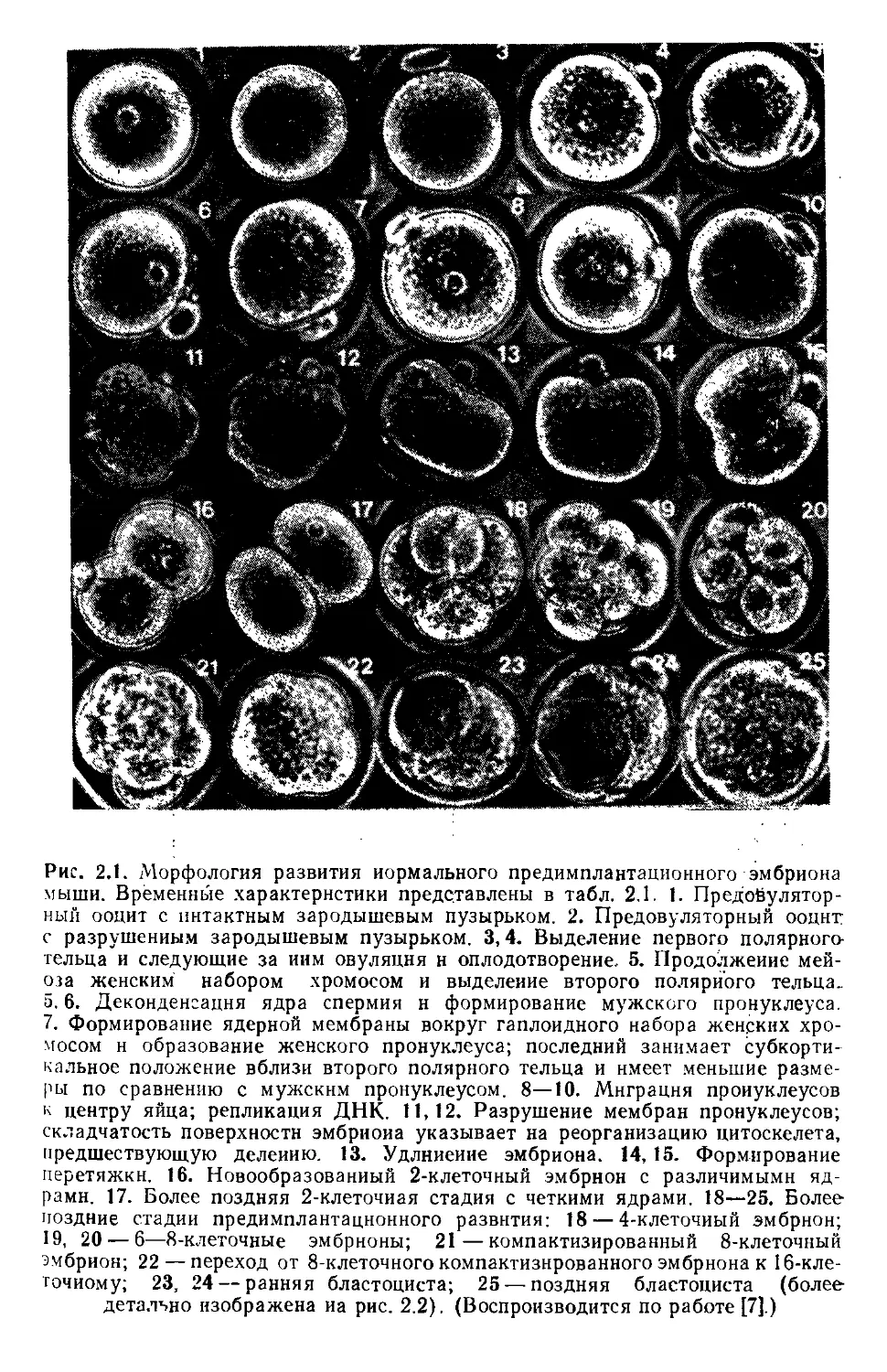
Рис. 2.1. Морфология развития нормального предимплантационного эмбриона
мыши. Временные характеристики представлены в табл. 2.1. 1. Предойулятор-
ный ооцит с интактным зародышевым пузырьком. 2. Предовуляторный ооцнт
с разрушенным зародышевым пузырьком. 3,4. Выделение первого полярного-
тельца и следующие за иим овуляция н оплодотворение. 5. Продолжение мей-
оза женским набором хромосом и выделение второго полярного тельца,
а. 6. Деконденсацня ядра спермия н формирование мужского пронуклеуса.
7. Формирование ядерной мембраны вокруг гаплоидного набора женских хро-
мосом н образование женского пронуклеуса; последний занимает субкорти-
кальное положение вблизи второго полярного тельца и имеет меньшие разме-
ры по сравнению с мужским пронуклеусом. 8—10. Миграция проиуклеусов
к центру яйца; репликация ДНК. 11,12. Разрушение мембран пронуклеусов;
складчатость поверхности эмбриона указывает на реорганизацию цитоскелета,
предшествующую делению. 13. Удлинение эмбриона. 14, 15. Формирование
перетяжки. 16. Новообразованный 2-клеточный эмбрион с различимыми яд-
рами. 17. Более поздняя 2-клеточиая стадия с четкими ядрами. 18—25. Более
поздние стадии предимплантационного развития: 18 — 4-клеточиый эмбрион;
19, 20 — 6—8-клеточные эмбрионы; 21—компактизированный 8-клеточный
эмбрион; 22 — переход от 8-клеточного компактизнрованного эмбриона к 16-кле-
точиому; 23, 24 — ранняя бластоциста; 25 — поздняя бластоциста (более
детально изображена иа рис. 2.2), (Воспроизводится по работе [7].)
Таблица 2.1. Временная последовательность событий при развитии1)
Временная характеристика развития
Часы после осеменения Часы после инъекции чХГ
Первый клеточный цикл [8] (1-клеточная стадия) Завершение мейоза II Разрыв зародышевого пузырька и овуляция Период G1 Выделение второго ПТ Формирование мужского проиук- леуса Формирование женского (меньше- го) пронуклеуса Миграции проиуклеуса к центру Период S Период G2/M Растворение пронуклеарных мемб- ран Цитокинез 2—И 1—2 4—7 5-8 8—10 11—17 17—20 16,5—19,5 18,5—21,5 -12 -32
Часы после деления Часы после инъекции чХГ
Второй клеточный цикл [11] (2-клеточиая стадия) Период G1 1-я транскрипция эмбриональных генов Период S 2-я транскрипция эмбриональных генов Период G2 Период М Цитокинез 0—1 1—1,5 1—6 7—10 6—18 18—20 18—20 -48—50
Третий клеточный цикл [3] (4-клеточная стадия) Период G1 Период S Период G2+M 0—1 1-8 8—13 —58—60
Четвертый клеточный цикл [3, 12, 13] (8-клеточная стадия) Период G1 Период S поляризация цитоплазмы уплощение клеток (компактизация) поляризация поверхности Период G2/M Цитокинез (декомпактизация в ходе митоза у иитактиых эмбрионов) 0—2 2—9 2—7 3,5—7,5 5—9,5 9—12 11—14 -72
Э к с п ер именты с предимплантациониыми эмбрионами мыши
33
Продолжение табл. 2.1.
Временная характеристика развития
Часы после осеменения Часы после инъекции чХГ
Пятый клеточный цикл [13—15] (16-клеточная стадия) Рекомпактнзация после деления (интактные эмбрионы) Цитокинез (наружные клетки) (внутренние клетки) 0,5 11 — 12 13—14 -84
Шестой клеточный цикл [13, 14] (32-клеточная стадия) Начало кавитации (интактные эмбрионы) 0,2 -90—98
Последующие клеточные циклы Полностью сформированная бласто- циста Вылупившаяся бластоциста -110 — 120
') В этой таблице приводится перечень четко наблюдаемых морфологических собы-
тнй, имеющих место в преднмплантацнонный период развития мыши in vitro. Указана
их связь с клеточными циклами (если она известна), а также время, отделяющее эти
события от предшествующего деления дробления и инъекции чХГ.
2.2. Стадии дробления (от 2 до 8-клеточной)
Развитие от 2-клеточной стадии до поздней 8-клеточной —
ранней 16-клеточной стадии (морулы) (рис. 2.1) происходит в
яйцеводе примерно за 47 ч. Второй клеточный цикл, как и пер-
вый, относительно продолжителен (около 20 ч), в то же время
третий и последующие циклы приближаются по своей длитель-
ности к 12-часовому циклу соматической клетки. Таким обра-
зом, популяции на нужной стадии должны извлекаться из яй-
цеводов через определенное время после инъекции чХГ или
оплодотворения in vivo. Однако эта точность весьма относи-
тельна, если учитывать межзародышевые и внутризародыше-
вые вариации темпов развития, выражающиеся в морфологи-'
чсских особенностях стадий или в стадиях клеточного цикла.
Возрастная гетерогенность эмбрионов объясняется асинхрон-
ностью оплодотворения [8], а вариации стадий клеточного
Цикла, по-видимому, связаны с внутренними, генетическими
различиями между бластомерами [16]. Чтобы понять меха-
низмы развития, нужно иметь точные представления о после-
3-171
34
Глава 2
довательности событий п их временных характеристиках. Вог
почему так необходимо совершенствовать методы получения
синхронизированных эмбрионов и составляющих их клеток.
Эти методы обсуждаются в разд. 3.1 н 3.2.
Наиболее чистый способ извлечения эмбрионов из яйцевода
заключается в вымывании их при помощи шприца с тонкой
иглой. Он требует определенного навыка, но овладевший этим
способом всегда предпочтет его более традиционному — разру-
шать яйцевод тонким часовым пинцетом и затем выбирать за-
родыши среди клеточных скоплений.
Процедура извлечения эмбрионов из яйцевода заключается
в следующем.
1. Извлеките яйцевод. Для этого вначале перережьте вер-
хушку матки так, чтобы небольшая часть ее осталась с
яйцеводом, затем отсеките яйцевод от яичника. Это нуж-
но сделать очень тщательно, чтобы не повредить отвер-
стие яйцевода, иначе потом будет очень трудно ввести
иглу. Для промывания яйцевода наиболее пригодны ме-
таллические иглы 30-го калибра, отрезанные и отполиро-
ванные так, чтобы конец был прямой и гладкий.
2. Поместите вырезанный яйцевод в каплю (она должна по-
крывать яйцевод) М2+БСА (табл. 2.5) в пластиковой
чашке Петри на нагревательном столике бинокулярного
микроскопа. Если капля слишком велика, яйцевод будет
плавать, затрудняя дальнейшие манипуляции.
3. Воспользуйтесь глазным пинцетом, чтобы удержать яйгс-
вод и найти его конец (имеющий у мышей многих штам-
мов вид ребристого рукава). Затем введите в него иглу,
соединенную со шприцем объемом 1 мл, наполненным сре-
дой для вымывания: М2 + БСА.
4. Инъецируйте 0,1—0,2 мл среды. В случае удачи яйцевод
раздуется и среда вместе с эмбрионами выльется из пе-
ререзанного конца матки.
5. Отберите эмбрионы из клеточной массы при помощи пи-
петки, отмойте их, перенеся через каплю теплой М16+БСА
(табл. 2.5), и культивируйте в той же среде под парафи-
новым маслом при 37°C (в воздухе должно быть 5% при-
меси СО2).
2.2.1. Двуклеточный блок
Здесь необходимо сделать одно важное замечание. Яйца и
эмбрионы большинства мышиных линий, помещенные в куль-
туральную среду до середины 2-клеточной стадии, останавли-
ваются в развитии на стадии G2 второго клеточного цикла,—
явление, названное двуклеточным блоком [16]. Если эмбрионы
Э кс 11 ер и менты с предимплантационными эмбрионами мыши
35
Таблица 2.2. Линии мышей, не подверженные
«двуклеточному блоку»1’
Лииня Литератур- ный источ- ник
(C57BL/10ScSn/Ola а X [18]
xCBA/Ca/Ola-^F,
(C57BLXCBA Тб Тб^ 119]
(C57BLXCBA—LAC)F1 [20]
(C57BL/10JxSJL/J)FI [21]
В6А Fi [17]
(C57XSJL) F, [17]
') 50% нлн более оплодотворенных яиц развиваются до
ггадни бластоцисты.
извлечь из яйцевода на поздней 2-клеточной стадии (примерно
48 ч после чХГ), развитие in vitro в простой среде не наруша-
ется. Есть, однако, несколько линий (главным образом инбред-
ных или гибридов между ними), яйца и эмбрионы которых на
2-клеточНой стадии не блокируются (табл. 2.2). Мы в своей
работе используем гибридных самок Fj от скрещивания
(C57BLXCBA/Ca).
Сравнение линий мышей, имеющих и не имеющих двукле-
точный блок, показало, что свойство эмбрионов останавливать-
ся в развитии на стадии двух клеток определяется исключи-
тельно генотипом яйца и не зависит от отцовского или эмбрио-
нального генома [18]. Наличие феномена двуклеточного блока
делает предпочтительным использование в экспериментах яиц
неб локирующихся линий (хотя в качественном отношении пе-
реход от одноклеточной к двуклеточной стадии у линий с бло-
ком и без него не отличается) [18]. Если же в опыте все-таки
участвует блокирующаяся линия, следует обратить особое вни-
мание на то, чтобы эмбрионы 2-клеточной стадии извлекались
из яйцевода в достаточно позднем периоде второго клеточного
цикла и их нормальное развитие в бластоцисту in vitro было
обеспечено. Этот момент соответствует приблизительно 36—
40 ч после чХГ, но его лучше определить экспериментально для
каждой линии, с которой проводится работа.
2.3. Морулы
Термин «морула» неточен, но обычно используется для
обозначения компактного агрегата бластомеров (рис. 2.1, 21,
22). Понятие объединяет все стадии развития от компактпзиро-
36
Глава 2
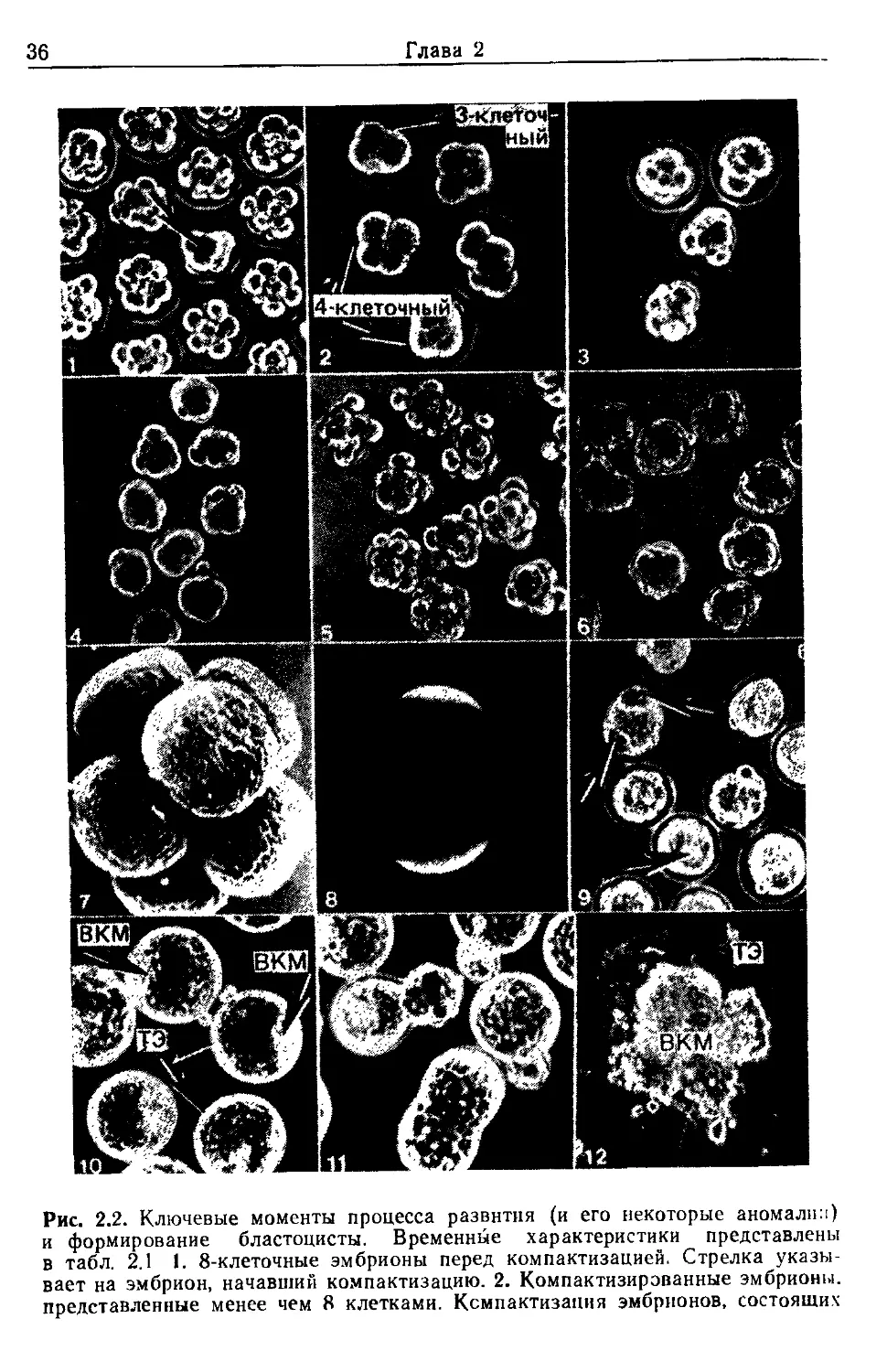
Рис. 2.2. Ключевые моменты процесса развития (и его некоторые аномалии)
и формирование бластоцисты. Временные характеристики представлены
в табл. 2.1 1. 8-клеточные эмбрионы перед компактизацией. Стрелка указы-
вает на эмбрион, начавший компактизацию. 2. Компактизирэванные эмбрионы,
представленные менее чем 8 клетками. Ксмпактизания эмбрионов, состоящих
Эксперименты с предимплантационными эмбрионами мыши 37
ванной 8-клеточной до 16- и 32-клеточной стадий и начала фор-
мирования бластоцеля. Наблюдения над развитием эмбрионов
in vitro в течение этого периода позволяют представить процесс
более детально: на 8-клеточной стадии бластомеры уплощают-
ся в местах контакта друг с другом (компактизируются), но в
течение митоза, ведущего к 16-клеточной стадии, декомпактизи-
руются и затем компактизируются снова (рис. 2.2, 4—6, перио-
дизацию см. в табл. 2.1). Поздние морулы обычно соответству-
ют 16—32-клеточным эмбрионам, в которых еще нет полости;
именно на этой стадии развития (около 84 ч после чХГ) оии
проходят место соединения яйцевода с маткой. Если эмбрионы
извлекаются именно в этот момент, целесообразно промывать
не только яйцеводы, но также и оба рога матки (см. разд. 2.4).
2.4. Бластоцисты
У эмбрионов, входящих в матку, кавитация (формирование)
бластоцеля начинается спустя 94 ч после инъекции чХГ. В это
время они состоят примерно из 32 клеток (т. е. большинство
клеток достигло конца своего пятого клеточного цикла, табл.
2.1). По мере того, как клетки пройдут следующие два цикла,
бластоциста окончательно формируется (рис. 2.1, 23—25,
рис. 2.2; 9, 10). Когда число клеток достигает приблизительно
128 (120 ч после чХГ), блестящая оболочка утоньшается, блас-
из меньшего числа клеток, чем обычно, свидетельствует о том, что это явление
не приурочено к определенному клеточному циклу. Несмотря на возможную
жизнеспособность этих эмбрионов, их лучше изъять из основной популяции.
3. Неиитегрированные бластомеры компактизированных эмбрионов. Компакти-
зация продолжается в течение 4—5 ч и не происходит одновременно со всеми
бластомерами, в результате эмбрион может иметь бластомеры, не включенные
в основную клеточную массу. Все эмбрионы, у которых компактизация не за-
вершилась через 8 ч после наступления 8-клеточиой стадии, должны быть
отброшены. 4—6. Декомпактизация компактизированных 8-клеточных эмбрио-
нов при переходе к 16-клеточной стадии и последующая рекомпактизация
после деления (см. разд. 3.1). Блестящая оболочка удалена, поэтому измене-
ния поверхности видны более четко (фотографии любезно предоставлены Джу-
лией Чишолм): 4 — компактизированные 8-клеточные эмбрионы; 5 — деком-
пактизация 8-клеточиых эмбрионов; 6 — рекомпактизированные 8-клеточные
эмбрионы. 7. Поляризация поверхностных микроворсинок на отдельных бла-
стомерах 8-клеточного эмбриона, наблюдаемая в сканирующем электронном
микроскопе (см. разд. 4.1.2). 8. Поляризация поверхностных микроворсинок
на паре бластомеров 8-клеточной стадии, выявляемая окрашиванием
ФИТЦ—КоиА (см. разд. 4.1.1). 9. Эмбрионы через несколько часов после
начала скапливания жидкости. У нормальных эмбрионов может иметь место
формирование более чем одной полости (указано стрелкой), ио по мере раз-
вития они сливаются в одну. Эмбрионы, у которых этого не происходит, долж-
ны быть отброшены. 10. Ранние бластоцисты. Указаны ВКМ и ТЭ. Отбросьте
все эмбрионы без отчетливо выраженной ВКМ. 11. Различные морфологиче-
ские картины вылупляющихся бластоцист. 12. Эмбриональные разрастания
в виде неправильных выростов ТЭ с узелком ВКМ.
38
Глава 2
тоциста вылупляется (рис. 2.2,//), прикрепляется к эпителию
матки и начинает имплантироваться. На этой стадии эмбрионы
уже не вымываются. Поэтому целесообразно собрать бласто-
цисты заблаговременно и наблюдать их дальнейшее развитие
in vitro в простой синтетической солевой среде (т. е. М16 + БСА,
табл. 2.5) или дать им вылупиться и культивировать в более
сложной среде, т. е. в среде Игла, модифицированной Дуль-
бекко (DMEM) с 10-ной эмбриональной телячьей сывороткой
(ЭТС), см. разд. 3.5.2.
Бластоцисты извлекают следующим образом.
1. Отсеките каждый рог матки от подлежащего мезентерия
и отрежьте в местах соединения матки с трубой и ее шей-
ки с влагалищем. Поместите каждый рог на фильтроваль-
ную бумагу, увлажненную М2+БСА (табл. 2.5), и удали-
те лишнюю кровь.
2. Введите инъекционную иглу 25-го калибра, соединенную
со шприцем, в просвет рога матки со стороны яичника и
промойте 0,2—0,5 мл М2 + БСА. Эту процедуру делайте
в пластиковой чашке Петри или в стеклянной камере, без
применения микроскопа.
3. Соберите бластоцисты в теплую (37 °C) М2 + БСА, про-
мойте в теплых и сбалансированных СОг каплях М16 +
+ БСА (табл. 2.5) и инкубируйте в М16+БСА под мас-
лом при 37 °C в атмосфере с 5% 'СОг.
4. Бластоцисты сформируются, вылупятся и прилипнут к со-
ответствующим образом покрытой поверхности пластико-
вой чашки Петри (т. е. типа Falcon), однако в среде
М16+БСА они как следует не прикрепятся и роста троф-
эктодермы не будет. Для последующего развития нужна
более сложная среда, содержащая сыворотку (т. е.
DMEM+10% ЭТС); см. разд. 3.5.2.
3. Манипуляции с эмбрионами и клетками
3.1. Сортировка эмбрионов по стадиям
Клеточные и молекулярные события предимплантационного
развития осуществляются в строгой последовательности. О вре-
менном их контроле известно очень мало, однако можно пред-
положить существование по меньшей мере двух «часов»: один
из них — сам клеточный цикл [2]. Анализ механизмов времен-
ного контроля требует синхронизированной популяции клеток
и эмбрионов.
Как уже обсуждалось в разд. 2.1.1, популяция эмбрионов,
полученная в результате осеменения in vivo, будет гетероген-
ной. поэтому время после инъекции чХГ и время, прошедшее с
Эксперименты с предимплантационными эмбрионами мыши 39
момента оплодотворения, не могут быть признаны адекватными
критериями начала развития. Более разумно выбрать морфоло-
гический критерий, анализировать культуры через определен-
ные промежутки времени (например, 30 минут или каждый
час) и отбирать те эмбрионы, которые в выделенный интервал
времени достигли данной стадии. Таким образом можно ото-
брать группы эмбрионов с одинаковым темпом развития. Ста-
дии, которые приведены ниже, хорошо различаются в интакт-
ных эмбрионах и могут быть использованы в качестве крите-
рия синхронизации (последовательность прохождения стадий
см. табл. 2.1).
1. Дробление (рис. 2.1, 11—21). Число клеток легко подсчи-
тать вплоть до стадии компактизации (8 клеток), затем
клеточные границы перестают быть четкими. Оплодотво-
рение in vitro способствует синхронности дробления до 2-
клеточной стадии, однако внутренняя гетерогенность вре-
мени дробления, вероятно, обусловливает десинхрониза-
цию каждого следующего деления дробления.
2. Домпактизация (рис. 2.1, 21, 22, рис. 2.2, 1—4). Процесс
уплощения клеток обычно происходит через 3,5—7,5 ч
после начала 8-клеточной стадии (4-клеточный цикл).
Его можно считать удобной отправной точкой для опре-
деления последовательности стадий развития; при неко-
торой тренировке компактизация выявляется вполне объ-
ективно.
3. Деление, приводящее к 16-клеточной стадии (конец 4-го
и начало 5-го цикла) (рис. 2.2, 5, 6). Эта стадия характе-
ризуется декомпактизацией перед митозом и рекомпакти-
зацией по завершении деления. Регулярные наблюдения
над поздними 8-клеточными эмбрионами дают, таким об-
разом, возможность выявить субпопуляцию новообразо-
ванных 16-клеточных эмбрионов.
4. Формирование бластоцеля (6-й клеточный цикл) (рис. 2.2,
9, 10). Наиболее раннее свидетельство транспорта жид-
кости можно заметить на 32-клеточной стадии (табл. 2.1),
когда образуется наполненная жидкостью вакуоль (иног-
да две вакуоли, которые увеличиваются и затем слива-
ются). Следовательно, отбор по этому признаку даст по-
пуляцию эмбрионов, начавших формировать бластоцель и
проходящих свой шестой клеточный цикл.
3.2. Сортировка изолированных клеток по стадиям
Дробление — процесс асинхронный, и по мере продвижения
предимплантационного развития степень асинхронности возрас-
тает. Морфологические критерии, описанные в разд. 3.1, могут
40
Глава 2
Таблица 2.3. Состав кислого раствора Тироде (г/100 мл) [22]
NaCl 0,80
КС1 0,02
СаС12 или (СаС12-2Н2О) 0,02(0,0265)
MgCl2-6H2O 0,01
D-глюкоза 0,10
PVP 0,40
Приготовление
Доведите pH до 2,6 при помощи 1 М НС1 (только Analar-HCl). Профильтруйте раствор
через миллипоровый фильтр и храните аликвотами в 1 мл при 4 °C. Используйте в те*
чение 2 суток.
быть достаточными для сортировки и синхронизации эмбрионов,
однако точный анализ механизмов развития требует синхрони-
зации материала на клеточном уровне. Один из способов полу-
чения клеток известного возраста заключается в диссоциации
эмбриона на отдельные клетки, наблюдении за культурами
этих клеток (как описано в разд. 3.1) ив отборе тех из них,
которые разделились в определенном временном интервале.
Эта процедура требует владения навыками снятия блестящей
оболочки и методикой диссоциации клеток.
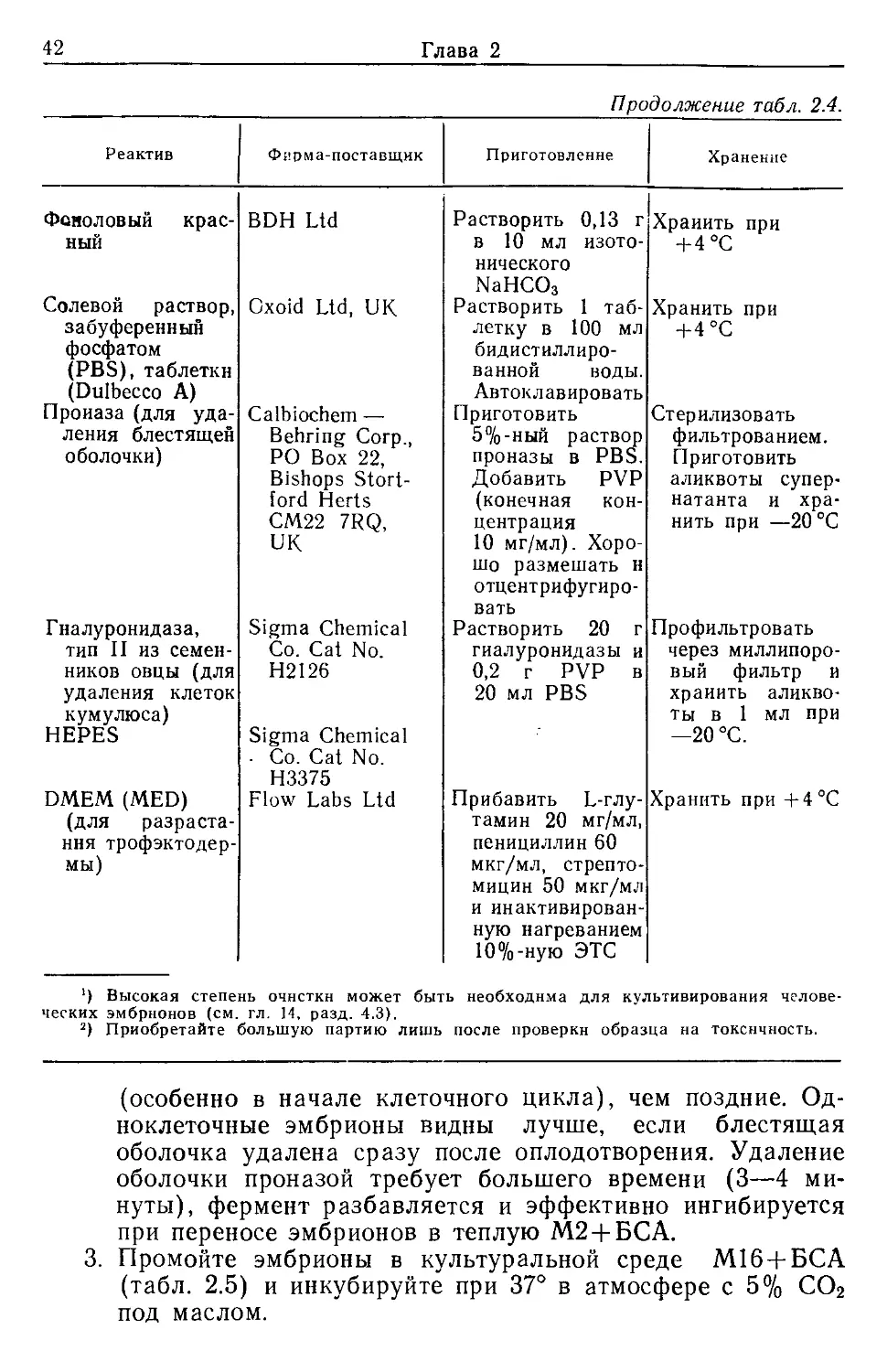
3.2.1. Уданение бнестящей обоночки (zona pellucida)
Блестящую оболочку можно удалить механически, но при
работе с большим числом эмбрионов лучше использовать для
этой цели химические воздействия. Наиболее часто применяют
два реактива — подкисленный раствор Тироде [22] и раствор
проназы [23]. В обоих случаях успех операции определяется
составом и осмолярностью, поэтому рекомендуется испытать
действие реактива на небольшом числе эмбрионов, прежде чем
обрабатывать всю партию.
1. Перенесите эмбрионы небольшими группами в теплый
(37 °C) кислый раствор Тироде (табл. 2.3) или раствор
проназы (табл. 2.4). Температура (37 °C)—наиболее
важное условие успешного удаления оболочки в растворе
Тироде.
2. Осторожно втягивая и выталкивая эмбрионы через пи-
петку, контролируйте изменение поверхности под микро-
скопом. В кислом Тироде процесс идет быстро — в тече-
ние секунд блестящая оболочка сжимается и истончается.
Рекомендуется до ее полного растворения перенести эмб-
рионы в теплую М2 + БСА (табл. 2.5). Передержка
эмбрионов в среде с низким pH приводит к лизису неко-
торых, а то и всех клеток. Ранние стадии (вплоть до ран-
ней 4-клеточной) более чувствительны к кислому Тироде
Таблица 2.4. Реактивы
Реактив Фирма-поставщик Приготовление Хранение
Вода (Analar)» Гормоны: гсжк BDH Ltd Poole, Dorset, UK Intervet 1 флакон Хранить при
чХГ PO Box 5830AA, Boxmeer, The Nether- lands Та же, что и для ГСЖК Sigma Chemical Со. Fancy Road, Poole, Dorset BH17 7NH UK Cat No. A4378 Flow Labs Ltd, (1000 ME) раз- вести до 20 мл 0,9 %-ным сте- рильным соле- вым раствором (50 МЕ/мл) 1 флакон —20 °C аликво- ты по 5 мл. Раз- мораживать при +4 °C. Препарат негоден после не- дельного хране- ния при +4 °C Как ГСЖК
Бычий сывороточ- ный альбумин (БСА)2> (кри- сталлизованный и лиофилизиро- ванный) Эмбриональная те- (1500 ME) раз- бавить до 30 мл 0,9 %-ным сте- рильным соле- вым раствором (50 МЕ/мл) Хранить при Хранить сухим при + 4 °C Хранить при
лячья сыворотка PO Box 17, Se- — 20 °C. Размо- — 20 °C аликвоы
(ЭТС)2» cond Avenue розить, инактиви- по 10 мл
Масло (легкое, Industrial Estate Irvine, Ayrshire KA12 8NB, UK BDH Ltd Product ровать нагрева- нием при 56 °C в течение 30 мин Автоклавировать Хранить при ком-
бесцветное, жид- No. 29436 20—30 мин при натной темпера-
кое, парафино- вое) Пируват (натрие- вая соль) кри- сталлический, тип II Лактат (натриевая соль) 60%-кый сироп Стрептомицина сульфат Бензилпенициллин (Ciystapen) Полнвинилпирро- литон Sigma Chemical Co., Cat No. P2256 Sigma Chemical Co., Cat No. L1375 Glaxo Labs, Ltd Ltd BDH Ltd 0,8 атм, остудить, профильтровать через фильтро- вальную бумагу (Whatman) туре или при 37 °C для ис- пользования в культуральных чашках Хранить сухим при +4 °C Хранить при + 4 °C
42
Глава 2
Продолжение табл. 2.4.
Реактив Фирма-поставщик Приготовление Хранение
Феноловый крас- ный BDH Ltd Растворить 0,13 г в 10 мл изото- нического NaHCO3 Хранить при +4 °C
Солевой раствор, забуференный фосфатом (PBS), таблетки (Dulbecco А) Oxoid Ltd, UK Растворить 1 таб- летку в 100 мл бидистиллиро- ванной воды. Автоклавировать Хранить при +4 °C
Проиаза (для уда- Calbiochem — Приготовить Стерилизовать
ления блестящей Behring Corp , 5%-ный раствор фильтрованием.
оболочки) PO Box 22, Bishops Stort- lord Herts CM22 7RQ, UK проназы в PBS. Добавить PVP (конечная кон- центрация 10 мг/мл). Хоро- шо размешать и отцентрифугиро- вать Приготовить аликвоты супер- натанта и хра- нить при —20 °C
Гиалуронидаза, Sigma Chemical Растворить 20 г Профильтровать
тип II из семей- Co. Cat No. гиалуронидазы и через миллипоро-
ников овцы (для удаления клеток кумулюса) H2126 0,2 г PVP в 20 мл PBS вый фильтр и хранить аликво- ты в 1 мл при
HEPES Sigma Chemical Co. Cat No. H3375 —20 °C.
DMEM (MED) (для разраста- ния трофэктодер- мы) Flow Labs Ltd Прибавить L-глу- тамин 20 мг/мл, пенициллин 60 мкг/мл, стрепто- мицин 50 мкг/мл и инактивирован- ную нагреванием 10%-ную ЭТС Хранить при +4 °C
’) Высокая степень очнсткн может быть необходима для культивирования челове-
ческих эмбрионов (см. гл. 14, разд. 4.3).
2) Приобретайте большую партию лишь после проверки образца на токсичность.
(особенно в начале клеточного цикла), чем поздние. Од-
ноклеточные эмбрионы видны лучше, если блестящая
оболочка удалена сразу после оплодотворения. Удаление
оболочки проназой требует большего времени (3—4 ми-
нуты), фермент разбавляется и эффективно ингибируется
при переносе эмбрионов в теплую М2 + БСА.
3. Промойте эмбрионы в культуральной среде М16 + БСА
(табл. 2.5) и инкубируйте при 37° в атмосфере с 5% СО2
под маслом.
Эксперименты с предимплантационными эмбрионами мыши 43
Следует обратить внимание на два важных обстоятельства.
Во-первых, удаление оболочки может повлиять на компоненты
клеточной поверхности и, возможно, цитоскелета [24J, поэто-
му необходимо некоторое время на регенерацию. Во-вторых,
эмбрионы, лишенные оболочек, становятся очень липкими, скле-
иваются и прилипают к дну культуральных чашек. Их культи-
вируют по отдельности в маленьких каплях М16 + БСА в спе-
циальных неадгезивных чашках Петри (Sterilin).
3.2.2. Диссоциация клеток
Чтобы диссоциировать эмбрионы на отдельные клетки эф-
фективно и с наименьшим повреждением клеток, необходимо
пользоваться микропипеткой, у которой конец оплавлен, а вну-
тренний диаметр чуть больше диаметра клеток. Микропппетки
легко изготовить вручную, вытягивая капиллярную трубку
1,2 мм (разд. 5.3.6), хотя можно воспользоваться электродным
вытягивателем, а конец пипетки подвергнуть микроковке. Ка-
пиллярные пипетки при помощи специальных переходников с
держателями присоединяют к мундштуку. Для диссоциации
эмбрионов на стадиях раннего дробления (от 2-клеточной до
поздней 8-клеточной) достаточно поместить их в бескальциевую
среду, которая нарушает Са2+-зависимую адгезионную систему
[2J-
1. Инкубируйте эмбрионы (без оболочки) приблизительно
10 мин в бескальциевой среде М2 + 6 мг/мл БСА (табл.
2.5) под маслом при 37 °C.
2. Небольшие группы эмбрионов (3—4) пипетируйте. втяги-
вая и выталкивая их через микропипетку, отверстие кото-
рой чуть меньше диаметра эмбрионов. Все манипуляции
проводите в культуральных чашках на нагревательном
(37°C) столике препаровального стереомикроскопа. Еди-
ничные клетки, отделяющиеся в процессе этой манипуля-
ции, отбирайте, а оставшиеся клеточные агрегаты и эм-
брионы продолжайте пипетировать до полной диссоциа-
ции. В некоторых случаях отделяются пары клеток, со-
единенных цитоплазматическим мостиком, — это резуль-
тат незавершенного цитокинеза; мостики маркируют мес-
то контакта между клетками и указывают на направле-
ние плоскости дробления [12J.
3. Изолированные клетки промойте в теплых каплях М16 +
+ БСА (табл. 2.5) в неадгезивных чашках (Sterilin) и
культивируйте их группами или поодиночке в маленьких
каплях. Если клетки культивируются группами, нужно
позаботиться о том, чтобы они не соприкасались (во из-
бежание слипания).
44
Глава 2
Таблица 2.5. Составление культуральной среды (№16, М2)
1. Исходный раствор для приготовления среды М16 и М2
Состав Г/100 мл
Исходный раствор А, 10 X NaCl 5,534
(десятикратный) КС1 0,360
КН2РО4 0,162
MgSO4-7H2O 0,294
Лактат натрия 60%-ный сироп 2,608(3,2 мл)
Глюкоза 1,000
Пенициллин (104 s ME) 0,060
Стрептомицин 0,050
Исходный раствор Б, 10Х NaHCO3 2,106
Исходный раствор В, 10X Пируват натрия 0,36
Исходный раствор Г, 10Х СаС12-2Н2О 2,52
Исходный раствор Д, 10 X HEPES перед разведением до 5,957
100 мл довести pH до 7,4
добавлением 2М NaOH
БСА 0,400
Приготовление
Профильтруйте через миллипоровый фильтр (размер пор 0,22 мкм) все исходные
растворы и храните при +4-°C в пластиковых пробирках. Растворы А, Г, Д можно хра-
нить 3 месяца, растворы Б, В — 2 недели.
2. Приготовление 10 мл М16 + БСА или М2+БСА из исходных растворов
Исходный раствор А Б В г д Феноловый красный Вода (Ап alar) БСА (мг) pH
(мл) М16 1,0 1,0 0,1 0,1 (мг) 0,1 (мл) 7,8 40 7,(
М2 1,0 0,16 0,1 0,1 0,84 — 7,8 40 7,4
Установите pH. Профильтруйте через миллипоровый фильтр (размер пор 0.22 мкм).
Храните удобными аликвотами в пластиковых пробирках при +4 °C. Для бескальциевых
М16 и М2 опустите исходный раствор Г, добавьте 0,1 мл 15 мг/мл NaCl. Приготовьте
6 мг/мл БСА.
4. Если требуются клетки известного возраста, культиви-
руйте изолированные клетки и ежечасно или каждые пол-
часа проверяйте их на предмет делений. Соберите пары
клеток, разделившихся в течение выбранного промежут-
ка времени, и культивируйте, пока они не достигнут нуж-
ного возраста. Если требуются единичные клетки, то об-
разовавшиеся пары бластомеров разделите еще раз, как
было описано.
Как прежде, цитоплазматический мостик служит топогра-
фическим маркером предыдущей борозды дробления.
Стадия 16 клеток — первая стадия, на которой различаются
два клеточных фенотипа (полярные и аполярные) (см. обсуж-
Эксперименты с предпмплантационными эмбрионами мыши 45
дение в разд. 4.1). Отбирая потомков единичных бластомеров
8-клеточной стадии in vitro, можно разделить эти две субпо-
ляции, так как бластомеры 16-клеточной стадии имеют разные
размеры: более крупные клетки — полярные (презумптивные
клетки трофэктодермы, ТЭ), а более мелкие — аполярные (пре-
зумптивная внутренняя клеточная масса, ВКМ) [26].
Начиная с середины 16-клеточной стадии, эмбрионы все
труднее поддаются диссоциации из-за формирования фокаль-
ных плотных контактов и адгезионных контактов между на-
ружными клетками. Эти контакты не так чувствительны к бес-
кальциевой среде, поэтому для диссоциации эмбрионов поздних
стадий применяются методы более жесткие и не всегда сов-
местимые с длительным выживанием диссоциированных клеток.
Например, инкубация в течение 15 мин при 37 °C в 25 мг/мл
трипсина и 10 мг/мл ЭДТА в бескальциевой М2 + 6 мг/мл БСА,
сопровождаемая интенсивным пипетированием, достаточна для
диссоциации 16-клеточных эмбрионов и бластоцист [13, 25].
Для поздних бластоцист используют предварительную инкуба-
цию в цитохалазине Д, разрушающем микрофиламенты (ЦХД
0,5 мкг/мл при 37°C в течение 10 мин). Этот препарат, вызы-
вая округление клеток и ослабевание межклеточной адгезии,
повышает чувствительность к воздействию раствором тринси-
на/ЭДТА. Успешная диссоциация бластоцист зависит от пол-
ного удаления ЦХД перед воздействием трипсином.
Диссоциация изолированных ВКМ описана в разд. 3.4.
3.3. Агрегация эмбрионов и изолированных клеток
Одним из методов создания химерных (или «аллофенных»)
мышей служит агрегация двух или более генотипически разли-
чающихся морул и реимплантация получившегося эмбриона в
матку псевдобеременной приемной матери ([23], см. также
гл. 6, разд. 2.2). Для получения агрегата два или более 8- или
16-клеточных эмбрионов без блестящей оболочки культивиру-
ют вместе, подталкивая их друг к другу до тех пор, пока они
не слипнутся. Адгезии способствует инкубация во время кон-
такта в лектине — фитогемагглютинине (ФГА, исходный раст-
вор Gibco 1 : 20).
В некоторых экспериментах требуется агрегация индивиду-
альных, парных клеток или групп синхронно или асинхронно
делящихся клеток. Чтобы получить такие агрегаты, нужно вос-
пользоваться присущей бластомерам «липкостью» и просто с
помощью пипетки привести их в контакт друг с другом в теп-
лой (37°С) М2 + БСА в культуральной чашке на нагреватель-
ном столике микроскопа. Для стимуляции адгезии можно в
среду добавить ФГА. Он не вызывает дефектов развития, но
46
Глава 2
может способствовать нужной ориентации клёток при их адге-
зии [12]. 1—2-минутная экспозиция ФГА прХ 37°С в М2 + БСА
обычно достаточна для стимуляции адгезий, после чего клетки
можно отмыть от лектина, сполоснуть и чистой пипеткой пере-
нести в маленькие капли М16+БСА. Инкубировать под маслом
при 37 °C в атмосфере с 5% СО2, где они будут агрегировать.
Анализ морфологии образовавшихся клеточных кластеров (че-
му способствует окрашивание ядер 4,6-диамидино-2-фенилин-
долом (ДАФИ), разд. 3.6.2) используется для изучения влия-
ния межклеточных взаимодействий на судьбу клетки в ходе
развития [26].
3.4. Выделение внутренних клеток морул
или внутренних клеточных масс бластоцист
Многочисленные способы выделения кластеров жизнеспо-
собных внутренних клеток морулы (>16 клеток) или бласто-
цисты (>32 клеток) основаны на применении цитотоксических
реагентов, повреждающих слой наружных клеток, но не прони-
кающих к внутренним (благодаря плотным контактам, которые
создают барьер проницаемости). Чаще всего для этого исполь-
зуют цитотоксические антитела, связанные с комплементом
(иммунохирургический способ), но пригоден и ионофорез.
Иммунохирургическая процедура основана на методе, впер-
вые описанном Солтером и Ноулсом [27, 28].
1. Эмбрионы с блестящей оболочкой или без нее инкуби-
руйте в теплой (37 °C) кроличьей антимышиной сыворот-
ке (инактивированной нагреванием при 56°C в течение
30 мин и разбавленной 1 : 10 средой М2), 2—4 мин (мо-
рулы и ранние бластоцисты) или 10 мин (поздние бласто-
цисты).
2. Трижды промойте эмбрионы переносом через теплую
М2+БСА (табл. 2.5), затем инкубируйте 5—7 мин с ком-
плементом в разведении от 1 : 5 до 1 : 10 солевым раство-
ром, забуференным фосфатом (PBS). Источник комп-
лемента — сыворотка крови; большое значение имеет тип
сыворотки. Наименее токсична сыворотка крысы [29], но
можно пользоваться и сывороткой морской свинки. Без-
условно токсична сыворотка кролика. Комплемент адсор-
бируйте агарозой [30]. Его следует хранить при —70 °C,
размораживать только перед употреблением.
3. После инкубации в /.омплементе перенесите эмбрионы в
свежую М2 и оставьте их в ней на 20—30 мин при 37°C;
в течение этого времени можно наблюдать, как на на-
ружных клетках появляются пузыревидные вздутия и на-
чинается их лизис.
Эксперименты с предимплантационпыми эмбрионами мыши 47
4. Пипетируя эмбрионы микропипеткой с оплавленным кон-
цом и внутренним диаметром чуть больше внутренней
клеточной массы (ВКМ), удалите наружные клетки.
Чтобы диссоциировать внутренние клетки или ВКМ, следу-
ет вначале разрыхлить агрегаты, для чего их инкубируют 20 мин
в теплой бескальциевой среде М2 + БСА или М2+БСА, содер-
жащей 0,5 мкг/мл ЦХД. Затем клетки диссоциируют в этой же
среде при помощи пипетки с оплавленным концом и диаметром
чуть меньше кластера диссоциируемых клеток [14].
Другой способ отделения продвинутой в развитии ВКМ за-
ключается в том, что бластоцисте дают вылупиться из блестя-
щей оболочки, прикрепиться и образовать трофэктодермальные
выросты (см. разд. 3.5.2). Затем при помощи ротовой пипетки
удаляют узелок клеток ВКМ, образовавшийся на верхушке
слоя трофэктодермального выроста.
3.5. Выделение трофэктодермы
Трофэктодерму (ТЭ) трудно отделить от ВКМ, не прибегнув
к микрохирургической методике, предложенной Гарднером
[31]. Однако можно воспользоваться двумя другими способами.
3.5.1. Выделение трофэктодермальных пузырьков
или отделение ТЭ от бластоцист
ВКМ мыши значительно более чувствительна к летальным
повреждениям рентгеновским облучением или антиметаболита-
ми, чем ТЭ, поэтому многими из таких воздействий пользуются
для получения трофэктодермальных пузырьков, лишенных ВКМ
[28]. Кроме того, бластомеры ранних стадий дробления, куль-
тивируемые после диссоциации поодиночке или небольшими
группами, часто дают начало «ложным бластоцистам», состоя-
щим, по всей видимости, только из ТЭ [26]. Однако чистота
ТЭ, полученной этими способами, вызывает сомнение.
Другой способ дает клеточные массы трофэктодермального
эпителия мурального района бластоцисты и связан с получени-
ем «гигантских» бластоцист в результате агрегации до 10 мо-
рул, как описано в разд. 3.3. Эти «гигантские» бластоцисты на-
столько велики, что можно бритвенным лезвием, скальпелем
или острой стеклянной иглой под контролем микроскопа отде-
лить муральную ТЭ от ВКМ и полярной ТЭ [32]. При перено-
се в культуру такие трофэктодермальные массы могут объеди-
няться, разрастаться и приобретать форму пузырьков ТЭ.
48 Глава 2
3.5.2. Выделение из трофэктодермальных разрастаний
Эмбрионы культивируют до стадии вылупившейся бласто-
цисты в М16 + БСА (табл. 2.5) и затем переносят в пластико-
вые чашки Пегри (Falcon) или на пластиковые покровные стек-
ла в более сложную среду (т. е. DMEMJ, в которую добавлена
инактивированная нагреванием 10%-Ная ЭТС, пенициллин
(60 мкг/мл) и стрептомицин (50 мкг/м^).
В течение 24 ч культивирования эмбрионы прикрепляются
ко дну культуральной чашки и на второй день образуют троф-
эктодермальные выросты, стелющиеся по субстрату (рис. 2.2,
12). Такие культуры сохраняют жизнеспособность примерно в
течение недели. Смена среды может продлить этот период. На
вершине выроста закладывается ВКМ в виде клеточного узел-
ка, который удаляют при помощи ротовой пипетки.
Осторожно подбирая субстраты и среды, можно получить и
более поздние постимплантационные стадии развития ([33],
см. также гл. 3, разд. 6).
3.6. Подсчет клеток
Число клеток может быть подсчитано прямо, если эмбрионы
или клеточные агрегаты легко диссоциируют. В противном слу-
чае ядра клеток эмбрионов или клеточных агрегатов окрашива-
ют, а затем производят диссоциацию, как описано ниже.
3.6.1. Окрашивание ядер по Гимза
1. Целые эмбрионы (с блестящей оболочкой или без нее),
ВКМ или другие кластеры клеток поместите в 0,9%-ный
раствор цитрата натрия. Инкубируйте от 1 до 10 мин.
В течение этого времени клетки должны округлиться
вследствие ослабления контактов (чем более продвинута
стадия развития эмбриона, тем большим должно быть
время действия цитрата натрия).
2. Перенесите эмбрионы или клетки на обработанные аце-
тоном предметные стекла, фиксируйте каплей этанол : ук-
сусная кислота (3:1 по объему) и высушите на воздухе.
3. Профильтруйте краску Гимза (5% в PBS, табл. 2.4) че-
рез миллипоровый фильтр непосредственно перед окраши-
ванием. Время инкубации препаратов с красителем —
10 мин до подсчета ядер.
Этот метод не подходит для поздних бластоцист (разд. 3.6.2).
3.6.2. Окрашивание ДАФИ
1. Окрашивайте ядра в флуоресцентном красителе ДАФН,
растворенном в культуральной среде 100 мкг/мл, в тече-
ние 30 мин при 37 °C [26].
Эксперименты с предимплаитациоииыми эмбрионами мыши 49
2. Диссоциируйте эмбрионы (вплоть до ранней бластоцис-
ты) или клеточные агрегаты в бескальциевой среде или
в 0,9 % -ном цитрате натрия, высушите на воздухе, фик-
сируйте 70%-ным этанолом и подсчитывайте ядра в флу-
оресцентном микроскопе, снабженном соответствующим
источником света и фильтрами.
Для подсчета ядер ТЭ и ВКМ поздних бластоцист выпол-
ните следующие процедуры.
1. Следуйте иммунохирургической методике (как описано в
разд. 3.4) до тех пор, пока клетки ТЭ не начнут разбу-
хать в комплементе.
2. Тщательно промойте бластоцисты в М2 + БСА (табл. 2.5),
инкубируйте в ДАФИ (100 мкг/мл в М2 + БСА в течение
30 мин при 37°C), снова промойте, поместите бластоцис-
ты в маленькую каплю М2 + БСА на обработанное ацето-
ном предметное стекло.
3. Отделите ВКМ от ТЭ оплавленной микропипеткой и пе-
ренесите в каплю 0,9%-ного цитрата натрия.
4. Дайте ТЭ подсохнуть и через 15 мин частично диссоци-
ируйте ВКМ; дайте подсохнуть.
5. Фиксируйте клетки, нанеся пипеткой 70%-ный спирт и
высушите на воздухе.
6. Подсчитайте флуоресцирующие ядра ТЭ и ВКМ, как опи-
сано выше.
В литературе имеются данные о возможности дифференци-
ального подсчета клеток ВКМ и ТЭ в интактной бластоцисте с
использованием полинуклеотидспецифических флуорохромов
[35].
3.6.3. Микроденситометрия ядер, окрашенных по фёльгену
О числе клеток можно судить, подсчитывая ядра, окрашен-
ные по Фёльгену. Содержание ДНК в окрашенных ядрах опре-
деляют на интегрирующем микроденситометре (Vickers М85)
путем сравнения с клеточными ядрами различной плоидност
в высушенных на воздухе препаратах печени мыши [3, 11].
1. Инкубируйте эмбрионы или клеточные агрегаты в тече-
ние 15 мин в среде Хэнкса (Gibco) без Са2+ и Mg2+ при
37 °C, высушите их на воздухе на чистых предметных
стеклах, фиксируйте в смеси этанол : уксусная кислота (3:1
по объему) в течение 5 мин, азатем в течение 1ч — в сме-
си этанол : уксусная кислота: 40%-ный формалин (85:5:
: 10 по объему), высушите на воздухе и храните при
—20 °C.
2. Приготовьте основной краситель Шиффа для реакции
Фельгена следующим образом. К 800 мл кипящей биди-
4—171
50
Глава 2
стиллированной воды прибавьте 4,0 г основного фуксина
и охлаждайте 30 мин. Внесите 12 г метабисульфита ка-
лия, растворенного в 120 мл IM НС1, и оставьте раствор
на ночь. Обесцветьте раствор перемешиванием с активи-
рованным древесным углем в течение часа, профильтруй-
те и храните в темной бутыли при 4 °C.
3. Окрасьте препараты реактивом Шиффа. Сначала погру-
зите в 5М НС1 на 55 мин при 26 °C, затем промойте биди-
стиллированной водой в течение 5 мин и окрасьте свеже-
приготовленным и профильтрованным реактивом Шиффа
2 ч в темноте.
4. Промойте трижды по 10 мин в свежеприготовленной сер-
нистой кислоте (т. е. смеси равных объемов 1%-ного мета-
бисульфита калия и 0,1М НО). Промойте водопроводной
водой.
5. Проведите обезвоживание в спиртах возрастающей кре-
пости и в ксилоле.
6. Заключите препараты в Depex, храните при 4 °C и спус-
тя 2 недели анализируйте их на интегрирующем микро-
денситометре (Vickers М85), измеряя поглощение света
с длиной волны 570 нм. Фиксированные и окрашенные
препараты мышиной печени, анализируемые параллельно
’ во всех экспериментах, дадут контрольные значения 2С и
4С количеств ДНК.
4. Применение меченых лигандов для
изучения организации клеток и эмбрионов
Для изучения организации, состава и функционирования
клеток мышиного эмбриона на разных стадиях развития приме-
няются многочисленные антитела, лектины и другие лиганды,
меченные флуоресцирующим веществом, пероксидазой или ра-
диоизотопами (соответствующие обзоры [36—39]).
4.1. Мечение клеток и морфология клеточной поверхности
Лектины и антитела используются для получения количест-
венных сведений о деталях поверхностной топографии и моле-
кулярной организации эмбриональных мембран [40], но чаще
они выступают в качестве мембранных маркеров, выявляющих
асимметрию в расположении поверхностных микроворсинок на
отдельных бластомерах [12]. У нормально развивающихся
эмбрионов эта поверхностная полярность появляется на 8-кле-
точной стадии развития. Она служит свидетельством глубокой
реорганизации бластомеров и клеточной дифференцировки [2].
В зависимости от возраста эмбрионов, времени их диссоциа-
ции (до или после окрашивания) мы можем получить разную
Эксперименты с предимплантационными эмбрионами мыши 51
информацию. Общие поверхностные маркеры (лектины или ан-
тимышиные антитела), применяемые до 8-клеточной стадии,
дают гомогенное окрашивание всех бластомеров до компакти-
зации и поляризованное — после компактизации независимо ог
того, были эмбрионы диссоциированы перед окрашиванием или
нет. На 16-клеточной стадии развития компактизация эмбрио-
нов обусловливает присутствие красителя лишь в наружных
клетках, поэтому в случае окрашивания интактного эмбриона,
после его диссоциации можно идентифицировать меченые (на-
ружные) и немеченые (внутренние) клетки. Если окрашивание
проведено после диссоциации, выявляются два типа клеток —
полярные и неполярные. Если использовать оба подхода и ок-
расить интактный эмбрион одним флуорохромом, например
изотиоцианатом флуоресцеина (ФИТЦ), а затем, после диссо-
циации, реактивом, связанным с другим флуорохромом, напри-
мер изотиоцианатом тетраметилродамина (ТРИТЦ), полярная
популяция (проспективные клетки ТЭ) будет соответствовать
наружному положению, а неполярная популяция (проспектив-
ная ВКМ) — внутреннему положению в интактном эмбрионе
[2, 25J.
Соответствующая процедура окрашивания описана ниже,
в разд. 4.1.1. Окрашенные эмбрионы или клетки можно изучать
с помощью сканирующей электронной микроскопии (разд. 4.1.2).
При этом получают информацию о клетках наружного или
внутреннего слоев, их фенотипе, количестве. Кроме того, окра-
шивание позволяет рассортировать наружные и внутренние
клетки (разд. 3.2, 3.3, 3.4).
4.1.1. Окрашивание эмбрионоа антителами и лектинами
Окрашивание может быть одноступенчатым или «прямым»
(с использованием меченого антитела и лектина) или двусту-
пенчатым (или «непрямым»). Используются два взаимодейст-
вующих реактива (обычно антитела), из которых первое мече-
ное, а второе немеченое. Интернализацию и другие формы пе-
рераспределения метки можно предотвратить включением
0,02%-ного азида натрия либо в разведения антисыворотки, ли-
бо в промывающую и заключающую среды. Та же цель дости-
гается при фиксации клеток в 3,7 %-ном формалине в PBS
(глутаральдегидная фиксация вызывает автофлуоресценцию
клеток). После фиксации клетки следует тщательно отмыть.
Важно помнить, однако, что после воздействия азидом натрия
и фиксации клетки уже не жизнеспособны.
1. Прямое мечение
а) нефиксированные эмбрионы или клетки инкубируйте при
комнатной температуре 15—30 мин в маленьких каплях
4
52
Глава 2
ФИТЦ- или родамин-конъюгированного лектина или ан-
титела. Нужные разведения антитела (в М2, см. табл.
2.5) необходимо определить экспериментально. Использу-
ется цельная сыворотка и фракции IgG [125]. Конкана-
валин А (КонА) — наиболее распространенный лектин —
применяется в концентрациях 0,5—1,0 мг/мл в М2 + БСА
(рис. 2.2, S);
б) промойте эмбрионы в нескольких каплях среды М2 + БСА
(феноловый красный не следует использовать из-за его
флуоресценции). Клетки и эмбрионы можно наблюдать
прижизненно или фиксировать немедленно после окра-
шивания;
в) для заключения эмбрионов наиболее удобны предметные
стекла, применяемые для типирования (разд. 5). Каждое
стекло поместите в контейнер адекватной глубины и по-
грузите в жидкое парафиновое масло. Расположите
контейнер со стеклом под бинокулярным микроскопом и с
помощью пипетки введите под парафиновое масло в
центр каждого углубления небольшой объем М2 + БСА.
В эти капли поместите небольшие группы меченых эмб-
рионов. Уменьшите объем капли, снимите стекло с мас-
ла, слегка подсушите и закройте покровным стеклом.
Уберите избыток масла и изучайте препарат под флуо-
ресцентным микроскопом, снабженным источником и
фильтрами, соответствующими использованным флуорох-
ромам.
2. Непрямая иммунофлуоресценция
Препарат обработайте первичным немеченым антителом,
продолжайте в соответствии с пп. «а» и «б» (см. выше).
Затем инкубируйте эмбрионы при комнатной температу-
ре 15—20 мин со вторым антителом (т. е. наслоите
ФИТЦ- или родамин-конъюгированное антитело или
IgG, связывающийся с первым антителом), соответст-
венно разбавленным М2. После этого следуют пп. «б»
и «в».
4.1.2. Сканирующая электронная микроскопия
Сканирующая электронная микроскопия (СЭМ) применяет-
ся для изучения морфологии клеточной поверхности (рис. 2.2,
7 и [4]), а также для топографического распределения мем-
бранных детерминант, выявленных иммунохимически [42]. Свя-
зывающиеся с поверхностью лиганды можно комбинировать с
видимым маркером (например, Staphylococcus aureus, который
присоединяется к Fc-части некоторых иммуноглобулинов через
свой компонент-белок А [42, 43].
Эксперименты с предимплантациоиными эмбрионами мыши 33
Основные процедуры СЭМ — те же, что используются при
работе с объектами большого размера; специфические детали,
разработанные для микроскопии эмбрионов или составляющих
их клеток, приведены ниже. В ходе работы объекты приклеива-
ются к покровным стеклам, поэтому нужно принять все меры
предосторожности против пыли и других загрязняющих частиц,
для чего непосредственно перед употреблением следует фильт-
ровать все растворы (Millipore, размер пор 0,22 мкм) [41].
1. У покровных стекол (удобный размер 9X9 мм) пометьте
сторону, несущую эмбрионы. Очистите стекла, погрузив
их в 100%-ный спирт, промакните бумагой без ворсинок
и высушивайте на воздухе не менее 1 ч. Погрузите чис-
тые покровные стекла в свежеприготовленный и профиль-
трованный раствор поли-Ь-лизииа (PLL, Sigma, тип 1b,
1 мг/мл в бидистиллированной воде) примерно на 1 ч.
Непосредственно перед употреблением дважды или триж-
ды сполосните покровные стекла в 0,1 М какодилатном
буфере pH 7,4 и поместите их в индивидуальные лунки
пластиковых культуральных плат (Nunc), содержащие
0,1 М какодилатный буфер pH 7,4.
2. Фиксируйте эмбрионы с удаленной блестящей оболочкой
или бластомеры в 6%-ном глутаральдегиде (Sigma) на
0,1 М какодилатном буфере с pH 7,4 в течение 1 ч при
комнатной температуре.
3. Поместите эмбрионы в 1%-ный глутаральдегид на 0,1 М
какодилатном буфере pH 7,4 и перенесите их с помощью
пипетки в центр приготовленного покровного стекла. Пла-
ты с лунками хорошо устанавливаются на предметном
столике бинокулярного микроскопа, и если сделать изог-
нутую пипетку, всю процедуру можно проводить под
контролем микроскопа. Применение 1%-ного глутаральде-
гида способствует плотному прикреплению клеток к по-
верхности покровного стекла, обработанной поли-Ь-лизи-
ном. Не прикасайтесь к клеткам концом пипетки и не да-
вайте им прикасаться друг к другу, иначе будет невоз-
можно отделить их, не повредив.
4. Немедленно обезводьте препараты проведением через
спирты возрастающей крепости, профильтрованные через
миллипоровые фильтры непосредственно перед употреб-
лением (по 30 мин в 20-, 40-, 60%-ном спиртах, на ночь в
70 %-ном спирте при 4 °C, затем по 30 мин в 70-, 90-, 95 % -
ном и две экспозиции до высыхания в 100%-ном Analar-
этаноле).
5. Высушивание до критической точки, напыление золотом и
заключение для рассматривания — как обычно [42].
54
Глава 2
4.1.3. Замораживание — скалывание
Эта методика, используемая в сочетании с агентами, кото-
рые связываются со специфическими компонентами мембраны
и вызывают идентифицируемые нарушения в мембранных реп-
ликах (например, связывающийся со стеролом антибиотик фи-
липин [32]), позволяет изучать топографию мембраны с раз-
решением, примерно в 10 раз превышающим разрешение СЭМ
[32, 44]. Проводка материала не отличается от обычной [45].
Разница состоит только в том, что малые размеры
эмбрионов вынуждают заключить их в специально сконструиро-
ванные держатели [44] или матрикс (например, желатину
[32]). В удалении блестящей оболочки нет необходимости.
4.2. Мечение внутриклеточных компонентов
4.2.1. Иммуноцитохимия на светооптичаском уровне
Мышиными эмбрионами ранних стадий и составляющими
их клетками наиболее легко манипулировать при фиксации и
иммуноцитологическом окрашивании в том случае, когда они
прикреплены к покровным стеклам. Методика, первоначально
разработанная для хромосом типа ламповых щеток, была мо-
дифицирована для выявления клеточных, органоидов и цитоске-
лета [46]. Она предусматривает использование антител к ак-
тину [46], тубулину [41], центрам сборки микротрубочек
[47], пластинам ядерного матрикса [48], клатрину и лизосо-
мам [48].
В случае необходимости соотнести внутриклеточную органи-
зацию с поверхностной полярностью, эмбрионы с удаленной
блестящей оболочкой или клетки метят ФИТЦ- или родамин-
КонА и затем отмывают (разд. 4.1.1) перед иммуноцнтологиче-
ским окрашиванием.
Для фиксации, экстрагирования и окрашивания эмбрионов
или клеток сконструированы специальные стеклянные камеры,
состоящие из стеклянного промывателя (толщиной 1,5 мм и
наружным диаметром 25 мм), к которому парафиновым воском
прикрепляется покровное стекло [46]. Цитохимическая про-
цедура заключается в следующем.
1. На покровное стекло нанесите при комнатной температу-
ре на 5 мин 0,1 мг/мл КонА или, если клетки уже окра-
шены КонА для определения поверхностной полярнос-
ти,— ФГА в разведении 1:20 (Gibco).
2. Сполосните покровное стекло М2 (см. табл. 2.5)+4 мг/мл
поливинилпирролидона (ПВП) и наполните камеру
смесью М2 + ПВП.
Эксперименты с предимплантационными эмбрионами мыши 55
3. Пипеткой нанесите на покровное стекло клетки, они
должны сразу прочно прикрепиться.
4. Закройте камеры другим покровным стеклом и поставь-
те в 50-мл поликарбонатную центрифужную пробирку с
отверстием в основании и Регрех-поршнем в нижней
трети. В пробирку можно поместить до 4 камер, переме-
жающихся 25-мм миллипоровыми префильтрами.
5. Приклейте клетки к покровным стеклам, центрифугируя
их при 500g в течение 10 мин при 20 °C в НВ-4 роторе
центрифуги Sorvall RC-5.
6. После центрифугирования через отверстие в основании
пробирки введите стержень и осторожно выталкивайте
поршень, пока не появятся камеры, которые можно из-
влечь.
7. Снимите верхнее покровное стекло, служащее крышкой.
Фиксируйте клетки 3,7%-ным формалином в PBS в те-
чение 35—40 мин при 20°C, промойте 4 раза в PBS,
экстрагируйте 0,25%-ным Тритоном Х-100 в PBS в те-
чение 10 мин при 20 °C и, наконец, дважды промойте в
PBS. Эта процедура обеспечивает наилучшую сохран-
ность клеток и проникновение антител и, кроме того,
сводит до минимума перестройку клеточных белков в
процессе экстрагирования фиксированных клеток [46].
Однако последовательность фиксации и пермеабилиза-
ции нужно устанавливать для каждого детерминанта от-
дельно. Например, при окрашивании центра сборки мик-
ротрубочек необходимо сначала провести экстрагирова-
ние, а затем фиксацию для уменьшения цитоплазматиче-
ского фона [47].
8. Выньте покровные стекла из их камер и перенесите в
PBS с 0,1%-ным твином-20 (PBS-твин) до инкубирова-
ния их со специфическим антителом или другим реаген-
том.
9. Инкубируйте экстрагированные клетки при комнатной
температуре около 30 мин в соответствующем разведе-
нии специфических антител в PBS-твине + З мг/мл БСА,
вторыми (мечеными) антителами, разведенными в PBS-
дважды промойте в PBS-твине и окрашивайте 20 мин
твине + 3 мг/мл БСА.
10. Снова промойте клетки в PBS-твине, удалите покровные
стекла из камер, заключите в Cittifluor (City Univer-
sity London), снижающий падение интенсивности метки,
и просматривайте в- флуоресцентном микроскопе. При
применении в качестве второго слоя антител, меченных
пероксидазой, пероксидазная активность выявляется ме*
тодом Грэма и Карновского [49].
56
Глава 2
4.2.2. Изучение внутриклеточного строения
с помощью просвечивающей электронной микроскопии
Приготовление препаратов для просвечивающей электрон-
ной микроскопии (ПЭМ) — многоэтапный процесс. Все манипу-
ляции в этом случае должны проводиться под контролем мик-
роскопа с использованием стеклянных блоков с лунками и стек-
лянных пипеток (если нужно, изогнутых). Для заключения в
смолу низкой вязкости (Spurr) следует применять ВЕЕМ-кап-
сулы (Polaron Equipment Ltd, Watford, Herts).
1. Фиксируйте эмбрионы или бластомеры в течение 15—
20 мин при комнатной температуре в свежеприготовлен-
ном 3%-ном растворе глутаральдегида в 0,1 М какоди-
лате натрия (pH 7,3). Дважды промойте в какодилатном
буфере.
2. Проведите постфиксацию в 1%-ном ОэОд в 0,1 М како-
дилате натрия (pH 7,3) в течение 30 мин при комнатной
температуре. Поскольку пары тетроксида осмия очень
токсичны, эту манипуляцию с эмбрионами лучше прово-
дить в маленьких каплях OsCh под маслом в пластико-
вых чашках Петри (в вытяжном шкафу).
3. Промойте эмбрионы или бластомеры в бидистиллирован-
ной воде, окрасые их в насыщенном водном растворе
уранилацетата в течение 30 мин при комнатной темпе-
ратуре.
4. Обезводьте проводкой через серию спиртов возрастаю-
щей крепости. По достижении 70 %-ного спирта эмбрионы
начинают прилипать к стеклянным поверхностям, поэтому
целесообразнее менять спирт, чем переносить эмбрионы.
5. В качестве заключающей среды наиболее пригодна смо-
ла Spurr ввиду ее низкой вязкости. Поскольку некото-
рые компоненты смолы лабильны, для каждой партии
сделайте пробную полимеризацию. Приготовьте смолу
Spurr согласно инструкции изготовителя. Храните при
—20 °C не более двух недель, перед использованием дай-
те нагреться до комнатной температуры.
6. Инкубируйте эмбрионы в смеси Spurr: 100%-ный спирт
1 : 1 (по объему) 1—2 ч при комнатной температуре, за-
тем в смеси, состоящей из тех же компонентов, но взя-
тых в соотношении 2 : 1 (по объему) 1—2 ч при комнат-
ной температуре. Чтобы расположить несколько эмбри-
онов в центре плоского основания ВЕЕМ-капсулы, вос-
пользуйтесь изогнутой пипеткой, наполните капсулы
Spurr и полимеризуйте при 60 °C 16 ч.
7. Сделайте ультратонкие срезы и поместите их на медную
сеточку (предпочтительнее использовать сетки Gilderg-
rids 200 HS).
Эксперименты с предимплаитациоиными эмбрионами мыши 57
8. Приготовьте красящие растворы:
а) спиртовой уранилацетат: насыщенный раствор в
50 %-ном этаноле, храните в темной посуде;
б) цитрат свинца: добавьте 2,33 г нитрата свинца и
1,76 г цитрата натрия к 30 мл бидистиллированной
воды без СОг в 50-мл мерной колбе. Постоянно
встряхивайте в течение 1 мин и время от времени
в течение 30 мин. Прибавьте 8 мл 1 М NaOH (Апа-
lar-раствор без хлопьев).
Долейте бидистиллированной воды до 50 мл.
9. Поместите нужное число капель (одна на сеточку) спир-
тового уранил-ацетата, профильтрованного через милли-
поровый фильтр, на розовый зубной воск в пластиковой
чашке Петри. На каждую каплю положите по одной
сеточке срезами вниз. Закройте крышкой и оставьте на
10—15 мин.
10. Сеточки извлекайте по очереди и смывайте краску сте-
кающими каплями бидистиллированной воды без СОг.
Высушите сеточки на плотной фильтровальной бумаге.
11. Центрифугируйте раствор цитрата свинца в течение
10 мин при 5000 об/мин. Нанесите капли чистого супер-
натанта на зубной воск в чашке Петри. По краю помести-
те несколько шариков NaOH и на каждую каплю поло-
жите по сеточке срезом вниз. Над каплями не дышите,
т. к. СО2 вызовет осаждение свинца. Окрашивайте 5 мин.
Смойте краситель бидистиллированной водой без СОг и
высушите.
12. Смотрите в просвечивающем электронном микроскопе.
4.3. Маркеры клеточкой линии
Идеальный маркер клеточной линии должен обладать ря-
дом особенностей: локализоваться во всех клетках данной ли-
нии, сохранять клеточную автономность, быть стабильным, лег-
ко выявляемым и нейтральным. В последнее время найдено
несколько маркеров, удовлетворяющих этим требованиям [50].
Опишем два приема мечения, которые с успехом используются
при работе с мышью (см. гл. 6). Изолированные бластомеры
или интактные эмбрионы инкубируют в среде с ФИТЦ
(0,5 мг/мл) в негазованной среде Мб, pH 7,7, содержащей
4 мг/мл ПВП, в течение 10 мин при комнатной температуре,
а затем промывают в М2 + БСА и культивируют в М16+БСА
[51]. Метящиеся клетки должны находиться в суспензии, ина-
че они прилипают к чашке и подвергаются лизису. Важно пом-
нить также, что клетки, выбранные для мечения, должны быть
отделены от соседей, вот почему важно подвергнуть их диссо-
58
Глава 2
циации. Идеальный маркер — это маркер, который был бы спо-
собен пометить всю клеточную популяцию в интактном эмбрио-
не без нарушения его целостности.
Этому требованию, по-видимому, отвечает монодисперсная
суспензия микрочастиц карбоксилированного латекса, покрытых
флуоресцеином (желто-зеленый Fluoresbrite; частицы 0,2 мкм
диаметром) [13]. Метка легко включается в дробящиеся блас-
томеры путем эндоцитоза, она четко выявляется на прижизнен-
ных и фиксированных препаратах, на срезах и не оказывает
видимого влияния на развитие. Более того, эти частицы могут
иметь различные размеры и могут быть покрыты другими
флуорохромами. Существует возможность пометить выделенные
бластомеры и проследить их судьбу после реагрегации с неме-
чеными клетками, можно произвести мечение наружных клеток
интактного эмбриона, начиная с 16-клеточной стадии.
Процедура мечения эмбрионов флуоресцирующими микро-
частицами заключается в следующем.
1. Эмбрионы, освобожденные от блестящей оболочки, или
клетки инкубируйте в разведении 1 :50 маточной суспен-
зии микрочастиц в М2 + БСА (табл. 2.5) в течение 5—
15 мин при 37 °C, промойте их переносом через 2—3 кап-
ли М2 + БСА, после чего их можно анализировать немед-
ленно или агрегировать с другими немечеными бластоме-
рами (см. разд. 3.3) и продолжать культивирование. При
необходимости произведите диссоциацию, как описано в
разд. 3.2.2.
2. Внутриклеточное мечение микрочастицами латекса, по-
крытыми флуоресцеином, можно проводить до или после
поверхностного мечения родамин-конъюгированным КонА
(Polysciences). Инкубирование эмбрионов или клеток в
ТРИТЦ-КонА при 1 мг/мл в М2 + БСА позволит иденти-
фицировать наружные клетки эмбриона со снятой обо-
лочкой (1 мин для 16-клеточного эмбриона, 5 мин для
бластоцисты при 37 °C) или определить полярность изо-
лированных клеток (5 мин, 37°C). После инкубации про-
мойте эмбрионы или клетки, сменив 2—3 капли М2 +
+ БСА.
3. Для анализа флуоресценции клетки или эмбрионы фик-
сируйте в 4%-ном формалине в PBS 10 мин, затем про-
мойте в М2 + БСА и заключите их в этой среде в лунках
стекла для тканетипирования (разд. 4.1.1). Для изучения
препаратов используйте флуоресцентный фотомикроскоп
и фотопленку Kodak Tri-X 35 мм.
4. Положение и судьбу меченных флуоресцеинлатексом
клеток в бластоцисте определите тремя способами:
а) анализируйте весь эмбрион целиком;
Эксперименты с предимплантационными эмбрионами мыши 59
б) поверхность интактного эмбриона пометьте ТРИТЦ-КонА
(в качестве маркера трофэктодермы), диссоциируйте
бластоцисту (см. разд. 3.2.2), фиксируйте клетки (4%-
ным формалином 10 мин) и выясните паттерн мечения
клеток ФИТЦ и (или) ТРИТЦ;
в) сделайте ультратонкие срезы для анализа в ПЭМ JB-4
(Polysciance), оптимальная заключающая среда — водо-
растворимая смола (детали соответствующей методики
см. в работе [13J).
5. Реактивы, среды и оборудование
5.1. Реактивы
Используйте лучшие из имеющихся у вас реактивов. Поль-
зуйтесь набором шпателей только для реактивов тканевой
культуры, следите за тем, чтобы после каждого употребления
они были тщательно вымыты.
В табл. 2.4 приводится список реактивов, которые мы можем
рекомендовать на основании опыта нашей лаборатории.
5.2. Среды
В табл. 2.5 описаны те среды, которые мы с успехом ис-
пользуем в своей работе. При извлечении эмбрионов и переносе
их из инкубатора применяем среду М2 с буфером HEPES
(pH 7.4 при 37 °C) [52]. Для культивирования наиболее эф-
фективна забуференная СО2 среда М16 [53]. Обе среды моди-
фицированы в соответствии с указаниями Гудэла и Маро [54]
и получены из исходных растворов, приведенных в табл. 2.5.
Осмолярность М2 и М16 находится в пределах 285—
290 мОсмоль. Для культивирования бластоцист используем
DMEM+10% ЭТО.
5.3. Оборудование
5.3.1. Стеклянная посуда
Для приготовления сред необходимо иметь специальный на-
бор стеклянной посуды, который сразу же после использования
споласкивается бидистиллированной водой, моется только в би-
дистиллированной воде и высушивается в сушильном шкафу.
5.3.2. Микроскопы
Важно, чтобы для манипуляций с эмбрионами и их культи-
вирования была отведена специальная комната. Рекомендуется
кондиционер воздуха для поддержания нормальной комнатной
Таблица 2.6. Возможные затруднения при экспериментировании
Вероятные причины
Способы устранения
Проблема: суперовуляция не удается или яйца дегенерируют в яйцеводе
1. Самки слишком стары Возьмите более молодых мышей
(3—4-иед)
2. Гормоны некачественные Сделайте свежие растворы
Купите новые препараты гормонов
Проблема: после спаривания яйца остаются неоплодотворенными
3. Самцы неплодовиты
4. Овуляция и спаривание не син-
хронизированы
Замените самцов-производителей
Проконтролируйте длительность све-
тового дня в виварии. (Спаривание
происходит в середине темного пе-
риода, обычно длящегося с 19.00
до 7.00.) Используйте мышей в со-
стоянии проэструса (3,5—4-иед).
Не инъецируйте чХГ ранее 14.00
Проблема: яйца или эмбрионы лизируют вскоре после их помещения в среду
5. Осмолярность неправильна (эм-
брионы разбухают или сморщи-
ваются)
6. Грязные стекла с лунками
7. Стерилизующие фильтры, шпри-
цы и другая пластиковая посуда,
использованная для приготовле-
ния сред, токсична
8. Среды и (или) масло токсичны
9. Летучие растворители
Проверьте осмолярность и, если нуж-
но, приготовьте свежие среды
Попробуйте пластиковые
Инкубируйте среды в контакте с со-
ответствующим материалом и за-
тем проверьте жизнеспособность
эмбрионов
Проверьте БСА и масло (см. [14])
Проверьте, ие пользовались ли не-
давно краской или растворителями
в помещении
Манипуляции с эмбрионами проведи-
те в другом месте и проверьте их
жизнеспособность
Проблема: яйца или эмбрионы лизируют вскоре после снятия блестящей
оболочки
10. Эмбрионы слишком долго под-
вергали воздействию растворов,
удаляющих оболочку (особенно
кислым раствором Тироде)
11. Растворы токсичны
Ускорьте процедуру. Используйте
меньшую партию эмбрионов. Убе-
дитесь в том, что кислый Тироде
применяется при 37 °C. Быстро
промойте эмбрионы М2+БСА и
вместо кислого Тироде возьмите
проназу (или наоборот)
Проверьте осмолярность
Убедитесь в том, что pH установлена
при помощи Апа1аг-НС1.
Растворы хранились слишком долго.
Не замораживайте кислый Тироде.
Выбросьте, и приготовьте новые.
Замените запас химикатов
Эксперименты с предимплантационными эмбрионами мыши
61
Продолжение табл. 2.6.
Вероятные причины
Способы устранения
Проблема: эмбрионы прекращают развитие на 2-клеточной стадии
12. Использована «блокирующаяся»
линия, эмбрионы извлечены нз
яйцевода слишком райо
13. Колебания температуры и (или)
pH. Проверьте пп. 5—9
Замените линию мышей. Не извле-
кайте эмбрионы раньше 32 ч после-
инъекции чХГ или позже
Проверьте состав и температуру всех
сред. Используйте камеры и на-
гревательные столики у микроско-
пов
Проблема: эмбрионы останавливаются в развитии при культивировании на
более поздних стадиях (часто 16-клеточная стадия) после нормального или
замедленного развития (эмбрионы без блестящей оболочки или диссоцииро-
ванные клетки особенно чувствительны к токсическим компонентам) [55]
Проверьте пп. 5—9 и 13
14. Среды токсичны
15. Пластиковые культуральные чаш-
ки загрязнены. См. также пп. 6
и 7
16. Неясные помехи по-прежиему
остаются
Проверьте БСА. Проанализируйте на.
токсичность разные партии сыво-
ротки и ее источники. Проверьте
масло иа токсичные примеси:
а) экстрагируйте масло средой в те-
чение ночи при 37 °C в инкуба-
торе с СО2. Сравните жизнеспо-
собность эмбрионов в среде под.
экстрагированным и неэкстрагиро-
ванным маслом;
б) сравните жизнеспособность эм-
брионов в каплях объемом 5—
10 и 50 мкл. Если масло имеет
примеси, в маленьких каплях эм-
брионы будут дегенерировать
быстрее. Проверьте другие партии
масла на 8-клеточных эмбрионах
без оболочки в маленьких каплях
среды и купите удачную партию
про запас. Смените реактивы для
сред, кислого Тироде и раствора
проназы
Проверьте другие упаковки и купите-
удачные про запас
Сравните эмбрионы и реактивы с та-
ковыми из другой лаборатории
температуры. Необходимы бинокулярные препаровальные мик-
роскопы (модель М5 Wild Heerbrugg) с увеличениями примерно
от Хб до Х50. Их следует держать в свободном от пыли боксе
(Регрех), желательно, чтобы они были снабжены нагреватель-
ными столиками и набором нагревающихся камер до 36±1°С.
62
Глава 2
5.3.3. Инкубаторы
Необходим инкубатор на 37 °C, содержащий 5% СО2 в воз-
духе. Система контроля концентрации СО2 может либо входить
в конструкцию инкубатора, либо придаваться к нему. Если со-
держание СО2 проконтролировать нельзя, культуральные чаш-
ки с эмбрионами помещают в герметически закрытые сосуды,
содержащие 5% СО2 в воздухе. Рекомендуется иметь отдельные
инкубаторы для длительного и краткосрочного инкубирования.
Последние следует использовать в тех случаях, когда в ходе
эксперимента необходимо часто анализировать культуры.
5.3.4. Ламинарный бокс
Все среды следует готовить в специальном ламинарном бок-
се (Slee Model HLF/H, поставщик: South London Electrical
Equipment Ltd.), который не должен находиться в одном поме-
щении с микроскопами и инкубаторами.
5.3.5. Чашки для манипуляций и культивирования
Для сбора эмбрионов из больших объемов жидкости реко-
мендуется пользоваться стеклянными чашками с углублением,
Hoslabs. Эмбрионы или клетки будут скатываться в это углуб-
ление.
Для стандартной культуры используются упакованные сте-
рильные пластиковые чашки Петри (Falcon, Becton Dickinson
and Co.) трубки и пипетки. Если требуется нелипкая поверх-
ность, рекомендуется пользоваться чашками Петри (поставщик
пластиковой посуды: Hawfell).
5.3.6. Стеклянные пипетки
Состав стекла и его толщина, диаметр продажных пастеров-
ских пипеток настолько варьируют, что неизбежно вызывают
затруднения при вытягивании микропипеток в пламени горел-
ки. По нашему мнению, наиболее подходящими являются пи-
петки «Volac» (Fisons Scientific). Ими пользуются для обыч-
ного переноса эмбрионов.
Чтобы изготовить микропипетки для манипуляций с диссо-
циированными эмбрионами и единичными клетками, необхо-
димы стеклянные капилляры точного диаметра (GC 100—15:
Clark Electromedical Instruments).
Микропипетки вытягивают и оплавливают в микрокузнице
(Microforge de Fonbrune Establissements Beaudoin). Воспользо-
вавшись пособиями для изготовления пипеток (AR Horwell,
Кат. № 7091 00 и 7091 10), их можно сделать самим.
Эксперименты с предимплантационными эмбрионами мыши 63
5.3.7. Иглы для вскрытия яйцеводов
Используют металлические иглы 30-го калибра (Holborn
Surgical Instruments Со. Ltd.), у которых скошенные концы
плоско срезаны и отполированы.
5.3.8. Стекла для типирования тканей
(Изготовитель: Baird and Tatlock). Кат. № 403/0522 Тип
Ji. Покровные стекла 70X46 мм, Кат. № 403/0523.
Благодарности
Большинство описанных в этой главе методических приемов
было разработано в течение последних 10 лет в лаборатории
Мартина Джонсона (Department of Anatomy, Cambridge). Мне
хотелось бы выразить благодарность всем его сотрудникам.
Особая признательность мисс Джин Флэч за методические и
[ организационные советы, докторам Мартину Джонсону и Тому
! Флемингу за полезные критические замечания, Шейле Эгго за
перепечатку рукописи. Работа субсидировалась MRC, CRC и
ЕМВО.
Литература
1. Johnson М. Н., McConnell Van Blerkom J. (1984). J. Embryol. Exp.
Morphol., 83, SuppL 1—6, 197.
2. Johnson M. H., Chisholm J. C., Fleming T. P., Houliston E. (1986). J.
Embryol. Exp. Morphol., 97, Suppl. 97.
3. Smith R. K. W., Johnson M. H. (1986). J. Reprod. Fertil., 76, 393.
4. Brinster R. L. (1971). In: Biology of the Blastocyst., Blandau R. J. (ed ).
University of Chicago Press, p. 303.
5. Biggers J. D., Whitten W. K., Whittingham D. G. (1971). In: Methods in
Mammalian Embryology. Daniel J. C. (ed.), W. H. Freeman and Co., San
Francisco, p. 86.
6. Gates A. H. (1971). In: Methods in Mammalian Embryology. Daniel J. C.
(ed.), W. H. Freeman and Co., San Francisco, p. 64.
7. Pratt H. P. M., Bolton V. N., Gudgeon K. A. (1983). Molecular Biology of
Egg Maturation. CIBA Foundation Symposium No. 98. Pitman Books, Lon-
don, p. 97.
8. Howlett S. K-, Bolton V. N. (1985). J. Embryol. Exp. Morphol., 87, 175.
9. Kaufman M. H. (1983). Early Mammalian Development: Parthenogenetic
Studies. Cambridge University Press.
10. Webb M., Howlett S. К., Maro B. (1986). J. Embryol. Exp. Morphol., 95,
131.
11. Bolton V. N., Cades P., Johnson M. H. (1984). J. Embryol. Exp. Morphol.,
12. Johnson M. H„ Ziomek C. A. (1981). J. Cell Biol., 91, 303.
13. Fleming T. P., George M. A. (1987). Wilhelm Roux’s Arch., 196, 1—11.
14. Chisholm J. C., Johnson M. H., Warren P. D., Fleming T. P„ Pickering S. J-
(1985). J. Embryol. Exp. Morphol., 86, 311.
64
Глава 2
15. MacQueen Н. A., Johnson М. Н. (1983). J. Embryol. Exp. Morphol., 77, 297.
16. Graham C. F., Lehtonen E. (1979). J. Embryol. Exp. Morphol., 49, 277.
17. Biggers J. D. (1971), In: Biology of the Blastocyst. Blandau R. J. (ed.),
University of Chicago Press, p. 319.
18. Goddard M. J., Pratt H. P. M. (1983). J. Embryol. Exp. Morphol., 73, 111.
19. Kaufman Al. H., Sachs L. (1976). J. Embryol. Exp. Morphol., 35, 179.
20. Kaufman M. H., Sachs L. (1975). J. Embryol. Exp. Morphol., 34, 645.
21. Whitten W. K., Biggers J. D. (1968). J. Reprod. Fertil., 17, 399.
22. Nicolson G. L., Yanagimachi R., Yanagimachi H. (1975). J. Cell Biol., 66,
263.
23. Mintz B. (1971). In: Methods in Mammalian Embryology, Daniel J. C. (ed.),
W. H. Freeman and Co., San Francisco, p. 186.
24. Sobel J. S. (1984). J. Cell Biol., 99, 1145.
25. Handyside A. H. (1980). J. Embryol. Exp. Morphol., 60, 99.
26. Johnson M. H., Ziomek C. A. (1983). Dev. Biol., 95, 211.
27. Solter D„ Knowles В. B. (1975). Proc. Natl. Acad. Sci. USA, 72, 5099.
28. Snow M. H. L. (1978). In: Methods in Mammalian Reproduction. Daniel J. C.
(ed.), Academic Press, New York, p. 167.
29. Nichols J., Gardner R. L. (1984), J. Embryol. Exp. Morphol., 80, 225.
30. Cohen A., Schlesinger M. (1970). Transplantation, 10, 130.
31. Gardner R. L. (1978). In: Methods in Mammalian Reproduction. Daniel J. C.
(ed.), Academic Press, New York, p. 137.
32. Pratt H, P. M. (1985). J. Embryol. Exp. Morphol., 90, 101.
33. Hsu Y.-C. (1978). In: Methods in Mammalian Reproduction. Daniel J. C.
(ed.), Academic Press, New York, p. 229.
34. Tarkowski A. K. (1966). Cytogenetics, 5, 394.
35. Handyside A. H., Hunter S. (1984). J. Exp. Zool., 231, 429.
36. Johnson M. H. (1981). Int. Rev. Cytol. Suppl., 12, 1.
37. Jacob F. (1979). Curr. Top. Dev. Biol., 13, 117.
38. Solter D„ Knowles В. B. (1979). Curr. Top. Dev. Biol., 13, 139.
39. Wiley L. M. (1979). Curr. Top. Dev. Biol, 13, 167.
40. Wolf D. E. (1983). In: Development in Mammals. Johnson M. H. (ed.),
Elsevier Amsterdam, Vol. 5, p. 187.
41. Maro B., Pickering S. (1984). J. Embryol. Exp. Morphol,, 84, 217.
42. Johnson M. H., Ziomek C. A. (1982). Dev. Biol., 91, 431.
43. Surolia A., Pain D., Khan M. J. (1982). Trends Biochem. Sci., 7, 74.
44. Magnuson T-, Detnsey A„ Stackpole C. W. (1977). Dev. BioL, 61, 252.
45. Navaratnam V., Thurley K. W., Skepper J. N. (1982). In: Progress in Anato-
my, Harrison R. J. and Navaratnam V. (eds.), Cambridge University Press,
Vol. 2, p. 201.
46. Maro B., Johnson M. H., Pickering S. J., Flach G. (1984). J. Embryol. Exp.
Morphol., 81, 211.
47. Maro B., Johnson M. H., Webb M., Flach G. (1986). J. Embryol. Exp. Mor-
phol, 92, 11.
48. Johnson M. H., Maro B. (1985). J. Embryol. Exp. Morphol, 90, 311.
49. Graham R. C„ Karnovsky M. J. (1966). J. Histochem. Cytochem, 14, 291.
50. West J. D. (1984), In: Chimeras in Developmental Biology. Le Douarin N.
and McLaren A. (eds.), Academic Press, New York, p. 39.
51. Ziomek C. A. (1982). Wilhelm Roux’s Arch., 191, 37.
52. Fulton В. P., Whittingham D. G. (1978). Nature, 273, 149.
53. Whittingham D. G. (1971). J. Reprod. Fertil. Suppl., 14, 7.
54. Goodall H„ Maro B. (1986). J. Cell Biol, 102, 568.
55. Fleming T. P., Pratt H. P. M„ Braude P. R. (1987). Fertil Steril, 47, 858—
860.
ГЛАВА 3
ЭКСПЕРИМЕНТЫ С ПОСТИМПЛАНТАЦИОННЫМИ
ЭМБРИОНАМИ МЫШИ
Роза Бэддингтон*
1. Введение
Дифференцировка и формирование плода у млекопитающих
происходят только после того, как эмбрион имплантируется в
матку. С процессом имплантации связано возникновение кле-
точных линий при сегрегации трех популяций клеток-предшест-
венников: одна даст трофобласт, другая — внезародышевую
энтодерму и третья, эпибласт (эмбриональная эктодерма), даст
начало всему плоду и внезародышевой мезодерме [1]. Все экс-
перименты с предимплантационными эмбрионами, ставящие
целью получить химерный зародыш — даже введение в бласто-
цисту единичной меченой клетки эпибласта, — приводят к то-
му, что химерными оказываются все ткани плода. И только в
процессе или после дифференцировки эпибласта эксперимен-
тальное вмешательство избирательно изменяет или выделяет
ту или иную ткань плода [2]. Отсюда следует, что для изуче-
ния развития частей плода нужно работать с постимплантаци-
онным эмбрионом.
До сих пор не разработаны сколько-нибудь точные методы
для манипуляций с эмбрионом in utero, поэтому эксперимен-
тальная работа ведется главным образом in vitro или на экс-
плантатах in vivo. Ни тот, ни другой способы не обеспечивают
оптимальных условий для нормально идущей дифференцировки
пли морфогенеза. В этой главе будут описаны наиболее часто
применяемые методики выделения, трансплантации и культи-
вирования, которыми пользуются для изучения происхождения
п развития зачатков раннего плода. Особый акцент будет сде-
лан на современной технике изучения дифференцировки клеток
в их нормальном окружении в интактном эмбрионе.
2. Анатомия гаструляции и раннего эмбриогенеза
Бластоциста начинает имплантироваться в стенку маткина
5-й день беременности. На 6-й день внутренняя клеточная мас-
са (ВКМ) врастает в бластоцель, а над ней сосредоточивается
1 R. Beddington. ICRF Developmental Biology Unit, Department of Zoology
South Parks, Oxford Oxi 3RE, UK.
5—171
66
Глава 3
Эксперименты с постимплантационными эмбрионами мыши 67
внезародышевая эктодерма, в результате эмбрион приобретает
цилиндрическую форму с четкой границей между зародышевой
и внезародышевой областями (рис. 3.1,Л). В эпибласте образу-
ется проамниотическая полость, которая, расширяясь, вдается
во внезародышевую область. Зародышевая область на этой
стадии представлена двумя эпителиальными слоями: висцераль-
ной энтодермой снаружи и эпибластом, граничащим с проам-
ниотической полостью, — внутри (рис. 3.1,Б).
Гаструляция начинается на 7-й день с появления первичной
полоски (рис. 3.1,В). Первичная полоска располагается по
средней линии зародыша. Она определяет передне-заднюю ось
плода. В месте первичной полоски происходит инвагинация
эпибласта, в результате чего образуется третий зародышевый
слой — рыхло лежащие клетки мезодермы. Мезодерма распро-
страняется между эпибластом и висцеральной энтодермой лате-
рально и вперед, формируя в зародышевой области непрерыв-
ный промежуточный слой. Клетки, происходящие из передней
части первичной полоски и располагающиеся по средней линии
переднего отдела цилиндра, приобретают эпителиеподобный
вид. Этот квазиэпителий, выявляющийся на 8-й день, называет-
ся головным отростком (рис. 3.1,Г). Вероятно, именно он слу-
жит предшественником нотохорда, прехордальной пластинки,
участвует в формировании наружного слоя висцеральной энто-
дермы, а возможно, и в образовании кишечной энтодермы [3].
Мезодерма задней части полоски растет во внезародышевую
область, при этом места соединения эпибласта и внезародыше-
вой эктодермы сдвигаются, образуя амниотические складки
(одна из них, задняя, выражена в наибольшей степени)
(рис. 3.1,В). Складки выпячиваются в проамниотическую по-
лость и затем сливаются. Тем временем во внезародышевой ме-
зодерме образуются лакуны, которые, сливаясь, формируют
следующую полость — экзоцелом, выстланный мезодермой и
Рис. 3.1. Схематическое изображение стадий развития мышиного эмбриона
с 6-го по 10-й день беременности. Возраст эмбриона измеряется числом дней,
прошедших после оплодотворении, и представлен следующими стадиями (см.
табл. 3.2): Л. Стадии 2. Б. Стадии 3. В. Стадии 5. Г. Стадии 7. Д. Стадия 10.
Е. Стадии 13.
Условные обозначении: Эп — эпибласт; Эвя — виезародышеэаи эктодерма;
ГК — гигантские клетки трофобласта; ВЭивя— висцеральная внезародышевая
энтодерма; ВЭнэм—висцеральная эмбриональная энтодерма; ПЭн — париеи-
тальнаи энтодерма; ОР — оболочка Рейхерта; АС — задний амииотическаи
складка; М — мезодерма; ПП — первичная полоска; ЭПК — эктоплацеитар-
ный конус; X — хорион; ЭЦ — экзоцелом; ЖП— желточная полость; АП —
амниотическая полость; ГО — голоиной отросток; Ам — амнион; НЭ — иейро-
эктодерма; К — кишка; С — сердце; Но — нотохорд; ГС — головная складка;
Со — сомит; ПБА — пупочно-брыжеечная артерии; Ал — аллантоис; ВЖМ—
висцеральный желточный мешок; ППК — почка передней конечности; СК —
слуховая капсула; ЗЯ — зрительная имка; ПЭ — поверхностиаи эктодерма.
68
Глава 3
отделяющий внезародышевую эктодерму от эпибласта (рис.
3.1, Г). Таким образом, яйцевой цилиндр подразделяется на
три четко различающиеся камеры, отделенные одна от другой
будущими амнионом и хорионом (рис. 3.1,7").
Гаструляция (в смысле продолжающейся миграции клеток
через первичную полоску) идет по 10-й день беременности
включительно, но первые признаки органогенеза появляются
уже к концу 8-го дня. Не останавливаясь на подробном описа-
нии органогенеза, сделаем краткий обзор формирования основ-
ных зачатков. Детальное описание дифференцировки различных
систем органов можно найти в других источниках [4, 5].
Эктодерма, расположенная перед первичной полоской, обра-
зует нервную пластинку, несколько выгнутую по средней линии.
Это углубление, называемое нервным желобком, становится бо-
лее выраженным при формировании на 9-й день нервных вали-
ков (рис. 3.1, Д). Валики сливаются, преобразуя нервную плас-
тинку в трубку, идущую по длинной оси эмбриона. Эта труб-
ка— зачаток центральной нервной системы. Поверхностная эк-
тодерма с обеих сторон нервной трубки уплощается, образуя
кубический или плоский эпителий — предшественник эпидерми-
са. На границе между поверхностной эктодермой и нейроэкто-
дермой появляются клетки нервного гребня. Они мигрируют в
вентральном направлении, рассеиваются по большей части те-
ла, давая начало разнообразно дифференцированным тканям,
таким, как меланоциты, элементы периферической • чувстви-
тельной и автономной нервной системы.
Под нервным желобком располагается нотохорд (рис. 3.1,
Д), а по сторонам его — параксиальная мезодерма. В паракси-
альной мезодерме головной области сегментация практически
ие заметна, но в туловищной области (особенно в хвостовой)
видна четкая метамерность в форме сомитов (рис. 3.1, Д, £).
Блоки сегментов параксиальной мезодермы последовательно
закладываются в краниокаудальном направлении начиная с
9-го дня, причем новые сомиты добавляются к уже сформиро-
ванным из иесегментированной пластинки мезодермы впереди
первичной полоски. Мезодерма, расположенная по бокам со-
митов, подразделяется на два слоя, одни из которых связан с эк-
тодермой, а другой — с энтодермой. Они отделяются друг от
друга зародышевым целомом.
Мезодерма, расположенная впереди, под головной складкой,
формирует сердце (рис. 3.1, Д), которое начинает ритмически
сокращаться к концу 9-го дня. Во внезародышевой мезодерме,
выстилающей висцеральный желточный мешок, появляются
кровяные островки, и на 10-й день устанавливается сложное
кровообращение желточного мешка, при котором кровь поступа-
ет в висцеральный желточный мешок через пупочио-брыжееч-
Эксперименты с постимплантациониыми эмбрионами мыши 69
иую артерию (рис. 3.1, Д) и возвращается к сердцу по пупочно-
брыжеечной вене. На заднем конце первичной полоски мезодер-
ма дает начало аллантоису (рис. 3.1, Д), который делается
заметным на 8-й день в виде отдельной почки. Аллантоис ра-
стет через экзоцелом и сливается с хорионом, устанавливая та-
ким образом прямую связь между эмбрионом и эктоплацентар-
ным конусом. Следующим этапом является дифференцировка
хориоаллантонсной плаценты, которая становится главным пи-
тающим и дыхательным органом эмбриона.
Точное происхождение и время деламинации кишечной эн-
тодермы остается невыясненным. Она возникает из эпибласта,
формирование начинается, вероятно, с ранних этапов гаструля-
ции, когда головной отросток и другие инвагинирующие клетки
эпибласта замещают всю или часть предсуществующей висце-
ральной зародышевой энтодермы. Длительность включения кле-
ток в этот новый наружный слой дефинитивной энтодермы не
известна. Листок кишечной энтодермы приобретает характерное
трубчатое строение на переднем и заднем концах эмбриона, но
в отличие от нервной трубки, формирующейся путем слияния
складок, кишечная трубка образуется в результате замыкания
на себя, отчего передняя и задняя кишка вначале слепо соеди-
нены. Поэтому кишка остается открытой в желточную полость
дольше в области туловища, чем в других местах (рис. 3.1, Д).
3. Извлечение эмбрионов
и классификация стадий их развития
3.1. Извлечение
Все манипуляции с ранними постимплантационными эмбри-
онами требуют их освобождения от децидуальной ткани и обо-
лочки Рейхерта, которые не растут в условиях культивирова-
ния. Применяемые для этого методы даны в табл. 3.1 и иллюст-
рированы рис. 3.2. Для препарирования эмбриона требуются
две пары заостренных часовых пинцетов.
Манипуляции с эмбрионами ведутся в среде РВ11 [6J, со-
держащей 10%-ную эмбриональную телячью сыворотку (ЭТС)
вместо бычьего сывороточного альбумина (БСА), при комнат-
ной температуре.
3.2. Классификация стадий развития
Для раннего постимплантационного развития характерны
быстрое образование новых тканей и значительные морфологи-
ческие изменения. Поэтому для интерпретации любого экспе-
1 Состав среды РВ1 приводится в гл. 13, разд. 3.1.1.
70
Глава 3
Таблица 3.1. Извлечение эмбрионов
Препарирование эмбрионов
1. Вырежьте матку и поместите ее в 50-мм чашку Петри со средой РВ1 +
+ 10% ЭТС.
2. При помощи часового пинцета надорвите матку по антимезометриальному
краю каждого места имплантации (рис. 3.2,4), обнажив децидуальную
капсулу. Скользищим движением пинцета вдоль мезометриальной стенки
матки отделите децидуальную капсулу с зародышем (decidua) (рис.3.2,Б).
Перенесите ее в чистую чашку Петри с такой же средой.
3. Захватите мезометриальный (тупой) конец капсулы пинцетом. Введите
сложенные концы пинцета по средней линии (примерно на треть длины
капсулы от мезометриального конца) и погружайте их в ткань, пока они
не коснутси дна чашки (рис. 3.2, В). Раздвиньте концы пинцета. Дециду-
альнаи капсула должна наполовину разорватьси (рис. 3.2,Г).
4. Осторожно выталкивайте эмбрион из децидуальной капсулы. Когда он
почти полностью освободитси, захватите пинцетом эктоплацентарный ко-
иус и извлеките зародыш из децидуальной капсулы.
Удаление оболочки Рейхерта
1. Фиксируйте положение эмбриона, введя сложенные концы пинцета в жел-
точную полость сразу под эктоплацентарным конусом. Поставив в то же
положение сложенные концы второго пинцета (рис. 3.2, Д) и прижав, ве-
дите их в направлении эмбрионального полюса (рис. 3.2, Е). При этом
разорвется оболочка Рейхерта, обнажив ийцевой цилиндр.
2. Оболочка Рейхерта остается у эктоплацентарного конуса, так что боль-
шую часть ее можно просто осторожно оторвать.
римента особую важность приобретает тщательная классифи-
кация стадий развития. Ошибкой многих эмбриологов прошло-
го было использование возраста эмбриона млекопитающего в
качестве критерия стадии его развития. Заметные вариации в
эмбриональном развитии можно наблюдать даже у новорож-
денных мышат, а при прохождении столь динамической фазы,
как ранняя предимплантационная, различия в возрасте всего
лишь в несколько часов могут сопровождаться очень значи-
тельными морфогенетическими изменениями. Поэтому в каче-
стве более точного и чувствительного способа стандартизации
экспериментов рекомендуется классифицировать эмбрионы на
основе явно выраженных морфологических признаков и опре-
деленных физических размеров. В табл. 3.2 дана возможная
схема классификации живых эмбрионов, но использованные
критерии можно уточнить или изменить в соответствии с при-
родой эксперимента.
Эксперименты с постимплантационными эмбрионами мыши
71
Рис. 3.2. Схема операции извлечения 8-дневного эмбриона и удаления оболоч-
ки Рейхерта. Соответствующие манипуляции описаны в тексте.
4. Выделение тканей или частей эмбриона
4.1. Микрохирургические операции на эмбрионах
Для операций используются силиконированные стеклянные
иглы (разд. 9.3), укрепленные в держателях лейцевского ма-
нипулятора, или заостренные вольфрамовые иглы. Этими инст-
рументами пользуются для нанесения прямых разрезов, разде-
ляющих эмбрион на отдельные области или фрагменты.
Бактериологические пластиковые чашки Петри (Sterilin)
следует предпочесть стеклянным, т. к. эмбрионы в них меньше
скользят. В табл. 3.3 в качестве примера описано препариро-
вание задней части первичной полоски и 8-дневного эмбриона
(рис. 3.3,Л), но основные приемы этой операции можно ис-
пользовать для получения любой части эмбриона на любой
стадии. Основной принцип, которого следует при этом придер-
72
Глава 3
Таблица 3.2. Схема классификации стадий развития эмбриона
мыши с 6-го по 11-й день1»
Л ни беременно- сти2) Ста- дия Морфологические признаки Размеры (ммр)
6-й день (У) 1 Сформирован яйцевой цилиндр. Чет- ко различается зародышевое/вне- зародышевое соединение 0,1x0,1
6-й день (П. П.) 2 Формирование проамниотической по- лости 0,2x0,2
7-й день (У.) 3 Проамниотическая полость внедряет- ся во внезародышевую область 0,25x0,25
7-й день (П. П.) 4 Формирование первичной полоски 0,3x0,4
7-й день (П. П.) 5 Появление амниотических складок 0,3x0,4
8-й день (У.) 6 Образование амниона 0,35X0,4
8-й день (У.) 7 Расширение амниотической полости. Головной отросток 0,4X0,4
8-й день (П. П.) 8 Почка аллантоиса. Нервный желобок 0,5X0,4
8-й день (П. П.) 9 Образование головных складок. За- чаток сердца 0,55X0,5
9-й день (У.) 10 1—7 сомитов. Нервные валики. Пе- редняя кишка. Ось не изогнута^ Сердце сокращается 1,0
9-й день (П. П.) И 8—12 сомитов. Воротные вены. Пе- редний и задняя кишка. Ось изги- бается. Слуховая плакода. Одна жаберная дуга. Кровообращение в висцеральном желточном мешке 1,5
10-й день (У.) 12 13—20 сомитов. Полный поворот оси. Замыкание переднего и открытый задний иейропор. Слияние голов- ных складок. Две жаберных дуги. Плакода хрусталика. Глазные бока- лы. Обонятельная плакода 2,0
10-й день (П. П.) 13 21—29 сомитов. Почка передней ко- нечности. Задний нейропор открыт. 3 жаберных дуги. Глазные бокалы. Утолщенная обонятельная плакода 3,0
11-й день (У) 14 30—34 сомита. Почка задней конеч- ности. Нервная трубка замкнута 4,0
11-й день (П. П.) 15 35—39 сомитов. Выросты почек ко- нечностей. Удлиненный хвост. Вы- деляются мозговые пузыри. Обоня- тельная ямка. Пупочная грыжа 5,0
’) См. [7].
s) (У.) — утро, (П. П.) — после полудня.
3) Размеры эмбрионов с 6-го по 8-й день оценивали, измеряя наибольший диаметр
(d) и высоту (h) эмбриональной области плода: величину эмбрионов с 9-го по 11-й день —
по краниокаудальному размеру.
Эксперименты с постнмплантацноннымн эмбрнонамн мыши
73
Таблица 3.3. Выделение заднего отдела первичной полоски
1. Расположите эмбрион прн помощи стеклянных нгл так, чтобы его длинная
ось была перпендикулярна осям нгл.
2. Удерживая левой иглой эмбрион от движения, положите другую иглу по-
перек места соединения зародышевой части с внезародышевой вдоль пред-
полагаемой лнннн разреза (рнс. 3.3, А, 1) Прижмите иглы, пока они не
коснутся дна чашки. Двигайте нглы вперед-назад, пока эмбрион не раз-
делится на две части. Если нужно, повторите эту манипуляцию несколько
раз, используя каждую иглу. Разъедините обе части, отделив, если тре-
буется, от дна чашки.
3. Ориентируйте зародышевую часть цилиндра так, чтобы первичная полоска
оказалась параллельной длинной осн нглы.
4. Сделайте продольный разрез так, как было описано выше, н как можно
ближе к заднему концу (рис. 3.3, А, 2).
5. Сделайте поперечный разрез на нужном уровне по оси первичной полоски
(рис. 3.3, А, 3). Отрезанный фрагмент станет плоским, и его боковые
крылья можно отделить, сделав еще два разреза по обе стороны от полос-
ки (рнс. 3.3, А, 4). Энтодерму н большую часть мезодермы можно удалить,,
осторожно подцепив ткань кончиком нглы.
6. Перенесите фрагмент первичной полоски в стеклянную камеру, воспользо-
вавшись тонкой, вручную вытянутой пастеровской пипеткой. Чтобы избе-
жать прилипания эмбрионов к стеклу, для переноса эмбрионов или нх
частей из одного раствора в другой рекомендуется пользоваться снлико-
нированными пастеровскими пипетками (Repelcote; Hopkin and Williams).
Их адгезивность можно уменьшить и применением охлажденной среды.
жпваться, — разделять эмбрион на все более мелкие фрагменты,
сохраняя при этом возможность идентификации того, что нуж-
но получить.
4.2. Ферментативное разделение тканей
Разделения зародышевых слоев можно добиться с помощью
протеолитических ферментов в сочетании с микрохирургичес-
кой техникой. Наиболее эффективная методика, предложенная
Левак-Свейджер [8], использует смесь трипсина с панкреати-
ном, позволяя не только чисто разделить ткани, но и уменьшить
их адгезивность, затрудняющую различные манипуляции [9].
Методика ферментативного разделения описана в табл. 3.4 и
проиллюстрирована рис. 3.3, 5; 8-дневный эпибласт, выделенный
этим способом, представлен на рис. 3.4. Метод может быть
использован для разделения многих тканей, даже из достаточ-
но далеко продвинутого в своем развитии эмбриона, если пе-
риод инкубации варьирует в зависимости от задачи эксперимен-
татора. Например, висцеральный желточный мешок может быть
разделен иа составляющие его энтодерму и мезодерму через
3 ч инкубации при 4 °C [10].
74
Глава 3
Рис. 3.3. А. Выделение задней части первичной полоски с помощью микрохи-
рургических методов. Подробное описание процедуры представлено в табл. 3.3.
Б. Микрохирургическое и ферментное отделение эпнбласта. Детали метода
описаны в табл. 3.4.
5. Культивирование
и эктопические трансплантации
изолированных тканей и фракций
5.1. Культивирование
Дифференцировку или дальнейшее развитие тех или иных
фракций части эмбриона можно изучать в культуре. При этом
используются разнообразные среды и субстраты, но здесь они
обсуждаться не будут, так как большинство соответствующих
экспериментов основано на обычной технике культивирования
тканей и органов, сведения о которой можно получить из спе-
циальных источников [11]. В общем условия культивирования
определяются задачами эксперимента. Стандартная техника
культивирования тканей и органов меняет взаимоотношения
тканевых пластов и, следовательно, применима в таких экспе-
риментах, где нарушения морфогенеза не имеют существенного
значения. Отклонения в морфогенезе могут быть даже исполь-
зованы при выделении тех или иных клеточных типов из экс-
плантата гетерогенной или полипотентной ткани. Например,
клетки нервного гребня могут быть выделены в результате экс-
плантации изолированной нервной трубки 9-дневного эмбриона
в стандартных тканевых культуральных чашках (Falcon), со-
держащих минимальную среду Игла + 15% ЭТС +2—4%-ный
экстракт зародыша цыпленка [12]. Спустя несколько дней
Эксперименты с постимплаитациоиными эмбрионами мыши 75
Таблица 3.4. Выделение эктодермы, мезодермы и энтодермы мышиного
эмбриона на стадии первичной полоски
1. С помощью микрохирургических инструментов отделите зародышевую об-
ласть яйцевого цилиндра (разд. 4.1).
2. Зародышевую часть (рис. 3.3, Б, 1) перенесите в профильтрованный (Milli-
pore, Sartorius, размер пор 0,45 мкм) 0,5%-ный (в/о) трипсин (BDH),
0,25%-ный (в/о) панкреатин (Difco) в бескальциевом и безмагниевом со-
левом растворе Тнроде — Рингера1', pH 7,6—7,7, при 4 °C. Инкубируйте при
4 °C 10—20 мнн.
3. Перенесите зародышевые фракции с минимальным объемом перевариваю-
щей смеси в большой объем РВ12>+10% ЭТС (т. е. 5 мл в 50-мм чашке
Петри).
4. Осторожно пипетнруйте эмбрионы, вводя н выталкивая их при помощи
вытянутой, оплавленной снлнконнрованной пипетки, внутренний диаметр
которой несколько меньше, чем зародышевый цилиндр (рис. 3.3, Б, 2). Каж-
дый раз вводите в пипетку эмбрион тем концом, где прошла плоскость
разреза. При этом энтодерма отслоится от цилиндра.
5. Обычно после удаления энтодермы подлежащая мезодерма уплощается
и выпячивается наподобие двух крыльев, связанных с эпибластом лишь
по длине первичной полоски. Отделите эти крылья разрезами, проходящи-
ми как можно ближе к первичной полоске (рис. 3.3, Б,3). Если мезодер-
мальные крылья после удаления энтодермы не выпячиваются, мезодерму
можно отделить нлн пипетнрованием, нлн осторожными прикосновениями
игл; она отслоится в передней н боковых областях.
6. Удалите оставшиеся рыхло лежащие вдоль полоски клетки кончиком иглы
(рнс. 3.3, Б, 4}. Учитывая природу первичной полоски, нельзя быть уверен-
ным, что в этой области эмбриона не осталось примеси мезодермальных
клеток, для этого потребовалось бы удалить всю полоску полностью (см.
рис. 3.4).
') Солевой раствор Тироде — Рингера (г/л): NaCl — 8, KCI — 0,3, NaH2PO4-5H2O —
0.093, КН2РО, — 0,025, NaHCOa — 1, глюкоза — 2.
2) Состав PBI см. в гл. 13, разд. 3.1.1.
эксплантат окружается популяцией выселившихся клеток с ад-
гезионной поверхностью, которую на основании ряда критериев,
в том числе по содержанию предшественников меланоцитов
[13J, можно определить как относительно чистую популяцию
клеток нервного гребня. В тех случаях, когда важно сохранить
какие-то черты нормального морфогенеза, фракции следует
инкубировать в суспензионной культуре. Чтобы предотвратить
рост ткани, поверхность чашки рекомендуется заливать 1%-ным
раствором агара в синтетической среде, выбранной для данной
культуры [14J. Например, специфические фракции 8-дневного
эмбриона, оставленные связанными с внезародышевой обла-
стью, растут в покрытых агаром чашках с модифицированной
Дульбекко средой Игла (DMEM) 4-50%-ная крысиная сыворот-
ка (разд. 6.1). В этих условиях фракции остаются в суспензии
и проявляют интенсивный морфогенез.
76
Глава 3
Рис. 3.4. Срез 8-дневного эпибласта, выделенного с помощью микрохирургии
в сочетании с ферментной обработкой. ПП — первичная полоска.
5.2. Эктопические трансплантации
Разнообразные фракции или ткани постимплантационных
эмбрионов мыши будут продолжать расти и дифференциро-
ваться, если их перенести в хорошо васкуляризованную область
иммунологически подходящего взрослого хозяина [2]. Исполь-
зовались и ксеногенные места имплантации, например, хорио-
аллантоис, но для оптимального развития наиболее предпочти-
тельна в качестве хозяина — сингенная мышь. Несмотря на то,
что морфогенез таких трансплантатов аномален, набор диффе-
ренцированных тканей, формирующийся в этих «эксперимен-
тальных тератомах», можно использовать в качестве критерия
гистогенетических потенций трансплантированной ткани. Следу-
ет отметить, что если в трансплантат входит трофэктодерма или
внезародышевая эктодерма, можно ожидать, что геморрагичес-
кий ответ, спровоцированный этими тканями, будет маскиро-
вать дифференцировку другого типа [15]. Особого интереса за-
служивает тот факт, что зародышевая область эмбриона (до
8 дней беременности), пересаженная под капсулу семенника
или почки, дает начало перевиваемой опухоли [16, 17]. Эта
опухоль характеризуется присутствием клеток эмбриональной
карциномы — слабо дифференцированных, сходных по морфо-
логическим и иным признакам с клетками эпибласта и при-
мордиальными половыми клетками. Процедура эктопической
имплантации под капсулу семенника описана в табл. 3.5.
Эксперименты с постимплаитациониыми эмбрионами мыши 77
Таблица 3.5. Эктопическаи трансплантация под капусу семенника
1. Отделите предназначенную для пересадки область илн ткань и поместите
ее в PBl^+lQo/o ЭТС.
2. Вручную вытяните силиконированную пастеровскую пипетку так, чтобы
ее диаметр был чуть больше размера кусочка ткани. Втяните в пипетку
столбик парафинового масла, затем пузырек воздуха и после немного
РВ1 + 10% ЭТС. Чтобы парафиновое масло эффективно контролировало
уровень жидкости в пипетке, пузырек воздуха должен находиться в тон-
кой части пипетки.
3. Анестезируйте сингенного самца (Avertin, 0,1 мл/5г массы тела).
4. Под контролем бинокулярного микроскопа сделайте ножницами попереч-
ный разрез кожи примерно 0,5 см длиной по средней линии живота над
передней границей селезенки. В том же положении сделайте разрез стен-
ки тела.
5. Топким пинцетом захватите жировую подушку семеиинка н вытяните его
через отверстие в стенке тела.
6. Заметив первоначальное положение пузырька воздуха в пастеровской пи-
петке, втяните в нее трансплантируемую ткань.
7. Удерживая в одной руке пипетку и инъекционную нглу (21-го калибра),
сделайте небольшое отверстие иглой в капсуле семенника и введите в от-
верстие кончик пнпеткн. Скользящим движением углубите пипетку под
капсулу семенника на расстояние около 4 мм.
8. Осторожно выведите трансплантат, слегка извлекая пипетку. Можно ви-
деть, как трансплантированная ткань появится на поверхности семенника.
Если нет, трансплантацию можно проконтролировать положением пу-
зырька воздуха в пипетке. Он должен переместиться к ее концу, но не
давайте ему выйти из пипетки в семенник. Извлеките пипетку полностью.
9. Возвратите семенник вместе с его жировой подушкой в брюшную полость.
Такую же операцию при необходимости можно проделать с другим, про-
тиволежащим, семенником.
10. Зашейте стенку тела, зашейте или наложите зажим на кожу.
1) Состав РВ1 см. в гл. 13, разд. 3.1.1.
6. Культивирование целых эмбрионов
Методы культивирования постимплантационных эмбрионов да-
ют возможность изучать клетки в их нормальном окружении.
Вот почему в высшей степени необходимо иметь в руках надеж-
ные и эффективные культуральные системы, позволяющие под-
держивать нормальное развитие эмбрионов в течение относи-
тельно длительного периода. К сожалению, было показано, что
мышиный эмбрион менее толерантен к эксплантации in vitro
во время гаструляции и органогенеза, чем эмбрион крысы [18,
19]. Использование режимов, поддерживающих почти нормаль-
ные рост и развитие эмбрионов крысы в течение нескольких
дней [20], в отношении мышиного эмбриона оказалось значи-
тельно менее успешным. Например, в отличие от крысы разви-
тие эмбриона мыши на стадии, предшествующей образованию
первичной полоски, является in vitro совершенно непредсказуе-
78
Глава 3
Таблица 3.6. Приготовление крысиной сыворотки
1. Анестезируйте крысу эфиром.
2. Сделайте веитарльиый, по средней линии, разрез кожи и етеики тела.
Извлеките внутренности и отведите их в сторону.
3. Спиииая аорта лежит по средней линии примыкания к полой вене.
Из двух кровеносных сосудов аорта меньше и бледнее. Введите иглу
(21-го калибра/Р/2 дюйма) от шприца на 10 или 20 мл в спиниую аорту
(скосом вниз и по направлению к сердцу).
4. Осторожно втяните кровь. Если делать это слишком быстро, может прои-
зойти гемолиз. В зависимости от размера крысы получают от 8 до 15 мл
крови.
5. Осторожно перенесите кровь в центрифужную пробирку. Немедленно от-
центрифугируйте при 1200 g в течение 5—10 мин. Кровяные клетки ося-
дут, и в слое плазмы сформируется фибриновый сгусток. (На этой стадии
разделенную кровь можно хранить при 4 °C 18 ч.)
6. Не касаясь эритроцитов, тонким пинцетом осторожно выньте фибриновый
сгусток. Центрифугируйте при 1200 g в течение 5 мии.
7. Пастеровской пипеткой соберите сыворотку и перенесите в другую цент-
рифужную пробирку.
8. Центрифугируйте при 1200 g 5 мии. Отберите сыворотку в следующую
пробирку. Приготовьте аликвоты удобного объема (т. е. по 2,5 или 5 мл).
9. Инактивируйте сыворотку нагреванием в водяной бане при 50 °C в тече-
ние 30 мии. Слегка приоткройте крышки пробирок для испарения эфира.
10. После охлаждения сыворотки внесите антибиотик (бензилпенициллин,
100 МЕ/мл; сульфат стрептомицина, 100 мкг/мл). Добавление антибиоти-
ка существенной роли ие играет. Если с сывороткой и средой обращаться
осторожно, загрязнение культуры и в отсутствие антибиотиков происходит
исключительно редко. Храните сыворотку при —20 °C до 3 месяцев.
мым. И хотя эксплантация эмбрионов с уже сформированной
первичной полоской идет значительно лучше, на сегодняшний
день невозможно поддерживать нормальные рост и развитие
мышиного эмбриона в культуре более 24 или 36 ч. Но даже
имеющиеся в нашем распоряжении 24 ч дают очень много для
изучения дифференцировки и морфогенеза, поэтому, несмотря
на существующую неадекватность метода, культивирование це-
лых эмбрионов остается ценнейшим источником информации о
постимплантационном развитии.
Описанная в этом разделе процедура культивирования пост-
имплантационных эмбрионов мыши основана на методах, раз-
работанных для крысиных эмбрионов [18J. Каждый исследова-
тель вправе усовершенствовать эти методические приемы.
Не следует рассматривать приведенные протоколы опытов как
законченные рекомендации.
6.1. Среды
Эмбрионы, эксплантируемые на 7, 8 или 9-й день бере-
менности, могут расти в крысиной сыворотке, разбавленной 1 : 1
DMEM (Flow Laboratories). Для эмбрионов более поздних сро-
Эксперименты с постимплантационными эмбрионами мыши
79
Таблица 3.7. Приготовление среды
1. Разморозьте нужный объем среды.
2. Добавьте равный объем DMEM (Flow Laboratories) с глутамином, если
синтетическая среда его не содержит. Перемешайте.
3. Профильтруйте среду через миллнпоровый фильтр (размер пор 0,45 мкм)
и поместите аликвоты по 25 или 3 мл в пробирки или флаконы, исполь-
зуемые под культуры. Культуральные сосуды должны отвечать пяти ос-
новным требованиям: они должны быть стерильными; иметь подходящий
размер для закрепления в системе вращения (разд. 6.2); объем позволяю-
щий содержать еще и газовую фазу, равную 20 мл; герметически закры-
ваться, и, наконец, эмбрионы не должны к ним прилипать. Учитывая все
эти требования, наиболее подходящими следует считать флаконы из силн-
конированного стекла со стеклянными пробками (не токсичными и скру-
пулезно очищенными) или универсальные культуральные пробирки (Steri-
Ип). Чтобы иметь гарантию герметичности, рекомендуется на пробку на-
носить слой вакуумной силиконовой смазки.
4. Выдержите среду на воздухе с 5% СО2 при 37 °C в течение нескольких
часов.
5. После переноса эмбрионов следует насытить среду соответствующей газо-
вой смесью (разд. 6.3), осторожно вдувая газ в течение 1 мин. Закройте
сосуды соответствующими пробками и поместите в роллеры при 37 °C.
ков рекомендуется 100%-ная сыворотка. Способ приготовления
крысиной сыворотки описан в табл. 3.6. Важно учесть следую-
щее: сыворотка должна быть получена из крови, где фибрино-
вый сгусток образовался в плазме, а не в цельной крови. Если
крови дать свернуться до отделения сыворотки, у ранних пост-
имплантационных эмбрионов крысы чаще наблюдаются анома-
лии в развитии и замедленный рост [21J. Ни пол, ни штамм до-
нора, по-видимому, значения не имеют [18].
Количество среды зависит от стадии и числа эксплантируе-
мых эмбрионов, а также от длительности культивирования.
В отношении эмбрионов мыши систематический анализ не про-
водился, но по грубой оценке 2,5 мл среды достаточно для под-
держания роста и развития четырех 8-дневных, трех 9-дневных
или двух 10-дневных эмбрионов в течение 24 ч.
Рекомендуется предварительно выдержать среду в течение
нескольких часов или оставить на ночь в увлажненном СОг-ин-
кубаторе (5% СО2 в воздухе) при 37°C. Это удалит из сыворот-
ки ббльшую часть эфира (см. табл. 3.6) и уменьшит время на-
сыщения среды СО2 вручную. В табл. 3.7 описана процедура
приготовления среды.
6.2. Роллерная система
При культивировании крысиных эмбрионов вращение куль-
туральных сосудов, обеспечивающее постоянный контакт всей
среды с газовой фазой и, следовательно, постоянное насыщение
80
Глава 3
Рис. 3.5. Основные детали роллерной культуральной системы. 1. Приводной
шкив. 2. Ось, соединенная с электродвигателем постоянного тока (30 об/мин).
3. Ременная передача, сопряженная с ведомой осью. 4. Ведомый шкив. 5. Вто-
рая ведомая ось. 6. Зубчатое колесо, закрепленное на второй ведомой оси.
7. Ведомое зубчатое колесо, закрепленное на оси барабана 8. Вращающийся
барабан (1,25 дюйма, длина 12 дюймов). 9. Пластина, на которой закреплены
оси барабана.
кислородом, создает условия для нормального развития в тече-
ние более длительного периода, нежели статическая культура
[18]. Ранние постимплантационные эмбрионы мыши развива-
ются в статической культуре, если эксплантацию произвели на
стадиях первичной полоски, однако нормальное развитие под-
держивается не более 24 ч [22]. Судя по опыту культивирова-
ния крысиных эмбрионов, можно ожидать, что система враще-
ния культуральных сосудов улучшит и условия развития эм-
брионов мыши на стадии первичной полоски, хотя сравнения
статических и ротационных культур именно для этой стадии
развития мыши не проводилось.
Ротация достигается помещением флаконов или пробирок
на параллельные роллеры или прикреплением флаконов к вра-
щающемуся диску. Чтобы культуральная среда полностью по-
крывала эмбрионы, флаконы или роллеры приподнимают под
небольшим углом, вследствие чего среда остается на дне фла-
кона. Скорость вращения должна быть порядка 30—60 об/мин.
Роллерную установку, заключенную в инкубаторе на 37 °C,
можно приобрести у В. Т. С. Engineering, Кэмбридж, Велико-
британия. Но несложно сконструировать и собственную систе-
му роллеров, подходящую к стандартному инкубатору. Приво-
Эксперименты с постимплантационными эмбрионами мыши
81
Рис. 3.6. Процентное содержание кислорода, необходимое для развития эмб-
рионов разных стадий. С — сомиты.
дящий мотор должен располагаться вне инкубатора, чтобы не
нарушать температурный режим. На рис. 3.5 показан основной
принцип устройства такой системы.
6.3. Газовый режим
По мере развития эмбрионы требуют все большего насыще-
ния среды кислородом, поэтому для разных стадий развития ис-
пользуют разные газовые смеси. Эксперименты на крысиных
эмбрионах указывают на то, что увеличение потребности в кис-
лороде объясняется двумя причинами: переходом от гликоли-
тического способа получения энергии к использованию цикла
Кребса и системы транспорта электронов и необходимостью
компенсировать отсутствие функциональной хориоаллантоисной
плаценты (служащей in vivo важнейшим органом газообмена)
[18J. Нужная концентрация кислорода в газовой фазе на раз-
ных стадиях развития представлена на рис. 3.6. На всех стади-
ях смесь должна содержать 5% СОг, конечный объем смеси
дополняется Ыг. На тех стадиях, где прослеживаются более
длительные сроки (более 24 ч), рекомендуется повысить про-
центное содержание кислорода.
6.4. Оценка развития
Очевидно, что ценность любого эксперимента, в том числе и
культивирования эмбрионов, определяется тем, насколько близ-
ко к норме происходит дифференцировка, морфогенез и рост
эмбрионов, развивающихся in vitro. Следовательно, важным
условием экспериментальной работы служит выбор надежного
6—171
«2
Глава 3
Таблица 3.8. Некоторые параметры оценки развития культивируемых
эмбрионов /
Общая морфология /
Степень осевого поворота
Число пар сомитов
Степень слияния нервных валиков
Состояние аллантоиса (слит или не слит с хорионом)
Присутствие илн отсутствие специфических структур (почек конечностей, слу-
ховых плакод и пр.)
Относительные размеры разных структур (головы, сомитов и др.)
Гистология
Присутствие, отсутствие и морфология специфических структур
Плотность упаковки клеток
Митотический индекс разных тканей
Индекс гибели клеток разных тканей
Рост и метаболизм
Кранио-каудальный размер (или другие удобные измерения физических раз-
меров)
Содержание белка, измеряемое колориметрическим методом по Лоури [23]
Включение радиоизотопов: [3Н]-тимидина, [35С]-метионииа
Физиология
Присутствие или отсутствие, скорость сердцебиения
Присутствие или отсутствие, степень циркуляции висцерального желточного
мешка
критерия правильности развития. В табл. 3.8 перечислены воз-
можные параметры. По ним сравнивают культивируемые эм-
брионы с эмбрионами того же возраста, развивающимися in
vivo. Кроме того, в любой эксперимент должен быть включен в
качестве стандарта неоперированный контроль.
7. Включение клеток или метки
в эмбрионы in vitro
Для того чтобы проследить за перемещением поверхностных
клеток, выстилающих полость, их маркируют, вводя соответст-
вующую метку в эмбриональную полость. Эта методика ценна
тем, что дает возможность изучать судьбу клеток эпибласта,
инвагинирующих через первичную полоску или миграцию кле-
ток нервного гребня [24]. Инъекция других веществ, таких,
как специфические ферменты или их ингибиторы, может ис-
пользоваться для оценки значения тех или иных молекул в раз-
личных морфогенетических или дифференцировочных событиях.
Инъекция меченых клеток в эмбрион (в эктопические или те-
Эксперименты с постнмплантационными эмбрионами мыши 83
теротопические участки) позволяет проследить развитие специ-
фических тканей или отдельных частей эмбрионов [25, 26]. Об-
щая стратегия получения «химер» in vitro представлена на
рис. 3.7.
Мы не ставили целью описать в этой главе все возможные
манипуляции с постимплантационными эмбрионами; ограни-1
чимся простейшей операцией инъекции клеток в эмбрионы на
стадии первичной полоски или ранней закладки сомитов, для
осуществления которой понадобятся всего лишь два инстру-
мента. Более сложные манипуляции требуют дополнительных
инструментов, таких, как прямые и закругленные иглы [27] или
электроды (например, для инъекции в отдельные клетки in situ
маркера — пероксидазы хрена) [28, 29]. На практике наилуч-
шую комбинацию инструментов для каждой конкретной опера-
ции экспериментатор подбирает сам методом проб и ошибок, но
если он овладел простой методикой наподобие инъекции с дву-
мя инструментами, принципы изготовления и применения бо-
лее сложных инструментов станут для него очевидными.
7.1. Оборудование
Для того чтобы контролировать инъекции растворов, кле-'
ток или плотных тканей, необходим микроманипулятор (Leitz,
Wetzlar, ФРГ). Основные компоненты этой установки представ-
лены на рис. 3.8. Манипуляции с эмбрионами на стадии пер-
вичной полоски или с зародышами более ранних стадий мож-
но проводить в капле среды, свисающей с силиконированного
покровного стекла, заключенного в камеру микроманипулятора
(Leitz), заполненную парафиновым маслом (детали см. гл. 12,
разд. 2.3.1). В этом случае наиболее приемлем микроскоп с фик-
сированным штативом, с выпрямляющей изображение оптикой
и длиннофокусными объективами (т. е. Microtec М2, Microin-
struments, Oxford). Манипуляции с эмбрионами более поздних
стадий лучше удаются в капле среды на залитом жидким пара-
фином дне бактериологической чашки Петри или в крышке от
чашки Петри. Здесь более уместен стандартный препароваль-
ный микроскоп. Таким микроскопом можно пользоваться и при
манипуляциях с эмбрионами на стадии первичной полоски, но
более слабое разрешение делает операции не очень точными.
Самое важное условие удачной микроманипуляции — хоро-
шие инструменты (см. разд. 9), точно приспособленные для ре-
шения конкретной задачи.
7.1.1. Подготовка микромамипупяторов и инструментов
Наиболее важные моменты, которые следует учесть при под-
готовке:
6«
«4
Глава 3
эктодермаль-
Контроль Контроль
in vivo
in vitro
ных клеток
Химера Меченый Контроль
in vitro контроль включения
Рис. 3.7. Общая стратегия получения химер in vitro. Контроль in vivo: эмб-
рионы того же возраста, извлекаемые из самки и используемые для оценки
степени развития. Контроль in vitro: нормальные, растущие в культуре эмб-
рионы; служат для оценки роста в условиях культивирования. Меченый конт-
роль: эмбрионы, меченные той же дозой [3Н]-тимидина, что н донорская ткань;
позволяют контролировать нетоксичность метки, однородность ее распределе-
ния и ожидаемое разведение в процессе культивирования. Химера in vitro:
инъецированный эмбрион. Контроль поглощения: эмбрионы, фиксированные
сразу после периода мечения; позволяют оценить процент меченых клеток.
а) в парафиновом масле, заполняющем систему, не должно
быть пузырьков воздуха;
6) инструменты должны быть совершенно прямыми, пересе-
кающими горизонтальное поле зрения микроскопа и ле-
жащими точно параллельно друг другу.
Рис. 3.8. Основные детали микроманипуляционной установки, используемой для инъекции в эмбрион клеток или метки.
1, 2. Правый н левый микроманипуляторы, закрепленные на платформе (Leitz). 1—используется с микроманипуляцион-
ной камерой Puliv, которая устанавливается на микроскопе с фиксированным штативом н выпрямляющей оптикой; 2 — ис-
пользуется для манипуляций в чашке Петри под стереомикроскопом. 3. Регулятор перемещения инструментов в верти-
кальной плоскости («вертикальный контроль»). 4. Регулятор наклона инструментов. 5. Регулятор перемещения инструмен-
тов в горизонтальной плоскости («латеральный контроль»), 6. Регулятор перемещения вперед. 7. Рычаг управления. 8. Ре-
гулятор высоты. 9. Регулятор угла наклона головкн инструмента. 10. Головка инструмента. 11. Микрометрический латераль-
ный контроль. 12. Микрометрический контроль перемещения вперед. 13. Держатель инструмента. 14. Шприц Agla с микро-
метром (Wellcome, Derbyshire, UK). 15. Насос де Фонбрюна, сообщающий системе положительное или отрицательное дав-
ление.
86
Глава 3
Таблица 3.9. Установка инструментов
1. Убедитесь в том, что трубки шприца (Agla) и иасоса (de Fonbrune) за-
полнены парафиновым маслом, что в них нет пузырьков воздуха и запас
масла в шприце и насосе достаточен.
2. Сияв колпачок и металлический промыватель, введите держатель инстру-
мента в каждый конец трубки и заполните парафиновым маслом так,
чтобы мениск масла выступал из отверстия держателя.
3. Поместите колпачки и промыватели в нужном положении на инъекцион-
ную и удерживающую пипетки. Введите инъекционную пипетку в ин-
струментальный держатель, прикрепленный к насосу, убедившись в том,
что капилляр удерживается промывателем. То же проделайте с удержи-
вающей пипеткой, прикрепив ее к шприцу. Оба инструмента заполните
парафиновым маслом.
4. Сфокусируйте микроскоп на каплю среды, в которой будет проводиться
инъекция.
5. Установите держатели так, чтобы инструменты (микропипетки) на мани-
пуляторе лежали точно один против другого горизонтально и находились
в поле зрения микроскопа. Убедитесь в том, что обе микропипетки после
окончательной установки могут перемещаться по всему полю зрения при
помощи рычага управления, оставаясь постоянно в фокусе.
6. Уберите микропипетки. Если используется манипуляционная камера, микро-
пипетки просто отодвигаются до границы горизонтального контроля; если
используется чашка Петри, удобнее поднять микропипетки из капли среды
с помощью «контроля наклона» манипулятора.
Процедура установки инструментов описана в табл. 3.9 (см.
также гл. 12, разд. 3.2).
В том случае когда вся процедура делается под препаро-
вальным микроскопом и инъекции проводятся в чашке Петри,
важно, чтобы концы инструментов располагались слегка под
углом к дну чашки, иначе плечо пипетки, образовавшееся при
вытягивании капилляра, будет мешать ее отверстию прикасать-
ся к дну чашки (см. разд. 9). Это достигается благодаря введе-
нию в инструменты двух изгибов (рис. 3.11 Г, 2) и наклону ин-
струментов и манипулятора, как показано на рис. 3.8. При ма-
нипулировании в висячей капле важно проследить за положени-
ем обоих инструментов — они должны быть параллельны пред-
метному столику микроскопа.
7.2. Приготовление метки или клеток для инъекций
7.2.1. Метка
Недавно было обнаружено, что инъекция агглютинина за-
родышей пшеницы (АЭП), связанного с такими метками, как
коллоидное золото или пероксидаза хрена (ПХ), дает эффек-
тивное мечение клеток, выстилающих амниотическую полость
[24J. Представляется вероятным, что и другие макромолекулы,
способные связываться с клетками эпибласта или эктодермы,
Эксперименты с постимплантационными эмбрионами мыши 87
например другие лектины или антитела (см. гл. 2, разд. 4), мо-
гут обеспечить эффективное избирательное маркирование этих
клеток или клеток, выстилающих иные полости эмбриона. Если
перед исследователем стоит задача пометить только выстилаю-
щие клетки, он должен иметь в виду, что к проникновению мет-
ки во внутренние ткани может привести повышение гидроста-
тического давления в полости. Чтобы избежать этого, следует
приготовить концентрированный раствор метящего вещества и
инъецировать его в минимальном количестве. Например, для
эффективного мечения всех клеток эпибласта эмбриона на ста-
дии поздней первичной полости достаточно приблизительно
2Х10~3 мкл раствора АЭП — ПХ в концентрации 1 мг/мл, при
этом метка останется различимой в течение 24 ч культивирова-
ния. Приготовив раствор метки, поместите его каплю в мани-
пуляционную камеру или чашку на достаточном расстоянии от
капли среды, в которой предполагается проводить инъекцию
(следите чтобы капли не слились, когда будете переносить пи-
петку из одной капли в другую).
7.2.2. Клетки
Для выявления инъецированных донорских клеток в окружа-
ющей ткани хозяина можно использовать генетические марке-
ры. Однако в настоящее время подходящие для этого маркеры
не подобраны.
Наиболее приемлемым является интенсивное мечение [3Н]-
тимидином. Благодаря короткому митотическому циклу клеток
раннего постимплантационного зародыша он быстро включается
в большинство клеток и не нарушает хода развития [25]. Про-
цедура мечения клеток [3Н]-тимидином описана в табл. 3.10.
На рис. 3.9 представлены радиоавтографы включения и меченый
контроль, а также две инъецированные химеры.
7.3. Инъекция
Процедура инъекции одинакова для растворов и для клеток,
поэтому мы ограничимся описанием инъекции клеток (табл.
3.11 и рис. 3.10). При введении растворов необходимо помнить,
что удерживающей пипеткой нельзя дотрагиваться до капли с
меткой. Каждая манипуляционная капля должна содержать
только один эмбрион и для каждой инъекции нужно брать све-
жую каплю. Введение материала в мезодерму или эктодерму
требует прохождения через эктодермальный слой. При инъек-
ции клеток это не имеет особого значения, между тем раствор
может попасть не по назначению. Чтобы решить эту проблему,
необходимо тщательно продумать путь инъекции, например вво-
дить в амниотическую полость через внезародышевую область.
88
Глава 3
Таблица 3.10. Приготовление для инъекции клеток, меченных [3Н]-тимидином
1. Внесите 10 мкКи/мл [3Н]- тимидина (с удельной активностью 10,5 Ки/мМ)
в модифицированную среду Игла (Flow Laboratories). Выдержите среду
в атмосфере с 5% СО2 при 37 °C.
2. Интактные эмбрионы с неповрежденной оболочкой Рейхерта инкубируйте
в среде не меиее двух часов. Этого срока достаточно, чтобы пометалось
90% клеток зародыша на стадии поздней первичной полоски, но эмбрионы
старшего возраста требуют более длительного времени инкубации, кото-
рое определяется приготовлением контрольных срезов (контроль включе-
ния, рис. 3.9, Д).
3. Промойте эмбрионы, трижды сменив РВ11>+10% ЭТС+Ю мкл/мл 10-3 М ти-
мидина. Немеченый тимиднн в этой концентрации (10~3М) должен присут-
ствовать во всех средах при всех дальнейших манипуляциях и культивиро-
вании.
4. Поместите 2—3 меченых эмбриона в роллерную культуру (меченый конт-
роль) (рис. 3.7). Присутствие таких эмбрионов в каждом опыте позволяет
проконтролировать все варианты мечения, разбавления метки и радиоавто-
графнческой процедуры (рис. 3.9,Б).
5. Отделите нужную для инъекции ткань или область эмбриона, как описано
в разд. 4.
6. Разрежьте ткань стеклянными иглами на фрагменты и перенесите эт»
кусочки в манипуляционную каплю, готовую для проведения в ней инъек-
ции в рецнпиентный эмбрион.
3) Состав РВ1 см. в гл. 13, разд. 3.1.1. 8
8. Введение клеток или метки
в эмбрионы in vivo
Более 10 лет тому назад было предложено несколько удач-
ных способов введения клеток или вирусов в эмбрион in utero
[30—32]. Преимущество этого подхода очевидно: он позволяет
получить эмбрион любой из последующих стадий развития и
взрослых химер. Наиболее впечатляющий пример — недавнее
сообщение о жизнеспособности мыши с четкой химерностью по
меланоцитам, что явилось результатом инъекции клеток нервно-
го гребня пигментированного донора альбиносному эмбриону
in utero [13]. Недостаток метода введения клеток в эмбрион
in vivo заключается в невозможности точно локализовать место
инъекции, а также в том, что в результате инъекций в висце-1
ральный желточный мешок или амниотическую полость лишь
ограниченное число клеточных типов может войти в состав
постимплантационного эмбриона. Более того, химерные эмбрио-
ны не удается получить при инъецировании до середины 9-го
дня беременности. Возможно, эмбрионы ранних стадий физиче-
ски еще слишком малы или слишком чувствительны к инъек-
циям. Прежде чем приступить к получению химер, следует по-1
практиковаться в проведении инъекций (табл. 3.12), используя
для этого хорошо идентифицируемые растворы или суспензии,
Рис. 3.9. Радиоавтографы контрольных н экспериментальных эмбрионов.
Л. Контроль поглощения. Б. Меченый контроль. В. Инъецированный эмбрион,
химерный в области первичной полоски. Г. Инъецированный эмбрион, химер-
ный в области нотохорда и кишки.
90
Глава 3
Таблица 3.11. Инъекция клеток в эпнбласт 8-дневного эмбриона
1. Поместите один или более эмбрионов, подлежащих инъецированию,
а также донорские клетки в манипуляционную каплю РВР’+К)^ ЭТС+
+ 10~5 М «холодного» тимидина. Введите в то же поле зрения предвари-
тельно установленные инъекционную и удерживающую пипетки
(разд. 7.1.1) и в каждую втяните немного среды.
2. Сфокусируйте микроскоп на кусочек донорской ткани и в той же фокаль-
ной плоскости расположите кончик инъекционной пипетки. Очень осторож-
но втягивайте клетки в пнпетку, пока они не окажутся на расстоянии 10—
20 мкм от ее кончика. Сильное всасывание может привести к тому, что
столбик парафинового масла диспергируется средой на отдельные капли
и гидравлический контроль будет нарушен.
3. Сфокусируйте эмбрион и поверните его предполагаемым местом инъекции
к концу инъекционной пипетки. Осторожно присосите эмбрион удерживаю-
щей пипеткой (рис. 3.10,Л). Приведите инъекционную пнпетку точно в ту
же фокальную плоскость, где находится предполагаемое место инъекции
(рис. 3.10,Л). Осторожно подталкивая эмбрион концом пипетки, убедитесь
в том, что они располагаются на одной линии.
4. Пользуясь только рычагом управления, придвиньте инъекционную пипетку
к боку эмбриона н отведите рычаг управления вперед до упора. При по-
мощи винта, контролирующего движение вперед («передний контроль»),
поставьте конец инъекционной пипетки на одну линию с наиболее удален-
ным краем амниотической полости (т. е. так, чтобы она, будучи продвину-
той рычагом управления до ее крайнего предела, не проникла в стенку
эмбриона на противоположной стороне) (рис. 3.10,Б).
5. При помощи рычага управления отодвиньте конец пипетки назад в преж-
нее положение — к месту предполагаемой инъекции (рис. 3.10,В). Ото-
двиньте ее слегка, а затем энергично переведите рычаг управления в его
крайнее положение. Пипетка должна проникнуть в эмбрион в нужном
месте и войти в амниотическую полость (рис. 3.10,Г).
6. Рычагом управления осторожно вытаскивайте пипетку, пока ее конец не
окажется как раз позади места предполагаемой инъекции. Медленно вы-
пустите клетки в пространство, образовавшееся при выдвигании пипетки
(рис. 3.10, Д). Как только клетки выйдут из пипетки, полностью извлеките
инъекционную пипетку, затем освободите эмбрион и от удерживающей
пипетки,
7. Поверните эмбрион и убедитесь, что трансплантат находится в нужном
положении.
8. Перенесите эмбрион в роллерную культуру.
9. В конце периода культивирования зафиксируйте опытные и контрольные
эмбрионы жидкостью Карнуа (60% абсолютного спирта, 30% хлороформа,
10% уксусной кислоты) и обрабатывайте обычными гистологическими н
радиоавтографическими методами. В каждую партию инъецированных эм-
брионов рекомендуется включать один меченый контроль.
’) Состав РВ1 см. в гл. 13, разд. 3.1.1.
такие, как протамин голубой и тушь, или клетки, меченные
флуоресцентными красителями. Процедуру следует повторять
до тех пор, пока инъекционная пипетка не будет безошибочно
достигать намеченной зоны. Можно делать инъекции и «вруч-
ную». В этом случае закрепление инъекционной пипетки в ма-
нипуляторе Сингера дает более надежное и точное движение.
Рис. 3.10. Техника инъекции трансплантируемых клеток в 8-дневный эмбрион. В верхней части схемы показано положение
рычага управления при каждом маневре. Детальное описание процедуры см. в табл. 3.11.
92
Глава 3
Таблица 3.12. Инъекции in utero
1. Чтобы следить за объемом инъецируемой жидкости, рекомендуется ка-
либровать инъекционную пнпетку (разд. 9) в соответствии с удобными
объемами (т. е. 0,5 мкл). Зная внутренний диаметр стеклянной трубки
и принимая ее за стандартный цилиндр, легко рассчитать длину, занимае-
мую данным объемом вдоль невытянутой колонки пипетки, и нанести
серию делений проксимальнее ее суженного конца.
2. Закрепите инъекционную пипетку в инструментальном держателе, при-
крепленном к насосу (de Fonbrune), и заполните ее парафиновым маслом.
3. Анестезируйте беременную мышь (Avertin: 0,1 мл/5 г веса тела) Поме-
стите ее на предметный столик препаровального микроскопа и осветите
нижнюю брюшную область (рекомендуется осветитель).
4. Сделайте средне-брюшной надрез (около 1,0—1,5 см длиной) через кожу
и стенку тела нижней части живота. Чтобы уменьшить кровотечение,
прижгите ранку, делая разрез очень горячими ножницами.
5. Осторожно извлеките матку через отверстие.
6. Наберите донорские клетки или метящее вещество в инъекционную пипет-
ку, всасывая достаточное количество среды или метки так, чтобы мениск
парафинового масла находился в пределах калиброванной части пнпетки.
Всасывайте осторожно, чтобы масло не образовало в среде мелких ка-
пель. Закрепите инструментальный держатель на манипуляторе Сингера.
7. Захватите матку пинцетом вблизи децидуальной капсулы зародыша.
Продвиньте инъекционную пипетку так, чтобы ее верхушка расположи-
лась напротив средней линии децидуальной капсулы, примерно на трети
расстояния до аитимезометриального конца.
8. Сделайте отверстие в мышце матки концом инъекционной иглы от шпри-
ца. Введите инъекционную пипетку через это отверстие в середину де-
цидуомы.
9. Следя за движением мениска парафинового масла, инъецируйте ~0,5 мкл
в эмбрион.
10. Извлеките инъекционную пипетку.
11. Повторите эту процедуру (п. 7—10) с оставшимися децидуомами или
с теми, которые удобно инъецировать. Если инъекционная пипетка заку-
поривается децидуальной тканью, замените ее.
12. Возвратите матку в брюшную полость и зашейте стенку тела. Кожу мож-
но зашить или соединить зажимами.
9. Изготовление стеклянных
микрохирургических инструментов
9.1. Стекло
Все инструменты делаются из стеклянных капиллярных тру-
бок. Инъекционные и удерживающие пипетки сделаны из «тон-
костенных» трубок (наружный диаметр—1,0 мм, внутренний —
0,8 мм), их можно получить от Drummond Scientific Company,
Pennsylvania. Используется натриевое стекло или Pyrex; и из
того, и из другого получаются хорошие инструменты, но стекло
Pyrex легче моется. Прочные микроиглы делает из «тонкостен-
ных» трубок (н. д. 1,0 мм, в.д. 0,5 мм) фирма Leitz. Все ка-
Эксперименты с постимплантациоиными эмбрионами мыши 93
пиллярные трубки следует предварительно очистить: на 24 ч по-
грузить в хромовую кислоту и тщательно сполоснуть несколько
раз дистиллированной водой. Чистые трубки высушите в су-
шильном шкафу и храните в пыленепроницаемом контейнере.
9.2. Оборудование
Все инструменты изготовлены вытягиванием постепенно су-
жающихся игл, которые модифицированы таким образом, что
составляют целый набор. Необходимо иметь два прибора —
электродный вытягиватель и микрокузницу. Электродные вытя-
гиватели имеются в продаже. Может понадобиться вариант
прибора, который приспособлен для работы со стандартными
микроэлектродными капиллярными трубками, имеющими диа-
метр, несколько больший, чем 1,0 мм.
Удобна микрокузница, сконструированная де Фонбрюном; се
можно получить от Beaudouin (Париж). V-образная нить пла-
тиновой проволоки (диаметр 0,3 мм) с небольшим стеклянным
шариком, сплавленным с ее вершиной, должна подходить к
держателю нити. Микроскоп снабжен окуляр-микрометром и
дает увеличение примерно Х100.
9.3. Инструменты
Лучшее описание способов изготовления различных' стек-
лянных инструментов дал сам де Фонбрюн [33J. Его микрркуз-
ница содержит инструкцию, включающую несколько основных
процедур. Процедуры, которыми мы пользуемся в нашей лабо-
ратории при изготовлении прочных препаровальных игл и удер-
живающих пипеток, описаны в табл. 3.13, а пипеток для инъ-
екций—в табл. 3.14. Рис. 3.11 иллюстрирует эти процедуры,
(см. гл. 12, разд. 2.4.)
Иглы можно приготовить простым вытягиванием лейцевско-
го капилляра на электродном вытягивателе, но такие иглы мо-
гут повредить ткань, если они сломаются в процессе работы.
Более предпочтительны сплошные (без полости) иглы; они
обладают большей механической прочностью.
Инъекционные пипетки делают точно так же, как и удержи-
вающие, за исключением того, что их отламывают при меньшем
внутреннем диаметре. Окончательный диаметр инъекционной
пипетки будет зависеть от того, что предполагается инъециро-
вать (в пределах от 1 мкм для внутриклеточных инъекций до
60 мкм для крупных кусочков ткани). Если нужно перенести
сравнительно крупные кусочки ткани, требующие внутреннего
диаметра более 35 мкм, удобно иметь пипетку с заостренным
концом. Приспособления для заострения можно приобрести.
«4
Глава 3
Таблица 3.13. Изготовление сплошных игл и удерживающих пипеток
Сплошные препаровальные иглы
1. Вращая стекло, чтобы диаметр трубки не уменьшился в пламени бунзе-
новской горелки, сплавьте центральную часть капиллярной трубки в сплош-
ной стеклянный стержень. Отодвиньте трубку от источника нагревания и
спустя 1 сек, в течение которой она охладится, вытяните трубку, не умень-
шая наружного диаметра сплавившейся части.
2. Поместите капилляр в электродный вытягиватель. Вытягивайте, чтобы
получить иглу, длина суженной области у которой не менее 5 мм.
3. Для того чтобы игла во время операции шла параллельно дну чашки,
в которой проводят опыт, в трубке целесообразно сделать два изгиба,
как показано на рис. 11, Г,1, с помощью микрогорелки. Для ее изготовле-
ния необходимо прикрепить к концу отводящей газовой трубки инъекци-
онную иглу от шприца. Если при сплавлении капилляра на одной стороне
инструмента образуется заметное плечо из сплошного стекла, первый изгиб
делайте к этому плечу, отчего в инструменте окончательной формы ои
будет направлен от дна чашки.
4. Наконец, силиконируйте иглу, погрузив ее конец в Repelcote (Hopkin and
Williams).
Удерживающая пипетка
1. Используя «тонкостенный» капилляр, вытяните иглу на электродном вытя-
гивателе. Введите иглу в лейцевский инструментальный держатель и уста-
новите ее горизонтально на микрокузнице.
2. Поместите нить и горизонтальный стержень иглы (в области, где ее внут-
ренний диаметр равен 80 мкм) в одну фокальную плоскость поля зре-
ния микроскопа.
3. Нагревайте нить до тех пор, пока не начнет плавиться стекло
(рис. 11, Л, /), после чего приподнимите ее так, чтобы кончик сплавился
с вижней стороной капилляра (рис. 3.11, А, 2). При этом старайтесь не
повредить или ие изогнуть капилляр.
4. Прекратите нагревание нити. Сжатие охлаждающейся нити вызовет пра-
вильный чистый разлом капилляра перпендикулярно его длинной оси
(рис. 3.11, А,3).
5. Оплавьте конец пипетки, вновь нагрев нить и приближая ее концом к пи-
петке до тех пор, пока поверхность разлома последней не начнет утолщать-
ся (рис. 3.11, А, 4). Выключите нагрев.
Вполне удовлетворительные инструменты можно приготовить,
разбивая кончик капилляра о холодную нить. Если пипетку не-
однократно упирать в холодный стеклянный шарик, получается
разлом примерно на уровне нужного внутреннего диаметра.
Обычно эти разломы проходят не перпендикулярно, а под уг-
лом, но, поскольку конец пипетки оплавляется, незначительные
неровности линии разлома не имеют особого значения.
Прямые держатели и пипетки для инъекций in vivo подхо-
дят для манипуляций, осуществляемых в манипуляционной ка-
мере. Для манипуляций с 9-дневными эмбрионами или старше
необходимо дважды изогнуть пипетки над микрогорелкой. Эти
изгибы подобны тем, что описаны для сплошных игл (табл.
Эксперименты с постимплантацноннымн эмбрионами мыши 95-
Таблица 3.14. Приготовление инъекционных пипеток для работы
Способ 1
1. Вытяните полую иглу из тонкостенного капилляра на электродном вытя-
гивателе.
2. Закрепите иглу в инструментальном держателе, присоединенном при по-
мощи трубки к источнику сжатого воздуха нли к наполненному воздухом
шприцу на 20 мл.
3. Поместите инструмент горизонтально на мнкрокузницу. Переломите пи-
петку в месте внутреннего диаметра 15 мкм (рис. 3.11,5,/).
4. Продувая воздух через пипетку, прикоснитесь нагретой нитью к ее вер-
хушке так, чтобы стекло с ней сплавилось, но сразу же за местом сплав-
ления осталось небольшое отверстие для продуваемого воздуха
(рис. 3.11,5, 2).
5. Не прекращая подачи воздуха, быстро отодвиньте нить от верхушки пипет-
ки, в результате этого образуется прямой, сплошной точечный конец
(рис. 3.11,5,3). Если точечная нерхушка слишком велика или изогнута,
отломите конец и повторите сначала (от сплавления нити со стеклом).
6. Ориентируйте инструмент вертикально, так, чтобы его отверстие было на-
правлено к нити. Поднесите нагретую нить к отверстию, чтобы оплавить,
его края (рис. 3.11,5,4).
Способ 2
1. Разломите полую иглу в месте нужного диаметра (т. е. 50 мкм).
2. Сплавьте нагретую нить с концом пипетки, затем быстро отодвиньте ее,,
чтобы получить сплошную заостренную верхушку (рис. 3.11,5,/, 2).
3. Ориентируйте инструмент вертикально. Сфокусируйте струю воздуха мик-
рокузницы сразу за сплошной верхушкой (это поможет локализовать на-
гревательный эффект нити). Продувайте воздух через инструмент, удер-
живая нагретую нить у места будущего отверстия пипетки, но не прика-
саясь ею к стеклу. Повышайте температуру ннтн до тех пор, пока не-
появится маленький пузырек расплавленного стекла (рис. 3.11,5,3). Когда
пузырек прикоснется к нити, быстро выключите нагревание (рис. 3.11,5,4).
Пузырек лопнет, и на его месте останется сравнительно большое выступаю-
щее отверстие (рис. 3.11,5,5).
4. Сплавьте нить с краями апертуры, которая будет уменьшаться в размере
(рис. 3.11, В, 6).
5. Когда апертура уменьшится достаточно, выключите нить и сдвиньте ее
вертикально вниз. Выступающее стекло отломится (рис. 3.11,5, 7).
6. Оплавьте края апертуры (рис. 3.11,5,3).
3.13), хотя точный угол изгибов будет зависеть от того, как
установлен микроманипулятор (рис. 3.10). Как было замечено-
ранее, важно, чтобы концы удерживающей и инъекционной пи-
петок были наклонены вниз под очень малым углом (рис.
4.11, Г, 2), иначе плечо вытянутой части пипетки будет мешать
ее контакту с эмбрионом или донорской тканью.
Пипетка для инъекций in vivo должна отвечать двум основ-1
ным требованиям. Первое — инструмент должен проникать в
ткань легко, второе — отверстие пипетки во время ее движения
в ткани не должно закупориваться прилипающими клетками..
96
Глава 3
Рис. 3.11. Изготовление микрохирургических стеклянных инструментов (по
де Фонбрюну [33]). Описание процедуры дано в табл. 3.13 и 3.14.
А. Удерживающая и инъекционная пипетки для операций in vitro. Б. Инъек-
ционная пипетка для операций in vivo (способ 1). В. Инъекционная пипетка
для операций in vivo (способ 2). Г. Изгибы на концах инструментов 1—ко-
нец сплошной иглы; 2 — конец инъекционной или удерживающей пипетки для
работы в чашке Петри.
В связи с этим де Фонбрюн рекомендует пользоваться пипет-
ками со сплошным заостренным концом, при этом отверстие
пипетки должно располагаться сбоку, сразу за верхушкой
(рис» 3.11, Б, В). В табл. 3.14 описаны два способа их изготов-
ления. Способ 1 подходит для пипеток с маленьким диаметром,
в то время как способ 2 — для пипеток большего диаметра. Ес-
Эксперименты с постимплантационными эмбрионами мыши 97
ли между операциями возникает необходимость заменить инъ-
екционную пипетку, можно ввести растворы при помощи обыч-
ной инъекционной пипетки для работы in vitro.
10. Заключение
Анализ обособления первичных тканей в ходе гаструляции и
раннего эмбриогенеза представляет собой трудную задачу. Од-
нако в последнее время найдены новые способы изучения эм-
бриональных клеток в их нормальном окружении в составе
эмбриона на этих стадиях развития. Некоторые из соответству-
ющих методик описаны в данной статье. Во многих отношениях
они .могут показаться весьма несовершенными. Перед исследо-
вателем открывается простор для работы над их улучшением,
особенно в области культивирования целых эмбрионов и точ-
ных манипуляций с эмбрионами как in vitro, та и in vivo. Не-
смотря на методические трудности, в течение последнего деся-
тилетия гистологические и морфологические описания раннего
посгимплантационного эмбриогенеза были обогащены ценной
информацией, касающейся динамических аспектов развития, на-
пример стало больше известно о нормальной судьбе и потенции
развития разных тканей и частей эмбриона. Такая информа-
ция обогащает наши представления о процессах, ведущих к на-
чальной организации и дифференцировке плода.
Благодарности
Мне хотелось бы поблагодарить профессора Гарднера, док-
торов Кокрофта и Тэма за обсуждение рукописи.
Литература
1. Gardner R. L. (1983). Int Rev. Exp. Pathol., 24, 63.
2. Beddington R. S. P. (1987). In: Experimental Approaches to Mammalian
Development. Pedersen R. A. and Rossant J, (eds.), Cambridge University
Press, New York, in press.
3. Jolly J., Ferester-Tadie M. (1936). Arch. Anat. Microsc., 32, 323.
4. Rugh R. (1968). The Monse. Burgess Publishing Company, USA.
5. Snell G. D., Stevens L. C. (1966). In: Biology of the Laboratory Mouse
Green L. (ed.), McGraw-Hill, New York, p. 205.
6. Whittingham D. G., Wales R. G. (1969). Aust. J. Biol. Sci., 22, 1065.
7. Theiler K. (1972). The House Mouse. Springer-Verlag, Berlin.
V Levak-Svajger B., Svajger A., Skreb N. (1969). E.xperientia, 25, 1311.
9. Snow M. H. L. (1978). In: Methods in Mammalian Reproduction. Daniel J.
C. Jr. (ed.), Academic Press, New York, p. 167.
10. Gardner R, L., Rossant J. (1979). J. Embryol. Exp. Morphol., 52, 141.
>1. Kruse P. F., Patterson M. K, (eds.) (1973). Tissue Culture: Methods and
Applications, Academic Press, New York.
7—171
98
Глава 3
12. Но К., Takeuchi Т. (1984). J. Embryol. Exp. Morphol., 84, 49.
13. Jaenisch R. (1985). Nature, 318, 181.
14. Snow M. H. L. (1981). J. Embryol. Exp. Morphol., 65 (Suppl.), 269.
15. Rossant J., Of er L. (1977). J. Embryol. Exp. Morphol., 39, 183.
16. Diwan S. B., Stevens L. C. (1976). J. Natl. Cancer Inst., 57, 937.
17. Damjanov I., Solter D., Skreb N. (1971). Wilhelm Roux’ Arch. Entwicklungs-
mech. Org, 173, 228.
18. New D. A. T. (1978). Biol. Rev., 53, 81.
19. Sadler T. W„ New D. A. T. (1981). J. Embryol. Exp. Morphol., 66, 109.
20. Buckley S. K. L., Steele С. E„ New D. A. T. (1978). Dev. Biol., 65, 390.
21. Steele С. E. (1972). Nature, New Biol., 237, 150.
22. Tam P. P. L., Snow M. H. L. (1980). J. Embriol. Exp. Morphol., 59, 131.
23. Lowry О. H., Rosebrough N. J-, Farr A. L., Randall R. J. (1951). J. Biol.
Chem, 193, 265.
24. Smits-Van Prooije A. E., Poelmann R. E„ Dubbeldam J. A., Mentink M. T„
Vermeij-Keers C. (1986). Stain Technol., 61, 97.
25. Beddington R. S. P. (1981). J. Embryol. Exp. Morphol., 64, 87.
26. Beddington R. S. P. (1982). J. Embryol. Exp. Morphol., 69, 265.
27. Gardner R. L. (1978). In: Methods in Mammalian Reproduction. Daniel J. C.,
Jr. (ed.), Academic Press, New York, p. 137.
28. Lawson K., Meneses J. J., Pedersen R. A. (1986). Dev. Biol., 115, 325.
29. Lo C. W„ Gilula N. B. (1979), Cell, 18, 399.
30. Weissman I. L., Papaioannou V. E., Gardner R. L. (1978). In: Differentiati-
on of Normal and Neoplastic Haemopoetic Cells. Clarkson B., Marks P. A.,
Till J. E. (eds.), Cold Spring Harbor Laboratory Press, New York, p. 33.
31. Jaenisch R. (1980), Cell, 19, 181.
32. Fleischman R. A., Mintz B. (1979). Proc. Natl. Acad. Sci. USA, 76, 5736.
33. De Fonbrune P. (1949). Technique de Micromanipulation (Monographies
d’Institut Pasteur). Masson et Cie., Paris.
ГЛАВА 4
АНАЛИЗ МЕЙОЗА
В ПОЛОВЫХ КЛЕТКАХ ЧЕЛОВЕКА И МЫШИ
Энн Чендлей'1
1. Введение
Методы получения мейотических хромосом весьма усо-
вершенствовались с той поры, когда их морфологию изучали с
помощью давленых препаратов. Уже давно широко использует-
ся лучший и более быстрый способ выделения метафазных
хромосом, в основе которого лежит высушивание половых кле-
ток на воздухе после кратковременного гипотонического воз-
действия. Кроме того, в последние годы для анализа профазы
мейоза была предложена новая эффективная методика так на-
зываемого «распластывания» хромосом.
Цель этой главы — детальное описание доступной в настоя-
щее время техники воздушного высушивания и распластывания
ооцитов и сперматоцитов человека и мыши.
2. Получение препаратов
мейотических хромосом
высушиванием на воздухе
Получить гистологический материал гонад мыши не состав-
ляет труда, между тем образцы ткани семенника человека для
анализа мейоза по вполне понятным причинам не так легко до-
ступны. Возможность получения этого материала появляется в
случаях орхидэктомии пациентов в старческом возрасте при ле-
чении рака предстательной железы или при изучении бесплодия
молодых мужчин.
Мейоз начинается у самцов в период их полового созрева-
ния. Он представляет собой интегральную часть сперматогене-
за— процесса, начинающегося серией митотических делений
сперматогониев. После предмейотической Эмфазы клетки уже в
качестве сперматоцитов вступают в длительную профазу мейо-
за и затем претерпевают два деления, ведущих к образованию
гаплоидных гамет. На препарате высушенной на воздухе сус-
пензии клеток из семенников половозрелого самца с нормаль-
ной способностью к оплодотворению можно различить три типа
1 А. С. Chandley M.R.C. Clinical and Population Genetic Unit, Western
General Hospital. Grewe Road, Edinburgh EH4 2XU, UK.
7’
100
Глава 4
делящихся клеток: сперматогониальные метафазы и метафа-
зы I и II делений мейоза. В случае бесплодия сперматогенез
подавлен или блокирован, о чем свидетельствует полное отсут-
ствие или недостаток на препаратах клеток, находящихся на
поздних стадиях сперматогенеза.
Метафаза I (MI)—стадия мейоза, обычно используемая
для определения числа бивалентов, частоты и положения хиазм
и установления хромосомных аномалий, таких, как реципрок-
ные транслокации, имеющие конфигурацию мультивалентов.
На стадии MI у человека X и Y хромосомы соединены конец-в-
конец двумя короткими плечами (р), а у мыши — двумя длин-
ными (q). У небольшой части клеток обоих видов половые хро-
мосомы в MI лежат раздельно, высокая частота встречаемости
таких клеток указывает на стерильность особи. Клетки, нахо-
дящиеся во втором делении мейоза (МП), анализируются с
трудом из-ва того, что плечи хромосом сдвигаются и перекры-
вают друг друга, но после распластывания они могут и в дей-
ствительности используются для выявления анеуплоидов, воз-
никающих в результате ошибок при расхождении хромосом во
время первого мейотического деления [1, 2J.
Методика воздушного высушивания, обычно используемая
для приготовления препаратов мейотических хромосом челове-
ка и мыши, принадлежит Ивансу [3J!). Этот способ в том виде,
как он был впервые описан, применяется для анализа мейоза
у мышей (разд. 2.1). Для материала человеческих семенников
этот метод модифицирован в нашей лаборатории (разд. 2.2).
2.1. Метод суховоздушных препаратов для мышей
1. Извлеките семенники из белочной оболочки (tunica albu-
ginea) и поместите их в изотонический раствор цитрата
натрия (2,2% в/о) при комнатной температуре.
2. Перенесите семенники в свежий 2,2%-ный раствор цитра-
та натрия в маленькой чашке Петри и осторожно вытяни-'
те канальцы. Придерживая массу канальцев тонким пря-
мым пинцетом, осторожно выдавливайте их содержимое
другим, тонким закругленным пинцетом. Когда канальцы
станут плоскими и матовыми, оставьте их и осторожно
перенесите надосадочную жидкость в 15-мл центрифуж-
ную пробирку.
3. Отцентрифугируйте полученную клеточную суспензию
5 мин при 50 g. При этом большинство спермиев останет-
ся в суспензии, а в осадке будут клетки большего разме-
1 Аналогичный метод несколько ранее предложен А. П. Дыбаном (Цито-
логия, 1979 г.). —Прим. ред.
Анализ мейоза в половых клетках человека и мыши
101
ра, включая сперматоциты. Удалите супернатант, а осев-
шие клетки ресуспендируйте приблизительно в 3 мл
1%-ного раствора цитрата натрия.
4. Оставьте клетки в гипотоническом растворе на 12 мин
при комнатной температуре. Отцентрифугируйте при 50 g
в течение 5 мин с медленным ускорением.
5. Удалите как можно больше супернатанта. Встряхните
осадок так, чтобы слой суспензии прилип к стенкам про-
бирки. Быстро прибавьте к суспензии клеток 0,25 мл
фиксатора метанол: ледяная уксусная кислота (3:1).
Тщательно перемешайте пастеровской пипеткой. Добавь-
те еще фиксатора по стенке, продолжая перемешивать,
пока пробирка не наполнится примерно на треть. Через
5 мин осадите клетки центрифугированием и ресуспенди-
руйте в свежем фиксаторе. Спустя 10 мин снова смените
фиксатор, контролируя вид суспензии; она должна быть
слегка мутной.
6. Сделайте пробный препарат, нанеся каплю суспензии на
обезжиренное (вымытое кислотой со спиртом) предмет-
ное стекло при комнатной температуре. Если стекло тща-'
тельно очищено и фиксация удовлетворительна, капля
равномерно растечется; осторожно подув на стекло, мож-
но ускорить высыхание капли. Изучайте при помощи фа-
зового контраста. Если клеточная суспензия слишком
разведена, отцентрифугируйте снова и добавьте меньший
объем фиксатора, если слишком густая — больше фикса-
тора.
2.2. Модификация методики при работе
с человеческим материалом
1. Соберите материал тестикулярной биопсии в гипотониче-
ский раствор 1%-ного цитрата натрия.
2. Спустя 30 мин очень мелко нарежьте семенные канальцы
ножницами в чашке Петри, слегка покачивая ее, чтобы
крупные куски канальцев осели на дно. Отсосите клеточ-
ную суспензию пипеткой и отцентрифугируйте при 450 g
(судя по нашему опыту, чем тоньше суспензия, тем лучше
фиксируются препараты).
3. Удалите большую часть надосадочной жидкости, так что-
бы оставшаяся едва покрывала клетки; перемешайте сус-
пензию, осторожно постучав по стенке пробирки.
4. Добавьте двойной объем фиксатора (метанол : ледяная
уксусная кислота, 3:1). Вносите медленно, по стенке про-
бирки. Энергично, но осторожно пипетируйте, стараясь
разрушить комки клеток. Добавьте фиксатора до 5 мл.
Центрифугируйте 8 мин при 450 g.
102
Глава 4
5. Удалите супернатант. Прибавьте около 5 мл фиксатора и
оставьте суспензию на час при комнатной температуре.
6. Центрифугируйте 8 мин при 450 g.
7. Внесите ~ 1 мл свежеприготовленного фиксатора. (До-
бавляйте фиксатор, пока суспензия слегка не помутнеет.)
8. Дайте клеточным сгусткам осесть на дно. Сделайте проб-
ный препарат, высушив на воздухе каплю суспензии на
предметном стекле. Просмотрите в фазово-жонтрастном
микроскопе и, если суспензия слишком разведена, снова
отцентрифугируйте и отбросьте часть фиксатора. Ресус-
пендируйте.
2.3. Рутинная окраска
Для рутинного окрашивания препаратов рекомендуется све-
жеприготовленный краситель Гимза (если используется Gurr
«R66», нужно к 1 мл добавить до 40 мл буфера с pH 6,8, приго-
товленного с помощью буферных таблеток Gurr). Окрашивай-
те 5 мин в сосуде Коплина.
В нашей лаборатории с успехом применяется окрашивание
карболовым фуксином [4].
Раствор А: основной фуксин 3 г
70%-ный этанол 100 мл
Раствор Б: раствор А 10 мл
5%чный фенол 90 мл
Краситель: раствор Б 45 мл
ледяная уксусная кислота 6 мл
37%-ный формальдегид 6 мл
Окрашивание срезов свежеприготовленным красителем за-
нимает не менее 30 мин, но по мере хранения краситель «созре-
вает» и время окрашивания укорачивается. Карболовый фук-
син особенно уместен при радиоавтографии: он не взаимодейст-
вует с эмульсией и не смывается в проявителе. Следовательно,
можно окрашивать клетки до нанесения эмульсии, например в
экспериментах по гибридизации in situ. На рис. 4.1 представле-
ны препараты клеток, полученных при биопсии семенника че-
ловека на стадиях сперматогониального митоза, MI и МП.
Препараты высушивали на воздухе и красили карболовым фук-
сином. Среди хромосом MI четко выделяется похожий на цепоч-
ку бивалент XY. Аутосомальные биваленты могут быть располо-
жены в порядке их размера, но идентификация хромосом в би-
валентах невозможна. В МП можно узнать хромосому 9 по вы-
тянутой области в длинном плече, соответствующей вторичной
перетяжке. Мышиные хромосомы в МП представлены на
рис. 4.1, Г.
Анализ мейоза в половых клетках человека и мыши
103
Рис. 4.1. Высушенные на воздухе метафазы клеток из суспензии семенников
человека и мыши (окрашены карболовым фуксином).
4. Митоз в сперматогонии человека. Б. Мейотическая стадия MI у человека
(стрелка указывает на бивалент XY). В. Мейотическая стадия МП у человека
(отмечена хромосома 9 с характерной вторичной перетяжкой). Г. Стадия МП
у мыши (отмечена хромосома 10).
2.4. С-окраска
С-окраска способствует идентификации мейотических би-
валентов. У мыши во всех хромосомах, кроме Y, С-сегмент рас-
положен вблизи центромеры (см. также гл. 5, разд. 5.2). На
рис. 4.2, Л представлены С-окрашенные хромосомы мыши (ста-
тин MI). Хорошо заметна транслокация Т( 14; 16)6 Са. В Х-хро-
мосоме и во всех аутосомах выделяется С-сегментный гетеро-
хроматин. Для стерильных самцов характерна ассоциация меж-*
ду центромерой Х-хромосомы и транслокационным квадрива-
лентом [5J. В Y-хромосоме четкий С-сегмент не выявляется.
На рис. 4.2,5 представлен С-окрашенный МП-юперматоцит
мыши. Рис. 4.2,5 дает представление о том, как выглядит
С-окрашенная MI-клетка человека, гетерозиготного по рецип-
рокной транслокации t(9; 22). Хорошо различаются биваленты
1. 16 и Y, а в квадривалентах (Q) можно видеть крупные
С-сегменты хромосомы 9. Метод выявления С-сегментов в
мейозе предложили Чендлей и Флетчер [6]. Он представляет
гобой модификацию методики Самнер [7]. (С-окрашивание ми-
тотических хромосом описано в гл. 5, разд. 5.2.)
104
Глава 4
Рис. 4.2. Сперматоциты, окрашенные С-методом.
А. Стадия MI в мейозе мыши. Хорошо видна транслокация (14;15)6СА. От-
мечена связь между квадривалентом реципрокной транслокации (CHIV) и би-
валентом XY. Б. МП нормального самца мыши. В. MI человека, гетерозигот-
ного по реципрокной транслокации (9;22). В квадриваленте (Q) видны четкие
С-сегмеиты хромосомы 9. Биваленты 1 и 16, так же как и Y-хромосому, мож-
но идентифицировать по крупным С-сегментам.
1. Поместите предметные стекла в 0,2 М НС1 при комнат-
ной температуре на 1 час.
2. Сполосните деионизированной водой.
3. Поместите стекла в 5%-ный гидроксид бария при 50°C
на 30 сек (человеческий материал), 4%-ный гидроксид
бария при 37 °C на 30 сек (мышь).
4. Сполосните деионизированной водой.
5. Поместите стекла в стандартный солевой раствор цитра-
та (2XSSC) при 60°C на 1 час.
6. Сполосните деионизированной водой.
7. Окрашивайте по Гимза в течение 45 мин—1 часа (см.
разд. 2.3).
8. Сполосните деионизированной водой, хорошо просушите
в течение нескольких минут, проведите через ксилол и
заключайте в бальзам.
Анализ мейоза в половых клетках человека и мыши 105
' 2.5. Q-окраска
Флуоресцентное окрашивание оказалось неинформативным
для анализа мейоза у мыши, однако этот же метод при изуче-
нии человеческого материала способствовал выявлению не-
флуоресцирующего короткого плеча Y-хромосомы, спаренного с
Х-хромосомой в метафазе I мейоза [8]. Применение флуоресци-
рующего АТ-специфического антибиотика дистамицина А в со-
четании с АТ-специфическим красителем 4',6'-диамидино-2-фе-
пилиндолом (ДАФИ) продемонстрировало у мейотических бива-
лентов человека яркое свечение хромосомных районов, содер-
жащих структурный гетерохроматин [9]. Флуоресценция обна-
руживается в парах хромосом 1, 9, 16 и У-рсромосоме, а иногда
п в биваленте 15 (рис. 4.3). Техника окрашивания заключается
в следующем.
1. Нанесите на стекло раствор дистамицина А (Sigma)
(0,1—0,2 мг/мл в буфере Мак-Илвина: лимонная кисло-
та— Na2HPC>4, pH 7,0), накройте покровным стеклом. Ин-
кубируйте при комнатной температуре 15 мин в темноте,
во влажной камере.
2. Смойте покровное стекло струей деионизированной воды.
3. Нанесите одну большую каплю раствора ДАФИ (Sigma)
(0,2—0,4 г/мл в буфере Мак-Илвина, pH 7,0) и опустите
на каплю свежее покровное стекло. Поместите предмет-
ное стекло во влажную камеру на 30 мин.
4. Смойте струей деионизированной воды.
5. Нанесите 2 капли буфера МакчИлвина.
6. Очень осторожно промакните фильтровальной бумагой.
7. Зафиксируйте границы покровного стекла раствором
каучука (Holdtite).
Дистамицин А теряет стабильность в водном растворе, по-
этому такой раствор хранить не рекомендуется. Окрашенные
препараты при первом просмотре могут обесцветиться. Для ста-
билизации их необходимо примерно в течение дня хранить при
температуре 4 °C в темноте.
2.6. Картирование пахитенных хромосом
Для получения качественных препаратов пахитенных хро-
мосом, которые могут быть использованы при картировании ин-
дивидуальных бивалентов, была предложена специальная ме-
тодика воздушного высушивания с длительным гипотоническим
воздействием при комнатной температуре или с более кратким
гипотоническим воздействием при повышенных температурах
[10—13]. На стадии поздней пахитены в каждом биваленте
становятся заметными линейно расположенные компактизиро-
106
Глава 4
Рис. 4.3. Препараты MI мейоза у человека, окрашенные последовательно кар-
боловым фуксином (А) и ДА/ДАФИ (Б). Флуоресценция позволяет иденти-
фицировать биваленты 1, 9, 15, 16 и Y-хромосому. Бивалент XY отмечен
стрелкой (на А).
ванные районы хроматина, или «хромомеры». Их число, размер
и последовательность для каждой хромосомы постоянны. Обна-
ружено поразительное соответствие в расположении хромомер
и митотических G-сегментов. Кроме того, С-окраска дает воз-
можность локализовать положение центромеры. Все это спо-
собствовало созданию пахитенных хромосомных карт человека
Анализ мейоза в половых клетках человека и мыши
107
Рис. 4.4. Человеческий сперматоцит на стадии пахитены; каждый из 22 ауто-
сомных бивалентов идентифицируется на основании паттерна хромомер (А)
и положения центромеры (Б). Я — ядрышко; ПП—половой пузырек. (Из ра-
боты [12], воспроизводится с разрешения.)
[12] и мыши [13] (рис. 4.4). Методика, используемая для при-
готовления препаратов пахитенных клеток человека, разрабо-
тана Лучиани [12].
108
Глава 4
1. Погрузите фрагменты семенника в 10 мл 0,88%-ного КС1
и инкубируйте при комнатной температуре в течение
8—10 ч.
2. Перенесите в фиксатор метанол: ледяная уксусная кис-
лота (3:1) и оставьте на ночь при комнатной темпера-
туре.
3. На следующий день измельчите фрагменты в фиксаторе.
4. Перенесите пипеткой клеточную суспензию в коническую
виалу и центрифугируйте 7 мин при 150 g.
5. Ресуспендируйте сгусток в 5 мл 45%-ной ледяной уксус-
ной кислоты, затем немедленно отцентрифугируйте при
150 g 5 мин.
6. Сделайте препараты, нанеся капли суспензии из пасте-
ровской пипетки на чистые, предварительно охлажденные
стекла и осторожно высушите над слабым пламенем газо-
вой горелки.
7. Окрасьте раствором Гимза с фосфатным буфером
(pH 6,8) (см. разд. 2.3).
8. Чтобы выявить центромеры, удалите краситель Гимза ме-
танолом и поместите стекла в 1 М НС1 на 5 мин при ком-
натной температуре. Промойте в воде и обработайте
5%-ным раствором гидроксида бария (3 мин при 58°C).
После промывания поместите стекла на 20 мин в 2XSSC
при 58 °C и доведите pH до 7,0. Снова сполосните стекла
и окрасьте по Гимза.
Для демонстрации рисунка расположения хромомер одни
и те же клетки должны быть сфотографированы дважды: после
этапа 7 и после завершения этапа 8.
Другой способ препарирования пахитенных хромосом — воз-
действие гипотоническим раствором КС1 в течение 1 ч при
37 °C — описан для человека Хангерфордом [10], а для мыши —
Фэнгом и Ягелло [13]. Полученные по этой методике препараты
после окраски по Гимза позволяют разглядеть в 9-й хромосоме
человека «парамеры» — тельца, различимые только на пахитен-
ной стадии [10, 14, 15]. Они соответствуют С-положительным
районам данной хромосомы, видимым в метафазе.
2.7. Использование высушенных на воздухе
мужских мейотических препаратов
для молекулярных анализов
Препараты пахитенных хромосом, приготовленные способом,
описанным в разд. 2.6, были использованы для локализации ге-
нов при гибридизации in situ [16, 17]. Сделан вывод, что раз-
решение, полученное в результате применения этого метода, зна-
чительно выше, чем в хромосомах соматических клеток даже
Анализ мейоза в половых клетках человека и мыши
109
при использовании высокоэффективного окрашивания [16, 17].
Локализовать последовательности в 9-й хромосоме человека
in situ удалось на высушенных метафазах MI и МП, приготов-
ленных по прописи, рекомендуемой в разд. 2.2 [15]. На рис. 4.5
показана последовательность Xbi вторичной перетяжки 9-й хро-
мосомы. Детали процедуры гибридизации in situ можно узнать
нз оригинального источника, а также из разд. 6 гл. 5 данного
руководства.
Недавно на мейотических клетках человека [18] и мыши
[19, 20] были выполнены эксперименты с ник-трансляцией in si-
tu в сочетании с радиоавтографией после воздушного высушива-
ния. В этих экспериментах препараты хромосом обрабатывали
на стекле ДНКазой I, ДНК-полимеразой I и радиоактивными
дезоксирибонуклеотидами. Районы хромосом, особенно чувстви-
тельные к нуклеазной атаке (с более открытой конформацией
хроматина), могут быть выявлены по распределению зерен се-
ребра на радиоавтографических препаратах. Детали этой мето-
дики (аналогичной той, что была предложена Керемом для со-
матических хромосом [21]) читатель найдет в оригинальных
статьях.
3. Приготовление препаратов женских мейотических хромосом
высушиванием на воздухе
3.1. Профаза мейоза
Первое мейотическое деление у самки начинается в эмбрио-
генезе и ооциты на стадии профазы вплоть до диплотены обна-
руживаются лишь в яичнике плода. Примерно ко времени рож-
дения ооциты вступают в стадию покоя (диплотена или диктио-
тена), которая заканчивается незадолго до овуляции. Ранние
цитогенетические исследования профазы мейоза в оогенезе при
помощи методик воздушного высушивания включают работы
Рерборна и Хэнсмана на мышах [22] и на человеке Лучиани
в Сталь [23], Курило [24].
У мыши профаза мейоза приходится на 13-й день беремен-
,ностщ максимальнее число пахитен наблюдается поТсле 16—
18 дня. У женщины мейоз начинается к концу первого тримест-
ра и далее, по мере развития беременности, все больше и боль-
ше клеток вступает в профазные стадии лептотены, зиготены
и пахитены. Изучение яичников абортированных человеческих
плодов показало, что клетки на стадии пахитены обнаружива-
ются с 16-й по 23-ю нед. беременности [25].
В последние годы техника приготовления препаратов феталь-
ных ооцитов человека и мыши благодаря методике микрорас-
J10
Глава 4
Рис. 4.5. Гибридизации in situ с зондом последовательности ХЬ] хромосомы
9 человека. Бивалент 9 отмечен стрелкой. (Из работы [15], воспроизводится
с разрешения )
пластывания намного продвинулась, качество препаратов стало
превосходным, возросла их разрешающая способность, поэтому
все дискуссии о целесообразности изучения хромосом в оогене-
зе на стадии профазы мейоза могут быть отставлены (разд. 4.3).
3.2. Препараты ооцитов Ml и МП,
полученные при культивировании in vitro
Ооциты в M.I получают при помощи культивирования in vit-
ro со стадии блокированной диктиотены зародышевого пузырь-
ка. Эта методика впервые была применена Пинкусом и Энцма-
ном [26] на ооцитах кролика. Продолжение мейоза было сти-
мулировано высвобождением ооцита из фолликула [27]. Наи-
более ранняя публикация, посвященная методике культивирова-
ния мышиных и человеческих ооцитов, принадлежит Эдвардсу
[28]. При успешном культивировании удается получить ооциты
на стадии МП; альтернативный способ заключается в сборе све-
жеовулированных ооцитов из фаллопиевых труб; соответствую-
щая процедура описана в разд. 3.3.
В нашей лаборатории препарирование яиц мыши, созревших
in vitro, с успехом производится по методу Спида [29].
Анализ мейоза в половых клетках человека и мыши
111
10 мкм
г»-.—«•, .
Рис. 4.6. Ооцит мыши, выделенный из фолликула, стрелкой отмечен его заро-
дышевый пузырек (А); после культивирования до MI (Б). (Из работы [29],
воспроизводится с разрешения.)
1. Вырежьте яичники и поместите их в физиологический рас-
твор (0,9%). Воспользовавшись препаровальным микро-
скопом, проколите крупные фолликулы стерильным скаль-
пелем.
2. Яйца с четко видимым зародышевым пузырьком (ЗП)
(рис. 4.6,А) перенесите стерильной пипеткой в культу-
ральную среду. В качестве среды используется эмбрио-
нальная телячья сыворотка (ЭТС 9,5 мл) и триптозофос-
фатный бульон (Oxoid, 0,5 мл), содержащий 0,25 мМ
пирувата натрия, 1,0 мМ глутамина и 5,56 мМ глюкозы.
3. Удалите приставшие к яйцу фолликулярные клетки, пипе-
тируя микропипеткой с узким отверстием.
4. Поместите 20—40 яиц в 1 мл среды в центральную лунку
чашки 60X15 мм для органной культуры (Falcon ЗОЮ)
и культивируйте под стерильным парафиновым маслом
(1 мл BDH) при 37°C в СОа-инкубаторе (5% СОг) с вы-
сокой влажностью. Через 5 ч яйца вступят в MI и через
17 ч —в МП.
5. Промойте яйца дважды и оставьте на 20 мин в свежем
1%-ном растворе цитрата натрия при комнатной темпера-
туре. Нанесите микрокаплю 1%-ного цитрата натрия с яй-
цом на обезжиренное стекло, воспользовавшись тонкой
пипеткой. На обратной стороне стекла отметьте место
кружком.
6. Фиксируйте по Тарковскому [30] (процедура также опи-
сана в гл. 5, разд. 4 этого руководства). Из другой тон-
кой пипетки, установив ее конец точно над поверхностью
капли с яйцом, нанесите несколько капель фиксатора (ме-
танол : ледяная уксусная кислота, 3:1). Оптимальное чис-
ло капель фиксатора — три.
112
Глава 4
7. Окрашивайте 5 мин 2%-ным раствором Гимза или дру-
гим рутинным красителем (см. разд. 2.3) или используйте
С-окраску, как описано в разд. 2.4.
Другой метод культивирования яиц до стадии MI и фикса-
ции описан Кемигучи [31], модификация для мыши предложе-
на Сугавара и Микамо [32]. Эта техника предполагает более
медленную фиксацию, что снижает возможность потери хро-
мосом.
1. Вырежьте яичники между 12.00 и 18.00 ч диэструса, т. е.
в середине астрального цикла, что контролируется осмот-
ром вагины. Поместите их на часовое стекло с раствором
Хэнкса и соберите яйца из крупных фолликулов, прока-
лывая их острой иглой.
2. Удалите прикрепившиеся клетки кумулюса, пипетируя
тонко оттянутой пипеткой.
3. Яйца с четким зародышевым пузырьком культивируйте
в 1 мл среды 3,5—6 ч при 37 °C в атмосфере с 5% СО2.
В качестве культуральной среды следует использовать
смесь, состоящую из 85% ТС199 (Gibco Biocult) и 15%
ЭТС. В этой среде 72% яиц достигает ML
4. Для получения яиц в МП требуется более длительный
период культивирования (см. выше). Но они могут быть
извлечены и из яйцеводов после овуляции (см. разд. 3.3).
5. Инкубируйте яйца мыши с 0,02%-ной проназой (Sigma)
в течение 15—20 сек для размягчения блестящей обо-
лочки.
6. Обработайте двумя гипотоническими растворами: снача-
ла 40%-ной ЭТС 10 мин, затем 1%-ным цитратом натрия
40 мин, в обоих случаях при 37 °C.
7. Осторожно перенесите яйца с небольшим количеством
раствора 1%-ного цитрата натрия в смесь абсолютный
метанол : ледяная уксусная кислота : дистиллированная
вода (5:1:4). Это воздействие растворяет оболочку и ща-
дяще фиксирует яйца. Вплоть до этого этапа чашку или
часовое стекло следует менять при каждой смене раство-
ра. Все этапы контролируют под микроскопом.
8. Спустя 5 мин после помещения яиц в указанный выше
раствор втяните одно из них вместе с небольшим объемом
фиксирующего раствора в шпеманновскую пипетку с тон-
ким концом и перенесите на обезжиренное стекло в кру-
жок (маркированный с обратной стороны). После растека-
ния фиксатора яйцо приклеится к стеклу недалеко от кон-
чика пипетки.
9. Немедленно вслед за этим еще раз фиксируйте яйцо раст-
вором Карнуа.
Анализ мейоза в половых клетках человека и мыши
113
10. Поместите стекло в сосуд Коплина с тем же фиксатором
и оставьте по меньшей мере на 20 мин.
11. Погрузите стекло в смесь абсолютный метанол: ледяная
уксусная кислота: дистиллированная вода (3:3:1) на
1 мин. Это способствует уплощению яиц прн высушивании.
12. Очень медленно извлеките стекло и высушите его возду-
хом, увлажненным прохождением через воду при 37 °C.
13. Окрашивайте по Гимза (разд. 2.3) или С-методом
(разд. 2.4).
Препараты, приготовленные по этой методике, демонстриру-
ют лучшее распластывание и сохранность хромосом [32], чем
полученные любым другим из упомянутых способов, включая
и по Тарковскому [30].
3.3. Препараты ооцитов в МП после овуляции
Сбору яиц в MI и МП предшествует стимуляция мейоза
гормональной инъекцией самки (гл. 1, разд. 5.4). Ооциты
в MI следует собрать из овариальных фолликулов спустя опре-
деленное время после введения фолликулостимулирующего гор-
мона гонадотропина сыворотки жеребой кобылы (ГСЖК) и че-
ловеческого хориогонадотропина (чХГ). Ооциты в МП извлека-
ют из ампул фаллопиевых труб после овуляции. Соответствую-
щая методика для мыши описана Рёборном и Хэнсманом [22].
1. Инъецируйте 2,5 ME ГСЖК и через 48 ч 2,0 ME чХГ
внутрибрюшинно.
2. Извлеките ооциты в MI из зрелых фолликулов через 5 ч
после чХГ, как описано в разд. 3.2.
3. Соберите ооциты в МП через 15 ч после инъекции чХГ нз
ампул фаллопиевых труб (см. гл. 2, разд. 2.1). Исполь-
зуйте для этого среду, предварительно нагретую до 37°C.
4. Обработайте клетки кумулюса, окружающие яйцо в МП,
гиалуронидазой 100 МЕ/мл (тип III, Sigma), в течение
2—5 мин (см. гл. 2, разд. 2.1).
Дальше продолжайте с п. 5 методики, описанной в разд. 3.2
для препарирования и окрашивания хромосом [29].
Яйцо мыши в МП после С-окраски представлено на рис. 4.7.
Применение этого способа окраски в большой степени способ-
ствует точному подсчету хромосом МП, что необходимо, напри-
мер, при изучении анеуплоидии.
Процедура, стимулирующая суперовуляцию у человека,
описана Ягелло [34], а недавно появилось сообщение о том,
как извлекать яйцеклетки для оплодотворения in vitro после
применения чХГ [35].
Следует заметить, что ни одному из исследователей не уда-
лось получить препараты яйцеклеток человека в MI такого ка-
е—171
1114
Глава 4
чества, чтобы можно было надежно подсчитать хиазмы. Другие
виды млекопитающих в этом отношении не представляют
серьезной проблемы, и совершенно неприятно, почему эта зада-
ча с таким трудом решается на человеческом материале.
4. Микрораспластывание
для профазного анализа
4.1. Поверхностное распластывание
для электронно-микроскопического анализа сперматоцитов
Разработка простого и быстрого метода поверхностного мик-
рораспластывания для выявления синаптонемного комплекса
в профазе мейоза сперматоцитов насекомых [36] революцио-
низировала наши представления о поведении хромосом в про-
цессе этой важной стадии мейоза. Применение этой методики
к сперматоцитам млекопитающих [37—39] продемонстрировало
ее возможности как для анализа аномалий в поведении хромо-
сом в профазе мейоза, так и для анализа нормального мейоза.
В современной цитогенетической литературе накопилось множе-
ство данных по «распластыванию», охватывающих все виды
эукариот от растений до человека.
Распластанные препараты синаптонемного комплекса (СК)
получены благодаря использованию поверхностного натяжения
Анализ мейоза в половых клетках человека и мыши
115
водного раствора. СК. к которому прикрепляются хроматино-
вые фибриллы, впервые появляется перед или во время синап-
са и исчезает при или после расхождения хромосом. Появление
признаков спаривания в СК точно отражает поведение конъю-
гирующих мейотических хромосом в течение профазы. Техника
распластывания не годится для клеток, вступивших в метафазу.
Существует большое разнообразие способов распластывания и,
конечно, было бы невозможно представить здесь точные детали
каждого из них. Тем не менее можно дать некоторые общие
рекомендации.
Существуют два принципиальных метода, два уровня на-
блюдений— светооптический (СМ) и электронно-микроскопиче-
ский (ЭМ). Первый подразумевает сбор распластанных клеток
с поверхности раствора соли или сахарозы на стекло и обычно
используется для распластывания сперматоцитов, в достаточ-
ном количестве имеющихся в суспензии. Второй включает осаж-
дение клеток на поверхность стекла через раствор сахарозы,,
соли или другого вещества и применяется главным образом для
ооцитов, но может быть использован и в случае суспензии спер-
матоцитов.
Методика поверхностного распластывания сперматоцитов
впервые была предложена Мозесом [37], она основана на спо-
собе Каунса и Мейера [36].
1. Поместите семенники или биоптат тестикул в минималь-
ную среду Игла (Gibco Biocult, МЕМ без глутамина),
осторожно мацерируйте канальцы закругленным тупым
пинцетом. Втяните клеточную суспензию в шприц 1-мил-
лилитровой иглы. Опустите шприц, чтобы крупные фраг-
менты ткани осели, подержите так в течение минуты.
Вылейте их и отбросьте. Оставшуюся чистую суспензию
несколько раз вылейте и втяните в шприц, чтобы разру-
шить комки клеток.
2. Снова доведите объем жидкости до 1 мл, добавив МЕМ,
вылейте в коническую стеклянную центрифужную про-
бирку и центрифугируйте 5 мин при 150g при комнатной
температуре.
3. Супернатант отбросьте, оставив объем, примерно вдвое
превышающий объем сгустка. Ресуспендируйте сгусток.
Держите суспензию на льду.
4. Осторожно возьмите микропипеткой каплю суспензии
и прикоснитесь ею к поверхности распластывающего
раствора. Последний состоит из профильтрованного
0,5%-ного раствора NaCl в черной эмбриологической
препаровальной чашке. Прежде чем внести клеточную
суспензию, протрите поверхность оптической или невор-
систой туалетной бумагой, отрегулируйте уровень жид-
8*
116
Глава 4
кости так, чтобы ее поверхность была слегка выпуклой.
После нанесения клеток дайте поверхности стабилизиро-
ваться в течение 1—2 мин.
5. Подготовьте медные сетки (Pelco: GC-100) следующим
образом. Нанесите на них пленку 0,3%-ного формвара,
затем напылите углем. Перед использованием сделайте
сетки гидрофильными путем ионизации их слабым раз-
рядом (2 мин, 1,3 кВ, 0,1 Torr). Пинцет для захвата се-
ток должен быть абсолютно чистым, после каждой ма-
нипуляции его нужно очищать, прокалывая концами ват-
манскую фильтровальную бумагу № 1.
6. Прикоснитесь сетками (каждой не более одного раза)
к разным районам поверхности, несущей клетки. Исполь-
зуйте примерно 10 сеток.
7. Фиксация. Фиксатором служит 10%-ный параформаль-
дегид в 0,1 М растворе сахарозы; нагрейте до 60—80 °C
с шестью каплями NaOH, помешивая, пока раствор не
станет прозрачным. При помощи боратного буфера с pH 9
доведите pH до 8,5. Профильтруйте и храните при 4 °C
не дольше 1 недели.
8. Быстро опустите сетки на чистую поверхность фиксатора
в маленькой чашке (по 5 сеток на чашку), прикройте
крышкой и оставьте по меньшей мере на 5 мин (но не
больше 10 мин). Можно поступить иначе: распределите
10 двойных капель фиксатора на восковой пластине (зуб-
ной воск или парафин, застывший на стеклянной поверх-
ности) и опустите каждую сетку на поверхность капли
(1 сетка на каплю, каждую каплю используйте один
раз). В качестве крышек при фиксации используют-
ся имеющиеся под рукой маленькие перевернутые чаш-
ки.
9. После фиксации выньте каждую сетку зажимающим
антикапиллярным пинцетом, удалите избыток фиксатора,
прикоснувшись краем сетки к поверхности фиксатора,
а затем положите сетку лицевой стороной вниз на по-
верхность 0,4%-ного Photoflo (Eastman Kodak, свеже-
приготовленного на боратном буфере, pH 8 и профиль-
трованного перед употреблением). Держите 20—30 сек,
осторожно покачивая. Промокните сетку, прикоснувшись
к краю фильтровальной бумаги, просушите под лампой
накаливания и поместите на фильтровальную бумагу
в чашку Петри для полного высыхания. Если Photoflo
перейдет на противоположную сторону сетки, возможны
артефакты. Сетки можно окрашивать в любое время
после высушивания (некоторые были окрашены год
.спустя).
Анализ мейоза в половых клетках человека и мыши
117
10. Окрашивание. В качестве красителя следует использо-
вать свежеприготовленную и профильтрованную смесь
4 %-ной фосфорновольфрамовой кислоты (ФВК) и
95%-ного этанола (1:3). Воспользовавшись антикапил-
лярным пинцетом, поместите сетку лицевой стороной
вниз на поверхность спиртовой ФВК на 1 мин, затем пе-
ренесите на поверхность 95%-ного этанола для сполас-
кивания в течение 15—30 сек, осушите фильтровальной
бумагой и дайте высохнуть на воздухе. Замечание: сле-
дует позаботиться о том, чтобы ни краситель, ни спирт не
затекли на обратную поверхность сетки, иначе в процес-
се высушивания поверхностное натяжение может разор-
вать угольно-формваровую пленку.
И. Поместите сетки на предметное стекло «лицом» вверх, за-
кройте покровным стеклом № 1 и смотрите с сухими фа-
зовыми объективами Х20 или Х40. На препарате хоро-
шо видны распластанные сперматоциты и синаптонемные
комплексы (СК)- Нужные сетки следует быстро ото-
брать.
12. Для анализа препаратов следует пользоваться малым
увеличением ЭМ (Х250—3000).
Вышеописанный метод был предложен .специально для элек-
тронно-микроскопического анализа СК. Однако, поскольку СК
п другие структуры, такие, как районы ядрышкового организа-
тора (ЯОР) и пластины прикрепления, видны и на уровне све-
тового микроскопа (СМ) после серебрения, многие авторы поль-
зуются этой методикой для СМ [40] или последовательно соче-
тая исследование в световом микроскопе с электронной микро-
скопией. Анализ в СМ занимает мало времени и может быть
использован для просмотра стекол и отбора хороших клеток,
которые затем будут изучаться в ЭМ. Один из таких приемов
описан в следующем разделе.
4.2. Распластывание для последовательного анализа
в световом и электронном микроскопах
Процедура, которой мы постоянно пользуемся в нашей лабо-
ратории, представляет собой модификацию метода Флетче-
ра [40].
4.2.1. Приготовление клеточной суспензии
1. Мышь. Извлеките семенники из белочной оболочки и по-
местите в среду F10, обогащенную 20%-ной ЭТС (Gibco
Biocult). Измельчите тонкими ножницами в наклонной
стеклянной чашке Петри. Дайте фрагментам канальцев
118
Глава 4
осесть в течение 20 мин. Отсосите суспензию пастеровской
пипеткой, оставив дебрис на дне, и перенесите в кони-
ческую центрифужную пробирку. Центрифугируйте при
150 g в течение 10 мин. Удалите большую часть надоса-
дочной жидкости, оставив количество, достаточное для ре-
суспендирования клеток.
2. Сперматоциты человека. Поместите материал тестикуляр-
ной биопсии в среду Дульбекко с фосфатным буфером
(PBS) и измельчите ножницами. Перенесите в колбу и
поставьте на магнитную мешалку примерно на 1 час.
Отцентрифугируйте при 150 g 10 мин в конической центри-
фужной пробирке. Отбросьте супернатант и ресуспенди-
руйте клетки в свежем буфере. Трижды промойте клетки
и оставьте в небольшом объеме PBS.
4ДД. Распластывание клеток
1. Прежде чем приготовить препараты, предварительно очи-
щенные и вымытые в спирте стекла покройте 5%-ным
раствором пластика Optilux (Falcon) в хлороформе [42].
Для погружения стекол используйте сосуд Коплина, за-
тем составьте их ребром на подставку и сушите в беспы-
левой атмосфере. Высушив, прикрепите пластиковое по-
крытие к краям стекол резиновым клеем Holdtite.
2. Распылите из пульверизатора черную краску «DEXION»
на 50-мм часовые стекла для создания водоотталкиваю-
щей поверхности. Подойдет и другая краска, дающая тем-
ный фон и водоотталкивающую поверхность.
3. Наполните черное часовое стекло свежеприготовленным
и профильтрованным раствором 0,2 М сахарозы; мениск
раствора должен выступать над краем часового стекла
(6—7 мл).
4. Втяните небольшое количество клеточной суспензии в
длинную пастеровскую пипетку и дайте одной капле
(примерно 0,02 мл) свеситься с конца. Осторожно при-
коснитесь этой каплей к поверхности распластывающего
раствора. Процесс можно контролировать в стереомикро-
скопе под малым увеличением, но в этом нет большой
необходимости, так как распластывание клеток можно
видеть невооруженным глазом.
5. Дайте клеткам распластаться по поверхности в течение
30 сек, затем соберите их, прикоснувшись к поверхности
раствора стеклом, покрытым пластиком.
6. Оставьте стекло в горизонтальном положении на 10 мин,
чтобы больше клеток осело, затем фиксируйте парафор-
мальдегидом.
Анализ мейоза в половых клетках человека и мыши
119
7. Спустя 5—40 мин промокните излишки фиксатора и про-
мойте стекла в течение 30 сек в 0,4 %-ном Photoflo.
8. Дайте стеклам высохнуть в горизонтальном положении
при комнатной температуре.
4.2.3. Окрашиввние
Наиболее удобный способ окраски — быстрый метод окра-
шивания коллоидным серебром Хаувелла и Блэка [43], перво-
начально предложенный для окрашивания ЯОР. Требуется два
раствора.
1. Приготовьте коллоидный проявитель, растворив 2 г по-
рошкообразной желатины в 100 мл деионизированной во-
ды и 1 мл чистой муравьиной кислоты. Помешивайте в те-
чение 10 мин, чтобы растворить желатину. Раствор стаби-
лен в течение 2 недель.
2. Приготовьте водный раствор нитрата серебра (4 г AgNOs
в 8 мл деионизированной воды). Этот раствор стабилен.
Храните коллоидный проявитель и раствор серебра в за-
крытых флаконах из коричневого стекла.
3. Нанесите пипеткой две капли коллоидного проявителя
и четыре капли водного нитрата серебра на предметное
стекло с распластанными клетками. Смешайте растворы
и закройте покровным стеклом.
4. Поместите предметное стекло на поверхность нагреватель-
ного столика для высушивания препаратов, отрегулиро-
ванного на 70 °C.
5. В течение 30 сек красящая серебром смесь пожелтеет,
а через 2 мин станет золотисто-коричневой. Возьмите пре-
парат, снимите покровное стекло и смойте красящую
смесь деионизированной водой.
6. Промокните препарат досуха и исследуйте немедленно
под микроскопом.
4.2.4. Приготовление сеток для электронной микроскопии
1. Найдите качественно окрашенные серебром клетки под
малым увеличением (Х25) светового микроскопа. Вокруг
клеток, пригодных для электронно-микроскопического ана-
лиза, проведите черту на пластиковом покрытии, восполь-
зовавшись для этого лейцевским алмазным карандашом.
2. Когда все нужные клетки будут локализованы, перенеси-
те круглые диски пластика, вырезанные алмазным каран-
дашом, на поверхность дистиллированной воды в большой
квадратной стеклянной чашке для окрашивания. Это до-
стигается медленным погружением наклоненного под уг-
120
Глава 4
Рис. 4.8. Сперматоциты иа стадии диплотены. Препарат получен путем поверх-
ностного распластывания н окрашивания серебром. А. Мышь (CM). X и Y
хромосомы лежат раздельно (стрелка). Б. Человек (ЭМ).
Анализ мейоза в половых клетках человека и мыши
121
лом стекла в воду, при этом поверхностное натяжение
воды снимет пластиковые диски со стекла.
3. Соберите диски один за другим следующим образом.
При помощи часового пинцета осторожно подведите ЭМ
сетки [G 200 HS Си (Gilder)] под пластиковые диски.
Осторожно поместите диск на поверхность сетки, восполь-
зовавшись для этого упругим волоском, прикрепленным
к концу тонкой деревянной палочки. Извлеките сетки
с дисками из воды. Дайте сеткам просохнуть, затем изу-
чайте в ЭМ.
На рис. 4.8, Л показаны распластанные диплотенные сперма-
тоциты мышн, полученные описанным выше способом, а на
рис. 4.8, Б — сперматоциты человека. На рис. 4.8, Л препарат
сфотографирован в СМ, на рис. 4.8,5 — в ЭМ. Следует заме-
тить, что ни центромеры, ни центральный элемент СК на препа-
ратах, окрашенных серебром, не различимы. Для выявления
этих структур их следует окрасить ФВК (разд. 4.1).
Результаты применения этой методики в нашей лаборатории
для изучения сперматоцитов у человека, гетерозиготного по
транслокации [44], нескольких инверсионных гетерозигот у мы-
ши [45] и для изучения спаривания X—Y хромосом у мужчи-
ны [46], можно найти в литературе. Распластывание широко
используется для анализа профазы мейоза у самцов. Эта мето-
дика действительно открывает новые перспективы исследования
мейоза.
4.3. Распластывание ооцитов
Материал (мышиные или человеческие яичники) должен
быть как можно более свежим. Через 24 ч морфология пахнтен-
ны.х клеток будет в большой степени искажена. У человека фе-
тальные яичники содержат максимальное число пахитен при-
мерно в возрасте 26 недель, хотя появляются они на 14-й неде-
ле и могут быть обнаружены уже на 30-й неделе беременности.
У мыши максимальное число пахитен выявляется около 18-го
дня после спаривания, хотя этот срок может варьировать в за-
висимости от ЛИНИН.
Простой способ распластывания ооцитов, впервые предло-
женный Спидом [47], заключается в следующем.
1. Поместите яичники в PBS. Кусочки яичннка человека
измельчите скальпелем на фрагменты около 2 мм (у мыши
для приготовления одного стекла используется весь яич-
ник).
2. Поместите один кусочек ткани яичника (илн целый яич-
ник, если это мышь) на чистое предметное стекло в 2—3
капли 0,2 М (4,5%) сахарозы (приготовленной на дистил-
лированной воде).
122
Глава 4
Рис. 4.9. Ооциты на стадии пахитены. Препарат получен путем распластыва
ння и окрашивания серебром. А. Мышь (СМ). Б. Человек (ЭМ).
Анализ мейоза в половых- клетках человека и мыши
123
3. Измельчите материал тупым концом скальпеля и препа-
ровальной иглой. Удалите крупные кусочки и диспергируй-
те клетки в сахарозе осторожным перемешиванием иглой.
Ооциты погрузятся в сахарозу и прилипнут к стеклу.
4. Если клетки предназначаются для изучения только в СМ,
оставьте стекло высохнуть как минимум в течение 30 мин
или даже на ночь. Если необходим ЭМ анализ, перенесите
клетки в сахарозе на покрытое пластиком стекло (см.
разд. 4.2.2) и осторожно, не прикасаясь к покрытию, рас-
пределите их на площади 1 —1,5 см2.
5. Фиксация, окрашивание и приготовление сеток для ЭМ
делаются так же, как опцеано в разд. 4.2.2, 4.2.3 и 4.2.4.
Распластанные ооциты на стадии пахитены, приготовленные
этим способом, представлены на рис. 4.9. Препарат мышиной
пахитены сфотографирован в СМ, а человеческий получен по-
средством ЭМ.
Использование техники распластывания для изучения ооци-
тов позволило, как и в случае сперматоцитов, выявить детали
конъюгации и других изменений конфигурации хромосом в про-
фазе мейоза. Анализ нормальных ооцитов, а также ооцитов
с разнообразными аномалиями у мыши представлен в работе
Спида [47]. Для человека такие данные получили Спид и Ченд-
лей [25, 48]. При изучении трисомии 21 эмбрионов человека
[49] и мышиных ХО [50] именно эта техника позволила обна-
ружить синаптические конфигурации, имевшие место в этих ано-
мальных случаях.
Благодарности
Автор благодарен Р. М. Спиду за советы при подготовке
разд. 3 и за рис. 6 и 9; а также Дж. Д. Бруку за представлен-
ные рис. 4.1 Г и 4.7.
Литература
1. Beatty В. A., Lim М.-C., Coulter V. J. (1975). Cytogenet. Cell Genet., 15.
256.
2. Laurie D. A., Firkett C. L., Hulten M. A. (1985). Ann. Hum. Gent., 49, 23.
3. Evans E. P., Breckon G., Ford С. E. (1964). Cytogenetics, 3, 289.
4. Carr D. H., Walker J. E. (1961). Stain Technol., 36, 233.
5. Forejt J., Gregorovd S. (1977). Cytogenet. Cell Genet., 19, 159.
6. Chandley A. C., Fletcher J. (1973). Humangenetik, 18, 247.
7. Sumner A. T. (1972). Exp. Cell. Res., 75, 304.
8. Pearson P. L„ Bobrow M. (1970). Nature, 226, 959.
9. Schweizer D., Arnbros P., Andrle M. (1978). Exp. Cell. Res., Ill, 327.
10. Hungerford D. A. (1971). Cytogenetics, 10, 23.
11. Luciani J. M., Morazzani M. R., Stahl A. (1975). Chromosoma, 52, 275.
12. Luciani J. M., Guichaoua M. R., Morazzani M. R. (1984). Hum. Genet., 66,
267.
124
Глава 4
13. Fang J.-S., Jagiello G. (1981). Chromosoma, 82, 437.
14. Page В. M. (1973). Cytogenet. Cell Genet., 12, 254.
15. Mitchell A. R., Ambros P., McBeath S., Chandley A. C. (1986). Cytogenet.
Cell. Genet., 41, 89.
16. Jhanwar S. C„ Neal B. S., Hayward W. S., Chaganti R. S. K. (1984). Cyto-
genet. Cell Genet., 38, 73.
17. Chaganti R. S. K.., Jhanwar S. C., Antonarakis S. E., Hayward W. S. (1985).
Somat. Cell Mol. Genet., 11, 197.
18. Chandley A. C„ McBeath S. (1986). In: Chromosomes Today. Stahl A. (ed.),
Geo. Allen and Unwin, London, Vol. 9, in press.
19. Sperling K-, Marcus M. (1984). In: Chromosomes Today. Bennet M. D.,
Gropp A., Wolf U. (eds), Geo. Allen and Unwin, London, Vol., 8, p. 169.
20. Richler C., Teitelboim E., Wahrman J. (1986), In: Chromosomes Today,
Stahl A. (ed.), Geo Allen and Unwin, London, Vol. 9, in press.
21. Kerem B., Goitein R-, Richler C_, Marcus M., Cedar H. (1983). Nature,
304, 88.
22. Rohrborn G., Hansmann I. (1971). Humangenetik, 13, 184.
23. Luciani J. M., Stahl A. (1971). Bull. Assoc. Anat. (Nancy), 15, 445.
24. Kurilo L. F. (1981). Hum. Genet., 57, 86.
25. Speed R. M. (1985). Hum. Genet., 69, 69.
26, Pincus G., Enzmann E. V. (1935). Anat. Rec., 75, 537.
27. Edwards R. G. (1962). Nature, 196, 446.
28. Edwards R. G. (1965). Lancet, i, 926.
29. Speed R. M. (1977). Chromosoma, 64, 241.
30. Tarkowski A. K. (1966). Cytogenetics, 5, 394.
31. Kamiguchi Y„ Funaki K., Mikamo K. (1976). Proc. Jap. Acad., 52, 316.
32. Sugawara S., Mikamo K. (1986). Chromosoma, 93, 321.
33. Wramsby H., Liedholm P. (1984). Fertil. Steril., 41, 736.
34. Jagiello G,, Karnicki J., Ryan R. J. (1968). Lancet, i, 178.
35. Angell R. R., Templeton A. A., Aitken R. J. (1986). Hum. Genet., 72, 333.
36. Counce S. J., Meyer G. F. (1973). Chromosoma, 44, 231.
37. Moses M. J. (1977). Chromosoma, 60, 99.
38. Moses M. J. (1977). In: Chromosomes Today, de la Chapelle A„ Sorsa M.
(eds), Elsevier/North-Holland, Amsterdam, Vol. 6, p. 71.
39. Moses M. J. (1980). In: Animal Models in Human Reproduction. Serio M.,
Martini L. (eds), Raven Press, New York, p. 169.
40. Fletcher J. M. (1979). Chromosoma, 72, 241.
41. Dresser M. E., Moses M. J. (1979). Exp. Cell Res., 121, 416.
42. Felluga B., Martinucci G. B. (1976). J. Submicrosc. Cytol., 8, 347.
43. Howell W. M„ Black D. A. (1980). Experientia, 36, 1014.
44. Chandley A. C., Speed R. M., McBeath S., Hargreave T. B. (1986). Cytoge-
net. Cell Genet., 41, 145.
45. Chandley A. C. (1982). Chromosoma, 85, 127.
46. Chandley A. C„ Goetz P., Hargreave T. B., Joseph A. M., Speed R. M. (1984).
Cytogenet. Cell Genet., 38, 241.
47. Speed R. M. (1982). Chromosoma, 85, 427.
48. Speed R. M., Chandley A. C. (1983). Chromosoma, 88, 184.
49. Speed R. M. (1984). Hum. Genet., 66, 176.
50. Speed R. M. (1986). Chromosoma, 94, 115.
ГЛАВА 5
КАРИОТИПИРОВАНИЕ И ОПРЕДЕЛЕНИЕ ПОЛА
ГАМЕТ, ЭМБРИОНОВ И ПЛОДОВ;
ГИБРИДИЗАЦИЯ ХРОМОСОМ
,Е. П. Эвансх
1. Введение
Важность применения цитогенетических методов для изуче-
ния развития млекопитающих не вызывает сомнения. Эти мето-
ды совершенно незаменимы в тех случаях, когда перед исследо-
вателем стоят задачи идентификации хромосом анеуплоидиых
зигот, определения пола эмбрионов, выявления инактивирован-
ной Х-хромосомы, анализа химеризма. Необходимы они и при
картировании природных и введенных путем гибридизации ге-
нов, и при изучении структуры хроматина. Для цитогенетическо-
го анализа иногда бывает достаточно всего лишь нескольких
клеток, что очень важно в тех условиях, когда биологический
материал ограничен.
Эмбриогенез млекопитающих служит важнейшим источни-
ком митотически делящихся клеток. Однако число качественных,
метафазных пластинок определяется возможностями современ-
ной техники приготовления препаратов хромосом. Цель этой
главы — описать некоторые методические приемы и пути ис-
пользования препаратов, полученных с их помощью, для реше-
ния разнообразных задач —от простого определения числа хро-
мосом до гибридизации in situ и выявления сегментной диффе-
ренцировки хромосом. Большинство методик, о которых будет
идти речь, разработано для мыши, однако многие из них могут
быть использованы и для экспериментирования на других мле-
копитающих, включая человека.
2. Основные методы
Одно из главных условий успешного цитогенетического ана-
лиза митотических клеток заключается в получении хорошо
расправленной метафазной пластинки, в которой хромосомы не
накладывались бы одна на другую. Для этого существует че-
тыре методических приема:
а) обработка клеток соединениями, останавливающими ми-
тоз;
1 Е. Р. Evans. William Dunn School of Pathology, South Parks Road,
Oxford OXI 3RE, UK.
126
Глава 5
б) воздействие гипотоническим раствором, вызывающее раз-
бухание клеток;
в) фиксация, сохраняющая морфологию хромосом;
г) распластывание и уплощение клеток на предметных стек-
лах.
Какой именно из этих четырех приемов следует применить
в каждом конкретном случае, зависит от исследуемой стадии
эмбриогенеза. В некоторых случаях полученные препараты под-
вергаются дальнейшей обработке. Такие ситуации будут также
описаны ниже.
3. Оборудование
Оборудование, необходимое для получения целых пред- или
постимплантационных эмбрионов или их фрагментов, описано
в гл. 2 и 3. Оно может быть использовано для получения образ-
цов материала и приготовления препаратов хромосом, причем
в этом случае требования к оборудованию гораздо меиее жест-
кие. Для цитогенетических целей подойдет любой препароваль-
ный микроскоп с падающим и проходящим светом, с хорошим
рабочим расстоянием и оптическим увеличением до Х50. Более
того, образцы можно брать достаточно тонкими, вытянутыми
вручную микропипетками. Поскольку этап культивирования
клеток очень краток, он не требует усложненной культуральной
среды с сывороткой и газом. Следует по возможности избегать
силиконирования оборудования, так как любой контакт с сили-
коном может значительно затруднять распластывание митотиче-
ских клеток.
Для получения и осаждения клеточных суспензий желатель-
но иметь центрифугу с плавным ускорением/замедлением, обес-
печивающую 200 g. Поскольку, согласно протоколам, обработка
препаратов производится при разных температурах, необходи-
мо иметь в распоряжении достаточное количество водяных бань
и печей, в которых поддерживается нужная температура. Ана-
лиз хромосом, особенно если они дифференциально окрашены
и несут зерна серебра, требует хорошего микроскопа. В иде-
альном случае он должен иметь фазово-контрастное устройство
для работы с неокрашенными препаратами, объективы Х16
и Х100 хорошего качества и фотонасадку 35 мм. Он должен
быть настроен таким образом, чтобы всегда мог дать макси-
мальное разрешение. Необходим набор фильтров для анализа
и фотографирования окрашенных хромосом; интерференцион-
ные фильтры, такие, как Balzer № 8, улучшают изображение
и увеличивают контраст. Для фотографирования хромосом
нужен набор 35-мм пленок. Каждая пленка имеет свои характе-
ристики. Наиболее широко применяются техническая пленка
Кариотипирование гамет, эмбрионов и плодов 127
Kodak Pan 2415 и проявитель Kodak НС110. Время проявления
указано в руководстве к заводскому раствору Д.
Все стекла, используемые для приготовления препаратов-
хромосом, должны быть чистыми. Хотя процесс очистки являет-
ся обязательным этапом при изготовлении стекол, их нельзя
считать абсолютно чистыми. Об этом свидетельствуют интерфе-
ренционные кольца, которые видны между сложенными стекла-
ми, когда они извлечены из упаковки. Их следует оставить на
ночь в смеси абсолютного спирта с концентрированной НС1
(1:1), на следующий день тщательно промыть водопроводной
водой, сполоснуть деионизированной водой и хранить в смеси
абсолютный спирт:диэтиловый эфир (1:1). Непосредственно
перед употреблением взять стекла пинцетом, не прикасаясь
к ним пальцами, и промокнуть досуха грубоволокнистым обез-
жиренным материалом, таким, как Kimwipes.
4. Препараты хромосом из эмбрионов и плодов
4,1. Препараты из зигот
Анализ митотических препаратов, полученных с пронуклеар-
ных стадий, позволяет выявить хромосомные наборы гамет.
Оплодотворенное яйцо позволяет судить о хромосомном вкладе
самца и самки, в неоплодотворенных яйцах можно выявить
хромосомы самки после активации партеногенеза целым рядом
воздействий ([1], гл. 12). Изучение пронуклеарного деления
оплодотворенных яиц дает информацию о хромосомных анома-
лиях, часть которых может быть причиной зиготических деталей
или проявиться в последующих делениях дробления. Гаплоид-
ные яйца с единственным пронуклеусом, полученные в резуль-
тате активации, незаменимы при анализе партеногенеза у млеко-
питающих [2].
Эмбриологический материал, полученный после естественной
овуляции и спаривания, обладает рядом недостатков, объясняю-
щихся асинхронностью оплодотворения. Чтобы избежать их,
обычно прибегают к процедурам, которые могли бы усовершен-
ствовать естественный процесс. Одна из таких процедур — супе-
ровуляция (гл. 1, разд. 5.4). Она увеличивает число получаемых
яиц и синхронность развития, а в комбинации с веществами-
цитостатиками в большой степени способствует успеху экспери-
ментов.
Для получения хромосомных препаратов предложены мно-
гочисленные методики in vivo и in vitro [3, 1, 4]. Они имеют
между собой много общего. Большинство из них предполагает
фиксацию и распластывание хромосом по методу Тарковско-
128
Глава 5
Таблица 5.1. Приготовление хромосомных препаратов из зигот
1. Индуцируйте суперовуляцию внутрибрюшинной инъекцией 5 ME СЖК
н через 40—48 ч 5 ME чХГ (см. гл. 1, разд. 5.4). После чХГ обеспечьте
спаривание.
2. Забейте самок вскоре после полудня следующего дня и извлеките яйца из
ампул яичников (гл. 2, разд. 2.1), поместите в культуральную среду (М2,
гл. 2, разд. 5.2).
3. Пронуклеарный митоз должен пройти следующей ночью между 28 и 32 ч
после инъекции чХГ. Обычно блокатор митоза добавляют в среду за не-
сколько часов до деления ядра, однако в данном случае, если эксперимен-
татор не ставит специальной цели получить неконденсированные хромосо-
мы, целесообразно внести блокатор (0,1% мкг/мл колцемида или винбла-
стина) в начале культивирования и оставить в нем яйца на всю ночь.
4. На следующий день удалите клетки кумулюса гиалуронидазой (гл. 2,
Йазд. 2.1) н промойте клетки чистой средой.
ока не появится опыт, проводите яйца партиями по три. Первую трой-
ку поместите в глубокое стекло с лункой, содержащее 1 мл гипотониче-
ского водного раствора 1%-ного трехзамещенного цитрата натрия или
0,56%-ного хлористого калия. В первом случае оставьте яйца в растворе
на 7—10 мин, во втором достаточно 6—8 мин.
6. Втяните яйца микропипеткой, отверстие которой чуть больше диаметра
яйца (следует заметить, что в гипотоническом растворе яйца разбухают,
поэтому оии могут закупорить пипетку, которая к ним подходила по
диаметру до воздействия), и поместите их в минимальном объеме гипото-
нического раствора на середину чистого предметного стекла. Тарковский
[5], автор методики, указывает, что капля остаточного гипотонического
раствора не должна превышать 2 мм в диаметре. Излишки его могут
вызвать разрыв и потерю хромосом, а недостаточное количество препят-
ствует их распластыванию.
7. Осторожно нанесите на яйца одну каплю (~0,02 мл) свежеприготовлен-
ного фиксатора (смеси метанол : ледяная уксусная кислота 3:1) так, что-
бы эта капля поглотила остаточный раствор. Между помещением янц на
стекло и их фиксацией не должно пройти более 1—2 сек, иначе яйца нач-
нут уплощаться преждевременно и не получится распластывания. Капайте
фиксатор с высоты не менее 2 см, в противном случае на яйца подейст-
вуют пары фиксатора и они также не распластаются.
8. Дайте капле фиксатора растечься. Это способствует высыханию. Подуйте
на стекло и подержите его над нагревательным столиком.
9. Продолжайте с другими партиями яиц. Количество яиц в партии может
быть увеличено, но при этом следите, чтобы яйца не располагались на-
столько близко друг к другу, чтобы их содержимое могло смешаться.
10. Полученные стекла могут быть окрашены рутинным способом (разд. 5.1),
С-методом (разд. 5.2) или G-методом (разд. 5.3).
го [5]. Один из методов получения хромосомных препаратов
представлен в табл. 5.1 и проиллюстрирован на рис. 5.1.
4.2. Приготовление препаратов
из зародышей предимплантационных стадий
Препараты хромосом могут быть получены из зародышей
всех предимплантационных стадий (описание см. в гл. 2). Успех
операции определяется числом миотически делящихся яиц и
Кариотипирование гамет, эмбрионов и плодов
129
Рис. 5.1. Пронуклеарные хромосомы
с удлиненным мужским комплемен-
том вверху, вблизи полярного тель-
ца. Клетка окрашена С-методом для
идентификации Y-хромосомы (отме-
чена стрелкой). Поскольку на пре-
парате хромосомные наборы разоб-
щены. они сфотографированы от-
дельно.
числом митозов, доступных
для анализа. Чтобы увеличить
их количество, разработаны
специальные методические
приемы. К ним относятся
суперовуляция и инъекция
самке блокатора митоза за не-
сколько часов до извлечения
эмбриона. Важно помнить, что
последняя процедура хотя и
увеличивает долю клеток с ми-
тотическими фигурами, но мо-
жет привести и к интенсивно-
му сокращению хромосом, за-
трудняющему анализ. Следует
не затягивать время действия
блокатора больше чем на 4 ча-
са. Этого достаточно чтобы
50—80% эмбрионов накопили
метафазные пластинки. Точ-
ные представления о скорости
дробления и отклонениях,
свойственных конкретной ли-
нии мышей, помогают экспери-
ментатору подобрать нужное
время воздействия блокато-
ром митоза.
Предимплантационное развитие может иметь место и в ус-
ловиях культуры (гл. 2), в этом случае блокатор митоза добав-
ляют в среду культивирования. При этом препараты готовят
точно так же, как и в случае извлечения эмбрионов из организ-
ма матери.
Следует заметить, что яйца с аномальным кариотипом часто
запаздывают в развитии по сравнению с нормальными [6]
и вследствие этого могут остаться невыявленными. Если в за-
дачу входит именно выявление анеуплоидии, требуется анализ
такого количества митотических клеток, которое обеспечит до-
стоверный результат.
Наиболее распространенный метод получения препаратов
хромосом предимплантационных эмбрионов разработан Тар-
ковским [5] (табл. 5.2).
Если действие гипотонического раствора и фиксация строго
контролируются, можно получить вполне удовлетворительные
препараты зародышей большинства предимплантационных ста-
дий (рис. 5.2), за исключением стадии очень поздней бластоци-
сты. Это можно сделать и при помощи «двойной фиксации и
9- 171
Таблица 5.2. Препараты зародышей предимплантационных стадий
1. Беременных самок, время спаривания которых обеспечивает нужную стадию
развития, инъецируйте внутрибрюшинно 0,25 мл 0,04%-ного водного рас-
твора колцемида или винбластина. Оставьте на 4 ч и извлеките эмбрионы,
как описано в гл. 2, разд. 5.2. Можно культивировать эмбрионы с добавле-
нием блокатора митозов в среду (0,1 мкг/мл).
2. Перенесите яйца в гипотонический водный раствор 1%-ного трехзамещен-
ного цитрата натрия или 0,56%-ного хлорида калия. Поскольку последний
является более эффективным гипотоническим агентом и может быстро
нарушить метафазу на такой чувствительной стадии, как раннее дробление,
рекомендуется, пока не появится опыт, работать с цитратом натрия.
3. Время воздействия гипотоническим раствором варьируйте в зависимости
от стадии развития: двуклеточиая стадия исключительно чувствительна, н
обработка в течение 2 мин может быть достаточной. Поздние бластоцисты
более устойчивы и могут потребовать до 15 мин. Чтобы сэкономить время,
поместите в серию стекол с лункой по одному эмбриону н инкубируйте
в гипотоническом растворе разное время (с интервалом в 1 мин). Если
в опыте находится партия асинхронно развивающихся эмбрионов, начните
с более ранних, так чтобы эмбрионы более поздних стадий оказались под
большим воздействием гипотонии.
4. Приготовьте препараты, как было описано для проиуклеариой стадии
(табл. 5.1), используя всякий раз по одному эмбриону, чтобы избежать
перекрывания хромосомных наборов при распластывании. Обычно одной
капли фиксатора достаточно для фиксации и распластывания ранних мо-
рул и бластоцист, поздние бластоцисты требуют нескольких капель.
5. Для окраски можно применить рутинный способ (разд. 5.1), С-метод
(разд. 5.2) или G-метод (разд. 5.3).
Таблица 5.3. Препараты хромосом нз зародышей пости мп лаитациониых стадий
А. Метод диссоциации уксусной кислотой
1. Приготовьте образец ткани, которым может быть весь плод или его часть
(гл. 3, разд. 3 и 4) и, если требуется, инкубируйте при 36—37 °C в тече-
ние 1 ч в среде (гл. 3, разд. 5 и 6), содержащей блокатор митозов, такой,
как колцемид или винбластин, в концентрации 0,1 мкг/мл. Предпочтитель-
нее пользоваться стеклянными, а не пластмассовыми контейнерами, так как
пластик в ходе этой процедуры может частично растворяться. Если объект
крупный (5 мм), для увеличения поверхности и лучшего проникновения
растворов разрежьте его на небольшие кусочки.
2. Поместите ткани в водный раствор 1%-иого трехзамещениого цитрата
натрия или 0,56%-иого хлорида калия. Оптимальное время обработки
должно быть установлено экспериментальным путем. Например, эмбрион
6-го дня развития очень чувствителен и требует всего лишь 3—4 мин
воздействия 0,56%-иым хлоридом калия или 4—6 мни 1%-ным трехзамещен-
иым цитратом натрия.
3. Фиксируйте ткань свежеприготовленной смесью метанол : уксусная кисло-
та 3 : 1 (фиксатор пригоден не более 3 ч после приготовления). Пинцетом
или пипеткой с широким отверстием перенесите ткань из гипотонического
раствора в 10-кратио превышающий объем фиксатора. Можно удалить
гипотонический раствор пипеткой и добавить к объекту нужный объем
фиксатора. Смешивание растворов вызывает движение жидкости, кроме
того, маленькие кусочки в фиксаторе часто становятся прозрачными и за
ними надо следить, чтобы не потерять. Фиксированный материал можно
хранить в достаточно большом объеме фиксатора до 3 мес при 4 СС.
Продолжение табл. 5.3
4. Дезагрегируйте в 5-кратном объеме водного раствора 60°/о-ной ледяной
уксусной кислоты при комнатной температуре. Контролируйте процесс
в препаровальном микроскопе.
5. Примерно через 5 мии сделайте пробный препарат, нанеся иа предметное
стекло 0,5 мл суспензии, поместите стекло на нагревательный столик при
40—60 °C. Поддерживайте суспензию в движении, т. е. покачивайте стекло,
чтобы капля по нему растекалась, или пипетируйте, перемещая каплю иа
свежую поверхность стекла. По мере того как капля перемещается по
стеклу и уменьшается, клетки будут распластываться на поверхности стекла.
Более высокая температура (60°C) быстрее высушивает препарат, однако
низкая (40 °C) часто дает препараты лучшего качества.
6. Просмотрите препарат под фазово-контрастным микроскопом Х128 и, если
качество его удовлетворительно, делайте следующие.
7. Если пробный препарат неудовлетворителен, из клеточной суспензии сде-
лайте еще один; если дезагрегация идет медленно, помогите часовым пин-
цетом илн пипетироваиием. Увеличение времени обработки 60%-иой уксус-
ной кислотой приводит к мацерации н повреждению митотических клеток.
8. Маленькие (^1 мм) кусочки ткани можно дезагрегировать в капле
60%-ной уксусной кислоты прямо на стекле. В этом случае следует обра-
тить внимание на то, чтобы с самого начала мацерации кусочек находил-
ся в центре капли, иначе он может подсохнуть до отделения, клеток.
9. Клетки можно окрасить рутинным способом (разд.. 5.1) нли использовать
С-метод (разд. 5.2).
Б. Метод клеточной суспензии
1. Приготовьте образцы целых плодов (размером 5—10 мм) или целых ор-
ганов, таких, как печень или селезенка 12—20-дневных эмбрионов, и сус-
пендируйте их, разрывая пинцетом и пипетируя в достаточном объеме сре-
ды (ХЮ), например ТС199 или МЕМ.
2. Наклоните чашку, чтобы дать осесть крупным фрагментам ткани до отде-
ления иадоеадочной жидкости со взвесью клеток. Если используется бло-
катор митоза (колцемид или винбластин 0,1 мкг/мл), инкубируйте клеточ-
ную суспензию около 1 ч при 36—37 °C.
3. Отцентрифугируйте клеточную суспензию в 10-мл конической стеклянной
центрифужной пробирке при 200 g в течение 5 мин, отбросьте надосадоч-
ную жидкость, ресуспендируйте клеточные сгустки в 10-кратном объеме ги-
потонического раствора 1%-иого трехзамещенного цитрата натрия или
0,56%-ного хлорида калия при комнатной температуре. Препараты эмбрио-
нальной печени и селезенки после обработки хлоридом калия становятся
более чистыми, поскольку это соединение вызывает лизис эритроцитов. Для
клеток молодых плодов время воздействия 0,56%-ным хлоридом калия
должно составлять 6—8 мии, в 1%-ном трехзамещенном цитрате натрия —
8—10 мин, для клеток плодов более поздних стадий время обработки уве-
личивается. Оптимальные сроки устанавливаются экспериментальным
путем.
4. Центрифугируйте клеточную суспензию при 200 g в течение 5 мин, уда-
лите надосадочиую жидкость и фиксируйте клеточные сгусткн, осторожно
добавляя пипеткой 5 мл свежеприготовленной смеси метанол: ледяная
уксусная кислота (3 : 1). Фиксация является критической стадией при из-
готовлении препарата, она получается лучше, если вначале внести фикса-
тор, не нарушая целостности клеточного сгустка, затем удалить его, при-
бавить следующую порцию, после чего энергично диспергировать сгусток,
постукивая по пробирке пальцем и одновременно добавляя еще одну пор-
цию фиксатора для ресуспенднровання клеток. О проникновении, фиксатора
в сгусток можно судить по изменению его окраски с кремовой на белую;
вся процедура фиксации должна занимать от 2 до 5 мин. Более длнтель-
9
132
Глава 5
П родолжение табл. 5.3
ное воздействие фиксатором может уплотнить сгусток и затруднить дис-
пергирование.
5. Центрифугируйте при 200 g 5 мин и ресуспендируйте в небольшом объеме
фвксатора.
6. Поместите 3 капли суспензии в ряд на чистое стекло. Дайте каплям пол-
ностью растечься, и когда они начнут высыхать (появятся интерференцион-
ные кольца), подышите на стекло и подержите его вблизи настольной
лампы (50 Вт). Сочетание потока влажного воздуха с мягким теплом
способствует хорошему распластыванию метафаз.
7. Просмотрите стекло под фазово-контрастным микроскопом при Х128 и,
если потребуется, нанесите следующие капли суспензии и повторите про-
цедуру высушивания.
8. Если митотические клетки не распластались нужным образом (из-за не-
достаточного воздействия гипотонии на предыдущем этапе или потому, что
хромосомы остались в цитоплазме) качество препаратов можно значи-
тельно улучшить добавлением трех капель фиксатора в тот момент, когда
капли клеточной суспензии начинают высыхать на стекле; пусть фиксатор
полностью растечется, а затем высушивайте, как описано выше.
9. Стекла можно окрасить рутинным способом (разд. 5.1), применить С-ме-
тод (разд. 5.2), G-метод (разд. 5.3) или использовать для гибридизации
in situ.
размягчения» по методу Дыбана и Баранова [7] или так, как
описано в табл. 5.3,Л.
4.3. Препараты зародышей постимплантационных стадий
Выбор технического приема определяется составом и разме-
ром объекта, а также целью исследования. Можно первоначаль-
но иметь дело с компактным материалом и диссоциировать за-
родыш после фиксации, можно диссоциировать его сразу
и дальше иметь дело с клеточной суспензией. Учитывая раз-
меры большинства эмбрионов (^5 мм), описанных в гл. 3, бо-
лее предпочтительной представляется работа с компактными
объектами. Плодные оболочки эмбрионов также лучше иметь
в виде компактного материала, так как они плохо поддаются
механической диссоциации. После гипотонического воздействия
и фиксации плотные ткани диссоциируют в уксусной кислоте
(способ описан в табл. 5.3, Л). Такие препараты обычно очень
информативны после С-окраски, но редко выявляют хорошую
сегментную дифференцировку хромосом после применения С-ме-
тода. Если нужны препараты целых эмбрионов старшего возра-
ста (Js5 мм) или более крупных органов, таких, как печень
или селезенка 12—20-дневных эмбрионов, материал сначала
переводят в суспензию, а затем проводят, как описано
в табл. 5.3, Б.
Кариотипирование гамет, эмбрионов и плодов
133
Рис. 5.2. А. Морула с пятью митотическими клетками. Вследствие того, что
отец был гетерозиготным по семи различным Робертсоновским транслокаци-
ям, клетки содержат метацентрические хромосомы. Б. Ранний триплоид с де-
вятью митотическими клетками, также содержащими метацентрические хро-
мосомы.
Хотя эта методика предполагает большие затраты времени,
препараты митотических клеток, полученные с ее помощью,
лучше по качеству и пригодны для выявления сегментной диф-
ференцировки хромосом (рис. 5.5) и гибридизации in situ
(разд. 6).
Как следует из гл. 3 (разд. 2), процессы роста и развития
раннего плода обеспечивают богатый источник митотических
делений. По мере их замедления и с переходом к дифференци-
ровке (после 14 дня) митотический индекс быстро падает как
в эмбрионе, так и в оболочках плода, затрудняя получение
хромосомных препаратов с поздних стадий, особенно незадолго
до рождения. После рождения митотическая активность возоб-
новляется, и в течение 5 дней можно получать превосходные
препараты из печени и селезенки. У раннего эмбриона высокий
митотический индекс позволяет обойтись без блокатора митоза
(колцемид, Sigma или винбластин, Lilly), но если митотический
индекс низок, как у поздних эмбрионов, применяют блокаторы.
Введение таких соединений матери не рекомендуется, так как
плацентарный барьер препятствует их проникновению в плод.
Блокатор можно инъецировать непосредственно в поздний
134
Глава 5
эмбрион, но эта процедура требует хирургического искусства от
экспериментатора и может травмировать мать. Более простой
способ введения блокатора заключается в извлечении ткани и ее
кратковременном инкубировании в культуральной среде (см.
гл. 3, разд. 5 и 6), содержащей 0,1% мкг/мл колцемида или
винбластина.
Эмбрионы мыши с несбалансированными хромосомными
наборами, как правило, погибают между 6 и 15 днем [8]. Хотя
митотическая активность у них падает за несколько часов или
даже дней до гибели, препараты митоза все-таки можно полу-
чить из оболочек плода. Их анализ позволяет установить, свя-
зана ли смерть с аномалиями хромосом. Пригодность внезаро-
дышевых оболочек для кариотипироваиия дает возможность
проводить исследования, в которых необходимо сохранить
эмбрион интактным независимо от того, нормален он или нет.
5. Окрашивание хромосомных препаратов
5.1. Хромосомы мыши после рутинной окраски
Нормальная мышь имеет 40 хромосом, все хромосомы акро-
центрические или с почти невидимыми короткими плечами. Раз-
мер хромосом в метафазе варьирует от 2 до 5 мкм. Уровень
разрешения, который обеспечивается рутинным окрашиванием,
не достаточен для анализа хромосом. Лишь некоторые аутосо-
мы имеют отличительные признаки (вторичные перетяжки)
(рис. 5.3,Л), а из половых хромосом удается идентифицировать
только Y-хромосому (рис. 5.3, А). Рутинное окрашивание позво-
ляет лишь подсчитать число хромосом в ситуациях, когда подо-
зревается анеуплоидия. Некоторые методики рутинного окра-
шивания представлены в табл. 5.4.
Однородная морфология мышиных хромосом, окрашенных
рутинным способом, препятствует выявлению химерности или
мозаицизма. Эта проблема может быть решена использованием
для мечения клеток маркерных хромосом. Маркерами могут
служить более длинные или более короткие хромосомы, полу-
ченные путем индуцированной неравной реципрокной трансло-
кации или метацентрических робертсоновских слияний, спонтан-
но возникающих в лабораторных или диких популяциях. При-
меры таких маркеров, в частности, наиболее часто используе-
мая Тб-хромосома, показаны на рис. 5.3, Б и В.
Идентификация Y-хромосомы после рутинного окрашивания
облегчается, если использовать линии мышей со спонтанно воз-
никшей измененной Y-хромосомой. Известны две такие линии,
их Y-хромосомы короткие и субметацентрические (рис. 5.3, Г)
Г9, 10].
Кариотипироваиие гамет, эмбрионов и плодов
135
Таблица 5.4. Рутинное окрашивание хромосом
Существуют многочисленные красители для хромосом, здесь же уместно
упомянуть два нз наиболее часто употребляемых: краситель Гимза и толуи-
диновый синий. Часто качество препаратов митотических клеток ухудшается
из-за прокрашивания цитоплазмы. Этого можно избежать, используя предвари-
тельный гидролиз препаратов.
I. Поместите стекла в сосуд Коплииа с 5 М НС1 при комнатной темпера-
туре, оставьте на 5 мин. Выньте стекла и промойте в проточной водо-
проводной воде, промокните лишнюю воду и окрашивайте по Гимза или
толуидиновым синим.
2. Окрашивание по Гимза
А. Имеющиеся в продаже красители Гимза имеют различные концентрации.
Концентрированный раствор Гимза (Merck), разведенный до 1%-иого фос-
фатным буфером pH 6,8 (таблетки буфера Gurr), дает удонлетворительиое
окрашивание в течение 15 мин. Для других растворов Гимза нремя окрас-
ки контролируется под микроскопом (Х128) на нлажных препаратах, прн
этом следует иметь в виду, что митотические клетки при нысыхаиии будут
более темными.
Б. После окрашивания быстро сполосните препараты буфером pH 6,8, про-
мокните, прикоснувшись фильтровальной бумагой, а остатки буфера уда-
лите струей воздуха из резиновой груши.
В. Для получения постоянных препаратов заключите в среду на основе кси-
лола, такую, как DePex (Gurr) или Eukitt (Riedel-de Наеп), и накройте
покровным стеклом. Если препарат не предназначен для длительного поль-
зования, иммерсионное масло можно наносить непосредственно на стекло.
Имейте в виду, что современные иммерсионные масла могут быстро вытя-
гивать краситель.
3. Окрашивание толуидиновым синим
Толуидиновый синий дает альтернативную по отношению к. Гнмза окрас-
ку. Этот краситель можно приобрести в виде готовой для употребления
заключающей среды (ASCO). Добавьте каплю красителя под покровное
стекло и осторожно выдавите его излишки. Это даст быстрое окрашива-
ние, интенсивность которого не увеличится. Такие препараты можно хра-
нить в защищенном от света месте в течение 4—6 иед. Если препарат
обесцветится, смойте покровное стекло водой при 60 °C и окрасьте снова
в свежей окрашивающей среде для заключения.
5.2. С-окрашенные препараты и их использование
для идентификации Y-хромосомы
С-методы выявляют структурный гетерохроматин, который
у мыши в норме представлен локализованной в центромерном
районе сателлитной ДНК- Существуют межлинейные различия
в количестве С-окрашенного материала у разных аутосом [11],
что используется в качестве клеточного маркера у химер [12 .
Что касается Y-хромосомы, то у всех известных линий она фак-
тически лишена С-сегмента. Это дает возможность легко и на-
дежно определять пол клетки. С-метод, применяемый в нашей
лаборатории (см. также гл. 4, разд. 2.4), описан в табл. 5 5
и иллюстрирован рис. 5.3, Д.
136
Глава 5
Рис. 5.3. А. Мужская метафаза. Препарат окрашен по Гимза, хорошо заметны
вторичные перетяжки (помечены маленькими стрелками) и Y-хромосома
{большие стрелки). Y-хромосома идентифицируется благодаря сближенным
параллельным хроматидам и маленькому короткому плечу. Б. Маркерная
хромосома Тб (указана стрелкой) в гомозиготном состоянии. Это результат
неравной реципрокной транслокации Т(14;15)6Са [17], но рутинное окраши-
Кариотипирование гамет, эмбрионов и плодов
137
Таблица 5.5. С-окраска хромосом
В литературе описаны многочисленные С-методы (см. также гл. 4, разд. 2.4);
большинство из них дает хорошие результаты, однако некоторые могут нару-
шать морфологию хромосом. Описанный ниже метод эффективен, прост н не
влияет на морфологию. Препараты, приготовленные этим способом, могут
быть использованы в течение 1—30 дней.
1. Поместите препараты в 5 М НС1 при комнатной температуре на 5 мин.
2. Отмойте препараты от кислоты в проточной воде и поместите в 2XSSC
(0,3 М хлорид натрия/0,03 М трехзамещенный цитрат натрии) в закрытый
сосуд Коплина на 15 мин при +65 °C.
3. Поместите в буферный раствор pH 6,8 (таблетки Gurr) при комнатной
температуре на 5 мин.
4. Окрасьте по Гимза (2—3% Merck Giemsa) в буферном растворе pH 6,8.
Просматривайте мокрые стекла при Х128.
5. Если контраст между С-сегментом и остальной хромосомой достаточен,
сполосните стекла буфером, промокните и высушите окончательно струей
воздуха из резиновой груши.
6. Сухне стекла для постоянных препаратов заключите в DePex или Eukitt
и накройте покровными стеклами; временные препараты смотрите с масля-
ной иммерсией, наноси масло прямо на поверхность стекла.
7. Если контраст между С-сегмеитом и остальной хромосомой неудовлетвори-
телен (она окрашена слишком темно), можно предположить, что время
выдержки в 2XSSC было недостаточным. Стекла без масла могут быть
возвращены для дальнейшей проводки и повторного окрашивания. Если
контраст остается слабым, несмотря на использование более концентриро-
ванного красителя, причина может заключаться в слишком длительном
воздействии 5М НС1 и (или) 2XSSC при 65 °C — такие препараты уже
нельзя улучшить. Это означает, что соответствующее время обработки
2XSSC нужно с самого начала сократить.
5.3. G-окрашенные препараты и их анализ
Индивидуальные хромосомы мыши, так же как и других мле-
копитающих, можно идентифицировать, применяя разные мето-
ды окраски, выявляющие сегментную дифференцировку хромо-
сом. Наиболее эффективный из них — G-метод. С его помощью
создается стандартная идиограмма кариотипа (рис. 5.4). Один
из способов такого окрашивания описан в табл. 5.6 и иллюст-
рирован рис. 5.5. На этом рисунке представлен традиционный
метод анализа кариотипа, заключающийся в расположении го-
мологичных хромосом парами в порядке уменьшения их линей-
ваиие не выявляет реципрокного продукта — длинного маркера. В. Одна из
Робертсоновских или метацентрических хромосом Rb (11.13)4Впг [17], кото-
рая также может быть использована в качестве маркерной (указана стрел-
кой). Г. Метафаза, пригодная для определения пола. Хорошо заметна малень-
кая субметацентрическая Y-хромосома (указана стрелкой). Д. Y-хромосома,
фактически лишенная С-сегмента, благодаря чему резко выделяется среди
других хромосом.
138
Глава 5
2
4
12
16
17
ГП0 Г5 'OJl >
Рис. 5.4. Стандартная
(Воспроизводится
идиограмма G-окрашеиных хромосом мыши [26].
с любезного разрешения редактора и издателя.)
Таблица 5.6. G-окрашивание хромосом
На свежеокрашенных препаратах сегментная дифференцировка выражена
слабо, и несмотря на то, что описан ряд способов устранения этого недо-
статка, препараты приходится выдерживать в коробках при комнатной тем-
пературе от 3 до 30 дней. Оптимального качества они обычно достигают при-
мерно через 10 дней.
I. Выдержанные в течение достаточного времени препараты поместите в за-
крытые сосуды Коплина в водный 2XSSC при 60—65°С. Оставьте на
1,5—2 ч. Этот период времени не является критическим и может быть уве-
личен до 3 ч.
2. Приготовьте 0,025%-иый трипсин (Difco 1 : 250) в 0,85%-ном нормальном
солевом растворе (солевые таблетки Oxoid) при комнатной температуре.
3. Остудите сосуды Коплина со стеклами до комнатной температуры в про-
точной водопроводной воде. Перенесите стекла в 0,85%-иый солевой рас-
твор и оставьте при комнатной температуре на 5 мии.
4. Извлеките одно стекло, уберите избыток солевого раствора, прикоснув-
шись к фильтровальной бумаге, положите стекло горизонтально (т. е. по-
перек двух стоящих ребром стеклянных пластинок) и поливайте из пасте-
ровской пипетки 0,025%-ным раствором трипсина. Время воздействия трип-
сином' существенно влияет на качество препарата: недодержка приводит
к слабому разрешению сегментов, а передержка нарушает морфологию
хромосом. Чувствительность хромосом к трипсину определяется нескольки-
ми факторами:
а) сократившиеся хромосомы более чувствительны, чем удлиненные;
б) недавно приготовленные препараты (2—7 дней тому назад) более чув-
ствительны, чем приготовленные раньше (за 7—30 дней);
в) «мягко фиксированные» хромосомы (оптически менее плотные под фазо-
вым контрастом) более чувствительны, чем «грубо» фиксированные хро-
мосомы (оптически плотные).
В нашей лаборатории, например, считают, что 10-дневиые препараты со сме-
шанной популяцией сокращенных и удлиненных «грубо» фиксированных хро-
мосом для удовлетворительного результата требуют обработки трипсином
в течение 20 сек. Рекомендуется сначала провести через всю процедуру воз-
действия трипсином и окрашивания одно пробное стекло, затем — остальные.
Раствор трипсина остается эффективным до 2 ч с момента приготовления.
5. После обработки дайте трипсину стечь, быстро наклонив стекло, затем по-
грузите его в 0,85%-ный солевой раствор, чтобы уменьшить активность
трипсина.
6. Сполосните буферным раствором с pH 6,8 (таблетки Gurr) и перенесите
в краситель Гимза, разбавленный тем же буфером (т. е. 1—2%-ный рас-
твор Merck Giemsa). Просмотрите мокрое стекло под микроскопам Х128.
7. Сполосните стекла в буфере с pH 6,8, промокните избыток буфера и вы-
сушите препарат струей воздуха из большой резиновой груши.
8. Анализируйте препарат с иммерсионным маслом. Это способствует дости-
жению наиболее высокого оптического разрешения.
9. Помните, что современные масла обесцвечивают (выщелачивают) краси-
тель. Если микроскопический анализ или фотографирование препарата не
предполагается в течение ближайших суток, рекомендуется сделать по-
стоянный препарат, заключив его в такую среду, как DePex или Eukitt,
и закрыть покровным стеклом.
Успех G-метода в большой степени зависит от качества растворов и чистоты
посуды. Растворы готовьте на дистиллированной в стеклянной посуде воде н
не храните их дольше 7 недель. Сосуды Коплина используйте только для
G-окрашивания, после чего просто споласкивайте ее горячей водопроводной
водой. Детергенты, крепкие кислоты и щелочи могут губительно сказаться
на сегментации.
139
Рис. 5.5. А. Кариотип инбредиой мыши C57BL/601a (G-окрашивание). Поло-
сы А1, соответствующие С-сегмеитам (стрелка), имеют одинаковые размеры
для каждой гомологичной пары хромосом (особь гомозиготна). Б. Полосы А1
(стрелка) в гомологах 7-й пары хромосом варьируют по длине (особь гетеро-
зиготна).
Карпотипироваине гамет, эмбрионов и плодов 141
кого размера, делающим очевидным любой выход из этого ряда.
По мере накопления опыта анализ стандартных микрофотогра-
фий позволяет обнаружить малейшие отклонения.
Стандартная идиограмма насчитывает в мышином кариоти-
пе 312 различных районов, но на одной метафазной пластинке
их можно наблюдать крайне редко. Достоинство принятой но-
менклатуры [13] заключается в том, что она оставляет место
для последующих данных о сегментной дифференцировке хро-
мосом, независимо от того, получены ли они в результате совер-
шенствования техники высокого разрешения деталей или про-
сто из-за большой проницательности наблюдателя. Некоторая
дополнительная информация уже получена, но, к сожалению,
техника высокого разрешения, предложенная для человеческих
хромосом, практически ничего не дала для анализа хромосом
мыши. Только в самое последнее время появилась публикация
идиограммы более высокого разрешения [14]. В настоящее
время стандартной считается идиограмма, принятая в 1974 г.,
но на ближайшее будущее планируется легализация ее послед-
ней версии.
G-окрашенный препарат находит применение во многих слу-
чаях, например для выявления отсутствующих, добавочных или
аберрантных хромосом, для выявления сбалансированных и не-
сбалансированных хромосомных сегментов после мейотической
сегрегации, для картирования локусов на хромосоме после
гибридизации in situ [5, 6] или традиционного iенетического
анализа с использованием маркерных генов, точек разрыва
п транслокации. При описании аномальных кариотипов необхо-
димо следовать правилам рекомендуемой номенклатуры [15].
Наиболее существенная вариация, обнаруженная в G-окра-
шенном кариотипе различных мышиных линий, — разное коли-
чество материала А1-сегмента (районы, являющиеся синонима-
ми С-сегментов), наблюдаемое в некоторых хромосомах. Раз-
мер Al-сегмента часто служит линейным маркером, а различия
по этому признаку могут быть использованы, в частности, для
выявления генетического вклада [16]. Некоторые различия
в А1-сегменте иллюстрирует рис. 5.5, Б.
5.4. Окрашивание неактивной Х-хромосомы
Если перед исследователем стоит задача убедиться в актив-
ности или,- наоборот, в отсутствии активности отцовской или ма-
теринской Х-хромосомы в клетках фетального происхождения,
прежде всего следует обеспечитьидентификациюэтих хромосом.
В качестве удобных маркеров можно рекомендовать X — ауто-
сомные реципрокные транслокации наподобие Т (Х;4)37Н
или X — аутосомные ипсерции Is (In7; X) 1 Ct, причем в обоих
142
Глава 5
Таблица 5.7. Окрашивание для выявления неактивной Х-хромосомы
1. Приготовьте клеточную суспензию плода в среде, как было описано в
табл. 5.3, А Отцентрифугируйте суспензию при 200 g в течение 5 мин и
отбросьте супернатант.
2. Ресуспендируйте клеточный сгусток в 0,56%-ном водном растворе хлори-
стого калия при 50 °C не более 30 мин. Длительное гипотоническое воздей-
ствие при этой температуре серьезно нарушает морфологию хромосом,
поэтому следует стараться сократить время воздействия, чтобы получить
адекватную дифференцировку и не допустить нарушений.
3. Центрифугируйте при 200 g в течение 5 мин, фиксируйте и делайте пре-
параты, как описано в табл. 5.3,5.
4. Окрашивайте стекла 2—5%-ным раствором Гимза в буфере pH 6,8 (таб-
летки Gurr). Просматривайте мокрые стекла при Х128, появление темного
окрашивания Х-хромосомы должно быть видно.
5. Если окраска удачна, сполосните стекло в буфере pH 6,8, высушите струей
воздуха.
6. Анализируйте препарат либо сразу с иммерсией, либо сделайте постоянный
препарат, заключив в DePex или Eukitt и закрыв покровным стеклом.
случаях речь идет о маркерной хромосоме, превосходящей по
длине самую большую нормальную хромосому мыши (см. в ра-
боте [17] список таких маркеров). Еще одно требование заклю-
чается в том, что неактивную Х-хромосому нужно уметь отли-
чить от активной. Существует ряд соответствующих методик,
включающих радиоавтографию и системы замещения 5-бром-
дезоксиуридином (БУДР). Мы приведем, по нашему мнению,
более простой метод Канда [18] (табл. 5.7). Он не требует
сложного оборудования и большой затраты времени.
К сожалению, метод Канда не лишен недостатков: многие
митотические клетки оказываются поврежденными, однако ос-
тавшихся интактными вполне достаточно, чтобы выявить актив-
ную Х-хромосому. На рис. 5.6, Л изображена митотическая
клетка, у которой нормальная Х-хромосома неактивна. Она ка-
жется короче обычной (в норме Х-хромосома представляет со-
бой пятую'из наиболее длинных хромосом), окрашена темнее,
чем остальные. Окраска другой Х-хромосомы, меченной
Is (1п7; X) ICt, остается неизмененной. Следует заметить, что
ожидаемое увеличение длины этой маркерной хромосомы не
всегда оказывается очевидным. Однако, если бы меченой хро-
мосомой была неактивная X, ее можно было бы увидеть как
наиболее темную и длинную хромосому клетки. В дифференци-
альном окрашивании проявляется различная реакция на рас-
кручивание хроматина, вызываемое длительным гипотоническим
воздействием (табл. 5.7). Вывод о том, что темное окрашивание
действительно отражает неактивность, получил подтверждение
в многочисленных цитогенетических и энзимологических иссле-
дованиях.
Кариотипирование гамет, эмбрионов и плодов
143
A Z Ж.. '
< -г Ж
*/'4 '
V
Рис. 5.6. А. Метафаза, в которой выявляется неактивная нормальная Х-хро-
мосома (большая стрелка) и маркированная (IslCt) активная Х-хромосома
(маленькая стрелка). Б. Интерфазное ядро клетки амниона, в котором выяв-
ляется тельце полового хроматина (большая стрелка) и хромоцентры (малень-
кие стрелки).
5.5. Окрашивание для выявления полового хроматина
Если в работе требуется определить пол эмбриона, а хромо-
сомные препараты неэффективны (Y-хромосома не является
маркерной, а применение С-метода нецелесообразно), самок от
самцов можно отличить по наличию или отсутствию полового
хроматина. Хотя половой хроматин в ядрах мыши не выделяет-
ся так четко, как в ядрах некоторых других видов млекопитаю-
щих, эта структура может быть использована для определения
пола; при этом источником ядер, как правило, служит
амнион [19]. Для приготовления препаратов амнион изолируют,
фиксируют в смеси метанол : ледяная уксусная кислота (3:1)
и распластывают ядра в 60 %-ном водном растворе уксусной
кислоты, как описано в табл. 5.3,Л. Распластанные ядра могут
быть окрашены в уксусно-кислом орсеине (Gurr) прямо под
покровным стеклом. Если окрашивание идет медленно или если
есть необходимость некоторое время хранить препараты, покров-
ное стекло окружают по краям специальным изолирующим
раствором (Weldite), препятствующим высыханию краски.
В ядрах женских плодов, половой хроматин выявляется в виде
темноокрашенного тельца (рис. 5.6,Б). У различных эмбрионов
женского пола число хроматин-положительных ядер варьирует
между 40 и 80%. Большинство вариаций объясняется техниче-
скими причинами, например числом ядер, у которых при упло-
щении на стекле тельце оказалось в хорошо просматриваемом
J44
Глава 5
положении. В этих ядрах тельце можно легко отличить от хро-
моцентров по более темной окраске и расположению в неболь-
шой «лунке» ядерной мембраны. В ядрах некоторых мужских
плодов выявляются ложные тельца полового хроматина, в дей-
ствительности представляющие собой крупные хромоцентры
периферической локализации. Число таких клеток редко дости-
гает 10%.
6. Гибридизация хромосом
В том случае, когда целью гибридизации in situ является
локализация в геноме неизвестных уникальных последователь-
ностей, необходимо использовать для анализа только те ткани,
которые могут обеспечить достаточное количество препаратов
с большим числом митотически делящихся клеток. Это ограни-
чение связано с тем, что часть материала будет потеряна по
техническим причинам, а при подсчете гранул серебра необходи-
ма выборка, достаточная для получения статистически досто-
верного результата. Другая проблема связана с трудностями
идентификации индивидуальных хромосом и выявления их
структурной дифференцировки, что вынуждает прибегнуть к не-
которым дополнительным приемам окрашивания. Если гибриди-
зация проводится с повторяющимися последовательностями,
для которых характерен сильный гибридизационный сигнал,
можно работать с меньшим числом стекол и митотических кле-
ток, но при этом необходимость окрашивания остается. С дру-
гой стороны, как уникальные, так и повторяющиеся последова-
тельности легче локализовать, если известно, в какой хромосо-
ме они расположены. В этом случае задача сводится к выявле-
нию данной хромосомы при помощи соответствующих хромосом-
ных маркеров. Более того, если мы знаем приблизительное рас-
положение интересующих нас последовательностей на хромосо-
ме, нужный район можно обнаружить при наличии подходящей
неравной реципрокной транслокации. Поскольку набор трансло-
каций, которые могут быть использованы в качестве маркеров,
ограничен [17], мы описываем более общий подход к гибриди-
зации in situ (табл. 5.8), основывающийся на допущении, что
предварительных сведений о положении уникальных последова-
тельностей нет. При наличии таких данных процедура упроща-
ется.
6.1. Культура эмбриональных фибробластов
и репликационная окраска
Проблема сохранения окраски в процессе гибридизации
in situ для клеток человека в значительной степени была реше-
на с введением репликационного G-метода и использованием
Таблица 5.8. Гибридизация хромосом in situ11
Предлагаемый нами метод представляет собой видоизменение методики, опи-
санной для человека [24].
1. Возьмите порцию третьей субкультуры мышиных эмбриональных фибро-
бластов в фазе роста [21], добавьте БУДР (Sigma) в концентрации
200 мкг/мл и продолжайте культивировать в течение 16 ч. Наличие
БУДРа в среде обеспечивает блокирование и синхронизацию многих клеток
в середине S-фазы. Защищайте культуру клеток, клеточную суспензию и
препараты клеток на стеклах от света во избежание спонтанных разры-
вов ДНК. Простой способ предохранения от света — завернуть все кон-
тейнеры в алюминиевую фольгу.
2. Спустя 16 ч промойте клетки средой четыре раза для полного удаления
БУДРа. Инкубируйте в свежей среде, содержащей 10 мкМ тимидина, и
соберите клетки через 6—6,5 ч.
3. Сбор клеток. Уменьшите объем жидкости до 10 мл. Отделите митотиче-
ские клетки, энергично постукивая флаконом о ладонь.
4. Если этот прием не дал результата, воспользуйтесь 0,25%-ным раствором
трипсина в PBS. Удалите среду, осторожно налейте 10 мл раствора трип-
сина и в течение минуты осторожно покачивайте жидкость над клетками,
чтобы ослабить их прикрепление, затем быстро вылейте трипсин, заменив
его средой. Легкое постукивание по флакону освободит митотические
клетки.
5. Отцентрифугируйте клеточную суспензию при 200 g в течение 5 мии.
Удалите иадосадочиую жидкость и ресуспендируйте клеточный сгусток
в 0,56%-ном водном растворе хлористого калия. Оставьте на 8 мии при
комнатной температуре.
6. Отцентрифугируйте при 200 g 5 мии и фиксируйте клетки; делайте пре-
параты, как было описано в табл. 5.3, Б. Для выявления сегментационно-
го рисунка хромосом рекомендуется делать препараты после того, как
суспензия клеток простоит в фиксаторе ночь прн +4 °C и затем ресуспен-
дируется в свежем фиксаторе. Суспеизни можно хранить в фиксаторе до
1 недели.
7. Для гибридизации in situ, начиная с этого пункта методики, все стекла
(предварительно окрашенные и предназначенные для постокрашнвания)
эквивалентны. В случае предокрашеииых препаратов сфотографируйте ми-
тотические клетки (разд. 5.3) и локализуйте нх положение при помощи
верньерного стекла или стекла со шкалой. Современные иммерсионные
масла трудно полностью удалить с поверхности стекла, поэтому для фо-
тографирования препараты должны быть заключены под покровным стек-
лом в водорастворимую заключающую среду (такую, как Hydramount,
Gurr), которую можно удалить проточной водой. Число необходимых фо-
тографий зависит от цели исследования. Прн картировании повторяющих-
ся последовательностей с помощью зондов, меченных с высокой удельной
активностью, можно обойтись наименьшим количеством (г£50); для кар-
тирования маленьких уникальных последовательностей зондом, меченным
с низкой удельной активностью, нужно максимальное количество (^100)
фотографий. Удобно работать с 10 стеклами одновременно и растворы
готовить заранее, за исключением тех случаев, когда требуются свеже-
приготовленные.
8. Перед гибридизацией in situ тщательно обесцветьте препараты в фикса-
торе (3 части метанола: 1 часть ледяной уксусной кислоты), так как кра-
сители типа Гимза характеризуются положительной хемографичностью в
отношении ядерных эмульсий.
9. Прокипятите раствор РНКазы (Sigma) в 2XSSC (100 мкг/мл) в течение
10 мин для удаления любых примесей ДНКазы. Охладите и нанесите на
каждый препарат по 200 мкл (под покровное стекло размером 1,64x 22 мм).
Препараты поместите в камеру, увлажненную 2XSSC на 1 ч при 37 °C.
10-171 145
Продолжение таб.1. 5.8
Подходящие камеры можно сделать нз пластиковых коробок для сэндви-
чей (Steward). Неиспользованную РНКазу можно хранить замороженной
около года.
10. Снимите покровные стекла и поместите препараты в стеклянный штатив,
промойте в четырех сменах средой 2XSSC, затем обезводьте в спиртах
возрастающей крепости: 10, 50, 75, 96 и 100%. Высушите стекла на воз-
духе и храните в эксикаторе. Рекомендуется держать стекла в штативах
(держателях), осторожно перенося нх в таком положении через раство-
ры, — это уменьшит потерю клеток. Можно приобрести соответствующие
наборы (Horwell).
11. Денатурируйте ДНК на стеклах в 70%-ном формамиде н 0,1 мМ ЭДТА
в 2xSSC при pH 7,0 в течение 4 мнн прн 65 °C. Установите pH добав-
лением 5 М НС1, Промойте, сменив четыре порцнн 2XSSC, обезводьте
проводкой через серию спиртов и храните в эксикаторе.
12. Если предполагаемый зонд помечен 1251, ацетилируйте стёкла в 0,25%-ном
уксуснокислом ангидриде в 0,1 М триэтаноламине с pH 2 прн комнатной
температуре в течение 10 мин. Ацетилирование уменьшает фон, но в нем
нет необходимости, если используется другая метка. Промойте, сменив
четыре раза 2XSSC, обезводьте в сернн спиртов н храните в эксикаторе.
13. Детали ник^грансляцнн, мечення н подсчета метки описаны в гл. 10.
Со следующего пункта пропнсн начинается работа с изотопом, напоми-
наем о необходимости соблюдать технику безопасности.
14. Для 10 стекол требуется 100—150 нг ДНК зонда, меченного згР (см.
гл. 10, разд. 2), растворенной в 300 мкл гибридизационного буфера, со-
стоящего нз 50%-ного формамида, х5 раствора Денхардта, х5 SSPE
pH 7,2 (0,9 М хлорида натрия/50 мМ однозамещенного фосфата нат-
рия/5 мМ ЭДТА), 10%-ного сульфата декстрана н 200 мкг/мл ДНК спер-
миев лосося (Sigma). Сульфат декстрана растворяется с трудом, его ре-
комендуется нагревать в кипящей воде и сильно встряхивать.
15. Перед использованием гибридизационную смесь с зондом денатурируйте
кипячением в течение 2—3 мнн, а затем погрузите в лед. Воспользуйтесь
закрытой пробиркой Эппендорфа, проколотой инъекционной иглой (21 ка-
либра), за которую можно держать пробирку в кипящей воде.
16. Пипеткой нанесите иа каждое предметное стекло по 30 мкл гибридиза-
ционной смесн. Этот объем примерно соответствует трем каплям. Их надо
расположить на равном расстоянии друг от друга, а затем осторожно
опустить на ннх покровное стекло (размером 64x22 мм) так, чтобы под
ним не образовались пузырьки воздуха. Края покровного стекла ограничьте
раствором резины (Weldtite) и поместите стекла во влажную камеру при
42 °C на ночь. Для этого стекла, укрепленные в стеклянном штативе,
помещают в плотно закрытые пластиковые коробки для сэндвичей (Ste-
ward) над увлажняющим раствором 2xSSC.
17. Снимите раствор резины пинцетом н осторожно удалите покровные стек-
ла в 5XSSC (0,75 М хлорида натрия/0,075 М трехзамещенного цитрата
натрия), погрузив в него стекло и покачивая. Промывайте в 2XSSC при
комнатной температуре в течение 1—2 ч.
18. Строгость, с которой следует относиться к последующей процедуре про-
мывания, зависит от типа использованного зонда; предлагаемые нами
рекомендации служат лишь общим руководством и могут быть изменены.
19. В случае уникальных последовательностей промойте стекла в 2xSSC при
65°C в течение 1 ч с одной сменой горячего 2XSSC. Затем один раз
промойте в 0.2XSSC прн комнатной температуре в течение 30 мин и один
раз в 0.1XSSC при той же температуре н то же время.
20. В случае повторяющихся последовательностей промывайте в (0,5—0,1) X
XSSC прн 60 °C в течение 1 ч с одной сменой горячего SSC. Затем про-
мойте в O.lxSSC прн комнатной температуре.
21. После промывания обезводьте препараты в серии спиртов и храните
в эксикаторе.
Продолжение табл. 5.8
22. Приготовьте эмульсию для погружения стекол. Радиоавтографии посвя-
щены многочисленные детальные обзоры (например, [25]), ниже приво-
дитси краткий перечень процедур с использованием Ilford Nuclear Emul-
sion L4. Эмульсия поступает в пузырьках из темного стекла объемом по
50 мл и имеет полужидкую консистенцию. Храните ее при 4 °C, вдали от
радиоактивных материалов. В темной комнате при красном свете (Kodak
№ 6В) разделите эмульсию примерно пополам, одну половину уберите
в темное место для использования в будущем, а другую растворяйте
в течение 30 мин в 25 мл дистиллированной воды в водяной бане при
55 °C. Растворять можно в 50-мл пластиковой центрифужной пробирке
(Nunc), она может быть использована и в качестве контейнера для по-
гружения стекол. Перед погружением стекол тщательно размешайте смесь
при помощи предметного стекла, удерживаемого пластиковым пинцетом
(металлическими инструментами пользоваться нельзя, они могут дать
зерна фона).
23. Медленно погружайте стекла; извлекая, вытирайте бумагой нижнюю
сторону каждого и устанавливайте их для высушивания в почти верти-
кальном положении. Оставьте на 30 мни. Разбавленной эмульсией можно
пользоваться 3—4 раза, если хранить ее в защищенной от света посуде
при 4 °C, при этом не теряется ее чувствительность и не увеличивает-
ся фон.
24. Поместите стекла в светонепроницаемые коробки для препаратов с осу-
шителем, заклейте коробки черной пластиковой лентой и храните для
экспозиции при 4 °C.
25. Учитывая вероятность появления технических проблем в ходе после-
дующего анализа, целесообразно обеспечить себя контрольными стек-
лами. Контроль должен включать препарат из предшествующего удач-
ного эксперимента с зондированием в качестве положительного контроля,
стекло, приготовленное в процессе текущего эксперимента, но без зонда
(в качестве отрицательного контроля), а также пустое стекло для опре-
деления фона эмульсии.
26. Время экспозиции зависит от нескольких факторов, в частности от при-
роды последовательности и активности зонда. Повторяющиеся последо-
вательности с зондом высокой активности требуют короткой экспозиции;
уникальные последовательности и низкая активность предполагают более
длительную экспозицию. Для 12SI оно должно составлять от 2 до 9 дней,
для 3Н— 10—30 дней. Чтобы определить время экспозиции, используйте
худшие стекла, например с небольшим числом метафаз; если они были
предварительно окрашены G-методом, возьмите те, с которых сделано
наименьшее число фотографий.
27. Нагревайте стекла до комнатной температуры в течение 1 ч, проявляйте
в полной темноте в D19 (Kodak) при 20 °C в течение 5 мин без движе-
ния жидкости. Сполосните их в 1%-иой уксусной кислоте и фиксируйте
в Amfix (May and Baker), разведенном дистиллированной водой 1:4
с добавленным отвердителем (Ilford). Промывайте стекла в слабой струе
воды в течение 1 ч в темноте, сполосните дистиллированной водой и вы-
сушивайте на воздухе, оставив их в вертикальном положении. Если
оборотная сторона стекол не была вытерта при их извлечении из эмуль-
сии, это можно сделать сейчас или после окрашивания.
28. Стекла, предварительно окрашенные G-методом, опустите в 2—4%-ный
раствор Гимза в буфере с pH 6,8. Контролируйте интенсивность окраски
на мокрых стеклах. Если она удовлетворительная, сполосните в буфере
и сушите на воздухе, установив в почти вертикальном положении. Лока-
лизуйте предварительно сфотографированные митотические клетки и срав-
ните нх с фотографическими отпечатками после G-окраски. Если они ока-
жутся информативными, сфотографируйте снова. Если стекла предвари-
тельно не были окрашены, попытайтесть сделать это после гибридизации,
используя краситель Райта [22].
10*
148
Глава 5
.Продолжение табл. 5.8
29. Защищайте стекла с дифференциальным окрашиванием от прямого света
в процессе работы н после высушивания. Окрашивайте высушенные стек-
ла в Hoechst 33258 (Sigma) (10 мкг/мл в 2XSSC) в течение 30 мни так,
чтобы под покровным стеклом был избыток красителя. Смойте покровные
стекла 2XSSC, положите стекла горизонтально в 2xSSC (в плоскую пла-
стиковую кювету) и, не закрывая, экспонируйте в течение 1 ч иа расстоя-
нии 15 см от источника длинноволнового ультрафиолетового света (Sylva-
nia). Сполосните стекла буфером pH 6,8 и окрашивайте в 5—10%-ном
растворе Гимза в буфере. Под микроскопом на мокрых стеклах контроли-
руйте появление дифференциальной окраски, промойте в буфере и сушите
на воздухе в вертикальном положении. Поскольку современные иммерсион-
ные масла могут обесцвечивать хромосомы, рекомендуется делать постоян-
ные препараты, заключив их под покровным стеклом в такие среды, как
DePex или Eukitt.
30. Если проявленные препараты удовлетворительны с точки зрения гибри-
дизационного сигнала н фона, проявляйте следующие. Если их качество
неудовлетворительно, их можно оставить для дальнейшей экспозиции.
Важно помнить прн этом, что дальнейшая экспозиция часто приводит
к повышению фона. Колебания в количестве зерен серебра и времени
экспозиции описаны [25].
См. также работы [27—29].
БУДРа [20]. Для мыши соответствующая методика только
начинает разрабатываться. Поскольку процедура требует про-
хождения клетками раунда репликации в присутствии БУДРа,
она может быть осуществлена только на культивируемых клет-
ках. Предложены многочисленные методики инициации культу-
ры эмбриональных фибробластов [21]. С их помощью исследо-
ватель может получить целый ряд стекол с большим количест-
вом митотических клеток, которые могут быть использованы
для нескольких различных зондов. Вместе с тем инициация
культуры на основе общего клеточного пула нескольких эмбрио-
нов имеет и отрицательную сторону, связанную с отсутствием
сведений об источнике клеточной линии. Кроме того, эта про-
цедура требует большой затраты времени.
6.2. Методы пред- и постокрашивания
Альтернативные способы идентификации хромосом на пре-
паратах заключаются в «предокрашивании» или «постокраши-
вании» (после гибридизации). Предокрашивание может быть
выполнено в соответствии с процедурой, описанной нами выше
(табл. 5.6). Оно не сохраняется после гибридизации in situ.
Поэтому митотические клетки необходимо сфотографировать,
локализовать их на препарате при помощи верньерного стекла
или стекла с сеткой и снова сфотографировать (после гибри-
дизации) для сравнения рисунка расположения сегментов и зе-
рен серебра.
Кариотипироваиие гамет, эмбрионов и плодов 149
Поскольку цели многих исследований предполагают фото-
графирование по меньшей мере 50 клеток, процедуру предокра-
шивания следует признать весьма трудоемкой. Для постокраши-
вания после гибридизации используют краситель Райта (Gurr
или Merck) [22]. Довольно ч?сто этот метод не дает желаемых
результатов: сегментная дифференцировка при этом не выяв-
ляется четко. Для пред- и постокрашивания не нужно культиви-
рование клеток, так как адекватное число митотических клеток
может быть получено прямо из плодов средних стадий раз-
вития (табл. 5.3,Б).
Итак, репликационное окрашивание хромосом эмбриональ-
ных фибробластов наиболее эффективно для локализации уни-
кальных последовательностей в немеченом мышином геноме.
Этот метод представлен в табл. 5.8 и на рис. 5.7,Л. Процеду-
ры пред- и постокрашивания также упоминаются в табл. 5.8
(см. также рис. 5.7,Б). Использование неравной реципрокной
транслокации для маркирования района хромосомы проиллюст-
рировано на рис. 5.7, В.
6.3. Оценка метки
Процедура анализа результатов определяется природой ис-
следования. Если локализация последовательности (последова-
тельностей) совершенно не известна и требует точного картиро-
вания G-сегмента (репликационные сегменты эквивалентны) на
хромосоме, возникает необходимость идентифицировать каждую
хромосому и отметить на ней положение каждого зерна сереб-
ра. Перед этим следует решить, нужно ли учитывать зерна,
прикасающиеся к концам хромосом. Удобно приготовить оценоч-
ные листы с изображением каждой мышиной хромосомы, сег-
ментированной G-методом (рис. 5.4) и прямо на них наносить
положение зерен. По мере подсчета на хромосомах будут
выявляться «горячие точки». Таким образом должны быть про-
считаны 50—100 митотических клеток. Результаты могут быть
выражены общим числом зерен, лежащих на хромосомах во
всех просчитанных митотических клетках, и общим числом зе-
рен, лежащих на каждой индивидуальной хромосоме, отнесен-
ным к ожидаемому числу зерен на этой хромосоме при условии,
что они распределены равномерно на единицу длины хромосо-
мы. Таблица процентной оценки длины гаплоидного набора
опубликована [23]. Например, если 265 зерен отмечены как ле-
жащие на хромосомах в 100 митотических клетках и если хро-
мосома 1 представляет 7,20% генома, тогда приблизительно
19 зерен (7,20% от 265=19,08) следует ожидать на гомологич-
ной паре. Получив ожидаемое число для каждой хромосомы
и сравнив его с наблюдаемым, можно зарегистрировать любое
Рис. 5.7. А. Метафаза после репликационного окрашивания и гибридизации
in situ с зондом человеческого гена 2'-5'-oligoA-CHHTeTa3H. На полосе А4 мы-
шиной хромосомы 11 (обозначена стрелкой) обнаружено статистически зна-
чимое число гранул. Б. Часть митотической клетки, предокрашенной для вы-
явления G-сегментов и сфотографированной до гибридизации in situ с зондом
ГФРТ человека. Зерно (указано стрелкой) на фотографии, сделанной после
гибридизации in situ, локализует последовательность, соответствующую по-
лосе А6 на Х-хромосоме. В. Метафаза в клетках мыши, гетерозиготной по не-
равной реципрокной транслокации Т (1; 17) 19ОН, расщепляющей хромосому 17
в сегменте В. В результате этой транслокации образуются две маркерные
хромосомы: длинная и короткая. После гибридизации in situ с зондом Тср-1
большее число зерен можно обнаружить над маркером меньшего размера
(показан стрелкой), что свидетельствует о проксимальном положении Тср-1
в сегменте 17В.
Кариотипирование гамет, эмбрионов и плодов 151
значительное отклонение. Если в какой-то хромосоме будет
обнаружено достоверное отличие от нормы, она может быть
исследована более детально.
Если используются маркерные хромосомы, анализ может
быть упрощен, так как ожидаемые по отношению к наблюдае-
мым значениям числа зерен нужно отнести только к общей про-
центной длине маркеров в геноме. Следует заметить, однако,
что, если маркеры используются в гетерозиготном состоянии,
ожидаемое число зерен уменьшится вдвое (можно полагать, что
другая их половина лежит на немаркированных и поэтому не
учтенных нормальных гомологах).
Благодарности
В этой главе сделана попытка изложить существующие ме-
тоды получения и использования препаратов хромосом эмбрио-
нов разных стадий развития. Некоторых авторов этих новых
методик мы упомянули в библиографических ссылках. Особую
благодарность я приношу доктору Веронике Бакл и Майку Бер-
теншоу за их помощь в разработке метода гибридизации in situ.
Литература
1. Kaufman М. И. (1982). J. Embriol. Exp. Morphol., 71, 139.
2. Robertsoon E. J., Kaufman M. FL, Bradley A., Evans M. J. (1983). In: Tera-
tocarcinoma Stem Cells. Cold Spring Harbor Conference on Cell Prolifera-
tion. Silver L. M., Martin G. R., Strickland S. (eds), Cold Spring Harbor
Laboratory Press, New York, Vol. 10, p. 647.
3. Donahue R. P. (1982). Proc. Natl. Acad, Sci. USA, 69, 74.
4. Maudlin I., Fraser L. R. (1977). J. Reprod. Fertil., 50, 275.
5. Tarkowski A. K- (1966). Cytogenetics, 5, 394.
6. Baranov V. S. (1983). Genetica, 61, 165.
7. Dyban A. P., Baranov V. S. (1978). «Nauka». Moscow, Russia.
8. Gropp A. (1982). Vichows Arch. (Pathol. Anat.), 395, 117.
9. Winking H. (1978). Mouse News Lett., 58, 53.
10. Burgoyne P. S. (1987). In: International Index of Laboratory Animals. 5th
Edition, Resting M. F. W. (ed.), Laboratory Animals Ltd, PO Box 101, New-
bury, Berkshire, UK.
11. Davisson M. T. (1981). In: Genetic Variants and Strains of the Laboratory
Mouse. Green M. C. (ed.), Gustav Fischer Verlag, Stuttgart and New York,
p. 357.
12. McLaren A. (1975). J. Embryol. Exp. Morphol., 33, 205.
13. Nesbitt M. N., Francke U. (1973). Chromosoma, 41, 145.
14. Sawyer J. R., Hozier J. C. (1986). Science, 232, 1632.
15. Rules for Nomenclature of Chromosome Anomalies. (1981). In: Genetic Va-
riants and Strains of the Laboratory Mouse. Green M. C. (ed.), Gustav
Fischer Verlag, Stuttgart and New York, p. 314.
16. Evans E. P., Burtenshaw M. D., Adler L-D. (1985). Genet. Res. Camb., 46,
353.
17. Searle A. G. (1981). In: Genetic Variants and Strains of the Laboratory
Mouse. Green M. C. (ed.), Gustav Fischer Verlag, Stuttgart and New York,
p. 324.
152
Глава 5
18. Kanda N. (1973). Exp. Cell Res., 80, 463.
19. Monk M., McLaren A. (1981). J. Embriol. Exp. Morphol., 63, 75.
20. Zabel B. U., Naylor S. L., Sagkaguchi A. Y., Bell G. T., Shows T. B. (1983).
Proc. Natl. Acad. Sci. USA, 80, 6932.
21. Freshney R. I. (1983). Culture of Animal Cells. Alan R. Liss, Inc., New
York.
22. Cannizzaro L. A., Emanuel В. E. (1984). Cytogenet. Cell Genet., 38, 308.
23. Roderick T. H. (1981). In: Genetic Variants and Strains of the Laboratory
Mouse. Green M. C. (ed.), Gustav Fischer Verlag, Stuttgart and New York,
p. 279.
24. Buckle V. J., Mondello V., Darling C., Craig I. W., Goodfellow P. N. (1985).
Nature, 317, 739.
2. 5. Rogers A. IF. (1979). Techniques of Autoradiography. Elsevier/North-Hol-
land Biomedical Press, Amsterdam/New York/Oxford.
26. Davisson M. T. (1981). In: Genetic Variants and Strains of the Laboratory
Mouse. Green M. C. (ed.), Gustav Fischer Verlag, Stuttgart and New York,
p. 317.
27. Buckle V. J., Craig I. W. (1986). In: Human Genetic Disease — A Practi-
cal Approach. Davies К. E. (ed.), IRL Press, Oxford, p. 85.
28. Malcolm S., Cowell J. K., Young B. D. (1986). In: Human Cytogenetics —
A Practical Approach. Rooney D. E., Czepulkowski В. H. (eds), IRL Press,
Oxford, p. 197.
29. Pardue M. L. (1985). In: Nucleic Acid Hybridisation — A Practical Appro-
ach. Hames B. D., Higgins S. J. (eds), IRL Press, Oxford, p. 179.
ГЛАВА 6
МЕТОДЫ МАРКИРОВАНИЯ
КЛЕТОК И ИХ ПРИМЕНЕНИЕ
Б. А. Дж. Пондер1
1. Введение
В этой главе описываются способы маркирования отдельных
клеток, групп клеток и их клональных потомков в организме
животного. Маркированные клетки используются при решении
задач двух типов: во-первых, когда необходимо выяснить вклад
той или иной клеточной линии в формирование органов или
тканей в ходе нормального или аномального развития. Так, изу-
чение судьбы маркированных клеток, введенных в морулу или
бластоцисту, показало возрастающее ограничение морфогене-
тических потенций клеток разных районов предимплантацион-
ного эмбриона мыши [1]; благодаря включению нейроэктодер-
мальных клеток перепела в куриный эмбрион были выявлены
все клеточные линии, происходящие из этой ткани [2]; анализ
мозаичного состава взрослых тканей химер, образованных в ре-
зультате агрегации ранних мышиных эмбрионов, дал возмож-
ность судить об истории клонов в связи с пролиферацией и пе-
регруппировкой клеток в процессе развития [3]. С помощью
маркированных клеток могут исследоваться и патологические
процессы: например, по клеточному составу опухолей мы можем
судить о том, от одной или нескольких клеток они произо-
шли [4]. Для всех этих исследований существенным условием
является идентичность поведения меченых клеток поведению
немеченых клеток той же ткани. Другими словами, один из кри-
териев выбора маркера заключается в том, чтобы он как мож-
но меньше нарушал нормальное поведение клеток.
Второй тип задач, для решения которых могут использовать-
ся клеточные маркеры, касается типа клеток, в котором экс-
прессируется мутантный ген. Например, дефект, связанный
с геном ped, вызывающим церебральную атаксию, экспрессиру-
ется в химерах pcdlpcd+-+ дикий тип (см. ниже) только в том
случае, когда клетки Пуркинье имеют мутантный генотип [5].
В принципе таким образом можно изучать клеточные основы
любого фенотипического различия между линиями мышей.
Маркирование клеток дает возможность ответить на вопрос,
1 В. A. J. Ponder. Institute of Cancer Research, Haddon Laboratories, Clifton
Avenue, Sutton, Surrey SM2 PX, UK.
153
154
Глава 6
свойственна ли высокая или низкая частота возникновения
опухолей клеткам-мишеням, или она является результатом
действия модифицирующих факторов в другой ткани (напри-
мер, в иммунной системе). При этом очень важно, чтобы мар-
керный генотип не вызывал в клетках плейотропный эффект,
т. е. не обусловливал дополнительных, усложняющих экспери-
ментальную ситуацию различий.
Идеальный клеточный маркер должен сохранять свою кле-
точную автономность (т. е. экспрессироваться только в клетках
соответствующего генотипа; это свойство не должно переда-
ваться соседним клеткам); он не должен влиять на поведение
маркированных клеток; клеточный маркер должен характеризо-
ваться устойчивой экспрессией во всех клетках исследуемой
ткани при различных физиологических или патологических усло-
виях; наконец, он должен технически легко выявляться. Жела-
тельно, чтобы клеточный маркер экспрессировался во всех тка-
нях на всех стадиях развития, но, конечно, в этом нет необхо-
димости, если эксперимент ограничен какой-то определенной
тканью. Что касается химерных тканей, то в идеальном случае
маркированными должны быть обе клеточные популяции, иначе
можно прийти к ошибочному заключению, что все немаркиро-
ванные клетки принадлежат другой популяции. Большинство
методик маркирования не эффективны на 100%. Если для задач
эксперимента наиболее существенна клеточная популяция, пред-
ставленная очень малым числом клеток, то именно ее и следует
пометить.
Идеальной маркирующей системы не предложено до сих
пор. Ниже обсуждаются достоинства и недостатки наиболее
распространенных маркеров. Экспериментальные системы, обес-
печивающие получение популяции дифференциально маркиро-
ванных клеток, представлены пятью обширными группами:
1) экспериментальные химеры;
2) Х-инактивационные мозаики;
3) индивидуальные клетки, маркированные инъекцией спе-
цифического соединения;
4) клеточные популяции, маркированные за счет интегра-
ции клонированных генов в некоторых клетках ранних
эмбрионов;
5) маркеры, индуцированные мутацией соматической клет-
ки.
Рассмотрим каждую из них.
Методы маркирования клеток и их применение 155
2. Химеры
2. 1. Определение понятия «химера»
Химера — животное, в состав которого входят клетки, про-
исходящие более чем от одной зиготы. Обзор химер млекопи-
тающих дан в работах [6, 7]. Различные клеточные популяции
могут быть объединены в одном организме на любой стадии
жизни животного. Химеры, известные биологам развития, обыч-
но создаются путем комбинации клеток двух или более предим-
плантацнонных эмбрионов, но они могут быть получены и вве-
дением в постимплантационные эмбрионы (см. гл. 3, разд. 7 и 8)
или во взрослый организм клеток иного генетического проис-
хождения. Пациент с трансплантированной почкой — химера.
В иммунологии и радиобиологии широко используются «радиа-
ционные химеры», созданные посредством разрушения красного
костного мозга и лимфоидной ткани животного-хозяина облу-
чением с последующей пересадкой костного мозга от генетиче-
ски отличающегося донора. Независимо от того, каким способом
получены химеры, принципы использования клеточных маркеров
остаются общими. В этой статье мы сосредоточим свое внима-
ние на химерах мышиных эмбрионов и их применении в биоло-
гии развития.
2. 2. Типы химер и их получение
2.2.1. Эмбриональные агрегационные химеры
Для получения химер этого типа 4—8-клеточные эмбрионы
от мышей разных линий агрегируют в условиях культуры,
после чего химерный эмбрион переносят в матку псевдоберемен-
ной «приемной матери», где он продолжает развиваться до
рождения. Процедура описана в табл. 6.1.
2.2.2. Химеры, полученные инъекцией в бластоцисту
Для получения химерных эмбрионов чаще пользуются инъ-
екцией одной или нескольких клеток в бластоцисту, чем агрега-
цией двух морул. (Методика описана в работах [8, 9].) Этот
способ хотя и более трудоемкий, но имеет некоторые существен-
ные преимущества. Он не требует удаления блестящей оболоч-
ки, поэтому может быть применен к получению химер у живот-
ных, не способных без нее развиваться, например у кролика.
Поскольку весь трофобласт представляет собой ткань хозяина,
не возникает препятствия для имплантации при некоторых
межвидовых комбинациях, для которых обычно отторжение
чужеродного эмбриона. При использовании для формирования
156
Глава 6
Таблица 6.1. Создание агрегационных химер1
1. Подготовьте получение в один и тот же день 4-клеточных эмбрионов от
мышей двух линий, которые будут использованы для создания химер.
Подготовьте псевдобеременную самку для трансплантации химерных эм-
брионов на следующее утро.
2. Приготовьте культуральные капли среды М164-БСА (гл. 2, рлзд. 5.2)
под маслом в пластиковой чашке (Falcon). Выдержите в инкубаторе
с 5% СО2 при 37°.
3. Приготовьте две стеклянные панели с лунками для промывания эмбрио-
нов, каждая с 1 мл М16+БСА под слоем масла (по одному стеклу для
каждой линии). Поместите в инкубатор.
4. Забейте самку (3-й день беременности) одной линии смещением шейных
позвонков. Следуя прописи в гл. 2, разд. 2.2, получите 4—8-клеточные
эмбрионы. Промойте в среде М2+БСА (гл. 2, разд. 5.2).
5. Удалите блестящую оболочку, поместив эмбрионы в проназу (гл. 2.
разд. 3.2). Когда оболочка станет тоньше и помутнеет (это произойдет
между 6-й и 15-й мин), перенесите эмбрионы обратно в М2+БСА. Осто-
рожно пипетируйте, чтобы удалить все фрагменты оболочки. Не обраба-
тывайте за один раз более 15 эмбрионов. (Раствор кислого Тироде — см.
гл. 2, разд. 3.2 — действует быстрее, ио его сложнее контролировать, и
риск повредить эмбрионы повышается. Судя но нашему опыту, после
проназы агрегация идет лучше.)
6. Перенесите эмбрионы в свежую среду М2+БСА.
7. Промойте эмбрионы в одном стекле для промывания средой М16+БСА
(другое используйте для второй линии).
8. Поместите по одному эмбриону в каждую культуральную каплю M16-F
-|-БСА в чашке (Falcon) и поставьте в инкубатор. (Чтобы избежать
изменения pH, старайтесь не держать М16+БСА, даже под маслом,
слишком долго вне инкубатора).
9. То же проделайте с мышью другой линии. Поместите по одному эмбрио-
ну в те же самые культуральные капли, где уже находятся эмбрионы
первой линии.
10. Подтолкните пары эмбрионов друг к другу концом вытянутой пипетки.
Повторите это в течение дня 2—3 раза.
11. Инкубируйте всю ночь.
12. На следующий день проверьте, сколько эмбрионов соединилось. Агрегаты
перенесите в псевдобеременную мышь-реципиент, как описано в гл. 13,
разд. 6.4.
*) Все детали, касающиеся извлечения эмбрионов, удаления блестящей оболочки,
среды культивирования, подготовки псевдобеременных приемных матерей, оборудования
и материалов, изложены в гл. 1 н 2. Перенос эмбрионов описан в гл. 13, разд. 6.4.
химер метода инъекции клеток в бластоцисту появляется воз-
можность анализировать судьбу более поздних эмбриональных
стадий, чем это позволяют агрегаты морул. И наконец, с по-
мощью инъекций в бластоцисту можно маркированные клетки
ввести в линию клеток зародышевого пути.
3. Клеточные маркеры в химерных системах
Набор используемых маркеров и их характеристики сумми-
рованы в табл. 6.2. (Описание потенциальных маркеров и мар-
керов для видов, не относящихся к млекопитающим, можно
Таблица 6.2. Маркеры, используемые при работе с химерными мышами1)
Маркер Метод Ткани Выбор линии2' (см. 1101) Литератур- ный источ- ник
А. Дающий информацию о топографии Пигмент
Меланин Лрямое наблю- дение Пигментный эпителий сет- чатки, мемб- рана лаби- ринта внут- реннего уха Широкий: альбиносы/не- альбиносы [6, 11]
Меланин/воло- сяиой фолли- кул (эффек- ты генов ок- раски покро- вов) Полимсрф/гм < Прямое наблю- дение ytc •~г(в Волос Широкий: ряд генов, действующих на меланоци- ты или воло- сяной фолли- кул [6]
р-глюкурэнпда- за Гистохимиче- ский3* Печень, другие тканн сме- шанного состава Широкий |11, 12]
₽-галактозида- за Яблочный фер- мент Гистохимиче- ский3’ Гистохимиче- ский3’ Клетки Пур- кинье в моз- жечке, ядра черепно-моз- говых, нервов, эпителий мо- лочной желе- зы Внезародыше- вые оболоч- ки, другие ткани Широкий ]П, 13] [11
Глюкозофосфа- тизомераза (ГФИ) Маркеры клето Гистохимиче- ский и имму- ногистохими- ческий мой поверхносп Многие Широкий (И, 14]
Н-2-антнгены Иммуногисто- химический Не у эмбриона или новорож- денного Широкий 115]
1а-антигены Иммуногисто- химический Лимфомиэло- ндные Широкий Данных нет
Thy-1-антигены Иммуногисто- химический Тимоциты (?), фибробласты, ЦНС, миоэпи- телиальные клетки; может не иметь кле- точной авю- ЬОМ’Ш (?) Ограничен [16]
Продолжение табл. 6.2
Ма.ркер Метод Ткаин Выбор линии-) (см. [101) Литератур- ный источ- ник
Полиморфизм углеводов, узнаваемый лектинами Гистохимия связывания лектинов Эпителий ки- шечника и эн- дотелий со- судов после 12-го дня бе- ременности Ограничен (см. табл. 6.7) 117—19]
Морфологичесы Мутация «Beige» ie маркеры Гистохимия внутрикле- точных гра- нул Гранулоциты, тучные клет- ки, остеокла- сты Ограничен (11, 20, 21]
.Специфическая дегенерация клеток
Дегенерация Морфология на Сетчатка Ограничен |22]
;сетчатки мы- гистологиче-
ши ских срезах
Дегенерация Морфология и а Клетки Пур- Ограничен |5]
клеток Пур- гистологиче- кинье моз-
кинье ских срезах жечка
Специфические различия в ДНК.
Различия в са-
теллитной
ДНК между
видами мы-
ши
Гибридизация
in situ на
срезах
Любые
Ограничен в на-
стоящее вре-
мя
Mus muscu-
lus/Mus caroli
123]
Б. He дгющйй информации о топографии
Полиморфизм, выявляемый с помощью электрофореза
Глюкозофосфа- Электрофорез Любые, в том Широкий (1, ю, 24]
тизомераза Изоцитратде- гидрогеназа Главный белок мочи (см. (10, И]) тканевого го- могената Электрофорез тканевого го- могената Моча числе ранний эмбрион Печень, почки, селезенка, мышцы Печень живой мыши Широкий Широкий ( 5] [26]
Хромосомные м Тб аркеры Морфология в клеточных культурах из ткани Любая митоти- ческая ткань; может потре- боваться кратковре- менное куль- тивирование Широкий [10, 11, Гл. 5 27]
Методы маркирования клеток и их применение
159
Продолжение табл. 6.2'
Маркер Метод Тканн Выбор линии-) (см. [10]) Литератур- ный источ- ник
Y-хромосома Морфология в клеточных культурах из ткани Любая митоти- ческая гкань; может потре- боваться кратковре- менная куль- тура [Н, 28]
Тетраплоидия Другие Морфология в клеточных культурах из ткани Морфология в клеточных культурах из ткани Любая митоти- ческая ткань; может потре- боваться кратковре- менная куль- тура (разьи- тие аномаль- ное) [29] [Ю]
Продолжение обсуждения клеточных маркеров у химер см. в работах [1, 6, 7..
И].
2) Сведения об имеющихся ннбредных линиях мышей можно найти в работе [101
и в периодических выпусках «Mouse News Letter». В любую пару линий, выбранных
для получения химер, удобно ввести маркер окраски шерсти для того, чтобы с легкостью
отобрать в потомстве химерных особей.
3) Эти ферменты не обязательно во всех клетках нормальной (не химерной) ткани
дают устойчивое позитивное окрашивание. Поэтому прежде, чем приступать к интер-
претации результатов, рекомендуется сделать соответствующий положительный и отри-
цательный контроль окрашивания (предпочитаются срезы, расположенные рядом с хи-
мерными срезами на одном и том же предметном стекле).
найти в обзоре Веста [11].) Для практического применения
наиболее существенное значение имеют маркеры двух типов:
выявляемые визуально на гистологических срезах или на то-
тальных препаратах, и те, для обнаружения которых необходи-
ма процедура электрофореза. Преимущества и недостатки мар-
керов каждого типа при анализе химерных тканей обсуждаются
в разд. 5. Мы изложим подробно лишь методики использования
Н-2-антигенов и углеводного полиморфизма. Способы примене-
ния других маркеров и краткие к ним комментарии представ-
лены в табл. 6.2.
3.1. Использование в качестве маркера Н-2-антигенов
Н-2-,антигены могут быть обнаружены иммуноцитохимически
при помощи моноклональных анти-Н-2 антител во многих тка-
нях мыши старше 7 дней [30] (см. табл. 6.3).
160
Глава 6
Таблица 6.3. Иммуногистохимическая локализация Н-2-антигенов у мыши
Постоянно присутствует Присутствует иногда Постоянно отсутствует
Эпителий Тонкая кишка Толстая кншка Матка Молочная железа Кожа Трахея Переходный эпителий почечной лоханкн Собирающие почечные канальцы Протоки слюнной желе- зы Околоушной ацинус Другие ткани Мозговой слой тнмуса Селезенка Лимфатические узлы Пейеровы бляшки Клетки моноцитарно-фа- гоцитарного ряда Эндотелий сосудов Клетки выстилки пече- ночных лакун Паренхима легких1’ Адипоциты Мочевой пузырь Бронхи Железистый желудок Желчный проток Проток поджелудочной железы Половой тракт (кроме маткн) Фолликулы щитовидной железы Ацннус слюнной желе- зы Почечные клубочки Интерстициальные клет- ки семенника Слущивающийся эпите- лий Язык Глотка Шейка маткн Влагалище Гепатоциты Надпочечники Клетки панкреатического островка Фолликул яйцевода Головной и спинной мозг Периферический нерв Скелетная мышца и мышца сердца
1) Диффузное окрашивание стенок альвеол, не приуроченное к определенному типу
клеток.
Хотя экспрессия Н-2-антигена у мышиного эмбриона может
-быть обнаружена серологически, только в клетках тимуса на
поздних этапах беременности присутствует такое количество
антигена, которое достаточно для проведения иммуноцитохими-
ческого анализа. Пока нет данных о том, что различия по
Н-2-антигену между линиями, составляющими химеру, оказы-
вают влияние на ее развитие, однако считать это доказанным
нельзя.
Преимущества мечения Н-2-антигенами заключаются в боль-
шом выборе тканей и линий мышей, а также в возможности
окрашивания клеток двух генотипов на одном и том же срезе
(при использовании антител к различным Н-2-антигенам).
Методы маркирования клеток и их применение 161
Основной недостаток этой методики — ее технические труд-
ности. Во-первых, многие эпитопы (антигенные детерминанты)
оказались лабильными в отношении фиксаторов и смол, исполь-
зуемых для заключения, вследствие чего можно пользоваться
только свежефиксированными криостатными срезами (см. ни-
же). Во-вторых, Н-2 антигены экспрессируются на клеточной
поверхности и разрешение окрашивания на уровне одной клет-
ки в смешанной ткани не всегда удовлетворительное.
Ниже перечислен ряд условий, которые необходимо соблю-
дать при организации эксперимента. Некоторые из них являют-
ся обязательными в любом эксперименте, основанном на имму-
ноцитохимии с моноклональными антителами.
1. Выберите подходящие для вашего опыта антитела (источ-
ники: коммерческий, American Type Culture collection, другие
лаборатории) и линии мышей. Гаплотипы мышей по Н-2-анти-
гену описаны у Грина [10].
2. Проверьте специфичность антител выбранных линий на
криостатных срезах тимуса молодых взрослых мышей. Срезы
от каждой линии, расположенные на одном стекле, нефиксиро-
ванные и фиксированные ПЛП (см. ниже, табл. 6.5), инкуби-
руют с серийными разведениями моноклонального антитела
(используют утраивающиеся разведения 1 : 10—>-1 :810). Моно-
клональное антитело может быть помечено прямо (см. ниже)
или обнаружено при помощи второго антитела — антимышино-
го иммуноглобулина, конъюгированного с пероксидазой или ще-
лочной фосфатазой. Если оптимальный титр второго антитела
еще не известен, его можно определить, воспользовавшись се-
рией утраивающихся разведений второго антитела с каждым
разведением первого антитела.
Оптимальной следует считать ситуацию, когда мозговой
слой тимуса одной линии сильно окрашивается разведением
его специфического моноклонального анти-Н-2-антитела, не даю-
щего окрашивания тимуса другой линии. Некоторое слабое
окрашивание может возникнуть благодаря второму (антимы-
шиному) иммуноглобулиновому антителу, — в таком случае
необходим контроль без анти-Н-2-антитела.
3. Если требуется, чтобы на одном и том же срезе химер-
ной ткани одновременно окрашивались клетки обоих феноти-
пов, необходимо выбрать соответствующую стратегию. Этого
можно добиться, используя моноклональные антитела разных
подклассов в качестве первых антител, а в качестве вторых —
антитела, специфические для подклассов IgG. С другой сторо-
ны, можно использовать непрямой метод гаптенизированных
антител (т. е. одно моноклональное антитело связывается с био-
тином, затем со стрептавидином, а другое моноклональное ан-
титело, конъюгированное с динитрофенолом (ДНФ), связыва-
11-171
162
Глава 6
Таблица 6.4. Приготовление конъюгатов фермента с антителом
(советы овладевающим методикой)
1. Для приготовления конъюгатов щелочной фосфатазы используйте фер-
мент высокой специфической активности (Boehringer или Sigma). Эти
конъюгаты могут быть использованы с 1 мМ левамязола (Sigma) в каче-
стве ингибитора эндогенной щелочной фосфатазы во всех тканях, кроме
кишечника ([33], см. табл. 6.5, и. 18).
2. Одинаковые ферменты из различных источников, по-видимому, отличаются
своей «липкостью», создавая проблему фона на гистологических препара-
тах. Если возникнут подобные затруднения, стоит попробовать фермент из
другого источника.
3. Убедитесь в том, что все буферы (Tris) со свободными аминогруппами
удалены диализом из препаратов ферментов и антител.
4. Может возникнуть необходимость испытать ряд пропорций антитело : фер-
мент, чтобы выбрать наилучшую.
5. Тестируйте свежеприготовленный конъюгат в разведениях, возрастающих
на 2^3 порядка, выберите дозу с наибольшей активностью при чистом
фоне на препарате контрольной ткани. Если появится окрашенный фон.
решению проблемы может помочь фракционирование конъюгата путем
гель-фильтрации (Sephadex G-200 или Sephacryl), особенно в случае пе-
роксидазных конъюгатов.
6. Не замораживайте конъюгат. Разделите на аликвоты и храните при 4 °C
в буфере при pH 7,5—8.0 с БСА (5 мг/мл) и азидом натрия (0,1%).
ется с анти-днф). Эти системы описаны в руководствах по
иммунологии. По нашим данным, наиболее воспроизводимые
результаты дает третий подход — прямая конъюгация каждого
моноклонального антитела с индикаторной молекулой. В этой
роли могут выступать фермент или флуорохром. Конъюгирован-
ные моноклональные антитела используются в процедуре двой-
ного окрашивания, описанной в табл. 6.5.
4. Некоторые моноклональные конъюгаты антитела анти-Н-2
(со щелочной фосфатазой, пероксидазой, флуорохромами) мож-
но приобрести. Этот выход из положения мы считаем наилуч-
шим, несмотря на дороговизну препаратов. Если конъюгаты
антител недоступны или предполагается использовать большие
их количества, приходится рассчитывать на собственные силы.
Стандартные методики приводятся в руководствах по иммуно-
логии и в работах [30] и [31]. Следует иметь в виду, что для
получения достаточного для работы количества антител вам
может понадобиться линия клеток гибридомы и концентриро-
ванный культуральный супернатант или асцитная жидкость.
Некоторые сведения, касающиеся приготовления конъюгатов
фермент — моноклональное антитело, можно почерпнуть из
табл. 6.4.
5. Тканевый препарат. Поскольку антигенные детерминанты,
узнаваемые различными моноклональными антителами
анти-Н-2, будут отличаться чувствительностью к разным фик-
Таблица 6.5. Двойное Н-2-антнген-окрашнвание гистологических срезов”-2’
Время одного окрашивания 5—6 ч.
1. Используйте криостатные срезы нефиксированной ткани толщиной 4 мкм
на покрытых желатиной стеклах.
2. Высушенные препараты храните при —20 °C, пока не понадобятся (до
нескольких недель).
3. Планируйте процедуру окрашивания. Пронумеруйте стекла (карандашом
или концами замороженного стекла), н для каждого из них составьте
схему конъюгатов антитела и окрашивания. Помните, что сбиться очень
легко, поэтому не делайте сразу слишком много препаратов (30 стекол,
если комбинация антнтела/красители ие слишком сложна, 15—20, если
она усложнена).
4. Для окрашивания поместите стекла на пластиковый лоток (рнс. 6.1).
Проконтролируйте при помощи уровня горизонтальность его положения.
Дайте стеклам высохнуть при комнатной температуре по крайней мере
20 мин.
5. Сполосните PBS. Для этого берите стекла одно за другим п поливайте
слабой струей из специальной пластиковой бутылки, стараясь, чтобы
струя не попала на срезы. Следите за тем, чтобы водоотталкивающие
части срезов были покрыты буфером (их можно осторожно смочить из
пастеровской пипетки). Стекла, покрытые PBS, снова поместите в лоток.
6. Фиксируйте ПЛП (0,5%-ный параформальдегид — 0,15 М лизни—10 мМ
перйодат натрия) в течение 2 мин (это время имеет существенное значе-
ние) при комнатной температуре. Для этого слейте со стекол PBS и на-
лейте из пастеровской пипетки ПЛП. Фиксатор должен быть свежеприго-
товленным: 1 часть 4%-ного формальдегида в дистиллированной воде,
3 части 0,2 М лизина и кристаллы перйодата натрия до 10 мМ. Маточ-
ный раствор лизина и параформальдегида можно хранить при 4 °C. Кри-
сталлы перйодата иатрня растворяются за несколько минут.
7. Смойте фиксатор со стекол слабой струей PBS.
8. Блокируйте эндогенную пероксидазу. Мы используем 0.1%-ный фенил-
гидразин-HCl в PBS, он не вызывает образования пузырьков (как мето-
ды, основанные на пероксидазе), которые могут разрушить замороженные
срезы. Слейте со стекол PBS и налейте феннлгндразин-HCl. Оставьте на
5 мин, затем высушите стекла н промойте PBS, как делали раньше.
9. Промойте стекла PBS, содержащим 0,5%-ный бычий сывороточный аль-
бумин (PBS+БСА).
10. Если не предполагается использование конъюгата антимышиного 1g,
инкубируйте стекла в течение 15 мин при комнатной температуре с PBS,
содержащим 10%-ную мышиную сыворотку (любой линии), для блокиро-
вания неспецифических сайтов связывания мышиных иммуноглобулинов.
Тем временем выполните следующие процедуры.
11. Приготовьте разведения конъюгата первого антитела в PBS, содержа-
щем 10%-ную мышиную сыворотку. Нанесите по 100 мкл разбавленного
конъюгата на каждое стекло (или больше, если на стекле несколько
больших срезов). Если срезы маленькие, а конъюгата в вашем распс.
ряжении не очень много, можно сократить покрываемую площадь, очер-
тив ее тушью. Всякий раз внимательно осматривайте пробирку с конъю-
гатом: на дне его обычно можно видеть небольшой осадок, но если его
количество заметно увеличилось — это может служить признаком дегра-
дации конъюгата. Агрегация конъюгата может вызвать образование фо-
на (см. «Помехи н их устранение», табл. 6.6).
12. Удалите со стекол PBS с мышиной сывороткой и осторожно промокните
жидкость бумагой, прикасаясь к поверхности стекла, где не срезов. Это
делается для того, чтобы не произошло разбавление конъюгата, который
вскоре предстоит нанести. Не давайте стеклам высохнуть — осушайте
их партиями (не более 2—3) и немедленно наносите на поверхность сре-
зов конъюгат (с помощью пипетки). Убедитесь в том, что он распреде-
11
П родолжение табл. 6.5
лился равномерно. Будьте внимательны, и, променивая стекла, не смажь-
те срезы. Инкубируйте в лотке под опущенной крышкой в течение 1 ч
прн комнатной температуре; количество жидкости должно быть достаточ-
ным для поддержания влажной атмосферы.
13. Тем временем приготовьте разбавления конъюгата второго антитела в
PBS+10%-ная мышиная сыворотка, как было указано выше. Если это
пероксидазный конъюгат, все разбавленные растворы не должны содер-
жать азида, который ингибирует пероксидазу.
14. Удалите со стекол конъюгат первого антитела и промойте PBS + БСА.
Осушите и промойте стекла снова; повторите с каждым стеклом цикл
трижды.
15. Промокните и нанесите второй (пероксидазный) конъюгат, как указано
выше. Инкубируйте 1 ч.
16. Осушите стекла и промойте PBS. Повторите цикл четырежды. Соберите
стекла в штатив для окрашивания, погрузив его в контейнер, содер-
жащий 200 мл 0,15 М трис с pH 7,6, такой же контейнер будет исполь-
зован для субстрата ДАБ при пероксидазном окрашивании.
17. Приготовьте субстрат ДАБ для пероксидазного окрашивания. (Замеча-
ние: все операции с участием ДАБ должны проводиться в перчатках и
под тягой.) Взвесьте 100 мг ДАБ. Поместите навеску в коническую пробир-
ку н добавьте 200 мл 0,15 М трис pH 7,6, 400 мкл концентрированной
перекиси водорода (храните при 4 °C н обновляйте через несколько ме-
сяцев). Осторожно покачайте пробирку до растворения. Если нужно,
добавьте NiCl2 (замечания относительно выбора субстрата см. в тексте).
Вылейте в сосуд, в котором будет проводиться окрашивание, и перенеси-
те туда из трис-буфера штатив со стеклами. Окрашивайте в течение 5 мин
при комнатной температуре, затем перенесите штатив со стеклами обрат-
но— в контейнер с трнс-буфером. Вылейте раствор субстрата ДАБ и
тщательно вымойте сосуд для окрашивания и остальную посуду. Про-
мойте стекла в трех порциях дистиллированной воды и возвратите их
в лоток для окрашивания, чтобы провести реакцию на щелочную фос-
фатазу.
18. Промойте стекла вероиал-ацетатным буфером с pH 9,2 (ВАБ) (но не
PBS, так как фосфат ингибирует реакцию кислой фосфатазы) н оставьте
их. Тем временем приготовьте раствор субстрата щелочной фосфатазы.
Взвесьте 5 мг Brentamine Fast Red TR (Sigma) н поместите навеску в
маленькую сухую стеклянную пробирку. 5 мг натриевой соли нафтола
As-BI (Sigma) внесите в универсальный пластиковый контейнер (объемом
на 20 мл) н добавьте 10 мл ВАБ, осторожно встряхните. Растворите
Fast Red TR в трех каплях диметнлформамида (храните защищенным
от света) и прибавьте к ВАБ/иафтол AS-BI, Остатки Fast Red TR нз
пробирки смойте раствором ВАБ. Прибавьте 200 мл левамизола (50 мМ
маточного раствора) (конечная концентрация левамизола 1 мМ) для ин-
гибирования эндогенной щелочной фосфатазы. Профильтруйте этот суб-
страт через бумагу Whatman № 1 в стеклянной воронке н немедленно
используйте.
19. Осушите стекла, промокнув ВАБ, и залейте раствором субстрата. Инку-
бируйте в течение 1 ч прн комнатной температуре. Интенсивность окраски
можно усилить, добавляя свежий субстрат с интервалами в 20 мни.
20. Промокните стекла, промойте несколько раз PBS или дистиллированной
водой, окрасьте гемалумом (слабо, если красный и коричневый цвета
предполагается демонстрировать иа черно-белых фотографиях) и заклю-
чите в водную среду. Замечание: продукт реакции щелочной фосфатазы
растворяется в спирте.
') Для окрашивания остальных препаратов делайте те же процедуры, но блокируй-
те эндогенную пероксидазу в течение 30 мин н точно выполняйте все указанные промы
вання.
!) См. ссылки [15] н [30].
Методы маркирования клеток н нх применение 165
саторам, лучший режим приходится искать методом проб и оши-
бок. Мы пришли к выводу о необходимости криостатных срезов
нефиксированной ткани; полученные срезы подвергают щадя-
щей фиксации в разбавленных белковых фиксаторах (напри-
мер, параформальдегид — лизин — периодит, табл. 6.5, п. 6) или
в очень холодном (—20 °C) ацетоне.
6. Последовательность инкубаций определяется опытным
путем. Мы получаем значительно лучшие результаты в том слу-
чае, когда конъюгат щелочной фосфатазы добавляется перед
пероксидазным конъюгатом, чем при их одновременном добав-
лении или внесении в обратной последовательности. Окраши-
вающая реакция пероксидазы должна предшествовать реакции
щелочной фосфатазы.
7. Выбор субстратов для контрастирующего двойного окра-
шивания. Несмотря на наличие многочисленных субстратов,
дающих различные цвета для щелочной фосфатазы и перокси-
дазы, мы остановились на том, что наибольшую интенсивность
и надежность обеспечивают Brentamine Fast Red TR (для ще-
лочной фосфатазы) и диаминобензидин (ДАБ) (для пероксида-
зы, коричневый цвет). Сочетание красного и коричневого пред-
ставляется не самым удачным. Многие пытаются усилить кон-
траст добавлением солей тяжелых металлов к раствору субстра-
та при пероксидазном окрашивании: например, добавление
50 мкл 8%-ного NiCl2 на 10 мл субстрата дает темно-пурпур-
ный продукт пероксидазной реакции, ^-галактозидаза (бирюзо-
во-голубой продукт) дает хороший цветной контраст в сочета-
нии с красным окрашиванием щелочной фосфатазой [32], но
у нас его интенсивность оказалась все же меньше, чем коричне-
вый цвет, даваемый пероксидазой/ДАБ.
8. Контроль. Необходимым условием надежности анализа
является присутствие на каждом стекле рядом со срезами хи-
мерной ткани срезов соответствующей ткани линий, составляю-
щих химеру. Это обеспечивает положительный контроль окра-
шивания и контроль специфичности. Кроме того, необходимо
иметь по крайней мере два стекла, каждое из которых инкуби-
ровано только с одним или только с другим конъюгатом анти-
тела, но окрашено обоими растворами субстрата. С их по-
мощью осуществляется контроль на неспецифическое окрашива-
ние и влияние одного конъюгата антитела на связывание
другого. Такой контроль особенно полезен, если вдруг возника-
ет проблема неспецифичности окрашивания в связи с тем, что
один из конъюгатов деградировал. Наконец, для контроля ка-
чества процесса окраски всегда включайте стекло со срезами
тимуса от каждой линии, содержащейся в химере.
Метод двойного окрашивания Н-2Ь- и Н-2к-антигенов на сре-
зах химерных тканей С.ВАч->С57 BL/6 описан в табл. 65 и про-
166
Глава 6
Вода, буфер и т. п.
Рис. 6.1. Лоток для иммуногистохимического окрашивания.
иллюстрирован рис. 6.2,А. Процедуры, с помощью которых
можно устранить помехи, представлены в табл. 6.6.
3.2. Другие антигены
1. Антигены 1а могут быть выявлены при помощи монокло-
нальных антител тем же способом, что и Н-2-антигены.
Они экспрессируются в костном мозге и лимфоидных тка-
нях, эндотелии и некоторых видах эпителия. Для каждого
антитела должен быть определен способ обработки ткани
и условия фиксации.
2. Антигены Thy-1 также образуют полиморфическую систе-
му, при этом AKR и другие А-линии мышей экспрессиру-
ют аллель Thy-1.1, а другие (преобладающее большинст-
во)— Thy-1,2. К соответствующим антигенам можно при-
обрести специфические моноклональные антитела. Есть
сведения о том, что Thy-1-антигены экспрессируются у мы-
ши на поверхности Т-клеток, клеток центральной нервной
системы, на фибробластах и, возможно, на миоэпители-
альных клетках; другие виды по этому признаку отлича-
ются. Паттерн окрашивания, демонстрируемый иммуноги-
стохимическим способом, в большой степени зависит от
использованного фиксатора, особенно в случае мозга.
Рис. 6.2. Визуальные маркёры хнмерности. 4. Криостатный срез эпителия
толстой кишки химеры В10а-<—i-BlOScSncc. Крипты В10а (А) окрашены
конъюгатом анти-Н2к щелочной фосфатазы в красный цвет, на отпечатке они
выглядят темными. Крипты BlOScSncc (В), окрашены конъюгатом пероксида-
зы с анти-Н2ь в желто-коричиевый цвет и кажутся светлыми. Б. Обработан-
ные ДВА парафиновые срезы толщиной 4 мкм эпителия тонкой кишки химе-
ры DDK*-»-C57BL16J(B6), окрашенные АДБ-пероксидазным конъюгатом.
На поперечном срезе ворсинок виден центральный кор (G), окруженный эпи-
телием (Е). В эпителии выявляются полосы клеток, происходящих от В6 (они
кажутся черными), н клетки, происходящие от DDK (они не окрашены).
Коры ворсинок содержат некоторое количество эндотелиальных клеток типа
DDK, также связывающих АДБ-пероксидазу. отчего они кажутся черными.
168
Глава 6
Таблица 6.6. «Помехи» при окрашивании Н-2-антигена и их устранение
1. Окрашивание слабое нли отсутствует.
Одно стекло/партня стекол.
Ошибка в процедуре разведении/добавления конъюгата на стекло.
Только одно антитело:
а) конъюгат антитела деградировал (это бывает довольно часто). Про-
верьте на срезах тимуса;
б) субстрат не тот/деграднровал. Проверьте эндогенное окрашивание на
«неблокироваиных» стеклах, обменяйтесь реактивами с коллегами или
попробуйте другую надежную систему антител, если она доступна;
в) вариации между линиями мышей, У некоторых экспрессия Н-2-антнгена
происходит не одинаково в разных тканях. Проверьте на срезах тимуса,
всегда дающего сильную реакцию.
Оба антитела.
а) если тимус также дает слабую окраску, можно полагать, что произо-
шла ошибка при фиксации или разбавлении конъюгатов;
б) если тимус окрашен хорошо, возможно, срезы были тоньше, чем обыч-
но, или неудача связана с особенностями линии мышей (см. выше).
Попробуйте другие срезы или ткани.
2. Неспецифическое окрашивание:
а) в общем, и особенно в отношении соединительной ткани, это указы-
вает на «грязный» конъюгат. Попробуйте более высокое разведение;
отцентрифугируйте агрегаты (5 мин в микроцеитрифуге); проведите
гель-фнльтрациоиную хроматографию для очистки фракции с меньшим
фоном (в этом случае вы скорее имеете дело с новым конъюгатом, чем
с деградирующим старым). Если вы полагаете, что этот конъюгат —
лучшее, что вы могли достать, попробуйте другие фиксаторы или сде-
лайте новый конъюгат;
б) специфические сайты. Решить проблему могут различные фиксаторы.
Можно эмпирически попробовать разные «блокирующие» агенты. Обыч-
но ситуация хуже в отношении одного нз конъюгатов, поэтому решение
следует искать в использовании ряда конъюгатов с разной степенью
конъюгации и в очистке гель-фильтрацией. Если это не поможет, по-
пробуйте сменить источник используемых реактивов (энзим, глутараль-
дегид) .
3. «Кристаллизация» окраски щелочной фосфатазы при хранении срезов.
Не исправить, фотографируйте наиболее важные результаты, пока препа-
раты свежие.
4. Низкая степень разрешения Н-2-антнгеиов соседних клеток. На уровне
светового микроскопа ситуацию можно улучшить, применив не фермент-
ные, а флуоресцентные конъюгаты. Возможна и электронная микроскопия
с Н-2-антигеиами [34].
Есть данные о том, что антигены Thy-1 могут отделяться
от одних клеток и захватываться другими, поэтому
Thy-1, по крайней мере в некоторых тканях, нельзя счи-
тать маркером, сохраняющим клеточную автономию.
3.3. Полиморфизм углеводов, выявляемый лектинами
По аналогии с полиморфизмом групп крови человека, мож-
но ожидать, что у мышей существует система полиформизма
Методы маркирования клеток и их применение 169
углеводов, тогда разные полиморфные варианты могут быть
использованы в качестве клеточных маркеров. Пока нашел при-
менение только один из них, он выявляется агглютинином Do-
lichos biflorus (АДБ) [17—19] (рис. 6.2,5), его использование
описано ниже, в разд. 3.4. Учитывая большую потребность
в маркерах, имеет смысл проверять соответствующие ткани мы-
шиных линий на присутствие нового маркера при помощи на-
бора лектинов, хотя, конечно, эта работа трудоемка и успех ее
ни в коей мере не гарантирован. Ниже приводится общая схема
скрининга для выявления полиморфизма углеводов при помощи
лектинов.
1. Приготовьте набор из 12 (или более) лектинов, конъюги-
рованных с ферментом или флуоресцирующим маркером,
в расчете на перекрывание как можно большего числа
специфических углеводов. Комплекты лектинов для подоб-
ного скрининга имеются в продаже. Если можно, титруй-
те лектины на ряде срезов тканей, с которыми они заве-
домо могут связаться. Ознакомьтесь с соответствующей
литературой. Убедитесь в том, что связывание является
специфическим. Для этого включите контроль, в котором
лектиновый конъюгат разбавлен до конечной концентра-
ции моно- или дисахарида 0,2 М.
2. Приготовьте криостатные срезы соответствующей ткани
от как можно большего числа животных генетически раз-
личных линий. Поместите срезы от трех или более разли-
чающихся линий на одно и то же стекло, это облегчит
процедуру окрашивания и сделает более удобным сравне-
ние. Вам понадобится вдвое большее число стекол по от-
ношению к числу лектинов, которыми вы располагаете
(чтобы иметь специфический контроль, включающий кон-
курирующий сахар), плюс несколько запасных. Для пер-
вого просмотра желательно взять нефиксированные сре-
зы; если это вызывает затруднения, можно использовать
слабый фиксатор (солевой раствор с 10% формалина
в течение 1 мин).
3. Инкубируйте лектиновые конъюгаты со срезами в течение
1 ч при комнатной температуре, промойте и окрасьте фер-
мент. Ищите бесспорные различия, по принципу «все или
ничего».
4. Если различия будут обнаружены, следует выяснить
устойчивость окраски к фиксации и заключению срезов
и изучить клеточную автономность полиморфного вариан-
та как маркера. Эта задача не простая. Если обнаружен-
ный полиморфизм удовлетворяет выбранным критериям,
следует убедиться в его стабильности при различных фи-
зиологических условиях (возраст эмбриона, а также
170
Глава 6
Таблица 6.7. Распределение аллелей Dlb-1 в линиях мышей
Dlb-1* Эндотелий+ DDK, GRS/A, LTS.AF, MAS, SM/Ja,
кишечный эпителий — STS, SWR, RHI-ro
Dlb-lb Эндотелий — кишечный+ эпителий AKR, A/Nimr, BALB/cHeA, C57L, C57BL/6J, C57BL/10ScSn, В.ЮА, C57BL/KS, C57BL/Nimr, CBA/Ca, CBA/HN, C3H/Bi, C3H/He, DBA/1, DBA/2, DBA/Af, GE, HTG, IF, LP/Nimr, LT, NZB, 020/A, SEA/J, SJL/J, 101/H, 129/RrJ
в условиях патологии (например, при неоплазии), если
они являются составной частью эксперимента. Помните,
что углеводы клеточной поверхности могут изменяться.
3.4. Использование в качестве маркера
лектина Dolichos (АДБ)
Этот маркер был открыт в результате систематического
скрининга, описанного в разд. 3.3, и применяется против мар-
кера Н-2. Его используют только в отношении некоторых сосу-
дистых эндотелиев и эпителия кишечника [8].. Он применяется
для изучения эмбрионального развития, начиная с 12-го дня,
и взрослых животных (детали анатомического описания см.
в [18, 19, 35]). Выявляемый полиморфизм имеет любопытный
реципрокный паттерн у двух аллелей — Dlb-1* и Dlb-lb
(табл. 6.7). У мышей Dlb-1* сайты связывания АДБ экспресси-
руются в эндотелии, но не в кишечном эпителии; у мышей
Dlb-lb распределение обратное.
Маркер АДБ обладает рядом преимуществ. Он исключи-
тельно стабилен. Сайты связывания лектина сохраняют устой-
чивость при многих процедурах фиксации (за исключением фик-
сации в 0,2%-ном глутаральдегиде) и заключения в парафин
или смолу (при условии, что температура не превышает 65°C).
Обусловленное этим маркером окрашивание —t сильное и чистое;
оно идеально для тотальных препаратов. Процедура окрашива-
ния проста, а удаление окраски инкубацией в 0,2 М N-ацетилга-
лактозамине служит эффективным контролем специфичности.
Недостатки этого маркера связаны с тем, что он имеется не
во всех тканях, а также с отсутствием положительной окраски
для другого компонента химеры. Экспрессия сайтов связывания
АДБ изменяется в диспластических центрах, индуцированных
в эпителии кишечника химическими канцерогенами. Она имеет
пятнистое распределение в эндотелии некоторых капилляров
Методы маркирования клеток и их применение 171
и в кишечных криптах, прилегающих к лимфоидным фоллику-
лам. Этот пример свидетельствует о том, с какой осторожно-
стью следует подходить к оценке любого нового маркера. Про-
цедура использования лектина Dolichos в качестве клеточного
маркера описана ниже.
3.4.1. Приготовление конъюгатов АДБ
Конъюгаты АДБ со щелочной фосфатазой, пероксидазой,
флуорохромами, биотином и частицами золота можно приобрес-
ти или приготовить стандартными способами, описанными в
оригинальной литературе [30, 31] и в учебных руководствах.
Конъюгацию осуществляйте в присутствии 0,2 М N-ацетилга-
лактозамина для защиты сайтов связывания лектина: впослед-
ствии сахар может быть удален диализом.
3.4.2. Подготовка тквни для получения срезов
Лучшим фиксатором является метакарн (метанол 60 : инги-
бисол 30: ледяная уксусная кислота 10). Этот фиксатор раство-
ряет пластик, поэтому нужно пользоваться стеклянной посудой.
Фиксируйте ткань в течение ночи и перенесите в 70%-ный эта-
нол. Можно использовать стандартную заливку в парафин, но
избегать при этом интенсивного нагревания.
3.4.3. Окрашивание срезов
Используйте по возможности свежеприготовленные парафи-
новые или криостатные срезы (толщиной 3 мкм), так как при
хранении срезов интенсивность окрашивания уменьшается.
Имейте в виду, что флуоресцирующие конъюгаты на парафино-
вых срезах дают неудовлетворительные результаты. Удалите
парафин или высушите криостатные срезы и промойте их соле-
вым раствором с фосфатным буфером (PBS).
1. При окрашивании АДБ, конъюгированным с пероксида-
зой, сначала блокируйте эндогенную пероксидазу 0,1 %-
ным фенилгидразином-HCl в PBS, затем промывайте
5 мин в PBS, после чего инкубируйте с PBS+ 0,5%-ный
БСА в течение 10 мин.
2. Осушите стекла, промокнув PBS + БСА (см. табл. 6.5,
п. 12) и прибавьте конъюгат АДБ, разбавленный PBS +
+ БСА.
3. Инкубируйте в течение 1 ч при комнатной температуре.
4. Промойте, сменив несколько раз PBS, и окрасьте соглас-
но методике, изложенной в табл. 6.5 (пп. 16 и 17).
172
Глава 6
Методы маркирования клеток н их применение 173
3.4.4. Контр о ли
Чтобы проконтролировать специфичность окрашивания,
включите один срез с конъюгатом АДБ, разведенным в PBS +
+ БСА до конечной концентрации N-ацетилгликозамина 0,2 М.
Для контроля качества окрашивания включите один срез стан-
дартной ткани (т. е. слюнной железы мыши линии SWR с ок-
рашиваемыми капиллярами).
3.4.5. Окрашнввние тотальных препврвтов
Процедура такая же, как и в случае срезов, со следующими
модификациями.
1. Фиксируйте приколотую ткань (разд. 6) в 10%-ном со-
левом формалине (метакарн будет растворять парафин
препаровальной ванночки, кроме того, он быстро фикси-
рует н уплотняет ткань, затрудняя удаление слизи).
2. В течение 30 мин блокируйте эндогенную пероксидазу
фенилгидразином-НС1.
3. Промойте тщательно после удаления конъюгата (особен-
но в случае кишечного эпителия).
Тотальные препараты химерной ткани представлены на
рис. 6.3.
4. Другие маркеры мозаичности
Мозаичность тканей может иметь естественное происхожде-
ние (инактивация Х-хромосомы) или вызываться внешними
причинами (индуцированная соматическая мутация или вклю-
ченные сегменты ДНК).
Рис. 6.3. Препарат химерной ткани, распределение клеток которой имеет кло-
нальный паттерн. А. Эндотелий аорты химеры C57BL/6J (В6)-м-DDK. Окра-
шен АДБ-пероксидазой. Эндотелиальные клетки, происходящие от DDK, свя-
зывают АДБ-пероксидазу и кажутся черными. Стрелка указывает на единич-
ную клетку. Расположение некоторых групп клонов выглядит волнообразным
в результате сокращения эндотелия (оно возможно из-за наличия в стенке
аорты эластичных волокон). Детальное обсуждение паттерна см. в рабо-
те [46]. Б. Слизистая тонкой кишки химеры C57BL/6J(B6) окрашен-
ная АДБ-пероксидазой; вид внутренней поверхности после удаления наруж-
ного мышечного слоя (см. разд. 6). Видны пять бляшек (1) крипт, происхо-
дящих от В6. Поскольку эта химера в значительной степени является «несба-
лансированной», компонент В6 оказался в меньшинстве, агрегация бляшек
минимальна и каждая бляшка, вероятно, представлена одним сцепленным
клоном. Сцепленные клоны могут быть частью одного потомственного клона
(обсуждается в работе [3]). Регулярный паттерн фона образован ворсинками
на противоположной стороне пласта, видны также кровеносные сосуды (2),
связывающие АДБ-пероксидазу.
174
Глава 6
4.1. Х-инактивация
Мозаичность по генам, сцепленным с Х-хромосомой, возни-
кает у самок в результате случайной инактивации в раннем
развитии одной из Х-хромосом. Любой ген, расположенный на
Х-хромосоме или оказавшийся там в результате транслокации,
является потенциальным кандидатом на роль маркера. Наибо-
лее широко используемый маркер при изучении развития мы-
ши— фермент 'фосфоглицерокиназа; полиморфные варианты
этого белка выявляются при электрофорезе. В последнее время
для маркирования клонов при изучении канцерогенеза широко
используется полиморфизм длины рестрикционных фрагментов
ДНК (ПДРФ) в сочетании с различиями между активной и
неактивной Х-хромосомами в отношении паттернов метилиро-
вания.
Преимущества Х-хромосомного мозаицизма для изучения
популяций маркированных клеток в организме заключается в:
а) отсутствии необходимости создавать химеры,
б) а также в том, что в этом случае нарушения нормально-
го роста и функционирования ткани минимальны (кле-
точные популяции мечены только генами, на которые
влияет лишь инактивация Х-хромосомы).
Недостатки системы мозаицизма Х-хромосомы приведены
ниже.
1. За исключением пигментированных тканей мышей «flec-
ked» с Х-аутосомной транслокацией, маркеры Х-инакти-
вации визуально не выявляются и, следовательно, не да-
ют информации о пространственном распределении. Воз-
можно, вскоре будут разработаны для практического ис-
пользования при изучении мышиных и человеческих се-
менников гистохимические методики, в основе которых
лежит «нулевой» аллель Х-сцепленного фермента глюко-
зо-6-фосфатдегидрогеназы.
2. X-ин активационные мозаики, как правило, «сбалансирова-
ны», что означает приблизительно равные популяции
клеток с активными материнскими или отцовскими X-
хромосомами. Даже при использовании генов Хее, обус-
ловливающих преимущественную активацию одной Х-хро-
мосомы, популяции мозаичных клеток образуют пропор-
цию 70 : 30 (в то время как при химеризме она составля-
ет 95:5). Это может усложнить анализ клонального
состава мозаичных участков (см. разд. 5).
4.2. Внешние маркеры единичных клеток
Для мечения клеток предимплантационных и ранних пост-
имплантационных эмбрионов используются маркеры, которые
Методы маркирования клеток и их применение
175
можно визуально распознать на гистологических срезах или
тотальных препаратах, — пероксидаза хрена, микросферы ла-
текса, [3Н]-тимидин и флуорохромы (см. гл. 2 и 3, а также
работы [1, 11]). Эти маркеры вводятся двумя путями: мечен-
ные in vitro клетки инъецируют в немеченый эмбрион или агре-
гируют с ним, другой путь заключается в прямой инъекции
маркера в отдельные эмбриональные клетки in situ. Маркеры,
введенные в отдельную клетку, наследуются ее потомками, од-
нако при этом происходит разбавление метки, поэтому метод
пригоден только для краткосрочных экспериментов.
4.3. Соматическая мутация
У Drosophila митотическая рекомбинация, индуцированная
облучением мух, гетерозиготных по рецессивной маркерной му-
тации, иногда дает гомозиготные мутантные маркированные
клетки. Эти клетки продолжают делиться, образуя клон, узна-
ваемый по мутантному фенотипу. Ценность методики очень
высока при клональном анализе развития мухи. В принципе,
подобную ситуацию можно смоделировать и у мыши, если у
животных, гетерозиготных по маркерному гену, индуцировать
соответствующую мутацию. Именно на этом основан spot-тест,
которым пользуются при анализе потенциальных мутагенов
[38]. Мутация «beige», которая в гомозиготном состоянии при-
водит к образованию четко аномальных меланоцитов сетчатки,
тоже может найти подобное применение [39]. В принципе, на
эту роль годится любой выявляемый визуально маркер, если
есть возможность получить его в гомозиготной форме. При этом
могут возникать следующие проблемы:
а) низкая частота и непредсказуемое место маркированных
клонов, что потребует для анализа большого объема тка-
ни;
б) нарушение нормального роста и развития, вызываемое
мутагенным воздействием;
в) необходимость большой популяции клеток-мишеней, обес-
печивающей достаточную вероятность маркирования
клеток в раннем эмбриональном развитии.
4.4. Трансгенные мыши
Трансгенные мыши описаны в гл. 11. Существует принципи-
альная возможность введения «маркерных» генов (продукт
которых может быть выявлен гистохимически, иммунологичес-
ки или гибридизацией in situ) практически на любой стадии
развития — от ооцита до взрослого организма. Для этого при-
меняют прямую микроинъекцию, инфицирование дефектными
ретровирусами или введение генетически маркированных кле-
176
Глава 6
ток. Насущной проблемой гистохимического или иммунологи-
ческого выявления маркера является получение генный конст-
рукций, обеспечивающих стабильную экспрессию выбранного
гена в широком спектре тканей эмбриона и взрослого организ-
ма. Передача маркированного гена по клеткам зародышевой
линии в некоторых случаях может привести к формированию
маркированной линии мышей, а интеграция гена в Х-хромосому
обеспечит к тому же и маркер Х-инактивации.
5. Интерпретация результатов маркирования
5.1, Анализ тканевых экстрактов
для выявления мозаичности
В этом случае маркер не дает топографической информации,
он лишь указывает на степень экспрессии генотипа в образце
ткани. Укажем проблемы, которые могут возникать при соот-
ветствующем анализе.
5.1.1. Чувствительность
Каковы минимальные размеры минорного компонента тка-
ни, достаточные для его выявления? Как правило, он составля-
ет 5—10%, хотя для глюкозофосфат-изрмеразы и фосфоглице-
раткиназы (гл. 7, разд. 3) продемонстрирована большая чув-
ствительность. Оценить чувствительность можно с помощью
экспериментов по реконструкции — смешивая тканевые экстрак-
ты от каждого генотипа и определяя активность обоих марке-
ров.
5.1.2. Специфичность
В настоящее время нет возможности определить, какие ти-
пы клеток представляют какие генотипы в тканевом образце.
Вследствие этого при оценке состава тканевой системы, состоя-
щей из нескольких клеточных типов (например, .опухоли), мо-
ноклональный состав одной клеточной популяции может быть
не распознан из-за примеси других, поликлональных популя-
ций.
5.1.3. Проблеме репрезентативности образцов
внвлизируемой тквни
Существует два аспекта этой проблемы.
1. Различия в составе ткани разных мышей были использо-
ваны для определения числа клеток-предшественников, от
Методы маркирования клеток и их применение 177
которых происходит ткань. Дедукция требует допущения,
что клетки-родоначальницы были выбраны случайно из
популяции и каждая из них внесла свой вклад в клеточ-
ный состав анализируемой популяции клеток [6]. Веро-
ятно, ни одно из этих допущений не верно.
2. Исходя из тех же соображений, по количеству клеток
каждого типа в образце химерной ткани судят о размере
соответствующего клона в этой ткани. Такой подход
справедлив, если клоны характеризуются одинаковыми
размерами и случайным распределением, однако это со-
вершенно не соответствует действительности [3].
5.2. Анализ видимых мозаичных паттернов
Использование маркеров, обеспечивающих визуальное опре-
деление генотипа индивидуальных клеток, позволяет обойти
многие проблемы, возникающие при работе с маркерами, не
обнаруживаемыми визуально. Но и при этом интерпретация
мозаичных паттернов не так проста, как можно заключить на
основании литературных источников. Ниже приведен краткий
перечень возникающих затруднений, подробности читатель най-
дет в цитированной литературе.
5.2.1. Некоторые определения
«Клон» — группа клеток, имеющих общего предка. «Сцеп-
ленный клон» — группа клеток, имеющих общего предка и ос-
тающихся пространственно объединенными; «потомственный
клон» •— группа клеток, имеющих общего предка, но разъеди-
ненных, так что по пространственному распределению их клас-
теров нельзя судить об их общем происхождении. «Бляшка» —
это группа пространственно объединенных клеток сходного ге-
нотипа, единица паттерна, реально наблюдаемая в мозаичной
ткани; несколько сцепленных клонов могут агрегировать, обра-
зуя бляшку.
S.2.2. Анализ
1. Измерение размера клона. При этом возникают две про-
блемы. Первая связана с агрегацией клонов. Для ее ре-
шения предложены различные математические методы,
позволяющие вычислить размер клона по наблюдаемому
размеру бляшки с учетом пропорций клеток каждого гено-
типа в ткани [40]. Условием применения этих мер является
правильная форма клонов, их одинаковые размеры и
случайное распределение [41]. Вторая проблема заклю-
12-171
178
Глава 6
чается в экстраполяции размеров двумерной бляшки из
одномерных паттернов, видимых на гистологических сре-
зах. И в этом случае экстраполяция основана на недока-
занном допущении регулярности или на основании рекон-
струкций по серийным срезам, что на практике представ-
ляет собой слишком утомительное занятие.
2. Пространственное расположение клонов. В результате
изучения пространственного распределения клонов может
быть получена дополнительная информация. Иногда чет-
кий паттерн выявляется визуально (например, в пигмент-
ном эпителии сетчатки [42] или коре надпочечника [43]).
В других тканях для определения потомственных клонов
необходим более тонкий анализ, например анализ диспер-
сии Грега — Смита. Он широко используется в экологии
растений и был применен к анализу тканей мыши Шмид-
том [3]. Частота встречаемости бляшек одного генотипа
определяется в каждом квадрате сетки, накладываемой
на ткань: случайное распределение бляшек даст пуассо-
новское распределение числа бляшек на квадрат; неслу-
чайное пространственное распределение («образование
кластеров», или «сверхдисперсия») узнается по отклоне-
нию от пуассоновского распределения. Этот подход поз-
волил выявить потомственные клоны в кишечном эпите-
лии и эндотелии аорты [3].
3. Применение тотальных препаратов для изучения «несба-
лансированных» химер. Анализ мозаичных паттернов
очень упрощается при использовании тотальных препара-
тов ткани (методику изготовления см. в разд. 6).
Они позволяют обойти проблемы экстраполяции от од-
номерной картины (эпителий на срезе) к двумерной (эпи-
телиальный слой). С помощью тотальных препаратов мы
получаем возможность изучать пространственное распре-
деление бляшек на большой площади и выявить паттерн,
не узнаваемый на меньшей шкале. Рисунок расположе-
ния бляшек будет даже более четким, если один из сос-
тавляющих генотипов находится в меньшинстве; каждая
бляшка при этом представляет индивидуальный клон, и
взаимное расположение этих клонов в потомственном кло-
не не затушевывается примесью клеток соседнего клона.
С такой ситуацией можно встретиться при работе с «не-
слабансированными» химерами или при введении единич-
ных клеточных маркеров на поздних стадиях развития.
Если используются несбалансированные эмбриональные
химеры, следует иметь в виду, что диспропорция обоих
составляющих химеру генотипов может быть случайной,
а может быть и следствием преимущественного роста од-
Методы маркирования клеток и их применение 179
ного из компонентов, что усложнит объяснение образо-
вавшегося паттерна. Если несбалансированные химеры
появляются часто и этот дисбаланс характеризуется од-
нонаправленностью, правдоподобность именно такой ин-
терпретации возрастает.
4. О чем свидетельствуют размеры клона в мышиной ткани?
Размеры сцепленных клонов отражают эффект клеточной
пролиферации (и соседство дочерних клеток), приводя-
щей к формированию клонов большого размера, и кле-
точного перемешивания в процессе роста. Относительный
вклад каждого эффекта определить не так просто, поэто-
му интерпретация окончательных паттернов, исходя из
истории клонов, оказывается весьма неопределенной.
Проблема обсуждается Вестом [40] и Шмидтом [3, 41].
В тканях, которые были проанализированы, распределе-
ние частоты размеров клона оказалось крайне неоднород-
ным, с преимуществом маленьких клонов и незначитель-
ным количеством очень крупных. Использование в каче-
стве характеристики данной ткани только клона среднего
размера недостаточно: гораздо лучше описывать ее
медианой и обеими квартилями, верхней и нижней. Асим-
метричность частоты распределения свидетельствует о
том, что одни клетки образуют клоны большего размера,
чем другие. Таким образом, даже если пренебречь воз-
можностью преимущественного роста одного из компо-
нентов данной ткани, на основании того, что, например,
минимальный вклад минорного генотипа составляет 1%,
нельзя сделать вывод о том, что вся ткань возникла из
100 клеток-предшественниц. Одна клетка из 100 не обя-
зательно даст 1% конечного объема ткани. Число потом-
ственных клонов также не укажет на число клеток-родо-
начальниц по двум причинам. Во-первых, предполагается,
что клоны происходят из клеточной смеси, в которой не
известны коммитированные клетки — предшественники
данной ткани. Во-вторых, общее число потомственных
клонов в ткани нельзя с уверенностью выводить из отно-
сительных пропорций минорного и мажорного генотипов
и числа клонов в минорном типе, так как существует воз-
можность того, что минорный генотип находится в не-
выгодных условиях роста.
12'
180
Глава 6
6. Приготовление тотальных препаратов
некоторых эпителиальных тканей
6.1. Оборудование
Пара тонких пинцетов.
Очень тонкие ножницы (т. е. ножницы профессора Кинмота,
Macarthy's Surgical Ltd. Dagenham, Essex, UK)-
Глазное лезвие (Beaver KB-225-063, Downs Surgical Ltd.,
Mitcham, Surrey, UK).
Энтомологические булавки (Watkins and Doncaster, Hawk-
hurst, Kent, UK).
Чашки Петри с 5-миллиметровым слоем воска на дне (Ral-
wax 2, Raymond A. Lamb, Sunbeam Road, London NW10, UK).
Препаровальный микроскоп с холодным источником падаю-
щего света.
6.2. Кишка
1. Отпрепарируйте кишку, отделив прилежащий мезенте-
рий и сосуды.
2. Перережьте в месте соединения подвздошной и слепой
кишки и восходящей толстой кишки.
3. Отрежьте слепую кишку.
4. Поместите в PBS на лед.
6.2.1. Окрашивание поверхности просвете толстой кишки
для выявления поверхностных пвттернов
ворсинок и эпителия
1. Начинайте с любого конца. Отрежьте фрагмент кишеч-
ника по длине, составляющей около 2/3 длины восковой
чашки (чтобы можно было расправить). Поместите на
восковое дно препаровальной чашки, покрытое холодным
PBS, и, слегка натянув, приколите ко дну каждый конец
булавкой.
2. Вскройте кишечник ножницами вдоль верхней границы
мезентерия на длину 2—3 см. Захватите перерезанные
концы тонким пинцетом (или кончиком булавки) и при-
колите к воску, осторожно и слегка натянув. Продолжай-
те прикалывать через каждые 8—10 мм, пока не прико-
лете полностью. Осторожным пипетированием удалите
с поверхности видимый фекальный материал. Если над
кишкой слой PBS не менее 5 мм, вы меньше рискуете
повредить ворсинки.
3. Проведите кратковременную фиксацию 10%-ным форма-
лином в солевом растворе (30 сек—1 мни) и промойте
PBS.
Методы маркирования клеток и их применение
181
4. Полностью закончив препарирование, проведите оконча-
тельную фиксацию формалином в солевом растворе в те-
чение 45 мин при комнатной температуре.
5. Затем инкубируйте в растворе: 20% этанола — 80%
150 мМ трис pH 8,2 с 25 мМ дитиотрейтола в течение
45 мин при комнатной температуре. Это способствует от-
делению слизи, которую можно удалить осторожным пи-
петированием под контролем микроскопа. Старайтесь не
повредить ворсинки.
6. Сполосните и перенесите в формол-солевой раствор. Хра-
ните до окрашивания в герметически закрытой посуде.
6.2.2. Окрашивание аблюминальной поверхности
кишечного эпителия с целью демонстрации мозаичных паттернов,
образуемых основаниями крипт
Поскольку у взрослой мыши крипты являются монокло-
нальными [44],— каждая крипта в мозаичном рисунке пред-
ставляет одну «клетку» [41].
1. Перед разрезанием инкубируйте кишку в 2 %-ном (в/о)
растворе гидрохлорида лигнокаина в PBS при 4 °C от
30 мин (тонкая кишка) до 1 ч (толстая кишка).
2. Приколите кишку к восковому дну препаровальной чашки
(лучше взять чашку с черным дном). Начиная с дуоде-
нального конца, пользуясь препаровальным микроскопом
и глазным лезвием, найдите плоскость, разделяющую
мышечные слои и слизистую оболочку. Тонкий прозрач-
ный мышечный слой можно медленно снять, отводя назад
пинцетом, как чулок. Весь адвентициальный жир или ме-
зентерий нужно предварительно осторожно удалить; в
местах вхождения крупных сосудов и лимфоидных фол-
ликулов плоскость разреза может быть потеряна, ее нуж-
но восстановить, осторожно обойдя эти структуры.
3. Закончив, вскройте продольно слизистую оболочку и при-
колите ворсинчатой поверхностью (поверхностью просве-
та) вниз.
4. Фиксируйте формалином в солевом растворе; удалять
слизь не надо. Обработка одной кишки занимает 2—3 ч
даже при наличии практики [45].
6.3. Аорта
1. Забейте мышь эфирной анестезией (смещение шейных
позвонков может порвать аорту).
2. Вырежьте аорту от дуги до диафрагмы. Поместите в хо-
лодный PBS в препаровальную чашку с черным воском
на дне.
182
Глава 6
3. Осторожно снимите весь жир и прилежащую ткань. За-
крепите один конец булавкой, осторожно вскройте аорту
продольно ножницами и приколите. Осторожно снимите
пипеткой прилипший кровяной сгусток.
4. Фиксируйте формалином в солевом растворе или соответ-
ствующим фиксатором.
5. Клеточные границы можно выявить серебряными краси-
телями (0,2%-ный нитрат серебра; экспозиция на ярком
свете), или окрасить ядра гемалумом для контроля цело-
стности эндотелиального слоя [46].
6.4. Эпидермис
1. Не забудьте до операции сфотографировать мышь, если
вы собираетесь сопоставлять паттерн окраски эпидермиса
и покровов.
2. Забейте мышь, очень осторожно выбрейте шерсть элект-
робритвой и вырежьте нужную площадь кожи. Для при-
готовления тотального «кожного препарата», на котором
будет изучаться средняя линия, сделайте разрез по одно-
му боку, вокруг грудной и тазовой областей и отметьте
средние линии и один бок стежком, прежде чем удалить
кожу (потом ориентация будет затруднена, так как кожа
потеряет форму).
3. Приколите кожу к деревянной планке и смажьте специ-
альным кремом для удаления волос, оставьте примерно на
20 мин и сполосните PBS. Пустым концом резиновой пер-
чатки на пальце осторожно размажьте смесь крема с PBS
по коже — это способствует удалению остаточных волос
с минимальным риском повредить эпидермис. Струей
PBS из пластиковой бутыли смывайте крем и волосы,
пока поверхность кожи не будет чистой и гладкой.
4. Уберите булавки и инкубируйте кожу в забуференном со-
левом растворе Хенкса с 20 мМ ЭДТА, pH 7,2 при 37 °C
в течение 20 ч, слегка взбалтывая. Это разрыхлит эпидер-
мис.
5. Прикрепите булавками на черное восковое дно чашки
эпидермисом вверх и покройте PBS. Под контролем пре-
паровального микроскопа осторожно отделите эпидермис,
удерживая одним пинцетом дерму и одновременно сло-
женными концами второго осторожно отталкивая назад
эпидермис. Интактный слой, соответствующий туловищной
области целиком, можно отпрепарировать за 1—2 ч.
6. Перенесите слой (воспользовавшись листом бумаги, стек-
лянной пластинкой или просто перелив из одной чашки
в другую) в восковую чашку с белым дном, где его при-
колите, базальной поверхностью вверх, для окрашивания.
Методы маркирования клеток н их применение
183
Литература
1. Gardner R. L. (1985). Phil. Trans. R. Soc. Lond. B, 312, 163.
2. Le Douarin N. M. (1985). Phil. Trans. R. Soc. Lond. B., 312, 153.
3. Schmidt G. H., Wilkinson M. M., Ponder B. A. J. (1985). J. Embriol. Exp.
Morphol., 88, 219.
4. Fialkow P. J. (1976). Biochim. Biophys. Acta, 458, 283.
5. Mullen R. J, (1977). Nature, 270, 245.
6. McLaren A. (1976). Mammalian Chimaeras. Cambridge University Press,
Cambridge.
7. Le Douarin N., McLaren A. (eds). (1984). Chimaeras in Developmental Bio-
logy. Academic Press, London.
8. Babinet C. (1980). Exp. Cell Res., 130, 15.
9. Papaioannou V. E., Dieterlan-Lievre F. (1984). In: Chimaeras in Develop-
mental Biology. Le Douarin N., McLaren A. (eds), Academic Press, London,
p. 3.
10. Green M. C. (ed.). (1981). Genetic Variants and Strains of the Laboratory
Mouse. Fischer Verlag, Stuttgart and New York.
11. West J. D. (1984). In: Chimaeras in Developmental Biology. Le Douarin N.,
McLaren A. (eds), Academic Press, London, p. 39.
12. Feder N. (1976). Nature, 263, 67.
13. Dewey M. J., Gervais A. G„ Mintz B. (1976). Dev. Biol., 50, 68.
14. Oster Granite M. L„ Gearhart J. (1981). Dev,. Biol., 85, 199.
15. Ponder B. A. J., Wilkinson M. M., Wood M. (1983). J. Embriol. Exp. Mor-
phol., 76, 83.
16. Enzine S., Weissmann I. L„ Rouse R. V. (1984). Nature, 309, 629.
17. Schmidt G. FL, Wilkinson M. M„ Ponder B. A. J. (1985). Cell, 40, 425.
18. Uiterdijk H. G., Ponder B. A. L, Festing M. F. W., Hilgers J„ Skow L.,
Van Nie R. (1986). Genet. Res. Camb., 45, 125.
19. Ponder B. A. Wilkinson M. M. (1983). Dev. Biol., 96, 535.
20. Kitamura Y., Matsuda H., Hatanaka K. (1979). Nature, 281, 154.
21. Oliver C., Essner E. (1973). J. Histochem. Cytochem., 21, 218.
22. Mintz B„ Sanyal S. (1970). Genetics, 64, Suppl. 43.
23. Rossant J. (1985). Phil. Trans. R. Soc. Lond. B„ 312, 91.
24. Peterson A. C. (1979). Ann. N. Y. Acad. Sci., 317, 630.
25. Mintz B., Baker W. W. (1967). Proc. Natl. Acad. Sci. USA, 58, 592.
26. Mintz B. (1974). Annu. Rev. Genet., 8, 411.
27. Ford С. E., Evans E. P., Gardner R. L. (1975). J. Embryol. Exp. Morphol..
33, 447.
28. McLaren A. (1975). J. Embryol. Exp. Morphol., 33, 205.
29. Lu T.-Y., Markert C. L. (1980). Proc. Natl. Acad. Sci. USA, 77, 6012.
30. Ponder B. A. J., Wilkinson M. M„ Wood M„ Westwood J. H. (1983). J. His-
tochem Cytochem., 31, 911.
31. Ponder B. A. J. (1983). In: Immunocytochemistry. Polak J. M., Van Noor-
den S. (eds), Wright, PSG, Bristol, p. 129.
32. Bondi A., Chieregatti G., Gusebi V., Fulcheri E., Bussolati G. (1982), Histo-
chemistry, 76, 153.
33. Ponder B. A. L, Wilkinson M. M. (1981). J. Histochem. Cytochem., 29, 981.
34. Van Ewijk W., Rouse R. V., Weissman I. L. (1980). J. Histochem. Cytochem.,
28, 1089.
35. Ponder B. A. J„ Festing M. F. W., Wilkinson M. M. (1985). J. Embryol. Exp.
Morphol., 87, 229.
36. Vogelstein B., Fearon G. R., Hamilton S. R„ Feinberg A. P. (1985). Science.
227, 642.
37. Wareham K. A., Howell S., Williams D., Williams E. D. (1983). Histochem
J., 15, 363.
38. Fahrig R., Neuhauser-Klaus A. (1985). J. Hered., 76, 421.
184
Глава 6
39. Searle A. G., Stephenson D. A. (1982). Mutat. Res., 92, 205.
40. West J. D. (1975). J. Theor. Biol., 50, 153.
41. Schmidt G. H., Garbutt D. J.t Wilkinson M. M„ Ponder B. A. J. (1985).
J. Embryol. Exp. Morphol., 85, 121.
42. Schmidt G. H., Wilkinson M. M., Ponder B. A. J. (1986). J. Embryol. Exp.
Morphol., 91, 197.
43. Weinberg W. C., Howard I. C., lannaccone P. M. (1985). Science, 227, 524.
44. Ponder B. A. J., Schmidt G. H., Wilkinson M. M„ Wood M. J., Monk M.,
Reid A. (1985). Nature, 313, 689.
45. Schmidt G. H., Wilkinson M. M., Ponder B. A. J. (1984). Anat. Rec., 210,
407.
46. Schmidt G. H., Wilkinson M. M„ Ponder B. A. J. (1986). J. Embryol. Exp.
Morphol., 93, 267.
ГЛАВА 7
БИОХИМИЧЕСКИЙ МИКРОАНАЛИЗ
СЦЕПЛЕННЫХ С Х-ХРОМОСОМОЙ ФЕРМЕНТОВ
ГФРТ И ФГК
Мэрилин Манк1
1. Введение
В этой главе описывается способ измерения активности фер-
ментов в небольших объемах биологического материала, напри-
мер, в яйцеклетках мыши, эмбрионах ранних стадий дробления,
небольших кусочках ткани, представляющих линии клеток-пред-
шественников, в небольшом числе стволовых или герминативных
клеток. В нашей лаборатории были разработаны методы экст-
ракции фермента и количественного определения его активнос-
ти в небольших объемах поддерживающей среды и на пре-
дельном уровне разрешения и чувствительности. Усилия сотруд-
ников были сконцентрированы на развитии методов микроана-
лиза для гипоксантин-фосфорибозилтрансферазы (ГФРТ) и
фосфоглицераткиназы (ФГК-1)—ферментов, сцепленных с X-
хромосомой. Однако основные принципы микроанализа, опи-
санные здесь, можно применять и для изучения других фермен-
тов.
Микрометод определения активности ГФРТ широко исполь-
зуется нами для выявления дозы гена ГФРТ, при изучении ак-
тивности Х-хромосомы в процессе развития женского эмбриона
мыши. Суть этого подхода заключается в следующем. Если
у самки на определенной стадии развития или в определенной
ткани активны обе Х-хромосомы, то в пробе женского материа-
ла активность ГФРТ будет вдвое выше, чем в мужской (Х+Х+:
X+Y=2: 1). Если одна Х-хромосома самки инактивируется, зна-
чение активности ГФРТ будет одинаковым в обоих случаях
(X+X-:X+Y=1 : 1).
Главным преимуществом методики является возможность
одновременно в одной пробе и в одной реакционной смеси из-
мерять активность Х-сцепленной ГФРТ и сцепленной с ауто-
сомой аденин-фосфорибозилтрансферазы (АФРТ). Ферменты
ГФРТ и АФРТ родственны в функциональном отношении — и
тот и другой вовлечены в метаболический путь утилизации пу-
ринов в связи с включением пуриновых оснований в ДНК.
Двойной микрометод позволяет вычислить отношение активно-
1 Marilyn. Monk. MRC Mammalian Development Unit, 4 Stephenson Way,
London NW1 2HE, UK.
186
Глава 7
сти Х-сцепленной ГФРТ к активности аутосомной АФРТ и, та-
ким образом, судить о дозе гена Х-сцепленного фермента. Со-
поставление отношений ГФРТ: АФРТ устраняет ошибки отбо-
ра проб и позволяет сравнивать пробы между собой. В том
случае, когда мы имеем дело со стандартными пробами, таки-
ми, как предимплантационные эмбрионы на определенной ста-
дии развития или известное число клеток, микрометод с доста-
точной точностью дает значения активности каждого индивиду-
ального фермента (выраженной на эмбрион или на клетку).
Результаты применения двойного микроанализа для ГФРТ :
: АФРТ можно представить следующим образом.
1. В ходе раннего развития мыши, до 8-клеточной стадии,
возрастание активности ГФРТ связано с трансляцией за-
пасенной материнской мРНК этого фермента [1, 2].
У ранних эмбрионов, происходящих из яиц матерей с
генотипом XX, активность ГФРТ вдвое выше, чем у эм-
брионов матерей ХО [3]. Использование ингибиторов мак-
ромолекулярных синтезов подтверждает представление о
запасенной материнской мРНК, обеспечивающей раннее
возрастание активности ГФРТ [2].
2 К стадии морулы активизируются ГФРТ-гены эмбриона.
Женские (XX) эмбрионы обладают вдвое большей актив-
ностью ГФРТ, чем мужские (XY). Следовательно, на этой
стадии развития женского эмбриона активны гены
ГФРТ, связанные с обеими Х-хромосомами [1, 4].
3. Инактивация одной из двух Х-хромосом происходит пос-
ледовательно: сначала во внезародышевой трофэктодер-
ме, затем в первичной энтодерме и, наконец, в ткани, из
которой разовьется эмбрион [5].
4. Реактивация инактивированной Х-хромосомы происходит
в линии женских половых клеток перед наступлением
мейоза [6].
5. В последнее время двойной микроанализ ГФРТ и АФРТ
используется для идентификации самок мыши, являющих-
ся мозаиками в отношении инактивации Х-хромосомы.
Гетерозиготные самки ГФРТ+/ГФРТ- оплодотворялись
химерным самцом, у которого половые клетки произошли
от ГФРТ-отрицательных эмбриональных стволовых кле-
ток; Половина мужского потомства гетерозиготных самок
оказалась ГФРТ-отрицательной [7]. У человека недоста-
точность ГФРТ служит причиной синдрома Леша — Най-
хана.
6. Чувствительность микрометода оказывается достаточной
для диагностики ГФРТ-дефицитных мужских эмбрионов
по анализу одного бластомера 8-клеточной стадии пред-
имплантационного эмбриона [8]. Прн этом сам зародыш
Биохимический микроанализ ферментов ГФРТ и ФГК 187
переносят в псевдобеременную приемную мать, чтобы
проконтролировать правильность диагноза. Определение
дозы ГФРТ в одном бластомере, полученном посредством
биопсии, позволяет судить и о поле эмбриона [9].
7. Мы использовали микрометод для анализа активности
клонированных генов ГФРТ, введенных в яйца путем
микроинъекции. Применение активности ГФРТ в ка-
честве «свидетеля» дает возможность сделать заключение
об эффективности различных премоторных функций.
В этом случае одновременное измерение уровня АФРТ
служит ценным контролем нормального развития инъеци-
рованного яйца.
Преимущество микрометода для анализа ФГК-1 заключает-
ся в том, что он позволяет не только различить две Х-хромосо-
мы, но и измерить уровни их активности. У мыши Х-зависимый
фермент ФГК-1 существует в двух различных электрофоретиче-
ских формах: ФГК-1А (обнаружена в популяции дикой мы-
ши Нильсеном и Чепменом [10]) и ФГК-1Б.
Изозпмы ФГК-1 могут быть разделены путем гель-электро-
фореза и использованы для количественного определения про-
порций клеток, характеризующихся активностью мужской или
женской Х-хромосомы в женском эмбрионе, гетерозиготном по
аллелям Pgk-Ia или Pgk-Ib. Количественная характеристика
обеих изозимных форм может быть получена путем повторного
измерения флуоресценции при реакции окрашивания. Этот ме-
тод в высшей степени чувствителен и выявляет активность
яйцеклетки или эмбриона мыши и даже нескольких клеток
различных клеточных линий, выделенных микрохирурги-
чески.
На основании количественного анализа изозимов ФГК были
сделаны следующие выводы относительно характера развития.
1. Х-хромосома, наследуемая по отцовской линии, инакти-
вируется преимущественно во внезародышевых клеточных
линиях — трофэктодерме и первичной энтодерме [11—13].
2. В примордиальных половых клетках наблюдается слу-
чайная инактивация Х-хромосомы мыши [14, 15].
3. Изучение мозаиков выявляет корреляции пропорций ма-
теринских и отцовских Х-хромосом, активных в дефини-
тивных герминативных слоях и в половых клетках. Сле-
довательно, эти ткани произошли из общего пула клеток
с инактивированной Х-хромосомой [16].
4. Преимущественная экспрессия транслоцированной Х-хро-
мосомы является следствием селекции клеток в раннем
развитии [17].
5. В ооцитах гетерозиготных самок, как правило, экспрес-
сируется аллель Pgk-P [18].
1S4
Глава 7
6. Индивидуальные крипты кишечника гетерозиготных са-
мок экспрессируют либо один, либо другой изозим, что
свидетельствует об их моноклональном происхождении
[19].
7. В случае химер по клеткам крови, образованных в ре-
зультате инъекции красного костного мозга летально об-
лученным мышам, изозимы ФГК-1 используются для
наблюдений над заселением красного костного мозга и
периферической крови [20].
8. Изозимы ФГК-1 дают ценную информацию при изучении
клональных взаимодействий в опухолях [21].
2. Двойной микроанализ ГФРТ и АФРТ
ГФРТ — Х-кодируемый фермент [3, 22], который катализи-
рует превращение гипоксантина и гуаиина в инозинмонофосфат
(IMP) и гуанозинмонофосфат (GMP). АФРТ закодирована в
аутосоме [23]; ее роль заключается в превращении аденина
в аденозинмонофосфат (АМР). Оба фермента используют фос-
форибозилпирофосфат (ФРПФ) в качестве источника фосфори-
бозилового остатка. С помощью метода двойного мечения Гар-
тлер [24] измерил уровни ГФРТ и АФРТ в волосяных фолли-
кулах, отделяя субстрат от продукта путем тонкослойной хро-
матографии (ТСХ). Другие исследователи разделяли субстрат
и продукт обеих реакций в процессе высоковольтного электро-
фореза [25]. Применяемый нами метод (он представляет собой
видоизменение метода, предложенного Бакеем [26]) использо-
вали также Мак-Берни и Адамсон [27]. Он заключается в том,
что эмбриональные экстракты добавляют в реакционную смесь,
содержащую меченые субстраты, [Н3]-гипоксантин (или гуа-
нин) и [С14]-аденин, и ФРПФ. Меченые нуклеотидные продук-
ты— IMP (или GMP) и АМР — осаждают хлоридом лантана,
собирают на стекловолокнистом фильтре и определяют связав-
шуюся радиоактивность с помощью сцинтилляционного счетчи-
ка (рис. 7.1).
Манк и Катуриа [28] разработали методику, оказавшуюся
достаточно чувствительной для измерения активности отдель-
ных предимплантационных эмбрионов; в течение последующих
лет нам удалось увеличить чувствительность метода, и сейчас
мы можем уловить активность менее чем 1 пмоль/ч в одной ре-
акционной смеси. Активность каждого фермента (или обоих)
можно измерять в отдельных клетках или даже в одном блас-
томере 16-клеточной стадии. После того как эксперимент отла-
жен и исследователь вполне овладел микрометодом, за день
можно сделать до 100 определений.
Биохимический микроанализ ферментов ГФРТ н ФГК
189
Фильтруйте
и считайте
Рис. 7.1. Схема, обобщающая двойной микрометод определения активности
ГФРТ и АФРТ в эмбриональном экстракте. Детали описаны в тексте. На схе-
ме представлен анализ единственного эмбриона. Эмбрион, помещенный в сре-
ду РВ1.ПВП, переносится в центр мнкрокапсулы, концы которой затем за-
паивают и хранят при —70 °C. Для анализа образец трижды подвергают про-
цедуре замораживания — оттаивания, затем центрифугируют и супернатант
(~5 мкмл) добавляют к реакционной смеси.
2.1. Приготовление образца
Способы получения яиц и предимплантационных эмбрионов,
а также методы работы с ними описаны в гл. 2 (разд. 2 и 3).
Выделение клеточных линий ВКМ и ТЭ на стадии бластоцисты
производят согласно методике, изложенной в разд. 3.4 и 3.5
гл. 2. Способ получения постимплантационных эмбрионов
описан в разд. 4, гл. 3. Выделение половых клеток может быть
осуществлено следующим образом.
1. Отпрепарируйте гонады у постимплантационных эмбрио-
нов. Промойте их и инкубируйте в фосфатно-солевом
буфере, содержащем вместо альбумина 0,4%-ный поливи-
нилпирролидон (РВ1.ПВП) (гл. 13, разд. 3.1.1) и 0,2%-
ный ЭДТА, при комнатной температуре в течение 30 мин.
2. Освободите половые клетки, разрывая гонады и надавли-
вая на них.
3. Очистите половые клетки от примеси соматических клеток
гонады и эритроцитов, слегка отгоняя их струей среды из
пипетки. Старайтесь при этом половые клетки собрать
вместе.
190
Глава 7
4. Перенесите половые клетки в микрочашки (см. ниже) и
храните при —70 °C.
В ходе всех процедур выделения эмбрионов и промывания
клеток или тканей в качестве среды следует использовать РВ1.
ПВП. Смысл включения ПВП в среду заключается в том, что
этот компонент увеличивает ее вязкость и снижает адгезивность
маленьких кусочков ткани или изолированных клеток, вследст-
вие чего они не пристают к стенкам стеклянных пипеток и мик-
рочашек, и, следовательно, не теряются. В случае предимплан-
тационных эмбрионов, окруженных нелипкой блестящей обо-
лочкой, опасность потери не столь велика. Альбумин не исполь-
зуется по той причине, что он может показать при микроанали-
зе ферментативную активность. Если проблема адгезивности
остается, можно посоветовать держать среду (и часовые стек-
ла) холодными и до помещения пробы в микрочашки не давать
жидкости с материалом отстояться.
Пробы собирают в микрочашки с 5 мкл РВ1.ПВП после
следующих манипуляций.
1. Наберите достаточное количество РВ1.ПВП в тонко от-
тянутую пастеровскую пипетку, соединенную резиновой
трубкой с мундштуком.
2. В ту же пипетку втяните образец. Образцом может быть
один бластомер, один предимплантационный эмбрион,
один постимплантационный эмбрион (или его часть)
или, например, 50 половых клеток.
3. Введите конец пипетки в микрочашку (Drummond) объ-
емом 10 мкл и выпустите эмбрионы с жидкостью так,
чтобы она заняла половину объема чашки (5 мкл). На-
блюдая в микроскоп, убедитесь в том, что эмбрионы,
ткань или клетки попали в чашку.
4. Удерживая микрочашку между большим и указатель-
ным пальцами, столкните образец в середину чашки мяг-
ким ударом основания ладони о поверхность стола.
5. Немедленно закупорьте края микрочашки, оплавив стек-
ло в пламени.
6. Проконтролируйте под микроскопом качество сплавле-
ния.
7. Храните образцы в маленьких пластиковых пробирках
(Falcon) при —70°C.
8. Перед анализом образцы трижды подвергните процедуре
замораживания — оттаивания, перенося их из —70°Св
комнатную температуру и обратно (не пользуйтесь жид-
ким азотом: микрочашка может лопнуть).
9. Центрифугируйте в контейнерах для пробирок типа Fal-
con (2000 об/мин в течение 5 мин при 4°C). (Если
Биохимический микроанализ ферментов ГФРТ и ФГК
191
образцы прилипнут к стенкам, можно отделить их центри-
фугированием в другом направлении).
10. Отберите супернатант для анализа.
2.2. Реакционная смесь
Для измерения активности ГФРТ и АФРТ эмбриональный
экстракт инкубируют при 37 °C в 50 мкл реакционной смеси,
которая состоит из следующих компонентов: натрий-фосфатный
буфер (38, 75 мМ с pH 7,4), хлорид магния (5 мМ), ФРПФ
(1 мМ), [3HJ-гипоксантин (10 мкМ) и [14С]-аденин (10 мкМ).
(В ранних экспериментах в качестве субстрата для ГФРТ мы
использовали вместо гипоксантина [3Н]-гуанин.) Чтобы чувст-
вительность была максимальной, следует использовать неразве-
денный Г14С] -аденин с самой высокой удельной активностью.
Прочие реактивы готовят и хранят в виде стерильных исходных
растворов небольшими объемами, чтобы не открывать лишний
раз пробирку во избежание загрязнений. Приготовление ис-
ходных растворов описано ниже.
2.2.1. Фосфатный буфер и [ Н]-гипонсантин
Приготовьте натрий-фосфатный буфер (50 мМ pH 7,4). Для
этого смешайте 1,75 мл 0,2 М NaH2PO4-H2O; 8,25 мл 0,2 М
Na2HPO4- 12Н2О и 30 мл воды. Доведите pH до 7,4. Стерили-
зуйте фильтрованием. Этим буферным компонентом реакцион-
ной смеси разведите [3Н]-гипоксантин (последний продается в
виде лиофилизированного препарата (TRA.74, 1—10 Ки/мМ,
Amersham) следующим образом.
1. Растворите лиофилизированный препарат в натрий-фос-
фатном буфере (50 мМ, pH 7,4) до концентрации
12,9 мкМ. Например, 1 мКи [3Н]-гипоксантина с актив-
ностью 6,2 Ки/мМ растворяется в 12,5 мл буфера.
2. Осторожно наберите раствор [3Н]-гипоксантина в шприц
и распределите его аликвоты примерно по 2 мл, пропус-
тив через стерилизующий фильтр (Millex, Millipore), по
небольшим пробиркам (Falcon). Храните при 4°С.
3. Если понадобится уменьшить удельную активность, сме-
шайте исходный радиоактивный гипоксантин с 12,9 мкМ
немеченого гипоксантина (Sigma), растворенного в фос-
фатном буфере (50 мМ). Немеченый исходный гипоксан-
тин стерилизуется фильтрованием через миллипоровый
фильтр и хранится порциями по 10 мл в пробирках (Fal-
con) при 4 °C.
4. Используйте по 38,75 мкл [3Н]-гипоксантина в буфере с
первоначальной удельной активностью (или, если потре-
192
Глава 7
буется, с более низкой удельной активностью) на 50 мкл
реакционной смеси (конечная концентрация гипоксанти-
на 10 мкМ и буфера 38,75 мМ).
2Л.2. Хлорид магния
Приготовьте хлорид магния (MgCl2-6H2O) в концентрации
50 мМ и пропустите через миллипоровый фильтр по 1 мл в
стерильные пробирки (Falcon). Храните при 4°C. Хлорид маг-
ния готовится отдельно и добавляется в процессе составления
реакционной смеси (во избежание осаждения при хранении).
На 50 мкл реакционной смеси следует брать по 5 мкл хлорида
магния (конечная концентрация 5 мМ).
2.2.3. Фосфорибозиллирофосфат
ФРПФ (м. в. 390,1; Sigma) храните в твердом состоянии
при —20°С, его раствор (10 мМ) готовьте непосредственно пе-
ред составлением реакционной смесн (конечная концентрация
1 мМ). Определите общий объем раствора ФРПФ, который по-
требуется, и растворите в концентрации 0,39 мг/0,1 мл в сте-
рильной дистиллированной воде.
2.2.4. [14С]-аденин
[U-I4C|-аденин (CFA-436; >220 мКи/мМ, Amersham) про-
дается в виде жидкости. Храните при 4 °C. Используйте одно-
родно меченный аденин, поскольку среди имеющихся препара-
тов аденина он.обладает наиболее высокой удельной активно-
стью. К 50 мкл реакционной смеси следует добавить объем,
обеспечивающий конечную концентрацию меченого аденина
10 мкМ при первоначальной удельной активности (т. е. к
50 мкл реакционной смеси нужно прибавить 2,35 мкл аденина
с удельной активностью 235 мКи/мМ в концентрации
50 мкКи/мл).
2.3. Реакция
2. 3.1. Контроль
Микрометод обладает исключительной чувствительностью:
он позволяет измерять активность ГФРТ до 0,1 пмоль/ч. Все
исходные растворы должны быть стерильны, при всех манипу-
ляциях следует стремиться к идеальной чистоте. Старайтесь
почти не прикасаться пальцами к пробиркам и микрочашкам
(но в перчатках работать неудобно).
Биохимический микроанализ ферментов ГФРТ н ФГК 193
Чтобы добиться максимальной чувствительности, реакцион-
ную смесь приходится инкубировать при 37 °C в течение многих
часов или даже оставлять на всю ночь. Необходимо исключить
риск бактериального или иного загрязнения и быть уверенным
в том, что активность каждого фермента с течением времени
будет оставаться линейной. Для того чтобы проследить эту ли-
нейность во времени, нужны предварительные эксперименты,
в которых решается еще одна задача — определение точки, по-
сле которой реакция пойдет на спад (графически это выража-
ется выходом кривой активности на плато). Без контрольных
экспериментов полученные отношения ГФРТ: АФРТ могут
оказаться артефактом.
Очень важную роль в реакции играет ФРПФ. Чтобы добить-
ся максимальной точности измерений, рекомендуется попробо-
вать разные концентрации этого соединения. Отсутствие ФРПФ
в реакционной смеси предотвращает появление радиоактивнос-
ти в осадке хлорида лантана. В эксперименте, предполагающем
анализ целого ряда экстрактов, образцы по очереди вносятся в
заранее приготовленные аликвоты (по 50 мкл) реакционной
смеси, при этом следует отмечать время внесения. Следует
включить в качестве «пустых» контролей поддерживающую сре-
ду (РВ1. ПВП) без экстракта в начале и конце загрузки образ-
цов. Если измерения, относящиеся к продукту в эксперимен-
тальных реакциях, даже незначительно превышают уровни
«пустых» контролей, абсолютно необходимо включить в экспе-
римент повторные «пустые» контроли с тем, чтобы убедиться в
постоянстве их счета. Результат будет зависеть от воспроизво-
димости экспериментальной техники запуска и остановки реак-
ции и от процедуры отмывки (см. ниже).
Рекомендуется также иметь «пустой» контроль среди
экспериментальных образцов для того, чтобы показать незави-
симость счета, полученного для одного образца, от счета преды-
дущего (т. е. «перекрывание») при последовательной фильтра-
ции и отмывке образцов. В идеальном случае этот «пустой»
контроль должен быть включен после образца, с которым свя-
зано ожидание высокого значения счета продукта.
2. 3.2. Постановив вксперимента
1. Составьте протокол опыта: например, «пустые» контроли
РВ1.ПВП), 1—3, образцы (допустим, их 20), 4—23; конт-
роль «перекрывания» (только 50 мкл реакционной смеси),
24; «пустые» контроли (РВ1.ПВП), 25—27.
2. Приготовьте реакционную смесь. В нашем примере
(см. п. 1), потребуется 27 объемов реакционной смеси,
по 50 мкл в каждом. Приготовьте общий объем реакци-
13—171
194
Глава 7
онной смеси следующим образом: 27X38,75 мкл [3HJ-
гипоксантина (12,9 мкМ) в фосфатном буфере (50 мМ,
pH 7,4)...1046,25 мкл.
27x5 мкл хлорида магния (50 мМ)...135 мкл.
27x5 мкл ФРПФ (0,585 мг в 0,15 мл)...135 мкл.
27x2,35 мкл [14С]-аденина (235 мкКи/мкМ)...63,45 мкл.
Общий объем = 1379,7 мкл.
3. Пронумеруйте реакционные пробирки (пластиковые тест-
пробирки, Falcon 2038, 10x75 мм) от 1 до 27 и налейте
в каждую из них по 50 мкл реакционной смеси.
4. В период подготовки эксперимента образцы в микрочаш-
ках трижды проведите через процедуру замораживания —
оттаивания и центрифугируйте (в двух направлениях,
если нужно) как предлагается в разд. 2.1.
5. Подготовьте микрочашки к вскрытию, нанеся царапину
алмазным ножом.
6. Пустите часы.
7. По очереди вскройте каждую микрочашку и, воспользо-
вавшись пипеткой с мундштуком, контролирующей тонко
оттянутую пастеровскую пипетку, извлеките надосадоч-
ную жидкость (~5 мкл), перенесите в соответствующим
образом пронумерованную реакционную пробирку и
ставьте инкубировать при 37 °C. Таким образом один за
другим все образцы поместите в водяную баню, отметив
стартовое время для каждой реакционной пробирки.
8. Оставьте при 37 °C на нужное для реакции время.
2.4. Прекращение реакции
Останавливающей смесью служит охлажденный во льду ра-
створ 0,1 М хлорида лантана (LaCl3-7H2O, Sigma), содержа-
щий 1 мМ немеченого гипоксантина (Sigma) и 1 мМ немече-
ного аденина (Sigma).
1. В нужное время вынимайте реакционные пробирки одну
за другой и добавляйте в каждую по 1 мл останавливаю-
щей смеси и так же по очереди ставьте в ледяную баню,
пока там не окажутся все образцы.
2. Оставьте на 30 мин после остановки последней реакции.
Тяжелый металл лантан вызовет осаждение нуклеотид-
монофосфатных продуктов (IMP и АМР).
2.5. Фильтрация и отмывка
Осажденные продукты собирают на стекловолокнистых
фильтрах (2,4 см, Whatman-CF/C), пользуясь аппаратом для
миллипоровой фильтрации. Остаточный радиоактивный субст-
Биохимический микроанализ ферментов ГФРТ и ФГК И®
рат удаляется интенсивным промыванием дистиллированной
водой.
1. Пронумеруйте стекловолокнистые фильтры, перенесите
в стеклянную чашку Петри (фильтр № 1 сверху) и залей-
те останавливающей лантановой смесью.
2. Установите аппарат миллипоровой фильтрации. Аппарат
помещается на пластиковый поднос (на случай возмож-
ного проливания) вместе с двумя большими стаканами —
один с дистиллированной водой для промывания воронки
после каждой фильтрации, другой с дистиллированной
водой и шприцем на 10 мл для промывания фильтров.
3. Поместите первый фильтр на стеклянный держатель, при-
крепите над фильтром воронку и включите миллипоро-
вый насос для создания вакуума.
4. Перемешайте содержимое первой пробирки с помощью
вортекса и осторожно налейте первый образец в воронку.
Несколько раз тщательно промойте водой пробирку (об-
щий объем воды 10 мл из 10-миллилитрового шприца) и
слейте все в воронку. Уберите пробирку.
5. Сполосните воронку струей дистиллированной воды (по
10 мл).
6. Возьмите воронку и промойте ее, погрузив сначала в
один, а затем в другой стакан с дистиллированной водой.
7. Промойте фильтр, которым пользовались, следующей
порцией воды (10 мл) и перенесите его на алюминиевую
фольгу для просушки.
8. Поставьте на место следующий фильтр, установите во-
ронку и повторите процедуру фильтрации и отмывки.
Продолжайте, пока все образцы не будут профильтрова-
ны и отмыты.
9. Высушите фильтры в горячем сушильном шкафу.
2.6. Подсчет и графическое изображение результатов
1. Перенесите каждый фильтр в пластиковую виалу и вне-
сите 5 мл сцинтилляционной жидкости (Liquiflour, New
England). Вставьте пластиковые наконечники и перенеси-
те в завинчивающиеся сцинтилляционные виалы.
2. Измерьте радиоактивность [3Н] и Г14С] в продуктах ре-
акции, содержащих [3HJ-IMP и [’4С]-АМР, используя
двухканальную настройку сцинтилляционного счетчика,
регистрирующую двойную метку.
Когда анализ производится впервые, а также при периоди-
ческих повторениях должны срабатывать выбросы счета 3Н,
появляющиеся в канале счета |4С, и наоборот.
196
Глава 7
Эта контрольная процедура делается с использованием ре-
акционных смесей, содержащих только 3Н (вместо [ИС]-аде-
нина берут воду) и только 14С (вместо [3Н]-гипосантина вно-
сят фосфатный буфер); этот экспериментальный материал ин-
кубируется, фильтруется и проводится как обычно. С каждой
из этих реакционных смесей («единичных») сделайте два пус-
тых контроля (только поддерживающая среда), чтобы вычесть
их возможный вклад из значений счета, полученных для об-
разцов. Для определения эффективности счета (числа импуль-
сов в минуту/пМ субстрата или продукта) для каждого изото-
па при использованной настройке сцинтилляционного счетчика
нанесите на стекловолокнистые фильтры 5 мкл реакционной
смеси с 3Н и реакционной смеси с 14С. Когда фильтры высох-
нут, просчитайте их прямо — без осаждения хлоридом лантана
и отмывки.
Чтобы узнать -активность каждого из ферментов при двой-
ном микроанализе, из общего значения следует вычесть сред-
ние для пустых контролей (представленных полной реакцион-
ной смесью и поддерживающей средой), а также значения
счета для образцов, инкубированных в «единичных» реакцион-
ных смесях. Затем число импульсов в минуту для каждого
фермента необходимо перевести в единицы активности (пМ/ч/
/образец) с учетом рассчитанной выше эффективности счета.
Типичные результаты представлены на рис. 7.2. Рис. 7.2, А
демонстрирует бимодальное распределение (у мужских и женс-
ких эмбрионов) активности ГФРТ и отношения ГФРТ : АФРТ
в индивидуальных морулах одного приплода. На рис. 7.2, Б
показаны активность фермента и распределения отношения
ГФРТ : АФРТ в бластомерах, полученных в результате биопсии
8-к-леточного эмбриона одного приплода. Значения отношений
ГФРТ : АФРТ в биоптатах позволяют четко идентифицировать
мужские и женские эмбрионы [9]. Анализ активности ГФРТ и
АФРТ в ходе предимплантационного развития продемонстриро-
ван на рис. 7.2, В.
2.7. Оценка двойного микрометода
Поскольку осаждение хлоридом лантана не позволяет пря-
мо идентифицировать продукты реакции, нельзя исключить воз-
можность утилизации или деградации субстратов или продук-
тов другими ферментами. Надежность метода подтверждается
рядом контрольных экспериментов.
1. Устранение ФРПФ исключает счет лантанового преципи-
тата.
2. ГФРТ-отрицательные клетки демонстрируют активность
АФРТ (но не ГФРТ).
Биохимический микроанализ ферментов ГФРТ и ФГК
197
А Морулы
I I ф ф
• ••
" I I I—Г1 I I I-----1 |—Г“"|—гт~
0,5 1 2 4
ГФРТ пМ/ч/эмбрион
АФРТ
—
" I I I I I I I I """ I । । ।
0,05 0,1 0,2 0,4
АФРТ пМ/ч/эмбрион
Отношение Г ФРТ/АФРТ
Б Биопсированные бластомеры
•SXmS«m •••••••• • •••Х» ••• •
““1 I 1'1 I II Г---г---1—I—ГТТТТ”
0,05 0,1 0,2 0,4
пМ ГФРТ/биопсия/ч
wU:: :a.LHL ..
0,005 0,01 0,02 0,04
пМ АФРТ/биолсия/ч
• • •
• ••• •
•••ее 9— •
I I I I I I I I г" | I ""“I “ Г' I"
5 10 20 40
Отношение ГФРТ/АФРТ
т—I—I I I I । ।--1---1—।—г
5 10 20 40
Отношение ГФРТ/АФРТ
Рис. 7.2. Активность ГФРТ и АФРТ у предимплантационных эмбрионов мыши.
А. Распределение ГФРТ, АФРТ и соотношение ГФРТ: АФРТ в индивидуаль-
ных морулах. В женских морулах активны обе Х-хромосомы, поэтому актив-
ность ГФРТ и соотношение ГФРТ: АФРТ имеет бимодальное распределение
в мужских и женских эмбрионах. У бластоцист Х-хромосома инактивируется
в большинстве клеток женского эмбриона и распределение ГФРТ и
ГФРТ: АФРТ становится унимодальным; соответствующие данные не пред-
ставлены. Б. ГФРТ, АФРТ и соотношение ГФРТ: АФРТ в бластомерах, полу-
ченных путем биопсии 8-клеточного эмбриона. В. Повышение активности
ГФРТ и АФРТ в процессе развития предимплантационного мышиного эмб-
риона. Суперовуляцию самок получали в соответствии с описанием, данным
в гл. 1 (разд. 5.4), сбор эмбрионов проводили в разные сроки после инъекции
чХГ. Первое повышение активности ГФРТ (до 8-клеточной стадии) опреде-
ляется материнской мРНК, а последующее кодируется геномом эмбриона.
198
Глава 7
3. Активность ГФРТ в яйцах от ХО-матерей вдвое ниже, чем
активность этого фермента у яиц от ХХ-матерей (в ходе
оогенеза активны обе Х-хромосомы), в то же время ак-
тивность АФРТ у таких яиц одинакова.
4. Мэри Харпер [20] использовала тонкослойную хромато-
графию (ТСХ) для разделения и количественного опре-
деления пуриновых субстратов и нуклеотидных продуктов
и затем сравнивала выход продуктов с тем, что мы име-
ем при работе с хлоридом лантана. Было бы неуместно
в этой главе подробно останавливаться на методах ТСХ
(см. [29]), достаточно заметить, что радиоактивность
субстрата действительно переходит в радиоактивность
продукта. Использованная система растворителя (60 г
сернокислого аммония в 100 мл 0,1 М фосфатного буфера
при pH 6,8, к которым прибавлены 2 мл п-пропанола)
разделяет субстраты и продукты двойного микроанализа
(см. рис. 7.3). Значения активности ГФРТ и АФРТ были
получены отдельно в эмбриональных экстрактах в «еди-
ничных» реакционных смесях (см. выше, разд. 2.6) при
повышении концентрации эмбриональных экстрактов.
В качестве субстрата ГФРТ использовали гуанин. Реак-
ции прекращали кипячением, изотопы субстрата и про-
дукта разделяли методом ТСХ, выявляли на бумаге и
измеряли их радиоактивность. Субстраты — [3Н]-гуанин
и [14С]-аденин—превращались в продукты [3H]-GMP и
[14С]-АМР соответственно (см. рис. 7.4, А). Для каждой
реакции суммарный счет субстрата и продукта составлял
более 90% общего значения, полученного путем хромато-
графии. Была сопоставлена оценка продуктов, полученная
в результате применения каждого метода. Радиоактив-
ность пятен [3Н]-GMP и [14С]-АМР сравнивали с радиоак-
тивностью GMP и АМР, осажденных на стекловолокнис-
тых фильтрах. Рис. 7.4,5 свидетельствует о том, что про-
цедура осаждения хлоридом лантана обеспечивает на-
дежное измерение продуктов реакций ГФРТ и АФРТ.
3. Микроанализ ФГК
Бютлер [30] описал разделение электрофоретических вари-
антов Х-сцепленного гликолитического фермента ФГК.-1 эрит-
роцитов человека путем электрофореза в крахмальном геле.
Соответствующие полосы обнаруживались в результате обрат-
ной реакции превращения 3-фосфоглицерата и АТР в 1,3-дифос-
фоглицерат и ADP (рис. 7.5, А). 1,3 дифосфоглицерат в этом
случае становится субстратом второй реакции, катализируемой
глицеральдегид-фосфат-дегидрогеназой (GAPDH, КФ 1.2.1.12),
Биохимический микроанализ ферментов ГФРТ и ФГК
199
Рис. 7.3. Разделение субстратов и продуктов двойного микрометода посред-
ством ТСХ. Растворитель помещают в вертикальный хроматографический ре-
зервуар и оставляют на 1 ч для эквилибрации. Образцы по 20 мкл наносят на
листы бумаги для ТСХ (тонкослойная хроматография) и высушивают. Раз-
деление проводят при комиатиой температуре в направлении, указанном
стрелкой, до тех пор, пока фронт растворителя не достигнет верхнего края
листа (~4 ч). Лист высушивают. Основания и нуклеотиды определяют ви-
зуально, затем их положение отмечают при коротковолновом УФ свете
(254 им). Положение адеиииа, гуанина, АМР и GMP, использованных от-
дельно или в виде смеси четырех соединений (смесь), показано в левой части
рисунка. В процессе анализа реакционную смесь с образцом высушивают
и разделяют в воде, содержащей четыре соединения (смесь+образец). Затем
определяют радиоактивность фрагментов каждой дорожки (дорожки обозна-
чены цифрами 1—7). (Фотография любезно предоставлена Мэри Харпер.)
в ходе которой флуоресцирующий NADH окисляется в нефлуо-
ресцирующий NAD. ФГК-1 выявляется с помощью длинноволно-
вого УФ-света (365 нм) в виде темной полосы на флуоресци-
рующем фоне.
200
Глава 7
Фракция эмбриона
Рис. 7.4. Конверсия субстратов в продукты в процессе двойного микроанали-
за. А. Экстракт постимплантационного эмбриона развели вдвое, по 5 мкл
разбавленных образцов добавили к 50-мкл аликвотам реакционной смеси.
Реакции были остановлены через 2 ч нагреванием пробирок в кипящей водя-
ной бане. В каждую пробирку внесли по 20 мкл дистиллированной воды, со-
держащей стандартные растворы гуаиииа, аденина, GMP и АМР (каждый
в концентрации 0,5 мМ); весь объем (по 5 мкл) нанесли на бумагу ТСХ и вы-
сушили. Процедура хроматографии проводилась согласно подписи к рис. 7.3.
По окончании разделения отметили положение маркерных оснований и нук-
леотидов и разрезали ТСХ иа полоски, соответствующие образцам. Целлюло-
зу снимали с учетом положения маркированных дорожек (на рис. 7.3 это 2,
3, 4 и 6), а также с остальной части полоски (1, 5, 7 на рис. 7.3) и измеряли
радиоактивность в сцинтилляционной жидкости (толуол: тритон Х-100: во-
да, 4:2:1). Конверсия субстратов (гуанина и аденина) в продукты (GMP
и АМР) показана в зависимости от количества эмбрионального экстракта
Счет каждой пары субстрат—продукт выражен процентом общего счета в по-
лоске образца. Б. Для анализа было взято несколько реакционных пробирок
с различным количеством эмбрионального экстракта, дублирующие образцы
использовали для разделения путем ТСХ и лантановой преципитации. Изме-
рения радиоактивности продуктов (GMP и АМР), полученных в результате
ТСХ (О), эквивалентны соответствующим значениям для GMP и АМР, осаж-
денных на стекловолокнистых фильтрах хлоридом лантана (ф).
Биохимический микроанализ ферментов ГФРТ и ФГК
201
А
Глицероп-Ф
Дигидроацетон-Ф
ТФИ
Г лицеральдегид-ЗФ
Б
Фруктозо-1,6-диФ
| АПД
Глицеральдегид-ЗФ + дигидроксиацетон-Ф
NAD J z^NADH->
ГАФДГ f ГДГ
NADH'f_______NAD-*
1,3 дифтлицерат
ADR >
Глйцерол-Ф
NAP^
NADH—'
ГАФДГ
1,3 дифтлицерат
А0Р-ч<
ФГК-1
АТР—'
ЗФ-глицерат
ФГК-1
ЗФтлицерат
Дополнительная система
Д-глюкоза
ГК
Г люкоза-бФ
NADP —
Г6ФД
NADPH-* .
бФ-глюконо-лактон
Рис, 7.5. Обратная [30] и прямая [32] окрашивающие реакции, используе-
мые для определения активности фосфоглицераткииазы (ФГК), представлены
соответственно в виде схем А и Б. А. Обратная реакция ФГК-1 связана
с превращением флуоресцирующего NADH в иефлуоресцирующий NAD под
действием глицеральдегид-3-фосфат—дегидрогеназы (ГАФДГ). Эффектив-
ность окрашивающей реакции можно повысить превращением глицеральде-
гцд-З-фосфата до а-глицеролфосфата триозофосфатизомеразой (ТФИ) и а-гли-
церофосфат-дегидрогеиазой (ГДГ). Б. 1,3-дифосфоглицерат, нестойкий суб-
страт ФГК-1, образуется из фруктозо-1,6-дифосфата с помощью альдолазы
(АЛД) и глицеральдегид-3-фосфат—дегидрогеназы (ГАФДГ). АТР, образую-
щийся под действием ФГК-1, связан с дополнительной ферментной системой
гексокиназы (ГК) и глюкозо-6-фосфат—дегидрогеназы (Г6ФД), приводящих
к превращению иефлуоресцируюшего NADP в флуоресцирующий NADPH.
ГК обеспечивает восстановление ADP; дигидроксиацетоифосфат и NADH эли-
минируются а-глицерофосфат-дегидрогеназой (ГДГ).
Мейра Хан [31] предложила проводить электрофорез не-
скольких ферментов, в том числе ФГК-1, на целлюлозно-аце-
татных гелях. Бючер [32] применила эту методику в сочетании
с окрашиванием прямой реакции (рис. 7.5, Б) для получения
изозимных полос ФГК-1А и ФГК-1Б в тканевых экстрактах ге-
терозиготных мышей.
202
Глава 7
Этот метод имеет ряд преимуществ:
а) лучшее разделение полос;
б) уменьшение времени электрофореза;
в) поступление 1,3-дифосфоглицерата — нестойкого субст-
рата ФГК-1—в результате его образования из фрукто-
зо-1,6-дифосфата.
Активность ФГК-1 может быть продемонстрирована посред-
ством дополнительной ферментной системы. Гексокиназа ис-
пользует АТР, образующуюся в реакции, катализируемой
ФГК-1, для фосфорилирования глюкозы, в результате чего вос-
станавливается ADP. Образующийся глюкозо-6-фосфат превра-
щается глюкозо-6-фосфат — дегидрогеназой (G6PD, КФ 1.1.49)
в глюконо-лактон с восстановлением нефлуоресцирующего
NADP в флуоресцирующий NADPH.
Флуоресценция, испускаемая NADPH, может быть измерена
фотоумножителем, который сконструировала Бючер [32]. Гель
помещается на движущийся конвейер (самописец представляет
собой вариант сканирующего устройства) и сканируется УФ-лу-
чами с длиной волны 365 нм, сфокусированными на геле через
щель. Фотоумножитель передает флуоресценцию NADPH на
усилитель, соединенный с самописцем, а тот в свою очередь
регистрирует пики, пропорциональные флуоресценции, источни-
ком которой является полоса на геле. Имеющийся в продаже
денситометр (Sigma FTP-20, Oriel Sci'entific) устроен принципи-
ально так же, с его помощью можно сканировать гели как при
просвечивании и отражении, так и при флуоресценции. Схемы
этих оптических устройств показаны на рис. 7.6.
С одной полосы можно получить несколько показаний ак-
тивности; таким образом по мере проявления краски на геле
области повышающихся пиков дадут соответствующую кривую.
Области под пиками в линейной части кривой, выражающие за-
висимости активности от времени, свидетельствуют об актив-
ности ФГК-1 в образце.
Мы проверили эффективность электрофореза в целлогеле и
количественного определения ФГК-1, использовав тканевые
экстракты линий мышей PGK-1A и PGK-1B. Значения активно-
сти ферментов в экстрактах сначала были установлены путем
спектрофотометрических измерений, затем ферменты смешива-
ли в разных соотношениях и наносили на гель. Регистрирова-
ли следующие параметры.
1. Калибровка. Гель-система может быть калибрована при
помощи образцов известного объема и активности и оп-
ределения количества изозимных полос. Это позволяет
прямо на геле оценить как общую активность ФГК-1, так
и пропорцию двух изозимов.
Биохимический микроанализ ферментов ГФРТ и ФГК
203
Рис. 7.6. Измерение флуоресценции в системе, описанной Бючер [32]. Носи-
тель образца — черная поливиниловая камера, в которую помешают полоску
целлогеля лицевой стороной вниз иа целлюлозно-ацетатную мембрану, про-
питанную красителем. Камера закрывается стеклянной пластинкой. Свет с дли-
ной волны 365 нм от источника—ртутной лампы — проходит через интерфе-
ренционный фильтр и через щель направляется сверху на движущуюся поло-
ску целлогеля. NADPH, образующийся в красителе под действием ФГК-1,
флуоресцирует в УФ свете. Флуоресценция проецируется оптическими линза-
ми через прерывающий фильтр на фотоумножитель, который направляет лу-
чи на компьютерный интегратор или самописец. Прибор Sigma FTR-20 снаб-
жен, кроме того, ртутной лампой, расположенной под образцом, что позво-
ляет сканировать гели не только в падающем, но и в проходящем свете.
2. Чувствительность. Оказалось возможным определять ак-
тивность ФГК-1 до 0,1 пмоль/ч.
3. Разрешение и точность. Метод позволяет выявить в сме-
си двух изозимов один процент каждого из них. В искус-
ственных смесях двух изозимов наблюдается хорошее со-
ответствие между пропорциями, предсказанными на ос-
нове спектрофотометрических измерений до смешивания
204
Глава 7
и значениями, полученными в результате измерения флу-
оресценции после электрофореза в целлогеле.
Методы описаны ниже (см. также работы [32] и [33]).
3.1. Приготовление образца
С помощью возвратных скрещиваний были получены ин-
бредные линии мышей СЗН и СВА, несущие аллель Pgk-P (об
их жизнеспособности см. в работе [34]). В качестве источника
аллеля Pgk-Ib можно использовать инбредные или случайно
скрещивающиеся линии.
Выделенные яйца и эмбрионы помещают в микрочашки, как
•описано в разд. 2.1, но объем поддерживающей среды на мик-
рочашку не должен составлять более 1 мкл. В качестве такой
•среды мы используем РВ1.ПВП (гл. 13, разд. 3.1.3) или буфер
для образцов (табл. 7.1).
1. Получите экстракты энзимов замораживанием — оттаива-
нием клеточных суспензий, гомогенизированной ткани или
образцов в микрочашках (эмбрионов на стадиях дробле-
ния, яиц или различных тканей, отсепарированных у
поздних эмбрионов).
2. Отцентрифугируйте экстракты при 4°C (5 мин,
1500 об/мин) для осаждения клеточных остатков. Отбе-
рите супернатант для анализа. Если требуется, разбавьте
образцы супернатанта.
3. В случае тканевых образцов малого объема (например,
отдельных ооцитов), которые не предназначаются для
разбавления, образцы сначала поместите в 1 мкл РВ1.
ПВП в 10 мкл-микрочашки и центрифугируйте в двух
направлениях, как описано в разд. 2.1.
3.2. Электрофорез
Состав различных буферов и красящих растворов, исполь-
зуемых при электрофорезе, приведен в табл. 7.1.
Буфер для проведения электрофореза и красящие растворы
можно хранить до 1 года при 4°C, но буфер для образца надо
готовить каждую неделю. Электрофорез проводится в целлю-
лозно-ацетатных полосках 5,7X14 см (Cellogel, Whatman), ко-
торые до употребления следует хранить в 30%-ном метаноле.
Для электрофореза используется ватмановский контейнер с
опорным мостиком, на который помещают гель; мостик охлаж-
дается водой при 20°, циркулирующей через опору из резерву-
ара, снабженного насосом и термостатом (рис. 7.7).
1. Возьмите полоску целлогеля и осторожно опустите на
поверхность буфера для проведения электрофореза
(150 мл которого налиты в пластиковый сосуд) на
Биохимический микроанализ ферментов ГФРТ и ФГК
205
Таблица 7.1. Приготовление растворов для электрофореза ФГК-1 в целлогеле
Буфер для проведения электрофоре- за pH 8,6 5,5-диэтилбарбитуровая кислота BDH 4,12 г/л
Цитрат натрия BDH Analar 2,94 г/л
MgSO47H2O BDH Analar 1,23 г/л
ЭДТА Sigma 0,74 г/л
1.4-дитиоэритритол Boehringer 25 мг, добавлен- ных непосредст- венно перед электрофорезом
АМР Boehringer 45 мг, добавлен- ных в катодный резервуар перед электрофорезом
Буфер для приготовления образцов
Триэтаноламин (50 мМ) pH 7,6 Sigma 20 мл
1,4-дитиоэритритол Boehringer 6 мг
Бычий сывороточный альбумин Miles 10 мг
Глицерол Aldrich 20 мл
Красящий раствор А
Триэтаноламин (100 мМ), pH 7,5 Sigma 14,92 г/л
MgSO4-7H2O BDH Analar 4,93 г/л
Красящий раствор Б
Триэтаноламин (50 мМ), pH 7,6 Sigma 10 мл
К2НРО4-ЗН2О BDH 91 мг
NAD Boehringer 9 мг
Ф руктозо -дифосфат Boehringer 220 мг
Красящий раствор Б
Триэтаноламин (50 мМ), pH 7,6 Sigma 5 мл
Глюкоза BDH 135 мг
ADP Boehringer 60 мг
NADP Boehringer 165 мг
MgSO;-7H2O BDH Analar 160 мг
Красящий раствор Г
Альдолаза Boehringer 20 мкл
Глюкозо-6-фосфат-дегидрогеназа Boehringer 20 мкл
Глине пол-фосфат-дегидрогеиаза Boehringer 10 мкл
Глицерзльдегид-З-фосфат—дегидро- геназа Sigma 10 мкл
Гексокииаза Boehringer 10 мкл
206
Глава 7
Рис. 7.7. Установка для электрофореза ФГК-1 в целлогеле. 1. Полоски целло-
геля в метаноле. 2. Полумикроаппликатор. 3. Сканирующий денситометр
Sigma FTR-20. 4. Ватмаиовский резервуар для электрофореза. 5. Опорный
мостик для геля. 6. Резервуар с водой при 20 °C, циркулирующей через опор-
ный мостик. 7. Черный пластиковый лоток для окрашивания и стеклянная
крышка. 8. Источник питания. (Фотография любезно предоставлена П. Мен-
дене.)
10 мин. Плавающий на поверхности гель должен равно-
мерно пропитаться буфером.
2. Промывайте гель еще 10 мин в 250 мл буфера для прове-
дения электрофореза, содержащего 25 мг дитиоэрнтрито-
ла. Поскольку это соединение нестойкое, его следует до-
бавлять непосредственно перед использованием. Дитио-
эритритол выполняет роль стабилизатора фермента. Ос-
торожно покачайте сосуд для лучшей гидратации геля.
3. Перелейте этот буфер после второго промывания в ватма-
новский контейнер для проведения электрофореза, накло-
нив его, распределите буфер равномерно ио двум отсе-
кам — анодному и катодному.
4. Внесите 45 мг АМР в катодный сосуд. (АМР, связываясь
с аденилаткиназой, заставляет ее перемещаться впереди
ФГК-1 и покидать гель; в противном случае аденплатки-
наза будет при окрашивании реагировать с ADP и АТР.)
5. Поместите гель на охлаждаемом мостике в контейнер,
присоедините электроды к источнику питания и проведите
преэлектрофорез при 20° и 200 В в течение 10 мин.
Биохимический микроанализ ферментов ГФРТ и ФГК
207
6. Разведите образец, если требуется, буфером для образцов
(табл. 7.1). Буфер для образцов содержит глицерол,
уменьшающий испарение.
7. Наберите аппликатором четыре пробы образца (Cellogel
semi-micro four-piece applicator, Whatman) и нанесите на
полоску целлогеля со стороны катода. В случае эмбрио-
нов в микрочашках и других микрообразцов маленькие
капли супернатанта по 5 мкл или менее наносите, вос-
пользовавшись пастеровской пипеткой с оттянутым кон-
чиком, прямо на гель. Объем наносимой пробы можно
определить, поместив пипеткой такой же объем жидкости
в микрочашку Drummond на 1 мкл.
8. Проводите электрофорез в течение 1,5 ч при 200 В при
20 °C.
3.3. Окрашивание
Следует подчеркнуть, что соблюдение чистоты и аккурат-
ности — необходимое условие получения хороших результатов
при окрашивании. К целлогелю и целлограмме нельзя прика-
саться руками, стеклянную пластинку и поднос нужно брать
только за края. Еще более важно очистить опорное и прикры-
вающее стекло от пыли нефлуоресцирующей бумагой (бумаж-
ным фильтром Whatman), так как пыль дает флуоресценцию.
Одним пинцетом берите гель до окрашивания, другим — окра-
шивающую целлограмму. Реактивы для приготовления крася-
щих растворов перечислены в табл. 7.1.
1. Приготовьте 660 мкл красителя за 5 мин до окрашива-
ния, смешав:
400 мкл красящего раствора А;
200 мкл красящего раствора Б;
50 мкл красящего раствора В;
10 мкл красящего раствора Г.
Вылейте краситель в черный пластиковый поднос, по раз-
меру соответствующий полоске целлогеля.
2. Вырежьте листок целлюлозно-ацетатной бумаги — цел-
лограмму (Cellogram; Shandon Southern, 57X127 мм) по
размеру черного пластикового подноса с красителем и
погрузите в краситель.
3. Отделите гель бритвенным лезвием от опорного мостика
в контейнере и положите лицевой стороной вниз на про-
питанную красителем целлограмму так, чтобы не было
пузырьков воздуха. Прикройте поднос чистым стеклом.
208
Глава 7
логеле. По данным спектрофотометрического анализа, процентное выражение
активности ФГК-1А до смешивания следующее: дорожка 1 —15%, дорож-
ка 2 — 30%, дорожка 3 — 40%, дорожка 4 — 50%. 1А и 1Б — указывают иа
миграции ФГК-1А и ФКГ-1Б. Б. Регистрация изменения флуоресценции (уве-
Биохимический микроанализ ферментов ГФРТ и ФГК
209
Время (мин)
2 3
4
личение площади пика по мере окрашивания) на дорожках, представленных
на А. В. Зависимость активности ФКГ от времени, основанная на данных Б.
Значения вклада активности ФГК-1А (в %) в смесях 1—4, полученные в ре-
зультате флуорометрической оценки геля, соответственно равны: 16,3+1.4;
31,8±1,8, 42,0±0,6 и 52,5±0,9. Таким образом, количественные характеристи-
ки, полученные в результате разделения в целлогеле, хорошо согласуются
с данными о составе искусственных смесей, полученными методом спектрофо-
тометрии.
3.4. Количественный анализ активности ФГК-1
и пропорции обоих ферментов
Гель-систему следует изначально калибровать на чувстви-
тельность, точность реагирования на активность внесенного
фермента, на разрешающую способность в отношении минорной
полосы в смеси, активность которой известна, и на воспроизво-
димость. Для этого приготовьте тканевые экстракты животных,
несущих разные аллели фосфоглицераткиназы и проведите
спектрофотометрический анализ фермента, чтобы с помощью
гель-системы тестировать смеси известных пропорций и извест-
ной активности. Спектрофотометрический анализ ФГК-1 описан
в разд. 3.5, а чувствительность, разрешение и точность можно
определить, руководствуясь разд. 3.6—3.8. В этом разделе опи-
саны сканирование и количественное определение.
1. Проинкубируйте гель в течение 8—10 мин до начала из-
мерений, чтобы проявились флуоресцирующие полосы.
Источник УФ-излучения также к этому времени должен
быть разогрет.
2. Поместите гель на движущийся конвейер и сканируйте
флуоресценцию вдоль каждой дорожки. Повторите ска-
14—171
210
Глава 7
нирование в течение различных интервалов времени—до
30 мин. Типичный гель и временная регистрация флуо-
ресценции для эмбриональных образцов представлены на
рис. 7.8 (А и Б).
3. Вклад обоих изозимов и активность ФГК-1 в эмбрио-
нальных образцах вычислены путем интеграции площадей
под пиками. Если используется самописец, пики можно
тщательно вырезать и взвесить; активность ФГК-1 в дан-
ное время для данной полосы изозима пропорциональна
весу пика. Графическое изображение веса пиков в зави-
симости от длительности окрашивания для четырех ис-
кусственных смесей (см. разд. 3.8, ниже) показано на
рис. 7.8, В. Временная зависимость выхода NADPH име-
ет линейный характер, для точной оценки пропорций при-
сутствующих энзимов достаточно 4—6 сканирований.
Вместо вырезания и взвешивания площадей пиков можно
воспользоваться денситометром FTR-20, который присо-
единяется к интегратору (Shimadsu C-R3A Chromatopac),
компьютер даст интегрированные значения областей под
пиками.
4. Общая активность ФГК-1 на образец получается путем
сравнивания с известными эталонами, как описано ниже.
3.5. Спектрофотометрический анализ активности ФГК-1
Метод спектрофотометрического анализа соответствует опи-
санному Харрисом и Хопкинсоном [35].
1. Для каждого анализа реакционная смесь содержит:
100 мкл 1,0 М трис-HCl буфера, pH 8,0 (Sigma);
270 мкл дистиллированной воды;
400 мкл 20 мМ АТР (Sigma);
100 мкл 0,1 М хлорида магния (BDH);
10 мкл ГФД (Sigma);
100 мкл 2 мМ NADH (Sigma);
Субстрат—фосфоглицероловая кислота — готовится в
виде 0,1 М раствора в дистиллированной воде.
2. Внесите 10 мкл экстракта в 980 мкл реакционной смеси
без субстрата в чистой кварцевой кювете и эквилибри-
руйте при 37 °C в течение 3 мин в спектрофотометре
(Cecil СЕ272), чтобы на самописце регистрировалась
прямая основная линия.
3. Внесите 10 мкл раствора субстрата, тщательно переме-
шайте пастеровской пипеткой и измерьте снижение опти-
ческой плотности (ДОП) при 340 нМ на самописце (Ser-
voscribe RE54) после двухминутной эквилибровки. Линей-
ная реакция длится 20—30 мин.
Биохимический микроанализ ферментов ГФРТ и ФГК
211
Рис. 7.9. Разрешающая способность электрофореза в целлогеле при разделе-
нии изозимов ФГК-1. Четыре искусственных смеси изозимов ФГК- 1А и ФГК-1 Б
тимоцитов мыши разделяли в целлогеле в соответствии с описанием, данным
в тексте. Активность ФГК-1А в смесях выражается следующим образом
(в %): 50% (дорожка 1), 20% (дорожка 2), 5% (дорожка 3) и 1% (дорож-
ка 4). Минорная полоса изозима ФГК-1А, четко выявляемая этим метолом,
составляет всего 1 % от общей активности.
4. Активность ФГК-1 в мкмоль/мин/мл получена делением
АОП/мин/мл на 6,22 (коэффициент молярной экстинкции
для NADH составляет 6,22хЮ3 при 340 нМ).
3.6. Чувствительность целлогель-электрофореза
в отношении ФГК-1
Чувствительность гель-электрофореза и процедуры окраши-
вания тестируется с использованием различных разведений тка-
невого экстракта.
1. Измерьте активность ФГК-1 в исходном образце на
спектрофотометре.
2. Рассчитайте активность в разбавленных образцах, нане-
сенных на гель. Например, экстракт мышиных фиброблас-
тов (ЗХЮ6 кл/мл) с известной активностью 500 нмоль/ч
на 10 мкл, разведен в 200 раз, и 0,5 мкл этого разведения
показали различимую активность на геле. Отсюда нане-
14 *
212
Глава 7
Рис. 7.10. Активность ФГК-1 в единственном ооците мыши, полученном от
самки, гетерозиготной по аллелям Pgk-la и Pgk-lb
сенная активность составит 0,125 нмоль/ч, что соответст-
вует активности, экстрагированной примерно из семи
клеток. Серия таких экспериментов позволила заключить,
что наименьшая из различимых активностей ФГК-1 со-
ставляет около 0,05 нмоль/ч [33].
3.7. Разрешение минорной полосы
Рис. 7.9 демонстрирует результаты анализа смеси экстрактов
из тимоцитов линий мышей, различающихся по вариантам
ФГК. Спектрофотометрический анализ показал, что исходная
активность ФГК-1 в каждом образце составляет 900 нмоль/ч
на 10 мкл. На гель были нанесены четыре искусственные смеси
с пропорцией ФГК-1 А соответственно 50, 20, 5 и 1%. В этом
случае пзозим ФГК-1А, являющийся минорным компонентом,
различим в виде довольно четкой полосы, несмотря на то что
представляет всего лишь 1 % общей активности. Реципрокный
гель, в котором активность ФГК-1Б составляет всего 1% общей
активности, должен дать такое же разрешение.
3.8. Определение точности
количественного анализа изозима
1. Проанализируйте спектрофотометрически тканевые экст-
ракты мышей, несущих разные аллели ФГК.
Биохимический микроанализ ферментов ГФРТ и ФГК
213
2. Сделайте серию разведений, содержащих разные пропор-
ции изозимных форм. В примере, показанном на рис. 7.8,
доля ФГК-1 А составляет 15, 30, 40 и 50%.
3. Проведите электрофорез, окрасьте гели (рис. 7.8, А) и
измерьте флуоресценцию сканированием в определенные
интервалы времени (рис. 7.8, .5).
4. Интегрируйте пики сканирования (рис. 7.8,В). Сопостав-
ление вклада ФГК-1 в эти смеси, оцененное данным ме-
тодом, демонстрирует хорошее соответствие результатам,
полученным при спектрофотометрическом анализе исход-
ных образцов (см. подпись к рис. 7.8, В), и доказывает
целесообразность использования системы для анализа не-
известных образцов.
Благодарности
Я благодарна Хасу Катуриа и Мэри Харпер за сотрудниче-
ство в разработке и использовании микроанализа ГФРТ/АФРТ,
Теодоре Бючер, Ингрид Линке, Энди Мак-Мэхон и Мэндри
Фостен за участие в работе по анализу ФГК-1.
Литература
1. Monk М., Harper М. (1978). J. Embryol. Exp. Morphol., 46, 53.
2. Harper M. I., Monk M. (1983). J. Embryol. Exp. Morphol., 74, 15.
3. Epstein C. J. (1972). Science, 175, 1467.
4. Kratzer P. G„ Gartler S. M. (1978). Nature, 274, 503.
5. Monk M„ Harper M. (1979). Nature, 281, 311.
6. Monk M., McLaren A. (1981). J. Embryol. Exp. Morphol., 63, 75.
7. Hooper M., Hardy K-, Handyside A„ Hunter S., Monk M. (1987). Nature,
326, 292.
8. Monk M., Handy side A., Hardy K„ Whittingham D. (1987). Lancet, in press.
9. Monk M., Handyside A. (1987). J. Reprod. Fert., submitted.
10. Nielsen J. T„ Chapman V. M. (1977). Genetics, 87, 319.
11. West J. D., Freis W. J., Chapman V. M., Papaioannou V. E. (1977). Cell,
12, 873.
12. Papaioannou V. E„ West J. D_, Bucher T., Linke I. M. (1981). Dev. Genet.,
2, 305.
13. Harper M. I., Fasten M., Monk M. (1981). J. Embryol. Exp. Morphol., 67,
14.
15.
Johnston P. G. (1981). Genet. Res., 37, 317.
McMahon A„ Fasten M., Monk M. (1981). J. Embryol. Exp.
Morphol., 64,
16' 207Aa!Wn A" F°Sten M" Monk M- <1983)- J- Embryol. Exp. Morphol., 74,
17. McMahon A„ Monk M. (1983). Genet. Res. Camb., 41 69
18. McMahon A. (1983). Genet. Res. Camb., 42, 77.
19. Ponder B. A. J„ Schmidt G. H„ Wilkinson M. M„ Wood M. J., Monk M
Reid A. (1985). Nature, 313, 689.
20. Ansell J. D., Micklem H. S. (1986). In: Handbook for Experimental Immu-
nology. Weis D. M. (ed.), Blackwell Scientific Publications, Oxford, Chap-
ter 56.
214
Глава 7
21. Woodruff М. F. A., Ansell J. D., Forbes G. M„ Gurdon J. C., Burdon D. 1.,
Micklem H. F. (1982). Nature, 229, 822.
22. Chapman V. M., Shows T. B. (1976). Nature, 259, 665.
23. Kozak C„ Nichols E„ Ruddle F. H. (1975). Somat. Cell Genet., 1, 371.
24. Gartler S. M., Scott R. C„ Goldstein J. L„ Campbell B., Sparkes R. (1971),
Science 172 572
25. Epstein C. J. (1970). J. Biol. Chem., 245, 3289.
26. Bakay B., Telfer M. A., Nyhan W. L. (1969). Biochem. Med., 3, 230.
27. McBurney M. W., Adamson E. D. (1976). Cell, 9, 57.
28. Monk M„ Kathurla H. (1977). Nature, 270, 599.
29. Harper M. I. (1981). Patterns of enzyme activity reflecting genetic expres-
sion in mouse embryogenesis. PhD Thesis, University of London.
30. Beutler E. (1969). Biochem. Genet., 3, 189.
31. Meera Khan P. (1971). Arch. Biochem. Biophys., 145, 470.
32. Bucher T., Bender W., Fundele R., Hofner H., Linke I. (1980). FEBS Lett.,
115, 319.
33. McMahon A. (1981). Cell differentiation and X-chromosome activity in the
definitive germ-layers and the germ-line of the mouse. PhD Thesis, Univer-
sity of London.
34. Festing M. F. W. (1987), International Index of Laboratory Animals. 5th
edition, Laboratory Animals Ltd, Newbury, Berkshire, UK.
35. Harris H., Hopkinson D. A. (1976). Handbook of Enzyme Electrophoresis in
Human Genetics. Elsevier/North Holland, Amsterdam.
fJlA&A d
КАЧЕСТВЕННЫЙ АНАЛИЗ БЕЛКОВ
НА РАННИХ СТАДИЯХ
ЭМБРИОГЕНЕЗА МЫШИ
Сара Хаулет1
1. Введение
В результате оплодотворения яйцо, находящееся в относи-
тельном покое, превращается в митотически делящуюся клет-
ку, способную дать начало целому организму. Логично ожи-
дать, что процессы дробления, роста и дифференцировки сопро-
вождаются изменениями в синтезе специфических белков. Пат-
терн транслируемых белков в клетках определяется характером
генной экспрессии, наличием мРНК, стабильностью белков и их
модификацией [1]. Каждый из указанных механизмов откры-
вает широкий простор для исследований. Решение этой задачи
для раннего мышиного эмбриона усложняется очень малыми
количествами белка, тем не менее для этого объекта удалось
разработать методики, позволяющие определять присутствие
белков и их синтез.
Электрофорез белков в полиакриламидных гелях (ПААГ) —
идеальное средство для анализа многих сотен генных продуктов
любой клетки (см. работы [2—4]). С помощью этой методики
очень удобно следить за изменением в ходе развития белковых
паттернов. Электрофорез дает возможность разделять белки на
основе их дифференциальной подвижности, обусловленной раз-
мером и/или зарядом молекул. В присутствии додецилсульфа-
та натрия (ДСН) электрофорез разделяет белки в соответствии
с их молекулярным весом. Изоэлектрическое фокусирование
(ИЭФ) обеспечивает разделение на основе изоэлектрической
точки полипептидов, отражающее их аминокислотный состав
и модификацию, например, ацетилирование, гликозилирование
и фосфорилирование. Комбинация этих методик позволяет раз-
делить белки в соответствии с их характеристиками в двумер-
ном плане. Теоретические основы и методические подробности
использования одномерных и двумерных гелей представлены в
работах [2] и [4].
Выбор стратегии мечения и определения белков с использо-
ванием ПААГ зависит от цели исследования. Одни методики
1 Sarah К. Howlett. AFRC Institute of Animal Physiology and Genetics
Research, Department of Molecular Embryology, Babraham, Cambridge CB2
4AT, UK.
216
Глава 8
Рис. 8.1. Фосфорилированные белки.
Яйца, меченные в первой S-фазе.
(«I», 10 ч после осеменения, дорож-
ки а и 6); неоплодотворенные яйца
(«М», 14 ч после овуляции, дорожки
в и г); меченные соответственно
[35S]-метионином (а и в) или [32Р]-
фосфатом (6 и г) в течение 1 или 2 ч.
ные группы белков — фосфопротеины по включению 32Р (разд.
2.3), гликопротеины по включению сахаров, меченных 14С или
3Н (разд. 2.4), либо с помощью лектинов. При наличии специ-
фических антител можно изучать индивидуальные белки. Так,
пригодны для изучения общих
или специфических белков,
другие позволяют выявить
лишь те белки, которые син-
тезируются в данное время.
О динамической картине
паттерна белков, находящих-
ся в процессе синтеза, можно
судить как по включению ме-
ченых аминокислот (они мо-
гут быть в виде смеси или
представлены индивидуально,
например [35S]-метионин,
разд. 2.1.1), так и визуаль-
но— при помощи флюорогра-
фии (рис. 8.1, 8.2 и 8.3). Судь-
бу новосинтезированных мече-
ных белков можно проследить,
отмыв клетки от метки и про-
должая инкубировать эмбри-
он в течение нескольких часов
(эксперименты с импульсным
мечением, разд. 2.1.3).
Для ' получения статичес-
кой картины тотальных бел-
ков используют либо окраши-
вание серебром немеченых
белков в геле (разд. 4.2 и
рис. 8.4), либо иодирование
лизированной смеси белков
(разд. 2.2) п выявление их
методом радиоавтографии. У
раннего эмбриона четко раз-
личаются паттерны белков,
синтезированных эмбрионом и
запасенных ооцитом (рис. 8.4).
Существует возможность
идентифицировать специаль-
при помощи антител можно определить присутствие антигена
среди белков, выделенных электрофоретически (иммуноблот-
тинг; разд. 4.3), или осадить меченый антиген из смеси мета-
болических белков (иммунопреципитация; разд. 2.5).
Качественный анализ белков ранних эмбрионов мыши
217
Рис. 8.2. Синтез белка в ходе предимплантационного развития. Одномерный
гель, демонстрирующий' паттерн белков, меченных [358]-метионином, кото-
рые были синтезированы в течение 1-часового импульсного мечения в неопло-
дотворенном яйце (НОЯ), 1-клеточном эмбрионе (1), 2-клеточном (2), 4-кле-
точном (-1), 8-клеточном (8), 16-клеточном (16), на стадии ранней кавита-
ции (32) и в поздней бластоцисте (БЛ). Каждая дорожка содержит материал
10 эмбрионов, за исключением последней, содержащей 6 бластоцист.
В этой главе описаны способы мечения разных классов
белков, их разделения и идентификации. Эти методики универ-
сальны, но, кроме того, их можно использовать в качестве мик-
рометодов для анализа развивающегося эмбриона.
218
Глава 8
Рис. 8.3. Разделение белков, меченных [358]-метцонином, в двумерном геле.
Двумерный гель демонстрирует паттерн белков, синтезированных в течение
2-часового периода мечения первых митозов (18—20 ч после осеменения).
Номера и буквы, обозначающие полипептиды, относятся к эталонным поли-
пептидам, использованным для локализации (объяснения см. в работах [15,
39]). Изоэлектрическое фокусирование направлено слева (~рН 7,0) направо
(~рН 4,5).
2. Мечение белков раннего эмбриона
Мечение синтезирующихся белков (метаболическое мечение)
производится посредством включения радиоактивных аминокис-
лот. Наиболее важное преимущество радиомечения заключает-
ся в том, что методики определения меченых белков после их
разделения в ПААГ характеризуются значительно более высо-
кой чувствительностью, чем методики окрашивания белков
(разд. 4.2). В роли меченой аминокислоты чаще всего выступа-
ет [35S]-метионин, что объясняется его высокой удельной ак-
тивностью. Важно помнить, однако, что число остатков метио-
нина в каждом белке будет влиять на интенсивность мечения;
с особой осторожностью нужно относиться к оценке белков,
имеющих низкое содержание метионина, например гистонам.
В качестве метки могут быть использованы и другие аминокис-
лоты — индивидуально или в смесях. Способы двойного мече-
Качественный анализ белков ранних эмбрионов мыши
219
Рис. 8.4. Сравнение аккумулированных белков с синтезированными в раннем
эмбрионе, показано на одномерном геле. Паттерн белков эмбрионов 2-клеточ-
ион и 4-клеточной стадий развития, меченных [MS]-метионином, представлен
па дорожках а и а, паттерн белков, окрашенных серебром (ОС), — на дорож-
ках биг.
220
Глава 8
ния (включение двух разных аминокислот, каждая из которых
содержит различные радиоизотопы) описаны в работах [5—7].
Двойное мечение применяется для определения различий по ти-
пам синтезированных белков в разное время или в разных
компартментах раннего эмбриона (см. также разд. 4.1). Ряд
посттрансляционных модификаций может быть выявлен по
включению меченых сахаров или фосфата. Использование спе-
цифических антител дает возможность определить синтез (по-
средством иммунопреципитации; разд. 2.5) или присутствие
(посредством иммуноблоттинга; разд. 4.3) анализируемых бел-
ков.
2.1. Метаболическое мечение общих белков
2.1.1. Прямое мечение
Яйца или эмбрионы инкубируют в культуральной среде
М16, содержащей БСА (см. гл. 2, разд. 5.2), в которую добав-
лены меченые аминокислоты, например, [35S]-метионин, [3Н]-
лейцин или смеси [3Н]- или [14С/-аминокислот. Культивирова-
ние эмбрионов в радиоактивной среде в течение 1—5 ч не влия-
ет на результаты анализа. Однако жизнеспособность эмбрионов
при длительном культивировании в присутствии даже такой
малой дозы, как"50 мкКи/мл [35S]-метионина, значительно сни-
жается [8]. Учитывая высокую удельную активность [35S]-
метионина ( — 1300 Ки/ммоль) достаточно добавления 1—3 мкл
меченой аминокислоты к 50 мкл (120—360 мкКи/мл) культу-
ральной среды (см. рис. 8.1, 8.2 и 8.3). Если в качестве метки
используют другие аминокислоты, целесообразно порцию мече-
ной аминокислоты лиофилизировать, а затем снова растворить
ь меньшем объеме культуральной среды. Этот прием обеспечит
более высокую концентрацию изотопа; например, 50 мкл
[3HJ-лейцина (удельная активность —100 Ки/мМоль) можно
лиофилизировать и снова растворить в 25 мкл культуральной
среды. Важно, чтобы мечение производилось в среде, лишенной
примесей немеченых аминокислот, так как последние снизят
удельную активность меченых. Кроме того, следует иметь в ви-
ду, что присутствие сыворотки в метящей среде тоже может
снизить концентрацию изотопа. Низкие эндогенные пулы метио-
нина, лейцина, фенилаланина и аланина в клетках раннего
эмбриона дают этим меченым предшественникам преимущество
при выборе [9].
2.1.2. Мечение двойным изотопом
Распознавание двух различных популяций новосинтезпро-
ванных белков можно осуществить на основе использования
двух различно меченых аминокислот (разные способы обсужда-
Качественный анализ белков ранних эмбрионов мыши 221
ются в работе [3]). Так, мечение одной популяции эмбрионов
смесью [|4С]-аминокислот, а другой популяции — смесью
[3Н]-аминокислот должно выявить любые различия в обоих
наборах новосинтезированных белков ([5, 6]; см. разд. 4.1).
Быстрый и эффективный способ двойного мечения эмбрионов
дает комбинация [35SJ-метионина и источника 7-излучения
[758е]-селенометионина — изотопов, обладающих высокой удель-
ной активностью. Каждой аминокислотой можно пометить
две группы эмбрионов и, вычислив радиоактивность (см.
разд. 2.7), загрузить гель смесями в соответствующих пропор-
циях. Чтобы избежать артефактов, следует использовать ре-
ципрокные комбинации обоих образцов, меченных каждым из
изотопов.
2.1.3. Импульсное мечение
Паттерн синтезируемых белков раннего эмбриона в значи-
тельной степени обусловлен посттрансляционной модификацией
первичных продуктов трансляции. Эксперименты с импульсным
мечением позволяют выявить «медленные» модификации (моди-
фикации, происходящие почти сразу после отделения первично-
го продукта от рибосомы, не определяются). Смысл этой мето-
дики заключается в том, чтобы пометить белки и проследить их
судьбу в ходе последующей инкубации. Перед внесением мече-
ной аминокислоты в среду важно снизить ее активность (путем
лиофилизации и растворения в большем объеме), иначе эмб-
риональное развитие в период инкубации может быть наруше-
но, в частности, затормозится дробление зародыша [8]. Обычно
используемая процедура [10] описана ниже.
1. Введите импульсную метку, инкубируя эмбрионы в тече-
ние 1 ч в культуральной среде М16+БСА (гл. 2, разд.
5.2), содержащей [358]-метионин и 1 —10 мкМ немечено-
го метионина. Метка должна обладать удельной актив-
ностью около 10 Ки/ммоль, но учитывая длительность по-
следующей инкубации, ее следует снизить до 1 Ки/ммоль.
2. Отмойте эмбрионы от метки, проведя их через несколько
капель среды М16 + БСА, в которую добавлены немече-
ные метионин, фенилаланин и лейцин (каждая аминокис-
лота в концентрации 100 мкМ).
3. Культивируйте эмбрионы в течение определенных проме-
жутков времени в той же среде. Обмен немеченых ами-
нокислот с эндогенными аминокислотами [9] способству-
ет выведению меченого метионина из пула свободных
аминокислот в клетках.
222
Глава 8
Таблица 8.1. Неметаболическое мечение белков
1. Отмойте эмбрионы (или единичный эмбрион) от БСА и поместите в 10 мкл
0,1 М боратного буфера с pH 8,5 в эппендорфовскне пробирки. Образцы
можно хранить при —70 °C.
2. Приготовьте реактив Болтона и Хантера. Фирма Amersham поставляет
реактив в бензине. Дайте бензину испариться, а затем растворите реактив
в боратном буфере. Каждая ампула содержит количество реактива, до-
статочное для иодирования 20—30 образцов.
3. Внесите реактив Болтона и Хантера в образцы и оставьте иа 20 мин при
комнатной температуре.1’ Поскольку белка в эмбрионах очень мало, объем
добавляемого реактива не имеет зйачения (в любом случае он окажется
в избытке).
4. Добавьте 9 объемов этанола температуры льда и оставьте белки для
осаждения на 2 ч при 4 °C.
5. Отцентрифугируйте образцы в микроцентрифуге (Eppendorf) в течение
10 мин, отбросьте супернатант и дайте осадку высохнуть. Растворите
сгусток в буфере для приготовления образца (табл. 8.4) и до электрофо-
реза храните при —70 °C (см. разд. 3.1).
]) См. работу [ 13].
4. Сравните путем электрофореза белки сразу после мече-
ния с образцами, полученными в конце периода наблюде-
ния. Изменения в мобильности белков указывают на их
посттрансляционную модификации?.
2.2. Неметаболическое мечение общих белков
Неметаболическое мечение существующих белков (в отли-
чие от метаболического мечения белков синтезирующихся)
представляет собой более чувствительный метод, чем окрашива-
ние белков в геле (см. разд. 4.2). Меченый материал отделяет-
ся путем гель-электрофореза и выявляется радиоавтографичес-
кп. Теоретически можно было бы пометить только белки кле-
точной поверхности, например, воспользовавшись непроникаю-
щей белковой меткой—[4 * * * * * * * * * 14С]-изоэтионилацетимидатом [И] —
или путем иодирования, катализируемого лактопероксидазой
[ 12J. Однако важнейшим условием эксперимента является со-
хранение нормальной жизнеспособности клеток, поэтому подоб-
ные процедуры требуют большой осторожности и включения
надежного контроля.
Для мечения общих белков при физиологических условиях
можно использовать [14С]-этилацетимидат, проникающий в
клетки без нарушения функций мембраны [И]. Другой метод
заключается в использовании N-сукцинимидильной группы ре-
актива Болтона и Хантера [Л^-сукцинимидил 3-(4-гидрокси-5-
[1251 (-иодофенил)пропионат], конденсирующейся со свободны-
Качественный анализ белков ранних эмбрионов мыши
223.
Таблица 8.2. Дефосфорилироваиие фосфопротеииов
1. Введите метку, ннкубнруя эмбрионы в течение 1 ч в среде с [358]-метио-
нином (как в разд. 2.1), затем отмойте нх, проведя через несколько ка-
пель среды М2 или Ml6 без БСА.
2. Контрольную группу меченых эмбрионов поместите прямо н буфер для
образцов (табл. 8.4).
3. Перенесите остальные три группы эмбрионов в 5 мкл бидпстиллировашюй
воды и оставьте на 30 мин для лизиса при —70 °C.
4. Разморозьте один лизированный образец и инкубируйте при 37 °C в тече-
ние 1 ч с фосфатазой (иапрнмер, со щелочной фосфатазой, Sigma) при
конечной концентрации 5 мкг/мл (0,3 едииицы/мл).
5. Разморозьте следующие два контрольных образца и инкубируйте — один
с 5 мкл 2 мМ феиилметилсульфонилфлуорида (ФМСФ, ингибитор протеаз),
а другой только с 5 мкл бндистиллированной воды.
6. Внесите в каждый образец буфер для образцов с удвоенной концентра-
цией ДСН. Храните образцы при —70°С до электрофореза (см. разд. 3.1).
ми аминогруппами полипептидов [13]. В этом случае информа-
тивен такой порядок нанесения образцов на гель, при котором
можно сравнивать на соседних дорожках паттерн синтезиро-
ванных белков (метаболически меченных Р58]-метионином) с
белками, существующими (неметаболически меченными125!) иа
данной стадии эмбриогенеза. Методика неметаболического ме-
чения белков представлена в табл. 8.1. При работе с 1251 не за-
бывайте о технике безопасности.
2.3. Фосфопротеины
Фосфопротеины можно идентифицировать по включению
меченого фосфата. Эмбрионы культивируют в течение 2 ч в
бесфосфатной среде (гл. 2, разд. 5.2, М16 без КН2РО4), содер-
жащей 1 мКи/мл [32Р]-ортофосфата без носителя (Amersham.
International pic.). Перед употреблением 32Р лиофилизируют и
растворяют. Фосфат включается слабо, особенно в период ип-
терфазы, поэтому для одномерного анализа требуется 100—
150 эмбрионов (см. рис. 8.1). Возможно, в будущем окажется
более удобным инъецировать метку непосредственно в цито-
плазму ранних эмбрионов по аналогии с экспериментами на
яйцах Xenopus [14].
Чтобы убедиться в том, что меченые белки были действи-
тельно фосфорилированы протеинкиназами, следует провести
обработку протеинфосфатазами, как описано в табл. 8.2. Тогда
фосфатные группы будут удалены и электрофорез обнаружит
сдвиг в положении полос, меченных [35S]-метионином. Этот
прием дополнит информацию, полученную в результате им-
пульсного мечения [15].
224
Глава 8
2.4. Гликопротеины
Гликозилированные белки можно осадить специфическими
лектинами. Однако эта методика требует большого числа эм-
брионов, поэтому мы ее не предлагаем [16]. Гликопротеины
могут быть также идентифицированы после электрофореза (см.
разд. 4.3) или путем метаболического мечения радиоактивными
сахарами. К сожалению, большинство сахаров плохо проника-
ет в эмбрионы вплоть до стадии бластоцисты. Ранние эмбрионы
можно пометить, например, [3Н]-глюкозамином, но анализ ме-
ченых белков потребует слишком большого числа эмбрионов
(см. работу [17]).
Гликопротеины, синтезированные бластоцистами, могут быть
помечены фукозой, маннозой или глюкозамином. Бластоцисты
следует отмыть в среде без глюкозы' (см. гл. 2, разд. 5.2, опти-
мальная среда для культивирования бластоцист), но с добавле-
нием альтернативных субстратов, таких как пируват, глутами-
новая кислота, аспарагиновая кислота [18]. Группы из 15—
60 бластоцист могут быть помечены в течение 3—6 ч в 50 мкл
среды без глюкозы, содержащей БСА и около 25 мкКи лиофи-
лизированных Гзн] -глюкозамина, [3Н]-маннозы или [14С] -фу-
козы (с удельной активностью 2—50 мкКи/мл). Затем эмбрио-
ны следует отмыть, проведя через несколько капель среды без
метки, несколько капель среды без БСА и до электрофореза
хранить в буфере для образцов (см. разд. 3.1). Важно удалить
сыворотку, так как большие количества немеченого белка бу-
дут нарушать структуру геля.
2.5. Выявление синтеза специфических белков
Особые белки, синтезируемые или накопленные ранним эмб-
рионом, могут быть обнаружены в результате комбинированно-
го применения специфических антител и иммунофлуоресценции,
иммунопреципитации или иммуноблоттинга. Для непрямой им-
мунофлуоресценции эмбрионы фиксируют и окрашивают (соот-
ветствующие методические приемы приведены в гл. 2, разд. 4).
К сожалению, количество белка, необходимое для анализа ме-
тодами иммунопреципитации и блоттинга, может быть обеспе-
чено только большим числом эмбрионов. Блоттинг описан в
разд. 4.3.
Иммунопреципитация предполагает идентификацию синте-
зированных специфических белков посредством их осаждения
антителами [16]. Наиболее часто используемый прием осажде-
ния комплекса антиген — антитело включает связывание с бел-
ком A Staphylococcus aureus. (В тех случаях, когда антитело
получено от вида, не связывающего белка А, преципитация ин-
Качественный анализ белков ранних эмбрионов мыши
225
Таблица 8.3. Иммунопреципитация
1. Пометьте эмбрионы [358]-метионииом, как рекомендуется в разд. 2.1.1.
2. Отмойте эмбрионы от среды, содержащей БСА, и соберите их в 200 мкл
свежеприготовленного буфера1*. Радиоактивность 1—2Х106 имп/мин иа
образец дает хорошие результаты. Для этого может потребоваться от 1
до 2000 бластоцист, меченных в течение 3 ч при удельной активности,
указанной в разд. 2.1.1.
3. Прокипятите образец в течение 1—2 мии и храните при —70 °C. Все
дальнейшие процедуры следует делать на льду.
4. Непосредственно перед использованием сонируйте образец, чтобы тща-
тельно разрушить клетки. Сохраните небольшой объем (10 мкл) в каче-
стве контроля при —70 °C. Оставшуюся часть образца разделите иа две
порции.
о. Центрифугируйте образцы, специфическую антисыворотку и контрольную
сыворотку при 15К в течение 15 мии.
6. Добавьте равные объемы специфической или контрольной сыворотки
к двум эмбриональным образцам. Оптимальное разведение специфической
сыворотки варьирует в зависимости от антитела и устанавливается в каж-
дом случае, ио оно должно быть таким же, какое используется для им-
мунофлуоресценции.
7. Подготовка гранул белкаА с сефарозой; промойте 500 мг гранул восемь
раз в 5 мл буфера для отмывки (100 мМ трис, pH 8,3; 2 мМ ЭДТА;
0,5% дезоксихолата натрия; 0,5% Nonidet Р-40 (NP-40); 0,1% ДСН).
Всякий раз центрифугируйте с небольшой скоростью и полученный оса-
док ресуспендируйте. В последний раз к осадку добавьте равный объем
буфера (~ 2 мл) — теперь гранулы готовы к работе.
8. Для каждого из образцов отцентрифугируйте по 100 мкл приготовленных
гранул, супернатант отбросьте, а смесь белок/аититело добавьте к об-
разцу.
9. Встряхивайте эту смесь на льду (энергично, чтобы гранулы не оседали)
в течение 2 ч. Затем четыре раза отмойте в 100 мл того же буфера. Для
этого центрифугируйте 2 мин иа малой скорости и ресуспендируйте оса-
док (использовать вортекс не нужно). Наконец, внесите 100 мМ трис
pH 8,3 для последней отмывки и осадите.
10. Соберите специфический белок-антиген с гранул кипячением с 50 мкл бу-
фера для образцов (табл. 8.4) в течение 3 мии. Отсосите раствор микро-
пипеткой (Eppendorf), опустив ее конец иа дно каждой пробирки. Хра-
ните при —70 °C до электрофоретического разделения (см. разд. 3.1).
’) 100 мМ трис pH 8,3; 2 мМ ЭДТА; 0,5% дезоксихолата натрия; 0,5% Nonidet Р-40
(NP-40); 0,5% ДСН. Перед употреблением добавить 10 мМ иодацетамида, 1 мМ ФМСФ,
1 мкг/мл пепстатина, 1 мкг/мл лейпептина, 10 мкт/мл апротинина. Процентное содержа-
ние ДСН в буферах для образцов и отмывки можно уменьшить, это даст возможность
получить больший специфический сигнал на фоне неспецифического связывания.
Аудируется вторичным антителом). Для контроля степени не-
специфического взаимодействия следует параллельно провести
«преципитацию» предиммунной или неиммунной сывороткой.
Антитела, полученные на природный белок, более уместно ис-
пользовать для этого метода, чем для блоттинга. В табл. 8.3
описана методика, разработанная для применения аутоиммунной
антисыворотки против ламина В. Другие процедуры подробно
изложены в работе [16].
15—171
226
Глава 8
Таблица 8.4. Исходные растворы для одномерных гелей
Буфер Хранение
Буфер для приготовления образцов: 4 °C
62,5 мМ трис-НС1, pH 6,8 2% ДСН;
5% 2-меркаптоэтанол; 10% глицерол;
0,002% бромфеиолоный синий Акриламид : бисакриламид 30%: 0,8% (А) 0,5 М трис-НС1, pH 6,8; 0,4% ДСН (ВГБ‘>) 1,5 М трис-НС1, pH 8,8; 0,4% ДСН (НГБ2) 10% персульфат аммония 4,°C, в темной посуде 4 °C 4 °C 4 °C, можно хранить до 1 месяца
Буфер для разделения: 25 мМ трис-НС1, pH 8,3; 192 мМ глицииа; 0,1% ДСН Удобно хранить в деся- тикратной концентра- ции при комнатной температуре, pH дово- дить глицином до 8,3
') ВГБ— верхний гелевый буфер.
2) НГБ—-ннжннй гелевый буфер.
2.6. Приготовление образцов
Промойте образцы, проведя через две капли культуральной
среды М2 или М16 + БСА (гл. 2, разд. 5.2), затем через нес-
колько капель культуральной среды М2 или Ml6 без БСА.
Перенесите эмбрионы группами в эппендорфовские пробирки,
содержащие по 10—30 мкл буфера для образцов (табл. 8.4;
[19]) для одномерного анализа или лизисного буфера (табл.
8.7; [20]) для двумерного анализа. Большее число эмбрионов
требует большего объема буферов. Следы БСА должны быть
минимальны: этот немеченый белок разрушающе действует на
гель. Проколите каждую пробирку иглой и кипятите в водяной
бане в течение 2 мин, затем храните при —70 °C. Что касается
меченых образцов, то существует зависимость между типом ис-
пользованного изотопа, длительностью периода мечения, стади-
ей эмбрионального развития и числом эмбрионов на дорожку
геля. Так, яйца и эмбрионы вплоть до 8-клеточной стадии ме-
тят [36S]-метионином в течение 1 ч группами по 10 штук для
одномерного анализа или по 30—50 для двумерного анализа;
применение флюорографии дает результаты через 4—7 дней.
Для одномерного анализа вполне можно использовать единич-
ный эмбрион, но при этом увеличивается и период мечения, и
время экспозиции.
Качественный анализ белков ранних эмбрионов мыши 227
2.7. Количественные измерения включенной метки
Может возникнуть необходимость определения количества
включенной в эмбрион метки и (или) количества меченого пред-
шественника, связанного эмбрионом. В этом случае можно ко-
личественно оценить синтез белка, в частности, скорость синте-
за индивидуальных белков. Процедура заключается в следую-
щем.
1. Отмойте эмбрионы, инкубировавшиеся в среде с мечены-
ми аминокислотами или сахарами, и соберите, как ука-
зано в разд. 2.6.
2. Отберите аликвоты по 1—3 мкл (при общем объеме 10—
30 мкл) из буфера для образцов или лизисного буфера
для количественного анализа.
3. Для оценки поглощения меченого предшественника алик-
воту поместите непосредственно в сцинтиллят.
4. Для оценки включения в белок аликвоту добавьте к сме-
си 500 мкл воды с 25 мкл 0,1%-ного БСА (в качестве
ко-преципитата) и осаждайте белки добавлением
500 мкл 10%-ной трихлоруксусной кислоты (ТХУ).
5. Оставьте образцы на 1—2 ч при 4 °C, затем профильтруй-
те через миллипоровый фильтр, дважды промойте 10%-
ной ТХУ, дважды 70%-ным этанолом и дайте высохнуть.
6. На каждом из фильтров определите связавшуюся радио-
активность с помощью сцинтилляционного счетчика.
При мечении [35S]-метионином в течение 1 ч с использова-
нием тех удельных активностей, которые рекомендованы в
разд. 2.1.1, можно ожидать значений порядка 400—500 имп/мин
па эмбрион.
3. Электрофорез в полиакриламидном геле
Электрофорез в полиакриламидном геле (ПААГ) служит
мощным аналитическим средством разделения и идентификации
белков. Присутствие ДСН обеспечивает белок отрицательным
зарядом, вследствие чего заряд на единицу массы остается при-
близительно постоянным. Поэтому для большинства белков
электрофоретическая подвижность комплекса практически пол-
ностью определяется их молекулярной массой. Технические де-
тали применения одно- и двумерных гелей приведены в работе
[2]. Кроме того, использование гель-электрофореза белков при-
менительно к эмбрионам млекопитающих обсуждается Ван
Блеркомом [4]. Здесь мы попытаемся изложить достаточно
подробно принципы вертикального ДСН-электрофореза.
В последнее время все чаще пользуются тонкими (0,1—
0,3 мм) и миниатюрными (мини-) гелями [4]. Для гелей тради-
15*
228
Глава 8
Таблица 8.5. Рецепт для приготовления одномерных гелей
Исходный раствор (см. табл. 8.4) Разделяющий гель Концентрирующий гель
А (мл) ВГБ (мл) 8 1,5 2,5
НГБ (мл) 8 —
Вода (мл) 8 6
Дезаэрация Персульфат аммония 45 30
(мкл) ТЭМЕД (мкл) 15 20
ционной толщины (0,8 мм) уменьшен объем растворов и сокра-
щено время процедуры. Последнее усовершенствование связа-
но с использованием системы, разделяющей белки в горизон-
тальных, а не вертикальных пластинах (приборы LKB); прив-
лекательность этой системы обусловлена тем, что она значи-
тельно сокращает время разделения и процессинга и позволяет
осуществлять как электрофорез в ПААГ, так и ИЭФ.
Источник и чистота ингредиентов, используемых при элек-
трофорезе, во многом определяют его разрешающую способ-
ность. Исходные растворы необходима готовить каждые 1—2
месяца и хранить как описано в табл. 8.4 и 8.7.
Недостаток одномерных гелей заключается в том, что число
белков, которые можно разделить с их помощью, весьма ог-
раниченно. Для двумерных гелей эта цифра значительно боль-
ше благодаря тому, что разделение полипептидов происходит
в соответствии и с их изоэлектрической точкой, и с молекуляр-
ной массой. Таким образом, двумерные гели могут идентифици-
ровать до 1000—1200 индивидуальных белков (см. рис. 8.3).
Однако и им свойственны недостатки. Они связаны с длитель-
ностью процедуры, трудностью интерпретации п плохой вос-
производимостью.
3.1. Одномерные гели
Распределение белков в результате гель-электрофореза за-
висит от концентрации акриламида, но, к сожалению, ни одна
его концентрация не может обеспечить равномерность распре-
деления. По-видимому, наилучшее разрешение дает гель с экс-
поненциальным градиентом. Подробности работы с таким гелем
(8—15%) описаны [4] и здесь не будут обсуждаться. С 10%-
ным акриламидным гелем работать проще, он дает более рав-
Таблица 8.6. Приготовление, загрузка и использование одномерных гелей
1. Ополосните две чистые пластинки 70%-иым спиртом и высушите непо-
средственно перед употреблением.
2. Поместите тефлоновые спейсеры (0,8 мм толщиной и ~8—10 мм ши-
риной) между пластинками с каждой стороны. По нижиему краю между
стеклянными пластинками пропустите узкий (отверстие 0,5 мм; стейка
0,25 мм) шланг нз силиконовой резины (ESCOLtd), который,изгибаясь
U-образио, должен подниматься по боковым граням пластинок снаружи
от спейсеров. Скрепите пластники зажимами. Убедитесь в том, что по-
верхность геля горизонтальна.
3. Приготовьте смесь нижнего, разделяющего геля (см. табл. 8.4 и 8.5)
из исходного раствора акриламида (А), буфера иижиего геля н биди-
стиллироваииой воды и дезаэрируйте. (Неполимеризованный акриламид
обладает нейротоксическими свойствами, обращаться с осторожностью.)
Добавьте 10%-ный персульфат аммония, затем ТЭМЕД.
4. Залейте гелевую смесь между стеклянными пластинками так, чтобы
уровень был на 15—25 мм ниже верхнего края меньшей из пластинок.
Наслоите сверху 1 мл 1-бутанола, насыщенного водой. В верхнюю часть
конструкции вставьте «гребенку» для формирования лунок, она должна
располагаться над уровнем покрывающего раствора, чтобы не нарушить
расположения пластинок. Оставьте гель на 1 ч для полимеризации. Если
гель не предназначается для непосредственного использования, замените
покрывающий раствор бидистиллироваиной водой или буфером нижиего
геля, разбавленным ,в четыре раза, заклейте липкой лентой, чтобы предот-
вратить испарение, и оставьте при 4 °C (до 24 ч).
5. Наполните нижний резервуар прибора буфером для разделения (табл. 8.4).
6. Удалите гребенку, покрывающий раствор и сполосните верх геля водой.
Осторожно осушите (ие повредив поверхности полимеризованного геля)
плотной фильтровальной бумагой.
7. Уберите шлаиг и поместите пластинки в сосуд, наклонив их и осторожно
погружая одним концом, чтобы свести к минимуму число пузырьков
воздуха под гелем.
8. Приготовьте концентрирующий гель (см. табл. 8.4 и 8.5) из исходного
раствора акриламида, буфера верхнего геля и бидистиллированиой воды,
дезаэрируйте. Добавьте 10%-иый раствор персульфата аммония, затем
ТЭМЕД, Наслоите эту смесь поверх иижиего, разрешающего геля и
вставьте тефлоновую гребенку, формирующую луики. Полимеризация
заканчивается через 30 мин.
9. Уберите гребенку. Удалите иеполяризоваииую смесь (это удобно делать
с помощью шприца с тонкой иглой). В верхний резервуар залейте буфер
для разделения, заполните луики.
10. Убедитесь в том, что удалены пузырьки воздуха в буфере для разделе-
ния под гелем; это делается с помощью струи такого же буфера, выпу-
щенной из шприца , снабженного загнутой на конце иглой.
11. Выпрямите стороны лунок и пометьте их маркирующим карандашом иа
передней пластинке. Загрузите образцы гамильтоновским шприцем. Перед
внесением очередного образца шприц должен быть хорошо отмыт от пре-
дыдущего.
12. Проведите электрофорез меченых и немеченых маркеров молекулярной
массы иа каждом геле. Загрузите по 10 мкл буфера для образцов в пус-
тые луики, фланкирующие образцы, для устранения краевого эффекта.
13. Несколько капель бромфенолового синего можно добавить и верхний
резервуар в качестве лидирующего красителя.
14. Проводите электрофорез при постоянном напряжении 150 В или при
постоянном токе 20 мА, пока фронт красителя ие окажется иа расстоянии
примерно 5 мм от дна геля. Это происходит примерно через 4 ч.
229
230
Глава 8
П родолжение табл. 8.6
15. Закончив электрофорез, немедленно фиксируйте гель, чтобы предупре-
дить диффузию белков (если гель не предназначается для блоттинга,
см. табл. 8.11). Для этого в течение 20—30 мин инкубируйте гель в сме-
си 45%-ного метанола с 10%-ной уксусной кислотой, вместо иее можно
использовать 50%-ную ТХУ.
16. Используйте систему маркирования углов геля. Этот прием позволит вам
различать гели между собой и ориентировать их при последующих про-
цедурах. (Если гель на этой стадии предназначен для хранения, его по-
мещают в 7%-ную уксусную кислоту.) Затем гель может быть исполь-
зован для флюорографии, прямой радиоавтографии (см. разд. 4.1) или
окрашивания серебром (см. табл. 8.10).
17. Очистите гель средством для снятия крема (которое не поцарапало бы
его поверхность) и маленькой губкой, хорошо сполосните водой, затем
70%-ным спиртом и осушите материалом, не оставляющим волокон.
номерное распределение белков и используется наиболее часто
(см. рис. 8.1, 8.2 и 8.4). В табл. 8.4, 8.5 и 8.6 подробно изложе-
на методика работы с таким гелем Он содержит 0,4% ДСН и
0,5 М трис-НС1 (pH 8,8). В систему входит концентрирующий
гель из 4,5%-ного акриламида с 0,4% ДСН и 0,125 М трис-НС1
(pH 6,8). Если количество метки известно, образцы с одинако-
вой (или известной) радиоактивностью лучше поместить на
соседние дорожки. Это позволит более точно оценить изменения
индивидуальных белков в популяции. Для полуколичественно-
го сравнительного анализа полипептиды из образцов, обычно
содержащих равное число эмбрионов на одной стадии развития,
могут быть разделены на соседних дорожках. Пример, иллюст-
рируемый рис. 8.2, показывает изменение паттерна белков, син-
тезированных в ходе предимплантационного развития. На геле
представлено равное число [10] меченных [35S]-метионином
неоплодотворенных яиц, эмбрионов на 1-, 2-, 4-, 8-, 16-клеточ-
ной стадиях, на стадии ранней кавитации и 6 эмбрионов на
стадии поздней бластоцисты.
3.2. Изоэлектрическое фокусирование и двумерные гели
Двумерные гели с высоким разрешением позволяют разде-
лять и различать до 1200 полипептидов. Метод был предложен
О’Фареллом [20], последующие модификации позволили повы-
сить разрешение, воспроизводимость и упростить процедуру [22,
23]. В основе этого метода лежит разделение белков в соответ-
ствии с их изоэлектрической точкой в цилиндрических полиак-
риламидных гелях (изоэлектрофокусирующий электрофорез,
14ЭФ). В результате различные белки оказываются сегрегиро-
ванными в виде дискретных узких зон [24]. Новая («flat-bed»)
система позволяет производить разделение электрофокусирова-
Качественный анализ белков ранних эмбрионов мышн
231
Таблица 8.7. Исходные растворы для приготовления
изоэлектрофокусирующих гелей
Компонент геля Хранение
Лизирующий буфер: 9,5 М мочевина; 5% 2-мер- каптоэтанол; 2% NP-40; 1,6% амфолииы 5—7; 0,4% амфолииы 3,5—10 Акриламид: бисакриламид 28,83% : 1,62% (В) 10%-ный NP-40 Амфолииы pH 3,5—10 и 5—7 10%-ный персульфат аммония 10 мМ Н3РО4 20 мМ NaOH Покрывающий раствор: 8 М мочевина Покрывающий раствор для образца: 9 М мочевина; 0,8% амфолииы, 5—7; 0,2% ам- фолнны, 3,5—10 ДСН-буфер для приготовления образца: 62,5 мМ трнс-НС1 pH 6,8; 2,3% ДСН; 5% 2-меркаптоэтанол; 10% глицерол Заморозить 4 °C в темной посуде Комнатная температура 4 °C 4 °C, до 1 месяца 1 М исходный раствор, комнатная температу- ра Комнатная температура Заморозить Заморозить 4 °C
Таблица 8.8. Рецепт приготовления изоэлектрофокусирующего геля
Ингредиент (см. табл. 8.7) Количество’)
Мочевина (г) Раствор В (мл) NP-40 Вода (мл) Амфолииы 3,5—10 (мл) Амфолииы 5—7 (мл) Персульфат аммония (мкл) Дезаэрация ТЭМЕД (мкл) 5,5 1,33 2,0 1,97 0,1 0,4 10 7
') Достаточное для приготовления 10 гелей.
нием нескольких образцов в одном пластинчатом геле, что по-
вышает воспроизводимость результатов [21J. ИЭФ может при-
меняться отдельно, но чаще сочетается с электрофорезом во
втором направлении, разделяющим полосы на серию пятен, из
которых каждое обычно соответствует индивидуальному поли-
пептиду (см. рис. 8.3). ИЭФ — не единственный способ разде-
Таблица 8.9. Процедура разделения с помощью изоэлектрофокусирования
и двумерных гелей
1. Залейте ИЭФ-гели в трубки, залепленные с одного конца парафилмом.
Важнейшее значение имеет чистота трубок (их следует выдержать в хро-
мовой кислоте, тщательно промыть бидистиллнроваиной водой, сполоснуть
спиртом и высушить).
2. Приготовьте в 125-мл колбе с боковым отводом гелевую смесь (доста-
точную для заполнения 10 трубок, с pH 4,5—7) из мочевины, исходного
раствора В акриламида (табл. 8.7), NP-40, бидистиллированной воды н
смеси амфолннов (LKB или Servolyte), как описано в табл. 8.8. Слегка
вращайте колбу (можно немного подогреть), пока мочевина не раство-
рится полностью. Добавьте персульфат аммония. Проведите дезаэрацию
смеси в вакууме в течение примерно 1 минуты. Внесите ТЭМЕД и не-
медленно загрузите смесь в трубки, воспользовавшись для этого шприцем
с длинной иглой. Загружая смесь через дно трубок, позаботьтесь о том,
чтобы не возникли пузырьки воздуха.
3. Наслоите сверху 20 мкл покрывающего раствора мочевины (табл. 8.7).
Спустя 1,5 ч замените его 24 мкл лизирующего буфера (табл. 8.7), а за-
тем 10 мкл воды. Чтобы избежать кристаллизации мочевины, поместите
трубки перед инфракрасной лампой.
4. Спустя еще 1,5 ч наполните иижний электродный резервуар 10 мМ фос-
форной кислотой. Удалите парафилм с каждой трубки и поместите по-
лимеризованные гели в верхний электродный резервуар.
5. Удалите раствор, покрывающий сверху каждый гель и замените его
20 мкл свежего лизирующего буфера.
6. Заполните верхнюю часть каждой трубки дезаэрированным 20 мМ NaOH.
Наполните верхний резервуар тем же раствором. Проведите предвари-
тельный электрофорез в течение 15 мнн при 200В, 30 мин при 300В и
30 мин при 400В.
7. Выключите питание и удалите лизирующий буфер и буфер из верхнего
резервуара. Теперь гели готовы для загрузки.
8. Убедитесь в том, что в образцах достаточно мочевины, трижды прове-
дите их через процедуру замораживания — оттаивания, а затем загрузи-
те в лунки при помощи гамильтоновского шприца или микропипетки
(Eppendorf).
9. Наслоите на образцы 10 мкл покрывающего раствора (табл. 8.7), свер-
ху каждую трубку осторожно заполните 20 мМ NaOH и снова напол-
ните верхнюю камеру. Разделение эффективно при суммарном напряже-
нии 6000В, причем в течение последнего часа оно должно составлять
800В. (Например, разделяйте при 325В в течение 16 ч плюс 800В в те-
чение 1 ч).
10. Извлеките гели из трубки, воспользовавшись наполненным водой шпри-
цем, соединенным с коротким куском шланга, второй конец которого
надевается на конец трубки. Осторожно увеличивая давление в шприце,
выдавите гель из трубки11.
11. Если предполагается анализировать белки только с помощью ИЭФ, сразу
высушите гели.
12. Для разделения во втором направлении уравновесьте гели 5 мл ДСН-бу-
фера для образцов (который следует менять каждые 15 мин) в течение
2 ч. Храните гели в ДСН-буфере для образцов при —70 °C или сразу ве-
дите электрофорез во втором направлении.
13. Для разделения во втором направлении положите трубчатый гель вдоль
края пластинки 10%-ного акриламидного геля (приготовленного точно по
рецепту, описанному в табл. 8.5 и 8.6), который отлит между соответст-
вующим образом измененными стеклами (см. работы [4, 20]). Приго-
товьте концентрирующий гель в соответствии с указанием табл. 8.6.
Гребенку для лунок замените куском тефлона, обеспечивающим плоскую
поверхность для укладки трубчатого геля. Удалите тефлоновую форму
232
Качественный анализ белков ранних эмбрионов мыши
233
Продолжение табл. 8.9
после полимеризации концентрирующего геля. Удалите весь неполиме-
ризованнын материал. Налейте около 1 мл 1%-ной расплавленной агаро-
зы поверх концентрирующего геля (чтобы прикрепить трубчатый гель) и
осторожно поместите трубчатый гель на агарозу (позаботьтесь о том,
чтобы под гелем не образовались пузырьки воздуха). Спустя приблизи-
тельно 2 мии наполните верхний резервуар буфером для разделения
(табл. 8.4). Разделяйте полипептиды в соответствии с молекулярной
массой, как описано в табл. 8.6.
’) Если требуется оценить градиент pH, незагруженный ИЭФ-гель нарезают на пла-
стинки, которые элюируют 1 мл дезаэрированной дистиллированной воды в течение не-
скольких часов и в каждой измеряют pH.
ления в первом направлении : например, при работе с основны-
ми белками полезно воспользоваться электрофорезом с несба-
лансированным градиентом pH [22J. Несмотря на громадные
возможности двумерных гелей, воспроизводимость и сопостави-
мость результатов, полученных с их помощью, остаются серь-
езной проблемой. Без компьютерных программ, позволяющих
стандартизировать данные, интерпретация результатов весьма
сложна.
Общее разрешение двумерных гелей в большой степени за-
висит от вклада ИЭФ. Процедура работы с ИЭФ-гелями в пре-
делах pH 4,5—7.0 детально описана в табл. 8.7, 8.8 и 8.9. За-
метьте, что смеси амфолинов могут обеспечить широкие интер-
валы pH (от 2 до 10) или особо узкие (при этом как лизирую-
щий буфер, так и буфер для приготовления образца, должны
иметь одно и то же значение pH).
4. Анализ белков, разделенных
с помощью электрофореза в геле
4.1. Обнаружение радиоактивно меченных белков;
сравнение методов радиоавтографии и флюорографии
Обнаружение радиоактивно меченных белков, разделенных
с помощью ПААГ, — метод значительно более чувствительный,
чем окрашивание немеченых белков. Стратегия выявления бел-
ков (радиоавтография высушенного геля или окраска флуорес-
цирующим веществом) определяется типом используемого изо-
топа [25, 26J. Прямая радиоавтография дает удовлетворитель-
ные результаты при использовании энергии излучения 32P/i25J.
Применение интенсифицирующего экрана повышает чувстви-
тельность, однако при этом снижается разрешающая способ-
ность метода. По сравнению с радиоавтографией флюорография
234
Глава 8
повышает эффективность выявления 3Н в 1000 раз, a 35S и 14С
в 15 раз с незначительной потерей разрешения. В последнее
время трудоемкий метод импрегнации 2,5-дифенилоксазолом в
диметилсульфоксиде ([2, 26J) в большой степени вытеснен ис-
пользованием готовых препаратов, таких как Enhance, Autof-
luor, Enlightening и Amplify. Мы обычно применяем Amplify
(Amersham), который оказался надежным и чувствительным.
В этом случае гели следует перенести из фиксирующей смеси
прямо в Amplify (достаточно его количество, покрывающее
гель, — это около 16 мл), встряхивать 15—20 мин, а затем
высушить в условиях постоянного вакуума при 60—80 °C на
фильтровальной бумаге. Важно, чтобы вакуум поддерживался
до тех пор, пока гель не высохнет полностью, иначе он может
треснуть. Чтобы не ошибиться в содержимом каждого геля,
воспользуйтесь радиоактивным маркированием (и/или системой
нумерации) гелей. Это можно сделать, обмакнув карандаш с
волокнистым концом в чернила, к которым добавлено немного
жидкости, содержащей 14С или 35S (такие «горячие» каранда-
ши можно приобрести у фирмы Amersham). Чтобы повысить
чувствительность медицинской фотопленки, ее засвечивают
вспышкой белого света длительностью около 1 мсек; это уве-
личивает поглощение на 0,1—0,2 А540 единиц по сравнению с
неэкспонированной проявленной пленкой. Оптимальное расстоя-
ние между фотопленкой и источником, вспышки и время вспыш-
ки определяют экспериментально [2].
Изображение, полученное на фотопленке после проявления,
может быть сфотографировано и/или сканировано с помощью
денситометра, что дает возможность количественно оценить
результаты. В продаже имеются различные сканирующие уст-
ройства, которые можно приспособить для сканирования одно-
и двумерных гелей прямо (после окрашивания или путем ска-
нирования при 280 нм) или получив их радиоавтографическое
изображение [3]. Применение сканирующего устройства для
двумерных гелей необходимо сопровождать компьютерным ана-
лизом результатов. Соответствующие программы можно приоб-
рести (например, приборы и программы Joyce — Loebl). При
работе с одномерными гелями вместо сканирования применяют
другую процедуру; влажный гель нарезают на пластинки и с
помощью сцинтилляционного счетчика определяют содержание
радиоактивного белка в каждой из них [2J. Теоретически этот
подход мог бы быть использован в экспериментах с двойным
мечением, если бы разрешение не было ограничено размером
пластинки. Более высокое разрешение достигается тщательным
выбором изотопов, выявляемых в интактном геле,
Как уже упоминалось в разд. 2.1.2, существует метод, по-
зволяющий различать 14С- и 3Н-меченные белки [5, 6]. Для об-
Качественный анализ белков ранних эмбрионов мыши 235
наружения обоих изотопов гель флюорографируют и экспони-
руют с предзасвеченной фотопленкой при —70 °C. Параллельно
проводят прямую радиоавтографию на неэкспонированной фо-
топленке при комнатной температуре для выявления только
14С. Этот метод не удобен в том отношении, что требует дли-
тельной экспозиции из-за низкой удельной активности изотопов
[3]. Сочетание [35S]-метионина и [765е]-селенометионина поз-
воляет быстро различить две популяции новосинтезированных
белков, включающих разные относительные количества каж-
дого из изотопов. Радиоактивность геля регистрируют на двух
отрезках предварительно засвеченной рентгеновскими лучами
пленки, разделенных засвеченной и проявленной (почерневшей)
пленкой. При такой экспозиции на первой пленке будет заре-
гистрировано излучение обоих изотопов, но на вторую, отде-
ленную засвеченной прокладкой, проникнет только более мощ-
ное 7-излучение 75Se (оно может быть усилено применением ин-
тенсифицирующего экрана). Если на первой пленке будет заре-
гистрирована полоса среди белков, меченных 35S, положение
этих белков на второй пленке можно установить путем сравне-
ния первого негатива со вторым.
4.2. Окрашивание общих белков
Наиболее известный краситель — кумасси ярко-голубой
R-250 способен выявлять 0,2—0,5 мкг белка в резкой полосе
(см. работу [2]); к сожалению, для обнаружения очень малых
количеств белка ранних эмбрионов он недостаточно чувствите-
лен. В связи с этим была усовершенствована методика окра-
шивания серебром [28, 29J. Авторы сообщают о ее высокой
чувствительности (в 100—200 раз выше, чем чувствительность
предыдущего метода) и пригодности для анализа белков ран-
него эмбриона. Вопрос о том, применима ли эта методика для
количественного анализа, оказался спорным [3]. По нашему
мнению, при достаточной осторожности в оценке сверхокрашен-
ных полос и при использовании эталонных белков, нормализую-
щих при разделении плотность других пятен, этот метод может
обеспечить количественный интегральный анализ денситометри-
ческим сканированием.
При выявлении белков методом окрашивания серебром для
приготовления белкового препарата в случае одномерного геля
требуется материал примерно 60 предимплантационных эмбрио-
нов на одну дорожку, а в случае двумерных гелей это количе-
ство возрастает до 250 эмбрионов. Метод (основанный па ра-
боте [29]) излагается в табл. 8.10.
236
Глава 8
Таблица 8.10. Окрашивание гелей серебром
1. После электрофореза и фиксации (в смеси: 45% метанол : 10% уксусная
кислота) отмывайте гель в 50%-ном метаноле не менее 30 мнн, затем
в дистиллированной воде (3x30 мин). Эта процедура обеспечивает пол-
ное удаление таких примесей, как глицерол и тритон.
2. Окрашивайте каждый гель в течение 15 мин в свежеприготовленном рас-
творе аммиачного нитрата серебра (0,8 г нитрата серебра в 4 мл биди-
стиллированной воды плюс 21 мл 0,36%-ного NaOH и 1,4 мл раствора
аммония; довести объем до 100 мл бидистиллированной водой).
3. Тщательно промойте гель бидистиллированной водой (3x2 мин).
4. Проявляйте в течение 8—15 мин в 500 мл раствора, содержащего
0,05 мг/мл лимонной кислоты и 0,18 мг/мл раствора формальдегида, за-
тем перенесите в 50%-ный метанол (30 мин), 20%-ный метанол или би-
дистиллнрованную воду (2X45 мин).
5. Высушите гель и сфотографируйте.
4.3. Выявление специфических белков
Для выявления специфических полипептидов было предло-
жено несколько методов окрашивания [2]. Так, использование
меченых лектинов дает возможность идентифицировать и час-
тично охарактеризовать гликопротеины, находящиеся в гелях.
Существуют разнообразные способы выявления лектинов —
прямые, если лектины конъюгированы с флуоресцентным краси-
телем [30] или пероксидазой хрена [31], или косвенные —
с помощью антилектпновых антител [32]. Эти методы пока не
находят широкого применения при анализе белков ранних эм-
брионов.
Для выявления специфических белков после электрофореза
могут быть использованы антитела, однако скорость диффузии
антител в гелевый матрикс очень низка. В связи с этим разра-
ботаны разнообразные способы переноса белков из полиакрил-
амидных гелей в нитроцеллюлозные [33]. Для иммуноблоттин-
га используют поликлональную антисыворотку и некоторые
моноклональные антитела, способные узнавать антиген, несмот-
ря на его денатурацию. Взаимодействие антиген — антитело
выявляется либо путем мечения антитела (обычно ,251), либо
посредством амплификации с вторичным или третичным анти-
телом. В настоящее время известно несколько систем ампли-
фикации:
а) вторичное антитело, связанное с пероксидазой хрена,,
выявляется при инкубации с 3,3-диаминобензидином и
перекисью водорода в виде окрашенной полосы F35J;
б) меченое вторичное антитело обеспечивает более высокую
чувствительность либо прямо (благодаря метке), либо
при связывании с первичным иммуноглобулином ,251-ме-
ченого белка А из S. aureus [36];
Качественный анализ белков ранних эмбрионов мышн 237
Таблица 8.11. Идентификация специфических белков методом вестерн-блот
Определение специфических белков требует большого числа яиц или ранних
эмбрионов (так, для приготовления исходного белкового препарата при
разделении в одномерном геле нужно взять 800—1000 иеоплодотворенных
яиц на одну дорожку геля).
1. Проведите предварительное вымачивание полоски бумаги Biodyne или
нитроцеллюлозы, вырезанной по размеру гелевой пластинки, и двух по-
лосок ватмановской фильтровальной бумаги большего размера в буфере,
содержащем 2 мМ ацетата натрия, 5 мМ MOPS и 20% этанола (если
используется биодииовая бумага) или в буфере, содержащем 25 мМ
трис (pH 8,3), 192 мМ глицина и 20% метанола (если используется нит-
роцеллюлоза) .
2. Сразу после электрофореза поместите гель на бумагу, предназначенную
для переноса белков (блот) и заключите между двумя полосками филь-
тровальной бумаги. Весь пакет поместите в аппарат для переноса (Bio-rad
Trans-Blot Cells), убедившись в том, что блот направлен к положительно
заряженному электроду. Наполните резервуар тем же буфером, в котором
вымачивались полоски бумаги. Перенос белков осуществляется при 50/60В
в течение 2 ч или при ЗОВ в течение ночи.
3. После переноса выньте блот (гель можно сохранить, чтобы проконтроли-
ровать полноту переноса белков с помощью окрашивания серебром, см.
табл. 8.10).
4. Блокируйте сайты неспецифического связывания белковых молекул путем
встряхивания препарата в течение ночи с 10% ЭТС, 3% БСА или
с 10 %-ным раствором обезжиренного молочного порошка в солевом рас-
творе, забуферениом фосфатом (PBS : 0,14 М NaCl, 2,7 мМ КС1, 1,5 мМ
К.Н2РО4, 8,1 мМ Na2HPO4, куда добавляется 0,02—0,1% азида натрия).
5. Споласкивайте блот в течение 10 мин в PBS, содержащем 0,1% Тритона
Х-100 или 0,1% твина. Эту процедуру следует проводить при каждом про-
мывании.
G. Разведите первичное антитело1* в соответствии с требованием (разведение
зависит от специфичности антитела; моноклональные антитела, например,
часто используются неразведеннымн) в 3%-ном БСА в PBS и инкубируйте
в течение 45—60 мни в закрытом контейнере небольшого объема (0,5—
2 мл). Тщательно отмойте блот (4X10 мин) прежде, чем инкубировать его
с вторичным антителом.
”. В качестве вторичного антитела используйте 5 мкл нужным образом био-
тинилированного анти-иммуноглобулина, разведенного в 100 мкл 3%-ного
БСА п PBS, доведите до 0,5 мл. Инкубируйте 45—60 мни.
' Тщательно промойте блот (4X10 мии), прежде чем инкубировать с 5 мкл
[!55]-стрептавидииа, разведенного в 50 мкл PBS с 3% БСА, в течение
60 мии в закрытом контейнере.
9. Тщательно промойте блот (4x10 мин), высушите на воздухе и экспонируй-
те с предварительно засвеченной пленкой при —70° в течение 3—20 сут.
’) К’.ждое разведенное антитело следует до использования центрифугировать при
1ёК в г. 2—5 мин.
в) биотинилированное вторичное антитело обнаруживается с
помощью третичного антитела—[355]-стрептавидина.
Последняя из перечисленных систем (соответствующие ре-
активы можно получить от фирмы Amersham) оказалась наибо-
лее чувствительной и с успехом применяется для электрофоре-
238
Глава 8
тического разделения белков с последующей амплификацией
антител. Эта система представлена в табл. 8.11.
5. Заключение
Для изучения белков раннего эмбриона применяется элект-
рофорез. Усовершенствование этого метода позволит иденти-
фицировать, описывать и выделять специфические белки. Осо-
бенно ценным представляется анализ с помощью двумерных ге-
лей высокого разрешения. Использование электроэлюции полос
специфических белков после электрофореза [37, 38] открывает
возможности анализа индивидуальных белков путем пептидно-
го картирования и секвенирования аминокислот [3]. Выделение
индивидуальных белков позволит получать антитела, с помо-
щью которых можно будет изучать белки эмбриона методами
иммунофлуоресценции, иммуноблоттинга или иммунопреципи-
тации. Субфракционирование и/или избирательная солюбилиза-
ция помогут связать присутствие специфических белков с опре-
деленными стадиями развития.
Благодарности
Я благодарю за помощь в написании этой главы Азима Су-
рани, Эвелин Хаулистон, Бернара Маро, Боба Моора и Марти-
на Джонсона.
Литература
1. Johnson М. Н., McConnell J., Van Blerkom J. (1984). J. Embriol. Exp. Mor-
phol., 83, suppl. 197.
2. Hames D., Richwood D., (eds). (1981). Gel Electrophoresis of Proteins —
A Practical Approach. IRL Press, Oxford.
3. Dunn M. J., Burghes A. H. M. (1983). Electrophoresis, 4, 173.
4. Van Blerkom J. (1978). In: Methods in Mammalian Reproduction. Dani-
el J. C„ Jr (ed)., Academic Press, New York, p. 68.
5. McConkey E. H. (1979). Anal. Biochem., 96, 39.
6. McConkey E. H. (1982). Proc. Natl. Acad. Sci. USA, 79, 3236.
7. Lecocq R. E., Hepburn A.t Lamy F. (1982). Anal. Biochem., 127, 293.
8. MacQueen H. A. (1979). J. Embryol. Exp. Morphol., 52, 203.
9. Schultz G. A., Kaye P. L., McKay D. J., Johnson Af. H. (1981). J. Reprod.
Fertil., 61, 387.
10. Bolton V. A'., Cades P. J., Johnson Af. H. (1984). J. Embryol. Exp. Morphol.,
79 139
11. Whiteley N. M„ Berg H. C. (1974). J. Mol. Biol., 87, 541.
12. Johnson L. V.., Calarco P. G. (1980). Dev. Biol., 77, 224.
13. Bolton A. E.. Hunter W. M. (1973). Biochem. J., 133, 529.
14. Taylor Af. A.. Robinson K. R., Smith L. D. (1985). J. Embryol Exp. Morphol.,
89, 35.
15. Howlett S. K. (1986). Cell, 45, 387.
Качественный анализ белков ранних эмбрионов мыши 239
16. Johnstone A., Thorpe R. (1982). Iminunochemistry in Practice. Blackwell
Scientific Publications, Oxford.
17. Van Blerkom J. (1981). Proc. Natl. Acad. Sci. USA, 78, 7629.
18. SuraniM. A. H. (1979). Cell, 18, 217.
19. Laemmli U. K. (1970). Nature, 227, 680.
20. O’Farrell P. H. (1975). J. Biol. Chem., 250, 4007.
21. Burghes A. H. Al., Dunn M. J., Dubowitz V. (1982). Electrophoresis, 3, 354.
22. O’Farrell P. Z., Goodman H. M, O’Farrell P. H. (1977). Cell, 12, 1193.
23. Garrells J. I. (1983). In: Methods in Enzymology. W. R., Grossmann L„
Moldave K. (eds.), Academic Press, New York, Vol. 100, p. 411.
24. Drysdale J. W. (1975). In: Methods of Protein Separation. Catsimpoolas N.
(ed.), Plenum Press, New York, p. 93.
25. Bonner IV. M„ Laskey R. A. (1974). Eur. J. Biochem., 46, 83.
26. Laskey R. A. (1984). Radioisotope Detection by Fluorography and Intensi-
fying Screens. (Review 23), Radiochemical Centre, Amersham, UK-
27. Laskey R. A., Mills A. D. (1975), Eur. J. Biochem., 56, 335.
28. Switzer R. C., Merril C. R., Shifrin S. (1979). Anal. Biochem., 98, 231
29. Wray W., Boulikas T., Wray V. P., Hancock R. (1981). Anal. Biochem., 118,
197.
30. Furlan M., Perret B. A., Beck E, A. (1979). Anal. Biochem., 96, 208.
31. Wood С. M., Sarinana F. O. (1975). Anal. Biochem., 69, 320.
32. Glass IV. F., Briggs R. C., Hnilica L. S. (1981). Anal Biochem., 115, 219.
33. Burnette IV. N. (1981). Anal. Biochem., 112, 195.
34. Anderson N. L„ Giometti C. S., Gemmell M. A„ Nance S. L., Anderson N. G.
S. Clin. Chem., 28, 1084.
K., Yamada К. M. (1977). Anal. Biochem., 78, 483.
36. Burridge K. (1978). In: Methods in Enzymology. Ginsburg V. (ed.), Aca-
demic Press, New York, Vol. 50, p. 54.
37. Wallis M. FL, Kramer G., Hardesty B, (1980). Biochemistry, 19, 789.
38. Niemann M. S., Volanakis J. E., Mole J. E. (1980). Biochemistry, 19, 1576.
39. Howlett S. K-, Bolton V. N. (1985). J. Embryol. Exp. Morphol., 87, 175.
ГЛАВА 9
СОЗДАНИЕ БИБЛИОТЕК кДНК
ДЛЯ ПРЕДИМПЛАНТАЦИОННОГО
ЭМБРИОНА МЫШИ
Кристина Уотсон, Джози МкКоннел'
1. Введение
Длительные клеточные циклы и «стойкость» мышиного эм-
бриона в отношении экспериментальных манипуляций позволи-
ли довольно детально изучить на клеточном уровне ранний
эмбриогенез мыши [1, 2] (гл. 2 и 3). Однако для анализа мо-
лекулярных событий, лежащих в основе наблюдаемых морфо-
логических изменений, предимплантационный эмбрион мыши
нельзя назвать подходящей системой. Только в последнее вре-
мя с усовершенствованием методов получения рекомбинантных
ДНК появилась возможность приступить к изучению эмбрио-
нального развития на молекулярном уровне.
Основная трудность, возникающая при биохимическом или
молекулярном анализе, связана с малыми размерами эмбрио-
нов и ограниченностью их числа: установлено, что каждый
ооцит мыши содержит около 0,35 пг тотальной РНК [3] и да-
же суперовуляция (гл. 1, разд. 5.4) не дает возможности по-
лучить от одной мыши более 30 ооцитов.
Несмотря на эти ограничения, накопился достаточный объем
биохимических данных о ранних стадиях развития [4]. Было
установлено, что в течение первых двух клеточных циклов
функционирует мРНК, унаследованная от матери, затем ее
большая часть (если не вся) разрушается и уступает место
мРНК, транскрибированной с генома эмбриона [5—8]. Более
тщательный анализ белков, кодированных материнскими транс-
криптами, показал, что их набор изменяется в зависимости от
клеточного цикла [9].
Один из подходов к пониманию того, как информация от
генов переходит к белкам, заключается в анализе популяций
мРНК на разных стадиях развития эмбриона. Избирательная
деструкция или активация индивидуальной мРНК, выявленная
в ходе этого анализа, может указать на ее роль в конкретных
событиях развития. Для такого анализа необходимо создание
1 Christine J. Watson. Department of Biochemistry and Microbiology, Uni-
versity of St Andrews, St Andrews KY 16 9AL, UK.
Josie McConnell. Department of Anatomy, University of Cambridge, Dow-
ning Street, Cambridge CB2 3DY, UK.
240
Библиотека кДНК Для предимплантационного эмбриона мыши 241
библиотек комплементарной ДНК (кДНК) на основе мРНК,
экстрагированных на разных стадиях развития. Однако, как
упоминалось выше, такой подход серьезно ограничивается ко-
личеством доступного исследователю материала. Не удобны в
качестве модельной системы и клетки эмбриональной карцино-
мы (эю [ioj. так как они не происходят от зиготы и, следо-
вательно, не могут имитировать раннее эмбриональное разви-
тие. Исходя из необходимости использовать РНК ооцита и РНК
поздних стадий эмбрионального развития, следует так усовер-
шенствовать все методики, чтобы компенсировать малый объем
экспериментального материала.
На основе векторной системы бактериофага может быть
обеспечена очень высокая эффективность клонирования — по-
рядка 2Х107 бляшкообразующих единиц (БОЕ) на микро-
грамм двухцепочечной ДНК [11], что дает возможность обой-
тись очень малым количеством исходной ткани. Число индиви-
дуальных клонов, которое обеспечивает 99%-ную вероятность
клонирования определенной мРНК в пределах популяции,
можно рассчитать по формуле:
N = ln(l —Р)/1п(1 — 1/п),
где N—число требуемых клонов, Р— вероятность получения
данной последовательности и 1/п — доля отдельных фракций в
популяции тотальной мРНК [12]. Например, в клеточной линии
фибробластов человека, трансформированных SV40, число раз-
личных последовательностей класса с низкой копийностью
(~ 14 копий на клетку) равно 10 670 и представляет 29% мРНК
[13]. В таком случае п—10 670/0,29 и равно 36 790. Следова-
тельно, чтобы обеспечить 99%-ную вероятность клонирования
низкокопийной мРНК требуется около 169 000 клонов. Если
допустить, что структура популяции РНК эмбриона такая же,
как в приведенном примере, можно заключить, что полную би-
блиотеку должны составить 200 000 клонов. В табл. 9.1 пере-
числено количество эмбрионов мыши различных стадий разви-
тия, которое дает минимальный объем РНК, необходимый для
создания полной библиотеки современными средствами. По ме-
ре усовершенствования каждой процедуры эти значения могут
быть уменьшены, но, скорее всего, незначительно.
Исключительная ценность библиотек эмбриона определяется
следующими причинами:
а) используя технику дифференциального скрининга, можно
выделить из библиотеки последовательности, экспресси-
рующиеся на разных стадиях развития [15];
б) можно количественно охарактеризовать экспрессию оп-
ределенных клонируемых последовательностей в ходе
16-171
.242
Глава 9
Таблица 9.1. Содержание РНК в предимплантационных эмбрионах
Стадия развития Всего РНК на эмбрион, иг* 1) Оценка ро!у(А) + РНК иа эмбри- он, пг2) Число эмбрио- нов для полной библиотек^)
Ооцит 0,35 7,0 14 286
1-клеточная 0,42 8,3 12 048
Поздняя 2-клеточная 0,24 2,6 38 461
8—16-клеточная 0,69 4,4 22 727
32-клеточная 1,47 14,2 7 042
‘) На основании данных Пико и Клегга |3|.
2) На основании данных Пико и Клегга (3|, заключивших, что содержание ро!у(А)+-
РНК в 10 раз выше содержания ро!у(А)+ эмбриона.
') Число клоиов для создания полной библиотеки оценено как 2ХЮ\ а количество
-пилиадеинлироваиной РНК приблизительно 0,1 мкг (в зависимости от эффективности каж-
дою шага процедуры). Детали см. в тексте.
раннего развития (это более чувствительный метод, чем
гибридизационный анализ при переносе);
в) клоны, выделенные из библиотек кДНК, можно исполь-
зовать для изучения мультигенных семейств; секвениро-
вание даст возможность безошибочно идентифицировать
члены семейства, экспрессирующиеся в данное время.
Таким образом, библиотеки кДНК эмбрионов представляют
собой мощное средство для изучения молекулярных событий,
связанных с ранним развитием.
2. Оценка РНК-носителя
Для преодоления технических трудностей, связанных с не-
обходимостью манипулировать очень малым объемом материа-
ла, должна быть применена система носителя, сводящая к ми-
нимуму потери мРНК и кДНК, которые в противном случае
окажутся непозволительно большими. Подходящий носитель,
используемый при селекции полиаденилированных РНК и по-
следующем синтезе кДНК, должен удовлетворять нескольким
требованиям. Среди них наиболее существенны следующие:
1) он должен быть полиаденилирован;
2) он должен быть представлен ограниченным числом ви-
дов— как можно меньшим;
3) он не должен иметь последовательностей, общих с ана-
лизируемой РНК;
4) вся последовательность РНК-носителя (ей) должна быть
известна;
5) носитель не должен иметь преимущества при клонирова-
нии.
Этим условиям отвечает геном вируса мозаики вигны
(CPMV). Это типичный представитель комовирусов, инфициру-
Библиотека кДНК дли предимплантационного эмбриона мыши 243
ет он только растения. Его геном состоит из двух одноцепочеч-
ных молекул РНК, обладающих положительной полярностью.
Каждая молекула заключена в капсид. Эти РНК имеют разные
размеры: большая состоит из 5889, а меньшая — из 3481 нук-
леотида. Обе РНК полиаденилированы [16], что необычно для
вирусов растений, определена их полная последовательность
[17, 18]. Таким образом, требования 1, 2 и 4, предъявляемые к
носителям, соблюдены. Пробный эксперимент клонирования,
в котором РНК CPMV была смешана с РНК печени мыши в
весовом отношении 9: 1, выявил, что с радиоактивным зондом
РНК CPMV было гибридизовано приблизительно в 9 раз боль-
ше бляшек, чем с зондом РНК печени мыши, и что ни одна
бляшка с обоими зондами не гибридизуется [19]. Следова-
тельно, требования 3 и 5 также соблюдены. Итак, РНК CPMV
по всем критериям подходит на роль носителя и должна быть
с успехом использована в экспериментах с клонированием
кДНК.
3. Сбор ооцитов и эмбрионов и выделение РНК
Суперовуляция самок мыши описана в разд. 5.4 гл. 1, а сбор
ооцитов и предимплантационных эмбрионов описан в разд. 2
гл. 2. Поэтому мы не будем подробно останавливаться на этих
процедурах, а сфокусируем все внимание на специальной про-
блеме выделения РНК.
Следует обратить внимание на необходимость соблюдения
стерильности при сборе ооцитов и эмбрионов. Важно также
предотвратить вероятность загрязнения рибонуклеазой.
3.1. Сбор ооцитов
Для получения количества РНК, достаточного для создания
полной библиотеки ооцита, требуется около 16000 ооцитов (от
~600 суперовулировавшпх, но не спаривавшихся самок мыши)
(см. табл. 9.1). Существенным условием получения хорошей
библиотеки служит предупреждение деградации РНК, поэтому
сбор ооцитов необходимо проводить очень быстро. Ооциты
с аномальными признаками следует отбрасывать. Особенно
важно отделить от ооцита клетки кумулюса, которые, являясь
по происхождению материнскими, могут внести в библиотеку
ненужную информацию. Ооциты, лишенные клеток кумулюса,
отмойте в среде М16 без БСА (гл. 2, разд. 5) и поместите их
2 мкл среды в пробирку (Eppendorf) на 1,5 мл. Судя по на-
шему опыту, удобно собрать 8000 ооцитов в одну пробирку,
немедленно заморозить в жидком азоте и хранить при —70 °C.
Исключительно важно, чтобы весь период времени между сбо-
16*
244
Глава 9
Таблица 9.2. Выделение тотальной РНК
1. Разморозьте образцы на льду и немедленно добавьте 100 мкл буфера
для экстракции (БЭ)1». Перемешайте пипетироваиием.— дважды втянув
и выпустив из пипетки.
2, Добавьте 10 мкл БЭ, содержащего 0,1% ДСН (в/о), затем 100 мкл забу-
ференного фенола2».
3. Добавьте еще по 0,5 мл БЭ и фенола. Воспользуйтесь вортексом (крат-
ковременно) и фракционируйте центрифугированием в микроцентрифуге
в течение 5 мин.
4. Отберите водную фазу (учтите, что поверхность раздела очень мала)
и вновь экстрагируйте фенольную фазу 100 мкл БЭ. Слейте водные фазы.
5. Удалите оставшиеся следы фенола встряхиванием с равным объемом
водонасыщенного эфира (трижды сменив его) или до просветления водной
фазы.
45. ДНК можно удалить на этом этапе путем воздействия РНКаза-свободиой
ДНКазой3». Добавьте 350 мкл трехкратного буфера RQ14», а затем
10 единиц ДНКазы. Проинкубируйте при 37 °C в течение 10 мин.
7. Внесите равный объем фенола и повторите экстракцию, как в пп. «4»
и «5».
8. Осадите РНК, перенеся водную фазу в стерильную силиконовую пробир-
ку Согех и добавив 2,5 объема холодного абсолютного этанола и 0,1 объ-
ема ЗМ ацетата натрия.
9. Храпите при —20 °C всю ночь.
.10. Соберите осажденную РНК центрифугированием при 10 000 об/мин в ро-
торе Sorvall SS 34 в течение 45 мин при 4 °C. Отмойте осадок холодным
70%-иым (о/о) этанолом, высушите под вакуумом и ресуспендируйте
в 0,5 мл стерильной бидистиллированной Н2О температуры льда.
11. Определите количество полученной тотальной РНК с помощью спектро-
фотометра, воспользовавшись стерильной автоклавируемой кюветой.
Измерьте отношение A2so: А280. Значение 1,8—2,0 указывает на то, что
препарат нуклеиновой кислоты свободен от белка, но значение отноше-
ния 1,7—1,8 также допустимо. По измерению Агео сделайте оценку то-
нальной РНК (для РНК 1 ОП=40 мкг/мл).
’> БЭ: 0.1 М NaCI; 0,001 М ЭДТА; 0,02 М трис-НС1 pH 7.4.
-> См. [26].
:‘) ДНКаза (продукт RQ1 поставляется Promega Biotech. I единица фермента
необходима для полной деградации I мкг ДНК в течение 10 мин при 37 °C.
4) Трехкратный буфер RQ1: 120 мМ трис-НС1 pH 7,9; 30 мМ MgCI2.
ром ооцитов и экстракцией из них РНК они оставались замо-
роженными, поскольку оттаивание клеток привело бы к их
лизису, высвобождению эндогенных рибонуклеаз и деградации
РНК.
3.2. Сбор предимплантационных эмбрионов
Этот материал соберите так, как описано в разд. 2, гл. 2.
Отбросьте все отстающие в развитии или морфологически ано-
мальные эмбрионы. Поскольку сбор образцов занимает много
времени, единовременно собранную порцию следует ограничить
примерно 1000 эмбрионов. Каждую порцию отмойте в среде
Библиотека кДНК для предимплантациоииого эмбриона мыши 245
М16 без БСА и поместите в пробирку (Eppendorf) в минималь-
ном объеме среды (как в случае ооцитов). Немедленно замо-
розьте в жидком азоте и храните при —70°C, пока не наберет-
ся количество материала, достаточное для экстракции РНК..
3.3. Выделение РНК
Нуклеиновую кислоту следует выделять из ооцитов и эмбри-
онов модифицированным методом Брауде и Пелхэма [20J
(табл. 9.2). При этом важно не добавлять тРНК-носителя, так
как он содержит небольшие количества двухцепочечной ДНК,
которая впоследствии будет клонирована.
3.4. Селекция poly (А)+РНК и добавление носителя
Исходя из уровня тотальной РНК (табл. 9.2), рассчитайте
количество poly (А)+-РНК (см. табл. 9.1). Внесите достаточное
количество РНК-носителя CPMV, чтобы в сумме получился
1 мкг полиаденилированной РНК.
Полиаденилированную РНК можно выделить либо хрома-
тографически на oligo (dt) -целлюлозе (Тип 3, Collaborative
Research), либо с помощью бумаги для аффинного связывания
РНК (Hybond™-mAP, Amersham). Последний метод рекомен-
дуется для малых количеств РНК. При этом следует изменить
пропись изготовителя, включив дополнительную отмывку в 0,1М
NaCl в течение 5 мин перед промывкой в 70%-ном (о/о) эта-
ноле.
4. Конструирование библиотек
Методы конструирования библиотеки кДНК детально опи-
саны в предыдущем томе этой серии [21] и поэтому здесь не
приводятся. Наше внимание будет сосредоточено на манипу-
лировании очень малыми объемами исходного материала.
4.1. Выбор вектора
В последнее время для создания библиотек кДНК вместо
плазмидных векторов начинают все шире применяться векторы
на основе фага лямбда, что объясняется следующими причи-
нами.
1. Лямбда-векторы обеспечивают очень высокую эффектив-
ность клонирования благодаря упаковке in vitro. В на-
стоящее время можно приобрести превосходные экстрак-
ты, улучшающие упаковку. Поскольку эффективность
клонирования кДНК зависит от эффективности упаковки,
246
Глава 9
при наличии очень малых количеств кДНК следует поль-
зоваться лучшими из имеющихся экстрактов.
2. Скрининг фаговых бляшек на нитроцеллюлозных фильт-
рах с помощью зондов нуклеиновых кислот является бо-
лее чувствительным и воспроизводимым методом, чем
гибридизация бактериальной колонии. Это обстоятельство
становится особенно существенным при проведении
дифференциального гибридизационного анализа.
3. Если используется иммунный вектор, несущий вставку,
такой как XgtlO или NM 607, нерекомбинантные молеку-
лы могут быть элиминированы из библиотеки введением
в соответствующий штамм Escherichia coli.
Для создания библиотек кДНК выбран фаг XgtlO [23].
Это хорошо размножающийся фаг, образующий крупные одно-
родные бляшки, что немаловажно для дифференциального скри-
нинга. С помощью этого вектора можно клонировать любые
фрагменты кДНК размером до 7,6 т. п. н. Большинство эукари-
отических мРНК относится именно к этому классу. Если тре-
буются копии очень длинных РНК, таких, как РНК ламина В2
(8 т. п.н.), лучше использовать другой фаговый вектор —
NM1149 [24], способный включать последовательности боль-
шего размера. В тех случаях, когда доступен только метод
скрининга, использующий зонды антител, узнающих белок, за-
кодированный кДНК, молекулы кДНК должны быть клониро-
ваны в таком фаговом векторе, как Xgtll [23].
4.2. Синтез и клонирование двухцепочечных кДНК
Для синтеза двухцепочечных кДНК предложено несколько
методик [И, 23, 25, 26]; можно пользоваться любой из них.
Мы рекомендуем метод Уотсон и Джексона [11], включающий
оригинальный способ синтеза второй цепи ДНК, который раз-
работан Окаямой и Бергом [27]. Он заключается в том. что
гибридный продукт мРНК/кДНК. полученный в результате
реакции синтеза первой цепи, обрабатывается РНКазой Н,
чтобы получить короткие фрагменты РНК, служащие затравкой
для ДНК-полимеразы 1 Е. coli, которая будет синтезировать
вторую цепь кДНК и замещать РНК на ДНК. Цепи сшиваются
ДНК-лигазой Е. coli. Этот подход благоприятствует синтезу
полноразмерной кДНК и снижает возможность потерь и арте-
фактов (таких, как присутствие кДНК, неколинеарных мРНК в
области б'-конца). Эти проблемы часто возникают при исполь-
зовании альтернативного подхода, включающего обработку
нуклеазой S1 [28], специфичной к однопепочечпым молекулам.
На рис. 9.1 представлена схема синтеза и клонирования
кДНК. Если количество исходных полиаденилированных РНК
меньше 1 мкг, в протокол следует ввести две модификации.
Библиотека кДНК для предимплантацнонного эмбриона мышн
247
КЛОНИРОВАНИЕ кДНК в Xgt 10
МРНК 5’-
<5*
Гибрид мРНК/кДНК
Репарация
ds-кДНК
---------Ап 3'
Обратная транскриптаза
oligo 18
---------An 3'
--------- Tn 5'
ДНК попимераза I, РНКаза H,
Лигаза Е.соП
---------Тп 5'
---------Ап 3'
ДНК полимераза Т4
— Тп
- ~~ Ап
Метилаза Есо R1
М
Линкеры £coR1
IHIIIII , 11ЛППП
М'
ООО 0 0000-1- £
£
Xgt 10
R1
EcoRl
М'
EcoRI
м
Удаление
линкеров
Лигирование
|Упаковка
-----
Рекомбинантный Xgt 10
Рис. 9д Синтез и клонирование двухцепочечных кДНК в векторе бактериофа
га XgtlO. Детали описаны в работе [1Ц.
1. Как уже отмечено в разд. 3.4, вирусный носитель (CPMV)
должен быть добавлен к образцу РНК перед отделением
poly (А)+-РНК.
2. Аликвота РНК CPMV в 1 мкг должна быть добавлена
к двухцепочечной ДНК с присоединенными линкерами,
перед хроматографией на колонке с ультрагелем (удаля-
ющей избыток линкеров), чтобы предотвратить потери
кДНК, обусловленные неспецифическим связыванием с
матриксом колонки.
Все манипуляции с ДНК необходимо проводить в силикони-
рованных микроцентрифужных пробирках. Кроме того, на ста-
дии колоночной хроматографии (для удаления линкеров fcoRl,
рис. 9.1) и колонка, и стекловолокнистая прокладка должны
248
Глава 9
Рис. 9.2. Гибридизационный паттерн, полученный в результате гибридизации
нитроцеллюлозного фильтра, несущего 65 бляшек библиотеки кДНК ооци-
та/CPMV (1 : 19), с радиоактивным зондом CPMV. Число гибридизоваиных
бляшек — 40, что очень близко к ожидаемому — 46 (65 минус 16 нерекомби-
нантных бляшек фона н 3 ооинт-спецнфическнх клона, см. работу [19]).
быть силиконированы. Для этого пробирки и стеклянное обору-
дование несколько раз споласкивают в 2%-ном растворе дихло-
родиметилсилана в 1,1,1-трихлороэтане (поставщики Hopkin
and Williams), а затем в стерильной дистиллированной воде,
после чего автоклавируют. Не забывайте о технике безопасно-
сти: этот раствор токсичен.
Упаковка гибридной ДНК фага in vitro должна производить-
ся в соответствии с указаниями изготовителя (Amersham, Giga-
pack), посев фага следует проводить в соответствии с рекомен-
дациями работы [23]. Для ценных образцов упаковочные
экстракты необходимо предварительно проверить.
Чтобы оценить представительность библиотек в отношении
интересующей нас РНК, следует предусмотреть ряд контроль-
ных процедур. Известно, что среди фаговых бляшек неизбежно
Библиотека кДНК для предимплантационного эмбриона мыши 249
присутствуют нерекомбинантные (фон). Для определения их
частоты необходимо лигировать вектор Xgt. Число клонов
CPMV можно оценить путем скрининга библиотеки радиоактив-
ным зондом, полученным нз РНК CPMV с использованием
обратной транскриптазы и случайных затравок из тимуса телен-
ка (см. табл. 9.3, В). Для подсчета эмбрионоспецифическпх
рекомбинантов необходимо из общего числа бляшек вычесть то
количество, которое соответствует фону и CPMV. На рис. 9.2
представлен нитроцеллюлозный фильтр, приготовленный из
библиотеки ооцита мыши и гнбридизованный с зондом CPMV
Исходя из среднего размера обеих РНК CPMV (4,7 т. п. н) (он
более чем вдвое превышает среднюю эукариотическую мРНК),
число молекул CPMV на грамм должно быть меньшим такового
для эмбриональной РНК, следовательно, число эмбриональных
клонов в действительности выше.
В следующем разделе описаны различные процедуры, кото-
рыми можно пользоваться для выделения интересующих клонов
из библиотеки кДНК.
5. Скрининг библиотеки
Обычно скрининг библиотек кДНК осуществляется гибриди-
зацией радиоактивных проб (зондов) нуклеиновой кислоты с
фаговой ДНК, нанесенной на нитроцеллюлозные или нейлоно-
вые мембранные фильтры. Гибридизация на фильтрах детально
описана в работе [26]. Наше внимание будет сосредоточено на
разнообразных зондах и методах мечения, а также на альтерна-
тивных способах выделения определенных клонов.
5.1. Тест на репрезентативность библиотеки
Очень важно убедиться в том, что эмбрионоспецифические
последовательности присутствуют в библиотеке. Это может быть
сделано путем скрининга одноцепочечной кДНК с высокой
удельной активностью, приготовленной из мРНК, которая была
использована для создания библиотеки. Поскольку данная про-
цедура требует очень ценную эмбриональную РНК, рекоменду-
ется сделать предварительный скрининг зондом, кодирующим
известную РНК, экспрессируемую в большинстве типов клеток
пли иа той стадии эмбриогенеза, на которой получен материал
библиотеки. На наш взгляд, целесообразно использовать зонды
для таких интенсивно экспрессируемых мРНК, как р-актиновая.
Скрининг клонами для редких РНК также информативен
По его результатам можно судить о репрезентативности (пред-
ставительности) библиотеки. На рис. 9.3 мы видим сигналы
гибридизации, полученные в результате зондирования одной
250
Глава 9
Рис. 9.3. Тест на представительность библиотеки. Пара фильтров с
—3000 бляшек библиотеки кДНК ооцита/CPMV была гибридизована с инсер-
ционным (вставочным) фрагментом рРЕ386, радиоактивномеченным с по-
мощью полимеризации со случайной затравкой (табл. 9.3.Л). Этот клон кДНК
содержит 1,1 т. п. н. последовательностей ламинина В1 [29]. Обнаружен 1 поло-
жительно гибридизованный клон. Высокий фон является следствием гибриди-
зации poly(А)-хвостов в клонах библиотеки с poly(А)-последовательностями
зонда.
пары фильтров из библиотеки ооцита мыши (высеянной при
высокой плотности) клоном кДНК, содержащим 1,1 т. п. н. 3'-
концевых последовательностей ламинина В1 [29]. Полипептид
В1 составляет примерно 0,06% белка ооцита [30]. Относитель-
ное содержание соответствующей РНК не известно, однако
число В1-гибридизующихся клонов ооцита примерно в 10 раз
ниже числа актин-гибридизующихся клонов, что хорошо согла-
суется с результатами экспериментов, выполненных на уровне
белков.
5.2. Скрининг библиотеки для выявления
специфических последовательностей
Существует пять основных подходов к скринингу библиотек
кДНК, в трех из них используются зонды нуклеиновых кислот.
Методики приготовления зондов представлены в табл. 9.3.
5. 2.1. Клонированные зонды
Зонды, состоящие из предварительно клонированных после-
довательностей, часто применяются для выделения специфиче-
ской последовательности из библиотеки кДНК разных видов
или разных стадий развития. Такие ДНК-зонды могут быть
Таблица 9.3. Приготовление зондов из радиоактивной НК
А. Полимеризация с использованием случайной затравки
Этот метод используется для мечения ДНК, плазмид, содержащих интере-
сующую последовательность, или специфических фрагментов ДНК, вырезан-
ных из агарозных гелей (LGT)1* (без предварительной очистки ДНК). В слу-
чае клонов кДНК инсерционный фрагмент должен быть вырезан до ме-
чения.
1. После рестрикции и электрофореза клонированной ДНК2* вырежьте из
геля нужную полоску, взвесьте этот гелевый фрагмент и поместите в про-
бирку (Eppendorf).
2. Растворите гель и денатурируйте ДНК, поместив пробирку в кипящую во-
ду на 10 мин.
3. Инкубируйте при 37 °C в течение 10—60 мин. Проведите реакцию мечения,
смешивая компоненты в указанной последовательности:
10,0 мкл буфера для мечения олигонуклеотидов3*
2,0 мкл бычьего сывороточного альбумина (10 мг/мл)
35,5 мкл раствора ДНК/агароза (не более 30 мкл от количества пунк-
та «2» + бидистиллированная Н2О)
2,0 мкл [32P]-dCTP (20 мкКи) (Amersham, уд. акт. 400 Ки/ммоль)
0,5 мкл фрагмента Кленова (2 единицы) (NBL Enzyme Ltd).
4. Инкубируйте при комнатной температуре в течение 5—16 ч и затем до-
бавьте 200 мкг останавливающего раствора [20 мМ NaCl; 20 мМ трис-НС1
pH 7,5; 2 мМ ЭД ТА; 0,25% (в/о) ДСН; 1 мкМ dCTP].
5. Проведите экстракцию фенолом один раз, затем водный слой трижды
экстрагируйте эфиром, добавьте 10 мкг ДНК сонированной спермы лосо-
ся и проведите экстракцию этанолом, прибавив 3 объема 96%-ного (о/о)
этанола и 0,1 объема 2М раствора ацетата натрия.
6. Отцентрифугируйте в течение 15 мин в микроцентрифуге Eppendorf, уда-
лите супернатант, высушите осадок и ресуспендируйте его в 200 мкл ТЭ11,
7. Прокипятите 5 мин перед гибридизацией.
Б. а'-концевое мечение синтетическими олигонуклеотидами
З'-концы олигонуклеотидов могут быть помечены с помощью полинуклеотид-
киназы Т4 и [у-32Р]-АТР. Этот энзим катализирует перенос ^-фосфата от
АТР к 5'-концам олигонуклеотида. Приготовьте 50 мкл метящего раствора
следующим образом.
1 Смешайте
1 мкг олигонуклеотида
5 мкл десятикратного буфера киназы5*
50 пмоль [у-32Р]-АТР (150 мкКи) (Amersham, уд. акт. 300 Ки/ммоль)
10 единиц полинуклеотидкиназы Т46>,
довести объем до 50 мкл бидистиллироваиной водой.
2 . Инкубируйте при 37 °C в течение 30 мин.
3 Добавьте 2 мкл 0,5 М ЭД ТА.
-1. Экстрагируйте один раз смесью фенол: хлороформ (1:1).
5. Удалите невключеиную [ч-32Р]-АТР путем хроматографии на колонке се-
фадекса G50 в 10 м.М трис-НС1 pH 7.5; 100 мМ NaCl; 1 мМ ЭДТА. Собе-
рите фракции, содержащие первый пик с, меченым олигонуклеотидом
В. Мечеиие олигонуклеотидов методом заполнения
Альтернативный способ заключается в мечении олигонуклеотидов путем син-
теза комплементарного олигомера с помощью короткого (обычно 8 осно-
ваний) олнгонуклеотида-затравки. Следует очень внимательно подойти
выбору радиоактивного нуклеотида, чтобы добиться наибольшего числа
Продолжение табл. 9.3
меченых оснований. Это в большой степени зависит от последовательности
олигонуклеотида. Проведите реакцию следующим образом.
1. Отжиг
1 мкл олигонуклеотида (30 нг) — матрицы
1 мкл олигонуклеотида (10 нг)—затравки
10 мкл ТМ7’-буфера.
2. Инкубируйте при 65 °C в течение 5 мин.
3. Охладите при комнатной температуре в течение 5 мин, затем кратко-
временно отцентрифугируйте.
4. Добавьте по 1 мкл каждого из трех холодных растворов dNTP (при
1 мМ) н 4 мкл (40 мкКи) (Amersham, уд. акт. 3000 Ки/ммоль) послед-
него «горячего» основания — [32P]-NTP.
5. Добавьте 4 единицы фрагмента Кленова (ДНК-полимеразы I).
6. Инкубируйте иа льду в течение 2 ч, затем добавьте 1 мкл того же само-
го, ио «холодного» основания (при 1 мМ) и инкубируйте иа льду 45 мин.
7. Добавьте 5 мкл деионизированного формамида, нагревайте в течение
5 мин при 65 °C и затем проводите электрофорез в 10%-ном разделяю-
щем акриламидном геле в течение приблизительно 3 ч или до тех пор,
пока краситель бромфеиоловый синий (загруженный в параллельную
лунку) не окажетси на расстоянии «10 см от дна.
8. Заверните гель в липкую ленту, сверху поместите рентгеновскую пленку
(при красном свете) и экспонируйте в течение 2 мии.
9. Проявите пленку, совместите ее с гелем и аккуратно вырежьте меченую
полосу.
10. Снова поместите гель на пленку, чтобы проверить правильность выреза-
ния полосы. Важно выделить только ту область геля, которая содержит
меченую полосу, чтобы избежать примеси немеченой олигонуклеотидиой
матрицы, снижающей эффективность гибридизации.
11. Внесите фрагмент геля в 500 мкл 1 мМ ЭДТА, содержащей 20 мкг ДНК
сонированной спермы лосося. Кратковременно гомогенизируйте, элюи-
руйте в течение ночи, добавьте непосредственно к гибридизационному
раствору.
Г. Одноцепочечная кДНК из популяций специфической РНК
Процедура получения такой кДНК представляет собой модификацию реак-
ции синтеза первой цепи для клонирования кДНК с использованием случай-
ных затравок, приготовленных из сонированной денатурированной ДНК ти-
муса теленка (Pharmacia).
1. Высушите под вакуумом по 50 мкКи (уд. акт. 3000 Ки/ммоль) ОСТР п
dTTP плюс 1 мкл актиномицина D8’.
2. Добавьте
5,0 мкл пятикратного буфера обратной транскриптазы9’
2,5 мкл случайной затравки (2 мг/мл)
0,5 мкл дитиотрейтола (1,0 М)
1,0 мкл dTTP (0,1 мМ)
1,0 мкл dCTP (0,1 мМ)
2,5 мкл dGTP (5,0 мМ)
2,5 мкл dATP (5,0 мМ)
1,0 мкл РНКазина (30 единиц)10’
1,0 мкл обратной транскриптазы (22 единицы)"’.
Доведите объем до 25,0 мкл бидистиллированной водой.
3. Инкубируйте при 37—42 “С в течение 2 ч.
252
Библиотека кДНК для предимплантационного эмбриона мыши 2аЗ
Продолжение табл. 9.3
4. Удалите иевключенные нуклеотиды путем хроматографии иа сефадексе
G-50. Кипятите пробу н течение 5 мни, прежде чем добавить к гибриди за-
ционному раствору.
') Поставщики SeaKem, Sigma.
2) См. работу (26).
3) Буфер для мечения олигонуклеотидов состоит из раствора О: 1,25 М трис-НСЛ
pH 8.0, 0,125 М MgCh; раствора А: 1 мл раствора O+I8 мкл 2-меркаптоэтанола+5 мгл
(1АТР+5 мкл dTTP+5 мкл dGTP (каждый 0,1 М в бидистиллированной Н2О); раствора Г>
2 М HEPES (Sigma) pH 6,6; раствора В: гексадезоксирибонуклеотиды (Phaimacia) в 1»
(см. сноску 4) при ОП 90 едиииц/мл. Смешайте растворы А: Б: В в отношении
100 : 250 : 150.
7) ТМ-буфер: 0.1 М трис-НС1 pH 8,0; 0,1 М MgCl2-
5) Десятикратный киназный буфер: 0,5 М трис-НС! pH 7,6; 0.1 М MgCl2; 50 мМ ди-
знотрейтол; 1 мМ спермидин; 1 мМ ЭДТА.
в) Поставляется BRL. Одна единица полинуклеотидкиназы Т4 переносит 1 пмочь
V фосфата от АТР к 5'-концу ДНК, обработанной микрококковой нуклеазой в течение-
30 мин при 37 °C.
7) ТМ-буфер: 0,1 М трис НС1 pH 8,0; 0.1 М MgCI2,
’) Поставляется фирмой Calbiochem. Исходный раствор 1 мг/мл в 80%-ном (о'о) де-
ноле.
9) Пятикратный буфер ОТ: 250 мМ трис-НС1 pH 8.3 (при 42 °C); 40 мМ МцО .
250 мМ КС1.
,0) Поставляется фирмой Biotech. 1 единица РНКазина ингибирует на 50% активность
5 нг рибонуклеазы А.
’*) Поставляется NBL Enzymes Ltd. 1 единица обратной транскриптазы включая г
1 нмоль dTMP в кислотонерастворимый продукт за 10 мин при 37 °C.
помечены либо ник-трансляцией [31], либо полимеризацией с
использованием случайной затравки [32]. Последней процедуре
отдается предпочтение в связи с тем, что она дает зонд с более
высокой удельной активностью. В случае межвидовой гибриди-
зации требования к отмывке (после процедуры гибридизации)
не такие жесткие.
5.2.2. Олигонуклеотидные зонды
Эти зонды используются для выделения индивидуальных чле-
нов мультигенных семейств или в тех случаях, когда известна (ча-
стично или полностью) только первичная структура белка.
Обычно достаточно олигомера размером 20 оснований, гомоло-
гичного интересующей последовательности. Он может быть,
помечен либо с 5'-конца с помощью полинуклеотидкиназы
(табл. 9.3,Б), либо методом «заполнения» с использованием
олигомера длиной в 8 оснований в качестве затравки для синте-
за комплементарной цепи (табл. 9.3,В).
5.2.3. Одноцепочечные кДНК-зонды
В тех случаях, когда возникает необходимость выделить
последовательности, уровень экспрессии которых на определен-
ной стадии развития выше, чем на других, следует воспользо-
254
Глава 9
Рис. 9.4. Фильтры из библиотеки кДНК клеток эмбриональной карциномы
(ЭК), несущие ~400 бляшек. Гибридизоваиы с одиоцепочечными зондами
кДНК (табл. 9.3, В), транскрибированными с РНК недифференцированных
(- РК) илн дифференцированных ( + РК) клеток ЭК. Стрелками отмечены
бляшки, наиболее активно гибридизовавшиеся с зондом недифференцирован-
ных клеток. РК — ретиноевая кислота.
ваться техникой дифференциального скрининга. Для этого
потребуются зонды кДНК с высокой удельной активностью
(приготовленные из двух популяций РНК, которые сравнивают)
п фаг, высеянный при низкой плотности (~400 бляшек на чаш-
ку 90 мм). Одноцепочечные зонды кДНК синтезируются в
соответствии с прописью табл. 9.3, Г, удельная активность
должна превышать 108 имп/мин/мкг. Типичный паттерн гибриди-
зации представлен на рис. 9.4. Эта пара фильтров с отпечатка-
ми из библиотеки кДНК клеток эмбриональной карциномы
(ЭК) была гибридизована с зондами, полученными для РНК
дифференцированных (дифференцировку индуцировали ретиное-
вой кислотой) и недифференцированных клеток ЭК- Между
двумя фильтрами четко видны различия; некоторые клоны эк-
спрессированы на более высоком уровне в недифференцирован-
ных клетках, для экспрессии других характерна обратная за-
кономерность. Описанный нами подход можно применить к
скринингу библиотек эмбриона, когда имеется достаточное
количество материала. Заметим, что зонды, полученные при ис-
пользовании случайной затравки, имеют в этом случае преиму-
щество перед кДНК с oligo (dt) -затравками. Во-первых, с их
помощью может быть получена более высокая удельная актив-
ность (~ в 10 раз), и во-вторых, могут быть обнаружены все
Библиотека кДНК для предимплаитациоииого эмбриона мыши 255-
гомологичные клоны (из-за отсутствия предпочтения З'-конце-
вых последовательностей, как в случае зондов с oligo(dt)-
затравкой).
5.1.4. Гибридный блок
При необходимости выделить клон, кодирующий белок,
который был идентифицирован путем трансляции in vitro,
используется процедура гибридного блока трансляции [33].
Ее успех зависит от способности мРНК к отжигу с клоном
комплементарной ей кДНК- Предложенные в последнее время
усовершенствования методики гибридного блока (см. детали в
работе [34]) делают ее более приемлемой для анализа систем
с ограниченным материалом, чем методы гибридной селекции и
трансляции мРНК.
5.2.5. Гибридизация in situ
Существует возможность идентифицировать ткане- или
клетка-специфическую экспрессию мРНК, кодируемой индиви-
дуальным клоном кДНК, посредством гибридизации in situ
(см. гл. 10). Эта техника вполне пригодна и для изучения пат-
терна экспрессии интересующей нас клонированной последова-
тельности. Она с успехом применялась в отношении ранних
стадий развития мыши [35]. Наиболее очевидное преимущество
этого метода перед процедурами скрининга заключается в том,
что он подразумевает использование небольшого числа эмбрио-
нов. Гибридизация in situ служит незаменимым инструментом
при изучении экспрессии генов у эмбрионов Drosophila. Можно
надеяться, что дальнейшее усовершенствование техники экспери-
мента даст возможность использовать гибридизацию in situ па
срезах мышиного эмбриона в качестве процедуры скрининга
клонов кДНК-
Благодарности
Мы выражаем благодарность Джиму Джексону за сотруд-
ничество и большой вклад в разработку методики клонирования
кДНК- Мы благодарны Дж. Дэвису и Д. Ломоносову за пре-
доставленную для работы РНК CPMV. Большое спасибо нашим
коллегам в Кэмбридже за их помощь в сборе ооцитов и бла-
стоцист мыши. Мы благодарим Мартина Джонсона, Питера
Ригби и Кейт Уиллисон за плодотворные обсуждения.
256
Глава 9
Литература
1. Johnson М. Н., McConnell J., Van Blerkom J. (1984). J. Embryol. Exp. Mor-
phol., 83 (suppl.), 197.
2. Howlett S. R„ Bolton V. N. (1985). J. Embryol. Exp. Morphol., 87, 175.
3. Piko L„ Clegg R. B. (1982). Dev. Biol., 89, 362.
4. Johnson M. H. (1981). Biol. Rev., 56, 463.
5. Flach G., Johnson M. H., Braude P. R., Taylor R. A. S„ Bolton V. Ar. (1982).
EMBO J., 1, 681.
6. Clegg R. B., Piko L. (1983). J. Embryol. Exp. Morphol., 74, 169.
7. Bolton V. N., Oades P. J., Johnson M. H. (1984). J. Embryol. Exp. Morphol.,
79 139
8. Giebelhaus D H., Heikkila J. J., Schultz G. A. (1983). Dev. Biol., 98, 148.
9. Howlett S. R. (1986). Cell, 45, 387.
10. Martin G. R. (1980). Science, 209, 768.
11. Watson C. J., Jackson J. F. (1985). In: DNA cloning — A Practical Approach.
Glover D. M. (ed.), IRL Press, Oxford, Vol. I, p. 79.
12. Clarke L„ Carbon J. (1976) Cell, 9, 91.
13. Williams J. G. (1977). Cell, 17, 903.
14. Rougeon F„ Mach B. (1977). J. Biol. Chem., 252, 2209.
15. Brulet P., Condamine H., Jacob F (1985) Proc. Natl Acad. Sci. USA, 82,
2054.
16. El Manna M. M., Bruening G. (1973). Virology, 56, 198.
17. Lomonossof G. P„ Shanks M. (1983). EMBO J., 2, 2253.
18. Van Weezenbeek P., Verver J., Harmsen J., Vos P„ Van Rammen A. (1983).
EMBO J„ 2, 941.
19. McConnell J., Watson C. J. (1986). FEBS Lett., 195, 199.
20. Braude P. R., Pelham H. R. B. (1979). J. Reprod. Fertil., 56, 153.
21. Glover D. M, (1985). DNA Cloning — A Practical Approarch. Volume I,
IRL Press, Oxford. [Имеется перевод: «Клонирование ДНК. Методы». Под
ред. Д. Гловера: Пер. с англ. — М.: Мир, 1988.]
22. Enquist L., Sternberg N, (1979). In: Methods in Enzymology. Wu R. (ed.),
Academic Press, New York, Vol. 68, p. 281.
23. Huynh T. V., Young R. A., Davis R. IV. (1985). In: DNA Cloning — A Prac-
tical Approach. Glover D. M. (ed.), IRL Press, Oxford and Washington, DC.
Vol. I, p. 49.
24 Murray N. E. (1983). In: Lambda II. Cold Spring Harbor Laborator}' Press.
New York.
25. Gubler U., Hoffman B. J. (1983). Gene, 25, 263.
26. Maniatis T., Fritsch E, F„ Sambrook J. (1982). Molecular Cloning — A La-
boratory Manual. Cold Spring Harbor Laboratory Press, New York. [Име-
ется перевод: Маниатис T., Фрич Э., Сэмбрук Дж. «Методы генетической
инженерии. Молекулярное клонирование»: Пер. с англ.— М.: Мир, 1984.]
27. Okayama Н„ Berg Р. (1982). Mol. Cell. Biol., 2, 161.
28. Land H., Grez M., Hauser H., Lindenmaier W., Schutz G. (1981). Nucleic
Acids Res., 9, 2251.
29. Barlow D. P., Green N. M., Rurkinen M., Hogan B. L. M. (1984), EMBO J.,
3 2355
30. Cooper A. R„ McQueen H. A. (1983). Dev. Biol., 96, 467.
31 Rigby P. W. J., Dieckman M„ Rhodes C., Berg P. (1977), J. Mol. Biol..
113, 237.
32 Feinberg A. P„ Vogelstein B. (1984). Anal. Biochem., 137, 266.
33 Paterson В. M„ Roberts B. E„ Ruff E. L. (1977). Proc. Natl. Acad. Sci.
USA, 74, 4370.
34. Minshull J., Hunt T. H. (1986). Nucleic Acids Res., 14, 6433.
35 Vasseur M., Condamine H., Duprey P. (1985). EMBO J., 4, 1749.
ГЛАВА 10
ГИБРИДИЗАЦИЯ IN SITU
С мРНК НА ГИСТОЛОГИЧЕСКИХ СРЕЗАХ
Ребекка Хаффнер и Кейт Уиллисон1
1. Введение
Возможность гибридизации проб (зондов) нуклеиновых
кислот с комплементарными им ДНК- или РНК-последователь-
ностями на цитологическом препарате обусловила появление
метода гибридизации in situ. Этот метод впервые был приме-
нен для локализации последовательностей клонированной ДНК
на индивидуальных хромосомах [1]. Гибридизация in situ вклю-
чает инкубацию меченых РНК- или ДНК-зондов с гистологиче-
ским срезом или клеточным препаратом и последующее удале-
ние негибридизованного или неспецифически связанного зонда
путем отмывки. Положение гибридизованного зонда определя-
ется либо с помощью радиоавтографии (в случае радиоактивно-
го зонда), либо с использованием иммуногистохимической тех-
ники (в случае зондов, меченных биотином).
Гибридизация in situ в последнее время стала рутинным',
методом картирования клонированных последовательностей
ДНК в определенных участках метафазных хромосом (.[2], а
также гл. 5, разд. 6). Этот метод все чаще применяется и для
выявления нуклеиновых кислот на гистологических срезах. При
изучении межвидовых химер мыши клональный анализ клеточ-
ных линий может быть осуществлен в тканях эмбриональных
или взрослых химерных животных с помощью зондов, специ-
фичных для клеток каждого из родительских организмов [3, 5].
Гибридизация с РНК на гистологических срезах позволяет ло-
кализовать последовательности мРНК на уровне отдельных
клеток. Можно утверждать, что эта методика стала мощным
орудием для изучения регуляции генов в ходе развития Drosop-
hila melanogaster [6], а в последнее время и мышей [7].
В этой главе описаны различные процедуры гибридизации
in situ с клеточными РНК в ткани и локализация этих РНК с
помощью радиоавтографии. Протоколы гибридизации in situ
с метафазными хромосомами здесь не приводятся, детальное
описание этой методики можно найти в работе [8], а также в
разд. 6 гл. 5 этой книги.
1 Rebecca Haffner and Keith. Willison. Institute of Cancer Research, Chester
Beatty Laboratories, Fulham Rood, London SW3 6YB, UK.
17—171 257
260
Глава 10
ления экспрессии индивидуальных членов мультигенных се-
мейств— иммуноглобулинов или антигенов класса I главного
комплекса гистосовместимости. Кроме того, короткие зонды да-
ют возможность идентифицировать точечные мутации, присутст-
вующие в активированных онкогенах или в их транскриптах в
ткани.
2.2. Выбор метки
Для мечения зондов при гибридизации наиболее часто ис-
пользуется тритий благодаря исключительно низкому уровню
энергии излучаемых им [3-частиц. Длина пробега частиц в ра-
диоавтографической эмульсии составляет менее 1 мкм, поэтому
положение зерна серебра на автографе точно указывает лока-
лизацию радиоактивной молекулы. Однако из-за низкой удель-
ной активности и низкой энергии излучения трития для получе-
ния заметного сигнала требуется экспозиция в течение несколь-
ких недель или даже месяцев.
Эту проблему можно обойти, используя вместо трития метку
35S. Зонды с высокой удельной активностью дают хорошее раз-
решение на уровне клетки. Несмотря на то что длина пробега
излучений в этом случае больше, чем у трития, и составляет
«20 мкм, радиоавтографическое разрешение можно повысить
до 4 мкм, используя тонкие эмульсии. Для получения четких
результатов потребуются дни, а не недели. Еще одна проблема
заключается в том, что применение зондов, меченных 3SS,
повышает уровень фонового мечения, однако система РНК-зон-
дирования позволяет использовать РНКазу для удаления негиб-
ридизованного зонда, разрешая тем самым и эту проблему.
В заключение заметим, что для точной локализации РНК
в клетке вполне подходят зонды, меченные тритием, но для
выявления в клеточной популяции клеток с соответствующими
молекулами лучше использовать 35S. Некоторые исследователи
включают следовые количества [32P]-UTP в реакцию синтеза
зондов, меченных 3Н или 3SS. В этом случае на срезах можно
обычным путем изучать гибридизацию с помощью рентгеновской
пленки, а затем на те из них, которые покажут положительную
гибридизацию, нанести жидкую эмульсию для лучшего разре-
шения.
Для локализации гибридизации in situ были разработаны и
нерадиоавтографические приемы исследования. Они подразу-
мевают использование зондов, меченных биотином, который вы-
является с помощью биотин-специфических антител, конъюги-
рованных с флуоресцентным или ферментным реагентом [14].
По мнению некоторых исследователей, биотинилированные зон-
ды обеспечивают большую чувствительность по сравнению с
Гибридизация in situ с мРНК на гистологических срезах 261
радиоактивными [5], однако такая точка зрения разделяется
далеко не всеми [8]. Очевидное преимущество зондов, меченных
биотином, заключается в том, что с их помощью сигнал гибри-
дизации можно регистрировать в тот же самый день, когда
сделана отмывка. Тем не менее все опубликованные результаты
гибридизации in situ с РНК эмбрионов Drosophila были полу-
чены на основе радиоавтографическбй техники. Детальное опи-
сание гибридизации с помощью биотин-меченвдх зондов приве-
дено в работе [15]. \-'г
-К?;
2.3. Приготовление радиоактивных зондов \-
Для того чтобы синтезировать одноцепочечные РНК-зонды
с использованием векторов Bluescript или SP6, нужная плаз-
мида должна быть переведена в линейную форму с помощью
фермента, расщепляющего ниже по ходу считывания от вставки
в сайте клонирования. Если не требуется зонд полной длины,
укороченный зонд можно получить с помощью рестрикта-
зы, разрезающей в пределах вставочной последовательности.
Продукт реакции экстрагируется фенолом, ДНК осаждается
этанолом и используется в качестве матрицы для транскрипции.
Совершенно необходимое условие получения зонда — такая
ориентация вставки, при которой синтезируемая цепь будет
комплементарна соответствующей молекуле мРНК- Для харак-
теристики синтезируемой молекулы используют метод нозерп-
блот (Northern blot analysis) (рис. 10.1).
Все реакции по получению РНК-зондов очень чувствительны
к рибонуклеазе, поэтому важно автоклавировать пробирки и
пипетки. Для приготовления всех буферов нужно использовать
бидистиллированную воду, обработанную 0,1%-ным диэтилпиро
карбонатом (ДЭПК). Работать следует только в перчатках,
чтобы не внести рибонуклеазу.
Процедура синтеза радиоактивного одноцепочечного РНК-
зонда представлена в табл. 10.1.
2.4. Расчет удельной активности зонда
Масса синтезированного зонда и его удельная активность
могут быть рассчитаны по процентному содержанию включен-
ных нуклеотидов. Включение определяется путем осаждения
трихлоруксусной кислотой (ТХУ). Фильтры, содержащие обра-
зец зонда (табл. 10.1, пп.3 и 6), отмойте последовательно
в 5 %-ной ТХУ в 5 %-ной ТХУ с 0,5 %-ным тетранатрийпирофос-
фатом и в 90%-ном этаноле (по 1 мин). Высушите и опреде-
лите радиоактивность в сцинтилляционном счетчике. Вклю-
чение обычно составляет от 30 до 70%. Масса получен-
264
Глава 10
2. Нейтрализуйте образцы внесением ацетата натрия pH 6,0
до 0,1 мМ и ледяной уксусной кислоты до 0,5% (о/о).
Нужное для гидролиза время рассчитывается по формуле
_ Lp — Lf
kL0- Lf
где t — время в мин, Lo — первоначальная длина фрагмента в
т. п. н., Lf — конечная длина фрагмента в т. п.н., k — постоян-
ная скорости гидролиза, составляющая около 0,11 т. п. н./мин.
Длина зонда может быть измерена прямо путем электрофо-
реза в 4%-ном агарозном геле (Nusieve, Miles Scientific, UK),
содержащем 0,1% ДСН, или в 10%-ном полиакриламидном геле
с 8 М мочевиной при 60 °C.
Укороченные зонды можно также получить, обрабатывая
плазмиду ферментом, который разрезает вставку в сайте, рас-
полагающемся на расстоянии требуемого числа оснований ниже
по ходу считывания от сайта инициации. Следует отметить, что
более короткие зонды включают радиоактивные нуклеотиды
менее эффективно.
3. Подготовка стекол
Наиболее существенная проблема, которая встает при при-
менении гибридизации in situ к тканям млекопитающих,—
вероятность потери среза с предметного стекла на некоторых
стадиях процедуры, в частности, при отмывке негибридизован-
ного зонда. Различные ткани обладают разной степенью адге-
зии к стеклу. Судя по нашему опыту, срезы семенников мыши
остаются в одном положении в течение всего воздействия. Сре-
зы мышиной печени или эмбрионы, напротив, сохраняются хуже.
В ряде случаев для удержания срезов на стекле бывает доста-
точно покрыть их раствором желатины с хромовыми квасцами,
однако наилучшая адгезия обеспечивается нанесением полили-
зина.
Тщательно очистите стекла в хромпике или спирте, затем
покройте слоем поли-Ь-лизина в концентрации 100 мкг/мл.
Наиболее эффективный способ покрытия стекол заключается
в использовании обычного распылителя краски (Badger Air
Brush Со., IL), создающего на поверхности стекол тонкий и ров-
ный слой. Другой способ — погружение стекол в сосуд Коплина
с этим раствором на 3 сек и затем высушивание в вертикаль-
ном положении на специальной подставке. Стекла, покрытые
желатиной, готовятся погружением в раствор 0,5 г желатины,
0,5 г хромовых квасцов (растворенных при 60 °C в 200 мл ди-
стиллированной воды).
Гибридизация in situ с мРНК на гистологических срезах 265
Таблица 10.2. Подготовка ткаии для получении парафиновых срезов
Число воз- действий1) Раствор Время (мин) Температура ГС) Цель воздействия
7 Изопропанол 10 40 Дегидратация
1 Ксилол 14 40 Промежуточная
1 Ксилол 20 40 среда
1 Парафин 14 60 Пропитывание
3 Парафин 20 60
’) Переносы в свежий раствор.
4. Фиксация и приготовление срезов
Парафиновые срезы хорошо сохраняют клеточную морфоло-
гию, но для гибридизации зондов in situ недостаточно чувстви-
тельны. Замороженные образцы обычно дают более сильный
сигнал, однако уровень разрешения морфологии клетки, обеспе-
чиваемый такими срезами, не всегда достаточен для окончатель-
ных выводов. Например, замороженные срезы не дают адекват-
ного разрешения, позволяющего различать промежуточные типы
клеток при сперматогенезе. Таким образом, исследователю в
зависимости от цели эксперимента необходимо выбирать между
четкой морфологией и гибридизационной чувствительностью.
4.1. Парафиновые срезы
1. Немедленно погрузите отпрепарированную ткань в 10 %-
ный формалин, 150 мМ хлорид натрия и фиксируйте не
менее, чем 5 ч до приготовления срезов.
2. Сделайте предварительную проводку (см. табл. 10.2) в
условиях меняющегося давления вакуумной системы (это
повышает эффективность пропитывания парафином).
3. После заливки сделайте на микротоме срезы толщиной
2—3 мкм и поместите их на поверхность теплой воды,
нанесенной на стекла, обработанные указанным способом.
4. Держите стекла в пыленепроницаемой коробке для пре-
паратов. Они могут храниться до использования по мень-
шей мере 6 мес.
4.2. Замороженные срезы
1. Заморозьте ткань в жидком азоте и погрузите в каплю
Tissue-Tek OCT (Lab-Tek Products, Illinois).
2. Повысьте температуру до —30°C и оставьте ткань в замо-
раживающем криостате на 1 ч.
268
Глава 10
кированию на срезах основных групп нерастворимых бел-
ков, способных связывать меченую РНК [17]. Неспецифи-
^ское связывание зонда может быть также снижено
погружением препаратов в раствор гепарина 50 мкг/мл на
5 мин непосредственно перед внесением зонда и путем
включения гепарина в гибридизационный буфер в концент-
рации 50 мкг/мл [18]. Гепарин проявляет способность к
стехиометрическому аффинному связыванию ДНК-связы-
вающих белков.
6. Гибридизация
Гибридизационную смесь, содержащую меченый зонд, сле-
дует наносить на гистологические срезы на стеклах согласно
указаниям табл. 10.4. Оптимальная концентрация зонда, даю-
щая лучший ответ на присутствующий в ткани сигнал, — это
концентрация, как раз достаточная для насыщения РНК-ми-
шеней. В случае большинства реакций зонд в концентрации
0,5 мкг/мл насытит клеточную РНК. Рассчитано, что насыщение
РНК происходит при 0,2—0,3 мкг/мл зонда на 1 т. п. н. длины
зонда [9].
7. Постгибридизационные отмывки
Процедуры отмывки описаны в таблице 10.5
8. Радиоавтография
После удаления негибридизованного зонда стекла покрыва-
ют тонким слоем жидкой эмульсии для радиоавтографии, как
описано в табл. 10.6. Тип использованной эмульсии зависит от
изотопа. 32Р для этого типа радиоавтографии не подходит из-за
высокой энергии излучения и длинного пути пробега частиц.
Для 3Н и 35S рекомендуются эмульсии Ilford К-2 или Kodak
NTB-2, имеющие размер зерен соответственно 0,2 и 0,26 мкм.
Эти эмульсии не накапливают фон так быстро, как другие [19],
однако их нельзя хранить более 3 мес при 4 °C. Вместо жид-
кой эмульсии можно использовать пленку Kodak AR-10. Ею
сложнее пользоваться, но эта пленка, уменьшает разброс чув-
ствительности благодаря постоянной толщине слоя эмульсии
над срезом [19].
Длительность экспозиции зависит от использованной метки.
Сигнал, источником которого служит тритий, должен быть виден
через ‘5 дней. 35S дает более быстрые результаты — сигнал по-
является через 3 дня или даже через 2, если относительное
содержание молекул-мишеней особенно велико. В начале эк-
спериментов по гибридизации рекомендуется при каждом
Таблица 10.4. Гибридизация зонда на срезах
1. Приготовьте гибридизационный буфер. Его конечная концентрация: 50%
формамид, 0,3 М NaCl, 10 мМ трис-НС1 pH 8,0, 1 мМ ЭДТА, раствор
Денхардта (0,02% БСА, 0,02% фиколл, 0,02% ПВП), 500 мкг/мл тРНК
дрожжей. Для повышения эффективности концентрации зонда можно
включить 10%-ный декстрансульфат, однако по некоторым данным разные
партии этого препарата вызывают значительные колебания в фоне иа
автографах.
2. Приготовьте аликвоты буфера в пробирках Eppendorf, достаточные для
того, чтобы покрыть одно стекло. 30 мкл гибридизационной смеси покроют
площадь в 700—1000 мм2. Добавьте зонд в нужной концентрации.
3. На подготовленные препараты (табл. 10.3), которые должны быть абсо-
лютно сухими, нанесите 30—40 мкл смеси, поместив каплю над центром
среза.
4. Сверху опустите покровное стекло минимального размера, достаточного
для того, чтобы прикрыть срез. Опуская, сначала поставьте край стекла
за каплей, затем осторожно, при помощи часового пинцета, положите его
па каплю так, чтобы пузырьки воздуха вытеснялись за пределы покров-
ного стекла.
5. Немедленно окружите края стекла полоской минерального масла, или ре-
зинового цемента, чтобы предотвратить испарение (Sigma поставляет ми-
неральное масло в удобных пластиковых пузырьках на 6 мл).
б. Пометьте стекла алмазным карандашом.
7. Гибридизуйте во влажной камере при температуре от 40 до 50 °C1’ всю
ночь. Влажную камеру легко сделать из пластиковой коробки для бу-
тербродов, поместив на ее дно бумажную салфетку, пропитанную водой.
Салфетка должна быть влажной, но не должна смачивать нижние края
стекол. Крышка коробки заклеивается изоляционной лентой.
’) В исследованиях по тепловой денатурации Коксом [9] приведен расчет, свиде-
тельствующий о том, что Тт для дуплексов РНК—РНК in situ примерно на 5 °C ниже,
чем Тт в растворе для зондов длиной 100—200 оснований. Максимум скорости гибриди-
зации in situ приходится на 25 °C ниже нормальной Тт. Снижение Тт для дуплексов
РНК—РНК составляет 0.235 °С/% формамида, в то время как для дуплексов ДНК—
ДНК, —0,65 °C. Кроме того, гибриды РНК—РНК примерно на 10 °C менее стабильны в
отсутствие формамида, чем дуплексы ДНК—ДНК. Обобщая эти данные, можно заклю-
чить. что оптимальные температуры для гибридизации РНК—РНК находятся между 40
и 50 °C при гибридизации в 50%-ном формамиде.
2) В большинстве случаев реакция гибридизации заканчивается примерно через 5 ч.
Как правило, реакцию проводят всю ночь, поскольку это допустимо и удобно для ис-
следователя.
Таблица 10.5. Процедура отмывки
1. По окончании гибридизации извлеките препараты из влажной камеры.
Удалите минеральное масло отмывкой стекол в двух порциях хлороформа
по 10 мин в каждой. Поверхностное натяжение препятствует отделению
покровных стекол, даже если их держать вертикально. Если был исполь-
зован резиновый цемент, снимите его.
2. Погрузите стекла в 4XSSC. Спустя минуту покровное стекло можно осто-
рожно снять, приподняв тонким пинцетом.
3. Промойте стекла в трех порциях 4XSSC, по 5 мин в каждой.
4. Удалите несвязавшийся зонд посредством инкубирования в 20 мкг/мл
РНКазы А в течение 30 мин прн 37 °C, как указано в п. 10 табл. 10.3.
5. Проинкубируйте в буфере РНКазы в течение 30 мии при 37 °C.
6. Проинкубируйте в 2XSSC в течение 30 мин при 45°С.
7. Проинкубируйте в 0.1XSSC в течение 45 мии при 45°C".
269
272
Глава 10
Рис. 10.2. Гибридизация зонда Тср-1 с областью спинного мозга 11-дневного»
эмбриона мыши; в качестве отрицательного контроля использована обратная'
нить Тср-1. А. Область спинного мозга, гибоидизованная с Тср-1, в светлом
Гибридизация in situ с мРНК на гистологических срезах
273
поле микроскопа. Б. То же в темном поле. Обратите внимание на отсутствие
четкого паттерна гибридизации в светлом поле. В. Фоновая гибридизация
с обратной нитью Тср-1. Г. То же в темном поле. Увеличение Х500.
18—171
276
Глава 10
Рис. 10.4. Артефакт при окрашивании парафиновых срезов семенника. Форми-
рующиеся головки сперматид создают впечатление положительного сигнала.
Увеличение Х4000.
связывания зонда. Некоторые клетки демонстрируют высокий
уровень неспецифического связывания, например, зрелые спер-
матиды в семеннике [22] (показаны на рис. 10.3). Окрашивание
может быть источником артефакта, когда специализированные
клетки реагируют с красителем таким образом, что появляются
структуры, подобные гранулам серебра (рис. 10.4). Однако
применение указанных выше контролей устранит возможность
неверных выводов.
Благодарности
Авторы благодарны Мартину Берджину за изготовление
гистологических срезов, Кену Меррифилду за помощь в публи-
кации и Кристине Уотсон за критический анализ рукописи. Эта
работа была обеспечена грантом, полученным Институтом ис-
следований рака от Объединенного комитета MRC/CRC. Ребек-
ка Хаффнер — стипендиатка Совета медицинских исследований
(MRC).
Литература
L Gall J. G„ Pardue М. L. (1969). Proc. Natl. Acad. Sci. USA, 63, 378.
2. Gall J. G., Pardue M. L. (1971). In: Methods in Enzymology. Academic
Press, New York, Vol. 21, p. 470.
Гибридизация in situ с мРНК на гистологических срезах
277
3. Siracusa L. D., Chapman V. М., Bennett К. L„ Hastie Н. D„ Pietras D. F.,
Rossant I. (1983). J. Embryol. Exp. Morphol., 73, 163.
4. Rossant J., Vijh M„ Siracusa L. D„ Chapman V. M. (1983). J. Embryol.
Exp. Morphol., 73, 179.
5. Lo C. №. (1986). J. Cell Sci., 87, 143.
6. Hafen E., Levine M., Garber R. L., Gehring №. J. (1983). EMBO J., 2, 617.
7. Awgulewitsch A„ Utset Л1. F., Hart С. P., McGinnis W„ Ruddle F. H. (1986).
Nature, 320, 328.
8. Pardue M. L. (1985). In: Nucleic Acid Hybridisation: A Practical Approach.
Hames B. D. and Higgins S. J. (eds), IRL Press, Oxford, p. 179.
9. Сох К. H„ DeLeon D. V., Angerer L. M., Angerer R. L. (1984). Dev. Biol.,
101, 485.
10. Akam M. E. (1983). EMBO J., 2, 2075.
11. Melton D. A., Krieg P. A„ Rebagliati M. R., Maniatis T., Zinn K., Green M. R.
(1984). Nucleic Acids Res., 12, 7035.
12. Uhl G. R„ Zingg H. H., Habener J. F. (1985). Proc. Natl. Acad. Sci. USA,
82 5555
13. Berger C. N. (1986). EMBO J., 5, 85.
14. Leary J. J., Btigati D. J., Ward D. C. (1981). Proc. Natl. Acad. Sci. USA,
78, 6633.
15. Brigati D. J., Myerson D., Leary J. J., Spalholtz B., Travis S. J., Fong С, K.,
Hsiung G. D., Ward D.C. (1983). Virology, 126, 32.
16. Zinn K„ DiMaio D„ Maniatis T. (1983). Cell, 34, 865.
17. Hayashi S., Gillam I. C„ Delany A. D., Tener G. M. (1978). J. Histochem.,
26, 677.
18. Singh L., Jones K. №. (1984). Nucleic Acids Res., 12, 5627.
19. Rodgers A. №. (1979). Practical Autoradiography. Review 20, Amersham
International pic, Amersham, UK.
20. Dormer P., Pachmann K., Lau B. (1978). Histochemistry, 59, 17.
21. Willison K. R„ Dudley K., Potter J. (1986). Cell, 44, 727.
22. Gizang-Ginsburg E„ Wolgemuth D. J, (1985). Dev. Biol., Ill, 293.
280
Глава 11
6. Вырежьте полосу ДНК из агарозы с низкой точкой плав-
ления п удалите излишки агарозы. Поместите гелевую
пластинку в соответствующую пробирку и инкубируйте
при 70 °C, пока агароза полностью не расплавится. Опре-
делите объем геля и добавьте 5 объемов 0,5 М NaCl
в ТЭ-буфере (10 мМ. трис-НС1 pH 7,5, 1 мМ. ЭДТА). Хо-
рошо перемешайте и продолжайте инкубировать еще
10 мин при 70 °C.
Смесь ДНК/агарозы очищается с помощью ион-обменной
колонки NACS-52 Prepac(BRL) следующим образом.
1. Проведите гидратацию колонки, промыв ее трижды 2,0 М
NaCl в ТЭ-буфере. Подготовьте колонку, промыв трижды
буфером для связывания ДНК (для фрагментов ДНК
больше 1 т. п.н. используйте 0,5 М. NaCl в ТЭ-буфере,
для фрагментов меньшего размера возьмите 0,2 М NaCl
в ТЭ-буфере).
2. Загрузите колонку теплой (40°C) смесью агарозы/ДНК
и отмойте связанную ДНК связывающим буфером (3—
5 мл при 42 °C) для освобождения от агарозы и любых
гелевых примесей.
3. Элюируйте связанную ДНК 3 объемами по 0,1 мл 2,0 М
NaCl в ТЭ-буфере.
4. Осадите ДНК внесением 0,6 мл холодного (—20 °C)
95%-ного этанола. Если присутствует небольшое количе-
ство ДНК (^Ю мкг), его нужно осадить вместе с
тРНК-носителем. Присутствие тРНК в конечном образце
ДНК не оказывает неблагоприятного воздействия на
частоту интеграции ДНК или на развитие зиготы.
5. Отцентрифугируйте осадок в течение 10 мин при
12 000 об/мин в микроцентрифуге, затем хорошо отмойте
сгусток 70%-ным холодным этанолом для удаления осаж-
денной соли. Высушите осадок и растворите в небольшом
объеме ТЭ-буфера.
6. Определите абсорбцию препарата при 260 нм и 280 нм
(1 ОП26о = 50 мкг ДНК, ОП2бо/ОП2зо= 1,8 для чистой
ДНК). На этой стадии препарат можно также проконт-
ролировать проведением электрофореза маленькой алик-
воты в агарозном мини-геле.
7. Разведите ДНК до нужной концентрации (см. ниже) бу-
фером для инъекции (10 мМ. трис-НС1 pH 7,6; 0,1 мМ
ЭДТА) и диализуйте при 4 °C, использовав большой объем
буфера для инъекции и сменив его несколько раз в тече-
ние 48 ч.
8. Извлеките ДНК и храните аликвотами по 20 мкл при
—20 °C. Для выделения фрагмента ДНК из агарозных ге-
лей можно воспользоваться и другими методами: электро-
Получение трансгенных мышей
281
элюцией, методом «сжимания и замораживания», связы-
вания ДНК стеклянной пудрой. Эти методы описаны
[9]. В любом случае необходим интенсивный диализ об-
разца.
2.1.3. Концентрация ДНК
В большинстве экспериментов используется ДНК в концен-
трации 1—2 нг/мкл; это соответствует 200—400 копиям фраг-
мента ДНК длиной 5 т. п. н., который и может быть инъециро-
ван в пронуклеус. В этих условиях частота интеграции ДНК до-
стигает оптимума, т. е. 20—40% [3]. ДНК, введенная в зароды-
шевую линию мыши путем микроинъекции в пронуклеус, имеет
тенденцию к интеграции в единственном сайте в виде различно-
го числа тандемно расположенных копий. Возможно, что подоб-
ная молекулярная организация интегрированной ДНК влияет
на ее экспрессию [10]. Число интегрированных копий можно
уменьшить снижением концентрации ДНК, например мыши с
единственной интегрированной копией были получены в резуль-
тате введения ДНК в очень низкой концентрации, хотя прямой
зависимости при этом не наблюдается. Однако, снижая концент-
рацию ДНК, надо помнить, что при этом значительно падает
общая частота интеграции [4]. С другой стороны, применение
слишком высоких концентраций ДНК (например, 10 нг/мкл)
резко снижает выживаемость эмбрионов [3].
В нашей лаборатории для каждой новой конструкции ДНК
мы ставим предварительные эксперименты, в которых просле-
живается развитие инъецированных яиц in vitro до стадии бла-
стоцисты (табл. 11.1). Если развитие значительной доли эмбрио-
нов блокируется на одноклеточной стадии (40% по сравнению
с обычными 20%), ДНК разводится в 2 или более раз. Блок
развития не обязательно связан с токсичностью ДНК per se,
он может быть следствием временной экспрессии введенной
ДНК- С другой стороны, высокая временная экспрессия генной
конструкции может служить хорошим тестом для оценки ее
интеграции, например, при измерении активности индикаторных
генов, таких, как гены тимидинкиназы [11] или р-галактозида-
зы у 2-клеточных эмбрионов.
2.2. Яйца и эмбрионы
2.2.1. Линии мышей
Выбор линии зависит от того, имеет ли ее генетический фон
прямое отношение к предполагаемому использованию трансген-
ных мышей. Например, анализ некоторых конструкций имму-
ноглобулиновых генов или генов главного комплекса гистосов-
284
Глава 11
Инъекционные иглы обоих типов мы оттягиваем на Model
Р-80, Brown Flaming Micropipette puller (Sutter Instrument Co.),
но существуют и другие приспособления, служащие для оттяги-
вания пипеток. Окончательная форма придается иглам с помо-
щью микрокузницы Beaudouin (см. гл. 12, разд. 2.3.2 и 2.4).
2.4. Изготовление микроинструментов
2.4.1. Удерживающая пипетка
Изготовление пипетки-присоски для удержания яиц описа-
но в разд. 2.4.1, гл. 12.
2.4.2. Инъекционные иглы
Инъекционная игла для обработки на микрокузнице де Фон-
брюна. Эти иглы делают из стандартных толстостенных капил-
ляров (из боросиликатного стекла), наружный диаметр которых
1,0 мм, внутренний диаметр 0,58 мм, длина 15 см (поставщик
Leitz, Clark Electromedical Instruments, Drummond Scientific
Company).
1. Силиконируйте стеклянные капилляры изнутри и снару-
жи, погружая их в раствор силикона (Sigmacote, Sigma)
и высушивая в сушильном шкафу.
2. Закрепите капилляр в приборе для оттягивания игл и
включите его. Модель Р-80 регулирует четыре параметра;
она снабжена регистраторами скорости, вытяжения, нагре-
вания и воздушного потока. Регистратор нагревания конт-
ролирует температуру платиновой нити, нагревающей
стеклянный капилляр, а поток воздуха охлаждает нить и
стекло. Полное описание всех регуляторов прилагается к
инструменту.
Когда регистраторы скорости, вытяжения и нагревания на-
ходятся в постоянном положении, контроль воздушного потока!
обеспечивает тонкую настройку инструмента, позволяющую'
придать игле нужную форму. Нужная настройка воздушного,
потока со временем может сбиться, поэтому качество первых
нескольких игл, оттянутых в течение данного дня, следует про-
контролировать в микроскопе, а затем приступить к изготовле-
нию следующих. Оптимальная форма и размеры иглы приведены
на рис. 11.1.
3. Из каждого стеклянного капилляра получаются 2 иглы.
Разрежьте его на фрагменты по 4—5 см алмазным ножом.
Мы рекомендуем оттягивать 20—30 игл для использова-
ния в тот же день. Можно было бы приготовить и больший
запас игл и хранить их, пока не понадобятся, но при этом1
Получение трансгенных мышей
285
< 45 мм
•10—13 мм - I
10 мкм
I .
~~—I----
-1___
I
<10 мкм
>10 мкм
| - — 250 мкм ..- I
Рнс. 11.1. Схематическое изображение инъекционной иглы (масштаб не соблю-
дается). А. Общая форма н оптимальные размеры инъекционной иглы: длина
иглы от плеча диаметром 1 мм до кончика должна составлять от 10 до 13 мм.
Как можно ближе к основной трубке иглы необходимо сделать изгиб 5°. Длина
основной трубки должна быть около 45 мм. Кончик инъекционной иглы может
быть представлен тремя вариантами (Б, В, Г). Б, В — концы пипеток прием-
лемой формы: диаметр последних нескольких микронов длины кончика доста-
точно мал, чтобы прн вхождении в яйцо не были необратимо повреждены
мембраны. Если игла слишком тонка, она не будет обладать нужной жестко-
стью для немедленной реакции на движения манипулятора. Чем меньше су-
жение к концу (Б), тем удобнее пользоваться пипеткой Г. Непригодная фор-
ма: игла слишком расширена у конца пипетки.
иглы собирают пыль, способную закупорить отверстие.
Кроме того, замечено, что со временем края их отверстия
становятся менее острыми.
4. Поместите иглы в свободный лейцевский инструментодер-
жатель. Погрузите кончик иглы в детергент NP-40 (Sig-
ma) и отмойте, опустив его затем в две порции дистилли-
рованной воды.
5. Поднимите иглу (в лейцевском инструментодержателе)
горизонтально на микрокузницу (см. в гл. 12, разд. 2.3.2’
описание микрокузницы и ее нити накаливания) и рас-
смотрите кончик иглы. Сломайте конец иглы о стеклянный:
288
Глава 11
37° по меньшей мере 15 мин. В каждую каплю серии,
находящейся в манипуляционной камере, поместите по
2 яйца. Число яиц, набранных в камеру, не должно пре-
вышать количества, которое вы можете спокойно проопе-
рировать за 20 мин. Более длительное выдерживание яиц
вне инкубатора может их повредить. Каждая группа
обычно ограничивается 15—20 яйцами.
3. Введите микроинструменты в камеру. Толстостенная иг-
ла должна быть наполнена раствором ДНК из первой
капли. Для этого введите иглу в каплю и отрегулируйте
высоту манипулятора так, чтобы кончик иглы оказался
в фокусе. Заполните иглу, сообщив шприцу де Фонбрюна
отрицательное давление. Наблюдая этот процесс с объек-
тивом X Ю, можно видеть, как отступает масло и в иглу
входит раствор ДНК; заполнение иглы маслом следует
закончить, когда мениск окажется на краю поля зрения.
Уравновесьте давление, затем, приложив небольшое по-
ложительное давление, убедитесь в том, что ДНК свобод-
но вытекает. Уберите иглу из капли ДНК-
•4. Передвиньте камеру так, чтобы в поле зрения оказалась
первая капля с яйцами. Поместите яйца в фокус.
5. Введите в каплю инструменты и, регулируя высоту ма-
нипулятора, поместите их в фокус. Будьте осторожны,
не сломайте инструменты. Замените объектив X10 на
объектив Х25 и отрегулируйте фокусное расстояние.
6. С помощью рычагов управления манипулятора ориенти-
руйте яйца так, чтобы оба пронуклеуса были в фокусе,
при этом мужской пронуклеус находился бы ближе к
инъекционной игле (рис. 11.2, А). Мужской пронуклеус
располагается дальше от полярного тельца, кроме того,
он имеет большие размеры, чем женский, и поэтому
более удобен для проведения микроинъекций.
7. Захватите яйцо удерживающей пипеткой, приложив к
шприцу Агла небольшое отрицательное давление.
8. Надежно удерживая яйца, поставьте мембрану пронук-
леуса (и ядрышки) в фокус регулировкой высоты мани-
пулятора (рис. 11.2,5).
9. Поместите кончик инъекционной иглы в фокус и с помо-
щью рычагов управления манипулятора введите в про-
нуклеус (рис. 11.2,5). Если конец инъекционной иглы
лежит в нужной плоскости, можно видеть деформацию
пронуклеарной мембраны. Если игла не прошла, после
попытки инъекции ДНК появляются «пузырьки» раство-
ра ДНК, окруженные вдавленными ядерной и яйцевой
мембранами (рис. 11.2,5). Для прокалывания мембран
часто требуется короткий, резкий укол.
Получение трансгенных мышей
289
Рис. 11.2. Инъекция в мужской пронуклеус. А. Идентификация мужского
и женского пронуклеусов (Ям — ядерная мембрана, Ядр— ядрышко, Пт —
2-е полярное тельце). Б. Захватите яйцо удерживающей пипеткой-присоской.
Кончик инъекционной пипетки расположите на одной линии с мужским про-
нуклеусом. В. Введите иглу в пронуклеус. Г. Инъецируйте ДНК. Игла еще
не проколола мембраны яйца и пронуклеуса, о чем можно судить по «пузырь-
ку» у кончика иглы и по ямке, образуемой мембраной яйца в результате
поступления в перивителлиновое пространство жидкости, предназначенной
для пронуклеуса. Д. Осторожно сделайте укол и инъецируйте еще порцию;
увеличившийся объем пронуклеуса свидетельствует об успешной инъекции.
Е. После удаления иглы объем пронуклеуса восстанавливается до первона-
чального размера.
19—171
290
Глава 11
Рис. 11.3. Неудачи, возможные при инъецировании. А. Одно лизировавшее
и два здоровых яйца через 30 мин после инъекции. Б. Яйцо с цитоплазмой,
излившейся через инъекционное отверстие под блестящую оболочку, 30 ми»
после ииъекцнн. Это яйцо не лизировало полностью, однако не будет дро-
биться. В. Ядрышко, вытолкнутое из яйца инъекционной иглой после инъек-
ции ДНК. Г. Ядрышко, вышедшее в цитоплазму через второе отверстие в про-
нуклеарион мембране.
10. Если нет сомнений в том, что кончик иглы попал в про-
нуклеус, приложите к инъекционной игле положительное
давление. Пронуклеус несколько разбухнет (рис. 11.2, Д).
Позже его объем сократится до обычных размеров
(рис. 11.2, Е).
И. Выведите инъекционную иглу и повторите процедуру с
другим яйцом.
12. После того как все яйца, находящиеся в камере, будут
инъецированы, соберите их в одну каплю РВ1 и культи-
вируйте в инкубаторе.
13. Возьмите следующую партию свежих яиц из тех, что на-
ходятся в РВ1 в инкубаторе, в капли камеры и повторите
процедуру. Для каждой партии яиц обычно используется
новая инъекционная игла.
14. В конце эксперимента отделите удачно прооперированные
яйца от лизировавших (рис. 11.3,Л). Промойте их про-
водкой по 6 каплям среды М16 + БСА и оставьте на ночь
в капле М16+БСА при 37°С с 5% СО2 в воздухе.
Получение трансгенных мышей
291
3.4. Возможные затруднения
Большинство проблем, возникающих при микроинъекциях,
связано с закупориванием инъекционной иглы. Это может быть
следствием многих факторов, например, присутствия пыли в
растворе ДНК, в случае толстостенной иглы это могут быть
маленькие капельки масла, прилипшие к внутренней поверхно-
сти кончика иглы, когда она заполнялась раствором ДНК-
Иногда иглы закупориваются белками, захватываемыми в про-
цессе инъекции, обычно это происходит при лизисе яиц. Часто
удается откупорить иглу, очень осторожно приблизив ее к от-
верстию удерживающей пипетки. Если удалить пробку не
удастся или кончик иглы обломится и образует слишком широ-
кое отверстие, следует взять новую иглу. Дефекты инъекцион-
ной иглы обычно приводят к лизису яйца. Степень лизиса
может различаться от полного (рис. 11.3,Л) до частичного, при
этом по ходу культивирования может произойти регуляция
дефекта (рис. 11.3,5), но большинство таких яиц не достигает
2-клеточной стадии.
У старой иглы ее наружная поверхность становится липкой
от белков яйца. Несмотря на то что такие иглы инъецируют
хорошо, они могут захватывать и извлекать ядрышки, когда
игла выводится из пронуклеуса (рис. 11.3, В). В результате
сквозного прободения ядра инъекционной иглой ядрышки вы-
талкиваются с противоположной стороны ядра под действием
давления введенного раствора ДНК (рис. 11.3,Г). Такие яйца
редко развиваются.
Использование удерживающих пипеток не вызывает столько
проблем, но и эти пипетки при длительном хранении иногда за-
купориваются крупными частицами пыли. Пузырьки воздуха на
границе масла могут привести к тому, что пипетка перестает
реагировать на изменение давления масла. Слишком сильное
присасывание яйца к пипетке может привести к его поврежде-
нию. Если блестящая оболочка лопнет или в ней образуется
отверстие, содержимое яйца может оказаться втянутым внутрь
пипетки.
3.5. Трансплантация эмбрионов
В некоторых лабораториях оперированные яйца трансплан-
тируют псевдобеременным самкам 1-го дня вечером в день инъ-
екции. Мы обычно переносим 2-клеточные эмбрионы на следую-
щее утро после операции (см. гл. 13, разд. 6.4). На это есть три
причины. Первая заключается в том, что эмбрион должен опра-
виться после микроинъекции прежде, чем будет подвергаться
переносу. Вторая, и более существенная, связана с тем, что у
19*
292
Глава И
значительного процента оперированных эмбрионов развитие
блокируется на 1-клеточной стадии (10—20%) и нужно время,
чтобы убедиться в отсутствии блока и целесообразности пере-
носа. Блок развития на 1-клеточной стадии может происходить
либо в результате свойств раствора ДНК, как обсуждалось вы-
ше, либо вследствие прямого физического повреждения яйца
при микроинъекции. Третья причина состоит в том, что асин-
хронный перенос больше соответствует замедленному развитию
яиц, которое обусловлено оперативным вмешательством.
4. Скрининг трансгенного потомства
4.1. Аутопсия хвоста и прищипывание пальца
После переноса эмбрионов в яйцевод приемной матери не-
обходимо время от времени наблюдать за течением беремен-
ности, в частности, в середине срока взвесить реципиента. Если
к вечеру 20-го дня не произойдет благополучных родов у явно
беременной мыши, следует произвести кесарево сечение, так
как число живых плодов может быть недостаточным для индук-
ции естественной родовой деятельности (см. гл. 1, разд. 5.6).
В нашей колонии мышей здоровое потомство отсаживается
на 21-й день после рождения. В это время мышата достаточно
велики для выявления инъецированных последовательностей
ДНК путем биопсии хвоста для последующего анализа ДНК-
Биопсия хвоста и нумерование животных прищипыванием паль-
ца должны быть сделаны одновременно для экономии времени
и во избежание возможных ошибок. Можно маркировать жи-
вотных отверстиями в ушах (гл. 1, разд. 3.2), но мы не советуем
этого делать в данном случае; этот метод следует оставить для
кратковременных экспериментов.
1. Анестизируйте животное авертином, возьмите его в руку,
брюшком вверх, и прищипните отдельные пальцы малень-
кими острыми ножницами. Убедитесь, что отрезана доста-
точная часть для идентификации мышонка. Удаляя не
более 2-х пальцев на конечность, можно индивидуально
пометить колонию из 10 000 животных, как показано на
рис. 11.4. В частности, обратите внимание, что порядок
прищипывания от крайнего пальца к среднему (начиная с
наименьшего пальца и двигаясь к большому) должен быть
сохранен на всех четырех конечностях. Мы находим эту
систему прищипывания более удобной, чем принятая во
многих лабораториях система только слева направо.
2. Сразу же после прищипывания удалите терминальную
треть хвоста острыми ножницами и немедленно прижгите
Получение трансгенных мышей
293
2 3 4 ®
в 7 в 9 О
Рис. 11.4. Способ нумерации мышей прищипыванием пальца. Метод иллюстри-
рован схемой для правой передней лапы. Для безошибочной идентификации
палец отрезается не меньше чем наполовину. Паттерн повторяется для всех
четырех конечностей в латерально-медиальном направлении (т. е. единицы
и сотни читаются слева направо, а десятки и тысячи — справа налево).
культю горячим бритвенным лезвием. Ткань хвоста раз-
режьте ножницами на маленькие фрагменты и погрузите
в 2 мл солевого раствора, забуференного фосфатом (PBS),
в полипропиленовой центрифужной пробирке (10 мл) на
лед.
294
Глава 11
3. С помощью политропного вращающегося ножа (Kinema-
tica, Luzern, Швейцария) гомогенизируйте ткань хвоста.
Регулятор скорости установите в положения 4—5. Введите
пестик в пробирку с образцом и медленно перемещайте
вверх и вниз, пока ткань хвоста не будет тонко изрублена.
Тщательно удалите следы ткани с лезвия и очистите пе-
стик прежде, чем гомогенизировать следующий образец.
4. Центрифугируйте при 1000—2000 об/мин в настольной
центрифуге в течение 10 мин, осторожно уберите суперна-
тант, ресуспендируйте осадок в 1 мл буфера А (75 мМ
NaCl, 25 мМ ЭДТА при pH 8) и добавьте 1 мл буфера
Б (10 мМ трис-НС1 pH 8, 10 мМ ЭДТА, 1% ДСН,
400 мкг/мл протеинкиназы К). Важно хорошо ресуспенди-
ровать сгусток в буфере А до добавления буфера Б, чтобы
обеспечить быстрый лизис клеток и предотвратить дегра-
дацию ДНК. Проинкубируйте лизаты при 37 °C, оставив
на ночь, или при 50 °C в течение 3 ч. По нашему опыту,
лизис при 50 °C дает ДНК лучшего качества. Экстрагируй-
те один раз фенолом и один раз хлороформом. Доведите
до 0,3 М NaCl и осадите, добавив равный объем изопро-
панола. Извлеките преципитат маленьким шпателем, от-
мойте в 70 %-ном изопропаноле и растворите в 300—
500 мкл ТЭ-буфера. Эта процедура даст выход ДНК в
концентрации 0,5—1 мг/мл. Поскольку предполагается, что
все животные относятся к одной возрастной группе, раз-
брос в концентрации ДНК должен быть незначительным.
Поэтому мы обычно не определяем оптическую плотность
образца.
4.2. ДНК из плодов
В некоторых случаях анализ на трансгенность необходимо
проводить на уровне плодов. Например, возможна ситуация,
когда определенная генная конструкция, экспрессируясь во вре-
мя эмбриогенеза, обусловливает пренатальную гибель. В этом
случае анализ уже родившихся животных продемонстрирует
низкую частоту трансгенных особей.
Количество ДНК, достаточное для- генотипирования индиви-
дуальных плодов, может быть получено начиная с 8-го дня бе-
ременности. Для обычного анализа мы выделяем ДНК начиная
с 10—11 дня (к 11 дню это составляет до 100 мкг).
1. Осторожно разорвите мышцу матки тонким пинцетом и
обнажите зародыш. Удалите децидуальную ткань и тро-
фобласт, если отсутствие примеси материнской ДНК
служит существенным условием эксперимента. Выделение
ДНК только из эмбриона имеет преимущество — клетки
Получение трансгенных мышей 295
легче диссоциируются, а полученная ДНК характеризует-
ся более высоким качеством.
2. Поместите эмбрион в пробирку Eppendorf, содержащую
0,5 мл PBS (эмбрион 10—12 дня), или в 10 мл-пропиле-
новую пробирку (с 13 дня), содержащую 2 мл PBS, и
гомогенизируйте ткань путем легкого пипетирования авто-
матической пипеткой (Gilson pipettman), при этом жел-
тыми концами пользуйтесь для гомогенизации 10—12
дневных эмбрионов, голубыми—для эмбрионов более
поздних стадий.
3. Центрифугируйте при 1000—2000 об/мин и дальше сле-
дуйте прописи для ткани хвоста с тем исключением, что
для эмбрионов концентрация протеиназы К должна быть
200 мкл/мл в буфере Б.
4.3. Анализ ДНК
Для первоначального анализа на трансгенность предложен
ряд разнообразных методик [3, 13, 14]. Обычно мы пользуемся
методом саузерн-блот [9], хотя дот-блот-гибридизация занимает
меньше времени. Дополнительная информация, источником ко-
торой служит первый из методов, в любом случае потребуется
на более позднем этапе. ДНК хвоста следует обработать рест-
риктазой, высвобождающей внутренний фрагмент из интегриро-
ванной конструкции ДНК, или ферментом, делающим в конст-
рукции один разрез. Поскольку тандемное расположение
превалирует, второй способ также даст фрагменты предсказуе-
мого размера. Если бы трансген содержал последовательности,
присутствующие также и в мышином геноме, можно было бы
использовать зонд ДНК, гибридизующийся как с трансгеном,
так и с собственным геном мыши при условии, что фрагменты,
полученные в результате рестрикции, четко различаются. Это
дает дополнительное преимущество при определении точного
числа копий по силе гибридизационного сигнала. Однако следу-
ет иметь в виду, что значительная доля (20—30%) мышей-
основательниц (Fo) представлена мозаиками [15], поэтому
оценка числа копий на геном возможна только в поколении Fj
или в следующих поколениях.
После рестрикции и электрофореза в агарозном геле ДНК
переносится из геля на мембрану. Мы находим, что наиболее
быстрый и надежный способ блоттинга и гибридизации — это
щелочной перенос [16] на нейлоновые мембраны (Gene screen
plus, Dupont Boston, США) и гибридизация с зондами, мечен-
ными олигонуклеотидной затравкой ([17], гл. 9, разд. 5.2).
296
Глава 11
4.4. Трансгенные мыши и замораживание эмбрионов
Важнейшим условием эффективного ведения линий мышей
в виварии служит четкая нумерация животных прищипыванием
пальцев (описано выше) и правильная регистрация спаривания
мышей для разведения. Любому виварию периодически начина-
ет угрожать перенаселенность. Решить проблему можно путем
замораживания эмбрионов. Этот прием позволяет сохранить
трансгенные линии, не используемые в данное время (см. гл. 13).
Еще один способ сохранения мышей ценных линий — внедрение
их в виварни родственных лабораторий в других институтах.
5. Перенос генов с помощью ретровирусов
Как указывалось выше, ретровирусы используются для полу-
чения трансгенных мышей путем воздействия инфекционным
вирусом на предимплантационных эмбрионов [1]. При этом
частота трансформации зародышевой линии близка к той, кото-
рую мы наблюдаем при микроинъекциях ДНК, а сама техника
эксперимента намного проще. Ранние морулы освобождаются
от блестящей оболочки, подвергаются кратковременному воз-
действию инфекционным вирусом и трансплантируются псевдо-
беременным приемным самкам. Различные способы ретровирус-
ного генного переноса основаны на получении высокого титра
клеточных линий, продуцирующих вирус. Подробные описания
соответствующих экспериментов можно найти в оригинальных
публикациях [13, 18, 19].
Укажем на несколько важных пунктов, отличающих эти
два пути трансгенеза. Трансгенные животные, полученные путем
интеграции ретровирусов, всегда являются мозаиками с одним
или более сайтов интеграции в различных клетках. Это обуслов-
лено тем, что эмбрионы обычно инфицируются на стадии мору-
лы, состоящей из 8—16 клеток [1, 20]. Вот почему для обнару-
жения потомства, несущего вставочную последовательность,
полученную с помощью ретровирусов, необходим скрининг.
В то же время трансгенные животные, полученные путем инъек-
ции ДНК в пронуклеусы, в большинстве случаев не мозаичны
и соответственно передают новый ген 50% потомков [15].
Ретровирусы обычно интегрируются в виде единственной копии
генов в разные сайты генома хозяина и не вызывают перестроек
ДНК в месте вставки, что часто наблюдается при инъекции
ДНК [21, 22]. Следовательно, они позволяют более четко ин-
терпретировать позиционные эффекты, обусловленные местом
интеграции чужеродной ДНК [23] и облегчают клонирование
сайта инсерции (вставки), что особенно важно для молекуляр-
ного анализа инсерционных мутаций [24]. Поскольку сигналы
Получение трансгенных мышей 297
контроля транскрипции самих вирусов в разнообразных тканях
и особенно в клетках ранних эмбрионов слабы, для обеспечения
как повсеместной, так и тканеспецифической экспрессии введен-
ных генов использованы ретровирусные векторы, несущие
соответствующие промоторы и энхансеры [25, 26]. Насколько
широко может быть применен этот подход, еще предстоит оце-
нить. Особенно интересно проследить, можно ли добиться с по-
мощью ретровирусных векторов высокой степени тканевой
специфичности (наблюдаемой при использовании различных
конструкций ДНК), несмотря на наличие вирусных последова-
тельностей контроля транскрипции в непосредственной близости
от введенного гена. Важно помнить также, что существует
связанное с упаковкой ограничение длины геномной РНК виру-
са, ее максимум предполагается в пределах 8—10 тысяч основа-
ний [27]. Во всяком случае, очевидно, что такие гены, как,
например, ген мышечной дистрофии Дюшенна с размером
мРНК 14 тысяч оснований [28], невозможно перенести с помо-
щью ретровирусных векторов, но легко инъецировать в яйца.
Следует также заметить, что конструирование ретровирусных
векторов не столь тривиальная процедура, как получение плаз-
мидных векторов для микроинъекции.
Важное преимущество использования ретровирусов заклю-
чается в возможности их введения в эмбрионы более поздних
стадий развития, чем предимплантационные, а также в специ-
фические ткани взрослого животного [29, 30]. При инфекции
клеток на разных этапах дифференцировки вирус может ока-
заться уникальным маркером потомков отдельных клеток,
позволяющим прослеживать судьбу клеточных линий [31—41].
Этот подход очень перспективен при анализе сложных линей-
ных отношений у эмбрионов млекопитающих и в тканях взрос-
лых животных, например кроветворной ткани.
Возможность избирательного введения генов с • помощью
ретровирусов в кроветворную ткань взрослых организмов поз-
воляет надеяться на реальность исправления дефектов, обуслов-
ленных единичным геном, таких, например, как синдром
комбинированного иммунодефицита [35]. В настоящее время
уже разработаны методики, обеспечивающие высокую эффектив-
ность трансформации стволовых клеток гемопоэза [32], однако
прежде, чем приступить к обсуждению практических вопросов
применения в клинике, необходимо решить ряд проблем, касаю-
щихся стабильности экспрессии и безопасности.
Другой подход к трансформации эмбриональных линий у
мыши в последнее время разрабатывается Эвансом с сотрудни-
ками [18, 36]. Культивируемые эмбриональные стволовые клет-
ки инфицируют ретровирусными векторами, поддерживают в
культуре, а затем вводят мыши путем инъекции в бластоцисту
298
Глава 11
[18]. Использование ЭС-клеток позволит успешно контролиро-
вать число ретровирусных инсерций и, чго более существенно,
выделять специфические вставки до реинтродукции в эмбрио-
нальную линию с помощью методов генетики соматических
клеток. Благодаря этому подходу недавно удалось сконструи-
ровать линию мышей, несущих мутацию в гене гипоксантин-
фосфорибозилтрансферазы [36].
Применение ретровирусной технологии может быть также
использовано для трансформации клеток зародышевого пути
других видов млекопитающих, у которых получение трансген-
ных животных невозможно из-за отсутствия разработанных сис-
тем культивирования эмбрионов или из-за затруднений с микро-
инъекцией в зиготу.
Благодарности
Мы благодарим Майкла Норриса за помощь в постановке
экспериментов, а Сару Хаулет за советы и обсуждение рукопи-
си. Мъ! признательны Майклу Дэвису и Джону Хейлу за пре-
красный уход за животными. Большое спасибо Линде Ноттон
за печатание рукописи. Работа Н. Аллена финансировалась
AFRC, работу В. Рейка отчасти финансировало ЕМВО.
Литература
1. Jaenisch. R. (1976). Proc. Natl. Acad. Sci. USA, 73, 1260.
2. Palmiter R. D., Brinster R. L. (1986). Annu. Rev. Genet., 20, 465.
3. Brinster R. L., Chen H. Y., Trumbauer M. E., Yagle M. K., Palmiter R. D.
(1985). Proc. Natl. Acad. Sci. USA, 82, 4438.
4. Chada K., Magram J., Raphael K., Radice G., Lacy E„ Costantini F. (1985).
Nature, 314, 377.
5. Townes T. M., Lingrel J. B., Chen H, Y., Brinster R. L„ Palmiter R. D.
(1985). EMBO, 4, 1715.
6. Michiels F., Burmeister M„ Lehrach H. (1987). Science, 236. 1305.
7. Lathe R.. Vilotte J. L., Clark J. (1987). Gene, in press.
8. Maniatis T., Fritsch E. F., Sambrook J. (1982). Molecular Cloning: A Labo-
ratory Manual Cold Spring Harbor Laboratory Press, New York.
9. Sealey P. G., Southern E. M. (1982). In: Gel Electrophoresis of Nucleic
Acids — A Practical Approach. Rickwood D, and Hames B. D. (eds.), IRL
Press, Oxford, p. 39.
10. Reik W7., Williams G., Barton S. C., Norris M. L., Neuberger M., Sura-
ni M. A. H. (1987). Eur. J. Immunol., 17, 465.
11. Brinster R. L„ Chen H. Y., Warren R„ Sarthy A., Palmiter R. D. (1982).
Nature, 296, 39.
12. Whittingham D. G., Wales R. G. (1969). Aust. J. Biol. Sci., 22, 1065.
13. Hogan B., Costantini F., Lacy E. (1986). Manipulating the Mouse Embryo:
A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York.
14. Palmiter R. D„ Chen H. Y„ Brinster R. L. (1982). Cell, 29, 701.
15. Wilkie T„ Brinster R. L., Palmiter R. D. (1986). Dev. Biol., 118, 9.
16. Chomczynski P., Qasba P. K. (1984). Biochem. Biophys. Res. Commun., 122,
340.
Получение трансгенных мышей
299
17. Feinberg А. Р„ Vogelstein В. (1984). Anal. Biochem., 137, 266.
18. Robertson Е., Bradley A., Kuehn М., Evans М. (1986). Nature, 323, 445.
19. Jaenisch R., Fan H., Croker B. (1975). Proc. Natl. Acad. Sci. USA, 72, 4008.
20. Jaehner D„ Jaenisch R. (1980). Nature, 287, 456.
21. Panganiban A. T. (1985). Cell, 42, 5.
22. Covarrubias L., Nishida Y., Mintz B. (1986). Proc. Natl. Acad. Sci. USA,
83, 6020.
23. Jaenisch R-, Jaehner D., Nobis P., Simon J., Loehler J., Harbers K, Grot-
kopp D. (1981). Cell, 24, 519.
24. Gridley T., Soriano P., Jaenisch R. (1987). Trends Genet., 3, 162.
25. Stewart C. L., Schuetze S., Vanek M., Wagner E. F. (1987), EMBO J., 6,
383.
26. Soriano P-, Cone R-, Mulligan R„ Jaenisch R. (1986). Science, 234, 1409.
27. Shimotohno K., Temin H. M. (1981). Cell, 26, 67.
28. Koenig M., Hoffman E P., Bertelson C. J., Monaco A. P., Feener C., Kun-
kel L. M. (1987). Cell, 50, 509.
29. Jaenisch R. (1980). Cell, 19, 181.
30. Joyner A., Keller G.t Phillips R. A., Bernstein A. (1983). Nature, 305, 556.
31. Soriano P., Jaenisch R. (1986). Cell, 46, 19.
32. Sanes J. R., Rubenstein J. L. R„ Nicolas J. F. (1986). EMBO J., 5, 3133.
33. Turner D. L., Cepko C. L. (1987). Nature, 328, 131.
34. Keller p., Paige C., Gilboa E., Wagner E. F. (1985). Nature, 318, 149.
35. Belmont J. W., Henkel-Tigges J., Chang S. M. W., Wager-Smith K, Kel-
lems R. E., Dick J. E., Magli M. C., Phillips R. A., Bernstein A., Caskey С. T.
(1986). Nature, 322, 385.
36. Kuehn M., Bradley A., Robertson E., Evans M. (1987). Nature, 326, 295.
ГЛАВА 12
ТРАНСПЛАНТАЦИЯ ЯДЕР
В ОПЛОДОТВОРЕННЫЕ И ПАРТЕНОГЕНЕТИЧЕСКИ
АКТИВИРОВАННЫЕ ЯЙЦА
Ш. Бартон, М. Норрис, А. Сурани1
1. Введение
Современные методы трансплантации ядер позволяют вме-
шиваться в генетическую организацию яиц и эмбрионов мыши.
Благодаря этим методам удалось достигнуть больших успехов
в ряде областей биологии развития, в частности в изучении:
а) вклада материнского и отцовского геномов в развитие;
б) ядерно-цитоплазматических отношений;
в) морфогенетических потенций ядер, трансплантированных
в яйца из эмбриональных клеток и возможностей их «кло-
нирования»;
г) репродукции млекопитающих путем партеногенеза.
Эти исследования позволили по-новому осмыслить функцио-
нальные различия между геномами родителей, однако точные
механизмы ограничения роста у яиц, получивших донорские
ядра с более поздних стадий развития, или у партеногенетиче-
ских яиц пока не известны.
Поскольку пронуклеусы и ядра яиц и ранних эмбрионов
имеют относительно крупные размеры, введение в яйцо пипетки
с диаметром отверстия, достаточным для захвата такого ядра,
обычно необратимо повреждает мембрану яйца и приводит к
лизису. В связи с этим предложен эффективный метод [1]
извлечения ядра в небольшом объеме цитоплазмы. Такие ну-
клеопласты могут быть слиты с другими яйцами или клетками
с помощью вируса Сендай. Указанный метод позволяет получить
в ходе одного четырехчасового эксперимента до 100 энуклеиро-
ванных яиц или 40—50 реконструированных яиц.
2. Основная экспериментальная техника
2.1. Культивирование
Яйца культивируют в стандартной среде (М16, [2], гл. 2,
разд. 5.2) с 0,4% БСА (Fraction В, Sigma) в пластиковых
чашках Петри (Sterilin) под маслом, в увлажненной атмосфере
1 5. С, Barton, М. L. Norris, М. А. Н. Surani. AFRC Institute of Animal
Physiology and Genetics Research, Department of Molecular Embryology, Babra-
ham, Cambridge CB2 4AT, UK.
Трансплантация ядер в яйца 301
с 5% СО2 при 37,5—38,5°C (см. гл. 2, разд. 1). Манипуляции
проводят в забуференной фосфатом среде с 0,4% БСА (РВ1,
[3], гл. 13, разд. 3.1.1), к которой в большинстве случаев добав-
ляют 1 мкг/мл цитохалазина D (ЦХД) (Sigma) и 0,1 мкг/мл
нокодазола (Sigma), растворенных в диметилсульфоксиде
(ДМСО). Разрушение микрофиламентов (воздействием ЦХД)
я микротрубочек (нокодазолом) делает яйца более пластичными
и устойчивыми к повреждению при манипуляциях. Исходные
растворы ЦХД в концентрации 2 мг/мл ДМСО и нокодазола
при 0,2 мг/мл ДМСО хранят в виде небольших аликвот при
—20 °C до 6 мес. Непосредственно перед употреблением по
1 мкл каждого из этих реагентов добавляют к 2 мл РВ1 (PNC).
Яйца небольшими группами переносят для манипуляций в PNC
и затем быстро отмывают, так как воздействие нокодазолом
свыше 4 ч может оказаться повреждающим.
Для культивирования следует использовать легкое жидкое
парафиновое масло (масса 1 мл при 20°С ~0,846 г), а для
манипуляционных камер и инструментов — тяжелое (масса на
мл при 20°C ~0,875 г). И то и другое поставляется фирмой
BDH. Перед употреблением каждую партию масла необходимо
проверять на токсичность. Стерилизовать масло мы не считаем
обязательным. Достаточно его профильтровать через ватманов-
скую бумагу № 1, разлить в чистые стерильные пузырьки и хра-
нить закрытым.
2.2. Приготовление препарата вируса Сендай
Вирус Сендай, или гемагглютинирующий вирус (японский
штамм), используется в качестве фузогена (фактора, вызываю-
щего слияние клеток). Процедура выращивания вируса в раз-
вивающихся куриных яйцах подробно изложена в работах [4,
5], мы приводим ее краткое описание.
1. Проинкубируйте свободные от вируса оплодотворенные
куриные яйца на боку при 30 °C в течение 11 дней.
2. Поместите яйца в просвечивающее устройство для обна-
ружения эмбриона, который выявляется в виде темной
тени в центре яйца, и на каждом яйце отметьте место
инъекции — между двумя основными венами вблизи воз-
душной камеры.
3. Слегка протрите яйца 70%-ным спиртом и подпилите
скорлупу в месте инъекции.
4. Стерильным шприцем с инъекционной иглой 25-го калиб-
ра введите 0,1 мл вируса, разведенного солевым раство-
ром Хенкса без глюкозы до концентрации 0,1 гемагглю-
тинирующих единиц (ГАЕ) на яйцо. Для этого погру-
зите иглу на глубину примерно 1 см, направив ее к заост-
302
Глава 12
ренному концу яйца, и медленно инъецируйте суспензию
вируса в яйцо.
5. Заклейте отверстие в скорлупе расплавленным парафи-
ном и инкубируйте яйца в течение 3 дней, установив их
на заостренных концах, при 30 °C.
6. После этого инкубируйте их в течение ночи при 4 °C, что-
бы убить эмбрионы и предотвратить кровотечение при
сборе жидкости из аллантоиса.
7. Быстро соберите вирус при 4 °C, чтобы получить высокий
титр. В стерильных условиях срежьте верхушку скорлупы
яйца. Разрежьте оболочку аллантоиса и извлеките пипет-
кой его жидкость. Жидкость должна быть мутной и блед-
но-желтого цвета; образцы, загрязненные желтком и
кровью, отбросьте.
8. Соедините жидкость, собранную из всех яиц, и поместите
на лед.
9. Центрифугируйте при 3000 об/мин в течение 10 мин.
10. Удалите супернатант и центрифугируйте снова при
15000 об/мин в течение 1 ч при 4 °C. Диспергируйте обра-
зовавшийся белый сгусток в 1 мл холодного раствора
Хенкса без глюкозы.
11. Возьмите небольшую пробу и определите число гемаг-
глютинирующих единиц [4]. Разведите препарат раство-
ром Хенкса без глюкозы до 20 000 ГАЕ/мл.
12. Заморозьте аликвоты по 1 мл на сухом льду и храните
при —70 °C. Препаратом вируса Сендай можно пользо-
ваться для слияния клеток в течение нескольких лет.
Прежде чем использовать вирус Сендай в экспериментах
по слиянию клеток, его нужно инактивировать. Это можно сде-
лать двумя способами. При инактивации ультрафиолетом у не-
которых вирусных частиц иногда сохраняется инфекционность,
что в высшей степени нежелательно. Гораздо эффективнее хи-
мический метод инактивации с помощью бета-проприолактона
(БПЛ) [6]. Обращаться с БПЛ следует с большой осторожно-
стью, во-первых, потому, что это потенциальный канцероген,
а во-вторых, потому, что при температурах выше 4 °C он крайне
лабилен.
1. Разморозьте одну из двух аликвот исходного раствора
препарата вируса и воспользуйтесь вортексом.
2. Отцентрифугируйте при 35000g" в течение 30 мин. Ресус-
пендируйте осадок в 0,9 мл раствора Хенкса без глюкозы
и диспергируйте с помощью вортекса.
3. Перенесите раствор в пробирку с хорошо подогнанной
пробкой и добавьте 5 мг БСА.
Дальше процедура ведется в холодной комнате, чтобы избе-
жать потери активности очень нестойкого БПЛ. Учитывая высо-
Трансплантация ядер в яйца 303
кую токсичность этого соединения, рекомендуем надеть две
пары перчаток и защитную одежду, включая маску. Все раство-
ры следует приготовить и проверить до переноса в холодную
комнату, чтобы до минимума сократить время работы с БПЛ.
Все растворы держать на льду.
4. В холодной комнате прежде всего добавьте 20 мкл БПЛ
к 162 мкл воды, дважды дистиллированной в стеклянной
посуде.
5. Быстро внесите 50 мкл этого раствора в 0,95 мл солевого
раствора (1,68 г ЫаНСОз+0,85 г NaCl + 0,2 мл 0,5%-ного
фенолового красного +100 мл воды, дважды дистилли-
рованной в стеклянной посуде).
6. Быстро добавьте 100 мкл полученного на предыдущей
стадии раствора к 0,9 мл препарата вируса (т. е. после
п. 3, см. выше).
7. Держите пробирку на льду; перемешайте содержимое с
помощью вортекса (частота интервалов 10—15 мин),
обеспечивающего тщательное перемешивание в плотно
закрытой пробирке.
<8. Поместите пробирку на 2 ч на медленно вращающийся
круг при 37 °C; инкубируйте всю ночь при 4 °C для обес-
печения полного гидролиза БПЛ.
9. Приготовьте аликвоты инактивированного вируса Сендай
объемом 20 мкл в пробирках Eppendorf, заморозьте в су-
хом льду и храните при —70 °C. При таком способе хра-
нения инактивированный вирус Сендай не теряет свойст-
во фузогенности в течение по меньшей мере 1 года.
10. Когда потребуется, разморозьте аликвоту вируса Сендай
и разведите ее раствором Хенкса без глюкозы до кон-
центрации 1000—10 000 ГАЕ/мл. Поскольку фузогенная
активность разных партий вируса Сендай может варьи-
ровать в довольно широких пределах, а также в связи
с отсутствием четкой зависимости между титром ГАЕ и
фузогенностью вируса, степень разведения определяется
в ходе предварительных экспериментов. Высокая кон-
центрация фузогенного вирусного препарата может вы-
звать интенсивный лизис клеток, низкая же, напротив,
не обеспечит эффективного слияния клеток. Судя по на-
шему опыту, разведенный препарат вируса Сендай в тече-
ние недели не теряет фузогенной активности, если пре-
парат хранится при 4 °C.
304
Глава 12
2.3. Оборудование
2.3.1. Микроманипупяционная установка
Мы пользуемся стандартным лейцевским микроманипулято-
ром [7] с фиксированным микроскопом Ergaval (Carl Zeiss,
Jena) и выпрямляющей изображение интерференционной опти-
кой Номарского. Микроскоп укомплектован объективами Х10
и Х32 с большим рабочим расстоянием и широкопольными
окулярами X 10 (см. также гл. 3; разд. 7.1).
Положительное и отрицательное давление в рабочем инст-
рументе регулируется микродозаторным насосом де Фонбрюна
(Beaudouin, Paris), а в удерживающем инструменте с помощью
простого 2 мл-шприца Wellcome Agla. Благодаря тому что пла-
стиковые трубки, входящие в систему, прозрачны, мы имеем
возможность обнаружить в ней пузырьки воздуха. Ригидность
трубок достаточна для того, чтобы рабочее давление подавалось
на кончик пипетки, не поглощаясь стенками трубки.
Стеклянные рабочие инструменты присоединяются к стан-
дартным лейцевским трубкам, которые фиксируются в инстру-
ментальной головке манипулятора.
Шприц и трубки заполняют тяжелым жидким парафиновым
маслом, при этом следует избегать попадания пузырьков воз-
духа в систему.
Микроманипуляции проводят в висячей капле в лейцевской
(Puliv) микроманипуляционной камере (внутренняя высота
3 мм), заполненной тяжелым жидким парафиновым маслом.
Камеры сразу после использования отмывают горячей водой, а
затем спиртом и хранят завернутыми в пыленепроницаемый
материал. Покровные стекла, образующие верхнюю поверхность
манипуляционной камеры, по размеру должны точно подходить
к ней. Перед употреблением их очищают спиртом, погружают в
силиконовый раствор (Sigmacote), высушивают на воздухе,
снова промывают спиртом и помещают в горячую печь для сня-
тия статического электричества.
Манипуляционную установку необходимо собрать на устой-
чивом рабочем столе на высоте, удобной для работы оператора.
Манипуляции желательно проводить в условиях кондициони-
рованного воздуха, так как высокая температура — выше 20°C
или (71° F) делает клеточные мембраны более липкими, тяжелое
парафиновое масло становится менее вязким, затрудняя работу
оператора и повышая число неудачных операций.
2.3.2. Другое оборудование
Для работы с яйцами, для загрузки и разгрузки манипуля-
ционных камер рекомендуется стандартный просвечивающий
микроскоп, например Wild М5.
Трансплантация ядер в яйца 305-
Чтобы получить тонко оттянутые микропипетки, нужен
простой электродный прибор для оттягивания пипеток. Приборы
различных конструкций можно приобрести или усовершенство-
вать в лабораторной мастерской. Для разламывания, изгибания
и полировки кончиков микропипеток требуется микрокузница де
Фонбрюна (Beaudouin, Paris), которая дает увеличение пример-
но в 100 раз и высокую точность. Микроскоп кузницы должен
быть снабжен окуляр-микрометром. В качестве нагревательной
нити используется платиновая или платино-иридиевая проволо-
ка длиной 2,5 см (диаметр 0,25—0,3 мм), изогнутая V-образно-
и горизонтально расположенная. Для придания заостренной
конической формы иглам, с помощью которых переносятся ядра,,
к нагревательной нити припаяны два стеклянных шарика-—
один на ее вершине, а другой на верхней поверхности вблизи
вершины (см. также гл. 3, разд. 9.2).
Кроме того, требуются некоторые приспособления для заост-
рения кончиков игл для переноса ядер. Их можно приобрести
или сделать в мастерской. В нашей лаборатории используется
шлифовальная поверхность, представляющая собой стеклянную-
пластинку, покрытую очень тонким алмазным диском, вращаю-
щимся со скоростью 200 об/мин (рис. 12.1).
2.4. Изготовление микроинструментов
Мы описываем ориентацию инструментов на микрокузнице
такой, как она есть в действительности, а не такой, какой мы ее
видим в микроскопе кузницы, так как инверсии в микроскопах
разных марок варьируют. Для приготовления стеклянных ми-
кроинструментов используют стандартные капиллярные трубки
из боросиликонового стекла, их наружный диаметр 1,0 мм,
внутренний диаметр 0,58 мм, длина 15 см; поставщики: Leitz,.
Clark Electromedical Instruments; Drummond Scientific Co.
Другие типы стекла, например Pyrex, при вытягивании и заост-
рении проявляют иные свойства.
2.4.1, Удерживающие пипетки
1. Вытягивайте капиллярную трубку вручную над малень-
кой бунзеновской горелкой, пока наружный диаметр не
уменьшится до 80—100 мкм, затем сломайте ее на рас-
стоянии 1,5—2 см от места сужения трубки. Оставьте ос-
новной стержень длиной около 4 см.
2. Укрепите пипетку на микрокузнице в горизонтальном по-
ложении и определите размер и характер разлома.
3. Если кончик абсолютно прямой, раскалите нить докрасна
и приблизьте к кончику, чтобы его оплавить и сократить
отверстие до 20 мкм в диаметре (рис. 12.2, Л).
20—171
306
Глава 12
Рис. 12.1. Шлифовальное устройство, изготовленное в лабораторной мастер-
ской. Пипетка зажимается клеммой, прикрепленной к видоизмененному шта-
тиву микроскопа. Для освещения используется осветитель. Пипетка устанав-
ливается над шлифовальной поверхностью с помощью ручной лупы. Шлифо-
вальная поверхность поддерживается влажной с помощью марлевого тампона,
смачиваемого дистиллированной водой.
4. На удерживающей пипетке должен быть небольшой изгиб
в месте ее сужения для того, чтобы при горизонтальном
положении пипетки ее кончик находился выше плеча.
Для этого поверните пипетку основанием вверх, нагрейте
нить и приблизьте ее к месту сужения пипетки; и без соприкос-
новения с нитью пипетка будет медленно изгибаться (рис.
12.2, Б).
2.4.2. Иглы для переноса ядер
1. Силиконируйте стеклянную капиллярную трубку из-
нутри и снаружи, погрузив ее в силиконовый раствор и
высушив в горячей печи.
2. Отрегулируйте оттягиватель игл таким образом, чтобы
вытянуть иглу, у которой вершина сужения расположена
на расстоянии 8-—9 мм от первого расширения трубки.
Оставьте толстый стержень иглы длиной около 4,5 см.
3. Укрепив иглу горизонтально на микрокузнице, очень
Трансплантация ядер в яйца
307
А
Рис. 12.2. А. Удерживающая пипет-
ка: схема готового конца пипетки.
Б. Придание изгиба плечу пипетки.
внимательно осмотрите
ее конец, чтобы убе-
диться в постепенности
конического сужения.
4. Сделайте прямой раз-
лом в том месте, где
наружный диаметр ра-
вен 20—30 мк. Чтобы
скол получился пра-
вильным, следует по-
ступить так. Нагревайте
нить накаливания, пока
она не начнет менять
цвет, и осторожно
сплавьте маленький
стеклянный шарик на
наружной поверхности
вершины нити с тонким
стержнем изготавлива-
емой иглы в нужной
точке. Когда стекло
плотно прикрепится,
но не изогнется и не су-
зится в месте прикреп-
ления, выключите на-
гревание; внезапное сокращение охладившейся нити нака-
ливания чисто расколет стекло (рис. 12.3, А).
5. Заостряйте пипетку в течение 30—60 сек на шлифоваль-
ном устройстве, сделав на ее конце скос под углом 35°.
6. Тщательно промойте в серной кислоте, сполосните ди-
стиллированной водой (ДВ), обработайте детергентом
NP-40 (он повышает неадгезивность силиконированной
поверхности) и, наконец, промойте дистиллированной
водой. Чтобы осуществить эти процедуры, пипетку с по-
мощью короткого пластикового шланга необходимо при-
соединить к игле маленького шприца и прогнать жидко-
сти через кончик пипетки вперед и назад. В нашей лабо-
ратории используется такой режим отмывки:
1. ДВ трижды
2. H2SO4 трижды
3. ДВ трижды
4. H2SO4 трижды
5. ДВ трижды
6. NP-40 трижды
7. ДВ 12 раз
8. ДВ (второй сосуд) 12 раз
В конце процедуры через пипетку для удаления остатков
воды продувается струя воздуха. Разумеется, сначала
следует убедиться в том, что заострение получилось удов-
летворительным, чтобы зря не тратить время на отмывку.
308
Глава 12
— ж
j
Рис. 12.3. Игла для переноса ядер.
А. Разламывание вытянутого капил-
ляра с внешним диаметром 25—
30 мкм. Показано положение двух
стеклянных шариков, сплавленных с
концом нити накаливания микрокуз-
ницы. Б. Положение конца пипетки
при его оплавлении. В. Положение
конца пипетки для получения острия.
Г. Заостренный конец пипеткн. Д.
Удаление шипа на конце для получе-
ния острого края. Е. Законченный
конец пипетки сбоку. Ж. Закончен-
ный конец пипетки сверху.
ней и нижней поверхностям
(рис. 12.3, Д,Е).
7. После промывания вер-
ните пипетку в горизон-
тальное положение и
ориентируйте кончик,
как показано на рис.
12.3, Б.
8. Нагревайте нить нака-
ливания, пока она не
начнет менять цвет, а
затем приблизьте конец
пипетки к стеклянному
шарику на расстояние,
достаточное для того,
чтобы сточенная по-
верхность оплавилась
до гладкости, но отвер-
стие не сузилось.
9. Поверните иглу так,
чтобы сточенное отвер-
стие было обращено в
противоположную от
наблюдателя сторону
(рис. 12.3, В).
10. Острый край на конце
пипетки позволяет про-
никнуть через блестя-
щую оболочку, не пов-
реждая мембраны яйца.
Слегка уменьшив нагре-
вание, приведите конец
пипетки в контакт со
стеклянным шариком
на вершине нити, затем
отодвиньте, оттянув на
гладком конце пипетки
тонкое острие (рис.
12.3, Г). Выключите на-
грев и уменьшите ост-
рие на конце, слегка
прикасаясь им к верх-
шарика на конце нити
11. Продолжая держать пипетку отверстием от оператора,
поверните ее в вертикальное положение на микрокузнице,
нагрейте нить до ярко-красного цвета и сделайте на
стержне пипетки, в том месте, где начинается четкое
Трансплантация ядер в яйца 309
расширение, изгиб под углом 5° (рис. 12.2,5). Это де-
лается для того, чтобы конец пипетки был на уровне ее
плеча, когда она находится в горизонтальном положении;
это позволит пипетке перемещаться по микрокапле в
вертикальной плоскости, не задевая плечом верхнюю
поверхность манипуляционной камеры. Вид окончательно
сформированного кончика пипетки сбоку (так она будет
ориентирована на микроманипуляторе) представлен на
рис. 12.3,5 и вид сверху — на рис. 12.3,Ж.
12. Тщательно высушите пипетки в сушильном шкафу и дер-
жите в чашке Петри прикрепленными к полоске Blu-tac,
расположенной на дне чашки.
2.5. Яйца и эмбрионы
2.5.1. Линии мышей
При выборе линии мышей для получения яиц следует
учесть два момента.
1. Часто возникает необходимость культивировать опериро-
ванные яйца одноклеточной стадии в течение нескольких
дней, прежде чем приступить к следующему этапу экспе-
римента. Однако известно, что у мышей большинства
линий развитие яиц in vitro блокируется на 2-клеточной
стадии (см. гл. 2, разд. 2.2.1). Исключение составляют
гибридные эмбрионы, полученные на основе C57BL. Вот
почему в качестве донора яиц мы обычно используем Fi
от скрещивания самок C57BL/6J с самцами СВА/Са (те
и другие от Bantin and Kingman).
2. При анализе результатов эксперимента удобно для дока-
зательства функциональности трансплантированного ядра
использовать такие маркеры, как окраска шерсти или
ферментативная активность. Например, животные Fi от
скрещивания C57BLXCBA имеют окраску черную агути
(АВ/аВ) и являются гомозиготами по В-форме глюкозо-
фосфатизомеразы (ГФИ-1В) [8]. Для обеспечения надле-
жащего контраста с ними скрещивают мышей аутбредной
альбиноской линии (CFLP), которые в результате тща-
тельного скрининга и скрещиваний стали гомозиготными
по изозиму А ГФИ. Анализ фермента проводят на цел-
люлозно-ацетатных пластинках с использованием Titan-
системы, поставляемой Helena Laboratories. Яйца от
самок Fj, спаренных с самцами CFLP используются для
получения андрогенетических или гиногенетических яиц.
310
Глава 12
2.5.2. Оплодотворение яйцв
Чтобы получить максимальное число яиц, у самок Fi (в воз-
расте примерно 28 дней) индуцируют суперовуляцию, инъецируя
внутрибрюшинно 7,5 ME ГСЖК, а затем через 46—48 ч 7,5 ME
чХГ. Сразу после этого самок подсаживают к самцам, одну
самку к одному самцу (см. гл. 1, разд. 5.4). Время инъекции
зависит от требующейся стадии пронуклеуса, например, инъек-
ция в 16.30 дает возможность получить яйца с мигрирующими
от поверхности пронуклеусами к середине следующего дня. На
следующее утро самок проверяют на наличие копулятивной
пробки.
Яйца с пронуклеусами получают от самок утром первого дня
беременности и освобождают их от клеток кумулюса гиалуро-
нидазой, как описано в гл. 2, разд. 2.1.
2.5.3. Активирование яиц
Яйца для активации обычно получают от гибридных самок
C57BL/CBA. Это объясняется, во-первых, отсутствием у таких
яиц блока на 2-клеточной стадии и, во-вторых, тем, что именно
для мышей этой гибридной комбинации отработаны надежные
условия активации яиц. Активацию неоплодотворенных яиц
можно стимулировать разнообразными воздействиями [9, 10].
Мы опишем метод получения гаплоидных и диплоидных парте-
ногенетических яиц, которым обычно пользуемся, — метод Кат-
бертсона. Он основан на использовании этанола в качестве
активирующего агента [11, 12].
1. Соберите яйца от суперовулировавших, но неспаривав-
шихся самок через 17—18 ч после инъекции чХГ.
2. Удалите клетки кумулюса обработкой гиалуронидазой
(гл. 2, разд. 2.1) в РВ1 (гл. 13, разд. 3.1.1) и отмойте
яйца в трех каплях М16+ВСА (гл. 2, разд. 5.2), а затем
в трех больших каплях 7%-ного этанола в М16 + БСА.
3. Держите яйца в третьей капле спирта, пока не пройдет
4,5 мин от момента помещения их в первую спиртовую
каплю. За минуту до конца этого периода захватите яйца
в пипетку с небольшим объемом спиртовой среды и по
прошествии 4,5 мин осторожно выпустите их в большую
каплю М16+БСА для отмывки. При смешивании двух
сред образуется воронка; попав в нее, яйца могут всплыть
на поверхность капли и прилипнуть, если не помочь им
опуститься на дно. Отмойте яйца проводкой по 6 каплям
М16 + БСА.
4. Если требуются гаплоидные партеногеноны, культиви-
руйте яйца в среде М164-БСА под маслом при
37,5°С, это приведет к формированию второго полярного
тельца. В течение 4 ч сформируется гаплоидный пронукле-
Трансплантация ядер в яйца
311
ус и второе полярное тельце полностью выделится.
5. Для получения диплоидных партеногенон необходимо по-
давить формирование второго полярного тельца, при этом
оба продукта второго мейотического деления останутся
в развивающемся яйце. Сразу после активации и отмывки
перенесите яйца в большую каплю под маслом среды
М16 + БСА+цитохалазин В (5 мкг/мл). (Исходный раст-
вор разводят в ДМСО до концентрации 2 мг/мл.) Инку-
бируйте в течение 3—4 ч. К этому времени в цитоплазме
будут четко видны два пронуклеуса. Очень осторожно
отмойте яйца. Для этого проводите их по девяти большим
каплям М16 + БСА и возвратите в культуру. Спустя 2 ч,
когда яйца оправятся от обработки, их следует тщательно
просмотреть и все аномальные отбросить.
2.5.4. Предимплантационные эмбрионы
Эмбрионы на стадиях дробления, используемые в экспери-
ментах по трансплантации ядер, могут быть получены либо
вымыванием их из яйцевода или матки, либо путем культиви-
рования начиная с 1- или 2-клеточной стадии in vitro (см. гл. 2,
разд. 2).
3. Перенос ядер
3.1. Установка камеры
1. Укрепите на манипуляционной камере покровное стекло с
помощью небольшого количества смеси вазелпн/парафин
(~5%-ный густой парафин), нанесенной шприцем на вы-
ступы камеры.
2. Под контролем наименьшего увеличения препаровального
микроскопа нанесите капли препарата вируса Сендай и
среды PNC на нижнюю сторону покровного стекла, вос-
пользовавшись для этого оттянутой пастеровской пипет-
кой. Сначала вблизи заднего выступа камеры поместите
большую каплю (полусфера диаметром около 3 мм) ви-
русной суспензии, затем примерно 12 маленьких капель
(полусферы диаметром около 1,5 мм) среды PNC, рас-
положив их в ряд по центру камеры (рис. 12.4,А).
3. Осторожно заполните камеру до боковых граней покров-
ного стекла тяжелым жидким парафином с помощью уко-
роченной пастеровской пипетки.
4. Введите оттянутой пастеровской пипеткой предназначен-
ные для этого яйца в капли. Во избежание путаницы по-
лезно всегда следовать одному и тому же порядку разме-
щения яиц по каплям: например, в первые четыре капли,
ближайшие к капле Сендай, поместить донорские яйца
312
Глава 12
Б
Рис. 12.4. А. Манипуляционная камера с каплями, нанесенными на нижнюю
поверхность покровного стекла. Б. Манипуляционная камера с инструмента-
ми, приготовленными к введению в камеру, внд сбоку.
группами по четыре, следующую каплю оставить пустой,
в нее можно отсаживать поврежденные ядра и дебрис и,
наконец, в оставшиеся капли поместите парами яйца-
реципиенты. При этом в инкубаторе уже должна быть
готова культуральная чашка с серией больших капель
М16 + БСА под маслом для приема оперированных яиц.
3.2. Установка инструментов
1. Поместите загруженную камеру на предметный столик
микроскопа и сфокусируйте объектив малого увеличения
на яйце. Затем сдвиньте камеру к задней границе столика.
Плоскость фокусного расстояния находится чуть выше дна
операционных капель; с этого момента не прикасайтесь
к микрометрическому винту, пока не установите инстру-
менты. Фокусирование инструментов производится подъ-
емом и опусканием Манипуляторов.
2. Установите оба манипулятора в положении, позволяющем
перемещаться в горизонтальной плоскости-—оно соответ-
ствует отметке «О» на шкале (см. гл. 3, рис. 3.8, правый
манипулятор).
3. Соедините удерживающую пипетку посредством трубки
со шприцем Agla (после того, как в системе не останется
пузырьков воздуха), затем закрепите трубку в инструмен-
тальной головке одного из манипуляторов (обычно лево-
го).
4. Расположите инструментальную головку так, чтобы удер-
живающая пипетка располагалась с небольшим накло-
ном— под углом <5°.
Трансплантация ядер в яйца
313
5. Введите кончик пипетки в поле зрения микроскопа и сфо-
кусируйте регулировкой высоты манипулятора. С помо-
щью шприца Agla заполните пипетку маслом до самого
кончика, а затем отодвиньте собранный блок на край мик-
роскопа.
6. Точно так же присоедините пипетку для трансплантации
ядер к шприцу де Фонбрюна на правом манипуляторе и
заполните маслом. Позаботьтесь о том, чтобы скос кон-
чика пипетки был виден в микроскопе абсолютно верти-
кально (см. рис. 12.3,Ж). Отодвиньте собранный блок в
сторону.
7. Наконец, поставьте в центр микроскопа манипуляционную
камеру с операционной каплей в поле зрения. Относитель-
ные углы и положения инструментов должны соответство-
вать рис. 12.4, Б.
8. Осторожно введите в камеру инструменты, сначала один,
затем другой, пока они не окажутся в капле. Каждый
инструмент после использования должен быть во избежа-
ние столкновения отодвинут в капле в свое крайнее поло-
жение вперед или назад.
3.3. Извлечение и перенос ядер
В качестве примера применения метода ниже будет деталь-
но описано получение гетерозиготных диплоидных андрогене-
тических яиц. В качестве доноров и реципиентов использованы
яйца на пронуклеарной стадии, полученные от скрещивания гиб-
ридных самок с самцами линии CFLP.
1. Соберите яйца примерно через 18 ч после инъекции чХГ и
после удаления клеток кумулюса (гл. 2, разд. 2.1), культи-
вируйте в течение 1 ч в М16+БСА (гл. 2, разд. 5.2).
2. Отберите ранние пронуклеарные яйца и рассортируйте их
в зависимости от положения пронуклеуса: те из них, у
которых пронуклеусы занимают четко периферическое по-
ложение, используйте в качестве реципиентов, яйца более
поздних стадий со сближенными и более крупными про-
нуклеусами— в качестве доноров. Примерно по 30 яиц
поместите в большие капли среды PNC под маслом и до
операции культивируйте в негазованном инкубаторе по
крайней мере 15 мин.
3. Приготовьте яйца-реципиенты, удалив женский пронукле-
ус. Ориентируйте яйцо таким образом, чтобы при удержи-
вании его за область полярного тельца оба пронуклеуса
были в фокусе и чтобы у прижатой к яйцу скошенной
пипетки мужской пронуклеус остался позади, а женский
лежал впереди. Это поможет более четко различать про-
314
Глава 12
нуклеусы после вхожде-
ния пипетки, когда мор-
А фология яйца изменится.
Чтобы сделать правильный
выбор между пронуклеусами,
следует учитывать, что жен-
Б ский пронуклеус обычно имеет
заметно меньшие размеры и
на ранних стадиях располага-
ется вблизи от второго поляр-
в ного тельца (см. гл. 2, рис.
2.1). Поскольку спермий мо-
жет войти в яйцо в любой точ-
Г|ке поверхности, мужской про-
нуклеус иногда может занять
аналогичное положение по
1
Вирус
Буферный
Мужской Масло
пронукпеус
отношению к полярному тель-
цу. Абсолютные размеры про-
нуклеусов на любой стадии
могут варьировать у разных
яиц, но женский пронуклеус
всегда меньше. С другой сто-
роны, у яиц более поздних ста-
дий, где пронуклеусы мигриро-
вали к центру и увеличились,
Рис. 12.5. Получение гетерозиготных
диплоидных андрогенетических яиц.
А—Г. Удаление женского пронук-
леуса из оплодотворенного яйца для
получения гаплоидного андрогеиети-
ческого яйца. Д—Ж. Перенос второ-
го мужского пронуклеуса в гаплоид-
ное андрогенетическое яйцо. 3. После
слияния.
различия в размерах иногда
перестают быть очевидными.
Яйца, у которых идентифика-
ция пронуклеусов вызывает
сомнения, следует отбросить.
(Число ядрышек для иденти-
фикации значения не имеет.}
4. Маневрируйте яйцом с
помощью удерживаю-
щей пипетки и пипетки
для энуклеации до тех:
пор, пока не появится-
возможность осторож-
но, но прочно захватить
его удерживающей пи-
петкой, при этом часть
блестящей оболочки и района полярного тельца засасы-
вается в удерживающую пипетку (рис. 12.5,А).
5. Приподнимите немного яйцо со дна капли, сфокусируйте
микроскоп на удаленном крае блестящей оболочки и по-
ставьте в фокус энуклеационную пипетку.
Трансплантация ядер в яйца
315
6. Осторожно прижмите энуклеационную пипетку к яйцу,
вдавив его блестящую оболочку, но не прорывая мембра-
ну (рис. 12.5,5).
7. Придвиньте кончик пипетки к женскому пронуклеусу и
осторожно сообщите пипетке небольшое отрицательное
давление. Мембрана яйца, деформируясь, втянется в пи-
петку, за ней последует пронуклеус.
8. Как только пронуклеус окажется внутри кончика пипет-
ки, осторожно выдвиньте пипетку из блестящей оболочки
(рис. 12.5,5). При этом мембрана яйца останется в уз-
кой трубке конца пипетки. Отщипните это выпячивание
мембраны, скользнув кончиком пипетки по краю отвер-
стия в блестящей оболочке. Разорванные края мембраны
яйца обычно сливаются по обе стороны разрыва
(рис. 12.5, Г).
9. Повторите операции, указанные в пп.З—8, со всеми яй-
цами-репициентами и перенесите женские пронуклеусы в
новую каплю.
10. Подвиньте камеру так, чтобы в поле зрения оказались
донорские яйца. Перемещайте яйцо, пока мужской про-
нуклеус не окажется в нужном положении, затем прочно
захватите яйцо удерживающей пипеткой. На этот раз не
обязательно фиксировать яйцо в районе полярного тель-
ца.
11. Пройдите пипеткой через блестящую оболочку и захва-
тите небольшой фрагмент цитоплазмы; отщипните от
яйца этот цитопласт. Он будет выступать в роли буфера.
12. Извлеките мужской пронуклеус. При вхождении в эну-
клеационную пипетку ядерная мембрана может слегка
деформироваться. Это не повредит ядро, но, если пипетка
слишком узка или просвет кончика ограничен прилипши-
ми капельками масла или частицами дебриса, некоторые
ядрышки могут сдвинуться в мембране и выскользнуть
из нее. В таком случае отбросьте ядро, постарайтесь
прочистить кончик пипетки или замените ее новой.
13. Выдвиньте из капли обе пипетки; в энуклеационной пи-
петке все еще находится мужской пронуклеус.
14. Подвиньте камеру так, чтобы в поле зрения оказалась
капля суспензии вируса Сендай. Введите энуклеацион-
ную пипетку в эту каплю.
15. Сообщите пипетке положительное давление, достаточное
для того, чтобы нуклеопласт частично вышел из кончи-
ка пипетки и омывался вирусной суспензией, затем втя-
ните его обратно на расстояние около 100 мкм так, чтобы
небольшой объем вирусного препарата занял кончик
пипетки.
316
Глава 12
16. Выведите пипетку из этой капли. Поставьте в поле зре-
ния каплю с яйцами-реципиентами и захватите яйцо так
же, как вы это делали при энуклеации (место легко уз-
нать по полярному тельцу и деформации блестящей
оболочки) (рис. 12.5,В).
17. Введите пипетку для переноса в первоначальное отвер-
стие в блестящей оболочке и осторожно выпустите вирус-
ный препарат и нуклеопласт в перивителлиновое прост-
ранство (рис. 12.5,£; Ж). Дополнительный цитопласт,
расположенный в пипетке проксимальнее кариопласта, во
время этой операции способствует выталкиванию карио-
г пласта из кончика пипетки, и, кроме того, играет роль
буфера, регулирующего равномерность движения масла.
Если пронуклеус в кариопласте сопровождается большим
количеством цитоплазмы, размеры кариопласта можно
уменьшить: как только пронуклеус окажется в блестящей
оболочке, выведите пипетку, отщипнув излишки цитоплаз-
мы, как уже было описано выше.
Слияние может произойти в любой момент примерно в тече-
ние следующего часа, а иногда и через несколько минут после
переноса (рис. 12.5,3).
18. Повторите процедуру переноса со всеми реципиентными
яйцами.
19. Перенесите камеру на препаровальный микроскоп и со-
берите все яйца вместе, быстро отмойте их и культиви-
руйте в М16+БСА.
20. В конце эксперимента промойте все яйца в девяти кап-
лях свежей М16+БСА, внимательно проверьте, произо-
шло ли слияние и продолжайте культивировать в этой
среде.
3.4. Другие варианты переноса ядер
Гетерозиготные диплоидные гиногенетические яйца могут
быть получены способом, описанным в разд. 3.3, или путем
использования гаплоидных партеногенетических яиц (рис. 12,6).
Обнаружено, что развитие гетерозиготных яиц с материнским
геномом не отличается от развития партеногенетических яиц,
которые были диплоидизированы путем подавления формирова-
ния второго полярного тельца. Подобным образом можно по-
лучить разнообразные анеуплоидные яйца. Перенос в периви-
теллиновое пространство в течение короткого времени одного
за другим нескольких кариопластов, может привести к их слия-
нию с плазматической мембраной яйца и к созданию триплоид-
ных или тетраплоидных яиц с разным числом материнских и
отцовских геномов.
Трансплантация ядер в яйца
317
Рис. 12.6. Получение гетерозиготного диплоидного гиногенетнческого яйца
путем использования в качестве донора оплодотворенного яйца, а в качестве
реципиента — гаплоидного партеногенетического яйца. А—Г. Извлечение жен-
ского пронуклеуса из оплодотворенного яйца. Д—Е. Перенос женского пронук-
леуса в гаплоидное партеногенетическое яйцо. Ж. Перед слиянием. 3. После
слияния.
Техника манипуляций с ядрами от эмбрионов более поздних
стадий развития в основном та же, что и описанная в разд. 3.3
[13, 14]. Размер пипетки для переноса должен быть немного-
изменен в соответствии с размером трансплантируемого ядра.
Ядра эмбрионов на 8-клеточной и более поздних стадиях эмбри-
онального развития часто бывают плохо видны, особенно трудно-
проследить за слиянием нуклеопласта с яйцом. Существует
318
Глава 12
Рис. 12.7. Пронуклеопласты. Муж-
ские пронуклеусы с минимальным ко-
личеством цитоплазмы, извлеченные
из оплодотворенных яиц.
возможность переноса ядер в
2-клеточные бластомеры. В
этом случае, как бы тщатель-
но ни была проделана опера-
ция, кариопласт имеет тенден-
цию проскользнуть в щель
между бластомерами, поэтому
имеет смысл оставить опери-
рованные эмбрионы в манипу-
ляционной камере и наблю-
дать за ними, пока не про-
изойдет слияние. Таким обра-
зом, нам удалось получить 2-
клеточные химеры: удалив
ядро из одной клетки 2-клеточ-
ного эмбриона FiXFi(F2), мы
заменили его ядром 2-клеточ-
ного эмбриона CFLPXCFLP.
Поскольку химерные эмбрио-
ны все еще находятся в блестящей оболочке, их можно перене-
сти на 2- или 4-клеточной стадии в яйцеводы приемной матери
1-го дня «беременности» (гл. 13, разд. 6.4.5). Из шести таких
эмбрионов, перенесенных на 4-клеточной стадии, дошли до мо-
мента родов и родились все шесть, из них два оказались явно
химерными (судя по окраске шерсти и типу глюкозофосфатизо-
меразы крови), один полностью типа CFLP, три относились к
типу F2.
3.5. Кариопласты с минимальным количеством цитоплазмы
Если требуются ядра или пронуклеусы, практически лишен-
ные цитоплазмы, в частности, для биохимического анализа, они
могут быть приготовлены в виде кариопластов с минимальным
объемом цитоплазмы. Существуют два способа уменьшения
объема цитоплазмы, следующей за ядром в энуклеационной
.пипетке.
1. Сделайте так, чтобы ядро выступило из кончика пипетки,
а затем проведите эту пипетку через границу разделения
среда/масло, пока кариопласт не оторвется от цитоплаз-
мы, оставшейся в пипетке.
2. Введите часть кончика энуклеационной пипетки в конец
удерживающей пипетки, немного выпустите ядро из эну-
клеационной пипетки, а затем к обеим пипеткам приложи-
те небольшое отрицательное давление; кариопласт разор-
вется сразу позади ядра. Эти приемы более эффективны
в случае больших излишков цитоплазмы, чем в случае
малых кариопластов. Описанные операции позволяют по-
Трансплантация ядер в яйца
319'
лучить кариопласты, в которых цитоплазма составляет
10% общего объема, но чем мельче ядра, тем труднее
получить это соотношение (рис. 12.7).
3.6. Разделение яиц
В тех случаях, когда обработка яиц цитохалазином или дру-
гими ядами нежелательна и требуются крупные кариопластьи
их можно получить, оперируя освобожденные от блестящей обо-
лочки яйца (гл. 2, разд. 3.2.1) в висячей капле РВ1 (гл. 13,
разд. 3.1.1). Для операции потребуется большая пипетка типа
присоски с внутренним отверстием около 40 мкм в диаметре
и сплошная стеклянная игла диаметром 10—12 мкм с тупым оп-
лавленным концом (см. гл. 3, разд. 9.3); оба инструмента
должны быть изготовлены из силиконированной капиллярной
трубки, обработаны NP-40 и хорошо отмыты. Яйцо осторожно,
втягивают в удерживающую пипетку и снова выпускают, вслед-
ствие чего оно принимает удлиненную форму; сплошную иглу
помещают поперек яйца в месте предполагаемого разделения
и опускают через границу среда/масло [15]. Тем же способом
можно удалить одно полярное тельце или оба. Степень выжи-
ваемости яиц, оперированных в РВ1, ниже, чем оперированных
в PNC, но не настолько, чтобы этой средой нельзя было поль-
зоваться.
4. Оценка результатов
Частота слияния во многом зависит от качества препарата
вируса Сендай. При хорошем качестве препарата и соблюдении-
всех условий частота слияния превышает 90%. Если реконст-
рукция яйца осуществилась, с ним можно обращаться как с лю-
бым другим яйцом. Такие яйца можно культивировать in vitro
для изучения предимплантационного развития (см. гл. 2) или
перенести псевдобеременным реципиентам для наблюдения над
развитием in vivo. Эмбрионы 1-, 2- или даже 4-клеточной стадии
могут быть перенесены в яйцеводы реципиента 1-го дня, а на
стадии поздней морулы/ранней бластоцисты — в матку реци-
пиента 3-го дня (см. 13, разд. 6.4.6). (При этом рекомендуется
в другой яйцевод или рог матки перенести нормальный эмбрион
для контроля эффективности псевдобеременности и оценки ин-
дивидуальных различий.) Реконструированные яйца можно
использовать при получении агрегационных химер или при кон-
струировании бластоцист [16], а также для анализа белков
и ферментов (см. гл. 6—8) или хромосом (см. гл. 5).
Развитие диплоидных партеногенетических, гиногенетичес-
ких или андрогенетических эмбрионов после их переноса псев-
320
Глава 12
добеременным реципиентам редко продолжается дольше 10 или
11 дня [17—20]. На этой стадии их морфологическая организа-
ция в большей или меньшей степени аномальна, но ткани живы
и достаточно здоровы, чтобы быть объектом биохимического
анализа. Анализ эмбрионов, прошедших половину срока беремен-
ности, описан в гл. 3. Химеры, полученные комбинацией ано-
мальных эмбрионов с нормальными, предоставляют возмож-
ность анализа на поздних стадиях беременности или после рож-
дения.
5. Перспективы и ограничения
техники трансплантации ядер яиц млекопитающих
Несмотря на некоторые успехи в применении вирусов и хи-
мических соединений (полиэтиленгликоль, ПЭГ), в качестве
агентов, вызывающих слияние [21, 22], следует признать, что
их использование в экспериментах на яйцах млекопитающих
имеет некоторые существенные ограничения. Мы обнаружили,
что ПЭГ, наиболее широко используемый химический фузоген
соматических клеток, очень ненадежен в исследованиях на ран-
них эмбрионах. Его активность может варьировать, кроме того,
он способен вызывать интенсивный лизис и препятствовать раз-
витию, поскольку с трудом удаляется с плазматической
мембраны.
Несмотря на то, что вирус Сендай оказался более удачным
фузогеном для яиц млекопитающих, применение и этого агента
имеет ряд недостатков. Всегда существует риск, хотя и незна-
чительный, что некоторые вирусные частицы в препарате оста-
нутся вирулентными, а это имело бы катастрофические послед-
ствия для колонии мышей. Мы обнаружили, что вирус Сендай
в качестве фузогена эффективен для кариопластов яиц и очень
ранних эмбрионов. При попытках слить клетки эмбрионов более
продвинутых стадий развития или тканей взрослых животных
степень слияния падает до 0—10%. Далее, если вирус Сендай
был применен для слияния кариопласта с яйцом, бластомеры,
образовавшиеся из такого реконструированного яйца, уже не
могут впоследствии служить донорами ядер, так как повторное
применение вируса не будет эффективным. Можно полагать, что
причиной этого феномена является потеря рецепторов клеточ-
ной поверхности, способных связывать вирус. Во всяком слу-
чае, это обстоятельство исключает возможность серийных ядер-
ных трансплантаций, осуществляемых только с помощью виру-
•са Сендай. Кроме того, свойства вируса, как и ПЭГ, варьируют
в зависимости от партии.
Наиболее перспективным нам представляется метод слияния
клеток, включающий использование электрического тока [23].
Трансплантация ядер в яйца 321
Этот метод в высшей степени эффективен. Его основное преи-
мущество заключается в возможности менять условия в зави-
симости от типа клетки и осуществлять серийные ядерные
трансплантации. В соответствии с этой методикой клетки сна-
чала получают импульс переменного тока, вызывающий обра-
зование тесных контактов и соединение, или «агглютинацию».
Вслед за этим следует импульс постоянного тока, приводящий
к разрушению клеточных мембран в месте контакта и в конеч-
ном счете к слиянию клеток. При этом силу тока, длительность
воздействия, число импульсов, а также среду, в которой осу-
ществляется слияние и затем культивируются клетки, темпера-
туру и предварительную обработку клеток можно варьировать,
создавая оптимальные условия для слияния клеток данного
типа. С помощью этого метода уже достигнуты определенные
успехи [24, 25], следовательно, можно считать, что некоторые
из параметров слияния уже установлены. Однако при значи-
тельных различиях в размере клеток, как, например, в случае
крупного яйца, сливающегося с маленьким кариопластом в опи-
санных выше экспериментах с ядерными трансплантациями, воз-
никает проблема точного соединения клеток. Эта проблема не
решена. Кроме того, значительная разница в стадии развития
и типе сливаемых клеток создает необходимость подбора спе-
циальных условий для слияния. Можно предположить, что, если
физические параметры для данной системы будут установлены,
слияние клеток с помощью электрического тока даст надежные
результаты и поможет избежать осложнений, связанных с ис-
пользованием вирусных и химических фузогенов.
Благодарности
Мы благодарны Саре Хаулет за полезное для нас обсужде-
ние, Катарине Струд за рисунки и диаграммы, а Полю Майлсу
и Джеффу Лисону за изготовление оттягивателя игл и прибора
для затачивания.
Литература
1. McGrath J., Seller D. (1983). Science, 220, 1300.
2. Whittingham D. G. (1971). J. Reprod Fert. (Suppl.), 14, 7.
3. Whittingham D. G„ Wales R. G. (1969) Aust. J. Biol. Sci., 22, 1065.
4. Harris H„ Watkins J. F. (1965). Nature, 205, 640.
5. Giles R. E., Ruddle R. H. (1973). In Vitro, 9, 103.
6. НеЦ J. M., Enders J. F. (1968). Proc. Soc, Exp. Biol. Med., 127, 260.
7. Gardner R. L. (1978), In: Methods in Mammalian Reproduction. Daniel J. C.
(ed.), Academic Press, New York, p 137.
8. Chapman V. M., Whitten W. K-, Ruddle F. H. (1971). Dev. Biol., 26, 153.
9. Kaufman M. H. (1978). In: Methods in Mammalian Reproduction. Daniel J. C.
(ed.). Academic Press, New York, p. 21.
21—171
322
Глава 12
10. Kaufman М. Н. (1983). Early Mammalian Development: Parthenogenetic
Studies. Cambridge University Press, Cambridge.
11. Cuthbertson K. S. R. (1983). J. Exp. Zool., 226, 311.
12. Kaufman M. H. (1982). J. Embryol. Exp. Morphol., 71, 139.
13. McGrath J., Setter D. (1984). Science, 226, 1317.
14. Surani M. A. H., Barton S. C„ Norris M. L. (1986). Cell, 45, 127.
15. Barton S. C„ Surani M. A. H. (1983). Exp. Cell Res., 146, 187.
16. Barton S. C., Adams C. A., Norris M. L„ Surani M. A. H. (1985). J. Embryol.
Exp. Morphol., 90, 267.
17. Surani M. A. H., Barton S. C. (1983). Science, 222, 1034.
18. Surani M A. H., Barton S. C., Norris M. L. (1984). Nature, 308, 548.
19. Mann J. R„ Lovell-Badge R. H. (1984). Nature, 310, 66.
20. Barton S. C., Surani M. A. H., Norris M. L. (1984). Nature, 311, 374.
21. Eglitis M. A. (1980). J. Exp. Zool., 213, 309.
22. Spindle A. (1981). Exp. Cell Res., 131, 465.
23. Zimmermann V., Vienken J. (1982). J. Membr. Biol., 67, 165.
24. Kubiak J. Z„ Tarkowski A. K. (1985). Exp. Cell Res., 157, 561.
25. Willadsen S. M. (1986). Nature, 320, 63.
ГЛАВА 13
НИЗКОТЕМПЕРАТУРНАЯ КОНСЕРВАЦИЯ
ЯИЦ И ЭМБРИОНОВ МЫШИ
Маури Вуд, Дэвид Уиттингхэм,
Уильям Ролл1
1. Введение
Методы хранения ооцитов и эмбрионов в жидком азоте (ЖА)
чрезвычайно важны для развития животноводства, биологии
и медицины. Исследователь, работающий в области биологии
развития млекопитающих, должен иметь в распоряжении обшир-
ный набор мутантных, инбредных и сингенных линий мышей.
Однако получение и поддержание таких линий обходится до-
рого и требует больших затрат времени. Наличие банка эм-
брионов нужных генотипов позволяет не опасаться случай-
ной утраты ценного генетического материала в результате оши-
бок скрещивания, генетических контаминаций или болезней и
дает возможность обойтись без дорогостоящего содержания
животных, не требующихся в данное время. Более того, упро-
щаются международные перевозки: транспорт эмбрионов дешев-
ле и гуманней, чем транспорт животных. Важно также, что
число болезней, переносчиками которых могут быть эмбрионы,
ниже, чем соответствующий показатель для животных. Это де-
лает менее жесткими карантинные ограничения.
Криоконсервация эмбрионов человека имеет особое значение
в связи с проблемой бесплодия. Методы стимуляции овуляции
и оплодотворения in vitro дают возможность получить большее
число эмбрионов, чем может быть немедленно имплантировано
матери. Консервация дополнительных эмбрионов для переноса
в последующих циклах повышает возможность удачной бере-
менности. Хранение неоплодотворенных яйцеклеток позволяет
обойти правовые и этические ограничения, связанные с консер-
вацией эмбрионов человека. Замороженные яйцеклетки могут
оказаться совершенно необходимыми, если женщина утрачивает
нормальную функцию яичников. Однако следует подчеркнуть,
что убедительные доказательства безопасности хранения полу-
чены только для замороженных эмбрионов, в отношении яиц,
консервированных при —196 °C, таких данных нет.
1 М. J. Wood, D. G. Whittingham. MRC Experimental Embryology and Tera-
tology Unit, Woodinansterne Road, Carshalton, Surrey SM5 4EF, UK. №. F. Rail.
Rio Vista International, Route 9, Box 242, San Antonio TX78227, USA.
21* 323
324
Глава 13
Метод консервации мышиных эмбрионов путем медленного
охлаждения (0,2—2°С/мин) в присутствии диметилсульфоксида
(ДМСО) и медленного оттаивания (4—25°С/мин) [1,2] вскоре
стал применяться для ооцитов мыши [3] и эмбрионов других
видов, включая человека [4, 5]. По мере того как создание
банков эмбрионов становилось частью повседневных медико-
биологических исследований и практического животноводства,
в методику был внесен ряд модификаций. В настоящее время
в мировой практике используется несколько вариантов, подра-
зумевающих применение разных криопротекторов, менее дли-
тельных процедур охлаждения и быстрого оттаивания. К сожа-
лению, ни в одном случае сравнительный анализ различных
модификаций методики замораживания не производился, поэто-
му мы не можем судить о преимуществах той или иной процеду-
ры. Не так давно живое потомство из эмбрионов мыши удалось
получить в результате их консервации при —196 °C без образо-
вания льда, т. е. не путем замораживания, а путем витрифика-
ции (превращения эмбрионов в «стеклянные») [6]; в настоящее
время разрабатываются различные модификации этой методи-
ки [7, 8].
Мы ограничимся описанием:
1) методов, используемых для замораживания ооцитов и эм-
брионов в нашей лаборатории (они были разработаны
в расчете на простые и недорогие приборы, однако впол-
не пригодны и для работы с современными биологически-
ми замораживателями);
2) метода витрификации, который позволяет нам получать
живых эмбрионов (правда, с меныней степенью выжи-
ваемости, чем при использовании стандартной техники
замораживания).
Компетентное обсуждение теоретических аспектов замора-
живания эмбрионов можно найти в работах [4, 9 и 10].
2. Стадии развития
Эмбрионы всех предимплантационных стадий развития мы-
ши после замораживания при —196 °C дают живое потомство.
Между тем после витрификации только 8-клеточные эмбрио-
ны [6], морулы и ранние бластоцисты [7] способны продол-
жать развитие до поздних плодов или живорожденного по-
томства.
Существует несколько причин, по которым для хранения
предпочтительно использовать эмбрионы именно 8-клеточпой
стадии развития.
1. Они менее чувствительны к манипуляциям in vitro, чем
эмбрионы 1- и 2-клеточной стадии.
Низкотемпературная консервация яиц и эмбриоион мыши 325
2. Их способность к дальнейшему развитию хорошо корре-
лирует с морфологией. Ранние аномалии легко выявля-
ются и все эмбрионы, отстающие в развитии или фраг-
ментирующиеся, сразу отбрасывают.
3. Процедура их замораживания несколько упрощена по
сравнению с таковой для бластоцист.
4. В случае повреждения одного или двух бластомеров ос-
тавшиеся могут обеспечить развитие жизнеспособного
плода.
5. Культивирование in vitro в течение 24 ч оттаявших эм-
брионов позволяет отобрать те из них, которые возобно-
вили дробление, и использовать их для переноса псевдо-
беременной самке.
Замороженные неоплод отворенные яйца мыши могут слу-
жить моделью 1) для разработки методов консервации ооцитов
человека; 2) для изучения клеточной реакции на низкие тем-
пературы; кроме того, их можно использовать для оплодотворе-
ния in vitro в тот момент, когда это удобно исследователю.
2.1. Источник ооцитов и эмбрионов
Неоплодотворенные или оплодотворенные яйца и эмбрионы
на стадиях дробления получают в результате стимуляции ову-
ляции у мышей инъекциями ГСЖК и чХГ (см. гл. 1, разд. 5.4).
Данных о том, что предварительная гормональная стимуляция
самок-доноров может повлиять на жизнеспособность яиц после
замораживания и оттаивания, нет. Однако известно, что неко-
торые линии мышей устойчивы к стимуляции гонадотропином,
в таких случаях оплодотворенные яйца и эмбрионы собирают
от половозрелых самок, овулировавших естественным путем.
Известно также, что у большинства гибридных самок Fi проис-
ходит надежная суперовуляция. Таким образом, в тех случаях,
когда генетический фон не имеет значения, затруднения с ин-
дукцией суперовуляции можно обойти, спаривая чувствительных
к гонадотропину гибридных самок Fi с самцами, несущими
нужный ген.
2.2. Сбор ооцитов и эмбрионов
Для освобождения недавно овулировавших ооцитов из яйце-
вода между 13 и 14,5 ч после инъекции чХГ стенки ампулы
разрывают глазным пинцетом (см. гл. 2, разд. 2.1.1). Ооциты
замораживают либо в интактных кумулюсных массах, либо
после удаления клеток кумулюса гиалуронидазой (гл. 2,
разд. 2.1.1). В последнем случае их следует собрать и отмыть
от фермента до того, как они будут помещены в пробирки для
замораживания.
22—171
326
Глава 13
Оплодотворенные яйца и эмбрионы на стадиях дробления
вымывают из репродуктивного тракта через определенные ин-
тервалы времени после овуляции или инъекции чХГ (гл. 2,
разд. 2.2). Эмбрионы всех стадий собирают в РВ1 или среду М2
(разд. 3.1) для замораживания или в среды РВ1, М2 или НВ1
(разд. 4.1) для витрификации.
3. Замораживание
Образование большого количества льда в любой клетке млеко-
питающих является основным фактором, приводящим к ее ги-
бели при оттаивании. Причиной летального исхода может быть
как механическое повреждение клетки кристаллами льда, так
и осмотический шок в результате образования или таяния льда.
Поэтому успех замораживания яиц и эмбрионов зависит от сте-
пени обезвоживания клеток, препятствующей формированию
внутриклеточного льда.
Удаление воды из клеток производится одним из двух спо-
собов.
1. При субнулевых температурах: во время образования
льда в суспензионной среде концентрация внеклеточных
растворов возрастает, давление паров воды внутри клетки
становится выше, чем снаружи, и вода устремляется из
клетки через мембрану для установления равновесия.
2. Перед охлаждением до субнулевых температур: прибав-
ление раствора сахарозы к суспензионной среде приводит
к частичному обезвоживанию бластомеров. Этот способ
удаления воды можно сочетать с выдерживанием при от-
носительно высоких субнулевых температурах (обычно
около —30 °C) (для дальнейшей потери воды) перед бы-
стрым охлаждением до —196 °C [11]. Возможен также
перенос эмбрионов из комнатной температуры прямо
в жидкий азот [12].
Процедуры, связанные с применением непроникающих рас-
творов для обезвоживания эмбриона перед охлаждением, отно-
сительно новы и недостаточно опробованы. Будущее покажет,
будут ли они включены в стандартный метод создания банка
эмбрионов. С другой стороны, постепенное обезвоживание
в процессе медленного охлаждения с успехом используется на
протяжении последних 14 лет во многих лабораториях. Описан-
ные здесь методы основаны на процедурах охлаждения, разра-
ботанных для консервации эмбрионов Уиттингхэмом, Лейбо
и Мейзуром [1] и Уилмутом [2], а также на модификациях
их методик [13]. Важнейшие характеристики данных методов:
1) выбор суспензионной среды;
2) присутствие криопротекторов в процессе охлаждения;
Низкотемпературная консервация яиц и эмбрионов мыши 327
Таблица 13.1. Схема опыта по замораживанию — оттаиванию ооцитов
и эмбрионов
1. Маркируйте ампулу и стерилизуйте ее нагреванием (разд. 3.1.2).
2. Внесите 0,15 мл среды (разд. 3.1.1.) в ампулу (разд. 3.1.3).
3. Внесите пипеткой ооциты/эмбрионы в ампулу (разд. 3.1.3).
4. Охладите образец до 0 °C.
5. Через 10 мин добавьте. к образцу 0,15 мл 3 М ДМСО (разд. 3.2.3).
6. Через 15 мин перенесите ампулу в баню для сидинга (разд. 3.3.1) при
—6 °C.
7. Через 2 мин индуцируйте процесс кристаллизации (сидинг).
8. Через 5 мин перенесите образцы с образовавшимся льдом в охлаждаю-
щую баню (разд. 3.4.2) при —6 °C.
9. Охлаждайте со скоростью ~0,5°С/мин до —40 °C либо ниже —70 °C
до помещения ампулы в ЖА (разд. 3.4.3).
10. Отогревайте образцы с соответствующей скоростью (разд. 3.5).
11. Разведите ДМСО (разд. 3.6).
3) индукция образования льда (сидинг) при температуре
ниже точки замерзания среды;
4) медленное охлаждение (—0,5°С/мин), заканчивающееся
при —40 °C или при температурах ниже —70° в резуль-
тате погружения в жидкий азот (ЖА);
5) хранение при —196 °C в ЖА;
6) осторожное удаление криопротектора после оттаивания
для предотвращения осмотического шока.
Факторы, влияющие на выживание, взаимозависимы, и из-
менение одного из них создает необходимость изменения осталь-
ных, чтобы выживаемость осталась на прежнем уровне.
Мы подчеркиваем, что для удачного замораживания яиц или
эмбрионов необходимо следовать только одной прописи.
В табл. 13.1 описана методика, которой пользуются двое из нас
(М. В. и Д. У.) для замораживания ооцитов и эмбрионов.
3.1. Приготовление образца
3.1.1. Выбор среды для замораживания
Чтобы яйца и эмбрионы не повредились в результате дли-
тельных воздействий неконтролируемых изменений pH среды,
забуференной СОг, материал следует собирать и замораживать
в средах со стабильным pH (от 7,2 до 7,4) в воздухе.
Перечень обычно используемых сред:
а) простой физиологический раствор (PBS);
б) забуференный фосфатом солевой раствор Дульбекко
(РВ1: [14], табл. 13.2);
в) эмбриональная культуральная среда, забуференная
HEPES (М2: [15]; табл. 13.2, см. также гл. 2, разд. 5.2).
22*
328
Глава 13
Таблица 13.2. Состав сред для замораживания ооцитов и эмбрионов
Компонент РВ1 М2
г/л | мМ r/л | мМ
NaCl 8000 136,87 5,533 94,66
КС1 0,200 2,68 0,356 4,78
СаС12-2Н2О 0,132 0,90 0,252 1,71
КН2РО4 0,200 1,47 0,162 1,19
MgCl2-6H2O 0,100 0,49 — —
Na2HPO4- 12Н2О 2,898 8,09 — —
MgSO4-7H2O NaHCOs — — 0,293 0,349 1,19 4,15
HEPES1’ .— — 4,969 20,85
Пируват Na2* Лактат Na3* 0,036 0,33 0,036 2,610 0,33 23,28
Глюкоза 1,000 5,56 1,000 5,56
БСА4) 3,000 — 4,000 —
Пенициллин G (соль ка- 0,060 — 0,060 —
лия) Сульфат стрептомицина — —. 0,050 —
Феноловый красный 0,010 — 0,010 —
') Calbiochem-Boehring: Ultrol.
2) Calbiochem-Boehring.
3) Sigma grade DL, 60%-ный сироп.
4) Кристаллический: Miles-Serovac.
Среды РВ1 и М2 обогащены пируватом, глюкозой и БСА;
М2, кроме того, содержит лактат. Эти источники энергии пред-
отвращают истощение метаболических пулов в эмбрионах в про-
цессе сбора.
3.1.2. Выбор и подготовка контейнере дпя образцов
Ооциты и эмбрионы можно замораживать в фиалах с за-
винчивающейся пробкой (Nunc, Kamstrup, Дания), в стеклян-
ных тест-пробирках (диаметром 10 мм, длиной 75 мм; Arthur
Н. Thomas Со., Philadelphia, США), в боросиликоновых стек-
лянных ампулах (на 1—2 мл, Wheaton Scientific, Millville,
США) или в пластиковых соломинках (пайетах) (IMV, L’Aigle,
Франция). Чтобы облегчить загрузку, интактные кумулюсные
массы часто замораживают в стеклянных тест-пробирках. На-
дежная процедура замораживания эмбрионов в пайетах описа-
на в работе [16]. Мы предпочитаем пользоваться стеклянными
ампулами на 2 мл и замораживать ооциты и эмбрионы, свобод-
ные от кумулюса. При этом мы руководствуемся следующими
соображениями.
1. Выживаемость эмбрионов, замороженных в стеклянных
пробирках или ампулах, выше, чем у эмбрионов, заморо-
Низкотемпературная консервация яиц и эмбрионов мыши 329
женных в пластиковых фиалах (по-видимому, это связа-
но с лучшей теплопроводностью стекла).
2. Стеклянные ампулы можно запаивать, чтобы предотвра-
тить внесение дебриса, накапливающегося при хранении
в ЖА. Учтите, что силиконовые пробки, которыми поль-
зуются для закрывания пластиковых пробирок, легко те-
ряются.
3. Вероятность случайного нагревания образца сравнительно
большого объема, который содержится в ампуле, ниже,
чем при использовании пайеты (ср. объемы: 0,2—0,3 мл
и 20—40 мкл).
4. Большой объем ампулы (по сравнению с пайетой) упро-
щает маркировку и последующую идентификацию образ-
цов.
Среди недостатков стеклянных ампул отметим следующие:
а) при хранении плохо запаянных ампул в них может по-
пасть ЖА, при размораживании ампул он начнет испа-
ряться, что в свою очередь может привести к взрыву;
б) в сосуд с ЖА можно загрузить сравнительно небольшое
число ампул.
Судя по нашему опыту, мыть ампулы не обязательно: следы
детергента на стекле могут послужить причиной гибели эм-
брионов. Перед стерилизацией маркируйте каждую ампулу спе-
циальными чернилами по стеклу (Gurr). Достаточно использо-
вать простой числовой код. Дайте чернилам высохнуть, стери-
лизуйте ампулы нагреванием при 150 °C в течение 90 мин.
Постоянными кодовыми номерами пользуются при длитель-
ном хранении, но их трудно разобрать на замерзшем стекле.
Чтобы упростить последующую идентификацию, непосредствен-
но перед загрузкой ампулы мы прикрепляем к ней клейкую
метку, наподобие тех, которыми пользуются электрики, чтобы
различать провода (маркеры проводов; W. Н. Brady and Со.
Ltd, UK).
3.1.3. Подготовка образца для охлаждения
1. Воспользовавшись имеющейся инъекционной иглой (ка-
либр 19, длина 50 мм), внесите в маркированную ампулу
0,15 мл среды М2.
2. Поместите пипеткой эмбрионы в среду (около 30 на обра-
зец). Прежде чем захватить их в пастеровскую пнпетку
с оттянутым кончиком, втяните 1—2 пузырька воздуха,
затем возьмите эмбрионы в наименьшем объеме среды М2.
Опустив конец пипетки в среду М2 в ампуле, осторожно
выпускайте эмбрионы, пока не выйдет пузырек воздуха.
Проконтролируйте под микроскопом, все ли эмбрионы
вышли из пипетки.
330
Глава 13
3. Охладите образцы до 0 °C в ледяной бане до внесения
криопротектора (разд. 3.2.3).
3.2. Криопротектор
Присутствие криопротектора во время охлаждения необхо-
димо для выживания эмбрионов. Механизм действия таких
соединений не совсем понятен, но можно предположить, что эти
вещества предохраняют эмбрион от повреждающего действия
высоких концентраций растворов, которые создаются в резуль-
тате вымораживания воды в суспензионной среде.
3.2.1. Выбор криопротектора
Защитное действие на замораживаемые неоплодотворенные
яйца и эмбрионы мыши оказывает целый ряд проникающих
соединений, включая ДМСО, глицерол, метанол, пропиленгли-
коль и полиэтиленгликоль [1,16,17,18,19]. Для неоплодотво-
ренных яиц эффективными криопротекторами служат ДМСО
или глицерол [3, 20]. В нашей лаборатории для заморажива-
ния ооцитов и эмбрионов по многим причинам используют
ДМСО.
1. До сих пор живых мышей удалось получить только из
тех ооцитов, которые были законсервированы с исполь-
зованием в качестве криопротектора именно ДМСО
[3, 21].
2. После замораживания в присутствии ДМСО эмбрионы
всех предимплантационных стадий развиваются нор-
мально.
3. Процедуры добавления и удаления ДМСО сравнительно
просты. Это обстоятельство особенно важно для новичков
и для использования метода в лабораториях, где замора-
живание требуется в редких случаях.
4. Высокий уровень выживаемости после оттаивания. Обыч-
но 50% эмбрионов от случайно скрещивающихся мышей
и до 30% эмбрионов, полученных от инбредных мышей,
нормально развиваются после замораживания на 8-кле-
точной стадии. Пропорция оттаявших ооцитов, способных
оплодотворяться in vitro, отчасти зависит от генотипа
спермия и варьирует от 1 до 60%• После переноса при-
мерно 50% оплодотворенных яиц дают нормальные
плоды.
5. Безопасность длительного хранения в ДМСО доказана
(разд. 5.1).
На выживаемость эмбрионов влияют:
а) концентрации криопротектора;
Низкотемпературная консервация яиц и эмбрионов мыши 331
б) скорость и температура, при которых криопротектор вно-
сится в образец, а также время экспозиции перед охлаж-
дением до субнулевых температур;
в) скорость и температура, при которых протектор удаляет-
ся после оттаивания (разд. 3.6).
3.2.2. Концентрация ДМСО
Высокая выживаемость эмбрионов наблюдается после за-
мораживания в среде, содержащей ДМСО в концентрации от
1,0 до 2,0 М. В концентрациях меньше 1,0 М ДМСО не проявля-
ет эффективного защитного действия, а в концентрациях выше
2,0 М ДМСО оказывается токсичным. На практике использует-
ся концентрация 1,5 М.
3.2.3. Добавление ДМСО
При смешивании ДМСО с замораживаемой средой выделя-
ется значительное количество тепла. Поэтому прямо в образец,
содержащий эмбрионы, неразведенный ДМСО не вносится;
вместо этого готовят его 3,0 М раствор (табл. 13.3) и к образцу
добавляется аликвота, достаточная для получения конечной
концентрации ДМСО 1,5 М. К образцам, представленным
неоплодотворенными яйцами или эмбрионами вплоть до стадии
морулы, весь необходимый объем добавляется сразу при 0°С
(табл. 13.3,Л). Если консервируются бластоцисты, их выживае-
мость повышается при постепенном увеличении концентрации
ДМСО при комнатной температуре (табл. 13.3,Б).
В настоящее время известно, что если перед заморажива-
нием создать условия, соответствующие проникновению гли-
церола, это приведет к увеличению выживаемости ооцитов
мыши [9]. Количество вещества, поступающего в эмбрион, оп-
ределяется его проницаемостью, временем и температурой
экспозиции перед замораживанием. При воздействии ДМСО
время п температура экспозиции не имеют столь критического
значения, как при действии глицерола (вероятно, это следст-
вие лучшей проникающей способности ДМСО). Оптимальный
уровень выживаемости мышиных эмбрионов 8-клеточной стадии
достигается после 5-минутной экспозиции при 0°С. Длительное
воздействие (до 30 мин) при 0°С не снижает выживаемости
и оказывается более удобным при установившейся практике
замораживания эмбрионов. После добавления ДМСО мы вы-
держиваем эмбрионы на льду в течение 15 мин. Тем временем
следует:
а) быстро извлечь ампулу из ледяной бани и, удерживая ее
длинным пинцетом, запаять;
332
Глава 13
Таблица 13.3. Приготовление ДМСО и добавление его перед замораживанием
Приготовление 3,0 М ДМСО
Предпочтительнее использовать ДМСО для спектроскопического анализа.
1. Наберите пипеткой 7,87 мл среды (РВ1 или М2) в пластиковую пробир-
ку для проб Falcon.
2. Добавьте 2,13 мл ДМСО.
3. Перемешайте осторожно, чтобы избежать денатурации БСА в среде.
4. Стерилизуйте, воспользовавшись миллипоровым фильтром с размером пор
0,22 мкм.
Добавление ДМСО к мышиным ооцитам и эмбрионам от 1-клеточной стадии
до морулы
1. Охладите 3,0 М ДМСО до 0 °C.
2. Пипеткой перенесите эмбрионы в ампулу, содержащую 0,15 мл М2, и ох-
лаждайте до 0°C (примерно 10 мин).
3. Добавьте 0,15 мл 3,0 М ДМСО.
4. Тщательно перемешайте.
5. Через 15 мии перенесите образец в баню для сидиига.
Добавление ДМСО к бластоцистам мыши
1. Дайте нагреться 3,0 М ДМСО до комнатной температуры.
2. Внесите пипеткой эмбрионы в ампулу, содержащую 0,15 мл среды М2 при
комнатной температуре.
3. Добавьте 0,05 мл 3,0 М ДМСО.
4. Тщательно перемешайте.
5. Подождите 5 мин.
6. Повторите пп. 3, 4, и 5.
7. Повторите пп. 3 и 4.
8. Охладите образец до 0 °C.
9. Подождите 15 мин, прежде чем перенести образец в баию для сидинга.
б) прикрепить образцы к держателям с помощью резиновых
полосок (рис. 13.1), подготовив их к переносу в баню
для сидинга;
в) отрегулировать температуру бани для охлаждения и ба-
ни для сидинга.
3.3. Индукция образования льда
в суспензионной среде: «сидинг»
В процессе охлаждения со скоростью, не снижающей жиз-
неспособности эмбриона, спонтанное замерзание суспензионной
среды при достижении точки замерзания (около —3,5 °C) про-
исходит редко, и образец может переохладиться до —21 °C.
При образовании льда, которое сопровождается выделением
латентного тепла, температура образца поднимается до точки
плавления, затем резко падает, пока не установится температур-
ное равновесие с охлаждающей баней. Точная скорость охлаж-
Низкотемпературная консервация яиц и эмбрионов мыши 333»
Рис. 13.1. Держатель ампул, изготовленный из медной проволоки (1), покры-
той пластиком и спаянной с медным винтом (2), ввинчивающимся в брусок из
перспекса (3). На бруске из перспекса крепится до четырех ампул с помощью
резиновой полоски.
дения зависит от разницы температур образца и охлаждающей,
бани и может быть порядка 10 °С/мин. Обезвоживание, необхо-
димое для выживания клетки, происходит только после обра-
зования льда в суспензионной среде. Если скорость охлажде-
ния после образования льда слишком высока, количество воды,,
покидающее клетку в процессе внутриклеточного заморажива-
ния, приведет ее к гибели.
Для того чтобы не допустить переохлаждения, в образце ин-
дуцируют образование льда (сидинг) при температурах, сле-
дующих сразу ниже точки замерзания суспензионной среды.
1. После обработки образца ДМСО перенесите запаянную-
ампулу из ледяной бани при О °C в баню с постоянной
температурой—5—6 °C (баня для сидинга; разд. 3.3.1).
2. Подождите 2 мин.
3. Индуцируйте сидинг в образце, прикоснувшись к ампуле
сразу над мениском концами тонкого пинцета (Watchma-
ker, № 5 подходит для этого идеально), предварительно
охлажденного в жидком азоте. Как только на стекле
появится изморозь, уберите пинцет. Замечание: будьте
осторожны, не переохладите образец.
4. Возвратите ампулу в баню для сидинга.
5. Через 5 мин, убедившись в том, что в образце сформиро-
вался лед, перенесите ампулу в охлаждающую баню при
той же температуре, какая была в бане для сидинга.
Другие способы сидинга описаны в работах [9, 10].
3.3.1. Баия для сидиига
Для сидинга можно использовать любую баню, в которой
может поддерживаться температура —5 или —6 °C дольше-
15 мин. Мы обычно пользуемся постоянно перемешиваемым
334
Глава 13
промышленным метиловым спиртом или этанолом, охлаждае-
мым в термоэлектрической бане (Virtis minifreezer: Technation,
Middlesex, Англия). Но может подойти и обычная смесь пова-
ренной соли со льдом. Другое оборудование описано Лейбо
и Мейзуром [9].
3.4. Охлаждение
Жизнеспособность эмбрионов зависит от эффективности
выведения внутриклеточной воды до того, как будет достигнута
температура замерзания. Если обезвоживание происходит
в процессе охлаждения, скорость охлаждения приобретает кри-
тическое значение. Если охлаждение идет слишком быстро,
эмбрион не успевает адаптироваться до того момента, когда
температура достигнет точки, при которой может произойти
инициация кристаллизации, в результате содержимое клетки
замерзает с образованием летальных количеств внутриклеточ-
ного льда. И напротив, если охлаждение идет слишком мед-
ленно, клетки подвергаются действию высоких концентраций
внеклеточных растворов слишком долго, в результате изменя-
ется pH и объем клеток уменьшается. В идеальном случае
клетки должны охлаждаться со скоростью, достаточно медлен-
ной для того, чтобы они продолжали оставаться в осмотическом
равновесии с внеклеточным раствором.
3.4.1. Скорость охлаждения
Для оптимальной выживаемости эмбрионы млекопитающих
требуют относительно низких, по сравнению с другими клетка-
ми, скоростей охлаждения (0,2—2,0°С/мин). На практике эм-
брионы мыши в 1,5 М ДМСО охлаждают со скоростью 0,3—
0,5сС/мпн. Скорость охлаждения измеряется в пределах от
—10 до —60 °C. Медленное охлаждение заканчивают при—40 °C
или при температурах —70 °C погружением образца в ЖА.
Выживаемость 8-клеточных эмбрионов в 1,5 М ДМСО снижа-
ется, если медленное охлаждение заканчивают при температу-
рах выше —35°C или в пределах от —45 до —70 °C. Бластоци-
сты мыши толерантны к прекращению медленного охлаждения
в широком интервале температур [13], но обычно их медленно
охлаждают до —40 или —70 °C. Ооциты до погружения в жид-
кий азот медленно охлаждают до температуры ниже —70 °C.
3.4.2. Охлаждающая баня
Охлаждающую баню наподобие той, которой мы пользуем-
ся в своей лаборатории (рис. 13.2), детально описали Лейбо
и Мейзур [9]. Она состоит из частично разреженного (10—
Низкотемпературная консервация янц н эмбрионов мыши
335
мм;
мм; т. е. Dilvac,
Ltd,
100 мм ртутного столба) не-
•серебреного сосуда Дьюара
(емкость 950 мл;
диаметр 70
высота 300
поставщик Day-Impex
Colchester, Англия), содержа-
щего 500 мл охладителя (про-
мышленный метиловый спирт @
или этанол). Охладитель дол-
жен постоянно перемешивать-
ся (для этого используют ме-
шалку с регулируемой ско- ®
ростью). Этот сосуд Дьюара
помещают в наружный (ем-
кость ~4 л), содержащий
ЖА. Быстрота охлаждения
зависит от объема охладителя,
степени разреженности внут-
реннего сосуда и глубины его
погружения в ЖА. В идеаль-
внутренний Регистратор
внутренняя теипвратуры
Регистратор
температуры
©-*
Рис. 13.2. Поперечное сечение охлаж-
дающей ванны. 1. Непосеребренный
сосуд Дьюара с частичным ваку-
умом. 2. Охладитель. 3. Мешалка с
меняющейся скоростью. 4. Сосуд
Дьюара посеребренный, с высоким
вакуумом. 5. Жидкий азот. 6. Ампу-
ла с эмбрионами. 7. Держатель об-
разцов с удобным доступом к нему
(см. рис. 13.1). 8, 9. Медь-константа-
новые термопары, регистрирующие
температуру образна и охладителя.
аом случае для наблюдения
за температурой используют
медь-константановые термопа-
ры и регистратор низких тем-
ператур (Speedomax 2500 mul-
tipoint; поставщик Zeeds and
Northrup LTD, Birmingham,
UK). Одна термопара регист-
рирует температуру охладите-
ля, другая помещена в «пу-
стой» образец (учтите, что
необходимо инициировать сидинг и в «пустом», контрольном
образце). Разница между температурами охладителя и образца
обычно составляет около 1 °C. Если подходящего регистратора
нет, температурные характеристики сосуда Дьюара можно по-
лучить заблаговременно, воспользовавшись низкотемператур-
ным термометром и секундомером. Поддержание постоянного
объема охладителя и погружение внутреннего сосуда на опре-
деленную глубину в ЖА обеспечивают высокую воспроизводи-
мость скорости охлаждения. Если глубина погружения в ЖА
остается постоянной все время, кривая охлаждения будет слег-
ка криволинейной. Для поддержания постоянной скорости ох-
лаждения между —10 и —60 °C можно постепенно углублять
внутренний сосуд в ЖА. Сравнение выживаемости эмбрионов
после охлаждения с постоянной скоростью с соответствующим
показателем для охлаждения с постепенно уменьшающейся ско-
336
Глава 13
ростью не проводилось. Мы пользуемся постоянной скоростью,
но, по мнению Лейбо и Мейзур [9], более эффективно посте-
пенное уменьшение скорости, так как при этом остается время
для выравнивания осмотических соотношений при более низких
температурах.
3.4.3. Охлаждение образца после инициации кристаллизации
После сидинга образец следует подвергнуть следующим про-
цедурам.
1. Проконтролируйте, чтобы температура охладителя отли-
чалась от температуры бани для сидинга в пределах
0,5 °C.
2. После уравновешивания температур (~5 мин, наблюдай-
те за температурой пустого образца) опустите ампулу
в охлаждающую баню так, чтобы содержимое было ниже
уровня охладителя.
3. Отрегулируйте наружный сосуд Дьюара, содержащий
ЖА, чтобы он давал скорость охлаждения порядка 0,3—
0,5 °С/мин.
4. Прекратите медленное охлаждение, когда температура
образца достигнет —40 °C, или продолжайте охлаждать
ниже —70 °C. По достижении выбранной температуры
извлеките ампулу из охлаждающей бани и погрузите ее
(по-прежнему прикрепленную к держателю) в ЖА.
5. К следующему пункту приступайте через 15 мин.
6. Охладите алюминиевый контейнер для хранения ампул
в ЖА.
7. Не извлекая ампул из ЖА, разрежьте резиновую полоску,
прикрепляющую ампулы к держателю.
8. С помощью длинного пинцета, концы которого предвари-
тельно охлаждены в ЖА, загрузите ампулы в контейнер
для хранения.
9. Перенесите контейнеры, погруженные в ЖА, в храни-
лище.
3.5. Размораживание
Температура, при которой заканчивается медленное охлаж-
дение, определяет скорость нагрева, необходимую для опти-
мального выживания эмбрионов. Если медленное охлаждение
заканчивается при относительно высоких субнулевых темпера-
турах (около —40 °C), эмбрионы содержат некоторое количест-
во внутриклеточной воды, которая витрифицируется при быст-
ром охлаждении до —196 °C [22]. Чтобы избежать девитрифи-
кации и роста летальных количеств внутриклеточного льда,
Низкотемпературная консервация яиц и эмбрионов мыши 337
необходим быстрый нагрев. И наоборот, эмбрионы, медленно
охлаждаемые до температур ниже —70 °C, нуждаются в мед-
ленном, а не быстром нагреве. Возможно, медленное насыще-
ние водой необходимо для постепенной реорганизации субкле-
точных структур. Для большинства клеток млекопитающих
оптимальная выживаемость достигается в результате быстрого
нагрева и не зависит от режима охлаждения.
При размораживании эмбрионов мыши, медленно охлаждав-
шихся (0,3—0,5°С/мин в 1,5 М ДМСО) перед быстрым охлаж-
дением до —196 °C, нужно выполнить следующие процедуры:
1. Если медленное охлаждение заканчивалось при —40 °C,
быстро нагревайте образец со скоростью 50°С/мин, опус-
тив его в воду при 40 °C; помешивайте воду ампулой.
2. Если медленное охлаждение продолжалось ниже —70 °C,
медленно нагревайте образец со скоростью от 4 до
20 °С/мин. Извлеките образец из ЖА и установите его на
решетке, обеспечивающей скорость нагрева около
20°С/мин. Другой способ: пробирку для кипячения (внут-
ренний диаметр 35 мм, наружная высота 200 мм) охла-
дите в ЖА, затем подвесьте ее на воздухе при комнатной
температуре и переместите в нее образец из ЖА; его на-
грев будет происходить со скоростью около 10°С/мин.
Независимо от скорости нагрева, после того как исчезнут
остатки льда, держите образец при комнатной температуре
в течение 5 мин до того, как приступите к удалению ДМСО.
3.6. Удаление ДМСО после оттаивания
Поскольку охлаждение образца ведется в присутствии про-
никающего криопротектора, возникает необходимость удалить
это соединение после размораживания клеток, чтобы избежать
их повреждения в результате осмотического шока. Адаптация
эмбрионов при возвращении в нормальные физиологические ус-
ловия (т. е. из среды с осмоляльностью в 1,5 осмоль в культу-
ральную среду с осмоляльностью около 0,3 осмоль) зависит от:
1) количества криопротектора, проникшего в бластомеры
перед охлаждением;
2) их относительной проницаемости для воды и протектор-
ного соединения;
3) температуры.
Вода обычно проникает легче, чем протектор, поэтому в ре-
зультате разбавления суспензии транспорт воды в клетку будет
превалировать над транспортом криопротектора из клетки
и бластомеры разбухнут. Контроль степени разбухания приня-
то осуществлять путем поэтапного удаления криопротектора из
мышиных эмбрионов при 0 °C или 20 °C [1, 2]. В последнее
время было показано, что эмбрионы мыши переносят быстрое
338
Глава 13
двухступенчатое удаление 1,5 М ДМСО при комнатной
температуре [13].
Процедура удаления ДМСО из ооцитов и эмбрионов после
оттаивания заключается в следующем:
1. После того как в образце растает лед, сделайте царапи-
ну на шейке ампулы и отломите верхушку.
2. Подождите 5 мин, чтобы образец приобрел комнатнук»
температуру.
3. Добавьте 1 мл М2 (при комнатной температуре) к образ-
цу объемом 0,3 мл и тщательно перемешайте.
4. Спустя 1 мин, воспользовавшись пластиковой пипеткой
на 1 мл (Falcon), перенесите содержимое ампулы в эм-
бриологическое часовое стекло.
5. Сполосните ампулу дважды средой М2, для каждого спо-
ласкивания берите по 1 мл, и затем переносите в часовое
стекло.
6. Соберите эмбрионы в хорошо оттянутую пастеровскую пи-
петку и отмойте, дважды сменив М2 (по 2 мл на от-
мывку).
Ооциты после удаления ДМСО оплодотворяются in vitro
(разд. 6.2), затем их переносят в псевдобеременную приемную
мать (разд. 6.4). Оплодотворенные яйца и эмбрионы можно
переносить мыши-реципиенту сразу же, но предварительный
период культивирования (разд. 6.3) повышает их выживае-
мость.
3.7. Техника безопасности
Совершенно необходимо знать и строго соблюдать правила
техники безопасности при работе с жидкими газами!
1. ЖА, заполняющий наружный сосуд Дьюара охлаждающе-
го устройства, кипит и разбрызгивается. Защищайте гла-
за и кожу.
2. Алюминиевый контейнер для хранения образца после
охлаждения до —196 °C начинает прилипать к коже. На-
девайте свободные перчатки.
3. Плохо запаянные ампулы после хранения содержат ЖА
и в результате при нагревании могут взрываться. Не рис-
куйте. Всегда надевайте маску или по крайней мере за-
щитные очки, если достаете ампулы из ЖА и начинаете
их размораживать. Позаботьтесь о безопасности окру-
жающих.
4. Витрификация
Цель исследований по витрификации состоит в упрощении про-
цедур криозащиты эмбрионов. Известно, что высококонцент-
рированные растворы криопротекторов способны охлаждаться
Низкотемпературная консервация янц н эмбрионов мыши ЗЗ^
до низких температур без кристаллизации [23]. В результате
сверхохлаждения вязкость этих растворов настолько увеличи-
вается, что они становятся стекловидно-твердыми; этот физи-
ческий процесс называется витрификацией. При использовании
витрификации эмбрионы уравновешиваются в смеси криопро-
текторов (витрификационном растворе) и, после того как цито-
плазма обезводится, суспензию эмбрионов (в пластиковой
пайете) витрифицируют путем помещения в ЖА. Скорость-
охлаждения практически не имеет значения, если она достаточ-
но высока для того, чтобы предотвратить кристаллизацию.
Нагревание также должно быть достаточно быстрым, чтобы
не произошла девитрификация (кристаллизация) в интервале
температур от —100 до —25 °C. Таким образом, витрификация
обеспечивает быстрый и простой способ криозащиты эмбрионов.
При этом отпадает необходимость сидинга эмбриональной
суспензии и контролируемого периода медленного охлаждения..
По-видимому, основными факторами, обусловливающими успех
витрификации, служат:
1) состав витрификационного раствора;
2) процедуры, применяемые для осмотического уравновеши-
вания (эквилибрации) эмбрионов в растворе.
4.1. Витрификационный раствор
Витрификационный раствор должен быть достаточно кон-
центрированным, чтобы в нем не наступила кристаллизация
при охлаждении с применяемыми на практике скоростями, и в
то же время он не должен обладать химической токсичностью
или вызывать осмотическое повреждение эмбриона при эквили-
брации или разбавлении. Витрификационный раствор (ВР1:
табл. 13.4), который предложили Роли и Фейхи [23], представ-
ляет смесь следующих криопротекторов в модифицированном
солевом растворе Дульбекко:
1) эквимолярные концентрации ДМСО и ацетамида, приме-
нение которого основано на способности некоторых ами-
дов снижать химическую токсичность растворов концент-
рированного ДМСО [24, 25];
2) пропиленгликоль благодаря его стеклообразующим свой-
ствам;
3) непроникающий полимер (полиэтиленгликоль), повышаю-
щий способность раствора витрифицироваться [26].
Воздействие исходным раствором ВР1 отрицательно сказы-
вается на развитии 8-клеточных эмбрионов мыши как in vitro,
так и in vivo [6], поэтому для витрификации эмбрионы суспен-
дируют в 90%-ном растворе ВР1 (табл. 13.5), концентрация
криопротекторов в котором менее токсична.
340
Глава 13
Таблица 13.4. Состав исходного витрификациоиного раствора ВР1
и HEPES-забуференного солевого раствора (НВ1)
Состав ВР11) НВ1
гУл мМ П/л мМ
Буферный раствор
NaCl 8,000 136,90 8,000 136,90
:КС1 0,200 2,68 0,200 2,68
MgCl2-6H2O 0,050 0,25 0,100 0,50
КН2РО4 0,120 0,88 0,136 1,00
СаС12-2Н2О 0,010 0,07 0,120 0,90
(Пируват Na — — 0,036 0,33
HEPES21 4,766 20,00 4,766 20,00
Глюкоза 1 ,000 5,56 1,000 5,56
БСА3> 0,750 — 3,000
'Пенициллин G (соль — — 0,060 —
калия)
Феноловый красный 0,010 — 0,010 —
Криопротекторы
.ДМСО 20,502 2,62 — —
Ацетамид 154,970 2,62 — —
Пропилеигликоль 100,00 1,30 — —
'Полиэтиленгликоль 60,00 6,0% в/о — —
(м. в. 8000)
pH витрификациоиного раствора — 7,8—8,0.
pH НВ1 —7,2.
1) Хранить в стеклянной посуде.
2) Calbiochem-Boehring: Ultrol.
3) Кристаллический: Miles-Serovac.
4.2. Осмотическое уравновешивание (эквилибрация)
эмбрионов в витрификационном растворе
Процедура эквилибрации разработана с целью получения
концентрированной обезвоженной цитоплазмы, способной к
витрификации при последующем охлаждении. Эта процедура
не должна вести к химической токсичности или значительному
осмотическому стрессу ни при эквилибрации, ни при последую-
щем разбавлении [23]. Возможность повреждения эмбрионов
отчасти зависит от степени проникновения криопротекторов,
содержащихся в витрификационном растворе, в цитоплазму.
Частичное проникновение, по-видимому, способствует успешной
консервации, однако полное — повышает вероятность поврежде-
ния клетки вследствие химической токсичности и осмотического
разбухания при разбавлении. На практике эмбрионы уравнове-
шивают при комнатной температуре в 25%-ном ВР1 и затем
в два приема подвергают воздействию повышающихся концент-
раций витрификациоиного раствора при 4 °C.
Соответствующая процедура описана в табл. 13.5.
Низкотемпературная консервация яиц и эмбрионов мыши
341
Таблица 13.5. Выдерживание 8-клеточных эмбрионов мыши
в внтрнфнкацнонном растворе
А. Растворы
1. Приготовьте три разведения исходного ВР1, используя для этого НВ1
(табл. 13.4):
1) 25%-ный ВР1: (1 часть ВР1 :3 части НВ1)
2) 50%-ный ВР1: (1 часть ВР1 : 1 часть НВ1)
3) 90%-ный ВР1: (9 частей ВР1 : 1 часть НВ1)
2. Охладите 50%-пый ВР1 п 90%-ный ВР1 до 4 °C.
Б. Подготовка пайеты
1. Отсосите 1-см столбик 90%-ного ВР1 в пластиковую пайету (трубочку)
на 0,25 см3; пробка из ваты/PVA должна увлажниться.
2. С помощью инъекционной иглы (№ 20, длина 3,5 см) п стеклянного шпри-
ца введите 2-см столбик (~40 мкл) 90%-ного ВР1 в центр пайеты.
3. Охладите пайету до 4 °C.
В. Процедура ступенчатой эквилибрации
1. Перенесите эмбрионы с помощью микропппеткн в 3 мл 25%-ного ВР1 и
тщательно перемешайте, осторожно вращая чашку.
2. Подождите 15 мин.
3. Перенесите эмбриональную суспензию в холодную комнату (2—4 °C).
Подождите 10 мин, а затем в холодной комнате проведите п остальные
этапы процедуры.
4. Перенесите эмбрионы в небольшом объеме 25%-ного ВР1 в 3 мл холод-
ного 50%-пого ВР1. Тщательно перемешайте суспензию микропипеткой.
(Если эмбрионы всплыли, смочите пипетку 50%-пым ВР1 и погрузите ею
эмбрионы на дно чашки.)
5. Через 10 мин перенесите эмбрионы в 90%-ный ВР1, загрузите сжавшиеся
эмбрионы в 2-см столбик 90%-ного ВР1 в пайсте.
6. Герметизируйте пайету путем термической сварки обеих ее концов (вос-
пользуйтесь прибором для склейки пластиковых пакетов).
Примечание: не нагрейте эмбриональную суспензию пальцами.
4.3. Охлаждение
Через 10 мин после переноса эмбрионов в 90%-ный ВР1
витрифицируйте эмбриональную суспензию охлаждением пайе-
ты в ЖА. Единственное требование касается скорости охлаж-
дения: опа должна быть достаточно высокой для предотвраще-
ния кристаллизации (>20°С/мин). Существуют три способа
охлаждения:
1) прямой перенос в ЖА;
2) перенос в холодные пары N2 (—150°C), т.е. в горловину
замораживателя (~200°С/мип);
3) перенос в постепенно охлаждающую изопентановую баню
(~20°С/мин) при температуре около —120°C, а затем
прямо в ЖА.
342
Глава 13
Иногда быстрый перенос в ЖА может приводить к раска-
лыванию остекленевшей суспензии, в результате оболочки и
бластомеры некоторых эмбрионов повреждаются.
4.4. Хранение витрифицированных образцов
Витрифицированные эмбриональные суспензии следует хра-
нить при температуре ниже той, которая вызывает изменение
физического состояния раствора (—120°C), чтобы избежать
повреждения эмбрионов в результате девитрификации. На дол-
говременное хранение витрификационные образцы лучше закла-
дывать в пайеты, погруженные в ЖА (см. разд. 5).
4.5. Нагрев
Чтобы избежать повреждающего действия девитрификации,
длительно хранившиеся витрифицированные суспензии должны
быть быстро нагреты (>200°С/мин) [8, 23]. Одним из удоб-
ных способов служит перенос образца в холодную (4 °C) воду.
При этом скорость нагрева составит около 2000°С/мин.
Не пользуйтесь нагревающей водяной баней; даже временное
повышение температуры суспензии может привести к химиче-
ской токсичности и поступлению избыточных количеств крио-
протекторов в клетку.
4.6. Разведение витрификационного раствора
Следует уделить серьезное внимание освобождению эмбрио-
нов от витрификационного раствора, поскольку известно, что
его высокие концентрации токсичны. Важно обеспечить условия,
при которых температура эмбриональной суспензии не подни-
мается выше 4 °C до тех пор, пока разведение криопротекторов
не достигнет по крайней мере 25% от исходной концентрации
ВР1. Быстрого разбавления путем переноса эмбрионов в изото-
нический раствор следует избегать: это может привести к по-
вреждению эмбриона в результате осмотического разбухания.
Среду, предохраняющую эмбрионы от разрушения, можно
создать разбавлением криопротекторов одним из указанных
способов.
1. Медленно, посредством ступенчатой процедуры ([23],
табл. 13.6,А). Концентрация витрификационного раствора
уменьшается в два приема при 4 °C, пока не достигнет
25% концентрации исходного ВР1. Затем оставшиеся
внутри- и внеклеточные криопротекторы медленно раз-
бавляются в несколько приемов при комнатной темпера-
туре.
Низкотемпературная консервация яиц и эмбрионов мыши
343
Таблица 13.6. Освобождение эмбрионов от витрификационного раствора
Нагрейте эмбрионы и проведите первоначальное разбавление при 4 °C. Сразу
после нагревания осушите пайеты салфеткой и срежьте оплавленные концы.
Удалите криопротекторы одним из приведенных ниже способов.
А. Процедура ступенчатого разбавления
1. Вылейте содержимое пайеты в маленькую чашку Петрн с 3 мл холодного
(4°C) 50%-ного ВР1 и сполосните пайету 50%-ным ВР1. Тщательно пе-
ремешайте содержимое чашки и подождите 10 мнн.
2. Перенесите эмбрионы микропипеткой в 3 мл холодного 25%-ного ВР1.
Тщательно перемешайте н подождите 10 мин.
3. Принесите суспензию эмбрионов нз холодной комнаты и подождите 10 мин.
4. Перенесите эмбрионы в 3 мл 12,5%-ного ВР1 (разведение 1:8 исходного
ВР1 буферным раствором НВ1, табл. 13.4). Смешайте, подождите 10 мин.
5. Перенесите эмбрионы в ВР1 или М2 и оставьте на 10 мин, прежде чем
перейти к следующей процедуре.
Б. Процедура «сахарозного» разбавления
1. Вылейте содержимое пайеты в маленькую чашку Петри с 3 мл раствора
холодной (4 °C) 1,08 М сахарозы в РВ1. Тщательно перемешайте (эмбрио-
ны будут плавать) н оставьте на 5 мин.
2. Принесите суспензию эмбрионов из холодной комнаты и подождите
10 мин.
3. Перенесите «сжавшиеся» эмбрионы в РВ1 илн М2.
2. Быстро, посредством «сахарозной» процедуры ([8],
табл. 13.6,5), основанной на методе, который предложи-
ли Лейбо и Мейзур [9]. Витрификационный раствор за-
меняется при 4 °C солевым раствором, в состав которого
входит 1,08М сахароза. Сахароза не проникает в бласто-
меры, создавая таким образом осмотический барьер, пре-
пятствующий излишнему разбуханию эмбрионов по мере
того, как криопротекторы выходят из цитоплазмы.
4.7, Техника безопасности
1. Точно следуйте инструкции изготовителя при работе
с ацетамидом.
2. Придерживайтесь правил работы с жидким азотом (см.
разд. 3.7).
5. Хранение образцов
Замороженные и витрификационные эмбрионы хранят при
—196 °C в любом доступном хранилище для ЖА. Следует убе-
диться в том, что уровень ЖА достаточен. Желательно контро-
лировать температуру или вес сосуда, чтобы не допустить паде-
344
Глава 13
ния уровня ЖА до опасного предела. Кроме того, для страхов-
ки от возможной потери лучше держать эмбрионы одной ли-
нии мышей более чем в одном хранилище.
5.1. Длительность хранения
При —196 °C могут происходить только фотофизические
реакции, т. е. ионизация вследствие радиации. При отсутствии
процессов ферментативной репарации в период длительного
хранения в эмбрионах могут накапливаться летальные количе-
ства повреждений. Однако до сих пор не получено каких-либо
аргументов в пользу этого предположения. Нормальные фер-
тильные мыши развились из 8-клеточных эмбрионов, хранив-
шихся в ЖА до 13 лет. Более того, длительное хранение замо-
роженных эмбрионов в условиях, имитирующих нормальную
фоновую радиацию, накапливаемую в течение 2000 лет, не выз-
вало никаких генетических отклонений [27].
5.2. Регистрация
Необходима правильная и полная регистрация данных о хра-
нящемся материале. Может оказаться так, что размораживать
эмбрионы будет кто-то другой. Минимально необходимая ин-
формация включает следующие данные: дата криоконсервации,
характеристика сохраняемой линии мышей, номер ампулы/пайе-
ты, число ооцитов/эмбрионов на ампулу/пайету, стадия разви-
тия, метод консервации, т. е. замораживание или витрификация,
местонахождение в хранилище Ж.А, необходимый способ раз-
мораживания и процедура разбавления.
В случае замороженных эмбрионов необходимо также за-
фиксировать сведения о замораживающей среде, объеме образ-
ца, конечной концентрации ДМСО, скорости охлаждения, ко-
нечной температуре перед погружением в ЖА.
Для витрифицированных эмбрионов обязательно укажите
характеристику витрификационного раствора.
5.3. Номенклатура
До сих пор не получено данных, свидетельствующих о том,
что мыши, происходящие из замороженных эмбрионов, оказа-
лись генотипически или фенотипически измененными в резуль-
тате процессов замораживания и оттаивания. Остается убедить-
ся в отсутствии каких-либо генетических изменений эмбрионов
после витрификации. Нужно учитывать, что фенотип может быть
модифицирован внутриматочным окружением, вследствие чего
Низкотемпературная консервация яиц и эмбрионов мыши 345
эмбрионы, развивающиеся в приемных мышах родительской
линии, могут отличаться от эмбрионов, полученных от реципи-
ентной линии. Важно знать историю подопытных животных;
вот почему вся информация, имеющая к ней отношение, также
должна быть включена в описание сохраняемого материала.
Описание сохраняемого материала инбредных линий мышей,
получаемых из замороженных эмбрионов, регламентируется
международными правилами [28]. Вопрос о номенклатуре ма-
териала, сохраняемого путем витрификации, еще не рассмат-
ривался.
6. Оценка жизнеспособности консервированного материала
Число сохраняемых яиц или эмбрионов, представляющих
каждую линию мышей, зависит от потребностей лаборатории.
Чтобы точно определить, сколько яиц/эмбрионов необходимо
для консервации, нужно оценить выживаемость каждой закон-
сервированной партии. Методика в данном случае идентична
той, что используется для живых мышей, за исключением того,
что приемная мать может быть забита на поздних стадиях
беременности (день 15—16) для подсчета числа нормальных
плодов. При оценке числа эмбрионов, обеспечивающих сохране-
ние линии, нужно учитывать и возможность постнатальной
потери животных.
6.1. Ооциты
После отмывки от ДМСО оттаявшие ооциты или кумулюс-
ные массы можно переносить в яйцеводы самок, предваритель-
но (за 2—4 ч) спаренных с фертильными самцами [3]. Выжи-
ваемость повышается, если ооциты оплодотворяют in vitro и пе-
реносят на 2-клеточной стадии в яйцеводы псевдобеременных
реципиентов дня 1 (разд. 6.4.5).
6.2. Оплодотворение in vitro
Процедуры, которые, судя по нашему опыту, оказались
наиболее удачными при оплодотворении оттаявших ооцитов in
vitro, обобщены в табл. 13.7 и детально описаны ниже.
6.2.1. Среды
Спермин собирают и диспергируют в модифицированном
растворе Тироде (Тб: 15, табл. 13.8). Для капаситации спер-
миев и оплодотворения среду обогащают 15 мл/мл БСА, Sigma,
Fraction V (Т6 + БСА).
23—171
346
Глава 13
Таблица 13.7. Оплодотворение in vitro оттаявших ооцитов мыши
(см. разд. 6.2)
Инкубацию проводите при 37 °C в увлажненной атмосфере с 5% СО;
в воздухе.
А. День накануне оплодотворения in vitro
1. Внесите пипеткой по 1 мл среды Тб (табл. 13.8) в две пластиковые чашки
Петри Falcon или в два эмбриологических часовых стекла. Поверх на-
слоите парафиновое масло. Инкубируйте в течение ночи. Эти камеры будут
использованы для начальной дисперсии спермы.
2. Добавьте БСА (Sigma, Fraction V, 15 мг/мл) к Тб (Т6-1-БСА), стери-
лизуйте фильтрованием и инкубируйте всю ночь.
Б. В день оплодотворения in vitro
1. Отрегулируйте pH среды Т6+БСА до 7,5—7,6, приготовьте капли среды
для оплодотворения (разд. 6.2.2) и инкубируйте.
2. Соберите сперму в капли Тб, приготовленные в п. А1 (выше), и инкуби-
руйте (разд. 6.2.3).
3. Через 25 мин в каждую каплю внесите пипеткой аликвоту хорошо диспер-
гированных подвижных спермиев, чтобы конечная концентрация состав-
ляла 1—2хЮб спермиев/мл. Инкубируйте.
4. Разморозьте ооциты, проведите разбавление ДМСО (разд. 3.6) и отмойте
в М2. Держите на воздухе на нагревательном столике при 37 °C.
5. Через 2 ч после сбора спермы промойте ооциты в Т6+БСА и перенесите
в капли для оплодотворения (разд. 6.2.4). Инкубируйте.
6. Примерно через 4 ч соберите ооциты, промойте в среде М16+БСА (см.
гл. 2, разд. 5.2) и инкубируйте всю ночь в каплях среды М16+БСА под
маслом (разд. 6.5.2).
7. Примерно через 20 ч инкубации имплантируйте эмбрионы на 2-клеточной
стадии в М2 псевдоберемеиным самкам дня 1 (см. разд. 6.4.5).
6.2.2. Приготовление капель для оплодотворения
Для оплодотворения ооциты, свободные от клеток кумулю-
са, или интактные кумулюсные массы инкубируют с капасити-
рованными спермнями в каплях Т6 + БСА под парафиновым
маслом. Приготовьте капли следующим образом.
1. Инкубируйте Т6+БСА в течение ночи при 37 °C в атмосфе-
ре с 5% СОг в воздухе.
2. На следующий день доведите pH среды до 7,5—7,6 с по-
мощью 0,1 М NaOH. Точное измерение pH необязатель-
но. Вполне достаточно реакции окрашивания с фосфат-
ным буфером Соренсена, где роль индикатора играет фе-
ноловый красный (табл. 13.9).
3. Внесите пипеткой 100 мкл среды в 60-мм пластиковую
чашку Петри (Falcon).
4. Окружите (но не закрывайте) каплю теплым, пре-газован-
ным парафиновым маслом.
5. Добавьте 300 мкл Тб + БСА к первой капле.
Низкотемпературная консервация яиц и эмбрионов мыши 347
Таблица 13.8. Состав среды Тб, используемой для оплодотворения in vitro
Компонент г/л мМ
NaCl 5,719 97,84
КС1 0,106 1,42
MgCl2-6H2O 0,096 0,47
Na2HPO4-12H2O 0,129 0,36
СаС12-2Н2О 0,262 1,78
NaHCO3 2,101 25,00
Лактат Na1’ 2,791 24,90
Пируват Na* 1 2) 3 4 5 0,052 0,47
Глюкоза 1,000 5,56
Пенициллин G (соль калия) 0,060 —
Сульфат стрептомицина 0,050 —
Феноловый красный 0,010 —
Т6 представляет собой модифицированный раствор Тнроде.
’) Sigma grade DI-V, 60%-иый сироп.
Calbiochem-Boehring.
6. Наслоите на каплю парафиновое масло.
7. Инкубируйте при 37 °C в воздухе с 5% СО2, пока не бу-
дет готова суспензия спермиев.
6.2.3. Источник и сбор спермы
Оплодотворяющая способность спермиев зависит от линии
мышей; обычно в качестве доноров спермы предпочитают гиб-
ридных самцов Fj от скрещивания C57BL/6XCBA/Ca. Исполь-
зуются половозрелые, проверенные самцы, находившиеся от-
дельно от самок в течение 2 нед. Готовят две суспензии спер-
миев. Соберите и диспергируйте спермин, как описано ниже.
Работайте быстро и пользуйтесь нагревательным столиком при
37 °C для поддержания нужной температуры среды.
1. Отпрепарируйте от каждого из доноров один хвост при-
датка яичка (cauda epididimis) и один семявыносящий
проток (vas deferens).
2. Положите вырезанную ткань на лабораторную салфетку,
чтобы промокнуть кровь.
3. Поместите ткани в 1 мл теплой Тб под маслом.
4. Вырежьте оставшиеся caudae+vasa deferentia и помести-
те во вторую аликвоту Тб.
5. Под контролем препаровального микроскопа осторожно
извлеките сперму из vasa deferentia, воспользовавшись
двумя тонкими пинцетами. Проколите ткань концами пин-
цета и выдавите сперму. Уберите ткань.
23'
348
Глава 13
Таблица 13.9. Приготовление фосфатного буфера Соренсена
для стандартного определения pH
1. Приготовьте исходный раствор А (1/15М КН2РО4): 9,08 г КН2РО4, 10 мг
фенолового красного, дважды дистиллированной в стеклянной посуде Н2О
до 1 л.
2. Приготовьте исходный раствор Б (1/15М Na2HPO4): 11,8 г Na2HPO4-2H2O,
10 мг фенолового красного, дважды дистиллированной в стеклянной посу-
де Н2О до 1 л,
3. Приготовьте растворы с известным pH, смешивая аликвоты исходных рас-
творов А и Б.
pH (при 18 °C) Исходный раствор А, мл Исходный раствор Б, мл
6,6 62,7 37,3
6,8 50,8 49,2
7,0 39,2 60,8
7,2 28,5 71,5
7,4 19,6 80,4
7,6 13,2 86,8
4. Проверьте pH с помощью pH-метра и, если нужно, доведите, внеся алик-
воты нужного раствора (А — для понижения pH, Б — для повышения pH).
5. Стерилизуйте аликвоты каждого раствора фильтрацией через мнллипоро-
вый фильтр в пробирки для проб Falcon.
6. Храните при 4 °C.
7. Растворы не меняют pH в течение ~6 мес.
6. Инкубируйте сперму при 37 °C во влажной атмосфере
с 5% СО2 в воздухе в течение 25 мин.
7. Выберите лучше диспергированную и с более активными
спермиями суспензию и оцените концентрацию спермиев
с помощью гемоцитометра.
8. Разведите суспензию спермиев в приготовленной капле
для оплодотворения (разд. 6.2.2) до конечной концентра-
ции 1—2ХЮ6 мл.
6.2.4. Инкубация оттаявших ооцитов
с капаситированными спермиями
Оттаявшие ооциты инкубируют с капаситированными спер-
миями в течение 4 ч. В описанных здесь условиях капаситация
спермиев мыши происходит примерно через 2 ч после сбора.
Мы рекомендуем следующую процедуру:
1. После удаления ДМСО (см. разд. 3.6) поместите оттаяв-
шие ооциты в М2 при 37 °C в воздухе.
2. Через 2 ч после сбора спермы промойте ооциты в T6-J-
4-БСА, чтобы удалить все следы буфера HEPES.
Низкотемпературная консервация яиц и эмбрионов мыши 34Э '
3. Перенесите ооциты в минимальном объеме среды в каплю
для оплодотворения (~ 100 ооцитов на каплю).
4. Инкубируйте в течение 4 ч при 37 °C с 5% СОг в воздухе.
6.2.S. Культивирование оплодотворенных ооцитов
и перенос эмбрионов
1. После 4 ч инкубации соберите ооциты из капли и отмойте
от спермиев, дважды сменив среду М16+БСА (гл. 2,
разд. 5.2).
2. Культивируйте в каплях среды М16+БСА под маслом.
3. На следующий день перенесите 2-клеточные эмбрионы
в яйцеводы псевдобеременных самок дня 1 (разд. 6.4.5).
6.3. Эмбрионы
Жизнеспособность эмбрионов, сохранявшихся в ЖА, в ко-
нечном счете оценивается по развитию из них живых мышей.
Однако морфологические признаки оттаявших эмбрионов, их
способность продолжить дробление in vitro хорошо коррелируют
с их жизнеспособностью после переноса реципиенту. Такая про-
межуточная оценка эмбрионов дает возможность сэкономить
время и средства, затрачиваемые на перенос дефектных
эмбрионов. Более того, эмбриональное развитие после оттаива-
ния, по-видимому, происходит с меньшей скоростью. Культиви-
рование in vitro способствует регуляции метаболизма перед пе-
реносом эмбрионов и повышает долю нормально разкивающих-
ся плодов. Замедления развития можно избежать и в том слу-
чае, если переносить реципиенту сразу же после оттаивания
8-клеточные эмбрионы. Между тем в случае бластоцист асин-
хронный перенос без предварительного культивирования (сразу
после оттаивания) не ускоряет замедленного темпа развития
[29, 30].
6.3.1. От 1-клеточиой до 8-клеточиой стадии
После удаления ДМСО просмотрите эмбрионы в препаро-
вальном микроскопе и классифицируйте их в соответствии со
следующими критериями.
1. Нормальные (внешний вид такой же, как у не замора-
живавшихся эмбрионов соответствующей стадии разви-
тия).
2. Бластомеры разбухли.
3. Один или более бластомеров подверглись лизису.
4. Дегенерировали.
5. Блестящая оболочка утрачена или разорвана.
350
Глава 13
Вероятность того, что нормальные эмбрионы возобновят раз-
витие в культуре, высока. Разбухание бластомеров свидетельст-
вует о том, что в клетках осталось некоторое количество
ДМСО, или транспорт через мембрану нарушен. Разбухшие
эмбрионы могут «оправиться», но некоторые лизируют. Способ-
ность поврежденных эмбрионов продолжить нормальное разви-
тие зависит от стадии, на которой они были заморожены, и от
числа поврежденных бластомеров. Потеря одного бластомера
на 2-клеточной стадии обычно оказывается летальной, и лишь
немногие из поврежденных 4-клеточных эмбрионов способны
сформировать нормальную бластоцисту. 8-Клеточные эмбрионы
после утраты одного или двух бластомеров сформируют бласто-
цисту с уменьшенным числом клеток внутренней клеточной
массы и смогут развиваться дальше после переноса. Эмбрионы
ранних стадий развития (<8 клеток) плохо развиваются
в культуре без блестящей оболочки, их не следует переносить
в яйцевод, пока не пройдет стадия компактизации. Эмбрионы
поздних стадий развиваются нормально без оболочки так же,
как и эмбрионы всех стадий, оставшиеся внутри разорванной
оболочки. Однако в отсутствие интактной блестящей оболочки
возрастает риск вирусной инфекции [31].
6.3.2. Бластоцисты
Сразу после оттаивания бластоцисты могут деформировать-
ся, затрудняя морфологическую оценку. Полностью дегенериро-
вавшие эмбрионы отбрасываются.
6.3.3. Развитие in vitro
Оттаявшие эмбрионы промывают в среде М16+БСА (гл. 2,
разд. 5.2) и культивируют в течение ночи в каплях той же сре-
ды под маслом. Эмбрионы, возобновившие нормальное дробле-
ние, и бластоцисты, восстановившие форму, переносят в яйце-
воды псевдобеременных реципиентов дня 1 (стадии от 2-клеточ-
ной до бластоцисты) или в матку (стадии морулы и бластоци-
сты) (разд. 6.4).
6.4. Перенос эмбриона
Для того чтобы развитие эмбрионов завершилось рожде-
нием живых детенышей, зародыши предимплантационных ста-
дий должны быть перенесены в организм псевдобеременной
приемной матери. Перенос можно осуществить с помощью хи-
рургических методов (в яйцевод или матку) и нехирургической
процедуры (см. работы [32, 33]). К сожалению, в последнем
Низкотемпературная консервация яиц н эмбрионов мыши 351
случае мы не можем предсказать, в каком именно роге матки
будет развиваться зародыш. К тому же этим методом трудно
овладеть. Здесь мы описываем суть операций по имплантации
эмбрионов в яйцевод (разд. 6.4.5.) и матку (разд. 6.4.6).
6.4.1. Приемные матери
Для обычного переноса эмбрионов мы предпочитаем брать
в качестве приемных матерей гибридных самок Fi (т.е.
C57BL/6JXCBA/Ca или C57BL/6XCBA/H), характеризующих-
ся более чем 90 %-ной псевдобеременностью после спаривания
со стерильным самцом и наступлением беременности после пе-
реноса. Аутбредных мышей (т.е. MFI; Harlan-Olac Ltd, Bices-
ter, Англия) можно использовать; инбредных линий следует
избегать. Мыши должны быть в возрасте по крайней мере
12 нед и весом 20 г и более.
6.4.2. Индукция псевдобеременности
Псевдобеременность индуцируется спариванием самок, на-
ходящихся в соответствующей стадии естественного цикла,
с вазэктомированным самцом (гл. 1, разд. 5.3) или с самцом,
обладающим естественной стерильностью, т.е. несущим транс-
локацию Т145Н [34]. Для того чтобы получить подходящего
реципиента в данный день, отберите для спаривания самок в
проэструсе или воспользуйтесь эффектом Уиттена (см. гл. 1,
разд. 5.2). Синхронизация с помощью экзогенных гонадотропи-
нов снижает пропорцию самок, у которых спаривание индуци-
ровало псевдобеременность.
6.4.3. Число эмбрионов
Обычно переносится от трех до шести эмбрионов в каждый
яйцевод или рог матки (т.е. общее число эмбрионов — 6—12 на
реципиент). Если использовать меньшее их число, беремен-
ность может не установиться, кроме того, у очень маленького
приплода меньше шансов выжить. Перенос слишком большого
числа эмбрионов приведет к замедлению их роста.
6.4.4. Анестетики
Реципиентов анестезируют за несколько минут до операции
1,2%-ным раствором авертина (гл. 1, разд. 5.5), инъецирован-
ным внутрибрюшинно в дозе 0,02 мл на г веса тела.
352
Глава 13
Таблица 13.10. Подготовка псевдобеременного реципиента
к переносу эмбрионов в яйцевод или матку
1. Анестезируйте мышь (разд. 6.4.4).
2. Положите мышь спиной вверх (головой «на 12 часов»), смочите спину
70%-ным этанолом и разделите шерсть по средней линии.
3. Тонкими заостренными ножницами (10,5 см) сделайте разрез длиной 2 см
вдоль средней линии.
4. Тонким зубчатым пинцетом (10,5 см) сдвиньте кожу влево и через стен-
ку тела найдите яичник в области поясничной впадины. Над этим райо-
ном ножницами разрежьте брюшные мышцы.
5. Извлеките яичник, яйцевод и часть рога матки, отведите в направлении
головы мыши и поместите на небольшой марлевый тампон.
6. Продолжайте, как описано в разд. 6.4.5 (перенос в яйцевод) или 6.5.6
(перенос в матку).
6.4.5. Перенос эмбрионов в яйцевод
Эмбрионы всех предимплантационных стадий (от яйца на
стадии пронуклеуса до поздней бластоцисты) могут быть пере-
несены в ампулы реципиента в 1-й день псевдобеременности
(день обнаружения копулятивной пробки). Подготовьте реципи-
ентов в соответствии с указаниями табл. 13.10 и продолжайте
процедуру дальше.
1. С помощью препаровального микроскопа (рекомендуется
Wild М5) исследуйте капсулу яичника и найдите бахром-
чатый конец яйцевода (обычно он свободно лежит под
яичником и направлен к средней линии).
2. Пользуясь парой глазных пинцетов, вскройте капсулу
яичника в месте наименьшей васкуляризации и помести-
те мышь в положение, облегчающее доступ к воронке яй-
цевода (головой «на 10 часов» для оператора-правши).
3. Под контролем другого микроскопа в пастеровскую пипет-
ку с тонко оттянутым концом введите несколько «маркер-
ных» пузырьков воздуха, а затем и эмбрионы в неболь-
шом объеме среды М2 (табл. 13.2).
4. С помощью тонкого пинцета (в левой руке) отведите яй-
цевод так, чтобы обнажился его бахромчатый конец, и
введите в него кончик пипетки; возвратите пинцетом яйце-
вод на место. Выпустите яйца и пузырьки воздуха (пу-
зырьки воздуха должны быть видны через стенку яйце-
вода), осторожно извлеките пипетку и с помощью пинце-
та на несколько секунд сомкните отверстие яйцевода.
5. Возвратите яичник и яйцевод в брюшную полость.
6. Сдвиньте кожу вправо, обнажите яйцевод, поверните
мышь (головой «на 5 часов») и повторите процедуру.
7. После возвращения правого яичника и рога матки
в брюшную полость соедините кожу одним или двумя за-
Низкотемпературная консервация яиц и эмбрионов мыши 353
Таблица 13.11. Синхронный и асинхронный перенос эмбрионов в матку
Тип переноса Стадия развития эмбриона Стадия псевдобере- менности реципи- ента1)
Синхронный 8-клеточная Морула/ранняя бластоциста День 3-й День 4-й
Асинхронный Морула/ранняя бластоциста Поздняя бластоциста/вылупившаяся бластоциста День 3-й День 3-й или 4-й
') День 1-й — день обнаружения копулятивной пробки.
жимами Michel. Накладывать швы на брюшные мышцы
нет необходимости.
6.4.6. Перенос эмбрионов в мвтиу
Эмбрионы на стадиях 8 бластомеров, морулы и бластоцисты
можно переносить в рога матки реципиента 3-го или 4-го дня
псевдобеременности (день 1-й — день обнаружения копулятив-
ной пробки). Эмбрионы ранних предимплантационных стадий
вскоре после переноса в матку дегенерируют. В случае свеже-
собранных эмбрионов синхронный и асинхронный перенос ока-
зывается успешным, если эмбрионы находятся на более про-
двинутых стадиях развития по сравнению с функциональным
состоянием эндометрия (табл. 13.11). Эмбрионы менее продви-
нутых стадий по сравнению с маткой (например, 8-клеточные
эмбрионы, трансплантируемые в матку 4-го дня беременности)
имплантируются редко. Если эмбрионы подвергались манипу-
ляциям in vitro, т. е. замораживанию или культивированию
(и развитие их в связи с этим замедлилось), асинхронный пере-
нос, дающий возможность адаптации перед имплантацией, по-
высит их выживаемость (табл. 13.11).
После подготовки мыши-реципиента, описанной в табл. 13.10,
продолжайте следующим образом.
1. С помощью препаровального микроскопа небольшого уве-
личения и с длинным рабочим расстоянием (например,
Prior Stereomaster 240; BDH) проконтролируйте наличие
в яичнике активных (красных) желтых тел.
2. Под другим микроскопом в тонко оттянутую пастеровскую
пипетку введите несколько маркерных пузырьков возду-
ха, затем захватите эмбрионы.
3. Держите пипетку и иглу (инъекционную иглу 25 калибра
или швейную иглу № 7) правой рукой; тонким тупым
пинцетом, наложив его на яйцевод, придерживайте матку
(не сдавливайте рог матки).
354
Глава 13
Таблица 13.12. Выживаемость ооцитов и эмбрионов мыши
после криоконсервации путем замораживания и витрификации
Способ кризкон- сервации Стадия развития Линия Выживае- мость1) Литература
Замораживание 1.5 М ДМСО Медленное охлаж- дение до —70 °C Медленное нагре- вание 1,5 М ДМСО Медленное охлаж- дение до —40 °C Быстрое нагрева- ние Ооциты 8-клеточная 8-клеточная Гибрид Fi2) Гнбрнд Fi4) Случайное скре- щивание Гнбрнд F25) 6 7 Инбредная СВА/СаН ХО Гнбрнд Fi4) Случайное скре- щивание FM1 Гибрид F25) 163) 42 50 20—30 21 14 36 51 54 35 [21] [13] [13] [35] [36] [36] [13] [23] [27]
Витрификация •90%-ный ВР1 8-клеточная Случайное скре- щивание MF1 18 [6]
1) Доля ооцитов или эмбрионов, которые после криоконсервации дали начало нор-
мальным плодам или живорожденным мышатам (%).
-') Собранные от гибридных доноров Fj (C57BL/6JLacXCBA/CaLac).
3) После оплодотворения спермой СВА/Сан-Тб.
J) Собранные от самок линии MF1, спаренных с самцами C57BL.
5) Собранные от гибридных самок Fi (СЗН/НеНХ101Н). спаренных с самцами той
же гибридной линии.
4. Проколите иглой рог матки с антимезометриальной сто-
роны у места соединения матки с трубой. Уберите иглу
(не спуская глаз с отверстия), введите конец пипетки (не
проколите стенку матки) и выпустите эмбрионы в неболь-
шом объеме среды (наблюдайте за движением пузырьков
воздуха, но не выпускайте их в матку).
5. Уберите пипетку и проверьте, не осталось ли в ней эм-
брионов.
6. Возвратите яйцевод и матку в брюшную полость, не тро-
гая матку.
7. Сдвиньте край разреза кожи вправо и повторите проце-
дуру-
s. Соедините края разреза кожи одним или двумя зажима-
ми (накладывать швы на брюшные мышцы не нужно).
Низкотемпературная консервация яиц и эмбрионов мыши 355
6.4.7. Послеоперационное содержание
После операции держите мышей в тепле, пока не закончится
действие анестетика. Их можно размещать небольшими группа-
ми вплоть до родов.
6.5. Общая выживаемость
Ожидаемый уровень выживаемости после криоконсервации
яиц и эмбрионов с использованием описанных методов приве-
ден в табл. 13.12.
7. Криоконсервация ооцитов и эмбрионов человека
Способы криоконсервации человеческих эмбрионов были
разработаны на основе методов, применяемых для криоконсер-
вации эмбрионов мыши и сельскохозяйственных животных.
Эмбрионы всех предимплантационных стадий развития челове-
ка (от яйца на стадии пронуклеуса до бластоцисты) после
криоконсервации дают начало нормальной беременности. Ис-
пользуемые методы различаются в деталях, их можно найти
в работах [5, 37—39]. Общая выживаемость низка, меньше 5%,
[40] от числа замороженных эмбрионов.
Есть одно сообщение о беременности у человека, наступив-
шей после оплодотворения in vitro ооцитов, прошедших про-
цедуру замораживания — оттаивания [41].
Об удачной витрификации ооцитов человека сведений нет.
Более того, мы убеждены в том, что потенциальная токсичность
витрификационного раствора делает его непригодным для крио-
консервации ооцитов и эмбрионов человека.
Литература
1. Whittingham D. G., Leibo S. Р„ Mazur Р. (1972). Science, 173, 411.
2. Wilmut I. (1972). Life Sci., II, part 2, 1071.
3. Whittingham D. G. (1977). J. Reprod. Fert., 49, 89.
4. Wilmut I. (1986). In: Manipulation of Mammalian Development. Gwat-
kin R. B. L. (ed), Plenum Press, New York.
5. Trounson A., Mohr L. (1983). Nature, 305, 707.
6. Rail W. F., Wood M. J., Kirby C., Whittingham D. G. (1987), J. Reprod. Fert.,
80, 499.
7. Scheffen B., Van Der Zwalmen P., Massip A. (1986). Cryo-Letters, 7, 260.
8. Rail W. F. (1986). Cryobiology, 23, 548..
9. Leibo S. P., Mazur P. (1978). In: Methods In Mammalian Reproduction. Da-
niel J. C„ Jr (ed ). Academic Press, New York, p. 179.
10. Whittingham D. G. (1980). In: Low Temperature Preservation in Medicine
and Biology. Ashwood-Sniith M. J., Farrant J. (eds), Pitman Medical, Tun-
bridge Wells, UK p. 65.
11. Renard J-P., Bui-Xuan-Nguyen., Garnier V. (1984). J. Reprod. Fert., 71,
573.
356
Глава 13
12. Takeda Т., Elsden R. P., Seidel G. E., Jr. (1984). Theriogenology, 21, 266.
13. Whittingham D. G., Wood M. J., Farrant J., Lee H., Halsey J. A. (1979).
J. Reprod. Fert., 56, 11.
14. Whittingham D. G. (1974). Genetics, 78, 395.
15. Quinn P„ Barros C.t Whittingham D. G. (1982). J. Reprod. Fert., 66, 161.
16. Renard J.-P., Babinet C. (1984). J. Exp. Zool., 230, 443.
17. Rail IV'. F„ polge C. (1984). J. Reprod. Fert., 70, 285.
18. Rail IV7. F., Czlonkowska M., Barton S. C„ Polge C. (1984). J. Reprod. Fert.,
70, 293.
19. Miyamoto H„ Ishibashi T. (1983). J. Exp. Zool., 226, 123.
20. Fuller B. J., Bernard A. (1984). Cryo-Letters, 5, 307.
21. Glenisler P. H., Wood M. Kirby C., Whittingham D. G. (1986). Gamete
Res 16 205
22. Rail W.’f., Reid D. S., Polge C. (1984). Cryobiology, 21, 106.
23. Rail W. F„ Fahy G. M. (1985). Nature, 313, 573.
24. Baxter S. J., Lathe G. H. (1971). Biochem. Pharmacol., 30, 1079.
25. Fahy G. M., MacFarlane D. R., Angell C. A., Meryman H. T. (1984). Cryo-
biology, 21, 407.
26. Mackenzie A. P. (1977). Phil. Trans. R. Soc. bond. Ser. B, 278, 167.
27. Glenister P. H., Whittingham D. G., Lyon M. F. (1984). J. Reprod. Fert.,
70, 229.
28. Staats J. (1976). Cancer Res., 36, 4333.
29. Whittingham D. G. (1977). In: The Freezing of Mammalian Embryos. Ciba
Foundation Symposium 52, Elliott K. and Whelan J. (eds), Elsevier/North
Holland, Amsterdam, p. 97.
30. Wood M. J. (1986). In: Proceedings of the Fourth World Congress on In
Vitro Fertilization. Johnston I. (ed.), Plenum Press, New York.
31. Carthew P., Wood M. J., Kirby C. (1985). J. Reprod. Fert., 73, 207.
32. Marsk L., Larson K. S. (1974). J. Reprod. Fert., 37, 393.
33. Moler T. L., Donahue S. E„ Anderson G. B. (1979). Lab. Anim. Sci., 29, 353.
34. Lyon M. F., Meredith R. (1966). Cytogen, 5, 335.
35. Whittingham D. G., Lyon M. F., Glenister P. H. (1977). Genet. Res., 29, 171.
36. Whittingham D. G., Lyon M. F., Glenister P. H. (1977). Genet. Res., 30, 287.
37. Zeilmaker G. H., Alberda A. T., van Gent I., Rijkmans С. M. P. M., Drogen-
dijk A. C. (1984). Fertil, Steril., 42, 293.
38. Cohen J., Simons R. F., Edwards R. G„ Fehilly С. B., Fishel S. B. (1985).
J. In Vitro Fert. Embryo Transfer, 2, 59.
39. Lassalle B., Testart J., Renard J.-P. (1985). Fertil. Steril., 44, 645.
40. Whittingham D. G. (1985). In: In Vitro Fertilization and Donor Insemina-
tion. Thompson W., Joyce D. N. and Newton J. R. (eds). Royal College of
Obstetricians and Gynaecologists, London, p. 269.
41. Chen C. (1986). Lancet, No. 8486, 884.
ГЛАВА 14
ОПЛОДОТВОРЕНИЕ ООЦИТОВ ЧЕЛОВЕКА
И КУЛЬТИВИРОВАНИЕ ЧЕЛОВЕЧЕСКИХ
ПРЕДИМПЛАНТАЦИОННЫХ ЭМБРИОНОВ
Питер Брауде^
1. Введение
Разработка метода оплодотворения in vitro (OIV) с после-
дующей имплантацией эмбрионов в материнский организм по-
зволила многим бесплодным парам иметь собственных детей.
Однако соответствующие методические процедуры нуждаются
в существенных усовершенствованиях, а для этого необходимо
лучше понимать механизмы оплодотворения у человека, анали-
зировать каждую неудачную попытку оплодотворения или
трансплантации.
В настоящей главе мы изложим технику получения сперма-
тозоидов и ооцитов, опишем процедуры оплодотворения и куль-
тивирования человеческих эмбрионов in vitro. Эти методы уже
применяются в клинике, однако очень велика их роль и в ис-
следовательской работе с человеческим материалом, которая
необходима для разработки новых направлений терапии беспло-
дия [1].
2. Источники ооцитов и эмбрионов человека
для проведения исследований
2.1. Получение ооцитов и эмбрионов
2.1.1. Оплодотворение in vitro в терапевтических целях
Вероятность начала беременности после оплодотворения in
vitro значительно возрастает, если в матку имплантировать не-
сколько эмбрионов [2]. Чтобы иметь их в достаточном количе-
стве, разработаны методы суперовуляции, позволяющие полу-
чать в результате лапароскопии сразу много ооцитов. Эти ме-
тоды основаны на использовании человеческих менопаузальных
гонадотропинов, представляющих собой смесь лютеинизирующе-
го (ЛГ) и фолликулостимулирующего гормонов (ФСГ), полу-
0 Peter R. Braude. Embryo and Gamete Research Group, Department of
Obstetrics and Gynaecology, University of Cambridge Clinical School, Rosie
Maternity Hospital, Cambridge, UK.
358
Глава 14
ченных из постменопаузной мочи; они применяются самостоя-
тельно или в комбинации с кломифеном — антагонистом рецеп-
торов эстрогенов [4]. Предовуляционная доза в 5000—10 000 ME
человеческого хорионического гонадотропина (чХГ Profasi
Serono Labs; Pregnyl, Organon Labs) за 34—36 ч до аспирации
фолликулов инициирует разрушение зародышевого пузырька
и окончательное созревание ооцитов.
С возрастанием числа имплантированных эмбрионов растет
и число случаев многоплодной беременности, для которой ха-
рактерен повышенный риск спонтанного аборта или недоношен-
ности [5]. В связи с этим обычно рекомендуется ограничивать
число имплантируемых эмбрионов тремя. Полученные «лиш-
ние» эмбрионы могут быть, с согласия супружеской пары, ис-
пользованы для исследовательской работы (разд. 8).
2.1.2. Перенос гамет в фаллопиевы трубы
Перенос гамет в фаллопиевы трубы (ПГФТ)—относитель-
но новое многообещающее терапевтическое средство преодоле-
ния бесплодия в тех случаях, когда патологии труб нет (бес-
плодие из-за недостаточного объема спермы или нарушения
транспорта спермиев) [6]. Ооциты извлекают из фолликулов,
а затем с помощью тонкого катетера возвращают в фаллопиевы
трубы с небольшой аликвотой отмытых сперматозоидов. Риск
получения многоплодной беременности вынуждает ограничить
число ооцитов, переносимых в фаллопиевы трубы, двумя-тре-
мя. Таким образом, в результате этой клинической процедуры
также окажутся «лишние» ооциты, которые можно использо-
вать для исследований.
2.2. Донорские ооциты
Донорами ооцитов для эмбриологических исследований мо-
гут быть пациентки, подвергающиеся лапароскопической стери-
лизации, если они согласятся на стимуляцию суперовуляции
[7]. Процедура сбора яйцеклеток не должна длиться дольше
времени анестезии, требующегося для операции стерилизации.
В таких случаях стимуляция суперовуляции может осуще-
ствляться одним из двух методов (рис. 14.1). Во-первых, можно
пользоваться фиксированным графиком стимуляции, при кото-
ром день менструации сдвигается в результате изменения дли-
ны предыдущего менструального цикла с помощью прогестеро-
на [8]; подходит и скользящий график, при котором день при-
менения чХГ приурочен к времени хирургической опера-
ции [9]. Оба метода дают возможность получить яйцеклетки,
Оплодотворение ооцитов человека
359
Оральная контрацепция
5 мг норэтистерона
7 11
Кломифен 50 мг
чХГ
.1__________
f
Операция
_____I---1__i f I I I I < I I _L L _ 1 > 1 » ! >
Дни цикла 1 5 11 12 13 14 15 16 17
5_________________9
| | Кломифен 150 мг/день| Ч^Г Б
| Менструация | wf
Операция
Рис. 14.1. Режимы, обычно применяемые для стимуляции суперовуляции у до-
норов, которым предстоит лапароскопическая стерилизация. А. Фиксирован-
ный день операции — день кровотечения регулируется применением гормонов.
Б. День операции варьирует в зависимости от дня менструации.
способные оплодотворяться in vitro, но в тех случаях, когда
пациентки в течение длительного периода принимали внутрь
контрацептивные средства, выход ооцитов очень низок.
3. Сбор ооцитов
Сбор ооцитов производится либо визуально путем лапаро-
скопии, либо трансвезикально, трансвагинально или трансуре-
трально с использованием ультразвукового просвечивания яич-
ников [10]. Лапароскопический сбор ооцитов применяют зна-
чительно чаще, чем ультразвуковой; вероятно, это объясняется
тем, что гинекологи в своей повседневной практике лучше зна-
комы именно с этой оперативной техникой. Однако методы, ис-
пользующие ультразвук, не требуют большого числа помощни-
ков, выполняются без общего наркоза, в любой удобный иссле-
дователю день. Не удивительно, что эти методы постепенно
приобретают все больше сторонников.
3.1. Лапароскопический сбор ооцитов
Детальное описание процедуры лапароскопии и аспирации
фолликулов не входит в задачу этой книги. Мы считаем целе-
сообразным дать лишь общие представления, которые помогли
бы эмбриологам, участвующим в оплодотворении in vitro, по-
нять хирургическую процедуру и свою роль в подготовке к опе-
рации. Более детальное описание сделано Крафтом [И].
Под общим наркозом делают небольшой надрез кожи ниже
пупка и в брюшную полось вводят иглу Veress с газовым кра-
ном в открытом положении. Газовую смесь (обычно это
100%-ный СО2, см. разд. 3.4) накачивают через иглу со ско-
ростью 1 л/мин. При этом регистрируется внутрибрюшинное
давление, которое не должно превышать давления покоя для
иглы более чем на 5 мм ртутного столба, свидетельствуя о том,
что игла правильно введена в перитонеальную полость. Брюш-
360
Глава 14
ную полость наполняют примерно 2,5 л газа. Иглу Veress уда-
ляют, разрез кожи увеличивают до 1,5 см и вводят в перитоне-
альную полость лапароскопический троакар и канюлю. Через
канюлю вводят лапароскоп с волоконным световодом. Если
место введения было выбрано правильно, можно различить со-
держимое брюшной полости. После осмотра органов таза для
облегчения доступа к яичнику над лобковой линией волос де-
лается второй разрез длиной 1 см, через который вводится
захватывающий пинцет. Этим пинцетом находят и захватывают
связку яичника. Манипулируя его положением, можно найти
оптимальный доступ к фолликулам. Если яичник подвижен, по-
воротом связки можно даже перевернуть его нижней стороной
вверх.
Как только будут обнаружены фолликулы и выбран наибо-
лее удобный способ подхода к ним, троакар и канюлю для от-
сасывающей иглы вводят латерально между двумя предыдущи-
ми разрезами. Ассистент захватывающим пинцетом удерживает
яичник в нужном положении, тем временем через канюлю про-
ходит отсасывающая игла и прокалывает соответствующий
фолликул. Фолликул не следует прокалывать в самом тонком
месте, так как быстрое падение внутреннего давления может
вызвать разрыв стенки фолликула, вследствие чего края отвер-
стия не будут достаточно плотно прилегать к игле. Отсасыва-
ние производится с помощью специального вакуумного насоса
(Craft Suction Unit, Rocket of London) при 80—100 мм ртутно-
го столба или вручную шприцем на 20 мл. Как только стенки
фолликула спадутся, отсасывание прекращают и пробирку
с аспиратом (фолликулярной жидкостью) передают эмбриологу
для исследования. Если ооцит обнаружен, иглу извлекают и от-
сасывают следующий фолликул. Если ооцита в первом аспира-
те нет, фолликул наполняют средой для вымывания ооцитов
(табл. 14.2), которую затем также отсасывают. При этом сле-
дует перемещать кончик иглы внутри фолликула, что повышает
вероятность соскабливания яйца с гранулезных клеток, к кото-
рым оно может быть все еще прикреплено. Подобные промыва-
ния фолликула делают до тех пор, пока не будет найден ооцит.
После аспирации всех фолликулов собирают жидкость из уте-
ро-ректальной ямки, так как яйцо может оказаться в излившей-
ся фолликулярной жидкости. И наконец, проверив, нет ли кро-
вотечения яичника, выпускают газ из брюшной полости и на-
кладывают швы на разрез кожи.
3.2. Инструменты для сбора ооцитов путем лапароскопии
К подготовке инструментов для лапароскопии следует отне-
стись с такой же тщательностью, как и к любой другой работе,
связанной с культивированием тканей. Необходимо объяснить
Оплодотворение ооцитов человека
361
Б Система игл с двумя просветами
промывания
Рис. 14.2. Типы игл, используемых для лапароскопического сбора ооцитов.
Игла с двойным просветом (Craft),
с выстилкой или без,градуирован-
ная на дистальном конце
медицинскому персоналу, что подготовка инструментов для
сбора ооцитов и культивирования эмбрионов выходит за рамки
обычной стерилизации. По нашему мнению, контроль за подго-
товкой инструментов должен осуществлять эмбриолог.
3.2.1. Типы игл, используемых для сбора ооцитов
Для фолликулярной аспирации используется целый ряд
игл. Они могут иметь один или два канала. В случае игл с един-
ственным каналом, после отсасывания фолликулярной жидко-
сти пробка, закупоривающая трубку для сбора, удаляется
и промывающая среда поступает по игле назад в фолликул.
Как только фолликул наполнится, трубка для сбора закрыва-
ется и жидкость из фолликула отсасывается снова (см.
рис. 14.2,4). Иглы с двойным каналом позволяют производить
постоянное промывание фолликула. Отсасывание осуществля-
ется через канал большего диаметра, канал меньшего диамет-
ра служит для наполнения фолликула без извлечения иглы
(рис. 14.2,5). Единственный недостаток этой системы заклю-
чается в том, что овальная форма иглы затрудняет получение
нужного отверстия в капсуле яичника. Можно приобрести круг-
лые иглы с двойными каналами (Go Medical Industries Castle-
24—171
362
Глава 14
med, UK). У игл некоторых типов (Renou, Craft, Mkl, Mk2;
Rocket Ltd) аспирационный канал выстлан тефлоновым шлан-
гом. Его гладкие стенки снижают вероятность потери в аспира-
ционной системе липких скоплений кумулюсных клеток. Одна-
ко, если не принять необходимых предосторожностей при под-
готовке игл и не производить частой смены тефлоновых вста-
вок, последние в результате повторных стерилизаций могут
деформироваться. У игл новых моделей (Craft МкЗ) такие
вкладыши отсутствуют.
3.2.2. Внутренние вкладыши игл
Мы производим замену тефлонового вкладыша игл после
одного или двух использований. Старую тефлоновую трубку
следует вытянуть из иглы, а скос острия иглы просмотреть под
препаровальным микроскопом. Обнаруженные зазубрины или
шероховатости можно устранить с помощью тонкозернистого
шлифовального бруска. После этого иглу тщательно прополас-
кивают ультрачистой водой для культуры тканей. Новую теф-
лоновую трубку протягивают через иглу. Поскольку при нагре-
вании трубка сокращается, ее конец обрезают на расстоянии
2 см от конца иглы. После стерилизации его срезают на уровне
скоса. Перед стерилизацией выстилающую трубку тщательно
споласкивают ультрачистой водой для культуры. Автоклавиро-
ванию мы предпочитаем стерилизацию двойных готовых игл
в горячем сушильном шкафу при 160 °C в течение 2 ч.
3.3. Подготовка инструментов для лапароскопии
Мы не советуем пользоваться общепринятым способом дез-
инфекции инструментов для лапароскопии—10-мин обработкой
в растворе глутаральдегида (Cidex, Surgicos Ltd). Эта про-
цедура не убивает спор, но является вирицидной и бактерицид-
ной. Она обеспечивает необходимую деконтаминацию инстру-
ментов между оперированием разных пациентов. Однако важно
учесть, что раствор глутаральдегида обладает высокой токсич-
ностью и адгезивностью. Он может оставлять следы на перчат-
ках хирурга или операционной сестры, на инструментах.
В связи с этим следует предпочесть стерилизацию путем авто-
клавирования.
3.4. Газы, используемые для лапароскопии
Существенное значение имеет контроль за постоянством pH
жидкости, окружающей яйцо. При стандартных процедурах
лапароскопии брюшная полость наполняется 100%-ным СО2.
Оплодотворение ооцитов человека
363
Учитывая высокую скорость реабсорбции, можно полагать, что
это наиболее безопасная из газовых смесей в случае неожидан-
ного повреждения крупного сосуда.
Было бы логично предположить, что длительный контакт
тонкостенного фолликула с СОг вызовет понижение внутрифол-
ликулярного pH, однако прямые свидетельства в пользу этой
гипотезы отсутствуют. С другой стороны, нет сомнения в том,
что отсасывание газа вместе с содержимым фолликула может
снизить pH отсосанной жидкости, особенно если газ замкнут
над фолликулярным аспиратом в плотно закрытых пробирках.
Исходя из этих соображений, 100%-ному СОг при аспирации
ооцитов предпочитают газовые смеси, содержащие физиологи-
ческий уровень СО2 (например, 5% СО2:5% Ог:90%\2) [12].
Сначала брюшная полость наполняется для безопасности СОг,
а затем, когда будет фиксировано положение канюли в пери-
тонеальной полости, накачивается смесь с уменьшенным коли-
чеством СОг-
Поскольку фолликулярная жидкость обладает слабыми бу-
ферными свойствами, следует как можно быстрее извлечь ооцит
из аспирационной жидкости и перенести в соответствующим об-
разом забуференную среду. Если лаборатория расположена не
в непосредственной близости от операционной, необходим пе-
редвижной столик со стереомикроскопом, снабженным нагрева-
тельным столиком и термостатически регулируемым нагрева-
тельным блоком (ВТ1; Grant Instrument) на 37 °C. Как только
будет обнаружен ооцит, его следует перенести в тест-пробирку
со средой, забуференной HEPES (табл. 14.2) при 37°C для
транспортировки в лабораторию.
4. Среды для культивирования эмбрионов
и вымывания ооцитов
Принципы, лежащие в основе приготовления сред для куль-
тивирования эмбрионов, детально изложены Праттом в гл. 2
(разд. 5). Мы опишем лишь некоторые модификации, связанные
со спецификой оплодотворения и культивирования эмбрионов
человека.
4.1. Ионный состав
Для оплодотворения ооцитов человека in vitro и культиви-
рования эмбрионов с успехом применяется ряд сред. Все они
представляют собой модификации сред, широко используемых
для культивирования in vitro предимплантационных эмбрионов
и для роста клеточных линий, — это сбалансированные солевые
растворы Эрла (EBS) [13], раствор Тб Уиттингхэма [14],
В2 Менезо [15], F10 Хэма [16]. Применение названных сред
24*
364
Глава 14
дает приблизительно одинаковые результаты по оплодотворе-
нию и скорости дробления.
Некоторые из основных сред можно приобрести в готовом
виде — в жидком (в десятикратной концентрации) или в форме
порошка (EBS, F10 Хэма, Flow Labs). Однако многие исследо-
ватели предпочитают готовить среды собственными руками, по-
скольку это дает возможность лично контролировать каждый
ингредиент [17]. Рецепты сред, основанных на EBS. представ-
лены в табл. 14.1—14.4. Следует тщательно проверить осмоляр-
ность среды, она должна находиться в пределах 280—
285 мОсмоль/л, иначе оплодотворение может не произойти.
4.2. Макромолекулы
Во многих лабораториях, где проводят в терапевтических
целях оплодотворение in vitro, к среде для оплодотворения
и роста добавляется сыворотка крови матери. Вопрос о необхо-
димости присутствия макромолекул в культуральной среде был
поставлен недавно [18], хотя соответствующие эксперименты
на мыши свидетельствовали об этом.
Концентрация сыворотки, используемой для оплодотворе-
ния у человека, варьирует от 7,5 до 10%, а для культивирова-
ния—-от 10 до 15% [13—17]. Приготовление сыворотки, необ-
ходимой для оплодотворения in vitro, культивирования и пере-
носа эмбрионов, описано в табл. 14.4. Следует учесть, что
добавление сыворотки крови к среде создает трудности при
сравнении результатов, поскольку среда не имеет определенно-
го состава. В попытках решить эту проблему в нашей лабора-
тории мы стандартизировали среду для оплодотворения, заме-
нив сыворотку крови матери 5 мг/мл фракции 5 БСА (BSA;
Sigma А9647). БСА можно приобрести в большом объеме и хра-
нить замороженным в течение, ряда лет. Оплодотворение
в этой среде, как правило, проходит успешно, и оплодотворен-
ные таким образом ооциты дают качало беременности.
Среды для роста ранних эмбрионов стандартизировать труд-
но, так как БСА может обеспечивать развитие до стадии бла-
стоцисты [19]. С успехом использовались как смешанная сыво-
ротка крови, так и смешанная сыворотка спинномозговой
жидкости, однако, поскольку существует риск переноса забо-
леваний, использовать эти эмбрионы для имплантаций не реко-
мендуется, за исключением случаев, когда сыворотка должным
образом экранирована.
4.3. Вода
Наиболее уязвимыми ингредиентами систем культивирова-
ния предимплантационных эмбрионов являются вода и масло.
По нашему убеждению, независимо от качества установки для
Таблица 14.1, Ингредиенты, входящие в состав сбалансированного
солевого раствора Эрла для оплодотворения in vitro
и культивирования эмбрионов человека
EBS, Х10
Вода
Бикарбонат натрия
Пируват натрия
Пенициллин
Гентамицин
Гепарин
БСА, фракция 5
HEPES
Сбалансированный солевой раствор Эрла, в
10-кратной концентрации, без бикарбоната
натрия (Flow Labs)
Подходящая для культуры ткани Analar (BDH)
или вода для инъекций, В. Р. (Boots Hospital
Products)
(BDH : Analar grade)
(Sigma Chemical Co.) основной раствор 1,1 мг/мл;
заморозьте аликвоты по 1 и 4 мл
Cristapen (Glaxo Ltd), виалы иа 600 мг. Храни-
те в морозильнике. Разводите 1 мл ультрачнс-
той воды для культуры тканей до 600 мг/мл
Garamycin (Schering), изготовитель Flow Labs,
поставляется в виде основного раствора
10 мг/мл
Mucous Heparin, без консерванта (Paynes and
Byrne Ltd). Ампулы 0,2 мл, содержащие
5000 ME. Храните в морозильнике
Bovine Serum Albumin Powder Fraction V (Sig-
ma, Кат. № A9647)
Свободная кислота; (Ultrol, Calbiochem). Исход-
ный раствор 21 мМ. (5,96 г/100 мл). pH дове-
сти 0.2 М NaOH до 7,35—7,4. После приготов-
ления можно хранить до 3 мес
Таблица 14.2. Среда для промывания фолликулов и работы
с эмбрионами (EBS, забуференный HEPES, для использования на воздухе)
Для приготовления 400 мл EBS, забуференного HEPES (HEBS) без анти-
биотика:
1. Разморозьте одну 4-мл аликвоту пирувата натрия (табл. 14.1).
2. Взвесьте 0,1348 г бикарбоната натрия. Добавьте 20 мл воды и рас-
творите.
3. Отмерьте 40 мл десятикратного EBS в коническую колбу. Добавьте
295 мл воды и оттаявшую аликвоту пирувата натрия.
4. Медленно добавьте пипеткой раствор бикарбоната натрия, осторожно
встряхивая. Делайте это очень медленно, тогда в растворе не обра-
зуется осадок карбоната кальция. Если все же раствор замутится, вы-
лейте и повторите сначала.
5. Введите 33,6 мл исходного раствора HEPES (табл. 14.1).
6. Проверьте осмолярность (она должна быть в пределах 280—
285 мОсмоль/л). Если значение слишком низкое, добавьте концентриро-
ванного EBS.
Среда для получения ооцитов
7. Добавьте 350 мкл гепарина (табл. 14.1) к 350 мл EBS, забуференного
HEPES. Стерилизуйте миллипоровой фильтрацией. Храните в холодиль-
нике аликвотами по 50 мл в культуральных флаконах.
8. Профильтруйте остальные 50 мл основного HEBS для приготовления
среды, в которую будете помещать эмбрионы (среды для переноса).
Продолжение табл. 14.2
Приготовление среды для переноса эмбрионов (содержащей сыворотку ма-
теринской крови (10%), инактивированную нагреванием):
9. Добавьте 2 мл инактивированной нагреванием сыворотки (табл. 14.4)
к 18 мл HEBS.
10. Желательная процедура. Стерилизуйте фильтрацией (миллипоровый
фильтр), отбросив первые 0,5—1 мл профильтрованного раствора (в филь-
трах могут содержаться увлажняющие агенты, способные ингибировать
нормальное эмбриональное развитие).
11. Храните в морозильнике в стерильных пробирках или флаконах.
Таблица 14.3. Среда для оплодотворения in vitro и культивирования
эмбрионов (EBS, забуферениый бикарбонатом)
Для получения 100 мл исходного раствора EBS, забуференного бикарбо-
натом:
1. Разморозьте одну аликвоту (на 1 мл) пирувата натрия (табл. 14.1).
2. Взвесьте 0,21 г бикарбоната натрия. Добавьте 10 мл воды и растворите.
3. Отмерьте 10 мл десятикратного раствора EBS в коническую колбу. До-
бавьте 77 мл воды и оттаявшую аликвоту пирувата натрия.
4. .Медленно внесите с помощью пипетки раствор бикарбоната натрия,
осторожно встряхивая. Медленное добавление не вызовет образования
карбоната кальция, который может замутить раствор. Если раствор все
же получился мутным — выбросьте его и начните сначала.
5. Добавьте 10 мкл раствора пенициллина (табл. 14.1) и 0,2 мл раствора
гентамицина (табл. 14.1).
6. Проверьте осмолярность (она должна быть в пределах 280—
285 мОсмоль/л). Если значение слишком высокое, добавьте воды, если
низкое — концентрированный раствор EBS.
Приготовление среды для оплодотворения in vitro:
7. Добавьте 0,4 г БСА (табл. 14.1) к 80 мл EBS и оставьте для постепен-
ного естественного растворения. Не встряхивайте, иначе раствор будет
пениться и образуются труднорастворимые хлопья. Профильтруйте через
миллипоровый фильтр и храните аликвотами по 10 мл в пластиковых
пробирках (Falcon) (№ 2001) в морозильнике.
8. Профильтруйте остальные 20 мл исходного раствора н храните аликво-
ты по 4 мл в фолконовских пробирках (№ 2002) в морозильнике (при-
готовление культуральной среды для эмбрионов см. ниже).
Среда для культивирования эмбрионов (содержащая сыворотку материнской
крови, инактивированную и нагреванием)1'!
9. Извлеките 0,4 мл EBS из одной 4-мл аликвоты, хранящейся в морозиль-
нике, и замените этот объем EBS 0,4 мл инактивированной нагреванием
сыворотки.
10. Желательаня процедура. Профильтруйте через миллипоровый фильтр,
первые 0.5—1 мл отбросьте.
11. Используйте каплями по 500—100 мкл под маслом для культивирования
эмбрионов, начиная со стадии пронуклеусов.
') Чтобы получить 15%-пую сыворотку, удалите 0.6 мл из 4-мл аликвоты EBS п 9
и замените их 0.6 мл сыворотки.
Оплодотворение ооцитов человека
367
Таблица 14.4. Приготовление сыворотки материнской крови
и среды для переноса
1. В асептических условиях возьмите 10—15 мл крови, воспользовавшись ие-
токсическим шприцем для культуры ткани (Steriseral, No. 1. Medical).
2. Извлеките иглу и опорожните шприц в 15-мл стерильную пластиковую
центрифужную пробирку Falcon 2095, Becton Dickinson).
3. Центрифугируйте немедленно в настольной центрифуге 5—10 мин при
1000 g (максимальная скорость, рекомендуемая для центрифугирования
пробирок № 2095—3600 об/мин).
4. Сразу же, как только закончится центрифугирование, перенесите плазму
в стерильную пластиковую пробирку (Falcon № 2001), воспользовавшись
пастеровской пипеткой.
5. Оставьте пробирку с плазмой для свертывания. (Если плазма находится
в гладкостенной пластиковой пробирке, это произойдет через несколько
часов. Процесс можно ускорить, использовав чистую стеклянную пробир-
ку или опустив в пластиковую пробирку конец стерильной стеклянной
пастеровской пипетки.)
6. Уберите фибриновый сгусток после перемешивания плазмы стерильной
стеклянной пастеровской пипеткой.
7. Инактивируйте нагреванием в водяной бане при 56 °C в течение
30 мии.
8. Профильтруйте образец через стерильный фильтр Millex-GS 0,22 мкм
(Millipore Ltd), не забывая отбросить первые несколько капель (~0,5 мл)
образца.
9. Распределите аликвоты в стерильные пластиковые пробирки. Заморозьте
для использования в будущем.
очистки воды, которой располагает лаборатория, ее лучше
покупать. Вода Analar (BDH) является химически чистой и в
принципе подходит для оплодотворения и культивирования, но
она не стерильна, хранится в больших объемах, поэтому может
содержать бактериальные токсины. С другой стороны, вода для
инъекций (BDH) стерильна и не обладает пирогенностью, упа-
кована в подходящих объемах и готовится в строго контро-
лируемых стандартных условиях. По-видимому, именно вода
для инъекций и промываний (Boots) удовлетворяет требовани-
ям культивирования человеческих эмбрионов.
4.4. Масло
В течение ряда лет мы используем легкое парафиновое мас-
ло (BDH). Метод приготовления основного запаса масла
описан Праттом (гл. 2, разд. 5.1). Небольшие его количества,
необходимые для немедленного употребления, сначала отмыва-
ют средой для тканевой культуры путем энергичного перемеши-
вания примерно 200 мл EBS с 500 мл масла в литровом сте-
рильном флаконе для культуры ткани (Nunc). Затем эмульсии
дают разделиться на две фазы. Масло хранят до употребления
при комнатной температуре в этом же контейнере (т. е. плаваю-
368
Глава 14
щим на поверхности среды). Как и в случае всех ингредиентов,
используемых для культуры ткани, необходим жесткий конт-
роль качества масла. Для проверки реактивов на токсичность
применяется культивирование мышиных эмбрионов в блестящей
оболочке и без нее до стадии бластоцисты (см. гл. 2, разд. 6).
Потенциальный риск токсичности масла [20] и время, тре-
бующееся для проверки его качества [13], заставили исследо-
вателей обратиться к поискам альтернативных путей работы
с небольшими объемами среды. Ведь масло используется для
предотвращения изменения осмолярности, которое имеет место
в результате испарения воды в ходе инкубации, но оплодотво-
рение и культивирование можно осуществлять и в тест-пробир-
ках (Falcon 2001, Becton Dickinson) в объемах около 1 мл
в увлажненной атмосфере.
4.5. Антибиотики
Антибиотики добавляют к большинству сред для ингибиро-
вания роста микроорганизмов. Последние могут быть занесены
из окружения, иногда они представлены патогенными форма-
ми, присутствующими в самом образце [21]. Поскольку пени-
циллин и стрептомицин наиболее часто используются в качест-
ве антибактериальных средств, имеет смысл специально оста-
новиться на двух вопросах, касающихся применения этих ан-
тибиотиков при оплодотворении ооцитов человека.
Аллергия на пенициллин встречается весьма часто, поэтому
мы советуем либо вообще исключить этот антибиотик из всех
сред для промывания фолликулов и процедур переноса эмбрио-
нов, либо иметь специальный запас сред без пенициллина для
склонных к аллергии пациентов. Применение бактериального
фильтра (Millex-GV, Millipore), предусмотренное в протоколе
процедуры вымывания ооцитов, защитит пациента при операции
в случае занесения бактерий в среду.
Грамотрицательные бактерии наиболее часто встречаются
в бактериальных контаминациях, возможных при оплодотворе-
нии in vitro [22]. Именно эти бактерии, как правило, инфици-
руют женский половой тракт при перенесении эмбрионов. Изве-
стно, что Escherichia coli прочно связывается со сперматозоида-
ми и может оказаться не удаленной в процессе приготовления
препарата для оплодотворения [21]. Хотя стрептомицин пока
эффективен в отношении многих грамотрицательных патогенов,
устойчивость микроорганизмов к этому антибиотику повышает-
ся. Более эффективным нам представляется применение гента-
мицина (Sigma-Shering) в концентрации 20 мкг/мл.
Оплодотворение ооцитов человека 369
4.6. Газы
Большинство сред, применяемых для оплодотворения и куль-
тивирования эмбрионов in vitro, содержит 25 мМ. бикарбона-
та Na, который обеспечивает pH 7,2—7,4 в результате газации
5% СОг. Пока еще нет определенности в отношении идеальной
концентрации кислорода, потребляемого человеческим эмбрио-
ном при развитии in vitro. В ранних работах, выполненных на
мышиных и коровьих эмбрионах, было показано, что развитие
до стадии бластоцисты in vitro лучше происходит в 5%-ном
О2 [23]. Между тем высокая частота формирования бластоцист
мышиными эмбрионами обычно наблюдается при культивиро-
вании в среде с 5% СО2 в воздухе (21% О2). Пока не получе-
но надежных сравнительных показателей для развивающегося
эмбриона человека, частота формирования бластоцист у которо-
го низка в обеих газовых смесях. Однако частота успешной
имплантации примерно одинакова для эмбрионов, растущих
в 5% СО2 в воздухе и в смеси 5% СО2:5% О2:90% N2.
4.7. Температура
Температура инкубаторов для оплодотворения ооцитов че-
ловека и культивирования предимплантационных эмбрионов
должна поддерживаться в пределах 37—37,5 °C. Температуры
ниже нормальных могут прерывать полимеризацию микротру-
бочек и влиять таким образом на формирование мейотического
веретена, поэтому среда для вымывания ооцитов из фолликулов
должна быть теплой и ооциты после обнаружения их в аспира-
те необходимо переносить в теплую среду как можно быстрее.
5. Ооциты
5.1. Манипуляции с ооцитами
Как только ооцит найден в аспирате, отсосанном из фолли-
кула, его следует немедленно перенести в среду для оплодотво-
рения in vitro (т. е. 1 мл EBS-j-БСА/сыворотка под маслом
в течение ночи 5% СО2 в воздухе). Оооцит переносят стериль-
ной стеклянной пастеровской пипеткой с гладким оплавленным
концом. Если в аспирате присутствует много эритроцитов, ооцит
следует отмыть в чистой капле среды. Перед осеменением ооцит
инкубируют в течение 4—5 ч. Показано, что эта процедура по-
вышает оплодотворяемость [24].
5.2. Градация ооцитов
Применяемые режимы суперовуляции приводят к появлению
популяции фолликулов, не отличающихся полной синхронно-
стью развития. Несмотря на разрушение зародышевого пузырь-
Рис. 14.3. А. Зрелый человеческий предовуляторный ооцит. Обратите внима-
ние на обширную массу кумулюсных клеток, окружающую ооцит и маски-
рующую его морфологию. Б. Незрелый человеческий ооцит с плотно упако-
ванными клетками кумулюса. В. Оплодотворенный человеческий ооцит; видны
два пронуклеуса (20 ч после осеменения). Г. Ооцит с тремя пронуклеуса-
ми (19 ч после осеменения). Д. 2-клеточиый эмбрион (40 ч после осеменения).
Е. 4-клеточный эмбрион (46 ч после осеменения). Ж. 8-клеточный эмбрион
(74 ч после осеменения). 3. Бластоциста (138 ч после осеменения).
Оплодотворение ооцитов человека 371
ка вследствие предоперационной инъекции чХГ, степень зрело-
сти цитоплазмы ооцита может оказаться недостаточной для
инициации нормального эмбрионального развития [25].
Зрелый предовуляторный ооцит (рис. 14.3,Д), полученный
при аспирации, находится в метафазе II второго мейотического
деления. Сам предовуляторный фолликул крупный (его сред-
ний диаметр 2,2—2,8 см) и содержит 3—10 мл жидкости [26].
Масса клеток кумулюса, окружающая ооцит, увеличена за счет
цементирующей их слизи. Незрелые ооциты (рис. 14.3,5)
обычно вымываются из фолликулов меньшего объема (<2 мл)
и еще могут иметь зародышевый пузырек. Клетки кумулюса,
окружающие такой ооцит, плотно прилегают к его поверхности
и с трудом отделяются, даже после контакта со сперматозоида-
ми в течение 18 ч и более. Однако накапливаются сведения
о том, что у части таких незрелых ооцитов после культивирова-
ния в течение 12—36 ч может произойти созревание in vitro
[27]. Несмотря на то что оплодотворяемость ооцитов, созрев-
ших in vitro, снижена, иногда происходит развитие in vitro до
стадии бластоцисты. Способность эмбрионов, развившихся из
ооцитов, которые прошли созревание in vitro, дать начало бере-
менности, снижена [28].
5.3. Удаление клеток кумулюса
Детальное изучение морфологии предовуляционного ооцита
затруднено из-за большой массы кумулюсных клеток, маски-
рующих ооцит (рис. 14.3,X). Хотя некоторые заключения о со-
стоянии зрелости ооцита можно сделать на основе характери-
стики кумулюсной массы, о точной стадии созревания невоз-
можно судить, не удалив клетки кумулюса. Последние работы
свидетельствуют о том, что удаление клеток кумулюса до осе-
менения не препятствует оплодотворению и позволяет оценить
развитие ооцита по выделению первого полярного тельца. Та-
ким образом можно определить время инкубации, необходимое
для полного созревания перед осеменением [27, 28]. Для уда-
ления клеток кумулюса можно воспользоваться гиалуронида-
зой (см. гл. 2, разд. 2.1). Следует заметить, что у некоторых
линий мышей обработка ооцитов гиалуронидазой вызывает
иногда партеногенетическую активацию.
Как правило, удаление кумулюсной массы перед добавле-
нием сперматозоидов не проводится. Если ооциты находятся
в присутствии сперматозоидов в течение 12 ч или более, клетки
кумулюса большинства ооцитов отделяются спонтанно под
действием гиалуронидазы сперматозоидов, кроме того, их от-
делению может способствовать осторожное пипетирование стек-
лянной пастеровской пипеткой, диаметр оттянутого конца у ко-
372
Глава 14
торой чуть больше диаметра ооцита. Оставшиеся кумулюсные
клетки удаляют механически с помощью инъекционных игл
27-го калибра.
5.4. Удаление блестящей оболочки
Для некоторых видов анализа строения эмбриона или ооци-
та (например, для кариотипирования или для иммуноцитохими-
ческой локализации) может потребоваться удаление блестящей
оболочки. Блестящая оболочка человека обладает большей
устойчивостью, чем мышиная, но и она может быть растворена
в теплой кислотной среде (например, в кислом растворе Тиро-
де) или при инкубации в течение 5 мин в растворе проназы
(концентрацией 0,2 мг/мл) (см. гл. 2, разд. 3.2.1).
6. Сперматозоиды
6.1. Сбор образца семени
6.1.1. Контейнеры
Сбор образца семени производится путем мастурбации в
широкогорлый стерильный пластиковый контейнер (Sterilin,
60 мл). На контейнере должно быть четко обозначено имя па-
циента, больничный номер, время получения и дата предыдуг
щей эякуляции или коитуса.
6.1.2. Инструкции
Каждый пациент должен быть снабжен инструктивным ли-
стом, в котором объясняется способ получения образца и до-
ставки его в лабораторию. Для сбора семени нельзя пользо-
ваться презервативами, так как они обычно содержат спермици-
ды. Нельзя пользоваться мазями, например «KY-желе», также
в связи с их спермицидностью. Пациент не должен пытаться
собрать образец, прервав коитус, так как при этом может быть
потеряна первая наиболее богатая сперматозоидами аликвота.
Наличие больших количеств плоских клеток в эякуляте часто
указывает на то, что он получен путем прерывания коитуса.
Если известно, что объем семенной жидкости велик (>6 мл),
эякулят следует разделить. При этом используются два контей-
нера для образцов: в один должна быть собрана первая пор-
ция, в другой — все остальные. Пациента нужно предупредить
о том, что первая, богатая сперматозоидами порция обычно не-
велика (пациенты часто стараются представить два образца
равных объемов).
Оплодотворение ооцитов человека
373
6.1.3. Доставив обрвзцв в лабораторию
Образец семени должен быть доставлен в лабораторию как
можно быстрее после его получения, в идеальном случае в те-
чение 2 ч. Образец при транспортировке должен находиться
при температуре не менее 15 °C и не выше 38 °C, но не обяза-
тельно при температуре тела человека. Более того, она может
оказать повреждающее воздействие на сперматозоиды, так как
является оптимальной для метаболической активности (в ре-
зультате повышается образование метаболитов и, вероятно,
снижается жизнеспособность) и, возможно, для роста бактерий.
Сперматозоиды, выделенные из семенной жидкости и помещен-
ные в соответствующую среду (табл. 14.2 и 14.3), сохраняют
жизнеспособность более недели, но, если спермин остаются
в семенной плазме, этот период ограничивается одним днем.
6.1.4. Воздержание
Время воздержания влияет на качество образца семени.
Хотя с увеличением времени воздержания повышается общее
количество сперматозоидов, главным образом в результате уве-
личения объемов эякулята, процент подвижных сперматозоидов
и их жизнеспособность снижается. По-видимому, оптимальный
период — 48—72 ч.
6.2. Анализ семени
Детальный анализ физико-химических свойств семени и его
клеточного состава выходит за рамки этой главы. Однако в по-
мощь эмбриологам, подготавливающим сперматозоиды к про-
цедуре оплодотворения, ниже приводится краткое обобщение
некоторых нормальных характеристик спермы. Подробные све-
дения можно найти в превосходной монографии Мортиме-
ра [29].
6.2.1. Семеннвя жидкость
Семенная жидкость составляет наибольшую часть эякулята
и является результатом секреции предстательной железы
и семенных пузырьков. Вскоре после эякуляции семя образует
коагулят, который в течение 30 мин после формирования раз-
жижается. Нормально разжиженный эякулят имеет водянистую
консистенцию и может выпускаться из пастеровской пипетки
в виде капель. Вязкость семени со временем увеличивается, что
можно определить по слизевидным тяжам, тянущимся за пасте-
ровской пипеткой, когда ее извлекают из семени. Повышение
374
Глава 14
- .0
. с J
□
1
Рис. 14.4. Разметка усовершенствованного гемоцитометра Нейбауэра. В каж-
дом гемоцитометре две камеры. Общее число спермиев в пяти квадратах
(1—5) подсчитывается для каждой камеры. При разведении 1:20 среднее
счета для обеих камер представит концентрацию спермиев в миллионах на
миллиметр.
вязкости не означает, что семя становится менее жидким: раз-
жиженная сперма может быть достаточно вязкой. Вязкость се-
мени для удобства подсчета сперматозоидов можно снизить
периодическим пипетированием через пастеровскую пипетку или
узкую инъекционную иглу (22-го или 23-го калибра). Высокая
вязкость может привести к неточному счету сперматозоидов,
так как маленькую аликвоту вязкого образца трудно распреде-
лить в счетной камере.
6.2.2. Счетные камеры
А. Гемоцитометр Нейбауэра. Стандартная камера для под-
счета сперматозоидов представлена усовершенствованным гемо-
питометром Нейбауэра (A. R. Horwell). Она имеет две счетные
сетки 3X3 мм. Центральная область (см. рис. 14.4) разделена
Оплодотворение ооцитов человека
375
вид А
у
3-Незрелое
И-Дефект: в средней части
В-Вакуоли-
‘зарезанный
Г-Конические
ила
грушевидные
Ж-Сросшиеся
Схематическое изображение
нормальных и аномальных,
сперматозоидов человека
Рис. 14.5. Схематическое изображение нормальных и аномальных форм спер-
матозоидов. Из: Mortimer D. (1985). Current Problems in Obstetrics, Gynaeco-
logy and Fertility. 7. Year Book Medical Publishers. Воспроизводится с раз-
решения.
на 25 больших квадратов (5X5) тройными линиями. Площадь
каждого большого квадрата равна 1 мм2 и в свою очередь под-
разделяется на 16 маленьких квадратов. При опущенном по-
кровном стекле глубина камеры составляет 0,1 мм. Таким об-
разом, общий объем над каждым большим квадратом состав-
ляет 1X1X0,1 мм=10-4 мл. Способ подсчета с помощью гемо-
цито.метра представлен в табл. 14.5.
Б. Специализированные счетные камеры. Две специализиро-
ванные счетные камеры позволяют сделать прямую оценку кон-
центрации подвижных сперматозоидов. Одна из них — камера
Меклера (Sefi Medical Instruments) очень удобна, но дорога.
Камера Хоруэлла (Horwell Fertility Counting Chamber,
A. R. Horwell) дешевле и основана на простых принципах.
При глубине камеры 10 мкм и учитываемой площади в 1 мм2,
разделенной на сотню квадратов 100ХЮ0 мкм, число сперма-
тозоидов, содержащихся в любых 10 маленьких квадратах,
представит концентрацию сперматозоидов в миллионах/мл.
3—5-мд аликвоту неразбавленного образца семени помещают
на счетную область камеры и закрывают покровным стеклом.
Таблица 14.5. Анализ семени
1. Перенесите образец семени из контейнера для сбора в градуированную
коническую пластиковую центрифужную пробирку (Falcon 2095). При
этом обратите внимание на вязкость образца. Объем и степень вязкости
зарегистрируйте.
2. Снабдив пипетку определенного объема стерильным пластиковым нако-
нечником, перенесите 50 мкл семени для счета в 950 мкл 10%-ного
забуференного формалина (в разведении 1:20). Воспользовавшись но-
вым стерильным наконечником, поместите 10 мкл семени на предметное
стекло и закройте стандартным покровным стеклом 22x22 мм для изу-
чения подвижности и морфологии (пп. 6 и 7).
3. Установите покровное стекло гемоцитометра над счетной камерой
(Improved Neubauer Haemocytometer). О правильном положении покров-
ного стекла свидетельствуют интерференционные кольца между проти-
волежащими поверхностями стекол.
4. Перенесите пастеровской пипеткой около 10 мкл заформалинениого об-
разца для подсчета в гемоцитометре — камера заполняется в результате
действия сил капиллярности.
5. Оставьте счетную камеру на 10 мин в увлажненной среде, чтобы спер-
матозоиды осели на счетную сетку.
6. Тем временем проанализируйте подвижность спермиев. На стекле, приго-
товленном в п. 2, под фазовым контрастом при увеличении Х250 про-
смотрите сто спермиев. Те из них, которые проявляют движение, учиты-
ваются как подвижные.
7. Оцените морфологию спермиев (см. рис. 14.5), изучив их с помощью фа-
зового контраста при увеличении Х400. Отметьте спермин с одним или
более дефектов как аномальные.
8. Теперь оцените концентрацию спермиев путем подсчета их числа в че-
тырех больших угловых квадратах и в центральном квадрате каждой
из двух сеток камеры гемоцитометра (одна сетка показана на рис. 14.4).
При этом спермин, касающиеся верхних или левых линий квадрата,
включаются в счет, а те, что прикасаются к нижним или правым ли-
ниям квадрата, не считаются.
9. При разведении 1 : 20 концентрация спермиев, присутствовавших в исход-
ном образце семени, является средним от общего числа в пяти квадратах
каждой из двух сеток камеры, выраженным в млн./мл. Если полученное
число оказалось меньше 20 млн./мл, это означает, что способ оценки
концентрации спермиев неточен. В таком случае рекомендуется просчи-
тать все 50 квадратов камеры (т. е. 25 квадратов каждой из сеток).
Концентрация спермиев в исходном образце, оцененная этим методом,
выражается общим числом, полученным для 50 квадратов, разделенным
на десять и выраженным в млн./мл.
10. Если число оказалось низким, подсчет следует повторить с меньшим
разведением спермиев, внеся соответствующие коррективы.
11. Подсчитайте также округлые клетки (лейкоциты и незрелые сперматоген-
ные формы) в 50 квадратах. Если их количество превысит 2 млн./мл,
окрасьте стекло на лейкоциты для выявления пиоспермии и культивируй-
те аликвоту для проверки на присутствие бактерий. Лучший краситель
для лейкоцитов—бензидин-цианозин-пероксидаза [30], однако бензидин
канцерогенен. Можно воспользоваться предокрашенными предметными
стеклами (Boehringer Manheim).
Оплодотворение ооцитов человека
377
Концентрация подвижных сперматозоидов оценивается с по-
мощью фазово-контрастного устройства. Эта камера особенно
удобна в тех случаях, когда число сперматозоидов в образце
невелико и требуется только приблизительный подсчет числа
подвижных сперматозоидов, например, в образце для осемене-
ния после разделения в перколле или применения метода
«флотации» (см. ниже).
6.2.3. Нормальные характеристики семени
Несмотря на недостаточную четкость в определении крите-
риев «нормальности» спермы, Всемирная организация здраво-
охранения рекомендует следующие стандарты, характеризую-
щие «нормальный» образец [30].
Объем 2—5 мл
Время разжижения В пределах 30 мин
Концентрация 20—200 млн./мл
Подвижность Более 40% подвижны
Морфология Более 40% нормальных форм
Лейкоциты Менее 1. млн./мл
6.2.4. Бактериологический скрининг
В некоторых лабораториях все образцы семени для оплодот-
ворения in vitro проходят бактериологический скрининг. При
всех достоинствах этой практики, позволяющей обнаружить
прежде не выявленные заболевания, переносимые половым
путем, трудность лечения некоторых инфекций (Clamydia
Ureaplasma), а также возможность заражения после скрининга
не гарантируют неинфекционности образца во время процедуры
оплодотворения. Образцы с высоким количеством круглых кле-
ток (табл. 14.5) обязательно надо подвергать бактериологиче-
скому контролю.
6.3. Подготовка сперматозоидов для оплодотворения in vitro
Целью соответствующих процедур является получение образ-
ца семени, обогащенного подвижными спермиями и свободного
от клеток и дебриса. Техника «флотации» (табл. 14.6) представ-
ляет собой простой метод получения фракции, обогащенной
подвижными спермиями. Разделение сперматозоидов центрифу-
гированием в самоформирующихся градиентах плавучей плот-
ности [31] (табл. 14.7 и 14.8) даст возможность не только от-
носительно быстро получать популяции спермиев с высокой
подвижностью; при этом отбираются спермин с нормальной
25—171
Таблица 14.6. Метод «флотации> сперматозоидов
1. Оставьте сперму для разжижения при комнатной температуре иа 30 мин»
Внесите примерно 2 мл в стерильную пластиковую пробирку (Falcon 2001).
2. Определите концентрацию и подвижность.
3. Добавьте 10 мл EBS+БСА или EBS+10%-ную сыворотку, инактивиро-
ванную нагреванием. Тщательно перемешайте осторожным пипетированием.
стерильной пастеровской пипеткой.
4. Центрифугируйте при 200 g в течение 5—10 мин. Удалите пастеровской,
пипеткой супернатант, оставив 0,5 мл. Ресуспендируйте сгусток осторож-
ным перемешиванием оставшегося супернатанта.
5. Прибавьте 1—2 мл свежей среды, очень осторожно спуская ее по внут-
ренней стенке пробирки так, чтобы оиа наслоилась на поверхность сус-
пензии спермиев, не перемешав ее. Если концентрация спермиев в об-
разце низка, пограничную область между семенем и средой можно уве-
личить, поместив небольшие объемы семени (0,5 мл) в ряд пробирок.
(Falcon 2002) и наслоив на них по 0,5—1 мл среды. Для получения нуж-
ной концентрации спермиев супернатанты из всех пробирок соединяются.
6. Оставьте пробирки в инкубаторе с 5% СО2 при 37° на 20—30 мин. В те-
чение этого времени подвижные спермин должны мигрировать в наслоен-
ную среду, оставив на дне пробирки мертвые спермин, дебрис н иные
клетки. Если пробирки будут установлены под небольшим углом, увели-
чится площадь контакта суспензии со средой и соответственно выход
подвижных спермиев.
7. Осторожно отсосите 0,5—1 мл чистого супернатанта. Для определения кон-
центрации и подвижности спермиев возьмите соответственно 50 н 10 мкл.
8. Доведите концентрацию суспензии путем разведения, центрифугирования
н ресуспендирования до нужной величины в пределах от 2 до 5 мли. по-
движных спермиев/мл.
Таблица 14.7. Градиенты перколла в EBS для центрифугирования'
в градиенте плавучей плотности
Для поддержания стерильности растворов работайте в ламинарном боксе
1. Растворите 1,25 г фракции V БСА в 25 мл EBS десятикратной концент-
рации (Х10 EBS+исходный раствор БСА). Стерилизуйте фильтрованием
(Millipore 0,22 мкм).
2. Поместите 90 мл перколла (Percoll, Pharmacia) в стерильный контейнер
и маркируйте этикеткой «90%-иый перколл в EBS + БСА».
3. Поместите 90 мл культуральной ультрачистой воды в отдельный сте-
рильный контейнер и пометьте: «EBS+БСА».
4. В каждый контейнер добавьте:
10 мл (ХЮ) EBS+исходный раствор БСА (см. п. 1),
100 мкл концентрированного (ХЮ) исходного раствора пирувата натрия
(11 мг/мл),
0.2 мл исходного гентамицина (табл. 14.1),
10 мкг исходного пенициллина (табл. 14.1).
5. В контейнер «EBS+БСА» добавьте 1,3 мл 7,5%-ного бикарбоната натрия
(Flow Labs или приготовьте).
Для приготовления 60%-ных градиентов перколла
6. Работая в стерильных условиях, поместите в автоклавированные поли-
карбонатные центрифужные пробирки (Sorvall, Du Pont, 03115):
6 мл 90%-ного перколла в- EBS+БСА,
3 мл EBS+БСА.
Оплодотворение ооцитов человека
379
Продолжение табл. 14.7
7. Тщательно перемешайте, переворачивая закрытые пробирки.
8. Центрифугируйте 15 мин при 20 °C (29 000 g) в центрифуге с угловым
ротором (Sorvall, head SS34 при 15 000 об/мин).
9. Храните центрифугированные колонки перколла в морозильнике.
10. Плотность разных слоев градиента можно тестировать в образце гра-
диента путем добавления смеси гранул — маркеров плотности (Pharmacia.
Кат. № 17-0459-01).
Таблица 14.8. Разделение спермиев в градиентах перколла [21]
1. Оставьте колонку перколла для нагревания до комнатной температуры.
2. Загрузите в колонку 1—3 мл семени.
3. Центрифугируйте в течение 20 мин в бекет-роторе при 400 g.
•4. Отсосите снизу 1 мл, опустив ко дну центрифужной пробирки иглу для
спинномозговой пункции 19G (Becton Dickinson), присоединенную к шприцу
на 1 мл. Если в вашем распоряжении есть полимерные пробирки, эту опе-
рацию можно сделать путем прокалывания основания пробирки после его
дезинфекции спиртом.
5. Самый нижний слой должен содержать высокоподвижные, морфологиче-
ски нормальные спермин в количестве, достаточном для оплодотворения
in vitro. Тем же способом можно получить аликвоты выше расположенных
слоев, но подвижность и морфология спермиев соответственно будет более
низкого качества. Неподвижные сперматогенные клетки, лейкоциты и деб-
рис обычно локализуются в верхних слоях, под границей раздела спер-
ма/перколл.
43. Сперматозоиды, приготовленные этим способом, могут быть использованы
прямо, в капле для оплодотворения, или отмыты путем добавления 10—
15 мл фертилизационной среды (среды для оплодотворения) и центрифу-
гирования.
морфологией. Процент морфологически нормальных спермиев
изменяется незначительно. Применение этой техники снижает
и бактериальное загрязнение образца [21].
7. Оплодотворение и культивирование эмбрионов
7.1. Осеменение
Ооцит, извлеченный из фолликулярного аспирата, переносят
в 1 мл среды для оплодотворения (табл. 14.3) под масло. Сте-
пень зрелости ооцита определяется по объему кумулюсной
массы (см. рис. 14.3) или, после удаления кумулюса гиалурони-
дазой, по наличию или отсутствию зародышевого пузырька
или полярного тельца (разд. 5). Ооциты выдерживают в инку-
баторе с 5% СО? при 37 °C в течение 4—5 ч. К капле, содер-
жащей яйцеклетку, добавляют кончиком стерильной микропи-
петки аликвоту, содержащую 100 000 подвижных спермиев, при-
25’
380
Глава 14
готовленных методом флотации или разделением в перколле.
Чашку возвращают в инкубатор, спустя 14—16 ч яйца извле-
кают и определяют, произошло ли оплодотворение.
7.2. Доказательство оплодотворения
Клетки кумулюса удаляются с поверхности блестящей обо-
лочки осторожным пипетированием стеклянной пастеровской
пипеткой, оттянутой таким образом, чтобы диаметр ее кончика
был чуть больше яйца, или путем отделения вручную с по-
мощью инъекционных игл (27 калибра). Учитывается наличие
и число пронуклеусов. Если оплодотворение произошло, через
14—20 ч после осеменения в ооплазме должны быть видны два
пронуклеуса (рис. 14.3,В). Трехъядерные яйца (рис. 14.3,Г)
отбрасывают ввиду потенциального риска беременности с пу-
зырным заносом [32].
7.3. Культивирование оплодотворенных яиц
Ооциты с двумя пронуклеусами переносят в капли культу-
ральной среды объемом 50—100 мкл (табл. 14.3) под маслом
и инкубируют в течение ночи с 5% СОг в воздухе. Если разви-
тие идет нормально, прохождения второй борозды дробления
и 2-клеточной стадии (рис. 14,3, Д) следует ожидать между 28
и 36 ч после осеменения, 4-клеточная стадия (рис. 14.3, £)
наступает между 32 и 48 ч. Оплодотворение in vitro предпола-
гает перенос эмбрионов в матку на этой стадии (табл. 14.9),
поэтому временные характеристики последующих стадий дроб-
ления человеческих предимплантационных эмбрионов изучены
хуже.
7.4. Развитие до стадии бластоцисты
Время удвоения числа клеток для индивидуальных эмбрио-
нов подвержено большим колебаниям (8,1—51,0 ч [33]) со сред-
ним значением в пределах между 18 и 23 ч в зависимости от
возраста пациентки и использованного режима стимуляции.
Возможно, эти вариации отражают качественные особенности
индивидуальных эмбрионов, так как понятно, что быстрее деля-
щиеся эмбрионы с большей вероятностью дадут начало бере-
менности после переноса в матку [34]. Развитие человеческих
эмбрионов до стадии поздней бластоцисты (рис. 14.3,3) про-
исходит примерно у трети оплодотворенных яиц [19, 35]. Боль-
шинство прекращает развитие на 4-клеточной стадии, однако
при этом следует помнить, что речь идет о «лишних» с точки
зрения терапевтических требований эмбрионах, поскольку «мор-
Оплодотворение ооцитов человека 381-
Таблица 14.9. Лабораторная процедура переноса эмбрионов
(техника «не-касания»)
1. Приготовьте примерно 20 мл среды для переноса, забуференной HEPES,
содержащей 10% сыворотки материнской крови, и нагрейте до 37 ;С.
2. Приготовьте крупную каплю среды для перемещения, забуференной
HEPES, под маслом и нагрейте в инкубаторе на воздухе (не СОд. Для
этого в пластиковую чашку Петрн (Falcon 3002) поместите около 500 мкл
среды и наслоите сверху около 8 мл масла. Затем добавьте еше 750—
1000 мкл среды, чтобы образовалась «крупная капля», облегчающая вса-
сывание в катетер для переноса.
3. Тем временем пациентку укладывают в «положение для камнесечения»
со слегка запрокинутой головой (или в положение колени к груди). Шей-
ку матки экспонируют с помощью двустворчатого влагалищного зеркала
и осторожно очищают от слизи тампоном, пропитанным средой для пе-
реноса. Затем шейку матки орошают из шприца с иглой еще 3—5 мл
среды. Избыток среды стекает к заднему своду влагалища п забуферива-
ет продукты его секреции.
4. Когда пациентка подготовлена для процедуры имплантации, на нагрева-
тельный столик стереомикроскопа поместите чашку с нагретой средой и
перенесите три «лучших» эмбриона в теплую «большую круглую каплю».
5. Вылейте содержимое одной пробирки среды для переноса в чашку Петри
(Falcon).
6. Заполните стерильный шприц на 1 мл этой средой и положите на бок,
проследив за тем, чтобы его конец не прикасался к нестерильной поверх-
ности.
7. Извлеките из стерильного пакета катетер для переноса (Bourn—Wallace;
HG Wallace Ltd), разрезав концы пакета чистыми ножницами. Сначала
разрежьте упаковку на конце, где находятся розовые коннекторы. Убе-
рите белую пластиковую пробирку и присоедините шприц, наполненный
средой. Разрежьте упаковку со стороны катетера и продвиньте канюлю
к новому отверстию в упаковке так, чтобы обнажился гибкий конец ка-
тетера.
8. Выпустите из шприца среду для замещения, оставив 0,2 мл.
9. Когда хирург будет готов, втяните небольшой пузырек воздуха в конец
катетера, затем эмбрионы в 10—20 мкл среды. Всосите еще один неболь-
шой пузырек для маркирования положения эмбрионов в катетере.
10. Извлеките шприц, катетер и интубатор из упаковки и осторожно все пе-
редайте хирургу, если нужно, помогите ему в обращении со шприцем.
11. Пользуясь наружным белым футляром как стабилизатором, хирург осто-
рожно вводит катетер через канал шейки матки, пока два розовых сочлене-
ния не сомкнутся. Медленно нажимая поршень шприца, следите, когда
уровень жидкости достигнет отметки 0,2 мл. Спустя примерно 1 мин
катетер извлекается полностью и передается эмбриологу.
12. Под контролем микроскопа вылейте остатки жидкости из шприца в чис-
тую чашку Петри н проверьте, не остались ли эмбрионы в катетере.
фологически лучшие» использованы для переноса и их репре-
зентативность в указанных характеристиках развития не нахо-
дит отражения.
Компактизация человеческих эмбрионов происходит на
16-клеточной стадии, полностью сформированные бластоцисты
обнаруживаются между 96 и 116 ч после осеменения. Это соот-
382
Глава 14
ветствует примерно тому же временному интервалу, который
наблюдается при развитии in vivo, т. е. между осеменением
и получением поздних бластоцист в результате промывания
матки у доноров [36].
8. Этические и правовые аспекты
В течение последних нескольких лет использование челове-
ческих гамет и эмбрионов для научных исследований находит
в центре внимания широкой общественности. Каждый, кто
намерен работать с человеческими гаметами и эмбрионами,
должен быть знаком с правилами использования этого материа-
ла, принятыми в его стране [37—42], и с историческими усло-
виями, которые привели к введению этих правил.
Поскольку автор лучше знаком с политическими и правовы-
ми дискуссиями в Великобритании и в связи с тем, что именно
на их основе выработаны многие правила, принятые в других
странах, именно они и обсуждаются ниже.
8.1. Законодательство
и научные исследования на человеческих эмбрионах
в Великобритании
В настоящее время человеческий эмбрион per se не имеет
правового статуса. В соответствии с английскими законами
ему не предоставляется тот же правовой статус, которым поль-
зуется ребенок или взрослый, закон не признает за человече-
ским эмбрионом права на жизнь. Согласно Закону о Личности
(Person Act) 1861 г. и Закону об абортах 1976 г., прерывание
беременности считается уголовным преступлением. Однако
эти законы приложимы к эмбриону in vivo и не относятся
к эмбриону in vitro. Некоторая защита эмбриона in vivo
обеспечивается и Законом о Гражданской ответственности
1976 г., который предусматривает компенсацию ущерба, нане-
сенного эмбриону или плоду in utero из-за преступной небреж-
ности третьей стороны. Какая-либо правовая защита эмбриона
in vitro не предусматривается.
8.2. Варнокский комитет
Несмотря на повторные запросы пионеров этих исследова-
ний [43], никаких официальных действий не предпринималось
до 1982 г., когда был создан специальный правительственный
комитет. Его целью было изучение вопросов, связанных с опло-
дотворением и эмбриологией человека, и разработка рекомен-
даций правительству в связи с необходимостью соответствую-
Оплодотворение ооцитов человека 383
щего законодательства [37]. Комитет, возглавляемый филосо-
фом Мэри Варнок (ныне баронесса Варнок), подготовил до-
клад, охватывающий большинство аспектов искусственного
воспроизводства. Ключевой рекомендацией комитета было
создание нового контролирующего органа, в полномочия кото-
рого входило бы регулирование и исследовательской работы,
и клиник, осуществляющих оплодотворение in vitro. Одна из
рекомендаций состоит в том, что «человеческий эмбрион в на-
стоящее время должен в определенной степени находиться под
охраной закона». По мнению членов комитета, «исследования,
проводимые на человеческих эмбрионах in vitro и культивирова-
ние таких эмбрионов должны проводиться только по снециаль-
ному разрешению, существование человеческого эмбриона, по-
лученного в результате оплодотворения in vitro, не должно под-
держиваться дольше четырнадцати дней, если он не перенесен
в женский организм». Культивирование человеческих эмбрио-
нов in vitro без разрешения или дольше четырнадцати дней
после оплодотворения следует классифицировать как уголовное
преступление. Более того, доклад ставил вопрос о существова-
нии «лишних» эмбрионов и о том, что «лишний эмбрион не
должен быть материалом для исследовательской работы без
официального согласия пары, от которой произошел такой
эмбрион». В докладе нашли свое отражение и разногласия.
По мнению некоторых членов комитета, экспериментирование
на человеческих эмбрионах не должно разрешаться ни при ка-
ких обстоятельствах.
8.3. Добровольная ассоциация экспертов
В связи с тем что за Варнокским докладом не последовало
никаких правительственных постановлений, Совет по медицин-
ским исследованиям и Королевский колледж акушеров и гине-
кологов совместно создали добровольную ассоциацию экспертов
по проблеме оплодотворения и эмбриологии человека in vitro.
Эта организация, созданная в духе рекомендаций Варнокского
комитета, в настоящее время представлена тремя учеными,
предложенными Советом по медицинским исследованиям, че-
тырьмя акушерами-гинекологами и семью непрофессионалами,
включая непрофессионала председателя. Члены ассоциации
ставят своей целью:
1) утвердить кодекс правил исследовательской работы, свя-
занной с оплодотворением in vitro и эмбриологией;
2) оценить работы всех научных центров, клиницистов и
ученых, занятых оплодотворением in vitro, и решить во-
прос о целесообразности выдачи им разрешения на тако-
го рода деятельность;
384
Глава 14
3) посетить каждый центр до предоставления ему разреше-
ния;
4) отчитываться перед Советом по медицинским исследова-
ниям и Королевским колледжем акушеров и гинекологов;
5) ставить в известность общественность о деталях как ут-
вержденных, так и неутвержденных работ.
Первое заседание ассоциации экспертов произошло 26-го
марта 1985 г., вскоре после чего было распространено руковод-
ство по клиническому и научному применению оплодотворения
человека in vitro. Первый отчет опубликован в апреле 1986 г.,
к этому времени было обследовано 25 центров, работы 24 цент-
ров были утверждены. Ко времени публикации второго отчета
(апрель 1987 г., см. табл. 14.10) разрешение получили еще
Таблица 14.10. Руководство по проведению оплодотворения
в клинике и для научных исследований
Введение
Цель этого руководства — изложить принципы, которыми, по мнению экспер-
тов Совета медицинских исследований и Королевского колледжа акушеров и
гинекологов, должны руководствоваться специалисты, использующие в ис-
следовательской работе человеческие гаметы и оплодотворение in vitro. Они
основаны на рекомендациях Варнокского комитета.
В процессе дискуссии эксперты пришли к выводу о необходимости уточ-
нения термина «предэмбрион», используемого в этом руководстве. Под пред-
эмбрионом следует понимать агрегат делящихся клеток до детерминации пер-
вичной полоски, т. е. до начала органогенеза.
Руководство
1. Научно обоснованные исследования, включающие опыты по оплодотворе-
нию in vitro и культивированию эмбрионов человека, допустимы с этиче-
ской точки зрения при соблюдении условий, изложенных в пунктах 2—
10 настоящего руководства.
2. В любой заявке на разрешение, представленной на рассмотрение экспер-
тов, должны быть обоснованы причины, по которым исследователь счита-
ет нужным работать именно на человеческом материале.
3. Цель исследования должна быть четко определена и соотнесена с таки-
ми практическими проблемами клиники, как диагностика и лечение бес-
плодия и генетических нарушений, а также совершенствование безопас-
ности и эффективности контрацептивных средств.
4. Предэмбрионы, полученные в результате эксперимента или использован-
ные в эксперименте, не должны имплантироваться в матку, за исключе-
нием клинических исследований, целью которых является повышение ве-
роятности успешной беременности у данного индивида.
5 В каждом случае донор должен дать соответствующим образом оформ-
ленное согласие на использование яйцеклеток и спермиев для исследова-
тельской работы; сперма из соответствующих банков также может быть
использована только с разрешения донора. Любой проект помимо эксперт-
ной ассоциации должен рассматриваться и утверждаться местным коми-
тетом по этике.
6. Если человеческие яйцеклетки были получены для оплодотворения
in vitro в терапевтических целях и больше для этого не нужны, нх ис-
пользование для научного исследования не противоречит этике, если оно
Продолжение табл. 14.10
достаточно серьезно обосновано и оба донора далн письменное согла-
сие, а также при наличии утверждения, упомянутого в предыдущем
разделе.
7. Человеческие яйцеклетки, оплодотворенные человеческими спермиями, не
должны культивироваться in vitro более 14 дней и не должны храниться
для использования в каких-либо иных исследованиях, кроме тех, которые
утвердил местный комитет по этнке н ассоциация экспертов.
8. В случаях крноконсервации предэмбриона (был ли он получен для целей
исследования при замораживании нли предназначен для этого впослед-
ствии) он может продолжать развитие до стадии, эквивалентной 14 дням
нормального развития, при условии, что на это имеется разрешение. Воп-
рос о хранении предэмбрионов должен перегматриваться спустя
два года, максимальный срок их хранения не должен превышать десяти
лет.
9. Использование предэмбрионов должно быть тщательно продумано до
осуществления каждого проекта. По окончании задуманного экспери-
мента развитие предэмбриона должно быть приостановлено. Его даль-
нейшее использование зависит от характера исследования, уже проведен-
ного на данном предэмбрионе. Учитывая ограниченность материала, ни
одни предэмбрион нельзя выбрасывать без тщательного изучения.
10. Изучение проникновения спермиев человека в яйцеклетки животных слу-
жит источником ценной информации о проникающей способности и хро-
мосомном наборе спермиев; оно допустимо с позиций этики при условии,
что развитие не пойдет дальше стадий раннего дробления.
И. При клиническом использовании яйцеклеток после криоконсервацин,
включающем оплодотворение in vitro, нельзя приступать к переносу эм-
бриона в матку до тех пор, пока не будет получено научно обоснованное
свидетельство безопасности этой процедуры.
12. Следует соблюсти предосторожности, связанные с тем, что повышение
вероятности начала беременности от переноса добавочных эмбрионов
может иметь следствием многоплодную беременность, вероятность кото-
рой нужно свести к минимуму. Вот почему:
а) если используется процедура оплодотворения in vitro, нельзя имплан-
тировать более трех эмбрионов в цикл, за исключением случаев, когда
специальные клинические причины требуют переноса четырех пред-
эмбрионов;
б) если используется процедура переноса яйцеклеток в фаллопиевы тру-
бы, нельзя переносить более трех, в исключительных случаях — че-
тырех ЯИЦ.
13. При организации клинических центров по изучению оплодотворения
in vitro, следует учесть некоторые общие положения:
а) каждый центр должен быть подотчетен комитету по этике, ни одна
процедура не должна проводиться без согласования с ним;
б) необходимо детальное описание исследований; обязательными следует
считать сведения о детях, рожденных в результате оплодотворения
in vitro; соответствующие записи должны быть легко доступны для
изучения экспертами с ориентацией на национальные исследования
в будущем;
в) в тех случаях, когда руководитель исследонания ие имеет статуса кон-
сультанта или не является клиницистом, полная ответственность мо-
жет быть возложена на консультанта-советника, который выполняет
роль активного наблюдателя за работой центра; весь остальной меди-
цинский, обслуживающий и технический персонал должен обладать
соответствующей подготовкой и опытом;
г) необходимо соответствующее медицинское, хирургическое н техниче-
ское оборудование;
336
Глава 14
Продолжение табл. 14.10
д) должны быть обеспечены все мероприятия, требующиеся в экстренных
случаях;
е) должны быть обеспечены условия, необходимые для перемещения
гамет и предэмбрионов между клиникой и лабораторией;
ж) каждый центр должен располагать независимыми и опытными кон-
сультантами;
з) все пациенты, поступающие в связи с программой оплодотворения
in vitro, должны пройти тест на гепатит В и антитела к вирусу им-
мунодефицита;
и) донорская сперма должна поступать только из банка, где прошла все
тесты, предусмотренные соответствующей инструкцией;
к) доноры яйцеклеток должны оставаться анонимными, поэтому следует
исключить донорство для клинических целей от близких родственников.
14. При оборудовании лаборатории, в которой предполагается проводить
оплодотворение in vitro, следует учесть следующее:
а) каждый центр должен быть связан с комитетом по этике (см. п. 13а);
б) протоколы опытов должны быть доступны для инспекции соответст-
вующими лицами;
в) лабораторный штат должен иметь необходимую квалификацию и опыт;
г) условия работы лаборатории должны быть на высоком техническом
уровне (это касается обеспечения культивирования, микроскопического
изучения, инкубаторов и владения методикой «не-касания»);
д) культивирование и хранение гамет и предэмбрионов должно быть
обеспечено очень высоким уровнем надежности, ведения протоколов и
маркирования.
шесть центров, при этом были изменены инструкции: в них бы-
ла включена и процедура переноса гамет в фаллопиевы трубы.
Из 30 утвержденных центров в девяти ведутся исследования
на человеческих эмбрионах.
Несмотря на то что ассоциация экспертов не облечена офи-
циальной властью, она способна оказать достаточно жесткое
давление. Это объясняется, во-первых, тем, что в штате практи-
чески всех центров работают сотрудники или стипендиаты Ко-
ролевского колледжа акушеров и гинекологов. Их деятельность
подчиняется кодексу профессиональных правил и в случае на-
рушений они подвергаются дисциплинарным взысканиям.
Кроме того, научный и медицинский авторитет экспертов так
велик, что финансирующие организации не поддерживают ис-
следования на человеческих эмбрионах, не утвержденные ассо-
циацией. Важно также, что публикация утвержденных и не-
утвержденных проектов научных центров, подотчетных ассоциа-
ции экспертов, позволяет знакомиться с ними широкой научной
общественности. Можно надеяться, что эта добровольная орга-
низация продемонстрирует обществу способность ученых со
всей ответственностью регулировать исследования на человече-
ских эмбрионах.
Оплодотворение ооцитов человека
387
Благодарности
Технические приемы, описанные в этой главе, основаны на
методах, разработанных и усовершенствованных в течение пос-
ледних пяти лет в Отделе акушерства и гинекологии клиниче-
ской школы при университете в Кембридже и в Лаборатории
доктора Мартина Джонсона в Отделе анатомии. Мне хотелось
бы выразить благодарность всем членам Группы эмбрионов
и гамет за поддержку и помощь, и в особенности доктору Вирд-
жинии Болтон и мисс Джин Флэч. Я признателен Шине Гле-
нистер за печатание рукописи и Мартину Джонсону за полез-
ные критические замечания.
Литература
1. Braude Р. R., Bolton V. N., Johnson М. Н. (1986). In: Embryo Research —
Yes or No? Bock G. and O’Connor M. (eds.), Tavistock Publications, p. 63.
2. Edwards R. G„ Steptoe P. (1983). Lancet, 8362, 1265.
3. Jones H. IF., Jones G. S., Andrews M. C. et al. (1984). Fertil. Steril., 38, 14.
4. Quigley Al. Al., Schmidt C. L„ Beauchamp P. J. et al. (1984). Fertil. Steril.,
42, 25.
5. Lancaster P. A. L. (1985). Br. Med. J., 291, 1160.
6. Asch R. H., Balmaceda J. P., Ellsworth L. R„ Wong P. C. (1985). Int. J. Fer-
til., 30, 41.
7. Ockenden K-, Bolton V. N„ Braude P. R. (1985). Lancet, 1, 452.
8. Templeton A., Van Look P„ Lumsden Al. A., Angell R., Aitken J., Dun-
can A. W., Baird D. T. (1984). Br. J. Obstet. Gynaecol., 91, 148.
9. Braude P. R., Bright M. V., Douglas С, P„ Milton P. J., Robinson R. E., Wil-
liamson J. G., Hutchinson J. (1984). Fertil. Steril., 42, 34.
10. Lenz S. (1983). Clin. Obstet. Gynaecol., 12, 785.
11. Craft I. (1985). In: In Vitro Fertilization and Donor Insemination. Thomp-
son W. et al. (eds), RCOG, London, p. 143.
12. Steptoe P. C., Webster J. (1985). Ann. N. Y. Acad. Sci., 442, 178.
13. Purdy J. Al. (1982). In: Human Conception in Vitro. Edwards R. G., Pur-
dy J. M. (eds), Academic Press, NY, p. 135.
14. Quinn P., Warnes G. M., Kerin J. F., Kirby C. (1984) Fertil. Steril., 41,
202.
15. Manezo Y. (1976). C. R. Acad. Sci. Paris, 282, 1967.
16. Dandeka P. V., Quigley Al. Al. (1985). Fertil. Steril., 42, 1.
17. Hillier S. G„ Dawson K. J-, Afnan M. et al. (1985). In: In Vitro Fertilization
and Donor Insemination. Thompson W. et al. (eds.), RCOG, London, p. 125.
18. Caro С. M., Trounson A. (1984). J. IVF and Emb. Trans., 1, 183.
19. Bolton V. N., Braude P. R. (1987). In: Current Topics in Developmental Bio-
logy Monroy A. (ed.), in press.
20. Fleming T. P., Pratt H. P. M„ Braude P. R. (1987). Fertil. Steril., 47. 858.
21. Bolton V. N., Warren R. E., Braude P. R. (1986). Fertil. Steril., 46, 1128.
22. Hewitt J., Cohen J., Fehilly С. B. et al. (1985). J. IVF and Embryo Trans
2, 105.
23. Wright R. W., Anderson G. B., Cupps P. T., Drost J. (1976) Biol Reprod
14, 157.
24. Trounson A., Mohr L., Wood C„ Leeton J. F. (1982). J. Reprod. Fertil., 64
285 *
25. Osborne J. C„ Moore R. M. (1985). In: In Vitro Fertilization and Artificial
Insemination. Thompson W. et al. (eds), RCOG, p. 101.
26. Bomsel-Helmreich O. (1985). Oxford Review of Reprod. Biol., 7, 1.
388
Глава 14
27. Templeton A. A., Van Look Р., Angell R. E. et al. (1986). J. Reprod. Fertil.,
76, 771.
28. Veeck L. L. (1985). Ann. N. Y. Acad. Sci., 442, 357.
29. Mortimer D. (1985). Current Prob. Obstet. Gynaecol., 7, 1.
30. WHO Laboratory manual for the examination of Human semen and semen —
cervical mucus interaction. (1987). Cambridge University Press.
31. Bolton V. N., Braude P. R. (1985). Arch. Androl., 13, 167.
32. Jacobs P. A., Szulman A. E., Funkhouser J. et al. (1982). Ann. Hum. Genet.,
46, 223.
33. Fishel S. B., Cohen J., Fehilly С. B. et al (1985). Am. N. Y. Acad. Sci., 442,
342.
34. Mohr L. R., Tounson A. 0., Leeton J. E, Wood C. (1983). In: Fertilization
of the Human Egg In Vitro. Beier H. M. and Lindner H. R. (eds), Springer -
Verlag, Berlin, p. 211.
35. Fehilly С. B., Cohen J-, Simons R. E. et al. (1985). Fertil. Steril., 46, 638.
36. Buster J. E., Bustillo M., Rodi I. A. et al. (1985). Am. J. Obstet. Gynaecol.,
153, 211.
37. Warnock M. (1984). Report of the Committee of Inquiry into Human Fertili-
zation and Embryology. HMSO. Cmnd. 9314.
38. The Second Report of the Voluntary Licensing Authority for Human In vil’o
Fertilisation and Embryology. (1987). Medical Research Council, London.
39. Ethics Committee of the American Fertility Society. (1986). Fertil Steril.,
46, Suppl.
40. Report and Conclusions of the Ethics Advisory Board of the Department oi
Health Education and Welfare: HEW Support of Research Involving Human
In Vitro Fertilization and Embryo Transfer. (1979). US Govt. Printing Of-
fice, Washington.
41. Senate Select Committee on The Human Embryo Experimentation Bill 1985:
Human Embryo Experimentation in Australia. (1986). Australian Govern-
ment Publishing Service, Canberra.
42. Council of Europe, Parliamentary Assembly. (1987). Human Reprod., 2, 67.
43. Edwards R. G. (1974). Quart. Rev. Biol., 49, 368.
44. Braude P. R., Pratt FL P. M., Johnson M. H. (1984). Bioessays, 1, 232.
ПРИЛОЖЕНИЕ
ФИРМЫ-ИЗГОТОВИТЕЛИ
Aldrich Chemical Со., Poole, Dorset, UK; Aldrich Chemical Co
Inc., 940 West St Pauls Avenue, PO Box 355, Milwaukee, WI
53201, USA.
Amersham International pic, White Lion Road, Amersham, Bucks
HP7 9LL, UK; Amercham Corp., 2636 S. Clearbrook Drive,
Arlington Heights, IL60005, USA.
Badger Airbrush Co., 150 Stanley Crescent Road, Poole, Dorset,
UK.
Baird and Tatlock (London) Ltd, PO Box 1, Romford, Essex RM1
1HA, UK.
Bantin and Kingman, The Field Station, Grimston, Hull HUH
4QE, UK.
BDH Chemicals Ltd, Broom Road, Poole, Dorset BH12 4NN, UK;
BDH Chemicals, Gallards Schlesinger Chemicals Mfg Corp.,
584, Mineola Avenue, Carle Place, NY 11514, USA.
Beaudouin, Paris, France. (Beaudouin microforge supplied in the
UK. by Microinstruments Ltd).
Becton Dickinson (UK) Ltd, Between Towns Road, Oxford 0X4
3LY, UK; Becton-Dickinson Labware, 1915 Williams Drive,
Oxnard, CA 93030, USA.
Boehringer Corp. (London) Ltd, Boehringer Mannheim House,
Bell Lane, Lewes, East Sussex BN7 1LG, UK; Boehringer
Mannheim Biochemicals, 7941 Castelway Drive, PO Box 50816,
Indianapolis, IN 46250, USA.
Boots Hospital Products pic., Nottingham, UK.
W. H. Brady and Co. Ltd, Daventry Road Industrial Estate, Ban-
bury OX16 7HU, UK.
Calbiochem-Behring (CP Laboratories Ltd), PO Box 22, Bishops
Stortford, Herts CM22 7RD, UK; Calbiochem-Behring Corp.,
10933 N. Torrey Pines Road, La Jolla, CA 92037, USA.
Casmed UK, 36 Kenley Walk, Cheam, Surrey SM3 8ES, UK.
Clark Electromedical Instruments, PO Box 8, Pangbourne, Rea-
ding RG8 7HU, UK.
Collaborative Research, 12—14 St Anns Crescent, London SW18
2LS, UK; Collaborative Research Inc., 128 Spring Street, Le-
xington, MA 02173, USA.
Crown Chemical Co. Ltd, Lamberhurst, Kent TN3 8DU, UK.
390
Фирмы-изготовители
Day-Impex Ltd, Station Works, Earls Colne, Colchester, Essex
CO6 2ER, UK.
Downs Surgical Ltd, Church Pass, Mitcham, Surrey CR4 3UE, UK.
Drummond Scientific Co., 500 Pkwy, Broomall, PA 19008, USA.
Dupont (New England Nuclear) UK Ltd, Wedgewood Way, Steve-
nage, Herts SGI 4QN, UK; Dupont, NEN Research Products,
549 Albany Street, Boston, MA 02118, USA.
Eastman Kodak Co., 343 State Street, Rochester, NY 14650, USA.
Falcon, supplied by R. L. Slaughter Ltd, 14 Bridge Close, Rom-
ford, Essex RM7 OAS, UK.
Fisons Scientific Equipment, Bishop Meadow Road, Loughborough,
Leics LEU ORG, UK.
Flow Laboratories Ltd, PO Box 17, Second Avenue, Industrial
Estate, Irvine, Ayrshire KA12 8NB, UK.
Gibco Ltd, PO Box 35, Trident House, Paisley РАЗ 4EF, UK;
GIBCO Laboratories, 3175 Staley Road, Grand Island, NY
14072, USA. .
Glaxo Laboratories, Greenford Road, Greenford, Middx UB6 OHE,
UK.
Th. Goldsmidt Ltd, York House, Station Road, Harrow, middx, UK.
Go-Medical Industries, 20 Denis Street, Subiaco, WA 6008, Aust-
ralia.
Gow-Mac Instrument Co., PO Box 32, Bound Brook, NJ 08805,
USA.
Grant Inst (Cambridge) Ltd, Barrington, Cambridge CB2 5QZ, UK.
Harlon-Olac Ltd, Shaw’ Farm, Blackthorn, Bicester OX60TP, UK.
Helena Laboratories, PO Box 752, 1530 Lindbergh Drive, Beau-
mont, TX 77704, USA.
Holborn Surgical Instruments Co. Ltd, Dolphin Works, Margate
Road, Broadstairs, Kent CT10 2QQ, UK.
A. R. Horwell Ltd, 73 Maygrove Road, London NW6 2BP, UK.
Hoslab Ltd, 12 Charterhouse Square, London EC1M 6BB, UK.
IMV, L’Aigle, France.
Intervet Laboratories Ltd, PO Box 5830AA, Boxmeer, The Nether-
lands.
Kinematica GmbH, Lucernestrasse 147A, Lucerne, Switzerland.
Raymond A. Lamb, Sunbeam Road, London NW10, UK-
Leeds and Northrup Ltd, Wharfedale, Tyseley, Birmingham Bll
2DJ, UK; Sumneytown Pike, North Wales, PA 19454, USA.
E. Leitz Inc., 24 Link Drive, Rockleigh, NJ 07647, USA.
Macarthy’s Surgical Ltd, Selina’s Lane, Dagenham, Essex, UK.
May and Baker Ltd, Liverpool Road, Eccles, Manchester M30
7RT, UK.
Merrell Dow Pharmaceuticals Ltd, Meadowbank, Bath Road, Ho-
unslow, Middx TW5 9Q7, UK.
Фирмы-изготовители
391
Microinstruments Ltd, 7 Little Clarendon Street, Oxford 0X1
2HP, UK.
Miles Scientific Labs (ICN Biochemical Ltd), Free Press House,
Castle Street, High Wycombe, Bucks HP 13 6RN, UK; West 475
North Aurora Road, Naperville, IL 60566, USA.
Millipore Co., 11 — 15 Peterborough Road, Harrow, Middx HA1
2YH, UK; Millipore Corp., 80 Ashby Road, Bedford, MA 01730,
USA.
NBL Enzymes Ltd, South Nelson Industrial Estate, Cramlington,
Northumberland NE23 9HL, UK.
Nunc (see Gibco).
Organon Laboratories Ltd, Cambridge Science Park, Milton Road,
Cambridge CB4 4BH, UK.
Oxoid Ltd, Wade Road, Basingstoke, Hampshire RG24 OPW, UK;
Oxoid (USA) Inc., 9017 Red Branch Road, Columbia, MD
21045, USA.
Paynes and Byrne, Greenford, Middx UB6 7HG, UK.
Pharmacia Fine Chemicals, PO Box 175, Uppsala 1, Sweden;
Pharmacia Inc., 800 Centenial Avenue, Piscataway, NJ 08854,
USA.
Polaron Equipment Ltd, Watford Business Park, Herts WD1 8XG,
UK.
Promega Biotech, 2800 S. Fish Hatchery Road, Madison, WI 53711,
USA.
Roche Products Ltd, Broadwater Road, Welwyn Garden City, Herts
AL7 3AY UK.
Rocket of London Ltd, Imperial Way, Watford WD2 4XX, UK.
Sefi Medical Instruments Ltd, PO Box 7295, Haifa, Israel.
Serono Chemical Co., 2 Tewin Court, Welwyn Garden City, Herts
AL7 1AU, UK.
Sigma Chemical Co., Fancy Road, Poole, Dorset BH17 7NH, UK;
Sigma, PO Box 14508, St Louis, MO 63178, USA.
South London Electrical Equipment Ltd, Lanier Works, Hither
Green Lane, London SE13, UK.
Sterilin Ltd, Sterilin House, Clockhouse Lane, Feltham, Middx
TW14 8QS, UK.
Steriseal, NI Medical, PO Box 3, 26/27 Thornhill Road, North
Moons Moat, Redditch, UK.
Stortz (Rimmer Bros), 18 Aylesbury Street, Clerkenwell, London
ECI ODD, UK.
Surgikos Ltd, Kirkton, Livingston, UK.
Sutte Instrument Co., PO Box 3592, San Rafael, CA 34912-3592,
USA.
Technation, 58 Edgeware Way, Edgeware, Middx, UK.
Arthur H. Thomas Co., Philadelphia, PA 19105, USA.
H. G. Wallace Ltd, Whitehall Road, Colchester CO2 8JH, UK.
392
Фирмы-изготовители
Watkins and Doncaster, PO Box 5, Cranbrook, Kent TN 18 5EZ,
UK.
Wellcome, Temple Hill, Dartford, Kent DAI 5AH, UK.
Whatman Ltd, Springfield Mill, Maidstone, Kent ME14 2LE, UK;
Whatman Chemicals Inc., 9 Bridewell Place. Clifton, NJ 07014,
USA.
Wheaton Scientific, 1000 North Tenth Street, Millville, NJ 08332,
USA.
Williton Box Co. Ltd, Williton, Somerset, UK-
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Агглютинин зародышей пшеницы,
инъекция в эмбрионы мыши 86—
87
— Dolichos biflorus (АДБ) как кле-
точный маркер 169, 170—173
Агрегация эмбрионов 45—46, 155, 156
Аденин-фосфорибозилтрансфераза
(АФРТ), двойной микрометод
185—186, 188—189
— измерение активности 186—198
Азот жидкий, хранение ооцитов и эм-
брионов 323, 327, 329, 334—335,
34—344
Анестетики 22—23
Анеуплоидия 129, 134
Антибиотики 368
Антигены 1а 166
— Thy-1 166, 168
Н-2-антигены в качестве маркеров
157, 159—166
Антитела моноклональные 161—162,
166
---- гаптенизированные 161
----конъюгированные 162
Банк консервированных эмбрионов
Белки, иодирование 216, 222
— окрашивание красителем кумасси
ярко-голубым R-250 235—236
---- серебром 235—236
— радиоактивно меченные, обнаруже-
ние методом радиоавтографии
233—235
----235------флюорография 233—
— раннего эмбриона, изоэлектриче-
ское фокусирование 215, 218,
230—233
-------качественный анализ 215—
238
-------мечение импульсное 216
221—222
26—171
----------метаболическое 220—222
----------неметаболическое 222—
223
-------электрофорез в полиакрил-
амидных гелях (ПААГ) 215,
227—233
— специфические, идентификация ме-
тодом вестерн-блот 237
Белок А из Staphylococcus aureus,
связывание комплекса антиген —
антитело 224, 236
Библиотеки кДНК, конструирование
на основе фага лямбда 245—246
-----скрининг 249—255
Биопсия хвоста мышей 292—293
— эмбрионов 187, 196, 197
Бластоцель, кавитации 37, 39
Бластоцисты 33, 37—38, 129
— выделение внутренних клеточных
масс 46—47
— «гигантские» 47
— подсчет ядер 49
— синтез гликопротеинов 224
Блокаторы митоза 133
Бляшка 177—178
Вазэктомия 20—21
Вестерн-блот, идентиифкация специ-
фических белков 237
Вирус Сендай, выращивание в кури-
ных яйцах 301—302
----- инактивации 302—303
----- слияние нуклеопластов (карио-
пластов) с яйцами 300, 317—318,
319—320
Витрификационный раствор 339—343
Витрификация 324, 338—343
Витрифицированные эмбрионы, хра-
нение 342
Гаметы, перенос в фаллопиевы трубы
358
Гаструляция 65—69
394
Предметный указатель
Гель-электрофорез 198, 201—213,
227—238
Геном вируса мозаики вигны (CPMV)
как носитель 242—243
Гены, перенос с помощью ретровиру-
сов 296—298
Гены, перенос с помощью ретровиру-
сов 296—298
'Гиалуронидаза, удаление клеток ку-
мулюса 29, 42, 371
Гибридизация, зонды радиоактивных
нуклеиновых кислот 249—255
— скрининг библиотек кДНК 249—
255
— in situ 255, 257—276
-----артефакты 274—276
-----зонд гена 2',5'-о11£оА-сиитета-
зы 150
-----зонды радиоактивной кДНК
258
-— — микроскопирование гистологи-
ческих срезов 271—274
----- подготовка гистологических
срезов 266—268
-----РНК-зонды 259—263
-----с клеточными РНК в тканях
257—258, 266—268
-----фиксация и приготовление сре-
зов 265—266
•----хромосом 109, 144—148, 150
— — — культура эмбриональных
фибробластов 144—149
—-------репликациоииый G-метод
144—148
Гибридный блок 255
Г ипоксаитин-фосфорибозилтранс-
фераза (ГФРТ), микрометод оп-
ределения активности 185, 186—
198
Гистерэктомия 23—24
Глитоп^ротеины, идентификация 216,
Диметилсульфоксид (ДМСО) как
криопротектор 324, 330—332
337—338
Дифференцировка 65, 69, 74
ДНК, анализ на траисгенность 295
— выделение из хвостов мышей 292—
294
------- эмбрионов 294—295
для микроииъекций, получение
278—281
— инъекция в проиуклеус 287—291,
-------яйца мышей 282, 288—291
— комплементарная (кДНК), синтез
и клонирование 246—249
----- создание библиотек иа основе
мРНК 241
ДНКаза I, обработка препаратов
хромосм 109
Замораживание — скалывание 54
Замораживание яиц и эмбрионов
326—336
Зонды, меченные биотином 257, 260
— оптимальная длина 263—264
— радиоактивных нуклеиновых кис-
лот 249—255, 258—264
----------приготовление 251—253,
261, 263
Иглы для переноса ядер 306—309
— •— сбора ооцитов 361—362
— инъекционные 283—286, 288, 290,
301
Идиограмма кариотипа стандартная
137, 138, 140, 141
Изоэлектрическое фокусирование, дву-
мерные гели 230—233
-----разделение белков 215, 218,
230—233
Иммуиоблоттинг белков 216, 224,
236—238
— — метод вестерн-блот 237
Иммунопреципитация белков 216,
224—225
Иммунофлуоресценция белков 224
— непрямая 52
Иммуиохирургическая методика вы-
деления внутренних клеток 46—
47, 49
Иммуноцитохимия 54—55, 159—166
Имплантация эмбриона 65
Х-инактивация, мозаичность тканей
174
Иодирование белков 216, 222
Кариопласты 316, 318—319
Кариотип 137—141
Карцинома эмбриональная 76
Клетки кумулюса, удаление 29, 371 —
372
— мечение 50—54, 57—59, 82—83,
153—179
Клеточные циклы 32—33
Клон, анализ 177—179
Предметный указатель
395
Клонирование кДНК 246—249
Коиканавалин А, окрашивание эмбри-
онов 52, 54, 58
Краситель Гимза, окрашивание хро-
мосом 102, 135, 137, 147
— карболовый фуксин, окрашивание
мейотических хромосом 102—103
Криоконсервация ооцитов и эмбрионов
человека 323, 355
Криопротекторы 324, 326, 327, 330—
332, 337
Крипты кишечника 181, 188
Культуральная система роллерная
79-81
Лактопероксидаза, иодирование бел-
ков 222
Лапароскопическая стерилизация 359
Лапароскопический сбор ооцитов
359_____352
Лапароскопия 357—358, 359, 362—363
— инструменты 362
Лектины как маркеры 50—53, 168—
173
Маркеры клеточной линии 57—59, 153
— клеточные, Н-2-антигены 159—166
— линии клеток зародышевого пути
156
Мезодерма 67
Мейоз 99—100
— профаза 109—ПО, 114—117, 121
Мейотические хромосомы, получение
высушенных на воздухе препара-
тов 100—102
Микроинструменты 92—97, 284—286,
287, 305-309
Микрокузница де Фонбрюна 93, 285,
287, 305
Мнкроманипуляции при проведении
экспериментов 82—92, 286—292,
300—319
/Микроскопия электронная просвечи-
вающая 56—57, 114—121
----сканирующая 52—53
Митоз, блокатор 128, 129, 130, 133
— пронуклеарный 128
Мозаичность по генам, сцепленным с
Х-хромосомой 174
— тканей, Х-инактивация 174
----маркеры 173—179
Морула 35—37, 133
Мутации ннсерционные 296
26’
Мутация соматическая 175
— «beige» 175
Мыши аксенические 14
— «барьерные» 13
— вазэктомия 20—21
— гистерэктомия 23—24
— дезинфекция помещений 16—17
— идентификация 17—18, 292—294
— ложная беременность (псевдобе-
ременность) 20—21, 351—353
— нумерация 18, 292—293
— оплодотворение in vitro 30, 345—
349
— оценка их здоровья 13
— разведение и содержание 13—17
— размножение 19
— спаривание 20—21, 28—29
— трансгенные 175
--- получение 278—296
---скрининг 292—296
— умерщвление 18—19
— усыновление 24—25
— эстральный цикл 20
Нейроэктодерма 68
Нервная трубка 68
Нитроцеллюлозные фильтры 246
Нозерн-блот, характеристика синте-
зируемых РНК-зондов 261, 262
Нотохорд 67, 68, 89
Нуклеиновые кислоты как зонды 246,.
250, 257
Нуклеопласты 300, 317
Оболочка блестящая, удаление 40—
43, 372
— Рейхерта 67, 69, 70, 71
Окрашивание гистологических срезов
271, 276
Ооцит человека предовуляторный
370—371
Ооциты оплодотворенные, культиви-
рование 380
— человека 369—372
---доноры 358—359
---получение 357—360
Оплодотворение in vitro ооцитов мы-
ши 28—29, 345—349
----------человека 357—358, 377,.
379
— —-------— подготовка спермато-
зоидов 377—379
396
Предметный указатель
------------- сбор и анализ семени
372—377
------------- руководство по про-
ведению в клинике и для научных
исследований 385—386
—------------этические и правовые
аспекты 382—384
Органогенез 68
Охлаждение эмбрионов мышей 334—
336, 341—342
Партеногееиз 127
Партеногеноны гаплоидные 310
— диплоидные 311
Первичная полоска 67, 69, 72, 73, 74,
83
Пипетки инъекционные 92, 93—96. 289
— удерживающие 92—93, 94, 96, 284,
' 305—306
Полиморфизм углеводов 168—173
Полиэтпленглпколь (ПЭГ) как фузо-
ген 320
Половые клетки, выделение 189—190
Проназа, удаление блестящей оболоч-
ки 42
Пронуклеопласты 318
Пронуклеус, инъекция ДНК 287—290
Пронуклеусы 30—31, 32, 127, 313—
3'18, 380
— перенос в яйца-реципиенты 313—
318
Протеинкпназы, фосфорилирование
белков 223
Псевдобеременность, индукция 351
Пуриновые основания, включение в
ДНК 185
Радиоавтография, использование при
гибридизации in situ хромосом
147—148
— локализация гибридизованных зон-
дов в гистологических срезах
268—270
Размораживание эмбрионов 336—337
Ретровирусные векторы 297
Ретровирусы, получение трангенных
мышей 296—298
РНК вируса мозаики вигны (CPMV)
как носитель 242—243
— выделение из эмбрионов 244, 245
— содержание в предимплантацион-
ных эмбрионах 242, 243—244
РНК-зонд радиоактивный одноцепо-
чечный, синтез 261, 263
РНК-зонды 259—263, 275
РНК-носитель 242—243
Серебро, окрашивание белков 119,
120, 122, 218, 235—236
Сидипг 332—334
Синаптонемный комплекс 114—117
Слияние клеток с помощью электриче-
ского тока 320—321
— нуклеопластов (кариопластов) с
яйцами 300, 317—318, 319—320
Сомиты 68, 83
Сперматозоиды человека 372—379
— — подготовка для оплодотворения
in vitro 377—379
— — получение методом «флотации»
377—378
----сбор и анализ 372—377
Сперматоциты насекомых 114—121
Спермин для оплодотворения in vitro
ооцитов мышей 345—349
— разделение в градиентах перколла
379
Среда для оплодотворения in vitro
366, 369
Среды культуральные 59, 78—79,
363-369
Суперовуляция 21—22, 28—29, 113,
127, 128, 129, 325, 357—358, 359,
369
— стимуляция 358
3Н-Тимидин, мечение клеток 87, 88,
88, 89, 90
Трансплантации эктопические 76—77
Трансформация эмбриональной линии
у мыши, использование эмбрио-
нальных стволовых клеток 297—
298
Трофобласт 65
Трофэктодерма, выделение 47
Углеводы, полиморфизм 168—173
УФ-свет длиноволновый, выявление
фермента ФГК-1 199
Ферменты, измерение активности
185—213
— конъюгаты с антителами 162, 165
— протеолитические, разделение за-
Предметный указатель
397
родышевых слоев 73
Фибробласты эмбриональные, культу-
ра 144—148, 149
Фитогемагглютинин, адгезия эмрио-
нов 45—46
Флуоресценция, измерение 187, 202,
203
Флюорография белков 216, 233—325
Фосфоглицераткииаза (ФГК), анализ
активности спектрофотометриче-
ский 202, 210—211, 212
— как маркер мозаичности 174
— микроанализ 198—213
— разделение изозимов путем элект-
рофореза 187, 204—207, 208, 211
Фосфопротеины, дефосфорилирование
223
— идентификация 216, 223
Фосфорибозилпирофосфат 192, 193
Ф'.‘Зиген 301, 320
Фузогенность 303
— из зародышей постимплантацнон-
ных стадий 130—134
—------предимплантационных ста-
дий 128—130
— маркерные 13, 136, 141—142, 144
— мейотические женские, высушива-
ние на воздухе 109—114
-----мужские, высушивание на воз-
духе 99—102
— — окрашивание 102—106, 119
— микрораспластывание ооцитов
121—123
— — сперматоцитов 114—119
— окрашивание 102—105, 119, 134—
144
— С-окрашнвание 103—104, 135, 137
— G-окрашиваиие 137—141
— Q-окрашивание 105, 137—141
— пахитенные, картирование 105—108
— пронуклеарные 129
Химерные («аллофенные») мыши 45,
155-159
— — клеточные маркеры 156—173
— эмбрионы, перенос ядер 318, 320
Химеры, инъекция клеток в бластоци-
сту 155—156
— клеточные маркеры 156—173
— «несбалансированные» 178—179
— эмбриональные агрегационные 155,
156
Хроматин половой, окрашивание 143—
144
Хроматография в крахмальном геле,
разделение электрофоретических
вариантов ФГК-1 198
— тонкослойная 198, 199—200
Хромомеры 106
Х-хромосома, выявление дозы гена
ГФРТ 185—186
— инактивация 174, 185—187
— мозаичность по сцепленным генам
174
— окрашивание 141—143
Y-хромосома. идентификация 129, 134,
135—136
Хромосомные наборы гамет 127—128
Хромосомы анеуплопдных зигот 125
— гибридизация in situ 109, 144—148,
150
— идентификация методами пред- и
постокрашивания 148—149
-----подсчет зерен серебра 144, 149—
151
Целлогель 202, 204—207, 208
Целлогель-электрофорез 211
Центрифугирование в градиентах
плотности, разделение спермато-
зоидов 377—379
Цитогенетический анализ 125—126
Эквнлибрацня 340—341
Эктодерма внезародышевая 68
Электрофорез в полиакриламидном
геле (ПААГ) 227—238
----------одномерные гели 228—
230
•---------разделение белков 215,
227—233
— ферментов в целлогеле 201—213
Эмбриональные стволовые клетки мы-
ши 278, 297—298
Эмбрионы мыши, охлаждение 334—
336, 341—342
---- перенос в яйцевод 352
----постимплантационные, введение
в них клеток пли метки 82—88
-------выделение половых клеток
189—190
—------культивирование 77—82
— ----- — изолированных тканей и
фракций 74—75
----- — мечение [3Н] -тимидином
-------микрохирургические опера-
ции 71, 73
398
Предметный указатель
------ — получение химер 83, 84, 88—
90
----— стадии развития 69—70, 72
— — предимплантациониые 27—59,
128—130
—------агрегация 45—46, 155, 156
— — — активность ферментов ГФРТ
и АФРТ 186—189, 196—197
------ — библиотеки кДНК 240—255
— — — внутренняя клеточная масса
46—47, 65
— — — двуклеточиый блок 34—35
---— диссоциация клеток 43—45
—------культивирование 27—38
------ — мечение внутриклеточных
поверхностей 54—57
— — — образование «гигантских»
бластоцист 47
--------- содержание РНК 242, 243—
244
—------стадии дробления 33—35, 39
----— удаление блестящей оболоч-
ки 40—43
----размораживание 336—337
— — трансплантация (перенос) 291 —
292, 319, 350—355
— человека, перенос в матку 380—
382
----- развитие до стадии бластоци-
сты 380—382
Энтодерма виезародышевая 65
Эпибласт 65, 67
Эпидермис 182
Эпителиальные ткаин, приготовление
тотальных препаратов 180—182
Ядра, микродеиситометрпя 49—50
— окрашивание 48—50
Яйца, активирование 310—311
— выбор линии мышей 309
— инъекции ДНК 282, 287-—291
— доноры и реципиенты при переносе
ядер 313—318
— культивирование 300—301
— оплодотворение 310
— оплодотворенные, пронуклеарное
деление 127
— разделение 319
Яйцевой цилиндр 68, 70, 72, 75
Яйцеклетки, получение 28—30
ОГЛАВЛЕНИЕ
Предисловие редактора перевода ......................... . . 5
Предисловие........................................................7
Список сокращений ' ... &
Введение.......................................................10
Глава 1. Разведение мышей. Колин Хедерингтон . . 13
1. Введение.........................................13
2. Разведение.......................................13
2.1. Источник мышей и общая оценка их здоровья . . 13
2.2. Клетки..........................................14
2.3. Подстилка . 14
2.4. Питание.........................................15
2.5. Вода............................................15
2.6. Среда обитания..................................15
2.7. Дезинфекция помещений......................... 16
3. Обращение с мышью..................................17
3.1. Подход..........................................17
3.2. Идентификация...................................17
3.3. Инъекции........................................18
3.4. Умерщвление.....................................18
4. Разведение..........................................19
5. Эксперименты.......................................19
5.1. Самцы-производители.............................19
5.2. Получение мышей с известным сроком беременности 20
5.3. Ложная беременность и вазэктомия................20
5.4. Суперовуляция...................................21
5.5. Анестетики......................................22
5.6. Получение потомства путем гистерэктомии ... 23
5.7. Усыновление.....................................24
6. Транспортировка.....................................25
7. Ввоз...............................................26
8. Правила работы с лабораторными животными ... 26
Литература...........................................26
Глава 2. Эксперименты с предимплантационными эмбрио-
нами мыши. Хестер Пратт................................27
1. Введение.......................... ... 27
2. Извлечение эмбрионов...........................27
2.1. Оплодотворенные и неоплодотворенные яйцеклетки 28
2.2. Стадии дробления (от 2 до 8-клеточной) ... 33
2.3. Морулы.....................................35
2.4. Бластоцисты................................37
3. Манипуляция с эмбрионами и клетками .... 38
400
Оглавление
3.1. Сортировка эмбрионов по стадиям..............38
3.2. Сортировка изолированных клеток по стадиям . . 39
3.3. Агрегация эмбрионов и изолированных клеток . . 45
3.4. Выделение внутренних клеток морул или внутренних
клеточных масс бластоцист.........................46
3.5. Выделение трофэктодермы......................47
3.6. Подсчет клеток...............................48
4. Применение меченых лигандов для изучения организации
клеток и эмбрионов.....................................50
4.1. Мечение клеток и морфология клеточной поверх-
ности ...............................................50
4.2. Мечение внутриклеточных компонетов .... 54
4.3. Маркеры клеточной линии.........................57
5. Реактивы, среды и оборудование......................59
5.1. Реактивы........................................59
5.2. Среды...........................................59
5.3. Оборудование....................................59
Литература...........................................63
Глава 3. Эксперименты с постимплантационными эмбрио-
нами мыши. Роза Бэддингтон....................................65
1. Введение...........................................65
2. Анатомия гаструляции и раннего эмбриогенеза . 65
3. Извлечение эмбрионов и классификация стадий их раз-
вития .................................................69
3.1. Извлечение.....................................69
3.2. Классификация стадий развития..................69
4. Выделение тканей или частей эмбриона...............71
4.1. Микрохирургические операции на эмбрионах . . 71
4.2. Ферментативное разделение тканей .... 73
5. Культивирование и эктопические трансплантации изо-
лированных тканей и фракций............................74
5.1. Культивирование............................74
5.2. Эктопические трансплантации....................76
6. Культивирование целых эмбрионов................77
6.1. Среды..........................................78
6.2. Роллерная система..........................79
6.3. Газовый режим..................................81
6.4. Оценка развития................................81
7. Включение клеток или метки в эмбрионы in vitro . . 82
7.1. Оборудование...................................83
7.2. Приготовление метки или клеток для инъекции 86
7.3. Инъекция.......................................87
8. Введение клеток или метки в эмбрионы in vivo . . 88
9. Изготовление стеклянных микрохирургических инстру-
ментов ................................................92
9.1. Стекло.........................................92
9.2. Оборудование...................................93
9.3. Инструменты................................. . 93
10. Заключение.........................................97
Литература..........................................97
Глава 4. Анализ мейоза в половых клетках человека
и мыши. Энн Чендлей ................................. 99
1. Введение............................................99
2. Получение препаратов мейотических хромосом высушива-
нием на воздухе........................................99
Оглавление
401
2.1. Метод суховоздушиых препаратов для мышей . . 100
2.2. Модификация методики при работе с человеческим
материалом......................................101
2.3. Рутинная окраска...........................102
2.4. С-окраска..................................103.
2.5. Q-окраска..................................105
2.6. Картирование пахитенных хромосом . . . . 105
2.7. Использование высушенных на воздухе мужских мей-
этических препаратов для молекулярных анлизов 108
3. Приготовление препаратов женских мейотических хромо-
сом высушиванием на воздухе.......................109
3.1. Профаза мейоза.............................109
3.2. Препараты ооцитов MI и МП, полученные при куль-
тивировании in vitro............................110
3.3. Препараты ооцитов в МП после овуляции . 113
4. Микрораспластывание для профазиого анализа . . . 114
4.1. Поверхностное распластывание для электронно-мик-
роскопического анализа сперматоцитов . . . 114
4.2. Распластывание для последовательного анализа в
световом и электронном микроскопах . . . . 117
4.3. Распластывание ооцитов.......................121
Литература........................................123
Глава 5. Кариотипирование и определение пола гамет, эм-
брионов и плодов; гибридизация хромосом.
Е. П. Эванс.....................................................125
1. Введение...........................................125
2. Основные методы....................................125
3. Оборудование.......................................126
4. Препараты хромосом из эмбрионов и плодов . . 127
4.1. Препараты из зигот.............................127
4.2. Приготовление препаратов из зародышей предим-
плантациопиых стадий .................... 128
4.3. Препараты зародышей постимплаитациониых стадий 132
5. Окрашивание хромосомных препаратов....134
5.1. Хромосомы мыши после рутинной окраски . . 134
5.2. С-окрашеииые препараты и их использование для
идентификации Y-хромосомы..........................135
5.3. G-окрашеиные препараты и их анализ .... 137
5.4. Окрашивание неактивной Х-хромосомы .... 141
5.5. Окрашивание для выявления полового хроматина 143
6. Гибридизация хромосом..............................144
6.1. Культура эмбриональных фибробластов и репликаци-
ониая окраска.......................................144
6.2. Методы пред- и постокрашиваиия.................148
6.3. Оценка метки...................................149
Литература..........................................151
Глава 6. Методы маркирования клеток и их применение.
Б. А. Дж. Пондер......................................153
1. Введение...........................................153
2. Химеры.............................................155
2.1. Определение понятия «химера»...................155
2.2. Типы химер и их получение......................155
3. Клеточные маркеры в химерных системах . . . 156
3.1. Использование в качестве маркера Н-2-аитигенов 159
402
Оглавление
3.2. Другие антигены................................166
3.3. Полиморфизм углеводов, выявляемый лектинами 168
3.4. Использование в качестве маркера лектнна Dolichos
(АДБ)...............................................170
4. Другие маркеры мозаичности ... .... 173
4.1. Х-ииактивацня.............................174
4.2. Внешние маркеры единичных клеток..........174
4.3. Соматическая мутация . ..... 175
4.4. Трансгенные мыши..........................175
5. Интерпретация результатов маркирования .... 176
5.1. Анализ тканевых экстрактов для выявления мозаич-
ности ..............................................176
5.2. Анализ видимых мозаичных паттернов . . . . 177
6. Приготовление тотальных препаратов некоторых эпители-
альных тканей.........................................180
6.1. Оборудование...................................180
6.2. Кишка..........................................180
6.3. Аорта..........................................181
6.4. Эпидермис......................................182
Литература..........................................183
Глава 7. Биохимический микроанализ сцепленных с Х-хро-
мосомой ферментов ГФРТ и ФГК. Мэрилин Манк 185
1. Введение......................................................185
2. Двойной микроанализ ГФРТ и АФРТ...................188
2.1. Приготовление образца..........................189
2.2. Реакционная смесь..............................191
2.3. Реакция........................................192
2.4. Прекращение реакции............................194
2.5. Фильтрация и отмывка...........................194
2.6. Подсчет и графическое изображение результатов 195
2.7. Оценка двойного микрометода....................196
3. Микроанализ ФГК................................... 198
3.1. Приготовление образца..........................204
3.2. Электрофорез...................................204
3.3. Окрашивание....................................207
3.4. Количественный анализ активности ФГК-1 н пропор-
ции обоих ферментов.................................209
3.5. Спектрофотометрический анализ активности ФГК-1 210
3.6. Чувствительность целлогель-электрофореза в отно-
шении ФГК-1.........................................211
3.7. Разрешение минорной полосы.....................212
3.8. Определение точности колнчественонго анализа нзо-
знма................................................212
Литература..........................................213
Глава 8. Качественный анализ белков на ранних стадиях
эмбриогенеза мыши. Сара Хаулет . .215
1. Введение......................................................215
2. Меченне белков раннего эмбриона...................218
2.1. Метаболическое мечение общих белков .... 220
2.2. Неметаболическое мечение общих белков . . 222
2.3. Фосфопротеины..................................223
2.4. Гликопротеины..................................224
2.5. Выявление синтеза специфических белков . . . 224
2.6. Приготовление образцов.........................226
Оглавление
403
2.7. Количественные измерения включенной метки . 227
3. Электрофорез в полиакриламидном геле..................227
3.1. Одномерные гели..................................228
3.2. Изоэлектрическое фокусирование и двумерные гели 230
4. Анализ белков, разделенных с помощью электрофореза в
геле..................................................233
4.1. Обнаружение радиоактивно меченных белков; сравне-
ние методов радиоавтографии и флюорографии 233
4.2. Окрашивание общих белков.........................235
4.3. Выявление специфических белков...................236
5. Заключение...........................................238
Литература............................................238
Глава 9. Создание библиотек кДНК для предимплантаци-
ного эмбриона мыши. Кристина Уотсон, Джози
МкКоннел.......................................................240
1. Введение............................................240
2. Оценка РНК-носителя.................................242
3. Сбор ооцитов и эмбрионов и выделение РНК . . . 243
3.1. Сбор ооцитов................................243
3.2. Сбор предимплантационных эмбрионов .... 244
3.3. Выделение РНК...............................245
3.4. Селекция ро1у(А)+-РНК и добавление носителя 245
4. Конструирование библиотек.............................245
4.1. Выбор вектора....................................245
4.2. Синтез и клонирование двухцепочечных кДНК . 246
5. Скрининг библиотеки..................................249
5.1. Тест на репрезентативность библиотеки .... 249
5.2. Скрининг библиотеки для выявления специфических
последовательностей ............................... 250
Литература............................................256
Глава 10. Гибридизация in situ с мРНК на гистологических
срезах. Ребекка Хаффнер и Кейт Уиллисон 257
1. Введение.........................................................257
2. Зонды................................................258
2.1. Выбор зонда.....................................258
2.2. Выбор метки.....................................260
2.3. Приготовление радиоактивных зондов .... 261
2.4. Расчет удельной активности зонда................261
2.5. Оптимальная длина зонда.........................263
3. Подготовка стекол...................................264
4. Фиксация и приготовление срезов.....................265
4.1. Парафиновые срезы...............................265
4.2. Замороженные срезы..............................265
5. Подготовка гистологических срезов для гибридизации 266
6. Гибридизация........................................268
7. Постгибридизационные отмывки........................268
8. Радиоавтография.....................................268
9. Проявление препаратов н окрашивание .... 270
9.1. Проявление......................................270
9.2. Окрашивание.....................................271
9.3. Заключение препаратов...........................271
10. Микроскопирование....................................271
И. Контроле и артефакты........................ . . 274
Литература......................................... 276
404
Оглавление
Глава 11. Получение трансгенных мышей. Николас Аллен,
Шейла Бартон, Азим Сурани, Вольф Рейк . . 278
1. Введение.......................................................278
2. Основная техника....................................278
2.1. Приготовление ДНК для микроннъекнии . . . 278
2.2. Яйца н эмбрионы.................................281
2.3. Оборудование....................................283
2.4. Изготовление микроинструментов..................284
3. Мнкроинъекцня ......................................286
3.1. Подготовка камеры...............................286
3.2. Подготовка инструментов.........................287
3.3. Инъекция в пронуклеус...........................287
3.4. Возможные затруднения...........................291
3.5. Трансплантация эмбрионов........................291
4. Скрининг трансгенного потомства.....................292
4.1. Аутопсия хвоста и прищипывание пальца . . . 292
4.2. ДНК из плодов............................... . 294
4.3. Анализ ДНК......................................295
4.4. Трансгенные мыши и замораживание эмбрионов 296
5. Перенос генов с помощью ретровирусов .... 296
Литература...........................................298
Глава 12 Трансплантация ядер в оплодотворенные и пар-
теногенетически активированные яйца. Ш. Бар-
тон, М. Норрис, А. Сурани.........................300
1. Введение............................................300
2. Основная экспериментальная техника..................300
2.1. Культивирование.................................300
2.2. Приготовление препарата вируса Сендай . . . 301
2.3. Оборудование....................................304
2.4. Изготовление мпкроинструментов..................305
2.5. Яйца и эмбрионы.................................309
3. Перенос ядер........................................311
3.1. Установка камеры................................311
3.2. Установка инструментов..........................312
3.3. Извлечение и перенос ядер.......................313
3.4. Другие варианты переноса ядер...................316
3.5. Кариопласты с минимальным количеством цито-
плазмы ............................................318
3.6. Разделение яиц..................................319
4. Оценка результатов..................................319
5. Перспективы и ограничения техники трансплантации ядер
яиц млекопитающих......................................320
Литература...........................................321
Глава 13. Низкотемпературная консервация яиц и эмбрио-
нов мыши. Маури Вуд, Дэвид Уиттингхэм,
Уильям Ролл......................................................323
1. Введение...........................................323
2. Стадии развития....................................324
2.1. Источник ооцитов и эмбрионов....................325
2.2. Сбор ооцитов и эмбрионов........................325
3. Замораживание ... 326
3.1. Приготовление образца...........................327
3.2. Криопротектор...................................330
Оглавление 405
3.3. Индукция образования льда в суспензионной среде:
«сидинг»............................................332
3.4. Охлаждение......................................334
3.5. Размораживание..................................336
3.6. Удаление ДМСО после оттаивания..................337
3.7. Техника безопасности............................338
4. Витрификация.......................................338
4.1. Внтрнфикационный раствор........................339
4.2. Осмотическое уравновешивание (эквнлибрация) эм-
брионов в витрификационном растворе .... 340
4.3. Охлаждение................................341
4.4. Хранение витрифицированных образцов . . . 342
4.5. Нагрев....................................342
4.6. Разведение витрификациоиного раствора . . 342
4.7. Техника безопасности......................343
5. Хранение образцов..................................343
5.1. Длительность хранения...........................344
5.2. Регистрация.....................................344
5.3. Номенклатура................................... 344
6. Оценка жизнеспособности консервированного материала 345
6.1. Ооциты..........................................345
6.2. Оплодотворение in vitro.........................345
6.3. Эмбрионы........................................349
6.4. Перенос эмбриона................................350
6.5. Общая выживаемость..............................355
7. Криоконсервация ооцитов и эмбрионов человека . 355
Литература...........................................355
Глава 14. Оплодотворение ооцитов человека и культивиро-
вание человеческих предимплантационных эмб-
рионов. Питер Брауде........................................357
1. Введение.........................................357
2. Источники ооцитов и эмбрионов человека для проведения
исследований ........................................ 357
2.1. Получение ооцитов и эмбрионов.................357
2.2. Донорские ооциты...............................358
3. Сбор ооцитов.......................................359
3.1. Лапароскопический сбор ооцитов.................359
3.2. Инструменты для сбора ооцитов путем лапароскопии 360
3.3. Подготовка инструментов для лапароскопии . 362
3.4. Газы, используемые для лапароскопии .... 362
4. Среды для культивирования эмбрионов и вымывания
ооцитов...............................................363
4.1. Ионный состав..................................363
4.2. Макромолекулы..................................364
4.3. Вода...........................................364
4.4. Масло..........................................367
4 5. Антибиотики..................................368
4.6. Газы...........................................369
4.7. Температура....................................369
5. Ооциты.............................................369
5.1. Манипуляции с ооцитами.........................369
5.2. Градация ооцнтов...............................369
5.3. Удаление клеток кумулюса.......................371
5.4. Удаление блестящей оболочки...................372
6. Сперматозоиды......................................372
406 Оглавление
6.1. Сбор образца семени..............................372
6.2. Анализ семени....................................373
6.3. Подготовка сперматозоидов для оплодотворения in
vitro.................................................377
7. Оплодотворение и культивирование эмбрионов . . 379
7.1. Осеменение.......................................379
7.2. Доказательство оплодотворения....................380
7.3. Культивирование оплодотворенных яиц .... 380
7.4. Развитие до стадии бластоцисты...................380
8. Этические и правовые аспекты.........................382
8.1. Законодательство и научные исследования на челове-
ческих эмбрионах в Великобритании .... 382
8.2. Варнокский комитет...............................382
8.3. Добровольная асоциация экспертов.................383
Литература............................................387
.'Приложение........................................................389
1Предметный указатель...............................................393
Научное издание
Колин Хедерннгтон, Хестер Пратт,
Роза Баддингтон и др.
БИОЛОГИЯ РАЗВИТИЯ МЛЕКОПИТАЮЩИХ.
МЕТОДЫ
Под ред. М. Манк
Заведующая редакцией М. Д. Гроздова
Ст. иаучн. редактор М. Р. Погосбекова
Мл. редактор О. В. Шагинян
Художник Л. В. Захаров
Художественные редакторы А. Я. Муснн. Л. М. Аленичева
Технический редактор О. Г. Лапко
Корректор Т. М. Подгорная
ИБ № 7127
Сдано в набор 21.03.90. Подписано к печати 4.09.90. Формат 60X90716. Бумага типограф-
ская № 1. Печать высокая. Гарнитура Литературная. Объем 12,75 бум. л. Усл. печ. л.
25.5. Усл. кр.-отт. 25,5. Уч.-изд. л. 27,19. Изд. № 4/6535. Тираж 5000 экз.- Зак. 171.
Цена 4 р. 90 коп.
Издательство «Мнр» В/О «Совэкспорткннга» Государственного комитета СССР по печати.
129820, ГСП, Москва, И-110, 1-й Рижский пер., 2.
Московская типография № И Государственного комитета СССР по печати.
113105, Москва, Нагатинская ул., д. 1.