/























































































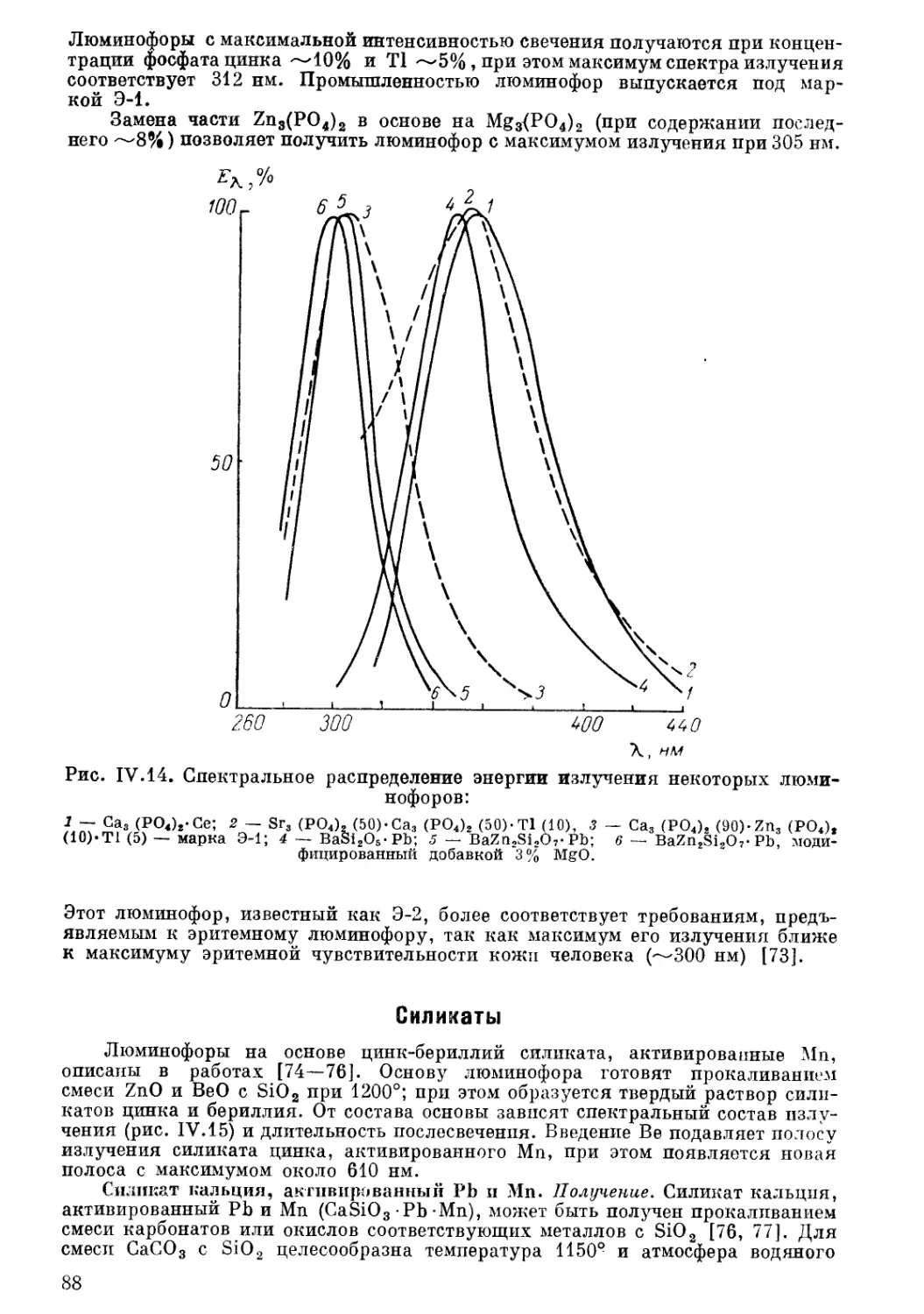









































































































Текст
неорганические
люминофоры
ИЗДАТЕЛЬСТВО „ХИМИЯ"
Ленинградское отделение* 1 9 75
' 4
УДК
621.3.032.35 : 546
Н52
Н52 Неорганические люминофоры. Л., «Химия», 1975.
192 стр., 43 табл., 118 рис., список литературы 572
ссылок.
Люминофоры — «светящиеся» вещества, широко применяемые
во многих отраслях народного хозяйства. В книге описаны методы
получения, физико-химические и электрофизические характеристики
неорганических люминофоров. Большое внимание уделено свойствам
химических веществ, применяемых для изготовления люминофоров.
Отдельно рассмотрены'люминофоры для люминесцентных ламп, катодо-
люминофоры для черно-белого и цветного телевидения, рентгенолюми-
нофоры и многие другие. Приведены рекомендации по способам изы-
скания новых люминесцирующих соединений.
Книга предназначена для инженерно-технических работников
химической и смежных отраслей промышленности, связанных с изго-
товлением или использованием люминофоров и изделий из них, и может
быть полезна студентам вузов соответствующих специальностей.
Н
31403—026
050(01) 75
26-75
Авторы: Казанкин О.Н., Марковский Л. Я.
Миронов И. А., Пекерман Ф. М., Петошина Л. Н.
© Издательство «Химия», 1975
ПРЕДИСЛОВИЕ
Настоящая книга посвящена химии и технологии наиболее важных
в практическом отношении люминофоров. В ней использованы мате-
риалы монографии «Люминофоры», выпущенной в 1966 г. Естественно,
за прошедшие годы как в Советском Союзе, так и за рубежом появились
новые направления в области химии и технологии люминофоров. Начаты
и успешно проводятся работы по синтезу порошковых электролюмино-
форов, возбуждаемых постоянным электрическим полем. Достигнуты
большие практические успехи в области инжекционной электролюмине-
сценции. Разработан ряд люминофоров, положивших начало новому
классу люминофоров, преобразующих инфракрасное излучение в види-
мое, благодаря чему исследования в области «антистоксовской» люмине-
сценции приобрели практический интерес. Ведутся работы по синтезу
катодолюминофоров, возбуждаемых электронами низкой энергии.
Большое внимание в последние годы стало уделяться повышению
эксплуатационной стойкости люминофоров. Успешно ведется поиск
новых люминофоров на редкоземельной основе. Все перечисленное в той
или иной форме нашло отражение в настояшем издании.
ВВЕДЕНИЕ
Люминофорами называют вещества, люминесцирующие * под действием
различного рода возбуждений. Способ возбуждения положен в основу класси-
фикации этих веществ, а именно:
фотолюминофоры — возбуждение ультрафиолетовым, видимым или
инфракрасным светом;
катодолюминофоры — возбуждение пучком электронов;
электролюминофоры — возбуждение электрическим полем;
рентгенолюминофоры — возбуждение рентгеновскими лучами;
радиолюминофоры** — возбуждение корпускулярным излучением
естественных или искусственных радиоактивных веществ.
В настоящее время люминофоры применяют в различных областях народ-
ного хозяйства, однако наиболее широко их используют в люминесцентных
лампах — самых экономичных источниках света. По ориентировочным данным,
мировой выпуск люминесцентных ламп достигает 1 млрд, в год. Производство
такого количества ламп требует ~10 ООО т люминофоров, что составляет не ме-
нее 90% от их общего количества.
Вторая по масштабам область применения люминофоров — цветное и черно-
белое телевидение. Кроме того, люминофоры используют: в экранах осцилло-
графических и радиолокационных трубок, а также электронно-оптических
преобразователей (приборов ночного видения); в приборах Индикации ядерных
излучений и в рентгеновских экранах. Люминофоры необходимы для изгото-
вления светящихся красок временного и постоянного действия, которые исполь-
зуют в световых аварийных и маскировочных указателях, а также для декора-
тивных целей. Можно указать еще не менее десятка других, более специальных
областей применения люминофоров, причем с каждым годом сфера применения
люминофоров расширяется. Так, люминофоры стали незаменимыми для визу-
ализации инфракрасного излучения.
Возникновение у неорганических веществ люминесцентных свойств связано
с образованием в кристаллической решетке соединения (основы люминофора)
в процессе синтеза структурных и примесных дефектов. Первые появляются
в результате термического диспропорционирования основы люминофора и пред-
ставляют собой вакансии и ионы или атомы, расположенные в междуузлиях
кристаллической решетки. Люминесценция, обусловленная такими наруше-
ниями, получила название неактивированнойз* (или самоакти-
вированной), поскольку для ее возникновения не требуется введения
активирующей примеси.
* Определение люминесценции см. на стр. 7.
** Радиолюминофоры не представляют собой специфического класса;
как иравтло, это соединения, обладающие эффективной люминесценцией при
катодном возбуждении.
к* Имеется в виду преднамеренно не активированная люминесценция.
Б ряде случаев образование центров самоактивпрованной люминесценции свя-
зано с присутствием неконтролируемых примесей или с наличием соактпваторов.
Примесные дефекты появляются за счет внедрения при высокой температуре
в кристаллическую решетку основы ионов или атомов посторонних элементов.
Люминесценция, вызванная такими нарушениями, получила название акти-
вированной, а активирующие примеси — активаторов. Большинство
наиболее важных в практическом отношении люминофоров являются активи-
рованными.
С активаторами связывают представление о центрах люминесценции. Хими-
ческое состояние активаторов в кристаллической решетке и структура центров
люминесценции до сих пор являются предметом многочисленных исследований.
Различие в валентном состоянии активатора, вошедшего в решетку основания,
-его положение в ней (замещение иона в узле, междуузельное положение), бли-
жайшее окружение активатора и наличие соактиваторов (примесей, внедря-
ющихся с активатором и связанных с ним) — все это определяет структуру
центра и его свойства.
Ультрафиолетовая энергия, возбуждающая люминофор, может поглощаться
как непосредственно люминесцентными центрами (активаторное или примесное
-поглощение), так и основным веществом люминофора (фундаментальное погло-
щение) .
В первом случае поглощение сопровождается Либо переходом электронов
внутри электронной оболочки активатора на более высокие энергетические
уровни, либо полным отрывом электрона от активатора и переходом активатора
в ионизованное состояние (образуется «дырка»). Во втором случае, при погло-
щении энергии основой, в основном веществе образуются дырки и электроны.
Дырки могут мигрировать по кристаллу и локализоваться на центрах люмине-
сценции. Излучение происходит в результате возвращения электронов на более
низкие (исходные) энергетические уровни Или при воссоединении (рекомбина-
ции) электрона с ионизованным центром (дыркой). Люминофоры, в которых
люминесценция (поглощение и излучение энергии) связана с электронными
переходами в пределах люминесцентного центра, получили название харак-
теристических. Активаторами в таких люминофорах являются ионы
переходных и редкоземельных элементов, а также ртутеподобные ионы. Кри-
сталлическая решетка основы, как правило, мало влияет на электронные пере-
ходы внутри центра, поэтому спектры возбуждения И люминесценции в основном
определяются природой активатора.
Относительная элементарность процесса люминесценции и слабое энергети-
ческое взаимодействие между центром и основой — причина, как правило,
высокого выхода люминесценции у характеристических люминофоров. Поэтому
именно их применяют в газоразрядных источниках света. Основой для таких
люминофоров служат вещества, которые не поглощают возбуждающую и излу-
чаемую энергию. Они имеют широкую запрещенную зону и поглощают в дале-
кой УФ-области. К таким соединениям относятся силикаты и различные фос-
фаты и подобные им соединения (арсенаты, германаты п др.). Концентрация
активаторов в характеристических люминофорах должна достигать нескольких
процентов; объясняется это необходимостью более полного поглощения актива-
тором возбуждающей УФ-энергии.
Люминофоры, в которых люминесценция связана с образованием и реком-
бинацией разноименных зарядов (электронов и дырок), получили название
реком би нацпонных. Основой для них служат соединения полупро-
водникового типа. В этих люминофорах кристаллическая решетка основы
является той средой, в которой развивается процесс люминесценции. Это дает
возможность, изменяя состав основы, широко варьировать свойства люмино-
форов. Изменение ширины запрещенной зоны при использовании одного и того же
активатора плавно в больших пределах Изменяет спектральный состав излучения.
Введение в основу примесей, образующих электронные ловушки разной
глубины, изменяет инерционные свойства люминофоров (время разгорания
и затухания люминесценции). Наличие глубоких ловушек позволяет коммули-
ровать энергию и высвечивать ее инфракрасными лучами (вспышечные люмино-
форы) пли теплом (термолюминесценция).
При поглощении энергии в основном веществе происходит ее передача
к люминесцентному центру, поэтому вероятность потери энергии в рекомбина-
ционных люминофорах больше, чем в характеристических, а выход люминесцен-
ции принципиально нпже.
5
Основой для наиболее важных в практическом отношении рекомбинацион-
ных люминофоров служат халькогениды металлов II группы периодической
системы. Наличие широких областей взаимной растворимости у этого класса
соединений позволяет получать многообразные люминофоры с различными
свойствами.
При возбуждении катодными лучами, корпускулярным и рентгеновским
излучением энергия поглощается в основном кристаллической решеткой люми-
нофора. Поэтому для получения высокого выхода люминесценции при указан-
ных возбуждениях необходимо эффективное энергетическое взаимодействие
между основой и активатором. В связи с этим халькогениды, представляющие
собой полупроводниковые соединения с высокой неравновесной проводимостью,
являются наиболее подходящей основой для синтеза к а т о д о -, рентгено-
и радиолюминофоров. С другой стороны, соединения, представля-
ющие собой хорошие основы для характеристических люминофоров при фото-
возбуждении, но не обладающие высокой проводимостью, не могут применяться
для синтеза указанных люминофоров.
Технология люминофоров относится к области тонкого неорганического
синтеза и характеризуется следующими особенностями:
использованием веществ высокой степени чистоты;
строгой регламентацией условий синтеза;
сравнительно небольшими масштабами производства.
В этой книге химический состав люминофоров обозначается формулами
типа MR-А, где MR — основа люминофора, а А — активатор, напри-
мер ZnS-Си *. При необходимости указать количественный состав люминофора
(в %) относительное массовое содержание компонентов приводится в скобках
после формулы компонента, например ZnS(60) -CdS(40) -Ag(0,02). Иногда в фор-
муле указывается также применяемый плавень и температура синтеза (в ?С),
например ZnS -Ag(0,02) -МаС1(2)800°.
* Иногда, кроме такого способа обозначения, применяют и такие: ZnS—Си
и ZnS : Си.
ГЛАВА I
ОСНОВНЫЕ ПОНЯТИЯ
Люминесценцией называется излучение, избыточное над тепловым излу-
чением тела, если это избыточное излучение обладает длительностью, превы-
шающей период световых колебаний.
Первая часть этого определения, данная Видеманом, подчеркивает отличие
люминесценции от свечения нагретых тел — свечение люминофоров происходит
без нагревания («холодный свет»). Вторая часть определения была введена
Вавиловым [1, с. 282] для отличия люминесценции от свечения при отражении
и рассеянии света, тормозного излучения заряженных частиц и излучения Вави-
лова — Черенкова. Все указанные виды свечения исчезают сразу же после пре-
кращения возбуждения, тогда как люминесценция продолжается в течение
некоторого времени после этого.
Некоторые авторы выделяют из люминесценции флуоресценцию,
под которой подразумевают свечение при возбуждении, и фосфоресцен-
цию — свечение, продолжающееся заметный промежуток времени после
прекращения возбуждения.
Рассмотрим сначала в самом общем виде основные характеристики люмино-
форов [1—9].
1.1. СПЕКТРЫ ПОГЛОЩЕНИЯ И ВОЗБУЖДЕНИЯ
Важная характеристика люминофоров — спектры поглощения, отража-
ющие зависимость величины поглощаемой энергии от длины волны падающего
на люминофор света.
Свет может поглощаться как решеткой основы (полоса поглощения
в этом случае называется основной или фундаментальной),
.так и центрами люминесценции (примесное поглощение).
Поэтому поглощение п спектральная область, в которой оно происходит, впре-
деляются свойствами активатора и кристаллической решетки. У боль-
шинства люминофоров основная полоса поглощения расположена в УФ-области
спектра (рис. 1.1). Как следует из рис. 1.1, граница основной полосы поглоще-
ния ZnS • Си соответствует А 334 нм. Полоса с максимумом при А 360 нм
обусловлена поглощением центрами люминесценции (ионами меди). Увеличе-
ние концентрации меди приводит к росту максимального поглощения.
При изменении состава основы люминофора, например за счет введения Cd
в основу ZnS-люминофоров, граница основной полосы поглощения смещается
в сторону более длинных волн. Замена меди, например, на марганец, приводит
к изменению полосы, соответствующей поглощению на уровнях активатора.
Для люминофоров на основе силикатов, фосфатов, германатов, арсенатов
(см. стр. 46, 51, 80) поглощение в области А = 200—400 нм обусловлено актива-
торами. Вызвано это электронными переходами внутри иона в последних;
энергетическое же состояние ионов активаторов может зависеть от окру-
жения. Так, центрами поглощения галофосфата кальция, активированного Sb
(см. рис. IV.8, стр. 80), являются ионы Sb, но максимум спектра поглощения
зависит от концентрации фтора и хлора, входящих в решетку галофосфата.
7
Иногда у люминофоров с двумя активаторами (например, галофосфат каль-
ция, активированный сурьмой и марганцем), энергия поглощается одним акти-
ватором (Sb), называемым сенсибилизатором, который передает
поглощенную энергию другому активатору (Мп).
Исследования, проведенные для выяснения вопроса: не определяются ли
спектры поглощения активатора спектрами поглощения атома активатора
в свободном состоянии, показали, что такое совпадение присуще люминофорам
с редкоземельными активаторами. Связано это с тем, что для указанных элемен-
тов оптические переходы протекают^
в защищенной /-оболочке [11]. Ана- !
логично ведут себя и ртутеподобные
активаторы [12].
Однако в большинстве случаев
центр люминесценции — это сложное
Рис. 1,1. Спектр поглощения люми-
нофора ZnS-Си [10].
Рис. 1.2. Спектры отражения люмино-
форов [13]:
1 — на основе галофосфата кальция, активи-
рованного Sb и Мп; 2 — MgWO4; 3 — CaSiO3.
• Pb-Mn; 4— Zn,SiO4-Mn.
образование, и на его свойства оказывают влияние окружающие ионы ре-
шетки основы люминофора. Наконец, поглощение может быть обусловлено
вакансиями в решетке основы люминофора, которые образуются в про-
цессе его формирования.
Область поглощения люминофора характеризуют спектрами отражения
(рис. 1.2). Величину поглощения рассчитывают по соотношению:
К по гл — 1 — Аотр
Спектры поглощения в ряде случаев характеризуют суммарное поглощение,
которое складывается из активного, вызывающего люминесценцию, и того,
которое не приводит к возникновению свечения. Кроме спектров поглощения
одной из важных характеристик люминофоров являются спектры воз-
буждения (см. стр. 80, 85, 91, 97), определяющие, в отличие от спектров
поглощения, только область активного поглощения энергии. Спектры возбужде-
ния показывают зависимость интенсивности свечения люминофора от длины
волны возбуждающего света.
1.2. СПЕКТРЫ ИЗЛУЧЕНИЯ
Фотовозбуждение
Спектры излучения люминофоров показывают распределение энергии люми-
несценции по длинам волн.
Согласно Правилу Стокса — Ломмеля, максимум спектра излучения смещен
по отношению к максимуму спектра поглощения в сторону длинных волн
(рис. 1.3). Это обусловлено тем, что часть энергии, поглощаемой люминофором!]
рассеивается в его решетке, переходя в тепло (так называемые ст оксо в|
скиепотери). *
Особое место занимают «антистоксовские» люминофоры, которые
при возбуждении в ИК-области спектра излучают в видимой области, что про-
тиворечит вышеуказанному правилу. Объяснение этого явления и свойств
подобных люминофоров приведены в главе IV.
У большинства описываемых далее люминофоров спектры излучения пред-
ставляют собой широкие полосы. Однако в некоторых случаях у люминофоров
с редкоземельными активаторами спектры люминесценции состоят из харак-
терных узких полос. Расширение полосы обусловлено воздействием ионов
основной решетки на энергетическое состояние ионов активатора.
Спектр излучения люминофоров зависит от химической природы актива-
тора и основы люминофора, их взаимодействия, а в случае люминофоров с не-
сколькими активаторами и от взаимодействия активаторов между собой.
Иногда один и тот же активатор в одной и той же основе дает полосы излу-
чения, расположенные в различных областях спектра. Например, люминофор
ZnS Си излучает в синей, зеленой и красной областях спектра; зависит это
от концентрации меди и условий приго-
товления люминофоров. Спектр люмине-
сценции характеристических
люминофоров также иногда состоит из
нескольких полос, соответствующих
различным электронным переходам дан-
ного центра свечения. Приме-
ром могут служить арсенат и фторгер-
манат магния, активированные Мп. Вве-
дение двух активаторов в одну основу,
например в случае сенсибилизирован-
ной люминесценции, может привести
к конкуренции полос излучения, соот-
ветствующих этим активаторам — возра-
стание интенсивности свечения в полосе одного активатора сопровождается
/00 г
60
20
°440 520 600 680
Лунм
Рис. 1.3. Спектры
поглощения (2)
100
60
S'
20
°200 280 360
излучения (2) и
люминофора
Zn2SiO4 Мп.
уменьшением интенсивности свечения в полосе другого.
Спектральный состав зависит от различий в валентном состоя-
нии активатора, вошедшего в решетку основы, его положения в этой решетке
(замещение иона в узле, междуузельное положение), а также от природы атомов
основы. В качестве примера влияния структуры центра свечения на спектр
излучения следует привести галофосфат кальция, активированный Sb и Мп.
Спектр излучения этого люминофора состоит из двух полос, соответствующих
Излучению Sb И Мп, положение и Интенсивность свечения которых зависит
от концентрации активаторов и соотношения F" и С1“, входящих в состав гало-
фосфата. Влияние галогенов, например С1~, на положение основной полосы Мп
обусловлено тем, что ионы Са2+ могут четырьмя различными способами окружать
Ионы Мп2+. В свою очередь, эти ионы Са2+ по-разному связаны с сурьмой, фто-
ром, хлором или кислородом.
Спектры излучения могут зависеть от интенсивности и длины
волны возбуждающего света, а также от температуры. Влияние интенсивности
возбуждающего света проявляется тогда, когда люминофор имеет несколько
полос излучения и интенсивность свечения каждой из них
по-разному зависит от интенсивности возбуждающего света, в частности, интен-
сивность одной Из полос быстрее достигает насыщения. Примером может служить
люминофор ZnS - Си [14], который при концентрации активатора 0,002—0,003%
в спектре излучения имеет две полосы, интенсивность свечения которых I зави-
сит от интенсивности возбуждающего света Е но закону I —Е^, где а, в свою
очередь, различно зависит от температуры для синей и зеленой полос (рис. 1.4).
Зависимость спектров излучения от длины волны возбуждающего света
может быть хорошо прослежена на характеристических люминофорах (Са,
Mg)3(PO4)2-Sn и (Са, Zn)3(PO4)2 -Sn. Спектры излучения последних состоят
Из широких полос, расположенных в красной и зеленой областях [15]. При
возбуждении излучением с X = 254 нм появляется, главным образом, красная
Полоса, а при X = 313 нм — зеленая (рис. 1.5).
9
Влияние температуры на спектры излучения люминофоров различных
классов подробно рассмотрено Студером и Гансом [16]. По данным этих авторов,
при повышении температуры для различных люминофоров наблюдаются сле-
дующие изменения в спектрах
излучения:
расширение спектра без
смещения положения его мак-
симума (ZnO, Zn2SiO4-Mn);
расширение спектра излу-
чения и смещение максимума
в длинноволновую область
(ZnS-Ag, ZnSCdS-Ag);
смещение спектра излуче-
ния в коротковолновую об-
ласть (MgWO4);
перераспределение интен-
сивности свечения отдельных
полос [(Zn, Be)2SiO4 Мп].
Зависимость спектра излу-
чения от температуры для
многополосных люминофоров
объясняется температур-
ным тушением люми-
несценции, которое различно
для различных полос.
а
Рис. 1.5. Зависимость спектров излучения
люминофора (Са, Zn)3(PO4)2-0,08Sn от темпе-
ратуры.
“ - Чозб = 254 нм: 6 - Чозб = 313 нм:
1 — 20°; 2 — 100°; 3 — 200°; 4 — 300°; 5 — 350°.
Рис. 1.4. Зависимость степени
нелинейности стационарной
яркости свечения голубой (7)
и зеленой (2) полос люмино-
фора ZnS Си от температуры.
Для характеристического люминофора (Са, Zn)3(PO4)a -Sn при повышении
температуры интенсивность зеленой полосы уменьшается, а красной — увели-
чивается (см. рис. 1.5).
Такое различное поведение полос, возможно, объясняется тем, что центры
красной люминесценции обусловлены ионами олова, расположенными в твердом
растворе фосфата цинка в фосфате кальция, имеющего ^-структуру, а зеленые
центры связаны с ионами олова в фосфате кальция, имеющем а-структуру [15].
Возбуждение катодными лучами
При возбуждении катодными лучами спектры излучения люминофоров
с одним активатором, как правило, не меняются с изменением условий возбу-
ждения, т. е. величин ускоряющего напряжения и плотности тока. Изменение
наблюдается только в том случае, если люминофор имеет несколько полос излу-
чения [7J. Так, у люминофора ZnS-Си [6, с. 227] при малых концентрациях
10
меди и повышении плотности тока уменьшается интенсивность длинноволновой
полосы излучения по отношению к коротковолновой (рис. 1.6).
У многополосных люминофоров спектры при фото- и катодовозбуждении
Иногда различаются из-за того, что при большей плотности тока возбуждения
в последнем случае длинноволновая полоса может быть подавлена. При фото-
возбуждении свет может проникать
глубже в кристаллы люминофора и
излучение в коротковолновой части
спектра может ослабляться в боль-
шей степени вследствие поглощения
центрами люминесценции.
Как и в случае фотовозбуждения,
спектр излучения при катодовозбуж-
дении зависит от температуры, если
Рис. 1.6. Зависимость спектра излу-
чения люминофора ZnS Си от плот-
ности тока:
1 <* 100 мкА; 2 — 250 мкА.
Л, нм
в нем есть несколько полос излучения, обусловленных, например, присут-
ствием нескольких активаторов.
Изменение спектра может быть результатом расширения или сужения его
или разрешения отдельных полос.
Так, в спектре ZnS-Tm, возбуждаемого катодными лучами [7] при —150®
проявляется линейчатая структура.
Возбуждение электрическим полем
Спектры излучения сульфидных электролюминофоров, возбу-
ждаемых переменным напряжением, определяются природой основы и актива-
торов, а также концентрацией последних (рис. 1.7). Кроме того, спектр э л е к -
т^р олюминесценции зависит от. частоты возбуждающего поля и для
Рис. 1.7. Спектральное распределение энергии излучения ZnS-электро-
люминофоров при частотах возбуждающего поля 400 Гц (а) и 5000 Гц (б):
1 — ZmS«Cu (0,03%); 2 — ZnS*Си (0,05%); 3 — ZnS-Cu-Al; 4 — ZnS-Си-Мп;
5 — (Zn.Cd) (S.Se)-Cu.
некоторых люминофоров от величины приложенного напряжения (см. ссылку
[36], гл. IV). Особенно резко эта зависимость проявляется в том случае,
когда электролюмйнофор имеет несколько полое излучения. Из рис. 1.8 следует,
11
что по мере повышения частоты в спектрах электролюминесценции увеличи-
вается доля коротковолнового излучения. Объяснение этому дано в рабо-
тах [17, 18].
Резкое изменение спектра излучения при изменении частоты наблюдается
и для электролюминофора ZnS-Си Мп (см. стр. 134). Связано это с тем, что
Рис. 1.8. Изменение спектраль-
ного распределения энергии
излучения электролюминофора
ZnS -Си при изменении ча-
стоты возбуждающего поля.
интенсивность излучения в разных полосах различным образом зависит от ча-
стоты и поэтому область насыщения может наступать при различных значениях
последней.
Электролюминофоры, возбуждаемые постоянным электрическим полем,
полученные на основе ZnS Или ZnS • ZnSe при активации марганцем, свинцом
и редкоземельными элементами, имеют спектр излучения, характерный для
электронных переходов внутри иона активатора [19].
1.3. ВЫХОД ЛЮМИНЕСЦЕНЦИИ
Фотовозбуждение
Энергетическим выходом люминесценции называют отно-
шение энергии люминесценции к поглощенной энергии:
Вэн = Ея/Ей
Поскольку часть поглощаемой люминофором энергии превращается в тепло,
постольку величина энергетического выхода характеризует полноту преобра-
зования энергии возбуждения в энергию люминесценции. Наибольший выход
следует ожидать в том случае, когда энергия поглощается непосредственно самим
центром люминесценции. Если же энергия поглощается основным веществом,
например в случае рекомбинационных люминофоров, то при этом образуются
электроны и дырки, которые при перемещении по решетке могут захваты-
ваться ловушками. Это, а также безызлучательная реком-
бинация дырок с электронами, приводит к уменьшению энергетического
выхода.
Кроме энергетического выхода, для характеристики эффективности транс-
формации поглощенной люминофором энергии возбуждения введено понятие
квантового выхода.
Квантовым выходом называют отношение числа квантов, излучаемых
люминофором (TVji), к числу поглощенных квантов возбуждающего света
Вкв — Nu/N п
Если обозначить частоты, соответствующие максимумам спектров излуче-
ния и поглощения через ул и vn, то:
_ Ел hv^Nn ул
^ЭН — ~р— в АТ -^кв ~
Еп “VnJVn Уп
12
В случае квантового выхода не учитывают энергетические потери при
стоксовском смещении спектра излучения по отношению к спектру поглощения.
Величина выхода существенно зависит от состава люминофора, технологии его
изготовления и, кроме того, от присутствия посторонних примесей и концентра-
ции активатора. Наконец, квантовый выход может зависеть от условий возбу-
ждения, длины волны и интенсивности возбуждающего света, а также от тем-
пературы.
Квантовый выход люминесценции характеристических люминофоров в пре-
делах длин волн, соответствующих полосе поглощения, не зависит от длины
волны возбуждающего света (рис. 1.9, кривая 7). Этого нельзя сказать о реком-
бинационных люминофорах (рис. 1.9, кривые 2 и 5): для самоактивированного
люминофора ZnS (кривая 2) в области основной полосы поглощения выход
постоянен; в области поглощения центров он возрастает и затем уменьшается.
При активации ZnS медью (кривая 3) появляется второй максимум.
Рис. 1.9. Зависимость выхода люминесценции
от длины волны возбуждающего света [20,21]:
1 — Zn2SiO4«Mn; 2 — ZnS-Zn; 3 — ZnS*Си.
Рис. 1.10. Зависимость выхода
люминесценции от интенсив-
ности возбуждающего света
[22].
Зависимость выхода люминесценции от интенсивности возбуждающего
света (рис. 1.10) связана, по-видимому, с соотношением между количеством
ионизованных центров при данной интенсивности возбуждающего
света и числом глубоких ловушек. Например, при малой интенсивности возбу-
ждающего света число ионизованных центров меньше или сравнимо с числом
ловушек, их действие становится заметнее и приводит к уменьшению выхода.
В 1947 г. Вавилов указал, что при возбуждении квантами с высокой энер-
гией можно получить квантовый выход больше 1. Впоследствии это было под-
тверждено экспериментально [23, 24]. Кроме того, оказалось, что генерация
одним фотоном нескольких электронно-дырочных пар приводит к излучению
нескольких квантов с меньшей энергией. Это явление получило название фо-
тонного умножения. Для люминофоров ZnS -Си и ZnS Мп оно
начинается в области 11 эВ. Для люминофора ZnS Мп абсолютный квантовый
выход при энергии возбуждения 21,2 эВ достигает 3. Фотонное умножение
наблюдается у люминофоров, для которых возможна эффективная передача
энергии от основы к центрам люминесценции и малые приповерхностные потери
энергии.
Катодное возбуждение
Величину энергии, поглощаемой люминофором при катодном возбуждении,
трудно определить из-за того, что часть электронов, падающих на люминофор,
рассеивается его поверхностью. Кроме того, в процессе возбуждения возникает
вторичная электронная эмиссия. Поэтому катодолюминофоры
характеризуют величиной свето- и энергоотдачи. Первая •— это отношение
величины светового потока, излучаемого люминофором, к подводимой энергий,
а вторая — отношением излучаемой энергии к подводимой. Энергоотдача
13
зависит от ускоряющего напряжения и плотности тока [7]. Из-за больших потерь
выход люминесценции при катодном возбуждении невысок и у лучших сульфид-
ных люминофоров не превышает 20—25% (см. табл. IX. 5, стр. 179).
Возбуждение электрическим полем
Эффективность электролюминесцентных изделий, в которых применяют
порошкообразные электролюминофоры (электролюминесцентные конденсаторы —
ЭЛК), оценивают величиной светоотдачи, т.е. отношением мощности
светового потока, излучаемого ЭЛК, к величине поглощаемой им мощности.
Светоотдача зависит от свойств используемого электролюминофора и свя-
зующего диэлектрика, а также от условий возбуждения (частоты и напря-
Рис. 1.11. Зависимость све-
тоотдачи электролюмино-
фора ЭЛ-510 от напряжен-
ности возбуждающего поля.
жения электрического поля). Типичная кривая
зависимости светоотдачи от напряженности воз-
буждающего поля показана на рис. 1.11. Вели-
чина напряженности, обеспечивающая макси-
мальную светоотдачу, определяется свойствами
используемого люминофора и методом приготов-
ления ЭЛК [25]. Резко выраженного максимума
на кривой зависимости светоотдачи от частоты
возбуждающего поля не наблюдается [25, 26].
При изменении частоты в пределах 2000—
4000 Гц, светоотдача остается постоянной. По-
вышение концентрации электролюминофора в
слое диэлектрика приводит к увеличению свето-
отдачи, при этом наблюдается смещение макси-
мума светоотдачи в сторону более низких частот
и напряженностей возбуждающего поля. При
увеличении размера частиц ЭЛК светоотдача
уменьшается и одновременно максимум ее сме-
щается в сторону более низких напряженностей возбуждающего поля [25,
27].
Существенное влияние на светоотдачу ЭЛК оказывают свойства применя-
емого диэлектрика [28]. При малых значениях диэлектрической проницаемости
(8 = 2,5) увеличение тангенса угла потерь может привести к увеличению яркости
без существенного уменьшения светоотдачи. Но в реальных случаях увеличение
тангенса угла потерь диэлектрика повышает яркость ЭЛК и при этом снижает
его светоотдачу.
При непрерывной работе ЭЛК наблюдается уменьшение светоотдачи, но
несколько меньшее, чем снижение светового потока. Объясняется это тем, что
при непрерывной работе ЭЛК уменьшается не только величина излучаемого
светового потока, но и поглощаемая ими мощность.
Формулы, связывающие светоотдачу ЭЛК с напряжением и частотой возбу-
ждающего поля, полученные на основании общих представлений о механизме
электролюминесценции, приводятся в работах [29—31].
Верещагин [31] измерял энергетический выход для ЭЛК, приготовленных
с различными фракциями электролюминофоров. Проведенный автором теорети-
ческий анализ позволил установить величину максимально возможного энер-
гетического выхода для отдельно взятого зерна электролюминофора в зависи-
мости от его размеров при фиксированной напряженности электрического поля.
Так, при диаметре частиц —7 мкм, энергетический выход оказывается рав-
ным 1,5%.
В ряде работ отмечается, что максимальная величина светоотдачи для ЭЛК
не может превышать 14—18 лм/Вт. Для промышленных ЭЛК величина свето-
отдачи не превышает 5—7 лм/Вт *.
* Для перехода от величины светоотдачи к величине энергетического вы-
хода следует перевести люмены в единицы мощности (Вт) с учетом коэффициента
видности для данного спектра излучения.
14
Выход излучения,
эффективность и яркость свечения
светодиодов
Выход излучения светодиода измеряют в световых единицах — люменах
или 1 кандела на 1 м-2 [(1 кд-м-2) = 1 нит], отнесенных к единице мощ-
ности (Вт). Величина выхода обычно имеет размерность — лм/Вт (с в е to-
ot д а ч а) или лм/А и кд-м-2/(А-см-2); отношение яркости свечения
к плотности тока позволяет сравнивать между собой диоды различной формы
и размера.
Энергетическая эффективность светодиода — безразмерная величина; ее
определяют как отношение мощности светового потока к мощности подводимой
электроэнергии.
Важным параметром является также квантовая эффектив-
ность, измеренная как отношение числа излученных квантов к числу элек-
тронов, прошедших через р—«-переход. Часто ее выражают в процентах [(число
квантов/число электронов)-100]. Энергетическая (т]₽) и квантовая (т]9) эффектив-
ности связаны между собой соотношением
где hv — энергия кванта, эВ;
U — напряжение смещения, В.
Различают внутреннюю и внешнюю квантовую эффективность.
Первая характеризует непосредственно излучательную рекомбинацию, а вто-
рая есть доля первой за вычетом потерь, связанных с выводом излучения из
кристалла.
В случае сильно поглощающегося излучения с энергией кванта, близкой
к Eg, величина внешней квантовой эффективности может составлять лишь сотую
долю от величины внутренней. Величина внешней квантовой эффективности
для различных светодиодов лежит в пределах 0,01—15%. Потери на поглощение
сильно увеличиваются при многократном отражении от внутренней поверх-
ности кристалла. Коэффициент преломления большинства ма-
териалов для светодиодов составляет 3,5 и угол полного внутреннего отражения
очень мал (16—17°), поэтому значительная часть излучения не выходит из кри-
сталла. Для улучшения световывода применяют покрытия с большим
коэффициентом преломления, а также придают кристаллу форму полусферы
или конуса. Это уменьшает долю излучения, падающего на внутреннюю по-
верхность кристалла под большим углом, и, следовательно, способствует
выводу света из кристалла.
Яркость свечения может быть рассчитана, исходя из величины
квантовой эффективности по уравнению:
В = 39300
^•тах 1’и3л
где В — яркость свечения, кд м-2;
L — средний коэффициент видности, лм/Вт, рассчитанный из спектра
излучения и кривой видности;
/ —плотность тока, А-см-2;
^гпах — длина волны в максимуме спектра, нм;
5Пер и >$и3л — площади р—«-перехода и поверхности, через которую выво-
дится излучение.
Приведенная формула пригодна в ограниченном интервале плотностей
тока, так как для большинства светодиодов характерны некоторые отклонения
зависимости яркость — ток от линейности.
15
1.4. ИНТЕНСИВНОСТЬ СВЕЧЕНИЯ ЛЮМИНОФОРОВ
Фотовозбуждение
Обычно на практике для технической характеристики люминофоров сравни-
вают интенсивности свечения исследуемого люминофора и люминофора, приня-
того за эталон (при одних и тех же условиях возбуждения).
В том случае, если люминофор излучает в видимой области спектра, интен-
сивность излучения может быть измерена в единицах яркости *.
У характеристических люминофоров интенсивность люминесценции воз-
растает пропорционально увеличению интенсивности возбуждающего света.
У люминофоров рекомбинационного типа зависимость между интенсивно-
стями излучения и возбуждения является более сложной [4, с. 19; 32]. Послед-
нее обусловлено тем, что при возбуждении подобных люминофоров центры све-
чения и элементы решетки основы ионизуются. При этом электроны могут за-
хватываться ловушками, освобождаться и рекомбинировать с центрами свече-
ния, дырками или повторно захватываться ловушками.
* 140
Температура, К
Рис. 1.12. Зависимость интенсивности свечения некоторых люминофоров от
температуры:
1 — ZnS (48)-CdS (52)-Ag (0,01); 2 — ZnS (60)-ZnSe (40); 3 — ZnS (60)-ZnSe (40)-Ag (0,005);
4 — ZnS (60)-ZnSe (40)-Cu (0,005); 5 — ZnS (88)-CdS (12)-Cu (0,008); 6 — ZnS-Ag (0,01) •
. Cu (0,005),
Антоновым-Романовским [32] показано, что при кратковременном (импульс-
ном) возбуждении люминофоров ZnS-Си интенсивность люминесценции I про-
порциональна квадрату интенсивности возбуждающего света Е : I — Е2.
Для некоторых люминофоров типа ZnS -CdS - Ag с добавками Ni характерна
сверхлинейная зависимость интенсивности люминесценции от интенсивности
возбуждающего света [33].
Интенсивность свечения люминофоров зависит от температуры (рис. 1.12).
Характер этой зависимости определяется составом основы люминофора, хими-
ческой природой активатора и присутствием так называемых гасителей
люминесценции. В определенном температурном интервале при повышении
температуры происходит уменьшение интенсивности свечения (температурное
тушение люминесценции).
Резкое уменьшение интенсивности свечения люминесценции при нагрева-
нии наблюдается у ZnS -CdS Ag -Ni [33]. Для целого ряда характеристических
* Метод измерения интенсивности и яркости излучения изложен на стр. 171.
Единица яркости в настоящее время имеет размерность —кд м-2, но, кроме
того, и нит : 1 нит = 0,1 миллистильба (мсб) = 10-4 стильба (сб) — 3,14 апо-
стильба (абс) = 0,314 миллиламберта (мламб) = 3,14 10~4 ламберта (ламб) =
= 0,296 фут-ламберта.
16
люминофоров (арсенат и германат магния, активированный Ni, кальций-цинк
фосфат, активированный Sn) при повышении температуры до 200—300° наблю-
дается повышение интенсивности свечения и затем только ее уменьшение.
Это свойство дает возможность использовать указанные люминофоры в лам-
пах высокого давления, где температура колбы, на которую наносится люмино-
фор, может достигать 200—300°.
Возбуждение катодными лучами
Зависимость интенсивности свечения I от плотности тока электронного
пучка у и приложенного напряжения V выражается формулой
1=/с/(у)(Г-У0)’
где к — константа, зависящая от природы люминофора;
/ (у) — функция, выражающая зависимость интенсивности свечения от плот-
ности тока пучка электронов;
Fo — «мертвое напряжение» (то минималь-
ное напряжение, которое необхо-
димо для прохождения электроном
поверхностного слоя).
Величину q большинство авторов счи-
тают близкой к единице, вообще же для раз-
ных люминофоров она лежит в пределах 1—3.
При определенных значениях плотности тока
интенсивность свечения достигает предела
(насыщение). Эти значения зависят от состава
люминофора [например, для люминофора
Zn2SiO4Mn насыщение достигается при
у = 10, а для ZnS -Ag при у = 200 мкА-см"* 2
(рис. 1.13)]. Эффект насыщения почти не за-
висит от энергии электронов. Насыщение при
Рис. 1.13. Зависимость интенсивности свече-
ния люминофоров от плотности тока элект-
ронного пучка (V = 15 кВ):
1 — Zn-Ag; 2 — ZnS-CdS-Lu; 3 — ZnS-CdS-Ag-A ;
4 — ZnS-Tm; 5 — Sr3 (PO4)s-Eu.
увеличении плотности тока электронного пучка обусловлено, во-первых, воз-
никновением на поверхности люминофора заряда, тормозящего электроны, и,
во-вторых, нагреванием экрана, следствием чего является температурное ту-
шение люминесценции.
Возбуждение электрическим полем
Исследование электролюминесценции цинксульфидных электролюмпно-
форов под действием переменного поля показало, что зависимость инте-
гральной* яркости электролюминесценции от возбуждающего
напряжения выражается формулой [8, с. 287; 9, с. 191; 34—38]
Л -b/VV
В = Ае
где А и Ъ — постоянные;
V — приложенное напряжение.
* Интегральной яркостью электролюминесценции называют среднее зна-
чение яркости свечения за период изменения приложенного напряжения; мгно-
венная яркость электролюминесценции — это Знаиение япкости свечения в дан-
ный момент времени.
2 Заказ 4 4
17
Согласно этой формуле, зависимость 1g В от 1/ \ V представляет собой
прямую линию, наклон которой определяется составом основы электролюмино-
фора, размером его кристаллов, а также природой и концентрацией активатора.
Леман {35J установил, что чем меньше размер кристаллов, тем круче идет кри-
вая зависимости яркости свечения от напряжения. Исследование изменения
мгновенной яркости электролюминесценции (так называемые волны
яркости) во времени [8, с. 190; 34, 36—38] показало, что в каждый полу-
период возбуждающего напряжения волны яркости состоят, как правило, из
двух пиков; первичного и вторичного (рис. 1.14). В большинстве случаев макси-
мум первичного пика несколько смещен относительно максимума приложенного
напряжения, вторичный пик появляется в тот момент, когда значение напря-
женности поля проходит через нуль. Форма волн яркости и фазовый сдвиг пер-
вичного и вторичного пгков зависят от амплитуды и частоты приложенного
напряжения и температуры. Из осциллограмм (рис. 1.14) видно, что при малых
напряжениях первичный пик больше вторичного. По мере возрастания напря-
жения изменяется соотношение амплитуд обоих пиков и появляются дополни-
тельные пики. Одновременно волны яркости все больше смещаются по фазе
по отношению к приложенному напряжению.
Рис. 1.14. Зависимость формы волн яркости от напряжения воз-
буждающего поля для электролюминофора ZnS Си [37].
60 В
Появление волн яркости объясняют следующим [34, 36, 38]. В течение каж-
дого полупериода на участке кристалла электролюминофора, который соприка-
сается с катодом, происходит ионизация центров свечения. Часть электронов при
этом не успевает рекомбинировать с центрами люминесценции и поле «отго-
няет» их к другому краю кристалла. В следующий полупериод электроны
возвращаются и наступает рекомбинация, сопровождающаяся излучением.
В этом видят причину появления вторичных пиков на волнах яркости.
Согласно работе [36], первичный пик на волнах яркости формируется
путем возвращения к ионизованным центрам свечения электронов, которые были
захвачены ловушками на противоположном краю кристалла в предыдущий
период. При этом электроны могут высвобождаться полем.
Помимо переменной составляющей на осциллограмме волн яркости электро-
люминесценции наблюдается постоянная составляющая, которая, возможно,
обусловлена излучением в объемной части кристалла [38].
При повышении частоты (рис. 1.15) интегральная яркость свечения вначале
увеличивается почти линейно, а затем наступает насыщение. Частотная зави-
симость интегральной яркости изменяется при введении в люминофор приме-
сей Fe, Со и Ni; при некоторой концентрации этих примесей она становится
сверх линейной. Люминофоры, содержащие большие количества Fe, Со и Ni,
фотолюминесценция которых почти потушена, обладают яркой электролюмине-
сценцией при высоких частотах.
Зависимость интегральной яркости электролюминесценции от температуры
описана в работах [9, с. 197; 39—41]. Для электролюминофоров на основе ZnS -Си
эта зависимость в большинстве случаев описывается кривой с двумя максиму-
мами, из которых один расположен в области отрицательных, а другой —
в области положительных температур (рис. 1.16). При увеличении частоты
последний перемещается еще дальше в область положительных температур.
Как следует из работы [41], кривая зависимости яркости электролюмине-
сценции от температуры не совпадает с таковой для фотолюминесценции и, сле-
довательно, в обоих случаях протекают различные процессы. Максимумы на
кривых температурной зависимости яркости электролюминесценции обычно
1S
не совпадают по положению с максимумами на кривых термического
высвечивания (КТВ) при возбуждении электрическим полем. В этом
случае КТВ имеют один максимум (—0,3 эВ). Таким образом, возрастание
яркости свечения электролюминесценции при повышении температуры нельзя
просто объяснить термическим освобождением электронов из ловушек.
Возможное объяснение температурной зависимости яркости электролюмине-
сценции следует из работы [9, с. 197]. Автор предполагает, что возбуждение
электролюминофоров ZnS-Си наступает в кристалле ZnS в области сильного
поля у барьера, соответствующего переходу между фазой Cu2S и ZnS
(см. главу VI). При повышении температуры число электронов у барьера в об-
ласти сильного поля возрастает; это увеличивает ток через кристаллы, число
ионизаций и яркость свечения. Однако при некоторой температуре рост тока
Рис. 1.15. Зависимость интегральной
яркости свечения ZnS-электролюми-
нофоров от частоты возбуждающего
поля:
1 — ZnS-Си-Мп; 2 — ZnS-Си (0,05); 3 —
ZnS-Cu (0,03); 4 —ZnS-Cu-Al; 5 — (Zn,
Cd) (S, Se)-Cu.
Рис. 1.16. Зависимость интенсивности
свечения люминофора ZnS - Си от тем-
пературы при фото- (7) и электровоз-
буждении (2).
вызывает перераспределение напряжения, подводимого к кристаллам: в толще
кристалла оно начинает увеличиваться, а на барьере — уменьшаться. Проте-
канием этих процессов объясняют основной максимум на кривой зависимости
яркости электролюминесценции от температуры.
Дополнительный же максимум на этой кривой, может быть, по мнению
автора, обусловлен двумя типами барьеров внутри кристаллов и между кристал-
лами (максимум при низких температурах зависит от гранулометрического со-
става люминофора).
1 5. РАЗГОРАНИЕ И ЗАТУХАНИЕ ЛЮМИНЕСЦЕНЦИИ
Фотовозбуждение |
Законы разгорания и затухания люминесценции различны для люминофоров
разных классов. При возбуждении характеристических люминофоров светом
происходит постепенное нарастание интенсивности люминесценции, которая
через некоторое время достигает стационарного значения (рис. 1.17). После
выключения возбуждения число возбужденных центров п в процессе затухания
начнет уменьшаться по закону
где а — вероятность уменьшения числа возбужденных центров; а = 1/т, при-
чем т — время жизни атома в возбужденном состоянии.
Отсюда:
п = noe~ai — пое~^т
2*
19
Интенсивность излучения изменяется по такому же закону
Л = ДГ'/Т
где It и Zo — интенсивности свечения в момент времени t и в начальный момент
после прекращения возбуждения.
Скорость затухания у характеристических люминофоров не
зависит от интенсивности возбуждающего света и температуры.
По экспоненциальному закону затухают люминофоры на основе силикатов,
фосфатов, арсенатов, германатов. При наличии у характеристических люмино-
форов двух активаторов (например, га-
лофосфат кальция, активированный Sb
и Мп), свечение каждого из них зату-
хает по экспоненциальному закону.
Следует отметить, что для люмино-
форов указанного типа не всегда точно
соблюдается экспоненциальный харак-
тер затухания. Иногда (например,
Zn2SiO4 - Мп) на начальных стадиях за-
тухание происходит по экспоненте,
Рис. 1.17. Кривые разгорания и зату-
хания свечения характеристических
люминофоров.
Рис. 1.18. Кривые разгорания и за-
тухания свечения люминофоров ре-
комбинационного типа.
а на последующих — по гиперболе.В этой области время затухания зави-
сит от температуры.
В случае рекомбинационных люминофоров процессы разгорания
и затухания происходят по другим законам. В данном случае в процессе раз-
горания число ионизованных центров и рекомбинаций электронов с ионизован-
ными центрами увеличивается.
Расчет кинетики такого процесса, проведенный Антоновым-Романов-
ским [4, с. 19] показывает, что интенсивность люминесценции I при разгорании
сложным образом зависит от времени $ и интенсивности возбуждающего света Е:
V^Ep® \2
/Epft /
где и — постоянная, пропорциональная коэффициенту поглощения;
р — вероятность рекомбинации.
Как следует пз рис. 1.18 свечение достигает стационарного значения через
некоторое время тем более длительное, чем меньше интенсивность возбужда-
ющего света. Насыщение достигается тогда, когда число актов рекомбинации
становится сравнимым с числом актов ионизации. Отметим, что время, в течение
которого интенсивность люминесценции рекомбинационных люминофоров дости-
гает стационарного значения, намного больше, чем в случае характеристических
люминофоров.
2ч
Исследование [42] разгорания люминофоров ZnS-Си и ZnS-Ag [43] пока-
,. зало, что на начальных стадиях оно протекает по закону, близкому к экспонен-
циальному
причем т зависит от интенсивности возбуждения и уменьшается при ее увели-
чении.
По выключении возбуждающего света затухание свечения люминофоров
рекомбинационного типа носит сложный характер [2, с. 339; 3, с. 40; 4, с. 13].
После возбуждения электрон покидает центр свечения и может либо рекомби-
нировать с каким-нибудь ионизованным центром, либо быть захваченным ло-
вушкой. Послесвечение обусловлено тем, что электроны могут теплом
освобождаться из ловушек и рекомбинировать с ионизованными центрами до
тех пор, пока не опустошатся все ловушки. Следует отметить, что послесвече-
ние у таких люминофоров как ZnS -Си, ZnS -Си -Со, SrS -Си - Bi может длиться
часами после выключения возбуждения.
Теоретическое рассмотрение [3, с. 30; 4, с. 28] даже простой модели с ло-
вушками одного сорта позволяет процесс свечения представить как бимолеку-
лярный. После выключения возбуждения число рекомбинаций в единице объема
в единицу времени равно
dn/dt = —рп2
где t — время;
р — постоянная.
Интегрирование этого уравнения дает
1 +
где п0 — начальная концентрация ионизованных центров люминесценции.
Поскольку интенсивность свечения I ~ рп2, то, принимая во внимание
результат интегрирования, находим:
(1-|-Pno02
Теоретически закон затухания определяется выражением, которое соответ-
ствует гиперболе второго порядка. У реальных люминофоров закон затухания
обычно отклоняется от указанной зависимости.
Антонову-Романовскому [4, 43] удалось показать, что затухание свечения
отдельных кристаллов ZnS Си в течение определенных промежутков времени
происходит точно по гиперболе второго порядка.
В общем случае кривые затухания свечения могут иметь начальный экспо-
ненциальный участок и участок, на котором интенсивность люминесценции
уменьшается согласно эмпирической формуле Беккереля:
/о
(1 -J- at )п
(1 < п ^2)
Исследование затухания люминофоров ZnS-Си и ZnS-Ag [42] показало,
что на начальных стадиях закон затухания отличается от закона Беккереля,
причем время, в течение которого наблюдается отклонение, уменьшается при
Увеличении интенсивности возбуждающего света. На дальних стадиях закон
затухания переходит в гиперболический. Отклонение закона затухания от про-
стого гиперболического объясняется тем, что в люминофорах существуют ло-
вушки различной глубины, и кинетика свечения зависит от распределения элек-
тронов между центрами люминесценции и ловушками. Из расчетов, проведенных
Фоком [3, с. 43], следует, что в том случае, когда большая часть электронов
из зоны проводимости попадает не на ловушки, а рекомбинирует
С ионизованными центрами, закон затухания будет экспоненциальным (это
соответствует начальному участку на кривой затухания). По мере затухания
люминесценции число ионизованных центров уменьшается и вероятность
21
локализации электронов на ловушках становится больше вероятности их рекомби-
нации с ионизованными центрами. В этом случае закон затухания будет гипер-
болическим (второй участок на кривой затухания).
Ход кривой затухания свечения зависит от интенсивности возбуждающего
света и температуры. Чем больше интенсивность возбуждающего света, тем
быстрее происходит затухание. С понижением температуры затухание заме-
дляется.
Как следует из вышесказанного, характер затухания определяется энерге-
тическим распределением ловушек для электронов и дырок, что, в свою очередь,
зависит от химической природы основы,
активатора, соактиватора, температуры
и длительности прокаливания люмино-
форов.
Зная законы разгорания и затуха-
ния свечения, можно определить так на-
зываемые светосуммы по разго-
ранию и затуханию (рис. 1.19) [2, с. 313;
Рис. 1.20. Зависимость длительности
затухания свечения люминофора
ZnS-Ag от температуры:
I — спад яркости свечения до 0,1% от
начальной величины; II— до 0,001%.
Рис. 1.19. Кривые нарастания и зату-
хания люминесценции.
3, с. 56]. За время возбуждения люми-
нофор высвечивает энергию, пропорцио-
нальную площади F. Площадь F меньше
площади прямоугольника О АВ С на
величину площади Е, которая пропорциональна энергии, запасаемой люмино-
фором. Эта энергия, не высвеченная люминофором при его возбуждении, назы-
вается светосуммой люминофора по разгоранию; она может быть высвечена лю-
минофором после прекращения возбуждения. Энергия, пропорциональная
площади D, высвечиваемая люминофором после прекращения возбуждения,
называется светосуммой люминофора по затуханию. Опыт показывает, что
светосумма по затуханию обычно меньше светосуммы по разгоранию. Это сви-
детельствует о существовании безызлучательных переходов, т. е. о неполном
превращении запасенной люминофором энергии в световую.
Катодное возбуждение
Разгорание и затухание катодолюминофоров имеет чрезвычайно большое
практическое значение. Так, для многих люминофоров, применяемых, например,
в телевизионных трубках и трубках с бегущим лучом, необходим короткий
период послесвечения. И наоборот, люминофоры, используемые в радиолока-
ционных устройствах, должны иметь длительный период послесвечения.
При возбуждении катодным пучком люминофоров, люминесценция кото-
рых вызвана переходом внутри центра, затухание подчиняется иногда экспо-
ненциальному закону. В ряде случаев этот закон сохраняется только на ранних
стадиях. Следует отметить, однако, что скорость нарастания свечения и затуха-
ния не зависит от условий возбуждения и температуры. На дальних стадиях
процесс затухания может подчиняться гиперболическому закону. Типичным
примером являются люминофоры ZnF2 Mn и Zn2SiO4Mn [6]. .
22
Затухание люминофоров на основе ZnS, ZnS-CdS, ZnS-ZnSe при акти-
вации Си, Ag, Аи подчиняется закону, близкому к гиперболическому (рис. 1.20).
Скорость затухания зависит от условий возбуждения и температуры и возрастает
при увеличении 'ускоряющего напряжения, нлотности тока и температуры.
Возрастание плотности тока, с одной стороны, приводит к увеличению коли-
чества электронов и дырок, захватываемых на ловушках, а с другой, — усили-
вает выброс уже локализованных на ловушках зарядов. В результате высвечи-
вание ускоряется.
Значительное снижение длительности послесвечения происходит при вве-
дении в основу сульфидных люминофоров метал лов-гасителей
люминесценции, в частности никеля. По данным работы |7], у сульфидных
люминофоров наблюдаются два типа кривых затухания: простые и сложные.
Первые подчиняются формуле Беккереля
В работе [7] на ряде люминофоров с различной длительностью послесвече>
ния показана непосредственная связь этого параметра с глубиной ловушек
в люминофоре. Вероятность теплового освобождения электронов резко умень-
шается с увеличением глубины ловушек, например, почти в 1000 раз при увели-
чении глубины уровня локализации от 0,25 до 0,52 эВ.
Некоторые данные по длительности послесвечения важнейших катодо-
люминофоров приведены в главе V (стр. 123, 125).
Возбуждение электрическим полем
Исследование разгорания электролюминесценции при возбуждении импуль-
сами синусоидального напряжения и импульсным напряжением прямоуголь-
ного типа показало, что после включения напряжения амплитуда волн яркости
и постоянная составляющей устанавливаются только через некоторое время.
Так, у электролюминофора Zn-Cu амплитуда переменной составляющей
волн яркости достигает постоянного значения через 5—15 циклов, а величина
постоянной составляющей — через 200—400 циклов [38].
Кинетика спада свечения в голубой и зеленой полосе люминофора
ZnS-Cu-Al исследована в работе [44]. Обнаружено, что затухание подчиняется
сложному закону, который только на некотором участке является экспонен-
циальным. На кинетику спада свечения, при возбуждении прямоугольным
импульсом влияет длительность импульсов приложенного напряжения. При
сокращении длительности скорость затухания увеличивается. Время затухания
волны яркости зависит от частоты приложенного напряжения сложным обра-
зом [45]. При / = 100 Гц т = 1,2 мс (т — время спада до уровня яркости,
равного 10% от начальной). Уменьшение частоты приводит к росту т; при / =
= 0,1 Гц, т = 1,8 мс, затем наблюдается резкий спад т.
Особый интерес представляют люминофоры ZnS Мп, возбуждаемые по-
стоянщям полем [19]. Спад излучения у них неэкспоненциальный. Время спада
излучения до уровня яркости, равного 5% от начальной, превышает 2 мс, но
зависит от амплитуды импульсов приложения напряжения и увеличивается
при уменьшении концентрации Мп.
1.6. КРИВЫЕ ТЕРМИЧЕСКОГО ВЫСВЕЧИВАНИЯ
Как известно, кинетика рекомбинационных процессов связана с наличием
в люминофоре ловушек (см. стр. 73). От числа ловушек, их энергетической глу-
бины и числа электронов, находящихся на них, зависит длительность после-
свечения.
Свойства электронных ловушек можно исследовать методом термического
высвечивания [46—49]. Суть его состоит в следующем. Люминофор охлаждают
До температуры жидких азота (—195 °C) или гелия (—270 °C), а затем возбу-
ждают светом, что локализует электроны на ловушках. Затем источник возбу-
ждения выключают и начинают нагревать люминофор с некоторой постоянной
23
скоростью. При определенных температурах интенсивность люминесценции
резко увеличивается. Это происходит тогда, когда люминофору сообщается
энергия, которая достаточна для того, чтобы освободить электроны с ловушек
определенной глубины. Освобожденные термическим путем электроны реком-
бинируют с центрами люминесценции, что приводит к вспышке. Таким
•образом, на кривых зависимости интенсивности свечения от температуры появ-
ляется ряд максимумов (рис. 1.21).
° Впервые такие периодические
подъемы и спады яркости свечения
/ 2 3 4 при нагревании люминофора наблю-
Рис. 1.21. Кривые термического высвечивания.
•а — ZnS-Си с различными соактиваторами (прокаливание в атмосфере HfS при 1150° С);
б — ZnS-Cu-А1, снятые обычным способом (А) и методом фракционирования (Б); в — ZnS-Си,
съемка при замораживании до температуры жидкого гелия:
1 — ZnS-Cu (5-IO-6)-Al (10-4); 2 — ZnS-Cu (5-10-6)-Sc (10-4); 3 — ZnS-Cu (5-10-*) •
• Ga (10-4); 4 — ZnS-Cu (3-10-6)-In (3-10-*).
расчет кривых термического высвечивания, допуская отсутствие явлений по-
вторного захвата электронов.
Более полный расчет был выполнен в работах [4, с. 236; 47]. Зная темпера-
туру, при которой наблюдается максимум вспышки, можно определить энерге-
тическую глубину ловушки. Согласно теории [46], значение энергии Е (соответ-
ствующей определенной глубине ловушки) можно вычислить по формуле
где Р — скорость нагревания;
s — параметр, связанный с природой люминофора (определяется экспери-
ментально).
Для люминофора ZnS Си при р — 0,01°/с связь между глубиной ловушки Е
и температурой Т, соответствующей максимуму КТВ, может быть с достаточным
приближением выражена формулой Урбаха:
Е = Т/400
24
Из теории следует, что площадь, ограниченная КТВ и осью абсцисс, про-
порциональна числу электронов, запасенных на ловушках. Форма КТВ зависит
от скорости нагревания, длительности возбуждения люминофора, промежутка
времени между прекращением возбуждения и началом нагревания, интенсив-
ности возбуждающего света.
В работе [48] КТВ определяли с большой точностью при помощи так назы-
ваемого метода фракционирования. Этот метод отличается от описан-
ного тем, что вместо плавного постоянного нагревания люминофор нагревают
так, что температура испытывает небольшие колебания, накладывающиеся
друг на друга. Это позволяет широкие максимумы на КТВ разрешить в виде
отдельных элементарных максимумов, соответствующих отдельным элементар-
ным уровням захвата (ловушкам).
На рис. 1.21, б для сравнения представлены КТВ для люминофора
ZnS-Cu-Al, снятые обычным методом (А) и методом фракционирования (Б).
Необходимо отметить, что часто даже для люминофоров одного и того же состава
КТВ отличаются друг от друга. Это обусловлено различием способов пригото-
вления и условий измерения.
Согласно имеющимся экспериментальным данным [49—53] природа лову-
шек в цинксульфидных люминофорах определяется нарушениями и примесями
в решетке основы люминофора, в частности: активаторами, соактиваторами,
вакансиями серы, ассоциациями вакансий.
При корректном подходе метод КТВ может рассматриваться так же как
один из чувствительных аналитических методов, позволяющих обнаруживать
в решетке люминофора посторонние примесные фазы [54].
1.7. ДЕЙСТВИЕ НА ЛЮМИНОФОРЫ
ИНФРАКРАСНОГО ИЗЛУЧЕНИЯ
Инфракрасные лучи могут оказывать как тушащее, так и стимули-
рующее действие на люминесценцию и фотопроводимость лю-
минофоров на основе ZnS, ZnS CdS, ZnSe, активированных различными эле-
ментами, а также люминофоров на основе сульфидов щелочноземельных металлов
[55—60]. Стимуляция обычно выражается в виде резкого, кратковремен-
ного увеличения интенсивности люминесценции (вспышки), после чего насту-
пает тушение.
Явление стимуляции для цинксульфидных люминофоров в самом общем
виде объясняется тем, что под действием ИК-лучей электроны, находящиеся
на ловушках, освобождаются и могут принять участие в рекомбинации с иони-
зованными центрами люминесценции.
Тушение люминесценции происходит в том случае, когда энергия ИК-лучей
оказывается достаточной для переноса электрона из валентной зоны
на уровни ионизованных активаторов. Это приводит к уничтожению положи-
тельного заряда на уровнях активатора и, следовательно, к уменьшению числа
переходов, сопровождающихся излучением света. Ослабление указанного
эффекта может возникнуть из-за того, что дырки, образовавшиеся в валентной
зоне, начнут перемещаться по ней и переходить на уровни активатора, вновь
создавая условия для излучательной рекомбинации. Если вводить в люмино-
фор Со, Ni и Fe, то эффект усиливается.
Согласно существующим представлениям [58], это обусловлено тем, что
введение указанных элементов способствует образованию дополнительных
глубоких электронных ловушек. В процессе тушения образовавшиеся дырки
могут захватываться ловушками, рекомбинировать без излучения с электронами.
Тем самым возникает препятствие для перехода дырок на уровни активатора.
Подробный разбор механизма действия ИК-лучей можно найти в рабо-
тах [3, 4].
Явление стимуляции у цинксульфидных люминофоров, например, таких,
как ZnS Си, в том случае, если одновременно включены источники возбужде-
ния и ИК-лучей, наблюдается при низкой температуре, причем понижение
температуры усиливает стимуляцию. Обусловлено это тем, что стимуляция
связана со срывом электронов с ловушек (см. ниже), а так как при понижении
25
температуры остаются заполненными даже самые мелкие ловушки, то естественно
ожидать увеличения эффекта стимуляции.
Так, в работе [60] было показано, что у люминофора ZnS -Си снижение тем-
пературы до 4,2 К позволило увеличить интенсивность стимуляции — в 80 раз
по сравнению с той, которая имела место при 77,4 К. При этом глубина ловушек
соответствовала 0,02—0,08 эВ. Для таких мелких ловушек авторы работы
[60] смогли получить стимуляцию ИК-лучами с длиной волны до 15 000 нм.
При 77,4 К предельная длина волны, вызывающей вспышку, соответствует
2000 нм.
Спектр стимуляции, т. е. зависимость интенсивности стимуляции от длины
волны ИК-лучей у люминофора ZnS Си, представляет собой кривую с двумя
максимумами, соответствующими 750 и 1250 нм [59].
При комнатной температуре стимуляция может возникнуть у цинксульфид-
ных люминофоров с длительным послесвечением, если ИК-лучи действуют на
люминофор в процессе послесвечения. В этом случае после выключения возбу-
ждения можно при каждом включении ИК-лучей наблюдать вспышки люмине-
сценции, интенсивность которых убывает со временем [55].
При комнатной температуре интенсивная стимуляция присуща люмино-
форам ZnS Си РЬ и ZnS Си - Мп [56]. В этом случае спектр излучения вспышки
у люминофоров соответствует спектру излучения РЬ или Мп. Предполагается,
что медь служит источником электронов, запасаемых на ловушках, образу-
ющихся при введении РЬ [56]. Спектральная область стимуляции этих люмино-
форов мало отличается от таковой для люминофоров ZnS -Си.
Люминофоры, которые дают наиболее интенсивную вспышку при облуче-
нии ИК-светом после прекращения возбуждения, относятся к классу сульфидов
щелочноземельных металлов, активированных редкоземельными элемен-
тами [51]. Эти люминофоры, называемые обычно вспышечными, нашли
широкое применение в ряде специальных приборов (дозиметры, приборы ноч-
ного видения и т. д.). К вспышечным люминофорам относятся, например,
SrS Ce-Sm, SrS-Eu-Sm, а также SrS-GaS-Eu-Sm. Спектр вспышки зависит
от присутствия Се или Ей; введение Sm увеличивает интенсивность вспышки
и определяет спектр стимуляции.
Тушение под влиянием действия ИК-лучей происходит при комнатной тем-
пературе тогда, когда на люминофор одновременно действуют возбуждающий
свет и ИК-лучи.
Спектральные области тушения и стимуляции обычно совпадают, но, как
показано в работе [59], зависимость коэффициента тушения р*
от X определяется составом люминофора. Величина коэффициента тушения тем
больше, чем меньше интенсивность возбуждающего света и концентрация акти-
ватора. Введение в люминофор некоторых тяжелых металлов, например, Ni,
Со, Fe, приводит к увеличению коэффициента тушения [58].
Тушение электролюминесценции инфракрасным светом было впервые
обнаружено Хекшером [61], но подробное исследование проведено в работах
[62, 63J.
Небольшой эффект тушения ИК-светом в случае электролюминофоров объ-
ясняется их особенностями и, в частности, тем, что они запасают очень малую
светосумму.
Чем выше напряжение и частота возбуждающего поля, тем меньше коэффи-
циент тушения. Это явление можно объяснить тем [63], что при увеличении
частоты возрастает вероятность повторной ионизации центров, с которых дырки
освобождаются ИК-светом, так как по мере увеличения частоты становится все
более затруднительным уход от центра освободившейся дырки.
Исследование влияния ИК-света на волны яркости электролюминесцен-
ции показывает, что наибольшему тушению подвергается вторичный максимум,
который появляется в результате рекомбинации с ионизованными центрами
электронов, первоначально отогнанных полем. В момент, когда электроны
и дырки разделены, вероятность освобождения дырок с ионизованных центров
увеличивается.
* Коэффициент тушения равен: р = (/0 — 1)11 i где 10 и I — интенсив-
ности люминесценции в отсутствие и при наличии ИК-лучей.
26
ЛИТЕРАТУРА
1. Вавилов С. И. Собрание сочинений. Т. 2. М., Изд. АН СССР, 1952.
547 с. — 2. Левшин В. Л. Фотолюминесценция жидких и твердых веществ. М.,
Гостехтеоретиздат, 1951. 456 с. — 3. Фок М. В. Введение в кинетику люмине-
сценции кристаллофосфоров. М., «Наука», 1964. 283 с.— 4. Антонов-Романов-
ский В. В. Кинетика фотолюминесценции кристаллофосфоров. М., «Наука»,
1966. 323 с. — 5. КюриД. Люминесценция кристаллов. М., ИЛ, 1961. 199 с. —
6. Leverenz Н. W. An Introduction to Luminescence of Solids. New York — Lon-
don, John Wiley and Sons, 1950. 569 p. — 7. Левшин В. Л., Арапова Э. Я.,
Блажевич А. И. и др. «Труды ФИАН им. П. Н. Лебедева», 1963, т. 23, с. 64—135. —
8. Хениш Г. Электролюминесценция. Пер. с англ. Под ред. Горина. М., «Мир»,
1964. 455 с. — 9. Верещагин И. К. Электролюминесценция кристаллов. М.,
«Наука», 1974, 278 с. — 10. Gisolf I. Н. «Physica», 1939, р. 295—355.
11. Антонов-Романовский В. В., Дубинин В. Т., Прохоров А. М. и др.
ЖЭТФ, 1959, т. 37, с. 1466—1467. — 12. Лущик Н. Е., Лущик Ч. Б. «Труды
ИФА АН ЭССР», 1957, № 6, с. 5—12. — 13. Bril A., Van Meurs-Hoekstra W.
Phil. Res. Repts, 1964, v. 19, № 3, p. 296—305. — 14. ВинокуровЛ. A., Фок M.B.
Опт. и спектр., 1958, т. 4, № 1, с. 118—121. — 15. Бабицкая Р. А., Пекер-
ман Ф. М., Успенская Е. М. Там же, 1967, т. 23, № 1, с. 121 —128. — 16. Stu-
der F. Y., Gans Z. In. Preparation and Characteristic of Solid Luminescent Mate-
rials. New York, 1948, p. 258—268. — 17. Фок M. В. Опт. и спектр., 1957, т. 2,
№ 4, с. 475—479. — 18. Бонч-Бруевич А. М. Изв. АН СССР. Сер. физ., 1962,
т. 26, с. 468—474. — 19. Vecht А., J. on Luminescence, 1973, № 7, р. 213—220. —
20. Антонов-Романовский В. В., Эпштейн М. И. ДАН СССР, 1949, т. 64,
с. 483—486.
21. Аленцев М. Н. Опт. и спектр., 1956, т. 1, № 2, с. 240—247. — 22. Ален-
цев М. Н., Антонов-Романовский В. В., Винокуров Л• А. ДАН СССР, 1954,
т. 96, с. 1133—1134. — 23. Бутаева Ф. А. «Светотехника», 1959, № 7, с. 17—
20. — 24. Илъмас Э. Р., Лийдъя Т. Т., Лущик Ч. Б. «Труды ИФА АН СССР»,
1967, № 26, с. 213—215. — 25. Lehmann W. Ilium. Eng., 1956, v. 51, p. 684—
688. — 26. Пекерман Ф. M., Петошина Л. Н. Опт. и спектр., 1964, т. 16, № 3,
с. 496—500. — 27. Lehmann W. J. Electrochem. Soc., 1958, v. 105, p. 585—
588. — 28. KotonaR. Ibid., 1962, № 109, p. 693—699. — 29. Георгобиани A. H.,
Львова E. Ю., Фок M. В. Опт. и спектр., 1962, т. 13, № 4, с. 564—568. —
30. Чукова Ю. П. Там же, 1967, т. 22, № 3, с. 450—455.
31. Верещагин И. К. Там же, 1966, т. 21, № 2, с. 204—210. — 32. Антонов-
РомановскийВ. В. «Труды ФИАН», 1937, т. 1, вып. 2, с. 35—93.— 33. Nail N. R.,
Urbach Р., Pearlman. JOSA, 1949, v. 39, p. 690—694. — 34. Zalm P. Phil. Res.
Repts, 1956, v. 11, p. 353—451. — 35. Lehmann W. J. Electrochem. Soc., 1960,
v. 107, p. 20—25. — 36. Георгобиани A. H., Фок M. В. Опт. и спектр. 1960,
т. 9, c. 775—781. — 37. Mattier J. J. Phys. Radium, 1956, v. 17, p. 725—728. —
38. Георгобиани A. H. «Труды ФИАН им. П. Н. Лебедева», 1963, т. 23, с. 3—
63. — 39. Нааке С. Н. Phys. Rev., 1955, v. 28, с. 1544—1545. — 40. Казан-
кин О. Н., Петошина Л. Я. Опт. п спектр., 1958, т. 4, с. 76—81.
41. Вергунас Ф. И., Карпухина В. Ф. В кн.: Электролюминесценция твер-
дых тел. Киев, «Наукова Думка», 1971, с. 142—148. — 42. Толстой Н. А.,
Феофилов П. П. Усп. физ. наук, 1950, т. 41, с. 44—107. — 43. Антонов-Рома-
новский В. В. ДАН СССР, 1936, т. 2 (11), с. 93—96. — 44. Бонч-Бруевич А. М.
Опт. и спектр.. 1961, т. 11, с. 87—92. — 45. Ребане К.-С. К., Талъвисте Е. К.
«Труды ИФА АН ЭССР», 1962, вып. 21, с. 257—274. — 46. Randall G., Wil-
lkins F. Proc. Rov. Soc., 1945, v. 184, p. 366—407. — 47. Лущик Ч. Б. «Труды
ИФА АН СССР», 1955, вып. 3. с. 1—230. — 48. Gobrecht Н., Nelkowski Н., Hof-
fmann D. е. a., Proceedings Л the International Conference on Luminescence.
Budapest, Akademia Klado, 1968, p. 1078—1085. — 49. Riehl N. Ibid., p. 974—
987. — 50. Левшин В. Л., Туницкая В. Ф., Черепнев Л. А. Опт. и спектр., 1956,
т. 1, № 2, с. 255—263.
27
51. Туницкая В. Ф. Там же, 1963, т. 14, № 3, с. 445. — 52. Hoogenstra-
aten W. Phil. Res. Repts, 1958, v. 13, p. 515—693. — 53. Вундель А. А., Жу-
ков Г. В. Опт. и спектр., 1965, т. 19, № 2, с. 247—251. — 54. Казанкин О. Н.,
Зимогляд Л. С. Ж. прикл. спектр., 1971, т. 15, № 2, с. 240—244. — 55. Вино-
куров Л. А., Фок М. В. Опт. и спектр., 1956, т. 1, № 2, с. 248—254. — 56. Лев-
шин В. Л., Орлов Б. М. Там же, 1959, т. 7, № 4, с. 530—536. — 57. Левшин В. Л
Антонов-Романовский В. В., Моргенштерн 3. Л. и др. ЖЭТФ, 1947, т. 17,
с. 949—963. — 58. Goldstein В., Dropkin J. J. Phys. Rev., 1962, v. 126, p. 966—
970. — 59. Рабане К.-С. К., Риттас В. И. Ж. прикл. спектр., 1965, т. 2,
с. 350—355. — 60. Baur G., Knobloch J., Riehl N. e. a. Proceedings of the Inter-
national Conference on Luminescence. Budapest, Academia Klado, 1968, p. 1012—
1014.
61. He.ksher H., JOSA, 1957, v. 47, p. 765. — 62. Вукке E. E., Виноку-
ровЛ. AФок M. В. Опт. и спектр., 1958, т. 5, с. 172—178. — 63. Ребане К.-С. К.,
Талъвисте Е. К. «Труды ИФА АН ЭССР», 1961, вып. 15, с. 161—171, с. 172—
183; 1962, с. 257—274.
ГЛАВА II
ХАРАКТЕРИСТИКА ОСНОВНЫХ КЛАССОВ
ХИМИЧЕСКИХ СОЕДИНЕНИЙ,
ПРИМЕНЯЕМЫХ ДЛЯ СИНТЕЗА ЛЮМИНОФОРОВ
Химия люминофоров изучает твердофазные системы, один или несколько
компонентов которых содержатся в малом количестве (10-4 — 1,0%). Особен-
ность этого раздела химии твердого тела заключается в том, что в качестве
компонентов системы рассматривают не только химические элементы или соеди-
нения, но также точечные дефекты кристаллической решетки (на-
пример, вакансии) и свободные носители тока (электроны
и дырки). Концентрация дефектов и свободных носителей тока (особенно
при высокой температуре, при которой происходит синтез люминофора) срав-
нима с содержанием примеси активатора в люминофоре.
На сегодняшний день данных о растворимости примесей, а также о природе
и концентрации точечных дефектов в решетках различных соединений, явно
мало. Сложность системы люминофор — активатор заключается и в том, что
в большинстве случаев при комнатной температуре она неравновесна и предста-
вляет собой пересыщенный твердый раствор.
Наиболее важные люминофоры, используемые промышленностью, отно-
сятся к следующим классам соединений:
халькогениды (сульфиды, селениды и теллуриды);
фосфаты (орто-, пиро-, галофосфаты);
силикаты;
окислы.
Помимо указанных соединений в промышленности применяют арсенаты,
германаты, вольфраматы и оксисульфиды.
Фториды, бораты, ванадаты, цирконаты и другие соединения представляют
практический интерес, но находятся в стадии исследования.
Катионами в указанных классах соединений почти исключительно являются
элементы второй группы периодической системы (цинк, кадмий, кальций, маг-
ний и стронций). В последние годы стали широко применяться соединения лан-
тана и иттрия.
11.1. ХАЛЬКОГЕНИДЫ ЭЛЕМЕНТОВ ВТОРОЙ ГРУППЫ
Для производства фото-, катодо- и электролюминофоров в основном исполь-
зуют сульфиды цинка и кадмия, а также селенид цинка. Некоторое значенпе
имеют теллуриды цинка и кадмия, применяемые в небольших количествах для
изготовления светодиодов. Сульфиды стронция и кальция пригодны для полу-
чения фото люминофоров с длительным послесвечением.
Физические свойства халькогенидов (табл. 11.1)
Все халькогениды кристаллизуются в двух модификациях: сфалерит-
ной и вюрцитной. Первая образуется путем заполнения катионами
половины тетраэдрических пустот в плотнейшей кубической упаковке анионов
29
р N О §3 n N Qu Q Qj Р гъ. *ч Н н QQ “ со СД ф Ф ф ф Соединение
S § g •§ S § g | | •§ 8 ®g®g®Bc.R15°a® тз-йтз-Етз-йтз Структура
О О о о о о о о р р о Ф "(IS ф 4S Ь is Сл rf* ел w сл ^СО^СОООСЗ^рр*- 00 --J О СО СЛ О СЭ СЮ № со о со оо со оо оз о о р -о 00 со Q ! Параметры решет- ки, нм
со р р р р CS , --J сз , оз . сз СО о СЛ со со ЦЗч 1 03 СЛ О 03 О со <3
о о о о р р со со со со со со 00 03 сз СП со О 4з- СО СП СО ЦЗч Длина связи, нм
1,26 1,42 1,15 1,30 0,99 1,12 Соотношение тетраэдриче- ских радиусов ионов
р X О 1 0° 1 00 я ° Диэлектрическая проница- емость
СО СО СЛ О СО 03 СО О 00 О О ЦЗч о СП о О о Температура плавления и равновесное давле- ние пара
о о о о р со СО 03 ЦЗч СЛ -"1 03 СО о со со со оз Па-10-в 3 3*
р р р р 00 р СО 03 ЦЗч СП 00 --J СО СО СО атм
3,7 2,4 2,7 1,7 2,2 1,5 Ширина запрещенной зо- ны, эВ
^3 8 8 8 ^8 8 8 8 Тип проводимости
1 102 10-2 10-2 (106) 10-2 (10-1) 10-2 10-2 10-1 Минимальное удельное электросопротивление, 0м-см
03 р СЛ р ЦЗч Р ЦЗч СО 00 00 ф' 00 о О НК со СО 00 о -q СО Плотность, г/см’
Физические свойства халькогенидов цинка и кадмия
(на один анйэн приходится одна октаэдрическая пустота и две тетраэдрические).
Вюрцитная модификация соответствует заполнению половины тетраэдрических
пустот в плотнейшей гексагональной упаковке. Октаэдрические пустоты в со-
единениях AnBVI не заполнены.
Любой катион в решетке сфалерита или вюрцита окружен четырьмя бли-
жайшими соседями-анионами, расположенными в вершинах тетраэдра. Однако
в случае вюрцита тетраэдр несколько искажен: один из анионов более удален
по сравнению с другими тремя.
Между структурами вюрцита и сфалерита (цинковой обманки) существует
тесная связь. Параметры решетки гексагональной элементарной ячейки с боль-
шой точностью можно вычислить из параметров кубической решетки одного
и того же соединения по соотношению: агеКС = 72 «.<уб V2; сгекс = 2/з «куб
Как геометрически, так и энергетически структуры сфалерита и вюрцита мало
отличаются друг от друга и поэтому легко образуют кристаллические сростки
и политипы.
Температура фазового перехода определена только для сульфида цинка
(1020°), причем кинетика перехода сильно зависит от наличия в кристаллах
примеси (например, меди) или от газовой среды, в которой происходит кристал-
лизация. Отметим, что хотя для всех соединений, кроме теллурида кадмия,
найдены обе кристаллические модификации, в обычных условиях синтеза лю-
минофоров сульфид кадмия имеет только вюрцитную модификацию, а селенид
цинка — только сфалеритную. Примесь сульфида кадмия способствует обра-
зованию гексагональной структуры твердых растворов ZnS—CdS даже при низ-
ких температурах, а примесь селенида цинка — образованию кубической струк-
туры. Селенид кадмия при температуре выше 700° имеет гексагональную струк-
туру. Сульфиды щелочноземельных металлов кристаллизуются в кубической
гранецентрированной структуре типа NaCl.
Почти все халькогениды представляют собой полупроводниковые соеди-
нения с проводимостью электронного типа. Для теллурида цинка характерна
проводимость дырочного типа, а для теллурида кадмия — как электронная,
так и дырочная. В условиях синтеза люминофоров сульфид и селенид цинка
имеют очень высокое удельное электросопротивление (1012 Ом см). Лишь при
специальных условиях легирования, обеспечивающих внедрение избыточного
цинка, удается получить сульфид и селенид цинка с низким удельным электро-
сопротивлением (10-2—102 Ом см). Концентрация носителей тока при этом
зависит как от содержания донорных примесей, так и от давления пара цинка.
Ширина запрещенной зоны халькогенидов цинка и сульфида кадмия
равна 2,2—3,7 эВ и поэтому излучать они могут в^широкой спектральной
области — от УФ- до ИК-диапазопа.
Способы получения
Сульфиды цинка и кадмия получают путем осаждения сероводородом из
водных растворов сульфатов. При этом образуются тонко дисперсные порошки.
Последние состоят из агломератов, содержащих еще более мелкие частицы.
Размер первичных частиц составляет десятки нанометров, а агломератов — еди-
ницы и десятки микрометров. Размер частиц исходных сульфидов цинка и кад-
мия во многом определяет гранулометрический состав порошков люминофоров.
Содержание микропримесей в продуктах удовлетворяет люминофорным требо-
ваниям (меньше 10~5% тяжелых металлов), но содержание основного вещества
значительно меньше 100%. Основные примеси — вода, окись и сульфаты цинка
и кадмия.
Селениды цинка и кадмия синтезируют из сульфидов по реакции, которая
в упрощенном виде может быть записана следующим образом [1]:
MeS-|~H2SeO3---> MeSe-f-SO2+H2O
Побочные продукты этой реакции — окислы металлов п селен, а также
нецрореагировавший исходный сульфид. Для удаления примеси окислов
Продукты обрабатывают уксусной кислотой. Известны и способы получения
31
селенидов из водных растворов путем восстановления селенитов гидразином
или другими восстановителями, а также селеносульфатный метод [2, с. 139].
Сульфиды щелочноземельных металлов получают сульфированием карбо-
натов металлов смесью серы и крахмала в присутствии различных солевых
смесей пли сульфированием окислов сероуглеродом [3, с. 290; 4]. Теллурид
цинка и кадмия синтезируют в основном методом сплавления компонентов
[5, с. 27]. Этим методом можно синтезировать сульфиды и селениды цинка
и кадмия, но это дорого и малопроизводительно [6]. Известен способ получения
бинарных соединений из паров компонентов, пригодный для всех халькогенидов
элементов II группы [7], но он еще не нашел широкого применения.
Физико-химические свойства
Сульфиды цинка и кадмия — соединения очень медленно гидролизующиеся
во влажном воздухе. Разложение сильно увеличивается при совместном дей-
ствии влаги и возбуждающего люминесценцию излучения (ультрафиолетового,
катодного или радиоактивного). Щелочноземельные сульфиды очень сильно
гидролизуются во влажном воздухе. Селенид цинка также подвержен действию
влаги и кислорода, особенно при освещении сильно поглощающимся излучением
(ультрафиолетовый и синий свет); при этом желтый порошок селенида цинка
приобретает красноватую окраску за счет выделения свободного селена.
Все халькогениды заметно окисляются при нагревании в присутствии воз-
духа. Температура начала окисления сильно зависит от размера и состояния
поверхности частиц порошка. Начиная с 550°, люминофоры из сульфида цинка
заметно окисляются с образованием сульфата, хотя спад интенсивности свечения
начинается после прогрева уже при 380—450°. Окисление при температуре
выше 800° идет до окиси цинка.
Сульфид кадмия окисляется легче, чем сульфид цинка; в результате реакции
возможно образование значительных количеств сульфата. Селенид цинка (начи-
ная с 450°) окисляется с образованием свободного селена и окиси цинка. При
полном окислении селенида цинка образуется окись цинка, содержащая 0,5—
1,5% селена и обладающая характерной желто-оранжевой люминесценцией
с ^тах = нм. Следует отметить, что при малых парциальных давлениях
кислород может не полностью окислять компоненты соединений AnBVI, изме-
няя тем самым их парциальные давления. В результате кислород выступает
в роли «восстановителя» металла вследствие большой устойчивости SO2. Восста-
новительное действие кислорода было экспериментально доказано, например,
в случае взаимодействия CdS с кислородом [8].
Сульфид цинка может растворять в себе значительные количества гекса-
гональной окиси цинка — до 1—1,5%. При введении в твердый раствор 0,7%
окиси цинка параметр кубической решетки уменьшается от 0,54093 до 0,54065 нм
(от 5,4093 до 5,4065 А) [3]. Растворимость кислорода в селениде цинка не пре-
вышает 0,1% при 1000° [10].
Все халькогениды цинка имеют заметное давление пара при температуре
синтеза люминофоров (—1000°). В парах халькогениды почти полностью диссо-
циируют по уравнению:
АВ (»в.) А (г.)Ч-1/2В2 (г.)
В связи с этим сублимация соединения AnBvl сильно зависит от парци-
ального давления пара того или иного компонента. Парциальные давления пара
компонентов (рА и связаны между собой соотношением
к=рл1>^:
где К — константа диссоциации соединения АВ.
Уравнение
связывает константу диссоциации с изменением энтальпии и энтропии в процессе
диссоциации. Величина К была определена многими исследователями и различ-
32
ними способами. Обзор работ по давлению и составу паров приведен в ра-
боте [И, гл. 2, 4].
Как следует из рис. II.1, при 1000° давление пара сульфида цинка ничтожно,
тогда как у теллурида кадмия — ~-10000 Па (нескольких десятков мм рт. ст.).
Фазовые диаграммы бинарных систем халькоген — металл изучены недоста-
точно; наиболее подробные данные имеются лишь для теллуридов кадмия [12]
и цинка [13]. Для остальных халькогенидов (кроме сульфида цинка) получены
кривые ликвидус [14, гл. 2]. Все халькогениды заметно растворяются в исход-
ных компонентах. Так, растворимость селенида цинка в цинке достигает ~-0,1%
при 1000° и ~1,0% в расплаве селена [15].
Рис. II. 1. Температурная зависимость давления паров
некоторых халькогенидов.
Растворимость компонентов в твердом соединении (границы солидуса)
определена только косвенно (по данным о высокотемпературной электропровод-
ности). Так, у CdTe отклонение от стехиометрии возможно в обе стороны, а ши-
рина области гомогенности его при 1000° составляет ~ 10“2 ат. % [16]. По дан-
ным масс-спектрометрического анализа отклонение от стехиометрии в CdTe
может достигать нескольких атомных процентов [17]. Отклонение от стехио-
метрии у ZnTe возможно только в сторону избытка теллура, растворимость
которого достигает 4,6 10-3ат. % при 1200° [18].
При обычных условиях синтеза люминофоров все халькогениды содержат
значительное количество точечных дефектов. Разупорядоченность кристалли-
ческой решетки неизбежна с термодинамической точки зрения. Сведения от сте-
пени разупорядоченности и природе дефектов получены в основном косвенными
методами — путем анализа результатов изучения электропроводности и данных
о самодиффузии в халькогенидах цинка и кадмия.
В теллуридах цинка и кадмия дефектность определяется термической
разупорядоченностью в подрешетке металла и наличием в междуузлиях ней-
тральных атомов теллура. В окпслах, сульфидах и селенидах цинка и кадмия
разупорядоченность связана с отклонением от стехиометрического состава
(избыток металла) и наличием в междуузлиях нейтральных атомов халькогена.
Природа дефекта, обусловленного избытком металла, наиболее четко устано-
влена для окиси цинка (междуузельный однократноионизованный цинк). В слу-
чае сульфида и селенида кадмия более вероятным дефектом, преобладающим
3 Заказ 4 4 33
по концентрации, является ионизованный междуузельный кадмий. В случае
окиси кадмия, селенида и сульфида цинка определенные выводы сделать
трудно.
Растворимость примесей
Cu2S ZnS
ZnS, мод %
Большинство элементов периодической системы растворимы в халькогени-
дах цинка и кадмия в концентрациях, оказывающих влияние на электрические
и люминесцентные свойства (10"4 ат. % и более). Однако количественные данные
о растворимости, особенно полученные в равновесных условиях, немного-
численны. Связано это с тем, что большин-
ство исследователей стремилось получить
сведения о концентрации примесей в кон-
кретных люминофорах, имеющих практи-
ческую ценность, которые в большинстве
случаев являются неравновесными систе-
мами. Сложность изучения растворимости
связана с тем, что люминофор даже в
простейшем случае — это трехкомпонент-
ная система (например, цинк, сера и медь).
Для полного термодинамического описа-
ния такой системы необходимо знать при
данной температуре парциальные давления
(активности) примеси и одного из компо-
нентов.
Наибольший интерес представляют
данные о растворимости меди, серебра,
марганца, алюминия, галлия, индия и га-
логенов. Детальное исследование выпол-
нено для систем ZnS—Си [19], ZnSe—Си
и CdSe—Си [20], а также ZnS—Ag,
CdS-Ag [21].
Как следует из рис. II.2, раствори-
мость Cu2S в сульфиде цинка достигает
большой величины ~1,0 мол. % при 1100° и резко падает с понижением темпе-
ратуры. Сопряженным раствором является сульфид меди, насыщенный сульфи-
дом цинка (до 25 мол. % при 1100°), который также сильно распадается при
охлаждении (уже при 700° остается несколько процентов ZnS в Cu2S).
Растворимость (в мол. %) халькогенидов меди в халькогенидах цинка И кад-
мия описывается уравнениями вида:
Рис. II.2. Диаграмма состояния
системы ZnS—Cu2S.
Cu2S в ZnS lgC = --^--[- 2,968
Cu2Se в ZnSe lgC = _ 2£±__i_ 3,369 1
Cu2Se в CdSe lgC = -^ + 3,362
Эти соотношения могут быть
обобщенным уравнением [20]
выражены с удовлетворительной
точностью
= 3,262
т /' ПЛ
где Гил — температура плавления кристалла растворителя.
Таким образом, природа твердых растворов меди имеет общий характер
для всех халькогенидов цинка и кадмия. Приведенные выше данные получены
при температуре выше 800°. Растворимость при мепыпих температурах может
34
быть оценена только экстраполяцией. Ряд экспериментальных данных подтвер-
ждает справедливость приведенных уравнений вплоть до 300°. Так, по данным
работы [22], растворимость меди в монокристаллах селенида цинка при 570
и 350° составляет 1,15-Ю-3 и 7,7-10~4 мол. %. Растворимость Cu2S в CdS
при 600° составляет 5 -10“4 мол. % [23], а в ZnS при 460? — 5 -10“ 3 мол. % [24].
Было показано, что в области равновесия двух сопряженных халькогенид-
ных фаз на основе меди и цинка растворимость Cu2S и Cu2Se не зависит от давле-
ния пара цИнка или кадмия в интервале от ^lO’1 Па до нескольких тысяч
(10-4 мм рт. ст. до десятков мм рт. ст.). Это позволяет предположить, что при
растворении меди не образуются электронные или собственные атомные дефекты
в количествах, сравнимых с концентрацией меди. Подобное есть следствие
внедрения электронейтральных относительно решетки пар, в которых один
из ионов меди располагается в катионном узле, а другой — в междуузлии кри-
сталлической решетки, что эквивалентно растворению Cu2S или Cu2Se [25].
Относительно растворимости галогенов известно, что при 10009 в сульфиде
кадмия растворяется до 0,1% F2 (взятого в виде CdF2) [26]. По различным дан-
ным, при 1000° в сульфид цинка может внедриться от 3 10" 2 до 0,2% С12. Повы-
шенные содержания хлора обнаруживаются при растворении его в виде
СпС1 [27]. В сульфиде кадмия растворяется при 8009 ~10"2% С12, взятого
в виде CdCl2. Содержание брома и иода в большинстве кристаллов, выращенных
в присутствии галогена, не превышает 10"2 ат. %.
Растворимость алюминия в сульфиде цинка составляет 0,5 ат. % при
1000° [28] и 1 ат. % при 1100° [29]. Путем диффузии в селенид цинка было
введено более 0,1% А1 при 1050° [30]. Максимальная растворимость алюминия
при легировании селенида путем диффузии из сплава Zn—Al достигала 1,5 ат. %
при 1000°. Но при комнатной температуре монокристаллы с концентрацией
даже 0,5 ат. % А1 были неоднородны и содержали фазу на основе селенида
алюминия, насыщенную цинком [31].
Растворимость галлия и индия в сульфидах, селенидах и теллуридах цинка
и кадмия достигает нескольких атомных процентов. Предполагается, что си-
стемы A12S3— ZnS и Ga2Se3—ZnSe должны образовывать непрерывный ряд
твердых растворов. Следует отметить, что алюминий и галлий совместно с медью
и серебром растворяются в значительно больших количествах, чем порознь, при
этом следует, видимо, говорить о растворимости соединений типа CuGaS2 и
AgGaS2 в сульфиде цинка. В присутствии алюминия распада твердого раствора
ZnS Си при концентрации меди до 1 ат. % не происходит [32].
Растворимость других элементов не определена. Имеются лишь отрывоч-
ные данные о концентрациях примесей в порошкообразных люминофорах и моно-
кристаллах халькогенидов. По данным спектрального и масс-спектрометриче-
ского анализов установлено, что щелочные металлы (Na, Li) часто встречаются
в концентрациях 10~4—10"2 ат. %. Концентрация примеси щелочноземельных
металлов примерно такая же, хотя растворимость, например магния, может
достигать 20 мол. % при 980° [33]. Переходные металлы и р. з. э. вводили в по-
рошки и монокристаллы в концентрациях до 1 ат. %. Железо обычно содер-
жится или вводится в количествах от 10-4 до 10"3 ат. %, но известно, что его
растворимость в сульфиде цинка достигает 40 мол. % (природные минералы —
железистые сфалериты). Марганец вводят обычно в количестве 1%, но раство-
римость его составляет десятки процентов как в ZnS, так и в CdS и CdSe [34].
Твердые растворы на основе халькогенидов
цинка и кадмия
Сульфиды л селениды, а также селениды и теллуриды образуют между
собой непрерывные ряды твердых растворов (рис. II.3). Это позволяет получить
материалы с разнообразными свойствами. Ограниченная растворимость наблю-
дается в системах сульфиды — теллуриды цинка и кадмия. В сульфиде цинка
растворяется не больше 8—10% ZnTe, а в сульфиде кадмия — до 3,5% CdTe.
Свойства твердых растворов изменяются почти во всех случаях линейно
с составом. Исключение представляет система ZnSe—ZnTe, где ширина запре-
щенной зоны проходит через минимум с изменением состава от ZnSe до ZnTe.
Помимо псевдобинарных систем известны и используются более сложные твердые
3*
35
растворы, например (Zn1_xCdx)(S1_ySe„). Эти твердые растворы кубиче-
ской и гексагональной структуры могут быть получены с неизменной шириной
запрещенной зоны при различных составах основы. Твердые растворы также
активируются в основном медью и серебром.
Как следует из рис. II.3, в системах ZnS—CdS и ZnS—ZnSe наблюдается
резкое изменение спектров излучения в неактивированных системах при неболь-
ших концентрациях второго компонента. Как селен, так и кадмий дают сначала
Рис. II.3. Зависимость положения максимума спектрального
распределения люминесценции от состава твердых растворов
ZnS—CdS (а) и ZnS—ZnSe (б):
1 — ширина запрещенной зоны; 2 — самоактивированное излучение; 3 — па-
раметр решетки; 4 — люминесценция, связанная с примесью меди; 5 —
самоактивированная люминесценция.
локальный уровень в запрещенной зоне, который постепенно сливается с валент-
ной зоной или зоной проводимости. Полосы меди и серебра почти линейно сме-
щаются е составом.
Диффузия примесей
Наиболее быстро диффундирующими примесями являются медь, серебро
и золото. Коэффициент диффузии меди при 500° равен:
D, см2/С
ZnS......................... 1.5.10-9
ZnSe..................... 3,4 • 10-9
CdS.......................(1,5- 3,0). 10-7
Примерно такие же величины коэффициента диффузии имеет серебро, а золото —
значительно меньше — 6-Ю-12 см2/с (500°).
Алюминий, галлий и индий диффундируют медленно. При 1050° коэффи-
циент диффузии алюминия в селениде цинка составляет 1,6-10-9 см2/с, а индия
в теллуриде кадмия при 1000° — 1,8-10"8 см2/с. Галогены диффундируют мед-
ленно. При 1000° коэффициент диффузии иода в сульфиде кадмия составляет
10"11 см2/с. Диффузия марганца и других переходных элементов также является
медленной.
Люминесцентные свойства
Халькогенидам цинка и кадмия присуща так называемая «самоакти-
вированная» люминесценция, обусловленная либо собственными дефек-
тами, либо их ассоциатами с примесью галогенов или трехвалентных катионов,
а также люминесценция, связанная с введением активирующих примесей.
36
Таблица 11.2
Длины волн максимума
спектрального распределения
для самоактивированных
люминофоров
Соединение Чах’ нм
300 к 77 к
ZnS (куб.) ZnS (гекс.) CdS (гекс.) ZnSe (куб.) 460 450 760 605—610 470 460 800 615-624
Практическое значение имеют такие активаторы, как Си, Ag, Аи и Мн, но интен-
сивная люминесценция получена и при активации другими примесями: As
И Р [35], О2 [36], Se и Те [37], Ег [38] и Тт [39]. Имеются сведения об акти-
вации In, Tl, Sn, РЬ [40], а также Fe, Ni, Со [41] и др.
С амоактивированная люминесценция. При прокаливании чистых порошков
сульфидов цинка и кадмия и селенида цинка без добавок активирующих при-
месей, но в присутствии хлоридного плавня, образуются люминофоры с интен-
сивным свечением (табл. II.2).
Особенностью этих полос излучения является то, что при охлаждении они
смещаются в длинноволновую область, тогда как полосы активаторов обычно
смещаются в коротковолновую. Природа
центров самоактивированной люмине-
сценции изучалась как на порошках, так
и на монокристаллах. Сопоставление
физико-химических условий возникно-
вения центров свечения и результатов
исследования поляризации излучения и
электронного парамагнитного резонанса
кристаллов, содержащих С1, Вт, I и
Al, Ga, позволило выдвинуть гипотезу
об образовании центров из дважды ио-
низированной вакансии цинка Vz“ и
однократно ионизированного примес-
ного донора А1^п или Cig. Предпола-
гается, что такой ассоциированный
центр [VZn—AlZn]- или [VZn—Cls]"
ведет себя, как компенсированный од-
нократноионизируемый акцептор,вводя-
щий в запрещенную зону заполненный
электроном локальный уровень вблизи
эопы проводимости.
Следует указать на ряд экспериментальных фактов, которые не согласуются
с моделью центра, составной частью которого является катионная вакансия.
Так, интенсивное синее свечение сульфида цинка (460—470 нм) и желто-оранже-
вое свечение селенида цинка (605—610 нм) возникает при прокаливании моно-
кристаллов в расплаве цинка (800—1100°) [42]. Характерно, что при этом кри-
сталлы становятся сильно проводящими (0,01—10 Ом см). Прогревание таких
образцов в вакууме, инертной атмосфере или парах халькогена приводит к исчез-
новению как люминесценции, так и электропроводности [43]. Таким образом,
люминесценция явно связана с внедрением цинка в кристаллы. Присутствие
Галогенов или алюминия в таких кристаллах мало влияет на спектр излучения,
но заметно повышает интенсивность свечения.
Помимо самоактивированной люминесценции у «чистых» халькогенидов
наблюдается так называемое краевое излучение,с энергией, близкой
к ширине запрещенной зоны: ультрафиолетовое в сульфиде цинка (380—390 нм),
синее в селениде цинка (460—470 нм) и зеленое (520 нм) в сульфиде кадмия.
Оно возникает только при низкой температуре (90 К и ниже) или при комнатной
температуре, но при очень высокой плотности возбуждения.
Активированная люминесценция. Медь, серебро и золото образуют почти
аналогичные центры свечения во всех халькогенидах цинка и кадмия. Для каж-
дого активатора характерно наличие нескольких полос излучения, которые
по-разному проявляются как в зависимости от условий синтеза, концентрации
активатора, вида основы люминофора, так и от условий возбуждения (табл. II.3).
При увеличении концентрации активаторов постепенно подавляется полоса
самоактивированной люминесценции и усиливаются полосы, свойственные
активаторам. В сульфиде цинка самоактивированная синяя полоса полностью
подавляется зеленой при 1 -10~2% Си, а в селениде цинка — при 1 •10-4% Си.
По мере увеличения концентрации меди в сульфиде цинка до (2—3) -10-а%
Появляется синяя полоса меди. Она совпадает по положению с самоактивиро-
ванной полосой, но, как уже отмечалось, смещается при охлаждении в коротко-
волновую область. При повышении концентрации меди до 0,05—0,1%, когда
37
Т а б л и ц а 11.3
Длины волн максимумов спектрального распределения
излучения халькогенидов, активированных медью,
серебром и золотом (фотовозбуждение, 365 нм)
Основа Активатор ^тах' нм
80 К 300 К
ZnS (гекс.) Си 430-440; 470; 520-540;
516-522; 670
690-700
Ag 390; 435 470; 500
Аи 470; 530 540
ZnSe (куб.) Си 530, 635 645
Ag 560 —
Аи 697(170 К) —
CdS (гекс.) Си 790; 1050 —
Ag 620; 730 716, 780
Аи 1180 800
ZnTe (куб.) Си 630; 844 (25 К) —
Ag 544; 646 (25 К) —
Аи 930 (25 К) —
хорошо заметно возникновение сульфида ее, появляются полосы в области
670 нм. Образование центров, ответственных за голубое и красно-оранжевое
свечение, связано с распадом пересыщенного твердого раствора ZnS—Cu2S [44].
Относительно природы центров свечения меди предполагается, что коротко-
волновое свечение обусловлено наличием ассоциатов типа [CuZn—Сиг]_, а длин-
новолновое — типов [CuZn]—[Cig] или [CuZn]— [AlZn].
Свечение марганца в сульфиде цинка наблюдается в узкой полосе 586 нм
при концентрациях активатора 0,1—1%. В селениде цинка подобное свечение
наступает только при низких температурах (77 К). Люминесценция марганца
в чистых халькогенидах очень слаба. Интенсивность свечения резко возрастает
при добавлении Cl, Al, Ga, In, Си и Ag. Возможно, что это связано с резонанс-
ной передачей энергии, поглощенной «самоактивированными» центрами или
центрами синего свечения Си и Ag, ионам Мп2+. Люминесценция редкоземель-
ных ионов (Pr, Nd, ТЬ, Ег, Тш) также возникает в присутствии специальных
добавок (Си, Ag или Li).
11.2. ФОСФАТНЫЕ И НЕКОТОРЫЕ БЛИЗКИЕ К НИМ СИСТЕМЫ
Фосфаты
Среди фосфатных люминофоров, используемых в люминесцентных лампах,
наибольшее значение имеют те из них, в основе которых лежат фосфаты каль-
ция и, в частности, галофосфаты состава Са3(РО4)2 Ca(F, С1)2 (апатит). Важное
значение приобрели и другие фосфатные люминофоры, главным образом на основе
двойных фосфатов металлов II группы (рис. II.4). Фосфаты цинка — основа
важного класса катод ©люминофоров с красным свечением (активатор Мп).
Фосфаты кальция, а также кальция и магния при активации Т1 дают хорошие
ламповые люминофоры с УФ-излучением; фосфаты стронция, активирован-
ные Ей, — эффективные малоинерционные катодолюминофоры. Синтезированы
и люминофоры на основе пирофосфатов некоторых металлов, например стронция
и бария, активированные Ti или Sn.
Фосфор в фосфатах образует $р3-связи с координационным числом четыре
и тетраэдрической симметрией. Основная структурная единица в фосфатахо—
тетраэдрическая группа РО4 с расстоянием Р—О, равным 0,156 нм (1,56 А).
38
Диаграммы состояния некоторых важнейших фосфатных систем (рис. II.5)
показывают, что последние весьма тугоплавки. Для фосфатов характерны
полиморфные модификации (табл. II.4); некоторые высокотемпературные моди-
фикации можно стабилизировать добавками других фосфатов. Так, высоко-
температурные р-Са3(РО4)2 и P-Sr(PO4)2 становятся стабильными при
комнатной температуре, если введены добавки фосфатов цинка, магния и алю-
миния, что существенно при синтезе двойных фосфатных люминофоров, активи-
рованных Sn(II) [45—52]. Сведения о фазовом составе основы фосфатных люми-
нофоров имеют большое практическое значение, так как различные фазы часто
резко различаются по люминесцентным свойствам. В фосфатных системах, на-
пример ZnO—СаО—Р2О6 или ZnO—SrO—Р2О5, образуются как твердые рас-
творы, так и индивидуальные химические соединения, в частности, с общей
формулой (МеМе')3(РО4)2 [45—52].
Рис. II.4. Спектральное распределение энергии излучения некоторых фосфат-
ных люминофоров:
1 — Са3(РО4)-Т1; 2 — BaP2O7-Ti; 3 — сс-Са3(РО4)2-Sn; 4 — P-Ca3(PO4)2Sn; 5 —
0-Zn3(PO4)2-Mn; в — (Sr, Mg)3(PO4)t-Sn; 7 — (Sr, Al)x (PO4V-Sn; 8 — (Sr, Zn)3(PO4)2.Sn;
9 — (Sr, Cd)3(PO4)2-Sn.
Большое значение в технологии фосфатных люминофоров имеет дикальций-
фосфат состава СаНРО4, являющийся основным сырьем для получения фосфата
и галофосфата кальция методом спекания в твердой фазе. Исследования, прове-
денные в СССР и за границей, показали, что размер и форма частиц порошка
СаНРО4 определяют размер и форму частиц люминофора и тем самым его каче-
ство. Поэтому технологии получения СаНРО4 уделяется большое внимание
[53—55].
Области образования различных фаз фосфата кальция для системы
СаО—Р2О6—Н2О представлены на диаграмме рис. II.6, по данным Ван Везера [56,
с. 395]. Кроме СаНРО4 и СаНРО4-2Н2О, в зависимости от условий (pH, темпе-
ратура, концентрации реагентов), могут осаждаться: Са4Р2О9; Са(Н2РО4)2
и его дигидрат; Са3(РО4)2-Н2О; СаН(РО4)3-ЗН2О; Са4Н(РО4)3-ЗН2О;
Са10(ОН)2(РО4)6. При наличии в растворе NH4OH могут образовываться
также кальций-аммоний фосфаты различного состава [CaNH4PO4-7Н2О;
Ca2NH4H7(PO4)4 -2H2O и др.] [53].
Изучение условий получения СаНРО4 из водных растворов хлорида каль-
ция и диаммонийфосфата, как это принято в производстве, показывает, что при
температуре до 50° и большой скорости смешения растворов образуется в основ-
ном гидроксилапатит [54]. В интервале 50—60° при осаждении получается
смесь СаНРО4 с дигидратом — СаНРО4-2Н2О.
Наиболее благоприятная температура для получения дикальцийфосфата
лежит выше 65°, так как при этом осаждается безводный СаНРО4.
Как показано в работе [57], при использовании периодического метода
осаждения (приливание раствора одного реагента в другой) образуются осадки
39
рг0<> CrtO^2P?0b Cocmat^Macc.
2СаО 3P,QS
Q
Рис. 11.5. Диаграммы со-
стояния системы CaO—Р2О5
(a), MgO—Р2О5 (б) и
ZnO—Р2О5 (в):
I —а-модификация 3ZnO- PtO4+
-U ^-модификация 2ZnO-P.O4;
II -a-модификация ZnO - PtO4 +
Ц- ^-модификация 2ZnO-PtO4.
Содержание P205,масс. %
T а б л и ц a 11.4
Некоторые характеристики важнейших фосфатов
Соединение Тип кристаллической решетки Температурная область сущест- вования, °C Добавки, стаби- лизирующие высокотемпера- турные модифи- кации при комнатной температуре
СаНРО4 • 2Н2О ♦ Моноклинная 69—110 ** —
СаНРО4» * Триклинная Ниже 400 —
a-SrHPO4 » — —
p-SrHPO4 Орторомбическая
cc-Ca2P2O7 » — —
р-Са2Р2О7 Тетрагональная —. —
у-Са2Р2О7 » — —
а-Са3(РО4)2 Моноклинная Выше 1180 Ва
а'-Са3(РО4)2 Выше 1430 —
0-Са3(РО4)2 (витколит) Ромбоэдрическая Ниже 1180 Mg, Al, Мп (при темпера-
турах выше
1180)
Sr3(PO4)2 » Ниже 1325 —
p-Sr3(PO4)2 » Выше 1325 Са, Mg, Zn, Al
a-Zn3(PO4)2 » Выше 942 Zn2P2O7
₽-Zn3(PO4)2 — —
ЗСа3(РО4)2 • CaF2 (фтор- Гексагональная — —
апатит)
Саю(ОН)2(РО4)в (гидро- » —
ксилапатит)
* Схема превращений: СаНРО4-2Н2О-------►СаНРО4.
“ В зависимости от давления водяного пара в газовой фазе.
400° 700°
** Схема превращений: СаНРО4----------► у-Са2Р2О7; СаНРО4----------► р-Са2Р2О»;
114D0
СаНРО4------> а-Са2Р2О7.
41
нестехиометрического состава, что связано с возможностью появления в реак-
ционной смеси локального избытка ионов Са2+ Или НРО^-. Строго стехиометри-
ческий состав СаНРО4 может быть получен при использовании непрерывного
метода осаждения (при регулировании процесса по pH), когда в заданном опти-
мальном режиме работы реактора непрерывного действия будут одновременно
сливаться растворы реагентов.
Однако в настоящее время в основном применяют двухстадийный периоди-
ческий метод синтеза СаНРО4 [55]. Сначала на холоду получают СаНРО4 -2НаО
из подкисленных растворов СаС12 и (NH4)2HPO4, а затем нагреванием в водной
среде его переводят в СаНРО4. Такой метод позволяет получать СаНРО4 с не-
обходимым размером кристал-
лов. Для получения средних
фосфатов существуют два ме-
тода: осаждение из водных рас-
творов, например по реакции
2(NH4)2HPO44-3MeCl2 ->
-> Me3(PO4)2+4NH4Cl+2HCI
и синтез в твердой фазе.
Метод осаждения из рас-
творов применяют, например,
для получения фосфатов цинка.
В случае же кальция при про-
ведении реакций в водных
растворах трудно подобрать
условия, полностью исключа-
ющие образование гидроксил-
апатита, соединения непостоян-
ного состава, отрицательно
влияющего на качество люми-
нофоров. Гидроксилапатиты
изоструктурны с галофосфа-
тами (фтор-и хлорапатитами) и
легко входят в их кристалли-
ческую решетку.
Фосфаты в твердой фазе можно синтезировать по-разному. В частности,
фосфаты кальция и стронция получают по одному из следующих вариантов:
Молярное
СаО '^отношение., Са ; Р - 2 1
СаТ=5 з
/ Ci P^J 2
са5он(ро4)3
L Са3(Р04)
апатита) X
Ca.P,0
60
Са:Р=/ /
80
Раствор
► Ок
5Са.:Р-/2
са(н2ро4)3_--'
Рис. II.6.
20 4<7 ео
Содержание Р2 05, масс. 7»
Фазовая диаграмма системы
Р2О5—СаО—Н2О при 25 °C.
1000°
Ме2Р2О7-]-МеСО3----> Ме3(РО4)2+СО2
1100°
3MeCO3+2(NH4)2HPO4------> Me3(PO4)2-|-4NH3-|-3CO2+2H2O
Структура фторапатита кальция, кристаллизующегося в гексагональной
решетке (рис. II.7), характеризуется наличием двух положений ионов металла.
Ионы, находящиеся в первом положении, связаны с ионами кислорода, а ионы
во втором положении связаны с ионами кислорода и фтора.
Структура хлорапатита несколько отличается от структуры фторапатита,
однако несмотря на это имеется непрерывный ряд твердых растворов между
фтор- и хлорапатитами. Как раз такими твердыми растворами являются про-
мышленные галофосфатные люминофоры, в которых при изменении соотношения
между фтором и хлором существенно изменяются люминесцентные свойства.
Для рыхлой структуры апатитов характерна возможность значительных откло-
нений от стехиометрии с образованием дефектных структур.
В соответствии с имевшимися ранее представлениями [58] химизм обра-
зования галофосфатов при использовании в качестве исходных компонентов
СаНРО4СаСО3 и CaF2 описывается следующими последовательно протека-
ющими при нагревании шихты реакциями:
450°
2СаНРО4-----► Са2Р2О7-|-Н2О
42
800
СаСО3---> СаО4-СО2
800°
СагРгС^-^-СаО • > Саз(РО4)2
1150°
Саз(РО4)24-СаР2 > ЗСаз(РО4)2 • CaF2
Более поздние данные [59] свидетельствуют о том, что представление о чисто
твердофазном ходе реакции неверно, и в образовании фторапатита большую
роль играют транспортные реакции с участием газообразного оксифторида
Рис. II.7. Структура фторапатита кальция.
Одинарные кружки изображают атомы, лежащие в плоскости рисунка (пе-
ресечение оси с); двойные кружки — два атома, лежащие ниже и выше
плоскости рисунка.
фосфора — POF3. По мнению авторов работы [59], POF3 образуется уже при
600—7009 в соответствии с реакцией
' 18Ca2P2O7+14CaF2-----► 5[ЗСаз(РО4)2«СаР2]4-6РОРз
и далее POFj реагирует
18СаСО3+6РОР3------> 3Cae(PO4)2«CaF24-18CO2+8CaF2
В работе [60] исследовано образование хлорапатита и показано, что на на-
чальной стадии нагревания смеси состава 6СаНРО4 + ЗСаСО3 + СаС1а появ-
ляется хлорсподиозит
500-700°
Са2Р2О7-[-СаСОз4-СаС12 > Са4(РО4)2С12-|-СО2
Который в дальнейшем при повышении температуры, взаимодействуя с Са2Р2О7
И СаСО3, превращается в хлорапатит
»оо°
Са4(Р О4)2С12+2Са2Р2O7—2СаСОз " ЗСа3(Р 04)2 • СаС12-[-2СО2
Результаты исследования, приведенные в работе [61], показывают, что
Представления о механизме образования фтор- и хлорапатита не распростра-
няются на фтор-хлорапатит.
43
Наличие хлорида кальция (или CaC03 + NHaCl, взаимодействующих
с образованием СаС12) в пихте галофосфатных люминофоров определяет химизм
и кинетику образования фтор-хлорапатита. Согласно работе [61], при 550—
6509 хлорид, карбонат и фторид кальция образуют эвтектический расплав.
Последний, взаимодействуя с Са2Р2О7 и СаСО3, при указанной температуре
дает фтор-хлорапатит. Благодаря участию в процессе жидкой фазы, скорость
формирования фтор-хлорапатита превышает таковую для фторапатита,
а отсутствие стадии образования хлорсподиозита объясняет более высокую
скорость образования фтор-хлорапатита, чем хлорапатита.
Близость размеров и сходство формы частиц исходного СаНРО4 и полу-
ченного из него (при прокаливании) люминофора показывают, что частицы
СаНРО4 или вернее образующегося из него Са2Р2О7 являются той основой,
к которой присоединяются остальные входящие в галофосфат вещества [62, 63].
Рис. II.8. Спектральное распределение энергии излучения
цинккальций фосфатных люминофоров различного состава,
активированных Sn.
Содержание' Zn3(PO4), (в мол. %) указано на кривых.
Наряду с многочисленными вариантами усовершенствования твердофазных
методов синтеза галофосфатов, появились также некоторые патенты, описыва-
ющие условия получения фтор(хлор)-апатитов кальция из водных растворов.
Однако практически ценных результатов в этом направлении пока не получено.
Наконец, следует остановиться на процессах, происходящих при прокали-
вании фосфатов в атмосфере, которая содержит примесь водорода. Этот вопрос
важен потому, что при синтезе двойных фосфатных люминофоров, активиро-
ванных Sn, а также других фосфатных люминофоров, требующих восстанови-
тельной атмосферы для перевода активатора в состояние низшей валентности,
применяют прокаливание в смесях азота с определенным количеством водорода.
В работах [64, 65] показано, что водород восстанавливает фосфаты щелочно-
земельных металлов и цинка до соответствующих фосфидов или элементарного
фосфора:
Ме3(РО4)24-5Н2----> ЗМеО4-5Н2О-]-Р2
Ме3(РО4)2+8Н2-----► Ме3Р2+8Н2О
Легче всего указанные процессы идут непосредственно с фосфатом цинка
или с теми системами, в которых он присутствует. Это вызывает необходимость
строгого контроля за количеством водорода, температурой и другими условиями
синтеза, чтобы свести к минимуму образование фосфора и фосфидов. С другой
44
стороны, если образовавшиеся сравнительно малолетучие фосфиды остаются
в люминофоре, необходимо дополнительное прокаливание люминофоров при
температуре начала улетучивания темных налетов фосфидов.
Разнообразие люминесцентных свойств фосфатных систем определяется
как наличием большого числа полиморфных модификаций, так и рыхлостью
соответствующих им кристаллических решеток. Последнее — необходимое
условие как образования твердых растворов, модифицирующих основу люмино-
фора, так и внедрения самых разнообразных активаторов, которые изоморфно
замещают соответствующие ионы металлов в решетке люминофора. В качестве
активаторов в фосфатных системах наиболее часто используют Мп (в случае
фотовозбуждения необходимы сенсибилизаторы, например, Sb, Sn или Се),
Се (III) или Т1, излучающие в УФ-области спектра; Sn(II) — дающее широкие
полосы излучения в красно-оранжевой части спектра, а также Ti.
В качестве примера влияния состава основы на люминесцентные свойства
на рис. II.8 в системе СаО — ZnO—Р2О5, активированной Sn, представлено изме-
нение спектральных характеристик полученных люминофоров.
В галофосфате кальция, активированном Sb и Мп, спектральные характери-
стики люминофора изменяются как при изменении состава галофосфата (соотно-
шения между F и С1 в решетке апатита), так и при изменении соотношения между
Sb и Мп) (см. рис. IV.9, стр. 82).
Описан ряд фосфатных люминофоров с использованием в качестве актива-
торов Cu2+, In, Ga, Ge, Pb [66, 67]. Количество вводимого активатора в фосфат-
ных системах колеблется от десятых долей процента до 1—2%.
Арсенаты
Соли мышьяковой кислоты (H3AsO4) — арсенаты по своей структуре и
химическим свойствам близки к фосфатам. Известны средние (Me3AsO4), кис-
лые (Me2HAsO4, MeH2AsO4) и основные арсенаты (п 4- 2) МеО -nAs2O6. Терми-
ческим разложением одно- и двузамещенные арсенаты могут быть переведены
в пироарсенаты (Me4As2O7). Основные соли образуются при взаимодействии
арсенатов с избытком окислов при высокой температуре. Арсенаты металлов
I группы сравнительно хорошо растворимы в воде, а ортоарсенаты металлов
II группы в воде не растворяются; однако они растворимы в минеральных ки-
слотах. При высокой температуре арсенаты легко восстанавливаются.
Нерастворимые арсенаты, в частности кальция и магния, могут быть полу-
чены взаимодействием водных растворов арсенатов щелочных металлов и соот-
ветствующих солей, а также спеканием окислов или карбонатов с As2O6. Для
получения арсенатов может быть также применен метод окисления арсенитов
(Me3AsO3) кислородом воздуха или некоторыми окислителями (растворами
гипохлоритов, перекиси водорода и др.).
Кристаллохимическпе свойства арсенатов исследованы недостаточно,
однако установлено, что они имеют много общего с соответствующими фосфатами.
В этой связи следует отметить возможность получения своеобразных соединений;
арсенофосфатов, в которых часть ионов POj“ замещается ионами AsO|“; гало-
арсенатов, имеющих по аналогии с галофосфатами структуру апатита; двойных
арсенатов, образуемых, например, арсенатами металлов 1 —111 групп периоди-
ческой системы. Некоторые из перечисленных соединений обладают люмине-
сцентными свойствами, например: основные арсенаты магния; магний-литий
и алюминий-лптий арсенат; арсенофосфаты металлов II группы; галоарсенаты
кальция.
Практическое значение в качестве основы люминофоров имеют только основ-
ные арсенаты магния и двойные арсенаты магния и лития. В системе MgO—As2O6,
кроме среднего арсената магния, установлено существование двух основных
солей — Mg3As2OP и Мц6А82О1х. В работе Травничека и Крегера [68] показано,
что наибольшую термическую стойкость при 1100—1200° имеет соединение
Mg6As2O11. Это же соединение является единственным в системе MgO—As2O6,
дающим люмпнесценпию при активации Мп. Избыток As2O5 приводит к сниже-
нию интенсивности люминесценции, так как образующийся при этом нелюмине-
сцируюшпй ортоарсеват махпия поглощает ультрафиолетовое излучение.
45
Кривые поглощения люминофоров 6MgO-As2O5 при различном содержа-
нии Мп (рис. II.9) показывают, что введение Мп приводит к появлению погло-
щения в области длин волн, больших 350 нм. В зависимости от валентности Мп
спектр излучения в MgO *As2O6 имеет либо широкую зеленую полосу с Хтах =
= 505 нм для Мп2+, либо пять узких полос в красной части спектра при X —
= 620, 630, 640, 650 и 658 нм для Мп4+. На практике используют лишь люмино-
форы с излучением в красной части
спектра, отличающиеся высокой темпера-
турной стабильностью [62J.
При частичном восстановлении Мп,
а это происходит при длительной работе
люминесцентных ламп, в спектре свече-
ния люминофора появляется зеленая по-
лоса излучения Мп2+.
Люминофор Mg6As2Oxx получают в ре-
зультате продолжительного спекания смеси
окислов при 1100—1200°. Благоприятное
действие оказывают минерализаторы —
небольшие количества борной кислоты, а
Рис. II.9. Спектры поглощения также фториды, вводимые в шихту для
люминофора 6MgO-As2O5-яМп. снижения температуры и длительности
прокаливания. Влияние указанных мине-
рализаторов проявляется и при синтезе двойного литий-магний арсената,
индивидуальной фазы, образующейся в системе MgO—Li20—As2O5 при со-
держании в арсенате более 0,8 моль Ы2О. Литий-магнийарсенат имеет
несколько уменьшенные параметры элементарной ячейки по сравнению
с MgeAs2Oxx.
Кривые спектрального распределения энергии излучения люминофоров,
содержащих литий, лишь немного изменяются, однако значительно возрастает
стабильность излучения при повышенной температуре.
11.3. СИЛИКАТНЫЕ И НЕКОТОРЫЕ БЛИЗКИЕ К НИМ СИСТЕМЫ
Силикаты
Из силикатов в производстве люминофоров наибольшее значение имеет
силикат цинка, используемый главным образом в качестве основы некоторых
катодолюминофоров (при активации Мп), этой же цели служат силикаты каль-
ция и магния, а также отдельные двойные силикаты (цинка и бериллия, магния
и кальция, кальция и алюминия и др.). Силикаты бария, активированные РЪ,
а также некоторые сложные силикатные системы (Zn—Ва или Zn—Sr) исполь-
зуют в качестве люминофоров с УФ-излучением. Описано применение тройного
силиката бария, стронция и лития, активированного Се и Мп, и ряда других
силикатных люминофоров в люминесцентных лампах высокого давления. Ранее
в люминесцентных лампах низкого давления широко использовали смеси
вольфрамата магния и двойных цинк-бериллии силикатов, активированных Мп.
Однако с появлением галофосфатных люминофоров использование много-
компонентных смесей люминофоров оказалось нецелесообразным. Известное
значение для ламп с улучшенной цветопередачей имеет силикат кальция,
активированный Мп и РЪ. Достоинство силикатов как основы люминофоров —
их сравнительно высокая химическая и термическая стойкость, а также ста-
бильность при действии электронного пучка, отсутствие окраски и способность
к образованию широких областей твердых растворов между собой.
Силикаты металлов II и III групп периодической системы хорошо изучены
в связи с их применением в технологии вяжущих веществ и керамики [69—73].
Характерная особенность всех силикатных структур — наличие в них
комплексного тетраэдрического иона [SiO4]4~. Расстояние Si—О в последнем
сохраняется почти постоянным у всех силикатов и равно 0,162 нм (1,62 А);
расстояние О—О колеблется в интервале 0,262—0,264 нм (2,62—2,64 А). Из пяти
групп силикатных структур (по классификации Махачки [см. 71, с. 1039]) наи-
большее значение для люминесцпрующих систем имеют две:
46
ортосиликаты с обособленными комплексами тетраэдров [SiO4]4-; в них
связь между тетраэдрами осуществляется только через катионы;
метасиликаты с тетраэдрами, соединенными в цепочки [SiO3]2“ через ионы
кислорода.
Однако при твердофазных синтезах силикатов в качестве промежуточных
могут быть и другие, не рассматриваемые здесь структуры. Следует также под-
черкнуть наличие у силикатов ряда полиморфных превращений, имеющих
большое значение при синтезе люминофоров (а-, 0- и y-Zn2SiO4; а-, а'-, 0- и
y-Ca2SiO4 и др.).
К структурам первой группы относятся ортосиликаты Са, Be, Zn, Mn2+,
а также алюмосиликаты состава Al2O3-SiO2. Типичная структура одного из
силикатов этого типа (форстерит) по-
казана на рис. 11.10, а в табл. II.5
приведены данные, характеризующие
свойства некоторых из них.
Все ортосиликаты являются ту-
гоплавкими соединениями, не рас-
творяются в воде, но, кроме фена-
кита, хорошо растворяются в мине-
ральных кислотах. Силикаты алюми-
ния отличаются высокой химической
стойкостью.
Рис. 11.10. Структура форстерита:
а — плотнейшая упаковка атомов; б — услов-
ное изображение.
Рис. 11.11. Схематическое изображе-
ние структур пироксенов (а) и
амфибол (б).
Во второй группе силикатных структур (метасиликатах) возможны два
типа соединений: пироксены и амфиболы (рис. 11.11), однако для люминофоров
имеют значение только первые. Наиболее характерные структуры пироксенов
(энстатита и диопсида) изображены на рис. 11.12. В них цепочки тетраэдров
[SiO4]4- связаны между собой ионами Mg и Са.
Свойства некоторых соединений группы пироксенов представлены
в табл. II.6.
Диаграммы состояния наиболее важных силикатных систем изображены
на рис. 11.13.
Самыми легкоплавкими из всех рассмотренных индивидуальных силикатов
являются силикаты цинка. Ввиду того что силикатные люминофоры синтези-
руют, как правило, в твердой фазе спеканием смесей окислов или чаще карбо-
натов с кремнеземом, большое значение имеет кинетика процессов взаимодействия
этих веществ. При этом существенную роль играет степень их дисперс-
ности. Как показали работы Келера с сотрудниками [74], в твердофазных реак-
циях этого типа в первую очередь появляются ортосиликаты; метасиликаты
образуются при более высоких температурах, причем их образованию способ-
ствует избыток двуокиси кремния.
47
Синтезу силикатов способствует присутствие в реагирующих смесях добавок
галогенидов щелочных и щелочноземельных металлов, действующих в качестве
минерализаторов *.
а
-*-0,520нм (5,20А}
Рис. 11.12. Структура энстатита (а) и диопсида (б).
б
0,524 нм (5,24А)
Механизм действия этих добавок пока нельзя считать твердо установленным,
но есть основание предполагать, что большое значение имеет перенос веществ
посредством летучих галогенидов кремния [73, 75]. В качестве минерализующего
2600
а
2400
2200
ж
СаО +Ж
§
2000
Ранкинит I
Ca3SiOs-'M
1800
\ /
^севдоволластонигт
1600
Кристобалит*
zCa3SiOs +
~ Са2 SiO4
Ca3SiO5+CaO
1400
1200
Тридимит +ж
Тридимит+псевдоволластонит
Ранкинит+псевдоволластонит -
1000
- Тридимит + волластонит
800
Кварц+волластонит
О 10
SiO2
20 30 у
CaO S1O
- Ранкинит
rf-Ca2SiO4
Ca3SiO5+d-Ca2SiO4
</~Са2 SiO4+CaO
Ранкинит-t- y-Ca.2 SiO4
r-Ca2SiO4 + caO
J-----I---1___
4(Г 50/^60 \70\в0 90 100
1O2 3CaO-2SiO2\ ЗСаО SiO2 CaO
&
I
я
о
2CaOSiO2
'лоста6,масс. %
* В некоторых случаях при введении в смеси МеО—SiO2 фторидов обра-
зуются сложные соединения — фторсиликаты. Фторсиликат магния может
служить основой для люминофоров (см. главу IV).
48
агента в силикатной технологии применяют также водяной пар. Оба эти приема
используют и при синтезе силикатных люминофоров.
Как уже упоминалось, в качестве активатора в силикатных системах исполь-
зуют главным образом Мп2+ в количестве 1—5%. В том случае, если при фото-
возбуждении отсутствует поглощение на центрах Мп2+, в решетку вводят сенси-
билизаторы: Се и др.
Марганец дает люминофоры с излучением от зеленой до оранжево-красной
областей спектра (рис. 11.14). Введение в качестве второго активатора РЬ позво-
ляет получить двухполосные люминофоры (CaSiO3-РЬ-Мп).
Церий дает свечение в ультрафиолетовой и синей части спектра с очень ма-
лой инерционностью. К таким люминофорам относятся катодолюминофоры:
кальций-алюминий силикат — 2СаО-А12О3-SiO2-Се (геленит), двойной каль-
ций-магний силикат — 2СаО MgO -2SiO2-Ce. Эти люминофоры имеют Zmax =
= 405-410 нм (рис. 11.15).
В качестве активатора используют также Ti4+, например в люминофоре
СаО -MgO SiO2 Ti, применяемом в экранах проекционных кинескопов
1900
1600
MgO+Mg2 S1O4
6
MgO+W
1890
1850
Форстерит + Ж
Mg2S104 +
+ MgSiO3
Жидкость ВI
1695
1557
MgjjSKV
+ ж
Жидкость А
Жидкость А+В \
Кристобалит*
+ жидкость В
1500'---
20
MgO
1543___________
кристобалит
40
Mg2 S1O4
60^ 80
MgSiO3 ' Клиноэстатит
Состав, масс. %
* Жидкость А
109
SiO2
1700
. 1600
Рис. 11.13. Диаграмма состоя-
ния систем:
Р
§ 1500
6
J 1695° \
4 —--------\
\
Кристобалит
+Ж
1470°
1975
/
/
/
I
’ZnO+Ж I
а — СаО—SiOjj б — MgO—SiO2;
е — ZnO—SiO2.
1400
1432
1512
Тридимит + Ж
1 1507
Zn2SiO4+#<
ж
Тридимит +Zn2SiO4
ZnO*Zn2SiO4
« т
1^
$
Ж
$
1№-----1----'---lJ--l_
0 20 40 60-80
SiO.
100
ZnO
Состав, мол. %
4 Заказ 44
Таблица 11.5
Некоторые характеристики ортосиликатов металлов II и III групп
периодической системы
Формула Минералоги- ческое название Тип кристал- лической решетки Плотность, кг/м3 Температура плавления или разло- жения, °C
2MgO • SiO2 Форстерит Ромбиче- ская 3,12-3,33 £пл = 1890
MgO • CaO -SiO2 Монтичел- лит » 3,2 ^разл —1493
2BeO • SiO2 Фенакит Ромбоэдри- ческая 2,96-3,0 ^разл = 1560
2ZnO • SiO2 Виллемит » 3,9-4,2 *пл = 1509
A12O3 • SiO2 Силлиманит Ромбиче- ская 3,2 ^пл —1860
3A12O3 -2SiO2 Муллит » 3,1 ^разл = 1810
(рис. 11.15, кривая 3). Можно предполагать, что активаторы в силикатных
люминофорах изоморфно входят в кристаллические решетки соответствующих
соединений. Однако прямые доказательства этого имеются только для случаев
активации Мп силикатов цинка и кадмия. Большое значение в технологии лю-
минофоров имеет также взаимная растворимость силикатов различных металлов,
Рис. 11.14. Спектральное распределение энергии излучения некоторых сили-
катных люминофоров, активированных Мп:
1 — Zn2SiO4«Mn; 2 — 8ZnO-CdO-5,5SiO2-Mn; 3 — CdSiO3-Mn; 4 — (Zn, Be)2SiO4-Mn;
5 — Mg2SiO4-Mn; 6 — MgSiO3-Mn.
T а б л и ц a 11.6
Свойства некоторых силикатов группы пироксенов
Формула Минералоги- ческое название Тип кристал- лической решетки Плотность, кг/м3 Температура плавления или разложения, °C
MgO • SiO2 Энстатит Ромбиче- ская 3,1—3,2 При 1557° распад на форстерит и кри- стобалит
CaO • SiO2 Волласто- нит Триклинная 2,78—2,91 При 1125° переход в псевдоволластонит, плавится при 1540°
CaO-MgO-2SiO, Диопсид Моноклин- ная 3,2-3,3 1391°
Рис. 11.15. Спектральное распре-
деление энергии излучения неко-
торых двойных силикатных лю-
минофоров:
1 — 2СаО-MgO-2SiO2-Се; 2 — 2СаО -
- А1,Оз-SiO2-Ce; 3 — CaO-MgO •
- 2SiO2-Ti.
Рис. 11.16. Спектры поглощения
люминофоров на основе различных
силикатов:
а — Ba2Si2Oe (—); BaSi2Os (— — —); б —
Sr2SiO4 (—); SrSiO8 (------------); в —
MgSiOj.
Активация: 1 — отсутствует; 2 — Мп;
3 — Се; 4 — Се и Мп.
4*
например, силикатов цинка и бериллия, магния и кальция и др. Силикат мар-
ганца дает твердые растворы с Zn2SiO4 до 50 мол. %, а с Cd2SiO4 — до 20 мол. %.
Существование широких областей твердых растворов в силикатных системах,
наряду с наличием индивидуальных соединений [76], позволяет в некоторой
степени управлять люминесцентными свойствами при синтезе люминофоров.
По данным Фонда [77], в силикатной основе увеличение ионного радиуса
катиона смещает полосу излучения в коротковолновую область. Собственное
поглощение силикатов лежит в коротковолновой УФ-области (рис. 11.16).
В качестве классического примера силикатных люминофоров можно рас-
смотреть ZnaSiO4 -Мп. Граница поглощения Zn2SiO4 -Мп лежит при К <[ 225 нм;
введение Мп, однако, смещает ее в длинноволновую область и одновременно
появляются полосы поглощения, присущие самому Мп. В этом люминофоре Мп
дает полосы излучения (Хтах == 525 и 610 нм). Однако для появления при ком-
натной температуре второй (красной) полосы необходимы особые условия
формирования основы, в частности, введение добавок силикатов бериллия
или кадмия, изоморфных виллемиту. Это обстоятельство используют на прак-
тике при синтезе люминофоров с излучением в оранжево-красной области
спектра (см. главу IV).
Германаты, бораты, вольфраматы
Кристаллохимическими аналогами силикатов являются германаты. Для
технологии люминофоров особое значение имеет система MgO—GeO2. Фазовая
диаграмма этой системы (рис. 11.17) очень похожа на таковую системы MgO—
SiO2 и показывает наличие соедине-
ний 4MgO-GeO2, 2MgO-GeO2 и
MgO -GeO2. Исследован также и ряд
других германатов металлов II груп-
пы периодической системы [78—81].
Как и при синтезе силикатов,
взаимодействие между окислами
металлов и GeO2 протекает медленно
и ускоряется при введении в шихту
минерализующих добавок, в част-
ности фторидов, а также при про-
ведении синтезов в атмосфере водя-
ного пара. При введении в реакци-
онные смеси фторидов возможно
также образование и фторгермана-
тов. Германаты и фторгерманаты
магния при активации Мп4+ дают
весьма эффективные ламповые лю-
минофоры (см. главу IV).
Бораты металлов II группы по
люминесцентным свойствам анало-
гичны силикатам и германатам. Си-
стемы ZnO—В2О3 и CdO—В2О3, ак-
тивированные Мп и другими метал-
лами, подробно исследованы в рабо-
тах [82—85]. Эти люминофоры дают
интенсивное свечение в оранжево-
красной области спектра. Бораты
кальция, стронция и бария при ак-
тивации Се, Pb, Sn или Т1 излучают
в УФ-области спектра [86—89]. По-
казана возможность практического [использования люминофора SrO B2O3 Ce
в эритемных лампах [90]. Описан барийоктаборат, активированный Еп2+,
излучающий в области 400 нм [91].
Большой интерес представляют вольфраматные системы [92, 93, 94, 95, 62];
примером могут служить такие практически важные люминофоры, как CaW04
и MgWO4 (см. стр. 125, 1С0). Описаны также люминофоры с интенсивным свече-
нием на основе вольфраматов редкоземельных элементов [96].
2000 Г 2доо°
1355 ±30 °
1800 :
'-1700 ±20
I+Ц
1600-
/4 95-10 п ° ° 2 °a .Me ид каст и
" « . 1555*5 5 n с '
^IV+Ж' 1483 ±10° g
П О П О /-I
5
1400 -
о
VII Uff rv±w
? 1099 ±5°
° 1007± 10°
1200
woo -
доо
О
MgO
Рис. 11.17.
1116'
о
825
vw ivvf
IV + VII
п+тш
20 4Q 60 60 100
мол. % Ge С
Диаграмма состояния си-
стемы MgO—GeO2:
I — MgO; II—2MgO-GeO2 (тип форсте-
рита); III—MgO-GeO2 (тип клиноэнста-
тита); IV — MgO*GeO2 (тип энстатита); V —
4 MgO- GeO2; VI — 2MgO- GeO2 (тип шпинели);
VII — GeO2 (тип кварца); VIII — GeO2 (тип
рутила).
Вольфраматы явЛяются типичными самоактивированными люминофо-
6ами; введение примесей, как правило, приводит к тушению люминесценции,
сключение представляет pjjt который при введении его в CaWO4 в количе-
стве до 1 /о спосооствует увеличению интенсивности люминесценции и смеще-
нию максимума в ДЛицН0В0ЛН0ВуЮ область. Аналогично действуют некоторые
редкоземельные элементы. 1
Вольфраматы магцая? цинка и кадмия кристаллизуются в моноклинной
системе, а вольфраматы калЬцИЯ и других щелочноземельных металлов —
в тетрагональной. п.р0Ме соединений MeWO4, в системах МеО—WO3 устано-
влено еще существовав^ соединений Me2WO5 и Me3WO6, однако люминесци-
руют только фазы соСтава MeWO4. '
При синтезе Mgvvo4 благоприятное действие на кинетику процесса оказы-
вает избыток Mgu в Пщхте. Благодаря тому, что MgO не поглощает возбужда-
ющее ультрафиолетовое излучение, наличие его избытка в люминофоре не
снижает люмпнесценцпи Отклонение от стехиометрии в сторону увеличения
количества М О3 ухуд!цает люминесцентные свойства.
Рис. 11.18. Спектральное распределение энергии
издУчения некоторых вольфраматов.
Вольфраматы возбуЖдаЮтся Уф-излучением, рентгеновскими и у-лучами,
а также катодными Лучами (рИС. 11.18). В вольфраматах люминесценцию при-
писывают дискретному центру — иону WO|“. Измерения показали, что
затухание люминесценции происходит по экспоненциальному закону, дли-
neJin-4CTb посл®с®еЛ®5йя у всех вольфраматов очень мала: у MgWO4 она равна
с’ а У ^aVv(J4 еще меньше — б-КМс.
На практике твердофазны^ синтез вольфраматов производится спеканием
меси люминофорно-чцСть1х ОКИСлов и WO3 при 1000—1200°; при более высокой
емпературе вольфраматы частично разлагаются, что приводит к уменьшению
Интенсивности люми5есценцИИ F 1
В литературе опцСана также люминесценция некоторых двойных вольфра-
атов цпнка и магния [97] и кальция и кадмия [98], причем показано, что вве-
Нзлуч<ми1н°Г° К0МП0Невта приводит к плавному перемещению максимума спектра
11.4. ОКИСНЫЕ СИСТЕМЫ
Почти все чистые окислы металлов II, III и IV групп периодической системы
ладают люминесцеццц^ ПрИ фото_, катодном и рентгеновском возбуждении,
писан также ряд окисных люминофоров, активированных различными метал-
лами, в частности Ми, сг, и др. r г
Однако из люмицОфОрОВ на основе окислов металлов практическое зна-
ние имеют лишь нел1цОгие, а именно: самоактивированная окись цпнка, окись
алюминия, активированная Сг или Се. В последние годы производятся много-
численные исследования для изыскания новых стойких к электронной бомбар-
дировке люминофоров на основе как чистых окислов, так и шпинельных систем.
В этой связи следует отметить работы по активации окислов некоторых р. з. э.
и иттрия [99], по разработке люминофоров на основе цирконатов, алюминатов,
ванадатов [100—106].
Активаторы в окисных люминофорах, по-видимому, изоморфно входят
в решетку основы. Для того чтобы такое внедрение активатора произошло,
требуется достаточно высокая температура. Так, люминофоры на основе А12О3
получают при 1300—1400°. Исходным сырьем для синтеза основы рассматрива-
емых люминофоров обычно служат различные специально очищенные соли,
разлагающиеся при высокой температуре до окислов. Окись алюминия очень
удобно получать из алюмоаммонийных квасцов, окись магния — из сульфата
или нитрата, а окислы щелочноземельных металлов — из карбонатов. При этом
Рис. 11.19. Спектральное распределение энергии
излучения некоторых окисных люминофоров:
1 — А12О3»А1 ; 2 — ZnO-Zn; 3 — ZnO (после прокалива-
ния в кислороде); 4 — ZnO-Se; 5—А12О3-Сг.
следует иметь в виду, что часто определяющим фактором в обеспечении люмине-
сцентных свойств препаратов является нужный фазовый состав окисла. Напри-
мер, для люминофоров на основе А12О3 требуется образование фазы а-А1аО8;
превращение у-А12О3 -► а-А12О3 происходит при 1250 9С.
Природа люминесценции безактиваторных окислов пока окончательно
не установлена. Согласно данным Риля и Ортмана [107], люминофор на основе
ZnO можно рассматривать как дефектную структуру с вакансиями в под-
решетке кислорода. Эти вакансии создаются в результате диспропорциониро-
вания основы при высокой температуре, аналогично тому, как это происходит
при формировании самоактивированного ZnS. Изменяя условия синтеза можно
получить, кроме основной полосы излучения этого люминофора (Хтах =
= 505 нм) и другие. Так, препараты, синтезированные путем сжигания метал-
лического цинка в кислороде, при интенсивном возбуждении катодным пучком
дают полосу с максимумом при 385 нм (рис. 11.19). Образцы же, синтезирован-
ные при 1000—1100р в избытке кислорода или из исходного сырья, содержащего
окислители, показывают полосы излучения в оранжевой области спектра.
Положение этих полос зависит от условий приготовления окиси.
Попытки активации ZnO металлами до сих пор оказались безуспешными,
но возможна активация его некоторыми металлоидами, например Se (рис. 11.19).
Последний дает интенсивную полосу излучения в оранжево-красной области
спектра (Хтах = 610 нм) [108].
Люминесценция ZnO температурно неустойчива; интенсивность ее резко
уменьшается при повышении температуры. Видимое и УФ-излучение окиси
цинка отличается очень коротким периодом послесвечения [93, 109].
Применяемые на практике методы синтеза люминофора на основе ZnO
основаны на сжигании люминофорно-чистого сульфида цинка на воздухе при
~900°. Вероятно, наличие сернистых соединений играет специфически благо-
приятную роль в процессе формирования решетки люминофора [110, 111].
54
Чистая окись алюминия в результате прокаливания при 1600° дает пре-
парат, излучающий в УФ-области спектра (рис. 11.19). При активации А1аО3
металлами (Мп, Gr, Ti, Ga, Fe, V, Pt, Ge) получаются люминофоры с излученпем
в различных областях спектра. Наиболее изучены люминофоры, активирован-
ные Мп и Ст. Люминесценцию А12О3-Мп приписывают ионам Мпа+. При акти-
вации хромом спектр излучения имеет большое число узких полос в красной
и ближней инфракрасной областях спектра, но наибольшую интенсивность
имеют две близкие полосы с Хтах = 692,7 и 694,2 нм. Люминесценция синтети-
ческого рубина (А12О3 • Ст) подверглась очень детальному исследованию в связи
с применением монокристаллов этого вещества в лазерной технике. Высокая
термическая и химическая стойкость люминофоров на основе А12О3 обусловли-
вает ряд преимуществ при использовании их в качестве катодолюминофоров
(см. стр. 124).
Важное практическое значение имеет окись иттрия, активированная Ен,
обладающая интенсивным красным свечением и применяемая в качестве катодо-
люминофора.
Алюминиевые шпинели — алюминаты типа МеА12О4 также обладают срав-
нительно интенсивной люминесценцией при активации многими металлами.
Наиболее известны люминофоры, активированные Мп и Сг с излучением в зеле-
ной и красной областях спектра как при фото-, так и при катодном возбуждении.
Описаны также люминофоры на основе алюминатов р. з. э.
Из люминофоров шпинельного типа следует упомянуть также самоакти-
вированные ванадаты металлов II группы и ванадаты металлов III группы,
в частности ортованадаты иттрия и гадолиния, активированные Ен и дру-
гими р. з. э.
ЛИТЕРАТУРА
1. Марковский Л. Я., Смирнова Р. И. ЖНХ, I960, т. 5, с. 2042—2047;
1961, т. 6, с. 948—956; 1962, т. 7, с. 540—548. — 2. Чижиков Д. М., Счастли-
вый В. П. Селен и селениды. М., «Наука», 1964. 319 с. — 3. Жиров И. Ф. Люми-
нофоры. М., Оборонгиз, 1960. 478 с. — 4. Фридман С. А., Черепнев А. А. Све-
тящиеся составы постоянного и переменного действия. М., Изд. АН СССР,
1945. 142 с. — 5. Абрикосов Н. X., Банкина В. Ф., Парецкая Л. В. и др., Полу-
проводниковые соединения, их получение и свойства, М., «Наука», 1967. 175 с. —
6. Андреев В. М., Гусаков В. В., Томсон А. С. и др. Авт. свид. 212238, 1964;
Бюлл. изобрет., 1968, № 9. — 7. Yocom С. W. J. Electrochem. Soc., 1972, v. 119,
№ 3, р. 381—392. — 8. Woodbury Н. Н. J. Phys. Chem. Solids, 1966, v. 27,
№ 7—9, p. 1257—1261. — 9. Stuckes A. P., Parrel G. Ibid., 1964, v. 25, № 5,
p. 477—482. — 10. Миронов И. А., Марковский Л. Я. ЖПХ, 1964, т. 37, № 3,
с. 492—497.
11. Герасимов Л. И., Крестовников А. Н., Горбов С. И. Химическая термо-
динамика в металлургии. Т. 6. М., «Металлургия», 1974. 312 с. — 12. Lo-
renz М. В. J. Phys. Chem. Solids, 1962, v. 23, p. 939—947. — 13. Carides G.,
Fischer A. C. Solid State Comm., 1964, v. 2, p. 217—219. — 14. Физика и химия
соединений А и BVI. Пер. с англ. Под ред. С. А. Медведева. М., «Мир», 1970.
623 с. — 15. Bubinstein М. J. Cryst. Growth, 1968, v. 3—4, р. 309—312. —
16. De Nobel S. S. Phil. Res. Repts, 1959, v. 14, p. 361—372. — 17. Ива-
нов Ю. M., Дмитриева H. В., Ванюков A . В. Изв. АН СССР. Неорг. материалы,
1972, т. 8, № 8, с. 1396 —1400. — 18. Jordan A. S., Zupp В. R. J. Electrochem.
Soc., 1969, v. 116, № 7, р. 1264—1268. — 19. Бунделъ А. А ., Вишняков А. В.,
Зубковская В. Н. Изв. АН СССР. Неорг. материалы. 1970, т. 6, № 7, с. 1248—
1251. — 20. Вишняков А. В., Хариф Я. Л. Там же, 1972, т. 8, № 2, с. 217—219.
21. Вишняков А. В., Иофис Б. Г. Там же, 1974, т. 10, № 7, с. 1184—1186. —
22. A ven М., Halsted R. Е. Phys. Rev., 1965, v. 137, № 1 А, р. А228—А234. —
23. Wilke К. Т. Z. Phys. Chem. (Leipzig), 1955, Bd. 205, № 1—2, S. 73—77. —
24. Morvinski H. Monatsver. Deutsch. Acad. Wiss. Berlin, 1962, Bd. 4, S. 202 —
207. — 25. Хариф Я. Л. Автореф. канд. дисс. М., МХТИ им. Д. И. Менделеева,
1973. — 26. Anselmo В. A., Woodbury Н. Н. Bull. Am. Phys. Soc., 1964, v. 9,
₽• 248. — 27. Kremheller A., Farie S., Le vine A. K. J. Electrochem. Soc., 1960,
55*
v. 107, № 9, р. 753—757. — 28. Wanmaker W. L., Так M. G. A. Extended Ab-
stracts, Electronics Division, Electrochem. Soc., 1962, v. 11, p. 54. — 29. Froe-
lich H. C. J. Electrochem. Soc., 1953, v. 100, № 11, p. 496—507. — 30. A ven M.
Segall В. Phys. Rev., 1963, v. 130, № 1, p. 81—91.
31. Миронов И. А., Строганова И. M., Рыжкин Ю. С. и др. В кн.: Мате-
риалы V совещания по электролюминесценции. Ставрополь, 1974, с. 27. —
32. A ven М. A., Parodi J. A. J. Phys. Chem. Solids, 1960, v. 13, p. 56—59. —
33. Smith A. L. J. Electrochem. Soc., 1952, v. 99, № 4, p. 155—158. —
34. Cook R. Gr. J. Am. Ceram. Soc., 1968, v. 51, № 9, p. 518—521. — 35. Гро-
мов JI. А., Николаева T. M. ЖФХ, 1968, t. 42, № 10, c. 2513—2518. — 36. Поль-
ских Э. Д., Галактионов С. С., Бунделъ А. А. Ж. прикл. спектр., 1973, т. 19,
№ 5, с. 877—881. — 37. Бунделъ А. А. «Труды МХТИ им. Д. И. Менделеева»,
1960, вып. 31, с. 8—19. — 38. Larach S., Schrader R. F., Yocom P. M. J. Elec-
trochem. Soc., 1969, v. 116, № 4, p. 411—474. — 39. Левшин В. Л., Ара-
пова Э. Я., Блажевич А. И. и др. «Труды ФИАН», 1963, вып. 23, с. 64—65. —
40. Wendel G. Acta Phys. Acad. Sci. Hung., 1962, v. 14, № 2, p. 145—153.
41. Бунделъ А. А. Изв. АН СССР. Сер. физ., 1951, т. 15, № 6, с. 737—
741. — 42. Aven M., Woodbury H. H. Appl. Phys. Lett., 1962, v. 1, p. 53. —
43. Марковский Л. Я., Миронов И. А., Рыжкин Ю. С. Изв. АН СССР. Сер.
физ., 1969, т. 38, № 6, с. 961. — 44. Presland М., Marshall R., Franks J. J. Elec-
trochem Soc., 1964, v. Ill, p. 168—172. — 45. Hummel F., Katnack F. Ibid.,
1958, v. 105, p. 528—533. — 46. Koelmans H., Cox A. Ibid., 1957, v. 104,
p. 442—445. — 47. Wanmaker W., Radielovic D. Electrochem. Technol., 1964,
v. 2, p. 16—20. — 48. Sarver J., Katnack F. J. Electrochem. Soc., 1959, v. 106,
p. 960—963. — 49. Sarver J., Hoffmann M., Hummel F. Ibid., 1962, v. 109,
p. 15—18. — 50. Hoffmann M. Ibid., 1963, v. 110, p. 1223—1227.
51. Brown F., Hummel F. Ibid., 1964, v. Ill, p. 660—665. — 52. Бабиц-
кая P. А., Марковский Л. Я. ЖПХ, 1962, т. 35, с. 1434—1441; 1962, т. 36,
с. 1186—1192. — 53. Comstock А., Jarnack S., Mooney R. Ind. Eng. Chem., 1959,
v. 51, p. 325—328. — 54. Aia M., Goldsmith R., Mooney R. Ibid., 1961, v. 53,
p. 55—57. — 55. Казанкин О. H., Ижевский М. Б., Александрова Р. А. и др.
Авт. свид. 196217. — 56. Ван-Везер Д. Фосфор и его соединения. Пер. с англ.
Под ред. А. И. Шершевского. М., ИЛ, 1962. 687 с. — 57. Вассерман И. MJ
Силантьева И. Н.Ж. неорг. хим., 1965, т. 10, с. 1320—1327. — 58. Wanmaker WJ
Hoekstra А., Так М. Phil. Res. Repts., 1955, v. 10, p. 11—15; 1956, v. 11,
p. 1—4. — 59. Rabatin JGillody G. J. Electrochem. Soc., 1965, v. 112, p. 489—
492. — 60. Hoekstra A. H. Phil. Res. Repts, 1968, № 1—2, p. 1—78.
61. Казанкин О. H., Гранатштейн В. Е. ЖПХ, 1974, т. 47, № 8, с. 1872;
Изв. АН СССР. Сер. физ., 1974, т. 38, № 6, с. 1136—1141. — 62. Ouweltjes J• L.
In: Modern Materials, Advances in Development and Application., v. 5. New
York — London, 1965, p. 161—257. — 63. Markowsky L. Ya., Kazankin O. N.,
Taushkanova L. B. In: Proceedings of the International Conference on Lumi-
nescence. Budapest, 1966, p. 1235—1239. — 64. Бабицкая P. А. Автореф. канд.
дисс. Л., ГИПХ, 1964. — 65. Штрихман Р. А., Шойхет Д. Н., МарковскийЛ. Я.
ЖПХ, 1961, т. 34, с. 1912—1920. — 66. Wanmaker W., Spier Н. J. Electrochem.
Soc., 1962, v. 109, р. 109—114. — 67. Лущик Н. Е., Мууга И. И. «Труды ИФА
АН ЭССР» 1961, вып. 17, с. 67—71. — 68. Travnicek М., Kroger F. «Phisica»,
1952, v. 18, р. 33—37. — 69. Августинник А. И. Физическая химия силикатов.
Л.—М., Госхимиздат, 1947. 324 с. — 70. Евстропьев К. С., Торопов Н. А.
Химия кремния и физическая химия силикатов. М., Госстройиздат, 1956. 340 с
71. Эйтелъ В. Физическая химия силикатов. Пер. с англ. Под ред.
Н. Н. Курцевой. М., ИЛ, 1962. 1055 с. — 72. Будников П. П. Физико-химия
силикатов. М., Госстройиздат, 1964. 231 с. — 73. Будников П. П., Гинст-
линг А. М. Реакции в смесях твердых веществ. Изд. 3-е. М., Стройиздат, 1971.
488 с. — 74. Келер Э. К., Глушкова В. Б. Ж. неорг. хим., 1956, т. 1, с. 2283—
2293; 1957, т. 2, с. 1254—1258; ЖПХ 1961, т. 34, с. 212—214. — 75. Новосе-
лова А. В., Орлова Ю. В., Соболев Б. П. ДАН СССР, 1964, т. 159, с. 1338—1341. —
56
16. Колпакова А. А., Марковский Л. Я. ЖПХ, 1963, т. 36, с. 530—536; 1965,
т. 38, с. 1432—1436; В кн.: Химия и технология люминофоров. Л., «Химия»,
1964, с. 102—106. — 77. Fonda G. JOSA, 1957, v. 47, р. 877—880. — 78. Eulen-
berger G., Witman A., Nowotny H. Monatsh., 1962, v. 93, S. 123—128, 1046—
1054. — 79. Koelmans H., Verhagen C. J. Electrochem. Soc., 1959, v. 106,
p. 677—682. — 80. Гребенщиков P. Г., Торопов H. А. ДАН СССР, 1963, т. 153,
с. 842—850.
81. Кузнецов В. Г., Тананаев И. В., Шпирт М. Я. Ж. неорг. хим., 1964t
т. 9, с. 1934—1938. — 82. Harrison A., Hummel F. J. Electrochem. Soc., 1956,
v. 103, р. 491—493. — 83. Subbarao Е., Hummel F. Ibid., 1956, v. 103, p. 29—
33. _ 84. Леонов Ю. С. ДАН СССР, 1957, т. 114, с. 976—979; Ж. неорг. хим.,
1958, т. 3, № 5, с. 1245—1253; Автореф. канд. дисс., МГУ им. М. В. Ломоносова,
1962. — 85. Казема К. Ю., Линдере А. К., Лутс Л. А. «Труды ИФА АН ЭССР»,
1958, вып. 7, с. 34—40. — 86. Witzmann Н. е. a. Naturwiss., 1962, Bd. 48,
S. 181—183; 1962, Bd. 49, S. 180; 1964, Bd. 51, S. 101—102, S. 239. — 87. Witz-
mann H. Zur Physik und Chemie der Kristallophosphore, 1960, Bd. 1, S. 150,
165, 179. — 88. Witzmann H., Herzog G. Z. Phys. Chem., 1964, Bd. 225, S. 197—
208. — 89. Вицман Г. Опт. и спектр., 1965, т. 18, с. 622—627. — 90. Кочу-
гова Е. И., Гуревич И. М. В кн.: Химия и технология люминофоров. Л., «Химия»,
1964, с. 83—87.
91. Blasse G., Bril A. J. de Uries. JECS, 1968, v. 115, p. 977—981. —
92. Левшин В. Л. Фотолюминесценция жидких и твердых веществ. М.—Л.,
Гостехиздат, 1951. 456 с. — 93. Leverenz Н. An Introduction to Luminescense
of Solids. New York — London, Wiley Sons, 1950. 569 p. — 94. Москвин A. В.
Катодолюминесценция. Ч. II. Катодолюминофоры и экраны, М.—Л., Гостех-
теоретиздат, 1949. 700 с. — 95. Garlick G. Luminescent Materials. Oxford,
Claredon Press, 1949. 255 p. — 96. Borchardt H. J. Chem. Phys. 1963, v. 38,
p. 1251 —1254; 1964, v. 39, p. 504—509. — 97. Шарупин Б. H., Кудряшова Г. H.
«Труды ГИПХ», 1966, вып. 53, с. 100—106. — 98. ДемъянецЛ. Н., Томбак М. И.
Изв. АН СССР. Неорг. материалы. 1965, т. 1, с. 758—763. — 99. Chang N.
Brit J. Appl. Phys., 1963, v. 34, p. 3500—3504. — 100. Roop R. J. Electrochem.
Soc., 1965, v. 112, p. 181—184.
101. Brixner L., A bramson E. A. Ibid., p. 70—75. — 102. Chem. Eng., 1964,
v. 71, № 25, p. 96. — 103. Ind. Eng. Chem., 1964, v. 56, № 12, p. 9—10. —
104. Bril A., Wanmaker W. J. Electrochem. Soc., 1965, v. 112, p. Ill—117. —
105. Levine A., Pallila F. Appl. Phys. Let., 1964, v. 5, p. 118—120. — 106. Jo-
nes S. J. Electrochem. Soc., 1964, v. Ill, p. 311—315. — 107. RielN.,Ortmann H.
Z. Electrochem., 1956, Bd. 60, S. 149—151. — 108. Марковский Л. Я., Оршан-
ская H. С. Опт. и спектр., 1960, т. 9, с. 77—82. — 109. Архангельская В. А.,
Толстой Н. А. Там же, 1958, т. 5, с. 415—422. — 110. Thomsen S. J. Chem.
Phys., 1950, v. 18, р. 77—80.
111. Kroger F., Vink H. Ibid., 1954, v. 22, p. 250—255.
ГЛАВА III
ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ СИНТЕЗА
И ПРОИЗВОДСТВА ЛЮМИНОФОРОВ
111.1. ОСНОВНЫЕ СТАДИИ ТЕХНОЛОГИЧЕСКОГО^ПРОЦЕССА
Неорганические люминофоры синтезируют при высоких температу-
рах [1—7]. Это обусловлено, с одной стороны, необходимостью получения
веществ с хорошо сформированной кристаллической структурой, а с другой, —
необходимостью создания в этой структуре дефектов, сообщающих веществу
люминесцентные свойства. Специфика синтеза люминофоров состоит в том,
что он является чрезвычайно тонким, поскольку готовый продукт не должен
содержать посторонних соединений или элементов, гасящих люминесценцию
или приводящих к появлению дополнительных нежелательных полос в спектре
люминесценции. Последнее обстоятельство требует строгого контроля за чисто-
той в производственном процессе на всех его стадиях.
Сказанное относится как к выбору материалов аппаратуры, применяемой
в производстве, из которой нежелательные примеси не должны попадать в сырье,
шихту и люминофор, так и к воздуху, подаваемому в рабочее помещение систе-
мой приточно-вытяжной вентиляции. Воздух следует тщательно очищать от
пыли. Повышенные требования предъявляются к воде, на которой готовят
растворы и шихту и которую используют для промывки готового люминофора.
Воду следует специально очищать либо на ионообменных колонках, либо на
дистилляционных установках. (Допустимое содержание примесей тяжелых
металлов в воде — не более 10"?—10-8%).
Практически процесс получения люминофоров состоит из следующих техно-
логических стадий:
очистка сырья, предназначенного для синтеза полупродуктов;
приготовление полупродуктов;
приготовление шихты люминофора;
прокаливание шихты;
измельчение спека люминофора;
обработка поверхности порошка люминофора специальными реагентами.
Очистка сырья преследует цель получения люминофорно-чистых
полупродуктов.
Примеси по-разному влияют на свойства люминофоров различных классов,
поэтому и допустимое содержание примесей в полупродуктах люминофорной
степени чистоты неодинаково. Особенно чувствительны к влиянию примесей
цинксульфидные люминофоры. Исходный сульфйд не должен содержать при-
месей Fe, Мп, Со и Ni более чем по 1 • 10"5% , а Си — более 5 -10"6%. Люмино-
форы на фосфатной и силикатной основе допускают содержание примесей не
более 2 10~4%. Самоактивированные люминофоры, например, вольфраматы
и окись цинка, требуют примерно такой же степени чистоты. Поэтому обычно
исходное сырье, используемое для изготовления полупродуктов, дополнительно
тщательно очищают. В большинстве случаев эту операцию, как и синтез полу-
продуктов, ведут непосредственно при производстве люминофоров.
Как правило исходным сырьем для полупродуктов служат растворимые
соли, для очистки которых применяют различные химические методы, детально
описанные ниже.
58
Приготовление полупродуктов — обычный технологиче-
ский процесс, в котором используют обычно применяемые в неорганической
технологии методы и аппаратуру.
В качестве наиболее важных примеров можно привести получение сульфи-
дов цинка и кадмия путем взаимодействия растворов сульфатов цинка
и кадмия с сероводородом, а также дикальцийфосфата путем взаимодействия
растворов хлорида кальция и диаммонийфосфата.
Сульфиды цинка и кадмия — полупродукты, лежащие в основе производ-
ства обширного класса сульфидных люминофоров различного назначения;
дикальцийфосфат является основным компонентом в рецептурах фосфатных
люминофоров. Синтез полупродуктов обычно складывается из стадий: осажде-
ния, отмывки от маточного раствора и сушки. Поскольку гранулометрический
состав полупродуктов в значительной мере определяет гранулометрический со-
став получаемых из них люминофоров, постольку на этой стадии важен контролы
за параметрами, определяющими дисперсность полупродуктов. К этим пара-
метрам относятся: концентрация и температура исходных растворов, pH среды,
в которой ведется осаждение, и интенсивность перемешивания. Большое внима-
ние при получении полупродуктов следует уделять предотвращению возмож-
ности их загрязнения. Это обстоятельство диктует выбор материала аппаратуры
и ее конструкции.
Из полученных полупродуктов готовят шихту, которую затем
прокаливают. В наиболее простом варианте шихта — это смесь точно рассчи-
танных количеств веществ, входящих в основу люминофора, и соли активатора.
Последнюю можно вводить в шихту как в жидком, так и в твердом виде. Во мно-
гих синтезах (особенно люминофоров сульфидного типа) для ускорения форми-
рования в шихту добавляют минерализаторы, обычно называемые плавнями.
По большей части в качестве последних используют хлориды металлов I
и II групп периодической системы. Подобно активаторам, плавни вводят в жид-
ком или твердом виде. Например, шихту люминофора ZnS (50) CdS (50) •
• Ag(l-10-2) составляют из 0,5 кг ZnS, 0,5 кг CdS И растворов, содержа-
щих 20 г NaCl и 0,158 г AgNO3 (0,1 г Ag).
В более сложном варианте шихту готовят из веществ, которые только при
прокаливании превращаются в соединения, служащие основой люминофора.
Например, галофосфатные люминофоры синтезируют не из готового галофосфата
кальция, а из смеси порошков кислого фосфата, карбоната, фторидов и хлоридов
кальция (с добавками солей марганца и сурьмы, служащими активаторами),
взаимодействующих при прокаливании с образованием галофосфата
(см. стр. 42, 79).
При составлении шихты надо иметь в виду, что обычно избыток одного из
компонентов способствует более быстрому и полному протеканию реакций.
Однако в случае отклонения от стехиометрического состава следует учитывать
также оптические свойства введенного в избыточном количестве вещества,
в частности для фотолюминофоров, степень поглощения им возбуждающега
излучения, поскольку такое поглощение снижает эффективность люминофоров.
Степень дисперсности исходных компонентов шихты, а также тщательность
перемешивания ее имеют большое значение, так как в значительной мере опре-
деляют скорость образования и качество люминофоров. От степени дисперсности
исходных компонентов шихты часто зависит также и гранулометрический состав,
получаемых люминофоров.
Шихту перемешивают смесителями различного типа, а также
измельчающими устройствами (мельницы). Если шихту составляют и перемеши-
вают во влажном состоянии, то перед прокаливанием ее сушат, а затем просеи-
вают через сито.
Непосредственно люминофор формируется во время прокаливания,
поэтому люминесцентные свойства зависят от строгого соблюдения заданного
режима прокаливания шихты, т. е. от точности поддержания нужной темпера-
туры и длительности, условий контакта шихты с окружающей атмосферой и т. д.
При этом следует иметь в виду, что оптимальный режим прокаливания даже
одного и того же люминофора в сильной степени зависит от величины загрузки
пшхты, от габаритов и инерционности применяемой печи. Как правило, замена
печи (на печь другого типа или размера) всегда требует внесения изменений
в режим прокаливания.
59
Для некоторых люминофоров, например сульфидного класса, необходимо
устранять возможность окисления их кислородом воздуха, внедрение которого
в решетку изменяет свойства люминофора. По этой причине применяют ней-
тральную атмосферу, но чаще используют защитное действие верхней части слоя
I
Сушка
, Н2О
(дистиллированная вода)
I
Отмывка от плавней
Отстой и деканта-
ция
Фильтрация
Сушка
Просев
Усреднение партии
Готовый продукт
NHiOH
Промывные
воды
Промывные
воды
I
4
I
1
Схема 1. Получение сульфидных люминофоров, активированных Ag.
шихты. В таком случае верхний слой полученного после прокаливания продукта
отбрасывают. Иногда шихту прокаливают под защитным слоем веществ, слу-
жащих геттером. В некоторых случаях в качестве дезоксидирующего агента
используют серу; в количестве 2—5% ее вводят в шихту. Известное защитное
действие оказывает также присутствующий в шихте плавень.
Синтез других люминофоров, в частности некоторых двойных фосфатов
и силикатов, ведут в восстановительной атмосфере, например, в смеси азота
с небольшим количеством водорода, необходимого для восстановления актива-
тора (см. стр. 44, 84). В отдельных случаях газообразные восстановители могут
быть заменены на твердые (углерод, металлы), вводимые в шихту в виде по-
рошка [8, 9].
60
Обычно люминофоры — это неравновесные системы, их люминесцентные
свойства могут зависеть и от режима остывания; поэтому последний также регла-
ментируется.
Полученные люминофоры обычно просматривают под ультрафиолетовой
лампой для отбраковки частей и включений с недостаточной интенсивностью
свечения или светящихся другим цветом. Такая операция необходима потому,
что в тиглях или кюветах, в которых прокаливают шихту, всегда в той или иной
степени происходит взаимодействие люминофора со стенками и с окружающей
.атмосферой.
После очистки полученные люминофорные спеки измельчают и
просеивают через сита, а некоторые люминофоры даже специально класси-
фицируют по размеру зерен с целью выделения тех фракций, которые обеспечи-
вают повышенную яркость свечения и хороший слой. При измельчении люмино-
форов всегда следует считаться с происходящим при этом снижением яркости
свечения, поэтому режим измельчения также должен быть строго регламенти-
рован.
И, наконец, последние операции промывка и обработка по-
рошков люминофора специальными растворами. Этим операциям подвергают
не все люминофоры, однако большую часть катод о люминофоров. Промывка
служит для удаления из люминофора примесей непрореагировавших веществ,
введенных в шихту. Некоторые из этих веществ могут оказать вредное действие
при использовании люминофоров в готовых изделиях, например при действии
катодного пучка в электроннолучевых трубках или УФ-излучения в газораз-
рядных ртутных лампах. Так, согласно имеющимся данным, примеси галогени-
дов уменьшают стабильность работы катодолюминофоров в процессе эксплуата-
ции; подобное же влияние оказывают избыточные количества Sb и Мп (не вошед-
шие в решетку люминофора) на галофосфатные люминофоры.
Порошки люминофоров обрабатывают различными реагентами и тогда,
когда необходимо создать на поверхности зерен люминофора тончайшие слои
яекоторых соединений, улучшающих адгезионные свойства люминофора
(см. стр. 112) или повышающих агрегативную устойчивость порошков в диспер-
сионных средах при нанесении экранов. Кроме того, иногда на поверхность
верен наносят специальные слог (путем обработки порошков различными рас-
творами), которые повышают стабильность в процессах эксплуатации и изгото-
вления люминесцйрующих экранов.
Перечисленные операции будут рассмотрены далее подробнее в связи с про-
изводством конкретных люминофоров. На схеме 1 показаны все стадии синтеза
некоторых сульфидных люминофоров.
111.2. АППАРАТУРНОЕ ОФОРМЛЕНИЕ ПРОЦЕССОВ
ПРОИЗВОДСТВА ЛЮМИНОФОРОВ
, Высокая чистота, необходимая при синтезе люминофоров, предъявляет
специальные требования к конструкционным материалам аппаратуры. При
этом, как уже упоминалось, должна быть исключена возможность попадания
Нежелательных примесей как в исходные, так и в промежуточные и конечные
Продукты.
Наиболее пригодными материалами аппаратуры для работы с растворами
являются: высококачественный фарфор, кварц или химически стойкое стекло *.
Однако вследствие плохой теплопроводности использование этих материалов
Затруднено в тех случаях, когда требуется нагревание **. Недостатком указан-
ных материалов является хрупкость: кроме того, аппаратура достаточной вме-
стимости в промышленных масштабах, к сожалению, не выпускается.
В связи с расширением масштабов производства люминофоров, были прове-
дены испытания чугунной и стальной эмалированной аппаратуры. При этом
сказалось, что специальные сорта эмали позволяют использовать покрытую
* В некоторых случаях применяют винипласт, оргстекло и фторопласт.
** В ГДР синтез люминофоров успешно ведут в аппаратах из химически
стойкого или жаростойкого стекла (если необходимо нагревание).
61
ею аппаратуру даже в кислых сульфатных растворах, например, в производстве
люминофорно-чистых сульфидов цинка и кадмия. Однако это оправдывается
(при условии надлежащего контроля) только при достаточно большом масштабе
производства, когда загрязнения, попадающие в растворы со стенок аппаратуры
в сильной степени разбавляются *.
В связи с увеличением производства люминофоров одна из важнейших
технологических задач — разработка технологических схем и аппаратов, при-
годных для организации непрерывного процесса изготовления люминофоров.
Кроме повышения производительности аппаратуры это должно значительно
улучшить качество люминофоров, так как уменьшается вероятность внесения
загрязнений, попадающих в материальные потоки на различных стадиях суще-
ствующих многостадийных периодических процессов. Однако вопрос об эконо-
мической целесообразности применения непрерывных процессов отнюдь не бес-
спорен и требует специального рассмотрения в каждом отдельном случае.
Сейчас прокаливание шихты ведут почти исключительно в сосудах из кварца.
Этот материал обладает термостойкостью, обусловленной малым коэффициен-
том линейного расширения [1-10-6 град-1 (в среднем) для интервала темпера-
туры 0—1300®], а также высокой температурой размягчения (~1400°). При
800—1300° (в этом интервале температур формируется большинство люмино-
форов) кварц наиболее удобен, потому что имеет сравнительно большую хими-
ческую стойкость по отношению к реакционным смесям веществ различной
природы, применяемым при синтезах, а также высокую степень чистоты по со-
держанию примесей. Тигли, пробирки, кюветы, трубы и другие предметы делают
из двух сортов кварца: «прозрачного» И «непрозрачного». Изделия из первого наи-
более дороги и дефицитны, поэтому их используют в производстве люминофоров
наиболее высокой степени чистоты. Несмотря на высокую химическую стойкость
кварца вследствие значительной агрессивности некоторых твердых, жидких
Или газообразных продуктов, образующихся при прокаливании шихты, он срав-
нительно быстро разрушается. Поэтому в некоторых случаях кварцевые тигли
выдерживают только одно прокаливание и затраты на кварц — существенная
статья расхода в производстве люминофора.
Многочисленные попытки заменить кварц другими материалами оказались
пока почти безуспешными. Показано, например, что некоторые люминофоры
можно прокаливать в тиглях из высококачественного корундиза или алунда,
спеченной окиси магния и т. п. Однако эти материалы мало пригодны для мас-
сового использования, так как не выдерживают (или плохо выдерживают)
перепада температуры, возникающего при внесении наполненных шихтой изде-
лий в нагретую до высокой температуры зону и при извлечении из нее.
Для прокаливания люминофоров применяются печи различных типов.
Так, приводятся [7] отдельные примеры использования в заводском масштабе
вращающихся печей. Однако практически чаще (например, в производстве
галофосфатных люминофоров) прокаливание ведут в печах, в которых кюветы
или тигли с шихтой непрерывно перемещаются в туннели печи.
В СССР применяют исключительно электрические печи**, причем в боль-
шинстве случаев с силитовыми или карборундовыми нагревателями. Этот тип
печей удобен, во-первых, тем, что материал нагревателей не содержит металли-
ческих примесей, которые могли бы при прокаливании в результате испарения
попасть в люминофор, и, во-вторых, вследствие возможности создания доста-
точно мощных печных агрегатов, обеспечивающих быстрый подъем температуры
в реакционной зоне. В настоящее время разработаны конструкции печей с нагре-
вательными стержнями из дисилицида молибдена, обеспечивающими нагрева-
ние до 1700°.
Все промышленные печи обязательно должны быть снабжены автоматически
действующими пуско-регулирующими устройствами, поддерживающими необхо-
димый температурный режим при прокаливании шихты с точностью не ме-
нее ± 10°.
* В ЧССР очистку растворов и осаждение сульфидов цинка и кадмия ведут
в гуммированных аппаратах.
** Имеются данные, что на некоторых зарубежных заводах до сих пор
используют также газовые печи.
62
111.3. ПРИГОТОВЛЕНИЕ ЛЮМИНОФОРНО-ЧИСТОГО СЫРЬЯ
Основные компоненты шихты люминофоров — это твердые вещества, кото-
рые могут быть очйщены, если их температуры кипения и возгонки сравнительно
невелики. В этом случае успешно применяют методы дистилляций или субли-
мации, как, например, при очистке селенистого ангидрида, необходимого для
синтеза сульфид-селенидных люминофоров, или элементарной серы и селена.
Большинство полупродуктов для синтеза люминофоров получают, как пра-
вило, из растворов соответствующих солей, подвергнутых специальной тонкой
очистке. Требуемая чистота в этом случае изредка достигается перекристалли-
зацией из водных растворов или дробной кристаллизацией. Поэтому для тон-
кой очистки растворов применяют методы выделения примесей в осадок при по-
мощи неорганических или органических реагентов, образующих с примесью
нерастворимые соединения. В некоторых пока еще редких для люминофорной
практики случаях используют экстракцию микропримесей при помощи органи-
ческих экстрагентов [10, с. 199].
Наибольшее распространение получили следующие методы тонкой очистки:
осаждение (соосаждение) микропримесей в виде сульфидов, карбонатов,
фосфатов, органических комплексов и др.;
хроматографическое отделение примесей на специальных колонках с приме-
нением органических комплексообразователей.
Кроме того, в отдельных случаях применяют методы цементации (например,
высаживание примеси меди из сульфатных растворов на порошке металличе-
ского цинка), а также электрохимическое осаждение примесей металлов из рас-
твора [2, 4].
Осаждение примесей
Эффективность этого процесса определяется соотношением произведений
растворимости соответствующих соединений основного вещества и микропри-
меси, однако всегда следует учитывать возможность их совместного осаждения.
Нйже приведены произведения растворимости при комнатной температуре
наиболее технически важных веществ [11]:
HgS................3 • 10-54
AgoS ..............1 • 10-50
CuS................3,5-10-42
CdS................5-10-29
PbS................3,4-10-28
NiS................1,1-10-27
CoS................1,9-10-27
ZnS................7-10-26
FeS................3 • 10-19—
—1 • 10-18
BaCO3
CaCOo
MgCO3
BaSO4
PbSO4
SrSO4
CaSO4
SrCO3...............1,6-10-9
7,0 10-9
1,2-10-8
2,0 • 10-4
1 • 10-ю
1,0-10-8
2,8-10-7
6,1 • IO-5
CaF2 -.............3,5 • 10-11
Mg(OH)2 .... 1,2-10-11
Рассмотрим в качестве примера некоторые известные способы очистки рас-
творов хлорида кальция и фосфата аммония (получаемый из них двузамещенный
фосфат кальция используют в производстве галофосфатных люминофоров).
В растворы обеих солей при постоянном перемешивании вносят суспензию
свежеосажденного карбоната бария в растворе сульфида аммония. При этом
МйкропрИмеси тяжелых металлов осаждаются в виде сульфидов и выделяются
Из раствора вместе с суспензией карбоната. Раствор сульфида аммония готовят
Насыщением водного раствора аммиака сероводородом. Карбонат бария полу-
чают из раствора хлорида путем осаждения карбонатом аммония. Для осажде-
ния микропримесей, содержащихся в 1 л раствора, в него добавляют 1 мл насы-
щенного раствора (NH4)2S и 0,5 г ВаСО3. Операцию выполняют в фарфоровых
Или эмалированных сосудах с мешалками. Перемешивание продолжается 8 ч.
После этого суспензии дают отстояться в течение 10—15 ч, затем раствор филь-
труют. Содержание микропримесей Fe, Си и Ni в растворе снижается
До (1—2)-10-4%.
В некоторых случаях вместо постороннего носителя, способствующего
Наиболее полному выделению микропримесей, целесообразно получать его
63
непосредственно в очищаемом растворе путем введения в него необходимых для
этого реактивов. Так, при очистке раствора (NH4)2HPO4 наряду с (NH4)2S
вводят СаС12. Образующийся обильный осадок фосфата кальция способствует
более полному удалению из раствора высокодисперсных осадков сульфидов
тяжелых металлов, выпадающих одновременно в результате действия (NH4)2S.
Схема 2. Очистка сульфата цинка.
Иногда при очистке растворы приходится подвергать ряду последователь-
ных операций. В качестве примера одной из таких методик приведена схема
химической очистки сульфата цинка (схема 2). Этот метод раньше применялся
в промышленности, в настоящее время он заменен хроматографическим методом
(см. ниже).
Хорошие результаты дает разработанная Ангеловым [12, 13] методика
очистки растворов сульфатов, хлоридов и некоторых других солей при помощи
диэтилдитиокарбамата натрия, осаждающего большинство тяжелых металлов.
Так, очистка растворов сульфатов цинка и кадмия, по Ангелову, состоит в сле-
64
дующем: к 12—15% раствору соли прибавляют NH4OH до начала выпадения
гидроокисей, затем вводят слабощелочной 1% раствор диметйлглиоксима до
полного осаждения никеля. Через 30 мин добавляют 3% водный раствор диэтил-
дитиокарбамата натрия для удаления меди и кобальта. После этого раствор
обрабатывают активированным углем марки «щелочной А», размешивают
И фильтруют. Примесь марганца удаляется окислением его до двуокиси при дей-
ствии на маточный раствор персульфатом в аммиачном растворе. Описанная
методика позволяет получать растворы с содержанием примесей меньше 1 • 10"5 % ,
она удобна для производства не слишком больших масштабов.
Хроматографическое отделение примесей
Преимущество хроматографических методов заключается в том, что они
при сравнительной простоте дают большую производительность и могут быть
использованы в непрерывных технологических процессах. В производстве
Рис. III.1. Схема ионообменной очистки воды:
1 — катионообменный фильтр; 2 — анионообменный фильтр; 3 — декарбонизатор; 4 — сбор-
ник декарбонизированной воды; 5 — вентилятор; 6 — насос; 7 — бак для сбора отмывочных
вод после катионообменника; 8 — цистерна; 9 — мерник; 10 — эжектор; 11 — бак для сбора
отмывочных вод после анионообменника; 12 — бак для растворов кальцинированной соды
или бикарбоната натрия; 13 — насос.
люминофоров используют два принципа очистки: ионообменный [14] и адсорб-
ционно-комплексообразовательный [15, 16]. В первом—используют ионо-
обменные свойства ионитов, во втором — адсорбционную способность неорга-
нических поглотителей (например, активированного угля или окиси алюминия).
В качестве практического примера использования ионообменного метода
на рис. II 1.1 приведена схема ионообменной-очистки воды [17]. Этот метод дает
хорошие результаты также при тонкой очистке растворов сульфатов цинка
и кадмия [14], однако по своей производительности он уступает адсорбционно-
комплексообразовательному методу.
В основе очистки растворов сульфатов цинка и кадмия на так называемой
угольно-диметилглиоксимовой колонке [16, 18—20] (рис. III.2) лежит адсорб-
ционно-комплексообразовательный метод. В колонке используют активирован-
ный уголь БАУ, специально обработанный при нагревании соляной кислотой
для удаления из него примеси минеральных солей, и смесь порошка диметил-
глиоксима с углем. Диметилглиоксим дает внутрикомплексные соединения не
только с никелем, но и с кобальтом, медью и Fe3+ *. Эти комплексные соедине-
ния, образующиеся в растворе при pH = 5,8—6,0, далее адсорбируются слоем
Угля и таким образом извлекаются из раствора. Постоянство pH раствора под-
держивается введением 1—2% ацетата натрия.
* Для окисления Fe2+ -> Fe3+ в раствор предварительно добавляют пере-
кись водорода.
О Заказ 44
65
Применение угольно-диметилглиоксимовых колонок позволяет снизить
концентрацию Fe в очищаемых солях до (2—4)-10“5%, ,\(и Со — до 1 -10"5%
и менее, а Си даже до (1—5)-10"б%. Производительность колонок в расчете
на 1 кг диметилглиоксима составляет: 1500 кг ZnSO4 и 1300 кг CdSO4.
Следует отметить, что описы-
ваемые колонки могут быть исполь-
зованы и для очистки растворов
плавней (NaCl, КС1 и др.).
В качестве комплексообразова-
телей могут также выступать а-нит-
розо-Р-нафтол, 8-оксихинолин, 5,7-
дибромоксихинолин, рубеановодо-
родная кислота, дитизон и некото-
рые другие органические реагенты
[21, 22]. С помощью этих веществ
практически можно очищать до нуж-
ной степени чистоты все соли, ис-
пользуемые в качестве сырья для
синтеза различных классов люмино-
форов.
В известной степени недостат-
ком описанного адсорбционно-комп-
лексообразовательного метода явля-
ется возможность попадания в ра-
бочие растворы примесей органичес-
ких реактивов, особенно в конце ра-
бочего периода угольной колонки,
вблизи предела ее насыщения. Орга-
нические примеси, попадая в шихту,
могут влиять на качество люминофора, например, давая восстанавливающие
летучие продукты при прокаливании. Поэтому при работе на очистительных
колонках необходим строгий контроль на проскок не только минеральных
примесей, но и комплексообразователя.
Рис. II 1.2. Схема очистки растворов
сульфидов цинка и кадмия:
1 — мерник; 2 — слой смеси диметилглиок-
сима с углем; 3 — слой угля; 4 — приемник.
II 1.4. ОБЩАЯ ХАРАКТЕРИСТИКА ТЕХНОЛОГИЧЕСКИХ СХЕМ
ПОЛУЧЕНИЯ ОСНОВНЫХ ВИДОВ СЫРЬЯ
В синтезе люминофоров используют различные полупродукты. На свойства
сульфидных люминофоров существенно влияет качество исходных сульфидов
цинка и кадмия. При этом играют роль не только примеси тяжелых металлов,
но и сульфат-иона, который при прокаливаний является источником окисных
соединений. Большое значение имеет также степень дисперсности применяемых
порошков сульфидов, в значительной степени определяющих гранулометриче-
ский состав люминофоров [23].
В СССР принят метод осаждения сульфидов сероводородом из кислых
сульфатных растворов; за границей применяют также методы осаждения из рас-
творов хлоридов и из щелочных растворов [1—3]. При осаждении из подкислен-
ных сульфатных растворов получаются лучше сформированные и более чистые
осадки. Степень дисперсности агрегированных частиц сульфидов, осаждаемых
из растворов, зависит от pH, содержания буфера, концентрации и температуры
раствора, скорости подачи и давления сероводорода, а также от скорости пере-
мешивания [5, 24—26].
Сероводород получают разложением серной кислотой раствора сульфида
натрия (схема 3). Сульфид цинка получают по реакции
ZnSO4 + H2S----►ZnS + H2SO4
(см. также схему 4). В качестве исходного сырья в этом случае используют
окись цинка, получаемую сжиганием металлического цинка. Далее окись цинка
растворяют в серной кислоте и полученный раствор сульфата цинка очищают;
затем сульфид осаждают сероводородом. Как следует из приведенного уравнения
реакции, в процессе осаждения ZnS выделяется серная кислота и кислотность
раствора повышается.
66
Для получения сульфида кадмия обычно используют очищенные растворы
сульфата кадмия.
Кроме сероводородного метода получения сульфидов цинка и кадмия нужно
упомянуть также тиосульфатный метод [27, 28], основанный на реакциях,
которые могут быть упрощенно изображены следующим образом:
MeSO44"2Na2S2O3----->• MeS2Og-]-Na2SO4
MeS2O3-4-H2O х MeS-j-H2SO4
Каг^гОз-]-H2SO4 —> NEI2SO4-I—SO2~|~ S-I-H2O
Получаемые по этому методу сульфиды всегда содержат значительную
примесь (5—10%) элементарной серы и, как показано в работах [28, 29], при-
годны для получения люминофоров, к которым не предъявляется особых требо-
ваний в отношении малой длительности послесвечения. Необходимо отметить
также и тиомочевинный метод осаждения ZnS [30, 31].
Схема 3. Получение сероводорода.
При синтезе фосфатных люминофоров основными компонентами шихты
служат соответствующие фосфаты и карбонаты. Для получения их используют
очищенные растворы аммонийных солей. Приводим технологические схемы
получения дикальцийфосфата и карбоната кальция (схемы 5 и 6). Для очистки
раствора карбоната аммония представлен вариант комплексообразовательно-
хроматографпческой очистки с использованием рубеановодородной кислоты [22],
хотя в промышленности для этой цели по большей части используется метод
осаждения примесей в виде сульфидов.
При синтезе силикатных, германатных и арсенатных люминофоров в ка-
честве сырья применяют карбонаты металлов и ангидриды соответствующих
кислот. Приводим технологическую схему получения двуокиси кремния
5*
67
Схема 4. Получение люминофорно-чистого сульфида цинка.
люминофорной степени чистоты из технического силиката натрия (схема 7). Полу-
чаемая двуокись кремния содержит не более 10% влаги, Fes^ 1 -10-3%; Си
5 -10-4% и Pbs^ 5 -10“3% . В тех случаях, когда требуется двуокись кремния
более высокой степени чистоты, обычно используют метод гидролитического
разложения кремнеэтилового эфира [2, 32].
Схема 5. Получение дикальцийфосфата.
Производство полупродуктов для синтеза люминофоров требует тщатель-
ного аналитического контроля содержания как основного вещества, так и микро-
примесей. Для этой цели разработаны соответствующие аналитические мето-
дики, на которых здесь нет возможности останавливаться. Следует только ска-
зать, что при определении содержания микропримесей по большей части исполь-
зуют колориметрические [33, 34] и спектральные [35—39] методы. Кроме того,
в необходимых случаях применяют метод концентрирования микропримесей.
Визуальные колориметрические методы позволяют надежно определять содер-
жание микропримесей до 10"5—10"6 % , спектральные методы без обогащения
при анализе сульфидов цинка и кадмия имеют следующую чувствительность
(в %): Fe — 1 -10_5; Си — 10-6; Ni и Со — 5 -10-4. Химическое или термическое
69
Схема 6. Получение карбоната кальция.
Силикат натрия (технический)
Разбавление и перемешивание Н2О (водопроводная)
I
Нагревание до 70° С
__________________I__________________ Активированный уголь
Длительное отстаивание (7 суток) <
Н2о2 (30% р-р)
Фильтрация на нутч-фильтре
SO 2 Сатурация Н2О (водопроводная)
Перемешивание, отстаивание и промывка
HNO3
HNO3 (реактивная)
НС1 (реактивная)
Отжим на центрифуге
1
Измельчение
1
-> Обработка HNO3 при перемешивании
1
Промывка
1
—> Длительная промывка
1
Промывка в котле
1
Промывка на нутч-фильтре
1
Отжим
1
—> Обработка смесью кислот при переме-
—> шивании
1
Промывка многократная
1
Отжим под вакуумом
1
Сушка в алюминиевых противнях
при 300—400° С
1
Готовый продукт
Н2О (водопроводная)
Н2О (дистиллирован-
ная)
Схема 7. Получение люминофорно-чистой двуокиси кремния.
71
обогащение позволяет повысить чувствительность на один-два порядка [36].
Разработаны также полярографические методы определения микропримесей
в люминофорно-чистых веществах [40, 41].
ЛИТЕРАТУРА
1. Ward R., Froelich Н. In: Preparation and Characteristic of Solid Lumi-
nescent Materials. Symposium Held at Cornell University, 1946. Nev York, 1948,
p. 22—86. — 2. Москвин А. В. Катодолюминесценция. 4. 2. Катодолюмино-
форы и экраны. М.—Л., ГИТТЛ, 1949. 700 с. — 3. Leverenz Н. An Introduction
to Luminescence of Solids. New York — London, Wiley Sons, 1950. 569 p. —
4. Тиде Э. В кн.: Руководство по препаративной неорганической химии. Под
ред. Г. Б. Брауэра. М., ИЛ, 1956. 205 с. — 5. Бунделъ А. А. Автореф. докт.
дисс. МГУ им. М. В. Ломоносова, 1955. — 6. Жиров Н. Ф. Люминофоры. М.,
Оборонгиз, 1940. 477 с. — 7. Ranby Р., Rice A. Ind. Chem., 1952, № 12, р. 545—
549. — 8. Lendvay О., Szabo I. Венгр, пат. 149991, 1961. — 9. Казанкин О. Н.
Авт. свид. 186593. — 10. Witzmann Н., Oeser W., Piesche L. In: Reinstoffe
in Wissenschaft and Technik Internationaler Symposium. Berlin, 1963, S. 199.
11. Колътгоф И. M., Сендел Е. Б. Количественный анализ. Изд. 3-е. Пер.
с англ. Под ред. Ю. Ю. Лурье. М.—Л., Госхимиздат, 1948. 824 с. — 12. Анге-
лов И. И. В кн.: Материалы V совещания по люминесценции. Тарту, Изд. ТГУ,
1957, с. 338—346. — 13. Ангелов И. И., Пятницкая Г. Н. «Труды ИРЕА», 1958,
вып. 22, с. 155—158. — 14. Божеволънов Е. А., Трусов В. В. В кн.: Материалы
V совещания по люминесценции. Тарту, Изд. ТГУ, 1957, с. 347—362. — 15. Tide
Е., Schikore W. Вег., 1938, v. 75, s. 582—589. — 16. Гурвич А. МГапон Т. Б.,
Рабинович М. С. Хим. пром., 1956, № 1, с. 31—34. — 17. Кастальский А. А.
Проектирование установок для химического обессоливания воды. Изд. 2-е.
М., Стройиздат, 1964. 212 с. — 18. Гурвич А. М., Гапон Т. Б., Рабинович М. С.
В кн.: Материалы V совещания по люминесценции. Тарту, Изд. ТГУ, 1957,
с. 363—372. — 19. Гурвич А. М. Автореф. дисс. МГУ им. М. В. Ломоносова,
1957. — 20. Гапон Т. Б., Гурвич А. М., Р абинович М. С. Авт. свид. 99924,
101183 и 101671.
21. Рак-Раевская А. А. В кн.: Химия и технология люминофоров. Л., «Хи-
мия», 1964, с. 4—8; «Труды ГИПХ», 1966, вып. 53, с. 131—134. — 22. Рак-Раев-
ская А. А. Сборники рефератов научно-исследовательских работ по химии и
технологии люминофоров за 1960—1964 гг. Л., изд. ГИПХ, 1961—1965. —
23. Марковский Л. Я., Таушканова Л. Б., Сапожников Ю. П. «Труды ГИПХ»,
1966, вып. 53, с. 72—77. — 24. Бунделъ А. А., Кочугова Е. И., Чаплина И. М.
Сборники рефератов научно-исследовательских работ по химии и технологии
люминофоров за 1959 и 1960 гг. Л., изд. ГИПХ, 1960, 1961. — 25. Ortmann Н.,
Hartmann Н. Z. Naturforsch., 1961, Bd. 16А, S. 903—908. — 26. Ortmann H.
Z. phys. Chem., 1961, Bd. 216, S. 75—83. — 27. Guntz A. Compt. rend., 1948,
v. 226, p. 80—83. — 28. Томбак M. И., Бунделъ А. А. Ж. неорг. хим., 1959,
т. 4, с. 1968—1973. — 29. Попова А. В.ГР ак-Раевская А. А., Хорошкова М. Н.
«Труды ГИПХ», 1960, вып. 43, с. 135—148. — 30. Попов В. П., Шульман В. М.,
Варанд В. Л. В кн.: Люминесцентные материалы и особо чистые вещества.
Вып. 4. Ставрополь, изд. ВНИИЛюминофоров, 1970, с. 86—90.
31. Крамарева Т. В., Косарева Л. А., Шульман В. М. В кн.: Халькогениды.
Под ред. Г. В. Самсонова. Киев, «Наукова Думка», 1967, с. 86—93. — 32. Сте-
пин Б. Д., Блюм Г. 3., Шварц М. М. Хим. пром., 1963, № 2, с. 158—160. —
33. Шафран И. Г. В кн.: Материалы V совещания по люминесценции. Тарту,
Изд. ТГУ, 1957, с. 373—389. — 34. Шафран И. Г. «Труды ИРЕА», 1959, вып. 23,
с. 88—95. — 35. Васильева В. Н., Дворжецкая Л. А., Хлебникова Л. Я. «Труды
ГИПХ», 1960, вып. 43, с. 151—160. — 36. Дворжецкая Л. А., Хлебникова Л. Я.
Там же, с. 161. — 37. Хлебникова Л. Я., Дворжецкая Л. А., Шванева М. К.
В кн.: Химия и технология люминофоров. Л., «Химия», 1964, с. 107—110. —
38. Калинина Л. Б. Там же, с. 117. — 39. Хлебникова Л. Я., Дворжецкая Л. А.
В кн.: Сборник рефератов научно-исследовательских работ по химии и техноло-
гии люминофоров за 1964 г. Л., изд. ГИПХ, 1965, с. 21; «Труды ГИПХ», 1966,
вып. 53, с. 142—144; 145—147. — 40. Денисов Е. И., Надеждина Л. С. ЖАХ,
1965, т. 20, с. 187—191.
41. Методы анализа веществ высокой чистоты. М., «Наука», 1965. 111 с.
ГЛАВА IV
ФОТОЛЮМИНОФОРЫ
Фотолюминофоры представляют собой обширный класс разнообразных
химических соединений, люминесцирующих под действием ультрафиолетового,
видимого и инфракрасного света. Этот класс включает в себя люминофоры как
характеристического, так и рекомбинационного типа.
IV.1. МЕХАНИЗМ ЛЮМИНЕСЦЕНЦИИ
Механизм люминесценции рекомбинационных люминофоров объясняют
с привлечением основных представлений зонной теорип твердого тела.
Подробное изложение этой теории можно найти в работах [1, 2], мы же рассмо-
трим лишь простейшую зонную схему электронных переходов в люми-
нофорах рекомбинационного полупроводникового типа.
В кристаллической решетке твердого тела вследствие взаимодействия
электронные уровни атомов расщеплены на столько близко расположенных
подуровней, сколько атомов вступает во взаимодействие. Совокупность таких
подуровней образует энергетическую зону.
В 1928 г. Блох показал, что в периодическом поле идеальной кристалли-
ческой решетки перемещающиеся электроны можно рассматривать как свобод-
ные, но не с любым значением энергии. Зоны разрешенных энергети-
ческих состояний, которые определяются энергетическими уровнями атомов,
входящих в кристаллическую решетку, разделяются запрещенными
зонами. Каждая зона разрешенных энергетических состояний имеет N
уровней. Согласно принципу Паули, на уровне размещаются 2N электронов.
Для обычных люминофоров предполагается существование двух зон — запол-
ненной электронами (валентная зона) и незаполненной, в которой элек-
троны могут свободно перемещаться (зона проводимости). Зоны разде-
лены промежутком — запрещенной энергетической областью (запрещенная
зона). Ширина запрещенной зоны у сульфидных люминофоров составляет
несколько электрон-вольт. Введение примесей (активаторов), а также наличие
примесей и дефектов в решетке создают условия для образования энергетических
Уровней, которые располагаются в запрещенной зоне.
Простейшая зонная схема для люминофоров полупроводникового типа
показана на рис. IV.1. Энергетические уровни Ах и А2, возникающие при вве-
дении активатора, располагаются в запрещенной зоне II. Наряду с уровнями
активатора в запрещенной зоне существуют уровни захвата электро-
нов (Л), обусловленные различными дефектами (в частности, примесными).
Так как природа ловушек различна, то уровни захвата могут иметь различную
глубину. Уровень А2 соответствует невозбужденному состоянию активатора
(основной уровень) и в этом состоянии заполнен, а уровни А2 (возбу-
жденный уровень) и уровень Л свободны.
При возбуждении люминофора светом энергия может поглощаться как на
Уровнях активатора, так и в основном веществе. В первом случае поглощение
света сопровождается переходом электрона с основного уровня активатора Ах
73
на возбужденный уровень А2 1, а излучение света возникает при переходе 2,
который соответствует возвращению электрона на основной уровень. В этом
случае возникает флуоресценция, длительность которой составляет
10-8—10-9 с.
Электроны, вырванные возбуждающим светом, могут перейти в зону прово-
димости 3 и локализоваться на ловушках 4. С ловушек 5 электроны могут ос-
вободиться только в том случае, если им будет сообщена необходимая энергия
(например, при нагревании люминофора или при действии ИК-лучей). При этом
электроны либо повторно захватятся ловушками, либо через зону проводимости
перейдут на уровень активатора 6 и рекомбинируют с центром свечения. Это
приводит к возникновению длительного свечения (фосфоресценции),
которое продолжается до тех пор, пока все электроны, захваченные ловушками,
не освободятся и не прорекомбинируют с ионизованными центрами.
При поглощении света в основе люми-
нофора электроны переходят из валентной
зоны в зону проводимости 7. Образовав-
шиеся в валентной зоне дырки мигрируют
и могут локализоваться на уровнях акти-
ватора. В этом случае излучение происхо-
дит в результате рекомбинации электронов
из зоны проводимости с дырками на уров-
нях активатора. Помимо образования
электронно-дырочных пар
при поглощении света в основе люмино-
фора, могут образоваться экситоны*,
способные ионизовать центры свечения и
привести к возникновению люмине-
сценции.
Следует отметить, что энергия, погло-
щенная другими примесями в решетке,
может передаваться активатору, например
в том случае, если спектр излучения при-
меси совпадает со спектром поглощения
активатора.
Помимо рассмотренной классической
модели процесса люминесценции [1—3]
редложенной Пренером и Вильямсом [4].
Согласно этой модели (рис. IV.2), основной уровень I располагается вблизи
валентной полосы, а возбужденный уровень II — ниже зоны проводимости.
После возбуждения светом и образования электронов и дырок для уровня I
более вероятен захват электрона из полосы проводимости, а для уровня II —
захват дырки из валентной полосы. Люминесценция возникает в результате
перехода электрона с уровня II на уровень I. Такая модель называется до-
норно-акцепторной; ее применяют в ряде случаев для объяснения
процессов люминесценции. В люминофоре ZnS -Си, согласно этой модели,
медь создает акцепторный уровень, а хлор — донорный.
Для характеристических люминофоров, когда электронные переходы совер-
шаются внутри самого центра свечения, энергетическое состояние центра и его
свойства могут быть описаны двухмерной энергетической
моделью. В этом случае невозбужденное состояние центра описывается
потенциальной кривой (рис. IV.3), показывающей зависимость его энергии
от конфигурационного параметра, который в случае двухатомной молекулы
есть расстояние между двумя ядрами. Кривая а2 характеризует возбужденное
состояние. Точки Е10 и Е20 принадлежат невозбужденному и возбужденному
состояниям центра при ОК, а горизонтальные отрезки соответствуют темпера-
туре выше нуля, когда ядра совершают колебания относительно положения
равновесия. Возбуждение системы на рис. IV.3 опишется переходом Е10 -> Е2.
Переход в равновесное состояние Е20 сопровождается передачей части энергии
* При поглощении света возможно также возбуждение электрона валент-
ной зоны, при котором он не переходит в зону проводимости, а образует с дыркой
связанную систему, получившей название экситона.
74
Рис. IV. 1. Зонная схема для лю-
минофоров полупроводникового
типа.
следует остановиться на модели,
в виде фононов решетке, а излучение описывается переходом Е20->£1.При
этом: Е2—Е2о—Ег. Последнее характеризует стоксовские потери, обу-
словливающие смещение спектра излучения в длинноволновую область по
отношению к спектру поглощения (см. стр. 8).
Если температура настолько ве-
лика, что в возбужденном состоя-
нии система оказывается вблизи точки
пересечения кривых С, то она мо-
жет спуститься по кривой а без из-
лучения. Такое тушение, прикотором
Рис. IV.2. Зонная схема
по Пренеру и Вильямсу.
Рис. IV.3. Двухмерная энергети-
ческая модель для характеристи-
ческих люминофоров.
поглощенная энергия внутри центра превращается в тепло, называется
внутренним.
Однокоординатная модель центра позволяет в ряде случаев объяснить форму
спектров поглощения и излучения и ее зависимость от температуры.
IV.2. ЛЮМИНОФОРЫ ДЛЯ ЛЮМИНЕСЦЕНТНЫХ ЛАМП
Требования, предъявляемые к люминофорам
Люминесцентное освещение — наиболее широкая область применения лю-
минофоров [5—9]. Поэтому их производство для люминесцентных ламп является
самым крупнотоннажным: на его долю приходится —90% от всего количества
производящихся люминофоров.
, Основное требование, предъявляемое к люминофорам этого класса —
высокая эффективность преобразования ультрафиолетовой энергии ртутного
разряда в видимую световую энергию. Этому требованию отвечают люминофоры
с внутрицентровым механизмом люминесценции. Квантовый выход
У лучших люминофоров такого типа близок к 100%, что позволяет, например
на основе галофосфатных люминофоров, выпускать люминесцентные лампы
со светоотдачей 80 лм/Вт (у ламп накаливания максимальная светоотдача со-
ставляет 25 лм/Вт).
Преимущество люминесцентных источников света состоит, кроме того,
еще и в возможности изменять спектральный состав излучения путем
Применения люминофоров (или их смесей) с различным цветом свечения. Одно
из основных требований при этом — приближение распределения энергии
в спектре излучения этих ламп к распределению энергии в спектре дневного
света, особенно в тех случаях, когда требуется правильная цветопередача.
Тип ламп— низкого или высокого давления — определяет тип исполь-
зуемых в них люминофоров.
Лампы низкого давления
Люминесцентная лампа низкого давления (рис. IV.4) — это
Полый стеклянный цилиндр, на внутреннюю поверхность которого нанесен
люминофор. В оба конца цилиндра впаяны электроды — обычно биспирали
Из вольфрамовой проволоки, покрытые тонким слоем окислов щелочноземельных
75
металлов, обладающих хорошей электронной эмиссией. После откачки
и обезгаживания в лампу вводят небольшое количество ртути, затем ее запол-
няют аргоном. В процессе газового разряда происходит возбуждение атомов
ртути. По данным Фабриканта и Бутаевой [10], для возбуждения люминофора
в лампах низкого давления основное значение имеют линии излучения ртути
с % = 254 и % = 185 нм. Люминофорный слой поглощает энергию излучения
ртути и излучает в требуемой области спектра.
Люминофоры для люминесцентных ламп низкого давления должны обладать
следующими свойствами:
способностью возбуждаться в области 254 и 185 нм;
высоким коэффициентом поглощения и квантовым выходом при возбужде-
нии в этой области;
излучением в требуемой области спектра;
пригодностью для нанесения на баллон лампы;
высокой стабильностью на стадии изготовления ламп;
устойчивостью в процессе эксплуатации ламп.
Рис. IV.4. Внешний вид люминесцентной лампы
низкого давления.
Совершенствование методов синтеза и химического состава люминофоров
с целью улучшения указанных характеристик, а также технологии производства
ламп привело к тому, что современные люминесцентные лампы обладают высо-
кой светоотдачей и большой долговечностью. Широкие возможности в варьиро-
вании спектрального состава излучения люминофоров позволяют в настоящее
время выпускать большой ассортимент ламп.
Однако целесообразными признаны следующие цветовые типы люминесцент-
ных ламп низкого давления:
лампы дневного света (ЛД), спектр излучения которых близок к таковому
стандартного источника белого света С (цветовая температура ТЦв = 6500 К)
и люминесцентные лампы (типа ЛДЦ) с улучшенной цветопередачей;
лампы холодно-белого света (ЛХБ), имеющие распределение энергии по
спектру, соответствующее источнику В (Тцв = 4800 К);
лампы белого света (ЛБ), имеющие распределение энергии по спектру,
соответствующее ТЦв = 3500 К;
лампы тепло-белого света (ЛТБ) (ТЦв — 2700 К), имеющие распределение
энергии, близкое к распределению энергии источника А (7\в = 2854 К).
Лампы указанных типов выпускают и в варианте с улучшенной цвето-
передачей.
Координаты цветности (см. стр. 174) указанных люминесцент-
ных ламп представлены на цветовом треугольнике (рис. IV.5). Пунктирная ли-
ния на нем характеризует цвет излучения абсолютно черного тела при различ-
ных температурах.
До начала 50-х годов в люминесцентных лампах низкого давления приме-
няли в основном цинк-бериллий силикат, активированный Мп, с излучением
в желтой области спектра и вольфрамат магния, излучающий в синей области.
Смеси этих люминофоров в разных пропорциях позволяли получать различные
спектры излучения — в зависимости от требований.
Дальнейшие поиски люминофоров для люминесцентных ламп привели
к созданию люминофоров на основе галофосфатов, которые полностью заменили
применявшиеся ранее смеси. Спектры излучения галофосфатных люминофоров
могут изменяться в широких пределах, в зависимости от содержания актива-
торов (Sb и Мп) и входящих в основу галофосфата галогенов. Таким образом,
при использовании галофосфатов можно получать все указанные выше типы
ламп низкого давления (ЛД, ЛХБ, ЛБ и ЛТБ).
76
Однако из-за сравнительно небольшой доли излучения в красной области
спектра цветопередача ламп с этими люминофорами неудовлетвори-
тельна. Поэтому одной из актуальных задач является создание люминофоров,
спектр излучения которых позволил бы улучшить цветопередачу ламп.
Успехом на этом пути следует считать синтез люминофора CaSiO3-Pb-Mn
(стр. 88), который обладает большой долей излучения в красной области
спектра. Для приготовления ламп с правильной цветопередачей к этому люмино-
фору добавляют люминофоры с излучением в зеленой области спектра (например,,
фторсиликат магния, активированный Ti). Позднее для ламп с улучшенной
цветопередачей стали применять также фосфаты элементов II группы периоди-
ческой системы, активированные
Sn [(Са, Zn)3(PO4)2 • Sn и (Са,
Sr)3(PO4)2-Sn, например]. Они
возбуждаются в области X =
= 200—365 нм, имеют широкие
полосы излучения и высокий
квантовый выход. К недостаткам
их следует отнести сравнительно
сложную технологию приготовле-
ния. Необходимо отметить, что в
качестве голубой компоненты для
ламп с улучшенной цветопереда-
чей применяется люминофор со-
става: 2ВаО TiO2 Р2О5, разра-
ботанный Хендерсоном [11], или
активированный сурьмой галофос-
фат.
Применение новых люмино-
форов в сочетании с ранее раз-
работанными позволяет осущест-
влять выпуск большого ассорти-
мента ламп. Так, только фирма
«Сильвания» для изготовления
люминесцентных ламп выпускает
18 различных люминофорных
композиций. Растущие требова-
ния по спектральному составу
излучения люминесцентных ламп в последнее время поставили задачу созда-
ния люминофоров со спектром излучения, идентичным спектру солнеч-
ного света по всему спектральному диапазону, включая и УФ-область.
Люминесцентные лампы сейчас начинают широко применять в сельском
хозяйстве для облучения растений. Оптимальный, с точки зрения биосинтеза,
спектральный состав излучения люминофоров способствует росту растений
и увеличивает урожайность. В лампах указанного типа в основном используют
люминофоры типа (Са, Zn)3(PO4)2-Sn и (Са, Mg)WO4-Pb, обеспечивающие не-
обходимое излучение в красной и синей областях спектра.
Для нужд техники разработаны люминофоры, возбуждающиеся в области
254 нм и излучающие в УФ-области спектра. Из этого класса люминофоров
практически важны те, максимум излучения которых соответствует областям
350—360 и 300—310 нм. Первые применяются в газоразрядных лам-
пах У ФО, которые пригодны для возбуждения сульфидных люминофоров,
нанесенных на шкалы приборов. Наиболее эффективны для ламп УФО люмино-
форы BaSi2O5 Pb и (Sr, Са)3(РО4)2-Т1 (стр. 87). Люминофоры с максимумом
излучения в области 300—310 нм используют вэр и темных лампах (область
эритемного действия лежит в интервале 290—300 нм). Из них высоким кван-
товым выходом обладают (Са, Zn)3(PO4)2-TI; (Са, Mg),(PO4)„-Т1;
(Ba, Zn)2SiO4Pb и (Ba, Mg)2SiO4 Pb.
Лампы высокого давления
В отличие от ртутного разряда при низком давлении разряд при в ы с о -
к о м^ давлении сопровождается значительным излучением в видимой
области спектра, а излучение в УФ-области спектра, помимо излучения при
77
длине волны 254 нм, имеет высокую интенсивность при длинах волн 313 и
365 нм.
В люминесцентных лампах высокого давления с исправленной
цветностью (рис. IV.6) УФ-излучение ртутного разряда горелки трансформи-
руется в видимый свет при помощи люминофора, нанесенного на внутреннюю
поверхность колбы, внутри которой находится горелка лампы. Поскольку доля
энергии, излучаэмая горелкой в УФ-области, невелика в сравнении с долей
энергии, излучаемой в видимой области спектра, люминофорное покрытие не
дает существенного повышения светоотдачи в данном типе ламп. Его роль сво-
дится в основном к тому, чтобы исправить цветность ламп, дополняя спектр
Рис. IV.6. Внешний
вид ртутной лампы
высокого давления с
исправленной цветно-
стью.
Рис. IV.7. Сравнительная диаграмма энергии излуче-
ния ртутной лампы высокого давления с исправлен-
ной цветностью в колбе, покрытой изнутри люмино-
фором (заштрихованная область), ив колбе без люми-
нофора (незаштрихованная область).
излучением в красной области. На рис. IV.7 представлено распределение энер-
гии излучения такой лампы без люминофора и с ним.
Люминофоры для ламп высокого давления должны обладать следующими
свойствами:
способностью возбуждаться в широкой области УФ-спектра (от 254 до
365 нм);
высоким поглощением и квантовым выходом при возбуждении в указанной
области;
преимущественным излучением в красной области спектра;
устойчивостью свечения до 300°, так как в рабочем режиме лампы люминофор
имеет приблизительно эту температуру;
высокой устойчивостью на стадии изготовления ламп.
Число люминофоров, которые более или менее удовлетворяют указанным
требованиям, невелико. К ним относятся: фторгерманат магния, активирован-
ный Мп: арсенат махния, активированный Мп; цинк-стронций, кальций-магний
и кальций-цинк фосфаты, активированные Sn. В последние годы были разрабо-
таны и нашли широкое применение в лампах ортованадат и фосфатованадат
иттрия, активированные Ей. Последние применяют либо отдельно, либо в смеси
с указанными выше фосфатными люминофорами. Обладая интенсивным красным
свечением, ванадаты обеспечивают высокую долю излучения ламп в красной
области спектра.
78
Галофосфаты
Состав и способы получения. Люминофоры этого класса были синтези-
рованы в 1942 г. [12, 13] и описаны во многих работах. Общая формула гало-
фосфатных люминофоров: ЗСа3(РО4)2-Ca(F, Cl)2-Sb-Мп. Они, как уже упоми-
налось, имеют структуру апатита.
Галофосфатные люминофоры готовят прокаливанием шихты, состоящей
обычно из СаНРО4, CaGO3, CaF2, Sb2O3, МпСО3 и NH4G1 при 1100—13009.
При этом к чистоте исходных продуктов предъявляются следующие требо-
вания: содержание Fe, Со, Ni, РЬ, Си не должно превышать 2-10"4%. Химизм
процессов образования основы люминофора описан на стр. 39.
Указанные соединения активаторов, вводимые в шихту люминофора,
неустойчивы и при нагревании шахты в присутствии кислорода воздуха пре-
вращаются в более устойчивые соединения. Карбонат марганца разлагаясь
дает ряд окислов; с повышением температуры до 1200° происходят сле-
дующие превращения:
MnCOg —► МпО —► МпО2 —► Мп2О3 —► МП3О4 —> МпО
Поскольку в апатите растворим только Мп2+, активирующий основу, целесо-
образно прокаливать шихту в инертной среде (N2, Аг), что позволяет снизить
температуру с 1200 до 1100° [13], или в слабовосстановительной среде. Оправ-
дано, по-видимому, также введение марганца в шихту в виде таких его двух-
валентных соединений, как например MnNH4PO4 и Мп2Р2О7 [15], которые
более устойчивы к окислению, чем МпСО3.
Окись Sb3+ при нагревании шихты в окислительных условиях переходит
в окислы более высокой степени окисления. Последние, взаимодействуя с дру-
гими компонентами шихты, дают такие соединения, как GaSb2O7 [16, 17],
CaSb2Oe [16] Ga4Sb4O1]LF2 [16] и ряд других антимонатов.
Необходимая для активации галофосфата кальция Sb3+ [18] может обра-
зоваться при температуре выше 1000° по реакции:
GaSb2Oe-|-Ga2P2O7 ► Са3(РО4)2-|—Sb2O3-[~O2^
Проведение синтеза в инертной среде (например, в атмосфере N2) позволяет
удержать сурьму в необходимом для активации трехвалентном состоянии.
Часть вводимой в люминофор Sb3+ теряется на начальной стадии нагрева-
ния в виде летучего продукта SnCI3, образующегося в результате взаимодей-
ствия окиси сурьмы с хлористым водородом, появляющимся при термической
диссоциации NH4G1. Поэтому количество сурьмы, вводимой в шихту люмино-
фора, всегда значительно превышает то ее оптимальное количество (0,7—1,0%),
которое необходимо для активации галофосфата. Применение при синтезе
более инертных в отношении взаимодействия с HG1 соединений сурьмы (таких,
как антимонаты и фторантимонаты кальция и кадмия [15, 19])
позволяет уменьшить ее потери при прокаливании и снизить, таким образом,
содержание в исходной шихте. Другим приемом, позволяющим также снизить
потери сурьмы при прокаливании, является введение хлор-иона в виде более
устойчивых соединений, например МпС12, СаС12, Са4(РО4)2С12 [20] и др.
Однако следует иметь в виду, что сам процесс активации фторхлорапатита
кальция сурьмой неизбежно сопровождается потерей части сурьмы в виде
SbGl3 [18].
Важное значение для получения высококачественных люминофоров имеет
молярное отношение суммы вводимых металлов (Са, Sb, Мп) к иону РО|~. Для
стехиометрического галофосфата это отношение равно 1,67. При больших зна-
чениях галофосфат не активируется, поскольку сурьма образует антимонаты
кальция, а марганец — окисные соединения, в которых он имеет валентность
выше 2. Поэтому во всех рецептурах указанное отношение меньше 1,67; по дан-
ным Догерти [21], оптимальная величина его равна 1,63-
Таким образом, галофосфатный люминофор после прокаливания всегда
содержит примесь таких фаз, как Са2Р2О7 и Са3(РО4)2. Последний появляется
еще и потому, что протекает вторичное взаимодействие галофосфата с влагой
атмосферы при прокаливании:
ЗСа3(РО4)2« СаС12-|-Н2О--► ЗСа3(РО4)2. СаО-|-2НС11
ЗСа3(РО4)2• СаО-|-Са2Р2О7 > Са3(РО4)2
79
В окрашенном твердом растворе с Са3(РО4)2 марганец находится в трех-
валентном состоянии [22].
Наличие Са3(РО4)2 и Са2Р2О7, активированных Sb и Мп [18], поглощающих
возбуждающую радиацию, существенно снижает эффективность гало-
фосфатного люминофора. Поэтому в большинстве способов получения гало-
фосфатных люминофоров присутствует стадия обработки люминофора химиче-
скими реагентами, растворяющими указанные фазы. К таким реагентам
относятся водные растворы лимонной, этилендиаминтетрауксусной и других
органических кислот, а также слабые растворы минеральных кислот. Одно-
временно при таких обработках извлекаются и избыточные соединения марганца
и сурьмы. Их удаление существенно повышает стабильность гало-
фосфатных люминофоров.
Рис. IV.8. Спектры поглощения (а) и возбуждения (б) галофосфата кальция:
1 — неактивированный галофосфат; 2 — активатор Мп; 3 — активатор Sb; 4 — активаторы
Sb и Мп; 5 — 9Са3 (PO4)2-CaCl2-2CaF2-Mn (5)-Sb (5); 6 — 9Са3 (PO4)2-CaCt2-2CaF2-Sb (5);
7 — ЗСа3 (РО4)2-СаС12-Мп (2)-Sb (5); 8 — ЗСа3 (РО4)2-CaF2-Mn (2)-Sb (5).
Часто в галофосфатных люминофорах присутствует и метаантимонат каль-
ция, активированный Мп, являющийся неэффективным люминофором. Однако
он присутствует в количестве менее 1% , поэтому его наличие не сказывается
ощутимо на яркости свечения галофосфата кальция [23, 24].
Как было показано Батлером [25], способность люминофора поглощать
возбуждающую радиацию существенно зависит от степени дисперс-
ности люминофора. Особенно нежелательна фракция с размером зерен
менее 3 мкм, поскольку ей присуще сильное диффузное отражение
возбуждающего излучения. Поэтому гранулометрический состав люминофора
существенно влияет на яркость свечения.
Для удаления из люминофора мелкой фракции в свое время применяли
фракционирование. Позднее было показано [26, 27], что грануло-
метрический состав люминофора определяется гранулометрическим составом
исходного СаНРО4. Это заставило разработать методы получения кристалли-
ческого СаНРО4 с необходимым размером и формой кристаллов [28, 29]. В на-
стоящее время экономически невыгодное фракционирование не используют,
а гранулометрическим составом люминофора управляют, задавая определенный
гранулометрический состав СаНРО4.
Люминесцентные характеристики. Спектры поглощения (рис. IV.8, а)
сняты для единичного кристалла при помощи УФ-микроскопа. Из рис. IV.8, а
следует, что ни основа галофосфата (кривая 7), ни основа, активированная Мп
80
(кривая 2), не имеют поглощения в области X = 254 нм. Только при актива-
ции Sb (кривая 3) появляется поглощение в этой области.
Спектры возбуждения (рис. IV.8, б) имеют два пика, положение которых
определяется соотношением между фтором и хлором. Из сравнения спектров
возбуждения следует, что люминофоры, близкие по составу к фторапатиту,
имеют максимум возбуждения в области резонансной линии ртути с Л = 254 нм.
Поэтому именно эти составы имеют наибольшую яркость свечения.
Спектры, излучения (рис. IV.9) зависят от концентрации активаторов и соот-
ношения между содержаниями фтора и хлора. При постоянной концентрации Мп
в люминофоре по мере повышения концентрации Sb интенсивность излучения Мп
возрастет. Вызвано это тем, что вероятность возбуждения Мп увеличивается,
поскольку сурьма является сенсибилизатором для Мп. При увеличении концен-
трации Мп полоса Sb подавляется. Это дает возможность получать с примене-
нием галофосфатных люминофоров лампы с различными оттенками белого
света.
Замена фтора на хлор смещает спектр излучения в красную область, интен-
сивность люминесценции при этом уменьшается. Содержание Sb и Мп в люмино-
форах, предназначенных для использования в люминесцентных лампах различ-
ной цветности, приведено в табл. IV.1 (стр. 84). По данным различных авторов,
величина квантового выхода галофосфатных люминофоров колеблется в преде-
лах 0,71—0,85. В работе [30] исследована роль линии 185 нм в светоотдаче
ламп и измерен абсолютный квантовый выход галофосфата при возбуждении
его линиями ртути с длиной волны 185 и 254 нм; он оказался равным 2,2 и 0,74.
Однако по данным работы [31], квантовый выход при возбуждении линией
с длиной волны 185 нм не превышает 1. Таким образом, количественный вклад
в люминесценцию указанной линии остается невыясненным.
Использование галофосфатов в люминесцентных лампах. Галофосфаты
Используют в лампах типа ЛД, ЛХБ, ЛБ и ЛТБ. На стадии изготовления ламп
И в процессе их эксплуатации люминофор подвергается внешним воздействиям,
снижающим интенсивность свечения. К последним относятся: измельчение на
стадии приготовления суспензии; нагревание на стадиях выжигания биндера
и вакуумной обработки слоя; взаимодействие с парами ртути и остаточными га-
зами в процессе эксплуатации ламп; радиационное разрушение под действием
излучения с длиной волны 185 нм при горении ламп.
В настоящее время, когда квантовый выход и коэффициент поглощения
возбуждающей радиации близки к предельным, повышение устойчивости к ука-
занным воздействиям является, по-видимому, основным направлением работ
в области повышения светоотдачи и стабильности светового потока люминесцент-
ных ламп. Это не исключает необходимости проведения работ по совершенство-
ванию технологии изготовления ламп с целью ослабления воздействия техно-
логических факторов на люминофор.
Основной причиной снижения яркости свечения на стадии размола, не-
видимому, следует считать увеличение диффузного отражения
возбуждающего излучения в результате уменьшения размера зерен люмино-
фора. Показано, что измельчение люминофора не изменяет квантовый выход.
Поэтому увеличение поглощательной способности люминофора путем повышения
в нем содержания активной сурьмы, ответственной за поглощение возбужда-
ющего излучения, является одним из приемов снижения потерь яркости све-
чения на стадии Измельчения. Однако наиболее рациональным решением пред-
ставляется разработка люминофора с оптимальным гранулометрическим соста-
вом, не требующим размола на стадии приготовления суспензии.
При нагревании люминофора на стадии выжигания биндера протекают
Два независимых друг от друга процесса порчи люминофора. Окончательно при-
рода этих процессов не изучена. Первый процесс протекает в самом люмино-
форе при нагревании его на воздухе и сопровождается появлением темной
окраски [32—35]. Это явление не имеет отношения к равновесному состоянию
системы, так как при увеличении длительности прогрева в окислительных усло-
виях (на воздухе) яркость свечения почти полностью восстанавливается, серая
окраска исчезает [35]. Применение некоторых специальных обработок позво-
ляет существенно снизить потерю яркости люминофора при нагревании; это
свидетельствует о поверхностном характере данного явления.
Можно предполагать, что в основе процесса лежит обугливание
6 Заказ 44
81
адсорбированных люминофором органических соединений или восстяповлептте
ими соединений сурьмы, расположенных на поверхности зерен люминофора. При
нагревании люминофора в вакууме, по-видимому, протекает тот же самый про-
цесс, но из-за отсутствия кислорода яркость свечения восстанавливается при
Рис. IV.9. Зависимость спектрального распределения энергии излучения гало-
фосфата кальция от концентрации сурьмы (а), марганца (б) и относительного
содержания хлора и фтора (в).
Содержание активаторов (в масс. %): 1 — 1,0; 2 — 3,0; з — 6,0; 4 — 0,0; 5 — 0,1; 6 — 0,5;
7 — 1,0; 8 — 2,0; 9 — 3,0; 10 — 4,0; 12—5,0.
Соотношение хлвр/фтор: 1 — без хлора; 2 — без фтора; з — C1/F = 1/1; 4 — Cl/F_= 1/3;
5 — C1/F = 3/1.
более высоких температурах и сопровождается отгонкой из люминофора метал-
лической сурьмы [35, 36].
Причина второго процесса порчи люминофорного слоя при нагревании —
взаимодействие люминофора с окислами щелочных металлов, диффундиру-
ющими из стекла в люминофор [37]. Этот процесс зависит от химического состава
как стекла, так и поверхности зерен люминофора.
82
На основании результатов изучения поведения люминофора в лампе при
горении определены законы спада яркости свечения, отражающие различную
природу процессов деградации люминофора в лампе. Наиболее быстро
протекает деградация, обусловленная радиационным разрушением люмино-
фора под действием излучения % = 185 нм [38—41J. Она сказывается уже на
начальной светоотдаче ламп (перед измерением начальной светоотдачи лампы
тренируются). Данные измерения ЭПР [40, 41] и результаты исследования
природы запасания светосуммы под действием излучения с X = 185 нм позво-
ляют сделать вывод, что в основе этого процесса лежит ионизация Мп2+ до Мп3+.
Повышение в люминофоре содержания активной сурьмы, поглощающей коротко-
волновое УФ-излучение [40, 41], стабилизирует люминофор. Этому же способ-
ствует введение в люминофор кадмия.
Рис. IV.10. Изменение во времени светоотдачи лампы
мощностью 40 Вт с галофосфатным люминофором.
Показано, что под действием излучения с X = 185 нм протекают процессы
и в безактиваторной основе люминофора, приводящие к появлению центров
окраски.
Взаимодействие люминофора с остаточными вредными газами изучено в ра-
ботах [42—45]. Однако химизм этого взаимодействия до настоящего времени
окончательно не выяснен.
На рис. IV. 10 представлена типичная кривая спада во времени светоотдачи
люминесцентных ламп, изготовленных с применением галофосфата кальция.
В табл. IV. 1 приведены данные по светоотдаче люминесцентных ламп раз-
личного типа.
6*
83
Таблица IV.1
Технические характеристики различных типов галофосфатных люминофоров
Марка люминофора Тип лампы (мощность 40 Вт) Светоотдача, лм/Вт Содержание актива- торов, масс. % \пах’ нм Соотношение полос в мак- симумах ^480/^тах1 %
началь- ная после 100 ч Мп Sb
ЛГ-1-2 ЛБ-40 83 80 1,4 0,7—1,0 578-584 22 ±3
Л Г-5 ЛТБ-40 77 75 2,4 0,7-1,0 579-585 12 ±4
ЛГ-6 ЛХБ-40 77 75 1,0 0,7—1,0 579-585 40 ±3
ЛГ-2К Л Д-40 65 63 0,4 0,7—1,0 570—580 85 ±5
Фосфаты
Состав и люминесцентные свойства. Исследование люминесцентных свойств
систем на основе различных фосфатов привело к разработке некоторых практи-
чески важных смешанных люминофоров [46—50]. Замена в люминофоре
Sr3(PO4)2, который обладает очень слабой люминесценцией с Лтах = 370 нм,
части стронция на алюминий, цинк, магний или кальций позволяет получить
люминофоры, возбуждающиеся в УФ-области спектра, с весьма интенсивным
излучением в красной области (см. рис. II.4, стр. 39). Квантовый выход этих
люминофоров близок к 0,9. Олово в фосфатных люминофорах должно нахо-
диться в двухвалентном состоянии. По этой причине их синтезируют или в вос-
становительной атмосфере строго дозированной смеси N2 + Н2 (—1,5 объ-
емн. % Н2), или с введением в шихту восстанавливающих агентов в условиях
предотвращения попадания кислорода в люминофор на стадиях прокаливания
и остывания [51].
Практическое примененье в люминесцентных лампах нашли следующие
люминофоры: (Zn, Sr)3(PO4)2-Sn; (Са, Sr)3(PO4)2 • Sn; (Ca, Zn)3(PO4)2 • Sn;
(Ca, Mg)3(PO4)2-Sn.
Исследование фазового состава и люминесцентных свойств указанных
систем проведено в работах [48, 50].
В состав шихты люминофора (Zn, Sr)3(PO4)2-Sn входят SrHPO4, SrCO3,
Zn3(PO4)2 (или ZnO), SnO (или SnO2). Люминофоры с наибольшей интенсив-
ностью свечения получаются при содержании Zn3(PO4)2, равном 15 мол. %;
их структура соответствует области твердого раствора фосфата цинка в фосфате
стронция.
Спектр излучения люминофора (Zn, Sr)3(PO4)2 • Sn (рис. IV.11) состоит—
из двух полос с максимумами 300—400 нм (малоинтенсивная полоса) и 620—
630 нм. Квантовый выход составляет 84%. Люминофор температурно устойчив,
интенсивность излучения его возрастает при повышении температуры до 200ч
и затем постепенно начинает уменьшаться (рис. IV.12).
Система (Zn, Са)3(РО4)2-Sn по фазовому составу аналогична системе
(Zn, Sr)3(PO4)2 - Sn. В области твердых растворов этих систем получены люмино-
форы, которые при возбуждении резонансной линией ртути (X = 254 нм) имеют
три полосы излучения: 390 нм (очень слабая), 500 и 610 нм (рис. IV.13). Соотно-
шение интенсивностей полос зависит от содержания олова. При Хнсзо = 365 нм
в спектре излучения этого люминофора остаются две полосы, интенсивность
которых становится одинаковой. Наибольшей интенсивностью свечения обладает
люминофор, содержащий 10 мол. % Zn3(PO4)2. Квантовый выход такого люми-
нофора равен 0,9. При нагревании люминофора (Zn, Са)3(РО4)2-Sn до 300°
яркость свечения его при возбуждении X, 03б — 365 нм увеличивается и начи-
нает уменьшаться при температуре выше 350°.
В последние годы были разработаны новые рецептуры ламповых люмино-
форов на основе различных соединений редкоземельных элементов [52—61].
С целью поиска новых составов ламповых люминофоров изучалось влияние
84
некоторых редкоземельных элементов и ванадия на свойства фосфатных люмино-
форов. Показано [61—64], что при введении р. з. э. могут быть получены эффек-
тивные ламповые люминофоры с широким спектром возбуждения и излучения.
Так, при введении Y или La в люминофор (Са, Sr)3(PO4)2 - Sn максимум его излу-
чения сдвигается в длинноволновую область, при этом улучшаются и темпера-
турные характеристики излучения. Использование ТЬ в качестве активатора
(в сочетании с Sn) приводит к появлению в спектре излучения зеленой полосы
с Хщах = 540 нм, особенно эффективной при возбуждении длинноволновым
Рис. IV.11. Спектры возбуждения (7)
и излучения (2) люминофора
(Zn, Sr)3(PO4)2 • Sn.
Рис. IV.12. Зависимость спектраль-
ного распределения энергии излуче-
ния люминофора (Zn, Sr)3(PO4)2-Sn
от температуры.
УФ-светом. Интегральная яркость излучения этих люминофоров в 2—2,5 раза
превосходит таковую для промышленного люминофора марки Л-42Дв.
Введение ванадия в фосфатные люминофоры в сочетании с р. з. э. позволило
синтезировать новые люминофоры на основе Са, Zn- и Са, Sr-фосфатов. Наиболее
эффективные из полученных люминофоров
при возбуждении А, = 365 нм при комнатной
температуре по интегральной яркости излу-
чения в 5—6 раз превосходят промышлен-
ные люминофоры с аналогичным цветом све-
чения (марки Л-43 и Л-50, фосфатованадаты
иттрия).
Состав и некоторые люминесцентные ха-
рактеристики новых люминофоров приведены
в табл. IV.2.
Применение. Люминофоры на основе фос-
фатов цинка и кальция применяются в лам-
пах низкого и высокого давления типа ДРЛ.
Прп этом светоотдача ламп повышается при-
мерно на 10% в сравнении со светоотдачей
кварцевой горелки лампы и достигает для
ламп мощностью 400 Вт 58 лм/Вт. Отметим,
Рис. IV. 13. Спектральное рас-
пределение энергии излучения
люминофора(гп,Са)3(РО4)2 -Sn
при АБ03б = 254 нм (7) и Авозб =
= 365 нм (2).
что в случае арсената или фторгерманата она понижается на 10—15%. Доля
излучения ламп типа ДРЛ в красной области спектра при использовании цинк-
кальций фосфата составляет 3—4% , что иногда ниже требуемого уровня. По-
этому в настоящее время указанный люминофор применяют в лампах ДРЛ
в смеси с ортованадатом иттрия, активированном Ей, что позволяет повысить
излучение в красной области спектра до 10—12%.
Применение цинк-кальций фосфата в лампах низкого давления позволило
Улучшить цветопередачу этих ламп, так как доля энергии, излучаемая этим
люминофором в красной области спектра, гораздо больше, чем у галофосфатных
люминофоров, и обеспечивает светотехнические требования по спектральным
зонам излучения для ламп с улучшенной цветопередачей.
В табл. IV.3 приведены значения светоотдачи ламп различного типа с улуч-
шенной цветопередачей, полученных при использовании фосфатных люмино-
форов в сочетании с другими люминофорами.
85
Таблица IV.2
Люминесцентные характеристики люминофоров на основе фосфатов
с использованием р. з. э. и ванадия
Состав люминофора Характеристика спектраль- ного распределения энер- гии излучения при воз- буждении Цвет свечения при воз- буждении
Х= 365 нм Х = 254 нм Х = 365 нм % = 254 нм
(Са, Sr, Y)3(PO4)2« Sn 655 640 Оранжево- красный Малиново - красный
(Са, Sr, Y)3(PO4)2. Sn-Tb 490, 540. 585 625, 650 490, 540, 645 Желто- заленый Красный
(Са, Zn, Y)3(PVO4)2- •Eu-Bi 595,615, 620 652, 700, 705 595, 615, 620, 652, 700, 705 Красный »
(Ca, Sr, Y)3(PVO4)2- Sm. Bi 495,525, 565 603, 645, 652 708 565, 603, 618, 645, 652, 708 Желто- оранжевый Белый
Таблица IV.3
Координаты цветности и светоотдача различных типов
люминесцентных ламп с улучшенной цветопередачей
Тип лампы Цветовая темпера- тура, К Координаты цвет- ности * Светоотдача лампы мощностью 40 Вт через 100 ч, лм/Вт Компоненты люминесциру- ющего покрытия
X У
лдц 6500 0,317 (0,313) 0,334 (0,323) 55,0 (Са, Zn)3(PO4)2 • Sn, люми- нофоры Л-29 или ЛГ-ЗК
ЛХБЦ 4300 0,370 (0,368) 0,370 (0,370) 55,0 (Са, Zn)3 • (РО4)2 • Sn, фтор- силикат, активированный Ti; люминофор ЛГ-ЗК
ЛБЦ 3500 0,405 (0,405) 0,390 (0,390) 42,6 (Са, Zn)3(PO4)2 • Sn, люми- нофоры К-35 и Л-35
ЛТБЦ 2900 0,440 (0,442) 0,400 (0,402) 42,5 (Са, Zn)3(PO4)2 • Sn люми- нофоры Л-35 и К-35
* В скобках для соавнения приведены координаты цветности абсолютно черного тела
при той же цветовой температуре.
Люминофор цинк-кальций фосфат, активированный Sn, выпускается в на-
стоящее время в промышленном масштабе под маркой Л-42 Дв для ламп высо-
кого давления и под маркой Л-42 Дн для ламп низкого давления. Цинк-строн-
ций фосфат, активированный Sn, выпускается под маркой ЛО-2, а кальций-
стронций фосфат, активированный Sn — под маркой ЛО-3.
Характеристики перечисленных промышленных люминофоров приведены
в табл. IV.4.
Люминофоры (Са, Mg)3(PO4)2 • Sn описаны в работах [65, 66]. Люминофор
Р-Са3(РО4)2- Sn при возбуждении коротковолновым УФ-излучением имеет
основную полосу излучения с максимумом ~630 нм и малоинтенсивную полосу
около 500 нм. Добавление фосфата магния (оптимальная концентрация Mg
соответствует отношению MgO : Р2О6 = 0,15) приводит к появлению новой
86
Таблица IV.4
Люминесцентные характеристики люминофоров на основе
двойных фосфатов, активированных Sn
Люминофор Характеристика спектрального распределения анергии
марка состав Лвозб=254 нм Чозб=365 нм
Л О-2 (Zn, Sr)3(PO4)2. Sn Ащах = 620-630 нм, ши- Хщях = 620—630 НМ,
Л-42ДН Л-42Дв Л О-З (Zn, Ca)3(PO4)2 • Sn (Zn, Ca)3(PO4)2 • Sn (Ca, Sr)3(PO4)2 • Sn рокая полоса излуче- ния от 500 до 750 нм Хтах — 500 и 610 нм; Лоо/^вю = 0,45 Хтах = 380 и 640 нм широкая полоса излучения от 500 до 750 нм Хтах = 495 и 605 нм А9бДвОб2> 1 ^тах = 640 нм
полосы с максимумом около 380 нм. Однако при увеличении концентрации Sn
от 0,01 до 0,05 г-ат на 1 моль Р2О5 интенсивность этой полосы уменьшается
и значительно увеличивается интенсивность полосы с максимумом излучения
около 500 нм; при этом получается очень эффективный двухполосный темпера-
туроустойчивый люминофор. При повышении температуры яркость свечения
его возрастает и достигает максимума при 250°, причем значительно увеличи-
вается интенсивность излучения красной полосы (630 нм). Благодаря этому
его можно применять как в лампах низкого давления с улучшенной цветопере-
дачей, так и в лампах высокого давления. Получают люминофор прокаливанием
шихты из смеси СаНРО4, СаСО3, MgO и SnO2 при 1100—1250° в течение 2 ч
в восстановительных условиях.
Люминофоры на основе фосфатов с излучением в УФ-области спектра.
Люминофор Са3(РО4)2 - Се описан в работах [67, 68]. Его основа
может быть получена спеканием СаНРО4 и СаСО3, взятых в стехиометриче-
ском соотношении. Церий должен находиться в трехвалентном состоянии,
поэтому прокаливание производят в восстановительной атмосфере. Наиболь-
шая интенсивность излучения имеет место при концентрации Се, равной 10%
(рис. IV.14, кривая 1). Максимум излучения расположен при 360 нм. Область
возбуждения начинается от 300 нм и простирается в область коротковолнового
УФ-света. Квантовый выход достигает 70%.
Люминофор Са3(РО4)2-Се применяли ранее в лампах УФО, в настоя-
щее время он заменен более стабильным люминофором Л-33 (BaSi2O5Pb)
(см. стр. 90).
Люминофор (Са, Sr)3(PO4)2 -Т1 [69] получается при прокаливании
смеси SrHPO4, SrCO3, СаНРО4, СаСО3 и T12SO4. Люминофор Sr3(PO4)2 Т1
имеет очень малую интенсивность излучения; добавление к этой основе Са3(РО4)2
приводит к резкому ее повышению и к сдвигу максимума излучения в коротко-
волновую область.
Оптимальный состав люминофора: Са3(РО4)2(50)-Sr8(PO4)2 (50)-Т1 (10);
максимум его излучения расположен около 350 нм (рис. IV. 14, кривая 2). Лю-
минофор возбуждается резонансной линией ртути 254 нм. Применяется он
в люминесцентных лампах низкого давления, баллон которых готовится из
стекла УФС-4 (максимум пропускания в области 365 нм). Такая лампа не тре-
бует дополнительных светофильтров для выделения УФ-излучения и может быть
Применена для подсветки шкал приборов и люминесцентного анализа.
Люминофоры (Са, Zn)3(PO4)2 Т1 и (Са, Mg)3(PO4)2 Т1 для эри-
темных ламп. Свойства и приготовление эритемных люминофоров описаны
в работах [70—73]. Спектр излучения люминофора представлен на рис. IV.14
(кривая 5). Интенсивность и спектр излучения люминофоров (Са, Zn)3(PO4)2 -Т1
зависят от содержания цинка в основе люминофора и концентрации Т1.
87
Люминофоры с максимальной интенсивностью свечения получаются при концен-
трации фосфата цинка ~10% и Т1 ~5% , при этом максимум спектра излучения
соответствует 312 нм. Промышленностью люминофор выпускается под мар-
кой Э-1.
Замена части Zn3(PO4)2 в основе на Mg3{PO4)2 (при содержании послед-
него ~8%) позволяет получить люминофор с максимумом излучения при 305 нм.
Рис. IV.14. Спектральное распределение энергии излучения некоторых люми-
нофоров:
1 — Са3 (РО4)2-Се; 2 — Sr3 (РО4). (50)-Са3 (РО4)2 (50)-Т1 (10), 3 — Са3 (РО4)2 (90)-Zna (P04)t
(10)-Т1 (5) — марка Э-1; 4 — BaSi2O6-Pb; 5 — BaZn2Si2O7-Pb; 6 — BaZn2Si30v Pb, моди-
фицированный добавкой 3% MgO.
Этот люминофор, известный как Э-2, более соответствует требованиям, предъ-
являемым к эритемному люминофору, так как максимум его излучения ближе
к максимуму эритемной чувствительности кожи человека (~300 нм) [73].
Силикаты
Люминофоры на основе цинк-бериллий силиката, активированные Мп,
описаны в работах [74—76]. Основу люминофора готовят прокаливанием
смеси ZnO и ВеО с SiO2 при 1200°; при этом образуется твердый раствор сили-
катов цинка и бериллия. От состава основы зависят спектральный состав излу-
чения (рис. IV.15) и длительность послесвечения. Введение Be подавляет полосу
излучения силиката цинка, активированного Мп, при этом появляется новая
полоса с максимумом около 610 нм.
Силикат кальция, активированный РЬ и Мп. Получение. Силикат кальция,
активированный РЬ и Мп (CaSiO3 РЬ-Мп), может быть получен прокаливанием
смеси карбонатов или окислов соответствующих металлов с SiO2 [76, 77]. Для
смеси СаСО3 с SiO2 целесообразна температура 1150° и атмосфера водяного
88
пара, который оказывает минерализующее действие. При прокаливании в от-
крытых кюветах шихта люминофора должна иметь следующий состав (в моль):
СаСО3........................... 1
SiO2-H2O....................... 1-2
PbF2 ......................... 0,008
MnCO3 ....................... 0,0066
Наибольшую интенсивность свечения имеют люминофоры со структурой
метасиликата. Оптимальная концентрация Мн лежит в пределах 0,04—0,13 моль
Мп на 1 моль СаО. Концентрация вводимого в шихту РЬ очень мала (0,003—
0,008 моль на 1 моль СаО) в том случае, если прокаливание ведут в закрытых
тиглях. При прокаливании в открытых тиглях в связи с летучестью солей
свинца, концентрация РЬ должна быть значительно повышена. Поэтому мето-
дика приготовления люминофора зависит от состава газовой среды, формы и гео-
метрических размеров тиглей и печей для прокаливания.
Рис. IV.15. Спектральное распределение
энергии излучения цпнк-бериллий сили-
катных люминофоров различного состава
Кривая Состав люминофора, моль Квантовый выход
ZnO ВеО Мп О S1O,
1 2,0 0 0,122 1,18 0,77
2 1.8 0,2 0,058 1,11 0,89
3 1,8 0,2 0,181 1,11 0,89
4 1,8 0,2 0,1 11 1,17 0,84
5 1,8 0,2 0,138 1,20 0,84
6 1,6 0,4 0,132 1,19 0,84
В работе [78] приведены данные, показывающие целесообразность исполь-
зования минерализующих добавок CaSO4 и CaF2 при прокаливании шихты
люминофора CaSiO3 PbjMn. В этом случае эффективные люминофоры полу-
чаются при более низкой температуре (1050°) и, кроме того, уменьшается дли-
тельность процесса. Применение в качестве минерализатора NH4F позволяет
получить хороший мелкокристаллический люминофор.
Люминесцентные свойства. Люминофор CaSiO3-Pb-Mn относится, как
и галофосфат кальция, к разряду сенсибилизированных. Излучение в области
254 нм поглощается на уровнях активатора РЬ, который передает поглощенную
энергию Мп. Спектр люминесценции (рис. IV. 16) состоит из двух полос, одна
из которых, расположенная в УФ-области, имеет максимум излучения около
350 нм и обусловлена активирующим действием РЬ, другая имеет максимум
при 610 нм, и определяется наличием Мп.
Квантовый выход люминофора превышает 80%, благодаря чему этот люми-
нофор с успехом может быть применен в лампах с улучшенной цветопередачей.
В этом случае для компенсации недостатка излучения в синей и зеленой областях
спектра к люминофору CaSiO3 РЬ Мп добавляют вольфрамат магния и силикат
цинка, активированный Мп. В Советском Союзе люминофор CaSiO3PbMn
выпускается под маркой Л-25.
Фторсиликат магния, активированный Ti. Изучение фазового состава
и люминесцентных свойств этого люминофора [79] позволило разработать со-
став ЛФ-1 для люминесцентных ламп. Для целей газосветного освещения при-
меняют люминофор марки Л-14 на той же основе.
Готовят фторсиликат магния, активированный Ti [80] прокаливанием шихты
состава (в моль): MgO — 1; SiO2— 1; MgF2 — 2; TiO2 — 0,015 в закрытых квар-
цевых тиглях при 1100—1200° на воздухе в течение ~2ч. Спектр излучения
89
этого люминофора показан на (рис. IV.16). Люминофор возбуждается в области
254 нм и имеет квантовый выход, близкий к 90%. Широкая полоса излучения
и высокий квантовый выход позволяют применять этот люминофор для испра-
вления цветности люминесцентных ламп низкого давления.
Тройной силикат бария, стронция и лития, активированный Се и Мп
[81—83]. Оптимальный состав люминофора соответствует формуле
2,4ВаО-0,6SrO • l,0Li2O • l,9SiO2, концентрация Се составляет 10%, а концен-
трация Мп — 2% . Прокаливание ведут в восстановительной атмосфере при 900°.
Люминофор относится к классу сенсибилизированных: при возбуждении его
энергия поглощается Се и передается Мп. Значительное поглощение в области
365 нм делает этот люминофор в случае применения его в лампах высокого да-
вления низкой мощности эффективнее арсената магния, активированного Мп,
Рис. IV. 16. Спектральное распределе-
ние энергии излучения люминофоров
на силикатной основе:
Рис. IV. 17. Спектральное распреде-
ленпе энергии излучения некоторых
люминофоров:
1 — CaSiO3>Pb-Mn; 2 — фторсиликат магния,
активированный Ti.
1 — тройной силикат бария, стронция и
лития, активированный Се и Мп; 2 — фтор-
германат магния, активированный Мп; 3 —
арсенат магния, активированный Мп.
и цинк-кальций фосфата, активированного Sn. Спектр люминесценции
(рис. IV. 17, кривая 7) состоит из двух полос с максимумами при 420 и 620 нм *.
При повышении температуры до 200° интенсивность люминесценции умень-
шается, поэтому этот люминофор не может конкурировать с фторгерманатом
магния, арсенатом магния и цинк-кальций фосфатом в мощных люминесцентных
лампах высокого давления. Уменьшая концентрацию Мп и увеличивая длитель-
ность прокаливания, можно улучшить стабильность люминофора в 1,5 раза [83].
В промышленном масштабе этот люминофор в СССР не изготовляется.
Люминофоры на силикатной основе, излучающие в ультрафиолетовой
области спектра. Люминофор BaSi2O5 • РЬ весьма эффективен при излу-
чении в УФ-области спектра (Хмах = 350 нм). Готовят его прокаливанием
шихты, состоящей из смеси ВаСО3, SiO2, РЬО2 и добавок минерализаторов —
BaSO4 и ВаС12 в течение 2 ч при 1100 9С [84]. Примерная рецептура шихты (в г):
SiO2 ..........................390
ВаСО3.........................500
BaSO4...........................78
ВаС12..........................19
РЬО2 ...........................28
Спектр излучения люминофора представлен на рис. IV. 14. Область воз-
буждения находится около 254 нм. Этот люминофор, выпускаемый промышленно-
стью под маркой Л-33, применяется в лампах У ФО для подсветки шкал приборов.
* На рис. IV. 17 показана только основная полоса излучения.
90
При исследовании ряда тройных силикатных систем, которые содержат
один из окислов щелочноземельных металлов вместе
с MgO или ZnO, активированных РЬ [85], было установлено, что некоторые
из них являются хорошими основаниями для люминофоров, излучающих в об-
ласти 300—400 нм. В частности, для эритемных ламп весьма эффективным
оказался люминофор BaZn2SiaO7-РЬ, выпускаемый под маркой Э-3. Этот люми-
нофор имеет максимум излучения в области 300—305 нм, а его квантовый выход
составляет 0,70—0,75. Люминофор применяют в эритемных лампах, позволя-
ющих повысить выход витамина D в процессе производства на 25—30%. Для
приготовления люминофора шихту, состоящую из SiO2, ВаСО3, ZnO, BaF2
и РЬО2, прокаливают в закрытых тиглях при НОО*5 около 2 ч. Спектр излуче-
ния люминофора BaZn2Si2O7 РЬ представлен на рис. IV.14. Добавление 3—
5% MgO (вместо ZnO) приводит к смещению спектра излучения в коротковолно-
вую область, что способствует увеличению эритемного действия.
Германаты и арсенаты
Фторгерманат магния, активированный Мп. Первые люминофоры на основе
орто- и метагерманатов магния были получены в 1936 г. Леверенцем [86],
однако они обладали слабой люминесценцией. Вильямс [87] добился заметного
повышения яркости свечения этих люминофоров, использовав при синтезе зна-
чительный избыток MgO над окисью германия. Дальнейшее усовершенствова-
ние свойств германата магния, активированного Мп, было осуществлено Торинг-
тоном [88]. Он, заменив часть MgO на MgF2, повысил яркость свечения и терми-
Рис. IV.18. Спектры возбуждения
некоторых люминофоров, активиро-
ванных Мп:
1 — фторгерманат магния: 2 — арсенат
магния.
ческую устойчивость люминофора, что сделало его пригодным для использова-
ния в лампах высокого давления. Для приготовления чистых германатов были
выбраны составы: 4MgO; l,0GeO2 и 0,01Мп. Наиболее эффективные фторгер-
манаты имеют следующий состав: 3,5MgO; 0,5MgF2; l,0GeO2 и 0,01Мп. Прокали-
вание ведут при 1100—1200° в течение 30 мин, после чего препараты размалы-
вают в мельнице и вторично прокаливают при 1100°. Опыт показал, что увели-
чение длительности вторичного прокаливания до 2—3 ч повышает яркость све-
чения люминофора. Магний-фторгерманатный люминофор (IV.18, кривая 1)
возбуждается в широкой области спектра от 200 до 480 нм. Введение фтора
увеличивает поглощение люминофора, что и объясняет соответствующее повы-
шение яркости свечения. Спектр излучения люминофора (см. рис. IV. 17, кри-
вая 2) состоит из узких полос с максимумом при 626; 634; 642,5; 653,5 и 659,5 нм.
При нагревании люминофора до 350° яркость свечения возрастает (рис. IV. 19),
а затем начинает уменьшаться. Квантовый выход при Хгозб = 365 нм дости-
гает 90%. Промышленностью люминофор выпускается под маркой Л-40 и пред-
назначен для исправления цветности ртутных ламп высокого давления.
Арсенат магния, активированный Мп. Люминофоры на основе арсената
Магния, активированного Мп, подобно уже описанному фторгерманату магния,
имеют спектр излучения, расположенный в красной области, состоящий из не-
скольких полос. Приготовление люминофора заключается в многократном дли-
тельном прокаливании смеси MgO, As2O6 и МпСО3 в окислительной атмосфере
'На воздухе) при 1100°. Введение плавней NH4C1 и NH4F [89] позволяет снизить
91
общую длительность прокаливания до 4 ч. Показано, что добавление в шихту
карбоната лития увеличивает стабильность свечения люминофора [90].
Люминофор возбуждается в области 254—500 нм. Спектр излучения пока-
зан на рис. IV.17 (кривая 5), а спектр возбуждения — на рис. IV.18 (кривая 2).
Кривые температурной зависимости (рис. IV. 19) свидетельствуют, что излуче-
ние люминофора является температурноустойчивым и поэтому его можно при-
менять в ртутных лампах высокого давления для исправления их цветности.
Недостаток арсенатного люминофора — сравнительно малая химическая
стабильность, в частности чувствительность к воздействию восстановительных
агентов. При этом изменяется цвет и уменьшается интенсивность свечения.
Стабильность свечения арсенатного люминофора может быть повышена при
замене части магния литием, однако при этом несколько уменьшается яркость
свечения. Оптимальную яркость свечения имеет состав 6MgO • As2O5 Мп (0,2).
Рис. IV.19. Влияние температуры на интенсивность
излучения фторгерманата магния (7) и арсената маг-
ния (6 — х) MgO -As2O5 -.гЫ-гО -0,01Мп:
2 — х = 0,01; 3 — х — 0,2; 4 — х — 0,6; 5 — х = 1,0.
Этот лкшинофор предназначался для улучшения цветопередачи ламп низкого
давления и выпускался под маркой Л-37. Для ламп высокого давления выпу-
скается люминофор с литием марки Л-41 следующего состава: 5,4 MgO,
0,3 Li2O • AsaO5 Мп (0,2); его используют в лампах ДРЛ в атмосфере£СО2.
IV.3. ЛЮМИНОФОРЫ ВРЕМЕННОГО ДЕЙСТВИЯ
Цинксульфидные люминофоры, активированные Си и Си совместно с Со,
прокаленные при 1200°, а также люминофоры на основе сульфидов щелочно-
земельных металлов обладают способностью при фотовозбуждении запасать
большую светосумму и высвечивать ее после прекращения возбужде-
ния. Длительность послесвечения подобных люминофоров оказывается доста-
точной для практического использования их вместо светосоставов постоянного
действия тогда, когда применение последних невозможно или недопустимо.
Эти люминофоры наносят на различные сигнальные устройства, шкалы прибо-
ров, часов и т. п. Для возбуждения люминофоров используют дневной свет,
лампы накаливания, газоразрядные источники света и лампы ультрафиолето-
вого облучения (УФО).
Способность к запасанию значительных светосумм связана с наличием
в люминофорах глубоких электронных ловушек. Наиболее глубокие из них
в цинксульфидных люминофорах дают Си и Со, а в люминофорах на основе
сульфидов щелочно-земельных металлов — Bi. Для каждого активатора харак-
терно свое собственное распределение электронных ловушек. Ротшильд еще
в 1963 г. показал на примере сульфидов щелочноземельных металлов, что
эффективность фосфоресценции может быть значительно улучшена добавкой
второго активатора. Так, в случае SrS - Bi и CaS - Bi благоприятны добавки Sm,
а также Си, а для ZnS Си — добавка Со.
92
Различие между этими классами люминофоров заключается в том, что на-
чальная яркость и яркость свечения на первой стадии затухания у цинксульфид-
ных люминофоров выше. Однако уже через короткое время яркость послесве-
чения люминофоров на основе сульфидов щелочноземельных металлов выше,
чем у цинксульфидных люминофоров. Первые имеют послесвечение, видимое
в темноте хорошо адаптированным глазом даже через 20—30 ч, послесвечение
цинксульфидных люминофоров не обнаруживается уже через 6—8 ч.
Недостаток люминофоров на основе сульфидов щелочноземельных метал-
лов — их малая стойкость по отношению к влаге воздуха вследствие гидро-
литического разложения с выделением серо-
водорода. Это обстоятельство значительно
ограничивает возможность их практического
использования, так как нет хороших средств
защиты люминофоров от влаги. В частности,
для этой цели оказались совершенно непри-
годны лаковые покрытия. Наилучшие резуль-
таты дает использование пластмасс. Однако
и в этом случае падение яркости свечения,
особенно в полевых условиях, довольно зна-
чительно.
Цинксульфидные люминофоры
временного действия получаю т прока-
ливанием смеси люминофорно-чистого ZnS
с плавнями при —1200° в течение 1—2 ч.
Концентрация активатора (Си) — от 5 -10_ 3
до Увеличение С1 уменьшает дли-
тельность послесвечения. Плавнем служит
смесь MgCl2 и NaCl (до 6%). Для повышения
длительности послесвечения в светосоставы
временного действия вводят —1 -10-3% Со.
Кобальт, однако, повышает чувствительность
Рис. IV.20. Спектральное рас-
пределение энергии излучения
некоторых люминофоров:
7 — ФК-106; 2 — ФКП-ОЗк; 3 —
ФКП-04; 4 - ФКП-05; 5 — ФКП-06.
люминофора к ИК-излучению, которое ока-
зывает тушащее действие. Вследствие этого люминофоры, активированные Со,
нельзя возбуждать лампами накаливания, свет которых содержит значитель-
ную долю красного и ИК-излучения.
Наша промышленность выпускает три марки цинксульфидных люминофоров
временного действия: ФКП-ОЗк, ФКП-04 и ФКП-05; все они дают зеленое све-
чение (табл. IV.5—IV.7, рис. IV.20).
Люминофоры на основе сульфидов щелочноземельных металлов
синтезируют как из заранее приготовленных сульфидов, так и путем
прокаливания смесей соответствующих карбонатов с активатором, серой, плав-
нями и восстановителем. В качестве восстановителя чаще всего применяют
крахмал. Основная реакция, которая протекает при синтезе люминофоров
может быть представлена уравнением:
4МеСО3 + 4S 3MeS + MeSO4 + 4СО2
Однако наряду с ней идут и другие, побочные реакции. Существует большое
количество рецептов для изготовления щелочноземельных люминофоров. Неко-
торые из них приведены в табл. IV.8.
В большинстве существующих рецептов в качестве активатора исполь-
зуют Bi (1-10‘3%), однако существуют рецептуры, в которых применяют РЬ,
Sn, Си, Мп и некоторые р. з. э.
По данным ГИПХ, оптимальный состав шихты для синтеза люмино-
фора (в %):
SrCO3 .......................
S............................
MgO..........................
Крахмал .....................
Bi ..........................
Си...........................
Li2CO3+ K2SO4(1 : 1).........
16
И
5
1 •10-2
1 •10-з
10
93
Таблица IV.5
Сравнительная характеристика светотехнических свойств люминофоров
с длительным послесвечением
я к" “к S <» о § Возбуждение лампой дневного света Возбуждение лампой накаливания
ФКП-ОЗк ФКП-06 соотно- ФКП-ОЗк ФКП-06 соотно-
в ° 2 и яркость свечения, кд«м~* (асб.) шение яркостей ФКП-06 яркость свечения, кд*м-* (асб.) шение яркостей ФКП-06
Дли' туха мин ФКП-ОЗк ФКП-ОЗк
1 0,27 0,84 0,22 0,6900 0,8 0,057 0,1820 0,51 0,1600 0,9
60 0,017 0,0051 0,105 0,0330 6 0,0007 0,0020 0,034 0,0106 5
120 0,005 0,0015 0004 0,0120 8 0,0002 0,0006 0,00116 0,0050 8
180 0,0003 0,0007 0,002 0,0072 10 0,0001 0,0004 0,001 0,0032 8
240 0,0001 0,0004 0,0014 0,0046 12 — 0,0002 0,0006 0,0020 10
480 0,00003 0,00003 0,0004 0.0014 14 — 0,0000 0,0002 0,0006 —
Таблица IV.6
Изменение яркости послесвечения люминофоров ФКП-04 и
ФК П-05 со временем
Время, мин Относительная яркость послесвечения (по сравнению с ФКП-ОЗк), %
Чозб = 365 нм при возбуждении лампой накаливания
ФКП-04 ФКП-05 ФКП-04 ФКП-05
5 100 100 6 50
30 148 237 29 100
60 185 327 59 145
120 233 483 125 200
Таблица IV.7
Характеристика люминофоров
с длительным зеленым послесвечением
(^тах = 520 нм: ДЛЯ ФКП-06 ^-щах =
= 500 нм)
Марка люмино- фора Состав Ориентиро- вочная дли- тельность послесвече- ния *
ФКП-ОЗк ZnS-Си 30—60 мин
ФКП-04 ZnS•Си• Со 1—3 ч
ФКП-05 ZnS•Си-Со 1,5—5 ч
ФКП-06 SrS • Bi • Си 8—10 ч
* Различимость светящейся поверхности 20X20 см на расстоянии
1 —2 м при кратковременной адаптации.
94
Таблица IV.8
Состав смесей для синтеза люминофоров на основе сульфидов
щелочноземельных металлов, активированных Bi [91, гл. II]
Содержание компонентов, г Плавень Продолжи- тельность
Цвет термической
свечения О и св о и о и св о и м св А „ св Ч а св эстав F 1-1 go Ч Д © s обработки при 1100° мин
О со ffl S о К S О x 0
Фиолето- 30,4 - . 6 8 1 Na2S О4 1 60
вый K2SO4 1
Синий 30,4 17,0 — 12 16 2 Na2SO4 2 30
K2so4 2
Голубой 20,0 23,0 11,0 16 22 2 Na2S O4 2 30
Ko,S 04 2
Зеленый 30,4 25,5 8,5 12 16 2 K2SO4 2 30
Na2S 2
Шихту прокаливают в кварцевых или неглазурованных фарфоровых тиглях
при 950° в течение 2 ч. Для люминофора готовят шихту сухим путем — смеши-
ванием компонентов в шаровой мельнице с последующим просеиванием через
сито. Активаторы вводят в виде водных растворов соответствующих солей
в смесь солей, используемых в качестве плавня. Люминофор SrS-Bi-Cu с зеле-
ным свечением был освоен промышленностью под маркой ФКП-06 (см. табл. IV.7,
рис. IV.20).
Для рассмотренного класса люминофоров характерен гиперболический
закон затухания свечения, т. е. резкий спад яркости свечения в первые минуты
затухания. Многочисленные попытки улучшения яркостных характеристик
этих люминофоров не дали существенных результатов. Поэтому они, конечно,
не могут заменить светосоставы постоянного действия. Тем не менее, значение
люминофоров временного действия велико, так как они позволяют обеспечить
минимальный уровень яркости светознаков, обеспечивающий ориентировку
в случае внезапного выключения света в помещениях и подземных сооруже-
ниях.
Методы нанесения светосоставов временного действия весьма разно-
образны. В качестве связующих веществ могут быть использованы те же
вещества, что и для светосоставов постоянного действия. При этом следует иметь
в виду, что из связующих веществ нужно полностью удалить влагу и примеси
свободных кислот, которые могут разрушить люминофор.
Иногда целесообразно добавлять к светосоставу в качестве наполни-
теля (до 10%) белые инертные вещества (ZnO, TiO2, BaSO4); это способствует
улучшению кроющей способности. Светящиеся пластмассы готовят на основе
полиметилметакрилата или полистирола; содержание люминофора в них дохо-
дит до 20%. Цинксульфидные люминофоры с успехом вводят в стекла и эмали.
IV.4. ЛЮМИНОФОРЫ ДЛЯ СВЕТЯЩИХСЯ КРАСОК
Люминофоры довольно широко применяют в производстве художественных
и декоративных изделий. Укажем, например, на так называемую люминесцент-
ную живопись, которая позволяет очень интересно решать некоторые художе-
ственные задачи при использовании УФ-подсветки. Люминесцирующие краски
применяют также в картографическом деле. В данном случае используют глав-
ным образом цинксульфидные и цинк-кадмийсульфидные люминофоры, активи-
рованные Ag и Си, с применением высококачественных прозрачных связующих
веществ.
95
На основе двойных сульфидов цинка и кадмия можно создавать краски
самых разнообразных оттенков. В светящихся красках применяют также воль-
фраматные и силикатные люминофоры. Использование их, однако, менее целесо-
образно, поскольку они возбуждаются только коротковолновым УФ-излуче-
нием, оказывающим заметное вредное биологическое действие на человека.
В составе светящихся красок используют преимущественно люминофоры
без видимого послесвечения, но иногда и рассмотренные ранее люминофоры
с послесвечением.
Часто для всех упомянутых целей находят применение и флюоресцирующие
органические красители, такие, например, как родамин, ауремин, антрацен,
хризен, антраниловая кислота и др. Однако органические люминофоры отли-
чаются пониженной стабильностью. Неорганические люминофоры нашли при-
менение также для изготовления флюоресцирующих и фосфоресцирующих
пластмасс, эмалей и стеклянных изделий, используемых как для художествен-
ных, так и для технических целей. При варке эмали и стекла пагубное действие
на цинксульфпдные люминофоры оказывают окислительные процессы, происхо-
дящие при высокой температуре на воздухе. Поэтому для этой цели используют
главным образом легкоплавкие стекла и эмали; иногда стекло варят в инертной
атмосфере.
При использовании светящихся красок цинксульфидного типа в изделиях,
подвергающихся прямому действию солнечного света, следует иметь в виду уже
упомянутое ранее фотолитическое действие коротковолновой части УФ-излу-
чения, приводящего к потемнению люминофоров.
Основные марки люминофоров для светящихся красок, выпускаемых оте-
чественной промышленностью, приведены в табл. IV.9. Изготовляются они
теми же методами, что и другие сульфидные люминофоры этого типа.
Таблица IV.9
Люминофоры для светящихся красок
Марка лю- минофора Цвет свечения Состав Марка лю- минофора Цвет свечения Состав
ФК-1 Синий ZnS • Ag ФК-5 Желтый ZnS•CdS • Ag
ФК-2 Голубой ZnS • Ag ФК-6 Оранжевый ZnS • CdS • Си
ФК-3 Зеленый ZnS • Cu ФК-7 Красный ZnS•CdS • Cu
ФК-4 Желто- зеленый ZnS • CdS • Cu ФК-8 Белый Смесь ZnS • Ag n ZnS•CdS•Ag
Для нанесения светящихся покрытий на металлические, стеклянные или
деревянные предметы можно пользоваться бесцветными лаками, например,
лаком БМК-5, полиметил- или полибутилметакрилатом, добавляя в них люми-
нофор в соотношении примерно 2 : 3.
IV.5. ЛЮМИНОФОРЫ ДЛЯ ПРЕОБРАЗОВАНИЯ
ИНФРАКРАСНОГО ИЗЛУЧЕНИЯ В ВИДИМОЕ
По закону Стокса длина волны возбуждающего излучения должна быть
меньше, чем длина волны люминесценции. Однако известны случаи, противо-
речащие этому постулату. В 1959 г. появились сообщения об антисток-
сов с к о й люминесценции в ноликристаллических сульфидах цинка и кад-
мия [92, 93J. В последнем случае наблюдалось зеленое свечение в полосе 525 нм
(2,35 эВ) при 80 К и возбуждении ИК-излучением с длиной волны 960 нм
(1,3 эВ). Эти результаты были подтверждены в 1962 г. на сульфидах и селе-
96
видах цинка и кадмия 194]. Эффективность преобразования ИК-излучения
в видимое достигала значительной величины. При этом интенсивность свечения
была довольно малой из-за слабого поглощения ИК-излучения в люминофоре.
Одновременно* Бломберген [95]
для целей детектирования ИК-излу-
чения предложил так называемый
счетчик квантов, в кото-
ром видимая люминесценция воз-
буждалась при последовательном по-
глощении двух фотонов различной
длины волны ионами р. з. э. Веро-
ятность двойного поглощения одним
ионом очень мала, поэтому антисток-
совое излучение было очень слабым.
В 1966 г. Овсянкин и Феофилов
[96] при изучении люминесценции
фторида бария и независимо от них
Озель при изучении натрий-иттер-
бий вольфрамата [97] обнаружили,
что видимая люминесценция Ег3+,
Но3+ и Тш3+ возрастает почти на
два порядка при введении в основу
ионов Yb3+.
С 1969 г. работы по люминофо-
рам, возбуждаемым инфракрасным
излучением, приобрели практиче-
Рис. IV.21. Спектры возбуждения лю-
минесценции:
1 _ NaYF4-Yb-Er; 2 — La2O2S-Yb-Er; 3 —
спектр излучения диода GaAs—Si.
ское значение, поскольку оказа-
лось, что спектр излучения ИК-источников из арсенида галлия близок
к спектру возбуждения подобных люминофоров (рис. IV.21). Это привело
к разработке нового класса светодиодов с зеленым, красным и голубым све-
чением на основе ИК-диодов из арсенида галлия, покрытых слоем люмино-
фора [98].
Механизм возбуждения
Для объяснения явления антистоксовской люминесценции редкоземельных
ионов привлекаются механизмы сенсибилизации люминесценции путем пере-
дачи энергии через промежуточные возбужденные состояния, а также коопера-
Рис. IV.22. Схема переноса энергии при сенсибилизации:
а — резонансный перенос энергии; б — перенос энергии с участием фоно-
нов; в — резонансная передача энергии через возбужденное состояние; г —
перенос энергии через возбужденное состояние с участием фонона.
тивные процессы, когда в элементарном акте участвуют одновременно несколько
ионов.
Многоступенчатое возбуждение [99]. Обычный механизм сенсибилизации
показан на рис. IV.22, а и б. Энергия, поглощенная сенсибилизатором S, резо-
нансно передается активатору А, находящемуся в основном состоянии. Возбу-
жденный уровень активатора может находиться либо на одном уровне с возбу-
жденным состоянием сенсибилизатора (рис. IV.22, а), либо несколько ниже
7 заказ 4 4
97
(рис. IV.22, б). В последнем случае часть энергии передается решетке в виде
фононов. Возможен и другой механизм передачи энергии, когда в начальный
момент в возбужденном состоянии находится не только сенсибилизатор, но и
активатор (рис. IV.22, в и г). В этом случае второе возбужденное состояние
активатора может оказаться выше возбужденного уровня сенсибилизатора
(рис. IV.22, в и г). Переход возбужденного иона в основное будет сопровождаться
излучением кванта с энергией, близкой к суммарной энергии поглощенных
квантов. Такой многоступенчатый механизм возбуждения антистоксовской
660 нм
'‘‘"/г
"‘К
Рис. IV.23. Схема многосту-
пенчатого механизма воз-
буждения антистоксовской
1 ^люминесценции.
люминесценции был предложен Озелем [97].
На рис. IV.23 показана схема возникновения
зеленого и красного свечения при возбуждении
ИК-излучением. Ион иттербия поглощает квант
ИК-излучения (970 нм) с энергией 1,28 эВ и пере-
ходит в возбужденное состояние с термом
затем происходит резонансная передача энергии
иону эрбия, который при этом также переходит
в возбужденное состояние а сенсибилиза-
тор — в основное После этого либо тот
же самый, либо другой ион иттербия поглощает
и передает возбужденному активатору энергию
второго кванта с Х = 970 нм. Возбужденный ион
теряет некоторую часть энергии в виде фононов
и переходит на излучательный уровень, а затем
в основное состояние с излучением кванта с энер-
гией почти вдвое большей, чем энергия квантов
возбуждения.
Пара Yb3+—Ег3+ позволяет получить не
только зеленое, но также красное, голубое и
УФ-излучения. Для красного и голубого свече-
ния возможны разнообразные варианты возбуж-
дения в соответствии с богатым набором уров-
ней иона эрбия [99].
Для возникновения зеленого излучения не-
обходимо два кванта возбуждающего ИК-излу-
чения. Для красного излучения возможен как
двух-, так и трехквантовый процесс возбужде-
ния. Для голубого и УФ-излучения необходимы
трех- и четырехквантовые процессы возбужде-
ния. Следствием многоквантового процесса возбуждения является степенная
зависимость яркости свечения от интенсивности возбуждения. Реально наблю-
дались квадратичная зависимость для зеленого излучения, степенная зависи-
мость с показателем 2,5 для красного* излучения и близкая к кубичной — для
голубого свечения.
Следует отменить, что необходимым условием для осуществления много-
ступенчатого процесса возбуждения является достаточно большое врем;я жизни
возбужденного состояния активатора (—40 мкс). Поэтому антистоксовские лю-
минофоры являются довольно инерционными.
Из рис. IV.23 видно, что двухступенчатое возбуждение эрбия возможно
и без сенсибилизатора. Но эффективность такого процесса значительно ниже.
Это связана с тем, что для ионов эрбия концентрационное тушение наблюдается
уже при малых концентрациях, тогда как содержание иттербия может быть
доведено до нескольких десятков процентов без заметного тушения зеленого
излучения эрбия. Кроме того, поглощение ИК-излучения на ионах иттербия
примерно на порядок эффективнее, чем на ионах эрбия.
Кооперативные явления. Кооперативные процессы заключаются в таком
взаимодействии нескольких возбужденных ионов, при котором энергия, запа-
сенная ими,, в едином акте передается одному иону, переводя его в возбужден-
ное ’состояние. Подобный механизм был предложен Овсянкиным и Феофило-
вым [96]. Конверсию инфракрасного излучения в видимое свечение в системах,
содержащих два сорта ионов (Yb3+ в паре с Ег3+, Но3+ и Тш3+), они объяснили
на i основе механизма кооперативной сенсибилизации люминесценции, схема
08
которого показана на рис. IV.24. В начальной стадии два иона сенсибилиза-
тора Sj и S2 находятся в возбужденном состоянии, а ион активатора А в основ-
ном. На конечной стадии ионы сенсибилизатора передают свою энергию акти-
ватору, переходя в основное состояние. Ион активатора приобретает суммарную
энергию от двух йоиов, минуя какое-либо промежуточное состояние.
Рис. IV.24. Схема коопера-
тивной сенсибилизации люми-
несценции туллия иттербием.
На рис. IV.25 приводится схема возбуждения иона эрбия излучением с дли-
ной волны 980 и 1550 нм при участии двух и трех квантов. Эффективность такого
Рис. IV.25. Схема кооперативного возбуждения ионов
эрбия:
а, в — зеленое излучение (540 нм); б — красное излучение
(670 нм).
кооперативного процесса может быть очень высокой. При большой плотности
возбуждения энергетическая эффективность преобразования ИК-излучения
в видимое теоретически может достигать 90% [100].
Технология изготовления люминофоров
Антистоксовские люминофоры получены на основе фторидов, оксисульфи-
Дов, оксихлоридов и различных окисных систем р. з. э. Лучшими основами
оказались: a-NaYF4, YF3 и La2O2S для зеленого свечения, a Y3OCI7 и YbOCl
Для красного. В качестве активаторов используют Ег (зеленое и красное све-
чение) и Тш (голубое свечение). Концентрация активаторов составляет несколько
тысячных долей в случае Тш и —1% для Ег. Во всех случаях в качестве сенси-
билизатора применяют Yb в концентрации 15—60 мол. %. Исходным сырьем
служат окислы р. з. э. высокой степени чистоты (99,999%). Недопустимы при-
меси некоторых р. з. э. Активными тушителями являются диспрозий или эрбий,
если активатором служит тулий. Заметно сказывается на люминесценции при-
месь железа в обычных для люминофоров концентрациях (—10-б%).
7*
99
Фториды редкоземельных элементов. Для получения фторида иттрия, акти-
нирогайного Ег и Yb, применяют осаждение с помощью плавиковой кислоты
из растворов хлоридов в азотнокислой среде. Образующиеся при этом фториды
прокаливают в инертной среде, а затем во фторирующей (HF, NH4F) для удале-
ния воды и кристаллизации аморфных частиц порошка. Иногда применяют
дегидратацию в высоком вакууме при высоких температурах.
По данным работы [101], шихту из фторидов готовят следующим образом.
Смесь окйслов в требуемом молярном соотношении растворяют в горячей кон-
центрированной соляной кислоте. Раствор разбавляют водой примерно в 2 раза,
охлаждают и добавляют смесь (1 : 1) концентрированных HNO3 и HF. Образо-
вавшийся студенистый осадок выпаривают при 70° и постоянном перемешивании
в течение суток, что способствует его кристаллизации. Затем осадок отделяют
от маточного раствора и многократно промывают смесью 10%-ных HNO3 и HF
до полного удаления хлоридов. Окончательно осадок промывают метанолом
и сушат при 110°. Полученный порошок затем медленно нагревают до 800° в по-
токе инертного газа в течение 2 ч, а затем в потоке безводного фтористого водо-
рода при 900° в течение 4—8 ч. Прокаливание ведут в лодочке из стеклоуглерода
пли платины, помещенной в трубу из этих же материалов. Для приготовления
a-NaYF4 смешивают фториды, осажденные по методу, описанному выше, с фтор-
силикатом натрия, взятом в некотором избытке, ц прокаливают смесь в токе
инертного газа при 600—660° с последующим медленным охлаждением. 'Режим
прокаливания и соотношение компонентов подбирают таким образом, чтобы
образовалась гексагональная фаза NaYF4, так как примесь кубической фазы
резко снижает эффективность люминофоров.
Оксисульфиды редкоземельных элементов. Оксисульфиды лантана и иттрия
с добавкой иттербия и эрбия синтезируют прокаливанием смеси соответству-
ющих окислов с содой, серой и фосфатом калия при температуре до 1100° [102].
Прокаливание ведут в закрытых тиглях на воздухе. Полученный спек обраба-
тывают водой для отделения от растворимых солей и полисульфидов. При син-
тезе очень существенным оказывается правильный подбор соотношения компо-
нентов и отсутствие подсоса воздуха.
Оксихлориды редкоземельных элементов. Оксихлориды получают прокалива-
нием соответствующих окислов в среде хлористого водорода. Для синтеза ис-
пользуют окисли, полученные разложением оксалатов, поскольку такие про-
дукты имеют повышенную химическую активность. Иногда для получения
оксихлоридов применяют гидролиз плавленных в токе влажного (пропущенного
через холодную воду) азота при 500—700° хлоридов р. з. э. Применяют также
метод, основанный на прокаливании окислов в газовой среде, содержащей хло-
рид аммония [103].
Основные свойства антистоксовских люминофоров
Эффективность. Уже отмечалось, что яркость свечения антистоксовских
люминофоров пропорциональна квадрату (или кубу) интенсивности возбуждения.
Насыщение не достигается даже при плотности возбуждения 170 Вт/см2, когда
эффективность зеленого излучения достигает 7,3%, а красного — 15% [104].
При умеренных плотностях возбуждения энергетическая эффективность не
превышает 1%. В табл. IV.10 приведены данные для различных люминофоров
при возбуждении их излучением диодов из арсенида галлия. Для сравнения
здесь же приведены данные для светодиодов на основе соединений AniBv.
Энергетическая эффективность люминофоров оценивалась при допущении, что
ПК-излучение полностью поглощается люминофорным слоем.
Спектры излучения и возбуждения люминесценции. Спектры излучения
(рис. IV.26) редкоземельных люминофоров состоят из узких полос, соответству-
ющих переходам в ионах эрбия (зеленое и красное свечение) и тулия (голубое
свечение). Соотношение интенсивности полос и их ширина зависят до некоторой
степени от основы люминофора. Для фторидов характерны спектры излучения
с преобладающей долей свечения в зеленой области. Для оксисульфидов и окис-
ных систем, наоборот, наблюдается преобладание интенсивности полос в красней
части спектра.
100
Т а б л и ц а IV.10
Эффективность преобразования электрической энергии
в видимую для светодиодов на основе арсенида
« галлия с люминофорным покрытием [99]
Светодиод Цвет свечения Энергетическая эффективность, % Условия возбуждения и эффективность ИК-диода (полусфериче- ского)
диода люмино- фора
GaP-ZnO GaAs — Si-j- Y0.74Yb0,25Er0,oiOCl Красный » 3—15 1,0 10,0 7= 1А/см2 0,ЗА; 1,5В; т] = Ю%
GaP-N GaAs— Si 4- 4- Bao,5Ybo,45Ero,O5p5 Зеленый » 0.6 0,1 0,8 Импульсный режим; 7 = 80 А/см2 0,ЗА; 1,5В; т)- 12%
GaN GaAs—Si4" 4" Yq, 65Yb0,35T то.оогРз Голубой » 0.005— 0,0015 0,011 0,1 Сублинейная зависи- мость (15—400 мВт) О,ЗА; 1.5В; г| ---10%
Рис. IV.26. Спектры излучения люминофоров с зеленым (а) и голубым
(б) свечениями:
1 — La2O2S-Yb-Er (Л-44); 2 — NaYF«-Yb-Ег (ИКВ-1); з — YF3-Yb-Tm.
101
Характерной особенностью люминофоров, содержащих иттербий и эрбий,
является перемена цвета свечения в зависимости от условий возбуждения. Это
связано с тем, что красная полоса разгорается пропорционально степени 2,5
от интенсивности возбуждения, а зеленая — пропорционально квадрату. При
импульсном возбуждении цвет свечения зависит также от длительности импуль-
сов ИК-возбуждения. При корот-
ких импульсах (-—100 мкс) преоб-
ладает зеленая полоса, а при длин-
ных (— 500 мкс) — красная.
В принципе, возможно получить
любой цвет, используя смесь из
трех люминофоров с голубым, зе-
леным и красным свечением и
комбинируя условия возбужде-
ния (интенсивность и длитель-
ность).
Спектры возбуждения люми-
несценции определяются в основ-
ном поглощением ИК-излучения
ионом иттербия, поэтому макси-
мальное возбуждение фосфоров
происходит вблизи 975 нм. Это
справедливо для всех люминофо-
ров с использованием Yb3+ в ка-
честве сенсибилизатора. Однако
решетка основы оказывает большое влияние на вероятности переходов в про-
Рис. IV.27. Спектр поглощения люмино-
фора ИКВ-1 (NaYF4 • Yb-Er).
цессе излучения и поглощения, на время жизни различных уровней и предель-
ные эффективности преобразования. В связи с этим спектры возбуждения не-
сколько различаются в зави-
симости от вида основы люми-
нофора. На рис. IV.21 пока-
заны спектры возбуждения
люминесценции для оксисуль-
фида лантана (Л-44) и натрий-
иттрий тетрафторида (ИКВ-1);
здесь же приведен спектр излу-
чения диода из арсенида гал-
лия, легированного кремнием.
Видно, что излучение GaAs —
Si может быть использовано
не больше чем на 30% в случае
фторидов и на 60% в случае
оксисульфида.
Среди других возможных
источников возбуждения сле-
дует отметить лампы накалива-
ния (особенно «иодные») и ксе-
ноновые дуговые лампы высо-
кого давления, значительная
1 — LaFs-Yb-Er; 2 — YFa-Yb-Er; 3 — Y.O.S-Yb-Er:
4 — NaYbWOs-Er.
часть излучения которых находится в ИК-области. У лампы накалива-
ния с иодным циклом примерно 3% всей мощности приходится на ИК-из-
лучение в области 900—1000 нм, т. е. примерно столько же сколько при-
ходится на излучение в видимой области спектра. Использование даже части
этой энергии для преобразования в видимое излучение заметно повысило бы
светоотдачу ламп накаливания. Сложность решения подобной задачи заклю-
чается в том, что антистоксовские люминофоры заметно поглощают в той же
области, где и излучают (рис. IV.27). Поэтому слой люминофора, нанесенный
на колбу лампы, будет сильно ослаблять излучение. Кроме того, поверхность
колбы лампы нагревают до 200°, а при этом происходит тушение люминесценции
большинства антистоксовских люминофоров.
Температурная зависимость люминесценции. Зависимость яркости свечения
антистоксовских люминофоров от температуры определяется главным образом
102
Природой соединения, и в меньшей степени соотношением концентраций акти-
вирующих добавок.
На рис. IV.28 показаны зависимости яркости свечения от температуры для
различных основ [99]; кривые нормированы к величине яркости при комнатной
температуре. Для фторидов подобная функция имеет максимум в районе (—60)—
(—80°); в случае оксисульфидов — при (—160°) и для вольфраматов — при 120°.
Применение антистоксовских люминофоров
Как уже отмечалось, интерес к антистоксовским люминофорам резко возрос
после того, как было обнаружено совпадение спектров возбуждения люмине-
сценции с ИК-излучением арсенида галлия. Практическое применение их в на-
стоящее время целиком связано с изготовлением светодиодов. Антистоксовские
люминофоры эффективно излучают только при высокой плотности возбуждения,
поэтому для концентрирования ИК-излучения применяют диоды очень малень-
кого размера. Поглощение редкоземельных люминофоров в ИК-области не-
велико, и значительная доля излучения проходит через слой люминофора без
поглощения. Поэтому подбирают оптимальную толщину слоя люминофора
и его гранулометрический состав таким образом, чтобы максимально использо-
вать ИК-излучение и избежать потерь на самопоглощение. Спектр поглощения
люминофоров в видимой области спектра полностью соответствует их спектру
излучения (см. рис. IV.27). Для увеличения степени использования ИК-излуче-
ния применяют органические связки с показателем преломления, промежуточ-
ным между полупроводником (3,5) и люминофором (1,4). Важно, чтобы в люми-
нофорном покрытии отсутствовали воздушные прослойки.
Антистоксовские люминофоры применяют также для визуализации ИК-излу-
чения лазеров, например с длиной волны 1,06 нм [105]. Предложена система
для визуализации излучения 10 нм, основанная на суммировании фотонов с раз-
ной энергией в системах, содержащих Sm2+ (сенсибилизатор) и Еп3+ (актива-
тор). В этом случае излучение в видимой области возникает под действием
двух квантов разной энергии (10 и 0,5 нм) [106]. Монокристаллы, содержащие
иттербий и эрбий, могут быть использованы для лазеров видимого излучения,
работающих при ИК-накачке. Так, система (BaY2F8- Yb, Er или Но) генерирует
в полосе 670 и 550 нм при 77 К при накачке в области 960 нм [107].
ЛИТЕРАТУРА
1. Фок М. В. Введение в кинетику люминесценции кристаллофосфоров,
М., «Наука», 1964. 283 с. — 2. Антонов-Романовский В. В. Кинетпка фото-
люминесценции кристаллофосфоров. М., «Наука», 1966. 323 с. — 3. К1а-
sens Н. A., Wise М. Е. «Nature», 1946, v. 158, р. 483—485. — 4. Prener J. S.,
Williams F. E. J. Electrochem. Soc., 1956, v. 103, p. 342—346. — 5. Вави-
лов С. И. О «теплом» п «холодном» свете (тепловое излучение и люминесценция).
М.—Л., Изд. АН СССР, 1949. 78 с.; Собрание сочинений. Т. 2. М., Изд.
АН СССР, 1952. 548 с. — 6. Фабрикант В. А. Усп. физ. наук, 1945, т. 27, № 2,
с. 8—10. — 7. Нилендер Р. А. Люминесцентные лампы и их применение. М.,
Госэнергоиздат, 1948. 98 с. — 8. Вознесенская 3. С., Скобелев В. М. Электри-
ческие источники света. М.—Л., Госэнергоиздат, 1957. 216 с. — 9. Федо-
ров В. В. Производство люминесцентных ламп. Изд. 2-е. М., «Энергия», 1969.
.224 с. — 10. Бутаева Ф. А., Фабрикант В. А. Изв. АН СССР. Сер. физ., 1949,
т. 13, с. 271 — 274.
11. Henderson S., Ranby Р. W. J. Electrochem. Soc., 1951, v. 98, № 8,
р. 932—934. — 12. McKeag А. Н., Ranby Р. W. Brit. Pat. 578192, 1942. —
13. McKeag A. H., Jenkins H. C., Ranby R. W. J. Electrochem. Soc., 1949,
v. 96, p. 1 — 12. — U.'Parodt J. A. Ibid., 1967, v. 114, p. 370-377. — 15. Wan-
maker W. L. Chem. Weekbl., 1971, v. 67, № 27, p. 14—17. — 16. Butler К. H.,
Bergin M. J., Hannaford V. M. B. J. Electrochem. Soc., 1950, v. 97, p. 117—
122. — 17. Mitsuishi T., Emoto M. Hitachi Bev., 1967, № 16, p. 230—233. —
18. Hoekstra A. H. Phil. Res. Repts, 1968, № 1 —2, — p. 1 —78. —19. Mo-
oney R. W., Ala M. А. Пат. США 3157603^ 1962. — 20. Hickok R. L. Пат. США
3378499, 1965.
103
21. Doherty M., Harrison W. Brit. J. Appl. Phys., 1955, № 4, p. 11 —15. —
22. Rabatin J., Gilloly G. J. Electrochem. Soc., 1964, v. Ill, № 5, p. 542—
546. — 23. Казанкин О. H., Зимогляд Л. С. «Светотехника», 1970, № 5, с. 23. —
24. Зимогляд Л. С., Казанкин О. Н. Там же, 1973, № 9, с. 16. — 25. But-
ler К. Н., Homer Н. Н. Ilium. Eng., 1960, р. 396—400. — 26. Mooney R. W., Con-
stock A. L. Пат. США 3099627, 1963. — 27. Marko vsky L. Ya, Kazankin O. N.„
Taushkanova L. B. In: Proceedings of the International Conference on Lumi-
nescence. V. 1. Budapest, 1966, p. 1235—1239. — 28. McKeag A. H. Англ,
пат. 927354, 1963. — 29. К азанкин О. Н., Ижевский М. Б., Александрова Р. А.
Авт. свид. 196217. Бюлл. лзобрет., 1967, № 11. — 30. Бутаева Ф. А. «Свето-
техника», 1959, № 7, с. 17—20.
31. Johoson Р. D. JOSA, 1961, V. 51, р. 1235—1238. — 32. Гуревич Э. М.
«Светотехника», 1968, № 6, с. 7—10. — 33. Казанкин О. Н., Белов М. П., Зимо-
гляд Л. С. — Изв. АН СССР, Сер. физ., 1969, т. 33, № 5, с. 908—910. — 34. Бу-
таева Ф. А., Гуревич Э. М. «Светотехника», 1969, № 1, с. 3—6. — 35. Бе-
лов М. П., Казанкин О. Н., Карасев В. С. Там же, 1973, № 5, с. 18. — 36. Скре-
блюков А. Е., Гришенков О. П. Там же, 1967, № 5, с. 7—10. — 37. Ерашо-
ва Э. М. Там же, 1970, № 1, с. 7—9; 1972, № 1, с. 16. — 38. Singleton J., Suc-
how L. J. Electrochem. Soc., 1963, v. 110, № 1, p. 36—41. — 39. Vrenken L. E.,
De Vette T. H., Van Der Wolf R. W. Ilium. Eng., 1964, v. 59, № 1, p. 59—66. —
40. Apple E. F., Aicher J. F. In: Proceedings of the International Conference
on Luminescence. V. 2. Budapest, 1966, p. 2013—2026.
41. Казанкин О. H., Зимогляд Л. С. «Светотехника», 1971, № 8, с. 14. —
42. Гугелъ Б. М., Шапочник М. С. Там же, 1959, № 4, с. 18—23. — 43. Гу-
гелъ Б. М. Люминофоры для электровакуумной промышленности. М., «Энергия»,
1967, 344 с. — 44. Гугелъ Б. М. «Светотехника», 1963, № 7, с. 9—13. —
45. Lowry Е. F. Ilium. Eng., 1948, v. 44, № 2, р. 98—102. — 46. Butler К. Н.
J. Electrochem. Soc., 1953, v. 100, p. 250—255. — 47. Koehnans H., Кох A.P .
Ibid. 1957, v. 104, p. 442—445. — 48. Шпгрихман P. А., Шойхет Д. H., Пекер-
ман Ф. M. Изв. АН СССР. Сер. физ., 1959, т. 23, с. 1387—1392; ЖПХ, 1961,
т. 34, с. 1912—1920. — 49. Горбачева Н. А., Гугелъ Б. М., Константинова-
Шлезингер М. А. Изв. АН СССР. Сер. физ., 1961, т. 25, с. 455—458. — 50. Ба-
бицкая Р. А., Марковский Л. Я. ЖПХ, 1962, т. 35, с. 1434—1441; 1963, т. 36,
с. 1186—1192.
51. Казанкин О. Н. Авт. свид. 186593. Бкил. пзобрет., 1966, № 19. —
52. Ropp R.C. Ilium. Eng., 1965, № 9, т. 530—535. — 53. Pallila F.C.
Electrochem. Technol., 1968, v. 6, № 1—2, p. 39—49. — 54. Aia M. J.
Electrochem. Soc., 1967, v. 114, p. 367—370. — 55. Bril A., Wanmaker W. L.
Ibid., 1964, v. Ill, № 12, p. 1363—1370. — 56. Pallila F. C., O'Reily В. E.
Ibid., 1968, v. 115, № 10, p. 1076—1080. — 57. Wanmaker W. L., ter VrugtJ. W.
Phil. Res. Repts, 1967, v. 22, p. 355—358. — 58. Mizuno H., Masuda M. Bull.
Chem. Soc. Japan, 1964, v. 37, p. 1239—1240. — 59. Wanmaker W. L., Bril A., ter
VrugtJ. W. Appl. Phys. Let., 1966, v. 8, № 10, p. 260—265. — 60. Toma S. L.,
Palumbo D. T. J. Electrochem. Soc., 1970, v. 117, № 2, p. 236—241.
61. Datta R. K. Ibid., 1967, v. 114, p. 1058—1060. — 62. Бабицкая P. A.,
Городина 3. Ф., Зытнер Г. Г. и др. Авт. свид. 312864. Бюлл. пзобрет.,
1971, № 26. — 63. Бабицкая Р. А., Городина 3. Ф., Зытнер Г. Г. и др. В кн.:
Материалы XIX совещания по люминесценции. Ч. 2. Рига, 1970, с. 16. —
64. Бабицкая Р. А., Городина 3. Ф., Зытнер Г. Г. и др. Химия и техноло-
гия люминофоров. Л., изд. ГИПХ, 1974, с. 3—10. — 65. Wanmaker W. L.,
RadielovicD. Electrochem. Technol., 1964, v. 2, p. 16—20. — 66. Бабицкая P. A.,
Городина 3. Ф., Марковский Л. Я. В кн.: Сборник рефератов научно-исследова-
тельских работ по химии и технологии люминофоров. Л., изд. ГИПХ, 1964,
с. 8. — 67. Froelich Н. С. Trans. Electrochem. Soc., 1947, v. 91, p. 241—245. —
68. Зелинский В. В., Тимофеева Т. В. ДАН СССР, 1949, т. 66, с. 187—189. —
69. Вайнберг Б. И., Пекерман Ф. М., Данилов В. П. ЖТФ, 1954, т. 24, с. 1707—
1710. — 70. Nagy R., Wallentine R. С., Lui С. К. J. Electrochem. Soc., 1949,
v. 96, р. 1—3.
104
71. Калиниченко И. И., Пекерман Ф. М., Трофимов А. К. ДАН СССР,
1951, т. 78, с. 887—890. — 72. Аленцев М. Н., Букштейн С. М., К алиничен-
цоИ.И. Изв. АН СССР. Сер. физ., 1951, т. 15, с. 824—828. — 73. Лазарев Д. Н.
Ультрафиолетовая радиация и ее применение. Л.—М., Госэнергоиздат, 1950.
120 с. — 74. Fonda G. Trans. Electrochem. Soc., 1949, v. 95, p. 304—309. —
75. Schulman J. H. In: Preparation and Characteristics of Solid Luminescent
Materials. Simposium Held at Cornell University 1946. New York, 1948, p. 407—
409. — 76. Merril J. B., Schulman J. H. JOSA, 1948, v. 38, p. 5—8. —
77. Froelich H. C. J. Electrochem. Soc., 1948, v. 93, p. 101—106. — 78. Бун-
делъ А. А., Пантелеева E. П. «Труды ГИПХ», 1960, вып. 43, с. И—21. —
79. Колпакова А. А., Марковский Л. Я. В кн.: Химия и технология люмино-
форов. Вып. 60. Л., «Химия», 1968, с. 46—53. — 80. Ranby Р. W., Hender-
son S. Т. J. Electrochem. Soc., 1955, v. 102, т. 631—635.
81. McKeag А. Н., Steward В. G. Brit. J. Appl. Phys., 1954, Suppl., № 4,
p. 526—528. — 82. McKeag A. H. J. Electrochem. Soc., 1958, v. 105, p. 78—81.—
83. Колпакова А. А., Марковский Л. Я., Пекерман Ф. М. В кн..‘ Сборник рефе-
ратов научно-исследовательских работ по химии и технологии люминофоров
за 1960 г. Л., изд. ГИПХ, 1962, с. 6 — 7. — 84. Бунделъ А. А., Пантелеева Е. П.
В кн.: Сборник рефератов научно-исследовательских работ по химии и техноло-
гии люминофоров за 1958 г. Л., изд. ГИПХ, 1959, с. 4. — 85. Klasens Н. А.,
Hoekstra А. И., Сох А. Р. J. Electrochem. Soc., 1957, v. 104, р. 93—100. —
86. Le verenz H. Пат. США 2066044, 1936. — 87. Patten S., Williams F. JOSA,
1949, v. 39, p. 702—710. — 88. Thorington L. Ibid., 1950, v. 40, t. 579—
582. — 89. Штрихман P. A., Шойхет Д. H., Пекерман Ф. M. В кн.: Сборник
рефератов научно-исследовательских работ по химии и технологии люминофоров
за 1958 г. Л., изд. ГИПХ, 1959, с. 18. — 90. Klasens Н. A. Phil. Res. Repts,
1954, v. 9, р. 377—380.
91. Фридман С. А., Черепнев А. А. Светящиеся составы постоянного и вре-
менного действия. М., Изд. АН СССР, 1945. 142 с. — 92. Halsted R. Е., Ap-
ple Е. F., Prener J. S. Phys. Rev. Lett., 1959, v. 2, p. 420. — 93. Potter R. M.,
Aven M. Bull. Am. Phys. Soc., 1959, v. 4, p. 227. — 94. Broser J., Broser-War-
minsky R. In: Luminescence of Organic and Inorganic Materials. Proceedings
of International Conference Held in New York in 1962. New York, John wiley
and Sons, 1962, p. 402—407. — 95. Bloembergen N. Phys. Rev. Lett., 1959,
V. 2, p. 84. — 96. Овсянкин В. В., Феофилов П. П. ЖЭТФ (письма), 1966, т. 4,
С. 317—318. — 97. Auzel F. Compt. rend., 1966, v. 262, p. 1016—1019; v. 263,
p. 819—821. — 98. Galgtnaitis S. V., Fenner G. E. In: Proceedings of the 2nd
International Conference on Gallium Arsenide. (Dallas, 1968). Inst. Phys, and
Phys. Soc. Conf., 1968, Ser. 7, p. 131. — 99. Озелъ Ф. ТИИЭР, 1973, т. 61, № 6,
с. 87—106. — 100. Овсянкин В. В. Опт. и спектр., 1970, т. 28, с. 206—208.
101. Parker S. G., Johnson R. Е. J. Electrochem. Soc., 1972, v. 119, p. 610—
614. — 102. Yocom P. N., Wittke J. P. W., Ladany J., Met. Trans., 1971, v. 2,
p. 763—767. — 103. Matsubara T. Mater. Res. Bull., 1972, v. 7, № 9, p. 963—
970. — 104. Rich T. C., Pinnow D. A. J. Appl. Phys., 1972, v. 43, p. 2357—
2365. — 105. Seki Y., Furukawa Y. Japan J. Appl. Phys., 1971, v. 10, p. 1294. —
106. Wright J. C., Fond F. K., Miller M. M. J. Appl. Phys., 1971, v. 42, № 10,
p. 3806—3811. — 107. Johnson L. F., Guggenheim H. J. Appl. Phys. Lett., 1971,
v. 19, № 2, p. 44—47.
ГЛАВА V
КАТОДОЛЮМИНОФОРЫ
V.1. ОБЩИЕ СВЕДЕНИЯ
По масштабам мирового производства катодолюминофоры занимают второе
место после ламповых люминофоров. Наибольшее значение среди них имеют
люминофоры, применяемые в кинескопах для черно-белого и цветного
телевидения. Кроме того, большое военное (и промышленное) значение имеют
люминофоры, предназначенные для радиолокационных установок
(люминофоры с длительным послесвечением), и малоинерционные люминофоры
для экранов электронно-оптических преобразователей. Некото-
рые катодолюминофоры используют в измерительной технике — в различного
рода электронно-лучевых трубках.
Такое большое разнообразие областей применения катодолюминофоров
естественно привело к тому, что их основой служат различные химические соеди-
нения. Однако наибольшее значение имеют сульфиды, которые используют
в кинескопах черно-белого и цветного телевидения, а также в радиолокации.
Далее идут силикатные люминофоры, применяемые в осциллографических
трубках-и проекционных кинескопах; окисные люминофоры с коротким после-
свечением, нашедшие применение в трубках с бегущим лучом, и наконец, фто-
ридные люминофоры с большой длительностью послесвечения и используемые
в радиолокационных трубках. В последнее время все более широкое применение
находят редкоземельные катодолюминофоры, в первую очередь YVO4-Eu,
Y2O3-Eu и Y2O2-S-Eu, которые служат в качестве красной компоненты при
изготовлении цветных электронно-лучевых трубок. Длительное время для этой
цели использовали фосфат цинка, активированный Мп.
На рис. V.1 изображена электронно-лучевая трубка. Трубка откачана
до остаточного давления порядка 0,133 Па (10'6 мм рт. ст.). Основные элементы
трубки: электронный прожектор, создающий пучок электронов, фокусирующие
и отклоняющие системы и люминесцирующий экран. Источником электронов
служит подогреваемый изнутри никелевый цилиндр (катод), покрытый слоем
термоэмитирующего вещества. Испускаемые с катода электроны при помощи
системы ускоряющих и управляющих электродов * приобретают необходимую
скорость и стягиваются в узкий пучок, направляемый на экран, покрытый слоем
люминофора определенной (оптимальной) толщины. С целью повышения яркости
и контрастности изображения, а также для уменьшения влияния вторичной
эмиссии, экраны часто металлизируют. В трубках, дающих цветное изображение
(рис. V.2), применяют мозаичное люминофорное покрытие в виде набора мель-
чайших, расположенных по углам треугольника, точек (из люминофоров с крас-
ным, синим и зеленым свечением). Эти разноцветные точки раздельно возбу-
ждаются электронным пучком (одним или тремя, в зависимости от конструкции
трубки), проходящим сначала через теневую маску — металлическую пластинку
с круглыми отверстиями, число которых равно числу элементов цветного изобра-
жения. Диаметр отверстшт маски составляет ~ 0,3 мм, таков же размер цветных
люминофорных точек; общее число точек на экране превышает миллион. Ре-
* На последние подают положительное относительно катода напряжение
(например, несколько тысяч вольт на втором аноде).
106
зультирующий цвет изображения определяется соотношением яркости отдельных
светящихся точек.
Возбуждение люминофоров электронным пучком — сложный процесс, в ко-
тором различают следующие стадии [1, ч. 1, с. 42—94; 2. 3, 4, р. 172—200;
5, р. 427-463; 6]:
1’ Проникновение электронов в кристаллическую решетку люминофора
и образование в ней в результате неупругих столкновений каскада в т о -
Рис. V.I. Принципиальная схема устройства электронно-лучевой трубки:
1 — нить накала; 2 — катод; 3 — управляющий электрод; 4 — первый анод; 5 — ускоря-
ющий электрод; 6 — второй анод; 7 — проводящее покрытие; 8 — катушки вертикального
отклонения; 9 — катушки горизонтального отклонения; 10 — электронный пучок; 11 —слой
люминесцирующего вещества (экран трубки).
Рис. V.2. Принципиальная схема устройства электронно-лучевой трубки с трех-
цветным мозаичным экраном:
1 — три электронных прожектора с электростатической фокусировкой; 2 — общая откло-
няющая система; 3 — теневая маска; 4—в — элементы «синего», «зеленого» и «красного» люми-
нофоров (сильно увеличены).
р и ч н ы х электронов, часть из которых теряется в результате вторичной
эмиссии;
2. Возбуждение электронами центров люминесценции;
3. Выделение поглощенной энергии в виде излучательных (люминесценция)
Или безизлучательных (потеря энергии на нагревание люминофора) переходов;
соотношение между вероятностями этих переходов с учетом потери первичных
И вторичных электронов в результате вторичной эмиссии характеризует эффек-
тивность люминофора; по оценке Левшина [2, 3], потери энергии на отражение
И рассеяние электронов составляют не более 5%.
107
Отметим следующие особенности катодного возбуждения:
электроны проникают в люминофоры на небольшую глубину (1—5 мкм
при энергии 10—40 кэВ), что приводит к увеличению в люминесценции роли
поверхностных слоев люминофора и повышению плотности воз-
буждения; это, в свою очередь, влияет на люминесцентные характеристики
люминофоров, а также способствует заметному их разогреванию;
люминофор поглощает энергию катодных лучей неселективно,
причем поглощение происходит не на. центрах люминесценции, а в кристал-
лической решетке в целом.
V.2. ЭКСПЛУАТАЦИОННЫЕ ХАРАКТЕРИСТИКИ
Изготовление экранов электронно-лучевых трубок сопряжено с рядом
технологических операций (приготовление люминофорной суспензии, отжиг
экрана при 400—450° для удаления связующего, отжиг в вакууме примерно
при той же температуре), которые существенно влияют на их люминесцентные
свойства [1, ч. 2, с. 536—594; 7].
Изменение свойств люминофора за время проведения перечисленных опе-
раций зависит от состава основы, рода активатора и условий приготов^ния
люминофоров. При оценке стабильности катодолюминофоров учи-
тывают:
химическую стойкость по отношению к действию различных хими-
ческих агентов, используемых в технологических операциях; в частности, окис-
лителей при отжиге в окислительной атмосфере;
термическую стойкость *, т. е. стойкость к температурной обработке
в вакууме;
радиационную стойкость, т. е. сопротивляемость к воздействию
электронного пучка;
примесную стойкость, т. е. стойкость к воздействию микропримесей
различных элементов, тем или иным путем попадающих в люминофорный экран
и влияющих после термообработки на его люминесцентные характеристики.
Имеющиеся в литературе экспериментальные данные, характеризующие
химическую, термическую и примесную стойкость люминофоров, весьма огра-
ничены, а порой и недостаточно надежны.
Химическая стойкость
Наиболее изучена химическая стойкость сульфидных люминофоров [7, 8, 9,
с. 117—124; 10, 11]. Показано [8], что отжиг ZnS-Ag на воздухе при 350—400°
не изменяет ни яркости свечения, ни спектральных характеристик люминофора.
Не происходит также никакого смещения спектров излучения у безактиваторных
цинк-кадмий сульфидных люминофоров. Если последние активированы сереб-
ром, то отжиг при тех же условиях приводит к снижению яркости свечения
примерно па 10—20%, причем снижение тем больше, чем выше содержание ZnS
в основе. Отжиг на воздухе люминофоров ZnS-CdS-Ag, кроме того, смещает
спектр излучения в коротковолновую область примерно на 5 нм, причем чем
выше содержание в люминофоре CdS, тем при более низкой температуре это
происходит. Смещение спектра излучения у окисленных ZnS-CdS-Ag люмино-
форов наблюдали также Де Бур и Бросс (табл. V.1) [10].
Данные [11] термогравиметрического изучения химической стойкости
сульфидных люминофоров позволяют сделать выводы, что она:
зависит от состава основы, возрастая с увеличением содержания CdS;
повышается при нанесении на поверхность зерен люминофора защитных
слоев из различных неорганических соединений;
снижается в результате гомогенизации в водном растворе поливинилового
спирта, используемого при нанесении экранов кинескопов; при этом особенно
большое влияние оказывает наличие в суспензии бпхромата аммония, что, веро-
ятно, связано с окислительным действием бихромат-иона.
* Иногда под термином термическая стойкое ть исдразулк 11 к т такие и хи
мическую стойкость.
108
Таблица V.1
Изменение оптических характеристик люминофора
после отжига
Оптические характеристики Люминофор
неото- жженный отожженный на воздухе при 440°
Координаты цветности: X ............ У • • ^max, НМ Край поглощения, нм ... . Световой эквивалент, лм/Вт Энергетический выход .... 0,230 0,550 515 400 425 0,18 0,260 0,645 522 (несим- метричная) 490 495 0,125
Термическая стойкость
Изучению изменения свойств сульфидных люминофоров в результате про-
каливания их в вакууме посвящены работы [7—9, с. 117—124; 12—15]. Впервые
термическая стойкость люминофоров ZnS • Ag и ZnS - CdS • Ag с различным содер-
жанием CdS была исследована Коровкиной и Бахманом с сотрудниками [8].
Эти авторы наблюдали уменьшение яркости катодолюминесценции при вакуум-
ной обработке указанных люминофоров, а также изменение их спектров свече-
ния, которые они связывали с перераспределением интенсивности свечения
отдельных полос. По данным, приводимым Гугелем [9, с. 117—124], отжиг
в вакууме при 300—500° ухудшает люминесцентные свойства люминофора
ZnS-Ag пропорционально удельной поверхности порошков люминофоров и за-
висит от температуры и длительности вакуумной обработки. Предварительный
отжиг слоя люминофора на воздухе при температуре выше 350° резко ухудшает
термостойкость. Автор связывает это с тем, что наличие фазы окиси цинка,
образующейся при окислении, способствует образованию вакансий цинка в ре-
шетке люминофора. По данным работ [12—15], лучшую термостойкость имеют
сульфидные люминофоры, активированные Си.
Радиационная стойкость
В процессе эксплуатации электронно-лучевых трубок люминофорный слой
подвергается разрушению. При этом снижается яркость свечения и даже наблю-
дается заметное потемнение экранов [1, с. 248—261; 16, с. 204; 17—22].
Интенсивность этого процесса зависит от длительности эксплуатации тру-
бок, величины приложенного напряжения, а также от плотности тока электрон-
ного пучка, увеличиваясь с повышением последней. Следует отметить, что при-
рода процессов, происходящих при низких и высоких напряжениях, различи а.
В частности, выгорание, происходящее при низком напряжении, обра-
тимо, а при высоком — необратимо.
Повышение напряжения приводит к уменьшению выгорания, что связано
с уменьшением плотности возбуждения вследствие увеличения глубины проник-
новения электронов. Имеющиеся экспериментальные данные показывают, что
при выгорании катодолюминофоров имеют значение:
физическое разрушение — иод действием пучка электронов;
физическое и химическое разрушение — под действием пучка положитель-
ных ионов, образующихся в электронно-лучевой трубке.
Скорость протекания обоих процессов в сильной степени зависит от физико-
химической природы основы люминофора, в частности от прочности соотве;-
109
ствующих кристаллических решеток. Наибольшую стойкость имеют люмино-
форы окисного типа, например: на основе окиси алюминия, оксисульфидные
и силикатные, применяемые для экранов проекционных кинескопов.
Очень низка стойкость фторидных люминофоров (например, ZnF2-Mn,
MgF2-Mn), что ограничивает возможности их широкого применения.
Сульфидные люминофоры, применяемые для кинескопов черно-белого
телевидения, отличаются сравнительно высокой стабильностью к действию
электронного и ионного пучков и за 10 000 ч эксплуатации снижают яркости
свечения не более чем на 30—40%.
Теоретическое объяснение процессов выгорания катодолюминофоров почти
отсутствует. Однако предполагается, что снижение яркости свечения связано
с появлением дислокаций в решетке основы, а потемнение экранов свя-
зано с выделением металла (падающие на люминофор электроны фактически
действуют как восстановители). Разрушение фторидных люминофоров связы-
вается с образованием F-центров.
Т а б л и ц a V.2
Характеристики выгорания люминофоров при возбуждении электронами
(7—10 кВ)
Люминофор Количество электричества, приводящее к снижению 1 в 2 раза, Кл 1 Константа выгорания с-10* 1’, см»
воздействие электронов воздействие ионов
ZnS-CdS-Си 11,9 0,140
Смесь ZnS • CdS • Ag и ZnS • Ag* 4,5 0,370 0,43 • 107
ZnS • Ag ]** 9,9 0,170 —
ZnS • CdS • Cu J 10,0 0,167 ,—
ZnS • Ag 20,0 0,083 0,35 • 107
Zn2S iO4 • Mn 104,0 0,016 0,02 • 107
CaO-MgO • SiO2-Ti 55,5 0,030 —
ZnO • BeO • SiO2 • Mn 83,3 0,020 0,02 • 107
(Zn, Mg, Cd)SiO3-Mn 16,6 0,100 —
ZnO • Zn 33,5 0,050 0,02 • 107
CaWO4 16,6 0,100 0,004 • 107
* Для экранов телевизионных кинескопов.
** Для двухслойных экранов (см. стр. 121).
Некоторые данные по стойкости различных катодолюминофоров приведены
в табл. V.2 [16, с. 204; 17]. За критерий стабильности катодолюминофоров
в табл. V.2 принята константа с в соотношении
/=———
14-су
где 10 — начальная интенсивность свечения;
I — интенсивность свечения «состаренного» экрана;
с — параметр выгорания, см2;
А' — общее число электронов, попавших на 1 сма поверхности экрана.
По данным ряда авторов, это соотношение наилучшим образом описывает
процесс выгорания люминофоров. Однако, по данным Князатова и Шерст-
нева [18], приведенное выше выражение не объясняет экспериментальных
результатов, полученных ими при исследовании свойств экранов на основе
«белой» телевизионной смеси. Авторы считают, что при старении экранов играют
роль как объемные, так и поверхностные процессы. При этом значение последних
столь велико, что разрушение люминофорного слоя больше зависит от сорбции
НО
и дальнейшей диффузии остаточных газов с поверхности экрана в глубь решетки,
чем от количества электронов, попавших на единицу площади экрана.
Из имеющихся экспериментальных данных следует, что на выгорание люми-
нофоров влияют, кроме структуры основы (гексагональный ZnS более стабилен,
чем кубический), также гранулометрический состав (мелкозернистые люмино-
форы чувствительнее к выгоранию) и условия возбуждения (при ионном выго-
рании снижение яркости свечения в 107 раз больше, чем при электронном).
Особенно губительно повышение температуры экрана и ухудшение вакуума.
Возрастание напряжения в пределах от 3 до 5 кВ не увеличивает ни электрон-
ного, ни ионного выгорания.
Примесная стойкость
Данные по экспериментальному исследованию примесной стойкости не-
многочисленны [7, 9, 23, 24]. Примесная стойкость сульфидных катодолюмино-
форов связана с диффузией на стадии отжига экранов ионов тяжелых металлов
(главным образом, меди, железа и никеля) в решетку основы и конкуренцией
полос, появляющихся при попадании микроколичеств примесных ионов с основ-
ной полосой излучения. Наряду с этим возможно и гасящее действие примесей.
Они могут попадать в люминофор в процессе изготовления экранов из воды
и растворов, применяемых при этом, а также вместе с пылью.
При рассмотрении вопросов примесной стойкости необходимо учитывать,
что сульфидные люминофоры активно сорбируют примеси из растворов, и по-
этому примесная стойкость зависит как от удельной поверхности порошка люми-
нофора, увеличиваясь с ростом последней, так и от длительности контакта с рас-
твором. На скорость диффузии примеси в люминофор влияет температура отжига,
состояние поверхности зерна люминофора, наличие в ней дефектов, а также
специально нанесенных защитных поверхностных пленок. Диффузия ионов
Тяжелых металлов зависит и от степени сформированности решетки основы.
Пути повышения стабильности
Термогравиметрическими исследованиями показано, что температура начала
окисления люминофоров на сульфидной основе зависит от сформированности
решетки порошкообразных люминофоров и от степени дисперсности порошков.
Таким образом, температурный режим синтеза люминофора может существенно
влиять на его эксплуатационную стабильность. Можно предположить, что изве-
стное влияние на этот параметр должна оказывать химическая природа тех
минерализующих или модифицирующих добавок, которые вводятся в шихту
при синтезе, так же как и чистота используемого сырья. Повысить эксплуата-
ционную стабильность люминофоров можно за счет дополнительной обработки
их поверхности с целью создания защитных поверхностных пленок различного
типа, например пирофосфата цинка, двуокиси титана и кремния, окиси алюминия
или силикатов металлов II группы периодической системы [7].
Известно также благоприятное влияние поверхностных фосфатных и фос-
фитных пленок на примесную стойкость, а именно, чувствительность к загряз-
нениям Си и Ni. По данным японских авторов [25], повышение примесной стой-
кости по отношению к Ni, Fe, Со, РЪ, Си и другим ионам тяжелых металлов
может быть достигнуто дополнительной термической обработкой люминофора
при температуре ниже 600° с плавнями (MgCl2, СаС12, NH4C1) и активаторами.
Рекомендуется [26] для повышения устойчивости к образованию пятен выгора-
ния в электронно-лучевых трубках вводить в шихту при синтезе люминофоров
соединения Si, Ge, Sn, As, Sb, Bi или P.
Большое внимание в патентной литературе [27] уделяется также извлече-
нию из готового люминофора избыточного количества солей, вводимых в каче-
стве активаторов. В результате соответствующих обработок достигается не
только повышение яркости свечения люминофоров, но и повышение стабиль-
ности люминофора к действию загрязнений тяжелыми металлами.
111
V.3. ЛЮМИНОФОРЫ ДЛЯ ЧЕРНО-БЕЛОГО ТЕЛЕВИДЕНИЯ
Люминофоры для черно-белого телевидения можно разделить на исполь-
зуемые в кинескопах прямого видения и применяемые в проек-
ционных кинескопах.
В первом случае годятся сульфидные люминофоры, обеспечивающие доста-
точную яркость свечения, стабильность свечения экранов во времени (до не-
скольких тысяч часов) и достаточно короткое послесвечение [28]. В проекцион-
ных трубках (в них плотность тока электронного пучка достигает 100 мкА/см2)
применяют люминофоры на оксисульфидной [29] или силикатной основе [28].
Сульфидные люминофоры
для кинескопов прямого видения
Экраны кинескопов прямого видения состоят из смеси люминофоров ZnS •
• Ag [голубое свечение (БЗ-С)] и ZnS-CdS-Ag [желтое свечение (БЗ-Ж)].
Применявшиеся ранее рецептуры однокомпонентных сульфидных люминофоров,
400 . 500 600 700
Рис. V.3. Спектры излучения
люминофоров БЗ-С (7) и БЗ-Ж
(2).
sl'Hv БЗ-Ж.
например ZnS-CdS-Ag-Auи ZnS-CdS-P-As
оказались малоэффективными и не нашли
практического применения. Недавно разра-
ботан и прошел производственные испытания
однокомпонентный «белый» оксисульфидный
люминофор [29] (см. также стр. 121).
К катодолюминофорам, входящим в со-
став «белых» смесей, предъявляются вполне
определенные требования по гранулометри-
ческому составу (табл. V.3) и цветности све-
чения (табл. V.4). На рис. V.3 приведены
спектры излучения люминофоров БЗ-С и
Состав шихты для приготовления люми-
нофоров БЗ-С и БЗ-Ж и режим прокали-
вания их приведены ниже:
БЗ-С (голубая
компонента)
Состав шихты, кг:
ZnS.................................... 30
CdS.................................. —
NaCl ............................... 0,6
MgCl2............................... 0,3
Раствор AgNO3 (0,1 н), мл .... 150
Температура прокаливания, °C . . 900—1000
Длительность, 4j........................ 2
БЗ-Ж (желтая
компонента)
16,5
19,5
0,18
0,54
108
I 700—720
II 760-800
I 2
II 1
Шихту люминофоров до загрузки в тигли тщательно сушат на алюминиевых
противнях до пыления, после чего просеивают через сито. Прокаливание произ-
водят в закрытых тиглях, которые по окончании процесса извлекают из печи.
После просмотра и очистки люминофоров под лампой порошки подвергают
специальной обработке с целью создания на частицах поверхностных фосфатных
пленок и пленки двуокиси кремния. Обработку производят разбавленными
(0,5—0,1%) растворами соответствующих солей для придания порошкам люмино-
фора адгезионных свойств, необходимых при нанесении люминофоров на экраны
методом осаждения из разбавленных растворов силиката калия. После про-
мывки и сушки при 150° люминофоры просеивают через сито и смешивают
в определенных пропорциях в специальных смесителях.
В СССР применяют «белые» смеси люминофоров КТБ-1а, КТБ-16 и КТБ-2,
координаты цветности которых расположены в пределах допусков, показанных
на рис. V.4. Люминофоры КТБ-1 и КТБ-2 отличаются, главным образом, по
гранулометрическому составу (КТБ-2 является более крупнозернистым, чем
112
Т а б л и ц a V.3
Требования к гранулометрическому
составу для люминофоров БЗ-С
и БЗ-Ж
Таблица V.4
Требования к координатам цветности
для люминофоров БЗ-С и БЗ-Ж
Координаты цветности БЗ-С БЗ-Ж
X 0,145-0,165 0,440-0.470
У 0,105-0,125 0,500—0,530
строгие тре-
Линейные раз-
меры частиц
люминофоров,
мкм
Менее 4,7 . .
От 4,7 до 6,6
От 6,6 до 16.
Более 16 . .
Содержание частиц, %
БЗ-С БЗ-Ж
>ю >20
>25 >30
>50 >40
>14 >10
Таблица V.5
Весовые соотношения между
люминофорами БЗ-С и БЗ-Ж,
входящими в состав «белых» смесей
КТБ-1 и КТБ-2
Фракция с раз- мером частиц, мкм КТБ-1 КТБ-2
Более 16 . . . >2,5 >2
От 16 до 6,6 . >1,5 >1,5
Менее 6,6 . . . — >1
КТБ-1). Кроме того, к люминофору КТБ-2 предъявляются менее
бования по равномерности цветности свечения в центре экрана кинескопа и по
его краям. Весовые соотношения люми-
нофоров БЗ-С и БЗ-Ж в различных фрак-
циях, необходимые для изготовления
смесей КТБ-1 и КТБ-2, приведены
в табл. V.5.
Люминофоры КТБ-1 и КТБ-2 удов-
летворяют всем основным требованиям,
связанным с их использованием в эк-
ранах кинескопов; им присущи:
достаточно высокая яркость свече-
ния (не менее 170 кд м~2 в экранах ки-
нескопов);
достаточно короткое время после-
свечения (отношение яркости свечения
люминофора через 40 мкс после прекра-
щения возбуждения при плотности тока
0,2 мкА-см-2 к начальной яркости све-
чения — 0,6—0,8); поэтому быстро сме-
няющиеся изображения на телевизион-
ном экране не накладываются друг
на друга;
строго регламентированный гранулометрический состав.
Последнее обстоятельство имеет существенное значение
_____________ _____________ __ v . для обеспечения
равномерного свечения, хорошей разрешающей способности экранов кинескопов
и адгезии. Следует отметить, что некото-
рые иностранные фирмы характеризуют
адгезию порошка к стеклянной поверх-
ности
Так,
ввела
Рис. V.4. Требования, предъяв-
: , t КТБ-1 И
КТБ-2 по цветности свечения.
8 3 аказ 44
ляемые к люминофорам
экрана специальным параметром,
американская фирма «Сильвания»
показатель «мокрой прочности».
100
95Н
90 ----1---1----1---1----1---1----
0 500 1000 1500
Время эксплуатации , ч
Рис. V.5. Изменение яркости свечения
экрана Р-4 со временем.
113
Он определяется по размеру диаметра пятна, которое создается в иевысушенном
и стандартным образом приготовленном экране из данного люминофора, если
направлять на него струю воды определенного диаметра при определенном
давлении.
В качестве важных технологических параметров люминофоров КТБ-1
и КТБ-2 введены такие показатели как:
способность к нанесению;
сухая прочность;
термическая и примесная стойкость.
Стабильность свечения сульфидных люминофоров для черно-белого телеви-
дения в процессе эксплуатации может быть охарактеризована кривой (рис. V.5)
для экрана Р-4, представляющего собой смесь ZnSreKc - Ag и (l,3ZnS Сй8)гекс -Ag.
Ход кривой показывает, что через 1750 ч уменьшение яркости свечения не пре-
вышает 7%.
Силикатные люминофоры для проекционных кинескопов
и осциллографических трубок
В связи с тем, что в проекционных трубках плотность тока электронного
пучка значительно больше, чем в кинескопах прямого видения, к люминофорам,
используемым в них, предъявляются требования высокой устойчивости при бом-
бардировке электронным пучком,
а также отсутствия насыщения
при увеличении плотности тока.
Этим требованиям удовлетворяют
люминофоры на силикатной и
оксисульфидной основах.
Роль желтой компоненты «бе-
лого» экрана для проекционных
трубок выполняет люминофор
К-57 (Zn, Be)2SiO4-Мп, содержа-
550' щий—13 мол. % ВеО. Активатор
Мп (МпСО3) вводят в коли-
честве 1,3—1,4%. Люминофор
готовят из смеси люминофорно-
чистых окислов, предварительно
размолотых в шаровой мельнице
и просеянных через сито. Для
большей полноты прохождения
реакции смесь двукратно прокали-
вают при 1250° с промежуточным
измельчением.
Синей компонентой является
люминофор К-58 (Са, Mg) (SiO3)2 •
• Ti. Активатор вводят в виде TiO2,
в количестве, соответствующем
100
50
0
X, нм
Рис. V.6. Спектральное распределение
энергии излучения некоторых катодолю-
минофоров:
Кривая
Марка
Состав
1
2, з
4
К-58
К-35, К-36
К-57
(Са, Mg)(SiO3)2 • Ti
Zn2SiO4-Mn
(Zn, Be)2SiO4-Mn
1,2% Ti. Для ускорения реакции целесообразно применять хлоридно-сульфат-
ные плавни или гель кремневой кислоты. Последний получают путем гидро-
лиза кремнеэтилового эфира в концентрированной соляной кислоте. Люмино-
фор прокаливают в открытых кварцевых кюветах при —1250° не менее 3 ч.
Полученный спек измельчают в шаровой мельнице.
Технические показатели люминофоров К-57 и К-58 представлены в табл. V.6,
а кривые спектрального распределения энергии излучения — на рис. V.6.
У алюминированных экранов, изготовленных с использованием смеси этих люми-
нофоров, светоотдача составляет 3,5 кд/Вт (при V — 6 кВ и/' = 10 мкА см-2)
и через 1000 ч работы снижается не более чем на 5%. Оба люминофора имеют
экспоненциальное затухание: для К-57 т = 10-2 с, для К-58 т = 10" 3 с.
В экранах для осциллографических трубок применяют люминофор К-35
на основе виллимита (Zn2SiO4), активированного Мп; он имеет максимум на
кривой спектрального распределения энергии при 525 ± 5 нм (рис. V.6). Этот
люминофор получают прокаливанием в закрытых кварцевых тиглях в течение
114
Таблица V.6
Технические характеристики силикатных люминофоров
для проекционных кинескопов черно-белого телевидения
Люминофор Цвет свечения Координаты цветности Средний раз- мер зерен, мкм
марка состав X У
К-57 К-58 (Zn, Be)2SiO4-Mn (Са, Mg)(SiO3)2 • Ti Желтый Голубой 0,489 ± 0,015 0,171 ±0,010 0,520 ±0,015 0,147 ±0.010 3-5 4-7
2 ч при 1200—1240° тщательно перемолотой в шаровой мельнице смеси 5 масс. ч.
люминофорно-чистой ZnO с 2,9 масс. ч. SiO2 (в расчете на безводную) и с 1%
активатора (в виде МпСО3). Применение более высокой температуры недопустимо
вследствие возможного при этом спекания люминофора. В случае недостаточно
полного протекания реакции применяют повторное прокаливание.
V.4. ЛЮМИНОФОРЫ ДЛЯ ЦВЕТНОГО ТЕЛЕВИДЕНИЯ
Для наилучшей передачи цвета координаты цветности люминофоров для
цветного телевидения должны быть возможно более близки к углам цветового
треугольника (см. рис. IV.5, стр. 77). Особенно большое значение имеет качество
«красного» люминофора: последний должен иметь при хорошем выходе люминес-
ценции возможно более узкую полосу излучения с резким обрывом в сторону
длинных волн вблизи А, — 610 нм.
В современных промышленных кинескопах для цветного телевидения
в качестве синей и зеленой компонент используют люминофоры на сульфидной
основе, а в качестве красной — люминофоры на основе соединений иттрия,
активированные Ен. Технические характеристики наиболее эффективных катодо-
люминофоров для цветного телевидения, выпускаемых в СССР, приведены
в табл. V.7 и на рис. V.7, а в табл. V.8 дано сравнение некоторых характеристик
люминофоров марок К-74 и К-75 с аналогичными образцами зарубежных фирм.
Ниже приведены состав шихты и условия прокаливания люминофоров К-74
и К-75:
К-74 (зеленая компонента) К-7 5 (синяя компонента)
Состав шихты, %:
ZnS 62 100
CdS 38 —
Ag 8 • 10-3 2•10-2
NaCl 1 1
MgCl2 — 2
S — 4
Температура прокаливания, °C . I 730—750 I 750—770
II 830—850 II 850-870
Длительность, ч . I 1 I 1.5
II 1 II 1,5
После прокаливания и очистки под лампой порошки люминофоров подвер-
гают специальной обработке с целью нанесения на их поверхность защитной
поверхностной пленки, состоящей из пирофосфата цинка, наносимого методом
осаждения из раствора. Помимо пирофосфата цинка могут быть использованы
и другие вещества. В зависимости от обработки поверхности люминофоров,
которая может быть «алюминатной», силикатной или цинксиликатной, разли-
чают модификации люминофоров К-74а, К-74с, К-74ц, К-75а и К-75ц. В табл. V.9
8* 115
Таблица V.7
Технические характеристики люминофоров для цветного телевидения
Марка Химический состав Цвет свечения Координаты цветности Светоотдача в алюминиро- ванном экране, кд/Вт
X У
К-75 ZnS • Ag Синий 0,152 ±0,010 0,050 ± 0,010 2
К-74 ZnS-CdS-Ag Зеленый 0,260 ± 0,020 0,575 ± 0,025 12
К-83 ZnS - CdS • Cu • Al » 0,300 ± 0,020 0,595
К-73 YVO4 • Eu Красный 0,670 ± 0,010 0,330 ± 0,010 2,4—2,5
К-77 Y2O3•Eu » 0,650 ±0,010 0,350 ± 0,010 4,0
К-78 Y2O2S • Eu • Tb » 0,660 ± 0,010 0,340 ± 0,010 4,0-4,4
Таблица V.8
Сравнение люминофоров марок К-74 и К-75
с зарубежными образцами
Люминофор
Относитель- ная яркость свечения, % Координаты цветности
X У
Люминофоры с синим цветом свечения
К-75 100 0,154 0,045 12
Японский; фирма «Киокко» 92 0,156 0,046 13
Американский; фирма «Сильвания» 96 0,153 0,044 19
Английский; фирма «Дер- би» 84 0,155 0,039 36
Люминофоры с зеленым цветом свечения
К-74 100 0,260 0,570 20
Японский; фирма «Киокко» 107 0,300 0,590 23
Американский; фирма «Сильвания» 94 0,263 0,565 43
Английский; фирма «'Дер- би» 84 0,216 0,543 40
Рис. V.7. Спектральное рас-
пределение энергии излучения
некоторых катодолюминофоров
для цветного телевидения:
Кривая Марка Состав
1 К-75 ZnS-Ag
2 К-74 ZnS-CdS-Ag
3 К-83 ZnS-CdS-Cu-Al
116
Таблица V.9
Влияние состава поверхностных пленок на характеристики катодолюминофоров
Люминофор Пленки Снижение яркости свечения при от- жиге, % Температура начала окисления, °C
на воз- духе в вакууме
Исходный (зеле- —. 7* 10 450
ный)
К-74 Zn2p2C7 11 11 470
К-74с Zn2?2О74- K2S1O3 8 9 520
К-74а AI2O3 7 8 500
К-74ц ZnSiO3 5 5 540
Исходный (синий) — 10 — 430
К-75 3 -— 490
К-75с Zn2P 2O 7—|— K2S iO3 5 — 510
К-75а A12O3 6 — 515
К-75ц ZnS iO3 6 — 545
* Экран желтеет.
приведены данные, показывающие влияние различных защитных пленок на
характеристики катодолюминофоров. Практика использования люминофоров
в кинескопах для цветного телевидения показала, что для люминофора К-75
наиболее подходящей является «алюминатная» обработка, а для люминофора
К-74 — цинксиликатная или силикатная. Использование такого сочетания обра-
боток предотвращает взаимное загрязнение люминофорных точек экрана.
В ряде работ [12, 14, 30, 31] отмечалась возможность использования в каче-
стве активатора для получения катодолюминофора с зеленым цветом свечения
меди. Люминофор ZnS-CdS-Си-Al имеет более высокую химическую стойкость
и повышенную яркость свечения по сравнению с люминофором ZnS-CdS Ag.
Кроме того, люминофор ZnS-CdS Си -Al обладает более насыщенным зеленым
цветом свечения.
Разработанные в последнее время люминофоры с красным цветом свечения
[29, 32—39] на основе р. з. э., о которых более подробно будет сказано дальше
(см. стр. 119), существенно превосходят по яркости свечения, цветовым характе-
ристикам и стойкости применяющиеся ранее для этой цели люминофоры
ZnS20-CdS80-Ag и Zn3(PO4)-Мп.
V.5. КАТОДОЛЮМИНОФОРЫ РЕДКОЗЕМЕЛЬНОГО ТИПА
Катодолюминофоры на основе соединений р. з. э. находят в последнее время
все более широкое применение. Указанные соединения могут выполнять роль
как активаторов, так и основы люминофоров. Соединения р. з. э. — это не только
важные материалы для создания катодолюминофоров цветного телевидения
[29, 32—42 J и препаратов с малой длительностью послесвечения, но и перспектив-
ные материалы в плане разработки новых катодолюминофоров, предназначен-
ных, главным образом, для использования в электронно-лучевых трубках,
работающих при высоких плотностях электронного возбуждения.
Первым редкоземельным элементом, получившим широкое практическое
применение в качестве активатора для катодолюминофоров, был Се. В электрон-
но-лучевых трубках с бегущим лучом люминофор возбуждается в течение 10-8 с
(пли меньше), а частота возбуждения может приближаться к 1000 МГц. Поэтому
люминофоры для подобных целей должны обладать высокой эффективностью
и исключительно малой инерционностью. Именно длительность послесвечения
117
лимитирует частоту возбуждения экранов электронно-лучевых трубок с бегу-
щим лучом.
Преимущество иона Се3+ как активатора малоинерционных катодолюмино-
форов состоит в том, что излучение его связано с разрешенными bd 4/ пере-
ходами, время жизни возбужденных состояний которых очень мало. Так как
затухание послесвечения окисных люминофоров, активированных Се3+, экспо-
ненциальное или близкое к нему, обычно длительность послесвечения т опреде-
ляют при спаде интенсивности свечения в е раз (табл. V.10).
Таблица V.1O
Характеристики некоторых катодолюминофоров редкоземельного типа
с коротким периодом послесвечения
Люминофор Энергетическая эффективность, % Длительность послесвечения, нс 1 Интенсивность послесвечения через 80 мкс, % 1 Максимум по- лосы излучения, нм Яркость свече- ния, отн. ед. Яркость свече- ния после облу- чения, % от начальной *
Ca2MgSi2O7 • Се 4 45 3 370, 395 120 58
Ca2Al2S Ю7 • Се 4,5 50 5-10 405 150 65
А120д • Се 400 100 95
Y2SiO5.Ce 6 30 0,1 415 200 92
Y2Si2O7 • Се 8 40 0,1 385 — —
* Время облучения 30 мин;
плотность тока 10 мкА/см2; энергия
электронов 10 кэВ.
Энергетический выход малоинерционных катодолюминофоров, синтезиро-
ванных с применением соединений р. з. э. ниже, чем у сульфидных. Однако их
преимуществом является очень малая длительность послесвечения и высокая
стойкость к действию электронного пучка, что особенно характерно для люмино-
форов А12О3-Се, Y2Si2O7 Се и Y2SiOs-Ce.
Большое число люминофоров, активированных Се3+ , синтезировано авторами
работ [43—45J. В частности, был предложен относительно эффективный мало-
инерционный катодолюминофор Y3AI5O12 Се [43], привлекший к себе внимание
как за границей, так и в СССР. Он не уступает по яркости свечения люминофору
ZnO • Zn (К-56), но выгодно (на порядок) отличается от него меньшей длитель-
ностью послесвечения. Из других малоинерционных люминофоров следует
отметить ZnS-Tm [2, 46], который имеет узкополосный спектр излучения (см.
рис. V.15, стр. 126) и высокую яркость свечения, а также люминофор Y2O3Bi
[47]. Малую длительность послесвечения (10-4 с) имеют также фосфатные люми-
нофоры, активированные Ей, в частности Sr3(PO4)2 -Ен [48]. На основе активи-
рованного Се фосфата иттрия с небольшими добавками лантана и гадолиния
(Y0;80Cd0,10Laa,loPO4 • Се0106) был предложен [42] катодолюминофор с очень
коротким послесвечением (т = 40 нс). Максимум излучения люминофора распо-
ложен в ультрафиолетовой области. По данным работ [44, 45], у люминофоров
YPO4 -Се, а также YOC1 Се и YA13B4O]2 -Се т = 25 нс. По стойкости к электрон-
ной бомбардировке фосфатные люминофоры аналогичны 2СаО -MgO-2SiO2-Се
и 2СаО А12О3 • SiO2 Се.
Такие свойства катодолюминофоров на основе р. з. э. как насыщенность
цвета, стабильность цветовых характеристик и возможность использования их
при высоких плотностях тока электронного возбуждения, позволяют применять
эти люминофоры в кинескопах для цветного телевидения. Особенно широкое
распространение для этих целей получили люминофоры с красным цветом све-
чения. До появления катодолюминофоров с р. з. э. в качестве красной компо-
ненты использовали малоэффективный препарат Zn3(PO4)2-Мп, который позже
был вытеснен более эффектным ZnS CdS Ag [32, 42]. Однако несмотря на высо-
118
2
кую энергетическую эффективность катодолюминесценции сульфидный «крас-
ный» люминофор был лимитирующим в системе компонент для цветного теле-
видения.
В 1964—1966 гг. в литературе появилось описание нового люминофора
с красным излучением, разработанного Левиным и Пал&йлг^ [32—37], ванадата
иттрия YVO4, активированного Ей. Этот люминофор готовят спеканием при
1300—1400° смеси окисей иттрия и ванадия высокой степени чистоты с добавкой
5 ат. % окиси европия. На катодолюминесценцию YVO4-Eu существенно вли-
яют условия синтеза и чистота исходных материалов [49—51].
Люминофор YVO4Eu имеет максимум
на кривой спектрального распределения
при л = 619 нм (рис. V.8). Преимущест-
вом его является узкая полоса на кривой
излучения, что обеспечивает большую чи-
стоту цвета; хороший выход люминесцен-
ции и повышенная светоотдача (табл. V.11).
Стойкость этого люминофора также выше,
чем у ZnS-CdS-Ag, а спад послесвечения
экспоненциальный ст 525 мкс. Длитель-
ность послесвечения при спаде яркости до
10% от начальной величины приблизи-
тельно равна 800 мкс. Зависимость яр-
кости свечения от плотности возбуждаю-
щего тока линейна в широком диапазоне
вплоть до 10 мкА/см2. К недостаткам
люминофора YVO4;Eu следует отнести его
невысокую энергетическую эффективность,
что вынуждает сохранять неравноточный
режим работы электронных прожекторов
в масочных кинескопах для цветного теле-
видения.
Серьезные конкуренты «красного» ва-
надатного люминофора Y2O3 • Eu и
Gd20g-Eu[34, 52—54]. Основное их преи-
мущество перед YVO4-Eu состоит в более
высокой энергетической эффективности
люминесценции и значительно более высо-
кой светоотдаче. Световой эквивалент излу-
чения Y2O3 - Eu при катодном возбуждении
более чем в 3 раза выше, чем у ZnS • Cd S • Ag,
что вполне компенсирует меньшую энерге
тическую эффективность первого (см.
табл. V.11). Яркость люминесценции
Y2O3 • Eu линейна в широком диапазоне
плотностей возбуждающего тока. По сравнению с YVO4 - Eu, излучение Y2O3 Eu
и Gd2O3 Eu является более оранжевым (см. рис. V.8), поэтому применение их
в качестве красной компоненты приводит к некоторому ухудшению пере-
дачи красных цветов. При этом, однако, увеличение яркости белого (9300 К)
поля достигает 33%. Цвет катодолюминесценции Y2O3-Ей и Gd2O3-Ей можно
сделать более красным, увеличив концентрацию Ей, но при повышении концен-
трации Ей наблюдается концентрационное тушение. В качестве недостатков
люминофора Y2O3 Eu можно отметить его низкую гидролитическую стойкость
и более высокую стоимость, по сравнению с YVO4-Eu.
В последнее время в качестве красной компоненты для цветного телевидения
был разработан люминофор Y2O2S-Eu [29, 39, 55]. Наиболее интенсивными
в спектре катодолюминесценции этого люминофора являются линии около 616
и 626 нм. Цвет и яркость свечения Y2O2S-Eu зависят от концентрации Ей.
Максимальная яркость достигается при 0,5 мол. % Ей. При низких концентра-
циях Ей цвет излучения люминофора Y2O2S-Eu оранжевый; при концентра-
циях выше 4 мол. % — красный, причем он незначительно меняется с дальней-
шим увеличением содержания Ей в люминофоре. Длительность послесвечения
при спаде яркости свечения до 10% составляет 175 мкс [56]. Насыщения яркости
3
700
500
600
Рис. V.8. Кривые спектрального
распределения энергии излучения
люминофоров:
1 — YVOi-Eu; 2 — Y2O3-Eu; 3 —
ZnS-CdS-Ag; 4 — Zn3 (PO4)2-Mn.
119
Таблица V.11
Свойства катодолюминофоров с красным цветом свечения
Люминофор Энергетиче- ская эффек- тивность, % Световой эквивалент, лм/Вт Светоотдача лм/Вт Координаты цветности Литература
X У
Zn3(PO4)2 • Мп 0,055 140 7,7 0,670 0,330 34, 41, 42
(Cd0>8- Zn0,2)S • Ag 0,17 80 13,6 0,660 0,340 52, 34
YV04 • Eu 0,062 250 15,5 0,670 0,330 52, 34
0,06 245 15,0 0,667 0,332 53
YVO4-Eu-Bi (0,2 ат. %) — —. — 0,637 0,352 69
Y2o3 • Eu 0,071 305 21,6 0,640 0,360 52, 34
0,075 300 23,0 0,658 0,342 53
Gd2O3 • Eu 0,08 330 26,0 — — 42
0,09 300 27,0 0,654 0,346 53
Y2O2S - Eu 0,073 255 18,6 0,660 0,340 52
0,10 290 29,0 0.660 0,340 29
Y2O3-Eu-Bi 0,078 390 26,5 0,648 0,350 29
при увеличении плотности возбуждающего тока до 10 мкА/см2 у люминофора нет
[57]. Замена YVO4 Eu на Y2O2SEu увеличивает яркость свечения в белом
•свете по меньшей мере на 35—40% [56], при этом улучшается цветопередача,
а отношения токов лучей «синего», «зеленого» и «красного» электронных прожек-
торов кинескопа приближаются к 1 [29]. Следует также отметить высокую гидро-
литическую стабильность оксисульфидного люминофора.
Как уже отмечалось (см. стр. 115) в качестве зеленой компоненты для кине-
скопов цветных телевизоров используют весьма эффективные сульфидные люми-
нофоры ZnS CdS-Ag и ZnS CdS-Си Al. Необходимость поиска новой более
эффективной зеленой компоненты возникла лишь в связи с разработкой эффек-
тивных «красных» люминофоров редкоземельного типа, в результате чего для
получения белого цвета в трехцветных приемных трубках большая часть тока
луча стала приходиться на «зеленый» люминофор. Рассмотрение схем энергети-
ческих уровней трехвалентных ионов р. з. э. показывает, что лишь ТЬ3+, Но3+
и Ег3+ могут служить активаторами для катодолюминофоров с излучением
в зеленой области. Поэтому были исследованы люминофоры, полученные путем
активации YVO4 и YPO4 этими элементами. Некоторые характеристики их при-
ведены в табл. V.12. Рядом исследователей были изучены люминофоры, полу-
Таблица V.12
Свойства катодолюминофоров, активированных р. з. э.
с зеленым цветом свечения
Люминофор Координаты цветности Относитель- ный световой эквивалент
Л' У
ZnS•CdS • Ag 0,260 0,610 0,700
YVO4- Er 0,248 0.730 0,894
YVO4«Ho 0,373 0,621 0,913
YPO4 - Tb 0,351 0,588 0,773
Y2O2S • Eu • Tb 0,340 0,560 —
120
ченные путем введения ТЬ3+ в фосфаты Y, La и Gd [58, 59], а также большое
число других соединений, активированных ТЬ3+ [30, 60—63].
Разработке катодолюминофоров с синим излучением на основе р. з. э.
уделялось и уделяется до сих пор относительно небольшое внимание, в основном
из-за того, что имеющийся стандартный «синий» люминофор для цветного теле-
видения— ZnS-Ag— является одним из лучших катодолюминофоров. Его
энергетическая эффективность близка, согласно теоретическим оценкам [2],
к предельной величине, а координаты цветности — к требуемым значениям
[52,-64]. Из катодолюминофоров с синим цветом свечения был исследован
YV04-Tm [52]. Световой эквивалент его излучения при катодном возбуждении
приблизительно вдвое выше, чем у ZnS -Ag. Но, наряду с существенно более
низкой энергетической эффективностью катодолюминесценции, координаты
цветности YV04Tm хуже, чем у ZnS Ag.
В синей области спектра излучают также люминофоры, активированные
Еи2+. Была исследована катодолюминесценция фосфатов стронция, боратов
кальция, стронция и бария, борофосфатов кальция, стронция и бария, а также
многочисленных силикатов и силикатных систем, активированных Еи2+ [56].
Эти люминофоры, как правило, при катодном возбуждении имеют низкие (реже —
средние) энергетические эффективности. Наиболее эффективным из них является
Sr3(PO4)2-Eu [2, 48].
Из катодолюминофоров на основе р. з. э. для черно-белого телевидения
следует упомянуть YV04 Tm и YVO4Dy [56]. Первый из них может быть ис-
пользован в качестве синей компоненты, а второй — в качестве желтой. Прямая,
соединяющая их координаты цветности на цветовом треугольнике, проходит
непосредственно через точку, соответствующую излучению с цветовой темпера-
турой 9300 К. Основной недостаток как «желтого» люминофора YV04-Tm, так
и «синего» YVO4-Dy — низкая энергетическая эффективность.
Практическое применение в экранах кинескопов черно-белого телевидения
нашел оксисульфид, активированный ТЬ [29], спектр свечения которого предста-
вляет собой группу узких полос в синей, желто-зеленой и оранжевой частях
спектра, что создает интегральное белое излучение с цветовой температурой,
близкой к 8500 К. Однокомпонентный состав этого люминофора позволяет
полностью исключить цветовую неоднородность экрана, что особенно суще-
ственно при формировании крупногабаритных широкоформатных кинескопов.
Другое преимущество оксисульфидного «белого» катодолюминофора — его
высокая радиационная и примесная стойкости, а также отсутствие насыщения
яркости свечения при увеличении плотности тока до 100 мкА/см2.
V.6. ЛЮМИНОФОРЫ ДЛЯ ЭКРАНОВ С ДЛИТЕЛЬНЫМ ПОСЛЕСВЕЧЕНИЕМ
В радиолокационных и некоторых осциллографических трубках применяют
экраны с длительным послесвечением (с «памятью»). Выбор люминофоров для
таких экранов затруднителен вследствие того, что при катодном возбуждении
большинство люминофоров (за исключе-
нием фторидных) не имеет длительного
послесвечения. Для обеспечения этого каче-
ства в большинстве случаев применяют
двухслойные экраны (рис. V.9), в которых
первый слой возбуждается электронным
пучком и дает излучение в синей области
спектра. Это излучение возбуждает второй
слой люминофора послесвечение которого
при фотовозбуждении достаточно продол-
жительно. Наиболее распространены двух-
слойные экраны типа Р-7 и Р-14. По дан-
ным Леверенца [5], экран Р-7 состоит из
слоя люминофора ZnS(86) -GdS(14) •
• Gu(0,007), синтезированного при~1200°,
и осажденного поверх него слоя люминофора ZnSreKC-Ag(0,015). Электронный
пучок, попадая на слой ZnS-Ag, возбуждает свечение (Хтах == 430 нм), кото-
рое возбуждает фотолюминофор ZnS CdS - Си с желто-зеленым послесвечением,
хорошо воспринимаемое глазом. Подбирая соотношение между ZnS и CdS в
Люминофор
Стеклянная
подлоокка
Фильтр
Электронной
пучок
наблю
дателю
ZnS-Ag-7'-ZnS CdS Cu
Рис. V.9. Устройство двухслой-
ного экрана.
121
основе люминофора, можно изменять длительность послесвечения в требуемых
пределах (для экрана Р-7 она равна 3 с).
Фотолюминофор в экране Р-14 имеет состав ZnS (75)-CdS (25)-Cd (0,008)
и длительность послесвечения ~1 с. Плотность слоя ZnS - Ag составляет 8—
10 мг/см2, фотолюминофора — 12 мг/см2. Кривые затухания для экранов Р-7
и Р-14 показаны на рис. V-10.
Характеристики аналогичных люминофоров, выпускаемых нашей промыш-
ленностью, представлены в табл. V.13 и на рис. V.11.
Указанные люминофоры обеспечивают высокую стабильность и надежность
работы экранов. Кроме них в последние годы разработаны новые люминофоры
для однослойных экранов с длительным послесвечением различного цвета.
Рис. V.10. Кривые затухания свече-
ния люминофоров, используемых в
двухслойных экранах Р-7 и Р-14
(7 = 4—9 кВ; 7 = 3 мкА/см2).
Рис. V.11. Спектральное распределение
энергии излучения люминофоров, ис-
пользуемых в двухслойных экранах с
длительным послесвечением:
1 _ к-430; 2 — К-14; 3 — Л-15; 4 — Л-20.
Возможность использования люминофоров с разным цветом свечения имеет
большое значение для экранов с памятью, где в ряде случаев необходимо полу-
чать сигналы разного цвета. Для этой цели могут быть использованы люмино-
форы на основе фторидов.
Фториды цинка, магния, алюминия и двойные фториды этих металлов,
имеющие перовскитную структуру, при активации главным образом Мп, а также
Nb и Та [65, 66] являются почти уникальным классом соединений, дающим при
катодном возбуждении длительное послесвечение, которое затухает по экспо-
ненциальному закону. Цвет и длительность послесвечения определяются соста-
вом основы. В частности, показано, что в ряде случаев введение в основу третьего
компонента способствует увеличению длительности послесвечения [65]. Фторид-
ные люминофоры получают при взаимодействии рассчитанных количеств люмино-
форно-чистых окислов или карбонатов с дважды перегнанной HF. Полученную
таким образом основу прокаливают при 800—1000° (в зависимости от состава)
в платиновых или графитовых тиглях в условиях, предотвращающих возмож-
ность окисления шихты. Количество активатора составляет —1%.
Недостаток фторидных люминофоров — быстрое выгорание их под действием
электронного пучка, что ограничивает длительность эксплуатации экранов
с этими люминофорами (не более 100—200 ч). Стабильность люминофоров суще-
ственно зависит от состава основы.
Характеристики отечественных марок фторидных люминофоров предста-
влены в табл. V.13 и на рис. V.12.
Из других фторидных люминофоров следует отметить ZnF2-YF3 Mn,
который имеет повышенную стойкость по отношению к электронному лучу,
и тройной фторид CaF2 A1F3 -LiF-Mn с длительностью послесвечения порядка
—40 с [67].
В электронно-лучевых трубках со знаковой индикацией используют люми-
122
Таблица V.13
Технические характеристики люминофоров для экранов
с длительным послесвечением
Люминофор Цвет свечения ^тах’ нм Средний размер зерна, мкм Длительность послесвече- ния при спаде яркости до 1% от начальной, с Применение
марка состав
К-14 Zn'SKy6' Ag Голубой 455 ±5 6-8 В двухслой-
К-430 ZnSгекс • Ag » 430 ±5 8—20 ных экранах в комбинации с Л-15 или Л-20
Л-15 ZnS • CdS -Cu Желтый 556 ±5 5—15 4—10 В двухслой-
Л-20 » Оранжевый 580 ±5 5—20 2—3 ных экранах в комбинации с К-14 или К-430
К-61 KMgF3 • Mn » 600 ±5 1—3 8
К-62 CaF2- A1F3 • Mn Зеленый 525 ±5 1-3 5 В однослойных
К-63 MgF2 • Mn • Cd Оранжевый 590 ±5 1-3 4 экранах
Лотос | ZnF2 • MgF2 • Mn » 590 ±5 — — То же
нофор марки КГ-2 (ZnS Cu-Ag), обладающий достаточно длительным после-
свечением (~0,3 с). Послесвечение, длящееся 3—4 с, имеет люминофор «Лимит»,
Рис. V.12. Спектральное распределение
энергии излучения фторидных люмино-
форов:
Кривая Марка Состав
7 К-62 CaF2-AlFa-Mn
2 Лотос ZnF2-MgF2’Mn
3 К-63 MgF2-Mn-Cd
4 К-61 KMgF3-Mn
Рис. V.13. Спектральное распреде-
ление энергии излучения фторси-
ликата магния, активированного Ti.
представляющий собой фторсиликат
магния, активированный титаном
(рис. V.13). Люминофор «Лимит» по-
лучают двойным прокаливанием
шихты, состоящей из 1,7 моль MgSO4;
0,3 моль MgO; 1 моль MgF2; 1 моль
SiO2 и 0,02 моль TiO2, в течение
49Q
1 ч при 1050—1100° с промежуточным помолом. Следы не вошедшего в реакцию
сульфата магния удаляют, размалывая люминофор в воде с последующей про-
мывкой горячей водой.
Стойкость люминофора Mg3SiO4F2 -Ti позволяет использовать его при боль-
ших токовых нагрузках и получать значительные яркости послесвечения.
V.7. ЛЮМИНОФОРЫ ДЛЯ ЭКРАНОВ С КОРОТКИМ ПОСЛЕСВЕЧЕНИЕМ
Люминофоры этого типа необходимы для приборов, с помощью которых
ведут наблюдение или измерение параметров процессов, протекающих с большой
скоростью. Последнее относится, например, к электронно-оптическим преобра-
зователям изображения (приборах для ночного видения), к некоторым осцилло-
графическим трубкам, к трубкам с разверткой бегущим лучом и к некоторым
другим приборам. В указанных случаях применяют, главным образом, люмино-
форы с зеленым или желто-зеленым и синим свечением. Большое значение имеет
также изыскание малоинерционных люминофоров с излучением в оранжевой
и красной областях спектра. Кроме высокой яркости свечения, люминофоры
этого типа должны обладать высокой степенью дисперсности, что обеспечивает
хорошую разрешающую способность экранов.
Высокую степень дисперсности порошков достигают либо использованием
мелкозернистого исходного сырья, в сочетании с максимально возможным сни-
жением температуры синтеза люминофоров (без ущерба для яркости свечения),
либо специальной обработкой поверхности уже готовых люминофоров, приводя-
щей к дезагрегации первично образующихся при синтезе агрегированных частиц.
Такие методы применяют, например, к сульфидным и сульфид-селенидным люми-
нофорам. При этом важно, чтобы указанная обработка в минимальной степени
влияла на яркость свечения.
Люминофоры сульфидного типа активируют Си, но чаще всего Ag. В послед-
нем случае длительность послесвечения может быть снижена до 10“ 3 с (при спаде
яркости свечения до 5% от начальной). Сокращение длительности послесвечения
достигается введением в основу Cd или Se. В некоторых сульфидных люмино-
форах длительность послесвечения снижают путем введения добавки Ni, но
так, чтобы при этом не снижалась существенно яркость свечения. Большое прак-
тическое применение среди малоинерционных люминофоров имеет окись цинка,
дающая широкую спектральную полосу излучения с Хгпах = 505 нм и очень
короткое послесвечение [5, 68].
По сравнению с перечисленными люминофорами, значительно более низкий
энергетический выход (3—5%) имеют люминофоры 2СаО А12О3 SiO2 Се п
А12О3 - Се. Однако последние обладают очень малой длительностью послесвече-
ния (5 -10-7 с) и высокой стойкостью к действию электронного пучка (см. стр. 118).
Из других малоинерционных люминофоров нужно упомянуть самоактивиро-
ванный пирофосфат циркония ZrP2O7 с излучением в УФ-области спектра и суль-
фид магния MgS, активированный Sb (0,01%) с излучением в желто-зеленой
области спектра (Хтах = 530 нм). Оба эти люминофора имеют длительность
послесвечения ~10-6с.-Их свечение затухает по экспоненциальному закону.
Однако в то время как первый из них отличается удовлетворительной химиче-
ской стойкостью и стабилен при действии электронного пучка, MgS • Sb, несмотря
на высокую яркость свечениц, мало пригоден для практического использования,
так как легко разрушается влагой воздуха и отличается недостаточной стой-
костью при катодном возбуждении.
Технические характеристики наиболее важных люминофоров с коротким
послесвечением представлены в табл. V.14, а кривые спектрального распределе-
ния энергии излучения — на рис. V.14 и V.15.
Методы приготовления этих люминофоров не отличаются существенно от уже
описанных методов синтеза люминофоров аналогичного состава. Следует только
отметить, что сульфид-селенидные люминофоры изготовляются в промышленном
масштабе по так называемому «мокрому методу» (см. стр. 59), т. е. путем взаимо-
действия люминофорно-чистого ZnS с соответствующим количеством раствора
селенистой кислоты и активатора (~0,01%). Образующуюся при этой реакции
смесь ZnS с ZnSeO3, серой и селеном прокаливают при 800—850°. В результате
получается люминофор, представляющий собой твердый раствор ZnSe в ZnS,
124
Т аблица V.14
Технические характеристики катодолюминофоров с коротким послесвечением
Люминофор Цвет свечения ^тах* нм Средний размер зерна, мкм Послесвечение Назначение
марка состав
К-9 ZnS • Ag Голубой 450 ±5 5-7 Короткое (<<5 • 10-3 с) Осциллография, фотогра- фирование
К-48 ZnS-CdS • Ag Зеленый 535 ±5 2-5 Короткое (0,1 с) Осциллография
УР-1 CaWO4 Синий 430 ±5 Очень короткое (1 -10-6 с) Осциллография, фотогра- фирование
К-56 ZnO-Zu Зеленый 505 ±5 4-6 Очень короткое (2-10-6 с) Трубки с разверткой бегу- щим лучом
А-1 A12O3 • Co Синий 405 ±5 1-2 Очень короткое (5 • 10-7 с) То же
Геленит 2CaO • A12O3 • •SiO2-Ce » 405 ±5 1-2 Очень короткое (5 • 10-7 с) »
Кристалл ZnS • ZnSe • Cu Желто-зеле- ный 550 ±5 1-2 Короткое (0,05 с до 5% от 70) Электронно-оптические преобразователи
Теллур ZnS•ZnSe•Cu То же 540+5 — Короткое То же
К-67 ZnS•CdS • Ag » 545 ±5 — Короткое (5 • 10-3 с до 5% от Го) »
К 71 ZnS• Ag•Ni Голубой 450 ±5 — Короткое »
ФС-1 ’ ZnS • Ag Синий 460 ±5 1—2 Короткое (0,05 с) »
Рис. V.14. Спектральное распределение энергии излучения малоинер-
ционных катодолюминофоров для электронно-оптических преобразо-
вателей:
Кривая Марка Состав
1 К-71 ZnS- Ag-Ni
2 ФС-1 ZnS-Ag
3 Теллур ZnS-ZnSe-Cu
4 К-67 ZnS-CdS-Ag
5 Кристалл ZnS-ZnSe-Cu
Рис. V.15. Спектральное распределение энергии излучения
некоторых малоинерционных катодолюминофоров:
Кривая Марка Состав
1 ZrP2O7
2 Геленит 2CaO-Al2O3-SiO2-Ce
3 A-l А12О3-Се
4 УР-1 CaWO4
5 К-9 ZnS-Ag
6 — ZnS-Tm
7 К-56 ZnO-Zn
8 К-48 ZnS-CdS-Ag
126
причем гранулометрический состав его зависит от режима прокаливания. В тех-
нических условиях на люминофоры этого типа строго регламентируется содержа-
ние рабочей фракции («процент отбора») в люминофоре.
Люминофор на основе геленита — 2СаО-А12О3-SiO2-Се (1,5) — готовят
путем длительного (5—8 ч) высокотемпературного (1250—1300°) спекания тща-
тельно гомогенизированной смеси окислов. При этом получается сильно спек-
шийся и очень твердый продукт с трудом поддающийся измельчению. Для луч-
шего прохождения реакции прокаливание люминофора производят в две стадии,
с промежуточным растиранием полученного в первый раз спека.
Из малоинерционных люминофоров с оранжевым и красным свечением
нужно указать на ZnS-Ag (Xmax = 560—580 нм) и CdS-Ag (Xmax = 750 нм)
[48], длительность послесвечения которых составляет —1-10-бс.
ЛИТЕРАТУРА
1. Москвин А. В. Катодолюминесценция. Ч. 1. Общие свойства явления.
М.—Л., ГИТТЛ, 1948, 348 с.; ч. 2, Катодолюминофоры п экраны. М.— Л.,
ГИТТЛ, 1949, 700 с. — 2. Левшин В. Л., Арапова А. Я., Блажевич Л. И. и др.
«Труды ФИАН им. П. И. Лебедева», 1963, т. 23, с. 64—135. — 3. Левшин В. Л.
Изв. АН СССР. Сер. физ., 1965, т. 29, с. 346—354. — 4. Garlik G. Luminescent
Materials. Oxford, Claredon Press, 1949. 255 p. — 5. Leverenz H. An Intro-
duction to Luminescence of Solids. New York — London. Wiley sons, 1950.
569 p. — 6. Ouweltjes J. F. In: Modern Materials. Advances in Development
and Applications. V. 5. New York — London, 1965, p. 161—257. — 7. Марков-
ский Л. Я. Неорганические люминофоры прикладного назначения. Вып. 1.
Катодолюминофоры. Л., изд. ГИПХ, 1972, с. 52—82. — 8. Коровкина Т. В.
В кн. Материалы V Совещания по люминесценции (кристаллофосфоры). Тарту,
Изд. АН ЭССР, 1957, с. 299—313. Bachman С. Н., Sawner М. L., Allen W.
J. Electrochem. Soc., 1956, v. 103, p. 117—122. — 9. Гугелъ Б. M. Люминофоры
для электровакуумной промышленности. М., «Энергия», 1967. 344 с. —
10. De Boor F., Broos J. Electrochem. Technol., 1966, v. 4, p. 38—41.
11. Марковский Л. Я., Пронъ Г. Ф. В кн.: Сборник рефератов по химии
и технологии люминофоров за 1969 г. Л., изд. ГИПХ, 1971, с. 35—37. —
12. Toshiba Review, 1969, р. 1316—1322. — 13. Гурвич А. М., Пивнева С. П.
Изв. АН СССР, Сер. физ., 1969, т. 33, с. 971—974. — 14. «Новости зарубежной
электроники», 1970, № 9, с. 3. — 15. Марковский Л. Я., Пронъ Г. Ф., Таушка-
нова Л. Б. В кн.: Химия и технология люминофоров. Вып. 69. Л., «Химия»,
с. 25—30. — 16. Pfanhl A. Advances in Electron Tube Technology. Oxford,
Pergamon Press, 1961. 235 p. — 17. Pjanhl A. Bell. System. Technol., 1963,
v. 42, p. 181—201. — 18. Князатов E. H., Шерстнев Л. Г. В кн.: Электронная
техника. Сер. 4, вып. 3. М., изд. ЦНИИЭлектроника, 1968, с. 204—213. —
19. Scharmann A., Grasser R. In: International Lumineszenz Symposium uber die
Physik und Chemia der Szintillatoren. Munchen, Verlag Karl Thiemig K. G.,
1966, S. 209—222. — 20. Бланк Ю. С., Завьялова T. А., Румянцева T. Я. В кн.:
Электронная техника. Сер. 4, вып. 3. М., изд. ЦНИИЭлектроника, 1968,
с. 177—183.
21. Москвин А. В., Рыжкин Ю. С. Сборник трудов I Всесоюзной научно-
технической конференции по электроннолучевым приборам. Т. 2. М., 1966,
изд. электронной пром. СССР, с. 104—120. — 22. Гугелъ Б. М. В кн.: Сборник
научных трудов ВНИИЛ. Вып. 5. 1971, с. 52—61. — 23. Гугелъ Б. М., Крупе-
нина А. Я. Опт. и спектр., 1968, т. 25, с. 465. — 24. Гугелъ Б. М. см. [22, с.43—
51]. — 25. Цудзи, Тампа. Лион. пат. 13007, 1965; РЖХим, 1970, 14Л233. —
26. Хатвара, Кумогая. Лион. пат. 7696, 1965; РЖХим, 1969, 4Л259. — 27. U tu-
ber ger J. Пат. США 3026750, 1962; РЖХим, 1964, 11Л132. — 28. Ларах С.,
Харди А. ТИИЭР, 1973, т. 19, № 7, с. 144—158. — 29. Сощин Н. П. Электрон,
пром., 1973, № 2, с. 100—105. — 30. Марковский Л. Я. В кн.: Химия и техно-
логия люминофоров. Вып. 69. Л., «Химия», 1972, с. 10—24.
31. Electr. Weekly, 1970, № 491, р. 28. — 32. Levine А. К., Palilla F. С.
Appl. Phys. Lett., 1964, v. 5, p. 118—120. — 33. Burdick G., Miller R., Bar-
121
tels В. Electrochem. Technol., 1966, v. 4, p. 12—15. — 34. Levine A. K. Pa-
lilla F. C. Ibid., p. 16—20. — 35. Ind. Eng. Chem., 1964, v. 56, № 12, p. 9. —
36. Chem. Eng., 1964, v. 71, № 25, p. 96. — 37. Chem. Week, 1965, v. 96, № 16,
p. 45. — 38. Trond S. S., Martin J. S., Stanavage J. P. e. a. J. Electrochem.
Soc., 1969, v. 116, p. 1047—1050. — 39. Haynes J. W., Brown J. J. Ibid., 1968,
v. 115, p. 1060—1066. — 40. BrilA., Wanmaker W. L. Ibid., 1964, v. Ill,
p. 1363—1368.
41. Levine A. K., Palilla F. C. In: International Lumineszenz Symposium
fiber die Physik und Chemie der Szintillatoren. Munchen, Verlag Karl Thie-
mig K. G., 1966, S. 317—324. — 42. BrilA ., Wanmaker W. L., De Laat C.D. J. C.
J. Electrochem. Soc., 1965, v. 112, p. 111.. — 43. Blasse G., Bril A. Appl. Phys.
Lett., 1967, v. 11, p. 53. — 44. Bril A., Blasse G., De Boortes J. A. J. Electro-
chem. Soc., 1970, v. 117, p. 346—348. — 45. Bril A., Blasse G., De Boortes J. A.
Ibid., 1969, v. 116, p. 123 c. — 46. Ibuki S., Langer D. Appl. Phys. Lett., 1963,
v. 2, p. 95—97. — 47. Колпакова А. А., Кауфман С. А., Тарасова JI. E. и dp.
В кн.: Сборник рефератов по химии и технологии люминофоров за 1969 г. Л.,
изд. ГИПХ, 1971, с. 24—26. — 48. Назарова В. П. Изв. АН СССР. Сер. физ.,
1961, т. 25, с. 332—335. — 49. Капо Т., Otomo J. J. Electrochem. Soc., 1969,
v. 116, р. 64—68. — 50. Mehalchik F. J., Mikus F. F., Mathers J. E. Ibid.,
p. 1017—1019.
51. Шапочник M. M. Изв. АН СССР. Сер. физ., 1969, т. 33, с. 1053—1057. —
52. Palilla, F. C. Electrochem. Technol., 1968, v. 6, p. 39—49. — 53. Bril A.,
De Laat C. D. J. C. Ibid., 1966, v. 4, p. 21—24. — 54. Амирян A. M., Котля-
ров P. M., Никонов В. H. и др. Изв. АН СССР. Сер. физ., 1969, т. 33, с. 1067—
1071. — 55. Сощин Н. П., Амирян А. М. Авт. свид. 283463; Бюлл. изобрет.,
1970, № 31, с. 83. — 56. Балодис Ю. Н. В кн.: Неорганические люминофоры
прикладного назначения. Вып. 1. Катодолюминофоры. Л., изд. ГИПХ, 1972,
с. 4—52. — 57. Meyer V. D., Palilla F. С. J. Electrochem. Soc., 1969, v. 116,
p. 535-539. — 58. Aia M. A. Ibid., 1967, v. 114, p. 367—370. — 59. Bril A.,
Wanmaker W. L., Schuil R. E. In: International Lumineszenz Symposium iiber
die Physik und Chemie der Szintillatioren. Miinchen, Verlag Karl Thiemig K. G.,
1966, S. 310—316. — 60. Blasse G., BrilA. Phil. Res. Repts, 1967, v. 22,
p. 481—504.
61. Blasse G. J. Chem. Phys., 1969, v. 51, p. 3529. — 62. Avella F. J.,
J. Electrochem. Soc., 1966, v. 113, p. 1225. — 63. Avella F. J., Sovers O. J.,
Wiggins C. S. Ibid., 1967, v. 114, p. 613—616. — 64. Palilla F. C., Levine A. K.,
Rinkevics M. Ibid., 1965, v. 112, p. 776—779. — 65. Векшина H. В., Марков-
ский Л. Я. В кн.: Химия и технология люминофоров. Вып. 43, Л., Госхимиздат,
1960, с. 22—45. — 66. Wilke К., Albers К., Mannheim R. Z. phys. Chem., 1960,
Bd. 213, S. 191—224.—67. Марковский Л. ЯЛесина Э- Я., Кириченко Т. М.
В кн.: Химия и технология люминофоров. Л., изд. ГИПХ, 1974, с. 51—56. —
68. Зеликин Я. М. Изв. АН СССР. Сер. физ., 1961, т. 25, с. 461—463. —
69. Dalta R. К. J. Electrochem. Soc., 1967, v. 114, р. 1057—1063.
ГЛ А В A VI
ЭЛЕКТРОЛЮМИНОФОРЫ
Электролюминофоры — это вещества, светящиеся при возбуждении элек-
трическим полем. Явления электролюминесценции обусловлены либо эффек-
том Лосева, либо эффектом Дестри о.
В первом случае электролюминесценция возникает под действием зарядов,
инжектируемых в кристаллы. Впервые такого рода свечение
твердых веществ в электрическом поле наблюдал в 1923 г. Лосев [1] на карбиде
кремния.
Во втором случае люминесцируют порошкообразные люминофоры на основе
сульфида цинка. Это свечение, которое впервые наблюдал Дестрио в 1936 г.
[2—7], некоторыми свойствами отличается от свечения карбида кремния: оно
возникает и тогда, когда кристаллы люминофоров помещены в диэлектрик,
а возбуждение инициируется переменным электрическим по-
лем. В отечественной литературе описываемое явление иногда называют
предпробойной электролюминесценцией.
Свечение в постоянном электрическом поле возникает лишь у люминофоров,
подвергшихся специальной обработке (см. стр. 138). Суть последней заклю-
чается в том, что на поверхности зерен кристаллов создаются условия для гетеро-
перехода, например Cu2S -» ZnS. Интенсивное свечение в этом случае возможно
лишь в отсутствие диэлектрика (или при минимальном его содержании).
VI.1. ПОРОШКООБРАЗНЫЕ ЭЛЕКТРОЛЮМИНОФОРЫ
Порошкообразные электролюминофоры широко используют в промышлен-
ности (индикаторные устройства, подсветка шкал, преобразователи изображе-
ния и т. и.) [8—10]. Кроме того, в намечающемся в настоящее время создании
Рис. VI.1. Схема устройства элек-
тролюминесцентного конденса-
тора:
1 — стекло; 2 — токопроводящий слой;
3 — слой электролюмпнофора е ди-
электриком; 4 — второй электрод.
нового типа плоских безвакуумных телевизионных экранов предполагается при-
менение электролюминофоров [8, с. 136; 10, с. 243J. Порошкообразные элек-
тролюминофоры возбуждаются в электролюминесцентном конденсаторе (ЭЛК),
устройство которого показано на рис. VI. 1. Электролюминофор в смеси с ди-
электриком наносят на электрод конденсатора, представляющий собой прозрачное
стекло с проводящим слоем (SnO2). Второй электрод обычно делается металли-
ческим.
9 Заказ 44
129.
Диэлектриками служат заполимеризованные эпоксидные смолы, цианэтил-
цртлюлоза и ее производные, цианэтиловый эфир поливинилового спирта [10,
с. 154; И, 12], а также эмали [13].
Методы синтеза электролюминофоров
и их люминесцентные характеристики
Исследование физических процессов, происходящих при электролюминес-
ценции, шло параллельно с исследованием химизма формирования электролю-
минофоров и усовершенствованием их свойств. На основании экспериментальных
данных было показано, что эффективная электролюминесценция в цинксуль-
эффективная электролюминесценция в цинксуль-
фидных электролюминофорах обусловлена
внедрением больших количеств меди в крис-
таллы ZnS и образованием фазы Cu2S. По-
этому специфика всех методов приготовления
электролюминофоров, по сравнению, напри-
мер, с фото- или катодолюминофорами, за-
ключается в том, чтобы при синтезе создать
условия, способствующие возникновению фазы
Cu2S. Для этого концентрация активатора Си,
вводимого в ZnS, должна быть в 5—10 раз
выше, чем в случае приготовления фотолюмп-
нофоров.
Кроме того, образованию фазы Cu2S спо-
собствует также прокаливание в атмосфере,
содержащей сернистые соединения. Как сле-
дует из работ [14, 15], электролюминофоры
ZnS - Си на стадии остывания представляют со-
бой пресыщенные твердые растворы Cu2S в
ZnS. Исследование, приведенное в работе
[15], показало, что фаза Cu2S образуется на
стадии остывания в результате распада пре-
сыщенного раствора Cu2S в ZnS.
Электролюминофоры ZnS • Си. Впервые
электро люминофоры на основе ZnS Си
были синтезированы Дестрио прокаливанием
на воздухе [16]. Однако их свечение было
слабым и наблюдалось при сравнительно вы-
соких напряжениях. Более эффективные
Хомер и др. в атмосфере инертного газа [17].
Рис. VI.2. Спектральное рас-
пределение энергии излучения
электро люминофоров ZnS - Си
при различной концентрации
Си.
Атмосфера прокаливания 40 объемн.
% НС1 + 60 объемн. % H2S.
электролюминофоры получили
Аналогичную среду при прокаливании электролюминофоров использовали и
другие исследователи [13—20].
Вахтель [20] готовил шихту из ZnS с добавками хлорида аммония, порош-
ковой серы, водного раствора ацетата меди и окиси цинка. Эту шихту прокали-
вали в атмосфере очищенного азота.
Прокаливание в инертной атмосфере с добавками соединений брома, иода
и серы применяют в синтезах некоторых промышленных марок электролюмино-
форов [21]. Таким способом готовят промышленные электролюминофоры марок
ЭЛС-460, ЭЛС-510.
Эффективным оказалось прокаливание в среде газообразных H2S и НС1
[3, 22]. Впервые электролюминофоры путем прокаливания в H2S были получены
Фрелихом [23].
Казанкин [15], исходя из установленного им механизма действия газообраз-
ных H2S и НС1, нашел, что все основные характеристики электролюминофоров —
спектр излучения, гранулометрический состав, стабильность — зависят от соот-
ношения между H2S и НС1. Свойства электролюминофоров зависят также от
концентрации Си, структуры основы и температуры прокаливания.
Учитывая специфику синтеза, оценим влияние различных технологических
факторов на люминесценцию получаемых цинксульфидных электро люминофоров.
Рассмотрим вначале действие различных факторов на свечение элек-
тролюмпнофоров, полученных в атмосфере H2S + НС1. При повышении концен-
130
•грации Си интенсивность электролюминесценции сначала увеличивается, про-
ходит через максимум, а затем уменьшается. Последнее объясняется в известной
степени выделением на поверхности кристаллов значительных количеств Cu2S,
что приводит, во-первых, к ослаблению свечения вследствие образования серого
слоя и, во-вторых, к уменьшению напряженности поля в кристаллах вследствие
шунтирующего действия пленки Cu2S.
Из рис. VI.2 и VI.3 следует, что при одинаковом составе атмосферы прокали-
вания повышение концентрации Си приводит к увеличению интенсивности излу-
чения в зеленой полосе. Наиболее эффективные электролюминофоры с голубым
Рис. VI.3. Спектральное распределение энер-
гии излучения электро люминофоров ZnS Си
(0,1%) при различном составе атмосферы про-
каливания:
Состав атмосфе-
ры, объемн. %
Состав атмосфе-
ры, объемн. %
НС1 H.S
НС1
1 10
2 20
3 30
4 40
5 50
90 6
80 7
70 8
60 9
50 10
60 40
70 30
80 20
90 10
100 —
свечением получаются при содержании 0,03—0,05% Си, а с зеленым — при
0,10—0,15% Си. Дальнейшее повышение концентрации Си может снижать интен-
сивность электролюминесценции, причем максимум спектра излучения смещается
еще больше в длинноволновую область.
Увеличение количества НС1 в атмосфере прокаливания приводит к возра-
станию интенсивности свечения в зеленой полосе. Наиболее мелкозернистые
электролюминофоры получаются при прокаливании в атмосфере H2S. Увеличе-
ние содержания НС1 в смеси газов приводит к укрупнению кристаллов.
Вендель [24] при синтезе электролюминофоров использовал двухстадий-
ное прокаливание: первая стадия — 1250° и атмосфера H2S + HG1; вторая
стадия — 450° на воздухе; подобная операция обеспечивала резкое возрастание
интенсивности электролюминесценции. Авторы работы [26] нашли, что при
таком способе получения электролюминофоров можно отказаться от применения
токсичного сероводорода и прокаливать электролюминофоры в атмосфере НС1.
В результате синтезируются крупнозернистые электролюминофоры со смешан-
ной структурой сфалерита и вюрцита, наиболее эффективны они при низких
напряженностях электрического поля.
Анализ многих работ показывает, что эффективность электролюминесценции
определенным образом связана со структурой рещетки. Так, наибольшей интен-
сивностью свечения обладают электролюминофоры, кристаллическая рещетка
которых имеет определенные нарушения, возникающие при переходе от вюр-
цита к сфалериту [26]. Баллентайн [27] указывает, что во всех случаях интен-
сивная электролюминесценция связана со смешанной структурой основы.
Однако эффективные электролюминофоры могут быть получены и в том случае,
если их кристаллическая решетка имеет структуру сфалерита [15]. При этом
подчеркивается, что основное значение при формировании электролюминофоров
имеет состояние фазы Cu2S.
К недостаткам технологии приготовления электролюминофоров в атмо-
сфере H2S + НС1 следует отнести: необходимость применения токсичного H2S;
усложнение процесса при работе с газами.
9*
131
Поэтому были предложены безгазовые методы синтеза электролюминофо-
ров [28—30]. Один из них состоит в том, что вместо H2S НС1 в зону прока-
ливания при помощи специального дозатора непрерывно вводят хлорид аммо-
ния и серу. При высокой температуре в зоне прокаливания образуются пары HG1,
серы и воды (в малых количествах), т. е. создается атмосфера, по действию ана-
логичная атмосфере H2S + НС1. Вместо NH4G1 можно использовать NH4Br
и NH4I.
Все основные характеристики электролюминофоров, полученных безвизо-
вым методом (яркость свечения, спектр излучения, стабильность, гранулометрп-
Рис. VI.4. Спектральное распределение энергии излучения электролюмино-
форов ZnS-Си при различных содержаниях S в смеси NH4Br-f- S:
а — Концентрация Си==0,05 б — Концентрация Си = 0,1
кривая S, % кривая S, %
1 90 Z 90
2 80 2 80
3 70 3 70
4 60 4 30
5 40
6 30
ческий состав), зависят от соотношения количеств серы и соответствующего
галогенида в шихте (рис. VI.4). Наиболее эффективные электролюминофоры
в случае использования NH4G1 получены при весовом соотношении между
NH4C1 и S, равном 3:7, и температуре прокаливания 900°. При содержании
меди 0,1% получаются эффективные электролюминофоры с зеленым цветом
свечения. Добавление алюминия увеличивает интенсивность зеленой полосы.
Снижение концентрации меди до 0,05% приводит к повышению интенсив-
ности синей полосы.
Применение NH4Br вместо NH4C1 позволяет получать самые эффективные
электро люминофоры с излучением в зеленой области спектра. Для них повыше-
ние содержания серы до 90% в смеси NH4Br + S приводит к росту интенсив-
ности свечения, после чего она начинает уменьшаться. Поэтому в промышлен-
ных условиях синтеза содержание серы в смеси близко к указанной величине.
132
Увеличение содержания меди приводит к сильному росту интенсивности
свечения. Максимальная интенсивность электролюминесценции достигается
при 0,1% Си, в дальнейшем происходит ее снижение. Спектральный состав
излучения также существенно зависит О'
При использовании смеси NH4I + S
зеленое свечение возникает только при
очень малых содержаниях серы и значи-
тельных меди. В интервале от 0,01 до
.0,03% Си происходит резкое нарастание
интенсивности; затем она остается посто-
янной вплоть до —0,2% Си. Эффективные
электролюминофоры с синим цветом све-
чения (рис. VI.6) могут быть получены
при использовании смеси, содержащей
20% NH4I и 80% S, при концентрации
меди 0,03—0,05%.
Суть усовершенствованного варианта
безгазового метода [30] состоит в том, что
необходимая для прокаливания электро-
люминофоров среда создается за счет
испарения галогенидов аммония и серы,
вводимых в шихту перед прокаливанием.
концентрации меди (рис. VI.5).
/,%
100 г
Для поддержания в течение синтеза задан-
ного состава газовой среды, прокаливание
ведут в сосудах с длинной горловиной,
выступающей из печи. Поскольку темпе-
ратура горловины низка, постольку гор-
ловина является зоной конденсации серы
и галогенидов аммония. Из горловины сера
стекает, захватывая галогениды аммония,
в зону прокаливания, где эти вещества
испаряются и таким образом регенери-
Л, нм
Рис. VI.5. Спектральный состав
излучения электролюминофора
ZnS - Си при различной концентра-
ции меди (атмосфера прокалива-
ния — NH4Br — 80% , S — 20%):
1~ 0,03%; 2 — 0,05%; 3 — 0,1%;
4 — 0,2%.
руется нужная атмосфера. Такой прием позволяет не только упростить техно-
логию, но и увеличить яркость электролюминесценции и уменьшить размер
кристаллов электролюминофора.
Рис. VI.6. Спектральное распределение
энергии излучения электролюминофо-
ров ZnS Си (0,03%) при различном
составе атмосферы прокаливания:
Состав
атмосферы,
масс. %
Состав
атмосферы,
масс. %
NH<I S
NH«I S
1 90 10
2 80 20
3 70 30
4 60 40
5 50 50
6 30 70
7 20 80
8 10 90
Наиболее простой метод получения электролюминофоров изложен в ра-
боте [31]. По этому методу шихту прокаливают в тиглях на воздухе. Для пред-
отвращения окисления в нее вводят серу, а сверху покрывают слоем угля. Ука-
занный метод позволяет получить электролюминофоры с зеленым и желтым
цветами свечения с повышенной яркостью и нужным гранулометрическим соста-
вом (промышленные электролюминофоры марок ЭЛ-525 и ЭЛ-590).
133
При непрерывной работе ЭЛК наблюдается уменьшение яркости свечения
со временем (старение). Для предотвращения этого электролюминофоры подвер-
гаю гея низкотемпературному отжигу на воздухе, что позволяет значительно
увеличить стабильность. На рис. VI.7 показано изменение яркости свечения
во врамени электролюмпнофоров зеленого цвета свечения различных промышлен-
ных марок. Наиболее стабильным является выпускаемый промышленностью
Рис. VI.7. Изменение во времени яркости
свечения ЭЛК, приготовленных с элек-
тролюминофорами различных марок
Кривая Марка электро- люминофора Время полуспада яркости свече- ния, ч
1 ЭЛС-510 80
2 ЭЛ-525 400
3 ЭЛ-516 5000
элек гролюминофор ЭЛ-516, который имеет время полуспада яркости более
5000 ч.
Электролюминофоры ZnS Си • Мн. Фрелих [32] показал, что электролюмино-
форы с интенсивным излучением в желто-оранжевой области спектра могут быть
получены при активации ZnS медью и
марганцем. Их можно синтезировать как
в атмосфере H2S + НС1, так и безгазовым
способом [30, 33]. Интенсивность свечения
и спектр излучения люминофоров зависит
от концентрации Си и Мп.
Как следует из рис. VI.8, при концен-
трации 0,1% Си и различном содержа-
нии Мп можно получить люминофоры, в
спектрах излучения которых имеются по-
лосы в синей, зеленой и оранжевой обла-
стях спектра. При увеличении концентра-
ции Мп до 1 % свечение люминофоров сос-
редоточивается в полосе с максимумом из-
лучения при 585 нм. Спектры излучения
элекгролюминофоров ZnS-Си-Мп зависят
также от условий возбуждения, поскольку
интенсивности свечения желтой, зеленой и
синей полос по-разному зависят от частоты
возбуждающего поля. В частности, при по-
вышении частоты сначала насыщается жел-
тая полоса . Особенно четко это видно на при-
мере электролюминофора ZnS-Си-Mn-I,
синтезированного «безгазовым» способом
[301 (в шихту вводили серу и иодид аммо-
Рис. VI.8. Спектральное распре-
деление энергии излучения элек-
тролюминофоров ZnS -Си -Мп при
различной концентрации мар-
ганца (указано на кривых).
Концентрация Си — 0,1%.
ния). При частоте возбуждения от 2 до 5 кГц этот люминофор имеет интенсив-
ное фиолетовое свечение, а при частоте 400 Гц — желтое [34]. Введение иодида
аммония способствует подавлению зеленой полосы [15]. Эффект перемены цвета
свойственен этому люминофору лишь при определенных концентрациях актива-
торов — Си и Мп, ответственных за синюю и желтую полосы излучения. Ха-
рактеристики ЭЛК, приготовленных с электролюминофорами, изменяющими
цвет свечения при изменении частоты возбуждающего напряжения, приведены
в работе [35]. Практическая ценность таких люминофоров в том, что их можно
использовать в многоцветных устройствах сигнализации и индикации.
134
На основе сульфида цинка, активированного Си и Мп, разработан одно-
компонентный электролюминофор с белым цветом свечения [36]. Белый цвет
излучения имеют электролюминофоры, содержащие от 0,04 до 0,07% Си и от
0,15 до 0,27% Мп; при этом заданной концентрации марганца должна соответ-
ствовать вполне определенная концентрация меди. В процессе непрерывной
работы эти электролюминофоры изменяют цвет свечения. Более стабильные
и яркие электролюминофоры с белым цветом свечения были получены на основе
смеси двухполосного электролюминофора с сине-зеленым цветом свечения,
который подвергался стабилизации [37], с электролюминофором с желтым
цветом свечения (ZnS-Си-Мп).
Электролюминофоры ZnS ♦ CdS • Си. Введение более 5% CdS в основу ZnS
электролюминофоров приводит к резкому спаду яркости свечения. Объясняется
это тем, что при добавлении Cd, во-первых, образуется структура вюрцпта,
и, во-вторых, выделяется фаза CdS-Cu2S (соединение темного цвета).
Рис. VI.9. Спектральное распределение
энергии излучения электролюминофоров
ZnS ZnSe-Си при различном составе ос-
новы:
Кривая Состав основы, % Кривая Состав основы, %
ZnSe ZnS ZnSe ZnS
1 10 90 5 80 20
2 30 70 6 90 10
3 50 50 7 100 ——
4 70 30
Леман [38] синтезировал эффективные электролюмипофоры на основе
ZnS-CdS-Си при добавлении в шихту подидов и бромидов. Полученные им
электролюминофоры, излучающие в зеленой области спектра, содержали
—-10% CdS; яркость свечения их на 20—30% превышала яркость свечения
обычных электролюминофоров. Препараты прокаливали при 720° в атмосфере
азота с добавлением серы. Эффективные электролюминофоры с содержанием
CdS )> 10% получали при добавлении иода и галлия. Первый препятствует
образованию CdS-Cu2S и способствует росту кристаллов, а второй смещает
спектр излучения в длинноволновую область. Таким способом удалось получить
электролюминофоры, содержащие 14% CdS, интенсивность свечения которых
не уступает хорошим образцам электролюминофоров с излучением в зеленой
области спектра.
Электролюминофоры на основе смешанных сульфидселенидов цинка и кад-
мия [19, 39—43]. На рис. VI.9 показано изменение спектров излучения при
постепенном замещении серы селеном в основе электролюмпнофора. При уве-
личении отношения Se : S спектры электролюминесценции сдвигаются в длинно-
волновую область.
Электролюминофоры на смешанной сульфидселенидной основе с интенсив-
ным свечением могут быть получены методом двухстадийного прокаливания [40],
по которому на первой стадии прокаливание ведут в атмосфере азота с добав-
кой HG1, а на второй — в атмосфере азота. Лодочку с исходной шихтой поме-
щали в кварцевую трубку, предварительно продутую смесью инертного газа
и НС1; затем вели прокаливание. Наилучшие по яркости свечения образцы полу-
чают при содержании в основе 30% ZnSe и концентрации Си, равной 0,08%.
Спектры излучения таких электролюминофоров приведены на рис. VI.10.
При увеличении содержания ZnSe происходит резкое уменьшение яркости
свечения. Однако при содержании 30% ZnSe может быть получен электро-
люминофор с зеленым цветом свечения, обладающий яркостью, не меньшей,
чем электролюминофор ZnS-Си. Достоинство рассмотренных электролю-
минофоров состоит в том, что они обладают большей яркостью свечения при
импульсном возбуждении, чем электролюминофоры ZnS-Си. Их высокая
135
яркость свечения обусловлена увеличением проводимости основы и, следова-
тельно, большей величиной поглощаемой энергии. Преимущество этих люмино-
форов при импульсном возбуждении, по сравнению с люминофорами на суль-
фидной основе, вызвано уменьшением глубины ловущек и их количества.
А, нм
Рис. VI. 10. Спектральное распределе-
ние энергии излучения различных элек-
тролюмпнофоров:
1 — ZnS (70)*ZnSe (30)-Cu; 2 — ZnS (80)*ZnSe
(20)• Cu; 3 — ZnS*Cu; 4 — ZnS (60)*ZnSe(40)*Cu.
ление энергии излучения электро-
люминофора с желтым цветом све-
чения на основе ZnS(40) • ZnSe(60) •
• Си *А1 (/ = 3000 Гц; U = 180 В).
Рис. VI.12. Зависимость яр-
кости свечения (7) и макси-
мума на кривой спектраль-
ного распределения энергии
излучения (2) от концентра-
ции серы в основе люминофора.
В последнее время эффективные электролюминофоры с желтым цветом
свечения были получены при прокаливании под углом шихты на основе
ZnS(40) • ZnSe(GO) • Cu • Al (рис. VI.11) [41]. Эти электролюмппофоры доведены
до промышленного внедрения и могут превы-
шать по яркости свечения электролюминофор
ЭЛ-580 — в 1,5 раза. В работе [43] детально
исследованы селенидные п цинккадмийсуль-
фоселенидные системы. Как следует из указан-
ной работы и работы [42], электролюмино-
' форы с ярким свечением могут быть получены
на основе Cdx)(Se] где х =
, = 0,05—0,15, а у = 0,05—0,5.
В качестве примера приведем электролю-
минофор с интенсивным свечением в красной
области спектра (Хтах = 650—680 нм), со-
держащий от 0,1 до 0,15 г-ат/моль Cd и от
0,2 до 0,45 г-ат/моль S. Яркость свечения и
спектры излучения электролюминофора опре-
деляются соотношением серы п кадмия
(рис. VI.12). На основе указанной системы
был разработан промышленный электролю-
минофор ЭЛ-650 [44]. Увеличить интенсив-
ность свечения этого люминофора удалось за
счет введения галлия [45]. Рецептура с при-
менением галлия была положена в основу
промышленного электролюмпнофора ЭЛ-670.
Спектр излучения последнего сдвинут еще
больше в красную область. Возрастание яркости электролюминесценции при
введении галлия, по-видимому, связано с увеличением растворимости меди
в основе. Влияние добавок серы на увеличение эффективности свечения селе-
нидных систем ряд последователей объясняют возникающими нарушениями
в кристаллической структуре электролюминофора, способствующими усилению
электролюминесценции [46]. Однако существенным для выяснения роли ZnS.
136
введенного в описанную систему, является то, что при этом увеличивается
наклон частотной характеристики электролюминофора и уменьшается темпе-
ратурное тушение при высоких частотах.
Стабильность свечения
электролюминесцентных конденсаторов
Яркость свечения ЭЛК в процессе их непрерывной работы уменьшается.
Общее число циклов действия
поля * 10 s
Наиболее сильно это выражено на начальных стадиях, а затем процесс замед-
ляется (рис. VI.13) [47, 48].
Величина спада яркости ЭЛК определяется свойствами электролюмино-
фора, способом приготовления ЭЛК и условиями возбуждения. Улучшение
стабильности электролюминофоров — весьма актуальная проблема и ей посвя-
щено значительное количество работ.
В интервале напряженностей поля
(1—3)-104В/см время полуспада яркости
ЭЛК не зависит от напряженности воз-
буждающего поля, но резко уменьшается
при увеличении частоты. Как следует из
экспериментальных данных, в диапазоне
частот 400— 10000 Гц изменение яркости
свечения можно представить «универсаль-
ной» кривой (рпс. VI.13), если по оси
абсцисс отложить общее число циклов
действия поля (величину, пропорциональ-
ную произведению времени действия на
частоту возбуждающего поля), а по оси
ординат — яркость свечения (в % от на-
чальной величины). Используя получен-
ную кривую и задавшись определенной ча-
стотой возбуждающего поля, можно найти
тот промежуток времени, за который яр-
кость свечения ЭЛК достигнет того или
Рис. VI. 13. Влияние общего чи-
сла циклов действия возбуждаю-
щего поля на стабильность элек-
тролюминесцентных конденсато-
ров.
иного уровня.
Старение ЭЛК сильно ускоряется при повышении температуры окру-
жающей среды и в присутствии влаги [49]. С повышением влажности воздуха
от 0 до 70% стабильность ЭЛК ухудшается незначительно; ее резкое снижение
приходится на интервал от 70 до 100%.
Стабильность электролюминофоров зависит от условий их синтеза. В ра-
боте [49] показано, что если электролюминофоры получены путем закаливания
на разных стадиях остывания, то стабильность их ухудшается по мере пониже-
ния температуры, до которой остывал образец. Справедливо это до темпера-
туры 400—500°; с повышением температуры остывания стабильность улучшается.
Стабильность повышается и при увеличении размеров кристаллов люминофора,
а также концентрации активатора [48].
На стабильность влияет природа вводимого галогена [50, 51]. Так, она
существенно улучшилась при введении брома. Подобным образом действует
и перецрокалцвание готовых электролюминофоров на воздухе [52, 53] и в парах
серы. Это было показано на примере ZnS-Си-Вт или ZnS-CdS-Си при обра-
ботке их в парах серы.
Отличительная черта электролюминофоров, полученных подобным обра-
зом,— независимость их стабильности от частоты. Результаты работы [54]
также подтверждают, что отжиг электролюминофоров в строго регламентиро-
ванных условиях температурного режима и состава атмосферы улучшает их ста-
бильность.
Процессы старения электролюминофоров сопряжены с изменением их физи-
ческих свойств [48, 50, 51, 55—58]. Эти изменения обусловлены:
образованием глубоких ловушек, что следует из кривых термического вы-
свечивания и изменения волн яркости состаренных люминофоров;
изменением наклона вольтяркостпой характеристики; как правило этот
наклон увеличивается;
137
изменением электрических свойств электролюминофоров; как правило,
при старении уменьшается проводимость фазы.
Глубокие ловушки для электронов в процессе непрерывной работы ЭЛК
в кристаллах электролюминофора могут создаваться за счет диффузии кисло-
рода. Возможно, что появляются они также из-за серных вакансий, диффунди-
рующих с поверхности кристаллов электролюминофора в объем. При отжиге
на воздухе серные вакансии заполняются кислородом, а это повышает стабиль-
ность. Однако в работах [55, 58] на КТВ при старении не обнаружено появле-
ния дополнительных максимумов. Веревкин [57] объясняет старение перемеще-
нием ионов меди из включений сульфида в объем кристалла. Миграция ионов
меди должна приводить к уменьшению напряженности поля около включений
фазы Cu2S. В работе [59] это объясняется «затуплением концов» включений
фазы Cu2S из-за образования экранирующего облака из ионов меди (см. стр. 139).
Возможно также, что старение обусловлено переходом фазы Cu2S в процессе
работы ЭЛК в менее проводящую фазу GuS. Экспериментально это подтверждено
данными работы [58].
Спектры ЭПР, снятые для состаренного, несостаренного электролюмино-
форов и для солей двухвалентной меди, подтверждают наличие в состаренном
электролюминофоре ионов двухвалентной меди. Электронно-микроскопическое
изучение ZnS-Си [56] подтверждает, что в процессе старения проводящие вклю-
чения, состоящие из Cu2S, превращаются в GuS.
Посерение электролюминофоров в процессе работы может быть объяснено
выделением элементарного цинка в результате восстановительно-окислитель-
ной реакции [60]. Для предотвращения этого предлагают покрывать зерна
электролюминофора окисью цинка или сульфидом кадмия.
Свечение порошкообразных люминофоров
при возбуждении постоянным полем
Интенсивное свечение при возбуждении постоянным электрическим полем
получено для электролюминофоров ZnS-Си-Мп, содержащих на поверхности
зерен фазу Cu2S. Свечение это было нестабильным и наблюдалось только в мар-
ганцевой полосе [33].
В работах английских исследователей [61, 62] указывается, что при стро-
гом контроле содержания активаторов и хлора можно получить весьма эффек-
тивные электролюминофоры, если осаждать на их поверхности медь химическим
способом. Согласно данным указанных работ, наибольшая яркость свечения
достигается при ~0,1—0,2% Си и —0,5% Мп. Лучшие образцы получаются
при одновременном осаждении судьфида цинка с активаторами, что способствует
более равномерному распределению их в объеме зерна люминофора.
Исследование осаждения меди на поверхности зерен [63] показало, что
наиболее эффективное свечение может быть получено тогда, когда слой Cu2S
наносят химическим способом, а не получают на поверхности зерен при форми-
ровании люминофоров. Интенсивность свечения зависит от концентрации меди
при данном гранулометрическом составе люминофора и от термической обработки
его после осаждения фазы Cu2S. Свечение при возбуждении постоянным полем
получено и для люминофоров ZnS-ZnSe. Mn, ZnS-GdSe-Mn, содержащих
фазу Cu2S.
Свечение возникает также при активации свинцом и редкоземельными акти-
ваторами — Er, Ти, однако в этом случае оно ослабляется в 10—100 раз. На-
иболее эффективные ЭЛК получены Вечтом [61]. Особенность таких ЭЛК —
пониженное (не более 5%) содержание органического связующего.
Наложение поля на ЭЛК создает условия для его формовки. Суть последней
заключается в том, что ток, идущий через ЭЛК, падает, а яркость свечения уве-
личивается. Экспериментальные данные показывают, что при этом происхо-
дит обеднение медью слоя, расположенного вблизи положительного элек-
трода. На основе наиболее эффективных электролюминофоров созданы ЭЛК,
которые при U = 100 В и / = 5 мкА-см-2 имеют яркость свечения —1000 кд/м2;
светоотдача составляет 0,5 лм/Вт, энергетический выход — 0,1—0,3%; время
спада яркости до 50% от начальной — 1000 ч.
138
Рассматриваемые электролюминофоры эффективны прн импульсном возбу-
ждении и при длительности импульса, равной нескольким микросекундам,
обладают яркостью большей, чем обычные. При этом коэффициент
дискриминации* достигает 500. Все сказанное позволяет эти электро-
люминофоры использовать в электролюминесцентных телевизионных экранах.
Механизм электролюминесценции
Электролюминофоры, возбуждаемые переменным полем. Возникновение
электролюминесценции впервые было объяснено Дестрио [64], который пред-
положил, что центры люминесценции могут возбуждаться за счет соударений
с электронами, ускоряемыми электрическим полем. Теория этого явления была
подробно развита Кюри [65], но она не смогла объяснить того обстоятельства,
что электролюминесценция возникает уже при сравнительно небольших напря-
женностях поля (порядка десятков киловольт на 1 см).
Пайпер и Вильямс [66] предполагают, что ударная ионизация центров
люминесценции происходит около барьера обеднения вблизи отрицательного
электрода, где возникает необходимая для этого большая величина напряжен-
ности поля. Электроны, участвующие в процессе ударной ионизации, освобо-
ждаются полем с уровней захвата. Однако эта теория рассматривает явления,
относящиеся к монокристаллам.
Для объяснения процессов, которые происходят в порошкообразных люми-
нофорах, помещенных в диэлектрик, Залм [3] предположил, что источником
электронов служит поверхностный слой Cu2S, покрывающий кристаллы элек-
тролюминофоров. При возбуждении электрическим полем электроны перехо-
дят из Cu2S к положительному концу кристалла и, соударяясь с центрами лю-
минесценции, ионизуют последние. При этом часть электронов может отгоняться
полем из области ионизации и захватываться на ловушках. Выключение поля
или перемена знака приводит к возврату электронов и рекомбинации их с цен-
трами излучения, в результате чего происходит излучение. Этим объясняются
волны яркости, о которых говорится на стр. 18.
В работах [67, 68] механизм электролюминесценции объясняется процес-
сом туннельного проникновения электронов, которое осуще-
ствляется из фазы Cu2S, находящейся на поверхности кристаллов ZnS.
Дальнейшее развитие представлений о механизме электролюминесценции
связано с исследованием под микроскопом свечения кристаллов электролюмино-
форов. В работах [59, 69—72] показано, что это свечение сосредоточено в отдель-
ных точках (или линиях). Предполагается [69], что светящиеся линии, наблю-
даемые под микроскопом, обусловлены линейными дефектами в кристаллах ZnS.
Так как свечение по длине линии неравномерно (ярче всего светится «голова»
линии), то, цо-видимому, начало линии находится в плоскости р—п-перехода.
Механизм электролюминесценции определяется двумя стадиями. На первой —
стадия активации — положительное напряжение приложено к п-области,
а отрицательное — к p-области. Это приводит к миграции электронов и дырок
из области р—n-перехода. Вторая стадия начинается при изменении знака
напряжения: дырки инжектируются в n-область, захватываются на линейных
дефектах и переносятся к центрам люминесценции. При рекомбинации электро-
нов с дырками происходит излучение.
Фишер [59] так же объясняет электролюминесценцию инжекцией носи-
телей. Используя представления Лемана [72], он предполагает, что проводя-
щие включения в кристаллах ZnS имеют линейчатую иглообразную форму
и основные явления «разыгрываются» около этих включений. При этом вводится
представление о биполярной природе инжекции носителей зарядов,
сущность которого заключается в следующем. При наложении поля определен-
ной полярности из одного конца проводящего включения в объем кристалла ZnS
выходят дырки, а из противоположного — электроны. Дырки захватываются
* Коэффициентом дискриминации называют отношение яркости свечения
при определенном напряжении к яркости свечения при напряжении, равном
половине данного.
139
центрами люминесценции, а электроны ловушками. При изменении полярности
знаки носителей, выходящих из концов проводящих включений, меняются.
Конец, из которого выходили дырки, при изменении знака поля будет поста-
влять электроны; последние могут рекомбинировать с дырками, находящимися
на центрах люминесценции. Предложенная модель позволила Фишеру интер-
претировать основные явления люминесценции: зависимость яркости свечения
от напряженности поля, величину светоотдачи, стабильность, изменение цвета
свечения электролюминофора при повышении частоты возбуждающего поля.
Механизм электролюминесценции, предложенный Верещагиным [7], осно-
вывается на следующих основных положениях:
1. Процессы электролюминесценции обусловлены ударной ионизацией,
которая приводит к умножению
еи зарядов.
2. На поверхности кристаллов
ZnS и внутри его образуются
энергетические барьеры, связан-
ные с возникновением нарушений
и выделением фазы Cu2S. Эти
барьеры появляются так же при
соприкосновении зерен электро-
люминофора друг с другом или
с электродами. При этом воз-
можна дополнительная инжекция
носителей, так как яркость све-
чения в этих случаях возра-
стает. Возбуждение центров лю-
минесценции происходит около
барьера; однако при рассмотре-
нии процесса следует принимать
во внимание падение напряжения
на барьере и в толще кристалла.
Это позволяет интерпретировать
ряд явлений электролюмин^рцен-
ции, например зависимость от
температуры (см. стр. 19).
Энергетическая модель зерна
электролюминофора, предложен-
ная Верещагиным [7, с. 108] в
отсутствие поля, показана на
рис. VI.14, а. При включении
поля (рис. VI.14, б) барьер у
катода включен в запирающем
Рис. VI. 14. Последовательность процессов
ионизации и рекомбинации в кристалле
с двумя барьерами [7].
Энергетическая схема кристалла в отсутствие
внешнего напряжения (а), после включения на-
пряжения (б) и после изменения его полярности
(в).
Внизу показана форма импульсов напряжения V
и временное положение световых пиков L (t —
время).
направлении. Вошедшие в него из электродов (или кристаллов) электроны за
счет ударной ионизации создают электроны, которые отводятся в правую часть
кристалла, и дырки. Последние отводятся в левую часть кристалла и могут
захватываться ловушками. После выключения поля дырки могут при возвра-
щении в область возбуждения захватываться центрами люминесценции. Элек-
троны после выключения поля также возвращаются в область возбуждения
и их рекомбинация с дырками дает вспышку около катода. Если знак поля
изменяется, то аналогичный процесс ударной ионизации протекает в правом
барьере (рис. VI.14, в).
Влияние диэлектрика. Как показано Робертсом [73], диэлектрическая
постоянная связующего диэлектрика в ЭЛК (сх) существенным образом влияет
на напряженность электрического поля Е2, приложенного непосредственно
к зерну электролюминофора, а значит и на яркость свечения ЭЛК. Формула,
устанавливающая это влияние, связывает величину Е2 с величиной напряжен-
ности внешнего поля Ег
р р ___________Sei________
2 1 26x4-62—у (е2—Sj)
где е2 — диэлектрическая проницаемость электролюмипофора;
v — доля объема, занятого электролюминофором.
Из приведенного соотношения следует, что при увеличении напряжен-
140
ность поля на зерне возрастает. Справедливость этой формулы в случае малого
содержания электролюминофора в диэлектрике (а) подтверждалась неодно-
кратно (см. например [74]): опытным путем было найдено, что при изменении 11
от 5 до 1000 [74] основное повышение яркости свечения приходится только на
промежуток до значений от 5 до 50. При увеличении от 50 до 1000, яркость
электролюминесценции увеличивалась всего на 30%.
Вебером [75] была предложена конструкция ЭЛК, состоящего из слоив
электролюминофора и диэлектрика. Автор показывает, что в этом случае интен-
сивность свечения ЭЛК гораздо больше, чем в случае однослойных ЭЛК. Для
повышения пробивного напряжения во многих ЭЛК создают дополнительный
слой диэлектрика. Расчетные формулы, позволяющие определить диэлектри-
ческие характеристики и распределение поля для таких гетерогенных систем,
приведены в работе [76]. Установлено, что максимальная яркость свечения ЭЛК
с дополнительным слоем может быть получена в том случае, когда связующее
имеет меньшую диэлектрическую проницаемость, чем дополнительный слой.
В работе [77] в качестве дополнительного слоя применяли рутил. При измене-
нии диэлектрическо!! проницаемости дополнительного слоя от 100 до 1000 яр-
кость свечения ЭЛК возрастала примерно в 2 раза.
Значительное повышение яркости свечения при низких напряжениях [78]
удалось получить у ЭЛК со стеклообразным диэлектриком, если в дополнитель-
ный (грунтовый) слой ввести соединение, повышающее его диэлектрическую
проницаемость. Вопросы, связанные с технологией формирования электролю-
минесцентных слоев, освещены в работе [79].
Электролюминофоры, возбуждаемые постоянным полем. Относительно
механизма электролюминесценции ЭЛК, светящихся в постоянном поле,, в на-
стоящее время нет установившегося представления. Основные предположения
сводятся к тому, что фаза Cu2S на поверхности кристаллов ZnS-Ми служит
источником носителей зарядов,"а это создает условия для гетероперехода Cu2S—
ZnS. Такие гетеропереходы для монокристаллов были исследованы в работе
Айвена и Кузано [80] и для пленок Cu2S-ZnS-Mn Власенко и Гергелем [81].
В последнем случае при включении поля в прямом направлении, когда элек-
троны идут в Cu2S, излучения не возникает, поскольку Cu2S не люминесцирует.
При включении поля в обратном направлении носители заряда из Cu2S пере-
ходят в ZnS-Мп, в котором в результате ударной ионизации решетки и марган-
цевых центров появляется излучение.
Вечт [62] так же считает, что в случае возбуждения люминофора ZnS-Мп
постоянным полем, фаза Cu2S на поверхности кристаллов ZnS-Мп обеспечивает
инжекцию носителей, а Мп, по-видимому, играет в процессе проводимости
ограниченную роль и участвует только в процессе излучения. Этот вывод
делается на основании того факта, что люминофоры, неактивированные Мп,
имели такую же вольт-амперную характеристику, что и люминофоры с Мп.
В этой же работе рассматривается вопрос о возможном механизме возбуждения
марганцевого центра. Автором было установлено, что люминофоры без Мп
также обладают слабой люминесценцией с максимумом свечения 575 нм, близ-
ким к излучению марганца (580 нм). Это позволило Вечту принять следующий
механизм. Излучение в люминофоре без Мп обусловлено рекомбинациец до-
норно-акцецторной пары, в которой акцептором является медный центр, распо-
ложенный на расстоянии 1,2 эВ от валентной полосы, а донором — вакансия
серы; энергетический уровень ее находится на расстоянии 0,3 эВ от полосы
проводимости. При ширине запрещенной зоны ZnS, равной 3,7 эВ, расстояние
между энергетическими уровнями донора и акцептора составит 2,2 эВ, что соот-
ветствует длине волны ~570 нм. Излучение донорно-акцепторной пары очень
слабое, но добавление больших количеств марганца приводит к передаче энер-
гии о т этой пары к марганцевым центрам и свечение усиливается.
Совместное действие электрического поля
и света на фото- и электролюминофоры
При совместном действии поля и света во многих фото- и электролюмино-
форах происходит как тушение, так и усиление свечения. Эти явления называют
электрофотолюминесценцией и фотоэлектролюми-
несценцией.
141
Из работ [82, 83] следует, что приложение сильного электрического поля
к некоторым фотолюминофорам в процессе затухания люминесценции приводит
к вспышке света. Например, приложение поля 20000 В/см при затухании люмп-
нофора вызывает вспышку, которая быстро спадает и процесс затухания про-
должается по обычному закону.
Дестрпо [84] наблюдал при наложении поля усиление свечения фотолюми-
нофора ZnS-CdS-Мп, помещенного в диэлектрик, возбуждаемого рентгеновыми
лучами. Увеличение или уменьшение интенсивности свечения при включении
поля оценивают величиной
= —^412—
где £?фэ — интенсивность свечения при совместном действии поля и света;
Вф — интенсивность фотолюминесценции;
Вэ — интенсивность электролюминесценции.
Для люминофора ZnS-CdS-Мп значение R возрастает при увеличении ин-
тенсивности возбуждающего света и напряжения и не зависит от частоты при-
ложенного напряжения. Длинноволновое излучение в спектре этого люмино-
фора усиливается, а коротковолновое — тущится. Дестрио [841 усиление объ-
ясняет тем, что поле ускоряет вторичные электроны, которые образуются при
возбуждении рентгеновыми лучами, а это приводит к дополнительному возбу-
ждению Мп-центров.
Усиление люминесценции при одновременном действии поля и света уста-
новлено также и для сублимированных пленок [85]. В этом случае люминофор
непосредственно соприкасался с электродами. Авторы интерпретировали уси-
ление свечения увеличением числа электронов, участвующих в процессе возбу-
ждения ионов Мп при облучении светом. Установлено усиление свечения при
облучении светом электролюминофоров ZnS ‘Си • G1 с зеленым и синим свечением.
Для получения эффекта необходимо присутствие хлора. Максимальная вели-
чина R составляла при возбуждении переменным полем 1,3, а при возбуждении
постоянным полем около 6. Однако при малых напряжениях как для постоян-
ного, так и для переменного поля наблюдалось тушение.
В работах [86, 87] эффект усиления отмечен для промышленных электро-
люминофоров с синим, зеленым и желтым цветами свечения при возбуждении
УФ-светом и переменным полем. Влияние света характеризовалось величиной:
“АВ = ВфЗЛ—(Вфп-^В^)
Исследование электролюминофора ZnS -Си с зеленым цветом свечения пока-
зало, что при малых напряжениях, когда нет электролюминесценции, происхо-
дит только тушение люминесценции,'а при повышении напряжения А5 пере-
ходит через нуль и яркость свечения начинает увеличиваться.
Авторы работы [87] нашли, что в области тушения АВ изменяется с напря-
жением по закону: В ~ e~b^v, т. е. процесс обусловлен действием электро-
нов, ускоряемых полем, как и в случае электролюминесценции. Тушение, по-
видимому, объясняется тем, что при действии света могут освобождаться пз
валентной полосы электроны, которые переходят на ионизованные центры све-
чения и препятствуют процессам излучательной рекомбинации.
Усиление электролюминесценции также подчиняется указанной зависи-
мости от напряжения и обусловлено тем, что освещение усиливает число носи-
телей, принимающих участие в процессе электролюминесценции.
Значение АВ зависит от интенсивности света: при малых интенсивно-
стях АВ возрастает линейно, а после перехода через максимум начинает умень-
шаться. Это объясняется тем, что при малых интенсивностях света растет число
электронов и, следовательно, увеличивается АВ. Однако одновременно с увели-
чением интенсивности падающего света возрастает падение напряжения в толще
кристалла и снижается падение напряжения на барьере, что, в конце концов,
приведет к снижению АВ.
Эта точка зрения подтверждает механизм фотоэлектролюминесценции,
предложенный Айви [88], который предположил, что кристалл электролюмино-
фора состоит из области, обладающей фотопроводимостью (занимает основной
142
объем кристалла), и области, включающей барьер, который обусловливает
электролюминесценцию. При освещении кристалла, к которому приложено
электрическое поле, сопротивление его падает (фотопроводимость) и напряже-
ние на этой части кристалла уменьшается, а на части, включающей барьер,
увеличивается, что и приводит к возрастанию интенсивности свечения.
VI.2. СВЕТОДИОДЫ
(НИЗКОВОЛЬТНЫЕ ЭЛЕКТРОЛЮМИНЕСЦЕНТНЫЕ ИСТОЧНИКИ СВЕТА)
В предыдущих разделах было показано, что порошковые электролюмпно-
форы позволили создать эффективные источники света, излучающие во всем
диапазоне видимого спектра. Однако для их возбуждения требуется напряже-
ние 100—220 В с частотой 400—3000 Гц. Развитие полупроводниковой техники
в направлении микроминиатюризации и снижения рабочих напряжений до еди-
ниц Вольт стимулировало работы по созданию инжекционных электролюмине-
сцентных источников света.
Интенсивное исследование инжекционной электролюминесценции началось
в 60-х годах, и спустя десятилетие в продажу поступили светодиоды, изгото-
вленные на основе фосфида и арсенида галлия, которые работают при напряже-
нии 1,5—3,0 В и токе ~10 мА.
В отличие от электролюминесцентных панелей с порошковыми электро-
люминофорами светодиоды — это точечные источники света специального на-
значения. Их используют в качестве дискретных и алфавитно-цифровых инди-
каторов и устройств для воспроизведения изображений, способных работать
в полупроводниковых схемах. Особенность светодиодов — их надежность (срок
службы 104—106 ч), небольшая потребляемая мощность (10-3Вт) и малая
инерционность (10~9—10~7 с) в сочетании с высокой яркостью свечения
(102—104 кд м-2) в зеленой и красной областях спектра.
Механизм возбуждения
инжекционной электролюминесценции
Основой светодиода является полупроводниковый диод (р—п или другой
инжектирующий переход). При пропускании тока через диод в электронную
и дырочную области перехода инжектируются неосновные носители тока (дырки
и электроны). Рекомбинация этих носителей с основными носителями тока (элек-
троны в n-области и дырки в p-области) сопровождается излучением.
Рассмотрим природу р—n-перехода. При легировании полупроводника
донорными и акцепторными примесями образуются области с электронной и ды-
рочной проводимостью. Например, при введении в кристалл фосфида галлия
примеси серы, замещающей фосфор, образуются донорные уровни, поскольку
у серы на один валентный электрон больше, чем у фосфора. Когда вводится
примесь цинка, замещающего галлий, то образуется акцепторный уровень,
так как у цинка на один валентный электрон меньше, чем у галлия. Вносимые
этими примесями избыточные электроны или дырки (недостающие электроны)
при комнатной температуре являются свободными, т. е. переходят с примесных
уровней в зону проводимости (электроны) или валентную зону (дырки). Стре-
мление носителей тока к равномерному распределению по кристаллу приводит
к тому, что часть электронов переходит на ближайшие акцепторы; это обедняет
электронами n-область и дырками p-область. На границе раздела возникает
отрицательный заряд со стороны р-области и положительный — со стороны
n-области (рис. VI.15). Эти заряды создают поле, препятствующее дальнейшему
движению электронов из п- в р-область. Собственно р—n-переход находится
в том месте обедненного слоя, где уровень Ферми пересекает середину запре-
щенной зоны.
Величина барьера А£, возникающего между п- и p-областями, определяется
уровнем легирования обеих частей кристалла, и при сильном легировании АЕ
приближается к величине Eg. Протяженность обедненного слоя называется
«толщиной перехода» и также определяется концентрацией легирующих при-
месей; толщина обедненной части уменьшается с ростом уровня легирования.
143
Электрическое поле, которое создается в слое обеднения, может быть очень
большим — от 104 до 106 В/см —при толщине 10"4—10"6 см. При прямом сме-
щении напряженность этого поля снижается, при обратном — увеличивается
пропорционально квадратному корню из напряжения до наступления пробоя.
Когда прямое смещение достаточно велико, чтобы позволить электронам
распределяться по зоне проводимости или дыркам по валентной зоне над пере-
ходом, возникает инжекционный механизм протекания тока, который возрастет
экспоненциально с напряжением.
Рис. VI.15. Схема р—«-перехода:
а — без внешнего напряжения (нулевое смещение); б — прямое смещение,
При пропускании через р—«-переход тока с плотностью ~10 А/см2 не-
основные носители инжектируются со скоростью 1024 см-3-с-1 в узкой области
вблизи перехода (1—2 мкм). При продолжительности жизни 10~9 с, такая ско-
рость генерации может обеспечить концентрацию носителей 1024-10- 9 =
= 1015 см-3, что значительно выше равновесной концентрации неосновных
носителей тока при комнатной температуре. Избыточные носители тока реком-
бинируют в пределах толщины перехода; при этом генерируется оптическое
излучение, а концентрация носителей снижается до равновесного уровня.
Материалы для светодиодов
Рис. VI.16. Спектры излучения светодиодов:
1 ~ GaP, N; 2 — SiC; 3 — GaAs0,eeP0,„; 4 — GaP—
Zn, О; 5 — ZnSe—Al, Mn.
Типы полупроводниковых соединений. Для создания эффективных свето-
диодов необходимы хорошо Люминесцирующие полупроводниковые соединения,
обладающие проводимостью как «-, так и р-типа.
Видимое излучение соответствует энергии квантов 1,8—2,7 эВ, поэтому
ширина запрещенной зоны этих полупроводниковых соединений должна быть
больше 1,8 эВ. Этим требова-
ниям отвечают соединения типа
AUiBv и AHBVI. Первые—ти-
пичные полупроводники, но
люминесциругот хуже, чем вто-
рые. Соединения AnBVI, на-
оборот, — прекрасные люмино-
форы, но обладают проводи-
мостью только одного типа
(либо электронной, либо ды-
рочной), что не позволяет со-
здать р—«-переход. Основные
свойства материалов для свето-
диодов приведены в табл. VI. 1.
Указанные в табл. VI. 1
соединения обеспечивают со-
здание светодиодов, излучаю-
щих во всем спектральном диа-
пазоне видимого излучения.
В табл. VI.2 и на рис. VI.16
144
Материалы для светодиодов [89]
Т а б л и ц а VI.1
Материал эВ Состояние разработки Особенности
GaN 3,5 Стадия исследования Нет p-типа. высокое напряжение возбуж- дения (20—100 В)
SiC 2,8—3,2 По л уп ромыш ленное производство Сложная технология из-за высокой темпе- ратуры (>2000°)
ZnSe 2,7 Стадия исследования Нет p-типа, среднее на- пряжение 5—15 В
ZnTe 2,23 То же —
AlxGaj_xP 2,26—2.45 » Сложность создания п-тппа; нестабиль- ность, высокая хими- ческая активность
GaP 2,26 Промышленное про- изводство Низкая эффективность в зеленой области
AlAs 2,16 Стадия исследования Нестабильность; высо- кая химическая актив- ность
In!_xGaxP 1,34-2,26 Первые успешные результаты Механические напряже- ния, плохая совме- стимость с подложкой по параметру кристал- лической решетки
Ini-XA1XP 1,34-2,45 Стадия исследования Механические напряже- ния, несовместимость с подложкой по пара- метру кристалличе- ской решетки
GaAsi-дРх 1,44-2,26 Промышленное про- изводство
AI^Gai -x As 1,44—2,16 То же —
приведены спектральные характеристики светодиодов. Фосфид галлия
и твердые растворы Gaj.xAl.^As и GaAsj.xPv применяют для изготовления
светодиодов с красным цветом свечения; карбид кремния, селенид цинка
и твердые растворы In^xGaxP — для получения желтого и зеленого излуче-
ний. Для светодиодов с зеленым свечением используют также теллурид цинка
и нитрид галлия; голубое, синее и ультрафиолетовое излучения получают
в основном с помощью нитрида галлия, хотя возможно также получить синее
и зеленое свечение с помощью редкоземельных люминофоров, возбуждаемых
ИК-излучением арсенида галлия.
Легирующие добавки. Донорные и акцепторные примеси, необходимые
для создания р—n-перехода, определяют не только величину и тип проводи-
мости, но и участвуют также в самом процессе излучательной рекомбинации.
В качестве доноров для соединений AJIiBv используют элементы VI группы —
Те, S и Se, а в качестве акцепторов — элементы II группы — Zn, Cd, Mg. В ка-
честве доноров и акцепторов (в зависимости от подрешетки замещаемого узла)
иногда используют элементы IV группы — Sn, Si и Ge.
Ю Заказ 44 115
Таблица VI.2
Спектральные характеристики различных светодиодов [90]
Материал Параметры основных полос излучения Средний коэф- фициент вид- ности, лм/Вт Характеристика цвета
S к >< СО ^Е полу- ширина, нм средняя энергия фотонов, эв Я А« ® Я Д’ as й dfg О О НЙКЯ о & S И О й Я Я д
GaP—Zn, О (красный) 698 93 1,76 20,7 632 100
GaP— Zn, О (только крас- ная полоса) 698 93 1,76 19,8 637 100
GaP, N (зеленый) 565 30 — 392 574 99,3
GaP, N (только зеленая полоса) 565 30 2,18 618 570 99,6
GaP, Zn (зеленый) 553 23 • • 284 563 98.0
GaP, Zn (только зеленая полоса) 553 23 2,24 647 ' 556 98,4
GaAsi-xPx (красный) 640 28 1,91 97,9 679 100
Gaj-xAlx-As (красный) 688 28 1,78 7,1 679 100
SiC (желтый) 590 155 1,97 293 582 98,3
ZnSe (желтый) 588 2,12 475 588 100
ZnTe (зеленый) 565 50 2,20 620 570 99,5
Оптимальная концентрация примеси определяете^ эмпирически. При этом
исходят из следующих соображений: концентрация легирующей добавки должна
обеспечить достаточно высокое количество излучательных центров и концен-
трацию носителей тока, способствующую высокой плотности возбуждения
и малым омическим потерям. Однако концентрация примеси не может быть очень
высокой, так как высокая концентрация носителей тока (1018—1019см"3) зна-
чительно повышает безызлучательные потери за счет передачи энергии рекомби-
нирующих пар свободному электрону или дырке (эффект Оже [91, с. 176—179]).
Кроме того, при высоком содержании примеси возможно выделение ее в виде
отдельной фазы. Обычно концентрацию доноров выбирают в пределах 1017—
101&, акцепторов 1017—1019см~3.'
Иногда для легирования используют «нейтральные» примеси. В случае
фосфида галлия вероятность излучательной рекомбинации может быть заметно
повышена, если межзонный переход будет идти через промежуточный нейтраль-
ный центр. Таким центром может быть азот, который является изоэлектронной
примесью, и пара Zn : О, которая также нейтральна. Концентрация азота в фос-
фиде галлия может достигать 1019 см-3, тогда как концентрация пар Zn : О не
превышает 1017 см-3. Малая концентрация центров свечения приводит к насы-
щению зависимости яркости от плотности тока уже при 10 А -см"2 в случае леги-
рованного цинком и кислородом фосфида галлия с красным свечением. Свето-
диоды с зеленым свечением, легированные азотом, не насыщаются вплоть до
100 А -см" 2.
Технология изготовления светодиодов
Процесс изготовления светодиодов складывается из следующих основных
этапов:
выращивание легированных монокристаллов в виде крупных слитков;
резка слитка на пластины, ориентированные по определенному кристалло-
графическому направлению; шлифовка, полировка и травление пластин;
создание р—n-перехода (легирование);
сборка светодиода (обеспечение надежности контактов и максимального
световывода, теплоотвода, герметизации и т. д.).
146
г
Все стадии отличаются от традиционных для полупроводникового производ-
ства, например кремниевых приборов. Особенно заметно отличаются две послед-
ние стадии.
Для создания р—n-перехода применяют два принципиально различающихся
способа: диффузию и эпитаксиальное наращивание *. Суть первого состоит во
внедрении, например, акцептора (цинка) из газовой фазы в поверхность моно-
кристаллической подложки с проводимостью гг-типа. В настоящее время диффу-
зию не используют как самостоятельный метод легирования, а применяют для
изготовления светодиодов только в
Последние заключаются в наращива-
нии слоя вещества с проводимостью,
например p-типа, на подложку (или
предварительно выращенный на ней
слой) с проводимостью n-типа. Для
этого используют кристаллизацию ве-
щества как из газовой, так и из жид-
кой фазы. Газофазная эпитаксия слу-
жит в настоящее время для получения
твердых растворов типа Gai-xAs.rP
и In1_AGaxP, а жидкофазная—
для выращивания GaP и Al^^Ga^As.
Именно применение жидкофазной
эпитаксии позволило резко улуч-
шить качество светодиодов и удеше-
вить их: с 1969 по 1972 гг. стои-
мость диодов уменьшилась в 40 раз,
их эффективность выросла в 20 раз,
а надежность — почти на 5 порядков
[90, с. 10].
Жидкофазная эпитаксия. Жид-
кофазная эпитаксия заключается в
кристаллизации вещества из раствора
сочетании с эпитаксиальными методами.
расплава в процессе его охлаждения
(рис. VI.17,а). Монокристаллическая
подложка 4 закреплена в одном
конце графитовой лодочки 2, а рас-
плав 3, например, галлия с легирую-
щими добавками 1 — в другом конце.
После насыщения расплава послед-
ними лодочку поворачивают; при
этом подложка заливается распла-
вом. Затем печь охлаждают с не-
большой скоростью (от долей градуса
до нескольких градусов в минуту).
Рис. VI. 17. Схема аппаратуры для
жидкофазной эпитаксии.
а — Однослойное наращивание: 1 — навеска
фосфора, мышьяка или легирующих добавок;
2 — графитовая лодочка; 3 — расплав галлия;
4 — монокристаллическая подложка; 5 —
держатель подложки; 6 — кварцевая труба,
б — Многослойное наращивание: 1 — непод-
вижные графитовые пластинки; 2 — ячейки
с расплавами различного состава; 3 — под-
ложка, укрепленная в подвижной графито-
вой пластине.
На этой стадии и начинается процесс кристаллизации, по окончании которого
лодочку снова поворачивают и подложка отделяется от расплава.
В случае выращивания фосфида галлия в качестве растворителя исполь-
зуют галлий с добавками цинка для получения p-слоя и теллур — для полу-
чения n-слоя. Кристаллизация начинается при 1060°. Процесс ведут в токе
водорода.
Эффективность светодиодов из фосфида галлия, легированного цинком
и кислородом, была повышена при наращивании двух слоев последовательно
на одной подложке: сначала кристаллизовали слой n-типа, а затем слой р-типа,
легированный цинком и кислородом из расплава галлия с добавками цинка
и окиси галлия. Схема устройства для многослойного наращивания показана
на рис. VI.17, б. Подобную технологию используют и для других материалов.
* Эпитаксия — это полное воспроизведение кристаллографической ориен-
тации подложки в выращенном на ней слое вещества. Различают автоэпитак-
сию — рост на одноименной подложке — и гетероэпитаксию — рост на хими-
чески отличающейся подложке.
10* к;
В ряде случаев при жидкофазной эпитаксии применяют методы погружения
и градиента температур. В первом случае подложка, укрепленная на держателе,
перемещающемся вертикально, может погружаться в ванну с раствором-распла-
вом. Вертикальное расположение позволяет вводить донорные и акцепторные
добавки последовательно в процессе кристаллизации и тем самым получить
р—n-переход путем одностадийного процесса. Метод градиента температур
заключается в том, что подложку и расплав поддерживают при неизменных,
но различных температурах (£ПОдл < красил) в течение всего процесса. За счет
температурного градиента идет перенос вещества из расплава к подложке, и про-
исходит рост слоя при фиксированной температуре. Это позволяет выращивать
слои сложных многокомпонентных
Рис. VI. 18. Схема аппаратуры для газо-
фазной эпитаксии:
1 — смеситель; 2 — лодочка с галлием; 3 — по-
дложка; 4 — кварцевый реактор.
соединений, состав которых резко
зависит от температуры кристал-
лизации.
Газофазная эпитаксия. Про-
цесс газофазного наращивания
вещества основан на реакциях
синтеза или перекристаллизации
за счет химического переноса, а
также на переносе вещества путем
испарения и конденсации. Один
из способов носит название сэн-
двич-метода (метод малых проме-
жутков). Он заключается в том,
что исходный порошкообразный
материал помещают на малом рас-
стоянии (0,1—1,0 мм) от моно-
кристаллической подложки, тем-
пература которой на 20—40° ниже,
чем у исходного вещества. В при-
сутствии/аза-носителя (например,
водяной пар, галогены) происхо-
дит кристаллизация вещества на
подложке с очень малыми потерями (коэффициент переноса достигает 90% от
массы исходного вещества).
Более совершенным и универсальным является метод, основанный на син-
тезе твердого вещества из летучих компонентов или их соединений. Легирующие
добавки вводят в виде газообразных соединений. Применение последних позво-
ляет очень точно и легко управлять дозировкой компонентов соединения и леги-
рующих добавок. В результате удается получать слои твердых растворов с пе-
ременным по толщине составом, что необходимо тогда, когда подложка и выра-
щиваемый материал плохо совместимы (по параметрам кристаллической решетки
и коэффициенту термического расширения). Например, в настоящее время мето-
дом газофазной эпитаксии синтезируют многие светодиоды с красным свечением
на основе твердых растворов GaAsr_xPA, причем по толщине слой может иметь
состав от х = 0, что соответствует подложке, до х — 0,4.
Принципиальная схема аппаратуры для газофазной эпитаксии за счет реак-
ций химического переноса показана на рис. VI. 18. Галлий транспортируется
в виде субхлорида, образующегося при пропускании хлористого водорода над
расплавом металла. Мышьяк и фосфор — в виде арсина и фосфина. Донорную
примесь (селен) вводят в виде селеноводорода. Иногда применяют теллур или
кремний в виде теллурорганических соединений и силанов. Акцептор (цинк)
поступает обычно за счет диффузии из пара уже после выращивания эпитакси-
ального слоя. Газом-носителем служит водород, очищенный пропусканием через
нагретый палладиевый фильтр. Скорость выращивания достигает 40 мкм/мин.
К достоинствам этого метода относится высокая чистота конечного продукта
и большая степень его однородности; кроме того, этот метод отличается про-
стотой, надежностью, производительностью, и, следовательно, экономичностью.
Недостаток метода — низкая степень использования исходных продуктов
(^3%), а также необходимость работы с токсичными веществами (гидриды
мышьяка, фосфора, селена и теллура). Схему, показанную на рис. VI.18, обычно
используют в лабораторных условиях. Для повышения производительности
148
в промышленных условиях сконструирован вариант с вертикальным располо-
жением аппаратуры, что позволяет поместить в реакционную зону большое
количество подложек, которые медленно вращаются для создания равноценных
условий кристаллизации.
В последние годы получены обещающие результаты по применению газо-
фазной эпитаксии для выращивания твердых растворов типа In1_xGa.rP («жел-
тые» и «зеленые» светодиоды).
Следует отметить, также, что подобная технология с успехом применена
для выращивания нитрида галлия на подложках из сапфира, который обладает
прозрачностью и хорошей теплопроводностью.
Свойства светодиодов различного типа
Фосфид галлия. Наибольшей эффективностью обладают светодиоды из фос-
фида галлия, полученные при жидкофазном выращивании. Освоено производ-
ство светодиодов типа GaP—ZnO с красным свечением с внешней квантовой
эффективностью 3%; рекордные величины эффективности (7—15%) были полу-
чены в лабораторных условиях [92]. Однако эти диоды были изготовлены при
низкой концентрации акцептора-цинка, за счет чего уменьшился коэффициент
внутреннего поглощения света, но появилось более раннее насыщение яркости
от плотности тока — ниже 1,0А-см~2. Диоды с 3%-ной эффективностью насы-
щаются при 10А -см-2. Внутренняя квантовая эффективность по расчетам дости-
гает 10—20% , и в дальнейшем можно ожидать как снижения стоимости, так и усо-
вершенствования технологии, но не повышения эффективности. Диоды на основе
GaP с зеленым цветом свечения находятся в стадии разработки. Эффективность
промышленных светодиодов не выше 0,1% при плотности возбуждения 10А -см-2.
Максимально достигнутые величины эффективности — 0,2% при 33 А-см-2
и 0,7% при 200 А-см-2 [93]. Следует отметить, что эффективность 0,1% может
быть получена также и у диодов, изготовленных методом газофазной эпитаксии.
Нитрид галлия. Нитрид галлия имеет запрещенную зону 3,5 эВ с прямыми
переходами и в принципе позволяет получить излучение во всем спектральном
диапазоне видимого излучения. В 1969 г. слои нитрида галлия были пригото-
влены путем эпитаксиального осаждения на сапфировую подложку [94]. До на-
стоящего времени получен только нитрид галлия низкого удельного электро-
сопротивления n-типа; попытка легировать нитрид галлия до р-типа (например,
цинком) приводит к образованию материала с высоким сопротивлением. Элек-
тролюминесценция была получена при приложении поля между двумя точеч-
ными контактами к высокоомному слою нитрида галлия, легированного цинком.
Излучение наблюдалось в ультрафиолетовой, голубой и зеленой областях
спектра.
Изготовлены также светодиоды, работающие на основе лавинно-инжек-
ционного механизма в i—«-переходах, при этом получена зеленая и голубая
люминесценция [94]. При комнатной температуре квантовая эффективность
для зеленого излучения выше 1% , но энергетическая эффективность значительно
ниже (0,1% для зеленого излучения и 0,005% для голубого) из-за большого
падения напряжения на переходе (20 В). Величина выхода, рассчитанная из
квантовой эффективности, достигала 12000 (кд -м-2)/(А -см-2) для зеленого излу-
чения. Интересное преимущество нитрида галлия заключается в том, что он
может быть выращен на сапфировой подложке, что значительно упрощает кон-
струкцию прибора, обеспечивает хороший теплоотвод и вывод излучения.
Диоды из нитрида галлия имеют высокую стабильность.
Твердые растворы на основе соединений AinBv. В табл. VI.1 (стр. 145)
приведены 6 типов твердых растворов на основе фосфидов алюминия, галлия
и индия, а также арсенидов алюминия и галлия. В настоящее время достигнуты
значительные успехи в разработке светодиодов на основе GaASj.x Рх — самого
дешевого материала для светодиодов, так как он легко получается методом газо-
фазной эпитаксии на подложках из арсенида галлия, который, в свою очередь,
является наиболее качественным и доступным материалом. Промышленный
выпуск светодиодов освоен в большом масштабе.
Светодиоды с %П1ах = 660 нм имеют максимальную эффективность 0,5%
(при 500 (кд-м~2)/(А-см-2)); оптимальное же значение — только 0,2%.
140'
Появились сообщения, что при легировании азотом можно получить эффективные
светодиоды с желтым и оранжевым цветами свечения с выходом 200 (кд •
• м~2)/(А-см~2) [95]. Ожидается, что дальнейшее исследование твердых рас-
творов в системе GaP — GaAs будет развиваться в направлении разработки
монолитных многоэлементных структур.
Значительные успехи достигнуты при исследовании арсенида алюминия
в качестве компонента твердого раствора. Несмотря на то, что это соединение
сильно подвержено гидролизу, в монолитном виде твердые растворы с составом
Al^^Ga^-As обладают стабильностью, вполне достаточной для практического
использования. Этот материал отличается полной совместимостью по параметру
решетки с GaAs. При х = 0,3 получено излучение с Хтах = 675 нм с эффектив-
ностью 1,3% и выходом 550 (кд-м“2)/(А-см-2); для А,тах= 750нм получен
выход—6%, а для гетероструктур типа GaAs — Gaj _ vAlvAs — даже
10% [96].
Наиболее перспективным материалом для получения светодиодов с зеленым
цветом свечения является твердый раствор In1_rGarP, так как он имеет прямо-
зонные переходы при Eg = 2,2 эВ. Светодиоды с желтым свечением на основе
In1-xGarP имеют светоотдачу 0,31 лм/Вт [97]. Для изготовления этого мате-
риала используют как жидкофазную эпитаксию, так и газофазную, причем
в последнем случае достигнуты заметные успехи: получена эффективность 0,2%
в красной области спектра (659 нм). Основная сложность, с которой приходится
сталкиваться, — это плохая совместимость с материалом известных подложек.
Твердые растворы типа In1_xAlvP и AlrGa^rP также весьма перспек-
тивны, но пока только показано, что они имеют высокую эффективность катодо-
люминесценции. Эффективность электролюминесценции для излучения 550 нм
в настоящее время не превышает 10-3% .
Светодиоды на основе арсенида галлия с покрытием из люминофоров, воз-
буждающихся ИК-излучением [98]. Арсенид галлия — технологически наи-
более разработанный материал, а диоды на его основе имеют самый высокий
к. п. д., однако они излучают в невидимой области (900—1000 нм).
ИК-излучение арсенида галлия может быть преобразовано в видимое с по-
мощью так называемых «антистоксовских» люминофоров (см. раздел IV.4),
фторидов и оксисульфидов р. з. э. Эти люминофоры возбуждаются в области
900—1000 нм и излучают в красной, зеленой и голубой частях спектра. Таким
образом, используя диоды из арсенида галлия, покрытые «антистоксовскими»
люминофорами, можно получить видимое излучение во всей спектральной
области. Такие светодиоды имеют ряд особенностей. Они излучают в узкой
полосе, характерной для редкоземельных ионов Ег3+ (зеленое и красное свече-
ние) и Тт3+ (синее), и имеют степенную зависимость яркости свечения от плот-
ности тока. Поэтому высокая эффективность может быть достигнута только
при очень больших плотностях тока.
При использовании диода GaAs, Si, имеющего эффективность 10%, были
получены светодиоды, излучающие в красной области с к. п. д. — 1% при плот-
ности тока 300 А-см-2, светодиоды, излучающие в зеленой области с к. п. д.
— 0,1%, и в синей — с к. п. д. — 0,01%. При плотности тока—10 А-см-2
эффективность светодиодов с люминофорным покрытием очень низка.
Карбид кремния. На основе карбида кремния (Я-политип) при легировании
бором получены светодиоды с излучением в желто-зеленой области спектра
(520 и 590 нм) с яркостью свечения — 200 (кд • м~2)/(А см-2), что соответствует
эффективности в несколько сотых процента. Максимальная величина внешней
квантовой эффективности достигает 0,1—0,3% .
Кристаллы карбида кремния выращивают при 2500° в графитовых тиглях;
при легировании азотом получается материал и-типа. р — n-Переход создается
путем диффузии акцепторов (бор, алюминий) при 2200°. Разработаны также
методы жидкофазной эпитаксии карбида кремния из растворов-расплавов в раз-
личных металлах (например, в кремнии) при температуре выше 1500°. Сложная
и трудоемкая высокотемпературная технология получения кристаллов и
р — п-переходов на карбиде кремния делает этот материал очень дорогим, по
сравнению с соединениями AmBv. Однако карбид кремния отличается очень
высокой химической и механической стойкостью, а светодиоды на его основе —
отсутствием спада яркости в процессе эксплуатации в течение 25 000 ч даже
при 200—400° при плотности тока 20 А -см~2 [90, с. 61 — 67].
150 »
Светодиоды на основе соединений AnBVI. Халькогениды цинка и кадмия
обладают широкой запрещенной зоной с прямыми переходами, а также имеют
высокую квантовую эффективность фотолюминесценции (0,3—0,9%) при ком-
натной температуре даже при малых плотностях возбуждения и довольно высо-
кую для широкозонных соединений концентрацию и подвижность носителей
тока; поэтому соединения AnBVI интенсивно исследуют как материал, перспек-
тивный для изготовления светодиодов.
Величина и тип проводимости соединений AnBVI. Величина и тип прово-
димости халькогенидов цинка и кадмия, в отличие от соединений AinBv, опре-
деляются не только концентрацией и видом примесей, но в значительной мере
и степенью отклонения их состава от стехиометрического. Так, сульфид и селенид
цинка могут быть получены с высокой концентрацией носителей тока только
при давлении пара цинка, превышающего равновесное давление диссоциации
халькогенидов при данной температуре.
Для халькогенидов цинка и кадмия характерна проводимость только одного
типа. Исключением является теллурид кадмия, который получен с высокой
проводимостью как п-, так и p-типа. Теллуриду цинка присуща проводи-
мость только p-типа, хотя сообщалось о получении при легировании галлием
в неравновесных условиях высокой проводимости п-типа[99, гл. 3—4]. Осталь-
ные материалы имеют проводимость только п-типа.
Невозможность изменения типа проводимости соединений AnBVI с широ-
кой запрещенной зоной путем легирования соответствующими примесями объ-
ясняется явлением электрической самокомпенсации, т. е. захватом носителей
внедренной примеси противоположно заряженным решеточным дефектом (на-
пример, вакансией [100, гл. 5]). Если энергия, выигрываемая при захвате-
носителей, больше энергии образования вакансий, то процесс внедрения при-
меси будет сопровождаться образованием вакансии. Энергия образования пос-
ледних оказывается тем больше, чем выше энергия атомизации кристалла,
величина которой, в свою очередь, уменьшается с ростом ионности связи и раз-
мера атома. С другой стороны, энергия, выигрываемая при компенсации, тем
больше, чем шире расстояние между уровнями донора и акцептора (т. е. чем
больше ширина запрещенной зоны и чем меньше глубина уровня компенсиру-
ющего дефекта). Отсюда ясно, что самокомпенсация более вероятна в соедине-
ниях с широкой запрещенной зоной и с ионной связью и менее вероятна в кова-
лентных соединениях с узкой запрещенной зоной.
Для арсенида галлия (Eq = 1,42 эВ, связь — ковалентная) явление само-
компенсации практически отсутствует; для сульфида цинка (Eq = 3,7 эВ и связь
преимущественно ионная) самокомпенсация очень сильно выражена.
Кроме явления самокомпенсации необходимо учитывать и другие факторы,
мешающие проявлению электрической активности вводимых примесей. Напри-
мер, растворимость ряда электрически активных примесей может быть очень
низкой. Некоторые акцепторные примеси могут, видимо, перераспределяться
между донорными и акцепторными состояниями, как это наблюдалось для
серебра в сульфиде кадмия и лития в теллуриде цинка [101]. Отмечалось, что
галогены, которые должны бы проявиться как доноры, оказываются неактив-
ными из-за образования комплексов со случайно присутствующими примесями
(например, с натрием, стронцием, железом) [102].
В обзоре по самодиффузии в соединениях AnBVI [103] показано, что при
обычных температурах прокаливания (^ 700°) большинство соединений типа
AnBVI содержит большие количества междуузельных атомов металла и халько-
гена. Эти междуузельные образования электрически неактивны при комнатной
температуре, но могут, по-видимому, взаимодействовать с вводимыми донорами
и акцепторными примесями, образуя атомнодиспергированные включения вто-
рой фазы.
Теллурид цинка [99]. Для изготовления р—«-перехода использовали моно-
кристаллы ZnTe p-типа с избытком теллура, а также легированные Li, Cs, Р.
Концентрация носителей тока в р- ZnTe достигала 3 -1018 см-3. Область с про-
водимостью n-типа создавали путем введения комплексов галлий-кислород
«неравновесным» методом: резким охлаждением расплавленного слоя ZnTe
с примесью окисных соединений галлия. Для этого на поверхность кристалла
p-типа наносили коллоидный слой гидроокиси галлия, а пластину устанавли-
вали на хладопровод и нагревали электронно-плазменным лучом, образованным
151
с помощью полого катода. В качестве рабочей среды использовали смесь газов
из кислорода и аргона с добавками или чистый аргон. Под воздействием луча
мощностью 105 Вт/см2 почти мгновенно происходит растворение окислов
галлия в расплавленном слое ZnTe. Это состояние «закаливается» при резком
охлаждении после выключения луча.
Концентрация носителей тока в n-области достигала 101" и 1018см~3 с по-
движностями 1200 и 800 см2/(В -с). Контакты к р-стороне готовили вплавлением
сплава Ан (60%), РЬ (38%), Те (2%) в атмосфере водорода при 500° в течение
3—5 мин; контакты к «-области были из эвтектического сплава А1 — In.
Спектр излучения при прямом
565 нм (2,20 эВ), а при 77 К — из од
Рис. VI. 19. Спектры фото- п элек-
тролюминесценции ZnSe—Al, Mn:
1 — (298 К), фотолюминесценция; 2 —
<77К); 3 — (298 К), 4 (77 К) — электро-
люминесценция.
смещении (300 ) состоял из узкой полосы
ой основной полосы 535 нм (2,32 эВ) и двух
дополнительных (512 и 547 нм). Внешняя
квантовая эффективность светодиодов воз-
растала с концентрацией акцепторов вплоть
до 5 • 10_ 8 см~3 и достигала 0,22% при ком-
натной температуре и 1,5% при 77 К; внут-
ренняя квантовая эффективность состав-
ляла 4,9% при 300 К и 50—70% при
70 К (при 20 А-см-2). При коэффициенте
видности 650 лм/Вт для полосы 565 нм это
соответствует светоотдаче 1,4—1,5 лм/Вт
или 30 лм/А.
Селенид цинка. В 1972 г. появились
три сообщения об изготовлении эффектив-
ных светодиодов на основе селенида цинка
[109—111]. Сам селенид цинка обладал
высокой проводимостью «-типа (удельное
электросопротивление 0,03—0,1 Ом-см-1)
и был легирован Мп и А1. Такой материал
получали путем выращивания слоя селе-
нида цинка методом жидкофазной эпи-
таксии из раствофа в жидком спла-
ве Zn (87%) — Sn(13%) с добавками
Мп и А1.
Запорный контакт создавали либо ва-
куумным напылением, либо химическим
осаждением слоя золота. В качестве омического контакта использовали впла-
вленный индий. Свечение наблюдалось у золотого контакта, когда он был отри-
цательным.
Спектры фото- и электролюминесценции селенида цинка, легированного А1
и Мп, приведены на рис. VI. 19. Для фотолюминесценции характерна полоса
510 нм. В отличие от фотолюминесценции, электролюминесценция проявляется
только в полосе марганца (588 нм).
Спад интенсивности свечения в полосе 588 нм после прекращения возбужде-
ния происходит до 50% за 10“4 с, что характерно для марганца. Коэффициент
видности для полосы излучения светодиодов составляет 475 лм/Вт; чистота
цвета — 100%.
Вольт-амперные и вольтяркостные характеристики светодиодов могут быть
«писаны степенной функцией с показателем — 8 в области 15—20 В. Напряже-
ние «пробоя» может изменяться в пределах от 7 до 20 В в зависимости от типа
контакта и уровня легирования кристалла. С ростом тока яркость свечения
изменяется в этом интервале напряжений почти линейно. Максимальная вели-
чина квантовой эффективности достигает 0,12%. При напряжении 15 В это
соответствует энергетической эффективности 0,017% и светоотдаче
0,082 лм/Вт.
Высокая яркость свечения может быть получена при довольно низкой плот-
ности тока — 800 кд м-2 при 3 мА-см“2 и — 2400 кд м-2 при 10 мА с.хг2
(1,2 лм/А). Эти величины вполне сравнимы с лучшими данными для зелено-
излучающего фосфида галлия и превышают его средний уровень [1600
(кд • м~2)/(А - см-2)].
Как уже отмечалось, величина энергетической эффективности поверхно-
стно-барьерных светодиодов не превышает сотых долей процента. Теоретическая
152
Основные параметры светодиодов [89]
Т а б л и ц а VI.3
Материал Промышленное производство Цвет S я * со ^Е Коэффициент вид- ности, лм/Вт о ч го И св «2 9 д’ О о «а «>ь ₽ й в, (кд .м-2)/(А .см-2)
GaP —ZnO Да Красный 690 20 3 —(15?) 1 200
А1о,зОао>?А8 Нет » 675 16 1,3 480
GaAs0,6P0,i Да » 660 42 0,5 400
GaAs, Si YOC1 — YbEr Нет » 660 30 2/1,0 25
Ill0,42Ga0>58P Оранжевый 617 284 0,1 1 06Q
GaAso,25Po,75 • N Да Оранжево- желтый 610 342 0,04 32а
SiC » Желтый 590 515 0,003 34
ZnSe —Al, Mn Нет » 588 475 0,12 2 40а
ZnTe —P, Ga » Зеленый 565 650 0,2 эзоа
GaP — N Да » 550 677 0,05-0,7 1 610
GaN Нет » — .— 1,0 12ооа
GaAs—Si+ ] » 550 660 0,1 70
YFo—Yb, Er) Голубой 470 60 0,01 0.3
величина, полученная на основе ряда допущений и предположений, составляет
10% в случае селенида цинка и 1—2% для сульфида цинка [105].
Сравнение свойств различных светодиодов (табл. VI.3). Величины выхода
и эффективности светодиодов взяты из оригинальных работ, а также рассчитаны
по уравнению, приведенному на стр. 15, если приводимые авторами данные
трудно непосредственно сравнивать между собой.
Из приведенных в табл. VI.3 данных видно, что в настоящее время достиг-
нуты значительные успехи в создании светодиодов с красным цветом свечения.
Энергетическая эффективность светодиодов в зеленой области низка и не пре-
вышает долей процента, но за счет коэффициента видности яркость этих диодов
превышает величину, характерную для высокоэффективных красноизлучающих
светодиодов. Разработка светодиодов с голубым свечением находится только
в начальной стадии.
Следует обратить внимание на светодиоды из нитрида галлия и селенида
цинка. Они имеют низкую энергетическую эффективность, так как работают
при напряжении 10—20 В, что значительно выше величины кванта излучения
(~ 2 эВ), но при плотности тока 0,1—1 А см-2 их яркость свечения в зелено-
желтой части спектра не уступает «зеленым» светодиодам GaP, N.
В табл. VI.3 включены данные для селенида и теллурида цинка, которые
являются представителями соединений AnBVI. Эти соединения рассматриваются
в настоящее время как перспективные основы для светодиодов, излучающих
в видимой области спектра при комнатной температуре.
В табл. VI.4 приведены основные параметры светодиодов на основе халько-
генидов цинка и кадмия.
Яркость свечения большинства светодиодов при комнатной температуре
достигает 10—20 кд/м2 при плотности тока 1 А/см2, но при этом эффективность
очень низка « 10" 3%).
При температуре жидкого азота квантовая эффективность резко возрастает
и достигает нескольких процентов (2—18%). Основная причина низкой эффек-
тивности при комнатной температуре — неудачи в создании переходов, обеспе-
чивающих высокую плотность возбуждения, достаточную для преодоления
температурного тушения.
153.
Таблица VI.4
Свойства светодиодов на основе соединений AnBVI при 77 К
Материал и ‘хвшу Напряжение, В при 0,1 А "См-2 Эффективность Светоотдача, лм/Вт Литература
(квант/электрон)х X ю* f)' /V
CdTe 850 1,5 12 12 10-5 109
Znx-xCdxTe 700 1,8 6,0 6,0 0,15 110
Mgi-xCd/Te 680 2,0 - ' —. - —и 108
ZnSei_xTex 620 50,0 18.0 1,0 2,0 107
ZnTe —Ga, Li,P 535 2,3 1,5-2 1,5—2 14 99
ZnTe—Ga, Li, P,(300 K) 565 (при 20 А - см'2) 0,22 0.2 1,4
ZnSe —Al, Mn, (300 K) 588 15 0,12 0,017 0,08 104
Можно выделить два пути решения этой проблемы:
разработка методов, обеспечивающих высокий уровень легирования Ar*BVI
па основе новых технологических приемов;
разработка светодиодов, в которых высокая плотность возбуждения дости-
гается за счет лавинно-инжекционного или ударно-ионизационного механизма
возбуждения.
Первые результаты в этих направлениях получены при создании р — п-пере-
хода на ZnTe и поверхностно-барьерного светодиода Ап — (ZnSe : Al, Mn).
Стабильность светодиодов [90]. Эффективность большинства светодиодов
уменьшается в процессе эксплуатации при плотности тока — 10 А -см~2. Заме-
чено также «старение» в процессе хранения (без эксплуатации). Оба процесса
«деградации» связывают с внешними факторами — с наличием поверхностных
загрязнений, трещин, царапин, а также с разогревом при эксплуатации. Вну-
тренние факторы, влияющие на процесс «деградации», не совсем ясны. Уста-
новлено, что «старение» связано с появлением безизлучательных центров ре-
комбинации в области объемного заряда. Природа этого безызлучательного
канала неясна. Кроме того, в процессе «деградации» мало изменяется число
центров люминесценции и не появляются новые центры свечения с энергией
переходов более 1 эВ. Заметная деградация наблюдается, как правило, при
прямом смещении.
Спад яркости свечения возрастает с увеличением температуры диода, при-
чем степень спада пропорциональна полному заряду, прошедшему через диод.
Время полуспада экспоненциально зависит от температуры; энергия активации
процесса старения, полученная из такой зависимости, заметно уменьшается
в присутствии ионов меди.
Деградацию удается сильно замедлить с помощью пассивации (окислением
при химическом травлении перекисью водорода или хлорноватокислым на-
трием). Известно, что начат выпуск светодиодов на основе GaP и GaAs^^P^
со сроком службы 105—106 ч. Наилучшей стабильностью обладают светодиоды
на основе карбида кремния.
Потребность в светодиодах постоянно возрастает и предполагается, что
объем их производства в ближайшие годы составит ~ 10% от общего коли-
чества всех индикаторных устройств различного типа (электронно-лучевые
трубки, плазменные и жидкокристаллические индикаторы).
154
Параметры светодиодов за ближайшие 25 лет должны быть значительно
улучшены и по прогнозам их светоотдача должна приблизиться к 70 лм/Вт.
В табл. VI.5 приведены для сравнения величины светоотдачи различных свето-
диодов и порошковых электролюминофоров.
Таблица VI.5
Выход электролюминесценции (лм/Вт) для различных материалов [111]
Материал Лучшие величины на 1972 г. Практический предел за 1980 г. для подложек
непро- зрачная плоская прозрач- ная плоская кониче- ская форма
ZnS • Си (зеленый) 14 14 14 14
ZnS • Cu • Mn 1.5 — — —
GaP (красный) 2,1 2,1 5,25 15,8
GaP (желто-зеленый) 1,5 1,5 25 75
GalnP (желтый) 0,31 7,8 23 70
GaAl, As (красный) 0,13 0.80 2,3 7,0
GaAs, P (красный) 0,10 2,5 7,5 22,0
Из табл. VI.5 следует, что разработка светодиодов находится только в на-
чальной стадии, тогда как выход порошковых люминофоров близок к пределу.
Теоретическая величина энергетической эффективности «предпробойной» элек-
тролюминесценции составляет .10—30, тогда как реально получено 3—5%.
При инжекционном механизме возбуждения электрическая энергия может быть
превращена в световую без потерь, и энергетическая эффективность электро-
люминесценции может достигать 100%.
Реально, однако, существует ряд принципиальных ограничений. Например,
из-за большого коэффициента преломления материала светодиодов, значитель-
ная доля излучения отражается от границы кристалл — воздух и поглощается
внутри кристалла.
ЛИТЕРАТУРА
1. Лосев О- В. «Телеграфия и телефония», 1923, т. 18, с. 61—65. — 2. De-
striauG., Ivey Н. Proc. IRE, 1955, v. 43, p. 1911—1940. — 3. Zalm P. Phil. Res.
Repts, 1956, v. 11, p. 353—451. — 4. Пайпер В., Вильямс Ф. Усп. физ. наук,
1960, т. 70, с. 621—677. — 5. Фок М. В. Там же, 1961, т. 75, с. 259—261. -
6. Хениш Г. Электролюминесценция. Пер. с англ. Под ред. М. М. Горшкова.
М., «Мир», 1964. 455 с. — 7. Верещагин И- К. Электролюминесценция кри-
сталлов. М., «Наука», 1974. 279 с. — 8. Веревкин Ю. Н. Электролюмине-
сцентные устройства в судовой автоматике. Л., «Судостроение», 1966. 150 с. —
9. Деркач В. П., Корсунский В. М. Электролюминесцентные устройства. Киев,
«Наукова Думка», 1968. 302 с. — 10. Казанкин О. Н., Лямичев И. Я., Нико-
лаев Ю. И. и др. Прикладная электролюминесценция. М., «Советское радио»,
1974. 413 с.
11. Агранат Б. Л., Вайсберг Ф. Я., Гуревич Л. А. Электролюминесцентные
покрытия и их применение в технике. Л., изд. ЛДНТП, 1965. 26 с. — 12. Со-
лодкин В. Е., Талъвисте Э. К. Изв. вузов. Физика, 1968, № 1, с. 139—143. —
13. Варгин В. В., Гуторова Л. Л., Мазурин О. В. и. др. Стальные эмалирован-
ные электролюминесцентные панели. Вып. 8. Л., «Знание», 1968. 20 с. —
14. Бунделъ А. А., Вишняков А. В. ФТТ, 1966, т. 8, № 1, с. 115—120. —
15. Казанкин О. Н. Автореф. канд. дисс. Л., ГИПХ, 1964. — 16. Destriau G.f
Soddy J. J. Phys. Radium, 1945, v. 6, p. 12—16. — 17. Homer H. H., Ru-
lon R. M., Butler К. H. J. Electrochem. Soc., 1953, v. 100, p. 566—571. —
18. Орлов И. H. Изв. АН СССР. Сер. физ. 1957, т. 21, с. 731—741. — 19. Heg-
gyi I. I., Larach S., Shrader R. E. — J. Electrochem. Soc., 1957, v. 104, p. 717—
721. — 20. Wachtel A. Ibid., v. 107, p. 602—608.
155
21. Данилов В. П., Меркулова В. В. Авт. свид. 258500; Бюлл. изобрет.,
1972, № 5, с. 64. — 22. Казанкин О. Н., Пекерман Ф. М., Петошина Л. Н,
Изв. АН СССР. Сер. физ., 1957, т. 21, № 5, с. 721—731. — 23. Froelich Н. С.
J. Electrochem. Soc., 1953, v. 100, р. 280—288; р. 496—507. — 24. Wendel G.
Z. phys. Chem., 1960, Bd. 215, S. 80—91. — 25. Кочугова E. И., Пекерман Ф. M.,
Гуревич И. М. В кн.: Химия и технология люминофоров. Под ред. Л. Я. Мар-
ковского. Вып. 53. Л., «Химия», 1966, с. 35—45. — 26. Short М. A., Ste-
ward Е. G., Tomlinson Т.В. «Nature», 1956, v. 177, р. 240. — 27. ВallentyneD. Н.
J. Electrochem. Soc., 1960, v. 107, р. 807—810. — 28. Казанкин О. Н., Сер-
геев В. П. Авт. свид. 154341; Бюлл. изобрет., 1963, № 1, с. 40. — 29. Казан-
кин О. Н., Сергеев В. П., Григорьева Т. Н. Авт. свид. 156635; Бюлл. изобрет.,
1963, № 16, с. 41. — 30. Осипов В. А., Казанкин О. Н., Григорьева Т. Н. и др.
Авт. свид. 390126; Бюлл. изобрет., 1973, № 30, с. 84.
31. Казанкин О. Н., Дихтер М. А., Паранин Г. А. и др. В кн.: Материалы
5-го Всесоюзного совещания по электролюминофорам. Ставрополь, изд. ВНИИЛ,
1974, с. 74—77. — 32. Froelich Н. С. JOSA, 1953, V. 43, р. 320. — 33. Казан-
кин О. Н., Пекерман Ф. М., Петошина Л. Н. Опт. и спектр., 1959, т. 7,
с. 776—779. — 34. Дихтер М. А., Казанкин О. Н., Петошина Л. Н. и др.
В кн.: Химия и технология люминофоров. Под ред. Л. Я. Марковского. Вып. 70.
Л., «Химия» 1974, с. 24—30. — 35. Ведехин А. В.. Ковалев Б. А., Дани-
лов В. П. В кн.: Люминесцентные материалы и особо чистые вещества. Вып. 3.
Ставрополь, изд. ВНИИЛ, 1970, с. 52—56. — 36. Петошина Л. И., Дих-
тер М. А., ДаниловВ. П. идр. «Светотехника», 1966, № 9. с. 14—17. — 37. Па-
ранин Г. А., Казанкин О. Н., Петошина Л. Н. и др. В кн.: Сборник рефератов
по химии и технологии люминофоров. Под ред. Л. Я. Марковского. Л., изд.
ГИПХ, 1972, с. 22—23. — 38. Lehmann W. J. Electrochem. Soc., 1963, v. 110,
p. 753—766. — 39. Казанкин О. H., Пекерман Ф. М., Петошина Л. Н. Опт.
и спектр., 1959, т. 6, с. 672—677. — 40. Кужелев Л. П., Максимова И. А.,
Миронов И. А. и др. В кн.: Электролюминесценция твердых тел и ее применение.
Киев, «Наукова думка», 1972, с. 134—140.
41. Паранин Г. А., Рыжкин Ю. С., Певцова Н. И. др. В кн.: Химия
и технология люминофоров. Под ред. О. Н. Казанкина и Л. Н. Петошиной.,
изд. ГИПХ. 1974 с. 33—36. — 42. Миронов И. А., Марковский Л. Я. Опт.
и спектр., 1964, т. 16, с. 285—289. — 43. Миронов И. А. Автореф. канд. дисс.
Л., ГИПХ, 1964. — 44. Миронов И. А., Марковский Л. Я. Авт. свид. 186057;
Бюлл. изобрет., 1966, № 18, с. 81. — 45. Кужелев Л. 77., Максимова И. А.,
Миронов И. А. и др. Авт. свид. 276294; Бюлл. изобрет., 1970, № 23, с. 89. —
46. Pallila F. G., Baird Р. Н. J. Electrochem. Soc., 1962, v. 109, р. 1162—
1166.— 47. Roberts S. J. Appl. Phys., 1957, v. 28, p. 262—265. — 48.
Thornton W. A. J. Electrochem. Soc., 1960, v. 107, p. 895—907. — 49.
Казанкин О. H., Дихтер М. А. В кн.: Сборник рефератов научно-исследова-
тельских работ по химии и технологии люминофоров за 1964 г. Под ред.
Л. Я. Марковского. Л., изд. ГИПХ, 1965, с. 3. — 50. Jaffe Р. М. J.
Electrochem. Soc., 1960, v. 107, р. 895—907.
51. Петошина Л. Н., Красноперов В. А., Пекерман Ф. М. В кн.: Химия
и технология люминофоров. Под ред. Л. Я. Марковского. Л., «Химия», 1968,
с. 5—11. — 52. Thornton W. A., Granford N. Official gazette U. S. Patent Office,
1963, v. 78, patent 3082344. — 53. Lehmann W. J. Electrochem. Soc., 1966,
v. 113, p. 40—43. — 54. Казанкин О. H., Осипов В. А., Дихтер М. А. и др.
В кн.: Электролюминесценция твердых тел. Киев, «Наукова думка», 1971,
с. 226—231. — 55. Пекерман Ф. М., Королев А. Л., Петошина Л. Н. и др.
В кн.: Электролюминесценция твердых тел и ее применение. Киев, «Наукова
думка», 1972, с. 192—197. — 56. ЗаплешкоН. Н., ДаниловВ. П., Веревкин ТО. Н.
Там же с. 197—200. — 57. Веревкин ТО. Н. Автореф. канд. дисс., Л., ЛЭТИ,
1970. — 58. Hahn D., Mimkes J. J. Phys. Chem. Solids, 1968, v. 29, p. 1287—
1292. — 59. Fisher A. G. J. Electrochem. Soc., 1963, v. 110, p. 733—747. —
80. Сощин H. П. Автореф. канд. дисс. Тарту, ТГУ, 1971.
61. Vecht A., Werring N. J., Smith P. J. Brit. J. Appl. Phys., 1969, Ser. 2,
v. 2, № 7, p. 953—966. — 62. Vecht A. J. on Luminiscence. 1973, v. 7, p. 213—
156
220. — 63. Казанкин О'. Н., Пекерман Ф. М., Паранин Г. А. и др. В кн.: Элек-
тролюминесценция твердых тел и ее применение. Киев, «Наукова думка», 1972,
с. 140—148. — 64. Destriau G. Phil. Mag., 1947, v. 38, p. 700—739, 774-793,
880—888. — 65. Curie D. J. Phys. Radium, 1953, v. 14, p. 510—524. — 66.Pi-
per W. W., WilliansF. E. Phys. Rev., 1952, v. 87, p. 151—152. — 67. Фок M. B.,
Георгобиани A. H. Усп. физ. наук, 1960, т. 72, с. 467—478. — 68. Георгоби-
ани А. Н. «Труды ФИАН им. П. Н. Лебедева», 1963, т. 23, с. 3—63. —
69. ОрановскийВ. Е., Хмелинин Б. А. Опт. И спектр., 1959, т. 7, с. 542—546. —
70. Gilson J. L., Darnell F. J. Phys. Rev., 1962, v. 125, p. 149—158.
71. Бонч-Бруевич A. M., Кариес Я. Э., Молчанов В. А. Опт. и спектр.,
1961, т. 11, с. 87—92. — 72. Lehmann W. J. Electrochem. Soc., 1957, v. 104,
p. 45—50. — 73. Roberts, JOSA, 1952, v. 42, p. 850—856. — 74. Katona R.
J. Electrochem. Soc., 1962, v. 109, p. 693—699. — 75. Weber K. Ilium. Eng.,
1964, v. 5, p. 329—335. — 76. Кюлъмоя, Г. % ., Талъвисте Э. К., Т аммиак А. А.
В кн.: Труды по люминесценции. Т. 1. Тарту, изд. ТГУ, 1971, с. 6—13. —
77. Ковалев Б. А., Ведехин А.Ф., Шикунова С. Т. и др. В кн.: Тезисы докладов
на совещании по электролюминофорам. Ставрополь, изд. ВНИИЛ, 1971, с. 23. —
78. Варгин В. В., Пекерман Ф. М., Липин А. Л. и др. «Светотехника», 1972,
№ 2, с. 5—8. — 79. КыласовВ. А., Орлов И. Н. В кн.: Семинар «Физико-техни-
ческие вопросы кибернетики». Вып. 4. Киев, изд. Ин-та кибернетики АН УССР,
1967, с. 65—67. — 80. Aven М., Cusano D. A. J. Appl. Phys., 1964, v. 35,
р. 606—611.
81. Vlasenko N. A., Gergel A. N. Phys. Stat. Sol., 1968, v. 26, p. 77. —
82. Gudden B., Pohl R. W. Z. Phys. 1924, Bd. 21, S. 1—8. — 83. Shmidt F.
Ann. Phys., 1923, Bd. 70, S. 161—198. — 84. Destrian G. J. Phys. Radium,
1956, v. 17, p. 734—736. — 85. Cusano D. A., WilliamsF. E. Ibid., p. 742—747. —
86. Коломейцев Ф. И., Сидоренко С. М. Ж. прикл. спектр., 1968, т. 8, с. 975—
977. — 87. Верещагин И. К., Хавруняк В. Г. Там же, 1969, т. 11, с. 560, 749;
1972, т. 17, с. 81—86. — 88. Ivey Н. F. Solid State Physics in Electron, a. Tele-
comm., 1960, v. 4, p. 611. — 89. Nuese C. J., Kressel H., Ladany J. J. Vac. Sci.
Technol., 1973, v. 10, № 5, p. 772—775. — 90. Берг А., Дин П. Светодиоды.
Пер. с англ. Под ред. А. Э. Юновича. М., «Мир», 1973. 98 с.
91. Панков Ж. Оптические процессы в полупроводниках. Пер. с англ.
Под ред. Ж. И. Алферова. М., «Мир», 1973. 456 с. — 92. Solomon R., Defe-
vere D. Appl. Phys. Lett., 1972, v. 21, № 6, p. 257—260. — 93. Ladany J.,
Kussel H., R. C. A. Rev., 1972, v. 33, p. 517—536. — 94. Pankove J. I. J. Lu-
minescence, 1973, v. 7, p. 114—126. — 95. Burmister A., Pighini, Jr., G. P.,
Greene P. E., Trans. Met. Soc. AIME, 1972, v. 245, p. 587—593. — 96. Blum J. M.,
Shih, Proc. IEEE, 1971, v. 59, № 10, p. 1498—1502. — 97. Kressel H., La-
dany J., J. Appl. Phys., 1968, v. 39, № 11, p. 5339—5340. — 98. Auzel F. E.
Proc. IEEE, 1973, v. 6, № 6, p. 758—786. — 99. Радауцан С. И., Цуркан A. E.
Теллурид цинка. Кишинев, 1972. 204 с. — 100. ВинецкийВ. Л., Холодарь Г. А.
Статистическое взаимодействие электронов и дефектов в полупроводниках.
Киев, «Наукова думка», 1969. 185 с.
101. Aven М. J. Appl. Phys., 1967, v. 38, № 11, р. 4421—4430. — 102. Wo-
odbury Н. Н. Bull. Am. Phys. Soc., 1967, v. 12, p. 43. — 103. Woodbury H. H.
In: II—VI Semiconductor Compounds, 1967. Ed. by D. G. Tohmas. N. Y. —
Amsterdam, W. A. Benjamin. Inc., 1968, p. 244. — 104. Миронов И. А., Стро-
ганова И. M., Кужелев Л. П. В кн.: Тезисы докладов Международной конфе-
ренции по люминесценции. (Ленинград, август, 1972 г.) Черноголовка, «Наука»,
1972, с. 300. — 105. Allen J. W., Livingstone A. W., Turvey К. Solid State
Electron., 1972, v. 15, № 12, p. 1363. — 106. Park J. S., Geesner C. R.,
Shin В. K. Appl. Phys. Lett., 1972, v. 21, № 12, p. 567—569. — 107. Aven M.,
Garvacki W. J. Appl. Phys., 1967, v. 38, № 5, p. 2302. — 108. Jamamoto R.,
Ito K., Japan. J. Appl. Phys., 1967, v. 6, № 4, p. 537—543. — 109. Mandel G.,
Morehead F. F. Appl. Phys. Lett., 1964, v. 4, p. 143—148. — 110. Morehead F. F.,
Mandel G. Ibid., v. 5, p. 53—56.
111. Лебнер, ТИИЭР, 1973, т. 61, № 7, с. 46—49.
157
ГЛАВА VII
РЕНТГЕНОЛЮМИНОФОРЫ
Специфика возбуждения рентгеновскими лучами, по сравнению с фото-
возбуждением, заключается в том, что на люминофор действуют фотоны
со значительно большей энергией. При этом свечение люминофора вызывается
не непосредственным действием самих рентгеновских лучей, а воздей-
ствием электронов, вырываемых из атомов или ионов основы люмино-
фора рентгеновскими лучами. Вследствие этого рентгенолюминесценция имеет
многие общие черты с катодолюминесценцией. Различие заклю-
чается в том, что эффективность возбуждения рентгеновскими лучами возрастает
с увеличением коэффициента поглощения рентгеновских лучей веществом люми-
нофора, который, как известно, растет с увеличением атомного номера элемен-
тов. Поэтому, в качестве рентгенолюминофоров наиболее целесообразно при-
менять соединения, содержащие тяжелые элементы, например, Cd, Ba, W.
Рентген о люминофоры применяют в экранах двух типов:
для рентгеноскопии и флюороскопии с непосредственным наблюдением
видимого изображения;
в усиливающих экранах, используемых для фотографирования просвечи-
ваемого объекта.
В последнем случае относительно слабое действие на фотоэмульсию самих
рентгеновских лучей усиливается за счет излучения люминофора, что способ-
ствует сокращению экспозиции *. Максимум на спектральных кривых излучения
экранов для рентгеноскопии должен быть близок к максимуму чувствительности
человеческого глаза, т. е. лежать между 520 и 560 нм, для усиливающих экранов
максимум излучения должен находиться в области наибольшей чувствитель-
ности применяемого фотографического материала.
Качество рентгеновского изображения на экране определяется:
яркостью свечения экрана;
контрастностью и резкостью изображения;
длительностью послесвечения.
Яркость свечения зависит от качества люминофора. На контра-
стность влияют как материал экрана, так и характеристики применяемого
рентгеновского излучения. Резкость изображения зависит главным обра-
зом от величины зерна люминофора. Длительность послесвечения
для рентгенолюминофоров желательна наименьшая, так как в противном слу-
чае изображения движущихся объектов искажаются. По данным Бунделя и По-
пова [1], максимальная допустимая величина послесвечения через 0,05 с после
прекращения возбуждения составляет 7% от начальной яркости свечения.
Для флюоресцирующих экранов как в советской, так и в зарубежной прак-
тике [1—3, с. 421; 4] применяют цинк-кадмийсульфидные люминофоры (30—
60 мол.% CdS), активированные Ag. Наличие кадмия в основе уменьшает дли-
тельность послесвечения. Разработаны также весьма эффективные электронно-
* Для характеристики свойств люминофоров, применяемых в рентгенов-
ских экранах, обычно используют величину так называемой относительной
плотности почернения стандартной рентгеновской фотографической пленки.
158
оптические преобразователи рентгеновского излучения [4, с. 423; 5, 6]. В них
также используют экраны с рентгено люминофорами сульфидного типа. Крупно-
кристаллический (40. мкм) цинк-кадмийсульфидный люминофор ZnS (55) •
. CdS (45)-Ag (0,006) дает яркость свечения 0,006—0,06 кд-м"* 2 при интенсив-
ности рентгеновского излучения (0,645—258) -10-7 Кл/кг (0,25—100 мр/с) |3,
с. 423).
Для изготовления усиливающих экранов, предназначенных для фотома)е-
риалов, чувствительных к синему излучению, применяют либо люминофор
ZnS-Ag (для рентгеновских лучей с энергией менее 100 кВ), либо, главным
образом, люминофор CaWO4, или CaSO4 - РЬ, а в последние годы также BaSO4
• РЬ (для рентгеновских лучей с энергией более 100 кВ).
По данным Гурвича [7, 8], приведенным ниже, наибольший абсолютный
энергетический выход при рентгеновском возбуждении дают сульфидные люмино-
форы:
Энер гетическ ий
выход, %
CaWO4 ............................. 4—6
BaSO4-Pb .......................... 4,8
ZnS-Ag............................ 20,7
ZnS (55) • CdS (45) • Ag (0,006):
мелкозернистый (средний раз-
мер зерна 12 мкм)............. 20,3
крупнозернистый (средний раз-
мер зерна 37 мкм)............. 20,6
Установлено, что энергетический выход практически не зависит от энергии
квантов возбуждающего излучения.
Технические данные изготавливаемых у нас рентгенолюминофоров при-
ведены в табл. VII.1.
Следует отметить, что рентгенолюминофоры применяют не только в экранах,
используемых для медицинских диагностических целей, но и в промышленных
установках для дефектоскопии с применением жестких рентгеновских лучей
и у-лучей.
Методы изготовления сульфидных рентгенолюмино-
форов принципиально ничем не отличаются от уже ранее описанных методов
синтеза, например катодолюминофоров. Различие заключается в том, что для
получения большой яркости изображения применяют более высокие темпера-
туры синтеза, что приводит к существенному укрупнению порошка люминофора.
Применение крупнозернистых люминофоров для рентгеновских экранов оказы-
вается возможным вследствие большой проникающей способности рентгеновских
лучей.
При синтезе вольфрамата кальция обычно исходят из очищен-
ного вольфрамового ангидрида WO3 и карбоната кальция люминофорной сте-
пени чистоты. Эквимолекулярную смесь порошков этих веществ тщательно
гомогенизируют, просеивают через сито и прокаливают в кварцевых тиглях
при 1100° около 1 ч, после чего охлаждают на воздухе. Люминофор возбуждается
коротковолновым УФ-светом и рентгеновскими лучами. Кривая спектрального
распределения энергии излучения люминофора изображена на рис. VII.1. Дли-
тельность послесвечения люминофора составляет ~ 10" 5 с.
Технология люминофора BaSO4 РЬ, по данным Бунделя и Булан-
ковой [9], заключается в следующем: сначала из 20% горячих растворов, очи-
щенных ВаС12 и РЬС12 *, действуя на них 10% раствором Na2SO4 получают
сульфаты BaSO4 и PbSO4. Сухие соли после просева через сито тщательно пере-
мешивают в соотношении 10:1 (по массе) при одновременном растирании; затем
смесь загружают в кварцевые тигли и прокаливают 3 ч в муфельной печи при
1150°. Полученный крупнозернистый порошок измельчают, просеивают через
сито и повторно прокаливают в течение 15 мин при 900°. Таким путем может
быть получен светосостав, по гранулометрическому составу не отличающийся
ст светосоставов на основе вольфрамата кальция.
* Допускается содержание железа и других тяжелых металлов не более
2 • 10"5 % в пересчете на сухое вещество.
159
Таблица VII.1
Технические характеристики рентгенолюминофоров
Люминофор Цвет свечения ин *хви\ Полуширина полосы, нм Средний раз- мер зерна, мкм Назначение
марка состав
P-535 ZnS - CdS - Ag Зеленый 535 85 10-12 Усиливающие экра- ны типа УС (при- меняются с сенси- билизированной пленкой типа РМ-6)
P-530 ZnS - CdS - Ag Желто- зеленый 530 85 30 Экраны для рентге- носкопии и флюо- рографии
P-450 Zn S • Ag Синий 450 53 8—10 Усиливающие экра- ны
P-420M CaWO4 » 420 100 5—10 Усиливающие экра- ны) применяются в медицине и про- мышленности)
P-420K CaWO4 » 420 100 20—25 Усиливающие экра- ны (применяются при работе с Гу- излучением и рент- геновским излуче- нием большой же- сткости)
P-420 CaW04 « 420 100 10 • Усиливающие экра- ны
P-355 BaSO4-Pb Фиолетово- синий 355 18—20 Усиливающие экра- ны типа СБ, ис- пользуемые при напряжении 80— 100 кВ
По технологии, предложенной Гурвичем и Дубовицкой [10], качество
люминофоров BaSO4Pb можно улучшить, если ввести в шихту плавни (смесь
NaHSO4 с Na2SO4 в соотношении 1:5 — 1 :,10) в количестве до 45% от массы
шихты. В этом случае температура прокаливания может быть снижена до 900—
-10000.
Рис. VII.1. Спектральное
распределение энергии из-
лучения рентгенолюмино-
форов:
Состав
7 Р-355
2 Р-420м
3 Р-450
4 Р-535
5 Р-530
BaSCU-Pb
CaWO4
ZnS-Ag
ZnS-CdS-Ag
ZnS-CdS-Ag
160
Максимум на кривой спектрального излучения люминофора BaSO4-Pb
лежит при 350—360 нм (см. рис. VII.1, кривая/). Длительность послесвечения
близка к 10"7 с. Люминофор возбуждается даже мягкими рентгеновскими лу-
чами, а также катодными лучами, но не возбуждается УФ-лучами с X = 253,7 нм.
Фотографическое действие люминофора BaSO4-Pb превышает фотографи-
ческое действие стандартного люминофора CaWO4 на 65—70%, что позволяет
соответственно уменьшить экспозицию.
В последние годы разработаны и находят все более широкое применение
рентгенолюминофоры на оксисульфидной основе [11—15].
ЛИТЕРАТУРА
1. Бунделъ А. А., Попов М. Ф., Чижуноеа Ю. А. Труды института рент-
геноскопии и радиологии, 1955, т. 9. с. 137—145. — 2. Бунделъ А. А., Руса-
нова А. И. Там же, с. 4—6. — 3. Leverenz Н. An Introduction to Luminescence
of Solids. New York — London, Wiley Sons, 1950. 569 p. — 4. Outwelties J. F.
In: Modern Materials Advances in Development and Application. V. 5. London,
1965, p. 161—257. - 5. Teves M. C., Tol T. Phil. Techn. Rev., 1952-1953,
v. 14, p. 33—43. — 6. Гурвич A. M., Ильина M. А. Опт. и спектр., 1964, т. 17,
с. 893—900. — 7. Гурвич А. М., Катомина Р. В. Новости мед. техники, 1962,
№ 3, с. 40—57. — 8. Гурвич А. М., Катомина Р. В. Изв. АН СССР. Сер.физ.,
1961, т. 25, с. 506—508. — 9. Бунделъ А. А., Буланкова Т. Г. В кн.: Химия
и технология люминофоров. Вып. 43. Л., Госхимиздат, 1960, с. 7—10. —
10. Гурвич А. М., Дубовицкая В. Б. Новости мед. техники, 1961, № 5, с. 61—67.
И. Тота S. Z., Shaffer Е. N. J. Electrochem. Soc., 1971, v. 118, р. 151. —
12. Wang S. P., Landi O., Lucks H. e. a. IEEE Trans. Nucl. Sci., 1970, v. 17,
p. 49—56. — 13. Wicker shetm K. A., Alves R. V., Buchanan R. A. Ibid., p. 57—
60. — 14. Ludvig G. W., Prener J• S. Ibid., 1972, v. 19, p. 7. — 15. Ratinen H.
Phys Stat. Sol., 1972, v. 12a, p. 447—451.
11 Заказ 44
ГЛАВА VIII
СВЕТОСОСТАВЫ ПОСТОЯННОГО ДЕЙСТВИЯ
Свойство люминофоров возбуждаться радиоактивным излучением исполь-
зуется в технике уже давно для изготовления самосветящихся радиоактивных
красок, которые часто называют светосоставами постоянного действия (СПД).
Области их применения весьма разнообразны и определяются необходимостью
в различного рода сигнальных и индикаторных устройствах, световых знаках,
которые не требуют источников внешнего возбуждения. До открытия искус-
ственных радиоактивных изотопов в самосветящихся красках в качестве источ-
ника возбуждения использовали исключительно соли естественных радио-
активных препаратов — радия и мезотория (вернее продукта его распада —
радиотория). Однако такие светосоставы имеют два существенных недостатка:
первый — большая биологическая вредность, обусловленная радиоактивным
излучением; второй — быстрое снижение яркости свечения радиоактивных
светящихся красок с течением времени. Оно объясняется разрушающим дей-
ствием а-частиц на вещество основы люминофора, в результате которого уже
примерно через год яркость свечения радиоактивных красок снижается на-
половину.
В связи с этим в последнее время для изготовления СПД успешно приме-
няют искусственные радиоактивные препараты, в особенности такие чистые
Р-излучатели, как 14?Рш и 3Н(Т). Преимущество использования р-излучателей
заключается в том, что их излучение (электроны с различной энергией) легко
задерживается даже тонкими защитным^ слоями и экранами и в значительно
меньшем разрушающем действии их на основу люминофора.
VIII.1. СВЕТОСОСТАВЫ С ИСПОЛЬЗОВАНИЕМ
ЕСТЕСТВЕННЫХ РАДИОАКТИВНЫХ ВЕЩЕСТВ
В этих светосоставах в качестве возбудителя люминесценции применяют
радий (период полураспада наиболее долгоживущего изотопа 226Ra составляет
1617 лет) или смесь мезотория (7\^ = 6,7 года) с радиоторием (Л/, =
= 1,9 года).
Под действием а-частиц люминесцирует большое количество различных
светосоставов, но наилучшие результаты дает сульфид цинка, активированный
Си. В СССР применяют главным образом светосостав ФК-106, представляющий
собой крупнозернистый люминофор с зеленым свечением (см. рис. IV.20,
стр. 93).
Приготовление светосостава заключается во внесении радиоактивного
вещества в люминофор. Соли радия вводят в люминофор в количестве 5—200 мг
на 1 кг ZnS. Для этой цели обычно употребляют водные растворы бромидов
радия. После перемешивания люминофора с раствором соли (следует помнить,
что от тщательности проведения этого простого процесса зависит качество СПД)
бромиды переводят в нерастворимые сульфаты путем обработки раствором суль-
фата аммония. Затем полученную массу выпаривают и сушат. Все указанные
операции должны проводиться в специальных боксах при соблюдении всех
необходимых мер радиационной защиты.
162
В СССР выпускаются три марки СПД — г, з и ФК-102П. В соответствии
с техническими условиями, светосоставы «г» и «з» должны отвечать следующим
требованиям:
быть зеленовато-желтого цвета с зеленым свечением в темноте;
быть сухими (влажность — не более 0,15%) и без комков;
яркость свечения светосоставов должна составлять (в кд-м-2), не менее
Марка СПД
г
3
Начальная
0,02—0,03
0,06—0,08
Через 1 год
0,018
0,034
Через 2 года
0,017
0,027
Через 3 года
0,017
0,019
Свечение ФК-102П — красновато-оранжевое.
При использовании СПД для изготовления самосветящихся знаков и над^-
писей используют различные прозрачные лаки, как, например, даммаровый
лак (раствор светлой даммаровой смолы в ксилоле), метилметакрилат, полисти-
роловый лак, лак БМК, лаки на основе эпоксидных смол и т. д. Так как с уве-
личением количества связующего существенно снижается яркость свечения
краски, то связующего следует брать как можно меньше, но достаточно для
получения прочного покрытия. Так, на 1 г даммарового лака берется — 2 г
СПД. Интенсивность свечения покрытий на 10—20% меньше, чем свечение
самого порошка светосостава. Количество СПД, необходимое для изготовления
светящихся покрытий, составляет ~ 0,5 г на 1 см2 поверхности.
VIII.2. СВЕТОСОСТАВЫ С ИСПОЛЬЗОВАНИЕМ
{ИСКУССТВЕННЫХ ^-ИЗЛУЧАТЕЛЕЙ (ТАБЛ. VIII.1)
Исследования, проведенные в различных странах, показали, что все пере-
численные изотопы пригодны для изготовления светосоставов постоянного
действия. Однако широкое распространение получили только прометий и тритий.
Т аб лица VIII.1
Наиболее практически важные р«излучатели
Изотоп Период полураспада, годы Максималь- ная энергия 0-частиц, МэВ Толщина необходимой защиты (стекло), мм
eosr/eoy 29 2,6 9,0
85Кг 9,4 0,80 3,0
204Т1 4,0 0.76 3,0
147pm 2,65 0,22 1,5
14С 5568 0,154 1.5
зн (тритий) 12,43 0.018 0,24
Изотопы стронция применяют ограниченно из-за сильной биологической вред-
ности. При использовании криптона возникают трудности, связанные с тем, что
практически всегда ему сопутствуют примеси, обладающие интенсивным у-излу-
чением. Соли таллия образуют темноократпенные сульфиды при контакте с люми-
нофором на основе ZnS, а изотоп 14С пригоден только для источников света
низкой интенсивности, так как его удельная активность мала из-за большого
периода полураспада.
Источники света с газообразным тритием
Источник света с газообразным тритием представляет собой стеклянный
баллон, на внутренней поверхности которого закреплен слой порошка люмино-
фора. Баллон заполнен газообразным тритием, p-излучение которого возбуждает
И* 163
свечение люминофора. Свечение наблюдается либо сквозь полупрозрачный слой
люминофора, либо через прозрачное окно. Тритиевые источники света выпу-
скаются во многих странах [1—3]. В СССР Всесоюзная контора «Изотоп» поста-
вляет разнообразные по форме и цвету свечения светознаки.
Для изготовления газонаполненных тритиевых источников света в СССР
используют в основном люминофоры типа ФК-106 и ФК-3 с высокой квантовой
эффективностью (94%) при фотовозбуждении и энергетической эффективностью
~ 18—20% при катодном возбуждении (10 кВ). Кроме этих люминофоров с успе-
хом могут использоваться и катодолюминофоры типа К-74, К-78 и др.
Тритий 3Н — изотоп водорода, в составе ядра которого имеется два ней-
трона и один протон. Его молекулярный вес равен шести. Тритий распадается
по реакции 3Н1—> 3Не2+ 0+гс периодом полураспада 12,43 года. Максималь-
ная энергия |3-частиц достигает 18,6 кэВ, средняя энергия — 5,54 кэВ. Только
15% от всех частиц имеют энергию больше 10 кэВ. Средняя длина пробега (3-ча-
стиц трития в воздухе при нормальных условиях составляет 0,8—0,9 мм, а в тка-
нях — 1 мкм. Средняя длина пробега 0-частиц трития в среде трития — 4,5 мм
при нормальных условиях. Данные о поглощении и глубине проникновения
0-частиц трития в сульфиде цинка противоречивы; считается, что электроны
с энергией меньше 10 кэВ проникают на глубину 0,1—1 мкм. Из-за столь малой
глубины проникновения для возбуждения 3Н очень существенным фактором
оказывается состояние поверхности частиц люминофора. Известно, что объем-
ная люминесценция, как правило, является более эффективной, чем поверхно-
стная. Так, показано, что при уменьшении энергии пучка электронов (и, следо-
вательно, глубины их проникновения) от 10 до 5 кэВ эффективность катодо-
люминесценции снижается на 40—50%. Для лучших катодолюминофоров энер-
гетическая эффективность составляет 0,18—0,22 при 10 кэВ, поэтому можно
ожидать, что при тритиевом возбуждении (средняя энергия электронов 5 кэВ)
эффективность будет не больше 0,1, а светоотдача для люминофоров с желто-
зеленым излучением 30—50 лм/Вт. Следует ответить, что, несмотря на высокую
светоотдачу, тритиевые источники света не могут обеспечить получение высокого
уровня яркости, так как повышение интенсивности возбуждения ограничи-
вается самопоглощением излучения трития. Яркость свечения люминофора,
возбуждаемого 0-излучением трития, возрастает пропорционально его давлению
только в ограниченном интервале давлений, а затем изменяется очень слабо.
Величина давления, при котором наблюдается насыщение, зависит от габаритов
баллона.
Самосветящиеся краски
Существует много патентов [4—10],<цосвященных использованию трития
для изготовления самосветящихся красок. В этом случае тритий добавляют
к люминофору в виде соответствующего тритированного органического соеди-
нения. Описано применение для этой цели различных тритированных жирных
кислот, смол, поверхностно-активных соединений, точный состав которых по
большей части держится в секрете. Яркость свечения таких красок достигает,
а иногда даже превышает таковую радиоактивных красок с использованием
радия и колеблется от 0,05 до 0,2 кд-м~3. Количество трития, приходящегося
на 1 г светосостава, сравнительно велико и соответствует активности ^ЮЭ ’с"1
(несколько сотен милликюри). Преимуществом тритированных красок является
их меньшая биологическая вредность, по сравнению с радиевыми, так как даже
тонкая пленка защитного лака предотвращает проникновение 0-лучей наружу.
Практически такие краски безвредны.
В настоящее время во многих странах разрабатываются новые, более эффек-
тивные самосветящиеся тритированные краски. Основные проблемы, которые
приходится при э г > vr решать, связаны, во-первых, с наиболее целесообразным
совмещением тритированного соединения с порошком люминофора и со связу-
ющим, что обусловлено малой длиной пробега 0-излучения трития (доли мик-
рона), и, во-вторых, с необходимостью использования наиболее радиационно-
и химически стойких веществ [11].
Самосветящиеся краски различного цвета свечения, использующие в каче-
стве возбудителя люминесценции 0-излучение l47Pm, выпускаются в промыш-
ленном масштабе в Чехословакии. Технические данные некоторых марок этих
светосоставов представлены в табл. VIII.2.
164
Таблица VIII.2
Яркостные характеристики СПД с использованием 147Рт
Цвет свечения Тип Содержание 147Рт Яркость свечения, кд-м~2«10_3 Средний попереч- ник зерен, мкм Плотность, г/см3
1 7 о мкюри/г начальная через 2 ме- сяца через 6 ме- сяцев через 2 года через 4 года
Зеленый PSP1 3.7 1 20,6 19.7 17.5 10,8 5,4
PSP5 18,5 5 79.5 75,0 67 41,5 20.6 12 4,4
PSP20 74 20 222 210 188 115 61
Желтый PSP1 3,7 1 23,8 22,6 20 12,1 6,4
PSP5 18,5 5 89 84,5 75,5 45,8 23,6 27 4,2
PSP20 74 20 255 236 215 131 67,5
Красный PSP1 3,7 1 6,4 6,1 5,4 3,5 1,6
PSP5 18,5 о 28,6 27,0 24,2 14,6 7,6 21 4,3
PSP20 74 20 92.5 87,5 81,1 47,8 24,5
Данные работы [12] показывают, что использование светящихся красок
с г47Рт совершенно безопасно для здоровья человека.
Изотоп 14С в связи с большим периодом полураспада применяют для изго-
товления источников излучения со стабильной яркостью — так называемых
эталонов яркости, необходимых для ряда светотехнических измерений. В СССР
выпускают эталоны яркости зеленого, синего и оранжевого цветов с яркостью
свечения от 0,005 до 0,01 кд-м-2. Уменьшение яркости свечения источника
за 3 года не превышает 10%. В настоящее время разработаны эталоны яркости
с излучением в области от ультрафиолетового излучения до красного со стабиль-
ной величиной интенсивности (105—1011 квант/с) в широком интервале темпера-
тур. Подробные данные приведены в обзоре [13].
VIII.3. ЛЮМИНОФОРЫ ДЛЯ ДОЗИМЕТРИИ ЯДЕРНЫХ ИЗЛУЧЕНИЙ
В связи с расширением применения атомной энергии в технике большое
значение имеет использование люминофоров для целей обнаружения и реги-
страции а-, Р-, у- и нейтронных излучений. Люминофоры, применяемые для
дозиметрии, должны обладать следующими свойствами:
наибольшей конверсионной эффективностью (отношение энергии, излуча-
емой в виде сцинтилляций, к поглощенной энергии);
спектральным составом излучения, соответствующим области максимальной
чувствительности фотокатода сцинтилляционного счетчика;
малой длительностью сцинтилляций, что необходимо для обеспечения высо-
кой разрешающей способности счетчика.
Кроме того, люминофоры, применяемые для регистрации излучений с боль-
шой проникающей способностью (рентгеновское и у-излучение), должны быть
прозрачными для собственного излучения и, желательно, содержать элементы
с большим атомным номером.
Для регистрации а- и Р-частиц (последних — лишь частично) применяют
цинксульфидные люминофоры, активированные Ag или Си. Конверсионная
эффективность их достигает 28%, длительность сцинтилляций у ZnS-Ag равна
10_5 с. Люминофоры используют в виде тонких слоев порошка (5—10мг/см2).
у-Излучение регистрируют монокристаллическими щелочногалогенидными
люминофорами, активированными Т1, например Nal-Tl или KI-T1. Максимум
на кривой спектрального распределения этих люминофоров лежит при 410 нм,
длительность послесвечения составляет 0,25-10“ в с. Конверсионная эффектив-
ность кристаллов Nal-Tl может достигать 8%. Недостаток указанных люмино-
165
форов — большая гигроскопичность, что вызывает необходимость применения
герметичной аппаратуры. В дозиметрии у-излучений применяются кристаллы
некоторых органических люминофоров, например, антрацена, стильбена и др.
Для дозиметрии медленных нейтронов пригодна ядерная реакция ’оВ (па)
7Li, которая приводйт к образованию частиц, регистрируемых по сцинтилляциям
на экране из люминофора ZnS • Ag. На практике для этой цели обычно применяют
специальный люминофор, в основу которого введено некоторое количество
бора [14]. Последний (в виде борной кислоты) вводят в шихту при синтезе люми-
нофора. Такой люминофор выпускается под маркой Т-1. Еще лучше, если в лю-
минофор введена борная кислота, обогащенная изотопом 10В, у которого попе-
речное сечение захвата тепловых нейтронов равно 3999 • 10_ 24 см2 (по сравнению
с 750-10"24 см2 для естественной смеси изотопов).
Медленные нейтроны регистрируют монокристаллами галогенидов лития,
активированными Т1 и другими элементами. В этом случае при действии нейтро-
нов идет ядерная реакция 6Li(na)3H. Поперечное сечение захвата нейтронов
у eLi равно 900• 10"24 см2.
Применяют люминофоры Lil-Tl, LiF-Tl, Lil-Eu [15] и др. Длительность
сцинтилляций у Lil-Tl равна 1-10"в с. Для регистрации быстрых нейтронов
находят применение порошки люминофора ZnS-Ag, запрессованные в прозрач-
ные пластмассы. При этом источником возбуждения люминофора являются
протоны, выделяющиеся из содержащих водород органических соединений под
действием нейтронов (так называемые протоны отдачи).
Для целей индивидуальной дозиметрии большое значение имеет использо-
вание явления термолюминесценции некоторых люминофоров, способных за-
пасать энергию, поглощенную под действием рентгеновского, у- или Р-излуче-
ния. При нагревании до 200—300° запасенная энергия высвечивается в форме
видимого излучения. Для этой цели пригодны многие люминофоры, например
CaF2-Mn [16], CaSO4-Mn [17], CaSO4-Sm [18], а также люминесцирующие
алюмофосфатные стекла, активированные Мп [19].
ЛИТЕРАТУРА
1. Бальхаузен Д. Мирное применение атомной энергий. Т. 15. М., Изд.
АН СССР, 1956. 365 с. — 2. Wilson Е., Hughes J. «Engineering», 1950, v. 187,
р. 497—499. — 3. Марковский Л. Я., Курюкин А. Е., Прокофьева Г. П. и др.
В кн.: Тезисы докладов XIV совещания по люминесценции. М., «Наука», 1965,
с. 75. — 4. Schweizer W. Uhr, 1962, Bd. 16, № 13, S. 20—22. — 5. Wahl R.
Ibid., № 1, S. 20. — 6. Chem. Nevjs, 1961, v. 39, № 1, p. 56. — 7. Вильсон E.
РЖХим, 1961, 8K138. — 8. Chem. Abs., 1963, v. 58, p. 9750. — 9. Fujimure R.,
A ton Y. Chem. Zbl., 1963, Bd. 134, S. 19484. — 10. Deleo F., Shapiro E. РЖХим,
1963, 20 л. 117.
11. Heusinger H., Rau H. Kerntechnik, 1961, Bd. 3, S. 67. — 12. Veit V.,
Ibid., 1963, Bd. 5, S. 221. — 13. Михальченко Г. А. Приборы и техника эксп.,
1965, № 1. с. 157. — 14. Голиков В. В. Там же, 1963, № 2. с. 59—64. —
15. Карпов И. К., Михальченко Г. А. Ж. прикл. спектр., 1966, № 3, с. 178—
181. — 16. Grinter R. J. Electrochem. Soc., 1957, v. 104, № 6, p. 365—369. —
17. Носенко Б. M., Ревзин Л. С., Ясколко В. Л. ЖТФ, 1956, т. 26, № 9,
с. 2046—2048. — 18. Peter Н. Atomkernenergic, 1960, Bd. 5, S. 453—457. —
19. Бочвар И. А., Васильева А. А., Кеирим-Маркус И. Б. «Атомная энергия»,
1963, т. 15, с. 48-52.
ГЛАВА IX
МЕТОДЫ ИЗМЕРЕНИЯ
ОСНОВНЫХ ФИЗИЧЕСКИХ ХАРАКТЕРИСТИК ЛЮМИНОФОРОВ
Данная глава посвящена методикам измерения люминесцентных характери-
стик [1, с. 39-91; 2, с. 54-84; 3, с. 595-666; 4, с. 221—457; 5, с. 21-34; 6,
с. 37—78 и 111—132; 7, с. 106—142], из которых наибольшее значение имеют
спектры поглощения и излучения, выход люминесценции, длительность после-
свечения, а также некоторых других физических свойств люминофоров. Кроме
того, описаны применяемые в измерительной практике методы возбуждения
фото-, катодо- и электролюминофоров.
IX.1. ВОЗБУЖДЕНИЕ ЛЮМИНОФОРОВ
Фотолюминофоры
Основными источниками возбуждения фотолюминофоров служат ртутные
лампы низкого, высокого и сверхвысокого давления (рис. IX.1).
Ртутные лампы низкого давления (бактерицидные лампы ДБ-30-1, изве-
стные ранее под маркой БУВ-30) мощностью 30 Вт выпускаются в трубках из
увиолевого стекла, пропускающего излучение дальней УФ-области спектра.
Около 70% энергии этих ламп излучается в области резонансной линии ртути
(Л = 254 нм), поэтому они пригодны для возбуждения всех люминофоров, ис-
пользуемых в лампах низкого давления. Распределение энергии в спектре излу-
чения этих ламп приведено в табл. IX. 1. Схема включения бактерицидной лампы
доказана на рис. IX.2. Для возбуждения ламповых люминофоров в области
254 нм удобно пользоваться выпускаемым нашей промышленностью ультра-
химископом марки УИ-1, который состоит из трех бактерицидных ламп, фильтра
УФС-1 и пульта управления.
Ртутные лампы высокого давления ДРТ, выпускавшиеся ранее под маркой
ПРК, имеют вид кварцевых трубок. Схема включения этих ламп показана на
рис. IX.3, а распределение энергии излучения в этих лампах приведено
в табл. IX.2. Наибольшее количество энергии в ртутных лампах высокого давле-
ния излучается в ближней УФ-области спектра (Л = 365 нм). Поэтому
указанные лампы обычно служат для возбуждения сульфидных, селенидных
и других люминофоров, область возбуждения которых расположена в ближ-
ней УФ-области.
Максимум энергии излучения ртутно-кварцевых ламп сверхвысокого давле-
ния, как и ламп высокого давления, приходится на ближнюю УФ-область, но
по мере повышения давления линии ртутного спектра расширяются, и в излуче-
нии ламп увеличивается доля сплошного спектра. Промышленность выпускает
осветители марки КП-1Н с ртутной лампой высокого давления и фильтром
УФС-6, пропускающим, главным образом, излучение с X = 365 нм.
Все ртутные лампы, применяемые для исследования фотолюминесценции,
заключены в светонепроницаемые кожухи. Для выделения требуемых областей
спектра служит набор светофильтров. Кроме того, для выделения линии ртути
254 нм используют фильтр УФС-1, а для 365 нм — фильтры УФС-4 и УФС-6.
Кривые пропускания этих светофильтров представлены на рис. IX.4.
167
a
6
&
Рис. IX.l. Ртутные лампы, используемые для воз-
буждения люминесценции.
а — Лампа низкого давления ДБ-30- 1; б — лампа высокого
давления ДРТ; в — лампа сверхвысокого давления СВДШ:
1,2 — электроды; 3 — вспомогательный электрод для зажи-
гания.
Таблица IX.1
Спектральное распределение энергии излучения
бактерицидной лампы мощностью 30 Вт [8, с. 368]
Длина вол- ны, нм Энергия излучения на расстоя- нии 1 м, мкВт/см2 Относитель- ная энергия излучения, 0/ /0 Длина вол- ны, нм Энергия излучения на расстоя- нии 1 м, мкВт/см2 Относитель- ная энергия излучения, %
254 43,0 100,0 313 1,0 2,3
265 0,06 0,14 365 0,85 1,98
280 0,02 0,046 405 3,7 8,6
289 0,08 0,18 436 6,1 14,2
297 0,18 0,42 546 5,8 13,5
303 0,10 0,23
1G8
Рис. IX.2. Схема включения
бактерицидной лампы:
Л — лампа; Э — электроды; С —
стартер; Д — дроссель.
Рис. IX.3. Схема включе-
ния лампы ДРТ:
Л — лампа; Д — дроссель; С,—
конденсатор 300—500 мкФ; Ct —
конденсатор 2—3 мкФ; КП —
конденсаторная полоса; Кп К2—
ключи.
Таблица IX.2
Спектральное распределение энергии излучения
ртутных ламп высокого давления
Длина волны, нм Относитель- ная энергия излучения, % Длина волны, нм Относитель- ная энергия излучения, %
248 10,5 313 68,0
254 26,1 365 100,0
265 23,4 405 35,9
280 10,3 436 62,4
297 14,3 546 71,7
303 31,2 577 70,4
Следует иметь в виду, что указанные фильтры обладают значительным
пропусканием в красной области спектра. Поэтому кроме указанных фильтров,
перед источником возбуждения необходимо помещать светофильтр C3C-23,
который не пропускает излучение в этой области. Область спектра, в которой
происходит возбуждение люминофоров, можно выделять также при помощи
монохроматоров с кварцевой оптикой.
Пропусканиях
Рис. IX.4. Кривые спектрального пропускания некото-
рых светофильтров (толщина светофильтров 5 мм).
Рис. IX.5. Схема уста-
новки со скрещенными
фильтрами для иссле-
дования свечения лю-
минофоров.
При наблюдении и измерении люминесценции надо учитывать, что воз-
буждающий свет, не поглощенный люминофором, добавляется к свету люми-
несценции и искажает получаемый результат. Чтобы избежать ошибок при изме-
рении, используют так называемые скрещенные фильтры. Свет от источника
возбуждения S (рис. IX.5) падает через фильтр Ф15 выделяющий область воз-
буждения, на люминофор Л. Свечение последнего воспринимается каким-либо
приемником излучения, например, фотоэлектронным умножителем пли глазом,
через фильтр Ф2, который не пропускает возбуждающий свет, но пропускает
свет люминесценции. Фильтры Фх и Ф2 называются скрещенными. Для люмино-
форов, излучающих в зеленой области спектра, скрещенными являются фильтры
УФС-6 и СЗС-22, кривые спектрального пропускания которых показаны на
рис. IX.4.
Катодолюминофоры
Катодолюминофоры возбуждаются в разборной электронно-лучевой трубке,
принципиальная схема которой показана на рис. IX.6. Пучок электронов,
испускаемых электронной пушкой П, проходит внутри корпуса трубки, которая
169
соединена с металлической камерой 9 при помощи вакуумного сочленения.
Электронная пушка П имеет катод 1 и модулятор 2; последний обычно заземлен,
а управляют током луча путем подачи на катод напряжения положительной
полярности. Система катушек 3 позволяет осуществлять развертку, фокусировку
и центровку электронного луча. Сфокусированный и развернутый в растр луч
подают на стеклянные экраны с испытуемым катодолюминофором 4. Экраны
Рис. IX.6. Принципиальная схема установки для наблюдения за разгоранием
и затуханием катодолюминофоров:
1 — катод; 2 — модулятор; 3 — фокусирующие катушки; 4 — экран с люминофором;
5 — зеркало; 6 — светофильтр; 7 — фотоэлектронный умножитель; 8 — катодный повтори-
тель; 9 — металлическая камера; 10 — металлический диск; 11 — рукоятка; 12, 13 — окна;
14 — патрубок; 15 — генератор прямоугольных импульсов типа Г5-6А; 16 — источник
постоянного напряжения; 17 — осциллограф типа С1-15; 18 — универсальный источник
питания УИП-1.
размещены в металлическом диске 10, который можно поворачивать при помощи
рукоятки 11. Окно 12 служит для наблюдения и измерения люминесценции
со стороны падающего электронного луча («на отражение»), а окно 13 — для
измерений «на просвет». Откачку всей системы производят через патрубок 14
сначала форвакуумным, а затем диффузионным парамасляным насосом до давле-
ния 1,33 • 10~3 Па (10~б мм рт. ст.). В выпускаемых промышленностью установ-
ках, предназначенных для измерения параметров катодолюминофоров, ускоря-
ющее напряжение изменяется в пределах от 2,5 до 30 кВ, а плотность тока луча
может достигать 25 мА см-2.
Для возбуждения катодолюминофоров электронным пучком, не развернутым
в растр, может быть использована установка типа ПРСЭЛ. На этой установке
помимо измерения характеристик катодолюминофоров, нанесенных на стеклян-
ные экраны, при помощи впециальной приставки можно исследовать порошки
катодолюминофоров в кюветах [9].
Для исследования свечения катодолюминофоров при изменении темпера
туры может быть применена приставка, сконструированная в Физическом ин-
ституте АН СССР [10].
170
Электролюминофоры
При возбуждении электролюминофоров переменным электрическим полем
источниками синусоидального переменного напряжения могут служить гене-
раторы типа ГЗ-ЗЗ или ГЗ-34. Для повышения напряжения на выходе звукового
генератора следует применять усилитель мощности. Кроме того, может быть
использован генератор повышенной мощности марки ИСН-1.
Напряжение определенной частоты подводят к двум электродам разборного
конденсатора, между которыми помещают слой электролюминофора в смеси
с диэлектриком (рис. IX.7). Один из электродов конденсатора должен быть
прозрачным, чтобы через него можно было наблюдать свечение электро люмино-
фора. Обычно для этого используют стекло с нанесенным на него токопроводя-
щим слоем. Другой электрод, как правило, делают металлическим. В качестве
диэлектрика может быть использовано касторовое или силиконовое масло.
Рис. IX. 7. Схема разборного элек-
тролюминесцентного конденсатора:
1 — диск; 2 — латунный электрод; з —
латунное кольцо; 4 — токопроводящее
стекло; 5 — крышка; 6 — резиновая
прокладка; 7 — люминофор с диэлектри-
ком.
Электролюминофоры возбуждают постоянным электрическим полем в раз-
борных ячейках подобного типа, только в этом случае в качестве источника
возбуждения служит не звуковой генератор, а универсальный источник питания
УИП-1 или какой-либо другой источник постоянного тока.
IX.2. ИЗМЕРЕНИЕ ИНТЕНСИВНОСТИ ЛЮМИНЕСЦЕНЦИИ
Определение относительной интенсивности
свечения люминофоров,
имеющих одинаковый спектр излучения
Обычно при оценке различных люминофоров у них измеряют относитель-
ную интенсивность свечения, т. е. интенсивность свечения относительно интен-
сивности свечения эталонного люминофора (определения ведут в одинаковых
условиях).
Относительную интенсивность свечения люминофоров измеряют при помощи
фотометров различных типов. Наиболее распространен фотометр марки ФМ-58
(фотометр Пульфриха) (рис. IX.8). Кюветы с исследуемым люминофором Лг
и эталонным люминофором Л2 располагают перед объективами фотометра Ог
и О2. Источник возбуждения устанавливают так, чтобы освещение кювет было
одинаковым. Свет люминесценции можно ослаблять при помощи диафрагм Д1
и Д2, раскрытие которых регулируется барабанами Бх и Б2. В поле зрения
окуляров видны соприкасающиеся полями светящиеся поверхности в форме
двух полукругов. Интенсивность свечения люминофора Лх может быть опре-
делена относительно интенсивности свечения эталона Л2 при уравнивании свето-
вых полей фотометра путем раскрытия диафрагм. Если — отсчет по бара-
бану Б1? а а2 — отсчет по барабану Б2, то относительная интенсивность свечения
исследуемого люминофора (в %) определяется по формуле:
171
В более поздних моделях фотометров световые потоки, идущие от люмино-
форов, можно регистрировать при помощи двух сурьмяноцезиевых фотоэлемен-
тов. В работе Рамазанова и Шапошникова [11] описана фотоэлектрическая
приставка к фотометру, которая позволяет повысить чувствительность и точ-
ность измерений примерно в 10 раз.
Относительную интенсивность свечения люминофоров можно измерять и при
помощи объективного фотометра, принципиальная схема которого показана
на рис. IX.9. На неподвижном прочном основании 6 укреплены в специальном
кожухе 2 источник возбуждения 1, фотоэлемент 3 и вращающийся диск 5 с от-
верстиями для кювет с люминофорами 4. Сначала перед приемником излучения
устанавливают кювету с эталонным лю-
минофором, освещают ее светом от ис-
точника возбуждения и определяют ин-
тенсивность люминесценции, для чего
производят отсчет аг по гальванометру,
соединенному с приемником излучения.
Затем поворотом вращающегося диска 5
перед приемником излучения устанав-
ливают кювету с испытуемым люмино-
фором и получают соответствующий
отсчет по гальванометру а2. Относи-
тельная интенсивность свечения иссле-
дуемого люминофора определяется из
Рис. IX.8. Схема установки для из-
мерения интенсивности свечения лю-
минофоров при помощи фотометра
Пульфриха.
соотношения (o^/aj • 100%. Для ус-
транения влияния возбуждающего
Рис. IX.9. Принципиальная схема объ-
ективного фотометра для измерения
относительной интенсивности свечения:
1 — источник возбуждения; 2 — кожух; 3 —
фотоэлемент; 4 — кювета с люминофором; 5 -»-
вращающийся диск; в — основание; 1 — сте-
клянная пластинка.
света, отраженного люминофором,
перед приемником излучения должйЬ стоять светофильтр Ф2, скрещенный со
светофильтром Фг на источнике возбуждения.
При возбуждении люминофоров бактерицидной лампой (X = 254 нм) без
фильтра УФС-1 относительная интенсивность люминесценции определяется
следующим образом. Кювету с эталонным люминофором освещают бактерицид-
ной лампой и снимают показание ccj гальванометра, соединенного с приемником
излучения. В этом случае пропорционально интенсивности люминесценции
и той части возбуждающего света, которая отражается люминофором. Для того
чтобы учесть последнюю, бактерицидную лампу закрывают стеклянной пла-
стинкой 7, которая не пропускает свет с длиной волны 254 нм. При этом люмино-
фор не возбуждается, так что показание гальванометра а2, деленное на коэффи-
циент пропускания стеклянной пластинки т, пропорционально величине интен-
сивности света, отраженного люминофором. Затем производят такие же
измерения для исследуемого люминофора. Относительную интенсивность люми-
несценции (в %) вычисляют по формуле.
а4
а-5—----
Т СС3Т — «4
ССо СС-|Т—CU
«1— ——
X
172
где а3 и а4 — показания гальванометра для исследуемого люминофора соответ-
ственно при открытой и закрытой стеклянной пластинкой бактерицидной лампе.
Аналогичным образом проводят измерения и в том случае, когда фильтр
УФС-1 имеет значительное пропускание в видимой области спектра.
Сравнение интенсивности свечения люминофоров,
имеющих различные спектры излучения
Описанные ранее методы определения относительной интенсивности люми-
несценции не могут быть применены к люминофорам с различными спектраль-
ными составами излучения, так как все перечисленные приемники излучения
[человеческий глаз, фотоэлемент, фотоэлектронный умножитель (ФЭУ)] обла-
дают селективной чувствительностью к различным длинам волн. В этом случае
у исследуемого и эталонного образцов в одних и тех же условиях измеряют
спектры излучения. Энергия, излученная люминофором, пропорциональна
площади, ограниченной осью абсцисс и кривой спектрального распределения.
Для измерения относительной интенсивности следует определить указанные
площади для исследуемого и эталонного образцов и взять их отношение.
IX.3. ИЗМЕРЕНИЕ ЯРКОСТИ СВЕЧЕНИЯ
Для измерения яркости свечения люминофоров может быть использован
селеновый фотоэлемент с фильтром, который «приводит» кривую спектральной
чувствительности фотоэлемента к кривой видности человеческого глаза (фото-
элемент с корригирующим фильтром). Если у гальванометра, соединенного
с таким фотоэлементом, цена деления шкалы определена в кд-м~2, то при по-
мощи этой установки можно измерять яркость люминесценции в абсолютных
единицах. Измерение абсолютной яркости свечения люминофоров можно произ-
водить также при помощи фотометров для визуальных измерений (типа АФМ,
ФПИ, ВФМ-57). Фотометром ВФМ можно измерять малые яркости свечения
в пределах от 5 до 3-10~& кд-м-2. Верхний предел измерения больших яркостей
составляет 1 Об кд-м“2 для цветного света и 5-10«кд-м-2 для белого. Яркомер
ЭЯ-67, разработанный во ВНИСИ, позволяет производить измерения светящихся
поверхностей с размерами от 0,25 мм2 и более в широком диапазоне яркостей
(от 1 до 1000 кд -м-2).
При измерении относительной интенсивности или абсолютной яркости
свечения катодо- и электролюминесценции могут быть использованы те же самые
установки и приборы, что и в случае измерений аналогичных характеристик
фотолюминофоров. Разница состоит лищь в способе возбуждения.
IXA ИЗМЕРЕНИЕ СПЕКТРОВ ИЗЛУЧЕНИЯ
Распределение энергии по спектру излучения люминофора измеряют при
помощи монохроматора и фотоэлектронного умножителя, помещенного у выход-
ной щели монохроматора и соединенного с гальванометром. Предварительно
при помощи светоизмерительной лампы накаливания с известным распределе-
нием энергии по спектру (например, источника с цветовой температурой 2854 °К)
градуируют установку по относительным значениям энергии через каждые 5 нм
в требуемом диапазоне длин волн. Для этого перед входной щелью монохрома-
тора устанавливают кювету с окисью магния, которую освещают светоизмери-
тельной лампой и для каждой длины волны определяют отклонения ctj гальвано-
метра, соединенного с ФЭУ. Таким образом находят коэффициенты Ку = Эх/ах,
учитывающие спектральную чувствительность ФЭУ и дисперсию монохроматора
(Эх — относительное значение энергии светоизмерительной лампы накаливания
для данной длины волны, взятое из таблиц распределения энергии излучения
источника А) [2].
Для измерения распределения энергии по спектру излучения люминофора
последний устанавливают перед входной щелью монохроматора и соответству-
ющим образом возбуждают. Затем для каждой длины волны в требуемом интер-
вале длин волн определяют по гальванометру отклонение а{. Энергию излучения
173
люминофора Ех в относительных единицах для данной длины волны определяют
по формуле:
В установке по измерению спектров люминесценции гальванометр может
быть заменен записывающим устройством. Затруднения, возникающие при авто-
матической записи спектров излучения, связаны главным образом с тем, что,
во-первых, дисперсия призменного монохроматора различна для различных
областей спектра и, во-вторых, чувствительность ФЭУ зависит от длины волны.
Поэтому при автоматической записи спектров излучения необходимо устройство,
корректирующее спектр.
В более ранних моделях автоматических спектрофотометров корректировка
спектров производилась механическим способом. Барабан длин волн был связан
с синхронным мотором через эксцентрик, который обеспечивал вращение бара-
бана со скоростью, пропорциональной длине волны. Поправка на спектральную
чувствительность ФЭУ производилась при помощи диска с переменным радиусом,
который помещался перед выходной щелью монохроматора.
В Тартусском университете разработан автоматический спектрофото-
метр [12] на базе монохроматора типа УМ-2 или ЗМР-З. Запись спектров люми-
несценции производится при помощи электронного потенциометра. Поправка
на спектральную чувствительность ФЭУ осуществляется при помощи фото-
сопротивления, освещаемого софитной лампой через щель, высота которой
изменяется в зависимости от поворота призмы монохроматора.
Прибор для автоматической записи спектров излучения, созданный на базе
спектрофотометра, описан в работе [13].
IX.5. ИЗМЕРЕНИЕ КООРДИНАТ ЦВЕТНОСТИ
Цвет свечения люминофоров, в частности люминофоров для люминесцент-
ных ламп и экранов кинескопов, характеризуется при помощи координат цвет-
ности. Известно [2, с. 54—84; 7, с. 106—142], что какой-либо цвет Цо количе-
ственно может быть выражен некоторым линейным уравнением:
Цо = "Ь у¥о z%o
Величины Хо, Yo, Zo связаны определенными соотношениями с тремя основ-
ными цветами: синим (Л = 435,8 нм), зеленым (X = 546,1 нм) и красным (К =
= 700 нм), а величины х, у, z, называемые координатами цветности, связаны
уравнением плоскости:
x-J-y-]-z = l
Г
Следовательно, только две из этих величин независимы, и поэтому цветность
обычно характеризуется двумя величинами — х и у.
Геометрически уравнение плоскости означает, что все цвета Цо лежат в одной
плоскости — секущей трехгранного угла xyz. Эта плоскость, называемая цвето-
вым треугольником, обычно изображается в прямоугольном цветовом гра-
фике х, у (см. рис. IV.5, стр. 77). На этом графике могут быть нанесены точки
цветности: монохроматических излучений, излучения черного тела, излучений,
получаемых сложением двух каких-либо цветов во всевозможных соотношениях..
Измерение координат цветности может быть произведено при помощи раз-
работанного во ВНИСИ универсального фотоэлектрического колориметра.
Внутри колориметрической головки последнего расположены селеновый фото-
элемент и два поворотных диска. Каждый диск имеет пять отверстий. Три отвер-
стия первого диска (именно он служит для измерения координат цветности)
закрыты фильтрами х, у, z, четвертое — свободно, а пятое — закрыто ширмой.
Ширма служит для закрывания фотоэлемента при проверке нуля гальванометра,
с которым соединен фотоэлемент. При введении фильтра у производят все свето-
вые измерения. Второй диск предназначен для измерения цветовой температуры
источника. Три отверстия этого диска закрыты красным, зеленым и синим свето-
фильтрами, одно — свободно и одно — закрыто сеткой.
174
При измерении координат цветности фотоколориметрическую головку
устанавливают перед светящимся люминофором и перед открытым фотоэлементом
вводят последовательно фильтры х, у, z. По полученным величинам трех фото-
токов гх, iy, i2, пользуясь соответствующими градуировочными уравнениями,
приведенными в паспорте прибора, определяют координаты цветности испыту-
емого люминофора.
При отсутствии универсального фотоколориметра ВНИСИ координаты
цветности могут быть вычислены по спектрам излучения. Для этого у исследу-
емого люминофора измеряют распределение энергии Ех (см. стр. 173) по длинам
волн через каждые 5 нм. Затем полученное значение Ех для каждой длины волны
умножают на удельные координаты а?х, и zx, значения которых можно найти,
например в работах [2, 7J, и, суммируя указанные произведения, иолучают:
х' = У Е^-, у' = 2 z 1=
Координаты цветности х, у, г определяют по формулам:
х' у' z'
X — —Т~, 7~, 7" I У = Г~, z = П 7"
х +у +z х +z х -y-у ~rz
IX.6. ИЗМЕРЕНИЕ СПЕКТРОВ ПОГЛОЩЕНИЯ
Поглощение энергии возбуждения такими рассеивающими веществами,
как люминофоры, определяется большей частью путем измерения величины
отражения падающего света от поверхности люминофора [14, с. 388—434].
Прямые количественные измерения коэффициента поглощения были произ-
ведены лишь в некоторых частных случаях [151.
Методика измерения спектров поглощения люминофоров заключается
в следующем. Перед входной щелью монохроматора устанавливают кювету
с окисью магния, освещаемую, например, водородной лампой, которая имеет
непрерывный спектр излучения в УФ-области. Свет, отраженный от окиси маг-
ния, попадает на ФЭУ, находящийся у выходной щели монохроматора, и реги-
стрируется гальванометром, соединенным с ФЭУ. Измерения производятся для
различных длин волн в требуемой области спектра. Затем кювету с окисью
магния заменяют кюветой с исследуемым люминофором, для которого также
записывают показания гальванометра. Пусть при некоторой длине волны —
отклонение гальванометра, когда перед ФЭУ стоит кювета с люминофором,
а а2 — отклонение гальванометра при измерениях с окисью магния, тогда вели-
чина отражения для люминофора определяется формулой
где -Kjjfgo — величина отражения для окиси магния (значение XMg0 по данным
различных авторов приведены в табл. IX.3).
Величина поглощения может быть получена из следующего выражения:
ТГпогл =1 — Хотр
Таблица IX.3
Величина отражения для MgO (в %)
Длина волны, нм
254 297 313 365 400 436 500 600 700
93 95 96 96 97
93 93 — 94 — 95-97 95-97 .— .—
93—96 94-97 *— 94—98 95—98 96—98 97—98 97 97
175
IX.T. ИЗМЕРЕНИЕ СПЕКТРОВ ВОЗБУЖДЕНИЯ
Сцектры возбуждения люминофоров определяют следующим образом.
Источник возбуждения располагают перед входной щелью монохроматора,
при помощи которого выделяется требуемая область спектра. У выходной щели
помещают кювету с люминофором, на который проектируется та или иная
область возбуждающего света. Излучение люминофора принимает ФЭУ, рас-
положенный над кюветой. Перед ним — для устранения влияния рассеянного
света, должен быть установлен светофильтр, не пропускающий возбуждающего
света. Для каждой длины волны определяют отношение
ахЖ
где ах — отклонение гальванометра, соединенного с ФЭУ, которое пропорци-
онально интенсивности люминесценции при данной длине возбужда-
ющего света Л;
Еу — величина, пропорциональная энергии возбуждающего света при той
же длине волны.
Для определения величины £х вместо кюветы с люминофором ставят кювету
с окисью магния и определяют отклонение гальванометра а{, тогда
ах
AXAMgO
где Ку — спектральная чувствительность ФЭУ для данной длины волны;
^Mg0 — величина отражения от MgO для данной длины волны.
IX.8. ОПРЕДЕЛЕНИЕ ЭНЕРГЕТИЧЕСКОГО, КВАНТОВОГО ВЫХОДА
И СВЕТООТДАЧИ
Фотолюминофоры
Монохроматор -г X
В литературе [16—19] описано несколько методов определения энергети-
ческого и квантового выхода, но и сейчас это определение представляет собой
очень сложную задачу, которая экспериментально решена лишь для частных
случаев.
Ниже описывается метод определения квантового выхода, в котором прием-
ником излучения служит фотоэлектронный умножитель с известной спектраль-.
ной чувствительностью. Схема уста-
новки показана на рис. IX.10. Свет
от источника возбуждения проходпт
через монохроматор, который выде-
ляет требуемую область возбужде-
ния. Перед выходной щелью моно-
хроматора помещается кювета либо
с окисью магния, либо с люминофо-
ром. Когда перед ФЭУ находится
кювета с окисью магния, отклонение
гальванометра, соединенного с ФЭУ,
будет пропорционально энергии воз-
буждения. Если обозначить спек-
тральную чувствительность ФЭУ для
данной длины волны падающего
света через Ку, то энергия возбуж-
дения Ев равна
_ «1
в KkKMgO
Люминофор
или Mg'O
Рис. IX.10. Схема установки для опре-
деления энергетического выхода.
1 де cbj — отклонение гальванометра при измерениях на MgO;
7?Mgo ~ величина отражения от MgO для данной длины волны (см. табл. IX.3).
176
Для того чтобы получить величину поглощенной энергии, нужно энергию
возбуждения Ев умножить на величину поглощения, способ измерения которой
был описан ранее. Тогда:
г, а1Апогл
С-ПОГЛ — р р
AXAMgO
Для определения энергии люминесценции вместо кюветы с окись'ю магния
перед выходной щелью монохроматора помещают кювету с исследуемым люми-
нофором. На ФЭУ в этом случае падает свет люминесценции и та часть воз-
буждающего света, которая не поглощена люминофором. Показание гальвано-
метра а2 в этом случае складывается из а3, пропорционального интенсивности
люминесценции, и а4, пропорционального интенсивности отраженного воз-
буждающего света, т. е. а2 = а34- а4. Тогда а3 = а2 — а4 = а2 — а^Аотр»
так как а4 = осмотр. Для определения величины, пропорциональной энергии
люминесценции Ел, нужно а3 умножить на некоторый коэффициент А, учиты-
вающий различие в спектральной чувствительности фотоэлектронного умножи-
теля для разных длин волн. Зная спектр излучения люминофора и спектральную
чувствительность ФЭУ, коэффициент А можно вычислить из отношения
л рА
~ f hKA
где /х — интенсивность люминесценции для данной длины волны;
Ку — коэффициент спектральной чувствительности ФЭУ, воспринимающего
свечение люминофора.
Таким образом, Ел = а3А и
Ел _ a3^£x£Mg0 лл
Оэн = -р------, Пкв — -Оэн -7---------
Zin а1«-П0ГЛ Ап
Таблица IX.4
Энергетический и квантовый выходы различных люминофоров
при фотовозбуждении
Состав и марка люминофора •Вэн BK3 Литера- тура
CaSiO3-Pb-Mn 0,29 0,63 18
(MgO)je(As2O5)p-Mn 0,29 0,73 18
ZnS•ZnSe «Си 0,31 0,45 20
ZnS • CdS • Си 0,32 0,50 20
ЗСа3(РО4)2 • Ca(F, Cl)2-Sb-Mn 0,34 0,71 18
Zn2SiO4-Mn 0,35 0,70 • 18
3Ca3(PO4)2 Ca(F, Cl)2-Sb-Mn 0,36 0,82 20
Zn2SiO4 Mn (K-35) 0,36 0,74 20
CaWO4-Pb 0,42 0,75 18
MgWO4 (Л-29) 0,42 0,79 20
(Zn, Ba)2SiO4-Mn 0,42 0,94 20
MgWO4 0,44 0,84 18
ZnS • CdS • Cu 0,44 0,67 20
(Ca, Mg)3(PO4)2 • T1 0,47 0,58 20
Ca3(PO4)2-Tl 0,49 0,56 18
ZnS • Си (ФКП-ОЗк) 0,50 0,73 20
BaSi2O5-Pb 0,55 0,75 18
BaSi2O5-Pb (Л-33) 0,56 0,78 20
ZnS • CdS • Си (Л-15) 0,57 0,86 20
(Ca, Zn)3(PO4)2 • T1 0,60 0,73 20
£nS • Ag 0,67 0,83 20
ZnS • Си (ФК-106) 0,69 0,96 20
12 Заказ 44
177
Недостаток этого метода заключается в том, что необходимо вносить по-
правку на спектральную чувствительность фотоэлектронного умножителя,
воспринимающего свет люминесценции. Для устранения этого недостатка Ба-
уэр [19] предложил перед ФЭУ, принимающим разложенное в спектр свечение
люминофора, помещать специальную маску, которая ослабляет интенсивность
люминесценции при различных длинах волн обратно пропорционально чув-
ствительности ФЭУ к этим длинам волн.
В табл. IX.4 приведены значения ВЭн в ВКв, полученные различными мето-
дами. Следует отметить, что для сульфидных люминофоров величина выхода
люминесценции зависит от интенсивности возбуждающего света, так что этот
параметр необходимо указывать, если требуется сравнение измеряемых величин.
Кроме энергетического и квантового выходов важной технической харак-
теристикой люминофоров является величина их светоотдачи. Она представляет
собой отношение величины излучаемого люминофором светового потока к пада-
ющей на него мощности возбуждения, измеряют ее в лм/Вт.
Катодолюминофоры
Энергетический выход люминесценции при возбуждении катодными лучами
представляет собой отношение энергии, испускаемой люминофором, к энергии
электронного пучка, возбуждающего люминофор. Методика измерения энерге-
тического выхода катодолюминофоров описана в работе Бриля и Клязенса [21].
Катодолюминофоры возбуждаются в разборной электронно-лучевой трубке.
Выход катодолюминесценции равен
В = ЕЛ]Р
где Ел — энергия, излучаемая люминофором;
Р = IV — мощность, затрачиваемая на возбуждение люминофора (I — ток
луча; V — напряжение на аноде).
Энергия излучения может быть измерена при помощи термоэлемента. Пусть
А —чувствительность термоэлемента, (Вт/В-см2); г — расстояние от термо-
элемента до люминофора; Т — пропускание окна разборной трубки; и — пока-
зание прибора, соединенного с термоэлементом. Тогда энергия излучения равна:
лг^иА # лг2иА
£л“ Т ’ В~ TIV
Величина В практически всегда получается уменьшенной вследствие того,
что часть света люминесценции может поглощаться в слое фосфора. Поэтому
для получения истинного значения выхода катодолюминесценции нужно внести
поправку, умножив В на величину 2/(Ято + 1), где — отражение собствен-
ного света люминесценции от «бесконечно толстого» слоя люминофора. Тогда:
лг^иА 2
- TIV
В практических целях для катодолюминофоров часто важнее знать не вели-
чину энергетического выхода, а величину светоотдачи т), равную
F
П ~ Р IV
где F — световой поток, излучаемый люминофором;
Р — мощность, затраченная на возбуждение люминофора.
Световой поток может быть измерен при помощи селенового фотоэлемента
с корригирующим фильтром, если известна его чувствительность.
В табл. IX.5 приведены значения энергетического выхода и светоотдачи
для некоторых катодолюминофоров, по данным различных авторов.
178
Таблица IX.5
Эиергетический выход и светоотдача некоторых катодолюминофоров
Состав и марка люминофора Вэн» % T), лм/Вт Литература
ZnS • Ag 19,6 22
21,0 18
20—25 4—10 24
15,0 7,5 23
ZnS • Ag-Cl 21,0 7,0 23
ZnS CdS • Ag 14,9 18
21,0 22
ZnS • CdS • Ag (зеленый) 14,0 46,0 23
ZnS • CdS • Ag (красный) 12,0 8,6 23
0,5ZnS • O,5CdS • Ag 19,5 98 23
18-20 100 24
0,2ZnS -0,8CdS • Ag 12-15 10 24
ZnS•Cu 20-25 90 24
ZnS • Cu • Al 23,0 92 23
ZnS - CdS - Cu-Al 13,0 65,1 23
ZnS • Mn 4,5 18 24
ZnSe•Cu 14 19 24
0,97ZnSe • 0,03CdSe • Cu 14 13,5 24
Zn2SiO4 • Mn 8,5 42,0 23
6,0 31,1 23
8,5 42,0 24
Zn2SiO4 • Ti 8,5 2,5 24
CaO • MgO • 2SiO2 • Ti 7,0 5,0 24
0,86ZnO • 0,12BeO • 0,50SiO2 • 0,02MnO 7,0 31,0 24
CdSiO3 • Mn 5,0 17,0 24
Mg2SiO4 • Mn 3,0 3,5 24
MgSiOa• Mn 3,0 1,0 24
Zn3(PO4)2 • Mn 5,0 7,0 23
5,5 8,5 24
ZnO • Zn 7,0 25,0 24
CaWO4 3,0 2,0 24
Y2O3- Eu 6,5 22
YVO4 • Eu 6,0 9,5 23
,3,4—-6,7 22
Y2O2S • Eu 6,0 17,1 23
Электролюминофоры
Для измерения светоотдачи электролюминесцентных конденсаторов необ-
ходимо знать две величины: поглощенную ЭЛК энергию и излучаемый им свето-
вой поток. Последний может быть измерен теми же методами, как и в случае
катодолюминесценции. Измерения мощности, потребляемой ЭЛК, представляют
значительные трудности.
В ранних работах для этой цели использовались различные варианты мосто-
вых схем. Однако, вследствие нелинейности процесса электролюминесценции,
подобные методы определения потребляемой мощности при возбуждении элек-
тролюминесцентных конденсаторов синусоидальным напряжением недостаточно
корректны, а в случае возбуждения несинусоидальным напряжением и совсем
непригодны. Кроме того, мосты переменного тока серийного изготовления для
подобного рода измерений не подходят из-за того, что создаваемый в них элек-
трический режим по частоте и напряжению сильно отличается от режима работы
электролюминесцентных конденсаторов: напряжение на последних в таких
12* 179
мостах не превышает 10—30 В, и измерения, как правило, проводятся на одной
или двух фиксированных частотах.
В ряде работ [25, 26] предложены методы измерения мощности, поглоща-
емой ЭЛК с помощью катодного осциллографа. В работе [27] описан метод
и приведены результаты измерений мощности по-
0-
терь с помощью калориметра. Ряд авторов для из-
мерения мощности, потребляемой ЭЛК, использо-
вали различные типы ваттметров. В настоящее время
промышленность выпускает ваттметры марки Ф-530,
предназначенные для измерения малых величин
мощности в цепях переменного тока с частотой
20—20 000 Гц. Недостаток этого прибора состоит
в том, что при измерении малых величин мощности
(например, при низких значениях напряженностей
и частот возбуждающего поля, а также при изме--
рении электролюминесцентных панелей малого раз-
мера) вследствие чрезвычайно высокой чувствитель-
ности прибора собственное потребление ваттметра
значительно превосходит величину измеряемой мощ-
ности, что приводит к увеличению погрешности из-
мерений.
Описана [25] методика измерения емкости и
тангенса диэлектрических потерь, а следовательно,
и мощности, потребляемой электролюминесцентными
Рис. IX. 11. Схема уста-
новки для измерения
мощности, потребляемой
электролюминесцентным
конденсатором:
конденсаторами, в широком диапазоне частот и на-
пряженностей возбуждающего поля. Принципиаль-
ная схема установки, которая представляет собой
уравновешенный мост переменного тока, собран-
ный по схеме Вина-Соти, приведена на рис. IX.11.
Уравновешивание моста производится на основ-
ной и вспомогательных схемах попеременно. При-
Со и С, — магазины емко-
стей типа Р-513; Ro, R, и
г2— магазины сопротивлений
типа Р-32, МСР-60 и Р-58;
R2 и г, — магазины со-
противлений типа Р-33; V —
нуль-индикатор (анализатор
напряжений типа С5-3),
включенный в диагональ
моста; V, — вольтметр типа
С-50; Сх — электролюмине-
менение в качестве нуль-индикатора селективного
прибора позволяет учитывать нелинейность электро-
люминесцентного конденсатора, так как в случае
синусоидальности напряжения мощность опреде-
ляется по первой гармонике тока.
IX.9 ИЗМЕРЕНИЕ ДЛИТЕЛЬНОСТИ ПОСЛЕСВЕЧЕНИЯ
сцентный конденсатор. для измерения длительности послесвечения
люминофоров служат специальные приборы, одним
из которых является фосфороскоп Беккереля
(рис. IX.12), состоящий из двух дисков N и М, смонтированных на одной оси.
Исследуемый люминофор помещают между дисками, которые установлены таким
образом, что, когда возбуждающий свет проходит через отверстия первого диска
и попадает на образец, непрозрачный сектор второго диска закрывает его от
наблюдателя. Когда люминесцирующее вещество становится видимым через
отверстие во втором диске, непрозрачным сектором первого диска закрыт путь
для возбуждающего света, что позволяет наблюдать процесс затухания люмине-
сценции. Меняя угол между секторами в обоих дисках и скорость вращения,
в известных пределах можно изменять время, проходящее между окончанием
возбуждения и моментом наблюдения. Количественные определения интенсив-
ности фосфоресценции для различных промежутков времени между возбужде-
нием и наблюдением могут быть сделаны с помощью фотометра или каким-либо
другим способом (см. стр. 171). При помощи двухдискового фосфороскопа можно
измерять длительности послесвечения от 0,1 до 10“4 с. В более широком времен-
ном интервале можно измерять длительности послесвечения при помощи одно-
дискового фосфороскопа. Подробное описание фосфороскопов и их характеристик
дано Левшиным [1, с. 75—86].
Для исследования процессов нарастания и затухания свечения может быть
применен тауметр, разработанный Толстым и Феофиловым [29]. Достоинство
180
этого прибора в том, что он удобен при исследовании процессов, происходящих
по экспоненциальному закону. При помощи тауметра можно измерять длитель-
ность затухания от 10“1 до 10“7 с.
Измерение длительности послесвечения катодолюминофоров в диапазона
от 10 до 10“5 с можно осуществить при помощи установки, работающей на сле-
дующем принципе.
В разборной электронно-лучевой трубке на люминофор подаются импульсы
напряжения строго прямоугольной формы с достаточной крутизной фронта.
Свечение экрана воспринимается ФЭУ, сигналы с которого после усиления
подаются на одну пару пластин осциллографа, тогда как на другую пару по-
дается временная развертка, синхронная с отпирающими импульсами. При этом
на экране осциллографа получается пол-
ная кривая разгорания и затухания лю-
минофора.
Принципиальная схема установки для
наблюдения кривых разгорания и зату-
хания катодолюминофоров показана на
рис. IX.6 (стр. 170). Для модуляции элек-
тронного
к входу
луча модулятор 2 подключается
генератора
15 прямоугольных
Рис. IX.13. Кривые разгорания и затуха-
ния катодолюминофоров.
Рис. IX. 12. Схема двухдиско-
вого фосфороскопа.
импульсов типа Г5-7А. Излучение люминофора 4 попадает на фотокатод ФЭУ 7.
Импульсы фототока ФЭУ, соответствующие импульсам излучения люминофора,
регистрируются на экране осциллографа 17 типа С1-15, который работает
в режиме ждущей развертки. Запуск осциллографа осуществляется генерато-
ром 15. На рис. IX.13 показаны кривые разгорания и затухания люминофора
(пунктир) и импульсы, отпирающие трубку (сплошная линия).
Длительность послесвечения люминофоров при спаде яркости свечения до
заданной величины (до 1, 5 или 10% от начальной яркости) определяется по
кривым затухания с использованием меток времени. При измерении длитель-
ности послесвечения катодолюминофоров удобно вместо осциллографа 17 и ге-
нератора 15 использовать прибор типа А-65901.
IX.10. ИЗМЕРЕНИЕ ГРАНУЛОМЕТРИЧЕСКОГО СОСТАВА
Для определения гранулометрического состава полидисперсных порошков
могут быть применены различные способы: седиментационный,
микроскопический, по определению насыпной массы, ад-
сорбционный, а также разные варианты электрических мето-
дов (кондуктометрический, импульсный и т. д. [30, с. 9—69]). Однако в практике
производства люминофоров в настоящее время используются только два первых
метода.
Седиментационный метод [31, с. 44—121] основан на наблюде-
нии скорости оседания порошка из суспензии. С этой целью могут быть исполь-
зованы торзионные весы, прибор Андреазена или седиментационные весы любого
типа, например весы фирмы Сарториус. Суть метода заключается в том, что через
определенные промежутки времени определяют массу осевшего порошка люми-
нофора и по полученным данным строят график, на котором по оси ординат
откладывают массу, а по оси абсцисс — время. По полученной на основании
181
экспериментальных данных кривой рассчитывают процентное содержание фрак-
ций, осевших к любому заданному моменту времени. Для этого в заданных точ-
ках к кривой проводят касательные, которые продолжают до пересечения с осью
ординат. Отношение массы, соответствующей точке пересечения к общей массе
осевшего люминофора дает (при умножении на 100) процентное содержание
фракции, осевшей к данному моменту времени. Линейные размеры частиц d
(в мкм) вычисляют, в соответствии с законом Стокса, по формуле
где ц — вязкость жидкости;
— плотность люминофора;
уж — плотность жидкости, в которой оседает люминофор;
h — высота осаждения люминофора;
t — время осаждения.
При микроскопическом методе особенно удобно использовать проекционные
микроскопы со сменной оптикой, дающие увеличение 300—800. Гранулометри-
ческий состав порошка люминофора определяется простым подсчетом в поле
зрения числа зерен, максимальный размер которых находится в определенном
диапазоне. Чтобы результат не зависел от случайного распределения зерен по
предметному стеклу, подсчитывается не менее 300—500 зерен. На предметное
стекло люминофор обычно наносится с водой, спиртом или иной жидкостью.
На основании полученных данных строят кривую распределения по величине
зерен, откладывая по оси ординат число зерен, а по оси абсцисс — их размер
в мкм. При измерении частиц меньше 0,5 мкм с помощью микроскопического
метода встречаются принципиальные трудности. Точность измерения величины
отдельных зерен ограничена тем, что размеры элементарных кристалликов
по разным направлениям различны. Для получения статистически точных ре-
зультатов при небольшой величине отбираемых проб и исключения случайных
ошибок, допускаемых в процессе отбора проб, требуется многократное повторе-
ние измерений. Кроме того, существенное влияние на результаты оказывают
субъективные ошибки наблюдателя.
IX.11. ИЗМЕРЕНИЕ СТАБИЛЬНОСТИ ЭЛЕКТРОЛЮМИНОФОРОВ
Для ускоренного определения стабильности электролюминофоров, которая,
как уже было отмечено ранее (см. стр. 137), является одной из важнейших харак-
теристик, было предложено несколько методов [32—34]. Метод, описанный
в работе [32], основан на использовании универсальности кривой старения (35,
36], согласно которой стабильность электролюминофоров определяется общим
числом циклов действия электрического поля, т. е. произведением частоты
возбуждающего поля на время его приложения. По этому методу измерение
стабильности электролюминофоров производят в разборных ячейках без при-
менения какого-либо специального диэлектрика (им служит воздух). Испытуемые
люминофоры равномерно наносят на электроды 2 (см. рис. IX.7, стр. 171) раз-
борных ячеек с плотностью ~ 15 мг/сма. Ячейки с нанесенными люминофорами
надевают на токоподводящие контакты, укрепленные на вкладыше эксикатора.
Так как стабильность электролюминофоров в сильной степени зависит от влаж-
ности среды, в которой производят испытания, то для создания'определенной
влажности на дно эксикатора насыпают слой свежего хлорида кальция. Экси-
катор закрывают крышкой из органического стекла, края которой для лучшей
герметизации смазывают вакуумной смазкой. Ячейки с испытуемыми люмино-
форами выдерживают в эксикаторе с хлоридом кальция в течение не менее 10 ч.
Затем через клеммы, имеющиеся на крышке эксикатора, к ячейкам подают от
звукового генератора переменное напряжение (200 В и 10 кГц) и несколько раз
измеряют интенсивности свечения всех люминофоров. Средние значения при-
нимают за 100%. Через определенные промежутки времени измерения интенсив-
ности свечения повторяют и полученные значения интенсивности для каждого
люминофора выражают в процентах от соответствующей начальной величины^.
Из графика зависимости: интенсивность свечения люминофора в % от начальной
величины — длительность испытаний, находят величину остаточной яркости
182
свечения через любой промежуток времени. Предложенный метод позволяет
более чем в 10 раз сократить время, необходимое для определения стабильности
электро люминофоров, так как старение проводится на повышенной частоте
возбуждающего поля. Применение этого метода на практике показало, что*
существует четкая корреляция между стабильностью порошков электролюмино-
форов, измеренной по ускоренному методу, и стабильностью их в готовых изде-
лиях при обычных рабочих режимах.
ЛИТЕРАТУРА
1. Левшин В. Л. Фотолюминесценция жидких и твердых веществ. М.—Л.г
Гостехиздат, 1951. 456 с. — 2. Гуревич М. М. Цвет и его измерение. М.—Л.,
Изд. АН СССР, 1950, 268 с. — 3. Москвин А. В. Катодолюминесценция. Ч. 2.
Катодолюминофоры и экраны. М.—Л., ГИТТЛ, 1949. 700 с. — 4. Шишлов-
ский А. А. Прикладная физическая оптика. М., Физматгиз, 1961. 822 с. —
5. Пр ингсгейм П. Флуоресценция и фосфоресценция. Пер. с англ. Под ред.
С. И. Вавилова. М., ИЛ, 1951. 622 с. — 6. Эпштейн М. И. Спектральные изме-
рения в электровакуумной технике. М., «Энергия», 1970. 143 с. — 7. Зер-
нов В. А. Цветоведение. М., «Книга», 1972. 239 с. — 8. Справочная книга по-
светотехнике. Т. 1. Световые приборы и источники света. М., Изд. АН СССР,
1958. 454 с. — 9. Почтарев Б. И. Изв. АН СССР. Сер. физ., 1961, т. 25, с. 514—
516. — 10. Левшин В. Л., Арапова Э. Я., Блажевич А. И. и др. «Труды ФИАН
им. П. И. Лебедева», 1963, т. 23, с. 64—135.
11. Рамазанов Л. Е., Шапошников Л. Н. Изв. вузов. Физика, 1971, № 12,
с. 155. — 12. КийсВ. И., Нымм У. X., Паз Л. Я. и др. Приборы и техн. эксп.,.
1960, № 4, с. 145. — 13. Ишунин В. К., Кузнецов В. Б. В кн.: Сборник науч-
ных трудов ВНИИЛ. Вып. 8. Ставрополь, изд. ВНИИЛ, 1973, с. 128—130. —
14. Иванов А. А. Оптика рассеивающих сред. Минск, «Наука и техника», 1969г
592 с. — 15. Брумберг Е. М., Пекерман Ф. М. Изв. АН СССР. Сер. физ., 1949,
т. 13, с. 218—223. — 16. Аленцев М. Н., Винокуров Л. А. Там же, 1951, т. 15г
с. 725—729. — 17. Gergely G. Z. phys. Chem., 1957, Bd. 207, S. 81—85. —
18. Bril A. In: Luminescence of Organic and Inorganic Materials. International
Conference. New York — London, J. Wiley Sons, 1962, p. 479—493. —
19. Bauer G. T. Acta Phys. Acad. Sci. Hungaricae, 1960, v. 11, p. 225—234. —
20. Пекерман Ф. M., Петошина Л. H. В кн.: Сборник рефератов научно-иссле-
довательских работ по химии и технологии люминофоров за 1957 г. Л.,
изд. ГИПХ, 1958, с. 29—31.
21. Bril A., Klasens Н. A. Phil. Res. Repts, 1952, v. 7, p. 401—420.—
22. Ludwig G. W., Kingsly I. D. J. Electrochem. Soc., 1970, v. 117, p. 348—
353. — 23. Ларах С., Харди А. ТИИЭР, 1973, t. 19, № 7, c. 144—158. —
24. Гугелъ Б. M. Люминофоры для электровакуумной промышленности. M.r
«Энергия», 1967. 344 с. — 25. Чукова Ю. П. В кн.: Оптика и спектроскопия.
Вып. 1. М.—Л., Изд. АН СССР, 1963, с. 339—347. — 26. Петошина Л. Н.,
Пекерман Ф. М. В кн.: Химия и технология люминофоров. Вып. 51. Л., «Химия»,.
1964, с. 66—74. — 27. Букке Е. Е. В кн.: Оптика и спектроскопия. Вып. 1.
М.—Л., Изд. АН СССР, 1963, с. 335—339. — 28. Фомина А. М. В кн.: Химия
и технология люминофоров. Вып. 60. Л., «Химия», 1968, с. 54—58. — 29. Тол-
стой Н. А., Феофилов П. П. Усп. физ. наук, 1950, т. 41, с. 44—107. — 30. Мяз-
дриков О. А. Электрические способы объемной гранулометрии. Л., «Энергия»,.
1968. 136 с.
31. Фигуровский Н. А. Седиментометрический анализ. М.—Л., Изд.
АН СССР, 1948. 332 с. — 32. Пекерман Ф. М., Петошина Л. Н., Дихтер М. А.
В кн.: Химия и технология люминофоров. Л., изд. ГИПХ, 1974, с. 57—59. —
33. Бляхман Э. Я., Лапин А. П. В кн.: Сборник научных трудов ВНИИЛ.
Вып. 7. 1972, с. 40—43. — 34. Ковалев В. А., Шикунова С. Т., Ведехин А . Ф.
Там же. Вып. 5. 1971, с. 98—107. — 35. Thornton W. A. J. Electrochem. Soc.r
1960, v. 107, р. 895—907. — 36. Петошина Л. Н., Пекерман Ф. М., Казан-
цева Н. Н. «Светотехника», 1964, № 12, с. 15—18.
183
предметный указатель
Активаторы 5, 7, 37, 59
Активация 37, 45, 49 и сл.
Алюминаты 54
Алюминия окись 54
активированная
Мп 55
Сг 55
Се 55, 118, 124
Алюминия фосфаты 39, 84
Алюмосиликаты 47, 50, 125 и сл.
Антистоксовские люминофоры 96, 99,
103
механизм возбуждения 97
применение 103
свойства 100 и сл.
технология изготовления 99
Арсенат
кальция 45
магния 45
активированный Мп 46, 91
магния и лития 46, 92
Бария и магния двойной силикат,
активированный РЬ 77
Бария и цинка двойной силикат,
активированный РЬ 77, 91
Бария пирофосфат, активированный
Sn 38
Ti 38
силикаты 46, 51
активированные РЬ, Се и Мп 51,
77, 88, 90
стронция и лития силикат, акти-
вированный Се и Мп 46, 90
сульфат 158 и сл.
Бериллия силикаты 47, 50, 52, 88
Ванадатные люминофоры 55, 78,
Видности коэффициент 14
Висмут, активатор 53, 93 и сл.
Возбуждение
инжекционной люминесценции,
механизм 143
люминофоров
антистоксовских, механизм 97
катодным пучком 169
рентгеновскими лучами 158
фотолюминофоров 167 и сл.
электролюминофоров 129, 130,
171
многоступенчатое 97
Волны яркости 18
Вольфраматные люминофоры 52,
125, 159 и сл.
Восстановительная среда при про-
каливании 44, 60, 84, 87, 92, 110,
ИЗ, 118
Вспышка люминесценции 25
Выгорание люминофоров 109 и сл.
Выход люминесценции 12 и сл.,
176 и сл.
Галлий
активатор 45, 55, 136
арсенид 150
нитрид 145, 149
фосфид 145, 149
Галофосфат кальция, активирован-
ный Sb и Мп 9, 61, 79 и сл.
Галофосфаты 38, 42 и сл., 79
Гасители люминесценции 26, 58, 111
Германат магния, активированный
Мп 91
Германатные люминофоры 52, 91
Германаты 52
Германий 145
активатор 45
Гетеропереход 129, 141
Гиперболический закон затухания
свечения 20 п сл., 95
Гранулометрический состав люми-
нофоров 112, 131, 159
измерение 181
Дикальций фосфат 39 и сл., 69, 80
Диэлектрики для электролюминес-
центных конденсаторов 130, 14*
484
Длительность послесвечения 23,
117, 121 и сл., 180
Европий, активатор 55, 85, 118 и сл.
Железо
активатор 37, 55
гаситель люминесценции 26, 58,
111
Закон Стокса-Ломмеля 8
Затухание люминесценции 19 и сл.,
121 и сл.
измерение 180
рекомбинационное 21
экспоненциальное 20
Золото, активатор 37, 112
Зонная схема люминесценции 73
и сл.
Излучатели а и (3 163 и сл.
Излучения
выход 15
спектры 8 и сл.
измерение 173
Излучения спектры при
возбуждении электрическим по-
лем 11, 131 п сл.
катодовозбужденпи 10
фотовозбуждении 8 и сл., 81, 100
Измельчение люминофоров 61
Изотопы радиоактивные, возбужде-
ние люминофоров 162 п сл.
Индий, активатор 45
Интенсивность
возбуждения, влияние на люмине-
сценцию 9, И, 16 и сл.
вспышки 25
свечения 16 п сл., 171 и сл.
Инфракрасное излучение 25
Источники света с газообразным
тритием 163
Иттербий, активатор 98
Иттрия
оксисульфпд, активированный Ей
119
окись, активированная Ей 119
ортованадат, активированный Ей
78, 119
фосфатованадат 78, 85
Кадмия
борат 52
вольфрамат 53
селенид 30, 35
силикаты 50, 52
сульфид 30 и сл.
теллурид 30, 33, 35
Кальция
вольфрамат 52, 125, 159 и сл.
галофосфат 9, 61, 79
карбонат, схема получения 70
Кальция
окись — двуокись кремния, си-
стема 48
окись — фосфорный ангидрид, си-
стема 40
ортофосфатные люминофоры 84
и сл.
ортофосфаты 39 и сл., 84 и сл.
силикаты 46 и сл.
люминофоры 38, 45, 79 и сл.
сульфид активированный Bi 92
фосфат кислый, получение 39 и сл.
фосфаты 38 п сл., 42
Карбид кремния 150
Катодолюминесценция 106 и сл.
Катодолюминофоры
длительность послесвечения 22
для осциллографии 114
для проекционных кинескопов 114
для радиолокационных трубок 121
и сл.
для цветного телевидения 115 и
сл., 118 и сл.
для черно-белого телевидения 112
и сл., 121
для экранов с длительным после-
свечением 121 и сл.
для экранов с коротким после-
свечением 124 и сл.
для электронно-оптических пре-
образователей 124 и сл.
интенсивность свечения 17
промышленные марки 112 и сл.,
122, 125
редкоземельного типа 117 и сл.
спектры излучения 10
эксплуатационные характери-
стики 108 и сл.
энергетический выход 13, 179
Классификация люминофоров 4
Коактиваторы 25
Кобальт
активатор 37, 92
гаситель люминесценции 11, 26, 5&
Конденсатор электролюминесцент-
ный 129, 171
Конструкционные материалы в про-
изводстве люминофоров 61
Кооперативные явления 98
Координаты цветности 76, 86, 109 и
сл., 174
Коэффициент видности 14
Краски
радиоактивные 162, 164
светящиеся 92, 95
Криптон-85, возбудитель люминес-
ценции 163
Лаки для
нанесения люминофоров 95
СПД 163
Ламповые люминофоры 38, 75 п сл.
185.
Лампы бактерицидные, ДРТ, СВДШ
167 и сл.
Люминофоры
антистоксовские 96 и сл.
временного действия 92 и сл.
для газосветных ламп 89
для дозиметрии ядерных излуче-
ний 165
для люминесцентных ламп 38, 46,
75 и сл.
высокого давления 77, 84, 91
низкого давления 75 и сл.,
79 и сл., 86 и сл.
для радиолокационных трубок
121 и сл.
для рентгеновских экранов 158
и сл.
для светящихся красок 95
для цветного телевидения 106, 115
и сл., 118 и сл.
для черно-белого телевидения 106,
112, и сл., 121
для электронно-лучевых трубок
106 и сл.
для эритемных ламп 87, 91
особенности синтеза 58 и сл.
с ультрафиолетовым излучением
46, 77, 87, 90
Магния
арсенат, активированный Мп 46, 91
вольфрамат 52, 76
германаты 52
окись — двуокись кремния, си-
стема 49
окись — фосфорный ангидрид, си-
стема 40
ортофосфаты 38, 84, 86
силикаты 46
активированные Мп 46, 50, 114
активированные Се и Мп 51
сульфид 124
фторгерманат, активированный
Мп 90
фторсиликат, активированный Ti
77, 89, 90, 123
Малоинерционные люминофоры 117,
118, 124 и сл.
Марганец, активатор 38, 45, 49, 52,
55, 79 и сл., 88 и сл., 114, 122,
134, 138, 152
Медь, активатор 37, 92 и сл., 112,
115 и сл., 121 и сл., 130 и сл.
Меди сульфид 130
Минерализующее действие 46, 48,
52, 89 и сл.
Нанесение люминофоров 95, 163
Насыщение свечения 17
Никель
активатор 37
Никель
гаситель люминесценции 23, 111,
124
Ниобий, активатор 122
Обработка поверхности люминофо-
ров 61, 111, 115
Окисные люминофоры 54, 118 и сл.,
124 и сл.
Окисные системы 53 и сл.
Олово, активатор 38, 44, 84 и сл.
Ортофосфатные люминофоры 84 и сл.
активир ованные
Sn 84 и сл.
Т1 87
Ортофосфаты
кальция 39 и сл., 84 и сл.
магния 39 и сл., 84 и сл.
стронция 39 и сл., 84 и сл.
цинка 39 и сл., 84 и сл.
Переход р—п 143
Плавни 6, 91, 93, 111
Платина, активатор 55
Послесвечение 21
катодолюминофоров 22, 117, 121
и сл.
методы измерения 180
Примеси 34, 58, 111
Прокаливание люминофоров 59, 62
Прометий-147, возбудитель люми-
несценции 162 и сл.
Радий, соли для возбуждения люми-
несценции 162
Радиоактивные препараты для
возбуждения люминофоров 162 и
сл.
Радиолюминофоры 4, 162 и сл.
Разборная электронно-лучевая труб-
ка 169
Разборный электролюминесцентный
конденсатор 171
Разгорание люминесценции 19 и сл.
. Разрушение люминофоров 80, 108
и сл., 162
Редкоземельные активаторы 38, 55,
78, 85, 99, 117 и сл.
Рекомбинационные люминофоры 5,
20, 73
Рентгенолюминофоры 4, 158 и сл.
Светодиоды 143 и сл.
на основе соединений
АП BVI 150
Ain bv 149
Светоотдача 14, 178 и сл.
Светофильтры 167, 169, 172
Свинец, активатор 46, 52, 88 и сл.,
159 и сл.
Селен, активатор 54
Селенидные люминофоры 37, 125
186
Селениды кадмия и цинка 29 и сл.
Сенсибилизация 8, 45, 49, 81, 89
Серебро, активатор 37, 108, НО,
112 пел. 117, 123, 125, 127, 159,
160
Силикатные люминофоры 46,50 и сл.,
60, 88 и сл., 114,118,124и сл.
для газосветных ламп 89
для ламп с ультрафиолетовым из-
лучением 90
для люминесцентных ламп 88
и сл.
для черно-белого телевидения
114
с коротким послесвечением 118,
124 и сл.
Соактиваторы 25
СПД 162 и сл.
Спектры
возбуждения 8, 176
вспышки 26
излучения 8 и сл., 173
измерение 173 и сл.
поглощения 7, 175
стимуляции 26
Среда восстановительная при
прокаливании люминофоров 44,
60, 84, 87, 92, НО, 113, 118
Стабильность
измерение 182
катодолюминофоров 113
ламповых люминофоров 83
светодиодов 154
светосоставов постоянного дей-
ствия 162 и сл.
электролюминофоров 134, 137
Стойкость
примесная катодолюминофоров
111
радиационная
катодолюминофоров 109 и сл.
фотолюминофоров 83
термическая
катодолюминофоров 109
фотолюминофоров 81
химическая катодолюминофоров
108 и сл.
Стронций-90, возбудитель люми-
несценции 163
Сульфидные люминофоры
временного действия 92 и сл.
вспышечные 26
для дозиметрии 165
для рентгеновских экранов 158
и сл.
для светосоставов постоянного дей-
ствия 162 и сл.
для светящихся красок 95
для цветного телевидения 115 и сл.
для черно-белого телевидения
112 и сл.
электролюминофоры 130 и сл.
Сульфид-селенидные люминофоры
35, 124 и сл., 135 и сл.
Сульфид-селениды 35
Сульфиды 29 и сл.
щелочноземельных металлов 26,
93 и сл.
Сурьма, активатор 7, 9, 45, 79 и сл.
Сцинтилляция 165
Сырье для синтеза люминофоров 58,
63 и сл.
Таллий, активатор 45, 52, 77, 87
Таллий-204, возбудитель люминес-
ценции 163
Тантал, активатор 122
Тауметр 180
Твердые растворы 35
АШ Bv 149
Теллурид
кадмия 33, 35
цинка 33, 35, 153
Тербий, активатор 85, 121
Термического высвечивания кривые
23 и сл.
Тип проводимости соединений AnBvl
30, 151
Титан, активатор 38, 45, 49, 55, 89,
123
Тритий, возбудитель люминесцен-
ции 162 и сл.
Тритиевые источники света 163
Тритиевые краски самосветящиеся
164
Тулий, активатор 38, 99, 121
Тушение
концентрационное 119
коэффициент 26
под действием инфракрасного излу-
чения 25
температурное 16 и сл., 84, 90
Углерод-14, возбудитель люминес-
ценции 163, 165
Фосфатные люминофопы 67. 79 и сл.,
106, 118, 121
активированные Т1 87
галофосфатные 79 и сл.
для люминесцентных ламп 79
и сл.
для электронно-лучевых трубок
106, 118, 121
ортофосфатные, активированные
Sn 84 и сл.
с ультрафиолетовым излучением 87
Фосфатные системы 38 и сл.
Фосфаты
двойные 38, 44
кальция 38 и сл., 42
магния 38 и сл.
стронция 38
цинка 38, 41, 44
187
•Фосфаты
щелочноземельных металлов 44
Фосфороскоп 180
Фотолитическое разрушение люмино-
форов 96
Фотолюминесценция 7 и сл., 12, 16,
19 и сл. 25
Фотолюминофоры 71
Фотометры 171 и сл.
Фотонное умножение 13
Фракционирование люминофоров
61, 80
Фторгерманат магния, активиро-
ванный Мп 52, 91
Фторидные люминофоры 106, 122
Фторсиликаты 89
магния, активированные Ti 89,
123
Характеристические люминофоры
5, 13, 19, 73
Хром, активатор 53 и сл.
Цветовые характеристики люмино-
форов 76, 77, 85, 87, 118
Центры люминесценции 5, 7, 9, 74
Церий, активатор 45, 49, 54, 87, 90,
117, 124 и сл.
Цинка
вольфрамат ы 53
окись
активированная Se 54
люминофор 10, 53, 124 и сл.
селенид 30, 32 и сл., 152 и сл.
силикаты 47, 50, 52
сульфат 64 и сл.
сульфид 29 и сл.
теллурид 30, 33, 35, 153
фосфаты 38, 41, 44
фторид, активированный Мп 122
Цинкбериллий силикатные люми-
нофоры 10, 50, 76, 88, 110, 114,
115
Цинккадмий сульфидные люмпно- ,
форы 10, 36, 110,112,115 исл.,
120 и сл., 125, 135, 158 и сл.
Цинккадмий сульфидселенидные
люминофоры 135 и сл.
Цинккальций фосфатные люминофо-
ры 84 и сл.
Цпнкортофосфатные люминофоры
39, 106, 117
Цинкселенидные люминофоры 37
Цинксиликатные люминофоры 10,
46, 50, 110, 114
Цинкстронций фосфатные люмино-
форы, активированные Sn 39, 84
и сл.
Цинксульфидные люминофоры 25,
36 исл., 92 исл., 112 и сл., 121
и сл., 130 и сл., 158 исл., 162
временного действия 92 и сл.
для цветного телевидения 115 и сл.
для черно-белого телевидения
112 и сл.
постоянного действия 162
рентгенолюмпнофоры 158 и сл.
электролюминофоры 130 и сл.
Цинксульфидселенидные люминофо-
ры 35, 36, 124 и сл., 135 и сл.
Циркония пирофосфат 124
Шихта для люминофоров 59
Щелочногалогенные люминофоры
165
Щелочноземельные люминофоры
временного действия 93 и сл.
Электролюминесцентный конденса-
тор 129, 171
светоотдача 14
стабильность 134, 137
Электролюминесценция 11, 14 и сл.,
23, 139 и сл.
инжекционная 143 и сл.
механизм 139 и сл., 143
применение 129
Электролюминофоры 4, 129 и сл.
инжекционные 144 и сл.
светоотдача 14
спектры излучения 11, 130 и сл.
стабильность 134, 137
сульфидселенидные 135 и сл.
цинксульфидны е 13 0 и сл.
Эпитаксия 147
Эрбий, активатор 97, 120
Эритемного действия люминофоры
87, 91
Эталоны яркости с p-из л учителями
165
Яркость 15 п сл., 173
ОГЛАВЛЕНИЕ
Предисловие ................................................... 3
Введение ........................................................ 4
Гла ва I. Основные понятия....................................... 7
1.1. Спектры поглощения и возбуждения...................... 7
1.2. Спектры излучения ..................................... 8
Фотовозбуждение ......................................... 8
Возбуждение катодными лучами............................ 10
Возбуждение электрическим полем......................... 11
1.3. Выход люминесценции .............................. . 12
Фотовозбуждение .................................... . 12
Катодное возбуждение ................................... 13
Возбуждение электрическим полем......................... 14
Выход излучения, эффективность и яркость свечения свето-
диодов ................................................. 15
1.4. Интенсивность свечения люминофоров.................... 16
Фотовозбуждение ........................................ 16
Возбуждение катодными лучами............................ 17
Возбуждение электрическим полем......................... 17
1.5. Разгорание и затухание люминесценции.................. 19
Фотовозбуждение ........................................ 19
Катодное возбуждение ................................... 22
Возбуждение электрическим полем......................... 23
1.6. Кривые термического высвечивания...................... 23
1.7. Действие на люминофоры инфракрасного излучения........ 25
Литература ..................................................... 27
Глава II. Характеристика основных классов химических соединений,
применяемых для синтеза люминофоров............................. 29
II.1. Халькогениды элементов второй группы................. 29
Физические свойства халькогенидов....................... 29
Способы получения ...................................... 31
Физико-химические свойства ............................. 32
Растворимость примесей ................................. 34
Твердые растворы на основе халькогенидов цинка и кадмия 35
Диффузия примесей ...................................... 36
Люминесцентные свойства ................................ 36
II.2. Фосфатные и некоторые близкие к ним системы.......... 38
Фосфаты ................................................ 38
Арсенаты ............................................... 45
II.3. Силикатные и некоторые близкие к ним системы......... 46
Силикаты ............................................... 46
Германаты, бораты, вольфраматы.......................... 52
II.4. Окисные системы...................................... 53
Литература ..................................................... 55
189
Глава III. Общая характеристика методов синтеза и производства
люминофоров .............................................. 58
Ш.1. Основные стадии технологического процесса................ 58
Ш.2. Аппаратурное оформление процессов производства люмино-
форов ............................................... 61
III .3. Приготовление люминофорно-чистого сырья.............. 63
Осаждение примесей ........................................ 63
Хроматографическое отделение примесей...................... 65
II I.4. Общая характеристика технологических схем получения
основных видов сырья.......................................... 66
Литература ................................................... 72
Глава IV. Фотолюминофоры *.................................... 73
1 V.1. Механизм люминесценции........................... 73
IV .2. Люминофоры для люминесцентных ламп............... 75
Требования, предъявляемые к люминофорам............... 75
Лампы низкого давления ............................... 75
Лампы высокого давления.................................... 77
Галофосфаты .......................................... 79
Фосфаты .............................................. 84
Силикаты ............................................. 88
Германаты и арсенаты....................................... 91
IV .3. Люминофоры временного действия........................ 92
I V.4. Люминофоры для светящихся красок . ............ 95
IV .5. Люминофоры для преобразования инфракрасного излучения
в видимое............................................... 96
Механизм возбуждения.................................. 97
Технология изготовления люминофоров........................ 99
Основные свойства антистоксовских люминофоров......... 100
Применение антистоксовских люминофоров................ 103
Литература ................................................... 103
Глава V. Катодолюминофоры .................................... 106
V.I . Общие сведения ................................... 106
V.2. Эксплуатационные характеристики........................ 108
Химическая стойкость...................................... 108
Термическая стойкость .................................... 109
Радиационная стойкость.................................... 109
Примесная стойкость ...................................... 111
Пути повышения стабильности............................... 111
V. 3. Люминофоры для черно-белого телевидения............... 112
Сульфидные люминофоры для кинескопов прямого видения 112
Силикатные люминофоры для проекционных кинескопов и
осциллографических трубок ............................ 114
V .4. Люминофоры для цветного телевидения.................. 115
V. 5. Катодолюминофоры редкоземельного типа................. 117
V.6 . Люминофоры для экранов с длительным послесвечением . . 121
V.7. Люминофоры для экранов с коротким послесвечением . . . 124
Литература ..................................................... 127
Глава VI. ЭлектролюмиДООЩДО ...................................... 129
VI. 1. Порощкообразцые электЛолюминофоры ........... 129
Методы синтеза плектуолгаминофоров и их люминесцентные
характер йстййи ................................. 130
Стабильность све^ншявеж'ролюминесцентных конденсаторов 137
Свечение порошкообразных люминофоров при возбуждении
постоянным полем ................................. 138
Механизм электролюминесценции ............................ 139
Совместное действие электрического поля и света на фото-
и электролюминофоры .............................. 141
VI .2. Светодиоды (низковольные электролюминесцентные источ-
ники света) ............................................. 143
190
Механизм возбуждения инжекционной электролюминесценции 143
Материалы для светодиодов............................. 144
Технология изготовления светодиодов................... 146
Свойства светодиодов различного типа.................. 149
Литература ................................................... 155
Глава VII. Рентгенолюминофоры ................................ 158
Литература ................................................... 161
Глава VIII. Светосоставы постоянного действия................. 162
VII 1.1. Светосоставы с использованием естественных радиоактив-
ных веществ.............................................. 162
VIII .2. Светосоставы с использованием искусственных (3-излучате-
лей ..................................................... 163
Источники света с газообразным тритием................ 163
Самосветящиеся краски................................. 164
VIII. 3. Люминофоры для дозиметрии ядерных излучений .... 165
Литература ................................................... 166
Глава IX. Методы измерения основных физических характеристик
люминофоров ............................................ 167
IX. 1. Возбуждение люминофоров.......................... 167
Фотолюминофоры ....................................... 167
Катодолюминофоры ................................... 169
Электролюминофоры .................................... 171
IX .2. Измерение интенсивности люминесценции............ 171
Определение относительной интенсивности свечения люмино-
форов, имеющих одинаковый спектр излучения.......... 171
Сравнение интенсивности свечения люминофоров, имеющих
различные спектры излучения......................... 173
IX. 3. Измерение яркости свечения....................... 173
IX.4 . Измерение спектров излучения..................... 173
IX.5. Измерение координат цветности...................... 174
IX.6 . Измерение спектров поглощения.................... 175
IX. 7. Измерение спектров возбуждения................... 176
IX.8 . Определение энергетического, квантового выхода и свето-
отдачи .................................................. 176
Фото люминофоры ...................................... 176
Катодолюминофоры ..................................... 178
Электролюминофоры .................................... 179
IX.9. Измеренйе длительности послесвечения.............. 180
IX.10. Измерение гранулометрического состава............. 181
IX.И . Измерение стабильности электролюминофоров...... 182
Литература ................................................... 183
Предметный указатель.......................................... 184
Олег Николаевич Казанкин,
Лев Яковлевич Марковский,
Игорь Алексеевич Миронов,
Фаня Михайловна Пекерман,
Людмила Николаевна Петошина
НЕОРГАНИЧЕСКИЕ ЛЮМИНОФОРЫ
Редактор В. А. Станкевич
Технический редактор Е. М. Соболева
Корректор В. Б. Генгут
М-18336. Сдано в наб. 21/1 1975 г. Поди, в печ. 7/V 1975 г.
Формат бумаги 60 х 90*/1в. Бумага тип. № 1. Усл. печ. л. 12.
Уч.-изд. л. 16,73. Тираж 2800 экз. Заказ 44.
Изд. № 268. Цена 1 р. 05 к.
Издательство «Химия», Ленинградское отделение,
191186, Ленинград, Д-186, Невский пр., 28.
Ленинградская типография № 6 Союзполиграфпрома при
Государственном комитете Совета Министров СССР по делам
издательств, полиграфии и книжной торговли.
196006, г. Ленинград, Московский пр., 91.