/






















































































































































Автор: Мастерс Кристофер
Теги: химия физическая химия химическая физика металлы химические вещества химические процессы
Год: 1983




Текст
К. МАСТЕРС
Гомогенный катализ
переходными металлами
Rh'"
Москва «Мир»
HOMOGENEOUS TRANSITION-METAL CATALYSIS
A gentle art
CRISTOPHER MASTERS
Formerly of Shell Chemicals UK Ltd
(pesently with Christian Salvesen Ltd, Edinburgh)
London New York
Chapman / л and Hall
К. МАСТЕРС
Гомогенный катализ
переходными металлами
Перевод с английского
д-ра хим. наук, профессора Ю.И. Ермакова
и канд. хим. наук В.А. Семиколенова
Москва «Мир»
1983
WUbbZV
г
ББК 24.5
М32
УДК 541.128.12
Мастерс К.
М32 Гомогенный катализ переходными металлами: Пер. с
англ.—М.т Мир, 1983.— 304 с., ил.
В книге автора из Англии удачно сочетается доступное
изложение основ катализа металлокомплексами с глубоким
рассмотрением механизма практически всех типов реакций,
протекающих в присутствии комплексов переходных метал-
лов.
• Для широкого круга химиков — научных и инженерно-техни-
ческих работников, а также студентов и аспирантов, специали-
зирующихся в области катализа.
1У11805000000—332 .33, ч. 1
041(01)—вЗ
ББК 24.5
541
Редакция литературы по химии
© 1981 Christopher Masters
© Перевод на русский язык,
"Мир", 1983
От переводчика
К основному названию своей книги "Гомогенный катализ ком-
плексами переходных металлов" Кристофер Мастерс добавил " a gentle
ап " , что можно перевести как "тонкое искусство". Действительно.,
лучшие исследования в области гомогенного катализа оставляют впе-
чатление изящества - по их замыслу, по тщательности проведения экс-
перимента, по тем тонким эффектам, которые наблюдаются при изу-
чении катализа комплексами металлов в растворах0 Благодаря иссле-
дованию механизма гомогенного катализа мы стали глубже понимать
сущность каталитического действияо Для многих каталитических реак-
ций, катализируемых металлокомплексами, достоверно установлены
многие стадии каталитического цикла и выделены промежуточные сое-
динения, образующиеся в ходе каталитического процесса. Сейчас воз-
можности тонкого исследования детального механизма реакций, ката-
лизируемых металлокомплексами в растворах, расширяются благода-
ря все большим возможностям физических методов исследования (в
первую очередь ЯМР-спектроскопии).
Но гомогенный катализ - это не отвлеченно прекрасное искусст-
во, оно уже принесло зримые практические результаты. В последние
два десятилетия на основе применения металлокомплексных катали-
заторов были созданы многие промышленные процессы, продукция ко-
торых исчисляется миллионами тонн. В книге Мастерса подробно опи-
сан механизм реакций, лежащих в основе таких процессов, и приведе-
ны сведения о технологии их проведения.
Гомогенный катализ — в настоящее время еще не установившая-
ся область: необходимо уточнить отдельные детали действия катали-
тических систем и усовершенствовать технологию применения метал-
локомплексных катализаторов,. Интенсивные исследования катализа
металлокомплексами, которые проводятся во всем мире, непрерывно
приводят к открытию новых каталитических реакций и расширению
класса металлокомплексных катализаторов (например, путем приме-
нения кластеров и закрепленных металлокомплексов), Некоторые перс-
пективные области применения металлокомплексного катализа (акти-
вация азота, активация парафинов, гидрирование окиси углерода) опи-
саны Мастерсом в разделе о разрабатываемых гомогенно-каталитических
системах.
6
ОТ ПЕРЕВОДЧИКА
Эту книгу интересно прочитать и новичку, и специалисту, постоян-
но работающему в области гомогенного катализа. Те, кто только на-
чинает интересоваться этой областью (в том числе студенты и аспи-
ранты), найдут в книге очень четкое изложение основ катализа метал-
локомплексами. Исследователи, давно увлеченные этой областью, най-
дут в книге достаточно полное и стройное изложение результатов по-
следних исследований большинства важных гомогенно-каталитических
реакций. Конечно, специалист, глубоко знакомый с какой-либо узкой
областью гомогенного катализа, может отметить, что изложение его
области в книге недостаточно полнов Однако более детального изло-
жения практически всех областей гомогенного катализа нельзя тре-
бовать от такой небольшой книги. Краткость и четкость изложения —
большие достоинства данного издания.
Мы считаем необходимым отметить тот большой вклад, который
внесли советские ученые в развитие гомогенного катализа. Такие
исследования, как работы школы Н.М.Эмануэля в области катализа
окислительных реакций, протекающих с участием радикальных стадий,
работы Б.А.Долгоплоска по катализу металлоорганическими соедине-
ниями (прежде всего полимеризации диенов и метатезису олефинов),
И.И.Моисеева, открывшего и детально исследовавшего каталитические
реакции окисления олефинов в присутствии комплексов палладия, ра-
боты НаМоВольпина в области фиксации азота и работы школы А.Е.Ши-
лова в различных областях гомогенного катализа (активация пара-
финов, активация азота, превращение олефинов), явились важнейши-
ми этапами становления области катализа металлокомплексами. При-
знанные исследования проведены советскими учеными в области по-
лимеризации и олигомеризации олефинов, гидроформилирования и кар-
бонилирования, фотокатализа. При подготовке русского издания мно-
гие работы советских авторов были включены нами в списки дополни-
тельной литературы.
Ю.И.Ермаков
сентябрь 1982 г.
Посвящается Джилл и Джемиме
\ Предисловие
I . « Л
Растворимые катализаторы широко используются во многих об-
ластях химии и являются жизненно важным элементом в осуществле-
нии многих процессов, протекающих в природе, Они нашли широкое
применение в химической промышленности, где с их помощью ежегод-
но производится несколько миллионов тонн химических веществ, По-
скольку гомогенные каталитические системы, особенно содержащие
переходные металлы, часто эффективно действуют при более мягких
условиях, чем их гетерогенные аналоги, их важность постоянно воз*
растает в настоящее время, когда человечество озабочено поиском
наиболее экономичных путей расходования энергии и улучшения ис-
пользования доступных источников сырья, ♦
Основная цель, которую ставил автор при написании этой книги
возбудить у.читателей стремление начать (или продолжить) привно-
сить свой собственный вклад в наши познания о гомогенном катализе.
После изложения некоторых основных закономерностей в книге
рассмотрены сушествуюшее положение дел и перспектива развития
рассматриваемой области химии. При этом были использованы данные
как академических исследований, так и промышленности. Такой под-
ход возникает в связи с*глубоким убеждением автора, что прогресс
может быть существенно ускорен более интенсивным диалогом меж-
ду химиками, работающими в промышленности, и исследователями,
работающими в академических исследовательских институтах. Там,
где возможно, приведены примеры промышленного применения гомо-
генных каталитических систем. Во всяком случае автор не делал по-
пыток замаскировать то множество нерешенных проблем и волнующих
вопросов, которыми изобилует эта быстро развивающаяся область.
По-видимому отдельным читателям рассмотрение предмета в кни-
ге может показаться до некоторой степени предвзятым; однако следу-
ет принять во внимание энтузиазм автора и воинствующую позицию ав*»
тора против сведения науки до обезличенного рассмотрения физических
данных.
Краткий оксфордский толковый словарь [ The Shorter Oxford English
Dictionary] дает общее определение понятия искусство как мастерства,
8
ПРЕДИСЛОВИЕ
являющегося результатом знаний и опыта, далее переходит к обсуж-
дению искусства как ’’рода занятий, в котором мастерство использу-
ется для доставления удовольствия или создания чего-либо прекрас-
ного” в Именно это и имелось в виду, когда выбрано было название
книгио ’’Гомогенный катализ переходными металлами — тонкое искус-
ство”
Я хочу воспользоваться возможностью, чтобы выразить мою ис-
креннюю благодарность профессору Бернарду Шоу за то, что он по-
знакомил меня с предметом гомогенного катализа, и за большую по-
мощь и поддержку, которую он мне оказывал в ранние годы0 Я также
благодарен администрации исследовательского отдела компании Shell
за прекрасные условия для исследования, предоставленные мне в те-
чение шести лет пребывания в Амстердаме, а также за разрешение
на создание этой книги.. И наконец, мне особенно хочется поблагода-
рить мисс Хильду Харвей за ее терпеливость, аккуратность и добро-
совестность при подготовке к набору зачастую неразборчивой руко-
писно
Кристофер Мастерс
Эдинбург, апрель 1980 г.
1. Общее рассмотрение
1.1. Искусство катализа, основные принципы
Краткий оксфордский толковый словарь дает общее определение
понятия искусства как "мастерства, которое является результатом
знаний и опыта"., и дцлее переходит к обсуждению искусства как "ро-
да занятий,, в котором мастерство используется для доставления удо-
вольствия или создания чего-либо прекрасного";, Хотя лишь .немногие
могли бы считать пропаналь "прекрасным", вероятно, имеется опре-
деленное удовольствие в конструировании вещества;, которое могло
бы заставить оксид углерода, водород и этилен вступать в реакцию
между собой с образованием пропаналя с 85%-ной селективностью, осо-
бенно когда содержание этого вещества в реакционной смеси состав-
ляет лишь несколько сотых от концентрации олефина [ 1]0 Гидридо-
карбонилтрис (трифенилфосфин) родий (I). Rh (Н ) (СО) (PPh ) яв-
ляется одним из таких веществ,, т.е. катализатором., и процесс., в ко-
тором он действует, относится к области катализа. Однако, прежде
чем перейти к обсуждению катализа, мы должны рассмотреть основы
термодинамики химических реакций.
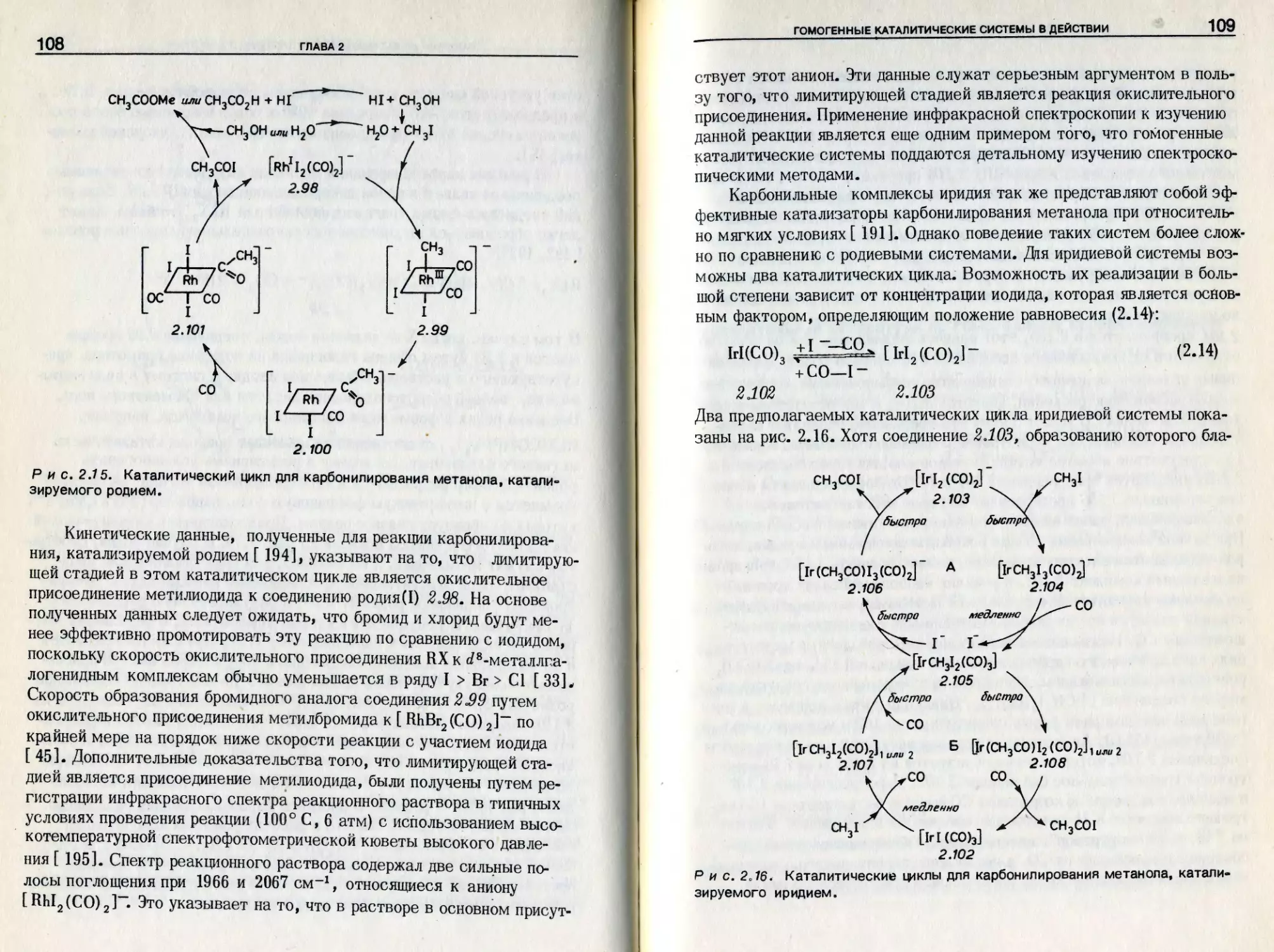
Химическая реакция - это процесс, в котором одно или несколь-
ко химических веществ превращаются в одно или несколько других
химических веществ. В общем случае это система, в которой два ве-
щества, А и В, реагируют между собой с образованием двух новых ве-
ществ, С и D [ уравнение (1.1 )L Согласно представлениям термо-
динамики, положение равновесия зависит от разности в свободной энер-
гии A G между реагентами А и В и продуктами С и D.
А + В - С + D (1Л)
A G = G (C,D) - G (А, В)
Для удобства обычно используют понятие стандартной свободной энер-
гии^, которая для соединения определяется как свободная энергия, необ-
ходимая для образования этого соединения из его элементов, когда
все реагенты и продукты находятся в стандартных условиях (25° С,
или 298,15 К, при 1 атм).
Для состояния равновесия
A G = —R ThKp (k2)
10
ГЛАВА 1
где Кр — константа равновесия, выраженная в терминах парциальных
давлений реагентов и продуктов. .Для реакции образования воды
Н2(г.) + У2 О2 (г.) —> Н2О(г.) Д6° = — 228э6 к/1ж/моль и
Кр = 1,19 •104Оатм"^-; следовательно,зравновесие этой системы
практически полностью сдвинуто вправОо Однако если водород и кис-
лород тщательно перемешаны в чистом состоянии, то ничего не про-
исходит - это именно тот случай, когда требуется катализатор^ «До-
бавьте немного тонко раздробленной платины к этой смеси — и реак-
ция пройдет так быстро, что платина разогревается до белого каления,
а смесь газов обычно взрывается. Аналогично, несмотря на то что
Д G °'298 для гидрирования этилена составляет —101 кДж/моль и
Кр = 5,1640 17 атм"1 при 298 К, в отсутствие подходящего катали-
затора смесь чистого водорода и чистого этилена практически не из-
меняется в течение неограниченного временно
Термодинамика может много рассказать нам о равновесном сос-
тоянии смеси, но она ничего не может сказать нам о скорости, с ко-
торой достигается это равновеснее Катализаторы не могут оказывать
влияния на положение окончательного равновесия. Однако они могут
коренным образом влиять на скорость, с которой это равновесие до-
стигается, или, согласно определению В^Оствальда [2], "катализа-
тор - это такое соединение, которое ускоряет химическую реакцию,
не влияя на химическое равновесие". Определение Оствальда примени-
мо только к обратимым реакциям и не охватывает никаких форм авто-
катализа, обнаруженного в реакциях окисления (см. разд. 2.7). Со-
гласно общему определению, предложенному ГКСабатье [3], катали-
затор — это вещество или система, которые изменяют скорость реак-
ции, участвуя в последовательности реакций, но не превращаясь в про-
дукты* о Это определение отчасти вводит в заблуждение, поскольку
для того, чтобы выполнить свою функцию эффективно, катализатор
должен постоянно возобновляться в течение реакции., Это свойство
.может быть использовано для того, чтобы охарактеризовать катали-
* Приведем определение катализа, данное академиком Г.К.Боресковым,
(Боресков Г.К. Катализ. 4.1 и 2. - Новосибирск: 1971О): "Феноменологичес-
ки катализ можно определить как возбуждение химических реакций или изме-
нение их скорости под влиянием веществ — катализаторов, многократно всту-
пающих в промежуточное химическое взаимодействие с участниками реакции
и восстанавливающих после каждого цикла промежуточных взаимодействий
свой состав” (см. также: Краткая химическая энциклопедия. — М.: Советская
энциклопедия, 1963). — Прим, перев.
ОБЩЕЕ РАССМОТРЕНИЕ
затор как вещество, одновременно являющееся и реагентом, и продук-
том химической реакции.
Не имеет значения., как мы определили катализатор, более важно
очертить рамки катализа. Катализатор может только ускорить реак-
цию, но он не в состоянии изменить окончательное положение равно-
весия. Прежде чем приступить к созданию какого-либо катализатора,
следует проверить термодинамическую возможность проведения необ-
ходимой реакции. Другими словами, если мы хотим найти катализатор
для реакции
А + В -* С + D (1.3)
то должны убедиться, что при разумных условиях (температура и
давление) термодинамически предсказанные равновесные концентра-
ции продуктов С и D являются более чем минимальными. Мало тол-
ку искать катализатор, если концентрация продуктов при равновесии
составляет, скажем, 10“4 моль/лс Никакое количество катализатора
не приведет к увеличению этого выхода. Из уравнения (1.2) следует,
что если А С° < 0, то равновесие будет сдвинуто вправо, и если
Д С ° > 0, то оно будет сдвинуто влево. Таким образом, реакция, пред-
ставленная уравнением ( L3 ), термодинамически осуществима, если
она сопровождается обшим уменьшением свободной энергии* Заман-
чиво упростить критерий и рассматривать реакции с положительным
A G° как неосуществимые. Но это было бы ошибкой, поскольку реак-
ции с небольшим положительным A G° могут давать заметные вы-
ходы нужного продукта при разумных экспериментальных условиях,
и наоборот, реакции с небольшим отрицательным А С° могут давать
очень низкие выхода. Практическое использование критерия A G°
может быть обобщено следующим образом:
АС° < 0 реакция возможна;
0 < A G° < 40 кДж реакция сомнительна, но, воз-
можно, стоит провести иссле-
дование;
А С° > 40 кДж можно и не трудиться искать
катализатор0
Трудно переоценить важность проверки термодинамической выполни-
мости, и, несомненно, одним из наиболее важных орудий в арсенале
химика - разработчика катализаторов является широкий набор термо-
динамических данных (см., например, [ 4]).
12
ГЛАВА 1
Рис. 1,1. Изменение свободной энергии для некаталитической и каталити
ческой реакций
— некаталитический путь реакции. .. в э каталитический путь реакции.
При условии, что реакция разрешена, возникает вопрос : каким
образом катализатор ее ускоряет? По существу, катализатор предла-
гает реагентам другой, низко энергетический путь к продуктам: он об-
легчает для системы путь от некоторого начального к некоторому ко-
нечному состоянию.
Эффективность катализатора определяется разницей в легкости,
с которой система проходит реакционный путь в присутствии катали-
затора и без него. Каждый путь обычно имеет некоторую особую ста-
дию, которая определяет обшую скорость реакции, т.е. лимитирующую
стадию. Скорость этой стадии определяется ее свободной энергией
активации, которая соответствует наивысшему энергетическому барь-
еру по пути следования от реагентов к продуктам реакции. Назначение
катализатора — уменьшить свободную энергию активации настолько,
насколько это возможно, или по крайней мере уменьшить ее по срав-
нению с некаталитическим механизмом. Графически эта ситуация
представлена на рис. 1.1.
До настоящего времени мы обсуждали катализ в общем смысле,
лишь мимоходом упоминая о действительной физической природе ка-
тализатора. Пока наш подход к.катализу был, если так можно выра«
зиться, подходом администратора, и единственное, .что нужно было
ОБЩЕЕ РАССМОТРЕНИЕ
13
делать, чтобы катализировать реакцию А + В -* С , только напи-
сать магическое слово "катализатор" или "кат," над стрелкой. Что
на самом деле представляет собой "катализатор" или "кат.", оста-
вим ученым. В конце концов им платят за то, чтобы они понимали та-
кие вещи. Действительно, катализатор может быть чем-то таким же
простым, как протон, или чем-то настолько сложным, как фермент.
Он может быть .чисто органическим веществом по природе, чисто не-
органическим веществом или смесью обоих. Он может присутство-
вать в той же фазе, что и реагенты (гомогенный катализатор), или
в разных фазах (гетерогенный катализатор). Если он присутствует
в той же фазе, то мы можем говорить о газовых реакциях или реак-
циях, протекающих в растворе. Очевидно, что для того, чтобы огра-
ничить предмет нашего рассмотрения разумными пределами, нам необ-
ходимо очертить те типы систем, которые мы собираемся рассматри-
вать. Ключевыми словами в определении наших граничных условий
являются "переходный металл" и "гомогенный".
1.2. Почему "переходные металлы"?
Можно выделить пять основных причин, по которым переходные
металлы вносят существенный вклад в столь широкую область ката-
литических систем: 1) способность к образованию связей; 2) широкий
выбор лигандов; 3) влияние лигандов; 4) способность к изменению сте-
пени окисления; 5) способность к изменению координационного числа.
Переходные элементы отличаются от элементов основной груп-
пы тем, что имеют частично заполненные d- или /-уровни. Основная
переходная группа, или «/-элементы, — это те элементы, у которых
частично заполнен «/-уровень, и это именно та группа, с которой глав-
ным образом мы будем иметь дело и которую будем иметь в виду,
используя термин переходный металл.
1С2ЛО Способность к образованию связейс Ион «/-металла име-
ет девять валентных орбиталей - ^,Px,pyfp2, d22> dx2 ~.у2, dxz> dy z>
Л*?, на которых размещаются его валентные электроны и с помощью
которых образуются гибридные молекулярные орбитали при связыва-
нии с другими группами. Доступность этих валентных орбиталей пе-
реходного металла позволяет ему образовывать как сигма (ст )- , так
и пи (тг)- связи с другими молекулами или лигандами. Именно эта
14
ГЛАВА 1
способность к образованию ст- и тр-связеи и является ключевым фак-
тором в определении каталитических свойств переходных металлов и
их комплексов.
Рассмотрим случай связывания олефина с переходным металлом
и возьмем, например, одно из первых распознанных металлоорганичес-
ких соединений, т.е. соль Нейзе К[ C2H4PtCl 3] [ 5,6]. Структура
иона [ C^H4PtCl 3]~ показана ниже (1Л )•; по существу, это плоский
квадрат в котором двойная связь этилена перпендикулярна координа-
ционной плоскости.
z СН2
С!
1Л
Если в таком плоскоквадратном комплексе мы расположим ось х так,
чтобы она совпадала со связью Cl — Pt - Cl, тогда четыре валентные
орбитали иона металла, включая сферически симметрическую 6 s-орби-
таль (6s, 5t/%2 _ у2> п 5ру), окажутся лежащими вдоль осей
связей металл-лиганд. Эти орбитали или гибридная комбинация из
них подходят для образования ст-связей путем перекрывания (взаимо-
действия) с орбиталями лиганда, имеющими подходящую симметрию
и энергию. .Для хлоридных лигандов ст-связь образуется путем пере-
крывания с 3 s-орбиталями хлора. Заполненная тг-орбиталь молекулы
этилена имеет подходящую симметрию и энергию для образования ст-свя-
зи с одной из пустых орбиталей металла, лежащих вдоль оси у. Схе-
ма ст-перекрывания металл - олефин показана на рис. 1.2, а (орбитали
металла представлены в виде гибридных орбиталей типа ds р2). Из
оставшихся орбиталей металла d?z лежит в координационной плос-
кости, определенной фрагментом Pt (С ,Н4), и имеет подходящую сим-
метрию для образования тг-связей с лигандом, который бы имел под-
ходящие лигандные орбитали. Незанятые антисвязываюшие т^-орбита-
ли олефина соответствуют указанной симметрии, и, таким образом,
r-связь формируется путем взаимодействия заполненных </ -орби-
талей металла с незанятой антисвязываюшей орбиталью этилена. Это
тг-связывание между металлом и олефином представлено на рис. 1.2, б,
а картина комбинированного связывания показана на рис. 1.2, в [7,8].
В принципе тесвязывание так же возможно между металлом и хлором
ОБЩЕЕ РАССМОТРЕНИЕ
15
Р и с> /.2. Молекулярно-орбитальное представление взаимодействия этилена
с переходным металлом0
с использованием вакантных 3 (/-орбиталей хлора. Однако энергия
последних орбиталей по сравнению с орбиталями металла слишком вы-
сока для того, чтобы происходило какое-нибудь заметное перекрывание
и связь хлор — металл имеет в основном о-характер. -
Рассмотренные выше компоненты связи металл - олефин облада-
ют свойством синергизма, т.е. они взаимно усиливают или дополняют
друг друга. В о-компоненте электронная плотность перетекает от свя-
зывающей орбитали к металлу, тогда как в случае тт-компоненты элек-
тронная плотность перемещается от металла на антисвязывающие ор-
битали олефина. Оба этих,перемещения приводят к ослаблению или
уменьшению порядка С-С-связи олефина [ 9,10].
Координация олефина на металлическом центре изменяет электрон-
ную плотность С-С-связи и во многих случаях делает ее более чув-
ствительной к нуклеофильной атаке такими агентами, как ОН~ и Н~.
Кроме того, вероятно, не менее важно, что координация с металлом
фиксирует олефин в определенном месте, и, таким образом, атакующие
группы "знают*’, где его найти. К этому вопросу мы вернемся при рас-
смотрении взаимодействия близко расположенных групп в разд. 1.5.
В карбонильных комплексах переходных металлов найден тип свя-
зывания, аналогичный типу связывания в металл-олефиновых ком-
плексах (т.е. вовлечение как а- , так и тг-компоненты, как это пока-
зано на рис. 1.3). а-Компонента в этом случае образуется путем вза-
имодействия между вакантной ст-орбиталью металла и заполненной
s р-орбиталью уг лерода. тг-Компонента образуется путем взаимодей-
ствия между заполненной dг- или гибридной dp к-орбиталью металла
и одним из наборов вакантных антисвязывающих р т-о'рбиталей СО .
16
ГЛАВА 1
а - компонента связи тг - компонента связи сг/тг - связь
а д' в
Р и Со 1оЗв Молекулярно-орбитальное представление взаимодействия моно-
ксида углерода с переходным металлом.
Так же, как и в случае олефина, о-компонента приводит к переносу
электронной плотности от лиганда к металлу, а т₽-компонента — к
переносу только в обратном направлении. Это связывание также об-
ладает эффектом синергизма и приводит к уменьшению порядка С—0-
связи [11].
-Связывание частицы СО с переходным металлом не ограничивает-
ся только монодентантными (или линейными) структурами М-СО.
Известны комплексы, в которых СО является мостиком между дву-
мя, как в Со2 (С0)8, ]
центрами. В случае комплекса [ Rh 7 (СО)~6 ]3
зываюшего взаимодействия присутствуют в одной и той же молекуле
[ 12]. Во всех представленных выше случаях связывание металл —
карбонил происходит исключительно через карбонильный углерод. Эти
три способа связывания представлены схематически формулами 1.2 -
8, или тремя, как в Rh6 (СО )j
б, металлическими
все три типа свя-
м
линейное
связывание
м м
&идентатнов
связывание
И I М
М
тридентатное
связывание
М
М
Недавно было найдено, что СО может также связывать два катали-
тических центра за счет тр-связываюших орбиталей моноксида угле-
рода, т.е. по типу 7.5 [ 13-15].^Хотя этот последний тип связыва-
ния встречается относительно редко, он может оказаться очень важ-
ным при разработке систем, катализирующих восстановление СО
[ 16] (см. разд.3.2).
Соединения трехвалентного фосфора и в меньшей степени трех-
валентного мышьяка являются исключительно важными лигандами во
многих каталитических системах на основе комплексов переходных
ОБЩЕЕ РАССМОТРЕНИЕ
17
металлов. Рассмотрение симметрии показывает, .что такие лиганды
могут в принципе координироваться с переходными металлами с ис-
пользованием как ст-, так и т^орбиталей почти таким же образом, как
и С2Н4 и СО.
Картина связывания фосфинового лиганда, по существу, аналогич-
на той, которая была ранее описана для моноксида углерода. Основ-
ным отличием в случае фосфора является то, что тг-компонента свя-
зи образуется путем перекрывания заполненной rf-, или с/р-гибридной,
орбитали металла с незанятой 3rf-, или Зс/Зр- гибридной, орбиталью
фосфора, а не с незанятой р тьантисвязываюшей орбиталью, как в слу-
чае с СО. Вопрос об относительной важности тг-компоненты в связях
металл — трифенилфосфин является спорным. Мейсон и Мик [ 17 ] пред-
положили, .что важность т^связывания в определении молекулярных па-
раметров таких комплексов была ранее преувеличена. По их мнению,
разнообразные проявления электронных свойств третичных фосфино-
вых лигандов обусловлены в основном ст-эффектом, так как при изу-
чении длин связей металл — триарцлфосфин они почти не обнаружили
эффектов тъсвязывания.
Из вышеизложенного следует, что (/-элементы могут легко об-
разовывать прочные связи с веществами, содержащими системы тг-элек»
тронов или имеющими орбитали подходящей симметрии и энергии для
образования с? тт-связей. Не менее важной с точки зрения катализа
является способность переходных металлов образовывать прочные
связи, в основном ст-типа, с некоторыми весьма реакционноспособны-
ми соединениями [ 18]. В связи с этим комплексы переходных метал-
лов облегчают получение этих соединений при относительно мягких
условиях ( часто при комнатной температуре и атмосферном давлении)
делают их последующее поведение более спокойным и часто заставля-
ют их реагировать очень специфичным образом. Наиболее часто в
.каталитических реакциях встречаются две активные частицы — гидрид
(Н ~) и алкил ( R ~).
Например, хлоротрис(трифенилфосфзин) родий RhCl(PPh ) (1.б)в
бензоле легко реагирует с молекулярным водородом с разрывом Н—Н-свя-
зи и образованием дигидридного комплекса R ИН 2 Cl (PPh ) , в кото-
ром два атома Н непосредственно связаны с металлом [ 19 J.
Металл-гидридная связь стабильна в том смысле, что ее можно
наблюдать, применяя обычные методы спектроскопии, например ЯМР-
ХН или ИКС. Однако при наличии олефина RhH2Cl (РГЦ )3 легко
отдает свои гидридные лиганды для присоединения по двойной связи
олефина:
18
ГЛАВА 1
RhCl(PPh,)3 + Н2 - RhH Cl(PPh ) г* RhH,CI (PPh,)2 + PPh.
RhH2Cl(PPh3)2 + RCH-CH2 "RhCl(PPb3)2” + RCH2CH3 (1.4)
последовательность реакций подробно обсуждается в разд. 2.1.1»
RhCl( PPh3 )3 реагирует и с такими молекулами, как CHJ , приводя
к образованию соединений, в .которых метильная группа непосредст-
венно связана с металлическим центром: RhCl (PPh л )з + Ме! ->
-> RhMeCH (PPh3)3. Реакция же с ацетилхлоридом5приводит к об-
разованию металлацетильногр комплекса типа Rh(COMe) Cl (PPh3)3 [ 20].
Имеется лишь несколько примеров способности переходных металлов
стабилизировать реакционноспособные частицы, а, как мы увидим в
последующих главах, такие частицы играют ключевую роль в широком
круге каталитических реакций, включающих много различных субстра-
тов и каталитических систем.
1.2.2. Широкий выбор лигандов В рамках координационной хи-
мии переходных элементов лиганд может быть определен как элемент или
комбинация элементов, которые образуют химическую связь с переходным
элементом. Во многих случаях наиболее предпочтительным лигандом пере-
ходного элемента служит он сам, что подтверждается тем фактом, что все
элементы являются металлами. Кроме того, переходные элементы
легко образуют связи почти со всеми элементами периодической сис«,
темы и практически со всеми органическими молекулами. Именно это
свойство делает столь богатой координационную химию переходных
элементов и, что самое важное, определяет их роль в катализе.
Можно выделить две основные группы .лигандов: формально ион-
ные лиганды [ Cl ~, Н ОН -CN(алкил) ~, (арил) ~, СОСНу ]
и формально нейтральные .лиганды [ СО, алкен, третичные, вторичные
и первичные фосфины, арсины или фосфиты, Н2О , амины].Разделение
лигандов на ионные и нейтральные, хотя и является полезным при опре-
делении степени окисления и при написании уравнений реакций, до не-
которой степени формально, поскольку в большинстве комплексов пе-
реходных металлов ионные лиганды образуют с металлическим центром
ковалентные, а не ионные связи. Во многих случаях разделение заряда
в связи металл — лиганд незначительно, и в отдельных случаях для
"ионных” лигандов оно меньше, чем для "нейтральных1’. Измерение
дипольных моментов и методы рентгеновской спектроскопии показали,
что в квадратных плоских платиновых комплексах ’’ионные" .лиганды
ОБЩЕЕ РАССМОТРЕНИЕ
19
Н~ и СН~ почти нейтральны в смысле разделения заряда по связи
металл - лиганд, тогда как ’’нейтральные” лиганды, например пири-
дины или третичные фосфины, могут нести довольно большой положи-
тельный заряд (<-0,3 е) [ 21].
Эти результаты затрагивают одну из проблем, возникающих при
обсуждении реакций комплексов переходных металлов, которая заклюй
чается в том, что -многие теоретические положения были выведены из
набора формальных определений. Как правило, согласно ожиданиям,
эти формальные концепции служат хорошо, и, таким образом, боль-
шинство реакций гидридных комплексов переходных металлов могут
быть объяснены при допущении, что Н реагирует как Н ~ и анало-
гично в металлалкильных соединениях алкил реагирует как R~ . Тем
не менее важно помнить, что такие объяснения скорее умозрительны,
чем реальны. Они исключительно полезны и часто могут служить оо
новой рабочих гипотез. Не следует, однако, ограничивать ими наше
воображение. Связанный с металлом метил обычно реагирует так, как
если бы он был R“, но нет таких причин, по каким не может быть
изобретена система, в которой он реагирует как R или R+ . Дей-
ствительно, как мы увидим далее, известны и такие системы.
До сих пор мы в основном рассматривали такие лиганды, как ал-
.кены, алкил, карбонил, гидрид, которые действительно или потенциаль-
но способны принять активное участие в каталитическом цикле в том
смысле, что входят в состав продукта каталитического никла, как на-
пример, гидриды и алкены в цикле каталитического гидрирования или
СО в реакции каталитического карбонилирования. Мы будем называть
такие лиганды "лигандами-участниками". Что касается катализатора,
то не менее важной группой лигандов являются .лиганды, .которые не
принимают прямого участия в каталитическом цикле в том смысле,
что остаются связанными с переходным металлом и не включаются
в состав продукта реакции. Примером таких "неучаствуюших" лиган-
дов могли бы быть хлор и трифенилфосфиновые группы в реакции (1.4).
Хотя эта последняя группа .лигандов не превращается физически в
прямые продукты катализируемой реакции, они играют ключевую роль,
определяя активность и селективность каталитической системы [ 22,23].
1и2зЗо Влияние лигандов, -Способность катализаторов на основе
переходных металлов одновременно располагать внутри своей коорди-
национной сферы как лиганды-участники, так и неучаствующие лиган-
ды, позволяет им направлять протекание реакции между лигандами-
20
ГЛАВА 1
участниками путем модификации структурных и электронных свойств
неучаствуюших лигандов.
Формально лиганд может оказывать влияние на поведение ката-
лизатора, содержащего переходный металл, модифицируя стеричес-
кое или электронное окружение активного центра, т.е. места, на .ко-
тором взаимодействуют лиганды-участники. Практически со всеми
простейшими лигандами результирующий эффект является комбина-
цией электронных и стерических факторов. Во многих случаях вклад
каждого из параметров является нерешенным вопросом, так как до
сих пор нет хорошо развитых теорий, учитывающих оба эффекта. Од-
нако существует ряд факторов, которые могут оказаться полезными
для интерпретации, а в некоторых случаях и для предсказания дей-
ствия лигандов-участников. Тремя такими факторами являются "транс-
эффект", "донорно-акцепторные электронные свойства лиганда" и
"конический угол". Первый в принципе касается всех ,лигандов, спо-
собных образовывать связи с переходным металлом. Два вторых от-
носятся в основном к фосфиновым и в меньшей степени к арсиновым
.лигандам.
Л. транс-Эффект. Вследствие того что большинство орбиталей
переходных металлов, образующих связи металл — лиганд, имеют на-
правленную природу, а также из-за того, что орбиталь-орбитдльное
взаимодействие максимально, .когда угол между орбиталями равен ну-
лю, электронные эффекты, осуществляющиеся в основном .через свя-
зывающие орбитали, максимальны в том случае, когда лиганды нахо-
дятся в яфш/оположении. Таким образом, максимальный электронный
эффект можно получить, располагая неучаствующие лиганды в транс-
положении к участвующему лиганду. Наиболее широко цитируемым при-
мером //фотоэффекта является изменение скорости замещения лиган-
да в плоских квадратных комплексах платины(П) [ 24]. Так, скорость
замещения хлора пиридином в комплексах типа 1.7
увеличивается в.отношении 1?: 30>: 200?: 10000 при переходе от X = С1
к Х= С Я59СН3,Н . Согласно привёденным данным, эти четыре лиган-
ОБЩЕЕ РАССМОТРЕНИЕ
21
да по величине дораи#-эффекта могут быть расположены в следующем
порядке: Cl < С6Н < СН 3 .< Н. Механизм взаимодействия двух транс-
лигандов, очевидно, зависит от природы рассматриваемых лигандов.
Простые о-связывающие лиганды, такие, как Н“, взаимодействуют
с транс*лигандом через сг-связывающую орбитальную систему, тогда
как тт-акцепторные лиганды, такие,как СО и третичные фосфины, мо-
гут взаимодействовать как через о-, так и через тр-связывающую
орбитальную систему. Хотя вопрос об относительной величине вклада
0-- и тт-составляющих в эффект является дискуссионным [17],
нет никаких сомнений в том, что лиганды, стоящие во главе серии
транс-эффектов, такие, как Н~ и SnCl“ , эффективно лабцлизируют
дор амо группы в комплексах металлов. В случае когда диссоциация .ли-
ганда или субстрата считается .лимитирующей стадией реакции [ 19,25],
введение в каталитическую систему .лигандов с высоким транс-эффек-
том может оказать благотворное воздействие на активность системы.
В приведенном выше обсуждении мы подразумевали, что транс-
эффект .лигандов является в основном функцией его электронных ха-
рактеристик. В представленных примерах электронные эффекты почти
всегда преобладали, однако для объемистых лигандов стерические фак-
торы могут также вносить значительный вклад в доряис-эффект. Пы-
таясь разграничить электронные и стерические эффекты фосфиновых
лигандов, Толман [ 26 ] предложил ввести два параметра; первый из
них основан на донорно-акцепторных электронных свойствах лигандов,
а второй — на размере лиганда.
Б9 Электронные донорно-акцепторные свойства — электронный
параметр v [26,27]. Частоты колебаний СО в ИК-спектре карбониль-
ной группы, связанной с переходным металлом, изменяются в зависи-
мости от природы и количества других .лигандов, входящих в состав
комплекса. В 1970 г. Толман [ 27] предположил, что это может быть
использовано для определения электронного фактора v, отражаюше- .
го электронные донорно-акцепторные свойства любого данного фосфи-
нового лиганда. Поскольку комплексы типа Ni (СО )3 L образуются
с высокой скоростью при смешивании в отношении 1?: 1 Ni (СО )4 и L
при комнатной температуре и в ИК-спектре таких комплексов наблю-
дается очень узкая полоса, соответствующая карбонильной группе (по-
22
ГЛАВА 1
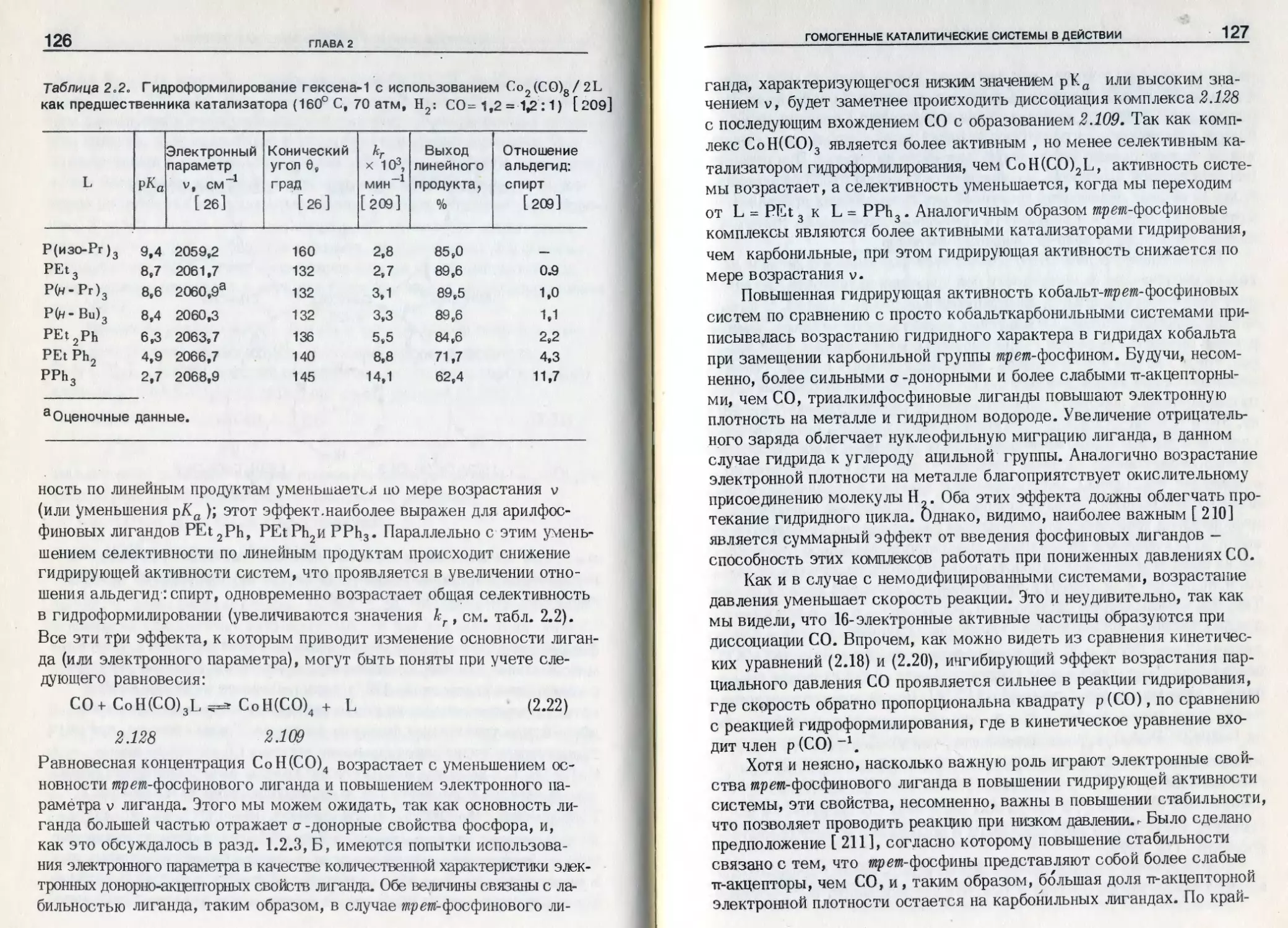
Таблица 1.1. Значения параметра х Для некоторых заместителей [ 26]
Группа X в лиганде типа Р’Х X- X 1 Z О X, см~1 Группа X в лиган- де типа РХ X X 2 2 3 Хз см*1
треп Б ути л 060 м-Т оли л 3.7
Циклогексил 0,1 п-Толил 3;5
Изопропил 1,0 Фенил 4,3
Бутил 1,4 ОМе 7.7
Этил 1.8 Н 7.3
Метил 2,6 ОВ1 97
М"олил Зв5 C6F5 11?2
Л CF- 3 19,6
.лоса A j h Толман предложил выбрать частоту этой полосы Av в ка-
честве меры донорно-акцепторных свойств .лиганда. Он обнаружил, что
эта частота является в основном аддитивной функцией, зависящей от
природы трех групп, связанных с атомом фосфора, и .что электронный
параметр v,.который является синонимом частоты полосы А}9 вы-
ражается уравнением v = 2056,1 + т X. (см*1), где 2056,1 -
частота А г для комплекса карбонила никеля с три-wp ед?-бутилфосфи-
ном (наиболее основной лиганд в исходной серии), ах- вклад замео
тителя, соответствующий каждой группе, присоединенной к фосфору
в лиганде РХхХ2Х3. В табл. 1.1 приведены некоторые характерные
значения х > а в табл. 1.2 сопоставлены отдельные рассчитанные зна-
'чения с экспериментально определенными /частотами А .
В. Конический угол - стерический параметр 0. В дополнение
к электронному параметру Толман [ 26,29] предложил ввести стери-
ческий параметр 6, применимый к лигандам на основе соединений трех-
валентного фосфора. Необходимость введения стерического парамет-
ра 6 вытекает из того, что положение равновесия, которое описыва-
ется уравнением (1.5 ) и выражает способность фосфорных лигандов
конкурировать за координационное место в:комплексе Ni( 0), не мо-
полностью объяснено только их электронными свойствами [ 29 ].
„U«NiL4_„ Lj + nL
Положение этого равновесия, по-видимому
объемом фосфорного лиганда L. Для —
(1.5)
, определяется физическим
1 наглядного представления раз-
ОБЩЕЕ РАССМОТРЕНИЕ
23
Таблица 1023 Значения электронного параметра v для некоторых ли*
гандов РХ^Х а также соответствующие значения частот полосы Лхдля
комплексов №(CO)3L (L = PX^jXj) [ 2б]
2063,9
2061,5
2059,1
2056,1
2066,6
2066,6
2067,3
2064,0
2072,4
2085,2
РХ,Х,Х,
1 х Э
2064,1
2061,7
2059,2
2056,1
2066,7
2066,6
2065,3
2063,7
2072,0
2085,3
РМе
РЕ.’
Р( изо-R- )3
Р( изо-Bu),
Р( п-ТЫ),
Р(о-То1),
ЖеРЬ2
PEt 2Ph
P(OMe)Ph2
P(OPh)3
меров лигандов были построены их молекулярные модели *. Для всех
заместителей, расположенных таким образом, чтобы они занимали как
можно меньший объем, был измерен конический угол. Этот угол опре-
дёлялся как угол цилиндрического конуса с вершиной, удаленной на
2,28 А (2,85 см при использовании молекулярных моделей со шкалой
1,25 см/ А) от центра и касающийся вандерваальсовых радиусов самых
отдаленных атомов модели (см. рис. 1.4).
Было найдено, .что способность лиганда конкурировать за коорди-
национные места в комплексах Ni (0) коррелирует с его коническим
углом — чем больше конический угол, тем слабее конкурирующая спо-
собность лигандов. Аналогично было найдено, что степень замещения
СО на L в карбониле никеля исключительно хорошо коррелирует с
измеренным коническим углом (в этом случае Ni( СО) обрабатывал-
ся восьмикратным избытком L и смесь выдерживалась до установле-
ния равновесия). Эти первые работы Толмана, впоследствии flonpjH
ные измерениями константы равновесия [ 30] для реакции NiL4==
*
Так называемые пространственно заполненные модели, в которых
размер каждого атома соответствует действительному объему, занимаемо-
му им в пространстве, т.е. его вандерваальсову радиусу.
24
ГЛАВА 1
Рис. 1О4О "Конический угол” (по Толману) для симметричного фосфинового
лиганда PR3 о
— [ NiL^] + L легли в основу таблицы значении 0 для широкого ряда
трехвалентных фосфорных ,лигандов [ 26]. Некоторые выбранные зна-
чения 0 приведены в табл. 1.3. Поскольку существует хорошая линей-
ная корреляция между степенью замещения (с.з.) карбонильных групп
Ni(CO)4 и коническим углом 6 замещающих лигандов, значения О
для новых лигандов или лигандов, в которых 0 трудно измерить по
модели, могут быть установлены на основании измерения величины
с.з. В экспериментальном отношении метод сравнительно прост. Ni(C0)4
обрабатывается восьмикратным избытком лиганда L в запаянной труб-
ке до установления равновесия. .Степень замещения определяется по
относительной интенсивности карбонильных полос в ИК-спектре обра-
зовавшейся смеси. Исходя из графика зависимости значений с.з. от
О, построенного для известных .лигандов, можно найти значение 0 для
нового лиганда. Конические углы исключительно полезны для оценки
относительных размеров различных лигандов, однако следует прини-
мать определенные меры предосторожности при использовании их для
Таблица ТЗ* Значение конического угла 0 для некоторых лигандов
РХ1Х2Х3, содержащих трехвалентный фосфор [ 26]
Лиганд Овград
Рнз 87
РМе 118
PHe Ph 122
PEt , J 132
PPh 145
Лиганд 6,град
P(изопропил)3 160
P( циклогексил)3 170
P( трет-бутил) 182
P(о-толил) 3 194
ОБЩЕЕ РАССМОТРЕНИЕ
25
оценки абсолютных размеров лигандов. Необходимость таких мер свя-
зана с тем, что оценка относительных размеров лигандов основана на
предположении, что длина связи металл - фосфор остается постоянной
(2,28 А) и фосфор сохраняет тетраэдрическую симметрию. Кроме то-
го, не учитывается возможность "совмещения" друг с другом фосфор-
ных пигандных групп в молекуле комплекса. Например, основываясь
на том, .что конический угол лиганда Р( циклогексил} составляет
170°, трудно ожидать, что комплекс Pt { Р (циклогексил будет
устойчивым или по крайней мере его можно будет выделить. Однако
при кристаллизации комплекса Pt { Р( циклогексил) } из гептана
в присутствии избытка трициклогексилфосфина при * ft°C легко вы-
деляется сольват Pt {Р(циклогексил)3 1,5 C7HJ6 [28]. Соглас-
но данным исследования структуры этой молекул^ рентгеновскими ме-
тодами, трициклогексилфосфиновые лиганды ориентируются таким об-
разом, что циклогексильные группы могут совмещаться друг с другом,
а это существенно уменьшает стерические затруднения и стабилизиру-
ет молекулу.
Очевидно, использование толмановских конических углов для
предсказания абсолютной стабильности данного соединения может
привести к ошибке. Однако не вызывает сомнения, что разумное при-
менение электронных и стерических параметров Толмана к данным о
влиянии лиганда в реакциях, катализируемых переходными металлами,
может облегчить понимание относительной важности обоих факторов,
а также может помочь в создании новых систем .лигандов и комбина-
ций металл - лиганд.
В приведенном выше обсуждении влияния лигандов главное вни-
мание уделяли тому, каким образом лиганд может влиять на электрон-
ные и стерические свойства комплекса. ^Лиганд может также влиять
и на физические свойства металлокомплекса, что в некоторых слу-
чаях может оказаться весьма важным при создании каталитической
системы. Растворимость комплексов, содержащих алкилфосфиновые
лиганды в органических растворителях, может быть повышена путем
увеличения длины алкильной цепи. Например, растворимость в бензо-
ле комплекса металла с третичным фосфином увеличивается при пе-
реходе от РМе к PEt3, РВ*3 и РВи^ ..Как правило, алкилфосфино-
вые .комплексы более растворимы, чем арилфосфиновые, хотя раство-
римость последних может быть увеличена введением алкильных групп
в арильные кольца. Растворимость комплексов в полярных раствори-
телях, таких, как вода, может быть повышена путем введения в.лиган-
26
ГЛАВА 1
дную систему полярных групп, например карбоксильных. Летучесть дан-
ного комплекса может быть изменена правильным подбором лиганда.
Так, например, летучесть .карбонилфосфиновых комплексов кобальта
Со (СО )3 Р[( CH 2 )п СН 3 ] 3 можно уменьшить путем увеличения числа
п, причем электронные свойства фосфинового лиганда практически
не изменяются.
1о2о4п Способность к вариации степени окисления., Теорети-
.чески переходный металл может иметь столько положительных сте-
пеней окисления, сколько валентных и s-электронов у него име-
ется. Например, Сг (3J5 4 s 1) 'в принципе может существовать в по-
ложительных состояниях окисления от I до VI , и действительно из-
вестны комплексы всех шести положительных состояний окисления
хрома, а также комплексы Сг (0) и Cr (-II).
Для переходных элементов характерна способность к образова-
нию стабильных комплексов металлов в различных состояниях окис-
ления, хотя не все элементы образуют стабильные комплексы во всех
доступных для них степенях окисления. тьСвязываюшие лиганды, та-
кие, как СО, третичный фосфин и амины, образуют комплексы с ме-
таллами предпочтительно в низких степенях окисления, тогда как a-свя-
зывающие .лиганды, например Н~ и F~, дают комплексы с метал-
лами предпочтительно в высоких степенях окисления. Возможность
существования переходного металла в различных степенях окисления
позволяет ему образовывать комплексы с широким набором разнооб-
разных элементов и соединений. Однако, по-видимому, более важной
является способность к быстрому переходу одного состояния окисле-
ния в другое в ходе каталитической реакции. Рассмотрим реакцию гид-
рирования (1.4), катализируемую родием. В течение каждого катали-
тического цикла родий проходит состояния окисления I ->Ш->1 в окис-
лительно-восстановительном цикле
И Р.С’Щ—*сн
“Rh'ClL2” —RhinH2ClL2 ----------------Rh,,1H(RCH2CH2)ClL2
-RCH2CH3
В типичных условиях гидрирования при комнатной температуре и ат-
мосферном давлении родий проходит этот цикл примерно один раз в
минуту. Аналогично механизм реакций гидрирования, катализируемых
RuCIH (PPh3) 3 , включает окислительно-восстановительный цикл с
ОБЩЕЕ РАССМОТРЕНИЕ
27
участием рутения с изменением степени окисления II ->IV ->П . Эта
способность легко входить в окислительно-восстановительные циклы
является отличительной чертой металлов VIII группы, благодаря че-
му их каталитическая, активность изменяется в широких пределах.
1 о2а5з Способность к вариации координационного числа0 Хо-
рошо известны комплексы переходных металлов, содержащие в коорди-
национной сфере девять лигандов, например ReH 2*, WH6(PR3) 3> Бо-
лее обычны координационные числа от 4 до 6. Способность переходного
металла объединять в своей координационной сфере несколько различ-
ных,лигандов бесспорно важна, если от этого комплекса требуется ка-
тализировать реакцию между одним или несколькими субстратами. На-
пример, в реакции гидроформилирования, где альдегид образуется из
олефина, СО и водорода, переходный металл должен обладать способ-
ностью координировать в ходе реакции олефин, карбонил и гидрид наря-
ду с поучаствуют ими лигандами, которые также могут присутствовать
в комплексе. Чрезвычайно важным свойством комплекса является спо-
собность иметь различные координационные числа и, следовательно,
принимать различные конфигурации и проявлять способность быстро-
го обмена между ними. Например, в ходе реакции гидрирования, ката-
лизируемой RhCl(PPh3) 3, можно считать , что в течение одного ката-
литического цикла родий изменяет свое координационное число от 4
(плоский квадрат) до 6 (октаэдр), а затем до 5 (квадратная пирами- .
да), 6 (октаэдр), далее до 5 ( квадратная пирамида), 4(треугольник)*
и снова до 4 (плоский квадрат или квадратная пирамида). Эти измене-
ния представлены на рис. 1.5.
Причина, по которой мы ранее ограничились обсуждением ката-’
лиза только комплексами переходных J-металлов, может быть выра-
жена одним словом - "многогранность". Комплексы переходных ме-
таллов являются по своим свойствам одними из наиболее многогран-
ных соединений, которые известны только в химии.
1.3. Почему "гомогенный?"
Прежде чем дать ответ на этот вопрос, мы должны рассмотреть
оба типа каталитических систем — гомогенные и гетерогенные — бо-
лее детально.
• * По-видимому9 автор подразумевает, что четвертое координационное
место занято молекулой растворителя,, — Прим, перев.
28
ГЛАВА 1
треугольник
квадратная
пирамида
октаэдр
Рис. 1.5. Изменение стереохимии комплекса в ходе гидрирования, катали»
зируемого RhClL ( L = PPh ).
э 3
По определению гомогенная реакция - это реакция, все составные
части которой находятся в одной и той же фазе. При обсуждении гомо-
генного катализа обычно подразумевается, что все составные части
реакции, включая, .конечно, катализатор, присутствуют в обычной жид-
кой фазе, т.е. в растворе. Другим допущением является то, :что катали-
затор представляет собой отдельную частицу, т.е. в случае гомогенно-
го катализатора - либо один комплекс переходного металла, либо
комбинацию отдельных комплексов. Гетерогенная реакция - это реак-
ция, один или более реагентов которой находятся в различных фазах.
В гетерогенном катализе катализатор обычно является твердым, а
реагенты жидкие или, более часто, газообразные. В этом случае ка-
тализируемая реакция протекает на границе раздела фаз, т.е. на по-
верхности катализатора. Благодаря тому:что гетерогенный катализа-
тор является твердым (или массивным), он имеет определенные пре-
имущества по сравнению с гомогенным аналогом с практической точки
зрения. Основное преимущество гетерогенного катализатора состоит
в том, что он легко может быть отделен от продуктов реакции при
условии, конечно, что это жидкие или газообразные продукты. До нас-
ОБЩЕЕ РАССМОТРЕНИЕ
29
тояшего времени именно это преимущество бцло исключительно важ-
ным для широкого применения гетерогенных катализаторов в промыш-
ленности. Будучи твердыми, гетерогенные катализаторы обычно отли-
ваются более высокой термической стабильностью по сравнению с го-
могенными системами.
Таким образом, повышения скорости реакции (при условии равен-
ства каталитической активности гомогенного и гетерогенного ката-
лизаторов ) можно достичь, проводя реакцию при белее высокой тем-
пературе. Это даст явные преимущества, особенно если энергия де-,
шевая, а время дорого.
Термическая стабильность может быть также преимуществом при
регенерации катализатора. К сожалению, ни один катализатор не ве-
чен. В гомогенных.каталитических системах побочные реакции умень-
шают концентрацию активных комплексов, превращая их в катдлитичес-
\ки неактивные формы. Поэтому после проведения нескольких циклов
реакции необходимо утилизировать металл или дезактивированный
комплекс и заново синтезировать активные комплексы. Обычно это
не удается сделать непосредственно in situ в ходе реакции, и за-
частую такая регенерация представляет собой очень дорогой процесс.
В гетерогеннокаталитических системах дезактивация обычно обус-
ловлена отложением на поверхности катализатора побочных продук-
тов реакции и регенерация может быть проведена путем простого вы-
жигания этих продуктов. Большинство гетерогенных каталитических
систем может быть просто и быстро регенерировано in situ , что обес-
печивает очевидное преимущество при промышленном осуществлении
процесса. Преимущества гетерогенного катализатора обусловлены
тем, что он является твердым веществом, но этим же обусловлены
его недостатки/Конкретная реакция катализируется определенными
центрами или комбинацией центров катализатора независимо от того,
гомогенный он или гетерогенный. В твердом катализаторе, особенно
в том, который приготовлен нанесением "коктейля” из элементов на
носитель, имеется множество различных центров. «Даже в чистом ме-
таллическом состоянии индивидуальные атомы металла могут нахо-
диться в различном окружении. Из всего этого набора различных цент-
ров, возможно, только один тип центров катализирует интересующую нас реак-
цию; в лучшем случае другие центры не принимают никакого участия в проведе-
нии реакции, в худщем — они катализируют нежелательные побочные процессы.
Гомогенный катализатор обычно имеет только один тип активных центров.
Таким образом, в единицах активности, отнесенной к атому ме-
30
ГЛАВА 1
тцлла*, гомогенные катализаторы зачастую более активны. Они так-
же способны быть намного более селективными. Эти свойства приобре-
тают важность в настоящий период, когда все общество и химичес-
кая промышленность, в частности, заняты поиском путей сохранения
энергии и улучшения использования доступных источников сырья.
Главная, требующая неотложного решения проблема в понимании
поведения твердых катализаторов заключается в нехватке физических
приборов, которые бы давали возможность точно контролировать реак-
ции, протекающие на поверхности катализатора, или которые бы по-
зволили получить сведения о природе активных центров.
Для изучения протекания реакций на поверхности и исследования
природы активных центров твердых катализаторов использовались
спектроскопические методы, такие, как ИК-спектроскопия и в послед-
нее времяЕЕЕО*%ЭСХА, оже-спектроскопия [ 31 ]. Однако из-за слож-
ности строения поверхности катализатора возникают существенные
трудности в однозначной интерпретации полученных результатов. Боль-
шинство рассматриваемых гомогенных реакций поддается изучению
с помощью методов ИК-спектроскопии и ЯМР (в случае диамагнитных
комплексов), так как они протекают в растворе в присутствии опре-
деленных комплексов металлов. Оба этих метода достаточно доступ-
ны и могут быть использованы для изучения катализируемых реакций.
Так, например, методы ЯМР^Н и 31Р были использованы для то-
го, чтобы охарактеризовать большую часть металлоорганических со-
единений, участвующих в реакции гидрирования олефинов, катализиру-
емой комплексом RhCl(PPhp з [ 19]. Недавно с помощью ИК-спек-
троскопии было показано, что в условиях образования этиленгликоля
из СО .и водорода, катализируемого родием, присутствуют карбониль-
ные кластерные соединения родия. [ 16]. Возможность такого деталь-
ного изучения гомогенных каталитических систем позволяет точно
проследить влияние изменений в лигандах и условий реакции и должно
привести к более глубокому пониманию гомогенных каталитических
систем по сравнению с гетерогенными системами. Очевидно, что эти
два типа систем связаны между собой тесным образом и что оба они
включают реакции, протекающие на металлических центрах, и зачастую
* Активность, отнесенная к одному атому металла, названа ПК.Борео
ковым "атомной каталитической активностью" (см. Boreskov G.K ‘Catalytic
activity of transition metal compounds in oxidation reactions. In; Catalysis, ed.
by J.W.Hightouer,North--Holland Publishing Co., Amsterdam, London, New York,
1973,, p, 981 986).— ГТрим. перев.
Дифракция медленных электронов. — Прим0 перево
ОБЩЕЕ РАССМОТРЕНИЕ
31
понимание действия гомогенных катализаторов помогает объяснить
результаты, полученные при изучении гетерогенных катализаторов.
Возвращаясь к исходному вопросу, заключаем,.что причины, по
которым для рассмотрения были выбраны гомогенные системы, могут
быть сведены к следующим четырем их особенностям: а) высокая се-
лективность, б) высокая активность, в) легкость модификации, г) лег-
кость изучения. Кроме того, в аспекте проблем эффективного исполь-
зования источников сырья гомогенные катализаторы будут играть все
более важную роль, поскольку в мире постоянно возрастает .необходи-
мость увеличения экономической эффективности производства.
1.4. Активация молекул
Обосновав выбор предмета рассмотрения, мы возвращаемся к
основным положениям катализа. Как отмечено в разд. 1.1, катализа-
тор ускоряет реакцию, предлагая другой, более низкий по энергии путь
к продуктам. Он осуществляет это, активируя один или несколько ре-
агентов и затем предоставляя им возможность реагировать между
собой. Если мы рассматриваем .катализ с точки зрения активации мо-
лекул, то можем выделить два основных процесса активации: путем
координации- и путем присоединения.
1.4.1» Активация путем координации. Активация путем коор-
динации — это процесс, в котором субстрат взаимодействует с.катали-
тическим центром таким образом, что сохраняется целостность суб-
страта XY.
Несмотря на то что распределение электронов по связи (связям)
может быть в корне изменено, в таком процессе X и Y формально
остаются связанными друг с другом, и в любом обменном процессе
субстрата XY целиком обменивается быстрее, чем по отдельности
X или Y..Среди гомогенных систем примерами процессов такого ти-
па могут служить .координация СО, алкена или аллильной группы на
отдельном металлическом центре. .Соответствующий процесс в гете-
рогенном катализе известен как "недиссоциативная адсорбция".
. В реакции окисления этилена, катализируемой палладием в ук-
сусной кислоте, координация этилена на центре Pd (II) уменьшает
электронную плотность на олефине и делает его более чувствительным
к нуклеофильной атаке ацетата, т.е.
СН2
\\ 2 СН2СН2ОАс
xpd^$H> —- ^Pd"^
^ОАс
32
ГЛАВА 1
Аналогично координация олефина с циклопентадиендикарбонцлжеле-
зом, сокращенно Fp+, делает олефин чувствительным к атаке широко
го набора нуклеофилов при мягких условиях:
^-NiT ___/Nu
Fp+-y-|[ ----* Fp
Типичными нуклеофилами являются СН (СО Et) . R”, X" и т.д.
2 2 о
Хотя эта последняя реакция и не является каталитической, она слу-
жит для иллюстрации принципа активации путем координации и сама
по себе очень полезна в органических синтезах [ 32].
В обоих указанных выше случаях активация носит в основном элек-
тронный характер» В процессах активации путем координации сте-
рические факторы могут быть в равной мере, а в некоторых случаях
и более важны, чем электронные. Это особенно справедливо для асим-
метрических синтезов, катализируемых или регулируемых комплекса-
ми переходных металлов ( см. разд. 2.1.3 и 2.4.5 ), и в стереоспеци-
фической олигомеризации (см. разд. 2.5.4).
1.4.2. Активация путем присоединения. Активация путем при-
соединения — это процесс, при котором субстрат взаимодействует с
металлическим центром таким образом, что нарушается целостность
XY, т.е. связь (связи), удерживающая вместе X и Y, формально
разрывается. В этом процессе активации .либо X, либо Y, либо они
оба оказываются связанными с металлическим центром, и в каком-ли-
бо обменном процессе X и Y обмениваются либо по отдельности, ли-
бо вместе. «Среди гомогенных систем примерами могут служить при-
соединение водорода к металлическому центру с образованием ди-
или моногидридных соединений или присоединение (HCN с образова-
нием гидридоциановых комплексов. «Соответствующий процесс в ге-
терогенном катализе известен как "диссоциативная адсорбция". В
гомогенных системах могут быть выделены три типа активации пу-
тем присоединения [ 18]: а) окислительное присоединение, г) гомо-
литическое присоединение, в) гетеролитическое присоединение.
Л. Окислительное присоединение. Окислительное присоеди-
нение [ 20, 33] определяется как присоединение субстрата ХУ к ме-
таллическому комплексу таким образом, что формальная степень
окисления и координационное число образующегося комплекса увели-
ОБЩЕЕ РАССМОТРЕНИЕ
33
чиваются на два*: M^L у + XY —> М х ~ 2 (X)(Y)Ly ..Обратная
реакция известна как восстав"вителъное элиминирование. Примерами
окислительного присоединения, которые имеют особое значение для
гомогенного катализа, являются присоединение водорода к комплек-
сам родия(I) или платины (И) и присоединение метцлиодада к родию (I)
цли иридию (I}:
Rh1 Cl{PPh 3) * + Н 2 -► RhinClH2 (PPh^)^ х = 2 или 3
PtnCl(Sna3)(PPh3)2 + Н2 -> PtIVClH2(SnCl3)(PPh3)2
[MhjCO),]- + Mel -> [ МШ1,.Ме (СО),]“ М = Rh или 1г
(При определении степени окисления металлов в этих или других гид-
ридо- или алкильных комплексах металлов условно считается, что ал-
кильная или гидридная группа является лигандом, несущим один отри-
цательный заряд.)
Очень подходящим примером восстановительного элиминирования
является образование алкана из алкилгидридного комплекса:
RhniClH(R)(PPh3 )х -> Rh1 Cl (PPh^) + RH х = 2 или 3
Во всех этих примерах металлы претерпевают переход из состояния
^8(RhI4J8, 5d8, Ir1 5d8) в состояние d6(RhIII4delPtIV5cf6,IrIII5ti6)<
что характерно для реакций окислительного при соедан ения или восста-
новительного элиминирования. Реакциям такого типа обычно наибо-
. лее подвержены последние элементы J-ряда, особенно металлы VIII
группы. Типичными окислительно-восстановительными переходами яв-
ляются d 10 d8 (например, Pd° ** Pd11 ), d8 d6 (например,
Rh1 Rh111) и d 6 ** d 4 (например, Ru11 < > Ru). В дополнение
к сказанному важно отметить, :что эти реакции в действительности
не ограничены элементами VII] группы цли переходными металлами.
Хорошо известной-реакцией, которая может служить примером окис-
лительного присоединения с участием металла главной группы, явля-
ется присоединение алкцлгалогенида к магнию с образованием реак-
тива Гриньяра: Mg° + RX -> Mgn(R)X.
* Часто реакция окислительного присоединения ускоряется или происхо-
дит одновременно с диссоциацией одного или нескольких лигандов, так что
координационное число конечного продукта превышает координационное чис-
ло исходного комплекса меньше чем на два. Поэтому обычно считают, что
увеличение1 формальной степени окисления на два вместе с сохранением X
и Y внутри координационной сферы металла является достаточным условием
для того, чтобы охарактеризовать реакцию как окислительное присоединение.
ИСАКОВ.
34
ГЛАВА 1
Поскольку окислительное присоединение приводит к увеличению
формальной степени окисления металла на два, то лиганды, которые
увеличивают электронную плотность на металлическом центре, т.е.
основные лиганды, обычно увеличивают и скорость окисления. Так,
комплексы с алкилфосфиновыми лигандами, например PEt , обычно
более легко подвергаются окислительному присоединению по сравнению
с аналогичными комплексами с арилфосфиновыми лигандами, такими,
как РРЬ3 [34]. Поскольку реакция окислительного присоединения
приводит к увеличению координационного числа, то лиганды, увеличи-
вающие стерические затруднения на металлическом центре, например,
три-^р^бутилфосфин, обычно уменьшают скорость каталитических
реакций, содержащих стадии окислительного присоединения [ 35]. Ре-
зультаты такого влияния лигандов на скорость каталитических реак-
ций, содержащих стадии окислительного присоединения, обсуждаются
в гл. 2, особенно в части, посвященной гидрированию.
Б, Гомолитическое присоединение, Гомолитическое присоединение
[ 18,19] определяется как присоединение субстрата XY к двум метал-
лическим центрам таким образом, что формальная степень окисления
каждого металлического центра повышается на единицу:
2M*L • + XY -» 2М* + 1 (X)(Y )L
У у
Этот тип присоединения встречается в реакциях, катализируемых ком-
плексами переходных металлов, и представлен на примере двух соеди*
нений кобальта. Присоединение водорода к водному раствору цианида
кобальта приводит к образованию аниона гидридопентацианокобальта
[ Со Н (CN) ]3~, который активен в восстановлении как неоргани-
ческих, так й органических соединений [19].
гСо’УсН)’- + Н ₽» 2Со ШН (CN)
5 2 5
Было сделано предположение, что эта реакция может протекать и че-
рез присоединение водорода к дикобальтовым соединениям, которые
образуются путем димеризации CO(CN)3“ [ 361:
2Con(CN)’“ Co^fCN)6!-
Co^fCN)?- + Н ** 2CoinH(CN)3“
Хотя современные данные не позволяют четко различить эти два мар-
шрута реакции, кажется несомненным, что активной ;частицей в по-
следующих реакциях восстановления является Со ш Н (CN) 3*~. -Слу-
чаем, когда, по-видимому, в реакцию вовлекаются димерные соедине-
ОБЩЕЕ РАССМОТРЕНИЕ
35
ния, является образование СоН (СО ) из дикобальтоктакарбонила
при гомолитическом присоединении к нему водорода [ 37}:
Со°(СО)8 + Н2 2Со1Н(СО)4
Что касается промышленного использования гомогенных каталитичес-
ких систем, то, как описано в разд. 2.4, эта реакция является одной
из наиболее важных реакций активирования присоединением, посколь-
ку Со2( СО )8 служит источником кобальтового катализатора для реак-
ции гидроформилирования олефинов:
Со?(СО)§
RCH - СН? + СО + Нэ —RCH СНОСНО + RCH(CHO)CH,
2 2 100 - 160°С 2 2
100 — 300 атм
Е 1976 г. более 3,5 млн. т альдегидов было произведено с использо-
ванием этих или близких систем.
Б представленных выше примерах гомолитическое присоединение
приводило к образованию двух мономерных частиц. Однако это не яв-
ляется неизбежной необходимостью процесса, поскольку присоединение
по связи металл — металл с сохранением полиметаллической структу-
ры могло бы также представлять собой гомолитическое присоединение.
Хотя еше не было никаких сообщений о примерах такого присоединения
' в каталитически важных системах, стехиометрическое присоединение
НХ по связи молибден — молибден в [ М° X ] 4 ~ (X = С1 или Вг ) яв-
ляется надежно установленной реакцией [ 38 ]>:
MoVx^ + НХ -* Мо’п(Н)ХЗ- + X-
2 о 2 о
Этот тип активации может быть весьма важным в каталитических сис-
темах, содержащих кластерные соединения металлов.
Как и окислительное присоединение, гомолитическое присоединение при-
водит к формальному окислению металла и точно также имеет склонность
ускоряться электродонорными лигандами. Так, гомолитическое присоедине-
ние водорода к Со2(СО)6(РВи3)2 происходит быстрее, чем к Со2(СО)8.
В.Гетеролитическое присоединение. Гетеролитическое при-
соединение определяется как присоединение субстрата XY к металли-
ческому центру таким образом, что не происходит общего изменения
формальной степени окисления или координационного числа металла,
и только .либо X, либо Y становится формально связанным с метал-
лом , .
М *L + XY —> М L , X + Y + + L “
У у — 1
36
ГЛАВА 1
В основном этот процесс может рассматриваться как замещение од-
ного анионного лиганда на другой при металлическом центре. Наибо-
лее распространенным примером такого типа активации является при-
соединение водорода к гексахлороаниону рутения (III) Ru С13 ~ :
RuinCl3“ + Ru111 Cl НЗ- + Н + + С1 6
6 2 5;
.Образующиеся гидридохлоридные соединения легко восстанавливают
Fe111 до Fe11 или Ru до Ru111 . Так хлорид рутения (III) в вод-
ной соляной кислоте катализирует восстановление водородом соедине-
ний железа(III) и рутения(IV) при 1 атм и 80°С [39].
Комплексы рутения(П) также могут принимать участие в гете-
ролитическом присоединении [ 40}:
RunCl (PPh,) + Н, RunCl(H)(PPh \ +Н+ + С1"
Z Э D X J J
(1.6)
Этому типу реакции благоприятствует присутствие соответствующего
основания: так, добавление этанола к бензольному раствору RuCl2(PPh3)
существенно увеличивает гидрирующую способность этой каталитичес-
кой системы. Другие основания, такие, как триэтиламин или, конечно,
гидроксид калия, также эффективно промотируют присоединение во-
дорода к RuCl (PPh ) , образуя RuCl (Н ) (PPh ) плюс (основа-
ние)-ЦС1 [ 19]. 33
На практике зачастую исключительно трудно различить гетероли-
тическое присоединение и окислительное присоединение, сопровожда-
ющееся восстановительным элиминированием. Так, в качестве альтер-
нативы уравнения (1.6) мы могли бы в равной мере написать
RunCl2(PPh3)* +Н2 RulvCl2(H)2(PPh3)x
RunCl(H)(PPh,) +НС1
3 л
Действительно, имеются доказательства этого пути реакции [ 41 ].
Аналогично присоединение водорода к цис- Pt Cl ( PEt ) на
первый взгляд включает гетеролитическое присоединение:
^Mc-PtnCl2(PEt ) +н2 mpaHC-PtnCl(H)(PEt ) + НС1
Однако на самом деле равновесие включает образование промежуточ-
ного соединения — дигидрида платины (IV) Р* Cl Н (PEt ) [42].
2 2 3 2
Остается установить, являются ли^все примеры гетеролитического
присоединения особыми случаями реакций окислительного присоеди-
нения и восстановительного элиминирования. Отметим, что этому про-
цессу активации присоединением обычно благоприятствует присутст-
ОБЩЕЕ РАССМОТРЕНИЕ
37
е в исходном металлическом комплексе .легкоионизируемого лиган-
да, а в случае водорода - присутствие подходящего основания. На сле-
ющей стадии каталитического цикла активированные молекулы суб-
страта должны прореагировать друг с другом. Этот процесс мы рао
смотрим в разд. 1.5.
1.5, Близкое взаимодействие
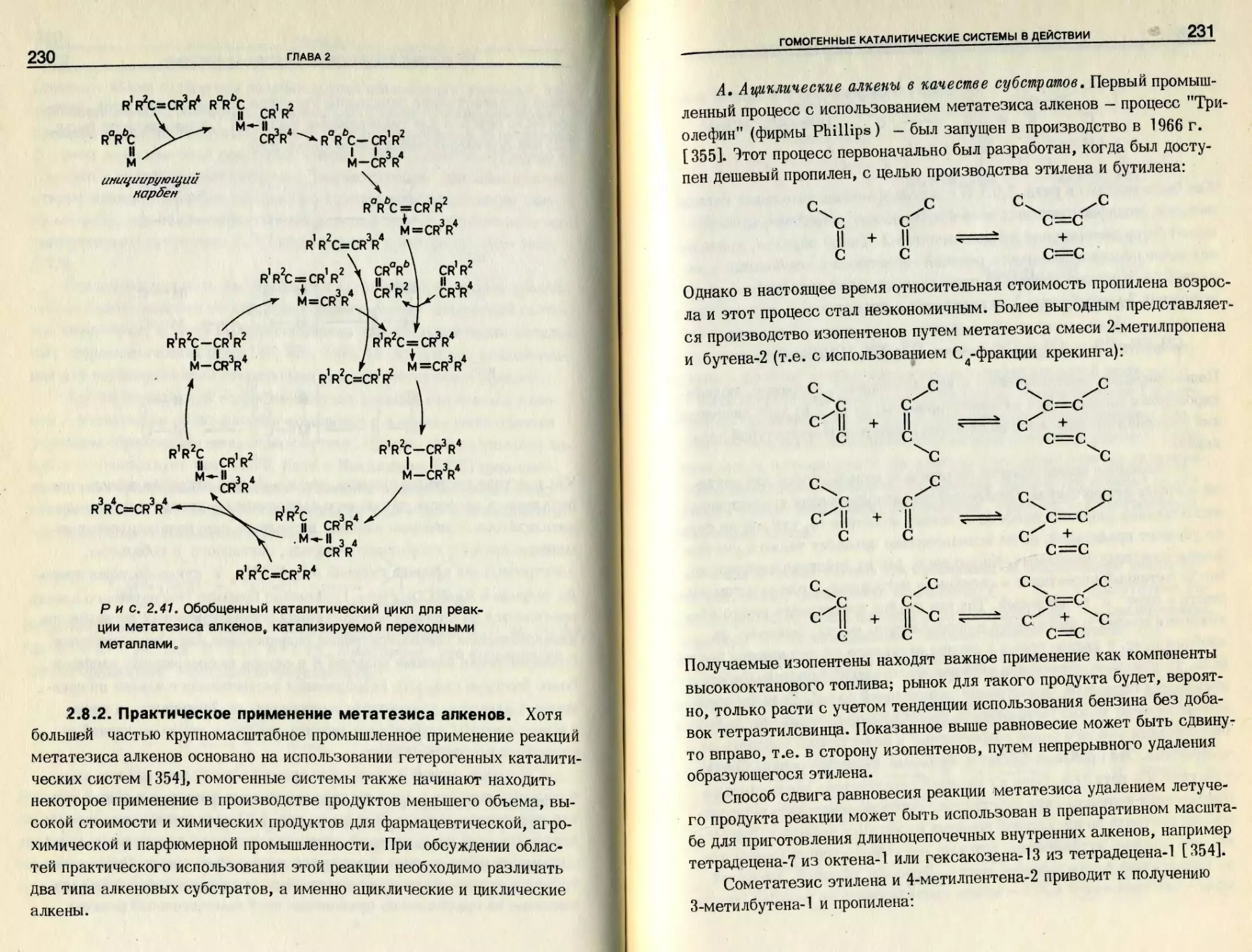
Термин "близкое взаимодействие" относится .к процессам, в кото-
рых активированные субстраты, находящиеся на каталитическом цен-
тре, взаимодействуют между собой или с внешним субстратом, обра-
зуя либо активированный интермедиат, либо продукт (продукты) ка-
талитического цикла. Можно выделить два процесса близкого взаимодействия:
1) внедрение и миграцию внутренних лигандов, 2) элиминирование.
1с5 1 Внедрение и миграция внутренних лигандов, В этом
процессе два субстрата, X и Y, присутствующие в каталитической
системе и связанные с одним или несколькими металлическими цен-
трами, взаимодействуют с образованием интермедиата XY,.который
остается связанным с металлическим центром (или центрами):
X
м------Y-----------XY (1-7)
или
X Y XY
М1-----М2---*М1------М2 (1.8)
Мы ограничиваем наше рассмотрение системами (1.7), включающими
один металлический центр, поскольку такого типа системы достаточ-
но хорошо изучены. .Системы, включающие более одного металличес-
кого центра [ например, (1.8) ], недостаточно хорошо изучены, хотя,
как это предполагается в разд. 3.3 и гл. 4, они представляют значи-
тельный интерес.
Примеры реакций внедрения и миграции внутренних .лигандов на
монометаллическом центре могут быть найдены почти во всех .ката-
литических системах; они представляют тему гл. 2. В этом разделе
для иллюстрации принципа мы описываем только один такой процесс.
’В бензольном растворе в атмосфере СО метилпентакарбонил
марганца,(1) находится в равновесии с ацетцлпентакарбонилом мар-
38
ГЛАВА 1
ганца (I) [ 43]:
Мп (Ме)(СО)5+-^* Мп(СОМе)(СО)
Повышение парциального давления СО сдвигает равновесие вправо
На этом примере мы наблюдаем внедрение СО по связи металл - мУ-
тадл. Изучение этой реакции с использованием 14 СО показало, :что
разрыв связи металл - металл происходит путем внедрения карбониль-
ной группы, связанной с марганцем, а не входящей извне, а именно
Мп(Ме)(СО)5 + 14СО —* Мп (СОМе ) (СО ) ( 14СО)
.Кинетические данные, полученные для этой реакции, .лучше всего со-
гласуются с последовательностью, показанной в уравнении (1.9 >:
R-MnfCO), =. RCO-Mn(CO)Й ПСО-Мп(СО) L (где L = СО)
(1.9)
Реакция протекает ;через 5-координированное промежуточное соедини
ние 1.8, к .которому присоединяется входящий .лиганд L. Таким об-
разом, эту реакцию внедрения более правильно рассматривать как
реакцию миграции внутреннего лиганда, в которой метильная группа
мигрирует к связанному С металлом карбонильному углероду-
Вакантное координационное место, образовавшееся в ходе реакции
может быть занято растворителем или входящим.лигандом и как ’
правило, таким реакциям миграции благоприятствует, присутствие рас-
творителя, способного к координации (например, эфира), цли подхо-
дящих лигандов (например, СО или третичного фосфина). В катали-
тических системах, где этот процесс играет важную роль (например,
в реакциях карбонилирования или гидроформилирования), входящим
лигандом обычно является СО. В стехиометрических реакциях такие
ЛИГ?’ ЛЫ, как Р(ОРЬ) и PPh R niP.mii vcr.n aj.
з и 1 %» в Равнои мере эффективны в промо-
тировании миграции алкильной группы. В ходе реакции миграции лиган-
да не происходит изменения формальной степени окисления металла
и соответственно электронные требования металла к ассоциациировк
ОБЩЕЕ РАССМОТРЕНИЕ
39
Нь1М .лигандам менее строги, :чем, например, в реакциях окислитель-
ного присоединения. Большинство J- металлов вступает в различные
типы реакций миграции внутренних .лигандов, и, как отмечалось ранее,
этот процесс, вероятно,-является одним из наиболее важных типов
реакций гомогенного :катдлиза. В дополнение к миграции алкила :к кар-
бонилу известны другие примеры, включающие миграцию гидрида к
координированному олефину в реакции каталитического гидрирования,
миграцию ацетата к олефину в реакции окисления, катализируемого
соединениями палладия, и миграцию алкила :к олефину во многих по-
лимеризационных системах. В процессе миграции алкила к карбонилу
реакция, вероятно, протекает :через трехцентровое промежуточное сос-
тояние: _
R
R
М
М
М
Миграция внутреннего .лиганда, включающая координированный алкен,
по-видимому, может протекать через четырехцентровое переходное
состояние [ 44]?:
t
СН2
М СН2
,СН2
! хсн2
м
СН2СН2Н
м
Следующим примером близкого взаимодействия, который может быть
рассмотрен-в разделе "Миграция внутренних .лигандов",-является окис-
.лительная димеризация, представленная на примере реакций типа
М
Процессы такого типа играют исключительно важную роль в систе-
мах олигомеризации и полимеризации алкенов и детально обсуждают-
ся в разд. 2.5 и 2.6.
В процессах миграции внутренних .лигандов продукт взаимодей-
ствия остается связанным с металлическим центром. В следующем
типе процессов близкого взаимодействия - элиминировании — это не
является необходимым уеловием, и элиминирование зачастую означа-
ет конец каталитического цикла.
40
ГЛАВА 1
1.5.2. Элиминирование. В этом процессе,либо два субстрата,
X и Y, присутствующие в каталитической системе, связанные с од-
ним или несколькими металлическими центрами, взаимодействуют и
образуют продукт XY,.который покидает координационную сферу ме-,
талла, .либо субстрат XY, связанный с одним или несколькими метал-
лическими центрами, претерпевает изменения, приводящие к образо-
ванию стабильного продукта, который может отделяться от металла.
Оба случая представляют конечную реакцию каталитического цикла.
Первый тип процессов элиминирования может быть определен как вос-
становительное элиминирование, и, .как указывалось ранее, он, по су-
ществу, противоположен процессу окислительного присоединения [ 18,
20, 33].
Примером восстановительного элиминирования в гомогенном катализе
•является гидрирование, в котором ^лкцлгидридный комплекс претер-
певает серию превращений, приводящих к образованию алкана:
Н
—> м*-2 + RH
Поскольку этот процесс противоположен окислительному присоединению,
то ему благоприятствуют .лиганды, понижающие электронную плотность
на металле. Однако, как правило, эта стадия не яцляется .лимитирующей
в.каталитическом процессе, другим примером восстановительного
элиминирования может быть элиминирование ацетцлиодида из комплек-
са 1.9 в реакции карбонилирования метанола, катализируемой родием
[ 45 k
1.9
[Rh,I2(CO)2]~ + СН3С^
ч
сн3он
/О
СН3С^ + HI
Х)СН3
Еше один пример — восстановительное элиминирование альдегида
в реакции гидроформилирования, катализируемой кобальтом [ 46]?:
ОБЩЕЕ РАССМОТРЕНИЕ
41
Co^CORH'COJjL +Н2 -> [ConiH2(COR)(CO)3L]
-> Co^CCHL +
2 \ н
В случае второго типа процессов элиминирования субстрат претерпе-
вает изменения, приводящие к образованию стабильного продукта.
Этот тип реакций известен как р-элиминирование, потому что во вза-
имодействие вступает обычно группа, находящаяся в субстрате в р-по-
ложении. Так, металлалкцльные комплексы превращаются в металл-
.гидридные комплексы и алкен путем переноса водорода от р-атома
углерода алкильной группы к металлу:
Н R
ХН Н CHR
М / ----- / + //
сн2 м сн2
Этот процесс особенно важен в реакции изомеризации, катализируемой
переходными металлами, а также как конечный процесс реакций поли-
меризации, катализируемых металлами. По существу, этот тип про-
цессов элиминирования является обратным реакции миграции внут-
ренних .лигандов, которая обсуждалась ранее. Может также происхо-
дить элиминирование с участием группы в «-положении субстрата,
хотя указания на этот процесс менее известны. Показано, :что такой
процесс «-элиминирования может происходить на вольфрамметиль-
ных комплексах [ 47}:
W-CH, ** ?
3 I
w==ch2
.Он может быть весьма важным для образования активных промежу*
точных соединений в реакциях диспропорционирования, катализиру-
емых вольфрамом и молибденом [ 48 ] (см. разд. 2.8).
Рассмотрев различные типы реакций, которые могут происходить
в ходе каталитического процесса, перейдем к следующему этапу —
объединению этих отдельных элементарных стадий в каталитический
цикл.
1.6. Каталитический цикл
Каталитическую систему можно рассматривать как серию реак-
ций, связанных циклическим образом так, что при прохождении одного
круга субстрат (субстраты) превращается в продукт (продукты). Что
42
ГЛАВА 1
касается катализатора, то он после прохождения цикла остается неиз-
менным. Мы можем построить каталитический цикл ,либо ретроспек-
тивно, пытаясь усовершенствовать известный каталитический процесс, ли-
бо перспективно, имея целью создание или открытие нового катализа-
тора. В обоих случаях полезным ориентиром является правило ”16 и
18 электронов”, впервые предложенное Толманом в 1972 г. [ 49].
1а6Х Правило ”16 и 18 электронов”. Правило ”16 и 18 электро-
нов” основано на наблюдении [ 50], :что в основном все хорошо оха-
рактеризованные диамагнитные комплексы элементов, расположенных
в конце с£-ряда, имеют 16 или 18 валентных электронов метадлц*. На
основе этих наблюдений Толман предложил два ’правила для метадлоор»
ганических комплексов и их реакций: 1) "Диамагнитные металлооргани-
.ческие комплексы переходных метадлов могут существовать в замет-
ных концентрациях при умеренных температурах только в том случае,
если валентные уровни метадла содержат 16 или 18 электронов. Зна-
чительной концентрацией считается концентрация, которая может быть
создана в газовой, жидкой или твердой фазе и определена спектроско-
пически или кинетически”. 2) "Реакции металлоорганических соедине-
ний, включающие каталитические реакции, протекают через элемен-
тарные стадии, содержащие интермедиаты только с 16 или 18 валент-
ными электронами".
•Следствием второго правила является то, что в последователь-
ности стадий реакции оказывается невозможной такая стадия, в .ко-
торой число валентных электронов (ч.в.э.) изменялось бы больше
чем на два, т.е. Д ч.в.э. = 0, ±2. Хотя известны некоторые исключе-
ния из этих правил (например, для Pt0 [ Р (циклогексил )3 ]2 и для
Rh*Н [ Р( циклогексил )3] 2 ч.в.э.= 14), они широко применимы ^чрез-
вычайно полезны в предсказании механизмов реакции и создании .ка-
талитических циклов. Ранее мы обсуждали миграцию метильной груп-
пы в метилпентакарбонцлмарганце, которая облегчается введением
лиганда. 'Суммарную реакцию можно записать так:
СО
Мп(Ме)(СО)5~---------* Мп(СОМе)(СО)
*При определении числа валентных электронов предполагается, что все
лиганды, ковалентно связанные с металлом, вносят два электрона.Считает-
ся, что количество электронов,вкладываемых металлом соответствуют его
формальной степени окисления, Так, RlJcHPHi ) имеет 8 +(4*2) “ 16,
а Мп (Ме)( СО )5 имеет 6 +(6*2) = 18 валентных электронов.
ОБЩЕЕ РАССМОТРЕНИЕ
43
и при отсутствии кинетических данных мы могли бы написать две воз-
можные последовательности элементарных стадий реакций:
Мп(Ме)(СО)5 — С.~—- Мп(МеНСО) -------------> Мп (Me СО) (СО
ч.в.э. 18 20 18
СО
Мп (Me) (СО)5------- Мп (МеСО) (СО )4 ——* Мп (МеСО') (СО)$
|.в.э. 18 18 18
Подсчет чидла валентных электронов, находящихся в индивидуальном
.комплексе, показывает, что вторая последовательность должна быть
более предпочтительным маршрутом; это предсказание подтверждает-
ся экспериментальными данными. Аналогично эти правила могут быть
использованы для оценки возможности осуществления предполагаемо-
го каталитического цикла. На рис. 1.6 представлен основной катали-
НСо’(СО)4
(18)
^СО
(18)
Ркс, 1.6. Каталитический цикл для гидроформилирования а-о лофинов, ка-
тализируемого комплексами кобальта.
44
ГЛАВА 1
тический цикл для реакции гидроформилирования а-олефинов, ката-
лизируемой .кобальтом. Число валентных электронов, относящихся к
каждому предполагаемому соединению, показано в скобках.
1.7. Заключение
В первом разделе мы попытались представить основные положе-
ния, позволяющие ознакомиться с некоторыми уже использующимися
в настоящее время (гл.2) или активно изучаемыми (рлв 3) гомоген-
ными каталитическими системами.
Как мы старались подчеркнуть, потенциальные возможности ком-
плексов переходных металлов как гомогенных катализаторов огром-'
ны и разнообразны. Тем не менее мы всегда должны осознавать гра-
ницы их возможностей. .Катализатор может только повлиять на про-
цесс, который прежде всего должен быть осуществимым, но он не мо-
жет заставить "реку течь вспять". Поэтому первым шагом в поиске
какого-.либо подходящего катализатора всегда должна быть проверка
термодинамической возможности осуществления процесса. На рис. 1.7
мы показали гипотетический каталитический цикл, все отдельные ста-
дии которого имеют определенную возможность экспериментального
Р и с‘ 1JQ Каталитический цикл гипотетической реакции RH + СО -> RCOH
ДС ° кДж/моль
К = СН3 56
С2Н5 39v8
С Н 46
э I
ОБЩЕЕ РАССМОТРЕНИЕ
45
осуществления [46, 51]. Ниже представлены в общем виде пред-
полагаемая реакция и изменения &G;°. Очевидно, что шансы
создать систему, катализирующую превращение смеси алкена и
СО в альдегиды, очень малы! Это краткое упоминание о термо-
динамике опять привело нас к теме, с которой мы начали этот
раздел, и это означает, что пора перейти к рассмотрению неко-
торых существующих гомогенных каталитических систем в действии.
Дополнительная литература
А. Термодинамика
Сох J.DO, Pilcher G. Thermochemistry of Organic and Organometallic
Compounds, l* London: Academic Press, 1970.
Janz GJ. Thermodynamic Properties of Organic Compounds. <— New York1:
Academi c Press, 1967.
Сталл Д., Веетрам Э., Зинке Г* Химическая термодинамика органи-
ческих соединений. — М.>: Мир, 1971.
Б. Переходные металлы и координационная химия*
Cotton FoA<,,t Wilkinson G> Advanced Inorganic Chemistry.3rd Edn.
London?: John Wiley, 1972 [ Есть русский Перевод раннего изда-
ния: Коттон Ф.А., Уилкинсон Дж. .Современная неорганическая
химия, т. 1, 2, 3. ~М.?: Мир, 1969].
Барнард А.К. Теоретические основы неорганической химии: Пер. с
англ. ~Му: Мир, 1968.,
Coates G°Ea, Green M.LoHe, Wade К. Organometallic Compounds Volume
Two?: The Transition Elements,by ML.IL ‘Green, 3rd End. London:
Chapman and Hall, 1968,
Heck RaF- Organotransition Metal Chemistry, A Mechanistic Approach,
New York?: Academic Press, 1974,
Pidcock Ao, McAuliffe C.A. Transition Metal Complexes of Phosphorus,
Arsenic and Antimony Ligands. — New York?: Wiley, 1973.
В. Гомогенный катализ
Schrauzer G.N (ed. ) Transitipn_Metals in Homogeneous Catalysis.
New York?: Marsel Dekker, 1971.
t/go R (edo) Aspects of Homogeneous Catalysis, vv 1, 29 3, — Dordrecht:
* См Также to ₽Грин.Металлоорганические соединения переходных
элементов. — М^: Мир, 1972. — Прим0 перев^
46
ГЛАВА 1
Reidel, 1970, 19^4, 1977. [:Есть русский перевод т. 1?: У го Р. Ас-
пекты гомогенного катализа. -М,»:.Мир, 1973].
Tsuji J. Organic Synthesis by Means of Transition Metal Complexes. —
Berlin?: Springer- Verlag, 1975.
Alpет Ц (ed.) Transition Metal Organometallic in Organic Synthesis, v
1,2 — New York»: Academic Press, 1976, 1978.
Catalysis, Chem. Socc Specialist Periodical Report, v. 1,2, — London»:
Chemical Society, 1977, 1978»
Parshall G IT. Industrial applications of homogeneous catalysis. — J. Mol
Catal, 1978, v. 4, 243.
Kochi J.K. Organometallic Mechanism and Catalysis. — New York»: Aca-
demic Press, 1978.
2. Гомогенные каталитические системы
в действии
В последние тридцать лет наблюдался неуклонный рост использо-
вания в химической промышленности гомогенных каталитических сис-
тем. Хотя процессы, в которых используются гетерогенные катализа-
торы, еще преобладают над процессами с участием гомогенных ката-
лизаторов, последние вносят существенный вклад в общий объем хими-
ческого производства,. По недавней оценке [52] в настоящее время
имеется более двадцати промышленных процессов, в которых в качес-
тве катализатора используются растворимые соединения металлов;
в 1975 г. только в США с применением таких процессов было произве-
дено 8 млн, т химических соединении. Некоторые соединения, в основ-
ном альдегиды, производятся почти исключительно с помощью гомо-
генных катализаторов,, Из 613 000 т ацетальдегида, потребленного в
США п 1976 г., свыше 90% было получено путем окисления этилена в
присутствии палладиевого катализатора [ 53 ]0 В западных странах
ежг "одно производится с использованием растворимых металлкарбо-
нильных катализаторов от 3,5 до 4,5 млн.т альдегидов или их произ-
водных [ 1 ]в Кроме того,- в США три крупнейших процесса производ-
ства уксусной кислоты, продукция которых, должнаг'была превысить
к 1980 г. 1,5 млНоТ (в США), основаны на гомогенном катализе [541
В разработанном недавно процессе получения уксусной кислоты кар-
бонилированием метанола используется гомогенный родиевый катали-
затор0 Разработчик этого процесса, фирма Monsanto, сообщает, что
этот процесс запатентован во всех странах, и предполагает, что пос-
ле ввода заводов в строй они будут способны ежегодно производить
более 1 млНоТ уксусной кислоты [ 451 Успехи в получении двух основ-
ных термопластичных материалов — полиэтилена и полипропилена -
обязаны тому, что в начале 1950-х годов Циглер нашел, что смесь три»
этилалюминия и тетрахлорида титана является активным катализато-
ром полимеризации этилена, В 1976 го мировой сбыт полипропилена
превысил 3 млн,тв, причем полимер целиком был получен с использо-
ванием модифицированных катализаторов Циглера (или катализаторов
типа Циглера - Натта), Хотя остается открытым вопрос о гомогенное-
48
ГЛАВА 2
ти этих полимеризационных систем*, нет никаких сомнений в том,
что изучение растворимых комплексов сыграло важную роль в раз-
витии современного поколения промышленных катализаторов [53 ].
Эти примеры служат для иллюстрации масштаба процессов, в ко-
торых гомогенные катализаторы играют жизненно важную роль» Не
менее важным по значению (хотя значительно меньшим по масштабам
применения) является использование гомогенных катализаторов для
осуществления высокоспецифичны к превращений органических соеди-
нений» Растворимые системы на основе никеля катализируют олиго-
меризацию олефинов, давая широкий набор линейных и циклических
продуктов» Модификация никелевого комплекса добавлением соответ-
ствующих лигандов приводит к высокоселективным каталитическим
системам для получения химических соединений, которые могут заин-
тересовать фармацевтическую промышленность [ 551 1-Дигидрокси-
фенилаланин (1-ДОФА) является одним из лекарств, использующихся
при лечении болезни Паркинсона» Ключевая стадия в его синтезе —
асимметрическое гидрирование ацетамида коричной кислоты — может
быть эффективно осуществлена при использовании растворимого ро-
диевого катализатора, имеющего оптически активные фосфиновые ли-
ганды [56, 57 L В этом разделе мы рассмотрим восемь основных об-
ластей, в которых гомогенные каталитические системы оказались наи-
более эффективными» Из них, за исключением гидрирования, изоме-
ризации и метатезиса, пять областей использования гомогенных ка-
талитических систем уже нашли промышленное применение, причем
мощность большинства действующих промышленных предприятий пре-
вышает 10 000 т продукции в год» Гидрирование и изомеризация
включены в рассмотрение постольку, поскольку это два наиболее тща-
тельно изученных процесса с использованием гомогенных катализа-
торов; кроме того, эти реакции имеют существенное значение в боль-
шинстве других рассматриваемых областей» Метатезис включен пото-
му, что это одна из наиболее интригующих каталитических реакций,
открытых в последнее время; эта реакция является основой для важ-
ного нового процесса производства С12 _ С18-а-олефинов (однако при
этом применяются- гетерогенные катализаторы)» Кроме того, хотя эти
конкретные гомогенные каталитические системы еще не могут быть
использованы в крупномасштабном химическом производстве, в ряде
*
Все производство полипропилена основано на применении гетерогенного
катализатора (фиолетовая модификация TiCl3). — Прим, перев.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
49
случаев они, как это показано на примере получения 1-ДОФА, имеют
большое значение в маломасштабном производстве специфичных со-
единений, представляющих интерес для фармакологической и агрохи-
мической промышленности»
2.1. Гидрирование
Гидрирование, твев присоединение водорода (Н2) к ненасыщенному
соединению, например олефину, является одной из наиболее глубоко
изученных гомогенных каталитических реакций. После того как в
1939 г. Игути открыл, что некоторые комплексы родия(Ш) катализи-
руют восстановление водородом органических соединений, таких, как
фумаровая кислота [58], были опубликованы данные [ 19] о получении
гомогенных катализаторов гидрирования с использованием практи-
чески всех J-металлов. Основное внимание уделялось элементам VIII
группы, поскольку при их применении получаются наиболее активные
каталитические системы. Мы будем в основном касаться комплексов
именно с этими металлами. Гомогенные катализаторы гидрирования
демонстрируют многие черты, которые мы обсуждали в гл» 1: ненасы-
щенный субстрат обычно активируется путем координации, а водород -
путем присоединения (известны системы, включающие окислительное,
гомолитическое и гетеролитическое присоединение Н2); путем тща-
тельного выбора каталитической системы возможно достичь весьма
высокой селективности, причем многие катализаторы активны в мяг-
ких условиях. В последующем обсуждении сделана попытка рассмот-
реть примеры каталитических систем для иллюстрации этих различных
аспектов.
В разд. "Простое гидрирование", посвященном гидрированию оле-
финовых соединений, описаны гомогенные катализаторы гидрирова-
ния, являющиеся примером трех типов процессов активации водорода.
В разд. "Селективное гидрирование" обсуждаются системы, способ-
ные селективно восстанавливать определенный тип ненасыщенных
связей или одну ненасыщенную связь в ди- или полиненасыщенной моле-
куле, и, наконец, в разд. "Асимметрическое гидрирование" мы рассмат-
риваем катализаторы, способные обеспечивать получение оптически
активных молекул путем стереоспецифического присоединения водо-
рода к оптически неактивному субстрату.
Для глубокого ознакомления со всей областью гомогенного гид-
рирования, катализируемого переходными металлами, читателю ре-
комендуются отличные работы Джеймса [19, 591
50
ГЛАВА 2
2.1.1. Простое гидрирование. А. Активация водорода путем
окислительного присоединения* В 1956 г0 две группы исследователей
[60, 61], работающие независимо, сообщили о том, что в органичес-
ком растворителе трис(трифенилфосфин)хлорородий(1) RhCl(PPh3)3
является активным катализатором гидрирования алкенов и алкинов
при 25°С и 1 атм газообразного водородао Вслед за этим было затра-
чено много усилий для того, чтобы охарактеризовать эту систему.
Принятый в настоящее время [59] каталитический цикл показан на
рис. 2.L Внутренний круг представляет собой главный каталитичес-
кий цикл. Оказалось, что каталитически активные соединения содер-
жат два третичных фосфиновых лиганда, но до сих пор остается от-
крытым вопрос, как такие соединения образуются из трифосфинового
комплекса 2Л. Первоначальное предположение [62] о том, что 2Л
быстро диссоциирует в растворе, образуя сольватное соединение 2.2,
было до некоторой степени подвергнуто сомнению, когда с использо-
ванием измерений молекулярной массы и ЯМР-31Р было показано, что
в бензольном растворе в отсутствие кислорода 2Л практически не
диссоциирует (К^ 3 • 10-3М)0 Это полностью соответствует правилу
Pte. 2.1. Каталитический цикл для гидрирования олефинов в присутствии
RhClL3(L«PPh3)u S — растворитель или слабосвязанный третичный фосфин.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
51
Толмана ”16 - 18 электронов” (см. разд. 1.6) в том отношении, что
предполагаемое равновесие подразумевает переход 16-электронного
соединения в 14-электронное:
Rh1 C1L, = "Rh1 C1L " + L
-J
ч.в.э. 16 14
В присутствии подходящего растворителя, способного к координации,
например, EtOH (при наличии примесей О ), это осложнение может
быть преодолено, если протекают следующие процессы:
Rh1 C1L„ + S ~= Rh1 C1L S =*= Rh1 C1L,S + L
2.2
ч.в.э. 16 18 16
В этом смысле примечательно, что скорость гидрирования циклогек-
сена в присутствии катализатора RhCl(PPh3)3 в смеси бензола с эта-
нолом (3:1) примерно в два раза выше скорости реакции в чистом бен-
золе [65, 66]. Кроме того, на практике трудно полностью исключить
кислород. Окислительное присоединение водорода к сольватированно-
му соединению 2.2 приводит к образованию дигидридного комплекса
родия(Ш) 2.3. Данные ЯМР1!! и 31Р показали, что два гидридных ли-
ганда находятся в г^с-положении между собой и в цис-положении к
третичным фосфиновым лигандам [ 671 Альтернативный путь к соль-
ватированным дигидросоединениям идет через последовательность
Rh1 C1L + Н, = Rhin CIH L„ —L RhII[ C1H.L, 2 Rh111 C1H.L.S
О « О — £ £ L 2
+L -S
ч.в.э. 16 18 16 18
Эта последовательность изображена в верхней половине внешнего ка-
талитического цикла на рис. 2.1, Однако несомненно, что формально
октаэдрические цис-дигидридные соединения родия(Ш) активно вклю-
чаются в каталитический цикл, Гидридные лиганды обладают высоким
тряис-эффектом (см. разд. 2.1.3,А;таким образом, ягрбшс-лиганд как
в 2.3, так и в 2Л лабилизирован и легко замещается олефиновой
группой субстрата, образуя 2.5, в котором все реагенты, необходимые
для завершения цикла, собираются вместе в ’’активированной” форме.
Последует ли реакция по внешнему или внутреннему правому полук-
ругу (см. рис, 2.1), будет до некоторой степени зависеть от природы
52
ГЛАВА 2
каталитического предшественника, тоео от комплекса родия, введен-
ного первоначально в раствор»
Обычно ди-трг^-фосфиновый комплекс родия(1) образуется при
добавлении четырех мольных эквивалентов третичного фосфина (или
два эквивалента на один родий) к легко получаемому димерному хло-
ридному комплексу родия(1) , содержащему лабильные олефиновые ли-
\ ганды, например [RhCl(C2H4)2 ]2 или [RbCl(C8H14)2 ]2. В случае та-
кого комплекса предпочтительным является внутренний цикл. При ис-
пользовании Ш1С1Ц предварительная активация, вероятно, протека-
ет по внешнему циклу. Следующей стадией в каталитическом цикле
является ’’близкое взаимодействие”, включающее миграцию гидрида
к олефину в 2.5 с образованием гидридоалкильного соединения ро-
дия(Ш)в Такая миграция, протекающая через четырехцентровое состо-
яние, легче всего может проходить, когда гидрид и алкен находятся
в ^-положении [ 68 ] друг к другу и лежат в одной плоскости [ 69 ],
Таким образом, начиная с 2.5, мы будем иметь последовательность
Как в 2.7, так и в сольватированных частицах 2.8 оставшиеся гидрид-
ная и алкильная группы находятся взаимно в ??фос-положении, и преж-
де чем может осуществиться завершающая стадия цикла, они должны
переместиться в ^с-положение, Идет ли эта изомеризация через 2.8
или прямо через 2.7, неясно, так как образующиеся в результате ал-
килгидцилные частицы 2.6 быстро претерпевают реакцию восстанови-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
53
тельного элиминирования, давая алкан в качестве продукта и завершая ка-
талитический цикл сольватированным комплексом родия(1 )2.2.
Считается, что лимитирующей стадией превращения 2.5 в 2.6 являет-
ся реакция миграции гидрида к олефину [70 L Как показано в верхнем
левом углу рис» 2.1, сольватированное ди-^ргт-фосфиновое соедине-
ние 2.2 может также димеризоваться с образованием хлорсвязанного
диродиевого комплекса 2.9, который быстро может присоединять водо-
род, образуя 2.10. Формально это дигидридное соединение содержит
родий в состоянии окисления (I) и (III). Разрыв мостика растворителем
2.10
превращает 2.10 в смесь 2.2 и 2.3', это объясняет тот наблюдаемый
факт, что 2.10 является также активным предшественником катализа-
тора. Концентрация димерных соединений в данной системе зависит
от конкретных условий реакции, т.е. от природы растворителя и типа
фосфинового лиганда. Тем не менее обычно в реальных условиях их
концентрация мала.
Как упоминалось выше, когда L = РРЬ3, считается, что лимити-
рующая стадия при переходе от дигидроолефинового комплекса 2.5
к гидридоалкильному комплексу 2.6 включает миграцию лиганда, т.е.
миграцию гидрида к олефину. Такая миграция может рассматриваться
как нуклеофильная атака гидридного лиганда по активированной двой-
ной связи [44]. Тогда, если пренебречь стерическими факторами, ли-
ганды, увеличивающие электронную плотность на гидридном лиганде
и уменьшающие ее на олефиновой двойной связи, будут увеличивать
скорость реакции. Так как оба лиганда - гидридный и олефиновый -
присутствуют в одном и том же комплексе, то подбор лиганда, кото-
рый осуществлял бы два противоположных эффекта, является чрез-
вычайно трудной или вообще невыполнимой задачей. Результаты изу-
чения комплексов с замещенными арилфосфиновыми лигандами пока-
зали, что увеличение электронной плотности на металле увеличива-
ет скорость гидрирования. Так, скорость гидрирования циклогексена
в присутствии катализатора RhClL3> где L = Р(п-метоксифенил) в
два раза выше, чем в присутствии катализатора, в котором L = PPh3.
Замещение PPh на Р(я-фторфенил) уменьшает скорость этой реакции
о о
54 ГЛАВА 2
в пять раз [71]. Аналогично замещение хлоридного лиганда на бро-
мидный или иодидный увеличивает общую скорость реакции. Скорость
каталитического гидрирования циклогексена в бензол в присутствии
RhX(PPh3)3 увеличивается в отношении примерно 1:2:3 при переходе
от Х = С1 к Х= Вг и I [ 62 ]. Эти результаты показывают, что в
данной частной реакции миграции лиганда увеличение электронной
плотности на металлическом центре, т.е. эффективное возрастание
гидрид но го характера лиганда Н, является основным фактором уско-
рения реакции. Однако таким путем невозможно достаточно сильно
увеличить каталитическую активность системы, поскольку аналогичные
комплексы с алкиларилфосфиновыми (типа PRnAr3 _ п) или с триалкил-
фосфиновыми лигандами, которые, как и следует ожидать, оба увели-
чивают электронную плотность на металлическом центре, имеют очень
низкую каталитическую активность [71, 72]. Это выдвигает на первый
план одну из проблем, возникающих при попытке модифицировать ка-
тализатор, который принимает участие в сложном каталитическом цик-
ле (как показано на рис. 2.1): отлаживая одну часть этого цикла, вы
часто приводите В беспорядок другие его части. В этом случае увели-
чение основности третичного фосфинового лиганда увеличивает ста-
бильность комплексов типа 2Л до такой степени, что они более не при-
нимают участия в каталитическом цикле. Так, RhClH2(PEtPh2)3, 2Л
с L = PEtPh2, является стабильным легко выделяемым комплексом
[711 При условиях, когда 2Л не может образовываться, например
когда отношение Rh:L = 1:2, образуется димерное соединение 2.9, и,
если L = PR3 или PRnAr3 _ п(я > 1), эти соединения показывают не-
значительную каталитическую активность. Таким образом, в данном
случае, как и в большинстве систем, которые мы будем рассматривать,
часто наученные горьким оптытом, мы понимаем, что к модифициро-
ванию каталитической активности следует подходить очень осторожно.
Рассматривая соединение 2.5, в котором L - триарил фосфин, мож-
но видеть, что этот комплекс стерически сильно стеснен (конический
угол PPh3 составляет 145°, см. разд. 1.2.3,Б).Таким образом, сле-
дует ожидать, что эта реакция является очень чувствительной к сте-
рическим параметрам. Это и наблюдается в действительности; так, нап-
ример, увеличение стерических препятствий при связи С=^С сущест-
венно уменьшает скорость ее гидрирования. Например, 1-метилцик-
логексен гидрируется в 50 раз медленнее, чем циклогексен, транс-оле-
фины восстанавливаются медленнее, чем цнс- олефины, и олефины с
внутренней двойной связью восстанавливаются намного медленнее, чем
олефины с концевой двойной связью [71, 73]. Аналогично увеличение
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
55
терических размеров триарилфэсфина уменьшает активность ката-
лизатора в гидрировании. Система, в которой L - три-о-толилфосфин (коничес-
кий угол 194°), в 700 раз менее активна, чем cncTeMaj в которой L -
три-ft толилфосфин (конический угол |45°) [19, 261 Мы затронули
вопрос о важности влияния стерических параметров олефинового суб-
страта. Электронные параметры также играют важную роль; замести-
тели, оттягивающие электроны с двойной связи, обычно увеличивают
скорость гидрирования, если их не слишком много и они не очень велики.
Так, акрилонитрил и аллилацетат восстанавливаются несколько быст-
рее (примерно на 30%), чем незамещенные а-олефины, такие, как гек-
сен-1 [74]. Этот электронный эффект объясняется тем, что уменьше-
ние электронной плотности на двойной связи делает ее более чувст-
вительной к нуклеофильной атаке в лимитирующей стадии реакции миг-
рации гидрида к олефину.
Важное преимущество системы на основе RhCl(PPh3)3 состоит
в том, что в мягких условиях, обычно при комнатной температуре,
в бензольном или бензольно-этанольном растворе при атмосферном
давлении водорода эта система не катализирует восстановление дру-
гих функциональных групп, находящихся в олефиновом субстрате. Та-
кие группы, как кето-, циано-, нитро-арильная, карбоксильная и дру-
гие обычно встречающиеся органические функциональные группы,
не затрагиваются при гидрировании каталитической системой
RhCl(PPh3)3/H2. Этот факт вместе с отмеченными ранее стерически-
ми ограничениями делает RhCl(PPh3)3 исключительно селективным
катализатором, имеющим большие возможности в синтезе сложных
органических молекул. Тем не менее, прежде чем перейти к более
подробному обсуждению этих возможностей в разд. 2.1.2, мы должны
рассмотреть второй тип процесса активации водорода, а именно гомо-
литическое присоединение.
Б .Активация водорода путем гомолитического присоединения.
Впервые об этом типе процесса активации на примере пентацианида
кобальта сообщил Игути в 1942 г. Водный или водно-спиртовой (МеОН
или EtOH) раствор цианида кобальта при обычных условиях легко
поглощает водород, образуя соединения, способные катализировать
гидрирование активированных алкенов типа
z
где А - это СН-СН2,—СО.,||,^СО- , - CN, — C(O)NH —C(O)R,
56
ГЛАВА 2
(
арил, a-C5H4N [ 17 ]. По отношению к органическим субстратам эта
система высокоселективна в том смысле, что совершенно не реаги-
рует с простыми моноолефинами. Так, сопряженные диены могут-быть
селективно восстановлены до моноенов, например бутадиен до бутена»
Основным соединением, отвечающим за катализ, является анион
пентацианогидрида кобальта, который образуется путем гомолитичес-
кого расщепления водорода:
2Co(CN)3~ + Н9->2HCo(CN)3~ (2.1)
2Л1
Использование правила "16 — 18 электронов” в том виде, как оно
применялось для диамагнитных комплексов, для этой частной систе-
мы ограничено, поскольку Con(CN)|- является d7, 17-электронным
парамагнитным ионом.
Образование гидридокомплекса 2.11 из цианида кобальта и водо-
рода изучалось целым рядом исследовательских групп [ 79], и было
предложено два механизма, включающих димерные соединения типа
[Со1’ (CN)10]6- и [(CN)5CoH2Co(CN)5 ]6~ [36, 77]. Однако в боль-
шинстве работ [ 19] отдается предпочтение простой межмолекулярной
реакции типа (2.1). Поскольку координированный Н условно принимает-
ся за Н“, степень окисления кобальта в 2Л1 составляет три. Однако
на основании полярографических исследований было предположено
[78, 79], что более правильно полагать, что кобальт в 2.11 присутст-
вует в степени окисления два, а водород присутствует как стабилизи-
рованный атом. Это означает, что 2.11 следует представлять как
[Со11 (CN)5( -Н) ]3“, а не как [Со111 (CN)5(: Н) ]3~ Именно это пред-
ставление соответствовало бы описанию такого процесса активации,
как "гомолитическое” присоединение. Но, как будет ясно при рассмот-
рении каталитического цикла, более важно то, что это представление
хорошо согласуется с природой реакций, протекающих с участием
HCo(CN)3~. Проблема ’’действительной" и "формальной" степени
окисления часто возникает в металлоорганической химии переходных
металлов. В большинстве случаев оправданием выбора формальных
степеней окисления является достаточная схематичность их исполь-
зования. Тем не менее в данном конкретном случае, вероятно, лучше
отказаться от формализма и рассматривать 2.11 как [Со11 (CN)5(-И) J3
Для того чтобы в дальнейшем избежать путаницы с ранее предложен-
ным формальным определением гомолитического присоединения (разд.
1.4.2,Б), мы в последующем обсуждении воздержимся от написания
степени окисления кобальта
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
57
Co(CNL3’
Рис. 2.2. Каталитический цикл для гидрирования активированных олефинов
в присутствии Co(CN)3~ (А = СН=*=СН9, СО~ C09H, CN, Ph, а- С -H4N).
о Zi Zi D “
Каталитический цикл для гидрирования активированных олефинов,
катализируемых [Co(CN)5H]3~ , показан на рисв 2.2, Можно пред-
ставить, что начальное взаимодействие 2Л1 с активированным олефи-
ном происходит через четырехцентровое переходное состояние 2.12,
в котором атом водорода перемещается к активированному группой
А фатому углерода, Изучение дейтерирования [77, 80] вмэсте с вы-
делением кобальторганических комплексов типа 2.13, где А - PECO,
°C“C5H4N, подтвердило направление присоединения [80 - 821 Ката-
литический цикл, которому теперь будет следовать система, зависит
от природы активированного олефина. Сопряженные диены и полиены
следуют циклу (I), тогда как большинство других активированных суб-
стратов гидрируется по циклу (II) [83]. Ниже мы рассматриваем гид-
рирование двух характерных субстратов - бутадиена и коричной кис-
лоты.
1) Гидрирование по циклу (I) на примере бутадиена. При взаимо-
действии с сопряженными диенами, такими, как бутадиен-1,3, алкил-
кобальтовое соединение типа 2.13 формально образуется путем цис-
58
ГЛАВА 2
CoH (CN)3’
4 '8
/CH3
НС —Co(CN)3
\
CH 2.14
II
ch2
CH3
CoH(CN)s» '
"сн
II
сн2
бутен ~1
2Co(CN)3
Э
КС
J
CH
нс£—Co(CN)*' +
CH 2.15
2
CN
нэс
^сн
- CoH(CN)s3‘ н/ .
СН3
транс - бутен-2
2Co(CNL
W
СН
нс*'
ХСН2—Co(CN)53’ CoH(CN)53,
2.15
2Co(CN)3'
5
CK
/CH2
CH
^CH2
бутен -1
Рис. 2.3. тт-а-равновесие в ходе гидрирования бутадиена-1,3, катализируемо-
го CoH'CN)3"
ь
присоединения гидрида кобальта к активированной двойной связив
Природа моноолефинового продукта, образующегося по циклу (I), за-
висит от концентрации свободного цианида, присутствующего в реак-
ционной среде» В случае бутадиена, когда гидрирование проводится в
присутствии избытка цианида, т.е. когда отношение цианидгкобальт
выше чем 5:1, основным продуктом ( ~90%) является бутен-L В при-
сутствии небольшого количества свободного цианида, т.е. когда отно-
шение цианид:кобальт меньше 5, основным продуктом (~80%) является
траис-бутен-2. В обоих случаях образуется лишь небольшое количест-
во (^5%) г^е-бутена-2 [ 84 ]< Эту зависимость продуктов от концентра-
ции цианида можно объяснить, используя последовательность, пока-
занную на рис. 2.3 [77, 82, 84]. Присоединение CoH(CN)|~ к бутади-
ену через 2.12 (А = —СН=СН2) приводит к образованию сг-комплек-
с&2-14 (2.13,А=—СН=«СН2). Потеря цианида комплексом 2.14 создает
вакантное координационное место в октаэдрическом комплексе, коор-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
59
дотированная двойная связь начинает взаимодействовать с этим ва-
кантным координационным местом, и фрагмент С занимающий одно
координационное место, превращается из о-лиганда в.тг-связывающий
лиганд, занимающий два координационных места в комплексе 2.15.
Метильная группа в этом комплексе направлена в противоположную
сторону от кобальта, т.е, находится в смм-положении. Этот тип а- тг-
превращений, широко распространенный среди комплексов металлов
VIII группы, содержащих аллильную группу (мы можем рассматривать
С4-фрагмент в комплексе 2Л5 как метилзамещенный аллил), обсуж-
дается в разд, 2,2 при рассмотрении изомеризации. Присоединение #
CN'k 2.15 может привести к образованию либо снова 2.14, либо 2.16,
в котором кобальт теперь о-связан с С-4, Хотя детальный механизм,
по которому CoH(CN)3- взаимодействует с о-и и тг-соединениями
2.14 и 2.16, неизвестен, подробные • спектроскопическое [82] и ки-
нетическое [80, 85] исследования показали, что ^рос-бутен-2 пред-
почтительно образуется из тт-аллильного комплекса 2.15 а бутен-1 —
из о-комплекса 2.16. В реакционных условиях в обоих случаях присут-
ствует лишь небольшое количество 2.14, но именно этот комплекс,
вероятно, и приводит к образованию бутена-1, Очевидно, что низкая
концентрация цианида будет благоприятствовать образованию и, что
очень важно для распределения продуктов, присутствию заметных ко-
личеств 2.15. Это согласуется с тем, что при отношении CN° Со ме-
нее 5:1 в основном образуется яфбше-бутен-2, Эта система высокосе-
лективна в восстановлении бутадиена, поскольку дальнейшего гидри-
рования или изомеризации получающихся бутенов не происходит,
В рассмотренном выше случае все предполагаемые интермедиа-
ты — кобальторганические соединения, в которых субстрат ковалент-
но связан с кобальтом. Для кобальтцианидного катализатора гидриро-
вания это справедливо только в том случае, когда субстратом явля-
ется сопряженный полиен или диен. Как отмечалось ранее, в случае
любых других активированных субстратов интермедиатами являются
свободные радикалы, как это показано в цикле II (рис. 2,2).
2) Гидрирование по циклу II на примере коричной кислоты
(транс-1-фенил-2-карбоксилэтилен). В катализируемой кобальтом реак-
ции гидрирования коричной кислоты в £-фенилпропионовую кислоту
первоначальный перенос атома водорода, вероятно, происходит через
четырехцентровое переходное состояние типа 2.17, 2.12 (где А - кар-
боксил, см. рис, 2.2), давая фенилкарбоксильный радикал 2.18. Отрыв
или перенос водорода от второй молекулы CoH(CN)^~ приводит к
образованию продукта [уравнение (2.2) ]:
60
ГЛАВА 2
CoH(CN)53“ + PhCH = CHCO2"
H---Co(CN)5
2.17
PhCH2CHCO2~ + Co(CN)53“
2.18
PhCH2CHCO2" + CoH(CN)53'-----► PhCH2CH2CO2~ + Co(CN)53
(2.2)
Изучение кинетики [80, 87 ] вместе с экспериментами по захвату ра-
дикалов на этой и подобных системах дали достаточное количество
доказательств этого радикального маршрута. Радикальный интерме-
диат 2.18 может образовывать кобальторганический комплекс типа
2.19. Такие соединения непосредственно не участвуют в каталитичес-
РЬСН.СИСОт
Co(CN)3-
ь
2Л9
ком цикле; действительно, субстраты типа анионов акриловой, мале-
иновой и фумаровой кислот, которые могут образовывать стабиль-
ные о-комплексы, эффективно связывают кобальт и ингибируют вос-
становление [ 81 ].
В отличие от родиевой системы, упоминавшейся при рассмотре-
нии активации водорода путем окислительного присоединения, свойст-
ва кобальтцианидного катализатора гидрирования не удается так лег-
ко изменить путем модификации лигандами. Однако этот его возмож-
ный недостаток сторицей окунается высокой селективностью по отно-
шению к активированным олефинам.
Третий тип процесса активации водорода, т.е. активация путем
гетеролитического присоединения, рассмотрен на примере поведения
некоторых каталитических систем на основе рутения(П).
В. Активация водорода путем гетеролитического присоединения. Вод-
ные солянокислые растворы хлорида рутения(П) катализируют гидри-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
61
Рис. 2.4. Каталитический цикл для гидрирования активированных олефинов
в водных растворах соляной кислоты, катализируемого комплексами руте-
ния(П).
рование а, [3-ненасыщенных карбоновых кислот или амидов, например
малеиновой, фумаровой, акриловой кислот или акриламида, однако
они неактивны в восстановлении простых олефинов и большинства
других органических субстратов [ 88 ]. Несмотря на то что еще не яс-
на природа хлоридного комплекса рутения(И), активного в каталити-
ческом цикле, вполне разумно предположить, что активация водоро-
да происходит путем гетеролитического присоединения к олефиновому
комплексу рутения(П)в1 Каталитический цикл, согласующийся с извест-
ными в настоящее время экспериментальными данными, показан на
рис. 2.4. На этом рисунке не делается никаких попыток точно уста-
новить природу соединений рутения(П). Присутствие заместителя, силь-
но оттягивающего электронную плотность, например —СОЭН или
—CONH2, необходимо для активации алкена. Незамещенные олефины,
такие, как этилен или пропилен, в водной соляной кислоте легко обра-
зуют комплексы 2.20 с рутением(П)в Однако эти комплексы относитель-
но стабильны и не гидрируются [19].-Предполагается, что стадия II
в каталитическом цикле обратима и что миграция лиганда (Н к олефи-
ну) в стадиях III и IV может происходить только тогда, когда олефин
сильно активирован для нуклеофильной атаки. В этой системе, в про-
тивоположность рассмотренной ранее кобальтовой системе, очень ве-
62
ГЛАВА 2
роятно, что лиганд Н реагирует как Н“. Изучение дейтерирования по-
казано, что присоединение Н2 к олефину происходит стереоспецифи-
чески в ^с-положение, Это указывает на то, что электрофильная ата-
ка Н+ рутенийалкильного соединения происходит с сохранением кон-
фигурации углеродного атома, связанного с атомом металла.
К сожалению, из-за экспериментальных сложностей, возникающих
при работе с этой специфической системой, оказалось трудно охарак-
теризовать большинство индивидуальных промежуточных соединений,
и поэтому подробности каталитического цикла остаются в значитель-
ной мере невыясненными. Другой системой на основе рутения(П), в
которой существенную роль играет гетеролитическое присоединение
водорода, является система, образующаяся при реакции комплекса
рутения(П) типа RuCl2(PPh3)n (п = 3 или 4)с водородом при комнатной
температуре в бензольно-этанольном раствэре [40]. Образующийся
гидридокомплекс RuCl(H) (PPh3)3 является одним из наиболее актив-
ных из до сих пор найденных гомогенных катализаторов гидрирования
алкенов-1. Он также катализирует восстановление нитросоединений в
амины [89 - 91 ]„и альдегидов в спирты [92]. Несмотря на то что эта
система по сравнению с системой RuC12/HC1/H2O намного легче под-
дается изучению, например, методом ЯИР-1!! и 31Р, детали каталити-
ческого цикла •• участием RuCl(H) (PPh3)3 остаются до некоторой
степени еще невыясненными. Цикл, представленный на рис. 2.5, слу-
Рис. 2.5. Каталитический цикл для гидрирования олефинов в присутствии
Ru11 Cl'HHPPh )QU
О О
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
63
жит просто для объяснения известных в настоящее время данных.
Полагают, что активным соединением является комплекс гид-
ридохлоротрис(трифенилфосфин)рутений(П) 2.22, образующийся путем
гетеролитического присоединения водорода к RuCl2(PPh„) :
Ru** Cl, (PPh,)„ + H„ -> Ru11 Cl(H)(PPh )q + H+ + Cl-
Z о о Z о о
2.21
2.22
Формально в этом процессе не происходит изменения ни координаци-
онного числа, ни степени окисления, и как 2.21, так и 2.22 остаются
пентакоординированными ^-соединениями, имеющими 16 валентных
электронов. Однако, как отмечалось в разд. 1.4.2,В, зачастую оказы-
вается чрезвычайно трудно экспериментально разграничить гетероли-
тическое присоединение и окислительное присоединение с последую-
щим восстановительным элиминированием. Комплекс 2.22 мог бы с
таким же успехом образовываться по реакции
Ru11 Cl (PPh,)_ + Н - RuIVCl (H)2(PPh ) Ru11 Cl(H) (PPL )
Z о о Z Z Z О о о о
+ Н+ + С1-
В обоих случаях реакция должна ускоряться в присутствии основа-
ния (например, триэтил амина), способного связывать ПСЕ Действи-
тельно, оказалось, что независимо от способа получения комплекс
2.22 играет важную роль в каталитическом процессе. Вначале предпо-
лагалось, что до координации с олефиновым субстратом 2.22 теряет
третичный фосфиновый лиганд, образуя 14-электронное соединение
"RuCl(H)(PPh )2". Это 14-электронное соединение до сих пор еще не
было выделено [93, 94] и непосредственно не наблюдалось спектрос-
копически. И поскольку спектрофотометрические измерения [ 94 ] по-
казали, что в бензоле при 25°С RuCl(H)(PPh3)3 при каталитической
концентрации 10 ~3М частично диссоциирован, то 16^18-*16-электрон-
ный процесс присоединения алкена непосредственно к 2.22 с последую-
щей потерей третичного фосфинового лиганда кажется слишком сом-
нительным [ 59 ]. В сильно координирующих растворителях до коорди-
нации алкена вполне может происходить процесс замещения PPh3
растворителем аналогично тому, как это описано для каталитической
системы на основе RhCl(PPh3)3. Миграция гидрида к олефиновому ли-
ганду, протекающая, вероятно, через четырехцентровое переходное
состояние 2.25, превращает гидридоолефиновый комплекс 2.23 в руте-
нийалкильное соединение 2.24, вновь связанное с молекулой третич-
ного фосфина или растворителя. Гидрогенолиз комплекса 2.24 приво-
64
ГЛАВА 2
дит к образованию продукта (алкена) и регенерации каталитически ак-
(PPh3)2ClRu-C
2.25
тивного соединения RuClH(PPh3)3 (2.22). Эта последняя часть ката-
литического цикла, включающая лимитирующую стадию реакции, мо-
жет протекать путем гетеролитического расщепления водорода:
Ru11 Cl (алкил) (PPh )о I- Н. - Ru11 (П) (алкил) (PPh, )„ + ц+ + С1~ •
О < J Z о О
-> RunCl(H)(PPh ) 4 Н(алкил)
о о
Однако более вероятно, что она протекает через реакции окислитель-
ного присоединения и восстановительного элиминирования, как пока-
зано на рис. 2.6 [ 19], причем лимитирующей стадией является окисли-
тельное присоединение к комплексу рутения(П) 2.24.
Эти три рассмотренные системы были выбраны для того, чтобы
показать три формальных типа активации водорода, встречающиеся
в гомогенных реакциях гидрирования, катализируемых комплексами
J-элементов. Дополнительное обсуждение ряда других важных ката-
литических систем приведет лишь к поверхностному рассмотрению
бесчисленного множества известных в настоящее время гомогенных
катализаторов [19]. До этого момента мы в основном старались оста-
новиться на аспектах механизма каталитических реакций; в следую-
щем разделе мы будем больше внимания уделять их практическому
использованию;
н
п -PPh3 рЬзН-^7 н
Ru Cl(«M«w) (PPh) н * / Ru/
3 3 + Н2 Ph,Pz I ZC-CH
2-24
+ PPh, - CH-CH
RunCl(H)(PPh3)3
Рис 2.6. Последовательность "окислительное присоединение — восстанови-
тельное элиминирование", приводящая к выделению алкана из хлоро(алкил)-
трис(трифенилфосфин) рутени я(П).
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
65
2.1.2. Селективное гидрирование. В настоящее время извест-
ны [19, 95] гомогенные системы, способные при относительно мяг-
ких условиях катализировать восстановление большинства обычно
встречающихся ненасыщенных органических групп. Однако именно се-
лективность таких систем, т.е. их способность восстанавливать толь-
ко один определенный вид ненасыщенности в присутствии других чув-
ствительных или потенциально восстанавливаемых видов ненасыщен-
ности, а не их универсальность делает гомогенные каталитические сис-
темы исключительно полезными в синтетической органической химии.
Мы уже кратко затрагивали этот аспект при обсуждении системы
RhCl(PPh3)3. Далее мы несколько более подробно рассмотрим неко-
торые типы возможных селективных превращений.
А. Гидрирование простых алкенов и алкинов. Гомогенными ката-
лизаторами, способными селективно восстанавливать алкены и алки-
ны в присутствии других функциональных групп, являются RhCl(PPh3)3,
RuCl2(PPh3)3 и RhH(CO) (PPh3)3. В мягких условиях - при 25°С и
давлении водорода 1 атм - все три катализатора являются очень вы-
сокоселективными по отношению к алкеновой или алкиновой функцио-
нальным эунпам, причем другие ненасыщенные группы, такие, как
—СНО, СО Н, —CN или —NO2, не затрагиваются. Путем
тщательного выбора условий реакции можно селективно восстановить
алкин в соответствующий алкен в присутствии другого алкина и алке-
на. Так, в фенол-бензольном (1:1) растворителе RhCl(PPh3)3 катали-
зирует восстановление смеси 1:1 гексина-1 и октена-1 таким образом,
что только после полного восстановления гексина-1 начинает восста-
навливаться либо откен-1, либо продукт гексен-1 [74]. Хотя абсолют-
ные скорости гидрирования алкина обычно ниже скоростей гидрирова-
ния соответствующих алкенов, в смеси алкен - алкин обычно более
предпочтительно восстанавливается алкиновая функциональная груп-
па благодаря ее более высокой способности образовывать связь с
металлическим центром [ 19, 85]. Все три каталитические системы
исключительно чувствительны к стерическим изменениям в субстра-
те и отдают определенное предпочтение терминальным алкенам или
алкенам. Это особенно заметно для RuCl2(PPh.,)3 — самой активной
их трех указанных систем, в присутствии которой олефины с конце-
вой двойной связью гидрируются примерно в 10 000 раз быстрее, чем
соответствующие внутренние олефины [96]. RhCl(PPh3)e слегка ме-
нее селективен, чем RuCl2(PPh3)3, хотя он еще предпочтительнее гид-
рирует терминальные алкены: скорость гидрирования октена-2 в че-
тыре раза ниже скорости гидрирования октена-1 [74]. Скорость вое-
66
ГЛАВА 2
становления имеет следующую тенденцию: алкен-1> г^с-алкен-2» транс-ал-
ке&2>траяс- алкен-3. Что касается циклических субстратов, то экзоцик-
лические двойные связи обычно восстанавливаются быстрее эндоцик-
лических. Высокая селективность также наблюдается при примене-
нии RhH(CO)(PPh3)3 для восстановления терминальных олефинов, вклю-
чая несопряженные диолефины [971.
Этот эффект селективности, который можно объяснить в рамках
стерических размеров третичных фосфиновых лигандов, с большим
успехом используется в селективном гидрировании. Например, при
использовании RhCl(PPh3)3 в бензоле пои 25°С 2.26 может быть про-
гидрирован в 11,12-дигидронооткатон (2.27) с селективностью 95%
[98] , и при использовании RuCl2(PPh3)3 в бензоле при 40°С стероид
андростадиен-1,4-Дион-3,17 (2.28) может быть восстановлен в соот-
ветствующий 4-ен (2.29) [ 99 ] с селективностью выше 90%.
RuCl2(PPh3)3
н2/с6нб
Бо Гидрирование сопряженных диенов, полимеров и активирован-
ных мономеров. Как отмечалось ранее, каталитическая система на ос-
нове раствора цианида кобальта в водной соляной кислоте является
высокоспецифичной по отношению к гидрированию сопряженных дие-
нов, полиенов и отдельных активированных моноенов. Однако в дейст-
вительности эта система имеет один недостаток, связанный с тем,
что ее обычно приготавливают в водном растворе, в котором большин-
ство органических субстратов слабо растворимо. Другие гомогенные
катализаторы, селективно восстанавливающие диены в моноены, обыч-
но менее активны , однако три каталитические системы — на основе
Fe(CO)5 [ 100],4г)5-СгНг ]М(СО)3Н(М=Сг, Мо или W? [101 ] и транс -
Pt(SnCl3) Н (PPh3)2 [102] - могут успешно работать в органических
растворителях. Обычно реакцию проводят при температуре выше 100°С
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
67
и давлении водорода порядка 30 атм. Каталитические системы на ос-
нове карбонилов железа и металлов VIA группы активируются при об-
лучении ультрафиолетовым светом [103 - 108]. При ультрафиолетовом
облучении Сг(CO)3(MeCN)3 катализирует гидрирование сопряженных
иенов в гж-моноены при комнатной температуре и атмосферном дав-
лении [ 107 ]. Для работы платинооловянной системы необходимы дав-
ление водорода выше 30 атм и температура порядка 90 - 110°С. При
таких условиях диены восстанавливаются в моноены с хорошей селек-
тивностью; например, гептадиен-1,5 в гептены с селективностью 91%,
гексадиен-1,5 в гексены - 70% [ 109 ]. Однако такая высокая селектив-
ность не проявляется в случае всех диеновых систем [110]. Хотя сис-
тема на основе цианида кобальта и ограничена использованием ее в
водных или водно-спиртовых средах, тем не менее, когда остро воз-
никает проблема селективности, выбирают именно ее.
В. Гидрирование ароматических и гетероциклических соединений.
Большинство гомогенных каталитических систем неэффективно в вос-
становлении ароматических углеводородов. Промышленное примене-
ние имеют некоторые циглеровские системы, т.е. комбинации солей
переходных металлов, обычно галогенидов, с алкилалюминием или с
алкилалюминийхлоридами. Некоторые из этих систем являются очень
высоко активными, например Со/2-этилгексаноат)2/А1Е13 в гексане
при 25°С и давлении водорода 40 атм восстанавливает фенол в цикло-
гексанол с селективностью выше 90% [111 ]. К сожалению, как мы уви-
дим в разд. 2.6 при обсуждении процессов полимеризации, эти системы
трудно охарактеризовать, и не всегда ясно, имеем ли мы дело с гомо-
генной или гетерогенной каталитической системами. В жестких уело*
виях (например, 200°С и 150 атм) в присутствии СО, т.е. в условиях
гидроформилирования (см. разд. 2.4.1), Со2(СО)8, вероятно, перехо-
дит в комплекс НСо(СО) который катализирует восстановление во-
дородом широкого ряда ароматических и гетероциклических соедине-
ний [112]. Полагают, что так же, как реакция гидрирования активи-
рованных олефинов, катализируемая Co(CN)|~ , эти реакции гидриро-
вания ароматических соединении протекают с участием свободноради-
кальных интермедиатов, а не кобальторганических комплексов [113].
Сообщалось [114], что в мягких условиях аллильные комплексы ко-
бальта(1) типа q-C H5CoL3 (L - третичный.фосфин или фосфит) ката-
лизируют восстановление ароматических соединений в циклогексаны.
Такие катализаторы обычно имеют очень ограниченное время жизни
и низкую активность (примерно 15 каталитических циклов за 24 ч).
Бэлее перспективная система образуется при обработке
68
ГЛАВА 2
[RuCl2(r|6 -С6Ме6) ]2 раствором карбоната натрия в пропаноле-2. Об-
разующийся димер, содержащий гидридные и хлоридные мостики (т|6-
С6Ме6) Ru(u-H )2(ц-С1 )Ru(t]6- С6Меб)С1, катализирует гидрирование
бензола и замещенных ароматических соединений при 50°С и 50 атм,
причем число каталитических циклов превышает 9000 [115].
Го Гидрирование альдегидов и кетонов. Катализаторы гидрирова-
ния кетогруппы на основе карбонила кобальта изучены наиболее под-
робно благодаря тому, что они используются в реакции гидроформи-
лирования (см. разд. 2.4). В типичных условиях реакции гидроформи-
лирования, т.е. при 130°С и давлении СО/Н2 порядка 200 - 350 атм,
СоН(СО)4, образующийся из Со2(СО)8, катализирует превращение ал-
кенов в альдегиды. При более высокой температуре порядка 160 -
350°G альдегиды гидрируются в спирты [1161. Вероятный механизм
[117] этого гидрирования включает нуклеофильную атаку гидридно-
го лиганда по карбонильному углероду через переходное состояние
2.30.
RCH—О
I •
I •
й—do(co)4
2.30
При таком переходном состоянии повышение гидридной приро-
ды водорода при кобальте заменой карбонильных лигандов кобальта
на более основные лиганды должно увеличить легкость гидрирования.
Оказалось, что это предположение справедливо; так, СоН(СО)3(РВиз)
Вил - w-бутил) катализирует гидрирование альдегидов в спирты при-
мерно при 150°С и давлении СО/Н2 30 атм. Активность катализатора
зависит от электронодонорной способности третичного фосфинового
лиганда, причем СоН(СО)3(РРЬ3) обладает заметно меньшей актив-
ностью, чем СоН(СО)3РВи3 [118, 119]. Подобным же образом ведут
себя аналогичные системы на основе карбонила родия, хотя в общем
они более активны, чем кобальтовые катализаторы. Обе эти системы
более подробно обсуждаются в разд. 2.4 при рассмотрении гидроформи-
лирования.
Катионные комплексы родия типа [ВЬ(диен)Ь2 ]+, где L - третич-
ный фосфин, существенно более активны в гидрировании кетогруппы.
Считают, что каталитически активным соединением является
[RhH2L2Sx]+ (где S - растворитель), который образуется путем гид-
рирования диенового лиганда и окислительного присоединения водоро-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
69
да к образующемуся комплексу родия(1). В том случае, когда L -
pMe2Ph, комплекс катализирует восстановление ацетона в пропанол-2
при 25°С и 1 атм 1120]. Сообщают, что комплексы родия типа
RhCl2(BH4) (ДМФ)ру3 в ДМФ также катализируют гидрирование кето-
нов в мягких условиях [ 121 ].
До Гидрирование нитрогрупп. Так же как и в случае ароматичес-
ких и гетероциклических соединений, гидрирование нитрогрупп в при-
сутствии гомогенных катализаторов протекает в относительно жест-
ких условиях, т.е. при температуре выше 100°С и давлении водорода
выше 40 атм. Однако упомянутый выше боргидридный комплекс родия
восстанавливает нитробензол в анилин при комнатной температуре и
1 атм Н2 [122]. Исключительно селективным катализатором гидри-
рования алифатических и ароматических нитросоединений в амины
является BuCl2(PPh3)3. В присутствии основания этот комплекс ката-
лизирует гидрирование ароматических нитросоединений, но не гидри-
рует ароматическое кольцо и не затрагивает другие заместители, та-
кие, KaxCN, CO2R, OR или галогенид [89, 90].
В предыдущем кратком обсуждении мы пытались проиллюстриро-
вать как область применения, так и селективность гомогенных ката-
лизаторов гидрирования. Однако обсуждение селективности не было
бы полным без рассмотрения таких систем с точки зрения наибольшей
их селективности - их способности образовывать хиральный продукт
из прохирального субстрата путем асимметрического гидрирования.
2.1.3. Асимметрическое гидрирование. Асимметрическим син-
тезом [ 59, 123, 124] является процесс, в котором ахиральное соединение или
совокупность таких соединений превращается в хиральное таким образом, что
образующиеся стереоизомерные продукты или энантиомеры получаются в неси
динаковых количествах. Эффективность асимметрического синтеза из-
меряется величиной преобладания (избытка) одного энантиомера над
другим, выраженной в процентах, т.е. процентом энантиомерного из-
бытка (% э.и. =%R - %S). Процент энантиомерного избытка - это
синоним обычно более часто используемого названия ’’оптическая чис-
тота” [123]. Асимметрический синтез является исключительно важ-
ным в фармацевтической и агрохимической промышленности, посколь-
ку зачастую два оптических изомера одной и той же молекулы имеют
заметную разницу в фармакологической и биологической активности
[ 125, 126]. Мы уже рассматривали пример использования 1-ДОФА
[56, 57] для лечения болезни Паркинсона. Аналогичный пример опти-
70
ГЛАВА 2
ческого предпочтения недавно был найден для синтетических пирет-
роидных пестицидов [127].
Имеются два способа осуществления асимметрического синтеза.
Первый из них - создание второго хирального центра под влиянием
присутствующего в молекуле хирального центра, например:
О ОН
Ph—С—С—о—C*RR'R2 ' McN*81 Ph—С —С—о—C*RR'R2
II 2Н I II
О Me О
Другой путь - создание нового хирального центра в прохиральном
субстрате под действием хирального реагента или катализатора. Асим-
метрическое гидрирование, как это показано в уравнении (2.3), явля-
ется наиболее широко распространенным примером второго способа
проведения асимметрического синтеза с участием катализатора.
ПСН_СН-В! + Нг RC^CWIP. '2.31
Если хиральным катализатором является комплекс переходного ме-
талла, то хиральность может быть введена в основном двумя способа-
ми: либо сам металл, если он окружен различными лигандами, может
быть центром хиральности, либо хиральными могут быть один или
несколько лигандов, связанных с металлом. В случае асимметричес-
кого гидрирования более важным является именно' второй способ. Поч-
ти все известные в настоящее время гомогенные катализаторы асим-
метрического гидрирования основаны на комплексах родия с третич-
ными фосфиновыми лигандами. В настоящее время приняты два под-
хода к введению оптической активности в такие системы, а именно
использование третичных фосфиновых лигандов с асимметрическим
фосфором типа PRR1R2 или использование лигандов, содержащих
асимметричный атом в одном из заместителей при атоме фосфора,
например PR2R*, где R = Ph и R* - ментил или неоментил. Попыткой
объединения обоих подходов было приготовление лигандов типа
p*R*R]R2 [ J28], однако до сих пор основное внимание уделялось от-
дельным подходам, и ниже мы ограничимся их рассмотрением.
А. Асимметрия при фосфоре. О первых примерах асимметричес-
кого гидрирования с использованием оптически активных фосфиновых
лигандов типа P*RRXR2 сообщалось в 1968 г. (в качестве катализато-
ра использовали комплексы родия, содержащие лиганд (8)(+)-метил-
фенил-«-пропилфосфин Р*МеРЬРгл)[ 129, 130 L Первоначально экспери-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
71
менты проводились с исходным комплексом типа RhCl3L3, который,
как полагают, под действием водорода в присутствии основания прев-
ращался в RhClL3 (ср. с разд, 2 Л Л, А), В последующих эксперимен-
тах катализатор готовили in situ добавлением третичного фосфино-
вого лиганда к лабильному олефиновому комплексу родия(1), например,
ц_дихлоробис(1,5-гексадиен)диродию(1) [ RhCl (1,5-ГД) 12 (этот прием
исключительно полезен при приготовлении in situ широкого круга го-
могенных /яр^-фосфиновых комплексов родия(1), поскольку димерные
комплексы типа [РИХ(олефин)2 ]2 легко приготовить и просто хранить).
Если в качестве субстрата используется атроповая кислота, то преоб-
ладание энантиомера составляет порядка 15% [129], Использование
вместо свободной атроповой кислоты ее соли с триэтиламином приво-
дит к повышению оптического выхода свыше 28% [22].
ph
JX=CH2
НООС
RhCIgLg*
HPh/EtOH/EtgN
60°С 20агмН2
ph ф сн3
НООС н
ж =• 15%
Без сомнения, для любого асимметрического синтеза важно, чтобы
в течение всего хода реакции функция, регулирующая хиральность,
оказывала как можно более сильное влияние на прохиральный суб-
страт. В случае асимметрического каталитического гидрирования
это означает, что ненасыщенная молекула должна быть как можно бо-
лее тесно связана с каталитической системой, поскольку гидрирова-
ние алкена с одной стороны двойной связи дает один энантиомер, тог-
да как гидрирование с другой стороны дает другой энантиомер. Тер-
мин "тесная связь" вовсе не является синонимом термина "прочная
связь". Более важным является то, чтобы субстрат имел предпочти-
тельную ориентацию по отношению к системе катализатора, нежели
он был бы прочно связан в этой ориентации, хотя зачастую это взаи-
мосвязано. При создании подходящего лиганда выполнить это требова-
ние можно путем целенаправленного изменения пространственных
или электронных свойств лиганда или, что еще лучше, их комбинацией.
При пространственном связывании координационное место или
полость, в которой будет располагаться субстрат, имеет такую форму,
которой соответствует субстрат, ориентированный определенным об-
разом. При электронном связывании электронные свойства координа-
ционного места или полости таковы, что сочетаются со свойствами
субстрата. Эти два типа связывания, которые схематично представле-
ны на рис. 2.7, означают, что для достижения оптимальной эффектив-
72
ГЛАВА 2
пространственное
соответствие
электронное
соответствие
Рис. 2.7. Пространственное и электронное соответствие между субстратом
и системой лигандов. ED — электронодонорный центр; ЕА — электроноакцеп-
торный центр.
ности необходимо, чтобы каталитическая система как можно лучше
соответствовала индивидуальным требованиям субстрата. При этом
следует стремиться использовать как можно больше свойств субстра-
та. Лучшими примерами соответствия субстрата и катализатора явля-
ются ферментативные системы, которые известны своей высокой сте-
реоселективностью. Однако повышение оптической чистоты при пере-
ходе от атроповой кислоты к ее соли отчасти объясняется тем, что
карбоксилатная группа координируется с металлическим центром, уве-
личивая таким образом близость взаимодействия. Хотя целенаправ-
ленное изменение свойств катализатора посредством использования
специально синтезированных лигандных систем находится в ранней
стадии своего развития, тем не менее был разработан лиганд 2-меток-
сифенилциклогексилметилфосфин (АСМР) [56, 57, 131 - 134] для вос-
становления а-ациламиноакриловой кислоты (<2оЗ£). При этом предпо-
лагалось, что, с одной стороны, олефиновая и карбоксилатная группа
кислоты связываются металлическим центром, а с другой — ацилами-
ногруппа образует водородную связь с метоксигруппой (группами)
АСМР-лиганда (лигандов). Происходит ли действительно такое взаимо-
действие в ходе каталитического цикла - спорный вопрос. Тем не ме-
нее не возникает никаких сомнений относительно оптической эффек-
тивности этих особых лигандных систем, о чем свидетельствуют ре-
зультаты, представленные в табл. 2.1 [131 — 134].
2.31
соон
RCH=C^
NHCOR'
2.32
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
73
Таблица 2.1. Асимметрическое гомогенное гидрирование а-ациламиноак-
риювых кислот 2.32 в присутствии каталитической системы родий-2-мето-
ксифенилциклогексиметилфосфина
Оптическая чистота
полученной аминокислоты,
% э.ио
м-ОМе, п-ОН-фенил Ph • 90
м-ОМе, п-0 Ас -фенил Me 88
Ph Me 85
Ph Ph 85
п-С1-фенил Me 77
а[(АСМР)2КЬ(диен)Г Х“; диен - 1,5-циклооктадиен, X”=6F4)PF6 или
BPh“ в метаноле при 50°С и давлении Н9 около 3 атм.
4 2
Основное ограничение, связанное с практическим использованием
хиральных фосфиновых лигандов с асимметрическим фосфином, обус-
ловлено сложностью стадий выделения в процессе получения таких
лигандов. Для получения хирального фосфина типа P*PhCH3R, исхо-
дя из РЬРС12, необходимо провести синтез из семи стадий, и даже
исходя из индивидуальной окиси фосфина Ph(Me) (О-ментил)РО, для
получения АСМР с общим выходом примерно 25% необходимо провести
три стадии [123]. Другой тип хиральных фосфиновых лигандов - это
лиганды, в которых асимметрия находится в группе, связанной с фос-
фином, а не на самом атоме фосфора; эти лиганды имеют то преиму-
щество, что могут быть приготовлены из легкодоступных и недорогих
хиральных предшественников.
Б. Лиганды с асимметрическим центром, связанные с атомом
фосфора. Спустя три года после первого сообщения об асимметричес-
ком гидрировании с использованием хиральных третичных фосфиновых
лигандов было обнаружено, что наличие хирального фосфора в таких
лигандах не является необходимым условием; введение асимметрии
в одну из групп, связанных с фосфором, также эффективно. Так, ро-
диевая система, имеющая в своем составе неоментилдифенилфосфин
[ (+)-NMDPP, 2,33] катализирует гидрирование р-метилкоричной
кислоты, причем образуется (Б)-(+)-3-фенилбутановая кислота с оп-
74
ГЛАВА 2
Me
Pr-UJO
2.33
PPh2
CH
CH
CH2PPh2
2.34
CH2PPh2
оческой чистотой 61% L135 J:
М"'-С_С^ 1 WC^-CH^CIVNMDPP^ %ICH C00H
n'' "COOH р|/ 61%э.и,
NMDPP имеет значительное практическое преимущество в том, что
его легко приготовить из дифенилфосфина и хирального предшествен-
ника [(—)-ментола] - соединений, не очень дорогих и коммерчески
доступных. Аналогично, по-видимому, наиболее часто упоминаемый
хиральный фосфиновый лиганд 2,3-о-изопропилиден-2,3-дигидрокси-1,4-
бис(дифенилфосфин)бутан (2.34), обычно известный как (-)-DIOP,
о котором впервые сообщил Каган с сотр. [136 ], может быть довольно
просто приготовлен из продажной (+)-винной кислоты. Родиевая ката-
литическая система, приготовленная добавлением этого бидентатного
лиганда к [НЬ(циклооктен)2С1 ]2 (1 моль на 1 моль родия) в бензоль-
но-этанольном растворе, содержащем триэтиламин, катализирует гид-
рирование 2.35 с образованием насыщенной кислоты 2.36 с оптической
чистотой 72% 11361:
РК NHCOCH
ХС-С <
Нz хсоон
2.35
^^—NHCOCIL
-> PhCH СН
^-—соон
72% э.и.
2.36
После этих первых работ было обнаружено множество хиральных
лигандных систем, в которых хиральность не локализована непосред-
ственно на атоме фосфора; некоторые из наиболее удачных лигандов
показаны формулами 2.37 - 2.43 (где Ви£ - трш-бутил). При исполь-
зовании в качестве катализаторов комплексов родия, содержащих эти
OPPh2
2.33
[138]
OPPh2
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
75
2.42
[И1]
СО2Ви-глре/п
2.43
[142]
или очень близкие к ним системы лигандов для гидрирования ряда
прохиральных субстратов, были получены продукты с высокой опти-
ческой чистотой. Энантиомерный избыток более 90% при использова-
нии в качестве лиганда 2А2 был достигнут в синтезе (Щ-фенилалани-
на, (Щ-лейцина, (П)-аланина и тирозина. Даже в случае относительно
простых олефинов, таких, как а-этилстирол, который может эффектив-
но связываться с металлическим центром только посредством двой-
ной связи, при использовании каталитической системы, состоящей из
комплекса родия в комбинации с 2Л0, оптический выход составил
60%[124L
Недавним достижением, которое может открыть путь к осущест-
влению широкого спектра реакций, катализируемых переходными ме-
таллами, является использование металлоорганических комплексов
в качестве лигандов. Так, замещенный ферроценовый комплекс (R)-
а-[(8)-К,2-бис(дифенилфосфино)ферроценил]этанол {2Л4)> известный
2.44
как (R)-(S)-BPPFOH, может координироваться с родием через два
фосфорных центра с образованием исключительно эффективного ка-
тализатора асимметрического гидрирования, который способен прово-
76
ГЛАВА 2
дить асимметрическое гидрирование даже прохиральных карбониль-
ных соединений [143] , например:
МеО о
МеО CCH2NH2.HC1
МеО
МеО
C*CH2NH2.HC1
з.к 90%
ОН
В этих и большинстве других систем, которые появились вслед за
DIOP, основное внимание уделялось бидентатным хелатирующим ли-
гандам, Такие крупные гдос-координирующие лиганды эффективно
связывают одну сторону комплекса и, как можно ожидать, создают
плотное асимметрическое окружение для входящего субстрата. Одна-
ко что касается оптической эффективности, то она немного увеличи-
вается по сравнению с эффективностью монодентатной системы типа
2.31. Это указывает на то, что высокой оптической эффективности
можно достичь и в том случае, когда хиральный центр значительно уда-
лен от металлического центра, например, в комплексе с (-)-DIOP хи-
ральные углеродные центры удалены от металла на расстоянии трех
длин связей.
Имеется еще много оставленных без ответа вопросов в области
асимметрического гидрирования, связанных с взаимодействием меж-
ду лигандом и субстратом. Ясно одно, что значительные изменения в
оптическом выходе могут вызываться относительно незначительными
изменениями в субстрате или лигандах. Аналогично из работ исследо-
вателей фирмы Monsanto [131 — 134] следует, что путем сочетания
лиганда и субстрата можно добиться многого. То, что незначительные
изменения в каталитической системе могут приводить к таким боль-
шим изменениям в оптическом выходе, не слишком удивляет, если при-
нять во внимание, что оптическому выходу в 99,9% соответствует раз-
ность свободной энергии переходных состояний диастереоизомеров
всего лишь 13 кДж/моль [ 1241 Как и большинство гомогенных катали-
тических реакций, каталитическое асимметрическое гидрирование тре-
бует от экспериментатора высокого мастерства,
2.1.4. Гидрирование путем переноса водорода. Все реакции
гидрирования, которые мы обсуждали до настоящего времени, были
реакциями типа
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
77
А + Н -» АН,
Л £
(2.4)
причем источником водорода служил молекулярный водород,, Другой
тип гидрирования [144, 145] представлен реакцией
A + DH-AH, +D (2.5)
где источником водорода служит донорная молекула ВН2> которая
сама претерпевает дегидрирование в ходе реакции» Обычно донором
водорода служат молекулы растворителя, поскольку присутствие их
в избытке по отношению к ненасыщенным молекулам акцептора А бла-
гоприятствует протеканию реакции» Тем не менее это не является не-
обходимым условием протекания данной реакции» Подходящими моле-
кулами донора, по существу, могут быть любые молекулы, способные
отдавать водород» Практически наилучшие результаты были получе-
ны при использовании спиртов (например, изопропанола ), кислот (на-
пример, муравьиной кислоты), циклических эфиров (например, диокса-
на) и ароматических циклических аминов (например, пирролидина и
пиперидина)» В этом классе реакций хорошо изучено гидрирование цик-
лопентена в присутствии RhCl(PPh3)3? где в качестве донора водоро-
да используется диоксан [ 146 - 148 ]:
Поскольку имеется много общего между гидрированием путем пере-
носа водорода и гидрированием с участием молекулярного водорода,
то большинство из обсуждавшихся ранее каталитических систем ак-
тивны и в гидрировании путем переноса водорода» Уже было отмече-
но , что соединения RhCl(PPh3)3 и RuCl2(PPh3)3 также активны и
в целом ряде реакций гидрирования путем переноса водорода [149,
150].
Основным отличием между гидрированием собственно водородом
и гидрированием путем переноса водорода, представленных уравнени-
ями (2»4) и (2»5) соответственно, является то, что в случае гидрирова-
ния путем переноса водорода не происходит изменения общей ненасы-
щенности в системе; в простейшей форме это выглядит следующим
образом:
"алкан (1)" + алкен (1) -> алкен (2) + "алкан (2)"
78
ГЛАВА
Это накладывает определенные ограничения на реакции гидрирования
путем переноса водорода. Донор водорода ’’алкан (1)” должен не толь-
ко обладать способностью взаимодействовать с металлическим цент-
ром с образованием по крайней мере одного атома водорода, но также
в разумных пределах конкурировать с алкеном (1) за вакантное коор-
динационное место катализатора. Практически это означает, что мо-
лекула донора должна содержать функциональную группу (например,
донорный кислород или азот), способную координироваться с метал-
лическим центром. Кроме того, образовавшийся продукт дегидрирова-
ния донора [алкен (2)] должен покидать координационную сферу, ос-
вобождая место для акцептора водорода [алкена (1)1 Эти ограничения
проявляются в сложной кинетике таких систем; обычно происходит
ингибирование реакции продуктом [алкеном (2) ] [148 1
Гидрирование путем переноса водорода является, по существу,
реакцией образования двух продуктов, и поэтому оно менее привлека-
тельно с практической и коммерческой точек зрения. Тем не менее
оно может быть полезным, если, например, учесть большую безопас-
ность по сравнению с процессом в присутствии газообразного водоро-
да или если в используемой реакционной среде уже присутствуют под-
ходящие доноры и акцепторы, например, в нефтяной фракции, получа-
емой в процессах нефтепереработки и риформинга. Однако в послед-
нем случае гетерогенная каталитическая система более предпочти-
тельна, чем гомогенная.
В разд. 2.1, посвященном гидрированию, мы пытались осветить
основные аспекты данной реакции. Нельзя считать охват этой темы
дочерпывающим, но по крайней мере освещена сущность проблемы.
Как указывалось в начале раздела, пока еще нет крупномасштабных
промышленных процессов гидрирования, использующих гомогенные ка-
талитические системы, хотя они широко используются в специаль-
ных химических Производствах, например в фармацевтической промыш-
ленности. Кроме того, некоторые черты реакции гидрирования встре-
чаются во многих каталитических реакциях, таких, как гидроформили-
рование, где гомогенные каталитические системы получили промыш-
ленное применение. Прежде чем перейти к обсуждению таких систем,
мы должны рассмотреть другой тип реакций, которые, подобно гидри-
рованию, в настоящее время хотя и не применяются в широком масшта-
бе с использованием гомогенных катализаторов, тем не менее играют
ключевую роль во многих гомогенных каталитических процессах. Мы
имеем в виду реакцию изомеризации.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
79
Дополнительная литература по гомогенному гидрированию
James B.R. Homogeneous Hydrogenation.— Adv. Organometal Chem.,
1979, v. 17, p. 319.
McQuillin F.J. Homogeneous Hydrogenation in Organic Chemistry. —
Dordrecht: Reidel, 1976.
Kwiatek J. — In: Transition Metals in Homogeneous Catalysis (ed.
G.N. Schrauzer ).—New York: Marcel Dekker, 1971, p. 13.
heck R.F. — In: Organotransition Metal Chemistry, A Mechanistic
Approach (ed. R.F.Heck). — New York: Academic Press, 1974, p. 55.
Webb G. — In: Catalysis, Vol. 2, London: Chemical Society, 1978, p. 145.
Kieboom A.P.G., Rantwijk F. Hydrogenation and Hydrogeneolysis in
Synthetic Organic Chemistry. — Delft: Delft University Press, 1979.
Rylander P.N. Organic Synthesis with Noble Metal Catalysts. — New
York: Academic Press, 1973, Ch. 2.
Harman R.E., Gupta S.K., Brown D.h — Chem. Soc. Rev., 1973, v.73,
P-21-
Marko L., Heil B. Asymmetric llomoheneous hydrogenation and related
reactions. — Catal. Rev., 1974, v. 8, p. 269.
Morrison J.D., Master W.F., Newberg M.K. Asymmetric homogeneous
hydrogenation. — Adv. Catal, 1979, v. 25, p. 81.
-Pearce R. — In: Catalysis, Vol. 2, Specialist Periodical Report London:
Chemical Society, 1978 p. 176.
2.2. Изомеризация
Изомеризация - это процесс, в результате которого происходит
перераспределение связей или групп внутри данной молекулы. В ре-
зультате этого процесса не происходит ни присоединения, ни отрыва
атомов от молекулы субстрата. В отличие от гидрирования и всех
других каталитических реакций, которые мы будем рассматривать,
этот процесс приводит к изменению только структуры молекулы, не
изменяя формулы субстрата. К двум наиболее хорошо изученным ти-
пам изомеризации, катализируемым гомогенными комплексами пере-
ходных металлов, относятся изомеризация алкенов, которая сопровож-
дается перераспределением двойных связей углерод — углерод внут-
ри молекулы, и скелетная изомеризация, которая сопровождается пе-
рераспределением одинарных или двойных связей углерод — углерод
внутри карбоциклической, обычно разветвленной молекулы.
Мы рассмотрим последовательно оба этих процесса изомеризация.
80
ГЛАВА 2
2.2.1. Изомеризация алкенов. Почти все (7-элементы, способ-
ные координировать алкены, в определенной степени катализируют изо
меризацию алкенов» Однако, как и большинство других гомогенных
каталитических реакций, наибольший интерес представляют комплексы
металлов VIII группы, т»е» Ru, Os; Со, Rh, Ir; Ni , Pd, Pt. В ходе
изомеризации, катализируемой этими металлами, алкен претерпевает
два процесса активации, т»е» активацию путем координации и актива-
цию путем присоединения» Процесс активации путем присоединения не-
посредственно отвечает за перемещение двойной связи или, другими
словами, за перемещение атомов водорода» Миграция двойной связи,
по существу, происходит путем сдвига водорода алкильной группы,
расположенной рядом с двойной связью, к а-атому углерода двойной
связи
—с—с=с—
—с=с—с—
Именно этот процесс катализирует комплекс переходного металла»
Формально можно выделить два основных типа реакций изомеризации,
а именно: реакции,протекающие через образование металлалкильных
интермедиатов, и реакции, протекающие через образование металлал-
лильных интермедиатов»
А. Изомеризация, протекающая через образование металлалкилъ-
ных интермедиатов» Весьма важными частицами в осуществлении этой
реакции являются металлгидридные комплексы» Координация алкена
приводит к образованию гидридоолефинового комплекса металла, кото-
рый быстро претерпевает реакцию миграции гидридного лиганда с об-
разованием металлалкильного соединения» Эта миграция, вероятно,
протекает через четырехцентровое переходное состояние и идентична
той, которая была описана ранее для процесса каталитического гидри-
рования (разд» 2»1»1)»
rch2
CH=CHR'
rch2
сн—ch2r’
RCH2
СН—CHR
М- -Н
(2-6)
2А5
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
81
Почти во всех гидридоолефиновых комплексах переходных металлов в
случае, когда алкен и гидридная группа находятся взаимно в цис-по-
ложении, миграция гидридного лиганда протекает исключительно быст-
рое Образовавшееся металлалкильное соединение 2А5 может претер-
певать ^-элиминирование по пути как А, так и Б»
RCH
CH=CHR’
М—Н
rch2
^СН—CHR
.....А
R
^сн—СН—CH2R
А-Л
RCH=CH—ch2r
н—м
Путь А -это просто процесс, обратный реакции (2»6), и он не приводит
к перемещению двойной связи» Путь Б, в котором происходит отрыв
не того атома водорода, который присоединился, а другого, приводит
к смещению двойной связи к соседнему атому углерода» В случае ес-
ли координированный с металлом алкен может обмениваться с алке-
ном, находящимся в растворе, представленная выше последователь-
ность реакций составляет каталитический цикл изомеризации» Хотя
путь А не приводит к перемещению двойной связи, он вполне может
вызвать реакцию водородного обмена, поскольку в металлалкильных
соединениях, таких, как,2Л5, в отсутствие серьезных стерических зат-
руднений может происходить свободное вращение вокруг С—С-связи,
и Н, отщепляемый по схеме А, не обязательно тот, который присоеди-
нился по реакции (2»6)» Так, в присутствии дейтерида металла зачас-
тую наблюдается внедрение дейтерия в молекулу неизомеризованного
олефина» Пути А и Б могут приводить к образованию как цис-, так и
юрлис-алкена, и если принять, что система достигает равновесия, то
в результате образуется стабильная смесь изомеров»
Металлгидридный комплекс может быть либо добавлен в реакци-
онную среду как таковой, либо образован in situ из комплекса метал-
82
ГЛАВА 2
2.48
Рис. 2.8. Каталитический цикл для изомеризации алкенов, катализируемой
RhH{CO){PPh )
О о
ла и подходящего источника водорода, такого, как газообразный водо-
род или водный раствор кислоты, RhH(CO) (РРЬ3)3 [152] является при-
мером первого способа, а Со2(СО)8/Н2 [153] и Ni [P(OEt)3]4i/H2SO4
[154] - примерами образования металлгидридного комплекса in situ.
Каталитический цикл для Rh(H)(CO)(Ph 3)3 показан на рис, 2Х Как
отмечалось, каталитически активным является соединение с 16 ва-
лентными электронами - wp^c-RhH(CO)(PPh3)2, которое образуется
из 18-электронного бипирамидального комплекса родия(1) при диссоци-
ации третичного фосфинового лигандао В этом каталитическом цикле
показано, что водород присоединяется (через переходное состояние
2.47 с образованием 2.48) к менее замещенному атому углерода двой-
ной связи, тое0 присоединение происходит по правилу Марковникова,
ГОМОГЕННЫЕКАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
83
Противоположный способ присоединения, против правила Марковнико-
ва (через 2.49 с образованием 2.50), не должен приводить к перемеще-
нию двойной связи, поскольку [3-элиминирование водорода от 2.50 мо-
жет привести только к образованию олефинового комплекса 2.46.
PPh3
ch2ch2ch2r
2.50
Согласно электронным представлениям, следует ожидать, что направ-
ление присоединения зависит от заряда на атоме водорода. Увеличе-
ние локализации отрицательного заряда на атоме водорода должно
благоприятствовать присоединению против правила Марковникова, и
наоборот. По стерическим соображениям линейный алкильный комплекс
2.50 более предпочтителен, чем разветвленный 2.48. Это связано с
тем, что поскольку оба комплекса содержат два объемистых трифенил-
фосфиновых лиганда, то существенным оказывается любое уменьше-
ние стерических затруднений, В случае этого комплекса родия оба фак-
тора облегчают присоединение против правила Марковникова и поэто-
му ни этот, ни большинство других комплексов родия(1) не являются
достаточно хорошими катализаторами изомеризации. Фактически ско-
рость изомеризации в присутствии газообразного водорода пренебре-
жимо мала по сравнению со скоростью гидрирования,
С другой стороны, в каталитической системе Сс2(СО)8/Н2 ката-
литически активным соединением служит СоН(СО)4; это соединение
образуется путем гомолитического присоединения водорода:
Со (СО)8 + Н2 -2СоН(СО)4
Водород в СоН(С0)4 кислотный, а не гидридный: в водном растворе
СоН(СО), ведет себя как сильная кислота, диссоциируя с образовани-
84
ГЛАВА 2
ем карбонилатного иона [Со(СО)4]~ Таким образом, СоН(СО)4 бо-
лее склонен присоединяться к терминальным олефинам по правилу
Марковникова, чем RhH(CO)(PPh ) •
О Ал
RCH2CH—сн2 1
1 • W-
I • ""
(СО)хСо—-Н
rch2ch—сн3
(СО)хСо
2.51
х = 3 или 4
Фактически при определенных условиях проведения реакции гидрофор-
милирования (см0 гл. 4), катализируемой СоН(СО) благодаря изо-
меризующей способности кобальтового катализатора образующаяся
смесь альдегидов по существу не зависит от изомерного состава ис-
ходного олефина [ 153 L Нг основе изучения реакции гидроформилиро-
вания с использованием дейтерированных олефинов было предположе-
но, что в случае кобальтовой системы присоединение - элиминирова-
ние водорода протекает по синхронному (согласованному), а не ста-
дийному механизму через переходное состояние 2.52 таким образом,
что полная изомеризация в п-олефиновом кобальткарбонильном сое-
динении происходит без элиминирования свободного олефина:
rch2ch=ch2
I
(СО)хСо—Н
RCH—СН—СН2
RCH=CH—СН
I
Н—Со(СО),
В случае системы Ni[P(OE$3]4/H2SO4 каталитически активным сое-
динением, по-видимому, служит 16-электронное соединение NiHtP(OEt) ]
содержащее формально Ni(IT), которое образуется путем протонирования
и отщепления лиганда L (где L = P(OEt) );
Ni°L + Н+ -> [ Ni11 (H)L J+ - [Ni11 (H)L ] + + L
4 4 о
чоВоЭо 18 18 J 6
Лимитирующей стадией является диссоциация лиганда, и образующе-
еся четырехкоординированное соединение никеля представляет собой
весьма эффективный катализатор изомеризации в растворе кислоты,
В случае малоосновного фосфинового лиганда 1по шкале электронных
параметров Толмана (смв разд, 1.2.3.Б) v для P(OEt)3 составляет
2077 см-*, ср. v(PPh3) 2088,9 см”1 и v(PBu3)= 2060,3 см-i ] гид-
ридный лиганд по своей природе занимает промежуточное положение
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
85
между гидридным лигандом в СоН(СО)4 и гидридным лигандом в
RhH(CO) (РРЬ3)3, и поэтому он может вполне присоединяться как по
правилу Марковникова, так и против правила Марковникова. Из-за
стерических препятствий так же, как и в случае родиевой системы,
преобладает в основном присоединение против правила Марковникова.
Хотя конический угол лиганда P(OEt)3 (109°) значительно меньше ко-
нического угла РРЬ3 (145°), в каталитически активном соединении
никеля присутствуют три лиганда P(OEt)3? тогда как в комплексе
родия присутствуют лишь два лиганда РРЬ3. При изучении изомериза-
ции 1,2-с?2-1-пентена в бензоле в присутствии очень сходной системы
Ni[P(OEt)3 ]4/CF3COOH были получены серьезные доказательства
гидридного механизма присоединения и элиминирования [ 155 L
Ввиду того что гидридоолефиновые комплексы металлов, как пра-
вило, нестабильны, имеется лишь несколько работ, в которых сообща-
лось о прямом наблюдении равновесия:
Н
М—/алкен) —(алкил)
В одной из работ изучался комплекс [(r]5«=C5H5)Rb(H)(C2H4)(PMe3) ]+,
который, согласно данным ЯМР-^-спектроскопии, в растворе CD3NO2
находится в равновесии с соответствующим этильным комплексом
[(T]5-C5H5)Rh(C2H5)(PMe3)]+[156]0 При -20°С обмен протекает мед-
ленно и могут быть определены сигналы, соответствующие обоим сое-
динениям. При комнатной температуре сигналы протонов С5И5 и РМе3
групп острые, тогда как сигналы гидридного атома Н и протонов С2Н4
широкие, что указывает на установление равновесия (2.7):
[ h и ) RhH(C„H.) (РМе,) 1+ [ (т]5’С.Н ) RhC2H,(PMe,) ]+ (2.7)
Во всех указанных выше изомеризующих системах активное металл-
гидридное соединение или вводилось как таковое, или образовывалось
in situ из подходящего источника водорода. В следующем классе реак-
ций изомеризации, катализируемой металлами, металлгидридное сое-
динение образуется из исходного олефина.
Б. Изомеризация через металлаллилъные интермедиаты. Другой
путь, приводящий к изомеризации алкена, лежит через образование
металлаллильного комплекса. В этой последовательности реакций,
которая аналогична описанному ранее механизму гидрирования бута-
диена-1,3 (см. разд. 2.1.1,Б), катализируемого CoH(CN)^-, после ко-
О
86
ГЛАВА 2
ординации алкена происходит отрыв водорода от примыкающего к
двойной связи sp 3-гибридного атома углерода и образуется гидрид-
ное металлаллильное соединение типа 2,54. В таком соединении,
обычно называемым аллильным комплексом, фрагмент С3 связан
тг-связью с металлом, причем все три атома углерода находятся на
расстоянии длины связи от металлического центра и этот фрагмент
эффективно занимает два координационных места в комплексе.
Н—cr,r2
"'"сн
Мх—1| -
CHR
2.53
2.54
HCRjR2
^сн
NT—1|
CHR
CR|R2
HCHR
(2o7a)
Будет ли происходить миграция,двойной связи, зависит от того, каким
образом гидрид вновь присоединяется к аллильной группе, Повторное
присоединение к месту отрыва (путь А) не приводит к перемещению
двойной связи. Присоединение к другой части фрагмента С3 (путь Б)
сдвигает двойную связь в углеродном скелете на одно положение0
Как процесс отрыва, так и процесс присоединения могут происходить
через пятицентровое переходное состояние типа 2.55. При переходе
тт-олефинового комплекса 2.53 к гидридо-тт-аллильному комплексу
2.54 происходит увеличение на две единицы как формальной степени
окисления, так и координационного числа. Таким образом, эта после-
довательность реакций представляет собой очередной пример везде-
сущей реакции "окислительного присоединения".
В противоположность обсужденной ранее металлалкильной изоме-
ризующей системе каталитический цикл изомеризации, включающий
тг-аллильные соединения металла, содержит окислительно-восстанови-
тельную последовательность Мх -* Мх+2 -> Таким образом, этот
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
87
механизм применим только для комплексов металлов в низких степе-
нях окисления %, которые могут их легко увеличивать на два до х +2а
Действительно, наиболее хорошо охарактеризованными примерами изо-
меризации с участием тг-аллильных интермедиатов является изомери-
зация, катализируемая карбонильными комплексами Fe (0). Различие
между металлаллильным и -алкильным механизмами заключается в
расстоянии, на которое перемещается атом водорода в молекуле суб-
страта, В тг-аллильном механизме водород субстрата перемещается
между атомами С-1 и С-3 в фрагменте С3? т,е, процесс включает 1,3-
водородный сдвиг. Это хорошо видно из приведенной выше схемы. Ме-
таллалкильный механизм включает 1,2-водородный сдвиг. Более по-
нятным 1,2-сдвиг становится после рассмотрения двух металлалкиль-
ных соединений в указанной ниже последовательности:
R2CH*—СН—СН3 R2C=CH—сн3 r,c—снн*—сн3
* I J z I J I 3
л I
м —* н — М * м
1,2- водородный, сдвиг I
Это различие в природе водородного сдвига позволяет с использова-
нием дейтерированных олефинов экспериментально различить эти
два механизма. Так, тг-аллильный металлгидридный механизм был
надежно установлен для изомеризации алкенов (в качестве алкена ис-
пользовали смесь, состоящую из 3-метилбутена-1 и 3-этилпентена-1-3(/1
(-2.55), катализируемую FeQ(C0)19) [ 157],
СИ2СН3 СН2СН3
сн.сн^.:—сн=сн9 сн,сн9—с=спсн,
О Сл L О Zj о
э
2.56 2.57
При 80°С 3-метилбутен-1 изомеризуется в смесь 2-метилбутена-1 и
2-метилбутена-2 (первоначально образуется 2-метилбутен-1, который
с течением времени превращается в термодинамически более стабиль-
ный 2-метилбутен-2) и З-этилпентен-1 изомеризуется в З-этилпентен-2
(2.57). После изомеризации было обнаружено, что дейтерий присутст-
вует только в трех метильных группах продукта З-этилпентена-2. Это
указывает на то, что изомеризация 2.56 в 2.57 сопровождается исклю-
чительно 1,3-водородным сдвигом, т.е. протекает через тг-аллильный
металлгидридный интермедиат. Кроме того, поскольку не было обна-
88
ГЛАВА 2
2.59
Рис. 2.9. Каталитический цикл для изомеризации З-этилпентена-1 в присутст-
вии Fe3(CO)12 в качестве катализатора.
ружено внедрения дейтерия в изомеры бутенов, можно заключить, что
этот сдвиг является внутримолекулярной реакцией и описывается пос-
ледовательностью (2.7а). Дальнейший анализ этих результатов пока-
зывает, что изомеризация протекает многократно, другими словами, в
течение того времени, пока олефин связан с металлом, двойная связь
может переместиться несколько раз, т.е. изомеризация закомплексо-
ванного алкена протекает быстрее, чем обмен между свободным алке-
ном и алкеном в комплексе. Ряд каталитических циклов, иллюстрирую-
щих образование трех изомеров, показан на рис. 2.9. Активным катали-
затором является, по-видимому, четырехкоординированное 16-электрон-
ное соединение Fe°(CO)p которое образуется при термическом расщеп-
лении кластера Fe3(CO)]2. Полагают, что лимитирующей стадией явля-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
89
ется отрыв СО от 18-электронного соединения Fe°(CO)4 (алкен) с
образованием 16-электронного соединения Ре°(СО)3(алкен) (2.58), ко-
торое далее быстро претерпевает внутримолекулярное окислительное
присоединение, образуя тт-аллильный гидридный комплекс 2.59.
В очень близкой реакции изомеризации, катализируемой Fe(CO)5
при облучении светом, координационно ненасыщенный карбонильный
комплекс образуется путем диссоциации лиганда под действием света.
Считают, что активным катализатором является тт-аллилгидридный
комплекс типа Ре(п-аллил) ( Н)(СО)3 [158].
Полагают, что тт-аллильный механизм также играет важную роль
при изомеризации алкенов, катализируемой бис(бензонитрил)дихлоро-
пал л ад ием(П)0 Однако в случае системы на основе палладия(П) экс-
периментальные данные не столь ясны, как в случае системы на осно-
ве Fe (СО)19.
В. Изомеризация через другие интермедиат. Рассмотренные вы-
ше два механизма изомеризации алкенов удовлетворительно объясня-
ют основную часть известных в настоящее время экспериментальных
данных. Общей чертой этих механизмов является присутствие металл-
гидридных соединений на определенных стадиях каталитического цик-
ла. Также возможно, что миграция водорода внутри алкена может про-
исходить фактически без координации водорода с металлическим цент-
ром. Полагают, что этот тип "внеметаллической" миграции происходит
в ходе реакции перестройки оптически активных комплексов железа
типа 2.60 [161]. Механизм, альтернативный тт-аллильной металлгид-
2.60
ридной последовательности, включает "сигматропный 1,3-надплоскост-
ный водородный сдвиг":
90
ГЛАВА 2
Предполагается, что водород мигрирует в плоскости, проходящей
через олефиновый лиганд, с противоположной стороны от металла, и,
таким образом, водород непосредственно не взаимодействует со свя-
зывающими орбиталями металла. Роль, которую играет такой процесс
в изомеризации простых олефинов, катализируемой карбонилом желе-
за, требует экспериментальных уточненийв Однако не вызывает сом-
нений, что он предполагает энергетически возможный путь, альтерна-
тивный п-аллильному металлгидридному механизму.
Другой последовательностью, не обязательно включающей метал-
лгидридный комплекс, является последовательность, содержащая ме-
таллкарбеновые соединения типа 2£1\
RCH CH-CIL
I
м
RCH„CCH,
2II 3
м
RCH=CHCH,
I
м
Считается, что образование 2£1 происходит через переходное состо-
яние типа 2£2, включающее 1,2-надплоскостный водородный сдвиг
[162]. В этом случае также нет прямых доказательств протекания та-
,Н
RCH2C5^CH2
м'
2.62
кого процесса при изомеризации алкенов, катализируемой металлами,
хотя были получены некоторые данные, свидетельствующие о том, что
карбеновый лиганд в 2£3 легко превращается в соответствующий оле-
финовый лиганд PhCH=CHR [163].
Ph CH2R
XCZ PhCH=CHR
|| ----- I (2-8)
w w
(CO), (CO),
2.63
В настоящее время считается, что металлкарбеновые комплексы иг-
рают важную роль в реакциях метатезиса (см. разд. 2.8), и процесс,
обратный процессу, описанному уравнением (2.8), вполне мог бы быть
одним из путей образования таких соединений в этих реакциях.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
91
Во всех описанных выше изомеризующих системах изомеризация
происходила в результате миграции двойной связи или водорода» Не
менее важным классом реакций изомеризации, катализируемых пере-
ходными металлами, являются реакции, включающие перестройку ос-
новного каркаса молекулы субстрата, в результате которой молеку-
ла продукта содержит другое число и распределение простых и крат-
ных связей»
2.2.2. Скелетная изомеризация. Существует целый ряд орга-
нических молекул, которые несмотря на то, что имеют более напря-
женное строение по сравнению с одним или несколькими своими ва-
лентными изомерами, тем не менее вполне стабильны при комнатной
температуре [164, 166 L Так, квадрициклен 2.64, хотя и сильно нап-
ряжен по сравнению со своим валентным изомером норборнадиеном
2.65, вполне стабилен при комнатной температуре, и даже при 140°С
время его полураспада (т%) превышает 14 ч [ 167 ];
2.64
(2.9)
Аналогично для сильно напряженной молекулы три-»гр^»г-бутилпризмана
(2.66) время полураспада в пиридине при 110°С составляет 18 ч [166].
(Ви‘ = я7ре/77-Ви)
Высокий энергетический барьер при изомеризации таких систем объяс-
няется необходимостью сохранения орбитальной симметрии в ходе сог-
ласованного процесса, поскольку, согласно подходу Вудворда - Хоф -
фмана [ 168], представленный выше тип превращений "запрещен по сим-
метрии". В конце шестидесятых годов было обнаружено, что некоторые
комплексы переходных металлов исключительно эффективно катализи-
руют такую запрещенную по симметрии скелетную изомеризацию» В
одном из первых сообщений рассматривалось раскрытие цикла квад-
92
ГЛАВА 2
рициклена [уравнение (2.9)L В присутствии определенных гомоген-
ных комплексов родия, палладия или платины (например, [ Rh( норборна-
диен)2С1]2) время полураспада квадрициклена при -26°С составляет
45 мин [169], Аналогично в метанольном растворе в присутствии нит-
рата серебра время полураспада замещенного призмана < 1 мин, при
этом он быстро превращается в смесь изомеров 2.67 - 2.70 (здесь
и ниже Вал - mpew-бутил) [170]. В ходе дальнейшей изомеризации 2.67
превращается в 2.68 и 2.69 - в 2.70. Для объяснения этих и близких к
ним реакций скелетной изомеризации, катализируемых металлами, бы-
ли предложены два основных механизма; первый включает согласован-
ный процесс, второй — последовательность стадий.
А. Скелетная изомеризация, протекающая по согласованному ме-
ханизму. Металл может катализировать запрещенные по симметрии
превращения, предлагая системе путь, обходящий ограничения по сим-
метрии [ 171 ]. Однако, чтобы понять, каким образом система с помо-
щью металла может обойти эти ограничения, следует -вкратце рассмот-
реть природу ограничений. Рассматривая изомеризацию квадрицикле-
на [уравнение (2.9) ], можем представить эту реакцию как превращение
циклобутана в две молекулы этилена:
Рассмотрение симметрии [171] позволяет нам выделить комбинации
связывающих а-орбиталей циклобутана SS и SA и комбинации антисвя-
'зывающих сг-орбиталей AS и АА, где S и А относятся к симметричным
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
93
СТ- связи
циклодутана
СТ- связи
циклобутана
ТГ- комбинация
.молекул этилена
ПТ- комбинация
молекул этилена
Рис. 2.10. Корреляционная диаграмма для превращения одной молекулы цик-
лобутана в две молекулы этилена.
и антисимметричным орбиталям относительно выбранных определен-
ных элементов симметрии в молекулярной комбинации. По отношению
к тем же элементам симметрии комбинации тг-связывающих орбиталей
двух образовавшихся молекул этилена могут быть представлены как
SS и AS, а антисвязывающие комбинации - SA и АА0 Корреляцион-
ная диаграмма для этого процесса показана на рис. 2.10.
Сохранение симметрии молекулярных орбиталей требует, чтобы
молекулярные орбитали сохраняли свою симметрию относительно выб-
ранных элементов симметрии при прохождении вдоль координаты ак-
ции. Так, комбинация циклобутана SA-cr превращается в олефиновую
комбинацию SA-тт* и комбинация AS-cj* — в комбинацию AS-тт. Пересе-
чение образующихся орбиталей указывает на то, что реакция запреще-
на по симметрии. Для преодоления ограничений по симметрии необхо-
димо удалить электронную пару с заполненной комбинации SA и "изме-
нить" ее симметрию на AS так, чтобы она могла образовать в продук-
те-связывающую комбинацию тт-AS. Именно здесь может сыграть свою
роль переходный металл с его частично заполненными с?-орбиталями.
Вакантные d-орбитали правильной симметрии могут взаимодействовать
с комбинацией SA и принимать пару электронов. Если в это же время
заполненные с?-орбитали с симметрией, близкой к AS, находятся в ори-
ентации, подходящей для образования комбинации AS-тт продукта, тог-
да после того, как вакантная с?-орбиталь примет пару электронов суб-
страта, заполненная d-орбиталь сможет отдать свою пару электронов
образующемуся продукту. Другими словами, переходный металл мо-
жет предложить системе путь, позволяющий выйти из тупика, обус-
ловленного симметрией. В случае квадрициклена это воздействие на
основе расчетов Манго [ 171 ] может быть схематически представлено
схемой, показанной на рис. 2.11.
94
ГЛАВА 2
Рис. 2.11. Схематическое изображение катализируемой металлокомплексами
изомеризации квадрициклена. Заштрихованные области отвечают заполненным
d-орбиталям мет алл ао •
Альтернативной последовательностью по отношению к описанно-
му выше согласованному механизму является "несогласованный" путь,
или стадийный механизм, включающий первоначально разрыв только
одной связи углерод - углерод посредством реакции окислительного
присоединения или процесса гетеролитического присоединения.
Б. Скелетная изомеризация, протекаюгцая через стадийный про-
цесс. а-Связи в напряженных карбоциклических системах, таких, как
циклопропан или циклобутан, имеют ярко выраженный р-характер и
поэтому предрасположены к взаимодействию с системой d-орбиталей
комплекса переходного металла. Связывание, по существу, аналогич-
но тому, которое было описано (разд. 1.2.1) для металлолефиновых
комплексов; оно включает а-компоненту, образующуюся путем подачи
электронов с напряженной связи углерод - углерод на вакантную (/-ор-
биталь металла подходящей симметрии, и слабую к-компоненту, об-
разующуюся путем обратного допирования с- заполненной орбитали ме-
талла на антисвязывающую а*-орбиталь карбоцикла [166].
Это состояние, которое, очевидно, очень близко к переходному
состоянию, существующему в рассмотренном согласованном процессе,
может привести к продуктам окислительного присоединения, которые
далее могут являться интермедиатами в стадийных процессах скелет-
ной перестройки. Серьезные доказательства в поддержку такого пути
окислительного присоединения были получены путем выделения ациль-
ных комплексов родия(Ш) в ходе целого ряда реакций изомеризации,
катализируемых родием(1). Так, добавление димерного карбонилхло-
рида родия, содержащего хлоридные мостики [RhCl (CO)2]Q, к квадри
циклону приводит (помимо изомеризации квадрициклена в норборнади-
ен) к образованию комплекса ацилродия(Ш) (2.7?), выделенного в виде
полимерного материала, связанного хлоридными мостиками, в кото-
ром число мономерных звеньев (п), вероятно, равно четырем 1172].
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
95
Считают, что вначале окислительное присоединение одной из циклопро-
пильных о-связей квадрициклена дает 2.72, которое затем может пре-
терпевать миграцию лиганда (алкила к карбонильному углероду) с об-
разованием стабильного ацильного производного (2.71). Очевидно,
комплексы родия(Ш) типа (2.72) могут играть важную роль в стадий-
ной изомеризации квадрициклена, катализируемой родиемс
Аналогичный путь окислительного присоединения был предложен для
изомеризации кубана, катализируемой родием(1) [ 173]
и для изомеризации гомокубанов в диены, катализируемой родием(1)
или палладием(П) [174]
В обоих случаях при стехиометрической реакции между напряженным
карбоциклом и [ RhCl (СО)2]2 были выделены [173, 175] соответствую-
щие ацильные комплексы 2.73 и 2.74.
Кроме указанного выше механизма (согласованное окислительное
присоединение) переходные металлы могут также катализировать
скелетную перестройку по стадийному механизму, включающему об-
96
ГЛАВА 2
273
разование металлокарбкатионов. По этому механизму напряженный
карбоцикл активируется на металлическом центре посредством вну-
тримолекулярного гетеролитического присоединения. Так, при пере-
стройке бициклопропенила в бензол Дьюара [реакция (2.12)] наиболее
вероятным начальным интермедиатом является карбкатион серебра
2О75, образующийся, по крайней мере формально, путем гетеролити-
ческого присоединения а -связи циклопропенила к металлическому
центру. Перестройка посредством миграции связи дает карбкатион
2О76, который далее замыкается в цикл с образованием бензола Дьюа-
ра [ 176]. Доказательство существования обоих карбкатионов было
2.75
2.76
(2.12)
получено путем "сольватного захвата”, т.е. использования подходя-
щего нуклеофила для связывания карбкатиона. Так, соединение 2,78
было выделено [ 177 ] при изомеризации 2.77, катализируемой перхло-
ратом серебра в метаноле при —20° С, а при изомеризации 2 79, ка-
тализируемой серебром в аналогичных условиях, был получен мето-
ксикарбоцикл 2.80 [ 178]. Катализ процессов скелетной перестройки
не ограничивается только переходными металлами. Во многих слу-
чаях простое протонирование напряженного карбоцикла, за которым
следуют разрыв связи и перестройка образующегося карбониевого
иона, предлагает простой путь для обхода ограничений по симметрии.
Так, эффективным катализатором перестройки три-^р^-бутилпризма-
на 2.66 [реакция (2.10)] служит 0,003 М соляная кислота (т1/2 = 35 мин),
однако продуктом является только 1,2,4-замещенный бензол, тогда
как при использовании в качестве катализатора нитрата серебра
(см. выше) получаются четыре продукта. Для перестройки призмана
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
97
2.80
эффективны самые разнообразные катализаторы, включая Hg(II),
Sn(II), Zn(II\ РЬ (II) и даже 1,3,5-тринитробензол. Предполагается,
что ключевым фактором, определяющим каталитическую активность
на первой стадии процесса, которая представляет собой образование
промежуточных комплексов с переносом заряда, является кислотность
по Льюису [170].
Как и для большинства реакций, катализируемых переходными
металлами, протекание определенных стадий механизма (даже среди
многообразия точно установленных механизмов) зависят от природы
субстрата и металла катализатора; однако, несмотря на неопределен-
ность некоторых механизмов, нет никаких сомнений относительно
способности комплексов переходных металлов катализировать широ-
кий круг процессов изомеризации и перестройки.
В двух последних разделах мы концентрировали наше внимание
на гомогенных каталитических реакциях, которые хотя сами по себе
и важны, но не находят прямого крупномаштабного промышленного
применения. Теперь мы перейдем к рассмотрению гомогенных ката-
литических систем, которые используются для получения органичес-
ких соединений в количестве более 1000 т в год.
Дополнительная литература по изомеризации,
катализируемой переходными металлами
Bird С.IF. Transition Metal Intermediates in Organic Synthesis.- New
York: Academic Press, 1968, p. 69.
98
ГЛАВА 2
Davies N.R. The isomerization of olefins catalysed by palladium and
other transition-metal complexes. — Rev. Pure Appl. Chem. 1967,
v. 17, p. 3.
Tsutsui M., Courtney /Lott- Rearrangements of organ otrans it ion metal
compounds. — Adv. Organometal Chem., 1977, v. 17 po 241.
Davidson J\LO — In: Inorganic Reaction Mechanism. Vol. 5. — London:
Chemical Society, 1977, p. 346.
Mango F'D* Mol ecular orbital symmetry conservation in transition metal
catalysis0 — Adv. Catal., 1969, v. 20, po 291.
Paquette L,A,9 Preparative aspects of silver ion catalysed rearrange-
ments of polycyclic systems. — Synthesis, 1975, p. 347.
Bisop K.C., Transition metal catalysed rearrangements of small ring
organic molecules. — ChemoRevc, 1976, v. 76, p. 461.
2.3. Карбонилирование
Два основных выдающихся события в области гомогенных ката-
литических реакций карбонилирования произошли в конце 1930-х и на-
чале 1940-х годов в Германии. В 1930-х годах Отто Роелен (фирма
Ruhrchemie), изучая химию процесса Фишера — Тропша, обнаружил
[ 179, 180], что при взаимодействии алкенов с СО и Н2 в присутствии
кобальтового катализатора при повышенной температуре и давлении
могут образовываться альдегиды
Этот процесс, получивший название "гидроформилирование”, посколь-
ку он формально включает присоединение формальдегида (Н—СНО) к
алкену, за сорок с ‘лишним лет своего развития достиг такого уровня,
что в настоящее время играет ведущую роль среди промышленных
процессов с участием гомогенных каталитических систем.
Другое выдающееся открытие произошло примерно в то же время.
В.Реппе, работавший в компании LG.Farben, открыл [ 181],» что кар-
бонильные комплексы металлов VIII группы способны катализиро-
вать целую серию реакций карбонилирования,, в которых в качестве
субстрата используются алкины, алкены или спирты, а продуктами, в
зависимости от используемого сореагента, являются карбоновые кис-
лоты (если сореагент - вода) или производные карбоновых кислот
(если сореагент — спирт или амин). Некоторые типичные примеры
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ 99
’’реакций Реппе” представлены ниже. Реакции с участием СО и Н2О
обычно называются реакциями гидрокарбоксилирования:
НС===СН + СО + Н9О -^Н2С==СНСООН
HC-zClI + СО + ROH —»H,C=CHCOOR
АЛ
R'C==CH + СО + ROH -~H,C=C(R')COOR
Ал
rch=ch2 + со + н2о —rch2ch2cooh
rch=ch2 + со + R'OH ->rch2ch2coor
rch=ch2 + CO + OH2 — rch2ch2conhr
rch2oh + co — rch2cooh
В этом разделе мы последовательно рассмотрим каждую из трех ти-
пов ’'реакций Реппе”, т.е. реакции, в которых в качестве субстратов
используются ацетилены, олефины и спирты. В разд. 2.4 мы рассмот-
рим реакции гидроформилирования, катализируемые гомогенными
системами. Реакции Реппе представляют большой самостоятельный
раздел каталитической химии СО, и на следующих примерно десяти
страницах мы сможем только в небольшой степени рассмотреть эту
область; перечень более подробных работ дан в конце этого раздела.
2.3.1. Карбонилирование алкинов. При взаимодействии водно-
органического раствора ацетилена с СО при 15(Р С и 30 атм в присут-
ствии каталитических количеств тетракарбонила никеля с высокой
селективностью (> 90%) образуется акриловая кислота [ 182]:
Н(Ь=СН 4- Н2О
Ni (СО)4 _
150° С/?0 атм
СН2=СНСО2Н
При проведении этой реакции в водном спирте (ROH) образуются соот-
ветствующие акриловые эфиры CH2CHCO2R. В случае метилацетиле-
на при проведении реакции в водном метаноле основным продуктом
(> 80%) является метилметакрилат:
СН3С==СН 4 СО + МеОН ^СН2==С(Ме)СО2Ме
Хотя карбонильные комплексы других металлов VIII группы, напри-
мер Fe(CO)5, также катализируют эти реакции, наиболее предпочти-
тельным металлом с точки зрения как активности, так и селективно-
сти, несомненно, является никель. В промышленности никель обычно
вводят в виде бромида или иодида никеля, которые оба в условиях
реакции (как правило, 200° С и 100 атм) быстро превращаются в карбо-
нильные соединения никеля. В тех случаях, когда вода является един-
100
ГЛАВА 2
СО (CO)2XNi-C=CH2
Р и Со 2J2O Каталитический цикл для изомеризации алкина, катализируемой
никелем.
ственным сореагентом, т.е# когда необходимо получить в качестве про-
дукта кислоту, растворителем часто служит тетрагидрофуран [ 53].
Обобщенный каталитический цикл для процесса карбонилирования ал-
кина показан на рис. 2.12.
Считают [ 183, 184 ], что каталитически активным соединением
является гидридокарбонильное соединение никеля типа 2.81, которое
образуется путем окислительного присоединения НХ к тетракарбониль-
ному комплексу никеля. Поскольку Ni (СО)4 является 18-электронным
комплексом, то реакции окислительного присоединения всегда должна
предшествовать диссоциация по крайней мере одного карбонильного
лиганда. При введении никеля в виде галогенида никеля(П) соответ-
ствующий галогеноводород (НХ) образуется в ходе восстановления
исходного NiX2 в Ni(CO)4:
NiX^ 5C0bH20^Ni(C0)4+C02 + 2НХ
В отсутствие галогенида соединение 2.81 может образоваться (хотя и
намного медленнее) из сореагента — спирта или воды, в этом случае
X =OR или ОН. Наивысшая скорость реакции карбонилирования дости-
гается, когда X = I [ 185],< однако в промышленности, в основном из-
за проблем коррозии, обусловленной HI, обычно предпочитают исполь-
зовать бромидную систему (Х= Вг ). Образовавшееся гидридокарбониль-
ное соединение может координировать субстрат (алкин RC—CH), да-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
101
вая 18-электронный интермедиат никеля(II) 2.82. Присоединение по
правилу Марковникова никельгидридного лиганда к координированно-
му алкину, протекающее, вероятно, через четырехцентровое переход-
ное состояние, аналогичное описанному в разд. 2.1 для катализато-
ров гидрирования, дает ст -алкеновый комплекс 2.83. Миграция ли-
ганда, т.е. С-алкена к карбонилу, которой способствует входящий ли-
ганд (СО), превращает 2Л83 в 2.84. Каталитический цикл завершает-
ся образованием акриловой кислоты или соответствующего R '-эфира,
а также никельгидридного комплекса 2.81 в результате гидролиза
(R' = Н ) или алкоголиза (R' = алкил) соединения 2.84.
Хотя в промышленных условиях обычно давление составляет поряд-
ка 100 атм, а температура выше 200° С [ 53], в случае синтеза метил-
метакрилата селективность выше 80% была достигнута при относи-
тельно низких температурах (132° С) и давлении (13,5 атм) [ 184].
Основным побочным продуктом реакции является метилкротонат, ко-
торый образуется путем присоединения гидрида к алкину в 2.82
против правила Марковникова, давая линейный ст -алкеновый интерме-
диат 2,85 (R = Me}:
(COLXNi-CH =CHR 2.85
В большинстве работ основное внимание уделялось использованию ни-
келевых катализаторов, тем не менее сообщается, что Pd Br2[ P(OPh)3]2
в присутствии хлорной кислоты в мягких условиях является активным
катализатором метоксикарбоксилирования ацетилена, приводящим
к образованию метилакрилата с селективностью 95% [ 186 L Так же
как и в случае никеля, каталитически активным соединением, по-види-
мому, является гидридный интермедиат. В отсутствие кислоты, но в
присутствии тиомочевины (NH2CSNH2) и следов кислорода хлорид
палладия при комнатной температуре и атмосферном давлении катали-
зирует реакцию между ацетиленом, СО и метанолом, приводящую к
образованию с 90%-ной селективностью диметилмалеата 2.86 [187].
PdCl2/NH2CSNH2
СН—СН + СО + МеОН ------------------МеО2ССН=СНСО2Ме
°2 2.86
Предполагаемый каталитический цикл для этой системы показан на
рис. 2.13. Считают, что активным соединением является карбониль-
ный катион палладия 2.87, содержащий в дорбшс-положении два лиган-
да тиомочевины. Нуклеофильная атака метанола или метоксианиона
координированного карбонила дает 16-электропное карбоксисоедине-
ние 2.88. Координация ацетилена с последующей миграцией карбо-
102
ГЛАВА 2
2.89
Рис. 2.13. Каталитический цикл для катализируемого палладием синтеза
диметилмалеата из ацетилена, СО и метанола.
ксилыюги лиганда через четырехцентровое переходное состояние
дает сг-карбоксиалкеновый интермедиат 2.89, который может затем
координировать СО, после чего претерпевать следующую, реакцию
миграции лиганда - карбоксиалкена к карбонильному углероду, обра-
зуя 2.90, Нуклеофильная атака метанола по связанному с металлом
карбонильному углероду приводит к выделению продукта (малеата) и образо-
ванию палладийгидридного соединения 2.91. В присутствии кислорода,
да у НС 1 и СО соединение 2.91 каким-то образом превращается в ак-
тивное соединение 2.87. Механизм этой последней стадии еще не ясен,
и образование воды в последней стадии никла является просто умозри-
тельным.
В настоящее время карбонилирование алкинов, катализируемое
палладием, если и нашло, то очень ограниченное промышленное при-
менение, однако эта реакция демонстрирует потенциальные возмож-
ности этой многогранной области химии. Катализируемые никелем
системы для производства акриловой кислоты и эфиров, главным обра-
зом метилметакрилата, еще используются в промышленности, в ос-
новном в Западной Германии и в Восточной Европе. Однако этот способ
теряет свое значение по сравнению с другими процессами, основан-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
103
ними на использовании более дешевого сырья, такого, как пропилен
и ацетон. Большая часть потребляемого западными странами метил-
метакрилата — важного мономера для производства акриловых поли-
меров (т.е. прозрачных полимерных материалов, широко используемых
в качестве заменителей стекла) — производится циангидринным спосо-
бом из ацетона:
СН3СОСН3 + HCN ^(CH3)2C(OH)CN
(СН3)2С(ОН) CN + сн3он + h2so4
-»СН2=С(СН3 ) СООСНз + nh4hso4
В последние пять лет реакция Реппе, в которой в качестве сырья ис-
пользуется ацетилен, вновь привлекла к себе внимание в основном
для малотоннажного производства специальных химических веществ,
таких, как бутиролактон и пирролидон [ 53]..
2.3.2. Карбонилирование алкенов. Хотя наиболее подходящим
металлом для карбонилирования алкинов является никель, для карбонилиро-
вания алкенов часто используют катализаторы на основе кобальта, родия,
железа, рутения или палладия. Общим интермедиатом в большинстве, если
не во всех реакциях карбонилирования алкенов, катализируемых переходны-
ми металлами, являются ацильные комплексы металле®, образующиеся в при-
веденной ниже последовательности реакций миграции лиганда:
Н
I
м
о
с
м—с—с—н
Можно представить, что эти реакции протекают через нуклеофильную
атаку по карбонильному атому ацилметаллического соединения с од-
новременной регенерацией металлгидридного соединения в случае
нуклеофила типа HNu:
HNu
I
м— Н + Н—С—С—(2.13)
I I ^Nu
104
ГЛАВА 2
Очевидно, что вид продукта этой реакции существенным образом за-
висит от природы нуклеофила, например, если HNu = НОН, то обра-
зуются кислоты, если HNu = HOR - эфиры , если HNu = H2NR, то ос-
новными продуктами являются амиды. В том случае, когда в качестве
нуклеофила выступает водород, происходит образование альдегидов
(эту реакцию мы рассмотрим в разд. 2.4, посвященном гидроформили-
рованию).
Реакция карбонилирования алкенов протекает не всегда так, как
это следует из схемы реакции (2.13). Так, карбонилирование этилена
в спирте ROH в присутствии палладиевого или никелевого катализато-
ра кроме ожидаемого продукта CH3CH2CO2R приводит к образованию
кетоэфира CH3CH2C(O)CH2CH2CO2R, содержащего две молекулы
этилена и две молекулы СО [ 188]:
О
сн2=сн2 + со + вон —ch3ch2co2r + ch3ch2£ch2ch2co2r
Образование эфира у-кетокапроновой кислоты (2.93) можно объяс-
нить на основании представленной ниже последовательности реакций
миграции лигандов. Эта последовательность аналогична описанной
ранее последовательности реакций синтеза диметилмалеата (рис. 2.13),
катализируемого палладием.
и сн2сн2 ч 2
М—ССН2СН3 -----2—* *1—ССН2СН
II
о
СО'
—> М-СН2СН2ССН2СН3 сн сн,ссн2сн, -
2 II 3 2 2 // 2 3
о 0 о
— м-ссн2сн2ссн2сн3М-Н +
о
2.92
СН3СН2ССН2СН2С
2.93
В отсутствие внешнего нуклеофила дикетокомплекс 2.92 может под-
вергаться внутримолекулярной атаке или реакции циклизации, давая
лактон 2.94. Гели М = Pd, то циклизация, которая происходит в ре-
зультате нуклеофильной атаки карбонильного кислорода, находящегося
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
105
в 4-положении по связанному с палладием ацильному углероду, сопро-
вождается миграцией металла от первого атома углерода к четверто-
му в Металлзамещенный лактон (2.94) может распадаться путем
р-элиминирования водорода, находящегося в положении 3 или 5, давая
продукты - два ненасыщенных лактона 2.95 и 2.96
2.92
м—н +
2.95
СН2СН
снсн3
Такого типа реакция циклизации происходит при нагревании раствора,
содержащего этилен и СО, в присутствии каталитических количеств
PdCl2(PPh3) 2 [ 189 ]- Каталитический цикл этой системы представлен
на рис. 2.14. Полагают, что каталитически активным соединением яв-
ляется палладийгидридный комплекс 2.97, который первоначально об-
разуется в результате реакции между PdCl 2(PPh3) 2 и этиленом
[ 1901:.
PdCl2(PPh3)2+ СН2СН2 — PdCl(H) (PPh3) 2 + СН2=СНС1
2.97
Аналогичные реакции циклизации наблюдались также в случае никеле-
вых и кобальтовых катализаторов [ 190 L Что касается крупнотоннаж-
ного промышленного использования реакции Реппе, то основным про-
цессом карбонилирования алкенов является процесс производства про-
пионовой кислоты из этилена с использованием в качестве катализа-
тора карбонила никеля при рабочем давлении 200 - 250 атм [ 53].
2.3.3. Карбонилирование спирта. Несомненно, наиболее важ-
ным для промышленного развития реакции Реппе в последние годы бы-
ло открытие Паулика и Рота (компания Monsanto), что в присутствии
иода определенные растворимые карбонильные комплексы родия или
106
ГЛАВА 2
Р и с. 2.14. Каталитический цикл для катализируемой палладием социкли-
зации этилена и С0о
иридия при низком давлении (30 — 40 атм) способны катализировать
карбонилирование метанола с образованием уксусной кислоты:
СН3ОН +- СО —* СН3СООН. Другой процесс, в котором используется
промотированный иодом карбонильный кобальтовый катализатор, был
ранее разработан фирмой BASF, тем не менее процесс, разработан-
ный Monsanto, с использованием родиевого катализатора имеет опре-
деленные преимущества, поскольку он проводится в значительно бо-
лее мягких условиях (180° С и 35 атм по сравнению с 230° С и
600 атм для кобальта)- Для этого процесса требуется более низкая
концентрация катализатора (~10~3 М по сравнению с 10”1 ЭД), и он
позволяет получать продукт с более высокой селективностью (> 99%
по сравнению с 90%) [ 45]. Высокая стоимость энергии требует бо-
лее эффективного использования сырья, и поэтому более низкие энер-
гетические затраты, т.е. более мягкие рабочие условия и повышенная
селективность родиевой системы, с избытком компенсируют высокую
стоимость родия по сравнению со стоимостью кобальта. Аналогичная
ситуация возникает в отношении кобальтового и родиевого катализа-
торов гидроформилирования, которые мы будем рассматривать в сле-
дующем разделе. Фирма Monsanto начала крупнотоннажное производ-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
107
ство уксусной кислоты с использованием родиевой системы в 1970 г.,
и предполагается, что к середине 1980-х годов с помощью этого про-
цесса ежегодно будет производиться более 1 млн. т. уксусной кисло-
ты [ 45].
В реакции карбонилирования метанола каталитически активным
соединением является анион дикарбонилдииодродия(1) 2.98. Если ро-
дий вводится в форме тригалогенидной соли RhX3, то анион может
легко образоваться по реакции восстановительного карбонилирования
[ 192, 193].*
RhX34- ЗСО + Н2О—*[RhX2(CO)2]- + СО2 + 2Н+ + Х~
2,99
В том случае, когда X не является иодом, соединение 2.99 превра-
щается в 2.98 путем обмена галогенида на иодид-ион промотора, при-
сутствующего в растворе. Обычно иод вводят в систему в виде метил-
иодида, водной иодистоводородной кислоты или элементного иода.
Введение родия в форме mp^m-фосфинового комплекса, например
RhX(CO) (РРЬ3)2, существенно не изменяет природы каталитически
активного соединения, поскольку в реакционных условиях связь
родий — фосфор разрывается и ??фе^-фосфиновый лиганд быстро пре-
вращается в четвертичную фосфиновую соль, например [ P(Ph)3Me]+I“",
которая не образует связи с родием. Предполагаемый каталитический
цикл для этого процесса показан на рис. 2.15. В случае родия, субстра-
том служит метилиодид, а не метанол, и первая стадия цикла пред-
ставляет собой окислительное присоединение метилиодида к 16-элек-
тронному соединению родия (I) 2.98, в результате чего образуется
18-электронный октаэдрический комплекс родия(Ш)2в99. В результате миг-
рации лиганда (метила к карбонильному углероду) 2.99 превращается
в 16-электронное ацильное соединение 2.100, которое быстро присое-
диняет молекулу СО, вновь превращаясь в 18-электронное соединение
родия (III) 2.101. Восстановительное элиминирование ацетилиодида из
2.101 завершает каталитический цикл. Ацетилиодид быстро подверга-
ется сольволизу метанолом или водой, которые присутствуют в реак-
ционной среде, давая метилацетат или уксусную кислоту и HI. Обра-
зовавшийся HI реагирует с метанолом, давая метилиодид, который
далее реагирует с родием. В процессе карбонилирования равновесие
сильно сдвинуто вправо, так что в рабочих условиях концентрация
свободного HI очень мала: СН3ОН + HI СН31 + Н2О. С практи-
ческой точки зрения это важно для создания реакторов и заводов,
поскольку HI из-за коррозионных свойств является одним из наиболее
нежелательных веществ в химической технологии.
108
ГЛАВА 2
2.100
Рис. 2.15. Каталитический цикл для карбонилирования метанола, катали»
зируемого родием.
Кинетические данные, полученные для реакции карбонилирова-
ния, катализируемой родием [ 194], указывают на то, что лимитирую-
щей стадией в этом каталитическом цикле является окислительное
присоединение метилиодида к соединению родия (I) 2.98. На основе
полученных данных следует ожидать, что бромид и хлорид будут ме-
нее эффективно промотировать эту реакцию по сравнению с иодидом,
поскольку скорость окислительного присоединения RX к с/8-металлга-
логенидным комплексам обычно уменьшается в ряду I > Вг > С1 [33],
Скорость образования бромидного аналога соединения 2.99 путем
окислительного присоединения метилбромида к [ RhBr2(CO) 21~ по
крайней мере на порядок ниже скорости реакции с участием иодида
[45]. Дополнительные доказательства того, что лимитирующей ста-
дией является присоединение метилиодида, были получены путем ре-
гистрации инфракрасного спектра реакционного раствора в типичных
условиях проведения реакции (100° С, 6 атм) с использованием высо-
котемпературной спектрофотометрической кюветы высокого давле-
ния [ 195]. Спектр реакционного раствора содержал две сильные по-
лосы поглощения при 1966 и 2067 см-1, относящиеся к аниону
[RhI2(CO)2]~ Это указывает на то, что в растворе в основном присут-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
109
ствует этот анион. Эти данные служат серьезным аргументом в поль-
зу того, что лимитирующей стадией является реакция окислительного
присоединения. Применение инфракрасной спектроскопии к изучению
данной реакции является еще одним примером того, что гомогенные
каталитические системы поддаются детальному изучению спектроско-
пическими методами.
Карбонильные комплексы иридия так же представляют собой эф-
фективные катализаторы карбонилирования метанола при относитель-
но мягких условиях [191]. Однако поведение таких систем более слож-
но по сравнению с родиевыми системами. Ддя иридиевой системы воз-
можны два каталитических цикла. Возможность их реализации в боль-
шой степени зависит от концентрации иодида, которая является основ-
ным фактором, определяющим положение равновесия (2.14)*:
IrI(CO)3 +1 [1г12(СО)2Г (2.14)
+ СО—I-
2Л02 2.103
Два предполагаемых каталитических цикла иридиевой системы пока-
заны на рис. 2.16. Хотя соединение 2ЛОЗ, образованию которого бла-
[1гСН312(СО)2]1ида, Б [lr(CH3CO)I2(CO)2]1w,„2
2.107 2.108
2.102
Рис. 2.16. Каталитические циклы для карбонилирования метанола, катали-
зируемого иридием.
110
ГЛАВА 2
гоприятствует высокая концентрация иодида, является полным анало-
гом дикарбонилдииодродия(1) 2.98, каталитический цикл А, по кото-
рому следует этот иридиевый комплекс, заметно отличается от цик-
ла, который проходит его родиевый аналог (ср. с рис. 2.15). Присое-
динение метилиодида к 2.103 к образованием октаэдрического
метильного комплекса иридия (III) 2.104 протекает быстро, тогда как
в случае родия это присоединение является лимитирующей стадией.
Однако 2.104 превращается в ацильный комплекс 2.106 медленно,
и, как полагают, это превращение осуществляется через 18-электрон-
ный 1 трикарбонилдииодометильный интермедиат 2.105, Соединение
2.105 не было обнаружено спектроскопически, и предположение о его
присутствии, хотя бы в исключительно низкой концентрации, основа-
но на кинетических данных. Независимо от того, каким образом
2.104 превращается в 2.105, этот процесс медленный, чем он заметно
отличается от аналогичного превращения комплекса родия. В реакци-
онных условиях основным соединением, определенным с помощью
инфракрасной спектроскопии, является 2.104, и при отсутствии избы-
точного давления СО (выше 5 атм) это соединение проявляет незна-
чительную склонность к перестройке в ацильный комплекс. Кроме то-
го , присутствие избытка иодида, которое способствует образованию
2.103, ингибирует превращение 2.104 в 2.106. Образовавшееся ациль-
ное соединение 2.106 претерпевает быстрое восстановительное
элиминирование, давая ацилиодид и завершая каталитический цикл.
При низкой концентрации иодида основным соединением иридия, кото-
рое наблюдается спектроскопически, является нейтральный трикарбо-
нилиодидный комплекс 2.102, и реакция карбонилирования протекает
по нижнему каталитическому циклу Б. В этом случае лимитирующей
стадией является присоединение метилиодида с одновременным от-
щеплением СО. Независимые эксперименты, проведенные не в усло-
виях каталитического карбонилирования, показали [45], что 1г1(С0)3
реагирует с метилиодидом, давая в качестве основного продукта ди-
мерное соединение [ 1гСН312(СО)2]2- Однако остается неясным, в мо-
номерной или димерной форме присутствует 2.107 в условиях реакции
(^10 атм, 125° С). Аналогичная ситуация наблюдается для ацильного
соединения 2.108, которое легко образуется из 2.107 через 18-элек-
тронное трикарбонильное соединение 2.105. Перегруппировка 2.107
в ацильное соединение до координации СО вызвала бы превращение 16-элек-
тронного комплекса в 14-электронный, которое, согласно правилу Толма-
на ”16 и 18 электронов”, маловероятно. Образовавшееся ацильное
соединение координирует СО, и после восстановительного элиминиро-
вания апилиодида происходит завершение каталитического цикла.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
111
Хотя в общем родиевые и иридиевые катализаторы имеют сравни-
мую активность при допустимых промышленных условиях, из-за слож-
ности иридиевой системы (без учета других факторов) маловероятно,
чтобы она в значительной мере смогла заменить родиевую гомогенную
каталитическую систему карбонилирования метанола в уксусную кис-
лоту.
Этот раздел мы начали с замечания, что в основном родиевые го-
могенные катализаторы потеснили кобальтовые катализаторы процес-
сов получения уксусной кислоты. Теперь мы переходим к рассмотре-
нию других процессов, в которых родиевые системы начинают конку-
рировать с традиционными кобальтовыми катализаторами, а именно к
рассмотрению процессов гидроформилирования.
Дополнительная литература по гомогенному карбонилированию
Bird CcW., Transition Metal Intermediates in Organic Svnthesis. —
New York: Academic Press, 1967, Ch. 7 and 8.
Falbc /• Carbon Monoxide in Organic Synthesis, Berlin: Springer-Ver-
lag, 1970. [Есть русский перевод: Фалъбе И, Синтез на основе
окиси углерода: Пер. с нем. Богорадовской П.М. и Рахлиной Е.Н./
Под ред. Имянитова Н.С. - Л.: Химия, 1971].
Ryang Мо Organic synthesis with metal carbonyls. — Organometal Chem.
Rev., Section A. 1970, v. 5, p. 67.
Thompson Whyman R. — In: Transition Metals in Homogeneous Ca-
talysis (ed. G.N.Schrauzer), New York, Marcel Dekker, 1971 , p. 147.
Maitlis The Organic Chemistry of Palladium, — London: Academic
Press, v. II, p. 18.
Heck R'F, Organotransition Metal Chemistry, A Mechanistic Approach. —
New York: Academic Press, 1974, Ch. 9.
He nr ici-Olive S., Olive S. Reactions of carbon monoxide with transition
metal-carbon.— Transition Met. Chem., 1976, v. 1, p. 77.
Davidson P*J*9 Hignett R.R., Thompson D.T. — In: Catalysis, vol. I,
London: Chemical Society, 1977, p. 369.
Forster D. Mechanistic pathways in the catalytic carbonylation of met-
hanol by rhodium and iridium complexes. — Adv. Organometal Chem.,
1979, v. 17, p. 255.
f
2Л« Гидроформилирование
По объему производимой продукции гидроформилирование, т.е.
присоединение частиц Н и СНО к двойной связи, является примером
самого крупного промышленного использования растворимых катали-
112
ГЛАВА 2
заторов на основе переходных металлов. В настоящее время от 3,6
до 4,5 млн. т альдегидов или их производных получается ежегодно с
использованием процессов, основанных на применении гомогенных ко-
бальтовых или родиевых каталитических систем, при этом некоторые
заводы имеют мощность свыше 300 000 т в год [ 196]. Как упомяну-
то во введении к разд. 2.3, эта реакция была открыта в 1938 г. О.Рое-
леном,работавшим в то время в компании Ruhrchemie в Германии
[ 179, 180]. Он нашел, что при рециркуляции олефиновой фракции про-
дуктов синтеза Фишера — Тропша над гетерогенным кобальтсодержа-
щим катализатором синтеза в присутствии синтез-газа, т.е. смеси
СО и водорода, наблюдается заметное возрастание концентрации аль-
дегидов в рециркулирующем потоке [ 185]. Вначале думали, что реак-
ция обусловлена присутствием гетерогенного кобальтового катализа-
тора, однако последующие работы показали, что истинным катализа-
тором являются растворимые'карбонилы кобальта, при этом реакция
протекает как гомогеннокаталитическая в жидкой фазе. Процесс час-
то называют "оксопроцессом", где "оксо” — сокращение от термина
"оксонирование”, обозначающего присоединение кислорода к двойной
связи. Однако термин "гидроформилирование" более точен и, веро-
ятно, более полезен при характеристике этого типа реакции.
Первичным продуктом гидроформилирования является альдегид,
содержащий на один атом углерода больше, чем исходный алкен:
RCH=CH2 + со 4- Н2 RCH2CH2CHO + RCH(CH3)CHO (2.15)
нормальный мзо-продукт
продукт
В отсутствие изомеризации в зависимости от ориентации присоеди-
нения Н—СНО к двойной связи образуются два возможных продук-
та из несимметричного исходного алкена. В том случае, когда суб-
стратом служит терминальный алкен, получаются нормальный и изо-
продукт, согласно схеме реакции (2.15). За исключением особых слу-
чаев, предпочтительно образуется нормальный альдегид, и относи-
тельный выход нормальных продуктов (отношение н/изо) является
важным параметром промышленного процесса гидроформилирования;
в общем случае, чем выше этот параметр, тем лучше. Основным
сырьем для гидроформилирования служит пропилен. Из более чем
5 млн. т пропилена, использованного в 1976 г. в Европе на производ-
ство химических продуктов, около 1 млн. т было израсходовано для
гидроформилирования [ 53]. В западных странах ежегодно с исполь-
зованием этого процесса получается около 2,7 млн. т бутаналя [ 1Ь
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ системы в действии
113
Большая часть «-бутаналя либо превращается в 2-этилгексанол пу-
тем последовательного проведения альдольной конденсации и гидриро-
вания (так называемый ”альдокс”-процесс), либо непосредственно
гидрируется в «-бутанол. Так как продукт изостроения находит зна-
чительно меньшее применение и, следовательно, обладает намного
меньшей коммерческой ценностью, то достижение высокого отноше-
ния н! изо имеет особую важность при гидроформилировании пропиле-
на. Аналогично при производстве высших (С7+)*спиртов гидроформили-
рованием с последующим (или одновременным) гидрированием селек-
тивность по «-изомерам является важным фактором при разработке
процесса, так как продукты нормального строения особенно полез-
ны в производстве биоразлагаемых моющих средств и высококачест-
венных пластификаторов (пластификаторами являются химические
продукты, добавляемые к синтетическим смолам, например поливи-
нилхлориду, для улучшения прежде всего способности к формованию).
В промышленном процессе гидроформилирования используется
три типа гомогенных комплексов переходных металлов. Отметим их в
порядке их современного промышленного значения (и исторического
развития процесса): а) простые карбонильные (скорее гидридокарбониль-
ные) комплексы кобальта; б) гидридокарбонильные комплексы кобальта
с третичными фосфинами в качестве лигандов; в) гидридокарбонильные
комплексы родия с ягр^-фосфиновыми лигандами. В следующих раз-
делах мы рассмотрим поочередно каждую из этих систем, затем срав-
ним системы на основе кобальта и родия, кратко опишем другие ком-
плексы переходных металлов, которые катализируют эту реакцию, и,
наконец, затронем проблему совершенствования катализаторов асим-
метричного гидроформилирования.
Так как реакция имеет большое промышленное значение, в пер-
вых четырех подразделах определенное внимание уделено промыш-
ленному применению процесса.
2о4а1. Кобальтовые катализаторы0 Ао Не модифицированные ко-
бальтовые карбонильные системы. Процессы, использующие немо-
дифицированные , т.е. без специально введенных инертных лигандов,
кобальтовые карбонильные катализаторы, были впервые пущены в
эксплуатацию в конце 1940-х годов; до сих пор эти процессы по мощ-
* Часто используемый в западной литературе символ означает сум-
му веществ с числом углеродных атомов п и выше^
114
ГЛАВА 2
(CO)3CoCCHR (Снз) (C0)4CoCHR(CH3)
2.116 2.114
RCH(CHO)(CH3) /
CoH(CO)4 ^co fV H
(2.109) -coSf ’ 2
RCH,CH,CHO „ CoH(CO)3
2 2 Vх* 2.110
О AH2
(CO)3CoCCH2CH2R
2.775
A A
CH2=CHR (CO)3CoCHR (CH
2.772
H CHR у/
(CO)3Co-||
I CH2
3^
(CO)4CoCH2CH2R
(CO)3CoCH2CH2R
2.7/7
P и c. 2.77. Каталитические циклы для гидроформилированияs катализируемо-
го родием» Цикл А приводит к продуктам нормального строения, цикл Б-к
продуктам изо-строения»
ности составляют около 80% от всех ныне существующих производств
гидроформилирования. Типичные температуры, при которых проводят-
ся эти процессы, лежат в интервале ПО — 180° С, давления — в интер'-
вале 200 — 350 атм; кобальт первоначально вводится в реактор в ви-
де кобальтовых солей (например, в виде ацетата кобальта), карбони-
лов кобальта [ например, Со2(СО) 8] или даже металлического кобаль-
та. Независимо от того, как введен кобальт, в условиях реакции он
превращается в истинный предшественник катализатора СоН(СО)4
(2.109). Перед вхождением в каталитический цикл, показанный на
рис. 2.17, эта 18-электронная частица теряет СО (лиганд) с образова-
нием четырехкоординированной 16-электронной частицы Со(Н)(СО)3
(2.110), которая и является катализатором в этом процессе:
СоН(С0)4 СоН(СО)з + со
(2 Л 6)
2.109 2.110
Следующий этап в каталитическом цикле включает координацию ал-
кена (с переходом 18-электронной частицы в 18-электронную) с пос-
ледующей быстрой миграцией гидрида с образованием алкильного
производного кобальта (I). В отсутствие миграции двойной связи (мы
вернемся к вопросу изомеризации в условиях гидроформилирования
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
115
несколько позже) водород, связанный с металлом, может присоеди-
няться либо к а-атому углерода (присоединение по правилу Марковни-
кова), либо к р -атому углерода (присоединение против правила Мар-
ковникова) с образованием либо г/зо-алкильного продукта 2,112, либо
продукта нормального строения (2.111). Учитывая электронные фак-
торы, мы можем ожидать присоединения водорода от металла к «-уг-
леродному атому с образованием разветвленного продукта, так как
металл является центром, богатым электронами. Однако по стеричес-
ким причинам большая металлсодержащая группа, очевидно, предпо-
читает быть связанной с наименее замещенным атомом углерода с
образованием линейного алкильного производного. При низких тем-
пературах (0° С) в отсутствие СО электронные факторы, по-видимо-
му, доминируют [ 198] и алкильный комплекс нормального строения
2.111 является основным продуктом стадии миграции гидрида. Появ-
ляется соблазн предположить, что направлением первичного присоеди-
нения водорода, связанного с металлом, полностью определяется
протекание реакции с точки зрения линейности получаемого альдеги-
да, т.е. отношения и/изо. То, что это не так, показывает наблюдение,
что при использовании такого внутреннего алкена, как гептен-3, при
обычных условиях гидроформилирования получается в основном нор-
мальный октаналь. Аналогично при 110° С и давлении СО около
90 атм отношение н/изо, достигаемое при использовании пентена в
качестве субстрата, не слишком отличается при использовании пен-
тена-1 или пентена-2 в качестве сырья (4,5 и 3,1 соответственно).
При низком парциальном давлении СО (1,7 атм) получается одинако-
вое отношение н/изо, равное 1,3 [ 153].
В обычных условиях гидроформилирования изомеризация коорди-
нированного алкена протекает быстро по сравнению с гидроформили-
рованием, и, таким образом, направление начального присоединения
гидрида имеет только кажущееся значение в определении отношения
н/изо. Мы умышленно использовали выражение "координированный
алкен", так как из изучения [ J] с использованием дейтерированных
алкенов-1, по-видимому, следует, что полная изомеризация может
происходить с участием только кобальталкенового комплекса, вероят-
но, через последовательность стадий 1,2-водородного сдвига (см.
разд. 2.4) без выделения свободного изомеризованного алкена. Что
касается кобальта, то по стерическим причинам для тг-алкенового
комплекса предпочтительна структура, которая образуется с алкено-
вым субстратом, имеющим двойную связь на конце молекулы. Таким
образом, в условиях, когда скорость изомеризации велика, преобла-
дает образование а- и [3-альдегидов.
116
ГЛАВА 2
Образовавшийся алкилтрикарбонилкобальт 2ЛИ или 2.112, име-
ющий 16 валентных электронов, присоединяет молекулу СО с образо-
ванием 18-электронного тетракарбонильного интермедиата 2.113 или
2 Л14. При миграции алкильной группы к карбонилу образуются ациль-
ные производные 2Л15 или 2Л16. Полагают, что именно эта послед-
няя стадия оказывает наибольшее влияние на линейность образующего-
ся продукта [199],
Для немодифицированных кобальткарбонильных каталитических
систем наиболее важным параметром в определении отношения и/изо
для данного алкена является парциальное давление СО. Когда субстра-
том служит пропилен, возрастание давления СО от 2,5 до 90 атм при
100° С приводит к увеличению отношения н/изо почти в 3 раза, от
1,6 до 4,4 [ 153]. Парциальное давление водорода оказывает лишь не-
большое влияние на линейность продукта, а отношение н!изо также
не зависит существенно от температуры реакции для температур вы-
ше 50° С. Зависимость селективности реакции от парциального дав-
ления СО может быть понята с точки зрения стерических затрудне-
ний в переходном состоянии при превращении алкильной группы в
ацильную [ 199]. В каталитическом цикле, изображенном на рис. 2.17,
показано, что это превращение связано с превращением 16-электрон-
ной частицы в 18-электронную:
(CO)3Co’CH2CH2R -CCL- (CO)4CoiCH2CH2R —
2.111 2.113
ч.в.э. 16 18
О
(СО) Со1 CCH,CH9R
2Л15
16
Переходное состояние для конечной стадии этого процесса, т.е. для
стадии 2Л13 -> 2Л15, может быть представлено формулой 2Л17.
Комплексы 2ЛИ w 2 Л15 — это 16-электроиные координационно-нена-
сыщенные частицы, поэтому они обладают явной тенденцией присоеди-
нять любые свободные донорные молекулы. Таким образом, повыше-
ние парциального давления СО бу детине только благоприятствовать
превращению 2ЛИ в 2Л13, но также и превращению 2Л15 в 18-элек-
тронный пятикоординированный комплекс (СО) .Со 1 СОСЩСПЛ1
I Ал Ал
(2.118).
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
117
(CO)+Co'COCH2CH2R
2.113
ч.в.з,- 1 8
О
(CO)3Co,CCH2CH2R
2.7/5
16
О
---- (CO)4ColCCH2CH2R
2.118
18
При высоком парциальном давлении СО независимое существование
16-электронных частиц 2Л15 маловероятно; можно представить, что
превращение 2.113 в 2Л18 в этом случае будет протекать по согла-
сованному механизму с образованием переходного состояния типа
2Л19, при этом образование связи кобальт - карбонил протекает
одновременно с разрывом связи кобальт — алкил и образованием свя-
зи алкил - карбонильный лиганд. Благодаря присутствию дополнитель-
2.119
118
ГЛАВА 2
кого, хотя и только начинающего входить в координационную сферу,
карбонильного лиганда частица 2.119 имеет несколько более запол-
ненную координационную сферу, и, следовательно, обладает несколь-
ко большей энергией. Это различие в энергии становится более оче-
видным, когда алкильная частица - диалкильное производное, как в
случае комплексов, приводящих к образованию замещенного продук-
та, показанного в цикле Б на рис. 2.17. Таким образом, повышение
давления СО благоприятствует протеканию цикла А по сравнению с
циклом Б, что приводит к возрастанию селективности по линейным
продуктам.
Следующим этапом в каталитическом цикле после образования
ацильных комплексов кобальта - как трикарбонильных (2 Л 5 или
2Л16), так и тетракарбонильных (типа^ЛЗ) - является гидрогено-
лиз с образованием конечного продукта (альдегида). В условиях гид-
роформилирования этот гидрогенолиз, почти без сомнения, протекает
через окислительное присоединение водорода к кобальт (I)-ацильному
комплексу [где FC = CH2CH2R или CHR(CH3)]:
(СО) Со1 COR'+ Н
1V
2.120
2 -ИСО)пСош (H)2COR'
2.121
Когда n = 3, то для протекания такого процесса препятствий нет, так
как это процесс перехода 16-электронного комплекса в 18-электрон-
ный между обычными начальной и конечной степенями окисления. Од-
нако, когда п = 4, как в случае 2.118, мы имеем дело с переходом
18-электронного комплекса в 20-электронный, что маловероятно. Та-
ким образом, в случае тетракарбонилацильного комплекса кобальта
стадии присоединения водорода должна предшествовать стадия дис-
социации карбонила:
(C0)4CoTC0R'(СО)Со1 COR+-CO
о
(2.17)
При высоком давлении (СО •: Н2== 1:1) равновесие реакции гидрофор-
милирования (2.17) сдвинуто влево и стадией, определяющей скорость
каталитического никла, является гидрогенолиз ацильного комплекса.
Действительно, тетракарбонилацильный комплекс типа 2.118 в слу-
чае R = С6Н13 был обнаружен методом ПК-спектроскопии в системе
кобальт/октен-1 при 150° С и 250 атм [200]. Альтернативный про-
цесс гидролиза заключается в реакции между трикарбонилацилко--
бальтом и СоН(СО)4 :
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
119
(CO)3CoCR' + (СО)4СоН
I
о
(СО)зСо-—С—R'
(СО)4Со—-Н
+ Со2(СО)7
При этом карбонилгидрид кобальта регенерируется либо по реакции
Со2(СО) + Н, — СоН(СО)3 к СоН(С0)4
либо по реакциям
Со2(СО)7 + СО -*Со2(СО)8
Со,(СО)8 кН2 —>2СоН(СО)4
Хотя и было установлено, что восстановление ацильных комплексов
кобальта при действии СоН(С0)4 протекает в условиях стехиоме-
трической реакции [ 201], кинетическое рассмотрение показывает,
что такая последовательность реакций не может играть главной роли
в каталитическом процессе.
Как мы уже видели, возрастание парциального давления СО
оказывает благоприятное давление на выход линейных продуктов. Од-
нако возрастание давления СО неблагоприятно сказывается на об-
щей скорости гидроформилирования. Кинетические измерения [ 202]
показали, что скорость обратно пропорциональна парциальному дав-
лению СО:
d[ альдегид] f'dt- &Набл [алкен] [ кобальт]р(Н2)р(СО)-1 (2.18)
Замедляющее влияние возрастания давления СО может быть понятно
при рассмотрении равновесия, представленного уравнениями (2.16) и
(2.17). В обоих случаях тетракарбонильные частицы не являются ка-
талитически активными, и в обоих случаях их образованию благопри-
ятствует высокое парциальное давление СО. Ингибирующее действие
СО вызывается также протеканием следующих реакций:
СоН(СО)3 + СоН(СО)4 Со2(СО)7 4- н2
Со2(СО)7 + СО -Со2(СО)8 (2.19)
2.122
Для регенерации активных гидридокарбонильных частиц октакарбонил-
дикобальт(О) 2.122 должен потерять карбонильный лиганд в соответ-
120
ГЛАВА 2
ствии с равновесной реакцией (2.19)- Очевидно, что протеканию про-
цесса но способствуют высокие давления СО. Какая из трех причин
ингибирующего действия СО преобладает, зависит от типа процесса
гидроформилирования и условий реакции, однако все равно высокое
давление р(СО) — это плохо с точки зрения скорости реакции, прав-
да , при одном существенном исключении. Определенное минимальное
парциальное давление СО, которое зависит от температуры, требует-
ся для стабилизации карбонилгидридокобальта в растворе. При 120° С
требуется примерно 10 атм, при 200° С - около 100 атм [185]. Ниже
этих значений давления металлический кобальт выпадает в виде осад-
ка, а это еще хуже для скорости реакции. Именно необходимость под-
держивать кобальт в растворе определяет те высокие давления, ко-
торые используются в промышленных процессах гидроформилирова-
ния с немодифицированными кобальтовыми катализаторами.
Мы уже вкратце упоминали (разд. 2.1.2, Г), что СоН(СО)4 может
действовать как катализатор гидрирования альдегидов в спирты. Это
не очень активный катализатор: при температурах ниже 180° С толь-
ко примерно 10% альдегида превращается в спирты. Аналогично толь-
ко небольшая доля (^1%) исходного алкена гидрируется в алкан.
При температурах выше 250' С (давление СОн-Н2 около 250 атм) дости-
гается более эффективное превращение альдегидов в спирты. Актив-
ной частицей цикла гидрирования (см. рис. 2.18) является, вероятно,
как и в случае цикла гидроформилирования, трикарбонилгидридный
комплекс 2.110. Предполагается [ 203], что начальная координация
альдегида протекает с образованием комплекса 2.123 с несимметрич-
ной связью тг-связью. Миграция гидридного лиганда к карбонильному
атому углерода в альдегиде через образование четырехцентрового
переходного состояния типа 2.126 может приводить затем к получе-
нию ст-связанных алкоксипроизводных 2.124. Окислительное присоеди-
нение водорода к этому 16-электронному cP-комплексу дает
электронный промежуточный дигидридокобальт (III) 2.125, который
затем может распадаться в результате восстановительного элимини-
рования с образованием спирта, чем и завершается каталитический
цикл. Учитывая кислотные свойства гидридного комплекса кобальта
(в водном растворе СоН(С0)4 - такая же сильная кислота, какНС!),
мы могли бы ожидать , что присоединение фрагмента Н— Со по аль-
дегидной группе С=9 произойдет в противоположном направлении,
т.е. с образованием гидроксиалкилкобальтового комплекса типа
2.127.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
121
рис. 2.18. Каталитический цикл для катализируемого кобальтом гидриро-
вания альдегидов в спирты.
RHC—О
I I
Н—Со(СО)3
2.126
ОН
RHC — Со(СО)3
(2.127)
Окислительное присоединение водорода к 2.127 с последующей мигра-
цией гидрида и восстановительным элиминированием полученного спир-
та, как это описано для алкоксильного цикла (рис. 2.18), завершило
бы каталитический цикл. По стерическим причинам комплекс 2.124,
очевидно, предпочтительнее, чем 2J27. Учитывая это, а также данные
[204] о том, что из альдегидов в условиях гидроформилирования мо-
гут образовываться эфиры муравьиной кислоты, можно утверждать,
что алкоксильный маршрут является предпочтительным циклом [ .190] j
Как и в случае реакции гидроформилирования, катализируемое
кобальтом гидрирование альдегидов ингибируется СО. Однако в пос-
леднем случае скорость обратно пропорциональна квадрату парциаль-
ного давления СО [ 203}:
d[ спирт1 /с/т = &набл [ альдегид] [< кобальт]р(Н2) р (СО) ~2 (2.20)
Это отчасти объясняет низкую гидрогенизационную активность в уело-
122 ГЛАВА 2
виях гидроформилирования, когда парциальное давление СО часто пре-
вышает 100 атм.
На крупнейших действующих заводах с производительностью око-
ло 350 000 т/год используются немодифицированные кобальтовые ка-
тализаторы. Такие процессы разработаны фирмами Ruhrchemie
или BASF [ 196]. Упрощенная схема процесса Ruhrchemie для по-
лучения /z-бутаналя, //-бутанола и изобутанола показана на рис. 2.19.
В реактор 1 подаются исходный пропилен, синтез-газ (смесь СО и Н2)
и кобальтовый катализатор. Температура поддерживается в интерва-
ле 130 - 175° С, а общее давление - в интервале 200 - 300 атм; кон-
центрация катализатора (в процентах металла к исходному алкену)
составляет от 0,1 до 1%. Продукты реакции, включая кобальтовый
катализатор, выводятся сверху реактора 1 и проходят в сепаратор 2f
в котором смесь подвергается термической или химической обработ-
ке с образованием органической фазы, не содержащей кобальта, и
неорганической фазы, обогащенной кобальтом. Эта стадия
процесса известна под названием "декобальтизации”. В реакторе 3
фазу, обогащенную кобальтом, подвергают обработке с целью ее пере-
вода в форму, удобную для возвращения в реактор гидроформилирова-
ния 1.
Рис. 2.19. Упрощенная технологическая схема для процесса гидроформи-
лирования с использованием немодифицированных кобальтовых катализато-
ров (фирма Ruhrchemie) (1 — 7 СМ. В тексте.)
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
123
Большое число методов было разработано для удаления и рецик-
ла кобальта, в большинстве из них используется стадия кислотной
обработки. Схема, наиболее приемлемая для процесса Ruhrchemie,
включает начальную нейтрализацию кислого гидридокарбонилкобальта
водным раствором бикарбоната: СоН(СО)4 + NaHCO3 ->Na[Co(CO)4] +
4- Н2О + СО2 с последующим подкислением водной неорганической
фазы серной кислотой с целью регенерации гидридокарбонилкобальта:
2Na[Co(CO)4] + H2SO4 —>2CoII(CO)4 + Na2SO4. Органическая фа-
за, из которой выделен кобальт и которая содержит в основном н-
и «зо-бутаналь приблизительно в отношении 4:1, направляется в рек-
тификационную колонну 4, где альдегиды отделяются от других орга-
нических продуктов реакции, например спиртов и продуктов альдоль-
ной конденсации. Если спирты являются целевым продуктом, тогда
альдегиды направляются в реактор гидрирования 6, содержа-
щий гетерогенный катализатор гидрирования. Полученные спирты за-
тем направляются во вторую ректификационную колонну 7. Кубовые
остатки, т.е. продукты альдольной конденсации, из колонны 4 могут
быть направлены на крекинг в аппарат 5, где они подвергаются терми-
ческому или каталитическому разложению на составляющие их альде-
гиды, которые затем направляются в реактор 6.
Процессы, в которых используются немодифицированные кобаль-
товые катализаторы, характеризуются большими энергетическими
расходами, связанными с высокой температурой и давлением, при
которых проводится процесс. Отметим также, что целевым продуктом
является w-изомер, а лучший показатель отношения н!изо в случае
этих систем составляет от 3:1 до 4: 1, что указывает на возможность
улучшения процесса. Первый значительный успех в преодолении отме-
ченного недостатка был достигнут в начале 60-х годов, когда Слог
и Муллинэ [ 205 - 207 ] (компания Shell Oil, Калифорния) открыли,
что добавление трет- фосфиновых лигандов к карбонилкобальтовым
системам приводит к образованию комплексов, для стабилизации ко-
торых не требуется высоких давлений СО и которые способны катали-
зировать превращение алкенов-1 в «-спирты с селективностью выше
90%.
Бо Кобалъткарбонилъные системы, модифицированные третич-
ными фосфинами. По сравнению с немодифицированными системами
катализаторы гидроформилирования на основе карбонилов кобальта
[208] и третичных алкилфосфинов, например Р(Ви)3> более стабиль-
ны. При температурах реакции от 100 до 180° С необходимо давление
124
ГЛАВА 2
смеси Н2 + СО всего 5—10 атм (в случае СоН(СО)4 необходимо дав-
ление 100 — 320 атм). Новые каталитические системы имеют повышен-
ную активность в гидрировании, что приводит к образованию в основ-
ном спиртов, а не альдегидов в условиях гидроформилирования. При
использовании в качестве субстратов линейных а-олефинов отношение
н!изо составляет обычно 8 : 1, что примерно вдвое превышает то, ко-
торое достигается в случае немодифицированных катализаторов. Впро-
чем, у новых систем есть и недостаток: их активность значительно
ниже, чем в случае карбонилов кобальта, не содержащих фосфиновых
лигандов: при 145 ° С гидроформилирование на немодифицированном
катализаторе протекает в пять раз быстрее, чем на модифицированном
при 180° С.
Механизм реакции очень близок к тому, который описан в пре-
дыдущем разделе для СоН(СО)^. Активной частицей является
CoH(CO)2L (2.127), который образуется при удалении карбонильного
лиганда из CoH(CO)3L (2.128) [ср. схему реакции (2.16)]:
CoH(CO)3L CoH(CO)2L +- СО (2.21)
2.128 2.127
Кобальт часто добавляют в форме стабильного диоктакарбонила Со 2(СО) 8; в
этом случае 2.128 образуется по следующим реакциям:
Со2(СО)8 + 2L^Co2(CO)6L2 4- 2СО
Co2(CO)6L, + Н2 2CoH(CO)3L
"Двойной” каталитический цикл в образовании первоначального аль-
дегида, а затем спирта, показан на рис. 2.20. Что касается немодифи-
цированных кобальтовых катализаторов, то для них оба цикла вклю-
чают стадии перехода 16-электронных частиц в 18-электронные (и об-
ратно), что и показано на внутренних циклах на рис 2.20.
Из-за возросших стерических затруднений, вызванных присутст-
вием громоздкого тр^-фосфинового лиганда (конический угол для
РВи3, т.е. для лиганда, который часто используется в системах гидро-
формилирования, составляет 132° по сравнению с 95° для карбониль-
ного лиганда [ 26]), комплекс СоН(СО)2Ь предпочтительнее реагиру-
ет с концевыми олефинами, чем с олефинами с внутренними двойными
связями, причем образуются тт-комплексы типа 2.129. Точно так же
переходные состояния, связанные с образованием наименее замещенных
алкильных или ацильных комплексов, оказываются более выгодными и в хо-
де всех последующих реакций миграции лигандов. В модифицирован-
ных кобальтовых системах именно стерический эффект третичного
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
125
L(CO),CoCH,CH,R
Р и Со 2о20 Каталитические циклы для процесса гидроформилирования/гид»
рирования, катализируемого кобальткарбонильными комплексами, модифици-
рованными трет-фосфиновыми лигандами.
фосфинового лиганда определяет высокие отношения н/изо. Поэтому
максимальное влияние, по-видимому, достигается в случае лигандов
с коническим углом около 130 °; лишь небольшое (если оно вообще
есть) улучшение отношения н/нзо достигается при переходе к более
объемистым триалкилфосфиновым лигандам. Таким образом, при ка-
талитическом гидроформилировании гексена-1 с использованием
СоН(СО)3 L в качестве исходного соединения выход линейных продук-
тов не зависит существенно от того, используется ли в качестве L
Р(изопропил)3 (6= 160°) или Р(и-пропил)3 (0 = 132°). Как показыва-
ют результаты, представленные в табл. 2.2, линейность продуктов по-
чти не зависит от конического угла трет -фосфиновых лигандов, но
в то же время она заметно зависит от основности лигандов (мерой ко-
торой служит величина рКа ) или от электронного фактора. Селектив-
126
ГЛАВА 2
Таблица 2.2. Гидроформилирование гексена-1 с использованием Co2(CO)8/2L
как предшественника катализатора (160° С, 70 атм, Н2: СО = 1,2 =1,2 :1) [209]
L рКа Электронный параметр v, см-1 [ 26 ] Конический угол 6, град [ 26 ] х 103, мин-1 [209] Выход линейного продукта, % Отношение альдегид: спирт [209]
Р(изо-Рг )3 9,4 2059,2 160 2,8 85,0
PEt о О 8,7 2061,7 132 2,7 89,6 0.9
P(w-Prk о 8,6 2060,9а 132 3,1 89,5 1,0
P(K-Bu), Л 8,4 2060,3 132 3,3 89,6 1,1
PEt2Ph 6,3 2063,7 136 5,5 84,6 2,2
PEt Ph, 4,9 2066,7 140 8,8 71,7 4,3
PPh. о 2,7 2068,9 145 14,1 62,4 11,7
aОценочные данные.
ность по линейным продуктам уменьшается ио мере возрастания v
(или уменьшения рКа ); этот эффект,наиболее выражен для арилфос-
финовых лигандов PEt2Ph, PEtPh2n PPh3. Параллельно с этим умень-
шением селективности по линейным продуктам происходит снижение
гидрирующей активности систем, что проявляется в увеличении отно-
шения альдегид: спирт, одновременно возрастает общая селективность
в гидроформилировании (увеличиваются значения кг, см. табл, 2.2).
Все эти три эффекта, к которым приводит изменение основности лиган-
да (или электронного параметра), могут быть поняты при учете сле-
дующего равновесия:
СО-ь CoH(CO)3L СоН(СО)4 + L
(2.22)
2.128 2.109
Равновесная концентрация СоН(СО)4 возрастает с уменьшением ос-
новности wp ^-фосфинового лиганда и повышением электронного па-
раметра v лиганда. Этого мы можем ожидать, так как основность ли-
ганда большей частью отражает ст-донорные свойства фосфора, и,
как это обсуждалось в разд. 1.2.3, Б, имеются попытки использова-
ния электронного параметра в качестве количественной характеристики элек-
тронных донорно-акцепторных свойств лиганда. Обе величины связаны с ла-
бильностью лиганда, таким образом, в случае трет-фосфинового ли-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
127
ганда, характеризующегося низким значением рКа или высоким зна-
чением v, будет заметнее происходить диссоциация комплекса 2.128
с последующим вхождением СО с образованием 2.109. Так как комп-
лекс СоН(СО)а является более активным , но менее селективным ка-
тализатором гидроформилирования, чем CoH(CO)2L, активность систе-
мы возрастает, а селективность уменьшается, когда мы переходим
от L = PEt3 к L = РРЬ3. Аналогичным образом дор^до-фосфиновые
комплексы являются более активными катализаторами гидрирования,
чем карбонильные, при этом гидрирующая активность снижается по
мере возрастания v.
Повышенная гидрирующая активность кобальт-дорвдо-фосфиновых
систем по сравнению с просто кобальткарбонильными системами при-
писывалась возрастанию гидридного характера в гидридах кобальта
при замещении карбонильной группы дор^до-фосфином. Будучи, несом-
ненно, более сильными ст -донорными и более слабыми тг-акцепторны-
ми, чем СО, триалкилфосфиновые лиганды повышают электронную
плотность на металле и гидридном водороде. Увеличение отрицатель-
ного заряда облегчает нуклеофильную миграцию лиганда, в данном
случае гидрида к углероду ацильной группы. Аналогично возрастание
электронной плотности на металле благоприятствует окислительному
присоединению молекулы Н Оба этих эффекта должны облегчать про-
текание гидридного цикла. Однако, видимо, наиболее важным [ 210]
является суммарный эффект от введения фосфиновых лигандов —
способность этих комплексов работать при пониженных давлениях СО.
Как и в случае с немодифицированными системами, возрастание
давления уменьшает скорость реакции. Это и неудивительно, так как
мы видели, что 16-электронные активные частицы образуются при
диссоциации СО. Впрочем, как можно видеть из сравнения кинетичес-
ких уравнений (2.18) и (2.20), ингибирующий эффект возрастания пар-
циального давления СО проявляется сильнее в реакции гидрирования,
где скорость обратно пропорциональна квадрату р(СО), по сравнению
с реакцией гидроформилирования, где в кинетическое уравнение вхо-
дит член р(СО)“\
Хотя и неясно, насколько важную роль играют электронные свой-
ства дорадо-фосфинового лиганда в повышении гидрирующей активности
системы, эти свойства, несомненно, важны в повышении стабильности,
что позволяет проводить реакцию при низком давлении, г Было сделано
предположение [211], согласно которому повышение стабильности
связано с тем, что доргдо-фосфины представляют собой более слабые
тт-акцепторы, чем СО, и, таким образом, большая доля тт-акцепторной
электронной плотности остается на карбонильных лигандах. По край-
128
ГЛАВА 2
ней мере настолько же важным фактором должно быть то, что трет-
фосфиновый лиганд обладает большими ст-донорными свойствами. Не
совсем ясно, является ли повышенная, электронная плотность на ко-
бальте результатом того, что фосфиновый лиганд меньше оттягивает
или же больше подает электронную плотность на металл. При повыше-
нии электронной плотности, -независимо от причины этого повышения,
большая ее доля становится доступной для связывающих орбиталей
металл - карбонил, и в результате комплексы значительно более ста-
бильны при введении ^-фосфиновых лигандов.
Возрастание стерического объема фосфинового лиганда не
только увеличивает селективность при снижении активности, но ме-
няет также природу стадии, лимитирующей скорость реакции. В слу-
чае немодифицированных кобальтовых катализаторов стадией, лимити-
рующей скорость, является гидрогенолиз кобальтацильных комплек-
сов, тогда как при использовании модифицированных систем самая
медленная стадия каталитического цикла - это начальная координация олефи-
на или, возможно, перенос гидридного лиганда и координированному олефи-
ну. Это в основном связано со стерическими помехами, создаваемыми
в координационной сфере дор^-фосфиновым лигандом, в результате че-
го модифицированные системы проявляют заметное предпочтение по
отношению к концевым (незамещенным) алкенам по сравнению с ал-
кенами с внутренней двойной связью. Так как комплексы являются
относительно хорошими катализаторами изомеризации, то селектив-
ность по отношению к образованию продуктов нормального строения
(но не обязательно общая скорость реакции) часто не зависит от то-
го, используются ли алкены с внутренней или концевой двойной связью.
Так, при использовании системы Со 2’(СО)8/2Р(С4Н9)3 в качестве
предшественника катализатора гидроформилирование гексена-1 или
гексена-2 при 195° С и 27 атм дает практически одинаковую (80 и 81%)
селективность по нормальным продуктам, хотя общая скорость реак-
ции в 2 раза медленнее с гексеном-2 [212]. При условии, что отноше-
ние кобальт : яф#ж-фосфин> 1, т.е. при образовании комплексов ти-
па СоН(СО)пР(С4Н9)3> как активность, так и селективность систе-
мы не зависят от дальнейшего введения фосфинового лиганда. При
соотношении Со : Р < 1 селективность системы снижается из-за
присутствия СоН(СО)4> а при низких давлениях СО снижается и ак-
тивность из-за разложения комплекса с образованием металлического
кобальта [ 118, 119].
При получении спиртов преимущество использования модифициро-
ванного катализатора состоит в том, что процесс может быть осущест-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
129
Рис. 2.21. Упрощенная технологическая схема для процесса гидроформили-
рования с использованием кобальтовых катализаторов, модифицированных
трет-фосфиновыми лигандами (фирма Shell),
в лен в одном реакторе, и нет необходимости проводить отдельную ста-
дию гидрирования. К сожалению, модифицированные системы на обла-
дают 100%-ной селективностью при восстановлении альдегидов.. Неко-
торая часть (обычно около 15%) исходного алкена также подвергается
гидрированию. Это представляет прямые потери, как экономические,
так и энергетические, поскольку алканы обладают значительно мень-
шей коммерческой ценностью, чем алкены, а превращение алканов в
алкены требует затрат энергии.
На рис. 2.21 показана упрощенная схема процесса гидроформили-
рования фирмы Shell с использованием модифицированных кобальто-
вых катализаторов. Обычно реакции проводятся при температурах
160 - 200° С и давлении смеси СО + Н2 50 — 100 атм. Спирты явля-
ются основным (^80%) продуктом, отношение н/изо обычно выше
8 : 1. По сравнению с процессом, в котором используется немодифи-
цированный кобальтовый катализатор (ср. рис, 2.20), требуется мень-
ше капитальных вложении, хотя из-за пониженной каталитической ак-
тивности модифицированных систем необходимо использовать реак-
торы большего объема для достижения той же конверсии за проход.
Однако из-за большей стабильности катализатора нет необходимости
в стадии декобальтизации, и отделение катализатора может происхо-
дить в первой ректификационной колонне после того, как реакционная
130
ГЛАВА 2
смесь, выходящая сверху реактора 1, отделяется от газа в сепарато-
ре 2. Катализаторный раствор возвращается в реактор через аппарат
4 для подготовки катализатора, в котором заменяются дезактивиро-
ванные компоненты катализатора. Окончательное разделение продук-
тов происходит в системе ректификации, представленной колоннами
5,6 и 7.
Хотя каталитическая система кобальт - до/шя-фосфины способна
эффективно работать в более мягких условиях, чем немодифицирован-
ный катализатор, модифицированная система проявляет пониженную
активность и увеличенную гидрирующую способность.
Следующим шагом в развитии промышленных каталитических про-
цессов гидроформилирования было использование родиевых катализа-
торов, модифицированных жре^-фосфинами.
2.412. Родиевые катализаторы. Потенциальные преимущест-
ва катализаторов гидроформилирования на основе родия по сравнению
с кобальтовыми синтезами были впервые установлены в середине
50-х годов, когда было обнаружено [213], что родий по сравнению с
кобальтом значительно более активен в гидроформилировании и спо-
собен эффективно работать при значительно более мягких условиях.
Как в случае немодифицированных кобальтовых катализаторов, родий
может быть введен в реакционную среду в различных формах [46],
т.е. в виде родия на носителе, в виде хлорных или карбоксильных со-
лей или в виде карбонильных комплексов, таких, как Rh4(CO)12 или
Ш12(СО)4С12 ; все эти соединения в условиях гидроформилирования
(70 - 150° С, общее давление 50 - 150 атм, Н2 : СО= 1:1) образуют
каталитически активные частицы RhH(CO)3. Здесь наблюдается пол-
ная аналогия с кобальтовым комплексом 2,110, и общий каталитичес-
кий цикл в основном тот же, что и в случае СоН(СО)3 (рис. 2.17).
Как и для кобальтовой системы, гидрогенолиз металл-ацильной свя-
зи в комплексе типа 2в115, Rh(COR) (СО)3, является стадией, опре-
деляющей скорость реакции. Но в противоположность кобальтовым
родиевые системы почти не проявляют активности в гидрировании аль-
дегидов, и, таким образом, конечная реакционная смесь почти не со-
держит спиртов, ^модифицированные родийкарбонильные катализа-
торы гидроформилирования - в 102 — Ю4 раз более активны, чем их
кобальтовые аналоги [ 196]. Они являются также высокоактивными
катализаторами изомеризации, но при всем этом такие системы ха-
рактеризуются слишком низким отношением н/изо^ которое редко
превосходит 1:1 даже в случае таких простых алкенов, как пропилен
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
131
(напомним, что для кобальтовой системы н/изо = 4 •: 1). Эта низкая
селективность вместе с высокой ценой родия, превышающей примерно
в 3500 раз цену кобальта, перевешивает преимущества высокой актив-
ности родия, в результате в настоящее время в промышленности нет
процесса, использующего немодифицированные родийкарбонильные ка-
тализаторы.
Основной успех в практическом применении систем гидроформи-
лирования на основе родия достигнут в начале 60-х годов, когда Слог
и Муллинэ [ 214] (из компании Shell Oil) использовали триалкил фосфи-
новые и арсиновые комплексы родия в качестве катализаторов гидро-
формилирования. Независимо от этого* Осборн, Янг и Уилкинсон
(1965 г.) в Имперском колледже в Лондоне обнаружили [215], что в
атмосфере Н2 + СО в растворе бензола из RhCl3(PPh3)3 образуется
каталитическая система, способная катализировать гидроформилиро-
вание пентена-1 и гексена-1 при нормальных условиях.
Некоторые дор£?я-фосфиновые комплексы родия, например
RhCl(CO)L2 (L = PBu3 или РРЬз), RhH(CO)(PPh3)3, RhCl(PPh3)3
были использованы в качестве предшественников катализаторов
[ 206]•• В условиях гидроформилирования, по-видимому, во всех слу-
чаях образуется гидридодикарбонилди-(^гт-фосфин)родий,
RhH(CO)2L2 (2.130). Это один из ключевых промежуточных комплек-
сов в этих системах. В случае хлорсодержащих предшественников об-
разование этих интермедиатов — медленный процесс, и реакция гидро-
формилирования часто характеризуется индукционным периодом, кото-
рый устраняется при добавлении органического основания (например,
триэтиламина) в реакционную среду. Когда предшественником катали-
затора является RhCl(CO)L2, комплекс 2Л30 может быть образован
путем последовательных реакций окислительного присоединения и
восстановительного элиминирования по следующей схеме:
Rh1 C1(CO)L, + Н2 -> RhmCl(H)2(CO)L2 -RhWOlL, ч-НС1
Rh1 H(CO)L2 + СО _RhIH(CO)2L2
Триэтиламин удаляет НС1; таким образом, реакция происходит по
схеме [152]:
R^CKCOIL, + Н2 + СО + Et,N — Rh1 H(CO),L2+ Et3NHCl
Если в качестве предшественника катализатора используется
RhH(CO)L3, то индукционный период не наблюдается и добавление
Et3N не оказывает влияния на скорость гидроформилирования. Ис-
132
ГЛАВА 2
СО
н
। -РРН3
,Rh 3
Ph3P।
3 СО
„ Н
Ph3R I
>h-CO
Ph3P^ I
3 CO
2.131
ОС I
>'Rh-|l
Ph3P^ I 4R
3 CO
RCH2CH2CHO 2.137
о Н
rch2ch2c-4..-h
2.136
2.134
P и Co 2.22o Диссоциативный каталитический цикл для реакции гидроформили-
рования алкенов в присутствии RhH(CO)2(PPh3) 2-
следование методом инфракрасной спектроскопии, проведенной в ус-
ловиях гидроформилирования, показало, что один и тот же (впрочем,
окончательно не охарактеризованный) комплекс образуется из
RhH(CO)(PPh3)3 и RhCl(CO) (PPh3)2/Et3N при повышенном давле-
нии СО + Н2 [217].
Два каталитических цикла были предложены для реакции гидро-
формилирования, катализируемой модифицированными родиевыми сис-
темами [ 152]. Они показаны на рис. 2.22 и 2.23. Первый цикл отчас-
ти аналогичен каталитическому циклу, который реализуется при полу-
чении альдегидов на модифицированных кобальтовых катализаторах
(нижний цикл на рис. 2.20); он включает начальную диссоциацию три-
фенилфосфинового лиганда из комплекса 2J.31 с образованием комп-
лекса 2О132, имеющего 16 валентных электронов. Координация алкена
с последующей быстрой миграцией гидридного лиганда приводит
к комплексу 2J.33, который быстро присоединяет третичный фосфино-
вый лиганд с образованием 18-электронных частиц 2Л34. Миграция
аллильного лиганда с последующим окислительным присоединением
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
133
со
н
PhqP^ I
3 jRh-CO
Ph3P*|
3 СО
2.131
CH^CHR
РЬ^Р
СНО
СН2
сн2
со
2.137
CH2CH2R
Rh-CO
Ph3p
РКР
3 со
2.134
Рис. 2.230 Ассоциативный каталитический цикл для реакции гидроформи-
лирования алкенов в присутствии RhII(CO)2(PPh3)2o
н
водорода дает комплекс 2.136, из которого путем восстановительного
элиминирования выделяется альдегид. Присоединением СО к комплек-
су цикл завершается. Медленной стадией, вероятно, является окисли-
тельное присоединение водорода к 2.135. То, что алкен присоединяет-
ся к дикарбонильному комплексу 2.132, а не к ди-wp ^-фосфиновым
комплексам 2О137, было подтверждено данными ЯМР-Ш-спектроскопии,
показавшими, что, если 2.137 и 2.131 присутствуют вместе в раство-
ре, только последний из них реагирует с этиленом при 25° С и 1 атм
[218].
В цикле, предполагающем ассоциативный механизм (рис. 2.23),
рассматривается присоединение алкена непосредственно к пятикоор-
динированной 18-электронной частице 2.131 без предварительной дис-
социации игрет-фосфина или карбонильного лиганда. Если это происхо-
дит по стадийному механизму с участием шестикоординированных ком-
плексов родия(1), то в таком процессе происходит изменение числа
валентных электронов от 18 до 20.
Можно отметить, что, хотя и нет теоретических оснований для
исключения такого процесса, все же допущение о его протекании про-
тиворечит опыту, лежащему в основе правил Толмана "16 — 18 элект-
ронов". При отсутствии прямых спектроскопических данных о сущест-
вовании комплекса 2.138 предпочтительным ( по сравнению с недиссо-
134
ГЛАВА 2
циативным превращением 2,131 в 2,134) является согласованный ме-
ханизм процесса, при котором образование связи с алкеном сопро-
вождается разрывом гидридной связи, т.е. с образованием переходно-
го состояния типа 2,139, Диссоциативный путь предполагает образо-
2.138
2.139
вание комплекса 2Л37 при удалении СО с последующим присоедине-
нием алкена, миграцией лиганда и вхождением СО в координационную
сферу с образованием 2,134. Этот путь был отвергнут на основе дан-
ных ЯМР, упомянутых выше, хотя в соответствии с правилом "16 -
18 электронов” он является предпочтительным перед рассмотренны-
ми стадийными или согласованными схемами.
Основное различие между ассоциативным и диссоциативным ка-
талитическими циклами реакции гидроформилирования заключается в
том, что в ассоциативном цикле у иона родия никогда не бывает мень-
ше двух яфет-фосфиновых лигандов. Ассоциативный цикл стерически
более "требователен"; можно ожидать, что это будет благоприятство-
вать образованию линейных продуктов. Действительно, эксперимен-
тальное наблюдение показывает, что повышение отношения 0ф<?^-фосфин:
родий в катализаторном растворе приводит к увеличению селективности
системы по отношению к линейным альдегидам. Таким образом, при
использовании RhH(CO)(PPh3)3 в качестве предшественника катали-
затора гидроформилирования пропилена при 100° С, 35 атм и СО : Но =
Л»
= 1:1 отношение н/изо только немного превосходит 1 : 1. При деся-
тикратном избытке трифенилфосфина достигается селективность по-
рядка 70% (отношение н/изо = 2 : 1) [ 219], а когда реакция проводит-
ся в трифенилфосфине как растворителе, что соответствует 600-крат-
ному избытку лиганда по отношению к родию, отношение прямоцепо-
чечных и разветвленных продуктов достигает 15,3 : 1 при 125° С и
12,5 атм [ 220]. В отсутствие специально введенного яфеде-фосфина
уменьшение парциального давления СО также приводит к повышению
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
135
селективности реакции. Оба этих эффекта могут быть объяснены на
основе равновесных стадий уравнения (2.24) [221, 222]. Высокие кон-
центрации лиганда и (или) низкое парциальное давление СО благопри-
ятствует преимущественному образованию частиц, имеющих более од-
ного /яр^-фосфинового лиганда.
+СО +СО
RhH(CO) (PPh3)3 RhH(CO)2(PPh3)2 RhH(CO)3(PPh3)
-PPh. -PPh3
з °
+ CO
RhH(C0)4
-PPh,
О
(2.24)
Хотя повышение отношения PPh3 : Rh увеличивает селективность
системы, это, к сожалению, снижает скорость гидроформилирования:
при использовании пропилена в качестве сырья скорость образования
бутаналя при 100° С и^35 атм снижается с 13 г/мин до 2,5 г/мин
по мере того, как отношение PPh3 • Rh возрастает с 5 : 1 до 50 : 1
[ 215]. Таким образом, при практическом проведении процесса часто
необходим компромисс между скоростью реакции и селективностью.
В дополнение к благоприятному влиянию на селективность увеличение
отношения PPh3 : Rh оказывает также благоприятный эффект на гид-
роформилирование терминальных алкенов потому, что при этом увели-
чении заметно снижается изомеризующая активность катализатора.
При 150° С и 35 атм при использовании RhCl(CO)(PPh3) в качест-
ве катализатора изомерный состав получаемых альдегидов не зависит
существенно от того, используется ли в качестве сырья гексен-1,
гексен-2 или гексен- 3 ; при этом около 20% продукта составляет 2-
этилпентаналь. В то же время при 20-кратном избытке PPh3 и при
гидроформилировании гексена-1 не обнаруживается 2-этилпентаналя,
а селективность по 1-гептаналю составляет 76,2% [ 216].-
Что касается влияния тр^-фосфинового лиганда, то из испытан-
ных фосфинов трифенилфосфин, по-видимому, оптимален с точки зре-
ния скорости реакции, селективности, а также стоимости использова-
ния. Катализаторы на основе триалкилфосфинов, как правило, облада-
ют пониженной активностью и селективностью. Так, при одинаковых
условиях родиевый катализатор, содержащий трибутилфосфин, был в
шесть раз менее активен и обладал на 10% меньшей селективностью,
чем катализатор, содержащий трифенилфосфин [221]. Эта ситуация
136
ГЛАВА 2
резко отличается от той, которая была найдена для модифицированных
кобальтовых систем. Однако в случае родия при низких давлениях
СО трифенилфосфин может эффективно конкурировать с СО за коор-
динационные места, и даже в отсутствие избытка PPh3 при давлении
СО менее 10 атм из RhH(CO)(PPh3) 3 образуется мало RhH(CO)^ или
он не образуется совсем. Пониженная активность в случае системы
Rh/P(C4Hg)3 не совсем понятна, хотя она и может быть обусловле-
на трибутилфосфиновым лигандом, который является лучшим ст-доно-
ром, чем трифенилфосфин, и поэтому стабилизирует дигидридоациаль-
ные комплексы типа 2.136 настолько, что это приводит к снижению
общей скорости реакции.
Одним из недостатков модифицированных кобальтовых систем яв-
ляется их склонность гидрировать алкены. Потенциально это может
быть проблемой и в случае родиевых систем, так как в отсутствие СО
комплекс RhH(CO)(PPh3)3 является эффективным катализатором гид-
рирования алкенов. Однако в присутствии избытка трифенилфосфиново-
го лиганда и (или) при возрастании парциального давления СО гидри-
рующая активность подавляется. Эти же факторы также снижают ско-
рость гидроформилирования, снижение этой скорости на порядок мень-
ше , чем снижение скорости гидрирования. Так, при использовании в
качестве исходного комплекса RhCl(CO) (PPh3) 2 в случае гексена-1
при 50-кратном избытке трифенилфосфина, 100° С, 35 атм, Н2 : СО =
=1: 1в течение 17 ч образуются только следы гексана. Аналогичным
образом в случае промышленного процесса, который будет описан ни-
же, при использовании, пропилена в качестве сырья, как указано в
[ 223], менее 2% олефина гидрируется в пропан.
Промышленная разработка процесса гидроформилирования с ис-
пользованием растворимых родиевых катализаторов, обычно с исполь-
зованием RhH(CO) (PPh3) в качестве исходного соединения, была
осуществлена совместно фирмами Union Carbide Corporation, Davy
International Ltd. и Johnson Matthey. Первые заводы: ОДИН (в Пуэрто
Рико) производительность 130 000 т/год и-бутаналя из пропилена и
два (в Тексас-Сити) производительностью 45 000 т/ год пропаналя из
этилена — были пущены в середине семидесятых годов. Заканчивает-
ся строительство завода для получения бутаналя (в Швеции) произво-
дительностью 100 000 т/год. В общих чертах упрощенная схема этого
процесса идентична той, которая описана для процессов, использующих
кобальтовый катализатор, модифицированный тр^-фосфином (рис. 2.21).
Из-за высокой стоимости катализатора необходимо введение дополни-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
137
тельной стадии очистки сырья (пропилена, СО и Н2) для снижения кон-
центрации серусодержащих каталитических ядов. В результате значи-
тельно большей активности родиевой системы (примерно в 103 раз)
может достигаться большая конверсия за проход, что позволяет либо
использовать реактор меньшего объема, либо же совсем исключить
рецикл субстрата. Использование пониженных температур и давлений
(примерно 100° С и 20 атм вместо 180° С и 80 атм) упрощает проекти-
рование завода и снижает капитальные затраты.
Прежде чем перейти к рассмотрению катализаторов, основанных
на использовании иных металлов, чем кобальт и родий, сопоставим
описанные выше три процесса.
2.4.3. Сравнение кобальтовых и родиевых систем. В табл.
2.3 приведены некоторые данные о промышленном использовании трех
каталитических систем, а именно: немодифицированной кобальтовой
системы, системы на основе кобальта, модифицированного юр^-фос-
финами, и системы Rh/PPh3. Хотя истинные условия работы катали-
заторов и значения селективностей являются хорошо охраняемым ко-
мерческим секретом, все же из данных табл. 2.3 можно сделать неко-
торые заключения. При переходе от кобальтовой системы к системе
(Со + L) и далее к системе (Rh + PPh3) наблюдается заметное сниже-
ние энергетических затрат как благодаря более мягким условиям
процесса, так и большей селективности по конечному продукту. Отно-
сительно высокие потери сырья за счет образования алкана в системе
Co/L компенсируются до некоторой степени возможностью получения
спиртов в одну стадию. С другой стороны, если целевым продуктом
является альдегид, т.е. бутаналь для производства 2-этилгексанола,
то образование спирта может быть недостатком процесса. Более бла-
гоприятные энергетические показатели при применении модифициро-
ванных систем способствовали возрастанию их использования в послед-
ние годы, когда промышленность стала ощущать все возрастающий
рост энергетических затрат. Однако экономия энергии оплачивается
в данном случае ценой за родий, которая выше цены кобальта прибли-
зительно в 3500 раз. Затраты на родий компенсируются до некоторой
степени большей (102 — 103) активностью родиевой системы, что позво-
ляет использовать более низкие концентрации катализатора. Однако
для того, чтобы избежать экономического ущерба, потери катализато-
ра должны быть минимальны. Даже потери 3 - 5 частей на миллион бу-
Таблица 2.3. Данные о процессах гидроформилирования с использованием кобальта и родия
о со
аз
ФЯЗОе i-cccq.
Н d о О < О < EI
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
139
дут соответствовать потерям от 10 до 15 центов на 1 кг бутаналя, по-
лученного из пропилена. При цене родия 30 000 долл./кг завод произ-
водительностью 100 000 т/год, работающий при загрузке мощности на
85%, потеряет от 87 до 127 тыс. долл, за счет потери катализатора.
Существует еще проблема доступности металла. В 1974 г. производст-
во родия во всем мире доходило до 6 т по сравнению с 22 000 тонн ко-
бальта только в западном полушарии [ 224 ] (в 1976 г. из 4,29 тонн
добытого родия около 25% приходилось на долю Советского Союза). Не-
смотря на эти трудности, гомогенные процессы, основанные на исполь-
зовании родия, успешно развиваются [225, 226 ] благодаря тому, чта
пониженные капитальные затраты и меньшие расходы энергии, по-види-
мому, перевешивают высокую стоимость катализатора. Высокая ста-
бильность системы Rh — трифенилфосфин часто позволяет отгонять
продукт непосредственно из катализаторного раствора. Для производ-
ства бутаналя был описан барботажный реактор, в котором газообраз-
ный пропилен, водород и СО барботируют через раствор катализатора,
а целевой бутаналь непосредственно удаляется в виде паров с отходя-
щими газами [ 227 ].
2.4.4. Катализаторы на основе других металлов. Сравнение
относительной гидроформилирующей активности переходных металлов,
которые легко образуют карбонильные комплексы, было опубликовано
в 1977 г. [ 228]. Как и ожидалось, родий оказался непревзойденным:
Металл
Относительная
активность
Rh > Со > Ru > Мп > Fe > Сг,Мо, W, Ni
103 - 104
10-2 10-4 10-6
0
Наиболее изученный рутениевый катализатор гидроформилирования
образуется из Ru(CO) 3(PPh3)2 [ 229]. В относительно жестких уело*,
виях (120° С, 100 атм, СО •: Н2 = 1:1) достигается 99%-ная селектив-
ность получения альдегида из алкена, хотя отношение и/изо было не-
удовлетворительным, так как редко превышало 3 •: 1. Полагают, что
каталитически активной частицей является дигидрокомплекс 2Л41, ко-
торый образуется при окислительном присоединении водорода к 2Л40
с одновременной или предшествующей потерей СО. Стадией, определя-
ющей скорость реакции в цикле гидроформилирования, показанном на
рис. 2.24, по-видимому, является окислительное присоединение водо-
рода, и если комплекс 2Л41 не добавляется сам по себе в реакционную
140
ГЛАВА 2
rch2ch2cho
PPh3
ОС. I X.CO
RCH2CH2(O)C' I 4H
2 2 PPh,
PPh3
। ло
ОС- Ru
I 40
PPh3
C0 2.140 CO
н2
СО
PPh3
ОС* । zH
^Ru_
OC^I H
PPh3
2.141
PPh3
ОС-J H
.Ru
OC^ I ^CHjCHjR
PPh3
2.142
PPh3
PPh3
ОС- ’ x- H
. Ru
н
R
PPh3
P и Co 2o24o Каталитический цикл для реакции гидроформилирования
алкенов в присутствии Ru(CO)3 (PPh3)3. Показан только каталитичес-
кий цикл, приводящий к образованию линейных продуктов.
среду, то наблюдается индукционный период (ср. наблюдения для ро-
диевой системы при использовании RhCl(CO) (PPh3)2 в качестве ис-
ходного компонента катализатора). Цикл для рутениевой системы от-
личается как от циклов для кобальта, так и для родия в том, что присо-
единение алкена происходит после присоединения водорода. Алкен пред-
почитает координироваться прежде всего с частицами, содержащими
формально рутений(Ц), чем с богатым электронами комплексом руте-
ния(О) (2,140). Как и модифицированные кобальтовые или родиевые
катализаторы, рутениевые системы проявляют заметное предпочтение
по отношению к терминальным алкенам, хотя конечное отношение
н/изо значительно ниже (около 2 : 1 по сравнению с 8 : 1 и 10 : 1 для
С© и Rh соответственно). Введение избытка иф^до-фосфина повышает
отношение н! изо до 5 : 1, но заметно снижает скорость конверсии
(около 10% за 20 ч в расплавленном трифенилфосфине как раствори-
теле). В отсутствие введенного яф^яг-фосфина селективность по нор-
мальным продуктам в основном та же, что была найдена для
RhH(CO)2(PPh3)2 в отсутствие избытка трифенилфосфина. Вероятно,
в обоих случаях стерический контроль реакции осуществляется на
стадии превращения гидридного лиганда, т.е. на стадии образования
комплекса 2.133 в родиевом цикле, показанном на рис. 2.22, и на ста-
дии образования комплекса 2.142 (рис. 2.24). На этих стадиях только
один яф^яг-фосфиновый лиганд связан с ионом металла, и стерические
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
141
препятствия, которые, по-видимому, являются наиболее важным фак-
тором в определении отношения н/изо, сводятся к минимуму.
Недавним пополнением арсенала гомогенных катализаторов гид-
роформилирования является платино-оловянная система, сообщения о
которой появились в конце 70-х годов [ 203, 231 ]. В отсутствие хло-
рида олова хлоридные комплексы платины, содержащие трт-фосфино-
вые и дор^т-арсиновые лиганды, обладают пренебрежимо малой актив-
ностью в гидроформилировании. Когда галогенид олова(П) (предпочти-
тельно SnCI 2) вводится в систему, получаются высокоактивные и вы»
сокоселектйвные катализаторы гидроформилирования. Эта ситуация
напоминает то, что было найдено в случае гидрирования, когда добав-
ление SnCl2 к PtHCl(PPh3)2 превращает почти инертный комплекс в
активные катализаторы гидрирования [19].
K2Pt С14 обладает пренебрежимо малой активностью в гидрофор-
милировании. Добавление пятикратного избытка SnCl2 приводит к по-
лучению системы, способной к гидроформилированию гептена-1 в
смесь октана (60%), изомеризованных гептенов (13%) и w-гептенов
(10%) при 66° С, 100 атм и отношении СО : Н2 = 1 • 1- Добавление
двух полярных эквивалентов трифенилфосфина к системе, т.е. исполь-
зование Pt С12 (РРЬ3)2 вместо К2 Pt С16, повышает как активность,
таки селективность системы. При использовании 200 молей гептена-1
на 1 моль платины и при соотношении Pt : PPh3 ; SnCl2, = 1:2:5
получена 100%-ная конверсия гептена при 60° С и 100 атм. Селектив-
ность по октаналю была 85%, отношение н/изо = 9 : 1, менее 5% изо-
меризованных гептенов или гептена было обнаружено в реакционной
смеси [ 231]. Полагают, что активной каталитической частицей в
этом последнем случае является PtH(SnCl 3)(СО) (РРЬ3) (2.143). Ка-
талитический цикл гидроформилирования с участием этого комплек-
са показан на рис. 2.25. Комплекс 2.143 аналогичен предложенному
для гидрирующей системы, содержащей PtHCl(PPh3)2SnCl2, в кото-
рой образуется Pt H(SnCl 3) (РРЬ3)2. Таким образом, различие между
двумя системами заключается в том, что в промежуточном комплек-
се трифенилфосфиновый лиганд замещен на одну молекулу СО.
Что касается селективности в гидроформилировании с получени-
ем м-октаналя, то не наблюдается какой-либо заметной зависимости
как от стерических (конический угол 0), так и от электронных (элек-
тронный параметр v) свойств фосфорсодержащего донорного лиган at
при условии , что он обладает основностью, достаточной для сохране-
ния координации с платиной во время всего хода реакции. При исполь-
142
ГЛАВА 2
RCH(CH3)C(O)Pt(SnCI3)(CO)L RCH (CH3) Pt(SnCl3)(CO)L
H2
RCH(CH3)C(O)Pt(SnCI3)(CO)L
H
RCH2CH2CHO^ "
V
RCH2CH2C(O) Pt (SnCt3)(CO)L
H
PtH(SnCL)(CO)L
Г 2.143
rch=ch2
HPt(SnCI3)(CO)L
ch2=chr
HPt(SnCl3)(CO)L
I
RCH=CH
H?-* \ t
RCH2CH2C(O)Pt(SnCl3)(CO)L RCH2CH2Pt(SnCl3)(CO) L
Рис. 2.25. Каталитические циклы для реакции гидроформилирования алке-
нов в присутствии Pt H(SnCl3) (СО) (PPh3) для образования продуктов нор-
мального (цикл А) и изостроения (цикл Б)в
Б
А
2
зовании очень слабо основных лигандов, таких, как трифенилфосфит
(рКа = 0), селективность снижается до уровня найденного для систе-
мы K2PtCl4/SnCl2, что указывает на то, что фосфорсодержащий
лиганд более не связан с ионом платины (ср. эффект PPh3 по сравне-
нию с РВи3 в случае модифицированной кобальтовой системы). Так
же, как и в случае других рассмотренных каталитических систем, при
отсутствии изомеризации скорость гидроформилирования алкена сни-
жается при возрастании степени замещенности алкена, т.е. в ряду
RCH=CH, > RR1C=CH, > RCH=CHR1
X X
Этот ряд отражает способность алкенов к координации. Аналогично скорость
ГИД
рмилирования снижается при возрастании концентрации РРЬ3. Внро-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
143
чем, в случае олово-платиновой системы это может происходить
благодаря либо полному удалению оловосодержащего лиганда с обра-
зованием неактивных частиц Pt С1х(РРЬ3)4_х, либо связыванию
pph3 с одним из олово-платиновых комплексов в каталитическом
цикле (рис. 2.25) с образованием стерически затрудненного 18-элек-
тронного комплекса типа Pt11 (Н)х (SnCl3) A(CO)(PPh3)2, где А -
~ алкен, х = 1 или же А- производное алкена, например RCH2CH2
или RCH2CH2CO, их=0. В любом случае будет наблюдаться
пониженная каталитическая активность.
Причина промотирующего действия хлорида олова и в меньше и
степени хлоридов германия и свинца как в гидроформилировании, так
и в гидрировании в присутствии платиновых комплексов пока еще пол-
ностью не ясна. Общепринято, что лиганды, содержащие элементы
группы IVB, такие, как SnCl^ , являются слабыми о-донорами и
сильными тг-акцепторами. Они имеют незаполненные орбитали подхо-
дящей симметрии и энергии для акцептирования электронной плотнос-
ти от частично заполненных 5с?- или гибридных с?р-орбиталей. Таким
образом, они могут эффективно снижать электронную плотность на
ионе платины, что, как предполагается, благоприятствует и образова-
нию исходного гидрида платины [ 232], и последующей атаке алкена
или СО [ 233], при этом одновременно комплекс стабилизируется по
отношению к восстановлению до платины(О) с образованием металли-
ческой платины. Лиганд SnCl “ проявляет сильное дарам с-лабилизирую-
щее влияние (дарамс-эффект того же порядка, которым обладает Н~),
Таким образом, при условии соответствующего стереохимического
строения промежуточных продуктов будут облегчаться стадии мигра-
ции лигандов, протекание которых предполагается в цикле гидрофор-
милирования. Относительное значение этих двух близкосвязанных
электронных эффектов пока не ясно. Можно ожидать, что лиганд SnCl~
в дополнение к электронному влиянию на протекание реакции оказыва-
ет также стерическое влияние, так как это относительно объемистый
лиганд, имеющий конический угол выше 120° ( ср. 6 = 124° для РС13).
Таким образом, присутствие SnCl“B каталитической системе, по
крайней мере по стерическим условиям, будет благоприятствовать
образованию линейных продуктов. Относительную роль стерических и
электронных эффектов в этих системах еще необходимо выяснить, хо-
тя и ясно как из результатов изучения гидроформилирования, так и
из предыдущих данных по изучению гидрирования, что SnCl “ как
лиганд обладает значительными возможностями в гомогенном ката-
лизе.
144
ГЛАВА 2
Что касается каталитической активности, то платиногидридный
комплекс PtH(SnCl 3) (СО) (PPh3) примерно в 5 раз более активен,
чем модифицированная кобальтовая система при аналогичных услови-
ях [ 230]. В недавней работе [ 234] было показано, что система плати-
надихлорид - тр ^-фосфин - оловодихлорид, где тр^-фосфин [ а имен-
но: 1,2- бис (дифенилфосфинометил)циклобутан ] катализирует гидро-
формилирование пентена с большей скоростью, чем это наблюдается
для RhH(CO)(PPh3)3. При 100° С, 100 атм и СО : Н2= 1:1 платино-
вые комплексы в 1,3 раза более активны, чем комплексы родия. При
гидроформилировании пентена-1 при 100%-ной конверсии продукты
состоят из гексаналя (79%), «-пентана (6,4%), пентена-2 (13,4%) и по-
лимерного продукта (0,2%). Примечательно, что отношение н : изо =
99 : 1, что приписано стерическим ограничениям из-за объемистого
хелатного дифосфинового лиганда. Наконец, прежде чем закончить
рассмотрение гидроформилирования, — несколько слов об асимметри-
ческом гидроформилировании.
2.4.5. 1 Асимметрическое гидроформилирование. Как и при
каталитическом гидрировании (см. разд. 2.1.3), стереоселективное
добавление частицы АВ (НСНО в случае гидроформилирования) к про-
хиральной двойной связи дает возможность появления оптической ак-
тивности в продукте:
+ НСНО RlR2CHCH,CHO + R1R2C(CHO)CH,
* W
„ • * *
или R1CH=CHR2+ НСНО —> R1CH2CH(CHO)R2+ RiCH(CHO)CH,R
Каталитические циклы гидроформилирования включают большее чис-
ло стадий миграции лигандов, так что появляется больше возможно-
стей для рацемизации, чем в случае каталитических циклов гидриро-
вания. Оптические выходы в асимметрических реакциях гидроформи-
лирования пока еще не достигли того уровня, который был получен
для реакций асимметрического каталитического гидрирования. Один
из лучших результатов был получен при использовании стирола как
субстрата в присутствии Rh2Cl2(CO) /4L, где L = 2О145. В случае
этой системы сообщалось о получении оптических выходов выше 44%
[235]. С неарильными прохиральными алкенами, например бутеном-2,
оптические выходы редко превышают 10%, и было сделано предположе-
ние, что более высокая селективность в случае стирола может быть
связана с образованием тт-бензильных интермедиатов типа 2о1460
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
145
Rh(CO)3_nP*„
2.146
Такой тип связей повышает конформационную жесткость диастерео-
изомерного интермедиата, облегчая асимметрическую индукцию. Оло-
во-платиновая система, содержащая (—)DIOP как лиганд, также ката-
лизирует асимметрическое гидроформилирование м-бутена [ 237 ]. При
использовании этой системы тип продуктов и оптический выход не
зависят от используемого бутена, что позволяет предполагать, что
асимметрическая индукция происходит после образования платиноал-
кильного интермедиата, т.е. комплекса 2О144, в каталитическом цик-
ле, показанном на рис. 2.25.
Каталитическое асимметрическое гидроформилирование облада-
ет значительными потенциальными возможностями при синтезе биоло-
гически или фармакологически активных молекул. Однако промышлен-
ное применение этой реакции, вероятно, потребует разработки бо-
лее селективных каталитических систем. Затронув вопрос о биологи-
чески активных молекулах или соединениях, встречающихся в при-
роде, мы переходим к другой области гомогенного катализа, которая
оказалась очень полезной в синтезе натуральных продуктов, а имен-
но к области олигомеризации алкенов, катализируемой комплексами
переходных металлов.
Дополнительная литература по гомогенному гидрофор-
милированию*.
Bird С.IF. Transition Metal Intermediates in Organic Synthesis. —
New York: Academic Press, 1967, Ch. 6, p- H7«
Thompson DtTif Whyman R» Carbonylation. — In: TransitionJMetals in
Homogeneous Catalysis (ed. G.N. Schranz er), New York: Marcel Dek-
ker, 1971, Ch. 5, p. 147о
* См. Также: В.Ю.Ганкин, Г.С.Гуревич. Технология оксосинтеза. — Л.:
Химия, 1981 . — Прим. перев.
146
ГЛАВА 2
Orchin М., Rupilius IF. On the mechanism of the oxo reaction. — Catal.
Rev., 1972, v. 6, p. 85.
P aulik F.E. Recent developments in hydroformylation catalysis. — CataL
Rev., 1972, v. 6, p. 49O
Heck R.F. Organotransition Metal Chemistry, A Mechanistic Approach.—
New York: Academic Press, 1974, Ch. 9, p. 201.
Marko L.9 Hydroformylation of olefins with carbonyl derivatives of the
noble metals as catalysts. — In: Aspects of Homogeneous Cataly
sis (edo R.Ugo), Dordrecht: Reidel, 1974, Vol. 2, Ch. 1.
Falbe J• Technical reaction of carbon monoxide with metal carbonyls as
catalysts. — J. Organometal Chem., 1975, v. 94, p. 213 (text in
German).
Henrici* Olive G., Olive S. Reactions of carbon monoxide with transition
metabcarbon bonds. — Transition Met. Chem., 1976, v. 1, 770
Davidson Hignett R*R'9 Thompson D.T* Homogeneous catalysis
involving carbon monoxide. — In: Catalysis, vol. 1, London: Chemi-
cal Society, p. 369.
Pino PPiacenti FBianchi M* — In: Organic Synthesis via Metal Car-
bonyls (eds. I.Wender, P,Pino), vol. 2, New York: Wiley, 1977,
p. 43.
Pruett R.L. Hydroformylation. — Advan. Organometal Chem., 1979, v. 17,
po 1 a
2.5. Олигомеризация
Олигомеризация - это общее название класса реакций, в которых
субстрат А или смесь субстратов А и В превращаются в высокомоле-
кулярные продукты большой молекулярной массы в результате серии
реакций взаимного сочетания:
пК А
п
хК + уВ А В
j х1у
Простейшим случаем олигомеризации является димеризация, когда
п = 2. Когда п велико (> 100), процесс называют полимеризацией
(см. следующий раздел). В случае олигомеризации практический инте-
рес представляют системы, для которых п или х + у меньше 15 - 20.
Реакция олигомеризации с участием двух различных молекул субстра-
та называется соолигомеризацией или гетероолигомеризацией. Для
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
147
описания реакции каталитической соолигомеризации приходится учи-
тывать большое число важных факторов* Например, степень олигоме-
ризации (т.е* значение п или х + у) будет определяться относительны-
ми скоростями стадии роста и обрыва; способ, которым происходит
внедрение субстратов в растущую цепь, определяет стереохимию ко-
нечного продукта*; в случае некоторых субстратов, например диенов,
имеется возможность образования как линейных, так и циклических
олигомеров; при использовании двух различных субстратов порядок и
способ, которым осуществляется внедрение, имеют первостепенное
значение в определении'природы олигомерного продукта; наконец, пос-
кольку мы имеем дело с процессом каталитического образования но-
вых связей, имеется возможность получения асимметричных оптичес-
ки активных молекул*
В последующих шести подразделах мы выбрали для рассмотрения
различные индивидуальные системы с целью проиллюстрировать поло-
жения, отмеченные выше* Мы начнем с рассмотрения двух простейших
алкеновых субстратов, т*е0 этилена и пропилена, и одного из наиболее
активных гомогенных катализаторов олигомеризации - никеля.
2.5.1. Никелевые комплексы как катализаторы олигомериза-
ции алкенов. Известны многочисленные каталитические системы
на основе комплексов переходных металлов, которые превращают ал-
кены в олигомеры* Активными системами, которые проводят реакции
олигомеризации при температурах до 100°С, и наиболее изученными
являются системы, содержащие никель [23, 239].
Одним из простейших методов приготовления таких катализато-
ров является так называемый метод Циглера (см. разд. 2*6), при ко-
тором соединение никеля(П) типа NiX2 или NiX2L2 (X - ацильный ли-
ганд, L — донорный лиганд, например, wpm-фосфин) обрабатывается
избытком алкилалюминия или алкилалюминийгалогенида, например
А1(С2Н5)хС13 _ х (х = 1, 2 или 3) или (С2Н5)3А12С13. Для введения
никеля в систему может быть использовано также никельорганичес-
кое соединение типа NiRxX2 _ xLn (где х и п обычно равны 1 или 2).
Из систем этого типа чаще всего использовались [ 240 ] димерные
тг-аллилникельгалогениды 2.147 или их моноаддукты с ^р^^-фосфина-
Если только субстрат не является полностью симметричным. — Прим,
перев.
148
ГЛАВА 2
ми 2.148; их взаимодействие с алюминийорганическим соединением
достаточно хорошо изучено, что будет показано ниже.
Рентгеноструктурные данные, полученные на монокристаллах,
подтвердили стереохимию комплекса 2.150, где L = Р(циклогексил)
AlRxX3 _х = А1Ме3 [23]. Алюминийорганические соединения обычно
приводят к образованию наиболее активных катализаторов. Однако в
качестве компонентов каталитических систем в сочетании с никелем
могут использоваться и другие кислоты Льюиса, такие, как ВС13,
TiCl4 или SbF5; могут также использоваться и бренстедовские кис-
лоты, такие, как H2SO4 и CF3COOH [23]. Во всех случаях независи-
мо от деталей метода приготовления, по-видимому, активными ката-
литическими частицами являются гидридные комплексы типа ,¥NiHY(L)n
(2.151, рис. 2.26), где Y — либо сложный анион кислоты Льюиса, на-
примёр XA1R%X3 _ х, как в 2.149 и 2.150, либо анион кислоты Бренс-
теда, например CF3COp Присутствие дозорного лиганда L не явля-
ется строго обязательным, хотя, как мы увидим вскоре, этот лиганд
может оказывать значительное влияние на направление протекания ре-
акции олигомеризации.
--------* 2(n-C3H5)NiX.A!RxX3_x
2.149
2.148
XAlRxX3_x
2.150
Конкретный механизм образования "NiHY(L)" зависит от приро-
ды предшественника катализатора. При использовании комбинации
Ni(0) - бренстедовская кислота происходит окислительное присоеди-
нение кислоты (HY) к комплексу Ni(0), при котором непосредственно
образуется комплекс (2.151) [ 154, 241 ]:
NiL4 + HY -> HNi(L)4 _ п Y + nL (n = 0 или 1)
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
149
Рис. 2.26. Каталитические циклы для олигомеризации этилена, катализиру-
емой никелем.
При использовании тг-аллильного комплекса никеля в сочетании с лью-
исовской кислотой тг-аллильный фрагмент часто оказывается соединен-
ным с алкеном, при этом образуются гидридные комплексы никеля, как
в результате последовательности реакций, показанных ниже для случая,
когда субстратом является циклооктен [23]:
‘HNiY’
150
ГЛАВА 2
Первой стадией реакции олигомеризации является координация алкена
с 2.151. При этом в отсутствие лиганда L образуются промежуточные
алкенникельгидридные комплексы 2.152, которые, по-видимому, имеют
г^£-строение; в присутствии L образуются аналогичные промежу-
точные частицы 2.153 или 2.154, в которых алкен находится либо в
цис-, либо в яфяис-положении к лиганду L:
2.152
2.153 2.154
Последующие стадии превращения этих комплексов определяют при-
роду продуктов олигомеризации.
А. Олигомеризация этилена', получение димеров и олигомеров.
Каталитические циклы для реакции олигомеризации этилена в присут-
ствии никеля показаны на рис. 2.26. После образования алкенгидрид-
ных частиц следует стадия миграции гидрида к алкену с образованием
никельэтильного интермедиата 2.156. Дальнейшая координация этиле-
на с последующей миграцией этильной группы приводит к образованию
никельбутильных частиц 2.157. Далее 2.157 может по реакции [3-гид-
ридного элиминирования образовывать бутен как конечный продукт и
никельгидридный интермедиат 2.151, завершая цикл димеризации А.
Другой путь превращения 2.157 - координация молекулы этилена с об-
разованием 2.158, при этом начинается цикл тримеризации Б. Ана-
логично с комплексом 2.159 может происходить либо реакция [3-элими-
нирования с завершением цикла Б, либо координация молекулы этиле-
на, что приводит к началу цикла тетрамеризации. Важным фактором,
определяющим степень олигомеризации, является относительная ско-
рость стадий элиминирования алкильной группы и ее миграции к коор-
динированному алкену, т.е. внедрения. При рассмотрении упрощенной
схемы
-------► RCH,CH2CH2CH,Ni
-------- RCH=CH, + HNi—1|
СН2
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
151
степень олигомеризации будет зависеть от отношения к-г /kd. Если kd » 'к{ ,
то преимущественно будет протекать цикл А и бутены будут основным продук-
том реакции. Если kd<Z' к{ ' то будут протекать цикл Б и последующие циклы,
что приведет к образованию олигомерных или полимерных продуктов. Мож-
но воздействовать на отношение к^ /к& и таким образом изменить нап-
равление реакции путем вариации природы донорного лиганда L, что
показано в табл. 2.4 [23].
Эти результаты показывают, что отношение /к& возрастает с
увеличением стерического объема дор^-фосфинового лиганда, т.е.
наблюдается несколько удивительное явление: повышение стеричес-
кой затрудненности в никелевой системе благоприятствует росту це-
пи по сравнению с реакцией переноса. Этот результат был объяснен
при предположении [23], что реакция элиминирования протекает че-
рез образование пяти (или более) координированных никельгидридных
интермедиатов, имеющих две молекулы алкена, координированные на
ионе металла, в то время как миграция алкильной группы к алкену (т.е.
внедрение) протекает через менее стерически затрудненный тетрако-
ординированный интермедиат. Если справедливо это объяснение, то
переходные состояния для двух процессов могут быть изображены как
2.160 и 2.161, в которых ясно видно различие в стерическом строении.
2.160 2.161
Это может означать, что в каталитическом цикле, изображенном на
рис. 2.26, комплекс 2.157 превращается непосредственно в 2.155 без
перехода в никельгидридный интермедиат 2.151. Это представляется
вполне вероятным, тем более что в присутствии избытка алкена и в
отсутствие других сильно координирующихся молекул вряд ли возмож-
но существование координационно-ненасыщенных частиц 2.151.
При использовании симметричного алкена, например этилена, нап-
равление присоединения И или алкильной группы не имеет значения в
отсутствие изомеризации. Однако, когда мы переходим к несимметрич-
ному алкену, такому, как пропилен, направление присоединения стано-
Таблица 2.4, Влияние трет-фосфиновых лигандов на димеризацию и олигомеризацию этилена3
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
153
вится важным фактором в определении распределения олигомерных
продуктов.
Б. Олигомеризация пропилена; региоселективностъ. При катали-
зируемой никелем олигомеризации пропилена или любого алкена типа
rch=ch2 начальная миграция гидрида может происходить с об-
разованием линейной (присоединение Н к С2, Ni к CJ или разветвлен-
ной (Н к Clf Ni к С2) алкильной группы. Аналогично стадия миграции
алкильной группы к алкену, являющаяся неизбежным следующим эта-
пом в цикле олигомеризации, также может происходить по двум раз-
личным направлениям. Возможность миграции лиганда по двум раз-
личным путям приводит к образованию набора изомерных продуктов.
На рис. 2.27 показаны пути образования шести возможных изомерных
продуктов при димеризации пропилена [239]. Ситуация несколько напо-
минает (хотя и является более сложной) ту, которая была рассмотрена
при гидроформилировании при обсуждении изменений отношения н/изо
(см. разд. 2.4). Для гидроформилирования было найдено, что на направ-
ление реакции значительное влияние оказывает природа донорного ли-
ганда, присутствующего в каталитической системе. В табл. 2.5 пред-
ставлены данные, иллюстрирующие эффект яфе^-фосфиновых лигандов
в димеризации пропилена, катализируемой никелем [23, 242]. Предпо-
лагая, что «-пропилникелевые комплексы 2Л62 обладают той же реак-
ционной способностью по отношению к пропилену, что и комплексы
2Л63 с изопропильной группой, и что изомеризация отсутствует, из
данных о распределении продуктов можно рассчитать относительную
роль присоединений Ni -> Сг и Ni -* С2 на стадиях 1 и 2 (см. рис. 2.27).
Для стадии 1:
i
%Ni -> С j _ %(2-метилпентен-1 * гексен-1 + гексен-2
%Ni-> С %(4-метилпентен-1 + 4-метилпентен-2 + 2,3-диметилбутен-1)
Для стадии 2:
%Ni~>C1 . %(2-метилпентен-1 + 2,3-диметилбутен-1)__________
%Ni-» С " %(4-метилпентен-1 + 4-метилпентен-2 + гексен-1 + гексен-2)
2
Данные табл. 2.5 указывают, что, за исключением Р(трет-Ви) 2(изо-
Рг), природа 0ф0^-фосфинового лиганда оказывает мало влияния на
Стадия 7
Стадия2
Ni — СНСН3
CH * CHjFCH (ch2)3 ch3
I 3 krLJ / р-элиминированиеот^^
Ni-CH(CH2)3CH3 ~NiH / , * 6
f алхил—*~£\ x.
" / Ni------>C2 CH3CH=CH (CH2)2 CH3
CH2=CHCH3 / 2.164 ft-элиминирование om№
3--------ch3 2
C",-«CH,KCHP!CH3 ,
Ni--->C!
2.165
гексены
Н ->Ci
Ni-*C2
2.763
CH^CHCH
ch3 ch3
Ni-CHCH2CHCH3
алхил—>с,
Ni—>C2
2.166
CH3CH3
Ni-CH2CH-CHCH3
алкил —► C?
CH2=CHCH2CH(CH3)CH3
^элиминирование от CH 3
CH3CH=CHCH(CH3)CH3
P-элиминирование pm CH2
метилпентены
* СН2=С(СН3)СН(СНз)СН3
2,3- диметилбутен
Ni —
2.167
Рис. 2.27. Пути образования шести возможных изомерных продуктов при димеризации пропилена.
О
Таблица 2.5. Влияние трет-фосфиновых лигандов на димеризацию пропилена, катализируемую никелем
трет-фосфины L Электронный параметр v, СМ"1 Стерический параметр 0, град Г ексены f Метилпентены 2,3-Диметил- бутены Cooti (Ni-^Cp : на 1-й стадии ношение (Ni-»C2) на 2-й стадии
рМе3 2064,1 118 9,9 80,3 9.8 15:85 15:85
Pet 2061,7 132 9,2 69,7 21,1 17:83 29:71
PBu3 2060,3 132 7,1 69,6 23,3 18:82 34:66
Р(ИЗО-Рг)3 2059,2 160 1,8 30,3 67,9 20:80 86:14
Р(циклогексил)3 2056,4 170 3,3 37,9 58,8 26:74 83:17
Р{ трет-Bu )2(изо-Рг) 2056,1 — 175б 0,6 70,1 29,1 69:31 98:2
а Каталитическая система (n-C3H5NiX)2/Et3Al2Cl3/L, — 20°С, 1 атм.
6Оценочные данные.
156
ГЛАВА 2
направление миграции первого лиганда (Н). Стадия присоединения
приводит преимущественно к образованию изо-продукта 2.163, обра-
зующегося при миграции гидрида к а-углероду пропилена, что соответ-
ствует присоединению по правилу Марковникова. Только в случае очень
объемистого яфояг-фосфина Р(трет-Ви) 2(изо-Рг) направление присоеди-
нения значительно меняется в сторону образования н- пропильной
группы, оказывающей меньше стерических препятствий, т.е. в сторо-
ну продукта 2.162. Однако именно на второй стадии юр ^-фосфиновые
лиганды оказывают влияние на строение продукта [23]. Возрастание
размера лиганда L заметно благоприятствует образованию стеричес-
ки менее затрудненных алкильных групп, имеющих заместители у С2
(по отношению к Ni), а не у Ср т.е. предпочтительно образуются про-
дукты 2.165 и 2.167 по сравнению с продуктами 2.164 и 2.166. Это в
особенности заметно для юрею-фосфина типа Р(юрею-Ви)2Н, где со-
отношения между вышеуказанными продуктами равны 85:15, 97:3 и
98:2 в случае R = Me, Et и («зо-Рг) соответственно [243 ], и в слу-
чае Р(юрею-Ви)2(«зо-Рг) выход «-гексена практически равен нулю. Ис-
следования [23, 239], проведенные с использованием большого набо-
ра юрею-фосфиновых лигандов, подтвердили, что влияние лигандов на
направление миграции никельалкильной группы к пропилену обуслов-
лено скорее стерическим, а не электронным влиянием лигандов (т.е.
изменение величины v играет небольшую роль).
До сих пор мы рассматривали субстраты, содержащие только од-
ну двойную связь. Но не менее важным классом реакций олигомериза-
ции, к тому же позволяющим получать как циклические, так и линей-
ные продукты, являются олигомеризация диеновых молекул. Для ил-
люстрации этого типа реакций мы рассмотрим олигомеризацию бута-
диена-1,3 - простейшего и наиболее доступного из диенов.
2.5.2. Олигомеризация бутадиена. В зависимости от катали-
тической системы и условий реакции при каталитической олигомери-
зации бутадиена может быть получено большое число разнообразных
продуктов, как это показано на рис. 2.28. Эти продукты подразделяют-
ся на два существенно различных типа - циклические олигомеры и оли-
гомеры с линейной цепью; В отсутствие изомеризации циклические
олигомеры могут рассматриваться как простые комбинации из бута-
диеновых звеньев, что показано штриховыми линиями на рис. 2.28.
Рассмотрение структуры продуктов с открытой цепью показывает,
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
157
Рис. 2.28. Набор продуктов, получаемьТй при каталитической олигомеризации
бутадиена.
что они не могут быть расчленены на простые бутадиеновые звенья и
что их синтез должен быть связан с реакциями переноса атома водоро-
да. Как и для олигомеризации алкенов, известно несколько эффектив-
ных каталитических систем для олигомеризации бутадиена [244]. Од-
нако и в этом случае наибольшее внимание привлекали никелевые сис-
темы [55]; именно эти системы лучше всего охарактеризованы [239],
в особенности для циклоолигомеризации. Что касается олигомеризации
до продуктов с открытой цепью, то в этом случае особенно полезными
оказались палладийсодержащие системы [ 245 ].
Л. Системы, содержащие никелъ(й)\ циклоолигомеризация» В про-
тивоположность тому, что было найдено для олигомеризации алкенов,
катализируемой никелем, где активными образованиями являются ком-
плексы никеля(П) (см. выше), в реакциях олигомеризации бутадиена
активными частицами служат комплексы никеля(О) с бутадиеном. Они
могут образовываться либо при восстановлении солей никеля(П), напри-
мер ацетилацетоната никеля в присутствии бутадиена, либо в результа-
те лигандного обмена с участием бутадиена и соединений нульвалент-
ного никеля, таких, как М(циклооктадиен)2, М(тг-С3Н5) 2 и
Ni(CH2=CHCN)2. В отсутствие донорных лигандов, таких, как ^рет-фос-
158
ГЛАВА 2
фин, и растворителей, содержащих активный водород, например спир-
тов или вторичных аминов, основным продуктом олигомеризации бу-
тадиена в присутствии никеля является циклический тример транс,
транс, яфаис-циклооктадодекатриен2Л6£в Образуются также неболь-
шие количества транс, транс, цис-2.169 и транс, цис, цис-2.170 изоме-
ров наряду с циклическими димерами и высшими олигомерами. Однако
в общем случае селективность по продукту 2.168 выше 80%, а в слу-
чае некоторых систем, например Ni(acac)2/(C2H5)2A1OC2H5, превы-
шает 90% [239]. Детальные сведения о механизме циклотримериза-
2.170
ции бутадиена были получены в элегантном и тщательном исследова-
нии, проведенном Вилке и сотр. [239 L Ими был выделен ключевой
интермедиат 2.171, образующийся при сочетании трех бутадиеновых
звеньев на атоме никеля. Последующее присоединение бутадиена к
этому интермедиату приводит к образованию циклододекатриена 2.168
и к регенерации 2.171. Возможный каталитический цикл с участием ин-
термедиата 2.171 показан на рис. 2.29.
2.171
Р(ОС6Н4-о-С6Н5)3
2.177
2.178
По-видимому, первая стадия включает координацию двух или бо-
лее молекул бутадиена на никеле с образованием, вероятно, комплек-
сов никель(О) - алкен типа 2.172. Восстановительное сочетание двух
диеновых звеньев приводит к образованию частиц 2.173 типа ди-(тт-ал-
лил)никель(П), которые существуют в равновесии с изомером 2.174,
содержащим п-аллильную и ст-аллильную группы. Координация третьей
молекулы бутадиена в ^-конфигурации приводит к образованию комп-
лекса 2.175, в котором координированный бутен может внедряться по
связи никель-тг-аллил с образованием комплекса 2.171 или одного из
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
159
Рис. 2.29. Каталитический цикл для тримеризации бутадиена, катализируе-
мой никелем(О).
его изомеров, например 2.176. При окислительном сочетании продук-
та С12 с образованием 2.171 или 2.176 с последующим восстановлени-
ем никеля(И) до никеля(О) получаются конечные продукты, на чем и
замыкается каталитический цикл. На основе стереохимии 2.171 можно
ожидать образования из этого комплекса транс, цис, ф/с-цик ло до де-
катриена 2.170. Из комплекса 2.176, который имеет син, ^«-располо-
жение тг-аллильных групп, должен образовываться транс, транс, транс-
изомер 2.168. Так как именно этот изомер является основным продук-
том реакции, то это показывает, что 2.171 может быстро изомеризо-
ваться в условиях реакции (т,е. в присутствии избытка бутадиена) с
образованием 2.176 или же со, со-изомера, При добавлении подходя-
щих лигандов, например Р(ОС6Н4-о-С6Н5)3 и Р( циклогексил) 3, ока-
зывается возможным изомеризовать как (2.173), так и (2.174) после
их стабилизации взаимодействием с лигандами, т.е. выделить продук-
ты 2.177 и 2.178. Кроме того, при добавлении триэтилфосфина к
2.171 возможно осуществить стехиометрическую реакцию, имитирую-
160
ГЛАВА 2
щую стадию каталитического цикла:
+ Ni(PEt3)4
Использование ягредо-фосфиновых лигандов не только может обеспе-
чить стабилизацию интермедиатов, которые могут быть затем вы-
делены, но также дает возможность направить реакцию олигомериза-
ции на комплексах никеля в нужную сторону.
При добавлении трет-фосфиновых или wpew-фосфитовых лигандов
к вышеописанным катализаторам тримеризации бутадиена (обычно в
соотношении L : Ni = 1:1) получаются каталитические системы, способ-
ные селективно катализировать циклодимеризацию бутадиена до сме-
си 1,5-циклооктадиена (2Л79), 4-винилциклогексена (2.180) и гдос-1,2-ди-
винилциклобутана (2.181). Относительный выход этих продуктов зави-
2.179 2.180 2.181
сит от природы фосфорсодержащего лиганда. В случае объемистых
слабоосновных лигандов, таких, как Р(ОС6Н4-о-С6Н5)3 (0 = 152°, v =
= 2085 см-1), в качестве главного (96%) продукта димеризации обра-
зуется 1,5-циклооктадиен (2.179). При использовании более сильноос-
новных лигандов приблизительно того же размера, например Р(цикло-
гексил)3 (0 = 170°, v = 2056,4 см-1), примерно в равных количествах
образуются 1,5-циклооктадиен и 4-винилциклогексен (41 и 40% соответ-
ственно); при этом главным побочным продуктом (14%) является циа-
1,2-дивинилциклобутан [ср. 0,2% в случае Р(ОС6Н4-о-С6Н5)3 ] [246].
Эти результаты были объяснены на основе последовательности реак-
ций, показанных на рис. 2.30 [239]- Использование основных лиган-
дов (т.е. с низкими значениями параметра v) благоприятствует
образованию частиц 2.184, содержащих тг-аллильную и ст-аллильную
группы. С другой стороны, использование менее основных лигандов (т.е. с вы-
сокими значениями v) благоприятствует образованию (мс-тт-аллильных
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
161
Рис. 2.30. Последовательность реакций, приводящая к образованию 1,5-цик-
лооктадиена, 4-винилциклогексена и цис-1,2-дивинилциклобутана при цикло-
олигомеризации бутадиена.
комплексов 2.182 и 2.183, и, таким образом, возрастание основ-
ности (электронодонорных свойств) лиганда должно способство-
вать образованию винилциклогексеновых продуктов. Это подтвержда-
ется результатами, полученными с использованием в качестве лиган-
доб Р(ОС6Н5)3 (v =2085,3 см-1), р<сбН5)3 (v = 2068,5 см-1) и Р(цик-
логексил) (v = 2056 ,4 см-1); при этом выход 4-винилциклогексена воз-
растает от 7,4 до 27 и 40%, тогда как выход 1,5-циклооктадиена умень-
шается от 81 до 64 и 41%. Как упоминалось ранее, при использовании
пониженных температур (реакции димеризации обычно проводятся при
80°С) оказалось возможным выделить 2Л83 [L = Р(ОС,Н.-о-С,НЛ„]
и 2Л84 [L = Р(циклогексил)3]. Наряду с электронными факторами,
влияние которых уже обсуждалось, стерические свойства лигандов так-
162
ГЛАВА 2
же играют значительную роль в изменении направления реакции, В
случае слабоосновных лигандов, например дорв^-фосфинов, размер ли-
ганда влияет на относительные концентрации комплексов 2.185 и
2.186 и, следовательно, на относительный выход 1,5-циклооктадиена
и ф/с-1,2-дивинилциклобутана, Увеличение размера лиганда L бла-
гоприятствует образованию стерически менее затрудненного интерме-
диата 2.186 и, следовательно, повышает выход циклооктадиена. Анало-
гично при использовании лигандов Р(ОС6Н4=о-С6Н5)3 и Р(ОС6Н5)3,
характеризующихся практически одинаковым параметром v (2085 и
2085,3 см-1 соответственно), относительные выходы циклооктадие-
на (и дивинилциклобутана) составляют соответственно 96% (0,2%) и
81% (9,2%), что отражает уменьшение размера лиганда при переходе
к Р(ОС6Н5)3 (конические углы 152 и 128° соответственно).
Дальнейшая модификация каталитических систем на основе нике-
ля^), включая введение сокатализаторов - доноров водорода (напри-
мер, спиртов, фенолов, вторичных аминов), дает дополнительные воз-
можности для регулирования направления реакции, на этот раз в сторо-
ну образования олигомерных продуктов с открытой цепью. Впрочем,
для того чтобы разнообразить наше обсуждение, мы рассмотрим неко-
торые катализаторы для линейной олигомеризации бутадиена, которые
включают палладий, являющийся одним из ближайших соседей никеля по
периодической системе.
Б. Системы, содержащие Pd(0)-, линейная олигомеризация [245,
247, 2481. В апротонных растворителях, таких, как бензол или тетра-
гидрофуран, примерно при 100°С комплексы палладия(0фтипа PdLnA
(где L = РРЬ3 или PEt3, п = 2 или 3, А - слабокОординирующийся ли-
ганд, например малеиновый ангидрид или бензохинон) катализируют
линейную димеризацию бутадиена до 1,3,7-октатриена 2.187. Когда в
качестве предшественника катализатора используется димер тт-аллил-
палладийацетата (тг-С3Н5Р(1ОАс)2, то основным (78%) продуктом ре-
акции является линейный тример 1,3,6,10-додекатетраен 2.188. В при-
сутствии протонных растворителей, таких, как спирты, фенолы, кар-
p. 187
1188
Н
2.189
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ 163
боновые кислоты или амины, и при использовании РсЦРРЦ) (ма-
леиновый ангидрид) как предшественника катализатора образуется
яфш/с-2,7-октадиен 2.189, т.е» продукт присоединения водорода. Для
описания реакции димеризации, катализируемой палладием, были пред-
ложены два механизма. Первый из них [240, 245] предполагает, что
начальная стадия реакции в основном протекает так же, как и в случае нике-
ля (см. рис. 2.30), т.е. две молекулы бутадиена соединяются на ме-
таллическом центре с образованием ^с-тг-аллильных комплексов
2.190 (здесь М = Ni или Pd). В случае никеля происходит дальнейшее
‘ML’ 2
2.190
4- ‘ML’
образование связей углерод - углерод с получением циклических ди-
мерных продуктов. При М = Pci с интермедиатом 2.190 происходит бы-
страя реакция переноса водорода от атомов С-4 или С-6 с образовани-
ем линейного октатриена. Считается, что различие в поведении меж-
ду никелем и палладием связано с большим атомным размером пал-
ладия по сравнению с никелем. Сделано предположение, что атомы уг-
лерода, которые должны связываться с образованием циклических про-
дуктов, не могут сблизиться на достаточное расстояние в случае пал-
ладия, что приводит к протеканию альтернативной реакции, т.е. пере-
носу водорода. Если так, то эта реакция является одним из немногих
примеров, когда размер атома металла, а не связанных с ним лиган-
дов, определяет направление реакции. В соответствии с этими пред-
ставлениями о механизме было сделано предположение [250, 251 ]
что в случае олигомеризации, катализируемой ацетатом палладия
когда основным продуктом являются тримеры, в каталитическом ^ик-
ле участвуют металлические димерные комплексы типа 2.191, содер-
жащие мостиковые ацетатные группы. Другая гипотеза о механизме
2.191
164
ГЛАВА 2
Pd°Ln
Рис. 2.31. Каталитический цикггкатализируемой палладием ступенчатой ди- •
меризации бутадиена.
[247] рассматривает в качестве активных частиц гидридиые комплек-
сы палладия(П); каталитический цикл из последовательных стадий,
изображенный на рис. 2.31, весьма близок к циклу олигомеризации ал-
кенов, катализируемой никелем (см. рис. 2.26), который был предложен
для объяснения образования линейных продуктов.
Экспериментальные данные, доступные к настоящему времени, не
позволяют сделать определенного выбора между двумя рассмотренны-
ми механизмами, хотя результаты изучения системы с использовани-
ем дейтериевой метки [252], в которой при димеризации бутадиена в
C3H7OD, катализируемой комплексом Pd(PPh3)2 (малеиновый ангид-
рид), был получен 1,3,7-октатриен, монодейтерированный в положении
С-6, говорят в пользу реализации в протонных растворителях меха-
низма второго типа (стадийного).
. Заканчивая описание гидроформилирования, с тем чтобы занять-
ся рассмотрением каталитической олигомеризации, мы упомянули, что
эта реакция может быть полезной при синтезе природных продуктов.
Это в особенности справедливо, когда диеном является изопрен, так
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
165
как региоспецифическая димеризация типа ’голова к хвосту” или те-
ломеризация, т.е. реакция с включением содержащего гетероатом за-
местителя в димерную молекулу, дает возможность синтезировать боль-
шой ряд монотерпенов. Напомним, что природный каучук также явля-
ется линейным полимером, состоящим из изопреновых звеньев, сое-
диненных по типу "голова к хвосту". Исследования этой области ка-
талитической димеризации диенов находятся еще на ранней стадии
развития [ 245 ]. Пока описано немного каталитических методов полу-
чения монотерпенов, встречающихся в природе, хотя цитронеллол
(2Л92) был приготовлен с удовлетворительным выходом при использо-
вании последовательности реакций, показанной ниже [253]:
Потенциальные возможности использования реакций этого типа вели-
ки, но при этом среди реакций димеризации содимеризация этилена и
бутадиена с образованием -гексадиена-1,4 является реакцией,
практическая полезность которой продемонстрирована однозначно.
2.5.3. Содимеризация этилена с бутадиеном, транс -Гекса-
диен-1,4 производится в промышленном масштабе (примерно 2000 т/год,
главным образом в США) с.использованием растворимого родиевого
катализатора; этот процесс разработан фирмой Du Pont. Этот продукт
в основном используется как сомономер в производстве синтетическо-
го этиленпропилендиенового каучука, производство которого составля-
ло в 1975 г. около 100 тыс.т. Введение молекулы диена в этилен-про-
пиленовый сополимер совершенно необходимо, поскольку молекула
такого сополимера представляет собой насыщенное соединение, что,
хотя и является преимуществом с точки зрения устойчивости к окис-
166
ГЛАВА 2
лению и фотостарению, тем не менее крайне затрудняет вулканиза-
цию, т.е. поперечное связывание цепей полимера. Осуществление со-
димеризации этилена и бутадиена, катализируемой родием [254 — 256],
— это своего рода триумф с точки зрения селективности, так как в
этом процессе легко достигается селективность порядка 80% по транс-
гексадиену-1,4 при использовании RhCl3 как предшественника ката-
лизатора в этаноле. Среди других продуктов реакции - ^-гексади-
ен-1,4 (^10%) и гексадиен-2,4 (^10%), а также небольшие количества
З-метилпентадиена-13 и высших олигомеров этилена. Удивительно,
что в этом процессе достигается такая высокая селективность, так
как та же каталитическая система в отсутствие бутадиена активна в
димеризации этилена. Предполагается, что высокая селективность свя-
зана с термодинамическим контролем благодаря образованию относи-
тельно стабильного комплекса родия типа 2Л93, где L — слабый до-
норный лиганд или молекула растворителя. Считается, что активными
каталитическими частицами служат гидридные комплексы родия(Ш),
4 RhinCl2L„
£ (вероятно, п ~ 2 )
2.193
которые, возможно, образуются в результате следующей серии реак-
ций:
Rh(III)Cl + CJL + И90 -> Rh(I)Cl + 2НС1 + CILCHO
о £ 4? Л о
Rh(I)Cl +НС1 - Rh(III) (Н) Cl
Это предположение поддерживается тем фактом, что в случае, когда
в качестве предшественника катализатора используется RhCl3* ЗН2О,
появляется индукционный период, который может быть устранен при
использовании комплексов родия(1), например Rh(C2H4)2(acac) в при-
сутствии НС1 [11]. Каталитические циклы, которые приводят к обра-
зованию как транс-, так и ^с-гексадисна, показаны на рис. 2.32 [256].
Так, реакция обычно проводится либо в растворителе, являющемся
потенциальным донором (например, в этаноле), либо в присутствии
специально введенных донорных лигандов, и так как считается, что
эти доноры оказывают регулирующее' влияние и па активность, и на се-
лективность каталитических систем, то участие таких донорных лиган-
дов предусмотрено в рассмотренных каталитических циклах. Участие
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ 167
Рис, 2.32. Каталитические циклы для катализируемой родием содимеризации
этилена и бутадиена с образованием цис- и транс-гексадиена-1,4 (L — слабо-
связанный лиганд или молекула растворителя).
этих лигандов позволяет также представить цикл как серию реакции
с превращением 16-18-электронных частиц.
Первой стадией в обоих каталитических циклах является коорди-
нация молекулы бутадиена с последующей быстрой миграцией гид-
ридного лиганда с образованием тг-кротильных частиц в форме либо
аио-изомера 2.194, либо со-изомера 2,195. В этих циклах возможно
взаимопревращение этих двух изомеров, и отношение транс/цис в ко-
нечных гексадиенах-1,4 определяется как соотношением первоначально
образующихся продуктов 2.195 и 2.194, так и степенью изомеризации
2.194 с образованием более стойкого продукта 2.195 перед координа-
цией этилена как следующей стадии цикла. Отношение транс/цис чув-
ствительно к присутствию и природе донорной молекулы. В отсутст-
168
ГЛАВА 2
ствие специально введенных доноров, т.е. в растворителях, молекулы
которых не координируются или координируются очень слабо (напри-
мер, в толуоле), отношение транс/цис менее 5. В присутствии боль-
шого (1000-кратного) избытка спирта (МеОН или EtOH) это отношение
повышается до 20. При использовании сильных доноров, таких, как три-
бу ти л фосфиноксид Ви3РО или [ (CH3)2N ]3РО, отношения донор/родий,
равного 5:1, достаточно для того, чтобы получить такое же возраста-
ние ^awc-селективности. Координируясь на металле, молекула (моле-
кулы) донора повышает стерическую затрудненность координационной
сферы, что благоприятствует трансоидной координации бутадиена и
образованию стерически менее "требовательного" сгш-тг-кротильного
изомера 2.195, Кроме того, благодаря эффективной конкуренции с
этиленом, т.е. благодаря замедлению образования как 2.196, так и
2.197,, присутствие донорного лиганда оставляет больше времени для
изомеризации 2.194 в 2.195. Однако роль конкуренции может быть
двойной, так как если она очень велика, то донорная молекула будет
препятствовать образованию 2.197, что остановит или по крайней мере
значительно замедлит реакцию содимеризации. Например, добавление
пиридина отравляет катализаторы, а добавление более чем двукратно-
го избытка Вп3РО уменьшает скорость содимеризации.
В обычных условиях проведения реакции, т.е. когда этилен и бута-
диен присутствуют в большом избытке, скорость процесса определяет-
ся миграцией аллильного лиганда к алкену (внедрением этилена) с об-
разованием либо 20_/98,либо 2J99.. Реакция p-гидридного переноса
с выделением гексадиена-1,4 и регенерацией гидридного комплекса
родия(Ш) завершает цикл.
Как упомянуто выше, высокая селективность по гексадиену-1,4
связана с относительной стабильностью тт-кротильного комплекса,
либо 2.194, либо 2.195, по сравнению с другими алкильными производ-
ными родия, образование которых можно предположить в условиях ре-
акции. Так как стадия внедрения алкена определяет скорость реакции,
то состав продукта зависит от относительной концентрации интерме-
диатов типа 2.200. Образование комплексов, в которых R — тг-кротил,
значительно более предпочтительно, чем образование комплексов, в
которых R - этил, и, таким образом, внедрение этилена в тг-кротиль-
ную связь происходит значительно быстрее, чем внедрение бутадиена.
Следовательно, образование гексадиена-1,4 контролируется термоди-
намически. Другие данные о стабильности тт-кротильных частиц были
получены при выделении 2.201 в результате реакции бутадиена с этил-
родий(Ш)хлоридом [254]. Хотя гексадиены-1,4 — первичные продукты
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
169
Г
R— RhCl2L„
2.200
2.201
содимеризации, они не являются наиболее стабильными изомерами
гексадиена, и, если только реакция не проводится при очень большом
избытке бутадиена в присутствии соответствующего количества эти-
лена, родиевый катализатор будет вызывать изомеризацию 1,4-изоме-
ров в сопряженные 2,4-изомеры. С практической точки зрения это не-
желательно, и условия реакции выбираются так, чтобы уменьшить изо-
меризацию (до степени менее 10%).
Для этой реакции олигомеризации были разработаны также ката-
литические системы на основе соединений никеля, палладия, кобаль-
та и железа [256]. Однако по селективности образования транс- гек-
садиена-1,4 родиевая система наилучшая, хотя сообщалось, что с ко-
бальтовой системой была достигнута селективность 100% по отно-
шению к -гексадиену-1,4 при почти 100%-ной конверсии.
Раздел, посвященный гидроформилированию, был закончен крат-
ким рассмотрением применения этого каталитического процесса для
асимметрического синтеза. Рассмотренная выше реакция олигомери-
зации, катализируемая комплексами металлов, также имеет значитель-
ные потенциальные возможности для каталитического хирального син-
теза. Мы рассмотрим эти возможности на двух примерах, прежде чем
перейти к рассмотрению реакций полимеризации.
2.5.4. Асимметрическая олигомеризация. Что касается опти-
ческой чистоты, то наибольший успех был достигунт для реакций соди-
меризации этилена с напряженными карбоциклическими алкенами, так-
кими, как норборнен 2.202 или норборнадиен 2.203 [23, 235, 257]. При
2.203
170
ГЛАВА 2
использовании в качестве предшественника катализатора (тг-ал-
лил) NiX2/(C2H5)3Al2Cl3/L, где L = ( - )-(ментил)2Р(изопропил), (2.204)
были получены оптические выходы 80,6% при -97°С и 77,5% при -65°С.
Оптические выходы резко повышаются при снижении температуры, и
если при 20 ° С превышение энантиомера в синтезе продукта 2.202
составляет только 30%, то при -97°С оно достигает 80,6%. Высокая
активность никелевых систем позволяет проводить реакцию при столь
низких температурах; способствует этому и использование напряжен-
ных алкенов. Однако возможность синтеза не ограничивается такими
напряженными алкенами; оптическая чистота в 64% была получена при
катализируемой никелем содимеризации этилена и бутена с получени-
ем (+)-(S)-CH2 =СНСН(СН3)СН2СН3 при —40°С и при использовании
(-)-(ментил)2РСН3 как асимметрического лиганда. При содимеризации
этилена и стирола при -10°С с образованием продукта 2.205 был полу-
чен оптический выход 37% в присутствии той же каталитической сис-
темы. Хотя все возможности этих реакций еще предстоит исследовать,
2.204
2.205
данные, полученные к настоящему времени, показывают, что эти ре-
акции могут быть чрезвычайно ценными для каталитического хираль-
ного синтеза.
Дополнительная литература по гомогенной олигомеризации
Wilke G. Cyclooligomerization of butadiene with transition metal
it- complexes* — Angew Chem* Internal* Edit* Eng* 1963, v* 2, p.105
Cramer R. Transition metal catalysis exemplified by some rhodium
promoted reactions of olefins* — Acc* Chem* Res*, 1968, v* 1, p,186
Bogdanovic, Непс B., Karmann H.G., MusselH.G. Walter D., Wilke G.
Olefin transformations catalysed by organonickel compounds* — Tnd*
Eng. Chem., 1970, v* 62, p.34.
Heimbach P., J ollv P.WWilke G. Allylnickel intermediates in organic
synthesis. — Adv* Organometal. Chem*, 1970, v* 8, p* 29.
Keim W. тг-allyl systems in catalysis. — In: Transition Metal in Homoge-
neous Catalysis (ed* G*N* Schrauzer), New York, Marcel Dekker, 1971,
p* 59
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
171
dolman С. A. Role о£ transition metal hydrides in homogeneous catalysis,
— In: Transition Metal Hydrides (ed* E*L* Muetterties), New York:
Marcel Dekker, 1971 p* 271.
Maitlis P.M. The Organic Chemistry of Palladium. Vol II, Catalysis
Reactions* — London: Academic Press, 1971, p. 33.
Bogdanovic В., Непс B., Losler A., Maister B., Pauling H., Wilke G.
Asymmetric synthesis with the aid of homogeneous transition-metal
catalysts. — Angew. Chem. Internal. Edit* Engl., 1973, v. 12, p. 954
Heck R.F. Organotransition Metal Chemistry, A Mechanistic Approach,
— New York: Academic Press, Ch* 5 and 8.
Jolly P.W., Wilke G. The Organic Chemistry of Nickel. Vol. II Organic
Synthesis* — New York: Academic Press, 1975.
Bogdanovic B. Selectivity control in nickel-catalysed olefin oligomerisa-
tion. — Adv. Organometal* Chem*, 1979, v, 17, p. 105.
Chiusoli G.P., Salerno G. Synthetic applications of organonickel
complexes in organic chemistry* — Adv* Organometal. Chem*, 1979,
v. 17, p. 195.
Su A.C.L. Catalytic codimerisation of ethylene and butadiene. — Adv.
Organometal. Chem*. 1979, v. 17, p. 269.
Tsuji J. Palladium catalysed reactions of butadiene and isoprene.—Adv,
Organometal Chem., 1979, v. 17, p. 141.
2.6. Полимеризация
Полимеризация — один из наиболее важных классов реакций, ис-
пользуемых в современной нефтехимической промышленности. Сей-
час коммерчески доступны примерно тридцать различных основных
типов полимеров, получаемых при связывании молекул одного ти-
па (мономера), и сополимеров, получаемых при связывании друг с
другом двух или более мономеров. Полимеры находят самое широкое
применение - от производства пластмассовых корзин до получения
искусственных почек. В 1976 г. мировое потребление (за исключением
СССР, Восточной Европы и Китая) полимерных продуктов, т.е. пласт-
масс, смол, синтетических каучуков и нецеллюлозных синтетических
волокон, превысило 45 млн. т [258], при этом производство синтети-
ческого каучука, в основном сополимера стирола с бутадиеном и по-
либутадиена, более чем в два раза превышало производство природ-
ного материала (8 млн.т по сравнению с 3,5 млн.т) [53]. В наиболь-
шем объеме выпускаются два полимера — полиэтилен (мировое произ-
водство 12,5 млн.т в 1976 г.) и полипропилен (мировое производство
172
ГЛАВА 2
в 1976 г. около 3 млн. г). Именно открытие новых катализаторов для
получения этих двух продуктов, сделанное профессором Циглером
(ФРГ) и профессором Натта (Италия), оказалось наиболее важным.
2.6.1. Катализаторы Циглера — Натта. При изучении реакции мно-
гократного внедрения этилена по связи алюминий — алкил с образова-
нием длинноцепочечных алюминий - алкилов типа 2.206 К.Циглер и
его сотрудники (ФРГ) обнаружили, что небольшое количество никеля
ингибирует эту реакцию полимеризации:
Al—R + nCH СН9-» A1(CH9CH9)„R (п< 100 при R = СН0СНо)
2.206
В присутствии никеля реакция обрыва происходит после каждого акта
внедрения, при этом основным органическим продуктом является бу-
тен-1 [239]. Это наблюдение привело не только к разработке многих
никелевых катализаторов олигомеризации, которые обсуждались в
предыдущем разделе, но также стимулировало интенсивное изучение
влияния других переходных металлов на полимеризационные системы,
содержащие связь алюминий - алкил. Исследования в этом направле-
нии привели в начале 1950-х г» к открытию [259] того, что некоторые
комплексы переходных металлов, такие, как ацетилацетонат циркония,
в присутствии алюминийорганических соединений, например AlEt2Cl,
катализируют полимеризацию этилена в мягких условиях (при 50°С и
10 атм) с образованием материала, имеющего молекулярную массу
свыше 50 000. Открытие имело громадное теоретическое значение,
так как в то время этилен считался молекулой, которая трудно поли-
меризуется — при давлениях свыше 1000 атм и температурах около
200°С. В 1952 г. Циглер передал лицензию на процесс полимеризации
при низком давлении итальянской химической компании Montecatini.
Научный консультант этой фирмы профессор Миланского политехни-
ческого института Натта распространил подход Циглера на полимери-
зацию пропилена и высших а-олефинов [260]. В 1955 г. Натта сообщил
[261, 262] о приготовлении, а также (что не менее важно) об определе-
нии характеристик стереорегулярных полимеров а-олефинов, получае-
мых с использованием катализаторов циглеровского типа на основе
титана. Он установил, что в этих высококристаллических полимерах
имеются длинные последовательности из мономерных единиц, облада-
ющих одинаковой стереохимией. Именно это открытие привело к сов-
ременным процессам получения полипропилена.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
173
В ходе дальнейших исследований было установлено, что многие
комбинации соединений металлов способны катализировать полимери-
зацию алкенов. Под определением "катализатор Циглера" стали под-
разумевать любую комбинацию алкилов, гидридов или галогенидов
элементов групп I — III с солями или комплексами переходных метал-
лов групп IV - VIII , включая Sc, Th и U, способную катализировать
полимеризацию этилена через стадию координации, а не по ионному
или радикальному механизму* [263 — 265 ]. Хотя к настоящему време-
ни известны и истинно гомогенные каталитические системы Циглера
- Натта, наиболее эффективными и наиболее используемыми на прак-
тике, особенно для получения изотактического (см. раздел 2.6.3) по-
липропилена, являются гетерогенные системы на основе соединений
титана. Строго говоря, эти системы, будучи гетерогенными, формаль-
но не должны были бы рассматриваться здесь. Однако из-за того, что
по механизму действия они близки к гомогенным системам, и учиты-
вая их большое практическое и научное значение, было бы неправиль-
но исключить их по формальным причинам.
Промышленные катализаторы Циглера - Натта обычно готовятся
обработкой трихлорида или тетрахлорида титана алюминийорганичес-
кими соединениями, например AlEt3 или Et2AIC1. Трихлорид титана
может существовать в четырех различных кристаллических модифи-
кациях а, р, 6 и у. Состояние получаемого катализатора зависит от
деталей метода приготовления, например от того, проводится ли оно
в растворе или в суспензии, а также от температуры реакции, скорос-
ти добавления алюминийалкила, скорости перемешивания и т.д. Актив-
ность и стереоспецифичность непосредственно связаны с физическим
состоянием катализатора, но детали метода приготовления засекрече-
ны фирмой-изготовителем. Предполагают, что реакция полимеризации
*
Под катализаторами Циглера — Натта обычно понимают каталитические
системы, в которых образование активных центров протекает через стадию
взаимодействия алкильного или гидридного соединения (сокатализатора) с сое-
динением переходного металла (чаще всего галогенида). Полимеризация оле-
финов на катализаторах Циглера — Натта относится к классу каталитической
полимеризации. Каталитическая полимеризация — процесс, в котором в обра-
зовании активированных комплексов на стадии роста принимает участие ка-
тализатор (см. Ермаков Ю.И., Захаров В.А. — Усп.хим., 1972, т.51, с.377).
Существенной особенностью каталитической полимеризации является проте-
кание реакции роста как реакции встраивания мономера по активной связи
переходный металл-углерод в активном центре. — Прим, перев.
174
ГЛАВА 2
происходит на выступающих гранях кристаллов TiCl3 и механизм Кос-
се и Арлмана, первоначально предложенный в 1964 г. 1266 — 268], до
сих пор является общепринятым в своей существенной части.
Считается, что активным каталитическим центром служит окта-
эдрический ион титана типа 2.207, имеющий четыре атома хлора в
качестве лигандов, которые входят в кристаллическую решетку, а
также алкильную группу, которая первоначально образуется в резуль-
тате обмена иона хлора на алкил при реакции между TiCl3 и алюми-
нийорганическим соединением, и, наконец, вакантное координацион-
ное место. Первая стадия реакции заключается в координации алкена
на вакансии. Затем следует миграция алкильной группы к алкену, про-
текающая через четырехцентровое переходное состояние типа 2.208,
в результате чего образуется соединение 2.209 с удлиненной на одну
мономерную единицу алкильной группой; 2.209 содержит вакантное
место в смежной позиции. Дальнейшая координация этилена с повто-
рением описанной стадии составляет процесс полимеризации*. Обрыв
происходит по реакции ^-элиминирования с образованием длинноцепо-
чечных а-алкенов и титангидридного центра, который либо участвует
в реинициировании полимеризации, либо взаимодействует с алюминий-
органическим соединением с образованием титаналкильного соедине-
ния 2.207, Таким образом, реакция роста полимера представляет собой реак-
цию миграции алкильной группы, при этом растущая алкильная цепь попере-
менно занимает одно из двух координационных мест, находящихся в ^zzc-no-
ложении друг к другу в октаэдрическом окружении титана. В этом меха-
низме алюминийорганическое соединение не играет существенной ро-
ли на стадии роста, и его связь с титановым центром во время ката-
Октаэдрическая координация и наличие свободного координационного мес-
та не являются обязательными свойствами активного центра полимеризации
олефинов. Обоснованна и другая точка зрения на механизм реакции роста,
согласно которой координационная сфера иона титана представляет собой ис-
каженную тригональную бипирамиду, при этом алкильная группа занимает
положение, промежуточное между двумя октаэдрическими вакансиями. Рас-
четы показывают, что такая структура энергетически более выгодна, чем
октаэдрическая [273 ] (см. также В.И.Авдеев, И.И.Захаров, В.А.Захаров,
Г.Д.Букатов, Ю.И.Ермаков. — Ж.структ.хим., 1977, т.18, с.525). При коорди-
нации мономера вновь образуется октаэдрический комплекс. Стадия коорди-
нации мономера определяет скорость реакции встраивания. Энергия актива-
ции этой стадии обусловлена необходимостью перестройки координационной
сферы иона титана при образовании тг-комплекса с олефином. — Прим.перев.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
175
2.209
литической реакции не обязательна. Основная роль алюминийоргани-
ческого соединения заключается в том, что оно алкилирует переход-
ный металл, выступает в качестве агента переноса цепи, способст-
вует очистке реакционной среды от каталитических ядов и является
восстанавливающим агентом в том случае, когда в качестве исход-
ного соединения при получении катализаторов используется тетра-
хлорид титана. Реакция роста происходит только на ионе титана.
Предложены и другие механизмы, в которых рассматривается учас-
тие в активном центре двух ионов металла (двух ионов титана или
титана и алюминия) [263], но по опубликованным данным более пред-
почтителен монометаллический механизм.
Известно несколько гомогенных каталитических систем на основе со-
единений титана и алюминия. В этих системах алюминий связан с ти-
тановым центром и в ходе всего процесса олигомеризации или поли-
меризации алкена выступает скорее в качестве лиганда, чем реаген-
та. Одна из таких систем готовится обработкой (r]5-C5H5)2TiCl2
этилалюминийдихлоридом [269]. Предполагают, что активным ката-
лизатором является комплекс титана(1У) 2.211, образующийся в ре-
зультате следующих реакций:
С1
Cp2TiCl2
Ср
Et^ ХЛ
Cl ^С1
-А1С13
176
ГЛАВА 2
Cp2Ti(Et)Cl
С2Н4/Е1А1С12
IF
Ср
—7 Cl Et
yCI“~ ^C1
Cp
2.210
2.211
В отсутствие алюминийорганического соединения этилтитановый
комплекс 2.210 стабилен в растворе и каталитически неактивен» Ме-
ханизм полимеризации, в котором рассматривается участие 2.211 в
реакции роста, в основном такой же, как и описанный выше для гете-
рогенных катализаторов. Входит ли алюминийорганическое соедине-
ние в качестве лиганда в активный центр гетерогенных систем, не
вполне ясно, хотя и предполагалось [270], что присутствие А1С13
в кристаллической решетке TiCl3 приводит к возрастанию доступнос-
ти и, возможно, реакционной способности поверхностных титановых
центров.
Хотя из-за ограниченной активности (мя вернемся к этому ниже)
гомогенные Ti — Al-катализаторы полимеризации, описанные до сих
пор, не имеют практического значения, их изучение позволило устано-
вить интересные детали возможного механизма полимеризации. Рас-
четы [271, 272 ], проведенные для гипотетической системы
Ti(OMe) 4/AlEt 3 олигомеризации этилена (реальной системой являет-
ся Ti(OC6Hz|-n*Me)3O(«- Bu) /AlEt3, но расчет для этой системы
потребовал бы слишком большого времени ЭВМ), показали, что наибо-
лее стабильной конфигурацией первоначального катализатора являет-
ся не октаэдричекий комплекс, имеющий вакантное место, как это
рассмотрено в механизме Коссе и Арлмана, а комплекс типа 2.212
(рис. 2.33), имэющий конфигурацию тригональной бипирамиды. Расче-
ты также показывают, что неспаренный электрон титана сильно лока-
лизован (с коэффициентом 0,742) на занятой молекулярной орбитали
(d ) с наиболее высокой энергией; этот электрон вносит значитель-
ный вклад в образование титаналкильной связи. Присоединение этиле-
на приводит комплекс в октаэдрическую конфигурацию. М,ежду входя-
щим этиленом и титаном образуется лишь слабая связь с небольшим
обратным донированием на тг*-антисвязывающие орбитали этилена. Как
только этилен входит в связывающую сферу титана, тут же образует-
ся слабая связь между алкильной группой и алкеном с участием d -
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
177
2.216
2.215
н/
СН2
2.217
/A1Et3
рис. 2.33. Димеризация этилена с использованием системы Ti(OMe)
в качестве предшественника катализатора.
орбиталей титана. Это может рассматриваться как плановый переход
к образованию четырехцентрового переходного состояния 2.214 на ран-
ней стадии при движении системы вдоль координаты реакции, при этом
^Х2-орбитали титана выступают в качестве ’’агента переноса”. Этот
тип механизма, по существу, очень близок к механизму ’’прямого внед-
рения” алкена по связи металл — алкил, в котором принимается, что
взаимодействие алкен — алкил начинается, как только алкен входит в
связывающую сферу металла. При завершении реакции миграции этиль-
ной группы к этилену (или реакции сочетания) образуется комплекс
2.215, содержащий бутильную группу и имеющий тригонально-бипира-
мидальную структуру. Этот комплекс при координации последующей мо-
лекулы этилена превращается в октаэдрический алкеналкильный комп-
лекс 2.216, аналогичный соединению 2.213. В полимеризационной сис-
> теме повторение этой последовательности реакций приводит к росту
178
ГЛАВА 2
цепи. Однако в случае рассматриваемой системы, вызывающей олиго-
меризацию, происходит реакция ограничения цепи с образованием в
качестве продуктов бутена-1. Предполагаемая природа реакции необыч-
на в сравнении с большинством реакций, которые мы обсуждали до
сих пор. Была высказана гипотеза, что по мере того, как молекула
этилена приближается к иону титана в комплексе 2.215, атомы угле-
рода молекулы этилена становятся все более отрицательно заряжен-
ными благодаря все большему взаимодействию с титаном. В то же
время В -атомы водорода в бутильной группе становятся все более
электроположительными по мере того, как они сближаются с этилено-
вым звеном. Этот процесс заканчивается ионным взаимодействием
между (3-атомом водорода в бутильной группе и атомом С-2 этилена,
при этом возникает своего рода водородная связь с образованием пе-
реходного комплекса типа 2.217, из которого при удалении бутена-1
вновь образуется первоначальный титанэтильный комплекс 2.212. Та-
кая реакция ограничения представляется очень привлекательной в про-
цессе полимеризации Циглера-Натта, так как в этом процессе непо-
средственно регенерируются первоначальные частицы, содержащие
этильные группы. При этом нет необходимости предполагать (как это
делается обычно в случае рассмотрения реакции элиминирования [3 -во-
дорода), что образуются промежуточные титангидридные комплексы.
В механизме, первоначально предположенном Коссе, предполага-
лось, что стабилизация d-орбиталей титана благодаря взаимодействию
с вакантными антисвязывающими орбиталями алкена играет ключевую
роль в каталитической реакции [266]. Результаты теоретического изу-
чения, рассмотренные выше, в основном согласуются с предположени-
ем о том, что стабилизация "промежуточной” орбитали (d ) является
ключевым фактором. Впрочем, теоретические доводы в пользу обрат-
ного допирования d -> тг* как источника этой стабилизации (как это
было первоначально предложено Коссе) немногочисленны. Основной
вывод (в соответствии с тем, что предлагает Коссе) заключается в
том. что "активация" связи Ti — а-С, т.е. активация по отношению
к миграции, обусловлена стабилизацией dxz-орбиталей октаэдрическо-
го комплекса при координации этилена и что эта орбиталь действует
затем как "промежуточная" орбиталь, облегчая протекание миграции
алкильной группы к алкену [271 ].
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
179
В результате некоторых ранних расчетных работ [273] с исполь-
зованием комплекса 2.218 как модели активного центра при учете
идеи, что d-орбитали иона титана могут эффективно действовать как
’’агенты переноса” при образовании пропильного комплекса 2.219, бы-
С1 С1
СК /С1\ I /Me Ск /СК ।
.Ti—Рг
СГ ^С1 I У/ СГ ^СГ I
С1 С1
2.218 2.219
ло установлено, что стабилизирующий эффект d-орбиталей пренебре-
жимо мал. Однако в этих ранних работах не было сделано четких пред-
положений относительно состояния окисления иона титана, что, вероят-
но, может объяснить различие точек зрения. Хотя сейчас, по-видимому,
преждевременно делать окончательные заключения об относительном
значении стабилизирующих эффектов этих орбиталей, все же несом-
ненно, что теоретические расчеты типа рассмотренных выше будут иг-
рать все более важную роль в получении детальных знаний как о рас-
сматриваемой, так и о других каталитических системах. При исполь-
зовании все более сложных теоретических подходов и увеличении воз-
можностей счета на ЭВМ расчет орбиталей полной системы, несомнен-
но, приобретет большое значение в будущем как при конструировании,
так и при выяснении механизма действия каталитических систем.
Как уже упоминалось во введении к данному разделу, катализато-
ры Циглера — Натта приобрели особенное значение в получении поли-
этилена и полипропилена, и, прежде чем оставить тему полимериза-
ции, мы рассмотрим несколько иолее подробно сведения об этих двух
материалах и о практических процессах их получения.
2.6.2. Полиэтилен. Приблизительно половина общего объема произ-
водства полиэтилена в настоящее время до сих пор получается с ис-
пользованием процессов высокого давления аналогичных тем. которые
были первоначально разработаны компанией Imperial Chemical Indust-
ries в конце 1930-х г. [53]. В этих процессах реакция проводится
обычно в трубчатом реакторе под высоким давлением (1500 — 3000 атм)
в интервале температур 100 — 300°С. Полимеризация, которая в этом
случае протекает по свободнорадикальному механизму, инициируется
либо введением небольших количеств кислорода в исходный этилен,
180
ГЛАВА 2
либо добавлением органических пероксидов в реактор. Предпочтитель-
но использование пероксидов, имеющих время полураспада около од-
ной минуты при температурах ПО - 210°С. Реакция высокоэкзотер-
мична (106 кДж/моль при 25°С); таким образом, необходимы отвод
тепла и тщательный контроль за температурой. На выходе из реактора
высокого давления продукт обычно отделяется от газа на двух стадиях
сепарации при давлениях 300 и 1-3 атм соответственно. Расплав поли-
мера охлаждается водой с получением гранул, подходящих для дальней-
шей переработки,, например для производства пленки [274]. Сообщает- .
ся, что в процессе, разработанном фирмой BASF [275], для получения
100 кг полиэтилена требуется 103 кг этилена. Механизм процесса сво-
боднорадикальный, поэтому во время полимеризации происходит значи-
тельное разветвление цепи. В результате плотность получаемого поли-
мерного материала несколько ниже той, которой обладал бы материал,
имеющий в основном линейные цепи и получаемый с использованием
циглеровских систем при низком давлении (0,92 — 0,93 г/см3 по срав-
нению с 0,94-0,97 г/см3). По этой причине полиэтилен, получаемый
с использованием свободнорадикальных процессов при высоком дав-
лении, называется полиэтиленом низкой плотности (ПЭНП), тогда как
материал, получаемый с использованием процессов нерадикального
типа при низком давлении, т.е. с использованием катализатора Циг-
лера, называется полиэтиленом высокой плотности (ПЭВП). Для про-
изводства ПЭВП в настоящее время используются в основном два ти-
па процессов.
А. Процессы в растворе или в суспензии. В этих процессах этилен
непрерывно подается вместе с катализатором и углеводородным раз-
бавителем д относительно большие (по сравнению с трубчатыми реак-
торами для производства ПЭНП) реакторы полимеризации. В раствор-
ных процессах используется углеводород (например, циклогексан) в
качестве растворителя для полиэтилена. Содержание полимера в раст-
воре ограничено вязкостью, при которой с ним еще можно работать,
и, таким образом, существует практический предел степени полиме-
ризации в подобных процессах . В нашедших более широкое примене-
*
Степень полимеризации определяется соотношением констант скорос-
тей реакции роста и реакции преобладающего процесса переноса полимерной
цепи. Время роста одной полимерной молекулы при полимеризации этилена
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
181
ние суспензионных процессах* используется углеводородный разба-
витель (например, гексан [276], легкие углеводородные фракции [277],
которые являются плохими растворителями для полиэтилена) и реакция
проводится в суспензии с образованием порошка полимера. Типичными
условиями проведения таких процессов являются 10 — 30 атм и темпера-
тура 80 - 105°С. С использованием суспензионных процессов легко
получается высокомолекулярный полиэтилен, при этом часто исполь-
зуется водород для регулирования степени полимеризации. Обычно от
1,03 до 1,07 т этилена необходимо для получения.1 т ПЭВП [276-278].
Применение более низких давлений в этих процессах получения поли ••
этилена значительно упрощает технологические проблемы, присущие
процессам высокого давления: нет необходимости использовать доро-
гостоящее компрессионное оборудование. С другой стороны, при этом
необходимы большие объемы реакторов, и, кроме того, необходимо
иметь оборудование для регенерации растворителя. Недавно фирмой
” Union Carbide” разработан процесс получения ПЭВП полимеризаци-
ей в газовой фазе [279].
Бо Газофазная полимеризация этилена. В этом процессе этилен и по-
рошкообразный катализатор** непрерывно подаются в реактор с псевдо-
ожиженным слоем, где происходит полимеризация. Углеводородный
растворитель не применяется. Процесс проводится при давлении око-
ло 18 атм и температуре 85 - 100°е в зависимости от того, какой про-
дукт необходимо получить. В этом процессе получается полиэтилен с
порядка 1 мин (см. В.А. Захаров, Г.Д. Букатов, Ю.И. Ермаков. - Усп. хим.,
1980, т.59, с.2213). Степень полимеризации (или молекулярная масса) поэто-
му не может определяться временем пребывания катализатора в реакторе
(обычно порядка 1 ч и более) .или количеством накопленного полимера в ре-
акторе (и, следовательно, вязкостью раствора). — Пгиг*. перев.
*
Подавляющая частьПЭВПпроизводится суспензионной полимериза-
цией. Основные катализаторы для этого процесса: a) CrO3/SiO2; б) различ-
ные варианты титано-магниевых катализаторов, например TiCl4/MgCl 2+ A1R3O
— Прим, перев.
**
В качестве катализаторов для процесса газофазной полимеризации
фирмы Union Carbide используются Cr(r|5“( 5H5)2/SiO2 и [(C6H5)3Si - О]2х
х CrO2 /SiO2, активированный обработкой (С2Н5)2 А1(ОС2П5). В промышлен-
ности процесс проводится при давлении 25 атм. — Прим, перев.
182
ГЛАВА 2
плотностью от 0.94 до 0,966 г/см3; утверждается, что достигается
выход полиэтилена 98% (от этилена) [279]. Существенной проблемой
при использовании этого процесса является тщательный контроль тем-
пературы при проведении высокоэкзотермической реакции в отсутст-,
вие какого-либо жидкого разбавителя. Однако утверждается, что вви-
ду отсутствия оборудования для регенерации растворителя и сушки
полимера достигается экономия 15% капитальных затрат и 10% теку-
щих затрат [279].
Необходимым условием проведения всех процессов получения
ПЭВП с использованием катализаторов типа Циглера является их вы-
сокая каталитическая активность. Для того чтобы избежать дорого-
стоящего процесса удаления твердого катализатора из полимера пос-
ле реакции (так называемого процесса обеззоливания), необходимо
достижение выхода более 10000 кг/(моль титана). С этой точки зре-
ния гомогенные катализаторы уступают гетерогенным, так как их
активность на несколько порядков меньше, чем у гетерогенных ката-
лизаторов. Теоретически не существует причин, по которым благода-
ря подходящему подбору лигандов не могли бы быть сконструированы
высокоактивные гомогенные катализаторы. Однако до сих пор сведе-
ния о таких системах не опубликованы. Общей проблемой для большин-
ства гетерогенных катализаторов является присутствие в них цент-
ров с различной активностью; чрезвычайно трудно получить поверх-
ность гетерогенного катализатора с однородной активностью. Кроме
того, благодаря частичному разрыву кристаллов катализатора в усло-
виях полимеризации обычно каталитическая активность изменяется в
ходе реакции. Оба этих эффекта приводят к получению полимера, име-
ющего широкое распределение по молекулярным массам. Можно ожидать,
что разработка высокоактивного гомогенного катализатора даст воз-
можность преодолеть эту проблему и позволит осуществить селек-
тивный синтез ПЭВП с узким молекулярно-массовым распреде-
лением, что привлекательно с коммерческой точки зрения. Другой об-
ластью, где открытие высокоактивных гомогенных систем могло бы
сыграть значительную роль, является синтез изотактического поли-
пропилена.
2.6.3. Полипропилен. При получении полипропилена с использовани-
ем катализаторов Циглера — Натта (а не катализаторов, работающих
по катионному или свободнорадикальному механизму) мономерные еди-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
183
Атактический
полимер
। »• ,
н н н- н А
Синдиотактический
полимер
изотактический
полимер
Me Me Me Me Me
Рис. 2.34. Конфигурация различных полимерных молекул, образующихся
при линейной, полимеризации пропилена по типу "голова к хвосту"о
ницы могут соединяться между собой с образованием трех существен-
но различных типов макромолекул: а) атактических макромолекул, в
которых метильные группы ориентированы случайно по отношению к
углеводородному скелету; б) синдиотактических макромолекул, в ко-
торых происходит поочередное изменение положения метильных групп
по отношению к углеводородному скелету; в) изотактических макро-
молекул, в которых все метильные группы располагаются с одной сто-
роны углеводородной цепи. Эти три типа расположения метильных групп
(все из них возможны) при связывании мономерных звеньев по типу
"голова к хвосту" показаны на рис. 2.34. С практической точки зрения
необходим изотактический полимер. Высокомолекулярный изотактичес-
кий полипропилен по многим свойствам напоминает ПЭВП и находит
широкое применение в таких областях, как при производстве изделий,
получаемых литьем (например, сейчас на рынок поступают яхты с кор-
пусами из полипропилена), при производстве пленки, используемой для
упаковки и тары, а также для получения волокна, которое использует-
ся для производства ковров и драпировочных тканей. Недавно была
создана синтетическая бумага, основанная на использовании полипро-
пиленового волокна. Атактический полипропилен является аморфным,
имеет низкую прочность и небольшую коммерческую ценность. Пер-
вые процессы получения полипропилена включали стадию удаления
184
ГЛАВА 2
атактической структуры; в настоящее время с использованием сов-
ременных катализаторов может быть получена селективность по изо-
тактическому полипропилену выше 95%, что устраняет необходимость
иметь специальную стадию удаления атактики. Далее, разработка вы-
сокоактивных катализаторов позволила также избавиться от стадии
удаления остатков катализатора, что значительно упрощает процесс.
Типичные условия полимеризации: температура 50-100°С и давление
5-30 атм. При непрерывной подаче каталитической системы, углево-
дородного растворителя и мономера в реактор полимеризации около
1060 т пропилена необходимо для производства 1000 т пропилена
[280, 281 ].
Замечательная стереоселективность катализаторов полимериза-
ции пропилена может быть связана со стерическим окружением метал-
ла в активном центре. Внедрение мономера в растущую полимерную
цепь (а вернее, миграция полимерной цепи, т.е. алкильной группы, к
полностью или частично координированному алкену) может происхо-
дить с образованием либо фрагмента а-СН2 (2.220), либо а-СН(СН)3
(2.221). Это так называемые акты первичного и вторичного внедрения
[282] соответственно.
М-СН2СН(СН3)-Р (первичное
М - Р + ОН = СН(СН ' Ш внедрение)
2 3
^^М-СН(СН3)СН2-Р (вторичное
2.221 внедрение)
Очевидно, что продукт (2.220) менее стерически "требователен" по
отношению к иону металла, и практически для всех каталитических
систем Циглера - Натта такая ориентация мономера при внедрении
более предпочтительна, что приводит к образованию полимера, содер-
жащего пропиленовые звенья, связанные по типу "голова к хвосту".
Стерическое строение полученной полипропиленовой цепи (см.
рис. 2.34) также зависит от стерического окружения иона металла,
но эта зависимость является более сложной. Для изотактического
роста цепи стерическое окружение активного центра должно быть та-
ким, чтобы стерические взаимодействия между входящей молекулой
пропилена (точнее, между метильной группой пропилена) и лигандами,
которые составляют окружение металла, обеспечивали единственно
возможную ориентацию алкена (пропилена) в переходном комплексе,
содержащем алкен (пропилен) и алкильную группу (растущую полимер-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
185
рис. 2.35. Возможное строение переход-
ного комплекса при изотактической поли-
меризации пропилена0
ную цепь). 1тот переходный комплекс образуется в реакции роста це-
пи (реакции внедрения алкена, представляющей собой миграцию алкиль-
ной группы). На рис. 2.35 показан один из таких комплексов, который
приводит к изотактической полимеризации. Стерические препятствия
с двух сторон металлического центра, которые обусловлены лиганда-
ми Хи Y, должны быть различными, с тем чтобы обеспечивалась за-
данная ориентация мономера, и, таким образом, входящий мономер
стерически был бы вынужден приближаться к активированной связи
металл — углерод растущей полимерной цепи всегда с одной и той же
стороны [282]. Согласно рис. 2.35, при приближении мономера со сто-
роны лиганда Y возникает больше стерических препятствий, чем со
стороны лиганда X. Макромолекулы изотактического полипропилена
имеют спиральную структуру с третичной симметрией (т.е. наблюда-
ется повторение структурной единицы через каждые три мономерных
звена) с периодом идентичности 6,5 А [283]. Если мы рассмотрим
случай, когда возможны два типа ориентации алкена при координации
перед присоединением к ^с-алкильной группе (эти два пути представ-
лены формулами 2.222 и 2.223), то оказывается, что для образования
стереорегулярного полимера (содержащего, скажем, 98% изотактичес-
кой фракции) необходимо, чтобы различие в свободной энергии двух
возможных реакций миграции алкильной группы к алкену (т.е. двух
направлений при росте цепи) составляло 10,5 кДж/моль при 25°С [284].
Для получения стереорегулярного полимера именно такое различие
должно быть обеспечено особенностями стерического окружения актив-
ного центра.
2.222
2.223
186
ГЛАВА 2
Сообщалось, что гомогенная каталитическая система VCl4/AlEtoCl
полимеризует пропилен в синдиотактический материал, если темпера-
тура реакции поддерживается ниже -40°С [285]. Стереорегулирование
в этом случае бы ло приписано сильному взаимодействию метильных
групп мономера и растущей цепи, которое заставляет входящий пропи-
лен всегда принимать конфигурацию, противоположную конфигурации
последнего встроившегося мономерного звена [264]. Однако актив- .
ность этих систем слишком мала, чтобы они представляли коммерчес-
кий интерес. Что же касается высокоактивных изотактических гетеро-
генных систем на основе TiCl3, то активные центры, образующиеся
на поверхности кристаллической решетки, обеспечивают постоянную
ориентацию мономера по отношению к растущей полимерной цепи, что
приводит к получению изотактического полимера. Каким образом и в
каких местах возникают такие центры на кристаллической решетке,
является пока предметом дискуссии [265], хотя почти нет сомнений
в том, что повышенная жесткость кристаллической решетки по срав-
нению с жесткостью системы лигандов в моноядерных титановых комп-
лексах имеет первостепенное значение в обеспечении стереорегулиру-
ющих свойств гетерогенных систем.
Потенциально катализ по Циглеру — Натту должен был бы быть
плодотворной и благодатной областью гомогенного катализа. При ус-
ловии достижения подходящей активности (>250 кг полимера на 1 г ка-
тализатора) катализатор может в большинстве случаев быть оставлен
в полимерном продукте, что устраняет один из обычных недостатков
гомогенных систем — трудность разделения катализатора и продукта.
Однородность активных центров, характерная обычно для гомогенных
систем, дает возможность получить материал с узким молекулярно-
массовым распределением, что привлекательно с коммерческой
точки зрения. Но что касается стереорегулирующих свойств, то, по-
видимому, необходимо конструировать активные центры с более жест-
ким, но в то же время более открытым лигандным окружением, если
ставится . задача достижения высокой стереоселективности и актив-
ности гетерогенных систем.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
187
Дополнительная литература по полимеризации и
катализу Циглера — Натта
Reich L.,‘Schindler A. Polymerization by Organometallic Compounds.» -
New York: Wiley , 1966.
Poor J, ;The nature of the active site in the Ziegler-type catalysts. —
MacromoL Rev., 1967, v. 2, p. 115.
Youngman E.4 /t Poor J. Syndiotactic polypropylene. — Macromol Rev.,
1967, v. 2, 33.
Ketley A.I). (ed.) The Stereochemistry of Macromolecules., vol 1- London:
Arnold, 1967.
Henrici* Olive G., Olive S. The active species in homogeneous Zigler —
Natta catalysts for the polymerization of ethylene. — Angew. Chem.
Internat. Edit.,' 1967, v. 6,p. 790.
Bikales /V.4- Homogeneous catalysis of polymerization. — Advances in
Chemistry Series, N° 70, p. 233, Washington: American Chemical
Society, 1968»
Raff R.4. Polyethylene. — In: Ethylene and its Industrial Derivatives
(ed. S.A. Miller), London: E. Benn, 1969, p. 335»
Valvassori A., Longi P.,tParini P. Polypropylene. — In: Propylene and
its Industrial Derivatives (ed. E.G. Hancock), London: Е» Benn,
1973, p. 155.
Chien J.C.W. (ed.), Co-ordination Polymerization.— New York: Acade-
mic Press, 1975»
Cesca S. The chemistry of unsaturated ethylene-propylene based terpoly-
mers. — MacromoL Rev», 1975, v. 10, p. 1.
Tsutsui M ,'Courtney A, p — тг rearrangements of organotransition metal
compounds. — Adv. Organometal. Chem.,’ 1977, v. 17, p. 241; Olefin
polymerization. The Ziegler - Natta process. — Adv. OrganometaL
Chem.,* 1977, v. 17, p. 256»
См также: Чирков H .M., Матковский П,Б., Дьячковский Фа С.
Полимеризация на комплексных металлоорганических катализаторах. М.:•
Химия, 1976; Захаров В.А., Букатов Г.Д., Ермаков Ю.И. Механизм каталити-
ческой полимеризации олефинов на основе данных о числе активных центров
и константах скоростей отдельных стадий. Усп. химии, 1980, т. 59,
С. 2213—2240;Zakharov V.A., Yermakov Yu. I. Supported organometallic cata-
lysts for olefin polymerization. — Catal. Rev. Eng., 1978, v. 19 (1), p. 67 103.
— Прим, перев.
188
ГЛАВА 2
Caunt A.Q., Ziegler polymerization. — In: Catalysis, voL 1, London:
Chemical Society, 197Z, p» 234»
2.7. Окисление
При выборе систем для рассмотрения в этом разделе мы следо-
вали классическому определению, принятому еще в девятнадцатом
столетии для процессов окисления, а именно : ’’окисление соответ-
ствует присоединению кислорода и потере водорода” [286]. Далее,
для того чтобы ограничить наше изложение приемлемым объемом,
мы будем рассматривать только такие реакции, которые приводят к
присоединению или включению одного или более атомов кислорода, и,
таким образом, не будем рассматривать реакции окислительного со-
четания, такие, как сочетание бензола, катализируемое палладием
[287], или фенола, катализируемое медью [288].
Как это было предложено Шелдоном и Коти [287], гомогенные
реакции окисления, катализируемые соединениями металлов, могут
быть условно разделены на два типа — гомолитические и гетеролити-
ческие реакции.
Гомолитические системы — это те, в которых образуются свобод-
ные радикалы как промежуточные продукты и в которых металл в ка-
талитическом цикле участвует в серии одноэлектронных окислитель-
ных или восстановительных стадий. Жидкофазные процессы гомолити-
ческого окисления (обычно называемые процессами автоокисления)
происходят по радикальному цепному механизму, и априори нет необ-
ходимости предполагать участие металлического центра на всех ста-
диях реакции. Действительно, в большинстве реакций автоокисления,
катализируемых металлами, химическое превращение в основном про-
исходит вне координационной сферы металла.
Гетеролитические процессы гомогенного окисления, катализиру-
емые комплексами металлов, более близки к большинству систем, рас-
смотренных в предыдущих разделах. В этих процессах органический
субстрат или кислородсодержащий реагент активируется путем коорди-
нации (или соединения) с металлическим центром. Если каталитичес-
кий цикл включает ион металла, который меняет степень окисления,
то такие изменения происходят путем протекания последовательности
двухэлектронных стадий. Свободные радикалы в этом случае не явля-
ются интермедиатами, и ион металла остается близко связанным с
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
189
субстратом (реагентом) в ходе большинства (если не всех) химических
превращений, входящих в каталитический цикл.
Системы, которые обсуждаются ниже, были выбраны для иллюстра-
ции этих двух основных типов реакций. Однако нужно иметь в виду,
что процессы окисления, как и большинство явлений природы, не так
просто однозначно классифицировать. В некоторых системах, особен-
но с пероксидами в качестве интермедиатов и с ионами металлов, по-
тенциально способными к участию в обоих типах катализа, не всегда
возможно проводить ясное различие между двумя типами окисления
(и это не всегда нужно делать). Так, окисление циклогексана, которое
обсуждается в разделе, посвященном гомолитическому окислению, мо-
жет при высоких концентрациях катализатора включать образование
комплекса между алканом и центром, содержащим кобальт(Ш) [299].
Аналогично при эпоксидировании пропилена (см. раздел о гетеролити-
ческом окислении) может происходить радикально-цепное, т.е. гомоли-
тическое, разложение гидропероксида, которое приводит к ухудшению
селективности эпоксидирования [287 ].
2.7.1. Гомолитическое окисление. Реакция между органически-
ми соединениями и кислородом, которая протекает по свободноради-
кальному цепному механизму, может быть описана как совокупность
трех процессов, а именно: инициирования, роста цепи и обрыва. Основ-
ная схема автоокисления, включающая эти три процесса, представле-
на уравнениями (2.25)~(2.31):
Инициирование:
In -> In’ (где In - инициатор) (2.25)
In’ + RH -> InH + R’ (2.26)
Рост цепи:
R’ + o2 - RO; (2.27)
RO’ + RH -> RO2H + R’ (2.28)
Обрыв:
2R’^R2 (2.29)
R’ + RO^ - RO2R (2,30)
2R(T II04R -> нерадикальные продукты + O2 (2.31)
В принципе ион металла может участвовать в любой, если не во
всех из этих основных стадий, оказывая таким образом влияние на
190
ГЛАВА 2
направление и скорость реакции автоокисления. На практике ключевая
роль комплексов металлов, добавленных к таким жидкофазным систе-
мам, заключается в катализе разложения гидропероксидов RO2H или
в меньшей степени пероксидов RO2R. Такие соединения могут присут-
ствовать в исходной системе. В этом случае добавление комплекса
металла может вызывать их разложение и, следовательно, значитель-
но уменьшать индукционный период, который обычно наблюдается в
случае некатализируемых реакций автоокисления. Если же такие со-
единения образуются, например, в результате стадий (2.28) и (2.30),
то присутствие металлокомплексного катализатора может ускорить
их разложение и, таким образом, увеличить общую скорость реакции
или изменить направление процесса автоокисления.
Две основные стадии катализируемого процесса разложения гид-
ропероксида, которые были впервые предложены [291 ] для объяснения
разложения пероксида водорода, катализируемого железом(П) (так
называемый реактив Фентона), могут быть изображены следующим
образом:
R02H + Мп+ — RO' + ОН"+ М(п + 1) + (2.32)
RO Н + М^” + 1)+ —* ^2 + Н+ + М 2 + (2.33)
Если данный ион металла способен влиять только на одну из этих ре-
акций, то происходит скорее стехиометрическое, а не каталитическое
разложение гидропероксида, если под каталитическим подразумевать
процесс, в котором ион металла возвращается в свое первоначальное
состояние окисления. Так, хотя соединения меди(1) и разлагают гид-
ропероксиды, например, тир^-бутилгидропероксид по уравнению (2.32),
однако только в присутствии некоторых органических субстратов RH
процесс становится каталитическим благодаря протеканию после-
довательности реакций, показанных ниже [287 ]:
Ви'О2Н + Си1 ВигО‘ + ОН- + Си"
Ви'О' + RH — Ви'ОН + R*
R' + Си11 Н-^-с ROAc + Н+ + Си1
(где Bi? означает дор^-бутил).
В общем случае, когда ион металла является сильным восстанав-
ливающим агентом (например, Си1, Сг ”), разложение гидропероксида
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
191
происходит по уравнению (2.32), тогда как в случае сильных окислите-
лей (например, Pb,v, Ce,v) преобладают реакции типа (2.33). Когда
ион металла с одинаковой вероятностью может находиться в состоя-
нии окисления п и п + 1, т.е. оба состояния окисления имеют сравни-
мую стабильность, тогда обе реакции (2.32) и (2.33) могут происходить
параллельно и образуется каталитический цикл с участием металла,
меняющего степень окисления от п до п + 1. Общая реакция [резуль-
тат сложения уравнений (2.32) и (2.33) ] представляет собой каталити-
ческое разложение гидропероксида до алкокси- и'алкилперокси-ради-
калов:
2RO2H _~ RO2 + RO* + И2О
Именно эта реакция лежит в основе большинства содержащих ионы
металла систем автоокисления. Как соединения кобальта (Со 11 /Со1,1),
так и соединения марганца (Мп11 /Мп111) способны принимать участие в
таком цикле. Однако наиболее широко применяются кобальтовые сис-
темы для катализа жидкофазных каталитических реакций окисления.
Особое значение для нефтехимической промышленности имеют реак-
ции жидкофазного окисления алкилароматических соединений, в част-
ности п-ксилола, и жидкофазного окисления алканов, в частности цик-
логексана.
А. Автоокисление ароматических соединений на примере п-кси-
лола. Терефталевая кислота — продукт, получаемый окислением обе-
их метильных групп п-ксилола, — обычно используется в виде димети-
лового эфира для приготовления полиэфирных волокон. Полиэфирные
волокна (сополимер этиленгликоля и терефталевой кислоты) — одни
из наиболее важных синтетических волокон; в 1976 г. они занимали
41,5% рынка синтетических волокон (для сравнения - доля найлона
составляла 30%) [53]. Первоначально процесс окисления п-ксилола
производился с использованием азотной кислоты как окислителя под
давлением и при температуре 150 —250°С. Однако этот процесс, хотя
он еще иногда и используется, заменен в настоящее время процесса-
ми, в которых в качестве окислителя применяется воздух или кисло-
род в присутствии гомогенных кобальтовых или марганцевых катали-
заторов. Обычно окисление проводится в уксусной кислоте с использо-
ванием ацетата кобальта (III) либо марганца (III) как катализатора,
часто в присутствии промоторов, таких, как бромиды металлов (см.
ниже).
192
ГЛАВА 2
Типичными условиями проведения процесса являются температура
от 100 до 200°С и давление в пределах от 15 до 30 атм. Например, в
процессе прямого окисления, разработанного фирмой Amoco, окисле-
ние проводится при 200°С, 15-30 атмгс использованием воздуха как
окислителя, в присутствии катализатора, представляющего «собой со-
ли кобальта в уксусной кислоте, промотированные соединениями бро-
ма. В процессе, разработанном фирмой Mobil, в котором используется
чистый кислород, применяются температура около 130°С и давление
15 атм [53]. Первая стадия окисления, т.е. получение п-толуиловой
кислоты из n-ксилола, легко происходит в присутствии небольших ко-
личеств солей кобальта или марганца. Для получения же дикислоты
необходимы более высокие концентрации катализатора или промоторов.
Другой процесс (так называемый процесс Виттена) заключается в эте-
рификации первоначально образующейся толуиловой кислоты с образо-
ванием, например, метилтолуилата. В присутствии избытка и-ксилола
метилтолуилат легко окисляется в метилтерефталат [52].
Реакция окисления алкилароматических соединений под действи-
ем катализаторов, содержащих кобальт(Ш), по-видимому, протекает
исключительно по механизму с переносом электрона [287]. Большин-
ство экспериментальных данных согласуется с предположением о на-
чальном образовании катион-радикала типа 2.224 при окислении метил-
ароматического соединения кобальтом (III) с переносом электрона:
АгСН + Со 111 [АгСН,]+ + Со 11 (2.34)
2.224
Потеря протона соединением 2.224 приводит к образованию бензиль-
ного радикала 2.225:
[ArCH3]i- _ АгСН; +Н* (2.35)
2.225
В присутствии кислорода 2.225 образует пероксисоединение 2.226, ко-
торое затем превращается в обычные продукты окисления:
АгСН‘2 + о2 — АгСН2О2 —- продукты
2.226
Соединения кобальта(III) могут быть регенерированы либо реакцией
(2.36) с соединением 2.226, либо реакцией (2.37) с гидропероксисоеди-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
193
нениями 2.227, образующимися на стадии продолжения цепи (2.38).
АгСН2Оз + Со” АгСНО + Со ,н + ОН” (2.36)
АгСН2О2Н + Со " АгСН2О* + Со + ОН” (2.37)
2.227 I___Аг СНО
АгСН2О2 + АгСН3 — АгСН2О2Н + АгСН* (2.38)
Обе эти реакции регенерации катализатора приводят к образованию
ароматических альдегидов, которые являются первичными продукта-
ми окисления метилбензола. Соответствующие кислоты получаются
при последующем окислении альдегида через перокси-кислоту как ин-
термедиат. Было высказано предположение [289], что в случае очень
реакционноспособных катион-ра дика лов, т.е. моноалкилбензолов, мо-
жет происходить в основном быстрый перенос протона по реакции
(2.35) внутри клетки растворителя таким образом, что свободные ка-
тион-радикалы никогда не образуются как дискретные интермедиаты,
например:
4- Со11 4- НОАс
Как упоминалось выше, бромиды металлов часто добавляются как про-
моторы при окислении n-ксилола, катализируемом кобальтом. В отсут-
ствие таких промоторов основным продуктом является п-толуиловая
кислота, при этом последующие процессы окисления происходят очень
медленно. Однако в присутствии бромида натрия последующее окисле-
ние происходит очень быстро и образуется терефталевая кислота вы-
сокой чистоты с почти количественным выходом. Разработка промыш-
ленных процессов, например процесса фирмы Amoco [292], привела к
получению доступной терефталевой кислоты высокой чистоты, что в
свою очередь вызвало тенденцию использовать в производстве поли-
эфирного волокна кислоту, а не ее диметиловый эфир. Промотирую-
щий эффект бромида приписан образованию атомов брома при реакции
окисления бромидом кобальта (III) * с переносом электрона
194
ГЛАВА 2
Вг~+ Со'" —* Со" + Вг‘
Атом брома является чрезвычайно эффективной ловушкой для водоро-
да. Таким образом, в данном случае быстро образуется бензильный
радикал 2.225, при этом инициируется процесс автоокисления:
АгСН3 + Вг* — АгСН^ + НВг (2.39)
2.225
АгСН‘ + О —— ArCFLO’ и т.д.
Соединения кобальта(Ш) регенерируются по уравнению (2.36) или (2.37),
а гидроксильные ионы или гидроксидные соединения кобальта, кото-
рые образуются параллельно, могут регенерировать бромид-ион из
НВг, который образуется по уравнению (2.39):
Со1П(ОН) + НВг — Со 111 Вг + Н2О
В противоположность тому, что наблюдалось в случае реакций авто-
окисления, катализируемых кобальтом в отсутствие бромида, в дан-
ном случае нет доказательств промежуточного образования катион-
радикалов и, по-видимому, не происходит прямой реакции между комп-
лексом кобальта(Ш) и углеводородом.
Хотя терефталевая кислота, несомненно, является одним из глав-
нейших химических продуктов, используемых в производстве синтети-
ческих волокон, на рынке с ней конкурируют адипиновая кислота
2.228 и капролактам 2.229, используемые в производстве найлона-
6/6 и найлона-6-соответственно. Автоокисление алканов, в даИном
случае циклогексана, катализируемое металлокомплексами, играет
ключевую роль в производстве обоих этих химических продуктов. “
ССООН
сн2 сн2
СООН HI|j--
2.228 2.229
Б. Автоокисление алканов на примере циклогексана. Реакции
окисления углеводородов обычно сильно экзотермичны, и когда они
проводятся в большом масштабе, важной практической проблемой яв-
ляется отвод тепла. Для обеспечения отвода тепла такие реакции
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
195
обычно проводятся в жидкой фазе. В отсутствие подходящих катализа-
торов автоокисление алканов обычно требует высоких температур, и
тогда, по крайней мере в случае нормальных алканов, разница в ре-
акционной способности различных С—Н-связей по отношению к ради-
кальной атаке становится незначительной, что приводит к образова-
нию широкого набора продуктов окисления. Однако все же имеется
пример некаталитической реакции автоокисления алкана, которая с
успехом применяется в промышленности, а именно окисление бутана
или фракций парафинов С4 — С8 в уксусную кислоту. С помощью это-
го процесса получается почти вся уксусная кислота, производимая в
Великобритании; этот процесс так же широко используется в других
странах мира. Обычно эта реакция проводится при температурах от
160 до 200°С и давлении около 50 атм. Строгий контроль за темпера-
турой необходим для предотвращения полного окисления до СО2 и Н2О,
Распределение продуктов зависит от условий проведения реакции и ис-
ходного сырья; при окислении фракции парафинов отношение муравьи-
ная кислота: уксусная кислота: пропионовая кислота составляет около
1 : 6 : 0,8. Таким образом, на 1 т исходного алкана получается от 1 до
1,4 т кислот как конечных продуктов [193]. Хотя этот процесс имеет
очевидное преимущество - получение в одну стадию из исходного уг-
леводородного сырья требуемых продуктов,— его недостатком являет-
ся образование значительного количества побочных продуктов.
Другой реакцией автоокисления алканов,которая широко использу-
ется в промышленном масштабе, на этот раз в присутствии гомоген-
ного кобальтового катализатора, является жидкофазное окисление
циклогексана кислородом воздуха с образованием циклогексанона и
циклогексанола как главных продуктов. Обычно используется раст-
воримая соль кобальта в циклогексане, например, нафтенат кобальта.
Для того чтобы избежать избыточного образования побочных продук-
тов, конверсию циклогексана ограничивают приблизительно 10% за про-
ход, при этом селективность по циклогексанолу и циклогексанону сос-
тавляет около 70%. Как и в случае реакции автоокисления алкиларома-
тических соединений, описанных выше, главная роль кобальта, по-ви-
димому, состоит в ускорении разложения циклогексилгидропероксида,
образующегося в результате радикальной реакции. На рис. 2.36 пред-
ставлены каталитические циклы этого процесса [52]. Цикл
Со (III) /Со (II), показанный внизу рисуйка, аналогичен тому, который
196
ГЛАВА 2
Со
продукты
Р и с. 2.36. Каталитические циклы для автоокисления циклогексана, катали-
зируемого кобальтом.,
описан ранее уравнениями (2.32) и (2.33). Кетоны могут образовывать-
ся непосредственно из гидропероксиднэго интермедиата 2.230 при по-
тере воды; в отсутствие катализаторов образуется примерно в 2 раза
больше кетона, чем спирта. Однако в присутствии солей кобальта(Ш)
образуется больше циклогексанола, чем циклогексанона. Опыты со стеара-
тами железа и никеля показали, что в присутствии этих соединений
пероксирадикалы могут непосредственно превращаться в спирт и ке-
тон без образования гидропероксидного интермедиата [294]. Как и в
случае катализируемого кобальтом автоокисления алкилароматичес-
ких соединений, скорость окисления циклогексана, катализируемого
ацетатом кобальта(Ш) в уксусной кислоте, ускоряется при добавлении
бромид-ионов [295]. По аналогии с окислением алкилароматических
соединений это ускорение, вероятно, связано с участием брома в ра-
дикальной цепной реакции. Относительно процесса инициирования
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
197
(т.е. стадии 1 на рис. 2.36) получены некоторые указания [287], что
алкильные радикалы могут образоваться при прямой реакции между
алканом и комплексом кобальта(Ш):
RH + Com R’+ Сон + Н+
Хотя природа взаимодействия между кобальтом(Ш) и алканом пока
еще не ясна, было высказано предположение [ 296 ], что такое взаимо-
действие может включать реакцию электрофильного замещения метал-
лом водорода у насыщенного атома углерода с образованием трехцен-
трового переходного состояния типа 2.231.
—С— Н ч- СоХ
—С
СоХ2
2.231
—С—СоХ2
—С* + СоХ2
Такое взаимодействие, если бы протекание его было доказано, мог-
ло бы рассматриваться как играющее важную роль в области акти-
вации алканов, катализируемой переходными металлами.
Важной областью применения для смеси циклогексанола с цикло-
гексаноном, получаемой при автоокислении циклогексана, является
производство адипиновой кислоты. Это превращение обычно проводит-
ся в отдельном реакторе с использованием в качестве окислителя
60%-ной азотной кислоты в присутствии катализатора медь(Ц)/ванадий(У)
при 60-80°С и слегка повышенном давлении [52, 53]. Очевидно, что
одностадийный процесс получения адипиновой кислоты из циклогекса-
на представлял бы значительный практический интерес. Хотя и поя-
вилось сообщение [297], что при наличии относительно высоких кон-
центраций ацетата кобальта(Ш) и 80—90°С циклогексан может быть
непосредственно окислен воздухом (предпочтительно кислородом) в
адипиновую кислоту с селективностью около 75% при 80%-ном превра-
щении, однако, по-видимому, в промышленных условиях проведения ре-
акции двухстадийный процесс пока еще остается предпочтительным
[53]. При окислении азотной кислотой достигается выход адипиновой
198
ГЛАВА 2
кислоты выше 90%, тогда как в процессе прямого окисления в резуль-
тате значительного образования побочных продуктов необходимо в
схему процесса включать дорогостоящие стадии очистки продуктов
при проектировании заводов.
Образование побочных продуктов при одностадийном процессе
приводит нас к рассмотрению одной из главнейших проблем реакции го-
молитического окисления — проблемы селективности. Участие ката-
лизатора в рассмотренных системах в основном ограничено стадией
разложения гидропероксида, а во многих других стадиях цикла авто-
окисления катализатор мало участвует или совсем не принимает учас-
тия, в результате чего возможности повышения селективности, осно-
ванные на подборе катализатора, ограничены. Большего успеха в
этом направлении можно ожидать в случае реакций окисления, ката-
лизируемых комплексами металлов, в которых металл остается тес-
но связанным либо с субстратом, либо с окислителем (предпочтитель-
но с обоими этими реагентами) во многих или во всех стадиях окис-
ления. Маловероятно, чтобы реакции окисления этого типа происходи-
ли с участием свободнорадикальных интермедиатов. Такие процессы
классифицируются обычно как процессы гетеро литического окисления.
2.7.2. Гетеролитическое окисление. Л. Эпоксидирование, ка-
тализируемое переходными металлами, на примере эпоксидирования
пропилена. В обсуждаемых выше реакциях гомолитического окисле-
ния, катализируемых металлами, взаимодействие между металлом и
гидропероксидом играет ключевую роль. Такое взаимодействие име-
ет также первостепенную важность в гомогенной реакции эпоксидиро-
вания, катализируемой переходными металлами. Однако в этом пос-
леднем случае происходит преимущественно координация гидроперок-
сида, а не его гомолитическое разложение. Суммарный процесс
эпоксидирования алкена заключается в присоединении атома кислоро-
да по двойной связи алкана:
Наиболее важным эпоксидом с точки зрения объема его промышлен-
ного производства является этиленоксид. Производство этого продук-
та в Западной Европе и Америке в 1980 г. должно было превысить
4 млн. т [53]. При этом почти весь этиленоксид получается прямым
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ 199
окислением этилена воздухом или кислородом с использованием ге-
терогенных катализаторов на основе серебра. При температурах око-
ло 250°С и давлениях от 10 до 30 атм селективность по этиленоксиду
(т.е. число молей этиленоксида на каждый моль прореагировавшего
этилена) может легко достигать 70%. Когда вместо этилена исполь-
зуется пропилен, то селективность эпоксидирования резко падает;
наблюдается образование лишь небольшого количества пропиленокси-
да, а основная часть пропилена полностью окисляется до СО2 и воды.
Большая часть пропиленокси да (в 1976 г. продукция США и западно-
европейских стран составляла около 1,5 млн.т) получается с исполь-
зованием хлоргидринного метода, первоначально разработанного для
получения этиленоксида:
СН.СН = СН9 + CL + И90 — СН3СН(0Н) СН С1 + НС1
2СН,СН(ОН) СН2С1 + Са(ОН), — 2СН3СНСН, + СаС12 + Н2О
Ofc fc ** f
о
Этот процесс, однако, имеет несколько недостатков, существенным
из которых является необратимый расход дорогостоящего и коррози-
онноактивного хлора. Относительно новый процесс, впервые нашед-
ший применение в промышленности в 1969 г. [298], основан на том,
что в присутствии некоторых гомогенных металлокомплексных ката* *
лизаторов (в основном содержащих молибден) органические гидропе-
роксиды реагируют с алкенами с образованием эпоксидов с высоким
выходом [287]:
О
'с = С + RO.H CZ ROH (2.40)
/\ / \
Предполагают, что важной стадией в реакции селективного эпоксиди-
рования* является недиссоциативная координация молекул гидроперок-
сида с металлическим центром через кислород, связанный с ал-
кильной группой, с образованием комплекса типа 2.232. В таком
комплексе гидропероксид "активирован координацией” таким об- /
разом, что происходит снижение электронной плотности на атомах
1
к
— "
/
* Механизм эпоксидирования олефинов гидропероксидами изучен Моисе-
евым И.И. и сотр.г см.. Гавриленко В.А., Евзерихин Е.И., Моисеев И.И. -
Изв.АН СССР, сер.хим., 1977, с. 34-38. Прим, перев.
\
200
ГЛАВА 2
2.232
кислорода пероксида благодаря взаимодействию с металлическим
центром, что делает атомы кислорода более чувствительными к нуклео-
фильной атаке реагирующим алкеном. Подходящие катализаторы вклю-
чают в состав переходные металлы в высшем состоянии окисления, на-
пример Mo(VI), W(VI), Ti(IV), и основной процесс эпоксидирования мо-
жет быть описан следующими реакциями:
мп+ + ro2h [Mn+R02H]
(2.41)
\ / \А/
[Мп+И02Н] + С = С —С—С +ROH + мп+
(2.42)
В случае когда комплекс металла содержит фрагмент М=О (напри-
мер, молибденил, вольфрамил, ванидил или титанил), предполагает-
ся [299, 300], что перенос кислорода от комплекса металл-гидропе-
роксид к алкену происходит через циклическое переходное состояние
типа 2.233 по уравнению (2.43).
ОН О
+ ROH
I OR I
(2.43)
2.233
Кроме гетеролитического процесса, представленного уравнениями
(2.41) и (2.42), имеется также возможность гомолитического разло-
жения гидропероксида, катализируемого металлом по реакциям
(2.44) и (2.45), т.е.. аналогично описанному в разд. 2.7.1.
[Mn+RO2H] — М(п ~ 1,++ RO2' + Н+ (2.44)
М<" ~ 1 >+ и. пп и быстР° Mn+ + RO' + ОН" (2.45)
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
201
Селективность образования эпоксидов определяется относительными
скоростями реакций (2.42) и (2.44).
Сильные окислители, такие, как кобальт(Ш) и марганец(III), окисли-
тельно-восстановительные потенциалы перехода которых в двухва-
лентные ионы составляют 1,82 и 1,51 эВ соответственно, благоприят-
ствуют протеканию реакции (2.44) и таким образом снижают селектив-
ность эпоксидирования. Очень слабые окислители, такие, как молиб-
ден(У1), вольфрам(У1) и титан(1У), окислительно-восстановительные
потенциалы которых составляют около 0,2, —0,03 и —0,37 эВ соответ-
ственно, являются плохими катализаторами для гомолитического раз-
ложения гидропероксида, что благоприятствует протеканию реакции
по уравнению (2.42).
Так как основная функция катализатора заключается в увеличе-
нии электрофильности координированного гидропероксида, активность
катализатора должна возрастать с увеличением кислотности по Льюи-
су. Действительно,это видно из сравнения Мо(У1) с W(VI). Мо03 явля-
ется значительно более сильной кислотой, чем WO3, и катализаторы
эпоксидирования, основанные на использовании Mo (VI), значительно
активнее катализаторов на основе W(VI). Можно ожидать также, что
увеличение электроноакцепторных свойств лигандов, связанных с ме-
таллом, будет повышать активность катализатора. Такой случай на-
блюдается при рассмотрении начальных стадий реакций эпоксидирова-
ния. Например,Мо02 (Ас )2 является'первоначально более активным ка-
тализатором эпоксидирования, чем соответствующий комплекс с дио-
лом 2.234. Однако в условиях эпоксидирования первоначально присут-
2.234
ствующие в комплексе лиганды быстро замещаются или разрушаются
молекулами субстрата, т.е. алкеном или гидропероксидом, и после
начального периода скорость эпоксидирования практически не зависит
от природы введенного первоначально молибденового комплекса
[300].
202
ГЛАВА 2
Рис. 2.37. Каталитический цикл для эпоксидирования алкенов, катализируе-
мого молибденом(У1 )о
На рис. 2.37 представлен * общий каталитический цикл эпоксиди-
рования, катализируемого комплексами молибдена. Необходимо под-
черкнуть, что этот цикл отчасти предположителен. Несмотря на ин-
тенсивное изучение этой реакции, имеется еще несколько неясностей
относительно точного механизма реакции эпоксидирования алкенов,
катализируемой металлокомплексами.
В промышленности при окислении пропилейа используется либо
гидропероксид, получаемый при окислении воздухом этилбензола, ли-
бо 0ф£яг-бутилгидропероксид, получаемый при окислении воздухом
изобутана. Когда используется гидропероксид этилбензола, то получа-
ющийся спирт дегидратируется с образованием стирола, который от-
деляется как побочный продукт. Суммарная реакция изображается
следующим образом:
гиен СН, + сн3сн = СН2 + О, — PhCH = сн, +
о о z z х
/°\
+ СН СН—сн, + н, о
’J Z Z
При использовании wp^-бутилгидропероксида тр ^-бутанол, образую-
щийся на стадии эпоксидирования, либо отделяется как таковой, либо
дегидратируется в изобутилен и возвращается в рецикл.
г
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ 203
Во всех реакциях окисления, катализируемых комплексами метал-
лов, которые мы рассматривали до сих пор, истинным окисляющим
агентом был гидропероксид, либо введенный в реакцию, либо образо-
вавшийся в ходе окисления. Ниже мы рассмотрим гомогенное окисле -
ние, катализируемое переходными металлами, причем окислителем
является сам металл.
Б, Окисление алкенов, катализируемое палладием, на примере окис-
ления этилена. В 1894 г. Ф.Филлипс [301] сообщил, что при обработ-
ке этиленом Pd Cl 2 в водном растворе восстанавливается до металли-
ческого палладия с одновременным образованием ацетальдегида. Эта
реакция соответствует стехиометрическому окислению этилена палла-
дием:
СН =СН9 + РЬС19 + Н90 — СНСНО + Pd + НС1
Долгое время к этой реакции не проявлялось интереса с точки зрения
ее практического использования, но в конце 1950-х г. группа исследо-
вателей, работающая по контракту с фирмой Wacker Chemie под руко-
водством Смидта, сообщила, что в присутствии некоторых окислите-
лей, таких, как хлориды меди или железа, металлический палладий мо-
жет быть легко реокислен в хлорид палладия [302, 303 ]. Так как в слу-
чае хлорида меди продукт ее восстановления (CuCl) может быть легко
реокислен воздухом или кислородом, то сочетание СиС12 и PdCl2 при-
водит к каталитической системе, в которой суммарная реакция соот-
ветствует окислению этилена в ацетальдегид воздухом или кислородом:
PdCl* * ~ + С2Н4 + Н2О — СН3СН0 + Pd + 2НС1 + 2С1 ~ (2.46)
2СГ + Pd + 2CuC12 —^PdCl4~ + 2CuCl
2CuCl + — О, + 2HC1 —>-2CuCl, + H,0
2 2 2 2
1
—O2 + C2H4------CH3CHO
Эта совокупность реакций служит химической основой для вакер-про-
цесса, с помощью которого в настоящее время производится пример-
но 1,5 млн. т ацетальдегида в год.
• Хотя имеются еще некоторые сомнения относительно деталей, как
именно палладий проводит окисление этилена в ацетальдегид, в общем
204
ГЛАВА 2
реакция может быть представлена такой последовательностью стадий
[304]:
PdCl 2.~ + С,НД
4 2 4
[PdCl3(C2H4)]- + Cr
(2-47)
2.235
[pdcl3(C2H4)]- + Н2О — PdCl2(C2H4)(H2O) + Cl" (2.48)
2.236
PdCl (С2н4)(H20) + Н,0 —[PdCl,(С Н,СН,ОН) (Н,О) Г + н+
2.237 (2.49)
[PdCl2(CH2CH2OH)(Н2О) ]“—>СН3СНО + Pd + 2СГ+ Н3О+
(2.50)
При низких концентрациях палладия(П) эта последовательность реак-
ций приводит к кинетическому уравнению:
4СН3СН01/Л = MPdCl*~][C н ]/[СП]2[Н+]
О Н Л Я-
Начальная стадия окисления, представленная равновесиями (2.47) и
(2.48), включает и координацию этилена с одновременным или последо-
вательным удалением хлорида с образованием 16-электронного анио-
на палладия (II) (2.235), который в свою очередь участвует в реакции
обмена лигандов с образованием нейтрального комплекса палладия(П)
(2.236). Экспериментально найдено [302, 303], что общий процесс ин-
гибируется ионами хлора, что согласуется с наличием реакций (2.47)
и (2.48). Координация этилена с ионом палладия(П) приводит к умень-
шению электронной плотности на двойной связи, что делает ее более
чувствительной к нуклеофильной атаке при действии ОН“или Н2О,
как это показано уравнением (2.49).
Пока полностью не ясно, происходит ли нуклеофильная атака на
координированный алкен внутри координационной сферы металла или
же она является внешнесферной, т.е. не ясно, требуется ли предвари-
тельная координация ОН“или Н2О [304, 305]. Первоначально предпола-
галось, что гидроксипалладирование включает стадию миграции лиган-
да, т.е. координированной частицы ОН-к координированному СН2—СН
аналогичную той, которая рассматривалась ренее в случае гидрирова-
ния (миграция координированной частицы Н“ к координированному ал-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
205
Кену, см. разд. 2.1). Для протекания такой миграции необходимо, что-
бы и гидроксигруппа, и этилен находились в гдос-положении, что при-
ведет к ео-присоединению через образование четырехцентрового про-
межуточного состояния типа (2.238).
2.238
Альтернативный механизм, рассматривающий внешнесферную
атаку ОН ““без предварительной координации (который бы приводил
к анти -присоединению), был отвергнут [306] на основе того, что он
требовал бы константы скорости большей, чем это возможно для про-
цесса, контролируемого диффузией. Этот аргумент, впрочем, не при-
меним к внешнесферной атаке водой и, таким образом, на основе
стереохимических данных, полученных с использованием частично
дейтерированного алкена [307, 308], которые указывают на а«шг-при-
соединение гидроксигруппы, были независимо предложены две опи-
санные ниже последовательности .реакций для протекания стадий
гидроксипалладирования.
Последовательность /
Н2О
СН2СН2+ОН2
(2.51)
Н2О +
СН2СН2+ОН2
(2.52а)
206
ГЛАВА 2
СН2СН2ОН
2.237а
медленно С1\
----* / Pci — СН2СН2ОН + С!"
Н2О
(2.53а)
Последовательность 2
медленно С1\
----* .Pd—СН2СН2ОН + СГ
Н2О
2.237$ (2.536)
Единственное существенное различие между двумя этими схемами
заключается в стереохимии промежуточных палладиевых комплексов.
Существенной чертой обоих механизмов является то, что лимитирую-
щая стадия реакции не есть гидроксипалладирование (как это пред-
полагалось ранее [306]), а скорее удаление иона хлора из 2.237 с обра-
зованием трехкоординированной четырехэлектронной частицы
PdCl(H2O) (СН2СН2ОН). Таким образом, реакция гидроксипалладиро-
вания является результатом установления быстрого равновесного
процесса [уравнения (2.51) и (2.52) в последовательности 1 или урав-
нение (2.526) в последовательности 2]. Хотя Смидт и др. [309] перво-
начально рассматривали гидроксипалладирование как равновесный
процесс (2.54), они предположили, что при этом происходит предваритель-
ная координация гидроксила как нуклеофильной частицы:
PdClJCH.)(Н,О) й* tPdCl2(CH2CH2OH)(H2O)]- <2-54>
Стереохимические данные, полученные при использовании частично
дейтерированного этилена, по-видимому, являются решающими для
суждения о том, происходит ли присоединение к координированному
алкену типа цис- или транс- (т.е. как происходит атака некоординиро-
ванной воды). Однако важно подчеркнуть, что эти стереохимические
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
207
эксперименты были проведены при высокой концентрации ионов хло-
ра (около 3 М), что затрудняет предварительную координацию нуклео-
филов ОН“ или Н2О. При других экспериментальных условиях (напри-
мер, при низких концентрациях С1“) может происходить предваритель-
ная координация этих чартиц, и, вероятно, сейчас преждевременно
утверждать, что механизмы, предложенные либо Штилле и др. (после-
довательность 1), либо Бэквалом и др. (последовательность 2), имеют
общее значение при всех экспериментальных условиях.
Хотя имеется некоторое различие во взглядах относительно путей
образования р-гидроксиэтилпалладия(П) (2.237), все же общепринято,
что этот комплекс образуется в результате реакции гидроксипаллади-
рования. Далее обсудим вопрос о том, как происходит его разложение
с образованием ацетальдегида и палладия (0).
Из экспериментальных результатов изучения рассматриваемой
реакции наиболее интересно то, что если этилен окисляется в D2O,
то при этом не образуется дейтерированный ацетальдегид [310]:
[PdCl (CH2CH2OD)(D2O)]- —* СН,СНО + СГ + Pd(0)
Таким образом, любое рассмотрение механизма разложения р-гидро-
ксиэтильного комплекса должно по необходимости включать быструю
реакцию 1,2-переноса водорода в комплексе с потерей гидроксильно-
го водорода.
Первоначально для этого процесса был предложен согласованный
механизм гидридного сдвига, катализируемый ионами палладия(П) с
образованием переходного состояния типа 2.239 [306]. В свете экспе-
риментальных данных, полученных позже, теперь представляется бо-
лее вероятным стадийный механизм, включающий образование палла- I
дийгидридных интермедиатов [306, 308, 309, 311, 312]. Этот тип про-
цесса представлен на рис. 2.38.
2.23Э
СН3СНО 4- Pd(O) + 2СГ 4- Н+
208
ГЛАВА 2
[Cl2(H2O)PdCH2CH2OH]^o CKHjOPd^l
Н
?ХНОН
сн2
быстро
н
| снон
Ct(H9O)Pd’*“||
2 сн2
ЛЬ
н-—сн,
! :1
Cl(H20)p'd — СН(ОН)
2.240
2.241
сн3_
CI2(H2O)Pd-^CH ->-|Cl2(H2O)PdH] + СН3СНО
Н — 6 J (2.243)
2242 ।► Pd°+ 2Cl” + Н3О+
Рис. 2.38. Стадийный процесс с участием палладийгидридных интермедиатово
Существенно, что в этом механизме рассматривается изомериза-
ция р-гидроксиалкильного комплекса 2.237 в а-гидроксиалкильные произ-
водные 2.242 через стадию удаления и присоединения гидрида, пол-
ностью аналогичную той, которая описана в разд. 2.2.1,А при рас-
смотрении изомеризации алкенов, катализируемой металлами. Для
того чтобы это согласовалось с результатами исследований, получен-
ных с использованием дейтерированных продуктов [304], необходимо,
чтобы виниловый спирт в комплексе 2.240 диссоциировал перед тем,
как произойдет'присоединение гидрида с образованием продуктов
2.242. Другими словами, присоединение гидрида в 2.240 должно проис-
ходить значительно быстрее, чем диссоциация из него алкена или же
реакция обмена. Это находится в соответствии с известной химией
гидридных комплексов металлов с алкенами. Кроме того, имеется воз-
можность взаимодействия между d-орбиталями алкена и гидроксилов,
что также будет препятствовать диссоциации или обмену. Удаление
ацетальдегида из комплекса 2.242 происходит путем необратимой ре-
акции удаления р-водорода из гидроксильной группы. В отсутствие
стабилизирующих лигандов образующиеся гидридные комплексы пал-
ладия(П) 2.243 быстро разлагаются с восстановительным удалением
НС1 с образованием нульвалентного металлического палладия.
Последовательность реакций, описанная выше, приводит к стехио-
метрическому окислению этилена или восстановлению палладия(П).
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
209
В присутствии системы, способной реокислять палладий(О), становит-
ся возможным осуществлять каталитический процесс по палладию.
В вакер-процессе почти всегда используется хлорид меди в качестве
окислителя палладия, так как получающийся хлорид одновалентной
меди может быть легко реокислен воздухом или кислородом:
Pd + 2d’ + 2Cu Cl _PdCL2' + 2CuCl
2 4
(2.55)
2CuCl + — O2 + 2HC1 — 2CuC12 + H2O (2.56)
2
В общем каталитическом цикле скорость реакции лимитируется ста-
дией реокисления меди(1) уравнение (2.56). Имеются два варианта
промышленного процесса — один с использованием кислорода, другой
с использованием воздуха [313]. Процесс с использованием воздуха
проводится в двух различных реакторах. Первый реактор содержит
раствор PdCl2 и СиС12. В этот реактор подается смесь этилена с воз-
духом (возможно использование воздуха, обогащенного кислородом)
до тех пор, пока большая часть Си (II) не восстановится до Си (I).
После этой стадии катализаторный раствор поступает во второй ре-
актор, где медь реокисляется воздухом. Путем переменного проведе-
ния этих двух стадий происходит непрерывный процесс. В одностадий-
ном процессе с использованием кислорода собственно реакция и реге-
нерация катализатора проводятся в одном реакторе. Этилен и кисло-
род поступают в вертикальный реактор, содержащий раствор катали-
затора. Окисление проводится при повышенном давлении (около 10 атм)
при температуре кипения водного раствора. Полученный ацетальде-
гид отгоняется из реакционной смеси и после конденсации отделяет-
ся от воды и порочных продуктов обычно путем двухстадийной дис-
тилляции. Общая схема одностадийного процесса использования кис-
лорода [313] показана на рис. 2.39. На рис. 2.40 изображен возмож-
ный каталитический цикл, учитывающий данные о механизме, изло-
женные выше.
В практических условиях использования процесса концентрация
меди намного превышает концентрацию палладия. И хотя в каталити-
ческом цикле,- представленном на рис. 2.40, роль меди заключается
только в реокислении палладия, все же, вероятно, хлорид меди участ-
вует не только в стадии реокисления. Действительно, в присутствии
уксусной кислоты, которая часто применяется в качестве сораствори-
210
ГЛАВА 2
пар пар
реактор конденсатор дегазатор дистилляция
Рис. 2.39. Схема одностадийного процесса получения ацетальдегида из
этилена.
Рис. 2.40. Каталитический цикл для реакции окисления этилена в ацетальде-
гид, катализируемой системой палладий(П )/медь(П)ь
1
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
211
теля или же получается при последующем окислении ацетальдегида,
использование разных окислителей, в том числе хлорида меди, при-
водит к получению насыщенных эфиров из этилена в присутствии' пал-
ладия(П) [314, 315]:
НОАс
Си + PdCl, +CuCl,------ С1СН,СН,ОАс, НОСН,СН,ОАс,
2 4 7/ 2 2 Z 7 z z 7
АсОСН2СН2ОАс
Такие продукты всегда образуются в малых количествах в ходе про-
ведения вакер-процесса в водном растворе. По-видимому, они появ-
ляются в результате взаимодействия между окислителем и ацетокси-
палладиевым интермедиатом* 2.244.
Результаты кинетических исследований показали, что хлорид
меди также участвует в основном процессе окисления этилена при
высоких концентрациях Си2+ [316, 317]. Было высказано предполо-
жение [318], что при этом образуются интермедиаты типа 2.245 и
что соответствующие гидроксиэтильные комплексы 2.246 могут
разлагаться без образования Pd(O).
^PdCH2CH2OAc
2.244
СН2
613Cu—Ck JI
Vch.
СЦСи—с/ ОН,
и. j 2
2.245
Была предложена последовательность реакций, в которой два
электрона переходят по мостику из палладия и иона хлора (по одному
электрону к центру, содержащему Си11) таким образом, что хлорид
меди восстанавливается внутрикомплексно, а палладий возвращает-'
ся в реакционную среду в виде палладия(П).
*
Механизм образования производных гликолей при окислении этилена
комплексами палладия изучен еще недостаточно. Возможные пути образова-
ния ацетатов и нитритов гликолей рассмотрены в работе: Лихолобов В.А.,
Ермаков Ю.И. О некоторых аспектах подбора металлокомплексных катали-
тических систем. — Кинетика и катализ, 1980, т.21, с.904 - 914. — Прим, перев.
212
ГЛАВА 2
2.246
сн3сно + н+ + Pdci2 ч- гс^сЛз2-
Кроме того 2.246 (или аналогичный комплекс) может перестроить-
ся в а-гидроксиэтильные частицы 2.247 при протекании процесса
удаления - присоединения гидрида, описанного ранее. Разложение
этого комплекса, катализируемое медью, приводит к выделению
ацетальдегида и возвращению палладия в цикл в виде палладия(П).
2.247
СН3СНО 4- Н3О+ 4- PdCi2 4-
4- гси’сц2"
Эта последовательность реакций показана пунктирными линиями на
рис. 2.40. Хотя, по-видимому, нет сомнений в том, что в промышлен-
ных условиях проведения вакер-процесса, т.е. при высоких концентра-
циях Си2+ относительно Pd2+, медь участвует не только в процессе
окислительной регенерации в каталитическом цикле, и хотя привлека-
тельно предположение о синергическом ее участии с образованием
комплекса 2.246 или 2.247, точная природа этого участия, так же как
и природа многих интермедиатов в вакер-процессе, еще ожидает более
детального исследования механизма. Тем временем вакер-процесс
остается одним из наиболее важных в применении гомогенного ката-
лиза в промышленной химии.
Хотя во всех окислительных системах, которые мы описали, участ
вует кислород, ни в одном из описанных случаев он не присоединяется
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
213
к субстрату непосредственно с помощью металлического центра. Во
всех случаях молекулярный кислород вводится апосредованно, т.е. ли-
бо через гидропероксид при окислении алкана и эпоксидировании алке-
на., либо через посредство воды в вакер-процессе. Известны многие
комплексы дикислорода [319], содержащие координированный симмет-
ричный или несимметричный мостиковый кислород (например, 2.248 —
2.250).
PPh,
I **
PPh3
I^CN
Со
I CN
CN
NH3
ч I ^NH
’Со ’
NH3
I /NH3
Со
I ^NH3
NH3 NH3
2.250 . ’
Однако до настоящего времени не сообщалось о каталитических реак-
циях, в которых происходит прямой перенос одного атома кислорода
или связанного дикислорода к органическому субстрату*. Тем не ме-
нее многие из химиков, работающих в промышленности, а также неко-
торые исследователи в академических учреждениях все еще заворо-
жены призрачной целью добиться прямого окисления пропилена в про-
пиленоксид
сн3сн= сн2 +
СИ—сн,
dC О
Дополнительная литература**
Уотер с В. А. Механизм окисления органических соединений. — М.:
Мир, 1966.
Berezin I.V., Denisov Е.Т.,Emanuel N.M. The Oxidation of Cyclohe-
xane. — Oxford: Pergamon, 1966.
*
В 1980 г. опубликована хорошая обзорная статья по процессам пере-
носа связанного дикислорода к органическим субстратам: Minoum н. -
J. Molec., Cat., 1980, № 7,p.l. Прим, перев.
См. также: Моисеев И.И. п-Комплексы в жидкофазном окислении
олефинов. — М.: Наука, 1970; Моисеев И.И. — ЖВХО им. Менделеева, 1977,
Т. 22, С. 30—45;Henry Р.М.,^Palladium catalyzed oxidation of hydrocarbons. —
Dorderect a.o., Reidel. 1980, 435 p. (Catalysis by metal complexes, Vol. 2.) —
Прим, перев.
214
ГЛАВА 2
Szonyi G. Recent homogeneously catalysed commercial processes* —
Advances in Chemistry Series, v. 70, Washington: American Chemi-
cal Society, 1968 p. 53.
Reich L., Stivala S.S. Autoxidation of Hydrocarbons and Polyolefins.
Kinetics and Mechanisms. — New York: Marcel Dekker, 1969.
Stem E.W. Homogeneous metal catalysed oxidation of organic compo-
unds — In: Transition Metal in Homogeneous Catalysis (ed. G.N.
Schrauzer), New York, Marcel Dekker, 1971, p. 93.
Maitlis P.M. The Organic Chemistry of Palladium, vol. II, Catalytic
Reactions Academic Press, 1971, Ch. 11, p. 77.
Hucknall D.J. Selective Oxidation of Hydrocarbons. — New York: Acade-
mic Press, 1974.
Lyons J.E. Oxidation of olefins in the presence of transition metal
complexes. — Adv. Chem. Series., v. 132, Washington: American
Chemical Society, 1974, p. 64.
Henry P.M. Palladium-catalysed1 organic reactions. — Adv. Organometal
Chem., 1975, v. 13, p.363.
Penson D. Mechanisms of Oxidation by Metal Ions. — Amsterdam!
Elsevier, 1976.
Sheldon R.A., Kochi J.K. Metal-catalysed oxidations of organic com-
pounds in the liquid phase: a mechanistic approach. — Adv. Catal.,
1976, v. 25, p. 272.
Lyons J.E. Transition metal complexes as catalysts for the addition of
oxygen to reactive organic substrates. — In: Aspects of Homoge-
neous Catalysis (ed. R. Ugo), vol. 3,.Dordrecht, Reidel, 1977, p. 1.
Alper H. Oxidation, reduction rearrangement, and other synthetically
useful processes. — In: Transition Metal Organometallic in Organic
Synthesis, (ed. H. Alper), vol. 2, New York, Academic Press, 1978,
p. 121.
Davidson J.M. Homogeneous catalytic oxidation. — In: Catalysis, vol.2,
London, Chemical Society, 1978.
2c8d Метатезис
Мы начали эту главу об использующихся гомогенных катализа-
торах рассмотрением реакции гидрирования — реакции, давно откры-
той и, вероятно, наиболее изученной среди реакций, катализируемых
переходными металлами. Мы закончим эту главу рассмотрением од-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
215
ной из недавно открытых и, несомненно, одной из наиболее интригую-
щих реакций - реакции метатезиса алкенов.
В соответствии с определением, данным Руни и Стьюардом [320],
метатезис алкенов — это процесс, в котором разрываются связи уг-
лерод — углерод в алкене и вновь образуются, причем происходит ста-
тистическое перераспределение алкилиденовых фрагментов таким об-
разом,.что в стационарных условиях устанавливаются равновесные
концентрации реагентов и продуктов. Следовательно, катализаторы
метатезиса алкена ускоряют установление равновесий типа (2.57) —
(2.59):
2R1CH — CHR2 R’CH^zCHR1 + R2CHz=CHR2 (2.57)
(2.58)
(2.59)
Первые сообщения об установленном случае метатезиса алкенов, ка--
та лизируемого соединениями металлов, появились в конце 50-х и в
начале 60-х годов, в этих случаях использовались гетерогенные ката-
литические системы [321, 322]. Например, оксид молибдена на оксиде
алюминия, "промотированный" триизобутилалюминием, катализирует
превращение пропилена в этилен и бутен [321].
сн_сн—сн9 сн сн сн2
+ =₽=-=]!+ (2.60)
СН,СН=СН_ сн,сн сн2
о Z °
216
ГЛАВА 2
В 1967 г. было опубликовано сообщение о первом гомогенном катали-
заторе* метатезиса алкенов — комбинации WC16, С2Н5ОН и С2Н5А1С12
[ 323], и с тех пор наблюдался взрывной характер увеличения экспери-
- ментальных исследований, посвященных этой теме. Сейчас известны
катализаторы метатезиса с участием большинства переходных метал-
лов, но наиболее активные системы основаны на использовании воль-
фрама, молибдена и в меньшей степени рения [48]. 1
Одним из наиболее интересных аспектов этой реакции является 1
установление пути, по которому металлический центр действительно
осуществляет разрыв старых и образование новых связей в алкенах. ]
Ранние эксперименты [328, 329] с использованием алкенов, меченных
14С [например, 2.251 в уравнении (2.61)], показали, что образование
продуктов является следствием разрыва связей С=С, а не переноса
алкильной группы. Только в последние 4 — 5 лет исследователи приш-
ли к общему согласию относительно вероятного механизма реакции
2СН — сн=14сн == 14сн = 14сн2 + сн3—сн=сн—сн3
О о о
2.251
(2.61)
2.8.1. Механизм метатезиса алкенов. С первого взгляда наибо-
лее привлекательным представляется механизм, отвечающий равнове-
сиям (2.58) — (2.60), т.е. согласованный механизм, включающий одно-
временную .координацию двух реагирующих молекул с последующим об-
разованием переходного состояния металл — циклобутан типа 2.252:
*
До сих пор продолжается дискуссия [324 — 32б], являются ли эта част-
ная каталитическая система и системы, основанные вообще на использова-
нии WC16, действительно гомогенными. Эта проблема часто возникает при
использовании сложных каталитических систем, т.е. систем, содержащих
несколько каталитических компонентов. В общем случае хорошим указани-
ем на гомогенность катализатора является получение в случае ’’гомогенной"
каталитической системы существенно отличающейся селективности по про-
дуктам по сравнению с гетерогенным аналогом. Хотя в отношении системы
WC16/C2H5OH/C2H5A1C12 возникают некоторые сомнения, заметно иные
селективности были получены [327 J с другими катализаторами метатезиса,
основанными на использовании соединений вольфрама, и, по-видимому, ис-
тинно гомогенные катализаторы метатезиса алкенов действительно сущест-
вуют.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
217
R'RsC
r5r6c
cr’r4
cr’r8
R'R’C -y-CR3R4
I M^l
r5r6c—cr’r8
2.252
r'r2c cr3r4
-M-
r5r6c cr’r8
\ (2.62)
Такой согласованный, или парный, механизм, который полностью ана-
логичен 'описанному для скелетной изомеризации в разд. 2.2.2Ъ А,
как первоначально считалось, применим и к метатезису алкенов
[330-332]. Однако сейчас общепринято*[48, 320, 333—335], что реак-
ция происходит как цепной процесс с участием металлкарбенов как
ключевых интермедиатов:
R,R2C=M RIR2C_J4 R'R^ М
r5r6c=cr7r8 r5r6c—cr’r8 rsR6C cr’r8
(2.63)
А. Карбены металлов в качестве интермедиатов. Большинство
экспериментальных доводов в пользу образования металлкарбеновых
частиц как промежуточных продуктов при метатезисе алкенов получе-
но при анализе продуктов, образующихся в начальных стадиях реакции
[335]. В условиях метатезиса смесь бутена-2, октена-4 и циклооктена
дают алкены С6, С12,С14, С16, состав которых показан ниже:
СН3СН=СНСН3 4- С3Н7СН=СНС3Н7 +
СН3СН=СНС3Н7 +
—снсн3 Г >=снсн3 г ^=СНС3Н7
-=снсн3 I >=СНС3Н7 I >=СНС3Н7
С|б
*
Карбеновый механизм метатезиса олефинов предложен и обоснован в
работах Б.А. Долгоплоска (см., например, Долгоплоск Б.А., Маковецкий К.Л.,
Коршак Ю.В., Орешкин И.А., Тинякова Ё.И., Яковлев В.А. Природа активных
центров и механизм реакции раскрытия циклоолефинов и метатезиса олефи-
нов. — Высокомол. соед., 1977, № 19, с. 2464—2477). — Прим, перев.
218
ГЛАВА 2
Если метатезис происходит по "парному” механизму, показанному
уравнением (2.62), то в начальных стадиях реакции концентрация С14
должна быть намного ниже, чем концентрация С12 или С1б, поскольку
наличие продукта С14 будет зависеть от присутствия продукта С6,
тогда как продукты С12иС16 могут непосредственно образоваться
из субстрата С4 и алкенов С8. Если же, с другой стороны, происходит
цепной процесс, изображенный уравнением (2.63), то концентрация С14
будет по крайней мере в два раза выше, чем концентрация как С12,
так и С16.
При использовании MaCl2(NO)2(P(C6H5) 3]3/А12 Ci3(CH3)3 как
предшественника катализатора в хлорбензоле было установлено, что
на первых стадиях метатезиса С1 всегда образуется в значительно
больших количествах, чем С12 или С16 [336]. Если мы примем, что
обмен алкенов не является стадией, определяющей скорость общего
процесса метатезиса [335], тогда результаты этих ’’перекрестных"
экспериментов надежно исключают координационный механизм, изо-
браженный уравнением (2.62).
Одно из первых прямых свидетельств о промежуточном образовании
металлкарбеновых частиц было получено с использованием замещен-
ных алкенов типа 2.253 [333, 337]. В присутствии RhCl(PPh3)3B кси-
лоле при 140°С в течение 2 ч из смеси 2.253а и 2.2536 образуется
приблизительно равновесное количество 2.253в. В таких условиях мо-
гут быть выделены монокарбеновые комплексы типа 2.254; при этом-
2.254а превращается в 2.2546 и оба комплекса катализируют вышеопи-
санную реакцию диспропорционирования.
Cl(PPh3)2Rh
2.254
2.253
(a) R1 = R2 = Ph
W R1 = R2 =п -tol
W R1 = Ph; R2 =/?-tol
(a) R1 - Ph
(£) Rl = n-tol
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ 219
Выводы о механизме, полученные при изучении этих систем, пред-
ставляются сомнительными, поскольку такие богатые электронами ал-
кены несколько отличаются от обычных алкенов, используемых в боль-
шинстве реакций метатезиса. Это является общей проблемой, когда ис-
пользуются модельные и обычно более стабильные комплексы для
целей выяснения механизма каталитической реакции. Однако несмотря на
эти ограничения, те выводы о механизме, которые были получены при
изучении этих систем, подтверждены в ходе недавних исследований.
В 1973 г. Кейси и Беркхардт [338] сообщили о синтезе и выделе-
нии металлкарбенового комплекса 2.255 (где R=Ph).
(СО) Bw=c
о
Этот комплекс намного ближе к типу карбеновых частиц, которые при-
нимают участие в реакции метатезиса, в том смысле, что он не содер-
жит гетероатомов, связанных с карбеновым атомом углерода. Значе-
ние этого комплекса для механизма метатезиса алкенов стало более
ясным в последующие годы, когда было показано протекание реакции
(2.64) [339]:
Ph Ph
с
W
(СО)
о
сн2
сн2
сн9
100°С , ч / \
----* (СН,),С —CPh, +
2 ч 32 2
Ph Ph
снз сн,
*5 О
W(CO)e
(2.64)
С использованием последовательности изображенных ниже реакций
Кейси рассмотрел образование этих продуктов, предполагая образо-
вание метилциклобутанового комплекса 2.256 (R1 = Ph, R2 = Me) в
качестве ключевого интермедиата (см. схему реакции на стр. 220).
В случае этой частной системы было выделено только дифенилдиме-
тилциклопропильное производное 2.257 (R1 = Me, R2 = Ph), т.е. в
этом случае имеется только косвенное свидетельство присутствия
Диметилкарбеновых частиц. Однако когда используется ди-n- толил-
карбеновый комплекс 2.255 (R = n-толил) в сочетании со стиролом
вместо изобутилена, то образуются оба ожидаемых циклопропильных
соединения, т.е. 2.258 и 2.259, что указывает на присутствие как ди-
220
ГЛАВА 2
я-толил-, так и монофенилкарбеновых интермедиатов [ 340].
сн2
(То1), с —CHPh
2.258
СН2
PhHC—СНРЬ
2.259
Хотя реакции с участием вольфрама, описанные выше, в основном
являются стехиометрическими, все же (дифенилкарбен)пентакарбо-
нилфольфрам 2.255 (где R — фенил) катализирует, хотя й в неболь-
шой степени, реакцию метатезиса ^с-пентена-2 [341] и некоторых
гдое-циклоалкенов [342] (см. разд. 2.8.2.). Дальнейшие свидетельства
образования промежуточных металлкарбеновых частиц были получе-
ны при пересмотре некоторых ранних факторов, обнаруженных при
использовании гетерогенных каталитических систем. В 1964 г. Бэнкс
и Бейли [322] сообщили о присутствии значительных количеств цик-
лопропана и метилциклопропана в продуктах метатезиса этилена на
катализаторе Мо03/А1203. Когда ’’парный” механизм был в моде,
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
221
это наблюдение игнорировалось [342]. Однако в свете работы Кейси
о нем вспомнили. Последующая работа [344] с гетерогенными сис-
темами, полученными при нанесении Re2O7 на оксид алюминия, пока-
зала, что во время метатезиса циклогептатриена и этилена с образо-
ванием главным образом бензола смесь продуктов С - С4 примерно
на 18% (от общего содержания фракции С3/С4, образующейся перво-
начально) состоит из циклопропана. В настоящее время, по-видимому,
почти нет Сомнений в том, что метатезис и циклопропанирование —
это близко связанные реакции; накоплено значительное число данных,
свидетельствующих о промежуточном образовании металлкарбеновых
частиц в метатезисе алкенов. Однако ни в одной из систем метатези-
са, имеющей реальное значение с точки зрения каталитической актив-
ности, не используются металлкарбеновые комплексы как таковые.
Мы попытаемся проанализировать причины этого.
Б, Генерирование металлкарбеновых комплексов. Большинство
активных гомогенных катализаторов метатезиса алкенов готовится
добавлением алкилалюминия или алкильных производных олова, цин-
ка или лития к галогенидным комплексам вольфрама, молибдена или
рения в инертных растворителях, таких, как хлорбензол, часто в при-
сутствии небольших количеств этанолц. Типичными примерами ката-
литических систем являются WCl6/RxAlCly (х + у = 3), WC16/R4Sп,
WCl6/R2Zn, WCl6/RLi,MoCl5/R3Al, ReCl5/Et3Al [48, 320]. С
использованием этих, систем алкильные производные переходных ме-
таллов могут легко образоваться при переносе алкильной группы, на-
пример, с алюминийалкила к хлориду вольфрама:
WCle + EtAICU —EtWCL + A1CL
о z о о
а-Гидридный перенос от таких алкильных соединений вольфрама мо-
жет приводить к образованию карбеновых частиц 2.260, которые затем
могут участвовать в последовательности реакций метатезиса. Хотя
более известна реакция (3 -элиминирования с образованием гидридных
комплексов алкена (см., например, разд. 2.2) в случае метилвольфра-
мовых комплексов,образующихся при o6pa6oTKe[W(rj5-C5H5)2CH3(C2H4)]PF6
Диметилфенилфосфором [47], происходит реакция а-элиминирования
типа (2.65)О Когда WC16 реагирует с Sn(CH3)4 или при взаимодействии
MoC12(NO)2 (PPh3)2 с (СН3)3 А12С13, в хлорбензоле получаются
222
ГЛ ACS A t.
C1,W— C—CH3
H
1
CLW=CC
CH3
(2.65)
2.26»
метан и этилен. При использовании Sn(CD3)4 как метан, так и эти-
лен получаются полностью дейтерированными [345]. Образование
метана может быть объяснено последовательностью реакций, анало-
гичной тем, которые были впервые предложены Мьютертезом [346]
для объяснения образования метана в системе WCL/Zn(CH )9, напри-
мер,
-ZnCt /СНз ?
WC16 + Zn(CH3) 2 —-—- C14W =± Cl4w = сн2-*
СНз снз
—» C14W=CH2 + сн4
и(или)
н
-ZnCl СН, I
WC1 + Zn(CH3)2 -------- Cl W—СН3 = CLW=CH,
CH3Zn+
-ZnCl+
Cl. W=CH? + снл
4 2 4
В обоих случаях существенно, что интермедиатами являются гидри-
докарбеновые частицы, образующиеся при а -элиминировании гидрида
из метильных производных вольфрама. В системе WCl6/Sn(CH3) 4
предположено, что этилен образуется при реакции димеризации карбена:
2LnW=CH2 — 21^ W + СН2=СН2
Когда каталитическая система, полученная при добавлении Sn(CH3)4
к WC16, используется,для метатезиса декадиена-2,8 (2.261},
2.261
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
223
то в начальных стадиях реакции, кроме обычных продуктов метатези-
са, т.е. бутена-2 и циклогексена, образуется пропилен. При использо-
вании декадиена-2,8, меченного’.дейтерием в двух терминальных ме-
тильных группах, получающийся пропилен дейтерирован исключитель-
но в положении С3; когда же для активации вольфрамовых комплек-
сов используется Sn(CD3)4, то метатезис 1,1,1,10,10,10-2Н6-декадие-
на-2,8 приводит к получению в дополнение к нормальным продуктам
1,1,3,3,3-2Н5-пропилена [345]. Эти очень элегантные эксперименты
с применением меченых соединений согласуются с последовательно-
стью реакций, показанной ниже, и являются существенным подтверж-
дением представлений о промежуточном образовании металлкарбено-
вых частиц через стадию а - элиминирования.
Гсн2=*сн2 + L„M
(СН2)6
**сн3
обычные продукты
метатезиса
♦*сн3
Когда в качестве катализаторов применяют алкильные производные
металлов, то описанные выше процессы алкилирования переходных
металлов с последующим а-элиминированием представляют собой
вероятный путь образования первоначальных карбеновых частиц. Од-
нако существует значительное число активных катализаторов мета-
224
ГЛАВА 2
тезиса, включая почти все гетерогенные системы, которые не содер-
жат в своем составе сокатализаторов, имеющих алкильные группы.
Если имеется источник гидридного водорода, то образование
алкильных комплексов металлов может происходить в результате хо-
рошо известной последовательности реакций:
Н
I
м +
I I
М—С—с—н
I I
2.262
Последующее а -элиминирование приводит к образованию карбеновых
частиц:
Н . Н I
I I I /С—н
м—с—с—н ------► м=с |
Поэтому, видимо, следовые количества протонсодержащих раство-
рителей, например этанола, часто способствуют реакции метатезиса
[320]. В отсутствие источника как гидридных, так и алкильных групп
мы можем представить себе еще два пути образования карбенов из
координированного алкена. Первый путь [уравнение (2.66)] включает
в качестве промежуточных продуктов гидридо-тт-аллильные комплек-
сы типа 2.263.
Первый этап в этой последовательности реакций — образование тг-ал-
лильных комплексов - надежно установлен (см. разд. 2.2.1,Б). Было
найдено [347 ], что комплексы типа 2.264 могут легко превращаться
в металлциклобутановые частицы 2.265 путем нуклеофильной атаки
гидрида на центральный атом углерода тг-аллильной группы.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
225
Такой тип инициирования требует наличия по крайней мере трех ато-
мов углерода в исходном алкене. Отметим, что в случае гетероген-
ной системы Re 2О7/А12О3необходим предварительный контакт либо
с пропиленом, либо с бутеном перед протеканием каталитической реак-
ции метатезиса этилена (т.е. смеси CH2=CH2/CD2==CD2) [348].
Второй путь образования металлкарЬеновых частиц из координи-
рованного алкена (см. разд. 2.2.1, Б) - через переходное состояние
типа 2.266, т.е. через 1,2-надплоскостный сдвиг водорода:
RCH=CH2
М
Для протекания такой реакции нет необходимости наличия трех угле-
родных фрагментов, т.е. радикал R может быть водородом, и таким
образом эта последовательность реакций может составлять универсаль-
ный процесс начальной генерации карбенов в метатезисе алкенов в от-
сутствие источника как гидридных, так и алкильных групп. К сожале-
нию, пока нет прямых экспериментальных доказательств протекания
этого процесса, хотя и была показана [163] возможность обратной ре-
акции - превращения 2.63 в 2.267:
Ph ^сн2и
с
W
(СО)5
2.63
PhCH=CHR
4
W
(CO)S
2.267
За исключением последнего процесса, предложенного для начального
образования карбенов, все другие процессы зарождения карбенов на
226
ГЛАВА 2
одной из стадий включают образование связи металл - гидрид, и к
настоящему времени накапливаются данные [ 320 ], подтверждающие,
что гидриды металлов играют важную роль в реакциях метатезиса ал-
кенов, так же как и в большинстве других гомогенных каталитических
реакций, которые мы рассматривали. !
Участие металлгидридных комплексов в реакциях метатезиса ал-
кенов, катализируемых с помощью гомогенных катализаторов, может
считаться дискуссионным, но почти не приходится сомневаться в том,
что металлкарбеновые комплексы участвуют в таких реакциях. Далее
рассмотрим, какое участие они принимают в каталитическом цикле.
В. Возможный каталитический цикл. После образования началь-
ного металлкарбенового комплекса последующей стадией является
соединение карбеновых частиц с исходным алкеном с образованием
металлциклобутанового интермедиата 2.268.
R’ R2
'с/ R1R2C—CR3R4
Al + R3R4C=CR5R6 —~ M—CR5R6
2.268
С точки зрения электронных факторов протекание этой реакции будет
в значительной мере определяться очевидным разделением заряда в
металл карбеновой связи. Такое разделение может быть представле-
но двумя экстремальными каноническими формулами, а именно 2.269
и 2.270.
LnM— CR’R2 — LnM=CR1R2--------- LnM—CR1R2
2.269 2.270
Таким образом, формально карбеновая частица может выступать ли-
бо как нуклеофил 2.269, либо как электрофил 2.270. Известные реак-
ции характерны для обоих возможных типов поведения, и, как в случае
гидридов металлов (см. разд. 2.1), относительное разделение зарядов,
по-видимому, зависит от природы металла М и связанных с ним лиган-
дов. Так, в карбеновых комплексах тантала типа 2.271, где R = СН2С(СН3)3,
карбеновые частицы выступают в качестве нуклеофила и легко реагируют с
кетонами с образованием алкенов типа 2.272, например для случая,
когда R1 = R2 = CFL [349]:
о
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
227
Н О R1 н
R3Ta=CZ + R1CR2 — ^С=С^ + [R3Ta(0)lx
ХС(СНЧ)Ч R2 С(СН )
О о о о
2.271 2-^2
Аналогично (т]5-С5Н5) ТаС12[=СНС(СН3)3] взаимодействует с про-
пиленом с образованием 2-метил-4,4-диметилпентена-1 с выходом 86%,
при этом протекает показанная ниже последовательность реакций,
опять-таки с участием нуклеофильного карбена [350] (происходит ата-
ка наиболее замещенного углерода в алкене):
Та=СНС(СН3)3 + СН2=СН(СН3)
Та—СНС(СН3)3
снгснсн3
2.273
[Та] + СН2=С(СН3)СН2С(СН3)3
Н—Та СНС(СН3)3
Н2С—ССН3
С другой стороны, в комплексах типа (CO)^M=CR1R2, где М = Сг,
Mo, W; R1 = арил; R2= арил или алкоксил, совокупность эксперимен-
тальных данных [351, 352], по-видимому, свидетельствует о разделе-
нии зарядов по типу 2.270.
Стерический фактор также влияет на направление начального при-
соединения. Если рассматривать только углеродсодержащие лиганды,
то можно считать, что металлциклобутановый продукт 2.273 будет,
по-видимому, более стерически затруднен, чем альтернативный продукт
электрофильного присоединения 2.27к.
Та—СНС(СН3)3
(сн3) nd— сн9
*5 Л.
2.274
228
ГЛАВА 2
Однако в общем случае при наличии других объемистых лигандов, ко-
ординированных с металлом, комплекс 2.273 может оказаться более
предпочтительным продуктом с точки зрения стерических затруднений.
В самом деле, имеются некоторые данные [48], свидетельствующие о
том, что в вольфрамовых системах "неучаствующие" лиганды препят-
ствуют вхождению любых объемистых групп в координационную сфе-
ру металла, что более эффективно благоприятствует образованию ме-
таллбутанового цикла типа 2.273 по сравнению с интермедиатом типа
^74.
Относительная роль электронных и стерических факторов оконча-
тельно будет зависеть от детальных характеристик конкретной систе-
мы метатезиса, и, хотя было рассмотрено значительное число деталь-
ных стереохимических схем [48, 320, 335], до сих пор нет полной тео-
рии для объяснения всей стереохимии реакций метатезиса алкенов.
Другой нерешенный вопрос механизма реакций метатезиса алке-
нов — является ли образование тг-комплекса с алкеном необходимым
условием образования металлциклобутана. Приняв, что разделение за-
рядов соответствует типу 2.270, Катц и Макджиннис [353] предполо -
жили, что образование металлциклобутанового кольца включает атаку
поляризованной металл карбеновой связи на алкен без необходимости
предварительной координации алкенов:
L„M—CR’R2 +
L„M—CR’R2
Напротив, данные, полученные с модельными системами (СО)5W=^CR1R2,
где R1 = R2 = Ph или R1 = Н; R2 = Ph, показывают, что координация
алкена предваряет, образование металлоцикла:
R’CR2
W *
W—С—
В свете данных о других реакциях алкенов, катализируемых комплек-
сами переходных металлов, которые мы рассмотрели в предыдущих
разделах, этот путь представляется наиболее привлекательным. Обра-
зование металлциклобутанового цикла может рассматриваться в основ-
ном как реакция миграции лиганда, происходящая через четырехцент-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
229
ровое промежуточное переходное состояние, аналогичное тому, кото-
рое было описано для гидрирования (разд. 2.1.), изомеризации (разд.
2.2) или полимеризации алкенов (разд. 2.6):
Как в случае миграции гидрида, так и в случае миграции алкила на-
правление и скорость начального присоединения зависят от природы
катализатора. Примером влияния лигандного окружения является из-
менение природы гидридного водорода, связанного с кобальтом:
электрофильная природа гидрида в СоН(С0)4 и нуклеофильцая приро-
да гидрида в СоН(СО) 3(РВи3). Влияние природы реагирующего алкена
проявляется на примере использования асимметрических лигандов при
каталитическом асимметрическом гидрировании или олигомеризации.
Отметим также влияние природы R в случае полимеризации олефинов,
более быструю скорость гидрирования терминального алкена по срав-
нению с алкеном, содержащим внутреннюю двойную связь, а также
влияние природы карбенового лиганда, очевидно тесно связанной со
строением исходного алкена.
Состояние знаний в настоящее время не позволяет сделать окон-
чательных выводов о влиянии каждого из этих факторов в общем слу-
чае метатезиса алкенов. Все же на рис. 2.41 представлен возможный
каталитический цикл, который согласуется со всей совокупностью эк-
спериментальных данных, имеющихся в настоящее время. Рассмотрев
тонкости механизма реакции метатезиса, мы можем теперь обратить
внимание на практическое применение этой замечательной реакции.
230
ГЛАВА 2
r'r2C=CR3R4 R°RbC cr)r2
cr3r4
инициирующий
нарйен
rVc-cr’r2
I I 3 4
M-CR R
RORbC = CR1 R2
Рис, 2.41. Обобщенный каталитический цикл для реак-
ции метатезиса алкенов, катализируемой переходными
металлами 0
2.8.2. Практическое применение метатезиса алкенов. Хотя
большей частью крупномасштабное промышленное применение реакций
метатезиса алкенов основано на использовании гетерогенных каталити-
ческих систем [ 354], гомогенные системы также начинают находить
некоторое применение в производстве продуктов меньшего объема, вы-
сокой стоимости и химических продуктов для фармацевтической, агро-
химической и парфюмерной промышленности. При обсуждении облас-
тей практического использования этой реакции необходимо различать
два типа алкеновых субстратов, а именно ациклические и циклические
алкены.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
231
Л. Ациклические алкены в качестве субстратов. Первый промыш-
ленный процесс с использованием метатезиса алкенов — процесс ’’Три-
олефин” (фирмы Phillips) - был запущен в производство в 1966 г.
[355]. Этот процесс первоначально был разработан, когда был досту-
пен дешевый пропилен, с целью производства этилена и бутилена:
Однако в настоящее время относительная стоимость пропилена возрос-
ла и этот процесс стал неэкономичным. Более выгодным представляет-
ся производство изопентенов путем метатезиса смеси 2-метилпропена
и бутена-2 (т.е. с использованием Сд-фракции крекинга):
Получаемые изопентены находят важное применение как компоненты
высокооктанового топлива; рынок для такого продукта будет, вероят-
но, только расти с учетом тенденции использования бензина без доба-
вок тетраэтилсвинца. Показанное выше равновесие может быть сдвину-
то вправо, т.е. в сторону изопентенов, путем непрерывного удаления
образующегося этилена.
Способ сдвига равновесия реакции метатезиса удалением летуче-
го продукта реакции может быть использован в препаративном масшта-
бе для приготовления длинноцепочечных внутренних алкенов, например
тетрадецена-7 из октена-1 или гексакозена-13 из тетрадецена-1 [354].
Сометатезис этилена и 4-метилпентена-2 приводит к получению
З-метилбутена-1 и пропилена:
232
ГЛАВА 2
С
Как было описано в разд. 2.5.1,Б с использованием никелевых катали-
заторов, модифицированных тр^-фосфиновыми лигандами, пропилен
может быть димеризован в 4-метилпентен-2. Таким образом, приведен-
ная выше последовательность реакций метатезиса в комбинации с ре-
акцией димеризации пропилена представляет собой каталитический путь
получения 3-метилбутена-1 из пропилена и этилена:
кат.
СЩСН-СН, + СН?-СН7--------- CH (CHJCHCH=CHO
О л. О О /
Полимеризация 3-метилбутена-1 с использованием, например, модифи-
цированных циглеровских катализаторов (см. разд. 2.6) дает пластичес-
кий материал специального назначения с высокой температурой плав-
ления.
В последнее время реакции метатезиса используются для синте-
за половых аттрактантов. Проблемой при использовании традиционных
инсектицидов широкого профиля действия является то, что они не толь-
ко убивают вредителей, но их использование приводит также к уничто-
жению полезных насекомых. Кроме того, для их действия необходимо,
чтобы активный инсектицид и насекомое находились в одном и том же
месте в одно и то же время. Так как трудно предсказать место на-
хождения вредного насекомого в определенном месте, например на
хлебном поле, в данное время в период активного существования пести-
цида, то обычно в применении общераспространенных пестицидов ис-
пользуется стратегия "избыточной смертельной дозы". Если бы вред-
ных насекомых можно было собрать на небольшой площади, тогда был
бы возможен намного более эффективный и селективный контроль их
количества. Эта проблема близка к проблеме взаимной реакции двух
субстратов в катализе. Одна из функций катализатора заключается в
обеспечении контакта двух субстратов. Потенциальными катализатора-
ми для контроля за популяцией вредных насекомых являются феромо-
ны, или половые аттрактанты, которые могут быть использованы для
их привлечения в определенное место. Феромон обычной домашней
мухи — ^с-трикосен-9 2.275 - можно синтезировать по реакции сомета-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
233
тезиса децена-1 и тетрадецена-1 [356]:
Ме(СН2)7СН=СН2
Ме(СН2).7СН
Ме(СН2)7СН
Ме(СН2)12СН
Ме(СН2)7СН
Ме(СН2)12СН=СН2
2.275
Ме(СН2)12СН
Ме(СН2)12СН
сн2
сн2
Метатезис дает смесь как цис-, так й ягрлис-изомеров. Фракция цис,
транс-ч\мкосекъг$ выделяется с выходом 29%, а затем необходимый
г^с-изомер выделяется с 95%-ной чистотой. Аналогично 2-метилгеп-
тадекан — половой аттрактант различных видов тигровой моли — мо-
жет быть приготовлен с 30%-ным выходом из 4-метилпентена-1 и тет-
радецена-1 реакцией метатезиса с последующим гидрированием. Как
должно быть ясно из вышеприведенного краткого рассмотрения, суще-
ствует много возможностей для синтеза с помощью реакции каталити-
ческого метатезиса ациклических алкенов. Однако эта реакция не ог-
раничена только ациклическими алкенами, в ней могут участвовать и
циклические алкены.
Б. Циклические алкены как субстраты. За исключением циклогек-
сена (см. ниже), циклические моноалкены С4 — С12 в присутствии ка-
тализаторов метатезиса алкенов образуют полимеры — полиалкеномеры..
Например, с использованием каталитической системы
№СЦ/С2Н5ОН/(С2Н5)2А1С1 циклооктен дает полимерный материал с
молекулярной массой порядка 200 000 — 300 000 [334]. Как и в слу-
чае метатезиса ациклических алкенов, рассмотренного выше, носите-
лями цепи являются металлкарбеновые частицы.
234
ГЛАВА 2
На основе "парного” механизма можно ожидать образования только
небольших количеств макроциклических лигандов:
Можно считать, что и те макроциклы, которые образуются, будут полу-
чаться в результате "перекусывания" растущей полимерной цепи на
активных карбеновых частицах:
Введение циклических алкенов в полимеризационную систему приводит
к обрыву цепи, например в результате реакций (2.67), и таким образом
может быть использовано для регулирования степени полимеризации.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
235
В реакциях метатезиса ациклических алкенов стереоспецифичность,
т.е. селективное получение либо цис-, либо доряяс-алкеновых продуктов,
в общем случае (но всегда) мала. При использовании циклических алке-
нов как субстратов высокая стереоспецифичность, напротив, является
обычной. Так, когда дифенилкарбен(пентакарбонил)вольфрам использу-
ется для инициирования полимеризации циклооктена, то свыше 97%
двойных связей полученных полиоктеномеров имеют ^с-структуру
[ 342]. Аналогично когда в качестве субстрата используется циклопен-
тен, то оказывается возможным при тщательном подборе как катализа-
тора, так и условий полимеризации получить либо в основном цис- [357],
либо в основном дораяс-полипентеномер [358]. г^яс-Полимер имеет хо-
рошие низкотемпературные характеристики, тогда как дораяс-полимер
может быть использован в качестве заменителя природного каучука
в производстве шин. Промышленный процесс для получения такого
дор ляс-полипентеномера был разработан фирмой Bayer [359]. Полиал-
кеномеризация 2-метилоктадиена-1,5 (2.276) дает полимерный матери-
ал, аналогичный тому, который мог быть получен при альтернантной
сополимеризации изопрена и бутадиена-1,3 и как таковой может быть ис-
пользован в качестве модельной системы для промышленной системы
сополимеризации [360]:
СН3
2.276
<?Н3
2-т-СН2—СН=)
! /2(бутадиен)
1 (изопрен)
(= СН — СН2 -7- сн2 — с =сн—СН
г/? (бутадиен) ; 1 (изопрен)
Как было упомянуто в начале этого подразделаv циклогексен не уча-
ствует в реакциях полиалкеномеризации. Однако его производные при-
нимают участие в реакциях метатезиса. Одно из таких производных,
а именно 4-винилциклогексен-1, при метатезисе над гетерогенным мо-
либденовым или вольфрамовым катализатором [361 - 363] при удале-
нии образующегося этилена с высоким выходом дает 1,2-(дициклогек-
сен-3)этилен (2.277).
Так как 4-винилциклогексен-1 может быть легко приготовлен из бута-
диена при димеризации, катализируемой никелем (см. разд. 2.5.2,А),
то эта вышеописанная реакция составляет часть каталитического
236
ГЛАВА 2
2.277
процесса получения 2.277 из бутадиена- Продукт 2.277 находит боль-
шое число специальных прменений, включая получение пламягасите-
лей после его бромирования, получение стабилизаторов для эфиров
целлюлозы против ультрафиолетового облучения после его гидрокси-
лирования, а после превращения в кетон он используется для изготов-
ления парфюмерных продуктов. Рассмотренные выше примеры были
выбраны для иллюстрации существующих и потенциальных синтетичес-
ких применений в реакции метатезиса алкенов. Набор возможных про-
дуктов почти столь же широк, как и набор доступных субстратов. Хо-
тя наше внимание было сконцентрировано на использовании простых
замещенных алкенов, не содержащих интермедиатов, известны так же
каталитические системы (впрочем, малоактивные), которые проводят
метатезис алкенов, содержащих галогены, азот, кислород и даже крем-
нийсодержащие заместители [354, 367, 368]. В настоящее время мета-
тезис — одна из наиболее быстро развивающихся областей катализа.
В заключение обсуждения уместно перейти от рассмотрения применяе-
мых в настоящее время гомогенных каталитических систем к рассмот-
рению систем, которые находятся еще в стадии разработки. Впрочем,
прежде чем сделать это, рассмотрим коротко один из новейших круп-
номасштабных промышленных процессов, в котором используется не
менее трех каталитических реакций, из числа тех, что мы обсуждали
во втором разделе (олигомеризация, изомеризация и метатезис).
2.8.3. Каталитическое получение высших олефинов. Процесс
получения высших олефинов был разработан группой компаний концер-
на Shell. Он заключается в превращении этилена в алкены С11 - С14,
необходимые для производства детергентов [369], и состоит из трех
различных каталитических стадий. На первой стадии этилен олигоме-
ризуется с образованием смеси а-олефинов от С4 — С20+. После выде-
ления алкенов-1 (С,0 - С20) оставшаяся фракция алкенов направляет-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В ДЕЙСТВИИ
237
ся на гетерогенный катализатор изомеризации. Глубокий процесс изо-
меризации приводит к образованию почти статистического (термодина-
мического) распределения изомеров w-алкенов (см. разд. 2.2.1). Эта
смесь, обогащенная низшими алкенами, затем направляется в реактор,
содержащий гетерогенный катализатор метатезиса. Благодаря преоб-
ладающему содержанию низших алкенов продукт, получающийся при
метатезисе, содержит избыток а-олефинов, которые образуются при
протекании реакций типа
Целевые продукты - С14, пригодные для получения детергентов,
разделяются обычной дистилляцией. Другие алкены вновь направляют-
ся в реактор метатезиса, причем продукты С10направляются непосред-
ственно, а продукты С1б+ - через стадию изомеризации. Благодаря
тщательному контролю условий проведения процесса достигаются высо-
кие выходы высокочистых (обычно 99%) моноалкенов, содержащих
94-97% а-олефинов нормального строения. Благодаря возможности
регулировать большое число параметров, можно обеспечить селектив-
ное производство широкого ряда продуктов. Одной из важнейших облас-
тей использования алкенов С - С14 является производство поверх-
ностно-активных спиртов. Эта стадия осуществляется с использовани-
ем гомогенных кобальтовых катализаторов гидроформилирования — гид-
рирования, модифицированных лигандами (см. разд. 2.4.1). Таким обра-
зом, при получении из этилена поверхностно-активных спиртов исполь-
зуется половина каталитических реакций, которые мы описали до сих
пор. Кроме того, при проведении различных стадий этого процесса до-
ля в использовании гомогенных и гетерогенных каталитических систем
составляет 50/50.
238
ГЛАВА 2
Дополнительная литература по метатезису алкенов* **
Bailey G.G. Olefin disproportionation- — CataL Rev-, 1969, v. 3, p- 37.
Khidekel M.L., Shebalova A.D.> Kalechits l.V. Catalytic disportionation
of unsaturated hydrocarbons- — Russ- Chem- Rev-, 1971, v. 40, p-
669.
Banks R.L. Catalytic olefin disproportionation— Topics in Current
Chem., 1972, v. 25, p. 39.
Calderon N. The olefin metathesis reaction. — Acc. Chem. Res-, 1972,
v. 5- p. 127.
Cardin D.J., Cetinkaya B., Doyle M.J., Lappert M.F. The chemistry of
transition-metal carbene complexes and their role as reaction inter-
mediates. — Chem. Soc, Rev., 1973, v. 2. p- 99-
Evans D.A., Green C.L. Insect attractants of natural origin. — Chem.
Soc. Rev., 1973, v. 2, p. 75.
Haines R.J., Leigh G.J> Olefin metathesis and its catalysis. — Chem.
Soo- Rev-, 1975, v- 4, p- 155-
Mol J.C., Moulijn J.A., The metathesis of unsaturated hydrocarbons cata-
lysed by transition metal complexes. — Adv. CataL, 1975, v. 24 p.
131-
Streck R. Olefin metathesis, a versatile tool petro- and polymerchemist-
ry. — Chem. Ztg-, 1975, v. 99, 397 (German text).
Calderon N., Ofstead E.A., Judy W.A. Mechanistic aspects of olefin
metathesis. — Angew. Chem. Int. Ed. EngL, 1976, v- 15, p. 401»
Katz T.J. The olefin metathesis reaction. — Adv. Organometal. Chem.*
1977, v. 16, p- 283-
Rooney J.J., Steward A. Olefin Metathesis.—In: Catalysis, vol- 1,
Specialist Periodical Report, Chem. Soc», London, 1977, p. 277.
Calderon N., Lawrence J.P., Ofstead E.A. Olefin metathesis. — Adv.
Organometal. Chem., 1979, 17, p- 449.
Jr
Как следует из названий некоторых ранних обзорных статей, для ме-
татезиса алкенов первоначально широко использовался термин диспропорци-
онирование алкенов.
*★
Фельдблюм В.Ш. Димеризация и диспропорционирование олефинов. —
М.: Химия, 1978. — Прим, перев.
3. Гомогенные каталитические системы
в стадии разработки
Различие между гомогенными каталитическими системами, кото-
рые находятся в стадии использования и в стадии разработки, в боль-
шой мере условно, так как большинство каталитических процессов,
которые мы обсуждали в гл. 2, непрерывно совершенствуются. Одна-
ко в настоящее время все рассмотренные нами гомогенные катали-
заторы могут найти практическое применение. В этой главе мы скон-
центрируем внимание на трех областях катализа, в которых, несмот-
ря на четкое понимание проблем, не найдено пока ее решения в виде
практически пригодных гомогенных катализаторов. Все три проблемы
могут быть объединены под общим названием " Активация малых
молекул", где малые молекулы — это азот, СО и метан, а сами проб-
лемы — это фиксация азота, восстановительная олигомеризация (по-
лимеризация) СО и активация алканов.
3JO Фиксация азота
Азот является существенным элементом для жизни всех расте-
ний и животных. Он составляет около 80 % воздуха, которым мы ды-
шим. Однако эффективно использоваться он может, только если
" фиксирован", т.е. превращен в форму, которую могут лекго усваи-
вать растения и животные. В биологических системах процесс такой
фиксации проводится некоторыми бактериями при помощи фермента
нитрогеназы. В природном азотном цикле полученные соединения
сохраняются в растениях, которые в свою очередь используются жи-
вотными в качестве пищи. Животные разлагают эти соединения, неко-
торая часть накопленного азота превращается в продукты, необхо-
димые для питания, тогда как часть азота возвращается в почву в
виде отходов жизнедеятельности животных и после превращения
под действием бактерий используется затем растениями. В начале
этого века с развитием техники интенсивного земледелия, необхо-
димого для обеспечения .пищей все возрастающего населения, стало
чрезвычайно актуальным повышение производства азотсодержащих
соединений. Это привело к развитию процесса Габера — Боша, в кото-
ром аммиак получается из азота и водорода на гетерогенном желез-
240
ГЛАВА 3
ном катализаторе при температурах 400 — 540°С и давлениях в интер-
вале 80 — 350 атм [ 370]. После начала использования этого процесса
в 1913 г. мировое производство аммиака этим методом достигло бо-
лее 70 млн. т в год. Во всем мире имеются сотни заводов, работаю-
щих по методу Габера - Боша, и в 1979 г. дополнительно вступили в
действие более 30 заводов общей производительностью около 10 млн.
т., строительство которых потребовало капитальных вложений более
2500 млн. долл. [371]. Около 80% аммиака, полученного таким обра-
зом, используются непосредственно или косвенно в качестве удобре-
ний, а остальные 20 % используются для производства синтетических
волокон, термореактивных смол, взрывчатых веществ.
Хотя за многие годы эффективность процесса Габера — Боша
возросла в результате применения более совершенных катализаторов
и улучшения технологии, все же этот процесс остается высокотем-
пературным, проводится при высоком давлении и требует как высо-
ких капитальных затрат, так и больших затрат энергии. При постоян-
но возрастающем дефиците каждого из этих ресурсов поиски процес-
са фиксации азота, имеющего пониженную стоимость, который по
возможности осуществлялся бы при атмосферном давлении и низкой
температуре, в последние годы стали особенно интенсивными. При-
родная система, основанная на нитрогеназе, — один из таких процес-
сов, и он до сих пор являлся главнейшим источником азота для сель-
ского хозяйства, с помощью которого "фиксируется" более 90 млн. т
азота в год [372]. В основном два подхода были использованы в изу-
чении фиксации азота, а именно биологический и химический. В пер-
вом подходе исследовались природные азотфиксирующие системы,
найденные в бактериях, с целью выяснения природы и способа действия
нитрогеназы. В другом подходе делались попытки смоделировать
природные системы и таким образом выяснить, как связывается
азот и каким образом реагирует металлический центр. Несомненно,
оба подхода тесно связаны между собой, и хотя в этом разделе мы
ограничимся обсуждением только химического подхода, значитель-
ное число ссылок на работы, посвященные биологическому подходу,
включено в список дополнительной литературы.
3.1.1. Азот, связанный с металлическим центром. Истори-
чески молекулярный азот (или диазот) всегда рассматривался как
инертный газ. Действительно, он и сейчас широко используется в
препаративной химии для обеспечения "инертной" атмосферы. Таким
образом, одной из первых проблем в активации диазота переходными
металлами было "заставить" его взаимодействовать с металличес-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
241
ким центром. Впервые это было сознательно осуществлено в 1965 г.,
когда Аллен и Сеноф выделили [Ru(NH3)5 (N2)] 2+ из продуктов реак-
ции между RuCl3 . ЗН2О и гидразином в воде [373]. С тех пор ком-
плексы диазота были получены с большинством переходных металлов
[ 374].
Беглое рассмотрение электронной молекулы диазота показыва-
ет, что возможны два основных пути, с помощью которых эта моле-
кула может связаться с металлическим центром, — путем использова-
ния: 1) свободных электронных пар, локализованных на атомах азо-
та, с образованием концевых комплексов типа 3.1 либо 3.2 или же
2) тройной связи азот — азот с образованием симметричных комп-
лексов типа 3.3 и 3.4.
М—- N=N-*M
концевое мостиковое”
” связывание
M*-N=N
„ концевое ”
связывание
„ боковое”
связывание
3.3
N
М*- ||—* М
N
боковое мастиковое °
связывание
3.4
Известны все четыре типа комплексов [374], хотя комплексы,
содержащие концевой связанный азот, более распространены. Элект-
ронная структура диазота аналогична структуре СО, и, как и в слу-
чае СО, связывание с металлическим центром может рассматривать-
ся как состоящее из двух компонент: ст-компоненты, образованной
переносом электронной плотности от свободной электронной пары
ст-орбитали диазота на свободную ст-орбиталь металла, и тг-компонен-
ты, образованной переносом электронной плотности от заполненных
dir- или гибридных б/рп-орбиталей металла на свободную ртг-анти-
связывающую орбиталь диазота. Обе компоненты связи, схематичес-
ки представленные на рис. 3.1, приводят к ослаблению связи азот —
азот в результате уменьшения электронной плотности на связывающих
орбиталях молекулы N—N и увеличению электронной плотности на
антисвязывающих орбиталях этой молекулы. Однако по сравнению
с СО, а также с другими аналогичными ст-донорными/ тг-акцепторными
лигандами азот является чрезвычайно слабым ст-донором. Его иони-
зационный потенциал выше примерно на 1,6 эВ, чем у СО, и состав-
ляет 15,6 эВ, что почти равно ионизационному потенциалу аргона
242
ГЛАВА 3
Рис. 3.1. а- и тг-«компоненты связи между диазотом и переходным металлом0
(15,7 эВ). Что же касается тг-акцептороной способности, то спектрос-
копические данные показывают, что молекула диазота является уме-
ренным тг- акцептором, располагающимся в ряду акцепторов примерно
между СО и RCN [374]. Теоретические расчеты с использованием
расширенного метода Хюккеля [375] согласуются со спектроскопи-
ческими данными и позволяют считать, что по крайней мере для ком-
плексов, содержащих одну молекулу диазота, его связывание имеет
преимущественно ст-акцепторный характер. Таким образом, коорди-
нация диазота с металлическим центром приводит только к переносу
электронной плотности от металла к диазотному лиганду и,, следо-
вательно, к активации диазота по отношению к электрофильной атаке.
3.1.2. Гидрирование связанного азота. Если координирован-
ный азот активирован по отношению к электрофильной атаке, выбор
протона в качестве электрофила является вполне очевидным, когда
ставится цель гидрирования N2. Известно несколько систем, в ко-
торых протонирование диазотных комплексов приводит к образова-
нию восстановленной молекулы азота, т.е. N2H4 или NH3. Так, об-
работка w^-W(N2)2(PMe2Ph)4 15-кратным избытком серной кислоты
в метаноле при 20° С приводит к почти количественному (98 %) обра-
зованию аммиака в соответствии с уравнением (3.1) [376].
H2SO4/MeOH
Цис-W(N2)2(PMe2Ph)4-------------
3.5
2NH3 + N2+ 4[PMe2PhH]HSO4 +
+ продукты окисления металла
(3.1)
Примерно то же наблюдается в случае молибденового комплекса,
хотя выход аммиака вдвое меньше того, который получается при
использовании комплексов вольфрама. В обоих случаях образуются
небольшие количества гидразина.
Как уже упоминалось, координация азота с металлическим центром повы-
шает электронную плотность диазота. Однако в случае моногаптокомплексов
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
243
типа 3.5, в которых только один конец азотного лиганда связан с метал-
лом, повышенная электронная плотность, как и можно ожидать, не-
одинаково распределена в молекуле азота и преимущественно лока-
лизована на одном из атомов азота. Четкое разделение двух атомов
азота в рентгеновских спектрах [374], а также высокая интенсив-
ность колебаний v(N2) в инфракрасном спектре подтверждают нали-
чие зарядовой асимметрии. Экспериментальные данные указывают,
что тот атом азота, который не координирован с металлическим цент-
ром, несет больший отрицательный заряд [377] . Таким образом, в
реакциях, протекающих по уравнению (3.1), первая стадия включает про-
тонирование концевого атома азота с образованием частиц N2H
(3.6), которые при дальнейшем протонировании концевого азота дают
гидразидные частицы (= N—NH2) (3.7). По-видимому, на этой ста-
дии реакция может идти по одному из двух путей в зависимости от
того, является ли металл молибденом или вольфрамом. В случае мо-
либдена может образовываться аммиак благодаря реакции диспропор-
ционирования (3.2), тогда как в случае вольфрама, для того чтобы
объяснить образование почти двух молей аммиака на один моль ком-
плекса, нужно допустить, что по крайней мере часть аммиака должна
образовываться путем дальнейшего протонирования комплекса 3.7
через образование продукта 3.3 [376; 378].
+НХ
VI(N2)2L4 —
+2НХ
MX(N2H)L4 ——- MX2(NNH2)L3
3.6
нх
+2НХ
-(LH)X
(М = W)
(М = WuwMo)
нх
2NH3 + —---- MX3(NHNH2)L2
окисления
металла
3.8
MoEEEN—NH2—*‘Mo’ + jN2 + jNH3
(3.2)
Комплексы, содержащие группы N2H hNNH2 , были выделены из
этой системы. Кристаллографическим методом с использованием
монокристаллов был охарактеризован целый ряд соединений, содержа-
щих фрагменты NHH2 и близкие к ним фрагменты NNH(Me).
Когда диазотный лиганд находится в симметричном мостике меж-
ду двумя атомами металла в комплексе, в таком, как 3.9 [379, 380],
то для атаки протоном нет выбора между двумя концами мостикового
244
ГЛАВА 3
диазота. Однако восстановление все же протекает стадийным путем
с образованием мономерных частиц ЗЛО на первой стадии реакции
[3811. N
II!
N
I;
(»j5-C5Me5)2Zr—N=N—Zr(45-C5Me5)2— Zr(«j5-C5Me5)2Cl2 + N2
11
I
11
N
II 5 /N2H
I + (^-C5Me5)2Zr^
jo 3 10 N*H
u.J U. /и
Дальнейшее протонирование происходит аналогично тому, как это
описано для ЗД WX(N2H)(PMe2Ph)4 (т.е. М= W, L= Р(СН3)2РЬ),
хотя основным продуктом гидрирования азота является в этом слу-
чае гидразин, а не аммиак.
ЗЛО —- Zr(r]5-C5Me5)2Cl2 + N2H4 + N2
Эксперименты, в которых в качестве метки использовался 15 N [381],
показали, что примерно в одинаковом количестве гидразин образует-
ся как из мостиковых, так и из немостиковых азотных лигандов.
Таким образом, по крайней мере в этой системе, дикоординирован-
ный азот, по-видимому, не обладает повышенной реакционной способ-
ностью к восстановлению по сравнению с монокоординированными ато-
мами. Протонирование (Ph3P)2HU5o-Pr)Fe(N2)Fe(«/so-Pr)(PPh3)2
(с мостиковым N2) при обработке НС1 при — 50°С дает небольшие
количества гидразина (0,1 моля на моль N2). Было сделано предпо-
ложение [382], что и в этом случае протекает симметричное про-
тонирование с образованием диазенового интермедиата,
L„Fe—NH— NH—FeL„ ,хотя эта система еще не охарактеризована
полностью.
Идея о двойной координации как необходимом условии активации
диазота, основанная на представлении о разрыве молекулы азота на
две половинки, подтверждается лишь данными, полученными при изу-
чении небольшого числа модельных систем. Напротив, Чатт и др. [374]
предложили каталитический цикл с участием монометаллических комп-
лексов, показанный на рис. 3.2, который может объяснить каталитичес-
кое восстановление диазота. По-видимому, нет оснований сомневать-
ся в том, что в биологической фиксации азота участвует более одного
атома металла [372]: нитрогеназа содержит 6 атомов металла, два
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
245
Рис. 3.2. Гипотетический каталитический цикл для восстановления молеку-
лярного диазота.
из них — атомы молибдена, а четыре — атомы железа. Такая же ситу-
ация характерна для большинства известных в настоящее время гомо-
генных катализаторов (см. ниже). Функция такого множества метал-
лических центров может, впрочем, заключаться в обеспечении элект-
ронами, необходимыми для процесса восстановления, и не иметь от-
ношения, к способу связывания азота. Как показано на рис. 3.2, для
осуществления превращения N2 в NH3 необходимо 6 электронов.
3.1.3. Гидрирование атмосферного азота. Во всех вышеопи-
санных системах, за исключением биологической системы, о которой
мы упоминали, перед гидрированием ди азот находится в виде вполне
определенного комплекса. Ни одна из этих реакций не является ката-
литической, таким образом, восстановление прекращается сразу же,
как только металл достигает своего высшего валентного состояния,
т.е. M(VI) для М - Мо и W, Zr (IV) в случае циркониевых систем. Для
того чтобы получить каталитическую систему, очевидно, необходимо
использовать восстанавливающий агент (проблема здесь обратна той,
которую надо решить для осуществления вакер-процесса, описанного
в разд. 2.7.2, Б, где необходимо было использовать окисляющий агент
для превращения неактивного палладия(О) в каталитически активный
палладий(П). В случае молибденовых комплексов были использованы
химические восстанавливающие агенты с целью моделирования био-
логической системы. Так, в водно-метанольном растворе моли-
бденовый комплекс, полученный либо из Na2 [Мо2 О4 (cys)2 ] (где
246
ГЛАВА 3
cys - это [S CH2CH(NH2) CO2 ] 2“), либо из K2[MoO(CN)4(H2 О) ], в
присутствии избытка боргидрида натрия осуществляет восстановле-
ние диазота в аммиак [383, 384]. Хотя выходы чрезвычайно низки и
редко превышают 0,3 моля азотсодержащего продукта гидрирования
на атом Мо, эти системы обладают рядом свойств, характерных для
нитрогеназы. Например, они восстанавливают ацетилен, этилен и нит-
рилы в углеводороды и аммиак [ 383]. Были найдены также молибде-
новые системы, в которых в качестве восстанавливающего атома ис-
пользуется либо титан(Ш ) [ 385, 386], который легко оксиляется в
титан(1У), либо амальгама натрия. В мягких условиях по-прежнему
наблюдались только низкие степени коверсии. Однако сообщалось
[386], что в более жестких условиях (при 90 — 110°С и 100 - 150 атм
N2 ) достигаются выходы около 100 молей NH3 или N2H4 на моль Мо.
Эти системы часто называют "супом", так как они обычно вклю-
чают несколько компонентов в дополнение к молибденовому комплек-
су и восстанавливающему агенту. Во избежание нежелательных по-
бочных реакций часто необходимо работать при очень низких концент-
рациях комплекса, обычно 10 ~5 Мо,и вследствие этого системы очень
чувствительны к следам загрязнений, в результате чего возникает
очень, серьезная проблема воспроизводимости экспериментов. Заклю-
чения о механизме действия таких систем являются малообоснованными.
Однако результаты, полученные с использованием такого "супа0, в
сочетании с данными, полученными при использовании определенных
комплексов переходных металлов, описанных выше, однозначно и не-
сомненно продемонстрировали, что' восстановление азота в мягких
условиях является химически возможным процессом, который при ис-
пользовании подходящего активного центра, содержащего переходный
металл, может быть проведен достаточно эффективно. Разработка
системы "фиксации" азота, пригодной для практического использова-
ния, пока еще остается проблемой. Пока еще трудно решить, сможет
ли такая система (по крайней мере в течение ближайших 20 лет)
успешно конкурировать с процессом Габера — Боша в крупномасштаб-
ном производстве аммиака. Несомненно, что такой процесс мог бы
иметь значительное применение при маломасштабном производстве
связанного азота в районах, где потенциальный рынок не оправдыва-
ет строительство заводов производительностью 1000 т/ сутки (обыч-
но приемлемый минимальный размер заводов [ 371], использующих
процесс Габера — Боша), или же в таких местах, где транспортные рас-
ходы очень высоки.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
247
Дополнительная литература по фиксации азота*,
Chart ]., Leigh G.H. Nitrogen fixation. — Chem. Soc. Rev., 1972, v. 1, p. 121
Allen AoD9, Harris Ro0o, Loescher B.R., Stevens ].RO, Whiteley R.N.
Dinitrogen complexes of the transition metals. — Chem* Rev., 1973,
v. 73, p. 11.
Шилов А.Е. Фиксация азота в растворе в присутствии комплексов пе-
реходных металлов.— Усп. химии, 1974, т. 43, с. 378.
Sellmann De Dinitrogen-transition metal complexes: synthesis properties
and significance. — Angew. Chem. Intemat. Ed. Engl., 1974^ v. 13, p.639.
Schrauzer C.N. Nonenzymatic simulation of nitrogenase reactions and
the mechanism of biological nitrogen fixation. — Angew. Chem.
Internet. Ed. Engl., 1975, v. 14, p. 514.
Wentworth R.AaDo Mechanisms for the reactions of molybdenum in
enzymes. — Co-ord. Chem. Rev., 1976, v. 18, p. 1.
Stiefel E.L The co-ordination and bioinorganic chemistry of molybdenum.
— Prog- Inorg. Chem., 1977, v. 22, p. 1.
Chart J., Dilworth J.R., Richards R,Le Recent advances in the chemistry
of nitrogen fixation. — Chem. Rev., 1978, v. 78, p. 589.
Richards R.L. Nitrogen fixation. — Educ. in Chem., 1979, v. 16, p. 66.
3o2o Восстановительная олигомеризация (полимеризация) CO
За последние годы произошло быстрое возрастание цены на
сырую нефть, кроме того, источники нефти по техническим или поли-
тическим причинам могут стать ограниченными в сравнении с потенци-
альными потребностями на 1985 — 1995 гг. [387]. Эти факторы возро-
дили интерес к углю как к источнику энергии и сырья для нефтехими-
ческой промышленности. При современном уровне потребления энергии
известных запасов угля хватит более чем на 250 лет [ 388].
Наиболее прямой метод использования угля в качестве первичного
источника энергии - его сжигание. Однако, если необходимо повысить
роль угля или полностью заменить им нефть, например, в производстве
моторных топлив и органических химических продуктов, необходимо
*См. также: Бородько Ю.Г., Шилов А.Е. Комплексы молекулярного азота. —
Усп. химии, 1969, т.38, и0 761; Шипов А0Е0 Фиксация азота в растворах в присутст-
вии комплексов переходных металлов. — Усп. химии, 1974, т. 43, с. 863 — 902;
Шипов А.Е. Координационный катализ. Активация кинетически и термодина-
мически стабильных молекул комплексами переходных металлов. — ЖВХО
им. Д.И. Менделеева, 1977, т. 22, с. 521 — 531; Проблемы фиксации азота:
Перо с англ0/Под редо Харди Ро и др, — Мо: Мир0 1982; Новое в химичес-
кой фиксации азота: Перо с англс/Под редо Чатта Джо и дро — Мо: Мир, 1983.
—»Примо перево
248
ГЛАВА 3
разработать эффективные пути превращения угля в газ, жидкие угле-
водородные продукты и предпочтительно в индивидуальные органичес-
кие соединения. Одним из методов, с помощью которого, по крайней
мере частично можно достичь этих целей, является синтез Фишера —
Тропша, при котором синтез-газ (смесь СО и водорода, полученная
при сгорании угля в присутствии кислорода и водяного пара) превра-
щается в широкий набор углеводородных продуктов.
Синтез Фишера — Тропша, который в общем случае представляет
собой восстановительную полимеризацию СО, может быть схематичес-
ки представлен реакцией
и катализатор
со + н2----------продукты "СНО" . (3.3)
где продукты "СНО” - это органические молекулы, содержащие угле-
род, водород и кислород, образование и существование которых термо-
динамически возможно в условиях синтеза. При использовании боль-
шинства гетерогенных катализаторов первичными продуктами являют-
ся неразветвленные алканы, тогда как вторичные продукты включают
разветвленные углеводороды, алкены, спирты, альдегиды и карбоновые
кислоты [389]. Со времени открытия этой реакции в 1920 г. был
достигнут значительный прогресс в разработке гетерогенных ката-
литических систем для синтеза Фишера - Тропша (в 1974 г. в Южной
Африке, например, более четверти миллиона тонн первичных продук-
тов — от метана до высокомолекулярного парафина — было произве-
дено с использованием этого процесса [ 390]).
Что касается традиционного процесса Фишера - Тропша, то раз-
работка гомогенных каталитических систем для этого процесса не
имеет смысла. Сложная смесь продуктов, получаемая на традицион-
ных катализаторах, требует проведения дорогостоящих процессов раз-
деления и очистки. Однако представляет значительный интерес раз-
работка катализаторов для селективной восстановительной полимериза-
ции и, вероятно, если говорить более реалистически, для олигомери-
зации СО в присутствии водорода. Можно ожидать, что именно в этой
области гомогенные системы могут внести значительный вклад в
решение проблемы использования СО.
3.2.1. Модельные системы и возможный механизм реакции.
С учетом двух основных процессов активации молекул, которые об-
суждались в разд. 1.4, каталитическая реакция с участием СО и во-
дорода может рассматриваться как процесс, в котором происходит ак-
тивация и путем координации, и путем присоединения, при этом СО
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
249
участвует в процессе первого типа, а водород - в процессе второго
типа. Гидридный фрагмент в гидридных комплексах переходных ме-
таллов, которые получаются при активации водорода путем его присо-
единения, обычно реагирует так, как если бы он находился в форме
анионного гидридного лиганда(:Н*“).Если при координации СО проис-
ходит разделение зярядов по типу $+ $-
:С=О
то начальная стадия реакции может быть представлена как атака гид-
рида на атом углерода карбонильной группы. Таким образом, в гид-
ридокарбонильном комплексе переходного металла, в котором СО и
Н находятся в ^z/c-положении по отношению друг к другу, мы можем
ожидать взаимодействия двух лигандов, например, может происходить
миграция гидрида в соответствии с уравнением
Н Н
М -.СО------ М -С — О (3.4)
3.11.
Возможность гидридной атаки на координированный карбонил в настоя-
щее время продемонстрирована в случае многих модельных систем.
Получено большое число анионных металлформильных комплексов
при обработке карбонильных соединений металлов триалкоксиборгид-
ридами [см. уравнение (3.5)] [391]. Аналогичным образом могут быть
получены нейтральные соединения, исходя из катионных карбонилов
металлов, например, по уравнению (3.6) [392, 393]:
[HB(OR) ]“ LM(CO)—-B(OR)3 + [L,M(CHO)] ~ (3.5)
О Аг О Аг
L =PPh3, P(OPh)3, СО; R. СН3, СН(СН3)2 ; М =Fe, Cr, W
[(n5-C2H5)Re(CO)(PPh3)(NO)]++ [HB(C2HS)3 ] ~----*
— (г|5-С2Н5) Re(CHO)(PPh3)(NO) + В(С2Н5)3 (3.6)
Хотя протекание внутримолекулярного переноса гидрида, приводя-
щего к образованию металлформильного комплекса, хорошо уста-
новлено, до сих пор нет сообщений о "простой” межмолекулярной миг-
рации гидрида к карбонильной группе в соответствии с уравнением
(3.4). Вероятно, есть некоторые основания предполагать, что такой
процесс будет, по крайней мере для большинства металлов, сильно
эндотермичным (около 125 кДж/моль). Это основано на наблюдении,
что в первом приближении аналогичный процесс
250
ГЛАВА 3
СНз СН3
м -СО----*М -С = О
который мы обсуждали в разд. 1.5, во многих случаях приблизитель-
но термонейтрален (+ 20 кДж/ моль) и что для заданного металла
связь металл - гидрид примерно на 125 кДж/ моль прочнее, чем ана-
логичная связь металл - метил [394]. Учитывая, что связи металл -
формил и металл - ацил сравнимы по энергиям, мы придем к заклю-
чению, что эндотермичность межмолекулярной миграции гидрида в
соответствии с уравнением (3.4) составит около 125 ± 20 кДж/моль.
Хотя и преждевременно рассматривать эту реакцию как эндотерми-
ческую во всех случаях, ни для одного из множества известных мо-
ноядерных гидридокарбонильных комплексов не было обнаружено
склонности подвергаться этому процессу без посторонней помощи.
Одним из путей облегчения миграции могло бы быть введение акцеп-
тора электронов, который способен координировать карбонильный кис-
лород. Это может а) понизить общую плотность электронов в карбо-
нильном лиганде и таким образом сделать карбонильный углерод
более подверженным гидридной атаке и б) снизить эндотермичность
процесса благодаря энергетическому вкладу связи между карбонильным
кислородом и акцептором электронов. Общая схема процесса может
быть выражена следующим образом:
М :С=О 4- А
Пока не ясно, осуществляется ли такая схема действительно в реак-
циях (3.5) и (3.6) (в обоих случаях побочным продуктом реакции явля-
ется акцептор электронов, или льюисовская кислота, т.е. B(OR)3 или
В(С2Н5)3). Однако очевидно, что введение акцептора электронов в
систему, содержащую гидридный водород, приводит к быстрому восстановле-
нию координированной формильной группы до координированной метильной
группы. Так, добавление ВН3 в тетрагидрофуране к (rj5-C5H5)Re(CHO)(CO)(NO)
(3.12) приводит к быстрому (за несколькЛлину'Гпри комнатной тем^'
пературе) образованию (ц{-С5Н5) ReCH3(CO)(NO) (3.13) [392, 395].
Суммарная реакция - восстановление координированной карбониль-
ной группы в координированную метильную — осуществлялась с ис-
пользованием NaBH4 . Так, обработка [(ry5-C5H5)Re(CO)2(NO)] +
раствором NaBH4 в тетрагидрофуране (ТГФ) при 25° С приводит к.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
251
почти количественному (95%) образованию 3.13 [393]. Предполагается,
что реакция проходит через образование аддукта с ВН3 типа 3.14 „
так как в присутствии воды образуется формильный (а не метильный)
комплекс 3.12 с выходом 93% и выделяется газообразный водород.
Сделано предположение [393], что промежуточный аддукт с ВН3
(3.14) гидролизуется раньше, чем произойдет,последующий гидридный
перенос с образованием метильного комплекса.
вн3
[(п5-Сдае(СО)^О]+ £
+NaBH4 ||
——^*[(T]5-C5H5)Re(CH)(CO)NOb(n5-C5H5)ReCHs(CO)NO
11 Ф, ZD Vz,
60 мин 3.14 3.13
н20
ТГФ/Н-О, V
——-------* (П 5-С Н 5) Re(CHO) (СО) NO
•1“С, 15 мин
3.12
Дальнейшая обработка 3.12 эквивалентным количеством NaBH4 или
аналогичная обработка [ (rj5- С2 Н5) Re (СО) 2 (NO) ]+ двумя молями
Na ВН в водном растворе ТГФ дает гидроксиметильные комплексы
(n5-C5H5)Re(CH2OH)(CO)NO (3.15)[ 393]. Схематически эта последо-
вательность реакций может быть представлена следующим образом:
Н
ВН4~ I
[М —СОГ -----* м—С=О:—*ВН3 -------> м— СН3
н2о
Н Н
I вн4“ I
м—с=о ------* м—с—о- —вн3
н
н2о
м—СН2ОН
м—сн3
252
ГЛАВА 3
MnCH,CK +OMhH 7
Ci £ J L/ Д с.
или
CO
H20 + MbHx_4
я 12H2
и т.п. 1
и т.д.
Рис. ЗоЗ. Возможный механизм восстановительной олигомериза-
ции (полимеризации) СОо
Эта и аналогичные модельные реакции [16, 396] доказывают, что в
суммарном процессе восстановления фрагмента М-СО в М-СН3 ком-
плексы, содержащие некоординированный кислород, т.е. "свободные”
формильные или гидроксиметильные производные, не являются актив-
ными интермедиатами. В результате можно предложить последователь
ность реакций, показанную на рис. 3.3, как один из возможных
механизмов восстановительной олигомеризации (полимеризации)СО.
Важно подчеркнуть, что это только одна из нескольких возмож-
ностей. Единственный существенный признак этой схемы — наличие
связывания как карбонильного углерода, так и карбонильного кисло-
рода. Это условие может соблюдаться в присутствии двух металличес-
ких центров, один из которых, Нх , является электронодефицитным.
Аналогично и центр, содержащий один металл, может осуществлять
обе функции, как, например, в комплексе 3.16,
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
253
Н
(ij5-C5Me5)2Zr ||
I
Н
3.16
который рассматривался как промежуточное соединение при
превращении (ц5- С5Ме5)2 Zr(CO)H2 в (ц5~ c5Me5)2Zr(OCH3)НГ397].
Нет необходимости, чтобы водород присутствовал на lVlfe, Ма также
может выступать в качестве центра, активирующего водород t или же
это действие могут осуществлять и Mfll и Mfc. Нет априорных причин,
по которым центр должен быть обязательно металлом; весьма
привлекательной возможностью с точки зрения конструирования ката-
литической системы является случай, когда = Н+ • В этом случае
начальный восстановительный этап может быть представлен как ге-
теролитическое присоединение водорода к металлическому центру с
одновременным протонированйем кислорода карбонила:
М"—со + н2 -------> мя+1—*сон+ —* Мя+1С=ОН+
н Н (3.7)
За исключением случая (rj5-C5H5)2NbH3 , все модельные системы, о
которых сообщалось до настоящего времени, включают восстанавли-
вающие агенты гидридного типа с участием элементов, которые могут
образовывать очень прочные связи с кислородом, т.е. В, Z? и А1
(мы кратко рассмотрим систему, в которой используются соединения
алюминия). Так, было установлено образование окисленного побочно-
го продукта типаОхМ>,Нг , который не может быть легко восстанов-
лен молекулярным водородом, и восстановление СО является в луч-
шем случае стехиометрическим при использовании М& Нх в качестве
восстановителя. Очевидно, что при подборе катализатора с двойной
координацией необходимо выбрать М& среди металлов, не подвержен-
ных легкому окислению при координации кислорода, т.е. М6 должен
быть благородным металлом, возможно в высшем состоянии окисле-
ния, или же протоном; в последнем случае не возникает проблемы сте-
хиометричности по отношению к восстановителю. Механизм реакции
(3.1) может быть описан как стадийная реакция роста, протекающая
через стадию восстановления — внедрения, при этом прекращение рос-
та происходит при гидрировании мет ал л алкильных частиц, а в случае
254
ГЛАВА 3
частиц MCH^CHj и частиц, содержащих высшие алкильные группы, —
путем р- элиминирования. При конструировании селективной катали-
тической системы для реакции СО с Н2 важно иметь в виду возмож-
ный путь протекания реакции роста, который следует из рассмотре-
ния продуктов, полученных при реакциях между некоторыми карбонила-
ми переходных металлов и аланом [ 398].
Обработка Ru3 (СО)12 15-кратным избытком А1Н3 в тетрагидро-
фуране при 25° С приводит к быстрому выделению метана, этилена,
этана, пропилена и пропана в приблизительно мольном отношении
1: 1,7 : 0,5 : 0,2 : 0,1. Хотя только примерно 10% СО, присутствую-
щего в Ru3 (СО) 12, превращается в углеводороды и дополнительное
введение А1Н3 или СО не оказывает, видимо, влияния на ход реакции,
все же эта реакция является одним из первых примеров восстанови-
тельной полимеризации (хотя и стехиометрической) координированного
СО в мягких условиях, т.е. представляет собой нечто вроде низкотем-
пературной реакции Фишера — Тропша. Однако, возможно, большее
значение имеет распределение продуктов: этилен является главным
продуктом (выход 52%). Концентрация других углеводородов снижает-
ся при переходе от Ct к С3 примерно в геометрической прогрессии,
соответствующий стадийной последовательности реакций, показанной
уравнением (3.8) [ 16, 399].
Н2 С° и Н2
восстановление внедрение восстановление
МСО ----------------* МСН ---------► МССН3 ---------------->
- ^2® - Н2О
сн,
4
—* МСН2СН3 -> и т.д. (3.8)
I
с2не
При использовании карбонилов металлов группы VIБ селектив-
ность по отношению к этилену заметно возрастает. Так, обработка
М(СО)б> где М - Сг, Мо или W, шестикратным избытком А1Н3 в тетра-
гидрофуране при 22°С приводит к селективному (95%) образованию
этилена. В случае Сг(СО)с превращение карбонильных лигандов в эти-
лен возрастает при увеличении концентрации А1Н3 , достигая макси-
мума (17 ± 3%), когда мольное отношение Сг(СО)6 к А1Н3 составля-
ет 1: 6. При шестикратном избытке алана выделение этилена закан-
чивается полностью в течение 15 мин при 22°С. В случае A1D3 вместо
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
255
А1Н„ образуется C2D4. Аналогичные результаты получены при исполь-
зовании Мо(СО)6 и W(CO)e . Однако, хотя селективность по этилену
высока, максимальное превращение СО в этилен в этом случае зна-
чительно ниже (4 % для молибдена и 6 % для вольфрама).
Эти результаты являются веским свидетельством о наличии аль-
тернативного механизма реакции роста, или процесса димеризации,
который приводит исключительно к образованию этилена. Эксперимен-
тальные данные согласуются с промежуточным образованием карбе-
ноидных производных металлов (3.17). Промежуточные частицы будут
участвовать либо в последовательности восстановление -» внедрение
карбонильного лиганда -» восстановление с образованием продуктов
Cj, С2 и С8 (путь А), либо в реакции димеризации с образованием
этилена (путь Б).
СН4
(СО)ХМ=СН2
3.17
(СО)ХМ—сн3
3.18
(СО)Х_|М—с—сн3
3.19
СН2.
\ с2н6
(СО)Х_,М—С2Н
(СО)ХМ. ! М(СО)Х
'СН^
2М(СО)Х + С2Н4
Гидрирование карбенового фрагмента и восстановление координиро-
ванной ацетильной группы до этильной, как это предполагается в схе-
ме А, было продемонстрировано при использовании модельных сис-
тем [400, 401], так же как была продемонстрирована и возможность
димеризации карбенов, подразумеваемая в схеме Б [ 402 - 404]. Вы-
сокая селективность по этилену, очевидно, достигается, когда преоб-
ладает механизм Б; и то, что это было найдено с использованием кар-
бонилов металлов группы VIB, по крайней мере на первый взгляд, не
слишком удивительно, так как именно эти металлы наиболее часто
участвуют в реакциях образования карбенов и в реакциях с участием
карбеновых промежуточных продуктов (см. метатезис алкенов в
разд. 2.8).
Возможное участие первичных карбеновых частиц как промежу-
точных продуктов в реакциях олигомеризации (полимеризации) СО
открывает другой путь реакции роста цепи. В стадийной реакции рос-
та по схеме А на первой стадии после образования первичного карбе-
на происходит восстановление до промежуточного метильного произ-
256
ГЛАВА 3
СО
I
м
+2Н2
-Н/Г
сн3
сн,
I
М М -► U /77. д.
I
СаН6
-Н2О +2Н2
Н2С— СН2
MZ М -^С->Н
3.21 2
СД
Рис. 3.4. Альтернативный механизм восстановительной олигомеризации
(полимеризации) СО.
водного 3.18, а далее может происходить либо миграция метильной
группы в карбонил с образованием 3.19, т.е. рост цепи, либо дальней-
шее восстановление с образованием метана (ограничение цепи). Имеются
некоторые данные, свидетельствующие о том, что первичные карбе-
новые частицы, координированные с переходными металлами, могут
действовать цак нуклеофилы [349] (см. раздел 2.8.1,В) и в таком
виде они атакуют карбонильный углерод координированного СО [405].
В этом случае может быть предложен механизм роста цепи, показан-
ный на рис. 3.4 (на рисунке представлен биметаллический механизм,
однако, как отмечалось выше, a priori не существует причины, по ко-
торой необходимо рассматривать по крайней мере два различных
типа координационных мест в каталитической системе). Здесь первая
стадия роста - образование звена С2 - заключается в атаке карбо-
нильного атома углерода, а не миграции метильной группы к карбо-
нильному углероду (рис. 3.3). Процесс образования метана, приведен-
ный на схеме рис. 3.4, также отличается от образования других
углеводородов тем, что он происходит при гидрировании связи металл
карбен, а не дегидрировании связи металл - алкил. Так как можно
ожидать, что скорость гидрирования карбенового комплекса металла
будет отличаться от гидрирования металлалкильной группы, то в ме-
ханизме, показанном на рис. 3.4, образование метана будет сильно за»
висеть от'природы М и не обязательно будет соответствовать статис-
тической прогрессии [16, 339], которая ожидается для образования
углеводородов С\ — Сп. Такой случай действительно наблюдается
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
257
для большинства гетерогенных каталитических систем, и содержание
метана почти всегда превышает то, которое ожидается в соответствии
со статистическими законами [14]. Согласно схеме, рассмотренной
на рис. 3.4, предполагается, что повышенное образование метана не
является обязательным, более того, из этой схемы следует, что при
правильном выборе каталитической системы (по-видимому, на основе
переходных металлов, стоящих в начале соответствующих рядов) обра-
зование метана может быть резко уменьшено или даже полностью
устранено. Если эта схема действительна, то создание каталитичес-
ких систем, способных вызывать селективную восстановительную ди-
меризацию СО, является достижимой целью.
3-2.2. Гомогенные катализаторы. С экономической точки зре-
ния этиленгликоль - весьма желательный продукт реакции между
СО и водородом. Первое сообщение о его получении непосредственно
из синтез-газа с использованием гомогенного катализатора появилось
в 1961 г., когда Грешэм [406, 407] из компании Du Pont сообщил,
что некоторые комплексы кобальта, например ацетат кобальта, в под-
ходящем растворителе, например в уксусной кислоте, катализируют
превращение смеси СО + Н2 до полифункциональных кислородсодер-
жащих органических соединений. Этиленгликоль, высшие полиолы и
эфиры этих спиртов были основными продуктами. Для протекания этой
реакции потребовались давление выше 1000 атм и температура
180 — 300°С. В типичном эксперименте из 2*: 1-смеси СО и Н2 , вы-
держанной 1 ч при 225 - 246° С и 3000 атм в присутствии уксусной кис-
лоты, содержащей ацетат кобальта (0,035 г/см3), получалось 13,7 г
смеси этиленгликольдиацетата и триацетата глицерина. В 1974 г.Р.Л. Прюэт и
В.Е. Уолкер (Union Carbide Corporation) обнаружили, ЧТО в аналогичных
условиях высокого давления, т.е. при 3000 атм СО + Н2 , некоторые
растворимые родиевые комплексы способны катализировать реакцию
между СО и Н2 с образованием этиленгликоля в качестве основного
продукта (около 70%) [408].
Предшественником катализатора, обычно используемым в реак-
ции, является дикарбонилацетилацетонат родия. Однако детальное
изучение методом ИК-спектроскопии в условиях реакции (при давле-
нии смеси СО + Н2 1000 атм и 200°С) показало, что в системе присут-
ствуют анионы как [Rh(C0)4 ] ~ , так и [Rh12 (СО)34 _ Зб] 2~ в различ-
ных концентрациях на различных стадиях реакции [409, 410]. Было
высказано предположение, что карбонильные кластеры родия, харак-
теризующиеся тремя интенсивными полосами поглощения в ПК-спектре
258
ГЛАВА 3
при 1868 ± 10, 1838 ± 10 и 1758 + 10 см"1, являются ответственными
за протекание катализа [ 409}; считается, что реакция зависит от
установления следующего равновесия:
2[Rh6(CO)15H]-— [Rh12(CO)30]2- — [Rh12(CO)34]2"
Н2 (3.9)
Введение органических азотсодержащих лигандов, таких, как 2-гидрок-
сипиридин или пиперидин, облегчает установление равновесия (3.9),
возможно, в результате образования противоионов (при протонирова-
нии) для анионных металлических кластеров [409].
Большой набор комплексов переходных металлов был проверен в
’ качестве возможных катализаторов для реакции, но до сих пор толь-
ко системы, содержащие родий, обладали заметной активностью в
образовании полиолов. Главные продукты в реакции, катализируемой
родием, — метанол и этиленгликоль; соотношение между ними, по-види-
мому , зависит от природы органического лиганда и давления СО + Н2.
При давлении СО + Н2 (мольное отношение 65 ; 35) в интервале
1260 - 1400 атм при использовании 2-гидроксипиридина в качестве ли-
ганда или в качестве противоина (после протекания протонирования)
при температуре реакции 230°С за 2 ч было установлено, что отноше-
ние этиленгликоля к метанолу составляет около 7:1, тогда как при
использовании 3-гидроксипиридина в качестве лиганда (противоиона)
при тех же температуре и давлении это отношение снижается до 2:1.
Аналогично уменьшение давления СО + Н2 от 1300 - 1400 до 650 атМ
вызывает снижение отношения гликоль •: метанол. Другими продукта-
ми реакции, которые образуются в меньшем количестве, являются ме-
тилформиат, этанол, глицерин, в некоторых случаях пропиленгликоль.
Утверждают [ 408], что в этом процессе практически отсутствует реак-
ция метанирования:
СО+ н2 —СН4 + Н2О (3.10)
Хотя никаких подробностей о механизме этого процесса не было опуб-
ликовано, все же заманчиво, учитывая высокую селективность по эти-
ленгликолю, предположить, что в качестве промежуточного продукта
образуются гидроксикарбеновые производные типа 3.22, которые пос-
ле димеризации и последующего гидрирования могут давать С2- гликоль:
СН(ОН) +Н9 СН2ОН
2М=С ----------* || --— |
ХОН СН(ОН) СН2ОН
3.22
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
259
Что касается промышленного использования, эта гомогенная катали-
тическая система имеет два недостатка: а) необходимость работы при
высоком давлении; б) большие затраты на катализатор. До сих пор нет
пока сообщений о планах производства этиленгликоля с использовани-
ем таких гомогенных кластерных родиевых катализаторов. Тем не ме-
нее эти системы однозначно демонстрируют возможность селективного
получения химических продуктов путем восстановительной олигомери-
зации СО.
Сообщалось [16, 441], что другие кластерные производные пере-
ходных металлов, например Ir 4 (СО) 12, Ru 3 (СО) 12 и Os3 (СО)12 ,
катализируют восстановление СО водородом.. Однако при мягких ус-
ловиях (140°С, давление СО + Н2 2 атм) скорости чрезвычайно низки
(конверсия около 1 % за 3 - 5 суток при числе оборотов катализатора
3 — 5), а при более жестких условиях (250°С, давление СО + Н2 100 атм)
гомогенность этих катилитических систем вызывает значительные
сомнения [ 412]. Как должно быть ясно из ряда предыдущих разделов,
СО и Н2 — две наиболее широко изученные молекулы в гомогенном
катализе, например в гидрировании, гидроформилировании, карбонили-
ровании, и конструирование систем, способных заставить эти моле-
кулы взаимодействовать селективно между собой, остается волную-
щим вызовом для химиков. В данном разделе мы сделали попытку
осветить исследования, проводимые в настоящее время, и рассмотреть
некоторые предположения о механизме, а также ряд умозрительных
заключений, которые к 1979 г. стали характерными для этой области.
Дополнительная литература по гетерогенным и гомогенным
реакциям СО и И*
£
СторчГ., Голамбик Н., Андерсон Р. Реакция Фишера - Тропша и
родственные синтезы: Пер. с англ./ Под ред. проф. А.Н. Башки-
рова. — М.: ИЛ, 1954.
Нефедов Б.К^Эйдус Я.Т. Разработка методов каталитического
синтеза органических соединений с окисью углорода и водородом. —
Усп. химии, 1965, т. 34, с. 272.
*См. также: Muetterties E.La, Stein J. Mechanistic Features of Catalytic
Carbon Monoxide Hydrogenation Reactions.— Chem. Rev., 1979,v. 79, p. 479 — 490;
Bell A, Catalytic Synthesis of Hydrocarbons over Group VIII Metals.
A Discussion of the Reaction Mechanism. — Catal. Rev. — Sci. Eng-, 1981,
v. 23,p. 203 — 232; King D.LO, Cusuman JOA. Garten R.LO A Technological
Perspective for Catalytic Processes Based on Synthesis gas. — Catal. Rev. —
260
ГЛАВА 3
Vannice М.А. The catalytic synthesis of hydrocarbons
from carbon monoxide and hydrogen* — Catal* Rev* Sci* Eng*, 1976,
v* 14, p- 153*
Henrici-Olive Ge, Olive S. The Fischer-Tropsch synthesis: molecular
weight distribution of primary products and reaction mechanism —
Angew* Chem* Internal* Et* Engl*, 1976*, v* 15, p* 136*
Denny Whan D»A. The heterogeneously catalysed hydrogena-
tion of carbon monoxide. — In: Catalysis, v. 2, London, Chemical
Society, 1978, p* 46*
Masters C* The Fischer-Tropsch reaction. — Adv. Organometal.
Chem*, 1979, v. 17, p. 61.
3.3. Активация алканов
Алканы - наиболее распространенные в природе углеводороды.
Они составляют важную часть большинства сырых нефтей, их содер-
жание зависит от типа нефти. Природный газ, мировое производство
которого соответствует примерно 44 % общей потребности в сырой неф-
ти, состоит почти исключительно из алканов Ц — С4 . Изобилие ал-
канов в природе свидетельствует об их химической инертности, и
большинство их химических реакций, которые проводятся при высоких
температурах, обычно имеют, к сожалению, низкую селективность.
Разработка катализаторов, способных селективно активировать алка-
ны, была бы значительным вкладом в наиболее эффективное исполь-
зование мировых ресурсов углеводородного сырья.
Некоторый прогресс был достигнут в разработке гомогенных
металлокомплексных систем, способных повышать селективность ре-
акций окисления алканов (см. разд. 2.7.1, Б}; известно также несколь-
ко гомогенных катализаторов, которые эффективно катализируют
селективные перегруппировки напряженных углеводородов (см. разд.
2.2.2). Тем не менее мы еще далеки от понимания факторов, сущест-
венных для взаимодействия простых ненапряженных алканов с метал-
лическими центрами. При изучении взаимодействия sp3-C — Н с
Sci. Eng., 1981, v. 23, р. 233 — 263; Dry М.Е», Hiogendoom J,CO Technology
of the Fischer-Tropsch Process — Catal. Rev. — Sci. Eng., 1981, v. 23^
p. 265 - 278. - Прим. перево
Из перечисленных работ только статья Мастерса посвящена специаль-
но гомогенным системам, тогда как остальные в основном посвящены ге-
терогенным катализаторам реакции СО и Н2.
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
261
металлом были использованы в основном два подхода; один из подхо-
дов базируется на использовании углеводородных молекул, содержа-
щих активированные связи sp3- С - Н; другой подход, который явля-
ется более увлекательным, включает использование простых алканов,
например метана и этана.
3.3.1. Взаимодействие металлов с активированными алифа-
тическими СН-связями. А. Стерическая активация. Стерическая
активация алифатической СН-связи заключается в обеспечении частого
и близкого соседства связи sp3- С — Н с металлическим центром
путем закрепления этой связи на металлическом центре через коор-
динирующий фрагмент, который в принципе может быть любой частью
молекулы, способной связываться с металлическим центром, т.е.
атомом фосфора, или двойной связью, или атомом кислорода, или
или ароматическим кольцом. Один из первых примеров этого типа ак-
тивации был описан [413] в 1965 г., когда было показано, что руте-
ний (0) в Ru(dmpe)2 (3.23) (где dmpe — это (СН3)2 РСН2СН2Р(СН3)2)
может взаимодействовать со связью sp3- С — Н лцганда. Позже было
показано, что получаемый продукт является димерным гидри-
доалкильным комплексом рутения(II) (3.24) [414]. Формаль-
где —СН2СН2—
Ru(dmpe)2
3.23
где dmpe =
(СН3)2РСН2СН2Р(СН3)2
но эта реакция представляет собой окислительное присоеди-
нение связи sp3 - С - Н к центру, содержащему рутений(0): •
С
С—H + Ru(0) -»Ru(II) -Н (3.11)
и в этом случае эта реакция напоминает окислительное присоединение
диводорода н /
Н-Н+ М(п)-Л(п+2)~Н. (3.12)
Что касается энергии диссоциации, то эта энергия для sp3-C — Н-
связи та же (например, для СН4 , £)(С~Н) = 435 кДж/моль), если не
меньше (как в случае С2Н6 , £)(С-Н)= 410 кДж/моль), что и энергия
262
ГЛАВА 3
диссоциации связи Н- Н (£)(Н- Н)= 435 кДж/ моль). Однако необхо-
димо проявлять осторожность в распространении аналогии слишком далеко,
так как энергосодержание связи М — С не обязательно приблизительно
равно этой величине для связи М — Н (см. разд. 3.2). Для реакции типа
(3.11) энтропийные факторы могут иметь большее значение, чем для
реакции (3.12).
В последние несколько лет было найдено много комплексов, со-
держащих алкилфосфиновые лиганды, которые участвуют в реакции
внедрения со связью С — Н, аналогичной реакции для комплекса
Ru(dmpe)2 [415, 416]. Увеличение стерического объема трет-фосфино-
вого лиганда облегчает реакцию внедрения. Так, в кипящем 2-меток-
сиэтаноле wzpawc-Pt С12 [P(«p^-Bu)2((«-Pr)]2 образует продукт внед-
рения 3.25 за несколько часов [417], тогда как mpaH0-PtC].2 [Р(и-Рг)3]2
не образует продуктов внедрения по связи С — Н даже в течение
нескольких недель. Полагают, что промотирующее влияние объемис-
L\ /С1
^Pt .Pt
СГ ^СК 4L
3.26
(t- третичный, п -нормальный)
тых заместителей связано как с кинетическими, так и с термодинами-
ческими факторами; они не только заставляют С-Н-связь алкильной
группы сближаться с металлом, но также обеспечивают заданную кон-
формацию связанной алкильной цепи (например, пропильного звена в
примере, рассмотренном выше), при этом потеря свободы (например,
уменьшение внутренней энтропии вращения), связанная с образовани-
ем циклического продукта, меньше,чем та, которая наблюдалась бы
в случае менее жесткой системы, например Pt С12 [Р (я-Рг)3]2 [ 418].
В примерах, рассмотренных выше, доказательством взаимодействия
связи С — Н с металлом служило выделение продуктов внедрения —
комплексов 3.2к и 3.25. Другим, менее прямым, но потенциально более
универсальным методом является обмен водорода с дейтерием. Когда
димерные комплексы типа 3.26 (где L — целый ряд алкил-wpетгфосфи-
новых лигандов) нагреваются в смеси D20 + СН3СО2В, то атомы во-
дорода алкильных групп обмениваются на дейтерий [419]. Этот обмен
является региоспецифическим. Так, при L = РРг 3 обмен происходит
исключительно у терминального углерода пропильной группы; в слу-
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
263
чае L = РВи3 , хотя обмен происходит и у терминального (С4), и пред-
последнего (С3) атомов углерода,все же обмен у атома С3 в 11 раз
быстрее, чем у С4; при L = PEt3 не наблюдается обмена на дейтерий,
в течение продолжительного времени реакции. Как считают, обмен
происходит при окислительном присоединении sp3-C — 11-связи к ме-
таллическому центру, что сопровождается частичным разрывом хлор-
ных мостиков с образованием /интермедиатов типа 3.27 в случае •
S = CH3COODmm d2o
L = PPr3 . Восстановительное удаление НС1 из 3.27 с последующим
окислительным присоединением DC1 приводит к обмену Н на D, а пос-
ледующее восстановительное удаление DC1 приводит к внедрению дейте-
рия в алкильную цепь. Считается, что преимущественный обмен у ато-
ма С 3 указывает на преимущественное образование в системе пяти-
чденных циклов 3.27 а (ср. 3.25). Хотя в этой частной системе нали-
чие объемистых групп в «гр^я-гфосфиновом лиганде не является обя-
зательным условием активации С — Н-связи, т.е. H-D-обмена, нали-
чие таких групп оказывает значительное влияние на скорость реак-
ции обмена. Так, оказалось, что константа скорости реакций перво-
го порядка для H-D-обмена у атома С3 пропильной группы возрастает
в отношении 1 •: 2,4 •: 26,7 по мере того, как лиганд L изменяется в
ряду РРг3 -> Р(»гргяг-Ви)Рг2 -> Р(ягргю-Ви)2 Рг. В случае L=P(w?p^-Bu)2Pr
внедрение дейтерия происходит также в ягрея-бутильные группы, что
указывает на образование трехчленных циклических промежуточных
продуктов.
Стерическая активация не ограничена использованием фосфора
как координирующего центра с металлом. Действительно, в одном из
первых примеров Н — D- обмена у sp 3 -атома углерода использовалось
ароматическое кольцо как координирующийся фрагмент [ 420]. Так, в
присутствии гомогенного катализатора на основе Pt (II) (например,
[Pt С14 ]2 - ) в водном (D2O) растворе уксусной кислоты (CH3COOD)
1,1-диметилпропилбензол подвергается дейтерированию в боковую
цепь исключительно в положении С3. Аналогичные результаты, т.е.
преимущественное дейтерирование в положении Сб , получены с ал-
264
ГЛАВА 3
кенами типа 3.28, когда R = Н или СН3 [ 421]. В этой системе геми
3.29
нальные метильные группы у атома С3 были введены для того, чтобы
препятствовать образованию п-алильных частиц при р-элиминировании водо-
рода, что привело бы к изомеризации алкена (см. разд. 2.2.1, Б). Однако в
свете более поздней работы [418], в которой использовались металлхе-
латные системы с большими циклами , представляется более вероят-
ным, что эти группы в действительности промотируют активацию
углерода С5, обеспечивая особую конформацию алкильной цепи в зна-
чительной мере таким же образом, как это происходит в случае
тр^-бутильных групп в системах,, содержащих трет-фосфиновые ли-
ганды, которые обсуждались выше. Считается, что активация связа-
- на с окислительным присоединением sp3- С— Н-связи к металлическо-
му центру. В случае системы Pt2 Cl4 L2 образующиеся димерные пла-
тиновые комплексы типа 3.29 в соответствии с кинетическими дан-
ными играют значительную роль в реакции обмена [421]. В отличие
от систем, содержащих трет-фосфиновые лиганды, системы, содер-
жащие алкены, являются каталитическими по отношению к платине,
так как в присутствии избытка алкена происходит быстрый обмен
между свободными и координирующимися алкенами.
Другая последовательность реакций, которая может быть сущест-
венной для разработки гомогенных катализаторов активации алканов,
наблюдалась с иридиевым аналогом комплекса 3.23, т.е. с [Ь (dmpe)2]+o
В растворе этот комплекс находится в равновесии с продуктом при-
соединения по связи С— Н (3.30Y, 3.30 в присутствии СО2 приводит
к образованию карбоксилатного комплекса 3.31 [422]. Суммарно
последовательность реакций представляет собой превращение насы-
щенной группы С — Н в карбоксилатный анион и с первого взгляда
указывает на легкость протекания реакции (3.13). Однако существу*'-
ет небольшая проблема: изменение энергии Гиббса (A G ) в реакции
(3.13), когда RH — молекула наиболее легко доступных алканов, ве-
лико и положительно при экспериментальных условиях реакций. Так,
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
265
[Ir(dmpe)2]+
для RH = СН4 величина AG0 возрастает с 80,5 кДж/моль при 400 К
до 150,2 кДж/ моль при 1000 К. Термодинамическая возможность
является, по-видимому, главным ограничением в разработке практи-
чески полезных катализаторов активации алканов. Многие последова-
тельности реакций, которые кажутся привлекательными на основе
известной химии металлалкильных и металлгидридоалкильных комплек-
сов, например с использованием реакции (3.14) или (3.15), оказывают-
ся неприемлемыми, когда рассматривается термодинамическая воз-
можность протекания реакций (AG °00 = 67,2 кДж/моль для RH = СН4
в уравнении (3.14); AG°9g = 101 кДж/моль для R = H в уравнении
(3.15». R R Q
I СО I II
RH + М — М—Н-*М— СО-*М—С—R— M + RC^ (3.14)
I I хн
н н
ch2ch2r ch2=chr
rch2ch3 + м — м—Н --------♦ м—н—*М + СН2—CHR + н2
н (3.15)
Итак, чрезвычайно важно в катализе активации алканов, как и в случае
любых каталитических реакций, убедиться в термодинамической воз-
можности реакции прежде, чем предпринимать попытки сконструиро-
вать каталитическую систему.
Б. Электронная активация.' Если предположить, что активация
алканов протекает через окислительное присоединение, то возраста-
ние электронной плотности на металлическом центре и понижение
электронной плотности на фрагменте 5р3-С — Н должны способство-
вать активации. Возрастание электронной плотности может достигать-
ся при использовании координационно-ненасыщенных комплексов ме-
266
ГЛАВА 3
талла в низкой степени окисления, предпочтительно нулевой, и этот
металл должен быть окружен электронодонорными лигандами, т.е.
основаниями. Уменьшение электронной плотности подразумевает при-
сутствие электроноакцепторных заместителей, таких, как -CN,
—NO2 , — CO2R, — О — , в молекулах алкана.
Спектроскопические измерения [ 423] подтверждают ожидаемую
высокую электронную плотность во фрагменте M(dmpe)2 комплексов
типа HMNp(dmpe)2 (3.32), где М — железо, рутений или осмий, Np -
2-нафтил и dmpe-бис-1,2-диметилфосфиноэтан. Fe(dmpe)2 — железосо-
держащий аналог комплекса 3.23, образующийся при восстановитель-
ном удалении HNp из 3.32, легко реагирует с активированными соеди-
нениями типа СН3 Х(где X=CN,-COR,-CO2R, -SOR, -SO2R) с
образованием комплексов HFe(CH2 X)(dmpe)2 [424]. В общем случае
комплекс, который имеет группы Н и СН2 X в wzpawc-положении по от-
ношению друг к другу, является конечным продуктом присоединения.
Однако кинетическое изучение с использованием спектроскопии ЯМР
показало, что реакция протекает через образование комплекса
tywc-HFe(CH2CN) (dmpe)2 (3.33) с последующей изомеризацией с обра-
зованием термодинамически более выгодного ягранс-изомера 3.34.
CHqCN
Fe(dmpe)2 -------*
3.33
3.34
Действительная природа взаимодействия связи СН с металлом пока
неясна. Заманчиво на основе того, что кинетика благоприятствует
образованию tywc-изомера, предположить трехцентровое переходное сос-
тояние типа 3.35. Впрочем, для некоторых, комплексов, для которых
(dmpe)2Fe'
'"'CH2CN
3.35
—С~Н“—м
3.36
3.37
взаимодействие С — Н с металлом не приводит к присоединению, было
установлено, что это взаимодействие, по-видимому, происходит через
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ
267
линейную трехцентровую "связь", т.е. с образованием 3.36, а не треу-
гольного аналога 3.37 [425 — 427].
Далее, некоторые данные свидетельствуют о том, что переходное
состояние реакции металла с С— Н аналогично тому, которое реали-
зуется при реакции внедрения синглетного карбена по связи С — Н
[51]. Теоретические расчеты [428, 429] показали, что внедрение яв-
ляется реакцией, протекающей по синхронному механизму, при этом
группа СН2 атакуется почти параллельно оси СН. Если выводы из
этих расчетов могут быть распространены на систему металл — СН,
то образование переходного состояния типа 3.38 будет предпочтитель-
ным по сравнению с 3.35, при этом окислительное присоединение может
рассматриваться как стадийный гомолитический процесс, при котором
вначале происходит перенос атома водорода с образованием 3.39 и
радикала *СН2СН, а затем радикал быстро подвергается реакции ради-
кальной рекомбинации с образованием 3.33.
[(dmpe)2Fe—Н — CH2CN]*— (dmpe)2Fe'—Н + CH2CN— (3.33)
3.38 3.39
На практике трудно различить эти два альтернативных варианта. Впро-
чем, все больше накапливается данных, свидетельствующих о значи-
тельной роли одноэлект^нных процессов в реакциях с участием ком-
плексов переходных металлов [430].
Реакция между Fe(dmpe)2 и CH3CN не ограничивается образова-
нием продукта присоединения (3.34). Если к раствору, содержащему
HFe(CH2 CN)(dmpe)2, добавляют СО2 и получаемую смесь либо нагре-
вают, либо обрабатывают бромом или иодом, при этом выделяется
свободная циануксусная кислота [ 424}:
Fe’(drape) 2
CHqCN + C02 --------* CNCH2C02H
Известна и другая реакция активации алканов — гидрирование с пере-
носом водорода (см. разд. 2.1.4), в котором активированный алкан
(например, диоксан) превращается в соответствующий алкен. Эта реак-
ция может привести к потенциально интересным органическим про-
дуктам и также является каталитической. Термодинамическая возмож-
ность превращения алкана в алкен обеспечивается сочетанием реакции
дегидрирования с подходящей реакцией гидрирования [ 146 — 148].
268
ГЛАВА 3
3.3.2. Взаимодействие металла с неактивированными али-
фатическими СН-связями. Хотя все вышерассмотренные системы
показывают, что подходящим образом активированная связь С — Н
может эффективно взаимодействовать с металлическим центром в
мягких условиях, все же решающим экспериментом в этой области
был бы, несомненно, тот, который бы недвусмысленно продемонстрировал
активацию простого неактивированного алкана с помощью металлокомплек-
са. Такой эксперимент был описан в 1969 г., когда было опубликовано сооб-
щение, что в присутствии [PtС14 ] 2“ простые алканы подвергаются
обмену водорода на дейтерий в смеси D2 О - СН3СО2 D -DC1 в ка-
честве растворителя при температурах 80 — 100°С [431]. Первоначаль-
но существовало некоторое сомнение относительно гомогенности
этой системы благодаря частичному присутствию металлической пла-
тины в условиях реакции из-за протекания реакции диспропорциони-
рования (3.16). Однако последующие работы [51, 415] устранили эту
неопределенность, и сейчас нет сомнений, что активным катализа-
тором являются растворимые комплексы платины (II ), а не метал-
лическая платина или комплексы платины (IV).
2Pt (II)—>Pt(O) +Pt(IV) (3.16)
На рис. 3.5 показан каталитический цикл, приложенный для реакции
в случае, когда основной каталитически активной частицей служит
РиСо 3.5„ Каталитический цикл для катализируемого платиной Н—D- об-
мена в алкан ах .
ГОМОГЕННЫЕКАТАЛИТИЧЕСКИЕ СИСТЕМЫ В СТАДИИ РАЗРАБОТКИ 269
ptCl2S2 (S-растворитель), образующийся в равновесных сольволи-
тических реакциях:
[PtCl4]2- +S^[PtCl3S]-+Cl~ (3.17)
[PtCl3S]“+S — PtCl2S2 + Cl" (3.18)
Существенным этапом в активации алканов является окислитель-
ное присоединение RH к 3.40 с образованием гидридоалкильных ком-
плексов платины(1У) 3.41. Как обсуждалось выше, сейчас представ-
ляется вероятным, что "окислительное присоединение" происходит
при двух одноэлектронных этапах, а не по согласованному двухэлект-
ронному механизму. Последующее восстановительное элиминирование
НС1 с дальнейшим окислительным присоединением DC1 приводит к
обмену Н на D в алкильном фрагменте.
Анализ дейтированных продуктов этой реакции показывает, что
алканы подвергаются множественному обмену, т.е. в течение единич-
ного контакта с металлическим центром алкан обменивает более од-
ного атома водорода. В случае этана и высших алканов этот множест-
венный обмен может быть объяснен в предположении, что 3.42
(R = СН2 CHR1) подвергается р-элиминированию водорода с образо-
ванием алкенового интермедиата 3.43, который затем далее может
обменивать Н на D. По-видимому, когда R = СН3 , множественный
обмен протекает с образованием промежуточного продукта типа
3.44, т.е. через образование первичных карбеновых частиц, получа-
ющихся при элиминировании а-атома водорода. Альтернативой об-
разованию ir-алкенового интермедиата 3.43 является возможность
образования комплекса 3.45 — непосредственного аналога частицы
3.44. В самом деле, совокупность доступных данных [51], включая
тот факт, что введение свободного алкена в среду реакции обмена
эффективно отравляет активность алканов в Н — D-обмене, говорит
о промежуточном образовании 3.45, а не
Н CHR* Н
Pt^CH.; Pt^
3.43 3.44
^chch2r
Pt^
Альтернативная гипотеза [415] о механизме множественного обмена
рассматривает образование промежуточных димерных платиновых
частиц типа 3.46 путем присоединения С — Н на обоих атомах платины.
270
ГЛАВА 3
С,,^
Дс1А
Pt /Pt\
| ^cr I
Cl Cl
3.46
Хотя в настоящее время мало прямых экспериментальных данных в
поддержку этого предположения, участие димерных платиновых ком-
плексов в системах Н — D-обмена, содержащих алкмл-трет-фосфин и
алкилалкен (см. выше), служит некоторым косвенным доказательст-
вом этой идеи.
Поиски решения проблемы активации алканов прошли длинный
путь с тех пор, как впервые примерно 10 лет назад было продемон-
стрировано взаимодействие sp3-C— Н-связи с металлом. Однако,
как и в случае большинства гомогенных обсуждаемых в гл. 3 ката-
литических систем, находящихся в стадии разработки, по-прежнему
остается сделать шаг от тех академических исследований, которые
в основном проводились до сих пор, к разработке практических
каталитических систем для реальной активации не только алканов,
а также азота и синтез-газа.
Дополнительная литература по активации алканов*
Parshall G.JV. The homogeneous catalytic activation of C — H bonds. —
Accounts Chem. Res., 1970, v. 3, p. 139*
Parshall G.JK — In; Catalysis, vol. 1, London, Chemical Society, 1977
p. 335.
Webster D, Е» Activation of alkanes by transition metal compounds. — Adv.
Organometal. Chem., 1977, v. 15, p. 147.
Shilov A.E., Shteinman A. A. Activation of saturated hydrocarbons by metal
complexes in solution. — Co-ord. Chem. Rev., 1977, v. 24, p. 97.
Shilov A.E. Activation of alkanes by transition metal complexes. — Pure
and Appl. Chem., 1978, v. 50, p. 725.
* См. также: Мои сеев И.И. Окисление и окислительное сочетание,—
ЖВХО им. ДоИ. Менделеева, 1 977, т. 22, №1, с. 30 - 45; Шилов А.Е.,
Штейман А.А. Активация насыщенных углеводородов металлокомплексами
в растворах. - Кинетика и катализ, 1977, т. 18, № 5, с. 1129 - 1145;
Рудаков Е.С., Луцык А.И. Окислительная активация насыщенных углево-
дородов в сернокислых средах под действием Металлокомплекс©в и окис-
лителей. - Нефтехимия, 1980, т. № 2, с. 163 - 179. - Прим, перев.
4. Что же дальше?
Разнообразие гомогенных катализаторов и наше понимание этих
систем заметно возросли за последние десятилетия, большинство рас-
смотренных в книге систем было найдено в последние тридцать лет,
однако все еще много таких областей, где наши знания весьма скуд-
ны и исследователей ждут волнующие открытия.
Гомогенные катализаторы, которые мы рассматриваем, содержат
активные центры из одного переходного металла (одно возможное ис-
ключение — родиевый катализатор для синтеза этиленгликоля из
СО + Н2, разд. 3.2.2). Как мы видим, большинство каталитических
циклов включают ион металла, способный к участию w многих процес-
сах. Например, в серии реакций, составляющих процесс гидроформи-
лирования, активный центр должен быть способным разорвать молеку-
лу водорода, координировать алкен, способствовать миграции атома
водорода к алкену, координировать СО, обеспечивать перенос алкиль-
ной группы к углероду СО-^руппы и, наконец, приводить к реакции
ацильной группы и водорода с образованием альдегида как конечного
продукта. Недостаточно, чтобы ион металла играл ведущую роль в
большинстве (если не во всех) из этих стадий; нужно также, чтобы он
проводил эти стадии с высокой специфичностью, хотя бы с помощью
лигандов как второстепенных участников действия. Хотя часто и на-
блюдается сходство между индивидуальными стадиями (например, ми-
грация Н к алкену или алкила к СО может рассматриваться как слу-
чаи нуклеофильной атаки), было бы натяжкой утверждать, что данный
металл, например родий в случае гидроформилирования, оптимален
для всех индивидуальных стадий каталитического цикла. Возможно,
он является лучшим для такой стадии, как миграция алкила к СО, но,
вероятно, какая-то другая система могла бы быть лучшей для стадии
активного водорода. Такого рода рассуждения приводят к тому, что
можно ожидать дальнейшего усовершенствования каталитических сис
тем введением двух различных металлических центров в каталитичес
кий цикл. Сейчас хорошо установлено, что такие лиганды, как гидрид
[432], алкил [433, 434] и карбонил [435] - важнейшие промежуточны
272
ГЛАВА 4
образования в каталитических реакциях, — могут свободно перемещать-
ся между различными металлическими центрами. Например, в хлоро-
форме при 0°С происходит быстрый обмен хлора и метильной группы
между PtMe2(COD) (COD - циклооктадиен) и PdCl2(PhCN) 2:
Pt Me (COD) + PdCUPhCN), PtCl(Me) (COD) + PdCl(Me) (PhCN),
2 CDC13
Этот обмен может быть обнаружен не только спектроскопически (по
ЯМР на протонах), но может наблюдаться и по протеканию реакции,
характерной для одного из продуктов реакции обмена. В рассматри-
ваемом случае добавление стирола к реакционной смеси приводит к
количественному образованию 13- метилстирола [433]. В отсутствие
PdCl 2(PhCN) 2 ни Pt Me2(COD), ни Pt Cl(Me) (COD) не приводят к реакции
метилирования стирола. Аналогично Pt Me (COD) в присутствии МеОН
и [Rh(CO)2Cl]2 вызывает образование метилацетата благодаря пере-
носу метильной группы от платины к родию с последующей миграцией
метильной группы к карбонилу на родии и метанолизом [436]. !
Сочетание свойств двух различных переходных металлов в ката-
литическом цикле открывает новые возможности. Например, гидридо-
хлоробис(циклопентадиенил)цирконий 4 J взаимодействует с изомер-
ной смесью линейных алкенов с селективным образованием а -Замещен-
ных алкильных производных:
Н
(45-C5H5)2Zr
421
4.1
~ (rf'-C^Zr^
Cl
4.2
Если бы эту реакцию удалось провести в комбинации с родиевой сис-
темой гидроформилирования, как это показано на гипотетическом ка-
талитическом цикле, изображенном на рис. 4.1, можно было бы осу-
ществить селективное превращение алкенов с внутренней двойной
связью в концевые альдегиды.
Сочетание двух различных металлов в одном комплексе также
может дать возможность селективной активации нужных участков би-
функционального субстрата. Например, если бы стояла проблема ка-
ЧТО ЖЕ ДАЛЬШЕ?
273
Р и 0. 4.1. Гипотетические циклы гидроформилирования в присутствии ката-
литической системы, содержащей цирконий и родий.
талитического гидрирования только замещенной двойной связи в не-
котором субстрате типа 4.3, 'го, вероятно^ этот процесс мог бы быть
проведен на каталитической системе типа 4.4, в которой Ма образует
сильную связь с алкенами, но является малоактивным катализатором
гидрирования (например, платина(П) с ^рш-фосфиновыми лигандами),
а не только может координировать алкены, но является также эф-
фективным катализатором гидрирования (например, родий(1) с трешг
фосфиновыми лигандами). Конструированием электронных и стеричес-
ких свойств системы лигандов можно было бы создать центр 4.5 и
осуществить селективное гидрирование (по крайней мере гипотетичес-
ки). Конечно, нет ничего проще химии на бумаге, но все же рассмот-
ренная бигетерометаллическая система — всего лишь допустимое, до-
вольно очевидное следствие из достигнутого уровня "искусства ка-
тализа".
274
ГЛАВА 4
R1R2C=C(CH2)xCH=CH2
LflMa
CH2=CH(CH2)xC==CR1R2
LeMa.„M,L*
4.5
Участие больше одного металлического центра в каталитической
реакции является центральной идеей катализа кластерами [435, 438].
Кластерные соединения переходных металлов, т.е. дискретные моле-
кулы или ионы, в которых два или более атома металла участвуют в
связывающем взаимодействии, обладают некоторыми свойствами,
обычно приписываемыми поверхности металла [435, 439]. Примером
могут служить карбонильные кластеры. Карбонильные лиганды часто
способны к быстрому обмену между различными атомами металла в
кластере; это явление аналогично подвижности хемосорбированных
частиц на поверхности металлов. Средние энергии связи металл —
металл в биядерных металлокомплексах близки к средним энергиям
связи металл - металл для кристаллического состояния металла. Од-
нако более важное потенциальное значение имеет то, что возможно
подобие различных типов активации молекул на би- йли полиметалли-
ческих системах. Некоторые из различных типов взаимодействий, воз-
можных при связывании СО, показаны в формулах 4.5 - 4.8.
мм м | м м / мм м
М м
4.5 4.6 4.7 4.8
Как упоминалось в разд. 3.2, взаимодействие типа 4.7 и 4.8 может
быть важным при активации СО в случае восстановления водородом.
Другая возможность в случае комплексов, содержащих кратные свя-
зи металл - металл, заключается в активации субстратов при присо-
единении к кратной связи с разрывом или без разрыва связей в суб-
страте [38] , например:
А В
I I
М=М + АВ -------► м—М (4.1)
ЧТО ЖЕ ДАЛЬШЕ?
275
М=м 4- А:
.А.
* М-—-М
(4.2)
Считают, что эти процессы очень напоминают процессы активации на
поверхности гетерогенных катализаторов. Так как реакции, происхо-
дящие на кластерах, должны быть более удобны для детального изуче-
ния, чем реакции на поверхности, то есть надежда, что детальное изу-
чение кластеров переходных металлов приведет к лучшему пониманию
действия гетерогенных каталитических систем. Кроме того, в ряде
случаев гетерогенные катализаторы заметно превосходят гомогенные,
например, в активации С-Н- ' и С-С-связей в насыщенных углеводо-
родах, поэтому было высказано предположение [ 435 ], что нужным об-
разом сконструированные кластерные системы могут лишить гетеро-
генные катализаторы указанного превосходства.
Хотя металлические кластеры и могут служить растворимыми
аналогами гетерогенных катализаторов, все же возможность ^'мо-
билизации гомогенных катализаторов на поверхности с сохранением
преимуществ в селективности и активности гомогенных систем пред-
ставляет значительный практический интерес. Предложен ряд
подходов к иммобилизации металлокомплексов [440, 441 ]*.
И хотя многие результаты, описанные к настоящему времени, были
отчасти разочаровывающими с точки зрения сохранения селектив-
ности и активности, присущих гомогенному катализу, все же за по-
следнее десятилетие был сделан значительный прогресс в этой облас-
ти. Очевидны потенциальные возможности таких систем сочетать луч-
шие свойства гомогенных и гетерогенных катализаторов. По-видимо-
му, в этом, а также и в других смежных областях можно ожидать
значительных результатов в будущем.
По мере накопления химических знаний мы были свидетелями
возникновения большого числа химических дисциплин (областей), та-
*
Дополнительные сведения о каталитических свойствах закрепленных
металлокомплексов читатель может найти в следующих работах: Yermakov Yu. I.
Vfl International Congress on Catalysis, Tokyo, 1980 Preprints of Plenary Lec-
tures, P5° Anchored Complexes in Fundamental Catalytic Research; Ермаков Ю.И,
Захаров В.А., Кузнецов Б.Н. Закрепленные комплексы на окисных носителях
в катализе. — Новосибирск: Наука, Сибирское отделение 1980; Yermakov Yu.,-
Kuznetsov B.N., Zakharov V.A. Catalysis by supported complexes, Elsevier
Scientific Publishing Company, 1981; Лисичкин Г.В., Юффа А.Л. Гетерогенные
металлокомплексные катализаторы. — М.? Химия, 1981. *- Прим, перев.
276
ГЛАВА 4
ких, как электрохимия, бионеорганическая химия, фотохимия, теоре-
тическая химия, гомогенный и гетерогенный катализ. В каждой из
областей сложились собственная литература и терминология, которые,
помогая внутриобластному общению, мало способствуют переносу
идей и подходов между разными областями химии. За последнюю де-
каду произошло значительное взаимное обогащение гетерогенного и
гомогенного катализа, и в результате многие из появившихся новых
направлений в катализе, по-видимому, лежат как раз посередине меж-
ду двумя этими областями. Электрохимия предлагает химику, работающему
с металлокомплексами, возможность приготовления и стабилизации
металлов в такой степени окисления, которая не может быть легко
получена более традиционным применением окислительно-восстанови-
тельных реакций [442, 443]. Ионы металлов в таких состояниях мо-
гут проявлять новые каталитические свойства. Кроме того, электрод-
ные реакции могут быть использованы для возвращения восстановлен-
ных или окисленных ионов металлов в каталитически активное состоя-
ние, например, Рd(0) в вакер-процессе или Мо (х) в азотфиксирующей
системе [372]. Выше уже была продемонстрирована плодотворность тес-
ного сотрудничества между биохимиками и специалистами в области
металлокомплексов при конструировании систем, способных катализи-
ровать восстановление молекулярного азота [ 347 ].
Необходимость распространения такого сотрудничества на область
селективного окисления алканов является очевидной. В нескольких пуб-
ликациях рассмотрены возможности использования фотовозбужденных
металлокомплексов в катализе [444—446]. Вероятно, быстрое увели-
чение возможностей ЭВМ, которое произошло в последние годы, в со-
четании с усовершенствованием методов теоретической химии позво-
ляет подойти к математическому моделированию сложных каталитиче-
ских реакций и к более глубокому пониманию процессов, происходящих
на атомном уровне. Мы до сих пор рассматривали каталитические реак-
ции, которые происходят с помощью наиболее употребительных в ката-
лизе переходных элементов. Но даже беглый взгляд на периодическую
систему позволяет увидеть почти такое же число других элементов —
лантанидов и актинидов, о которых наши знания в области их каталити-
ческих свойств минимальны. Однако и то немногое, что известно об
этих элементах, дает основание полагать, что их ожидает блестящее
будущее в катализе [447, 448].
ЧТО ЖЕ ДАЛЬШЕ?
277
Успехи гомогенного катализа за последние тридцать лет выглядят
впечатляющими. Однако в течение этих лет мы только начинаем разра-
батывать поверхность потенциально богатого пласта. Мы прошли боль-
шой путь по дороге, конца которой еще не видно. Наука катализа непре-
рывно развивается, будучи ограничена только физическими законами
термодинамики и воображением химиков.
Литература
1. Pruett RL., Adv* organometaL Chemo, 17, 1 (1979).
2. Ostwald Wo, Phys. Zo, 3, 313 (1902).
3. Emmett P.H., Sabatier P,, Reid E.E-, Catalysis Then and Now,
Englewood Cliffs, New Jersey: Franklin, 1965.
4. Сталл Вестрам Э9, 9инке Г, Химическая термодинамика орга-
нических соединений: Пер. с англ. — М.: Мир, 1971.
5. Zeise W.C-, Progg. Ann., 9, 632 (1827).
6. Zeise W.C-, Progg. Ann., 21, 497 (1831).
7. Dewar M.J..S.,,Bull. Soc. Chine France, 18, C79 (1951).
8o Chatt J., Duncans on L.A., J. chem. So c, 2939 (1953b
9. Aiderman P.R.H., Owston P.G., Rowe J.M.. Acta. Cryst., 13, 149
(1960).
10. Фишер Э9, Вернер Г, тг-Комп лексы металлов: Пер. с англ./Под ред.
Луценко И.Ф. - М.: Мир, ^968.
И. Cotton F.А.» Inorg. Chem., 3, 702 (1964).
12. Albano V.G-Bellon PoL»> Ciani G., J. *chem. Soc., Chem. Commun.,*
1024 (1969).
13. Commons Co J., Hoskins P.E., Austral. J. Chem., 28, 1663 (1975).
14. Colton Ro, Commons CaJ*, Austral. J. Chem. , 28, 1673 (1975).
15. Manassero Mo, Sansoni M., Longoni Gof J. rhem. Soc./, Chem.
Commun., 919 (1976).
16. Masters C., Adv. organdmetal. Chem., 17, 61 (1979).
17. Mason Ro, Meek D.W., Angew. Chem. Int. Ed. Engl., 17, 183 (1978).
18. Halpern Ja, Advances in Chemistry Series No. 70, p. 1, Washington:
American Chemical Sqciety, 1968.
19. Джеймс Б. Гомогенное гидрирование: Пер. с англ./Под ред. Шило-
ва А.Е. - М.: Мир, 1976.
20. 'Collrmn J.P., Roper W.R., Adv. organometal Chem.,7, 53 (1968).
21. Chatt J*, Leigh G.J., Angew. Chem. Int. Ed. Engl., 17, 400 (1978).
22. Knowles VLS., Sabacky M.JVineyard P.D., Ann. N.Y. Acad. Sci.,
172(9), 232 (1970).
Литература
279
23. Bogdanovic В., Adv* organometal Chem., 17, 105 (1979).
24. Basolo В.* Chatt J., Gray H.В9, Pearson R.G., Shaw B.L., J. chem.
Soc., 2207(1961).
25. Cramer R., Inorg. Chem., 4, 445 (1965).
26. Tolman C\Aoy Chem. Revs., 77, 313 (1977).
27. Tolman C.A., J. Amer. chem. Soc., 92, 2953 (1970).
28. Immirzi A., Muse о A., Mann BOE., Inorg. Chem. Acta , 21, L37 (1977).
29. Tolman COA., J. Amer. chem. Soc., 92, 2956 (1970).
30. Tolman C.A., Seidel 1EG>, Gosser L.W., J. Amer. chem. Soc., 96,
53 (1974).
31. Ugo R., Catal. Rev-Sci Eng., 11, 225 (1975).
32. Birch A.J„ Jenkins IODO, In: Transition Metal Ogranometallics in
Organic Synthesis (ed. Alper H.)*, Vol. 1, p. 1, New York: Academic
Press, 1976.
33. Cramer R»v Accounts chem. Res./1, 186 (1968).
34. Deeming A.]Shaw B.L., J. Chem. Soc. (A), 1(1969).
35. Shaw B.L., Stainbank R.E°, J. chem. Soc.,-Dalton, 223 (1972).
36. de Vries B., J. Catal., 1» 489 (1962).
37. Chalk A. J., Harrod J.F., Adv. organometal. Chem.,- 6, 119 (1968).
38. Chisholm MoH*, Transition Metal Chem., 3, 321 (1978).
39. Harrod J«F°, Ciccone S^ Halpern J,, . Canada J. Chem.,-39 1372
(1961).
40. Evans D°, Osborn J. A., Jardine F.H., Wilkinson G., Nature, 208
1203 (1965).
41. James B.RO, Rattray AaDo, Wang D.K.W., J. chem. Soc.,-Chem.
Commun., 792 (1976).
42. Belluco U°, Giustiniani M°, Graziani Mo, J. Amer. chem. Soc.,-89,
6494 (1967).
43. Calderazzo F°, Angew. Chem. Int. Ed. Engl., 16, 299 (1977).
44. Tsutsui Ms, Courtney A., Adv. organometal. Chem., 16, 241 (1977).
45. Forster Ds, Adv. organometal. Chem., 17, 255 (1979).
46. Marko Lo, Ini Aspects of Homogeneous Catalysis (ed. Ugo R.),
Vol. 2. Dordrecht: Reidel, 1973.
47. Cooper N.J,, Green M.L.HO, J. chem. Soc., Chem. Commun., 761,
(1974).
48. Calderon N„ Lawrence J.Ps, Of stead E^A*, Adv. organometal. Chem.,
17, 449 (1979).
280
ЛИТЕРАТУРА
49. Tolman С.А., Chem. Soc. Revs., 1, 337 (1972)*
50* Coates G.E., Green M.L.!J.,;Wade K.> Organometallic Compounds
(3rd edition), Vol. 2, The Transition Elements, London:
Chapman and Hall, 1968.
51- Shilov A.E.^Shteinman Coord- Chem- Rev-, 27, 97 (1977)-
52. Parshall G.W., J. mol- CataL, 4, 243 (1978)-
53. Waddams A. L*> Chemicals from Petroleum (4th edition), London:
John Murray, 1978.
54- Lowery R.P., Aguilo A., Hydrocarbon. Proc-, 53(11), 103 (1974).
55. Chuisoli G.P., Salerno G., Adv.organometal Chem-/17, 195 (1979).
56. Knowles W.S.., Sabasky M.J., Vineyard P.D., Homogeneous Cata-
lysis—11 (eds. Forster D.,Roth J-E-), Washington: America Chemical
Society, 1974, p.274.
57- Knowles Ж.5., Sabacky M.J., U-S- Patent, 3849480 (1974).
58- Iguchi M., J. chem- Soc. Japan-, 60, 1287 (1939).
59- James B.R., Adv- organometal Chem-, 17, 319 (1979)-
60- Young J.E., Osborn J. A- Jardine F.R., Wilkinson G,, J. Chem- Soc-,
Chem. Comm-, 131 (1965).
61- Coffey R.Sj., Imperial Chemical Industries, Brit- Pat- 1121642 (1965).
62- Osborn J-Ач Jardine F.R., Young J.F., Wilkinson G-, J- Chem. Soc.,
(A) 1711 (1964)-
63- Lehman D.H., Shriver D-F-, Wharf L, J. rhem Soc-, Chem- Commun.,
1486 (1970)-
64- Eaton D.R., Suart S.R., J- 'Amer- chem- Soc-, 90, 4170 (1968).
65- Hussey A.S,., Takeuchi У., J- Amer, chem- Soc-, 91, 672 (1969).
66. Hussey A.S,. Takeuchi Y., J- org- Chem-, 35, 643 (1970)-
67. Tolman C. A., Meakin P.Z., Lindner D.L-, Jesson J.P., J. Amer-
chem. Soc-, 96, 2762 (1974)-
68- Clark H.C., Jablonski C., Halpern J., Mantovani A., Weil T.A.,
Inorg. Chem., 13, 1541 (1974)-
69. Crabtree R.R., Felkin H., Khan T.N., Morris G.E.,. J- organometal
Chem-, 168, 183 (1979).
70. Halpern J., In: Organotransition Metal Chemistry (ed- Ishida Yo,
Tsutsui M-), New York: Plenum, 1975, p- 109.
71. Montelatici S., van der Ent A., Osborn J.A., Wilkinson G., J. chem.
Soc., (A), 1054, (1968).
72. Horner L., Puthe H., Siegel H., Tett. Letts, 4023 (1968).
ЛИТЕРАТУРА
281
73. Jardine F.M., Osborn J-A., Wilkinson G., J. chem. Soc. (A) 1574
(1967).
74. Candlin J.P., Oldham A./?., Discuss* Faraday Soc., 46, 60 (1968)i
75. Iguchi M., J* chem* Soc. Japan, 63,634 (1942).
76. Iguchi M>, J* chem* Soc. Japan, 63, 1752 (1942).
77. Kwiatex J., Mador I.C., Seyler J.Y., Adv. Chem*, 37, 201 (1963)*
78. Simandi L.,Nagy F., Proceedings of a Symposium on Coordination
Chemistry in Tihany, Hungary, Budapest: Akademiani Ki ado, 1965,
p* 83.
79. Vlcek A.4м Proceedings of the 7th International Conference on
Coordination Chemistry, Stokholm, 1962, p* 286
80* Halpern J., Wong L.Y., J. Amer, chem., Soc.,-90, 6665 (1968).
81. Kwiatek. J., Catal. Rev., 1, 37 (1967).
82. Kwiatek J., Seyler J.K., J* organometal Chem*, 3, 421 (1965).
83. Kwiatek J., In: Transition Metals in Homogeneous Catalysis (ed*
Schrauzer G.N.) New York: Marcel Dekker, 1971, p- 13.
84. Kwiatek J., Seyler J.%., Adv* Chem*, 70, 207 (1968).
85. Burnett M.Q., Conolly P. J., Kemball C., J* chem. Soc* (A), 991 (1968).
86. Kwiatek J., Mador I.K., Seyler I. %., J* Amer* chem* Soc.,-84, 304
(1962).
87. Simandi L., Nagy F., Acta* Chim. Akad. Sci* Hung*, 46, 137 (1965).
88. Halpern J., Harrod J. FJames B.ft., J* Amer* chem* Soc., 88,5150
(1966).
89. Knifton J.F., J. org* Chem., 40, 519 (1975)*
90. Knifton J.F., J. org. Chem*, 41, 1200 (1976).
91. Knifton J.F., Tett. Letts, 2163 (1975).
92. Tsuji J., Suzuki H.,;Chem* Letts, 1085 (1977)*
93. Friends R.Q.J., van Koten G.^Vrieze K., Inorg* Chim* Acta, 26,
L29 (1978).
94. James B.R., Markham L.F).9lWang D.ft., J* chem* Soc*,-Chem.
Commun: 439 (1974).
95. McQuillin F.J., Homogeneous Hydrogenation in Organic Chemistry,
Dordrecht: Reidel, 1976.
96. Hallman Р.$., McGarvey B. ft., Wilkinson G., J. chem* Soc. (A) ,
3143 (1968).
97. O'Connor G., Wilkinson G. J* chem* Soc. (A), 2665 (1968)*
98. Odom HoQ.^Pinder A. ft.,]]* chem* Soc. Perkin I, 2193 (1972).
282
ЛИТЕРАТУРА
99. Nishimura S., Tsuneda К. Pull. chem. Soc. Japan, 42, 852 (1969),
100. Frankel Е.Ц., Emken E.4-, Peters H.tyh, Davison V.L., Petter-
field R.O. J. org. Chem., 29, 3292 (1964).
101. Miyake A., Kondo H., Angew. Chem. Int. Ed. Engl. 7, 880 (1968).
102. Frankel E.N°» Emken E.A., Itatani H., Pailar J.C., J. org. Chem.,
32, 1447 (1968).
103. Wrighton M.S., Ginley D.S., Schroeder M.A., Morse D.L., Pure appl.
Chem., 41,671 (1975).
104. Schroeder M.A.,Wrighton M.S., J. Amer. chem. Soc., 98,551 (1976).
105. Na,sielski J., Kirsch P., Steinert L.W., J. organometal. Chem., 27,
C13 (1971).
106. Wrighton M.S., Schroeder M.A. J. Amer. chem. Soc., 95, 5764
(1973).
107. Schroeder M. A., Wrighton M.S. J. organometal. Chem., 74, C29,
(1974).
108. Platbrood Go, Steinert LANa> J. organometal. Chem., 70ff 393 (1974).
109. Tayim H°A^ Railar J.Cai> J. Amer. chem. Soc., 89, 4330 (1967),
110. Adams RoW0, Ratley G.E., Railar J.C.* J. Amer. chem. Soc., 90,
6051 (1968).
111. Rressan G,» Proggi R., Chim. Ind. (Milan), SO, 1194 (1968).
112. Friedman S., Metlin S., Svedi A., Wender I., J. org. Chem., 24,
1287 (1959).
113. Feder H.M., Halpern J., J. Amer. chem. Soc., 97, 7186 (1975).
114. Stuhl L.S., Dubois M.R., Hirsekorn F.J., Blecke J.R., Stevens A.E.,
Muetterties E.L., J. Amer. chem. Soc., 100, 2405 (1978).
115. Bennett M.A., Huang T.-N., Turney T.W., J. chem. Soc., Chem.
Commun., 312 (1979).
116. Wender I./ Levine R., Orchin M. J. Amer. chem. Soc., 72, 4375
(1950).
117. Goetz R.W., Orchin M., J. org. Chem., 27, 3698 (1962).
118. Tucci E.R., Ind. Eng. Chem. Prod. Res. Dev., 7, 32 (1968).
119. Tucci E.R., Ind. Eng. Chem. Prod. Res. Dev., 7, 125 (1968).
120. Schrock R.R.j Osborn J.A., J. chem Soc., chem. Commun., 567
(1970).
121. Love C.J., McQuillin F.J., J. chem. Soc. Perkins I, 2509 (1973).
122. Jardine I., McQullm F.J., J. chem. Soc., Chem. Commun., 626 (1970).
ЛИТЕРАТУРА
283
123- Morrison J.D., Master W.F., Newberg M.K> Adv- Catal-, 25, 81
(1976)-
124, Pearce R. In: Catalysis, Vol- 2, p- 176, London: Chemical Society, 1978.
125- Brittain R.TO, Jack D., Ritchie A.C., Adv- Drug Res-, 5, 197 (1970).
126- Patil P.N., Miller D.D., etal. Pharmocol. Rev-, 26, 323 (1975).
127. Elliot M., Janes N.F.,, Chem- Soc. Revs, 7, 473 (1978)-
128. Fischer C., Mosher H.S. Tett. Letts, 2487 (1977).
129- Knowles Ж-S., Sabacky chem. Soc., Chem. Commun-, 1445
(1968)-
130. Horner L., Siegel H., Buthe Ho Angew. Chem. Int. Ed. Engl., 7,
942 (1968).
131. Knowles IV.S., Sabacky M.J. German Patent 2,123,063 (1971).
132. Knowles ILS., Sabacky M.J., Vineyard B.D. German Patent 2,210,938
(1972).
133. Knowles ILS., Sabacky M.J», Vineyard B.D. J- chem. Soc., chem(J
Commun., 10 (1972).
134. Knowles ILS., Sabacky M.J., Vineyard BoD. Ann. N.Y. Acad. Sci.,
214, 119 (1973).
135. Morrison J.D., Burnett R.E., Aguiar A.M., Morrow C.J., Phillips C.
J- Amer. chem. Soc., 93, 1301 (1971).
136. Kagan H.B., Dang T.P. J. Amer. chem. Soc., 94, 6429 (1972).
137, Benes J., Hetflejs J. Coll. Czech, chem. Commun., 41P 2264 (1976).
138. Tanaka Mo, Ogata I. J. chem. Soc., chem. Commun., 41, 735 (1975),
139. Dang ToPo, Poulin JoCo, Kagan H.B° J. organometal Chem., 91,
105 (1975).
140. Hayashi To, Tanaka Mo, Ogata L Tett. Letts, 295 (1977).
141. Fryzuk MoDo» Bosnich Bo J. Amer. chem. Soc., 99, 6262 (1977).
142» Achiwa K.J. Amer. chem. Soc., 98, 8265 (1976).
143. Hayashi T , Katsumura A°, Konishi M.s Kumada Mo Tett. Letts,
425 (1979)-
144. Briegen (1, Nestrick T°Jo Chem. Revs, 74, 567 (1974).
145. Коломников И.)С., Куколее В Л., Волъпин M.E. Перенос водоро-
да от органических соединений, катализируемый комплексами
переходных металлов. Успехи химии, 43, № 5, 903—932 (1974).
14бо Nishiguchi То, Fukuzumi Ко J. Amer. chem. Soc., 96, (1893(1974).
147. Nishiguchi T., Tachi К., Fukuzumi К. J. org. Chem-, 40, 237 C1975).
148, Masters C., Kiffen A. A., Visser J.P. J- Amer, chem- Soc-, 98,
1357 (1976)-
284
ЛИТЕРАТУРА
149. Sasson Y., Blum Ja J. org. Chem., 40, 1887 (1975).
150. Descotes GOil Sinou Do Tett. Letts, 4083 (1976).
151. The Petroleum Handbook. London: Shell International Petroleum
Company Ltd. (1966).
152. Evans D., Osborn J.A., Wilkinson G. J. chem. Soc. (A), 3133(1968).
153. Piacenti F., Pino P., Lazzaroni R., Bianchi M. J. chem. Soc. (C),
488 (1966).
154. Tolman C.A., J. Amer. chem. Soc., 94, 2994 (1972).
155. Bingham D., Webster D.W.,Wells P.B. J. chem. Soc., Dalton, 1928
(1972).
156. Werner H., Feser R. Angew. Chem. Int. Ed. Engl., 18, 157 (1979).
157. Casey C.P.f Cyr C.R. J. Amer. chem. Soc., 95, 2248 (1973).
158. Schroeder M.A., Wrighton M.S. J. Amer. chem. Soc., 98,551 (1976).
159. Harrod J.Fo, Chalk A.JO J. Amer. chem. Soc., 88, 3491 (1966).
160. Maitlis P.M. The Organic Chemistry of Palladium, Vol, 2, p. 128,
London: Academic Press, 1971.
161. Green M., Hughes R.P. J. chem. Soc. Dalton, 1907 (1976).
162. Davies N°R* Rev. pure appl. Chem., 17, 83 (1967).
163. Fischer E.O., Held IT. J. organometal Chem., 112, C59 (1976).
164. Cardin Cetinkaya; Doyle M.J., Lappert M.F, Chem. Soc.
Revs, 2, 132 (1973).
165. Paquette L.A. Synthesis, 347 (1975).
166. Bishop K.C. Chem. Revs, 76, 461 (1976).
167. Hammond G.S., Turro N.J., Fischer A. J. Amer. chem. Soc., 83e
4674 (1961).
168. Hoffmann R., Woodward R.P. Accounts chem. Res., 1, 17 (1968).
169. Hogeveen H^Volger H.C. J. Amer. chem. Soc., 89, 2486 (19671
170. Kaiser K.L., Childs R.F., Maitlis P.M. J. Amer. chem. Soc., 93,
1270 (1971).
171. Mango F.D. Adv. Catal., 20, 291 (1969).
172. Cassar L., Halpern J. J. chem. Soc., chem. Commun., 1082 (1970).
173. Cassar L., Eaton P.E., Halpern J. J. Amer. chem. Soc., 92, 3515
(1970).
174. Paquette L.A., poggs R.A., Ward J.S. J. Amer. chem. Soc., 97, 1118
(1975).
175. Plum J., Zlotogorski C., Zogan A. Tett. Letts, 1117 (1975).
176. R,, Schlierf C. Angew. Chem., 83, 887 (1971).
ЛИТЕРАТУРА
285
177. Peelen F.C., Rietveld G.G.A., Landheer I.JO, de Wolf W.H.,
Bickelhaupt F. Tett. Letts, 4187 (1975).
178. Landheer I. J., de Wolf W.H., Bickelhaupt F. Tett.- Letts, 349 (1975).
179. Roelen O. German Patent 849548 (1938).
180. Roelen 0. Angew. Chem., 60, 62 (1948).
181. Reppe W. Liebigs Ann. Chem., 582, 1 (1953).
182. Bird C.W. Chem. Revs, 62, 283 (1962).
183. Kunichika S.fSabakibara Y°,.Nakamura T. Bull. chem. Soc. Japan,
41, 390 (1968).
184. Rappel J., Umemura S., Sabakibara Y., Blanc IL, Kunichika So
Ind. Eng. Chem. Prod. Res. Dev., 14э 44 (1975).
185. Фалъбе Ю. Синтез на основе окиси углерода (пер. с нем. Богора-
довской Н.М. и Рахлиной Е.Н. под редакцией Имянитова Н. С.).
Л., Химия, 1971.
186. Калия О.Л., Темкин О.Н., Мехрякова Н.Г., Флид Р.М. О новом
механизме карбонилирования ненасыщенных соединений в растворах
комплексов палладия (II). ДАН СССР, 199, № 6, 1321 (1971).
187. Cassar L°, Chiusoli G.P., Guerrieri F. Synthesis, 509 (1973).
188. Thompson D.T., Whyman R. In:Transition Metals in Homogeneous
Catalysis (ed. Schrauzer G.N.), p. 147, New York: Marcel Dekker
(1971).
189. Hayden P. British Patent 1148043(1969).
190. Heck R.F. Organotransition Metal Chemistry, A Mechanistic
Approach, p. 201, New York, Academic Press,1974.
191. Paulik F.EO, Roth J.F. J. chem. Soc., chem. Commun., 1578 (1968).
192. Foster D. Inorg. Chem., 8, 2556 (1969).
193. James B.R., Rempel G.L. J. chem. Soc., chem. Commun., 158 (1967).
194. Roth J.F., Craddock J.H., Hershman A., Paulic F.E., Chem.
TechnoL, 23, 600 (1971).
195. Forster D. J. Amer. chem. Soc., 98, 846 (1976).
196. Cornils B., Payer R., Traenckner K.C. Hydrocarbon Processing,
54 (6), 83 (1975).
197. Heck R.F., Breslow D.S. J. Amer. chem. Soc., 83, 4023 (1961).
19&. Wender L, Feldman J., Metlin S., Gwynn B.H., Orchin M. J. Amer,
chem. Soc., 77, 5760 (1955).
199. Orchin M., Rupilius 1У. Catal. Rev., 6, 85 (1972).
200. Whyman R. J. organometal Chem., 94, 303 (1975).
286
ЛИТЕРАТУРА
201. Heck R., Preslow D.S. Chem. Ind. (London), 467 (1960).
202. Natta G. Brennstoff Chem., 36, 176 (1955).
203. M^rko L. Proc. chem. Soc. (London), 67, (1962).
204. Marko L.} Szabo Pl Chem. Tech. (Leipzig), 13, 482 (1961).
205. Slaugh L.H., Mullineaux R.D U.S. Patent 3239569 (1966).
206. Slaugh L.H., Mullineaux R.D. U.S. Patent 3239570 (1966).
207. Slaugh L.H., Mullineaux R.D. J. organometall. Chem., 13, 469(1968).
208. Paulik F.E. Catal. Rrv., 6, 49 (1972).
209. Tucci E.R. Ind. Eng. Chem. Prod. Res. Dev., 9, 516 (1970).
210. Ugo R., Pregaglia G.F.,Andretta A., Ferrari G.F. J. Organometal.
Chem., 30, 387 (1971).
211. HenricTOlive G.,-Olive S. Transition Metal Chem., 1, 77 (1976).
212» Hershman A., Craddock J.H. Ind. Eng. Chem. Prod. Res. Develop.,
7, 226 (1968).
213. Schiller G. German Patent 953,605 (1956).
21^Slaugh L.H., Mullineaux R.D. German Patent Appl., 1,186,455
(1961).
Slaugh L.H., Mullineaux R.D. U.S. Patent 3239566 (1966).
215. Osborn J.A., Young J.F., Wilkinson G. J. chem. Soc., chem.
Commun., 17 (1965).
216. Craddock J.H., Hershman A., Paulic F.E., Roth J.F. Ind. Eng. Chem.
Prod. Res. Dev., 8, 291 (1969).
217. Morris D.E., Tinker H.B. Chem. Technol., 24, 554 (1972).
218. Evans D., Yagupsky G., Wilkinson G. J. chem. Soc. (A), 2660
(1968).
219. Oliver K.L., Booth F.B. Hydrocarbon Processing, 49 (4), 112 (1970).
220. Wilkinson G. U.S. Patent 4108905 (1978).
221. Pruett R.L., Smith J.A. J. org. Chem., 34, 327 (1969).
222. Yagupsky G., Brown C.K., Wilkinson G. J. chem. Soc. (A), 1392
(1970).
223. Brewster E.A.V. Chem. Eng., 83 (24), 90 (1976).
224. Falbe J. J. organometal. Chem., 94, 213 (1975).
225. Fowler R., Connor H., Paehl R.A. Hydrocarbon Processing,55(9),
248 (1976).
226. Chem. Eng., 84(26), 110 (1977).
227. Hershman A., Robinson K.K., Craddock J.H., Roth J.F. Ind. Eng.
Chem. Prod. Res. Dev., B, 372 (1969).
ЛИТЕРАТУРА
287
228- Алексеева К.А., Высоцкий МД., Имянитов М.С., Рыбаков В.А. -
ЖВХО им. Д.И. Менделеева, 22, 45 (1977).
229- Sanchez-Delgado R.A., Bradley J.S., Wilkinson G. J. chem, Soc,,
Dalton, 339 (1976).
230- Hsu C.Y., Orchin M.J. J. Amer, chem, Soc», 97, 3553 (1975),
231» Schwager I.. Knifton J.F. J, Catal,, 45, 256 (1976).
232, Bailar J.C., Itatani H. J, Amer, chem, Soc,, 89, 1592 (1967)»
233. TayimH.A., Bailar J.C. J, Amer, chem, Soc,, 89, 3420 (1967),
234» Kawabata Y., Hayashi T., Ogata I. J, chem, Soc», chem, Commun.,
462 (1979),
235» Tanaka M., Ikeda Y., Ogata I. Chem, Letts, 1115 (1975),
236, Davidson P.J., Hignett R.R., Thompson D.T. In:Catalysis, Vol, 1,
p, 369, London: The Chemical Society, 1977.
237, Consiglio G, Pino P. Helv. Chin, Acta, 59, 642 (1976),
238, Lefebvre G., Chauvin Y. Aspects Homog. Catal,, 1, 107 (1970),
239, Jolly P.W., Wilke G. The Organic Chemistry of Nickel, Vol, 2,
New York: Academic Press, 1975*
240, Wilke G. Angew, Chem. Int, Ed. Engl,, 5, 151 (1966),
241. Tolman C.A. J, Amer, chem. Soc., 92, 4217 (1970).
242. Bogdanovic В., Непс B., Kermann H.G., Nussel H.G., Walter D.,
Wilke G. Ind. Eng. Chem., 62, 34 (1970).
243» Rarmann H. G. Ph.D. Thesis, University of Bochum-41970).
244, Keim Ж. InlTransition Metals in Homogeneous Catalysis (ed.
Schrauzer G.N.), p, 59, New York: Marcel Dekker (1971),
245, Tsuji J., Adv. organometal Chem., 17, 141 (1979),
246.эBrenner W., Heimbach P., Hey H., Muller E.W., Wilke G. Justus
Liebigs Ann. Chem., 727, 161 (1969).
247, Maitlis P.M. The Organic Chemistry of Palladium, Vol. 2, p. 40,
London: Academic Press, 1971.
248* Barker R. Chem. Revs, 73, 487 (1973),
249. Heimbach P., Jolly P.W., Wilke G. Adv. organometal Chem., 8, 29
(1970).
250. Medema D., van Helden R. Rec, Trav. Chim. Pays-Bas,, 90,
304 (1971),
Medema D., Helden R. Rec. Trav. Chim.Pays-Bas,, 90, 324 (1971).
252. Takahashi H., Yamazaki H.> Hagihan N. Bull, chem, Soc, Japan,
Liebigs Ann. Chem., 41, 254(1968).
288
ЛИТЕРАТУРА
253- Hidai М., Ishiwatari Н., Yagi Н., Tanaka Е., Onozawa К., Uchida у.
J- chem- Soc-, chem- Commun-, 170 (1975)-
254- Cramer R. J- Amer- chem- Soc-, 89, 1963 (1967)-
255. Cramer R. Accounts chem- Res-, 1, 186 (1968)-
256- Su A.C.L. Adv- organometal- Chem-, 17, 269 (1979)-
257- Bogdanovic В., Непс B., Losler A., Meister B., Pauling H., Wilke G.
Angew- Chem- Int- Ed- Engl-, 12, 954 (1973)-
258- Chemicals Information Handbook,1977 — 78, London: Shell Interna-
tional Chemical Company Ltd-, 1979-
259- Ziegler К.» Holzkamp E., Briel H., Martin H. Angew- Chem-, 67,
541 (1955)-
260- Wilke G. In:Coordination Polymerization (ed. Chien J-C-W-), p. 1,
New York: Academic Press, 1975.
261. Natta G. J- Polymer Sci-, 16, 143 (1955)-
262- Natta G., Pino P., Corrandi P., Danusso F., Mantica E., Mazzanti G.,
Moraglio G. J- Amer- chem- Soc-, 77, 1708 (1965)-
263- Boor J. Macromol- Revs, 2, 115 (1967)-
264- Cault A.D. In: Catalysis, Vol- 1, p- 234, London: Chemical Society,
1977-
265- Chiert J.C.W. Coordination Polymerization, New York: Academic
Press, 1975-
266- Cossee P. J- Catal-, 3, 80 (1964)-
267- Arlman E.J. J- Catal-, 3, 89 (1964)-
268. Arlman E.J., Cossee P. J. Catal., 3, 99 (1964)-
269- Henrici-Olive G., Olive S. Angew- Chem- Int- Ed- Engl-, 6, 790
(1967).
270- Armstrong D.R.,Perkins P.G.,Stewart J.J.P. Rev- Roumaine Chim-,
19, 1695 (1974)-
271. Novaro 0., Chow S., Magnouat P. J- CataL, 41, 91 (1976)-
272. Novaro 0., Chow S., Magnouat P. J- CataL, 41, 131 (1976)-
273. Armstrong D.R., Perkins P.G., Stewart J.J.P. J- chem- Soc-, Dalton,
1972 (1972)-
274- Miller S.A. Ethylene and its Industrial Derivatives, London: E- Benn
Ltd-, 1969-
275- Hydrocarbon Processing, 54(11), 183 (1975)-
276- Hydrocarbon Processing, 54 (11), 189 (1975)-
277. Hydrocarbon Processing, 54 (11), 191 (1975)-
ЛИТЕРАТУРА 289
278- Hydrocarbon Processing, 54 (11), 188 (1975)-
279- Rasmussen D.M. Chemo Eng., 79, 104 (1972)-
280- Hydrocarbon Processing, 54 (11), 195 (1975)-
281. Hydrocarbon Processing, 54 (11), 196 (1975)-
282. Zambelli A. In:Coordination Polymerization (ed- Chien J-C-W-), p-
15, New York*. Academic Press, 1975-
283- Natta G., Corradina P. Chim. Ind- (Milan), 45, 299 (1963)-
284. Pino P., Oschwqld A.,Ciardelli F., Carlini C.,Chiellini E., In:
Coordination Polymerization (ed- Chien J.C-W-), p- 25, New York:
Academic Press, 1975.
285- Natta G., Pasquon L.f Zambelli A. J- Amer- chem- Soc-, 84, 1488
(1962)-
286- Уотерс У.Л. Механизм окисления органических соединений.
(Пер. с англ. Бутина К.П. под ред. Несмеянова А.Н.), М., Мир,
1966, ^5 с.
287- Sheldon R.A., Kochi J.K. Adv- Catal-, 25, 272 (1976)-
288- Stern E.W. In:Transition Metals in Homogeneous Catalysis, ed.
Schranzer G-N-, p- 93, New York: Marcel Dekker, 1971.
289- Sheldon R.A., Kochi J.K., Adv- Catal-, 25, 272 (1976)-
290- Tanaka K. Amer- chem- Soc-, Petrol Chem- Div-, Prepr-, 19(1),
103 (1974)-
291- Haber F., Weiss J. Naturwiss, 20, 948 (1932)-
292- Towle P.H., Baldwin R.H. Hydrocarbon Processing, 43, 149 (1964)-
293- Creen H.S. In: Basic Organic Chemistry, Part 5, Industrial Products
(ed- Tedder J-M-, Nechvatal A-, Jubb A-H-), p- 61, London: John
Wiley, 1975-
294- Reich L., Stivala S.S. Autoxidation of Hydrocarbons and Polyole-
fins, p- 271, New York: Marcel Dekker, 1969.
295- Kamiya У. J- Catal-, 33, 480 (1974)-
296- Olah G.A. Chem- Brit-, 8, 281 (1972)-
297. Tanaka K. Chem- Technol-, 555 (1974)-
298. Stobaugh R.B., Calarco KA., Morris R.A., Stroud L. Hf. Hydrocar-
bon Processing, 52, 99 (1973).
299- Sheldon R.A., van Doom J. A. J. Catal., 31, 427 (1973)-
300- Sheldon R.A. Rec. Trav- Chim. Pay-Bas, 92, 253 (1973)-
301- Phillips F.C. Amer- chem- J-, 16, 255 (1894)-
302. Smidt J., Hafner W., Jira R.. Sedlmeier J., Sieber R., Rutlinger R.,
290
ЛИТЕРАТУРА
Kojer Н. Angew* Chem*, 71(5), 176 (1959)*
303* Smidt J., Hafner E., Jira R., Sieber R., Sedlmeier J., Sabel A.
Angew* Chem*, 74(3), 93 (1962)*
304* Henry P.M. Adv* organometal* Chem*, 13, 363 (1975).
305* Bdckvall J.E.,Akermark B., Ljunggren S.GL J * Amer* chem* Soc*,
101, 2411 (1979)*
306. Henry P.M. J* Amer* chem* Soc*, 86, 3246 (1964)*
о
307* Bdckvall J.E., Akermark B., Ljunggren S.O. J* chem* Soc*, chem*
Commun, 264 (1977)*
308* Stille J.K., Divakaruni R. J* organometal* Chem*, 169, 239 (1979)*
309* Jira R., Sedlmeier J., Smidt J. Ann* Chem*, 693, 99 (1966)*
310* Smidt J., Jira R., Sedlmeier J., Seiber R., Ruttinger R., Kojer H.
Angew* Chem* Int* Ed* Engl*, 1, 80 (1962).
311. Maitlis P.M. The Organic Chemistry of Palladium, Vol* 2, p* 141,
London! Academic Press, 1971 о
312* Henry P.M. Adv* organometal* Chem*, 13, 363 (1975)*
313. Hydrocarbon Process, 54, (11), 100 (1975)*
314* Henry P.M. J* org* Chem*, 32, 2575 (1967)*
315* Schultz R.G.,Cross D.E. Adv* chem., 70, 97 (1968)*
316. Francois Po Ann* Chim. (Paris), 4(14), 371 (1969)*
317* Francois P., Trambouze Y. Bull* Soc* Chim. Fr*, 51 (1969)*
318* Maitlis P.M. The Organic Chemistry of Palladium, Vol* 2, p* 89,
London! Academic Press, 1971 о
319* Lyons J.E. In: Aspects of Homogeneous Catalysis (ed* Ugo R*),
Vol* 3, p* 1, Dordrecht: Reidel (1977)*
320* Rooney J.J., Stewart A, In:Catalysis, Vol* 1, p* 277, London!
Chemical Society (1977)»
321* Peters E.F.,Evering B.L. U.S* Patent, 2,963,447 (I960),
322. Banks R.L., Bailey G.C. Ind. Eng* Chem* Prod* Res* Dev*, 3, 170
(1964)*
323* Calderon N., Chen R.Y., Scott K.W. Tett. Letts, 3327 (1967)*
T
324* Muetterties E.L., Busch M.A. J* chem* Soc*, chem* Commun*, 754
(1974)*
323* Grubbs R.H., Burk P.L., Carr D.D. J* Amer* chem* Soc*, 97, 3265
(1975)*
326- Wolovsky R. , Nir Z. J* chem* Soc*, chem* Commun*, 302 (1975)*
327* Bilhou J.L., Basset J.M. J* organometal* Chem*, 132(3), 395 (1977)
ЛИТЕРАТУРА
291
328» Mol J.С., Moulijn J.A., Boelhouwer C. ;J. chem* Soc», chem. Common.,
633 (1968).
329. Clark A.,Cook C. J. Catal., 15, 420 (1969).
330. Bradshaw C.P.C., Howmann E.J., Turner L. J.Catal., 7, 269 (1967).
331. Lewandos G.S., Pettit R, J. Amer. chem. Soc., 93, 7087 (1971).
332. Grubbs R.H., Brunck T.K., J. Amer. chem. Soc., 94, 2538 (1972).
333. Cardin D.J., Cetinkaya B., Doyle M.J., Lappert M.F. Chem. Soc.
Revs, 2, 139 (1973).
334. Calderon N., Ofstead E. A., Judy W.A. Angew. Chem. Int. Ed.
Engl., 15, 401 (1976).
335. Katz T.J. Adv. organometal. Chem., 16, 283 (1977).
336. Katz T.J., McGinnis J.J. J. Amer. chem. Soc., 99, 1903 (1977).
337. Cardin D.J., Doyle M.J., Lappert M.F. J. chem. Soc., chem. Com-
mun., 927 (1972).
338. Casey C.P., Burkhardt T.J. J. Amer. chem. Soc., 95, 5833 (1973).
339. Casey C.P., Burkhardt T.J. J. Amer. chem. Soc., 96, 7808 (1974).
340. Casey C.P.f Tuinstra H.E., Saeman M.C. J. Amer. ohem. Soc. 98,
608 (1976).
341. Katz T.J., Hersh W.H. Tett. Letts, 585 (1977).
342. Katz T.J., Lee S.J., Acton N. Tett. Letts, 4247 (1976).
343. Mol J.C., Moulijn J. Adv. Catal., 24, 131 (1975)..
344. Archibald J.I.C., Rooney J.J., Steward A., J. chem. Soc., chem.
Commun.,547 (1975).
345. Grubbs R.H , Hoppin C.R. J. chem. Soc., chem. Commun., 634 (1977).
346. Muetterties EOL> Inorg. Chem., 14, 951 (1975).
347. Ephritikhine M.»: Green MOL<>,- MacKenzie R.E. J. chem* Soc., chem.
Commun., 619 (1976).
348. Olsthoom AaA., Boelhouwer Co J. Catal., 44, 207 (1976).
349. Schrock R.R. J. Amer. chem. Soc., 98, 5399 (1976).
350. Schrock RoRo Paper presented at the Central Region Meeting of
the American Chemical Society (cited in 48), 1977.
35L Krusic P0Jo, Klabunde IL, Casey COPO> Block T.Fo, J. Amer. chem.
Soc., 98, 2015 (1976).
352. Brookhart M., Nelson GO. J. Amer, jzhem. Soc., 99, 6099 (1977).
353. Katz T.J.,; McGinnis J. J. Amer. chem. Soc., 97, 1592 (1975).
354. Streck R. Chem. Ztg., 99p 397 (1975).
355. Hydrocarbon Processing, 46(11), 232 (1976).
292
ЛИТЕРАТУРА
356.Rossz R, Chimica e Industria, 57, 242 (1975).
357. Haas F., Niltzel К,/ Pampus G./ Theisen D. Rubber Chem. Tech.,
43, 1116 (1970).
358. DalFAsta G, Rubber Rev., 47, 511 (1974).
359. Graulich WSwodenk W,» Theisen D,< Hydrocarbon Processing,
51(11), 71 (1972)»
360. Ofstead EoA0 Abstracts of the 4th International Rubber Symposium,
2, 42 (1969).
361. Crain D.L. U.S. Patent 3,463,828 (1969).
362. Harrison R.C. U.S. Patent 3,567,789 (1971).
363. Kubicek D0H, U.S. Patent 3,728,407 (1973)»
364. Hallum A, U.S. Patent 2, 723,495 (1973).
365. Solomon P,WO U.S. Patent 3,763,240 (1973).
366. Gray ROAO/ Brady D.G. U.S. Patent 3 , 755,227 (1973).
367. van Dam P.P,, Mittelmeijer M,C,, Poelhouwer C. J. chem. Soc.,
chem. Commun., 1221 (1972).
368. Mol J.C'j Woerlee EoF.G, J. chem. Soc., chem. Commun., 330 (1979).
369. Freitas E0R,, Gum C,RO Chem. Eng. Progs., 75(1), 73 (1979).
370. Andrew SOPOSO Education in Chem., 15(4), 114 (1978).
371. Kovaly ЕСА» Chem. Eng., 345<? 417 (1979).
372. Richards R.L, Education in Chem., 16(2), 66 (1979).
373. Chatt J,/ Leigh G0J3 Chem. Soc. Revs5 % 121 (1972).
374. Chatt J,/Dilworth J,R^ Richards RaL. Chem. Revs, 78(6), 589
(1978).
375. Hoffman R,/Chen M,M°Lo/ Thorn D,L> Inorg. Chem., 16, 503 (1977).
376. Chatt J,/Pearman A,J./Richards FLL/ J. chem. Soc., Dalton ^853
(1977).
377. Chatt Crabtree R^H,/ Jeffery E,A0, Richards RaLo/ J* chem. Soc.,
Dalton, 1167 (1973).
378. Chatt Jo/ Richards ROL> J. Less Common Metals, 54, 477 (1977).
379. Manriquez J, Mo, Per caw JE, J. Amer. chem. Soc., 96, 6229 (1974).
380. Manriquez J,Ma/.Sanner R.D,, Marsh RaEo, Percaw JOEO J. Amer,
chem. Soc., 98, 8351 (1976).
381. Manriquez J.Mo, Sanner ROD./Marsh R.E», Percaw J.E. J. Amer,
chem. Soc., 98, 3042 (1976).
382. Borodko YuoGo/ Proitman М,О,/ Kachapina K.L. /Shilov A.E./Ukin
L. Yuo J. chem. Soc., chem. Commun, 1185 (1971).
ЛИТЕРАТУРА
293
383. Schrauzer GONO Angew. Chem. Int. Ed. Engl., 14, 514 (1975).
384. Schrauzer Go/yoji Robinson PORO, Moorehead E.L., Vickrey T.M. J
Amer, chem, Soc., 98, 2815 (1976).
385. Shilov ABE., Denisov N. To, Efimov ОoN*,. Shuvalov N*R0/ Shuva-
lova N.I., Shilova A.K. Nature (London), 231, 460 (1971).
386. Denisov N*T°>‘.Shilov AoEoy Shuvalova NoE? Panova ToP° React.
Kinet. Catal. Letts, 2e 237 (1975).
387. Oil in Perspective. London: Shell International Petroleum Co. Ltd.,
1978.
388. The Coal Option. London: Shell International Petroleum Company
Ltdb, 1978.
389. Сторч Г., Голамбик H., Андерсон Р. Синтез углеводородов из
окиси углерода и водорода (пер. с англ. Кагана Ю.Б. и др. под
ред. Башкирова А» Н\), М., Изд-ве*, иностр, лит-ры, 1954, 516 с.
390. Bussemeier ро,Frohning C.Do, Cornlis Ро Hydrocarbon Processing,
55(11), 105 (1976).
391. Casey CoP,? Neumann SoM° J. Amer. chem. Soc., 98, 5395 (1976).
392. Tam Wo, Wong Wo°Ko? Gladysz J,AQ J. Amer. chem. Soc., 101, 1589
(1979).
393. Sweet JOR.? Graham IL/l.Go J. organometal. Chem., 173, C9 (1979).
394. Connor JoAa Topics in Current Chem., 71, 71 (1977).
395. Casey C 0P о, Andrews M.AO, Rinz J0E0 J. Amer. chem. Soc., 100, 741
(1979).
396. Labinger JOAO, Wong KoS ° ^Scheldt WORO J. Amer. chem. Soc., 100ff
3254 (1978).
397. Manriquez JOMO, McAlister DoRo, Sanner R°DO> Bercaw JOE. J. Amer,
chem. Soc., 100. 2716 (1978).
398. Masters Co, van der Woude Co, van Doorn J0A0 J. Amer. chem. Soc.,
101, 1633 (1979).
399. Fischer Fo? Tropsch FE Brennstoff Chemie, 4, 276 (1923).
400. Casey CoPo, Neumann SoMo J. Amer. chem. Soc., 99, 1651 (1977).
401. van Doorn JoAo, Masters Co? Volger FECo J. organometal.Chem., 105,
245 (1976).
402. Fischer Е.О, Plabst D„ Chem. Ber., 107, 3326 (1974).
403. Casey C.P,, Anderson R.L J. chem. Soc., chem. Commun., 895
(1975).
404. Schrock R.R„ Sharp P,R, J. Amer. chem. Soc., 100, 2389 (1978).
294
ЛИТЕРАТУРА
405. Herrmann Plank Jo, Ziegler Mob.: Weidenhammer Ko, J. Amer,
chem. Soc., 101, 3133 (1979).
406. Gresham WOFO British Patent, 655,237 (1951).
407. Gresham W.F, US Patent 2,636,046 (1953).
408. Pruett R.b. Walker W.E. US Patent, 3,833,634 (1974).
409. Pruett Rob. Walker W.E. US Patent, 3,957,857 (1976).
410. Kaplan b US Patent, 3,944,588 (1976). ;
411. Thomas Peier BOFO. Muetterties E.b J. Amer. chem. Soc.,
98, 1296 (1976).
412. Doyle MoJa, Kouwenhoven AoPa, Schaap C.A.. van Oort Po J. organo-
metal. Chem., 174r C55 (1979).
413. Chatt ]a. Davidson J.Mo J. chem. Soc., 843 (1965).
414. Cotton FoAo, Frenz poAo, Hunter DoEo J. chem. Soc., chem. Commun.,
755 (1974).
415. Webster Dob Adv. organometal. Chem., 15, 147 (1977).
416. Parshall G.U7. In: Catalysis, Vol. 1, p. 335, London: Chemical
Society, 1977.
4Л7. Cheney AoJ0, Mann B.Eo. Shaw R.b, Slade RoMoJo J. chem. Soc.
(A), 3833 (1971).
418. Shaw Pob J. Amer. chem. Soc.., 97, 3856 (1975).
419. Kiffen AoA., Masters Co. Raynand b J. chem. Soc., Dalton, 853
(1975).
420. Garnett Job. Kenyon R0Sa J. chem. Soc., chem. Соштшц 1227 (1971).
421. Kramer PoAb Masters Co J. chem. Soc., Dalton, 849 (1975).
422. English AoDo, Herskovitz To J. Amer. chem. Soc., 99, 1648, (1977).
423. Tolman CoAo, Ittel Sob, English AoDo, Jesson JoPo, J. Amer. chem. Soc.,
100, 4080 (1978).
424. Ittel SoDa. Tolman Co A^ English Aob. Jesson JoPo J. Amer. Chem. Soc.,
100, 7577 (1978).’
425. Cotton FoA..\ LaCour To, Stanislowski A.G. J. Amer. chem. Soc.,
96, 754 (1974).
426. Cotton bA,, Stanislowski A.G. J. Amer. chem. Soc., 96, 5074 (1974).
427. Wolliams J°Mo. Prown RoK., Schultz Ao J Stucky Gob, Ittel SaDB
J. Amer. chem. Soc., 100, 7407 (1978).
428. Dobsbn R Co, Hayes DoMo. Hoffman Ro J. Amer. chem. Soc., 93O
6188 (1971).
429. Podor No^Dewar M,J.S..: Wasson JOSO J. Amer, chem, Soc., 94, 9095
(1972).
ЛИТЕРАТУРА
295
430. Kochi J0K. Organometallic Mechanisms and Catalysis, New York:
Academic Press, 1978.
431. Голъдшлегер Н.Ф^Тябин Ч.Б., Шилов A.E., Штейкман Л.Л. Акти-
вация насыщенных углеводородов. Дейтероводородный обмен в
растворах комплексов переходных мета л лов Ж. физич. химии,
1969, 43, 2174-2175.
432. van Dongen ].Р.С.М., Masters С., Visser J.P. J. organometal.
Chem., 94, С29 (1975).
433. Visser J. P., Jager W.W., Masters C. Rec. Trav. Chim. Pays-Bas,
94, 70 (1975).
434. Puddephatt R.J., Thompson P.J. J. organometal.Chem., 166, 251
(1979).
435. Muetterties E.L. Bull. Soc. Chim. Belg., 84(10), 959 (1975).
436. Visser J. P., Masters C. Seventh International Conference on Organo-
metallic Chemistry, Venice, Abstract paper Nr. 245, 1975.
437. Schwartz J,, Hart D.W. J. Amer. chem. Soc., 96, 8115 (1974).
438. Smith A.K., Passet J.M. J. molec. Catal., 2, 229 (1977).
439. Muetterties E.L., Rhodin T.N., Band E.> Brucker C.F., Pretzer W.R.
Chem. Revs, 79(2), 91 (1979).
440. Hartley F.R., Vezey P.N- Adv. organometal.Chem., 15, 189 (1977).
441. Yermakov Y.l. Catal. Rev., 13, 77 (1976).
442. Lehmkuhl Н» In:Organic Electrochemistry, p. 621, New York: Dek-
ker, 1973.
443. Gilet M., Mortreux A., Nicole J., Petit F. J. chem. Soc., chem,
Commun., 521 (1979).
444. Sprintschnik G., Sprintschnik N.W., Kirsch P.P., Whitten D.G.
J. Amer. chem. Soc., 98, 2337 (1976).
445. Okwra I., Kim-Thuan N. J. molec. Catal., 5, 311 (1979).
446. Brown G.M., Brunschwig B.S., Creutz C., Endicott J.F., Sutin N.
J.Amerochem. Soc., 101, 1298 (1979).
447. Manriquez J.M., Fagan P.J., Marks T.J. J. Amer. chem. Soc., 100,
3939 (1978).
448. Manriquez J,M., Fagan P.J., Marks T.J., Day C.S., Day V.W.
Jo АшеГо chem. Soco, 100, 7112 (1978)о
Содержание
V7 1 '1У11ХС1 оооооооооооэоооооеоооооооооееоо
ПрСДИСЛОВИО оооооооооооооооо.оо.ооооо.ео....о 7
1 о Общее рассмотрение . . . . . . <> . . . . . о . . . . . . . . . » . . , 9
1.1. Искусство катализа, основные принципы................ 9
1.2. Почему "переходные металлы"?........................ 13
1.2.1. Способность к образованию связей............... 13
1.2.2. Широкий выбор лигандов......................... 18
1.2.3. Влияние лигандов............................... 19
А. wpawe-Эффект.................................... 20
Б. Электронные донорно-акцепторные свойства —
электронный параметр v....................... 21
В. Конический угол - стерический параметр 0 . . . . 22
1.2.4. Способность к вариации степени окисления .... 26
1.2.5. Способность к вариации координационного числа 27
1.3. Почему "гомогенный"?................................ 27
1.4. Активация молекул................................... 31
1.4.1. Активация путем координации................ 31
1.4.2. Активация путем присоединения.............. 32
А. Окислительное присоединение..................... 32
S. Гомолитическое присоединение.................... 34
В. Гетеролитическое присоединение.................. 35
1.5. Близкое взаимодействие.............................. 37
1.5.1. Внедрение и миграция внутренних лигандов .... 37
1.5.2. Элиминирование................................. 40
1.6. Каталитический цикл................................. 41
1.6.1. Правило « 16 и 18 электронов»................... 42 .
1.7. Заключение................г......................... 44
Дополнительная литература............................. 45
2 . Гомогенные каталитические системы в действии.... 47
2.1. Гидрирование........................................ 49
2.1.1. Простое гидрирование........................... 50
СОДЕРЖАНИЕ
297
А. Активация водорода путем окислительного присо-
единения ........................................ 50
Б. Активация водорода путем гомолитического при-
соединения ...................................... 55
В. Активация водорода путем гетеролитического
присоединения................................... 60
2.1.2. Селективное гидрирование...................... 65
А. Гидрирование простых алкенов и алкинов..... 65
Б. Гидрирование сопряженных диенов, полимеров и
активированных мономеров......................... 66
В. Гидрирование ароматических и гетероцикличес-
ких соединений................................... 67
Г. Гидрирование альдегидов и кетонов............. 68
Д. Гидрирование нитрогрупп....................... 69
2.1.3. Асимметрическое гидрирование.................. 69
А. Асимметрия при фосфоре........................ 70
Б. Лиганды с асимметрическим центром, связанным
с атомом фосфора................................. 73
2.1.4. Гидрирование путем переноса водорода....... 76
Дополнительная литература по гомогенному гидрирова-
нию........................................ 79
2.2. Изомеризация...................................... 79
2.2.1. Изомеризация алкенов................^. . . 80
А. Изомеризация, протекающая через образование
металлалкильных интермедиатов.................... 80
Б. Изомеризация через металлаллильные интерме-
диаты ........................................ 85
В. Изомеризация через другие интермедиаты ... 89
2.2.2. Скелетная изомеризация........................ 91
А. Скелетная изомеризация, протекающая по согла-
сованному механизму.............................. 92
Б. Скелетная изомеризация, протекающая через
стадийный процесс.............................. 94
Дополнительная литература по изомеризации, катализи-
руемой переходными металлами......................... 95
2.3. Карбонилирование.................................. 98
2.3.1. Карбонилирование алкинов...................... 99
298
СОДЕРЖАНИЕ
2.3.2. Карбонилирование алкенов.................... 103
2.3.3. Карбонилирование спирта..................... 105
Дополнительная литература по гомогенному карбонилиро-
ванию .................................................. 111
2.4. Гидроформилирование............................. 111
2.4.1. Кобальтовые катализаторы.................... 113
А. ^модифицированные кобальтовые карбонильные
системы......................................... 113
Б. Кобальткарбонильные системы, модифицированные
третичными фосфинами........................ 123
2.4.2. Родиевые катализаторы....................... 130
2.4.3. Сравнение кобальтовых и родиевых систем..... 137
2.4.4. Катализаторы на основе других металлов...... 139
2.4.5. Асимметрическое гидроформилирование......... 144
Дополнительная литература по гомогенному гидроформили-
рованию ................................................. 145
2.5. Олигомеризация......................................... 146
2.5.1. Никелевые комплексы как катализаторы олигомери-
зации алкенов...................................... 147
А. Олигомеризация этилена; получение димеров и оли-
гомеров ................................... 150
Б. Олигомеризация пропилена; региоселективность . . 153
2.5.2. Олигомеризация бутадиена........ 157
А. Системы, содержащие никель(О); циклоолигомери-
зация ...................................... 162
Б. Системы, содержащие Pd(0); линейная олигомери-
зация ..................................... 165
2.5.3. Содимеризация этилена с бутадиеном.......... 166
2.5.4. Асимметрическая олигомеризация.............. 169
Дополнительная литература по гомогенной олигомеризации . . 170
2.6. Полимеризация.......................................... 171
2.6.1. Катализаторы Циглера - Натта................ 172
2.6.2. Полиэтилен.................................. 179
А. Процессы в растворе или в суспензии................ 180
Б. Газофазная полимеризация этилена................... 181
2.6.3. Полипропилен....................................... 182
СОДЕРЖАНИЕ 299
Дополнительная литература по полимеризации и катализу
Ниглера — Натта......................................... 187
2о7о Окисление ..............
2.7.1. Гомолитическое окисление . .................. 189
А. Автоокисление ароматических соединений на-при-
мере п-ксилола.................................... 191
Б. Автоокисление алканов на примере циклогексана . 194
2.7.2. Гетеролитическое окисление.................... 198
А. Эпоксидирование, катализируемое переходными
металлами, на примере эпоксидирования пропи-
лена ........................................ 198
Б. Окисление алкенов, катализируемое палладием, на
примере окисления этилена....................... 203
Дополнительная литература......................... 213
2.8. Метатезис.......................................... 214
2.8.1. Механизм метатезиса алкенов.................. 216
А. Карбены металлов в качестве интермедиатов .... 217
Б. Генерирование металлкарбеновых комплексов ... 221
В. Возможный каталитический цикл................ 226
2.8.2. Практическое применение метатезиса алкенов. . 230
А. Ациклические алкены в качестве субстратов .... 231
Б. Циклические алкены как субстраты............... 233
2.8.3. Каталитическое получение высших олефинов .... 236
Дополн ительная литература по метатезису алкенов .... 238
3. Гомогенные каталитические системы в стадии разработки . 239
3.1. Фиксация азота..................................... 239
3.1.1. Азот, связанный с металлическим центром..... 240
3.1.2. Гидрирование связанного азота................. 241
3.1.3. Гидрирование атмосферного азота . ............ 245
Дополнительная литература по фиксации азота.......... 247
3.2. Восстановительная олигомеризация (полимеризация) СО . 247
3.2.1. Модельные системы и возможный механизм реакции 248
3.2.2. Гомогенные катализаторы...................... 257
Дополнительная литература по гетерогенным и гомоген-
ным реакциям СО и Н2................................. 259
3.3. Активация алканов ................................. 260
3.3.1. Взаимодействие металлов с активированными али-
300
СОДЕРЖАНИЕ
фатическими СН-связями........................ 261
А. Стерическая активация....................... 261
Б. Электронная активация........................ 265
3.3.2. Взаимодействие металла с неактивированными али-
фатическими СН-связями ........................... 268
Дополнительная литература по активации алканов.... 270
4. Что же дальше?....................................... 271
Литература.............................................. 278
Уважаемый читатель!
Ваши замечания о содержании книги, ее оформлении, качестве пе-
ревода и другие просим присылать по адресу: 129820, Москва,
И-110, ГСП, 1-й Рижский пер., д. 2, издательство «Мир».
К: Мастерс
ГОМОГЕННЫЙ КАТАЛИЗ
ПЕРЕХОДНЫМИ МЕТАЛЛАМИ
Научный редактор И. С. Беленькая
Мл. научный редактор И. А. Колчин
Художник Н. Н. Дронова
Художественный редактор М. Н. Кузьмина
Технические редакторы Т. А. Алюлина, Л. А. Тихомирова
Корректор В. И. Киселева
ИБ № 3395
Подписано к печати 18.08.83. Формат 60 х 9046. Бумага офсетная № 1.
Гарнитура тайме. Печать офсетная. Объем 9,50 бум. л. Усл. печ. л. 19,00.
Усл. кр.-отт. 19,35. Уч.-изд. л. 17,22. Изд. № 3/2213. Тираж 3000 экз. Зак.' 8267. Цена 2 р. 70 к.
Набрано в редакции по подготовке оригинал-макетов изд-ва «Мир».
129820, Москва, И-110, ГСП, 1-й Рижский пер., 2.
Типография В/О „Внешторгиздат"
Москва, 127576, Илимская, 7
Издательство "Мир”
выпускает в свет в 1984 году книги по хиыии
Мейтис Л.
Введение в курс химического равновесия и кинетики*: Пер. с англ. —
42 л., 4 р. 50 к.
Учебное пособие по аналитической химии, в котором автор ото-
шел от традиционного изложения материала, учитывая современное
состояние этой области науки — интенсивное развитие ее теории и
практики, тесное переплетение с другими областями химии. В данном
руководстве изложение материала по аналитической химии тесно свя-
зано с основными положениями физической химии.
Для преподавателей и студентов химических и химико-технологи-
ческих вузов.
Сланина 3.
Теоретические аспекты явления изомерии в химии: Пер. с чешек. —
10 л., 1 р. 80 к.
Книга из серии монографий по теории химии. В небольшом по
объему труде чешского ученого явление изомерии, являющееся основ-
ным понятием органической и неорганической химии, рассматривает-
ся с единых позиций термодинамики. Большим достоинством книги
является ее оригинальность, доступность изложения и широта охвата
материала.
Для научных работников — теоретиков и экспериментаторов саг
мых различных химических специальностей, а также для преподавате-
лей и студентов химических вузов.
Заблаговременно оформляйте заказы на интересующие Вас кни=
ги. Заказы принимают магазины, распространяющие научно-техни-
ческую литературу. Своевременно сданный заказ гарантирует приоб-
ретение нужных Вам книг.