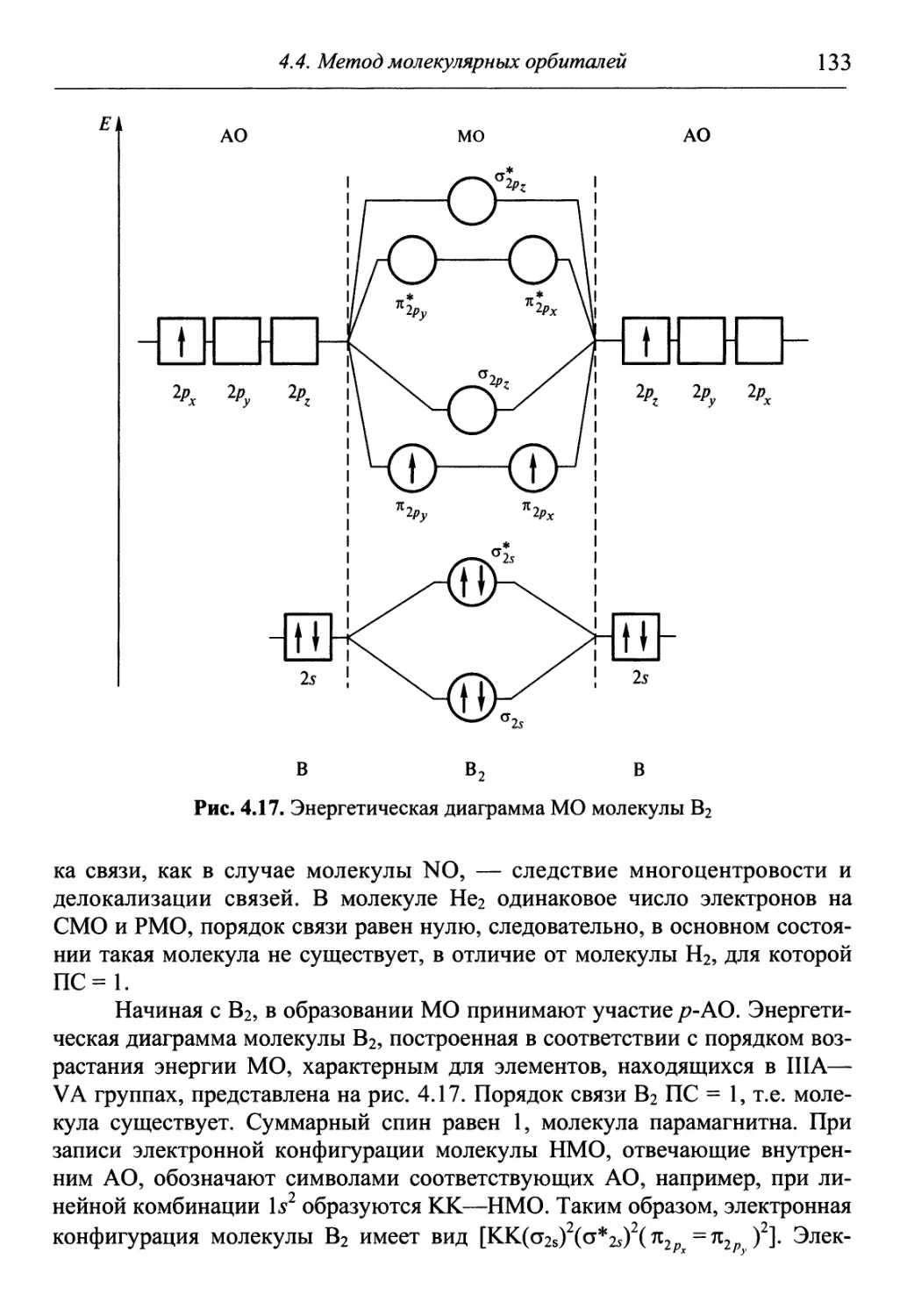
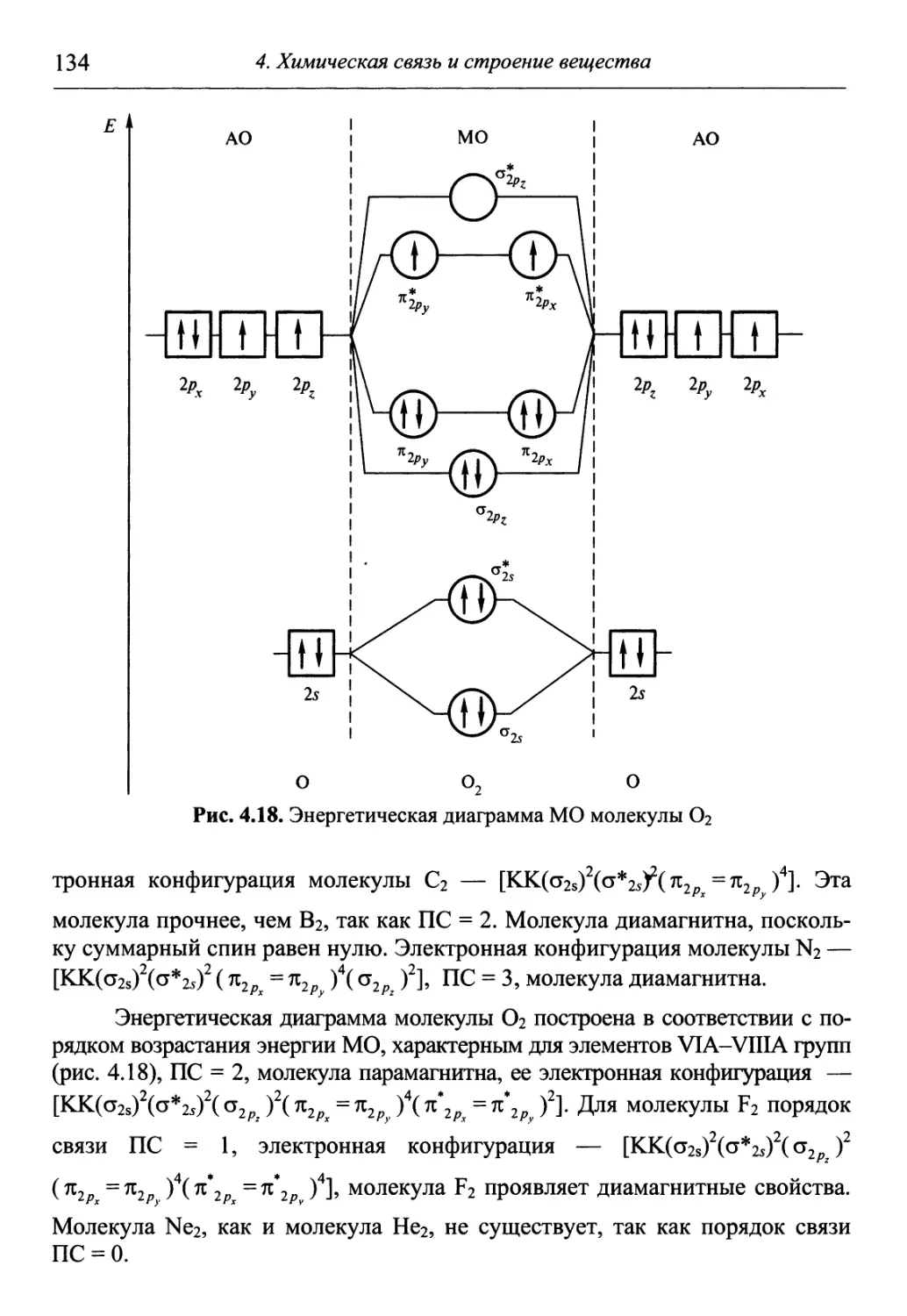
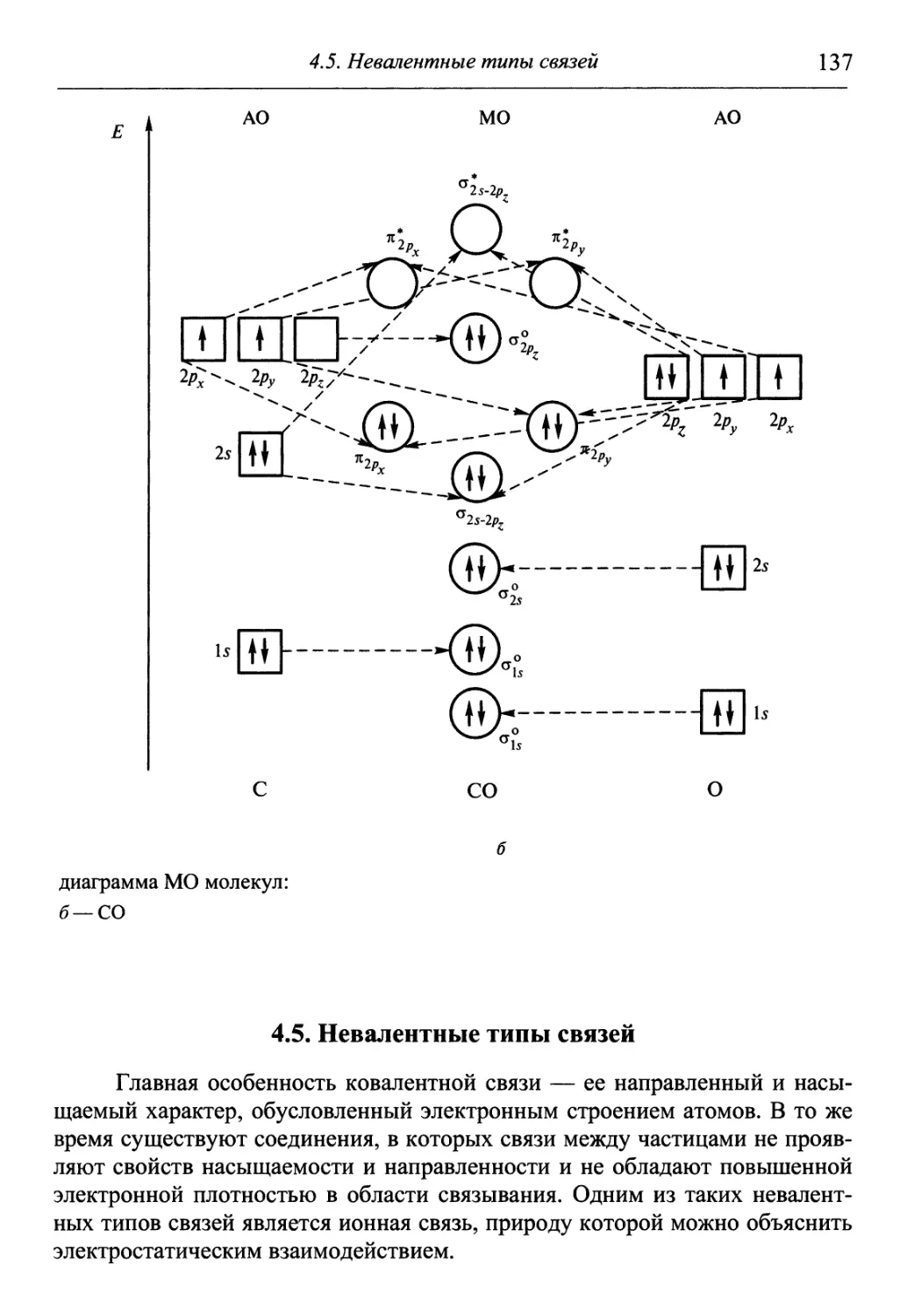
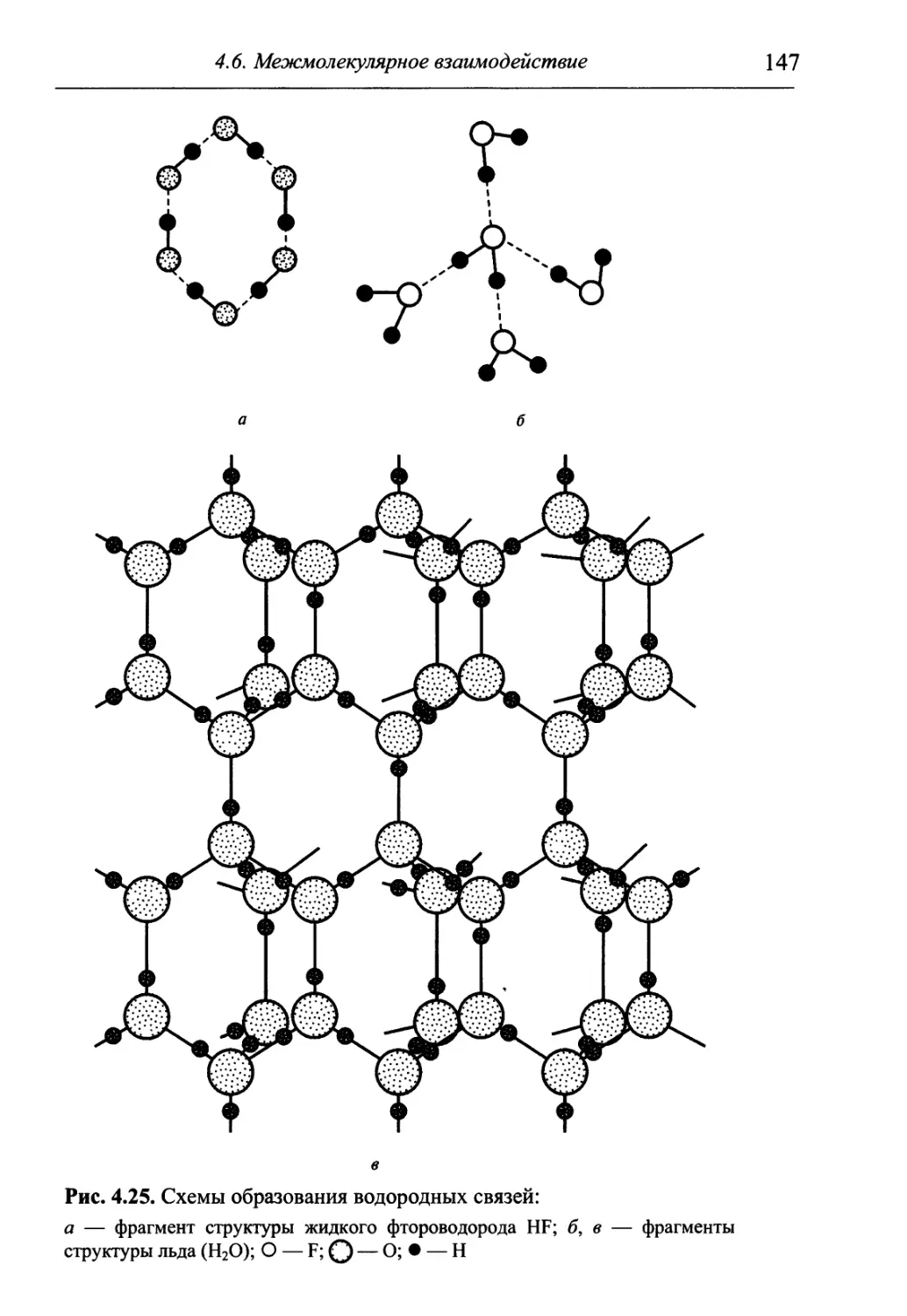
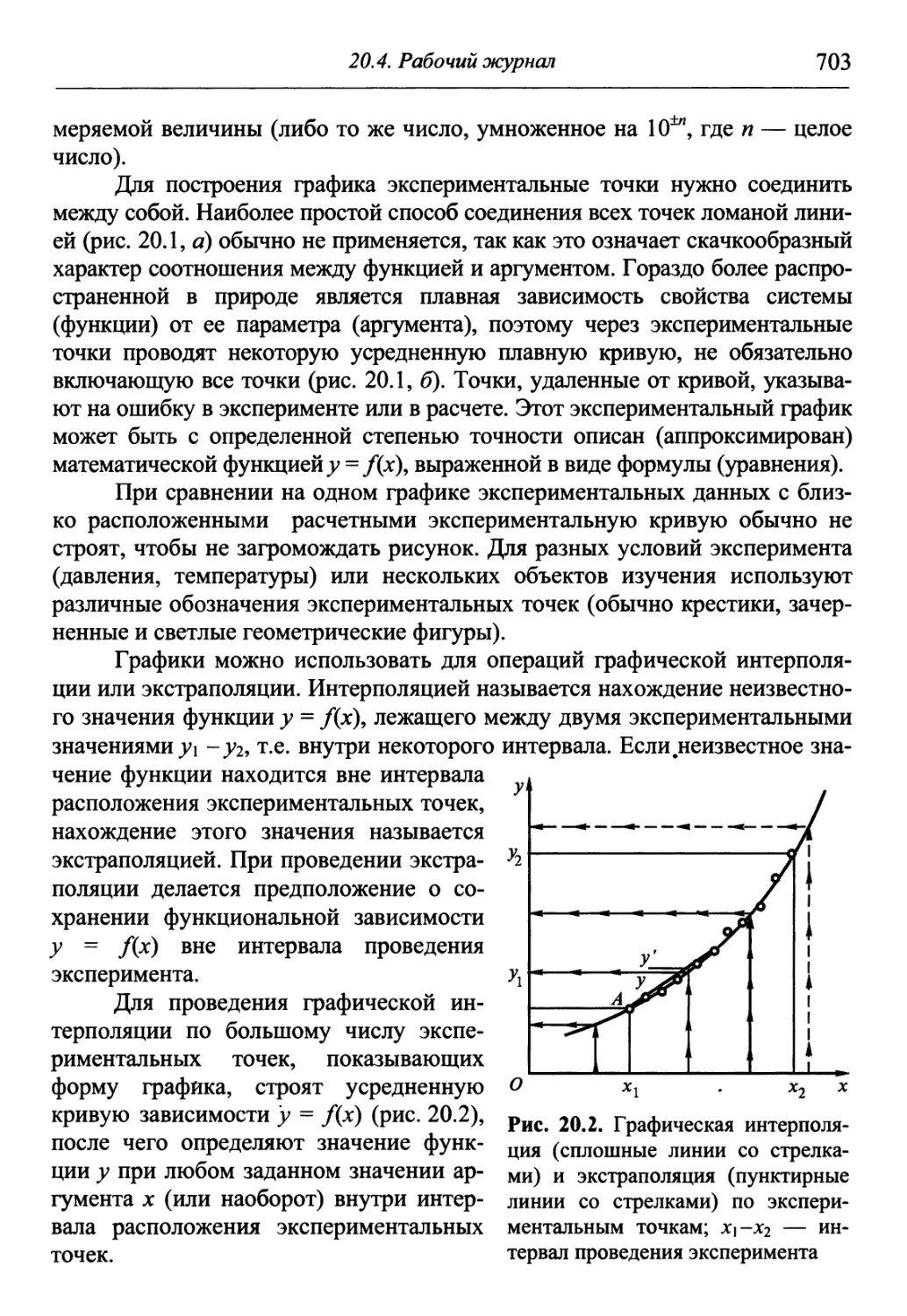
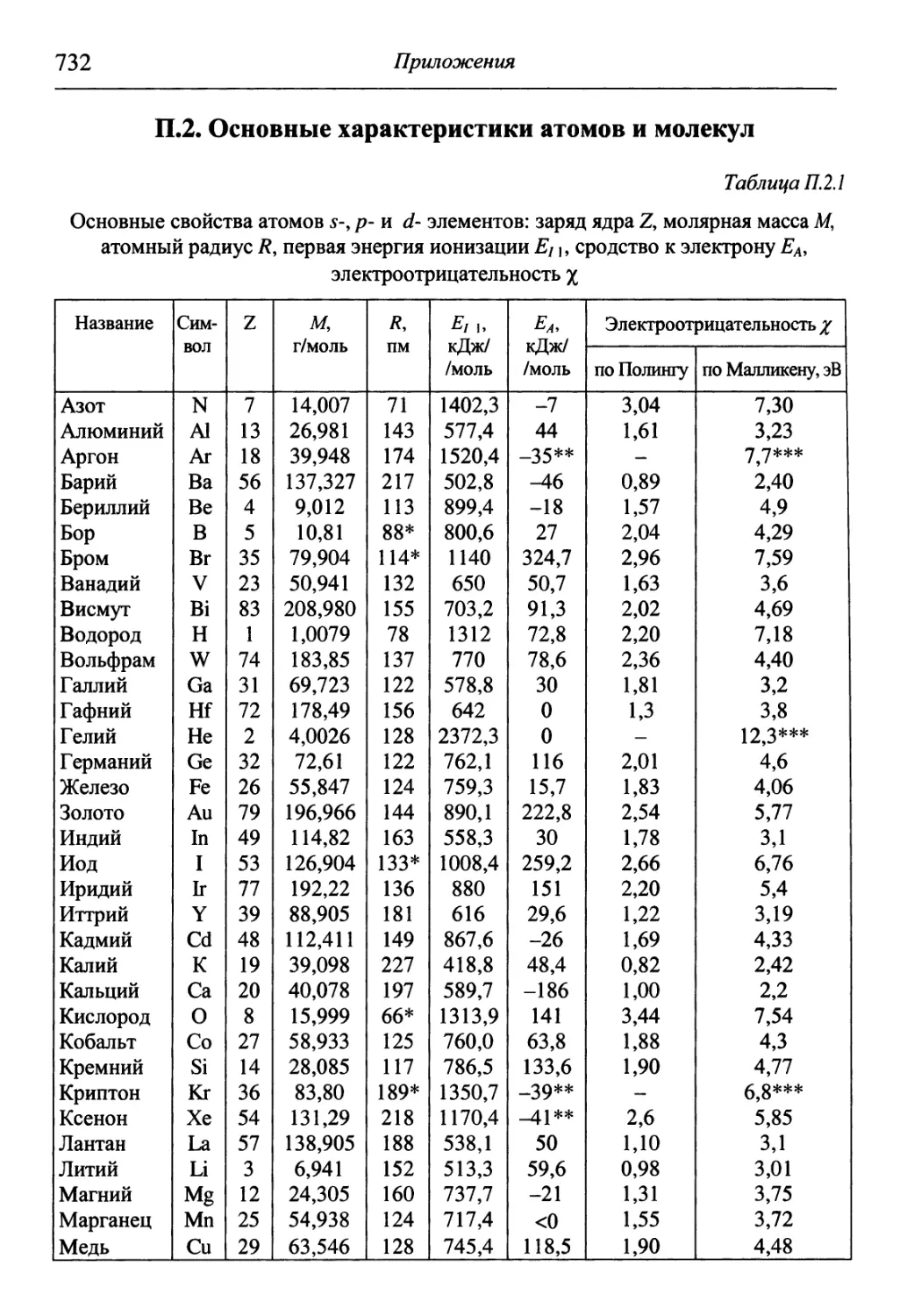
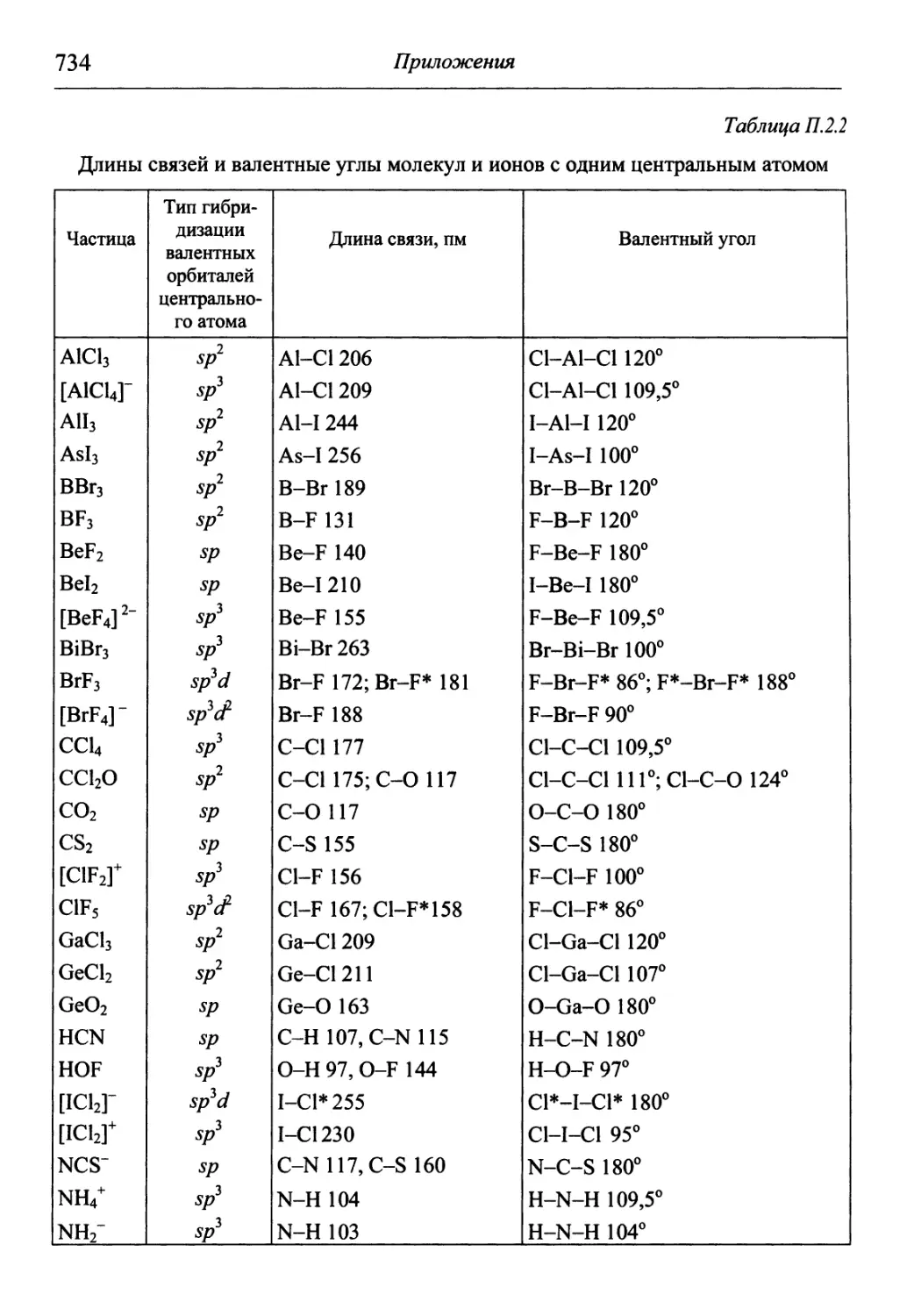
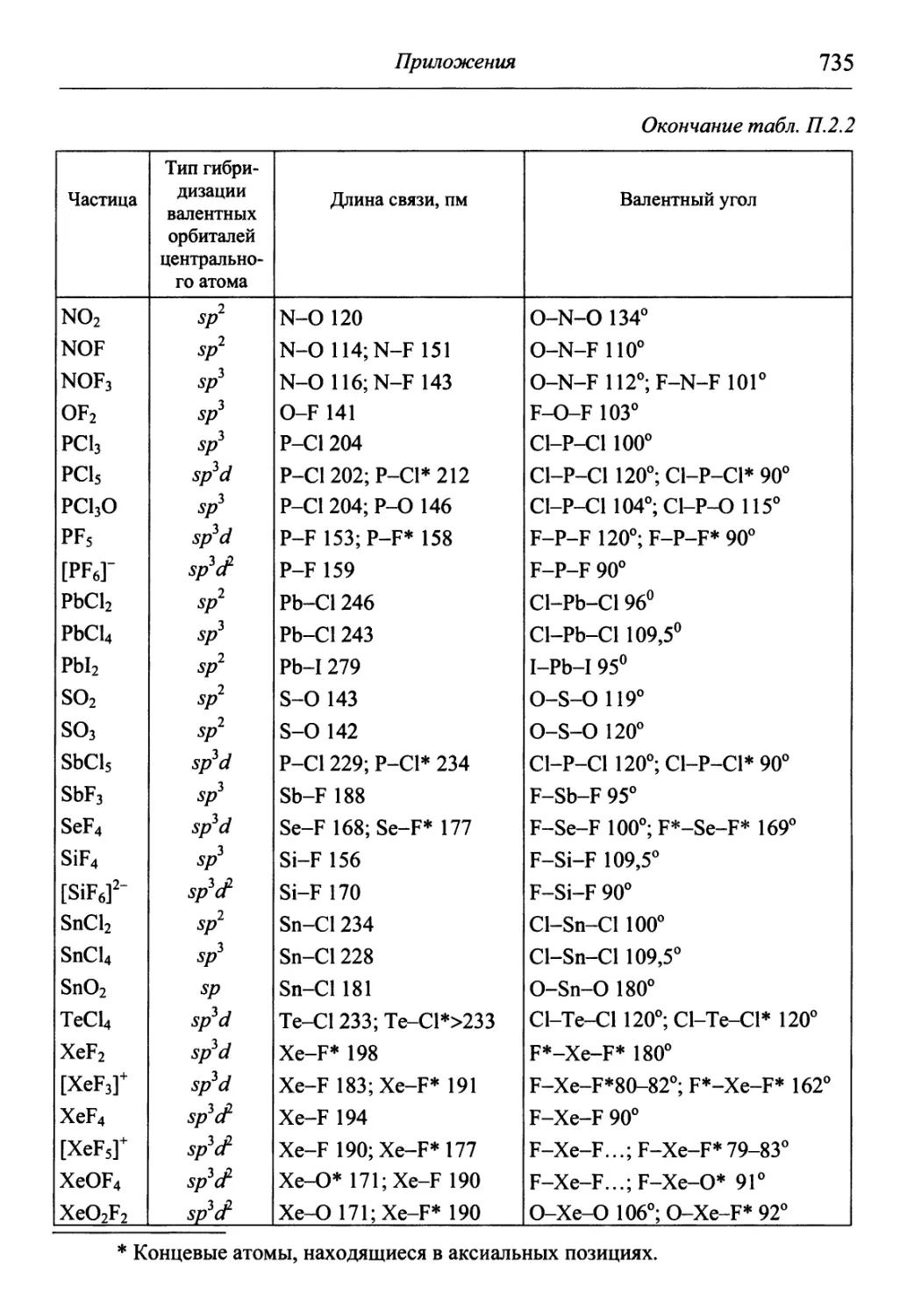
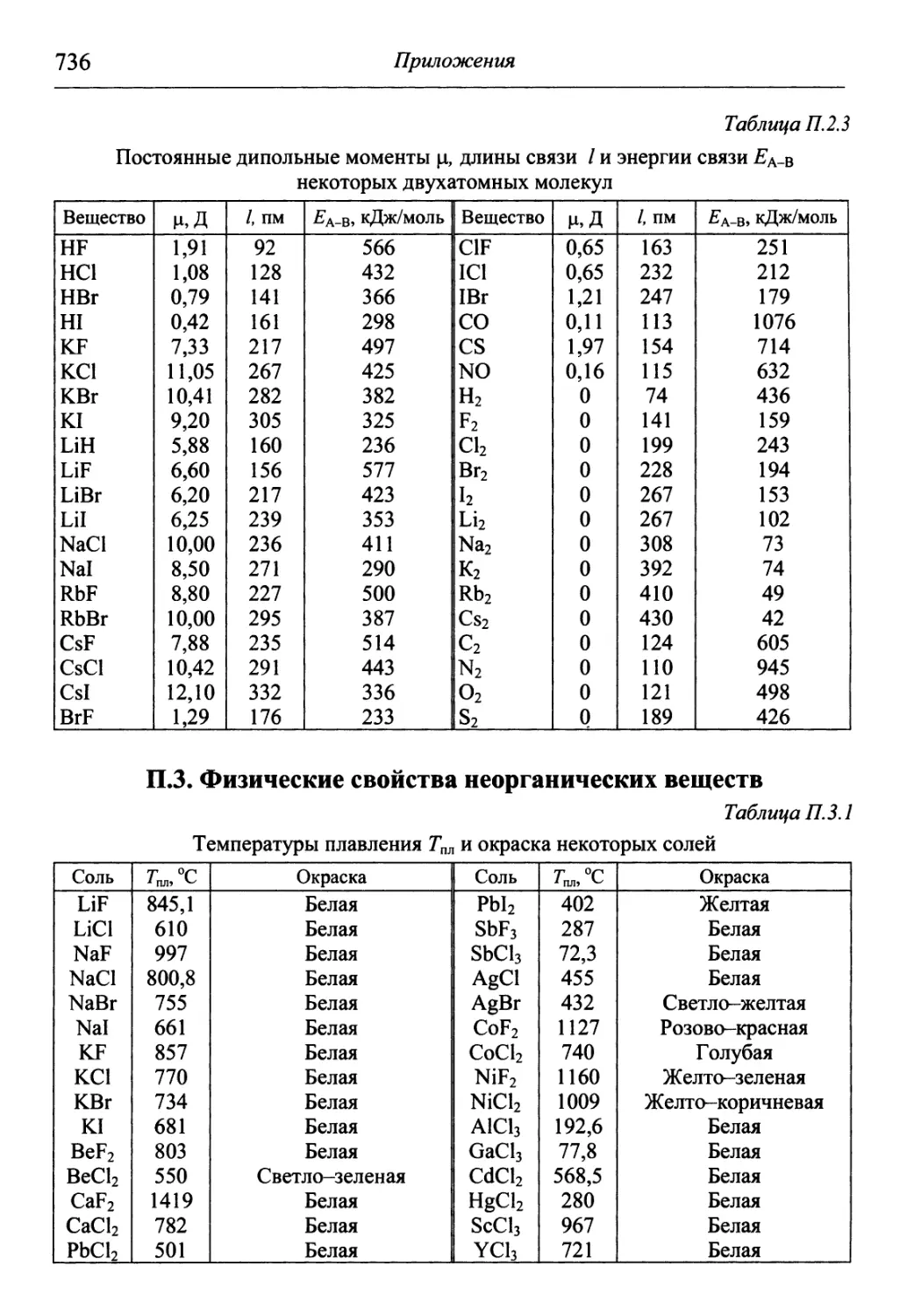
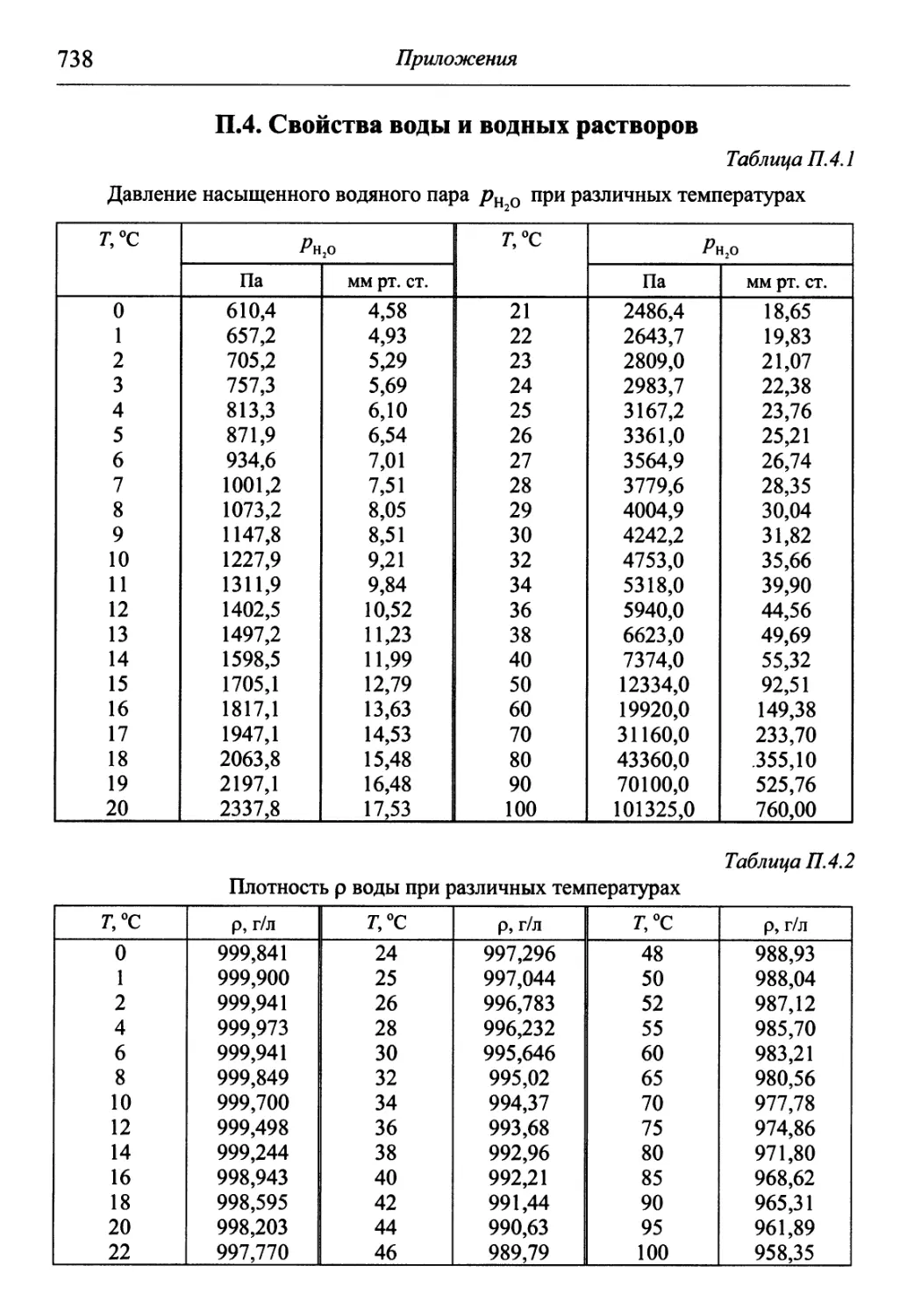
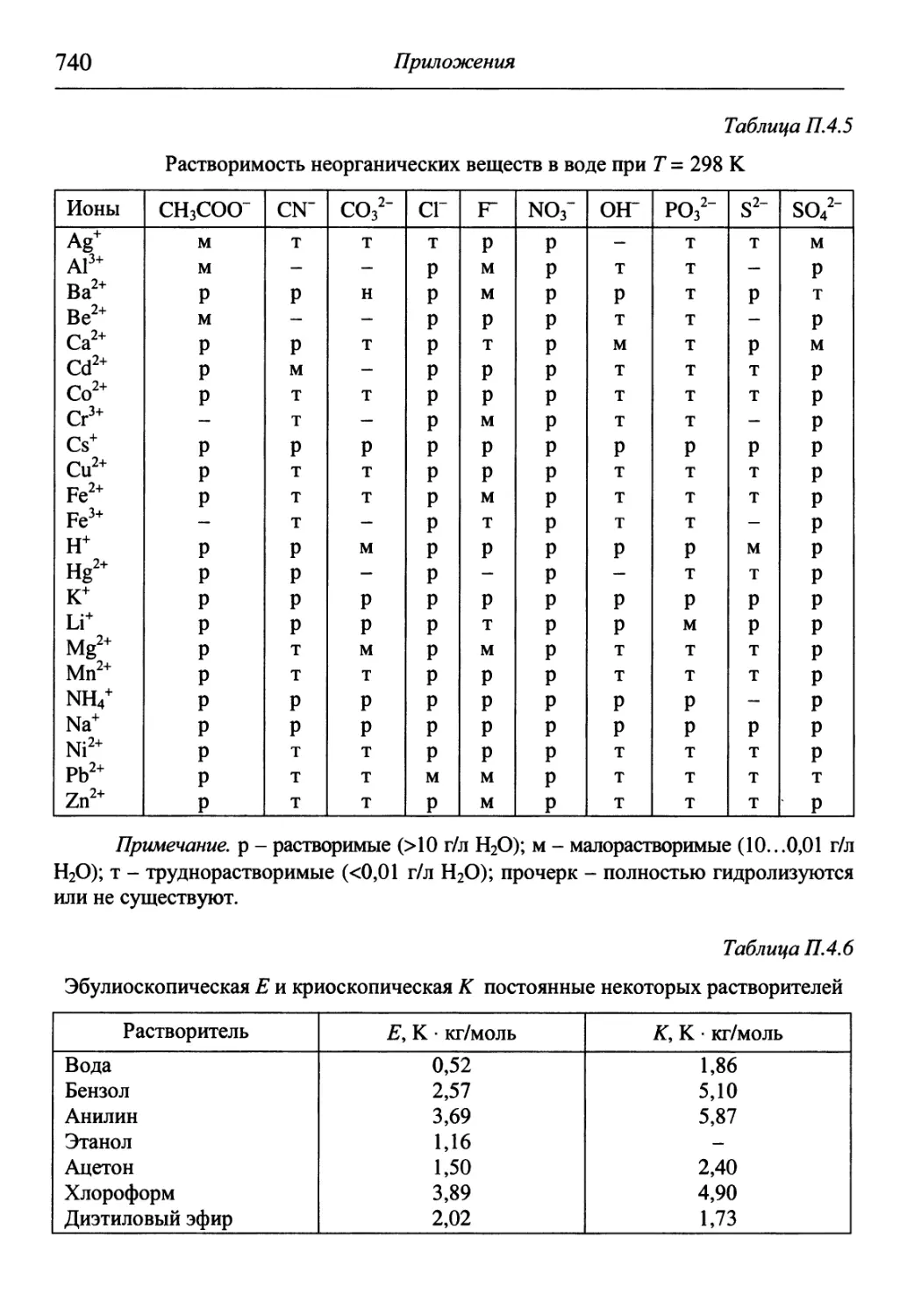
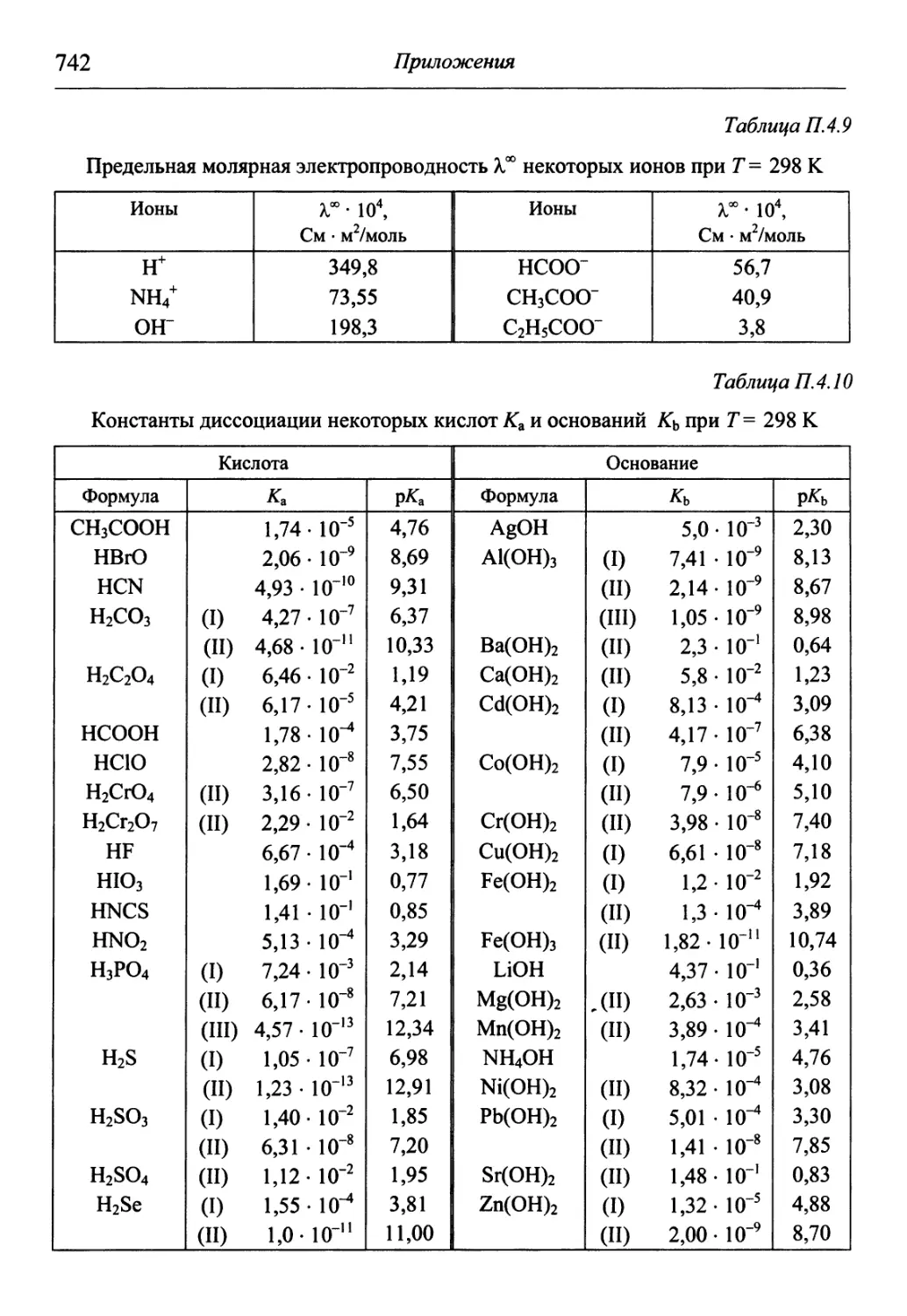
/
















































































































































































































































































































































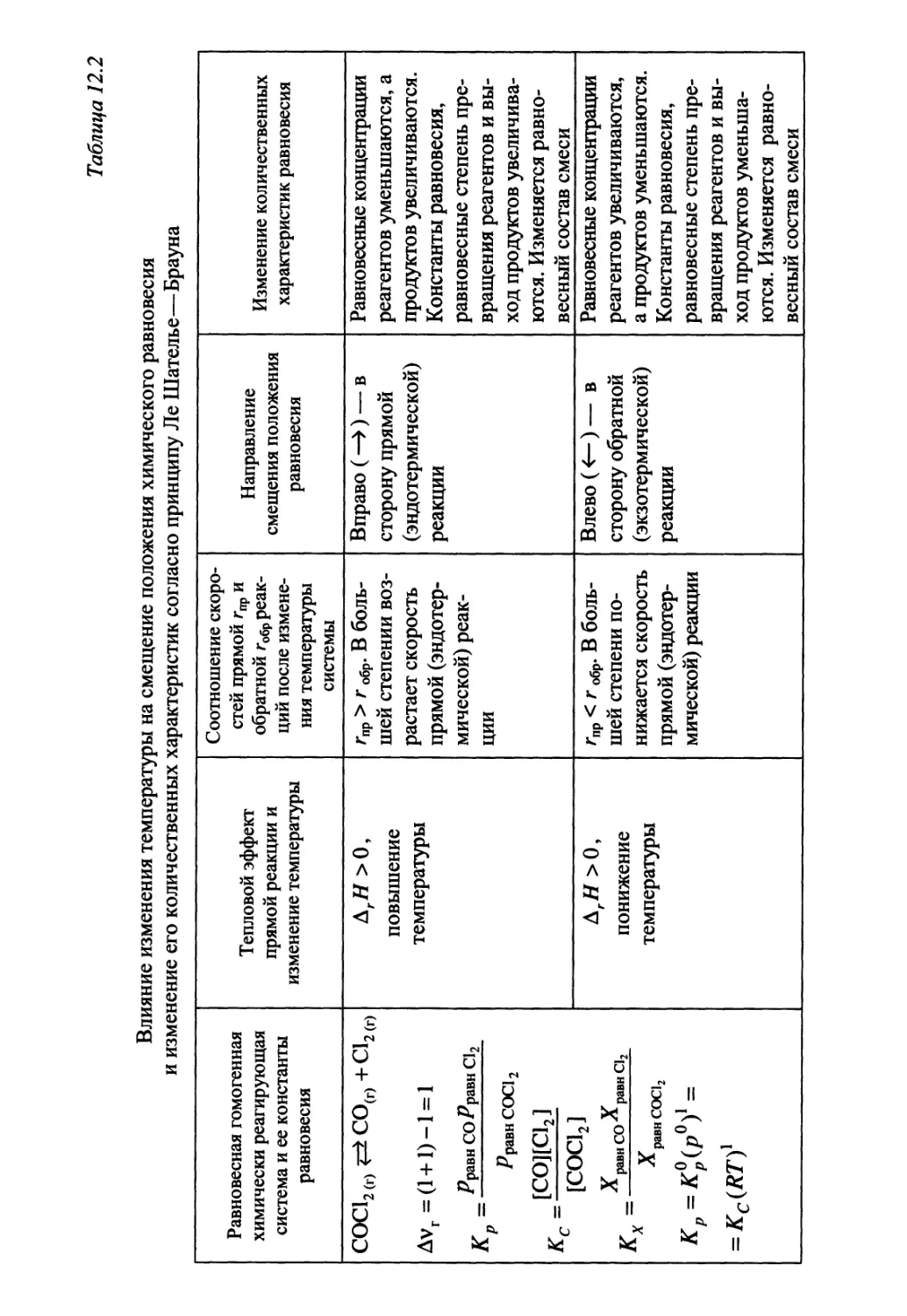



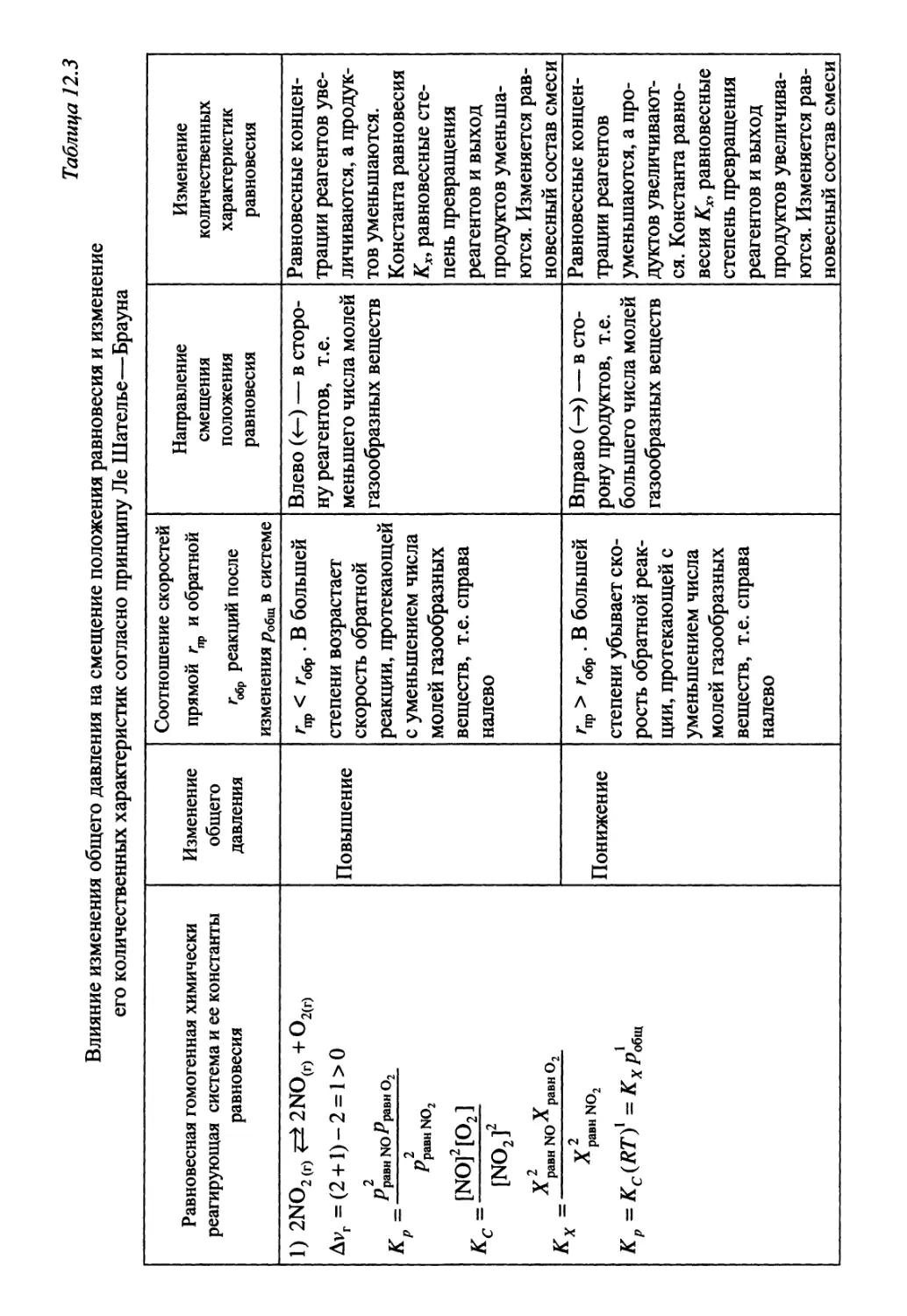
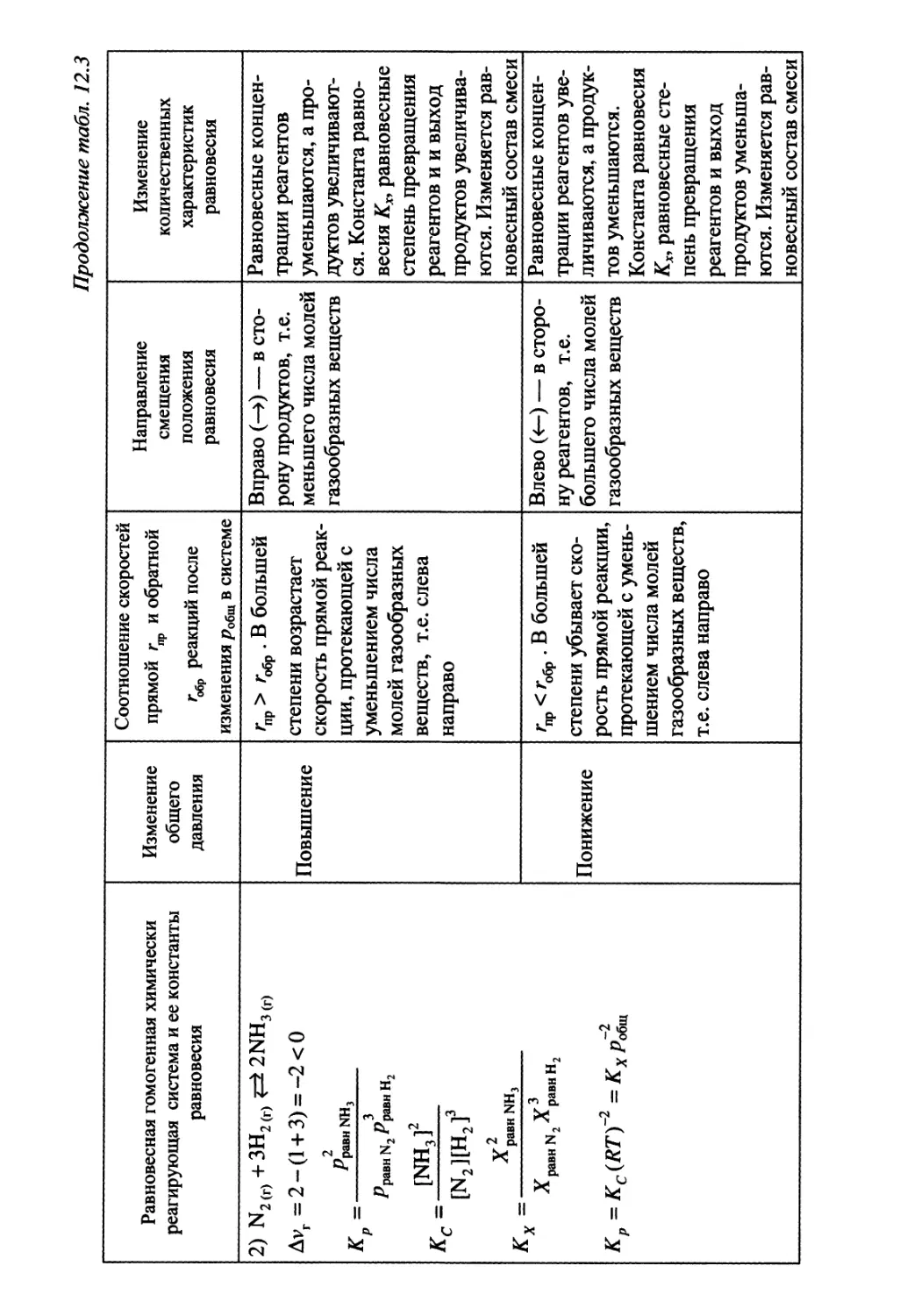























































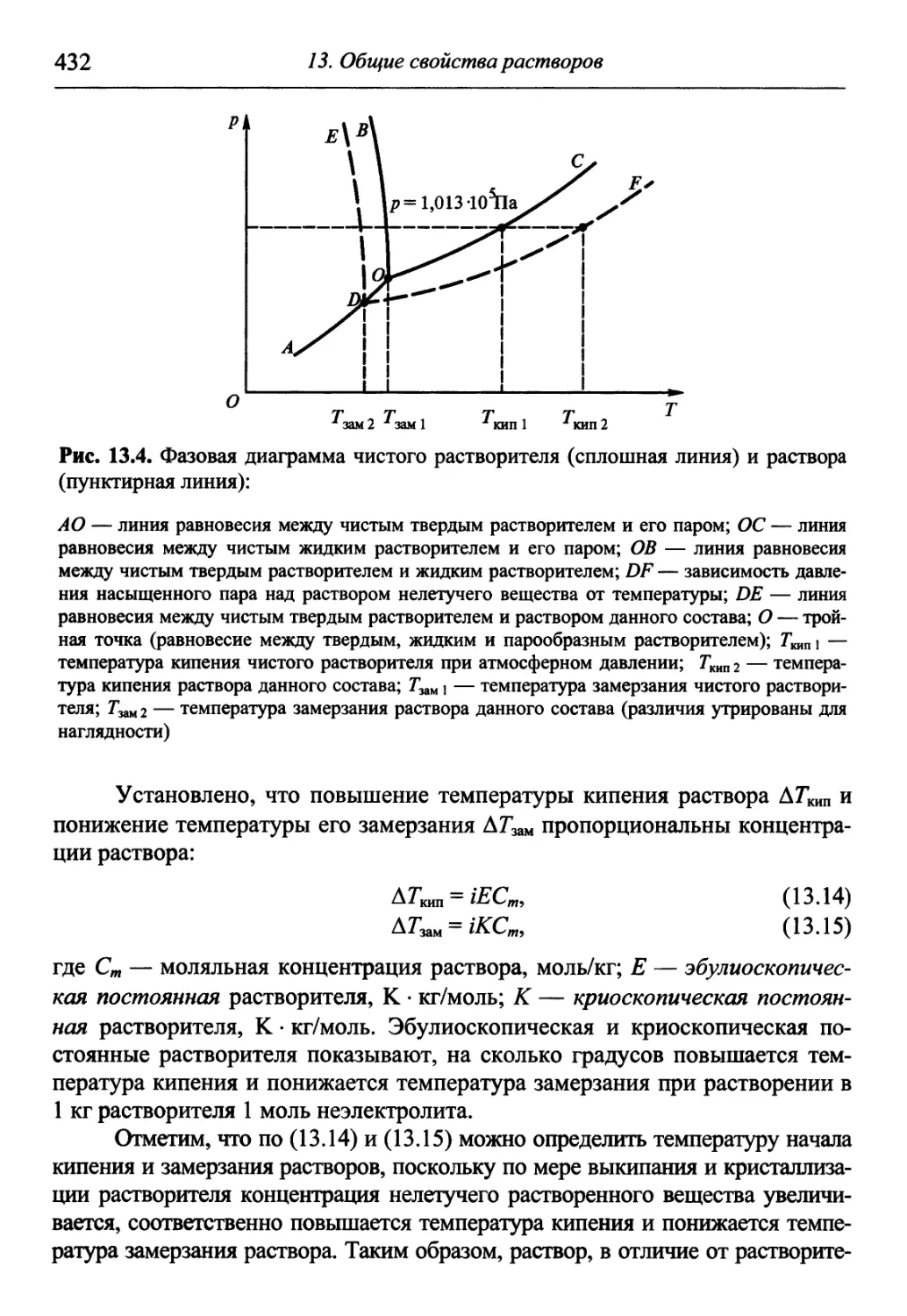








































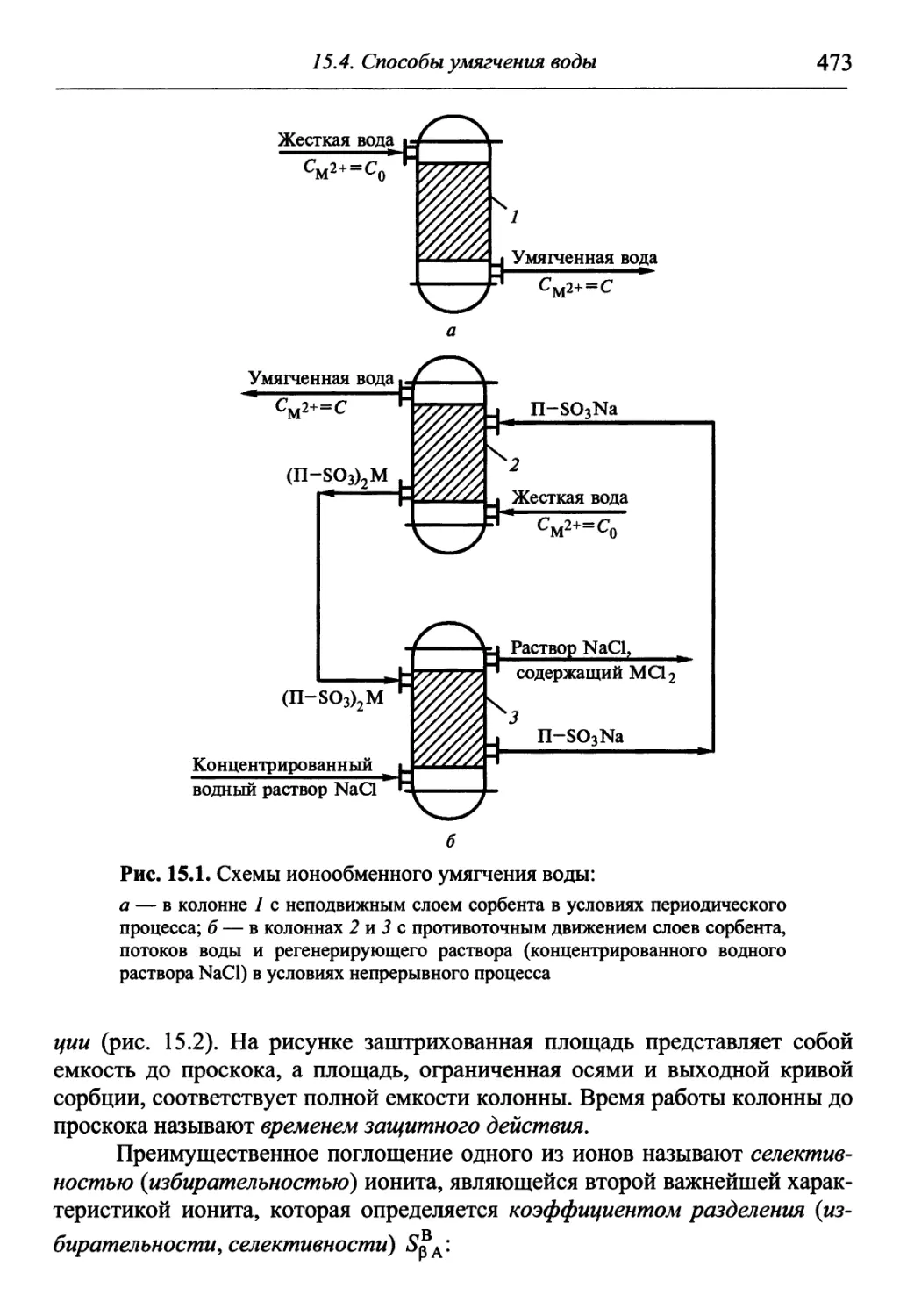



















































































































































































































































































Автор: Гуров А.А. Бадаев Ф.З. Овчаренко Л.П. Шаповал ВН.
Теги: химия общая и неорганическая химия учебник задачи по химии
ISBN: 5-7038-2445-1
Год: 2004




Текст
А.А. Гуров, Ф.З. Бадаев,
Л.П. Овчаренко, В.Н. Шаповал
Химия
Допущено Министерством образования
Российской Федерации
в качестве учебника для студентов
высших учебных заведений,
обучающихся по техническим направлениям
и специальностям
Москва
Издательство МГТУ имени Н.Э. Баумана
2004
УДК 54@75.8)
ББК24.1
Х465
Выпуск учебника осуществлен при финансовой поддержке
ЗАО БАСФ (Германия)
Рецензенты:
д-р хим. наук, проф. С.Н. Соловьев;
кафедра химии Московского инженерно-физического института
(государственного университета),
зав. кафедрой д-р хим. наук, проф. В.В. Сергиевский
Химия: Учебник / А.А. Гуров, Ф.З. Бадаев, Л.П. Овчаренко,
Х465 В.Н. Шаповал. — М.: Изд-во МГТУ им. Н.Э. Баумана, 2004. —
748 с: ил.
ISBN 5-7038-2445-1
Учебник состоит из девяти разделов, первые восемь включают в себя теорети-
теоретический материал и практические занятия. В девятом разделе изложены основы тех-
техники лабораторных работ. Такая структура учебника позволяет рассматривать его
как совокупность учебника, практикума и задачника.
Содержание учебника соответствует курсу лекций, который авторы читают в
МГТУ им. Н.Э. Баумана.
Для студентов технических университетов и вузов. Может быть полезен пре-
преподавателям и специалистам-химикам.
УДК 54@75.8)
ББК24.1
© Коллектив авторов, 2004
ISBN 5-7038-2445-1 © Изд-во МГТУ им. Н.Э. Баумана, 2004
Предисловие
Настоящий учебник охватывает полный объем материала первого об-
общетеоретического этапа обучения, наиболее сложного для студентов. Он
написан в соответствии с действующей в технических университетах про-
программой по курсу химии и государственным образовательным стандартом
по этой дисциплине для технических направлений и специальностей.
Аналогов этого учебника нового поколения нет ни среди отечествен-
отечественной, ни среди переводной литературы. Весь материал в нем разбит на девять
разделов. Первый служит связующим звеном между школьным и универси-
университетским курсами химии, второй-восьмой являются основными, и в них
представлена большая часть университетского курса химии, в девятом из-
изложены основы практических занятий. Каждый из первых восьми разделов
содержит теоретический материал и практические занятия, которые вклю-
включают в себя примеры решения задач, задачи для самостоятельного решения
и лабораторные работы. Такая структура является главным отличием учеб-
учебника от ранее издававшихся и позволяет рассматривать его как совокуп-
совокупность учебника, практикума и задачника.
В разделе «Приложения» содержится обширный справочный матери-
материал, необходимый для решения задач и обработки результатов опытов. В
конце книги приведены список рекомендуемой литературы, предметный и
именной указатели. В предметный указатель входят все выделенные в тек-
тексте светлым курсивом термины с указанием страниц, на которых они строго
определены или описаны. Аннотированный именной указатель содержит
краткие сведения об упоминаемых в книге ученых.
Данный учебник предназначен студентам 1-3-го курсов технических
университетов и вузов, изучающим химию. Может быть полезен аспиран-
аспирантам, преподавателям и специалистам в области химии.
В работе над учебником принимал участие коллектив авторов, подго-
подготовивших следующие материалы: предисловие, от авторов, главы 10, 11, 12,
15, 18, 19, задачи к разделам V, VIII и лабораторные работы ЛР 8, ЛР 10,
ЛР 12-ЛР 13 — Гуров А.А; главы 1, 2, 4, 13, 14, задачи к разделу I и лабора-
лабораторные работы ЛР 1-ЛР 3, ЛР 9 — Овчаренко Л.П.; задачи к разделу VI и
приложения — Овчаренко Л.П. и Гуров А.А; главы 3, 7, 8, 9, задачи к раз-
разделам IV, VII и лабораторные работы ЛР 5-ЛР 7 — Бадаев Ф.З., задачи к
разделу II — Бадаев Ф.З. и Овчаренко Л.П.; главы 5, 6, 16, 17, 20-22, задачи
к разделу III и лабораторные работы ЛР 4, ЛР 11 — Шаповал В.Н.
Предисловие
Авторы с благодарностью примут отзывы по данному учебнику и бу-
будут признательны всем, кто в той или иной форме выскажет конструктив-
конструктивные критические замечания и пожелания, направленные на его улучшение и
совершенствование.
Все замечания и пожелания просьба направлять по адресу: 105005,
Москва, 2-я Бауманская ул., 5, Издательство МГТУ им. Н.Э. Баумана.
От авторов
Химия как одна из фундаментальных естественнонаучных дисциплин
играет важную роль в подготовке высококвалифицированных специалистов.
Преподавание химии в технических вузах, как правило, проводится на пер-
первых годах обучения, а количество часов ограничено. Студенты младших
курсов еще не имеют необходимой физико-математической подготовки для
изучения современной химии, которая накопила большой объем информа-
информации и стала не описательной, а количественной наукой со сложным матема-
математическим аппаратом. Соответственно многие фундаментальные вопросы
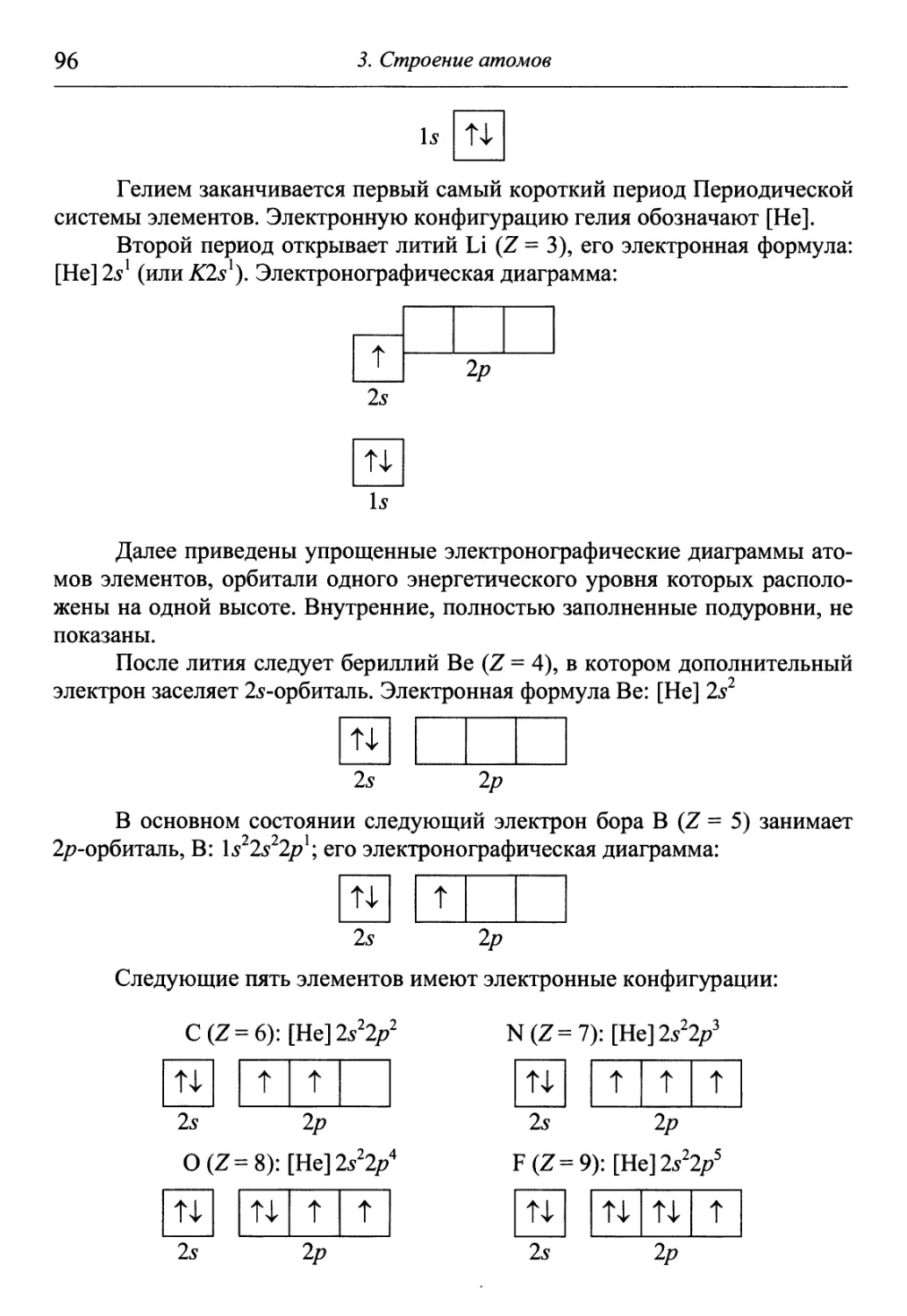
излагаются упрощенно, основные закономерности даются в виде готовых
положений и формул, без выводов и строгого обоснования. Перед препода-
преподавателем химии, таким образом, стоит проблема доступно объяснить концеп-
концептуально сложные и важные понятия, такие как волновая функция, химиче-
химическая связь, термодинамические функции и многие другие, которые форми-
формируют общее научное и техническое мировоззрение студента.
Курс химии в высших учебных заведениях тесно связан с другими
дисциплинами, например, физикой, математикой, биологией, геологией, по-
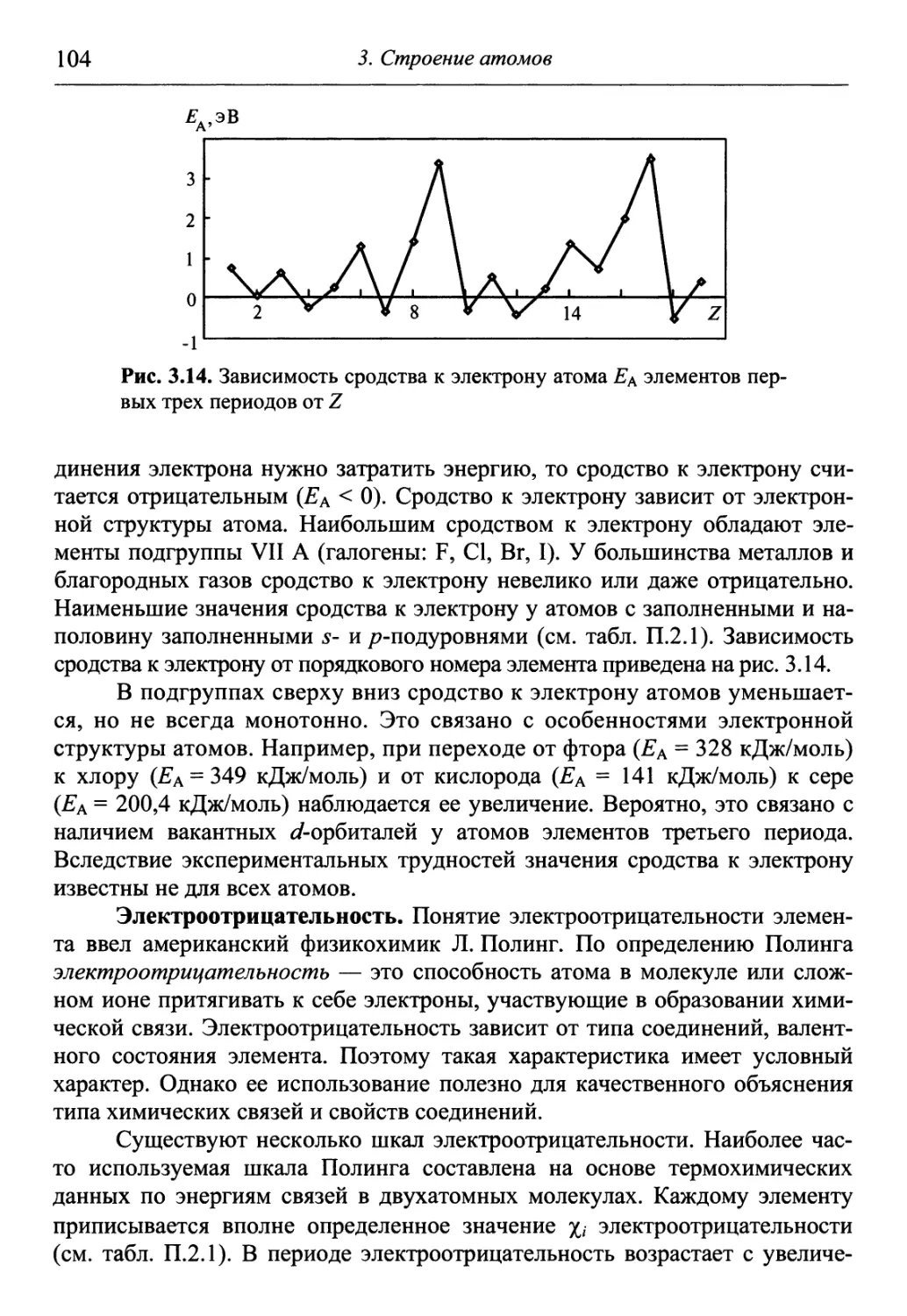
поэтому его преподавание в большинстве технических вузов подразделяют на
два этапа. На первом, независимо от будущей специальности, профиля фа-
факультета, учебного заведения излагается большая часть материала (до 80 %),
которая является общетеоретической и включает такие разделы, как строе-
строение вещества, закономерности протекания химических процессов, свойства
растворов, электрохимические явления и процессы и др. Второй этап связан
с изучением химических свойств конкретных элементов (чаще всего метал-
металлов) и их соединений, а также рассмотрением прикладных химико-
технологических вопросов и формируется уже с учетом профиля и специа-
специализации вуза.
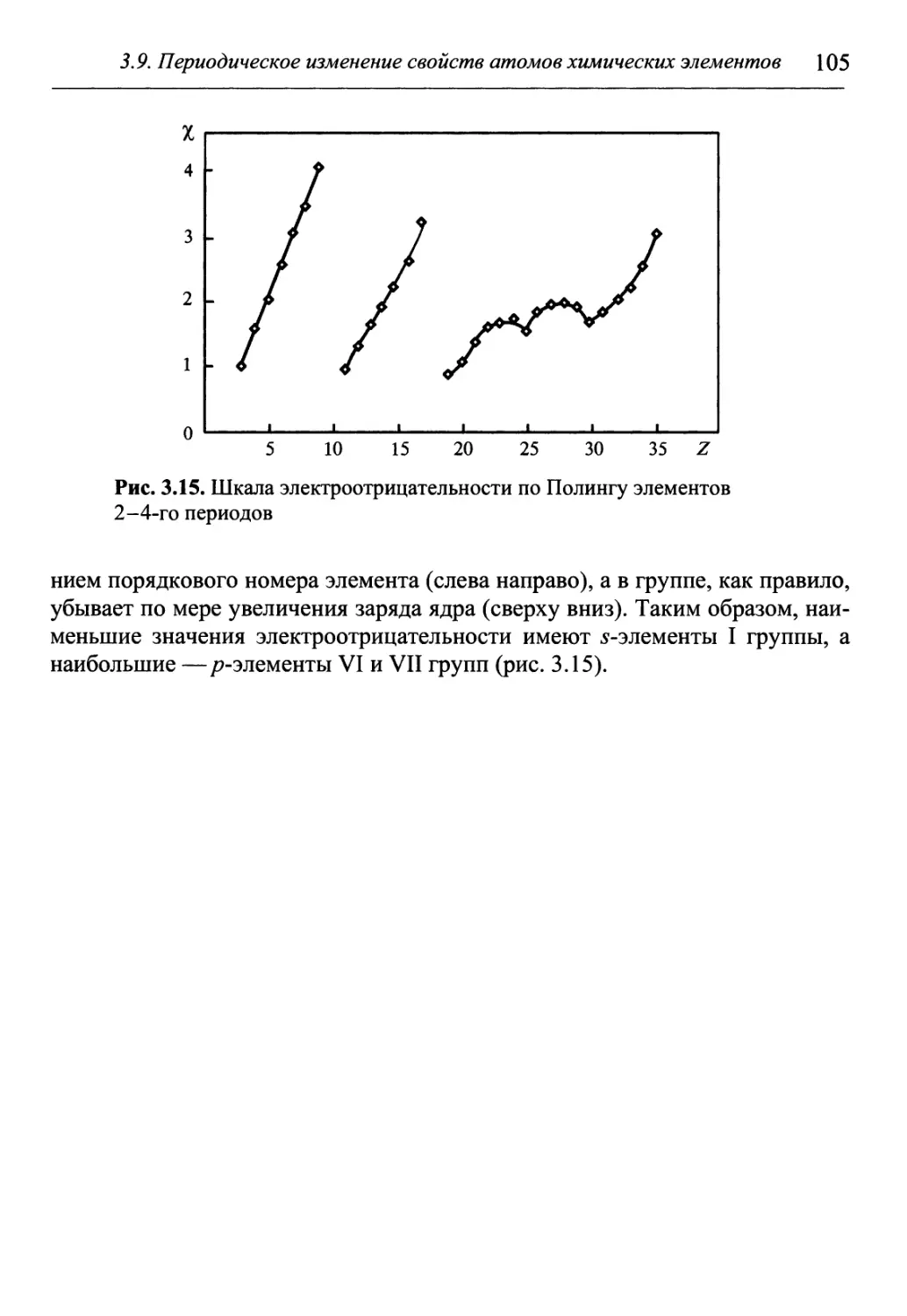
Теоретический материал в учебнике излагается кратко и доступно, с
учетом современных физико-химических представлений и последних дос-
достижений физики и химии. Большое внимание в нем уделяется практиче-
практическим занятиям, которые позволяют лучше усвоить теоретический материал.
Практические занятия включают в себя примеры решения задач, достаточ-
достаточное количество разнообразных задач различного уровня сложности, лабора-
лабораторные работы. Условия большинства задач содержат наборы из различных
вариантов, что существенно расширяет объем фактологических знаний. Не-
Необходимые для решения справочные данные представлены в приложениях.
От авторов
Лабораторная работа — самая эффективная форма организации само-
самостоятельной учебной и научной деятельности студентов. В предлагаемом
учебнике каждая лабораторная работа содержит опыты различного уровня
сложности и технического обеспечения, охватывающие основные теорети-
теоретические аспекты. Как правило, они просты в исполнении, наглядны, не тре-
требуют длительных затрат времени. После проведения опыта предлагается
обработать его результаты, т.е. написать уравнения протекающих реакций,
объяснить причину и сущность наблюдаемых явлений, рассчитать различ-
различные характеристики, а также сделать практические выводы. Такой подход к
выполнению лабораторных работ помогает студенту внимательно изучить и
проанализировать химические явления.
В учебнике используются, насколько это возможно, определения и
обозначения, рекомендованные Международным союзом теоретической и
прикладной химии, имеются обозначения, принятые в учебной и справоч-
справочной литературе, а также в исключительных случаях — старые устоявшиеся
обозначения.
Мы полагаем, что обширный теоретический материал, многообразие
опытов и большое количество задач позволит преподавателю подходить к
обучению дифференцированно, с учетом специализации вуза, объема часов
программы, технического и материального обеспечения лабораторий, а так-
также уровня подготовки студентов. Надеемся, что изучение химии для них
будет интересным и полезным, а приобретенные при этом знания помогут
им в их будущей профессии.
В заключение хотим поблагодарить всех, кто способствовал выходу
нашего учебника в свет.
В качестве приятного долга выражаем глубокую благодарность рецен-
рецензентам — коллективу кафедры химии Московского инженерно-физического
института и ее заведующему проф., д-ру хим. наук В.В. Сергиевскому,
проф. кафедры общей и неорганической химии РХТУ им. Д.И. Менделеева,
д-ру хим. наук С.Н. Соловьеву за обстоятельный анализ материалов рукопи-
рукописи, ценные замечания и полезные советы, а также различные методические
рекомендации.
Неоценимую помощь в подготовке рукописи оказали А.Н. Гончаров,
М.С. Гречкин, А.З. Бадаев и А.А. Шарков.
И, наконец, мы благодарим руководство Российского подразделения
всемирно известной химической фирмы БАСФ (Германия) в лице его Гене-
Генерального директора г-на Х.-Э. Рольманна и руководителя отдела химии
канд. хим. наук A.M. Гальминаса. Активная творческая поддержка и финан-
финансовая помощь возглавляемого ими подразделения фирмы способствовали
изданию учебника. Краткая информация о фирме БАСФ дана в конце пред-
предлагаемого читателю учебника.
I
ВВЕДЕНИЕ В ХИМИЮ
¦ Понятия вещества
и химической
реакции
¦ Стехиометрия.
Закономерности
изменения и способы
определения
количества вещества
Химия — наука, изучающая вещества, их свойст-
свойства, строение и взаимные превращения, — пред-
представляет собой обширную систему знаний о
зависимости свойств веществ от их состава и
строения, влиянии условий на возможность и
скорость их взаимодействия, процессах, воз-
возникающих при прохождении электрического
тока, под действием света и радиации, проте-
протекающих в неорганических, органических, кол-
коллоидных системах, живых организмах, в зем-
земной коре, космосе и др. Современная химия —
это комплекс научных дисциплин: неоргани-
неорганической, органической, аналитической, физиче-
физической, коллоидной, квантовой химии, электро-
электрохимии, геохимии, кристаллохимии, биохимии,
фотохимии, космохимии, радиационной и ра-
радиохимии, лазерной химии и др.
Фундамент химической науки — общая химия
исследует основные законы и формулирует
понятия химических процессов и явлений,
показывает логические связи между различ-
различными областями знаний о веществах и их пре-
превращениях, позволяет ориентироваться в мно-
многообразном мире современной химии. Пони-
Понимание основных химических концепций необ-
необходимо во всех областях науки, техники и
технологии, поскольку почти вся практическая
деятельность человечества связана с примене-
применением тех или иных веществ.
X 4 Понятия вещества
Г 1 и химической реакции
Объектом изучения в химии является вещество — материальное обра-
образование, состоящее из элементарных частиц (протонов, нейтронов,
электронов и др.). Индивидуальные вещества, характеризующиеся оп-
определенными свойствами, могут превращаться в другие соединения,
состоящие из тех же частиц, посредством химических реакций (от лат.
re — приставка, обозначающая обратное действие, и action — дейст-
действие), для осуществления которых часто требуются определенные усло-
условия — температура, давление, облучение, протекание электрического
тока, наличие растворителя и др. Химические реакции могут сопро-
сопровождаться выделением или поглощением теплоты, изменением агре-
агрегатного состояния веществ, световыми и шумовыми эффектами и т.п.
Химическая активность веществ, т.е. их реакционная способность, за-
зависит от природы соединений и условий протекания реакции. Чтобы
ориентироваться в многообразии химических веществ и процессов,
необходимо изучить основные понятия и законы химии.
1.1. Основные определения
Молекула — наименьшая частица индивидуального вещества,
обладающая его основными свойствами и способная к самостоятель-
самостоятельному существованию. Она состоит из атомов — наименьших частиц
вещества, которые нельзя разделить на составные части химическим
путем. Атом представляет собой электронейтральную систему, со-
состоящую из положительно заряженного ядра и движущихся вокруг
него отрицательно заряженных электронов. Совокупность атомов с
одинаковым зарядом ядра называют химическим элементом. Каж-
Каждый известный химический элемент имеет в Периодической системе
химических элементов Д.И. Менделеева свой символ и порядковый
номер, соответствующий заряду его ядра. В свободном состоянии
химические элементы находятся в виде простых веществ.
Простыми называют вещества, состоящие из атомов одного
элемента. Их обозначают химической формулой А„, где А — символ
химического элемента; п — число атомов в молекуле. Среди про-
простых веществ выделяют металлы, составляющие большинство из-
известных элементов, и неметаллы. К неметаллам относятся простые
вещества таких элементов, как водород Н, бор В, углерод С, крем-
кремний Si, азот N, фосфор Р, мышьяк As, кислород О, сера S, селен Se,
1.1. Основные определения 1 1
теллур Те, фтор F, хлор С1, бром Вг, йод I, астат At, гелий Не, неон Ne, аргон
Аг, криптон Кг, ксенон Хе, радон Rn. Химические элементы могут сущест-
существовать в виде нескольких простых веществ, различающихся по количест-
количественному составу или кристаллическому строению. Например, кислород су-
существует в виде дикислорода О2 (обычно называемого кислородом) и три-
кислорода Оз (называемого озоном), а углерод встречается в виде графита а-С,
алмаза 0-С, карбина (С2)„, фуллеренов С6о, С7о. Такое явление называют ал-
аллотропией, а различные виды одного и того же элемента — аллотропными
модификациями.
Сложные вещества, или химические соединения, состоят из атомов
двух и более элементов. Их химические формулы, записанные с помощью
символов соответствующих элементов и числовых индексов, несут инфор-
информацию о качественном и количественном составе соединения. Например,
Н2О — формула воды, молекулы которой состоят из двух атомов водорода и
одного атома кислорода, a H2SO4 — серной кислоты, поскольку ее молекулы
образованы двумя атомами водорода, одним атомом серы и четырьмя ато-
атомами кислорода.
Химические формулы сложных веществ можно составить различными
способами. Так, эмпирическая формула, рассчитанная на основании данных
о массовых соотношениях элементов в соединении, отражает количествен-
количественный состав молекулы, и, в частности, для этилового спирта может быть за-
записана как С2Н6О. Однако такая же эмпирическая формула может принад-
принадлежать и другому веществу, например С2Нб0 соответствует диметиловому
эфиру. Поэтому на основании дополнительных сведений эмпирические
формулы уточняют, т.е. выделяют реально существующие фрагменты моле-
молекулы и составляют молекулярные формулы. Для этилового спирта молеку-
молекулярная формула записывается как С2Н5ОН, а для диметилового эфира —
СН3ОСН3. Для того чтобы показать взаимное расположение атомов в моле-
молекуле, часто используют графические формулы, в которых черточкой обозна-
обозначают связь между атомами:
Н Н Н Н
м II
н—с—с—о—н н—с—о—с—н
II II
н н н н
этиловый спирт диметиловый эфир
С помощью химических формул в компактной форме можно описать хи-
химические реакции — процессы взаимодействия веществ, приводящие к образо-
образованию новых соединений. Вещества, подвергающиеся превращению (измене-
(изменению химического состава), называют реагентами (исходными веществами), а
12 I- Понятия вещества и химической реакции
образующиеся вещества — продуктами реакции. Запись химической реакции с
использованием символов элементов и формул соединений называют уравне-
уравнением химической реакции, или химическим уравнением. Химическое уравнение,
в котором указано относительное количество реагентов и продуктов реакции,
называют сбалансированным, или стехиометрическим. Например, взаимодей-
взаимодействие кальция с водой приводит к образованию гидроксида кальция и выделе-
выделению водорода. Эту реакцию можно записать в виде химического уравнения:
Са + Н2О -> Са(ОНJ + Н2
или стехиометрического уравнения, включающего стехиометрические ко-
коэффициенты, т.е. относительное количество участников реакции:
Са + 2Н2О = Са(ОНJ + Н2
При этом часто буквенными индексами указывают физические состояния реа-
реагентов и продуктов: т — твердое, ж — жидкое, г — газообразное, р — раство-
растворенное, а вертикальными стрелками — выпадение осадков и выделение газов:
Са(т) + 2Н2О(Ж) = Са(ОНJ(р) + H2(r)t
При составлении уравнений химических реакций требуется правильно
и однозначно записывать молекулярные формулы реагирующих веществ и
продуктов их взаимодействия, что позволяет точно отражать происходящие
изменения и рассчитывать количественные соотношения участников реак-
реакции. Международным союзом теоретической и прикладной химии разрабо-
разработана химическая номенклатура ИЮПАК (IUPAC) — правила составления
химических формул и способы наименования индивидуальных химических
веществ. При кратком знакомстве с этими правилами ограничимся рассмот-
рассмотрением основных классов неорганических соединений, исключив органиче-
органические соединения (т.е. большинство соединений углерода) вследствие слож-
сложности состава и строения большинства из них.
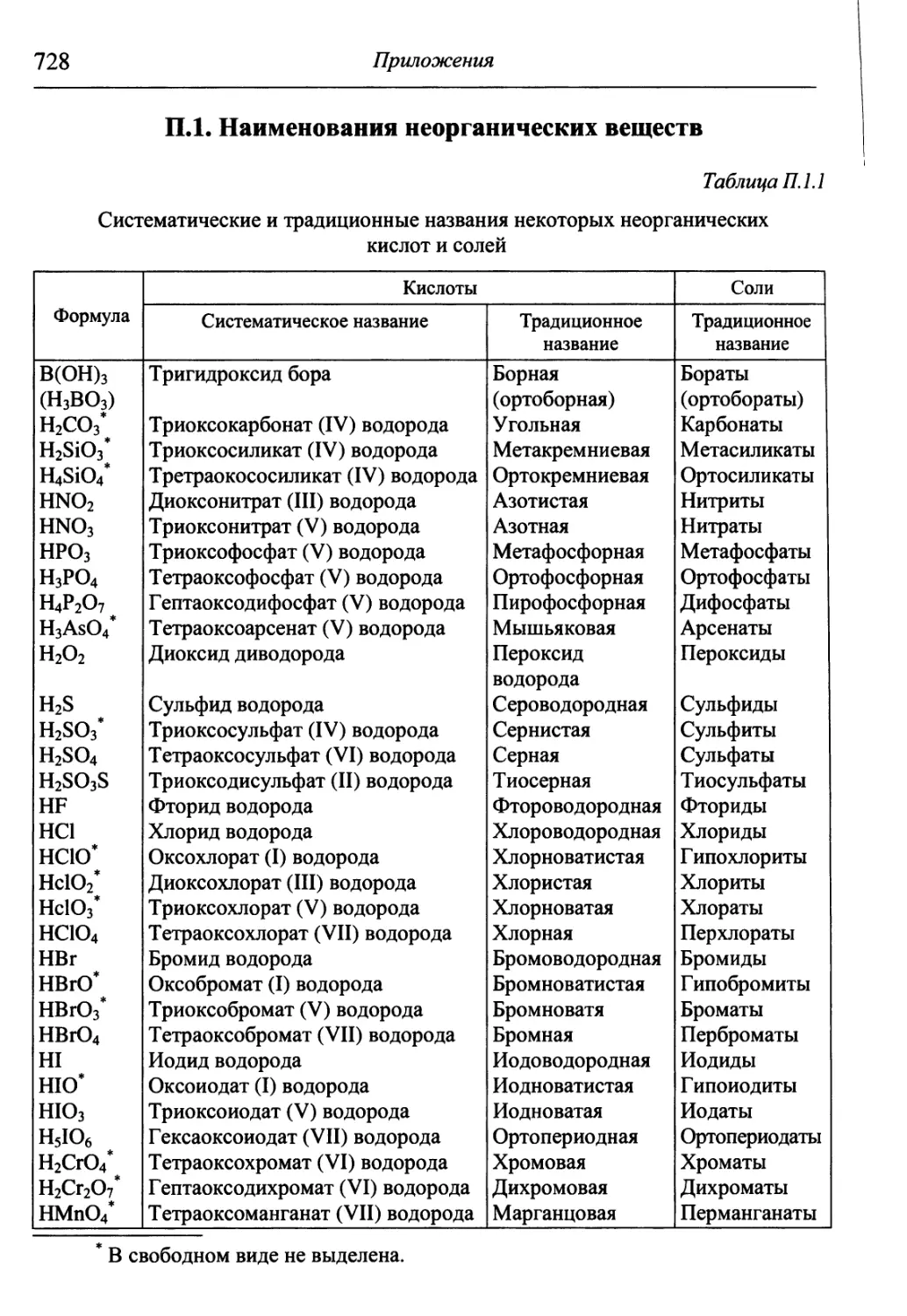
1.2. Классификация и номенклатура неорганических соединений
Неорганические соединения различают по составу (бинарные и мно-
многоэлементные) и функциональным признакам. К бинарным соединениям от-
относят соединения элементов с кислородом (оксиды), галогенами (галогени-
ды — фториды, хлориды, бромиды, иодиды), халькогенами (халькогениды —
сульфиды, селениды, теллуриды), азотом (нитриды), фосфором (фосфиды),
углеродом (карбиды), кремнием (силициды), а также соединения металлов
друг с другом (интерметаллиды) и водородом (гидриды). Среди многоэле-
многоэлементных соединений выделяют гидроксиды (вещества, содержащие гидро-
1.2. Классификация и номенклатура неорганических соединений
13
ксидные группы -ОН), производные гидроксидов — соли, а также ком-
комплексные соединения, гидраты и кристаллогидраты.
Наименование любого вещества должно однозначно указывать на его
состав, поэтому в основу систематических (т.е. составленных на основе сис-
системы ИЮПАК) названий неорганических веществ положены названия эле-
элементов, входящих в их состав.
Название бинарного соединения образуется из латинского корня на-
наименования более электроотрицательного элемента с окончанием -ид и рус-
русского наименования менее электроотрицательного элемента в родительном
падеже. При написании формулы вещества менее электроотрицательный
элемент стоит левее, например, А12О3 — оксид алюминия, Agl — иодид се-
серебра, OF2 — фторид кислорода. Для большинства элементов корни их рус-
русских названий совпадают с корнями латинских, за исключением элементов,
представленных ниже:
Ag
As
Аи
С
Си
Fe
Н
Hg
Мп
Серебро
Мышьяк
Золото
Углерод
Медь
Железо
Водород
Ртуть
Марганец
Аргент-
Арс-, арсен-
Аур-
Карб-, карбон-
Купр-
Ферр-
Гидр-, гидроген-
Меркур-
Манган-
N
Ni
О
РЬ
S
Sb
Si
Sn
Азот
Никель
Кислород
Свинец
Сера
Сурьма
Кремний
Олово
Нитр-
Никкол-
Окс-, оксиген-
Плюмб-
Сульф-, тио-
Стиб-
Сил-, силиц-, силик-
Станн-
Абсолютную величину степени окисления менее электроотрицатель-
электроотрицательного элемента, имеющего различные степени окисления, указывают в скоб-
скобках римскими цифрами, например, СО — оксид углерода (II), СО2 — оксид
углерода (IV). Для обозначения количественного состава используют грече-
греческие числительные в качестве приставки, например, Hg2Cl2 — дихлорид
диртути, СО — монооксид углерода, СО2 — диоксид углерода, а именно:
1
2
3
4
Моно-
Ди-
Три-
Тетра-
5
6
7
8
Пента-
Гекса-
Гепта-
Окта-
9
10
И
12
Нона-
Дека-
Ундека-
Додека-
Название многоэлементного соединения обусловлено его функциональ-
функциональными признаками. Так, гидроксиды, которые можно рассматривать как соеди-
соединение оксидов с водой, подразделяют на основные, проявляющие в химических
реакциях свойства оснований, кислотные — проявляющие свойства кислот, и
амфотерные — способные проявлять как кислотные, так и основные свойства.
К классу оснований, согласно теории электролитической диссоциации,
относят вещества, способные в водном растворе диссоциировать с образо-
образованием гидроксид-ионов ОН":
14 1- Понятия вещества и химической реакции
КОН = К+ + ОЬГ
Са(ОНJ = Са2+ + 2ОН"
Наименование основного гидроксида (или основания) образовано из
слова «гидроксид» и названия элемента в родительном падеже, после кото-
которого при необходимости указывают степень окисления элемента, например,
NaOH — гидроксид натрия, Fe(OHJ — гидроксид железа (II), или дигид-
роксид железа. Общую формулу основания можно записать как M(OH)W, где
М — металл, т — число гидроксидных групп, или кислотность основания.
Вещества, способные диссоциировать в растворе с образованием ио-
ионов водорода Н+, в соответствии с теорией электролитической диссоциации
относятся к классу кислот. Среди них есть не только кислотные гидрокси-
ды, такие как
но и бинарные соединения неметаллов с водородом, например
НС1 = Н+ + СГ
Кислоты в зависимости от наличия или отсутствия в их составе кис-
кислорода подразделяют на кислородсодержащие (или оксокислоты) и бескис-
бескислородные. К классу кислот также относятся и продукты замещения в моле-
молекулах оксокислот атомов кислорода на группу -О—О- (пероксокислоты),
например H2SO3(O2), или на атомы серы (тиокислоты), например H2SO3S. В
общем виде формулу кислоты можно записать как Н„А, где А — кислотный
остаток, п — число атомов водорода Н в молекуле, или основность кислоты.
Систематическое название кислоты включает в себя наименования
двух частей: электроположительной (атомы водорода) и электроотрицатель-
электроотрицательной (кислотный остаток, или анион). В названии аниона вначале указывают
атомы кислорода (-оксо-), затем кислотообразующего элемента с добавлением
суффикса -ат, далее в скобках абсолютную величину степени окисления это-
этого элемента, например, Н2СО3 — триоксокарбонат (IV) водорода, H2SO4 —
тетраоксосульфат (VI) водорода. При наличии в анионе других атомов наз-
название аниона составляют из латинских корней названий соответствующих
элементов и соединительной гласной -о- в порядке их размещения в форму-
формуле справа налево, например, H2SO3(O2) — пероксотриоксосульфат (VI) во-
водорода, H2SO3S — тиотриоксосульфат (VI) водорода. Систематические и
соответствующие им традиционные наименования наиболее употребитель-
употребительных кислот представлены в Приложениях, табл. П. 1.1. Традиционное назва-
название также состоит из двух слов — прилагательного, производного от корня
названия кислотообразующего элемента, и слова «кислота», например,
H2SO4 — серная кислота, HNO3 — азотная кислота.
1.2. Классификация и номенклатура неорганических соединений 15
Амфотерные гидроксиды способны диссоциировать в водных раство-
растворах как по типу оснований, так и по типу кислот, например,
Zn(OHJ = Zn2+ + 2OH"
Zn(OHJ + 2Н2О = 2Н+ + [Zn(OHL]2~
При взаимодействии с кислотами они проявляют свойства оснований, а при
взаимодействии с основаниями — свойства кислот. Их названия составляют
по схеме, соответствующей основным гидроксидам.
Соли представляют собой продукты замещения атомов водорода ки-
кислоты на металл или гидроксидных групп основания на кислотный остаток.
В зависимости от полноты замещения атомов водорода или гидроксидных
групп соли подразделяют на средние (или нормальные), например K2SO4,
кислые (или гидросоли), например NaHCO3, и основные (или гидроксосоли),
например FeOHCl. Различают также двойные соли, образованные двумя ме-
металлами и одним кислотным остатком (KA1(SO4J), и смешанные соли, обра-
образованные одним металлом и двумя кислотными остатками (CaClOCl). На-
Названия солей обусловлены систематическими названиями соответствующих
кислот, например, K2SO4 — тетраоксосульфат (VI) калия, NaHCO3 — триок-
сокарбонат (IV) водорода-натрия, FeOHCl или, точнее, FeClOH — гидро-
ксид-хлорид железа (II).
При наличии числовых приставок A, 2, ...) в названии вещества во
избежание неверного понимания формулы применяют умножающие при-
приставки (например, КА1з(8О4J(ОН)б — гексагидроксид-бис(сульфат) три-
триал юминия-калия) :
1
2
3
4
Монокис-
Бис-
Трис-
Тетракис-
5
6
7
8
Пентакис-
Гексакис-
Гептакис-
Октакис-
9
10
И
12
Нонакис-
Декакис-
Ундекакис-
Додекакис-
Традиционные наименования солей также содержат названия анионов
в именительном падеже и названия катионов в родительном падеже (см.
табл. П. 1.2), например, K2SO4 — сульфат калия, NaHCO3 — гидрокарбонат
натрия, FeOHCl — гидроксохлорид железа (II).
Оксиды в зависимости от характерных функций, выполняемых в хи-
химических реакциях, подразделяют на солеобразующие (среди них выделяют
основные, кислотные и амфотерные) и несолеобразующие (безразличные).
Основные оксиды образуют соли при взаимодействии с кислотами или кис-
кислотными оксидами, им соответствуют основания, например СаО — Са(ОНJ.
Кислотные оксиды образуют соли при взаимодействии с основаниями или
основными оксидами, они могут быть получены путем отделения воды от
соответствующей кислоты, поэтому их называют также ангидридами кис-
16 l- Понятия вещества и химической реакции
лот, например SO3 — ангидрид H2SO4. Амфотерные оксиды образуют соли
как при взаимодействии с кислотами, так и при взаимодействии с основа-
основаниями, например, ZnO, A12O3 и др.
Существуют соединения элементов с кислородом, по составу относя-
относящиеся к классу оксидов, но по своим свойствам принадлежащие к классу
солей. Это пероксиды — соли пероксида водорода Н2О2, например Na2O2
или СаО2, характерная особенность которых — наличие в их структуре двух
связанных между собой атомов кислорода -О—О-.
Комплексные соединения представляют собой сложные вещества, мо-
молекулы которых содержат центральный атом или ион (комплексообразова-
тель), связанный с несколькими способными к самостоятельному сущест-
существованию молекулами или ионами, называемыми лигандами. Согласно коор-
координационной теории Вернера A878), различают внутреннюю и внешнюю
сферы комплексного соединения. Более прочно связанные частицы внут-
внутренней сферы называют комплексным ионом или комплексом. При написании
координационной формулы эту часть комплексного соединения заключают
в квадратные скобки, например K3[Fe(CNN] или [Co(NH3N]Cl3. Комплекс
выступает как самостоятельная единица в химических реакциях, процессах
растворения, структуре кристалла. Частицы внешней сферы, связанные в
соединении менее прочно, при диссоциации в растворе отделяются:
K3[Fe(CNN] = ЗК+ + [Fe(CNN]3"
[Co(NH3N]Cl3 = [Co(NH3N]3+ + ЗСГ
В качестве комплексообразователя может выступать любой элемент,
но наиболее характерна эта роль для d- и/^металлов. Лигандами могут быть
простые (СГ, Г, Вг~ и др.) и сложные (ОН~, СО]~, N0^ и др.) ионы, а также
электронейтральные молекулы, такие как Н20, NH3, CO, C2H5NH2 и др.
Число химических связей, которые образует лиганд с комплексообра-
зователем, называют дентатностью. Так, однозарядные анионы (например,
СГ, N02) относятся к числу монодентатных лигандов, двухзарядные анио-
анионы (например, S2O3~~, С2С>4 ) —к числу бидентатных.
Число химических связей, которые комплексообразователь образует с
лигандами, называют координационным числом. Если комплексообразова-
комплексообразователь связан с монодентатными лигандами, то координационное число равно
числу лигандов. Значение координационного числа комплексообразователя
зависит от его природы, степени окисления, природы лигандов и условий
протекания реакции комплексообразования и может изменяться в пределах
от 2 до 12. Наиболее распространенными являются координационные числа
6, 4 и 2, соответствующие симметричной геометрической конфигурации
комплекса. Между значением координационного числа и степенью окисле-
1.2. Классификация и номенклатура неорганических соединений 17
ния комплексообразования существует определенная связь. Часто коорди-
координационное число равно удвоенному значению степени окисления комплек-
сообразователя: например, [Ag(NH3J]+, [Zn(NH3L]2+, [Сг(Н2ОN]3+. Ней-
Нейтральные лиганды могут присоединяться к комплексообразователю в боль-
большем количестве: [Со(Н2ОN]2+ и [СоС14]2~. Координационное число зависит
и от размеров лиганда: [А1СЦ]~ и [АШб]3~ (радиус иона С1~ больше радиуса
иона F").
Известно, что комплексных соединений значительно больше, чем всех
других неорганических соединений, их роль в природных и технологиче-
технологических процессах чрезвычайно велика. Единой классификации комплексных
соединений вследствие их многообразия не существует. Помимо разделения по
функциональным признакам {комплексные кислоты, например H2[Pt(CNN],
комплексные основания, например [Ag(NH3J]OH, комплексные соли, напри-
например K3[Co(CNN]), соединения классифицируют исходя из особенностей
комплексов.
Так, в зависимости от знака заряда комплекса выделяют соединения,
содержащие катионные комплексы, например [Zn(NH3L]2+, анионные ком-
комплексы, например [А1Н4]~, и нейтральные комплексы, например [Fe(COs)].
По природе лигандов различают соединения, в состав которых входят
аквакомплексы (лиганды — Н2О), например [Си(Н2ОL]2+, амминокомплексы
(лиганды — NH3), например [Ag(NH3J]+, гидроксокомплексы (лиганды —
ОН"), например [Zn(OHL]2~? ацидокомплексы (лиганды — анионы кислот),
например [Fe(NCSN]3~, комплексы смешанного типа, например
[Co(H2O)(NH3LCl]2+ и др. Комплексы классифицируют и по внутренней
структуре: моно- и полиядерные, простые и циклические.
Номенклатура комплексных соединений утверждена комиссией ИЮПАК
с учетом особенностей, принятых в русском языке: первым называют анион,
затем катион; лиганды перечисляют в следующем порядке: анионные, ней-
нейтральные, катионные; нейтральные группы называют так же, как молекулы,
за исключением аква-(Н2О) и аммин-(>Щ3); число лигандов указывают соот-
соответствующей приставкой; степень окисления комплексообразователя обо-
обозначают в скобках после его названия; окончание названий комплексов за-
зависит от их классификации по функциональному признаку: [Zn(NH3L]Cl2 —
хлорид тетраамминцинка (II), Li[AlH4] — тетрагидроалюминат (III) лития,
[Cu(H2OL]SO4 — сульфат тетрааквамеди (II), H2[Pt(CNN] — гексацианопла-
тинат (IV) водорода, или гексацианоплатиновая (IV) кислота, [Ag(NH3J]OH —
гидроксид диамминсеребра (I).
Гидраты и кристаллогидраты — соединения, содержащие в своем
составе воду, например, NH3H2O, Fe2O3«H2O, CuSO4-5H2O. Как система-
систематические, так и традиционные названия таких соединений начинаются со
слова «гидрат» с соответствующей приставкой: NH3H2O — гидрат аммиа-
18 I- Понятия вещества и химической реакции
ка, Fe2O3wH2O — полигидрат оксида железа (III), CuSO4-5H2O — пентагид-
рат тетраоксосульфата меди (II), или пентагидрат сульфата меди (II).
В технической литературе и лабораторно-заводской практике часто
применяют тривиальные (технические) названия неорганических веществ,
использование которых в номенклатуре ИЮПАК не рекомендуется (см.
табл. П. 1.2).
13. Окислительно-восстановительные реакции
Основные понятия и определения. Классификация химических ре-
реакций построена на формальных признаках: термодинамических, кинетиче-
кинетических, специфических. Так, по знаку теплового эффекта различают эндотер-
эндотермические и экзотермические реакции, по фазовому состоянию реакционной
среды — гомогенные и гетерогенные реакции, по наличию катализатора —
каталитические и некаталитические реакции, выделяют также электрохи-
электрохимические, фотохимические и другие виды реакций. Кроме того, различают
реакции по типу частиц, участвующих в процессе (молекулярные, ионные,
радикальные), по типу реагента (галогенирование, нитрование, гидрирова-
гидрирование, гидратация), по изменению числа реагентов и продуктов реакции (ре-
(реакции соединения, разложения, замещения).
Все многообразие типов химических реакций можно свести к двум
классам, учитывая изменение степеней окисления элемецтов, которые вхо-
входят в состав реагирующих веществ. Если в результате реакции степени
окисления элементов не изменяются, такие реакции называют обменными, в
противном случае — окислительно-восстановительными реакциями.
Протекание химических реакций обусловлено обменом частицами
между реагирующими веществами. Например, в реакции нейтрализации
происходит обмен между катионами и анионами кислоты и основания, в
результате чего образуется слабый электролит — вода:
H2SO4 + 2NaOH = Na2SO4 + 2Н2О
Часто обмен сопровождается переходом электронов от одной частицы
к другой. Так, при вытеснении цинком меди в растворе сульфата меди (II)
Zn (т) + CuSO4 (р) = ZnSO4 (р) + Си (т)
электроны от атомов цинка переходят к ионам меди:
или
1.3. Окислительно-восстановительные реакции 19
Zn(T)+ Си* = ZnBp+} +Cu(T)
Процесс потери электронов частицей называют окислением, а процесс
приобретения электронов — восстановлением. Окисление и восстановление
протекают одновременно, поэтому взаимодействия, сопровождающиеся пе-
переходом электронов от одних частиц к другим, называют окислительно-вос-
окислительно-восстановительными реакциями. •
Передача электронов может быть и неполной. Например, в реакции
вместо малополярных связей С-Н появляются сильнополярные связи Н-С1.
Для удобства описания окислительно-восстановительных реакций исполь-
используют понятие степени окисления, характеризующее состояние элемента в
химическом соединении и его поведение в реакциях. Степень окисления —
величина, численно равная формальному заряду, который можно приписать
элементу, исходя из предположения, что все электроны каждой из его связи
перешли к более электроотрицательному атому данного соединения.
Используя понятие степени окисления, можно дать более общее опреде-
определение процессов окисления и восстановления. Окислительно-восстановитель-
Окислительно-восстановительными называют химические реакции, которые сопровождаются изменением
степеней окисления элементов участвующих в реакции веществ. При восста-
восстановлении степень окисления элемента уменьшается, при окислении — увели-
увеличивается. Вещество, в состав которого входит элемент, понижающий степень
окисления, называют окислителем; вещество, в состав которого входит эле-
элемент, повышающий степень окисления, называют восстановителем:
Шкала степени окисления
-4 -3 -2 -1 0 +1 +2 +3 +4 +5 +6 +7 +8
Окисление
Восстановление
Степень окисления элемента в соединении определяют в соответствии
со следующими правилами:
степень окисления элемента в простом веществе равна нулю;
алгебраическая сумма всех степеней окисления атомов в молекуле
равна нулю;
алгебраическая сумма всех степеней окисления атомов в сложном ио-
ионе, а также степень окисления элемента в простом одноатомном ионе равна
заряду иона;
отрицательную степень окисления проявляют в соединении атомы
элемента, имеющего наибольшую электроотрицательность;
20 1. Понятия вещества и химической реакции
максимально возможная (положительная) степень окисления элемента
соответствует номеру группы, в которой расположен элемент в Периодиче-
Периодической системе Д.И. Менделеева.
Степени окисления атомов элементов в соединении записывают над
символом данного элемента, указывая вначале знак степени окисления, а
+1 +7 -2
затем ее численное значение, например КМ11О4.
Ряд элементов в соединениях проявляет постоянную степень окисле-
окисления, что используют при определении степеней окисления других элементов:
фтор, имеющий наивысшую среди элементов электроотрицатель-
электроотрицательность, имеет степень окисления -1;
водород проявляет степень окисления +1, кроме гидридов металлов (-1);
металлы IA подгруппы имеют степень окисления +1;
металлы ПА подгруппы, а также цинк и кадмий имеют степень окис-
окисления +2;
степень окисления алюминия +3;
степень окисления кислорода равна -2, за исключением соединений, в
которых кислород присутствует в виде молекулярных ионов: О2 — катиона
диоксигенила, О2 — надпероксидного аниона, О2~ — пероксидного анио-
аниона, О3 — озонидного аниона, а также фторидов кислорода OJF2.
Окислительно-восстановительные свойства атомов различных эле-
элементов проявляются в зависимости от многих факторов, важнейшие из ко-
которых — электронное строение элемента, его степень окисления в веществе,
характер свойств других участников реакции. Соединения, в состав которых
входят атомы элементов с максимальной (положительной) степенью окис-
+7 +6 +5 +4
ления, например KMnO4, K2Cr2O7, HNO3? PbO2, могут только восста-
восстанавливаться, выступая в качестве окислителей. Соединения, содержащие
-3 -2 -1
элементы с минимальной степенью окисления, например NH3, H2S, HI,
могут только окисляться и выступать в качестве восстановителей. Вещества,
содержащие элементы с промежуточными степенями окисления, например
+3 -10 0 +3 +4
HNO2, H2O2, S, Ь, СгС13, МпО2, обладают окислительно-восстанови-
окислительно-восстановительной двойственностью. В зависимости от партнера по реакции такие
вещества способны и принимать (при взаимодействии с более сильными
восстановителями), и отдавать (при взаимодействии с более сильными окис-
окислителями) электроны.
Состав продуктов восстановления и окисления также зависит от мно-
многих факторов, в том числе среды, в которой протекает химическая реакция,
1.3. Окислительно-восстановительные реакции 21
концентрации реагентов, активности партнера по окислительно-восстанови-
окислительно-восстановительному процессу.
Чтобы записать уравнение окислительно-восстановительной реакции,
необходимо знать, как изменяются степени окисления элементов и в какие
другие соединения переходят окислитель и восстановитель. Рассмотрим
краткие характеристики наиболее часто употребляющихся окислителей и
восстановителей.
Важнейшие окислители. Среди простых веществ окислительные
свойства характерны для типичных неметаллов: фтора F2, хлора С12, брома
Вг2, йода 12, кислорода О2.
Галогены, восстанавливаясь, приобретают степень окисления -1, при-
причем от фтора к йоду их окислительные свойства ослабевают (F2 имеет огра-
ограниченное применение вследствие высокой агрессивности):
2Н2О + 2F2 = O2+ 4HF
2Р(Красный) + ЗС12 сжи1?ние 2РС1з
SO2+ Br2 + 2Н2О = H2SO4 + 2HBr
H2S(Hacbiiu) + Ьссуспензия)= S* + 2HI
Кислород, восстанавливаясь, приобретает степень окисления -2:
4NH3 + ЗО2 сг°=ние 2N2 + 6Н2О
4Fe(OHJ + О2 + 2Н2О = 4 Fe(OHK
К наиболее важным окислителям среди кислородсодержащих кислот
и их солей относятся азотная кислота HNO3 и ее соли, концентрированная
серная кислота H2SO4, кислородсодержащие кислоты галогенов ННа1Ох и их
соли, перманганат калия КМпО4 и дихромат калия К2Сг2С>7.
Азотная кислота проявляет окислительные свойства за счет азота в
степени окисления +5. При этом возможно образование различных продук-
продуктов восстановления:
NO3 + 2Н+ + е = NO2 + Н2О
NO3 + 4Н+ + Зе = N0 + 2Н2О
NO3 + 5Н+ + 4е = 0,5N2O + 2,5Н2О
NO3 + 6Н+ + 5е = 0,5N2 + ЗН2О
NO3 + ЮН+ + 8е = NH; + ЗН2О
Глубина восстановления азота зависит от концентрации кислоты, а также от
активности восстановителя, определяемой его окислительно-восстанови-
окислительно-восстановительным потенциалом:
22 1. Понятия вещества и химической реакции
Концентрация кислоты
NO2 NO N2O N2 NH4+
Активность восстановителя
Например, окисление цинка (активный металл) азотной кислотой сопро-
сопровождается образованием различных продуктов восстановления, но при концен-
концентрации HNO3 примерно 2 % (масс.) преимущественно образуется NH4NO3:
нагревание
4Zn + 10HNO3 = 4Zn(NO3J + NH4NO3 + ЗН2О
при концентрации HNO3 приблизительно 5 % (масс.) — N2O:
4Zn + 10HNO3 = 4Zn(NO3J + N2Ot + 5H2O
при концентрации HNO3 около 30 % (масс.) — NO:
нагревание
3Zn + 8HNO3 = 3Zn(NO3J + 2NOt + 4H2O
а при концентрации HNO3 примерно 60 % (масс.) преимущественно образу-
образуется NO2:
нагревание
Zn + 4HNO3 = Zn(NO3J + 2N02t + 2H2O
Окислительная активность азотной кислоты усиливается с ростом
концентрации, поэтому концентрированная HNO3 окисляет не только ак-
активные, но и малоактивные металлы, такие как медь и серебро, образуя пре-
преимущественно оксид азота (IV):
ЗСи + 8HNO3(pa36)= 3Cu(NO3J + 2NOT + 4Н2О
Си + 4HNO3 (К0Нц) = Cu(NO3J + 2N02t + 2Н2О
Ag + 2HNO3 (конц) = AgNO3 + NO2T + H2O
а также и неметаллы, такие как сера и фосфор, окисляя их до кислот, со-
соответствующих высшим степеням окисления:
кипячение
S + 6HNO3 (конц) = H2SO4 + 6NO2T + 2Н2О
кипячение
5HNO3 (конц) = Н3РО4 + 5NO2T + Н2О
Соли азотной кислоты (нитраты) могут восстанавливаться в кислот-
кислотной, а при взаимодействии с активными металлами и в щелочной средах, а
также в расплавах:
1.3. Окислительно-восстановительные реакции 23
нагревание
4Zn + NaNO3 + 7NaOH + 6Н2О = 4Na2[Zn(OHL] + NH3
сплавление
Zn + KNO3 + 2K0H = K2Zn02 + KNO2 + H2O
Царская водка — смесь концентрированных азотной и соляной ки-
кислот, смешанных в соотношении 1 : 3 по объему. Название этой смеси свя-
связано с тем, что она растворяет даже такие благородные металлы, как золото
и платина:
Аи + HNO3(kohu) + 4НС1(ко„ц)= ЩАиСЦ] + NOt+ 2H2O
Протекание этой реакции обусловлено тем, что царская водка выделяет нит-
розилхлорид NOC1 и свободный хлор С12:
HNO3 + ЗНС1 = С12 + NOC1 + 2Н2О
под действием которых металлы переходят в хлориды.
Серная кислота проявляет окислительные свойства в концентриро-
концентрированном растворе за счет серы в степени окисления +6:
SO^ + 4Н+ + 2е = SO2 + 2Н2О
SO^ +8H+ + 4e = S + 4H2O
SO4" + 10Н+ + 8е = H2S + 4Н2О
Состав продуктов восстановления определяется главным образом ак-
активностью восстановителя и концентрацией кислоты:
Концентрация кислоты
H2S S SO2
Активность восстановителя
Так, взаимодействие концентрированной H2SO4 с малоактивными ме-
металлами, некоторыми неметаллами и их соединениями приводит к образо-
образованию оксида серы (IV):
Си + 2H2SO4(koh4) = CuSO4 + SO2T+ 2H2O
нагревание
2H2SO4 (конц) = CO2t + 2SO2T + 2Н2О
нагревание
2KBr(T) + 3H2SO4 (конц) = Br2 + SO2t + 2Н2О + 2KHSO4
Активные металлы восстанавливают концентрированную серную кис-
кислоту до серы или сероводорода:
3Zn + 4H2SO4(Korai)= 3ZnSO4 + S4- + 4H2O
4Mg + 5H2SO4(KOHU) = 4MgSO4 + H2St+ 4H2O
24 l- Понятия вещества и химической реакции
при этом одновременно образуются H2S, S и SO2 в различных соотношени-
соотношениях. Однако и в этом случае основным продуктом восстановления H2SO4 яв-
является SO2, так как выделяющиеся S и H2S могут окисляться концентриро-
концентрированной серной кислотой:
кипячение
S + 2H2SO4 (конц) = 3S02t + 2Н2О
H2S + H2SO4 (конц) = Si + SO2t + 2Н2О
Кислородсодержащие кислоты галогенов и их соли (см. табл. П. 1.1)
часто используют как окислители, хотя многие из них проявляют двойствен-
двойственный характер. Как правило, продукты восстановления этих соединений —
хлориды и бромиды (степень окисления -1), а также йод (степень окисления 0):
нагревание
1) MnS + 4НС1О = MnSO4 + 4НС1 2) 3S + 2КСЮ3 = 3S02t + 2КС1
нагревание
3) 4Zn + LiC104 = 4ZnO + LiCl 4) H2O2 + НВЮ = O2t + H2O + HBr
нагРевание нагревание
5) 2NH3 + NaBrO3 = 6) Н1(конц)+ КВЮ4(насыщ) = KIO4+ HBr
= N2+NaBr + 3H2O
7) 10НС1(КОНЦ) + 2Н1Оз(КО„ц) = 8) 5Na2SO3+ 2НЮ3= 5Na2SO4 +12 + H2O
= 5C12T +12 + 6H2O
Однако и в этом случае состав продуктов восстановления зависит от усло-
условий протекания реакции, концентрации окислителя и активности восстано-
восстановителя:
h + 2НС103(К0Нц) = 2ШО3 + Cl2t
h + 2НВгО4(конц)+ 4Н2О Нагре=НИе 2Н5Ю6 + Вг2
ЗН2О2 + NaIO3 = ЗО2Т + Nal + ЗН2О
Перманганат калия проявляет окислительные свойства за счет мар-
марганца в степени окисления +7. В зависимости от среды, в которой протекает
реакция, он восстанавливается до разных продуктов: в кислотной среде —
до солей марганца (II), в нейтральной — до оксида марганца (IV) в гидрат-
ной форме МпО(ОНJ, в щелочной — до манганат-иона МпО^":
кислотная среда
5KNO2 + 2КМпО4 + 3H2SO4(pa36) = 5KNO3 + 2MnSO4 + ЗН2О + K2SO4
10FeSO4 + 2KMnO4 + 8H2SO4(pa36) = 5Fe2(SO4K + 2MnSO4 + 8H2O + K2SO4
5Na2SO3 + 2KMnO4 + 3H2SO4(pa36) = 5 Na2SO4 + 2MnSO4 + 3H2O + K2SO4
1.3. Окислительно-восстановительные реакции 25
нейтральная среда
ЗН2 + 2KMnO4 = 2KOH + 2МпО(ОНJ4
3H2S + 2KMnO4= 3SI + 2MnO(OHJi + 2К0Н
3Na2SO3 + 2KMnO4 + 3H2O = 3Na2SO4+ 2MnO(OHJl + 2K0H
щелочная среда
KCN + 2KMnO4 + 2КОН(КОНЦ) = KOCN + 2K2Mn04 + H2O
KIO3 + 2КМпО4(коНц) + 2КОН(КОНЦ) = КЮД + 2K2Mn04 + H2O
Na2SO3 + 2KMnO4 + 2KOH = Na2SO4 + 2K2MnO4 + H2O
Дихромат калия, в состав молекулы которого входит хром в степени
окисления +6, является сильным окислителем при спекании и в кислотном
растворе
спекание
2С(КОКС) + К2Сг207 = Сг2О3 + К2СО3 + COt
6KI + K2Cr207+ 7H2SO4(pa36)= 3I2 + Cr2(SO4K + 7Н2О + 4K2SO4
проявляет окислительные свойства и в нейтральной среде
3H2S + K2Cr2O7 + Н2О = 3S>1 + 2Cr(OHKl + 2KOH
В щелочной среде равновесие между хромат- и дихромат-ионами
Cr2O72 -+2OH" ^± 2СЮ42+Н2О
2СЮ42+ 2Н+ ?± Сг2О?'+ Н2О
смещено в сторону образования СЮ42", поэтому в щелочной среде окисли-
окислителем является хромат калия К2Сг04:
3K2S + 2К2СЮ4 + 8Н2О = 3Si + 2K3[Cr(OHN] + 4KOH
однако К2Сг04 более слабый окислитель по сравнению с К2Сг207.
Среди ионов окислительные свойства проявляют ион водорода Н+ и
ионы металлов в высшей степени окисления. Ион водорода Н+ выступает
как окислитель при взаимодействии активных металлов с разбавленными
растворами кислот (за исключением HNO3)
Mg + H2SO4(pa36)= MgSO4 + H2T
Ионы металлов в относительно высокой степени окисления, такие как
Cu2+, Hg2+,
пени окисления
Fe3+, Cu2+, Hg2+, восстанавливаясь, превращаются в ионы более низкой сте-
SnCl2 + 2HgCl2 = SnCl»
H2S + 2FeCl3 = Si + 2FeCl2 + 2HC1
26 1. Понятия вещества и химической реакции
или выделяются из растворов их солей в виде металлов
2А1 + ЗСиС12 = 2А1С13 + ЗСи
Важнейшие восстановители. К типичным восстановителям среди
простых веществ относятся активные металлы, такие как щелочные и ще-
лочно-земельные металлы, цинк, алюминий, железо и другие, а также неко-
некоторые неметаллы (водород, углерод, фосфор, кремний).
Металлы в кислотной среде окисляются до положительно заряжен-
заряженных ионов:
Zn + 2HC1 = ZnCl2 + H2t
Pb + 3H2SO4(> so «/о масс.) = Pb(HSO4J + S02t + 2H2O
В щелочной среде окисляются металлы, проявляющие амфотерные
свойства; при этом образуются отрицательно заряженные анионы или гид-
роксокомплексы:
сплавление
2 Al + 2(NaOH • Н2О) = 2NaA102 + ЗН2Т
Sn + 2NaOH(KOHU) + 2H2O = Na2[Sn(OHL] + H2t
Неметаллы, окисляясь, образуют оксиды или соответствующие ки-
кислоты:
С + 4HNO3 (конц. гор.) = СО2Т + 4NO2T + 2Н2О
51(аморфн) + 6HF(K0Hy) = H2[SiF6] + 2Н2Т
Р4 + 6H2SO4(kOhu) = 4H2(PHO3) + 6SO2t
Восстановительными функциями обладают бескислородные анио-
анионы, например СГ, Вг~, Г, S2", Н~ и катионы металлов в низшей степени
окисления.
В ряду галогенид-ионов, которые, окисляясь, обычно образуют галогены:
нагревание
4НС1(КО„Ц) + РЬО2 = РЬС12^ + С12Т + 2Н2О
2НВг(К0Нц) + Н2О2(КОНц)= Вг2 + 2Н2О
2HI + NO2 = 12 + NOT + Н2О
восстановительные свойства усиливаются от СГ к Г.
Гидриды металлов проявляют восстановительные свойства за счет
окисления связанного водорода (степень окисления -1) до свободного водо-
водорода:
спекание
2СаН2 + ТЮ2 = 2СаО + Ti + 2Н2Т
1.3. Окислительно-восстановительные реакции 27
Катионам металлов в низшей степени окисления, таким как Sn2+,
Fe2+, Cu+, Hg22+ и другим, при взаимодействии с окислителями свойственно
повышение степени окисления:
5SnCl2 + 2КМпО4 + 26НС1(к0Нц) = 5H2[SnCl6] + 2МпС12 + 2КС1 + 8Н2О
нагревание
2FeSO4 + Н2О2(КОНЦ)+ H2SO4(Pa36) = Fe2(SO4K + 2Н2О
Окислительно-восстановительная двойственность. Среди простых
веществ окислительно-восстановительная двойственность характерна для
элементов VIIA, VIA и VA подгрупп, которые могут как повышать, так и
понижать свою степень окисления.
Часто используемые как окислители галогены под действием более
сильных окислителей проявляют восстановительные свойства (за исключе-
исключением фтора). Их окислительные способности уменьшаются, а восстанови-
восстановительные способности увеличиваются от С12 к 12:
Окислительная способность
Cl2 Br2
Восстановительная способность
Эту особенность иллюстрирует реакция окисления йода хлором в водном
растворе:
h + 5С12 + 6Н2О = 2ШО3 + ЮНС1
В состав кислородсодержащих соединений, проявляющих двойствен-
двойственность поведения в окислительно-восстановительных реакциях, также входят
элементы в промежуточной степени окисления. Кислородсодержащие ки-
кислоты галогенов и их соли, в состав молекул которых входит галоген в про-
промежуточной степени окисления, могут быть как окислителями
нагревание
S + NaC102 = NaCl + SO2t
так и восстановителями
5NaC102 + 2КМпО4 + 3H2S04(pa36) = 5NaC103 + 2MnSO4 + ЗН2О + K2SO4
Пероксид водорода, содержащий кислород в степени окисления -1, в
присутствии типичных восстановителей проявляет окислительные свойства,
так как степень окисления кислорода может понижаться до -2:
2KI + Н2О2 = h + 2KOH
PbSDepHblii) + 4Н2О2 = PbSO4FejIblii) + 4Н2О
28 I- Понятия вещества и химической реакции
(Последнюю реакцию используют при реставрации картин старых мастеров,
краски которых, содержащие свинцовые белила, чернеют из-за взаимодей-
взаимодействия с сероводородом воздуха.)
При взаимодействии с сильными окислителями степень окисления ки-
кислорода, входящего в состав пероксида водорода, повышается до 0, и Н2С>2
проявляет свойства восстановителя:
Н2О2 + 2Hg(NO3J = О2Т + Hg2(NO3J + 2HNO3
Азотистая кислота и нитриты^ в состав которых входит азот в сте-
степени окисления +3, также могут выступать как в роли окислителей
2HI + 2HNO2 = 12 + 2NOt + 2Н2О
так и в роли восстановителей
2NaNO2(pa36,rop)+ O2 = 2NaNO3
Классификация. Различают четыре типа окислительно-восстанови-
окислительно-восстановительных реакций.
1. Если окислитель и восстановитель разные вещества, то такие реак-
реакции относятся к межмолекулярным. Примерами служат все рассмотренные
ранее реакции.
2. При термическом разложении сложных соединений, в состав кото-
которых входят окислитель и восстановитель в виде атомов разных элементов,
происходят окислительно-восстановительные реакции, называемые внут-
внутримолекулярными:
+5 -2 нагревание -1 О
2КС1Оз = 2KCl + 3O2t
-3 +6 нагревание о +3
(N Н4 J Сг2 О7 = N2 t + Cr2 O3 + 4Н2О
+7 -2 нагревание +6 +4 О
2КМпО4 = К2Мп04 + МпО2 + О2 Т
3. Реакции диспропорционирования {дисмутации или, согласно уста-
устаревшей терминологии, самоокисления—самовосстановления) могут проис-
происходить, если соединения, содержащие элементы в промежуточных степенях
окисления, попадают в условия, где они оказываются неустойчивыми (на-
(например, при повышенной температуре). Степень окисления этого элемента
и повышается, и понижается:
С12 + 2К0Н= КС1 + К СЮ +Н20
+4 нагревание +6 -2
4К2 S 03 = ЗК2 S 04 + К2 S
-1 нагревание о -2
2Н2О2 = О2Т+2Н2О
1.3. Окислительно-восстановительные реакции 29
4. Реакции контрпропорционирования (коммутации) — это процессы
взаимодействия окислителя и восстановителя, в состав которых входит один
и тот же элемент с разными степенями окисления. В результате продуктом
окисления и восстановления является вещество с промежуточной степенью
окисления атомов данного элемента:
Na2 S О3 + 2Na2 S + 6НС1 = 3S + 6NaCl + ЗН2О
+5 -1 О
НВгО3 (конц)+ 5НВг(конц)= ЗВг2 +ЗН2О
Существуют также реакции смешанного типа. Например, к внутримо-
внутримолекулярной реакции контрпропорционирования относится реакция разло-
разложения нитрата аммония
-3 +5 нагревание +1
NH4NO3 = N2O+2H2O
Составление уравнений. Уравнения окислительно-восстановительных
реакций составляют, основываясь на принципах равенства числа одних и тех
же атомов до и после реакции, а также учитывая равенство числа электронов,
отдаваемых восстановителем, и числа электронов, принимаемых окислителем,
т.е. электронейтральность молекул. Реакцию представляют в виде системы двух
полуреакций — окисления и восстановления, суммирование которых с учетом
указанных принципов приводит к составлению общего уравнения процесса.
Для составления уравнений окислительно-восстановительных реакций
наиболее часто используют метод электронно-ионных полуреакций и метод
электронного баланса.
Метод электронно-ионных полуреакций применяют при составлении
уравнений реакций, протекающих в водном растворе, а также реакций с
участием веществ, степень окисления элементов которых трудно определить
(например, KNCS, СН3СН2ОН).
Согласно этому методу, выделяют следующие главные этапы состав-
составления уравнения реакций.
а) записывают общую молекулярную схему процесса с указанием вос-
восстановителя, окислителя и среды, в которой протекает реакция (кислотная,
нейтральная или щелочная). Например
SO2 + K2Cr207 + H2SO4(pa36) -> ...
б) учитывая диссоциацию электролитов в водном растворе, данную
схему представляют в виде молекулярно-ионного взаимодействия. Ионы,
степени окисления атомов которых не изменяются, в схеме не указывают, за
исключением ионов среды (Н+, ОН~):
SO2+ Сг2О^ +Н+->...
30 1- Понятия вещества и химической реакции
в) определяют степени окисления восстановителя и окислителя, а так-
также продуктов их взаимодействия:
окисление восстановителя восстановление окислителя
S О2 -> (S О4 J" (С г2О7 J~ -> 2Сг3+
(Данный этап не является обязательным и его можно опустить, если
определение степеней окисления затруднительно. Скобки при записи анио-
аниона обычно не используются, но в данном случае необходимы, чтобы избе-
избежать путаницы при определении степени окисления элемента и заряда
аниона.)
г) записывают материальный баланс полуреакции окисления и восста-
восстановления:
окисление восстановителя восстановление окислителя
SO2 +2H2O-2e= (SO4J- + 4H+ (Сг2О7J"+ 14Н++ 6е = 2Сг3++ 7Н2О
(Согласно правилам ИЮПАК, полуреакцию окисления восстановите-
восстановителя записывают следующим образом: SO2 + 2Н2О = SO4~ + 4Н+ + 2е, однако,
по мнению авторов настоящего учебника, используемая выше запись более
наглядна.)
д) суммируют полуреакции, учитывая принцип равенства отданных и
принятых электронов:
SO2 + 2Н2О - 2е = SO24~ + 4Н+
Сг2О^+ 14Н++ бе = 2Сг3++ 7Н2О
3SO2 + 6Н2О + Сг2О^" + 14Н+ = 3SO^" + 12Н+ + 2Сг3++ 7Н2О
и, сокращая одноименные частицы, получают общее ионно-молекулярное
уравнение
3SO2 + Сг2О?" + 2Н+ = 3SO^" + 2Сг3++ Н2О
е) добавляют ионы, не участвовавшие в процессе окисления—восста-
окисления—восстановления, уравнивают их количество слева и справа, и записывают молеку-
молекулярное уравнение реакции
3SO2 + К2Сг207 + H2SO4(pa36) = Cr2(SO4K + K2SO4 + Н2О
Наибольшие трудности возникают при составлении материального
баланса полуреакций окисления и восстановления, когда изменяется число
атомов кислорода, входящих в состав частиц окислителя и восстановителя.
1.3. Окислительно-восстановительные реакции
31
Следует учитывать, что в водных растворах связывание или присоединение
кислорода происходит с участием молекул воды и ионов среды.
В процессе окисления на один атом кислорода, присоединяющийся к
частице восстановителя, в кислотной и нейтральной средах расходуется од-
одна молекула воды и образуются два иона Н+; в щелочной среде расходуются
два гидроксид-иона ОН" и образуется одна молекула воды (табл. 1.1).
Для связывания одного атома кислорода окислителя в кислотной сре-
среде в процессе восстановления расходуются два иона Н+ и образуется одна
молекула воды; в нейтральной и щелочной средах расходуется одна молеку-
молекула Н2О и образуются два иона ОН" (табл. 1.2).
Таблица 1.1
Присоединение атомов кислорода к восстановителю в процессе окисления
Среда
Кислотная,
нейтральная
Щелочная
Частицы,
участвующие
в присоединении
одного атома
кислорода
Н2О
2ОН"
Образую-
щиеся
частицы
2Н+
Н2О
Примеры полуреакций
окисления
SO2' + Н2О -2е = SO4" + 2Н+
SO2 + 2Н2О -2е = SOf + 4Н+
SO2" + 2ОН" -2е = SO^~ + Н2О
SO2 + 4ОН" -2е = SO|" + 2Н2О
Таблица 1.2
Связывание атомов кислорода окислителя в процессе восстановления
Среда
Кислотная
Нейтральная,
щелочная
Частицы,
участвующие
в связывании
одного атома
кислорода
2Н+
Н2О
Обра-
Образующиеся
частицы
Н2О
2ОН"
Примеры полуреакций
восстановления
Сг2Оу~ + 14Н+ + бе = 2Сг3+ + 7Н2О
МпО4 + 8Н+ +5е = Мп2+ + 4Н2О
СЮ^+4Н2О + Ъе = [Сг(ОНN]3" + 2ОН"
MnOJ + ЗН2О + Ъе = MnO(OHJ + 4OH"
Достоинства метода электронно-ионных полуреакций заключаются в
том, что при составлении уравнений окислительно-восстановительных ре-
реакций учитываются реальные состояния частиц в растворе и роль среды в
протекании процессов, нет необходимости использования формального по-
понятия степени окисления.
32 1- Понятия вещества и химической реакции
Метод электронного баланса, основанный на учете изменения степе-
степеней окисления и принципе электронейтральности молекулы, является уни-
универсальным. Его обычно используют для составления уравнений окисли-
окислительно-восстановительных реакций, протекающих между газами, твердыми
веществами и в расплавах.
Последовательность операций, согласно этому методу, такая:
1) записывают формулы реагентов и продуктов реакции в молекуляр-
молекулярном виде:
FeCl3 + H2S -> FeCl2 + S + НС1
2) определяют степени окисления атомов, меняющих ее в процессе
реакции:
+3 -2 +2 О
FeCl3 + Н2 S -> FeCl2 + S + НС1
3) по изменению степеней окисления устанавливают число электро-
электронов, отдаваемых восстановителем, и число электронов, принимаемых окис-
окислителем, и составляют электронный баланс с учетом принципа равенства
числа отдаваемых и принимаемых электронов:
+3 +2
Fe +le= Fe
-2 О
S -2e= S
4) множители электронного баланса записывают в уравнение окисли-
окислительно-восстановительной реакции как основные стехиометрические коэф-
коэффициенты:
2FeCl3 + H2S -> 2FeCl2 + S + HC1
5) подбирают стехиометрические коэффициенты остальных участни-
участников реакции:
2FeCl3 + H2S = 2FeCl2 + S + 2HC1
При составлении уравнений следует учитывать, что окислитель (или
восстановитель) может расходоваться не только в основной окислительно-
восстановительной реакции, но и при связывании образующихся продуктов
реакции, т.е. выступать в роли среды и солеобразователя.
Примером, когда роль среды играет окислитель, служит реакция
окисления металла в азотной кислоте, составленная методом электронно-
ионных полуреакций:
1.3. Окислительно-восстановительные реакции
3 3
ЗСи
3
NOJ + 4H+ + Зе = NO + 2Н2О 2
ЗСи + 2NOJ + 8Н+ = ЗСи2+ + 2NO + 4Н2О
6HNO3(cpe«a)= 3Cu(NO3J + 2NO + 4Н2О
ИЛИ
ЗСи + 8ЮЮ3(разб)= 3Cu(NO3J + 2N0 + 4Н2О
Примером, когда восстановитель является средой, в которой протекает ре-
реакция, служит реакция окисления соляной кислоты дихроматом калия, со-
составленная методом электронного баланса:
С12 + КС1 + Н20
НС1 + К2Сг2О7 -> СгС13
-1 +6 +3
НС1 + К2Сг207 + НС1->СгС13
-1 О
Cl-le= C1
+6 +3
Сг + Зе = Сг
6НС1 (восстановитель) + К2Сг207 + НС1 (среда) ~'
6НС1 + К2Сг207 + 8HC1 = 2СгС13 -»
КС1
О
С12 +КС1
Н20
6
2
• 2СгС13 + ЗС12 + КС1 + Н2О
ЗС12 + 2КС1 + 7Н2О
или
14НС1 + К2Сг207 = 2СгС13 + ЗС12 + 2КС1 + 7Н2О
При расчете количественных, массовых и объемных соотношений
участников окислительно-восстановительных реакций используют основ-
основные стехиометрические законы химии и, в частности, закон эквивалентов.
Для определения направления и полноты протекания окислительно-восста-
окислительно-восстановительных процессов используют значения термодинамических парамет-
параметров данных систем, а при протекании реакций в водных растворах — значения
соответствующих электродных потенциалов.
2 - 9795
Стехиометрия.
2 ¦ Закономерности изменения и способы
~" определения количества вещества
Раздел химии, в котором рассматривают количественные (массовые,
объемные) соотношения между реагентами и продуктами реакций, а
также количественный состав веществ, называют стехиометрией (от
греч. stoicheion — основа, элемент, metreo — измеряю). Она базиру-
базируется на законах, обусловленных строением вещества. Именно опре-
определение массовых соотношений элементов в соединениях и веществ
в химических реакциях привело к открытию Периодического закона
химических элементов Д.И. Менделеева и созданию атомно-молеку-
лярной теории строения вещества. Стехиометрия имеет фундамен-
фундаментальное значение, так как является основой количественного анализа,
позволяет вычислить расход реагентов, выход продуктов и эффек-
эффективность химических процессов.
2.1. Основные определения
В стехиометрии используют следующие понятия.
Формульная единица вещества — реально существующие час-
частицы: атомы (S, С, Fe), молекулы (Н2О, СОг), ионы ( Са2+, СО2), ра-
радикалы (ОН, NO2) и другие частицы вещества.
Моль — количество вещества, содержащее столько формульных
единиц, сколько атомов содержится в 0,012 кг изотопа углерода Х\С.
Постоянная Авогадро NA — число частиц в 1 моль любого ве-
вещества; NA « 6,022 • 1023 моль.
Молярная масса М— масса 1 моль вещества. Молярная масса
численно совпадает с массами атомов и молекул, выраженными в
атомных единицах массы, и измеряется в граммах на моль (г/моль).
Молярный объем VM — объем 1 моль газа, измеряется в литрах
на моль (л/моль).
Химический эквивалент Э — реальная или условная частица,
которая может присоединять, высвобождать или быть каким-либо
другим способом равноценна одному атому (иону) водорода в об-
обменных (кислотно-основных) реакциях или одному электрону в
окислительно-восстановительных реакциях.
Число эквивалентности (эквивалентное число) z3KB показыва-
показывает, сколько химических эквивалентов содержит одна формульная
единица вещества.
2.2. Количественные законы протекания химических реакций 3 5
Фактор эквивалентности /экв — величина, обратная числу эквива-
эквивалентности; показывает, какая доля формульной единицы вещества прихо-
приходится на один химический эквивалент /экъ = 1/ z3KB-
Молярная масса химического эквивалента Мэкв вещества — масса
1 моль химического эквивалента вещества; выражается в граммах на моль
(по правилам ИЮПАК указание на эквивалент при записи размерности Мэкв
опускается) и связана с молярной массой вещества соотношением:
М
Мэкв= = М/ЭКВ. B.1)
Молярный объем химического эквивалента Уэкв вещества — объем
1 моль химического эквивалента газа; выражается в литрах на моль и связан
с молярным объемом соотношением:
Ужв = ^- = VMf3KB. B.2)
2.2. Количественные законы протекания химических реакций
Теоретической базой количественных расчетов химических реакций
являются стехиометрические законы.
Закон сохранения массы (MB. Ломоносов, 1756; А. Лавуазье, 1777).
Общая масса веществ, вступивших в химическую реакцию, равна общей
массе продуктов реакции.
Закон постоянства состава (Ж. Пруст, 1797). Всякое чистое вещест-
вещество независимо от способа его получения, имеет постоянный качественный и
количественный состав.
Закон кратных отношений (Дж. Дальтон, 1803). Если два элемента
образуют между собой несколько молекулярных соединений, то массовые
количества одного элемента, приходящиеся на одно и то же массовое коли-
количество другого, соотносятся между собой как небольшие целые числа.
Закон объемных отношений (Ж. Гей-Люссак, 1808). Объемы всту-
вступающих в химическую реакцию газов и газообразных продуктов реакции
относятся друг к другу как небольшие целые числа.
Закон эквивалентов (И. Рихтер, 1792). Химические элементы входят в
состав соединений в строго определенных отношениях масс, поэтому веще-
вещества реагируют и образуются в эквивалентных количествах.
При расчетах реакций, протекающих с участием газообразных ве-
веществ, опираются на основные газовые законы.
2*
36 2. Стехиометрия. Закономерности изменения количества вещества
Закон Авогадро (А. Авогадро, 1811). В равных объемах идеальных
газов при одинаковых давлении и температуре содержится одинаковое
число молекул. Согласно этому закону, 1 моль идеального газа при нор-
нормальных условиях (н.у.), Го = 273,15 К; ро = 101,3 кПа, занимает объем
Км= 22,414 л/моль.
Закон Бойля—Мариотта (Р. Бойль, 1662; Э. Мариотт, 1676). При по-
постоянной температуре объем данного количества газа обратно пропорцио-
пропорционален давлению, под которым он находится:
Закон Гей-Люссака (Ж. Гей-Люссак, 1802). При постоянном давлении
изменение объема газа прямо пропорционально температуре:
Последние два закона часто выражают формулой объединенного газо-
газового закона:
B.3)
Здесь /?, V и Т — давление, объем и температура данного количества газа в
состояниях 1 и 2 соответственно. Если в B.3) подставить значения /?, V и Г,
соответствующие 1 моль идеального газа при нормальных условиях (часто
pV
эти величины обозначают р0, Vo и Го), то соотношение —— станет постоян-
постоянным. Его обозначают R и называют универсальной газовой постоянной
(R = 8,314 Дж/(мольК)). С учетом этого обозначения объединенный газо-
газовый закон для 1 моль газа записывают в виде
pV=RT B.4)
и называют уравнением состояния идеального газа. Если количество газа
отлично от 1 моль, то B.4) принимает вид
pV= nRT, B.5)
где п — число молей данного газа, которое может быть определено по от-
( т\
ношению массы т данного вещества к его молярной массе М\п-— . Со-
v М)
отношение B.5) называют уравнением Клапейрона—Менделеева.
Закон парциальных давлений или закон Дальтона (Дж. Дальтон, 1801).
Общее давление смеси газов, химически не взаимодействующих между со-
собой, равно сумме их парциальных давлений:
2.2. Количественные законы протекания химических реакций 37
Парциальное давление р{ — давление /-го газа, входящего в состав смеси,
которое создавал бы этот газ, если бы он один при той же температуре за-
занимал объем, равный объему всей смеси.
На основе стехиометрических и газовых законов по известному коли-
количеству одного из веществ рассчитывают значения масс и объемов остальных
участников реакции. Наиболее часто при количественных расчетах исполь-
используют закон эквивалентов, который позволяет во многих случаях обойтись
без составления уравнения химической реакции. С учетом современного
понятия химического эквивалента, закон эквивалентов можно сформулиро-
сформулировать следующим образом: один химический эквивалент одного вещества
всегда взаимодействует с одним химическим эквивалентом другого. Поэто-
Поэтому для условной реакции
vAA + vBB = vDD + vFF,
где А, В, D, F — участники реакции; vA, vB, vD, vF — соответствующие сте-
хиометрические коэффициенты, числа молей химических эквивалентов пэкъ
участников реакции одинаковы:
Для расчета массовых соотношений участников химической реак-
реакции удобно использовать другую формулировку закона эквивалентов:
массы реагирующих между собой веществ, а также массы продуктов ре-
реакции пропорциональны молярным массам химических эквивалентов
этих веществ.
Обозначая массу вещества через т, для условной химической реакции
можно записать
тА =эквА^ гпА =эквА^ тА =МжвА ^^
тВ ^эквв' mD M3Kbd' Щ M3kbF
Если реакция протекает в газовой фазе, то можно использовать
соответствующие объемные соотношения:
г/ V г/ V т/ V
УА _ экв А . 1А___эквА_. V К _ экв А итп ^ J\
V V ' V V ' V V V • /
В случае, когда А — твердое вещество, а В — газ, справедливо выра-
выражение
тА _ ^эквА /9 о\
V V
УВ У экв В
38 2. Стехиометрия. Закономерности изменения количества вещества
Отметим, что объемы газов, а также молярные объемы их химических
эквивалентов следует привести к одинаковым условиям (как правило, к
нормальным условиям), используя B.3) или B.5).
В стехиометрических расчетах, основанных на законе эквивалентов,
главным является установление чисел эквивалентности z3KB участников ре-
реакции. В обменных реакциях число эквивалентности вещества определяется
стехиометрическим уравнением реакции. Если известно число эквивалент-
эквивалентности z3KB а вещества А в реакции
vAA + vBB = vDD + vFF,
то число эквивалентности z3KBb вещества В можно найти из соотношения:
2эквВ = 2эквА • B-9)
VB
Эмпирическим путем было установлено, что числа эквивалентности
многих веществ при протекании реакций, происходящих без изменения степе-
степеней окисления элементов, можно рассчитать, используя состав формульной
единицы вещества (табл. 2.1). Очевидно, что только одноосновные кислоты,
однокислотные основания и их соли и оксиды имеют постоянное значение чис-
числа эквивалентности (z3KB = 1) в обменных реакциях. Для многоосновных кислот,
многокислотных оснований, их солей и оксидов значение числа эквивалентно-
эквивалентности зависит от стехиометрического уравнения и условий протекания реакции.
В окислительно-восстановительных реакциях число эквивалентности
окислителя равно числу электронов, которые принимает одна формульная
единица окислителя, а число эквивалентности восстановителя равно числу
электронов, которые отдает одна формульная единица восстановителя. На-
Например, в зависимости от среды окислительно-восстановительные реакции с
участием перманганат-иона протекают по-разному, соответственно изменя-
изменяется и число эквивалентности этого окислителя (табл. 2.2). Числа эквива-
эквивалентности остальных участников окислительно-восстановительных реакций
определяются стехиометрией реакции B.9).
Стехиометрические законы справедливы только для соединений с мо-
молекулярной структурой, так называемых дальтонидов, химический состав
которых постоянен и не зависит от способа их получения. Соединения с не-
немолекулярной структурой (бертоллиды) часто имеют переменный состав,
зависящий от способа их получения. Примерами таких веществ служат со-
соединения металлов между собой (интерметаллиды), а также многочислен-
многочисленные оксиды, сульфиды, селениды, теллуриды, нитриды, карбиды, фосфиды,
силициды и др. Следовательно, для таких соединений применять законы
постоянства состава, кратных отношений и эквивалентов некорректно.
2.2. Количественные законы протекания химических реакций
39
Таблица 2.1
Определение числа эквивалентности z3KB по составу формульной единицы вещества
Класс неорганических
соединений
Кислоты Н„А:
одноосновные, п = 1
многоосновные, п > 1
Основания М(ОН)т:
однокислотные, т = 1
многокислотные, т > 1
Кислотные оксиды 3Jdy
(соответствующие
кислоте Н„А)
Основные оксиды Э^
(соответствующие
основанию M(OH)W)
Вода Н2О
Соли МХКУ
*экв
^экв кисл
Z3KB= П
*экв ^ Л
^экв осн
z3KB= m
*экв ^ т
z3KB =
=**эквкисл
Z3KB =
—XZ3KB осн
?экв= 1
Z3KB= 2
^экв =
—xz3KB осн
Примеры
ДляНС1 z3KB=l
Для H2SO4 z3KB = 2 в реакции образования
средней соли
H2SO4 + 2NaOH = Na2SO4 + 2Н2О
и z3KB = 1 в реакции образования кислой соли
H2SO4 + NaOH = NaHSO4 + Н2О
Для NaOH z3KB = 1
Для Cu(OHJ z3KB= 2 в реакции образования
средней соли
Си(ОНJ + 2НС1 = СиС12 + 2Н2О
и z3KB < 1 в реакции образования основной
соли
Cu(OHJ + HC1 = Си(ОН)С1 + Н2О
Для N2O5 z3KB = 2, так как для HNO3 z3KB = 1:
N2O5 + 2NaOH = 2NaNO3 + H2O
Для СО2 z3KB = 2 в реакции образования сред-
средней соли
СО2+ 2NaOHKOHU = Na2CO3 + Н2О
и z3KB = 1 в реакции образования кислой соли
СО2 + NaOH разб. = NaHCO3
Для Na2O z3KB = 2, так как для NaOH z3KB = 1:
Na2O + Н2О = 2NaOH
Для MgO z3KB = 2 в реакции образования
средней соли
и z3KB = 1 в реакции образования основной
соли
2MgO + Н2О + СО2 = Mg2(OHJCO3
В кислотно-основных реакциях:
нейтрализации NaOH + НС1 = NaCl + Н2О
гидролиза Na2CO3 + Н2О = NaHCO3 + NaOH
В других обменных реакциях
Для A12(SO4K z3KB = 6, так как для А1(ОНK
z3KB=3
Для Cu(OH)Cl z3KB= 1, так как для Си(ОНJ
Для NaHCO3 z3KB = 1, так как для NaOH
40
2. Стехиометрия. Закономерности изменения количества вещества
Таблица 2.2
Число эквивалентности перманганат-иона
Среда
Кислотная
Нейтральная
Основная
Уравнение полуреакции восстановления
перманганат-иона
MnOJ + 8Н+ + 5е = Мп2+ + 4Н2О
MnOi + 3 Н2О + Зе = MnO(OHJ>L + 4OH"
MnOJ+ le = MnO4~
Значение
^экв
5
3
1
Законы идеального газа и, в частности закон Авогадро, объединенный
газовый закон, уравнение Клапейрона—Менделеева для реальных молеку-
молекулярных газов выполняются достаточно точно при нормальных температуре
и давлении. Ассоциированные газы, например пары органических кислот,
не подчиняются этим законам даже при очень низком давлении.
Закон сохранения массы, будучи универсальным, верен лишь в прак-
практическом смысле. Согласно уравнению Эйнштейна (Е = тс2), энергия и мас-
масса эквивалентны, поэтому при любом химическом взаимодействии, сопро-
сопровождающемся изменением энергии системы, изменяется и масса системы.
Однако энергетические эффекты химических реакций незначительны, по-
поэтому изменения массы столь малы, что не могут быть измерены современ-
современными инструментальными методами.
2.3. Растворы. Общие понятия
Многие химические реакции протекают в растворах. Растворы —
это гомогенные (состоящие из одной фазы) многокомпонентные смеси
переменного состава. Растворы могут быть газообразными, жидкими и
твердыми. Среди соединений, образующих раствор, условно выделяют
растворенные вещества и растворитель. Растворителем принято считать
компонент, который в чистом виде существует в том же агрегатном со-
состоянии, что и образовавшийся раствор, или компонент, содержание ко-
которого в растворе выше содержания остальных компонентов, называемых
растворенными веществами. Наибольшее практическое значение имеют
жидкие растворы, в частности водные, в которых растворителем является
вода.
Для выражения состава раствора и содержания растворенного вещест-
вещества применяют различные способы.
1. Массовая доля компонента со, — отношение массы /-го компонента
/и,- к массе раствора
2.3. Растворы. Общие понятия 41
B.10)
где со, — безразмерная величина, принимающая значения от 0 до 1 или от 0
до 100 % (в последнем случае говорят о процентной концентрации).
2. Молярная {мольная) доля компонента Xt — отношение числа молей
/-го компонента щ к сумме молей 5л всех компонентов, образующих рас-
раствор, безразмерная величина
^. BЛ1)
3. Моляльная концентрация, или моляльность, Ст — число молей п
растворенного вещества, приходящихся на 1 кг растворителя, моль/кг:
Ст= -=- = » B.12)
ms Mms
где т — масса растворенного вещества, г; М — молярная масса растворен-
растворенного вещества, г/моль; ms — масса растворителя, кг.
4. Молярная концентрация, или молярность, С — число молей п рас-
растворенного вещества в 1 л раствора, моль/л, или сокращенно М:
С=^- = -^-, B.13)
V MV }
где V— объем раствора, л; т — масса растворенного вещества, г; М— мо-
молярная масса растворенного вещества, г/моль.
5. Молярная концентрация химического эквивалента, или нормальная
концентрация, или нормальность, Сэкв — число молей химического эквива-
эквивалента иЭкв растворенного вещества в 1 л раствора, моль экв/л, или сокращен-
сокращенно н.:
МЖЪУ MV
B.14)
где Мэкв — молярная масса химического эквивалента растворенного вещест-
вещества, г/моль экв; z3KB — число эквивалентности растворенного вещества; V —
объем раствора, л; т — масса растворенного вещества, г; М— молярная мас-
масса растворенного вещества, г/моль.
6. Массовая концентрация, СмаСс — масса растворенного вещества т в
1 л раствора, г/л:
у, B.15)
где V— объем раствора, л.
42 2. Стехиометрия, Закономерности изменения количества вещества
Можно применять и другие способы выражения состава раствора. На-
Например, в справочниках растворимость данного вещества, т.е. состав насы-
насыщенного раствора, выражают как отношение массы растворенного вещества
к массе растворителя.
При проведении химических реакций часто требуется использовать
растворы определенной концентрации. Для приготовления раствора задан-
заданной концентрации взвешивают необходимую массу (отмеряют необходи-
необходимый объем) чистых компонентов раствора или используют более концен-
концентрированные растворы известной концентрации, в частности, фиксаналы —
растворы, помещенные в герметичные ампулы заводского производства,
содержащие строго определенное (обычно 0,1 моль) количество химическо-
химического соединения.
При расчете массы (объема) компонентов раствора учитывают соот-
соотношения между различными способами выражения его состава, например,
представленные в табл. 2.3 соотношения концентраций бинарного раствора,
т.е. раствора, состоящего из двух компонентов — растворителя и раство-
растворенного вещества.
Таблица 2.3
Соотношения между различными способами выражения
состава бинарного раствора
Вели-
Величина
со
С
ст
(-'масс
СО
СМ
Р
СэквМ
СМ
СО — ¦'
СтМ +1000
смасс
р
с
сор
м
Г1— экв
с Ст9
СтМ + 1000
/-> ^масс
м
(-экв
С «Ргэкв
м
с-ft.
экв CwM + 1000
/^ экв масс
Сэкв" м
^ ЮООоо
с 1000С
р-СМ
г юоосэкв
с _ юоосмасс
(-масс
(-масс ~ ^P
(-масс "~ С^*
с =с м
^—масс ^^экв
с СтЛф
масс с„л/+юоо
Примечание, р — плотность раствора данного состава, г/л.
2.3. Растворы. Общие понятия 43
Установление состава раствора — одна из задач аналитической хи-
химии. Количественный анализ выполняют физическими, химическими и
физико-химическими методами. К наиболее простым среди них относят-
относятся денсиметрия и титриметрия.
Денсиметрия — метод количественного анализа, основанный на из-
измерении плотности исследуемого раствора и сравнении полученной величи-
величины с табличными данными (значениями градуировочной функции р = /(со)).
Для приближенного определения плотности раствора применяют ареометр —
прибор, принцип действия которого основан на законе Архимеда. Жидкость
наливают в стеклянный цилиндр и подбирают такой ареометр, который сво-
свободно плавает в растворе, не касаясь дна и стенок цилиндра. Плотность опре-
определяют по делению шкалы ареометра, до которого он погружается в раствор.
Для более точного определения плотности раствора используют пикнометр —
небольшой стеклянный сосуд, объем и масса которого точно известны.
Плотность находят путем взвешивания на аналитических весах пикнометра,
заполненного исследуемым раствором. Значения плотности растворов
большинства широко используемых кислот, щелочей, солей в зависимости
от концентрации раствора и температуры табулированы (табл. П.4.4).
Титриметрия — аналитический метод, основанный на измерении
объема раствора реагента, взаимодействующего с определяемым веществом.
Растворы реагентов известной концентрации, используемые в титриметрии,
называют титрованными (стандартными) растворами, или титрантами.
Принцип титрования заключается в следующем: к раствору анализи-
анализируемого вещества неизвестной концентрации {пробе) добавляют небольши-
небольшими порциями раствор титранта до тех пор, пока не будет получен сигнал
индикатора, свидетельствующий о прекращении реакции (достижении ко-
конечной точки титрования). Момент окончания титрования можно устано-
установить при помощи химической реакции или по изменению некоторого физи-
физического свойства. Часто используют органические красители, окраска которых
изменяется по достижении конечной точки титрования. Такие индикаторы
должны проявлять свойства, положенные в основу соответствующей титри-
метрической реакции. Индикацию растворов можно проводить как визуально,
так и с использованием инструментальных методов. Если проба или титрант
окрашены, специальные индикаторы могут и не потребоваться.
Концентрацию исследуемого раствора в соответствии с законом экви-
эквивалентов рассчитывают по формуле
СЭкв1К1 = Сэкв2К2, B.16)
где Сэкв 1 и СЭкв 2 — молярные концентрации химического эквивалента пробы
и титранта соответственно; V\ — отмеренный объем пробы; V2 — объем
титранта, израсходованный на титрование.
44 2. Стехиометрия. Закономерности изменения количества вещества
Экспериментально установленная конечная точка титрования вслед-
вследствие погрешностей измерений и наблюдений отличается от истинной точ-
точки эквивалентности, соответствующей стехиометрическим соотношениям
реагирующих веществ. Погрешность измерения зависит от многих факто-
факторов: класса точности применяемых приборов, методики измерения, индиви-
индивидуальных особенностей наблюдателя и др. Погрешность измерения, которая
при повторных измерениях остается постоянной или закономерно изменяет-
изменяется, называют систематической погрешностью. В титриметрии главную
роль играет погрешность измерения объема, определяемая точностью счи-
считывания показаний шкалы бюретки, которая обычно составляет 0,2-0,3 %.
Точность определения концентрации титранта также является условием вы-
высокой точности определения концентрации пробы.
Концентрацию кислот и оснований вычисляют, используя метод ки-
кислотно-основного титрования, в основе которого лежит реакция нейтрали-
нейтрализации
Н+ + ОН" = Н2О
В кислотной среде концентрация ионов водорода Н+ превышает кон-
концентрацию гидроксид-ионов ОН", а в щелочной среде наоборот. Для удоб-
удобства оценки характера (реакции, кислотности) среды вводят понятие водо-
водородного показателя рН, численно равного отрицательному десятичному ло-
логарифму молярной концентрации ионов водорода в растворе
PH = -lgCH+ B.17)
В водных растворах произведение молярных концентраций ионов во-
водорода и гидроксид-ионов
Сн* Сон- = *н2о B-18)
называемое ионным произведением воды, — постоянная величина и при
Т= 298 К КН20 = 10~14, поэтому в нейтральной среде рН = 7, в кислотной
рН < 7, в щелочной рН > 7.
Истинная точка эквивалентности кислотно-основного титрования со-
соответствует равенству молярных концентраций ионов водорода Н+ и гидро-
гидроксид-ионов ОН":
С = С
Реакция нейтрализации не сопровождается видимыми изменениями,
например переменой окраски раствора. Поэтому для фиксирования точки
2.3. Растворы. Общие понятия
45
эквивалентности используют органические красители, структура и окраска
которых зависит от величины водородного показателя рН. Изменение окра-
окраски кислотно-основных индикаторов происходит внутри определенного уз-
узкого интервала значений рН растворов (табл. 2.4). Этот интервал зависит
только от свойств данного индикатора, следовательно, перемена окраски
индикатора происходит, как правило, не строго в точке эквивалентности, а с
некоторым отклонением от нее. Значение рН, при котором заканчивается
титрование с данным индикатором, называют показателем титрования и
обозначают рТ. Значение рТ соответствует рН индикатора промежуточной
окраски и находится внутри области перехода.
Таблица 2.4
Области перехода окраски некоторых рН-индикаторов
Индикатор
Фенолфталеин
Лакмус
Метиловый
оранжевый
Метиловый
красный
Окраска
кислотной
формы
Бесцветная
Красная
Розовая
Красная
Окраска
щелочной
формы
Красная
Синяя
Желтая
Желтая
Область
перехода,
рН
8,0-10,0
5,0-8,0
3,1-4,4
4,2-6,2
Показатель
титрования,
рТ
9,0
7,0
4,0
5,5
Очевидно, что от выбора индикатора зависит индикаторная ошибка
титрования (систематическая погрешность), значение которой может коле-
колебаться в широких пределах в зависимости от того, какой взят индикатор и
какие кислота и основание реагируют между собой. Так, в случае титрова-
титрования кислоты основанием при рТ < 7 имеет место недотитрованность, а при
рТ > 7 — перетитрованность.
Для сильных кислот и оснований относительная систематическая по-
погрешность кислотно-основного титрования равна
С -Г
сон- сн+
100%,
B.19)
где С , С + и Со — молярные концентрации ионов ОН , Н+ и титруемого
вещества соответственно. Например, при титровании 0,01 М раствора силь-
сильной кислоты раствором сильного основания в присутствии фенолфталеина
1<Г5-10-9
10
,-2
100% = 0,1%.
Практические занятия
Фундаментальные понятия и законы химии
Примеры решения задач
Задача 1. Определить, какой металл массой тм = 3,24 г образует оксид мас-
массой тмо = 3,48 г.
Решение. Процесс взаимодействия металла М с кислородом О2 можно вы-
выразить следующим уравнением:
тМ + 0,5лО2 = МтО„.
Масса О2, израсходованного на образование оксида, согласно закону
сохранения массы, т0 = тм 0 - тм = 3,48 - 3,24 = 0,24 г.
В соответствии с B.6) молярная масса эквивалента металла
эквМ "" iKf3KBO2
то2
Число эквивалентности О2 z3KB = 4, так как в реакциях окисления ме-
металлов степень окисления кислорода изменяется от 0 до -2, т.е. О2 прини-
принимает четыре электрона. Молярная масса эквивалента О2 согласно B.1)
АЛ -зо
о, 32 _ . *
Mwa п = = — =8 г/моль экв.
2экв 4
Следовательно,
3,24
0,24
Мэкв м = — 8 = 108 г/моль экв.
Молярная масса металла Мм = ^экв^Эквм- Полагая z3KB = 1 (другие
значения не подходят), по Периодической таблице элементов Менделеева
определяют искомый металл Ag (MAg= 107,9 г/моль).
Задача 2. При растворении металла массой /им = 0,32 г в кислоте выделился
водород Н2, который при температуре Т = 294 К и давлении р = 745 Торр
(мм рт.ст.) занимает объем VH = 120,4 мл. Определить молярную массу
эквивалента металла, установить металл.
Авторы используют обозначение «моль экв» вопреки правилам ИЮПАК в учебных
целях.
Примеры решения задач 47
Решение. Процесс растворения металла в кислоте можно выразить уравнением
Согласно B.8), молярная масса эквивалента металла
шэквМ ~ v кэквН2*
Ч
Число эквивалентности Н2 z3KB = 2, так как степень окисления водорода изме-
изменяется от +1 до 0, следовательно, молярный объем химического эквивалента Н2 при
нормальных условиях в соответствии с законом Авогадро и уравнением B.2) равен
22 4
^экв н2 = ~у~ = ! Ь2 л/моль экв.
Объем выделившегося водорода приводят к нормальным условиям (н.у.), ис-
используя уравнение B.3),
Л&Г = 120,4.745.273 _
рТ0 294-760
Следовательно,
0,32
AC
11200 = 32,7 г/моль экв.
109,6
Молярная масса металла Мм = z3KB Мэкв м. Полагая z3KB = 2 (другие значения не
подходят), по таблице элементов определяют искомый металл: Zn (MZn= 65,4 г/моль).
Задача 3. Какой объем водорода (н.у.) потребуется для восстановления оксида мас-
массой тм 0 =159 г, содержащего 79,87% (масс.) металла. Определить молярную
массу эквивалента металла, установить металл.
Решение. Процесс восстановления оксида металла можно выразить уравнением:
= пМ + иН2О
Согласно B.6), молярная масса эквивалента металла
мэквм = —мэкво = —1-^~ 8 = 31,76 г/мольэкв.
эквМ ^ эквО2 100_79?87
Молярная масса металла Ми = z3KB Мэкв м. Полагая z3KB = 2 (другие значения
не подходят), по таблице элементов Менделеева определяют искомый металл: Си
(Мси = 63,55 г/моль).
Объем водорода (н.у.), необходимый для восстановления оксида, находят,
используя B.8). Поскольку масса металла в оксиде тм = 159 • 0,7987 = 127 г, необ-
необходимый объем водорода
= тм у =-^_.и,2=44,8л(н.у.).
Н2 д г ЭКВ Г12 л л пг ' "> \ J /
М 31,76
48 Фундаментальные понятия и законы химии
Задача 4. Измеренная с помощью ареометра плотность водного раствора хлорида
натрия NaCl p = 1095,0 г/л. Определить массовую долю раствора, рассчитать его
молярную концентрацию, молярную концентрацию эквивалента соли, моляльную
концентрацию и массовую концентрацию раствора.
Решение. Заданное значение плотности раствора NaCl в табл. П4.4 отсутствует, по-
поэтому для определения массовой доли соли в растворе применяют метод интерпо-
интерполяции. Выписывают ближайшие меньшее и большее значения плотности и соответ-
соответствующие им массовые доли раствора данной соли. Считая в указанных пределах
зависимость между плотностью и массовой долей раствора линейной, рассчитыва-
рассчитывают массовую долю со растворенного вещества в приготовленном растворе по про-
пропорции:
Рб-Рм _Рб~Р
где индекс «б» означает большее, «м» — меньшее.
По табл. П.4.4 находят рб= 1100,9 г/л; соб= 0,14; рм= 1085,7 г/л; сОм= 0,12. Тогда
1100,9-1085,7 1100,9-1095,0
0,14-0,12 " 0,14-со
Решая уравнение, получают со = 0,132 (или 13,2%). Используя табл. 3.1 и
учитывая, что молярная масса NaCl M= 58,5 г/моль, а число эквивалентности NaCl
^экв= U рассчитывают:
молярную концентрацию (см. B.13)) С= — = — — = 2,48 моль/л;
молярную концентрацию эквивалента (см. B.14)) Сэкв=гэквС= 1 • 2,48 = 2,48 моль экв/л;
г „ ™^ 1000@ 10000,132
моляльную концентрацию (см. B.12)) Ст= = = 2,60 моль/кг;
МA-со) 58,5 A-0,132)
массовую концентрацию (см. B.15)) Смасс = сор = 0,132 • 1095,0 = 144,54 г/л.
Задача 5. Какой объем 0,1н. раствора серной кислоты H2SO4 необходимо отмерить
для приготовления 100 мл 0,02н. раствора?
Решение. Количество молей эквивалентов H2SO4 в исходном и приготовленном рас-
растворе одинаково, поэтому в соответствии с B.16) объем исходного раствора
т/ Сэкв2У2 0,02-100 ол
V\ = = = 20 мл.
Сэкв, 0,1.
Задача 6. Какой объем 12%-ного раствора серной кислоты H2SO4 потребуется для
приготовления 1 л 0,1 н. раствора этой кислоты?
Решение. Масса H2SO4, согласно B.14), равна:
»4so4 = М^щ%0СжгУ =49 0,11 =4,9 г.
Примеры решения задач 49
Такая масса H2SO4 содержится в 40,83 г 12%-ного раствора в соответст-
соответствии с B.10):
wh2so4 4,9
Шр~ со ~0,12
Плотность исходного раствора (см. табл. П.4.4) р = 1080,0 г/л. Следователь-
Следовательно, для приготовления 1 л 0,1н. раствора H2SO4 потребуется
V = — = —!— = 0,0378 л или 37,8 мл 12%-ного раствора,
р 1080
Задача 7. Какой объем 12%-ного раствора серной кислоты H2SO4 необходимо доба-
добавить к 100 мл 35%-ного раствора H2SO4, чтобы получить ее 20%-ный раствор?
Решение. Для вычисления массовых соотношений между смешиваемыми раствора-
растворами используют графический прием, называемый правилом смешения:
При составлении схемы слева записывают одну под другой процентные кон-
концентрации исходных растворов, а в центре — конечную концентрацию получаемой
смеси. Справа по противоположным концам диагоналей помещают разности между
каждой из начальных концентраций и конечной, причем из большего числа вычитают
меньшее. Отношения полученных разностей соответствуют отношениям масс раство-
растворов, процентная концентрация которых написана на той же горизонтальной строке:
ти% 15
Плотность 35%-ного раствора H2SO4 (см. табл. П.4.4) р = 1260,0 г/л, поэтому
масса 100 мл данного раствора тЪ5% = 1260,0 • 0,1 = 126 г. Следовательно, масса
12%-ного раствора той же кислоты т\2% = 126 • — = 236,25 г.
8
Плотность 12%-ного раствора H2SO4 (см. табл. П.4.4) р = 1080,0 г/л, поэтому
236,25
необходимый объем этого раствора VX2%= =0,219 л = 219 мл.
1080
Задача 8. Рассчитать объемы 12%-ного и 52%-ного растворов уксусной кислоты
СН3СООН, необходимые для приготовления 1 л ее 20%-ного раствора.
Решение. В соответствии с правилом смешения
12 N У2
тп% 32
получают соотношение масс исходных растворов = — = 4.
т52% 8
50 Фундаментальные понятия и законы химии
Масса 1 л 20%-ного раствора СН3СООН (см. табл. П.4.4) тХ2% = 1026,1 г/л.
Массу 12%-ного раствора обозначают тп% = х, тогда масса 52%-ного раствора
#*52 % = х - 1026,1. Решая уравнение х = 4A026,1 - jc), находят х = 820,88.
Используя значения плотностей заданных растворов (см. табл. П.4.4), полу-
получают необходимые объемы исходных растворов:
V\2% = —— = — = 0,808 л (808 мл);
Рп% 1015>4
р52О/о 1059,0
•
Задача 9. Для нейтрализации 1 л 12%-ного раствора кислоты, плотность которого
р= 1080,0 г/л, потребовался 191 мл 39%-ного раствора едкого натра NaOH, плот-
плотность которого р = 1420,0 г/л. Рассчитать молярную массу эквивалента кислоты и
определить кислоту.
Решение. Масса кислоты в 1 л данного раствора: тк = FKpKcoK = 1 • 1080,0 0,12 =
= 129,6 г.
Масса едкого натра, израсходованного на нейтрализацию кислоты, wNa0H =
= ^NaOH pNaOHCONaOH = 0,191 • 1420,0 • 0,39 = 105,8 Г.
Молярная масса эквивалента NaOH Мжв Na0H = —M?L = — = 40 г/моль экв.
Z 1
Согласно B.6), молярная масса эквивалента кислоты
Мэквк= МэквНа0Н тк = -^-129,6 = 49,0 г/моль экв.
1058
Тогда кислота — H2SO4 (z3KB = 2), так как Мн so = Мэквгэкв = 49 • 2 = 98 г/моль.
Задача 10. Рассчитать объем 0,5н. раствора соляной кислоты, необходимый для
растворения гидроксида железа (III) Fe(OHK массой wFe(OH) =15 г.
Решение. В соответствии с законом эквивалентов число молей эквивалентов НС1 и
Fe(OHK: «эквНС1 = "экв Fe(OHK •
Поскольку
WFe(OHK 2экв Fe(OHK WFe(OHK 3-15
пт ?еЮт = — = = = 0,42 моль экв;
эквЬе(инK д, ,, 1/V7
M3KBFe(OHK MFe(OHK 107
"эквНС1 = ^НаСэквНСЬ
объем 0,5н. раствора НС1
_ KBFe(OHK _ 0,42 _
Уна ~ — — - 0,841 л.
Сэкв НС1 U> ^
Примеры решения задач 51
Задача 11. Рассчитать объем 0,1н. раствора едкого натра NaOH, необходимый для ней-
нейтрализации 10 мл 0,1 М раствора серной кислоты H2SO4. Будет ли нейтрализован рас-
раствор H2SO4 при титровании раствором NaOH, если показатель титрования с индикато-
индикатором метиловым оранжевым рТ = 4? Какова относительная систематическая погреш-
погрешность (индикаторная ошибка) титрования данного раствора H2SO4 с этим индикатором?
Решение. Так как Сэкв н so = гэкв н S(>4 Сн so = 2 • 0,1 = 0,2 моль экв/л, объем 0,1н. рас-
раствора NaOH, необходимый для нейтрализации данного объема H2SO4, согласно B.16)
маин „
При нейтрализации водородный показатель конечного раствора рН = 7. Оче-
Очевидно, что при титровании с метиловым оранжевым среда остается кислотной, так
как показатель титрования рТ < 7. В конечной точке титрования рН = - lgC + =
н
10~14
= рТ = 4, следовательно, С + = 10 моль/л, аС. = = 10~10 моль/л в соот-
Н ОН /~*
н+
ветствиисB.18).
Систематическая погрешность титрования, согласно B.19),
С -С +
он- н+
С,
о
100 =
\о-10-ю-4
0,1
100 =0,1%.
Задача 12. Вычислить массу йода 12, выделившегося при взаимодействии йодида
калия KI массой тК1 =16,6 г с 0,5М раствором дихромата калия К2Сг207 объемом
V= 100 мл, подкисленного серной кислотой H2SO4.
Решение. Рассчитывают количество молей эквивалентов окислителя, учитывая, что
число эквивалентности окислителя равно числу электронов, принятых одной его
формульной единицей (Сг2Оу" + 14Н+ + бе = 2Сг3+ + 7Н2О):
"экв K2Cr2O7 = ^С^экв К2Сг207 = °'] " 0,5 ¦ б = 0,3 МОЛЪ ЭКВ
и восстановителя, учитывая, что число эквивалентности восстановителя равно чис-
числу электронов, отданных одной его формульной единицей A~-е = 0,512):
WlKi 16,6
«эквК1= ТГ"^эквК1 =Т7Г1 =0' 1 М0ЛЬЭКВ-
мК1 166
Поскольку окислитель взят в избытке, массу выделившегося 12 рассчитывают
по восстановителю. Согласно B.6),
12
Задача 13. Рассчитать массу перманганата калия КМпО4, необходимую для замены
дихромата калия К2Сг207 массой тКСт0 = 10 г в окислительно-восстановительной
реакции в кислотной среде.
52
Фундаментальные понятия и законы химии
Решение. Для замены одного окислителя другим необходимо взять КМЮ4 в таком
количестве, чтобы числа молей эквивалентов взаимозаменяемых окислителей были
равны: пэкв КМпо4 = пжъ к2сг2о7 •
Процесс восстановления данных окислителей в кислотной среде протекает
следующим образом:
Сг2С>7~ + 14Н+ + бе = 2Сг3+ + 7Н2О
МпО; + 8Н+ + 5е = Мп2+ + 4Н2О
поэтому молярные массы эквивалентов этих окислителей равны соответственно:
158
Z3KBKMnO4
М
— = = 31,6 г/моль экв;
294
К Сг О Zy4
Л/ к с п = 2 2 7 = = 49,0 г/моль экв.
эквК2Сг2О7 , »
2эквК2Сг207 ^
Используя B.6), находят массу КМ11О4:
тК2Сг207
г*КМпО.
М.
М
эквКМп04
экв К,Сг9О7
Задачи для самостоятельного решения
1-10. Рассчитайте молярную массу эквивалента металла и с помощью Периодиче-
Периодической системы элементов, определите металл, при взаимодействии 1 г которого с
разбавленной соляной кислотой выделяется водород объемом V, измеренным при
давлении р и температуре Т.
№
1
2
3
4
5
К я
1,364
2,689
1,020
0,220
0,469
р, мм рт. ст.
744
750
735
740
755
Г,°С
20
18
19
21
22
№
6
7
8
9
10
0,436
0,376
0,520
0,414
0,419
р, мм рт. ст.
742
752
756
477
738
Г,°С
17
23
20
19
18
11-20. Рассчитайте объем водорода (н.у.), который потребуется для восстановления
100 г оксида, массовая доля металла в котором со. Вычислите молярную массу эквива-
эквивалента металла и с помощью Периодической системы элементов, определите металл.
№
11
12
13
14
15
со,%
74,39
82,71
69,41
78,77
92,83
№
16
17
18
19
20
00,%
83,48
89,70
88,83
79,90
93,10
Задачи для самостоятельного решения
53
21-30. Для нейтрализации 100 г 20%-ного по массе раствора одноосновной кислоты
необходим раствор едкого натра массой т, в котором массовая его доля составляет
со. Рассчитайте молярную массу эквивалента кислоты и определите ее формулу.
№
21
22
23
24
25
т, г
160,0
55,0
62,5
65,8
70,5
со,%
25
40
10
15
18
26
27
28
29
30
т9 г
29,6
28,4
20,7
127,0
23,5
со,%
32
28
22
12
35
31-40. Рассчитайте объем 0,1н. раствора серной кислоты, необходимый для раство-
растворения гидроксида металла массой т.
№
31
32
33
34
35
Гидроксид
Mg(OHJ
А1(ОНK
Мп(ОНJ
Zn(OHJ
Сг(ОНJ
т, г
10
15
20
25
30
№
36
37
38
39
40
Гидроксид
Fe(OHJ
Со(ОНK
Sn(OHJ
Pb(OHJ
Cu(OHJ
т, г
35
40
45
50
55
41-60. Рассчитайте молярную концентрацию, молярную концентрацию эквивален-
эквивалента, моляльную концентрацию и массовую концентрацию раствора данного вещест-
вещества, используя значение плотности данного раствора (см. табл. П.4.4).
№
41
42
43
44
45
Раствор
NaCl
р, г/л
1010,0
1050,0
1080,0
1120,0
1170,0
№
46
47
48
49
50
Раствор
H2SO4
р, г/л
1030,0
1050,0
1090,0
1190,0
1250,0
№
51
52
53
54
55
Раствор
НС1
р, г/л
1015,0
1035,0
1055,0
1080,0
1125,0
№
56
57
58
59
60
Раствор
СНзСООН
р, г/л
1006,0
1010,0
1018,0
1025,0
1032,0
61—75. Рассчитайте массу соли и объем воды, необходимые для приготовления 1 л
раствора NaCl заданного состава со (см. табл. П.4.4).
№
61
62
63
64
65
со
0,01
0,02
0,03
0,04
0,06
№
66
67
68
69
70
со
0,08
0,10
0,12
0,14
0,16
№
71
72
73
74
75
со
0,18
0,20
0,22
0,24
0,26
54
Фундаментальные понятия и законы химии
76-95. Используя данные табл. П.4.4, рассчитайте объем раствора данной массовой
доли со, необходимый для приготовления 1 л 0,1 н. раствора кислоты.
H2SO4
№
76
77
78
79
80
со
0,350
0,326
0,302
0,277
0,252
№
81
82
83
84
84
со
0,227
0,201
0,174
0,147
0,091
НС1
№
86
87
88
89
90
со
0,262
0,243
0,223
0,204
0,184
№
91
92
93
94
95
со
0,165
0,145
0,125
0,105
0,085
96-115. Рассчитайте объемы исходных растворов, которые необходимо смешать для
приготовления 1 л раствора заданной концентрации (см. табл. П.4.4).
№
96
97
98
99
100
№
106
107
108
109
ПО
Водные растворы НС1
исходные
СО]
0,262
0,223
0,243
0,145
0,204
со2
0,064
0,044
0,085
0,004
0,024
конечный
со
0,184
0,165
0,204
0,105
0,125
Водные растворы NaCl
исходные
СО!
0,26
0,22
0,24
0,20
0,18
со2
0,10
0,01
0,02
0,04
0,06
конечный
со
0,18
0,12
0,08
0,16
0,14
№
101
102
103
104
105
№
111
112
ИЗ
114
115
Водные растворы СН3СООН
исходные
со.
0,56
0,52
0,48
0,44
0,40
со2
0,08
0,04
0,12
0,16
0,04
конечный
со
0,40
0,28
0,36
0,20
0,32
Водные растворы H2SO4
исходные
СО!
0,350
0,326
0,302
0,277
0,302
со2
0,091
0,120
0,032
0,147
0,062
конечный
со
0,227
0,252
0,174
0,201
0,147
116-125. Составьте уравнение окислительно-восстановительной реакции и рассчи-
рассчитайте массу восстановителя, необходимую для взаимодействия с окислителем, объ-
объем V и концентрация С раствора которого заданы:
№
116
117
118
119
120
121
Восстановитель
KI
KI
KI
FeSO4
FeSO4
FeSO4
Окислитель
KMnO4
KMnO4
КЮ3
KMnO4
K2Mn04
KC1O3
V, мл
100
150
200
250
300
350
С, моль/л
0,80
0,75
0,65
0,50
0,40
0,30
Среда
НС1
Н2О
H2SO4
КОН
КОН
H2SO4
Задачи для самостоятельного решения
55
№
122
123
124
125
Восстановитель
Na2S
Na2S
Na2SO3
Zn
Окислитель
KIO3
K2Cr207
К2Сг207
K2Cr207
К,мл
400
450
500
550
С, моль/л
0,25
0,20
0,15
0,10
Среда
H2SO4
H2SO4
H2SO4
НС1
126-135. Рассчитайте массу дихромата калия К2Сг207, которая потребуется для за-
замены заданного окислителя массой т в окислительно-восстановительной реакции,
протекающей в кислотной среде.
126
127
128
129
130
Окислитель
КМпО4
К2Мп04
МпО2
РЬО2
FeCl3
т, г
10
15
20
25
30
№
131
132
133
134
135
Окислитель
КС1О3
КЮ3
NaCIO
КВЮ
Н2О2
т, г
35
40
45
50
55
136-145. Составьте уравнение окислительно-восстановительной реакции и рассчи-
рассчитайте массы продуктов окисления и восстановления, если восстановитель массой 10 г
взаимодействует с окислителем, объем V, массовая доля со и плотность р раствора
которого заданы.
№
136
137
138
139
140
141
142
143
144
145
Восстановитель
H2S
Си
Fe
KI
S
Н2О2
K2S
FeSO4
HI
Na3SO3
Окислитель
AgNO3
FeCl3
Fe2(SO4K
H2O2
KC1O4
K2Cr207 *
KIO3*
KMnO4*
HIO3
NaBrO3
К,мл
250
150
200
100
2000
300
100
35
10
40
CO
0,20
0,30
0,25
0,28
0,01
0,10
0,06
0,06
0,30
0,24
p, г/л
1194,2
1291,0
1241,0
1104,0
1005,4
1070,3
1051,5
1041,4
1321,8
1219,3
В кислотной среде (H2SO4).
ЛР 1. Окислительно-восстановительные реакции
Цель работы — ознакомление с основными закономерностями процессов
окисления и восстановления, освоение методик составления уравнений окислитель-
окислительно-восстановительных реакций, изучение окислительно-восстановительных свойств
металлов, неметаллов и их соединений.
Опыты в работе сгруппированы в соответствии с классификацией окислительно-
восстановительных реакций (см. гл. 1): межмолекулярные — опыты 1-5; внутримоле-
внутримолекулярные — опыт 6; реакции диспропорционирования — опыт 7; реакции контрпро-
порционирования — опыт 8.
56
Фундаментальные понятия и законы химии
Опыт 1. Сравнение восстановительных свойств металлов. Предлагается
исследовать процессы окисления металлов разбавленными растворами кислот. В
качестве окислителя используется разбавленная соляная (или серная) кислота, в
качестве восстановителей — магний, цинк, алюминий, железо, свинец, медь (по
указанию преподавателя).
Последовательность проведения:
1) в шесть пробирок, стоящих в штативе, налейте по 2-3 мл 1М раствора соляной
НС1 или серной H2SO4 кислот;
2) внесите в первую пробирку кусочек магния, во вторую — цинка, в третью —
алюминия, в четвертую — железа, в пятую — свинца, в шестую — меди;
3) наблюдайте выделение пузырьков газа. Если газ не образуется или образуется
медленно, слегка нагрейте пробирку в пламени горелки.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 1.1;
Таблица Л. 1.1
Результаты эксперимента
Исходные данные
№
про-
пробирки
1
2
3
4
5
6
Металл, реаги-
реагирующий с рас-
раствором кислоты
Магний Mg
Цинк Zn
Алюминий А1
Железо Fe
Свинец РЬ
Медь Си
Наблюдения
Выделение пузырьков газа
без
нагревания
с
нагреванием
Выводы
Ряд восста-
восстановительной
способности
металлов
Электрохи-
Электрохимический ряд
напряжений
металлов
Уравнение
полуреакции
окисления
полуреакции
восстановления
суммарной окислительно-
восстановительной реакции
Mg + HC1 ->
Zn + НС1 ->
Al + HC1 ->
Fe + HC1 ->
Pb + HC1 ->
Cu + HC1 ->
2) определите, какой газ образуется в результате взаимодействия металлов с раство-
растворами кислот. Сравните интенсивность выделения пузырьков газа в пробирках. Сде-
Сделайте выводы об активности металлов как восстановителей;
3) напишите уравнения протекающих окислительно-восстановительных реакций,
используя метод электронного баланса;
4) на основании сделанных выводов расположите металлы в порядке убывания их
восстановительной способности (активности). Сравните составленный ряд активно-
активностей металлов с электрохимическим рядом напряжений металлов (табл. П.6.1).
JIP L Окислительно-восстановительные реакции
57
Опыт 2. Вытеснение металлов из растворов их солей. Известно, что более
активные металлы способны восстанавливать менее активные металлы из растворов
их солей. На основании выводов опыта 1 предположите, какие металлы могут вос-
восстановить медь из растворов ее солей. Проверьте свои предположения.
Последовательность проведения:
1) налейте в пробирки по 2-3 мл 1М раствора хлорида меди (II) СиС12 и внесите в
каждую кусочек одного из выбранных вами металлов;
2) наблюдайте выделение меди по образованию осадка красно-коричневого цвета.
Если медь не выделяется, слегка нагрейте пробирку.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 1.2;
2) сравните интенсивность выделения меди в пробирках и сделайте выводы о вос-
восстановительной активности металлов;
3) напишите уравнения протекающих окислительно-восстановительных реакций,
используя метод электронного баланса;
4) расположите металлы в порядке убывания их активности в реакции с хлоридом
меди (II) и сравните составленный вами ряд с рядом электрохимических напряже-
напряжений металлов (см. табл. П.6.1);
5) на основании табл. Л Л. 1 предположите, какие другие металлы могут быть вытес-
вытеснены из растворов их солей. Напишите уравнения соответствующих окислительно-
восстановительных реакций. Проверьте свои предположения, выбрав соответст-
соответствующие реагенты: металл и раствор соли (п. 5 выполняется по указанию
преподавателя).
Таблица Л. 1.2
Результаты эксперимента
Исходные данные
№
про-
пробирки
1
2
3
4
5
Металл, реаги-
реагирующий с раство-
раствором соли меди
Магний Mg
Цинк Zn
Алюминий А1
Железо Fe
Свинец РЬ
Наблюдения
Выделение осадка меди
без
нагревания
с
нагреванием
Выводы
Ряд восста-
восстановительной
способности
металлов
Электрохи-
Электрохимический
ряд напря-
напряжений
металлов
Уравнение
полуреакции
окисления
полуреакции
восстановления
суммарной окислительно-
восстановительной реакции
Mg + CuCl2 ->
Zn + CuCl2 ->
Al + CuCl2 ->
Fe + CuCl2 ->
Pb + CuCl2 ->
58
Фундаментальные понятия и законы химии
Опыт 3. Сравнение окислительной активности галогенов. В данном опы-
опыте предлагается сравнить окислительную активность галогенов, используя реакции
взаимодействия галогенов с соответствующими галогенидами.
Внимание! Опыт выполняется в вытяжном шкафу
Последовательность проведения:
1) налейте в одну пробирку 2-3 мл раствора йодида калия KJ, а в другую — 2-3 мл бро-
бромида калия КВг;
2) добавьте в пробирки с помощью капельной пипетки несколько капель хлорной
воды (Cl2 aqua). Наблюдайте изменение окраски растворов;
3) добавьте в каждую из пробирок по 2-3 мл бензола;
4) закройте пробирки пробками и энергично встряхните их. Дайте слоям жидкости в
пробирках разделиться. Сравните их окраску. Подтвердите присутствие свободных
йода и брома, добавив в пробирки по 2-3 капли крахмального клейстера или полос-
полоску крахмальной бумаги. Бром образует с крахмалом оранжево-красный продукт, а
йод — продукт темно-синего цвета;
5) повторите пп. 1-4, заменив КВг на КС1, а хлорную воду бромной водой (Вг2 аква);
6) повторите пп. 1-4, заменив KI на КС1, а хлорную воду раствором йода A2 аква).
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 1.3;
Таблица Л 1.3
Результаты эксперимента
Исходные данные
Реагирующие вещества
Галогенид
Формула
KI
КВг
KI
КС1
КВг
КС1
Окраска
водного
раствора
Галоген
Формула
С12
С12
Вг2
Вг2
12
h
Окраска
водного
раствора
Наблюдения
Окраска
водного
раствора
после взаи-
взаимодействия
реагентов
Уравнение
полуреакции
окисления
полуреакции
восстановления
суммарной окислительно-
восстановительной реакции
KI + С12 ->
КВг + С12->
К1 + Вг2->
КС1 + Вг2 ->
КВг + 12->
КС1 +12 -»
Окраска
бензольного
раствора
после рас-
расслоения
жидкостей
Окраска
крах-
крахмальной
пробы
Вывод
Ряд окислительной
активности галогенов
ЛР 1. Окислительно-восстановительные реакции
59
2) объясните наблюдаемые изменения окраски водных растворов и растворов в бен-
бензоле после взаимодействия с соответствующими солями. По результатам крахмаль-
крахмальной пробы отметьте присутствие в растворе йода и брома;
3) сделайте выводы об окислительной активности галогенов;
4) напишите уравнения протекающих окислительно-восстановительных реакций,
используя метод электронного баланса;
5) на основании выводов расположите галогены в порядке убывания их окисли-
окислительной активности;
6) укажите, какое место в ряду окислительной активности галогенов занимает фтор
и почему в данном эксперименте он не исследуется.
Опыт 4. Восстановление перманганата калия КМпО4 в различных средах.
Состав продуктов восстановления перманганат-иона МпО4 зависит от реакции сре-
среды (кислотная, нейтральная, щелочная), в которой протекают окислительно-
восстановительные процессы. В данном опыте предлагается исследовать влияние
среды на реакцию взаимодействия сульфита натрия Na2SC>3 и перманганата калия
КМпО4. Следует учитывать, что водные растворы, содержащие ионы МпО4, окра-
окрашены в фиолетовый цвет; содержащие ионы МпО^" — в зеленый; содержащие ио-
ионы Мп2+ — практически бесцветны, точнее, имеют слабо-розовую окраску, которую
растворам придают аквакомплексы [Мп(Н2ОN]2+; а осадок МпО(ОНJ — бурого
цвета.
Последовательность проведения:
1) налейте в три пробирки по 2-3 мл 0,1 М раствора перманганата калия КМпО4;
2) в первую пробирку прилейте 1-2 мл 1М раствора серной кислоты H2SO4, во вто-
вторую — 1-2 мл дистиллированной воды, в третью — 1-2 мл 2М раствора едкого ка-
кали КОН. Отметьте окраску раствора в пробирках;
Таблица Л. 1.4
Результаты эксперимента
Исходные данные
Окислитель
Формула
КМпО4
Окраска
водного
раствора
Восстановитель
Формула
Na2SO3
Окраска
водного
раствора
Среда
H2SO4
Н2О
КОН
Наблюдения
Окраска
водного
раствора
после взаи-
взаимодействия
Выводы
Продукт
восстанов-
восстановления
окисли-
окислителя
Глубина
восстанов-
восстановления
окисли-
окислителя
Уравнение
полуреакции
окисления
полуреакции
восстановления
суммарной окислительно-
восстановительной реакции
Na2SO3 + KMnO4 + H2SO4 ->
Na2SO3 + KMnO4 + Н2О ->
Na2SO3 + KMnO4 + КОН ->
60 Фундаментальные понятия и законы химии
3) в каждую пробирку прилейте по 2-3 мл 0,2М раствора сульфита натрия
Na2SO3. Отметьте происходящие изменения (изменения окраски, образование
осадка и др.).
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 1.4;
2) объясните наблюдаемые изменения окраски растворов, сопоставляя с известными
окрасками осадка и растворов соответствующих ионов;
3) напишите уравнения протекающих окислительно-восстановительных реакций,
используя метод электронно-ионных полуреакций;
4) сделайте вывод, при какой реакции среды глубина восстановления перманганат-
иона больше.
Опыт 5. Окислительно-восстановительные свойства пероксида водорода
и нитрит-иона. Пероксид водорода Н2О2 и нитрит-ион NO2 в окислительно-восста-
окислительно-восстановительных реакциях проявляют двойственный характер: при взаимодействии с
сильными окислителями выступают в роли восстановителя, а при взаимодействии с
сильными восстановителями — в роли окислителя. Предлагается исследовать двой-
двойственность поведения Н2О2 и NO2 в реакциях взаимодействия с КМпО4 и KI в ки-
кислотной среде.
Последовательность проведения:
1) приготовьте две пробирки. В первую налейте 2-3 мл 0,1 М раствора йодида калия
KI и 1-2 мл 1М раствора серной кислоты H2SO4, во вторую — 2-3 мл 0,1М рас-
раствора перманганата калия КМпО4 и 1-2 мл 1М раствора H2SO4. Отметьте окра-
окраску растворов;
2) в каждую пробирку прилейте по 2-3 мл концентрированного раствора Н2О2. От-
Отметьте наблюдаемые изменения окраски растворов. Обратите внимание на выделе-
выделение пузырьков газа во второй пробирке;
3) докажите образование в первой пробирке свободного йода 12, добавив в про-
пробирку несколько капель крахмального клейстера или воспользовавшись полоской
крахмальной бумаги. Йод образует с крахмалом продукт темно-синего цвета;
4) повторите пп. 1-3, заменив раствор Н2О2 0,1М раствором нитрита натрия
NaNO2.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 1.5;
2) объясните происходящие изменения окраски растворов и результат крахмаль-
крахмальной пробы. Укажите, какие газы выделяются при взаимодействии Н2О2 и NaNO2
с КМпО4;
3) напишите уравнения протекающих окислительно-восстановительных реакций,
используя метод электронно-ионных полуреакций;
4) сделайте вывод, в каких реакциях Н2О2 и NaNO2 являются окислителями, а в ка-
каких — восстановителями.
ЛР 1. Окислительно-восстановительные реакции
61
Результаты эксперимента
Таблица Л. 1.5
Исходные данные
Реагирующие вещества
Форму-
Формула
Н2О2
NaNO2
Окраска
водного
раствора
Формула
КМпО4
KI
КМпО4
KI
Окраска
водного
раствора
Среда
H2SO4
H2SO4
H2SO4
H2SO4
Наблюдения
Окраска вод-
водного раство-
раствора после вза-
взаимодействия
Выводы
Продукты
реакции
Роль Н2О2
(NaNO2)
в реакции
Уравнение
полуреакции
окисления
полуреакции
восстановления
суммарной окислительно-восстановительной
реакции
Н2О2 + КМпО4 + H2SO4 ->
H2O2+KI + H2SO4->
NaNO2 + KMnO4 + H2SO4 ->
NaNO2+KI + H2SO4->
Опыт 6. Разложение дихромата аммония. Дихромат аммония (NH4JCr2O7
является неустойчивым соединением и при нагревании разлагается: (NH4JCr207 —>
—> Сг2Оз + N2 + Н2О. Процесс разложения сопровождается изменением степеней
окисления элементов, составляющих соединение. Предлагается исследовать данную
внутримолекулярную окислительно-восстановительную реакцию.
Внимание! Опыт выполняется в вытяжном шкафу
Последовательность проведения:
1) в пробирку положите 2-3 г соли (NH4JCr207;
2) осторожно нагрейте пробирку в небольшом пламени горелки. Наблюдая процесс
термического разложения соли, обратите внимание на изменение окраски реактива
и выделение газов;
3) поднесите к открытому концу пробирки, не закрывая ее, часовое стекло. Обрати-
Обратите внимание на появление на нем капель конденсированной воды;
4) поднесите к открытому концу пробирки тлеющую лучину. Обратите внимание на
прекращение горения лучины.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 1.6;
2) объясните наблюдаемые явления, учитывая образование продуктов разложения соли;
3) составьте уравнение реакции разложения дихромата аммония, используя метод
электронного баланса;
4) сделайте вывод, какие атомы в данном соединении выступают в роли окислителя
или восстановителя при протекании внутримолекулярной окислительно-восстано-
окислительно-восстановительной реакции.
62
Фундаментальные понятия и законы химии
Таблица Л. 1.6
Результаты эксперимента
Исходные данные
Реагент
Формула
(NH4JCr207
Цвет кристаллов
Условия проведения
эксперимента
Наблюдения
Изменение окра-
окраски кристаллов
Вывод
Уравнение
полуреакции
окисления
полуреакции
восстановления
суммарной окислительно-
восстановительной реакции
(NH4JCr207->
Опыт 7. Самоокисление—самовосстановление галогенов. Степень окис-
окисления галогенов в простых веществах является промежуточной, что обусловливает
их неустойчивость и склонность к реакциям диспропорционирования (дисмутации).
Например, реакция дисмутации хлора в воде протекает по уравнению:
С12 + Н2О = НС1 + НС1О
Как и окислительная активность, склонность галогенов к самоокислению — са-
самовосстановлению зависит от положения элемента в Периодической системе
элементов.
Предлагается исследовать реакции диспропорционирования галогенов. На-
Наличие продуктов дисмутации галогена можно установить по изменению окраски
универсальной индикаторной бумаги, так как продукты данной реакции — кислоты.
Универсальный индикатор относится к группе кислотно-основных индикаторов,
изменяющих окраску в зависимости от характера среды (см. § 2.2). Чем больше в
водном растворе галогена продуктов дисмутации, тем больше окраска универсаль-
универсального индикатора смещена к красной области спектра.
По количеству капель щелочи, необходимых для нейтрализации растворов,
также можно судить о степени дисмутации галогенов (табл. Л. 1.7а).
Таблица Л. 1.7а
Цвет универсального индикатора (раствора или бумаги)
Среда
кислотная
Направление уси-
усиления кислотности
среды
t
1
Цвет
индикатора
Красный
Оранжевый
Желтый
нейтральная
Цвет
индикатора
Зеленый
щелочная
Направление
усиления
щелочности среды
1
1
Цвет
индикатора
Бирюзовый
Голубой
Фиолетовый
JIP L Окислительно-восстановительные реакции
63
Внимание! Опыт выполняется в вытяжном шкафу
Последовательность проведения:
1) в три пробирки налейте по 2 мл водных растворов галогенов: в первую — С12, во
вторую — Вг2, в третью —12;
2) определите реакцию среды каждого раствора, используя указатель окраски универ-
универсального индикатора. Для этого пипеткой нанесите каплю раствора на полоску индика-
индикаторной бумаги;
3) по каплям с помощью пипетки в каждую пробирку приливайте 1М раствор едко-
едкого кали КОН (или едкого натра NaOH). Необходимое количество капель раствора
КОН контролируйте по цвету универсальной индикаторной бумаги для нейтраль-
нейтральной среды, а также по исчезновению характерного запаха или окраски водного рас-
раствора галогена.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 1.76;
Таблица Л. 1.7б
Результаты эксперимента
Исходные
данные
Водный
раствор
галогена
С12
Вг2
h
Наблюдения
Цвет
универсального
индикатора в
водном растворе
галогена
Характер
среды
в водном
растворе
галогена
Количество
капель 1М рас-
раствора КОН,
необходимое для
нейтрализации
раствора
Вывод
Ряд галогенов
по увеличению
интенсивности
реакции
дисмутации
Уравнения
полуреакций
окисления
полуреакций
восстановления
суммарной окислительно-
восстановительной реакции
С12 + Н2О->
Вг2+Н2О->
12+Н2О->
реакции
нейтрализации
2) укажите реакцию среды в растворах галогенов и количество капель щелочи, не-
необходимое для нейтрализации раствора;
3) сделайте вывод, какой из галогенов в большей степени подвергается реакции дис-
пропорционирования. Расположите галогены в порядке увеличения степени дисму-
дисмутации;
4) сравните ряд интенсивности реакций самоокисления—самовосстановления галоге-
галогенов с рядом их окислительной активности, составленным по результатам опыта 3;
64
Фундаментальные понятия и законы химии
5) составьте уравнения реакций диспропорционирования галогенов в воде, исполь-
используя метод электронно-ионных полуреакций;
6) составьте уравнения реакций нейтрализации водных растворов галогенов;
7) поясните, почему в данном опыте не исследуется водный раствор фтора F2. Подвер-
Подвергается ли этот галоген дисмутации?
Опыт 8. Взаимодействие соединений йода. При взаимодействии соедине-
соединений, содержащих один и тот же элемент с различными степенями окисления, воз-
возможно протекание реакций, которые сопровождаются образованием продукта, со-
содержащего данный элемент в промежуточной степени окисления. Предлагается ис-
исследовать реакции контрпропорционирования на примере взаимодействия йодида
калия KI и йодата калия
Внимание! Опыт выполняется в вытяжном шкафу
Последовательность проведения:
1) в пробирку налейте 1-2 мл 0,1М раствора KI. Добавьте 2-3 капли 1М раствора
H2SO4. Обратите внимание на окраску исходного раствора;
2) прилейте в пробирку 1-2 мл 0,1 М раствора КЮ3. Отметьте происходящие изме-
изменения окраски раствора;
3) прилейте в ту же пробирку 1-2 мл бензола. Закройте пробирку пробкой и энер-
энергично встряхните. Дайте слоям жидкости в пробирке разделиться. Отметьте окраску
бензольного слоя, сравните ее с аналогичной окраской бензольного раствора йода,
установленной вами в опыте 3 (см. табл. Л. 1.3);
4) подтвердите образование свободного йода 12 с помощью крахмальной пробы, как
указано в опыте 3.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 1.8;
Таблица Л. 1.8
Результаты эксперимента
Исходные данные
Реагирующие вещества
Йодид
Формула
KI
Окраска
водного
раствора
Йодат
Формула
КЮ3
Окраска
водного
раствора
Наблюдения
Окраска
водного
раствора
после взаи-
взаимодействия
реагентов
Окраска
бензольного
раствора
после рас-
расслоения
жидкостей
Уравнение
полуреакции
окисления
полуреакции
восстановления
суммарной окислительно-
восстановительной реакции
KI + КЮ3 +
H2SO4->
Окраска
крах-
крахмальной
пробы
Вывод
Характер реагентов
Окислитель
Восстановитель
ЛР 2. Химический эквивалент
65
2) объясните наблюдаемые изменения окраски раствора и результат крахмальной
пробы;
3) составьте уравнение реакции контрпропорционирования соединений йода, ис-
используя метод электронно-ионных полуреакций;
4) сделайте вывод, какое соединение йода выступает в качестве окислителя или вос-
восстановителя.
ЛР 2. Химический эквивалент
Цель работы — ознакомление с понятием химического эквивалента вещества
с помощью простых методов определения молярной массы химического эквивален-
эквивалента и расчетов на основе закона эквивалентов.
Опыт 1. Определение молярной массы химического эквивалента металла.
Предлагается определить молярную массу химического эквивалента металла кос-
косвенным методом, основанным на измерении объема водорода, выделяющегося в
результате взаимодействия избытка соляной кислоты с металлом известной массы:
М + иНС1 = МС1„+ -Н2
Используемый для этой цели прибор (рис. Л.2.1) представляет собой две ук-
укрепленные на штативе и соединенные резиновой трубкой бюретки емкостью 50 мл,
одна G) из которых плотно соединена с реакционным сосудом, а другая B) откры-
открыта. В эвдиометр наливают воду приблизи-
приблизительно до половины высоты бюреток. Сис-
Система сообщающихся сосудов позволяет
выравниванием уровней воды в бюретках
создать в приборе атмосферное давление.
Объем выделившегося Н2 определяют по
разности уровней воды в одной из бюреток
до и после проведения опыта при давле-
давлении в закрытой бюретке, равном атмо-
атмосферному. В качестве реакционного сосу-
сосуда используют пробирку с боковой труб-
трубкой C).
Молярная масса химического экви-
эквивалента металла Мэквм согласно B.8)
М
кэквН2
Металл
НС1
где тм - масса металла; Ущ - измерен- рис л 2 д Эвдаометр:
ный объем Н2; Уэкв н — молярный объем 1,2 — бюретки; 3 — реакционный сосуд
3 - 9795
66 Фундаментальные понятия и законы химии
химического эквивалента водорода при условиях опыта.
В соответствии с B.3)
V
г э
экв Н2 Т 0 экв Н2
Р
о Рщ
где /?„ — парциальное давление Н2 в закрытой бюретке; Т — температура, при
которой проводится эксперимент; Vo экв н — молярный объем химического эквивален-
эквивалента водорода при нормальных условиях (Го= 273,15 К;ро= 101,3 кПа = 760 мм рт. ст.).
22 414
Поскольку число эквивалентности водорода z3KB = 2, то в силу B.2) Vo экв н = — =
= 11,207 л/моль.
В результате выравнивания уровней воды в эвдиометре общее давление в за-
закрытой бюретке равно атмосферному, поэтому парциальное давление водорода
рИ равно разности атмосферного давления /?атм и давления насыщенного водяного
пара рио при температуре опыта (даже при комнатной температуре давление пара
над поверхностью воды значительное и им нельзя пренебрегать):
Рщ = Ратм - РщО
Следовательно, расчетная формула молярной массы химического эквивален-
эквивалента металла имеет вид
mM tPqV0 3kbu2
Последовательность проведения:
1)в реакционный сосуд налейте до одной четверти его объема 20...30%-ный
раствор соляной кислоты, а в боковую трубку, досуха вытертую фильтровальной
бумагой, поместите полученную у лаборанта навеску металла. Запишите массу
металла;
2) отверстия реакционного сосуда тщательно закройте пробками и присоедините
сосуд к прибору;
3) проверьте герметичность эвдиометра. Для этого поднимите или опустите откры-
открытую бюретку на 10... 15 см. Если при этом уровень воды в закрытой бюретке прак-
практически не изменяется, прибор герметичен. Если уровни в бюретках выравнивают-
выравниваются, следует плотнее вставить пробки в бюретке и пробирке;
4) убедившись в герметичности эвдиометра, давление в приборе приведите к атмо-
атмосферному, установив уровни воды в бюретках на одной высоте. Запишите положе-
положение нижнего края мениска в закрытой бюретке до начала реакции hH&4;
5) поверните реакционный сосуд так, чтобы металл оказался в растворе кислоты;
6) водород, выделяющийся в результате реакции, вытесняет воду из закрытой бю-
бюретки. Чтобы давление внутри прибора было близко к атмосферному, при прохож-
ЛР 2. Химический эквивалент
67
дении реакции перемещайте открытую бюретку вниз, поддерживая уровни воды в
бюретках на одной высоте;
7) после окончания реакции и охлаждения реакционного сосуда до комнатной тем-
температуры вновь тщательно установите одинаковые уровни воды в бюретках, пере-
перемещая бюретку 2. Запишите положение нижнего края мениска в бюретке 1 после
окончания реакции Акон;
8) откройте пробку реакционного сосуда и установите бюретки в исходное положение;
9) для получения более точных результатов опыт повторите еще раз.
Обработка результатов:
1) исходные данные, условия проведения опыта, результаты измерений и расчетов
занесите в табл. Л.2.1;
Таблица Л. 2.1
Результаты эксперимента
№ опыта
1
2
№ опыта
1
2
Условия проведения опыта
Г, К
Ратм,
мм рт. ст.
мм рт. ст.
Результаты измерений
тМуг
/*нач, МЛ
hK0H, мл
Результаты расчетов
мм рт. ст.
\-
МЛ
¦***экв М эксп>
г/моль
-***экв М эксп»
г/моль
-Л^экв М теор»
г/моль
л
2) рассчитайте объем выделившегося водорода по разности конечного и начального
уровней воды в закрытой бюретке VH = hK0H- hm4;
3) определите парциальное давление ри 0 водяного пара при температуре опыта
(табл.П.4.1);
4) рассчитайте молярную массу химического эквивалента металла МэквМэксп;
5) вычислите среднее значение молярной массы химического эквивалента МэквМэксп»
если результаты имеют расхождение в пределах 1 г/моль. Если разность значений
молярной массы химического эквивалента имеет большее значение, следует опыт
повторить вновь;
6) используя Периодическую систему элементов, рассчитайте теоретические значе-
значения молярной массы химического эквивалента МЭКвМтеор магния, алюминия и цинка
по формуле B.1);
7) сравнивая результат эксперимента с теоретическими значениями молярной массы
химического эквивалента металлов, определите, какой из металлов (Mg, A1 или Zn)
был взят для исследования;
8) проверьте точность результатов опыта, вычислив относительную ошибку экспе-
эксперимента по формуле
68 Фундаментальные понятия и законы химии
1 экв М теор 1V1 экв М эксп
экв М теор
100%;
9) укажите, какие операции эксперимента вносят наибольшие погрешности в значе-
значение молярной массы химического эквивалента металла;
10) напишите уравнение изученной реакции.
Опыт 2. Определение молярного объема химического эквивалента и числа
эквивалентности углекислого газа. Предлагается определить молярный объем
химического эквивалента углекислого газа СО2, а также рассчитать число эквива-
эквивалентности.
Масса одного из реагирующих веществ (Na2CO3) известна. Определив объем
выделившегося в результате реакции
Na2CO3 + H2SO4 = Na2SO4 + Н2О + CO2t
газа, можно рассчитать молярный объем химического эквивалента СО2 в условиях
опыта
_экв№2СО3
КэквСО2 ~
СО2
mNa2CO3
где тш со — масса прореагировавшего карбоната натрия; Мэкв Na со — молярная
масса химического эквивалента Na2CO3; Vco — объем образовавшегося в резуль-
результате реакции СО2.
Молярный объем химического эквивалента СО2 следует привести к нормаль-
нормальным условиям в соответствии с B.3)
_7^Рсо2_
у 0 экв СО2 ~ j, экв СО2 >
где ^о экв со — молярный объем химического эквивалента СО2 при нормальных
условиях; Т — температура проведения опыта; рсо — парциальное давление СО2.
Зная молярный объем идеального газа VqM= 22,414 л/моль (н.у.), можно рассчитать
число эквивалентности СО2 по B.2):
М)А/
к0эквСО2
Для определения объема СО2 используют тот же прибор, что и в предыдущем
опыте (см. рис. Л.2.1). В качестве жидкости, наливаемой в сообщающиеся сосуды, в
данном опыте применяют насыщенный водный раствор NaHCO3. Реакционным сосу-
сосудом в этом опыте является двухколенная пробирка Оствальда (рис. Л.2.2), в одно коле-
колено которой наливают заданный объем (от 5 до 10 мл) 0,2М раствора соды Na2CO3, а в
ЛР 2. Химический эквивалент 69
другой — 5 мл 10... 15%-ного раствора H2SO4. Для прове-
проведения химической реакции раствор кислоты переливают в
раствор соды поворотом пробирки. Na2CO3 / \ H2SO4
Объем выделившегося углекислого газа Ксо оп-
определяют по разности уровней жидкости в одной из
бюреток до и после проведения опыта при давлении в
закрытой бюретке, равном атмосферному. Рис* Л#2*2# пРобиРка Ост"
„ вальда
Парциальное давление углекислого газа рсо
равно разности между атмосферным давлением paTM и давлением насыщенного во-
водяного пара рн 0 над насыщенным водным раствором NaHCO3 в закрытой бюретке:
РСО2 = Ратм ~ Рн2О
Давление насыщенного водяного пара при температуре опыта можно определить по
табл. П.4.1.
Учитывая, что серную кислоту берут в избытке, массу прореагировавшего
карбоната натрия определяют по формуле:
WNa2CO3 = CFMNa2CO3
где С — молярная концентрация раствора соды, моль/л (в данном опыте С = 0,2 моль/л);
V— объем раствора соды, л; MNa со — молярная масса Na2CO3, г/моль.
Последовательность проведения:
1)отмерьте с помощью мерного цилиндра 5 мл 10... 15%-ного раствора H2SO4 и
перелейте в одно из колен пробирки Оствальда;
2) отмерьте указанный преподавателем E, 7 или 10 мл) объем 0,2М раствора соды
Na2CO3 и осторожно перелейте в другое колено пробирки Оствальда;
3) присоедините пробирку Оствальда к эвдиометру;
4) проверьте герметичность прибора. Для этого поднимите или опустите бюретку 2
на 10... 15 см. Если при этом уровень жидкости в бюретке 1 практически не изменя-
изменяется, прибор герметичен. Если уровни в бюретках выравниваются, следует плотнее
вставить пробки в бюретке и пробирке;
5) убедившись в герметичности эвдиометра, давление в приборе приведите к атмо-
атмосферному, установив уровни жидкости в бюретках на одной высоте. Запишите по-
положение нижнего края мениска в бюретке 1 до начала реакции /гнач;
6) поворотом пробирки осторожно перелейте раствор серной кислоты в раствор со-
соды (Почему нельзя раствор соды переливать в раствор серной кислоты?);
7) по мере выделения СО2 поддерживайте уровни жидкости в открытой и закрытой
бюретках в одной горизонтальной плоскости;
8) после окончания реакции и охлаждения реакционного сосуда до комнатной тем-
температуры вновь тщательно установите одинаковые уровни жидкости в бюретках,
перемещая бюретку 2. Запишите положение нижнего края мениска hK0H в бюретке 1
после окончания реакции;
9) отсоедините пробирку, установите бюретки в исходное положение;
10) для получения более точных результатов опыт повторите еще раз.
70
Фундаментальные понятия и законы химии
Обработка результатов:
1) исходные данные, условия проведения опыта, результаты измерений и расчетов
занесите в табл. Л.2.2;
Таблица Л. 2.2
Результаты эксперимента
№
опыта
1
2
№
опыта
1
2
Условия проведения опыта
CNa2CO3 '
моль/л
FNa2CO3> Л
Г, К
/7атм, ММрТ. СТ.
рно,ммрт. ст.
Результаты
измерений
/*нач, МЛ
hKOH, мл
Результаты расчетов
"Чсо3> г
рсо , мм рт. ст.
^эквСО2> Л/М^Ь
Результаты расчетов
РэквСО2, Л/МОЛЬ
К0эквСО2> Л/МОЛЬ
^экв эксп
^экв расч
Л
2) рассчитайте объем выделившегося СО2 по разности конечного и начального
уровня воды в бюретке 7: Vco = hK0H - hHa4;
3) рассчитайте массу соды тш со , участвовавшей в реакции, и молярную массу
химического эквивалента соды Мэкв Na со (см. B.1) и табл. 2.1);
4) рассчитайте молярный объем химического эквивалента углекислого газа Ужв со ;
5) сравните результаты расчета для двух опытов и, если они отличаются не более
чем на 1 л/моль, определите среднее значение Ужъ со2;
6) по табл. П.4.1 определите парциальное давление ря 0 водяного пара при темпе-
температуре опыта и рассчитайте парциальное давление рсо углекислого газа;
7) используя среднее значение Кэкв со2, рассчитайте молярный объем химического
эквивалента углекислого газа Vo экв со при нормальных условиях;
8) рассчитайте экспериментальное значение числа эквивалентности СО22ЭКВЭксп;
9) по составу формульной единицы вещества (см. табл. 2.1) или в соответствии с фор-
формулой B.9) вычислите число эквивалентности СО2 гЭКврасч и сравните с 2ЭКВЭКСП;
10) проверьте точность результатов опыта, вычислив относительную ошибку экспе-
эксперимента по формуле: г| =
экв эксп экв расч
ы экв расч
100%;
ЛР 3. Приготовление раствора и определение его концентрации
71
11) укажите, какие операции эксперимента и допущения вносят наибольшие по-
погрешности при определении молярного объема химического эквивалента и числа
эквивалентности углекислого газа.
ЛР 3. Приготовление раствора и определение его концентрации
Цель работы — ознакомление с понятием «растворы» и способами
выражения содержания растворенного вещества, приобретение навыков
приготовления растворов различных концентраций, определение концентраций
растворов денсиметрическим и титриметрическим методами.
Опыт 1. Приготовление водного раствора соли заданного состава и оп-
определение его концентрации методом денсиметрии. В данном опыте отрабаты-
отрабатываются навыки приготовления раствора из чистых компонентов и определения его
концентрации с помощью ареометра. Качественный и количественный состав
раствора соли задается преподавателем.
Последовательность проведения:
1) рассчитайте массу соли и объем воды, необходимые для приготовления раствора
заданного состава;
2) взвесьте на лабораторных весах чистый сухой стакан, запишите массу стакана. Рас-
Рассчитайте массу стакана вместе с необходимой массой соли; шпателем насыпьте в
стакан соль. Контролируйте массу соли взвешиванием на лабораторных весах;
3) мерным цилиндром отмерьте необходимый объем дистиллированной воды; пере-
перелейте воду в стакан. Растворите соль, помешивая ее стеклянной палочкой;
4) ознакомьтесь с устройством ареометра (рис. Л.3.1). Каждый ареометр пред-
предназначен для измерения плотностей в определенных пределах. Для выбора ареомет-
ареометра установите приближенно плотность раствора заданной массовой доли соли по
табл. П.4.4;
5) перелейте раствор соли в мерный цилиндр; осторожно опустите в раствор вы-
выбранный ареометр так, чтобы он не касался стенок цилиндра. Отметьте по шкале
ареометра его показания;
6) раствор вылейте обратно в стакан. Ареометр вымойте.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов занесите в табл. Л.3.1;
Таблица Л.3.1
Результаты эксперимента
Исходные
данные
со3
Результаты
измерений
р, г/см3
Результаты расчетов
со
с,
моль/л
с
моль экв/л
ст,
моль/кг Н2О
с
г/л
72
Фундаментальные понятия и законы химии
2) с помощью табл. П.4.4. определите массовую долю рас-
растворенного вещества со в приготовленном растворе по зна-
значению измеренной плотности р раствора;
3) если показания ареометра не совпадают с табличными
данными, то массовую долю определите методом интерпо-
интерполяции (см. задачу 4);
4) определите погрешность г| приготовления раствора соли,
сравнив заданное значение массовой доли со3 соли в раство-
растворе и значение массовой доли со соли в приготовленном рас-
творе: г| =
со,
100%;
Рис. Л.3.1. Ареометр
5) рассчитайте массовую, моляльную, молярную концен-
концентрации и молярную концентрацию эквивалента приготов-
приготовленного раствора соли, используя табл. 2.3.
Опыт 2. Приготовление раствора кислоты из более концентрированного
раствора и определение его концентрации методом титрования. В данном опыте
приготовление раствора проводят из более концентрированного раствора той же
кислоты (H2SO4, HC1 или СН3СООН). Молярную концентрацию эквивалента (или
плотность) исходного раствора кислоты, а также концентрацию и объем E0 или
100 мл) конечного раствора указывает преподаватель.
Концентрация определяется методом титрования стандартным (титрованным)
раствором щелочи (NaOH или КОН). Титриметрической реакцией является реакция
нейтрализации раствора кислоты раствором щелочи. Конечную точку титрования уста-
устанавливают по изменению окраски какого-либо кислотно-основного индикатора (фе-
(фенолфталеина, метилового оранжевого, лакмуса или универсального индикатора).
Последовательность проведения:
1) рассчитайте объем исходного раствора кислоты известной концентрации, необ-
необходимый для приготовления данного объема раствора кислоты заданной концен-
концентрации (см. задачу 5). Если указана только плотность исходного раствора кислоты,
определите массовую долю кислоты в растворе (см. опыт 1, пп. 1, 2). Для перерасчета
массовой доли в молярную концентрацию эквивалента используйте соотношения,
представленные в табл. 2.3;
2) приготовьте мерную посуду: колбы, цилиндры, пипетки, бюретки; налейте в мер-
мерную колбу (емкостью 50 или 100 мл) примерно половину необходимого количества
дистиллированной воды;
3) отмерьте необходимый объем исходного раствора кислоты с помощью мерного
цилиндра или мерной пипетки;
4) налейте в мерную колбу с водой отмеренный объем кислоты. После охлаждения
раствора долейте в колбу дистиллированную воду до метки;
5) закройте колбу пробкой и перемешайте раствор, переворачивая колбу вверх
дном;
6) в две конические колбы емкостью 100 мл с помощью мерной пипетки перенесите
по 10 мл приготовленного раствора;
ЛР 3. Приготовление раствора и определение его концентрации
73
7) приливая дистиллированную воду, доведите объем раствора в каждой колбе до
100 мл;
8) добавьте по 2-3 капли индикатора фенолфталеина. Отметьте окраску растворов;
9) мерную бюретку, закрепленную в штативе, заполните стандартным (титрован-
(титрованным) раствором щелочи так, чтобы в носике бюретки не было пузырьков воздуха.
Запишите молярную концентрацию эквивалента раствора шелочи СЭКВщ;
10) определите уровень титранта в бюретке с точностью до 0,1 мл. При этом глаз
наблюдателя и нижний край мениска жидкости должны находиться на одной гори-
горизонтальной плоскости (рис. Л.3.2). Запишите начальный уровень /гнач титранта;
11) проведите титрование раствора кислоты, добавляя небольшими порциями рас-
раствор щелочи в одну из конических колб (см. рис. Л.3.2). Раствор кислоты при этом
непрерывно перемешивайте, вращая колбу;
12) конец титрования определите по появлению едва заметной, не исчезающей в
течение 30 с, малиновой окраски фенолфталеина (Окраску раствора удобнее наблю-
наблюдать на белом фоне, поэтому подложите под колбу лист белой бумаги.). Отметьте и
запишите конечный уровень hK0H титранта в бюретке;
13) добавьте еще одну каплю раствора щелочи. Если окраска раствора стала более
заметной, значит, конечная точка титрования определена правильно;
14) проведите титрование кислоты, находящейся во второй колбе, выполняя пп. 10-13;
15) повторите пп. 6-11, заменив фенолфталеин другим индикатором, например ме-
метиловым оранжевым. Конец титрования определите по изменению окраски индика-
индикатора (см. табл. 2.4).
Обработка результатов:
1) исходные данные, результаты измерений и расчетов занесите в табл. Л.3.2;
Таблица Л.3.2
Результаты эксперимента
Исходные данные
Проба
Титрант
Уравнение титримет-
рической реакции
Заданные значения
VK, мл
^ экв к»
моль экв/л
с
моль экв/л
Результаты измерений и расчетов
Индикатор —фенолфталеин
№
опыта
1
2
/гнач, мл
/*кон, МЛ
Кщ, мл
с
^экв to
моль экв/л
л
Индикатор — метиловый оранжевый
№
опыта
1
2
/гнач, мл
hK0H, мл
Кщ, мл
V* мл
^экв ю
моль экв/л
Л
74
Фундаментальные понятия и законы химии
Бюретка с\
Мениск
Раствор
R>=i| щелочи
2) по разности конечного /*кон и начального hm4
уровней жидкости в бюретке рассчитайте объ-
объем Кщ титранта, израсходованного на титрова-
титрование. Сравните два значения этой величины,
полученные при титровании с использованием
в качестве индикатора фенолфталеина, и при
расхождении более чем на 0,2 мл, повторите
титрование;
3) вычислите среднее значение Ущ и рассчи-
рассчитайте молярную концентрацию эквивалента
приготовленного раствора кислоты Сэквк по
формуле:
Гк=10мл;
Раствор
кислоты
Рис. Л.3.2. Титрование раствора
кислоты стандартным раствором
щелочи к
4) сравните заданное значение концентрации раствора С*кв к кислоты и определен-
определенное методом титрования значение концентраций Сэкв к приготовленного раствора:
С -С
экв к ЭКВ К
100%;
5) рассчитайте концентрации раствора кислоты при титровании с другим индикато-
индикатором, например метиловым оранжевым, повторив пп. 1-4;
6) объясните, чем обусловлено расхождение значений концентрации одного и того
же раствора, оттитрованного в присутствии разных индикаторов? Укажите, при ка-
каких значениях рН раствора происходит изменение окраски использованных индика-
индикаторов и какова относительная систематическая погрешность титрования;
7) напишите уравнение титриметрической реакции в молекулярной форме.
II
СТРОЕНИЕ ВЕЩЕСТВА
¦ Строение атомов
¦ Химическая связь
и строение вещества
Для изучения процессов химических превра-
превращений и их кинетических закономерностей
необходимо понимать природу элементарных
химических реакций, в ходе которых разру-
разрушаются и создаются молекулы. В основе этого
лежит теория строения атомов, молекул и хи-
химической связи. Сведения о структуре моле-
молекул позволяют получать определенные пред-
представления о механизмах химических реакций
и соответственно управлять скоростью хими-
химического процесса. Знание основ теории строе-
строения вещества необходимо при проведении
работ по созданию новых материалов: сверх-
сверхпроводников, полупроводников, конструкци-
конструкционных сплавов, керамических и полимерных
материалов.
В настоящее время для изучения веществ и их
превращений применяют различные физичес-
физические методы (спектральные и радиоспектральные,
рентгеноструктурный анализ, электронография,
электронная микроскопия и др.). Чтобы пра-
правильно понимать результаты исследований и
уметь интерпретировать их при использовании
на практике, также требуется знать законы,
описывающие движение элементарных частиц
в атомах и молекулах. Таким образом, изуче-
изучение теории строения вещества — важный этап
в подготовке современных исследователей и
инженеров.
3 ¦ Строение атомов
Современная теория строения атомов и молекул базируется на за-
законах движения микрочастиц, т.е. объектов, обладающих очень ма-
малой массой. Эти законы были сформулированы в 1923-1927 гг. и
привели к созданию новой науки — квантовой механики. Установ-
Установлено, что поведение микрочастиц принципиально отличается от по-
поведения макрообъектов, изучаемых классической физикой.
Применение законов квантовой механики к химическим явлениям
привело к созданию квантовой химии, которая является основой со-
современной теории строения вещества и химической связи.
ЗЛ. Основные положения квантовой механики
Квантовая механика базируется на трех основных положениях:
энергетические изменения, происходящие в микросистемах,
носят дискретный характер (принцип квантования энергии);
поведение микрочастиц (в том числе и электронов) определяется
двойственностью их природы (корпускулярно-волновой дуализм);
законы микромира обусловлены статистическим (вероятност-
(вероятностным) характером.
Квантование энергии. Для объяснения особенностей излуче-
излучения нагретых тел М. Планк в 1900 г. выдвинул предположение, что
энергия излучается и поглощается не непрерывно, а дискретно от-
отдельными порциями — квантами. Энергия Е кванта зависит от час-
частоты электромагнитного излучения v:
E = hv, C.1)
где h — постоянная Планка; v = с/Х (к — длина волны излучения);
с— скорость света. Идея о квантовании энергии позволила объяс-
объяснить фотоэлектрический эффект (А. Эйнштейн, 1905) и линейчатую
структуру атомных спектров (Н. Бор, 1913).
Корпускулярно-волновой дуализм характеризует способность
любого объекта проявлять одновременно волновые и материальные
свойства. Двойственная природа микромира была впервые установ-
установлена для излучения. С одной стороны, для излучения характерны
такие явления, как интерференция и дифракция, которые характери-
характеризуют его волновую природу. С другой стороны, излучение проявляет
свойства частиц, о чем свидетельствует явление фотоэффекта.
Эйнштейн предположил, что излучение испускается в виде частиц,
3.1. Основные положения квантовой механики 77
названных фотонами или квантами. Каждый фотон обладает энергией, оп-
определяемой C.1).
В силу корпускулярной природы излучения фотоны должны обладать
определенной массой. Масса покоя фотона равна нулю, а при движении фо-
фотон приобретает динамическую массу. Для вычисления этой массы Эйн-
Эйнштейн обосновал применение уравнения эквивалентности массы и энергии:
Е = тс2.
В то же время Е= hv= h(c/X). Отсюда следует
л=А=А. (з.2)
тс рф
Здесь рф = тс — импульс фотона. Французский физик Л. де Бройль в 1923 г.
предположил, что двойственной природой обладает не только свет, но и лю-
любой материальный объект (в частности, электрон). Длина волны любого
движущегося объекта
Х=—9 C.3)
mv
где т — масса частицы; v — скорость движения частицы; X — длина волны
объекта, называемая длиной волны де Бройля. Из этого выражения следует,
что чем меньше масса частицы, тем больше длина ее волны и тем значи-
значительнее проявляются ее волновые свойства.
Уравнение де Бройля C.3) в 1927 г. было подтверждено эксперимен-
экспериментально. Американские физики Дж. Дэвиссон и А. Джермер обнаружили ди-
дифракцию электронов на монокристаллах никеля Ni. Позднее способность к
дифракции была обнаружена у других микрообъектов (атомов гелия Не, мо-
молекул водорода Нг, нейтронов).
В макромире волновые свойства не проявляются — перемещение тел
хорошо описывается как движение частиц. Причина заключается в том, что
постоянная Планка очень мала (см. Приложения), поэтому длины волн ока-
оказываются соизмеримыми с размерами частиц только в микромире.
Принцип неопределенности. Волновые свойства микрочастиц выра-
выражаются также в ограниченности применения к ним некоторых понятий
классической механики, а именно координаты и импульса. Например, один
из способов наблюдения за объектом — воздействие на него электромаг-
электромагнитного излучения (свет, радиоволны) и регистрация отраженного сигнала,
что широко используется в радиолокации, эхолокации. Причем чем сильнее
воздействие на наблюдаемый объект, тем сильнее отраженный сигнал. Если
ведется наблюдение за макрообъектами, то действие на них электромагнит-
электромагнитного излучения не изменяет ни их положения, ни их скорости. В случае на-
78 3. Строение атомов
блюдения за объектами микромира (например, электронами) ситуация вы-
выглядит иначе. При действии кванта света (фотона) на микрочастицу ее ско-
скорость не остается без изменения. Зная положение микрочастицы в какой-то
момент времени, нельзя в это же мгновение определить ее скорость, по-
поскольку она уже изменилась.
В. Гейзенберг в 1927 г. предложил соотношения, которые получили
название принципа неопределенности. Согласно этому принципу, невоз-
невозможно одновременно точно определить координаты частицы и ее импульс.
Математически принцип неопределенности формулируется следую-
следующим образом:
АА^А АА ^ h h
АхАрх> —-, АуАру> —-, AzApz> —,
4я 4я An
где Ajc, Ay, Az — погрешность определения координат х, у, z соответствен-
соответственно; Арх, Ару, Apz — погрешность определения проекций импульса на оси
координат. Из этих соотношений следует, что чем точнее определены коор-
координаты электрона, тем с меньшей точностью будет найден его импульс, и
наоборот. Другими словами, говорить о траектории движения электрона не
имеет смысла, так как для ее описания необходимо точно знать координаты
электрона и его импульс в каждый момент времени. Из соотношений неоп-
неопределенностей следует, что точно описать движение такой микрочастицы,
как электрон, невозможно. Движение электрона можно описать, используя
вероятностный подход, причем есть лишь определенная вероятность нахож-
нахождения электрона в заданной области пространства.
Движущейся частице можно поставить в соответствие волновой про-
процесс с длиной волны C.3), тогда этот процесс будет характеризоваться час-
частотой v и волновой функцией Ч? — величиной, определяющей волнообразно
распространяющееся возмущение. Аналогом служит смещение частицы от
положения равновесия при распространении волны, которое периодически
изменяется во времени по гармоническому закону, т.е. по закону синуса или
косинуса. В этом случае для волн де Бройля функция Ч* является гармони-
гармонической функцией времени и имеет вид:
^(х,у9z,/) = \|/(x, y,z)sin2nvt или x?(x,y,z,t) = \\f(x,y,z)cos2nvt.
Здесь \|/(jc, у, z) — амплитуда волн де Бройля или координатная волновая функ-
функция, зависящая только от пространственных координат. Одним из постулатов
квантовой механики является вероятностное толкование волны де Бройля. Как
показал М. Борн, волны де Бройля — это волны вероятности. Они не матери-
материальны, т.е. не связаны с переносом энергии. Волнообразно распространяется
лишь вероятность нахождения частицы, которая пропорциональна |\|/|2.
3.2. Стационарное уравнение Шрёдингера 79
3.2. Стационарное уравнение Шрёдингера
Квантово-механическая модель атома не обладает наглядностью. Ма-
Математический аппарат квантовой механики довольно сложен. Поэтому ос-
основные положения квантово-механической модели строения атома рассмот-
рассмотрены качественно, без использования математического аппарата. Многое из
того, что изложено в этом и следующих параграфах, придется принять «на
веру», без доказательств.
Законы движения частиц в квантовой механике выражаются уравне-
уравнением Шрёдингера, которое играет в ней ту же роль, что и законы Ньютона в
классической механике. Уравнение Шрёдингера представляет собой диффе-
дифференциальное уравнение в частных производных. Э. Шрёдингер в 1926 г.
предложил использовать волновое уравнение в качестве модели для описа-
описания поведения электрона в атоме — уравнение, связывающее энергию сис-
системы с ее волновым движением. Стационарное уравнение Шрёдингера для
одной частицы можно записать в следующей форме:
iTfff^ff
8л т \ дх ду dz
где U — потенциальная энергия частицы; Е — ее полная энергия; х9 y9 z —
декартовы координаты. Входящую в C.4) переменную величину \|/ называ-
называют волновой функцией. Эта функция описывает все свойства системы в ста-
стационарном состоянии — состоянии, которое не изменяется во времени.
Функция \|/ зависит от координат частиц и может зависеть от времени. Каж-
Каждая частица (или набор частиц) характеризуется квантово-механической
волновой функцией, которая описывает состояние данной системы. Функ-
Функцию \|/ можно рассматривать как амплитуду волнового процесса, следова-
следовательно, она может быть больше и меньше нуля, а также мнимой величиной.
Квадрат ее абсолютной величины |\|/|2 характеризует вероятность нахожде-
нахождения частицы в данном месте пространства. Величина |\|/|2<iF равна вероятно-
вероятности нахождения рассматриваемой частицы в элементе объема dV= dxdydz.
В соответствии с физическим смыслом волновая функция конечная,
непрерывная и однозначная, а также обращается в нуль там, где частица не
может находиться. Например, при рассмотрении движения электрона в ато-
атоме \|/ становится равной нулю на бесконечно большом расстоянии от ядра.
Конечность означает, что нигде \|/ и ее квадрат |\|/|2 не могут стать бесконеч-
бесконечно большими. Однозначность соответствует тому, что вероятность нахож-
нахождения частицы в данном единичном объеме всегда строго определенная.
Непрерывность функции означает, что нет такого элемента объема, где
нельзя определить вероятность нахождения частицы.
80 3. Строение атомов
Рассмотрим, как можно получить уравнение Шрёдингера. Поскольку
атом представляет собой замкнутую систему, то за модель поведения элек-
электрона в атоме Шрёдингер предложил использовать уравнение стоячей вол-
волны. Из теории колебаний и волн известно, что для одномерной стоячей вол-
волны дифференциальное уравнение имеет вид
где/— смещение волны в точке х. Поскольку атомная система трехмерная,
волновое движение в трехмерном пространстве описывается уравнением
сферической стоячей волны:
д\ д\ д\ 4п2
дх2 ду2 dz2 X2
Здесь \|/(jc, у9 z) — трехмерный аналог величины/(х). В оптике и акустике это
уравнение определяет стоячие волны и показывает пространственное рас-
распределение амплитуд, если процесс ограничен в пространстве. Примером
могут быть волны в жидкости, заполнившей целиком сферическую емкость.
Если в это уравнение подставить величину X из C.3), то можно получить
д\ д\ д\ 4n2m2v2 Л _ _ч
дх2 8у2 dz2 h2
Полная энергия системы равна сумме потенциальной U и кинетиче-
кинетической Тэнергий:
Отсюда
2_2(E-U)
т
Подставим это выражение в C.5) и получим
—Т" +—т + —т + ^ (E-U)\\f = 0. C.6)
дх2 ду2 dz2 h2
Уравнение C.6) и есть стационарное уравнение Шрёдингера для од-
одной частицы. Очевидно, что его легко привести к виду C.4). Используя опе-
оператор Лапласа
3.3. Уравнение Шрёдингера для атома водорода 81
А д2 д2 д2
дх2 ду2 dz2
уравнение C.4) можно представить в виде
h2
—Avi/ + U\\f = E\\f.
8n2m
Уравнение Шрёдингера часто записывают в операторной форме:
H\\f = Е\у,
где Н — оператор Гамильтона:
Sn2m{dx2 ду2 dz
Квантово-механическое решение задач в теории атома и молекулы
сводится к нахождению удовлетворяющих уравнению Шрёдингера волно-
волновых функций, обладающих указанными свойствами, и значений энергии.
Вообще говоря, может быть несколько различных функций \|/ь ц/2, \|/з,..., *|/„,
которые являются решениями уравнения Шрёдингера, причем каждой соот-
соответствует свое значение энергии ЕиЕ2,..., Еп.
Волновые функции, для которых уравнение Шрёдингера имеет реше-
решение, называют собственными функциями; значения энергии, вычисленные
из таких решений — собственными значениями.
Таким образом, согласно представлениям квантовой механики, веро-
вероятность пребывания электрона в различных областях пространства неодина-
неодинакова. Современным представлениям отвечает понятие об электронном облаке,
плотность которого в различных точках определяется |\|/|2. В научной лите-
литературе используют понятие «орбиталь», которое характеризует совокуп-
совокупность положений электрона в атоме. Каждой орбитали соответствует опре-
определенная волновая функция.
3.3. Уравнение Шрёдингера для атома водорода
Уравнение Шрёдингера можно решить точно только для очень про-
простых систем. Одна из таких систем — атом водорода. Решение уравнения
Шрёдингера для него позволило определить стационарные состояния атома,
рассчитать его спектр и объяснить на основе этого его химическое поведе-
82
3. Строение атомов
ние. Обобщение полученных выводов
позволило понять физическую сущ-
сущность периодического закона и объ-
объяснить химические свойства элементов.
Атом водорода состоит из ядра с
зарядом {+ё) и одного электрона с за-
зарядом (-е).
Потенциальная энергия электро-
электрона в поле ядра
и- *
4тге0г
Рис. 3.1. Электрон в сферической
системе координат
где г — расстояние от электрона до
ядра; б0 — электрическая постоянная.
Поскольку поле ядра обладает сферической симметрией, для удобства пере-
переходят от декартовых координат к сферическим, г; 0; ф, (рис. 3.1). Соотноше-
Соотношения между декартовыми и сферическими координатами следующие:
jc = rsin9coscp, j> = rsin0sinq), z = rcos9.
Уравнение C.6) в сферических координатах приобретает вид
Решая это уравнение, получают
где R(r) — радиальная составляющая волновой функции; 0F) и Ф(ф) — уг-
угловые составляющие волновой функции (получаемые решения в явном виде
не приводятся). Следовательно, волновые функции в C.7) зависят от трех
целочисленных параметров л, / и mh которые называют квантовыми числа-
ми. Волновую функцию C.7), описывающую состояние электрона, называ-
называют атомной орбиталъю (АО).
3.4. Квантовые числа
В квантовой механике каждая АО определяется тремя квантовыми
числами.
Главное квантовое число п. Может принимать целочисленные зна-
значения от 1 до оо. Главное квантовое число определяет:
3.4. Квантовые числа 83
номер энергетического уровня;
интервал энергий электронов, находящихся на данном уровне;
размеры орбиталей;
число подуровней данного энергетического уровня (первый уровень
состоит из одного подуровня, второй — из двух, третий — из трех и т.д.);
в Периодической системе элементов максимальному значению глав-
главного квантового числа соответствует номер периода.
Иногда используют буквенные обозначения главного квантового чис-
числа, т.е. каждое численное значение п обозначают соответствующей буквой
латинского алфавита:
Численные значения п 1 2 3 4 5
Буквенное обозначение К L М N О
Главное квантовое число определяет также узловые свойства АО, у
которой имеются узловые поверхности. Узловая поверхность — геометри-
геометрическое место точек, для которых \|/ = 0и|\|/|2 = 0 соответственно. На такой
поверхности волновая функция меняет фазу, подобно изменению фазы при
колебаниях струны. Число узловых поверхностей равно значению главного
квантового числа п, причем одна из этих поверхностей соответствует гра-
граничному условию для бесконечного расстояния г. Поэтому п принимает
значения чисел натурального ряда.
Орбитальное квантовое число /. Определяет орбитальный момент
количества движения (импульс) электрона, точное значение его энергии и
форму орбиталей. Может принимать значения 0, 1,2, 3,..., (и - 1).
Уточним понятие «орбиталь», которое имеет совершенно иной смысл,
чем термин «орбита», используемый в классической механике. Атомная
орбиталь — геометрический образ одноэлектронной волновой функции \|/,
представляющий собой область наиболее вероятного пребывания электрона
в атоме. Она ограничивает область пространства, в которой вероятность на-
нахождения электрона имеет определенное значение (90...99 %). Иногда ор-
биталью называют граничную поверхность этой области, а на рисунках, как
правило, изображают сечение этой области плоскостью, проходящей через
начало координат и лежащей в плоскости рисунка. В начало координат по-
помещают центр ядра атома. Понятие «орбиталь», в отличие от «орбита», не
подразумевает знания точных координат электрона.
Значения орбитального квантового числа также связаны с узловыми
поверхностями. Они показывают, сколько существует узловых поверхно-
поверхностей, зависящих от углов и проходящих через ядро. Поскольку всего имеет-
имеется п узловых поверхностей, то должно быть еще (п - I) узловых поверхно-
поверхностей без угловой зависимости, т.е. сферически симметричных. Поэтому /
всегда должно быть меньше я, по крайней мере на 1, т.е. / = 0,1,2,...,(«- 1).
84
3. Строение атомов
Например, если п = 2, то / может быть равно либо 0, либо 1. При зна-
значении / = О зависящих от угла, узловых поверхностей не будет, а обе ради-
радиальные узловые поверхности — сферы. Одна из этих поверхностей
характеризуется г = оо. При / = 1 одна из узловых поверхностей зависит от
угла — это плоскость. Другая узловая поверхность («-/ = 2-1 = 1)
представляет собой сферу, которая также уходит в бесконечность. Таким
образом, орбитальное квантовое число определяет форму атомной орбитали.
При / = 0 это сфера, при / = 1 — объемная восьмерка (гантель), при 1 = 2 —
четырехлепестковая розетка.
Каждому значению главного квантового числа соответствует п значе-
значений орбитального квантового числа / (см. табл. 3.1). Например, если п = 1,
то / принимает только одно значение (/ = 0), при п = 2 — два значения: 0 и 1
и т.д. Каждому численному значению / соответствует определенная геомет-
геометрическая форма орбиталей и приписывается буквенное обозначение. Первые
четыре буквы обозначения имеют историческое происхождение и связаны с
характером спектральных линий. s,p, d,f— первые буквы английских слов,
использованных для названия спектральных линий: sharp (резкий), principal
(главный), diffuse (диффузный), fundamental (основной). Обозначения дру-
других орбиталей приведены в алфавитном порядке: &/*,...
Таблица 3.1
Значения главного и орбитального квантовых чисел
Орбитальное
квантовое
число /
Значение
Буквенное
обозначение
Главное квантовое число п
1
0
s
0
s
1
1
Р
3
0
s
1
Р
2
d
4
0
s
1
Р
2
d
3
/
5
0
s
1
Р
2
d
3
/
4
g
Отметим также, что орбитальный момент количества движения L —
вектор, в квантовой механике его абсолютная величина принимает строго
определенные (квантованные) значения:
Обозначение любого подуровня определяется двумя квантовыми чис-
числами — главным (при записи указывается численное значение) и орбиталь-
орбитальным (при записи указывается буквенное обозначение). Например, энергети-
энергетический подуровень, для которого п = 2 и / = 1, следует обозначить так:
2р-подуровень. Все орбитали с одинаковым значением / имеют одинаковую
3.4. Квантовые числа
85
геометрическую форму и в зависимости от значений главного квантового
числа различаются размерами. Например, все орбитали, для которых 1 = 0
(s-орбитали) являются сферически симметричными, различаются размерами
в зависимости от значения главного квантового числа. Чем выше значение
и, тем больше размеры орбиталей.
Магнитное квантовое число mt. Определяет возможные значения
проекции орбитального момента количества движения электрона на фикси-
фиксированное направление в пространстве (например, на ось z). Движение элек-
электрона вокруг ядра можно сравнить с движением тока по замкнутому конту-
контуру. При этом возникает магнитное поле, вектор напряженности Н которого
направлен перпендикулярно плоскости вращения электрона. Если атом на-
находится во внешнем магнитном поле, то, согласно квантово-механическим
представлениям, его электроны должны располагаться так, чтобы проекции
их магнитных моментов на направление этого поля были целочисленными
(рис. 3.2). При этом они могут принимать как отрицательные, так и положи-
положительные значения, включая нулевое.
Проекция орбитального момента количества движения в квантовой
механике определяется соотношением
т h
Lj7 =m,—.
Численное значение проекции магнитного момента Lz и представляет
собой магнитное квантовое число. Оно принимает отрицательные и поло-
положительные значения /, включая нуль. Общее число значений равно 2/ + 1:
пц= -/,-(/- IX --I, 0, +1,..., (/-1),/.
От значения магнитного квантового числа зависит взаимодействие магнит-
магнитного поля, создаваемого электроном, с внешним магнитным полем. Если нет
внешнего магнитного поля, то энергия электрона в атоме не зависит от т/. В
этом случае электроны с одинаковыми значениями п и /, но с разными зна-
значениями mi обладают одинаковой энергией. Если существует внешнее маг-
н
Рис. 3.2. К пояснению физического смысла магнитного квантового числа
86
3. Строение атомов
нитное поле — энергия электронов с разными mi различается (разное взаи-
взаимодействие с полем). Поэтому в магнитном поле происходит расщепление
некоторых атомных спектральных линий (эффект Зеемана).
В общем случае магнитное квантовое число характеризует ориента-
ориентацию АО в пространстве относительно внешней силы. Магнитное квантовое
число определяет ориентацию орбитального углового момента относитель-
относительно некоторого фиксированного направления.
Общее число возможных значений W/ соответствует числу способов
расположения орбиталей данного подуровня в пространстве, т.е. общему
числу орбиталей на данном подуровне (табл. 3.2).
Таблица 3.2
Число орбиталей на подуровне
Главное квантовое
число п
1
2
3
Число значений W/
1
1
3
1
3
5
Квантовое число
/
0E)
0E)
\<Р)
0E)
1(Р)
2(d)
т,
0
0
-1,0,+1
0
-1,0,+1
-2,-1,0,+1,+2
Орбитальному квантовому числу / = 0 отвечает единственное значение
магнитного квантового числа mt = 0. Эти значения / и т{ характеризуют все
5-орбитали, которые имеют форму сферы. Так как в этом случае магнитное
квантовое число принимает только одно значение, то каждый ^-подуровень
состоит только из одной орбитали. Рассмотрим любой /^-подуровень. При / = 1
орбитали имеют форму гантелей (объемные восьмерки), магнитное квантовое
число принимает следующие значения: /и/ = -1, 0, +1. Следовательно,
/^-подуровень состоит из трех АО, которые располагаются вдоль осей коорди-
координат, их обозначаютрх, ру, pz соответственно (рис. 3.3). Для J-подуровня / = 2,
mi = -2, -1, 0, +1, +2 E значений), и любой ^/-подуровень состоит из пяти орби-
z z z z
s р р р
Рис. 3.3. Пространственная форма s- и /7-атомных орбиталей
3.4. Квантовые числа
87
Рис. 3.4. Пространственная форма d-орбиталей
талей, которые определенным образом расположены в пространстве (рис. 3.4),
и обозначаются
^ dyz, d 2 _ 2, d 2 соответственно. Четыре из пяти
биталей имеют форму четырехлепестковых розеток, каждая из которых обра-
образована двумя гантелями, пятая АО представляет собой гантель с тором в эква-
экваториальной плоскости (d 2 -орбиталь) и расположена вдоль оси z. Лепестки
орбитали d 2_ 2 расположены вдоль осей хиу. Лепестки орбиталей d^ d^ dyz
х у
расположены симметрично между соответствующими осями.
Четвертый энергетический уровень состоит из четырех подуровней —
s9 р, d и / Первые три из них аналогичны описанным выше, а четвертый
/¦подуровень состоит из семи АО, пространственная форма которых доста-
достаточно сложна и в данном разделе не рассматривается.
С. Гаудсмит и Дж. Уленбек для объяснения некоторых тонких эффек-
эффектов в спектре атома водорода в 1925 г. выдвинули гипотезу о наличии соб-
собственного момента импульса электрона, который назвали спином. Спин
нельзя выразить через координаты и импульсы, у него нет аналога в класси-
классической механике. Спин электрона может принимать только одно значение,
равное —. Проекция вектора спина на определенное направление внешнего
поля (например, на ось z) называется спиновым квантовым числом и может
принимать только два значения: ms= ±— (рис. 3.5).
88
3. Строение атомов
+1/2
~= -1/2
Рис. 3.5. Квантование спи-
спина электрона
Понятие «спин» введено для характеристики
специфического квантового свойства электрона.
Наличие спина не следует из уравнения Шрёдинге-
ра, в котором не учитываются требования теории
относительности. Согласно релятивистской кван-
квантовой теории Дирака, у электрона существуют соб-
собственный момент импульса, а следовательно, и
собственный магнитный момент. Можно сказать,
что спин — это проявление релятивистских эффек-
эффектов на микроскопическом уровне.
Электрон имеет четыре степени свободы;
переменные х, у, z изменяются непрерывно, спино-
спиновое квантовое число принимает только дискретные
1 1
—. Таким образом, состояние
значения:
или
электрона в атоме определяется набором значений
четырех квантовых чисел: п, /, w/ и т$.
Обозначение и структура электронных энергетических уровней.
Определим некоторые термины, которые используются для разъяснения фи-
физического смысла квантовых чисел. Группа орбиталей, имеющих одинако-
одинаковое значение орбитального квантового числа, образует энергетический по-
подуровень. Совокупность всех орбиталей с одинаковым значением главного
квантового числа образует энергетический уровень.
Структуру атомных электронных уровней можно изобразить двояко: в
виде электронных формул и электронографических диаграмм. При написа-
написании электронных формул используют два квантовых числа п и /: первый
уровень — Is; второй уровень — 2s, 2р; третий — 3s, Зр, 3d; четвертый —
4s, Ар, 4d, 4/и т.д. (табл. 3.3).
Таблица 3.3
Структура электронных энергетических уровней атома
Главное квантовое
число п
1
1
3
Л
4
Обозначение
подуровня
15
25
2/7
35
Ър
3d
45
Ар
Ad
4/
Квантовое число
/
0
0
1
0
1
2
0
1
2
3
mi
0
0
-1,0,+1
0
-1,0,+1
-2,-1,0,+1,+2
0
-1,0,+1
-2,-1,0,+1,+2
-3,-2,-1,0,+1,+2,+3
5.5. Атомные орбитали водорода 89
Более полно строение электронных уровней описывается с использо-
использованием трех квантовых чисел: п, /, W/. Каждая АО условно изображается в
виде квантовой ячейки, около которой ставится номер уровня и символ под-
подуровня.
3.5. Атомные орбитали водорода
Рассмотрим основные особенности решения уравнения Шрёдингера
для атома водорода и обсудим их физический смысл. Атомную орбиталь
V/i / т УД°бно представить в виде произведения двух функций — радиаль-
радиальной (Rnl) и угловой (Ylmi =&1тФт{):
Решая уравнение Шрёдингера для атома водорода, получают следую-
следующее выражение для энергии электрона:
те4 ( 1
Важная особенность атома водорода состоит в том, что энергия элек-
электрона в нем зависит только от главного квантового числа. Это означает, что
энергия S-, р-, J-орбиталей, относящихся к одному и тому же энергетиче-
энергетическому уровню, одинакова и не зависит от значений орбитального квантового
числа. Атомные орбитали с равными значениями энергии называют вырож-
вырожденными. Энергия атома отрицательна, поскольку за нулевой уровень вы-
выбрана энергия электрона, расположенного на бесконечно большом расстоянии
от ядра. Энергия электрона принимает наименьшее значение при п = 1 и
возрастает до 0 с увеличением п. Энергетическая диаграмма атома водорода
представлена на рис. 3.6.
Атомная орбиталь для основного состояния атома водорода \s (n = 1,
/ = 0, т/ = 0) имеет наименьшую энергию:
Радиальная и угловая функции равны:
4o.2f4'W-4 _„
'у
где а0 = ° =0,0529 нм — величина, соответствующая радиусу первой
пте
90
3. Строение атомов
»-*? ггп ггггп I I I I I m
45 4р 4d 4/
п = Ъ U ГГП МММ
35 Зр 3d
25 2р
15
Рис. 3.6. Энергетическая диаграмма атома водорода
орбиты в предложенной ранее Н. Бором модели атома водорода. Ясно, что
угловая функция Уоо и квадрат ее модуля |Уоо|2 для состояния s (/ = 0, m/ = 0)
не зависят от углов Э и ф, т.е. обладают сферической симметрией. Это озна-
означает, что вероятность обнаружения электрона на всех направлениях одина-
одинаковая (см. рис. 3.2).
Функции R\0 и \R\o\2 экспоненциально понижаются с удалением от яд-
ядра (рис. 3.7). Поэтому уже на расстоянии г = 0,3 нм от ядра вероятность об-
обнаружения электрона очень мала. Если вокруг ядра очертить такую сферу,
что за ее границами значение квадрата модуля волновой функции |\|/|2 не
превысит, например, одной сотой максимального значения, то получим гра-
граничную поверхность для Is-АО, внутри которой вероятность нахождения
электрона составит 99 %. Радиус такой сферы для Is-АО атома водорода
будет приблизительно равен 0,24 нм.
Один из способов наглядного представления АО заключается в по-
построении графика функции радиального рас-
распределения вероятности. Вероятность нахож-
нахождения электрона в элементарном объеме dV
равна |\|/|2с/К Если за элементарный объем
принять объем шарового слоя толщиной dr,
где г — расстояние от ядра атома до шарово-
шарового слоя, то dV = Anfdr. Тогда вероятность на-
нахождения электрона в шаровом слое на рас-
расстоянии от г до г + dr в любом направлении
Рис. 3.7. График радиальной равна
составляющей волновой функ- Р( \-л 21 I2 л
-АО v ' m
3.5. Атомные орбитали водорода
91
О rm r
Рис. 3.8. График функции ради-
радиального распределения вероят-
вероятности для Ь-АО
Функцию Р{г) называют функцией радиаль-
радиального распределения вероятности нахожде-
нахождения электрона в атоме (рис. 3.8). Из рисунка
следует, что Is-АО имеет один максимум,
соответствующий наиболее вероятному
расстоянию гт электрона от ядра. Для атома
водорода гт(Щ=а0.
Как и все s-AO, 2^-АО обладает сфе-
сферической симметрией (рис. 3.9). При г = 2а0
она проходит через нуль, вследствие чего
внутри электронного облака образуется уз-
узловая поверхность, на которой v|/2oo= 0 и
вероятность обнаружения электрона равна нулю. Поэтому радиальная
функция распределения Р2о имеет два максимума: малый, очень близко рас-
расположенный от ядра и большой на расстоянии 5,24я0 от ядра (рис. 3.10). В
общем случае число максимумов равно (п - /).
График функции радиального распределения для 2р-АО имеет такой
же вид, как и для Is-АО, но более протяженный. Угловая функция 2р-АО
определяется квантовыми числами / = 1, ю/ = -1, 0, +1. Трем значениям
квантового числа W/ соответствуют три орбитали. Они обладают одинаковой
энергией, определяемой главным квантовым числом л, т.е. /^-состояние триж-
трижды вырождено, 2р-АО обладают осевой симметрией и имеют вид объемных
восьмерок, ориентированных по осям х, y9 z (см. рис. 3.3). Через начало ко-
координат проходит узловая плоскость, поэтому одна из долей восьмерки име-
имеет знак «+», а другая знак «-». Так как угловая функция Y не зависит от п, то
симметрия любых пр-АО аналогична 2р-АО, а различаются они только
энергией и размером.
На третьем энергетическом уровне появляются пять Зс/-орбиталей (/ = 2),
соответствующих пяти значениям квантового числа mi = -2, -1, 0, +1, +2.
R
Р(г)
о
Рис. 3.9. График радиальной со-
составляющей волновой функции
для 2^-АО
О
Рис. ЗЛО. График функции ра-
радиального распределения веро-
вероятности для 2s- АО
92
3. Строение атомов
Атомные орбитали 3d также не имеют узловых поверхностей, при
этом у функции Р(г) только один максимум, так как {п-1)= 1. Угловая со-
составляющая, как и ее квадрат, имеет вид объемных лепестков (см. рис. 3.4).
Знак волновой функции \|/ меняется при переходе из одного квадранта в
другой. Обозначения этих орбиталей обусловлены видом соответствующих
формул для волновых функций.
3.6. Принципы построения электронной структуры
атомов элементов
В многоэлектронных атомах, как и в атоме водорода, состояние каж-
каждого электрона можно характеризовать квантовыми числами и, /, w/, ms.
Межэлектронное отталкивание приводит к тому, что энергия электронов,
имеющих одно и то же значение и, но разные значения /, становится различ-
различной. Последовательность заполнения электронами подуровней в многоэлек-
многоэлектронных атомах определяется принципом наименьшей энергии, принципом
Паули и правилом Хунда.
Принцип наименьшей энергии гласит: заполнение электронами АО про-
происходит в порядке возрастания их энергии. На основании изучения спектров
атомов и квантово-механических расчетов установлена энергетическая диа-
диаграмма (рис. 3.11) для различных АО в многоэлектронных нейтральных ато-
атомах, находящихся в основном состоянии (состоянии с наименьшей энергией).
¦
4/
Рис. 3.11. Энергетическая диаграмма АО для многоэлектронных атомов
3.6. Принципы построения электронной структуры атомов элементов 93
Последовательность АО в порядке возрастания их энергии имеет вид
\s < 2s < 2р < 3s < Ър < As < 3d< Ар < 5s < Ad< 5p <
<6s< Af< 5d< 6p<ls< 5/« 6d< Ip.
Порядок возрастания энергии АО в сложных атомах описывается пра-
правилом (п + /) (или правилом Клечковского): энергия АО возрастает в соот-
соответствии с увеличением суммы п + / главного и орбитального квантовых
чисел. При одинаковом значении суммы энергия меньше у АО с меньшим
значением главного квантового числа (табл. 3.4).
Таблица 3.4
Сумма значений главного и орбитального квантовых чисел
Энергетический
уровень п
1
2
3
А
4
5
Энергетический подуровень
/
0
0
1
0
1
2
0
1
2
3
0
1
2
3
4
обозначение подуровня
Ь
2s
2р
3s
Зр
3d
As
Ар
Ad
Af
5s
5р
5d
5/
5g
v* A- 1
Yl • I
1
2
3
3
4
5
4
5
6
7
5
6
7
8
9
Согласно принципу Паули, в атоме не может быть двух электронов с
одинаковым набором четырех квантовых чисел. Каждый электрон в атоме
имеет свой набор четырех квантовых чисел, который полностью определяет
энергетическое состояние электрона. Так, для АО-Is существует два набора
квантовых чисел:
п
1
т1
ms
... 1
... 0
... 0
1
2
1
0
0
1
2
94
3. Строение атомов
Следовательно, на s-AO может находиться только два электрона с различ-
различными значениями спинового квантового числа.
Для каждой из трех 2р-АО также возможно только два набора кванто-
квантовых чисел. Например, для одной из 2р-АО:
п
I ..
щ ..
. 2
. 1
. 0
1
+ 2
2
1
0
1
2
Значит, нар-подуровне любого уровня может находиться шесть электронов.
Таким образом, на одной орбитали могут находиться только два элек-
электрона с противоположными по знаку значениями спинового квантового
числа. Два электрона, находящиеся на одной орбитали, т.е. имеющие одина-
одинаковые значения квантовых чисел ю, /, т/, но различные значения спинового
квантового числа ms, называют спаренными. Принцип определения макси-
максимального числа электронов, которые могут находиться на уровне с главным
квантовым числом, показан в табл. 3.5.
Таблица 3.5
Максимальное число электронов на первых четырех энергетических уровнях
Энергетический
уровень п
1
2
3
4
1
0E)
0E)
1(/>)
0E)
1(р)
2(</)
0E)
1(Р)
2i4)
3(/)
/П/
0
0
-1, 0, 1
0
-1, 0, 1
-2,-1,0,1,2
0
-1,0,1
-2,-1,0,1,2
-3,-2,-1,0,1,2,3
ms
±1/2
±1/2
±1/2
±1/2
±1/2
±1/2
±1/2
±1/2
±1/2
±1/2
Число электронов
на подуровне
2
2
6
2
6
10
2
6
10
14
на уровне
2
8
18
32
Общее число орбиталей на энергетическом уровне со значением глав-
главного квантового числа п равно п2. Так как на одной орбитали может нахо-
находиться лишь два электрона, получаем, что максимальная электронная ем-
емкость энергетического уровня составляет 2п2.
Правило Хунда определяет последовательность заполнения АО электро-
электронами в пределах одного подуровня и формулируется следующим образом: при
3.7. Электронные конфигурации атомов элементов Периодической системы 95
данном значении квантового числа / (т.е. в пределах одного подуровня) в ос-
основном состоянии электроны располагаются таким образом, что значение сум-
суммарного спина атома максимально. Это означает, что на подуровне должно
быть максимально возможное число неспаренных электронов.
3.7. Электронные конфигурации атомов элементов
Периодической системы
Распределение электронов по различным АО называют электронной
конфигурацией атома. Электронная конфигурация с наименьшей энергией
соответствует основному состоянию атома, остальные конфигурации отно-
относятся к возбужденным состояниям.
Электронную конфигурацию атома изображают двумя способами — в
виде электронных формул и электронографических диаграмм. При написа-
написании электронных формул используют главное и орбитальное квантовые
числа. Подуровень обозначают с помощью главного квантового числа (циф-
(цифрой) и орбитального квантового числа (соответствующей буквой). Число
электронов на подуровне характеризует верхний индекс. Например, для ос-
основного состояния атома водорода электронная формула: Is1.
Более полно строение электронных уровней можно описать с помо-
помощью электронографических диаграмм, где распределение электронов по по-
подуровням представляют в виде квантовых ячеек. Орбиталь в этом случае
принято условно изображать квадратом, около которого проставлено обо-
обозначение подуровня. Подуровни на каждом уровне должны быть немного
смещены по высоте, так как их энергия несколько различается. Электроны
обозначают стрелками Т или i в зависимости от знака спинового квантового
числа. Электронографическая диаграмма атома водорода:
Is
Принцип построения электронных конфигураций многоэлектронных
атомов состоит в добавлении протонов и электронов к атому водорода. Рас-
Распределение электронов по энергетическим уровням и подуровням подчиня-
подчиняется рассмотренным ранее правилам: принципу наименьшей энергии, прин-
принципу Паули и правилу Хунда.
С учетом структуры электронных конфигураций атомов все извест-
известные элементы в соответствии со значением орбитального квантового числа
последнего заполняемого подуровня можно разбить на четыре группы:
^-элементы, /^-элементы, d-элементы,/-элементы.
В атоме гелия Не (Z = 2) второй электрон занимает Ь-орбиталь, его
электронная формула: Is2. Электронографическая диаграмма:
96
3. Строение атомов
Is
п
Гелием заканчивается первый самый короткий период Периодической
системы элементов. Электронную конфигурацию гелия обозначают [Не].
Второй период открывает литий Li (Z = 3), его электронная формула:
[Не] 2sl (или K2sx). Электронографическая диаграмма:
2р
2s
и
Is
Далее приведены упрощенные электронографические диаграммы ато-
атомов элементов, орбитали одного энергетического уровня которых располо-
расположены на одной высоте. Внутренние, полностью заполненные подуровни, не
показаны.
После лития следует бериллий Be (Z = 4), в котором дополнительный
электрон заселяет 25-орбиталь. Электронная формула Be: [He] 2s2
и
2s
2р
В основном состоянии следующий электрон бора В (Z = 5) занимает
2/?-орбиталь, В: \s22s22px\ его электронографическая диаграмма:
п
т
2* 2р
Следующие пять элементов имеют электронные конфигурации:
С (Z = 6): [Не] 2s22p2 N (Z = 7): [Не] 2s22pz
п
т
t
п
т
t
t
2s 2p
O(Z=8):[He]2sV
2s
2p
F(Z=9):[He]2s2
и
7
и
t
t
2p
и
~2s~
и
п
t
2p
3.7. Электронные конфигурации атомов элементов Периодической системы 97
Ne(Z=10):[He]2522/76
п
и
п
п
2s
2р
Приведенные электронные конфигурации определяются правилом
Хунда.
Первый и второй энергетические уровни неона полностью заполнены.
Обозначим его электронную конфигурацию [Ne] и будем использовать в
дальнейшем для краткости записи электронных формул атомов элементов.
Натрий Na (Z = 11) и Mg (Z = 12) открывают третий период. Внешние
электроны занимают Зя-орбиталь:
Na(Z=ll):[Ne]351
т
35
Mg(Z=12):[Ne]3s2
Ър
и
3s
3d
Зр
3d
Затем, начиная с алюминия (Z= 13), заполняется 3/>-подуровень. Тре-
Третий период заканчивается аргоном Аг (Z = 18):
Al(Z=13):[Ne]3sV
t
Зр
3d
Ar(Z=18):[Ne]3523p6
и
п
и
3s
Зр
3d
Элементы третьего периода отличаются от элементов второго тем, что у них
имеются свободные Зб/-орбитали, которые могут участвовать в образовании
химической связи. Это объясняет проявляемые элементами валентные со-
состояния.
4 - 9795
98
3. Строение атомов
В четвертом периоде, в соответствии с правилом (п + /), у калия
К (Z = 19) и кальция Са (Z = 20) электроны занимают 4?-подуровень, а не 3d.
Начиная со скандия Sc (Z = 21) и кончая цинком Zn (Z = 30), происходит
заполнение Зс/-подуровня:
Sc: [Аг] 4s23dt -> Zn: [Ar] 4s23dx\
Электронные формулы rf-элементов можно представить в ином виде:
подуровни перечисляются в порядке возрастания главного квантового чис-
числа, а при постоянном п — в порядке увеличения орбитального квантового
числа. Например, для Zn такая запись будет выглядеть так: [Ar] 3dw4s2. Обе
эти записи эквивалентны, но приведенные ранее электронная формула цин-
цинка правильно отражает порядок заполнения подуровней.
В ряду 3 ^-элементов у хрома Cr (Z = 24) наблюдается отклонение от
правила (п + /). В соответствии с этим правилом электронная конфигурация
Сг должна выглядеть так: [Аг] 3d4 As2. Установлено, что его реальная конфи-
конфигурация — [Ar] 3d54sl. Иногда этот эффект называют «провалом» электрона.
Подобные эффекты объясняют повышенной устойчивостью наполовину (р3,
d5,/1) и полностью (р6, d10,/14) заполненных подуровней.
Отклонения от правила (п + /) наблюдаются и у других элементов
(табл. 3.6). Это связано с тем, что с увеличением главного квантового числа
различия между энергиями подуровней уменьшаются.
Таблица 3.6
Исключения из (п + /) — правила для первых 86 элементов
Элемент
Cr(Z=24)
Cu(Z=29)
Nb(Z=41)
Mo(Z = 42)
Tc(Z=43)
Ru(Z = 44)
Rh(Z = 45)
Pd(Z = 46)
Ag(Z = 47)
La(Z=57)
Ce(Z=58)
Gd(Z=64)
Ir(Z=77)
Pt(Z=78)
Au(Z=79)
Электроннаяi
по правилу (и + /)
[Ai]4s23d4
[Ai]4s23d9
[Kr]5i24c/3
[Ki]5s24d4
[Ki]5s24d5
[Ki]5s24d6
[Ki]5s24d7
[Ki]5s24d*
[Kr]5s24d9
[X<i]6s24fi5d0
[Xe]6s24f25d0
[Xe]6s24f5d°
[Xe]6s24fw5d1
[Xe]6s24fu5d*
[Xe]6s24f45d9
конфигурация
фактическая
4sl3d5
4s]3dt0
5s* 4 d4
5sUds
5sl4d6
5sx4dn
5sl4d8
5s°4dw
5sl4dw
6s24f°5di
6s24fl5dl
6s24f5dl
6s°4fH5d9
6sl4fl4Sd9
bsx4fX45dw
3.7. Электронные конфигурации атомов элементов Периодической системы 99
Далее происходит заполнение 4р-подуровня (Ga—Кг). В четвертом
периоде содержится всего 18 элементов. Аналогично происходит заполне-
заполнение 5s-, Ad- и 5/7-подуровней у 18-ти элементов пятого периода. Отметим,
что энергии 5s- и 4^-подуровней очень близки, и электрон с 5^-подуровня
может легко переходить на 4я?-подуровень. На 5^-подуровне у Nb, Mo, Tc,
Ru, Rh, Ag находится только один электрон. В основном состоянии 55-под-
уровень Pd не заполнен. Наблюдается «провал» двух электронов.
В шестом периоде после заполнения 6^-подуровня у цезия Cs (Z = 55)
и бария Ва (Z = 56) следующий электрон, согласно правилу (п + /), должен
занять 4/-подуровень. Однако у лантана La (Z = 57) электрон поступает на
5й(-подуровень. Заполненный наполовину D/7) 4/-подуровень обладает по-
повышенной устойчивостью, поэтому у гадолиния Gd (Z = 64), следующего за
европием Eu (Z = 63), на 4/-подуровне сохраняется прежнее количество
электронов G), а новый электрон поступает на 5й?-подуровень, нарушая пра-
правило (п + /). У тербия Tb (Z = 65) очередной электрон занимает 4/-под-
уровень и происходит переход электрона с 5</-подуровня (конфигурация
4/9б/). Заполнение 4/-подуровня заканчивается у иттербия Yb (Z = 70).
Следующий электрон атома лютеция Lu занимает 5^-подуровень. Его элек-
электронная конфигурация отличается от конфигурации атома лантана только
полностью заполненным 4/-подуровнем.
В настоящее время в Периодической системе элементов Д.И. Менде-
Менделеева под скандием Sc и иттрием Y располагают иногда лютеций (а не лан-
лантан) как первый ^-элемент, а все 14 элементов перед ним, включая лантан,
выносят в особую группу лантаноидов за пределы Периодической системы
элементов (помещена на форзаце).
Химические свойства элементов определяются, главным образом,
структурой внешних электронных уровней. Изменение числа электронов на
третьем снаружи 4/-подуровне слабо отражается на химических свойствах
элементов. Поэтому все 4/-элементы схожи по своим свойствам. Затем в
шестом периоде происходит заполнение 5я?-подуровня (Hf—Hg) и бр-под-
уровня (Tl—Rn).
В седьмом периоде 7^-подуровень заполняется у франция Fr (Z = 87) и
радия Ra (Z = 88). У актиния наблюдается отклонение от правила (п + /), и
очередной электрон заселяет 6^-подуровень, а не 5/. Далее следует группа
элементов (Th—No) с заполняющимся 5/-подуровнем, которые образуют
семейство актиноидов. Отметим, что 6d- и 5/- подуровни имеют столь близ-
близкие энергии, что электронная конфигурация атомов актиноидов часто не
подчиняется правилу (п + /). Но в данном случае знание точной конфигура-
конфигурации 5/ n5dm не столь важно, поскольку она довольно слабо влияет на хими-
химические свойства элемента.
100 3. Строение атомов
У лоуренсия Lr (Z = 103) новый электрон поступает на 6*/-подуровень.
Этот элемент иногда помещают в Периодической системе под лютецием.
Седьмой период не завершен. Элементы 104-109 неустойчивы и их свойства
мало известны. Таким образом, с ростом заряда ядра периодически повторя-
повторяются сходные электронные структуры внешних уровней. В связи с этим сле-
следует ожидать и периодического изменения различных свойств элементов.
Отметим, что описанные электронные конфигурации относятся к изо-
изолированным атомам в газовой фазе. Конфигурация атома элемента может
быть совершенно иной, если атом находится в твердом теле или растворе.
3.8. Периодический закон и Периодическая система
химических элементов Д.И. Менделеева
Периодический закон был открыт великим русским ученым Д.И. Мен-
Менделеевым в 1869 г. и сформулирован следующим образом: свойства простых
тел, а также форма и свойства их соединений находятся в периодической
зависимости от атомного веса элементов.
До появления сведений о сложном строении атома основной характе-
характеристикой элемента служил атомный вес (относительная атомная масса).
Развитие теории строения атома привело к установлению того факта, что
главной характеристикой атома служит заряд его ядра. В настоящее время
периодический закон формулируется так: свойства химических элементов, а
также форма и свойства образуемых ими соединений находятся в периоди-
периодической зависимости от величины заряда ядер их атомов.
Графическим изображением периодического закона является таблица
Периодической системы элементов. Формы такого изображения различны.
Наиболее широко используются три: 1) короткая, 8-клеточная; 2) полудлин-
полудлинная, 18-клеточная; 3) длиннопериодная, 32-клеточная.
Принципиальный подход к построению таблиц единый — элементы
располагают в порядке возрастания заряда ядер их атомов. Физическая ос-
основа структуры Периодической системы элементов состоит в определенной
последовательности формирования электронных конфигураций атомов по
мере роста порядкового номера элемента. Элементы со сходной электрон-
электронной конфигурацией внешних энергетических уровней обладают и сходными
химическими свойствами.
Периодом в Периодической системе элементов называют последова-
последовательный ряд элементов (расположенных в порядке возрастания заряда ядер
их атомов), электронная конфигурация внешнего энергетического уровня
которых изменяется от nsl до ns2np6 (для первого периода s{ и s2). При этом
номер периода совпадает со значением главного квантового числа внешнего
энергетического уровня.
3.8. Периодический закон и Периодическая система Д.И. Менделеева 101
Каждый из периодов (исключая первый) начинается типичным
металлом (металл щелочной группы) и заканчивается инертным газом. В
периоде с увеличением заряда ядра атомов наблюдается постепенное из-
изменение свойств от металлических к типично неметаллическим, что свя-
связано с увеличением числа электронов на внешнем энергетическом уров-
уровне. Первые три периода содержат только s- и /^-элементы. Четвертый и
последующие периоды включают в свой состав также элементы, у кото-
которых происходит заполнение d- и /-подуровней соответствующих внут-
внутренних энергетических уровней. При этом /-элементы объединяют в се-
семейства, называемые лантаноидами D/-элементы) и актиноидами
E/элементы).
В длиннопериодном варианте Периодической системы отражается
вся последовательность элементов в каждом периоде. В короткопериод-
ном и полудлинном вариантах лантаноиды и актиноиды вынесены за ее
пределы.
В вертикальных колонках, называемых группами, объединены эле-
элементы, имеющие сходное электронное строение внешнего энергетиче-
энергетического уровня. В короткопериодном варианте таблицы всего 8 групп, каж-
каждая из которых состоит из главной и побочной подгрупп. У элементов
главной подгруппы последними заполняются s- и р-подуровни внешних
энергетических уровней. У элементов побочных подгрупп происходит
заполнение внутренних (п - 1) d- и (п - 2) /подуровней при наличии на
внешнем энергетическом уровне («^-подуровень) одного или двух
электронов.
Элементы-аналоги (т.е. элементы, расположенные в одной подгруппе)
имеют одинаковое строение внешних электронных оболочек атомов при
различных значениях главного квантового числа и поэтому проявляют
сходные химические свойства.
Таким образом, при последовательном увеличении зарядов атомных
ядер периодически повторяется конфигурация электронных оболочек и, как
следствие, периодически повторяются химические свойства элементов. В
этом заключается физический смысл периодического закона.
Элементы главных и побочных подгрупп различаются своими хими-
химическими свойствами, однако им присуще и общее, что объединяет их в одну
группу, — номер группы. Он, как правило, указывает на число электронов,
которые могут участвовать в образовании химических связей (валентные
электроны). В этом состоит физический смысл номера группы.
Основное отличие между элементами главных и побочных подгрупп
состоит в том, что у элементов главных подгрупп валентными являются
электроны внешнего энергетического уровня, а у элементов побочных под-
подгрупп — и электроны предпоследних уровней.
102
3. Строение атомов
3.9. Периодическое изменение некоторых свойств
атомов химических элементов
Наиболее ярко периодичность свойств элементов выражена в элек-
электронной структуре атомов. Периодически изменяются и свойства атомов
элементов, определяемые их электронным строением. К важнейшим харак-
характеристикам атомов относятся их размеры, энергия ионизации, сродство к
электрону, электроотрицательность. В этом параграфе рассмотрено перио-
периодическое изменение лишь этих свойств атомов.
Атомные радиусы. Атомы не имеют строго определенных границ.
Если исходить из квантово-механических представлений, то за радиус атома
можно принять теоретически рассчитанное расстояние от ядра до максиму-
максимума радиальной функции распределения вероятности. Так определяется ор-
орбитальный радиус атома.
Орбитальные радиусы атомов элементов изменяются периодически
(рис. 3.12):
1) в периодах по мере роста заряда ядер орбитальные радиусы атомов
уменьшаются. Это связано с тем, что при одинаковом числе энергетических
уровней в периоде возрастает заряд ядра, а следовательно, и притяжение
электронов к ядру;
2) в группах с ростом заряда ядер орбитальные радиусы атомов уве-
увеличиваются. При этом в главных подгруппах (группах А) такое увеличение
происходит в большей степени, чем в побочных подгруппах (группах В).
Это связано с увеличением числа энергетических уровней и экранирующим
действием внутренних электронов.
Иногда наблюдаются отклонения от монотонной зависимости, что
объясняется особенностями электронной структуры атомов.
Энергетические характеристи-
характеристики атомов. Для химии особый интерес
представляет энергетическое состоя-
состояние электронов внешних уровней, так
как именно они ответственны за обра-
образование химических связей, определя-
определяют прочность этих связей. Количест-
Количественные характеристики необходимы
для оценки реакционной способности
атомов.
Рис. 3.12. Зависимость орбитальных ЭнеРгия ™низацш Е, - мини-
радиусов г атомов от порядкового мальная энергия, которую требуется
номера Z элементов для первых трех затратить на то, чтобы удалить данный
периодов электрон с АО невозбужденного атома
г, пм
200 ¦
3.9. Периодическое изменение свойств атомов химических элементов 1 03
Рис. 3.13. Зависимость первой энер-
энергии ионизации Ej атомов элементов
первых трех периодов от Z
на бесконечно большое расстояние от ?/>мдж/моль
ядра без сообщения ему кинетической
энергии. Энергия ионизации соответст-
соответствует следующему процессу:
Э + Ej -» Э+ + е,
где Ej — в кДж/моль. Энергия иониза-
ионизации количественно характеризует спо-
способность атома удерживать электроны,
что является важной характеристикой
его химической активности. Для много-
многоэлектронных атомов можно рассмат-
рассматривать несколько энергий ионизации,
соответствующих энергиям отрыва 1, 2, 3, ... электронов, считая от наибо-
наиболее удаленного. Периодичность изменения первой энергии ионизации от-
отчетливо видна на рис. 3.13.
Энергия ионизации возрастает в периоде по мере увеличения поряд-
порядкового номера элемента. Наименьшее ее значение имеют щелочные метал-
металлы, находящиеся в начале периода. Наибольшие значения энергии иониза-
ионизации характерны для инертных газов (Не, Ne, Ar, Кг, Хе), находящихся в кон-
конце периодов.
Отклонения от монотонной зависимости наблюдаются при переходе в
пределах периода от s-элементов (Be, Mg) к ^-элементам (В, А1) или от
d-элементов (Zn, Cd, Hg) к/?-элементам (Ga, In, Tl).
В группе элементов энергия ионизации уменьшается с повышением
порядкового номера элемента. Это обусловлено увеличением размеров ато-
атомов и экранированием внешних электронов внутренними.
Для характеристики способности атома отдавать электроны иногда
используют понятие «потенциал ионизации». Под потенциалом ионизации
понимают разность потенциалов, под воздействием которой электрон при-
приобретает энергию, соответствующую энергии ионизации, и измеряют в
вольтах. Численное значение потенциала ионизации в вольтах равно энер-
энергии ионизации в электронвольтах на атом.
Сродство к электрону атома ЕА — энергия, которая выделяется (или
затрачивается) при присоединении к нейтральному атому электрона с обра-
образованием отрицательного иона:
Э + е -> Э" + ЕА,
где ЕА — в кДж/моль.
Сродство к электрону считают положительным, если присоединение
электрона сопровождается выделением энергии (ЕА > 0). Если для присое-
104
3. Строение атомов
Рис. 3.14. Зависимость сродства к электрону атома ЕА элементов пер-
первых трех периодов от Z
динения электрона нужно затратить энергию, то сродство к электрону счи-
считается отрицательным (ЕА < 0). Сродство к электрону зависит от электрон-
электронной структуры атома. Наибольшим сродством к электрону обладают эле-
элементы подгруппы VII А (галогены: F, С1, Вг, I). У большинства металлов и
благородных газов сродство к электрону невелико или даже отрицательно.
Наименьшие значения сродства к электрону у атомов с заполненными и на-
наполовину заполненными s- и ^-подуровнями (см. табл. П.2.1). Зависимость
сродства к электрону от порядкового номера элемента приведена на рис. 3.14.
В подгруппах сверху вниз сродство к электрону атомов уменьшает-
уменьшается, но не всегда монотонно. Это связано с особенностями электронной
структуры атомов. Например, при переходе от фтора (ЕА = 328 кДж/моль)
к хлору (ЕА = 2>49 кДж/моль) и от кислорода (ЕА =141 кДж/моль) к сере
(ЕА = 200,4 кДж/моль) наблюдается ее увеличение. Вероятно, это связано с
наличием вакантных J-орбиталей у атомов элементов третьего периода.
Вследствие экспериментальных трудностей значения сродства к электрону
известны не для всех атомов.
Электроотрицательность. Понятие электроотрицательности элемен-
элемента ввел американский физикохимик Л. Полинг. По определению Полинга
электроотрицательность — это способность атома в молекуле или слож-
сложном ионе притягивать к себе электроны, участвующие в образовании хими-
химической связи. Электроотрицательность зависит от типа соединений, валент-
валентного состояния элемента. Поэтому такая характеристика имеет условный
характер. Однако ее использование полезно для качественного объяснения
типа химических связей и свойств соединений.
Существуют несколько шкал электроотрицательности. Наиболее час-
часто используемая шкала Полинга составлена на основе термохимических
данных по энергиям связей в двухатомных молекулах. Каждому элементу
приписывается вполне определенное значение X/ электроотрицательности
(см. табл. П.2.1). В периоде электроотрицательность возрастает с увеличе-
3.9. Периодическое изменение свойств атомов химических элементов 1 05
10
15
20
25
30
35
Рис. 3.15. Шкала электроотрицательности по Полингу элементов
2-4-го периодов
нием порядкового номера элемента (слева направо), а в группе, как правило,
убывает по мере увеличения заряда ядра (сверху вниз). Таким образом, наи-
наименьшие значения электроотрицательности имеют ^-элементы I группы, а
наибольшие —р-элементы VI и VII групп (рис. 3.15).
4 ¦ Химическая связь и строение
ЕЯ вещества
Понятие химической связи является основополагающим в современной
химии, поскольку физико-химические свойства веществ определяют-
определяются их химическим и кристаллохимическим строением. Все химиче-
химические процессы обусловлены преобразованием связей между частица-
частицами (атомами, молекулами, ионами, их группировками и комплекса-
комплексами), происходящими в результате перераспределения электронной
плотности между двумя и более центрами. Изучение природы взаи-
взаимодействий атомов позволяет представить механизм образования и
строение молекул и других частиц, что, в свою очередь, дает воз-
возможность предсказать их реакционную способность, определить ус-
условия синтеза веществ с заданными свойствами.
4.1. Основные характеристики химической связи
Химическая связь — совокупность взаимодействий атомов,
приводящая к образованию устойчивых систем (молекул, комплек-
комплексов, кристаллов и др.). Химическая связь возникает, если в результате
перекрывания электронных облаков атомов происходит уменьшение
полной энергии системы. Мерой прочности химической связи между
атомами А и В служит энергия связи ЕА-в, которая определяется ра-
работой, необходимой для разрушения данной связи. Так, для атомиза-
ции 1 моль газообразного водорода требуется затратить энергию Е =
= 436 кДж, следовательно, образование молекулы Н2 из атомов
н + н = н2
сопровождается выделением такого же количества энергии, т.е.
?н-н = 436 кДж/моль.
Для молекул типа АВ„, имеющих равноценные связи, напри-
например, СН4, NH3 и Н2О, используют понятие средней энергии связи
ЕА_В. Поскольку для атомизации 1 моль метана
СН4 = С + 4Н
требуется затратить Е = 1647 кДж, то ЕС_И = 1647 :4 » 412 кДж/моль.
Если в состав молекул входит большее число элементов, то каждому
типу связи в первом приближении приписывают значения, опреде-
определенные для молекул с однотипными связями. Значения энергии свя-
связи для органических молекул определены точнее, чем для неоргани-
неорганических, так как большее разнообразие органических молекул харак-
характеризуется меньшим числом типов связей.
4.1. Основные характеристики химической связи 107
Важной характеристикой связи является ее длина, т.е. расстояние
между центрами атомов А и В в молекуле. Энергия и длина связей зависят
от характера распределения электронной плотности между атомами. На
распределение электронной плотности влияет пространственная направ-
направленность химической связи. Если двухатомные молекулы всегда линейны,
то формы многоатомных молекул могут быть различны. Так, трехатомная
молекула может иметь линейную или угловую форму. Угол между вообра-
воображаемыми линиями, которые можно провести через центры связанных ато-
атомов, называется валентным.
Распределение электронной плотности между атомами зависит также
от размеров атомов и их электроотрицательности — способности атомов к
оттягиванию на себя электронной плотности партнеров. В гомоатомных
(т.е. состоящих из одинаковых атомов) молекулах электронная плотность
распределена равномерно между атомами. В гетероатомных (состоящих из
атомов разных элементов) молекулах электронная плотность смещается в
том направлении, которое способствует уменьшению энергии системы. На-
Например, в молекуле АВ электронная плотность смещена в сторону атома В.
Следовательно, выделившаяся при этом энергия
где ЕА(В) — сродство к электрону атома В; ?/(А) — энергия ионизации ато-
атома А, превышает энергию, выделяющуюся при смещении электронной
плотности в обратном направлении:
Е2 = ЕА(А)-Е1{В\
где ЕА(А) — сродство к электрону атома А; ?/(В) — энергия ионизации ато-
атома В. Это означает, что
Р. Малликен предложил электроотрицательность % — способность
атома элемента притягивать электронную плотность при образовании хими-
химической связи — рассчитывать как среднее арифметическое энергии иониза-
ионизации и сродства к электрону
EA). D.1)
Очевидно, что электронная плотность повышается вблизи ядра атома
более электроотрицательного элемента. Связь в гетероатомных молекулах
всегда в той или иной мере полярна, так как электронная плотность в них
распределена несимметрично.
Для оценки степени полярности связи Л. Полинг A932) использовал
разность между энергией связи молекулы АВ и средним геометрическим
энергий связи молекул А2 и В2:
108 4. Химическая связь и строение вещества
AEB_B, D.2)
где ?а-в, ?а-а и ЕВ-в — энергии связи в молекулах АВ, А2 и В2 соответст-
соответственно. Чем существеннее АЕ отклоняется от нуля, тем более полярной явля-
является связь между атомами А и В. Величина АЕ зависит от разности (хв - Ха)
электроотрицательностей элементов, с увеличением которой растет степень
полярности связи
АЕ=С(хв-Ха)\ D.3)
где С = const.
Как указывалось (см. гл. 3), электроотрицательность как количествен-
количественная мера степени полярности связи — величина приближенная. Строго го-
говоря, элементам нельзя приписать постоянную электроотрицательность.
Она зависит от типа взаимодействия атомов данного элемента в конкретной
молекуле. Поэтому кроме шкалы абсолютной электроотрицательности,
рассчитываемой по формуле D.1), существует много других шкал, в основу
которых положены различные свойства веществ. Например, в широко рас-
распространенной системе Полинга D.3) значения электроотрицательностей
элементов подобраны таким образом, что их разность соответствует АЕ в
электронвольтах при С = 1. А. Оллред и Э. Рочоу рассчитывают электроот-
электроотрицательность, исходя из значений эффективного заряда ядра Z* (Z* — заряд
ядра относительно электронов валентной оболочки, создаваемый протонами
и уменьшенный в результате экранирования остальными электронами) и
ковалентного радиуса атома г:
Формула Шомакера—Стивенсона объединяет длину связи гА_в с ко-
валентными радиусами атомов гА, Пз и их электроотрицательностями Ха, Хв-
>а-в = гА + гв - 0,091 Ха - Хв I •
Положение некоторых элементов относительно их места в Периоди-
Периодической системе и значений их электроотрицательностей по Малликену и
Полингу показано на рис. 4.1 и 4.2. Несмотря на различия в подходах к рас-
расчету электроотрицательности, расположение элементов в порядке возраста-
возрастания х в целом одинаково: наиболее электроотрицательные элементы распо-
расположены в верхнем правом углу таблицы Д.И. Менделеева (максимальным
значением электроотрицательности обладает фтор F), а элементы, имеющие
низкие значения электроотрицательности, — в левом нижнем углу. Очевид-
Очевидно, что наибольшая полярность связи наблюдается в галогенидах щелочных
4.1. Основные характеристики химической связи
109
Группы
0 2 4 6 8 10 X
Рис. 4.1. Шкала абсолютной электроотрицательности s- и/7-элементов
Группы
0 1 2 3 X
Рис. 4.2. Шкала электроотрицательности по Полинту s- и/?-элементов
металлов, т.е. в соединениях элементов VIIA и IA групп. Таким образом,
при оценке полярности связи важны не значения электроотрицательностей
элементов, а их разности, сопоставимые в различных шкалах.
110 4. Химическая связь и строение вещества
Вследствие неравномерности распределения электронной плотности в
гетероатомной молекуле электрические центры положительных зарядов
ядер и отрицательных зарядов электронов не совпадают, и в молекуле воз-
возникает постоянный электрический диполь — система двух равных по вели-
величине и противоположных по знаку зарядов: +д и -q.
Дипольный момент связи Д — вектор, условно направленный в сто-
сторону более электроотрицательного атома, его величину можно рассчитать
по формуле
Д = qh D.4)
где q = \+q\ = \-q\, I — радиус-вектор, условно направленный от центра тя-
тяжести положительного заряда (+q) к центру тяжести отрицательного заряда
(-q). Модуль радиус-вектора соответствует длине диполя /, которую не сле-
следует отождествлять с длиной связи, так как центры тяжести зарядов не сов-
совпадают с центрами ядер. Дипольный момент обычно выражают в дебаях
A Д = 3,33564- 1(Г30Кл.м).
Для многоатомных молекул различают понятия электрического мо-
момента диполя отдельных связей и дипольного момента молекулы в целом,
который равен результирующему вектору, определенному по правилу па-
параллелограмма. Так, дипольный момент молекулы СО цсо = 0,11 Д, а ли-
линейная молекула СО2 неполярна (цсо = 0), поскольку дипольные моменты
связей С = О равны по величине и противоположно направлены:
М-с=о И-с=о
Для молекулы НгО экспериментально определено, что |iH о = 1,86 Д, а
цо_н=1,65 Д. Согласно правилу сложения векторов, валентный угол должен
составлять 104,5°, что соответствует угловому строению молекулы:
Н Н
Следовательно, дипольный момент позволяет определить не только
меру полярности химической связи, но и сделать выводы о геометрической
структуре молекул.
Изучение строения вещества возможно как физическими, так и хими-
химическими методами. A.M. Бутлеров в 1861 г. на основании исследований хи-
химических превращений органических молекул выдвинул теорию химиче-
химического строения, сущность которой заключается в следующих положениях:
4.2. Квантово-механические представления о природе химической связи \\\
атомы в молекуле соединяются между собой в определенном порядке;
соединение атомов происходит в соответствии с их валентностью;
валентность атома определяется числом образуемых им химических
связей;
свойства соединений зависят от природы составляющих атомов и от
их взаимного расположения.
Теория химического строения показала, что по свойствам веществ
можно выявить их строение, а по строению молекул — предсказать их хи-
химическое поведение. Например, как уже упоминалось в гл. 1, одной и той же
эмпирической формуле С2Н6О соответствуют два изомера {изомеры —
одинаковые по составу, но различные по строению вещества): этанол
СН3—СН2—ОН и диметиловый эфир СН3—О—СН3. Химическое строение
определяет различные свойства этих соединений. Так, этанол при обычных
условиях реагирует с металлическим натрием и кислотами, а диметиловый
эфир нет.
В теории Бутлерова впервые молекула была представлена как система,
в которой существует строгий порядок химической связи между атомами.
Однако установить физические причины возникновения химической связи
стало возможным лишь после создания квантовой механики, описывающей
законы движения микрочастиц.
4.2. Квантово-механические представления
о природе химической связи.
Основные методы описания ковалентной связи
Через год после опубликования уравнения Шрёдингера появилась ра-
работа, положившая начало применению квантовой механики для решения
химических проблем. В. Гейтлер и Ф. Лондон в 1927 г. представили кванто-
во-механический расчет молекулы Нг, основанный на определенных допу-
допущениях. При рассмотрении механизма образования молекулы они опира-
опирались на теорию парно-электронной связи Г. Льюиса A916).
Льюис считал, что при взаимодействии атомы стремятся приобрести
конфигурацию внешней электронной оболочки инертного газа, находящего-
находящегося в одном периоде с данным элементом. Это достигается за счет общей
электронной пары, на образование которой каждый из атомов отдает по од-
одному электрону. Например, образование молекулы водорода происходит за
счет неспаренных электронов в атомах Н (Is1)
Н + Н = Н:Н
112
4. Химическая связь и строение вещества
в результате чего каждый из атомов
приобретает электронную конфигура-
конфигурацию гелия Не, и возникает одинарная
ковалентная связь. При образовании
молекулы азота N2 возникновение трой-
тройной ковалентной связи в соответствии с
методом Льюиса можно представить
следующим образом:
Каждый атом азота N (Is22s22p3)
Рис. 4.3. Схема молекулы Н2
имеет три неспаренных электрона, которые и образуют три парно-электрон-
парно-электронные связи, и в результате электронная конфигурация атомов соответствует
неону Ne. Такой способ осуществления ковалентной связи, когда каждый из
атомов отдает по одному электрону для образования общей электронной
пары, называют обменным.
Гейтлер и Лондон также исходили из предположения, что ковалентная
связь осуществляется электронной парой, находящейся в общем владении
двух атомов. Молекула Н2 представляет собой систему из двух протонов и
двух электронов (рис. 4.3), потенциальная энергия которой определяется их
электростатическим взаимодействием:
?/ = ¦
1
4тге0
rA-B
2
ГВ1
D.5)
где 80 — электрическая постоянная (см. Приложения). Для построения вол-
волновой функции электронов в молекуле Н2 Гейтлер и Лондон приняли за ос-
основу собственные волновые функции V|/A(l) и \|/вB) электронов
изолированных атомов НА и Нв соответственно. Каждая из этих функций,
согласно решению уравнения Шрёдингера для атома водорода,
определяется соотношением
D.6)
где ao — радиус первой боровской орбиты; г — расстояние электрона от
ядра.
Вероятность одновременного нахождения электрона I у ядра А и
электрона 2 у ядра В, согласно теории вероятности, равна произведению
частных вероятностей \|/А2A)\|/в2B), поэтому волновую функцию системы
можно выразить уравнением ц/ = \|/АA)\|/вB).
4.2. Квантово-механические представления о природе химической связи 113
Электроны в молекуле абсолютно неразличимы, поэтому можно счи-
считать, что электрон 1 движется в поле ядра В, а электрон 2 — в поле ядра А.
Тогда волновую функцию можно записать в виде V|/ = i|/aB)\|/bA). Оба эти вы-
выражения равнозначны, но являются плохим приближением к действительному
виду волновой функции электронов молекулы Н2, поскольку движение элек-
электронов в молекуле отличается от их движения в свободных атомах.
Гейтлер и Лондон предложили волновую функцию молекулы Н2 счи-
считать линейной комбинацией равнозначных функций:
V = VaA)VbB) ± V|/aB)v|/bA). D.7)
При этом возможны два варианта представления волновой функции:
симметричная
щ = \|/A(l)v|/BB) + i|/AB)yB(l),
в которой перестановка индексов 1, 2, А, В не ведет к изменению знака
функции, и
антисимметричная
\|/_ = \|/АA)\|/вB) - \|/АB)\|/вA),
перестановка индексов которой изменяет знак i|/.
Плотность электронного облака определяется квадратом волновой
функции, поэтому симметричной волновой функции отвечает увеличение
электронной плотности между ядрами
i|/+2 = i|/A2(l)WB) + 4>a2B)WA) + 2VaA)VbB)vaB)VbA),
что соответствует образованию химической связи, а для антисимметричной
волновой функции наблюдается уменьшение электронной плотности между
ядрами:
V-2 = X|/A2(l)v|/B2B) + i|/2W
и, следовательно, химическая связь не
возникает.
Согласно принципу Паули, элек-
электроны в молекуле Н2, состояние кото-
которых выражается симметричной функци-
функцией \|/+, должны иметь различные спино-
спиновые квантовые числа ms, т.е. антипарал-
антипараллельные спины, и наоборот, антисим-
антисимметричная функция соответствует тако-
такому состоянию, когда электроны имеют
параллельные спины (рис. 4.4).
а б
Рис. 4.4. Электронная плотность ато-
атомов водорода:
а — антипараллельные спины (атомы со-
соединяются в молекулу); б — параллельные
спины (атомы отталкиваются)
114
4. Химическая связь и строение вещества
\
Рис. 4.5. Зависимость энергии Е мо-
молекулы водорода от межъядерного
расстояния гА_в:
1 — экспериментальная кривая; 2 — кри-
кривая, рассчитанная с помощью сим-
симметричной волновой функции; 3 — кри-
кривая, рассчитанная с помощью антисим-
антисимметричной волновой функции
Гейтлер и Лондон рассчитали
энергию Е электронов для различных
расстояний между ядрами гА_в, исполь-
используя для решения уравнения Шрёдингера
соотношения D.5)-D.7). Результаты рас-
расчета представлены на рис. 4.5 пунктир-
пунктирными линиями. Линия 2 соответствует
симметричной волновой функции, а ли-
линия 3 — антисимметричной. Экспери-
Экспериментальным данным, основанным на
спектральных исследованиях, соответст-
соответствует сплошная линия 7, участки которой
характеризуют притяжение атомов водо-
водорода (ab) и их отталкивание (be). Точка Ъ
отвечает устойчивому состоянию систе-
системы, для которого энергия связи атомов
?о(опыт) = 4,505 эВ, а расстояние между
атомами г0(ОПыт) = 74,142 пм. Вычислен-
Вычисленное по методу Гейтлера и Лондона зна-
значение равновесного расстояния между
атомами гО(Расчет)= 86,9 пм, а энергия молекулы Н2 ?Ь (расчет) = 3,14 эВ. Расхо-
Расхождение опытных и расчетных данных можно считать небольшим, принимая
во внимание приближенный характер волновых функций, составленных из
волновых функций изолированных атомов и не учитывающих взаимное от-
отталкивание электронов.
Работа Гейтлера и Лондона имеет очень большое значение. В ней до-
доказано, что уравнение Шрёдингера является фундаментальным, поскольку
справедливо не только для атома, но и для молекулы. Показано, что химиче-
химическая связь имеет электрическую природу и возникает за счет перекрывания
атомных орбиталей. При образовании химической связи электронная плот-
плотность между ядрами взаимодействующих атомов увеличивается, а энергия
всей системы понижается, т.е. образование молекулы происходит, когда
энергетически выгодно пребывание электронов в поле двух (и более) ядер.
Квантово-механический расчет молекулы водорода, выполненный
Гейтлером и Лондоном с помощью приближенной волновой функции, впо-
впоследствии уточнялся другими учеными, использовавшими более сложные
выражения \|/ и возможности современных компьютеров. Проводились тру-
трудоемкие и точные измерения энергии диссоциации Н2. В настоящее время
расхождение рассчитанного значения энергии связи в молекуле Н2 и экспе-
экспериментального составляет не более 0,001 %. Расчеты Гейтлера и Лондона
положили начало использованию приближенных квантово-механических
4.3. Метод валентных связей 115
методов для описания химической связи, два из которых — метод валент-
валентных связей и метод молекулярных орбиталей — находят наибольшее прак-
практическое применение.
4.3. Метод валентных связей
Представления Гейтлера и Лондона о механизме образования химиче-
химических связей составляют основу метода валентных связей (МВС), значи-
значительный вклад в развитие которого внесли Дж. Слейтер и Л. Полинг. Этот
метод базируется на следующих основных положениях:
каждая пара атомов в молекуле удерживается вместе при помощи од-
одной или нескольких общих электронных пар;
одинарная ковалентная связь образуется двумя электронами с антипа-
антипараллельными спинами, расположенными на валентных орбиталях связы-
связывающихся атомов;
при образовании связи происходит перекрывание волновых функций
электронов, ведущее к увеличению электронной плотности между атомами
и уменьшению общей энергии системы;
связь образуется в том направлении, при котором возможно макси-
максимальное перекрывание волновых функций;
угол между связями в молекуле соответствует углу между образую-
образующими связи электронными облаками;
из двух орбиталей атома более прочную связь образует та, которая
сильнее перекрывается орбиталью другого атома.
В данном методе валентность, как и в теории Бутлерова, рассматрива-
рассматривается как возможность атома образовывать химические связи с другими ато-
атомами. Валентность в определенной мере зависит от числа неспаренных
электронов, которые могут находиться на внешних (валентных) орбиталях
атома. Так, водород Н (Is1) всегда одновалентен. Для гелия Не (Is2) распари-
распаривание электронов, т.е. промотирование одного электрона на более высокий
энергетический уровень Is2 > \sl2s\ требует большой затраты энергии
A672 кДж/моль), которая не окупается выигрышем энергии при образова-
образовании связи. Поэтому Не не образует химических соединений. Литий Li
(Is22sl) одновалентен, так как переход Is22sl > 1.у12лг12р1 также требует
большой затраты энергии. Промотирование одного электрона атома берил-
бериллия Be с 2s на 2р (т.е. осуществленное в пределах одного энергетического
уровня) требует затраты энергии 259 кДж/моль, поэтому Be в отличие от Не
способен образовывать две связи с другими атомами (\s22s2 > Is22sl2pl),
энергия образования которых компенсирует энергию возбуждения электро-
электрона. Для бора В также характерно не одновалентное, а трехвалентное состоя-
116
4. Химическая связь и строение вещества
ние (Is22s22pl > Is22sl2p2). У атома углерода С в основном состоянии
имеется два неспаренных электрона, но для него характерно четырехвалент-
четырехвалентное состояние: \s22s22p2 > Is22sl2p3 или с использованием квантовых
ячеек Хунда,
К2у
t
t
K2s
t
2p
t
t
t
Но нередко валентность элемента превышает число неспаренных
электронов в его атомах. Происходит это в случае, когда один из атомов
представляет в общее пользование пару электронов, а другой — свободную
орбиталь. Первый называют донором, второй — акцептором, а механизм
образования такой связи — донорно-акцепторным; его схематически можно
представить следующим образом:
Д: + ПА = Д:А
Донорно-акцепторный механизм образования связи отличается от об-
обменного только происхождением общей электронной пары, во всем осталь-
остальном оба эти механизма тождественны. Например, молекула СО2 образуется
по обменному механизму:
О K2s
С K2s
и
t
2р
2р
и
t
1
1
t
I
1
t
О К2р
i
i
и
2s
П
а при образовании молекулы СО связь осуществляется и по обменному, и по
донорно-акцепторному механизмам:
О K2s
С K2s
п
и
2р
2р
и
1
1
т
1
1
т
В молекуле СО2 атом углерода образует четыре ковалентные связи, следова-
следовательно, он четырехвалентен, а атомы кислорода — двухвалентны, так как
каждый из них образует по две связи (О=С=О). В молекуле СО валентность
атомов углерода и кислорода одинакова и равна 3 (:С = О:).
Часто один и тот же атом может выступать как в роли донора, так и в
роли акцептора электронов. Механизм образования связи между такими
атомами называют дативным. Например, у атома хлора Cl (KL3s23p53d°) на
4.3. Метод валентных связей
117
внешнем энергетическом уровне, помимо одного неспаренного Зр-элек-
трона, имеются пары электронов и свободные З^-орбитали, поэтому в моле-
молекуле СЬ связь осуществляется и за счет обменного, и за счет дативного ме-
механизмов:
Cl
Cl 3d
3s
n
1
3p
U
1
U
1
ъР
i
1
t
3d
1
u
1
u
3s
1
u
Образование кратной (двойной, тройной и т.д.) связи упрочняет мо-
молекулу, поэтому энергия, необходимая для разрыва связи молекулы С12,
больше энергии связи молекулы F2, атомы которой (F — K2s22p5) не имеют
свободных орбиталей на внешнем энергетическом уровне и связаны оди-
одинарной обменной связью СЕЪ-а = 243 кДж/моль, ?F-f = 159 кДж/моль).
Валентные возможности атомов ограничены, они зависят от их элек-
электронного строения и определяются энергетической выгодой образования
химических связей. Поэтому важным свойством ковалентной связи является
ее насыщаемость.
Геометрическая форма молекул зависит от направленности химиче-
химической связи. Атомы, у которых валентные электроны находятся на s-AO, спо-
способны образовывать одинаково прочные связи в любых направлениях. Общее
электронное облако в таких случаях сосредоточено вдоль линии связи атомов
(а-связь). Для р-АО максимальное перекрывание возможно и по линии, со-
соединяющей атомы (а-связь), и по обе стороны от нее (я-связь), для d-op-
биталей возможно и а-перекрывание, и тс-перекрывание, и 5-связь (рис. 4.6).
Объяснение направленности химических связей основано на учете
формы различных орбиталей. Так, в молекуле воды Н2О атом кислорода
р-АО р-АО
s-AO s-AO
р-АО р-АО
а-связь
а-связь
s-AO р-АО
d-AO d-AO
71-СВЯЗЬ
d-AO d-AO
а-связь
d.A0
d-AO
n-связь
5-связь
Рис. 4.6. Перекрывание АО
118 4. Химическая связь и строение вещества
имеет два неспаренных электрона, которые занимают^ две р-орбитали, рас-
]). При образо-
положенные под углом 90° (K2s22/?4 или K2s |it| 2p
вании связей с двумя атомами водорода И (Is или 1
t
) угол между двумя
общими электронными парами должен быть близок к 90°. Однако валент-
валентный угол в молекуле Н2О, как отмечалось ранее, составляет 104,5°. Увели-
Увеличение валентного угла можно объяснить двумя причинами.
1. Связь О-Н является полярной ковалентной связью (рис. 4.1, 4.2 и
табл. П. 1.4), электронная плотность которой смещена к атому кислорода.
Вследствие этого у атома водорода появляется некоторый положительный за-
заряд. Отталкивание этих зарядов приводит к увеличению угла между связями.
2. Электроны, принадлежащие двум связям О-Н, имеют одинаково
направленные спины, что также приводит к отталкиванию связей.
Показательно, что уменьшение полярности связи приводит к умень-
уменьшению валентного угла в однотипных соединениях. Например, угол между
связями в изоэлектронных молекулах (молекулах, содержащих одинаковое
число электронных пар в валентной оболочке) H2S и H2Se составляет 92,2° и
91,0° соответственно, поскольку с увеличением размеров атомов уменьша-
уменьшается их электроотрицательность (рис. 4.1. и 4.2 и табл. П. 1.4).
Аналогично можно объяснить экспериментальное значение валентно-
валентного угла в молекуле аммиака NH3, равное 107,3°. В соответствии с электрон-
электронным строением атома азота N (K2s22p3 или K2s IT
1 связи меж-
между атомами N-H, обусловленные перекрыванием /?-АО атома азота с s-AO
атомов водорода, должны расположиться под углами, близкими к 90°. Одна-
Однако отталкивание положительных зарядов, появившихся в области атомов
водорода вследствие смещения общей электронной плотности к более элек-
электроотрицательному атому N, приводит к увеличению валентного угла. В
соединениях РН3, AsH3 и SbH3 углы между связями составляют 93,3°, 91,8°
и 91,3° соответственно, поскольку связь в этих изоэлектронных молекулах
менее полярна, чем в молекуле NH3.
Часто атомы формируют связи за счет электронов различных состоя-
состояний. Так, у бериллия Be два валентных электрона: 2sl2p\ у бора В — три:
2sl2pl2p\ а у углерода С — четыре: 2sl2pl2pl2pl. Хотя формы «s-AO и/?-АО
различны, установлено, что обе связи в молекуле ВеС12 равноценны и рас-
расположены под углом 180°. Также и в молекуле ВС13 связи равноценны и
расположены под углами 120°. В молекуле ССЦ равноценные связи распо-
расположены под углом 109°28\
Для объяснения подобных фактов в рамках МВС используют понятие
гибридизации валентных орбиталей: при образовании химических связей
исходные атомные орбитали смешиваются, взаимно изменяются, образуя
равноценные гибридные орбитали (ГО), которые отличаются от АО значи-
значительным увеличением электронной плотности в определенном направлении
4.3. Метод валентных связей 119
180°
-СЮ-
5-АО+р-АО sp-YO Cl Be C1
Рис. 4.7. Образование sp-ГО молекулы ВеС12
пространства. Волновая функция гибридной орбитали составляется из вол-
волновых функций валентных электронов, умноженных на соответствующие
коэффициенты. Так, при гибридизации s-AO и одной р-АО образуются две
sp-гибридные орбитали, волновые функции которых различаются значения-
значениями коэффициентов а и Ь:
\\rsp = a\\fs+b\\fp.
Математический расчет показывает, что sp-ГО являются диагональ-
диагональными орбиталями, т.е. угол между ними составляет 180° (рис. 4.7):
(
поэтому молекула ВеСЬ имеет линейную структуру.
Комбинация s-AO и двух р-АО приводит к образованию трех триго-
налъных sp2-YO, расположенных под углом 120° (рис. 4.8), поэтому в моле-
молекуле ВС13 связи направлены к вершинам равностороннего треугольника.
При гибридизации s-AO и трех р-АО образуются четыре тетраго-
тетрагональные 5^3-ГО, направленные к вершинам тетраэдра (рис. 4.9), поэтому ва-
валентный угол в молекуле ССЦ соответствует 109°28\
В атомах с rf-орбиталями гибридизация приводит к образованию более
сложных конфигураций электронных облаков. Геометрические модели ГО в
зависимости от типа гибридизации атома и числа ГО представлены ниже:
Прямая линия sp 2
Треугольник sp2, dp2, sd2 3
Тригональная пирамида pd2 3
Тетраэдр sp3 4
Квадрат dsp2 4
Тригональная бипирамида sp3d 2 5
Квадратная пирамида sp dxi_yi 5
Октаэдр sp3d2 6
120
4. Химическая связь и строение вещества
s-AO+/>-AO+/?-AO
Рис. 4.8. Образование з/?2-ГО молекулы ВС13
s-AO+/>-AO+/>-AO+/>-AO sp2-TO
Рис. 4.9. Образование sp'-YO молекулы СС14
Экспериментальные данные показывают, что существуют молекулы,
например, рассмотренные ранее ВеСЬ, ВСЬ и СС14, валентные углы которых
соответствуют правильным геометрическим формам. Однако чаще встреча-
встречаются молекулы с несколько иными, чем можно было бы ожидать, валент-
валентными углами. Рассмотрим образование молекул метана СН4, аммиака NH3 и
воды Н2О с позиций гибридизации валентных орбиталей центрального атома.
Центральный атом углерода в молекуле СН4 окружен четырьмя ато-
атомами водорода (периферические атомы), каждый из которых имеет по од-
одному неспаренному электрону (Is1). Следовательно, для образования хими-
химической связи по обменному механизму атом углерода должен иметь четыре
неспаренных электрона, что возможно при промотировании валентных
электронов С: K2s22p2—*—> K2s]2p3 (K2s [Т| 2р |Т] [Т] |Т|). В молекуле
СН4, как и в молекуле СС14, валентные орбитали центрального атома угле-
углерода находятся в состоянии з/?3-гибридизации, поэтому образование связей с
атомами водорода (перекрывание валентных орбиталей) происходит в на-
направлении к вершинам тетраэдра, в центре которого расположен атом углерода
(рис. 4.10). Гибридные орбитали атома углерода в молекуле метана равно-
равноценные (эквивалентные), соответственно валентный угол СН4 — тетраэдри-
ческий, т.е. равен 109°28\
4.3. Метод валентных связей
121
Н
NH,
Н
Рис. 4.10. Геометрические конфигурации молекул СН4, NH3, H2O
Центральный атом азота в молекуле NH3 окружен тремя атомами во-
водорода. Для образования химической связи по обменному механизму атом
азота должен иметь три неспаренных электоона, что соответствует основ-
основному состоянию N: K2s22/?3 (K2s [it] 2p _f_ _T_ Q_ ). При образовании хими-
химической связи валентные орбитали центрального атома образуют четыре sp3-ГО,
на которые приходится пять электронов. Значит, из четырех электронных
пар в молекуле NH3 три пары являются связывающими, одна — несвязы-
вающей (неподеленной), т.е. яр3-ГО — неэквивалентными. Это ведет к ис-
искажению формы тетраэдра и изменению валентных углов. Как уже упоми-
упоминалось, молекула NH3 представляет собой тригональную пирамиду, а ва-
валентный угол H-N-H A07,3°) меньше тетраэдрического (рис. 4.10).
Полагают, что в молекуле_НЬО валентные^орбитали центрального ато-
атома кислорода (K2s22/?4 или
T
) также находятся в состоя-
состоянии sp -гибридизации. На четыре sp -ГО здесь приходится шесть электронов,
таким образом, в молекуле имеются две связывающие и две несвязывающие
электронные пары. Молекула Н2О имеет угловую форму (см. рис. 4.10), а ва-
валентный угол равен 104,5°, что можно объяснить образованием неэквива-
неэквивалентных гибридных орбиталей и искажением формы тетраэдра.
Образование кратных связей по обменному механизму происходит,
если в основном состоянии атома число неспаренных электронов больше,
чем необходимо для образования а-связей. Так, например, в молекуле эти-
этилена С2Н4, в которой каждый атом углерода имеет три а-связи (с двумя ато-
атомами водорода Н и соседним атомом углерода С), наибольшее перекрыва-
перекрывание орбиталей возникает, если одна .s-AO и две р-АО центрального атома С
образуют три sp2-TO, а третья р-АО не подвергается гибридизации. Гибрид-
Гибридные орбитали образуют три а-связи, лежащие в одной плоскости под углами
120°, а негибридные р-АО — я-связь, лежащую в плоскости, перпендику-
перпендикулярной плоскости а-связей (рис. 4.11). Такое расположение орбиталей соот-
122
4. Химическая связь и строение вещества
Рис. 4.11. Геометрические конфигурации молекул С2Н4 и С2Н2
ветствует их максимальному удалению и минимальной энергии молекулы.
В молекуле С2Н4 между атомами углерода формируется двойная связь за
счет одной а-связи и одной я-связи.
В молекуле ацетилена С2Н2 только две орбитали атома углерода обра-
образуют 5/?-ГО, участвующие в образовании а-связей. Эти sp-ГО расположены
под углом 180° друг к другу, а под углами 90° к ним расположены две не-
негибридные р-АО, участвующие в образовании тг-связей (см. рис. 4.11). В
итоге одна а-связь и две тс-связи формируют тройную связь между атомами
углерода в молекуле С2Н2.
Образование комплексных соединений (см. гл. 1) можно рассматривать с
позиций донорно-акцепторного механизма взаимодействия комплексообра-
зователя и лигандов: комплексообразователь выступает в роли акцептора, а
лиганд (анион или молекула, имеющая неподеленные электронные пары,
например, NH3 или Н2О) — в роли донора электронов. Геометрическая фор-
форма комплексных частиц определяется типом гибридизации АО комплексо-
образователя. Например, комплексные ионы [Cu(NH3J]+, [Zn(NH3L]2+,
[Co(NH3N]3+ имеют одинаковые лиганды — молекулу NH3, строение кото-
которой характеризуется наличием одной неподеленной электронной пары (см.
рис. 4.10). В образовании комплекса [Cu(NH3J]+участвуют две свободные
АО иона Си+: одна 4s-AO и одна 4р-АО, которые образуют две sp-ГО; соот-
соответственно геометрическая конфигурация комплекса линейная. В образова-
образовании комплекса [Zn(NH3L]2+ участвуют четыре свободные АО иона Zn2+: од-
одна 4.У-АО и три 4р-АО, которые образуют четыре 5^3-ГО; соответственно,
геометрическая конфигурация комплекса тетраэдрическая. В образовании
комплекса [Co(NH3N]3+ участвуют шесть свободных АО иона Со3+: одна
4s-AO, три 4/?-АО и две 3t/-AO, которые образуют шесть cfsp3-TO; соответ-
соответственно геометрическая конфигурация комплекса октаэдрическая (табл. 4.1).
В комплексных соединениях возможно образование и кратных связей.
Например, при координации монооксида углерода СО атомом железа Fe,
образование двойной связи происходит за счет дативного механизма, т.е.
4.3. Метод валентных связей
123
передачи, с одной стороны, пары электронов молекулы СО на вакантную
d-AO Fe, а с другой стороны, пары электронов с заполненной d-AO Fe на
свободную орбиталь лиганда.
Таблица 4.1
Геометрическая конфигурация некоторых комплексных ионов
Комплекс
Комплексо-
образователь
АО комплексообразователя,
участвующие в образовании
химической связи
Тип
ГО
Геометрическая
конфигурация
комплекса
[Cu(NH3J]+
[Zn(NH3Lf
[Co(NH3N]3+
Cu+: KL3s23p63ct°
Zn2+:KL35V3</10
Co3+: KL3s23p63d6
sp
sp>
cfsp*
Линейная
Тетраэдрическая
Октаэдрическая
Примечание. Ц — свободная АО комплексообразователя и 4^Т — неподелен-
ная электронная пара лиганда, участвующая в образовании донорно-акцепторной
связи |If|.
Отметим, что гибридизацию валентных орбиталей нельзя считать ре-
реальным физическим явлением, она представляет собой формальный матема-
математический прием, позволяющий предсказывать и объяснять структуру соеди-
соединений. Этот прием основан на принципе максимального перекрывания элек-
электронных облаков и содержит условия устойчивой гибридизации орбиталей: в
гибридизации участвуют орбитали с близкими значениями энергии, т.е. s- и
р-АО внешнего и d-AO внешнего и предвнешнего энергетических уровней и с
достаточно высокой электронной плотностью. Для выполнения этого прин-
принципа необходимо также, чтобы ГО были ориентированы и вытянуты в про-
пространстве так, чтобы энергия их отталкивания была минимальна, а перекры-
перекрывание с орбиталями соседних атомов наиболее полно. Учитывается, что ГО
вследствие их асимметрии в образовании 7г-связей участвовать не могут.
Существование большого числа молекул, геометрическая форма кото-
которых точно описывается в рамках представлений о гибридных орбиталях,
свидетельствует о важном теоретическом значении такого подхода. В то же
время известны довольно простые молекулы, которые невозможно описать с
помощью этого метода. Например, трудно интерпретировать с позиций гиб-
гибридизации АО уменьшение валентного угла в ряду изоэлектронных молекул
Н2О( 109,5°) — H2S(92,2°) — H2Se(91,0°) — Н2Те(89,5°). Предположение, что
с увеличением порядкового номера уменьшается гибридный характер ва-
валентных орбиталей, входит в противоречие с тем фактом, что энергии 3^-АО
и 3/7-АО более близки, чем энергии 2s-АО и 2/?-АО, поэтому гибридизация в
первую очередь должна осуществляться у более тяжелых элементов.
124 4. Химическая связь и строение вещества
Другой подход к описанию геометрической формы молекул был пред-
предложен Н. Сиджвиком и Г. Пауэллом A940 г.) и разработан Р. Гиллеспи. В
его основе лежит теория отталкивания электронных пар валентных орби-
талей (ОЭПВО), базирующаяся на следующих положениях:
конфигурация связей многовалентного атома обусловливается числом
связывающих и несвязывающих электронных пар в валентной оболочке
центрального атома;
ориентация облаков электронных пар валентных орбиталей определя-
определяется максимальным взаимным отталкиванием заполняющих их электронов.
Теория ОЭПВО построена на следующих основных допущениях: 1) не-
неразличимости электронов; 2) действия кулоновских сил; 3) действия сил от-
отталкивания Паули. Силы отталкивания возникают вследствие действия
принципа Паули, согласно которому электроны с одинаковым спином
должны находиться на возможно большем расстоянии друг от друга, в то
время как электроны с противоположными спинами притягиваются друг к
другу. Поэтому наиболее вероятное пространственное расположение элек-
электронных пар в молекуле соответствует наиболее вероятному расположению
электронов с одним и тем же спином в атоме.
Определение расположения центров облаков отталкивающихся элек-
электронных пар эквивалентно решению задачи размещения нескольких частиц
на поверхности сферы при их взаимном максимальном удалении. Каждая
конфигурация данного числа электронных пар может привести к несколь-
нескольким молекулярным формам в зависимости от числа связывающих и неподе-
ленных пар. Гиллеспи ввел для центрального атома обозначение А, для свя-
связывающих электронных пар обозначение X, а для несвязывающих — Ей
показал, что форма молекулы АХШЕ„, не содержащей кратные связи, будет
определяться наиболее вероятным расположением (т+п) электронных пар
(табл. 4.2).
Развитие теории ОЭПВО приводит к следующим стереохимическим
обобщениям.
Правило 1. Неподеленная (несвязывающая) электронная пара за-
занимает больший объем, чем пара электронов, участвующих в образовании
одинарной связи, поскольку находится под влиянием только одного поло-
положительно заряженного атомного остова и, следовательно, более диффузна,
чем связывающая электронная пара, локализованная в поле двух положи-
положительных остовов атомов. Следует ожидать, что отталкивание электронных
пар будет усиливаться в следующем порядке: Х-Х < Х-Е < Е-Е. Именно
увеличением числа неподеленных пар объясняют уменьшение валентных
углов в ряду таких изоэлектронных молекул, как СН4э NH3, H2O. Это прави-
правило объясняет, почему неподеленные пары электронов предпочтительно за-
занимают экваториальные, а не аксиальные положения в тригональной бипи-
рамиде молекул АХ4Е, АХ3Е2, АХ2Е3 (см. табл. 4.2).
4.3. Метод валентных связей
125
Таблица 4.2
Геометрическая форма молекул АХШЕИ, не содержащих кратные связи
Общее число
электронных
пар
Тип молекулы
АЗД,
Геометрическая
форма
Примеры
Молекула
Валентный
угол
АХ2
АХ3
АХ2Е
АХ4
АХ3Е
АХ2Е2
АХ5
АХ4Е
ВеСЬ
BF3
SnCb
СН4
NH,
Н2О
РС15
SF4
180°
120°
100°
109°28'
107° 18'
104°30!
120°, 90°
104°, 89°
126
4. Химическая связь и строение вещества
Окончание табл. 4.2
Общее число
электронных
пар
Геометрическая
форма
Примеры
Молекула
Валентный
угол
AX3E2
AX2E3
AX6
AX5E
AX4E2
BrF3
86°, 188°
XeF2
180°
SF6
IF5
XeF4
90°
82°
90°
Правило 2. Объем связывающих электронных пар изменяется в
зависимости от электроотрицательности периферического атома: чем боль-
больше электроотрицательность последнего, тем сильнее он притягивает общее
электронное облако и тем меньше размер последнего. Поэтому увеличение
валентных углов в ряду PF3(97,8°)—РС13A00,3°)—РВг3A01,5°)—Р13A02°)
можно объяснить уменьшением электроотрицательности соответствующих
галогенов.
Правило 3. Кратные связи фактически не влияют на геометрию мо-
молекулы. Конфигурацию молекул с кратными связями предсказывают, основы-
основываясь на уже известных принципах, выделяя для рассмотрения электронные
пары а-связей и неподеленных электронов, т.е. орбиталь кратной связи счи-
считается условно одинарной, поскольку кратные связи расположены вместе в
пространстве и не могут разойтись на максимально возможное расстояние.
4.3. Метод валентных связей 127
При этом длина связи уменьшается с увеличением порядка связи, что объ-
объясняет изменение длины связи в ряду С2Н6 A54 пм)—С2Н4 A34 пм)—С2Н2
A20 пм). В то же время в кратной связи наблюдается повышенная элек-
электронная плотность и, следовательно, электронное облако кратной связи за-
занимает больший объем, чем электронное облако одинарной. Последнее об-
обстоятельство приводит к увеличению угла между кратной и одинарной свя-
связями и вызывает уменьшение угла между одинарными связями. Поэтому в
молекуле COF2, относящейся к типу АХ3, наличие двойной связи С=О при-
приводит к тому, что валентные углы отличаются от 120° и для связей O-C-F
составляют 126°, а для связей F-C-F — 108°.
Правило 4. Отталкивание между электронными парами заполнен-
заполненного уровня больше, чем между электронами незаполненного уровня, по-
поскольку в заполненном уровне орбитали занимают все доступное пространст-
пространство вокруг атома. Валентный уровень атомов элементов второго периода за-
заполняется четырьмя электронными парами, поэтому в молекулах АХ*, АХ3Е
и АХ2Е2, образованных элементами этого периода, угол между связями не
должен существенно отличаться от значения 109°28\ У атомов следующего
периода теоретический внешний уровень должен быть заполнен девятью
электронными парами. Это приводит к уменьшению валентных углов до
значений, близких к 90°. Таким образом, уменьшение валентных углов в ряду
изоэлеюронных молекул Н2ОA04,5°)—H2S(92,20)—H2Se(91,O0)—H2Te(89,5°)
объясняется тем, что в этих молекулах, относящихся к типу АХ2Е2, начиная
с H2S, уменьшение доступного объема приводит к сжиманию валентных
орбиталей.
Правило 5. Если атом с заполненным валентным уровнем, в кото-
котором есть одна или несколько неподеленных электронных пар, связан с дру-
другим атомом, у которого валентный уровень не заполнен, неподеленные пары
первого атома стремятся перейти с заполненного уровня на незаполненный.
Иными словами, наличие неподеленных пар у одного атома и незаполнен-
незаполненных орбиталей у другого может привести к частичному образованию крат-
кратных связей за счет донорно-акцепторного механизма, что обусловливает
отклонение валентного угла от ожидаемого. Так, например, согласно прави-
правилу 2, можно ожидать, что валентный угол молекулы РН3 окажется больше
валентного угла молекулы PF3, так как электроотрицательность водорода
меньше электроотрицательности фтора. Однако наличие у атома F неподе-
неподеленных электронных пар, а у атома Р — незаполненных орбиталей приводит
к образованию частично кратных связей, которые усиливают отталкивание
связывающих пар. Поэтому валентный угол РН3 равен 93,3°, a PF3 — 97,8°.
Правило 6. Если электронные пары валентного уровня имеют разное
число ближайших соседей, то те из них, у которых это число наибольшее,
будут расположены на большем расстоянии от ядра. В тригональной бипи-
128 4. Химическая связь и строение вещества
рамиде аксиальные электронные пары имеют большее число ближайших сосе-
соседей, чем экваториальные, поэтому аксиальные связи длиннее экваториаль-
экваториальных. Например, в молекуле РСЬ аксиальные связи Р-С1* имеют длину 212 пм,
а экваториальные связи Р-С1 — 202 пм.
Отметим, что МВС построен на аддитивном подходе к описанию со-
соединений: молекула рассматривается как сумма отдельных связей, а свойст-
свойства молекулы — как сумма свойств ее связей. Этот метод, основанный на
представлении о локализованной паре электронов, обеспечивающей хими-
химическую связь, наглядно и логично описывает структурные формулы многих
соединений и их свойства, позволяет предвидеть главные детали молеку-
молекулярной структуры без проведения трудоемких расчетов. Модели гибридиза-
гибридизации валентных орбиталей и ОЭПВО можно экстраполировать на сложные
молекулы, если выделить фрагмент, содержащий центральный атом, коор-
координирующий вокруг себя другие частицы.
Однако МВС, как и всякая приближенная теория, сталкивается с ря-
рядом трудностей. Распределение облаков электронных пар в пространстве
относительно центрального атома не подчиняется точным расчетам. Откло-
Отклонения особенно заметны при значительном количестве электронов, как, на-
например, в электронных оболочках d-элементов. Участие d-AO в связях, об-
образуемых элементами больших периодов, также приводит к отклонениям от
геометрии молекул, ожидаемой на основании представлений гибридизации
и ОЭПВО. Так, например, если молекулы галогенидов бериллия имеют
ожидаемую линейную форму (sp-TO центрального атома; молекулы типа
АХг), то галогениды бария — угловую. Метод валентных связей не объясня-
объясняет существования молекул с дефицитом электронов, таких как диборан
В2Н6, где на восемь связей приходится 12 электронов. Не объясняет МВС и
наличие парамагнитных свойств жидкого и твердого кислорода, явление
электрической проводимости металлов, не описывает ароматические и дру-
другие сложные соединения.
4.4. Метод молекулярных орбиталей
Более универсальным квантово-химическим методом описания хими-
химической связи является метод молекулярных орбиталей (ММО), идея которо-
которого развита в трудах Ф. Хунда, Дж. Леннарда—Джонса и Р. Малликена
A927-1929). В этом методе состояние электронов в многоатомной системе
описывается молекулярными орбиталями (МО) подобно тому, как состояние
электронов в атоме характеризуется АО. Метод основан на следующих
принципах:
молекула рассматривается как единое целое, каждый электрон при-
принадлежит молекуле в целом и движется в поле всех ее ядер и электронов;
4.4. Метод молекулярных орбиталей 129
состояние /-го электрона описывается одноэлектронной волновой
функцией у,, характеризуемой определенным набором квантовых чисел;
функция 1|//9 называемая молекулярной орбиталью, в отличие от АО
является многоцентровой и делокализованной;
квадрат модуля волновой функции |\|/|Д как и для электрона в атоме,
определяет плотность электронного облака;
полное описание состояния электрона характеризует молекулярная
спин-орбиталь, выражаемая произведением МО и спиновой функции \\fS;
каждой МО соответствует определенная энергия, которая слагается из
кинетической энергии электрона, потенциальной энергии притяжения элек-
электрона ко всем ядрам и усредненной потенциальной энергии отталкивания
данного электрона от всех остальных электронов;
совокупность МО, называемая электронной конфигурацией молекулы,
строится на основе фундаментальных положений квантовой механики:
принципа наименьшей энергии, принципа Паули и правила Хунда (см. гл. 3);
движение электронов взаимно независимое, и общая волновая функ-
функция основного состояния молекулы задается как произведение одноэлек-
тронных волновых функций v|/ = Пу,;
образование МО упрощенно рассматривается как линейная комбина-
комбинация АО.
Способ построения МО путем линейной комбинации АО, так назы-
называемый метод МО ЛКАО, наиболее часто используют для приближенных
расчетов электронного строения и реакционной способности молекул. При
сложении волновых функций двух валентных АО разных атомов (А и В),
имеющих близкие значения энергии, электронная плотность между ядрами
атомов увеличивается. Это ведет к образованию связывающей молекуляр-
молекулярной орбитали (СМО), имеющей более низкое значение энергии, чем ис-
исходные АО:
где \|/ — симметричная волновая функция СМО; \|/А и \|/в — волновые функ-
функции АО атомов А и В соответственно; а и Ъ — коэффициенты, учитывающие
вклад соответствующих АО в образование МО, при этом а = Ь, если А и В
— атомы одного элемента, и Ъ > я, если электроотрицательность атома В
выше. При вычитании волновых функций двух валентных АО разных ато-
атомов, имеющих близкие значения энергии, электронная плотность между яд-
ядрами атомов уменьшается, что ведет к образованию разрыхляющей молеку-
молекулярной орбитали (РМО), имеющей более высокое значение энергии, чем
исходные АО:
у* = a\\fA - 6\|/в.
Здесь i|/ — антисимметричная волновая функция РМО (рис. 4.12).
5 - 9795
130
4. Химическая связь и строение вещества
м2
в
смо
РМО
'в б
Рис. 4.12. Распределение электронной плотности при образовании:
а — связывающей МО; б — разрыхляющей МО
Линейная комбинация невалентных АО, как показывают квантово-
механические расчеты, приводит к образованию МО, имеющих вид и энер-
энергию, близкую к виду и энергии исходных АО, поэтому такие орбитали ус-
условно считают несвязывающими (НМО).
Для возникновения МО требуются определенные условия:
энергии АО должны быть соизмеримы, иначе, если энергия АО атома
А значительно ниже энергии АО атома В, электрон атома А не перейдет в
область атома В и МО не образуется;
электронные облака взаимодействующих атомов должны максималь-
максимально перекрываться, чем значительнее перекрывание, тем прочнее связь;
максимальное перекрывание возможно для АО, обладающих одинако-
одинаковыми свойствами симметрии относительно оси молекулы.
В соответствии со свойствами симметрии (осью молекулы условно
выбрана ось z) максимальное перекрывание АО и образование МО возмож-
возможно при линейной комбинации двух s-AO, когда образуются <т5-СМО и
а*5-РМО, при линейной комбинации двух pz-AO, когда образуются о>2-СМО
и а*/?2-РМО, при линейной комбинации двух рх-АО, когда образуются
прх-СМО и л;*/?х-РМО, и при линейной комбинации двух ру-АО, когда обра-
образуются пру-СМО и п*ру-?МО (рис. 4.13). Возможна комбинация s-АО и/?г-АО,
перекрывание которых происходит вдоль оси молекулы, а при наложении
s-AO и рХ'АО возникают две области перекрывания, равные по величине и
противоположные по знаку, и в результате суммарное перекрывание ока-
4.4. Метод молекулярных орбиталей
131
Рис. 4.13. Схема образования МО:
а — ст-перекрывающие; б — я-перекрывающие
зывается равным нулю (рис. 4.14). Ниже представлены разрешенные в ММО
комбинации АО (разрешенные комбинации ру определяют по аналогии с рх,
a dyz vidxy — по аналогии с d^):
А s pz px dxz di ^x2-y2
В s,pz,dz2 s,pz,dz2 px,dxz Px,dxz s9pz,dz2 ^2_/
Порядок возрастания энергии МО устанавливается с использованием
экспериментальных методов, например, исследованием молекулярных спек-
спектров, и квантово-механических расчетов. Для гомоатомных двухатомных
молекул элементов первого и второго периодов, выявлена такая последова-
последовательность:
для легких молекул (от Н2 до N2)
<?ls < C7*i5 < G2s < CT*2s< П2рх =
для более тяжелых молекул (от О2
< a*i, < g2s < а*2s < <*2pz < n2p
_ ¦ _ * *
~ к2Ру < п 2Рх ~ п 2Ру < а 2Рг -
4.14. Влияние симметрии АО
на образование МО
132
4. Химическая связь и строение вещества
АО
МО
Н
Рис. 4.15. Энергетическая диа-
диаграмма МО молекулы Н2
Энергетическую диаграмму МО мо-
молекулы Н2 можно построить следующим
образом (рис. 4.15): линейная комбинация
двух ls-AO приводит к образованию а^-СМО
и сг*ь-РМО, оба электрона в соответствии с
принципом наименьшей энергии и принци-
принципом Паули находятся на связывающей МО.
Электронная конфигурация молекулы Н2 в
основном состоянии — [(стьJ], суммарный
спин равен нулю.
Характер распределения электронов
по МО позволяет объяснить магнитные
свойства частиц. Молекулы, суммарный
спин которых равен нулю, проявляют диа-
диамагнитные свойства, т.е. во внешнем магнитном поле их собственные маг-
магнитные моменты ориентируются против направления поля. Молекулы, сум-
суммарный спин которых отличен от нуля, проявляют парамагнитные свойст-
свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты
ориентируются в направлении поля. Таким образом, молекула Н2 диамаг-
диамагнитна.
Молекула Не2 (рис. 4.16) в основном состоянии [(crisJ(a*i5J] существо-
существовать не может. Нахождение электронов на СМО означает увеличение элек-
электронной плотности в межъядерном пространстве, что приводит к уменьше-
уменьшению длины связи и увеличению энергии связи. Нахождение электронов на
РМО означает уменьшение электронной плотности в межъядерном про-
пространстве, что приводит к увеличению длины связи и снижению энергии
связи.
. Показателем прочности молекулы
может служить порядок связи (ПС), кото- ^
рый определяется разностью чисел элек-
электронов, находящихся на СМО, и электро-
электронов, находящихся на РМО:
ПС =
лв(СМО)-ив(РМО)
D.8)
Не
Не
где пе(СМО) — число электронов на связы-
связывающих МО; Яе(РМО) — число электронов
на разрыхляющих МО. По существу, поря-
порядок связи — другая форма понятия валент- Рис. 4.16. Энергетическая диа-
ности, а нецелочисленные значения поряд- грамма МО молекулы Не2
4.4. Метод молекулярных орбиталей
133
АО
МО
АО
1
2s
t
2s
В
Рис. 4.17. Энергетическая диаграмма МО молекулы В2
ка связи, как в случае молекулы N0, — следствие многоцентровости и
делокализации связей. В молекуле Не2 одинаковое число электронов на
СМО и РМО, порядок связи равен нулю, следовательно, в основном состоя-
состоянии такая молекула не существует, в отличие от молекулы Н2, для которой
ПС=1.
Начиная с В2, в образовании МО принимают участие р-АО. Энергети-
Энергетическая диаграмма молекулы В2, построенная в соответствии с порядком воз-
возрастания энергии МО, характерным для элементов, находящихся в ША—
VA группах, представлена на рис. 4.17. Порядок связи В2 ПС = 1, т.е. моле-
молекула существует. Суммарный спин равен 1, молекула парамагнитна. При
записи электронной конфигурации молекулы НМО, отвечающие внутрен-
внутренним АО, обозначают символами соответствующих АО, например, при ли-
линейной комбинации Is2 образуются КК—НМО. Таким образом, электронная
конфигурация молекулы В2 имеет вид [KK(a2sJ(a*25J( n2p =п2р J]. Элек-
134
4. Химическая связь и строение вещества
АО
АО
И
1
¦ ¦
о
о
Рис. 4.18. Энергетическая диаграмма МО молекулы О2
тронная конфигурация молекулы С2 — [KK(o2sJ(G*2sf(n2px = n2p L]. Эта
молекула прочнее, чем В2, так как ПС = 2. Молекула диамагнитна, посколь-
поскольку суммарный спин равен нулю. Электронная конфигурация молекулы N2 —
[KK(a2sJ(cr*25J (п2рх = п2 L( c2pz J], ПС = 3, молекула диамагнитна.
Энергетическая диаграмма молекулы О2 построена в соответствии с по-
порядком возрастания энергии МО, характерным для элементов VIA-VIIIA групп
(рис. 4.18), ПС = 2, молекула парамагнитна, ее электронная конфигурация —
[КК(а28J(а*25J(а2/?гJG12^=я2л )\п2рх=п2р J]. Для молекулы F2 порядок
связи ПС = 1, электронная конфигурация — [KK(a2sJ(a*25J( o2p J
(п2рх = п2Р.)\п*2рх =71*2/7,L]' молекула F2 проявляет диамагнитные свойства.
Молекула Ne2, как и молекула Не2, не существует, так как порядок связи
ПС = 0.
4.4. Метод молекулярных орбиталей 135
Гетероатомные двухатомные молекулы рассмотрим на примере мо-
молекул N0 и СО. Атомы N и О имеют неодинаковые заряды ядер (ZN = 7, Zo = 8),
квантовые уровни атома кислорода лежат ниже соответствующих уровней
атома азота. На рис. 4.19, а представлена энергетическая диаграмма молеку-
молекулы N0, построенная в соответствии с экспериментальными данными. Поря-
Порядок связи ПС = 2,5, молекула парамагнитна, ее электронная конфигурация —
[KK(G2sJ(a*2,J( п2Ря = п2ру )\ а2р J( п2рх = к2ру I]. Сохраняется порядок рас-
расположения МО, характерный для гомоядерных молекул, в частности N2. Это
связано с тем, что электроотрицательности, заряды ядер и размеры атомов N
и О близки.
Валентные орбитали атомов, электроотрицательность, заряды ядер и
размеры которых сильно различаются, существенно различаются и значе-
значениями энергии, что необходимо учитывать при построении энергетических
диаграмм МО. Различия в энергии, а также в свойствах симметрии валент-
валентных АО приводят к появлению НМО, расположенных выше или ниже свя-
связывающих МО. Энергетическая диаграмма молекулы СО представлена на
рис. 4.19, б. Порядок связи молекулы ПС = 3, молекула диамагнитна, ее
электронная конфигурация — [(о°иJ(о0иJ(о\J( а25_2р J( к2рх = п2ру L
(с°2р J], где индекс «о» указывает на несвязывающий характер МО.
В рамках метода МО ЛКАО для описания сложных молекул применяют
различные полуэмпирические подходы. Волновая функция многоатомной мо-
молекулы построена из многоцентровых делокализованных МО (ДМО), однако
часто удобным является использование двуцентровых локализованных МО
(ЛМО). Они представляют собой гибридные орбитали, в которых смешаны
волновые функции двух взаимодействующих атомов. Такое приближение воз-
возможно, поскольку, хотя электроны принадлежат всей молекуле, т.е. делокали-
зованы, общее распределение электронной плотности практически близко к
характерному для двуцентровых связей. Этот прием позволяет объяснить
структурные формулы и изомерию молекул, удобен для описания основных
свойств связи — длины, прочности, дипольного момента, поляризуемости.
Квантово-механические расчеты показали возможность выделения
типовых ЛМО при построении молекул, что объясняет схожесть их химиче-
химического поведения. В то же время такие свойства, как энергия ионизации мо-
молекулы или энергия возбуждения электрона, зависящие от состояния элек-
электрона в молекуле, подтверждают реальность ДМО. Для описания сложных
соединений используют различные подходы. Так, для ароматических угле-
углеводородов применяют метод Хюккеля, для координационных соединений —
теорию поля лигандов. Расчеты молекулярных характеристик этими мето-
методами основаны на ряде допущений, упрощающих решение уравнения Шрё-
дингера, но в целом сложны и выходят за рамки данного учебника.
136
4. Химическая связь и строение вещества
АО
t
t
АО
И
t
¦ I
Ч 2рх
2s
N
NO
О
Рис. 4,19. Энергетическая
а —N0;
Методы валентных связей и молекулярных орбиталей, будучи при-
приближенными способами описания молекул, не исключают, а дополняют
друг друга. Каждый из них обладает своими достоинствами и недостатками.
Описывая отдельные связи и фрагменты молекул, МВС позволяет предска-
предсказывать реакционную способность, свойства и особенности поведения молекул.
Универсальный, физически адекватный ММО, рассматривающий молекулу
в целом, точнее интерпретирует опытные данные, объясняет существование
молекул, не укладывающихся в рамки представлений о парно-электронных
связях.
4.5. Невалентные типы связей
137
АО
МО
2s-2p7
АО
шиш
со
О
диаграмма МО молекул:
б —СО
4.5. Невалентные типы связей
Главная особенность ковалентной связи — ее направленный и насы-
насыщаемый характер, обусловленный электронным строением атомов. В то же
время существуют соединения, в которых связи между частицами не прояв-
проявляют свойств насыщаемости и направленности и не обладают повышенной
электронной плотностью в области связывания. Одним из таких невалент-
невалентных типов связей является ионная связь, природу которой можно объяснить
электростатическим взаимодействием.
138
4. Химическая связь и строение вещества
Рис. 4.20. Кристаллическая решетка хлорида натрия:
О — ионы Na+; •— ионы СГ
Ионная связь — предельный случай ковалентной полярной химической
связи. Как отмечалось, наибольшая полярность связи характерна для галоге-
нидов щелочных металлов — соединений между элементами, имеющими
наибольшие и наименьшие значения электроотрицательности. Вследствие
поляризации связи под действием соседних полярных молекул такие соедине-
соединения в кристаллическом состоянии представляют собой упорядоченное распо-
расположение отрицательно и положительно заряженных ионов (анионов и катио-
катионов). Электрическое поле, создаваемое ионом, имеет сферическую симметрию,
и все находящиеся в этом поле частицы испытывают его действие. Заряды
ионов обусловливают их притяжение и отталкивание и в целом определяют
стехиометрический состав соединения. Ненаправленный и ненасыщаемый
характер электростатического взаимодействия ионов обусловливает структу-
структуру ионных соединений. Энергетически выгодно, чтобы каждый ион был ок-
окружен максимальным числом ионов противоположного знака, но вследствие
отталкивания одноименных ионов устойчивость системы достигается лишь
при определенной взаимной координации частиц. Поэтому строение ионных
соединений определяется соотношением размеров ионов (принципом плотной
упаковки) и требованием электронейтральности всего кристалла (рис. 4.20).
Отметим, что чисто ионных соединений нет. Полное разделение заря-
зарядов А+В~ не может осуществиться из-за волновых свойств электрона (вероят-
(вероятность нахождения электрона возле ядра атома А может быть мала, но отлична
от нуля). Кроме того, электростатическое взаимодействие ионов вызывает
смещение в них электрических зарядов. Поляризация ионов — процесс дву-
двусторонний, в нем сочетаются поляризуемость ионов (способность электрон-
электронной оболочки ионов к деформации) и их поляризующее действие (способ-
(способность деформировать, поляризовать другой ион).
Поляризуемость ионов зависит от электронной структуры, заряда и
размера иона:
4.5. Невалентные типы связей 139
при одинаковых зарядах и близких радиусах поляризуемость мини-
минимальна у ионов с конфигурацией инертного газа (например, Na+, Ca2+, Ва2+),
больше у ионов с незавершенным электронным слоем (Ti2+, Fe2+, Pb2+) и
максимальна у ионов с 18-электронной оболочкой (Zn2+, Hg2+, Ag+);
поляризуемость ионов с аналогичным электронным строением возрас-
возрастает с увеличением ионного радиуса (F~ < СГ < Вг~ < Г и Li+ < Na+ < K+ <
<Rb+<Cs+);
если элемент образует ионы разного заряда, то поляризуемость иона
тем меньше, чем больше его заряд (Fe3+ < Fe2+);
в ряду изоэлектронных ионов с конфигурацией инертных газов по-
поляризуемость растет с увеличением отрицательного заряда (Mg2+ < Na+ <
<F~<02).
Поляризующее действие ионов также зависит от их электронной
структуры, заряда и размера. Оно тем значительнее, чем больше заряд,
меньше радиус и устойчивее электронная оболочка иона. Наибольшее поля-
поляризующее действие оказывают ионы, которые сами слабо поляризуются.
Поэтому многоатомные ионы больших размеров (анионы) сильно поляри-
поляризуются и оказывают незначительное поляризующее действие. В то же время,
если катион легко деформируется, то возникающий в нем диполь усиливает
его поляризующее действие на анион, что в свою очередь приводит к появ-
появлению дополнительного поляризационного эффекта. Поэтому суммарное
поляризующее действие особенно велико у 18-электронных ионов.
Поляризуемость и поляризующее действие ионов оказывают влияние на
свойства ионных соединений. Это вызвано тем, что в результате поляризации
длина диполя оказывается меньше межатомного расстояния, дипольныи момент
уменьшается, что превращает ионную молекулу в полярную ковалентную.
Известно, например, что в водных растворах ВаС12 диссоциирует пол-
полностью, a HgCl2 незначительно, температура плавления ВаС12 ТПЛ = 1235 К, а
HgCl2 — Тш = 550 К. Очевидно, что чем выше поляризация ионов, тем силь-
сильнее деформируется кристаллическая решетка и тем ниже температура плав-
плавления ионных кристаллов. Температура плавления HgCl2 значительно ниже
температуры плавления ВаС12, поскольку поляризуемость и поляризующее
действие Hg2+ больше, чем у Ва2+, и следовательно, кристаллическая
решетка HgCl2 сильнее деформирована в результате поляризации. При дис-
диссоциации солей в водном растворе также проявляется характер связи в кри-
кристалле. Чем больше доля ковалентности связи, тем меньше степень диссо-
диссоциации молекул. Связь в молекулах HgCl2 имеет большую долю ковалент-
ковалентности, что объясняет малую степень диссоциации HgCl2 в водном растворе.
Поляризация ионов сказывается и на оптических свойствах молекул.
Окраска соединений зависит от поглощения света и связана с возбуждением
внешних электронов. Если для их возбуждения требуется большое количе-
140 4. Химическая связь и строение вещества
ство энергии (ультрафиолетовые лучи), молекула оказывается бесцветной.
Если электронные переходы характеризуются меньшей энергией, то им со-
соответствует видимая часть спектра, — и молекула окрашена. Таким образом,
чем более поляризуема частица, тем меньше энергии требуется при возбужде-
возбуждении внешних электронов и тем больше вероятность окраски соединения.
Именно большей поляризацией иона Г объясняется тот факт, что HgCl2 имеет
белую окраску, a Hgl2 — красную, в то время как катионы Hg2+ бесцветны.
Другим невалентным типом связи является металлическая связь. Как
и ионная, металлическая связь проявляется в конденсированном состоянии
вещества (в данном случае, металла) и не обладает свойствами направлен-
направленности и насыщаемости. При этом металлическая связь имеет некоторое
сходство с ковалентной связью — она основана на обобществлении валент-
валентных электронов, возможность которого обусловлена невысокими значения-
значениями энергии ионизации атомов металлов. Полностью делокализованные
электроны не принадлежат конкретным соединениям атомов, а обслужива-
обслуживают весь кристалл. Следовательно, металлическая связь — это многоцентро-
многоцентровая связь, в которой электронная плотность распределена равномерно по
всему объему кристалла во всех направлениях. Метод валентных связей, как
уже отмечалось, не может интерпретировать металлическую связь. С пози-
позиций ММО эта связь характеризуется дефицитом электронов относительно
обычной ковалентной связи, поэтому порядок связи в металлах и интерме-
таллидах (соединениях металлов друг с другом) — дробное число.
Рассмотрим кратко основные представления зонной теории твердых
тел с координационной структурой, базирующейся на ММО. При сближе-
сближении двух одинаковых атомов происходит квантово-механическое взаимо-
взаимодействие их валентных орбиталей, и в результате из двух АО, имеющих
одинаковое значение энергии, образуются две молекулярные орбитали —
СМО и РМО (рис. 4.21). Взаимодействие 4, 6, ... атомов приводит к расще-
^т рмо
нмо
СМО
0 1 2 4 6 1023 N
Рис. 4.21. Образование энергетической зоны:
Ео — энергия валентного электрона в изолированном атоме; N— число атомов
4.5. Невалентные типы связей 141
плению валентных АО на такое же число МО. Образование кристалла в ре-
результате взаимодействия большого числа атомов (~Na = 6,02 • 1023) приводит
к появлению практически сплошного спектра энергий МО. Общая ширина
энергетической зоны, т.е. разность между самым низким и самым высоким
уровнями, практически не зависит от числа атомов, участвующих в образо-
образовании кристалла. Понятие об энергетической зоне, характеризующей со-
состояние электронов в твердом теле, — основополагающее в зонной теории.
Подобно тому, как в изолированном атоме существуют разрешенные и за-
запрещенные уровни энергий, в кристалле имеются разрешенные и запрещен-
запрещенные зоны энергий. Зону, которую занимают электроны, осуществляющие
химическую связь, называют валентной. Свободную зону, расположенную
выше валентной, называют зоной проводимости.
В качестве примера рассмотрим возникновение и перекрывание энер-
энергетических зон в кристаллах натрия и кремния в зависимости от межатомно-
межатомного расстояния г (рис. 4.22, а и 4.22, б). При сближении атомов натрия Na
(Is 2s22p63sl) в первую очередь расщепляется вакантный 3/?-подуровень
(чем ближе атомы, тем больше расщепление), затем наполовину заполнен-
заполненный валентный Зя-подуровень (см. рис. 4.22, а). При дальнейшем сближении
(г') происходит перекрывание 35- и 3/>-разрешенных зон. Сближение атомов
заканчивается при го (межатомное расстояние в кристалле), таким образом,
валентные Зя-электроны Na могут занимать любое состояние в пределах зо-
зоны, образовавшейся в результате перекрывания валентной зоны и зоны про-
проводимости. Чем больше ширина этой разрешенной зоны, тем слабее связаны
электроны с ядром и тем больше они делокализованы в пространстве. Сво-
Свободное перемещение электронов по всему пространству кристалла приводит
к появлению электрической проводимости первого рода. Поэтому делокали-
зованные электроны металлов называют электронами проводимости.
Характерная особенность зонной структуры кристалла кремния
(Is22s22p 3s23p2) заключается в том, что вакантная зона 4s не перекрывается
с валентной на межатомных расстояниях г0, а отделена от последней зоной
запрещенных энергий АЕ (см. рис. 4.22, б). Поскольку в валентной зоне
кремния все состояния заняты (у каждого атома по четыре электрона на че-
четырех АО), изменение состояния электрона возможно только путем перево-
перевода его в следующую разрешенную зону (зону проводимости). Для возбуж-
возбуждения электрической проводимости необходимо сообщить электронам энер-
энергию, равную или превышающую АЕ.
Ширина запрещенной зоны АЕ представляет собой важную характе-
характеристику кристаллического вещества. В зависимости от ширины запрещен-
запрещенной зоны все кристаллические вещества подразделяют на три класса: метал-
металлы, полупроводники, диэлектрики. В металлах ширина запрещенной зоны
равна нулю, поэтому валентные электроны способны к свободному переме-
перемещению по всему объему кристалла. Если ширина запрещенной зоны очень
142
4. Химическая связь и строение вещества
El
\ \ \ \ \ \ \ v
Зона проводимости
Рис. 4.22. Энергетические зоны кристалла:
а — натрия Na; б — кремния Si
велика (АЕ > 4 эВ), то возбудить электрическую проводимость практически
невозможно. Такие вещества называют диэлектриками или изоляторами,
например, для алмаза АЕ = 5,1 эВ, для кварца АЕ = 5,2 эВ. Промежуточные
значения АЕ характерны для полупроводников, например, для кремния
4.6. Межмолекулярное взаимодействие 143
АЕ= 1,1 эВ, для германия АЕ = 0,75 эВ. Полупроводники под влиянием не-
некоторых внешних воздействий (усиления электрического поля, нагревания,
освещения) способны проводить электрический ток, так как валентные
электроны, попадая в зону проводимости, становятся делокализованными.
При устранении внешних воздействий электроны возвращаются на низший
энергетический уровень, и вещество вновь становится непроводником.
Делокализация валентных электронов обусловливает высокую тепло-
и электропроводность металлов, приводит к тому, что кристаллические
структуры металлов характеризуются высокими координационными числа-
числами, а формульный состав интерметаллидов не соответствует классическим
представлениям о валентности.
Металлическая связь, как и ионная, не исключает некоторой доли ко-
валентности. В наиболее чистом виде она характерна только для щелочных
и щелочноземельных металлов. Для d- и/- металлов установлено, что не бо-
более 20 % валентных электронов находятся в состоянии обобществления, а
остальные осуществляют направленные ковалентные связи между атомами.
4.6. Межмолекулярное взаимодействие
Между молекулами, валентно-насыщенными в обычном понимании,
на расстояниях, превышающих размеры частиц, могут проявляться электро-
электростатические силы межмолекулярного притяжения, или так называемые силы
Ван-дер-Ваалъса. Как показывают квантово-механические расчеты, энергия
притяжения Епр определяется суммой энергий ориентационного Еор, индук-
индукционного ЕШД и дисперсионного Етсп взаимодействий:
р = р + р + р
Ориентационное взаимодействие (эффект Кьезома, или диполь-
дипольное взаимодействие) проявляется между полярными молекулами,
которые при приближении поворачиваются (ориентируются) друг к другу
разноименными полюсами так, чтобы потенциальная энергия системы стала
минимальной (рис. 4.23, а). Энергия ориентационного взаимодействия тем
существеннее, чем больше дипольный момент молекул ц и меньше расстоя-
расстояние / между ними:
F
где 80 — электрическая постоянная.
Индукционное взаимодействие (эффект Дебая) связано с процессами
поляризации молекул окружающими диполями. Если неполярную молекулу
поместить в электрическое поле, то электрические центры тяжести положи-
144
4. Химическая связь и строение вещества
(О (Ф
B)
Рис. 4.23. Схемы межмолекулярного взаимодействия:
а — ориентационное; б — индукционное; в — дисперсионное
тельных и отрицательных зарядов, совпадавшие до этого, смещаются отно-
относительно прежнего положения. При этом происходит деформация непо-
неполярной молекулы и индуцируется диполь (рис. 4.23, б). Взаимодействие по-
постоянного диполя одной молекулы и индуцированного диполя другой по-
понижает потенциальную энергию системы. Индукционное взаимодействие
тем значительнее, чем выше поляризуемость а неполярной молекулы и ди-
польный момент ц полярной молекулы
?инд = ~8^/'
Поляризуемость а неполярной молекулы называют деформационной,
поскольку она связана с деформацией частицы и характеризует смещение
электронного облака и ядер относительно прежних положений. Ее опреде-
определяют как отношение индуцированного (наведенного) диполя (динд молекулы
к напряженности F электрического поля
а =
Дисперсионное взаимодействие (эффект Лондона) возникает у любых
молекул независимо от их строения и полярности. Вследствие мгновенного
несовпадения центров тяжести зарядов электронного облака и ядер образу-
4.6. Межмолекулярное взаимодействие
145
ется мгновенный диполь, который индуцирует мгновенные диполи в других
частицах (рис. 4.23, в). Движение мгновенных диполей становится синхрон-
синхронным (согласованным). В результате соседние частицы испытывают взаим-
взаимное притяжение, и энергия системы понижается. Энергия дисперсионного
взаимодействия зависит от энергии ионизации Е/ и поляризуемости а молекул:
_ 3 ЕтлЕт>у otiOto
2 Еп
Г
Дисперсионные силы играют большую роль не только при взаимодействии
отдельных молекул, но и макроскопических частиц, например коллоидных.
Силы притяжения Ван-дер-Ваальса — дальнодействующие. На не-
небольших расстояниях между молекулами заметными становятся близкодей-
близкодействующие силы отталкивания (силы Паули), которые возрастают при сбли-
сближении частиц. Они являются причиной несжимаемости веществ, находящих-
находящихся в конденсированном состоянии. Энергию отталкивания аппроксимируют
эмпирической формулой
где В — константа, определяемая опытным путем. Равновесное состояние
между молекулами в жидкостях и кристаллах устанавливается в результате
баланса сил притяжения и отталкивания.
Для неполярных молекул суммарную энергию межмолекулярного
взаимодействия часто описывают соот-
соотношением, называемым потенциалом
Леннарда-Джонса U:
U= -—+ —
где а и Ъ — постоянные, зависящие от
природы вещества.
Зависимость U от расстояния /
между молекулами выражается потенци-
потенциальной кривой с минимумом (рис. 4.24),
глубина которого С/о соответствует
энергии взаимодействия молекул на
равновесном расстоянии /0 D00...600 пм
в жидкости или кристалле). Эта энер-
энергия обычно не превышает значений
1...5 кДж/моль, т.е. по сравнению с ко-
валентной связью межмолекулярное
взаимодействие очень слабое.
О
Притяжение
Рис. 4.24. Зависимость энергии меж-
межмолекулярного взаимодействия U
двух молекул от расстояния между
ними /
146 4. Химическая связь и строение вещества
При образовании растворов электролитов большую роль играют не-
неспецифические ион-молекулярные взаимодействия, близкие по природе к
электростатическому взаимодействию и имеющие черты донорно-акцеп-
торной связи. Энергия связи ЕИ_Д ионов и полярных молекул растворителя
(ион-диполъное взаимодействие) определяется ориентационной составляю-
составляющей сил Ван-дер-Ваальса и зависит от заряда z иона и дипольного момента
ц молекулы растворителя:
^и-д "~
4яе0/
Между ионом и неполярной молекулой растворителя осуществляется
в основном индукционное и дисперсионное взаимодействия. Кроме того, в
растворах электролитов между катионами и полярными молекулами,
имеющими одну или несколько неподеленных электронных пар, как, на-
например NH3 или Н2О, или неполярными молекулами с легко поляризуемыми
электронами, например, С6Н6, возникает донорно-акцепторная связь за счет
переноса электронной плотности молекул на свободную орбиталь катиона.
Это приводит к образованию устойчивых соединений растворенных частиц
с молекулами растворителя — солъватов.
Промежуточный характер между валентным и межмолекулярным
взаимодействием имеет водородная связь. Она характерна для жидкостей, в
состав молекул которых (вода, спирты, кислоты) входит положительно по-
поляризованный атом водорода. Малые размеры и отсутствие внутренних
электронов позволяют атому водорода вступать в дополнительное взаимо-
взаимодействие с ковалентно с ним не связанным отрицательно поляризованным
атомом другой (или той же самой) молекулы:
я- я+ я- я+
х-н-х-н.
Такая специфическая связь имеет черты электростатического и донорно-
акцепторного взаимодействия и приводит к образованию ассоциатов моле-
молекул (рис. 4.25).
Энергия водородной связи невелика (8...80 кДж/моль), но больше энер-
энергии взаимодействия Ван-дер-Ваальса, поэтому физические свойства веществ с
водородной связью отличаются от свойств их аналогов. Ассоциация молекул
приводит к уменьшению летучести, повышению температуры кипения и тепло-
теплоты испарения, увеличению вязкости и диэлектрической проницаемости жидко-
жидкостей. Например, температура кипения этанола С2Н5ОН, ассоциированной жид-
жидкости, в состав молекул которой входит положительно поляризованный атом
водорода, равна 351 К, в то время как температура кипения его изомера — ди-
метилового эфира СН3ОСН3 — 249 К.
4.6. Межмолекулярное взаимодействие
147
Рис. 4.25. Схемы образования водородных связей:
а — фрагмент структуры жидкого фтороводорода HF; б, в
структуры льда (Н2О); О — F;Q — О; • — Н
фрагменты
148 4. Химическая связь и строение вещества
Высокий дипольный момент молекул воды (цН2О = 1,86 Д) и способ-
способность образовывать четыре водородные связи (две — как донор протонов и
две — как акцептор протонов) (см. рис. 4.10) приводит к формированию мо-
молекулярных ассоциатов в жидкой и особенно твердой воде. Лед имеет тетра-
эдрическую кристаллическую решетку. В центре тетраэдра расположен
атом кислорода одной молекулы воды, в четырех вершинах находятся ато-
атомы кислорода соседних молекул, которые соединены водородными связями
с ближайшими соседями (рис. 4.25, б, в). Ажурная структура и наличие
внутренних пустот определяют рыхлость и меньшую плотность льда (р =
= 0,92 г/см3) по сравнению с жидкой водой.
В жидкой воде водородные связи частично разрушены, в ее структу-
структуре наблюдается динамическое равновесие между ассоциатами молекул и
свободными молекулами, перемещающимися в полостях между ассоциа-
ассоциатами. С повышением температуры одновременно происходят два процес-
процесса: увеличение степени заполнения полостей свободными молекулами во-
воды, что приводит к увеличению плотности; увеличение размеров полостей
и уменьшение размеров ассоциатов, что приводит к уменьшению плотно-
плотности системы. В интервале температур от 0 °С до 4 °С преобладает первый
процесс, поэтому плотность воды максимальна при 4 °С (р = 0,999973 г/см3),
при более высоких температурах — второй, и плотность воды уменьшает-
уменьшается (см. П.4.2).
Особенности строения (наличие внутренних пустот), высокая диэлек-
диэлектрическая проницаемость (е = 78,5 • 10~12 Ф/м), способность проявлять про-
тонодонорные и протоноакцепторные, а также электронодонорные и элек-
троноакцепторные свойства, небольшая вязкость (г| = 9,14 • 10 Па • с при
Т= 298 К) обусловливают уникальные свойства воды как растворителя.
Мерой энергии межмолекулярного взаимодействия может служить
разность между теплотой испарения жидкости Д#исп (или теплотой возгонки
кристалла) и работой расширения одного моля идеального газа (определяе-
(определяемой по уравнению состояния идеального газа: pV - RT) при атмосферном
давлении. В табл. 4.3 представлены температуры кипения Гкиш теплоты ис-
испарения А//°исп при температуре кипения и энергии межмолекулярного
взаимодействия Емол некоторых веществ, рассчитанные по формуле:
Очевидно, что энергия межмолекулярного взаимодействия ассоции-
ассоциированных веществ (таких как Н2О, Н2О2, HF, НС1О4) выше, чем неассоции-
рованных (например, СО, СО2, СНД энергия межмолекулярного взаимодей-
взаимодействия ртути Hg выше, чем аргона Аг, водорода Н2, кислорода О2 или хлора
С12 за счет вклада металлической связи, а высокие температуры кипения
4.7. Строение вещества в конденсированном состоянии
149
ZnF2 и СаС12 обусловлены ненасыщаемым и ненаправленным характером
ионной связи, преобладающей в этих соединениях.
Таблица 4.3
Температура кипения Гкип, теплота испарения Д#°исп,
энергия межмолекулярного взаимодействия Емол некоторых веществ
Вещество
Аг
н2
о2
С12
Hg
СО
со2
сн4
^кип» К
87,29
20,397
90,188
239,18
629,81
81,64
185,2
111,66
АЯ°ЙСШ
кДж/моль
6,50
0,916
6,82
20,42
59,23
6,07
18,21
8,19
¦^мол>
кДж/моль
5,77
0,746
6,07
18,43
53,99
5,39
16,67
7,26
Вещество
H2S
Н2О
Н2О2
HF
НС1
НС1О4
ZnF2
СаС12
212,87
373,15
424,5
292,7
188,1
312
1778
2230
АН исп,
кДж/моль
18,67
41,11
46,15
25,20
16,16
30,78
185,0
235,3
J7
^МОЛэ
кДж/моль
16,90
38,01
42,62
22,76
14,59
28,18
170,22
216,77
4.7. Строение вещества в конденсированном состоянии
В зависимости от условий (давления и температуры), а также характе-
характера и энергии взаимодействия частиц вещества могут находиться в различ-
различных агрегатных состояниях, отличающихся физическими свойствами
(табл. 4.4). В газообразном состоянии частицы вещества находятся на рас-
расстояниях, значительно превышающих размеры молекул, вследствие чего
силы межмолекулярного взаимодействия между ними очень малы. Кинети-
Кинетическая энергия молекул газа значительно превышает их потенциальную
энергию, поэтому силы Ван-дер-Ваальса не могут удержать молекулы газа
друг возле друга, которые свободно перемещаются в пространстве и зани-
занимают весь предоставленный объем. Характерной особенностью газов явля-
является их способность сжиматься и расширяться, а также самопроизвольно
смешиваться друг с другом в любых отношениях.
Состояние газа определяется его температурой, давлением и объемом.
Вследствие слабого межмолекулярного взаимодействия при небольших
давлениях и высоких температурах различные газы ведут себя приблизи-
приблизительно одинаково, и их поведение достаточна хорошо описывается класси-
классическими уравнениями идеального газа (см. B.4)-B.5)). Повышение внешне-
внешнего давления и понижение температуры (т.е. в условиях, при которых газы
обычно используются в технике) приводит к усилению действия сил меж-
межмолекулярного притяжения и влияния природы составляющих частиц. В
этих случаях применяют уравнения состояния реальных газов, например
уравнение Ван-дер-Ваалъса:
150
4. Химическая связь и строение вещества
где р — давление; V — молярный (мольный) объем; Т — температура, К;
R — универсальная газовая постоянная; а и Ъ — постоянные для данного
газа величины, учитывающие влияние сил притяжения и собственный объем
частиц соответственно.
Таблица 4 А
Характерные свойства газов
Кинетические
и физические
характеристики
Притяжение между
частицами
Движение частиц
Расстояния между
частицами
Упорядоченность
структуры
Объем
(при Т = const)
Форма
Сжимаемость
Плотность
Газ
Слабое
Сильное
Большое
Отсутствует
Совпадает с объе-
объемом сосуда
Нефиксирован-
Нефиксированная — заполняет
сосуд, принимая
его форму
Высокая
Низкая
, жидкостей и твердых тел
Жидкость
Умеренное
Умеренное
Малое
Невысокая
(агрегаты частиц)
Фиксирован-
Фиксированный
Нефиксиро-
Нефиксированная — пол-
полностью или
частично за-
заполняет сосуд
Малая
От умеренной
до большой
Твердое вещество
Сильное
Малое
Малое
Высокая (кристал-
(кристаллическая решетка)
Фиксированный
Фиксированная
Практически
отсутствует
Относительно
большая
В жидкостях средняя кинетическая энергия частиц примерно равна
их средней потенциальной энергии, поэтому в них существует ближний по-
порядок в расположении молекул. Он проявляется в том, что число соседних
молекул у каждой частицы и их взаимное расположение в среднем одинако-
одинаково во всем объеме жидкости. Частицы жидкости образуют относительно
упорядоченные агрегаты, не имеющие четких границ и разделенные облас-
областями беспорядочного расположения молекул. Поэтому для жидкостей ха-
характерна самодиффузия (непрерывное перемещение молекул), текучесть и
изотропность (одинаковость физических свойств во всех направлениях), с
одной стороны, и вязкость (сопротивляемость течению), сравнительно вы-
4.7. Строение вещества в конденсированном состоянии 151
сокая плотность и малая сжимаемость, с другой. Подобно газам, жидкости
не имеют определенной формы и принимают форму того сосуда, в котором
находятся, занимая, однако, ту часть объема, которая соответствует массе и
плотности жидкости. Подобно твердым телам, жидкости обладают опреде-
определенной структурой и индивидуальными физическими свойствами, завися-
зависящими от характера сил межмолекулярного взаимодействия. Например,
структура жидкой воды напоминает структуру льда (рис. 4.25, б, в). Моле-
Молекулы жидкой воды соединены посредством водородных связей и для боль-
большинства из них сохраняется тетраэдрическое окружение.
Характерная особенность жидкого состояния — наличие поверхност-
поверхностного натяжения. На молекулу, находящуюся в глубине жидкости, со всех
сторон равномерно действуют силы межмолекулярного притяжения. Однако
на поверхности эти силы не сбалансированы, поэтому поверхностные моле-
молекулы испытывают действие результирующей силы, направленной внутрь
жидкости. Поверхность жидкости стремится сократиться — оказывается в
состоянии натяжения. Поверхностное натяжение жидкости — это сила, не-
необходимая для преодоления стремления жидкости к сокращению поверхно-
поверхности. Существованием поверхностного натяжения объясняется сферическая
форма свободнопадающих капель жидкости.
В то же время относительно свободное перемещение молекул приво-
приводит к испарению частиц с открытой поверхности жидкости, скорость кото-
которого тем выше, чем больше площадь поверхности, выше температура и ни-
ниже внешнее давление.
Твердое аморфное состояние, как и жидкое, характеризуется только
ближним порядком, что проявляется в изотропии его свойств и отсутствии
определенной температуры плавления. Однако энергия межмолекулярного
взаимодействия в аморфных телах достаточно велика, чтобы препятствовать
их самодиффузии и текучести. При образовании аморфных тел часто сказы-
сказывается влияние водородных связей. Так, например, органические соедине-
соединения, содержащие много гидроксидных групп, в частности глицерин, затвер-
затвердевают в виде аморфной массы. Типичные аморфные тела — силикатные
стекла, большая вязкость которых препятствует переориентации молекул.
Однако в целом аморфное состояние вещества является нестабильным и при
благоприятных условиях переходит в кристаллическое.
Для кристаллического состояния характерно строго определенное
расположение частиц во всем объеме — дальний порядок. Это обусловлива-
обусловливает анизотропию, или векторность свойств, кристаллов — различие физи-
физических свойств, таких как теплопроводность, сжимаемость, прочность на
разрыв, коэффициент преломления света — в разных направлениях. Распо-
Располагаясь в кристалле определенным образом, частицы образуют кристалли-
кристаллическую решетку — трехмерное упорядоченное геометрическое расцределе-
152 4. Химическая связь и строение вещества
ние в пространстве точек, называемых узлами. Это определяет внешнюю
форму вещества в виде кристалла, ограниченного плоскими гранями, схо-
сходящимися в точечных вершинах, и прямолинейными ребрами.
В соответствии с природой составляющих частиц кристаллические
решетки могут быть ионными, атомными (ковалентными или металличе-
металлическими) и молекулярными. Различие в типе химической связи определяет и
существенное отличие физических и химических свойств веществ с разны-
разными типами кристаллической решетки.
Ионные кристаллические решетки построены из катионов и анионов,
между которыми действуют электростатические силы притяжения. Ионы
могут быть простыми, как, например, в кристалле NaCl (см. рис. 4.20), или
сложными, как в кристалле (NH4JSO4. Строение таких кристаллов, как уже
отмечалось, определяется соотношением радиусов ионов и принципом элек-
электронейтральности кристалла. Вследствие ненаправленного и ненасыщае-
мого характера ионной связи, для ионных кристаллов характерны высокие
температуры плавления и большая твердость. Однако ионные кристаллы
отличаются повышенной хрупкостью, так как механическое воздействие
приводит к нарушению правильного расположения разноименно заряжен-
заряженных ионов, что уменьшает энергию их взаимодействия.
В узлах атомно-ковалентной кристаллической решетки находятся ато-
атомы одинаковых или различных элементов, осуществляющие направленные ко-
валентные связи, количество которых определяется валентными возможностя-
возможностями атомов. Большая прочность ковалентных связей приводит к еще более вы-
высоким, чем в ионных кристаллах, значениям температуры плавления, к более
высокой твердости. Вместе с тем электро- и теплопроводность таких кристал-
кристаллов невелики. Механическое воздействие приводит к разрыву направленных
ковалентных связей, что обусловливает их хрупкость. Типичными примерами
веществ, кристаллизующихся в атомно-ковалентных решетках, являются алмаз
(рис. 4.26) и кварц.
В металлических кристаллических решет-
решетках атомы удерживаются металлической связью,
которая определяет построение решетки по прин-
принципу плотной упаковки и такие характерные свой-
свойства металлов, как высокая тепло- и электропро-
электропроводность. Делокализованное электронное облако
обусловливает также пластичность металлов, по-
поскольку сдвиг одного слоя кристаллической ре-
решетки относительно другого не приводит к разры-
разрыву металлической связи.
Рис. 4.26. Фрагмент В узлах молекулярной кристаллической ре-
структуры алмаза шетки расположены молекулы, связанные между
4.7. Строение вещества в конденсированном состоянии
153
собой слабыми межмолекулярными силами. От природы молекул зависит
строение кристаллов. Так, если основной вклад в межмолекулярное взаимо-
взаимодействие вносят силы Ван-дер-Ваальса, кристаллы характеризуются высо-
высокими значениями координационных чисел. Для кристаллов йода Ь, напри-
например, координационное число (число ближайших соседей) равно 12
(рис. 4.27). Если между молекулами действуют направленные водородные
связи, координационное число меньше и зависит от электронного строения
отрицательно поляризованного атома. У льда, как уже отмечалось, каждая
молекула тетраэдрически связана с соседними, и координационное число
соответственно равно 4 (см. рис. 4.25, б). Так как энергия межмолекулярно-
межмолекулярного взаимодействия невелика, для веществ с молекулярной кристаллической
структурой характерны низкие температуры плавления, летучесть, невысо-
невысокая твердость.
Форму кристаллов изучает кристаллография. Кристаллические ре-
решетки описываются с помощью кристаллографических осей а, Ъ и с, кото-
которые в отличие от обычных координатных осей имеют конечный размер и
могут располагаться под углами, отличными от 90° (рис. 4.28). Кристалло-
Кристаллографические оси и углы между ними задают длину ребер и их взаимную
ориентацию в элементарной ячейке кристаллической решетки. Элементар-
Элементарная ячейка — это мысленно выделенная часть кристаллической решетки,
включающая все элементы симметрии данного кристалла, параллельная
трансляция которой во всех направлениях создает тело кристалла. Всего
существует 14 типов элементарных ячеек, которые систематизированы на
основе их симметрии (рис. 4.29).
Важнейшими характеристиками элементарной ячейки, помимо типа
кристаллической решетки, являются: кратчайшее расстояние I между час-
частицами, образующими данный тип элементарной ячейки; координационное
число К — число одинаковых частиц, расположенных на кратчайшем рас-
расстоянии от данной частицы (число ближайших соседей); число частиц п,
необходимое для построения элементарной ячейки.
Рис. 4.27. Кристаллическая ре-
решетка йода
Рис. 4.28. Система кри-
кристаллографических осей
154
4. Химическая связь и строение вещества
Триклинная
аФЪФс
в
Ромбоэдрическая
а-Ь=с
в
Гексагональная
а-ЪФс
а=р=90°; y=120
Рис. 4.29. Основные системы кристаллов
У
/
а
У
/¦
/
с
yt
У
У
с
/а
)
С
/а
Кубическая
а=Ь-с
а=р=у=90°
Тетрагональная
а=ЬФс
a=p=Y=90e
Орторомбическая
аФЬФс
a=p=Y=90°
Моноклинная
аФЪФс
a=Y=90°; pi^90
Одно и то же вещество в зависимости от условий может образовывать
различные кристаллические формы. Это явление называют полиморфизмом,
а различные кристаллические формы одного и того же вещества — поли-
полиморфными модификациями. Примером полиморфизма служат полиморфные
модификации углерода — алмаз, графит, карбин, фуллерены. Существова-
Существование различных веществ в одной и той же кристаллической форме называют
изоморфизмом, а такие вещества — изоморфными.
При совместной кристаллизации различных веществ могут формиро-
формироваться смешанные кристаллы. Так, при кристаллизации изоморфных ве-
веществ образуются твердые растворы замещения, когда частицы одного ве-
вещества заменяются в его кристаллической решетке однотипными частицами
другого вещества. Примером таких однородных смешанных кристаллов яв-
являются алюмокалиевые и хромокалиевые квасцы: KA1(SO4J* 12H2O и
KCr(SO4J* 12H2O. Если частицы вещества-примеси существенно меньше
частиц основного вещества, они размещаются в междоузлиях кристалличе-
кристаллических решеток {твердые растворы внедрения). При проникновении в моле-
молекулярную кристаллическую решетку одного вещества молекул другого об-
образуются клатраты. Примерами клатратов являются включения в междоуз-
междоузлия льда молекул газов (H2S, Cl2 и др.).
4.7. Строение вещества в конденсированном состоянии
155
Большинство кристаллических тел плавятся
при определенной температуре, которая остается
постоянной, пока не расплавится весь кристалл.
Встречаются, однако, такие кристаллические
вещества, которые при определенной, свойст-
свойственной им температуре перехода образуют мут-
мутную жидкую фазу, а затем окончательно плавят-
плавятся, переходя в прозрачную жидкость. Мутную
жидкую фазу сближает с жидкостью явление те-
текучести и наличие поверхностного натяжения,
но в ней, как в кристалле, сохраняется упорядо-
упорядоченное положение молекул, что приводит к про-
проявлению анизотропии. При прохождении света
через эту фазу, называемую мезоморфной (ме-
зофазой), возникает двойное лучепреломление и
интерференция плоскополяризованного света.
Электрические и магнитные свойства мезофазы
также зависят от направления.
Такие вещества были открыты Ф. Рейнит-
цером и О. Неманом A888) и названы жидкими
кристаллами. Все жидкие кристаллы — орга-
органические вещества, имеющие длинные молекулы
с полярной группой на конце: -CN, -OR, -NO2,
-NH2. Для них характерно наличие двойной свя-
связи, препятствующей вращению и обеспечивающей жесткость молекулы по
отношению к ее оси. Дипольные моменты этих молекул велики, поэтому
межмолекулярные силы диполь-дипольного взаимодействия выстраивают
молекулы так, что их длинные оси оказываются параллельными. В зависи-
зависимости от характера расположения молекул различают три основных типа
жидких кристаллов — смектический, нематический и холестерический
(рис. 4.30). Своеобразная структура жидкокристаллических соединений,
обеспечивающая сочетание упорядоченности в расположении молекул с их
высокой подвижностью, определяет широкие области практического ис-
использования. Для изменения ориентации молекул жидких кристаллов доста-
достаточно очень слабого изменения электрического поля или температуры, что
создает возможность управления оптическими, электрическими и магнит-
магнитными свойствами, а также позволяет регистрировать указанные воздействия.
Рис. 4.30. Структура смек-
тических (а), нематических
(б) и холестерических (в)
жидких кристаллов
Практические занятия
Строение атомов и ионов.
Периодический закон и периодическое изменение
свойств элементов
Примеры решения задач
Задача 1. Сколько орбиталей включает в себя af-подуровень?
Решение. Для ^-подуровня орбитальное квантовое число 1 = 2. Число орбиталей
на подуровне определяется числом значений магнитного квантового числа.
Магнитное квантовое число mi принимает значения от -/ до +/. При / = 2 mt
принимает значения: -2, -1, 0, +1, +2. Следовательно, d-подуровень состоит из
пяти d-орбиталей.
Задача 2. Какова последовательность заполнения электронами подуровней,
для которых п + / = 6?
Решение. Запишем возможные значения п и /:
п 4 5 6
/ 2 1 О
Подуровень Ad 5p 6s
Исключены комбинации п = 1, / = 5; п = 2, / = 4; п = 3, / = 3, так как
они не удовлетворяют условию / < (и — 1). В соответствии с правилом (и + /),
при одинаковой сумме п + / энергия подуровней повышается в порядке уве-
увеличения главного квантового числа. Таким образом, данные энергетические
подуровни располагаются в следующем порядке: EDd) < ЕEр) < EFs).
Задача 3. Требуется написать электронные формулы атома железа и иона Fe2+.
Покажите для них распределение электронов внешнего энергетического уровня
по квантовым ячейкам. Укажите количество неспаренных электронов.
Решение. Порядковый номер атома железа 26. В соответствии с порядком
заполнения АО электронная формула имеет вид:
Ь22522р63523/764523^.
Упрощенная электронографическая диаграмма для внешних элек-
электронов выглядит так:
п
t
т
t
t
3d
4s
Лр
В соответствии с правилом Хунда электроны заполняют АО с одина-
одинаковой энергией таким образом, чтобы абсолютное значение суммарного
спина системы было максимальным. В основном состоянии атом железа
имеет 4 неспаренных электрона.
Примеры решения задач
157
2+:
При написании электронной формулы иона Fe2+ следует учитывать, что вна-
вначале удаляются электроны с внешней 4$-орбитали. Электронная формула иона Fe2+
\з22522рв3з23р63а*4з0.
Упрощенная электронографическая диаграмма имеет вид:
п
t
т
t
t
3d
As
Ар
В основном состоянии ион Fe имеет четыре неспаренных электрона.
Задача 4. Требуется написать электронную формулу и электронографическую диа-
диаграмму внешних энергетических уровней атома меди в основном состоянии.
Решение. Порядковый номер Си Z = 29. Согласно правилу (и + /), электронная фор-
формула должна иметь вид Is22s22p63s23p63a^4s2. Однако экспериментально установ-
установлено, что электронная формула атома Си: Is22s22p63s23p63dxo4s\ Это связано с
особой устойчивостью электронных конфигураций подуровней, занятых наполови-
наполовину или полностью, т.е. d5 или dxo. В этом случае говорят, что у Си происходит «про-
«провал» электрона с 4у-орбитали на 3^-орбиталь.
Электронографическая диаграмма для внешних электронов имеет вид:
п
и
и
и
п
3d
Ар
В основном состоянии атом Си имеет один неспаренный электрон. Анало-
Аналогичные «провалы» электронов наблюдаются у атомов Cr, Ag, Au, Mo, Pd и дру-
других (см. табл. 3.6).
Задача 5. Требуется написать электронные формулы атома фосфора Р в основном и
возбужденном состояниях, показать распределение валентных электронов по кван-
квантовым ячейкам и определить число неспаренных электронов.
Решение. Порядковый номер атома фосфора 15. В соответствии с порядком запол-
заполнения АО электронная формула атома Р в основном состоянии: \s22s22pe3s23p2'.
Внешними орбиталями в этом атоме являются 3s-, Зр- и незаполненные Зб/-орби-
тали. Электронографическая диаграмма для внешних электронов имеет вид:
т
t
t
3s
Зр
3d
В основном состоянии атом фосфора имеет три неспаренных электрона. При
затрате некоторого количества энергии один из Зя-электронов атома Р может быть
переведен на вакантную Зб/-орбиталь (Is22s22p63sl3p33dl). Электронная конфигура-
конфигурация атома фосфора в возбужденном состоянии (Р *) соответствует следующей схеме:
158
Строение атомов и ионов. Периодический закон
t
т
t
т
3s
3d
В этом возбужденном состоянии атом Р имеет пять неспаренных электронов.
Задача 6. Требуется написать электронные формулы атомов азота N и кислорода О,
показать распределение электронов внешнего уровня по квантовым ячейкам и срав-
сравнить орбитальные радиусы, первые энергии ионизации и сродство к электрону этих
элементов.
Решение. Атомы N и О расположены во втором периоде. Порядковый номер атома
N — 7, а О — 8. Поскольку заряд ядра у атома О больше, чем у ядра атома N, то
ядро атома О сильнее электростатически взаимодействует с электронами. Поэтому
размер (орбитальный радиус) атома О меньше размера атома N. В периоде с увели-
увеличением порядкового номера орбитальный радиус атома закономерно уменьшается.
Первые энергии ионизации атомов элементов в периоде с увеличением по-
порядкового номера имеют тенденцию к возрастанию в связи с уменьшением разме-
размеров атомов. Однако в ряде случаев наблюдаются отклонения от этой тенденции, что
связано с тонкой электронной структурой атомов. Именно это имеет место в случае
NhO.
Электронная формула атома N: Is22s22p2. Строение внешнего электронного
уровня в соответствии с правилом Хунда выражается следующей схемой, из кото-
которой ясно, что на каждой/7-орбитали имеется по одному неспаренному электрону:
п
t
t
t
2s 2p
У атома О (Z = 8) электронная формула: \s22s22pA. Следующий электрон по-
поступает на/7-орбиталь, уже занятую одним электроном:
и
и
t
t
2s
2р
Два электрона, находящиеся на одной и той же орбитали, сильно отталкива-
отталкиваются. Поэтому, чтобы удалить электрон с 2/7-орбитали атома О, нужно затратить
меньше энергии, чем для удаления электрона с 2/7-орбитали атома N. Таким обра-
образом, первая энергия ионизации атома О несколько меньше энергии ионизации атома N.
Чтобы сравнить сродство к электрону атомов NhO, надо учесть, что атом N
имеет наполовину заполненный 2/?-подуровень, который отличается особой устой-
устойчивостью. В случае приема дополнительного электрона эта устойчивая конфигура-
конфигурация будет нарушаться, что энергетически невыгодно. Следует учитывать, что у ато-
атома О более положительный заряд ядра, следовательно, выше энергия электростати-
электростатического взаимодействия с электронами. В случае приема атомом О еще одного
электрона конфигурация 2р4 перейдет в 2ръ. Поэтому сродство О к электрону боль-
больше (см. табл. П.2.1).
Примеры решения задач 159
Задача 7. Рассчитать эффективный заряд ядра атома магния, используя одноэлек-
тронное приближение и табл. П.2.1.
Решение, Одноэлектронное приближение — рассмотрение каждого электрона в от-
отдельности в поле ядра и усредненном поле остальных электронов. С помощью этого
приближения можно определить эффективный заряд ядра, действующий на внеш-
внешний электрон, который легче всего удалить.
Для водородоподобного атома энергия электрона
/7- т^ 7*2
2 2 2~ '
где Z* — эффективный заряд ядра. Для 1 моль электронов (при NA = 6,022 • 1023 моль)
1312,1 *2
где — Ев кДж/моль.
Таким образом, энергия ионизации
1312,1 *2
Ei=—T-Z •
где — Ei в кДж/моль.
Из этого уравнения получаем формулу для эффективного заряда ядра атома
Z =
'1321,1
Порядковый номер магния 12, его электронная формула: \s22^2pe3s2. Внешние
электроны находятся на ^-подуровне и для них главное квантовое число п = 3. Вычис-
Вычисления приводят к следующему значению эффективного заряда ядра атома магния:
Задача 8. Разница в энергиях 3s- и 3/?-подуровней для атома магния составляет
260,5 кДж/моль. Определить длину волны излучения при переходе атома магния из
возбужденного состояния в основное (переход Зр —> 3s).
Решение. Энергия излучения связана с длиной волны соотношением
Отсюда длина волны излучения
Е
Вычисления приводят к следующему результату:
. 6,022 • 1023 моль • 6,626 • 10~34 Дж • с • 2,998 • 108 м/с _9
X = 459,2 10 м = 459,2 нм.
260500 Дж/моль
160
Строение атомов и ионов. Периодический закон
Задача 9. Используя значения энергии ионизации (см. табл. П.2.1), определить воз-
возможность протекания процесса
F(r) + Cs+(r) = F+(r)+Cs(r)
Решение. Первые энергии ионизации атомов F и Cs составляют:
F = F+ + e,
Cs = Cs+ + е,
Еи = 1680,4 кДж/моль;
Е12 = 375,6 кДж/моль.
Перепишем второе уравнение в противоположном направлении, просумми-
просуммируем с первым и вычислим изменение энергии при протекании этого процесса:
F(r) + Cs+(r) = F+(r)+ Cs(r); АЕ = Еп-Еп= 1680,4 - 375,6 = 1304,8 кДж/моль.
Положительное значение свидетельствует о поглощении энергии и невоз-
невозможности самопроизвольного протекания процесса. Противоположный процесс
F+(r) + Cs(r) = F(r) + Cs+(r); AE = -1304,8 кДж/моль,
сопровождающийся выделением энергии, возможен.
Задачи для самостоятельного решения
1-5. Электрон в атоме находится на данной атомной орбитали. Опишите его со-
состояние квантовыми числами я, /, т{.
№
АО
1
3d
2
4/
3
Ър
4
6d
5
6-10. Определите, на какой орбитали находится электрон, если его состояние ха-
характеризуется данными квантовыми числами.
№
6
7
8
9
10
п
3
5
4
6
6
/
2
1
3
0
2
11-15. Определите последовательность заполнения электронами подуровней, для
которых сумма (п + /) равна данному значению.
№
я + /
И
5
12
7
13
4
14
8
15
3
Задачи для самостоятельного решения
161
16-21. Напишите электронные формулы данного атома в основном и возбужденном
состояниях, покажите распределение валентных электронов по квантовым ячейкам,
определите число неспаренных электронов.
№
Атом
16
S
17
As
18
А1
19
Мп
20
С1
21
Ti
22-26. Укажите значения квантовых чисел и, /, mi для электронов, находящихся на
внешнем энергетическом уровне в данных атомах.
№
Атомы
22
Si,Mn
23
Ca,Ti
24
Sc,Pb
25
Se, Mo
26
Co,Pd
27-31. Определите порядковый номер элемента, у которого заканчивается заполне-
заполнение электронами орбиталей указанного подуровня.
№
Подуровень
27
3d
28
Ар
29
4/
30
5s
31
5d
32-35. Определите порядковый номер элемента, у которого начинается заполнение
электронами орбиталей данного подуровня.
№
Подуровень
32
Ad
33
5р
34
Is
35
4р
36-^46. Напишите электронные формулы указанного атома и его иона. Покажите для
них распределение электронов внешнего энергетического уровня по квантовым
ячейкам. Укажите количество неспаренных электронов.
№
36
37
38
39
40
41
Атом
S
Ti
А1
Sn
Mn
Со
Ион
s2-
Ti3+
Al3+
Sn2+
Mn2+
Со3+
№
42
43
44
45
46
Атом
Си
Сг
Аи
Sc
Pd
Ион
Си2+
Сг3+
Аи3+
Sc3+
Pd2+
47-51. Напишите электронные формулы атомов указанного ряда. Покажите распреде-
распределение электронов по квантовым ячейкам. Предскажите изменение орбитального радиу-
радиуса, энергии ионизации, сродства к электрону и электроотрицательности в этом ряду.
№
Ряд атомов
47
Cl-Br-I
48
N-O-F
49
N-P-As
50
O-S-Se
51
Al-Si-P
52-56. Напишите электронные формулы атомов указанного ряда. Покажите распре-
распределение электронов по квантовым ячейкам. Предскажите изменение орбитального
радиуса, энергии ионизации и электроотрицательности в этом ряду.
6 - 9795
162
Строение атомов и ионов. Периодический закон
№
Ряд атомов
52
Ga-Ge-As
53
In-Sn-Sb
54
Be-C-F
55
Ве-Са-Ва
56
B-N-F
57-62. Напишите электронные формулы указанных атомов. Покажите распределе-
распределение электронов внешнего уровня по квантовым ячейкам. Сравните орбитальные
радиусы и первые энергии ионизации этих элементов.
№
Атомы
57
P,S
58
Si, Ge
59
Mg,Al
60
Sn,Pb
61
Ca, Zn
62
Sr,Cd
63. Сродство к электрону атома углерода значительно больше, чем у атома азота.
Объясните причину этого, рассмотрев электронные конфигурации атомов.
64-73. Используя одноэлектронное приближение и табл. П.2.1, определите эффек-
эффективный заряд ядра для атомов следующих элементов.
№
64
65
66
67
68
Элемент
Не
Li
Be
В
С
№
69
70
71
72
73
Элемент
N
О
F
Na
Са
74-80. Зная длину волны излучения атомов элемента для указанного перехода элек-
электрона с одного подуровня на другой, рассчитайте разность энергий между этими
подуровнями.
№
74
75
76
77
78
79
80
Элемент
Li
К
Rb
Cs
Са
Sr
Ва
Переход
2p->2s
4р—>4s
5/?-> 5s
6p^>6s
4p->4s
5p->5s
6/7 —» 6s
Длина волны Л, нм
670,8
404,4
420,2
455,5
422,7
460,7
553,6
81-85. Определите, какое количество энергии выделяется при протекании или не-
необходимо затратить для протекания следующих процессов в газовой фазе, исполь-
используя табл. П.2.1.
№
81
82
83
84
85
Н + Li+ = Н+ + Li
Н + Na+ = Н+ + Na
Cl + Rb+ = СГ + Rb
F + K+ = F+ + К
H" + F = F + H
Процесс
Примеры решения задач 1 63
Химическая связь и строение вещества
Примеры решения задач
Задача 1. На основании табл. П.2.3, в которой приведены значения постоянного
дипольного момента \х молекул, длины связи / и энергии связи ЕА_В, предположить,
как будет изменяться характер связи в ряду галогеноводородов и в ряду галогени-
дов калия.
Решение. Связь в гетероатомных молекулах всегда в той или иной мере полярна, и
электронная плотность в них распределена несимметрично. Вектор дипольного мо-
момента Д связи условно считают направленным в сторону более электроотрицатель-
электроотрицательного атома, его величину рассчитывают по формуле D.4): ц = qU где q — заряд ди-
диполя, / — длина диполя.
По дипольному моменту можно судить о степени полярности связи. Однако
более наглядно это показывает распределение зарядов в молекуле. Вследствие раз-
разностей электроотрицательностей атомов в молекулах галогеноводородов и галоге-
нидов щелочных металлов электронная плотность всегда смещена к атому галогена.
Распределение зарядов в молекулах относительно элементарного электрического
заряда 8 = — приближенно можно рассчитать, принимая длину диполя
1,60 109Кл
равной длине связи.
Для решения задачи следует выписать из табл. П.2.3 необходимые данные: ц,
/, ?а-в- При расчете относительного заряда учитывают, что размерность ц в таблице
представлена в дебаях (Д = 3,33564 • 10~30 Кл • м), а / — в пм A пм = 10~12 м), и, сле-
следовательно,
3,33564- 10V Л1ц
о *
1,60-109/-102 /'
Относительное распределение заряда в галогеноводородах и галогенидах ка-
калия следующее:
HF 5н=+0,44; 8F=-0,44, KF 8K=+0,71; 8F=-0,71,
НС1 8н=+0,18; 8С1=-0,18, КС1 8к=+0,87; 8а=-0,87,
НВг 8н=+0,12; 6*=-0,12, КВг 8к=+0,76; 8Вг=-0,76,
HI 8H=+0,05; 8i = -0,05, KI 8K = +0,63; 8i = -0,63.
Отметим, что рассчитанные значения относительных электрических зарядов
атомов (часто называемых эффективными зарядами) не отвечают действительному
распределению зарядов в молекулярном диполе. При расчете не учитывали разли-
различия в размерах атомов, расположения в пространстве неподеленных электронных
пар галогенов, несовпадения центров зарядов и центров атомов и др. Однако значе-
значения эффективных зарядов могут служить ориентирами для оценки степени поляр-
полярности связи. Очевидно, что связь в галогенидах калия более полярна, чем в галоге-
6*
164 Химическая связь и строение вещества
новодородах. Среди галогеноводородов наиболее полярной является молекула HF, а
среди галогенидов калия — молекула КС1. При этом в ряду галогеноводородов на-
наблюдается тенденция увеличения энергии связи с увеличением эффективного заря-
заряда диполя, а в ряду галогенидов калия эту тенденцию нарушает KF.
Меру полярности связи можно оценить по критерию Полинга D.2):
^В-В '
где Еа-в, Еа-а и ?в-в — энергии связи в молекулах АВ, А2 и В2 соответственно. Чем
больше отклонение АЕ от нуля, тем более полярной является связь между атомами
А и В. Для заданных соединений получены следующие значения АЕ, кДж/моль:
HF ае = 566-л/436 159 = 303, KF Д? = 497-л/57-159 =402,
НС1 Д? = 432-л/436-243 = 106, KC1 Д? = 425-л/57-243 = 307,
НВг Д? = 366-л/436 194 = 75, КВг Д? = 382-л/57-194 =277,
HI АЕ = 298 -л/436 153 =40, KJ АЕ = 325 -л/57 -153 = 234.
Критерий Полинга также показывает, что полярность связи больше у галоге-
галогенидов калия и максимальна у KF. Расхождение в оценке меры полярности связи KF
и КС1 обусловлено допущениями, принятыми в расчетах.
Задача 2. Рассмотреть с позиций МВС образование молекул SF2, SF4 и SF6, опреде-
определить полярность молекул. При решении использовать табл. П.2.1 и П.2.2.
Решение. Центральным атомом в данных молекулах является атом серы, электрон-
ное строение которого в основном состоянии: КЬЗЛ/?4 (КЬЗз1 |^t| Ър |it| |_t_ |_tj).
Периферическими атомами являются атомы фтора, в основном состоянии которого
имеется один неспаренный электрон: K2s22/?5 (или K2s [tf] 2р |Щ[Или).
В методе валентных связей (МВС) образование молекул можно рассматри-
рассматривать с позиций теории гибридизации валентных орбиталей и теории отталкивания
электронных пар валентных орбиталей (ОЭПВО). Для образования молекулы SF2 по
обменному механизму центральный атом S должен предоставить два неспаренных
электрона, что соответствует основному состоянию атома. Следовательно, при об-
образовании химической связи, в соответствии с теорией гибридизации, валентные
орбитали центрального атома образуют четыре sp3-TO, на которые приходится
шесть электронов. Таким образом, молекула SF2 имеет угловое строение, валентный
угол (равен 98°) значительно меньше тетраэдрического, поскольку гибридные орбитали
не эквивалентны: в молекуле имеются две связывающие и две несвязывающие элек-
электронные пары. Геометрические конфигурации молекул SF2, SF4, SF6 приведены ниже.
Согласно теории ОЭПВО, молекула SF2 относится к типу АХ2Е2 (см. табл. 4.3) и
имеет угловое строение. Отличие валентного угла от тетраэдрического вызвано от-
отталкиванием несвязывающих электронных пар друг от друга и от связывающих
электронных пар молекулы (см. правило 1). Уменьшение валентного угла по срав-
сравнению с молекулой Н2О (см. табл. 4.3) можно объяснить уменьшением пространст-
пространства, занимаемого валентными электронными парами центрального атома серы (см.
Примеры решения задач
165
правило 4). В то же время валентный угол SF2 оказывается больше, чем в молекуле
H2S (92,2°), несмотря на то, что электроотрицательность фтора выше, чем водорода.
Это вызвано возможностью образования частичных кратных связей в молекуле SF2 (см.
правило 5).
Электронная плотность связывающей пары направлена в сторону перифери-
периферического атома (связь S—>? полярна). Молекула SF2 также полярна в соответствии с
правилом сложения векторов — дипольных моментов связей и неподеленных пар.
166 Химическая связь и строение вещества
Для образования молекулы SF4 по обменному механизму центральный атом S
должен предоставить четыре неспаренных электрона, что возможно при промотировании
валентных электронов S: КЬЗ^Зр4 —^-> KL3s23p33dl (KL3s Щ Ър \Т\ \Т\ \Т\ 3d [Т]).
При комбинации одной s-AO, трех р-АО и одной d-AO образуются пять sp3d-TO,
направленных к вершинам тригональной бипирамиды. Поскольку одна из ГО занята
несвязывающей электронной парой, молекула SF4 имеет форму искаженного тетра-
тетраэдра (валентные углы 104° и 89°).
По классификации теории ОЭПВО молекула SF4 относится к типу АХ4Е, не-
поделенная электронная пара находится в экваториальном положении, углы между
связывающими парами, находящимися в экваториальном положении, меньше 120°
вследствие отталкивания от неподеленной пары (см. правило 1), углы между эквато-
экваториальными и аксиальными парами близки к 90°. Длина аксиальных связей S-F*
A64 пм) больше экваториальных S-F A54 пм) по правилу 6.
Молекула SF4 полярна в соответствии с правилом сложения векторов.
Для образования молекулы SF6 по обменному механизму центральный атом
S должен предоставить шесть неспаренных электрона, что возможно при промоти-
промотировании валентных электронов S: KL3s23p4 —*—> КЬЗ^З^З*/ (KL3s |t] Зр \f] |t] [t|
^Щш)- При комбинации одной s-AO, трех/?-АО и двух d-AO, образуются шесть
sp3cr-TO9 направленных к вершинам октаэдра (квадратной бипирамиды). Соответ-
Соответственно молекула SF6 имеет октаэдрическое строение (валентный угол 90°) и по
правилу сложения векторов неполярна. По классификации теории ОЭПВО относит-
относится к типу АХ6.
Задача 3. Рассмотреть с позиций МВС образование молекул СО2 и SO2. При реше-
решении использовать табл. П.2.1 и П.2.2.
Решение. В молекуле СО2 центральный атом углерода находится в состоянии
K2sl2/?3 (или K2s [_TJ 2p Qj |_TJ [_TJ), поскольку ковалентно связан с двумя атомами
кислорода, имеющими по два неспаренных электрона: K2s22p4 (K2s |IT| 2р [И] Щ щ).
Согласно теории гибридизации, в образовании ст-связей, определяющих геометрию
молекулы, участвуют два электрона атома С, находящиеся на двух 5/?-гибридных
орбиталях. Остальные 2 электрона, находящиеся на р-АО, не подвергшихся гибри-
гибридизации, осуществляют тг-связи с атомами кислорода. Молекула СО2 соответствен-
соответственно имеет линейную форму, валентный угол составляет 180°, связи эквивалентны,
поэтому молекула неполярна. По классификации теории ОЭПВО молекула относится
к типу АХ2. Геометрические конфигурации молекул СО2, SO2 приведены ниже:
О с О
со2 со2
Примеры решения задач
167
Для образования молекулы SO2 центральный атом S также должен предоста-
предоставить четыре неспаренных электрона, что возможно при промотировании валентных
электронов S: KL3s23/?4—'-+ KL3s23p33d] (или KL3s |И| Ър \f\\f\\f\ 3d\f\). Одна-
Однако из четырех неспаренных электронов S только два электрона необходимы для об-
образования а-связей, так как число а-связей определяется количеством перифериче-
периферических атомов. Полагают, что валентные орбитали центрального атома подвергаются
частичной гибридизации, образуя три sjr-ГО с четырьмя электронами, из которых
одна орбиталь занята несвязывающей электронной парой, а две образуют а-связи с
атомами кислорода. Не подвергшиеся гибридизации р-АО и а1-АО участвуют в об-
образовании я-связей. Поэтому молекула SO2 имеет угловое строение. По классифи-
классификации теории ОЭПВО молекула относится к типу АХ2Е. Валентный угол лишь не-
немного меньше 120° (равен 119°), несмотря на наличие несвязывающей электронной
пары. Это объясняется тем, что связи S=O двойные и занимают в пространстве
больше места, чем одинарные (см. правило 3). Молекула полярна.
Задача 4. Рассмотреть с позиций МВС образование молекулы BF3 и иона [BF4]~.
При решении использовать табл. П.2.1 и П.2.2.
Решение. Центральному атому В для образования молекулы BF3 требуется три не-
неспаренных электрона: K2s22p] —*—> K2sl2p2 (K2s [Tj 2/? [_Tj [t] LJ)- Валентные ор-
орбитали бора находятся в ^/?2-гибридном состоянии, поэтому форма молекулы пло-
плоская, треугольная, валентный угол 120°, молекула неполярна. Геометрические кон-
конфигурации молекулы BF3 и иона [BF4]~ представлены ниже:
168
Химическая связь и строение вещества
Образование иона [BF4] можно рассматривать как процесс: BF3 + F = [BF4] . В
образовании связей участвуют все четыре валентные орбитали центрального атома, три
из которых образуют связи с атомами фтора по обменному механизму, а одна (свобод-
(свободная) — с ионом F~ по донорно-акцепторному механизму (ион F" — донор электронной
пары). Следовательно, валентные орбитали бора находятся в ^-гибридном состоянии,
форма иона — тетраэдр, валентный угол 109,5°. Положение F" в структуре [BF4]~ на
рисунке показано условно, отрицательный заряд принадлежит всей частице [BF4]~, так
как донорно-акцепторный механизм образования связи отличается от обменного только
происхождением общей электронной пары. По классификации теории ОЭПВО молеку-
молекула BF3 относится к типу АХз, а ион [BF4]~ — к типу АХ^
Задача 5. Рассмотреть с позиций ММО молекулу О2, супероксид-ион Оз и перок-
сид-ион О22'. Сравнить прочность связи в частицах и их магнитные свойства.
Решение. Метод молекулярных орбиталей (ММО) рассматривает образование мо-
молекул как результат формирования делокализованных, принадлежащих всей части-
частице, молекулярных орбиталей (МО). Одной из его основных задач является построе-
построение основанных на квантово-механических расчетах энергетических диаграмм МО.
Электронная конфигурация атома кислорода в основном состоянии: \s2ls22pA
или K2s22/?4, электронные конфигурации соответственно:
молекулы О2 [КЩст*J(а**J( а1р J( я2, = п2р> L( л*2„ = п\р> J];
супероксид-иона
[КК(сьJ(с1*2*J( а2р )\ л2р = я2р L( п' 1р< = к
K];
2~
пероксид-иона О2 [КК(а2,) (а*25) (a2,s) (n2Ps = п2р>) {п 2Рх = п 2р ) ].
Энергетические диаграммы МО данных частиц в упрощенном виде пред-
представлены ниже:
О,
¦Н-.
•V,
-м-
оГ
¦-Н- -И-
¦I
Примеры решения задач
169
Для молекулы О2 порядок связи в соответствии с B.8) ПС =
\-4
8-5 2-
супероксид-иона О2 ПС = =1,5; для пероксид-иона О2 ПС =
= 2,0; для
= 1,0.
,; д рд 2
Следовательно, связь в молекуле О2 прочнее.
Магнитные свойства частиц определяет наличие или отсутствие неспаренных
электронов. Молекула О2 и супероксид-ион О2 — парамагнитны, так как имеют
неспаренные электроны; пероксид-ион О2~ — диамагнитен, так как не имеет не-
неспаренных электронов.
АО
Е
Н
МО
¦t-°
HF
АО
2рх 2ру 2pz
\W\2s
Задача 6. Рассмотреть с позиций ММО
молекулу HF.
Решение. При построении энергетиче-
энергетических диаграмм МО гетероатомных мо-
молекул необходимо учитывать, что энер-
энергетические уровни АО атома, имеющего
большее значение электроотрицатель-
электроотрицательности, расположены ниже соответст-
соответствующих энергетических уровней парт-
партнера. Абсолютные, т.е. рассчитанные
по B.1), электроотрицательности ато-
атомов, образующих молекулу HF: хн =
= 7,18 эВ; Xf = Ю,41 эВ (см. табл.
П.2.1). Электронные конфигурации
атомов: Щ1*1]; F[ls22s22p5].
Образование МО HF можно уп-
упрощенно представить как линейную
комбинацию Is-АО атома водорода и
2pz-AO атома фтора, имеющих близкие значение энергии и одинаковые свойства
симметрии относительно оси молекулы. В результате образуются o]s_2p -CMO и
G*\s-2P -РМО. Остальные МО (а°ь; а°25; к°2р и п°2р ) являются несвязывающими.
На рисунке показана энергетическая диаграмма МО молекулы HF. Электронная
конфигурация молекулы HF может быть записана в виде: [(goijJ(go25J (ои_2р J
(п°2р = к°2р L]. Таким образом, основной вклад в химическую связь в молекуле HF вно-
вносит электронная пара на Gi5_2/?z-CMO. Порядок связи равен 1, молекула диамагнитна.
Задача 7. Рассчитать эффективные радиусы атомов алюминия, хрома и цинка. Для
решения использовать табл. П.3.2.
Решение. Поскольку металлическая связь не проявляет характерных для ковалентной
связи свойств направленности и насыщаемости, металлы кристаллизуются в соответст-
170 Химическая связь и строение вещества
вии с принципом плотной упаковки, и для них характерны объемно-центрированная
кубическая (ОЦК), гранецентрированная кубическая (ГЦК) и гексагональная плотно-
упакованная (ГПУ) кристаллические решетки. Элементарные ячейки металлических
решеток показаны ниже.
Для расчета эффективного радиуса атома металла гэфф следует определить
кратчайшее расстояние / между частицами в элементарной ячейке кристаллической
решетки, так как гэфф = 0,5/. Очевидно, что кратчайшее расстояние между атомами в
простой кубической (ПК) ячейке равно величине ребра куба: / = а; в ячейке ОЦК —
половине диагонали куба: / = —а\ в ячейке ГЦК — половине диагонали грани
куба: / = а; в ячейке ГПУ — стороне правильного шестиугольника основания
призмы: 1= а.
Чтобы определить величину а, необходимо определить объем элементарной
ячейки V. В случае ячеек ПК, ОЦК и ГЦК V= я3, а для ячейки ГПУ V= 4,24а3, так
как при плотной упаковке шаров высота призмы h = 1,63а. В то же время V = —,
Р
где т — масса элементарной ячейки; р — плотность металла.
Массу элементарной ячейки можно рассчитать по формуле т = , где п —
NA
число частиц, необходимое для построения элементарной ячейки, М — молярная
масса металла, NA — число Авогадро.
Для ячеек ПК, ОЦК и ГЦК а = з/-—, для ячейки ГПУ а = з/
Число частиц, необходимое для построения элементарной ячейки ПК, «=—8=1,
о
так как каждая частица в вершине куба принадлежит одновременно восьми кубам,
сходящимся в данной точке. Для построения элементарной ячейки ОЦК необходи-
необходимо я = — 8 +1=2 частицы, поскольку одна частица находится внутри элементарной
о
ячейки; а для ячейки ГЦК п= — 8 + — 6=4 частицы, так как на шести гранях куба
8 2
расположено по одной частице, принадлежащей одновременно двум кубам. Для
ячейки ГПУ п = —12 + — 2 + 3 = 6, поскольку на каждой из двенадцати вершин
призмы расположена частица, одновременно принадлежащая шести сходящимся в
одной точке ячейкам, в центре верхнего и нижнего основания призмы лежат части-
частицы, половина которых принадлежит данной ячейке и внутри призмы находятся еще
три частицы:
Примеры решения задач
171
ПК
ОЦК
ГЦК
ГПУ
Эффективный радиус атомов алюминия, кристаллизующегося в ГЦК решетке:
4 • 26,98 г/моль
_ V2 \AM
эффА1 4 \pNA ' \ 2698 -103г/м3- 6,02 -1023моль"!
= 1,43;1ООм =
= 0,143 нм.
Эффективный радиус атомов хрома, кристаллизующегося в ОЦК решетке:
\гм
эффСг:
= 0,433
2-51,996 г/моль
7190-103г/м3- 6,02-1023моль"
= 1,25- 1О-1Ом =
= 0,125 нм.
Эффективный радиус атомов цинка, кристаллизующегося в ГПУ решетке:
6М
= 0,5з
6 • 65,39 г/моль
4,24 -7133-103г/м3 • 6,02 • 1 О^моль
= 1,39- 100m = 0,139 нм.
172
Химическая связь и строение вещества
Задачи для самостоятельного решения
1-15. В соответствии с основными положениями ММО постройте энергетические
диаграммы МО, определите магнитные свойства, сравните порядок и энергию связи
в ряду частиц.
№
1
2
3
4
5
Частицы
Н2 , Н2, Н2
Не2+, Не2, Не2"
Li2+, Li2, Li2"
Ве2+, Ве2, Ве2"
В2 , В2, В2
№
6
7
8
9
10
Частицы
Сг , С2, С2~
N2+,N2,N2"
о2+,о2,о2-
F2+, F2, F2"
Ne2+, Ne2, Ne2"
№
11
12
13
14
15
Частицы
B2, C2, N2
C2,N2,O2
N2, O2, F2
O2, F2, Ne2
NO+, NO, NO"
16-30. Используя табл. П.2.2, опишите с позиций МВС молекулы и ионы, имеющие
один и тот же центральный атом, определите геометрическую конфигурацию час-
частиц и полярность молекул, укажите наличие несвязывающих электронных пар, а- и
я-связей, отметьте механизм образования связей (обменный или донорно-
акцепторный), объясните причины отличия валентных углов от характерных для
данного типа гибридизации валентных орбиталей центрального атома.
№
16
17
18
19
20
Частицы
А1С13, [А1С14Г
BeF2, [BeF4]2"
BrF3, [BrF4r
ССЦ, CC12O
GeCl2, GeO2
№
21
22
23
24
25
Частицы
[C1F2]+, C1F5
[IC12]-, [IC12]+
NH4+, NH2"
NOF, NOF3
PF5, [PF6]~
№
26
27
28
29
30
Частицы
PbCl2, PbCl4
so2, so3
SiF4, [SiF6]2~
SnCl2, SnO2
XeF2, XeF4
31-45. Используя табл. П.2.2, опишите с позиций МВС молекулы и ионы, состоя-
состоящие из одинакового числа атомов, определите геометрическую конфигурацию час-
частиц и полярность молекул, укажите наличие несвязывающих электронных пар, а- и
я-связей, отметьте механизм образования связей (обменный или донорно-акцеп-
торный), объясните причины различия в строении данных частиц, а также отличия
валентных углов от характерных для данного типа гибридизации валентных орби-
орбиталей центрального атома.
№
31
32
33
34
35
Частицы
Bel2, NO2
HCN, SnCl2
HOF, SnO2
NCS", Pbl2
CS2, OF2
№
36
37
38
39
40
Частицы
A1I3, Asl3
BBr3, BiBr3
GaCl3, PC13
BF3, BrF3
SbF3, [XeF3]+
№
41
42
43
44
45
Частицы
SnCl4, TeCl4
SiF4, SeF4
PC13O, XeO2F2
SbCl5, [XeF5]+
PCI5, XeOF4
Задачи для самостоятельного решения
173
46-60. Укажите ион-комплексообразователь и лиганды, определите геометриче-
геометрическую конфигурацию комплексного иона, используя МВС.
№
46
47
48
49
50
Комплексные
ионы
[Ag(CNJ]"
[Cd(H2ON]2+
[Co(CNN]3-
[Au(NH3J]+
[Cr(H2ON]3+
[Zr(OHL]2-
[CuCl4]2-
[Zn(H2ON]2+
[Fe(CNN]3-
[Hg(NH3L]2+
№
51
52
53
54
55
Комплексные
ионы
[ZnCl4]2"
[Au(CNJ]"
[MoCl6]3-
[Ag(NH3J]+
[CdCy4"
[Hg(NCSL]2"
[МпСЦ]2"
[Ir(H2ON]3+
[Cu(CNJ]"
[OsBr6]2"
№
56
57
58
59
60
Комплексные
ионы
[Rh(H2ON]3+
[Ptld2'
[Pd(NH3N]4+
[Ni(CNL]2-
[ЩОНN]2-
[Ag(NH3J]+
[Pt(NH3N]4+
[HgCL,]2"
[V(H2ON]3+
[CoCl4]2"
61-75. Рассчитайте эффективные заряды 5 атомов и критерий Полинга Д?, ис-
используя табл. П.2.2; сравните полученные значения с рассчитанными в задаче 1
аналогичными значениями для соответствующих галогеноводородов и галоге-
нидов калия (например, LiF, LiBr, HF, HBr, KF, KBr) и сделайте выводы о по-
полярности молекул.
№
61
62
63
64
65
Молекулы
LiF, LiBr
LiH, Lil
NaCl, Nal
RbBr, RbF
CsF,CsCl
№
66
67
68
69
70
Молекулы
Lil, Csl
NaCl, CsCl
LiF, RbF
LiBr, RbBr
RbF, CsF
№
71
72
73
74
75
Молекулы
BrF, RbF
C1F, BrF
IBr, ICl
NO, CO
CO,CS
76-90. Используя табл. П.3.1, объясните изменение температуры плавления и окра-
окраски солей, учитывая явление поляризации ионов.
№
76
77
78
79
80
Соли
LiF, LiCl
NaF, NaBr
KF, KC1
BeF2, BeCl2
CaF2, CaCl2
№
81
82
83
84
85
Соли
PbCl2, Pbl2
SbF3, SbCl3
AgCl, AgBr
CoF2, CoCl2
NiF2, NiCl2
№
86
87
88
89
90
Соли
A1C13, GaCl3
CdCl2, HgCl2
ScCl3, YC13
NaCl, Nal
KBr, KI
174
Химическая связь и строение вещества
91-105. Рассчитайте эффективные радиусы атомов металлов, используя табл. П.3.2.
№
91
92
93
94
95
Металлы
Ag,W,Be
Аи, Ва, a-Hf
a-Yb, V, La
Ir, Eu, a-Ho
Си, а-Fe, a-Y
96
97
98
99
100
Металлы
a-Co, К, a-Cd
p-Li, a-Mn, a-Gd
Ni,Nb,a-Na
a-Sr, Mo, Mg
Pd, Rb, a-Sc
101
102
103
104
105
Металлы
Pt, Та, Os
Rh, Cs, Re
a-Th, W, a-Ti
Ge, K, a-Tl
a-Sn, V, a-Zr
Ill
ЗАКОНОМЕРНОСТИ
ПРОТЕКАНИЯ
ХИМИЧЕСКИХ ПРОЦЕССОВ
¦ Тепловой эффект
химической реакции
¦ Направление
химической реакции
При изучении и анализе закономерностей проте-
протекания любых химических реакций в современной
химии применяют два основных метода — тер-
термодинамический и кинетический. Познаваемый
объект в обоих методах один, но принципиаль-
принципиальные подходы и исследуемые свойства различные.
Соответствующие области науки, называемые
химической термодинамикой и химической ки-
кинетикой, в широком смысле являются составны-
составными частями термодинамики и кинетики как раз-
разделов физики.
Термодинамика — наука о наиболее общих свой-
свойствах макроскопических (состоящих из очень
большого числа частиц) систем, находящихся в
состоянии термодинамического равновесия, и
процессов перехода между равновесными со-
состояниями. Коротко ее определяют как науку о
теплоте, работе и температуре. Базис всей термо-
термодинамики составляют три основных закона (на-
(начала), с помощью которых может быть выведено
любое ее положение. Подобно математическим
аксиомам, термодинамические начала имеют ха-
характер постулатов — это фундаментальные зако-
законы природы, многократно подгвервденные и ни
разу не опровергнутые всем опытом развития
человечества. Исторически термодинамика воз-
возникла в первой половине XIX в. при разработке
теории паровой машины (С. Карно) и исследова-
исследовании зависимости между теплотой и работой
(Ю. Майер, Д. Джоуль, Г. Гельмгольц). Позже
основные термодинамические положения и
приемы были применены к описанию химиче-
химических реакций и физико-химических явлений (фа-
(фазовые переходы веществ, процессы растворения
и др.), вследствие чего возникла новая наука —
химическая термодинамика.
5 ¦ Тепловой эффект химической реакции
Химическая термодинамика позволяет решать следующие важные
задачи:
1) рассчитывать тепловые эффекты процессов (химических реакций,
фазовых переходов, процессов в растворах и др.) и энергий связей на
основе первого закона термодинамики;
2) определять направления самопроизвольного протекания процесса
при фиксированных внешних условиях на основе второго закона
термодинамики;
3) рассчитывать константы равновесия химических реакций и равно-
равновесный состав систем, а также определять оптимальные условия про-
проведения реакций (максимизация выхода продукта) на основе третьего
закона термодинамики.
5.1. Основные понятия и определения
химической термодинамики
Термодинамическая система (далее система) — часть про-
пространства, выделенная для рассмотрения и отделенная от окружаю-
окружающей среды реальной (межфазовой) или условной границей (так на-
называемой контрольной поверхностью). Системы могут быть изоли-
изолированными, закрытыми (замкнутыми) и открытыми. Изолированная
система характеризуется постоянством массы т, объема V и энергии
U (m= const, V = const, U = const), она не обменивается с окружаю-
окружающей средой ни веществом, ни энергией. Закрытая система обмени-
обменивается с окружающей средой только энергией и не обменивается ве-
веществом (т = const, V * const, U * const). В открытой системе
осуществляются оба указанных вида обмена с окружающей средой
(т * const, V # const, U * const).
Состояние системы определяется ее физическими и химиче-
химическими свойствами (объем, давление, температура, химический
состав, внутренняя энергия, энтальпия, энтропия и др.), которые под-
подразделяют на параметры состояния и функции состояния. Парамет-
Параметры состояния — свойства системы, выбранные в качестве независи-
независимых переменных. Функция состояния — величина, определяемая
этими параметрами, однозначно характеризует систему и не зависит
от пути ее перехода из одного состояния в другое. Например, для
1 моль идеального газа в качестве параметров состояния можно произ-
произвольно выбрать два параметра из трех: давление /?, объем V, абсолют-
абсолютную температуру Т. Третий параметр можно рассчитать по уравнению
5.7. Основные понятия и определения химической термодинамики \ 77
состояния Менделеева—Клапейрона pV = RT, где Л = 8,31 Дж/(моль • К) —
универсальная газовая постоянная.
Различают экстенсивные и интенсивные параметры состояния. Экс-
Экстенсивными называют параметры состояния, пропорциональные количеству
вещества системы (объем, масса, заряд, внутренняя энергия, энтальпия, эн-
энтропия и т.д.). Интенсивными — параметры состояния, не зависящие от ко-
количества вещества (температура, давление, электрический потенциал и др.).
Удельные значения экстенсивных параметров являются интенсивными (на-
(например, молярная концентрация и молярный объем вещества).
Важным в термодинамике является понятие процесса. Процессом на-
называют любое изменение параметров системы. Различают следующие виды
процессов: изотермический (Т = const), изобарный (р = const), изохорный
(V= const), изохорно-изотермический (V = const, T= const), изобарно-изо-
термический (р = const, T = const) и др. Самопроизвольные процессы проис-
происходят сами по себе, без какого-либо воздействия извне. Одни из них закан-
заканчиваются быстро, другие требуют для своего завершения большего времени.
Химическая реакция может быть сколь угодно медленной, но самопроиз-
самопроизвольной. Цикл — замкнутый процесс, параметры состояния которого изме-
изменяются, но в конце возвращаются к своим исходным значениям (например,
цикл Карно, состоящий из двух изотерм и двух адиабат).
В качестве идеализированного химического процесса в термодинами-
термодинамике, как правило, рассматривают реакцию, протекающую в изобарно-изо-
термических условиях. Равновесным называют такое состояние системы,
при котором ее свойства постоянны во времени и пространстве (отсутству-
(отсутствуют потоки вещества и энергии). Стационарным называют такое состояние
системы, при котором ее свойства постоянны во времени, но изменяются в
пространстве по координатным осям (имеются потоки вещества и энергии).
Примером системы, находящейся в стационарном состоянии, служит бас-
бассейн с проточной водой.
Произвольное изменение одного или нескольких исходных парамет-
параметров системы, находящейся в состоянии равновесия, приводит к смещению в
ней положения равновесия в том или ином направлении. Через некоторое
время система приходит к новому равновесному состоянию, из которого ее
можно вернуть в прежнее, приведя параметры к исходным значениям (так
называемый обратимый процесс). Равновесным (квазистатическим) назы-
называют процесс, при котором система непрерывно проходит через ряд равно-
равновесных состояний вследствие бесконечно малых изменений параметров. Не-
Неравновесным (нестатическим) — процесс, при котором система выводится
из состояния равновесия вследствие конечного изменения параметров, по-
после чего стремится вернуться к нему. При равновесности процессов в окру-
окружающей среде квазистатический процесс можно провести обратимо без ка-
178 5. Тепловой эффект химической реакции
ких-либо изменений вне системы. Обычно в химической термодинамике
термины «равновесный», «обратимый», «квазистатический» — синонимы.
Реальные природные и технологические процессы не могут быть равновес-
равновесными. В равновесном процессе работа W9 совершаемая системой против сил
окружающей среды при переходе из начального состояния в конечное, мак-
максимальна (^Гравн > %еравн)> а работа, совершаемая силами окружающей среды
над системой по переводу ее из конечного состояния в начальное, мини-
минимальна (|*Гравн| < |^неравн|).
Термодинамические системы подразделяют на гомогенные и гетеро-
гетерогенные в зависимости от числа составляющих фаз. Фаза — однородная часть
системы, ограниченная поверхностью раздела и характеризующаяся в отсут-
отсутствие внешних сил постоянством физических и химических свойств во всех
точках. Обычно понятие фазы относят к системе, у которой объемы гомоген-
гомогенных частей не очень малы, чтобы исключить влияние молекул, расположен-
расположенных вблизи поверхности раздела и обладающих избыточной энергией, на
свойства системы. Гомогенной называют систему, составляющие части кото-
которой образуют одну фазу, например, смесь идеальных газов, водный раствор
соли, сплав серебра и золота. Гетерогенной — систему, составляющие части
которой образуют несколько фаз, отделенных одна от другой поверхностями
раздела, например, смесь двух кристаллических веществ, насыщенный вод-
водный раствор соли с ее осадком, смесь ограниченно растворимых друг в друге
жидкостей, сплав кадмия и висмута, представляющий в твердом состоянии
смесь отдельных кристаллов этих веществ.
Фазовым переходом называют равновесный изотермический переход
вещества из одного фазового состояния в другое. Примерами фазовых пере-
переходов служат взаимные превращения твердого, жидкого и газообразного
состояний вещества. Плавление — равновесный переход вещества из твер-
твердой фазы в жидкую, кристаллизация — обратный процесс. Если вещество
равновесно переходит из жидкого состояния в газообразное, процесс назы-
называют испарением, обратное превращение — конденсацией. Для равновесного
перехода вещества из твердого состояния в газообразное прямой процесс —
сублимация {возгонка), а обратный — конденсация (десублимация). К фазо-
фазовым переходам относятся также аллотропные (полиморфные) превращения
кристаллических веществ, например, переход серы из орторомбической моди-
модификации в моноклинную, углерода — из графита в алмаз.
Внутренняя энергия U характеризует общий запас энергии в системе.
Она включает все виды энергии движения и взаимодействия составляющих
структурных единиц (молекул, атомов, ядер, электронов), за исключением
кинетической энергии системы как целого и ее потенциальной энергии в
поле внешних сил. Внутренняя энергия представляет собой функцию со-
состояния. Абсолютную величину U нельзя определить даже для простейших
5.2. Первый закон термодинамики и его приложение 179
систем, однако в этом нет необходимости, так как в теории и на практике
определяют изменение этой величины Ш = Ui-U\ при переходе системы
из состояния 1 в состояние 2. Значение АС/ положительно, если в ходе про-
процесса внутренняя энергия возрастает.
Энергия может передаваться от одной системы к другой в виде теплоты
Q или работы W. Теплота Q — микроскопическая форма передачи энергии
при столкновении хаотически движущихся молекул и атомов соприкасаю-
соприкасающихся тел. Энергия передается от более нагретых частей системы к менее
нагретым. Химические реакции, идущие с выделением теплоты, называют
экзотермическими (от греч. е'хо — вне, снаружи), при этом система нагре-
нагревается и отдает энергию в окружающую среду, Q < 0. Химические реакции,
идущие с поглощением теплоты, называют эндотермическими (от греч.
e'ndon — внутри), система охлаждается и забирает энергию из окружающей
среды, Q > 0. При размещении системы в вакууме теплообмен отсутствует,
6=0.
Работа W обусловлена действием системы против внешних сил, на-
нарушивших равновесие. Работа есть макроскопическая форма передачи энер-
энергии от упорядоченного поступательно движущегося потока большого числа
частиц системы к частицам окружающей среды с созданием в ней аналогич-
аналогичного потока. Система совершает работу расширения (W> 0), окружающая
среда совершает над системой работу сжатия {W< 0).
В качестве одного из видов работы в химической термодинамике рас-
рассматривается механическая работа расширения (или сжатия) системы реа-
реагирующих веществ WMex. При постоянном внешнем давлении
где AV— изменение объема системы. В отличие от внутренней энергии, те-
теплота и работа не являются функциями состояния, они зависят от вида про-
процесса. Об изменении внутренней энергии в процессе судят по количеству
переданной теплоты и количеству совершенной работы. Единицы измере-
измерения теплоты и работы совпадают с единицами измерения энергии. В СИ
[Q] = [W] = Дж или кратные ему единицы.
5.2. Первый закон термодинамики и его приложение
к процессам в идеальном газе
Первый закон термодинамики вытекает из более общего закона сохране-
сохранения энергии. Для термодинамической системы он формулируется следующим
образом: количество теплоты Q, сообщенное системе, расходуется на увеличе-
увеличение ее внутренней энергии At/и на совершение работы ^системой, т.е.
180 5. Тепловой эффект химической реакции
Q=AU+W. E.1)
Для элементарных процессов с бесконечно малыми изменениями па-
параметров E.1) принимает вид:
bQ = dU + bW= dU+pdV+ bW\ E.2)
rnepdV— работа расширения системы; bW— сумма других видов работ (элек-
(электрической, сил поверхностного натяжения и др.). Внутренняя энергия соответ-
соответствует функции состояния системы, поэтому в E.2) перед символом U постав-
поставлен знак полного дифференциала Теплота и работа не являются функциями
состояния, и их бесконечно малые количества обозначены буквой 8.
Уравнения E.1) и E.2) описывают первый закон термодинамики в ин-
интегральной и дифференциальной формах соответственно. В термомехани-
термомеханических системах при протекании процессов совершается только работа
расширения или сжатия, т.е. bW'= 0. Тогда E.2) принимает вид:
bQ=dU+pdV. E.3)
Рассмотрим, используя уравнение E.3), применение первого закона
термодинамики к так называемым изопроцессам в идеальном газе.
1. Изотермический процесс (Т = const). В идеальном газе силы меж-
межмолекулярного взаимодействия равны нулю. Внутренняя энергия идеально-
идеального газа зависит от его температуры, количества вещества и не зависит от
давления и объема, поэтому для данных условий U = const, dU = 0. Уравне-
Уравнение E.3) имеет вид:
bQT=bW=pdV, E.4)
Qt=W. E.5)
Теплота, сообщенная системе, в изотермическом процессе полностью
расходуется на совершение работы расширения. Для 1 моль идеального газа
RT
р = . Подставив значение давления в E.4) и проинтегрировав, получим
выражение для работы изотермического расширения 1 моль идеального газа
W = RT\n^- = ЛПп^Ц E.6)
У\ Pi
так как по закону Бойля—Мариотта (pV)T= const.
2. Изохорный процесс (V= const). При постоянном объеме dV= 0, зна-
значит, работа расширения газа bW= pdV= 0. Уравнение E.3) принимает вид:
E.7)
QV=U2-U] = AU. E.8)
5.3. Понятие теплового эффекта химической реакции 181
В изохорном процессе теплота, сообщенная системе, полностью рас-
расходуется на увеличение ее внутренней энергии и характеризует изменение
состояния системы.
3. Изобарный процесс (р = const). Постоянную величину/? можно вне-
внести под знак дифференциала, поэтому работа расширения bW= pdV= d(pV).
Тогда E.3) имеет вид
8QP= dU + d(pV) = d(U+ pV) = dH, E.9)
QP=H2-HX=AH. E.10)
Величины pV и U характеризуют состояние системы. Их сумма
Н= U + pV также соответствует функции состояния, которую называют
энтальпией (от греч. enthalpo — нагреваю). По физическому смыслу эн-
энтальпия есть энергия расширенной системы.
В изобарном процессе теплота, сообщенная системе, расходуется на
увеличение ее внутренней энергии и на совершение работы расширения
против сил внешнего давления и характеризует изменение состояния систе-
системы. Работа расширения идеального газа от объема V\ до объема V2 в равно-
равновесном изобарном процессе определяется уравнением
v2
W= jpdV = p(V2-Vl). E.11)
5.3. Понятие теплового эффекта химической реакции
Выделение или поглощение теплоты является одним из основных
признаков химической реакции. Для газофазных реакций причина этого за-
заключается в различии энергий химических связей, существующих между
атомами в молекулах исходных веществ и продуктов реакции (известно, что
отдельные молекулы веществ присутствуют только в газовой фазе). Реакция
образования 1 моль хлороводорода НС1 из простых веществ, в которой ис-
исходные вещества (Н2, С12) и продукт реакции (НС1) являются газами:
~ Н 2 (г) + "Г С1 2 (г) = НС1 (Г)
Для превращения исходных веществ в продукты формально необхо-
необходимо затратить энергию на разрыв химических связей Н—Н в 0,5 моль во-
водорода и С1—С1 в 0,5 моль хлора:
182 5. Тепловой эффект химической реакции
Езатр =±?дн2 + -ЕЛс\2 =1D36+ 242,6) = 339,3 кДж,
где Ед — энергия диссоциации молекул, кДж/моль. При этом образуется по
1 моль атомов Н и С1, которые реагируют между собой с образованием 1 моль
НС1. При образовании связей Н—-С1 выделяется энергия EBbm = -EJlHc\ =
= -431,6 кДж.
Суммарный тепловой эффект реакции ?Сумм = Езптр + ЕЕЪЩ = 339,3 -
- 431,6 = -92,3 кДж. Энергия выделилась из системы реагирующих веществ,
поскольку химические связи в продукте реакции в среднем прочнее, чем в
исходных веществах.
Аналогично образование 1 моль йодоводорода из простых веществ в
газовой фазе
Т Н 2 (г) + "Г I 2 (г) = HI (Г)
сопровождается поглощением энергии
436 + A48,8 + 62,4) =
= 323,6 кДж,
где Есу6л1г — энергия сублимации йода; Евъщ = -ЕлШ = -298,3 кДж. Сум-
Суммарный тепловой эффект реакции Есумм = 323,6 - 298,3 = 25,3 кДж. Энергия
поглотилась системой реагирующих веществ, так как химические связи в
продукте реакции в среднем слабее, чем в исходных веществах. Тепловым
эффектом химической реакции называют количество теплоты, выделяемое
или поглощаемое в результате осуществления химического процесса в тер-
термомеханической системе {bW = 0) при постоянном давлении или объеме
(р = const или V = const) и равенстве температур исходных веществ и про-
продуктов (Гисх = Гпрод).
Раздел химической термодинамики, изучающий тепловые эффекты ре-
реакций и фазовых превращений, называют термохимией. Термохимические
измерения позволяют определить исходные величины для расчета энергии
химических связей в молекулах, кристаллической решетки, энтальпию гидра-
гидратации ионов в кристаллогидратах и др. Стехиометрическое уравнение реак-
реакции, записанное с указанием агрегатных состояний исходных веществ и про-
продуктов, а также теплового эффекта, называют термохимическим.
Различают тепловой эффект реакции при постоянном объеме (Qv) и
при постоянном давлении (Qp). Согласно первому закону термодинамики
(см. E.8) и E.10)), Qv = A,-f/(тепловой эффект при постоянном объеме равен
5.3. Понятие теплового эффекта химической реакции 183
изменению внутренней энергии), Qp = АГН (тепловой эффект при постоянном
давлении равен изменению энтальпии). Тепловые эффекты АГ1/^ и Дг#298>
приведенные к стандартным термодинамическим условиям (р = 101325 Па =
= 1 атм = 760 мм рт. ст., что означает индекс «0» справа вверху; Т= 298,15 К,
что указывает индекс «298» справа внизу), называют стандартной внут-
внутренней энергией и стандартной энтальпией реакции соответственно. Знак А
показывает, что речь идет об изменениях внутренней энергии и энтальпии в
результате реакции, но для краткости формулировок слово «изменение»
опускают.
Исторически сложились термохимическая и термодинамическая фор-
формы записи термохимических уравнений реакций, характеризующиеся вза-
взаимно обратными знаками тепловых эффектов. Выделяемая теплота в термо-
термохимическом обозначении имеет знак «+» (Qp > 0) (рассматривается возрас-
возрастание энергии окружающей среды), а в термодинамическом — знак «-»
(Дг#298 < 0) (убывание энергии системы). При рассмотрении в термодина-
термодинамической системе знаков величины Дг#298 и Qp совпадают по модулю и
знаку. В настоящее время термохимическая система устарела и употребля-
употребляется редко.
С учетом вышесказанного запишем термохимическое уравнение реак-
реакции полного окисления (горения) углерода (р9Т= const) в термохимической
и термодинамической формах соответственно:
СAТафит)+ О2(Г)= СО2(Г)+ 393,5 кДж (или Qp = 393,5 кДж),
О2 (г) = СО2 (D (Дг#2°98 = Qp= -393,5 кДж).
Нижний индекс, стоящий у формулы вещества, обозначает его агрегатное
состояние: (г) — газообразное, (ж) — жидкое, (к) — кристаллическое или
(т) — твердое.
Тепловой эффект может быть отнесен ко всей реакции (измеряется в
кДж) или к 1 моль любого /-го участника реакции (измеряется в кДж/моль /-го
вещества). Например, для реакции
N2(r) + 3H2(r)=2NH3(r)
Дг#298 = -92,4 кДж, или Д,.#298 = -46,2 кДж/моль NH3, или Дг#298 =
= -30,8 кДж/моль Н2. Указанное значение, теплового эффекта относится к
стандартным термодинамическим условиям: Гисх = ГпрОд= 298,15 К; парци-
парциальное давление каждого газообразного вещества, участвующего в реакции,
поддерживается постоянным и равным 1 атм A01325 Па) (общее давление
равно 3 атм). Если все участники реакции находятся в конденсированном
состоянии, то внешнее атмосферное давление поддерживается равным 1 атм.
184 5. Тепловой эффект химической реакции
Поскольку Н = U + pV, то для реакции, проведенной в изобарно-изо-
термических условиях,
где A VT = (Е Рпрод. г - ? )^исх. г) — разность объемов газообразных веществ в правой
и левой частях уравнения реакции. Полагая, что газообразные вещества-
участники реакции подчиняются уравнению состояния идеального газа, имеем
pAVr=AvrRT,
где Avr = (ХУпрод. г - ?vHCX. г) — разность между количеством (числом молей)
газообразных веществ в правой и левой частях уравнения реакции. При од-
одних и тех же условиях объем газа на три порядка превышает объем жидко-
жидкости или твердого вещества, поэтому при термодинамических расчетах рабо-
работы расширения не учитываются объемы конденсированных веществ.
Для приведенной выше реакции горения углерода Avr = vco? ~ v0? =
= 1-1=0, для реакции синтеза аммиака Avr = vNH -vN-vH =2-1-
- 3 = -2 моль.
С учетом проведенных рассуждений стандартные тепловые эффекты
при постоянном давлении и постоянном объеме связаны уравнением
E.12)
где Т= 298 К.
5.4. Стандартные энтальпии образования
и сгорания веществ
Сложные вещества можно гипотетически или реально синтезировать
из соответствующего количества простых веществ в стандартных термоди-
термодинамических условиях, например:
С(графит) + О2(Г)= СО2(Г) (Дг#2°98 = -393,5 кДж), E.13)
6С(графит) + ЗН2(Г)= С6Н6(Ж) (Аг#2°98 = 49 кДж), E.14)
2Na(T)+S(,p0M6)+ l,5O2(r)=Na2SO3(T) (Artf2°98 =-1090кДж). E.15)
Простые вещества, участвующие в приведенных реакциях, должны на-
находиться в стандартных состояниях. В термодинамике стандартное состоя-
состояние вещества может быть выбрано при любой температуре (она не входит в
5.4. Стандартные энтальпии образования и сгорания веществ 185
понятие стандартного состояния), но чаще всего принимают Т= 298,15 К, что
указывают подстрочным индексом.
За стандартное состояние твердого вещества при Т = 298 К принима-
принимают его чистый кристалл под давлением/? = 101,3 кПа. При наличии у веще-
вещества двух или нескольких аллотропных модификаций стандартным состоя-
состоянием считают наиболее термодинамически стабильную форму. Например,
для углерода, имеющего четыре аллотропные модификации (алмаз, графит,
карбин и фуллерен), наиболее термодинамически устойчив графит. Исклю-
Исключение составляют фосфор и олово. Для них за стандартное состояние при-
принимают реакционноспособный белый фосфор и белое Р-олово, а не более
термодинамически устойчивые черный фосфор и серое а-олово.
За стандартное состояние жидкого вещества (бром, ртуть) при Т = 298 К
принимают чистую жидкость под давлением р = 101,3 кПа. Для газообраз-
газообразного при этой же температуре вещества (водород, гелий, кислород, азот
и др.) стандартным является состояние гипотетического идеального газа,
имеющего летучесть (фугитивность) / = 101,3 кПа и свойства бесконечно
разреженного газа.
Стандартной энтальпией образования вещества (Ду//298) (f0T англ-
formation — образование) называют тепловой эффект (стандартную энталь-
энтальпию Дг#298) реакции образования 1 моль данного вещества из соответст-
соответствующего количества простых веществ, находящихся в стандартных условиях.
Стандартные энтальпии реакций E.13)—E.15) есть стандартные эн-
энтальпии образования диоксида углерода, бензола и сульфита натрия соот-
соответственно. Стандартные энтальпии приведенных ниже реакций не могут
быть энтальпиями образования диоксида углерода,
С(алмаз) + Оад = СО2(г) (Дг#2°98 = -395,3 кДж); E.16)
СО(Г) + 0,5О2(г) = СО2(Г) (АГЯ2°98 ^ -283 кДж), E.17)
так как в первом случае углерод не находится в стандартном состоянии, а во
втором — одно из исходных веществ (монооксид углерода) не является про-
простым.
Для реакций
Н2(г) + 0,5О2(г) = Н2О(Ж) ( АГЯ2°98 ^ -285,8 кДж); . E.18)
Над + 0,5О2(г)= Н2О(Г) (АГЯ2°98 s-241,8 кДж). E.19)
тепловые эффекты при постоянном давлении — это энтальпии образования
жидкой и газообразной (гипотетический пар) воды соответственно. Разность
величин Аг#298 этих реакций равна тепловому эффекту изобарно-изо-
186 5. Тепловой эффект химической реакции
термического фазового перехода НгО(Г) = НгОдо при/? =101,3 кПа и Г= 298 К
(-44,0 кДж).
Согласно определению, стандартная энтальпия образования любого
простого вещества в термодинамически стабильном состоянии равна нулю
при любой температуре: Ду#? (простого вещества) = 0. Это начальная точ-
точка на шкале отсчета энтальпий образования сложных веществ и термодина-
термодинамически неустойчивых модификаций простых веществ. Стандартную эн-
энтальпию образования катиона водорода Н+ (точнее, катиона гидроксония
НзО+) в бесконечно разбавленном водном растворе принимают равной нулю
(условно, поскольку нельзя определить энтальпии образования отдельно для
Н+(р) и ОН~(р)). Энтальпии образования ионов в водном растворе используют
для расчетов тепловых эффектов реакций, протекающих в этой среде.
Стандартные энтальпии образования определены для большого числа
веществ и сведены в таблицы (см. табл. П.5.1).
Стандартной энтальпией сгорания вещества (Дс#298) (от англ. com-
combustion — горение) называют тепловой эффект (стандартную энтальпию
Дг#298) реакции окисления 1 моль вещества кислородом с образованием
соответствующих продуктов в стандартных условиях. Обычно эта величина
определяется для органических соединений, поскольку многие неорганиче-
неорганические вещества не горят. Для углеводородов продуктами сгорания являются
газообразный диоксид углерода и жидкая вода. Вид и агрегатное состояние
других продуктов реакции оговаривается специально. Как правило, азот,
входящий в состав сжигаемого соединения, превращается в N2(r), галогены
Cl, Br, I — в НС1(Г), НВГ(Г) и Н1(Г), сера — в SO2(r), но не в 8Оз(Г), так как окис-
окисление диоксида серы в триоксид характеризуется большой энергией актива-
активации и осуществляется только каталитическим путем.
По определению, стандартная энтальпия сгорания кислорода, воды,
диоксида углерода и других высших оксидов равна нулю при любой темпе-
температуре: Дс//298 (^2(г)> Н2О(Ж), СС>2(г)) = 0, так как они не окисляются кислоро-
кислородом. Это начальная точка на шкале энтальпий сгорания любых веществ.
Стандартную энтальпию Дс#298 рассчитывают по теплоте сгорания точной
навески вещества в избытке кислорода. Стандартные энтальпии сгорания
определены для многих веществ (см. табл. П.5.2).
При образовании сложного вещества из простых теплота может выде-
выделяться или поглощаться: при Д/#298 < 0 образуется термодинамически бо-
более стабильное соединение с меньшим запасом энергии, при Aj-H^ > 0 —
вещество с большим запасом энергии, менее стабильное с термодинамиче-
термодинамической точки зрения. Таким образом, стандартная энтальпия образования ве-
5.5. Закон Гесса и следствия из него
187
щества может быть отрицательной, равной нулю (для простых веществ в
стандартных состояниях) и положительной величиной. При горении ве-
веществ всегда выделяется теплота, поэтому стандартная энтальпия сгорания
отрицательна для всех веществ, способных окисляться в кислороде, или
равна нулю для негорючих соединений.
5.5. Закон Гесса и следствия из него
Русский ученый Г.И. Гесс в 1840 г. экспериментально установил ос-
основной закон термохимии: тепловой эффект химической реакции определя-
определяется только видом и состоянием исходных веществ и продуктов, но не зави-
зависит от пути процесса.
Проиллюстрируем закон Гесса, который можно рассматривать как
приложение первого начала термодинамики к химической реакции, простой
схемой. Процесс окисления аммиака до оксида азота (II) может быть осуще-
осуществлен в одну стадию (именно так проходит реакция каталитического окис-
окисления аммиака в промышленности):
4NH3(r) + 5О2(Г)= 4NO(r) + 6Н2О(Ж), Дг#2°98 = А,^ = -1166 кДж. E.20)
Эту же реакцию можно провести в две стадии: окислить аммиак в кис-
кислороде с образованием газообразного азота и жидкой воды (некаталитичес-
(некаталитическое горение), а затем азот окислить до оксида азота (II):
4NH3(r) +ЗО2(Г) = 2N2(r) +6Н2О(Ж)
Дг#2°98 = Д,#2=-1531,2кДж; E.21)
2N2(r) + 2O2(r) = 4NO(r)
Дг#2°98 = ДГЯ3 = 365,2 кДж. E.22)
Из сопоставления тепловых эф-
эффектов реакций E.20)-E.22) следует,
что
Исходные
вещества
4NH3(r),5O2(r)
Продукты
реакции
4NO(r),6H2O(x)
Дг#3.
В простейшем случае, когда
одна стадия заменяется двумя, полу-
получается так называемый треугольник
Гесса (рис. 5.1). При увеличении
числа промежуточных стадий число
сторон многоугольника возрастает.
III
Промежуточные
вещества
2N2(r)NH2O(x)JO2(r)
Рис. 5.1. К пояснению закона Гесса
188
5. Тепловой эффект химической реакции
Рис. 5.2. К пояснению первого следствия из
закона Гесса
Для расчета тепловых эффек-
эффектов реакций большое практиче-
практическое значение имеют следствия
из закона Гесса, которые мож-
можно рассмотреть в применении к
необратимой (протекающей
практически со стопроцентным
выходом в одном направлении)
химической реакции, записан-
записанной в общем виде:
где V/ и v- — стехиометрические коэффициенты исходных веществ и про-
продуктов реакции соответственно.
Первое следствие. Тепловой эффект реакции равен сумме энтальпий
образования продуктов за вычетом суммы энтальпий образования исходных
веществ с учетом стехиометрических коэффициентов (рис. 5.2).
Как исходные вещества, так и продукты реакции можно синтезиро-
синтезировать из простых веществ в стандартных условиях. На цикле Гесса линия,
обозначающая суммарный тепловой эффект, утолщена. Из рис. 5.2 опреде-
определяем, что
E.23)
Второе следствие. Тепловой эффект реакции равен сумме энтальпий
сгорания исходных веществ за вычетом суммы энтальпий сгорания продук-
продуктов с учетом стехиометрических коэффициентов (рис. 5.3).
Как исходные вещества, так и продукты гипотетически можно сжечь в
избытке кислорода в стандарт-
стандартных условиях. На образующем-
образующемся цикле Гесса линия, символи-
символизирующая суммарный тепловой
эффект, утолщена. Из рис. 5.3
определяем, что
^298/• E.24) Рис. 5.3. К пояснению второго следствия из
закона Гесса
Исходные
вещества
\<*
Продукты
+ Л Оад +П О2(г)
Продукты
сгорания
5.5. Закон Гесса и следствия из него
189
Непосредственно из закона Гесса следует, что тепловые эффекты двух
взаимно обратных реакций равны по модулю и обратны по знаку:
СаСОз(Т) = СаО(т) + СО2(г), ДГЯ2°98 = +177,9 кДж;
СаО(т) + СО2(Г) = СаСО3(Т), ДГЯ2°98 s -177,9 кДж.
Наиболее важное практическое значение закона Гесса состоит в воз-
возможности расчета на его основе теплового эффекта реакции, эксперимен-
экспериментальное проведение которой в чистом виде затруднительно или невозможно.
Вычисления проводят алгебраическим комбинированием стехиометриче-
ских уравнений и тепловых эффектов реакций, изученных эксперименталь-
экспериментально, т.е. построением соответствующих циклов Гесса или термохимических
схем.
Например, тепловой эффект реакции
2Fe
(T)
= 2FeO(T), Д,Я2°98 =
E.25)
экспериментально измерить невозможно, так как часть навески железа обя-
обязательно окислится до БезО/цт) и Fe2O3(T), но его можно вычислить, согласно
закону Гесса, по опытным значениям энтальпий реакций окисления железа
и оксида железа (II) до оксида железа (III):
о _
2Fe(T) + 1,5Оад = Fe2O3(T), ДгЯ2и98 = ДГЯ2 = -822,2 кДж;
2FeO(T) + 0,5О2(г) = Fe2O3(T), АГЯ2°98 = А,Я3 = -292,6 кДж.
E.26)
E.27)
С использованием этих данных можно составить цикл Гесса для реак-
реакции получения оксида железа (III) из металлического железа при разном
числе промежуточных стадий окисления (рис. 5.4). Из рис. 5.4 следует, что
АГЯ, = АГН2 - АГЯ3 = -822,2 - (-292,6) = -529,6 кДж. На основе материально-
материального баланса E.25) можно получить путем вычитания из E.26) уравнения
E.27). Таким же образом будут связаны между собой тепловые эффекты
2Fe(T)+l,5O2(r)=Fe2O3(T)
2Fe(T)+O2(r)=2FeO(T)
2FeO(T)+0,5O2(r)=Fe2O3(T)
Рис. 5.4. К пояснению расчета согласно методу термохимических схем
190
5. Тепловой эффект химической реакции
Я,кДж
2Fe(T);l,5O2(r)
2FeO(T);0,5O2(r)
Рис. 5.5. Энергетическая диаграмма для реакций окисления железа кислородом
этих реакций: АГНХ = АГН2 - Лг#3. Согласно E.25), Ay#298FeoT = 0,5Ar#i =
= -264,8 кДж/моль.
Для вышеописанных процессов (см. E.25)-E.27)) на рис. 5.5 приве-
приведена так называемая энергетическая диаграмма, на которой нулевой уро-
уровень соответствует простым веществам, взятым в стандартных термодина-
термодинамических условиях. На таких диаграммах тепловые эффекты экзотермиче-
экзотермических реакций и фазовых переходов отмечаются стрелками, направленными
вниз (уменьшение энергии системы), эндотермических процессов — стрел-
стрелками, направленными вверх (увеличение энергии системы).
В соответствии с методом термохимических схем можно рассчитать
энтальпии образования органических соединений, используя данные по эн-
энтальпиям сгорания. Например, прямой синтез ацетона в стандартных усло-
условиях осуществить нельзя:
ЗН2(Г) + 0,5О2(Г) = СзН6О(Ж), АГН\ = А/Я298СзНбО(ж), E.28)
но каждое из простых веществ и ацетон можно сжечь в кислороде:
ЗС(графит) + ЗО2(Г) = ЗСО2(Г), АГН2 = 3(-393,5) = -1180,5 кДж; E.29)
ЗН2(Г) + 1,5О2(Г) = ЗН2О(Ж), АГН2 = 3(-285,8) = -857,4 кДж; E.30)
С3Н6О(Ж) + 4О2(Г) = ЗСО2(Г) + ЗН2О(Ж), ДЛ = -1789,8 кДж. E.31)
Алгебраической комбинацией уравнений E.29)—E.31) можно полу-
получить E.28), а такая же комбинация тепловых эффектов дает энтальпию об-
образования ацетона:
= -1180,5 - 857,4 - (-1789,8) = -248,1 кДж/моль.
6 ¦ Направление химической реакции
Все химические реакции можно подразделить на две большие груп-
группы: самопроизвольные, протекающие самостоятельно без подвода
энергии от внешнего источника, и несамопроизвольные (энергозави-
(энергозависимые), для осуществления которых необходима постоянная энерге-
энергетическая подпитка. Примерами самопроизвольных процессов служат
реакция нейтрализации:
Н (Р) + ОН (р) —> Н2О(Ж)
а также окислительно-восстановительные реакции, идущие в гальва-
гальванических элементах или в реакционных сосудах. К несамопроиз-
несамопроизвольным процессам относятся, например, фотосинтез, протекающий
в листьях и хвое растений под действием солнечной энергии
6СО2 + 6Н2О -> С6Н12О6+ 6О2
и окислительно-восстановительные реакции на аноде и катоде при
электролизе.
6.1. Второй закон термодинамики.
Энтропия как функция состояния системы
Вопрос о направлении и условиях самопроизвольного проте-
протекания процесса решается в рамках второго закона термодинамики.
Одна из первых формулировок второго начала термодинамики —
постулат Клаузиуса A850) гласит: теплота не может самопроиз-
самопроизвольно переходить от менее нагретого тела к более нагретому.
Рассмотрев работу тепловой машины, Клаузиус ввел новую, не
известную ранее функцию состояния системы, названную энтропией
(от греч. entropia — внутреннее превращение), для полного диффе-
дифференциала которой в применении к элементарному обратимому про-
процессу получил выражение
</S=^L5 FЛ)
где 5??обР — алгебраическая сумма теплоты, полученной и отданной
рабочим телом (идеальным газом) при совершении процесса, Дж;
Т— абсолютная температура, К.
Интегрируя F.1), получают
AS = S2 - S, = |
^обр
. T
192 6. Направление химической реакции
где индексы 1 и 2 соответствуют начальному и конечному состояниям сис-
Q (TGI
темы. Величину —, имеющую размерность теплоемкости =
называют приведенной теплотой. Следовательно, по Клаузиусу, энтропия
есть функция состояния, изменение которой равно алгебраической сумме
приведенной теплоты всех элементов обратимого процесса.
В силу первого закона термодинамики для термомеханической систе-
системы (8W = 0) 8Q = dU + pdV. Так как работа, совершаемая системой в необ-
необратимом процессе, всегда меньше работы, производимой в обратимом
Процессе (^необр< ^обр),ТО
dS> ^=&. F.2)
Объединяя F.1) и F.2), получают
dS > -^S или TdS > 8Q. F.3)
Неравенство F.3) представляет собой математическое выражение второго
закона термодинамики, где знак равенства относится к обратимым процес-
процессам, а знак неравенства — к необратимым.
Из F.1)—F.3) следует, что энтропия, как и приведенная теплота, имеет
Дж
размерность теплоемкости: [S] = , или в применении к 1 моль вещества
га.
моль•К
Подставляя в F.3) значение 8Q из E.3), получают неравенство
TdS^dU+pdV, F.4)
представляющее собой обобщенное выражение первого и второго законов
термодинамики, в соответствии с которым энтропия является функцией
внутренней энергии и объема системы: S = f(U, V).
При использовании F.4) в изолированной системе, в которой U = const,
V= const, т.е. dU= 0, dV= 0, имеют TdS > 0, но Т > 0, поэтому
dS> 0, илиАЯ^ 0. F.5)
Следовательно, в изолированной системе знак изменения энтропии
является критерием направленности самопроизвольного процесса:
а) если AS = 0 (S = Smax, энтропия достигла своего максимального зна-
значения), то система находится в состоянии термодинамического равновесия;
6.1. Второй закон термодинамики 193
б) если AS > О (S -> Smax, энтропия возрастает), то процесс самопроиз-
самопроизвольно протекает в прямом направлении, т.е. термодинамически возможен;
в) если AS < О (S -> Sm\n, энтропия убывает), то самопроизвольно про-
протекать может лишь обратный процесс, связанный с увеличением энтропии,
прямой процесс термодинамически невозможен.
Только возрастанием энтропии можно объяснить самопроизвольное
протекание в изолированных системах таких процессов, как передача тепло-
теплоты от более нагретого тела к менее нагретому или взаимная диффузия (сме-
(смешение) двух или нескольких химически не взаимодействующих газов. Для
химии как науки о веществах, изучающей их в том числе на уровне состав-
составляющих частиц (молекул, атомов, ионов), более актуально определение эн-
энтропии через термодинамическую вероятность, выведенное Л. Больцманом.
Химическая термодинамика изучает системы реагирующих веществ,
состоящие из очень большого числа частиц. В таких системах различают
макро- и микропараметры, или макро- и микрохарактеристики. К макропа-
макропараметрам относятся независимые переменные, свойственные системе в це-
целом, например, давление (р), объем (F), температура (Г), молярные концен-
концентрации веществ (С,), которые можно определить экспериментально или
рассчитать теоретически. Наряду с этим каждая частица имеет свои непре-
непрерывно изменяющиеся микрохарактеристики (координаты jc, у, z в простран-
пространстве, скорость, энергию), расчет которых для отдельных частиц затруднен.
Совокупность микропараметров каждой частицы характеризует ее микросо-
микросостояние.
Термодинамической вероятностью w называют число микросостоя-
микросостояний, через которое можно реализовать данное макросостояние системы. В
отличие от математической вероятности, равной или меньшей единице,
термодинамическая вероятность представляет собой очень большую вели-
величину: значение w имеет порядок 104 для системы, состоящей всего из десят-
десятка частиц, в то время как 1 моль вещества содержит NA = 6,02 • 1023 струк-
структурных единиц.
Связь энтропии с термодинамической вероятностью установил
Больцман:
S= kB\nw, F.6)
уравнение F.6) названо его именем, где кБ — константа Болъцмана (уни-
(универсальная газовая постоянная, рассчитанная на одну молекулу), Дж/К,
R 8 31
кБ = = —— = 1,38 • 10~23 Дж/К. Таким образом, по Больцману, энтро-
энтропия есть функция термодинамической вероятности того или иного состояния
индивидуального вещества или системы. Согласно уравнению F.6), второе
начало термодинамики можно сформулировать так: всякая изолированная
7 - 9795
194
6. Направление химической реакции
Ш2222Щ222222Щ
система самопроизвольно стремится
принять состояние, характеризую-
характеризующееся максимальной термодинамиче-
термодинамической вероятностью.
На основании уравнения Больц-
мана можно показать, что любой не-
необратимый процесс, самопроизвольно
протекающий в изолированной сис-
системе, характеризуется увеличением
энтропии. Пусть в изолированной
системе находятся два химически не
взаимодействующих газа, например
гелий и неон, при одинаковых темпе-
температуре и давлении, разделенные пере-
перегородкой (рис. 6.1, а). В этом состоя-
состоянии термодинамическая вероятность
системы равна w\. При удалении пе-
перегородки (рис. 6.1, б) газы начинают
самопроизвольно диффундировать друг в друга до тех пор, пока молекулы
каждого газа равномерно не распределятся по всему объему. В конечном
состоянии термодинамическая вероятность равна w2. Система самопроиз-
самопроизвольно перешла из менее вероятного состояния в более вероятное (w2 > w\).
Энергетический обмен системы с внешней средой отсутствует, следователь-
следовательно, единственная причина протекания этого процесса — увеличение энтропии:
AS = S2 - Sx - кБ In w2 - кБ In Wj = кБ In—,
Рис. 6Л. Схема смешения двух хи-
химически не взаимодействующих газов
но w2 >wu ^- > 1; ln^- > 0, т.е. AS> 0.
i i
Термодинамическая вероятность системы непосредственно связана со
степенью беспорядка (хаотичности) в расположении частиц. Из трех агре-
агрегатных состояний вещества (кристаллическое, жидкое, газообразное) наи-
наибольшая степень хаотичности характерна для газа, а наименьшая — для
кристалла. Молекулы идеального газа не влияют друг на друга и имеют все
возможные степени свободы. В жидкости существуют межмолекулярное
взаимодействие и ближний порядок в расположении частиц. Атомы, моле-
молекулы или ионы, составляющие кристаллическую решетку, «зафиксированы»
в ее узлах и совершают только колебательные движения. В кристалле суще-
существуют ближний и дальний порядки в расположении структурных единиц.
Следовательно, при прочих равных условиях вещество обладает минималь-
6.2. Изменение энтропии в некоторых процессах 195
ной энтропией в кристаллическом (твердом) состоянии, а максимальной —
в газообразном: S(T) < 5(Ж) < 5(Г>, вследствие чего газы называют носителями эн-
энтропии.
Все процессы, сопровождающиеся увеличением степени молекуляр-
молекулярного беспорядка в системе, ведут к увеличению энтропии (AS > 0): плавле-
плавление и сублимация твердых веществ, испарение жидкостей, расширение газов,
растворение кристаллов, химические реакции, протекающие с увеличением
количества вещества газов (объема системы). В обратных процессах (за-
(затвердевание жидкостей, кристаллизация веществ из растворов, конденсация
паров, сжатие газов, химические реакции, протекающие с уменьшением
объема системы) степень молекулярного беспорядка уменьшается, следова-
следовательно, энтропия системы убывает (AS < 0).
Энтропия любого вещества увеличивается с ростом температуры, а у
газов, кроме того, незначительно уменьшается с ростом давления.
6.2. Изменение энтропии в некоторых процессах
Рассмотрим расчет изменений энтропии в некоторых равновесных
процессах: при фазовых переходах индивидуальных веществ (A trS) (от англ.
transition — переход), а также при изохорном (ASV) и изобарном (ASP) нагре-
нагреве и при изотермическом (АЗУ) расширении идеального газа.
При фазовых переходах первого рода энтропия и объем системы из-
изменяются скачкообразно. Примерами таких процессов являются полиморф-
полиморфные превращения (переход вещества из одной кристаллической модифика-
модификации в другую, например Р-олова в а-олово), а также взаимные переходы
между твердыми, жидкими и газообразными фазами вещества (плавление —
кристаллизация, сублимация — кристаллизация, кипение — конденсация).
Эти переходы происходят в изобарно-изотермических условиях по схеме:
Фаза 1 <=> Фаза 2; AtrH.
Изменение энтропии при фазовых переходах первого рода вычисляют,
интегрируя уравнение F.1) при/?, Т= const (Q = Qp):
F.7)
где AtrH — молярная теплота (стандартная энтальпия) фазового перехода,
Дж/моль; Т— температура фазового перехода, К.
196 б. Направление химической реакции
Для определения изменения энтропии в процессах, происходящих в
идеальном газе, вводят понятие теплоемкости. В применении к веществу
или системе теплоемкость (с, Дж/К) есть отношение количества сообщен-
сообщенной теплоты 8Q к наблюдаемому повышению температуры dT в отсутствие
химических реакций, фазовых переходов и bW = 0:
c = 5Q/dT.
При расчете на единицу массы /-го вещества получают удельную теп-
теплоемкость cmi= Cilntb Дж/(кг-К), на единицу количества A моль) — моль-
мольную (молярную) теплоемкость с„/=с//и/, Дж/(моль-К). В зависимости от ус-
условий нагревания различают теплоемкости при постоянном давлении cPi и
при постоянном объеме сУг В соответствии с определением теплоемкости и
первым законом термодинамики для изобарного и изохорного процессов
нагревания /-го вещества
cPi = —1- , или при cPi= const в интервале температур Т\...Т2
V и± J р
Р
cVi= —L , или при cVi = const в интервале температур Т\...Т2
V дТ )у
cVi(T2-Tx). F.7")
Так как для одного моля идеального газа Я = U+ pV= U+ RT, то ср = су+ i?.
Для большинства конденсированных веществ ср = cv.
Рассмотрим изменение энтропии 1 моль идеального газа в различных
равновесных процессах:
а) изохорный (V= const, dV= 0) нагрев от Т\ до Г2. Неравенство F.4)
для равновесного изохорного процесса принимает вид
5Qy= dU= cvdT, dSv= cv—\ ASV = ]cv—.
T TT
Если принять cv= const в интервале температур Т\...Т2 •> то
Д^=сукД; F.8)
б) изобарный (р = const) нагрев от Т\ до Т2:
r=cpdT,dSp=cp-f; ASp=jcp-^.
г,
6.2. Изменение энтропии в некоторых процессах 197
Принимая ср = const в интервале температур Т\... Т2, получают
ASp=cp]n^ F.9)
Так как зависимости внутренней энергии U = cvT и энтальпии Н = срТ от
температуры справедливы для любого агрегатного состояния вещества,
уравнения F.8) и F.9) можно использовать для расчета изменений энтропии
в процессах изохорного и изобарного нагрева и охлаждения конденсирован-
конденсированных веществ;
в) изотермическое (Г = const) расширение газа, т.е. увеличение объема
от V\ до V2 (напомним, что для идеального газа при постоянной температуре
[/= const, dU=0):
TdST=8QT=pdV,
RT
но для 1 моль идеального газа р = , тогда
AST= [R— = R\n^ = Rln-^-, F.10)
так как, согласно закону Бойля—Мариотта, (pV)T= const.
По формулам F.7)-F.10) рассчитывают молярные изменения энтро-
энтропии веществ в процессах. При количестве вещества в системе, равном п
моль, соответствующие молярные значения AS умножают на п и получают
изменение энтропии системы в джоулях на кельвин.
Рассмотрим изменение энтропии при смешении химически не взаимо-
взаимодействующих идеальных газов Am^S (от англ. mix — смешение). Предста-
Представим изолированную систему в виде цилиндрической капсулы, состоящей из
к различных по объему Vt ячеек, разделенных перегородками. В каждой
ячейке содержится и,- моль идеального инертного газа, давление и темпера-
температура у всех газов одинаковы (р,Т= const). При удалении всех (?-1) перего-
перегородок, разделяющих чистые газы, самопроизвольно начинается диффузия
газов, происходящая до тех пор, пока молекулы каждого газа не распреде-
распределятся равномерно по объему всей капсулы.
В отсутствие химической реакции этот процесс можно рассматривать
как изотермическое расширение щ моль каждого газа от объема ячейки Vt до
объема капсулы V = ^Vt. В соответствии с F.10)
198 б. Направление химической реакции
Inf. F.11)
У идеальных газов, находящихся в смеси при одинаковых давлении и
температуре, объемные ср, и молярные Xi доли совпадают:
При этом для любой газовой смеси ^Ф/ = ^-У,- = 1. Уравнение F.11) мож-
но преобразовать следующим образом:
Умножая и деля правую часть F.11) на суммарное количество вещества
всех газов п = ^и, = const для данной системы, получают
,., Дж/К, F.12)
значит, при образовании 1 моль смеси идеальных газов изменение энтропии
(Дж/(моль • К))
F.13)
В соответствии с F.13) Am^S > 0, так как молярная доля любого /-го
компонента смеси Д < 1, IilY) < 0, т.е. при смешении газов энтропия системы
возрастает.
6.3. Третий закон термодинамики.
Абсолютные значения стандартных энтропии веществ
Абсолютные значения внутренней энергии и энтальпии невозможно
вычислить даже для простых систем. Абсолютные значения энтропии рас-
рассчитываются как для простых, так и для сложных веществ, поскольку у них
имеется начальная точка в шкале отсчета, устанавливаемая третьим законом
термодинамики.
6.3. Третий закон термодинамики 199
Третье начало термодинамики, или постулат Планка, формулирует-
формулируется следующим образом: при температуре абсолютного нуля (Г= О К) энтро-
энтропия идеального кристалла любого простого или сложного вещества равна
нулю: lim S = О, или So = 0. Действительно, в бездефектном кристалле ве-
щества существует абсолютный порядок в расположении частиц. При абсо-
абсолютном нуле возможно единственное состояние системы, при котором час-
частицы «застывают» в узлах кристаллической решетки. Термодинамическая
вероятность равна минимальному значению (w = 1), поэтому для 1 моль ве-
вещества 5о = R In w = R In I = 0. У неидеальных кристаллов, смесей, твердых
растворов, стеклообразных структур всегда имеется нулевая энтропия EЬ > 0),
связанная с дефектами кристаллической решетки, возможностью различной
ориентации частиц в пространстве и другими причинами.
Стандартную энтропию вещества обозначают символом S® (р =
= 101,3 кПа), ее можно определить при любом значении температуры, однако
для удобства сравнения величин S® для различных веществ их определяют
обычно при стандартных термодинамических условиях (р = 101,3 кПа,
Т = 298 К) и обозначают S^g (Дж/(моль • К). Для газообразного вещества
при Г= 298 К стандартное значение энтропии S%9S имеет 1 моль идеального
газа при собственном давлении;? = 101,3 кПа = 1 атм. Для конденсирован-
конденсированного (жидкого или кристаллического) вещества значением энтропии S%9S
обладает 1 моль этого вещества при внешнем давлении р = 1 атм. Для рас-
растворенных веществ и ионов в растворах стандартное состояние и значение
энтропии S^ соответствует моляльной концентрации Cmh равной 1 моль/кг
НгО, но при этом предполагается, что раствор обладает свойствами беско-
бесконечно разбавленного (идеального) раствора.
Стандартная энтропия любого вещества всегда положительная вели-
величина ( S^ > 0). Стандартные энтропии образования ионов в водных раство-
растворах (А/5Г298(иона)) определяются для процессов образования и гидратации
ионов, они могут быть как положительными, так и отрицательными. Изме-
Изменения энтропии (AS) в процессах могут быть положительными, отрицатель-
отрицательными или равными нулю.
Приведем схему расчета стандартных энтропии 5^8 • Вблизи абсо-
абсолютного нуля все вещества находятся в твердом состоянии. Если при стан-
стандартных термодинамических условиях вещество находится в кристалличе-
кристаллическом состоянии, то в интервале температур от 0 до 298 К энтропия изменя-
изменяется только за счет повышения температуры, значит, в соответствии с F.3)
при постоянном давлении
200 б. Направление химической реакции
т т р т
298 ^т 298 dj
или Д5= \ср—, следовательно, 5298=50н- \ср—, но, согласно треть-
третьей о Т
ему закону термодинамики, 50 = 0. В случае неидеального кристалла So =
= const. Таким образом,
298
со _ г ai
^298 - J Cp — •
Если при Т = 298 К вещество является газом, то при расчете его стан-
стандартной энтропии необходимо учитывать не только изменения энтропии,
связанные с изобарным нагревом твердой, жидкой и газообразной фаз, но и
изменения энтропии, связанные с фазовыми переходами (плавление, испа-
испарение):
29
АЯИСП 29г dT
^+ J —
г dT /Г1.Ч
J ср(г)— - F.14)
Если при Т = 298 К вещество находится в жидком состоянии, то при
расчете стандартного значения энтропии в формуле F.14) ограничиваются
тремя первыми слагаемыми. Величины S^s рассчитывают, используя зави-
зависимость теплоемкости вещества от температуры методом аналитического
или графического интегрирования.
Применение метода графического интегрирования для расчета S^g
этилена показано на рис. 6.2. Значение S^g численно равно площади фигу-
ры, заключенной между кривыми — = /(Г), отрезком оси абсцисс @.. .298 К)
и перпендикуляром, опущенным из точки на графике — = /(Г) на ось абс-
абсцисс при Т = 298 К, с учетом величин А5 фазовых переходов. Значения
стандартных энтропии веществ S%9S приведены в таблицах термодинамиче-
термодинамических величин (см. табл. П.5.1).
Энтропия представляет собой функцию состояния системы, поэтому
стандартное изменение энтропии в результате осуществления химической
реакции (стандартная энтропия реакции АГ5Г298) можно вычислить как раз-
6.4. Критерии направленности самопроизвольного процесса
201
0
50
150 200
250
Г=15К Г =104К Г =169К
пл кип
300 Г, К
= 298К
Рис. 6.2. К расчету абсолютного значения стандартной энтропии эти-
этилена (S^) графическим способом (точки на графике — эксперимен-
экспериментальные данные)
ность между суммой стандартных энтропии продуктов реакции и суммой
стандартных энтропии исходных веществ с учетом стехиометрии процесса:
7
298
-Vv
/°298 •
F.15)
6.4. Критерии направленности самопроизвольного процесса
в закрытой системе
Реальные процессы проводятся, как правило, в закрытых системах в
изобарно-изотермических (/?, Т= const) или изохорно-изотермических (F, Т=
= const) условиях. Критерием направленности самопроизвольного процесса
в этих случаях является знак изменения энергии Гиббса AG или энергии
Гельмгольца АА в системе.
По рекомендациям ИЮПАК энергии Гиббса G и энергии Гельмгольца
А соответствуют следующие выражения:
G=H-TS= U+pV-TS\
A=U- TS;
F.16)
F.17)
202 б. Направление химической реакции
при этом G= flp, T); A=f(V, T).
Уравнения F.16) и F.17) можно представить в виде
H=G+ TS; F.18)
U=A+ TS, F.19)
где величина TS характеризует связанную с частицами системы энергию,
т.е. ту часть полной энергии системы, которая рассеивается в окружающей
среде в виде теплоты (так называемая потерянная работа). Согласно F.18) и
F.19), энтропия есть связанная энергия системы, приходящаяся на 1 К.
Энергия Гиббса (или энергия Гельмгольца) характеризует ту часть
полной энергии системы, которая может быть превращена в работу в изо-
барно-изотермическом (или изохорно-изотермическом) процессе (так назы-
называемая полезная работа, совершаемая системой). Энергия Гиббса и энергия
Гельмгольца являются функциями состояния системы, их абсолютные зна-
значения не поддаются вычислению.
Для выяснения критерия направленности самопроизвольного процесса
в закрытой системе дифференцируют F.16)
dG=dU+ pdV + Vdp - TdS - SdT. F.20)
В силу F.4), TdS > dU+pdV, т.е.
dU^TdS-pdV. F.21)
Подставляя в F.20) вместо dU большую либо равную ей величину (TdS -pdV)
из F.21), получают неравенство
dG<-SdT+ Vdp. F.22)
Для изобарно-изотермического процесса (р, Т = const, dp = dT = 0)
выражение F.22) принимает вид dGpj < 0, или для конечных изменений
энергии Гиббса
Авр,т<0;в^>вщп. F.23)
Следовательно, в закрытой системе знак изменения энергии Гиббса является
критерием направленности самопроизвольного процесса при проведении
его в изобарно-изотермических условиях:
а) при AG = 0 (G = Gm\n, энергия Гиббса имеет минимальное значение)
система находится в состоянии термодинамического равновесия;
б) при AG < 0 (G -> Gmin, энергия Гиббса убывает) процесс самопроиз-
самопроизвольно протекает в прямом направлении, т.е. термодинамически возможен;
в) при AG > 0 (G -> Gmax, энергия Гиббса возрастает) самопроизвольно
протекает только обратный процесс, прямой процесс термодинамически не-
невозможен.
6.4. Критерии направленности самопроизвольного процесса 203
Изохорно-изотермический процесс (F, T= const) в закрытой системе
термодинамически возможен при АА < 0, термодинамически невозможен
при АА > 0, система находится в термодинамическом равновесии при А А = 0.
Для химической реакции, проводимой при/? = 101,3 кПа и постоянной
температуре Г, изменение энергии Гиббса (эту величину называют также
стандартной энергией Гиббса реакции) рассчитывают по формуле F.24),
аналогичной F.16):
ArGQT = ArH* - TArS°. F.24)
Выражение F.24) называют уравнением Гиббса—Гелъмголъца. Оно свя-
связывает стандартную энергию Гиббса химической реакции с ее стандартными
энтальпией, энтропией и температурой, при которой проводится процесс.
Анализ F.24) показывает, что знак и величина ArGj, а значит, термо-
термодинамическая возможность самопроизвольного протекания реакции зависят
от двух факторов: энтальпийного (энергетического) ArHj и энтропийного
TArSj. С одной стороны, в ходе химической реакции система стремится
прийти к минимальному уровню энергии, выделив часть ее в виде теплоты
или работы (ArHj < 0). С другой стороны, система стремится занять наибо-
наиболее вероятное состояние, характеризующееся максимумом молекулярного
беспорядка, т.е. максимумом энтропии (ArSj > 0). В этом случае энталь-
пийный и энтропийный факторы действуют в направлении, благоприятст-
благоприятствующем протеканию реакции.
Рассмотрим варианты, возможные при проведении химических про-
процессов:
а) ArHj < 0; ArSj > 0; в этом случае ArGj < 0 при всех значениях
температуры, процесс термодинамически возможен при любой температуре;
А Н°
б) АГН* < 0; ArS° < 0; в этом случае ArG* < 0 при Т< =^9 т.е.
ArST
реакция термодинамически возможна при сравнительно низкотемператур-
низкотемпературном режиме;
А Н°
в) ArHj > 0; ArSj > 0; тогда ArGj < 0 при Т> г [, значит, про-
ArST
цесс термодинамически возможен при сравнительно высокотемпературном
режиме;
г) ArHj > 0; ArSj < 0 — оба фактора действуют в неблагоприятном
направлении, реакция термодинамически невозможна при любых значениях
температуры.
204 6. Направление химической реакции
По знаку ArHj можно определить направление только таких процес-
процессов, в которых энтропия практически постоянна (ArSj = 0). Тогда тепловой
эффект является мерой химического сродства: термодинамически возможны
экзотермические реакции {ArGj = ArHj < 0) и невозможны эндотермиче-
эндотермические (ArGj = ArHj > 0). Это утверждение называется принципом Бертло—
Томсена.
При ArGj = 0, т.е. ArHj = TArSj, в системе устанавливается под-
подвижное термодинамическое равновесие, положение которого может сме-
смещаться в любую сторону при изменении внешних факторов (температура,
давление, концентрации реагирующих веществ). Температуру равновероят-
равновероятности протекания прямой и обратной реакций можно вычислить по выражению
Как следует из F.25) и физического смысла абсолютной температуры, Гравн
существует только для реакций, имеющих одинаковые знаки ArHj и ArS®.
Стандартную энергию Гиббса химической реакции при Т = 298 К
можно рассчитать двумя способами: используя уравнение Гиббса—Гельм-
гольца F.24), в которое нужно подставить указанное значение Г, и по стан-
стандартным энергиям Гиббса образования исходных веществ и продуктов ре-
реакции Ду^298*
Стандартную энтальпию реакции Дг#298 Haxo^T по первому или
второму следствию из закона Гесса (см. E.23) и E.24)), а стандартную эн-
энтропию реакции Д^^з — по F.15). При расчете Afi^ необходимо учи-
учитывать, что единицей измерения энтальпии является килоджоуль, а энтропии
— джоуль на кельвин.
Энергия Гиббса является функцией состояния системы, поэтому по
аналогии с E.23) и F.15) рассчитывают A^G^s с использованием таблич-
табличных значений стандартных энергий Гиббса образования исходных веществ
и продуктов реакции A^G^s1
А/<&8. F-26)
Стандартной энергией Гиббса образования вещества Aj-G^ назы-
называют стандартную энергию Гиббса реакции образования 1 моль данного со-
6.5. Химический потенциал 205
единения из простых веществ, находящихся в термодинамически устойчи-
устойчивых модификациях, которая проведена в стандартных термодинамических
условиях. Стандартная энергия Гиббса образования вещества ДуСг? может
быть определена при любой температуре, но чаще всего принимают Т= 298 К.
Для простых веществ, находящихся в термодинамически устойчивых
состояниях, а также для гидратированного протона в водном растворе
Д/^298 = О- Значения Л/С/298 %*** раз™4111»1* индивидуальных веществ и ио-
ионов в водном растворе приводятся в справочной литературе (см. табл. П.5.1).
В отсутствие справочных данных по A/G298 какого-либо сложного
вещества эту величину можно вычислить по уравнению
A7G2°98 = А/Я2°98 -298А/52°98, F.27)
где Л/#298 и А^?298 — стандартные энтальпия и энтропия соответственно
реакции образования 1 моль этого соединения из простых веществ, находя-
находящихся в термодинамически устойчивых состояниях, проведенной при стан-
стандартных термодинамических условиях.
6.5. Химический потенциал
Для открытой системы возможен обмен с окружающей средой не
только энергией, но и веществом: m * const. Следовательно, внутренняя
энергия этой системы может изменяться за счет передачи теплоты Fg ), со-
совершения работы расширения (pdV) и переноса некоторого количества ве-
вещества dnt из системы в окружающую среду или обратно. В открытой сис-
системе состав образующих ее веществ изменяется, поэтому энергию Гиббса
можно представить соотношением
G =/(/?, Г, л,), F.28)
где rii — количество вещества /-го компонента, моль.
Выражение F.28) справедливо также для закрытых систем, в которых
совершаются химические реакции. Масса системы при этом постоянна, но
ее состав изменяется: в ходе прямой (идущей слева направо) реакции коли-
количество исходных веществ (л,) уменьшается, а количество продуктов реакции
(л/) увеличивается.
Для рассмотренных случаев полный дифференциал энергии Гиббса
dG = -SdT+ Vdp + У [ — I dni9 F.29)
206 б. Направление химической реакции
где индекс и, означает, что частная производная энергии Гиббса по количе-
количеству вещества /-го компонента берется при постоянном количестве всех ос-
остальных веществ j системы. Эта частная производная, учитывающая зави-
зависимость энергии Гиббса от состава системы, называется химическим потен-
потенциалом \it /-го компонента
ffl ¦ (б.зо)
По физическому смыслу величина ц,- означает изменение энергии
Гиббса системы в изобарно-изотермическом процессе при добавлении
1 моль /-го компонента к бесконечно большому количеству смеси, чтобы ее
состав был постоянным (w,- = const). Зависимость химического потенциала от
состава системы для идеальных растворов выражается следующими уравне-
уравнениями:
F31)
где |i° — стандартный химический потенциал i'-го компонента (при относи-
относительном давлении р., = ^~ = 1), X, — мольная доля /-го компонента. Второе
Р
уравнение справедливо для любых идеальных растворов, а первое — для
газовых. Для индивидуального вещества стандартный химический потенци-
потенциал ц? равен стандартной энергии Гиббса образования вещества AfG^sr
Отметим некоторые свойства химического потенциала. Из физики из-
известно, что поток электрических зарядов всегда направлен в сторону
уменьшения электрического потенциала ср. По аналогии поток /-го вещества
в неравновесной системе при изобарно-изотермических условиях направлен
в сторону убывания энергии Гиббса (от точки с большим значением ц,), по-
поэтому величину Ц/ называют химическим потенциалом.
Энергия Гиббса является экстенсивным свойством системы. Согласно
определению, химический потенциал — интенсивное свойство системы.
Таким образом, направление самопроизвольного протекания реакции опре-
определяется разностью химических потенциалов продуктов реакции и исход-
исходных веществ:
При проведении реакции в изобарно-изотермических условиях выра-
выражение F.29) принимает вид
6.5. Химический потенциал 207
A- F.32)
Для газофазной реакции, протекающей в закрытой системе при
р,Т = const,
vAA(r) + vBB(r) ^± vDD(r) + vFF(r) F.33)
выведем общие условия химического равновесия и самопроизвольного про-
протекания процесса. Пусть в начальный момент времени в реакционной смеси
присутствуют как исходные вещества А и В, так и продукты реакции D и F.
В ходе прямой реакции количество исходных веществ уменьшается (Аи, < 0),
количество продуктов реакции увеличивается (Ап\ > 0), где Ди, и Ап\ — из-
изменение количества исходного вещества или продукта реакции соответст-
соответственно.
Вследствие имеющейся стехиометрии реакции F.33) справедливо со-
соотношение
АпА _ Апв _ AnD _
vD vF
F.34)
__ M: Alt': fc „ „
Величину L = —т- = ? называют химической переменной (глуои-
V V
I I
ной, степенью протекания реакции, числом пробегов реакции). Химическая
переменная ? характеризует изменение количества веществ-участников в
процессе реакции: ? = 0 при / = 0 (реакция еще не началась); ? = 1, если из vA
и vB моль исходных веществ получили vD и vF моль продуктов реакции (ис-
(исходные вещества полностью прореагировали в количествах, равных их сте-
хиометрическим коэффициентам). Говорят, что при ? = 1 реакция соверши-
совершила один пробег.
Например, если при окислении диоксида серы в триоксид
Z0U2 (p) т vy2 (г) ^—? Z0U3 (г)
за какое-то время прореагирует 2 моль SO2 и 1 моль О2, то ? = 1 (реакция
совершит один пробег), а если прореагирует 0,5 моль SO2 и 0,25 моль О2, то
? = 0,25 (реакция совершит четверть пробега).
Переходя к бесконечно малому изменению количества вещества dnt и
dn'i, из F.34) получают
или для исходных веществ и продуктов реакции соответственно
dni = -\idb dni' = \i'd?r F.35)
208 6. Направление химической реакции
Подставляя F.35) в F.32), получают соотношение, которое при дос-
достижении химического равновесия в системе равно нулю, а для самопроиз-
самопроизвольного протекания процесса — отрицательной величине:
Химическая реакция проходила от момента смешения реагентов (/ = 0) до
момента установления химического равновесия (t = tpaBH), значит, d% > 0.
Следовательно,
5>ti, < 0. F.37)
Уравнение F.37) выражает общие условия химического равновесия и
самопроизвольного протекания реакции, идущей в закрытой системе при р,
Т= const. В F.37) слагаемые для продуктов реакции должны иметь знак «+»,
а для исходных веществ — знак «-».
6.6. Температурная зависимость стандартных энергии Гиббса,
энтальпии и энтропии химической реакции
Часто необходимо вычислять стандартные энтальпию, энтропию и
энергию Гиббса процесса при температуре, не равной 298 К (например, при
синтезе аммиака из простых веществ Т= 723...773 К). Стандартную энер-
энергию Гиббса химической реакции рассчитывают по формуле F.24)
ArG°T=ArH°-TArS°T,
где ArHj и ArSj — функции температуры. Следовательно, F.24) задает
явную зависимость ArGj =f(T) через энтропийную составляющую TArS^
и неявную — через энтальпийный ArHj и энтропийный TArSj факторы.
Зависимость стандартной энтальпии (теплового эффекта) необрати-
необратимой реакции
v2A2 -> v^A'j + v'2A'2; Ar#?. F.38)
от температуры описывается уравнением Кирхгофа в дифференциальной
форме:
^l^,, F.39)
6.6. Температурная зависимость энергии Гиббса, энтальпии и энтропии 209
где c'pi и cpi — истинные мольные теплоемкости при постоянном давлении
для /-го продукта и реагента соответственно, Дж /(моль • К). Величина Аср
играет роль температурного коэффициента в зависимости ArHj =f(T): она
представляет собой изменение энтальпии реакции при изменении темпера-
температуры на 1 К.
Для практического применения интегрируют F.39) в температурном
интервале Т\... Т2 и используют уравнение Кирхгофа в интегральной форме:
r#?=ArtfJ + \AcpdT.
F.40)
В качестве нижнего предела интегрирования обычно выбирают Т\ = 298 К,
так как ArH29S легко вычислить по следствиям из закона Гесса. Для расчета
ArHj2 необходимо знать аналитический вид зависимости Аср = f(T), т.е.
теплоемкость каждого продукта и реагента как функцию температуры.
Если теплоемкость каждого индивидуального вещества, участвующе-
участвующего в реакции F.38), задать степенным рядом вида cpi=ai+biT + ciT2 для
исходных веществ и crpi =a\ + b-Т + с\Т2 для продуктов, то из уравнения
F.39) можно получить зависимость Аср от температуры
F.41)
где Да = v\a[ + v'2a2 - vxax - v2a2; выражения для АЬ и Ас аналогичны.
Подставляя значение Аср из F.41) в F.40), после интегрирования по-
получают
АГН°Т =АгН0+АатА2 +^-Т3, F.42)
где АГНО — константа интегрирования для данной реакции, имеющая раз-
размерность энергии и физический смысл гипотетического теплового эффекта
при 0 К.
На практике при расчете ArHj применяют следующие приближения.
1. Пренебрегают температурной зависимостью энтальпии реакции по
уравнению Кирхгофа и считают АГН^ = Дг#298 = cons^
2. Принимают Аср = Ас°рШ = const для данной реакции. Величину
^ вычисляют по стандартным значениям теплоемкостей. В этом случае
210 б. Направление химической реакции
ArH°=ArH?>9S+Acp(T-298). F.43)
Стандартная энтропия реакции при Тф 298 К
АА°=ЛЛ°98+ \^dT9 F.44)
Т
298 i
где Аср рассчитывают по F.41).
Подставляя Аср из F.41) в F.44), после интегрирования получают
ArSr =ArS0+AalnT + AbT +—T2, F.45)
где ArS0 — константа интегрирования для данной реакции, имеющая раз-
размерность теплоемкости и физический смысл гипотетической энтропии ре-
реакции при 0 К.
На практике при расчете ArS^ применяют следующие приближения:
1) пренебрегают температурной зависимостью стандартной энтропии
реакции и считают ArS% = A^^s = const>
2) принимают Аср = Ас^298 = const для данной реакции. В этом случае
F.46)
При расчете ArGj используют следующие приближения.
1. Принимают стандартные энтальпию и энтропию реакции не зави-
зависящими от температуры: ArHj = Ar#298 = const' ^г$т ~ ^^298 = const. При
этом F.24) примет вид
Д^^=АГЯ2°98-ГАГ52°98. F.47)
Эта зависимость в координатах Т - ArGj представляет собой в отсут-
отсутствие фазовых переходов в системе прямую линию. Роль температурного
коэффициента выполняет стандартная энтропия реакции A^S^s, взятая с
обратным знаком: чем больше величина Д,.*^? тем сильнее зависимость
ArGj =f(T); при Л,.*^ w 0 ^г^т * ^r^298 = const — температурная зави-
зависимость ArGj отсутствует.
Графики ArGj = /(Г), построенные по уравнению F.47), для различ-
различных реакций приведены на рис. 6.3 (четыре типа химических реакций, вы-
6.6. Температурная зависимость энергии Гиббса, энтальпии и энтропии 211
кДж
Уг \S=0;ArII>0
ArS>0
Рис. 6.3. Зависимость энергии Гиббса различных химических реакций
от температуры в предположении ArHj = Ar#298 = const' ^г$т =
= АГ5298 = const (заштрихованы области термодинамически возмож-
возможного самопроизвольного протекания реакций бив)
деленные при анализе выражения F.24), отмечены буквами а-г). Тангенс
угла наклона прямой (tg a = -Ar529g) равен коэффициенту при температуре
в F.47); отрезок, отсекаемый прямой на оси ординат при Т = О К, равен сво-
свободному члену Дг#298. Из рис. 6.3 ясно, что с изменением температуры
возможно изменение знака ArG®, следовательно, изменение направления
самопроизвольного протекания процесса (точки Т\ и Т2 на оси абсцисс). Об-
Область под осью абсцисс соответствует самопроизвольным реакциям: ArGj <0.
2. С учетом Аср = Ас°р29% вычисляют по уравнениям F.43) и F.46) ве-
величины ArHj и ArS® соответственно, а затем по уравнению F.24) рассчи-
рассчитывают ArGj.
Практические занятия
Энергетика химических реакций и фазовых переходов
Примеры решения задач
Задача 1. Рассчитать стандартную энтальпию (стандартный тепловой эф-
эффект при постоянном давлении) реакции
Fe3O4(T) + 4CO(r) = 3Fe(T) + 4CO2(r)
по стандартным энтальпиям образования исходных веществ и продуктов.
Решение. Согласно первому следствию из закона Гесса, стандартная энтальпия
данной реакции
-^/^298Fe3O4(T) " 4AfH298 СОД =
,5) = -14,9 кДж (Qp< 0).
Данная реакция является экзотермической.
Задача 2. По термохимическому уравнению рассчитать стандартную эн-
энтальпию образования продукта реакции:
2Fe3O4(T) + 9SO2(r) + 5O2(r) = 3Fe2(SO4K(T), Л,#298 = -2845,7 кДж.
Решение. Согласно первому следствию из закона Гесса, стандартная энтальпия
данной реакции
АГЯ298 = 3A/#298Fe2(SO4K(T) - 2Л/#298резО4(т)- 9Д/#2988О2(г)- 5Л/#
следовательно, стандартная энтальпия образования
AfH29S Fe2(SO)(x) = " (А^Я298 + 2А/Н 2 + 9AH+ 5АЯ
fH29S Fe2(SO4K(x) = " (А^Я298 + 2А/Н 2Шс3О4(т) + 9AfH2ШО2(г) + 5А/Я298 О2(г)
= - [-2845,7 + 2(-1117,1) + 9(-296,9) + 5 • 0] = -2584 кДж/моль.
Задача 3. Для реакции
BaSO4(T) + 4C(rpa4)HT) = BaS(T) + 4CO(r)
рассчитать стандартные тепловые эффекты при постоянных давлении (Qp) и
объеме (Qy).
Примеры решения задач 213
Решение. Согласно первому следствию из закона Гесса, стандартный тепловой эф-
эффект при постоянном давлении
Qp = АГН298 = А///298Ва5(т) + 4А///298СО (г) - А/Я298BaSO4(T) ~ ^/
= -460,5 + 4(-110,5) - (-1465,0) -40 = 562,5 кДж = 562500 Дж.
Стандартные энтальпия АГЯ298 и внутренняя энергия ArU°29S реакции связаны
уравнением
АГЯ298
где Avr — разность числа молей газов в продуктах и исходных веществах.
Следовательно, Qv = Аг?/298 = АГЯ298 -AvrRT. Для приведенной реакции
Avr = 4-0 = 4 моль.
Отсюда стандартный тепловой эффект при постоянном объеме
Qv = ArU°m = 562500 - 4 • 8,31 • 298 = 552594 Дж s 552,6 кДж.
Задача 4. Рассчитать стандартную энтальпию образования ацетальдегида по его
стандартной энтальпии сгорания: ДС#298С н О(г) = -1192,4 кДж/моль.
Решение. По определению стандартной энтальпии сгорания, уравнение реакции
сгорания 1 моль ацетальдегида имеет вид
С2Н4О(Г) + 2,5О2(Г) = 2СО2(Г) + 2Н2О(Ж), АГЯ298 = Ас#
Согласно первому следствию из закона Гесса,
- AfH29S С2н4о (г) - 2,5А/Я298 ^ (г),
следовательно, стандартная энтальпия образования ацетальдегида
=^^ + ^^298 Н2О (ж) ~ 2,5А/Я298 ^ (г) - Ас#
= 2(-393,5) + 2(-285,8) - 2,50 - (-1192,4) = -166,2 кДж/моль.
Задача 5. Вычислить стандартную энтальпию реакции синтеза метанола
СО(Г) + 2Н2(Г) = С
с использованием стандартных энтальпий сгорания веществ-участников реакции,
если стандартная энтальпия сгорания метанола АсЯ298 сн 0Н(ж) = -726,6 кДж/моль.
Решение. В соответствии со вторым следствием из закона Гесса, тепловой эффект
данной реакции
214 Энергетика химических реакций и фазовых переходов
_ д jjrO , ^д т/О д ггО
Ос/7 29g С0(г) -Г ?1АСП 29g Нг(г) 1АСП
Первые два слагаемых в правой части этого уравнения находят с использованием
стандартной энтальпии сгорания:
1)СО(г) + 0,5О2(Г) = СО2(г)
Ar#298 (i) = Дс#298СО(г) = A/#298Co2(r) ~А/Я298со(г) = -393,5-(- 110,5) =
= -283 кДж/моль;
2) Над + 0,5О2(г) = Н2О(Ж)
Ar#°98 B) = Дс# °98 Н2(г) = AfH°29S Н20(ж) = -285,8 кДж/моль.
Отсюда искомая величина
Дг#298 = -283 + 2(-285,8) - (- 726,6) = - 128,0 кДж.
Задача 6. Вычислить стандартную энтальпию сгорания ацетилена С2Н2 по его стан-
стандартной энтальпии образования А/Я298С н (г) = 226,8 кДж/моль. Рассчитать стан-
стандартный тепловой эффект сгорания 7,33 л С2Н2 (объем измерен при температуре
17 °С и р = 98,6 кПа) при постоянном давлении.
Решение. Запишем уравнение реакции сгорания одного моля ацетилена:
С2Н2(Г) + 2,5О2(Г) = 2СО2(Г) + Н2О(Ж)
Согласно первому следствию из закона Гесса и определению стандартной эн-
энтальпии сгорания вещества,
^#•#298 = Ас#298С2Н2(г) = ^/^298 СО2(г) + ^/^298 Н2О (ж) ~ ^/^298 С2Н2(г) ~
- 2,5Д/#298О2(Г) = 2(-393,5) + (-285,8) - 226,8 - 2,5 • 0 = -1299,6 кДж/моль С2Н2.
Для вычисления количества вещества ацетилена приводят объем газа к нор-
нормальным условиям (ро = 101,3 кПа, Го = 273 К) по объединенному газовому закону:
То Т
р0Т 101,3B73 + 17)
Учитывая, что 1 моль газа при н.у. по закону Авогадро занимает объем 22,4 л,
определяют
*с,н, 6,72
ис,
VM 22,4
/0
Отсюда Qp = АГЯ298«С н = -1299,6 • 0,3 s -389,9 кДж.
Примеры решения задач 215
Задача 7. Рассчитать стандартную энтальпию образования озона из молекулярного
кислорода, используя стандартные энтальпии реакций окисления оксида меди (I)
кислородом и озоном:
Cu2O(T) ч
Cu2O(T) -i
Уравнение реакции
- - 02(r) = 2CuO(T)
- -O3(r) = 2CuO(T)
Ar#29g, кДж
-150,8
-198,2
Решение. В соответствии с методом термохимических схем вычитают из первого
уравнения второе и получают третье уравнение реакции:
- Оад = Т °з(г)
Стандартные энтальпии (тепловые эффекты) реакций будут алгебраически
связаны между собой так же, как их материальные балансы, т.е.
Аг3Н°ш =ДГ,#298 -Аг2Я^98 =-150,8-(-198,2) = 47,4 кДж.
Приведя уравнение реакции путем умножения обеих его частей на 3 к 1 моль
озона, получают уравнение реакции, стандартная энтальпия которой отвечает иско-
искомому значению:
1,5О2(г) = О3(г); Лг4#298 = А/Я^98Оз(г) = 3 . 47,4 = 142,2 кДж/моль.
Задача 8. Рассчитать энергию химической связи в молекуле воды, если известны
стандартная энтальпия образования газообразной воды и энергии диссоциации мо-
молекул водорода и кислорода: Д/#298н о(г) = —241,8 кДж/моль; ЕДН = 436 кДж/моль;
ЕЛ02 = 498 кДж/моль.
Решение. Реакцию образования 1 моль газообразной воды записывают через стадии
диссоциации соответствующих количеств веществ водорода и кислорода на атомы
и соединения их в молекулу:
1) Н2(Г) = 2Н(Г) (Дг|Я=?ДН2 = 436кДж);
2) 0,5О2(г) = О(г) (Д,2Я = 0,5 • ЕаОг = 0,5 • 498 = 249 кДж);
3) 2H(r) + O(r) = H2O(r) (Дг3#).
Суммируя уравнения, получают реакцию образования газообразной воды из
простых веществ:
4)H2(r) + 0,5O2(r) = H2O(r)
216 Энергетика химических реакций и фазовых переходов
тепловой эффект которой Ar4#= Аг1# + Аг2#+ Аг3Я = A/H°29S н 0(г) = -241,8 кДж.
Следовательно, АгЪН = Аг4Н - АгХН - Аг2Н = -241,8 - 436 - 249 = -926,8 кДж.
Так как в одной молекуле воды имеется две связи О—Н и при разрыве химических
А М
связей энергия поглощается, то ?свн-о = — — = 0,5 • 926,8 = 463,4 кДж/связь.
2
Таким образом, энергию химической связи можно рассчитать, используя таб-
табличные значения энтальпий химических реакций и энергий диссоциации.
Задача 9. Рассчитать энергию кристаллической решетки хлорида кальция, если из-
известны следующие термодинамические величины:
стандартная энтальпия образования хлорида кальция А/#298СаС1 (т) =
= -795 кДж/моль;
энтальпия сублимации кальция Asub#298Ca(T) = 177,3 кДж/моль;
первая и вторая энергии ионизации атома кальция ?лСа = 589,8 кДж/моль,
Erica = 1145 кДж/моль;
энергия диссоциации молекулы хлора ЕД С1 = 242,6 кДж/моль;
сродство к электрону атома хлора ЕАС\ = -348,8 кДж/моль.
Решение. Представляют процесс образования кристаллического хлорида кальция из
простых веществ как первый путь и из ионов — второй путь.
Первый путь: Са(т) + С12(Г) = СаС12(Т); А/^298Саа (т) = ~795 кДж/моль.
Второй путь рассматривают как сумму последовательных стадий.
1. Сублимация кристаллического кальция (переход из твердого состояния в
газообразное):
Са(т) = Са(г); Аг1#= А8иЬ#298Са(т) = 177,3 кДж/моль;
2. Отрыв двух электронов от атома кальция (сумма первой и второй энергии
ионизации):
Са(г) - 2е = Са2+(г); Ar2H = EnCa + Епс* = 589,8 +1145 = 1734,8 кДж/моль;
3. Диссоциация молекул хлора на атомы:
С12 (Г) = 2С1 (г); Аг3# = ?д cl = 242,6 кДж/моль;
4. Присоединение двух электронов к двум атомам хлора (удвоенная энергия
сродства к электрону):
2С1 (г) + 2е = 2СГ(Г); Аг4Я = 2ЕАСХ = 2(-348,8) = -697,6 кДж;
5. Взаимодействие ионов Са2+ и СГ:
Са (Г) + 2С1 (Г) = СаС12(Т); ArsH= —EK?m реш caci2
Энергия кристаллической решетки Екр. реш — энергия, которую необходимо
затратить на разрыв ионных связей в решетке и последующее удаление ионов на
Задачи для самостоятельного решения
217
достаточно большое расстояние, на котором не проявляются силы межионного
взаимодействия. Согласно этому определению, ?кр. реш равна взятой с обратным зна-
знаком энтальпии реакции 5. Непосредственный синтез хлорида кальция из простых
веществ и сумма реакций 1-5 приводят к одному и тому же продукту при одинако-
одинаковых исходных веществах (можно убедиться в этом, сложив уравнения реакций 1-5).
В соответствии с законом Гесса,
298СаС.2(т)
Аг3Я+
следовательно, ?кр.решСаа2 = -Аг5Я= Аг1Я + Аг2Я + Аг3Н + Аг4Я -
= 177,3 + 1734,8 + 242,6 - 697,6 -(-795) = 2252,1 кДж/моль.
Задачи для самостоятельного решения
При решении задач используйте данные, приведенные в табл. П.5.1. и П.5.2.
1-14. Рассчитайте стандартную энтальпию (стандартный тепловой эффект при по-
постоянном давлении) приведенных реакций, предварительно подобрав стехиометри-
ческие коэффициенты уравнений.
№
1
2
3
4
5
6
7
Реакция
NH3(r) + О2(Г) = NO(r) + Н2О(Г)
Fe2O3(T) + Mg(T) = Fe(T) + MgO(T)
H2S(r) + О2(Г) = SO2(r) + Н2О(Ж)
WO3(T) + H2(r) = W(T) + H2O(r)
HNO3(«) = NO2(r) + Н2О(Ж) + О2(Г)
FeSO4(T) = Fe2O3(T) + SO2(r) + O2(r)
Na2CO3(T) + SiO2(T) =
= Na2SiO3(T) + CO2(r)
№
8
9
10
11
12
13
14
Реакция
SiH4(r) + C>2(r) = SiO2(T) + Н2О(Ж)
ZnS(T) + O2(r) = ZnO(T) + SO2(r)
MgCO3(T) = MgO(T) + CO2(r)
SO3(») + Н2О(Ж) = H2SO4(»)
СбН6(Ж) + О2(Г) = СО2(Г) + Н2О(Ж)
TiCl4(r) + Mg(T) = Ti(T) + MgCl2(T)
CuO(T) + NH4Cl(T) =
= Си(т) + Н2О(ж) + НС1(г) + Ы2(Г)
15-24. Рассчитайте стандартные энтальпии (АГЯ298) и стандартные внутренние
энергии (ArU^9S) приведенных реакций.
№
15
16
17
18
19
Уравнение реакции
N2(r, + 3H2(r) = 2NH3(r)
H-C8Hi8(r) = H-C4Hio(r) + С4Н8(Г)
бутен-1
4НС1(Г) + О2(г) = 2С12(Г) + 2Н2О(Ж)
Fe2O3(T) + 3CO(r) = 2Fe(T) + ЗСО2(г)
2SO2(r) + O2(r) = 2SO3(r)
№
20
21
22
23
24
Уравнение реакции
С2Н2(Г) + 2Н2(Г) = С2Н6(Г)
СН4(Г) + 2Н2О(Г) = СО2(Г) + 4Н2(Г)
СН4(г) + Н2О(Г) = СО(Г) + ЗН2(г)
СО(Г) + Н2О(Г) = СО2(Г) + Над
М2О4(Г) + О3(г) = N2O5(K) + Оад
218
Энергетика химических реакций и фазовых переходов
25-34. По термохимическим уравнениям рассчитайте стандартную энтальпию обра-
образования исходных веществ (А/Я^, кДж/моль), если известны стандартные энталь-
энтальпии химических реакций.
25
26
27
28
29
30
31
32
33
34
Уравнение реакции
2AgNO3(T) = 2Ag(T) + 2NO2(r) + О2(г)
2Pb(NO3J(T) = 2РЬО(Т) + 4NO2(r) + O2(r)
4NaC103(T) = 3NaC104(T) + NaCl(T)
4FeSO4(T) = 2Fe2O3(T) + 4SO2(r) + O2(r)
2A1(OHK(T) = A12O3(T) + ЗН2О(Ж)
2Fe(OHK(T) = Fe2O3(T) + ЗН2О(ж)
Fe(COM(HC) = Fe(T) + 5CO(r)
4HNO3(HC) = 4NO2(r) + 2Н2О(Ж) + O2(r)
NH4NO3(T) = N2O(r) + 2H2OEK)
(NH4JCr207(T) = Cr2O3(T) + N2(r) + 4Н2О(Ж)
АГЯ298, кДж
317,4
601,6
-97,9
886,0
96,6
-26,4
211,5
261,6
-124,6
-484,7
35-44. По термохимическим уравнениям рассчитайте стандартную энтальпию обра-
образования продуктов (А/Я 298, кДж/моль), если известны стандартные энтальпии хи-
химических реакций.
№
35
36
37
38
39
40
41
42
43
44
Уравнение реакции
К2О(Т) + Н2О(Ж) + 2СО2(Г) = 2КНСО3(Т)
К2О(Т) + СО2(Г) = К2СОз(Т)
2Сг2О3(Т) + 6SO2(r) + 3O2(r) = 2Cr2(SO4K(T)
2А12Оз(т) + 6SO2(r) + 3O2(r) = 2A12(SO4K(t)
4СаО(т) + Р40,о(т) + 2Н2О(ж) = 4СаНРС>4(Т)
6СаО{т) + Р4О10(Т) = 2Са3(РО4J(т)
2FeO(T) + 3SO2(r) + 2O2(r) = Fe2(SO4K(T)
2Fe2O3(T) + 6SO2(r) + 3O2(r) = 2Fe2(SO4K(T)
Fe3+(p) + 6CN-(p) = [Fe(CNN]3-(p)
Fe2+(p) + 6CN-(p) = [Fe(CNN]4-(p)
ДЛ298, *Д*
-482,6
-389,4
-2553,4
-1751,0
-1164,8
-1450,2
-1148,5
-1742,2
-225,7
-289,9
45-56. Вычислите стандартные энтальпии образования A/H°29S следующих веществ
по их стандартным энтальпиям сгорания АсЯ^.
Задачи для самостоятельного решения
219
№
45
46
47
48
49
50
51
52
53
54
55
56
Вещество
Ацетилен
Этилен
Этан
Бензол
Этанол
Ацетон
Глицерин
Этилацетат
Бензойная кислота
Метил хлорид
Дисульфид углерода
Анилин
Химическая формула
и агрегатное состояние
С2Н4(г)
С2Н6(Г)
СбНб(ж)
С2Н6О(Ж)
СзНбО(ж)
С3Н8Оз(Ж)
С4Н8С>2(ж)
С7Н6О2(т)
СНзС1(Г)
CS2(hc)
C6H7N(«)
АСЯ298? кДж/моль
-1299,6
-1411,0
-1559,9
-3267,7
-1366,9
-1789,8
-1664,4
-2247,7
-3227,5
-689,1
-1075,0
-3396,2
57-64. Рассчитайте стандартную энтальпию перечисленных реакций с использова-
использованием стандартных энтальпий сгорания веществ-участников.
№
57
58
59
60
Уравнение реакции
/~ч ТТ | ТТ /~* ТТ
^2П4(г) "Т" (г) — ^2Пб(г)
С2Н2(Г) + Н2(Г) = С2Н4(Г)
(J2Al2(r) ~^~ 2ri2(r) = ^-/2Иб(г)
\^6аА6(ж) ~r"Jil2(r) ~~ ^6^12(ж)
№
61
62
63
64
Уравнение реакции
ЗС2Н2(Г) = C6H6(r)
2С2Н6О(Ж) = С4НюО(Ж) + Н2О(Ж)
с2н6о(ж) = с2н4(г) + н2о(ж)
СН3СООН(Ж) + С2Н6О(Ж) =
= СНзС'ООС2Г15(ж) "Ь Г12О(Ж)
65-68. Вычислите стандартный тепловой эффект сгорания вещества или смеси при
постоянном давлении.
Вещество (смесь), масса (количество, объем, состав)
65
66
67
68
300 г этанола С2Н5ОН(ж)
5 моль бензола С6Н6(Ж)
5,02 л ацетилена С2Н2 (объем измерен при давлении 745 мм рт. ст. и тем-
температуре 27 °С)
100 л пропан (С3Н8)-бутановой (С4Ню) смеси, содержащей 70 % (об.) про-
пропана (объем измерен при давлении 750 мм рт. ст. и температуре 20 °С)
69-72. Используя стандартные энтальпии реакций окисления оксидов металлов ки-
кислородом и озоном, рассчитайте стандартную энтальпию образования озона из мо-
молекулярного кислорода.
220
Энергетика химических реакций и фазовых переходов
№
69
70
71
72
Уравнение реакции
GeO(T) + '/Юад = GeO2(T)
GeO(T) + ЙОз<г) = GeO2(T)
SnO(T) + l/2O2(r) = SnO2(T)
SnO(T) + I/3O3(r) = SnO2(T)
MnO(T) + '/2O2(r)= МпОад
MnO(T) + УзОз(Г) = MnO2(T)
3CoO(T) + ЙОад = СозОад
3CoO(T) + '/зОзм = СозОад
Дгя"98, кДж
-299,7
-347,1
-294,8
-342,2
-136,4
-183,8
-161,1
-208,5
73. Вычислите энергию химической связи в молекуле оксида азота (И), если извест-
известны его стандартная энтальпия образования и энергии диссоциации молекул азота и
кислорода:
А/^298 NO(r) = 91,3 кДж/моль; Еа N = 946 кДж/моль; Ело = 498 кДж/моль.
74. Определите энергию химической связи в молекуле оксида углерода (II), если
известны его стандартная энтальпия образования, энтальпия сублимации (атомиза-
ции) графита и энергия диссоциации молекул кислорода:
А/Я298 со (г) = -1 Ю,5 кДж/моль; Asub#
) = 712,5 кДж/моль;
ЕД 0 = 498 кДж/моль.
75. Рассчитайте энтальпию образования фторида серы (VI) из атомов и среднюю
энергию связи S—F, если известны его стандартная энтальпия образования, эн-
энтальпия сублимации (атомизации) серы и энергия диссоциации молекул фтора:
= 273,0 кДж/моль;
=-1219кДж/моль;
?flF =159 кДж/моль.
76. Вычислите энергию кристаллической решетки фторида калия, если известны
следующие термодинамические величины: стандартная энтальпия образования
фторида калия A/#298kf(t) = ~567,4 кДж/моль; энтальпия сублимации (атомизации)
калия А8иЬЯ298К(т) = 89,2 кДж/моль; первая энергия ионизации атома калия Епк =
= 418,8 кДж/моль; энергия диссоциации молекул фтора Ел F = 159 кДж/моль; срод-
сродство к электрону атома фтора EAF = -327,9 кДж/моль.
77. Рассчитайте энергию кристаллической решетки бромида бария, если известны
следующие термодинамические величины: стандартная энтальпия образования
бромида бария A/#298BaBr (т)= -756,5 кДж/моль; энтальпия сублимации (атомиза-
Примеры решения задач 221
ции) бария Asub#298Ba(T) = 174,6 кДж/моль; первая и вторая энергии ионизации ато-
атома бария ?лва = 502,8 кДж/моль; Епва = 965,2 кДж/моль; энергия диссоциации мо-
молекул брома ЕЛВт = 190,1 кДж/моль; сродство к электрону атома брома ЕАВг =
= -324,6 кДж/моль; энтальпия испарения брома Дуар#вг (ж) = 30,9 кДж/моль.
Направление самопроизвольного протекания
химических реакций
Примеры решения задач
Задача 1. Вычислить изменение энтропии при кристаллизации 10 кг фторида кальция,
если для этой соли известны молярная теплота кристаллизации АНщ, = -29,7 кДж/моль
и температура кристаллизации Т^ = 1691 К при давлении/? =101,3 кПа.
Решение. Изменение энтропии при фазовом переходе (кристаллизации) 10 кг фторида
кальция рассчитывают с учетом количества вещества wCaF :
V'aF —
2 Т М
7кр MCaF2
где m и М— масса и молярная масса соли соответственно; MCaF — 78 г/моль.
Подставляя числовые значения величин, получают
s - 2252 Дж/К s -2,25 кДж/К (AS™ < 0).
кр
1691 78
При кристаллизации вещества энтропия уменьшается.
Задача 2. Определить изменения энтальпии и энтропии при изобарном нагреве 1,6 кг
сульфата меди (II) в интервале изменения температуры от 300 до 700 К. Средняя
молярная теплоемкость ср соли при постоянном давлении в указанном интервале
температур является постоянной величиной, равной 114,4 Дж/(моль-К).
Решение. Подставляя в уравнения
АН=ср(Т2-Т{) и ASp = cpln^
числовые значения величин и учитывая количество вещества сульфата меди (II),
вычисляют изменения энтальпии и энтропии при изобарном нагреве соли:
ДЯ= ср(Тг - Г,)«Си5О4 = ср(Т2-
CaSO4
222 Направление самопроизвольного протекания химических реакций
= 114,4 • G00 - 300) • ^- = 457600 Дж = 457,6 кДж;
lou
300ll60j
При нагреве вещества энтальпия и энтропия увеличиваются.
Задача 3. Рассчитать изменения энтальпии, внутренней энергии и энтропии при
изобарном и изохорном охлаждении 420 г азота в интервале температур 1200...300 К.
Средняя молярная теплоемкость ср азота при постоянном давлении в указанном ин-
интервале температур постоянна и равна 31,05 Дж/(моль • К).
Решение. В уравнения
с/7(Г2Г1) и ASp cp\n
Т\
подставляют числовые значения величин и учитывая количество вещества азота
»Ч 420
иы = — = = 15 моль,
2 Мщ 28
вычисляют изменения энтальпии и энтропии при изобарном охлаждении газа:
АН= ср(Т2 - ТОпщ =31,05C00 - 1200)-15 = -419175Дж s - 419,2 кДж;
7; зоо
ASP = nN cp 111-2-= 15 . 31?05 In = -645,7 Дж/К (AS, < 0).
2 7] 1200
При охлаждении вещества энтальпия и энтропия уменьшаются.
Соответствующие изменения внутренней энергии и энтропии при изохорном
охлаждении газа рассчитывают по уравнениям
т
AU=cv(T2-T]) и ASV
учитывая количество вещества азота «N =15 моль и соотношение для 1 моль иде-
идеального газа
су = ср - R = 31,05 - 8,31 = 22,74 Дж/(моль • К);
ДС/= су(Т2- Тх)пщ = 22,74C00 - 1200)-15 = -306990 Джs-307,0 кДж;
ASV= nN cv\n— = 15 • 22,74 In -^L = -472,9 Дж/К (ASP > 0).
2 7] 1200
Примеры решения задач 223
При этом I AiSV I < I ASP I, так как при изобарном охлаждении газа энтропия
дополнительно убывает за счет уменьшения объема системы.
Задача 4. Вычислить изменение энтропии при изобарно-изотермическом смешении
140 г азота, 44 г оксида углерода (IV) и 80 г неона для всей системы и для 1 моль
смеси. Газы химически не взаимодействуют.
Решение. При смешении химически не взаимодействующих газов изменение энтро-
энтропии вычисляют по формуле
Количества веществ газов равны:
"Ч 140 гпсо 44
Их, = —— = = 5 моль; пгп = — = — =1 моль;
2 Мщ 28 С°2 MCOi 44
WN 80 л
= — = 4 моль.
MNe 20
Общее количество веществ газов в смеси: пш = ^и,. =5 + 1+4=10 моль.
Молярные доли газов в смеси равны:
у - ЛЬ-- у -OS- X -0 1- Г -П4- Уг -1
Изменение энтропии при изобарно-изотермическом смешении газов
AmixS = -10 • 8,31@,5 In 0,5 + 0,1 In 0,1 + 0,4 In 0,4) = -10 • 8,31 (-0,9433) =
= 78,4Дж/К,
или для 1 моль смеси
Amix5 = -Д]>]Х/1п*/ =7,84Дж/(моль-К) (AmixS > 0).
у
Следовательно, при смешении газов энтропия системы возрастает.
Задача 5. Рассчитать энтропию 1 моль этанола СгНбО в идеальном водном раство-
растворе, в котором массовая доля спирта равна 0,23.
Решение. Энтропия 1 моль этанола S°29S = 160,7 Дж/(моль * К) при образовании иде-
идеального раствора с водой увеличится на величину энтропии смешения
за счет распределения молекул спирта в объеме всего раствора и возрастания бес-
беспорядка в системе. Известно, что идеальные жидкие и твердые растворы подчиня-
подчиняются законам идеальных газов, т.е.
224 Направление самопроизвольного протекания химических реакций
V п
mixu = ПД 1П -^ ? nfi In -S*- = - W/Д In Д,
где Д = —— — молярная доля /-го компонента. Тогда энтропия смешения для всей
"см
системы Лт1х5 = ХЛ1™А-
Таким образом, решение данной задачи сводится к определению молярной
доли этанола в растворе. Масса раствора, согласно B.10),
т - *с2Н6о Мс2н6о 12-
0
С^О ^С^О °>23
Масса воды тно = т- гпсно = 200 -46= 154 г.
тн о 154 ^
Количество вещества воды пи п = —^— = = 8,556 моль.
2 МНг0 18
Молярная доля этанола в растворе Хс н 0 = — = = 0,1046.
«с2н6о+«н2о 1 + 8,556
Энтропия этанола в растворе
^С2Н6О = ^298С2Н6О +AmixS'c2H6O = ^298 С2Н6О ~ ^'П ^С2Н6О =
= 160,7 - 8,31 In 0,1046 = 179,46 Дж/(моль • К).
Задача 6. Рассчитать стандартную энтропию (изменение энтропии в процессе, про-
протекающем в стандартных термодинамических условиях) реакции
2H2S(r) + ЗО2(Г) = 2SO2(r) + 2Н2О(Ж)
Является ли эта реакция термодинамически возможной в изолированной системе?
Решение. Стандартную энтропию данной реакции определяют, используя таблич-
табличные значения стандартных энтропии исходных веществ и продуктов:
А С0 =99° +9^° —99° —Ч^° =
L\го 298 АО 298 Sq2(г) -г АО 298 Н2о(ж) АО 298 ц2$(Г) Jo 298 q2(г)
= 2-248,1 +2-70,08-2-205,07-3-205,04 = -388,9 Дж/К (АГ5'298<0)'
следовательно, данная реакция термодинамически не возможна в изолированной
системе при стандартных условиях.
Задача 7. Рассчитать стандартную энергию Гиббса (изменение энергии Гиббса в
процессе, протекающем в стандартных термодинамических условиях) реакции
Примеры решения задач 225
2H2S(r) + ЗО2(Г) = 2SO2(r> + 2Н2О(Ж).
Является ли эта реакция термодинамически возможной в закрытой системе?
Решение. Стандартную энергию Гиббса данной реакции рассчитывают:
а) по уравнению Гиббса—Гельмгольца
ArG°29S =ArH°29S -298ArS298,
где стандартная энтальпия реакции
д tjO _ лд гтО лд ттО 9Л Л® —
^г** 298 — LL*fn 298SO2(r) "*" ^^f*2 298Н2О(ж) ^V« 298H2S(r) ~
= 2(-296,9) + 2(-285,83) - 2(-21) = - 1123,46 кДж.
Стандартная энтропия реакции ArS298 = -388,9 Дж/К (см. задачу 6).
Подставляя числовые значения стандартных энтальпии и энтропии процесса
в уравнение Гиббса—Гельмгольца, получают
ArG 298 = -1123460 - 298(-388,9) = -1007192 Дж = -1007,6 кДж (ArG2098 < 0),
следовательно, данная реакция является термодинамически возможной в закрытой
системе при стандартных условиях;
б) по стандартным энергиям Гиббса образования веществ-участников реакции
ArG298 =2A/G +2AG 2AG
298 =2A/G298SO2(r)
= 2(-300,2) + 2(-237,24) - 2(-33,8) = -1007,3 кДж < 0.
Задача 8. По значениям стандартных энтальпий образования и энтропии соответст-
соответствующих веществ рассчитать стандартную энергию Гиббса образования сульфата
натрия при температуре Т = 298 К.
Решение. Реакция образования 1 моль сульфата натрия из соответствующего коли-
количества простых веществ имеет вид
2Na(T) + S(Tp0M6) + 2О2(Г) = Na2SO4(T)
По определению, стандартная энергия Гиббса образования сульфата натрия
Стандартная энтальпия данной реакции равна стандартной энтальпии обра-
образования сульфата натрия, так как энтальпии образования простых веществ в их тер-
термодинамически наиболее устойчивых состояниях равны нулю:
ДГЯ°98 =A/H^98Na2SO4(T) =-1384,6 кДж.
Стандартная энтропия реакции
8 - 9795
226 Направление самопроизвольного протекания химических реакций
~^298Na2SO4(T) ~ ZO 298 Na(T) ~ ^ 298 S(t ^^ ~ Zt> 298 О2 (г)
= 149,5-2-51,5-31,9-2-205,04 =-395,8 Дж/К.
Используя уравнение Гиббса—Гельмгольца, рассчитывают стандартную
энергию Гиббса образования сульфата натрия:
A/G298Na2so4(T) = 384,6 - 298(-0,3955) = -1266,7 кДж.
Задача 9. Применяя критерий самопроизвольного протекания процесса в закрытой
системе, необходимо ответить на вопрос о термодинамической устойчивости нит-
нитрата меди (II) при температуре Т = 298 К и 1000 К (р= 101,3 кПа).
Решение. Записывают уравнение реакции термического разложения нитрата меди (II)
2Cu(NO3J(t) = 2CuO(T) + 4NO2(r) + О2(г)
Значения стандартной энергии Гиббса для данной реакции при Т = 298 К и
1000 К рассчитывают по уравнению Гиббса—Гельмгольца:
А^298 =А^298 ~ 298 Аг?298;
ArG°T s Аг#298 -TArS°29S.
Стандартные энтальпию Аг#298 и энтропию ArS°29S процесса определяют по
табличным значениям энтальпий образования и энтропии соответствующих веществ:
А'^298 = 2Д/^298 СиО(т) + ^fH298 NO2(г) ~ 2^fl298 Cu(NO3J (т) =
= 2(-162) + 4-34,2 - 2(-305,3) = 423,4 кДж;
A rO 298 ZO 298 СиО(т) ЧО 298 NO2 (г) ^ 298 О2 (г) Zu 298 Cu(NO3 J (т)
= 2-42,6 + 4-240,1 + 205,0 - 2-192,5 = 865,6 Дж/К.
^Отсюда при Т = 298 К
ArG298 = 423400 - 298-865,6 =165451,2 Дж^165,5 кДж (ArG2°98 > 0).
Следовательно, при Т= 298 К процесс термического разложения нитрата ме-
меди (II) не может протекать самопроизвольно — это соединение является термоди-
термодинамически устойчивым. Самопроизвольно в закрытой системе осуществляется об-
обратная реакция синтеза нитрата меди (II) из оксида меди (II), оксида азота (IV) и
кислорода, для которой ArG 298 =-165,5 кДж (A^G^ <0).
Стандартная энергия Гиббса при температуре 1000 К
ArG °mo = 423400 - 1000 • 865,6 = -442200 Дж = -442,2 кДж < 0.
Следовательно, при температуре 1000 К процесс термического распада нит-
нитрата меди (II) протекает самопроизвольно, соединение является термодинамически
неустойчивым.
Примеры решения задач 227
Задача 10. Рассчитать температуру начала реакции (равновероятности протекания
прямого и обратного процессов) термического разложения нитрата меди (II). Стан-
Стандартные значения энтальпии и энтропии реакции считать не зависимыми от темпе-
температуры.
Решение. При термодинамическом равновесии
ArG°T zArH°29S - ГрввнДг^в =0,
следовательно,
^ 423400
Д^, 423400 а489ми216
Значения стандартных энтальпии и энтропии реакции взяты из задачи 9. Та-
Таким образом, в закрытой системе при температуре выше Т = 489 К для данной реак-
реакции ArG\ < 0, т.е. идет процесс разложения нитрата меди (И), при температуре ни-
ниже Т = 489 К ArGQT > 0, т.е. идет процесс синтеза соли.
Задача 11. Без проведения термодинамических расчетов определить, высоко- или
низкотемпературный режим способствует проведению в закрытой системе экзотер-
экзотермической реакции
2NO(r) + O2(r) = 2NO2(r).
Энтальпийным или энтропийным фактором определяется термодинамическая воз-
возможность протекания этой реакции?
Решение. В первом приближении стандартная энергия Гиббса реакции
ArG°T s Ar#298 " т^^ш •
Для экзотермического процесса АГН^9В < 0.
Данная реакция гомогенная и гомофазная, так как все вещества-участники
являются газами. В продуктах реакции присутствуют 2 моль газа (NO2), в исходных
веществах — 3 моль газов B моль NO и 1 моль О2), следовательно, энтропия про-
продуктов меньше таковой для исходных веществ, и стандартная энтропия процесса
-Iv,.52°98,<0.
гО
Анализируя уравнение Гиббса—Гельмгольца с учетом знаков ArH29S и
А Н°
ArS°29S, приходят к выводу, что ArG298 < 0 при Т< ——~^-, т.е. проведению реак-
АЛ98
ции в закрытой системе способствует низкотемпературный режим. Термодинамиче-
228 Направление самопроизвольного протекания химических реакций
екая возможность протекания процесса (ArGT< 0) обеспечивается энтальпийным
фактором (Дг#298 < 0), действующим в благоприятном направлении. Энтропийный
фактор (-ГДГ? 298 > 0) действует в неблагоприятном направлений.
Задачи для самостоятельного решения
Для решения задач используйте данные, приведенные в табл. П.5.1 и П.5.2.
1-6. Вычислите изменение энтропии при фазовом переходе указанной массы т ве-
вещества, если известны молярная теплота (AtrH^) и температура (Ttr) фазового пере-
перехода при стандартном давлении.
№
1
2
3
4
5
6
Фазовый переход
NaCl(T) ** NaCiw
А1С13(Ж) i=t А1С13(Т)
MgO(T) <=± МёО(ж)
А12О3(ж) ±± А12О3(Т)
ИС14(Ж) <н> TiCV)
НгО(г) ^± Н2О(Ж)
т, г
585
200
400
500
150
180
А,гН°Т,
кДж/моль
28,7
-35,6
77,3
-118,5
36,0
-40,7
т,г,к
1074
466
3123
2345
410
373
7-16. Рассчитайте изменения энтальпии и энтропии при изобарном нагреве (охлаж-
(охлаждении) указанной массы m кристаллического вещества от начальной температуры Т\
до конечной температуры Гг. Средняя молярная теплоемкость ср вещества при по-
постоянном давлении в указанном интервале температур постоянна и приведена в ус-
условии.
№
7
8
9
10
11
12
13
14
15
16
Вещество
Оксид алюминия
Хлорид бария
Карбонат кальция
Сульфат кадмия
Сульфид меди (II)
Оксид железа (И)
Дисульфид железа (II)
Оксид марганца (И)
Гексафтороалюминат натрия
Оксид свинца (IV)
т, г
306
208
500
416
960
612
900
1065
735
956
300
600
300
1000
300
1200
300
500
300
900
Г2,К
800
300
700
300
1000
300
600
300
600
300
С^,Дж/(мольК)
107,2
77,40
103,1
127,6
51,55
56,57
70,17
47,28
241,18
72,68
Задачи для самостоятельного решения
229
17-32. Определите изменения энтальпии, внутренней энергии и энтропии при изо-
изобарном и изохорном нагреве (охлаждении) указанной массы т газообразного веще-
вещества от начальной температуры Т\ до конечной температуры Г2. Средняя молярная
теплоемкость ср вещества при постоянном давлении в указанном интервале темпе-
температур постоянна и приведена в условии.
№
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
Вещество
Бром
Хлор
Фтор
Водород
Йод
Азот
Кислород
Cepa(S2)
Трихлорид бора
Оксид углерода (II)
Оксид углерода (IV)
Бромоводород
Хлороводород
Вода
Аммиак
Оксид азота (II)
W, Г
800
568
456
30
508
280
320
352
470
420
880
891
219
360
510
900
300
800
300
900
300
1500
300
700
300
1000
300
1200
300
1500
300
2000
Г2,К
1000
300
700
300
600
300
800
300
500
300
900
300
1500
300
700
300
Ср, Дж/(моль • К)
40,00
36,23
34,02
29,42
37,28
31,71
31,76
34,98
68,47
30,92
46,44
30,83
30,92
39,66
41,71
33,72
33-42. Вычислите изменение энтропии при изобарно-изотермическом смешении
указанных масс трех химически невзаимодействующих газов для всей системы и
для 1 моль смеси.
33
34
35
36
37
38
39
40
41
42
Газ1
Азот
Азот
Азот
Гелий
Неон
Аргон
Азот
Азот
Гелий
Аммиак
Ш\, Г
84
182
70
40
160
80
308
252
60
170
Газ 2
Оксид углерода (II)
Неон
Гелий
Неон
Аргон
Криптон
Кислород
Оксид азота (II)
Оксид углерода (IV)
Неон
Ш2, Г
112
30
24
100
280
168
224
210
132
120
ГазЗ
Гелий
Аргон
Криптон
Аргон
Криптон
Ксенон
Криптон
Криптон
Неон
Аргон
w3, г
12
80
126
200
420
131
168
336
40
160
43-52. Рассчитайте энтропию одного моля вещества В при растворении его в рас-
растворителе А с образованием идеального раствора, если известна массовая доля со
растворенного вещества.
230 Направление самопроизвольного протекания химических реакций
№
43
44
45
46
47
48
49
50
51
52
Растворенное вещество В
Серебро
Графит
Графит
Никель
Метанол
Бензол
Уксусная кислота
Ацетон
Ацетилен
Тетрахлорид углерода
Растворитель А
Золото
Железо
Железо
Медь
Вода
Толуол
Вода
Вода
Ацетон
Диэтиловый эфир
00,%
4,32
0,60
2,40
23,48
32,00
19,50
0,30
2,90
32,50
30,80
53-64. Предварительно расставив стехиометрические коэффициенты, рассчитайте
стандартную энтропию приведенных реакций и определите направление самопро-
самопроизвольного процесса в изолированной системе (Г = 298 К).
№
53
54
55
56
57
58
Уравнение реакции
Mg(T) + CO2{r) ?:
CuS(T) + O2(r) &
Fe2(SO4K(T) <=*
Fe2O3(T) + CO(r)
N2O3(r) ^ N0:
CO(r) + O3(r) *±
* MgO(T)
: CuO(T) +
Fe2O3(T) +
*=* FeO(T)
2(r) + NO(r)
CO2(r) + (
+ co(r)
so2(r)
so3(r)
+ co2(r)
№
59
60
61
62
63
64
N2O(r) -
Cr2O3(T
Уравнение реакции
•" Оз(г) ^
) + А1(Т)
Pb(NO3J(T) *±
PbO(T)-
HBr(r) н
V2O3(T)
± NO(r) + NO2(r)
?=± Cr(T) + A12O3(T)
*=± PЮ(т) + NO2(r) + O2(r)
Юэд ?
hCl2(r) ?
+ Ca<T) 5
i PbO2(T) + O2(r)
=i HCl (r)+Вг2(ж)
с* CaO(T) + V(T)
65-76. Рассчитайте стандартную энергию Гиббса приведенных реакций при Г= 298 К и
определите направление самопроизвольного процесса в закрытой системе.
№
65
66
67
68
69
70
Mg(T) +
CuS(T) ¦+
Fe2(SO,
Fe2O3(T)
N2O3(r)
co(r) +
Уравнение реакции
CO2(r) ?± MgO(T) + CO(r)
¦O2(r) <=i CuO(T) + SO2(r)
0з(т> ^ Fe2O3(T) + SO3(r)
+ co(r) ^
*=* FeO(T) + CO2(r)
^ NO2(r) + NO(r)
Оз(г) ^ CO2(r) + O2(r)
№
71
72
73
74
75
76
N2O(r)J
Cr2O3(T
Pb(NO3
PbO(T)-
HBr(r) н
V2O3(T)
Уравнение реакции
Ю3(Г) <=
+ A1(T) ?
)ад ^
^O3(r) ?=
-ci2(r) ?=
+ Ca<T) ?
^ NO(r) +
=* Cr(TL
Ю 4 + NO
^ PbO2(T)
t HCl(r)
i CaO(T)
NO2(r)
A12O3(T)
+ O2(r)
+Вг2(ж)
+ V(T)
77-90. По табличным значениям стандартных энтальпий образования и энтропии
соответствующих веществ рассчитайте стандартную энергию Гиббса образования
следующих соединений при Т = 298 К.
Задачи для самостоятельного решения
231
№
77
78
79
80
81
82
83
Вещество
Сульфат калия (т)
Нитрат серебра (т)
Титанат бария (т)
Ортофосфат кальция (т)
Тетрагидрат нитрата кадмия (т)
Хлорная кислота (ж)
Гидроксокарбонат меди (II) (т)
№
84
85
86
87
88
89
90
Вещество
Гептагидрат сульфата железа (II) (т)
Гидрокарбонат калия (т)
Перхлорат аммония (т)
Серная кислота (ж)
Азотная кислота (ж)
Ортофосфорная кислота (т)
Гидроксид хрома (III) (т)
91-100. Расставив стехиометрические коэффициенты и используя критерий само-
самопроизвольного протекания процесса в закрытой системе, ответьте на вопрос о тер-
термодинамической устойчивости соединения, указанного в уравнении реакции слева
(исходное вещество), при значениях температуры 298 К и 1000 К (р = 101,3 кПа).
Агрегатное состояние воды при 298 К жидкое, при 1000 К — газообразное. Рассчи-
Рассчитайте температуру начала реакции (равновероятности протекания прямого и обрат-
обратного процессов). Примите стандартные значения энтальпии и энтропии реакции не
зависящими от температуры.
№
91
92
93
94
95
А1@Н)з(Т) ?
AgNO3(T) ?=
Ba(NO3J(T,
Сг(ОНK(т) ?
Реакция
^ A12O3(T)H
hH2O
> Ag(T) + N02(r) + 02(r)
<=>
+± BaO(T) 4
=> Cr2O3(T)H
[Cu(OH)]2CO3(T) ?±
<=* CuO(T)
N02(r) + Оад
hH2O
+ CO2(r)+H2O
№
96
97
98
99
100
СН4(Г) <=±
Fe(OHK(T)
MnCO3(T)
KNO3(T) <=
Pb(NO3J(T)
Реакция
С2Н2(Г) + Н2(Г)
^ Fe2O3(T)
^=± MnO(T) +
± KNO2(T) +
±PbO(T) + NC
+ H2O
co2(r)
O2(r)
V) +O2(r)
101—106. Без проведения термодинамических расчетов определите, высоко- или
низкотемпературный режим способствует проведению в закрытой системе указан-
указанных реакций в прямом направлении. Энтальпийным или энтропийным фактором
определяется термодинамическая возможность протекания каждой реакции?
№
101
102
103
104
105
106
Уравнение реакции
2СН4(Г) <г^ С2Н2(Г) + ЗН2(Г)
СгНб(г) <-^ СгН^г) + Н2(Г)
СбНб(г) + ЗН2(Г) <=^ СбН12(г)
2Zn(NO3J(T) ^ 2ZnO(T) + 4NO2(r) + O2(r)
ВаО(т) + СО2(Г) <=± ВаСОз(Т)
SO2(r) + С12 (г) *=$ SO2C12(»)
Знак теплового эффекта
АгЯ>0
АгЯ>0
АгЯ<0
АгЯ>0
АгЯ<0
АгЯ<0
107-112. На основании термодинамических расчетов определите реакции, имеющие
одинаковое направление самопроизвольного протекания в изолированной и закры-
232 Направление самопроизвольного протекания химических реакций
той системах при стандартных термодинамических условиях (р = 101,3 кПа;
Т = 298 К).
№
107
108
109
Уравнение реакции
4FeSO«T) <=t
^"(тбел) + -Э "^5(т) ^ ^"<-Лз(ж)
4СГ(Т) + ЗО2(Г) f=^ 2Сг2О3(т)
№
110
111
112
Уравнение реакции
С(графит) + СаСО3(т) <=*
*12(г) ^ ^А2(г) <—^ ^llv^l(r)
2N0(r) + 02(r) & 2NO2(r)
ЛР 4. Определение энтальпий химических реакций
и процессов растворения солей
к электродвигателю
6 7
Рис. Л.4.1. Схема калоримет-
калориметрической установки:
1 — внутренний стакан; 2 — внеш-
внешний стакан; 3 — теплоизолирую-
теплоизолирующие прокладки; 4 — крышка; 5 —
термометр; 6 — мешалка; 7 —
воронка; 8 — резиновое кольцо
Цель работы — проведение опытов по ка-
калориметрическому измерению энтальпий про-
процессов и выполнение относящихся к ним
термодинамических расчетов.
Большинство химических реакций сопро-
сопровождаются выделением (экзотермические процес-
процессы) или поглощением (эндотермические процессы)
теплоты. При этом происходят соответственно
нагрев или охлаждение системы, количественно
характеризуемые изменением температуры АГ,
которая измеряется экспериментально и использу-
используется при расчете энтальпии реакции.
Энтальпии (тепловые эффекты) различных
процессов экспериментально определяют с по-
помощью прибора, называемого калориметром.
Конструкции и техническое оформление калори-
калориметров могут быть самыми разнообразными. Их
выбирают в зависимости от типа процесса, вели-
величины теплового эффекта и требуемой точности
измерений.
Схема простейшей калориметрической ус-
установки приведена на рис. Л.4.1. Она состоит из
двух стаканов разного размера. Внутренний кало-
калориметрический стакан 1 объемом 0,3 или 0,5 л (в
зависимости от объемов смешиваемых растворов
кислоты и щелочи) является реакционным сосу-
сосудом. В нем осуществляется химическая реакция
или другой процесс, сопровождающийся выделе-
выделением или поглощением теплоты. Этот сосуд
вставляется во внешний стакан 2 объемом 0,5 или
ЛР 4. Определение энтальпий химических реакций 233
0,8 л. Для сведения к минимуму теплопотерь через стенки калориметрического стака-
стакана между внутренним и внешним сосудами вставляются пробковые или пенопласто-
пенопластовые прокладки 3.
Сверху калориметрический сосуд закрывают крышкой 4, изготовленной из мате-
материала с малой тепловой проводимостью (дерево, фанера, подходящие полимеры). В
крышке просверливаются отверстия для термометра 5, мешалки 6 и воронки 7. Для
фиксирования температурных скачков применяется чувствительный ртутный термо-
термометр с ценой деления 0,1 К. Глубина погружения термометра в стакан 7 регулируется с
помощью резинового кольца 8. Для увеличения точности измерения температуры воз-
возможно большая часть корпуса термометра должна быть погружена в жидкость, запол-
заполняющую сосуд 7. Лопасти мешалки 6 не должны касаться термометра.
Вал мешалки через простейшую муфту (кусок резиновой трубки) соединяет-
соединяется с электромотором, подключаемым в сеть через лабораторный автотрансформатор
(ЛАТР), который позволяет регулировать скорость вращения вала. Перемешивание
способствует выравниванию температуры во всех частях реакционной системы.
Цилиндрическая часть воронки 7 должна иметь не очень малый диаметр,
чтобы частицы растворяемых твердых веществ не застревали в ней. Количество
теплоты, выделяющейся или поглощающейся в калориметрической установке, рас-
рассчитывают по формуле
Q = ср сумм AT = срсуМм (Т2 - Т\),
где срсумм — суммарная изобарная теплоемкость калориметрической системы, Дж/К;
А Г — изменение температуры калориметра (температурный скачок), К; Гг и Т\ —
конечная и начальная абсолютная температура калориметра, К.
Величина срсуми складывается из теплоемкостей калориметрического стакана
и находящейся в нем жидкости:
WCT+Cp ж УПЖ = Ср ст 7WCT+ Cp y\2q ^ж?
где срст — удельная изобарная теплоемкость стекла, срст = 750 Дж/(кг • К); срж —
удельная изобарная теплоемкость жидкости (разбавленного раствора), срж = срН 0 =
= 4180 Дж/(кг • К); тст, тпж — массы калориметрического стакана и жидкости, кг.
Опыт 1. Определение энтальпии реакции нейтрализации сильной кис-
кислоты сильным основанием. Независимо от природы сильной кислоты и сильного
основания реакция нейтрализации описывается следующим сокращенным ионно-
молекулярным уравнением:
Н(+р) + ОН-р)=Н2О(ж) (АЯн°ейтр).
Процесс нейтрализации относится к реакциям ионного обмена и является
практически необратимым, так как вода — очень слабый электролит. Согласно тео-
теории электролитической диссоциации Аррениуса, вода ведет себя как идеальный
амфолит (вещество двойственной природы) и в равной степени проявляет как ки-
кислотные (донор Н+), так и основные (донор ОН") свойства.
234 Направление самопроизвольного протекания химических реакций
Поскольку сущность реакции нейтрализации сильной кислоты сильным основа-
основанием заключается в образовании воды из катиона водорода и аниона гидроксида (ос-
(остальные ионы не изменяют своего состояния в растворе), то энтальпия процесса будет
приблизительно постоянной величиной. Ее можно вычислить по закону Гесса:
= -285,8 -0 -(-230,2) = -55,6 кДж/мольН2О (АЯн°ей1р <0).
Реакция, обратная нейтрализации, представляет собой процесс электролити-
электролитической диссоциации воды:
Н2О(ж) = Нф + ОН(р)
Константа равновесия этого превращения, выраженная через равновесные
молярные концентрации участников,
Вследствие того что вода очень слабый электролит и лишь в малой степени
диссоциирует на ионы, можно считать равновесную молярную концентрацию воды
[Н2О] равной исходной ее молярной концентрации [Н2О]0 в себе самой как в инди-
индивидуальном веществе. При [Н2О] = const функцию константы равновесия процесса
диссоциации воды выполняет выражение Кно = [Н+][ОН~] = 104, называемое
ионным произведением воды. Диссоциация воды — эндотермический процесс, поэтому
ионное произведение воды КИ 0 увеличивается с ростом температуры (см. ПАЗ).
Зная два значения Кно A) и КИ 0B) при двух температурах Т\ и Г2, можно с
использованием уравнения Гиббса—Гельмгольца и стандартного уравнения изо-
изотермы (по второму закону термодинамики) рассчитать средние величины стандарт-
стандартных энтальпии и энтропии для реакции диссоциации воды в интервале температуры
Т\...Т2. Выберем табличные значения (см. ПАЗ) Кн^0 при Т\ = 288 К и Г2= 308 К:
Для рассматриваемого процесса AG® = АН® -TAS® =-RT\nKH 0. Считая
АН® и AS® постоянными величинами, не зависящими от температуры, получим
систему двух линейных уравнений с двумя неизвестными:
ГаЯ?-288А5?=-8,31.2881п@,45-104)
1ДЯ? - 308AS? = -8,31 • 308 lnB,09 • 104 ),
решение которой дает АН® = АН®ИСС = 53,8 кДж/моль Н2О; AS® = AS®KCC =
= -86,9 Дж/(моль Н2О • К).
ЯР 4. Определение энтальпий химических реакций 235
Процесс нейтрализации сильной кислоты сильным основанием протекает
практически мгновенно и сопровождается выделением большого количества тепло-
теплоты, поэтому является удобной модельной реакцией для лабораторного практикума.
Последовательность проведения:
1) с помощью мензурки или мерного цилиндра отмерьте равные объемы A00-200 мл)
1М раствора сильной одноосновной кислоты (хлороводородной НС1 или азотной
HNO3) и 1М раствора сильного однокислотного основания (гидроксида натрия
NaOH или гидроксида калия КОН);
2) на технохимических весах с точностью 0,1 г взвесьте сухой калориметрический
стакан и вставьте его в калориметрическую установку, записав значение массы в
лабораторный журнал;
3) в калориметрический стакан 1 собранной в соответствии с рис. Л.4.1 калоримет-
калориметрической установки через воронку 7 налейте отмеренный объем 1М раствора кисло-
кислоты и измерьте его температуру Т[ с точностью 0,1 К;
4) в мерном цилиндре измерьте температуру 1М раствора щелочи Т" с точностью 0,1 К;
5) включите электродвигатель мешалки и через воронку 7 быстро прилейте раствор
щелочи к раствору кислоты;
6) с помощью термометра 5 зафиксируйте с точностью 0,1 К максимальную темпе-
температуру Т2, наблюдаемую после сливания растворов;
7) по окончании эксперимента отключите мешалку.
Обработка результатов:
1) исходные данные, уравнение реакции и результаты измерений занесите в табл. Л.4.1;
2) по исходным данным рассчитайте следующие величины и результаты вычисле-
вычислений занесите в табл. Л.4.1:
а) начальную температуру смеси растворов кислоты и щелочи Гь К как сред-
среднее арифметическое значений 7]' и 7]! Она приблизительно равна начальной тем-
температуре калориметра;
б) температурный скачок (К) АТ=Т2-Т\;
в) массу жидкости тж (г) в калориметрическом стакане: тж = рж(У\ + V2).
Примите плотность жидкости равной плотности воды (рж = рн 0 = 1 г/мл), так как
растворы кислоты и щелочи являются разбавленными;
г) суммарную теплоемкость калориметрической системы (Дж/К) ср сумм;
д) количество теплоты Q (Дж), выделившееся в калориметрическом стакане
при реакции нейтрализации;
е) количество вещества образовавшейся воды пн 0 (моль), равное количеству
вещества вступившей в реакцию нейтрализации кислоты (или щелочи) при ее извест-
известных молярной концентрации Ск, (моль/л) и объеме раствора VK (л): пно = пк = CKVK;
ж) энтальпию реакции нейтрализации сильной кислоты сильным основанием,
измеренную в эксперименте (Дж/моль Н2О):
АГГо Q
которую запишите рядом с уравнением реакции в табл. Л.4.1;
236 Направление самопроизвольного протекания химических реакций
Таблица Л АЛ
Результаты эксперимента
Исходные данные
Масса
калори-
калориметр,
стакана
WCT, Г
Объем
раствора
кислоты
V\, мл
Объем
раствора
щелочи
К2,мл
Результаты измерений
Темпера-
Температура рас-
раствора
кислоты
Т}\ К
Темпера-
Температура рас-
раствора
щелочи
тх\ к
Макси-
Максимальная
темпера-
температура рас-
раствора Т2, К
Результаты расчетов
Масса жид-
жидкости в
калориметр,
стакане
Суммарная
теплоем-
теплоемкость кало-
калориметр,
системы
ср сумм? Дж/К
Количество
теплоты,
выделив-
выделившейся в
калориметр,
стакане Q,
Дж
Количество
вещества
образовав-
образовавшейся воды
ино, моль
Энтальпия
реакции
нейтрализа-
нейтрализации
нейтр эксп'
Дж/моль
Н2О
Результаты расчетов
Начальная
температу-
температура раствора
Г,, К
Темпера-
Температурный
скачок
АГ,К
Выводы
Термохим
уравнение
ическое
реакции
3) сравните полученное экспериментальное значение энтальпии нейтрализации с
теоретическим (А#дейтр теор =-55,6 кДж/моль Н2О). Рассчитайте абсолютную г|абс и
относительную г|отн погрешности эксперимента (Дж/моль Н2О):
Лабс^ А/
Лота =
_ дтг
нейтр.эксп
Лабе
АН0
^L^ нейтр. теор
нейтр.теор'
100%;
4) занесите в табл. Л.4.1 выводы по проделанной работе.
Опыт 2. Определение энтальпии процесса растворения соли в воде. Про-
Процесс растворения соли в воде, как правило, сопровождается изменением энтальпии
системы (тепловым эффектом) АЯраств. Например, безводные сульфат цинка ZnSO4
или хлорид кальция СаС12 растворяются с выделением теплоты, а нитраты калия
KNO3 и аммония NH4NO3 — с поглощением. Как и в случае химической реакции,
соответствующая величина изменения температуры АГ может быть измерена в опы-
опыте и использована при расчете энтальпии растворения соли. Упрощенно процесс
растворения кристаллической соли в воде можно представить в виде суммы двух
последовательных стадий.
ЛР 4. Определение энтальпий химических реакций 237
1. Постепенное разрушение ионной кристаллической решетки соли поляр-
полярными молекулами воды, атакующими поверхность кристалла, которое сопровожда-
сопровождается поглощением теплоты ( А#разр реш > 0).
2. Гидратация образующихся катиона и аниона соли молекулами воды с об-
образованием устойчивых гидратных оболочек, происходящая с выделением теплоты
В соответствии с законом Гесса, А#расгв = А#разр реш + ^^гадр* ^ зависимо-
зависимости от соотношения абсолютных величин энтальпий этих составляющих стадий
суммарный процесс растворения соли характеризуется определенным знаком
энальпии:
а) если кристаллическая решетка прочная, а энтальпия гидратации сравни-
сравнительно невелика (| А#разр реш | > | А//Г°идр |), соль растворяется с поглощением теплоты:
Траста ><>;
б) если прочность кристаллической решетки мала, а энтальпия гидратации боль-
большая (| А//разр реш | < | А#г° |), соль растворяется с выделением теплоты: А#раств < 0.
Калориметрическая установка для проведения данного опыта аналогична ис-
использованной в опыте 1, но в отверстие крышки 4 вместо воронки 7 вставляется
пробирка с навеской исследуемой соли.
При проведении опыта необходимо учесть, что физико-химический процесс
растворения соли в воде является гетерогенным (протекает на границе раздела
твердой и жидкой фаз), скорость его значительно меньше скорости гомогенной ио-
ионообменной реакции нейтрализации и сильно зависит от перемешивания. По этой
причине перемешивание в опыте 2 должно быть более интенсивным, чем в опыте 1.
Последовательность проведения:
1) с помощью мензурки или мерного цилиндра отмерьте 200 мл дистиллированной
воды и вылейте ее в калориметрический стакан, масса которого известна из опыта 1;
2) на лабораторных весах взвесьте навеску исследуемой соли E-10 г) с точностью
0,01 г и перенесите вещество в пробирку;
3) вставьте пробирку в отверстие крышки калориметрической установки. В течение
10 мин выдержите систему для уравнивания температуры всех ее частей;
4) измерьте начальную температуру установки Т\ с точностью 0,1 К;
5) включите электродвигатель мешалки, выньте пробирку с солью из отверстия в
крышке, вставьте туда воронку и высыпьте соль в воду;
6) с помощью термометра 5 зафиксируйте с точностью 0,1 К максимальную (мини-
(минимальную) конечную температуру 7г, наблюдаемую после высыпания навески соли;
7) по окончании эксперимента отключите мешалку.
Обработка результатов:
1) исходные данные и результаты измерений занесите в табл. Л.4.2;
238 Направление самопроизвольного протекания химических реакций
Результаты эксперимента
Таблица Л.4.2
Исходные данные
Масса ка-
лориметри-
лориметрического
стакана,
тст,г
Объ-
Объем
воды
К,,мл
Масса
навески
соли
wc, г
Результаты измерений
Началь-
Начальная тем-
температура
Г,, К
Конеч-
Конечная тем-
температура
Т2, К
Результаты расчетов
Темпе-
ратур-
ратурный
скачок
АГ,К
Масса
раст-
раствора
тр, г
Результаты расчетов
Количество выде-
выделенной (поглощен-
(поглощенной) теплоты Q, Дж
Количество
растворенного
вещества пс, моль
Энтальпия растворения,
Суммар-
Суммарная теп-
теплоемкость
Ср сумм»
Дж/К
Выводы
2) по исходным данным рассчитайте следующие величины и результаты расчетов
запишите в табл. Л.4.2:
а) температурный скачок (К) AT =T2-T\;
б) массу раствора тр, (г) в калориметрическом стакане: /wp = pH OVX + mc\
в) суммарную теплоемкость калориметрической системы (Дж/К):
сР сумм = ср ст юст + 4,18тр
(теплоемкость разбавленного раствора соли примите равной теплоемкости воды
4,18Дж/(г-К));
г) количество теплоты Q (Дж), выделенной или поглощенной в процессе рас-
растворения: Q = срсуммАТ;
д) количество растворенного вещества пс (моль) при его известных массе и
молярной массе: лс= тс/Мс;
е) энтальпию процесса растворения соли в воде, измеренную в опыте (Дж/моль):
Q
АН,
раств. эксп
3) сравните полученное экспериментальное значение энтальпии растворения с тео-
теоретическим при бесконечном разбавлении:
+°оН2О(ж)=
(ДЯ°аств);
При расчете воспользуйтесь данными табл. П.5.1. Рассчитайте абсолютную и
относительную погрешности эксперимента по формулам, аналогичным приведен-
приведенным в описании опыта 1;
4) занесите в табл. Л.4.2 выводы по проделанной работе.
IV
КИНЕТИКА
ХИМИЧЕСКИХ ПРОЦЕССОВ
¦ Кинетика гомогенных
химических реакций
¦ Особенности кинетики
гетерогенных реакций
¦ Основы катализа
Химическая кинетика — раздел химии, изу-
изучающий скорости протекания химических
процессов. Цель изучения — установление
механизма реакции и выяснение возможности
управления скоростью химической реакции.
Химическая кинетика наряду с термодинами-
термодинамикой является теоретической основой химиче-
химической технологии, поскольку позволяет интен-
интенсифицировать и оптимизировать промышлен-
но важные реакции.
Все химические процессы, наблюдаемые в
природе и осуществляемые человеком в его
практической деятельности, протекают во
времени с определенными скоростями. На-
Направление и глубина химических превраще-
превращений диктуются законами термодинамики. Од-
Однако термодинамическое рассмотрение хими-
химических процессов не дает ответа на вопросы о
том, с какой скоростью и через какие проме-
промежуточные стадии система перешла из началь-
начального состояния в конечное. Время протекания
химической реакции является предметом рас-
рассмотрения химической кинетики.
В настоящее время химическая кинетика про-
проникла во все разделы химии. Эта наука уни-
универсальна, ее законы могут быть использова-
использованы для описания широкого круга химических
реакций — от образования руд в геохимиче-
геохимических процессах с длительностью во много
миллионов лет до взрыва, происходящего за
миллионные доли секунды.
7 4 Кинетика гомогенных химических
~~ реакций
Химические процессы могут протекать в системах, состоящих из од-
одной или нескольких фаз. Гомогенные химические реакции протекают
в одной фазе (в смеси газов, жидком растворе или твердой фазе). При
протекании химической реакции с течением времени изменяются ко-
количества реагирующих веществ. Однако изменение количества веще-
вещества в реакционном пространстве может быть обусловлено как про-
протеканием реакции, так и обменом веществом с окружающей средой.
Поэтому отдельно рассматривают кинетику химических реакций в
закрытых и открытых системах.
В этой главе рассмотрены кинетические закономерности гомогенных
химических реакций в закрытых системах. В закрытой системе изме-
изменение количества вещества с течением времени происходит только за
счет протекания химической реакции.
7.1. Основные понятия и определения
Химическая кинетика — раздел физической химии, изучаю-
изучающий скорости протекания химических процессов и их механизм. Ме-
Механизмом реакции называют совокупность элементарных стадий, из
которых складывается химическая реакция. Элементарной называют
реакцию, которая осуществляется путем непосредственного превра-
превращения реагентов в продукты реакции (в одну стадию).
Каждая элементарная реакция (стадия) представляет собой со-
совокупность большого числа однотипных превращений исходных
частиц — элементарных актов химического превращения.
Элементарным химическим актом называют единичный акт
превращения частиц (молекул, атомов, радикалов, ионов) исходных
веществ в частицы продуктов реакции.
Для характеристики элементарных реакций, происходящих в
одну стадию, используют понятие молекулярности реакции. Число
молекул реагентов, участвующих в элементарном акте химического
превращения, называют молекулярностью реакции.
Для сложных реакций молекулярность устанавливается для
каждой элементарной стадии. Различают мономолекулярные, бимо-
бимолекулярные и тримолекулярные реакции.
В мономолекулярной реакции участвует одна молекула реагента.
Это может быть реакция изомеризации (А —» В) или реакция разложе-
разложения (А -> В + С), например:
7.1. Основные понятия и определения 241
CH3NC (метилизоцианат) -> CH3CN (ацетонитрил)
В бимолекулярных реакциях две молекулы реагентов взаимодействуют
с образованием одной или нескольких молекул продукта. Это могут быть
или реакции соединения (А + В -> АВ), или реакции обмена (А + В -> С + D),
например:
С2Н4 + HI -> C2H5I
Тримолекулярные реакции происходят при одновременном столкнове-
столкновении трех молекул с образованием продукта (А + В + С-»Р)и встречаются
относительно редко. Одновременное столкновение трех молекул менее ве-
вероятное событие, чем столкновение двух молекул. Например:
2NO + С12 -> 2NOC1
Тетрамолекулярные реакции экспериментально не наблюдаются.
Главная причина — очень малая вероятность одновременного столкновения
четырех молекул.
Во многих случаях продукты реакции могут взаимодействовать между
собой, образуя исходные вещества. Тогда считают, что химическая реакция
обратима. Примером обратимого процесса может служить реакция:
2NO + О 2 ?=± 2 NO2
Строго говоря, обратимыми являются любые химические реакции. На прак-
практике же обратная реакция может быть настолько медленной по сравнению с
прямой, что обратимостью реакции можно пренебречь. Такую реакцию
можно рассматривать как необратимую или одностороннюю. В дальнейшем
рассматриваются гомогенные односторонние химические реакции.
Гомогенными называют реакции, протекающие в одной фазе (в смеси
газов, жидком растворе или в твердой фазе). Скорость — важнейшая коли-
количественная кинетическая характеристика любой химической реакции. Наи-
Наиболее общим является определение скорости реакции г как скорости возрас-
возрастания степени завершенности реакции ?, которую называют химической
переменной.
Пусть в закрытой системе протекает необратимая химическая реакция
vAA + vBB -> vDD + vF F,
где А, В, D, F — участники реакции; vA, vB , vD, vF — соответствующие сте-
хиометрические коэффициенты. Тогда химическая переменная ?,-, характери-
характеризующая глубину протекания реакции по /-му компоненту, определяется как
242 7. Кинетика гомогенных химических реакций
о
$/=-*—Ч G.1)
где щ — число молей /-го компонента в момент времени t\ n® — число мо-
молей того же компонента в начальный момент времени (t = 0); v,- — стехио-
метрический коэффициент при /-м компоненте в уравнении реакции. Сте-
хиометрические коэффициенты у исходных веществ берутся со знаком «-»,
а у продуктов реакции — со знаком «+». Это связано с тем, что количество
исходных веществ убывает, а количество продуктов реакции увеличивается.
При таком выборе знаков ^ всегда положительна независимо от выбора
участников реакции. Скорость гомогенной химической реакции в целом оп-
определяется как скорость изменения глубины протекания реакции в единицу
времени в единице объема:
г = -—, G.2)
V А
где V — объем системы; t — время. Определенная таким образом скорость
реакции не зависит от выбора компонента и будет практически одинаковой
для разных веществ, участвующих в реакции. Дифференциальная форма
G.1) имеет вид
db -drli
отсюда из уравнения G.2) для г получают
1 dnt
r~v,F dt '
Величину
называют скоростью реакции по i-му компоненту. Она характеризует изме-
изменение количества /-го вещества пг (в молях) в единицу времени в единице
объема. Таким образом, скорость реакции в целом и скорости реакции по
отдельным компонентам связаны следующим соотношением:
7.1. Основные понятия и определения 243
Выражения G.2) и G.3) определяют скорости реакции для системы с
переменным объемом. На практике часто используют более простое уравне-
уравнение, пригодное для реакции в системе постоянного объема. Так как отноше-
отношение tti IV равно концентрации вещества С,-, то при постоянном объеме
г,-А
Вообще говоря, скорость реакции при постоянном объеме представ-
представляет собой изменение концентрации данного компонента в единицу време-
времени. Скорость реакции есть положительная величина. В выражении G.4)
используют знак «+», если скорость определяется по образующемуся в ре-
реакции компоненту (продукту), и знак «-», если скорость определяется по
расходующемуся в реакции компоненту (реагенту). Тем самым учитывают-
учитываются разные знаки стехиометрических коэффициентов реагентов и продуктов
реакции.
Например, для реакции 2HI = Н2 + h
r = dCH2=dCl2= \dCm
dt dt 2 dt '
Уравнение G.4) наиболее часто применяют для реакций, протекаю-
протекающих в растворах, и для газофазных реакций, осуществляемых в замкнутых
реакционных сосудах. Если концентрация измеряется в молях на литр, а
время в секундах, то скорость реакции имеет размерность [моль/(л • с)].
Уравнение G.4) характеризует мгновенную скорость реакции, т.е. ско-
скорость реакции в данный момент времени. На практике иногда используют
понятие «средняя скорость реакции». Средняя скорость реакции гср в интер-
интервале времени от t\ до ti определяется соотношением
где С\И С2 — молярные концентрации любого участника реакции в момен-
моменты времени t\ и h соответственно.
Кинетический эксперимент заключается в том, что при проведении
реакции по мере ее протекания определяют тем или иным методом концен-
концентрации исходных веществ и продуктов реакции. Результаты такого экспе-
эксперимента представляют в виде кинетических кривых образования и расходо-
расходования соответствующих веществ.
Под кинетической кривой понимают график зависимости концентра-
концентрации реагента или продукта реакции от времени.
244
7. Кинетика гомогенных химических реакций
О tx t
Рис. 7.1. Типичные кинетические
кривые для исходного вещества Л и
продукта реакции В (г = tg a)
Рассмотрим простейшую хими-
химическую реакцию превращения вещест-
вещества А в продукт В в одну стадию:
А->В.
В процессе реакции концентра-
концентрация реагента А уменьшается, а продук-
продукта В — возрастает (рис. 7.1).
Как следует из уравнения G.4),
скорость реакции по данному компо-
компоненту в момент времени t соответству-
соответствует тангенсу угла наклона касательной,
проведенной к кинетической кривой
этого компонента в точке, соответст-
соответствующей моменту времени /. На рис. 7.1 показан угол наклона касательной к
кинетической кривой, соответствующий начальному моменту времени (/ = 0).
Поскольку скорость расходования исходного компонента наивысшая в на-
начальный момент времени, то наклон касательной, соответствующий началь-
начальной скорости, максимален. По мере протекания реакции наклон касательной
к кривой в каждой следующей точке уменьшается.
Другой важной кинетической характеристикой реакции является вре-
время {период) полупревращения (tm) — время, необходимое для химического
превращения половины начального количества компонента.
На скорость гомогенной химической реакции могут оказывать
влияние следующие факторы: природа реагирующих веществ; концен-
концентрации реагирующих веществ; давление (если в реакции участвуют га-
газы); температура; наличие специфических веществ, ускоряющих реакцию
(катализаторы) или замедляющих ее (ингибиторы); световое или радиа-
радиационное излучения.
Влияние природы и концентраций реагирующих веществ определяет-
определяется основным законом химической кинетики.
7.2. Основной постулат химической кинетики
При постоянной температуре системы скорость реакции определяется
концентрациями веществ, составляющих систему. В первую очередь име-
имеются в виду концентрации исходных веществ (реагентов). Концентрации
продуктов также могут влиять на скорость, но при малых степенях превра-
превращения реагентов этим влиянием можно пренебречь, поскольку концентра-
концентрации продуктов еще малы. Уравнение, описывающее зависимость скорости
7.2. Основной постулат химической кинетики 245
химического процесса от концентрации участников реакции, называется
кинетическим уравнением процесса.
Основной постулат химической кинетики базируется на большом
числе экспериментальных данных и выражает зависимость скорости реак-
реакции от концентраций реагирующих веществ: скорость реакции в каждый
момент времени при постоянной температуре пропорциональна произве-
произведению концентраций реагирующих веществ, возведенных в некоторые
степени.
Если записать уравнение реакции в виде
то математическую формулировку основного закона можно представить так:
r = kC"ACg. ' G.5)
Коэффициент пропорциональности к называют константой скорости
химической реакции. Константы скоростей значительно отличаются для разных
реакций и сильно зависят от температуры. Константа скорости химической ре-
реакции численно равна скорости при концентрациях реагирующих веществ,
равных единице. Поэтому иногда ее называют удельной скоростью реакции. В
учебной и научной литературе основной постулат химической кинетики часто
называют законом действующих масс, который для элементарных реакций был
сформулирован К. Гульдбергом и П. Вааге A879).
Показатель степени, в которой концентрация реагента входит в кине-
кинетическое уравнение, называют порядком реакции по данному реагенту или
частным порядком реакции. Общий порядок реакции q есть алгебраическая
сумма показателей степеней при концентрациях всех реагентов, которые
входят в кинетическое уравнение:
q = п + т.
Частные порядки реакции определяются экспериментально. Размер-
Размерность константы скорости химической реакции определяется общим поряд-
порядком реакции q = n + m (см. G.5)): [к] = [время]'1 [концентрация]1"*.
Вид кинетического уравнения нельзя предсказать исходя из стехио-
метрического уравнения реакции, он зависит от механизма протекания ре-
реакции. Для элементарной реакции порядок по данному реагенту равен числу
частиц данного реагента, участвующих в элементарном химическом акте.
Например, для элементарной реакции:
2NO + С12 -> 2NOC1
молекулярность равна 3. Порядок реакции по N0 равен 2, а по СЬ — 1. Об-
Общий порядок реакции равен 3, всегда положителен и целочислен (#=1,2, 3).
246 7. Кинетика гомогенных химических реакций
Порядок сложной реакции в общем случае может быть целочисленным,
дробным и даже отрицательным, что обусловлено механизмом реакции.
Большинство встречающихся на практике химических реакций явля-
являются сложными, многостадийными. Каждая стадия сложной реакции проте-
протекает с определенной скоростью. Скорость любого многостадийного процес-
процесса при существенном различии скоростей отдельных стадий определяется
скоростью самой медленной стадии. Самую медленную стадию сложной
химической реакции называют лимитирующей стадией.
Все химические реакции по скорости их протекания условно можно
подразделить на медленные и быстрые. К медленным относят реакции, у
которых значения констант скорости лежат в интервале 10~8...1,0 (концен-
(концентрации выражены в молях на литр, время — в секундах). Реакции, кон-
константы скорости которых находятся в интервале значений 10... 1011, отно-
относят к быстрым реакциям. Примером быстрой реакции служит реакция
нейтрализации:
Константа скорости этой реакции к = 2 • 1011 л • моль 1 • с 1 (Г= 298 К).
7.3. Кинетические уравнения односторонних реакций
Рассмотрим результаты решения дифференциальных уравнений для
простых односторонних реакций нулевого, первого и второго порядков.
Реакции нулевого порядка. Кинетическое уравнение для реакции
нулевого порядка имеет вид
Л
Разделяя переменные (-dC = kdi) и интегрируя в пределах от началь-
начальной концентрации Со (t = 0) до текущей концентрации С в момент времени t:
с t
-jdC=jkdt,
с0 о
получают
C = C0-kt. G.6)
Реакции нулевого порядка имеют следующие особенности.
1. В соответствии с G.6) в реакциях нулевого порядка концентрация
исходного вещества линейно уменьшается во времени (рис. 7.2).
2. Константа скорости
7.3. Кинетические уравнения односторонних реакций
247
Ее размерность такая же, как и размерность
скорости реакции ([к] = [г]).
3. Время полупревращения tm, согласно
G.6), при С = Со/2:
о
'1/2 =
2*'
Рис. 7.2. Зависимость концен-
концентрации от времени для реак-
реакции нулевого порядка
т.е. для реакции нулевого порядка t\a пропорционально начальной концен-
концентрации исходного вещества.
Нулевой порядок имеет место в тех случаях, когда убыль вещества в
результате протекания химической реакции восполняется доставкой его из
другой фазы. Примером служит реакция разложения малорастворимых в
воде сложных эфиров на границе раздела эфир — вода. Нулевой порядок
наблюдается также, если скорость процесса лимитируется подачей энергии,
необходимой для активации реагирующих молекул. Например, при фотохи-
фотохимических реакциях определяющим фактором может служить количество
поглощенного излучения, а не концентрация веществ. Часто в каталитиче-
каталитических реакциях скорость зависит от концентрации катализатора, а не от кон-
концентрации реагирующих веществ.
Реакции первого порядка. Для реакции первого порядка кинетиче-
кинетическое уравнение имеет вид
dC
dt
G.7)
Разделяя переменные
и интегрируя полученное уравнение, находят
= lnC0-klt,
= Coexp(-V).
G.8)
G.9)
G.10)
248
7. Кинетика гомогенных химических реакций
In С
In С
01
02
In С,
03
tg а = - к
Реакции первого порядка
имеют следующие особенности.
1. Согласно G.9), график за-
зависимости In С от t выражается
прямой линией. Это означает, что в
полулогарифмических координатах
при разных начальных концентра-
концентрациях прямые будут параллельны
между собой (рис. 7.3).
2. Концентрация исходного
вещества G.10) стремится к нулю
при /—»оо. Полностью вещество
реагирует за бесконечно большой
промежуток времени.
3. Скорость реакции также
отличается экспоненциальной зави-
зависимостью:
О t
Рис. 7.3. Зависимость In С от / для реак-
реакции первого порядка при различных на-
начальных концентрациях
4. В соответствии с G.8)
*,=-•
следовательно, размерность константы скорости реакции первого порядка
соответствует обратному времени и ее можно выразить в обратных секун-
секундах, минутах, часах и т.д.
5. Время полупревращения с учетом G.8) при С = Со/2
In 2
т.е. f i/2 не зависит от начальной концентрации реагирующего вещества.
6. Кинетическая кривая реакции первого порядка инвариантна при
линейном преобразовании концентрации. Заменяя в G.7) С на пропорцио-
пропорциональную величину яС, получают
d(nC)
dt
= k]nC.
Поскольку п постоянная величина, то ее можно вынести за знак дифферен-
дифференциала и затем сократить:
7.3. Кинетические уравнения односторонних реакций
249
-п— = к{пС.
dt
Это означает, что кинетическое уравнение остается неизменным при умно-
умножении концентрации на какую-либо постоянную величину. Таким образом,
для оценки скорости реакции вместо концентрации можно использовать
любую другую пропорциональную ей величину, например, спектральную
оптическую плотность, электропроводность, объем раствора, израсходо-
израсходованного на титрование. Логарифм такой величины также меняется линейно
во времени, и наклон прямой определяется значением -к.
7. Скорость реакции в заданный момент времени определяется тан-
тангенсом угла наклона касательной к кинетической кривой. Начальная ско-
скорость превращения исходного вещества r0 = CJtx^ где tx — время пересече-
пересечения касательной к начальному участку кинетической кривой с осью време-
времени. Подставляя эту величину в кинетическое уравнение, получают
- ° -
е. гх -—.
Следовательно, при любых начальных концентрациях Со касательные к на-
начальному участку (начальные скорости) пересекут ось времени при одном и
том же значении tx (рис. 7.4).
8. Среднее время жизни молекул исходного вещества
1
т = —
и соответствует уменьшению ис-
исходной концентрации в е раз.
Например, для реакции тер-
термического разложения ацетона при
7=777 К E04 °С)
СНзСОСНз -> С2Н 4 + Н2 + СО
константа скорости к = 4,27-10~4 с.
Тогда при Т= 777 К средняя про-
продолжительность жизни молекул
ацетона
т=
= 2342с«39мин.
9. Согласно G.10), в началь-
начальный момент времени при малых
значениях t разложение экспоненты
01
-02
-03
Рис. 7.4. Касательные к кинетическим
кривым реакции первого порядка в точ-
точках, соответствующих начальным кон-
концентрациям
250 7. Кинетика гомогенных химических реакций
в ряд (при х -> 0 ехр х * 1 + х) дает приближенное равенство для количества
прореагировавшего вещества (Со - С):
Со - С = Со A - exp(-fy))« ед,
отсюда
где Со — постоянная величина. Тогда
dc~k
есть кинетическое уравнение нулевого порядка. Поэтому не имеет смысла изу-
изучать кинетику реакции при малых степенях превращения. Обычно трудно заме-
заметить отклонения от прямой линии при превращении исходного вещества менее
чем на 10... 15 %. Для однозначного отличия одного порядка от другого необ-
необходимо проследить за реакцией до глубины ее протекания на 60.. .70 %.
Примеры реакций первого порядка:
разложение ацетона
СНзСОСНз („ар) -> С2Н4 + СО + Н2
радиоактивный распад
28262Rn -» <а + ««Rn
инверсия тростникового сахара в кислотной среде
С12Н22О11 + Н2О -> 2C6Hi206 (фруктоза + глюкоза).
Реакции второго порядка. Рассмотрим три случая.
1. В реакции участвуют два реагента, начальные концентрации кото-
которых равны: А + В -> Р.
Дифференциальное уравнение для скорости реакции имеет вид
кС
л 2 •
Разделение переменных и интегрирование в пределах от Со до С приводит к
следующему результату:
— - — = k2t. G.11)
С Со 2
Из уравнения G.11) следует, что концентрация исходных веществ зависит
от времени следующим образом:
7.3. Кинетические уравнения односторонних реакций
251
1/С"
1 + *2С0/
Полученные уравнения позволяют от-
отметить следующие особенности реакций вто-
второго порядка.
а) Согласно G.11), для реакций второго
порядка при равенстве начальных концентра-
концентраций реагирующих веществ обратная концен-
концентрация линейно зависит от времени (рис. 7.5):
J__J_
c"q+ lL
б) Константа скорости
о t
Рис. 7.5. Зависимость обратной
концентрации от времени для
реакции второго порядка
где к — в л • моль • с.
в) Вычислим в соответствии с G.11) при С = Со/2 время полупревра-
полупревращения для реакции второго порядка
1 1 ,
отсюда
'1/2 =
к2С0
В отличие от реакций первого порядка время полупревращения реакций
второго порядка обратно пропорционально начальной концентрации.
2. В реакции начальные концентрации реагентов А и В не равны, и
кинетическое уравнение имеет вид
^С С
Л
Интегрирование приводит к следующему результату:
1 с с
Ч)В 0)А
G.12)
Если обозначить через В =
, то зависимость inD от времени
будет иметь вид прямой линии (рис. 7.6).
252
7. Кинетика гомогенных химических реакций
О
lnZ) '
Рис. 7.6. Линейная зависимость
от времени параметра D для ре-
реакции второго порядка
На протяжении всей реакции выполня-
выполняется уравнение материального баланса:
С учетом этого соотношения из уравнения
G.12) получают выражение для времени по-
полупревращения вещества А (Сд = Сод/2):
n
BС0В - Со А )
'1/2=
3. Реакция 2А -> Р, кинетическое уравнение которой имеет вид
dCA -
- -
Множитель 2 означает, что в каждой элементарной реакции превращают-
превращаются две молекулы вещества А. Интегрирование приводит к следующему
результату:
— — = 2*Л.
G.13)
Отсюда текущая концентрация
r _
+ 2k2C0At
Время полупревращения в соответствии с G.13) при СА = С 0д/2
1
Примеры реакций второго порядка:
Н2 + h -> 2HI
2HI -^ Н2 + h
2NO2 -> 2NO + О2
Аналогично можно получить решение для дифференциальных
уравнений реакций и-го порядков. Результаты интегрирования приведе-
приведены в табл. 7.1.
7.4. Методы определения порядка химических реакций
253
Таблица 7.1
Кинетические уравнения реакций различных типов
Порядок
реакции
Нулевой
Первый
Второй
Третий
п
Дифференци-
Дифференциальное
уравнение
-dc=k
dt
dt '
dc kc1
dt 2
Интегральное
уравнение
Co C-kt
с с
С С
—Ц- l—-(n-l)kt
гп-\ гп-\ v 7 п
с и0
Размерность
константы
скорости
моль
л с
с
л
моль•с
л
моль •с
/ \П-\ 1
f л )
V моль у с
Время
полупревращения
ha
Со
2к
In 2
I
3
1к С2
2"-I-l
^(if-l)^
Рассмотрим реакцию А + В -> Р, когда один из реагентов берется в
большом избытке. Если предположить, что вещество В присутствует в зна-
значительном избытке, тогда его концентрация остается практически постоян-
постоянной в ходе реакции, т.е. Св « Сов- В этом случае кинетическое уравнение уп-
упрощается:
г ~ ^2^а^в ~ ^2^аО)в * ^^а-
Здесь к!2 - к2С0в — эффективная константа скорости. Полученное уравне-
уравнение представляет собой уравнение скорости первого порядка. В этом случае
говорят, что реакция протекает в условиях псевдопервого порядка (по отно-
отношению к веществу А). Например, такой кинетике могут подчиняться реак-
реакции в водных растворах, происходящие с участием молекул воды (гидролиз
сложных эфиров).
7.4. Методы определения порядка химических реакций
Современные методы вычислительной математики (различные мето-
методы минимизации) позволяют с помощью ЭВМ по кинетическим кривым,
полученным из эксперимента, непосредственно найти порядки реакции по
отдельным реагентам. На практике часто используют более простые методы
определения порядков реакций по экспериментальным данным.
254 7. Кинетика гомогенных химических реакций
Рассмотрим лишь некоторые методы определения порядка реакции.
Метод подстановки основан на подстановке экспериментальных дан-
данных в какое-либо из известных уравнений реакций нулевого, первого и второго
порядков (см. табл. 7.1). Правильность выбора уравнения проверяется по тому,
как хорошо сохраняется константа скорости, вычисленная по уравнению соот-
соответствующего порядка. Постоянные значения к в пределах ошибок экспери-
эксперимента свидетельствует о правильности выбора порядка реакции.
Следует отметить, что этот способ довольно трудоемок. Если порядок
дробный, то, вообще говоря, трудно выяснить его истинную величину.
Метод Вант-Гоффа (определение порядка реакции по начальным
скоростям). Если протекает реакция А + В -> продукты, то в соответствии с
основным законом химической кинетики начальная скорость реакции
Для определения порядка реакции по веществу А проводят серию
экспериментов при постоянной начальной концентрации вещества В (Сов =
= const) и различных начальных концентрациях вещества А (Соа * const).
При этих условиях
где к' = кС™в. При различных значениях Соа измеряют начальные скорости и
строят график зависимости lg го от lg Соа? который выражается прямой линией.
Тангенс угла наклона прямой равен порядку реакции по веществу A (tg а = п)
(рис. 7.7). Аналогично по данным другой серии экспериментов, когда СОа =
= const, а Сов * const, можно определить порядок реакции по веществу В.
Порядок реакции можно оценить и расчетным путем, если известны
начальные скорости при двух начальных концентрациях вещества
v
Отношение этих скоростей лога-
логарифмируют:
Отсюда
О lg Сод lg
0 А
02
Рис. 7.7. Графическое определение
порядка реакции по начальным ско- 1
ростям (Со в = const, Со а * const) ^ ^02 А
7.4. Методы определения порядка химических реакций 255
Метод определения порядка реакции по периоду полупре-
полупревращения (частный случай метода Оствальда—Нойеса). Основан на
изучении зависимости периода полупревращения от начальной концентра-
концентрации вещества. Для реакции п-то порядка кинетическое уравнение записыва-
записывают в виде
Л " п '
Интегрирование этого уравнения приводит к следующему результату:
Отсюда время полупревращения при С = С0/2
hn =—=• ^т = -^т? G.14)
1/2 кA)сГ <Т1
где Q — постоянная величина для данной реакции. Логарифм периода по-
полупревращения линейно зависит от логарифма начальной концентрации ве-
вещества:
Тангенс угла наклона определяет порядок реакции п по данному ве-
веществу. В наиболее простом случае порядок реакции по веществу можно
оценить, определив периоды полупревращения для двух начальных концен-
концентраций. Тогда
Вычитая из второго уравнения первое:
^1/2J-^1/2I =("-l)(lgC01 -lgC02),
после преобразований получают
I/2/1 ^02
lgc
c
C02
Уравнение G.14) не применимо для реакции первого порядка (п = 1).
Для реакции первого порядка
256 7. Кинетика гомогенных химических реакций
In 2
и не зависит от начальной концентрации вещества.
7.5. Влияние температуры на скорость химических реакций
Основное влияние температура оказывает на константу скорости.
Опираясь на опытные данные, Вант-Гофф установил, что при повышении
температуры на 10 К скорость реакции увеличивается в 2-4 раза (правило
Вант-Гоффа). Коэффициент
_ Г7Ч10 *
кт
был назван температурным коэффициентом скорости реакции Вант-Гоффа.
Правило Вант-Гоффа является приближенным и было установлено для ре-
реакций в растворах, протекающих при сравнительно низких температурах
@...120 °С). Оно хорошо выполняется для таких реакций, которые идут с
удобной для измерения скоростью, т.е. не слишком быстро и не слишком
медленно. При повышении температуры у не остается постоянным, а
уменьшается и стремится к единице. Если известны скорость реакции при
температуре Т\ и температурный коэффициент, то скорость реакции при
температуре Т2 можно определить по уравнению
Т2-Тх
10 .
Более точную зависимость константы скорости реакции от температу-
температуры устанавливает уравнение Аррениуса:
jy G.15)
где Еа — аррениусовская или опытная энергия активации; Т — абсолютная
температура; R — универсальная газовая постоянная; ?0 — предэкспоненци-
альный множитель, мало зависящий от температуры.
Уравнение Аррениуса часто представляют в логарифмической форме:
In к = In к0 -EJRT, G.16)
удобной для графического определения энергии активации. Энергию акти-
активации (энтальпию активации) можно вычислить по значениям константы
7.5. Влияние температуры на скорость химических реакций
257
скорости, измеренным при несколь-
нескольких разных температурах. По экспе-
экспериментальным данным строят график
зависимости In А: от \1Т (рис. 7.8).
Тангенс угла наклона полученной
прямой линии равен (-EJR).
Энергию активации можно
также оценить, если известны кон-
константы скорости при двух температу-
температурах Т\ и Т2. Тогда для каждой темпе-
температуры можно записать
Ink
О
\/T
In ki =
-EJRTi,
Рис. 7.8. Графическое определение энер-
энергии активации
Вычитая из второго уравнения первое, получают
In кг - In kx= (In ко - EJRT2) - (In k0 - EJRT,).
Отсюда
Jb.).*.<r,-T,\
RT2T}
Тогда выражение для энергии активации имеет вид
'к,
Т2-Тх
G.17)
где Еа — в Дж/моль или кДж/моль.
Энергия активации представляет собой избыток энергии (в расчете на
1 моль) по сравнению со средней энергией молекул при данной температу-
температуре, необходимый для того, чтобы реагирующие частицы могли вступить в
химическую реакцию и определяется свойствами реагирующих частиц, их
энергетическим состоянием.
Чтобы написать уравнение Аррениуса в дифференциальной форме,
дифференцируют уравнение G.16) по температуре и получают
dink _ Ea
dT RT
2 *
G.18)
Из G.18) следует, что чем больше энергия активации, тем быстрее увеличи-
увеличивается константа скорости реакции с возрастанием температуры.
9 - 9795
258 7. Кинетика гомогенных химических реакций
7.6. Теоретические представления
о скоростях элементарных реакций
В настоящее время разработаны две основные теории, объясняющие
кинетику протекания элементарных реакций.
Теория активных столкновений. Эта теория базируется на двух об-
общих положениях:
реакция осуществляется в момент столкновения двух молекул А и В;
столкновение молекул приводит к химическому превращению только в
том случае, когда молекулы А и В обладают достаточно большим запасом
энергии. Эта энергия необходима для преодоления действия сил отталкивания,
проявляющихся при сближении любых валентно-насыщенных молекул. Если
относительная скорость движения молекул А и В (вдоль линии, соединяющей
центры) достаточно велика, молекулы могут сблизиться до таких малых рассто-
расстояний, на которых возможно перераспределение химических связей в реагентах.
Теория активных столкновений построена на использовании результа-
результатов, полученных в кинетической теории газов. Согласно кинетической тео-
теории газов, число двойных соударений
Здесь кь — постоянная Больцмана; ц — приведенная масса молекул
тАтв
тА+тв
где тд — масса молекулы A; wb — масса молекулы В; став = (<Та + <Тв)/2;
аА — диаметр молекулы А; ав — диаметр молекулы В; лА, ив — число соот-
соответствующих молекул в единице объема.
Из кинетической теории газов также известно, что доля молекул, об-
обладающих энергией больше некоторой величины Ео, выражается множите-
множителем exp (-EqIRT).
Для скорости реакции А + В —» D + F можно записать выражение:
Этому выражению соответствует константа скорости реакции второго
порядка
7.6. Теоретические представления о скоростях элементарных реакций 259
где кг в с • молекул • см3. В этой формуле параметр Ео называют истинной
энергией активации в отличие от опытной энергии активации в G.15). Ясно,
что G.19) очень похоже на G.15) и позволяет выразить константу скорости в
виде двух сомножителей: предэкспоненциального множителя Zo, слабо за-
зависящего от температуры (~Г1/2), и экспоненты exp (-Eo/RT), которая и вы-
выражает основную зависимость k2 от температуры.
Следует отметить, что с помощью теории активных столкновений не-
невозможно теоретически вычислить энергию активации. При расчете кон-
константы скорости необходимо использовать экспериментально полученные
данные энергии активации.
Установим связь между опытной и истинной энергиями активации.
Уравнение G.19) можно представить в виде
RT
где В — некоторая константа. Прологарифмируем это выражение:
-1пГ —^-.
2 RT
Дифференцируя полученное уравнение по температуре, получаем
d\nk2 = 1 1 Ео ^Eo+^RT
dT IT RT2 RT2
Сопоставим это соотношение с дифференциальной формой уравнения G.18)
и получим
а- 0+^ >
видим, что истинная энергия активации
E0=Ea--RT.
Она зависит от температуры, если считать Еа в первом приближении посто-
постоянной величиной. Однако различие между Ео и Ел обычно мало и часто на-
находится в пределах ошибок опыта.
Уравнение G.19) достаточно хорошо описывает к2 для ряда элемен-
элементарных реакций. Однако для реакций с участием довольно сложных молекул
опытная константа скорости меньше, чем рассчитанная по G.19). Различия
обусловлены несовпадением величин предэкспоненциального множителя.
Например, для реакции
9*
260 7. Кинетика гомогенных химических реакций
2NO2 -> 2NO + О2
отношение экспериментальной и расчетной констант скоростей кэ1кр& 0,05.
Это означает, что не все активные соударения приводят к химическому пре-
превращению.
Для того чтобы привести теорию в соответствие с экспериментом, в
уравнение G.19) вводят так называемый стерический множитель Р, учиты-
учитывающий необходимую ориентацию молекул реагентов при столкновении.
При этом уравнение для константы скорости записывают в виде
Для различных реакций Р имеет значения от 1 до 10~8. Установлено также,
что скорость многих мономолекулярных реакций в газовой фазе и многих
реакций в растворах больше предсказываемой теорией, т.е. множитель Р > 1.
Теория активных столкновений не позволяет предложить удовлетворитель-
удовлетворительного объяснения этих результатов.
Теория активированного комплекса. В теории активированного
комплекса для любой элементарной реакции предполагается, что начальная
конфигурация атомов переходит в конечную в результате непрерывного из-
изменения межъядерных расстояний.
Например, в ходе элементарной реакции
А + ВС -* АВ + С
сближаются атомы А и В. Расстояние А-В (R\) уменьшается, а расстояние
В—С (R 2) увеличивается (рис. 7.9).
В соответствии с этой теорией на первом этапе строят поверхность
потенциальной энергии системы. Установлено, что подобные реакции
осуществляются с наименьшей затратой энергии, если атомы располага-
располагаются на линии, соединяющей их центры. Тогда ход реакции можно опи-
описать, используя всего два межъядерных расстояния R\ и /?2- В процессе
непрерывного изменения межъядерных расстояний всегда образуется
промежуточная конфигурация А...В...С, в которой связь В—С уже ос-
ослаблена, но еще не полностью разорвана, а связь А—В уже начала обра-
образовываться. Такая конфигурация является критической для данной реак-
Рис. 7.9. Схема протекания реакции А + ВС -> АВ + С через образова-
образование активированного комплекса
7.6. Теоретические представления о скоростях элементарных реакций 261
А+ВС
АВ+С
Рис. 7.10. Карта поверхности потенциальной энергии реакции
А + ВС = АВ + С
ции. Продукты реакции могут появиться только при условии образования
этой конфигурации, которая называется переходным состоянием или ак-
активированным комплексом.
При изменении межъядерных расстояний изменяется и потенциальная
энергия Е системы за счет электронного взаимодействия. С помощью кван-
тово-механических методов можно рассчитать потенциальную энергию сис-
системы как функцию межъядерных расстояний Е = f(R\, R2) и представить
расчеты в виде поверхности потенциальной энергии. Для наглядности
обычно изображают карту поверхности на плоском чертеже. Поверхность
рассекают равноудаленными горизонтальными плоскостями (Е = const) и
проецируют контуры сечения на плоскость. На плоскости получаются ли-
линии равной энергии (изоэнергетические линии). Карта поверхности потен-
потенциальной энергии представлена на рис. 7.10. Цифры около линий обознача-
обозначают энергию в условных единицах. На энергетической карте можно выделить
«долину» Р\, в которой система (А + ВС) находилась до реакции, и «доли-
«долину» Р2, в которой находится система (АВ + С), образовавшаяся в результате
реакции. Путь реакции проходит по «дну долины» от Pi до «перевала» Р
(помечен точкой) и затем спускается по «долине» Р2.
Наиболее энергетически выгодный путь перехода от исходной систе-
системы к конечной — по линии, соединяющей точки, соответствующие мини-
минимуму потенциальной энергии (пунктирная линия на рис. 7.10), называют
координатой реакции. Высшая точка этого пути — точка «перевала» Р со-
соответствует активированному комплексу. Достигнув этой точки, система
может начать спуск в «долину» продуктов (АВ + С).
262
7. Кинетика гомогенных химических реакций
А+ВС
АВ+С
О R
Рис. 7.11. Изменение потенциальной
энергии системы вдоль координаты
реакции А + ВС -> АВ + С; Ео —
энергия активации; Е — потенциаль-
потенциальная энергия системы; R — координата
реакции
Для большей наглядности путь
реакции разворачивают в прямую ли-
линию и откладывают участки этого пути
по оси абсцисс, а на оси ординат нано-
наносят значения энергии, соответствующие
точкам пути. Тогда получается кривая с
максимумом, которую называют энер-
энергетическим профилем пути реакции
(рис. 7.11). Высота максимума Ео харак-
характеризует энергию активации. Она равна
разнице нулевых энергий активирован-
активированного комплекса и исходных молекул,
т.е. соответствует энергии активации
при температуре Т= О К.
Таким образом, построение по-
поверхности потенциальной энергии по-
позволяет теоретически рассчитать энергии активации и непосредственно най-
найти межъядерные расстояния в активированном комплексе. Однако такие
расчеты можно проводить для не очень сложных систем.
Принципиальная возможность теоретического расчета энергии акти-
активации открывает путь для расчета констант скоростей химических реакций.
Кинетическая схема для элементарной реакции имеет вид
А + ВС <=> [ABCf -> АВ + С .
исходные активированный продукты
вещества комплекс реакции
Основной постулат теории активированного комплекса — исходные веще-
вещества всегда находятся в равновесии с активированным комплексом. Кон-
Константа равновесия реакции образования активированного комплекса:
„ф _ [ABC]*
Скорость образования продуктов реакции:
Г ~
где kr = кКф — константа скорости реакции (без вывода):
G.20)
Здесь х — трансмиссионный коэффициент, учитывающий вероятность пе-
перехода активированного комплекса в продукты реакции. Константу К4" рас-
рассчитывают методами статистической физики.
7.6. Теоретические представления о скоростях элементарных реакций 263
Достоинство метода заключается в возможности связать константу
скорости с изменением термодинамических функций реакции образования
активированного комплекса. Из химической термодинамики известно, что
константа равновесия
^(^). G.21)
Используя выражение для изменения энергии Гиббса
AG0*=AH°*-TAS0*,
получают следующее выражение для константы скорости реакции, которое
называется уравнением Эйринга
0*) { АН0*) .....
НЬН' G-22)
где AS0* — стандартное изменение энтропии (энтропия активации); АН0* —
стандартное изменение энтальпии (энтальпия активации). Уравнение G.22)
справедливо для мономолекулярных реакций в газовой фазе и для реакций в
растворах. Уравнения G.21) и G.22) показывают, что, строго говоря, кон-
константу скорости определяет энергия активации Гиббса, а не опытная энер-
энергия активации, как это следует из G.15). Уравнение G.22) позволяет объяс-
объяснить существование быстрых процессов с большими энергиями активации и
медленных с малыми энергиями активации. Энтальпию активации обычно
находят экспериментально, используя зависимость наблюдаемой константы
скорости от температуры, а энтропию активации вычисляют теоретически
для разных моделей активированного комплекса.
Чтобы установить связь между опытной энергией активации и энталь-
энтальпией активации, следует прологарифмировать G.22):
, , - кБ , ^ AS0* АН0*
Ыкг =1пх—+ 1пГ +
К h R RT
и далее продифференцировать это уравнение по температуре:
d\nkr _ 1 АН0* _AH°*+RT
dT " ' Т RT2 ~ RT2
Сравнивая это выражение с уравнением G.18), получают следующие
соотношения:
Ea=AH°*+RT; AH°*=Ea-RT.
Рассмотрим в качестве примера результаты квантово-механического
264
7. Кинетика гомогенных химических реакций
расчета для достаточно простой реакции атома дейтерия D с молекулой во-
водорода Н2:
D + H2=DH + H
Для этой системы удалось с высокой точностью найти энергию системы как
функцию двух параметров — величин R\ и Л2, определяемых при располо-
расположении трех ядер на одной прямой:
D
Н
Н
Функция E(R\t R2) описывает некоторую поверхность. Приведем более про-
простой для рассматриваемой системы график, показывающий изменение энер-
энергии системы Е вдоль «пути реакции» (рис. 7.12).
Отметим две особенности. Во-первых, величина Ео — энергетический
барьер, который нужно преодолеть на пути от исходного состояния (D + Н2)
к конечному состоянию (DH + Н) системы, составляет небольшую величи-
величину G...8 %) от энергии диссоциации молекулы Н2. Тем самым с помощью
теоретического расчета можно объяснить известный из опыта факт, что
энергия активации реакции замещения обычно оказывается значительно
меньше энергии разрыва связей. Иными словами, акт замены одних химиче-
химических связей на другие протекает во много раз легче, чем разрыв самих свя-
связей. Во-вторых, он примечателен тем, что при значениях R\ = R2 » 0,85 А
(R2 = 0,74 А — равновесное межъядерное расстояние) для конфигурации
D...H...H движение по пути реакции на сравнительно большом отрезке
AR « 0,3 А происходит практически при постоянной энергии. Таким образом,
«активированное состояние», достигаемое после затраты энергии, равной
34 кДж/моль, в действительности представляет собой целый набор состояний
(D...H...H)
о
R
Рис. 7.12. Изменение энергии линейной
системы D...H...H от межъядерных рас-
расстояний (R — функция от R\ и R2, для
D + Н2 R2 = 0,74 А, Л, » R2, для DH + Н
Я, = 0,74 A, R2» R\;Eo* 34 кДж/моль
с примерно одинаковой энергией, но
с различными значениями R\ и R2.
Это означает также, что химическое
превращение в системе — уменьше-
уменьшение одних длин связей и увеличение
других — не требует какого-либо
изменения энергии, если только уда-
удается достичь одной из конфигура-
конфигураций, отвечающих активированному
состоянию. Считается, что анало-
аналогичным образом (качественно) про-
происходит перестройка и многих дру-
других химических систем.
Особенности кинетики гетерогенных
реакций
Химические реакции, протекающие на границе раздела фаз, назы-
называют гетерогенными. Гетерогенные процессы широко распростра-
распространены в природе и часто используются на практике. Примерами мо-
могут служить процессы растворения, кристаллизации, испарения,
конденсации, химические реакции на границе двух фаз, электрохи-
электрохимические процессы на границе твердое тело — раствор электроли-
электролита, гетерогенный катализ. Гетерогенные процессы могут протекать
на границе между разными фазами: твердая — твердая (ti-t2), твер-
твердая — жидкая (т-ж), твердая — газообразная (т-г), жидкая — жид-
жидкая (ж!-ж2), жидкая — газообразная (ж-г).
В этой главе рассмотрены гетерогенные реакции с участием твер-
твердых веществ. Кинетика реакций с участием твердых веществ в на-
настоящее время занимает одно из ведущих мест среди проблем хи-
химической кинетики. Химические процессы в гетерогенных системах
очень сильно отличаются от процессов, протекающих в гомогенных
системах. Природа этих процессов значительно сложнее.
Экспериментальное изучение кинетики гетерогенных реакций —
довольно трудная задача. Во-первых, это обусловлено природой
твердых веществ. Для одной и той же системы скорость процесса
зависит от структуры твердого вещества, наличия дефектов в кри-
кристаллической решетке и содержания различных примесей. В этом
заключается основная причина затруднений для получения удовлет-
удовлетворительной воспроизводимости результатов. Во-вторых, проблема
состоит в отделении особенностей химических реакций от законо-
закономерностей явлений переноса. Явления переноса могут проявляться
в большей или меньшей степени в зависимости от эксперименталь-
экспериментальных условий протекания процесса.
8.1. Реакции на границе раздела твердое тело — газ
и твердое тело — жидкость
Гетерогенные реакции этого типа широко используются в хи-
химической технологии. К ним относятся реакции горения твердого и
жидкого топлива, окисления металлов, восстановления металлов из
оксидов и сульфидов, реакции, идущие на поверхности твердых ка-
катализаторов.
Примерами некоторых типов гетерогенных реакций служат:
1) реакции окисления металлов (Ti + Г -> Т2), например
Ni(T)+io2(r)->NiO(T)
266 8. Особенности кинетики гетерогенных реакций
2) реакция разложения карбоната кальция (Ti -> Т2+ Г)
СаСОз (Т) = СаО(т) + СО2 (г)
3) реакции обжига и восстановления, играющие важную роль в метал-
металлургии (Ti + Г\ -» Т2 + Г2):
4FeS2 (т) + 11О2 (Г) = 2Fe2O3 (т) + 8SO2 (г)
3Fe2O3 (т) + СО(Г) = 2Fe3O4 (т) + СО2 (г)
Fe3O4 (т) + 4СО (г) = 3Fe (т, ж) + 4СО2(г)
4) реакции с участием трех твердых фаз (Ti + Т2 -> Т3), например
ВаО(т) + ТЮ2 (т) = BaTiO3 (т)
5) реакции двойного обмена в твердой фазе (Ti + Т2 -> Т3 + Т4), например
2CuI(T) + Ag2S(T) = 2AgI(T) + Cu2S(T)
6) взаимодействие металлического цинка с раствором соляной кисло-
кислоты (Ti + Ж -> Ж + Г):
Zn(T) + 2НС1(Р) = ZnCl2 (p) + Н2 (Г).
В гетерогенных реакциях, как правило, можно выделить по меньшей
мере три стадии: 1 — перенос реагирующих веществ к поверхности раздела
фаз, т.е. в реакционную зону; 2 — собственно химическое взаимодействие;
3 — перенос продуктов реакции из реакционной зоны. Могут быть и другие
стадии, например адсорбция и десорбция; дополнительные химические пре-
превращения продукта реакции у поверхности твердого тела: комплексообразо-
вание, димеризация, протонирование.
Скорость всего процесса определяется лимитирующей (наиболее мед-
медленной) стадией. Если скорость собственно химического взаимодействия
значительно больше скорости подвода реагентов к реакционной зоне и от-
отвода продуктов от нее, то общая скорость процесса будет соответствовать
скорости переноса реагентов и продуктов, и для самопроизвольных терми-
термически инициируемых реакций она будет определяться процессами диффузии
веществ. В этом случае говорят, что реакция протекает в диффузионном
режиме или находится в диффузионной области.
Перенос вещества к границе между фазами может осуществляться за
счет конвекции и диффузии. Конвекцией называют перемещение всей среды
в целом. Конвекция раствора на границе с твердой поверхностью может
происходить из-за разной плотности в объеме раствора и вблизи поверхно-
поверхности твердого тела, что вызывается неодинаковой концентрацией или темпе-
температурой раствора. Конвекцию можно создавать также перемещением твер-
твердого тела в растворе или раствора вблизи поверхности твердого тела (вра-
(вращение твердого тела, перемешивание раствора).
8.1. Реакции на границе раздела 267
Диффузия — это направленное перемещение вещества из области с
большей концентрацией в область с меньшей концентрацией. Подвод веще-
вещества диффузией определяется первым законом Фика, согласно которому:
количество dn вещества, продиффундировавшего через площадку S, пер-
перпендикулярную потоку в направлении от большей концентрации к меньшей,
пропорционально времени dt и градиенту концентрации (dC/dx)
dn = -DS—dt.
dx
Коэффициент D называют коэффициентом диффузии. Он зависит от
природы диффундирующего вещества и среды, а также от температуры.
В том случае, когда собственно химическое взаимодействие является
наиболее медленной стадией, т.е. диффузия протекает сравнительно быстро,
говорят, что реакция протекает в кинетическом режиме или находится в
кинетической области. В том случае, когда скорости реакции и диффузии
приблизительно равны, говорят о переходном режиме или переходной об-
области гетерогенной реакции.
При повышении температуры лимитирующая стадия и соответственно
механизм гетерогенной реакции могут измениться. Это связано с разным
характером зависимости константы скорости химической реакции k и коэф-
коэффициента диффузии от температуры. Константа скорости химической реак-
реакции зависит от температуры в соответствии с G.15):
ЕЛ Е
— или 1п? = 1п&0 —,
^ RTJ о RT>
где Ех — энергия активации гетерогенной реакции. Зависимость коэффици-
коэффициента диффузии от температуры определяется аналогичным выражением:
(ЕЛ Е
Z) = Z)oexp - или 1п?> = 1п?>0 -,
о ^ RTj о RT>
где ЕД — энергия активации процесса диффузии.
Предположим, что при низкой температуре лимитирующей стадией
гетерогенного процесса является химическая реакция (кинетическая об-
область). При повышении температуры константа скорости химической реак-
реакции быстро увеличивается и, начиная с некоторой температуры, когда ско-
скорость реакции станет больше скорости диффузии, лимитирующей стадией
становится диффузионный процесс (диффузионная область). При этом про-
происходит постепенный переход из кинетической области гетерогенного про-
процесса в диффузионную. Для гетерогенной реакции типичная форма зависи-
зависимости логарифма скорости In г от A/Г) приведена на рис. 8.1. При повы-
268
8. Особенности кинетики гетерогенных реакций
In ri
О
1/Т
Рис. 8.1. Зависимость логарифма ско-
скорости гетерогенной реакции от \1Т
шении температуры на графике изме-
изменяется тангенс угла наклона от
(-EJR) (прямая DC) до (-ED/R) (прямая
АВ). По величине энергии активации
можно судить о механизме процесса.
Для диффузионного процесса энергия
активации имеет значения в пределах
5...20 кДж/моль. В кинетической об-
области энергия активации обычно равна
50...200 кДж/моль. В промежуточной
области (прямая ВС) гетерогенная ре-
реакция контролируется как диффузией,
так и химической реакцией на границе
раздела фаз.
8.2. Скорость гетерогенных реакций
Для количественного описания гетерогенного процесса существуют
разные подходы к определению скорости реакции. В одном из них исполь-
используют понятие поверхностной концентрации. Скорость гетерогенной хими-
химической реакции измеряется изменением поверхностной концентрации одно-
одного из веществ (газа или жидкости), участвующих в реакции, за единицу вре-
времени
= ±-
dt
G.23)
Здесь г — в моль/(м2 • с) или моль/(см2 • с); Q — поверхностная концентра-
концентрация вещества, т.е. количество молей вещества, приходящееся на единицу
реакционной поверхности, моль/м2 или моль/см2.
Концентрации веществ в твердом состоянии постоянны, поэтому в
кинетическое уравнение гетерогенных реакций они не входят. Например, в
реакции
СаО(т) + СО2 (г) = СаСОз (т) G.24)
соударения между молекулами СО2 и твердого вещества могут происходить
только на поверхности раздела фаз. В этом случае, согласно основному за-
закону химической кинетики, в выражение скорости реакции будет входить
только поверхностная концентрация СО2:
-SCO?
8.2. Скорость гетерогенных реакций
269
Следует заметить, что определять поверхностную концентрацию ве-
вещества довольно трудно. Поэтому на практике часто вычисляют скорость
гетерогенной реакции, используя вместо поверхностной концентрации объ-
объемную концентрацию. Например, скорость реакции G.24) рассчитывают по
уравнению
где г1 — скорость реакции, моль/(л • с); к — константа скорости, с; ССОг —
объемная концентрация СОг в газовой фазе, моль/л.
Выражение скорости гетерогенных реакций г в моль/(л • с) приводит
к тому, что г' становится функцией площади реакционной поверхности S, так
как чем больше эта площадь, тем больше число столкновений молекул реа-
реагирующих веществ, находящихся в разных фазах гетерогенной системы.
Отметим, что при постоянной температуре на единице поверхности раздела
фаз число столкновений молекул постоянно. Поэтому при правильном тол-
толковании основного закона химической кинетики (см. § 7.2) применительно к
гетерогенным реакциям скорость гетерогенной реакции G.23) не зависит от
площади поверхности раздела фаз, так же как скорость гомогенной реакции
не зависит от объема системы.
В другом подходе скорость гетерогенной реакции с участием твердого
вещества определяют через степень превращения вещества.
Степень превращения вещества представляет собой относительное
изменение количества исходного реагирующего вещества к моменту време-
времени /:
ix —
_mo-mt
где п0 — начальное количество исходно-
исходного вещества, моль; nt — количество ис-
исходного вещества в момент времени t\
то— начальная масса исходного веще-
вещества; mt — масса исходного вещества в
момент времени t.
На рис. 8.2 показан типичный гра-
график зависимости степени превращения
от времени. Участок 1 соответствует пе-
периоду индукции, когда реакция протекает
очень медленно. После индукционного пе- Рис. 8.2. Кинетическая кривая для
риода следует участок ускорения 2, про- гетерогенной реакции с участием
должающийся до точки перегиба (обо- твердого вещества
270
8. Особенности кинетики гетерогенных реакций
значена крестиком). Участок 3 соот-
соответствует замедлению реакции. Ско-
Скорость реакции в данный момент опре-
определяется по углу наклона касательной
в точке кинетической кривой, соответ-
соответствующей этому моменту времени.
Можно отметить, что для многих про-
процессов кривая а = /(/) имеет 5-образ-
ный вид (сигмоидная кривая). Пересе-
Пересечение касательной, проведенной в точке
перегиба, с осью абсцисс дает значе-
значение времени, называемого периодом
индукции U.
Зависимость скорости реакции
от степени превращения твердого реа-
реагента представлена на рис. 8.3. На нем
отчетливо видно увеличение скорости
реакции, достигающей максимума в
момент времени, соответствующий точ-
точке перегиба на рис. 8.2.
В реакциях, ход которых описы-
описывается сигмоидной кривой, продукты
реакции не появляются равномерно по
всему образцу. Они возникают в виде отдельных островков, зародышей, что
схематично показано на рис. 8.4. Обычно процесс образования зародышей
происходит на поверхности твердого реагента.
° «max I a
Рис. 8.3. Зависимость скорости гете-
гетерогенного процесса от степени пре-
превращения твердого реагента
ооо
Рис. 8.4. Схема образования зароды-
зародышей при протекании гетерогенной ре-
реакции с участием реагента
83. Твердофазные реакции
Реакции между веществами в твердой фазе и реакции полиморфных
превращений называют твердофазными. Рассмотрим реакции, происходя-
происходящие между различными кристаллическими веществами на границе их раз-
раздела. Подобные реакции существенно отличаются от реакций, проходящих
в жидкой или газовой фазах. Различие обусловлено тем, что атомы, ионы
или молекулы кристаллических фаз значительно менее подвижны, и взаи-
взаимодействие возможно лишь в местах контакта реагирующих веществ. При
этом образуется слой продукта на границе раздела двух реагирующих ве-
веществ и дальнейшее прохождение твердофазной реакции зависит от скоро-
скорости диффузии взаимодействующих веществ через слой продукта. Заметим,
8.3. Твердофазные реакции 271
что продукт тоже участвует в процессах диффузии. Это обстоятельство де-
делает механизм протекания твердофазного процесса еще более сложным.
Твердофазная реакция термодинамически возможна, т.е. может
протекать самопроизвольно, если изменение энергии Гиббса ArG < 0. Из-
Изменения энтальпий твердофазных реакций очень малы по сравнению с
изменениями энтальпий реакций, в которых хотя бы один из реагентов
или продуктов не является твердой фазой. Твердофазные взаимодействия
сопровождаются также малыми изменениями энтропии, поскольку в
твердых фазах реагентов более высокая степень упорядоченности по
сравнению с газо- и жидкофазными веществами. В качестве примера в
табл. 8.1 представлены значения изменения энтальпии и энтропии неко-
некоторых твердофазных реакций.
Таблица 8.1
Стандартные изменения энтальпий и энтропии
некоторых гетерогенных реакций
Реакция
Fe + У20 2 = FeO
FeO + Fe2O 3 = Fe3O4
Si + O2 = SiO2
Si + С (графит) = SiC
Ar#298> кДж/М0ЛЬ
-265
-30
-911
-66
А А, Дж/(мольК)
-69,9
-1,6
-182
-7,9
Другой характерной особенностью твердофазных реакций является их
топохимический характер, т.е. локализация реакционной зоны на поверх-
поверхности раздела фаз реагентов и продуктов. В отличие от этого в газо- и жид-
кофазных системах при интенсивном перемешивании эффективные столк-
столкновения молекул реагентов могут происходить в любой точке пространства,
занимаемого системой.
Важнейшие типы твердофазных реакций можно выразить следующи-
следующими схемами:
1)Т,->Т2,
2)T!+T2->T3,
3)T!+T2->T3 + T4,
где Т/ — твердофазный реагент или продукт.
Типичным примером реакций типа 1 являются полиморфные превра-
превращения простых и сложных веществ, широко используемые при создании
неорганических материалов. Например,
С (графит) = С (алмаз)
a-Sn = P-Sn
272
8. Особенности кинетики гетерогенных реакций
Agl Agl
Agl
Agl
Agl
Agl
Agl Agl Agl
Hgl2 Hgl2
Hgl2
Hgl-
Hgb
Hgl 2 Hgl 2 Hgl 2
Agl Agl
Agl
Agl Agl
Ag +
Ag2[HgI<
Hg
2+
Hgb
Hgb
Hgb
Рис. 8.5. Схема, иллюстрирующая механизм твердофазной реакции
Реакции типа 2 широко используют для синтеза многочисленных
функциональных материалов. Примером может служить синтез ферритов —
магнитных диэлектриков, например, по реакции
xZnO + A -x)NiO + Fe2O3 = ZnJCNi1_^Fe204
Возможность протекания реакции типа 3 необходимо учитывать при
эксплуатации композиционных материалов, когда взаимодействуют матри-
матрица и наполнитель. Реакции этого типа необходимо учитывать также при по-
получении гетероструктур специальными методами (например, плазменным
или лазерным напылением), при этом возможно взаимодействие пленки с
подложкой, на которую эта пленка наносится. В обоих случаях реакции типа 3
нежелательны и их пытаются избежать, подбирая соответствующие компо-
компоненты и учитывая условия эксплуатации.
При протекании твердофазных процессов особую роль играют содер-
содержание дефектов в кристаллах реагирующих веществ и скорость диффузии,
которая обеспечивает подход реагентов друг к другу (т.е. скорость переноса
веществ через слой продукта). Для осуществления реакции между твердыми
веществами необходимо обеспечить доступ реагентов друг к другу и отвод
продуктов. Реакция практически не проходит, если исходные вещества не
способны перемещаться навстречу друг другу через слой продуктов.
Например, при взаимодействии йодидов серебра и ртути с образова-
образованием комплексной соли
= Ag2[HgI4]
обе частицы (катионы серебра Ag+ и ртути Hg2+) диффундируют через слой
продукта, и количество исходных веществ уменьшается за счет роста слоя
продукта (рис. 8.5). На поверхностях раздела слоя продукта и двух исход-
исходных веществ протекают реакции:
4AgI + Hg2+ = Ag2[HgI4] + 2 Ag+
2HgI2 + 2Ag+ = Ag2[HgI4] + Hg2+
Материалы, изложенные в данной главе, свидетельствуют о чрезвы-
чрезвычайной сложности гетерогенных процессов, при изучении которых необхо-
необходимо учитывать влияние множества факторов.
9 ¦ Основы катализа
Катализ — важный раздел химической кинетики, который имеет
практическое значение и динамично развивается в настоящее время.
Гомогенные и гетерогенные катализаторы широко используются в
промышленности основного органического синтеза, нефтехимиче-
нефтехимической, фармацевтической и пищевой отраслях промышленности. С
помощью каталитических процессов производится около 80 % про-
продукции.
Каталитические процессы составляют основу производства различ-
различных видов топлива, удобрений, пестицидов, фармацевтических пре-
препаратов, огромного числа химических продуктов. Каталитические
процессы широко распространены в природе и эффективно исполь-
используются в различных отраслях промышленности, науки и техники.
Например, в нефтехимической промышленности более половины до-
добываемой нефти перерабатывается с помощью каталитических про-
процессов в моторное топливо, различные мономеры для получения по-
полимерных волокон и пластмасс.
9.1. Основные понятия и определения
Термин «катализ» был введен шведским ученым Й. Берцелиу-
сом A835) в работе, обобщившей уже известные факты ускорения
реакций. К тому времени химики уже знали о влиянии на протекание
многих реакций веществ, участие которых в химических превраще-
превращениях явно не проявляется. В то время уже были известны такие
примеры каталитических реакций, как дегидратация спиртов на
глине (Дж. Пристли, 1783); этерификация уксусной кислоты в при-
присутствии минеральных кислот (К. Шееле, 1797); влияние оксидов
азота на окисление диоксида серы (Ш. Дезорм, Н. Клеман, 1806);
разложение аммиака и пероксида водорода под влиянием различных
твердых веществ (Л. Тенар, 1813-1818); окисление углеводородов до
оксида углерода и водорода при комнатной температуре в присутст-
присутствии платины (Г. Дэви, 1817; И. Доберейнер, 1821); быстрое горение
водорода в присутствии измельченной платины (И. Доберейнер, 1821).
После обобщений Берцелиуса были развернуты систематиче-
систематические исследования явлений катализа. Основой первого промыш-
промышленного процесса с использованием гетерогенного катализа явился
процесс Дикона — получение хлора окислением хлористого водо-
водорода при температуре 400 °С в присутствии солей меди, нанесен-
нанесенных на А12О3:
274 9. Основы катализа
2НС1 + -О2 = Н2О + С12
2
Катализом называют явление изменения скорости термодинамически
возможной химической реакции под влиянием катализаторов — веществ,
участвующих в реакции, но остающихся в неизменном количестве и составе
после ее завершения.
Все каталитические процессы подразделяют на две группы. К гомоген-
гомогенному катализу относятся процессы, в которых реагирующие вещества и ката-
катализатор находятся в одной фазе и образуют гомогенную систему, например,
некоторые газофазные каталитические реакции, многие каталитические ре-
реакции в растворах. Примером гомогенной каталитической реакции является
окисление оксида серы (IV) кислородом в присутствии оксида азота (IV):
2SO2+O2 N°2 >2SO3
При гетерогенном катализе катализатор представляет собой само-
самостоятельную фазу, граничащую с фазой реагентов. В этом случае каталити-
каталитическая реакция протекает на поверхности раздела фаз: газ — твердое тело,
жидкость — твердое тело, жидкость — газ. Наибольшее практическое зна-
значение имеют каталитические реакции, когда катализатор находится в твер-
твердой фазе, а реагенты — в жидкой или газообразной. Примером гетерогенно-
гетерогенного каталитического процесса служит окисление оксида серы (IV) кислоро-
кислородом в присутствии оксида ванадия (V):
2SO2+O2 V2°5 >2SO3
В особую группу выделяют ферментативные каталитические реак-
реакции, что связано с происхождением катализаторов, называемых фермента-
ферментами. Ферменты (или энзимы) — это специфические белки, выполняющие
каталитические функции в живых системах. Они отличаются от обычных
катализаторов высокой активностью и уникальной селективностью.
Можно выделить следующие общие закономерности каталитических
реакций.
1. Катализатор не влияет на положение термодинамического равнове-
равновесия, а только ускоряет наступление равновесия. Он не входит в состав исход-
исходных веществ и продуктов реакции и не может оказать влияние на изменение
энергии Гиббса реакции ArG, являющейся функцией состояния. Следователь-
Следовательно, и не может вызвать протекание реакций, для которых в данных условиях
ArG > О, а может увеличить скорость реакции в том случае, если ArG < 0.
2. Катализатор принимает химическое участие в реакции. Действие ката-
катализатора заключается в том, что он образует с реагирующими веществами про-
9.1. Основные понятия и определения 275
межуточный комплекс, который затем разрушается с образованием продуктов
реакции, а сам катализатор освобождается и переходит в исходное состояние.
3. Скорость каталитической реакции пропорциональна концентрации
катализатора (гомогенный катализ) или площади поверхности катализатора
(гетерогенный катализ).
4. Основной причиной увеличения скорости в результате катализа яв-
является значительное уменьшение энергии активации реакции Еа.
Например, пероксид водорода в водном растворе медленно разлагает-
разлагается на воду и кислород по уравнению:
Н2О2=Н2О+-О2
Энергия активации этой реакции Еа = 75,6 кДж/моль. В присутствии ио-
ионов трехвалентного железа Fe3+ энергия активации уменьшается до
54,6 кДж/моль. Наименьшее значение энергии активации соответствует катали-
каталитическому разложению под действием фермента каталазы — 20 кДж/моль. В
его присутствии скорость реакции увеличивается приблизительно в 108 раз
по сравнению с некаталитической реакцией.
Уменьшение энергии активации не единственная причина увеличения
скорости реакции в присутствии катализатора. В некоторых случаях ускоре-
ускорение процесса обусловлено увеличением предэкспоненциального множителя ко
(см. G.15)).
5. Катализатор по своему действию селективен, т.е. избирателен. Он
увеличивает скорость преимущественно одной из возможных реакций и не
влияет заметно на скорость других реакций.
Например, при пропускании паров этанола над оксидом алюминия (III)
при Т= 300 К образуется этилен, а с медным катализатором — ацетальдегид:
С2Н5ОН Al2°3 >С2Н4 + Н2О
С2Н5ОН—^->СН3СНО + Н2
Селективность — важное технологическое свойство катализатора, так
как при ее повышении уменьшается количество побочных веществ и повы-
повышается качество целевого продукта. Селективность катализатора может
быть охарактеризована долей реагента, превратившегося в целевой продукт
(интегральная селективность), или отношением скорости образования це-
целевого продукта к сумме скоростей химического превращения реагентов по
всем возможным направлениям (дифференциальная селективность). Наи-
Наибольшей селективностью (95... 100 %) обладают ферменты и некоторые го-
гомогенные катализаторы. Селективность гетерогенных катализаторов обычно
не превышает 70 %.
276 9. Основы катализа
Всем катализаторам в той или иной мере свойственна специфич-
специфичность. Специфичность каталитического действия заключается в том, что
реакции данного типа ускоряются катализаторами определенного химиче-
химического состава независимо от того, гомогенные они или гетерогенные. Так,
кислотно-основные реакции ускоряются кислотами или основаниями, а
окислительно-восстановительные реакции — переходными металлами и
их соединениями. Это явление обусловлено специфичностью химических
связей (ковалентной, донорно-акцепторной, водородной). В образовании
химической связи с реагентами участвуют лишь определенные группы
атомов катализатора. Такую группу атомов принято называть катали-
каталитическим или активным центром.
В гомогенном катализе каждую молекулу катализатора можно рас-
рассматривать как активный центр. Активные центры гетерогенных катализа-
катализаторов находятся на поверхности твердого тела и представляют собой атом
или группу атомов, ионов кристаллической решетки. Более сложное строе-
строение имеют активные центры ферментов. Специфичность действия того или
иного катализатора определяется химическим составом, строением и струк-
структурой его активных центров.
9.2. Механизм протекания каталитических реакций
Основным положением теории каталитических процессов является
представление об образовании неустойчивых промежуточных соединений
катализатора с реагирующими веществами, которые затем распадаются с
освобождением катализатора. Для описания механизма протекания катали-
каталитических реакций в настоящее время используют две концепции. Одна из
них соответствует слитной {одностадийной) схеме катализа, другая — ста-
стадийной (раздельной) схеме катализа.
Согласно слитной схеме катализа, некаталитическая реакция типа
в присутствии катализатора К проходит по схеме
А + В + К <=± (ABKf -> Р + К.
Здесь (АВК)* — активированный комплекс исходных веществ и катализато-
катализатора. Этот процесс в присутствии катализатора проходит в одну стадию и его
скорость описывается кинетическим уравнением:
r = kCACBCK.
9.2. Механизм протекания каталитических реакций
277
Е i
А,В,К
Р,К
Энергетическая диаграмма ка-
талитической реакции, осуществля-
осуществляемой по слитному механизму пред-
представлена на рис. 9.1. В присутствии
катализатора процесс идет с мень-
меньшей энергией активации (нижняя
кривая).
Чаще протекают каталитиче-
каталитические реакции, осуществляемые по
стадийной схеме. В соответствии с
этой схемой вещества-реагенты по-
последовательно взаимодействуют,
образуя на каждой стадии соответ-
соответствующий активированный ком-
комплекс:
1)А + К <z> (AK)*-*AK;
2) АК + В <=± (АВК) *-> Р + К.
Согласно записанным уравнениям,
промежуточное соединение АК мо-
может превратиться по обратной реак-
реакции с константой скорости к-\ в ис-
исходное вещество и катализатор или в продукты реакции Р и катализатор по
второй реакции (константа скорости к2). Соответствующая энергетическая
диаграмма представлена на рис. 9.2. Она характеризуется несколькими мак-
максимумами (активированные комплексы отдельных стадий) и минимумами
(промежуточные соединения).
Каталитические реакции протекают по слитной и стадийной схемам,
обычно в разных температурных интервалах. По слитной схеме часто осу-
осуществляются гомогенные каталитические реакции при температурах
300...400 К и ферментативные реакции. По стадийной схеме протекают, как
правило, гетерогенные каталитические реакции при температурах 600.. .800 К.
Обе приведенные схемы отражают общую и характерную особенность
механизма протекания каталитических реакций, а именно его цикличность.
В каталитических реакциях один и тот же активный центр или молекула ка-
катализатора может многократно A03-10п раз) вступать в химическое взаи-
взаимодействие с молекулами реагента.
Активность катализаторов. Мера каталитической активности есть
число оборотов катализатора. Число циклов, совершаемых на одном актив-
активном центре за единицу времени, называют числом оборотов катализатора.
Для гомогенных катализаторов число оборотов пк можно определить, если
разделить скорость реакции на молярную концентрацию катализатора:
о
Рис. 9.1. Энергетическая диаграмма го-
гомогенной каталитической реакции, про-
протекающей по слитному механизму:
1 — без катализатора; 2 — с катализатором;
Еа — энергия активации некаталитической
реакции; ЕаК — энергия активации катали-
каталитической реакции; (АВ)* — активирован-
активированный комплекс некаталитической реакции;
(АВК)* — активированный комплекс ката-
каталитической реакции; R — координата ре-
реакции
278
9. Основы катализа
Рис. 9.2. Энергетическая диаграмма
для гомогенной каталитической ре-
реакции, протекающей по стадийному
механизму:
1 — некаталитическая реакция; 2 — ка-
каталитическая реакция; (АВ)" — активи-
активированный комплекс некаталитической
реакции; (АК)*, (АВК)* — активиро-
активированные комплексы каталитической ре-
реакции
где гк — скорость реакции; Ск — кон-
концентрация катализатора, моль/л. На-
Например, для гомогенных кислотно-
основных катализаторов при Т = 298 К
число оборотов пк = 10~7...1(Г2 с, а для
ферментовпк= 102 ...105с~1.
Для гетерогенных катализаторов
число оборотов, как правило, не удается
определить, так как природа и число
активных центров на поверхности ката-
катализатора обычно неизвестны. Поэтому
их активность Ак характеризуют скоро-
скоростью реакции, отнесенной к единице
площади поверхности SK катализатора:
Активность катализатора зависит от условий протекания процесса:
температуры, концентрации реагентов, давления, от природы растворителя,
если реакция протекает в жидкой фазе. Поэтому сравнение каталитической
активности катализаторов проводят при одинаковых условиях.
Одним из важных отличий гетерогенных катализаторов от гомоген-
гомогенных является зависимость их свойств от условий приготовления и высокая
чувствительность к действию небольших количеств посторонних веществ.
Добавление некоторых каталитически неактивных веществ иногда значи-
значительно увеличивает эффективность катализатора. Такие вещества называют
промоторами или модификаторами. Например, промоторами железного
катализатора при синтезе аммиака являются примеси А12Оз и КгО.
Присутствие в реакционной системе некоторых веществ, даже в очень
малом количестве, способно понижать или полностью подавлять активность
катализатора. Подобные вещества называют каталитическими ядами, а са-
само явление — отравлением катализатора. К типичным каталитическим
ядам относятся соединения серы (H2S, CS2, тиофен, тиоспирты), соли ртути,
соединения фосфора, мышьяка, свинца. Например, железный катализатор
синтеза аммиака полностью теряет каталитические свойства в присутствии
0,1 % серы. Существенная особенность процессов промотирования, моди-
модифицирования и отравления состоит в том, что все многообразие действия
добавок часто проявляется при их очень малых концентрациях. Воздействие
9.3. Гомогенный катализ 279
микроколичества примесей — одна из важнейших характеристик гетероген-
гетерогенного катализа как физико-химического явления.
9.3. Гомогенный катализ
По характеру промежуточного взаимодействия можно выделить две
основные группы каталитических процессов, свойственных как гомоген-
гомогенным, так и гетерогенным каталитическим реакциям. К первой группе отно-
относятся процессы, осуществляющиеся по кислотно-основному механизму, а ко
второй группе — по окислительно-восстановительному механизму.
Кислотно-основный катализ. Многие гомогенные реакции в
растворах катализируются кислотами и основаниями. Примером служит
гидролиз сложных эфиров, который может ускоряться или кислотой или
основанием.
Гидролиз сложных эфиров в кислотной среде включает обратимую
стадию присоединения иона водорода к кислороду карбонильной группы за
счет донорно-акцепторного взаимодействия с образованием карбоний-ка-
тиона:
R-C + Н ^-R-
Карбоний-катион затем реагирует с молекулой воды:
R— С + Н2О ^ R— C^ +ROH + Н
В результате получаются спирт и кислота, а также ион водорода, ко-
который вновь вступает в реакцию.
Согласно теории Брёнстеда—Лоури, кислота — это вещество, спо-
способное отдавать ион водорода (протон):
Н А -> Н + + А"
а основание — вещество, склонное к присоединению иона водорода:
В этом случае говорят, что частица А~ является сопряэюенным основанием.
Рассмотрим вещество В, которое вступает в элементарные реакции с
кислотой, основанием или с ними обоими. Например, механизм кислотного
катализа можно представить следующей схемой:
280 9. Основы катализа
1)В + НА +
2) ВН+ -> Р! + Р2 + Н+
3)А" + Н+ <=> НА
Здесь НА — кислота (Н2О, СН3СООН); А" — сопряженное основание (Н2О,
ОН", СН3СОО"); Pi и Р2 — продукты реакции.
Скорость каталитической реакции
где kK — константа скорости каталитической реакции:
К =kr
kr
Здесь кг — константа скорости некаталитической реакции; кц+, ?QH_, &НА,
кА_ — константы скорости каталитических реакций с участием соответст-
соответствующих частиц.
Выделяют два типа кислотно-основного катализа: специфический и
общий кислотно-основной катализ.
Скорость некоторых реакций пропорциональна концентрации иона Н+
или ОН". Такие реакции относятся к реакциям со специфическим кислотно-
основным катализом. В этом случае одна или обе константы (k+ и ?. и_)
н он
велики по сравнению с кНА и к и уравнение (9.1) упрощается:
А
Константы скорости каталитических реакций определяют измерением ско-
скоростей в широком интервале значений водородного показателя рН.
Если реакция протекает в сильно кислотной среде, то (9.2) принимает вид
Логарифмируем это выражение и получаем, учитывая, что рН = -lgC +:
=lgkH+ +lgCH+ или lgkK =lg*H+ -pH (9.3)
Таким образом, в интервале значений рН, отвечающих кислотной среде, за-
зависимость lg kK от рН линейна с тангенсом угла наклона, равным (-1).
В сильно основном растворе уравнение (9.2) преобразуется к виду
У -Ег Г -V ^Н2О
9.3. Гомогенный катализ
281
где Кн 0 — ионное произведение воды. Ло- te К
гарифмирование этого выражения приводит
к уравнению
Следовательно, зависимость lg kK от рН ли-
линейная и имеет тангенс угла наклона, рав-
равный (+1). В области промежуточных значе-
и ' Рн
Рис. 9.3. Зависимость констан-
тт , , „ты скорости реакции гидролиза
нии рН \gkK не зависит от концентрации СЛ0ЖНых эфиров от рН среды
Си+ и Сли_. В этом случае кк зависит толь-
н он
ко от кп но не от рН. На рис. 9.3 представлена зависимость lg kK от рН для
гидролиза сложных эфиров. Из рисунка следует, что каталитическое дейст-
действие на эту реакцию оказывают и кислота и основание.
Реакции, катализируемые всеми кислотами и основаниями, относятся
к случаю общего кислотно-основного катализа. Если применяют такие ус-
условия, когда концентрации ионов Н+ и ОН" не влияют на скорость, т.е. если
константами кн+ и kQU_ можно пренебречь, скорость многих реакций зави-
зависит от концентраций недиссоциированной кислоты и сопряженного основа-
основания А~. Считая, что вклад некаталитической реакции в скорость процесса
мал, вместо выражения (9.1) получаем
Окислительно-восстановительный катализ. Определяющей стади-
стадией многих окислительно-восстановительных реакций (см. гл. 1) является
перенос электрона от одного реагента к другому. Этот процесс в ряде случа-
случаев протекает медленно. Тогда применяют катализаторы, ускоряющие ста-
стадию переноса электрона. К реакциям окислительно-восстановительного ти-
типа относят разложение пероксида водорода, гидрирование, окисление орга-
органических соединений молекулярным кислородом.
Рассмотрим характерные особенности окислительно-восстановитель-
окислительно-восстановительных каталитических реакций на примере разложения пероксида водорода.
Как уже упоминалось, пероксид водорода в водных растворах самопроиз-
самопроизвольно медленно разлагается на воду и кислород по уравнению
Н2О2 = Н2О+-О2
Энергия активации этой реакции ?а = 75,6 кДж/моль. В присутствии катали-
катализатора дихромата калия К2Сг2О7 скорость разложения резко увеличивается.
282 9. Основы катализа
Процесс разложения протекает в две стадии. На первой стадии (обратимой)
при взаимодействии К2Сг2О7 с Н2О2 быстро образуется промежуточное со-
соединение:
2Н2О2+К2Сг2О7 <=> К2Сг2О9+2Н2О
или в молекулярно-ионном виде
2Н2О2 + Cr2O?" <z± Сг2О^ + 2Н2О
Образовавшееся промежуточное соединение К2Сг209 распадается по
уравнению:
К2Сг2О9^К2Сг2О7+О2
или в молекулярно-ионном виде
В результате выделяется кислород и освобождается катализатор. Таким об-
образом, дихромат калия К2Сг207 отвечает двум важнейшим требованиям,
входящим в определение катализатора. Он ускоряет процесс распада перок-
сида водорода и в то же время после окончания процесса возвращается в
исходное состояние и присутствует в той же концентрации.
Скорость реакции в целом определяется скоростью лимитирующей
стадии последовательных реакций. В данном случае вторая стадия процесса
более медленная. Следовательно, она лимитирует процесс. Поэтому ско-
скорость всего процесса разложения пероксида водорода в присутствии катали-
катализатора К2Сг207 прямо пропорциональна концентрации промежуточного со-
соединения:
г = *[К2Сг209],
где к — константа скорости реакции. Чтобы найти концентрацию промежу-
промежуточного соединения, нужно записать выражение константы равновесия для
первой стадии в виде
К = ^k2i] , (9.4)
[Н2О2]2(С0К-[К2Сг2О9])
где Со к — начальная концентрация катализатора; (Со к - [К2Сг209]) — кон-
концентрация катализатора после установления химического равновесия.
Из уравнения (9.4) можно получить
чОп ] =
29 l K[H0
г- Сл к.
l + K[H202f
9.3. Гомогенный катализ 283
Скорость процесса разложения Н2О2
]22C0K, (9.5)
]2 °К
где [Н2О2] — концентрация неразложившегося пероксида водорода в дан-
данный момент времени; к — константа скорости реакции разложения проме-
промежуточного соединения; С0К — начальная концентрация катализатора.
Из уравнения (9.5) следует, что скорость процесса при гомогенном ка-
катализе прямо пропорциональна первоначальной концентрации катализатора,
а порядок реакции по пероксиду водорода может изменяться от нуля до
двух в зависимости от условий проведения эксперимента. Возможны сле-
следующие предельные случаи.
1.При^[Н2О2]2»1
г — kC0K,
т.е. реакция имеет нулевой порядок по Н2О2.
2. ПриА:[Н2О2]2«1
г = ЩН2О2]2С0К,
т.е. реакция имеет второй порядок по Н2О2.
Автокатализ. Явление ускорения реакции под действием продукта
самой химической реакции называют автокатализом. В этом случае кон-
концентрация катализатора в процессе химического превращения непрерывно
возрастает. Вначале скорость реакции очень низка, затем она резко увели-
увеличивается и лишь на заключительном этапе уменьшается, когда концентра-
концентрация реагентов становится малой. Начальный промежуток времени, в течение
которого реакция протекает с очень малой скоростью, называют индукцион-
индукционным периодом.
Примером автокаталитической реакции может служить реакция йоди-
йодирования ацетона, катализируемая в водных растворах ионами водорода. Ре-
Реакция протекает в три стадии.
На первой стадии в результате присоединения к молекуле ацетона ио-
иона водорода образуется промежуточное соединение:
СН3—СО—СН3 + Н+ ^ СН3 — С+—СН3
ОН
На второй стадии ион водорода отщепляется и промежуточное соеди-
соединение переходит в енольную форму:
284
9. Основы катализа
сн3 — с —сн3
он
сн3 — с=сн2
он
н
На третьей стадии происходит быстрое и необратимое взаимодействие
енольной формы с молекулой йода:
СН3 — С=СН2
ОН
i2 -*¦ сн3 — с —ch2i + н + г
О
Наиболее медленной, определяющей скорость, является первая ста-
стадия. Поэтому скорость реакции пропорциональна концентрациям ацетона и
ионов водорода:
/•=*садсн+.
В ходе реакции непрерывно возрастает концентрация ионов водорода. Сле-
Следовательно, йодирование ацетона происходит с ускорением, т.е. является
автокаталитическим процессом.
Гидролиз сложных эфиров при отсутствии в реакционной смеси ки-
кислоты также является автокаталитической реакцией. Продуктом гидролиза
является органическая кислота, поэтому по мере ее накопления в растворе
скорость реакции будет постепенно возрастать. Автокатализ наблюдается и
для окислительно-восстановительных реакций.
Зависимость скорости автокаталитической реакции от времени пред-
представлена на рис. 9.4. Реакция начинается с очень малой скоростью, но по
мере накопления продукта реакции скорость возрастает и проходит через
максимум. Достигнув максимального зна-
значения, скорость реакции уменьшается за
счет снижения концентрации исходного
реагента.
9.4. Адсорбция
Важное значение в гетерогенных ре-
реакциях играют процессы адсорбции. Ад-
Адсорбцией называют процесс самопроиз-
самопроизвольного увеличения концентрации вещест-
вещества на границе раздела фаз. Адсорбирую-
Адсорбирующееся вещество носит название адсорбата,
Рис. 9.4. Зависимость скорости
автокаталитической реакции от
времени
9.4. Адсорбция 285
адсорбирующее — адсорбента. Процесс, обратный адсорбции, называют
десорбцией.
Адсорбционное равновесие определяется двумя процессами: притя-
притяжением молекул к поверхности под действием межмолекулярных сил и теп-
тепловым движением, стремящимся восстановить равенство концентраций в
поверхностном слое и объеме фазы. Количественно адсорбцию характери-
характеризуют числом молей или массой вещества, накапливающегося на границе
раздела фаз, в расчете на единицу площади поверхности раздела.
Адсорбция является поверхностным процессом, который заключается
во взаимодействии молекул или ионов адсорбата (газа или растворенного
вещества) с поверхностью адсорбента за счет сил Ван-дер-Ваальса, водо-
водородных связей (см. гл. 4), электростатических сил.
Существует два вида адсорбции: адсорбция на твердой поверхности и
адсорбция в поверхностном слое жидкости.
Рассмотрим подробнее адсорбцию на твердой поверхности. Поверх-
Поверхность твердых тел, как и жидкостей, обладает избыточной энергией Гиббса.
Тенденция к уменьшению избыточной поверхностной энергии Гиббса про-
проявляется у твердых тел в способности удерживать на поверхности молекулы
газа или растворенного вещества.
Различают физическую и химическую адсорбцию. Адсорбцию, обу-
обусловленную действием сил Ван-дер-Ваальса, называют физической адсорб-
адсорбцией (физисорбцией). Физическая адсорбция — обратимый экзотермический
процесс (А#а < 0). Это явление можно объяснить следующим образом. Ког-
Когда частица адсорбируется на поверхности, ее поступательное движение ог-
ограничивается, и поэтому процесс сопровождается уменьшением энтропии.
Так как AGa = Д#а - ГА*^ < 0, это значит, что А#а должна быть меньше нуля,
чтобы AGa была отрицательной. Отрицательное изменение энтальпии соот-
соответствует экзотермическому процессу. При повышении температуры ад-
адсорбция уменьшается, а десорбция увеличивается. Значения энтальпии фи-
физической адсорбции невелики и обычно составляют -8... -20 кДж/моль. Фи-
Физическая адсорбция не носит специфического избирательного характера.
При физической адсорбции быстро устанавливается равновесие меж-
между адсорбированными частицами и частицами в газовой фазе. О поверхно-
поверхностной концентрации реагентов можно судить по равновесному распределе-
распределению молекул адсорбата между поверхностью твердого тела и газовой фазой.
Это распределение зависит от давления, температуры, химической природы
веществ, площади поверхности. Равновесное распределение оценивают по
изотермам адсорбции. Простейшим уравнением для изотермы адсорбции
служит уравнение изотермы Ленгмюра.
Вывод уравнения изотермы Ленгмюра основан на следующих допу-
допущениях:
286 9. Основы катализа
поверхность адсорбента однородна;
взаимодействие между адсорбированными молекулами отсутствует;
адсорбция протекает лишь до образования монослоя, т.е. каждый
центр может присоединить только одну частицу (мономолекулярная ад-
адсорбция);
процесс динамичен, и при заданных условиях устанавливается равно-
равновесие между адсорбцией и десорбцией.
При анализе гетерогенных процессов с участием адсорбции и де-
десорбции вместо закона действующих масс применяют закон действующих
поверхностей: скорость адсорбции пропорциональна количеству столкно-
столкновений частиц адсорбата со свободными адсорбционными центрами на по-
поверхности (оно пропорционально давлению газа и числу свободных цен-
центров); скорость десорбции пропорциональна числу занятых адсорбцион-
адсорбционных центров; скорость элементарной реакции между адсорбированными
частицами реагентов пропорциональна произведению долей покрытия по-
поверхности при адсорбции каждым из них в степенях, равных стехиометри-
ческим коэффициентам.
Рассмотрим газ А, находящийся при давлении рА в равновесии с по-
поверхностью:
А(Г) + S (поверхность) <i± AS.
Пусть No — общее количество центров на поверхности; NA — количество
центров, занятых адсорбированными молекулами А. Доля поверхности, за-
занятая адсорбированным газом, 6 = NA/N0. Тогда доля свободной поверхности
равна A -0).
При равновесии скорость адсорбции пропорциональна давлению и
доле свободной поверхности:
r*= kapA(l-Q).
Скорость десорбции зависит от количества адсорбированного газа и
поэтому пропорциональна 0:
гд= ?Д0.
При равновесии скорости адсорбции и десорбции равны:
Отсюда
Разделим числитель и знаменатель дроби на кД и обозначим ka/ka = Ь& (ад-
(адсорбционный коэффициент). Тогда
9.4. Адсорбция
287
е= ЬаРа
1 *
(9.6)
0=1
О р
Рис. 9.5. Изотерма адсорбции Ленг-
мюра
Выражение (9.6) называют уравнением
изотермы Ленгмюра. Оно определяет
долю площади поверхности, покрытой
молекулами в момент равновесия. За-
Зависимость адсорбции от давления газа
характеризуется кривой на рис. 9.5.
Приведем два предельных случая
уравнения изотермы Ленгмюра.
1. Если вещество адсорбируется слабо или давление в системе низкое,
то можно считать, что ЪАрА « 1. При этом уравнение (9.6) переходит в
уравнение изотермы Генри:
0 = ЪАрА. (9.7)
2. При высоких давлениях или при сильной адсорбции ЬАрА » 1, тог-
тогда 0 ~ 1, что отвечает полному заполнению поверхности. При этом адсорб-
адсорбция не зависит от давления (состояние насыщения).
Важное значение имеет ситуация, когда адсорбируются два газа А и В,
в связи с возможной реакцией между двумя веществами на поверхности.
Можно показать, что в этом случае
а _ ЬаРа . (9 8)
(9.9)
Химическая адсорбция (сокращенно хемосорбция) — процесс адсорбции,
который протекает в результате образования химической связи (обьино кова-
лентной). Энергия связи адсорбент — адсорбат достаточно велика и примерно
равна энтальпии образования химических соединений (80...800 кДж/моль).
Хемосорбция характеризуется высокой специфичностью, т.е. для определенно-
определенного адсорбата количество хемосорбированного вещества очень чувствительно к
химической природе адсорбента. Например, оксид углерода (II) удерживается
на поверхности меди и платины сравнительно слабо, а на никеле и палладии
довольно прочно из-за образования поверхностных соединений типа
с=о
NT
где М — атом металла на поверхности.
288 9. Основы катализа
Принципиальное отличие хемосорбции от физической адсорбции за-
заключается в том, что в результате образования более прочных связей хемо-
сорбированное вещество с трудом удаляется с адсорбента, причем
десорбция может сопровождаться химическими превращениями. Например,
при адсорбции кислорода на поверхности угля образуется настолько
прочная связь, что при десорбции в газовую фазу выделяются оксиды СО и
СОг. Это связывают с наличием так называемых «оксидов Шилова»,
образующихся при взаимодействии кислорода с углем:
^с —с=о
>> =с=о уо
=с^ —с=о
Особенностью таких соединений является то, что в них атомы углеро-
углерода сохраняют связь с остальными атомами, образующими кристаллическую
решетку угля. Между атомами углерода и атомами кислорода, образующи-
образующими поверхностные соединения, нельзя установить стехиометрического со-
соотношения. Невозможно провести физическую границу раздела между объ-
объемом твердого адсорбента и возникшим химическим соединением. Такое
химическое соединение не образует самостоятельной фазы, и его нельзя по-
получить в свободном виде.
Экспериментально установлено, что хемосорбция на оксидных метал-
металлических катализаторах (к ним относится большая часть реальных катализа-
катализаторов) является процессом, идущим во времени с измеримой скоростью.
Иногда состояние насыщения поверхности при данных температуре и дав-
давлении достигается в течение многих часов и даже дней. Замедленность ре-
реальных процессов адсорбции объясняют представлением об активированной
адсорбции, требующей, как и химические реакции, некоторой энергии акти-
активации. Поэтому хемосорбция с измеримой скоростью может осуществляться
в определенном температурном интервале. Гипотеза об активированной ад-
адсорбции позволила дать удовлетворительное объяснение многим аномали-
аномалиям, установленным при изучении процессов адсорбции. Например, теплоты
(энтальпии) адсорбции часто малы при низких температурах и имеют значи-
значительную величину при высоких температурах. Это обусловлено тем, что при
низких температурах преобладает физическая адсорбция, имеющая нулевую
энергию активации. При высоких температурах преобладает хемосорбция, ха-
характеризующаяся большим тепловым эффектом. Например, теплота (энталь-
(энтальпия) физической адсорбции водорода на вольфраме составляет -8,3 кДж/моль,
а теплота хемосорбции равна -154 кДж/моль.
Энергетическая диаграмма адсорбционного процесса (рис. 9.6) пред-
представляет собой зависимость энергии Е взаимодействия частиц адсорбата и
адсорбента от координаты процесса х (расстояния от поверхности адсор-
9.5. Гетерогенный катализ
289
бента). При больших значениях х опреде-
определяющую роль играют силы межмолеку-
межмолекулярного взаимодействия, происходит фи-
физическая адсорбция (АЯф), которая всегда
протекает с нулевой энергией активации.
При дальнейшем сближении частиц ад-
сорбата и адсорбента начинают преобла-
преобладать силы химического взаимодействия с
образованием химических связей, т.е. имеет
место хемосорбция (АЯХ). Подобно хими-
химической реакции хемосорбция (в некото-
некоторых ее формах) может осуществляться со Рис 9.6. Энергетическая диаграм-
значительной энергией активации (Ех). ма адсорбционного процесса
Если энергия активации равна нулю, то хемосорбцию называют неактиви-
неактивированной (например, адсорбция многих газов на очень чистых металличе-
металлических поверхностях). При наличии активационного энергетического барьера
хемосорбцию называют активированной (например, на загрязненной приме-
примесями поверхности металла). Такая активированная адсорбция будет проте-
протекать с заметной скоростью, но только выше определенной минимальной
температуры.
Десорбция хемосорбированных частиц всегда является активацион-
ным процессом (?д — энергия активации десорбции), так как частицам не-
необходимо подняться со дна «потенциальной ямы» хемосорбции.
Хемосорбция играет важную роль при протекании гетерогенных реак-
реакций, которые имеют сложный многостадийный механизм, включающий хе-
мосорбционные процессы.
9.5. Гетерогенный катализ
В гетерогенном катализе реагирующие вещества и катализатор нахо-
находятся в разных агрегатных состояниях. Наиболее часто катализатор пред-
представляет собой твердое вещество, а реагенты являются газами или жидко-
жидкостями. Реакция протекает на поверхности катализатора. Следовательно,
свойства поверхности (площадь, химический состав поверхностного слоя,
его структура) существенно влияют на активность катализатора.
Гетерогенные каталитические процессы очень сложны. Если катали-
катализатор имеет достаточно развитую поверхность, то можно выделить семь ста-
стадий каталитического процесса:
1) диффузия реагирующих веществ к поверхности катализатора
(внешняя диффузия);
10 - 9795
290
9. Основы катализа
О
R
Рис. 9.7. Условные энергетические
диаграммы реакций с гетерогенным
катализатором 2 и без него 7:
Еа и ?ак — энергии активации некатали-
некаталитической и каталитической реакций;
ЛДщс — энтальпия адсорбции, Д#д —
энтальпия десорбции
2) диффузия в поры катализатора
(внутренняя диффузия);
3) адсорбция реагентов на поверх-
поверхности катализатора;
4) химическое превращение на
поверхности катализатора (которое мо-
может происходить в несколько стадий);
5) десорбция продуктов с поверх-
поверхности катализатора;
6) диффузия продуктов реакции в
порах к внешней поверхности гранул
катализатора;
7) диффузия продуктов от внеш-
внешней поверхности катализатора.
В гетерогенном катализе значи-
значительную роль играет адсорбция, так
как каталитическая реакция протекает
в поверхностном слое. Адсорбирован-
Адсорбированные молекулы определенным образом
ориентированы к поверхности, причем уменьшение энергии активации
происходит за счет перераспределения связей и повышения энтропии ак-
активации.
На рис. 9.7 изображены энергетические диаграммы реакции, прохо-
проходящей с участием гетерогенного катализатора и без него. От аналогичной
энергетической диаграммы с участием гомогенного катализатора данная
диаграмма отличается наличием энтальпий адсорбции и десорбции.
Очень сильное адсорбционное взаимодействие может препятствовать
прохождению химического процесса. Молекулы продуктов должны быть
оторваны (десорбированы) от поверхности. На это затрачивается энергия
(энтальпия десорбции Д#д > 0). Слишком большое значение энтальпии
десорбции также может затруднять протекание процесса. Только некото-
некоторое оптимальное сочетание энергии активации, энтальпии адсорбции и
десорбции будет способствовать ускорению каталитического процесса.
В настоящее время нет единой теории катализа. Для объяснения
механизма гетерогенных каталитических реакций предложено несколько
теоретических подходов: мультиплетная теория катализа Баландина, тео-
теория активных ансамблей Кобозева, электронная теория катализа Воль-
кенштейна. Общим для этих теорий является представление о существова-
существовании активных центров — активных участков на поверхности катализатора.
Различие заключается в трактовке природы поверхностных соединений и
природы активных центров поверхности катализатора, участвующих в обра-
9.5. Гетерогенный катализ 291
зовании поверхностных соединений. Рассмотрение основных положений
этих теорий выходит за рамки данной книги.
Кинетика гетерогенных каталитических реакций. Для гетероген-
гетерогенных каталитических процессов роль одного из реагентов играют адсорбци-
адсорбционные центры твердой поверхности, которые следует включать в соответст-
соответствующие уравнения.
Рассмотрим элементарную химическую реакцию между адсорбиро-
адсорбированными частицами:
где А и В — реагенты; D и F — продукты; vA, vB, vD, vF — соответствующие
стехиометрические коэффициенты. Если продукты реакции занимают на
поверхности больше адсорбционных центров, чем исходные вещества на
величину Аи, то превращение в адсорбционном слое окажется возможным
при наличии недостающего числа свободных центров Q адсорбции. Поэто-
Поэтому стехиометрическое уравнение для химической реакции, протекающей в
адсорбционном слое, можно записать в виде
+ УВВадс + AnQ = VDDaw + УрРадс- (9.10)
Если процесс (9.10) протекает в одну стадию, то скорость его в прямом на-
направлении
гх = ^еААеввеАл (An > о) (9.11)
а для обратной реакции
^^-№8?. (9Л2)
Если в элементарном акте принимают участие молекулы вещества А,
не занимающие адсорбционных центров на поверхности катализатора (т.е.
поступающие из газовой фазы), то уравнение реакции принимает вид
А(г) + УвВадс + AnQ = УООадс +
а выражение для скорости реакции — вид
l*. (9.13)
Уравнения (9.10)—(9.13) относятся к элементарным процессам. Кине-
Кинетическое уравнение для процесса в целом должно выражать скорость реак-
реакции через измеряемые в опыте величины — парциальные давления веществ,
участвующих в реакции. Для составления таких уравнений необходимо
знать (или постулировать) механизм реакции и относительные скорости от-
отдельных процессов. Несмотря на то что уравнению изотермы Ленгмюра
ю*
292 9. Основы катализа
подчиняются немногие системы, оно полезно при определении формального
кинетического порядка реакции. В основе метода определения порядка гетеро-
гетерогенных каталитических реакций лежит закон действующих поверхностей.
Рассмотрим простейший случай элементарного химического превра-
превращения адсорбированных молекул вещества А:
Аадс -> Вадс + Dw (Аи = 0).
Считая, что адсорбция подчиняется уравнению изотермы Ленгмюра, урав-
уравнение для скорости реакции можно записать в виде
Существенное отличие гетерогенного процесса от гомогенного состоит в
том, что для данной элементарной реакции порядок и эффективная энергия
активации зависят от адсорбционной способности всех присутствующих в
системе веществ.
Предположим, что вещества В и D адсорбируются слабо, тогда Ьврв « 1
и 6qPd « 1 • При этом возможны следующие случаи.
1. Вещество А адсорбируется слабо, т.е. ЬАрА « 1. Уравнение для
скорости принимает вид
r = kbApA=k'pA. (9.14)
Реакция следует кинетике первого порядка. Уравнение (9.14) выполняется
для следующих реакций каталитического разложения:
2N2O Au >2N2+O2
2HI-
2. Вещество А адсорбируется со средней силой, тогда
Здесь показатель степени т обычно меньше единицы, что приводит к кине-
кинетике дробного порядка. Так как адсорбционный коэффициент зависит от
температуры, то порядок реакции также зависит от температуры. Например,
для реакции
2SbH3
9.5. Гетерогенный катализ 293
т = 0,6 при Т = 298 К. Вследствие уменьшения адсорбции при повышении
температуры m -» 1.
3. Вещество А адсорбируется сильно, тогда Ь^р^ » 1. В этих услови-
условиях доля поверхности, занятая реагентом, близка к единице, и кинетическое
уравнение имеет вид
r = k.
Реакция подчиняется кинетике нулевого порядка. Например, нулевой
порядок наблюдается при разложении аммиака на вольфраме при давлени-
давлениях, превышающих 2666 Па B0 Торр):
2NH3 W >N2+3H2
Рассмотренные здесь простейшие примеры наглядно показывают, как
разнообразны могут быть кинетические закономерности гетерогенных ката-
каталитических реакций.
¦ Практические занятия
Кинетика химических реакций
Примеры решения задач
Задача 1. Константа скорости реакции первого порядка в газовой фазе
С2Н6 -> С2Н4+Н2
при Т = 856 К равна к = 4,83 • КГ4 с. Определить, какой процент этана
разложится при температуре Т= 856 К за время t = 120 мин.
Решение. Реакция первого порядка подчиняется уравнению
С
которое в экспоненциальной форме имеет вид
С = С0 exp(-fe).
Количество распавшегося этана
х = С0-С.
Подставляя в уравнение С = С о - х, получают
С0~х = С0 exp(-fo), х = С0-С0 exp(-fo), х = Со A - exp(-fa))-
Доля распавшегося этана
у = — = l-exp(-fo),
у = 1 - ех0(-4,83 • 10 • 120 • 60) * 1 - ехр(-3,478)« 0,969.
Следовательно, за 120 мин разложится 96,9 % этана.
Задача 2. Реакция превращения вещества А протекает по следующей схеме:
ЗА -> 2В + D
и описывается кинетическим уравнением реакции 2-го порядка с констан-
константой скорости, равной 9,3 • 10~4 л/(моль • с). Исходная концентрация вещества
А Со а = 0,1 моль/л. За какое время прореагирует 30 % исходного количества
вещества А?
Решение. В соответствии с уравнением
Примеры решения задач 295
1 1
для нахождения времени необходимо предварительно определить текущую концен-
концентрацию вещества А (СА). По условию задачи, СА = 0,7С0а- Следовательно,
1 1 ,_ 0,3 0,3 з
0,7С0А С0А ' 0,1С0Ак
Задача 3. Для элементарной реакции А + В = D + F при начальных концентрациях
реагентов С0А = СОв = 0,6 моль/л через 20 мин после начала реакции концентрация
вещества А уменьшилась до значения С]А = 0,4 моль/л. Определить концентрацию
вещества А через 60 мин после начала реакции.
Решение. Поскольку данная реакция элементарная, то это реакция второго порядка.
При равных начальных концентрациях реагентов для реакции второго порядка ре-
решение дифференциального уравнения
Л
приводит к следующему результату:
1 1
Отсюда константа скорости и текущая концентрация равны
К , Од
1 Ч)АиА 1 + и0А^
По заданным условиям вычисляют константу скорости
л
20 0,6-0,4 моль-мин
Далее находят концентрацию вещества А через 60 мин после начала реакции:
С2А= ^ = 0.24-!™.
1 + 0,6-0,0417-60 л
Задача 4. Используя приближенное правило Вант-Гоффа, вычислить, на сколько
градусов нужно повысить температуру, чтобы скорость реакции возросла в 80 раз?
Температурный коэффициент скорости реакции равен 3.
Решение. Используя уравнение
296 Кинетика химических реакций
можно выразить из него отношение скоростей и прологарифмировать обе его части:
10
Т2{, ^39,9,
lgy ig3
Таким образом, чтобы скорость реакции возросла в 80 раз, необходимо повы-
повысить температуру примерно на 39,9°.
Задача 5. Воспользовавшись приближенным правилом Вант-Гоффа, вычислить,
при какой температуре некоторая реакция закончится за 25 мин, если при Т = 20 °С
на это требуется 2 ч. Температурный коэффициент скорости реакции равен 3.
Решение. Между константами скоростей и временем завершения реакции существу-
существует обратно пропорциональная зависимость
где /]И/2 — время завершения реакции при температуре Т\ и Т2 соответственно.
Тогда уравнение для логарифма отношения t\lt2 (см. задачу 3) можно запи-
записать так:
t2 10
Отсюда
120
101g-
2 J-1 — > 2 — 1 — т z<u = J4,j \^.
lgy lgy lg3
Задача 6. Для одной из реакций первого порядка были определены две константы
скорости: кх = 0,00670 с'1 при Г, = 443 °С и к2 = 0,06857 с при Т2 = 497 °С. Опреде-
Определить константу скорости этой же реакции при температуре Т= 508 °С.
Решение. По двум значения констант скорости реакции, используя уравнение G.17),
определяют энергию активации реакции
Е= ^-, 7]=716К, Г2=770К,
Т Т
Т —Т
12 1\
Ел = 0'00670 а 197400 Дж/моль.
770-716
Задачи для самостоятельного решения 297
Аналогичное выражение для энергии активации записывают через Т2 = 770 К и Г3 =
= 781 К E08 °С) и выражают логарифм отношения констант скоростей:
а Т -Т
Таким образом,
197400-11 ^ =
к
к2 8,314-770-781 к2
А:3 = А:2 • 1,54 = 0,06857 • 1,54 = 0,10560 с.
Задача 7. Определить порядок гетерогенной реакции А(т) + Т(г) —> С(г), если из-
известно, что при увеличении концентрации вещества Т в 3 раза скорость реакции
возросла в 5,2 раза.
Решение. Так как реакция гетерогенная, то в кинетическое уравнение не будет вхо-
входить концентрация твердого вещества, и оно будет иметь следующий вид:
Концентрацию вещества Т для первого случая обозначают через Cj\ = х, тогда по-
после увеличения концентрации она станет равной СТ2 = Зле. Подставляя эти концен-
концентрации в кинетическое уравнение, записывают выражение для отношения скоростей
г,-fa"; r2=*C*)";^^
гх кх
Логарифмируя это выражение, находят порядок реакции п:
0.716 , .
= 1,5.
g g,
г, Ig3 Ig3 0,447
В данном случае частный порядок реакции по веществу Т и общий порядок
реакции совпадают.
Задачи для самостоятельного решения
1-8. Для данной химической реакции при данных температуре, порядке п реакции,
начальных концентрациях Со реагентов, времени tm полупревращения определите
время, за которое прореагирует указанная доля у исходного вещества, %.
298
Кинетика химических реакций
№
1
2
3
4
5
6
7
8
Реакция
SO2C12 -> S02 + Cl2
2NH3 -> N2 + ЗН2
RBr + ОН" -> ROH + Вг"
С2Нб —^ С/2Г14 ^~ Н2
Н2О2-> Н2О + 1/2О2
С2Н5С1-^С2Н4+НС1
нсоон -> со2 + н2
НВг + О2 -> НО2 + Вг
п
1
0
2
1
1
1
1
2
Г, К
593
1129
293
823
293
873
413
700
/1/2, МИН
577,6
1035,0
78,25
462,0
13,6
8,7
21,0
0,03
Со, моль/л
0,6
0,2
0,1
0,3
0,3
0,5
0,2
. 0,1
у,%
60
30
60
90
99
96
90
99
9. Константа скорости реакции первого порядка в газовой фазе
SO2C12 -> SO2 + Cl2
при Т= 320 °С равна * = 2 • 10 с. Какой процент SO2C12 распадается при Т= 320 °С
в течение 90 мин?
10. Разложение хлорида фенилдиазония в водном растворе при Т= 50 °С протекает
как реакция первого порядка:
C6H5N2Cr +H2O ->C6H5OH + НС1 + N2
Константа скорости при этой температуре k = 0,0689 мин. Определите концентра-
концентрацию хлорида фенилдиазония в растворе через 60 мин после начала реакции, если
его начальная концентрация Со = 0,2 моль/л.
11. Реакция разложения аммиака на вольфрамовой проволоке при Г= 856 °С под-
подчиняется кинетике нулевого порядка:
2NH3 -> N2 + ЗН2
Константа скорости реакции к = 1,61 • 10 моль/(л • с). Определите период полу-
полупревращения этой реакции, если начальная концентрация аммиака Со= 0,2 моль/л.
12. Реакция нейтрализации нитроэтана в водном растворе описывается кинетиче-
кинетическим уравнением
dC
ОН"
dt
dt
"-^сдаю^он-
При Т= 0 °С и начальных концентрациях обоих реагентов 0,01 моль/л время полупре-
полупревращения составляет / = 150 с. Вычислить константу скорости А: реакции при Т= 0 °С.
13. Гидролиз алкилбромида в водно-спиртовом растворе протекает по схеме
RBr + ОН" -> ROH + Вг"
Это реакция второго порядка. Константа скорости гидролиза при Т = 20 °С равна
к = 2,13 • 10~3 л/(моль • с). Определите период полупревращения реакции, если на-
начальные концентрации обоих реагентов равны 0,1 моль/л.
Задачи для самостоятельного решения
299
14. Реакция триэтиламина с метилйодидом
(C2H5KN +CH3I -> CH3(C2H5KNI
в растворе четыреххлористого углерода - реакция второго порядка. Константа ско-
скорости при Т = 20 °С равна к = 2,48 - 10~2 л/(моль • с). Вычислите период полупре-
полупревращения при начальных концентрациях обоих реагентов 0,224 моль/л.
15-24. Для данной химической реакции п-то порядка известно, что при начальных
концентрациях реагентов Со (моль/л) при некоторой температуре за время t\ кон-
концентрация исходного вещества стала С\. Определите концентрацию исходных ве-
веществ С2 через время t2 после начала реакции.
№
15
16
17
18
19
20
21
22
23
24
Реакция
2NH3 -» N2 + ЗН2
A + B->D + F
Н2О2->Н2О+1/2О2
НСООН -> СО2 + Н2
С2Н5С1->С2Н4+НС1
SO2C12 -> SO2 + Cl2
С2Нб —> С2Н4 + Н2
А->В +D
2NO2->2NO + O2
2NOBr -> 2NO + Br2
Порядок
реакции, п
0
2
1
1
1
2
2
моль /л
0,1
0,5
0,4
0,2
0,4
0,4
0,5
0,2
0,4
0,2
/|,МИН
300
120
13,6
1,25
15
200
50
300
200
0,1
моль /л
0,071
0,215
0,2
0,1
0,2
0,3
0,4
0,08
0,15
0,05
t2, мин
500
180
80
3
30
600
150
600
400
0,5
25. Формальдегид, реагируя с пероксидом водорода, образует муравьиную кислоту.
Реакция протекает по уравнению НСНО + Н2О2 —> НСООН + Н2О и является реак-
реакцией второго порядка. Если смешать равные объемы одномолярных растворов Н2О2
и НСНО, то через 2 ч при Т = 60 °С концентрация муравьиной кислоты становится
равной 0,215 моль/л. Вычислите константу скорости реакции и определите, за какой
промежуток времени прореагирует 90 % исходных веществ.
26. Определите константу скорости элементарной газофазной реакции А + В -> D + F,
если при начальных концентрациях исходных веществ, равных по 0,04 моль/л, через
1 ч образовалось 0,01 моль/л продукта D.
27. Реакция газообразного аммиака с диоксидом азота на начальной стадии подчи-
подчиняется кинетическому уравнению второго порядка. Вычислите энергию активации и
предэкспоненциальный множитель. Константы скорости при Т= 600 К и Т= 716 К
равны к\ = 0,385 л/(моль • с) и к2 = 16,0 л/(моль • с) соответственно.
28. Константы скорости реакции второго порядка
А + В ->D
при Т= 298,2 К и Т= 328,2 К равны кх = 0,001 и к2 = 0,01 мин • моль • л соответ-
3 00 Кинетика химических реакций
ственно. Вычислите скорость этой реакции при Т = 343,2 К в начальный момент
реакции, если концентрации обоих веществ одинаковы и равны 0,01 моль/л.
29. Константы скорости реакции второго порядка при Т= 282,6 К и Т= 287,6 К рав-
равны к\ = 2,37 и к2 = 3,20 мин • моль • л соответственно. Вычислите скорость этой
реакции при Т = 300 К в начальный момент времени, если концентрации обоих ве-
веществ одинаковы и равны 0,01 моль/л.
30. Термическое разложение этана — реакция первого порядка. Вычислите энергию
активации и предэкспоненциальный множитель, если константа скорости при тем-
температуре Г, = 823 К равна кх = 2,5 • 10 с, а при Т2 = 863 К равна к2 = 23,1 • 10 с.
31. Константа скорости реакции второго порядка
2HI -> Н2 + h
при Тх = 647 К равна к\ = 8,96 • 10 л /(моль ¦ с), а при Т2 = 700 К равна к2 =
= 1,21 • 10 л/(моль • с). Вычислите энергию активации и предэкспоненциальный
множитель. Рассчитайте значение константы скорости к3 при температуре Т3 = 680 К.
32. Константа скорости реакции разложения диоксида азота NO2 при Т = 592 К рав-
равна *, = = 5,22 • 10~5 л/(моль • с), а при Т2 = 627 К — к 2 = 17,00 • 10 л/(моль • с). Вы-
Вычислите энергию активации.
33. В реакции первого порядка время, необходимое для уменьшения начальной
концентрации наполовину, при Т = 325 К составляет / = 5000 с, а при Т= 335 К рав-
равно t= 1000 с. Вычислите константы скорости при обеих температурах и определите
энергию активации.
34. Константа скорости реакции третьего порядка
2NO +2Н2 -> N2+ 2H2O
при Т = 1000 К равна к = 0,4 ^/(моль2 • с), а энергия активации — Ел= 184 кДж/моль.
Рассчитайте константу скорости при температуре Т= 973 К.
35. Константа скорости мономолекулярной реакции при Т= 404 К равна 4,14 • 10 с.
Известно, что энергия активации равна 108 кДж/моль. Вычислите значение кон-
константы скорости при Т = 440 К и определите температурный коэффициент скорости
реакции.
36. Некаталитическое разложение ацетальдегида СН3СНО имеет энергию активации
198 кДж/моль. Катализируемое йодом разложение ацетальдегида имеет энергию
активации 134 кДж/моль. Оцените, во сколько раз больше константа скорости ка-
каталитической реакции при Т = 800 К, если предположить, что предэкспо-
ненциальные множители этих реакций равны.
37. При изучении кинетики распада диэтилпероксида получены следующие кон-
константы скорости реакции при различной температуре:
Задачи для самостоятельного решения 301
Г,°С 140,0 147,8 156,5 184,5
к, с 1,75 • 10~4 3,6 • 10 6,67 • 10'4 7,88 • 10
Определите графически энергию активации и предэкспоненциальный множитель.
38. Реакцию окисления бромоводорода, протекающую по уравнению НВг + Ог -»
—> НО2 + Вг исследовали при различных значениях температуры. При этом была
получена следующая зависимость константы скорости реакции от температуры:
Г, К 700 762 800
к, л/(моль • с) 5,1 46,2 151,0
Определите графически энергию активации.
39. Константы скорости щелочного гидролиза этилйодида в температурном интер-
интервале 20.. .80 °С следующие:
Ы03,л/(моль-с) 0,100 0,335 1,41 3,06 8,13 21,1 50,1
Г,°С 20 30 40 50 60 70 80
Определите графически энергию активации.
40. Для некоторой реакции первого порядка период полупревращения при Т — 378,5 К
равен 363 мин. Энергия активации 217,36 кДж/моль. Определите, сколько времени
потребуется для разложения 75 % исходного вещества при Т= 450 К.
41. Разложение оксида азота (I) на поверхности золота при высоких температурах
протекает по уравнению N2O = N2 + О. Константа скорости данной реакции при
Т= 900 °С равна к = 5 • 10 с. Начальная концентрация N2O равна 3,2 моль/л. Оп-
Определите скорость реакции при указанной температуре в начальный момент време-
времени и за какое время произойдет разложение 78 % начального количества оксида.
42. Элементарная реакция между веществами А и В протекает по уравнению А + 2В = С.
Начальная концентрация вещества А равна Со а = 1,5 моль/л, а В — Сов = 3 моль/л.
Константа скорости реакции к = 0,4. Вычислите скорость химической реакции в
начальный момент времени и по истечении некоторого времени, когда прореагиру-
прореагирует 75 % вещества А. Укажите размерность константы скорости, если время измеряет-
измеряется в минутах.
43. Разложение пероксида водорода в водном растворе описывается кинетическим
уравнением первого порядка. Константа скорости этой реакции к = 0,051 мин.
Определите время, за которое пероксид водорода распадается на 50 и 99 %.
44. Константа распада радия А равна к = 3,79 • 10~3 с'1. Определите период полу-
полупревращения /j/2 и время, за которое радий А распадается на 90 %.
45. Через какое время активность актиния В составит 40 % от первоначальной, если
период полупревращения его t = 36,1 мин?
3 02 Кинетика химических реакций
46. Сколько граммов муравьиной кислоты образуется через 3 ч после начала реак-
реакции, если смешать 1лШ раствора муравьиного альдегида с 1 л 1М раствора Н2О2?
Константа скорости реакции второго порядка НСОН + Н2О2 = НСООН + Н2О при
Т= 60 °С равна k = 0,75 (концентрация выражена в молях на литр; время — в часах).
47. Константа скорости реакции второго порядка этилового эфира уксусной кисло-
кислоты с едким натром при Т— 10 °С равна к = 2,38 (концентрация выражена в молях на
литр, время — в минутах). Какое время потребуется на омыление 80 % эфира, если
смешать при температуре 10 °С 1 л 0,04М раствора уксусноэтилового эфира с 1 л
0,04М раствора NaOH? Как изменится время реакции, если исходные растворы бу-
будут разбавлены в 10 раз?
48. Вычислите, на сколько градусов необходимо повысить температуру, чтобы ско-
скорость реакции возросла в 50 и в 100 раз, если температурный коэффициент скорости
реакции равен 3.
49. Температурный коэффициент реакции разложения йодистого водорода в интер-
интервале температур 356...374 °С равен 2. Используя приближенное правило Вант-
Гоффа, вычислите константу скорости этой реакции при Т = 374 °С, если при
Т= 356 °С она равна к = 8,09 • 10~5 л/(моль • с).
50. Муравьиная кислота разлагается на диоксид углерода и водород на поверхности
золота. Константа скорости этой реакции при Т= 140 °С равна к\ = 5,5 • 10 с, а
при Т- 185 °С — к2 = 9,2 • 10~3 с. Вычислите температурный коэффициент скоро-
скорости реакции в указанном интервале температур.
51. При Т= 150 °С некоторая реакция заканчивается за t = 16 мин. Приняв темпера-
температурный коэффициент скорости 2,5, вычислите, через сколько минут закончилась бы
эта же реакция при Т= 180 и 130 °С.
52. Воспользовавшись приближенным правилом Вант-Гоффа, вычислите, при какой
температуре реакция закончится в течение t = 20 мин, если при Т = 20 °С на это тре-
требуется 3 ч. Температурный коэффициент скорости реакции принять равным 2.
53. Во сколько раз увеличится время, необходимое для завершения реакции, если
понизить температуру на 45°? Температурный коэффициент скорости реакции при-
принять равным 3.
54. Константа скорости омыления этилацетата едким натром при Г = 9,4 °С равна
к\ = 2,37 л/(моль • мин), а при 14,4 °С — к2 = 3,204 л/(моль • мин). Вычислите по
уравнению Аррениуса, при какой температуре константа скорости будет равна к2 =
= 15 л/(моль • мин).
55. Для реакции разложения паров уксусного альдегида (порядок реакции п = 1,5)
константа скорости при Т = 460 °С равна к\ = 0,035, а при Т = 518 °С — к2 = 0,343
(концентрация выражена в моль/л, а время — в секундах). Определите константу
скорости этой реакции при Т= 486 °С. Сравните полученный результат с результа-
результатом, найденным с использованием приближенного правила Вант-Гоффа.
Задачи для самостоятельного решения 303
56. При Т= 25 и 40 °С константы скорости реакции разложения гипохлорита натрия
в растворе равны к\ = 0,0093 с и к2 = 0,0342 с соответственно. Вычислите по
уравнению Аррениуса константу скорости этой реакции при 50 °С.
57. Термическое разложение оксида этилена является реакцией первого порядка.
При температуре Т = 378,5 °С период полупревращения оксида этилена равен
t = 363 мин. Определите константу скорости реакции при Г = 450 °С. Энергия акти-
активации данной реакции Еа = 217 кДж/моль.
58. Вещество А в водном растворе разлагается по уравнению первого порядка. Кон-
Константа скорости реакции при Т= 24,7 и 30 °С равна к\ = 9 • 10~3 и к2 = 1,3 • 10~2 мин
соответственно. Вычислите константу скорости реакции при Т = 35 °С и время, в
течение которого при этой температуре распадется 99 % вещества А.
59. В реакции первого порядка энергия активации равна Еа = 104,5 кДж/моль и пред-
экспоненциальный множитель в уравнении Аррениуса равен к0 = 5 • 1013 с . Опре-
Определите, при какой температуре период полупревращения равен: 1) 1 мин; 2) 30 суток.
60. Протекают две реакции. Энергия активации первой из них на 9 кДж/моль мень-
меньше, чем второй. Определите, во сколько раз скорость первой реакции больше скоро-
скорости второй при Т = 100 и 700 °С, если предэкспоненциальные множители их одина-
одинаковы.
61. Энергия активации реакции разложения хлористого этила (C2H5CI) в газовой
фазе Ег\ =237 кДж/моль, а энергия активации реакции разложения бромистого эти-
этила (C2H5Br) Е& = 225 кДж/моль. Определите, во сколько раз быстрее протекает раз-
разложение бромистого этила, чем хлористого этила при температуре Т = 70 °С. Счи-
Считать, что предэкспоненциальные множители этих реакций одинаковы.
62. Реакция разложения хлористого этила является реакцией первого порядка:
С2Н5С1 ->С2Н4
Энергия активации этой реакции равна Ей =237 кДж/моль, предэкспоненциальный
множитель к0 = Ю13'5 с. Рассчитайте константу скорости этой реакции при Т= 600 °С
и определите период полупревращения при этой температуре.
63. Напишите кинетическое уравнение для гетерогенной реакции М(т) + F(r) -^- Р(г),
если известно, что при увеличении концентрации вещества F в 2 раза скорость ре-
реакции возрастает в 1,4 раза.
64. Рассчитайте скорость гетерогенной реакции А(т) + D(r)—» ЩГ), если известно, что об-
общий порядок реакции п = 1,5, константа скорости к = 0,3, концентрация CD = 0,4 моль/л.
Укажите размерность константы скорости, если время измеряется в минутах.
65. Рассчитайте скорость гетерогенной реакции В(т) + N(r) -> C(r)+ D(r), если извест-
известно, что порядок реакции равен п = 0,5, константа скорости к = 0,02, концентрация
Cn = 0,3 моль/л. Укажите размерность константы скорости, если время измеряется в
минутах.
3 04 Кинетика химических реакций
66. Определите, как изменится скорость гетерогенной реакции второго порядка
Mg(T) + 2НС1(р) = MgCl2(p) + Н2(г)
если концентрацию НС1 увеличить в 4 раза?
67. Определите, как изменится скорость гетерогенной реакции
А(т)+ В(Р) -> С(р> + D(P),
если начальную концентрацию вещества В увеличить в 4 раза. Порядок реакции п = 1,5.
Укажите размерность константы скорости, если время измеряется в минутах.
68. Определите порядок гетерогенной реакции А(т) + В(г) -» Р(г), если известно,
что при увеличении концентрации вещества В в 3 раза скорость реакции возрас-
возрастает в 9 раз.
69-71. Энергия активации реакции 2SO2 + О2 = 2SO3 при температуре Т = 450 °С
равна Еа = 280 кДж/моль. При применении данного катализатора энергия актива-
активации уменьшается до указанного значения. Оцените, во сколько раз увеличивается
константа скорости каталитической реакции при Т = 450 °С, если предположить,
что предэкспоненциальные множители каталитической и некаталитической реакций
равны.
№
69
70
71
Катализатор
Pt
v2o5
Fe2O3
Энергия активации, кДж/моль
68
92
160
72. Некаталитическое разложение ацетальдегида при Т= 800 К по уравнению:
СНзСНО = СН4 + СО
имеет энергию активации Еа\ = 190,4 кДж / моль. Применение йода в качестве ка-
катализатора уменьшает энергию активации до значения Еа2 = 136,0 кДж / моль. Оце-
Оцените температуру, при которой скорость некаталитической реакции станет равна
скорости каталитической реакции при температуре Т = 800 К, если предположить,
что предэкспоненциальные множители каталитической и некаталитической реакций
равны.
73. Константа скорости жидкофазной реакции второго порядка А + В = D + F при
Т= 25 °С равна к\ = 3,5 • 10~8 л/(моль • с), а ее энергия активации Еа\ = 180 кДж/моль.
В присутствии катализатора константа скорости &2 = 1,6 • 10~2 л/(моль • с). Оцените
энергию активации каталитической реакции в предположении, что предэкспонен-
предэкспоненциальные множители каталитической и некаталитической реакций равны.
74. Определите время, за которое прореагирует 90 % вещества А, разлагающегося
по реакции первого порядка А —> В + D, в отсутствии и при наличии катализатора,
ЛР 5. Кинетика гомогенных химических реакций 3 05
если известно, что время полупревращения некаталитическои реакции составляет
(* 1/2I = 30 мин, а каталитической — (/1/2J = 4 мин.
75. Константа скорости жидкофазной реакции второго порядка А + В = D + F при
Т = 25 °С равна кх = 3,2 • 10" 5 л/(моль • с). В присутствии катализатора константа
скорости равна к2 = 1,6 • 10"{ л/(моль • с). Начальные концентрации равны С0А = СОв =
= 0,5 моль/л. Определите время, в течение которого начальная концентрация
уменьшится до С = 0,2 моль/л для некаталитической и каталитической реакций.
ЛР 5. Кинетика гомогенных химических реакций
Цель работы — знакомство с основными закономерностями протекания го-
гомогенных химических реакций и факторами, влияющими на их скорость, а также
определение их некоторых характеристик. У таких реакций реагенты и продукты
находятся в одной фазе, чаще всего, в жидкой или газовой. Скорость гомогенных
реакций определяется природой реагирующих веществ и зависит, главным образом,
от условий проведения: температуры, концентрации реагентов, давления и др.
Опыт 1. Зависимость скорости химической реакции от концентрации
реагирующих веществ. В данном опыте изучается влияние концентрации реаген-
реагентов на скорость гомогенной реакции, протекающей в растворе между тиосульфатом
натрия и серной кислотой:
Na2S2O3 + H2SO4 = Si + SO2T + Н2О + Na2SO4
Данная реакция протекает в две стадии. На первой стадии быстро образуется тио-
серная кислота H2S2O3:
Na2S2O3 + H2SO4 = H2S2O3 + Na2SO4
На второй стадии тиосерная кислота медленно разлагается с образованием свобод-
свободной серы:
H2S2O3 = Si + SO2T + Н2О
Таким образом, лимитирующей стадией всего процесса является вторая ста-
стадия реакции.
Гомогенная реакция сопровождается выделением коллоидной серы, что про-
проявляется в помутнении раствора. Момент исчезновения из поля зрения линий на
бумаге соответствует выделению определенного количества серы, одинакового во
всех экспериментах. Так как степень мутности зависит от толщины слоя раствора,
то все опыты следует проводить в одном и том же стакане, чтобы толщина слоя бы-
была одной и той же во всех опытах.
Началом реакции является момент сливания (смешения) растворов, услов-
условным окончанием реакции — выделение одного и того же количества серы (когда
становятся невидимыми линии на подложенной под стакан бумаге). Время, необхо-
необходимое для выделения этого количества серы, обратно пропорционально средней
скорости гср реакции.
306
Кинетика химических реакций
Последовательность проведения:
1) ополосните два стакана небольшим количеством дистиллированной воды;
2) налейте в один стакан 10 мл 0,05М раствора серной кислоты (Vu so ) с помощью
соответствующего цилиндра (см. этикетки на цилиндрах и стаканах). В другой ста-
стакан налейте 10 мл 0,ЗМ раствора тиосульфата натрия (FNaSO ) и 20 мл дистилли-
дистиллированной воды (FH 0);
3) поставьте стакан с раствором серной кислоты на лист линованной бумаги;
4) к раствору кислоты прилейте содержимое второго стакана и одновременно вклю-
включите секундомер;
5) круговым движением перемешайте содержимое стакана;
6) глядя сверху через раствор, отметьте момент исчезновения линий из поля зрения
и запишите время протекания реакции tt в секундах;
7) выполняя операции пп. 1-6, проведите эксперимент еще четыре раза с другими
объемами растворов H2SO4, Na2S2O3 и воды в соответствии с данными табл. Л.5.1.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов, а также выводы занесите в
табл. Л.5.1;
Таблица Л.5.1
Результаты эксперимента
Исходные данные
Объем V, мл
раствора реагента
Na2S2O3
10
20
30
20
10
10
H2SO4
10
10
10
10
20
30
среды
Н2О
20
10
10
10
Результаты
измерений
Время условного
окончания реак-
реакции th С
Результаты расчетов
Порядок реакции по веществу
Na2S2O3
п х
Иср
H2SO4
т2
Ю ср
расчетов
Начальная концен-
концентрация реагента
Со, моль/л
Na2S2O3
Кинетическое
уравнение
изучаемой
реакции
H2SO4
Вывод]
Условная
скорость
реакции rh
Реакция имеет:
а) порядок (общий, частные);
б) лимитирующую стадию
а) по Na2S2O3, no H2SO4,
общий;
б) уравнение этой стадии
ЛР 5. Кинетика гомогенных химических реакций 307
2) рассчитайте начальную концентрацию реагентов в реакционной смеси по формулам:
*Na2S2O3 . _ 05 FH2SO4
C0Na2S2O3 ~U'J F > C0H2SO4 ~U'UD „
см ' см
где FCM — объем реакционной смеси, VCM = 40 мл; KNa s 0 и FH so — объемы рас-
растворов реагентов Na2S2O3 и H2SO4 соответственно, взятые для эксперимента;
3) рассчитайте условную скорость реакции при различных начальных концентраци-
концентрациях реагентов по соотношению:
100
где U — время условного окончания реакции; 100 — коэффициент пропорциональ-
пропорциональности, принятый для удобства ведения расчетов и построения графической зависи-
зависимости.
4) по полученным данным постройте графическую зависимость в координатах:
условная скорость реакции — начальная концентрация тиосульфата натрия
5) напишите в общем виде кинетическое уравнение для изучаемой реакции, в кото-
котором п — порядок реакции по Na2S2O3, am — порядок реакции по H2SO4;
6) определите частные порядки реакции (по тиосульфату натрия и серной кислоте)
методом Вант-Гоффа по уравнениям:
> W2-—77ГТ' лср- ^~
'<!) "(I
7) приведите кинетическое уравнение реакции (см. п. 5) в логарифмическом виде;
8) зависимость условной скорости реакции от начальной концентрации тиосульфата
натрия (см. п. 4) представьте графически в координатах: lgr— lgC0Na s 0 . Из полу-
полученного графика определите порядок реакции по Na2S2O3. Сравните полученное
значение со средним значением, рассчитанным согласно методу Вант-Гоффа (см. п. 6);
9) по полученным результатам напишите кинетическое уравнение для исследуемой
реакции;
10) сделайте вывод относительно совпадения частных порядков реакции, опреде-
определенных различными методами, и общего порядка реакции;
308 Кинетика химических реакций
Опыт 2. Зависимость скорости реакции от температуры. В данном
опыте на примере реакции взаимодействия тиосульфата натрия с серной кислотой
проверяется справедливость правила Вант-Гоффа и оценивается энергия актива-
активации реакции.
Последовательность проведения:
1) по термометру определите температуру воздуха Гкомн в лабораторном помещении
и запишите в тетрадь;
2) трижды ополосните небольшими порциями дистиллированной воды стаканы для
растворов тиосульфата натрия и серной кислоты;
3) при помощи соответствующих мерных цилиндров (см. этикетки) налейте в один
стакан 20 мл 0,05М раствора серной кислоты, .а в другой — 20 мл 0,ЗМ раствора
тиосульфата натрия Na2S2O3;
4) поставьте стакан с кислотой на лист линованной бумаги;
5) из другого стакана быстро прилейте к нему раствор Na2S2O3, включив одновре-
одновременно секундомер;
6) круговыми движениями перемешайте смесь и следите сверху через раствор за его
помутнением;
7) остановите секундомер в момент исчезновения линий из поля зрения и запишите
время tj условного окончания реакции в секундах;
8) вылейте раствор в склянку для слива, ополосните оба стакана;
9) повторите эксперимент, нагрев реагирующие растворы на 10° выше температуры
первого эксперимента. Для этого оба стакана с растворами поместите одновременно
на горячую водяную баню. В одном из стаканов периодически измеряйте темпера-
температуру (не оставляя термометр в стакане). По достижении требуемой температуры
быстро смешайте растворы и отметьте по секундомеру условную продолжитель-
продолжительность течения реакции (см. пп. 4-8);
10) повторите этот же эксперимент, повысив температуру обоих растворов на 20°
выше температуры первого эксперимента.
Обработка результатов:
1) исходные данные, условия проведения опыта, результаты измерений и расчетов,
а также выводы занесите в табл. Л.5.2;
2) рассчитайте условную скорость реакции при разных температурах по формуле
100
3) полученные данные представьте в виде графической зависимости скорости в ко-
координатах: условная скорость реакции г,- — температура Г и In r — A/7);
4) приведите выражение закона (правила) Вант-Гоффа. По значениям условной ско-
скорости при разных температурах рассчитайте для данной реакции температурный
коэффициент Вант-Гоффа:
h гъ Yi + Y2
v — • v — ' v — •
Г\ Г2 2
ЛР 6. Гетерогенные химические реакции
309
Таблица Л. 5.2
Исходные данные
Объем раствора
реагента V, мл
Na2S2O3
20
20
20
H2SO4
20
20
20
Результаты
эксперимента
Условия проведения опыта
Концентрация
реагентов в смеси
Со, моль/л
Na2S2O3
H2SO4
Температура реакционной
смеси Т, К
Т
1 коми
Гкомн + 10
^ коми ~*~ ^У
Результаты расчетов
Условная скорость
реакции г,-, с1
Температурный коэф-
коэффициент Вант-Гоффа у/
Yi
Y2
Yep
Энергия
активации
Еа, кДж/моль
Результаты
измерений
Время условного
окончания реак-
реакции //, С
Выводы
Правило
Вант-Гоффа
соблюдается, нет
5) рассчитайте энергию активации этой реакции для всего интервала температур по
уравнению
тъ-тх
6) определите энергию активации графически и сравните ее значение с рассчитан-
рассчитанным по уравнению;
7) сделайте выводы о выполнении правила Вант-Гоффа.
ЛР 6. Гетерогенные химические реакции
Цель работы — ознакомление с основными факторами, влияющими на ско-
скорость гетерогенных реакций. На скорость гетерогенных реакций твердое тело —
жидкость (т-ж) большое влияние оказывают состояние твердой фазы (аморфное,
кристаллическое), площадь поверхности ее соприкосновения с жидкой фазой, кон-
концентрация и скорость движения жидкой фазы.
Опыт 1. Влияние площади поверхности раздела на скорость гетероген-
гетерогенной реакции твердое тело—жидкость. Влияние площади поверхности раздела
изучается на примере двух реакций: взаимодействия твердого простого (цинка) с
серной кислотой и сложного (карбоната кальция СаСО3) вещества с соляной ки-
310
Кинетика химических реакций
слотой. Скорость реакции оценивается по скорости выделения газа (соответст-
(соответственно Н2 и СО2) и определяется по тангенсу угла наклона касательной к кинети-
кинетической кривой, характеризующей зависимость общего объема выделившегося
газа от времени, в данный момент. Изучение проводят на установке, изображен-
изображенной на рис. Л.6.1.
Последовательность проведения:
1) заполните бюретку 2 водой (см. рис. Л.6.1). Для этого кран 6 поставьте в положе-
положение а и с помощью уравнительного сосуда 3 доведите уровень воды в бюретке до
верхней метки. Затем зафиксируйте уровень воды поворотом крана в положение б;
2) возьмите три гранулы цинка и поместите их в реакционную колбу 4 (объем 50 мл).
В эту же колбу до метки налейте раствор серной кислоты (H2SO4) (объем раствора
около 15 мл);
3) закройте колбу пробкой с газоотводной трубкой, переведите кран 6 в положение в
и включите секундомер;
4) непрерывно, аккуратно потряхивая, перемешивайте содержимое колбы;
5) через определенные промежутки времени (например, через каждые 2 мин) изме-
измеряйте положение уровня жидкости в бюретке по нижнему краю мениска с точно-
точностью до 0,1 мл. Проведите не менее семи
замеров. Полученные результаты занеси-
занесите в табл. Л.6.1;
6) повторяя операции пп. 1-5, проведите
аналогичный эксперимент с приблизитель-
приблизительно равной по массе навеской цинкового
порошка (три лопаточки порошка);
7) по указанию преподавателя проведите
эксперимент с СаСО3 в компактном и
порошкообразном состоянии и 3 %-ным
водным раствором НС1. Положение уров-
уровня жидкости в бюретке замеряется в этом
случае каждые 30 с.
Обработка результатов:
1) исходные данные, наблюдения, урав-
уравнения протекающих реакций, кинетиче-
кинетические уравнения, результаты измерений,
расчетов и выводы занесите в табл. Л.6.1;
2) приведите стехиометрическое уравне-
уравнение протекающей реакции и кинетиче-
кинетическое уравнение для нее (для компактного
и порошкообразного состояний твердого
вещества). Объясните различия;
3) по разности положений уровня воды в
бюретке между начальным йо и текущим
h показаниями вычислите общий объем V
выделившегося газа в соответствующий
момент времени для гранулы и порошка.
Данные занесите в табл. Л.6.1;
Рис. Л.6.1. Установка для определения
скорости гетерогенной реакции:
1 — штатив; 2 — бюретка; 3 — уравнитель-
уравнительный сосуд; 4 — реакционная колба; 5 —
резиновая пробка с газоотводной трубкой;
6 — трехходовой кран (а — заполнение
бюретки водой; б — фиксация уровня воды;
в — рабочее положение
ЛР 6. Гетерогенные химические реакции
311
4) постройте по полученным данным на миллиметровой бумаге кинетические кри-
кривые (в координатах общий объем газа V—время t) для реакций с гранулами веще-
вещества и порошком вещества;
5) определите графически начальные скорости (в момент времени t = 0) по водороду
(в мл/мин) для реакций с гранулами и порошком вещества, а также через 4 и 8 мин
после начала реакции. Сравните их, рассчитав отношения гпор ,- /Ггран ;. Результаты
занесите в табл. Л.6.1;
6) на основании полученных результатов сделайте вывод о влиянии площади по-
поверхности раздела фаз на скорость гетерогенной реакции.
Таблица Л.6.1
Результаты эксперимента
Исходные данные
Реагирующие вещества (формула,
агрегатное состояние, форма)
первое
второе
Наблюдения
Выделение
пузырьков газа
(интенсивное,
слабое)
Уравнение
протекающей
гетерогенной
реакции
Кинетическое
уравнение
Zn, тв., гранулы
Zn, тв., порошок
H2SO4, 20%-ный
водный раствор
Результаты измерений
Время t, положение уровня воды h в бюретке, V= ho-h выделившегося газа
при взаимодействии 20 %-ного раствора H2SO4
с гранулами цинка
с порошком цинка
t, мин
h
К,мл
0
0
0
К
о
Результаты графических определений и расчетов
Выводы
Время t,
мин
Скорость (мл/мин) реакции, с
с гранулами Zn
'гран /
с порошком Zn
'пор /
Отношение
скоростей
пор
/ ' *
Влияние площади
поверхности на ггет
гран/
Опыт 2. Влияние степени измельчения и смешения реагентов на ско-
скорость твердофазной реакции. Скорость гетерогенных твердофазных реакций
твердое тело — твердое тело (т-т) в большей степени зависит от площади поверх-
поверхности соприкосновения фаз (реагирующих веществ), чем для реакций т-ж. Послед-
Последнее определяется степенью измельчения и смешения реагентов. Важную роль в
твердофазных реакциях играет скорость диффузии частиц (атомов, ионов, молекул)
реагирующих твердых веществ.
В данном опыте на примере двух твердофазных реакций, протекающих между:
1) нитратом свинца (II) и йодидом калия; 2) хлоридом (или нитратом) кобальта (II) и
312
Кинетика химических реакций
роданидом аммония изучается влияние этих факторов на скорость взаимодействия.
Одним из продуктов первой реакции является йодид свинца (И) РЬЬ — вещество
ярко-желтого цвета, второй реакции — комплексное соединение синего цвета со-
состава (NH4J [Co(NCSL]. По интенсивности окраски и времени ее появления судят
соответственно о степени превращения реагентов в продукты и скорости протека-
протекания реакции.
Последовательность проведения:
1) на кончике шпателя возьмите нитрат свинца (II) Pb(NO3J и поместите его в чис-
чистую сухую ступку;
2) аналогичным образом возьмите приблизительно равное количество йодида калия
KJ и поместите его в ту же ступку рядом с нитратом свинца. Отметьте цвет, агре-
агрегатное состояние и форму веществ;
3) стеклянной палочкой тщательно перемешайте вещества в течение 1-2 мин, а за-
затем отметьте цвет смеси;
4) после этого энергично разотрите полученную смесь пестиком в течение 1 мин.
Отметьте произошедшие изменения;
5) в полученную смесь из капельницы добавьте 3^4 капли дистиллированной воды
и содержимое ступки тщательно разотрите пестиком. Запишите в лабораторную
тетрадь происходящие при этом изменения;
6) повторяя операции пп. 1-5 данного опыта, проведите по указанию преподавателя
аналогичный эксперимент с хлоридом (или нитратом) кобальта (II) и роданидом
аммония NH4 NCS.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих процессов и выводы
занесите в табл. Л.6.2;
Результаты эксперимента
Таблица Л.6.2
Исходные данные
Реагирующие вещества (формула,
агрегатное состояние, форма, цвет)
первое
Pb(NO3J, твер-
твердый, кристал-
кристаллический, бе-
белый порошок
СоС12, твердый,
кристалличе-
кристаллический
(NH4J[Co(NCSL]
второе
KI, твердый,
кристалличе-
кристаллический, белый
порошок
NH4NCS, твер-
твердый, кристал-
кристаллический, бес-
бесцветный
Н2О, ЖИДКОСТЬ
Наблюдения
Появление
окраски (какой):
при смешении,
растирании,
растирании с
водой
Уравнение
протекаю-
протекающей
гетерогенной
реакции
Выводы
Зависимость ггет от
площади поверхно-
поверхности раздела фаз (S)
Соотношение меж-
ДУ >*гет И Ггом
ЛР 7. Каталитические реакции 313
2) объясните причину появления окраски при интенсивном размешивании смеси,
подтвердите соответствующим уравнением протекающей реакции;
3) почему интенсивность окраски резко усиливается или изменяется на другую по-
после добавления воды? Ответ аргументируйте;
4) приведите стехиометрические уравнения протекающих в жидкой фазе реакций в
ионно-молекулярном виде и соответствующие им кинетические уравнения. Назови-
Назовите комплексные соединения;
5) сделайте выводы относительно: а) влияния площади поверхности раздела на ско-
скорость ггет гетерогенных реакций; б) соотношения между скоростью гомогенной ггом
и скоростью ггет гетерогенной реакций.
ЛР 7. Каталитические реакции
Цель работы — ознакомление с основными закономерностями каталитиче-
каталитических реакций.
Опыт 1. Активность различных катализаторов в реакции разложения
пероксида водорода. При гомогенном катализе реагирующие вещества и катализа-
катализатор находятся в одной фазе — газовой или жидкой. На практике часто приходится
иметь дело с гомогенными каталитическими реакциями, протекающими в раство-
растворах. Скорость таких реакций зависит от активности катализатора и его концентра-
концентрации. В данном опыте предлагается сравнить эффективность действия различных
катализаторов (K2Cr207, FeCl3, KI) на реакцию разложения пероксида водорода:
Н2О2 = Н2О+-О2
Скорость реакции обратно пропорциональна времени ее протекания. Последнее
определяется как разность между временем окончания и начала выделения пузырь-
пузырьков газа.
Последовательность проведения:
1) в две мерные конические пробирки налейте по 1 мл 3 %-ного водного раствора
пероксида водорода;
2) в одну пробирку добавьте две капли 0,ЗМ раствора дихромата калия (К2Сг207), а в
другую пробирку — четыре капли. С помощью секундомера измерьте время проте-
протекания реакции (от начала и до окончания выделения пузырьков газа) в обеих про-
пробирках, а также окраску раствора;
3) повторяя пп. 1-2, проведите аналогичные эксперименты с 0,ЗМ растворами FeCl 3 и
KI в качестве катализаторов.
Обработка результатов:
1) исходные данные, наблюдения, результаты измерений и расчетов, а также выво-
выводы занесите в табл. Л.7.1;
2) отметьте цвета растворов в начале реакции и после ее завершения;
314
Кинетика химических реакций
Таблица Л. 7.1
Результаты эксперимента
Исходные данные
Вид и состав раствора
реагента, изучаемая
реакция
3%-ный водный
раствор Н2О2,
Н2О2 = Н2О+ -О2
2
Катализатор
0,3M водный
раствор
1.К2Сг207
2. К2Сг207
3. FeCl з
4. FeCl з
5.KI
6.KI
Объем
или чис-
число капель
2
4
2
4
2
4
Наблюдения
Выделение
пузырьков газа
(да, нет,
быстрое, медленное)
Результаты
измерений
Катализатор
и количество
(в каплях)
К2Сг207B)
К2Сг207D)
FeCl3B)
FeCl3 D)
KIB)
KID)
Время
протекания
реакции t, с
расчетов
Условная скорость реакции
r^lOOO/^c
Изменение окраски
раствора
Выводы
Активность
катализаторов
убывает в ряду
3) вычислите условную скорость реакции по формуле
1000
г = -
t
где t — время протекания реакции;
4) сделайте вывод относительно эффективности действия различных катализаторов.
Расположите их в убывающий ряд по активности;
5) укажите, как влияет на скорость разложения Н2О2 концентрация катализатора.
Опыт 2. Каталитическое восстановление ионов железа (III). В данном
опыте в качестве примера гомогенного катализа рассматривается реакция восста-
восстановления ионов Fe3+ ионами S2C>3~
2Fe(NCSK + 2Na2S2O3 = Na2S4O6 + 2Fe(NCSJ + 2NaNCS
которая ускоряется в присутствии ионов Cu2+.
ЯР 7. Каталитические реакции
315
Водные растворы роданида железа (III) имеют кроваво-красную окраску. Ре-
Реакция его образования используется в аналитической химии для обнаружения ионов
Fe3+. По времени исчезновения окраски судят о скорости протекания реакции.
Последовательность проведения:
1) приготовьте раствор роданида железа (III) Fe(NCSK, смешав в колбочке 5 мл 0,1М
раствора хлорида железа (III) FeCl3 и 1 мл 0,1 М раствора роданида аммония
NH4NCS;
2) пипеткой отберите 1 мл приготовленного раствора в коническую пробирку;
3) в другую коническую пробирку налейте 1 мл 0,1 М раствора тиосульфата натрия
Na2S2O3;
4) содержимое обеих пробирок объедините, для чего раствор Na2S2O3 быстро пере-
перелейте в пробирку с раствором Fe(NCSK, включив при этом секундомер. Наблюдайте
за происходящими изменениями;
5) отметьте по секундомеру время до полного обесцвечивания раствора, которое
условно можно считать временем протекания реакции;
6) выполняя пп. 2-5, проведите аналогичный эксперимент, добавив в пробирку с
раствором Fe(NCSK одну каплю 0,05М раствора CuSO4;
7) повторите опыт с тремя каплями 0,05М раствора CuSO4.
Обработка результатов:
1) исходные данные, уравнение реакции, результаты измерений и расчетов, а также
выводы занесите в табл. Л.7.2;
Таблица Л.7.2
Результаты эксперимента
Исходные данные
Растворы реагентов
вид и состав
Первый: 0,1М водный
раствор Fe (NCS) 3
Второй: 0,1М водный
раствор Na2S2O3
объем, мл
1
1
Катализатор,
изучаемая реакция
0,05М водный раствор CuSO4,
2Fe(NCSK + 2 Na2S2O3 =
= Na2S4O6+2Fe(NCSJ +
+ 2NaNCS,
Результаты
измерений
Количество
катализатора
(число капель)
0
1
3
Время протекания
реакции t, с
расчетов
Условная скорость
реакции
Вы
Наблюдения
Скорость
обесцвечива-
обесцвечивания раствора
воды
2) приведите уравнения, описывающие появление и исчезновение кроваво-красной
окраски у растворов;
316 Кинетика химических реакций
3) рассчитайте условную скорость реакции г = ;
t
4) на основании полученных результатов сделайте выводы относительно:
а) влияния на скорость реакции восстановления Fe3+ присутствия ионов Си2+;
б) влияния на скорость этой реакции концентрации ионов Си 2 +.
Опыт 3. Кинетика каталитического разложения пероксида водорода в
условиях гомогенного катализа. Этот опыт рекомендуется для студентов с высо-
высоким уровнем теоретической и экспериментальной подготовки. В данном опыте изу-
изучение кинетики разложения пероксида водорода основано на измерении объема
выделяющегося при разложении кислорода. Работа проводится на установке (см.
рис. Л.6.1).
Последовательность проведения:
1) в уравнительный сосуд 3 налейте воду. Трехходовой кран б переведите в поло-
положение сообщения с атмосферой. Путем вертикального перемещения уравнительно-
уравнительного сосуда добейтесь положения уровня воды в бюретке (на 100 мл) на нулевой
отметке;
2) переведите кран в положение, отсекающее бюретку от сообщения с атмосферой;
3) отмерьте градуированной пипеткой 5 мл 3 %-ного раствора пероксида водорода и
перенесите в реакционную колбу 4;
4) наберите в шприц 0,5 мл 0,1М раствора дихромата калия К2Сг207. Герметично
закройте реакционную колбу пробкой с отводной трубкой и шприцем;
5) введите шприцем раствор катализатора в колбу, одновременно включите секун-
секундомер, отметьте положение уровня воды в бюретке по нижнему краю мениска h0 и
запишите в тетрадь;
6) кислород, выделившийся в результате разложения пероксида водорода, вытесня-
вытесняет из бюретки воду. Уравнительный сосуд при этом опускайте и во время опыта
старайтесь держать воду в нем и в бюретке на одном уровне, чтобы давление газа
внутри прибора все время было близким к атмосферному;
7) записывайте положение уровня воды h в бюретке каждые 30 с (с точностью
до 0,1 мл);
8) после кажущегося прекращения выделения кислорода нагрейте раствор, опустив
реакционную колбу в стакан с горячей водой (80...90 °С) и подержите ее там 5 мин.
Затем охладите колбу до комнатной температуры, приведите давление в приборе к
атмосферному с помощью уравнительного сосуда и запишите положение нижнего
края мениска h^ в бюретке;
9) повторите эксперимент, взяв 1 мл 0,1М раствора К2СГ2О7.
Обработка результатов:
1) вычитанием из записанных текущих показаний бюретки (И) начального положе-
положения уровня воды (ho) найдите объем выделившегося кислорода в каждый данный
момент времени (F,);
2) определите объем выделившегося кислорода, соответствующий полному разло-
разложению пероксида водорода (V^ = /*<» - ho)\
3) результаты измерений занесите в табл. Л.7.3;
ЛР 7. Каталитические реакции
317
Таблица Л. 73
Результаты измерений
№
/, мин
Показания бюретки {И)
Объем выделившегося кислорода, см3
(Vt = h-h0)
4) результаты экспериментов изобразите графически в координатах V (см )—/ (мин)
на миллиметровой бумаге;
5) определите графически начальную скорость реакции (в см3/мин) в каждом случае
(как тангенс угла наклона касательной к кинетической кривой);
6) определите графически скорость через 3 мин после начала реакции в каждом
случае;
7) определите графически время полупревращения пероксида водорода в каждом
эксперименте;
8) результаты расчетов занесите в табл. Л.7.4;
9) сделайте вывод о влиянии концентрации катализатора на скорость разложения
пероксида водорода.
Результаты эксперимента
Таблица Л.7.4
Количество
раствора
катализатора,
мл
0,5
1,0
Результаты расчетов
Начальная
скорость го,
см3/ мин
Скорость гъ
через 3 мин,
см3/мин
Отношение скоростей
fa^Ol)
(Г32/Г31)
Время полу-
полупревращения
t\l2, МИН
Опыт 4. Каталитическое разложение пероксида водорода в условиях ге-
гетерогенного катализа. Гетерогенный катализ характеризуется принадлежностью
реагирующих веществ и катализатора к разным фазам. Обычно катализатор являет-
является твердым веществом, а реагенты — газами или жидкостями. Непосредственное
химическое взаимодействие протекает на поверхности катализатора. Поэтому его
активность зависит от свойств поверхности: ее величины, химического состава,
структуры. Важную роль в гетерогенных каталитических процессах играет адсорб-
адсорбция. В данном опыте изучается влияние гетерогенных катализаторов (МпО2 и РЬО2)
на скорость разложения пероксида водорода. Предполагается, что реакция с участи-
участием диоксида марганца протекает через следующие стадии:
МпО2 + Н2О2 + 2Н+ = Мп2+ + 2Н2О + О2
Мп2+ + 2Н2О = Мп(ОНJ + 2Н+
Мп(ОНJ + Н2О2 = МпО2 + 2 Н2О
Последовательность проведения:
1) в две пробирки с помощью мерного цилиндра налейте по 3 мл 3 %-ного раствора
пероксида водорода;
318
Кинетика химических реакций
2) в первую пробирку добавьте немного (на кончике шпателя) порошка диоксида
марганца МпО2 и одновременно включите секундомер. Отметьте время протекания
реакции до окончания выделения пузырьков газа;
3) во вторую пробирку добавьте столько же порошка диоксида свинца РЬО2. От-
Отметьте время протекания реакции;
4) повторяя операции пп. 1-3, проведите аналогичные эксперименты с большими в
2 раза количествами диоксидов марганца и свинца.
Обработка результатов:
1) исходные данные, наблюдения, результаты измерений и расчетов, а также выво-
выводы занесите в табл. Л.7.5;
Таблица Л. 7.5
Результаты эксперимента
Исходные данные
Вид и состав раствора реагента,
уравнение изучаемой реакции
3 %-ный водный раствор Н2О2
Н2О2->Н2О+-О2
2
Катализатор
№
1
2
3
4
Вид
МпО2
РЮ2
МпО2
РЮ2
Результаты
Измерений
№ опыта
1
2
3
4
Время
протекания
реакции t, с
Расчетов
Условная скорость
реакции
г, = 100//,, с
Количество ус-
условных массовых
частей
1
1
2
2
Наблюдения (ско-
(скорость выделения
пузырьков газа)
Выводы
Влияние активности
катализатора и его количества
на скорость реакции
2) вычислите условную скорость реакции по формуле г -
100
3) сравните каталитическую активность диоксида марганца и диоксида свинца. За меру
каталитической активности принять скорость реакции разложения Н2О2;
4) сделайте вывод о влиянии количества катализатора на скорость разложения пе-
роксида водорода.
ЛР 7. Каталитические реакции 319
Опыт 5. Автокаталитическая реакция перманганата калия с щавелевой
кислотой. Для автокаталитических реакций характерно самоускорение за счет об-
образования в ходе процесса катализатора — конечного продукта. Автокаталитиче-
Автокаталитическая реакция перманганата калия с щавелевой кислотой выражается следующим
уравнением:
2КМпО4 + 5Н2С2О4 + 3H2SO4 = 2MnSO4 + K2SO4 + 10СО2 + 8Н2О
или в молекулярно-ионной форме
+ 5Н2С2О 4 + 6Н+ = 2Мп2 + + 10СО2 + 8Н2О
Реакция протекает очень медленно. По мере увеличения концентрации ионов Мп2+ воз-
возрастает скорость их взаимодействия с ионами МпО^, при этом образуются ионы Мп3+:
МпО; + 4 Мп2+ + 8Н+ = 5Мп3+ + 4Н2О
Затем ионы Мп3+ быстро взаимодействуют с щавелевой кислотой:
Н2С2О4 + 2Мп3+ = 2Мп2+ + 2СО2 + 2Н+
При этом ион Мп3+ вновь переходит в ион Мп2+. Если в реакционную смесь сразу
добавить ионы Мп2+, скорость реакции резко увеличивается. Таким образом, реак-
реакция КМпО4 с Н2С2О4 является автокаталитической.
Последовательность проведения:
1) в каждую из двух пробирок налейте по 1 мл 0,02М растворов перманганата калия
и ОДМ щавелевой кислоты;
2) прибавьте в каждую пробирку по шесть капель 2М раствора серной кислоты для
создания достаточной кислотности;
3) одну пробирку оставьте для сравнения, а в другую добавьте 3 капли 0,1М раство-
раствора соли двухвалентного марганца (МпС12 или MnSO4) и включите секундомер;
4) отметьте время, через которое обесцветились растворы в первой и во второй про-
пробирках.
Обработка результатов:
1) исходные данные, наблюдения, уравнение реакции, результаты измерений и рас-
расчетов, а также выводы занесите в табл. Л.7.6;
2) отметьте цвета растворов в начале реакции и после ее завершения;
1000
3) вычислите условную скорость реакции по формуле г = ;
t
4) сделайте вывод относительно эффективности действия ионов двухвалентного
марганца.
320
Кинетика химических реакций
Результаты эксперимента
Таблица Л. 7.6
Исходные данные
Растворы реагентов
Вид и состав
1. 0,02М раствор КМпО4
2. 0,Ш раствор Н2С2О4
3. 2М раствор H2SO4
Объем, мл
(или капли)
1 мл
1 мл
6 капель
Уравнение реакции
Результаты
измерений
Наличие катализатора
Нет
3 капли 0,Ш раствора
МпС12 или MnSO4
Время
протекания
реакции t, с
расчетов
Условная скорость
реакции
г, = 1000//;, с
Наблюдения
Скорость обесцвечи-
обесцвечивания раствора в
отсутствии и в при-
присутствии катализатора
Вывод
Влияние солей
двухвалентного
марганца на скорость
реакции
V
ХИМИЧЕСКОЕ РАВНОВЕСИЕ
¦ Виды, особенности
и характеристики
химического
равновесия
¦ Термодинамическое
описание химического
равновесия
¦ Влияние различных
факторов
на химическое
равновесие.
Особенности
равновесий
в гетерогенных
системах
В химии приходится сталкиваться не столько со
статическим, сколько с динамическим равновеси-
равновесием. Динамическое химическое равновесие — это
термодинамическое равновесие в химически реа-
реагирующей системе, т.е. в системе между компо-
компонентами которой осуществляется обратимая реак-
реакция. Термодинамическим равновесием называют
состояние термодинамической системы, не изме-
изменяющееся во времени и не сопровождающееся
переносом через систему вещества или энергии.
Химическое равновесие может устанавливаться в
различных системах и процессах: в растворах
электролитов, комплексных и малорастворимых
соединений, при экстракции, адсорбции и гидро-
гидролизе, в кислотно-основных и окислительно-вос-
окислительно-восстановительных реакциях и др. В зависимости от
внешних условий (температура, давление и др.)
его положение может смещаться в сторону ис-
исходных веществ или продуктов реакции. Смеще-
Смещение положения равновесия лежит в основе полу-
получения аммиака и полупроводников, очистки ме-
металлов, варки стали и приготовления пищи. Им
определяются также карстовые явления и боль-
большинство биохимических процессов, таких как ды-
дыхание, формирование и рост костной ткани и др.
На основе учения о химическом равновесии и
влиянии на его положение различных факторов
впервые удалось объяснить и предсказать направ-
направление большинства химических процессов, научно
обосновать условия, обеспечивающие максималь-
максимальный выход продуктов в них, разработать техноло-
технологии важнейших крупнотоннажных производств.
В данном разделе рассмотрены примеры равнове-
равновесия в обратимых гомогенных и гетерогенных ре-
реакциях, протекающих при высоких температурах
преимущественно с участием газов. Критерии,
закономерности и особенности установления та-
таких равновесий показаны как с кинетических, так
и с термодинамических позиций.
11 -9795
Виды, особенности
и характеристики
химического равновесия
В зависимости от способа классификации различают гомогенное и ге-
гетерогенное, истинное и заторможенное и другие виды химического
равновесия. Те или иные его разновидности имеют свои характерные
особенности и могут одновременно принадлежать к различным видам.
Важнейшей количественной характеристикой всех видов и разновид-
разновидностей химического равновесия является константа равновесия, кото-
которая указывает на степень завершенности (или степень продвинутости)
обратимой химической реакции. Константа равновесия может быть
размерной или безразмерной величиной.
Обратимые реакции, согласно кинетическому подходу, представляют
собой совокупность двух одновременно протекающих реакций (пря-
(прямой и обратной) и составляют значительную часть промышленных
процессов, таких как синтез аммиака из азота и водорода, окисление
диоксида серы в триоксид, реакции этерификации и др.
10.1. Обратимые и необратимые химические реакции
Сущность любой химической реакции состоит в том, что
исходные вещества (реагенты), записываемые в левой части урав-
уравнения, в результате взаимодействия их молекул, атомов или ионов
превращаются в продукты (конечные вещества), записываемые в
правой части уравнения. Реагенты и продукты часто объединяют
под одним общим названием участники, или компоненты реак-
реакции. Различают кинетически обратимые и кинетически необра-
необратимые химические реакции.
Кинетически необратимые реакции. В дальнейшем будем
называть их необратимые, или односторонние. Такие реакции про-
протекают самопроизвольно только (или преимущественно) в одном
направлении, которое в уравнениях указывают символом необра-
необратимости (односторонней направленности) процесса — стрелкой (—>)
вместо знака равенства (=). При этом происходит практически пол-
полное превращение взятых в стехиометрическом соотношении ис-
исходных веществ и теоретический выход продуктов в них составля-
составляет 100 %. О таких реакциях говорят, что они идут до конца, т.е. до
полного исчезновения реагентов.
Рассмотрим признаки необратимости химических реакций.
1. Образование устойчивого (одного или нескольких) в усло-
условиях проведения реакции продукта:
10.1. Обратимые и необратимые химические реакции 323
а) соединения удаляющегося из сферы реакции, — малорастворимого
вещества, выпадающего в осадок, и (или) газа, улетучивающегося из реак-
реакционной смеси;
б) прочного комплекса и (или) малодиссоциирующего вещества —
слабых электролитов.
В соответствии с этим признаком такие реакции, как
ВаС12 + K2SO4 -> BaSO4 I + 2КС1
CuBr2 +4NH3 ->[Cu(NH3L]Br2
HC1 + KOH->KC1 + H2O
протекают полно и служат примерами практически необратимых процес-
процессов, хотя и BaSO4 несколько растворим, и комплексный катион [Cu(NH3L]2+
не является абсолютно устойчивым, и молекулы Н2О немного диссоциируют.
2. Экзотермический характер (экзотермичность) реакции (АГН < 0),
сопровождающейся образованием большего числа молей продуктов, чем
исходных веществ, т.е. протекающей с увеличением энтропии (ArS > 0). Со-
Согласно этому признаку, реакции, в результате которых сложные вещества
экзотермически превращаются в более простые, например:
2KMnO4 -> K2MnO4 + MnO2 + O2 2NI3 -> N2 + 3I2
2С6Н6 + 15О2 -> 12СО2 + 6H2O Pb(N3J -> Pb + 3N2
2КСЮ3->2КС1 + ЗО2
относят к истинно, или совершенно, необратимым процессам независимо
от условий, так как непосредственно из продуктов таких реакций в обычных
условиях реагенты нельзя получить никаким известным способом.
На основании вышеизложенного можно сделать заключение, что
необратимыми (практически, совершенно) являются реакции нейтрали-
нейтрализации, комплексообразования, термического разложения сложных ве-
веществ, взаимодействия активных металлов с кислородом, водой и кисло-
кислотами и др.
Для необратимых реакций, а их в природе больше, чем обратимых, в
принципе, можно подобрать условия, при которых они будут протекать об-
обратимо. Например, если в открытой системе реакция протекает необратимо,
т.е. идет практически до конца, то в условиях закрытой системы во многих
случаях она может быть доведена только до состояния равновесия. Обычно
химики-практики стараются проводить процессы так, чтобы преобладала
реакция, идущая в одном направлении, т.е. выбирают такие условия их про-
проведения, при которых достигается максимально возможный выход продук-
продуктов при наибольшей скорости процесса их образования. Естественное или
324 10. Виды, особенности и характеристики химического равновесия
искусственное исключение возможности протекания обратной реакции по-
позволяет довести процесс практически до конца.
Кинетически обратимые реакции. Понятие «химическое равнове-
равновесие» применимо только к кинетически обратимым химическим реакциям.
Кинетически обратимыми (в дальнейшем обратимыми, двусторонними, или
противоположно направленными) называют реакции, протекающие само-
самопроизвольно при данных условиях одновременно и независимо в двух про-
противоположных направлениях: прямом (-») — слева направо, т.е. от реаген-
реагентов к продуктам, и обратном (<-) — справа налево, т.е. от продуктов к реа-
реагентам. В них наряду с превращением исходных веществ с заметной (срав-
(сравнимой) скоростью осуществляется и противоположно направленная реакция
превращения продуктов. В результате каждой реакции образуются исход-
исходные вещества, необходимые для осуществления противоположной реакции,
причем уменьшение скорости одной реакции сопровождается увеличением
скорости другой до тех пор, пока скорости обеих реакций не станут равны-
равными. В уравнениях таких реакций, чтобы подчеркнуть их обратимость, вме-
вместо знака равенства между левой и правой частями ставят символ обратимо-
обратимости (<=±).
Понятие обратимой реакции не соответствует термодинамическому
понятию «обратимый процесс». Обратимые химические реакции обратимы
в термодинамическом смысле только в непосредственной близости от со-
состояния равновесия, когда скорости прямой и обратной реакций различают-
различаются на бесконечно малую величину.
Обратимые (и необратимые) химические реакции бывают как гомо-
гомогенными, так и гетерогенными. Гомогенными называют реакции, проте-
протекающие в одной фазе — газовой или жидкой. Они характеризуются отсутст-
отсутствием поверхности раздела между реагентами и продуктами, взаимодействие
которых осуществляется во всем объеме реакционной смеси. У гомогенных
обратимых реакций все участники находятся в одном агрегатном состоянии
и составляют одну фазу, например:
реакции между газообразными веществами
CH4(r)+CO2(r)^2CO(r)+2H2(r) A0.1)
реакции этерификации, протекающие в растворах
СН3СООН(р) +С2Н5ОН(р) <=±СН3СООС2Н5(р) + Н2О(р) A0.2)
Равновесие, имеющее место у таких реакций, называют гомогенным.
Обратимые гетерогенные реакции протекают на поверхности (грани-
(границе) раздела между веществами (реагентами и продуктами), находящимися в
различных фазах: ti-t2, т-ж, т-г, Ж1-ж2, ж-г (т — твердое вещество, ж —
10.2. Виды и особенности химического равновесия 325
жидкость, г — газ), и равновесие в этом случае называют гетерогенным
(или фазовым) равновесием. Примерами кинетически обратимых гетероген-
гетерогенных реакций служат следующие превращения:
FeO(T) +CO(r) ?=*Fe(T) +CO2(r) A0.3)
MgO(T) +CO2(r) <=> MgCO3(T) A0.4)
NH4Cl(T)<=>NH3(r)+HCl(r) A0.5)
(r) <=iCO(r) +H2(r) A0.6)
При нахождении всех компонентов в одном агрегатном состоянии об-
обратимую гомогенную или гетерогенную реакцию называют гомофазной
(ЮЛ)—A0.2), если компоненты находятся в разных фазах — гетерофазной
(Ю.З)-(Ю.б).
Обратимые гомогенные и гетерогенные реакции в отличие от необра-
необратимых идут не до конца, т.е. не до полного исчезновения реагентов: они
«прекращаются» прежде, чем будут полностью израсходованы их исходные
вещества (если они были взяты в стехиометрических соотношениях), поэто-
поэтому в реакционной смеси у таких реакций всегда присутствуют (сосущест-
(сосуществуют) и исходные вещества, и продукты их взаимодействия. Максимальный
выход продуктов у них менее 100 % и соответствует равновесному составу.
Такие реакции протекают до установления в них определенного концентра-
концентрационного предела, общего для их прямого и обратного направлений, назы-
называемого состоянием химического равновесия. Именно с наступлением по-
последнего и связывают прекращение протекания реакции в целом.
10.2. Виды и особенности химического равновесия
Равновесие в системе наступает в результате стремления ее к мини-
минимальному значению энергии и максимальному хаотическому распределе-
распределению в пространстве, характеризующемуся энтропией. Достигнутое и неиз-
неизменное при состоянии химического равновесия соотношение достаточно
больших, отчетливо определяемых аналитически концентраций всех ве-
веществ называют положением химического равновесия. В состоянии равно-
равновесия нельзя указать, какие из компонентов реакционной смеси фактически
являются реагентами, а какие продуктами, поскольку все вещества, присут-
присутствующие в такой системе, а также процессы их взаимодействия участвуют
в его создании. В равновесных химических системах вещества, образующие
их, если и называют реагентами и продуктами, то формально, в соответст-
соответствии с местом расположения в уравнении протекающей обратимой химиче-
326 №. Виды, особенности и характеристики химического равновесия
ской реакции. Правильнее было бы их называть веществами-участниками
равновесия.
Под химическим равновесием понимают достигаемое с двух противо-
противоположных сторон и неизменное во времени при постоянных давлении, объ-
объеме и температуре состояние системы, содержащей вещества, способные к
химическому взаимодействию. Различают истинное и заторможенное (ка-
(кажущееся) химическое равновесие.
Истинное химическое равновесие (в дальнейшем — химическое рав-
равновесие) устанавливается только в закрытых системах и характеризуется
следующими основными особенностями (признаками):
термодинамической устойчивостью — постоянством (неизменно-
(неизменностью) во времени состояния (равновесного состава) системы в отсутствие
(или при неизменности) внешних воздействий (условий) в виде изменения
температуры, давления или концентраций веществ, участвующих в образо-
образовании системы. В силу этого признака истинное химическое равновесие
часто называют устойчивым. Его термодинамическая устойчивость обу-
обусловлена энергетической выгодностью такого состояния системы — мини-
минимальным значением энергии Гиббса (или энергии Гельмгольца) и отсутст-
отсутствием их изменения в состоянии равновесия;
подвижностью — способностью положения равновесия легко сме-
смещаться в ту или иную сторону при наличии внешних воздействий, сколь ма-
малы бы они ни были. Смещением (или сдвигом) положения равновесия назы-
называют изменение состояния равновесия в результате изменения условий, т.е.
переход системы из одного равновесного состояния в другое. После пре-
прекращения внешнего воздействия она вновь возвращается в исходное равно-
равновесное состояние. Для истинного равновесия характерна однозначная коли-
количественная связь между воздействием и его результатом;
возможностью достижения системой равновесного состояния с раз-
различным исходным соотношением компонентов при подходе к нему с двух
сторон — как со стороны реагентов (в результате самопроизвольного про-
процесса), так и со стороны продуктов (в результате несамопроизвольного про-
процесса), т.е. как по прямой (слева направо), так и по обратной (справа налево)
реакции. Следовательно, обе реакции — и прямая, и обратная — в зависи-
зависимости от условий могут протекать самопроизвольно;
динамическим характером (динамичностью), означающим непрерыв-
непрерывное протекание с одинаковой скоростью как прямой, так и обратной реак-
реакций, т.е. продолжение (не прекращение, не отсутствие) химического взаи-
взаимодействия на атомно-молекулярном уровне между веществами-участни-
веществами-участниками. При этом за единицу времени по прямой реакции образуется такое же
количество продуктов, какое по обратной реакции превращается в исходные
вещества. В результате концентрации веществ-участников равновесия оста-
10.2. Виды и особенности химического равновесия 327
ются постоянными во времени (так называемые равновесные концентра-
концентрации), а общая (результирующая) или наблюдаемая скорость всего процесса
становится равной нулю. Таким образом, установление химического равно-
равновесия не означает прекращение реакции.
Примером системы, пребывающей в состоянии истинного химического
равновесия, может служить находящаяся над катализатором при Г= 810 °С
смесь газов СО2, Н2, СО, Н2О в эквимолярном соотношении, которое сохра-
сохраняется сколь угодно долго во времени. При температуре выше Т = 810 °С
положение равновесия реакции смещается вправо:
СО2(Г)+Н2(Г)^>СО(Г)+Н2О(Г)
однако система вновь возвращается в исходное состояние, как только эта
температура восстанавливается.
Состояние равновесия сохраняется до тех пор, пока остаются неиз-
неизменными температура, давление и в систему не вводится извне или не вы-
выводится из ее реакционного объема какое-то количество (одного или сразу
нескольких) веществ-участников реакции.
Заторможенное химическое равновесие лишь по первому признаку
тождественно истинному, поэтому, в сущности, его нельзя назвать равнове-
равновесием. Так, азотно-водородную смесь можно нагревать до высоких темпера-
температур и сжимать до больших давлений, но образования аммиака происходить
не будет. В отсутствии возмущающих факторов сколь угодно долго могут
существовать гремучая смесь (Н2 с О2) и термит (смесь А1 с Ре2Оз) в услови-
условиях, когда пары этих веществ реакционноспособны. Но если в азотно-водо-
азотно-водородную и гремучую смеси ввести катализатор (мелкодисперсную платину),
а термит поджечь, то начинается энергичное взаимодействие, сопровож-
сопровождающееся выделением большого количества теплоты:
N2(r) + 3H2(r) =2NH3(r) (Artf2°98 =-92,38 кДж) A0.7)
2Н2(Г) +О2(Г) =2Н2О(Г) (А,Я2°98 =-483,64 кДж) A0.8)
2А1(Т) + Fe2O3(т) = А12О3(т) + 2Fe(T) (Дг#2°98 = -853,8 кДж) A0.9)
Вышеприведенные реакции не происходили из-за различных по сво-
своему характеру препятствий, возникающих на их пути и тормозящих взаимо-
взаимодействие веществ. Торможение может наблюдаться на различных этапах их
пути (в начале, середине и др.). После устранения причин торможения одни
из них приходят в состояние истинного равновесия A0.7), другие A0.8)-
A0.9) протекают практически до конца и уже не возвращаются самопроиз-
самопроизвольно в прежнее заторможенное состояние, так как оно для них было мета-
стабильным.
328 10. Виды, особенности и характеристики химического равновесия
Заторможенное равновесие в природе и технологических процессах
встречается довольно часто. Например, древесина, нефть и другие горючие
вещества находятся в контакте с воздухом и не горят, закаленная сталь так-
также пребывает в состоянии заторможенного равновесия и др.
10.3. Количественные характеристики химического равновесия
Количественно химическое равновесие характеризуют: равновесным
составом реакционной смеси; равновесной степенью превращения (конвер-
(конверсии) реагентов; равновесным выходом продуктов; константой химического
равновесия. Все эти характеристики взаимосвязаны, поэтому, зная одну,
можно рассчитать остальные.
Равновесным называют состав реакционной смеси, соответствующий
состоянию равновесия. Он зависит от природы веществ, участвующих в
равновесии, соотношения их исходных количеств, а также от внешних усло-
условий (температуры, давления). Изменение любого из этих факторов ведет к
изменению равновесного состава. Его обычно выражают через равновесные
молярные {мольные) доли (X ui) веществ-участников равновесия, являю-
являющиеся безразмерными величинами.
Пусть в закрытой системе в изобарно-изотермических условиях (робщ,
Т = const) протекает обратимая гомогенная реакция, описываемая урав-
уравнением
vAA(r) +vBB(r) ±± vDD(r) + vFF(r) A0.10)
реагенты (А и В) и продукты (D и F) которой газообразные вещества. Тогда,
согласно определению (см. § 2.2), равновесные молярные доли веществ-
участников этой реакции рассчитывают следующим образом:
__ Ppasni _
равн / ~~ "^ ~~ "^ ~"
2 ^равн /
где «равн/, Рравн! и [/] — равновесные количества вещества (число молей),
парциальное давление и молярная концентрация /-го участника соответст-
соответственно. При этом J]Рравн/ =Л)бщ — общее давление в равновесной системе
(смеси), а [/] = равн*, где V— ее объем (/ = А; В; D; F).
Равновесному составу смеси отвечают равновесные концентрации или
пропорциональные им равновесные парциальные давления (для реакций с
10.3. Количественные характеристики химического равновесия 329
участием газов). Равновесными концентрациями веществ, образующих сис-
систему, называют концентрации, которые устанавливаются в ней при наступ-
наступлении состояния равновесия. Они зависят от природы реагирующих ве-
веществ, температуры и общего давления, но не зависят друг от друга, и опре-
определяются только положением химического равновесия. При постоянных
температуре и давлении они не изменяются во времени, т.е. постоянны, но
могут принимать различные значения в зависимости от условий установле-
установления равновесия. Чаще всего равновесные концентрации выражают в молях
на литр и называют их равновесными молярными {мольно-объемными) кон-
концентрациями, для обозначения которых обычно используют квадратные
скобки с указанием внутри формулы соответствующего вещества, например
[HI], [H2], [Н+], [Н2О].
В отличие от равновесных, неравновесные — текущие (концентрации
в каждый момент времени до наступления состояния равновесия), а также
начальные {исходные) молярные концентрации веществ обозначают через С
и Со соответственно с индексом в виде формулы вещества: СН1, СНз, Сн+,
^н2о> Qhi? Q)h2> ^он+' он2о-
Равновесное состояние обратимых гомогенных и гетерогенных реак-
реакций, протекающих с участием газов, удобнее характеризовать равновесными'
парциальными давлениями газообразных веществ (ррпвн,-) или если эти реак-
реакции протекают еще и без участия растворителя (инертного газа), т.е. равно-
равновесная смесь состоит только из участников реакции, то — равновесными
молярными долями.
Используя уравнение Клапейрона—Менделеева B.5) и закон Дальто-
Дальтона (см. § 2.1) для смеси идеальных газов, получаем следующие соотношения
между равновесными количеством вещества (числом молей), молярной до-
долей, парциальным давлением и концентрацией:
равн /
у „ . (Ю.П)
,7W (Ю.12)
У реальных систем (газовых смесей, находящихся при общем давле-
давлении порядка десятков мегапаскаль, и обратимых реакций, протекающих в
растворах электролитов и концентрированных растворах неэлектролитов)
наблюдаются отклонения от идеального поведения, обусловленные наличи-
наличием в них сильного межмолекулярного и межионного взаимодействия. Для
более точного описания существующего в них равновесия используют
330 10. Виды, особенности и характеристики химического равновесия
предложенный Дж. Льюисом метод активностей A901). Сущность его со-
состоит в том, что в уравнениях концентрации и парциальные давления (рав-
(равновесные, неравновесные, начальные) заменяют соответствующими актив-
активностями (эффективными, действующими концентрациями) и фугитивностя-
ми (летучестями, исправленными давлениями).
Активностью (фугитивностью) называют величину, при подстановке
которой вместо концентрации (парциального давления) в выражения, выве-
выведенные для идеальных систем, можно применять их к реальным системам.
Определения активности и фугитивности равноценны по смыслу. Актив-
Активность а, и молярные концентрация С,- и доля Xh фугитивность/ и парциальное
давление р{ /-го компонента раствора (смеси) связаны соотношениями:
аа=УасГ> axi
где ус/, 4xi и у/ — молярный и рациональный коэффициенты активности и
коэффициент фугитивности соответственно. Указанные коэффициенты, яв-
являясь безразмерными величинами, учитывают все виды взаимодействий
между частицами в растворе, приводящих к отклонению от идеального по-
поведения. Фугитивность имеет такую же размерность, как и давление, а ак-
активность — такую же, как и концентрация.
Равновесный выход продукта Р есть отношение количества вещества
(числа молей) продукта в состоянии равновесия к его стехиометрическому
количеству, рассчитанному при условии необратимого протекания реакции,
либо отношение соответствующих концентраций. Так, для реакции A0.10)
R _"равнР_ [D] „Q _>VbhF_ [F]
Pd ~ "-"П и Pf ~~
^стех D ^стех F ^"стех F
характеризуют глубину протекания обратимой химической реакции по ве-
веществам D и F.
Положение равновесия может быть охарактеризовано и равновесной
степенью превращения реагента а — отношением количества вещества
превратившегося реагента к его начальному количеству, либо отношением
соответствующих концентраций:
_ ^превр А _ -, ^авн А _ ^превр А _ 1 [А]
аА - -1 или аА -—р; ~ ~г '
W0A W0A Ч)А Ч)А
_ ^преврВ _1 ГСравн в _ Чфевр В _ 1 [В]
CXg — — 1 ИЛИ IXg — — 1 .
Одна из важнейших количественных характеристик химического рав-
равновесия — константа равновесия, позволяющая судить о полноте протека-
10.3. Количественные характеристики химического равновесия 331
ния реакции при тех или иных условиях. В общем случае применительно к
любым химическим системам, как идеальным, так и реальным, константа
равновесия KvaBH обратимой химической реакции есть величина постоянная
при данных температуре, давлении и в данном растворителе, равная отно-
отношению произведения равновесных активностей (или фугитивностей для га-
газов) продуктов реакции, взятых в степенях, равных их стехиометрическим
коэффициентам, к аналогичному произведению для исходных веществ в со-
состоянии химического равновесия. Это определение представляет собой одну
из формулировок закона действующих масс Гульдберга—Вааге для химиче-
химического равновесия. В соответствии с определением константу равновесия
АГраВн выражают и через равновесные активности, и через равновесные фуги-
тивности исходных веществ и продуктов реакции. Таким образом, для обра-
обратимой реакции A0.10) можно записать следующие соотношения:
„ _иС равн D"C равн F _ „
раВН " IF* ~п^ ~ с'
"С равн А "С равн В
У Ра/равн F _ „
jr _ -'равн DJравн F _ ^
равн Va vB J9
JpaBH A ^ равн В
являющиеся термодинамическими выражениями основного закона химическо-
химического равновесия — закона действующих масс, где Кас, Ках, Kj- — термоди-
термодинамические константы равновесия. Они как для идеальных, так и для не-
неидеальных систем зависят от минимального числа факторов: температуры,
давления и природы растворителя, поэтому являются наиболее фундамен-
фундаментальными характеристиками равновесия и чаще всего приводятся в спра-
справочниках.
Для гомогенных химических равновесий, устанавливающихся в иде-
идеальных жидких и газообразных (газовых смесях) растворах, константу рав-
равновесия можно выразить и через равновесные молярные концентрации и
равновесные молярные доли, а для равновесий в газовых смесях — через
равновесные парциальные давления:
J L J =KC; A0.14)
Г вн =
равн [A]Va[B]Vb
^равн D^равн F _
равн А равн В
332 10. Виды, особенности и характеристики химического равновесия
vD vF
Увн Р/^равн F _
Vh va vb ^' A0Л6)
jKpaBH А/^равнВ
Коэффициенты активности и фугитивности компонентов таких систем рав-
равны единице, поэтому активности становятся равными концентрациям, а фу-
фугитивности — давлениям.
Соотношения (ЮЛ 4)—A0.16) представляют собой другие варианты
(формы записи) термодинамического выражения закона действующих масс.
Константы Кс, Кр и Кх иногда называют концентрационными, или эмпиричес-
эмпирическими константами равновесия, поскольку для их расчета используются экспе-
экспериментально определяемые значения [/],/?равн/ и-*раю/- И* размерности можно
вывести непосредственно из этих уравнений: [Кс] = [[/]AVr ] = (моль/м3)АУг или
(моль/л)д\ [Кр] = [р$И] = (Па)^ или (атм)А\ [КХ] = [Х$И] - безраз-
мерная величина, поскольку молярная доля безразмерна.
Взаимосвязь между эмпирическими константами легко установить,
если в выражения A0.14)—A0.16) подставить соотношения A0.11)-A0.13).
Тогда
где VM — мольный объем смеси (раствора); Avr =(vD +vF)-(vA +vB) —
изменение числа молей газообразных веществ, рассчитываемое по разности
между суммами стехиометрических коэффициентов продуктов и реагентов.
Стехиометрические коэффициенты конденсированных (жидких и твердых)
индивидуальных веществ при этом не учитываются (см. § 5.3).
На основании выше приведенного можно сделать следующие выводы.
1. Если газофазная реакция протекает без изменения числа молей газооб-
газообразных веществ-участников, т.е. Avr = 0, то все константы не зависят от обще-
общего (суммарного) давления в системе (при не очень высоких его значениях), без-
безразмерны и их значения равны между собой: Кр-Кс = Кх.
2. Кр = Кх при робщ = 1 атм. Это имеет место, если реакция прово-
проводится в открытой системе при постоянном давлении A атм). Например, в
потоке газов Кс = Кх не только при Avr =0, но и при VM = 1 л/моль.
3. Для смеси идеальных газов константы Кр и Кс не зависят от общего
давления и являются только функциями температуры. Константа равновесия Кх
зависит и от температуры, и от общего давления в системе, так как равновес-
равновесный состав реакционной смеси для реакций, идущих с изменением числа молей
газообразных веществ, как указывалось ранее, зависит от этих параметров.
10.3. Количественные характеристики химического равновесия 333
4. Математические выражения для эмпирических констант К& Кр
а также их числовые значения и размерности (только для Кс, Кр) зависят от
выбора пробега (степени протекания, степени продвижения) реакции ?, т.е.
от формы записи ее стехиометрического уравнения. Например:
?
КрХ = —/?paBHN3H3 = 0,016 МПа при Т= 673 К;
-Рравн N2 -Рравн Н2
2/3
Кр2= ,/равнШз =(^)!/3=0,252 МПа-2/3 приГ=673К;
Рравн N2 Рравн Н2
-Рравн N2 Рравн Н2
=0,126 МПа приГ=673К.
Из рассмотренного примера следует, что для одной и той же реакции в
зависимости от формы записи ее уравнения константа равновесия может
иметь различные размерность и числовые значения.
5. Числовые значения констант равновесия Кр и К^ рассчитанные че-
через равновесные парциальные давления и фугитивности компонентов и вы-
выраженные в паскалях и атмосферах соответственно, не совпадают.
6. Константы равновесия К/иКа при невысоких давлениях обладают
для реальных систем теми же свойствами, что и константы равновесия Кр и
Кс для идеальных систем, т.е. зависят только от температуры.
7. Для газа активность равна фугитивности {а =/), поэтому для реак-
реакций, протекающих в газовой фазе, Ка = Kf. Если еще и давление в системе
невелико, то Ка = К/= Кр, так как/ = ph поскольку у,- = 1. Это справедливо и
для растворов (жидких и твердых) невысокой концентрации а{ = С,-, Ка = Кс.
8. При высоком давлении фугитивность становится совершенно не-
несравнимой с р величиной (может отличаться в несколько раз). В этом случае
константы равновесия Кр и К/ значительно различаются и зависят не только
от температуры, но и от общего давления, даже когда реакция протекает без
изменения числа молей газообразных компонентов (Avr = 0). Зависимость
констант равновесия от давления обусловлена различной сжимаемостью
компонентов газовой смеси. Направление, в котором смещается положение
равновесия при высоких значениях давления, определяется исключительно
334 10. Виды, особенности и характеристики химического равновесия
знаком изменения объема AF, не всегда совпадающим со знаком изменения
количества вещества Avr.
Значения констант химического равновесия определяют его поло-
положение, т.е. относительное содержание исходных веществ и продуктов в
равновесной реакционной смеси. Они изменяются в пределах 0 < Kp2iBH < оо
и никогда не равны нулю (исходные вещества не взаимодействуют) и
бесконечности (исходные вещества практически полностью превращают-
превращаются в продукты). В этих крайних случаях реакции необратимы и к ним за-
закон действующих масс неприменим. Если Краш > 1, то в равновесной ре-
реакционной смеси преобладают продукты: их относительное содержание в
ней выше, чем исходных веществ, т.е. положение равновесия смещено
вправо (->). При ?равн < 1 в равновесной реакционной смеси преобладают
исходные вещества: их относительное содержание в ней выше, чем продук-
продуктов, т.е. положение равновесия смещено влево (<—).
10.4. Кинетический вывод закона Гульдберга—Вааге
Кинетический подход к описанию равновесия рассмотрим на примере
хорошо изученной обратимой газофазной реакции, протекающей в закры-
закрытом сосуде между водородом и йодом:
Н2(г)+12(г) <=*2Н1(г)
И прямая, и обратная реакции имеют общий второй порядок, поэтому
изменение их скоростей происходит в соответствии с параболическим зако-
законом. При этом константа скорости прямой реакции превышает константу
скорости обратной. Порядки реакций по компонентам в данном случае сов-
совпадают со стехиометрическими коэффициентами уравнения. Обратная ре-
реакция бимолекулярна.
В начальный момент времени
(сразу же после введения в сосуд газо-
газообразных Н2 и 12), когда концентрации
Н2 и 12 максимальны (Со Нз = Стах, Со, =
оор
= Стах), скорость прямой реакции мак-
максимальна (го пР = '"max), а обратной равна
г.
Опр
гообр 'равн t нулю (гообр = 0) (рис. 10.1). Затем, по
Рис. ЮЛ. Изменение во времени меРе уменьшения концентраций Н2 и 12
скоростей прямой гпр 1 и обратной и увеличения концентрации HI во вре-
гобр 2 реакций в процессе установ- мени, скорость прямой реакций умень-
ления химического равновесия шается, а обратной — возрастает, но в
(?пр > ?Обр) любой момент времени их значения оп-
10.4. Кинетический вывод закона Гульдберга—Вааге 335
ределяются уравнениями кинетического выражения закона действующих
масс (см. § 7.2):
где ?пр и ?обр — константы скорости прямой и обратной реакций соответст-
соответственно.
И, наконец, наступает такой момент времени, когда скорости прямой
и обратной реакций перестают меняться: гпр = гобр = гравн. Кривые 1 гпр =/@
и 2 гобр — f{t) сливаются в одну прямую линию, параллельную оси абсцисс.
Общая скорость обратимой реакции, т.е. наблюдаемая экспериментально
степень превращения веществ в единицу времени, определяемая как раз-
разность между скоростями прямой и обратной реакций (гОбЩ = гпр - гОбР), стано-
становится равной нулю (см. рис. 10.1). Такое состояние химически реагирующей
обратимой системы называют состоянием динамического равновесия. При
равновесии действие противоположно протекающих реакций взаимно ком-
компенсируется, концентрации всех их участников, называемые равновесными,
постоянны и не изменяются во времени, хотя продолжают одновременно
протекать и прямая, и обратная реакции.
Таким образом, кинетическим критерием {условием) химического
равновесия является равенство скоростей прямой и обратной реакций: г^ = гОбР.
Он указывает на динамический характер и подвижность химического равно-
равновесия: в системе непрерывно протекают и прямая (г„р > 0), и обратная (г^р > 0)
реакции, происходит непрерывный процесс разрушения одних и образова-
образования других частиц.
Для обратимой реакции в условиях равенства скоростей, т.е. при рав-
равновесии, справедливо соотношение
которое удобнее записать иначе:
*пр [HI]2
*обр [Н2][12]
так как константы скоростей не зависят от концентрации.
к
Отношение двух постоянных величин —— в левой части равенства
^обр
также постоянная величина, зависящая, как и сами константы скоростей, от
природы реагирующих веществ и температуры. Ее принято называть кон-
jr ^Пр
стантои равновесия К =——, а правую часть равенства, представляю-
336 10. Виды, особенности и характеристики химического равновесия
щую собой отношение равновесных концентраций участников, — выраже-
выражением константы равновесия и обозначать как Кс.
Это и есть кинетический вывод закона действующих масс для химического
равновесия, впервые предложенный норвежскими учеными К. Гульдбергом и
П. Вааге A867). (В литературе этот закон часто называют их именами.) «Дей-
«Действующими или активными массами» они называли молярные концентрации
реагирующих веществ. В их формулировке закон гласит: для идеальных сис-
систем в состоянии химического равновесия отношение произведения равновес-
равновесных концентраций продуктов реакции в степенях, равных стехиометрическим
коэффициентам, к аналогичному произведению для исходных веществ, есть
величина постоянная при данных температуре, давлении и в данном раство-
растворителе, называемая константой равновесия. В такой формулировке он спра-
справедлив и для реальных разбавленных (с концентрацией менее 0,01М) жидких
и газообразных растворов.
Константа равновесия Кс связывает равновесные молярные концен-
концентрации всех веществ, участвующих в обратимой химической реакции. По-
Поэтому нельзя изменить концентрацию ни одного из них, чтобы это не по-
повлекло за собой соответствующие изменения концентраций всех остальных
участников обратимой химической реакции. Если изменить концентрации
реагентов, то изменятся концентрации и продуктов реакции, но так, что зна-
значение константы равновесия Кс останется неизменным. Значение константы
определяет, каким должно быть соотношение концентраций всех реаги-
реагирующих веществ при равновесии.
Для сложной обратимой реакции, осуществляемой через ряд после-
последовательно протекающих стадий, константу равновесия можно выразить
через константы равновесия этих стадий. Так, например, реакция окисле-
окисления оксида азота (II) кислородом, описываемая суммарным стехиометри-
стехиометрическим уравнением
;f>2NO2
протекает следующим образом:
стадия 1
[NO]
10.4. Кинетический вывод закона Гульдберга—Вааге 337
стадия 2
( кпп^ rxr.ni ^
N2O2+O2^N2O4 \КС2=-^
1;
I W [N2O2][O2]J'
стадия 3
N2O4?=>2NO2 (ксз=^
Каждая из стадий представляет собой обратимую элементарную реак-
реакцию со своим частным равновесием, поэтому ее молекулярность и порядок
совпадают. После несложных преобразований получают соотношение меж-
между константой равновесия суммарной реакции и константами равновесия
отдельных стадий:
К.С — Kq^Kq 2&с 3 ^^^ —
а также выражение константы равновесия Кс сложной обратимой реакции,
записанной через равновесные концентрации участников реакции:
к [NO2]2
С [NO]2[O2]'
Константа скорости реакции в общем виде связана с энергией (Ел =
= АН* + A - Avr)RT) и энтропией (Sa = AS*) активации соотношением:
? = Zexp ехр —- ,
Ч RTJ P{R/
которое подобно уравнению Эйринга (см. § 7.6). Подставляя его вместо кон-
констант скоростей прямой и обратной реакций, находят
к __
{ RT )
RT ) \ R
Z
Принимая во внимание, что отношение —— есть величина постоянная, по-
Zo6p
еле несложных преобразований получают
338 Ю. Виды, особенности и характеристики химического равновесия
^а пр ^а обр | | ^а пр ^а обр
ехр
RT ) { R
¦7Г =еМ п„ » A0Л7)
lepf l exPl
RT ) Ч R ) Ч RT
где АГН = Еапр -Еаобр — изменение энтальпии (тепловой эффект), a ArS =
= ?а пр - Sa обр — изменение энтропии реакции.
В заключение приведем ряд выводов, вытекающих из кинетического
рассмотрения химического равновесия.
1. Существует некоторый общий для прямой и обратной реакций кон-
концентрационный предел, называемый состоянием истинного динамического
химического равновесия, при котором парциальные давления или концен-
концентрации всех участников постоянны во времени.
2. Истинное динамическое химическое равновесие может устанавливать-
устанавливаться только в том случае, если равновесны все элементарные стадии механизма
реакции, — так называемый принцип микроскопической обратимости.
3. Выражение константы равновесия не зависит от пути и механизма
реакции, т.е. сколь бы сложны ни были механизмы прямой и обратной реак-
реакций, в выражение константы входят равновесные концентрации исходных
веществ и продуктов в степенных показателях, равных стехиометрическим
коэффициентам суммарной обратимой реакции.
4. Константа равновесия не зависит от энергии активации реакции, т.е.
от механизма реакции, поэтому его изменение (например, при использова-
использовании катализатора, который понижает энергию активации) не влияет на ее
значение, но изменяет скорость достижения равновесного состояния.
5. Константа равновесия определяет соотношение только равновесных
концентраций реагентов и продуктов реакции, поэтому изменение их ис-
исходных или текущих концентраций, а также общего давления в случае их
невысоких значений не изменяет значение константы равновесия Кс (или
Кр)9 но приводит к смещению положения равновесия. Например, если при
каких-то условиях продуктов реакции оказалось больше их равновесных
значений, то реакция самопроизвольно будет протекать справа налево (в
обратном направлении) до тех пор, пока снова не установится новое поло-
положение химического равновесия, но с прежним значением константы.
6. Если для обратимой реакции кпр > &ОбР, то, независимо от соотноше-
соотношения скоростей гпр ^ гобр до наступления равновесия, в равновесном состоя-
состоянии концентрация продуктов в реакционной смеси будет выше концентра-
концентрации исходных веществ и положение равновесия, как принято говорить, бу-
будет смещено вправо, и наоборот. Увеличивая или уменьшая концентрации
10.4. Кинетический вывод закона Гульдберга—Вааге 339
участвующих в реакции веществ, можно изменять скорости прямой и об-
обратной реакций и смещать тем самым положение химического равновесия в
ту или иную сторону.
7. В соответствии с A0.17) константа равновесия сильно зависит от
изменения температуры: для эндотермических реакций (протекающих с по-
поглощением энергии, т.е. АГН > 0) повышение температуры приводит к уве-
увеличению константы равновесия, для экзотермических (протекающих с вы-
выделением энергии, т.е. АГН< 0) — ее уменьшению.
8. Соотношение A0.17) показывает, что при постоянной температуре
константа равновесия определяется изменениями энтальпии (тепловым эф-
эффектом) и энтропии реакции. Зависимость ее от этих двух факторов (эн-
тальпийного и энтропийного) свидетельствует о влиянии на нее природы
реагентов. Если АГН и ArS имеют разные знаки, то значение константы рав-
равновесия будет либо очень большим (АГН < 0, a ArS > 0), либо очень малым
(АГН> 0, a ArS < 0). Если же АгНи ArS имеют одинаковые знаки, то должна
существовать такая температура, при которой константа равновесия равна
единице.
Термодинамическое описание
химического равновесия
Как упоминалось ранее (см. § 10.2), химически реагирующая сис-
система может прийти в состояние равновесия в результате протекания
и прямой, и обратной реакций. Следовательно, обе реакции могут
протекать самопроизвольно. Однако, согласно законам химической
термодинамики, возможность самопроизвольного протекания пря-
прямой реакции означает невозможность самопроизвольного осущест-
осуществления обратной. Рассмотренный в данной главе термодинамиче-
термодинамический подход к описанию химического равновесия, являясь более
строгим и универсальным, объясняет это кажущееся противоречие.
11.1. Термодинамические критерии.
Термодинамический вывод закона Гульдберга—Вааге
Термодинамическими критериями (условиями) химического
равновесия могут быть различные термодинамические функции
состояния в зависимости от вида термодинамической системы и
условий, в которых протекает в этой системе обратимая химиче-
химическая реакция. Эти критерии вытекают из второго закона (начала)
термодинамики (см. гл. 6). Рассмотрим их для каждого вида тер-
термодинамической системы.
В изолированной системе (см. § 5.1) протекают лишь само-
самопроизвольные процессы, поскольку к ней нельзя подвести энер-
энергию. Все они адиабатны, так как не изменяются внутренняя энергия
(U= const), масса (т = const) и объем (V= const). Если в такой систе-
системе протекает нестатический процесс, в результате которого она при-
приходит в состояние термодинамического равновесия при заданных
условиях, то в таком состоянии энтропия системы в целом достигает
своего максимально возможного и неизменного во времени значе-
значения. В соответствии с этим в любой изолированной системе в со-
состоянии равновесия ее энтропия максимальна и постоянна, т.е.
S = Smax, (ArS)UtV = 0,
(dS)UyV=0, (d2S)UiV<0.
Первые два соотношения соответствуют критериям равновесия в
интегральной форме, вторые два — в дифференциальной.
Из равновесного состояния изолированная система самостоя-
самостоятельно, без вмешательства (сообщения ей энергии) извне, выйти
уже не может. Для этого потребуется нарушение ее изолированности.
ILL Термодинамический вывод закона Гулъдберга—Вааге
341
^нач
"нач
"min
\Аг^обр>0 ArGo6p<0
(г
^кон
^кон
А;В
D;F Z,
Рис. 11.1. Изменение энергии Гиббса
обратимой 1 и необратимой 2 хими-
химических реакций, протекающих в за-
закрытой системе при р, Т = const, в
зависимости от состава исходной ре-
реакционной смеси
Равновесное состояние сохраняется неиз-
неизменным сколь угодно долго, причем оно
не поддерживается каким-либо внешним
по отношению к системе процессом.
Свойства изолированной системы в со-
состоянии равновесия не изменяются.
На практике большинство процес-
процессов протекают в закрытых (замкнутых)
системах (см. § 5.1). Чаще они осуществ-
осуществляются в изобарно-изотермических (р,Т=
= const) и реже — в изохорно-изотер-
мических (V, Т = const) условиях. При
этом критериями направленности и хи-
химического равновесия служат изменения
других термодинамических функций со-
состояния. Если в такой системе, находя-
находящейся в изобарно-изотермических или
изохорно-изотермических условиях, протекает самопроизвольный процесс,
в результате которого она приходит в состояние термодинамического рав-
равновесия, то в таком состоянии энергия Гиббса (или энергия Гельмгольца)
системы в целом достигает своего минимально возможного и неизменного
во времени значения, характерного для данной системы.
Изменение энергии Гиббса обратимой 7 и необратимой 2 химических
реакций, описываемых A0.10) и осуществляемых в закрытой системе в изо-
изобарно-изотермических условиях, в зависимости от состава исходной реак-
реакционной смеси изображено на рис. 11.1. Точка Z отвечает состоянию равно-
равновесия в системе. Ей соответствует минимум на кривой 7 и равновесный со-
состав смеси на оси абсцисс, которому отвечает максимально возможный в
данных условиях выход продуктов. Как следует из рис. 11.1 (кривая 7), к
состоянию равновесия система может подойти как слева, со стороны исход-
исходных веществ, так и справа, со стороны продуктов, что определяется исход-
исходным составом смеси. При этом прямая и обратная реакции протекают само-
самопроизвольно, что отмечено стрелками, и (ArG)PtT < 0. В состоянии равнове-
равновесия энергия Гиббса минимальна, а ее изменение для обеих реакций равно
нулю, т.е. (ArG)pj = 0. После прохождения (слева направо) прямой реакцией
точки, соответствующей минимуму энергии Гиббса на кривой 7, (ArG)p>T для
нее становится положительной величиной, что свидетельствует о самопро-
самопроизвольном протекании в данной области уже обратной реакции, для которой
(ArG)PtT < 0. То же самое наблюдается и для обратной реакции.
Необратимая химическая реакция (рис. 11.1, кривая 2) самопроиз-
самопроизвольно протекает лишь в одном направлении (слева направо), отвечающем
342 И. Термодинамическое описание химического равновесия
превращению исходных веществ в продукты. При этом на протяжении всего
процесса энергия Гиббса уменьшается. Самопроизвольный обратный про-
процесс термодинамически невозможен.
Таким образом, химическое равновесие в закрытой системе характе-
характеризуется минимумом и постоянством энергии Гиббса в случае протекания в
ней изобарно-изотермического процесса или энергии Гельмгольца — в слу-
случае изохорно-изотермического процесса, т.е.
G = Gmin, (ArG)pJ=0 или Л = 4шп> 0MVr=0;
(drG)pJ=09 (d2G)pT>0 или(</гЛ)г,г=О, (d2A)VJ>0.
До сих пор критерии равновесия, устанавливающегося в различных
реагирующих системах, рассматривались с допущением, что его участники
являются индивидуальными веществами, не влияющими на свойства друг
друга. Такой подход для реальных систем — весьма неточен и поэтому для
них используют уже другие термодинамические функции в качестве кри-
критериев. Если в закрытой реальной системе, находящейся в изобарно-
изотермических условиях, протекает самопроизвольный процесс, в резуль-
результате которого она приходит в состояние термодинамического равновесия
при рассматриваемых условиях, то в таком состоянии алгебраическая сум-
сумма произведений стехиометрических коэффициентов v,- и химических по-
потенциалов ц/ для всех веществ, участвующих в процессе (реакции), дости-
достигает минимально возможного и неизменного во времени значения, харак-
характерного для данной системы. В соответствии с этим термодинамический
критерий равновесия для такой системы можно записать в виде соотно-
соотношения
В выражении суммы слагаемые для исходных веществ берутся со зна-
знаком «-», для продуктов — со знаком «+». Это соотношение является общим
условием химического равновесия в системе с переменным составом, так
как аналогичные соотношения получаются и для других функций (критери-
(критериев равновесия A,U,H) при постоянстве соответствующих условий.
Согласно этому критерию для реакции A0.10), все участники которой
газообразные вещества, подчиняющиеся законам идеальных газов, можно
записать:
=o- A1Л)
Для смеси идеальных газов химический потенциал индивидуального
/-го компонента определяется соотношением:
ILL Термодинамический вывод закона Гульдберга—Вааге 343
ц,=ц?+ЛГ1пр„ A1.2)
где ц^ — стандартный химический потенциал /-го компонента газовой сме-
смеси при его относительном парциальном давлении, равном единице (Д = 1);
Pi = -^ — относительное парциальное давление /-го компонента, равное
Р
отношению его парциального давления pt в газовой смеси к стандартному
давлению р° = 1 атм (Д — безразмерная величина, которую для равновес-
равновесной газовой смеси обозначают j?paBH t).
После подстановки выражения A1.2) для состояния равновесия в
уравнение A1.1) и последующего преобразования получают:
(
RT
Поскольку (if зависит только от природы /-го индивидуального ком-
компонента и температуры, то при Т = const левая часть A1.3) есть величина
постоянная, следовательно, постоянна и его правая часть. Левую часть
A1.3) называют стандартной константой равновесия К0:
К° =
правую — ее выражением:
„О _ ^равн Р/^равн F ,« . .,ч
Р ~ hVA nVB ' '
/^равн А^г равн В
Используя A1.4) и A1.4'), получают
°A -увц°в =-RT]nK°p. A1.5)
Как следует из A1.4), стандартная константа равновесия не зависит от
общего давления в системе и является функцией температуры. Поскольку
она безразмерна, то в ее выражение входят также безразмерные величины, в
данном случае относительные равновесные парциальные давления компо-
компонентов j?paBH/. Уравнение A1.4') описывает закон действующих масс Гулъд-
берга—Вааге для химического равновесия, т.е. является одним из термоди-
термодинамических выражений этого закона, вывод которого был сделан в 1885 г.
нидерландским физико-химиком Я. Вант-Гоффом.
344 П. Термодинамическое описание химического равновесия
Используя соотношения
ц,=(ц?)ЧДГ1пС,; iii=tf + RT]n(Xipo6m) = (tfy + RTlnXi, A1.6)
справедливые для идеальных (газообразных и жидких) систем, и соотношения
справедливые для реальных систем, можно получить другие выражения
стандартной константы равновесия:
[rj ^ о _ "равнР paBHF ^0 _ «/равн р/равн F
rO-,VB ' Q=^~A ~~?1 ' /"Т^А ?VB '
L°J ^*равн А равн В /равн А/равн В
где [/'], а /? /равн/ —относительные равновесные концентрация, активность
равн/, /равн/
-^; д= р
С
и фугитивность /-го компонента соответственно, [/] = -^; дравн/
С
/равн/ = paBQHI- ^ последних соотношениях: [/], apaBHhfp2LBHi — абсолютные
равновесные молярная концентрация, активность и фугитивность /-го ком-
компонента соответственно; С0 = 1 моль/л, а0 = С0 = 1 моль/л;/0 =р° = I атм —
стандартные молярная концентрация, активность и фугитивность соответст-
соответственно. Последние две величины принимают равными стандартной концен-
концентрации и стандартному давлению соответственно.
Согласно определению стандартного состояния для газов и конденси-
конденсированных (жидких и твердых) веществ, К® =К°р, К® =К^. Константы рав-
равновесия К\ и Кх равны, так как обе являются безразмерными величина-
величинами. В связи с тем, что (ц^)" в A1.6) зависит от температуры и давления, то
В учебной литературе часто стандартную константу равновесия как
безразмерную величину обозначают А'0. Ее значение вычисляют по термо-
термодинамическим данным: AG0, АА° или A//0, AS0.
Числовые значения стандартной и соответствующих эмпирической и
термодинамической констант равновесия совпадают. Взаимосвязь между
различными константами равновесия устанавливают, используя закон Даль-
Дальтона, уравнение состояния Клапейрона—Менделеева и соотношения между
относительными и абсолютными величинами, например:
Кс = К°С(С°Г'; Кр = K°p(p°)Av'; Кх =
11.2. Уравнения изотермы, изобары и изохоры химической реакции 345
N-Avr
Анализируя вышеприведенные соотношения, можно сделать два вы-
вывода:
если pi и р° измерены в атмосферах, то константы Кр и К°р численно
равны. В единицах СИ такого совпадения нет;
если газофазная реакция протекает без изменения числа молей ве-
веществ-участников (Avr = 0), то все константы безразмерны, а их значения
равны.
11.2. Уравнения изотермы, изобары и изохоры химической реакции
Связь между константами равновесия и термодинамическими харак-
характеристиками системы устанавливается уравнениями изотермы, изобары и
изохоры химической реакции. Эти уравнения впервые были выведены
Я. Вант-Гоффом A886), поэтому в литературе их часто называют его име-
именем, а именно: изотерма, изобара и изохора Вант-Гоффа. Каждое из них
имеет несколько разновидностей и используется либо в дифференциальной,
либо в интегральной формах. Уравнение изотермы в общем виде связывает
энергию Гиббса, константу равновесия и начальные (или текущие), т.е. не-
неравновесные концентрации (активности в случае реальных растворов) или
неравновесные парциальные давления (фугитивности в случае реальных
газовых смесей) всех компонентов химической реакции.
Рассмотрим смесь идеальных газов, находящуюся в закрытой системе
в изобарно-изотермических условиях, в которой квазистатически (см. § 5.1)
протекает реакция, описываемая уравнением A0.10). Предположим, что в
начальный момент времени (/ = 0) в реакционной смеси присутствуют и реа-
реагенты (А и В), и продукты (D и F), относительные неравновесные парциаль-
парциальные давления которых равны рА, рв, pD, pF соответственно. Если реак-
реакция совершает один пробег (^ = 1), то
i
Подставляя в последнее выражение уравнения A1.2) и A1.5), получают со-
соотношение
(ArG)D T =-RTlnK°D+ RTln^*P*
Pa Pb
346 П. Термодинамическое описание химического равновесия
которое и называют уравнением изотермы химической реакции в общем ви-
виде. Это уравнение имеет несколько разновидностей: для реакций, проте-
протекающих в идеальных растворах, в него входят константа равновесия К\
(или Кх) и относительные неравновесные молярные концентрации С, (или
неравновесные молярные доли Xt) компонентов; при описании реальных
систем (твердых, жидких, газообразных) неравновесные концентрации и
неравновесные парциальные давления заменяют на активности at и парци-
парциальные фугитивности /г соответственно.
По уравнению изотермы вычисляют энергию Гиббса реакции, а также
определяют направление и глубину ее самопроизвольного протекания при
данных условиях, если известны стандартная константа равновесия для дан-
данной температуры и исходный состав произвольно приготовленной реакци-
реакционной смеси, т.е. неравновесные относительные, например, парциальные
давления pi всех компонентов в начальный момент времени. Очевидно, что
чем больше абсолютное значение (ArG) T, тем менее равновесна система.
Проанализируем это уравнение. Если в начальный момент времени
компоненты газовой смеси взяты в таком соотношении, при котором:
а) в реакционной смеси присутствуют либо только исходные вещества
(А) = Р? ~ 0)> либо только продукты (рА = рв = 0), то в соответствии с
уравнением изотермы (ArG)pT ->±oo для любой реакции. При таких усло-
условиях величина (ArG)pT не может служить критерием самопроизвольного
протекания реакции, поэтому в термодинамике за исходное берут такое со-
состояние, при котором в системе присутствуют все реагирующие вещества;
б) ——— > К®, т.е. второе слагаемое по абсолютной величине боль-
Рк Рвв
ше первого в уравнении изотермы, тогда (ArG) т > 0, что соответствует
самопроизвольному протеканию реакции в обратном направлении (справа
налево) до установления равновесия;
в) р Л <^р> т-е. первое слагаемое по абсолютной величине боль-
РааРвв
ше второго в уравнении изотермы, тогда (ArG)p т < 0, что соответствует
самопроизвольному протеканию реакции в прямом направлении (слева на-
направо) до установления равновесия;
г) величины, стоящие под знаком логарифма у первого и второго сла-
слагаемых в уравнении изотермы становятся равными между собой, т.е.
11.2. Уравнения изотермы, изобары и изохоры химической реакции 341
К°р = в ^ , тогда (ArG)p T = О, что отвечает состоянию химического
Р1АРв*
равновесия в закрытой системе, так как исходная реакционная смесь в этом
случае имеет равновесный состав;
д) величина, стоящая под знаком логарифма во втором слагаемом, бу-
будет равна единице. Это возможно, например, при начальных относительных
парциальных давлениях всех компонентов, равных единице, т.е. когда ком-
компоненты находятся в стандартных состояниях. Тогда само слагаемое стано-
становится равным нулю, а уравнение изотермы трансформируется:
{ArG)pT =-RTlnK°p или ArG* =-RTlnK°p A1.7)
и его называют стандартным уравнением изотермы химической реакции,
или уравнением стандартного сродства. С помощью A1.7) можно вычис-
вычислить значение К® без экспериментального изучения равновесия. Подстав-
Подставляя в него уравнение Гиббса—Гельмгольца для стандартных условий в виде
получают
1п^о=_А^+А^ (И8)
р RT R
Соотношение A1.8) выражает зависимость константы равновесия от
температуры в интегральной форме. Входящие в него величины ArHj и ArSj
зависят от природы реагирующих веществ и температуры реакции в соответст-
соответствии с уравнениями Кирхгофа (см. § 6.6). Для невысоких значений Г и в узком
температурном интервале такой зависимостью можно пренебречь и считать
ArHj = const и ArSj = const. Тогда с учетом этих допущений зависимость
In К® от — A1.8) выражается прямой линией, тангенс угла наклона а к
( АгНт)
оси абсцисс которой равен —-—— , а отрезок Оу, отсекаемый ею на оси
I R )
(A S0]
ординат, равен г т (рис. 11.2). Угол наклона прямой зависит от знака и
V R )
величины теплового эффекта реакции: для экзотермической (Дг#? < 0) —
0° < а < 90°, a tg а > 0, для атермической (с нулевым тепловым эффектом
348
11. Термодинамическое описание химического равновесия
У1
У2
Уз
а=0°
(Дг#? = 0))—а = 0 и tg а = 0, для
эндотермической (АГН® > 0) —
90° < а < 180°, a tga<0 (см.
рис. 11.2, прямые 1,2,3 соот-
соответственно).
Из уравнения A1.8) и
рис. 11.2 следует, что
Такой метод вычисления
о х 1_ к теплового эффекта и энтропии
т' реакции называют расчетом по
Рис. 11.2. Зависимость натурального логариф- второму закону термодинамики,
ма стандартной константы равновесия от об- Обычно его используют, если
ратной абсолютной (термодинамической) тем- невозможно непосредственно
пературы для экзотермической 7, атермичес- экспериментально определить
кой 2 и эндотермической 3 реакций тепловой эффект (например, ре-
реакция осуществима только при
высоких температурах). Обратим внимание также на то, что в подобных
графических расчетах тангенс угла вычисляют из отношения длин соответ-
соответствующих катетов (Оу и Ох), взятых в определенных единицах измерения,
но не по углу наклона на графике, зависящему от произвольно выбранного
масштаба.
Уравнение A1.8) можно использовать и в относительно широком
интервале температур, если в него вместо величин Дг#? и ArS^ подста-
подставить их средние значения АгН®р и Аг^р для выбранного температурного
интервала.
Уравнение изобары химической реакции определяет зависимость кон-
константы равновесия К°р от температуры и в дифференциальной форме выво-
выводится на основании стандартного уравнения изотермы A1.7):
A1.9)
и дифференциального уравнения Гиббса—Гельмгольца для стандартных
условий:
dT
11.2. Уравнения изотермы, изобары и изохоры химической реакции 349
Последнее соотношение после несложных преобразований принимает вид
dT{ Т ) Т2
Используя A1.9) и A1.10), получают искомое уравнение изобары:
d\aK°D д Hi
dT RT2
Его можно также записать в виде
d{\IT) R
A1.11)
П112)
Анализируя A1.11), можно установить, что чем больше абсолютная
величина теплового эффекта и чем ниже температура, тем больше значение
производной, следовательно, сильнее сказывается изменение температуры
на константе равновесия и в большей степени смещается положение равно-
равновесия.
Уравнение A1.12), как было показано выше, полезно при вычислении
графическим путем тепловых эффектов реакций, если температурная зави-
зависимость константы равновесия выражена в координатах \пК°р .
Интегрируя уравнение A1.11) в узком интервале температур, в преде-
пределах которого можно считать Дг#° = const, получают уравнение изобары в
интегральной форме
1п
К°рХ RT2TX
С помощью этого уравнения можно рассчитать тепловой эффект реак-
реакции, если известны значения константы равновесия при двух температурах, и
пересчитать их с одной температуры на другую, зная тепловой эффект.
Если газофазная реакция A0.10) осуществляется в закрытой системе
при постоянном объеме (V = const), что характерно также и для реакций,
протекающих в растворах, то уравнения Гиббса—Гельмгольца и стандарт-
стандартной изотермы Вант-Гоффа для таких процессов имеют следующий вид:
350 11- Термодинамическое описание химического равновесия
Из последних выражений с помощью преобразований, аналогичных исполь-
использованным при выводе уравнения изобары, можно получить следующие раз-
разновидности уравнения изохоры химической реакции:
в дифференциальной форме
г/
с _
dT RT2
в интегральной форме
In
LC1
в виде
K°cl RT2TX
где Аги® — изменение стандартной внутренней энергии реакции; К^ —
безразмерная стандартная константа равновесия, выраженная через отно-
относительные равновесные молярные концентрации компонентов [/].
Из термодинамического рассмотрения химического равновесия выте-
вытекает ряд важных выводов.
1. Обратимая химическая реакция может самопроизвольно проте-
протекать как в прямом (ArGnp)pT < 0, так и в обратном (ArGo6p)pj < 0 на-
направлениях, все определяется исходными условиями, а именно темпера-
температурой и соотношением компонентов в реакционной смеси в начальный
момент времени.
2. Ив прямом, и в обратном направлениях реакция протекает не до
конца, а только до определенного предела, называемого состоянием дина-
динамического химического равновесия и определяемого равенством (ArGnp) T =
= (ArGo6p)p г= 0. Этому равенству удовлетворяет бесчисленное множество
парциальных давлений или концентраций компонентов.
3. В состоянии равновесия энергия Гиббса системы минимальна, а
парциальные давления или концентрации всех участников постоянны во
времени. Система из любого произвольно заданного состояния самопроиз-
самопроизвольно стремится к состоянию равновесия, что подчеркивает его термоди-
термодинамическую устойчивость и выгодность. При этом к равновесному состоя-
состоянию система может подойти как со стороны исходных веществ, так и со
стороны продуктов.
11.3. Правило фаз Гиббса 351
4. Самопроизвольно выйти из равновесного состояния система не мо-
может, так как произойдет увеличение концентраций или парциальных давле-
давлений продуктов одной из реакций — либо прямой, либо обратной. Это при-
приведет к тому, что значение (ArG) T такой реакции станет положительным
и система самопроизвольно вернется к состоянию равновесия.
11.3. Правило фаз Гиббса
Правило фаз (уравнение) Гиббса, известное также как закон фазового
равновесия, было сформулировано в 1876 г. Оно относится к одному из об-
общих законов физики и химии и является выражением второго закона термо-
термодинамики применительно к фазовым равновесиям. С его помощью можно
описать и гомогенные, и гетерогенные химические равновесия, а именно:
термодинамически установить (предсказать) возможность или невоз-
невозможность существования данной гомогенной или гетерогенной системы;
вычислить максимальное число фаз, которые одновременно могут на-
находиться в равновесии при данных условиях;
априори характеризовать фазовый состав сколь угодно сложной мно-
многокомпонентной системы при заданном числе независимых переменных,
определяющих равновесие;
предсказать по числу степеней свободы поведение исследуемой сис-
системы, т.е. установить варианты возможных фазовых превращений при изме-
изменении одного, двух и более внешних условий (при сохранении остальных
параметров состояния системы постоянными).
В общем виде правило фаз Гиббса формулируется следующим обра-
образом: число термодинамических степеней свободы, или вариантность равно-
равновесной термодинамической системы, С определяется как разность числа не-
независимых компонентов системы К и числа сосуществующих фаз Ф плюс
число внешних факторов и, влияющих на равновесие. Его математическое
выражение записывается в виде простого соотношения:
Анализируя это соотношение, можно сделать следующие выводы:
число степеней свободы равновесной системы возрастает с увеличе-
увеличением числа независимых компонентов и понижается с увеличением числа
сосуществующих фаз;
число степеней свободы может быть равно либо нулю, либо целому
положительному числу, т.е. С > 0;
352 11• Термодинамическое описание химического равновесия
так как число степеней свободы не может быть отрицательным, то
число сосуществующих фаз в равновесной термодинамической системе в
общем случае не может превышать суммы К + и.
Числом термодинамических степеней свободы, или вариантностью
системы С, называют максимальное число независимых интенсивных термо-
термодинамических параметров состояния фаз равновесной системы, произвольное и
независимое изменение которых в определенных пределах не влияет на качест-
качественный и количественный фазовый состав системы (т.е. на вид и число фаз), а
именно не вызывает исчезновения одних и образования других фаз.
По числу степеней свободы системы подразделяют: на ин- или безва-
безвариантные (С = 0); на одно- или моновариантные (С = 1); на двух- или дива-
риантные (С = 2) и т.д. У инвариантных систем изменение любого их пара-
параметра состояния ведет к изменению числа фаз. У моновариантных систем
один из их параметров состояния может быть изменен без изменения числа
фаз, находящихся в равновесии. Дивариантные системы позволяют изме-
изменять в них сразу два параметра состояния в определенных пределах без из-
изменения числа фаз.
Независимыми компонентами называют составляющие систему
вещества, наименьшее число которых необходимо и достаточно для об-
образования и однозначного выражения состава каждой фазы данной сис-
системы при любых условиях ее существования. Понятие «независимый
компонент» родственно, но не тождественно понятию «составляющее
вещество» и является химическим аналогом математического понятия
«независимая переменная». Независимые компоненты системы — это те
ее составляющие вещества, равновесные концентрации которых можно
изменять произвольно (в определенных пределах), не вызывая изменения
числа и вида фаз в системе. По их числу различают системы одно-, двух-,
трех- и т.д. компонентные.
Составляющими веществами (составными частями) или просто
компонентами системы называют индивидуальные химические вещества
(простые или сложные), которые входят в ее состав, могут быть выделены
из нее простыми препаративными методами (кристаллизацией, выпаривани-
выпариванием, осаждением и др.) и которые могут существовать вне ее самостоятельно,
т.е. в изолированном виде. Так в системе, образованной солью и водой (на-
(например, водный раствор хлорида натрия), обе ее составные части независи-
независимые компоненты, поскольку они могут быть выделены из нее и существо-
существовать в виде индивидуальных химических соединений. Их количества можно
изменять независимо друг от друга, пока раствор является ненасыщенным.
Ионы Na+ и СГ, содержащиеся в таком растворе, составляющими вещества-
веществами не являются, так как они не могут быть выделены в химических реакци-
реакциях и не могут существовать изолированно.
11.3. Правило фаз Гиббса 353
В системе, между составляющими веществами которой нет химиче-
химического взаимодействия, число независимых компонентов равно числу состав-
составляющих веществ (В), т.е. все вещества, образующие систему, являются ее
независимыми компонентами. Тогда эти понятия тождественны. Например,
механическая смесь Zn, C2H5OH и КЫОз представляет собой трехкомпо-
нентную систему, каждое вещество которой является одновременно незави-
независимым компонентом и ее составной частью. Можно произвольно менять
количество цинка, при этом количества этилового спирта и нитрата калия
останутся без изменения.
В системе, между составными частями которой осуществляется взаи-
взаимодействие и при этом протекает необратимая химическая реакция, число
независимых компонентов определяется состоянием системы в момент пре-
прекращения реакции и равно числу ее составных частей. Например, в закры-
закрытой системе, в которой протекает необратимая реакция между стехиометри-
ческими количествами цинка и серной кислоты в виде водного раствора,
после ее окончания будут присутствовать сульфат цинка, вода и водород,
т.е. три независимых компонента. Если же взять для реакции избыток цинка,
то число независимых компонентов при этом возрастает на единицу и ста-
становится равным четырем, так как в системе после окончания реакции будет
присутствовать еще и непрореагировавший цинк.
При установлении в системе равновесия в результате протекания обра-
обратимой химической реакции появляются дополнительные количественные свя-
связи, определяемые уравнением закона действующих масс и другими независи-
независимыми математическими соотношениями. В этом случае число независимых
компонентов всегда меньше числа всех реально присутствующих в системе
веществ и находится как разность между числом составляющих веществ сис-
системы (В) и числом независимых математических уравнений (соотношений)
(У), связывающих равновесные концентрации или равновесные парциальные
давления этих веществ: К = В - У. В зависимости от способа образования и
условий существования равновесной системы при одном и том же числе со-
составляющих ее веществ число независимых компонентов может быть различ-
различным. Например, равновесная смесь газов N2, H2 и NH3 может быть получена
двумя способами: либо по реакции, описываемой уравнением
N2(r)+3H2(r)^2NH3(r)
либо по реакции, описываемой уравнением
2NH3(r)?±N2(r)-f3H2(r)
Число составляющих веществ в обоих случаях равно трем, но в пер-
первом мы имеем дело с двухкомпонентной системой, а во втором — с одно-
12 - 9795
354 П. Термодинамическое описание химического равновесия
компонентной. Если азот и водород в первом процессе взять в произвольном
соотношении, то равновесные концентрации либо пропорциональные им
равновесные парциальные давления всех трех газов системы связаны между
собой только одним уравнением:
] Р
[NH3] РравнШз /111,14
f
р
поэтому число независимых компонентов равно двум, т.е. для получения
равновесного состава газовой фазы достаточно произвольно задать концен-
концентрации или парциальные давления любых двух из трех составляющих ве-
веществ системы. Концентрацию (парциальное давление) третьего вещества
при равновесии можно определить из уравнения A1.14), т.е. она будет иметь
строго определенное значение.
Во втором случае равновесные концентрации или равновесные парци-
парциальные давления трех газов системы связаны между собой уже двумя урав-
уравнениями:
л2
[NH3f или
[[H2] = 3[N2]
/7равнЫ2/7равнН2
~
Рравп Н2 -
A1.15)
поэтому число независимых компонентов равно единице, т.е. равновесный
состав газовой фазы может быть получен, если взять только один компонент
(NH3), при термическом разложении которого остальные (N2 и Нг) получаются
в таких соотношениях, которые удовлетворяют системе уравнений A1.15).
При подсчете числа независимых компонентов в системе необходимо
учитывать уравнения, связывающие только равновесные концентрации или
пропорциональные им равновесные парциальные давления, но не равновес-
равновесные количества составляющих веществ. Это особенно важно в случае гете-
гетерогенных систем. Например, в равновесных системах, образующихся в ре-
результате термического разложения СаСОз(Т) и NH4C1(T) по реакциям:
CaCO3(T)?±CaO(T)+CO2(r) (KC=[CO2] или Кр=рравиС02)
NH4Cl(T)^NH3(r)+HCl(r)
^Кс = [NH3 ] [НС1], _ I Кр = Рравн NH3 Рравн НС1 >
[[NH3]=[HC1]
при одинаковом числе составляющих веществ, равном трем, число незави-
независимых компонентов различно: для первой оно равняется двум, для второй —
единице.
11.3. Правило фаз Гиббса 355
Хотя в первой системе число молей СаО(Т) равно числу молей СОг (Г>,
но [СаО] Ф [СОг], />равн сю * Ррюн со2 > так как Ф^ы различны по своим объ-
объемам, а равновесная молярная концентрация СаО(Т) постоянна и равна отно-
отношению плотности вещества к его молярной массе (/?равнсао— равновесное
парциальное давление паров СаО над твердым СаО). Такая система может
быть построена из СаСОз и СаО, СаО и СОг, СаСО3 и СОг, но она не может
стать однокомпонентной, даже если число молей СаО окажется равным
числу молей СОг.
Во второй системе оба продукта реакции (NH3 и НС1) газообразные
вещества, поэтому появляется еще одно независимое дополнительное мате-
математическое уравнение, связывающее их равновесные молярные концентра-
концентрации или равновесные парциальные давления между собой. Благодаря ему
число независимых компонентов в этой системе по сравнению с первой
снижается на единицу.
Число независимых компонентов в системе зависит от характера
взаимодействия ее веществ и условий, в которых она находится. Так, при
нагревании достаточного количества карбоната аммония в закрытой системе
устанавливается при некоторой постоянной температуре гетерогенное рав-
равновесие, описываемое уравнением
(NH4JCO3(T) <=> 2NH3(r) +СО2(Г) + Н2О(г)
Число независимых уравнений, связывающих равновесные концентра-
концентрации или равновесные парциальные давления всех веществ системы, равно трем:
уравнение закона действующих масс:
Кс =[NH3]2[CO2][H2O] или Кр =?
уравнения, связывающие равновесные концентрации или равновесные
парциальные давления газообразных продуктов:
[СО2 ] = [Н2О], /?Равн СО2 = Л>авн Н2О >
[H2O] = 0,5[NH3] ИЛИ /7равнН2о =0,5/7равнШз.
Тогда число независимых компонентов К = 4 - 3 = 1.
Если смешать эти четыре вещества при очень низкой температуре, ко-
когда они все будут находиться в кристаллическом состоянии, и химического
взаимодействия между ними при этом происходить практически не будет,
то система окажется уже четырехкомпонентной.
Аналогичными рассуждениями можно показать, что при высоких
температурах гомогенная система Н2(г)- О2(г)- Н2О(г), в которой имеет ме-
место равновесие:
12*
356 11. Термодинамическое описание химического равновесия
2Н2(Г)+О2(Г)^2Н2О(Г)
является двухкомпонентной, а при низких (например, комнатных), когда
можно считать, что химического взаимодействия практически не происхо-
происходит, так как крайне низка скорость реакции, число независимых компонен-
компонентов в этой системе становится равным трем и совпадает с числом ее состав-
составляющих веществ.
Рассмотренные примеры показывают, что для ряда систем не всегда
удается установить, из каких конкретно независимых компонентов они со-
состоят. Для них можно определить лишь их число, которое всегда равно тому
наименьшему числу химически индивидуальных веществ, которое доста-
достаточно для образования всех фаз данной системы.
Число сосуществующих фаз Ф в системе определяется индивидуально
в каждом конкретном случае. При этом считают:
а) идеальные газы, неограниченно смешивающиеся друг с другом, об-
образуют однофазную систему, поэтому смесь индивидуальных газов, т.е. лю-
любое их число, составляет одну фазу;
б) конденсированные (твердые и жидкие) вещества, неограниченно
растворяющиеся друг в друге, образуют также одну фазу;
в) при их ограниченной взаимной растворимости или практически
полной нерастворимости друг в друге число фаз равно числу индивидуаль-
индивидуальных веществ в системе.
По числу фаз системы подразделяют на одно-, двух-, трех- и т.д. фаз-
фазные. Системы, состоящие из одной фазы, называют гомогенными, из двух и
более — гетерогенными.
Смесь газообразных азота, водорода и аммиака (N2(r)-H2(r)-NH3(r)),
равновесие в которой описывается уравнением реакции A0.7), представляет
собой гомогенную однофазную систему. Твердые и жидкие сплавы золота и
серебра, растворы воды и этилового спирта также являются однофазными
системами. Совокупность кристаллов какой-либо соли составляет одну фа-
фазу, так как каждый из кристаллов однороден по химическому составу и фи-
физическим свойствам и подобен любому другому кристаллу из их совокупно-
совокупности, хотя и отделен поверхностью раздела. Отсюда следует, что наличие по-
поверхности раздела — только один из признаков фазы.
Системы вода—анилин (Н2О(ж)-С6Н5>Ш2(ж)) и свинец—олово
(Pb(T)-Sn(x)) двухфазны, механическая смесь кристаллов CsNCb, C11SO4,
PbS, KC1 состоит из четырех твердых фаз. Неустойчивая смесь кристаллов
Na2SC>4, Na2SO4 • Н2О, Na2SO4 • 5Н2О представляет собой трехфазную систе-
систему, так как каждое вещество не только отличается от остальных по содер-
11.3. Правило фаз Гиббса 357
жанию воды, но и имеет определенную кристаллическую структуру, а сле-
следовательно, отличается и по физическим свойствам.
Системы, содержащие твердые и газообразные вещества, а также равно-
равновесия, устанавливающиеся в них, всегда являются гетерогенными, например:
С(т) - °2 (г) ~ С02 (г) С(т) + °2 (г) ?
СаСО3(т)-СаО(т)-СО2(г) СаСО3(т) <^СаО(т) +СО2(г)
Fe2O3(T)-Fe(T)-CO(r)-CO2(r) Fe2O3(T) + 3CO(r) ^ 2Fe(T) + 3CO2(r)
Первая из этих систем — двухфазная и состоит из одной твердой и
одной газовой фазы, вторая и третья — трехфазные и состоят из двух твер-
твердых и одной газовой фазы. Фазы могут существовать в любом из трех агре-
агрегатных состояний вещества. Примером многофазной системы, все фазы ко-
которой представляют собой различные агрегатные состояния веществ, служит
трехфазная система вида: пар воды—водный раствор соли—твердая соль.
К внешним факторам, влияющим на состояние равновесия любой
системы (гомогенной или гетерогенной), относятся температура, давление,
внешние поля (электрическое, магнитное, гравитационное и др.). Их общее
число обозначают п. Хотя влияние внешних полей на поведение химических
систем велико, о нем говорят лишь в тех случаях, когда их напряженности
существенно отличаются от нормальных значений. В обычных условиях
(температура комнатная, т.е. Т « 20 °С, давление атмосферное, т.е. р да
«101 кПа, ускорение силы тяжести g » 9,8 м • с, напряженность магнитно-
магнитного поля Н да 40 А/м, напряженность электрического поля Е да 130 В/м, осве-
освещенность видимым светом Ev да 500 лк) влияние этих полей на состояние
равновесия настолько незначительно, что его обычно не учитывают, и рас-
рассматривают воздействие только двух важнейших факторов: температуры и
давления, поэтому п = 2, а математическое выражение правила фаз для та-
таких условий принимает вид
С = К-Ф + 2.
Если в системе один из факторов (давление или температура) сохра-
сохраняется постоянным, тогда п = 1, а число степеней свободы, называемое в
этом случае условным, рассчитывают следующим образом:
Сусл = К-Ф+1.
Это же уравнение применяют в тех случаях, когда давление вообще не
влияет на смещение положения равновесия, например для реакции
2Н1(г) ^±Н2(г) + 12(Г), либо когда изучаемая система находится под атмо-
атмосферным внешним давлением и не содержит летучих веществ.
358
11. Термодинамическое описание химического равновесия
Если оба фактора (и температура, и давление) в системе поддержива-
поддерживаются постоянными, то п = 0, а
Сусл=К-Ф.
Для однокомпонентных систем, у которых составы двух равновесных
фаз, например жидкой и парообразной, одинаковы, при расчете условного
числа степеней свободы следует учитывать еще одно уравнение, связываю-
связывающее концентрации компонентов, выраженные в массовых или мольных до-
долях ю„япи> = сопяпН|, , Хплю!_ =ХЖ„,_. В этом случае следует пользо-
уравнением
Сусл = К-Ф+1.
Физический смысл правила фаз Гиббса прост: вариантность системы
С выводится как разность между числом независимых термодинамических
параметров состояния и числом связывающих их независимых уравнений.
Из линейной алгебры известно, что при равенстве числа независимых пере-
переменных и числа уравнений система имеет единственное решение, что соот-
соответствует инвариантной термодинамической системе (все параметры жестко
связаны друг с другом). Если число независимых переменных на единицу
больше числа уравнений, то одна переменная (при постоянных значениях
других) может принимать любые значения (в термодинамике — в опреде-
определенных пределах), что соответствует моновариантной системе и т.д.
В табл. 11.1 обобщены результаты рассмотрения по правилу фаз Гиббса
различных равновесных систем (гомогенных и гетерогенных, химически
реагирующих и нереагирующих).
Таблица 11.1
Характеристика различных равновесных систем
Равновесная система
Число
фаз Ф
независи-
независимых компо-
компонентов К
внешних
факторов
п
степеней
свободы
С
Гетерогенная реагирующая
С(Т)+СО2(Г)?±2СО(Г)
СаСО3(г)^СаО(т) + СО2(г)
2Fe(T) +ЗСО2(Г) *=* Fe2O3(T) +3CO(r)
NH4Cl(T)^NH3(r)+HCl(r)
(NH4JCO3(T) *± 2NH3(r) +CO2(r) +H2O(r)
2(т,г)
3Bт,г)
3Bт,г)
2(т,г)
2(т,г)
3-1=2
3-1=2
4-1=3
3-2=1
4-3 = 1
2(Р,Г)
2<Р,Т)
1(Г)
2<Р,Г)
2(Р,Т)
2
1
1
1
1
11.3. Правило фаз Гиббса
359
Продолжение табл. 11.1
Равновесная система
Число
фазФ
независи-
независимых компо-
компонентов К
внешних
факторов
п
степеней
свободы
С
Гомогенная реагирующая
Н2(г)+12(г)^2Н1(г)
N2(r)+3H2(r)^2NH3(r)
2NH3(r)?±N2(r)+3H2(r)
2Н2(Г)+О2(Г)?^2Н2О(Г)
1(г)
1(г)
3-2=1
3-1=2
3-1=2
3-2=1
3-1=2
ЦТ)
ЦТ)
2(р,Т)
2(Р,Т)
2{р,Т)
1
2
3
2
3
Гетерогенная нереагирующая
CsNO3 (т) -КС1(Т) -CuSO4 (T) -PbS(T)
Na2SO4(i)-Na2SO4 -H2O(T)-Na2SO4 -5H2O(T)
(NH4JCO3(T)-NH3(TL:O2(T)-H2O(T)
2(т,ж)
2(ж,г)
4Dт)
3Cт)
4Dт)
1-0=1
1-0=1
4-0 = 4
3-0 = 3
4-0 = 4
ЦТ)
2{р,Т)
ЦТ)
ЦТ)
ЦТ)
0
1
1
1
1
Гомогенная нереагирующая
Н2(г)"°2(г)~Н2°(г)
1(г)
3-0 = 3
2{р,Т)
4
Влияние различных факторов
на химическое равновесие.
12 ¦ Особенности равновесий
НИ в гетерогенных системах
Различные факторы по-разному влияют на равновесие: одни уско-
ускоряют либо замедляют его наступление, другие смещают его поло-
положение. И то, и другое оказывается полезным в химической техно-
технологии: ускорение наступления состояния равновесия сокращает
продолжительность технологического процесса (цикла), смещение
положения равновесия в прямом направлении увеличивает выход
продуктов и степень превращения реагентов.
12.1. Введение катализатора и изменение
концентрации компонентов
Катализатор — вещество (твердое, жидкое или газообраз-
газообразное, ионы, молекулы или радикалы), изменяющее скорость термо-
термодинамически возможной реакции. Он вступает во взаимодействие
на различных стадиях реакции, но в конце ее пути остается хими-
химически неизменным, т.е. катализатор не является ни реагентом, ни
продуктом обратимой химической реакции. Сущность каталитиче-
каталитического действия, как правило, состоит в уменьшении энергии акти-
активации реакции вследствие сильного взаимодействия катализатора с
активированным комплексом и слабого — с реагентами и продук-
продуктами (см. гл. 9).
Катализатор не влияет на константу равновесия, поэтому не
вызывает смещения его положения. Ускоряя в равной степени как
прямую, так и обратную реакции, он способствует более быстрому
установлению равновесия, т.е. сокращению времени, необходимого
для его достижения (рис. 12.1). Катализатор приводит реакцию к
тому же состоянию равновесия, в которое она приходит самостоя-
самостоятельно без него, но только за больший промежуток времени. Если
скорость реакции невелика, что чаще всего характерно для гетеро-
гетерогенных обратимых реакций, то равновесие в них устанавливается
за десятки, а иногда даже и тысячи часов. Напротив, гомогенные
обратимые реакции (в газовых системах или водных растворах)
протекают с очень большой скоростью, поэтому равновесие в них
устанавливается почти мгновенно. Придя в состояние равновесия,
термодинамическая система будет находиться в нем до тех пор, пока
12.1. Введение катализатора и изменение концентрации компонентов 361
внешние условия (концентрации ве-
веществ, температура, давление), оп-
определяющие это состояние, будут
оставаться неизменными.
Изменение состояния равно-
равновесия в результате изменения внеш-
внешних условий называют, как уже
упоминалось, смещением или сдви-
сдвигом положения равновесия. Чаще
всего приходится сталкиваться со
смещением положения равновесия:
а) в результате изменения концен-
концентраций либо парциальных давлений
'"обр
гОобр
'равн к
'равн
Рис. 12.1. Изменение во времени скоро-
(для обратимых гомогенных и гете- стей прямой гпр и обратной гОбР реакций
рогенных химических реакций, про- в процессе установления химического
текающих с участием газов) одного равновесия с катализатором 1 и без него 2
(^^)
или сразу нескольких веществ- (^пР
участников равновесия; б) вследствие изменения температуры системы; в) в
результате изменения общего давления в системе; г) за счет введения в газо-
газообразную равновесную систему инертного газа (газа, не участвующего в
реакции), т.е. разбавления ее. Иногда изменяются одновременно все внеш-
внешние условия.
Влияние изменения внешних условий на состояние равновесия уста-
устанавливается термодинамическим положением, впервые сформулированным
А. Ле Шателье в общем виде в 1885 г. и затем в 1887 г. теоретически обос-
обоснованным К. Брауном, которое называют правилом смещения положения
подвижного равновесия, или принципом Ле Шателье—Брауна: если на сис-
систему, находящуюся в состоянии истинного химического равновесия, оказы-
оказывать внешнее воздействие путем изменения какого-либо из условий (С,-, Г, ph
Робщ)? определяющих положение равновесия, то в системе происходит изме-
изменение равновесного состава и смещение положения равновесия в направле-
направлении того процесса, протекание которого ослабляет эффект (влияние) этого
воздействия.
. При изменении концентрации направление смещения положения рав-
равновесия определяется тем, какой из компонентов вводится в равновесную
систему, т.е. берется в избытке, а степень его смещения — стехиометриче-
скими коэффициентами в уравнении. Так, увеличение в равновесной систе-
системе концентрации одного или сразу всех исходных веществ путем введения в
нее дополнительных их количеств вызывает смещение положения равнове-
равновесия в сторону образования продуктов (т.е. расходования части дополнитель-
дополнительно введенных веществ) и, наоборот, повышение концентрации одного или
362 12. Влияние различных факторов на химическое равновесие
нескольких продуктов по той же причине смещает положение равновесия в
сторону исходных веществ. При этом чем больше стехиометрический коэф-
коэффициент при компоненте в уравнении реакции, тем существеннее смещается
положение равновесия.
Влияние этого фактора можно рассмотреть на примере обратимой го-
гомогенной реакции синтеза аммиака NH3 (табл. 12.1). Увеличение концен-
концентрации либо водорода, либо азота (либо обоих компонентов) выводит сис-
систему из состояния равновесия и приводит к увеличению знаменателя в кон-
константном отношении. Значит, в системе должна пройти такая реакция, кото-
которая ослабила бы этот эффект, т.е привела бы к увеличению числителя и
уменьшению знаменателя в этом отношении. Для этого положение равнове-
равновесия должно сместиться вправо, в сторону понижения концентрации введен-
введенного вещества. При переходе к новому равновесному состоянию концентра-
концентрации Н2 и N2 уменьшаются, a NH3 — увеличивается до тех пор, пока их от-
отношение вновь не будет равно Ко При добавлении Н2 равновесие смещается
в большей степени, чем при добавлении того же количества N2.
К аналогичному выводу можно прийти и на основании кинетического
подхода к рассмотрению равновесия. Например, увеличение концентрации
Н2 приведет к возрастанию скорости прямой реакции, не повлияв при этом
на скорость обратной. Следовательно, концентрация NH3 будет возрастать
до тех пор, пока скорости обеих реакций вновь не сравняются, а это про-
произойдет тогда, когда отношение новых концентраций всех веществ будет
равно Ко Уменьшение концентрации компонентов вызывает противопо-
противоположное смещение положения равновесия. Во всех случаях система стремит-
стремится либо восполнить убыль вещества за счет расходования других веществ
системы, либо превратить в другие вещества его избыток.
Изменяя состав системы, можно добиться протекания реакции в лю-
любом из двух противоположных направлений. Вводя в систему избыток того
или иного вещества-участника равновесия или, наоборот, выводя его из сис-
системы, можно регулировать выход продуктов реакции, увеличивая или
уменьшая его. Этим широко пользуются на практике. Так, в аналитической
химии при необходимости более полного перевода какого-либо иона в оса-
осадок прибавляют избыток реактива, осаждающего этот ион из раствора.
Таким образом, изменение в равновесной системе концентрации лю-
любого из компонентов или концентраций всех компонентов: не влияет на
константу равновесия Кс\ смещает положение равновесия в ту или иную
сторону, изменяя тем самым равновесные состав реакционной смеси, кон-
концентрации компонентов, выход продуктов реакции, степень превращения
исходных веществ.
Изменение парциальных давлений газообразных компонентов действует
на смещение положения равновесия аналогично изменению их концентраций.
Влияние изменения концентраций компонентов на смещение положения химического равновесия
и изменение его количественных характеристик согласно принципу Ле Шателье—Брауна
Таблица 12.1
Равновесная гомогенная
химически реагирующая
система и ее константы
равновесия
Изменение
концентрации
компонентов
Соотношение скоростей
прямой гп и обратной гоб
реакций после изменения
концентраций
Направление смещения
положения равновесия
Изменение количественных
характеристик равновесия
N2(r)+3H2(r)<z±2NH3(r)
Avr=2-(l + 3) = -2
2
„ _ Ррави NH3
Kp~ з
Рравн N2 Рравн Н2
r _ tNH3]2
C [N2][H2]3
Кх=-
X
равн NH3
L равн N2 л равн Н2
Повышение
концентрации
реагентов
гпр > гобр . Возрастает ско-
скорость прямой реакции, по
которой вводимое веще-
вещество расходуется
Вправо (—») - в сторону
прямой реакции, по
которой вводимое ве-
вещество расходуется
Равновесные выход и концен-
концентрации продуктов увеличива-
увеличиваются. Константа равновесия
не изменяется. Изменяется
равновесный состав смеси
Понижение
концентрации
реагентов
гпр
гобР • Уменьшается
скорость прямой реакции,
в которой реагентом явля-
является вещество, концентра-
ция которого уменьшается
Влево («—) - в сторону
обратной реакции, по
которой образуется
вещество, концентрация
которого уменьшается
Равновесные выход и концен-
концентрации продуктов уменьша-
уменьшаются. Константа равновесия
не изменяется. Изменяется
равновесный состав смеси
Повышение
концентрации
продуктов
гпр < гобР • Возрастает ско-
скорость обратной реакции,
по которой вводимое
вещество расходуется
Влево (<-) - в сторону
обратной реакции, по
которой вводимое ве-
вещество расходуется
Равновесная степень превраще-
превращения реагентов уменьшается, их
равновесные концентрации
увеличиваются. Константа рав-
равновесия не изменяется. Изменя-
ется равновесный состав смеси
Понижение
концентрации
продуктов
гпр > гобР • Уменьшается
скорость обратной реак-
реакции, в которой реагентом
является вещество, кон-
концентрация которого умень-
уменьшается
Вправо (-») - в сторону
прямой реакции, по
которой образуется
вещество, концентрация
которого уменьшается
Равновесная степень превраще-
превращения реагентов увеличивается,
их равновесные концентрации
уменьшаются. Константа рав-
равновесия не изменяется. Изменя-
Изменяется равновесный состав смеси
364 12. Влияние различных факторов на химическое равновесие
12.2. Изменение температуры
Чтобы предсказать направление смещения положения равновесия при
изменении температуры, необходимо знать знак теплового эффекта реак-
реакции. Большинство химических реакций сопровождаются либо поглощением
энергии в виде теплоты (эндотермические реакции, АГН > 0), либо ее выде-
выделением (экзотермические реакции, АГН < 0). При этом если прямая реакция
эндотермическая, то обратная экзотермическая и наоборот. Для прямой и
обратной реакций АГН (АГЯ°), а также ArS (ArS°), ArG (ArG°), In К (In K°)
равны по абсолютной величине и противоположны по знаку. Влияние тем-
температуры на эндотермические и экзотермические реакции, а следовательно,
на состояние и положение химического равновесия различно.
При повышении температуры положение равновесия всегда смещает-
смещается в направлении протекания эндотермической реакции, при понижении —
в направлении протекания экзотермической реакции. Для атермических ре-
реакций, не сопровождающихся тепловыми эффектами, АГН= 0, и изменение
температуры не нарушает равновесия. Этот вывод Вант-Гоффа является ча-
частным случаем принципа Ле Шателье—Брауна. Он применим ко всем хи-
химическим системам вне зависимости от агрегатного состояния реагентов и
продуктов в них.
При изменении температуры прежде всего изменяются константы
скоростей прямой и обратной реакций, причем в различной степени. Следо-
Следовательно, изменяется значение константы равновесия и равновесный состав
реакционной смеси.
Более строгое рассмотрение вопроса о смещении положения равнове-
равновесия при изменении температуры решается путем анализа дифференциально-
дифференциального уравнения изобары A1-11) или изохоры A1.13) химической реакции. Из
A1.11) и A1.13) следует, что направление температурного изменения кон-
константы равновесия определяется только знаком теплового эффекта реакции
ArHj (ArUj). Чем больше численное значение АГН® {ArUj) для одного и
того же температурного интервала, тем сильнее сказывается влияние изме-
изменения температуры на смещение положения равновесия.
Для эндотермических реакций ArHj > 0. В правой части выражения
A1.11) Т > 0, R > 0, следовательно, первая производная логарифма констан-
(d\nK°D } 0
ты равновесия по температуре положительна — >0 , т.е. 1пЛ^ и са-
v )
ма величина К° возрастающие функции температуры. При повышении
12.3. Изменение температуры 365
температуры значения К°р и Кр увеличиваются, что, согласно закону дей-
действующих масс A0.16), A0.140 соответствует смещению положения равно-
равновесия в сторону прямой (эндотермической) реакции.
Для экзотермических реакций АГН® < 0, что соответствует отрица-
отрицательной производной логарифма константы равновесия по температуре
(dlnK°D ) 0 0
— <0 , таким образом, 1пКр, Кр, Кр являются убывающими
функциями температуры, т.е., согласно закону действующих масс, при уве-
увеличении Т равновесие смещается в сторону обратной (эндотермической)
реакции.
0 (dlnK°D )
Для атермических превращений ArHj=0, - = 0 , следова-
l dT J
тельно, 1пК^9 К®р, Кр не зависят от температуры, т.е. положение химиче-
химического равновесия не смещается с изменением температуры.
Итак, по знаку АГН® можно точно предсказать характер изменения
константы равновесия и направление смещения его положения при измене-
изменении температуры, но невозможно указать в каком интервале значений будет
происходить ее изменение.
Примеры влияния изменения температуры на смещение положения
химического равновесия рассмотрены в табл. 12.2.
12.3. Изменение давления и разбавление системы
инертным газом
Влияние изменения давления на смещение положения равновесия, а
следовательно, и на значение равновесных концентраций компонентов про-
проявляется практически только при наличии в системе газов. Чтобы устано-
установить взаимосвязь между характером изменения давления и направлением
смещения положения равновесия, логарифмируют уравнение Кх = К°р^^
а затем полученное соотношение {\пКх = 1пА^-Дуг1пробщ) дифференци-
дифференцируют при постоянной температуре (при этом К°р = const, lnA^J = const):
d\rvKx =-Avrdlnp^=-Avr^ = -Avr^. A2.1)
А>бщ Робщ
Таблица 12.2
Влияние изменения температуры на смещение положения химического равновесия
и изменение его количественных характеристик согласно принципу Ле Шателье—Брауна
Равновесная гомогенная
химически реагирующая
система и ее константы
равновесия
СОС12(Г)^СО(Г)+С12(Г)
Avr =A + 1)-1 = 1
v Рравн СО Ррявн С12
КР ~
PpaBHCOClj
к [СО][С12]
С [СОС12]
у у
v __ лравнСОлравнС12
у
лравнСОС12
КР=КР(Р ) =
= KC(RTI
Тепловой эффект
прямой реакции и
изменение температуры
АгЯ>0,
повышение
температуры
АгЯ>0,
понижение
температуры
Соотношение скоро-
скоростей прямой Гпр и
обратной гобр реак-
реакций после измене-
изменения температуры
системы
гпр > г обр. В боль-
большей степении воз-
возрастает скорость
прямой (эндотер-
(эндотермической) реак-
реакции
гпр < г обр. В боль-
большей степени по-
понижается скорость
прямой (эндотер-
(эндотермической) реакции
Направление
смещения положения
равновесия
Вправо (—») — в
сторону прямой
(эндотермической)
реакции
Влево (<—) — в
сторону обратной
(экзотермической)
реакции
Изменение количественных
характеристик равновесия
Равновесные концентрации
реагентов уменьшаются, а
продуктов увеличиваются.
Константы равновесия,
равновесные степень пре-
превращения реагентов и вы-
выход продуктов увеличива-
увеличиваются. Изменяется равно-
равновесный состав смеси
Равновесные концентрации
реагентов увеличиваются,
а продуктов уменьшаются.
Константы равновесия,
равновесные степень пре-
превращения реагентов и вы-
выход продуктов уменьша-
уменьшаются. Изменяется равно-
равновесный состав смеси
Окончание табл. 12.2
Равновесная гомогенная
химически реагирующая
система и ее константа
равновесия
N2(r)+3H2(r)<=>2NH3(r)
Avr=2-(l + 3) = -2<0
v РравнШз
Р " п п3
Гравн N2 ^равн Н2
Г - [ШЗ]2
С [N2][H2]3
Y2
„ ЛравнМН3
Y у3
лравн1Ч2лравнН2
= ЛГС(Л7Т2
Тепловой эффект
прямой реакции
и изменение температуры
АгН<0,
повышение
температуры
АгН<0,
понижение
температуры
Соотношение скоро-
скоростей прямой гпр и
обратной гобр реак-
реакций после измене-
изменения температуры
системы
гпр<г обр- В боль-
большей степени воз-
возрастает скорость
обратной (эндо-
(эндотермической) ре-
реакции
гпр > ''обр- В боль-
большей степени по-
понижается скорость
обратной (эндо-
(эндотермической) ре-
реакции
Направление
смещения положения
равновесия
Влево D— ) — в
сторону обратной
(эндотермической)
реакции
Вправо (—>) - в
сторону прямой
(экзотермической)
реакции
Изменение количественных
характеристик равновесия
Равновесные концентрации
реагентов увеличиваются,
а продуктов уменьшаются.
Константы равновесия,
равновесные степень пре-
превращения реагентов и вы-
выход продуктов уменьша-
уменьшаются. Изменяется равно-
равновесный состав смеси
Равновесные концентрации
реагентов уменьшаются, а
продуктов увеличиваются.
Константы равновесия,
равновесные степень пре-
превращения реагентов и вы-
выход продуктов увеличива-
увеличиваются. Изменяется равно-
равновесный состав смеси
368 12. Влияние различных факторов на химическое равновесие
Преобразуя A2.1), получают:
*г; ^М*=_^г_ A2.2)
"Робщ РоЪщ
ИЛИ
AV
так как для реакций с участием газов, согласно уравнению состояния иде-
идеального газа, роъщАУ = AvrRT. Поскольку константа равновесия Кх зависит
и от температуры, и от давления, то в уравнениях A2.2) и A2.2*) полную
производную при Т= const заменяют на частную:
(зькЛ __av,. (аъьЛ _jy__ (|23)
Jr у фобщ )т Робщ { фобщ )т RT
Соотношения A2.3) называют дифференциальными уравнениями Планка—
Ван Лаара. Анализируя их, можно сделать следующие выводы:
чем меньше изменение числа молей газообразных веществ в обрати-
обратимой химической реакции, т.е. чем меньше изменение объема системы, тем
менее значительно общее давление влияет на смещение положения химиче-
химического равновесия;
если обратимая реакция протекает без изменения числа молей газооб-
газообразных веществ, т.е. если объем системы не меняется (Avr = О, AV = 0), то
(д\пКх) Л
— = 0; константа равновесия Кх не зависит от давления, изменение
V Фобщ )т
его практически не влияет на смещение положения химического равновесия
и его количественные характеристики. Например, ни увеличение, ни
уменьшение общего давления не сказывается на равновесном выходе HI в
реакции Н2 (г) +12 (г) ^ 2Н1(г), так как Avr = 2 - A +1) = 0;
для реакций, сопровождающихся увеличением числа молей газооб-
газообразных веществ, т.е. увеличением объема системы (Avr > 0, AV> 0), част-
частная производная логарифма константы равновесия Кх по общему давлению
(или логарифму общего давления) при постоянной температуре отрицатель-
(д\пКх) л ( д\пКх ) л - „
на, — < 0, — <0; \пКх и Кх являются убывающими
V Фобщ )т 151п^общЛ
функциями общего давления, следовательно, с его увеличением величины
12.3. Изменение давления и разбавление системы инертным газом 369
In Kx и Кх уменьшаются, положение равновесия смещается в сторону исход-
исходных веществ, т.е. обратной реакции, идущей с уменьшением объема, или
числа молей газов;
для реакций, протекающих с уменьшением числа молей газообразных
веществ, т.е. уменьшением объема системы (Avr< О, AV< 0), частная произ-
водная — положительна; с ростом общего давления значения In Kx
\ Фобщ )т
и Кх увеличиваются, положение равновесия смещается в сторону прямой
реакции, т.е. продуктов.
Эти выводы согласуются с принципом Ле Шателье—Брауна, в соот-
соответствии с которым при увеличении давления равновесие смещается в сто-
сторону той (прямой или обратной) реакции, которая сопровождается умень-
уменьшением объема (газообразных продуктов или реагентов соответственно), а
при уменьшении давления — в противоположную сторону.
Примеры влияния изменения общего давления на смещение положе-
положения химического равновесия в различных системах рассмотрены в табл. 12.3.
Если в реакциях участвуют только конденсированные фазы (твердые веще-
вещества и жидкости), то влияние изменения давления на смещение положения
равновесия подчиняется тем же закономерностям, что и для газофазных ре-
реакций, рассмотренных в табл. 12.3. Так как данное количество вещества,
находящегося в конденсированном состоянии, занимает объем примерно на
три порядка меньший, чем в газообразном, то относительное изменение объ-
объема у таких реакций очень незначительно. В связи с этим существенного
смещения положения равновесия таких процессов можно достигнуть лишь
при очень больших давлениях (порядка 102 МПа и выше). Поэтому в реаль-
реальных условиях считают, что изменение внешнего давления в таких системах
не приводит к заметному нарушению равновесия. При изменении давления,
как и при изменении концентрации компонентов, значения Кр и Кс не изме-
изменяются.
Как видно из табл. 12.1-12.3, максимальный выход аммиака дости-
достигается при увеличении концентраций азота и водорода, уменьшении кон-
концентрации образующегося аммиака, при высоком давлении и низкой
температуре.
Разбавление равновесной газовой смеси путем накачивания в нее
инертного газа, например аргона, можно осуществлять:
при постоянных давлении и температуре, т.е. в изобарно-изотермичес-
ких условиях (/?общ = const, Г= const);
при постоянных объеме и температуре, т.е. в изохорно-изотермичес-
ких условиях (V= const, T= const).
Влияние изменения общего давления на смещение положения равновесия и изменение
его количественных характеристик согласно принципу Ле Шателье—Брауна
Таблица 12.3
Равновесная гомогенная химически
реагирующая система и ее константы
равновесия
lJNO2(r)^2NO(r)+O2(r)
Avr =B + l)-2 = l>0
2
v РраънЫоРраънО2
к [NO]2[O2]
С [NO2]2
у2 у
лравнШлравнО2
X2
равн NO2
Изменение
общего
давления
Повышение
Понижение
Соотношение скоростей
прямой i^p и обратной
гобР Реакций после
изменения робщ в системе
гпр < гобР • в большей
степени возрастает
скорость обратной
реакции, протекающей
с уменьшением числа
молей газообразных
веществ, т.е. справа
налево
гпр > гобР • в большей
степени убывает ско-
скорость обратной реак-
реакции, протекающей с
уменьшением числа
молей газообразных
веществ, т.е. справа
налево
Направление
смещения
положения
равновесия
Влево (<—) — в сторо-
сторону реагентов, т.е.
меньшего числа молей
газообразных веществ
Вправо (—») — в сто-
сторону продуктов, т.е.
большего числа молей
газообразных веществ
Изменение
количественных
характеристик
равновесия
Равновесные концен-
концентрации реагентов уве-
увеличиваются, а продук-
продуктов уменьшаются.
Константа равновесия
Кх, равновесные сте-
степень превращения
реагентов и выход
продуктов уменьша-
уменьшаются. Изменяется рав-
равновесный состав смеси
Равновесные концен-
концентрации реагентов
уменьшаются, а про-
продуктов увеличивают-
увеличиваются. Константа равно-
равновесия КХ9 равновесные
степень превращения
реагентов и выход
продуктов увеличива-
увеличиваются. Изменяется рав-
равновесный состав смеси
Продолжение табл. 12.3
Равновесная гомогенная химически
реагирующая система и ее константы
равновесия
2)N2(r)+3H2(r)^2NH3(r)
Avr=2-(l + 3) = -2<0
кр з
Рравн N2 Рравн Н2
к _ [NH3]2
С [N2][H2]3
X2
v ЛравнМН3
&х — '
X X
лравнМ2лравнН2
Изменение
общего
давления
Повышение
Понижение
Соотношение скоростей
прямой г^ и обратной
г^ реакций после
изменения робщ в системе
гпР > гобР • в большей
степени возрастает
скорость прямой реак-
реакции, протекающей с
уменьшением числа
молей газообразных
веществ, т.е. слева
направо
ГпР<ГобРВб°ЛЬШеЙ
степени убывает ско-
скорость прямой реакции,
протекающей с умень-
уменьшением числа молей
газообразных веществ,
т.е. слева направо
Направление
смещения
положения
равновесия
Вправо (-») — в сто-
сторону продуктов, т.е.
меньшего числа молей
газообразных веществ
Влево («-) — в сторо-
сторону реагентов, т.е.
большего числа молей
газообразных веществ
Изменение
количественных
характеристик
равновесия
Равновесные концен-
концентрации реагентов
уменьшаются, а про-
продуктов увеличивают-
увеличиваются. Константа равно-
равновесия Кх, равновесные
степень превращения
реагентов и и выход
продуктов увеличива-
увеличиваются. Изменяется рав-
равновесный состав смеси
Равновесные концен-
концентрации реагентов уве-
увеличиваются, а продук-
продуктов уменьшаются.
Константа равновесия
Кх, равновесные сте-
степень превращения
реагентов и выход
продуктов уменьша-
уменьшаются. Изменяется рав-
равновесный состав смеси
Окончание табл. 12.3
Равновесная гомогенная химически
реагирующая система и ее константы
равновесия
3)N2(r)+O2(r)?=*2NO(r)
Avr=2-(l + l) = 0
у Ррэяп NO
КР
Ppam N2 Рравн О2
к [N0]2
C [N2][O2]
X2
v _ Л равн NO
К. у —
У У
л равн N2 л равн О2
Кр= К с = Кх
Изменение
общего
давления
Повышение
или
понижение
Соотношение скоростей
прямой г^ и обратной
гобР реакций после
изменения ро6ш в системе
Гпр=ГобРСк°РОСТИ
прямой и обратной
реакций изменяются
(увеличиваются или
уменьшаются) в оди-
одинаковое число раз
Направление
смещения
положения
равновесия
Изменение общего
давления не влияет на
смещение положения
химического равнове-
равновесия
Изменение
количественных
характеристик
равновесия
Количественные ха-
характеристики не изме-
изменяются, так как сме-
смещения положения
химического равнове-
равновесия не происходит
12.4. Особенности описания равновесия в гетерогенных системах 373
В первом случае объем системы увеличивается (он пропорционален
., nRT ч
числу молей п газов, V = ), что приводит к уменьшению молярных
Р
концентраций Ci, = — и парциальных давлений pt = X^^^ исходных ве-
веществ и продуктов реакции. Создается эффект уменьшения (понижения)
давления в системе, ведущий к смещению положения равновесия в соответ-
соответствующем направлении. Например, для обратимой реакции 2 (см. табл. 12.3)
в сторону исходных веществ, т.е. влево, так как в ходе прямой реакции про-
происходит уменьшение количества газообразных веществ и объема системы;
произведение pvm^lp]mni уменьшается сильнее, чем />равнШз, а это тре-
требует при Кр = const дополнительного уменьшения р^щ за счет разложения
аммиака. Поэтому разбавление инертным газом реакционной смеси в изо-
барно-изотермических условиях с целью увеличения выхода продуктов по-
полезно только для тех обратимых реакций, которые протекают с увеличением
объема и количества газообразных веществ (см. табл. 12.3, реакция 1). Дей-
Действие инертного газа в этом случае равносильно уменьшению давления в
системе. Если же Avr = 0, то система будет нечувствительна к присутствию
инертного газа (табл. 12.3, реакция 3).
Во втором случае при накачивании инертного газа в автоклав общее
давление в равновесной газовой смеси возрастает пропорционально увели-
увеличению количества вводимого инертного газа, но при щ - const молярные
концентрации С,- = — и парциальные давления pt = CtRT исходных ве-
веществ и продуктов реакции сохраняются постоянными, следовательно, разбав-
разбавление инертным газом в этом случае не влияет на химическое равновесие, т.е.
не смещает его положение.
12.4. Особенности описания равновесия
в гетерогенных химических системах
В гетерогенных системах вещества находятся в различных фазах. Если
вещества химически не взаимодействуют, то такую систему называют химиче-
химически нереагирующей системой. В гетерогенных химически реагирующих систе-
системах вещества чаще всего взаимодействуют на границе раздела фаз. Равновесие,
устанавливающееся в таких системах, называют гетерогенным химическим
равновесием. Основные закономерности и особенности его установления рас-
рассмотрим на трех важных в практическом отношении реакциях.
3 74 12. Влияние различных факторов на химическое равновесие
1. Гетерогенная реакция, протекающая при высокотемпературной кор-
коррозии железа в атмосфере углекислого газа
2Fe(T) + ЗСО2(Г) <=± Fe2O3(T) + ЗСО(г)
Чтобы вывести выражение стандартной константы равновесия К®
считают, что эта реакция протекает при рш, Т = const. Применяя к ней общее
условие равновесия АгЦ = ^\'1-ц/ =0, получают:
^Fe2O3 + 3^СО - 2HFe ~ 3Иш2 = °- О2'4)
Полагая, что газообразные вещества подчиняются законам идеальных
газов, находят их химические потенциалы для равновесного состояния:
Нсо =1^со + кт1пРРавнСо> цСОг = ц^ + RT]nppuBHC02. A2.5)
Химические потенциалы конденсированных (твердых и жидких) веществ (в
данном случае БегОз и Fe), если они не образуют твердых и жидких раство-
растворов, не зависят от парциальных давлений и являются в изобарно-изо-
термических условиях постоянными величинами, равными при р° = 1 атм
значениям их стандартной энергии Гиббса образования: ЦРе2о3 = Д/G? Fe2o3!
HFe = Ay-G°Fc (см. табл. П.5.1).
Подставляя A1.15) в A1.14), получают
A2.6)
РравнСО2
Левая часть этого равенства при Т = const — постоянная величина, поэтому
.Рравн СО г
и правая часть, а значит, и -^ , должны быть постоянными величина-
Рравн СО2
ми. Согласно определению стандартной константы равновесия примени-
применительно к рассматриваемой обратимой гетерогенной химической реакции,
K1 ) ^равнСОз РравнСО2
Кр-—Ъ -JiT). A2.7)
Уравнение A1.17) показывает, что в выражение константы равновесия
данной (и любой) гетерогенной реакции входят только парциальные давле-
давления газообразных компонентов.
2. Гетерогенная реакция, в которой участвует только одно твердое
вещество, а все остальные газы:
С(т)+ СО2(Г) <=± 2СО(Г); Дг#2°98 = 172,47 кДж; АГ52°98 = 175,66 Дж/К.
12.4. Особенности описания равновесия в гетерогенных системах 375
Данную реакцию используют дня получения генераторного газа СО. Она
является эндотермической, поэтому при нагревании углерода в атмосфере СО2
в условиях закрытой системы с ростом температуры положение равновесия
смещается вправо. При этом часть твердого углерода переходит в газовую фа-
фазу. Наоборот, при понижении температуры положение равновесия смещается
влево так, что часть углерода выделяется из газовой фазы. Таким образом, уг-
углерод транспортируется через газовую фазу. Подобные реакции называют
транспортными и широко применяют в технике для очистки цветных, редких
и рассеянных металлов, выращивания кристаллов нелетучих и неплавких ве-
веществ, получения металлических порошков и различных пленочных покрытий.
А Н°
При Т « г ™ » 918,84 К » 708,69 °С (температурной зависимостью
^/•^298
Аг//^ и AS® пренебрегают) в системе устанавливается состояние химиче-
химического равновесия, характеризуемое минимумом энергии Гиббса, равенством
скоростей прямой и обратной реакций и отсутствием изменений параметров
и функций состояния системы. При этой температуре движущая сила реак-
реакции равна нулю, так как ее энтальпийный и энтропийный факторы взаимно
компенсируют (уравновешивают) друг друга, а К*р = ^равн— = 1. В случае,
/*равн СО2
если Т < 918,84 К, то ArG^>0, K^<1, положение равновесия смещено
влево, самопроизвольно протекает обратная реакция, которая приводит сис-
систему к состоянию равновесия. Если Т> 918,84 К, то ArGj <0, Кр >1, по-
положение равновесия смещено вправо, самопроизвольно протекает прямая
реакция, которая приводит систему к состоянию равновесия.
3. Гетерогенная реакция, в которой участвует только одно газообраз-
газообразное вещество, а остальные находятся в твердых фазах:
СаСО3(т)^СаО(Т) + СО2(г)
(Дг#2°98 = 177,89 кДж; АГ52°98 = 160,48 Дж/К).
Данная реакция лежит в основе промышленного способа получения
негашеной извести СаО и углекислого газа СО2 из известняка СаСОз путем
его термического разложения. Она также является эндотермической, поэто-
поэтому с повышением температуры положение равновесия смещается вправо. В
этом же направлении оно смещается и при увеличении объема системы, При
этом часть СаСОз разлагается, но равновесная концентрация СО2 остается
. (псоЛ „
неизменной . При уменьшении объема, т.е. повышении давления,
)
3 76 12. Влияние различных факторов на химическое равновесие
часть С02 реагирует с СаО с образованием СаСО3. О равновесии в этой ре-
реакции можно говорить только в том случае, если она протекает в условиях
закрытой системы. Если карбонат кальция СаСОз нагревать в таких услови-
условиях, то с увеличением температуры давление СО2 будет расти до тех пор, пока
выдержат стенки аппарата (сосуда). Если его разлагать в условиях открытой
А Н°
системы, то при Г« r f8 «1108,49 К (~ 835,34 °С) ^=?равнсо2 =1> те-
давление СО2, называемое давлением диссоциации^ станет равным атмо-
атмосферному A01,3 кПа). Температуру, при которой давление диссоциации
равно атмосферному и установление равновесия в системе невозможно, на-
называют температурой разложения. Остальные выводы аналогичны выво-
выводам для реакции 2. Дальнейшее повышение температуры системы оказыва-
оказывается невозможным вследствие того, что равновесное давление СО2 должно
превысить 101,3 кПа. Но так как система открытая, то СО2 покидает ее, по-
поэтому положение равновесия не может сдвинуться вправо и вся подводимая
теплота расходуется на разложение СаСОз. Новое равновесное состояние в
такой системе установиться не может, процесс идет до конца, т.е. до полно-
полного исчезновения фазы СаСОз. Таким образом, при Т> 1108,49 К гетероген-
гетерогенная реакция протекает необратимо в прямом направлении вследствие того,
что один из продуктов (СО2) удаляется из сферы реакции и состояние сис-
системы в этом случае можно сравнить с состоянием кипящего чайника — вся
подводимая теплота идет на испарение воды и так продолжается до тех пор,
пока вся вода не выкипит.
Анализ уравнений A2.4)—A2.7) позволяет сделать следующие выводы
об особенностях равновесия в гетерогенных системах.
1. Конденсированные фазы (жидкие и твердые), не улетучивающиеся
в ходе реакции и не переходящие полностью в раствор и их количества не
оказывают влияния на равновесие и его положение.
2. В выражение константы равновесия Кр обратимых гетерогенных ре-
реакций входят только равновесные парциальные давления газообразных ве-
веществ, взятые в степенях стехиометрических коэффициентов, например, для
реакций 2 и 3 Кр-—— , и Кр= ppgLBilco2 соответственно. Давление
насыщенного пара чистых конденсированных веществ при данной темпера-
температуре постоянно, поэтому при записи выражения Кр его вводят под знак кон-
константы равновесия.
3. В выражение констант равновесия Кси К® гетерогенных обрати-
обратимых реакций не входят равновесные молярные концентрации чистых твер-
твердых и жидких веществ. Это связано с тем, что эти концентрации не зависят
12.4. Особенности описания равновесия в гетерогенных системах Ъ11
от количества конденсированной фазы и постоянны во всей области суще-
существования данных агрегатных состояний этих веществ. В соответствии с оп-
определением стандартного состояния конденсированных веществ их относи-
относительные молярные концентрации также постоянны, и их принимают равными
единице. Таким образом, значения абсолютных и относительных моляр-
молярных концентраций чистых твердых и жидких веществ, как молекулярные
константы, входят в неявной форме в значения констант равновесия Кс и
Кс, которые определяются в этом случае только значениями равновесных
концентраций газообразных веществ.
4. Константы Кр и К^9 Кс и К^ зависят от свойств всех веществ,
участвующих в гетерогенной реакции, а не только газов. Например, для ре-
реакций термического разложения СаСОз и ВаСОз выражения констант рав-
равновесия совпадают (Кр = /?равнСо2> ^с = [СО2]), но их значения при одной
и той же температуре будут разными, так как различаются химические по-
потенциалы веществ СаСО3, СаО, ВаСО3, ВаО.
5. Для обратимых гетерогенных реакций введение дополнительного
количества жидких или твердых компонентов (реагентов или продуктов) в
реакционную смесь или их выведение из нее не влечет за собой изменения
состава и, следовательно, не нарушает состояния равновесия, т.е. не смеща-
смещает его положения. Например, для обратимой гетерогенной реакции:
Си2О(т)+2НС1(Г) <=> 2СиС1(ж) + Н2О(г)
добавление в систему избытка хлороводорода смещает положение равнове-
равновесия в ней вправо, в то время как введение дополнительных количеств твер-
твердого оксида меди (I) на состояние равновесия в этой системе не влияет.
6. При изучении влияния изменения давления на смещение положения
равновесия в обратимых гетерогенных реакциях в отличие от гомогенных
стехиометрические коэффициенты при формулах жидких и твердых ве-
веществ во внимание не принимаются, например:
2N2O(r) + S(T) *± 2N2 (г) + SO2 (D (Avr = B +1) - 2 = 1 > 0);
N2 (г) + Над + 2С(Т) <=± 2HCN(r) (Avr = 2 - A +1) = 0);
2СО2(Г) + 2Н2О(Г) + 2НС1(Г) <=> 2СНзС1(г)+ЗО2(Г)
(Avr=B + 3)-B + 2 + 2) = -l<0).
7. Смещение положения равновесия в обратимых гетерогенных реак-
реакциях при изменении температуры вне зависимости от агрегатного состояния
реагентов и продуктов подчиняется общим закономерностям (правилам).
¦ Практические занятия
Химическое равновесие в гомогенных
и гетерогенных системах
Примеры решения задач
Задача 1. По равновесным концентрациям компонентов, равным, моль/л:
1,4 (NF3), 1,0 (Н2), 0,8 (N2) рассчитать начальные концентрации реагентов
(исходных веществ) и константу равновесия Кс реакции
2NF3(r)+3H2(r)^6HF(r)+N2(r)
протекающей при Т = const. Продукты в начальный момент времени отсут-
отсутствовали.
Решение. Чтобы определить неизвестную равновесную концентрацию про-
продукта — HF, необходимо проанализировать уравнение реакции. Равновес-
Равновесная концентрация HF в 6 раз превышает равновесную концентрацию N2,
следовательно,
[HF] = 6[N2 ] = 6 • 0,8 = 4,8 моль/л.
Зная равновесные концентрации всех компонентов, рассчитывают
константу равновесия Кс:
CHF]'[N;1 4^0,8 84ЗД
[NF,]2[H2]J !,4!-l,0!
Начальное количество реагентов равно сумме равновесных и прореа-
прореагировавших, следовательно, исходные концентрации реагентов составляют:
Сощ =[
ощ
Количество прореагировавших реагентов можно найти по количеству
образовавшихся продуктов, если учитывать стехиометрические коэффици-
коэффициенты в уравнении реакции. Для наглядности можно составить таблицу:
Концентрация, моль/л NF3 H2 HF N2
Равновесная [/] 1,4 1,0 6х 0,8 =х
Прореагировавшего вещест-
вещества Спрор/ 2х 3* - -
Начальная Со/ 1,4 +2л: 1,0 + 3* 0 0
Ясно, что Спрорщ = 0,8 • 2 = 1,6 моль/л, СпрорН2 = 0,8 • 3 = 2,4 моль/л.
Примеры решения задач 379
Таким образом, начальные концентрации реагентов равны:
Сощ = 1,4 +1,6 = 3,0моль/л; С0Нг =1,0 + 2,4 = 3,4 моль/л.
Задача 2. Для обратимой гомогенной реакции
СОС12(Г)^СО(Г)+С12(Г),
протекающей в изобарно-изотермических условиях при температуре Т= 1000 К и
общем давлении /?Общ = 36,6 атм, рассчитать равновесную степень превращения реа-
реагента, которого было взято 5 моль. Каким образом можно увеличить равновесный
выход продуктов? Температурной зависимостью стандартных энтальпии Аг#? и
энтропии А,.^ реакции пренебречь.
Решение. По определению, равновесная степень превращения есть отношение ко-
количества вещества превратившегося реагента к его начальному количеству:
_ ^превр СОС12
acoci2-— •
СОС12
Чтобы найти количество превратившегося СОСЬ, используют константу рав-
равновесия
кх=-
у
л равн СОС12
которая связана с константой равновесия К°р соотношением:
КХ=Кр(Ро6щ) '
где робщ = ро6щ/ р° — относительное общее давление, безразмерная величина (р° —
стандартное давление, р° = 1 атм = 1,013 • 105 Па); Avr — изменение числа молей
газообразных веществ в ходе реакции, Avr = vco + vcl - vC0Ci =1 + 1-1 = 1.
Стандартную константу равновесия К°р рассчитывают по стандартному
уравнению изотермы химической реакции
В последнем соотношении стандартную энергию Гиббса реакции можно вы-
вычислить по уравнению Гиббса—Гельмгольца:
380 Химическое равновесие в гомогенных и гетерогенных системах
Для того чтобы ответить на вопрос об увеличении равновесного выхода про-
продуктов, необходимо знать стандартный тепловой эффект Ar#298 реакции, который
согласно следствию из закона Гесса равен:
Л tJ® — Л fJ® л. Л 14® — Л И®
^гп 298 ~^/п 298 СО ¦*" **/ " 298 С12 **/ " 298 СОС12 '
где А/Я2Э98/ — стандартные энтальпии образования веществ (см. табл. П.5.1).
АгЯ2°98 = -110,52 + 0 -(-220,30) = 109,78 кДж/моль СОС12.
Так как Ar#298 > 0, то реакция эндотермическая — идет с поглощением теплоты.
Аналогично вычисляют АГ5298:
д с0 _ с0 , п0 _ ~0
ZAr°298 ~ °298 СО ^ °298 С12 °298 СОС12 ~
= 197,54 + 222,98 - 283,90 = 136,62 Дж/(моль СОС12 • К).
Отсюда ArG1°000=109,78 103 Дж/(моль СОС12) - 1000 К -136,62 Дж/(моль
СОС12 • К) = -26840 Дж/(моль СОС12) и, соответственно,
. ^о ArG7° 26840 Дж/моль ^^
In К = = = 3,23,
р RT 8,314 Дж/(мольКI000К
К°р =25,3, а Кх =25,3C6, б) =0,69.
Так как в условии задачи нет данных о количестве каждого из продуктов в
начальный момент времени, то принимают их равными нулю. Все стехиометриче-
ские коэффициенты в уравнении реакции равны единице, поэтому при установле-
установлении состояния равновесия исходное число молей СОС12 уменьшится, а число обра-
образовавшихся молей СО и С12 увеличится на одну и ту же величину, которую обозна-
обозначим х. Для наглядности можно составить таблицу:
Количество вещества, моль СОС12 СО С12
Начальное nOi 5 0 0
Прореагировавшее лпрор i х - -
Образовавшееся лобр _ х х
Равновесноеправн/ 5 -jc 0 + л: 0+х
Равновесное общее правн. общ E - х) + @ + х) + @ + х) = 5 + х
Молярные доли веществ-участников равновесия
Примеры решения задач 381
__ Яравн СОС12 _ 5 - X
2>рав„,
Аналогично для СО и С12
Отсюда
х х
2
Х =0,69.
E + хJE-л:) 25-jc2
Решая уравнение, получают х = 3,19 (отрицательный корень не имеет физи-
физического смысла). Такое число молей СОС^ распалось. Следовательно,
3,19
acoci = ~— = 0> 639 или 63,9 %.
Увеличить равновесный выход продуктов означает сместить положение рав-
равновесия реакции вправо. Это можно осуществить:
а) увеличением температуры, так как осуществляемая реакция является эндо-
эндотермической, Дг#? > 0;
б) снижением общего давления, поскольку реакция протекает с увеличением
числа молей газообразных веществ, т.е. с возрастанием объема;
в) выводом продуктов реакции из равновесной смеси;
г) разбавлением равновесной газовой смеси путем накачивания в нее инерт-
инертного газа — создается эффект понижения давления в системе и его действие равно-
равносильно уменьшению общего давления (п. б.).
Задача 3. В каком направлении будет протекать обратимая гомогенная реакция
при Т = 450 К и общем давлении ро6щ = 0,09 атм, если газовая смесь в начальный
момент времени состоит, моль: 0,2 (РС13), 0,1 (С12) и 0,1 (РС15)? Рассчитать объем-
объемный состав (в %) равновесной газовой смеси. Как повлияют на положение равнове-
равновесия увеличение температуры и давления?
Решение. Направление самопроизвольного протекания реакции определяют по зна-
знаку ее энергии Гиббса, которую рассчитывают, используя уравнение изотермы Вант-
Гоффа:
ArGT =-RT\nKx +ДГ1п—
^0РС1,А0С12
382 Химическое равновесие в гомогенных и гетерогенных системах
где Хо t — молярные доли компонентов в начальный момент времени.
Константу равновесия Кх определяют из величины константы равновесия
Последняя вычисляется по стандартному уравнению изотермы химической реакции
и связана с константой равновесия Кх соотношением:
где робщ — относительное общее давление, безразмерная величина; Avr — измене-
изменение числа молей газообразных веществ в ходе реакции (см. задачу 2).
Суммарное число молей компонентов в начальный момент времени
= О,4 моль.
Отсюда молярные доли равны:
"орд, ОД 1. "opci3 0,2 1 "от, 0,1 1
5 2, "о, °>4 4 Lwo, °>4 2 L"o, °'4 4
/ / /
Стандартная энергия Гиббса реакции (см. задачу 2)
Аг°Т = АгЯ298 -TArS29Z>
где
Дг^298 = **/** 298 РС15 "" ^/^298 РС13 ~ Д/^298 С12 ~
= -374,89 - (-287,02) - 0 = -87,87 кДж/моль,
А С° _ С° _ С° _ С° _
Ar^298 ~ ^298 РС15 °298 РС13 ^298 С12 ~
= 364,47 - 311,71 - 222,98 = -170,22 Дж/(моль • К),
Отсюда ArG450 = -87,87-103 - 450(-170,22) = —11271 Дж/моль < 0.
Затем рассчитывают К°р, Кхп ArG450:
о =__ArG450= 11271 ^3Ql или ко s 2029^
р R450 8,314-450 '
= 20,29-ОДО1 = 1,83,
ArG450 =-8,314-4501nl,83 + 8,314-4501n A/4) g332,34 Дж/моль>0.
г 45° A/2X1/4)
Примеры решения задач 383
Так как ArG450 > 0, то реакция в данных условиях самопроизвольно протека-
протекает в обратном направлении.
Для смеси идеальных газов при/?, Г= const составы, выраженные в объемных
ф, и молярных Xi долях, совпадают. Объемный состав можно рассчитать по схеме,
рассмотренной в задаче 2:
Количество вещества, моль РС13 Cl2 PCls
по, 0,2 0,1 0,1
прор/ л
"обр/ X X -
Лравн, 0,2 +Х 0,1 +JC 0,1 -X
Правнобщ @,2+*) +(ОД +JC) + @,1 -*) = 0,4+ X
Молярные доли компонентов в состоянии равновесия:
ПравнРС15 ОД-X 0,2 + Х 0Д + *
^равнРС15 ~ ^i ~ л /I ' равнРСЦ ~ Л ^ ' АравнС12 ~" Л . '
2^пРавн/ 0,4 + х 3 0,4 + д: 2 0,4 + л:
/
отсюда
^равнрсц = @Д-хХ0>4 + дс)@,4 + лс) = @Д-дс)@,4 + лг) = 83
Х @,4 + ^)@,2 + х)@,1 + JC> @,2 + х)@,1 + X)
Решая квадратное уравнение, получают х= 3,95 • 10~3 (отрицательный корень
не имеет физического смысла). Соответственно, объемный состав равновесной га-
газовой смеси:
равнга, 400-10 + 3,95 • 10 400 + 3,95
200 + 3 95
Фравн га, = ^равн рсЬ = 4(K 95 = 0,5049 или 50,49 %;
100 + 3,95
Фравнс2 = ^равна2 = ^ 95 = 0-2573 или 25,73 %.
Влияние изменения температуры на смещение положения равновесия определя-
определяется путем анализа дифференциального уравнения изобары химической реакции
dlnK°p Агн°
dT " RT2 '
3 84 Химическое равновесие в гомогенных и гетерогенных системах
Так как Аг#° < 0, то и первая производная логарифма константы равновесия
(dlnK°p )
— <0 . Следовательно, In AT и величи-
по температуре также меньше нуля ,
I dT )
шК°р является убьюающими функциями температуры, т. е. увеличение температуры
смещает положение равновесия реакции влево — в сторону исходных веществ.
Влияние изменения общего давления на смещение положения равновесия
определяется дифференциальным уравнением Планка—Ван Лаара в виде
I Фобщ )Т Робщ
Так как Avr=vPC1 -vpcl -vcl =1-1-1 = -1 < 0, то частная производная
> 0, значит, с ростом общего давления будет расти и величина Кх, т.е.
)
V Фобщ )т
положение равновесия реакции будет смещаться вправо — в сторону продуктов.
Задача 4. В реактор объемом 10 л поместили навеску железа, ввели газовую смесь,
состоящую, моль: 1,05 (СО) и 0,05 (СО2) и нагрели до температуры 1000 К. К мо-
моменту наступления равновесия количество диоксида углерода СО2 увеличилось на
0,50 моль. Рассчитать константы равновесия Кр,К®, Кси Кх обратимой гетероген-
гетерогенной реакции
3Fe(T) + 2CO(r)<z>Fe3C(T)+CO2(r)
а также общее давление в реакторе. Описать по правилу фаз Гиббса данную гетеро-
гетерогенную систему. Рассчитать новые равновесные концентрации газообразных ком-
компонентов реакции, если первоначально установившееся равновесие было нарушено
путем уменьшения концентрации СО на 4 • 10~3 моль/л. В каком направлении при
этом сместилось положение равновесия?
Решение. Известно, что в гетерогенных системах молярные доли, концентрации или
парциальные давления чистых конденсированных (жидких и твердых) фаз постоян-
постоянны и поэтому не входят в выражения констант равновесия Кх, Кс и Кр.
Согласно условию, число молей СО2 при равновесии составит 0,05 + 0,50 =
= 0,55 моль. Так как по уравнению реакции 2 моль прореагировавшего СО дают
1 моль СО2, то к моменту наступления равновесия прореагирует 0,50 • 2 = 1,00 моль
СО. Следовательно, в равновесии будет находиться 1,05 - 1,00 = 0,05 моль непро-
реагировавшего СО. Общее число молей газов в состоянии равновесия составит
0,05 моль + 0,55 моль = 0,60 моль. Отсюда константа равновесия Кх:
Примеры решения задач 385
равн СО2 Иравн С02 /X ^равн / 0,55 / 0,60
** =—5 =7 ~ г= г = 132.
Зная число молей газов в состоянии равновесия, рассчитывают их равновес-
равновесные концентрации:
ггугъ-1 ^Pa811 со ^,05 , 1Л-3 /
[СО] = — = = 5 10 моль/л;
V 10
[СО2] = ^^ = ^ = 5,5.Ю-2 моль/л.
На основании последних вычисляют константу равновесия Кс по соотношению:
Щ 0
[СО]2 E-ЮJ
Обе константы связаны между собой уравнением
= \?Щ = 5>510 = 2,2 • 103 л/моль = 2,2 м3/моль.
[СО]2 EЮJ
с *yRT)
где Avr — изменение числа молей газообразных веществ в ходе реакции, Avr =
Из последнего соотношения находят
Po6m=—RT = ^Г 8,314 ДЖ 1000Kg498,8 103 Па.
Кс 2,2 м /моль моль • К
Константы Кр9 К°р можно найти через константу Кх либо Кс, так как они
связаны уравнением
кр =
Тогда
ЛГр =Kc(RT)AVr = 2,2м3/моль(8,314Дж/(мольКI000К) ^2,65-10 Па,
К°р =Кр(р°уАУг =2,65-10 Па • 1,013 105 Па^26,8.
Исследуемая реакция является гетерогенной реагирующей системой, которая со-
состоит из одной газовой и двух твердых фаз. Таким образом, общее число фаз равно 3.
Число независимых компонентов К находят по разности между числом веществ,
присутствующих в системе и равных 4, и числом математических уравнений (соотно-
13 - 9795
386 Химическое равновесие в гомогенных и гетерогенных системах
шений), связывающих равновесные концентрации (или равновесные парциальные дав-
давления) этих веществ. В данном случае такое математическое уравнение одно:
Р 2
/VbhCO
Следовательно, К = 3.
Внешними факторами, влияющими на состояние равновесия в данной систе-
системе, являются давление и температура, т.е. п = 2.
Число степеней свободы, согласно правилу фаз Гиббса, С = К-Ф + л = 3-3 +
+ 2 = 2 — система дивариантна.
Парциальное давление СО2 является функцией температуры и парциального
давления СО. В данной системе можно одновременно в определенных пределах
произвольно изменять и температуру, и давление, при этом не произойдет исчезно-
исчезновения прежних и образования новых фаз.
Новые концентрации, которые уже не будут равновесными, составят
Ссо = 5 • 10 - 4 • 10 = 10 моль/л; ССОз = 5,5 • 10 моль/л.
Стехиометрический коэффициент СО больше, чем СО2, поэтому изменение
концентрации или парциального давления СО сильнее сказывается на смещении
положения равновесия. В соответствии с принципом Ле Шателье—Брауна, умень-
уменьшение концентраций или парциальных давлений исходных веществ смещает поло-
положение равновесия влево, т.е. в сторону реагентов.
Пусть к моменту наступления нового равновесия концентрация СО2 умень-
уменьшилась на х моль/л. Тогда концентрация СО увеличится на 2х моль/л (см. стехио-
метрическое уравнение) и составит A0~3 + 2х) моль/л, а равновесная концентрация
СО2 будет равна E,5 10~2 -х) моль/л.
Поскольку константа равновесия Кс не зависит от концентрации компонен-
компонентов, то для нового равновесия можно записать:
[СО]2 A0+2xJ
Решая это уравнение, получают х\ = 1,96 • 10~3, а *2 = -3,07 • 10~3. Отрица-
Отрицательный корень не имеет физического смысла. Таким образом, новые равновесные
концентрации будут составлять:
[СО] = 10+ 2 • 1,96 • 10 = 4,92 • 10 моль/л;
[СО2] = 55 • 10- 1,96 • 10 = 53,04 • 10 моль/л.
Задача 5. Как изменятся константа равновесия KQp и равновесные парциальные
давления компонентов при увеличении температуры на 100° у обратимой гомоген-
гомогенной реакции
Примеры решения задач 387
2NO2(r)?=*N2O4(r)
протекающей при температуре Т— 21Ъ К и общем давлении /?Общ = 1 атм?
Решение. Чтобы узнать, как изменится константа равновесия Кр, используют инте-
интегральную форму уравнения изобары Вант-Гоффа:
to*P373 =АГЯ?C73-273)
П
Кр272 Л-273-373
Пренебрегая температурной зависимостью теплового эффекта реакции, рас-
рассчитывают Дг#? для стандартной температуры согласно следствию из закона Гесса:
ДГЯ2°98 = AfH°29Sn2ot ~2A^2°98NO2 = 11,11 -2C4,19) = -57,27кДж,
отсюда
к -57270-100 К К
. л/,373 -Э/Z/UIUU л 373 t 1С 1Л_з лр273 о,, _
In—-— = ^-6,76, или —-—?1,15-10 , или ——= 866,7,
Крт 8,314.273-373 Кр213 КрШ
т.е. константа равновесия Кр реакции при увеличении температуры на 100° умень-
уменьшится в 866,7 раза. Константа равновесия Кр для заданной реакции
,О4 Робщ " ^равн NO2
Р Л 2
j2 -гравн NO2
Решая это уравнение относительно /?paBHN0 -> получают
^равнКОз = "TZ *
Зная Кр для двух значений температуры, находят отношения
Ррюн NO2 273
4 373 „ г, _ ^_^ т^
. Значения л^ для температуры Г= 273 К можно определить по термо-
N2O4 273
динамическим данным (см. задачу 2):
ДА°98 =S2°98N2o4 -252°98NO2 =304,35-2B40,06) = -175,77 ДжЛС;
ArG2°73 = АГЯ2°98 -273АГ52°98 = -57,27 • 103 -273(-175,77) = -9284,79 Дж;
In^o Mk= 9284>79 g4,09
р2?3 Л-273 8,314-273
13*
388 Химическое равновесие в гомогенных и гетерогенных системах
КР 273 = К27з(/>Тг = 59,8 • A атм) s 59,
Крт =59,8/866,7 s 6,9- 10-2атм-'.
Далее вычисляют Ррава 1Юг 273 и Ррти NO2 373:
-l + Vl + 4-59,81
/>равн NO2 273 = J^ = 0,12 ЗТМ;
+ 4-6,910-2!
отсюда
^равн NO2 373 0,94
— = 7,0
^12
и
Ррии}*2О4т Робщ /УвнШ.ЗТЗ 1-0,94 0,06 /?равнЫ2О4 273 ^
~~~~~~^~^ — ^—~~—~"~"~"~~*~ — "~^~~~~~~" — ~~~^~~ = 0,0ио ИЛИ —~~~*~~~~~~~~~ = 14,7.
Р равн N2O4 273 Робщ ~ Р равн NO2 273 1 ~ 0,12 0,88 /7 равн щО^ 373
Таким образом, при повышении температуры на 100° равновесное парциаль-
парциальное давление NO2 возрастет в 7,8 раза, а равновесное парциальное давление N2O4
уменьшится в 14,7 раза.
Задача 6. Определить температуру, при которой в начальный момент времени со-
состав газовой смеси /?0/, атм: 2(NO2), 1(N2O4) не изменяется и соответствует ее рав-
равновесному составу. В каком направлении протекает самопроизвольно обратимая
гомогенная реакция:
выше и ниже этой температуры? Рассчитать ее константу равновесия К°р.
Решение. По условию, равновесные парциальные давления компонентов равны их
начальным, т.е. рравнШз =Ажо2= 2 атм, рравнЩОл =A)n2o4 = * атм- Константа
равновесия Кр этой реакции равна:
^paBHNO2 .
Кр = — = 4 атм.
Рравн N2O4
Она связана с соответствующей стандартной константой К0 соотношением
Примеры решения задач 3 89
кр=кр(р ) >
где р° = 1 атм — стандартное давление; Avr — изменение числа молей газообраз-
газообразных веществ в ходе реакции, Avr = vN0 - vN 0 =2-1 = 1;
где Ррявн! =/7Равн///7° — относительное равновесное парциальное давление, без-
без7Равн
размерная величина.
Далее в уравнение изотермы химической реакции
ArGT=ArGj+RT\n
~2
подставляют данные для состояния равновесия и получают
ArGT =ArGj+RT\n \^r
Изменение стандартной энергии Гиббса связано с соответствующими изме-
изменениями энтальпии и энтропии реакции уравнением Гиббса—Гельмгольца:
После преобразований находят искомую температуру:
r____Mk___
1 — z г"
Числовые значения АГЯ298 и А^^ можно взять из предыдущей задачи, поменяв у
них знаки, поскольку данная реакция обратна реакции задачи 5.
Г = *Ш* = 348,7КG5,5*С).
A75,77-11,53)Дж/К
При Т = 75,5 °С исходная газовая смесь одновременно является равновесной
смесью. При Т > 75,5 °С реакция протекает преимущественно в прямом направле-
направлении (слева направо), при Т< 75,5 °С — в обратном направлении (справа налево).
390
Химическое равновесие в гомогенных и гетерогенных системах
Задачи для самостоятельного решения
1-10. Для обратимой реакции напишите выражения констант равновесия Кс, Кр, Кх
и установите связь между ними. Каким образом можно увеличить равновесный вы-
выход продуктов? Приведите все возможные способы. Ответ аргументируйте.
№
1
2
3
4
5
6
7
8
9
10
Уравнение обратимой реакции
N2(r)+3H2(r)^2NH3(r)
2C(T) + 2H2(r)?±C2H4(r)
2CO(r)+O2(r)?±2CO2(r)
ВаО(т)+СО2(г)?±ВаСОз(т)
CO(r)+H2O(r)^CO2(r) + H2(r)
NH3(r) + HCl(r)?=±NH4Cl(T)
РС15(Г)^РС13(Г)+С12(Г)
FeO(T)+H2(r)^Fe(T)+H2O(r)
2SO2(r)+O2(r)^2SO3(r)
NH4NO2(T)^N2(r)+2H2O(r)
Тепловой эффект реакции, АгН
ArH<0
ArH >0
ArH <0
АГЯ <0
АГЯ <0
АГЯ <0
АГЯ >0
АГЯ >0
АГЯ <0
АГЯ >0
11-14. Для следующих обратимых гомогенных и гетерогенных реакций напишите
выражения соответствующих констант равновесия. Назовите константы равновесия.
№
11
12
а)
б.
Уравнение обратимой реакции
NH4NO2(T)?±N2(r) + 2H2O(r)
hno2(p)^h;) + no2(p)
г) N02(p) + Н20(ж) *± HNO2(p) + 0Н-р)
а)
г)
CaCO3(T)^CaO(T)+CO2(r)
со^ + нр^нсо^+он-,
№
13
14
a)
6)
г)
a)
в)
Уравнение обратимой реакции
NH4Cl(T)?=>NH3(r) + HCl(r)
nhIw+h.o^nh.oh^+h;
NH4F(T)^NH3(r)+HF(r)
ту" i т т /^ \ LJp i (~\T T~
Задачи для самостоятельного решения
391
15-20. По известным равновесным концентрациям компонентов рассчитайте: а) не-
неизвестную равновесную концентрацию компонента; б) константы равновесия Кс и
Кр реакции при Т = 298 К; в) начальные концентрации исходных веществ, если на-
начальные концентрации продуктов были равны нулю.
№
15
16
17
Уравнение реакции и значения равно-
равновесных концентраций компонентов,
моль/л
4NO(r)+6H2O(r)^4NH3(r)+5O2(r)
0,20 0,30 0,04
CS2(r)+3O2(r)?±CO2(r)+2SO2(r)
1,00 0,06 1,20
2CH4(r)?^C2H2(r)+3H2(r)
0,10 0,50
№
18
19
20
Уравнение реакции и значения равно-
равновесных концентраций компонентов,
моль/л
CH4(r)+H2O(r)?±CO(r)+3H2(r)
0,30 0,10 0,09
2HCl(r)+F2(r)<=>2HF(r)+Cl2(r)
0,20 0,05 0,10
4HCl(r)+O2(r)F*2Cl2(r)+2H2O(r)
0,20 0,10 0,60
21-26. По термодинамическим данным рассчитайте значения констант равновесия
Кс и Кр обратимой гомогенной химической реакции, протекающей при стандартных
условиях. Определите их размерность. В каком направлении (реагентов или про-
продуктов реакции) сместится положение равновесия: а) при увеличении давления;
б) при увеличении температуры; в) при введении избытка кислорода; г) в случае
разбавления этой равновесной системы инертным газом (например, аргоном) в изо-
барно-изотермических условиях. Как при этом изменятся (увеличатся, уменьшатся)
или останутся постоянными значения констант равновесия и равновесного выхода
продуктов. Предложите такое одновременное изменение (увеличение, уменьшение)
термодинамических параметров р и Г, которое бы обеспечило увеличение равно-
равновесной степени превращения реагентов. Ответ аргументируйте.
№
21
22
23
Уравнение обратимой реакции
2H2(r)+O2(r)^2H2O(r)
2SO3(r)^2SO2(r)+O2(r)
2NO(r)^N2(r) + O2(r)
№
24
25
26
Уравнение обратимой реакции
CS2(r)+4O2(r)^CO2(r)+2SO3(r)
2С12(Г) + 2Н2О(Г)?±4НС1(Г)+О2(Г)
2CO(r)+O2(r)^2CO2(r)
27-34. Для обратимой гетерогенной химической реакции рассчитайте по правилу
фаз Гиббса число степеней свободы, а по термодинамическим данным значения
констант равновесия Кс и Кр при стандартных условиях. Какова их размерность?
Определите, в каком направлении (реагентов или продуктов реакции) сместится
положение равновесия: а) при уменьшении давления; б) при увеличении температу-
температуры; в) при введении избытка водорода; г) в случае разбавления этой равновесной
системы инертным газом (например, аргоном) в изобарно-изотермических услови-
условиях. Как при этом изменятся (увеличатся, уменьшатся) или останутся постоянными
значения констант равновесия и равновесного выхода продуктов. Предложите такое
392
Химическое равновесие в гомогенных и гетерогенных системах
одновременное изменение (увеличение, уменьшение) термодинамических парамет-
параметров р и Т, которое бы обеспечило повышение равновесного выхода продуктов. От-
Ответ аргументируйте.
№
27
28
29
30
Уравнение обратимой реакции
СО2(Г)+4Н2(Г)^СН4(Г)+2Н2О(Ж)
2Fe(T) +3H2O(r) ** Fe2O3(T) +3H2(r)
Fe3O4(T) +4H2(r) ?±3Fe(T) +4H2O(r)
Ni(T) + H2O(r)^NiO(T) + H2(r)
№
31
32
33
34
Уравнение обратимой реакции
МпО2(т) + 2Н2 (г) & Мп(т) + 2Н2О(Г)
Со(т) + Н2О(г)?±СоО(т) + Н2(г)
WO3(T)+3H2(r)^W(T)+3H2O(r)
2НВг(г)?=*Вг2(ж)+Н2(г)
35-44. Для обратимой гомогенной реакции по ее константе равновесия Кс и одина-
одинаковым начальным концентрациям Со исходных веществ рассчитайте равновесные
концентрации всех компонентов газовой смеси.
№
35
36
37
38
39
40
41
42
43
44
Уравнение обратимой гомогенной реакции
SO2(r)+CO2(r)^SO3(r)+CO(r)
CO(r) + H2O(r)?±CO2(T) + H2(r)
SO3(r)+H2(r)^SO2(r) + H2O(r)
SO3(r)+CO(r)^SO2(r)+CO2(r)
H2(r)+CO2(r)^CO(r)+H2O(r)
COCl2(r)^CO(r)+Cl2(r)
SO2F2(r)?=*SO2(r)+F2(r)
2HBr(r)<=±H2(r) + Br2(r)
2HCl(r)?=±H2(r)+Cl2(r)
C2H6 (г) *=* С2Н4 (г) + H2 (r)
Kc
3,0
2,0
1,6
2,0
3,0
1,2*
4,0'
0,4
0,3
2,2*
Co,
моль/л
0,5
1,0
0,5
1,0
1,0
3,0
2,6
0,8
0,5
2,0
Kc в моль/л.
45-54. Для обратимой гетерогенной реакции по ее константе равновесия Кс и на-
начальной концентрации Со газообразного реагента рассчитайте равновесные концен-
концентрации всех газообразных компонентов.
45
46
Уравнение обратимой гетерогенной реакции
2СиО(т) + CO(r) <=t Cu2O(T) + СО2 (г)
NiO(T)+CO(r)?±Ni(T)+CO2(r)
Кс
16,0
16,0
Со,
моль/л
3,0
2,0
Задачи для самостоятельного решения
393
№
47
48
49
50
51
52
53
54
Уравнение обратимой гетерогенной реакции
Fe2O3 (т) + CO(r) ^± 2FeO(T) + СО2 (г)
Fe2O3 (т) + Н2 {r) z± 2FeO(T) + H2O(r)
CoO(T)+H2(r)^Co(T)+H2O(r)
C(T)+CO2(r)?=*2CO(r)
С(т)+2Н2(г)^СН4(г)
2C(T)+O2(r)?^2CO(r)
c(T)+o2(r)^co2(r)
2C(T) +H2(r) ?±C2H2(T)
Kc
11,0
7,0
6,5
0,8*
0,8"
0,5*
0,8
0,7
Co, моль/л
3,0
2,0
2,0
0,8
2,0
0,5
1,0
1,4
Kc в моль/л.
Кс в л/моль.
55-60. В реактор объемом V л ввели по 2 моль реагентов. К моменту наступления
равновесия прореагировало 10% начального количества первого реагента. Рассчи-
Рассчитайте константу равновесия Кс обратимой гомогенной реакции.
№
55
56
57
Уравнение обратимой
гомогенной реакции
2SO2(r)+O2(r)^2SO3(r)
2NO(r)+Cl2(r)^2NOCl(r)
N2(r)+3H2(r)^2NH3(r)
К я
11
7
2
№
58
59
60
Уравнение обратимой
гомогенной реакции
4НС1(Г)+О2(Г)^2С12(Г) + 2Н2О(Г)
CS2(r)+3O2(r)^CO2(r)+2SO2(r)
CH4(r)+H2O(r)^CO(r)+3H2(r)
V, л
5
1
10
61-63. Выразите константу равновесия реакции 1 (КС\) через константы равновесия
реакций 2 и 3 (КС2 и КСъ).
№
61
62
63
2.
2.
2.
COCl2(r)
2CO2(r)<
2H2O(r) <
Уравнение реакции
1. 2C(T) + O2(r)+2C1
?±co(r)+ci2(r)(/:c2)
l.S03(r)+C0(r)?i
1. 4HCl(r)+02(r)?±
2(r) *
3.
SO;
3.
2H2
3.
=±2COCl2(r)(Kcl)
2CO(r)Fi2C(T)+O2(r)(^c3)
2SO2(r)+O2(r)?±2SO3(r)(^c3)
O(r)+2Cl2(r)(^cl)
Н2(г)+С12(г)^2НС1(г)(/:сз)
394
Химическое равновесие в гомогенных и гетерогенных системах
64-66. Рассчитайте константу равновесия реакции 1 (КС\\ если известны константы
равновесия реакций 2 и 3 {КСг и КСъ) при некоторой температуре.
№
64
65
66
2. 2H2(r)+O2(r)^
(КС2~ 1,3 л/моль)
1
2. 2CO(r)+O2(T)F*
(КС2 = 3,4 л/моль)
2-Fe(T)+H2O(r)?=*
(КС2 = 160,0)
2Н2О(Г)
2Н2О(Г)
so2(r)
2СО2(Г
• со2(г)
FeO(T) +
Уравнение реакции
+ 2С12(Г)
+ СО
2 (г)
)
+ н2(г)?
н
2 (г)
?±4Hci(r)+o2(r)(/:c.)
3. Н2(Г)+С12(Г)?=±2НС1(Г)
^±SO3(r)+CO(r)(^cl)
3. 2SO3(r)?±2SO2(r)+O2(r)
(KCi= 1,4 моль/л)
:±CO(r)+H2O(r)(KCi)
3. FeO(T)+CO(r)?=iFe(T)+CO2(r)
67-70. У какой из обратимых гетерогенных реакций A или 2) равновесный выход
диоксида серы (SO2) выше, если его равновесное количество в газовой смеси со-
составляет 0,7 моль, а начальный объем кислорода был равен 56 л (н.у.)? Рассчитайте
равновесную степень превращения кислорода в этих реакциях. Выведите соотно-
соотношение между равновесным выходом SO2 и равновесной степенью превращения О2
для этих реакций.
№
67
68
69
70
Уравнение обратимой гетерогенной реакции
1.4FeS(T) + 7O2(r)?=
2. 4ReS2(T)+15O2(r)
1. 2Bi2S3(T
2. 2MoS2(T
1. 4FeS2(T)
2- 2Sb2S3(T
1. 4ReS2(T)
2. 4FeS2(T)
+ 9О2(г,
)+7О2(г,
+по2(г)
)+9О2(г)
+ 15О2(г)
+ 1Ю2(гГ
±2Fe2O3(T)+4SO2(r)
?^2Re2O7{T)+8SO2(r)
?=?2Bi2O3(T)+6SO2(r)
^±2MoO3(T)+4SO2(r)
^2Fe2O3(T)+8SO2(r)
?^2Sb2O3(T)+6SO2(r)
^2Re2O7(T)+8SO2(r)
<=i2Fe2O3(T)+8SO2(r)
71-74. Рассчитайте равновесные концентрации газообразных компонентов обрати-
обратимой гетерогенной химической реакции, протекающей при некоторой температуре,
если известна ее константа равновесия Кс и их начальные концентрации Со.
Задачи для самостоятельного решения
395
№
71
72
73
74
Уравнение обратимой гетерогенной реакции
2Fe(T)+3CO2(r)?±Fe2O3(T)+3CO(r)
Fe2O3(T) + 3H2(r) ?± 2Fe(T) +3H2O(r)
2PbS(T) + 3O2 (r) <=L 2PbO(T) + 2SO2 (r)
LiH(T) + H2O(r)^LiOH()K)+H2(r)
Kc
io-3
8
101*
10
Co, моль/л
0,l(CO2);l,0(CO)
0,4(H2);0,2(H2O)
0,5(O2);7,4(SO2)
2,7(H2O);5,0(H2)
Kc в моль/л.
75-77. Рассчитайте новые равновесные концентрации газообразных компонентов
обратимой гетерогенной химической реакции, протекающей при некоторой темпе-
температуре, если первоначально установившееся равновесие с равновесными концен-
концентрациями было нарушено путем увеличения концентрации СО на 1,20 моль/л.
№
75
76
77
78
Уравнение обратимой гетерогенной реакции
Sb2S3 (т) + ЗСО(Г) <Л 2Sb(T) + 3CSO(r)
Fe3C(T)+CO2(r)^±3Fe(T)+2CO(r)
2CuO(T)+CO(r)^Cu2O(I)+CO2(r)
2FeO(T) + CO2 (r) *± Fe2O3 (T) + CO(r)
Равновесные концентрации,
моль/л
0,3 (CO); 0,3 (CSO)
0,15 (CO2); 0,45 (CO)
0,25 (CO); 1,50 (CO2)
2,40 (CO2); 0,24 (CO)
79-82. Для обратимой реакции, протекающей в стандартных условиях, найдены
значения константы равновесия К°р при разных температурах. Установите, каким
значениям К^ соответствует: а) преимущественное протекание реакции в сторону
продуктов; б) преимущественное протекание реакции в сторону реагентов; в) рав-
равновероятное протекание реакции в обоих направлениях; г) практически необрати-
необратимое протекание реакции в сторону продуктов: д) практически необратимое проте-
протекание реакции в сторону реагентов; е) преобладание продуктов в реакционной рав-
равновесной смеси; ж) преобладание реагентов в реакционной равновесной смеси;
з) отсутствие преобладания реагентов или продуктов в реакционной равновесной
смеси; и) наибольший равновесный выход продуктов; к) наименьший равновесный
выход продуктов. Определите знак изменения теплового эффекта реакции. Дайте
обоснованный ответ.
№
79
80
81
82
2 • 10~3;
3 • 109;
5 • 10;
4-1010;
2-
3-
6-
4-
10-
Ю2;
Ю-2;
Ю5;
Значение
l;
l;
l;
l;
3-
4-
3-
5-
Kp в порядке возрастания Т
Ю2;
Ю-2;
Ю3;
Ю-3;
2
6-
3
7-
•105
lO2
•107
ю-8
396
Химическое равновесие в гомогенных и гетерогенных системах
83-90. Опишите по правилу фаз Гиббса равновесия в системах, в которых протека-
протекают обратимые гетерогенные реакции, описываемые следующими уравнениями, а
именно: а) установите, какие фазы присутствуют и их число; б) приведите незави-
независимые уравнения (математические соотношения), связывающие равновесные кон-
концентрации или пропорциональные им равновесные парциальные давления всех
компонентов системы, и подсчитайте их общее число; в) найдите число независи-
независимых компонентов в системе; г) определите внешние факторы, влияющие на состоя-
состояние равновесия, и подсчитайте их общее число; д) рассчитайте число термодинами-
термодинамических степеней свободы, или вариантность системы. Полученные результаты све-
сведите в таблицу.
№
83
84
85
86
87
88
89
90
Уравнение обратимой гетерогенной реакции
1. 2NH4VO3(T) f± V2O5(T) +2NH3(r) +H2O(r)
2.V2O5(T) + 2NH3(r) + H2O(r)^ 2NH4VO3(T)
l.N2(r) + 2SO2(r) ^2NO2(r)+2S(T)
2. 2NO2(r) + 2S(T)^N2(r)+2SO2(r)
1.Na2CO3(T) + CO2(r) + H2O(r) ^ 2NaHCO3(T)
2. 2NaHCO3(T) <=± Na2CO3(T) +CO2(r) +H2O(r)
l.CS2(r)+2Cl2(r)F*CCl4(r)+2S(T)
2. CCl4(r)+2S(T)?=*CS2(r)+2Cl2(r)
1. PH4I(T)^PH3(r) + HI(r)
2. PH3(r) + HI(r) ^PH4I(T)
1. NH4HCO3(T) ^ NH3(r) +CO2(r) +H2O(r)
2. NH3(r) +CO2(r) +H2O(r) ^ NH4HCO3(T)
1. NH3(r) + HB!(r)^NH4Br(T)
2. NH4Br(T)?± NH3(r)+HB!(r)
1. TiO2(T) + 2C(T) +2Cl2(r) ^ TiCl4(r) + 2CO(r)
2. TiCl4 (r) + 2CO(r) ^ TiO2 (T) + 2C(T) + 2C12 (r)
91-97. По исходному составу реакционной смеси (щ,-, Со,, ро,) и значению констан-
константы равновесия (Кр, Кс, Кх) при температуре Т определите направление самопроиз-
самопроизвольного протекания обратимой гомогенной реакции в данных условиях. Рассчи-
Рассчитайте объемный состав (в %) равновесной газовой смеси. Напишите выражение ука-
указанной в таблице константы равновесия.
Задачи для самостоятельного решения
397
0
2
91
92
93
94
95
96
97
Уравнение обратимой
гомогенной реакции
СН4(Г) + Н2О(Г)?=*
<=±ЗН2(г)+СО(г)
2CH4(r) ?± C2H6(r) + H2(r)
H2(r) + I2(r)<=± 2HI(r)
РС13(Г)+С12(Г)?±РС15(Г)
N2O4(r)?=* 2NO(r)
6 12 (г} ^ 4 6 (г) 2 6 (г}
Н2(г) + С12(г)?=г2НС1(г)
Исходный состав
реакционной смеси
wo сн4 = по н2о = по н2 =
= посо =2 моль
"осн4=Ион2=1моль
"ос2нб=2моль
Со н = 2,2 моль/л
С01 = 5,0 моль/л
Сош = 12,1 моль/л
Р0РС'5 =00205МПа
Рою, = °'01 МПа
/'окл = 3,7 атм
иос6н|2 = 6 моль
иос2н6 = 2 моль
Г, К
1000
2000
700
500
1000
800
300
Константа
равновесия
АГд- = 40
Ал-=8,5 10
Ас =50
Ар=3,ЗМПа-'
<=2,13
i^=2,4 106Па
Кс = 6,8
98-103. Определите температуру, при которой в обратимой гомогенной реакции
устанавливается равновесие, принимая, что все газы находятся в стандартном со-
состоянии, а тепловой эффект и энтропия реакции не зависят от температуры. Сделай-
Сделайте вывод о направлении протекания реакции выше и ниже этой температуры.
№
98
99
100
Уравнение обратимой
гомогенной реакции
N2(r)+3H2(r)<=±2NH3(r)
2NO2(r)<i±N2O4(r)
2HBr(r)^H2(r)+Br2(r)
101
102
103
Уравнение обратимой
гомогенной реакции
2H2O(r)^2H2(r)+O2(r)
CO(r)+H2O(r)?±CO2(r) + H2(r)
2NO(r)+O2(r)^2NO2(r)
104-109. Определите температуру, при которой константа равновесия реакции Кр = 1.
Рассчитайте равновесный состав (в объемных долях, %) реакционной смеси, если
для реакции было взято щ моль реагентов. Все газы находятся в стандартном со-
состоянии, а тепловой эффект и энтропия реакции не зависят от температуры.
398
Химическое равновесие в гомогенных и гетерогенных системах
№
104
105
106
Уравнение обратимой
гомогенной реакции
2HI(r)^H2(r)+I2(r)
2НС1(Г)^Н2(Г)+С12(Г)
2HF(r) ?=* H2(r)+F2(r)
МОЛЬ
1
2
3
107
108
109
Уравнение обратимой
гомогенной реакции
со2(г)+н2(г)?=>
ТТ , т ч TLJT
Н2(г) + 12(г)?= 2Н1(г)
МОЛЬ
1(СО2);3(Н2)
3(Н2);5A2)
3(Н2);2(Вг2)
110-112. В закрытом сосуде смешано по п0 моль реагентов. Реакция протекает при
постоянной температуре. К моменту наступления равновесия прореагировало Z%
первоначального количества второго реагента. Определите давление газовой смеси
при равновесии, если исходное давление равнялось /?исх.
№
ПО
111
112
Уравнение обратимой
гомогенной реакции
O2(r)+2SO2(r)^2SO3(r)
С12(г)+СО(г)?±СОС12(г)
CO2(r) + H2(r)?±CO(r)+H2O(r)
«о,
моль
5(O2);8(SO2)
3(С12);4(СО)
5(СО2);5(Н2)
Z,%
80
50
80
Рисх*
кПа
300
100
100
113-120. Рассчитайте равновесную степень превращения (конверсии) реагентов
реакции, протекающей в закрытой системе в следующих условиях: температура Г,
общее давление ро6щ, исходные концентрации Со, или количество «0/ (моль) ве-
веществ, константа равновесия Кс или Кр. Температурной зависимостью теплового
эффекта и энтропии реакции пренебречь.
№
113
114
115
116
Уравнение обратимой
гомогенной реакции
F2(r) *=* 2F(r)
со2(г) + н2(г)^со(г) + н2о(г)
N2O4(r)^±2NO2(r)
СОС12(Г)?=*СО(Г)+С12(Г)
1500
const
const
550
Робщ.»
кПа
101,3
-
-
101,3
Константа
равновесия
-
Kc= 1
Кс=\
моль/л
-
Исходный состав
реакционной смеси
w0/, моль: 1(F2);
0(F)
nOi, моль:
0(СО); 0(Н2О);
1(СО2);2(Н2)
Со/, моль/л:
6(N2O4); 0(NO2)
Со/, моль/л:
0(СО);0(С12);
1(СОС12)
Задачи для самостоятельного решения
399
117
118
119
120
Уравнение обратимой
гомогенной реакции
НСОН(Г)^СО(Г)+Н2(Г)
С12(Г)^±2С1(Г)
2Н1(г) <=> Н2(г) + 12(г)
РС15(Г)^РС13(Г)+С12(Г)
Г, К
300
const
const
const
Робщ.»
кПа
-
-
-
-
Константа
равновесия
#р=2атм
Кс=
= 3 1<Г3
моль/л
?,= 0,02
Кс=0,04
моль/л
Исходный состав
реакционной смеси
Со/, моль/л:
0(СО);0(Н2);
1(НСОН)
С0/, моль/л:
0(С1);0,05(С12)
W0H2 =П0\2 =0
пот = 0,1 моль
Со/, моль/л:
0(РС1з);0(С12);
1(РС15)
121-124. Вычислите константу равновесия Кс обратимой гомогенной реакции и
равновесные концентрации реагентов, если известен исходный состав реакционной
смеси С0/ и равновесная концентрация продукта [/].
№
121
122
123
124
Уравнение обратимой
гомогенной реакции
Н2(г)+12(г) ?=* 2Н1(г)
N2(r)+O2(r)?±2NO(r)
РС13(Г)+С12(Г)?^РС15(Г)
H2(r)+cl2(r)^2HCl(r)
Исходный состав
смеси Со |, моль/л
0,8(Н2); 1,8A2)
0,7(N2); l,4(O2)
0,3(РС13); 0,5(С12)
0,1(Н2);0,1(С12)
Равновесная концентра-
концентрация продукта [('], моль/л
[HI] = 0,8
[NO] = 0,4
[РС15] = 0,2
[HCl] = 0,05
125-128, По известным равновесному составу газовой смеси, общему давлению /?общ
и температуре Т рассчитайте константы равновесия Кр и Кс обратимой гомогенной
реакции.
№
125
126
127
128
Уравнение обратимой
гомогенной реакции
Н2(г)+12(г) ** 2Н1(г)
2NO(r)+O2(r)?±2NO2(r)
со2(г)+н2(г)^
?=±CO(r)+H2O(r)
2CO(r) + O2(r)?=i2CO2(r)
Равновесный состав
реакционной смеси, % (об.)
32(Н2); 38A2); 30(Н1)
44(NO); 20(O2); 36(NO2)
12(СО2); 32(СО);
18(Н2); 38(Н2О)
85(СО2); Ю(СО); 5(О2)
Робщ,
кПа
101,3
101,3
101,3
Г, К
300
1300
2000
400
Химическое равновесие в гомогенных и гетерогенных системах
129-132. Используя стандартные термодинамические характеристики, найдите тем-
температуру, при которой равновесное давление газообразного продукта реакции рав-
равно стандартному давлению. Температурной зависимостью стандартных теплового
эффекта и энтропии реакции пренебречь. Сделайте выводы о направлении протека-
протекания реакции выше и ниже этой температуры. Приведите выражение константы рав-
равновесия Кр.
№
129
130
Уравнение обратимой
гетерогенной реакции
ВаСО3(т)<=±ВаО(т)+СО2(г)
CaCO3(T)F*CaO(T)+CO2(r)
№
131
132
Уравнение обратимой
гетерогенной реакции
SrCO3(T)^SrO(T)+CO2(r)
MgCO3(T)^MgO(T)+CO2(r)
133. Константа равновесия реакции: Н2(г) + 12(г) +± ^Ш^ при Т= 720 К равна 50.
Какое число моль водорода надо взять на 1 моль йода, чтобы 80 % последнего про-
прореагировало?
134-139. Рассчитайте при температуре Т константу равновесия Кр обратимой гомо-
гомогенной реакции и объемный состав (в %) равновесной газовой смеси, если реагенты
в исходной смеси были взяты в количествах (моль), соответствующих стехиометри-
ческим коэффициентам уравнения реакции. Укажите, в каком направлении (исход-
(исходных веществ или продуктов) будет смещаться положение равновесия при повыше-
повышении температуры. Для ответа используйте дифференциальное уравнение изобары
Вант-Гоффа. Температурной зависимостью стандартных теплового эффекта и эн-
энтропии реакции пренебречь.
134
135
136
Уравнение обратимой
гомогенной реакции
2HCl(r) + F2(r)^2HF(r)+Cl2(r)
CO2(r)+H2(r)^CO(r) + H2O(r)
2CH4(r) z± C2H6(r) +Н2(г)
Г, К
5000
1500
2000
№
137
138
139
Уравнение обратимой
гомогенной реакции
Н2(Г)+С12(Г)^2НС1(Г)
2NO(r)?±N2(r)+O2(r)
2HI(r) ?± H2(r) + I2(r)
Г, К
700
2000
450
140. Используя стандартные термодинамические характеристики, рассчитайте при
800 К константу равновесия Кр обратимой гетерогенной реакции: FeO(T) + СО^ <=2
3=± Fe^ + СО2(Г) и равновесные парциальные давления оксидов углерода, если об-
общее давление в системе составляет 2 атм.
141-146. Рассчитайте объемный состав (в %) равновесной газовой смеси, образо-
образовавшиеся в результате реакции, протекающей в изобарно-изотермических условиях
при температуре Т и общем давлении /7общ. Температурной зависимостью стандарт-
стандартных теплового эффекта и энтропии реакции пренебречь. Укажите, в каком направ-
ЛР 8. Химическое равновесие
401
лении (исходных веществ или продуктов) будет смещаться положение равновесия:
а) при увеличении давления в системе; б) при уменьшении ее объема? Для ответа
используйте дифференциальное уравнение Планка—Ван Лаара.
141
142
143
Уравнение обратимой
гомогенной реакции
N2(r)^2N(r)
2N(D^N2(r)
Н2(г) <=* 2Н(г)
г,к
6000
5000
4000
Робщ,
атм
1
10
2
№
144
145
146
Уравнение обратимой
гомогенной реакции
2Н(г) *=* Н2(г)
°2(D ^ 2О(Г)
2О(г)^О2(г)
Г, К
3500
4500
4000
Робщ,
атм
1
2
0,1
147. Рассчитайте константы равновесия Кр9 Кс и Кх реакции: 2Н2(г) + О2(г) <=?
<=> 2Н2О(г) при Т = 1400 К и /?общ = 1 атм, если при этой температуре известны
стандартные энергии Гиббса реакций:
1) СО(Г) + Н2О(Г) <=> СО2(г) + Н2(г) (Д^0 =9,6 кДж),
2) 2СО2(Г) <=> 2СО(Г) +O2(r) (ArG2° =316,6 кДж).
На основании найденных значений констант укажите, в каком направлении
смещено положение равновесия.
148. Вычислите константу равновесия Кс реакции СО(г) + Н2О(г) <=> СО2(г) + Н2(г)
при Т = 600 К, если при Т = 800 К ее константа равновесия Кр = 4. Считайте, что в
этом интервале температур ДГЯ? = const и ArS? = const.
149. Рассчитайте равновесный выход (мольную или объемную долю) NO2 в резуль-
результате диссоциации его димера N2O4 по уравнению N2O4(r) <=? 2NO2(r) при Т= 340 К
= 1 атм, если константа равновесия при этой температуре Кр= 1,3 атм.
ЛР 8. Химическое равновесие
Цель работы — определение основных характеристик химического равнове-
равновесия и изучение влияния различных факторов на смещение его положения.
Опыт 1. Определение количественных характеристик гомогенного хи-
химического равновесия. В данном опыте экспериментально предлагается опреде-
определить равновесные концентрации всех компонентов обратимой гомогенной химиче-
химической реакции, протекающей в растворе между гексаметокситрифенилкарбинолом
(ГМТФК) С25Н28О7 и соляной кислотой НС1:
402 Химическое равновесие в гомогенных и гетерогенных системах
Н2О(р)
бесцветный малиновый
а затем по ним рассчитать остальные характеристики равновесия: равновесные вы-
выход продуктов р и степень превращения реагентов а, константу равновесия Кс:
[С25Н27О6С1][Н2О]
[С25Н28О7][НС1]
где [...] — равновесные молярные концентрации компонентов.
Принять равновесную концентрацию воды приблизительно равной концен-
концентрации чистой воды, а именно 55,56 моль/л, так как в опыте используются разбав-
разбавленные водные растворы, т.е. растворы, близкие по составу к чистой воде.
Равновесная концентрация эфира С25Н27О6С1 определяется экспериментально
фотоколориметрическим методом по интенсивности малиновой окраски исследуе-
исследуемого раствора, обусловленной присутствием в нем этого соединения.
Равновесные концентрации реагентов ГМТФК и НС1 следует рассчитывать
по разности между их исходными концентрациями и равновесной концентрацией
эфира в растворе, так как, согласно стехиометрическому уравнению реакции, число
молей образовавшегося эфира равно числу молей каждого из прореагировавших
реагентов.
Последовательность проведения:
1)в мерную колбу на 50 мл слейте из бюреток заданные преподавателем объемы
растворов реагентов: ГМТФК A0~3 М) и НС1 A0~2 М). Отметьте происходящие при
этом изменения;
2) дистиллированной водой доведите объем раствора до метки и, закрыв мерную
колбу пробкой, тщательно его перемешайте;
3) налейте раствор в кювету на 3/4 высоты, протрите ее торцевые стенки фильтро-
фильтровальной бумагой и измерьте оптическую плотность раствора на фотоэлектроколо-
риметре. Значение запишите. Правила работы на фотоэлектроколориметре смотрите
в инструкции к прибору.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов, а также выводы занесите в
табл. Л.8.1;
2) объясните причину появления малиновой окраски при сливании растворов реа-
реагентов, подтвердив соответствующим уравнением реакции;
3) по объемам К растворов реагентов, взятых для эксперимента, с учетом их после-
последующего разбавления до 50 мл рассчитайте их исходные концентрации Со в иссле-
исследуемом растворе по формулам:
v
HC1
Ч)гмтфк
4) по измеренной оптической плотности определите из калибровочного графика,
полученного у лаборанта, равновесную концентрацию эфира [C25H27O6CI] в иссле-
исследуемом растворе;
ЛР 8. Химическое равновесие
403
Таблица Л. 8.1
Результаты эксперимента
Исходные данные
Объем раствора
реагента V, мл
ГМТФК,
1(Г3М
НС1,
1(Г2М
Результаты
измерений
Оптическая
плотность
исследуемого
раствора
Результаты расчетов
Исходная концен-
концентрация реагента Со,
моль/л
ГМТФК
НС1
Равновесная концентрация
компонента [...], моль/л
Эфир
Н2О
55,56
Результаты расчетов
Равновесная степень
превращения реа-
реагента а, %
ГМТФК
НС1
Равновесный
выход эфира Р,
%
Константа
равновесия
КсшК°с
Изменение
стандартной энергии
Гиббса ArG2°98,
кДж/моль
ГМТФК
НС1
Выводы
В равнове
смеси
преоблад
(реагент
продукт
сной
ают
ы,
ы)
5) рассчитайте равновесные концентрации реагентов (ГМТФК и НС1);
6) направление и глубину протекания исследуемой реакции оцените, рассчитав:
а) равновесную степень превращения каждого из реагентов
к ГМТФК
-[ГМТФК] Со на-[HCl]
100; аНС1= 100,
0 ГМТФК
0 НС1
где [...] — равновесные молярные концентрации реагентов;
б) равновесный выход эфира
Р
с25н27о6С1
_ [С25Н27О6С1]
25Н27О6С1
где Сстех с н о ci — концентрация эфира, рассчитанная при условии полного пре-
превращения взятого в недостатке реагента в соответствии со стехиометрическим
уравнением;
в) константу равновесия Кс (см. (Л8.1) и (Л.8.2));
г) изменение стандартной энергии Гиббса Д^^, равное в данном случае
изменению стандартной энергии Гельмгольца АГА29%, так как изучаемая реакция
протекает в растворе при /?, V = const да еще и без изменения количества вещества
(Av = 0). Величина Аг^298 связана с константой равновесия К^ соотношением
404 Химическое равновесие в гомогенных и гетерогенных системах
7) на основании полученных данных (результатов расчетов) сделайте выводы о том,
какие вещества (реагенты или продукты) преобладают в равновесной смеси и в ка-
каком напрвлении смещено положение равновесия в исследуемой системе.
Опыт 2. Влияние посторонних веществ и изменения концентраций ком-
компонентов на положение химического равновесия. В данном опыте влияние посто-
посторонних веществ и изменения концентраций компонентов на положение химического
равновесия изучается на примере обратимой гомогенной реакции, протекающей в
растворе между ионом трехвалентного железа Fe3+ (ионом-комплексообра-
зователем) и изотиоцианат (роданид)-ионами NCS" (анионами-лигандами):
Fe?} + /iNCS"} *± [Fe(NCS)w ]^ (Л.8.3)
желтый красный
где соответственно п = 1; 2; 3; 6; z = 2+; 1+; 0; 3-.
Продуктами реакции, окрашивающими раствор в красный цвет, являются
комплексы железа, состав которых зависит от концентрации реагирующих веществ.
Поскольку реакция обратима, то для получения окрашенных комплексов следует
брать избыток роданид-ионов. При их большом избытке красная окраска появляется
даже при ничтожно малых концентрациях ионов Fe3+, и продуктом, отвечающим за
ее появление, является комплекс [Fe(NCSN]3~. При средних концентрациях ионов Fe3+
и NCS" окрашенным соединением, преимущественно образующимся в растворе, явля-
является роданид железа [Fe(NCSK]°. По изменению интенсивности красной окраски су-
судят о концентрации комплексов, т.е. о направлении смещения положения равновесия.
Протеканию данной реакции мешают анионы-лиганды, образующие с ионом
Fe3+ более устойчивые, но бесцветные комплексы (см. табл. П.4.4). К их числу отно-
относятся фторид-, оксалат-, фосфат-ионы и некоторые другие:
Fe* + ЗС2О*"ф) ?=t [Fe(C2O4),fc (Л.8.4)
В их присутствии равновесие реакции (Л.8.3) смещается в сторону реагентов,
вследствие чего роданидные комплексы разрушаются, а интенсивность красной
окраски ослабевает. Объединяя (Л.8.3) и (Л.8.4), (Л.8.3) и (Л.8.5), запишем уравне-
уравнения, описывающие процессы разрушения одних и образования других (более проч-
прочных) комплексов железа:
[Fe(NCSK]^ + ЗС2О^} <±[Fe(C2O4K]^ + 3NCS"W (Л.8.6)
красный бесцветный
[Fe(NCSK4 + 6F-, ^ [FeF6]?) + 3NCS") (Л.8.7)
красный бесцветный
Положение равновесия реакций смещено вправо.
ЛР 8. Химическое равновесие
405
Последовательность проведения:
1)в химическом стакане на 50 мл смешайте по 10 мл 0,01М раствора FeCl3
(Fe2(SO4K или Fe(NO3K) и 0,02М раствора NH4NCS (или KNCS). Отметьте происхо-
происходящие при этом изменения;
2) полученный раствор разлейте поровну в 5 конических пробирок;
3) первую оставьте в качестве эталона — образца сравнения окраски, во вторую,
третью, четвертую и пятую добавьте на кончике шпателя кристаллическое вещест-
вещество: соответственно FeCl3, NH4NCS, NH4CI, NaF (или Н2С2О4), или по 2-3 капли их
концентрированных растворов;
4) содержимое пробирок тщательно перемешайте интенсивным встряхиванием или
с помощью стеклянных палочек до полного растворения солей. Отметьте изменение
интенсивности окраски растворов в каждой из пробирок в сравнении с эталоном.
Обработка результатов:
1) исходные данные, наблюдения и выводы занесите в табл. Л.8.2;
Таблица Л.8.2
Результаты эксперимента
Исходные данные
№
про-
пробирки
1
2
3
4
5
Добавляемое
RfillieCTRO
Dvil^vv L D\J
Эталон
FeCl3
NH4NCS
NH4C1
NaF(H2C2O4)
Наблюдения
Изменение
(ослабление,
усиление)
интенсивно-
интенсивности красной
окраски
раствора
Красная
Выводы
Изменение (увеличение, уменьше-
уменьшение) равновесной концентрации
FeCl3
NH4NCS
Fe(NCSK
NH4CI
Смещение
(вправо, влево)
положения
равновесия в
системе
Первоначально установившееся равновесие в сис-
системе FeCl3 +3NH4NCS <=t Fe(NCSK +3NH4C1
с равновесными концентрациями компонентов
2) объясните причину появления интенсивной красной окраски при сливании рас-
растворов FeCl3 и NH4NCS;
3) сформулируйте критерии химического равновесия, используя кинетическое и
термодинамическое выражения закона действующих масс. Напишите выражение
константы равновесия Кс\
4) на основании наблюдений изменения интенсивности окраски раствора сделайте
выводы об изменении равновесных концентраций компонентов и о смещении по-
положения равновесия в системе при добавлении: a) FeCl3; б) NH4NCS; в) NH4C1;
г) NaF (или Н2С2О4);
5) сместится ли положение равновесия в рассматриваемой системе при увеличении
ее объема, т.е. при разбавлении? Ответ аргументируйте;
406 Химическое равновесие в гомогенных и гетерогенных системах
6) как согласуются полученные результаты с принципом Ле Шателье—Брауна и
законом действующих масс?
Опыт 3. Влияние изменения температуры на смещение положения
гомогенного химического равновесия. В данном опыте действие температуры на
химическое равновесие изучается на примере обратимой гомогенной химической
реакции термической диссоциации N2O4, протекающей в газовой фазе:
N2O4(r) ^2NO2(r) (ДгЯ>0)
бледно-желтый красно-бурый
Используемый для этой цели прибор (рис. Л.8.1) представляет собой запаян-
запаянный стеклянный {/-образный сосуд 7, колена которого погружены: одно — в стакан
2 со снегом или колотым льдом, другое — в стакан 3 с горячей водой. Сосуд запол-
заполнен газовой смесью из N2O4 и NO2, находящихся в состоянии динамического равно-
равновесия. Для ее получения в лабораторных условиях используют реакцию термичес-
термического разложения нитрата свинца:
2Pb(NO3J(T) =2PbO(T) +4NO2(r) +O2(r)
Красно-бурая окраска NO2 обусловлена одним неспаренным электроном ато-
атома азота. Благодаря этому электрону две молекулы NO2 объединяются в димер N2O4 —
газ бледно-желтого цвета. Связь между атомами азота в нем осуществляется элек-
электронной парой. Предполагают, что атомы азота в N2O4 связаны я-связью.
При Г = 21... 135 °С и /? = 760 мм рт. ст. NO2 существует в равновесии со своим
димером N2O4. Состав равновесной смеси зависит от температуры:
Температура Г, °С 27 40 50 100 135
Массовая доля NO2
в равновесной смеси, % 20 31 40 89 98,7
Из приведенных данных видно, что нагревание газовой смеси приводит к увели-
увеличению равновесного содержания в ней NO2 и, как следствие, к усилению интенсивности
ее красно-бурой окраски. Наоборот, охлаждение увеличивает содержание в смеси N2O4
и тем самым ведет к ослаблению интенсивности окраски. Таким образом, процесс тер-
термической диссоциации N2O4 — эндотермический процесс (АГН > 0 ), обратный про-
процесс — процесс димеризации NO2 — экзотермический ( Дг# < 0).
Визуально оценивая интенсивность красно-бурой окраски газовой смеси в
обоих коленах {/-образного сосуда, можно судить о равновесной концентрации
NO2, т.е. о смещении положения равновесия.
Последовательность проведения:
1) на фоне листа белой бумаги сравните интенсивность красно-бурой окраски газо-
газовой смеси в обоих коленах G-образного сосуда в исходном состоянии 1 (при ком-
комнатной температуре). Результаты запишите;
2) затем, перевернув его концами вниз, поместите левое колено в стакан с горячей
водой, нагретой до температуры кипения, правое — в стакан со снегом или колотым
льдом (см. рис. Л.8.1);
ЛР 8. Химическое равновесие
407
3) через 3...5 мин сосуд приподнимите над
стаканами и на фоне листа белой бумаги
наблюдайте произошедшее изменение ин-
интенсивности окраски газовой смеси в обоих
коленах в сравнении с интенсивностью в
исходном состоянии 1. Результаты запи-
запишите;
4) затем, поменяв колена местами (исходное
состояние 2), снова опустите их в стаканы;
5) каждые 10... 15 с сосуд приподнимайте и
фиксируйте происходящие изменения интен-
интенсивности окраски в обоих коленах;
6) спустя 5 мин сосуд выньте из стаканов,
положите на лист белой бумаги, отметив ин-
интенсивность бурой окраски в коленах. Далее
наблюдайте ее постепенное изменение во
времени в естественных условиях в течение
10... 15 мин. Результаты запишите.
Обработка результатов:
1) условия проведения опыта, наблюдения и
выводы занесите в табл. Л.8.3;
Рис. Л.8.1. Прибор для изучения
влияния изменения температуры
на смещение положения равнове-
равновесия реакции термической диссоци-
диссоциации N2O4:
/ — стеклянный {/-образный сосуд,
заполненный газовой смесью из N2O4 и
NO2; 2 — стакан со снегом или колотым
льдом; 3 — стакан с горячей водой
Таблица Л.8.3
Результаты эксперимента
Условия проведения опыта
Колено
{/-образ-
{/-образного
сосуда
т,
°С
Окраска
газовой
смеси
Исходное состояние 1
Левое
Правое
Исходное состояние 2
Левое
Правое
т,
°С
Наблюдения
Выводы
Изменение (усиление, ослабление, увеличе-
увеличение, уменьшение)
интенсивно-
интенсивности красно-
бурой окрас-
окраски смеси
равновесной
концентрации
в газовой смеси
N2O4
NO2
величин
КхКръКс
реакции:
Смещение
(вправо, влево)
положения
равновесия
реакции:
N2O4 ±± 2NO2
Состояние 2 - после нагрева левого и охлаждения правого колена
Состояние 3 - после нагрева правого и охлаждения левого колена
2) с помощью уравнения реакции опишите равновесие, существующее в газовой
смеси между ее компонентами. Приведите выражения констант равновесия Кх, Кр и
Кс, согласно закону действующих масс, и установите взаимосвязь между ними для
данной системы;
3) объясните наблюдаемые эффекты и их причину;
408 Химическое равновесие в гомогенных и гетерогенных системах
4) как изменяются равновесные концентрации (молярные доли) компонентов газо-
газовой смеси и степень превращения реагента а в каждом колене? Укажите, в каком
направлении при этом смещается положение равновесия;
5) в какую сторону смещается положение равновесия: а) при увеличении общего
давления в системе; б) при увеличении объема системы? Как при этом изменяются
величины Кх, Кр, Krf При ответе используйте дифференциальное уравнение План-
Планка—Ван Лаара A2.3);
6) используя термодинамические данные, рассчитайте константы равновесия Кр и
Кс, а также степень превращения а реагента при Г= 0 и 100 °С процесса термиче-
термической диссоциации N2O4.
Опыт 4. Влияние кислотности (рН) среды на положение гомогенного
равновесия. Действие кислотности среды изучается на примере протекающих в
водных растворах солей и кислот реакций взаимного превращения хромат- (СгО4~)
и дихромат- (Сг2О^~) ионов. Подобные реакции по своей природе близки к реакци-
реакциям амфотерных переходов и гидролиза. Они протекают без изменения степени
окисления (валентного состояния) атомов при изменении рН раствора и являются
обратимыми. Равновесие в них очень подвижно и может наступать при подходе к
нему с обеих сторон.
Хромовый ангидрид СгО3 относится к кислотным оксидам, хорошо раство-
растворяется в воде, образуя хромовые кислоты Н2Сгп03л+1 (п = 1,2, 3). Им соответствуют
соли: хроматы, дихроматы и т.д. Чем концентрированнее раствор (соли, кислоты) и
выше его кислотность, тем больше степень поликонденсации в нем анионов. Обра-
Образующиеся полихроматные анионы (Сг2О^~, Сг3О^, Cr4Oj23~) имеют цепочечное
строение: у каждого атома хрома почти правильное тетраэдрическое окружение из
атомов кислорода, один из которых выступает в роли мостика, связывающего тет-
тетраэдры СЮ4 между собой.
Из хромовых кислот наибольшее значение имеют хромовая Н2СЮ4 и ди-
хромовая Н2Сг207 кислоты. В индивидуальном виде они не выделены, получены
лишь их водные растворы, в которых они ведут себя как сильные электролиты, под-
подвергаясь двухступенчатой диссоциации на ионы (см. табл. П.4.10):
Н2СЮ4 ->Н++ НСЮ4 Н2Сг2О7 -> Н
НСгО" <=* Н+ + СЮ4~ НСг2О; <=> Н+ + СгО'
При этом лишь вторые их ступени являются обратимыми, причем положение рав-
равновесия в обеих сильно смещено влево — в сторону образования соответственно
НСЮ4- и НСг^- ионов.
Водные растворы хроматов имеют щелочную, а дихроматов — кислотную
реакцию вследствие протекающих в них реакций гидролиза, которые могут быть
описаны следующими ионно-молекулярными уравнениями:
ЛР 8. Химическое равновесие 409
~
f Cr2C>7 (рН
оранжевый
2Н++2СЮ4~
>7)
(рн<
= 7)
(Л.8.8)
(Л.8.9)
желтый
Сг2О,' + Н2О i=t 2НСЮ4
оранжевый желтый
Ясно, что и в тех, и в других растворах всегда присутствуют оба аниона, находя-
находящиеся в кислотно-основном равновесии друг с другом. Положение равновесия в
обеих реакциях смещено влево, поэтому водные растворы хроматов имеют желтую
окраску вследствие большей концентрации в них СЮ^~ -ионов, а водные растворы
дихроматов — оранжевую, обусловленную преимущественным содержанием в них
Сг2Оу~-ионов. Хромат-ионы существуют преимущественно в нейтральных и ще-
щелочных средах, дихромат-ионы — в кислотных.
Равновесие, устанавливающееся в водных растворах хроматов и дихроматов,
может быть нарушено либо действием кислот и оснований, либо введением в них
катионов, образующих с хромат- или дихромат-ионами малорастворимые соедине-
соединения, выпадающие в осадок. Так, увеличение концентрации водных растворов хро-
хроматов и дихроматов, подкисление первых и подщелачивание вторых приводит к
смещению положения равновесия последних реакций вправо и превращению хро-
хромат-ионов в дихромат-, а дихромат-ионов, наоборот, в хромат-ионы по реакциям
соответственно:
2СгО24~ + 2Н+ <z> 2НСЮ" <=* Сг2О7~ + Н2О (Л.8.10)
Сг2О7~ + 2ОН" +± 2СтО24- + Н2О (Л.8.11)
Следовательно, реакции (Л.8.10) и (Л.8.11) обратны реакциям (Л.8.8) и (Л.8.9) и
близки к реакциям амфотерных переходов. О смещении положения равновесия в
реакциях (Л.8.8)-(Л.8.11) в ту или иную сторону при изменении рН можно судить
по изменению окраски раствора. При рН < 6 положение равновесия в реакции
(Л.8.8) смещено уже вправо и раствор окрашен в ярко-оранжевый цвет, присущий
дихромат-ионам. Положение равновесия в реакции (Л.8.9) при рН > 6 смещено так-
также вправо, и при рН > 13 в растворе обнаруживаются только хромат-ионы. В слабо-
слабокислотных растворах заметно присутствие НС1О4 -ионов.
При добавлении растворимых солей свинца, бария или серебра к водным рас-
растворам хроматов выпадают кристаллические осадки малорастворимых хроматов:
желтые — свинца (РЬСЮ4) и бария (ВаСЮ4) и красно-бурый — серебра (Ag2CrO4).
Положение равновесия в реакции (Л.8.8) при этом еще сильнее смещается влево.
Аналогичные по составу осадки образуются и при добавлении таких солей к вод-
водным растворам дихроматов. Этот факт подтверждает существование равновесия
между хромат- и дихромат-ионами в таких растворах, которое описывается уравне-
уравнением (Л.8.9). Хотя положение равновесия реакции (Л.8.9) и смещено влево, тем не
менее концентрация СЮ24~ -ионов в растворе достаточна для образования осадков
менее растворимых по сравнению с дихроматами хроматов свинца, бария и серебра.
410 Химическое равновесие в гомогенных и гетерогенных системах
По мере связывания ионов СгО24~ в малорастворимую соль происходит смещение
положения равновесия реакции (Л.8.9) вправо, что приводит к накоплению в рас-
растворе водородных ионов, т.е. увеличению его кислотности. В конце концов кислот-
кислотность раствора достигает такого значения, при котором образующаяся вначале ма-
малорастворимая соль становится растворимой, так что в системе устанавливается
новое равновесие (на примере ВаС12):
К2Сг207 + 2ВаС12 + Н2О <=> 2ВаСЮ4 + 2КС1 + 2НС1
(Л.8.12)
Сг2С>7~ + 2Ва2+ + Н2О <± 2ВаСЮ4 + 2Н+
Для смещения приведенного равновесия вправо и практически полного осаждения
ВаСЮ4 необходимо связать ионы Н+. Последнее достигается добавлением, напри-
например раствора ацетата натрия. Наоборот, увеличение концентрации водородных ио-
ионов смещает положение равновесия в реакции (Л.8.12) влево, не давая осаждаться
ВаСЮ4.
В данном опыте на примере водных растворов хромата и дихромата калия
предлагается проверить:
а) существование в таких растворах равновесия между хромат- и дихромат-
ионами;
б) влияние на положение этого равновесия кислотности растворов и присут-
присутствия в них посторонних веществ.
Последовательность проведения:
1) налейте в одну пробирку 2-3 мл 0,1 М водного раствора дихромата калия К2Сг207,
в другую — такой же объем 0,1М водного раствора хромата калия К2Сг04. Сравни-
Сравните окраску растворов, свои наблюдения запишите;
2) в пробирку с раствором К2Сг207 прибавьте по каплям 1М раствор NaOH, а в про-
пробирку с раствором К2Сг04 — аналогичным образом 1M раствор H2SO4. Отметьте
изменения окраски растворов;
3) к раствору, полученному в пробирке 1, прилейте по каплям 0,5М раствор H2SO4
до изменения его окраски, а к полученному в пробирке 2 — аналогичным образом
2М раствор NaOH. Сравните окраску растворов с первоначальной;
4) повторив пп. 1, добавьте в обе пробирки по каплям 1М раствор ВаС12 или Ba(NO3J.
Произошедшие изменения запишите;
5) осадок, полученный добавлением к раствору К2Сг2О7 раствора ВаС12, разделите
поровну по трем пробиркам. В первую добавьте по каплям 5М раствор HNO3, во
вторую и третью — растворы СН3СООН и CH3COONa соответственно такой же
концентрации. Отметьте наблюдаемые изменения;
6) повторив пп. 1, 4, 5, проведите аналогичные эксперименты с использованием вме-
вместо ВаС12 растворимых солей свинца и серебра: Pb(NO3J или Pb(CH3COOJ, AgNO3.
Обработка результатов:
1) исходные данные, уравнения протекающих реакций, наблюдения и выводы
занесите в табл. Л.8.4а, Л.8.46;
2) объясните, чем обусловлена различная окраска водных растворов К2Сг207 и
К2СЮ4? Какие ионы в них содержатся? Приведите в молекулярном виде уравнения
реакций, обусловливающих существование этих ионов в растворах;
ЛР 8. Химическое равновесие
411
3) объясните изменение окраски растворов К2Сг207 и К2Сг04 при добавлении со-
соответственно NaOH и H2SO4 с точки зрения смещения положения установившего-
установившегося в них равновесия. Подтвердите свои выводы уравнениями протекающих реак-
реакций в молекулярном виде. Как согласуются полученные выводы с принципом
Ле Шателье—Брауна?
4) укажите цвет и состав осадков, выпадающих из растворов хромата и дихромата
калия при добавлении в них ВаС12. Приведите в молекулярном виде уравнения со-
соответствующих реакций. Объясните происходящие процессы с точки зрения сме-
смещения положения равновесия. Почему в обоих случаях образуется нерастворимый
ВаСЮ4? Для ответа на этот вопрос используйте числовые значения ПР (табл. П.4.11);
Результаты эксперимента
Таблица Л 8.4а
№
п/п
1
2
3
4
№
п/п
1
2
3
4
Исходные данные
Исходный раствор
Состав
ОДМК2Сг207
0,1МК2СЮ4
ОДМК2Сг207
0,ШК2СгО4
Окраска
Добавляемый 1М
водный раствор
NaOH
H2SO4
ВаС12
ВаС12
Наблюдения
Изменение окраски
раствора
Образование, цвет и состав
осадка
Уравнение
протекающей реакции
Выводы
Смещение (вправо, влево)
положения равновесия
реакций (Л.8.8) и (Л.8.9)
Результаты эксперимента
Таблица Л. 8.46
Исходные данные
№
пробирки
1
2
3
Добавляемый 5М
водный раствор
HNO3
СНзСООН
CH3COONa
Наблюдения
Растворение, увеличе-
увеличение количества осадка
Выводы
Смещение (вправо, влево) положе-
положения равновесия реакции (Л.8.12)
5) почему осадок ВаСЮ4 растворяется в растворе HNO3, но не растворяется в рас-
растворах СНзСООН и CH3COONa? Объясните эти явления смещением положения рав-
равновесия реакции (Л.8.12). В каком направлении оно смещается при добавлении каждого
из этих веществ?
412 Химическое равновесие в гомогенных и гетерогенных системах
Опыт 5. Влияние изменения температуры на смещение положения рав-
равновесия реакции образования йодом и крахмалом нестехиометрического со-
соединения включения. Соединения постоянного состава в химии называют стехио-
метрическими, переменного — нестехиометрическими. К числу последних относят
и соединения включения, или клатратные соединения (сокращенно клатраты). Они
образуются в результате обратимого внедрения молекул одного вещества, называе-
называемого «гостем», в свободные полости кристалла (решетчатые клатраты) или одной
большой молекулы (молекулярные клатраты) другого вещества, называемого «хо-
«хозяином» X и условно обозначаются как X • тГ, где т — среднее число молекул
«гостя». Образование клатрата происходит, если размеры полостей «хозяина» дос-
достаточно велики для вхождения «гостя» и в то же время достаточно малы, чтобы его
не выпустить. Молекулы включаемых соединений должны обладать определенной
конфигурацией, соответствующей форме полости «хозяина» В зависимости от фор-
формы полостей различают клеточные, канальнве и слоистые клатраты. Между «гостя-
«гостями» и «хозяевами» существует ван-дер-ваальсово взаимодействие или связи типа
водородных.
Свободный йод и крахмал образуют подобный клатрат темно-синего цвета,
называемый йодокрахмалом. Йод 12 как простое вещество представляет собой серо-
черные молекулярные ромбические кристаллы с фиолетовым металлическим бле-
блеском и острым запахом. Он хорошо растворяется в этиловом спирте и плохо в воде.
Его растворимость в воде значительно увеличивается в присутствии иодидов ме-
металлов. Например, он легко переходит в бесцветные водные растворы KI, окраши-
окрашивая их в бурый цвет. Такие растворы получили название «йодной воды», а коричне-
коричневые спиртовые растворы — «йодной настойки». Увеличение растворимости 12 в
растворах йодидов металлов обусловлено процессом комплексообразования, проис-
происходящим по схеме
где п = 1, 2, 3 и т.д. Образующиеся в таких растворах полийодид-ионы [1(Ь)Г,
[1A2J] (или упрощенно I3, IJ) непрочны, поэтому 12 в них ведет себя как свобод-
свободный йод.
Крахмал — белое аморфное порошкообразное вещество, представляющее
собой смесь двух полисахаридов: амилозы A0...20 %) и амилопектина (80...90 %).
Первая хорошо растворяется в теплой воде, второй — с трудом даже в горячей воде,
при этом набухает в ней с образованием растворов, похожих на вязкие коллоидные
растворы крахмала (крахмальный клейстер).
Амилоза и амилопектин состоят из a-D (+) глюкопиранозных мономерных
единиц (a-D-глюкозных остатков C6Hi0O5), соединенных а-A->4)-гликозидными
связями в неразветвленные цепи у амилозы и многократно разветвленные — у
амилопектина. У амилопектина в точках разветвления a-D-глюкозные остатки со-
соединены а-A->6)-гликозидными связями. Ниже показаны структура и строение
a-D(+) глюкопиранозной мономерной единицы макромолекул амилозы и амилопек-
амилопектина:
ЛР 8. Химическое равновесие
413
Гликозидная связь
а-F->1) /у'
Гликозидная связь Н CHLOH
С6Н10О5 = С6Н7О2(ОНK
Гликозидная связь
ooodo
Макромолекула амилозы и отдельные участки полигликозидных цепочек ами-
лопектина свернуты в спираль. При добавлении к водным растворам амилозы или
амилопектина «йодной воды» полийодид-ионы, захватываясь во внутренний канал
спирали, распадаются там с выделением свободного йода. Последний образует с ами-
амилозой аддукт характерного темно-синего цвета, а с амилопектином — темно-
фиолетового. Это молекулярный канальный клатрат (кавитат), в котором макромоле-
макромолекулы амилозы или амилопектина являются «хозяином», а молекулы йода «гостями».
Общую формулу крахмала можно условно представить в виде (С6Ню05)и, а
уравнение реакции образования йодокрахмального соединения написать как:
(С6НШО5)Я +т\2
т\2 (ДгЯ<0).
темно-синий
Данная реакция обратима, и при комнатной температуре положение равновесия в
ней смещено в сторону образования йодокрахмала, поэтому водные растворы, со-
содержащие йод и крахмал, имеют темно-синюю окраску. Повышение температуры
смещает его в противоположную сторону, вследствие чего такая окраска у нагретых
растворов отсутствует. Охлаждение последних вновь приводит к ее появлению.
В данном опыте предлагается проверить изложенные факты и подтвердить
основные особенности истинного химического равновесия: термодинамическую
устойчивость, подвижность, динамический характер, возможность достижения сис-
системой равновесного состояния при подходе как со стороны исходных веществ, так и
со стороны продуктов.
Последовательность проведения:
1) в коническую пробирку налейте 4-6 мл йодной воды или водного раствора йода.
Добавьте затем туда 2-3 капли раствора крахмала (крахмального клейстера). От-
Отметьте происходящие изменения;
414
Химическое равновесие в гомогенных и гетерогенных системах
2) содержимое пробирки тщательно перемешайте интенсивным встряхиванием или
стеклянной палочкой;
3) полученный темно-синий раствор разлейте пополам в две пробирки;
4) одну оставьте в качестве контрольного образца (эталона сравнения окраски), а
содержимое второй нагрейте на пламени газовой горелки, не доводя его до кипения.
Отметьте при этом произошедшие изменения в сравнении с контрольным образцом;
5) осторожно охладите нагретую пробирку под струей водопроводной воды. Что
при этом наблюдаете?
Обработка результатов:
1) исходные данные, наблюдения и выводы занесите в табл. Л.8.5;
2) объясните причину исчезновения темно-синей окраски при нагревании раствора
и ее появление вновь при его охлаждении. Укажите, какая из особенностей истин-
истинного химического равновесия при этом проявляется;
3) сделайте выводы о направлении смещения положения равновесия реакции обра-
образования йодокрахмала при нагревании и охлаждении раствора. Как они согласуются
с принципом Ле Шателье—Брауна? Какая из реакций (прямая или обратная) явля-
является эндотермической (Аг# > 0)?
Таблица Л.8.5
Результаты эксперимента
Исходные данные
№
про-
пробирки
1
2
Исследуемый образец раствора
Вид
Эталон
После нагревания
После охлаждения
Температура, Т°С
Т =
1 коми
Г«100°С
гн2о*
Наблюдения
Появление или исчезновение
окраски раствора
Раствор темно-синей окраски
Выводы
Смещение (вправо, влево) положения равновесия реакции:
(С6Н10О5)и +ml2 ^(С6Н10О5)„ ml2 при
нагревании
охлаждении
Тепловой эффект реакции
(Дг# ? 0)
прямой
обратной
Опыт 6. Влияние изменения концентрации углекислого газа в растворе
на смещение положения гетерогенного равновесия. В данном опыте рассматри-
рассматривается гетерогенное химическое равновесие, устанавливающееся в водном растворе
между растворимым гидрокарбонатом и малорастворимым карбонатом кальция. В
его основе лежит обратимая гетерогенная реакция, описываемая уравнением:
СаСО3(т) +СО2(Г) +Н2О(Ж) ^Са(НСО3J(р)
(Л.8.13)
Это равновесие играет исключительно важную роль в природных процессах:
оно лежит в основе карстовых явлений, регулирует содержание ионов кальция в
ЛР 8. Химическое равновесие 415
океанской воде, дает возможность морским организмам строить раковины, а птицам —
нести яйца, защищенные скорлупой.
При контакте твердых карбонатных пород с водой, содержащей углекис-
углекислый газ атмосферы, происходит образование гидрокарбоната. Последний суще-
существует только в растворах в условиях равновесия. Повышение или понижение
парциального давления СО2 в атмосфере изменяет его содержание в воде, а сле-
следовательно, смещает положение равновесия реакции (Л.8.13) соответственно
вправо или влево.
Для получения гетерогенной системы, в которой твердая фаза в виде СаСО3
находится в равновесии с водным раствором, содержащим СО2 и Са(НСО3J, через
насыщенный водный раствор гидроксида кальция пропускают углекислый газ до
образования вначале осадка СаСО3 по реакции:
Са(ОНJ (р) + СО2 (г) = СаСОз (т) + Н2О(ж)
а затем его последующего полного растворения по реакции (Л.8.13). При этом сум-
суммарный процесс описывается уравнением
Са(ОНJ (р) + 2 СО2 (г) = Са(НСО3J <»
В полученный таким образом прозрачный раствор на кончике шпателя добавляют
свежеосажденного СаСО3 так, чтобы был его небольшой избыток в виде нераство-
рившейся твердой равновесной фазы. Содержимое, тщательно перемешав интен-
интенсивным встряхиванием, хранят в плотно укупоренной склянке.
Растворимость углекислого газа в воде и формы нахождения его в водных
растворах зависят от температуры, состава и кислотности его растворов.
Последовательность проведения:
1) содержимое склянки, представляющее собой водный раствор Са(НСО3J и СО2,
содержащий в равновесии твердую фазу в виде СаСО3, хорошо взболтайте;
2) налейте его в три пробирки по 3-5 мл;
3) одну оставьте в качестве контрольного образца — эталона сравнения. Во вторую
пропускайте углекислый газ из аппарата Киппа. Отметьте происходящие при этом
изменения в сравнении с контрольным образцом;
4) третью пробирку нагрейте на пламени газовой горелки. Вновь отметьте происхо-
происходящие изменения, сравнив с контрольным образцом.
Обработка результатов:
1) исходные данные, наблюдения, рассмотрение системы по правилу фаз Гиббса и
выводы занесите в табл. Л.8.6;
2) напишите выражения констант равновесия Кс и Кх для реакции (Л.8.13), проте-
протекающей в исследуемой гетерогенной системе;
3) основываясь на растворимости углекислого газа, объясните причину полного
растворения осадка при пропускании углекислого газа через раствор и увеличения
его количества при нагревании раствора;
4) сделайте выводы относительно направления смещения положения равновесия
реакции (Л.8.13) в зависимости от изменения концентрации СО2 в растворе. Укажи-
Укажите, как они согласуются с принципом Ле Шателье — Брауна;
416
Химическое равновесие в гомогенных и гетерогенных системах
Результаты эксперимента
Таблица Л.8.6
Исходные данные
№
проби
рки
1
2
3
Исследуемый образец раствора
Вид
Эталон: раствор, находяи
СаСО3: СаСО3(т)+СО2(г
После пропускания СО2
После нагревания
Содержание СО2
дийся в равновесии с ос
>+Н2О(ж)<Р±Са(НСО3
Выше, чем в эталоне
Ниже, чем в эталоне
Выводы
№
про-
пробирки
2
3
Положение равновесия
реакции (Л.8.13) смещено
(вправо, влево)
Преимуществен-
Преимущественно протекает сле-
следующая реакция
]
]
Наблюдения
Полное растворение или
увеличение количества
осадка
адком в виде твердого
Рассмотрение системы
по правилу фаз Гиббса
Число
фаз
Ф
независимых
компонентов К
степеней
свободы С
5) рассмотрите данную гетерогенную реагирующую систему по правилу фаз Гиббса:
перечислите присутствующие в ней фазы и подсчитайте их общее число;
определите число независимых компонентов;
рассчитайте число (условное) степеней свободы;
6) почему число степеней свободы для одной и той же системы, находящейся в про-
пробирках 2 и 3, одинаково? Чем это обусловлено? Приведите независимые математи-
математические уравнения (соотношения), связывающие равновесные концентрации (или
равновесные молярные доли) веществ, присутствующих в системе для обоих случа-
случаев (пробирки 2 и 3).
VI
РАСТВОРЫ
¦ Общие свойства
растворов
¦ Растворы
электролитов
¦ Свойства
природной воды
Химически чистое вещество представляет со-
собой предельное состояние, которое практически
не достигается. Даже особо чистые металлы,
получаемые методами вакуумной или зонной
плавки, содержат определенное количество
примесей, около 10~6 % (масс.) и по существу
являются твердыми растворами. Образование
растворов — фаз, не подчиняющихся стехио-
метрическим законам и обладающих перемен-
переменным составом, существенно изменяет условия
протекания химических реакций между компо-
компонентами. Растворы широко распространены в
природе (особенно водные) и играют важную
роль во многих отраслях промышленности и
техники.
Изучение физико-химических свойств раство-
растворов, условий их образования обогащает и уг-
углубляет представления о механизме и законо-
закономерностях протекания химических процессов,
поскольку непрерывность изменения состава
системы — наиболее общий случай химическо-
химического взаимодействия. Современная теория рас-
растворов играет важную роль в развитии неорга-
неорганической и органической химии, коллоидной
химии, биохимии, электрохимии, молекулярной
физики, химической термодинамики, химиче-
химической кинетики, науки о полимерах, тесно связа-
связана с проблемами разделения природных и про-
промышленных смесей, получения чистых ве-
веществ.
14 - 9795
13 ¦
Общие свойства растворов
При смешении двух и более веществ происходит образование либо
новых соединений в результате протекания химической реакции,
либо смеси, которую можно разделить на исходные составные час-
части с помощью простых физических или механических методов. Та-
Такие смеси, называемые дисперсными системами (от лат. dispergo —
рассеиваю, рассыпаю), могут быть неоднородными (гетерогенны-
(гетерогенными) или однородными (гомогенными). Обычно термин «дисперс-
«дисперсные системы» используют для указания на многофазность объекта.
Гомогенные смеси двух и более компонентов, относительное коли-
количество которых может непрерывно меняться в различных пределах,
называют растворами.
13.1. Основные понятия и определения
Дисперсные системы в зависимости от агрегатного состояния
компонентов (твердое, жидкое, газообразное) подразделяют на 9
типов (табл. 13.1). Такие системы состоят, как правило, из двух
фаз. Одна из них является сплошной и называется дисперсионной
средой. Другая — дисперсная фаза — раздроблена и распределена
в первой.
Свойства дисперсных систем зависят от размеров распреде-
распределенных частиц. Если частицы велики по сравнению с молекулами,
то дисперсные системы, называемые в этом случае взвесями, неус-
неустойчивы и со временем разделяются на отдельные фазы. Если рас-
распределенное вещество находится в виде отдельных молекул, то
дисперсные системы состоят из одной фазы, устойчивы и не разде-
разделяются при сколь угодно долгом стоянии. Такие системы называют
молекулярными (истинными) растворами, или просто растворами.
Промежуточную область занимают коллоидные растворы (или зо-
золи), в которых размеры распределенных частиц находятся между
размерами частиц взвесей и молекулярных растворов. К ним отно-
относятся микрогетерогенные, метастабильные системы с жидкой дис-
дисперсионной средой. Строго говоря, четких границ между рассмат-
рассматриваемыми областями не существует, условно принято считать
взвесями системы с размерами распределенных частиц более 100 нм,
а молекулярными растворами — с размерами менее 1 нм.
Дисперсные системы широко распространены в природе и
играют важную роль в технике и технологии (см. табл. 13.1, а так-
также табл. П.2.1). Так, природные воды всегда содержат взвешенные
13.1. Основные понятия и определения
419
частицы, к естественным эмульсиям относятся нефть, млечные соки расте-
растений, например каучуконосов. В живых организмах многие процессы проте-
протекают в коллоидных растворах. Примером сложной дисперсной системы мо-
может служить молоко, основными составными частями которого являются,
помимо воды, жир, казеин и молочный сахар. Жир находится в виде эмуль-
эмульсии и со временем постепенно поднимается кверху (сливки). Казеин содер-
содержится в виде коллоидного раствора и самопроизвольно не выделяется, но
может быть осажден при подкислении молока (творог). Молочный сахар
находится в виде молекулярного раствора и выделяется лишь при испарении
воды. В виде дисперсных систем выпускается большинство промышленных
продуктов и веществ бытового потребления. Широко применяют взвеси, в
частности суспензии, в химической, цементной, силикатной, горной, метал-
металлургической, бумажной, текстильной, пищевой, кожевенной и других об-
областях промышленности. Эмульсии используют в процессах эмульсионной
полимеризации, как составы для консервации изделий, смазочно-
охлаждающие жидкости, препараты для обработки кожи и тканей, буровые
растворы при проходке нефтяных и газовых скважин и т.д.
Таблица 13.1
Классификация дисперсных систем по агрегатному состоянию компонентов
Дисперсионная
среда
Твердая
Жидкая
Газообразная
Распределенное
вещество
Твердое
Жидкое
Газообразное
Твердое
Жидкое
Газообразное
Твердое
Жидкое
Газообразное
Условное
обозначение
системы
т-т
ж-т
г-т
т-ж
Л\.~~~Л\
г-ж
т-г
ж-г
г-г
Примеры гетерогенных
дисперсных систем
Твердые гетерогенные системы:
минералы, сплавы, композицион-
композиционные материалы, бетон
Капиллярные системы: жидкость в
пористых телах, адсорбенты в рас-
растворах, почвы, грунты
Пористые тела: адсорбенты и ката-
катализаторы в газах
Суспензии и золи: промышленные
суспензии, пульпы, пасты, илы.
Эмульсии: природная нефть, моло-
молоко, млечные соки растений, кремы.
Газовые эмульсии и пены: флота-
флотационные, противопожарные,
мыльные пены
Аэрозоли: пыли, дымы, порошки
Аэрозоли: туманы, в том числе
промышленные, облака
Гетерогенные системы отсутствуют
14*
420 13. Общие свойства растворов
Изучением процессов, протекающих в дисперсных системах, занима-
занимается коллоидная химия. В данном разделе рассмотрены основные свойства
растворов.
С точки зрения термодинамики, все компоненты раствора равноцен-
равноценны, однако на практике используют понятия «растворитель» и «растворен-
«растворенное вещество». Обычно растворителем называют компонент, находящийся
в избытке, или существующий в том же агрегатном состоянии, что и образо-
образовавшийся раствор. Остальные компоненты раствора относят к растворен-
растворенным веществам. Для выражения состава раствора используют различные
величины. Наиболее распространенные из них, такие как массовая доля, мо-
молярная доля, массовая концентрация, молярная концентрация, молярная
концентрация эквивалента и моляльная концентрация, рассмотрены в гл. 2.
По агрегатному состоянию различают газовые, жидкие и твердые
растворы. Как известно, газы всегда самопроизвольно смешиваются и обра-
образуют гомогенные системы. Примером газового раствора служит воздух. Об-
Образование однородных твердых фаз переменного состава возможно, если
растворенное вещество заменяет в кристаллической структуре растворителя
атом на атом, ион на ион или молекулу на молекулу (твердый раствор заме-
замещения), или атомы (молекулы) растворенного вещества располагаются в
междоузлиях кристаллической решетки растворителя (твердый раствор внед-
внедрения). К твердым растворам относятся многие металлические сплавы, на-
например сплав серебра и золота. Изучением твердых растворов занимается
кристаллохимия.
Образование жидких растворов обычно сопровождается процессом
сольватации. Под сольватацией понимают совокупность энергетических и
структурных изменений, происходящих в растворе. В результате межмоле-
межмолекулярных (ориентационных, индукционных, дисперсионных) и ион-молеку-
ион-молекулярных взаимодействий (см. гл. 4) в растворе возникают соединения —
сольваты, представляющие собой частицы растворенного вещества, окру-
окруженные определенным (или переменным) числом молекул растворителя
(сольватной оболочкой). Различают ближнюю и дальнюю сольватации, вы-
выделяя первичную и вторичную сольватные оболочки. В первичную сольват-
ную оболочку входят молекулы растворителя, находящиеся в непосредст-
непосредственной близости к частице и совершающие движение в растворе вместе с
ней. Число молекул растворителя в первичной сольватной оболочке назы-
называют координационным числом сольватации. Во вторичную сольватную
оболочку входят молекулы растворителя, находящиеся на больших расстоя-
расстояниях от частицы растворенного вещества, но ориентированные вокруг час-
частицы определенным образом.
Сольваты тем легче образуются и тем более устойчивы, чем более по-
лярны частицы растворенного вещества и растворителя. Так, сольватация
13.1. Основные понятия и определения 421
очень сильно проявляется в водных растворах (в этом случае ее называют
гидратацией) и особенно в водных растворах электролитов. Она является
причиной образования аквакомплексов и кристаллогидратов.
Процесс растворения тесно связан с диффузией. В результате диффу-
диффузии растворенное вещество равномерно распределяется по всему объему
растворителя. По мере растворения новых порций концентрация растворен-
растворенного вещества увеличивается, вязкость раствора возрастает, диффузия за-
замедляется, уменьшается число свободных (не участвующих в образовании
сольватов) молекул растворителя. Процесс растворения протекает самопро-
самопроизвольно до тех пор, пока химический потенциал (см. гл. 6) чистого компонен-
компонента ц^ превышает химический потенциал данного компонента в растворе ц,.
При ц? = ц/ в системе устанавливается равновесие:
чистый компонент <=t растворенный компонент.
Иными словами, больше вещества при данных условиях раствориться не
может. Такой раствор называется насыщенным, а концентрация (состав) на-
насыщенного раствора — растворимостью. В справочниках часто раствори-
растворимость выражают через коэффициент растворимости ks, равный отношению
массы растворенного вещества mt к массе растворителя ms: ks = w,7 ms.
Растворимость веществ определяется физическим и химическим срод-
сродством молекул растворителя и растворенного вещества, т.е. зависит от энер-
энергии межмолекулярного взаимодействия в чистых компонентах. Если значе-
значения энергии взаимодействия близки, растворимость, как правило, высока, а
если сильно различаются — мала. Отсюда следует эмпирическое правило,
установленное еще алхимиками: подобное растворяется в подобном. Так
вещества, состоящие из полярных молекул, и вещества с ионным типом свя-
связи хорошо растворяются в полярных растворителях (воде, этаноле, жидком
аммиаке), а неполярные вещества — в неполярных растворителях (бензоле,
сероуглероде). Например, наличие или отсутствие ион-дипольных, диполь-
дипольных взаимодействий или водородной связи обусловливает при сме-
смешении жидкостей:
неограниченную взаимную растворимость. Как правило, это смеси по-
полярных веществ, таких как вода Н2О—серная кислота H2SO4, вода Н2О—
этанол С2Н5ОН, или неполярных веществ, таких как бензол СбН6—толуол
С6Н5СН3;
полную взаимную нерастворимость. Обычно это смеси полярных и
неполярных жидкостей, например вода Н2О—бензол СбН6;
ограниченную взаимную растворимость. В таких системах, состоящих
из полярных и малополярных жидкостей, при определенных концентрациях
и в определенном интервале температур образуются гомогенные растворы,
422
13. Общие свойства растворов
HCb(w+m)H2O Н+»иН2О С1 •тЪр
Рис. 13.1. Схема гидратации молекулы НС1
однако изменение условий приводит к расслоению жидкостей. Примером
служит смесь воды Н2О и анилина C6H5NH2.
Растворенные вещества можно классифицировать по их исходному
агрегатному состоянию: газы, жидкости, твердые соединения, а также по
свойствам, проявляемым ими в растворах: электролиты и неэлектролиты.
Согласно теории электролитической диссоциации, электролитами на-
называют вещества, которые в растворе или расплаве самопроизвольно час-
частично или полностью распадаются на ионы — заряженные частицы, спо-
способные к самостоятельному существованию. К электролитам относятся
твердые вещества с ионной кристаллической решеткой или вещества, со-
состоящие из молекул с полярной связью, т.е. большинство кислот, оснований
и солей. При попадании электролита в полярный растворитель (в частности,
в воду) происходит ослабление и разрыв связей между атомами в молекуле
растворенного вещества, что приводит к образованию сольватированных
(гидратированных) заряженных частиц — положительно заряженных ка-
катионов и отрицательно заряженных анионов. Например, диссоциация газо-
газообразного хлороводорода НС1 (рис. 13.1) и кристаллического хлорида на-
натрия NaCl (рис. 13.2) в водном растворе может быть представлена следую-
следующим образом:
o©o©S
©о©
о©©
NaCl(T)*(p+0)H2O Na+*/?H2O d • tfH2O
Рис. 13.2. Схема гидратации кристаллического NaCl
13.2. Термодинамические характеристики процесса образования растворов 423
НС1 (г) + (п + m)H2O <=> Н+ • лН2О + СГ • wH2O
NaCl (Т) + (р + ?)Н20 <i± Na+ • /?Н20 + СГ • ?Н20
Процесс распада полярного вещества в растворе на ионы называют
электролитической диссоциацией (ионного — ионизацией). По способности
к электролитической диссоциации электролиты условно подразделяют на
сильные и слабые. К сильным электролитам обычно относят вещества, ко-
которые в растворе практически полностью диссоциированы на ионы. Слабы-
Слабыми электролитами считают вещества, степень диссоциации которых неве-
невелика. Понятие степени диссоциации электролита а как величины, равной
отношению числа распавшихся (диссоциированных) молекул Nmcc к общему
числу молекул Л^ электролита,
а =
было введено С. Аррениусом A887) — создателем первой количественной
теории растворов электролитов.
Теория электролитической диссоциации и основанная на ней класси-
классификация кислот и оснований (см. гл. 1) в полной мере применимы лишь к
водным растворам. Для процессов, протекающих в неводных средах или без
участия растворителя, разработано несколько обобщенных теорий кислот и
оснований. Так, согласно протонной теории Брёнстеда и Лоури A923), к ки-
кислотам относятся вещества — доноры протона, а к основаниям — акцепто-
акцепторы протона (см. § 9.3). В электронной теории Льюиса A923) кислотно-
основное взаимодействие есть результат образования донорно-акцепторной
связи, где кислота — акцептор, а основание — донор электронных пар. В
дальнейшем ограничимся рассмотрением процессов, протекающих в водных
растворах, и представлениями теории электролитической диссоциации.
Сольватация неэлектролитов не приводит к диссоциации их молекул.
Образование растворов электролитов и неэлектролитов, несмотря на разли-
различие электрохимических свойств, определяется общими термодинамически-
термодинамическими характеристиками.
13.2. Термодинамические характеристики процесса
образования растворов
Термодинамическим условием самопроизвольного образования рас-
раствора является уменьшение энергии Гиббса системы ASG < О, определяемой
по уравнению
424 13. Общие свойства растворов
ASG = ASH-TASS, A3.1)
где ASH — энтальпия (теплота) растворения; ASS — энтропия растворе-
растворения (от англ. solution — раствор).
Образование растворов протекает как с поглощением (ASH > 0), так и
с выделением (ASH < 0) теплоты, поскольку сопровождается эндотермиче-
эндотермическими процессами разрушения исходных связей растворителя и растворен-
растворенного вещества и экзотермическим процессом образования новых связей
(сольватацией). Различают интегральные, промежуточные и дифференци-
дифференциальные энтальпии растворения.
Интегральная энтальпия растворения А3Нит — это тепловой эффект
растворения 1 моль вещества в некотором количестве чистого растворителя.
Ее можно рассчитать по формуле
Д,#инт= ^, A3.2)
где и, — число молей растворенного вещества. В свою очередь,
AsH=-mcpAT, A3.3)
где т — масса раствора; ср — удельная изобарная теплоемкость раствора;
AT— изменение температуры.
В случае, если растворенное вещество кристаллизуется из раствора в
виде кристаллогидрата (например, C11SO4 • 5Н2О или, точнее, [Cu(H2OM]SO4),
можно рассчитать интегральную энтальпию гидратации AhHmT — теплоту,
выделяемую при образовании 1 моль кристаллогидрата (от англ. hydration —
гидратация). Так, процесс растворения в воде соли C11SO4 можно считать
состоящим из двух стадий:
гидратация безводной соли
CuSO4 (Т) + 5Н2О(Ж) = CuSO4 • 5Н2О(Т) А аЯ^;
растворение образовавшегося гидрата
CuSO4 • 5Н2О(Т) + иН2О(ж) = CuSO4 • 5Н2О(р) Д5Яинт2.
Количество теплоты А5ЯИНть выделяющееся при растворении безвод-
безводной соли, равно алгебраической сумме тепловых эффектов этих двух про-
процессов, если составы растворов, образующихся при растворении безводной
соли и кристаллогидрата, одинаковы:
Д5ЯИНТ, = ДАЯИНТ + А5Яинт2. A3.4)
Следовательно,
13.2. Термодинамические характеристики процесса образования растворов 425
Для данных растворителя и растворенного вещества можно пригото-
приготовить много различных по концентрации растворов, поэтому число инте-
интегральных энтальпий растворения в данной системе может быть велико. При
написании термохимических уравнений процессов растворения указывают
концентрацию раствора, которую часто выражают величиной разбавления и,
т.е. числом молей растворителя, приходящимся на 1 моль растворенного
вещества. Например,
NH3 (Г) + 5 Н2О(Ж) = NH3 (p) (АЛинт =-33,158 кДж/моль).
Особый интерес представляют первая и полная интегральные энталь-
энтальпии растворения. Первой интегральной энтальпией растворения называют
теплоту растворении 1 моль вещества в бесконечно большом количестве
растворителя. В результате процесса образуется бесконечно разбавленный
раствор. Полной интегральной энтальпией растворения называют теплоту
растворения 1 моль вещества в таком количестве растворителя, которое не-
необходимо для образования насыщенного раствора. Первая и полная инте-
интегральные энтальпии растворения, как правило, различаются по величине и
нередко — по знаку. Чем больше растворимость данного вещества, тем
больше разность между их значениями. Для малорастворимых веществ обе
величины практически совпадают.
Промежуточной энтальпией растворения называют теплоту раство-
растворения 1 моль вещества в растворе, уже содержащем некоторое количество
этого вещества. Если растворение 1 моль происходит в бесконечно большом
количестве раствора, тепловой эффект называют дифференциальной эн-
энтальпией растворения Д5#ДИф. В этом случае концентрация раствора прак-
практически не изменяется, или, точнее, возрастает на бесконечно малую вели-
величину, которой пренебрегают. Дифференциальная энтальпия растворения в
растворе, концентрация которого отличается от концентрации насыщенного
раствора на бесконечно малую величину, называют последней энтальпией
растворения.
Интегральная и дифференциальная энтальпии растворения связаны
между собой соотношением
П
Тепловые эффекты растворения веществ различны при разных зна-
значениях температуры. По уравнению Кирхгофа (см. гл. 6), зависимость эн-
энтальпии растворения от температуры определяется величинами теплоемко-
теплоемкости растворителя и заданного раствора
426 13. Общие свойства растворов
ет )Р
где Ascp — разность молярных изобарных теплоемкостей чистого раство-
растворителя и раствора конечной концентрации.
Для термодинамического описания реальных растворов используют
понятие избыточных термодинамических функций, определяемых как раз-
разность каких-либо термодинамических свойств реального и идеального рас-
растворов одного и того же состава. В идеальных растворах сила межмолеку-
межмолекулярного взаимодействия однородных и разнородных частиц одинакова (в
смесях идеальных газов межмолекулярное взаимодействие отсутствует),
поэтому выполняются следующие условия:
при образовании идеального раствора не изменяются объем, внутрен-
внутренняя энергия и энтальпия системы: As V= О, Д5?/= О, Д5#= 0;
зависимость химического потенциала компонента от состава идеаль-
идеального раствора имеет вид, аналогичный зависимости химического потенциа-
потенциала компонента идеальной газовой смеси от состава:
где ц^ — стандартный химический потенциал /-го компонента; Хг — мо-
молярная доля /-го компонента в растворе;
изменение энтропии при смешении щ молей растворителя и пг рас-
растворенного вещества можно найти по уравнению, аналогичному для смеси
идеальных газов:
ASS = -nxR In X\ - n2R In X2.
Понятие идеального раствора — абстрактное, поскольку свойства
идеального раствора не зависят от природы компонентов, а определяются
только его составом. Реальные растворы могут лишь в той или иной степени
приближаться к идеальным. Так, при смешении жидкостей, молекулы кото-
которых неполярны и сходны между собой по структуре и химическим связям,
тепловые и объемные изменения очень малы, как, например, при смешении
бензола СбН6 и толуола С6Н5СНз: А5 V » 0 и Д5# « 0. Близки к идеальным и
бесконечно разбавленные растворы.
Таким образом, определяя избыточные (отличные от нуля) энтальпии
растворения, можно получить важную информацию о протекающих в рас-
растворах процессах, а также на основании закона Гесса (см. гл. 5) рассчитать
энтальпии образования многих жидких и кристаллических веществ. Весьма
ценную информацию дают и избыточные энтропии растворения, характери-
характеризующие степень неупорядоченности системы.
13.2. Термодинамические характеристики процесса образования растворов 427
Растворимость газов, жидких и твердых веществ в жидкостях опреде-
определяется не только природой компонентов, но и изменяется в широких преде-
пределах в зависимости от давления и температуры. Влияние давления на раство-
растворимость количественно можно описать уравнением
Щ
\ др }Т RT'
где Xi — молярная доля /-го компонента в насыщенном растворе; р — дав-
давление; As Vi — изменение объема 1 моля /-го компонента при переходе из
чистого состояния в состояние насыщенного раствора. Для газов при отно-
относительно небольших давлениях AsVt < О, поэтому с увеличением давления
растворимость газов в жидкостях увеличивается. Эта закономерность выра-
выражается законом Генри A802):
Pi=KiXi9 A3.6)
где Xi — молярная доля /-го газа в растворе; pt — парциальное давление газа
над раствором; Kt — постоянная Генри для /-го компонента. Закон Генри не
выполняется, если растворение газа в жидкости сопровождается образова-
образованием новых химических веществ. Например, растворение в воде аммиака
приводит к образованию гидроксида аммония (NH3 + Н2О = NH4OH), а рас-
растворение углекислого газа — к образованию угольной кислоты (СО2 + Н2О =
= Н2СОз). Для жидкостей и твердых тел влияние давления на растворимость
незначительно, так как для них, как правило, As F,« 0.
Влияние температуры на растворимость количественно выражает
уравнение
I 1ГГ )р RT2
где AsHumi - энтальпия перехода 1 моль /-го компонента из чистого со-
состояния в состояние насыщенного раствора (полная интегральная энтальпия
растворения). Если А5Ншт1 > 0, то растворимость увеличивается с ростом
температуры, если Д5#*нт/ < 0 — уменьшается. Растворение газов в жидко-
жидкостях в основном экзотермический процесс, поэтому растворимость боль-
большинства газов уменьшается с ростом температуры. Например, при Т= 0 °С в
100 г воды растворяется 5,56 мл метана СН4, а при Т = 100 °С — 1,70 мл.
Для жидкостей и твердых тел наблюдается как увеличение, так и уменьше-
уменьшение растворимости с ростом температуры, а также встречаются случаи, ко-
когда растворимость практически не зависит от температуры (А5Я*НТ/ « 0). На-
428 13. Общие свойства растворов
пример, растворимость хлорида кальция СаСЬ увеличивается с ростом тем-
температуры (при Т = 20 °С она составляет 74,5 г на 100 г воды, а при
Т = 80 °С — 147,0 г на 100 г Н2О), а растворимость гидроксида кальция
Са(ОН)г — уменьшается (при Т= 20 °С она составляет 0,160 г на 100 г во-
воды, а при Т = 80 °С — 0,092 г на 100 г Н2О).
13.3. Коллигативные свойства растворов
Все растворы обладают некоторыми общими свойствами, которые за-
зависят от соотношения числа частиц компонентов, природы растворителя и
практически не зависят от природы растворенного вещества. Общие (колли-
(коллигативные) свойства раствора - это свойства идеального раствора.
Одно из важных явлений, характерных для жидких растворов, — пони-
понижение давления насыщенного пара компонентов по сравнению с давлением
насыщенного пара чистых веществ. Давление насыщенного пара при данной
температуре характеризует равновесие между жидким и газообразным состоя-
состоянием вещества в закрытой системе, возникающее на границе раздела фаз вслед-
вследствие выравнивания скоростей эндотермического процесса испарения и экзо-
экзотермического процесса конденсации. В растворе концентрация молекул данно-
данного компонента в поверхностном слое меньше, чем в чистом веществе, поэтому
равновесие достигается при меньшем давлении насыщенного пара.
Условие равновесия представляет собой равенство химических потен-
потенциалов компонента в растворе ц/ и в паре над раствором ц*: ц,= ц*. Пред-
Предположим, что раствор и пар над ним подчиняются законам идеального
раствора и идеального газа соответственно, тогда
где Xi— молярная доля /-го компонента в растворе; pt — давление насыщен-
насыщенного пара /-го компонента над раствором. Дифференцируя эти уравнения и
используя условие равновесия, получаем
Интегрируя это уравнение в пределах по давлению от р? (pf — дав-
давление насыщенного пара чистого компонента) до/?, и по молярной доле от 1
до Xh имеем
А
Pi
13.3. Коллигативные свойства растворов
429
или
A3.8)
Выражение A3.8) количественно отражает связь между концентрацией
летучего компонента в идеальном растворе и давлением его пара над рас-
раствором (не имеет значения, будет ли этот компонент растворителем или рас-
растворенным веществом). Его называют законом Рауля: давление насыщенного
пара летучего компонента раствора прямо пропорционально его молярной
доле в растворе.
В случае когда все компоненты раствора летучие, общее давление па-
пара над раствором, согласно закону Дальтона, равно сумме парциальных дав-
давлений его компонентов: /?Общ= Х#* Уравнение A3.8) характеризует линей-
ную зависимость давлений насыщенного пара компонента от его молярной
доли в растворе, что возможно только в идеальных растворах (рис. 13.3, а).
Для реальных растворов линейная зависимость общего давления от состава
раствора нарушается: наблюдаются положительные (рис. 13.3,6) и отрица-
отрицательные (рис. 13.3, в) отклонения.
Причина отклонений от закона Рауля заключается в межмолекуляр-
межмолекулярном взаимодействии частиц растворенного вещества и растворителя (соль-
(сольватации). Если энергия взаимодействия однородных частиц превышает
энергию взаимодействия разнородных частиц (?а-а > ?а-в и ?в-в > ^а-вХ что
характерно для эндотермических процессов растворения (ASH > 0), то необ-
необходимо меньше энергии для перехода в пар молекул А, окруженных моле-
молекулами В, и молекул В, окруженных молекулами А. В этом случае имеют
место положительные отклонения от закона Рауля. Если же энергия взаимо-
взаимодействия однородных частиц меньше энергии взаимодействия разнородных
частиц (?а-а < ?а-в и ?В-в < ^а-в), что характерно для экзотермических про-
о
XB I," 0 Хв 1,0 0 хь 1,0
а бе
Рис. 133. Зависимость парциальных давлений насыщенного пара лету-
летучих компонентов АрА и Врв и общего давления насыщенного пара/?обЩ
от состава смеси Хв для идеальных (а) и неидеальных (б, в) растворов
430 13. Общие свойства растворов
цессов растворения (ASH < 0), то для перехода в пар молекул А и молекул В
требуется больше энергии. Поэтому отрицательные отклонения имеют ме-
место, когда процесс растворения сопровождается выделением теплоты.
Если растворяемое вещество — нелетучее соединение, то давление
насыщенного пара над таким раствором понижается по сравнению с давле-
давлением насыщенного пара чистого растворителя, и это понижение давления в
соответствии с законом Рауля пропорционально молярной доле растворен-
растворенного вещества. Для растворов неэлектролитов его можно вычислить по
формуле
АР = Л°-Л=А0-А0^1=А00-^1) = А°^2. О3-9)
где индекс «1» относится к растворителю, а индекс «2» — к растворенному
веществу.
Выражение A3.9) можно записать в виде
^ =*2, A3.10)
А
и закон Рауля сформулировать следующим образом: относительное пони-
понижение давления пара над раствором равно молярной доле растворенного
вещества.
Понижение давления пара растворителя над раствором определяется в
основном количеством растворенных частиц. Однако количество раство-
растворенных частиц в растворах электролитов, в отличие от растворов неэлектро-
неэлектролитов, определяется не только концентрацией раствора, но и степенью дис-
диссоциации электролита, поскольку, как уже отмечалось, все молекулы или
часть молекул электролита в растворе распадаются на ионы. Поэтому в A3.10)
следует ввести коэффициент диссоциации (коэффициент ионизации) /, учиты-
учитывающий увеличение числа частиц в растворе в результате электролитиче-
электролитической диссоциации
%=iX2. A3.11)
А
Коэффициент диссоциации i показывает, во сколько раз число частиц
в растворе электролита больше числа частиц в растворе неэлектролита той
же концентрации, т.е. для растворов неэлектролитов / = 1, а для растворов
электролитов i > 1. Если в результате диссоциации одной молекулы элек-
электролита образовалось п ионов (например, при диссоциации уксусной кисло-
кислоты п = 2: СН3СООН ±± СН3СОО~+ Н+, а при диссоциации гидроксида же-
железа II п = 3: Fe(OHJ <p* Fe2++ 2ОН~), то число ионов в 1 л раствора
13.3. Коллигативные свойства растворов 431
а число недиссоциированных молекул растворенного вещества
AUhcc =(l-a)CYA,
N
где a = дисс — степень диссоциации электролита (No = Nmcc + NHemcc); С —
No
молярная концентрация раствора, моль/л; N& — число Авогадро, моль.
Таким образом, коэффициент диссоциации i связан со степенью дис-
диссоциации а электролита соотношением:
N +N (\ — n\CN 4-nnCN
i= *недисс vH0H=u a)^jyA+an^A= _ )+ 1+ ( _1} A3 12)
No CNA
Отсюда
<х = -Ц- A3.13)
n-\
и, значит, по относительному изменению давления пара растворителя над
раствором известной концентрации A3.11) можно определить степень дис-
диссоциации электролита.
Как следствие понижения давления пара растворителя над раствором
нелетучего вещества повышается температура кипения и понижается темпе-
температура замерзания (кристаллизации, отвердевания) раствора по сравнению с
чистым растворителем. Температуре кипения жидкости соответствует тем-
температура, при которой давление ее насыщенного пара равно внешнему дав-
давлению (так, при давлении 101,3 кПа температура кипения воды равна
100 °С). Температура замерзания — это температура, при которой давление
пара над жидкостью становится равным давлению пара над твердой фазой
(при давлении 101,3 кПа температура замерзания воды равна 0 °С).
Повышение температуры кипения и понижение температуры замерза-
замерзания раствора по сравнению с чистым растворителем иллюстрирует рис. 13.4.
Давление пара над раствором нелетучего вещества всегда ниже давления
насыщенного пара над растворителем при той же температуре, поэтому ли-
линия ?>F, характеризующая зависимость давления насыщенного пара над рас-
раствором от температуры, лежит ниже линии ОС, характеризующей равнове-
равновесие между чистым растворителем и его паром. Следовательно, температура
кипения, соответствующая точке пересечения линии атмосферного давления
с кривой равновесия «жидкость <z± пар», для раствора 7^ 2 будет больше, чем
для растворителя Т^^ \. Очевидно также (см. рис. 13.4), что температура замер-
замерзания раствора Тзам2 меньше температуры замерзания растворителя Тзш \.
432
13. Общие свойства растворов
Т Т
л зам2 ¦* зам 1
кип 1
?кип2
Рис. 13.4. Фазовая диаграмма чистого растворителя (сплошная линия) и раствора
(пунктирная линия):
АО — линия равновесия между чистым твердым растворителем и его паром; ОС — линия
равновесия между чистым жидким растворителем и его паром; ОВ — линия равновесия
между чистым твердым растворителем и жидким растворителем; DF — зависимость давле-
давления насыщенного пара над раствором нелетучего вещества от температуры; DE — линия
равновесия между чистым твердым растворителем и раствором данного состава; О — трой-
тройная точка (равновесие между твердым, жидким и парообразным растворителем); Tmn \ —
температура кипения чистого растворителя при атмосферном давлении; ТКИП 2 — темпера-
температура кипения раствора данного состава; Тзйм i — температура замерзания чистого раствори-
растворителя; Гзам 2 — температура замерзания раствора данного состава (различия утрированы для
наглядности)
Установлено, что повышение температуры кипения раствора АГКИп и
понижение температуры его замерзания ДГзам пропорциональны концентра-
концентрации раствора:
A3.14)
A3.15)
где Ст — моляльная концентрация раствора, моль/кг; Е — эбулиоскопичес-
кая постоянная растворителя, К • кг/моль; К — криоскопическая постоян-
постоянная растворителя, К • кг/моль. Эбулиоскопическая и криоскопическая по-
постоянные растворителя показывают, на сколько градусов повышается тем-
температура кипения и понижается температура замерзания при растворении в
1 кг растворителя 1 моль неэлектролита.
Отметим, что по A3.14) и A3.15) можно определить температуру начала
кипения и замерзания растворов, поскольку по мере выкипания и кристаллиза-
кристаллизации растворителя концентрация нелетучего растворенного вещества увеличи-
увеличивается, соответственно повышается температура кипения и понижается темпе-
температура замерзания раствора. Таким образом, раствор, в отличие от растворите-
13.3. Коллигативные свойства растворов
433
ля, не имеет постоянной температуры
кипения и температуры замерзания, а
кипит и отвердевает в определенном
интервале температур.
При измерении температуры на-
начала кипения и замерзания растворов
получают важную информацию о взаи-
взаимодействии растворителя и растворен-
растворенного вещества, о составе и свойствах
последнего, например, определяют ко-
коэффициент диссоциации электролита.
Используя A3.14) и A3.15), можно рас-
рассчитать моляльную концентрацию Ст
неэлектролита (/ = 1), а по B.12) — его
молярную массу М (г/моль):
Рис. 13.5. Схема осмоса:
1 — раствор; 2 — растворитель; 3 — полу-
полупроницаемая мембрана
т
тЕ
тК
A3.16)
если известны масса растворенного веществ т (г) и масса растворителя ms
(кг), что очень важно для сложных органических молекул.
Коллигативное свойство растворов — осмотическое давление. Если
раствор и растворитель разделить полупроницаемой перегородкой, через
которую могут проникать только молекулы растворителя, то вследствие
разности химических потенциалов (химический потенциал растворителя в
растворе |iip меньше химического потенциала чистого растворителя щ: щр< [i\)
растворитель будет диффундировать через перегородку (рис. 13.5). Процесс
односторонней диффузии растворителя через полупроницаемую перегород-
перегородку называется осмосом (от греч. osmos — давление).
Для того чтобы получилась полностью гомогенная система, весь рас-
растворитель должен перейти в раствор. Однако по мере диффузии растворите-
растворителя объем раствора возрастает, и давление на перегородку со стороны рас-
раствора увеличивается. Возникает гидростатическая сила, препятствующая
осмосу (рис. 13.5). Осмотическое давление равно избыточному внешнему
давлению, которое нужно приложить к раствору, чтобы осмос прекратился
и в системе установилось равновесие.
Как показал Я. Вант-Гофф A884), для разбавленных растворов осмо-
осмотическое давление л определяется молярной концентрацией раствора (урав-
(уравнение Вант-Гоффа):
к = iCRT,
A3.17)
434
13. Общие свойства растворов
где г — коэффициент диссоциации, названный Вант-Гоффом изотониче-
изотоническим коэффициентом'^ С — концентрация раствора, моль/м3.
Для идеальных бинарных растворов зависимость осмотического
давления от состава выражается уравнением Ван Лаара:
п =
VM\ Р\
A3.18)
где Vm\ — молярный объем чистого растворителя; р® — давление насы-
насыщенного пара растворителя; р\ — давление насыщенного пара над раство-
раствором при температуре Г. Уравнение A3.18) свидетельствует о связи осмоса с
законом Рауля и его следствиями.
Осмотические явления широко распространены в природе. Осмос имеет
большое значение для растительных и животных организмов, поскольку спо-
способствует необходимому обводнению клеток и межклеточных структур. Так,
осмотическое давление крови, лимфы и тканевых жидкостей человека при Т =
= 37 °С равно тс = 7,7 атм, поэтому концентрация физиологических растворов
должна соответствовать этому значению (физиологические растворы должны
быть изотоническими). Осмос — главная сила, обеспечивающая движение во-
воды в растениях и ее подъем от корней до вершин. У пустынных растений тс до-
доходит до 170 атм. Явлением осмоса обусловлено то, что пресноводные рыбы не
могут жить в морской воде, а морские — в речной.
В технике используют обратный осмос — явление перехода раство-
растворителя из раствора через полупроницаемую мембрану, возникающее, если
со стороны раствора приложить давление, превышающее осмотическое
(рис. 13.6). Используя мембранные методы разделения компонентов, осно-
основанные на явлении обратного осмоса, можно проводить опреснение морской
воды, очистку сточных и технологических вод при температуре окружаю-
окружающей среды, без фазовых превращений и применения химических реагентов,
т.е. создавать малоэнергоемкие и экологически чистые производства.
11111
i i i i i
i I i i i
I i i i I
i i i I i
2 -:-:->:
р<п
1
:::":н2о;
1,1,1,1,1,
i i i i !¦
1 1 1 1 1
1 1 1 1 1
1 1 1 1 1
2 -:-:-:-:
р=п
н2о
1
----------
2-:->>:
р>п
н2о
н2о
1
Осмос
Равновесие
Обратный осмос
Рис. 13.6. Схема возникновения обратного осмоса:
1 — раствор; 2 — растворитель; 3 — полупроницаемая мембрана; р — давле-
давление над раствором; к — осмотическое давление
Растворы электролитов
Раствор электролита представляет собой смесь молекул раствори-
растворителя и сольватированных молекул и ионов растворенного вещества.
Относительное количество молекул, распавшихся на ионы, харак-
характеризующее степень диссоциации электролита а, зависит от природы
растворителя, природы и концентрации электролита, температуры,
давления и наличия других электролитов в растворе. Соответствен-
Соответственно, и свойства растворов электролитов, и особенности их поведения
зависят от этих факторов. В данной главе мы ограничимся рассмот-
рассмотрением основных свойств разбавленных водных растворов электро-
электролитов, как имеющих наибольшее значение для общего понимания
проблемы, а также играющих существенную роль в природных и
технологических процессах.
14.1. Влияние различных факторов на свойства
растворов электролитов
Природа растворителя во многом определяет характер элек-
электролитической диссоциации. Но при одновременном влиянии на
физико-химические свойства электролита различных параметров
растворителя, таких как диэлектрическая проницаемость, вязкость,
способность выступать в качестве донора или акцептора протонов
или электронных пар и других, не существует прямой зависимости
между свойствами растворителя и свойствами раствора. Одни и те
же вещества в разных растворителях могут быть как сильными, так
и слабыми электролитами. В основном наибольшую степень дис-
диссоциации электролиты проявляют в растворах жидкостей, имею-
имеющих высокую диэлектрическую проницаемость, малую вязкость и
высокую сольватирующую способность. К таким веществам в пер-
первую очередь относится вода.
В случае когда растворитель обладает малой диэлектриче-
диэлектрической проницаемостью, создаются условия для электростатического
взаимодействия ионов противоположного знака, энергия которого
равна
2
где Z\ и Z2 — зарядовое число катиона и аниона, соответственно;
е — заряд электрона; 8 — диэлектрическая проницаемость раство-
436 14. Растворы электролитов
рителя; / — расстояние между ионами. При этом возможно образование так
называемой ионной пары — сложного агрегата, состоящего из двух проти-
противоположно заряженных ионов, окруженных молекулами растворителя. Рас-
Расстояние q между ионами в ионной паре, согласно теории ионной ассоциа-
ассоциации, разработанной В.К. Семенченко A924) и Н. Бьёррумом A926), опреде-
определяется соотношением
ZxZ2e2
4 2гкТ
где q — параметр Бьёррума; к - постоянная Больцмана. Из этого соотноше-
соотношения следует, что при увеличении заряда ионов, расстояние, на котором они
начинают взаимодействовать, образуя ионную пару, увеличивается, а при
повышении диэлектрической проницаемости растворителя сила электроста-
электростатического взаимодействия между ионами уменьшается в е раз. Если рас-
расстояние между ионами меньше q, то растворенную частицу можно считать
недиссоциированной. В то же время при расчете параметра Бьёррума необ-
необходимо учитывать не только электростатическое взаимодействие ионов, но
и их взаимную поляризуемость (см. гл. 4), а также влияние на характер меж-
межионного взаимодействия сольватной оболочки, возрастающее с увеличением
заряда и уменьшением размера ионов.
В растворах сильных электролитов, полностью диссоциирующих на
ионы, даже в растворителях, имеющих высокую диэлектрическую прони-
проницаемость, роль взаимодействия ионов высока. Согласно электростатиче-
электростатической теории Дебая—Хюккеля—Онзагера A923), сольватированные ионы в
растворах сильных электролитов окружены преимущественно сольватиро-
ванными ионами противоположного знака, так называемой ионной атмо-
атмосферой, радиус rd которой может быть рассчитан по формуле
2e2l000NAl'
где rd называется также дебаевским радиусом экранирования; е — диэлектри-
диэлектрическая проницаемость растворителя; So — электрическая постоянная; / —
ионная сила раствора.
В зависимости от концентрации электролита выделяют область раз-
разбавленных растворов, которые по своей структуре близки к структуре чис-
чистого растворителя, переходную область и область концентрированных рас-
растворов. Разбавленные растворы слабых электролитов по свойствам близки к
идеальным растворам и хорошо описываются классической теорией элек-
электролитической диссоциации. Свойства разбавленных растворов сильных
электролитов и идеальных растворов заметно различаются. Их удовлетвори-
14.2. Диссоциация слабых электролитов 43 7
тельное описание проводится в рамках электростатической теории Дебая—
Хюккеля—Онзагера. В переходной концентрационной области под влияни-
влиянием ионов происходит существенное изменение структуры растворителя, а в
области высоких концентраций почти все молекулы растворителя связаны с
ионами в сольватных оболочках, т.е. структура раствора приближается к
структуре кристаллосольватов.
В зависимости от температуры и давления выделяют низкотемпера-
низкотемпературную и высокотемпературную области свойств электролитов, а также об-
области нормальных и высоких давлений. Изменение температуры и сущест-
существенное изменение давления оказывает влияние на ион-молекулярную упо-
упорядоченность раствора и сольватационные процессы. Сложность описания
электролитических систем, зависимость их свойств от большого числа фак-
факторов проводит к тому, что в настоящее время не существует единой теории
растворов электролитов.
14.2. Диссоциация слабых электролитов
Процесс электролитической диссоциации, возникающий в результате
сольватации, обратим, т.е. наряду с распадом молекул растворенного веще-
вещества происходит их образование из ионов:
где КтА„ — молекула электролита; KZ]+ — катион; AZl~ — анион; Z\ и 2г —
зарядовое число катиона и аниона соответственно; тип — стехиометриче-
ские коэффициенты. (Для упрощения уравнения электролитической диссо-
диссоциации молекулы растворителя, участвующие в сольватации частиц, не за-
записывают.) Равновесие между ионами и молекулами электролита подчиня-
подчиняется закону действия масс (см. гл. 10). Поэтому важной характеристикой
процесса диссоциации является константа диссоциации (константа иониза-
ионизации) Kd(C^ вычисленная по равновесным концентрациям молекул и ионов:
где [KZl+], [AZ2~] — равновесные молярные концентрации катионов и
анионов соответственно; [КтА„] — равновесная молярная концентрация не-
диссоциированных молекул электролита.
Константу равновесия процесса диссоциации принято обозначать Кй в
случае слабых кислот (от англ. «acid» — кислота) и ^ь для слабых основа-
438 14. Растворы электролитов
ний (от англ. «base» — основание). Так, слабая цианистоводородная кислота
диссоциирует по уравнению
HCN <=> Н+ + CN- Ka = [H+][CN ] = 4,93 • 1(Г10 .
[HCN]
Примером слабого основания служит гидроксид аммония:
NH4OH <-> nh; + он- къ = [NH^][0H"] = 1,8.10-5.
4 [NH4OH]
Многоосновные кислоты и многокислотные основания диссоциируют
ступенчато. Например, процесс диссоциации щавелевой кислоты Н2С2О4
протекает следующим образом:
I ступень
тт r~* r\ ^ ur+ i иг^ гл~
rl2L,2\J4 <fHZ Г1 + rl\^2U4
П ступень
HC2U4 ?=± H + C2O4
общее уравнение диссоциации
H2C2O4 <=> 2Н+ + С20^
КаA) = 6,46-
^а(Н) = 6,17-
Ю-2;
Ю-5;
Аналогично для гидроксида свинца РЬ(ОНJ можно записать:
I ступень
РЬ(ОНJ <± РЬОН+ + ОН"
П ступень
РЬОН+ & РЬ2+ + ОН"
общее уравнение ионизации
АГьда = 9,55
•ИГ4;
10"8;
РЬ(ОНJ <=> РЬ2+ + 2ОН- tfb = Къа)КЬ(П).
Рассмотрим раствор слабого бинарного электролита (число ионов, на
которые диссоциирует электролит п = 2, зарядовые числа ионов равны Zx =
= Za = Z) с молярной концентрацией С, степень диссоциации которого а:
КА ^± Kz+ + Az".
В состоянии равновесия концентрации катионов Kz+ и анионов Az~
будут равны [Kz+] = [Az~] = ocC, а концентрация недиссоциированных моле-
молекул [КА] = (С-аС) = СA - а). Используя уравнение A4.1), получают
14.2. Диссоциация слабых электролитов 43 9
к =[Kz+][Az-] = aCaC
d(C) [KA] (l-a)C
или
^С. A4.2)
Это выражение описывает закон разбавления (или разведения) Оствальда. В
случае когда степень диссоциации электролита a « 1, что имеет место при
Q
> 100, величиной а по сравнению с 1 можно пренебречь и закон раз-
бавления Оствальда записать в упрощенном виде
Kd{C)*a2C. A4.3)
Константа диссоциации характеризует процесс диссоциации данного
электролита в данном растворителе, но не зависит от концентрации элек-
электролита и при постоянной температуре А^(С) = const. Очевидно, что степень
диссоциации а тем больше, чем ниже концентрация, т.е. чем сильнее раз-
разбавлен раствор
A4.4)
При введении в раствор электролита ионов KZ]+ или AZl~ произой-
произойдет сдвиг равновесия диссоциации влево (в сторону образования недиссо-
циированных молекул) в соответствии с принципом Ле Шателье—Брауна и
установится равновесие с другими концентрациями [KZl+ ], [ AZl~] и [KWAJ,
но с тем же значением K^q. Таким образом, добавление одноименных ионов
равносильно увеличению концентрации раствора слабого электролита и по-
подавляет его диссоциацию.
Константа диссоциации будет меняться при варьировании температу-
температуры согласно уравнению изобары химической реакции (см. гл. 9):
2
дт )р rt
где AdH — тепловой эффект процесса диссоциации (от англ. dissociation —
диссоциация). Для многих веществ зависимость Kd^C) =/(Г) проходит че-
через максимум, что связано с различным влиянием температуры на гидрата-
гидратацию ионов и нейтральных молекул. Очевидно, что при температуре, соот-
соответствующей максимуму, AdH = 0. Например, константа диссоциации му-
440 14. Растворы электролитов
равьиной кислоты НСООН максимальна при Т= 24,4 °С, а уксусной кисло-
кислоты СНзСООН — при Т = 22,5 °С.
Рассмотренные закономерности справедливы для разбавленных раство-
растворов слабых электролитов. Поведение концентрированных растворов слабых
электролитов и растворов сильных электролитов невозможно описать с пози-
позиций классической теории электролитической диссоциации, развитой в трудах
С. Аррениуса, В. Оствальда, В. Нернста, Л.В. Писаржевского, И.А. Каблукова
и др. В этой теории не учитываются силы ионного взаимодействия, прояв-
проявление которых изменяет константы диссоциации слабых электролитов уже в
области умеренных концентраций, определяет свойства растворов сильных
электролитов.
14.3. Растворы сильных электролитов.
Ионная сила и активность
Растворы сильных электролитов обнаруживают особенности в пове-
поведении, не соответствующие их полной диссоциации на ионы. Так, реальная
концентрация ионов оказывается значительно меньше концентрации, зада-
задаваемой при приготовлении раствора. Кажущаяся (или определяемая экспе-
экспериментально, например, по данным эбулиоскопических или криоскопиче-
ских измерений) степень диссоциации сильных электролитов в соответст-
соответствии с опытными данными меньше 1 даже в разбавленных растворах, что по-
показывают приведенные ниже значения степени диссоциации а сильного
электролита НС1 в водных растворах различной концентрации:
С, моль/л 1,0 0,1 0,01 0,001
а 0,79 0,92 0,97 0,99
Это связано с тем, что в растворах электролитов наблюдается некото-
некоторая степень упорядоченности взаимного расположения ионов, вызванная
электростатическим взаимодействием катионов и анионов. На небольших
расстояниях от каждого иона преимущественно располагаются ионы проти-
противоположного знака, т.е. вокруг каждого иона в растворе создается ионная
атмосфера.
Таким образом, для процессов диссоциации и химических реакций,
протекающих в растворах с участием сильных электролитов, а также в кон-
концентрированных растворах слабых электролитов, нельзя рассчитывать кон-
константы равновесия на основании концентраций свободных ионов, которых
нет в реальных системах. Кроме того, различная степень сольватации ве-
веществ, участвующих в реакции, по-разному изменяет скорости прямой и
обратной реакций, что также приводит к зависимости константы равновесия
14.3. Растворы сильных электролитов. Ионная сила и активность 441
от общего содержания ионов в растворе. Поэтому для описания свойств ре-
реальных растворов электролитов, как и других реальных систем (см. § 10.3),
используют метод активностей Льюиса, в котором для учета межионных и
межмолекулярных взаимодействий введено понятие эффективной концен-
концентрации, или активности. Подстановка активности вместо концентрации в
термодинамические соотношения, справедливые для идеальных растворов,
позволяет применять их для описания любых систем. Активность а электро-
электролита суммарно отражает все эффекты взаимодействия ионов между собой и
с молекулами растворителя:
а = уСт, A4.5)
где Ст — моляльная концентрация электролита; у — коэффициент актив-
активности, который можно рассматривать как меру различия поведения элек-
электролита в данном растворе и в растворе, который принимают за идеальный.
Для идеальных растворов у = 1. Бесконечно (предельно) разбавленные рас-
растворы по свойствам приближаются к идеальным, поэтому в таких растворах
полагают у «1.
Коэффициенты активности и, следовательно, сами активности опре-
определяют экспериментально, измеряя различные свойства раствора, например,
давление пара растворителя, температуру кипения или кристаллизации рас-
раствора и др. Методов определения коэффициентов активности катионов у+ и
анионов у_ нет, поскольку получить раствор, содержащий только катионы
или анионы невозможно. Поэтому используют понятия средней ионной ак-
активности а± и среднего ионного коэффициента активности у±, которые
связаны соотношением
а±=У±Ст±, A4.6)
где Cmt — средняя моляльная концентрация ионов электролита, определяе-
определяемая как среднее геометрическое из концентраций катиона Cw+ и аниона Ст_.
Среднюю концентрацию ионов электролита можно рассчитать по формуле:
где тип — стехиометрические коэффициенты в уравнении диссоциации
электролита:
Средний коэффициент активности электролита у± представляет собой сред-
среднее геометрическое из коэффициентов активности катиона у+ и аниона у_:
442 14. Растворы электролитов
а средняя ионная активность — среднее геометрическое из активности ка-
катиона а+ и аниона ал
Электростатическая теория сильных электролитов, развитая в трудах
П. Дебая, Э. Хюккеля, Л. Онзагера и других ученых, позволяет вычислить
средний коэффициент активности у± сильного бинарного электролита в раз-
разбавленных растворах. Сила электростатического взаимодействия ионов с их
окружением (ионной атмосферой) определяется плотностью заряда в этом
окружении, а плотность заряда, в свою очередь, зависит от того, сколько
ионов находится в единице объема раствора, т.е. от их концентрации, и от
того, какой заряд несут эти ионы. Мерой этого взаимодействия является
ионная сила раствора /, рассчитываемая по формуле
ад2, A4.7)
где С,,,- — моляльная концентрация /-го иона; Z,- — зарядовое число /-го ио-
иона. В очень разбавленных растворах (/< 0,1) средний коэффициент активно-
активности электролита зависит только от ионной силы раствора и не зависит от
природы присутствующих в растворе ионов. Его можно рассчитать по урав-
уравнению предельного закона Дебая—Хюккеля:
\gy±=-AZxZ24l, A4.8)
где А — коэффициент, вычисляемый теоретически и зависящий от темпера-
температуры и диэлектрической проницаемости растворителя (для разбавленных
водных растворов при Г =298 К А-0,509). При средних концентрациях
раствора G=0,5...0,8) коэффициент активности /-го иона у, можно рассчи-
рассчитать по уравнению Дэвис, также основанному на теории Дебая—Хюккеля:
- 0,2/1. A4.9)
Равновесие в растворах сильных электролитов характеризуется тер-
термодинамической константой диссоциации К^у.
атат rKZl + lwrAz2"lw vmvn vmvn
К =^g^ = LK J LA J УЛ- ~ Vjl A410)
d{a) a [KWAJ у d(C) у
где а и у — активность и коэффициент активности недиссоциированных мо-
молекул электролита КтА„ соответственно; K^q — концентрационная (или
14.3. Растворы сильных электролитов. Ионная сила и активность 443
аналитическая) константа диссоциации. Уравнение, связывающее концен-
концентрационную и термодинамическую константы диссоциации, можно полу-
получить, прологарифмировав A4.10) с использованием A4.8):
A4.11)
где V/ — стехиометрический коэффициент /-го иона в уравнении диссоциа-
диссоциации электролита.
Очевидно, что при уменьшении концентрации электролита и, следова-
следовательно, ионной силы раствора К^а) -> K^q, и в бесконечно разбавленных
растворах эти константы равны. В случае разбавленных растворов слабых
электролитов / « 0, поэтому можно использовать концентрационную кон-
константу диссоциации. В случае разбавленных растворов сильных электроли-
электролитов, как уже отмечалось, с хорошим приближением можно считать, что их
диссоциация происходит практически полностью, поэтому концентрацион-
концентрационная константа диссоциации не имеет физического смысла. Например, при
диссоциации серной кислоты H2SO4 по I ступени:
H2SO4 = Н+ + HSO4 Ka A) -> оо,
так как концентрация недиссоциированных молекул [H2SO4] -» 0, тогда как
для II ступени:
HSO4 <=> Н+ + SO2" Ka („) = 1,12 • 1(Г2.
Аналогично для диссоциации сильных оснований, например гидро-
ксида кальция Са(ОНJ:
I ступень
Са(ОНJ = СаОН+ + ОН" Къ A) -> оо,
II ступень
СаОН+ <=> Са2+ + ОН" Къ т = 4,0 • 1<Г2.
Отметим, что электростатическая теория электролитов опирается на
значительно упрощенную физическую модель раствора, в ней не учитывает-
учитывается ассоциация ионов и растворитель рассматривается как непрерывная сре-
среда, характеризующаяся только диэлектрической проницаемостью ?, без уче-
учета молекулярной структуры растворителя и его взаимодействия с ионами. В
реальных условиях с увеличением концентрации возрастает роль сил оттал-
отталкивания, изменяются ион-дипольные взаимодействия, диэлектрическая про-
проницаемость, вязкость. Учет этих и многих других факторов представляет
серьезную проблему современной теории растворов электролитов.
444 14. Растворы электролитов
14.4. Ионные равновесия в водных растворах электролитов
Поскольку диссоциация в большинстве случаев — обратимый про-
процесс, то для реакций, протекающих в растворах электролитов, справедливы
общие законы равновесия (см. гл. 10) и соответственно A4.1) и A4.10). Рас-
Рассмотрим некоторые примеры ионных равновесий в водных растворах элек-
электролитов.
Наиболее распространенный природный растворитель, вода, пред-
представляет собой слабый электролит. По упрощенной схеме ее диссоциацию
можно выразить следующим уравнением:
н2о(ж)^ н(+р)+он(-р)
С учетом гидратации ионов водорода одно из уравнений диссоциации мож-
можно представить так:
Н2О(Ж) + Н2О(Ж) ^ Н3О+р) + ОН("р)
хотя установлено, что, кроме иона гидроксония НзО+, в растворе существуют
и более сложные гидраты: диаквапротон Н5О2, триаквапротон Н7Оз и др.
Константу диссоциации воды можно записать в виде
[Н2О] '
Степень диссоциации воды очень мала, поэтому концентрацию недиссоции-
рованных молекул можно принять равной концентрации индивидуального
вещества, которая постоянна при Т= const. Поэтому константа равновесия
процесса диссоциации воды определяется произведением равновесных мо-
молярных концентраций ионов
[Н+][ОН"]=^(С)[Н2О] = const.
Произведение концентраций ионов [Н+] и [ОН~] называют ионным
произведением воды А^н2о и рассчитывают по термодинамическим данным
процесса диссоциации:
ArG° = Д/??+ + A/G?H- -A/C#2o =0 + (-157)-(-237) = 80кДж/моль;
( °] 80000 ^ 1Л_14
10
( ArG] ( 80000 ^ 1Л
К о о = ехр —-— = ехр = 10
н2о fi RT I yy 8,3b298j
14.4. Ионные равновесия в водных растворах электролитов 445
Таким образом, при температуре Г= 298 К:
+ = 10-14. A4.12)
Диссоциация воды есть эндотермический процесс, поэтому ионное
произведение воды Кп 0 увеличивается с ростом температуры (см. табл.
ПАЗ).
Для указания концентрации ионов водорода в растворе используют
водородный показатель
pH = -lg[H+], A4.13)
а для обозначения концентрации ионов гидроксида — гидроксидный пока-
показатель:
pOH = -lg[OH-], A4.14)
при этом в соответствии с определением стандартного состояния все вели-
величины молярных концентраций (активностей) отнесены к стандартной кон-
концентрации (активности), т.е. безразмерны. При температуре Г= 298 К
рН + рОН=14, A4.15)
поэтому в чистой воде [Н+] = [ОН~] = 1 • 10~7 моль/л, следовательно, рН =
= рОН = 7;
в кислотной среде [Н+] > [ОН ], рН < 7, а рОН > 7;
в щелочных растворах [Н+] < [ОН"], рН > 7, а рОН < 7.
При необходимости более точных расчетов следует вычислять не рН,
a pV :
ран+ =-lgtfH+ =pH-lgyH+ A4.16)
поскольку в растворах сильных электролитов, а также в концентрированных
растворах слабых электролитов вместо понятия концентрации используют
понятие активности.
Обменные реакции в растворах электролитов протекают в направле-
направлениях связывания ионов, приводящих к образованию малорастворимых ве-
веществ (осадков или газов) или молекул слабых электролитов. Например,
реакция
2КС1 + Na2SO4 <-> 2NaCl + K2SO4
не протекает, поскольку не образовалось ни малорастворимых веществ, ни
слабых электролитов (в растворе имеются сольватированные ионы Na+, K+,
СГ, SO'");
446 14. Растворы электролитов
реакция
ВаС12 + K2SO4 -> BaSO4^ + 2KC1
сопровождающаяся образованием осадка, идет практически до конца;
в реакциях нейтрализации равновесие смещено в сторону образования
слабого электролита — воды, и, если остальные участники являются силь-
сильными электролитами, реакции нейтрализации протекают до конца:
2NaOH + H2SO4 -> Na2SO4 + Н2О
если кислота или основание являются слабыми электролитами, равно-
равновесие смещено в сторону образования более слабого электролита (Н2О), но
реакция до конца не протекает, и в растворе образуется избыток ионов ОН"
или Н+. Так, при нейтрализации слабой уксусной кислоты сильным основа-
основанием
КОН + СНзСООН ^ СН3СООК + Н2О
в равновесной системе преобладают гидроксид-ионы [ОН"] > [Н+] и рН > 7.
Очевидно, что реакции нейтрализации, в которых участвуют слабые кисло-
кислоты или основания, обратимы, что является причиной гидролиза их солей.
В водных растворах многие катионы, имеющие вакантные орбитали,
например катионы d- и р-металлов, существуют в виде комплексов, в част-
частности аквакомплексов, так как молекулы воды, имеющие неподеленные
электронные пары, являются донорами электронов
CuSO4 + 4Н2О = [Cu(H2OL]2+ + SO^
Мерой прочности комплексного иона служит константа нестойко-
нестойкости Кн, соответствующая процессу полной диссоциации комплексного иона
на комплексообразователь и лиганды:
[Cu(H2OL]2+ ^> Cu2+ + 4H2O KH =
[Cu(H2O)f]
[Fe(CN)«f^Fe* + 6CN- ..-ЙЙ
Чем меньше значение константы нестойкости Кя, тем более устойчив ком-
комплекс. Поэтому реакции с участием комплексных ионов часто сопровожда-
сопровождаются образованием новых более устойчивых комплексных соединений:
[AgCl2]-+ 2Г -> [Agl2]" + 2СГ
К гд .,, ,_ =1,76 • 1(Г5; К Г4 = 1,4 • 104.
H[AgCl2] ' H[Ag2]
14.5. Гидролиз солей 447
Процесс образования прочного комплекса может привести к тому, что оса-
осадок малорастворимого электролита при добавлении лиганда полностью рас-
растворится:
Zn(OHJl + 2OH" -> [Zn(OHL]2"
Однако, если в раствор комплексного соединения ввести ионы, образующие
с продуктами его диссоциации соединения, очень плохо растворяющиеся в
воде, то комплекс разрушится:
[Ag(NH3J]+ + S2--» Ag2Sl + 2NH3
Рассмотрим подробнее процессы, протекающие в водных растворах
солей и малорастворимых электролитов.
14.5. Гидролиз солей
Большинство неорганических солей в водных растворах представляют
собой сильные электролиты, т.е. полностью диссоциированы. Процесс рас-
растворения и сольватации многих солей в воде сопровождается образованием
нейтральных растворов. Однако некоторые соли взаимодействуют с водой
так, что образуются кислотные или щелочные растворы. Это обусловлено
поляризующим взаимодействием ионов соли с их гидратной оболочкой.
Катионы связаны с молекулами воды донорно-акцепторной связью:
донор — атомы кислорода воды, имеющие две свободные электронные па-
пары, акцептор — катионы, обладающие свободными орбиталями. Чем боль-
больше заряд катиона и меньше его радиус, тем сильнее проявляется поляри-
поляризующее действие катиона на Н2О. Анионы связаны с молекулами воды во-
водородной связью. Донорная активность аниона будет тем значительнее, чем
больше заряд и меньше размер аниона. Равновесие электролитической дис-
диссоциации воды
Н2О <=> Н+ + ОН"
смещается в зависимости от силы поляризующего влияния катиона и аниона
на молекулы воды. Чем значительнее это взаимодействие, тем интенсивнее
протекает гидролиз соли — обменное взаимодействие ионов соли с молеку-
молекулами воды, приводящее к увеличению кислотности или щелочности раство-
раствора и образованию слабодиссоциированных соединений:
KZ]+ + Н2О ^± K(OH)(Zl+H + Н+ (рН < 7),
А72" + Н2О ^± HA(Z2")+1 + ОН" (рН > 7).
448 14. Растворы электролитов
Различают четыре случая взаимодействия соли и воды.
1. Если поляризующее воздействие и катиона, и аниона на молекулы
воды невелико, то равновесие диссоциации воды в присутствии ионов соли
почти не смещается (рН = 7), т.е. гидролиз практически не происходит. Это
относится к солям, образованным сильным основанием и сильной кислотой,
например КС1, NaNO3, CaSO4.
2. Если поляризующая активность аниона превосходит поляризующее
действие катиона, имеет место гидролиз по аниону. Соли в этом случае образо-
образованы сильным основанием и слабой кислотой: KCN, ЫазРО^ СН3СООК и др.
Например, гидролиз соли К2СО3:
I ступень
К2СО3 + Н2О <=> КНСОз + КОН
или сокращенное ионно-молекулярное уравнение
СО^ + Н2О <=± НСОз + ОН"
II ступень
КНСО3 + Н2О <=± Н2СО3 + КОН
или сокращенное ионно-молекулярное уравнение
НСОз + Н2О <=* Н2СО3 + ОН"
Ясно, что гидролиз по аниону приводит к появлению избытка ионов ОН",
т.е. раствор становится щелочным (рН > 7).
3. Если анионы — слабые доноры электронов, а катионы обладают
значительным поляризующим действием, происходит гидролиз по катиону.
В этом случае соль образована слабым основанием и сильной кислотой
(C11SO4, ZnCl2, NH4NO3 и др.). Например, гидролиз соли ZnCl2:
I ступень
ZnCl2 + Н2О <=> Zn(OH)Cl + HC1
или сокращенное ионно-молекулярное уравнение
II ступень
Zn2++H2O <± Zn(OH)++H+
Zn(OH)Cl +H2O <zi Zn(OHJ>l + НС1
или сокращенное ионно-молекулярное уравнение
Zn(OH)++H2O <i± Zn(OHJl + H+
14.5. Гидролиз солей 449
Избыток ионов Н+ в этом случае обусловливает кислотную среду рас-
раствора (рН < 7).
4. Если катионы и анионы обладают значительным поляризующим
действием, т.е. соли образованы слабым основанием и слабой кислотой, то
происходит гидролиз по катиону и аниону. Примером служит гидролиз соли
CH3COONH4:
CH3COONH4 + Н2О <=± NH4OH + CH3COOH
При этом протекают параллельно два процесса:
nh; +н2о <=> nh4oh+h+
СН3СОСГ + Н2О <± СНзСООН + ОН"
Растворы солей этого типа могут иметь слабокислотную или слабощелоч-
слабощелочную реакцию (рН » 7) в зависимости от соотношения констант диссоциации
образующихся при гидролизе кислоты или основания. Если эти величины
близки по силе, то гидролиз идет практически до конца.
Количественными характеристиками гидролиза являются степень
гидролиза и константа гидролиза. Степень гидролиза аГ равна доле гидро-
лизованных молекул и может быть выражена отношением концентрации
гидролизованной соли Сг к концентрации растворенной соли С:
Степень гидролиза определяется тремя факторами: ее значение тем больше,
чем ниже концентрация раствора, больше константа гидролиза и выше тем-
температура (гидролиз — эндотермический процесс). Константа гидролиза КТ
соответствует величине константы равновесия процесса гидролиза и в отли-
отличие от степени гидролиза аг, не зависит от концентрации раствора, поэтому
является более удобной характеристикой процесса.
Рассмотрим связь между константой гидролиза, степенью гидролиза и
рН растворов гидролизующихся солей. При гидролизе по аниону, например
СНзСОО" + Н2О <± СНзСООН + ОН"
выражение константы равновесия имеет вид
_ [СН3СООН][ОН~]
С [СН3СОО"][Н2О]"
Величину [НгО], являющуюся постоянной, можно перенести в левую часть
равенства, что даст новую постоянную величину — константу гидролиза:
15 - 9795
450 14. Растворы электролитов
[СН3СОСГ]
Но [ОН"] = —^-, тогда
[СН3СОО~][Н+] '
ш, [Н+][СН3С(ХГ]
Учитьшая, что константа диссоциации уксусной кислоты ла = ,
3 [СН3СООН]
получают
^ A4.17)
Поскольку [СНзСООН] = [ОН"] = arC, a [СН3СОО"] = A - аг)С, то констан-
константа гидролиза и степень гидролиза связаны между собой отношением, анало-
аналогичным закону разбавления Оствальда
^ A4.18)
1-аг
а при малых значениях аг:
АГГ« а]С, A4.19)
следовательно,
ar=J^k A4.20)
Концентрация [ОН"] = агС = у/КгС, поэтому при гидролизе по аниону
pH=14 + 0,51g(/«:rC). A4.21)
При гидролизе по катиону, например
NH4 + Н2О +± NH4OH + Н+
рассуждая аналогично, получают
_ [NH4OH][H+]
[NHJ]
14.5. Гидролиз солей 451
Кт=-^-. A4.22)
къ
Концентрация [Н+] = о^С = -jKrC, следовательно, при гидролизе по катиону
рН = -0,51ё(ВД. A4.23)
Если гидролиз протекает и по катиону, и по аниону, например
NHJ + СНзСОО" + Н2О ^± NH4OH + СН3СООН
константа гидролиза
_ [nh4oh][ch3cooh] [nh4oh] [ch3cooh]
[nh;][chcoo-] ~ H2°
Отсюда
K<=JJ-- A4-24>
Поскольку [NH4OH] = [СН3СООН] = arC, a [NHj] = [СН3СОО] = A - аг)С,
то степень гидролиза в этом случае не зависит от концентрации соли:
A4.25)
1-сц.
Не зависит от концентрации соли в случае гидролиза по катиону и аниону
также и рН раствора, так как концентрации [Н+] и [ОН"] определяются кон-
константами диссоциации слабых основания и кислоты:
\ A4.26)
[ОН"]
Поэтому в данном случае
[н+] &25L, A4.27)
V къ
pH = -0,51g^^. A4.28)
къ
Если гидролиз средней соли протекает в две ступени, то константу
гидролиза по I ступени рассчитывают по уравнению
15*
452 14. Растворы электролитов
или Кт=-^-, A4.29)
а по II ступени по уравнению
is ^Н2О „ ^Н2О /^олч
Кт= —*- или A^id= —*-, A4.30)
где АГаA) и ^а(И)9 ^b(i) иЛГЬ(и)— константы диссоциации по I и II ступеням соот-
соответственно для кислоты (в случае гидролиза по аниону) или для основания
(в случае гидролиза по катиону). Поскольку константы диссоциации кислот
и оснований по I ступени, как правило, значительно больше констант диссо-
диссоциации по II ступени, то К^щ « КТ^у Гидролиз солей слабых многоосновных
кислот или слабых многокислотных оснований протекает преимущественно
по I ступени, и при приближенных расчетах гидролизом по II ступени мож-
можно пренебречь.
Константы гидролиза кислых или основных солей рассчитывают как
константы гидролиза средних солей по II ступени.
Термодинамические характеристики процесса гидролиза — изменения
энергии Гиббса, энтальпии и энтропии — рассчитывают по формулам тер-
термодинамических характеристик процессов равновесия (см. гл. И). В случае
гидролиза:
ArG°=-RTlnKr, A4.31)
АгН° =*^Ы^<™9 A4.32)
Т Т V
Отметим, что гидролизу подвергаются не только соли, но и солеоб-
разные бинарные соединения, например галогенангидриды, реакции взаи-
взаимодействия с водой которых во многих случаях протекают необратимо:
РС13 + ЗН2О -* Н3РОз + ЗНС1
BF3 + ЗН2О -> Н3ВОз + 3HF
Гидролиз органических соединений широко применяют для получения
спиртов, альдегидов, карбоновых кислот из их производных или галогенза-
мещенных углеводородов, например
С2Н5С1 + Н2О ?=> С2Н5ОН + СГ + Н+
14.6. Произведение растворимости 453
причем подбор соответствующих условий и катализаторов позволяет прово-
проводить реакции избирательно и практически до конца. Гидролиз жиров ис-
используют для получения мыла и глицерина, ферментативный гидролиз — в
пищевой, текстильной, фармацевтической промышленности. Гидролизные
производства служат для получения пищевых, кормовых и технических
продуктов из отходов переработки сельскохозяйственных культур и древе-
древесины.
14.6. Произведение растворимости
Малорастворимые электролиты в очень разбавленных растворах дис-
диссоциируют практически полностью, т.е. их можно рассматривать как силь-
сильные электролиты. При постоянной температуре в насыщенном растворе ма-
малорастворимого электролита устанавливается термодинамическое равнове-
равновесие между твердой фазой и ионами в растворе
КтАл(т) +± КтАл(р)= (p)
или с учетом полной диссоциации электролита:
Термодинамическая константа равновесия
^efb^5L A4.34)
В случае очень низкой растворимости электролита образуется сильно
разбавленный раствор, который можно считать идеальным, и вместо термо-
термодинамической использовать концентрационную константу
КС= U^mf!X. A4.35)
Концентрация твердой фазы [KmAw (т)] = const, тогда
Кс[КтАп(т)] = [KZ'T[AZ2T = ПР = const. A4.36)
При постоянной температуре в насыщенном растворе произведение
концентраций (или активностей) ионов, на которые диссоциирует электро-
электролит, с учетом степеней, соответствующих стехиометрическим коэффициен-
коэффициентам процесса диссоциации, есть величина постоянная, называемая произве-
произведением растворимости ПР малорастворимого электролита.
454 14. Растворы электролитов
Чем меньше значение ПР, тем хуже растворяется соединение. При
внесении в раствор избытка ионов KZl+ или AZl~ выпадет дополнительное
количество осадка, изменятся концентрации ионов в растворе (сдвиг равно-
равновесия диссоциации влево по принципу Ле Шателье—Брауна), но ПР оста-
останется постоянной при неизменной температуре.
Условием образования осадка служит превышение произведения кон-
концентраций ионов над произведением растворимости: C"Zl+C™Z2_ > ПР. Под
концентрацией ионов в данном случае понимают не равновесную концен-
концентрацию ионов, установившуюся после образования осадка, а концентрацию
ионов в первоначальный момент образования системы. Например, если к 1 л
0,1М раствора СаС12 добавить 1 л 0,1 М раствора H2SO4, то в соответствии с
уравнениями диссоциации сильных электролитов
СаС12 = Са2+ + 2СГ
H2SO4 = 2H++ SO^~
в первоначальный момент образования системы концентрации ионов соста-
составят: Сп 2+ = 0,05 моль/л, Сс_ 2- = 0,05 моль/л (изменением объема системы
при приближенных расчетах можно пренебречь). В результате взаимодействия
СаС12 + H2SO4 = CaSO4i + 2HC1
образуется малорастворимый электролит CaSO4, произведение растворимо-
растворимости которого ПР = [Ca2+][SO4~] = 3,7-10~5. В рассматриваемом случае
Сп 2+Ссгл2- = 2,5 • 10~3 > ПР, поэтому осадок CaSO4 выпадет из раствора.
Если при образовании раствора C"zl+C™z2- < ПР электролит будет
растворяться до тех пор, пока произведение концентраций ионов не сравня-
сравняется со значением ПР.
Произведение растворимости можно рассчитать, если известна рас-
растворимость малорастворимого электролита. Равновесные молярные концен-
концентрации ионов насыщенного раствора связаны с растворимостью соотноше-
соотношениями
[KZl+] =mSu [AZ2~] =nS9
где S — концентрация насыщенного раствора (растворимость), моль/л. Сле-
Следовательно,
ПР = {mS)m{nS)\ A4.37)
Например, для малорастворимого электролита
14.7. Электропроводность растворов электролитов 455
CaSO4(T) <=> CaSO4(p)=Ca2+(p)H- S02"(p)
произведение растворимости связано с растворимостью соотношением
а для РЫ2 (т) <=> РЫ2 (р) = РЬ2+(Р) + 2Г(р)
ПР = [РЬ2+][Г]2 = 5B5J = 4S3.
Произведение растворимости зависит от температуры так же, как и
соответствующая константа равновесия. Зная хотя бы два значения произ-
произведения растворимости электролита при двух значениях температуры, мож-
можно рассчитать термодинамические характеристики процессов растворения
(см. гл. 13). Составляя систему уравнений:
A5d = -RTX In ПР! = ASH° - TiAsS°9
ASG2 = -ЛГ21п ПР2 = ASH° - T2ASS\ A4.38)
можно найти энтальпию ASH° и энтропию ASS° растворения, если считать,
что эти величины не зависят от температуры в рассматриваемом интервале
температур.
14.7. Электропроводность растворов электролитов
Характерная черта растворов электролитов — их способность про-
проводить электрический ток. Сольватированные ионы в растворе находятся
в беспорядочном тепловом движении. При наложении электрического
поля возникает упорядоченное движение ионов к противоположно заря-
заряженным электродам — электрическая проводимость второго рода. Ско-
Скорость движения ионов в электрическом поле определяется силой, дейст-
действующей на ион, которая равна произведению заряда иона и градиента
потенциала, и сопротивлением среды, зависящим от температуры, при-
природы иона и растворителя
eZJJ
Vi= —,
Rl
где vt — скорость /-го иона; е — элементарный электрический заряд; Z,- —
зарядовое число /-го иона; U — разность потенциалов между электродами;
R — сопротивление среды; / — расстояние между электродами.
456 14. Растворы электролитов
Для сравнения скоростей движения различных ионов устанавливают
градиент потенциала поля 1 В/м. В этих условиях скорость движения ионов
называют электрической подвижностью ионов и,:
eZ,
ы,-Т.
Движение сольватированного иона, мигрирующего в электрическом
поле вместе с сольватной оболочкой, можно приближенно описать форму-
формулой Сшокса для движения шарика в вязкой среде
где F — сила, действующая на ион; г| — коэффициент вязкости среды; г,- —
эффективный радиус частицы. В соответствии с A4.39) ионы с высокой сте-
степенью сольватации обладают меньшей электрической подвижностью, чем
слабо сольватированные ионы.
Мерой способности вещества проводить электрический ток является
электрическая проводимость (электропроводность) L — величина, обрат-
обратная электрическому сопротивлению R. Так как
ТО
Т IS S
р/ /
где р — удельное сопротивление, Ом • м; S — поперечное сечение, м2; / —
длина проводника, м; х — удельная электрическая проводимость, Ом'1 • м
или См/м.
Удельная электрическая проводимость х раствора электролита —
это электрическая проводимость объема раствора, заключенного между
двумя параллельными электродами площадью 1 м2, расположенными на
расстоянии 1 м:
*'Ts* A440)
/
где постоянная электролитической ячейки, которую определяют не-
S
посредственным измерением или в калибровочном опыте. Согласно закону
Ома,
14.7. Электропроводность растворов электролитов 457
где U— разность потенциалов; /— сила тока.
Отсюда
%=-— = А A4.41)
SU X V '
где у — плотность тока, А/м2; JF — градиент потенциала, В/м. Таким обра-
образом, удельная электропроводность х характеризует плотность тока при гра-
градиенте потенциала 1 В/м.
Величина х определяется количеством ионов, переносящих электри-
электричество, и скоростью их миграции. Для раствора бинарного электролита, за-
заряд катиона которого равен заряду аниона (Z+ =Z-=Z) при концентрации
С (моль/м3) и степени диссоциации а, имеем
х = aZFC(u+ + w_), A4.42)
где F — постоянная Фарадея, равная произведению заряда электрона и
числа Авогадро:
F=eNA = 96485 Кл/моль.
Для исследования поведения ионов в растворе удобно использовать
молярную электрическую проводимость X раствора. Она характеризует про-
проводящую способность всех ионов, образующихся при диссоциации 1 моль
электролита в растворе данной концентрации, помещенном между двумя
параллельными электродами одинаковой формы и площади, расстояние ме-
между которыми равно 1 м. Так как между электродами находится раствор
объемом V (м3), то соотношение между молярной и удельной электропро-
водностями имеет вид
А. = хГ=—, A4.43)
где X — молярная электрическая проводимость раствора, Ом • м2 • моль
или См • м2/моль; С — концентрация, моль/м3. Если концентрация раствора
выражена в моль/л, то соотношение A4.43) преобразуется:
Из уравнений A4.42) и A4.43) следует, что
X = aZF(u+ + м_). A4.45)
458
14. Растворы электролитов
При отсутствии взаимодействия между ио-
ионами удельная электропроводность должна линей-
линейно возрастать с ростом концентрации электролита,
а молярная электропроводность не должна зави-
зависеть от концентрации. Такая ситуация наблюдается
только в предельно разбавленных растворах, для
которых силами кулоновского взаимодействия
между ионами можно пренебречь. Опыт показыва-
показывает, что, начиная с некоторой концентрации, зави-
зависящей от природы электролита, линейная зависи-
зависимость х = /(С) начинает заметно нарушаться.
Зависимости удельной электропроводности х
от концентрации раствора С для сильных и слабых
электролитов характеризуются наличием макси-
максимума (рис. 14.1). Такой вид кривых можно объяс-
о с
Рис. 14.1. Зависимость
удельной электрической
проводимости растворов
сильных 1 и слабых 2
электролитов от концен-
концентрации
нить следующим образом. Для сильных электролитов с ростом концентра-
концентрации увеличивается число ионов в единице объема, и сначала наблюдается
увеличение электропроводности х. Однако при дальнейшем возрастании
концентрации увеличение вязкости раствора и взаимодействия между иона-
ионами приводят к снижению скорости движения ионов и соответственно элек-
электрической проводимости. Удельная электропроводность слабых электролитов
значительно ниже удельной электрической проводимости сильных электро-
электролитов той же концентрации, что обусловлено небольшой степенью диссо-
диссоциации слабого электролита. При повышении его концентрации одновре-
одновременно увеличивается число молекул и снижается
степень диссоциации.
Общий характер изменения молярной элек-
электропроводности X с концентрацией для сильных
и слабых электролитов выражен кривыми, пред-
представленными на рис. 14.2. Снижение молярной
электрической проводимости при переходе от
бесконечно разбавленного раствора к растворам
конечных концентраций, согласно электростати-
электростатической теории сильных электролитов, связано с
уменьшением скоростей движения ионов, окру-
окруженных ионной атмосферой. Под действием
внешнего электрического поля при измерении
электропроводности раствора ион частично вы-
выходит из окружающего его ионного облака. Это
о с
Рис. 14.2. Зависимость
молярной электрической
проводимости растворов
сильных 1 и слабых 2
облако снова формируется в новом положении электролитов от концен-
иона (явление релаксации), но не мгновенно, а трации
14.7. Электропроводность растворов электролитов 459
через 10~9...10~7 с. Поэтому при движении иона в электрическом поле число
ионов противоположного заряда позади иона больше, чем перед ним. Такая
асимметрия ионной атмосферы вызывает замедление движения иона, повы-
повышает электрическое сопротивление раствора и понижает его электропровод-
электропроводность {релаксационное торможение). Асимметрия ионной атмосферы обу-
обусловлена также тем, что ионы атмосферы под действием электрического
поля стремятся переместиться к противоположно заряженному электроду,
что также снижает электропроводность раствора (электрофоретическое
торможение). Следует учитывать и то, что вследствие образования вокруг
иона любого заряда сольватной оболочки перемещение иона в электриче-
электрическом поле замедляется.
С уменьшением концентрации молярная электрическая проводимость
как сильных, так и слабых электролитов возрастает и стремится к предель-
предельному значению А,00. Эта величина отвечает электрической проводимости ги-
гипотетического бесконечно разбавленного раствора, характеризующегося
полной диссоциацией электролита и отсутствием сил электростатического
взаимодействия между ионами. В соответствии с A4.45) молярная электри-
электрическая проводимость бесконечно разбавленного раствора
Г° = ZF(u+ + и") = ZFu+ + ZFu™ = А," + X™. A4.46)
Произведения ZFu™ = X™ и ZFu™ = X™ называют предельной моляр-
молярной электрической проводимостью иона (катиона и аниона соответственно).
Таким образом, молярная электрическая проводимость бесконечно разбав-
разбавленного раствора равна сумме двух независимых слагаемых. Соотношение
A4.46), установленное Ф. Кольраушем A879), называют законом независи-
независимого движения ионов, или законом Кольрауша. Предельная молярная элек-
электрическая проводимость — специфическая величина для данного вида ио-
ионов, зависящая только от природы растворителя и температуры.
В соответствии с A4.45) и A4.46) молярная электропроводность X и
предельная молярная электропроводность V0 связаны соотношением
A4.47)
х = — - коэффициент электрической проводимости. Для силь-
сильней!0
ных электролитов, диссоциирующих полностью, а = 1 и
¦~ =Л. A4-48)
460 14. Растворы электролитов
т.е. коэффициент электропроводности f% является мерой электростатических
взаимодействий ионов. В растворах слабых электролитов электрические
подвижности ионов практически не зависят от концентрации, так как меж-
межионные взаимодействия значительно слабее, чем в растворах сильных элек-
электролитов. Поэтому для них fx = 1 и
— =а. A4.49)
X00
Это соотношение, называемое уравнением Аррениуса, используют для опре-
определения степени диссоциации а слабого электролита по измерениям элек-
электропроводности.
Изучение электрической проводимости позволяет получить сущест-
существенную информацию о свойствах и особенностях растворов электролитов,
играет важную роль в установлениия механизма электрохимических реак-
реакций, действия химических источников тока, процессов электролиза.
Свойства природной воды
Вода относится к наиболее важным химическим соединениям на
нашей планете. Ее роль обусловлена рядом уникальных свойств,
благодаря которым вода является: обязательным компонентом
растений и всех живых организмов; главным компонентом среды
обитания, выполняющим важную роль в регулировании клима-
климатических условий, в частности, стабилизации температуры на
поверхности Земли; активным участником процессов жизнедея-
жизнедеятельности и неотъемлемым компонентом большинства техноло-
технологических процессов промышленного и сельскохозяйственного
производства.
По образному выражению Нобелевского лауреата А. Сент-Дьердьи,
вода — «матрица жизни».
15.1. Состав природной воды
Общие запасы воды как самого распространенного на Земле
вещества оцениваются приблизительно в 16,4 • 109 км3 и распреде-
распределяются следующим образом:
Объект Атмосфера Гидросфера Литосфера Мантия
Объем, км3 ... -1,5 • 104 -1,5 • 109 -1,2 • 109 -13,7 • 109
(-9,1%) (-7,3%) (-83,5%)
Вода покрывает 80 % поверхности Земли и содержится во
многих ее объектах: горных породах и минералах, почве, растени-
растениях, продуктах питания, живых организмах и др. Например, содер-
содержание воды в организме человека достигает 70 % (масс).
Принято воду гидросферы называть природной, 96 % которой
сосредоточено в Мировом океане, остальная часть распределена
среди рек, озер, ледников, подземных и почвенных вод. Лишь 3 %
природной воды являются пресными, среди которых только 20 %
доступны для использования. Пресную природную воду применя-
применяют в сельском хозяйстве (-82 %), быту (-10 %) и промышленности
(-8 %). Воду, расходуемую промышленными предприятиями, на-
называют технической и используют в качестве охлаждающего аген-
агента, транспортирующей среды для сыпучих материалов, энергоноси-
энергоносителя на атомных и гидроэлектростанциях, растворителя.
Природная вода находится в непрерывном взаимодействии с
окружающей средой. Контактируя с воздухом и почвой, раститель-
растительностью и горными породами, она растворяет различные вещества
462 15. Свойства природной воды
органического и неорганического происхождения. Поэтому природная вода
никогда не бывает чистой и представляет собой не индивидуальное соеди-
соединение, а динамичную, многокомпонентную систему, содержащую почти все
элементы Периодической системы. Значительная их часть присутствует в
ней в столь малых количествах (микрокомпоненты — Li, Be, В, Rb, Zn, Cu,
Bi, W, V, Br, I и др.), что не оказывает никакого влияния на ее органолепти-
ческие и физические свойства, однако крайне важна для жизнедеятельности
живых организмов. Макрокомпонентами природной воды обычно являются
Са, Mg, Na, К, Fe (катионогенные воды), Si, С, S, C1 (анионогенные воды).
Среди веществ, содержащихся в природной воде, имеются коллоид-
коллоидные и крупнодисперсные частицы, органические вещества и различные
микроорганизмы, минеральные соли и газы. Крупнодисперсные частицы в
воде находятся во взвешенном состоянии. Они неоднородны по форме и
различны по происхождению. К ним относятся ил, песок, остатки растений
и живых микроорганизмов и др. Среди органических веществ, растворенных
в природной воде, следует отметить гуминовые (HO-R'-COOH) и карбоно-
вые (R-COOH) кислоты, а также различные углеводороды. Наиболее чистой
является дождевая (снеговая) вода вдали от больших городов, но и в ней
содержатся растворенные вещества — газы воздуха, соли и др.
Соли, содержащиеся в природной воде, поступают в нее различными
путями в ходе ее круговорота в природе, и их подразделяют следующим об-
образом:
соли, непосредственно смытые водой или появившиеся в ней вследст-
вследствие сброса промышленных стоков: NaCl, Na2SC>4, СаСЬ, NH4NO3 и др.;
соли, поступившие в воду в результате ее контакта с горными поро-
породами (известняками, доломитами, магнезитами и др.) — гидрокарбонаты,
хлориды, сульфаты кальция и магния, например:
СаСО3 -МёСО3(т) +2СО2(р) +2Н2О(ж) ^Ca(HCO3J(p) + Mg(HCO3J(p)
доломит
Все растворенные в воде соли полностью диссоциированы, частично
гидролизованы и находятся в ней в виде гидрагированных ионов: катионов
и анионов. Ниже приведены часто встречающиеся в природной воде ионы в
порядке убывания частоты их присутствия в ней:
Катионы Са2+ Mg2+ Na+ K+ NH4+ Fe2+
Анионы НСО~ СГ SO2" NO" F" PO4"
Между частотой присутствия ионов и их концентрацией в воде не на-
наблюдается четкой корреляции (табл. 15.1).
15.1. Состав природной воды
463
Таблица 15.1
Количественный состав воды некоторых природных водоисточников
Водоисточник
Океан
Черное (море)
Волга (река)
Москва (река)
Рейн (река)
Байкал (озеро)
Мичиган (озеро)
Са2+
418,0
250,0
48,9
41,3
50,3
15,2
26,2
Содержание
Mg2+
1329,0
650,0
10,1
8,4
П,7
4,2
8,3
Na+ + К+
11428,0
5510,0
11,9
2,3
5,2
6,1
4,7
ионов, мг/л
НСОз
146,0
80,0
63,7
79,4
181,4
59,2
58,3
SO2"
2768,0
1310,0
61,9
7,7
24,6
4,9
7,1
СГ
19833,0
9630,0
14,4
4,4
8,0
1,8*
2,7*
*сг + вг
При загрязнении водоисточников бытовыми стоками в воде могут об-
обнаруживаться аммиак, соли аммония, нитраты, нитриты, фосфаты, сульфи-
сульфиды, сероводород, производственными стоками — ионы Cu2+, Zn2+, Pb2+, Hg2+,
As3+, радиоактивные вещества, фенол, нефтепродукты и другие примеси.
Большинство из них являются вредными веществами. По мере их накопле-
накопления в водоемах вода становится непригодной, поэтому требуется более глу-
глубокая и тщательная ее очистка перед использованием в питьевых и про-
промышленных целях. В настоящее время охрана водных ресурсов и очистка
сточных вод, ограничение потребления воды и борьба с загрязнением окру-
окружающей среды, а также воспроизводство пресной воды — наиболее акту-
актуальные экологические проблемы общества.
Основными газами, содержащимися в природной воде, являются: СО2,
N2, О2, СО. В подземных водах часто обнаруживаются еще СН4 и H2S, соз-
создающие восстановительную среду. Хорошая растворимость кислорода в воде
приводит к тому, что относительное его содержание в ней выше (~35 % (об.)),
чем в воздухе (~21 % (об.)), что обеспечивает существование жизни в водоемах.
Часть углекислого газа, содержащегося в воде, вступает в химическое
взаимодействие с ее молекулами:
СО2 + Н2О
Н2СО3
Положение равновесия этой реакции сильно смещено влево (в сторону ис-
исходных веществ) и при рН = 4,4 99 % всего растворенного газа находится в
форме СО2 и около 1 % — в форме Н2СО3. Обе эти формы называют сво-
свободной углекислотой. Кроме свободной углекислоты в воде присутствует и
связанная (гидрокарбонатная (НСО3) и карбонатная (СО2) формы) в
464 15. Свойства природной воды
виде соответствующих солей. Все эти формы могут переходить одна в дру-
другую в зависимости от рН воды:
(С02 + Н2СО3 + HCOJ)
рН
примерно до 4,4 приблизительно при 8,4 — примерно после 12,4
только в форме сво 99,7 % в гидрокарбонатной только в карбонатной
бедной углекислоты форме (исо;) форме (С02-}
(СО2, Н2СО3)
При подкислении карбонаты превращаются в гидрокарбонаты:
2СаСО3 + 2НС1 ^± Са(НСО3J + СаС12
которые при дальнейшем подкислении переходят в свободную углекислоту:
Са(НСО3J + 2НС1 <± СаС12 + 2Н2СО3
нсо;+н+<->н2со3
При подщелачивании воды происходят обратные реакции:
Н2СО3 + NaOH +± NaHCO3 + Н2О
н2со3 + он" <=± нсо;+н2о
NaHCO3 + NaOH <=± Na2CO3 + Н2О
НСО3 + ОН" f± СО^" + Н2О
Протекание таких реакций отражает смещение положения углекис-
лотного равновесия:
со2 + н2о +± н2со3 <=* нсо; + н+ <± 2Н++со^"
Подщелачивание смещает его вправо, подкисление — влево. В зависимости от
происхождения природная вода может иметь кислотный или щелочной харак-
характер. Все определяется ее составом и протекающими в ней процессами диссо-
диссоциации и гидролиза солей. Состав воды может меняться при хранении. Особенно
это характерно для подземных вод. Такие воды называют нестабильными.
Обычно химический состав воды характеризуется следующими показа-
показателями: ионным составом, общей щелочностью или общей кислотностью, ак-
активной концентрацией ионов водорода, общим солесодержанием, массой сухо-
сухого остатка, содержанием растворенного кислорода, свободной углекислоты,
15.2. Жесткость воды: виды, единицы измерения 465
активного хлора (при хлорировании), сероводорода, окисляемостью, жестко-
жесткостью. Отметим, что жесткость — один из главных технологических показате-
показателей, принятых для характеристики состава и качества природной воды.
15.2. Жесткость воды: виды, единицы измерения
Жесткостью называют совокупность свойств воды с растворенными
в ней солями (карбонатами, гидрокарбонатами, сульфатами, хлоридами,
нитратами кальция, магния, железа и др.). Согласно ГОСТ 6055-86, разли-
различают следующие виды жесткости:
а) карбонатную жесткость Нк — совокупность свойств воды, обуслов-
обусловленных присутствием в ней главным образом гидрокарбонатов кальция, маг-
магния, железа и незначительной части их карбонатов. Она отвечает той части со-
содержащихся в воде катионов Са2+, Mg2+, Fe2+, которая эквивалентна содержа-
содержащимся в ней анионам НСОз и СО2". Часть карбонатной жесткости, устра-
устраняемой кипячением, назьюают временной НВ9 или устранимой жесткостью.
Последняя меньше карбонатной жесткости на величину так называемой оста-
остаточной карбонатной жесткости Нок, обусловленной наличием карбонатов,
которые остаются в растворенном состоянии в воде, хотя и в малой концентра-
концентрации. Все соли, оставшиеся в растворенном состоянии в воде после кипячения,
определяют ее постоянную НП, или неустранимую жесткость;
б) некарбонатную жесткость Ннк — совокупность свойств воды,
обусловленных присутствием в ней сульфатов, хлоридов, силикатов и нит-
нитратов кальция, магния и железа. Она отвечает той части содержащихся в ней
катионов Са2+, Mg2+, Fe2+, которая эквивалентна содержащимся в ней анио-
анионам СГ, SO4", NO3 и др. Некарбонатная жесткость воды меньше постоян-
постоянной на величину остаточной карбонатной жесткости;
в) общую жесткость Яо, включающую карбонатную и некарбонат-
некарбонатную жесткость: #0=#к+#нк. Она равна суммарной (общей, аналитиче-
аналитической) концентрации содержащихся в воде катионов Са2+, Mg2+, Fe2+. Общую
жесткость также можно рассматривать как сумму постоянной и временной
жесткости. Ниже приведено соотношение между различными видами жест-
жесткости воды поверхностных водоисточников:
я0
Як = @,7-0,8)Яо
^ Дэ.к
Ян
*яп
Ян
466
75. Свойства природной воды
Количественно жесткость воды в России выражают единицами жестко-
жесткости. Согласно ГОСТ 6055-86, за единицу жесткости принимают жесткость
воды, в 1 л которой содержится 1 ммоль экв ионов Са2+ или Mg2+. Числовое
значение жесткости в ммоль экв/л совпадает со значением в моль экв/м3 и зна-
значением, выраженным в 10~3 н. Одна единица жесткости A ммоль экв/л) соот-
соответствует содержанию ионов кальция, равного 20,04 мг/л B0,04 г/м3) или ионов
магния, равного 12,15 мг/л A2,15 г/м3). В других странах для выражения жест-
жесткости воды используются градусы жесткости (табл. 15.2).
Таблица 15.2
Градусы жесткости и соответствующее им содержание веществ
Градус жесткости
Английский
Американский
Немецкий
Французский
Содержание вещества
г/л
0,014 (СаСОз)
0,017 (СаСОз)
0,010 (СаО)
0,010 (СаСОз)
ммоль экв/л (моль экв/м3)
0,2848
0,0200
0,3566
0,1998
Взаимосвязь между различными градусами жесткости показана в
табл. 15.3.
Таблица 15.3
Соотношение между градусами жесткости
Наименование единиц
1 английский градус
1 американский градус
1 немецкий градус
1 французский градус
Градус жесткости
английский
1
0,070
1,253
0,702
американский
14,253
1
17,847
10,000
немецкий
0,799
0,056
1
0,560
французский
1,426
0,100
1,785
1
В зависимости от общей жесткости природные воды подразделяют на
группы
#0, ммоль экв/л < 1,5 1,5-3,0 3,0-6,0 6,0-9,0 > 9,0
Группа очень средней очень
мягкая мягкая жесткости жесткая жесткая
Общая жесткость воды рек и озер в тайге и тундре составляет
ОД...0,2 ммоль экв/л, морей, океанов, подземных водоемов — 80... 100 ммоль
экв/л. В поверхностных водоисточниках преобладает, как правило, карбо-
карбонатная жесткость, составляющая 70...80 % от общей. Магниевая жесткость
75.3. Требования, предъявляемые к питьевой и технической воде
467
обычно не превышает 30 % от общей. Наибольшей жесткости вода достига-
достигает в конце зимы, наименьшей — в период паводка. Жесткость подземных
вод более постоянна и менее подвержена изменению в течение года.
15.3. Требования, предъявляемые к питьевой
и технической воде
Природная вода не всегда пригодна для использования ее в питьевых
и промышленных целях из-за присутствия вредных для объекта водоснаб-
водоснабжения веществ, поэтому различные примеси в ней нормируются. В соответ-
соответствии с ГОСТ 2874-82 к питьевой и технической воде предъявляются опре-
определенные требования (табл. 15.4).
Таблица 15 А
Допустимое содержание различных ионов в питьевой воде
Ионы, не влияющие
на органолептические
свойства воды
А13+
Ве2+
Мо2+
As3+
NO3~
Pb2+
Si*
Допустимое
содержание,
мг/л
0,5
2 10
0,25
0,05
45,0
0,03
7,0
Ионы, влияющие
на органолептические
свойства воды
Fe2+,Fe3+
Мп2+
Си2+
?of
сг
Zn2+
Mg2+
Допустимое
содержание,
мг/л
0,3
од
1,0
3,5
350
5,0
100
Так, жесткость питьевой воды во избежание ухудшения ее органолеп-
тических свойств не должна превышать 7 и в исключительных случаях
10 ммольэкв/л. При этом сухой остаток составляет до 1000 мг/л, сульфатов
не более 500 мг/л, хлоридов до 350 мг/л, а рН находится в пределах от 6 до 9.
Количество взвешенных веществ и мутность должны быть до 2 мг/л, цвет-
цветность до 20 градусов, прозрачность до 20 см по Снеллену, вкус, привкус и
запах до 2 баллов. По медицинским показаниям строго регламентируется и
содержание фтора @,7... 1,5 мг/л). Как избыток, так и недостаток фтора
влияет на состояние зубов у человека.
Вкус питьевой воды определяется в первую очередь присутствием
гидрокарбонатов кальция и магния. Содержание этих солей до 10 мг/л
делает воду вкусной, освежающей, что отличает родниковую воду от
дистиллированной. Однако присутствие в воде большего количества ка-
катионов магния придает ей горьковатый вкус. Катионы Са2+ образуют с
468 15. Свойства природной воды
белками пищевых продуктов нерастворимые соединения, поэтому в воде
с высокой жесткостью плохо развариваются мясо и овощи. Такая вода не
пригодна для питья и приготовления напитков. В ней плохо заваривается
чай и при этом ухудшаются его вкусовые качества. Регулярное употреб-
употребление жесткой воды вызывает нарушение осмотического давления жид-
жидких сред организма человека.
При добавлении мыла в жесткую воду образуются осадки и пленки.
Они представляют собой нерастворимые кальциевые и магниевые соли, яв-
являющиеся продуктами замещения ионов натрия и калия в растворимых со-
солях высших жирных кислот (RCOOH): стеариновой (R—С17Н35), пальмити-
пальмитиновой (R—С15Н31), миристиновой (R—С13Н27) и других, например:
Са?, + 2C17H35COONa(p) -> (C17H35COOJCa(T)i + 2Na^
стеарат Na (мыло)
По этой причине ухудшается качество тканей при их стирке в жесткой воде,
а мыло в ней плохо пенится.
Жесткость воды, используемой для производства целлюлозы, не
должна превышать 2 ммоль экв/л. Вода, применяемая в качестве охлаждаю-
охлаждающего агента в системах циркуляционного водоснабжения, не должна содер-
содержать ионов НСОз, СО^, ОН", SO^-, РО^, SiOf-, Ca2+, Mg2+, Fe2+,
Fe3+, Al3+, Zn2+, которые способствуют образованию солевых отложений.
Наиболее часто встречающимся компонентом солевых отложений является
СаСОз. Хлорид натрия и некоторые другие примеси в котлах переходят в
пар и затем, осаждаясь на лопатках турбин, изменяют их профиль и соответ-
соответственно снижают КПД. Вода, используемая в паровых котлах высокого дав-
давления, а также при производстве целлюлозы, полупроводников, лекарствен-
лекарственных препаратов, не должна содержать большого количества метакремниевой
кислоты. Растворенные в воде газы повышают ее коррозионную активность
(О2, СО2, H2S) и придают неприятный привкус и запах (H2S, CH4), поэтому и
питьевую, и техническую воду дегазируют. Содержание О2 в воде, из которой
генерируется водяной пар, не должно превышать 5...40 мкг/кг (в зависимости
от типа парогенератора).
Обработка воды или водоподготовка — это чаще всего комплекс тех-
технологических процессов по приведению ее качества в соответствие с требо-
требованиями потребителей. В него входят процессы осветления и обесцвечива-
обесцвечивания, обеззараживания, стабилизации, фторирования (или обесфторивания),
умягчения, опреснения, обессоливания, дегазации. Главным среди них явля-
является умягчение.
15.4. Способы умягчения воды 469
15.4. Способы умягчения воды
Под умягчением воды понимают либо устранение, либо уменьшение
(снижение) ее жесткости. Главным образом оно заключается в полном или
частичном удалении из нее катионов Са2+, Mg2+ и Fe2+. Существуют три ос-
основных способа умягчения воды: термическая обработка, химическая обра-
обработка, ионный обмен.
Термическая обработка. Суть способа заключается в предваритель-
предварительном нагревании воды до Т= 70...80 °С или ее кипячении. При этом катионы
Са2+, Mg2+ и Fe2+ осаждаются в виде малорастворимых соединений:
+СО2Т +Н2О
Распад гидрокарбонатов магния и железа протекает сложнее и сопро-
сопровождается гидролитическим разложением их карбонатов вследствие того,
что последние, в отличие от карбоната кальция, более растворимы, чем их
гидроксиды (табл. П.4.11). Если гидролиз карбоната магния протекает лишь
по I ступени:
2MgCO3 + НОН -> Mg2(OHJCO31 + СО2 Т
то карбонат железа гидролизуется до конца:
FeCO3 + НОН -» Fe(OHJ1 + CO21
Продукты гидролиза представляют собой основную соль Mg2(OHJCO3 и
гидроксид Fe(OHJ, который под действием кислорода окисляется до мета-
гидроксида
4Fe(OHJ + О2 -> 4FeO(OH) I + 2Н2О
Таким образом, суммарные процессы разложения гидрокарбонатов
магния Mg(HCO3J и железа Fe(HCO3J описываются уравнениями:
2Mg(HCO3J —^->Mg2(OHJCO3 i + ЗСО21 + Н2О
4Fe(HCO3J + О2 -^->4FeO(OH) i + 8CO2 T + 2H2O
Вышеприведенные процессы протекают при нагревании воды в паро-
паровых котлах, системах водяного отопления и охлаждения, бытовой металли-
металлической посуде и приводят к образованию на их поверхности слоя накипи из
малорастворимых соединений. При этом происходит снижение коэффици-
коэффициентов теплопередачи и ухудшение их теплотехнических характеристик, что
обусловливает перерасход топлива и перегрев металлических поверхностей.
Чем больше ионов железа содержится в воде, тем более бурым является
470 15. Свойства природной воды
цвет накипи. При термической обработке воды удается снизить также и со-
содержание в ней растворенных газов, так как их растворимость с увеличени-
увеличением температуры падает.
Химическая обработка воды (или реагентный способ). Данный
способ позволяет устранять как временную, так и постоянную жесткость.
Суть заключается в обработке воды специальными реагентами, образующи-
образующими с катионами жесткости (Са2+ , Mg2+ , Fe2+) малорастворимые соединения.
К числу таких реагентов относятся: гашеная (Са(ОНJ) и негашеная (СаО)
извести, сода (Na2CO3), различные фосфаты натрия (Na3PO4, (NaPO3N и др.
Процесс обработки воды известью называют известкованием или де-
декарбонизацией. При этом происходит устранение временной жесткости во-
воды и одновременное снижение ее щелочности, а также связывание и умень-
уменьшение содержания растворенного в ней углекислого газа:
СаО + Н2О-*Са(ОНJ
Са(НСО3J + Са(ОНJ -> 2СаСО31 + 2Н2О
Са(НСО3J + СаО -> 2СаСО31 + Н2О
Са(ОНJ +2СО2 ->Са(НСО3J
В отличие от извести сода и фосфаты натрия позволяют устранять и
временную, и постоянную жесткость воды:
Са(НСО3J + Na2CO3 -> СаСО31 + 2NaHCO3
СаС12 + Na2CO3 -> СаСО31 + 2NaCl
ЗСа(НСО3J + 2Na3PO4 -> Са3(РО4J1 + 6NaHCO3
ЗСаС12 + 2Na3PO4 -> Са3(РО4J1 + 6NaCl
Использование фосфатов предпочтительнее, так как образующиеся
фосфаты кальция, магния и железа менее растворимы, чем соответствующие
им карбонаты и гидроксиды (табл. П.4.11). Особенно эффективен гексаме-
тафосфат натрия (ЫаРО3)б. Сам он мало растворим в воде, а его кальциевые
и магниевые соли (Na4MP6Oi8 и Na2M2P6Oi8, где М — Са, Mg) еще менее
растворимы.
Для одновременного устранения карбонатной и некарбонатной жест-
жесткости в промышленности используют известково-содовый метод: обработ-
обработку воды смесью СаО и Na2CO3. Для повышения его эффективности процесс
ведут при нагревании, сочетая достоинства термического и химического
способов, так называемое термохимическое умягчение. Остаточная жест-
жесткость воды после такого умягчения не превышает 0,3 ммоль экв/л.
Подчеркнем — если в жесткой воде присутствуют ионы Mg2+ и Fe2+,
то малорастворимыми соединениями, выпадающими в осадок при ее хими-
15.4. Способы умягчения воды
471
ческой обработке различными реагентами, являются М§2(ОН)гСОз и
FeO(OH) или их фосфаты соответственно.
Ионный обмен. Этот способ позволяет не только уменьшить жест-
жесткость воды, но и осуществить ее глубокую очистку, называемую обессоли-
ванием. Вода, подвергшаяся обессоливанию, практически не содержит по-
посторонних ионов (катионов и анионов), поэтому широко используется в
электронной промышленности. Он основан на способности некоторых ве-
веществ, нерастворимых в воде, стехиометрически обменивать свои ионы на
ионы внешней среды (воды, растворов электролитов). Вещества, обладаю-
обладающие такими свойствами, называют ионообменниками {ионообменными сор-
бентами) или сокращенно ионитами. Они состоят из каркаса (матрицы) и
закрепленных на нем ионогенных {активных функциональных) или комплек-
сообразующих групп. Эти группы диссоциируют на полиионы (фиксирован-
(фиксированные ионы), ковалентно связанные с каркасом, и эквивалентное число под-
подвижных противоионов, способных к обмену и компенсирующих своими за-
зарядами заряды полиионов. Например, для одной ионогенной группы:
Катионит в
Н+-форме
П—SO
ионит
Подвижные
противоионы
П—СН
П— СН2— NFL—ОН
ч J
V
Анионит в
ОН"-форме
где П — полимерный каркас; -SO3H и -NR3-OH — ионогенные группы.
По знаку заряда подвижных противоионов, т.е. по знаку заряда обме-
обменивающихся ионов, иониты делятся на катиониты, аниониты и амфолиты,
по химической природе каркаса — на органические, неорганические и ми-
минерально-органические. Органические и неорганические иониты могут быть
природными (цеолиты, целлюлоза и др.) и синтетическими (AI2O3, силика-
гель, сульфоуголь и наиболее важные — ионообменные смолы).
Из неорганических ионитов наибольшее значение имеют цеолиты —
алюмосиликаты сложного состава, имеющие кристаллическое строение. На-
Например, алюмосиликат состава Na2O • AI2O3 • 4SiO2 • WH2O (NaAU^Oe • /W/2H2O)
имеет пространственную решетку, образованную атомами Al, Si и О. Ре-
472 15. Свойства природной воды
шетка пронизана полостями, в которых размещаются молекулы воды и ио-
ионы Na+. Последние, обладая определенной свободой перемещения, замеща-
замещаются на ионы Са2+ и Mg2+ при пропускании воды через слой зерен (гранул)
цеолита:
2NaAlSi2O6(T) +Ca?) ^Ca(AlSi2O6J(T)
Более совершенны ионообменные смолы — синтетические органиче-
органические иониты. Они обладают одновременно высокими эксплуатационно-
техническими характеристиками и разнообразными физико-химическими
свойствами.
Умягчение воды проводят в динамических условиях: пропусканием ее
через неподвижный слой ионита (сорбента) в периодических процессах
(рис. 15.1, а) либо противоточным движением воды и сорбента в непрерыв-
непрерывных процессах (рис. 15.1, б). При этом контакт воды с ионитом должен быть
повторен многократно, что осуществляется в ионообменных колоннах.
Реакции ионного обмена, происходящие при умягчении воды и реге-
регенерации сорбента (например, катионита), могут быть описаны следующими
уравнениями соответственно:
* + 2n-SO3Na(T) -> (n-SO3JM(T)
+ (n-SO3JM(T) -> 2n-SO3Na(T) + МС12(р)
где II-SCbNa — катионит в №+-форме; (П-ЗОз^М — катионит с М2+-иона-
ми; М2+ — ионы Са2+, Mg2+, Fe2+.
Для обессоливания, т.е. глубокого умягчения, необходимо последова-
последовательное пропускание воды через колонны, заполненные одна катионитом в
Н+-форме, другая анионитом в ОН~-форме. При этом из нее удаляются и ка-
катионы, и анионы, а в умягченную воду не поступают посторонние ионы.
Важнейшей количественной характеристикой всех ионитов служит
обменная емкость. Обычно ее относят к единице массы сухого или единице
объема набухшего ионита и выражают либо в ммоль экв/г, либо в ммоль
экв/л. В зависимости от условий определения обменная емкость бывает
статической и динамической. Различают также рабочую (до проскока ионов
жесткости с профильтрованной водой) и полную (до полного истощения ио-
ионита) емкости. Полная емкость, отнесенная к определенному количеству
ионита, есть постоянная величина. Рабочая емкость — переменная величи-
величина, зависящая от свойств и условий регенерации ионита, общего солесодер-
жания, скорости фильтрования, и может изменяться в пределах 40...70 % от
полной. График зависимости концентрации ионов жесткости См2+ в выте-
вытекающей воде от ее прошедшего объема V называют выходной кривой сорб-
15.4. Способы умягчения воды
473
Жесткая вода
Умягченная вода
См2+=С
Умягченная вода
(n-SO3JM
Концентрированный
водный раствор Nad
Рис. 15.1. Схемы ионообменного умягчения воды:
а — в колонне 1 с неподвижным слоем сорбента в условиях периодического
процесса; б — в колоннах 2 и 5 с противоточным движением слоев сорбента,
потоков воды и регенерирующего раствора (концентрированного водного
раствора NaCl) в условиях непрерывного процесса
ции (рис. 15.2). На рисунке заштрихованная площадь представляет собой
емкость до проскока, а площадь, ограниченная осями и выходной кривой
сорбции, соответствует полной емкости колонны. Время работы колонны до
проскока называют временем защитного действия.
Преимущественное поглощение одного из ионов называют селектив-
селективностью {избирательностью) ионита, являющейся второй важнейшей харак-
характеристикой ионита, которая определяется коэффициентом разделения {из-
{избирательности, селективности) ??А:
474
75. Свойства природной воды
ммоль экв
л
D.
а В
У/-/./,
• • • • /
• • • • /
• • • • • /
• • • /
д
а А
где DaA и DaB — коэффи-
коэффициенты распределения ионов
А и В соответственно; ZA и
ZB — зарядовые числа ионов
А и В соответственно. Коэф-
о \ ^ фициент распределения равен
отношению концентраций
иона в ионите [А]и к его кон-
Рис. 15.2. Условная выходная кривая сорбции цеН1рации в растворе [А]р в
М -ионов состоянии равновесия:
D -
\
Проскок
ионов М
2+
Аналогичное выражение можно записать и для иона В. Если S*A > 1,
то ион В обладает большим сродством к иониту; S^A = 1 — сродство ионов
А и В к иониту одинаково; S^A < 1 — ион А сорбируется (поглощается)
предпочтительнее.
Практические занятия
Общие свойства водных растворов
Примеры решения задач
Задача 1. Вычислить интегральную энтальпию растворения хлорида калия
КС1 в воде, если при растворении 25 г соли в 1 л Н2О температура понизи-
понизилась на 1,5 °С. Удельная изобарная теплоемкость полученного раствора ср =
= 4,18Дж/(гК).
Решение, Интегральную энтальпию растворения А5ЯинТ, т.е. тепловой эф-
эффект растворения 1 моль вещества в некотором количестве чистого раство-
растворителя, можно рассчитать по формуле A3.2):
_ ASH
щ
где rii — число молей растворенного вещества; ASH — энтальпия раство-
растворения данного количества вещества.
В соответствии с A3.3)
ASH =-mcpAT9
где m — масса раствора; ср — удельная изобарная теплоемкость раствора;
AT — изменение температуры.
В данном случае m - 1000 + 25 = 1025 г, поэтому количество погло-
поглощаемой теплоты
Д5# = -1025 • 4,18 • (-1,5) = 6427 Дж = 6,427 кДж.
Количество растворенного вещества
пкс, = —— = = 0,335 моль.
Мка 74,55
Следовательно,
As#hht= "
0,335
19,184 кДж/моль.
Задача 2. При растворении 4,0 г сульфата меди CuSO4 в 0,2 л воды темпе-
температура раствора повысилась на 2 °С. Вычислить интегральную энтальпию
гидратации безводного сульфата меди, если энтальпия растворения кри-
кристаллогидрата CuSO4-5H2O равна +11,72 кДж/моль. Удельная теплоем-
теплоемкость раствора ср составляет 4,18 Дж/(г • К).
476 Общие свойства водных растворов
Решение. Как уже отмечалось, процесс растворения CuSO4 состоит из двух стадий:
гидратация безводной соли
CuSO4 (Т) + 5Н2О(Ж) = CuSO4 • 5Н2О (т) (AhH)
и растворение образовавшегося гидрата
CuSO4 • 5Н2О(Т) + лН2О(ж) = CuSO4 • 5Н2О(р) (А5ЯИНТ 2)
Если при растворении CuSO4 (т) и CuSO4 • 5Н2О(т) образуются растворы одинакового
состава, энтальпию гидратации можно вычислить по формуле A3.4)
где А5#инт 1 — интегральная энтальпия растворения безводной соли.
При растворении 4 г безводной соли в 200 г воды @,2 л) выделяется количе-
количество теплоты, равное
А5#, = - тсрАТ=- 204 • 4,18 • 2= -1705 Дж = -1,705 кДж.
Следовательно, в соответствии с A3.2) интегральная энтальпия растворения
AI/ Mcuso4 1,705 -159,61 ЛО ^
А5ЯИНт1 = A5#j = » -68,03 кДж/моль.
WCuSO4 4
По условию тепловой эффект растворения гидрата CuSO4 • 5Н2О AsHHm2 -
= +11,72 кДж/моль, поэтому интегральная энтальпия гидратации
Дл#инт = А5ЯИНТ, - А,Яинт2 = -68,03 - 11,72 = -78,36 кДж/моль.
Задача 3. При температуре 10 °С давление водяного пара р,° = 1227,8 Па. Опреде-
Определить массу воды, необходимую для растворения 16 г метилового спирта СН3ОН,
чтобы давление пара понизилось до 1200 Па.
Решение. Понижение давления насыщенного пара над раствором, по условию,
должно составить Ар = 1227,8 - 1200 = 27,8 Па. В соответствии с законом Рауля
A3.11), понижение давления насыщенного пара Ар связано с составом раствора вы-
выражением:
где i — коэффициент диссоциации растворенного вещества; р® — давление пара
над чистым растворителем при данной температуре; Х2 — молярная доля раство-
растворенного вещества.
Метанол — неэлектролит, поэтому коэффициент диссоциации / = 1. Моляр-
Молярная доля метанола в его водном растворе должна составить:
хси он = ^- = 2?>8 « 0,0226.
СН3ОН о Л ОО1 Я
р 1227,8
Примеры решения задач 477
Так как молярная масса метанола Мсн он = 32 г/моль, то число молей спирта
в данном растворе псп он = — = 0,5. Необходимое число молей воды пн 0 можно
определить по формуле C.12)
у
^CHjOH
Тогда
«но=— -0,5 = 21,6.
Нз° 0,0226
Молярная масса воды составляет 18 г/моль, поэтому ее требуемая масса
Задача 4. Из скольких атомов состоят молекулы йода в спиртовом растворе, если
температура кипения этилового спирта Тшп\ = 78,30 °С повышается до ТКИп2 =
= 78,59 °С при растворении йода массой 12,7 г в спирте массой 200 г?
Решение. Молекулярная формула йода - 1„. Если рассчитать молярную массу, то
можно определить количество атомов п в молекуле.
Повышение температуры кипения раствора, по условию, составляет
АТШП = Ткип 2 - Ткнп, = 78,59 - 78,30 = 0,29 °С = 0,29 К,
и, согласно A3.14), равно:
где / — коэффициент диссоциации растворенного вещества; Ст — моляльная кон-
концентрация раствора, моль/кг; Е - эбулиоскопическая постоянная этилового спирта:
Е = 1,16 К • кг/моль (см. табл. П.4.6).
Так как йод — неэлектролит, то коэффициент диссоциации / = 1. Поскольку в
условии даны массы компонентов раствора, молярную массу йода можно рассчи-
рассчитать по формуле A3.16)
Щ 12 7
М, =Е != = 1,16 «254 г/моль,
" A7 0Л9 02
где тх — масса йода, г; тс н он — масса этанола, кг.
Молярная масса атомов йода — 127 г/моль. Следовательно, молекулы йода в
спиртовом растворе состоят из двух атомов: п = 2.
Задача 5. Вычислить кажущуюся степень диссоциации 2 %-ного водного раствора
хлорида натрия NaCl, который начинает кристаллизоваться при температуре
478 Общие свойства водных растворов
Решение. Кажущуюся степень диссоциации сильного электролита можно рассчи-
рассчитать по формуле A3.13):
j-1
<х=
и-1
где i — коэффициент диссоциации, который связан с понижением температуры за-
замерзания раствора соотношением A3.15):
АГзам = iKCm.
Число ионов, на которые диссоциирует одна молекула электролита, п - 2 (NaCl =
Понижение температуры замерзания раствора по сравнению с чистым рас-
растворителем, по условию,
АГзам= Тзш , - Гзам2 = 0 - (-1,26) = 1,26 °С= 1,26 К.
В 100 г раствора содержится 2 г NaCl и 98 г воды. Молярная масса MNaC1 = 53,5 г/моль.
Моляльность раствора, согласно B.12),
Cm= т™ = 1 = 0,38 моль/кг.
м^тщо 53,5 0,098
Коэффициент диссоциации /, с учетом криоскопической постоянной воды
К= 1,86 К • кг/моль (см. табл. П.4.6),
КСт 1,86 0,38
Следовательно, кажущаяся степень диссоциации
2-1
Задача 6. Давление насыщенного пара 10%-ного раствора нитрата магния Mg(NO3J
при температуре Т= 20 °С составляет/? = 22,9 • 102 Па. Каково осмотическое давле-
давление этого раствора при той же температуре?
Решение. Осмотическое давление к в соответствии с A3.17) связано с молярной кон-
концентрацией раствора С и коэффициентом диссоциации электролита / соотношением
n = iCRT.
Для определения осмотического давления заданного раствора необходимо
провести перерасчет массовой доли растворенного вещества (<о = 0,1) в молярную
концентрацию С. Для этого используют формулы, представленные в табл. 2.3, а
также табл. П.4.7 (для определения плотности раствора р):
Примеры решения задач 479
С = — = °?1'1076>2 = 0,726 моль/л = 726 моль/м3.
MMg(NO3J 148'3
Коэффициент диссоциации в соответствии с A3.11)
Ар ^А°-А
0у 0 у
Р\ *2 Р\ Х2
В 100 г заданного раствора содержится 10 г Mg(NO3J и 90 г Н2О, что составляет
_10_ =
148,3
тщо 90
ин п = —— = — =5 моль.
2 МЩо 18
Следовательно, молярная доля Mg(NO3J в растворе
х2=
По данным табл. П.4.1 можно определить давление насыщенного пара чистой
воды при заданной температуре: р® = 23,38 • 102 Па. Отсюда в соответствии с A3.11)
. B3,38-22,9)-102 _
/ = l,Do.
23,38-102.0,013
Осмотическое давление заданного раствора
к = 1,58 • 726 • 8,31 • 273 = 2,6 • 106Па.
Задача 7. Для приготовления раствора, имитирующего жесткую воду, в 5 л дистил-
дистиллированной воды растворили некоторое количество солей магния. При анализе ока-
оказалось, что концентрации ионов НСО7 и SO?" (С _, С 2_) равны 0,305 и
мэсс HCOj масс SO^
0,144 г/л соответственно. Рассчитать:
1) карбонатную Нк, некарбонатную Ннк и общую Но жесткость имитирующе-
имитирующего раствора;
2) концентрацию ионов Mg2+ в г/л, отвечающих за карбонатную Ск 2+ и
некарбонатную Снк . _ 2+ жесткость;
r J MaccMgz '
480 Общие свойства водных растворов
3) массу т осадков, образующихся при термической и химической (с исполь-
использованием Na2CC>3) обработках 1 л такого раствора;
4) объемы в мл 0,1н. раствора НО Vhci и 0,025М раствора трилона Б Vtb, из-
израсходованных на титрование двух проб этого раствора по 50 мл каждая;
5) число поглощенных катионитом ионов Mg2+ N 2+;
6) массу катионита с рабочей обменной емкостью Ev = 4 ммоль экв/г, необхо-
необходимого для поглощения всех ионов Mg2+.
Написать уравнения реакций всех процессов.
Решение. 1. Карбонатная жесткость в данном примере обусловлена гидрокарбонат-
ионами и эквивалентно связанными с ними ионами Mg2+, некарбонатная — суль-
сульфат-ионами и эквивалентно связанными с ними ионами Mg2+. Общая жесткость есть
сумма карбонатной и некарбонатной жесткости. По определению любой вид жест-
л3 y
вы
Н = Сэкв (ммоль экв/л = 10~3 моль экв/л).
кости измеряется в ммоль экв/л или моль экв/м3 и соответствует молярной концен-
концентрации химического эквивалента вещества Сэкв, выраженной в 10н.:
По данным табл. 2.3,
масс масс^экв
мэкв м
где Мэкв — молярная масса химического эквивалентна вещества, равная отношению
его молярной массы М к числу эквивалентности 7.экв. Число эквивалентности иона
равно его зарядовому числу. Таким образом,
масс НСО; Z3kb HCOJ _ 0,305 Г/Л • 1 МОЛЬ ЭКВ/МОЛЬ _
к~ эквНС°з *~ М . " 61,02 г/моль
HCOj
= 5 • 10" н. = 5 ммоль экв/л;
масс so24~ ^экв soj' 0,144 г/л • 2 моль экв/моль
«к- „soj-"" M - 96,06г/моль "
= 3 • 10~3 н. = 3 ммоль экв/л;
Яо = #к + Ннк = 5 + 3 = 8 ммоль экв/л.
2. Согласно закону эквивалентов, молярная концентрация химического эквивалента
ионов Mg2+, отвечающих за карбонатную жесткость Ск 2+, равна таковой для гидро-
экв Mg
карбонат-ионов Ск _ и соответствует карбонатной жесткости, выраженной в 10~3н.:
Ск 7
гк _г _И массМё2+ ^эквМ82+
Используя это выражение, можно рассчитать концентрацию ионов Mg2+, отвечаю-
отвечающих за карбонатную жесткость:
Примеры решения задач 481
_ *щ2+ 510 3 моль экв/л • 24,30 г/моль _ л ПА
С 2+ — — — О, "о * г/л.
MaccMg 2 2 МОЛЬ ЭКВ/МОЛЬ
экв Mg
Аналогично определяют массовую концентрацию ионов Mg2+, отвечающих за не-
некарбонатную жесткость:
TJ \Ж _л
" hk7WMg2+ 3 10 моль экв/л • 24,30 г/моль л л„ ^ ,
С 2+ = = = 0,036 г/л.
MaccMg z 2 МОЛЬ ЭКВ/МОЛЬ
экв Mg
3. При термической обработке раствора разложению подвергается только гидрокар-
гидрокарбонат магния. Процесс описывается следующим суммарным уравнением:
2Mg(HCO3J —^-»Mg2(OHJCO31 + ЗСО21 + Н2О
Массу Mg(HCO3J, содержащегося в 1 л раствора, находят из соотношения:
СэквМ6(НСО3J =CTKBMg2+ =СэквНСО3- =Як =
откуда
Mg(HCO3 J Z3kb Mg(HCO3 J _ WMg(HCO3 J Z3kb Mg(HCO3 J
5 • 10 3 моль экв/л 1л-146,35 г/моль
= °>366 г-
*экв Mg(HCO3J 2 М^ЛЬ ЭКВ/МОЛЬ
По стехиометрии реакции разложения Mg(HCO3J масса осадка Mg2(OHJCO3
'"MgCHCo, J ^Mg2 (онJсо3 0,366 г • 142,65 г/моль
WM /ОНч го = = = U, I /о Г.
м82(онJсо3 2MMg(HCOjb 2 • 146,35 г/моль
При химической обработке раствора содой протекают реакции, описываемые
уравнениями:
2Mg(HCO3 J + 2Na2CO3 + Н2О -> Mg2 (OHJCO31 + CO21 + 4NaHCO3
2MgSO4 + 2Na2CO3 + H2O -> Mg2(OHJCO3 i + CO21 + 2Na2SO4
При этом устраняется и карбонатная, и некарбонатная жесткость, поэтому масса
образующегося осадка больше, чем при термической обработке, на величину
WMg2(OHJCO3
2MMgSO4
Массу MgSO4, содержащегося в 1 л раствора, находят в соответствии с соотноше-
соотношением, аналогичным для Mg(HCO3J:
16 - 9795
482 Общие свойства водных растворов
Г -ГНК -Г - И WMgSO4Z3KBMgSO4
Чкв MgSO4 - Сэкв Mg2+ - Ьэкв SO2- ~Пш~
откуда
янк WMgso4 3 • 10 моль экв/л 1л-120,37 г/моль Л л о
wMgso4 = = " : = 0,181 г,
2экв MgSO4 ^ МОЛЬ ЭКВ/МОЛЬ
0,181 г -142,65 г/моль
/wM. /ПНч гп = = 0,107 г.
мё2(онJсо3 2.120,37 г/моль
Общая масса осадка, образующегося при химической обработке содой, со-
составляет
т = 0,178 + 0,107 = 0,285 г.
4. При титровании проб растворами НС1 и трилона Б (Na2H2Y) протекают реакции:
Mg(HCO3J + 2НС1 -> MgCl2 + 2СО2 t + 2Н2О
Mg(HCO3 J + Na2H2 Y = MgH2 Y + 2NaHCO3
MgSO4 + Na2H2 Y = MgH2 Y + Na2SO4
поэтому раствор НС1 используют для определения карбонатной жесткости, а рас-
раствор трилона Б — для определения общей жесткости.
Из выражения B.16) закона эквивалентов определяют объемы реагентов (НС1
и трилона Б), израсходованных на титрование:
^НС1СэквНС1=Рн2ОЯк>
у С —V И
'ТБ^эквТБ "" KH2O/io'
где Як и #0 выражены в моль экв/л. Из этих соотношений получают:
50 мл • 5 • 10~3 моль экв/л л _
Кнс, = = 2,5 мл,
0,1 моль экв/л
50 мл • 8 • 10~3 моль экв/л
VTB = = 8,0 мл.
0,05 моль экв/л
5. В соответствии с определением числовое значение молярной концентрации экви-
эквивалента всех ионов Mg2+ С 2+ bz 2+ раз больше их молярной концентрации
С 2+ и соответствует общей жесткости, выраженной в моль экв/л:
Отсюда
СэквМЕ2+ ~Z3KBMg2+CMg2+ ~J
Hn 8 • 10 3 моль экв/л _i , •,
С 2+= 2—= = 410 3 моль/л = 4-Ю'3М.
Mg z 2+ 2 моль экв/моль
экв Mg
Задачи для самостоятельного решения
483
В 5 л раствора содержится
ИЛИ
п 2+ = НУ = 8 10 моль экв/л • 5 л = 0,04 моль экв
экв Mg °
п 2+ = С 2+ V = 4 • 10 3 моль/л • 5 л = 0,02 моль ионов Ме2+.
Mg Mg v
Умножая последнее значение на число Авогадро, получают число поглощенных
катионитом ионов Mg2+:
NM 2+ = «м 2+NA = 0,02 моль • 6,02 • 1023 ионов Mg^/моль = 1,22 • 1022 ионов Mg2+.
6. Используя закон эквивалентов, рассчитывают массу т катионита, необходимого
для поглощения всех ионов Mg2+, т.е. 0,04 их моль экв:
Отсюда
«=¦2=-=-
0,04 МОЛЬ ЭКВ
0 3
Ep 4 10 J мольэкв/г
¦ = 10r.
При нахождении катионита в Н*-форме, процесс поглощения описывается уравнением:
2П—SO3H(T) + Mg2^ ~> (П—SO3JMg(T) + 2H(+p)
Задачи для самостоятельного решения
1-7. Считая удельную теплоемкость раствора равной удельной теплоемкости воды
ср= 4,18 Дж/(г • К), определите интегральную энтальпию растворения А5Нит элек-
электролита, если известно изменение температуры AT в процессе растворения электро-
электролита массой тэл_та в воде массой тн 0.
№
1
2
3
4
5
6
7
Электролит
NaOH
КОН
KNO3
NH4C1
NH4C1
C11SO4
CuSO4
^эл-та> г
10
7
10
25
10
8
15
тно, г
250
125
240
430
233
192
300
АГ,К
9,7
12,6
-3,4
-4,3
-2,8
3,9
5,4
16*
484
Общие свойства водных растворов
8-15. Считая удельную теплоемкость раствора равной удельной теплоемкости воды
Ср= 4,18 Дж/(г • К), определите, как изменится температура при растворении 10 г
безводного электролита в 100 г воды, а также рассчитайте интегральную энтальпию
гидратации электролита АиНИНТ9 используя значения интегральных энтальпий рас-
растворения соответствующих солей.
№
8
9
10
11
12
13
14
15
Электролит
SrCl2
СаС12
Na2CO3
CuSO4
MgCl2
Na2SO3
ZnSO4
Na2HPO4
АД.ГТ ь кДж/моль
-47,7
-75,7
-24,6
-66,1
-149,9
-11,3
-77,6
-23,6
Кристаллогидрат
SrCl210H2O
CaClr6H2O
Na2CO310H2O
CuSO4-5H2O
MgClr6H2O
Na2SOr7H2O
ZnSO4-7H2O
Na2HPO412H2O
АЛнт2, кДж/моль
+31,0
+19,1
+66,9
+11,7
-12,3
+46,9
+17,7
+95,1
16-35. Определите температуру начала кипения Гкип, температуру начала замерза-
замерзания Гзам и осмотическое давление я водного раствора электролита заданной массо-
массовой доли со, используя значения давления насыщенного пара и плотности растворов,
приведенные в табл. П.4.7.
№
16
17
18
19
20
21
22
23
24
25
Электролит
СиС12
СиС12
СиС12
СиС12
CuSO4
CuSO4
CuSO4
KI
KI
KI
CO
0,10
0,15
0,20
0,25
0,10
0,15
0,20
0,20
0,30
0,40
№
26
27
28
29
30
31
32
33
34
35
Электролит
Mg(NO3J
Mg(NO3J
Mg(NO3J
NaBr
NaBr
NaBr
ZnSO4
ZnSO4
ZnSO4
ZnSO4
CO
0,15
0,20
0,25
0,20
0,30
0,40
0,05
0,15
0,25
0,30
36-41. Антифризами называют растворы с пониженной температурой замерзания,
используемые в системе охлаждения автомобилей. Широкое применение в этом
качестве находят растворы этиленгликоля СгЕЦОН^. Рассчитайте, в каких объемных
соотношениях необходимо смешать этиленгликоль, плотность которого р = 1,116 г/мл,
и воду, чтобы получить антифриз, замерзающий при заданной температуре Т.
№
36
37
тх
-15
-18
№
38
39
ТХ
-20
-25
№
40
41
ТХ
-30
-35
Задачи для самостоятельного решения
485
42-51. Используя данные табл. П.4.6, определите молярную массу растворенного
неэлектролита, если известны масса растворителя ти масса растворенного вещества
т2 и температура начала кипения Гкип или начала замерзания Гзам раствора.
№
42
43
44
45
46
47
48
49
50
51
Растворенное
вещество
Глицерин
Сера
Йод
Фенол
Хлороформ
Фосфор
Нитробензол
Глицерин
Фенол
Камфора
И*2, Г
2,3
3,24
12,7
0,94
15
0,1155
6,15
2,76
2,5
0,04
Растворитель
Ацетон
Бензол
Этанол
Этанол
Диэтиловый эфир
Бензол
Бензол
Вода
Бензол
Бензол
оть г
100
40
200
50
400
19,3
400
200
91
20
Т °С
1 кит ^
56,73
80,82
78,59
78,622
35,135
—
—
—
—
—
т °с
1 зам» ^
5,15
4,86
-0,279
3,8
5,463
52. При перегонке 5 л воды ее масса уменьшилась на 1,44 г. Рассчитайте ее жест-
жесткость, обусловленную содержащимся в ней гидрокарбонатом кальция. Приведите
уравнение, описывающее происходящий процесс.
53. Жесткость воды, обусловленная содержащимся в ней гидрокарбонатом магния,
составляет 2 ммоль экв/л. Рассчитайте массу осадка, образующегося при термиче-
термической обработке 1 л такой воды. Приведите уравнение происходящей при этом сум-
суммарной реакции.
54. Вычислите карбонатную, некарбонатную и общую жесткость воды, в 10 л кото-
которой содержится по 0,81 г Са(НСО3J и MgCl2.
55. Рассчитайте концентрацию солей в г/л в воде, общая жесткость которой равня-
равняется 5 моль экв/м3 и обусловлена содержащимися в ней MgCl2 и Са(НСО3J, причем
магниевая жесткость составляет 30 % от общей.
56. Магниевая жесткость воды, составляющая 20 % от общей, равна 1 моль экв/м3.
Какую массу соды Na2CO3 следует добавить к 100 л такой воды, чтобы полностью
устранить ее жесткость? Приведите уравнения происходящих при этом процессов.
57. К 100 л воды с общей жесткостью 5 моль экв/м3 добавили 16,4 г Na3PO4. Приве-
Приведите уравнения происходящих при этом реакций. Рассчитайте остаточную жест-
жесткость воды.
58. Рассчитайте карбонатную, некарбонатную и общую жесткость воды, на титро-
титрование 50 мл которой расходуется по 4 мл 0,1 % (масс.) раствора НС1 (р « 1 г/мл) и
0,025М раствора трилона Б.
486 Общие свойства водных растворов
59. При последовательном пропускании 10 м3 воды через колонны, заполненные
одна — катионитом в Н+-форме (П—SO3H), другая — анионитом в ОН"-форме
(П—СН2—NR3OH), дополнительно образовывался 1 кг Н2О. Рассчитайте исходную
жесткость воды, обусловленную содержащимся в ней сульфатом магния. Приведите
уравнения процессов, происходящих при катионировании и анионировании воды.
60. Рассчитайте карбонатную жесткость воды, в 5 л которой содержатся 1,216 г
Са(НСОзJ и 0,732 г Mg(HCO3J. Какая масса гексаметафосфата натрия (NaPO3N
потребуется для ее устранения? Приведите уравнения соответствующих реакций.
61. Какое количество ионов Са2+ и СГ содержится в 620 мл морской воды, некарбо-
некарбонатная жесткость которой составляет 10 ммоль экв/л?
62. Как изменилась жесткость воды, 2 м3 которой подверглись обработке гашеной
известью Са(ОНJ массой 74,1 г?
63. Рассчитайте карбонатную, некарбонатную и общую жесткость воды, в 2 м3 кото-
которой содержится, мг: 48,6 (Mg2+), 80,2 (Са2+), 70,9 (СГ), 122,0 (НСО~).
64. После пропускания воды через колонну, заполненную катионитом в Ыа+-форме,
содержание ионов Na+ в ней увеличилось на 92 г/м3. Рассчитайте изменение ее
жесткости.
65. Какую массу негашеной извести СаО следует добавить к 20 м3 воды для устра-
устранения ее карбонатной жесткости, равной 2 ммоль экв/л? Опишите происходящие
при этом процессы уравнениями.
66. Жесткость воды, 100 л которой подверглись катионообменной обработке (ка-
тионированию), уменьшилась на 5 моль экв/м3 и составила 0,3 моль экв/м3. Рассчи-
Рассчитайте исходную жесткость, а также массу поглощенных катионитом ионов Са2+.
67. Рабочая обменная емкость катионита равна 3 моль экв/кг. Рассчитайте массу
ионов Mg2+, поглощаемых катионитом массой 5 кг.
68. Какой объем воды с жесткостью, равной 5 ммоль экв/л, можно умягчить с по-
помощью катиони
250 моль экв/л?
мощью катионита объемом 10 м3, имеющего рабочую обменную емкость, равную
69. Рассчитайте массу NaCl, необходимую для приготовления концентрированного
регенерирующего раствора с целью перевода 4 м3 анионита с рабочей обменной
емкостью, равной 200 ммоль экв/л в СГ-форму, если для этого требуется полутор-
полуторный избыток массы.
70. Какую массу Сг2О2~-ионов поглотят 5 м3 анионита в СГ-форме, (П—СН2—
—NR3C1) с рабочей обменной емкостью, равной 200 ммоль экв/л. Приведите урав-
уравнения процессов ионного обмена: поглощения ионов и регенерации анионита.
Примеры решения задач 487
71. Какую массу катеонита с рабочей обменной емкостью, равной 4 ммоль экв/г, следу-
следует взять для умягчения 2 м3 воды, общая жесткость которой составляет 8 моль экв/м3?
Свойства водных растворов электролитов
Примеры решения задач
Задача 1. Как изменится рН 0,1М раствора циановодородной кислоты HCN, если к
1 л раствора добавить 0,1 моль цианида натрия NaCN, кажущаяся степень диссо-
диссоциации которого aNacN = 0,85?
Решение. Слабая циановодородная кислота диссоциирует согласно уравнению
HCN ?=> Н+ + CN'
Учитывая, что константа диссоциации HCN Клцсы = 4,93 • 10~10 (табл. ПАЮ),
степень диссоциации кислоты в растворе без добавления соли можно вычислить по
упрощенному выражению закона разбавления Оствальда A4.4):
Отсюда равновесная молярная концентрация иона водорода
[Н+] = (XhcnChcn =V*aHCNCHcN = лМ,93 .100 • 0,1 = 7,02 • Iff* моль/л.
Следовательно, водородный показатель исходного раствора, согласно A4.13),
рН = -lg [Н+] = -lg G,02 • 10) = 5,15.
При добавлении в раствор соли NaCN равновесие диссоциации кислоты, со-
согласно принципу Ле Шателье—Брауна, сместится влево (в сторону образования
HCN) в результате появления в растворе большого количества ионов CN" за счет
диссоциации сильного электролита:
При этом уменьшится концентрация ионов водорода в растворе, т.е. диссо-
диссоциация слабой кислоты будет подавлена. Новую концентрацию ионов водорода
можно обозначить через х (моль/л). Концентрация ионов CN", вносимых солью с
концентрацией 0,1 моль/л:
CCN. = aKcNCKcN= 0,1 • 0,85 = 0,085 моль/л.
Общая концентрация цианид-ионов в новом растворе составит
[CN~] = х + CCN_ = x + 0,085 (моль/л).
488 Свойства водных растворов электролитов
Подставляя концентрации ионов Н+ и CN~ в выражение константы диссоциа-
диссоциации кислоты КЛ hcn и учитывая, что из-за очень незначительной диссоциации циа-
новодородной кислоты концентрация недиссоциированной кислоты [HCN] практи-
практически совпадает с исходной концентрацией, получают
[H+][CN1 *(* +0,085) х- 0,085 10
A-aHCN = = « = 4,93 * 10 .
[HCN] 0,1 0,1
Решая уравнение, находят концентрацию ионов водорода в растворе с добав-
добавленной солью: х = 5,80 • 10~10 моль/л. Следовательно, новое значение водородного
показателя раствора
рН = -lg E,80 10-10) = 9,24.
Таким образом, после добавления соли среда раствора изменилась и вместо
слабокислотной стала слабощелочной, что обусловлено не только подавлением дис-
диссоциации слабой кислоты HCN, но и гидролизом по аниону соли NaCN.
Задача 2. Рассчитать водородный показатель 0,005н. раствора хлороводородной
кислоты НС1, содержащего 0,01 моль/л хлорида натрия NaCl.
Решение. Данный раствор содержит сильные электролиты, которые диссоциируют
по уравнениям:
НС1 = Н+ + СГ
Водородный показатель такого раствора, согласно A4.16), определяется ак-
активностью иона Н+:
рн= рян+ = -igv
Активность иона водорода в соответствии с A4.5)
Q
Молярная концентрация раствора НС1 С = —— = 0,005 моль/л, следовательно,
*экв
концентрации ионов Сн+ = 0,005 моль/л; CNa+= 0,01 моль/л; С = 0,01 + 0,005 =
= 0,015 моль/л.
Плотность очень разбавленного раствора можно принять равной плотности
воды р « 1000 г/л, поэтому при вычислении ионной силы / раствора по уравнению
A4.7), а также активности иона, вместо моляльных концентраций Cmi можно ис-
использовать приблизительно равные им молярные концентрации С;:
= 0,5 • @,005 • I2 + 0,015 • I2 + 0,01 • I2) = 0,015.
Примеры решения задач 489
По уравнению предельного закона Дебая—Хюккеля A4.8) определяют сред-
средний коэффициент активности ионов у± в данном растворе:
lgy± =-A\Z+Z_\J7 =-0,509 • I2 • @,015I/2 = -0,622.
Следовательно, средний коэффициент активности ионов у+ = 0,867.
Считая коэффици
тивность ионов водорода
Тогда
Считая коэффициент активности ионов водорода у + = у±, вычисляют ак-
н
V = Ты* Сн* = 0'867 ¦ °'005 = °'0043-
н н м
Задача 3. Вычислить константу гидролиза Кг, степень гидролиза аг и рН 0,1М рас-
раствора цианида натрия NaCN.
Решение. Соль NaCN образована сильным основанием NaOH и слабой кислотой
HCN, поэтому гидролизуется по аниону. Чтобы научиться составлять уравнения
гидролиза, следует представить соль и воду (последнюю — условно) в диссоцииро-
диссоциированном виде, записать продукты обменного взаимодействия ионов, при этом силь-
сильные электролиты — в виде ионов, слабые — в виде молекул:
затем сократить одноименные ионы (в данном случае, Na+), а воду как слабый элек-
электролит вновь записать в виде молекулы. Получится сокращенное ионно-
молекулярное уравнение гидролиза:
CN'+H2O <=> HCN + OH"
Чтобы определить, какие кислота и основание являются сильными электро-
электролитами, а какие — слабыми, следует обратиться к справочным данным по констан-
константам диссоциации кислот и оснований (см. табл. П.4.10). Концентрационные кон-
константы диссоциации Кл и Кь для слабых электролитов приводятся для всех ступеней
диссоциации, а для сильных электролитов — только для второй ступени (если тако-
таковая имеется). Так, константа диссоциации HCN Яансы = 4,93 • 10~10, т.е. HCN —
очень слабый электролит, а константа диссоциации NaOH не приводится, так как
Значение константы гидролиза находят по формуле A4.17):
«.но. 4,93-10-'°
Поскольку Кг мала, степень гидролиза по A4.20)
\ГТ /2,03-lQ-5
ar= A—- = A » 1,42 •
V с v 0,1
490 Свойства водных растворов электролитов
Так как имеет место гидролиз по аниону, то согласно A4.21),
рН = 14 + 0,51g (KrQ = 14 + 0,51g A,42 • 10 • 0,1)« 11,09.
Среда — щелочная.
Задача 4. Определить значение рН 0,01М раствора хлорида цинка ZnCl2.
Решение. Соль ZnCl2 образована слабым основанием Zn(OHJ и сильной кислотой
НС1, поэтому гидролизуется по катиону:
I ступень
Zn2+ + Н2О ?± ZnOH+ + Н+ Кт = ^— согласно A4.29);
^b(II)Zn(OHJ
II ступень
ZnOH+ + Н2О <± Zn(OHJ + Н+ Кт = Щ°— согласно A4.30).
По табл. П.4.10
b(i)Zn(OHJ Ь , b(ii)Zn(OHJ , 10 .
Отсюда
204 10"8 ^ = — = 758 • 100
т = 2,04 • 10"8, ^П) = — = 7,58 • 100.
4,90 10 1,32-Ю
Концентрации ионов водорода, образующихся на I и II ступенях гидролиза,
соответственно равны:
[Ы], =у[к^С = л/2,04-10-8.10 = 1,43 • 10,
[Н+]„= ylKmC = л/7,58-Ю-10-10 = 2,75 • КГ6.
Следовательно,
+]„= 1,705-10
рН = -lg[H+] = 4g(l,705 • 10) = 4,77.
Среда — кислотная.
Согласно A4.23), для I ступени гидролиза
рН, = -0,5 lg^oQ = -0,51g B,04 • lO"8 • Ю-2) = 4,85,
значит, при приближенном расчете рН растворов солей II ступенью гидролиза мож-
можно пренебречь.
Задача 5. Определить значение рН насыщенного водного раствора гашеной извести
Са(ОНJ.
Решение. Гашеная известь Са(ОНJ относится к малорастворимым электролитам.
Произведение растворимости ПРСа@Н) = 6,3 • 10 (см. табл. П.4.11).
Примеры решения задач 491
В соответствии с уравнением равновесного процесса растворения сильного
электролита
Са(ОНJ (т) <=± Са(ОНJ (р) = Са2+ + 2ОН"
и формулой A4.36), можно записать
ПРСа(он,2=[Са2+][ОН-]2
Если принять равновесную концентрацию [Са2+] = х, то равновесная концен-
концентрация ионов [ОН"] = 2х, и тогда ПРСа@Н) = 4х3. Отсюда концентрация гидроксид-
ионов
щр
[ОН"] = 2 3J_2pL = з^2ПРСа(ОНJ = V2-6,3.1(T6 = 2,33 • 10.
Согласно A4.4) и A4.5), водородный показатель насыщенного раствора га-
гашеной извести при Т= 298 К
рН = 14 - рОН = 14 - (-lg [ОН"]) = 14 + lg B,33 • 10) = 12,37.
Задача 6. Рассчитать объем воды, необходимый для растворения 2,35 г йодида се-
серебра Agl при Т= 298 К.
Решение. По табл. П.4.11 произведение растворимости IlPAgi = 8,5 • 107 при Т= 298 К.
В соответствии с уравнением диссоциации соли
= Ag++r
жду вели
ением
nPAgI=[Ag+][r] = S2.
и формулами A4.36) и A4.37) связь между величиной ПР и растворимостью S, вы-
выраженной в моль/л, определяется выражением
Отсюда растворимость Agl
S= JnFTT = л/8,5-107 = 9,22 • 10"9 моль/л
Agl
и масса Agl в 1 л насыщенного раствора (массовая концентрация)
CMacCAgi = SAg,MAgI=9,22- Ю~9-235 = 2,17- КГ* г/л.
Учитывая, что основной объем раствора занимает вода, объем воды, необхо-
необходимый для растворения 2,35 г Agl, вычисляют по формуле:
vh о = "*AgI = 2>35 А = 1,083 • 106 л = 1083 м3.
2 Г 1 17 1П
о
2 Г 1 17
492 Свойства водных растворов электролитов
Задача 7. Определить, выпадет ли осадок карбоната никеля NiCO3, если к 1 л 0,1М
раствора хлорида гексаамминникеля (II) [Ni(NH3N]Cl2, содержащему избыток ам-
аммиака NH3, добавить 1,38 г кристаллического карбоната калия К2СО3?
Решение. Осадок NiCO3 выпадет, если произведение концентраций ионов Ni2+ и
СО2' больше произведения растворимости данного малорастворимого электролита:
CNi2+Cco?- > nP
Произведение растворимости nPNiC0 = 1,3 • 10 7 (см. табл. П.4.11).
Если пренебречь изменением объема при смешении 1л раствора
[Ni(NH3N]Cl2 и 1,38 г кристаллического К2СО3, то в соответствии с уравнением
диссоциации хорошо растворимого электролита:
К2СО3=2К++СО32'
молярная концентрация ионов СО32~ составит
С 2 = Ск гп = —!S?2i_ = _J— = jo. ю моль/л.
СО, N2CU3 1/ т/ по 1 '
3 JvLv гп V 13о • 1
К2СО3
Уравнение полной диссоциации комплексного иона
[Ni(NH3N]2+ <=> Ni2+ + 6NH3
Если обозначить за х (моль/л) равновесную концентрацию ионов [Ni2+] в исходном
растворе, то в соответствии с выражением и величиной константы нестойкости
комплексного иона [Ni(NH3N]2+ (см. табл. П.4.12) получают:
[Ni2+][NH3]6 _x-x6 9
Кн 1,оо • 10 ,
[[Ni(NH3N]2+] 0,1
[Ni2+] = л/1,86 10~9 0,1 = 4,07 • 10 моль/л.
NiCO3 выпадет.
Так как CN.2+Cc(J. = 4,07 • 10 • 1,0 • 10 = 4,07 • 10 > ПРК1СОз, осадок
Задача 8. Значения удельной электропроводности и предельной молярной электро-
электропроводности насыщенного раствора фторида магния MgF2 при Т= 298 К соответст-
соответственно равны: х = 2,54 • 10~2 См/м и ^°° = 2,17 • 10~2 См • м2/моль. Рассчитать произве-
произведение растворимости nPMgF и изменение энергии Гиббса ASG в процессе растворения.
Решение. В насыщенном растворе малорастворимой соли концентрация ионов
очень мала, поэтому молярная электропроводность практически совпадает с моляр-
молярной электропроводностью при бесконечном разбавлении, т.е. X = X00. Следователь-
Примеры решения задач 493
но, используя уравнение A4.44), по данным значениям х и Xе0 можно рассчитать
молярную концентрацию насыщенного раствора, или растворимость S, при Т= 298 К:
S= —?- = 2>54Л°2 « U7 • 10 моль/л.
10V 103-2,17 10~2
Исходя из уравнения равновесного процесса растворения соли
MgF2 (T) <=> MgF2 (p) = Mg2+ + 2F"
можно рассчитать произведение растворимости ПР по A4.36) и A4.37):
nPMgF2 = [Mg2+][F-]2 = SBSJ= 453 = 4A,17 • 1(Г3K= 6,4 • 10'9.
Изменение энергии Гиббса в процессе растворения в соответствии с A4.38)
ASG = -RTln ( nPMgF2 ) = -8,31 • 298 • In F,4 • 10"9) * 46731,9 Дж * 46,73 кДж.
Задача 9. Определить рН и константу диссоциации Ка 0,001М раствора муравьиной
кислоты НСООН, если удельная электропроводность этого раствора х =
= 1,41-КГ2 См/к
Решение. Для расчета значений рН и Ка необходимо определить степень диссоциа-
диссоциации а слабого электролита — муравьиной кислоты в данном растворе:
НСООН <=й Н+ + НСОО"
В соответствии с A4.49)
где X — молярная электропроводность раствора электролита данной концентрации;
Xх — молярная электропроводность электролита при бесконечном разбавлении. Вели-
Величина X связана со значением удельной электропроводности х соотношением A4.44):
103С
и равна соответственно
X = -—¦ = 1,41 • 10 См • м2/моль = 141 • 10" См • м2/моль.
10310
Величину А.00 можно определить, используя A4.46):
494
Свойства водных растворов электролитов
где Х+00 и А,_°° — предельные молярные электропроводности (предельные под-
подвижности) катионов и анионов, соответственно. Значения предельных подвиж-
ностей ионов (см. табл. П.4.9): Г°(Н+) = 349,8 • КГ4 См • м2/моль, Г^НСОО) =
= 56,7 • КГ4 См • м2/моль. Следовательно,
Г(НСООН) = Г(Н+) + Г(НСОСГ) = 349,8 • КГ4 + 56,7 • КГ4 =
= 406,5-1(Г4См-м2/моль.
Степень диссоциации муравьиной кислоты в данном растворе по A4.49)
<х =
14Ы(Г
406,5 10"
= 0,346.
Тогда
рН = - lg [H+]= -lg (<хС) = - lg@,346 • 0,001) = 3,46.
Константа диссоциации в соответствии с A4.2):
a" ^ 0,346"
1-а
-С =
1-0,346
:0,001 = 1,83 • КГ4.
Задачи для самостоятельного решения
1-10. Вычислите ионную силу / и активность ионов а, в растворе, содержащем два
сильных электролита заданных молярных концентраций С, считая плотность рас-
раствора равной плотности воды р = 1000 г/л.
№
1
2
3
4
5
6
7
8
9
10
Электролит 1
KNO3
KNO3
NaCl
Pb(NO3J
FeCl3
ВаС12
Na2SO4
HgCl2
NH4CI
MgSO4
С1, моль/л
0,005
0,004
0,003
0,002
0,001
0,001
0,002
0,003
0,004
0,005
Электролит 2
K2SO4
Ca(NO3J
ZnCl2
Pb(CH3COOHJ
MgCl2
Ba(NO3J
ZnSO4
A1C13
(NH4JSO4
A12(SO4K
C2, моль/л
0,001
0,002
0,003
0,004
0,005
0,004
0,003
0,001
0,005
0,002
11-20. Вычислите молярную концентрацию С, молярную концентрацию химиче-
химического эквивалента Сэкв и массовую концентрацию Смасс сильной кислоты или силь-
сильного основания, раствор которого имеет заданное значение рН.
Задачи для самостоятельного решения
495
№>
И
12
13
14
15
Сильная кислота
НС1
НС1
H2SO4
H2SO4
HNO3
рН
1,0
2,0
3,0
4,0
5,0
16
17
18
19
20
Сильное основание
NaOH
NaOH
КОН
Ва(ОНJ
Ва(ОНJ
рН
13,0
12,0
11,0
10,0
9,0
21-30. Используя табл. ПАЮ и принимая степень диссоциации сильного электро-
электролита равной 1, вычислите, как изменится рН раствора слабой кислоты или основа-
основания заданной концентрации С, к 1 л которого добавили соль массой т.
№
21
22
23
24
25
26
27
28
29
30
Исходный раствор
Слабый электролит
СНзСООН
НВЮ
НСООН
HNO2
H2S
NH4OH
Cd(OHJ
Со(ОН)г
Mg(OHJ
Mn(OHJ
С, моль/л
ю-'
ю-2
10-'
ю-2
ю-3
ю-'
ю-5
кг6
ю-4
ю-5
Соль
Сильный электролит
СНзСООК
NaBrO
НСООК
NaNO2
Na2S
NH4CI
CdCl2
СоС12
MgCl2
MnCl2
/и, г
9,8
1,2
8,4
6,9
7,8
5,3
1,8
1,7
1,0
1,3
31-40. Укажите, какие из приведенных ниже солей подвергаются гидролизу, напи-
напишите сокращенные ионно-молекулярные уравнения гидролиза, укажите реакцию
среды.
№
31
32
33
34
35
Соли
СиС12, ВаС12, Са(СН3СООJ,
Pb(NO3J
NaClO4,(NH4JSO4,(NH4JS,
KCN
Na3As04, COSO4, AICI3, CaSO4
CrCl3, K2SO4, Na2S, Zn(NO3J
KI, NH4Br, Na3PO4, ZnSO4
№
36
37
38
39
40
Соли
MnCl2, (NH4JCO3, NaHCO3,
Sr(NO3J
Rb2S, KNO3, Li2SO4, Na2HPO4
Mg(CH3COOJ, SnCl2, NH4NO3,
CsCl
NaHS, Li2SO3, Rb2SO4, Bi(NO3K
BaS, A12(SO4K, HgCl2, Cr(NO3K
41—50. Напишите уравнения гидролиза и, используя данные табл. П.4.10, вычислите
константу гидролиза КГ, степень гидролиза аг и рН водных растворов солей задан-
заданной концентрации С.
496
Свойства водных растворов электролитов
№
41
42
43
44
45
Соль
CH3COONa
НСООК
KF
NaHS
КНСОз
С, моль/л
ю-'
ю-2
ю-'
ю-'
ю-2
№
46
47
48
49
50
Соль
Pb(NO3J
K2SO3
К2С2О4
Na3PO4
ZnSO4
С, моль/л
10"'
10"'
ю-2
10"'
ю-'
51-65. Вычислите объем воды, необходимый для растворения 1 г малорастворимого
электролита, используя табл. П.4.11.
№
51
52
53
54
55
Малорастворимый
электролит
Ag2Cr207
BaSO4
СаСО3
Со(ОНJ
CuCN
№
56
57
58
59
60
Малорастворимый
электролит
Fe(OHJ
HgS
Mg(OHJ
МпСОз
Ni(OHJ
№
61
62
63
64
65
Малорастворимый
электролит
РЬС12
Pb(OHJ
Snl2
Sr(OHJ
Zn(OHJ
66-75. Используя табл. П.4.11 и П.4.12, определите, выпадет ли осадок при смеше-
смешении равных объемов двух растворов электролитов заданных концентраций.
№
66
67
68
69
70
71
72
73
74
75
Электролит 1
[Ag(NH3J]Cl
K2[Ag(OHK]
[Cd(NH3N]Cl2
[Co(NH3N]Cl2
[Co(C5H5NJ]Cl3
[Cu(NH3L]Cl2
[Hg(NH3L]Cl2
K3[Fe(CNN]
K[Pb(OHK]
[Zn(NH3L]Cl2
C\, моль/л
2 • 10"'
4 • 10
2 • 10
2 10
2 • 10"'
2 • 10"'
2 • 10
6 • 10"'
4 • 10'2
6IO-4
Электролит 2
KBr
K2SO4
K2CO3
K2C2O4
K2CO3
K2S
K2S
K2C2O4
K2SO4
K2CO3
C2, моль/л
2 • 10"'
1 • 10"'
4 • 10"'
2- 10
2--10"'
4 • 10
2 • 10
2 • 10~2
2 • 10"'
2 • 10"'
76-90. Определите рН насыщенного раствора малорастворимого электролита, ис-
используя табл. П.4.11.
№
76
77
78
79
80
Электролит
AgOH
Ва(ОНJ
LiOH
Са(ОНJ
Cd(OHJ
№
81
82
83
84
85
Электролит
Со(ОНJ
СиОН
Fe(OHJ
Mg(OHJ
Mn(OHJ
№
86
87
88
89
90
Электролит
Ni(OHJ
Pb(OHJ
Cr(OHJ
Sr(OHJ
Zn(OHJ
Задачи для самостоятельного решения
497
91-98. Вычислите произведение растворимости ПР и изменение энергии Гиббса ASG
в процессе растворения малорастворимого электролита при Т = 298 К, если извест-
известны удельная электропроводность х и предельная молярная электропроводность А,00
его насыщенного раствора (см. табл. П.4.8).
№
91
92
93
94
Электролит
Т1С1
SrSO4
CaF2
BaF2
№
95
96
97
98
Электролит
РЬС12
PbBr2
РЫ2
AgCl
99-106. Используя табл. П.4.9, определите рН и константу диссоциации Kd(Q слабо-
слабого электролита, молярная концентрация С и удельная электропроводность х водно-
водного раствора которого заданы.
№
99
100
101
102
103
104
105
106
Электролит
NH4OH
NH4OH
NH4OH
СН3СООН
СН3СООН
СН3СООН
С2Н5СООН
С2Н5СООН
С, моль/л
0,001
0,01
0,1
0,001
0,01
0,1
0,16
0,0312
х-102,См/м
28,0
9,6
3,3
41,0
14,3
4,6
5,65
2,43
107. При определении общей жесткости воды необходимо поддерживать рН в тит-
титруемой пробе равным 10. Рассчитайте концентрации ионов Н+ и ОН", а также гид-
гидроксидный показатель рОН пробы.
108. Рассчитайте рН производственных сточных вод, а также концентрации ионов
Н+ и ОН", если их гидроксидный показатель рОН = 2.
109. Водородный показатель рН дистиллированной воды, контактирующей с возду-
воздухом, при Т= 298 К меньше 7 и составляет 6,0. Объясните этот факт, а также рассчи-
рассчитайте гидроксидный показатель рОН и молярные концентрации Н+ и ОН+ в такой
воде.
498 Свойства водных растворов электролитов
ЛР 9. Растворы электролитов
Цель работы — ознакомление с процессами, протекающими в водных рас-
растворах электролитов, изучение таких явлений и понятий, как электропроводность,
гидратация, подавление диссоциации слабого электролита сильным электролитом,
ионное произведение воды, водородный и гидроксидный показатели, гидролиз,
произведение растворимости, сравнение свойств растворов сильных и слабых элек-
электролитов, гомогенных и гетерогенных равновесий в растворах электролитов и ком-
комплексных соединений.
Опыт 1. Гидратация сильного электролита. Как известно, образование
водных растворов сопровождается процессом гидратации. Гидратация особенно
сильно проявляется в водных растворах электролитов вследствие взаимодействия
ионов с полярными молекулами воды. Схематично процесс гидратации электролита
можно представить в виде:
KZl+ + А22" + (р + q)H2O <± KZl+ • рН2О + А72" • qH2O
В данном опыте предлагается убедиться в том, что гидратация электролита
протекает более интенсивно, чем неэлектролита, что приводит к выделению по-
последнего из раствора при добавлении к нему сильного электролита. Для сравнения
используется насыщенный раствор анилина C6H5NH2, гидратация которого обу-
обусловлена диполь-дипольным взаимодействием полярных молекул воды с полярны-
полярными молекулами C6H5NH2. Если к данному раствору добавить кристаллы сильного,
хорошо растворимого электролита, например NaCl, то в результате гидратации по-
последнего из насыщенного раствора будет выделяться анилин в свободном виде.
Процесс гидратации NaCl в насыщенном водном растворе анилина можно предста-
представить следующим образом:
C6H5NH2 • «Н2О + NaCl ^± C6H5NH2 + Na+ • pU2O + СГ • qH2O
где п = р + q.
Внимание! Опыт выполняется в вытяжном шкафу
Последовательность проведения:
1) налейте в пробирку 3.. .5 мл прозрачного насыщенного водного раствора анилина;
2) добавьте примерно 1 г измельченного кристаллического NaCl.
3) перемешайте содержимое пробирки, встряхнув ее, дайте отстояться;
4) отметьте происходящие изменения, наблюдайте образование и расслоение эмуль-
эмульсии (выделение на поверхности раствора свободного анилина).
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы
занесите в табл. Л.9.1;
2) объясните причины выделения анилина (C6H5NH2) на поверхности раствора. По-
Почему равновесие гидратации анилина в воде смещается влево (в сторону образова-
образования свободного C6H5NH2)?
ЛР 9. Растворы электролитов
499
Результаты эксперимента
Таблица Л.9.1
Исходные данные
Исследуемая равновесная
система
Насыщенный раствор
анилина в воде
Добавляемый сильный электролит
Кристаллический NaCl
Уравнения
гидратации
анилина в воде
гидратации NaCl
вводе
гидратации NaCl
в насыщенном водном
растворе анилина
Наблюдения
Выводы
Опыт 2. Зависимость электропроводности растворов от степени диссоциа-
диссоциации электролитов. Общим свойством растворов электролитов является их способность
проводить электрический ток. Количество электричества, переносимое в растворе элек-
электролита за единицу времени, определяется количеством ионов (носителей заряда) и их
подвижностью. Количество носителей заряда зависит от концентрации электролита и
степени его диссоциации. Очевидно, что растворы сильных электролитов, практически
полностью диссоциирующих на ионы, лучше проводят электрический ток, чем раство-
растворы слабых электролитов, степень диссоциации которых мала.
В данном опыте предлагается сравнить электропроводность растворов различ-
различных электролитов одинаковой концентрации @,1н.) и сделать соответствующие выво-
выводы. Для измерения электропроводности растворов используют установку (рис. Л.9.1),
V77/J
777/j
—0
-220 В
7777А
Рис. Л.9.1. Прибор для измерения электрической проводимости растворов:
1 — электроды; 2 — миллиамперметр; 3 — выпрямитель тока; 4 — трансформатор
500
Свойства водных растворов электролитов
СО
h2so4
сн3соон
Для
электродов
оо
0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9
ооооооооо
ооооооооо
ОДн. растворы
О О О О
НС1 СН3СООН NaOH NH4OH
и
Н2°диСТ
Рис. Л.9.2. Штатив с пробирками для измерения электрической про-
проводимости растворов (вид сверху)
состоящую из трансформатора 4, выпрямителя тока 3, миллиамперметра 2 и двух элек-
электродов 1. В штативе с пробирками (рис. Л.9.2) находятся водные растворы сильных и
слабых электролитов, занимающие 2/3 объема пробирки.
Последовательность проведения:
1) налейте в стакан дистиллированную воду, приготовьте фильтровальную бумагу;
2) включите источник питания установки для измерения электропроводности рас-
растворов (см. рис. Л.9.1);
3) поместите два электрода в пробирку с 0,1н. раствором соляной кислоты НС1.
(Электроды не должны касаться друг друга.);
4) запишите показания миллиамперметра в табл. Л.9.2;
Таблица Л.9.2
Результаты эксперимента
Исходные данные
Электролит
НС1
СНзСООН
NaOH
NH4OH
Сэкв,
моль экв/л
0,1
ол
0,1
0,1
Результаты измерений
и расчетов
Показания
амперметра
Сила тока
/, мА
Уравнение
диссоциации
Вывод
5) выньте электроды, опустите в стакан с дистиллированной водой, затем осушите
бумажной салфеткой;
6) повторите пп. 3-5, опуская электроды поочередно в пробирки с ОДн. растворами
СНзСООН, NaOH и NH4OH;
ЛР 9. Растворы электролитов 501
7) отключите источник питания установки, закройте штатив с пробирками стеклян-
стеклянной пластиной.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов, уравнения диссоциации
электролитов и выводы занесите в табл. Л.9.2;
2) учитывая размерность шкалы амперметра, рассчитайте силу тока /;
3) составьте уравнения диссоциации соответствующих электролитов;
4) объясните, почему электролиты, имеющие одинаковую молярную концентрацию
эквивалента, проводят электрический ток по-разному;
5) какие из изученных растворенных веществ можно отнести к сильным, а какие к сла-
слабым электролитам? Что можно использовать в качестве критерия силы электролита?
6) выпишите из табл. П.4.10 значения констант диссоциации Kd{C) исследованных
электролитов. Почему значения A^(q для НС1 и NaOH в справочниках не приводятся?
Опыт 3. Зависимость электропроводности раствора от концентрации
электролита. С увеличением числа носителей зарядов электропроводность раство-
растворов электролитов должна возрастать. Однако при увеличении концентрации степень
диссоциации слабых электролитов уменьшается, что приводит к снижению величи-
величины электропроводности в растворах значительных концентраций. В растворах
сильных электролитов увеличение концентрации приводит к усилению ионных
взаимодействий и снижению подвижности носителей заряда, вследствие чего, на-
начиная с некоторой концентрации (индивидуальной для каждого электролита) элек-
электропроводность начинает уменьшаться по мере роста концентрации раствора.
Влияние концентрации электролита на электрическую проводимость раство-
раствора предлагается исследовать на примере растворов сильной кислоты H2SO4 и слабой
кислоты СН3СООН различных концентраций (массовые доли со электролита в рас-
растворах соответственно равны 0,1; 0,2; 0,3; 0,4; 0,5; 0,6; 0,7; 0,8; 0,9) с помощью ус-
установки, использованной в опыте 2.
Последовательность проведения:
1) выполните пп. 1-5 опыта 2, погружая поочередно электроды в пробирки с рас-
растворами серной кислоты H2SO4 с массовыми долями со от 0,1 до 0,9;
2) повторите пп. 1-5 опыта 2, погружая поочередно электроды в пробирки с раство-
растворами уксусной кислоты СН3СООН с массовыми долями со от 0,1 до 0,9;
3) отключите установку для измерения электропроводности от источника питания;
4) закройте штатив с пробирками стеклянной пластиной.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов, уравнения диссоциации
электролитов, графики и выводы занесите в табл. Л.9.3;
2) учитывая размерность шкалы амперметра, рассчитайте величину силы тока /;
3) по результатам расчетов постройте графики зависимости силы тока от концен-
концентрации электролита /=/(со) для каждой из кислот;
4) объясните полученные зависимости силы тока от концентрации раствора сильно-
сильного электролита и от концентрации раствора слабого электролита. Какие явления
способствуют уменьшению электропроводности растворов сильных электролитов
при высоких концентрациях? Почему электропроводность растворов слабых элек-
электролитов при высоких концентрациях также уменьшается?
502
Свойства водных растворов электролитов
Результаты эксперимента
Таблица Л. 9.3
Исходные
данные
со
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
Результаты измерений
и расчетов
H2SO4
Показания
амперметра
/, мА
СНзСООН
Показания
амперметра
/,мА
Графиче-
Графические
зависимо-
зависимости
/= f(<o)
Выводы
Опыт 4. Образование слабодиссоциирующих веществ. Обменные реакции
в растворах электролитов протекают в направлениях связывания ионов, приводя-
приводящих к образованию слабодиссоциирующих веществ, осадков, выделению газов. В
данном опыте предлагается установить направление протекания реакций в раство-
растворах электролитов.
Последовательность проведения:
1) налейте в пробирку 2-3 мл раствора NH4C1;
2) прибавьте 1-2 мл раствора едкого натра NaOH;
3) перемешайте, определите по запаху, какой газ выделяется;
4) налейте в другую пробирку 5 мл 1н. раствора НС1 и поместите в нее кусочек Zn;
5) после достижения равномерного выделения водорода прибавьте в пробирку при-
примерно 0,5 г кристаллического CH3COONa (или СН3СООК) и тщательно перемешай-
перемешайте содержимое, встряхивая пробирку;
6) наблюдайте уменьшение интенсивности выделения водорода и появление нового
запаха.
Обработка результатов:
1) исходные данные, наблюдения, молекулярные и ионные уравнения всех процес-
процессов, а также выводы занесите в табл. Л.9.4;
Результаты эксперимента
Таблица Л.9.4
Исходные данные
Реагенты
NH4C1 + NaOH
HCl + Zn
НС1 + CH3COONa
Наблюдения
Уравнения реакций
Выводы
ЛР 9. Растворы электролитов
503
2) объясните причины появления запаха аммиака, замедления растворения цинка и
появления запаха уксуса. О чем свидетельствуют величины констант диссоциации:
?b(NH4OH) = 1,8 • 10-5;?а(СН3СООН) = 1,7 • 10?
Опыт 5. Определение рН растворов с помощью универсального индика-
индикатора. На основании приближенных измерений рН растворов кислот и оснований в
данном опыте предлагается сделать выводы о способности к диссоциации и рассчи-
рассчитать константы диссоциации различных электролитов.
В лабораторной работе 1 (опыт 7) для оценки кислотности и щелочности сре-
среды студенты уже использовали универсальный индикатор, меняющий свою окраску
во всем диапазоне значений рН. В табл. Л.9.5а приведены значения рН растворов,
которые соответствуют определенной окраске универсального индикатора.
Таблица Л. 9.5а
Цвет универсального индикатора (раствора или бумаги)
рН
1-3
4-5
6
7
Цвет
Красный
Оранжевый
Желтый
Зеленый
рН
8
9-10
11-13
Цвет
Бирюзовый
Голубой
Фиолетовый
Более точное соответствие значений рН оттенкам окраски индикатора приво-
приводится на цветовой шкале, прилагаемой к набору универсальной индикаторной бу-
бумаги.
Последовательность проведения:
1) налейте в чистую пробирку 1-2 мл 0,1 М раствора НС1;
2) добавьте в пробирку несколько капель универсального индикатора или нанесите
на полоску универсальной индикаторной бумаги каплю исследуемого раствора. От-
Отметьте полученный цвет раствора (или бумаги) в табл. Л.9.56;
Таблица Л. 9.56
Результаты эксперимента
Исходные данные
Электролит
НС1
СНзСООН
NaOH
NH4OH
с,
моль/л
Наблюдения
Окраска
индикатора
рН
Результаты расчетов
[Н+]
([ОН"])
Выражение и
значение K^q
—
Уравнение
диссоциации
Вывод
3) повторите пп. 1-2, наливая в чистую пробирку поочередно 0,1М растворы
CHjCOOH, NaOH, NH4OH.
504 Свойства водных растворов электролитов
Обработка результатов:
1) исходные данные, наблюдения, результаты расчетов, уравнения диссоциации
электролитов и выводы занесите в табл. Л.9.56;
2) сравните окраску исследованных растворов (или индикаторной бумаги) с цвето-
цветовой шкалой индикатора (или данными табл. Л.9.5а) и определите рН раствора;
3) составьте уравнения диссоциации данных электролитов;
4) сравните значение рН 0,1М раствора НС1 с величиной рН 0,1М раствора СН3СООН.
Сделайте вывод о силе уксусной кислоты;
5) зная рН раствора и исходную концентрацию кислоты, рассчитайте равновесную
молярную концентрацию ионов водорода [Н+] и константу диссоциации Kd(Q
СН3СООН, напишите ее выражение;
6) сравните значение рН 0,1М раствора NaOH с величиной рН 0,1М раствора NH4OH.
Сделайте вывод о силе гидроксида аммония;
7) зная рН раствора и исходную концентрацию основания, рассчитайте равновес-
равновесную молярную концентрацию гидроксид-ионов [ОЬГ] и константу диссоциации
Kd(c) NH4OH, напишите ее выражение;
8) сравните полученные вами значения Kd(Q Для СН3СООН и NH4OH с приведен-
приведенными в опыте 4 справочными данными.
Опыт 6. Подавление диссоциации слабого электролита. При внесении в
раствор слабого электролита одноименных ионов диссоциация слабого электролита
подавляется в соответствии с принципом Ле Шателье. Это явление предлагается
изучить, наблюдая изменение концентрации ионов Н+ и ОН". Изменение концен-
концентрации ионов можно определить по изменению окраски индикаторов метилового
оранжевого и фенолфталеина, для чего используйте данные табл. 2.4, приведенные
в гл. 2.
Последовательность проведения:
1) налейте в пробирку 2-3 мл 0,1 н. раствора уксусной кислоты;
2) прибавьте 1-2 капли индикатора метилового оранжевого. Отметьте окраску ин-
индикатора в растворе СН3СООН;
3) добавьте примерно 1 г кристаллического ацетата натрия CH3COONa, перемешайте
содержимое пробирки, встряхивая ее. Наблюдайте изменение окраски индикатора;
4) повторите пп. 1-3, используя в качестве слабого электролита 0,1н. раствор
NH4OH, в качестве индикатора фенолфталеин, в качестве сильного электролита
кристаллическую соль NH4C1 в тех же соотношениях.
Обработка результатов:
1) исходные данные, наблюдения, уравнения диссоциации электролитов и выводы
занесите в табл. Л.9.6а и Л.9.66.
2) на основании изменения окраски индикатора метилового оранжевого сделайте
вывод об изменении (увеличении или уменьшении) значения рН раствора
СН3СООН после добавления соли CH3COONa. Укажите, как изменилась концен-
концентрация ионов водорода;
3) на основании изменения окраски индикатора фенолфталеина сделайте вывод об
изменении (увеличении или уменьшении) значения рН раствора NH4OH после до-
добавления соли NH4C1. Укажите, как изменилась концентрация гидроксид-ионов;
ЛР 9. Растворы электролитов
505
Результаты эксперимента
Таблица Л. 9.6а
Исходные данные
Слабый
электролит
СНзСООН
Сильный
электролит
CH3COONa
Уравнение
диссоциации
СН3СООН
Наблюдения
Исходная
окраска
раствора
с метил-
метилоранжем
Выражение Kd{c)
СН3СООН
Конечная окра-
окраска раствора с
метилоранжем
Значение Kd^Q
СН3СООН
1,7- 10~5
Измене-
Изменение зна-
значения рН
Уравнение диссо-
диссоциации CH3COONa
Изменение
концентра-
концентрации ионов
Н+
Вывод
Результаты эксперимента
Таблица Л.9.66
Исходные данные
Слабый
электролит
NH4OH
Сильный
электролит
NH4C1
Уравнение
диссоциации
NH4OH
Наблюдения
Исходная окра-
окраска раствора с
фенолфталеи-
фенолфталеином
Выражение
Kd(Q NH4OH
Конечная окра-
окраска раствора с
фенолфталеином
Значение Kd{c)
NH4OH
1,8 1(Г5
Измене-
Изменение зна-
значения рН
Уравнение диссо-
диссоциации NH4C1
Изменение
концентра-
концентрации ионов
ОН"
Вывод
4) составьте уравнения диссоциации сильных электролитов, уравнения диссоциации
и выражения констант диссоциации слабых электролитов;
5) объясните причины изменения окраски индикаторов, используя принцип Ле Ша-
телье—Брауна и выражения констант диссоциации слабых кислоты и основания;
6) сделайте вывод о влиянии на равновесие диссоциации слабого электролита одно-
одноименных ионов сильного электролита.
Опыт 7. Гидролиз солей. Гидролиз соли — обменная реакция соли с водой,
в результате которой изменяется значение рН среды. Гидролиз протекает, если в
результате взаимодействия образуется слабый электролит (слабая кислота или сла-
слабое основание).
В данном опыте предлагается изучить гидролиз соли, образованной сильным
основанием и слабой кислотой, и соли, образованной слабым основанием и сильной
кислотой. Убедиться в протекании гидролиза и определить рН растворов солей
можно с помощью универсального индикатора (см. опыт 5).
506
Свойства водных растворов электролитов
Последовательность проведения:
1) налейте в пробирку 2-3 мл дистиллированной воды;
2) насыпьте в пробирку примерно 0,5 г кристаллического №2СОз, тщательно
встряхните;
3) добавьте 1-2 капли универсального индикатора (или нанесите на полоску инди-
индикаторной бумаги каплю исследуемого раствора). Отметьте окраску универсального
индикатора в растворе или цвет бумаги;
4) повторите пп. 1-3, прибавив в другую пробирку с дистиллированной водой при-
примерно 0,5 г кристаллического A12(SO4K.
Обработка результатов:
1) исходные данные, наблюдения, результаты расчетов, уравнения гидролиза солей
и выводы занесите в табл. Л.9.7;
Результаты эксперимента
Таблица Л.9.7
Исходные
данные
Соль
Na2CO3
A12(SO4K
Наблюдения
Окраска
универсального
индикатора
Характер
среды, рН
раствора
Результаты
расчета
Значение
Уравнение
реакции
гидролиза
Вывод
2) составьте молекулярные и ионные уравнения гидролиза солей;
3) укажите характер среды и величины рН водных растворов солей, определенные
по цветовой шкале индикатора (см. табл. Л.9.5а);
4) рассчитайте значения констант гидролиза солей по I и II ступеням, используя
значения констант диссоциации по I и II ступеням для соответствующих слабых
кислоты и основания, приведенные в табл. П.4.10;
5) на основании наблюдений и расчетов сделайте вывод, какая соль сильнее подвер-
подвергается гидролизу.
Опыт 8. Взаимодействие растворов солей, взаимно усиливающих гидро-
гидролиз. При смешении растворов солей, одна из которых гидролизуется по катиону, а
другая по аниону, происходит взаимное усиление гидролиза, в результате чего об-
образуются осадки слабых оснований и растворы слабых кислот (Н2СОз, H2S). В дан-
данном опыте предлагается убедиться в невозможности существования в водном рас-
растворе карбонатов и сульфидов алюминия и хрома.
Последовательность проведения:
1) налейте в пробирку 1-2 мл 0,1 М раствора хлорида или сульфата алюминия (А1СЬ
или A12(SO4K);
2) нанесите на полоску универсальной индикаторной бумаги каплю исследуемого
раствора. Отметьте окраску индикатора;
3) во вторую пробирку налейте 1-2 мл 0,1 М раствор Na2CO3 и также испытайте рас-
раствор универсальным индикатором;
ЛР 9. Растворы электролитов
507
4) аккуратно прилейте содержимое второй пробирки к первой до образования осадка;
5) повторите пп. 1-4, при этом в одну пробирку налейте 1-2 мл 0,1 М раствора хлорида
или сульфата хрома (СгС13 или Сг2(8О4)з), а в другую — 1-2 мл 0,1 М раствор Na2S;
6) экспериментально докажите, что в результате самоусиливающегося гидролиза
образуются амфотерные гидроксиды алюминия или хрома. Для этого разделите ка-
каждый из осадков на две части. К одной части прилейте избыток кислоты НС1 или
H2SO4, а к другой — избыток щелочи NaOH или КОН. Растворение осадков и в том,
и в другом случае свидетельствует об их амфотерности. (Выполняется по указанию
преподавателя.)
Обработка результатов:
1) исходные данные, наблюдения, уравнения реакций и выводы занесите в табл. Л.9.8;
Таблица Л. 9.8
Результаты эксперимента
Исходные
данные
Соль
А1С13
Na2CO3
СгС13
Na2S
Наблюдения
Окраска универ-
универсального индикатора
рН раствора
соли
Уравнение реакции
гидроли-
гидролиза соли
взаимодей-
взаимодействия солей
Вывод
2) составьте уравнения гидролиза реагирующих солей. О чем свидетельствует окра-
окраска универсального индикатора?
3) составьте уравнения реакций взаимодействия солей. О чем свидетельствует вы-
выпадение осадков?
4) объясните смещение равновесия реакции гидролиза в данных случаях, используя
принцип Ле Шателье—Брауна. Возможно ли усиление гидролиза, если обе соли
гидролизуются по катиону? (по аниону?) Рассмотрите смещение равновесия при
совместном гидролизе А1С13 и CrCl3, Na2CO3 и Na2S;
5) сделайте вывод, почему невозможно существование в водном растворе карбона-
карбонатов и сульфидов алюминия и хрома?
6) из табл. П.4.5 выпишите формулы солей, полностью гидролизующихся в водном
растворе, и предложите пары солей, взаимно усиливающих гидролиз;
7) составьте уравнения реакций, подтверждающих амфотерность гидроксидов алю-
алюминия и хрома.
Опыт 9. Сравнение характера гидролиза по катиону солей s- и */-метал-
лов. Поляризация (поляризуемость и поляризующее действие) ионов определяет
вероятность и характер протекания гидролиза солей. Чем больше взаимная поляри-
поляризация катионов и молекул воды, тем интенсивнее протекает гидролиз по катиону.
Также чем больше взаимная поляризация анионов и молекул воды, тем интенсивнее
протекает гидролиз по аниону.
508
Свойства водных растворов электролитов
Известно, что поляризующее действие катионов rf-металлов больше, чем по-
поляризующее действие катионов л'-металлов одинаковых зарядов и близких радиусов.
В особенности велико поляризующее действие катионов, имеющих 18-электронную
оболочку. В данном опыте предлагается выяснить, какие соли больше подвергаются
гидролизу: 5- или ^-металлов, и сделать соответствующие выводы.
Последовательность проведения:
1) налейте в чистую пробирку 2-3 мл дистиллированной воды;
2) насыпьте в пробирку несколько кристалликов Са(>ЮзJ, перемешайте содержи-
содержимое, встряхнув пробирку;
3) добавьте 2-3 капли универсального индикатора (или лакмуса) или нанесите на
полоску универсальной индикаторной бумаги каплю исследуемого раствора. От-
Отметьте окраску индикатора в растворе или цвет бумаги;
4) повторите пп. 1-3, добавив в другую пробирку с дистиллированной водой не-
несколько кристалликов Zn(NO3J.
Обработка результатов:
1) исходные данные, наблюдения, уравнения реакций гидролиза и выводы занесите
в табл. Л.9.9;
2) в соответствии с окраской индикатора определите значение рН и характер среды,
используя данные табл. Л.9.5а (или табл. 2.4, если в качестве индикатора применяли
лакмус);
3) составьте и запишите в табл. Л.9.9 уравнения гидролиза в ионной и молекулярной форме;
4) какая соль подвергается гидролизу? Объясните, почему одинаково заряженные
катионы s- и rf-металлов ведут себя по-разному. Сравните константы диссоциации
гидроксидов Са(ОНJ и Zn(OHJ, приведенные в табл. П.4.10. Сделайте вывод.
Результаты эксперимента
Таблица Л.9.9
Исходные
данные
Соль
Ca(NO3J
Zn(NO3J
Наблюдения
Окраска
индикатора
Характер среды,
рН раствора
Уравнение реакции
гидролиза
Вывод
Опыт 10. Сравнение гидролиза средних и кислых солей. Соли многоос-
многоосновных кислот и многокислотных оснований подвергаются гидролизу ступенчато,
при этом Кг{щ « Kr(ii)« КГ({у Исследуя растворы средних и кислых солей, можно
убедиться, что гидролиз по II (и III) ступени средних солей не вносит существенно-
существенного вклада в значение рН водных растворов солей.
Последовательность проведения:
1) налейте в одну пробирку 1-2 мл 1н. раствора NaH2PO4, в другую — такой же
объем 1н. раствора Na2HPO4, в третью — 1н. раствора Na3PO4;
ЛР 9. Растворы электролитов
509
2) добавьте в каждую пробирку по 2-3 капли универсального индикатора или нане-
нанесите на полоску универсальной индикаторной бумаги каплю исследуемого раство-
раствора. Отметьте окраску индикатора в пробирках или цвет бумаги в табл. Л.9.10.
Обработка результатов:
1) исходные данные, наблюдения, результаты расчетов, уравнения реакций гидро-
гидролиза и выводы занесите в табл. Л.9.10;
Результаты эксперимента
Таблица Л. 9.10
Исходные
данные
Соль
NaH2PO4
Na2HPO4
Na3PO4
Наблюдения
Окраска
индикатора
рН
раствора
Результаты
расчетов
Значение Кг
Уравнение
реакции гид-
гидролиза
Вывод
2) по цветовой шкале универсального индикатора (см. табл. Л.9.5а) определите зна-
значения рН и характер среды растворов солей;
3) составьте ионные и молекулярные уравнения гидролиза исследованных солей;
4) рассчитайте константы гидролиза солей и объясните наблюдаемые явления, ис-
используя значения констант диссоциации для Н3РО4, приведенные в табл. ПАЮ.
Сделайте вывод.
Опыт 11. Влияние температуры на степень гидролиза. В данном опыте
необходимо выяснить, как температура влияет на смещение равновесия реакции
гидролиза, и сделать вывод о знаке теплового эффекта процесса.
Последовательность проведения:
1) налейте в пробирку 1-2 мл 0,1н. раствора ацетата натрия CH3COONa;
2) прибавьте 1-2 капли фенолфталеина (или универсального индикатора). Переме-
Перемешайте содержимое раствора, встряхнув пробирку. Отметьте окраску индикатора в
растворе;
3) затем нагрейте раствор до кипения и вновь отметьте окраску индикатора.
Обработка результатов:
1) исходные данные, наблюдения, уравнение реакции гидролиза и вывод занесите в
табл.Л.9.11;
2) в соответствии с табл. 2.4 (или табл. Л.9.5а) по окраске индикатора определите
характер среды (и, если использовали универсальный индикатор, рН) раствора до и
после нагревания;
3) составьте ионное и молекулярное уравнения гидролиза;
4) по изменению характера среды (и рН раствора) сделайте вывод о направлении
смещения равновесия реакции гидролиза, о знаке теплового эффекта процесса и
о зависимости гидролиза от температуры в соответствии с принципом Ле Ша-
телье—Брауна.
510
Свойства водных растворов электролитов
Результаты эксперимента
Таблица Л.9.11
Исходные
данные
Соль
CH3COONa
Наблюдения
Окраска
индикатора
до на-
нагрева
после
нагрева
Характер среды
(или рН)
до на-
нагрева
после
нагрева
Уравнение
реакции
гидролиза
Знак
Дг#
Вывод
Опыт 12. Влияние продуктов гидролиза на смещение равновесия. Равно-
Равновесие реакции гидролиза можно сдвинуть в соответствии с принципом Ле Шателье
с помощью тех или иных реагентов. Предлагается выяснить направление смещения
равновесия гидролиза при добавлении кислоты к раствору соли, гидролизующейся
по катиону.
Последовательность проведения:
1) налейте в пробирку 2-3 мл дистиллированной воды;
2) добавьте немного кристаллического Pb(NO3J (или РЬ(СН3СООJ);
3) перемешайте содержимое, встряхивая пробирку. Обратите внимание на помутне-
помутнение раствора за счет образования малорастворимой основной соли РЬ2+;
4) к раствору добавьте несколько капель 0,1н раствора HNO3 (или СН3СООН). Об-
Обратите внимание на исчезновение мути.
Обработка результатов:
1) исходные данные, наблюдения, уравнения реакции гидролиза и выводы занесите
в табл. Л.9.12;
Таблица Л.9.12
Результаты эксперимента
Исходные данные
Соль
Кислота
Наблюдения
Раствор
соли
Раствор соли
с кислотой
Уравнение
реакции
гидролиза
Направление
смещения
равновесия
Вывод
2) напишите уравнение реакции гидролиза соли в ионном и молекулярном виде,
уравнение диссоциации кислоты;
3) объясните наблюдаемые изменения равновесия гидролиза с помощью принципа
Ле Шателье—Брауна. Укажите, в каком направлении смещается равновесие данной
реакции. Сделайте вывод.
Опыт 13. Влияние разбавления раствора на смещение равновесия гид-
гидролиза. С увеличением разбавления степень гидролиза соли увеличивается в соот-
соответствии с принципом Ле Шателье—Брауна. Предлагается убедиться в этом на
практике с помощью универсального индикатора.
ЛР Р. Растворы электролитов
511
Последовательность проведения:
1) налейте в мерный цилиндр 5 мл 0,5 н. раствора Bi(NO3K;
2) добавьте 2-3 капли универсального индикатора или нанесите на полоску универ-
универсальной индикаторной бумаги каплю исследуемого раствора. Отметьте окраску ин-
индикатора в растворе или цвет бумаги в табл. Л.9.13;
Таблица Л.9.13
Результаты эксперимента
Исходные
данные
Соль
Bi(NO3K
SbCl3
Наблюдения
Окраска
индикатора
ДО
разбав-
разбавления
после
разбав-
разбавления
Значение рН
ДО
разбав-
разбавления
после
разбавле-
разбавления
Уравнение реакции
гидролиза
Bi(NO3K+H2O <=±
BiONO3l + 2HNO3
SbCl3 + Н2О +=>
SbOCll + 2НС1
Направ-
Направление
смеще-
смещения
равнове-
равновесия
Вывод
3) доведите объем раствора до 10 мл, приливая в цилиндр дистиллированную воду.
Отметьте окраску индикатора после разбавления раствора;
4) повторите пп. 1-3, используя 0,5н. раствор SbCl3 (по указанию преподавателя).
Обработка результатов:
1) исходные данные, наблюдения, уравнения реакций гидролиза и выводы занесите
в табл. Л.9.13;
2) по цветовой шкале универсального индикатора (или по табл. Л.9.5а) определите
рН растворов до и после разбавления;
3) на основании значений рН сделайте вывод о смещении равновесия при разбавле-
разбавлении раствора соли, используя составленные вами уравнения гидролиза;
4) сделайте вывод о влиянии разбавления на степень гидролиза солей.
Опыт 14. Обменные реакции, протекающие с образованием осадков. Пред-
Предлагается изучить обменные реакции в растворах электролитов, протекающие с обра-
образованием осадков, и сравнить количества реагента, необходимые для выпадения
осадков солей с одноименными ионами.
При образовании осадков в растворах электролитов устанавливается гетеро-
гетерогенное равновесие, которое схематически можно представить в следующем виде:
77 (Т)
= mKz
kz,-
(p)
(р)-
Количественной характеристикой равновесия, возникающего на границе раздела
фаз твердого электролита и его насыщенного раствора, служит величина произведения
растворимости ПР, связанная с растворимостью S (моль/л) электролита соотношением:
ПР = [ KZl + П AZl~ ]" = (mS)m(nS) \
512
Свойства водных растворов электролитов
Последовательность проведения:
1) налейте в пробирку 2 мл 0,1н. раствора СаС12;
2) прибавьте в пробирку, считая капли, 0,1н. раствора H2SO4 до появления мути, т.е.
до образования осадка;
3) налейте в другую пробирку 2 мл 0,1н. раствора ВаС12;
4) повторите п. 2.
Обработка результатов:
1) исходные данные, наблюдения, результаты расчетов, уравнения протекающих
реакций и выводы занесите в табл. Л.9.14;
Таблица Л.9Л4
Результаты эксперимента
Исходные
данные
Реагенты
СаС12 +
H2SO4
ВаС12 +
H2SO4
Наблюдения
Количество
капель H2SO4
Уравнение
реакции
Выраже-
Выражение ПР
осадка
Значение
ПР осадка
(CaSO4)
3,7 • 1(Г5
(BaSO4)
1,8 - 100
Результа-
Результаты расче-
расчетов
5, моль/л
Вывод
2) составьте уравнения реакций и выражения произведений растворимости мало-
малорастворимых солей;
3) объясните наблюдаемое. Сформулируйте условия выпадения осадков. Сделайте
выводы, сравнив значения произведений растворимости обеих солей, приведенные
в табл. ПАИ.
4) рассчитайте растворимость CaSO4 и BaSO4 (в моль/л и г/л).
Опыт 15. Условия образования осадка. Осадок образуется, если произве-
произведение концентраций ионов, образующих малорастворимый электролит, превышает
величину произведения растворимости. В данном опыте исследуют условия образо-
образования осадка. Точные объемы растворов солей отмеряют с помощью мерного цилин-
цилиндра.
Последовательность проведения:
1) в два стакана налейте по 10 мл 0,1 М раствора нитрата свинца РЬ(ЫОзJ;
2) в первый стакан прилейте 10 мл 0,1 М раствора хлорида натрия (или калия);
3) во второй прилейте 9 мл дистиллированной воды и 1 мл 0,1 М раствора хлорида
натрия (или калия);
4) отметьте, в каком случае произошло образование осадка.
Обработка результатов:
1) исходные данные, наблюдения, результаты расчетов, уравнения реакции и выво-
выводы занесите в табл. Л.9.15;
ЛР 9. Растворы электролитов
Исходные данные
ста-
стакана
1
2
Реагенты
Pb(NO3J + NaCl
Pb(NO3J + NaCl
Результаты эксперимента
Наблюдения
Выпадение осадка
Уравнение реакции
513
Таблица Л.9.15
Результаты расчетов
моль/л
Выраже-
Выражение ПР
РЬС12
С ,
а
моль/л
Значение
ПР РЬС12
С 2+С2
рь2 с\~
Вывод
2) составьте уравнение реакции образования осадка и выражение ПР малораствори-
малорастворимого электролита;
3) рассчитайте концентрации ионов РЬ2+ и СГ в момент смешения. Вычислите про-
произведение концентраций ионов С 2+ С2 _ в обоих растворах и сравните их со значе-
значением ПР РЬС12, приведенным в табл. П.4.11;
4) сделайте вывод и объясните, почему выпадение осадка наблюдается в первом
стакане и не наблюдается во втором.
Опыт 16. Получение и растворение осадков. В данном опыте исследуют
обменные реакции в растворах электролитов, сопрововдающиеся образованием осадков
и слабодиссоциирующих веществ. Определяют направление протекания реакций, срав-
сравнивая константы диссоциации слабых электролитов и ПР малорастворимых солей.
Последовательность проведения:
1) в три пробирки налейте по 1-2 мл 0,1н. раствора хлорида кальция СаС12;
2) прилейте в каждую пробирку по 1-2 мл 0,1н. раствора карбоната натрия Na2CO3.
Наблюдайте выпадение осадка;
3) добавьте в одну пробирку несколько капель 0,1н. раствора НС1, в другую — 0,1н.
раствора СН3СООН, в третью — 0,1н. раствора H2SO4. Отметьте наблюдаемые яв-
явления.
Обработка результатов:
1) исходные данные, наблюдения, уравнения реакций и выводы занесите в табл. Л.9.16;
Таблица Л. 9.16
Результаты эксперимента
Исходные данные
Уравнение реакции
образования осадка
CaCl2+Na2CO3->
Кислота
НС1
СНзСООН
H2SO4
Наблюдения
Уравнение
реакции
Вывод
17 - 9795
514
Свойства водных растворов электролитов
2) составьте уравнение реакции образования осадка;
3) составьте уравнения реакций, протекающих после добавления к осадку растворов
кислот;
4) объясните наблюдавшиеся явления, используя значения констант диссоциации и
ПР соответствующих веществ. Сделайте вывод о влиянии исследованных кислот на
смещение равновесия, устанавливающего на границе раздела фаз твердого электро-
электролита и его насыщенного раствора, в соответствии с принципом Ле Шателье—
Брауна. Для ответа используйте константы диссоциации СН3СООН и Н2СО3 и про-
произведения растворимости CaSO4 и СаСО3, приведенные в табл. П.4.10 и П.4.11.
Опыт 17. Образование и разрушение комплексных соединений. В зависимости
от соотношений констант нестойкости комплексных ионов и произведений раство-
растворимости электролитов с одноименными катионами возможно как растворение осад-
осадков с образованием устойчивых комплексов, так и разрушение комплексных соеди-
соединений и выпадение осадков веществ, имеющих очень малое значение ПР.
Последовательность проведения:
1) налейте в пробирку 1 мл 0,1 М раствора CuSO4;
2) добавьте по каплям 1М раствор NH4OH до получения осадка соответствующего
гидроксида. Отметьте цвет осадка;
3) растворите полученный осадок в избытке NH4OH, добавляя его по каплям. От-
Отметьте окраску раствора;
4) добавьте по каплям 0,1М раствор Na2S до образования осадка. Отметьте цвет
осадка;
5) повторите пп. 1-4, используя поочередно в качестве исходных 0,1М растворы
NiSO4, ZnSO4, AgNO3;
6) повторите пп. 1-4, используя поочередно в качестве исходных 0,1М растворы
ZnSO4, Pb(NO3J, СгС13, а вместо NH4OH — 1М раствор NaOH.
Обработка результатов:
1) исходные данные, результаты наблюдений, уравнения соответствующих реакций
и выводы запишите в табл. Л.9.17;
Таблица Л. 9.17
Результаты эксперимента
Исходный раствор
Соль
CuSO4
NiSO4
ZnSO4
AgNO3
ZnSO4
Pb(NO3J
CrCl3
Окраска
раствора
Образование осадка гидроксида
Уравне-
Уравнение ре-
реакции
Цвет
осадка
ПР гид-
гидроксида
Растворение осадка гидроксида
Уравне-
Уравнение ре-
реакции
Окраска
раствора
Кн ком-
комплекса
ЛР10. Жесткость и умягчение воды
515
Окончание табл. Л.9.17
Исходный раствор
Соль
CuSO4
NiSO4
ZnSO4
AgNO3
ZnSO4
Pb(NO3J
CrCl3
Окраска
раствора
Образование осадка сульфида
Уравнение
реакции
Цвет осадка
ПР сульфида
Вывод
2) составьте уравнения реакций: получения гидроксидов; растворения гидроксидов
в избытке водного раствора аммиака, сопровождающихся образованием комплексов
[Ag(NH3J]+, [Cu(NH3L]2+, [Ni(NH3N]2+, [Zn(NH3L]2+, растворения гидроксидов в
избытке щелочи, сопровождающихся образованием комплексов [Zn(OHL]2~,
[Pb(OHK]~, [Cr(OHL]3"; образования сульфидов;
3) используя табл. П.4.11 и П.4.12, сравните значения констант нестойкости Кн комплекс-
комплексных ионов и ПР малорастворимых электролитов (гидроксидов и сульфидов) и сделайте
выводы о направлении протекания реакций с участием комплексных соединений.
ЛР 10. Жесткость и умягчение воды
Цель работы — определение различных видов жесткости и умягчение воды.
Опыт 1. Определение общей жесткости воды. Анализ воды на жесткость
обычно предполагает определение ее общей и карбонатной жесткости и вычисление
некарбонатной. Трилонометрический (стандартный) метод определения общей же-
жесткости воды основан на способности трилона Б (комплексона III, ЭДТА, дигидра-
тодинатриевой соли этилендиаминтетрауксусной кислоты — Na2H2Y • 2Н2О) обра-
образовывать с катионами жесткости (Са2+, Mg +, Fe2+) и многими другими достаточно
прочные окрашенные комплексные соединения сложного строения.
Исследуемый образец воды титруют раствором трилона Б в присутствии ин-
индикатора — эриохрома черного Т (хромогена черного ЕТ). С катионами жесткости,
присутствующими в воде, он образует окрашенные комплексные соединения при
рН = 10, придающие ей винно-красный цвет. Эти комплексные соединения менее
прочны, чем комплексные соединения трилона Б с ионами жесткости. В точке экви-
эквивалентности происходит разрушение комплексов с индикатором и образование
комплексов с трилоном Б, в результате чего окраска из вино-красного переходит в
сине-голубой цвет. Следует учитывать, что на результаты определения общей жест-
жесткости влияет присутствие в воде ионов Cu2+, Zn2+, Mn2+ и других, поэтому если они
содержатся в анализируемом образце воды (производственные стоки), то перед ана-
анализом их следует перевести в осадок добавлением небольшого количества раствора
Na2S. Выпавший осадок затем отделить.
17*
516
Свойства водных растворов электролитов
Последовательность проведения:
1) бюретку на 25 мл заполните 0,05н. (или 0,01н. при Но < 0,5 ммоль экв/л, или 0,1н. при
Яо > 20 ммоль экв/л) раствором трилона Б и запишите с точностью до 0,1 мл на-
начальное положение уровня жидкости по нижнему краю мениска (/*Нач);
2) мерной пипеткой на 100 мл отберите в две чистые конические колбы на 250 мл
по 100 мл водопроводной воды, добавьте в каждую из них по 5 мл аммиачного бу-
буферного раствора срН=10ипо4-5 капель раствора индикатора — эриохрома
черного (или кислотного хрома синего К при Но < 0,05 ммоль экв/л). Если раствор
индикатора отсутствует, добавьте на кончике шпателя сухой соли, после чего рас-
раствор обязательно перемешайте;
3) одну из колб отставьте в сторону на лист белой бумаги, используя ее в качестве
контрольного образца для сравнения окраски растворов;
4) вторую поставьте под бюретку на лист белой бумаги и по каплям, при непрерыв-
непрерывном вращательном перемешивании, приливайте в нее 0,05н. раствор трилона Б до
перехода окраски от одной капли из вино-красной в фиолетовую;
5) подождите 1-2 мин, перемешивая раствор: раствор изменит окраску на сине-го-
сине-голубую с зеленоватым оттенком. Если этого не происходит, добавьте еще несколько
капель раствора трилона Б из бюретки до получения указанной окраски. Сравните
окраски рабочего и контрольного растворов. Помните, что перетитрованный рас-
раствор имеет ту же окраску, что и правильно оттитрованный;
6) по достижении необходимой окраски титрование прекратите. Запишите конечное
положение уровня раствора трилона Б в бюретке по нижнему краю мениска (hKOli);
7) процесс титрования повторите еще два раза с новыми образцами воды, выполняя
пп. 1-6. При расхождении одного из результатов более чем на 0,1 мл, титрование
повторите.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов занесите в табл. Л. 10.1;
Таблица Л. 10.1
Результаты эксперимента
Исходные данные
100
100
100
Сэкв ТБ >
моль экв/л
0,05
0,05
0,05
Результаты измерений
hHsn, мл
hK0H, мл
Результаты расчетов
^тБ==Лко„-/г„ач,мл
УТБ, мл
Яо, ммоль экв/л
С 2+, г/л
Са„г/л
ЛР10. Жесткость и умягчение воды 517
2) по разности конечного и начального уровней раствора трилона Б в бюретке вы-
вычислите его объем КТБ, пошедший на титрование каждого из трех новых образцов
воды;
3) используя среднее значение объема трилона Б КТБ, рассчитайте общую жест-
жесткость воды по формуле
Самостоятельно выведите эту формулу. Какие законы химии использовали
при ее выводе?
4) по величине Но сделайте выводы о характере воды, т.е. установите, к какой груп-
группе она относится: очень мягкой, мягкой, средней жесткости, жесткой, очень жесткой?
5)рассчитайте массовые концентрации ионов Са2+ и Mg2+ (С 2+ и С 2+ в
7 Г г о v масс Са^ масс Mg^
граммах на литр) в этой воде, если ее магниевая жесткость составляет 30 % от най-
найденной общей жесткости.
Опыт 2. Определение карбонатной жесткости воды. Вода, обладающая
карбонатной жесткостью, имеет щелочную реакцию среды (рН > 7), что обусловле-
обусловлено протекающими по аниону процессами гидролиза содержащихся в ней гидрокар-
гидрокарбонатов и карбонатов кальция, магния и железа:
СО2" + НОН <=± НСО~ + OFT (рН > 7),
НСО; + НОН <=> Н2СО3 + ОНГ (рН > 7).
Определение карбонатной жесткости сводится к определению щелочности
воды. Это может быть осуществлено титрованием раствором кислоты известной
концентрации с использованием соответствующего индикатора (например, метило-
метилового оранжевого и др.).
При этом избыток гидроксид-ионов связывается кислотой ОН" + Н+ —» Н2О
и суммарные уравнения реакций, протекающих при титровании воды раствором
соляной кислоты, можно представить в виде
Са(НСО3J + 2НС1 +± СаС12 + Н2О + СО2
СаСО3 + 2НС1 ^ СаС12 + Н2О + СО2
Последовательность проведения:
1) пипеткой на 100 мл отберите в две чистые конические колбы на 250 мл по 100 мл
водопроводной воды;
2) в каждую из колб добавьте по 3 капли раствора индикатора — метилового оран-
оранжевого (метилоранжа), придающего щелочным и нейтральным растворам желтую
окраску, кислотным — красную;
3) одну из колб отставьте в сторону на лист белой бумаги: она будет служить кон-
контрольным образцом для сравнения окраски растворов;
518
Свойства водных растворов электролитов
4) бюретку на 25 мл заполните 0,1н. раствором соляной кислоты и запишите на-
начальный уровень жидкости hm4 по нижнему краю мениска с точностью до 0,1 мл;
5) вторую колбу поставьте под бюретку на лист белой бумаги и по каплям, при не-
непрерывном вращательном перемешивании, приливайте в нее 0,1н. раствор НС1 до
перехода окраски раствора от желтой до оранжевой (сравните с окраской контроль-
контрольного образца). Помните, что перетитрованный раствор имеет красную окраску;
6) по достижении необходимой окраски титрование прекратите. Запишите положе-
положение уровня раствора НС1 в бюретке по нижнему краю мениска (АКОн);
7) процесс титрования повторите еще два раза с новыми образцами воды, вы-
выполняя пп. 1-6. Результаты титрования не должны отличаться более чем на 0,1 мл.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов занесите в табл. Л. 10.2;
Таблица Л. 10.2
Результаты эксперимента
Исходные данные
V'MJI
100
100
100
Сжв НС1>
моль экв/л
0,05
0,05
0,05
Результаты измерений
/*„ач,мл
^кон > МЛ
Результаты расчетов
J^HCl = ^кон - hHsn, МЛ
FHC1, мл
Як, ммоль экв/л
Янк, ммоль экв/л
2) по разности конечного и начального уровней раствора НС1 в бюретке вычислите
его объем КНсь пошедший на титрование каждого из трех новых образцов воды;
3) используя среднее значение объема НС1 FHC1, рассчитайте карбонатную жест-
жесткость воды по формуле
Я =
к
4) самостоятельно выведите эту формулу. Какие законы химии использовали при ее
выводе?
5) рассчитайте некарбонатную жесткость Янк и ее долю в общей жесткости воды, ис-
используя данные опыта 1. Присутствием каких ионов обусловлен этот вид жесткости?
6) приведите уравнения реакций гидролиза карбонатов и гидрокарбонатов магния и
железа.
ЛР10. Жесткость и умягчение воды
519
Опыт 3. Снижение карбонатной и общей жесткости воды. Умягчение
воды заключается в полном или частичном удалении из нее главным образом
катионов Са2+, Mg2+, Fe2+. Вода считается мягкой, если ее общая жесткость ко-
колеблется в пределах 1,5...3,0 ммоль экв/л и очень мягкой, когда она не превы-
превышает 1,5 ммоль экв/л. Карбонатная жесткость воды может быть уменьшена пу-
путем ее термической обработки до величины остаточной карбонатной жесткости.
При этом уменьшается и общая жесткость до величины постоянной жесткости.
В данном опыте предлагается исследовать влияние длительности кипячения tKim
воды на величину ее карбонатной и общей жесткости и определить постоянную
жесткость воды.
Последовательность проведения:
1) в чистую термостойкую коническую колбу на 250 мл с помощью пипетки отбе-
отберите 100 мл водопроводной воды;
2) накрыв горлышко часовым стеклом, поставьте ее на асбестированную сетку, рас-
расположенную на треноге;
3) нагрейте до кипения и прокипятите в течение 30 с;
4) остудите колбу под струей водопроводной воды до комнатной температуры;
5) с помощью конической воронки и складчатого бумажного фильтра, содержимое
колбы отфильтруйте в другую чистую коническую колбу на 250 мл;
6) определите карбонатную жесткость фильтрата (отфильтрованной воды) (см. пп. 2,
4-6 опыта 2);
7) выполняя пп. 1-6 данного опыта, проведите аналогичные эксперименты с други-
другими образцами воды, увеличив время кипячения до 1, 2, 3, 5, 10 мин;
8) к осадку, оставшемуся на стенках колбы в последнем эксперименте, добавьте 3-5
капель разбавленной соляной кислоты. Запишите наблюдения.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов занесите в табл. Л. 10.3;
Таблица Л. 10.3
Результаты эксперимента
Исходные данные
VHfi9 мл
100
100
100
100
100
100
Сэкв НС1>
моль экв/л
о,
о,
о,
о,
о,
о,
'кип, МИН
0,5
1,0
2,0
3,0
5,0
10,0
Результаты
измерений
Акон,мл
Анач,мл
Результаты расчетов
^HCI = ''кон ~ Ьит>
МЛ
як,
ммоль
экв/л
2) по разности конечного и начального уровней раствора НС1 в бюретке вычислите его
объем Кнсь пошедший на титрование каждого из образцов отфильтрованной воды;
520 Свойства водных растворов электролитов
3) рассчитайте карбонатную жесткость фильтратов по формуле
V
4) постройте график зависимости величины карбонатной жесткости воды от време-
времени ее кипячения: Як =/(/КИп) и по нему экстраполяцией найдите жесткость воды при
'кип = 0. Полученное значение сравните с величиной карбонатной жесткости, опре-
определенной в опыте 2;
5) приведите уравнения процессов термической обработки воды с карбонатной же-
жесткостью, обусловленной присутствием в ней растворимых гидрокарбонатов каль-
кальция, магния и железа;
6) напишите уравнения процессов, происходящих при выполнении п. 8, считая, что
в состав осадка, образующегося после термической обработки воды, входят соот-
соответствующие соединения Са, Mg, Fe;
7) какой вид жесткости устраняется термической обработкой?
Опыт 4. Устранение некарбонатной жесткости воды. Некарбонатную же-
жесткость, обусловленную присутствием в воде растворимых сульфатов, хлоридов и
нитратов кальция, магния и железа, устраняют реагентным способом: обработкой
воды химическими реагентами (осадителями), образующими с катионами жестко-
жесткости осадки в виде малорастворимых соединений. В данном опыте на модельных
(имитирующих) растворах (водных растворах MgSO4, CaCl2, CaSO4) предлагается
исследовать эффективность действия различных осадителей.
Последовательность проведения:
1) в чистую пробирку с помощью пипетки отберите 5 мл 0,17М раствора MgSO4;
2) добавьте туда одну каплю 1,0М раствора Na2CO3. При образовании осадка, со-
содержимое пробирки тщательно перемешайте интенсивным встряхиванием до его
полного растворения;
3) повторяйте п. 2 до тех пор, пока образовавшийся осадок перестанет растворяться.
При этом подсчитайте число капель п раствора Na2CO3, пошедших на образование
осадка;
4) выполняя пп. 1-3, проделайте то же самое с растворами СаС12 и CaSO4;
5) аналогичный эксперимент проведите, взяв в качестве осадителя 0,7М раствор
Na3PO4.
Обработка результатов:
1) исходные данные, уравнения протекающих реакций, наблюдения и результаты
изнвфений, а также числовые значения произведений растворимости ПР соедине-
соединений, выпадающих в осадок, занесите в табл. Л. 10.4;
2) дайте характеристику осадков, указав цвет-(белый, желтый и т.д.), структуру
(кристаллический, аморфный) и состояние (творожистый, компактный, оседающий
на дно, находящийся во взвешенном состоянии и т.д.);
3) используя понятие произведения растворимости ПР (табл. П.4.11), сделайте вы-
выводы об эффективности действия различных осадителей.
ЛР 10. Жесткость и умягчение воды
521
Таблица Л. 10.4
Результаты эксперимента
Исходные данные
Состав модельного
раствора
0,17М водный
раствор MgSO4
0,17М водный
раствор СаС12
Насыщенный
водный раствор
CaSO4
Вид
осадителя
Na2CO3
Na3PO4
Na2CO3
Na3PO4
Na2CO3
Na3PO4
Уравнение
протекающей
реакции
Результаты
измерений
Число капель
п, шт.
Наблюдения
Цвет, структура и
состояние осадка,
значение ПР
Опыт 5. Обессоливание воды. Обессоливанием, или деминерализацией, на-
называют процесс удаления солей из воды до концентраций, соответствующих их со-
содержанию в дистиллированной воде (доли или несколько мг/л). Его осуществляют
выпариванием, ионным обменом, электродиали-
электродиализом и обратным осмосом. Выбор способа зави-
зависит от солесодержания исходной воды и тре-
требуемого остаточного содержания. В данном
опыте предлагается проверить высокую эффек-
эффективность ионного обмена. Процесс осуществля-
осуществляется на установке для обессоливания воды,
представленной на рис. Л. 10.1. Для наглядности
вместо жесткой воды используется ее имитатор
(модельный раствор) — насыщенный водный
раствор CaSO4.
Последовательность прове-
проведения:
1)с помощью качественных аналитических ре-
реакций обнаружения убедитесь в присутствии
ч2~ в растворе, подвергающемся
ионов Са2 и SO
процессу обессоливания. Для этого в две про-
пробирки отберите по 2-3 мл насыщенного раство-
раствора CaSO4, в одну добавьте 3-5 капель водного
раствора оксалата аммония (NH4JC2O4, в дру-
другую — хлорида бария ВаС12. Выпадение осадков
свидетельствует о присутствии ионов. Наблю-
Наблюдения и результаты запишите;
2) с помощью пипетки отберите 25 мл насыщен-
насыщенного раствора CaSO4, пропустите его черет ио-
Рис. Л.ЮЛ. Ионообменная ко-
колонка:
1 — стеклянная бюретка; 2 — слой
катионита или анионита; 3 — слой
стекловаты или стекловолокна; 4 —
кран для регулирования скорости
фильтрования; 5 — стакан для сбо-
сбора фильтрата; 6 — металлический
штатив с лапкой
522
Свойства водных растворов электролитов
нообменную колонку (см. рис. Л. 10.1), заполненную катионитом (КУ-2х8), со ско-
скоростью фильтрования не превышающей 15 капель в минуту (скорость регулируется
краном). Фильтрат 1 соберите в чистый сухой стакан. Проанализируйте его на на-
наличие ионов Са2+и SC>4~, выполняя п. 1. Результаты запишите;
3) остаток фильтрата пропустите через колонку, заполненную анионитом АН-1 (эс-
патит ТМ) или АВ-17х18, выполняя п. 2. Собранный при этом раствор (фильтрат 2)
подвергните анализу (см. п. 1) на наличие в нем ионов Са2+ и SO4 . Результаты за-
запишите.
Обработка результатов:
1) исходные данные, уравнения качественных аналитических реакций, наблюдения
и результаты анализов, а также числовые значения произведений растворимости ПР
соединений, выпадающих в осадок, занесите в табл. Л. 10.5;
Таблица Л. 10.5
Результаты эксперимента
Исходные данные
Исследуемый
раствор
Насыщенный
раствор CaSO4
Фильтрат 1
Фильтрат 2
Результаты анализов
Наличие (да, нет) в растворе
ионов Са2+
ионов SO4~
Уравнение
качественной
аналитической
реакций
Наблюдения
Цвет, структура и
состояние осадка,
значение ПР
2) дайте характеристику осадков (см. п. 2 обработки результатов эксперимента
опыта 4);
3) приведите уравнения процессов, протекающих при ионообменной обработке,
если катионит находился в Н+-форме (П—SO3H), а анионит — в ОН"-форме
(П—СН2—NR3OH);
4) объясните причину отсутствия осадков в фильтратах;
5) используя числовое значение ПР для CaSO4, рассчитайте массы ионов Са2+ и
SO2^, поглощенных катионитом и анионитом из 10 мл его насыщенного раствора.
VII
ЭЛЕКТРОХИМИЧЕСКИЕ
ЯВЛЕНИЯ И ПРОЦЕССЫ
¦ Основные определения
Законы Фарадея
Химические источники
тока и электролиз
Электрохимическими называют реакции (про-
(процессы), в которых происходит взаимное пре-
превращение электрической и химической форм
энергии. В основе этих процессов лежат окис-
окислительно-восстановительные реакции. Как и
любые термодинамические процессы, элек-
электрохимические превращения могут протекать
при бесконечно малом значении силы тока с
близкой к нулю скоростью (равновесный про-
процесс) или при измеримых значениях силы тока
с конечной скоростью (неравновесный про-
процесс).
Электрохимические процессы являются осно-
основой современных методов анализа веществ (ку-
лоно-, кондукто- и потенциометрии, поляро-
полярографии). Электролиз и электросинтез — основ-
основные методы промышленного получения и очи-
очистки щелочных, щелочно-земельных и цветных
металлов, неорганических окислителей и газов
(хлора, водорода, кислорода). В гальванотехни-
гальванотехнике наносят металлические слои на поверхность
металлических и неметаллических изделий.
Хемотроника занимается разработкой и созда-
созданием электрохимических преобразователей ин-
информации (датчики сейсмоколебаний, усилите-
усилители, выпрямители и др.). Важнейшие задачи
прикладной электрохимии — создание новых
химических источников тока и разработка эф-
эффективных методов защиты металлических ма-
материалов от коррозии.
16 4
Электродные процессы
Электродными {потенциалопределяющими) процессами называют
химические превращения, происходящие на электродах с участи-
участием заряженных частиц (ионов, электронов), которые переносятся
через границу раздела двух фаз. Эти процессы многостадийны, ге-
терогенны, имеют сложный механизм, их подразделяют на две
группы: процессы возникновения разности потенциалов и элек-
электрического тока в гальванических элементах; химические процес-
процессы при электролизе. Эти две группы процессов являются окисли-
окислительно-восстановительными реакциями и взаимно обратны по на-
направлению.
16.1. Основные определения. Законы Фарадея
Электрохимической системой (ЭХС) называют устройство, в
котором осуществляются взаимные превращения электрической и
химической форм энергии. Конструктивно ЭХС (рис. 16.1) состоит
из двух электродов 1 и 3, разделенных полупроницаемой пористой
перегородкой (диафрагмой) 2 и образующих внутреннюю цепь.
Каждый электрод состоит из взаимно контактирующих проводника
первого рода (металла или полупроводника) и проводника второго
рода (раствора электролита). Напомним, что в проводниках первого
рода переносчиками электрического заряда являются электроны, а
в проводниках второго рода — ионы.
В электрохимии в стандартной форме записи, когда на элек-
электроде протекает процесс восстановления, электроды изображают в
виде схемы (условной записи): слева от вертикальной черты, озна-
означающей границу раздела фаз, указывают вещества, находящиеся в
растворе; справа — вещества, образующие электродный материал
(другую фазу). Если фаза содержит несколько веществ, то их фор-
формулы разделяются запятыми. Например, электроды Zn2+1 Zn — цин-
цинковый; СГ | СЬ, Pt — хлорный; MnC>4, Mn2+, H+ | Pt — перманганат-
ный (в кислотной среде).
Относительные величины электродных потенциалов (их зна-
значения в вольтах по водородной шкале) обозначают греческой бук-
буквой ф с индексом в виде дроби. В числителе дроби через запятую
указывают исходные вещества (окисленная форма, Оф) электрод-
электродной реакции со стехиометрическими коэффициентами, в знамена-
знаменателе — таким же образом, продукты этого процесса (восстановленная
16.1. Основные определения. Законы Фарадея
525
Внешняя цепь
Внутренняя цепь
Рис. 16.1. Схема электрохимической системы:
1,3 — электроды; 2 — пористая полупроницаемая перегородка; 4 — внешний
источник постоянного тока или гальванометр; 5 — границы раздела фаз. Зна-
Знаки электродов указаны для гальванического элемента
форма, Вф). В сокращенной форме записи стехиометрические коэффициен-
коэффициенты не указывают. Электродную реакцию записывают как процесс восста-
восстановления (присоединения электронов). Например, для кислородного элек-
электрода Н2О, ОН~|С>2, Pt, электродный процесс которого
Ог + 2Н2О + 4е -> 4ОН", стандартный электродный потенциал Фо2,2н2о =
4ОН"
= 0,401 В (индекс «0» означает стандартные термодинамические условия).
Внешняя цепь обеспечивает прохождение тока между электродами и
представляет собой металлический проводник. В зависимости от вида ЭХС
во внешнюю цепь включают гальванометр или источник постоянного тока.
Устройство, осуществляющее превращение химической энергии
окислительно-восстановительной реакции в электрическую, называют хи-
химическим источником тока (ХИТ). В этом случае ЭХС генерирует постоян-
постоянный электрический ток, во внешнюю цепь включается гальванометр. Разли-
Различают три вида ХИТ: гальванические элементы, аккумуляторы, топливные
элементы. В источниках тока окислительно-восстановительные реакции про-
протекают самопроизвольно, следовательно, в закрытой системе ArGj < 0.
526 16. Электродные процессы
Устройство, превращающее электрическую энергию постоянного тока
в химическую, называют электролизером. В его внешнюю цепь включается
источник постоянного тока, на электродах протекают химические реакции
(например, анодное растворение или катодное осаждение металла, выделе-
выделение газа на аноде или катоде). Окислительно-восстановительные реакции на
электродах электролизера осуществляются принудительно, под действием
электрической энергии внешнего источника тока, т.е. в закрытой системе
A,G°>0.
В связи с противоположным направлением процессов в гальваниче-
гальваническом элементе (ГЭ) и в электролизере (Э) одноименные электроды (полюсы)
имеют разные знаки. На аноде (знаки «-» в ГЭ, «+» в Э) протекают реакции
окисления, на катоде (знаки «+» в ГЭ,«-» в Э) — процессы восстановления.
Для реакций, происходящих во всех ЭХС, справедливы законы Фара-
дея A833), химической термодинамики и химической кинетики. Великий
английский физик М. Фарадей открыл законы, связывающие массу или эк-
эквивалентное количество вещества, прореагировавшего на электроде, с заря-
зарядом, прошедшим через ЭХС.
Первый закон Фарадея. Масса mt вещества /, прореагировавшего на
электроде ЭХС, пропорциональна электрическому заряду q, прошедшему
через систему:
/и,- -q; mi = M3Xiq = M3XiIt, A6.1)
где Мэх t — коэффициент пропорциональности, называемый электрохимиче-
электрохимической эквивалентной массой, г/Кл или г/(А • ч); / — сила тока, А; / — время
пропускания тока, с; q — электрический заряд, Кл. Из A6.1) следует, что
А/эх/ численно равна массе вещества, прореагировавшего на электроде при
пропускании через систему заряда q = 1 Кл.
Второй закон Фарадея. При постоянном заряде, прошедшем через
ЭХС, массы веществ, прореагировавших на электродах, соотносятся между
собой как молярные массы их химических эквивалентов:
при q = const
^ ^1 A6.2)
т2 Мзкъ2
Второй закон Фарадея применим к веществам, реагирующим:
а) на аноде и катоде одной и той же ЭХС;
б) на электродах последовательно соединенных ЭХС, для которых / =
= const, / = const, а следовательно, и q = const;
в) на электродах любых ЭХС при q = const.
Применяя свойство пропорции к A6.2), можно получить другую фор-
формулировку второго закона Фарадея: при постоянном заряде, прошедшем
16.1. Основные определения. Законы Фарадея 527
через ЭХС, эквивалентные количества веществ, прореагировавших на элек-
электродах, равны между собой:
при q = const
а. = д. , т.е. пэкъ1=пэкъ2. A6.3)
С использованием A6.1) и A6.2) выводится следствие из закона Фара-
Фарадея, устанавливающее пропорциональность электрохимических эквивалент-
эквивалентных масс и молярных масс химических эквивалентов веществ:
^LL = -7T=i- A6.4)
Из второго закона Фарадея следует, что для электрохимического пре-
превращения 1 моль эквивалента вещества необходим постоянный заряд F, назы-
называемый константой (числом) Фарадея: F = eNA = 1,6022 • 10~19 • 6,022 • 1023 =
= 96484 Кл/моль экв = 26,8 А • ч/моль экв. Для расчетов принимают F =
= 96500 Кл/моль экв. Постоянная Фарадея равна заряду, переносимому 1 моль
элементарных носителей (электронов, однозарядных ионов). Число F пред-
представляет собой коэффициент пропорциональности
МЭКВ/ = МЭХ/К A6.5)
Объединяя уравнения A6.1) и A6.5), получают обобщенное выраже-
выражение законов Фарадея:
F z F F
г ^экв ir r
где Mi — молярная масса вещества, г/моль; z3KB,- —число эквивалентности,
равное заряду катиона металла или числу электронов, принимающих участие в
электродном процессе (так называемое зарядовое число); f-^i = фак-
тор эквивалентности.
В применении к газам, подвергающимся превращению на электродах
(выделение при электролизе водорода на катоде, хлора или кислорода на
аноде, поглощение кислорода на катоде при электрохимической коррозии),
обобщенное выражение законов Фарадея записывают чаще через объемы
реагирующих веществ:
%, A6.7)
528 16. Электродные процессы
где Vj — объем выделившегося или поглощенного газа; V3KBi — молярный объ-
объем химического эквивалента этого газа (при н.у. для О2 — 5,6 л/моль экв, для
Н2, С12 — 11,2 л/моль экв).
Впоследствии в выражение A6.6) был введен коэффициент выхода по
току г|/ вещества. Величина г|, в долях единицы равна отношению массы
вещества mnph практически подвергшейся превращению, к теоретически
вычисленной тТ ,- в соответствии с законами Фарадея:
mnpi
Л/=——
m
Ti
Коэффициент Tj/ равен доле от общего пропущенного заряда, пошедшей на
осуществление данного процесса. Тогда A6.6) принимает вид
=-^Л- A6.8)
Отклонения от законов Фарадея являются кажущимися, поскольку
вызваны протеканием параллельных реакций на электродах или другими
причинами.
16.2. Потенциалы электрохимической системы.
Двойной электрический слой
Электрохимическую систему называют правильно разомкнутой, если
к обоим концам ее внешней цепи присоединен один и тот же металл М2 ,
обычно — медь как самый распространенный из хороших проводников (см.
рис. 16.1).
В такой системе имеются следующие границы раздела двух фаз:
Вк — М2 (вакуум — металл 2); М2 — Вк (металл 2 — вакуум);
М2 — Mi (металл 2 — металл 1); М3 — М2 (металл 3 — металл 2);
в) Mi — Li (металл 1 — раствор электролита 1); L2 — М3 (раствор
электролита 2 — металл 3);
Li — L2 (раствор электролита 1 — раствор электролита 2).
При соприкосновении двух разнородных фаз система находится в не-
неравновесном состоянии. Согласно второму закону термодинамики, она
стремится к равновесию. Этот процесс сопровождается обменом заряжен-
заряженных частиц между фазами, в результате чего по одну сторону от границы
раздела появляется избыток заряда определенного знака, а по другую — его
недостаток. Возникает двойной электрический слой (ДЭС), напоминающий
плоский конденсатор с заряженными обкладками, следовательно, на грани-
16.2. Потенциалы электрохимической системы 529
це раздела фаз электрический потенциал ф резко изменяется и наблюдается
скачок потенциала Дф.
В правильно разомкнутой ЭХС имеются следующие скачки потенциа-
потенциалов, которые обозначают ф:
а) поверхностные потенциалы фВк и фМг; при данной температуре
М^ ~йк
они равны по модулю, противоположны по знаку и компенсируют друг друга;
б) контактные потенциалы фМз и фМз; их сумма равна контактному
потенциалу фм ;
м^
в) электродные потенциалы фМ] иф^ на границе металл — раствор
его соли;
г) диффузионный потенциал фь на границе двух растворов
V2
электролитов.
Контактный потенциал возникает на поверхности соприкосновения
двух металлов. Он обусловлен различной концентрацией электронов прово-
проводимости в фазах, определяется разностью работ выхода электрона, зависит
от температуры и для некоторых пар металлов достигает нескольких вольт.
Абсолютную величину контактного потенциала определить невозможно. На
практике используют его относительное значение фКОнт.
Диффузионный потенциал возникает на поверхности раздела двух
растворов электролитов с одинаковым растворителем, различающихся по
виду электролита или по его концентрации. Он появляется за счет различия
абсолютных скоростей (подвижностей) ионов электролита. Так, в растворах
нитрата серебра AgNO3 подвижность аниона NO3 выше, чем катиона Ag+.
Диффузия растворенного вещества происходит в направлении убывания его
концентрации (активности). Поэтому граница раздела двух растворов
AgNO3 заряжается отрицательно со стороны более разбавленного раствора и
положительно со стороны раствора с большей концентрацией электролита
(рис. 16.2). Возникает диффузионный ДЭС. По прошествии некоторого вре-
времени скорости диффузии катионов Ag+ и анионов NO3 выравниваются. На
границе растворов наступает стационарное состояние, при котором электро-
электролит диффундирует как единое целое (в виде AgNO3) и ток отсутствует. Ус-
Установившуюся между растворами разность потенциалов называют равно-
равновесным диффузионным потенциалом фд. Его абсолютное значение опреде-
530
16. Электродные процессы
Рис. 16.2. Схема возникновения диффузионного потенциала:
1 — раствор AgNO3 с активностью ах\ 2 — раствор AgNO3 с активностью а2 > ах\
3 — диффузионная полупроницаемая перегородка; 4 — диффузионный двой-
двойной электрический слой; © — катионы Ag+; © — анионы NO"; @ — катио-
катионы Ag+, перешедшие из раствора 2 в раствор 1; 0 — анионы NO~, пере-
перешедшие из раствора 2 в раствор 1
лить невозможно, поэтому на практике используют относительное значение.
Расчет и опытное определение <рд затруднены, в связи с чем его пытаются
максимально уменьшить, что достигается разъединением растворов в про-
пространстве и расположением между ними солевого мостика.
Солевой мостик представляет собой [/-образную стеклянную трубку,
заполненную насыщенным раствором электролита, у которого подвижности
ионов приблизительно одинаковы (КС1, NH4NO3). Электрическая проводи-
проводимость на границах контакта растворов с солевым мостиком обеспечивается
этими ионами. Возникают очень малые обратные по знаку диффузионные
потенциалы, суммой которых пренебрегают.
Наиболее важный электродный потенциал фм существует на грани-
Т
це металл — раствор электролита. При погружении металлической пластин-
пластинки, с поверхности которой удалена оксидная пленка, в раствор электролита
между металлом и раствором возникают ДЭС и скачок потенциала, назы-
называемый электродным потенциалом. Основная причина его существования —
движение ионов между поверхностью металла и раствором.
16.2. Потенциалы электрохимической системы 531
Важную роль играют специфическая адсорбция (концентрирование
вещества на границе раздела фаз) ионов и полярных молекул, а также выход
электронного газа за границы кристаллической решетки металла.
Рассмотрим образование ДЭС при погружении металлической пла-
пластинки в воду или другой полярный растворитель. При этом происходят
следующие процессы:
1) катионы, расположенные в кристаллической решетке металла на
поверхности, взаимодействуют с полярными молекулами воды, которые
ориентированы к катионам своими отрицательными полюсами. Взаимодей-
Взаимодействие может быть настолько сильным, что катионы отрываются от поверх-
поверхности металла и переходят в жидкую фазу:
Системой поглощается энергия, равная энергии связи катиона с кри-
кристаллической решеткой ?Mz+- Избыток электронов заряжает поверхность
металла отрицательно, а выходящие катионы заряжают граничащий с пла-
пластинкой слой воды положительно;
2) катионы металла гидратируются полярными молекулами воды в объе-
объеме жидкой фазы (в неводном растворителе сольватируются его молекулами)
Системой выделяется энергия, равная энергии гидратации Ен (или
сольватации Es). В соответствии с термодинамической системой знаков
Е 2+ > 0, ?/, и 2?5 < 0. Процессы 1) и 2) протекают одновременно и описыва-
описываются уравнением
М(т) + лН2О -» Mz+ • иН2О(р) + Ze;
3) концентрирование гидратированных катионов в граничащем с по-
поверхностью металла слое жидкости из-за электростатического взаимодейст-
взаимодействия. Таким образом, на границе металл — вода возникает обменный ДЭС и
соответствующий ему скачок потенциала — электродный потенциал (рм
Г
(рис. 16.3).
С ростом потенциала (рм все быстрее будут протекать обратные про-
Т
цессы — дегидратация (десольватация) ионов металла и их восстановление
до атомов:
532
16. Электродные процессы
Mz+ • иН2О(р) + Ze -> М(т) + иН2О
При некотором скачке потенциала
наступает равновесие:
М(т) + /iH2O
• иН2О(р)
Обменный двойной
электрический слой
Рис. 16,3. Схема возникновения об-
обменного двойного электрического слоя
и электродного потенциала на границе
металл—раствор электролита:
^^— катионы металла, расположенные в
узлах кристаллической решетки; \j —
пустоты в кристаллической решетке;
ft*
*0? — гидратированные катионы метал-
металла, перешедшие в раствор; © — избыточ-
избыточные электроны, оставшиеся в металле после
ухода катионов
при котором скорости прямого и об-
обратного процессов равны.
В упрощенной форме записи из
этого уравнения исключают молеку-
молекулы воды, записывают его в виде, ре-
рекомендованном ИЮПАК (как вос-
восстановление), и называют уравнением
электродного (потенциалопределяю-
щего) процесса
1М A6.9)
При погружении металла в
раствор его соли (электролита) на-
наблюдаются сходные явления, но рав-
равновесие наступает при другом скачке
потенциала.
Равновесным электродным по-
потенциалом cpL =9Mz+ называют
м "й~
скачок потенциала, установившийся
между металлом и раствором электролита в условиях равновесия. Металл в
растворе собственной соли может заряжаться как отрицательно (Al, Zn, Fe,
Ni), так и положительно (Си, Ag, Аи).
В закрытой системе знак заряда металла в растворе собственной соли
и знак соответствующего потенциала фм2+ определяются изменением энер-
м
гии Гиббса процесса A6.9):
1) если ArGj >0, самопроизвольно протекает обратный процесс, по-
поверхность металла заряжается отрицательно;
2) если ArG® < О, самопроизвольно протекает прямой процесс, по-
поверхность металла заряжается положительно.
В первом приближении можно пренебречь энтропийной составляю-
составляющей энергии Гиббса (ArS? =0). В этом случае в закрытой системе самопро-
16.3. Уравнение Нернста 533
извольно пойдет экзотермический процесс (Дг#? < 0) — принцип Бертло—
Томсена. Иными словами, направление процесса будет определяться соот-
соотношением абсолютных величин энергии сольватации Es катиона металла
молекулами растворителя и энергии связи EK.Z+ катиона в кристаллической
м
решетке:
1) при |/?J> p z+ экзотермическая сольватация преобладает над эн-
эндотермическим процессом удаления катионов из узлов решетки, самопроиз-
самопроизвольно происходит переход катионов с поверхности металла в раствор его
соли, поверхность металла заряжается отрицательно, 9Mz+ < 0;
м
2) при |/?,|<|2?2+ экзотермический процесс внедрения катионов в
узлы создаваемой решетки превалирует над эндотермической десольватаци-
ей, самопроизвольно осуществляется переход катионов из раствора соли
металла на его поверхность, которая заряжается положительно, <PMz+ > 0.
м
16.3. Уравнение Нернста
Это уравнение было получено немецким физикохимиком В. Нернстом,
оно связывает электродный потенциал с природой металла, активностью
(концентрацией) его ионов в растворе и абсолютной температурой. Строгий
вывод уравнения Нернста основан на применении второго начала термоди-
термодинамики к электродному процессу A6.9). Электрод, на котором происходит
данная реакция, представляет собой металлическую пластинку, опущенную
в раствор соли этого же металла (так называемый металлический электрод).
Электрическая работа по обратимому переносу 1 моль катионов с по-
поверхности металла в раствор или обратно равна произведению суммарного
перенесенного заряда ZF на разность потенциалов фм2+ между начальной и
м
конечной точками переноса
W^ZFy^ A6.10)
м
где Z — зарядовое число катиона, т.е. число электронов, участвующих в
электродном процессе, моль экв/моль; F = 96 500 Кл/моль экв — постоян-
постоянная Фарадея; ср. 2+ — электродный потенциал, В.
м
м
534 16. Электродные процессы
Согласно второму закону термодинамики, максимальная работа, со-
совершаемая закрытой системой в изобарно-изотермическом процессе
(р,Т= const), равна убыли ее энергии Гиббса: W3Jl = -AG.
Из уравнения изотермы химической реакции изменение энергии Гибб-
Гиббса для процесса A6.9) равно
%^-, A6.11)
где ам — относительная (т.е. отнесенная к стандартной) активность метал-
металла в его кристалле; a z+ — относительная активность ионов металла в рас-
растворе.
Умножая обе части уравнения A6.11) на -1, получают
^--. A6.12)
Приравнивая значения электрической работы из уравнений A6.10) и A6.12),
имеют
ZF(p..z+ = RTInK" -RTln-^r^-. A6.13)
м M
Разделив обе части выражения A6.13) на ZF, получают уравнение Нернста
для металлического электрода
RT, ггс\ RT, Лху|
MZ+ =—1пАГа -—1п-^_. A6.14)
"м" ZF ZF aMz+
Поскольку Kl - const при Т = const — стандартная термодинамическая
константа равновесия реакции A6.9), то при постоянной температуре для
данного электрода первое слагаемое в правой части выражения A6.14) явля-
является постоянной величиной, обозначаемой как <P°Mz+ и называемой стан-
станем"
дартным электродным потенциалом.
Активность металла ам в молях на литр в его кристалле при данной
температуре есть постоянная величина, равная частному от деления плотно-
плотности металла рм в граммах на литр на его молярную массу атомов Мам в
граммах на моль. Эта же величина является для индивидуального вещества
стандартной активностью, следовательно, ам = 1. Тогда уравнение Нернста
принимает вид, в котором его обычно применяют для расчета потенциалов
металлических электродов в нестандартных условиях:
16.3. Уравнение Нернста 535
RT
м м
Из уравнения A6.15) следует, что при относительной активности ка-
катионов металла в растворе, равной единице, ф z+ = ф^ z+ — стандартный
м ~мГ
электродный потенциал. В теории величина ф^2+ есть скачок потенциала на
м
границе раздела фаз металл — раствор его соли при относительной актив-
активности катионов металла в растворе, равной единице. Практические (таблич-
(табличные) значения ф^2+ (см. табл. П.6.1) отличаются от теоретических на фК0Нт-
.м
м
В разбавленных растворах электролитов коэффициенты активности
ионов несильно отличаются от единицы, поэтому в уравнении Нернста
A6.15) вместо относительной активности катионов металла Mz+ можно ис-
использовать их относительную молярную концентрацию С
м м
Отметим, что в учебной и научной литературе часто знак «~» над обо-
обозначениями относительных активностей, молярных концентраций ионов и
парциальных давлений газов не ставят, т.е. обозначают так же, как их абсо-
абсолютные значения, выраженные в молях на литр или в атмосферах. В даль-
дальнейшем для упрощения не будем использовать знак «~» над этими символа-
символами, однако необходимо помнить, что физическая величина, стоящая под
знаком логарифма, должна быть безразмерной.
Чтобы преобразовать уравнение Нернста A6.15), заменяют в нем на-
натуральный логарифм активности катионов металла на десятичный и под-
подставляют Т = 298 К; R = 8,31 Дж/(моль • К); F = 96500 Кл/моль экв. После
выполнения расчетов уравнение Нернста для металлического электрода
принимает вид
о 0,059
м^^^м- <1бЛ7>
м м
В общем случае уравнение электродного процесса записывается как
Оф + г^^Вф, A6.18)
где Оф — окисленная; Вф — восстановленная формы частиц, участвующих
536
16. Электродные процессы
Фоф
Фоф
Вф
Фоф Т,если
Вф
Фоф
Вф
Рис 16.4. Влияние температуры и активностей (концентраций) окис-
окисленной и восстановленной форм на электродный потенциал окисли-
окислительно-восстановительной системы
в реакции. Уравнение Нернста для этого процесса записывают следующим
образом:
11 A6.19)
офФоф11
if if ZF %Ф
или через десятичный логарифм отношения активностей
вф вф z аЩ
Зависимость электродного потенциала от природы окислительно-вос-
окислительно-восстановительной системы характеризуется величиной фоф (стандартный по-
Вф
тенциал, табличное значение), зависимость от абсолютной температуры и
активностей окисленной и восстановленной форм — вторым слагаемым
правой части A6.19).
Потенциал окислительно-восстановительной системы увеличивается с
ростом температуры и активности (концентрации) окисленной формы и
уменьшается с ростом активности (концентрации) восстановленной формы.
Уменьшение потенциала наблюдается при противоположном изменении
указанных факторов (рис. 16.4).
16.4. Стандартный водородный электрод.
Ряд стандартных электродных потенциалов металлов
Абсолютное значение электродного потенциала металлического элек-
электрода фМ2+ (скачок потенциала) определить невозможно, поскольку нельзя
м
16.4. Стандартный водородный электрод
537
подсоединить вольтметр к двойному электрическому слою. В связи с этим
на практике выбирают так называемый электрод сравнения, потенциал ко-
которого принимают равным нулю при любой температуре и относительно
него измеряют потенциалы всех других электродов.
Электродным потенциалом фм2+ называют величину, равную по моду-
м
лю электродвижущей силе Е (ЭДС) правильно разомкнутого гальванического
элемента, составленного из исследуемого электрода и электрода сравнения.
Согласно правилам ИЮПАК, электродвижущая сила Е равна макси-
максимальной разности потенциалов катода и анода (правого и левого электродов
соответственно) при равновесном проведении процесса в правильно ра-
разомкнутом гальваническом элементе
?' = Фк-фа. A6.21)
При этом сила тока в цепи равна бесконечно малой величине (в пре-
пределе — нулю). При конечном (измеряемом) значении силы тока в гальвани-
гальваническом элементе происходит неравновесный процесс. В этом случае раз-
разность потенциалов катода и анода называют напряжением
?/ = Фк/-фа/. A6.22)
За электрод сравнения принимают стандартный водородный электрод,
у которого активность ионов водорода в растворе равна 1 моль/л и давление
газообразного водорода над раствором составляет 101,3 кПа. Поскольку во-
водород при температуре 298 К является газом, из него невозможно, как из ме-
металла, изготовить пластинку. В
качестве твердой фазы в водород-
водородном электроде используют инерт-
инертный металл (например Pt).
Конструктивно водородный
электрод (рис. 16.5) состоит из
платиновой пластинки, покрытой
слоем мелкозернистой платины —
платиновой чернью, частично по-
погруженной в раствор, содержащий
ионы водорода. Обычно берут
раствор нелетучей кислоты —
серной H2SO4 или хлорной НСЮ4.
К пластинке подают чистый газо-
газообразный водород под постоян-
постоянным давлением.
t=^H+(p)"
Рис. 16.5. Схема водородного электрода
538 16. Электродные процессы
В кислотной среде водородному электроду Н3О+1 Н2, Pt соответствует
уравнение электродного процесса
которое в упрощенной форме записывают как
или в эквивалентном виде
Уравнение Нернста для водородного электрода имеет вид
По определению, стандартный потенциал водородного электрода равен ну-
нулю, тогда
Фн+ =0,0591gaH+ -0,02951g/?H2 = -0,059pH-0,02951g/7H2, A6.24)
нГ
где ян+ — активность ионов водорода в растворе; рц2 — давление газообразно-
газообразного водорода над раствором; pH = -lgtf + —водородный показатель раствора.
При/?н2 = 1 уравнение A6.24) принимает вид
Фн+ = -0,059рН. A6.25)
щ
Составив гальванический элемент из стандартных водородного и ис-
исследуемого металлического электродов, можно экспериментально измерить
стандартный потенциал <P°Mz+ металлического электрода. При расположе-
м
нии металлов в горизонтальный (вертикальный) ряд в порядке возрастания
слева направо (сверху вниз) значений (p^z+ получают ряд стандартных
м
электродных потенциалов (или электрохимический ряд напряжений) метал-
металлов, для которого справедливы следующие положения.
1. Потенциалу металла присваивается тот же знак, который имеет
стандартный металлический электрод в паре со стандартным водородным.
Металлы, стоящие левее (выше) водорода, имеют отрицательные значения
(p^z+, правее (ниже) — положительные.
м
16.4. Стандартный водородный электрод 539
2. При перемещении по ряду слева направо (сверху вниз) возрастает
окисляющая способность катионов Mz+ в водном растворе, при продвиже-
продвижении справа налево (снизу вверх) увеличивается восстанавливающая способ-
способность металлов М. Например, железо (фре2+ = -0,440 В) более сильный вос-
~Fe~
становитель, чем медь (ф^и2+ =0,337 В), но катионы Си2+ в водном растворе
Си
более сильный окислитель, чем Fe2+.
3. Отрицательные значения ф^2+ свидетельствуют о том, что данные
м
металлы окисляются катионами водорода, т.е. металлы вытесняют Нг из рас-
растворов минеральных кислот с активностью ионов водорода ан+ = 1 моль/л,
анионы которых не проявляют окислительных свойств, — так называемых
кислот-неокислителей (НС1, HBr, HI, H3PO4, разбавленная H2SC>4)
Fe + H2SO4 -> FeSO4 + H2 t; ф^2+ = -0,440 В
и разлагают воду
2Na + 2H2O->2NaOH + H2t; ф^а+ =-2,714 В.
~Na~
Потенциал водородного электрода в чистой воде при температуре
25 °С (рН = 7)
<р° =-0,0592 -7 = -0,414 В.
н
Воду с выделением водорода разлагают только те металлы, у которых
Ф^2+ < 0,414 В, но практически вследствие образования на металлических
м
поверхностях защитных оксидных пленок такой способностью обладают
только щелочные и щелочно-земельные металлы.
4. Положительные значения ф^2+ означают, что катионы Mz+ яв-
м
ляются окислителями по отношению к молекулам водорода. Эти металлы
не разлагают воду и не вытесняют водород из растворов кислот-
неокислителей, в которых ан+ = <*mz+ = 1 моль/л. Напротив, водород вы-
вытесняет металлы из растворов их солей.
540 16. Электродные процессы
5. Если данный металл химически не реагирует с водой, он вытесняет
все стоящие правее металлы из растворов их солей:
Fe + CuSO4 -> FeSO4 + Си i
Си + 2AgNO3 -> Cu(NO3J + 2Ag I
В обратном направлении эти реакции не протекают.
6. Если (p^z+ < 0, то стандартный металлический электрод в паре со
м
стандартным водородным является анодом (отрицательным полюсом галь-
гальванического элемента), на котором самопроизвольно происходит процесс
окисления М -» Mz+ + Ze.
7. Если ф^2+ > 0, то стандартный металлический электрод в паре со
м
стандартным водородным есть катод (положительный полюс гальваниче-
гальванического элемента), на котором самопроизвольно протекает процесс восстанов-
восстановления Mz+ + Ze -> М.
8. В гальваническом элементе, составленном из двух стандартных метал-
металлических электродов, катодом всегда является тот электрод, потенциал которо-
которого больше. Гальванический элемент — это система, совершающая электриче-
электрическую работу, следовательно, W3Jl = ZFE = ZF(<pK - фа) > 0, т.е. фк > сра. Цин-
Цинковый электрод (ф^ 2+ =-0,763 В) в паре с алюминиевым (Фд,з+ =-1,662 В)
Zn A1
является катодом, а в паре с никелевым (Ф?г2+ =-0,250 В) — анодом. При
Ni
этом чем больше различаются значения стандартных потенциалов двух
электродов, тем больше ЭДС полученного из них гальванического элемента.
Все перечисленные выше положения справедливы не только для ме-
металлических, но и для всех окислительно-восстановительных систем. При
проведении окислительно-восстановительных реакций окисленная форма
системы с большим потенциалом всегда является окислителем по отноше-
отношению к восстановленной форме системы с меньшим потенциалом.
Стандартные электродные потенциалы определяют экспериментально
компенсационным методом (рис. 16.6), но для неустойчивых электродов
(щелочные и щелочно-земельные металлы в водных растворах их солей и
оснований) важны также термодинамические расчеты. Например, в гальва-
гальваническом элементе (см. рис. 16.6) стандартный водородный электрод — это
анод, стандартный медный — катод.
16.4. Стандартный водородный электрод
541
Рис. 16.6. Схема установки для измерения электродных потенциалов
и ЭДС гальванического элемента:
1 — стандартный водородный электрод; 2 — исследуемый электрод; 3 — со-
солевой мостик; 4 — переменное сопротивление АВ с линейным законом изме-
изменения; 5 — гальванометр; 6 — источник компенсационного тока (элемент
Вестона)
Схема такого гальванического элемента имеет вид
Cu|Pt,H2|H+||Cu2+|Cu
ри2 = 1 a™j ан+ - #cu2+ = 1 моль/л.
Если падение внешнего потенциала от элемента Вестона на участке
АС равно ЭДС элемента, то гальванометр покажет отсутствие тока в цепи.
Для испытуемого элемента ЭДС Е° рассчитывают по формуле
Си Н2
где Ew — ЭДС элемента Вестона при температуре 25 °С.
Так как ф^+ =0 В, то
Си
542 16. Электродные процессы
16.5. Классификация электродов
Электроды подразделяют на обратимые и необратимые. Если изме-
изменить направление электрического тока во внешней цепи на противополож-
противоположное, то на обратимом электроде протекает тот же самый процесс в обратном
направлении, а на необратимом — другой процесс.
Серебряная пластинка, находящаяся в растворе нитрата серебра, пред-
представляет собой обратимый электрод. Электродный процесс
протекает в прямом или обратном направлениях.
Серебряная пластинка, находящаяся в растворе кислоты, служит при-
примером необратимого электрода. В зависимости от направления тока во
внешней цепи на электроде происходит восстановление катионов водорода
или окисление атомов серебра
В зависимости от свойств веществ и заряженных частиц, участвую-
участвующих в электрохимических процессах, и характера равновесия обратимые
электроды классифицируют на электроды первого и второго рода, окисли-
окислительно-восстановительные и ионообменные.
Электрод первого рода представляет собой пластинку, изготовленную
из простого вещества (металла или полупроводника) и погруженную в рас-
раствор, содержащий его ионы. В качестве примеров приведем серебряный и
селеновый электроды
Ag+|Ag;Se2-|Se
Для их электродных процессов характерно участие только одного ви-
вида ионов:
Ag+ + e <=> Ag
Уравнения Нернста для этих электродов имеют следующий вид:
к
Ag Ag Ag
4>Se =<p°Se +ЦШ—= Cp0Se -^Vse2- A6-27)
SF SF 2F V- iF 2
16.5. Классификация электродов 543
Выражения A6.26) и A6.27) показывают, что потенциалы серебряного
и селенового электродов зависят от активностей катионов серебра и селе-
нид-анионов в растворе, поэтому серебряный электрод обратим относитель-
относительно катиона, а селеновый — относительно аниона.
К электродам первого рода относят также амальгамные электроды, в
которых восстановленная форма представляет собой раствор металла в рту-
ртути M(Hg) (амальгаму). Примером служит амальгамный кадмиевый электрод
Cd2+ | Cd(Hg), применяемый в стандартном элементе Вестона. Этот элемент
имеет устойчивое значение ЭДС при данной температуре и используется
как эталон для измерения ЭДС электрохимических цепей (см. рис. 16.6).
Уравнение электродного процесса для амальгамного кадмиевого электрода
имеет вид
Cd2++2e<=iCd(Hg)
а уравнение Нернста —
о ЯТЛ <*Cd2+ 0 0,059, ^Cd2+
— активность кадмия в амальгаме.
Среди электродов первого рода в отдельную группу выделяют газовые
электроды, к которым относятся водородный, кислородный, хлорный и др.
Водородный электрод обратим относительно катиона, кислородный и хлор-
хлорный — относительно аниона. Все газовые электроды конструктивно устрое-
устроены одинаково. Они представляют собой инертный металл (Pt) с развитой
поверхностью, хорошо проводящий электрический ток и обладающий ката-
каталитическими свойствами по отношению к электродному процессу. Плати-
Платиновая пластинка электролитически покрывается слоем мелкодисперсной
платины с целью увеличения адсорбции газа поверхностью металла. Плати-
Платина одновременно контактирует с газом и раствором, содержащим соответст-
соответствующие ионы.
В стандартном кислородном электроде платиновая пластинка по-
погружена в раствор щелочи (NaOH, КОН) с активностью гидроксид-ионов,
равной 1 моль/л. Давление чистого кислорода (или его парциальное давле-
давление в смеси газов над раствором) составляет 101,3 кПа.
В щелочной среде кислородному электроду Н2О, ОН" | О2, Pt соответ-
соответствует уравнение электродного процесса
О2(Г) + 2Н2О(р) + 4е <z± 4ОН(Р)
или в эквивалентном виде
544 16. Электродные процессы
Уравнение Нернста для кислородного электрода при Т= 298 К имеет вид
о 0,0591 Р°Ло2 а°й2о /1^^оч
Фо2,н2о =Фо2>н2о +—J—*ё ~—• A6-28)
он" он" ао\г
В разбавленных растворах активность воды практически постоянна и
^н о 104
равна 1. Подставляя в уравнение A6.28) аОн- = —— = и учитывая,
что -lg ац+ = рН, а фо н 0 =0,401 В, после проведения преобразований и
ОН"
вычислений получают формулу для расчета потенциала кислородного элек-
электрода
^ 2 A6.29)
ОН"
Фо,,^ =1,229-0,059рН. A6.30)
ОН"
Сравнивая A6.24) и A6.29), можно сделать вывод, что потенциал во-
водородного электрода линейно увеличивается с уменьшением водородного
показателя рН (ростом кислотности) среды и уменьшением парциального
давления газообразного водорода над раствором. Потенциал кислородного
электрода линейно увеличивается с уменьшением рН раствора и увеличени-
увеличением парциального давления газообразного кислорода над ним.
Схемам водородного электрода в щелочной или нейтральной средах
Н2О, ОН" IН2, Pt и кислородного электрода в кислотной среде Н2О, Н+102, Pt
соответствуют уравнения электродных процессов
Уравнения Нернста для этих процессов будут отличаться от A6.23) и
A6.28), но после преобразований с учетом A4.12) и A4.15) также можно
получить для них выражения A6.25) и A6.30), пригодные для расчета
потенциалов при любом значении рН.
16.5. Классификация электродов
545
ф, в
1,6
1,2
0,8
0,4
0
-0,4
-0,8
I 5
¦••• ••••: л
.••Л'пЛ-з.
. • * • *
^—2^C*V 6
ш
1*
1#8. ".10 • 12*рН
/ • • • •
Ф, в
1,229
0,815
0,401
О
-0,414
-0,828
Рис. 16.7. Диаграмма электрохимической устойчивости воды
При увеличении рН раствора от 0 до 14 потенциал водородного элек-
электрода уменьшается от 0 до -0,828 В, а потенциал кислородного электрода —
от 1,229 до 0,401 В (рис. 16.7, прямые б и а соответственно).
Зависимости потенциалов различных окислительно-восстановитель-
окислительно-восстановительных систем от рН раствора называют диаграммами Пурбе. Их используют
для оценки термодинамической устойчивости этих систем в водных раство-
растворах, в частности при электрохимической коррозии металлов. Диаграммы
Пурбе учитывают три возможных типа равновесия в системе металл—вода:
равновесие, связанное с обменом электрическими зарядами:
Mz+ + Ze <=> М и определяемое только потенциалом, поэтому линии, харак-
характеризующие их, параллельны оси рН;
равновесие, не связанное с обменом электрическими зарядами:
M(OH)Z ^± Mz+ + ZOH~ и определяемое только значениями рН, поэтому
линии, характеризующие их, параллельны оси потенциалов;
равновесие, зависящее как от рН, так и от потенциала, например на
металл-оксидных электродах: M(OH)Z + Ze <=± М + ZOH". Потенциал таких
электродов описывается уравнением
Ф m(oh)z = Ф°м(онJ -0,059рН.
М, ZOH" M, ZOH"
Из рис. 16.7 ясно, что во всем диапазоне изменения значений рН по-
потенциал кислородного электрода приблизительно на 1,23 В больше, чем по-
потенциал водородного электрода, следовательно, в водных растворах моле-
18 - 9795
546 16. Электродные процессы
кулы кислорода более сильный окислитель, чем катионы водорода или мо-
молекулы воды.
На рис. 16.7 можно выделить три области.
I- Фоф >сРо2,н2о> т-е- окисленная форма окислительно-восстанови-
Вф ОН"
тельной системы термодинамически способна окислять воду в кислотных
или гидроксид-ионы в нейтральных и щелочных растворах с выделением
кислорода. Восстановленная форма кислородом, растворенным в воде,
окисляться не будет. Именно поэтому металлы с Ц>°мг+ > 1,229 В не корро-
м
дируют (не окисляются) в водных растворах в присутствии кислорода при
любом значении рН.
П. фн+ <фОф <(Ро2,н2о> т-е- восстановленная форма окислительно-
окислительному в* °н~
восстановительной системы будет окисляться растворенным в воде кисло-
кислородом.
III. фОф < фн+, т.е. восстановленная форма окислительно-восстанови-
ВФ 1н7
тельной системы, с точки зрения термодинамики, будет окисляться катио-
катионами водорода в кислотных или молекулами воды в нейтральных и щелоч-
щелочных растворах с выделением водорода в качестве продукта восстановления.
Молекулы растворенного в воде кислорода тоже могут быть окислителем.
Область И, в которой разложение воды в присутствии окислительно-
восстановительных систем термодинамически невозможно, называют обла-
областью электрохимической устойчивости воды.
Электроды второго рода представляют собой металл, покрытый сло-
слоем его малорастворимого соединения (соли, оксида или гидроксида) и по-
погруженный в раствор, содержащий анионы, одноименные с анионами
труднорастворимого соединения. Условная запись электрода второго рода
Az~ | MA, M. В качестве примеров можно привести хлоридсеребряный
СГ | AgCl, Ag и каломельный СГ | Hg2Cl2, Hg электроды, на которых про-
протекают процессы
Уравнения для расчета электродных потенциалов при температуре 298 К
имеют вид
16.5. Классификация электродов 547
<PAgci =<PQAga -0,0591g*cr,
Ag,CF Ag,CP
9Hg2ci2 =Фнё2а2 -0,0591gaa_.
Hg,cr Hg,cr
Электроды второго рода обратимы одновременно и по катиону, и по
аниону. Устанавливаются два типа гетерогенного равновесия:
между атомом металла в твердой фазе и катионом малорастворимого
соединения в растворе
между ионами малорастворимого соединения в растворе и его твердой
фазой
МА(Т) ?=* М(р5 + А^. A6.32)
Суммируя A6.31) и A6.32), получают уравнение электродного про-
процесса
";. A6.33)
В электродах второго рода окисленной формой является малораство-
малорастворимое соединение (МА), восстановленной — атом металла (М) и анион рас-
раствора (А7~).Уравнение Нернста для данного электрода имеет вид
Ф МА =Ф°МА +^[
При «ма = ^м = 1 это уравнение принимает вид
Ф ма =(Р ma -—lnaAZ.. A6.34)
M,AZ" M,AZ" Z
Для процесса, описываемого A6.31), электродный потенциал
о ЛГ,
м м
18*
548 16. Электродные процессы
Подставляя в A6.35) под знак логарифма вместо ам2+ ее значение, вы-
вычисленное из произведения растворимости малорастворимого соединения
MA, a z+ = —, получают
v-
о RT_, пр _Д7\
м м
Выражения A6.34) и A6.36) описывают потенциал одного и того же
электрода. Приравнивая их, получают соотношение между стандартными
потенциалами процессов A6.31) и A6.33), которое часто используют для
RT
расчета произведения растворимости ф°МА = 9^+ + 1пПРМА.
Окислительно-восстановительный электрод представляет собой пла-
платиновую пластинку, погруженную в раствор, содержащий вещества, в со-
состав которых входит один и тот же элемент в разной степени окисления или
нескольких веществ. Такой раствор называют окислительно-восстанови-
окислительно-восстановительной системой. В потенциалопределяющих процессах этих электродов не
участвуют материал электрода и атомы или молекулы простых веществ.
Платиновая пластинка в окислительно-восстановительных электродах
играет роль посредника при переносе электронов от восстановленной фор-
формы к окисленной. Непосредственный обмен электронами между окисленной
и восстановленной формами принципиально возможен, но он, если учиты-
учитывать гидратацию ионов в растворе, протекает с гораздо большей энергией
активации, поэтому скорость такого процесса существенно меньше.
Скачок потенциала, возникающий в состоянии равновесия на инерт-
инертном платиновом электроде, погруженном в окислительно-восстановитель-
окислительно-восстановительную систему, называют равновесным окислительно-восстановительным
потенциалом. Его относительную величину обозначают фОф.
Вф
Различают два типа окислительно-восстановительных электродов:
простые и сложные. Приведем несколько примеров таких электродов, запи-
записывая последовательно схему электрода, уравнение потенциалопределяю-
щего процесса и уравнение Нернста для расчета потенциала.
В электродных процессах простых электродов осуществляется пере-
перенос только электронов, например:
Sn4+, Sn2+ | Pt Snft + 2e *± Sn2+
p}
RT, <
In-
a
16.5. Классификация электродов 549
В электродных процессах сложных электродов происходит перенос
электронов и протонов, например:
no;, н+ | no, Pt
NO,H2O NO,H2O
i, Mn2+, H+ I Pt MnO;(p) + 8Нф} +5e<± Mn?, + 4H2O
(p)
^7\
Mn2+, H20 Mn2+, H2O Mn , л , ^ -ч
A6.37)
^
= Ф°мпо4-,н+ + 0,01181g^-0,0944pH.
Mn2+,H2O a™*l+
Потенциалы таких электродов могут, как это следует из A6.37), в
большей степени зависеть от рН раствора, нежели от активностей окислен-
окисленной и восстановленной форм. Это позволяет использовать их в качестве ин-
индикаторных электродов на катионы Н+.
Стандартным окислительно-восстановительным потенциалом на-
называют скачок потенциала, возникающий на границе раздела платина —
раствор, в котором активности окисленной и восстановленной форм равны
1 моль/л (или просто равны между собой при равенстве их стехиометриче-
ских коэффициентов), а в сложных электродах активность ионов водорода
также должна составлять 1 моль/л. Стандартные потенциалы окислительно-
восстановительных электродов являются константами при данной темпера-
температуре (см. табл. П.6.1).
Ионообменные (ионоселективные) электроды — это датчики, потен-
потенциалы которых линейно зависят от логарифма активности определяемого
иона в растворе. Их важнейшая составная часть — тонкая пленка (мембра-
(мембрана), отделяющая внутренний раствор электрода от внешнего (анализируемо-
(анализируемого) и пропускающая преимущественно ион одного вида. Эта пленка содер-
содержит ионогенные группы. Между мембраной и обоими растворами происхо-
происходит обмен ионами по схеме
где М+, N+ — вид обменивающихся катионов, а индексы «(м)», «(р)» отно-
относятся к мембране и каждому из растворов. Активность ионов N+(p) по обе
550 16. Электродные процессы
стороны пленки различна, поэтому возникают скачки потенциала, сумму
которых называют мембранным потенциалом.
Мембраны в ионоселективных электродах могут быть жидкими или
твердыми. Жидкие мембраны — это органические жидкости, нераствори-
нерастворимые в воде, но растворяющие ионогенные вещества. Твердые мембраны ча-
чаще всего изготавливают из стекла (стеклянный электрод), монокристаллов
или прессованных малорастворимых соединений.
В настоящее время созданы ионообменные электроды для нескольких
десятков катионов и анионов. Определение концентрации ионов сводится к
измерению ЭДС гальванического элемента, составленного из анализируемо-
анализируемого и стандартного растворов, в каждый из которых погружены одинаковые
ионоселективные электроды. Концентрацию определяемого иона в растворе
вычисляют по формуле
7F
lgC = —+ lgC0,
где Со — концентрация определяемого иона в стандартном растворе; 9 =
= 58,2 мВ при температуре 25 °С — изотермическая константа.
17 + Химические источники тока
и электролиз
Химическими источниками тока называют устройства, превра-
превращающие химическую энергию окислительно-восстановительных
реакций в электрическую. К ним относятся гальванические эле-
элементы, аккумуляторы и топливные элементы.
Электролиз — совокупность окислительно-восстановительных
процессов, протекающих на электродах при пропускании элек-
электрического тока, подаваемого от внешнего источника, через рас-
раствор или расплав электролита. Ячейка, состоящая из двух элек-
электродов, соединенных с внешним источником постоянного тока и
помещенных в электролит, называется электролизером. Продукты
электролиза, выделяющиеся на аноде и катоде, могут химически
взаимодействовать, поэтому в электролизерах анодное и катодное
пространства разделяют диафрагмами из асбеста, керамики или
пластмассы.
17.1. Химические и концентрационные гальванические
элементы. Элемент Даниэля—Якоби
Химические гальванические элементы состоят из двух элек-
электродов, различающихся потенциалопределяющими процессами.
Примером служит элемент Даниэля—Якоби (рис. 17.1), состоящий
из двух электродов — цинкового и медного. Цинковая пластина
погружена в раствор сульфата цинка, а медная — в раствор сульфа-
сульфата меди. В стандартных термодинамических условиях при аси2+ =
= tfzn2+ = 1 моль/л электроды имеют следующие потенциалы:
Ф^ 2+ =-0,763 В, ц>°Г 2+ = 0,337 В, следовательно, цинковый элек-
Zn Cu
трод заряжен отрицательно и является анодом, а медный — поло-
положительно, и это — катод. Анодное и катодное пространства разде-
разделены пористой полупроницаемой перегородкой 7. Электроды со-
соединены между собой металлическим проводником через гальва-
гальванометр 2.
На аноде происходит процесс окисления, и цинк переходит в
раствор в виде катионов
(—)A(Zn):Zn-»Zn2+ + 2e
Масса цинковой пластинки уменьшается, остающиеся на ней
электроны сообщают ей отрицательный заряд.
552
/ 7. Химические источники тока и электролиз
На катоде восстанавливаются
ионы меди, осаждающиеся на пластинке:
(+)K(Cu):Cu2+ + 2e->Cu
Масса медной пластинки увеличивает-
увеличивается, в результате расходования некото-
некоторого количества свободных электронов
она приобретает положительный заряд.
Токообразующая реакция пред-
представляет собой сумму анодной и ка-
катодной полуреакций:
Рис. 17.1. Схема гальванического эле-
элемента Даниэля—Якоби:
1 — пористая полупроницаемая перего-
перегородка; 2 — гальванометр
Zn + Cu2+
Zn2+ + Си A7.1)
или в молекулярном виде
Zn + CuSO4 -> ZnSO4 + Си A7.2)
В гальваническом элементе возникает направленное движение ионов
во внутренней цепи и электронов во внешней (электрический ток), поэтому
A7.1) и A7.2) называются уравнениями токообразующего процесса. Реак-
Реакции A7.1) и A7.2) могут протекать при погружении цинковой пластины в
стакан с раствором медного купороса (CuSO4 • 5Н2О), но в этом случае
анодный и катодный процессы пространственно не разделены, а движение
электронов в коррозионных гальванических микроэлементах происходит на
очень коротких отрезках и не является электрическим током.
Электродвижущая сила и напряжение гальванического элемента.
В случае соединения полюсов элемента Даниэля—Якоби медным провод-
проводником через гальванометр (см. рис. 17.1) его условную запись представляют
в виде
Си | Zn | ZnSO4 ! CuSO4 | Си
12 3 4
A7.3)
Цифры на схеме указывают границы раздела фаз с соответствующими скач-
скачками потенциалов. Граница раздела между двумя растворами (проводника-
(проводниками второго рода) изображена пунктирной вертикальной чертой.
Электродвижущая сила гальванического элемента равна алгебраиче-
алгебраической сумме всех скачков потенциалов на границах раздела фаз при наступ-
наступлении равновесия на каждой из границ
Е = ф конт + фа + фд + фк = ф1 + ф2 + фЗ + ф4,
A7.4)
17.1. Химические и концентрационные гальванические элементы 553
где фконт = 9i = Феи — контактный
Zn
потенциал; Фа = Ф2 = Ф zn — потен-
Zn2+
циал анода; фд = ф3 = ф2п2+ —
2+
Рис. 17.2. Схема гальванического эле-
элемента Даниэля—Якоби с элиминиро-
элиминированным диффузионным потенциалом:
1 — солевой мостик; 2 — гальванометр
Си
диффузионный потенциал; фк = ф4 =
= ФСи2+ — потенциал катода. Все
Си
потенциалы равновесные, сила тока
в цепи стремится к нулю.
В правильно разомкнутом ГЭ
его полюсы соединяют проводни-
проводником из третьего металла, например
серебра. Контактный потенциал
Фконт = Феи ПРИ этом не изменяется,
Zn
его величина, по определению, входит в алгебраическую сумму фк и фа (или
ф? и ф^ в стандартных условиях) и является постоянной величиной при
данной температуре. Диффузионный потенциал фд исключают из A7.4),
снижая его величину до пренебрежимо малого значения включением в цепь
солевого мостика, который на схемах изображают двумя сплошными верти-
вертикальными линиями.
Элемент Даниэля—Якоби, составленный в соответствии с указанны-
указанными изменениями, изображен на рис. 17.2. Говорят, что в этом случае кон-
контактный потенциал элиминирован (расщеплен). На рис. 17.1 и 17.2 направ-
направления электрического тока и движения электронов противоположны, как это
принято в физике.
Схема гальванического элемента Даниэля—Якоби с элиминирован-
элиминированным диффузионным потенциалом имеет вид
(—) Ag | Zn | ZnSO4 || CuSO41 Cu I Ag (+)
12 3 4 5
причем ф3 = 0. Тогда
Е = ф2 + ф4 = фа + фк = Ф_гп_ + ФСи2+ ' A7'5)
Zn2+ Tu~
В ряду стандартных электродных потенциалов значения фоф приве-
Вф
дены для процессов восстановления. При изменении направления реакции
554 17. Химические источники тока и электролиз
на противоположное (окисление) знак потенциала также изменяется, поэто-
поэтому потенциал катода записывают при алгебраическом сложении со своим
знаком, а анода — с противоположным. Таким образом, ЭДС элемента равна
разности равновесных электродных потенциалов катода и анода:
Е = фк - Фа = ФСи2+ - Ф2п2+ • A7.6)
Си Zn
В случае стандартных медного и цинкового электродов (аСи2+ =
= aZn2+ = 1 моль/л) имеет место стандартная ЭДС
= ф°Сц2+ - Ф°2п2+ =
Си Zn
= ф°Сц2+ - Ф°2п2+ = 0,337 - (- 0,763) = 1,10 В.
Если электроды в элементе Даниэля—Якоби нестандартные (аси2+ *
Ф 1 моль/л; я2п2+ * 1 моль/л), их потенциалы рассчитывают по уравнению
Нернста, в результате чего выражение A7.6) приобретает вид
^ A7.7)
Е Е+\п
или через десятичный логарифм
Выражения A7.7) и A7.8) аналогичны уравнению Нернста для электродного
потенциала, поэтому в литературе их часто называют уравнениями Нернста
для расчета ЭДС гальванического элемента.
В общем виде для любой окислительно-восстановительной реакции,
лежащей в основе работы гальванического элемента
аОф2 + ЬВфх <=> сВф2 + <Юф1 A7.9)
уравнение Нернста имеет вид
v 7
где Е° =Фоф2 "Фоф, — стандартная ЭДС при яОф2 = <*>h = ^вф2 = ^вф! = 1,
> Ф
Фоф2 > Фоф!' Z — число электронов, принимающих участие в токообра-
Вф2 Вф!
17.1. Химические и концентрационные гальванические элементы 555
зующей реакции. Для газообразных веществ в A7.10) активности заменяют
парциальными давлениями.
При работе гальванического элемента в необратимых условиях через
электрохимическую систему проходит электрический ток и электроды вы-
выходят из равновесного состояния (поляризуются).
Напряжение гальванического элемента представляет собой разность
неравновесных потенциалов катода и анода:
U =Фк/ ~Фа/ =(Pcu^7 "^Zn^/ (ПЛ1)
Си Zn
Напряжение гальванического элемента меньше чем его ЭДС на сумму
величин перенапряжения гальванического элемента Г|гэ и омического паде-
падения напряжения во внешней и внутренней цепях, равного Zt/ = I(R{+ R2)'.
где / — сила тока в цепи, А; Ru R2 — сопротивления внешней и внутренней
цепей ЭХС соответственно, Ом. Напряжение можно определить путем пря-
прямого измерения разности потенциалов на клеммах гальванического элемен-
элемента с помощью вольтметра. ЭДС определяют компенсационным методом (см.
рис. 16.6) или менее точно — путем прямого измерения напряжения с по-
помощью высокоомного вольтметра.
Частным случаем химических гальванических элементов являются
окислительно-восстановительные элементы. Из двух электродов хотя бы
один должен быть окислительно-восстановительным. Если в качестве вто-
второго электрода использовать стандартный водородный, то ЭДС элемента
(-) Pt, H21 H + II Fe3+, Fe2+ I Pt (+)
Ри2 = ан+ = 1
равна электродному потенциалу окислительно-восстановительной системы
где фр з+ — стандартный потенциал окислительно-восстановительной сис-
темы, равный ее потенциалу при яРез+ = aFe2+. Электродные и токообразую-
щий процессы в таком элементе описываются уравнениями
556
17. Химические источники тока и электролиз
(-)А:Н2(г)->2Н+)+2е
S:
2Fe?)+H2(r)
2Fe*;+2Hj
e;
(р)
xl
х2
Z = 2
Различают химические гальванические элементы с переносом (при на-
наличии в ЭХС границы раздела двух растворов электролитов) и без переноса
(в отсутствие такой границы, т.е. при соединении анодного и катодного про-
пространств солевым мостиком).
Концентрационные гальванические элементы состоят из двух одина-
одинаковых электродов, у которых различаются активности одного или несколь-
нескольких участников электродного процесса. Они генерируют электрическую
энергию за счет выравнивания химических потенциалов веществ в раство-
растворах. Существуют следующие концентрационные элементы:
а) элементы с различной активностью иона в растворах электролита
катодного и анодного пространств, например никелевый концентрационный
гальванический элемент
В потенциал каждого электрода входит константа ф^.2+, поэтому
^.2
уравнение Нернста для расчета ЭДС такого элемента имеет вид
RT '
? =—In-
2F
где а\ Ni2+ и а2 ы2+ — активности катионов никеля в анодном и катодном про-
пространствах соответственно.
Уравнения электродных процессов:
(-) A: Ni -> Ni2+ + 2e
(+) К: Ni2+ + 2е -> Ni
б) элементы с одним раствором электролита, у которых различаются
активности металла в составе сплавов катода и анода или давления газа в
газовых полуэлементах, например амальгамный концентрационный элемент
(-)Cd (Hg) | CdSO 41 Cd (Hg) (+)
Я1 Cd(Hg) > Я2 Cd(Hg)
и водородный концентрационный элемент
17.2. Термодинамика окислительно-восстановительных реакций 557
(-)Pt,H2|H+|H2,Pt(+)
/>1Н2>/?2Н2
Уравнения Нернста для расчета их ЭДС имеют вид соответственно
2F a2Cd(Hg)
? =—In
IF
где яi cd(Hg) и a2 cd(Hg) — активности кадмия в амальгамах анода и катода; р\ н2
и/?2 н2 — давления водорода на аноде и катоде.
Анодами в концентрационных гальванических элементах всегда являют-
являются электроды с меньшими значениями активностей (давлений) окисленной
формы при яВф ~ const (пример а) или с большими значениями активностей
(давлений) восстановленной формы при доф = const (пример б). Стандартная
ЭДС для концентрационных гальванических элементов равна нулю.
17.2. Термодинамика окислительно-восстановительных реакций,
протекающих в гальванических элементах
Работа гальванического элемента. Критерии равновесия и само-
самопроизвольного протекания процессов. При прохождении тока в гальвани-
гальваническом элементе осуществляется движение носителей зарядов (электронов
во внешней цепи и ионов во внутренней). Совершаемая при этом электриче-
электрическая работа равна произведению перенесенного заряда (ZF) и разности по-
потенциалов катода и анода (напряжение U)
Максимальную работу система совершает при равновесном проведе-
проведении процесса, когда (рк - сра = Е. В этом случае
A7.12)
В стандартных условиях для элемента Даниэля—Якоби
W3n max = ZFE° = 2 * 96500 * 1,Ю- 1(Г3 = 212,3 КДЖ.
В силу второго закона термодинамики максимальная работа, совер-
совершаемая системой при протекании реакции в изобарно-изотермических усло-
условиях, равна убыли энергии Гиббса реакции
558 17. Химические источники тока и электролиз
WMmm=-ArGpJ. A7.13)
Сравнивая выражения A7.12) и A7.13), получают
ZFE=-ArGpJ, A7.14)
или для стандартных условий
ZFE°=-ArG%. A7.15)
Уравнения A7.14) и A7.15) устанавливают связь между электрической
и химической формами энергии. По известным значениям энергии Гиббса
токообразующей реакции можно рассчитать ЭДС гальванического элемента
и наоборот. Величины Е и ArGpj> — критерии равновесия и самопроиз-
самопроизвольного протекания окислительно-восстановительных реакций в растворах
электролитов. Термодинамическим условием самопроизвольного протекания
процесса в закрытой системе в прямом направлении является положитель-
положительное значение ЭДС (Е> 0), соответствующее, согласно A7.14), ^rGpT < 0.
Термодинамическим условием равновесия является равенство ЭДС
гальванического элемента нулю, т.е. равенство потенциалов катодного и
анодного процессов, что соответствует ArGp T = 0.
При Е > 0 окислительно-восстановительная реакция в гальваническом
элементе в отсутствие кинетических затруднений протекает в прямом на-
направлении. Согласно уравнению Нернста, потенциал катодной полуреакции
Фоф2 уменьшается, а анодной
увеличивается вследствие соответ-
Вф2
ствующих изменений активностей окисленной и восстановленной форм
окислительно-восстановительных систем. При химическом равновесии по-
потенциалы обеих сопряженных окислительно-восстановительных пар равны
и ЭДС равна нулю. В этом случае A7.10) приобретает вид
Е° = 1пК°р, A7.16)
где К® — стандартная термодинамическая константа равновесия токообра-
зующей реакции A7.9). В качестве критерия для стандартных условий мож-
можно использовать стандартную ЭДС: при Е° > 0 прямой процесс термодина-
термодинамически возможен, а при Е° < 0 он термодинамически невозможен, при Е° = 0
в системе существует химическое равновесие.
При применении критерия самопроизвольного протекания процесса
для нестандартных условий можно отметить следующее.
17.2. Термодинамика окислительно-восстановительных реакций 559
1. Если для окислительно-восстановительной реакции Е° > 0,3 В
(ArG$ < -30 кДж) или Е° < -0,3 В (ArGj > 30 кДж), то направление реакции
определяется практически однозначно: в первом случае процесс термодина-
термодинамически возможен, а во втором — нет.
2. При -0,3 В < ?° < 0,3 В направление и глубину протекания процесса
невозможно определить однозначно. В этом случае при расчете потенциа-
потенциалов полуреакций по уравнению Нернста необходимо учитывать рН среды,
температуру, активности или давления исходных веществ и продуктов реак-
реакции, поскольку при изменении параметров процесса может изменяться на-
направление его протекания.
Например, окислительно-восстановительная реакция, по которой в ла-
лаборатории получают хлор
6NaCl + K2Cr207 + 7H2SO4 <=> ЗС12 + Cr2(SO4K + 7Н2О + 3Na2SO 4 + K2SO4
при стандартных условиях, т.е. при aQr q2_ = яСгз+ = aR+ = aQr = aQXi = 1 и
T- 298 К, протекает в обратном направлении (справа налево), так как
Сг3+,Н2О
В условиях, отличных от стандартных, она может изменить направление на
противоположное. Если в системе изменить только один параметр, напри-
например а з+ = Ю, то в соответствии с уравнением Нернста
о 0,059 асг2оГа1н+ . „, . Ода * nQ9R
УГЖ =(Рсг2оГ,н+ +~T"lg~^ = 1,333 + ——6 = 1,392 В.
Сг3+,Н2О Сг3+,Н2О Сг3+
Тогда
? = ФСГ2О2- н+ -9^=1,392-1,358 = 0,034 В>0
Сг3+,Н2О СГ
и реакция самопроизвольно протекает в прямом направлении (слева напра-
направо), т.е. дихромат-ион является в кислотной среде окислителем хлорид-иона,
превращая его в молекулы хлора и восстанавливаясь до катионов Сг3+.
При определении направления самопроизвольно протекающей окис-
окислительно-восстановительной реакции можно использовать простое правило:
если две полуреакции, записанные в соответствии с правилами ИЮПАК
(как восстановление), разместить так, чтобы потенциал верхней был меньше
потенциала нижней, то проведенная между ними буква Z укажет направле-
560
/ 7. Химические источники тока и электролиз
ние самопроизвольного протекания полуреакций. Например, для реакции
получения хлора
1+ + бе <z> 2 Сг3+ + 7Н2О
Уравнения анодной и катодной полуреакций и суммарное уравнение
токообразующей реакции имеют вид
(-) А: 2Сг3+ + 7Н
(+)К:С12+2е-)-2СГ
14Н+ + бе
: 2Cr3+ + 7Н2О+ЗС12
^ + 14Н+ + 6СГ
xl
хЗ
Z = 6
Связь ЭДС гальванического элемента с другими термодина-
термодинамическими функциями. Чтобы определить связь ЭДС гальванического
элемента со стандартными энтальпией и энтропией реакции, следует
воспользоваться уравнениями A7.14), обе части которого умножены на -1,
ArG = -ZFE A7.17)
и F.24), записанным для нестандартных условий (см. гл. 6):
ArG = ArH-TArS. A7.18)
Приравнивая правые части A7.17) и A7.18) и деля полученное выра-
выражение на -ZF, находят
AM ArS.
A7.19)
ZF ZF
Затем дифференцируют по температуре обе части уравнения A7.19) в пред-
предположении независимости энтальпии и энтропии реакции от температуры:
dE ArS A c __dE
— = —^, или ArS = ZF—.
dT ZF r dT
Подставляя это значение ArS в A7.19), получают
ZF dT
A7.20)
A7.21)
17.2. Термодинамика окислительно-восстановительных реакций 561
или
ИР
A7.22)
dT
Зависимость ЭДС гальванического элемента от температуры опреде-
определяется A7.21). Величину dEldT называют температурным коэффициентом
ЭДС. Измеряя величину Е при различных значениях Г, находят этот
коэффициент и вычисляют термодинамические характеристики (энтальпию,
энтропию, энергию Гиббса) токообразующего процесса.
Анализ выражения A7.22) позволяет утверждать, что электрическая
работа гальванического элемента не равна энтальпии (тепловому эффекту)
протекающей в нем реакции. При dE/dT< О работа меньше энтальпии, сле-
следовательно, гальванический элемент в закрытой системе при р,Т= const от-
отдает теплоту в окружающую среду или нагревается в изолированной систе-
системе. При dEldT > О электрическая работа больше энтальпии, значит, гальва-
гальванический элемент в закрытой системе при /?, Т = const поглощает теплоту из
окружающей среды или охлаждается в условиях тепловой изоляции. Заме-
Заметим, что ЭДС эталонных гальванических элементов практически не зависит
от температуры {dEldT» О).
Применяя законы термодинамики к ЭХС, можно рассчитать значения
ЭДС и равновесных электродных потенциалов. Уравнение изотермы для
токообразующей реакции A7.9), протекающей в изобарно-изотермических
условиях, имеет вид
&rG = -RT]nK%-RTln c 2 d ' , A7.23)
где К® — стандартная термодинамическая константа равновесия реакции;
а, — неравновесные активности веществ-участников реакции; а, Ь, с, d —
стехиометрические коэффициенты.
Объединив A7.17) и A7.23), можно получить уравнение Нернста для
ЭДС гальванического элемента
ЫКа+^\а^ф^. A7.24)
ZF a ZF асвф2а"оф|
При я, = 1
RT
Е = Е°=^-]пК°а, A7.25)
Zr
следовательно, уравнение A7.24) принимает вид
]nao*>^ . A7.25а)
ZF *вфХоф,
562
/ 7. Химические источники тока и электролиз
Уравнение Нернста A7.25а) позволяет рассчитать ЭДС гальваниче-
гальванического элемента в зависимости от температуры и активностей веществ-
участников токообразующей реакции.
17.3. Практическое применение химических источников тока
Среди химических источников тока, применяемых на практике, мож-
можно выделить сухие гальванические элементы (ГЭ), электрохимические ак-
аккумуляторы и топливные элементы.
Сухие гальванические элементы. Эти элементы широко применяют
как источники питания для радиоаппаратуры, карманных фонарей и др. Их
принцип работы и устройство сходны, различия заключаются в химической
природе электродов (см. табл. П.6.2). Анодами являются активные металлы
(Zn, Mg, Li), катодами — оксиды марганца, меди, серебра, хлориды свинца,
меди и другие вещества-окислители.
Самым распространенным среди сухих ГЭ является марганцево-
цинковый элемент, схема которого приведена на рис. 17.3. Его условную за-
запись и электродные реакции можно представить в виде
(-)Zn|NH4Cl|C,Mn02(+)
(-) A: Zn -> Zn2+ + 2e
(+) К: 2МпО2 + 2Н2О + 2е -> 2MnO(OH) + 2OH"
Анодом является цинковый электрод 7, катод 2
изготовлен из смеси диоксида марганца с графитом
(для увеличения электрической проводимости). Токо-
отвод 3 изготовлен из чистого графита. Паста-
электролит 4 состоит из раствора хлорида аммония и
загустителя (мука, крахмал).
При анодном окислении цинка образуется
пленка труднорастворимого гидроксида
,1
,2
,3
А
Рис. 17.3. Схема су-
сухого марганцево-цин-
кового элемента:
1 — анод (цинковый
стаканчик); 2 — катод
(смесь диоксида мар-
марганца с графитом); 3 —
токоотвод из графита с
металлическим колпач-
колпачком; 4 — электролит
Zn2++2OH~->Zn(OHJ
препятствующая дальнейшему протеканию анодного
процесса. Для растворения гидроксида цинка приме-
применяют хлорид аммония, поэтому анодный процесс
описывается уравнением
(-) A: 2Zn + 4NH4C1 +
[Zn(NH3L]Cl2 + ZnCl2
4ОгГ ->
+ 4Н2О + Ае
17.3, Практическое применение химических источников тока 563
Суммарное уравнение токообразующей реакции имеет вид
2Zn + 4MnO2 + 4NH4C1 -> 4Mn0@H) + ZnCl2 + [Zn(NH3L]Cl2
Заменяя цинк на магний, имеющий меньшее значение стандартного
электродного потенциала ф^ 2+ =-0,763 В; ср^ 2+ =-2,363 В , или исполь-
V Zn Mg )
зуя щелочной электролит (раствор КОН), можно повысить напряжение сухого
ГЭ. Высокие напряжение и удельную энергию имеет серебряно-цинковый эле-
элемент, но его стоимость высока вследствие дороговизны оксида серебра.
Сухие ГЭ сравнительно дешевы, маловосприимчивы к сотрясениям и
механическим воздействиям. На их основе выпускаются батареи напряже-
напряжением от 3 до 102 В, емкостью от 0,5 до 30 А • ч и массой от 100 г до 20 кг.
Главные недостатки всех сухих ГЭ — однократность использования и само-
саморазрядка, понижающая напряжение и емкость.
Электрохимические аккумуляторы. Аккумуляторами называют
устройства, в которых поэтапно происходит преобразование электрической
энергии в химическую, а химической — в электрическую. Конструктивно
они представляют собой агрегаты многоразового действия, сочетающие в
себе ГЭ и электролизер (см. табл. П.6.3). Процесс накопления химической
энергии под действием внешнего постоянного тока называют зарядкой ак-
аккумулятора, а процесс ее превращения в электрическую — разрядкой. В
первом случае аккумулятор работает как электролизер, а во втором — как ГЭ.
В настоящее время наиболее распространены свинцовые аккумулято-
аккумуляторы. Из свинца с примесью сурьмы (хартблея) отливают пластины ячеистой
структуры, собирают в батареи и помещают в корпусы из полипропилена. В
ячейки предварительно запрессовывают смесь оксида свинца (II) РЬО с гли-
глицерином, затвердевающую в виде глицерата свинца. Свинцовые аккумуля-
аккумуляторы называют кислотными, поскольку электролитом в них служит раствор
серной кислоты. Оксид свинца (II) при взаимодействии с H2SO4 превраща-
превращается в сульфат (PbSQO, а последний при зарядке восстанавливается до
свинца (РЬ) на катоде и окисляется до оксида свинца (IV) (РЮг) на аноде.
Кислотные аккумуляторы широко применяют на автомобилях, самолетах,
электростанциях и т.д.
При разрядке аккумулятор работает как ГЭ, на электродах которого
протекают следующие процессы:
(-) А: РЬ -> РЬ2+ + 2е
(+) К: РЬО2 + 4Н+ + 2е -> РЬ2+ + 2Н2О
С учетом связывания образующихся катионов РЬ2+ в сернокислотной
среде в малорастворимый сульфат (PbSC>4), оседающий на поверхности
564 17. Химические источники тока и электролиз
электродов, электродные полуреакции и токообразующую реакцию запи-
записывают в виде
(-) А: РЬ(Т) + SO^p) -> PbSO4(T) + 2е
(+) К: РЬО2 (т) + SO^p) + 4Н+р) + 2е -> PbSO4(T) + 2Н2О(р)
I: Pb (T) + PbO2 (T) + 2H2SO4(P) -> 2PbSO4(T) + 2И
ЭДС аккумулятора рассчитывают по уравнению
,,_ о о RT. а1о1- ДУ
PbSO4,H2O Pb.SOa ^
0,059. asoraH+
= 1,685-(-0,356)
н2о
С ростом активности (концентрации) H2SO4 увеличивается ЭДС, но
при массовой доле кислоты coH2so4 > 0,4 резко уменьшается электропровод-
электропроводность растворов и увеличивается растворимость свинца, поэтому оптималь-
оптимальными являются 32.. .39%-ные растворы H2SO4.
При зарядке аккумулятор работает как электролизер — электродные
процессы и токообразующая реакция имеют обратное направление. На ка-
катоде PbSO4 восстанавливается до РЬ, на аноде PbSO4 окисляется до РЬО2 —
происходит регенерация активных веществ. Достоинства свинцовых акку-
аккумуляторов — высокий КПД (до 80 %), большое число циклов (зарядка—
разрядка), высокая ЭДС, устойчивость в работе. К недостаткам можно отне-
отнести малый срок службы B-5 лет), большую массу, токсичность свинца, вы-
выделение водорода при зарядке.
В промышленности изготавливают щелочные аккумуляторы, среди
которых наиболее распространены никель-кадмиевые и никель-железные.
Они более долговечны (до 10 лет) и удобны в эксплуатации. В качестве
электролита используют раствор гидроксида калия КОН. Уравнения элек-
электродных и токообразующего процессов при разряде никель-кадмиевого ак-
аккумулятора имеют вид
(-) А : Cd(T) + 2ОН^ -> Cd(OHJ (т) + 2е
(+) К : 2NiO(OH)(T) + 2Н2О(Р) + 2е -> 2Ni(OHJ (т) ^
I: Cd(T) + 2NiO(OH)(T) + 2Н2О(р) -> Cd(OHJ (т) + 2Ni(OHJ (т)
Гальванические элементы называют первичными источниками тока, а
аккумуляторы — вторичными.
17.3. Практическое применение химических источников тока
565
-~f-
Топливные элементы. Это уст-
устройства непрерывного действия, в ко-
которых энергия сгорания топлива непо-
непосредственно превращается в электро-
электроэнергию. Они относятся к первичным
химическим источникам тока. Окисле-
Окисление топлива происходит на поверхно-
поверхности индифферентных электродов (гра-
(графит, Pt, Ag, Ni и др.), содержащих ката-
катализатор. Восстановителем (топливом)
могут быть водород, гидразин, уголь,
оксид углерода, углеводороды и другие
органические соединения, окислителем
чаще всего является чистый кислород
или воздух.
В настоящее время наиболее изучен водородно-кислородный топлив-
топливный элемент с щелочным электролитом C0-40%-ный раствор КОН), схема
которого приведена на рис. 17.4. Химическая энергия реакции окисления
водорода (Нг + 0,5Ог -> НгО) превращается в электрическую. Катод и анод
изготовлены из мелкодисперсных порошков угля и катализатора — металла
семейства платины и имеют пористую структуру для увеличения
поверхности. , К катоду подводится кислород (или воздух), восста-
восстанавливающийся до гидроксид-ионов, к аноду подается водород,
окисляющийся до воды. Уравнения электродных и токообразующего процес-
сов имеют вид () д . ^
Рис. 17.4. Схема водородно-кисло-
родного топливного элемента:
1 — анод; 2 — электролит; 3 — катод
(+) К : 02 + 2Н2О + 4е -> 40Н"
2: 2Н2 + 02 -> 2Н2О
При использовании раствора фосфорной кислоты (Н3РО4) в качестве
электролита вид электродных процессов изменяется, но суммарный процесс
остается прежним:
(-)А:2Н2->4Н+ + 4е
(+) К: О2 + 4Н + + 4е -> 2Н2О
X: 2Н2 + О2 -+ 2Н2О
ЭДС этого элемента не зависит от рН,
Е = Фк -Фа = 1,229-0,059рН -(-0,059рН) = 1,229В при Т= 298 К.
Для большинства топливных элементов значение ЭДС составляет
1,0...1,5 В.
566 17. Химические источники тока и электролиз
17.4. Электролиз. Потенциал разложения,
последовательность процессов на электродах
Суммарный процесс при электролизе противоположен по направле-
направлению процессу, протекающему в ГЭ. Поэтому в электролизере анодом явля-
является положительный электрод, на котором протекает реакция окисления, а
катодом — отрицательный (реакция восстановления). Потенциал (напряже-
(напряжение) разложения (?/н р) — минимальное значение внешней разности потен-
потенциалов, приложенной к электродам, при которой начинается электролиз
данного соединения. Для каждого вещества это константа, стандартное зна-
значение которой дано в справочниках.
Напряжение разложения превышает разность равновесных электрод-
электродных потенциалов анода и катода на сумму значений их электродной поляри-
поляризации (|т)а| + 1Лк|) и омического падения напряжения
Е4р = С/о + (hd + foJ) + ZC/, A7.26)
где С/о = фа - Фк — обратимое напряжение разложения электролита, числен-
численно равное ЭДС ГЭ, в котором протекает обратная реакция. Для увеличения
скорости электролиза к электродам прикладывают разность потенциалов,
превышающую напряжение разложения. При этом энергия, затрачиваемая
на компенсацию электродной поляризации и омических потерь, превраща-
превращается в теплоту. При электролизе тип электродного процесса зависит от мно-
многих факторов (состав электролита, материал электродов, температура, на-
напряжение, плотность тока и др.). Необходимо отметить, что электролиз воз-
возможен только для растворов и расплавов электролитов, когда в системе при-
присутствуют ионы.
Различают три типа электролиза:
с химическим разложением электролита;
с химическим разложением растворителя;
электролиз растворов солей металлов с растворимыми анодами, изго-
изготовленными из этих же металлов.
Первый и второй типы называют электролизом с нерастворимым
(инертным) анодом. К нерастворимым анодам относятся золото, платина
и металлы семейства платины, оксид свинца (IV), графит. В качестве
инертных анодов применяют металлы, которые практически не раство-
растворяются из-за большой анодной поляризации — титан, тантал, нержавею-
нержавеющая сталь, в щелочной среде железо и никель. Замедление скорости и по-
последующее прекращение процесса анодного растворения металла вслед-
вследствие образования защитных слоев называют пассивацией. Потенциал
инертного анода всегда больше, чем потенциал кислородного электрода
при данном значении рН.
17.4. Электролиз. Потенциал разложения 567
Примерами электролиза указанных типов служат:
электролиз водных растворов НВг, СиС12 с инертными электродами;
электролиз водных растворов КОН, Na2SO4, Са(Ж>зJ, A12(SC>4K с
инертными электродами;
электролиз водных растворов C11SO4 с медными и AgNC>3 с серебря-
серебряными электродами соответственно.
Наиболее просто протекает электролиз расплавов электролитов, так
как в них существует по одному виду катионов и анионов, которые разря-
разряжаются на электродах. Положительно заряженные ионы (катионы) подходят
к отрицательно заряженному электроду (катоду) и принимают от него элек-
электроны (восстанавливаются). Отрицательно заряженные ионы (анионы) дви-
движутся к положительно заряженному электроду (аноду) и отдают ему элек-
электроны (окисляются).
Хлорид кальция, имеющий ионную кристаллическую решетку, в рас-
расплаве существует в виде ионов
СаС12 (Т) »Са(ж) + 2С1(ж)
которые разряжаются на электродах
(+)А:2СГ->С12+2е
(-)К:Са2++2е->Са
xl
xl
Z = 2
I : СаС12->Са + С12
В водных растворах электролитов появляется второе вещество — во-
вода. В отсутствии гидролиза соли водородный показатель раствора рН = 7,
при гидролизе соли по аниону рН > 7 (избыток анионов ОН"), при гидролизе
по катиону рН < 7 (избыток катионов Н+). Молекулы воды, катионы водо-
водорода и гидроксид-анионы наряду с ионами растворенной соли могут участ-
участвовать в электродных реакциях.
Из электродных процессов наиболее вероятен тот, осуществление ко-
которого связано с минимальной затратой энергии. Весь ряд стандартных
электродных потенциалов окислительно-восстановительных систем заклю-
заключен между значениями (роф приблизительно от -3 до +3 В. К электродам
Вф
электролизера можно приложить любую постоянную разность потенциалов,
значительно превышающую U = 6 В, следовательно, катод — самый силь-
сильный восстановитель, а анод — самый сильный окислитель. Поэтому на ка-
катоде первым восстанавливается наиболее сильный окислитель (окисленная
форма системы с максимальным значением (роф )> на анОДе первым окисля-
Вф
568 17. Химические источники тока и электролиз
ется наиболее сильный восстановитель (восстановленная форма системы с
минимальным значением электродного потенциала).
В реальных процессах порядок разрядки ионов может быть нарушен
вследствие электродной поляризации и побочных процессов.
Рассмотрим возможные катодные процессы при электролизе водных
растворов:
а) восстановление катионов металла: (-) К: Mz+ + Ze —»М;
б) восстановление молекул воды в нейтральных и щелочных растворах:
(-) К: 2Н2О + 2е -> Н2 Т +2ОН" A7.27)
или ионов водорода в кислотных растворах:
И К: 2Н++2е->Н2Т A7.28)
в) восстановление других окислителей, имеющихся в растворе:
Уравнения A7.27) и A7.28) относятся к водородному электроду,
потенциал которого при Т = 298 К зависит от значения рН раствора по
уравнению <р + =ф Нг0 =-0,059рН.
н7 н2,он-
Все окислители в зависимости от значений стандартных потенциалов
подразделяют на три группы.
I- Фоф >(Рн+ =^ ®# ^™ окислители восстанавливаются на катоде в
ВФ щ
порядке уменьшения фоф, водород не вьщеляется. Например, при совместном
Вф
присутствии в растворе ионов серебра и меди с активностью а = 1 сначала вос-
восстанавливаются ионы Ag+ фд8+ =0,799 В , затем Си2+ ф^,и2+ =0,337 В без
v ^g" ) { "йГ )
выделения водорода.
П. Окислители (от Li+ до А13+ включительно), у которых ф^2+ суще-
м
ственно меньше нуля. В этом случае в нейтральной и щелочной средах вос-
восстанавливаются молекулы воды, а в кислотной среде — ионы водорода, т.е.
на катоде выделяется водород. Эти металлы можно получить электролизом
расплавов, но не растворов их солей.
17.4. Электролиз. Потенциал разложения 569
III. Окислители, у которых ф^3+ < ф^2+ < О В. На катоде одновремен-
~аГ ~м~
но выделяются металл и водород (параллельные реакции), однако при изме-
изменении условий электролиза коэффициент выхода по току можно существен-
существенно увеличить в пользу одного из процессов.
Возможные анодные процессы при электролизе водных растворов:
а) растворение металла анода: (+) А: М -> Mz+ + Ze\
б) окисление гидроксид-ионов в щелочных растворах:
(+) А: 4ОН" ->О2 Т+2Н2О + 4е A7.29)
или молекул воды в кислотных и нейтральных растворах:
(+)А:2Н2О->О2+4Н++4е A7.30)
в) окисление других восстановителей:
(+)А: Вф-^Оф
Уравнения A7.29) и A7.30) относятся к кислородному электроду, по-
потенциал которого при Г= 298 К зависит от рН раствора по уравнению
Фо2,н2о=Фо2,н+ =U29-0,059pH.
он- ~~Й^Г
Все восстановители в зависимости от значений стандартных потен-
потенциалов подразделяют на две группы.
I. Восстановители, у которых стандартный потенциал меньше потен-
потенциала кислородного электрода при данном значении рН. Если
<?°Mz+ <1,229-0,059рН, то анод, изготовленный из этого металла, раство-
м
ряется.
И. Восстановители, у которых стандартный потенциал превышает
Ф^ н+ =1,229 В. В этом случае идет электролиз с нерастворимым анодом
н2о
при следующей очередности разрядки ионов. Сначала окисляются простые
ионы по мере роста значений их стандартных потенциалов, не превышаю-
превышающих 1,5 В (S2~, Г, Вг", СГ), с выделением простых веществ S, I2, Br2, Cfe. На
аноде не окисляются ионы СО2", NO3, SO^", PO^", F". В этом случае
выделяется кислород по уравнению A7.29) в щелочных растворах или
A7.30) — в кислотных и нейтральных.
570 17. Химические источники тока и электролиз
П.5. Практическое применение электролиза
В химической промышленности электролизом раствора хлорида на-
натрия NaCl (поваренной соли) получают три ценных продукта: гидроксид
натрия NaOH (едкий натр, каустическую соду), хлор СЬ и водород Н2:
(+)А: 2СГ->С12 + 2е
(-)К: 2Н2О + 2е -> Н2 + 2ОН"
I : 2СГ + 2Н2О -> С12 + Н2 + 2ОН"
или в молекулярной форме 2NaCl + 2Н2О -> С12 + Н2 + 2NaOH
Для предотвращения взаимодействия (диспропорционирования) хло-
хлора, выделяющегося на аноде, со щелочью, образующейся в растворе вблизи
катода, анодное и катодное пространства разделяют диафрагмой.
Электролизом воды, подщелоченной гидроксидом натрия для сниже-
снижения омических потерь, в промышленности производят кислород и водород
высокой чистоты:
(+) А: 4ОН" -> О2 + 2Н2О + 4е х 1
Z = 4
(-) К: 2Н2О + 2е -> Н2 +2ОН"
х2
или в молекулярной форме 2Н2О —» О2 + 2Н2
Электрохимическим способом получают многие сильные окислители:
гипохлорит NaCIO и хлорат NaClCb натрия, хлорную кислоту НСЮ4 и ее
соли перхлораты, пероксид водорода Н2О2, пероксодисерную кислоту
H2S2O8, перманганат КМ11О4 и дихромат К2Сг20у калия, фтор F2 из раствора
фторида натрия в плавиковой (фтороводородной) кислоте HF.
Гипохлорит натрия, применяемый для отбеливания в текстильном и
бумажном производствах и для хлорирования воды, производят электроли-
электролизом раствора NaCl в электролизерах без диафрагмы:
(+) А: СГ + 2ОН" -> СЮ" + Н2О + 2е
При изменении технологических условий в этом процессе можно по-
получить хлорат-анион СЮ3 .
В металлургии электролизом получают металлы (электрометаллур-
(электрометаллургия). Электролиз водных растворов солей называют гидроэлектрометаллур-
гидроэлектрометаллургией, а расплавов солей или оксидов при высокой температуре — пироэлек-
трометаллургией. Из соединений ионного типа электролизом можно выде-
выделить металл любой активности. Электролизом водных растворов получают
17.5. Практическое применение электролиза 571
золото, серебро, медь, никель, кобальт, цинк, хром, кадмий, марганец и дру-
другие металлы. Металлы, имеющие (p^z+ < О В, выделяются на катоде, так как
м
вследствие поляризации электродов <p°Mz+ > Фи+ • (—) К: Mz+ + Ze —> М. На
м н^~
инертном аноде при электролизе сульфатов металлов выделяется кислород,
серная кислота регенерируется в растворе.
Электролиз с растворимым анодом используют при электролитиче-
электролитическом рафинировании (очистке) черновых металлов (меди, серебра, олова и
др.). В электролизере без диафрагмы анодом является металл, подвергаемый
очистке. При включении тока катионы основного металла и металлов-
примесей с меньшим потенциалом переходят в раствор. Металлы-примеси с
большим потенциалом оседают из анода в виде шлама. На катоде происхо-
происходит преимущественное выделение основного металла, поскольку концен-
концентрация ионов примесей в растворе мала и в соответствии с уравнением
Нернста их потенциалы уменьшаются.
При электролитическом рафинировании меди содержание основного
металла увеличивается от 98...99 % (черновая медь) до 99,92...99,96 %
(медь, используемая в электротехнике). Анодами являются отливки из чер-
черновой меди, катодами — помещаемые между ними тонкие листы электроли-
электролитической меди, электролитом — водный раствор сульфата меди с добавкой
серной кислоты для повышения электрической проводимости:
(+) А: Си -> Си2+ + 2е
(-)К: Си2++2е->Си
Примеси золота и серебра выпадают в виде шлама, а цинк, никель, железо,
сурьма и мышьяк переходят в раствор.
Электролизом расплавов солей и оксидов получают алюминий, магний,
натрий, литий, бериллий, кальций и некоторые сплавы. Технологические про-
процессы такого типа сопровождаются высокой температурой и большим расхо-
расходом электроэнергии. Для понижения температуры плавления и увеличения
электрической проводимости применяют смеси солей. Необходимо решать и
экологические проблемы, так как при получении алюминия из расплава АЬОз и
криолита Ыаз[А1Рб] в атмосферу выделяются токсичные соединения фтора, а в
производстве магния — очень ядовитый диоксин. В технике электролиз ис-
используется в таких процессах, как гальванопластика, гальваностегия, электро-
электрохимическая обработка металлов, электромеханическая заточка и шлифование,
очистка поверхностей от жировых и оксидных пленок.
Гальванопластика — процесс получения изделий путем электролиза.
На формы из алюминия электролитически наносят медный слой нужной
572 17. Химические источники тока и электролиз
толщины, а затем алюминий вытравливают раствором кислоты или щелочи.
Получают тонкостенные (порядка микрометров) медные изделия сложной
формы, используемые в радиотехнике и приборостроении. Гальваностегия —
процесс нанесения металлических покрытий (гальванопокрытий) путем
электролиза. Толщина покрытий составляет 1...100 мкм. Для получения по-
покрытий с хорошей адгезией (сцепляемостью) и эстетическим внешним ви-
видом поверхность изделия предварительно подвергают механической, хими-
химической и электрохимической обработке.
Выражая, с одной стороны, массу металла покрытия, согласно обоб-
обобщенному уравнению законов Фарадея, а с другой, — как произведение
плотности металла на его объем, можно определить толщину металлическо-
металлического покрытия 6:
где р — плотность; МА — молярная масса атомов; Z — зарядовое число ка-
катиона металла покрытия; F — постоянная Фарадея; — = i — катодная плот-
S
ность тока; S — площадь поверхности, на которую наносится металлическое
покрытие; t — время процесса; г| — коэффициент выхода по току.
Электролиз используют для анодирования алюминия. Алюминиевое
изделие является анодом, коррозионно-стойкая сталь — катодом, раствор
кислоты (серной, фосфорной и др.) — электролитом. При протекании тока
на катоде выделяется водород, а на аноде образуется мелкопористая и очень
прочная пленка оксида алюминия, которую пропитывают разными состава-
составами для улучшения внешнего вида и повышения коррозионной стойкости.
17.6. Кинетика электрохимических процессов
Кинетика электрохимических реакций изучает скорости и механизмы
электродных процессов и базируется на основных положениях химической
кинетики (см. § 7.1). При этом учитывается, что скорость электрохимиче-
электрохимической реакции определяется силой тока, протекающего через электрод. Для
характеристики скорости электродных процессов используют значение
плотности тока /, равной отношению величины силы тока / к площади по-
поверхности электрода S:
17.6. Кинетика электрохимических процессов
573
Поскольку электрохимические реакции представляют собой разновидность
гетерогенных процессов, скорость электродного процесса определяется как
количество вещества, реагирующего на единице площади поверхности элек-
электрода в единицу времени. С учетом законов Фарадея
г = -
т
i
A7.32)
MSt ZFS ZF
где г — скорость электродного процесса, моль^м2 • с); / — плотность тока, А/м2;
S— площадь поверхности электрода, м2. Из A7.32) следует, что / = ZFr.
В состоянии равновесия скорости катодного и анодного процессов
равны, следовательно, равны и плотности тока, протекающего при восста-
восстановлении /к и окислении /а:
h = h = *0>
где /0 — плотность тока обмена, зависящая от природы электродного про-
процесса, активности веществ-участников реакции и температуры.
При прохождении электрического тока равновесное состояние ЭХС
нарушается и значение электродного потенциала изменяется. Явление изме-
изменения равновесного электродного потенциала при прохождении тока через
ЭХС, а также значение изменения потенциала г| = Дер, называют поляриза-
поляризацией электрода. Катодную цк и анодную г|а поляризации рассчитывают, ис-
используя выражения
Ф, в
Лк = фк/-фК; Л а = фа/~ фа,
где фк, фк/, фа, фа/ — равновесные и неравновесные потенциалы катода и
анода соответственно. При поляризации по-
потенциал анода увеличивается, а катода —
уменьшается, поэтому катодная поляризация
имеет знак «-» (г|к < 0), а анодная — знак «+»
(Па > 0). С увеличением плотности тока элек-
электродная поляризация возрастает.
Связь между величинами поляризации и
плотности тока выражается с помощью кине-
кинетических уравнений или графических зависи- о /, а/м2
мостей, построенных в координатах/—ф7,/-Ф/ Рис# 17#5> поляризационные
или lg i—г|, называемых поляризационными кривые для идеально неполя-
кривыми. На рис. 17.5 приведены поляризаци- ризуемого G) и идеально по-
онные кривые, вырождающиеся в прямые ли- ляризуемого B) электродов
574
17. Химические источники тока и электролиз
Ф,в| нии, параллельные осям координат, для
идеально неполяризуемого (ф = const при
любой плотности или силе тока) и идеаль-
идеально поляризуемого (/ = const при любом
потенциале) электродов. Потенциал анода
плавно увеличивается с ростом плотности
тока, но по достижении некоторого значе-
значения /пред? называемого предельной плотно-
плотностью тока, на поляризационной кривой
наблюдается резкое увеличение потенциа-
потенциала (рис. 17.6). Предельная плотность тока
характеризует максимальную скорость
электродного процесса в данных условиях.
На рис. 17.7 изображены поляриза-
поляризационные кривые для гальванического эле-
элемента (а) и электролизера (б). В обоих случаях поляризация играет отрица-
отрицательную роль: в ГЭ снижает рабочее напряжение U по сравнению с ЭДС Е,
в электролизере увеличивает напряжение разложения t/H. Р электролита, что
является причиной перерасхода электроэнергии. Положительную роль по-
поляризация играет при выделении на катоде металлов, стоящих в ряду стан-
стандартных потенциалов до водорода, из водных растворов их солей.
Поляризация в гальваническом элементе Лгэ и в электролизере Лэ рав-
равна сумме катодной и анодной поляризаций:
Лгэ = 1Лк| + 1Ла|, Лэ=1Лк| + 1Па|.
Рис. 17.6. Анодная поляризацион-
поляризационная кривая (концентрационная
поляризация; /пред — предельная
плотность тока)
в
0
^Ч^ 'max
\>| 4 А/м2
У !
в
О
б
'Ла
/, А/м
л*
г-—
Рис. 17.7. Катодные и анодные поляризационные кривые для гальва-
гальванического элемента (а) и электролизера (б)
17.6. Кинетика электрохимических процессов 575
Учитывая омическое падение напряжения ZU = I(R\+R2), для галь-
гальванического элемента получают
U=E-4r3-I(Ri+R2),
для электролизера
При большем значении поляризации ГЭ получают меньший ток
Rl+R2
и при некотором значении плотности тока /тах в гальваническом элементе,
находящемся в полностью поляризованном состоянии, потенциалы катода и
анода могут стать равными друг другу, т.е. U = 0. В действительности пол-
полная поляризация невозможна, так как внутреннее сопротивление Ri*0.
Явление электродной поляризации обусловлено тем, что гетероген-
гетерогенный электрохимический процесс представляет собой ряд последовательных
стадий:
1) массоперенос реагентов из объема электролита к поверхности элек-
электрода и продуктов реакции в обратном направлении;
2) адсорбция реагентов на поверхности электрода и десорбция про-
продуктов реакции с его поверхности;
3) переход заряженных частиц через границу раздела фаз металл — рас-
раствор электролита, т.е. разряд или ионизация (электрохимическая стадия);
4) фазовые превращения, сопровождающиеся образованием или раз-
разрушением твердых веществ, выделением или поглощением газов;
5) химические реакции, предшествующие электрохимической стадии
или протекающие следом за ней.
При мгновенном протекании этих стадий поляризация как явление от-
отсутствовала бы, однако на практике они идут с конечными скоростями. Са-
Самая медленная стадия (лимитирующая) определяет скорость всего элек-
электродного процесса и является причиной поляризации. Если лимитирующая
стадия известна, вместо термина «поляризация» используют термин «пере-
«перенапряжение». При торможении электродного процесса стадиями массопе-
реноса перенапряжение называют диффузионным. Если самой медленной
стадией является стадия разрядки (ионизации), возникает электрохимиче-
электрохимическое перенапряжение. Различают также адсорбционное, фазовое и реакци-
реакционное перенапряжения. Каждый вид перенапряжения обусловлен специфи-
специфическим механизмом и описывается собственными кинетическими уравне-
уравнениями.
576 17. Химические источники тока и электролиз
Диффузионным перенапряжением (или концентрационной поляриза-
поляризацией) называют изменение потенциала электрода при прохождении тока,
обусловленное изменением концентрации (активности) реагента в приэлек-
тродном слое. Этот слой называют также диффузионным.
В элементе Даниэля—Якоби диффузионное перенапряжение обу-
обусловлено различием концентраций ионов меди и цинка в диффузионном
слое (С5) и в объеме раствора (Со): на катоде C5cu2+ < Cocu2+; на аноде
Cszn2+ > Cozn2+. Согласно уравнению Нернста, для электрода первого рода
концентрационная поляризация пропорциональна логарифму отношения
активностей (концентраций) ионов металлов в диффузионном слое и в объ-
объеме раствора:
Лд=^ьА A7.33)
ZF а0
и при температуре Т= 298 К для идеальной системы
„ 0,059, С,
Анализ выражения A7.33) показывает, что анодная концентрационная поля-
поляризация положительна, а катодная — отрицательна.
Диффузионное перенапряжение уменьшается с увеличением коэффи-
коэффициента диффузии, температуры и концентрации реагента, а также с умень-
уменьшением плотности тока и толщины диффузионного слоя (последнее дости-
достигается принудительным перемешиванием раствора).
Электрохимическим перенапряжением называют изменение потен-
потенциала электрода при прохождении тока, обусловленное замедлением элек-
электрохимического процесса.
В 1930-1940 гг. М. Фольмер, Т. Эрдей-Груз и А.Н. Фрумкин разрабо-
разработали для объяснения электрохимического перенапряжения теорию замед-
замедленного разряда, основанную на общих положениях химической кинетики,
связывающих скорость процесса с энергией активации. Электрохимические
реакции, как и химические, совершаются в том случае, если средняя энергия
реагирующих частиц больше или равна энергии активации. Скорость реак-
реакции можно увеличить повышением температуры (рост доли активных моле-
молекул) или применением катализатора (снижение энергии активации).
Энергия активации электрохимической реакции зависит также от зна-
значения электродного потенциала и соответственно электрохимического пе-
перенапряжения. При малых значениях плотности тока мало перенапряжение,
система незначительно отклоняется от равновесия. При этом энергия акти-
активации велика, а скорость процесса в обоих направлениях низкая. С увеличе-
17.6. Кинетика электрохимических процессов
577
нием потенциала константа скорости
процесса восстановления уменьшается, а
константа скорости процесса окисления
увеличивается.
Авторами теории замедленного
разряда было показано, что при прохож-
прохождении через ЭХС значительного тока за-
зависимость электрохимического перена-
перенапряжения г|эл от плотности тока i опреде-
определяется уравнением
RT
aZF
RT
aZF
In/, A7.34)
Рис. 17.8. Зависимость электро-
электрохимической поляризации элек-
электрода от логарифма плотности
тока
где плотность тока обмена /0 и коэффици-
коэффициент переноса а — кинетические параметры процесса. С введением обозна-
обозначении я = -
aZF
lg/0 и b = -
aZF
уравнение A7.34) преобразуется к виду
= а + big L
A7.35)
Первоначально уравнение A7.35) было экспериментально получено
Тафелем в 1905 г. и названо его именем. Величины а и Ъ называют тафелев-
скими константами. Константа а численно равна значению электрохимиче-
электрохимической поляризации при единичной плотности тока. Тафелевские константы
можно определить по графику зависимости A7.35), которая в координатах
lg /—г|эл представляет собой прямую линию (рис. 17.8). Значение а соответ-
соответствует отрезку, отсекаемому прямой на оси ординат при lg i = 0, величина Ъ —
тангенсу угла наклона а прямой к оси абсцисс. Уравнение Тафеля A7.35)
отражает влияние материала электрода на перенапряжение, так как посто-
постоянные а и Ъ неодинаковы для различных металлов (см. табл. П.6.4).
Очевидно, что для выбора оптимальных условий проведения электро-
электрохимических процессов необходимо определить наиболее медленную ста-
стадию, изучить ее природу. Выяснение механизма лимитирующей стадии
электродного процесса позволяет подобрать специальные вещества — депо-
деполяризаторы^ введение которых в электролит или материал электрода позво-
позволяет уменьшить величину электродной поляризации, т.е. приводит к ослаб-
ослаблению поляризационных явлений {деполяризации).
19 - 9795
¦ Практические занятия
Электрохимические процессы
Примеры решения задач
Задача 1. Вычислить электродный потенциал цинка при Т = 298 К в рас-
растворе хлорида цинка ZnCl2, в котором активность ионов Zn2+ равна 0,07.
Решение, Согласно уравнению Нернста, электродный потенциал металли-
металлического электрода при Т= 298 К
о 0,059
м м
Уравнение для заданного электрода имеет вид
cpZn2+ =Ф°2п2+ +^Zn ^
Zn Zn
Задача 2. Вычислить ЭДС серебряно-никелевого гальванического элемента
при Г= 25 °С, в котором активности ионов Ag+ и Ni2+ соответственно равны
0,1 и 0,005.
Решение. Потенциалы отдельных электродов, согласно уравнению Нернста,
(см. задачу 1) равны:
фа§+ =qV +^^~lg<V = 0,799 + 0,059 lg 0,1 = 0,799-0,059 = 0,740 В;
~Ag~ ~Ag~
(pN.2+ =(p°N.2+ +- lgaN.2+ =-0,250 + -^ lg 0,005 = -0,250-0,070 =
Ni Ni
= -0,318 B.
Из двух электродов катодом будет серебряный электрод, так как у
него потенциал больше. ЭДС равна разности потенциалов катода и анода:
? = ср + -<р 2+ =0,740-(-0,318) = 1,058 В.
Ag Nl
Ag Ni
Задача 3. Вычислить ПР Agi при Т = 298 К, если потенциал серебряного элек-
электрода, погруженного в насыщенный раствор иодида серебра, равен 0,325 В.
Решение. Потенциал серебряного электрода, согласно уравнению Нернста,
равен:
Примеры решения задач 579
+ =Ф°а + +0,0591gtf +.
g ^Ag ° Ag
Ag Ag
Логарифм активности ионов серебра
lga t=!^j= ^25-0,799 =
Ag 0,059 0,059
Следовательно, активность ионов серебра равна
а += Ю-8'034 =9,25-Ю-9.
Ag
В соответствии с уравнением диссоциации электролита Agl (т) <=± Ag+ + Г, такая же
активность ионов иода в растворе: а = 9,25-10~9.
Произведение растворимости (произведение активностей) иодида серебра
nPAgI =aAg+a_ =(9,25-10"9J =8,5610-17.
Табличное значение nPAgi = 8,50 • 10~17 (см. табл. П.4.11).
Задача 4. Определить направление самопроизвольного протекания реакции
Fe(NO3K + Ag = Fe(NO3J + AgNO3
если активности в водном растворе равны:
ас3+=0,01, а 2+=0,001, а + =0,01.
Fe Fe Ag
Решение. Значения стандартных электродных потенциалов взаимодействующих
электрохимических систем составляют:
Fe3++e = Fe2+, Ф°ре3+ =0,771 В;
Ag++e = Ag, ф°д+ =0,799 В.
Ag
Значения электродных потенциалов при указанных активностях ионов равны:
*- *L 1 a 2+ 0,001
Fe2+ Fe2+ Fe
,059 = 0,830 В;
Ф + =ф° + + V>w^lga + = 0,799 + 0,0591g0,01 = 0,799-0,059-2 = 0,681 B.
Ag T Ag j ° Ag °
Ag Ag
19*
580 Электрохимические процессы
В данном случае
Ag
Полуреакция, у которой потенциал больше, будет протекать в направлении восста-
восстановления окисленной формы окислителя, а полуреакция, у которой потенциал
меньше — в направлении окисления восстановленной формы восстановителя, что
соответствует процессам:
Fe3+ + e->Fe2+
Ag - е -> Ag+
Суммируя обе полуреакции, получают суммарное уравнение самопроизвольно про-
протекающей реакции в ионном виде:
Fe3+ + Ag->Fe2+ + Ag+
Таким образом, заданная реакция самопроизвольно будет протекать в прямом
направлении (слева направо).
Задача 5. Рассчитать при Г= 298 К константу равновесия реакции
SnCl2 + h + 2KC1 = SnCU + 2KI
Решение. Уравнение реакции в ионно-молекулярной форме имеет вид
В реакции участвуют две электрохимические системы:
Sn4+ + 2е = Sn2+, q>Jn4+ = 0,151 В;
2Г, ф, =0,621 В.
21"
Значение стандартной ЭДС элемента, построенного на этих полуреакциях
?0=Ф°,2 -ф^п4+ =0,621-0,151 = 0,470 В.
2Г ^"
Тогда константа равновесия реакции
2-96485
RT 8,31-298
0,470 = 36,62;
Задача 6. Написать уравнения электродных процессов, протекающих при электро-
электролизе водного раствора сульфата магния MgSO4 с инертными электродами.
Примеры решения задач 581
Решение, Стандартный электродный потенциал системы Mg2+ + 2е = Mg равен
Ф° 2+ =-2,363 В. Сульфат магния является солью, образованной слабым основа-
~mJT
нием и сильной кислотой, поэтому вследствие гидролиза среда в его водном
растворе будет слабокислотной (рН < 7). Следовательно, потенциал водородного
электрода
q> + >-0,059рН = -0,059- 7 «-0,413 В.
2Н
На катоде будет проходить процесс с наибольшим значением потенциала, т.е.
электрохимическое восстановление воды с выделением водорода:
а ионы магния Mg2+ будут накапливаться в прикатодном пространстве.
На аноде будет происходить электрохимическое окисление воды, приводя-
приводящее к выделению кислорода:
2Н2О-4е->О2 +
поскольку отвечающий этой системе электродный потенциал кислородного элек-
электрода в слабокислотной среде:
Ф + < 1,229-0,059рН = 1,229-0,413 «0,816 В.
О2»4Н
2Н2О
значительно меньше, чем стандартный потенциал, характеризующий систему:
S2O2" +2e= 2SO2"; фоад2. =2,01 В.
Ионы SO^" будут накапливаться в прианодном пространстве.
Умножая уравнение катодного процесса на 2 для подведения баланса по
электронам и складывая его с уравнением анодного процесса, получают суммарное
уравнение процесса электролиза:
6Н2О = 2Н2 + 4ОН~ + О2 + 4Н+
Принимая во внимание, что одновременно происходит накопление ионов
магнии в прикатодном пространстве и сульфат-ионов в прианодном пространстве,
итоговое уравнение процесса можно записать так:
6Н2О + 2MgSO4 = 2Н2 + 2Mg(OHJ1 + О2 + 4Н+ + 2SO4"
Следует учитывать, что гидроксид магния — малорастворимое соединение.
582 Электрохимические процессы
Задача 7. Написать уравнения процессов, протекающих при электролизе водного
раствора, содержащего смесь солей: Cu(NO3J и ZnBr2. Электроды графитовые.
Решение. На катоде возможно протекание следующих процессов:
Си2+ + 2е = Си ф^ц2+ = 0,337 В;
Си
Zn2+ + 2e = Zn ф^2+ =-0,763 В;
Zn
2Н2О + 2е -> Н2 + 2О1Г ф 2Н0 >-0,413 В (в слабокислотной среде).
Н2,2ОН"
Необходимо учитывать, что выделение водорода протекает со значительным
перенапряжением. Наибольший потенциал у первого процесса, поэтому на катоде
будут восстанавливаться ионы меди:
Си2+ + 2е = Си
В прикатодном пространстве накапливаются ионы цинка Zn2+. ,
На аноде возможно протекание следующих процессов:
2ВГ - 2е = Вг2 ф°Вг2 = 1,087 В;
2Н2О - 4е -> О2 + 4Н+ ф + < 0,81 В (в слабокислотной среде).
2Н2О
Нитрат-ионы NO3 в водных растворах на аноде обычно не окисляются. Не-
Несмотря на то, что потенциал второго процесса меньше, но из-за значительного пере-
перенапряжения выделения кислорода при больших плотностях тока осуществляется
окисление бромид-ионов:
2ВГ-2е = Вг2
В прианодном пространстве накапливаются нитрат-ионы NO3~.
Суммарное уравнение процесса электролиза можно получить сложением
уравнений катодного и анодного процессов:
Си2+ + 2Вг~ = Си + Вг2
Принимая во внимание, что одновременно происходит накопление ионов
цинка в прикатодном пространстве и нитрат ионов в прианодном пространстве,
итоговое уравнение процесса можно записать так:
Cu(NO3J + ZnBr 2 = Си + Br2 + Zn2+ + 2NO"
Задача 8. Серебрение изделий ведется в растворе азотнокислого электролита с
плотностью тока 3 А/дм2. Рассчитать толщину серебряного слоя, образующегося за
2 мин, если выход по току г\ = 0,90. Плотность серебра р = 10 490 кг/м3.
Задачи для самостоятельного решения
583
Решение. Согласно закону Фарадея, масса выделившегося серебра
ZF
где MAg — молярная масса серебра, г/моль; /—сила тока, А; /—время, с; т| — выход по
току; Z— число электронов, участвующих в электродном процессе; F — постоянная
Фарадея.
После преобразований находят толщину покрытия
т. = pS8 = -
ZF
'; 5 =
ZFpS ZFp
где / = I/S — плотность тока, А/м2; р — плотность металла, г/м3.
Учитывая соответствующие размерности, вычисляют:
1•96485•10490 1О3
Задачи для самостоятельного решения
1-5. Вычислите потенциал данного электрода при Г= 298 К в растворе электролита,
если дана активность потенциалопределяющего иона.
№
1
2
3
4
5
Электрод
Cu2+/Cu
Cd2+/Cd
Со2+/Со
Fe2+/Fe
Ni2+/Ni
Активность иона
аСц2, =0,003
acd,=0,01
ясо2+ =0,005
агг =0,007
V, =0,002
6-10. Вычислите активность указанных ионов в растворе электролита при Т= 298 К,
если дано значение потенциала электрода.
№
6
7
8
9
10
Электрод
Мп27Мп
Н+/Н2
Со27Со
РЬ2+/РЬ
С12/СГ (/?с,2 = 1 атм)
Электродный потенциал, В
-1,25
-0,24
-0,31
-0,18
1,20
Ион
Мп2+
Н+
Со2+
РЬ2+
сг
584
Электрохимические процессы
11-20. Вычислите ЭДС гальванического элемента при указанных активностях ио-
ионов в растворах. Напишите уравнения процессов, протекающих на аноде и катоде,
уравнение токообразующей реакции, составьте схему гальванического элемента.
№
11
12
13
14
15
16
17
18
19
20
Электрод 1
Pb2+/Pb, ярь2+=0,01
Ni2+/Ni, aN.2+
Zn27Zn, a 2+
Zn
Pb27Pb, anV
Fe2+/Fe, а?г
Co2+/Co, aCo2
Cu2+/Cu, аСц2
Fe2+/Fe, a^
A13+/A1, ад|
Ag+/Ag, aAg
= 0,01
= 0,001
= 0,01
= 0,004
.=0,01
. =0,01
.=0,1
.=0,1
.=0,5
Электрод 2
Zn2+/Zn, a
Zn
Pd2+/Pd, apd
2. =0,001
2. =0,005
Zn2+/Zn, a 2. =0,01
Zn
Ag+/Ag, a
Ni2+/Ni, a
Mn2+/Mn, a
AgVAg, a
^ =0,1
a,=0,2
м.-- °'2
^ =0'5
Ti3+/Ti, aTj3+ =0,005
Sn2+/Sn, aS(
Co2+/Co, a
,2. =0,05
co^0'1
21-25. По известным значениям ЭДС гальванического элемента при Т - 298 К, со-
составленного из указанных электродов, определите неизвестную активность ионов.
Составьте схему гальванического элемента.
№
21
22
23
24
25
Электрод 1
Ag7Ag, aAg+=0,l
Ni2+/Ni, aN., =?
Mn2+/Mn, a 2t =0,1
Mn
Zn2+/Zn, a&*=?
Fe2+/Fe, aFe2+ =0,01
Электрод 2
Cd27Cd, <JCd2t=?
Co2+/Co, a 2. =0,05
Co
рь2+/рь, v-=?
Ni2+/Ni, aN.2. =0,05
A13+/A1, яд1з+=?
ЭДС, В
1,16
0,00
1,10
0,60
1,30
26. Вычислите ЭДС гальванического элемента, составленного из двух водородных
электродов, погруженных в растворы с рН = 2 и рН = 4.
27. Пользуясь справочными данными о стандартных электродных потенциалах полуре-
полуреакций, рассчитайте при температуре 298 К константу равновесия реакции
МпО2 + 4Н+ + 2Fe2+ <i± Mn2+ + 2Fe3+ + 2H2O
Задачи для самостоятельного решения
585
28. Гальванический элемент составлен из двух электродов Fe3+, Fe2+ | Pt и Ag+ | Ag.
Условия стандартные. Определите катод и анод, напишите уравнение токообра-
зующей реакции и рассчитайте ее константу равновесия.
29-34. Пользуясь справочными данными о стандартных электродных потенциалах
полуреакций, определите направление окислительно-восстановительной реакции.
Рассчитайте значение константы равновесия.
№
29
30
32
33
34
Fe2+ + S
MnO;
2Fe2+ +
2MnO;
5Sn4+ +
Окислительно-восстановительная реакция
+ 2H+?=±Fe3+.
+ 5Co2+ + 8H+ ^
Sn4+^± 2Fe3+ +
+ 5PbSO4 + 2H2
2Mn2+ + 8H2O ^
HH2S
I Mn2++ 5Co3++ 4H2O
Sn2+
0 +± 2Mn2+ + 5PbO2 + 4H+ +
± 5Sn2++ 2MnO^ + 16H+
5SO2"
35. Гальванический элемент составлен из двух электродов, при работе которых
протекают полуреакции:
Fe3+ + e?=> Fe2+
MnO; + 8Н+ + 5е <± Мп2+ + 4Н2О
Пользуясь значениями стандартных электродных потенциалов, определите
катод и анод, напишите уравнение токообразующей реакции и рассчитайте кон-
константу равновесия.
36. Вычислите nPAgscN при Т = 298 К, если потенциал серебряного электрода, по-
погруженного в насыщенный раствор роданида серебра, равен 0,447 В.
37. Вычислите ПРРе(онJ при Т = 298 К, если потенциал железной пластины, погру-
погруженной в насыщенный раствор Fe(OHJ, равен (-0,595 В).
38-42. Напишите уравнения процессов, протекающих при электролизе водных рас-
растворов электролитов с указанными электродами.
№
38
39
40
41
42
43
44
Электролит
Fe(NO3K
NaCl
CuSO4
CuBr2
AgNO3
Ca(NO3J
MnCl2
Электроды
графитовые
Al
Cu
Cd
Ni
Pt
Ni
45-49. Напишите уравнения процессов, протекающих при электролизе водных рас-
растворов двух электролитов с графитовыми электродами.
586
Электрохимические процессы
№
45
46
47
48
49
Электролиты
Ca(NO3J,KI
ZnCl2, Cu(NO3)
MnCl2,NiSO4
CuBr2, K2SO4
A1(NO3K, PdCl2
50. Для оцинкования изделий электроосаждением можно воспользоваться раство-
раствором сульфата цинка ZnSO4. Рассчитайте время, за которое толщина покрытия дос-
достигнет 100 мкм, если плотность тока равна 2 А/дм2, а выход по току равен 0,90.
Плотность цинка принять равной 7140 кг/м3.
51. Электрохимическое лужение изделий проводится из раствора сульфата олова (II)
SnSO4 при плотности тока 2,5 А/дм2 до образования слоя олова толщиной 10 мкм.
Определите продолжительность процесса, если выход по току равен 0,95. Плот-
Плотность олова принять равной 7300 кг/м3.
52. Определите время, необходимое для электрохимического покрытия поверхности
изделия площадью 13,5 см2 слоем золота толщиной 10 мкм, если золочение ведется
из раствора K[Au(CNJ] при плотности тока 0,2 А/дм2 и выходе по току 0,70. Плот-
Плотность золота принять равной 19300 кг/м3.
53. Изделие необходимо покрыть слоем никеля толщиной 20 мкм. Рассчитайте пло-
площадь покрытия, если при выходе по току 0,90 и силе тока 1000 А процесс длится 1,5 ч.
Плотность никеля принять равной 8900 кг/м3.
54. Через водный раствор иодида бария пропускают в течение 18 мин ток силой 5,2 А.
Напишите уравнения процессов, протекающих на электродах. Определите количе-
количества веществ, выделившихся на электродах.
55. Напишите уравнения электродных процессов, протекающих при электролизе
водного раствора NiSO4, если оба электрода сделаны из никеля. Определите изме-
изменение массы анода после пропускания тока силой 3,2 А в течение 30 мин.
56. Металлический магний получают электролизом расплава MgCl2. Определите
количество магния, которое можно получить за t = 8 ч, если сила тока на электро-
электролизной установке равна 100 кА.
57. Серебрение изделий из растворов цианистых электролитов ведется при плотно-
плотности тока 0,3 А/дм2. Определите время, необходимое для покрытия изделий слоем
серебра толщиной 10 мкм, если выход по току равен 0,90. Плотность серебра при-
принять равной 10 490 кг/м3.
58. Сколько времени надо проводить электролиз 20 мл 0,15н. раствора сульфата
кадмия током силой 0,2 А до полного выделения кадмия, если выход по току со-
составляет 0,93?
ЛР11. Химические источники тока
587
59. Через соединенные последовательно растворы SnCl2 и SnCl4 пропускали в тече-
течение t = 10 мин ток силой 3 А. Вычислите количества олова и хлора, выделившихся
из каждого раствора в отдельности.
60. При электролизе раствора соли никеля (II) в течение 4,5 ч катод площадью
10 см2 покрылся слоем никеля толщиной 0,025 мм. Вычислите силу тока и катод-
катодную плотность тока, если выход по току составил 0,81. Плотность никеля принять
равной 8900 кг/м3.
61. При электролизе водного раствора Сг(МО3)з в течение t = 12 мин на катоде вы-
выделилось т = 0,29 г хрома. Вычислите силу тока.
62-70. Определите выход по току, если известна масса металла, выделившегося на
катоде при электролизе водного раствора электролита. Электроды графитовые.
№
62
63
64
65
66
67
68
69
70
Электролит
CoSO4
NiCl2
ZnSO4
AgNO3
CuSO4
SnCl2
Pb(NO3J
K[Au(CNJ]
CdSO4
Сила тока, А
5
4
2
2
7
2
6
0,5
8
Время электролиза,
мин
20
30
40
60
50
90
20
60
120
Масса выделивше-
выделившегося металла, г
i,6
1,8
1,2
5,4
5,3
5Д
6,9
2,9
23,4
ЛР 11. Химические источники тока
Цель работы — изучение устройства, принципа действия и влияния различ-
различных факторов на характеристики гальванических элементов.
Опыт 1. Измерение ЭДС гальванического элемента и расчет термодина-
термодинамических характеристик токообразующей реакции. В данном опыте предлагает-
предлагается на основании измерений ЭДС гальванического элемента рассчитать изменение
энергии Гиббса ArG® и константу равновесия К® сжислительно-восстановитель-
ной реакции.
Стандартная энергия Гиббса окислительно-восстановительной реакции
где Z — число электронов, участвующих в реакции (наименьшее общее кратное
числа электронов, участвующих в полуреакциях окисления и восстановления);
588 Электрохимические процессы
F= 96 500 Кл/моль экв — постоянная Фарадея; Е° — стандартная ЭДС гальваниче-
гальванического элемента; фк, фЦ — стандартные потенциалы катода и анода. Она связана с
константой равновесия К°а токообразующей реакции соотношением (частный слу-
случай изотермы химической реакции):
Экспериментально, с помощью высокоомного вольтметра, можно измерить
нестандартную ЭДС Е гальванического элемента при относительных активностях
ионов металлов, отличных от 1, и по A7.8) найти значение Е°. Например, в гальва-
гальваническом элементе, составленном из свинцового (РЬ и раствор Pb(NO3J известной
концентрации) и цинкового (Zn и раствор ZnSO4 известной концентрации) электро-
электродов, Zn | Zn2+ || Pb2+1 Pb протекают следующие реакции:
(-) A: Zn <=* Zn2+ + 2e
(+) К: РЬ2+ + 2е *=* РЬ
I: Zn + Pb2+ ^ Zn2+ + Pb (суммарное уравнение токообразующей реакции, Z=2).
Уравнения Нернста для цинкового и свинцового электродов при 298 К
имеют вид
о 0,059
Zn Zn
о 0,059
— — 2
Pb Pb
Соответственно уравнение Нернста для свинцово-цинкового гальванического
элемента имеет вид
„_ _ о . 0,059,_ I о . 0,059,_ I „ , 0,059,
РЬ V Zn / Zn
где а — активность, в данном случае ионов РЬ2+ и Zn2+. Активности отдельных ио-
ионов неизвестны, поэтому в практических расчетах используют значения средних
ионных активностей а±и средних ионных коэффициентов активностей у± (см. § 11.2):
а± «у±С,
где С — молярная концентрация иона. Использование молярной концентрации вме-
вместо моляльной (см. § 11.6) допустимо, поскольку плотность разбавленного водного
раствора мало отличается от 1 г/мл и масса растворенного вещества значительно
меньше массы растворителя. Средние ионные коэффициенты активности в водных
растворах сильных электролитов при Т— 298 К приведены в табл. П.4.13.
ЛР11. Химические источники тока
589
Последовательность проведения:
1) в ячейки установки (рис. Л.11.1) поместите
два сухих химических стакана объемом 100 мл
и в один из них налейте примерно 50 мл 0,1 М
водного раствора сульфата цинка ZnSO4, а в
другой — такой же объем 0,01М водного
раствора нитрата свинца Pb(NO3J;
2) опустите в раствор сульфата цинка цинко-
цинковую пластину или проволочку, в раствор
нитрата свинца — свинцовую. Если металли-
металлические пластины в результате окисления по-
покрылись слоями оксидов, их поверхность
необходимо предварительно очистить мелкой
наждачной бумагой и протереть салфеткой,
смоченной растворителем;
3) соедините электроды солевым мостиком и
измерьте ЭДС исследуемого гальванического
элемента с помощью высокоомного милли-
милливольтметра;
4) отключите милливольтметр, выньте соле-
солевой мостик из стаканов и поместите его в
раствор нитрата калия;
5) вымойте стаканы и металлические пласти-
пластины, последние протрите фильтровальной бу-
бумагой (рис. Л.11.1).
Обработка результатов:
1) исходные данные, результаты измерений и расчетов, а также выводы занесите в
табл. Л.11.1;
2) рассчитайте активности ионов Zn2+n Pb2+, для чего используйте табл. П.4.13;
Рис. Л.11.1. Схема установки для
измерения ЭДС гальванических
элементов:
1 — центральный химический стакан;
2— угловой химический стакан; 3 —
металлическая пластинка (электрод 1);
4 — металлическая пластинка (электрод
2); 5 — сборка (контейнер) для раз-
размещения стаканов; 6 — высокоомный
милливольтметр; 7 — солевой мостик
Таблица Л.11.1
Результаты эксперимента
Исходные данные
С, моль /л
ZnSO4
Pb(NO3J
Ф°г+,В
м
Zn2+/Zn
РЬ2+/РЬ
Результаты
измерений
?эксп>
в
Результаты расчетов
Активность иона
а, моль/л
Zn2+
Результаты расчетов
^хеорВ
*7абс> В
*7отн, %
ArGr°, Дж
<
РЬ2+
& эксп»
в
Выводы
590 Электрохимические процессы
3) по измеренному значению ЭДС Ежсп гальванического элемента рассчитайте его
стандартную ЭДС 2?°Эксп по уравнению Нернста
lg—;
4) рассчитайте теоретическое значение стандартной ЭДС ,Е°теоР> используя значения
стандартных электродных потенциалов:
Pb Zn
5) найдите абсолютную rja6c и относительную г|отн погрешности опыта по соотноше-
соотношениям:
м = /Г° _ F0
Лабе ^ эксп *5г теор?
„ -Лабе
•100%;
6) по экспериментальному значению ЭДС гальванического элемента рассчитайте
стандартную энергию Гиббса ArG° токообразующей ОВР по уравнению:
ArG0T=-ZFE\KCn;
7) рассчитайте стандартную константу равновесия токообразующей реакции по
уравнению:
8) сделайте вывод относительно типов исследованных электродов (первого, второго
рода), вида образованного из них гальванического элемента (концентрационный,
химический), направления протекания токообразующей реакции. Укажите, какие
причины, помимо погрешностей измерений ЭДС и приготовления растворов задан-
заданной концентрации определяют отличие экспериментального значения ЭДС от тео-
теоретического;
9) укажите, какие гальванические элементы можно составить из предложенных
шести металлических электродов (Ag, Cu, Pb, Ni, Cd, Zn), напишите их схемы. По
указанию преподавателя проведите аналогичное исследование одного из составлен-
составленных вами гальванических элементов.
Опыт 2. Зависимость ЭДС гальванического элемента от активности ио-
ионов металла в растворе. В данном опыте предлагается изучить зависимость ЭДС
свинцово-цинкового гальванического элемента от активности ионов свинца в ка-
катодном пространстве при постоянной активности ионов цинка в анодном простран-
пространстве. Согласно уравнению Нернста
ЛР11. Химические источники тока
591
зависимость Е = /(lg я 2+) должна быть линейной.
РЬ
Последовательность проведения:
1) в центральную и угловые ячейки установки (рис. Л.11.1) поместите четыре сухих
химических стакана объемом 100 мл. В центральный стакан налейте примерно 50 мл
0,1М водного раствора сульфата цинка ZnSO4, концентрация которого в опыте по-
постоянна. В три угловых стакана налейте такие же объемы 0,001М, 0,01М и 0,1М
водных растворов нитрата свинца Pb(NO3J в порядке возрастания концентрации
электролита по часовой стрелке;
2) опустите в раствор сульфата цинка цинковую пластину или проволочку, а в рас-
раствор нитрата свинца с концентрацией 0,001М — свинцовую;
3) соедините полуэлементы солевым мостиком и измерьте ЭДС гальванического
элемента с помощью высокоомного милливольтметра;
4) без отключения милливольтметра перенесите свинцовую пластину и колено со-
солевого мостика, находившиеся в 0,001М растворе Pb(NO3J, в раствор нитрата свин-
свинца с концентрацией 0,01М. Измерьте ЭДС гальванического элемента;
5) повторите п. 4, перенеся колено солевого мостика и свинцовую пластину в 0,1М
раствор Pb(NO3J;
6) отключите милливольтметр, выньте солевой мостик из стаканов и поместите его
в раствор нитрата калия;
7) вымойте стаканы и металлические пластины, последние протрите фильтроваль-
фильтровальной бумагой.
Обработка результатов:
1) исходные данные, результаты измерений и расчетов, а также выводы занесите в
табл. Л.11.2.
Результаты эксперимента
Таблица Л.11.2
Исходные данные
С, моль/л
ZnSO4
0,1
Pb(NO3J
0,001
0,01
0,1
<Р° „, В
м
Zn27Zn
Pb2+/Pb
Результаты
измерений
?эксп> В
Результаты расчетов
Активность
иона а, моль/л
Zn2+
Результаты расчетов
^эксп >
В
Е теор> В
Лабе, В
Чотн? '
/о
РЬ2+
Е°
*^ эксп»
в
Выводы
592 Электрохимические процессы
2) рассчитайте активности ионов Zn2+ и РЬ2+, стандартные экспериментальные ?°эксп
и теоретическую ?0теор ЭДС гальванического элемента, повторив пп. 2—4 обработки
результатов опыта 1;
3) рассчитайте среднее арифметическое значение стандартной экспериментальной
ЭДС Гт по формуле: Тт Щ
4) найдите абсолютную r|a6c и относительную т^отн погрешности опыта по соотноше-
соотношениям:
Лабе"
теор»
5) постройте графическую зависимость ЭДС гальванического элемента от десятич-
десятичного логарифма активности ионов свинца ?Эксп = /(lg я 2+ );
6) сделайте выводы о соответствии полученной экспериментальной зависимости
уравнению Нернста.
VIII
КОРРОЗИЯ И БОРЬБА С НЕЙ
Коррозия металлов
и сплавов
¦ Защита от коррозии
Проблемы коррозии определяются двумя ос-
основными аспектами:
1) экономическим, означающим снижение себе-
себестоимости получения металлов (путем эконом-
экономного использования их мировых ресурсов) и
уменьшение материальных потерь в результате
коррозии различных объектов. Подсчитано, что
только прямые потери железа от коррозии со-
составляют около 10 % его ежегодной выплавки в
мире;
2) экологическим, целью которого является пре-
предотвращение (путем повышения надежности
различных объектов) катастроф и аварий с че-
человеческими жертвами и загрязнением окру-
окружающей среды.
Объект (изделие, конструкция, сооружение) из
металла или сплава и контактирующая с ним
среда образуют сложнейшую физико-химичес-
физико-химическую систему, обладающую ярко выраженной
многоаспектностью способов воздействия на
нее и последствий от ее функционирования.
Изучение коррозии всегда преследует цель ее
предотвращения или, по крайней мере, замед-
замедления. Коррозия зависит от большого числа
разнообразных факторов, многие из которых
зачастую не поддаются учету. Именно поэтому
коррозия бывает непредсказуема, а данные ла-
лабораторных исследований иногда не подтвер-
подтверждаются результатами испытаний материалов в
реальных условиях их эксплуатации.
18 ¦
Коррозия металлов и сплавов
Абсолютно коррозионно-стойких металлов нет. Золото — стойкое в
обычных условиях — растворяется в растворах цианидов калия или
натрия вследствие образования устойчивых комплексных ионов. В
результате коррозии металлические изделия теряют свои ценные
технические свойства, поэтому важное значение имеет антикорро-
антикоррозионная защита металлов и сплавов.
Коррозия (от латинского corrodo) — это разрушение конструкций и
изделий из металлических материалов (металлов и сплавов), проис-
происходящее вследствие их физико-химического взаимодействия с ок-
окружающей средой, которую называют коррозионной (или агрессив-
агрессивной), а образовавшиеся химические соединения — продуктами
коррозии. Коррозия сопровождается выделением энергии и рассеи-
рассеиванием продуктов коррозии в окружающей среде. Процесс корро-
коррозии железа и его сплавов называют ржавлением.
В международном стандарте ИСО 8044-1986 под термином «корро-
«коррозия» понимают самопроизвольно протекающее физико-химическое
взаимодействие металла со средой, в результате которого изменя-
изменяются его свойства и ухудшаются функциональные характеристики
металла и среды или технической системы, содержащей их.
18.1. Классификация коррозионных сред, разрушений
и процессов. Показатели скорости коррозии
Коррозионные среды бывают жидкими и газообразными, то-
копроводящими и неэлектролитами, естественными и искусственно
созданными. К газообразным относятся природная атмосфера и газы,
образующиеся при сгорании топлива или выделяющиеся в различных
химических производствах. Жидкие — это жидкости-электролиты
(водные растворы солей, кислот, щелочей, морская вода) и жидкости-
неэлектролиты (сернистая нефть, бензин, керосин и др.). Естествен-
Естественными, кроме атмосферы, являются вода и почва, искусственными —
многие химические вещества.
По характеру разрушения поверхности различают коррозию:
а) сплошную (общую), при которой поражается вся поверх-
поверхность изделия. Она бывает равномерной и неравномерной;
б) локальную (местную), при которой поражаются лишь от-
отдельные участки поверхности. Она проявляется в виде пятен, язв и
питтинга (точечного разрушения на большую глубину). Перечислен-
Перечисленные коррозионные разрушения являются макроскопическими дефек-
дефектами.
18.1. Классификация коррозионных сред, разрушений и процессов 595
К микроскопическим дефектам относятся разрушения, происходящие
при коррозии:
а) селективной (избирательной), т.е. в случае преимущественного
разрушения одного или нескольких компонентов сплава, например обес-
цинкование латуни;
б) межкристаллитной (интеркристаллитной), т.е. при разрушении
по границам кристаллитов (или зерен), приводящем к ослаблению связи ме-
между ними, например окислении чугунов при переменном нагреве и охлаж-
охлаждении. Кристаллитами (или зернами) называют кристаллы поликристалли-
поликристаллического тела, имеющие неправильную форму в отличие от правильно огра-
ограненных кристаллов;
в) транскристаллитной, т.е. при разрушении, возникающем под дей-
действием механических напряжений и сопровождающемся появлением глубо-
глубоких транскристаллитных трещин.
Существуют и другие виды разрушений, происходящих вследствие
физических причин (например, эрозия — механическое истирание, износ),
либо сочетающих в себе воздействие коррозионной среды и механические
воздействия: кавитационная коррозия (разрушение при одновременном
ударном и коррозионном воздействиях среды — коррозия лопастей гребных
винтов), коррозионная эрозия, или фреттинг-коррозия (разрушение при од-
одновременном воздействии сил трения и коррозионной среды — коррозия
насосов, двигателей, турбин) и др.
По механизму процесса коррозию подразделяют на химическую и элек-
электрохимическую. Причина коррозии металлов и сплавов состоит в их термоди-
термодинамической неустойчивости, поэтому коррозионные процессы протекают са-
самопроизвольно и сопровождаются убылью энергии Гиббса: (ArG)PtT < 0. Чем
меньше (более отрицательно) значение (ArG)pj коррозионного процесса,
тем выше термодинамическая возможность (вероятность) его протекания.
Для химической коррозии изменение стандартной энергии Гиббса (ArG°)pT
связано с термодинамической константой равновесия KQa соотношением, по-
подобным A1.7). В случае электрохимической коррозии (ArG°)pr связана со
стандартной ЭДС Е° коррозионного ГЭ уравнением A7.15).
Химическая и электрохимическая коррозия относятся к гетерогенным
окислительно-восстановительным процессам, протекающим на поверхности
металлов и сплавов (на границе раздела фаз материал — коррозионная сре-
среда). При этом разрушаемый материал, являющийся восстановителем, непо-
непосредственно взаимодействует с окислителем коррозионной среды. По сте-
хиометрическим коэффициентам реакции коррозии довольно просты, но по
механизму и элементарным стадиям относятся к числу наиболее сложных
гетерогенных процессов.
596 IS. Коррозия металлов и сплавов
Гетерогенный процесс состоит из последовательно протекающих эле-
элементарных реакций (стадий): диффузии частиц окислителя к поверхности
металла, их адсорбции на ней, поверхностной химической реакции, десорб-
десорбции продуктов с поверхности, их переноса в объем коррозионной среды. Ско-
Скорость коррозии определяется скоростью наиболее медленной (лимитирующей)
в данных условиях стадии, которая может иметь как химическую (переход
электрона, окисление металла и др.), так и физическую (диффузия электролита
или газа) природу. Зная лимитирующую стадию, можно воздействовать на ее
ход и тем самым тормозить или ускорять коррозионное разрушение.
Наиболее часто для характеристики скорости коррозии используют
отрицательный массовый (показатель убыли массы) и глубинный (линейный)
показатели, реже — объемный и токовый (плотность коррозионного тока).
Отрицательный массовый (в дальнейшем массовый) показатель гшсс
указывает на потерю массы Am в единицу времени t с единицы поверхности
S испытуемого образца:
_ Am
Гмасс " —•
Чаще всего гмаСс выражают в г/(м2 • сут).
Глубинный показатель гглуб определяется отношением средней глуби-
глубины h разрушения металла в единицу времени /:
В справочной литературе гглуб обычно приводится в мм/год.
Связь между этими показателями устанавливается следующим соот-
соотношением:
365
Рм
где гГлуб — в мм/год; гмаСс — в г/(м2 • сут); 365 — число дней в году; рм —
плотность металла, кг/м3.
Все металлы, согласно ГОСТ 13819-68, по величине глубинного пока-
показателя подразделяют на шесть групп, каждой из которой присваивается со-
соответствующий балл десятибалльной шкалы коррозионной стойкости (см.
табл. П.6.6).
Объемный показатель гоб скорости коррозии равен объему V газа, по-
поглощаемого (чаще всего СЬ) или выделяемого (чаще всего Нг) единицей по-
поверхности S металла газа в единицу времени t:
V
18.2. Химическая коррозия: виды и разновидности 597
Плотность коррозионного тока iKopp характеризует только электрохи-
электрохимическую коррозию. Этот показатель связан с массовым и глубинным соот-
соотношениями:
i = v М Ifs й- i = v- ^мРм О? Я
'корр 'масс ~Лг? ' ^°>°9 'корР 'глуб ~АХЖ ' ^U'°»
24ММ 365 • 24ММ
где ZM — зарядовое число (формальный заряд) иона металла или число
электронов, отдаваемых одним его атомом при окислении; 24 — число ча-
часов в сутках; Мм — молярная масса атомов металла, г/моль; 26,8 — посто-
постоянная Фарадея, А • ч/моль экв.
Перечисленные выше показатели, как правило, используют для харак-
характеристики скорости равномерной коррозии.
В целом коррозия и ее скорость зависят от множества одновременно
действующих факторов: внутренних, связанных с природой, химическим и
фазовым составом, типом, структурой и технологией материала, состоянием
его поверхности, предысторией получения и обработки из него изделия, на-
наличием в последнем механических напряжений и деформаций и других, а
также внешних, определяемых видом и составом агрессивной среды, скоро-
скоростью ее движения, климатом, географическими и геологическими условия-
условиями эксплуатации, условиями протекания самого коррозионного процесса
(температурой, давлением и др.).
18.2. Химическая коррозия: виды и разновидности
Химическая коррозия характерна для сред, преимущественно не про-
проводящих электрический ток. В зависимости от вида этих сред различают:
химическую коррозию в жидкостях-неэлектролитах; химическую газовую
коррозию (в дальнейшем газовую). Они не сопровождаются возникновени-
возникновением электрического тока и протекают по механизмам гетерогенных окисли-
окислительно-восстановительных реакций.
Жидкости-неэлектролиты — это неэлектропроводные жидкие корро-
коррозионные среды неорганического (жидкий бром, расплавленная сера и др.) и
органического (нефть, керосин, бензол и др.) происхождения. В чистом виде
они малоагрессивны, однако присутствие в них даже небольших количеств
примесей (меркаптанов, сероводорода, воды, кислорода и др.) резко увели-
увеличивают их химическую активность. Так, меркаптаны (R-SH) и сероводород
(H2S), содержащиеся в сырой нефти, вызывают коррозию Fe, Cu, Ni, Ag,
Pb, Sn с образованием соответственно их меркаптидов (M(SR)Z) и суль-
сульфидов (MmS,,). Следы воды в тетрахлориде углерода усиливают его кор-
коррозионную активность из-за образования в результате гидролиза токо-
598 18. Коррозия металлов и сплавов
проводящих компонентов (НС1) и протекания коррозии по электрохими-
электрохимическому механизму:
СС14 + Н2О -> СОС12 + 2НС1
ССЦ + 2Н2О ^-> СО2 + 4НС1
Газовая коррозия является наиболее часто встречающимся видом хи-
химической коррозии и обычно протекает при высоких температурах в газах и
парах агрессивных веществ, когда исключена возможность их конденсации
на поверхности металла, поэтому ее называют высокотемпературной кор-
коррозией. Это коррозия сопел ракетных двигателей, лопаток газовых турбин,
элементов электронагревателей и др. К газовым коррозионным агентам от-
относятся О2, СО2, SO2, H2O, H2S, Cl2 и т.п. Их агрессивность по отношению к
различным металлам не является одинаковой, следовательно, и скорость
коррозии различается. Так, скорость окисления Fe, Co, Ni при температуре
900 °С возрастает в ряду Н2О —> СО2 —> О2 —> SO2. При этом металлы в зави-
зависимости от скорости коррозии в атмосфере данных реагентов располагаются
в возрастающий ряд: Ni —> Со—> Fe. На примере железа процессы окисления
можно описать следующими уравнениями:
Fe + Н2О -> FeO + H2 2Fe + О2 -> 2FeO
Fe + CO2 -> FeO + CO 3Fe + SO2 -> 2FeO + FeS
Fe + H2O + SO2 -> FeSO3 + H2
Состав продуктов окисления определяется главным образом температурой
газовой коррозионной среды.
Рассмотрим наиболее характерные и часто встречающиеся на практи-
практике случаи газовой коррозии.
Коррозия железа, чугуна и сталей в атмосфере О2, СО2, Н2О. При
нагревании этих материалов происходит их окисление, причем железа и
сталей — поверхностное, чугуна — внутреннее селективное, с образованием
окалины сложного строения на поверхности материала и границах зерен и
поверхности включений графита соответственно. Наряду с окислением, в
сталях и чугуне протекает процесс обезуглероживания — обеднения поверх-
поверхностного слоя углеродом вследствие взаимодействия карбида железа, со-
содержащегося в них, с кислородом и кислородсодержащими реагентами:
Fe3C + О2 -> 3Fe + СО2
Fe3C + CO2^3Fe + 2CO
Fe3C + Н2О -> 3Fe + СО + Н2
При этом ухудшаются их механические и антикоррозионные свойства.
Обезуглероживание может происходить и в атмосфере водорода:
18.2. Химическая коррозия: виды и разновидности 599
Fe3C + 2Н2 -^-> 3Fe + CH4t
Этот вид газовой коррозии называют водородной. Наряду с обезуглерожи-
обезуглероживанием одновременно осуществляется и наводороживание — проникнове-
проникновение атомарного водорода в материал и последующее его растворение в нем,
что ведет к резкому снижению пластичности материала.
Коррозия под действием продуктов сгорания топлива (СО2, Н2О,
СО). Продукты сгорания топлива (угля, мазута и др.) в большинстве случаев
содержат значительные количества соединений серы и ванадия в виде V2O5.
Взаимодействуя с оксидами, легкоплавкий V2O5 сначала разрушает защит-
защитные оксидные пленки
Fe2O3 + V2O5 -> 2FeVO4
а затем активно участвует в процессе окисления самого металла:
6FeVO4 + 4Fe -> 5Fe2O3 + 3V2O3
4Fe + 3V2O5 -¦ 2Fe2O3 + 3 V2O3
O2-+V2O5
причем сам V2O5 при этом практически не расходуется.
Под действием соединений серы (SO2, H2S) железоуглеродистые стали
подвергаются интенсивной межкристаллитной коррозии из-за большего
числа дефектов в кристаллических решетках сульфидов, чем оксидов. Это
ведет к интенсификации диффузионных процессов. С повышением содер-
содержания оксида углерода (II) в продуктах сгорания заметно снижается ско-
скорость газовой коррозии углеродистых и низколегированных сталей. Однако
очень высокая его концентрация ведет к науглероживанию поверхности:
3Fe + 2СО -> Fe3C + СО2
Коррозия в атмосфере хлора и хлороводорода. Ей подвергаются
практически все металлы. Это связано с тем, что большинство из них обра-
образует хлориды с низкой температурой плавления (см. табл. П.3.1). Некоторые
из хлоридов при плавлении легко возгоняются, например:
Ti + 2С12 -> TiCl4t
Zr + 2Cl2-»ZrCl4t
Реакция взаимодействия металла с хлором протекает при повышенной
температуре и с выделением большого количества теплоты. Скорость отвода
теплоты бывает ниже скорости самой реакции, вследствие чего металлы вос-
воспламеняются (или часто говорят, «горят») в атмосфере хлора. Ниже приведена
температура воспламенения некоторых металлов в атмосфере сухого хлора:
Металл Ti Pb Fe Cu Ni
Температура
воспламенения, °С < 20 90—100 -150 200 > 500
600 18. Коррозия металлов и сплавов
Наиболее стойкими к нему являются никель, свинец и хромоникеле-
вые стали.
В сухом хлороводороде при комнатной температуре удовлетворительно
стоек ряд металлов и сплавов. Максимальная допустимая температура приме-
применения в нем некоторых металлических материалов приведена ниже:
Материал Си Ag СтЗ 12Х18Н9Т Ni W Аи Pt
Максимальная
температура, °С ... 100-120 230 260-350 450-500 510-600 600 8701200
Таким образом, поведение металлов в среде газообразного хлороводо-
рода принципиально отличается от поведения в других агрессивных газовых
средах.
18.3. Термодинамика и кинетика газовой коррозии
На практике газовая коррозия чаще всего проявляется как коррозия
металлических материалов при высокой температуре в атмосфере кислород-
кислородсодержащих газов, поэтому ее помимо высокотемпературной коррозии не-
нередко называют высокотемпературным окислением. На поверхности изде-
изделий из металлов и сплавов в результате их взаимодействия с такой коррози-
коррозионной средой образуется пленка продуктов, представляющих собой различ-
различные оксиды металлов. Процесс может быть описан следующим уравнением:
()+^02(г)<=Шт0л(т) A8.1)
Реакция окисления обратима и ее константа равновесия
Положение равновесия в A8.1) сильно смещено вправо (в сторону
образования оксида) из-за высокого химического сродства большинства
металлов к кислороду и способности их оксидов (особенно технически
важных металлов) растворяться в самих металлах, покидая сферу (поверх-
(поверхность) реакции:
MWOW(T) <=> [МтОп](р)
Термодинамическую возможность протекания процесса окисления в
конкретных условиях можно оценить с помощью уравнения изотермы хи-
химической реакции (см. § 11.2), которое для A8.1) имеет вид
18.3. Термодинамика и кинетика газовой коррозии 601
\п/2
Ро2
где рравно2 и Ро2 — равновесное и неравновесное относительные парциаль-
парциальные давления кислорода соответственно (р = р/р°, р° = 1 атм — стан-
стандартное давление); Кр=Кр(р°)п/2 — стандартная константа равновесия.
Если pOi >ррат02 (р02 >/Vbho2)> то \°т <0 — процесс окисления возмо-
возможен и наоборот.
При стандартных условиях {р0^ = 1, Г= 298 К) уравнение изотермы
трансформируется и принимает вид
= ArG2°98 =-29ШпК°р =
В этих условиях
ArG298 =AfG29SMmOn wA/G298M
так как для простых веществ A^^G^g = 0. У большинства оксидов Ay^s < ^,
что говорит о неустойчивости и возможности окисления (химической кор-
коррозии) многих металлов в атмосфере кислорода при стандартных условиях.
Чтобы исключить возможность протекания коррозии, требуется обеспечить
очень низкие парциальные давления кислорода, не существующие в реаль-
реальных условиях. В особо важных случаях для предотвращения коррозии ис-
используют искусственную (обескислороженную) атмосферу.
С ростом температуры увеличиваются равновесное парциальное дав-
давление кислорода и стандартная энергия Гиббса образования оксидов, что
приводит к снижению термодинамической вероятности процесса окисления
(коррозии), несмотря на то, что повышение температуры ускоряет его про-
протекание. Если процесс окисления проводится при достаточно высоких тем-
температурах, парциальное давление паров оксида металла в газовой фазе зна-
значительно и им уже нельзя пренебрегать. С учетом этого уравнение A8.1),
выражение константы равновесия и уравнение изотермы принимают вид
+ ^О2(г) ^ МтОи(г); Кр =
где рравнМ о И Рм о„ — равновесное и неравновесное относительные пар-
парциальные давления паров оксида металла в газовой фазе соответственно.
602
18. Коррозия металлов и сплавов
В этом случае:
а) если р02 > />МЛ (Ро2 > Рмто„ )> то реакция протекает преимущест-
преимущественно в сторону образования оксида;
б) если р0 < рм 0 (Ро7 < рм о X то реакция протекает преимущест-
преимущественно в обратном направлении: оксид диссоциирует на металл и кислород;
в) если ро2 = рм 0 (pOi = рм о X то система находится в равновесии.
При окислении металлов в воздушной атмосфере парциальное давле-
давление кислорода постоянно и зависит от атмосферного давления. Таким обра-
образом, зная р0^ и рМтОп в газовой смеси, можно определить температурный
интервал протекания данного процесса окисления.
Окисление металлов — многостадийный гетерогенный процесс, бла-
благодаря которому вначале на поверхности образуется моно-, а затем и поли-
полимолекулярный слой оксида, так называемая оксидная пленка. Рост ее тол-
толщины может происходить либо в обоих направлениях одновременно (ме-
(металла и газовой среды), либо преимущественно в одном (рис. 18.1). Если
рост осуществляется в направлении газовой среды, то наблюдается сущест-
существенное увеличение размера детали, например при оксидировании, что необ-
необходимо учитывать в машиностроении.
Направление роста определяется соотношением между скоростями
к о,
Зона
роста
к
-'аде
•
е \
. • . е J
• •
• Л <о
К Оад
•;
А-!
0
о„'.
аде
е ^^ •
Рис. 18.1. Схема ионно-электронного механизма образования и роста тол-
толщины оксидной пленки 5 на поверхности металлической детали с первона-
первоначальным размером, равным /:
а — одновременно в обоих направлениях: металла и газовой среды (г 2_ «г z+);
дифф О дифф м
б — преимущественно в направлении металла (г 2_ > г z+); в — преимуще-
дифф к) дифф М
ственно в направлении газовой среды (г АА , <r xx.,z+)
r r v дифф (у дифф mz+ 7
18.3. Термодинамика и кинетика газовой коррозии 603
процессов встречной диффузии ионов металла Mz+ и кислорода О2~ внутри
оксидной пленки. Если скорости диффузии ионов металла и кислорода
сильно различаются, то рост оксидной пленки происходит преимуществен-
преимущественно в одном направлении, например, при г 2_ <г ,,кл2+ — в направлении
ДИфф U ДИфф М
газовой среды и наоборот (рис. 18.1, в, б).
В большинстве случаев скорости диффузии ионов металла и кислоро-
кислорода соизмеримы, но для ионов кислорода скорость несколько ниже из-за того,
что они больше по размерам, поэтому зона роста, находящаяся в толще са-
самой оксидной пленки, располагается ближе к ее внешней поверхности
(рис. 18.1, а). По мере утолщения оксидной пленки процессы встречной
диффузии ионов и электронов затрудняются. В результате протекания этих
процессов поверхность металла становится анодом (А), и на ней протекает
реакция ионизации атомов металла (М -> Mz+ + Ze\ а внешняя поверхность
оксидной пленки — катодом (К), и на ней адсорбированные атомы кислоро-
кислорода принимают электроны (О + 2е —> О2").
Оксидные пленки толщиной до 40 нм оптически прозрачны и невидимы.
Достигая толщины 40...500 нм, становятся видимыми как цвета побежалости
(красный, оранжевый, желтый, зеленый, голубой, синий, фиолетовый) на
поверхности металла за счет интерференции в них световых лучей. Цвет
пленки определяется ее толщиной, которая зависит, в основном, от природы
металла и свойств коррозионной среды, а также от температуры нагрева и
времени выдержки металла при этой температуре. Различают тонкие (до 40
нм), средней толщины D0...500 нм) и толстые (> 500 нм) пленки. Последние
можно обнаружить визуально, как, например, окалину на стали. Если обра-
образовавшаяся оксидная пленка препятствует дальнейшему проникновению
коррозионной среды к поверхности металла, то ее называют защитной. Ме-
Металл с защитной пленкой на поверхности становится химически неактив-
неактивным, т.е. пассивным. Начальная стадия образования защитной оксидной
пленки — исключительно химический процесс. Дальнейшее протекание
процесса определяется скоростью встречной диффузии ионов металла и ки-
кислорода внутри оксидной пленки.
В оксидных пленках определенной толщины и совершенной структу-
структуры (без трещин, пор, вакансий и др.) процессы встречной диффузии пре-
прекращаются. Такие пленки и являются защитными. Чтобы обладать защит-
защитными свойствами, оксидная пленка должна удовлетворять следующим тре-
требованиям: быть сплошной, беспористой, химически инертной к агрессивной
среде, иметь высокие твердость, износостойкость, адгезию (прилипаемость
к металлу) и близкий к металлу коэффициент термического расширения.
Главным требованием является условие сплошности Пиллинга—Бедвордса,
согласно которому объем образовавшегося оксида должен быть больше из-
604 18. Коррозия металлов и сплавов
расходованного на окисление объема металла — VM 0 > VM. Отношение
этих объемов называют фактором сплошности Пиллинга—Бедвордса а, ко-
который рассчитывают, используя молярную массу атомов Мм и плотность рм
металла, а также молярную массу Мм о и плотность рм 0 его оксида:
A8.2)
где т — число атомов металла в молекуле оксида. Если а > 1, то формирование
и рост толщины пленки при окислении происходит в условиях сжатия, поэтому
она является сплошной и может обладать защитными свойствами. Если а < 1,
то пленка в процессе своего формирования и роста испытывает растяжения,
которые способствуют ее разрушению и появлению трещин, различных дефек-
дефектов, вследствие чего кислород свободно проникает к поверхности металла.
По фактору сплошности Пиллинга—Бедвордса можно лишь прибли-
приблизительно оценить защитные свойства оксидных пленок, так как их состав
имеет широкую область гомогенности, а это отражается на их плотности. В
реальных условиях у сплошных пленок (а > 1) может и не быть защитных
свойств, как, например, у FeO, MoO3, WO3 и др. Поэтому ориентировочно
считают, что если 1,0 < а < 2,5, то пленка сплошная и может обладать за-
защитными свойствами. Причинами плохих защитных свойств у пленок с а > 2,5
могут быть летучесть оксида, напряжения, разрушающие оксидную пленку,
ее недостаточная пластичность и др.
Выполнение условия сплошности всегда является необходимым, но
недостаточным требованием. При формировании и росте защитной оксид-
оксидной пленки важным является также и условие ее ориентационного со-
соответствия металлу, т.е. максимального сходства кристаллических реше-
решеток металла и образующегося оксида при минимальном смещении атомов.
Защитными свойствами преимущественно обладают лишь тонкие
(< 40 нм) оксидные пленки, поскольку они являются сплошными, плотными и
эластичными. Их образование в сухом воздухе и существование на поверхно-
поверхности многих металлов (Al, Zn, Ti, Cr, Cu, Fe, Та, Be и др.) происходит уже при
комнатной температуре. Например, подобная защитная пленка практически
всегда присутствует на алюминии. Толщина ее составляет примерно 10 нм, она
не отстает при изгибе, проводит электрический ток, плавится лишь при темпе-
температуре 2050 °С, тогда как сам алюминий становится жидким при температуре
660 °С. При нагревании этих металлов на воздухе тонкие оксидные пленки,
увеличиваясь в толщине, становятся видимыми как цвета побежалости. Даль-
Дальнейшее нагревание приводит к образованию толстых, непрозрачных, рыхлых,
пористых, легко растрескивающихся при высоких температурах пленок из ок-
оксидов различного состава. Такие пленки имеют определенный постоянный цвет
18.4. Законы роста толщины оксидных пленок
605
и не обладают защитными свойствами. Состав и структура любых оксидных
пленок определяются главным образом природой металла, температурой на-
нагрева и видом газовой коррозионной среды.
Состав оксидных пленок может быть очень сложным, что характерно
чаще всего для металлов с переменной валентностью, которые образуют
сразу несколько оксидов с различными структурой и свойствами.
18.4. Законы роста толщины оксидных пленок
Обычно скорость г газовой коррозии, т.е. процесса окисления, выра-
выражают через скорость роста толщины 5 оксидной пленки во времени t:
Y =
dt'
Рост толщины пленки, т.е. окисление поверхности металла, может про-
проходить в соответствии с различными кинетическими зависимостями, или зако-
законами: линейным, параболическим, логарифмическим (рис. 18.2, табл. 18.1).
Согласно линейному закону, скорость процесса окисления постоянна
во времени. Этот закон выполняется как при полном отсутствии оксидной
пленки на поверхности, так и при наличии тонкой или незащитной (порис-
(пористой, несплошной) оксидной пленки, у которой а < 1. Во всех этих случаях
доступ кислорода к поверхности свободен и лимитирующей стадией про-
процесса является поверхностная химическая реакция, протекающая с постоян-
постоянной скоростью, т.е. окисление осуществля-
осуществляется в кинетическом режиме. По линейно-
линейному закону происходит окисление щелоч-
щелочных и щелочноземельных металлов, а так-
также ванадия, вольфрама и молибдена при
высоких температурах. У первых оно обу-
обусловлено их разогревом из-за плохого от-
отвода теплоты, вызванного образованием на
поверхности рыхлых оксидных пленок,
препятствующих ее оттоку, у вторых — ле-
летучестью их оксидов при высоких темпера-
температурах.
В соответствии с параболическим за-
законом скорость процесса окисления об-
обратно пропорциональна толщине оксидной
пленки. Этот закон соблюдается, когда на
поверхности металла при его окислении логарифмическим E = km In t + С)
О t
Рис. 18.2. Рост толщины 8 ок-
оксидной пленки во времени t9 опи-
описываемый законами:
1 — линейным E = k't + А)\ 2 — па-
параболическим E =v2k"t + 2B); 3 —
606
18. Коррозия металлов и сплавов
образуется пленка, обладающая защитными свойствами, т.е. сплошная и
непористая, для которой а > 1. Тогда при комнатной температуре окисление
протекает в кинетическом режиме и описывается законами химической кине-
кинетики, а при высокой — в диффузионном и подчиняется законам диффузии.
При этом диффузионный характер проявляется тем сильнее, чем толще и
качественнее пленка, т.е. чем выше ее защитные свойства. Эксперименталь-
Экспериментально было установлено, что лимитирующими стадиями в этом случае являют-
являются стадии встречной диффузии ионов кислорода и металла внутри оксидной
пленки. Согласно параболическому закону, окисляются вольфрам, кобальт,
никель (за исключением начальных участков), а также медь в интервале
температур 300.. .1000 °С и железо — 500... 1000 °С.
Таблица 18.1
Закон роста и соответствующие ему уравнения скорости процесса окисления
и координаты линейной зависимости толщины оксидной пленки
Закон роста
толщины пленки
Линейный
Параболический
Логарифмический
Уравнение скорости процесса окисления
дифференциальное
dt
db к"
dt 8
db к"
dt ~ t
интегральное
\db = k'\dt
8 = k't + A
jddd = k"$dt
b = -J2k"t + 2B
\db = k"\^
t
8 = k"]nt + C
Координаты линейной
зависимости
8-/
8 -ft
8-In?
Примечание. к\ к" и к'" — некоторые постоянные. Если в начальный момент
времени на поверхности металла не было оксидной пленки, то постоянные интегри-
интегрирования А = В= С = 0.
Логарифмический закон имеет место, когда происходит либо уплот-
уплотнение защитной оксидной пленки, либо появление в ней дефектов в виде
пузырей или расслоений, тормозящих процессы встречной диффузии ио-
ионов кислорода и металла. При этом наблюдается сильное затухание про-
процесса окисления, и рост толщины оксидной пленки осуществляется мед-
медленнее, чем по параболическому закону. В соответствии с логарифмиче-
логарифмическим законом, окисляются медь при температуре ниже 100 °С, тантал —
ниже 150 °С, железо — ниже 400 °С, а также алюминий, цинк и никель —
ниже 300 °С. Скорость процесса окисления в этом случае обратно пропор-
пропорциональна времени его протекания.
18.4. Законы роста толщины оксидных пленок 607
Процесс окисления большинства металлов с изменением условий
(температуры, состава газовой коррозионной среды, времени контакта) про-
протекает по различным законам. Например, для титана:
Температура, °С < 350 630—830 > 850
Закон роста толщины
оксидной пленки логарифмический параболический линейный
Из большого числа внутренних факторов существенное влияние на
скорость при газовой коррозии оказывает состояние поверхности металла,
связанное с образованием, устойчивостью и разрушением оксидных пленок.
Их защитные свойства в значительной степени определяются природой и
составом сплавов. Так, хром, алюминий, кремний и углерод очень сильно
замедляют процесс окисления стали вследствие образования пленок с высо-
высокими защитными свойствами. Аналогичная картина наблюдается и при ле-
легировании меди бериллием, оловом, алюминием и цинком. Наоборот, эле-
элементы, образующие легкоплавкие и летучие оксиды, например ванадий,
вольфрам, молибден, ускоряют процессы окисления.
Внешние факторы, такие как вид, состав, давление, температура и
скорость движения газовой среды, время ее контакта, режим нагрева оказы-
оказывают большее влияние на скорость коррозии, чем внутренние. При повыше-
повышении температуры, с одной стороны, понижается термодинамическая воз-
возможность газовой коррозии, с другой, — увеличиваются константа скорости
химической реакции и коэффициент диффузии, а также изменяются защит-
защитные свойства оксидной пленки. В целом с ростом температуры скорость
коррозии увеличивается в соответствии с зависимостью, близкой к экспо-
экспоненциальной. Колебания температуры, особенно попеременные нагрев и
охлаждение, вызывают быстрое разрушение защитной пленки из-за возник-
возникновения больших внутренних напряжений.
С повышением парциального давления кислорода скорость окисления
ряда металлов при высокой температуре увеличивается, а затем при дости-
достижении некоторого критического значения уменьшается и остается достаточ-
достаточно низкой в широком интервале давлений. Наблюдаемое явление получило
название высокотемпературной пассивации. Пассивное состояние металла
обусловлено образованием на его поверхности совершенной по структуре
оксидной пленки.
Для защиты от газовой коррозии применяют жаростойкое легирова-
легирование хромом, алюминием, кремнием, различные покрытия (в основном тер-
термодиффузионные хромом, алюминием, кремнием и жаропрочные эмали), а
также защитные атмосферы (Ar; N2; N2-CO2; N2-H2 и др.).
608 18, Коррозия металлов и сплавов
18.5. Электрохимическая коррозия:
причины и механизм возникновения
На практике чаще всего приходится иметь дело с электрохимической
коррозией. Она, в отличие от химической, сопровождается возникновением
электрического тока и протекает, как правило, в средах с хорошей ионной
проводимостью. По условиям осуществления различают: коррозию в элек-
электролитах, атмосферную коррозию, электрокоррозию, коррозию под напря-
напряжением и др. Причинами возникновения электрохимической коррозии слу-
служат различные виды неоднородностей как самой поверхности металла или
сплава, так и коррозионной среды. В результате вся поверхность, соприка-
соприкасающаяся с токопроводящей коррозионной средой, разделяется на катодные
и анодные участки, которые имеют очень малые размеры и чередуются друг
с другом. В такой среде они представляют собой совокупность огромного
числа короткозамкнутых коррозионных гальванических микроэлементов,
вследствие чего электрохимическую коррозию часто называют гальваниче-
гальванической коррозией. Обычно имеют место следующие основные случаи неодно-
неоднородностей в системе металл—электролит, которые приводят к возникнове-
возникновению и функционированию коррозионных микроэлементов:
1) неоднородность металлической фазы, обусловленная наличием за-
загрязнений, примесей в металле, пленок на его поверхности, различным хи-
химическим и фазовым составом сплава;
2) неоднородность жидкой фазы, обусловленная различной концен-
концентрацией в электролите (коррозионной среде) ионов данного металла и рас-
растворенного кислорода на отдельных участках контакта фаз, неодинаковым
значением рН отдельных зон объема электролита и др.;
3) неоднородность наложения внешних условий: неодинаковая темпе-
температура отдельных участков поверхности металла (более нагретые являются
анодами); различный уровень механических напряжений в одной и той же
детали; неравномерное наложение внешнего электрического поля и др.
В системах возможно возникновение коррозионных не только микро-,
но и макроэлементов, например, при контакте с электролитом двух соприка-
соприкасающихся деталей, изготовленных из металлов различной активности (так
называемая контактная коррозия).
Механизм электрохимической коррозии сводится к возникновению
и функционированию коррозионных гальванических макро- и микроэле-
микроэлементов, поэтому ее процессы аналогичны процессам, протекающим в хи-
химических источниках тока: гальванических и топливных элементах, ак-
аккумуляторах. Основное отличие коррозионных процессов — отсутствие
внешней цепи. Электроны в процессе коррозии не выходят из корроди-
корродирующего металла, а перемещаются внутри него от анодных участков к
катодным.
18.5. Электрохимическая коррозия: причины и механизм возникновения 609
Процесс электрохимической коррозии представляет собой совокуп-
совокупность двух сопряэюенных (взаимосвязанных) полуреакций (в дальнейшем
реакций), одновременно протекающих на поверхности металла:
а) анодной, сопровождающейся окислением атомов металла на анод-
анодных участках поверхности:
(-)A:M-»MZ+ + Ze A8.3)
б) катодной, сопровождающейся восстановлением окислителя (окис-
(окисленной формы компонента Оф) коррозионной среды (электролита) на ка-
катодных участках поверхности:
(+) К: Оф + Ze -> Вф
Окислители электрохимической коррозии называют деполяризатора-
деполяризаторами. К наиболее часто встречающимся деполяризаторам относятся молекулы
кислорода О2, воды Н2О и ионы водорода Н+. Основными катодными реак-
реакциями с их участием при электрохимической коррозии являются:
1) в аэрированных (насыщенных кислородом) коррозионных средах:
нейтральных и щелочных (рН > 7)
(+) К: О2 + 2Н2О + 4е -> 4ОН" I Фо2,2Н2о = °>401 в \ A8.4)
V 4ОН- /
кислотных (рН < 7)
(+)К: О2+4Н++4е->2Н2О ф^4Н+ = 1,229 В ; A8.5)
V 2Н2О )
2) в деаэрированных (несодержащих растворенный кислород) коррозионных
средах:
нейтральных и щелочных (рН > 7)
(+) К: 2Н2О + 2е -+ Н21+2OH" ф° 2Нг0 = -0,828 В ; A8.6)
V Н2,2ОН" )
кислотных (рН < 7)
(+)К: 2Н++2^-> H2t L°2H+=0B. A8.7)
Коррозию, сопровождающуюся восстановлением (ионизацией) моле-
молекул кислорода (см. A8.4) и A8.5)), называют коррозией с поглощением кис-
20 - 9795
610 18. Коррозия металлов и сплавов
лорода, или коррозией с кислородной деполяризацией. С кислородной депо-
деполяризацией протекают следующие виды электрохимической коррозии: ат-
атмосферная, подземная, в воде (пресной и морской), растворах солей, аэри-
аэрированных растворах органических кислот и др. Коррозию, сопровождаю-
сопровождающуюся восстановлением молекул воды и ионов водорода (см. A8.6) и A8.7)),
называют коррозией с выделением водорода, или коррозией с водородной
деполяризацией. В некоторых условиях электрохимическая коррозия может
протекать одновременно и с водородной, и с кислородной деполяризацией —
так называемый смешанный вид деполяризации.
Чтобы получить уравнения реакций, лежащих в основе электрохими-
электрохимической коррозии металлов в различных средах (коррозионные токообра-
зующие реакции), суммируют A8.3) поочередно с A8.4)—A8.7), предвари-
предварительно уравняв число отданных и принятых электронов:
4М + ZO2 + 2ZH2O -> 4M(OH)Z A8.8)
4M + ZO2 + 4ZH+ ->4MZ+ + 2ZH2O A8.9)
2M + 2ZH2O -> 2M(OH)Z + ZH2t A8.10)
2M + 2ZH+ -> 2MZ+ + ZH2T A8.11)
Схему (условную запись) коррозионного ГЭ записывают аналогично
схеме обычного ГЭ: слева — анод, справа — катод, между ними указывают
коррозионную среду, причем лишь те ее компоненты, которые принимают
участие в токообразующей реакции. Коррозионную среду от анода и катода
отделяют вертикальными линиями, обозначающими границу раздела фаз т-ж.
Иногда в ней указывают концентрации окислителей, парциальные давления
газов и температуру.
Процессы, описываемые уравнениями A8.4)—A8.7), называют первич-
первичными процессами, а их продукты — первичными продуктами коррозии. Кро-
Кроме первичных при электрохимической коррозии протекают еще и вторичные
процессы — химические взаимодействия первичных продуктов друг с дру-
другом, с компонентами электролитной коррозионной среды, с растворенными в
ней газами и др. При этом образуются пленки малорастворимых вторичных
продуктов коррозии, например, гидроксидов, фосфатов металлов:
Mz+ + ZOH" -> M(OU)zi
3Mz++ZPO^--> M3(PO4)z|
которые затрудняют доступ электролита к поверхности металла. В результа-
результате снижается скорость электрохимической коррозии, а иногда коррозия и
совсем прекращается.
Рассмотрим наиболее характерные и часто встречающиеся на практи-
практике случаи электрохимической коррозии.
18.5. Электрохимическая коррозия: причины и механизм возникновения 611
-:-:?>/>_-нлоЛ - нул:н;.- н,_- он;
Рис. 18.3. Диаграмма состояния (а) и соответствующие схемы возмож-
возможного селективного (б) и интеркристаллитного (в) коррозионного раз-
разрушения эвтектического сплава составов I и II соответственно
Коррозионное разрушение эвтектического сплава обусловлено его
неоднородностью по химическому и фазовому составу. При контакте с
электролитной средой может происходить либо его селективная коррозия
(рис. 18.3, б), либо интеркристаллитная (рис. 18.3, в). В первом случае про-
происходит вытравливание из поверхностного слоя более активного металла D
(р^21+ < (pgZ2+ (сплав состава I), во втором осуществляется разрушение эв-
v ~5~ ~в~ J
тектики в глубину из-за высокой плотности тока, обусловленной малой по-
поверхностью зерен D в эвтектике (сплав состава II). Этот случай наблюдает-
наблюдается, когда более активный металл весь входит в состав эвтектики, разделяю-
разделяющей кристаллические зерна металлов.
Контактная коррозия связана с конструктивными особенностями из-
изделий и машин и имеет место при их эксплуатации в реальных условиях.
Часто в одном узле контактируют детали из разных металлов. В возникаю-
возникающем в электролитной среде коррози-
коррозионном гальваническом макроэлемен-
макроэлементе катодом является деталь, металл
которой имеет больший потенциал,
анодом — деталь, изготовленная из
металла с меньшим потенциалом. На
рис. 18.4 показана схема контактной
коррозии двух листов железа, соеди- 2+
ненных медными заклепками. Функ- '' ' е'* е"^ е + е
ционирует коррозионный гальвани- (+)K(Cu): 2H +2е-»Н2Т
ческий макроэлемент Рис. 18.4. Схема контактной коррозии
рН<7|_ -_о-ь_- _2-оо_-_-_-_
20*
612
18. Коррозия металлов и сплавов
(-) Fe | Электролит | Си (+),
в котором железо \ ц>° 2+ = -0,440 В — анод, медь q>° 2+ = 0,337 В — ка-
V ^~~ ) V ~7Г~ )
тод. Анодный процесс представляет собой растворение железа, катодный же
определяется видом, составом и кислотностью коррозионной среды. Ско-
Скорость этой разновидности электрохимической коррозии в целом тем больше,
чем дальше отстоят друг от друга в ряду напряжений металлы, из которых
изготовлены детали, образующие макроэлемент.
Функционирование макроэлемента влияет на скорость коррозии каждо-
каждого из образующих его металлов: коррозия металла с меньшим потенциалом в
большинстве случаев усиливается, а с большим потенциалом — ослабляется
или иногда даже совсем прекращается. Скорость коррозии металла с мень-
меньшим потенциалом зависит от природы металла-партнера (металла с большим
потенциалом, называемого металлом катодного контакта), а точнее, от зна-
значения его потенциала и катодной поляризуемости, а также площади его по-
поверхности. Чем больше будет разница в значениях электродных потенциалов
контактирующих металлов, тем выше будет скорость коррозии более актив-
активного металла, а следовательно, и всего коррозионного процесса.
Коррозия под напряжением — разрушение деталей, находящихся в
электролитной среде в механически напряженном состоянии. Ее причиной
является различный уровень как внутренних, так и внешних механических
напряжений в пределах одной детали. При всех видах механической обра-
обработки затраченная энергия частично превращается в тепловую, а частично
идет на увеличение поверхностной энергии обработанного участка. В ре-
результате обработанная часть детали об-
обладает повышенным запасом энергии,
при этом более деформированные (на-
(напряженные) участки металла являются
анодными, поэтому если пластину из
стали, дюраля или титанового сплава
согнуть и в таком состоянии поместить,
например, в коррозионную среду с
рН < 7, то растянутая поверхность нач-
начнет корродировать и на ней через ко-
короткое время появятся трещины, а сжа-
сжатая будет оставаться без изменений
(рис. 18.5). Это объясняется следую-
следующим. Механические напряжения изме-
изменяют потенциал растворения металла:
потенциал металла растянутой поверх-
Рис. 18.5. Коррозионное разрушение
детали, находящейся в механически
напряженном состоянии:
1 — стальная пластина; 2 — тефлоновая
подставка; 3 — кислотная коррозионная
среда
18.6. Термодинамика электрохимической коррозии 613
ности будет меньше потенциала металла без напряжения, а потенциал ме-
металла сжатой, наоборот, больше потенциала металла без напряжения на ве-
величину Аф. В связи с этим растянутая поверхность будет являться анодной,
сжатая — катодной. Возникающий при этом коррозионный ГЭ будет иметь
ЭДС, равную 2Аф.
Коррозионные трещины могут при этом распространяться не только
межкристаллитно, но и транскристаллитно. Подвергая детали термической
обработке, снимают в них остаточные напряжения, а искусственно созданные
при сгибании упругие напряжения переводят в пластические. При этом раз-
разности потенциалов не возникает, а следовательно, и коррозии не происходит.
Рассмотренные случаи не исчерпывают всего многообразия электро-
электрохимических коррозионных процессов, но дают представления об основных
видах неоднородностей, характере взаимодействий изделий с коррозионной
средой и причинах возникновения потенциалов в коррозионных парах.
18.6. Термодинамика электрохимической коррозии
Возможность электрохимической коррозии, как и любого химическо-
химического процесса, определяют по изменению энергии Гиббса. Поскольку корро-
коррозия является самопроизвольно протекающим процессом, то сопровождается
ее убылью, т.е. ArGT < 0. Так как электрохимическая коррозия связана с
функционированием коррозионного ГЭ, то возможность ее протекания
можно оценить и по знаку ЭДС. Последняя связана с энергией Гиббса соот-
соотношением A7.15). Отрицательному значению ArGr соответствует положи-
положительное значение ЭДС, которое рассчитывают по разности значений элек-
электродных потенциалов катода и анода (или окислителя и восстановителя):
Вф м
Отсюда следует, что электрохимическая коррозия возможна, если потенциал
окислителя (деполяризатора) больше потенциала восстановителя (металла):
Вф М
Основные катодные реакции A8.4)—A8.7) соответствуют реакциям
кислородного и водородного электродов, потенциалы которых зависят от
кислотности (водородного показателя) раствора и парциальных давлений
газов (см. § 16.4, 16.5).
Полную картину термодинамической устойчивости металлов дают
компакт-диаграммы Пурбё. По ним можно определять вероятность протека-
614 18. Коррозия металлов и сплавов
ния тех или иных катодных и анодных реакций, т.е. возможность или не-
невозможность электрохимической коррозии. Каждая система металл—вода
имеет свой вид диаграммы Пурбе, большинство из которых достаточно
сложны, особенно если учитывать возможность образования всех видов час-
частиц, и не дают информации о скоростях реакций. Их анализ для разных ме-
металлов показывает, что в водных средах, содержащих Н+ и СЬ, металлы кор-
корродируют по-разному в зависимости от рН:
а) если потенциал металла больше потенциала кислородного электро-
электрода (рис. 16.7, область I), то коррозия металла практически невозможна (на-
(например, Аи);
б) в случае, если потенциал металла меньше потенциала кислородно-
кислородного, но больше потенциала водородного электродов (см. рис. 16.7, область II),
то коррозия возможна с поглощением кислорода и невозможна с выделени-
выделением водорода (например, Си, Ni, Cd, Sn);
в) если потенциал металла меньше потенциала водородного электрода
(см. рис. 16.7, область III), то его коррозия возможна как с поглощением ки-
кислорода, так и с выделением водорода (например, Zn, A1, щелочные и ще-
щелочноземельные металлы).
Как следует из рис. 16.7, коррозия с кислородной деполяризацией
термодинамически более вероятна и выгодна, однако реализация термоди-
термодинамической возможности определяется кинетическими факторами, которые
влияют на поляризуемость электродов, т.е. на перенапряжение электродных
реакций.
Приближенно о термодинамической устойчивости (стабильности) ме-
металлов можно судить по значению их стандартных электродных потенциа-
потенциалов ф^2+: чем меньше значение cp^z+, тем выше восстановительная способ-
ность металла — способность отдавать электроны, т.е. корродировать. По
величине q>^z+ все металлы подразделяют на 5 групп, каждая из которых
м
отделена друг от друга значениями потенциалов водородного и кислородно-
кислородного электродов в кислотной (рН = 0) и нейтральной (рН = 7) средах:
Ф^ =0,000 В; Ф 2н2о = -0,414В; 9^.= 1,229 В; фр2>2Н2О =0,815 В.
Н2 Н2,2ОН" 2Н2О 4ОН-
1. Металлы низкой термодинамической стабильности ф° z+ < -0,414 В
V м /
корродируют даже в нейтральных средах, не содержащих кислород и окис-
окислители: Na, Mg, Be, Al, Zn и др.
18.7. Кинетика электрохимической коррозии 615
2. Металлы термодинамически нестабильные -0,414 <cp°z+ <0B
V м /
устойчивы в нейтральных средах при отсутствии кислорода, но в кислотных
корродируют даже при его отсутствии: Cd, Ni, Sn, Pb и др.
3. Металлы промежуточной термодинамической стабильности
^z+ < 0,814 В устойчивы в нейтральных и кислотных средах при от-
сутствии кислорода и окислителей: Ag, Cu, Bi, Re, Rh и др.
4. Металлы высокой термодинамической стабильности 0,814 < ф°,+ <
1 JiL_
V м
< 1,229 В не корродируют в нейтральных средах при наличии в них ки-
кислорода: Pt, Pd, Ir и др.
5. Металлы практически полной термодинамической стабильности
[(p°Mz+ > 1,229 В устойчивы в кислотных средах даже при наличии кисло-
рода, однако могут корродировать в растворах комплексообразователей при
наличии окислителей. К этой группе относится золото, которое не корроди-
корродирует ни с поглощением кислорода, ни с выделением водорода во всей облас-
области значений рН при отсутствии комплексообразователей ф^ц3+ = 1,498 В .
V Аи У
Такое деление металлов коррелирует с их положением в Периодиче-
Периодической системе элементов: наиболее коррозионно-стойкие располагаются вни-
внизу групп переходных элементов (Os, Ir, Pt, Аи). В левых подгруппах (IVB,
VIB, VIIB) находятся легко пассивируемые металлы, причем с ростом по-
порядкового (атомного) номера их пассивность в подгруппе растет.
18.7. Кинетика электрохимической коррозии
Как уже указывалось в § 18.5, электрохимическая коррозия представ-
представляет собой совокупность двух сопряженно протекающих гетерогенных ре-
реакций: анодного окисления металла и катодного восстановления деполяри-
деполяризатора (окислительного компонента коррозионой среды). Каждая из них
является многостадийной, в которой одна из стадий лимитирующая. Поскольку
катодная и анодная реакции протекают взаимосвязанно, то замедление
одной тормозит другую. Таким образом, общая скорость всего коррозион-
616 18. Коррозия металлов и сплавов
ного процесса будет определяться скоростью лимитирующей стадии либо
катодной {катодный контроль), либо анодной {анодный контроль) реакций.
Поскольку коррозионные ГЭ являются короткозамкнутыми, а электрохими-
электрохимическая коррозия чаще всего протекает в средах с высокой ионной проводи-
проводимостью, то ни стадия перемещения электронов внутри металла, ни стадии
диффузии ионов в растворе не могут быть лимитирующими.
Коррозия металлов с лимитирующей катодной реакцией (катод-
(катодным контролем). Известно, что катодная реакция обычно представляет со-
собой восстановление либо Ог (кислородная деполяризация), либо Н+ (водо-
(водородная деполяризация). С кислородной деполяризацией протекает коррозия
большинства технически важных металлов и сплавов в условиях их экс-
эксплуатации. Растворимость кислорода в воде и водных растворах мала и при
температуре 25 °С, парциальном давлении в воздухе р0^ =21 кПа (ргтм= 101,3
кПа) составляет 2,6 • КГ4 моль/л. Поэтому катодное восстановление кисло-
кислорода по реакциям A8.4) и A8.5) лимитируется скоростью его диффузии к
поверхности и протекает с концентрационной поляризацией.
Скорость коррозии с кислородной деполяризацией возрастает при пе-
перемешивании раствора, что обусловлено уменьшением толщины диффузи-
диффузионного слоя и увеличением коэффициента диффузии и растворимости ки-
кислорода. Температурная зависимость скорости имеет максимум, наблю-
наблюдающийся при температуре 70...80 °С. Такой ее ход определяется тем, что с
одной стороны растет коэффициент диффузии кислорода, с другой —
уменьшается его растворимость в воде.
Коррозию с кислородной деполяризацией замедляют деаэрацией (сниже-
(снижением содержания О2, обескислороживанием) коррозионной среды, осуществ-
осуществляемой либо термической, либо химической обработками, либо уменьшением
давления кислорода над раствором. Изменение состава катодных участков мало
влияет на скорость коррозии с поглощением кислорода, поэтому степень чис-
чистоты металла практически не играет роли при этом виде коррозии. Если элек-
электродный потенциал металла меньше потенциала водородного электрода (см.
рис. 16.7, область III), то коррозия, как уже отмечалось, может протекать как с
выделением водорода, так и с поглощением кислорода (смешанная деполяри-
деполяризация). Если кислород в системе отсутствует, либо очень быстро расходуется и
при этом не восполняется, например при коррозии в закрытой системе, то име-
имеет место катодный процесс лишь с выделением водорода (водородной деполя-
деполяризацией). Такой вид коррозии возможен во всех типах сред (нейтральных, ки-
кислотных и щелочных), но чаще всего наблюдается в кислотных, например, при
транспортировке и хранении кислот, кислотной очистке металлической по-
поверхности и кислотном травлении проката. Несмотря на то что в таких раство-
растворах содержится кислород, скорость его восстановления мала по сравнению со
скоростью выделения водорода, поэтому в первом приближении пренебрегают
18.7. Кинетика электрохимической коррозии 617
скоростью коррозии за счет поглощения кислорода и учитывают только ско-
скорость коррозии за счет выделения водорода.
Ввиду малых размеров и большой подвижности ионов водорода их диф-
диффузия не лимитирует процесс катодного выделения водорода. Лимитирующи-
Лимитирующими стадиями являются либо восстановление ионов водорода, либо рекомбина-
рекомбинация его атомов в молекулы. В этом вопросе до сих пор нет единого мнения.
Скорость катодного выделения водорода практически не зависит от пе-
перемешивания раствора и возрастает как с повышением температуры в соот-
соответствии с уравнением Аррениуса, так и с увеличением концентрации ионов
водорода, поскольку равновесный потенциал водородного электрода зависит
от рН раствора (см. § 16.4). На скорость этого процесса влияет также природа
катодных участков, так как выделение водорода может происходить с боль-
большим перенапряжением (см. § 17.6). Чем выше перенапряжение, тем меньше
скорость выделения водорода при данном потенциале катодного участка, и,
следовательно, ниже скорость коррозии металла и наоборот. Выделение во-
водорода на таких металлах, как Pb, Zn, Cd, Hg, протекает со значительным пе-
перенапряжением. Их присутствие в составе сплава или не меняет скорости
коррозии основного металла, или снижает ее из-за уменьшения площади по-
поверхности, занимаемой основным металлом, на которой и происходят раство-
растворение металла и выделение водорода. Такие металлы, как Pt, Co, Ni, обладают
каталитическим действием в реакции катодного выделения водорода, по-
поскольку перенапряжение выделения Н2 на них мало.
Таким образом, коррозия с водородной деполяризацией может быть
замедлена путем снижения температуры, удаления из металла катализи-
катализирующих катодный процесс примесей, уменьшения содержания Н+-ионов,
изоляции поверхности металла.
Коррозия металлов с лимитирующей анодной реакцией (анодным
контролем). В анодной реакции принимают участие анионы, содержащиеся
в коррозионной среде, поэтому ее протекание определяется их природой и
концентрацией. Как и катодная, она является многостадийной гетерогенной
реакцией и состоит из последовательно протекающих стадий:
1) химической адсорбции анионов на анодных участках поверхности
металла
2) электрохимической реакции, определяющей скорость всего процесса
(MAXC^(MAJ№+Ze
3) распада образовавшегося на стадии 2 комплекса на простые ионы
618 IS. Коррозия металлов и сплавов
Процесс может тормозиться также диффузией анионов к поверхности
и ионизацией атомов металла вследствие возрастания концентрации его ио-
ионов вблизи поверхности, что может привести к образованию и выпадению в
осадок малорастворимых гидроксидов, например
Fe2++2OH->Fe(OHJi
так как при коррозии происходит повышение рН среды вследствие одно-
одновременного протекания катодных процессов — восстановления молекул ки-
кислорода и Н+-ионов. Возможно также образование малорастворимых солей,
например
3Fe2+ + 2РО^ -> Fe3(PO4Ji
Малорастворимые соединения, оседая на поверхности металла, экрани-
экранируют ее. Тем самым затрудняется диффузия частиц, образующихся при про-
протекании анодной реакции, а следовательно, тормозится коррозия в целом.
Торможение анодной реакции может происходить и по причине пас-
пассивности металлов, которая наблюдается в определенных условиях у таких
металлов, как Al, Cr, Ni, Mo, Ti, Zr, Та, Mg, Co, Fe, причем первые пять от-
относятся к легко пассивирующимся. Пассивацией называется процесс пере-
перехода металла или сплава в состояние, при котором наблюдается аномально
резкое уменьшение скорости их коррозии при контакте с сильными окисли-
окислительными жидкими или газообразными средами. В случае электрохимиче-
электрохимической коррозии это водные растворы веществ-окислителей, таких как Ог,
HNO3, H2O2, КМпО4, К2СГ2О7 и др. Для титана сильной окислительной сре-
средой является аэрированная вода.
Вещества, вызывающие наступление пассивного состояния у металлов,
называют пассивирующими реагентами, или пассиваторами. Они хемосор-
бируются на поверхности металла или образуют с ним самостоятельные
твердофазные химические соединения в виде пассивирующих защитных
пленок, которые препятствуют нормальному росту металлических кристал-
кристаллов, их растворению, окислению, дальнейшему взаимодействию с пассива-
пассиваторами и т.д. По природе соединений, образующих барьер (защитную плен-
пленку), различают оксидную, гидроксидную, солевую, гидридную и другие ти-
типы пассивности металлов. Наиболее важная среди них оксидная, поскольку
кислород — самый распространенный пассиватор.
Пассивирующим действием в водных растворах обладают и анионы
малорастворимых солей данного металла. Особенно эффективны анионы лег-
легко пассивирующихся металлов, например CrOJj", так как образующийся при
их восстановлении оксид также входит в защитный барьер. К пассивирую-
пассивирующим факторам относят также и анодную поляризацию, осуществляемую от
внешнего источника постоянного тока или возникающую вследствие функ-
функционирования ГЭ, в котором пассивирующийся металл является анодом.
18.7. Кинетика электрохимической коррозии 619
Пассивное состояние проявляется только в определенной области по-
потенциалов (при этом с термодинамической точки зрения металл в ней впол-
вполне реакционноспособен) и характеризуется значительным увеличением по-
потенциала металла (например, для хрома от -@,6...0,4) до + @,9... 1,0) В) и
аномально низкой и постоянной во времени скоростью коррозии. Возникно-
Возникновение пассивного состояния зависит от природы металла и его свойств, вида,
состава (концентрации окислителя и содержания растворенного кислорода),
температуры и скорости перемешивания окислительной среды. Пассивности
металла способствуют легирование легко пассивирующимися металлами,
повышение электродного потенциала до критического значения, увеличение
концентрации пассиватора вблизи поверхности металла. Механизм пасси-
пассивирования сложен и недостаточно изучен.
Согласно пленочной теории (М. Эванс, Г.В. Акимов, В.А. Кистяков-
ский и др.), торможение коррозии наступает вследствие образования на по-
поверхности металла плотной защитной пленки (другой фазы) толщиной до
нескольких десятков нанометров, которая представляет собой чаще всего
слой оксида или гидроксида. Она, изолируя металл от окружающей среды,
затрудняет диффузию, а следовательно, замедляет коррозию. В некоторых
случаях защитная пленка на поверхности не обнаруживалась и пассивное
состояние, имевшее все-таки место при этом, с позиций данной теории не
находило объяснения.
В соответствии с адсорбционной теорией (А.Н. Фрумкин, Я.М. Колотыр-
кин, Б.Н. Кабанов и др.), наступление пассивного состояния связано с пере-
перестройкой поверхностного слоя металла и обусловлено насыщением валентно-
валентностей его поверхностных атомов в результате возникновения химических связей
с адсорбированными атомами кислорода, образующимися из адсорбированных
молекул воды или гидроксидных ионов. Атомы кислорода, адсорбированные
на наиболее активных участках поверхности, блокируют процесс ионизации
атомов металла, в результате чего последние теряют свою реакционную спо-
способность и переходят в пассивное состояние. Установлено, что оно наступает
даже тогда, когда количество адсорбированного кислорода не соответствует
полному монослойному покрытию поверхности.
Почти все окислители в коррозионных средах с одной стороны вызы-
вызывают пассивность металла и резкое торможение коррозии, а с другой стороны,
в случае, если их концентрация недостаточна для достижения пассивного со-
состояния, восстанавливаясь на катодных участках, усугубляют процесс разру-
разрушения металла. Явление пассивности имеет большое практическое значе-
значение, поскольку коррозионная стойкость большинства металлов обусловлена
именно их пассивным состоянием. Часто оно нарушается при определенных
концентрации окислителя и значениях потенциала, превышение которых
приводит к возрастанию скорости коррозии. Это явление называют перепас-
620
18. Коррозия металлов и ставов
сивацией. Его причиной является образование в сильных окислительных
средах хорошо растворимых оксидов металлов высших валентностей. Перепас-
Перепассивации способствует также повышение температуры среды. Бороться с ней
можно, уменьшая потенциал металла. Это достигается снижением концентра-
концентрации окислителя, добавлением восстановителей либо путем катодной поляриза-
поляризации металла, осуществляемой от внешнего источника постоянного тока.
18.8. Влияние различных факторов на скорость
электрохимической коррозии
Изменение температуры может ускорять и замедлять процесс элек-
электрохимической коррозии. Это связано с ее различным влиянием на скорость
любой из стадий сопряженных реакций. Например, с увеличением темпера-
температуры ускоряется диффузия и уменьшается концентрация газообразных рас-
растворенных веществ (О2, С12), участвующих в электродных процессах, сни-
снижаются перенапряжение катодного восстановления окислителя (Н+, О2) и
защитные свойства пленок из вторичных продуктов (малорастворимых со-
солей, гидроксидов и др.), может измениться и полярность (катодные или
анодные) металлических защитных покрытий.
По влиянию кислотности раствора (рН среды) на скорость электро-
электрохимической коррозии все металлы подразделяют на пять групп, каждая из
которых имеет свой вид зависимости (рис. 18.6):
1) металлы с высокой коррозионной стойкостью в кислотных, ней-
нейтральных и щелочных растворах, такие как Ag, Au, Pt и др. Скорость их
коррозии не зависит от рН раствора (рис. 18.6, а);
2) металлы, устойчивые в кислотных растворах, но нестойкие в ще-
щелочных — Мо, Та, W и др. (рис. 18.6, б);
3) металлы, малостойкие в кислотных растворах, но устойчивые в ще-
щелочных — Ni, Cd и др. (рис. 18.6, в);
Sn
7
а
14 0
7
б
14 0
7
в
14 0
14 0 4
8,5 10
д
Рис. 18.6. Основные виды кривых (а-д) зависимости скорости элек-
электрохимической коррозии гмасс металлов от рН коррозионной среды
(добавки НС1 и NaOH)
рН
18.8. Влияние различных факторов на скорость коррозии 621
4) металлы, устойчивые в растворах, близких к нейтральным, но раз-
разрушающиеся в щелочных и кислотных из-за амфотерности — Zn, Al, Sn, Pb
(рис. 18.6, г). Каждый из них имеет свое определенное значение рН, при ко-
котором скорость коррозии минимальна: 7(А1), 8(Pb), 9(Sn), 10(Zn), 14(Fe);
5) металлы, малостойкие в кислотных растворах, в интервале значе-
значений рН 4...8,5 имеют постоянную скорость коррозии, которая при рН > 10
резко уменьшается вследствие образования на их поверхности малораство-
малорастворимых гидроксидов — Fe, Mg, Си, Мп и др. (рис. 18.6, д).
Влияние давления на скорость электрохимической коррозии связа-
связано, главным образом, с изменением растворимости газа, участвующего в
катодном процессе. Так, увеличение давления приводит к усилению кор-
коррозии (возрастанию ее скорости), протекающей с кислородной деполяри-
деполяризацией, и не влияет на коррозию, осуществляемую с водородной деполя-
деполяризацией.
Скорость движения электролита в большей степени влияет на кор-
коррозию, протекающую с кислородной деполяризацией, чем на коррозию с
водородной деполяризацией. Это влияние имеет сложный характер: вначале
скорость коррозии увеличивается (из-за возрастания скорости диффузии
растворенного кислорода и уменьшения толщины диффузионного слоя),
затем падает (за счет образования на поверхности защитной пленки), потом
вновь резко возрастает (вследствие механического удаления (эрозии) за-
защитной пленки движущимся потоком электролита).
Влияние состава (вида, числа и концентрации компонентов) электро-
электролитной среды также имеет сложный характер. С возрастанием концентра-
концентрации раствора скорость коррозии вначале увеличивается, а затем падает. Та-
Такая зависимость типична для коррозии металлов в нейтральных растворах
солей (Na2SO4, NaCl, KC1, LiCl), протекающей с кислородной деполяризаци-
деполяризацией. Возрастание скорости коррозии обусловлено увеличением электропро-
электропроводности раствора и, следовательно, активизацией анодного процесса,
уменьшение — снижением растворимости кислорода, которое становится
превалирующим фактором.
Электрохимическая коррозия значительно ускоряется в присутст-
присутствии небольшого количества веществ, называемых в соответствии с ха-
характером их действия активаторами (ускорителями, или стимулятора-
стимуляторами). К их числу относятся галогенид-ионы: СГ, Вг~, Г, которые, адсорби-
руясь на поверхности защитной пленки, вытесняют из нее кислород. При
этом образуются растворимые галогениды металлов, переходящие в рас-
раствор. В пленке возникают поры, которые, облегчая доступ коррозионной
среды, способствуют началу и дальнейшему усилению коррозии. Осо-
Особенно велико влияние хлорид-ионов на растворение таких металлов, как
железо, хром, никель, алюминий.
622 18. Коррозия металлов и сплавов
18.9. Коррозия в естественных условиях
Коррозия в естественных условиях (атмосферная, подземная, блуж-
блуждающими токами, морская) протекает по электрохимическому механизму и,
как правило, с кислородной деполяризацией. В ее основе лежат реакции,
описываемые уравнениями A8.8)—A8.9).
Атмосферная коррозия — самый распространенный и наиболее часто
встречающийся на практике вид электрохимической коррозии. Примерно 80 %
металлических конструкций эксплуатируются в атмосферных условиях. По-
Поэтому под атмосферной коррозией понимают процессы разрушения поверхно-
поверхности металлов и сплавов в условиях реальной земной атмосферы при влажности
воздуха более 70 %. На ее скорость и механизм влияют такие факторы, как ха-
характер атмосферы (промышленная, сельская, континентальная и др.) и ее влаж-
влажность, температура и продолжительность воздействия, состав материала и со-
состояние его поверхности. Так, близость моря ускоряет атмосферную коррозию.
Эффект ощущается на расстоянии до 2 км от него. В условиях промышленной
атмосферы (загрязненной СО2, SCb, N02, NH3, HC1, угольной пылью, частица-
частицами солей) коррозия может протекать и с водородной деполяризацией.
Влажность воздуха (атмосферы) является одним из главных факторов,
так как определяет степень увлажненности корродирующего металла, а сле-
следовательно, механизм и скорость атмосферной коррозии. По степени ув-
увлажненности поверхности различают следующие ее виды:
1) сухую атмосферную коррозию — коррозию при полном отсутствии
влаги на поверхности металлов и сплавов. Она описывается уравнением
A8.1), и ее механизм аналогичен механизму газовой коррозии. Образую-
Образующиеся на поверхности оксидные пленки обладают защитными свойствами,
поэтому данный вид коррозии не приводит к существенному разрушению
металлических конструкций;
2) влажную атмосферную коррозию — коррозию при наличии на по-
поверхности металлов и сплавов тончайшей, невидимой пленки влаги, которая
образуется в результате капиллярной, адсорбционной или химической кон-
конденсации из воздуха при относительной его влажности ниже 100 %. Эта плен-
пленка практически всегда имеется на поверхности металлических материалов,
находящихся в атмосферных условиях. В результате растворения в ней газов,
содержащихся в воздухе (главным образом СОг и О2), она становится элек-
электропроводной, что и обусловливает функционирование коррозионных галь-
гальванических микроэлементов, возникающих вследствие электрохимической
неоднородности поверхности материала. Его структурные составляющие, об-
обладающие меньшим электродным потенциалом, выполняют роль анодных
участков на поверхности, например участки феррита, а роль катодных участ-
участков — структурные составляющие или различные примеси и включения (не-
18.9. Коррозия в естественных условиях
623
(-)A(cc-Fe):Fe -» Fe +-
(+)K(Fe3C):2H++2e->
IFe+2H+-> Fe2++H2t
(-)A(a-Fe):Fe ^ Fe2++2e 2
(+)K(Fe3C):O2+2Hp+4e-> 40H~| 1
+0,5O2
S2Fe+(X, +2H-O -> 2Fe(OH
+0,5C
2FeO(OH) +H20 -> m?exOy - wH2O
Рис. 18.7. Схема коррозионного разрушения углеродистой стали под
пленкой влаги, происходящего с водородной (а) и кислородной (б)
деполяризациями
металлов, оксидов, нитридов, карбидов и др.), электродный потенциал кото-
которых больше, например, кристаллиты цементита Fe3C в стали, микровключе-
микровключения графита в чугуне. На рис. 18.7 приведена схема коррозионного разруше-
разрушения углеродистой стали под пленкой влаги с кислородной и водородной де-
деполяризациями;
3) мокрую атмосферную коррозию — коррозию при наличии на по-
поверхности металла или сплава видимой пленки влаги, наблюдающуюся при
относительной влажности воздуха около 100 %, когда имеет место капель-
капельная конденсация влаги либо ее непосредственное попадание на металл в ви-
виде дождя, струй и др. На рис. 18.8 приведена схема коррозии железа под ка-
каплей воды. Мокрая атмосферная коррозия вызвана функционированием
коррозионного ГЭ и обусловлена неоднородностью коррозионной среды, а
именно неравномерной ее аэрацией — неодинаковым доступом воздуха к
различным слоям капли. Со временем в центральных (внутренних) слоях
капли содержание растворенного кислорода становится ниже, чем в пери-
периферических (наружных) слоях, непосредственно соприкасающихся с возду-
воздухом. В образующемся концентрационном коррозионном ГЭ центральная
часть смоченной поверхности металла является анодным участком и под-
подвергается разрушению, а периферийная (в виде кольца) — катодным, на ко-
624
18. Коррозия металлов и сплавов
А: Fe->Fe2++2e
К: 02 + 2Н2О + 4е -> 40Н"
Z2Fe + 02 + 2H2O -> 2Fe(OHJ
2FeO(OH) + Н20
• пН2О
Рис. 18.8. Схема коррозионного разрушения железной пластины B)
под каплей воды G):
а — первоначальное беспорядочное образование на ней анодных (А) и
катодных (К) участков; б, в — окончательное (со временем) формирование на
ней анодного (А) и катодного (К) участков и зоны ржавчины (Р) между ними
18.9. Коррозия в естественных условиях
625
тором протекает процесс восстановления растворенного кислорода. После
высыхания капли в ее центре обнаруживается углубление, а иногда даже
отверстие (для пластин толщиной 0,1...0,2 мм). Такие процессы часто на-
наблюдаются при атмосферной и почвенной коррозии железных и стальных
изделий (троса, стопки листов и др.) и приводят к точечным разрушениям,
переходящим в питтинг.
Продукты электродных (анодного и катодного) процессов в толще
электролита или на поверхности металла в пленке влаги (в рассмотренном
выше случае) взаимодействуют друг с другом с образованием гидроксида
железа Fe(OHJ, который затем окисляется кислородом до мета-гидроксида
FeO(OH), отвечающего составу бурой ржавчины. Хотя конечный продукт
коррозии — ржавчина т?ех0у • иН2О и нерастворим, однако он не препятст-
препятствует процессу растворения металла, так как формируется за пределами
анодного участка (на границе соприкосновения его с катодным) в виде
кольца внутри капли. Для постоянной защиты от нее традиционно приме-
применяют сплавы и защитные покрытия, для временной — масла, летучие инги-
ингибиторы коррозии.
Подземная коррозия — это разрушение металлических конструкций
(трубопроводов, опор, свай и др.) в почвах и грунтах. Для нее характерны
язвенные точечные разрушения, располагающиеся в нижней части тех уча-
участков трубопроводов, к которым затруднен доступ кислорода (рис. 18.9).
Как правило, скорость подземной коррозии возрастает с увеличением глу-
глубины залегания металлических конструкций.
Почва и грунты представляют собой одну из наиболее сложных по хими-
химическому составу и структуре коррозионных сред. Их характеристиками являются
влажность, пористость, кислотность, солевой состав и др. Максимальное зна-
о2 о2 о2 о2
77 lllllltllir
(((
// / 7 7 / t-TlirtltTi
/^ 7 7 I i 7^7 7 7 I 7 /
Песок * .' А Суглинок
Рис. 18.9. Схема возникновения токов {S/, 4S) длинных линий (про-
(протяженных конструкций) и коррозионной пары в плохо изолированном
стальном трубопроводе вследствие дифференциальной (неравномер-
(неравномерной) аэрации почвы различной пористости
626 18. Коррозия металлов и сплавов
чение скорости коррозии наблюдается при влажности 15...20 %. Пористые
грунты агрессивнее глинистых, так как легко аэрируются и долго сохраняют
влагу. Особенно велика коррозия в торфянистых и болотистых грунтах,
имеющих рН = 3, в которых она протекает с водородной деполяризацией.
Трудно выделить влияние на скорость подземной коррозии какой-либо од-
одной характеристики.
Борьбу с подземной коррозией осуществляют посредством нанесения
защитных и изолирующих покрытий, а также путем катодной защиты про-
протектором или внешним потенциалом.
Коррозия под действием блуждающих токов {электрокоррозия) —
особый вид электрохимической коррозии в естественных условиях. Блуж-
Блуждающими называют токи, ответвляющиеся от своего основного пути. Это
токи утечки из электрических цепей или любые токи, попадающие в землю
от внешних источников (различных систем и устройств). Источниками
постоянных блуждающих токов являются пути электропоездов, заземления
линий постоянного тока, установки для электросварки (электросварочные
аппараты) и нанесения гальванических покрытий (электролизеры), системы
катодной защиты. Источники переменных блуждающих токов — это обыч-
обычно заземления линий переменного тока и электрические кабели, индуци-
индуцирующие токи в проложенных рядом трубопроводах.
Блуждающие токи вызывают коррозию газо- и нефтепроводов, элек-
электрокабелей, различных подземных металлических сооружений. Радиус их
действия исчисляется десятками километров, при этом постоянные блуж-
блуждающие токи вызывают большие разрушения, чем переменные, а действие
токов высокой частоты сильнее, чем токов низкой частоты. Обычно корро-
коррозионные разрушения бывают локального типа и располагаются в местах вы-
выхода токов в землю или воду. Для подземных трубопроводов и путей элек-
электропоездов это, как правило, места изолированных сочленений и плохого
контакта рельсов на стыках соответственно, а также места с недостаточной
изоляцией от земли. Они являются анодными зонами и подвергаются уси-
усиленной коррозии (рис. 18.10).
Поддержание в хорошем состоянии электрических контактов в источни-
источнике блуждающих токов (например, между рельсами на стыках), увеличение со-
сопротивления между ним и почвой (или водой), а также электродренаж — отвод
блуждающих токов к их источнику путем соединения его с подземным соору-
сооружением (например, трубопроводом) металлическим проводником с низким со-
сопротивлением — вот основные меры борьбы с этим видом коррозии.
Коррозии в морской воде подвержены суда, гидросамолеты, металли-
металлические сооружения портов и нефтепромыслов и др. Морская вода обладает
хорошей электрической проводимостью, ее кислотность колеблется от 7,2
до 8,6, содержание растворенного кислорода достигает 8 мг/л, значительна
18.9. Коррозия в естественных условиях
627
Контактный воздушный провод
• | (+) Катодная зона ^
Нейтральная зона # j(~) Анодная зона
О2 + 2Н2О + 4е -> 4ОН"
Fe -> Fe2+ + 2e
Рис. 18.10. Схема возникновения блуждающих токов от трамвайной
линии и механизм коррозионного разрушения ими рельсов и трубо-
трубопровода
концентрация в ней хлорид-ионов, являющихся активаторами коррозии и
препятствующих образованию пассивных пленок на поверхности металла.
Коррозия в морской воде протекает, как правило, с катодным контролем и
наиболее интенсивно развивается в зоне, располагающейся несколько выше
ватерлинии, где происходит периодическое смачивание поверхности изделия.
Для борьбы с морской коррозией широко используют катодную защи-
защиту протектором или внешним потенциалом, а также различные виды покры-
покрытий (кадмиевые, лакокрасочные на различных основах, специальные с
токсичными для микроорганизмов веществами).
19 ¦
Защита от коррозии
Защита от коррозии — это комплекс мероприятий, направленных
на предотвращение и замедление коррозионных процессов, сохра-
сохранение и поддержание работоспособности узлов и агрегатов машин,
оборудования и сооружений в требуемый период их эксплуатации.
Способы защиты от коррозии выбирают на стадии конструирова-
конструирования и осуществляют в процессе изготовления и эксплуатации объ-
объектов. Среди них условно выделяют методы воздействия на металл
и саму конструкцию (рациональное конструирование, легирование,
обработка поверхности, нанесение защитных покрытий), методы
воздействия на коррозионную среду и условия эксплуатации (инги-
бирование, обработка среды, электрохимическая защита, гермети-
герметизация, осушка воздуха, создание искусственной атмосферы), а так-
также комбинированные методы. Выбор способа определяется его эф-
эффективностью и экономической целесообразностью.
19.1. Основные факторы рационального конструирования.
Легирование металлических материалов
Рациональное конструирование изделий — первый и обяза-
обязательный этап борьбы с коррозией, на стадии которого учитывают
следующие обязательные факторы:
а) правильный выбор материалов (металлов, сплавов, гермети-
ков, диэлектриков, пропиток и др.) для изделий и конструкций:
стойких в данной коррозионной среде, не способных впитывать вла-
влагу, не выделяющих коррозионно-активных агентов при старении;
б) рациональные сочетание и компоновка в одном узле дета-
деталей, изготовленных из металлов, отличающихся значениями элек-
электродных потенциалов: предотвращение их непосредственного кон-
контакта друг с другом и с коррозионной средой путем изоляции со-
соприкасающихся поверхностей, применения различных прокладок,
уплотнительных мастик и герметиков, чтобы исключить возмож-
возможность возникновения контактной коррозии (рис. 19.1);
в) оптимальная форма деталей: с дренажными отверстиями и
проветриваемыми полостями, с минимумом коррозионно-опасных
участков (углублений, пазов, щелей, канавок, зазоров, застойных зон);
г) минимальная слитность сечения (отношение периметра
сечения к его площади) у конструкций, характеризующая поверх-
поверхность их соприкосновения с коррозионной средой;
19.1. Основные факторы рационального конструирования
629
д) характер соединения элементов в
сборке: сварные соединения предпочтитель-
предпочтительнее клепаных и болтовых, которые ведут к
возникновению больших внутренних напря-
напряжений и пор;
е) возможность нанесения и возобнов-
возобновления различных покрытий в процессе экс-
эксплуатации изделий и при их ремонте.
Легирование {модифицирование) метал-
металлических материалов — эффективный про-
процесс повышения их стойкости к воздействию рис# 19.I. Конструкция (изде-
агрессивных сред при обычной и повышенных Лие) из алюминиевого уголка
температурах. Сущность его состоит в том, G) и листа из медного сплава
что в материал (металл, сплав), из которого D) с защитой герметиком C) и
изготавливают изделия, вводят легирующие уплотнительной прокладкой B)
компоненты, вызывающие его пассивацию. Различают объемное (металлур-
(металлургическое) и поверхностное (ионное) легирование.
Объемное легирование применяют в основном тогда, когда другие ме-
методы защиты от коррозии для данного материала не приемлемы. Его осуще-
осуществляют на стадии выплавки конструкционных материалов. Считают, что
легирующие компоненты диффундируют из объема на поверхность сплава и
вместе с основным металлом окисляются (пассивируются) кислородом воз-
воздуха, образуя устойчивые смешанные оксидные слои (защитные пленки) со
структурой шпинелей М(П)О • М2(Ш)Оз, которые препятствуют дальнейше-
дальнейшему проникновению коррозионной среды. Шпинели — группа минералов под-
подкласса оксидов МA1)М2(Ш)О4, где М(П) — Fe, Mg, Zn, Mn и др.; М(Ш) —
Al, Fe, Cr, Ti, Mo, Ni и др. Железо, алюминий, титан, магний, кадмий, цинк и
их сплавы легируют хромом, никелем, молибденом, медью и др. В результа-
результате получают сплавы с более высокой коррозионной стойкостью, чем исход-
исходные материалы. Эти сплавы одновременно обладают жаростойкостью и жа-
жаропрочностью.
Жаростойкость — свойство материалов противостоять химическому
разрушению под действием воздуха при высокой температуре.
Жаропрочность — способность конструкционных материалов выдер-
выдерживать без существенных деформаций механические нагрузки при высоких
температурах в инертной среде.
Повышенной коррозионной стойкостью обладают, как правило, спла-
сплавы с определенным содержанием легирующего компонента, которое опре-
определяется некоторым его критическим значением {границей устойчивости),
выше которого начинается избирательная коррозия: растворению подверга-
подвергается легирующий компонент, а основной металл остается в виде «губки»
630 19. Защита от коррозии
или порошка. Граница устойчивости, зависящая также от вида коррозион-
коррозионной среды, — характерная для каждого типа сплава величина, например:
Тип сплава Со—Cr Fe—Сг Ni—Сг
Сг,%(масс.) 8 12 14
В промышленности широко применяют объемно-легированные спла-
сплавы на основе:
а) железа — стали углеродистые, низколегированные, содержащие до
3 % (масс.) легирующих компонентов, и высоколегированные, или коррози-
коррозионно-стойкие, среди которых хромистые и хромоникелевые самые распро-
распространенные (например, марки 12X13, 12X17, 30X13, 20X13 — первая цифра
показывает содержание углерода в сотых долях процента (масс), буква и
цифра после нее — содержание хрома в % (масс);
б) меди — бронзы обычные (оловянистые с 8... 10 % (масс) Sn) и спе-
специальные (например, алюминиевые до 10 % (масс.) А1); латуни (сплавы с
содержанием 10...40 % (масс) Zn); мельхиор (группа сплавов, содержащих,
% (масс): 5...33 (Ni), -I (Fe), ~1 (Mn));
в) никеля — нихром B0 % (масс) Сг); хромель Р A0 % (масс.) Сг);
инконель 600 (% (масс): 16 (Сг), 7 (Fe)); алюмель (% (масс): 2 (А1), 2 (Мп),
1 (Si)); хастеллой С (% (масс): 54 (Ni), 15 (Сг), 16 (Мо), 4 (W), 5 (Fe));
г) алюминия — дуралюмин (% (масс): 4 (Си), 0,6 (Mg), 0,6 (Мп), 0,7 (Si),
0,7 (Fe)).
Главным недостатком объемного легирования является его дорого-
дороговизна, так как зачастую используются легирующие компоненты высокой
себестоимости.
Поверхностному легированию подвергают уже готовые изделия. Его
осуществляют методом ионной имплантации. Этот метод позволяет вводить
любую модифицирующую добавку в любой металл при низкой температуре.
На практике железо легируют хромом, никелем, алюминием, танталом,
свинцом; различные сорта сталей — гелием, кислородом, фосфором, тита-
титаном, молибденом, хромом, никелем и др. Легированный слой формируется в
результате бомбардировки поверхности изделия ионами легирующих ком-
компонентов. Ускоренные ионы, проникая в глубь металла, тормозятся при
столкновении с атомами, а затем нейтрализуются их свободными электро-
электронами. В результате ионы встраиваются в кристаллическую решетку металла,
замещая узлы или располагаясь в междоузлиях. Поверхностное легирование
позволяет избежать излишних затрат ввиду модифицирования только по-
поверхностного слоя и, в отличие от объемного, получать сплавы с такими ме-
металлами, как тантал, свинец, вольфрам, палладий, рутений, платина. Слож-
Сложность и высокая стоимость оборудования, а также малая толщина модифи-
модифицированного слоя ограничивают его применение.
19.2. Изменение состава и свойств коррозионной среды 631
19.2. Изменение состава и свойств коррозионной среды
Изменение состава и свойств коррозионной среды, т.е. уменьшение ее
агрессивности, осуществляют либо введением в нее специальных веществ —
ингибиторов коррозии, либо соответствующей ее обработкой.
Защиту ингибиторами применяют в системах с постоянным или мало
обновляемым объемом коррозионной среды (в котлах, цистернах, химических
аппаратах). Ингибиторами называют химические соединения или компози-
композиции на их основе, введение которых в небольших количествах (до 1 % (масс.))
в коррозионную среду резко снижает скорость коррозии. По химическому
составу различают органические и неорганические ингибиторы, по условиям
применения — жидкофазные (ингибиторы для растворов) и летучие ингиби-
ингибиторы, дающие защитный эффект в условиях атмосферной коррозии, по меха-
механизму своего действия на процесс электрохимической коррозии — катодные,
анодные и экранирующие (изолирующие) ингибиторы. В свою очередь, жид-
жидкофазные ингибиторы подразделяют на кислотные, щелочные и нейтральные,
в соответствии с тем, в какой среде они применяются.
Механизм защитного действия большинства ингибиторов заключается
в адсорбции (концентрировании) их на корродирующей поверхности и по-
последующем торможении анодных (анодные ингибиторы) и катодных (ка-
(катодные ингибиторы) процессов электрохимической коррозии, а также в об-
образовании защитных и пассивирующих пленок (экранирующие ингибито-
ингибиторы). Анодные ингибиторы — это неорганические соединения, обладающие
окислительными свойствами (хроматы, нитриты, молибдаты и др.). Адсор-
бируясь на корродирующей поверхности, они легко восстанавливаются на
ее катодных участках при плотностях тока, обеспечивающих пассивацию
анодных участков (образованию на них защитных пленок из оксидов или
малорастворимых солей), поэтому нередко их называют пассиваторами.
К анодным ингибиторам относят и ряд веществ, не являющихся окис-
окислителями, но обладающих щелочными свойствами (гидроксид и бензоат на-
натрия, различные фосфаты, силикаты, тетрабораты и др.). Их ингибирующее
действие проявляется лишь в присутствии растворенного кислорода. Они
способствуют его избыточной адсорбции на корродирующей поверхности.
При избытке кислорода пленка продукта (например FeO в случае коррозии
железа) дополнительно окисляется с образованием другой пленки, имеющей
лучшие защитные свойства:
4FeO + О2 -+ 2Fe2O3
Кроме того, эти вещества сами образуют защитные пленки, тормозя-
тормозящие анодный процесс растворения и представляющие собой малораствори-
малорастворимые продукты взаимодействия с ионами переходящего в раствор металла
632 19. Защита от коррозии
(например, фосфаты на поверхности стали образуют с ионами железа за-
защитные пленки из смеси Y-Fe2O3 и FePO4 • 2Н2О).
Наилучший ингибирующий эффект достигается при содержании анод-
анодного ингибитора, превышающем определенное значение, называемое крити-
критической концентрацией. Для ионов СЮ^', NO^, МоО^' и \VO4~ она со-
составляет от 1(Г3 до КГ4 моль/кг. Ниже этого значения они ведут себя как ак-
активные деполяризаторы, увеличивая скорость коррозии, например:
Концентрация NaNO2 в водно-
бензиновой смеси Ст • 103, моль/кг 2,33 4,67 7,00 11,67
Скорость коррозии стали гглуб , мм/год 0,08 0,02 0,00 0,00
Катодные ингибиторы по защитному действию менее эффективны,
чем анодные, однако не вызывают усиления коррозии при их недостаточном
содержании в коррозионной среде. Они тормозят протекание катодных про-
процессов:
а) путем уменьшения концентрации растворенного кислорода связы-
связыванием его в химические соединения, например:
2Na2SO3 + О2 = 2Na2SO4
— при коррозии с кислородной деполяризацией;
б) за счет повышения перенапряжения катодного выделения водорода
добавлением в растворы таких солей, как AsCb, Bi2(SC>4K и др. — при кор-
коррозии с водородной деполяризацией;
в) в силу уменьшения площади катодных участков экранированием.
Анодные и катодные ингибиторы, являясь преимущественно неорга-
неорганическими веществами, оказывают эффективное защитное действие толь-
только в нейтральных и щелочных растворах. В сильнокислотных растворах
используют экранирующие ингибиторы — органические соединения, со-
содержащие в своем составе атомы серы, кислорода, азота (альдегиды, фено-
фенолы, меркаптаны, амины, соли ароматических каГрбоновых кислот и др.).
Адсорбируясь (за счет полярных групп или неподеленных пар электронов
у атомов О, S, N) по всей поверхности (как на катодных, так и анодных
участках), одни из них затрудняют протекание в большей степени катодной
реакции: (+) К: 2Н+ + 2е —> H2f, другие — анодной реакции: (-) А: М —>
—> Mz+ + Ze. Эти ингибиторы часто называют органическими катодными
замедлителями или ингибиторами травления. Так как анион «травильной»
кислоты непосредственно участвует в формировании адсорбционного слоя,
то одни и те же экранирующие ингибиторы обладают различной эффектив-
эффективностью в растворах разных кислот.
Атмосферная коррозия протекает, как правило, с кислородной деполя-
деполяризацией, поэтому для ее торможения пригодны все пассиваторы, используе-
19.2. Изменение состава и свойств коррозионной среды 633
мые в качестве анодных замедлителей в нейтральных и щелочных растворах.
Ингибиторы атмосферной коррозии подразделяют на нелетучие — контакт-
контактные и летучие — парофазные. Нелетучие (например NaNO2) применяют в
основном при хранении изделий на складе. Их наносят либо на поверхность
упаковочного материала, либо на поверхность изделий. Летучие используют
для защиты как в сухой, так и во влажной атмосфере. Как правило, это неток-
нетоксичные вещества, с достаточным для защиты (но невысоким) давлением па-
паров при обычных температурах. Испаряясь, они заполняют окружающую из-
изделие воздушную среду. Последующая адсорбция их паров на поверхности
металла приводит к образованию пленок с анодным и катодным механизмами
защитного действия. Эти пленки обладают и гидрофобными свойствами. К
летучим ингибиторам относятся нитриты и карбонаты замещенных аминов,
сложные эфиры карбоновых кислот и другие соединения.
Практическое использование того или иного ингибитора определяется
его эффективностью, токсичностью и ущербом, наносимым окружающей
среде промышленными сбросами, содержащими ингибитор.
Для защиты металлоизделий от атмосферной коррозии используют и
ингибированные смазки: нефтяные масла, воск, вазелин, содержащие в каче-
качестве ингибиторов амины, ланолин, продукты окисления нефти и др. Меха-
Механизм их защиты аналогичен ингибиторам травления с той лишь разницей,
что они эффективны в области рН, близкой к нейтральной. Антикоррозион-
Антикоррозионные смазки относительно дешевы, достаточно стабильны при хранении и
применении, их легко наносить, удалять, возобновлять.
Обработка газовой и жидкой неэлектролитной (а иногда и электро-
электролитной) коррозионных сред сводится к изменению их состава так, чтобы
была исключена термодинамическая возможность протекания химических
процессов между компонентами материала и среды (смеси). Газовые среды
(смеси) с таким составом называют защитными атмосферами. Для сталь-
стальных изделий это атмосферы, не вызывающие окисления, обезуглероживания
и науглероживания, а также наводороживания. Состав защитных атмосфер
рассчитывают по константам равновесия, устанавливающегося в системе
материал—газ. Рассмотрим это на примере стали, находящейся в атмосфере
углекислого газа. Между компонентами стали и углекислым газом при
температуре 900 °С протекают две реакции:
окисление железа
Fe(T) + CO2(r) <=± FeO(T) + СО(г) \Кр = Л?авнС0 = 2,2 ]
и обезуглероживание стали
Fe3C(T) + CO2(r) ±± 3Fe(T) + 2СО(г) \Кр = -3=?°. = 9,2 атм ].
634 19. Защита от коррозии
Если соотношение неравновесных парциальных давлений газов для
первой реакции >2,2, а для второй >9,2 атм, то положение
Рсо2 Рсо2
равновесия в обеих гетерогенных реакциях смещено влево (в сторону ис-
исходных веществ), так что окисления железа по первой реакции и обезугле-
обезуглероживания стали по второй не происходит и наоборот.
В качестве защитных атмосфер применяют следующие газовые смеси:
азот — оксид углерода (II) — водород; водород — водяной пар — азот; во-
водород — водяной пар — оксид углерода (II) — азот и др.
Обработка жидких электролитных коррозионных сред сводится к
уменьшению содержания в них деполяризаторов (Н+-ионов, О2). Это дости-
достигается нейтрализацией кислотных растворов, вызывающих коррозию с во-
водородной деполяризацией: (+)К: 2Н+ + 2е —> H2f, и деаэрацией (обескисло-
(обескислороживанием) нейтральных и щелочных, в которых она протекает с кисло-
кислородной деполяризацией: (+)К: О2 + 2Н2О + Ае —> 4ОН~.
Кислотные растворы нейтрализуют добавлением в них щелочных реа-
реагентов: едкого натра (NaOH), негашеной извести (СаО) и др.:
В основе химической деаэрации лежат окислительно-восстановитель-
окислительно-восстановительные процессы между растворенным в коррозионной среде, например воде,
кислородом (окислителем) и добавляемыми в нее восстановителями (гидра-
зингидрат N2H4 • Н2О, гидразинсульфат N2H4 • H2SO4, Na2S2O4, Na2SO3 и
др.). Они связывают его в различные химические соединения, например:
N2H4 • Н2О + О2 -> N2t + ЗН2О
При использовании гидразинсульфата и дитионита натрия (Na2S2O4)
одновременно снижается и щелочность коррозионной среды:
N2H4 • H2SO4 + О2 + 2NaOH -> N2| + Na2SO4 + 4H2O
2Na2S2O4 + 3O2 + 4NaOH -> 4Na2SO4 + 2H2O
Растворенный кислород удается связать и при пропускании воды че-
через колонну, заполненную сульфидом железа:
4FeS + О2 + 6 Н2О -> 4FeO(OH) + 4H2S
или стальной насадкой (стружкой):
3Fe + 2О2 -> Fe3O4
19.3. Электрохимическая защита: виды и механизм действия 635
Скорость процесса химической деаэрации зависит от рН коррозион-
коррозионной среды, концентрации в ней кислорода, температуры, а также от концен-
концентрации (или площади поверхности для последних случаев) восстановителя.
Термическая деаэрация основана на зависимости растворимости газов
от их парциальных давлений и температуры и проводится в паровых де-
деаэраторах. Воду, нагретую до кипения, противотоком пару подают в ко-
колонну, в которой осуществляется их максимальный контакт, вследствие чего
из нее испаряется кислород и большая часть других растворенных газов.
Сорбционное обескислороживание воды осуществляют пропусканием ее
через колонну, заполненную ионитом (ионообменной смолой в виде зерен
сферической или неправильной формы), в состав которого входят вещества,
быстро реагирующие с кислородом (сульфиты металлов, гидроксиды железа
(II), марганца (II) и др.).
Широко распространено в промышленности десорбционное обескис-
обескислороживание, осуществляемое в газоводяном эжекторе. Оно основано на
создании пониженного парциального давления кислорода над жидкостью,
что достигается интенсивным перемешиванием воды с инертным газом, в
ходе которого кислород, диффундируя в газ, покидает ее.
19.3. Электрохимическая защита:
виды и механизм действия
Электрохимическая защита эффективна в коррозионных средах с хо-
хорошей ионной электрической проводимостью и основана на снижении ско-
скорости коррозии торможением анодных или катодных реакций путем поля-
поляризации (изменения потенциала) защищаемой конструкции (катода или ано-
анода) постоянным током. В зависимости от вида поляризации различают
катодную и анодную защиты (о поляризации см. § 17.6).
В основе катодной защиты лежит катодная поляризация: смещение
потенциала металла защищаемой конструкции в отрицательную сторону.
Катодную поляризацию, а следовательно, и катодную защиту осуще-
осуществляют двумя способами:
1) подключением защищаемой конструкции к отрицательному полюсу
внешнего источника постоянного тока. Соответствующую разновидность
катодной защиты называют защитой внешним (наложенным) потенциалом
(рис. 19.2);
2) присоединением к защищаемой конструкции электрода («жертвен-
(«жертвенного» анода, или протектора), изготовленного из металла, имеющего мень-
меньший электродный потенциал, чем потенциал металла защищаемой конст-
конструкции.
636
19. Защита от коррозии
Рис. 19.2. Схема катодной защиты внешним потенциалом:
1 — выпрямитель тока; 2 — токопроводящая засыпка; 3 — вспомогательный
анод; 4 — защищаемая конструкция (трубопровод) — катод; 5 — стальной
провод с изоляцией; 6 — грунт
Разновидность катодной защиты в этом случае называют протекторной
(гальванической) защитой, или защитой «жертвенным» анодом (рис. 19.3).
Для защиты внешним потенциалом требуется вспомогательный анод 5,
подключаемый к положительному полю-
полюсу внешнего источника постоянного тока
и располагаемый на некотором расстоя-
расстоянии от защищаемой конструкции 4, кото-
которая является катодом (см. рис. 19.2). Ма-
Материалами вспомогательного анода могут
быть графит, чугун (в виде лома), сталь (в
виде старых труб и рельсов) и др. Для
снижения сопротивления коррозионной
среды, например грунта, непосредственно
окружающего вспомогательный анод,
последний помещают в токопроводящую
засыпку 2 — смесь кокса, NaCl и
CaSO4 • 0,5Н2О.
Ток от вспомогательного анода те-
течет к катодным и анодным участкам за-
777777
/
/
/
у
\
• *•• •• . •
о
о
О
о
ос
с
о о о сГ
\, \
1 ''
^2
У/////////
Рис. 19.3. Схема като/]
протектором («жертве
дом):
(+)JS
/////
he
f 4
[ной защиты
иным» ано-
1 — засыпка (суспензия бентонита и
алебастра, увеличивающая сопротив-
сопротивление грунта); 2 — протектор, или
«жертвенный» анод; 3 — защищаемая
конструкция (трубопровод) — катод;
4 — стальной наконечник; 5 — стальной
провод с изоляцией; 6—грунт
19.3. Электрохимическая защита: виды и механизм действия 637
щищаемой конструкции. Как только в результате поляризации потенциал
катодных участков достигает значения потенциала анода, вся поверхность
защищаемой конструкции становится эквипотенциальной (катодный и
анодный потенциалы равны), локальный ток больше не протекает и корро-
коррозионные гальванические микроэлементы не работают. Суммарный ток на
анодных участках течет из электролита в металл, благодаря чему ионы ме-
металла не могут перейти в раствор.
Применение тока выше требуемого (когда в результате поляризации
потенциал металла конструкции выше потенциала анода) не дает положи-
положительных результатов, кроме как излишних затрат электроэнергии и возмож-
возможности вспучивания органических покрытий из-за большого выделения водо-
водорода и связанного с ним водородного охрупчивания и последующего рас-
растрескивания стали. Данные отрицательные явления называют эффектом
«перезащиты».
При протекторной защите «жертвенный» анод (массивный протектор)
растворяется, после чего его заменяют новым. Материалами протектора ча-
чаще всего являются магний и его сплавы, реже — алюминий и цинк. В воде и
грунте А1 и Zn склонны к образованию на поверхности плотных оксидных
пленок, нарушающих токоотдачу. Для их растворения в засыпку вводят
хлориды или другие вещества, а также компоненты, увеличивающие сопро-
сопротивление грунта. Это приводит, с одной стороны, к уменьшению скорости
растворения протектора, а с другой, — к сокращению его радиуса защитно-
защитного действия (максимально возможного удаления от защищаемой конструк-
конструкции, на котором еще обеспечивается надлежащая защита), т.е. к снижению
эффективности этой разновидности защиты. Достоинство протекторной за-
защиты — отсутствие необходимости во внешнем источнике постоянного то-
тока, специальное сооружение которого часто бывает экономически нецелесо-
нецелесообразным.
В настоящее время катодную защиту широко используют для борьбы
с коррозией трубопроводов, кабельных установок и металлоконструкций в
воде (морской, речной), грунте и других нейтральных средах.
В основе анодной защиты лежит анодная поляризация: смещение по-
потенциала металла защищаемой конструкции в положительную сторону до
значений, лежащих в пассивной области анодной поляризационной кривой.
Ее осуществляют присоединением защищаемой конструкции к положитель-
положительному полюсу внешнего источника постоянного тока, а вспомогательного
электрода — к отрицательному, при этом конструкция — анод, а электрод —
катод. Анодная защита в отличие от катодной применима только к легко-
пассивируемым (покрывающимся пассивной пленкой оксида, например,
(-)А: 2Сг + ЗН2О —> Сг2О3 + 6Н+ + бе) при анодной поляризации металлам и
сплавам. Это большинство переходных металлов и сплавов на их основе,
638 19. Защита от коррозии
включая нержавеющие и углеродистые стали. При анодной защите ток про-
противоположен по направлению току при катодной защите. Достоинства
анодной защиты: высокая рассеивающая способность — возможность защи-
защиты на большом удалении от катода (большем, чем в случае катодной защи-
защиты); невысокая защитная плотность тока (ниже, чем в случае катодной за-
защиты), а следовательно, и небольшое потребление энергии; возможность
защиты электрически экранированных поверхностей. К недостаткам следует
отнести ограничение области применения (анодная защита не осуществима
для Mg, Cd, Ag, Си и медных сплавов), а также то, что скорость коррозии
при анодной защите, в отличие от катодной, никогда не падает до нуля.
19.4. Защитные покрытия: виды, методы нанесения
и области применения
Защитные покрытия предохраняют изделия от коррозии и одновремен-
одновременно придают их поверхности ряд ценных свойств: износостойкость, отража-
отражательную способность, паяемость и др. Их подразделяют на металлические и
неметаллические (неорганические, лакокрасочные, покрытия смолами и пласт-
пластмассами). Общими требованиями для всех видов покрытий являются: высокая
адгезионная способность («прилипаемость» к поверхности материала), изно-
износостойкость, твердость, герметичность (сплошность, беспористость и непро-
непроницаемость для агрессивной среды).
По механизму своего защитного действия и поведению изделия в це-
целом в электролитной коррозионной среде, т.е. в условиях электрохимиче-
электрохимической коррозии, металлические защитные покрытия подразделяют на катод-
катодные — коррозионно-стойкие и анодные — протекторные.
Металлы катодных покрытий имеют в данной среде большие (более
положительные) значения электродных потенциалов, чем значение потен-
потенциала металла, на который они нанесены. Это, например, серебряное
[Ф^ + =0,779 В или никелевое (р^.2+ =-0,250 В покрытие на железе
[фре2+ =-0,440 В . Изолируя поверхность от коррозионной среды, они за-
~fT )
щищают изделия лишь механически, поэтому основным требованием,
предъявляемым к ним, является герметичность. В противном случае проис-
происходит разрушение металла основы (изделия) вследствие возникновения и
функционирования коррозионного ГЭ, в котором покрытие выступает в ка-
качестве катода (рис. 19.4). В зависимости от вида и состава коррозионной
19.4. Защитные покрытия: виды, методы нанесения и области применения 639
Рис. 19.4. Поведение изделия из железа 3 с катодным (а) и анодным
(б) покрытиями 2 в кислотном растворе 1 в условиях электрохимиче-
электрохимической коррозии при нарушении целостности покрытия
среды на нем выделяется водород или поглощается кислород. Чем выше по-
пористость катодного покрытия и чем больше в нем различных дефектов (ца-
(царапин, вмятин, оголенных участков и др.), тем значительнее разрушения
основного металла.
Металлы анодных покрытий имеют меньшие (более отрицательные)
значения потенциалов в данной среде, чем потенциал металла, на который
они нанесены. Поэтому цинковое ф° 2+ =-0,763 В или в некоторых сре-
Zn2"
Zn
дах алюминиевое
= -1,662 В покрытие на железе
= -0,440 В
является анодным. Анодные покрытия защищают изделия не только механи-
механически, но и электрохимически. Это означает, что при повреждении или нали-
наличии пор в образующемся в коррозионной среде ГЭ покрытие будет выступать
в роли анода и подвергаться разрушению, поэтому требование герметичности
для анодных покрытий не существенно (рис. 19.4). При этом они защищают
изделие тем дольше, чем больше их толщина. Подвергаясь растворению в
процессе эксплуатации изделия, анодное покрытие генерирует контактный
ток, который, протекая между ним и изделием, катодно поляризует послед-
последнее, т.е. смещает потенциал металла основы в сторону меньших значений.
Поведение изделия с анодным покрытием аналогично организации катодной-
защиты протектором. Поэтому анодные покрытия часто называют протектор-
протекторными. Таким образом, в основе электрохимического механизма защитного
действия анодных покрытий лежит катодная поляризация от покрытия —
аналога протектора.
640 19. Защита от коррозии
Вид металлического покрытия (анодное или катодное), а следователь-
следовательно, и механизм его защитного действия (только изолирующий, т.е. механи-
механический, или изолирующий и электрохимический одновременно) зависит,
кроме природы контактирующих металлов, также от условий эксплуатации,
вида и состава коррозионной среды. Три последних фактора определяют
прежде всего величину потенциала металла покрытия. Так, олово
ф° 2+ =-0,136 В на железе ф° 2+ =-0,440 В в обычной атмосфере и в
I fl_ I I ?f_ J
V Sn / \ Fe /
растворах неорганических кислот и солей ведет себя как катодное покрытие,
а в растворах ряда органических кислот (пищевые среды) — как анодное
покрытие. Это связано с тем, что в таких растворах оно находится в виде
устойчивых комплексов. Вследствие этого его потенциал смещается в отри-
отрицательную сторону и становится меньше потенциала железа. Подобная кар-
картина наблюдается и для покрытий из кадмия ф° 2+ =-0,403 В , алюминия
Ф^3+ =-1,662 В и хрома ф°г3+ =-0,774 В . Кадмий имеет очень близкий
к железу, но все-таки несколько больший, потенциал. Во влажной атмосфере
и в присутствии СГ-ионов его потенциал становится меньше потенциала же-
железа и поэтому его покрытие, являясь в таких средах анодным, защищает же-
железные изделия и механически, и электрохимически. В мягкой воде потенци-
потенциал алюминия больше потенциала стали, поэтому его покрытие на ней являет-
является катодным. В морской и некоторых видах пресной воды, особенно содер-
содержащей СГ- и SOj' -ионы, все наоборот. В таких средах анодное алюминиевое
покрытие защищает сталь еще и электрохимически, катодно поляризуя ее. Из-
за высокой склонности хрома к пассивации в большинстве коррозионных
сред его покрытия на железе, сталях и некоторых других металлах практиче-
практически всегда являются катодными, а посему и защищают только механически.
В качестве металлов покрытий обычно используют металлы, образую-
образующие на своей поверхности защитные пленки. К ним относятся Zn, Al, Sn, Cd, Cr
и др. Известны различные методы их нанесения. Выбор того или иного метода
определяется назначением покрытия. Отметим лишь самые распространенные.
1. Электролиз — покрываемое изделие, являющееся катодом, погру-
погружают в гальваническую ванну с электролитом, содержащим ионы наноси-
наносимого металла, через который пропускают постоянный электрический ток.
2. Металлизация — расплавленный металл с помощью газовой струи
из шприц-пистолета наносят на поверхность мостов, деталей судов, боль-
больших баков и др.
19.4. Защитные покрытия: виды, методы нанесения и области применения 641
3. Горячий метод — защищаемое изделие, обработанное флюсом, по-
погружают в расплавленный металл. Покрытие (например, из Zn, Sn, Pb и др.)
образуется после извлечения его из ванны.
4. Химический — покрытия (например, из Си, Ni и др.) формируются
при восстановлении (водородом, гидразином, гипосульфитом и др.) метал-
металлов из растворов их солей.
5. Термодиффузионный — покрытия (например, из Al, Si, Cr, Ti и др.)
образуются в результате диффузии ионов металла из твердой или газооб-
газообразной фазы.
6. Газофазный — покрытие оседает на подложку в результате протекания
газофазного процесса: окислительно-восстановительного, разложения и др.
7. Плакирование — покрытие наносится совместной прокаткой ос-
основного и защищающего металлов. Метод применим для листов, лент и
других профилей.
В качестве примеров применения материалов с металлическими покры-
покрытиями можно привести следующие: железо с оловянным покрытием, получае-
получаемым электроосаждением, называют электролитической белой жестью и ис-
используют при изготовлении тары для жидких и твердых пищевых продуктов;
сталь с покрытием из сплава свинца и олова (до 30 % (масс.) Sn) называют лу-
женой жестью и используют в кровельных работах; никелевые покрытия (на
Al, Co, Pd, стали, стекле, пластмассах) широко применяют как для защиты от
коррозии, так и для декоративной отделки в машиностроении, приборострое-
приборостроении, автомобильной, медицинской и электронной промышленностях; цинковые
покрытия используют для защиты стальных листов, проволоки, водопроводных
труб, резервуаров и трубопроводов в реакторах АЭС; хромовые покрытия в
основном используют как износоустойчивые защитно-декоративные покрытия
из-за их высокой твердости и эстетического внешнего вида.
Среди неорганических покрытий можно выделить конверсионные,
портландцементные, силикатные эмали. Их защитное действие сводится, в
основном к изоляции металла от окружающей среды.
Портландцемент — многокомпонентная порошкообразная смесь из
основных (Na2O, CaO, MgO) и кислотных (Fe2C>3, AI2O3, SiO2) компонентов,
которая при смешивании с водой превращается в вязкую массу. Нанесенная
на изделия, она застывает, образуя покрытия до 25 мм толщиной.
Силикатные эмали, являющиеся соединениями кремния, представляют
собой тонкие непрозрачные покрытия с высокими защитными свойствами
(даже при высоких давлениях и температурах) на черных и цветных металлах.
Их получают погружением изделий в эмальную массу (водную суспензию,
содержащую SiO2, СаСОз, Na2CO3 • 10Н2О или Na2SC>4, уголь и SnO2) с после-
последующей их термообработкой — сушкой и плавлением эмальной массы.
Эмалированные металлы используют при производстве аппаратуры в
фармацевтической, химической, пищевой промышленностях.
21 - 9795
642 19. Защита от коррозии
Конверсионные покрытия на металлах образуются вследствие изменения
состояния их поверхности путем ее химической или электрохимической обра-
обработки, например, окисления или осаждения на ней малорастворимых соедине-
соединений. В зависимости от вида соединения, формирующегося на поверхности, раз-
различают оксидные, фторидные, сульфатные, фосфатные, хроматные и другие
конверсионные покрытия. Среди их многообразия наибольшее значение имеют
оксидные покрытия. Процесс их получения, т.е. создания на поверхности
металлов, носит название оксидирования. Различают: а) термическое оксидиро-
оксидирование — контролируемое высокотемпературное окисление поверхности на
воздухе; б) химическое — обработка изделия горячим концентрированным
раствором щелочи, содержащим персульфаты, нитраты и хлораты; в) электро-
электрохимическое — анодное окисление в кислотном растворе. Химическое оксиди-
оксидирование стали получило название «воронения», а электрохимическое алюминия
и его сплавов — анодирования (анодного окисления). Сульфатное покрытие на
свинце в виде малорастворимого PbSO4 образуется при его обработке разбав-
разбавленной серной кислотой. Фторидное покрытие на магнии в виде MgF2 получа-
получают анодированием металла в 10.. .30 % (масс.) растворе NH4HF2 при комнатной
температуре. Фосфатные и хроматные покрытия получают соответственно
фосфатированием и хроматированием изделий.
Органические защитные покрытия (лакокрасочные, полимерными ма-
материалами и др.), обладая высокой адгезией и сплошностью, обеспечивают
достаточно хорошую изоляцию изделий от окружающей среды.
Лак — раствор смолы (фенолформальдегидной, кремнийорганической,
эпоксидной) в летучем растворителе, например спирте. Покрытия из лаков ис-
используют в химической промышленности для защиты от коррозии емкостей,
трубопроводов и деталей. Краска — суспензия частиц пигмента (оксида метал-
металла или его соли — ZnO, РЬзО4, ZnCrO4, PbSC>4 и др.) в органическом или вод-
водном связующем (растительном масле (льняном, ореховом), синтетической смо-
смоле и др.). Перед нанесением лакокрасочных покрытий поверхность подвергает-
подвергается специальной обработке. Как правило, их изготавливают многослойными для
получения высокой сплошности, а для достижения хорошей адгезии наносят
фосфатный подслой. Полимерные материалы (резина, неопрен, 1,1-
полидихлорэтилен и др.) применяют для футеровки, изоляции и защиты от
коррозии. Клейкой лентой из вигошласта и полиэтилена изолируют трубопро-
трубопроводы и оборудование, расположенные под землей. Наиболее стойкий к химиче-
химическим средам политетрафторэтилен (тефлон) используют в химическом произ-
производстве для футеровки, изготовления прокладок и мембранных клапанов.
Большинство из рассмотренных способов защиты изделий используют и
при их консервации — защите на период хранения и транспортировки. Выбор
способа и средства консервации определяется климатическими условиями, ви-
видом, габаритами и конструктивными особенностями металлоизделий.
Практические занятия
Коррозия и защита металлов и сплавов
Примеры решения задач
Задача 1. На поверхности какого из металлов, Ni или А1, толщина оксидной
пленки увеличится в большее число раз, если время их окисления, прово-
проводимого при температуре 600 и 200 °С соответственно в атмосфере, содер-
содержащей кислород, увеличить с 3 до 9 ч. Привести уравнения окислительно-
восстановительных реакций, протекающих при этом. Оценить защитные
свойства образующихся оксидных пленок, полагая, что их в начальный мо-
момент времени на поверхности обоих металлов не было.
Решение. Уравнения протекающих реакций имеют вид
2Ni(T) + O2(r)=2NiO(T)
4А1(Т)+ЗО2(Г)=2А12О3(Т)
Окисление поверхности Ni в данных условиях протекает по парабо-
параболическому, а поверхности А1 — по логарифмическому законам.
Законы окисления описываются уравнениями в интегральной форме:
параболический для Ni
и логарифмический для А1
где В и С — константы интегрирования; В = С = 0.
Для двух промежутков времени
62№2=2Г/2и6А|2=А:"Ы2,
ИЛИ
8Ni2 \U 8A12
8А11
откуда
«^ 8Ai2 In 9
5Nil V3 8А|1 1пЗ
= 2,00.
644 Коррозия и защита металлов и сплавов
Таким образом, толщина оксидной пленки на А1 увеличится в большее число раз.
Чтобы оценить защитные свойства оксидных пленок, следует рассчитать для
них фактор сплошности Пиллинга—Бедвордса:
„ ^NioPNi 74,69-8,91
pNi0MNj 6,67-58,69
МА1ОрА1 101,96-2,70
*""¦ pMfi2MM 3,97-2-26,98
где р и М — плотность и молярная масса атомов металла и его оксида соответст-
соответственно; 2 — число атомов алюминия в молекуле его оксида. Значения а для обоих
оксидов попадают в интервал 1,0 < а < 2,5, следовательно, оксидные пленки на этих
металлах должны обладать защитными свойствами.
Задача 2. Магний и цинк корродируют в морской воде по электрохимическому ме-
механизму с плотностью коррозионного тока /корр = 0,034 А/м2. Какой из металлов
корродирует быстрее? Ответ подтвердить расчетами глубинного показателя гглуб
скорости коррозии. К какой группе коррозионной стойкости относятся эти метал-
металлы? Какой балл десятибальной шкалы коррозионной стойкости соответствует им?
Рассчитать для них массовый показатель гмасс скорости коррозии.
Решение. Плотность коррозионного тока связана с массовым и глубинным показа-
показателями скорости коррозии следующими соотношениями:
— г мРм *¦>/: о
"- га*365-24Мм"
где рм, Мм, ZM — плотность, кг/м3, молярная масса атомов, г/моль, и зарядовое чис-
число ионов металла соответственно.
Следовательно,
MaKMg 268ZMg 2682
Mg 26,8ZMg 26,8-2
365-24^1,» 365-24 24,30-0,034
лЛ ш = L-— =
гл>бМв 26,8ZMgpMg 26,8-2-1737
icop|, 24-65,390,034 , „
= 0,078 мм/год;
365.24MzXpp=365-24-65,39-0,034
26,8ZZnpZn 26,8-2-7133
26,8-2 ' ^
39-0,034 =
Примеры решения задач 645
Глубинный показатель скорости коррозии можно также рассчитать и через
массовый. Они связаны между собой соотношением
365
у —у
глуб масс
Рм
Таким образом, быстрее корродирует магний, так как гглу6щ >гглуб2п. По ве-
величине глубинного показателя скорости коррозии магний и цинк принадлежат к
группе стойких металлов, но магнию соответствует 4-й, а цинку 5-й балл десяти-
бальной шкалы коррозионной стойкости (см. табл. П.6.6).
Задача 3. Определить термодинамическую возможность газовой коррозии изделия
из железа при Т = 1500 К под действием кислорода с парциальным давлением
р0 =0,01 атм с образованием на его поверхности оксида состава FeO. Вычислить
парциальное давление кислорода, ниже которого коррозия прекращается при задан-
заданной температуре. Оценить температуру, выше или ниже которой коррозия стано-
становится невозможной при стандартных состояниях компонентов. Написать уравнение
соответствующей реакции.
Решение. Уравнение протекающей реакции и выражение ее константы равновесия
Кр имеют вид
2Fe(T)+O2(r)^2FeO(T) (*,= ——
V Л>авнО2
где /?равн 0 — абсолютное равновесное парциальное давление кислорода.
Далее, для определения термодинамической возможности газовой коррозии
используют уравнение изотермы Вант-Гоффа (см. § 11.2):
\Ро2
Ро2
где р0 =—— — относительное неравновесное парциальное давление кислорода,
2 Р
безразмерная величина (ро — абсолютное неравновесное парциальное давление
кислорода, р° = 1 атм — стандартное давление).
Стандартную энергию Гиббса реакции ArG,0500 можно приближенно рассчи-
рассчитать по уравнению Гиббса—Гельмгольца:
в котором Лг#2°98 вычисляют по следствию из закона Гесса с помощью справочных
термодинамических величин AfH^n (см. табл. П.5.1).
646 Коррозия и защита металлов и сплавов
2AfH02nFc-AfH°mO2 = 2(-264,8)-2-0-0 = -529,6 кДж.
Аналогично по справочным величинам 52°98 находят А^^.
Аг5298 = 252°98 Fe0 - 25298 Fe - S298 Ог = 2 • 60,75 - 2 • 27,15 - 205,04 = -137,84 Дж/К,
А^^ =-529,6-1500-(-137,84-10-3) =-322,84 кДж,
AG1500=-322,84-8,314150010ln0,01 = -265,41 кДж.
Так как ArG1500 < 0, то газовая коррозия в данных условиях термодинамиче-
термодинамически возможна. Она не будет протекать, если ArG1500 > 0, т.е.
\пр0 < г 150° и
°2 RT
I -322840 \ л12
ро <ехр или ро < 5,7-10 атм.
2 \8,314-1500/ °2
При температуре Т = 1500 К и давлении кислорода, превышающем
р0 =5,7 • 10~12 атм, коррозия будет происходить.
Температуру, выше которой коррозия невозможна при стандартных состоя-
состояниях компонентов можно найти из соотношения
откуда, подставив конкретные значения АгН°2П и Аг5*298, имеем, что
Т > ^™ > 3842,1 К.
137,84
Задача 4. Определить возможность электрохимической коррозии изделия из латуни
(сплава Си и Zn) при Т = 298 К в 0,03М водном растворе ZnSO4, находящемся в
контакте с газовой смесью, парциальные давления кислорода и водорода в которой
составляют соответственно 0,21 и 0,41 атм. При каких значениях рН прекратится
выделение водорода? Привести уравнения анодной и катодной реакций. Как изме-
изменится ЭДС коррозионного ГЭ в результате концентрационной поляризации анода,
если концентрация Zn2+ в растворе возрастет до 0,05 моль/л?
Решение. В водном растворе Z11SO4 подвергается гидролизу. По первой ступени,
процесс может быть описан следующим ионно-молекулярным уравнением:
Zn2+ + НОН <± ZnOH+ + Н+ (рН < 7).
Конкретное значение рН раствора рассчитывают по соотношению (см. § 14.23):
Примеры решения задач 647
*4zn(OHJ 2
С,
где Кн 0 — ионное произведение воды, Кн 0 = 10~14 (моль/лJ при 25 °С; KbZa(OH) 2 —
bZn@H) 2
константа диссоциации Zn(OHJ по второй ступени, KbZn@H) 2 = 2,0 • 10"9 моль/л при
25 °С; С — молярная концентрация соли.
Тогда
210"9
Предположительно будет протекать избирательная электрохимическая кор-
коррозия и растворению в сплаве будет подвергаться цинк, так как
ф^2+ = -0,763 В < ф°и2+ = 0,337 В. Далее по уравнению Нернста рассчитывают рав-
Zn Си
новесные потенциалы цинкового (предполагаемого анода), кислородного и водо-
водородного (вероятных катодов) электродов:
=-0,763+ O,O2951gO,O3 =-0,808 В,
Ф2п, Ф^+
~ziT ~zT
+ =1,229 -0,059рН + 0,0147 lgpo =l,229-0,059-3,41 + 0,01471g0,21 = l,018 В,
2Н2О
ф2н+ =-0,059pH-0,02951gpH2 =-0,059-3,41-0,02951g0,41 = -0,213 В.
ЭДС коррозионных ГЭ равны:
Ех =фо 4н+ -ф^ =1,018-(-0,808) = 1,826 В > 0, следовательно, коррозия с
2Н2О Zn
поглощением кислорода в данных условиях возможна.
Е2 =ф2н+ -ф^ =-0,213-(-0,808) = 0,595 В > 0, коррозия и с выделением
водорода в данных условиях также возможна.
Таким образом, осуществляется избирательная коррозия латуни с протекани-
протеканием одной анодной и двух катодных реакций:
(-) A: Zn -> Zn2+ + 2e
О2+4Н++4е-»2Н2О
648
Коррозия и защита металлов и сплавов
Процесс выделения водорода прекратится, если Е2 < О, т.е. -0,059рН -
- 0,0295 lg/?H + 0,808 < 0, откуда следует, что это произойдет при рН > 13,9. Кор-
Коррозия с поглощением кислорода будет иметь место при любых значениях рН.
При повышении концентрации ионов Zn2+ в растворе увеличивается потен-
потенциал анода:
q>'zn2+ =-0,763 + 0,0295 lg 0,05 = -0,801 В
Zn
и уменьшается значение ЭДС коррозионных ГЭ на величину
Задача 5. Рассмотреть электрохимическую коррозию изделия из алюминиевой
бронзы (сплава Си и А1), если оно попадет в водный раствор NaOH. Привести урав-
уравнения анодной, катодной и токообразующей реакций, а также схему коррозионного
ГЭ. Предложить анодное и катодное покрытия для защиты изделия от электрохи-
электрохимической коррозии.
Решение. Так как
= -1,662 В<ф°2+ =0,337 В, то кристаллиты алюминия яв-
Си
ляются анодными участками и будут подвергаться разрушению, т.е. происходит
избирательная электрохимическая коррозия — вытравливание более активного
компонента из сплава. Кристаллиты меди будут являться катодными участками.
Характер катодной реакции определяется видом и составом коррозионной среды.
Поскольку среда щелочная (рН > 7), то в таких средах коррозия алюминия возможна
и с кислородной — поглощением О2, и с водородной — выделением Н2 деполяриза-
деполяризациями. С учетом сказанного электродные и токообразующая реакции имеют вид
: А1->А13++Зе
t +20H"
4 + 2 = 6
12A1 + О2 + 4Н2О -> 2А1(ОНK1 + Н2 Т
Следовательно, продуктами коррозии являются водород и гидроксид алюми-
алюминия. Поскольку среда сильнощелочная, то А1(ОНK вследствие своей амфотерности
будет растворяться:
А1(ОНK + NaOH -» Na[Al(OH)J
С учетом этого анодная реакция описывается следующим уравнением:
(-) А (А1): А1 + 4ОН" -> [А1(ОНL]" + Ъе
Примеры решения задач 649
Так как коррозия алюминия в щелочных средах протекает очень интенсивно,
то содержащийся в них кислород быстро расходуется, так что в дальнейшем остает-
остается возможной лишь одна катодная реакция — реакция выделения водорода. Таким
образом, конечным продуктом коррозии является комплексное соединение
Na[Al(OHL].
Схему коррозионного ГЭ записывают аналогично схеме обычного ГЭ. Ос-
Основное ее отличие от последней состоит в отсутствии границы раздела между рас-
растворами обоих электродов, т.е. между анолитом и католитом. С учетом сказанного
схема коррозионного ГЭ такова:
(-) А11 NaOH, H2O, O2 | Си (+).
По определению, металлы анодных покрытий имеют меньшие (более отрица-
отрицательные) значения потенциалов, чем потенциал основного (защищаемого) металла.
В данном случае анодным будет покрытие, изготовленное из металла, у которого
потенциал меньше потенциалов компонентов сплава (Си и А1), например из Mg,
имеющего ф° 2+ =-2,363 В.
Mg
Mg
Металлы катодных покрытий, наоборот, имеют большие (более положитель-
положительные) значения потенциалов по сравнению с потенциалом металла, на который они
нанесены. Это, например, серебряное покрытие, у которого потенциал
(ф° + =0,799 В) больше потенциалов компонентов сплава (Си и А1). Если первые
Ag
Ag
защищают изделия и механически, и электрохимически, то вторые — только меха-
механически, поэтому при нарушении их герметичности коррозии будет подвергаться
более активный компонент сплава, из которого изготовлено изделие, т.е. алюминий.
Покрытия из Ni, Sn, Pb, у которых ф^3+ <ф° <ф° ц2+, будут являться катод-
AI Си
ными по отношению к алюминию (более активному компоненту сплава) и анодны-
анодными — по отношению к меди.
Задача 6. Медное изделие с цинковым покрытием в течение 10 мин подвергалось
электрохимической коррозии при Т= 298 К в водном растворе с рН = 11. Сколько и
какого металла прокорродировало, если в процессе коррозии поглотилось 280 мл О2
и выделилось 112 мл Н2, измеренных при н.у.? Чему равна сила коррозионного то-
тока? Привести уравнения анодной, катодной и токообразующей реакций. Кроме во-
водорода, какое вещество еще является продуктом коррозии? Рассчитать его массу.
Решение, Так как ц>°^2+ = -0,763 В < ф^2+ = 0,337 В, то цинковое покрытие на мед-
Zn Си
ном изделии является анодным и при электрохимической коррозии именно оно и
будет разрушаться. При этом само медное изделие не будет принимать участия в
анодном процессе, т.е. корродировать.
Анодная, катодная и токообразующая реакции коррозионного ГЭ имеют вид
650
Коррозия и защита металлов и сплавов
(-) A (Zn): Zn -> Zn2+ + 2е
2е->Н2Т+2ОН~
I 3Zn + 02 + 4H2O -> 3Zn(OHJ 4, + H21
Вторым продуктом коррозии является гидроксид цинка, который растворяет-
растворяется вследствие своей амфотерности в избытке щелочи с образованием комплексных
ионов [Zn(OHL]2~.
В соответствии с условием задачи число моль эквивалентов поглощенного О2
и выделившегося Н2
о2
г'эквО2
280 103л
5,6 л/моль экв
= 0,05 моль экв,
г'эквН2
— _ _
112-Ю л
Кэкв н 11,2 л/моль экв
= 0,01 моль экв,
гДе ^экво и ^эквн — молярные объемы химических эквивалентов при н.у. соответ-
соответственно кислорода и водорода (см. § 2.1). Суммарное число моль эквивалентов га-
газов, претерпевших изменение на катоде: пэкв^ = 0,05 + 0,01 = 0,06 моль экв. Соглас-
Согласно закону эквивалентов, такое же число моль эквивалентов цинка прокорродирова-
ло и такое же число моль эквивалентов его гидрооксида образовалось. Тогда
"э..1=^°>06 = 1>96г,
м
Zn(OHJ
в Zn(OHJ
99,39
0,06 = 2,98 г,
где Мэкв2п и М
экв2п(ОНJ
— молярные массы химических эквивалентов цинка и его
гидроксида соответственно; 2экв2п и 2:экв2п(ОН) —числа эквивалентности цинка и его
гидроксида соответственно (см. § 2.1).
Согласно закону Фарадея,
М
сила коррозионного тока при х\ = 1:
-Itr\,
1,96-2-96485,3
65,39-300.1
= 19,28 А.
Примеры решения задач 651
Задача 7. Рассчитать давление водорода, необходимое для прекращения электро-
электрохимической коррозии изделия из никеля, протекающей в деаэрированной воде при
Т = 298 К с образованием Ni(OHJ. Привести уравнения анодной, катодной и токо-
образующей реакций.
Решение. В деаэрированной воде коррозия протекает с водородной деполяризацией,
поэтому анодная, катодная и токообразующая реакции таковы:
(-)А: Ni -> Ni2+ + 2e
t +2OH"
I Ni + 2H2O -> Ni(OHJ + Н21
Коррозия прекращается, когда вся поверхность изделия становится эквипо-
эквипотенциальной, т.е. когда потенциалы катодных и анодных участков равны. Эти по-
потенциалы рассчитывают, используя уравнение Нернста:
о 0,059, .
Ф« = Ф^н^ = ф\^ +^Ъ^- = -0,059рН - 0,0259 lg Д,.
Н2,2ОН' Н2,2ОН~ ОН ~^Н2
Активности ионов Ni2+, ОН" и Н+ находят из выражений произведения
растворимости гидроксида никеля IlPNj@HJ и ионного произведения воды Кн 0
(см. § 14.4 и 14.6)
В соответствии с уравнением реакции выражение для произведения раство-
растворимости Ni(OHJ имеет вид
ПРЩОНJ
Из уравнения реакции следует также, что а . = 2а 2+. С учетом этого
ОН' Ni2+
ПР№@„); =4,, откуда а№, =^^f^ = f^f^- = 1,59-10 моль/л,
а _ = 2а ,+ = 2 1,59-КГ =3,18-КГ моль/л, Кно=а ,а _ =104 (моль/лJ
ОН Ni n2*j j^ он
Тогда
моль/л'
<ра=Фы.г, =-0!250 + -^^lg(l,5910-5) = -0,392 В.
Ni
652 Коррозия и защита металлов и сплавов
Приравнивая значения потенциалов анода и катода (водородного электрода),
можно найти искомое давление водорода:
-0,392 = -0,059рН - 0,0295 lg pH ;
-0.392-0,059рН, '
2 0,0295 2
рн = рн р° = 1,94 Ю-1,013-105 Па = 0,197 Па.
Таким образом, при Т = 298 К и давлении водорода, равном или превышаю-
превышающем 0,197 Па, электрохимическая коррозия никеля происходить не будет.
Задача 8. Электрохимическая коррозия изделия из железа в морской воде протекает
со скоростью 2,5 г/(м2 • сут). Определить минимальную плотность тока (А/м2), не-
необходимую для полной катодной защиты изделия. Привести уравнения анодной и
катодной реакций.
Решение. Коррозия железа в морской воде осуществляется с кислородной деполяри-
деполяризацией, поэтому электродные реакции таковы:
(-)А: Fe -» Fe2+ + 2e
(+)К: О2+2Н2О + 4е-> ВН-
ВНЕСЛИ при использовании катодной защиты внешним потенциалом плот-
плотность приложенного тока будет большей или равной плотности коррозионного
тока (/вн > /корр)» то электрохимическая коррозия прекратится, так как при этом на-
наступает полная катодная защита.
Плотность коррозионного тока связана с массовым показателем скорости
коррозии соотношением:
где Мм и ZM — молярная масса атомов металла и зарядовое число его ионов соот-
соответственно. Согласно условию задачи, получаем
/ =2,5 22М =0,1 А/м2.
корр 24-55,85
Таким образом, при /„„ > 0,1 А/м2 железное изделие не будет подвергаться
коррозии.
Задачи для самостоятельного решения
653
Задачи для самостоятельного решения
1-4. Рассчитайте глубинный гглуб (мм/год) и массовый гмасс (г/(м2 • сут)) показатели
скорости коррозии металла по значению плотности коррозионного тока /КОрР. К ка-
какой группе коррозионной стойкости относится металл и какой балл десятибалльной
шкалы коррозионной стойкости ему соответствует?
№
1
2
Металл
РЬ
Fe
Плотность коррозионного
тока гК0Рр, А/м2
0,097
0,022
№
3
4
Металл
Си
А1
Плотность коррозионного
тока/корр, А/м2
0,018
0,061
5-8. Рассчитайте плотность коррозионного тока /корр (А/м2) и массовый показатель
^масс (г/(м2 • сут)) скорости коррозии металла по значению глубинного показателя
^глуб- К какой группе коррозионной стойкости относится металл и какой балл деся-
десятибалльной шкалы коррозионной стойкости ему соответствует?
№
5
6
Металл
Mg
Ti
Глубинный показатель
скорости коррозии
гглуб, мм/год
0,210
0,081
№
7
8
Металл
Та
Zr
Глубинный показатель
скорости коррозии
гглуб, мм/год
0,022
0,056
9-13. Установите законы роста толщины оксидной пленки на поверхности металла в
ходе его окисления на воздухе при температуре Т\ и Т2. Во сколько раз увеличится
толщина пленки для каждой из температур, если время контакта металла с газовой
средой увеличить с 2 до 20 часов? В начальный момент времени оксидной пленки на
поверхности не было. Приведите уравнение реакции окисления металла кислородом.
№
9
10
И
12
13
Металл
Си
Ti
Fe
Ti
W
Температура окисления, °С
г,
70
300
300
200
800
т2
700
750
700
1200
1400
14-16. Установите закон роста толщины оксидной пленки на поверхности металла в
ходе его окисления на воздухе при температуре Т. В каком случае толщина пленки
возрастет в большее число раз, если время контакта металла с газовой коррозионной
средой увеличить с t\ до t2 часов? В начальный момент времени оксидной пленки на
поверхности не было. Приведите уравнение реакции окисления металла кислородом.
654
Коррозия и защита металлов и сплавов
№
14
15
16
Металл
Си
Ti
Fe
Температура
окисления Т, °С
50
50
300
300
250
250
Время контакта, ч
'i
20
40
15
10
5
10
h
40
80
45
30
25
50
17-19. Определите термодинамическую возможность газовой коррозии изделия из
металла при температуре Т под действием кислорода с парциальным давлением,
равным р0 . При каком парциальном давлении кислорода прекратится газовая кор-
коррозия при указанной температуре? Приведите уравнение соответствующей реакции.
Ответ аргументируйте расчетами.
№
17
18
19
Металл
Ni
Ag
Pd
Температура
коррозии Т, К
2000
800
600
Парциальное давление кислорода р0 , атм
0,4
0,1
0,3
20-23. Определите температуру, выше или ниже которой невозможна газовая кор-
коррозия изделия из металла под действием газообразного хлора при стандартных со-
состояниях компонентов. Приведите уравнение соответствующей реакции.
№
Металл
20
Ti
21
Zr
22
Al
23
Ag
24-27. Определите парциальное давление кислорода в газообразной среде, ниже
которого невозможна газовая коррозия металла при стандартных условиях. Приве-
Приведите уравнение соответствующей реакции.
№
Металл
24
Ni
25
Ag
26
Си
27
Со
28-31. Определите, возможна ли атмосферная коррозия металла при стандартных
условиях в соответствии с химическим и электрохимическим механизмами? Приве-
Приведите уравнения соответствующих реакций. Ответ аргументируйте термодинамиче-
термодинамическими расчетами.
Металл
28
Mg
29
Си
30
Zn
31
Аи
Задачи для самостоятельного решения
655
32. Термодинамическими расчетами подтвердите возможность или невозможность
электрохимической коррозии меди при стандартных условиях по уравнению:
2Си + О2 + Н2О + СО2 = (СиОНJСО3
Рассчитайте ЭДС коррозионного ГЭ. Приведите уравнения анодной и катодной ре-
реакций.
33-35. Рассмотрите термодинамическую возможность коррозии двухвалентного
металла при стандартных условиях по уравнению:
2М + О2 + 2СО2 = 2МСО3
Рассчитайте ЭДС коррозионного ГЭ. Приведите уравнения анодной и катодной ре-
реакций.
№
Металл
33
Mg
34
Zn
35
Pb
36-39. Подтвердите расчетами, какие металлы (см. ряд напряжений табл. П.6.1) мо-
могут корродировать при температуре 298 К в водном растворе с определенным зна-
значением рН по одному из механизмов электрохимической коррозии — с водородной
или кислородной деполяризациями. Водный раствор находится в контакте с возду-
воздухом, парциальное давление кислорода и водорода в котором равны 0,21 и 0,1 атм соот-
соответственно. Приведите уравнения анодной и катодной реакций.
№
36
37
38
39
рН водного раствора
8
2
2
9
5
7
10
10
Механизм электрохимической коррозии
С кислородной деполяризацией
С водородной деполяризацией
С кислородной деполяризацией
С водородной деполяризацией
С кислородной деполяризацией
С водородной деполяризацией
С кислородной деполяризацией
С водородной деполяризацией
З. Определите, возможна ли при температуре 298 К электрохимическая корро-
коррозия изделия из металла в водном растворе, имеющем определенное значение рН и
находящемся в контакте с газовой смесью, парциальные давления кислорода и во-
водорода в которой составляют соответственно р0 и рн . Приведите уравнения
анодной и катодной реакций. Рассчитайте, при каких значениях рН раствора будет
возможна коррозия металла с водородной деполяризацией.
656
Коррозия и защита металлов и сплавов
№
40
41
42
43
Металл
Pb
Sn
Ni
Fe
рН водного раствора
5
6
5
8
Парциальное давление, атм
кислорода р0
0,21
0,23
0,20
0,21
водорода рн
1,00
1,50
1,20
2,00
44-45. Определите, возможна ли при температуре 298 К электрохимическая корро-
коррозия металла с выделением водорода в деаэрированном водном растворе собствен-
собственной соли, в котором активность его ионов составляет я, а водородный показатель рН.
Водный раствор соли находится в контакте с газовой смесью, парциальное давление
водорода в которой составляет рн . Ответ подтвердите расчетами. Рассчитайте зна-
значение потенциала металла в данных условиях. Приведите уравнения анодной и катод-
катодной реакций.
44
45
Металл
Си
Fe
Вид и состав коррозионной среды
Водный раствор CuSO4:
acu2+ = ^ 2 М0ЛЬ//д и рН = 0
Водный раствор FeCl2:
а^ = 10~3 моль/л и рН = 2
Парциальное давление
водорода рн , атм
5
2
46. Цинковую пластину с окисленной поверхностью погрузили в водный раствор
СиСЬ. Уравнениями опишите четыре основных процесса, происходящих при этом.
Рассчитайте соотношение активностей ионов Zn2+ и Си2+, при котором в данной
системе установится равновесие.
47—52. Рассмотрите электрохимическую коррозию изделия из сплава в данной кор-
коррозионной среде: а) приведите уравнения анодной и катодной реакций; б) изобрази-
изобразите схему коррозионного ГЭ; в) установите, какие вещества являются продуктами
коррозии. Ответ аргументируйте.
№
47
48
49
Вид сплава
Cu —Zn
Ni —Си
Си — Zn
Вид и состав
коррозионной среды
Морская
вода
Деаэрированная
вода с рН = 8
Дистиллированная
вода
№
50
51
52
Вид сплава
Fe —Cd
Cu —Ni
Cu —Al
Вид и состав
коррозионной среды
Деаэрированная
вода с рН = 4
Водный раствор
NaOH
Водный раствор
НС1
Задачи для самостоятельного решения
657
53-56. Определите возможность электрохимической коррозии металла при темпе-
температуре 298 К в водном растворе собственной соли, находящемся в контакте с га-
газовой смесью, парциальные давления кислорода и водорода в которой составляют
соответственно ро и рн . Приведите уравнения анодной и катодной реакций.
Рассчитайте, как изменится ЭДС коррозионного ГЭ, если концентрация раствора
соли возрастает с С до С'.
№
53
54
55
56
Ме-
Металл
А1
РЬ
Zn
Fe
Вид и состав
коррозионной среды
Водный раствор А1С13:
Са1сц = 0,01 и СдЮ1з = 0,03 моль/л
Водный раствор Pb(NO3J:
CpbcNo,>2 = 0,01 и С^ = 0,02 моль/л
Водный раствор ZnSO4:
cznso4 =0,01 и C'ZnS0^ =0,04 моль/л
Водный раствор FeCl2:
CFeC] =0,01 и C'Ftcli =0,02 моль/л
Парциальное давление, атм
кислорода
0,20
0,75
0,21
0,20
водорода
1,1
1,4
1,0
1,0
57. Рассчитайте массовый показатель скорости коррозии, протекающей с образова-
образованием РегОз на нагретом участке площадью 120 м2 стального трубопровода, если
вода, поступающая в него со скоростью 60 л/мин, содержит 6,1
мл
при темпе-
температуре 298 К и давлении 0,1 МПа, а выходящая содержит 0,1
мл О,
58. Определите, будет ли никель корродировать при температуре 298 К в деаэриро-
деаэрированной воде (рН = 8) с выделением водорода и образованием гидроксида. Рассчи-
Рассчитайте ЭДС коррозионного ГЭ.
59. Серебро корродирует в водном растворе СиСЬ с образованием твердого AgCl.
Рассчитайте ЭДС коррозионного ГЭ, если в этом растворе ас^2+ = 0,04, a acf =
= 0,1 моль/л. Приведите уравнения анодной и катодной реакций.
60-62. Изделие из сплава в течение времени t подвергалось избирательной электро-
электрохимической коррозии при температуре 298 К в водном растворе с определенным
значением рН. Определите, какой металл и в каком количестве прокорродировал,
если в процессе коррозии поглотилось Vo мл кислорода и выделилось VH мл во-
водорода, измеренных при н.у. Чему равна сила коррозионного тока? Приведите
658
Коррозия и защита металлов и сплавов
уравнения анодной и катодной реакций. Предложите анодное и катодное покрытия
для защиты изделия от электрохимической коррозии. Ответ аргументируйте.
№
60
61
62
Вид
сплава
Ni —Pt
Fe —Pd
Cu —Zn
Время корро-
коррозии/, мин
5
3
2
рН
раствора
1
11
10
Объем при н.у., мл
поглощенного Ог, Vo
112
56
56
выделившегося Нг, Уц
44,8
112
22,4
63. Рассчитайте давление водорода, необходимое для прекращения электрохимиче-
электрохимической коррозии цинка, протекающей при температуре 298 К в кислотном (рН = 5,5)
растворе с а 2+ = 0,85 моль/л. Приведите уравнения анодной и катодной реакций.
Zn
64. Кадмий корродирует в деаэрированной воде при температуре 298 К с образова-
образованием Cd(OHJ. Рассчитайте давление водорода, при котором коррозия кадмия в ней
будет невозможна. Приведите уравнения анодной и катодной реакций.
65. В медном баке с кислотным (рН = 0) раствором создано давление водорода в
0,1031 МПа. Рассчитайте максимально возможное содержание ионов Си2+ (моль/л) в
этом растворе. Приведите уравнения анодной и катодной реакций.
66. Рассчитайте парциальное давление кислорода, ниже которого невозможна газо-
газовая коррозия платины при температуре 298 К с образованием РЮг. Приведите урав-
уравнение соответствующей реакции.
67. Рассчитайте температуру, выше которой невозможна газовая коррозия серебра в
атмосфере кислорода (ро = 1,013 " 105 Па) с образованием Ag2O. Приведите урав-
уравнение соответствующей реакции.
68. Медь и цинк корродируют в морской воде со скоростью, равной 0,9 г/(м2 • сут).
Рассчитайте минимальные начальные плотности токов, необходимые для их полной
катодной защиты.
69. Для полной катодной защиты железа, корродирующего в морской воде с кисло-
кислородной деполяризацией, требуется минимальная начальная плотность тока, равная
0,1 А/м2. Рассчитайте глубинный гглуб показатель скорости коррозии. Приведите
уравнения анодной и катодной реакций.
70. Рассчитайте минимальную начальную плотность тока, необходимую для полной
катодной защиты свинца, корродирующего с кислородной деполяризацией в аэри-
аэрированной дистиллированной воде со скоростью 0,29 мм/год. Приведите уравнения
анодной и катодной реакций.
Задачи для самостоятельного решения
659
71-76. Определите по фактору сплошности Пиллинга—Бедвордса, у какого из ме-
металлов оксидная пленка на поверхности обладает лучшими защитными свойствами
против коррозии.
№
Металл и
его оксид
71
Са—СаО
Zn—ZnO
72
Си—Cu2O
Ва—BaO
73
Sn—SnO
Mg—MgO
74
Nb—Nb2O5
Та—Та2О5
75
Al—A12O3
W—WO3
76
Pb—Pb3O4
Mo—MoO3
77. Два железных изделия покрыты: одно — кадмием, другое — свинцом. Опреде-
Определите, к какому виду (анодные, катодные) относятся эти покрытия по механизму за-
защитного действия? Приведите уравнения анодной и катодной реакций, протекаю-
протекающих при коррозии указанных изделий во влажном воздухе и в растворах соляной
кислоты в случае нарушения герметичности их покрытий. Какие продукты образу-
образуются в результате коррозии?
78. Как протекает атмосферная коррозия изделия из железа с никелевым покрытием
в случае нарушения герметичности последнего? Приведите уравнения анодной и
катодной реакций. Каков конечный состав продуктов коррозии? Какое это покры-
покрытие — анодное или катодное и почему?
79. Приведите примеры металлов катодных и анодных покрытий для изделий из
никеля и кобальта. Составьте уравнения анодных и катодных реакций, протекаю-
протекающих при коррозии этих изделий во влажном воздухе и в деаэрированной воде в слу-
случае нарушения герметичности покрытий.
80. Два изделия, одно из луженого железа, другое — из луженой меди находятся во
влажной атмосфере. Уравнениями опишите анодные и катодные реакции, происходя-
происходящие при их коррозии в случае нарушения герметичности покрытия. В каком случае
оловянное покрытие является катодным, в каком — анодным? Ответ аргументируйте.
81-85. Уравнениями опишите анодную и катодную реакции, протекающие при
электрохимической коррозии изделия с металлическим покрытием в случае нару-
нарушения герметичности последнего в данной коррозионной среде. Определите, к ка-
какому виду (анодное, катодное) относится это покрытие по механизму своего защит-
защитного действия. Ответ аргументируйте. Что является продуктами коррозии?
№
81
82
83
84
85
Материал
изделия
А1
Fe
Со
Fe
Pd
покрытия
Ni
Zn
Ni
Pb
Ni
Вид коррозионной среды
Деаэрированная вода
Аэрированная вода
Раствор соляной кислоты
Разбавленный раствор NaOH
Водный раствор MgCl2
660 Коррозия и защита металлов и сплавов
86. На железную пластину электролизом водного раствора SnCl2 в течение 30 мин
при плотности тока 0,2 А/м2 наносили покрытие из олова. Рассчитайте его толщину,
если выход по току при этом составил 87 %. Приведите уравнения анодной и катод-
катодной реакций, протекающих при этом. Каков механизм защитного действия (механи-
(механический, электрохимический) данного покрытия? Ответ аргументируйте.
87. Рассчитайте, при какой активности ионов Ni2+ в коррозионной среде (водный
раствор соли кадмия с активностью acd,+ = 0,1 моль/л при 298 К) никелевое покры-
покрытие на кадмии будет вести себя как анодное при электрохимической коррозии.
88. Железное изделие с никелевым покрытием поместили в коррозионную среду:
водный раствор солей с а? 2+ = 1 и а^2+ = 10~8 моль/л при температуре 298 К. Опре-
Определите вид (анодное, катодное) и механизм защитного действия (механический,
электрохимический) данного покрытия. Ответ аргументируйте расчетами. Приведи-
Приведите уравнения анодной и катодной реакций.
89. Какой из металлов (Mg, Zn или Fe) целесообразно выбрать в качестве материала
протектора, используемого для защиты свинцовой оболочки кабеля от атмосферной
коррозии. Ответ аргументируйте расчетами. Приведите уравнения анодной и катод-
катодной реакций. Какие вещества являются продуктами коррозии?
90. Какой из приведенных металлов (Mg, Zn, A1) может служить протектором для
защиты находящегося в растворе соляной кислоты изделия из алюминиевой (до 10 %
(масс.) А1) бронзы. Ответ аргументируйте расчетами. Приведите уравнения анодной
и катодной реакций.
91. Подберите металл для протектора, который должен служить для защиты желез-
железного изделия от электрохимической коррозии в аэрированном водном растворе с
рН = 8. Приведите уравнения анодной и катодной реакций. Ответ аргументируйте.
ЛР 12. Коррозия металлов и сплавов
Цель работы — изучение условий возникновения и протекания химической и
электрохимической коррозии, а также исследование влияния различных факторов
на ее скорость.
Опыт 1. Высокотемпературное окисление меди и железа. Устойчивость
их оксидных пленок. В данном опыте предлагается вначале получить оксидные
пленки различной толщины на поверхности медной и железной пластин путем их
высокотемпературного окисления на воздухе в пламени газовой горелки, а затем
оценить защитные свойства этих пленок по отношению к электрохимической кор-
коррозии в водном растворе CuSO4 или Cu(NO3J (рис. Л. 12.1).
ЛР 12. Коррозия металлов и сплавов
661
Рис. Л.12.1. Схема высокотемпературного окисления железной (медной) пластины:
1 — газовая горелка; 2 — железная (медная) пластина; 3 — оксидная пленка;
4 — капли 0,2 н. водного раствора Cu(NO3J или CuSO4; 5 — тигельные щипцы
При нагревании меди на воздухе вначале на ее поверхности образуется оксидная
пленка цвета побежалости, которая затем по мере роста ее толщины приобретает
черный цвет вследствие своей непрозрачности. Толстая непрозрачная пленка состо-
состоит из внутреннего, непосредственно прилегающего к поверхности металла, слоя
оксида состава С112О, имеющего решетку с недостатком катионов, и внешнего, со-
соприкасающегося с воздухом, а потому и наиболее богатого кислородом слоя оксида
меди состава СиО. Состав внутреннего слоя более точно выражается формулой
Cui,8O, так как оксид содержит наряду с ионами Си+ некоторое количество и двух-
зарядных ионов Си2+ в соответствующей пропорции.
Состав оксидной пленки на железе и обычной углеродистой стали сложнее и
зависит от температуры нагрева:
Температура, °С <400 400...575 575...730
Закон роста логарифмический параболический параболический
Состав пленки Fe2O3 Fe3O4 | Fe2O3 FeO | Fe3O4 \ Fe2O3
Уже при температуре 250...300 °С на их поверхности появляется видимая
пленка оксидов. При нагревании выше Т = 400 °С поверхность этих материалов по-
покрывается пленкой серого цвета, называемой окалиной. Она имеет сложное строение,
толщина слоев различных оксидов в ней зависит от условий коррозии: температу-
температуры, продолжительности нагревания и состава коррозионной среды. При температу-
температурах, превышающих Т = 575 °С, окалина состоит из трех основных слоев, разделен-
разделенных промежуточными слоями из твердых растворов соприкасающихся оксидов:
прилегающего к поверхности железа слоя вюстита FeO, затем идут слои магнетита
Fe3O4 и гематита Fe2O3. При температурах выше Т = 700 °С толщины этих слоев
соотносятся как 5FeO : 5Fe0 : 5Fe 0 « 100 : 10 : 1. Вюстит имеет кубическую решетку
с избытком анионов (ионов кислорода) и недостатком катионов (ионов железа). На-
Наличие незанятых катионных узлов облегчает диффузию. Поскольку окалина состоит
в основном из вюстита FeO, то она обладает плохими защитными свойствами. Кро-
Кроме того, она имеет трещины и поры, которые способствуют проникновению кисло-
кислорода к металлу и тем самым усиливают дальнейшее его окисление.
662 Коррозия и защита металлов и сплавов
Последовательность проведения:
1) поверхность медной пластины шириной 2-3 см и длиной 12-15 см тщательно
очистите от оксидной пленки и загрязнений с одной стороны наждачной бума-
бумагой по всей ее длине;
2) зачищенную поверхность обезжирьте протиркой ватным тампоном (или тка-
тканевой салфеткой), пропитанным одним из растворителей: ацетоном, уайт-
спиритом, бензином. Подождав 1-2 мин для испарения растворителя, опиши-
опишите внешний вид поверхности (укажите ее цвет и состояние — блестящая, ма-
матовая и др.);
3) взяв пластину зачищенной стороной вверх за один конец тигельными щипца-
щипцами, другой внесите в бесцветное пламя газовой горелки и оставьте нагреваться
неподвижно относительно него в течение 2-3 мин до появления цветов побежа-
побежалости (см. рис. Л. 12.1);
4) прекратите нагревание и дайте пластине остыть на воздухе до комнатной
температуры. После этого опишите внешний вид ее окисленной поверхности;
5) повторяя пп. 1 -4 данного опыта, проведите аналогичный эксперимент с же-
железной или стальной пластиной;
6) после охлаждения железной пластины до комнатной температуры на ее зачи-
зачищенную поверхность пипеткой быстро, начиная с конца, не подвергавшегося
нагреванию, нанесите равномерно по всей длине через 1-2 см друг от друга ма-
маленькие капли (приблизительно 4-6 шт.) 0,2н. водного раствора Cu(NO3J или
CuSO4(cm. рис. Л.12.1);
7) по окончании включите секундомер и, наблюдая за пластиной, отметьте вре-
время появления красно-бурых осадков меди в виде пятен по всей ее длине.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и результа-
результаты расчетов и коррозионных испытаний занесите в табл. Л.12.1;
2) почему при нагревании вначале появляющиеся цвета побежалости затем ис-
исчезают? Какой цвет приобретает поверхность медной и железной пластин после
их длительного нагревания (окисления) и чем он обусловлен?
3) приведите уравнения реакций окисления поверхности медной и железной пла-
пластин. Каков состав и закон роста толщины оксидных пленок, образующихся на
их поверхности?
4) используя A8.2), рассчитайте фактор сплошности Пиллинга—Бедвордса для
различных оксидов меди и железа и по нему оцените защитные свойства пленок
из них. Плотности металлов и их оксидов возьмите из табл. П.3.2 и П.3.3;
5) где и почему на поверхности железной пластины быстрее появляются красно-
бурые осадки меди? Объясните причину их образования, подтвердив соответст-
соответствующим уравнением реакции. Какой коррозионный процесс оно описывает?
Приведите уравнения анодной и катодной реакций;
6) сделайте выводы, касающиеся защитных свойств тонких и толстых оксидных
пленок различного состава на поверхности меди и железа.
ЛР 12. Коррозия металлов и сплавов
663
Таблица Л.12.1
Результаты эксперимента
Исходные данные
Оки
щ
об]
сляю-
ийся
эазец
Медная
пластина
Железная
(стальная)
пластина
Условия
проведения
окисления
Нагревание
на воздухе
в пламени
газовой
горелки
Результаты расчетов
Значение фактора сплошно-
сплошности Пиллинга—Бедвордса
оксида а
%
и
CuO
FeO
О
со
о
Наблюдения
Внешний вид (состояние,
цвет) поверхности после
очистки
длительного
окисления
Уравнения
протекающих
реакции
Си + О2->
Си + О2->
Fe + O2->
Fe + O2->
Fe + O2-»
Результаты коррозионных испытаний
Соотношение на различных
участках пластины между
темпера-
температурой:
Г, %Т2 %
ТОЛЩИНОЙ
пленки:
5,^52 ^
временем
появления
пятен Си:
Уравнения анодной,
катодной и токооб-
разующей реакций
(-)А:
(+Ж:
Опыт 2. Электрохимическая коррозия железа в различных электроли-
электролитах. Водные растворы кислот, щелочей, солей и газов, являясь электролитами, мо-
могут служить коррозионными средами для машин, аппаратов, трубопроводов и дру-
других изделий в различных химических производствах. Значение рН растворов элек-
электролитов зависит не только от их состава, но и от процессов гидролиза, гидратации,
диссоциации и др., протекающих между содержащимися в них компонентами. В
данном опыте изучается влияние вида и состава электролита (коррозионной среды)
на характер (с водородной, кислородной деполяризацией) и скорость катодной ре-
реакции, а следовательно, и на скорость электрохимической коррозии в целом. По-
Последняя оценивается по интенсивности и времени появления «турнбулевой сини»
(коллоидного раствора синей окраски или синего осадка), которая образуется при
взаимодействии ионов Fe2+ с индикатором — гексацианоферратом (III) калия
K3[Fe(CNN] (красной кровяной солью) по реакции:
3Fe2+ + 2K3[Fe(CN6)]
Fe3[Fe(CNN]2
6K+
или
Fe2+ + K3[Fe(CN6)] -> KFe [Fe(CN)J I + 2K+
664 Коррозия и защита металлов и сплавов
Эта реакция широко используется в аналитической химии для обнаружения Fe2+-
+2 +3 +2 +3
ионов. Состав «турнбулевой сини» Fe3 [Fe(CN)J2 или KFe [Fe(CN)J до сих пор
остается предметом дискуссий. Считают, что он идентичен составу «берлинской
+3 +2 +3 +2
лазури» Fe4 [Fe(CNN]3 или KFe [Fe(CNN], образующейся по аналогичным реак-
реакциям между Ре3+-ионами и гексацианоферратом (II) калия K4[Fe(CNN] (желтой кро-
кровяной солью). В щелочных средах и та, и другая разлагаются с образованием осад-
осадков гидроксидов железа (II) и (III), в кислотных и при добавлении избытка реагента
K4[Fe(CNN] растворяется лишь осадок «берлинской лазури».
В качестве коррозионных сред в опыте используют дистиллированную воду
и водные растворы NaCl, MgCl2, NaOH и С12. Водный раствор хлора (хлорная вода)
получают пропусканием последнего через воду. При обычных условиях 1 л Н2О
растворяет 2,3 л С12 (включая все формы галогена в воде, т.е. незначительное физи-
физическое и основное химическое растворение). Химическое растворение обусловлено
протеканием между хлором и водой окислительно-восстановительной реакции* дис-
пропорционирования:
С12+Н2О<=>НС1 + НС1О (рН<7).
Его часто рассматривают как реакцию гидролиза галогена. Она обратима и положе-
положение равновесия в ней сильно сдвинуто влево (в сторону исходных веществ). Таким
образом, растворы хлора в воде — это системы, содержащие галоген С12, кислоты
НС1 и НС1О, а также незначительные количества продуктов распада последней, об-
образующихся в водных растворах по нескольким параллельно протекающим реакци-
реакциям, скорости которых зависят от условий проведения эксперимента:
ЗНС1О-»2НС1 + НСЮ3
2НС1О->2НС1 + О2
Хлорноватистая кислота НСЮ — слабая, ее диссоциация подавлена более сильной —
соляной кислотой. Сильным электролитом является и водный раствор NaOH.
В водных растворах MgCl2 подвергается гидролизу по катиону, a NaCl нахо-
находится практически в негидролизованном состоянии (соль образована катионом
сильного основания и анионом сильной кислоты).
Дистиллированная вода без соблюдения особых условий хранения всегда со-
содержит растворенный кислород.
Последовательность проведения:
1) в пять пробирок налейте поровну по 2-4 мл следующих растворов: первую —
дистиллированной воды, вторую, третью и четвертую — 2н. водных растворов со-
соответственно NaCl, MgCl2 и NaOH, пятую — хлорной воды;
2) в первую-четвертую добавьте по 2-4 мл раствора красной кровяной соли
K3[Fe(CNN], в пятую — аналогичное число капель раствора желтой кровяной соли
K4[Fe(CNN], являющихся реагентами для обнаружения в растворе ионов Fe2+ и Fe3+
соответственно. Содержимое всех пробирок тщательно перемешайте интенсивным
встряхиванием;
ЛР 12. Коррозия металлов и сплавов
665
3) поместите в них одновременно по одному железному образцу приблизительно
одинаковой массы или длины в виде стружки, гранулы, полоски, стержня, гвоздя,
куска проволоки, канцелярской скрепки;
4) зафиксируйте время и, не трогая пробирки в штативе, наблюдайте происходящие в
них изменения (появление окраски, выделение пузырьков газа и др.) в течение 30 мин.
При этом отмечайте и записьгоайте № пробирки, время появления в ней изменений и их
вид;
5) по истечении времени отметьте интенсивность окраски растворов в пробирках, а
также пробирки, в которых наблюдается выделение пузырьков газа.
Обработка результатов:
1) исходные данные, наблюдения, результаты измерений, уравнения протекающих
анодной, катодной и токообразующей реакций коррозионного ГЭ и выводы занеси-
занесите в табл. Л. 12.2;
Таблица Л. 12.2
Результаты эксперимента
№
про-
пробирки
1
2
3
4
5
№
про-
пробирки
1
2
3
4
5
Исходные данные
Вид, состав и кислотность
коррозионной среды
Дистиллированная вода, рН
2н. водный раствор NaCl, рН
2н. водный раствор MgCl2, рН
2н. водный раствор NaOH, рН
Хлорная вода С12(аКва)> рН
Наблюдения
Выде-
Выделение
газа
(да,нет)
Окраска
раствора
Уравнения протекающих реакций
анодной
(-)А:
(-)А:
(-)А:
(-)А:
(-)А:
катодной
(+)К:
(+)К:
(+)К:
(+)К:
(+)К:
токообразующей
Результаты
измерений
Время появления
окраски, начала вы-
выделения газа, мин
Выводы
Возможность и
механизм коррозии
2) укажите кислотность коррозионной среды (рН > 7, рН < 7, рН « 7), приняв во
внимание, что между компонентами и водой имеют место различные взаимодейст-
взаимодействия (гидролиз, гидратация, диссоциация). Приведите уравнения, описывающие эти
взаимодействия;
3) основываясь на наблюдениях, напишите уравнения возможных анодной, катодной й
токообразующей реакций для каждого вида коррозионной среды;
4) на основании результатов наблюдений (времени появления и интенсивности ок-
окраски раствора, времени начала и интенсивности выделения газа) сделайте выводы
666 Коррозия и защита металлов и сплавов
о возможности (протекает, нет) и механизме (с кислородной, водородной деполяри-
деполяризацией) электрохимической коррозии железа;
5) в какой среде и почему скорость электрохимической коррозии выше? Располо-
Расположите электролиты (коррозионные среды) в порядке возрастания их коррозионной
активности, т.е. величины скорости коррозии {г\ ^ г2 Ч ... ^ /*,);
6) как связаны ЭДС коррозионного ГЭ и энергия Гиббса коррозионного процесса?
Опыт 3. Хлорид-ионы — активные стимуляторы электрохимической кор-
коррозии. Известно, что на поверхности алюминия в атмосфере, содержащей кислород,
имеется защитная оксидная пленка, которая предохраняет его от действия различ-
различных агрессивных сред. Однако хлорид-ионы быстро разрушают ее. Сульфат-ионы
производят подобное действие лишь при большой их концентрации и притом мед-
медленно. При этом алюминий начинает корродировать.
В данном опыте предлагается изучить влияние хлорид-ионов на скорость
электрохимической коррозии алюминия в водных растворах солей CuSO4 и СиСЬ.
Последние подвергаются в них гидролизу.
Последовательность проведения:
1) в первые две пробирки налейте по 2-4 мл слегка подкисленного серной кислотой
0,5н. водного раствора сульфата меди C11SO4. В одну из них добавьте 2—3 капли на-
насыщенного водного раствора NaCl;
2) во вторые две пробирки налейте те же объемы 0,5н. водных растворов: в одну —
сульфата меди CuSO4, в другую — хлорида меди СиСЬ;
3) в каждую из четырех пробирок поместите по образцу алюминия в виде стружки,
гранул, пластины, стержня или куска проволоки;
4) включите секундомер и наблюдайте за происходящими изменениями (выделение
пузырьков газа, образование осадка меди и др.). При этом отмечайте и записывайте,
в какой из пробирок интенсивнее выделяется газ по количеству образующихся его
пузырьков, быстрее появляется красно-бурый осадок меди и др.
Обработка результатов:
1) исходные данные, наблюдения, схему и уравнения протекающих анодной, катод-
катодной и токообразующей реакций коррозионного ГЭ, результаты расчетов и выводы
занесите в табл. Л. 12.3;
2) какой газ выделяется в пробирках и почему? Каким образом он получается? От-
Ответ аргументируйте соответствующими уравнениями анодной, катодной и токооб-
токообразующей реакций. Приведите схему образующегося коррозионного ГЭ;
3) рассчитайте значения потенциалов анода и катода по уравнению Нернста и вели-
величину ЭДС коррозионного ГЭ, если рН водных растворов CuSO4 и СиС12 составляет
4, а подкисленных серной кислотой — 2,0. При этом считайте что р0 = рн -1, а
САK+ =10~3 моль/л;
4) объясните причину (механизм) ускоряющего действия хлорид-ионов на электро-
электрохимическую коррозию алюминия. Каков состав защитной оксидной пленки на нем?
Приведите уравнения ее растворения в растворах НС1 и NaOH, а также уравнения
реакций взаимодействия алюминия с водой, сульфатом и хлоридом меди, соляной и
азотной кислотами, щелочами. Укажите условия протекания этих реакций;
ЛР12. Коррозия металлов и сплавов
667
Результаты эксперимента
Таблица Л.12.3
№
про-
пробирки
1
2
3
4
Схе
анод
токообр
КОР]
Исходные данные
Вид и состав
коррозионной среды
Подкисленный H2SO4 0,5н.
водный раствор CuSO4
Подкисленный H2SO4 0,5н.
водный раствор CuSO4,
содержащий NaCl
0,5н. водный раствор CuSO4
0,5н. водный раствор СиС12
ма и уравнения
ной, катодной и
шующей реакций
розионного ГЭ
(-)А( ):
(+Ж( ):
I
Корродирующий
образец
Алюминиевый
образец в виде
стружки, гра-
гранул, пластины,
куска проволо-
проволоки или стержня
Результаты расчетов
Потенциал анода (ра, катода
фк и ЭДС Е коррозионного ГЭ
Al
Фк=Ф^ =
н2
Наблюдения
Выделение
газа
Образование
осадка Си
(да сразу, через t мин, нет)
Выводы
Изменение гэх м в присутст-
присутствии ионов СГ и Н+
(увеличение, уменьшение)
5) на основании полученных результатов сделайте выводы о влиянии присутствия
СГ- и Н+-ионов на скорость электрохимической коррозии алюминия гэх дь
Опыт 4. Удаление защитной пленки с поверхности магния. На воздухе
магний практически всегда покрыт тонкой оксидной пленкой из MgO, которая при-
придает его поверхности матовый оттенок и не позволяет ему самому подвергаться
глубокому разрушению. В воде она растворяется, вследствие чего затем начинается
его взаимодействие с водой. Процесс протекает очень медленно, практически неза-
незаметно. Причиной тому является образование малорастворимого гидроксида
Mg(OHJ, оседающего на поверхности в виде защитной, изолирующей от действия
воды, пленки. Нагревание несколько ускоряет реакцию между магнием и водой, так
как при этом увеличивается растворимость Mg(OHJ. Добавление в воду солей ам-
аммония приводит к полному и быстрому растворению как защитной пленки из
Mg(OHJ, так и самого магния. В данном опыте предлагается проверить выше-
вышеизложенное.
Последовательность проведения:
1) в каждую из трех пробирок налейте поровну по 2-4 мл дистиллированной воды,
а затем одновременно внесите в них по кусочку или по несколько крупинок магния.
Интенсивным встряхиванием тщательно перемешайте содержимое пробирок;
668
Коррозия и защита металлов и сплавов
2) первую отставьте в сторону в качестве контрольного образца: она будет служить
эталоном — образцом сравнения. Отметьте и запишите происходящие в ней изме-
изменения (выделение пузырьков газа, образование осадка и др.);
3) во вторую добавьте 6-8 капель насыщенного водного раствора хлорида аммония,
содержимое тщательно перемешайте интенсивным встряхиванием и наблюдайте за
происходящими в ней изменениями в течение 5-7 мин по сравнению с контрольным
образцом. Отметьте, запах какого газа при этом чувствуется;
4) содержимое третьей пробирки нагрейте на водяной бане до кипения и кипятите в
течение 2-4 мин. Сравните с контрольным образцом происходящие при этом изме-
изменения.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций, результаты расче-
расчетов энергии Гиббса и ЭДС коррозионного ГЭ, а также выводы занесите в табл. Л. 12.4;
Таблица Л.12.4
Результаты эксперимента
№
про-
пробирки
1
2
3
Уравне
творени
и знач<
эн
MgO(T)
Mg(OH
Исходные данные
Вид и состав
коррозионной среды
Дистиллированная
вода при Гкомн
Водный раствор
NH4C1 при Гкомн
Дистиллированная
вода при Г=100°С
?ния реакций рас-
[я MgO и Mg(OHJ
гния стандартной
ергии Гиббса
»„„-„.с,„.
Корродирующий
образец
Магний в виде
стружки, ку-
кусочков или
крупинок
Коррозионный
ГЭ
Уравнения анод-
анодной, катодной и
токообразующей
реакций
«К:
I
Наблюдения
Выделение газа
(да, нет)
Результаты расчетов
Значения
стандартных ЭДС Е°
и энергии Гиббса &rGm
Е =Ф 2Н,0 "ФМ8- =
2ОН", Н2 Mg
Образование
или растворе-
растворение осадка
Выводы
Коррозия Mg
в Н2О и рас-
растворах солей
аммония
2) приведите уравнения, описывающие реакции растворения MgO в воде и Mg(OHJ
в растворе NH4C1. Какие из визуально наблюдаемых изменений говорят в их поль-
пользу? Почему чувствуется запах аммиака и раствор становится прозрачным? Подтвер-
Подтвердите термодинамическую возможность процессов растворения расчетом их стан-
стандартной энергии Гиббса;
ЛР 12. Коррозия металлов и сплавов 669
3) электрохимическую коррозию (растворение) магния в воде опишите уравнения-
уравнениями соответствующих реакций: анодной, катодной и токообразующей. Подтвердите
термодинамическую возможность ее протекания при стандартных условиях расче-
расчетами ЭДС коррозионного ГЭ и энергии Гиббса коррозионного процесса;
4) на основании полученных данных сделайте выводы о возможности или невоз-
невозможности коррозии магния в воде и водных растворах солей аммония. Если она
возможна, то укажите соотношение скоростей \rx ^ г2). Почему в присутствии иона
аммония NH* он активнее взаимодействует с водой? В какой среде, воде или вод-
водных растворах щелочей, и почему магний обладает более высокой коррозионной
стойкостью?
Опыт 5. Влияние природы электролита, состава и кислотности его рас-
растворов на скорость электрохимической коррозии цинка. Скорость электрохими-
электрохимической коррозии как гетерогенного процесса зависит от природы корродирующего
металла, площади его соприкосновения с коррозионной средой, а также очень силь-
сильно от вида, состава и значения рН последней. Чем меньше значение водородного
показателя рН коррозионной среды и больше содержание в ней окислителей, тем
быстрее протекает коррозия. Концентрация ионов водорода в электролите (корро-
(коррозионной среде), т.е. рН зависит от степени его диссоциации, которая определя-
определяется в первую очередь природой и концентрацией электролита и наличием в его
растворах веществ, усиливающих и подавляющих диссоциацию либо связы-
связывающих ионы водорода.
В данном опыте предлагается исследовать эти аспекты на примере электро-
электрохимической коррозии цинка в растворах НС1, H2SO4, CH3COOH одинаковой моляр-
молярной концентрации. Она протекает в них с водородной деполяризацией и описывает-
описывается следующим ионно-молекулярным уравнением:
которое является уравнением гетерогенного процесса типа твердое тело — жидкость.
Скорость последнего, согласно закону действующих масс, определяется соотношением:
где к — константа скорости; С + — молярная концентрация ионов водорода; SZn —
н
площадь поверхности цинка, соприкасающейся с коррозионной средой. Если цин-
цинковый образец дан в виде гранулы или пластины, то SZn мало меняется во времени и
ее можно считать постоянной. Тогда
где к' = kSZn; п — порядок реакции по Н+-иону.
Последовательность проведения:
1) в каждую из четырех пробирок налейте поровну приблизительно по 2 мл сле-
следующих растворов: в первую, вторую и третью — 1М водных растворов кислот со-
670
Коррозия и защита металлов и сплавов
ответственно НС1, H2SO4 и СН3СООН, в четвертую — 100 % (масс.) (ледяной) или
80 % (масс.) уксусной кислоты;
2) в каждую пробирку одновременно поместите по кусочку или грануле цинка при-
приблизительно одинаковой массы и поверхности растворения. Отметьте, в какой про-
пробирке быстрее начинается выделение пузырьков газа;
3) через 1-3 мин сравните скорость коррозии цинка в различных электролитах визу-
визуально по количеству выделяющихся в единицу времени пузырьков газа;
4) после установления равномерного вьщеления пузырьков газа добавьте на кончике
шпателя кристаллических солей: в первую пробирку NaCl, во вторую и третью —
CH3COONa. Содержимое четвертой пробирки разбавьте пополам дистиллирован-
дистиллированной водой;
5) тщательно перемешав интенсивным встряхиванием содержимое всех пробирок,
отметьте произошедшие в них изменения.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций, результаты
рассмотрения равновесий в различных коррозионных средах и выводы занесите в
табл. Л. 12.5;
Результаты эксперимента
Таблица Л. 12.5
про-
пробирки
1
2
3
4
№
про-
пробирки
1
2
3
4
Исходные данные
Вид и состав кор-
коррозионной среды
1М водный
раствор НС1
1М водный
раствор H2SO4
1М водный рас-
раствор СНзСООН
100 или 80%
СНзСООН
Корроди-
Корродирующий
образец
Цинк
в виде
гранул,
кусочков
Наблюдения
Выделение пузырьков
газа (да сразу, через /
мин, нет)
Уравнен
гетерог
ие протекающей
енной реакции
Zn(T) + HCl(p)->
Zn(T) + H2SO4 (p) —>
Zn(T) + CH3COOH(p) ->
Zn(T) + СН3СООН(Ж) ->
Результаты рассмотрения равновесий в различных коррозионных средах
Уравнение устанавливающегося
в коррозионной среде равновесия
НС1-»
H2SO4 <zi
СН3СООН ?±
СН3СООН <=>
Добавляемое
вещество
NaCl
CH3COONa
CH3COONa
Н2О
Смещение
положения
равновесия
(вправо, влево)
Изменение
концентрации
Н+-ионов
(увеличение,
уменьшение)
ЛР12. Коррозия металлов и сплавов
671
Окончание табл. Л. 12.6
№
про-
пробирки
1
2
3
4
Коррозионный ГЭ
Уравнения анод-
анодной, катодной и
токообразующей
реакций
(-)А:
(+)К:
I
Изменение интен-
интенсивности выделения
газа (увеличение,
уменьшение)
Уравнение реак-
реакции, изменяющей
интенсивность
выделения газа
Выводы
Коррозия (уси-
(усиливается, осла-
ослабевает)
2) в какой пробирке быстрее и интенсивнее начинают выделяться пузырьки газа и
почему? Ответ аргументируйте, используя понятие диссоциации электролита и рас-
рассматривая протекающие реакции как гетерогенные процессы. Какой газ выделяет-
выделяется? Приведите уравнения соответствующих реакций;
3) опишите уравнениями равновесия, существующие в различных коррозионных сре-
средах. Укажите, как изменяется концентрация Н+-ионов и в каком направлении сме-
смещается при этом положение равновесия при добавлении в них различных веществ;
4) объясните причину выделения пузырьков газа, основываясь на возникновении и
функционировании коррозионных гальванических микроэлементов. Приведите
уравнения анодной, катодной и токообразующей реакций. Укажите причину, под-
подтвердив соответствующими реакциями, изменения (увеличения, уменьшения) ин-
интенсивности выделения газа после добавления различных веществ в каждом кон-
конкретном случае.
Опыт 6. Коррозия стали в результате электрохимической неоднородно-
неоднородности ее поверхности и неравномерной аэрации. В данном опыте смоделированы
два вида атмосферной коррозии — влажная, или в пленке влаги (см рис. 18.7) и
мокрая — под каплей воды (см. рис. 18.8). На примере двух стальных образцов в
виде пластин рассмотрены основные закономерности ее протекания: причины, слу-
случаи возникновения коррозионных гальванических микроэлементов и возможные
анодная, катодная и токообразующая реакции. Токопроводящей коррозионной сре-
средой служит водный раствор ферроксил-индикатора. Входящие в его состав: красная
кровяная соль Кз|Те(С]Ч)б] является реактивом на ионы Fe2+ (см. опыт 2), фенолфта-
фенолфталеин служит индикатором на ОН"-ионы, т.е. используется для установления рН сре-
среды, NaCl добавляют для увеличения электропроводности и одновременно как ак-
активный стимулятор коррозии (СГ-ионы), агар-агар вводят для повышения вязкости
раствора. Последний представляет собой смесь полисахаридов, содержащую около
70 % агарозы и получаемую из красных морских водорослей. Это аморфный про-
продукт, не растворимый в холодной воде, но легко растворяющийся в кипящей. Вод-
Водные растворы, содержащие 0,5... 1,5 % (масс.) агар-агара, при охлаждении до темпе-
температуры 35...39 °С образуют плотный гель.
Капля раствора ферроксил-индикатора имитирует каплю воды, а бумажный
фильтр, смоченный им, выполняет роль тонкой пленки влаги.
672
Коррозия и защита металлов и сплавов
Последовательность проведения:
1) наждачной бумагой тщательно очистите от ржавчины и загрязнений поверхность
одной стороны каждой из двух стальных пластин шириной 2-3 см и длиной 12-15 см
по всей их длине;
2) далее промойте их под струей водопроводной воды, а затем осушите фильтро-
фильтровальной бумагой;
3) на зачищенную поверхность одной из пластин положите, плотно прижав, бумаж-
бумажный фильтр, смоченный ферроксил-индикатором, а другой — нанесите его боль-
большую каплю;
4) наблюдайте в лупу или микроскоп при малом увеличении за окраской капли и
состоянием поверхности под ней. Происходящие изменения фиксируйте в лабора-
лабораторном журнале;
5) через 2-3 мин опишите изменение цвета бумажного фильтра, форму, размеры и
распределение пятен.
Обработка результатов:
1) исходные данные, наблюдения, схему и уравнения анодной, катодной и токо-
образующей реакций коррозионного ГЭ, а также результаты расчетов значений
ЭДС, энергии Гиббса и электродных потенциалов занесите в табл. Л. 12.6;
Таблица Л. 12.6
Результаты эксперимента
Исходные данные
Корродирующий
образец
Стальная
пластина
Вид, состав
и кислотность кор-
коррозионной среды
Аэрированный
раствор феррок-
ферроксил-индикатора
Вид смоделиро-
смоделированной атмо-
атмосферной коррозии
Влажная
(в пленке влаги)
Мокрая
(под каплей воды)
Коррозионный ГЭ
Схема и уравнения анодной, катодной
и токообразующей реакций
(-)А:
(+)К:
Наблюдения
Цвет, форма и распределение
пятен на бумаге. Окраска и ее
распределение внутри капли
Результаты расчетов
Значения электродных потенциалов,
ЭДС и энергии Гиббса
фа= ;
Е = фк ~ фа
ArG = -ZR
фк =
2) чем обусловлено появление пятен на фильтровальной бумаге и различного окра-
окрашивания в капле ферроксил-индикатора? Какие составляющие (компоненты) стали
выполняют роль катодных и анодных участков при коррозии? Приведите схему и
уравнения анодной, катодной и токообразующей реакций коррозионного ГЭ;
3) объясните причину различного окрашивания у краев и в центральной части кап-
капли. Приведите уравнения соответствующих реакций. Что является анодом и что —
катодом? Связано ли оно с воздействием кислорода воздуха?
ЛР12, Коррозия металлов и сплавов
4) подтвердите возможность протекания атмосферной коррозии расчетами ЭДС
коррозионного элемента и энергии Гиббса коррозионного процесса. При этом при-
примите, что рН = 7, р0 = 0,21 атм, /?н =0,1 атм, а С^2+ = 10 моль/л.
Опыт 7. Роль кислорода воздуха в мокрой атмосферной коррозии железа.
Мокрая атмосферная коррозия железа и сплавов на его основе протекает преимуще-
преимущественно с катодным контролем (контролирующей катодной реакцией) и кислород-
кислородной деполяризацией, поэтому ее скорость зависит от содержания кислорода в пленке
влаги. Концентрация последнего в ней в свою очередь определяется его содержани-
содержанием в воздухе.
В данном опыте изучается влияние содержания кислорода во влажном
воздухе на скорость мокрой атмосферной коррозии железа. Для этого
используется установка, изображенная на рис. Л. 12.2. В плотно закрытой
резиновой пробкой 4 и закрепленной в штативе 1 пробирке 3, внутренняя
поверхность которой смочена раствором NaCl, находится предварительно
промытый и обезжиренный порошок железа. Вместо него можно использовать
также железные опилки, стружку или канцелярские скрепки. Пробирка соединена
газоотводной трубкой 5, изготовленной из пипетки с делениями, диаметром 3-3,5
мм со стаканом (или колбой) 7 емкостью 50 мл, в котором налито 25-30 мл
подкрашенной 4-6 каплями метилового оранжевого воды.
Коррозия железа в данных условиях протекает с поглощением кислорода, за
счет чего его содержание в пробирке уменьшается, что приводит к понижению дав-
давления в ней и, как следствие, к подъему уровня воды в трубке.
Последовательность прове-
проведения:
1) смочите внутреннюю поверхность пробирки
раствором NaCl: для этого на дно внесите 4-6
капель раствора и вращательными круговыми
движениями распределите жидкость по внут-
внутренней поверхности пробирки на 1/3 ее высоты;
2) стряхните в нее с сухого микрошпателя
приблизительно 10-15 мг порошка железа ли-
либо поместите другие, имеющиеся в распоряже-
распоряжении, его образцы (опилки, стружку, скрепки);
3) быстро и плотно закройте пробирку резино-
резиновой пробкой с газоотводной трубкой и одно-
одновременно включите секундомер. Пробку для
герметичности можно предварительно смазать
вазелином;
4) другой конец газоотводной трубки опустите
в стакан с подкрашенной водой;
5) за газоотводной трубкой укрепите линейку
либо миллиметровую бумагу (если трубка без
делений), совместив их начало отсчета с уров-
уровнем жидкости в трубке;
Рис, Л.12.2. Установка для изу-
изучения мокрой атмосферной кор-
коррозии:
1 — металлический штатив с лапкой;
2 — порошок железа; 3 — стеклянная
пробирка; 4 — резиновая пробка; 5 —
газоотводная стеклянная трубка; 6 —
линейка с делениями; 7 — стакан
22 - 9795
674
Коррозия и защита металлов и сплавов
6) отмечайте уровень воды в трубке каждые 10 с. Результаты заносите в лаборатор-
лабораторный журнал.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций (анодной, катодной
и токообразующей), результаты измерений и выводы занесите в табл. Л. 12.7;
Таблица Л. 12.7
Результаты эксперимента
Исходные данные
Корродирующий
образец
Железный обра-
образец в виде по-
порошка, стружки,
опилок
Вид, состав и ки-
кислотность корро-
коррозионной среды
Аэрированная
дистиллирован-
дистиллированная вода, рН = 7
Наблюдения
Повышение, пони-
понижение уровня воды
в трубке
Результаты измерений
Время коррозии
Г, мин
Высота подъема уровня воды
в трубке h, мм,
в зависимости от значения t
Уравнения анодной,
катодной и токооб-
токообразующей реакций
(-)А:
(+)К:
I
Выводы
Характер зависимости
скорости коррозии от
содержания О2
в атмосфере
2) объясните причину подъема уровня воды в трубке, подтвердив соответствующи-
соответствующими уравнениями анодной, катодной и токообразующей реакций;
3) по результатам измерений постройте график в координатах: высота подъема
уровня воды — время коррозии, который наглядно показывает изменение скорости
коррозии во времени;
4) каким показателем проще всего можно выразить скорость коррозии в данном
случае? Как он связан с объемом поглощаемого кислорода?
5) на основании графика сделайте вывод о характере изменения зависимости скоро-
скорости мокрой атмосферной коррозии от содержания кислорода во влажной атмосфере.
Опыт 8. Коррозия при контакте двух различных металлов. В данном опыте
на примере контакта цинкового / и медного 2 образцов в виде стержней, пластин,
полос или кусков проволоки смоделирован и рассмотрен один из случаев контактной
коррозии — в результате образования и функционирования коррозионного гальвани-
гальванического макроэлемента (рис. Л. 12.3). Образцы вначале находятся во внутреннем кон-
контакте (соприкасаются в растворе, т.е. замкнута внутренняя цепь), а затем во внешнем
(концы образцов соединены проводником вне раствора, т.е. замкнута только внешняя
цепь). В качестве коррозионной среды используется водный раствор серной кислоты,
находящийся в согнутой под углом около 90° стеклянной трубке 3. Последняя закре-
закрепляется в держателе или подставке 4.
ЛР12. Коррозия металлов и сплавов
675
7/
Рис. Л. 12.3. Установка для изучения
контактной коррозии:
1 — цинковый образец-анод; 2 — медный
образец-катод; 3 — стеклянная трубка; 4 —
подставка
Последовательность про-
проведения:
1) в согнутую под углом около 90° стек-
стеклянную трубку налейте приблизительно
до половины объема 0,01н. водного
раствора H2SO4;
2) наждачной бумагой зачистите цинко-
цинковый и медный образцы (в виде пластин,
полосок, стержней, кусков проволоки),
промойте под струей водопроводной во-
воды, а затем осушите их фильтровальной
бумагой;
3) в левое колено трубки поместите цин-
цинковый образец, в правое — медный так,
чтобы они не соприкасались;
4) подождав 1-2 мин, запишите наблюде-
наблюдения (выделение пузырьков газа и др.);
5) приведите образцы в соприкосновение
внутри раствора;
6) подождав опять 1-2 мин, запишите наблюдения (выделения пузырьков газа и
др.). При этом не забудьте отметить произошедшие изменения;
7) разъедините образцы металлов и, не вынимая из раствора, соедините находящие-
находящиеся вне раствора концы проводником (алюминиевой или медной проволокой). От-
Отметьте наблюдения.
Обработка результатов:
1) исходные данные, наблюдения, схему и уравнения анодной, катодной и токооб-
разующей реакций коррозионного ГЭ, а также результаты расчетов занесите в
табл. Л. 12.8;
2) почему при отсутствии контакта образцов пузырьки газа образуются только на
цинке? Что это за газ и какова причина его выделения? Ответ аргументируйте урав-
уравнениями анодной, катодной и токообразующей реакций;
3) объясните причину вьщеления водорода на меди в случае внутреннего контакта
образцов. Приведите соответствующие уравнения реакций. Почему Си не вытесняет
Н2 из кислотных растворов?
4) отличается ли электрохимическое поведение образцов при внешнем контакте от
их поведения при внутреннем контакте? Если да, то чем? Приведите уравнения
электродных и токообразующей реакций для этого случая. Укажите, какой образец
при этом анод, а какой — катод;
5) приведите схему коррозионного ГЭ. Рассчитайте по уравнению Нернста значения
электродных потенциалов, а по ним значение ЭДС. Примите рн =0,1 атм, а С 2+ =
= 0,01 моль/л. Объясните роль меди в изучаемом процессе;
6) что произойдет, если вместо меди взять графит, железо или алюминий? Сформу-
Сформулируйте выводы, указав в обобщенном виде, какой образец-электрод (металл, неме-
неметалл) будет растворяться, а на каком будет выделяться водород.
22*
676
Коррозия и защита металлов и сплавов
Результаты эксперимента
Таблица Л. 12.8
Исходные данные
Вид, состав и
кислотность
коррозионной
среды
0,01н. вод-
водный раствор
H2SO4,
рН<7
Схема
коррозионно
Исследуемая
электрохимическая
система
Zn и Си образцы
вне контакта
Zn и Си образцы во
внутреннем контакте
Zn и Си образцы во
внешнем контакте
гоГЭ
Наблюдения
Выделение
(на Zn, Си)
пузырьков газа
Уравнения анодной,
катодной и
токообразующей реакций
коррозионного ГЭ
(-)А( ):
(+Ж( ):
2
(-)А( ):
(+) К( ):
Е
(-)А( ):
(+Ж( ):
2
Результаты расчетов
Электродные потенциалы фа, фк
Фа=Ф^ =
Zn
Н2
ЭДС коррозионного ГЭ Е
? = ФК-Фа=ф?н1-Ф^: =
Н2 Zn
Опыт 9. Влияние природы металла-партнера на скорость контактной
коррозии железа. В данном опыте изучается контактная коррозия железа (стали) в
паре с медью и в паре со свинцом в кислотном растворе. Для этого используется
установка, которая представляет собой {Т-образную стеклянную трубку 1 с двумя
закрепленными на панели 2 и опущенными по одному в каждое ее колено металли-
металлическими образцами в виде пластин или стержней, один из которых железный или
стальной, а другой из меди или свинца (рис. Л.12.4). Образцы соединяются друг с
другом через милливольтметр 4. Коррозионной средой является 0,2н. водный рас-
раствор H2SO4, которым заполняется закрепленная в штативе 6 (/-образная трубка.
Последовательность проведения:
1) наждачной бумагой зачистите, а затем промойте под струей водопроводной воды
закрепленные на панели железный и медный образцы;
2) заполните наполовину (/-образную трубку 0,2н. водным раствором H2SO4;
3) осторожно погрузите стержни в раствор, соответственно стальной левого колена,
медный — правого;
4) замкните внешнюю цепь через милливольтметр и запишите значение напря-
напряжения;
5) выполняя пп. 1 -4 данного опыта, проведите аналогичный эксперимент с желез-
железным и свинцовым образцами в паре.
ЛР 12. Коррозия металлов и сплавов
677
Обработка результатов:
1) исходные данные, результаты измерений, схему и уравнения анодной, катодной и
токообразующей реакций коррозионного ГЭ занесите в табл. Л. 12.9;
Таблица Л. 12.9
Результаты эксперимента
Исходные данные
Вид, состав и
кислотность
коррозионной
среды
0,2н. водный
раствор
H2SO4,
рН<7
Исследуемая
электрохимическая
система
Fe и Си образцы во
внешнем контакте
Fe и РЬ образцы во
внешнем контакте
Результаты
измерений
Напряжение
коррозионного
ГЭС/, В
Схема и уравнения
анодной, катодной
и токообразущей реакций
коррозионного ГЭ
<-)А( ):
(+Ж( ):
I
(-)А( ):
(+)К( ):
I
2) что является причиной отклонения стрелки милли-
милливольтметра? Ответ аргументируйте соответствующи-
соответствующими уравнениями анодной, катодной и токообразую-
токообразующей реакций. Приведите схему коррозионного ГЭ;
3) рассчитайте теоретическое значение потенциала ка-
катода (катодной реакции), если рн = 0,1 атм, у+ = 0,256.
Объясните влияние природы металла катодного кон-
контакта на скорость контактной коррозии и величину
напряжения коррозионного ГЭ;
4) рассмотрите, как будет протекать контактная кор-
коррозия в кислотном растворе у следующих пар метал-
металлов: Fe-Al, Fe-Sn?
Опыт 10. Контактная коррозия как следствие
образования и функционирования коррозионных
микроэлементов. Образование и работа огромного числа
короткозамкнутых коррозионных гальванических мик-
микроэлементов является причиной и отличительной осо-
особенностью электрохимической коррозии. В данном опы-
опыте рассматривается еще один из случаев их возникнове-
возникновения — при контакте кристаллитов двух металлов друг с
другом, например цинка и меди. Кристаллиты меди обра-
образуются в виде осадка красно-бурого цвета на поверхно-
поверхности цинкового образца, находящегося в водном растворе
серной кислоты после добавления к последнему CuSO4.
В качестве восстановителя выступают атомы цинка.
!
j i
i
]
г
-и
i
в
7
-_
= :Ъ=Г::
:
":
)
Г/ ц=р
///////////////////
Рис. Л.12.4. Установка для
изучения контактной кор-
коррозии:
1 — стеклянная {/-образная
трубка; 2 — панель; 3 — же-
железный (стальной) образец-
анод; 4 — милливольтметр;
5 — медный или свинцовый
образец-катод; 6 — металли-
металлический штатив с лапкой
678
Коррозия и защита металлов и сплавов
Последовательность проведения:
1) в две пробирки налейте по 2-4 мл 2н. водного раствора H2SO4;
2) в каждую поместите по цинковому образцу приблизительно одинаковой массы и
поверхности растворения, лучше всего в виде гранул;
3) подождите некоторое время B-4 мин), пока не начнется выделение на них пу-
пузырьков газа;
4) одну пробирку отставьте в сторону в качестве контрольного образца: она будет
служить эталоном — образцом сравнения. Во вторую добавьте 2-4 капли 0,4н. вод-
водного раствора CuSO4, содержимое тщательно перемешайте, интенсивно встряхивая;
5) через 1-2 мин сравните скорость электрохимической коррозии цинка в этой про-
пробирке со скоростью в контрольном образце по интенсивности выделения пузырьков
газа. Отметьте и другие изменения, произошедшие в ней;
6) спустя 5 мин слейте раствор и промойте образец дистиллированной водой. Опи-
Опишите его внешний вид.
Обработка результатов:
1) исходные данные, наблюдения, схему и уравнения анодной, катодной и токооб-
разующей реакций коррозионного ГЭ занесите в табл. Л. 12.10;
Результаты эксперимента
Таблица Л. 12.10
Исходные данные
Корроди-
Корродирующий
образец
Цинк
в виде
гранулы,
кусочка
Вид, состав и кислотность
коррозионной среды
2н. водный раствор
H2SO4,pH< 7
2н. водный раствор
H2SO4, содержащий
CuSO4, pH < 7
Наблюдения
Выделение
(быстрое, медленное)
пузырьков газа,
образование осадка Си
Схема и уравнения
анодной, катодной и
токообразующей
реакций коррозион-
коррозионного ГЭ
(-)А( ):
(+)К( ):
S
(-)А( ):
(+)К( ):
Z
2) что является причиной образования и выделения пузырьков газа после помеще-
помещения цинкового образца в раствор H2SO4? Какой газ при этом выделяется? Ответ ар-
аргументируйте уравнениями анодной, катодной и токообразующей реакций. Что яв-
является анодом, что — катодом? Приведите схему коррозионного ГЭ;
3) с чем связано возрастание скорости выделения водорода после прибавления рас-
раствора сульфата меди? Почему его пузырьки теперь образуются и выделяются на
красно-буром осадке меди? Что представляет собой омедненный образец цинка?
Какова здесь роль кристаллитов меди, осевших на поверхности цинка? Свои ответы
подтвердите соответствующими уравнениями анодной, катодной и токообразующей
реакций. Приведите схему коррозионного ГЭ и уравнение реакции, протекающей
между цинком и раствором CuSO4.
ЛР 12. Коррозия металлов и сплавов 679
Опыт 11. Пассивирование алюминия и железа. Пассивирование исполь-
используют для создания на поверхности металлов и сплавов защитных (адсорбционных,
фазовых или адсорбционно-фазовых) пленок, чаще всего оксидных. В данном опыте
предлагается вначале осуществить пассивирование алюминиевого и железного (или
стального) образцов в виде пластин, отрезков проволоки и др. дымящей азотной
кислотой плотностью 1,5 г/см3 или концентрированной серной, а затем испытать их
на коррозионную стойкость в растворах различных кислот и солей, в частности в
HNO3, H2SO4, CuSO4, CuCl2, NaCl. Если в лаборатории нет дымящей азотной кисло-
кислоты, то можно использовать концентрированную с плотностью 1,4 г/см3, предвари-
предварительно добавив в нее несколько капель спирта или немного сахара для образования
оксидов азота, благодаря которым она приобретает желтую окраску и пассивирую-
пассивирующую способность.
Внимание! Опыт проводится только в вытяжном шкафу
Последовательность проведения:
1) в четыре пробирки налейте по 2-4 мл следующих 0,2н. водных растворов: пер-
первую — HNO3, вторую — H2SO4, третью — CuSO4, четвертую — СиС12;
2) тонкой наждачной бумагой тщательно очистите от оксидной пленки и загрязне-
загрязнений с одного конца поверхность четырех алюминиевых образцов;
3) в каждую пробирку зачищенным концом опустите по образцу и наблюдайте за
происходящими изменениями (выделение пузырьков газа, образование на поверх-
поверхности красно-бурого осадка меди и др.);
4) в две пробирки налейте по 2-4 мл дымящей азотной или по указанию преподава-
преподавателя концентрированной серной кислоты для проведения процесса пассивирования;
5) образцы с осадком меди отставьте в сторону, а два других извлеките из растворов
кислот (HNO3 и H2SO4), промойте дистиллированной водой, избытки воды аккурат-
аккуратно стряхните и затем погрузите их на 2-3 мин в пробирки с дымящей азотной ки-
кислотой. Что при этом наблюдаете?
6) по истечении времени образцы осторожно извлеките, промойте дистиллирован-
дистиллированной водой и снова верните их в соответствующие разбавленные первоначальные
растворы кислот @,2н. HNO3 и H2SO4). Что наблюдаете? Отметьте, как изменилась
при этом скорость коррозии, оценив ее по количеству выделяющихся в единицу
времени пузырьков газа;
7) в пробирку с 0,2н. раствором HNO3 добавьте несколько капель насыщенного рас-
раствора хлорида натрия, а в пробирку с 0,2н. раствором H2SO4 — аналогичное число
капель насыщенного раствора хлорида меди. К чему это привело?
8) повторяя пп. 1-7 данного опыта проведите по указанию преподавателя экспери-
эксперимент с образцами железа или стали.
Обработка результатов:
1) исходные данные, наблюдения, уравнения анодной, катодной и токообразующей
реакций занесите в табл. Л. 12.11;
2) объясните наблюдаемые явления: выделение пузырьков газа и образование крас-
красно-бурого осадка меди. Приведите уравнения соответствующих реакций: анодной,
катодной и токообразующей;
3) в растворе какой соли CuSO4 или СиС12 быстрее появляется осадок меди? Что
является тому причиной? Почему водные растворы этих солей имеют кислотную
680
Коррозия и защита металлов и сплавов
реакцию среды? Ответ подтвердите уравнениями протекающих реакций (диссоциа-
(диссоциации, гидролиза и др.);
Таблица Л. 11И
Результаты эксперимента
№
про-
пробирки
1
2
3
4
Исходные данные
Корродирующий
образец
Алюминиевый
или стальной
образец в виде
пластины, от-
отрезка проволоки
Вид, состав и ки-
кислотность корро-
коррозионной среды
0,2н. водный
раствор HNO3,
рН<7
0,2н. водный
раствор H2SO4,
рН<7
0,2н. водный
раствор C11SO4,
рН<7
0,2н. водный
раствор СиС12,
рН<7
Наблюдения
Выделение пузырьков
газа, образование крас-
красно-бурого осадка Си
-
Уравнения анод-
анодной, катодной и
токообразующей
реакций корро-
коррозионного ГЭ
(-)А():
(+)К():
2
(-)А( ):
(+)К():
(~)А( ):
(+Ж():
(-)А( ):
(+Ж():
I
4) какова причина более интенсивного выделения пузырьков газа в пробирках 3 и 4?
Почему он выделяется не только на алюминии (железе), но и на образующемся
осадке меди?
5) почему алюминий (железо) не взаимодействуют с концентрированной серной и
дымящей азотной кислотами? Укажите состав образующейся на поверхности за-
защитной пленки;
6) объясните причину более энергичного взаимодействия пассивированного и не-
пассивированного алюминия (железа) с раствором СиС12, чем с раствором CuSO4.
Какова здесь роль СГ-ионов? Ответ подтвердите уравнениями реакций, протекаю-
протекающих между Al (Fe) и СиС12, А12Оз (FeO) и НС1. Предскажите действие на алюминий
растворов FeSO4 и FeCl2.
Опыт 12. Поведение пассивированного и активированного образцов же-
железа в водном растворе хлорида натрия. В данном опыте предлагается сравнить
поведение пассивированного и активированного образцов железа (или стали) в ус-
условиях электрохимической коррозии — в водном растворе хлорида натрия. Активи-
Активирование осуществляют обработкой 0,5М водным раствором соляной кислоты, пас-
пассивирование — дымящей азотной кислотой плотностью 1,5 г/см3. Для исследования
используют установку, аналогичную установке на рис. Л. 12.4. На панели закрепле-
закреплены два образца — графитовый и железный в виде стержней или пластин.
Внимание! Опыт проводится только в вытяжном шкафу
ЛР 12. Коррозия металлов и сплавов 681
Последовательность проведения:
1) наждачной бумагой зачистите, а затем промойте под струей водопроводной воды
закрепленные на панели стальной и графитовый стержни;
2) стряхнув избыток влаги, погрузите железный стержень на 2-3 мин в стакан с
0,5М водным раствором соляной кислоты;
3) по истечении времени извлеките его из раствора, промойте под струей водопро-
водопроводной воды, а затем стряхните ее избыток;
4) заполните закрепленную в штативе (/-образную стеклянную трубку 0,5М водным
раствором NaCl. В раствор левого ее колена добавьте 3-4 капли 0,5М водного рас-
раствора K3[Fe(CNN], правого — такое же число капель фенолфталеина. Указанные
реагенты являются индикаторами соответственно на Fe2+- и ОН~-ионы;
5) осторожно погрузите стержни в раствор, соответственно железный левого коле-
колена, графитовый — правого;
6) замкните внешнюю цепь через милливольтметр и запишите значение напряжения;
7) отключив милливольтметр от стержней, замкните их, т.е. внешнюю цепь, медным
или алюминиевым проводником и наблюдайте за происходящими в обоих коленах
изменениями в течение 3-5 мин;
8) по истечении времени закрепленные на панели стержни извлеките из раство-
раствора, промойте их под струей водопроводной воды, избыток влаги аккуратно
стряхните, а затем железный стержень поместите на 2-3 мин в стакан с дымя-
дымящей азотной кислотой;
9) по окончании извлеките его из кислоты, промойте под струей водопроводной
воды, избыток влаги аккуратно стряхните;
10) вылейте раствор из (/-образной трубки и замените его новым (см. п. 4 данного
опыта);
11) погрузите железный и графитовый стержни в (/-образную трубку (см. пп. 5-7
данного опыта) и повторите измерения и последующие наблюдения.
Обработка результатов:
1) исходные данные, результаты измерений, наблюдения, уравнение анодной, ка-
катодной, токообразующей и качественных аналитических на Fe2+- и ОН"-ионы реак-
реакций, а также схему коррозионного ГЭ занесите в табл. Л. 12.12;
2) какую роль выполняют стержни в используемой установке? Что из них анод,
что катод? Ответ аргументируйте. Приведите схему коррозионного ГЭ;
3) на основании наблюдений приведите уравнения возможных анодных и катодных
реакций для случаев активированного и пассивированного железных образцов;
4) объясните причину изменения окраски растворов у анода и катода, подтвердив
соответствующими уравнениями качественных аналитических реакций;
5) чем обусловлена разница в коррозионном поведении железного образца, под-
подвергшегося пассивированию? Объясните также различное поведение катода в паре с
активированным и пассивированным железным образцом. Свои ответы подтвердите
расчетами потенциалов катодных процессов с учетом рН коррозионной среды и
давления газов, равного атмосферному;
6) приведите уравнения токообразующих реакций и объясните причину различ-
различного напряжения коррозионного ГЭ для обоих случаев.
682
Коррозия и защита металлов и сплавов
Результаты эксперимента
Таблица Л А 2.12
Исходные данные
Вид, состав и
кислотность
коррозион-
коррозионной среды
0,5М вод-
водный раствор
NaCl,
рН = 7
Исследуемая электро-
электрохимическая система
Графитовый образец
во внешнем контакте
с активированным
железным
Графитовый образец
во внешнем контакте
с пассивированным
железным
Результаты измерений
Напряжение
коррозионного ГЭ U, В
Схема и уравнения анодной, катодной
и токообразующей реакций коррозионного ГЭ
<
(
(
(
-)А( ):
+)К():
С
-)А( ):
+Ж( ):
г
Наблюдения
Появление (синей,
малиновой) окраски,
выделение пузырь-
пузырьков газа в (левом,
правом) колене
Уравнения качественных
аналитических реакций
на ионы Fe2+n ОН"
ЛР 13. Защита от коррозии
Цель работы — знакомство с основными способами защиты от коррозии и их
практической реализацией в лабораторных условиях.
Опыт 1. Сравнение коррозионной стойкости легированных и нелегиро-
нелегированных материалов. В данном опыте на примере электрохимической коррозии
образцов из железа и его объемно-легированного хромом и никелем сплава — стали
в кислотном растворе сравниваются скорости коррозионных процессов и соответст-
соответственно коррозионная стойкость металлов и сплавов.
Последовательность проведения:
1) в две пробирки налейте по 2-4 мл 0,2н. водного раствора H2SO4;
2) в каждую добавьте по 2-4 капли раствора красной кровяной соли K3[Fe(CNN],
являющейся реактивом для обнаружения ионов Fe2+ в растворе;
3) в первую поместите зачищенную наждачной бумагой и промытую под струей
водопроводной воды железную полоску (или пластину), во вторую — стальную, но
без зачистки;
ЛР 13. Защита от коррозии
683
4) в течение 10 мин наблюдайте за происходящими изменениями в пробирках, не
трогая их. Отмечайте время начала выделения пузырьков газа и появления окраски;
5) по окончании сравните интенсивность окраски растворов и количество выде-
выделяющихся в единицу времени пузырьков газа в обеих пробирках.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций, результаты
расчетов и выводы занесите в табл. Л. 13.1;
Таблица Л. 13.1
Результаты эксперимента
Исходные данные
Испытуемый
образец
Железная полоска
Стальная полоска
Вид и состав кор-
коррозионной среды
0,2н. водный
раствор H2SO4
Коррозионный ГЭ
Уравнения анодной, катодной
и токообразующей реакций
(-)А:
(+)К:
I
Наблюдения
Выделение
газа (да, нет)
Окраска
раствора
Выводы
Скорость кор-
коррозии (п ^ г2)
Результаты расчетов
Величины электродных потенциалов
и ЭДС коррозионного ГЭ
Фа =9^= Фк=Ф2н1 =
Fe H2
Е = фк - Фа =
2) приведите уравнения анодной, катодной и токообразующей реакций;
3) используя уравнение Нернста, рассчитайте величины электродных потенциалов,
а по ним величину ЭДС коррозионного ГЭ, если активности ионов Н+ и Fe2+ в рас-
растворе составляют соответственно 1,5 и 0,05 моль/л, а рн = 1 атм;
4) напишите уравнение реакции обнаружения Ре2+-ионов;
5) объясните причину различного поведения полосок в растворе. Какие компоненты
входят в состав стали, какие из них являются легирующими? Поясните механизм
повышения коррозионной устойчивости стали;
6) по времени появления и интенсивности окраски раствора, а также количеству
выделяющихся пузырьков газа сделайте вывод о скорости коррозии испытуемых
образцов (r\ ^ г2), т.е. об их коррозионной стойкости.
Опыт 2. Влияние присутствия ингибитора в растворе на скорость элек-
электрохимической коррозии железа. В данном опыте изучается ингибирующее (за-
(замедляющее) действие уротропина (гексаметилентетрамина — (CH2NN4 ) на процесс
электрохимической коррозии железа в кислотном растворе.
Последовательность проведения:
1) в две пробирки налейте по 2-4 мл 0,2н. водного раствора H2SO4;
2) в каждую опустите несколько кусочков (стружек) железа или 1-2 канцелярских
скрепки;
684
Коррозия и защита металлов и сплавов
3) содержимое пробирок нагрейте на пламени газовой горелки. Запишите наблюдения;
4) в одну из пробирок добавьте измельченную таблетку уротропина, содержимое
пробирки тщательно перемешайте интенсивным встряхиванием. Отметьте произо-
произошедшие изменения.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций, результаты
расчетов и выводы занесите в табл. Л. 13.2;
Результаты эксперимента
Таблица Л. 13.2
Исходные данные
Корродирующий
образец
Железо в виде
стружки, кусочков
или скрепки
Вид и состав
коррозионной среды
0,2н. водный раствор H2SO4
0,2н. водный раствор
H2SO4 с уротропином
Коррозионный ГЭ
Уравнения анодной, катодной
и токообразующей реакций
(-)А:
(+)К:
I
Наблюдения
Выделение
газа (да, нет)
Выводы
Скорость кор-
коррозии (г2 ^ Г\)
Результаты расчетов
Величины электродных потенциалов
и ЭДС коррозионного ГЭ
Фа =4>FjL= Фк=Ф2н1 =
Fe H2
Н2 Fe
2) приведите уравнения анодной, катодной и токообразующей реакций коррозион-
коррозионного ГЭ;
3) используя уравнение Нернста, рассчитайте величины электродных потенциалов,
а по ним величину ЭДС коррозионного ГЭ, если активности Н+- и Ре2+-ионов в рас-
растворе составляют соответственно 1,2 и 0,06 моль/л, а рн = 0,1 атм;
4) по составу, механизму защитного действия и условиям применения к какому
классу, виду, типу ингибиторов относится уротропин (CH2NN4? Каков механизм его
защитного действия?
5) по результатам испытаний сделайте вывод о скорости коррозии железа с ингиби-
ингибитором и без него (r2 ^ гх).
Опыт 3. Протекторная защита. В данном опыте рассматривается протек-
протекторная защита на примере электрохимической коррозии стали и свинца в кислотном
растворе. В качестве протектора используется образец из цинка.
Последовательность проведения:
1) в две пробирки налейте по 2-4 мл 0,2н. водного раствора H2SO4;
2) в каждую добавьте по 2-4 капли водного раствора красной кровяной соли
Кз[Ре(СИ)б], являющейся реактивом для обнаружения ионов Fe2+ в растворе;
ЛР 13. Защита от коррозии
685
3) в первую поместите стальную полоску, во вторую — сборку, состоящую из со-
соединенных (склепанных, сваренных, спаянных) вместе аналогичной стальной и
цинковой полосок, т.е. находящихся в контакте;
4) запишите наблюдения (выделение газа, появление окрашивания и др.) и измене-
изменения, произошедшие с течением времени (через 5-10 мин) в обеих пробирках;
5) аналогичный эксперимент (по указанию преподавателя) проведите с полосками
из свинца и свинца, находящегося в контакте с цинком, выполняя операции пп. 1 -4
данного опыта. В качестве коррозионной среды возьмите 0,5н. водный раствор
СН3СООН, а реактива для обнаружения ионов РЬ2+ — водный раствор KJ.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и расчеты зане-
занесите в табл. Л. 13.3;
Таблица Л.13.3
Результаты эксперимента
№
1
2
1
2
Исходные данные
Исследуемый
образец
Стальная полоска
Спаянные сталь-
стальная и цинковая
полоски
Вид и состав
коррозионной среды
0,2н. водный рас-
раствор H2SO4
Стандартный
потенциал (р°, В
...(Mg27Mg)
...(Fe2+/Fe)
...(Zn2+/Zn)
Коррозионный ГЭ
Схема
Уравнения электродных реакций
(-)А( ):
(+Ж( ):
(-Ж ):
(+Ж( ):
Наблюдения
Выделение
газа (да,нет)
Окраска
раствора
Расчеты
Стандартная ЭДС, В
Zn-Fe
Mg-Fe
Е°=
Е°=
2) объясните причину различий в поведении исследуемых образцов;
3) напишите уравнение реакции обнаружения Ре2+-ионов;
4) приведите схемы коррозионных гальванических элементов и напишите уравне-
уравнения электродных реакций;
5) какой из протекторов, цинк или магний, эффективнее для защиты изделий из желе-
железа? Ответ аргументируйте расчетами значений стандартной ЭДС пар Zn-Fe и Mg-Fe.
Опыт 4. Защита внешним потенциалом. В данном опыте рассматривается
способ защиты, в основе которого лежит катодная поляризация, осуществляемая от
внешнего источника постоянного тока. Используемая для этой цели установка
(рис. Л. 13.1) представляет собой стеклянный сосуд / с двумя электродами (желез-
(железным и угольным в виде стержней, полос или пластин), закрепленными на панели 2.
Роль защищаемой конструкции выполняет железный электрод 4, а вспомогательно-
вспомогательного анода — угольный 3, которые помещены в 0,5н. водный раствор NaCl 5, имити-
имитирующий токопроводящую коррозионную среду.
686
Коррозия и защита металлов и сплавов
Последовательность прове-
проведения:
1) соберите установку, изображенную на рис.
Л. 13.1, в которой железный стержень (катод)
защищен внешним потенциалом;
2) в пробирку (до V3 объема) и стакан (до V2
объема) налейте 0,5н. водный раствор NaCl,
добавьте туда по 2-4 капли раствора красной
кровяной соли K3[Fe(CNN], являющейся ре-
реактивом для обнаружения ионов Fe2+ в рас-
растворе;
3) железные стержни (один расположен на
панели вместе с угольным, другой — отдель-
отдельно) зачистите наждачной бумагой и затем
промойте под струей водопроводной воды;
4) электроды, расположенные на панели,
подсоедините: железный к отрицательному, а
угольный к положительному полюсам внеш-
внешнего источника постоянного тока, затем
опустите их в стакан с раствором;
5) отдельный железный стержень опустите в
пробирку с раствором. Он будет служить
образцом сравнения (без защиты). Одновременно включите рубильник источника
постоянного тока. Опишите происходящие изменения (выделение газа, окрашива-
окрашивание и др.). После этого рубильник отключите, электроды извлеките из раствора.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 13.4;
Рис. Л.13.1. Установка для осуще-
осуществления катодной защиты внеш-
внешним потенциалом:
1 — стеклянный стакан; 2 — панель; 3 —
угольный стержень-анод; 4 — желез-
железный стержень-катод; 5 — токопрово-
дящая коррозионная среда @,5н. вод-
водный раствор NaCl)
Результаты эксперимента
Таблица Л. 13.4
Исходные данные
Исследуемый
образец
Железный
стержень
Железный
стержень с
защитой
Вид и состав кор-
коррозионной среды
0,5н. водный
раствор NaCl
Наблюдения
Выделение
газа (да, нет)
Окраска
раствора
Выводы
Электрохимическая кор-
коррозия (протекает, нет)
Уравнения протекающих анодной, катодной и токообразующей реакций
при электрохимической коррозии
(-)А:
(+)К:
I
при защите внешним потенциалом
(+)А( ):
(~)К( ):
Z
ЛР 13. Защита от коррозии
687
2) где и почему быстрее появляется синяя окраска? Приведите соответствующее
уравнение реакции;
3) уравнениями опишите анодную, катодную и токообразующую реакции, проте-
протекающие при электрохимической коррозии железа в нейтральной среде;
4) какому электрохимическому процессу аналогична защита внешним потенциа-
потенциалом? Почему не появляется окраска в этом случае? Подтвердите это аналогичными
уравнениями;
5) сделайте выводы об эффективности защиты.
Опыт 5. Катодные и анодные металлические защитные покрытия. В дан-
данном опыте на примере электрохимической коррозии оцинкованного и луженого
(покрытого оловом) железа в кислотном растворе рассматриваются механизмы за-
защитного действия катодных и анодных покрытий.
Последовательность проведения:
1) в две пробирки налейте по 2-4 мл 0,2н. водного раствора H2SO4;
2) в каждую добавьте по 2-4 капли раствора красной кровяной соли K3[Fe(CNN],
являющейся реактивом для обнаружения ионов Fe2+ в растворе;
3) в первую поместите полоску луженого железа, во вторую — оцинкованного;
4) через десять минут сравните окраску растворов в обеих пробирках. Отметьте дру-
другие изменения, происходящие в них;
5) аналогичный эксперимент (по указанию преподавателя) проведите, взяв в качест-
качестве коррозионной среды 0,5н. водный раствор NaOH и выполнив пп. 1-4 данного
опыта.
Обработка результатов:
1) исходные данные, наблюдения, уравнения протекающих реакций и выводы зане-
занесите в табл. Л. 13.5;
Результаты эксперимента
Таблица Л. 13.5
№
1
2
№
1
2
Исходные данные
Исследуемый
образец
Полоска луже-
луженого железа
Полоска оцинко-
оцинкованного железа
Вид и состав
коррозион-
коррозионной среды
0,2н. вод-
водный раствор
H2SO4
Потенциал металла (р°, В
покрытия
...(Sn2+/Sn)
...(Zn2+/Zn)
Коррозионный ГЭ
Схема
Уравнения электродных
реакций
(-)А( ):
(+Ж( ):
(-)А( ):
(+Ж( ):
основы
...(Fe2+/Fe)
Наблюдения
Окраска
раствора
Выделение
газа (да, нет)
Выводы
Вид
покрытия
Механизм защит-
защитного действия
688
Коррозия и защита металлов и сплавов
2) объясните причину различий в поведении образцов;
3) напишите уравнение реакции обнаружения Ре2+-ионов;
4) приведите схемы и уравнения электродных реакций коррозионных ГЭ;
5) по значениям стандартных электродных потенциалов и на основании результатов
коррозионных испытаний определите вид покрытия (катодное, анодное) и механизм
его защитного действия (механический, электрохимический);
6) перечислите основные требования, предъявляемые к покрытиям.
Опыт 6. Цинкование меди. Цинкованием называют процесс нанесения по-
покрытий из цинка на поверхность изделий. В данном опыте химическим методом с
использованием щелочного цинкатного электролита предлагается изготовить такое
покрытие на медном изделии и определить его вид (катодное или анодное).
Внимание! Опыт проводится в вытяжном шкафу
Последовательность проведения:
1) в фарфоровый тигель, наполненный до половины объема 2,8н. водным раствором
NaOH, опустите медную пластину или монету;
2) насыпьте немного цинкового порошка и содержимое прокипятите в течение
10-15 мин;
3) затем образец тигельными щипцами извлеките из раствора, промойте под струей
водопроводной воды, осушите фильтровальной бумагой и опишите его внешний
вид: цвет и состояние (блестящая, матовая, шероховатая, пористая и др.) поверхности.
Обработка результатов:
1) исходные данные, условия проведения опыта, уравнения протекающих реакций и
выводы занесите в табл. Л. 13.6;
Результаты эксперимента
Таблица Л. 13.6
Исходные данные и условия проведения опыта
Обрабатываемый
образец
Медная пластина
или монета
Состав
раствора
2,8н. раствор
NaOH с Zn
порошком
l кип»
МИН
10-15
Г,°С
Т
1 кип
Стандартный
потенциал (р°, В
...(Zn27Zn)
...(Cu27Cu)
Выводы
Вид покрытия и
механизм защитного
действия
Уравнения протекающих анодной, катодной и токообразующей реакций
при растворении цинка
«А:
(+)К:
I
при цинковании
(-)A(Zn):
(+Ж(Си):
I
2) по значениям стандартных электродных потенциалов определите, к какому виду
относится цинковое покрытие на меди (анодное, катодное) и каков механизм его
защитного действия (механический, электрохимический);
ЛР 13. Защита от коррозии
689
3) объясните пути попадания цинка в раствор. Приведите уравнения соответствую-
соответствующих реакций. Помните, что цинк и его соединения (оксид, гидроксид) обладают
амфотерными свойствами;
4) с помощью уравнений опишите и реакции, протекающие при цинковании.
Опыт 7. Химическое никелирование стали. Никелированием называют
процесс нанесения покрытий из никеля на поверхность изделий. В данном опыте
предлагается изготовить химическим методом такое покрытие на образце из стали и
определить его вид (катодное, анодное) путем проведения коррозионных испыта-
испытаний. В основе метода лежит следующая окислительно-восстановительная реакция:
Ni(CH3COOJ
окислитель
2NaH2PO2 + 2Н2О
Nil + 2NaH2PO3
Н2Т
2СН3СООН
восстановитель
Внимание! Опыт проводится в вытяжном шкафу
Последовательность проведения:
1) стальной образец (в виде полоски или пластины) зачистите наждачной бумагой;
2) поместите его на 30 мин в ванну (стакан или какую-либо другую емкость) с
раствором для никелирования состава, г/л: 15(Ni(CH3COOJ), 30(NaH2PO2),
15(NH2CH2COOH — глицин), нагретым предварительно на песчаной бане до
температуры 97...98 °С. Опишите происходящие изменения (выделение газа,
изменение цвета и др.);
3) по окончании обработки образец тигельными щипцами извлеките из раствора,
промойте под струей водопроводной воды, а затем осушите фильтровальной бума-
бумагой. Опишите внешний вид обработанного образца: цвет и состояние (блестящая,
матовая, шероховатая, пористая и др.) его поверхности;
4) проведите коррозионные испытания никелированного и контрольного (без по-
покрытия) образцов по способу, описанному в опытах 3 и 5.
Обработка результатов:
1) условия проведения опыта и наблюдения занесите в табл. Л. 13.7;
Результаты эксперимента
Таблица Л.13.7
Условия проведения опыта
Обрабатываемый
образец
Стальная полоска
или пластина
/, мин
Т,°С
Наблюдения
Изменение внешнего
вида образца
Выделение газа
(да, нет)
2) результаты проведенных коррозионных испытаний представьте в форме, анало-
аналогичной табл. Л. 13.5;
3) ответьте на вопросы и выполните требования пп. 2—6 обработки результатов
опыта 5.
690
Коррозия и защита металлов и сплавов
Опыт 8. Воронение стали. В данном опыте химическим оксидированием
предлагается изготовить оксидное покрытие на стали. Свое название способ полу-
получил по цвету образующегося покрытия (цвета «воронова крыла» — черного, синего
или коричневого), которое состоит в основном из Fe3O4 (FeO • Fe2O3).
Внимание! Опыт проводится в вытяжном шкафу
Последовательность проведения:
1) стальной образец (в виде пластины или полоски) предварительно обезжирьте.
Для этого поместите его на 10 мин в ванну (стакан или какую-либо другую емкость)
с горячим раствором состава, г/л: 15(NaOH), 25(Na2CO3), 10(Na3PO4);
2) по истечении времени образец тигельными щипцами извлеките и промойте под
струей водопроводной воды. Опишите его внешний вид: цвет и состояние (блестя-
(блестящая, матовая, шероховатая, пористая и др.) поверхности;
3) после этого образец поместите на 10 мин в ванну с раствором для воронения со-
состава, г/л: 20(NaNO2), 50(NaNO3), 700(NaOH). Процесс проводите при температуре
138...142°С;
4) по окончании образец тигельными щипцами извлеките из раствора, промойте под
струей водопроводной воды, а затем осушите фильтровальной бумагой. Опишите
его внешний вид (см. п. 2);
5) проведите сравнительные испытания на коррозионную стойкость вороненого и
контрольного образцов. Для этого на их поверхность нанесите по одной капле 0,1н.
водного раствора C11SO4. Отметьте время появления на ней красно-бурых осадков
меди.
Обработка результатов:
1) исходные данные, результаты измерений, уравнения протекающих реакций и
выводы занесите в табл. Л. 13.8.
Результаты эксперимента
Таблица Л. 13.8
Исходные данные
Испытуемый
образец стали
Вороненый
Контрольный
Внешний вид
образца
Результаты измерений
Время появления
осадка Си t, мин
Выводы
Коррозионная стойкость
(выше, ниже)
—
Уравнения протекающих реакций
при воронении
при коррозионных испытаниях
2) по времени появления красно-бурого осадка меди на поверхности сделайте вывод
о том, у какого из образцов выше коррозионная стойкость. Объясните причину их
различного поведения;
3) приведите уравнения реакций, протекающих при воронении и испытании на кор-
коррозионную стойкость образцов. Определите степень окисления железа в Fe3O4.
ЛР13. Защита от коррозии
691
Опыт 9. Химическое оксидирование магния и его сплавов. В данном
опыте предлагается провести оксидирование образца из магния или его сплава в
растворе окислителя, в результате которого на его поверхности образуется оксидная
пленка, и сравнить коррозионную стойкость оксидированного и неоксидированного
(контрольного) образцов.
Последовательность проведения:
1) образец (в виде пластины или полоски) из магния или его сплава предварительно
обезжирьте двух- трехкратной протиркой ватным тампоном или матерчатой салфеткой,
пропитанных одним из растворителей: ацетоном, уайт-спиритом, бензином и др.;
2) после испарения растворителя образец промойте под струей водопроводной во-
воды. Опишите внешний вид образца: цвет и состояние (блестящая, матовая, шеро-
шероховатая, пористая и др.) его поверхности;
3) образец поместите в ванну с одним из двух водных растворов для оксидирования
состава, г/л: первый — 200(СЮ3), второй — 30...50(K2Cr2O7), 8...12(KA1(SO4J),
3...5(СН3СООН). Оксидирование ведут в течение 10 мин при температуре 50...60 °С в
растворе 1 и при температуре 20.. .30 °С в растворе 2;
4) по окончании процесса образец тигельными щипцами извлеките из раствора,
промойте под струей водопроводной воды, а затем осушите фильтровальной бума-
бумагой. Опишите внешний вид образца (см. п. 2);
5) проведите сравнительные испытания на коррозионную стойкость оксидирован-
оксидированного и неоксидированного (контрольного) образцов, поместив их в подкисленный
раствор CuSO4, содержащий NaCl. Отметьте происходящие изменения.
Обработка результатов:
1) исходные данные, результаты измерений, уравнения протекающих реакций и
выводы занесите в табл. Л. 13.9;
Таблица Л. 13.9
Результаты эксперимента
Исходные данные
Испытуемый образец магния
Вид
Оксидированный
Неоксидированный
(контрольный)
Цвет и состояние
поверхности
Результаты измерений
Время появления
осадка Си t, мин
Выводы
Коррозионная стойкость
(выше, ниже)
—
Уравнения протекающих реакций
при оксидировании
при коррозионных испытаниях
MgO + H2SO4 ->
Mg + H2SO4 ->
Mg + C11SO4 ->
2) по времени появления красно-бурого осадка меди сделайте вывод о том, у какого
из образцов выше коррозионная стойкость и почему?
692
Коррозия и защита металлов и сплавов
12...15В
т
К
2 2в
3) приведите уравнения реакций, протекающих при оксидировании и испытании на
коррозионную стойкость образцов.
Опыт 10. Анодирование алюминия. Анодное окисление алюминия прово-
проводят в растворах серной, фосфорной и др. кислот. При этом процесс описывается
уравнением:
(+) A (Al): 2A1 + ЗН2О -+ А12О3 + 6Н+ + бе
Образующееся на поверхности покрытие (пленка) из А12О3 достигает толщины до
500 мкм и состоит из двух слоев: плотного барьерного, толщиной 0,01-0,10 мкм и
расположенного непосредственно на поверхности металла, и внешнего пористого
толщиной до 200—400 мкм. Рост покрытия происходит со стороны металла, причем
увеличивается в толщине только пористый слой. Для повышения защитных свойств
покрытия изделия из алюминия дополнительно обрабатывают вначале паром или
горячей водой, а затем разбавленным горячим раствором солей-пассиваторов
(Na2Cr04, Na2Cr207). Такой процесс получил название наполнения покрытия (плен-
(пленки). При обработке паром образуется гидроксид
алюминия, а в растворах солей-пассиваторов — бо-
более устойчивое соединение типа (А1ОJСгО4. В ре-
результате происходит заполнение ими пор и закры-
закрытие внешнего слоя.
Введение в раствор для анодирования краси-
красителей позволяет получать оксидные покрытия раз-
различного цвета: от черного до золотого.
В данном опыте предлагается провести ано-
анодирование образца из алюминия, а затем испытать
его на коррозионную стойкость. Для анодирования
используют установку, аналогичную установке опы-
опыта 4 (рис. Л. 13.2).
Внимание! Опыт проводится в вытяжном
шкафу
Последовательность проведения:
1) обезжиренный (см. пп. 1-2 опыта 9) и промытый
под струей водопроводной воды алюминиевый об-
образец (в виде пластины или полоски) подсоедините
в качестве анода к положительному полюсу внешнего
источника постоянного тока с напряжением 12... 15 В,
а свинцовый — в качестве катода к его отрицатель-
отрицательному полюсу (см. рис. Л. 13.2);
2) погрузите электроды на 4-6 см в электролит Dн.
водный раствор H2SO4), которым заполнен электро-
электролизер объемом 100-300 мл — химический стакан
или ^/-образный сосуд (рис. Л. 13.2);
3) проверив контакты и включив рубильник, про-
Рис. JI.13.2. Установка для
анодного окисления (аноди-
(анодирования) алюминия:
1 — стеклянная ^/-образная труб-
трубка; 2 — алюминиевый образец-
анод; 3 — металлический шта-
штатив с лапкой; 4 — электролит
Dн. водный раствор H2SO4);
5 — свинцовый образец-катод;
6 — панель; 7 — источник по-
постоянного тока с напряжением
12...15В
ЛР 13. Защита от коррозии
693
пускайте в течение 15-20 мин ток при анодной плотности, равной 100...200 А/м2;
4) по истечении времени отключите рубильник, извлеките алюминиевый образец,
промойте его под струей водопроводной воды и опишите внешний вид: цвет и со-
состояние (блестящая, гладкая, шероховатая, пористая и др.) его поверхности;
5) анодированный образец опустите на 5... 10 мин в один из растворов анилинового
красителя (табл. Л. 13.10а);
Таблица Л. 13.10а
Состав растворов для окрашивания анодированного алюминия
Цвет покрытия
Синий
Желтый
Красный
Под «золото»
Черный
Состав раствора
1%-ный (масс.) водный раствор соответственно
анилинового голубого, желтого или красного
Водный раствор состава, г/л: 0,1 (красителя оран-
оранжевого марки «2Ж»), 0,001 (красителя кислотного
черного марки «М»), 0,05 (соды кальцинированной)
Водный раствор состава, % (масс): 0,5 (анилиново-
(анилинового черного красителя для шерсти), 0,5 (анилинового
черного красителя марки «ФФ»)
Температура
раствора Г, °С
20-25
60
80-90
6) по истечении времени образец тигельными щипцами извлеките, промойте под
струей водопроводной воды, осушите фильтровальной бумагой и снова опишите
внешний вид (см. п. 4);
7) проведите сравнительные испытания на коррозионную стойкость неанодирован-
ного, анодированного и анодированного с пропиткой анилиновым красителем об-
образцов, для чего образцы поместите на 10 мин в подкисленный серной кислотой
раствор C11SO4, содержащий NaCl. Отметьте различия в поведении образцов (начало
выделения газа, время появления красно-бурого осадка меди и др.).
Обработка результатов:
1) исходные данные, результаты измерений, уравнения протекающих реакций и
выводы занесите в табл. Л. 13.106;
Результаты эксперимента
Таблица Л. 13.106
Исходные данные
Испытуемый образец алюминия
Вид
Контрольный
(неанодированный)
После анодирования
После анодирования и
пропитки красителем
Цвет и состояние
поверхности
Результаты измерений
Время появления
осадка Си t, мин
Выводы
Коррозионная стойкость
(выше, ниже)
—
694
Коррозия и защита металлов и сплавов
Окончание табл. Л. 13.106
Уравнения протекающих реакций
при анодировании
(+)А(А1):
(-)К(РЪ):
S
при коррозионных испытаниях
А12О3 + H2SO4 ->
Al + H2SO4 ->
Al + CuSO4 -»
2) по времени появления осадка меди сделайте вывод о том, у какого из образцов
выше коррозионная стойкость и почему?
3) приведите уравнения процессов, происходящих при оксидировании и испытании
на коррозионную стойкость образцов.
Опыт 11. Хроматирование цинка. Хроматированием называют процесс
создания покрытий из малорастворимых хроматов и дихроматов на поверхности
металлов. В данном опыте предлагается получить покрытие в виде ZnCrO4 на по-
поверхности цинкового образца (гранулы или пластины) и оценить его защитные
свойства при электрохимической коррозии в кислотном растворе.
Обычно хроматирование цинковых и оцинкованных изделий осуществляют в
растворе состава, г/л: 12...17(H2SO4), 200(Na2Cr2O7 • 2Н2О) в течение 5-30 с. От
времени обработки зависит окраска пленки. При выдержке в течение 5 с — цвет
пленки радужный с зеленоватым оттенком. При дальнейшем увеличении времени
цвет меняется от желтого до коричневого.
Последовательность проведения:
1) налейте в пробирку 2-4 мл 0,1 н. водного раствора НС1;
2) поместите в нее гранулу или пластину цинка. Запишите наблюдения: выделение
пузырьков газа, изменение цвета и состояния (блестящая, матовая, шероховатая,
пористая и др.) поверхности;
3) когда начнется интенсивное выделение газа, добавьте несколько кристалликов
К2Сг207 или Na2Cr207 и содержимое пробирки тщательно перемешайте интенсив-
интенсивным встряхиванием. После этого отметьте произошедшие изменения.
Обработка результатов:
1) исходные данные, уравнения протекающих реакций и выводы занесите в табл.
Л.13.11;
2) какой газ выделяется при травлении образца? Почему он выделяется не сразу?
Приведите уравнения соответствующих реакций;
3) объясните причину замедления и последующего практически полного прекраще-
прекращения выделения водорода после добавления дихромата калия. Напишите уравнение,
иллюстрирующее процесс хроматирования. Обладает ли защитными свойствами
данное покрытие при электрохимической коррозии? Сделайте выводы о механизме
его защитного действия.
ЛР13. Защита от коррозии
695
Таблица Л. 13\11
Результаты эксперимента
Исходные данные
Вид и состав раствора
для травления
0,1н. водный
раствор НС1
для хроматирования
0,1н. водный
раствор НС1,
содержащий К2Сг207
Исследуемый образец цинка
Вид
После травления (до
хроматирования)
После хроматирова-
хроматирования
Уравнения протекающих реакций
при травлении
ZnO + HC1 ->
(-)А:
(+)К:
I
при хроматировании
К2Сг207 +Н2О ?=>
Zn +НС1 ->
Zn2++ СЮ2" -»
Zn + HC1 + К2Сг207 + Н2О ->
Цвет и состояние
поверхности
Выводы
Механизм защитного
действия
Опыт 12. Фосфатирование стали. Фосфатированием называют процесс
создания покрытий из малорастворимых фосфатов на поверхности металлов. В дан-
данном опыте предлагается получить покрытие из МНРО4 и М3(РО4J на стали, где
М — Fe, Mn, Zn. Для этого стальное изделие обрабатывают горячим раствором од-
нозамещенных фосфатов марганца, железа, цинка (М(Н2РО4J). В результате на по-
поверхности образуется пористое кристаллическое покрытие с высокой адгезией из
нерастворимых двух- и трехзамещенных фосфатов:
М(Н2РО4J ±± МНРО4| + Н3РО4
ЗМ(Н2РО4J +± М3(РСШ + 4Н3РО4
Для ускорения процессов разложения в раствор вводят ускорители в виде ионов
Си2+, С1Оз~, NO3~. Образованию покрытия предшествуют процессы ионизации желе-
железа на анодных участках: (-) A: Fe —>Fe2+ + 2e и выделения водорода на катодных:
(+) К: 2Н+ + 2е-»Н2|. Железо, переходящее в раствор, связывает фосфорную
кислоту, сдвигая тем самым равновесия приведенных выше реакций вправо (в
сторону продуктов). Фосфатные покрытия не обеспечивают надежной защиты
от коррозии, поэтому их используют преимущественно как грунт под лакокра-
лакокрасочные покрытия.
Последовательность проведения:
1) обезжиренный (см. пп. 1-2 опыта 8, обратите внимание на цвет и состояние его
поверхности) стальной образец (в виде пластины или полоски) поместите на 10 мин
в ванну (стакан или какую-либо другую емкость) с электролитом для фосфатирова-
696
Коррозия и защита металлов и сплавов
ния — в водный раствор состава, г/л: 3 (препарата «Мажеф» — Fe(H2PO4J +
+ 10Mn(H2PO4J), 2(CuO), нагретый до температуры 98 °С;
2) по истечении времени образец тигельными щипцами извлеките, промойте под
струей водопроводной воды, а затем осушите фильтровальной бумагой. Опишите
его внешний вид, указав цвет и состояние (блестящая, гладкая, шероховатая, порис-
пористая и др.) его поверхности.
Обработка результатов:
1) исходные данные и уравнения протекающих реакций занесите в табл. Л. 13.12;
Таблица Л13.12
Результаты эксперимента
Исходные данные
Вид и состав раствора
для фосфатирования
Водный раствор со-
состава, г/л: 3 (препа-
(препарата «Мажеф»),
2(СиО)
Исследуемый образец стали
Вид
После
обезжиривания
После
фосфатирования
Цвет и состояние
поверхности
Уравнения реакций, проте-
протекающих при фосфатировании
и предшествующих ему
2) какой газ выделяется при фосфатировании? Подтвердите соответствующими
уравнениями;
3) приведите уравнения, иллюстрирующие процесс фосфатирования. Какова роль
2+
ионов Си в растворе?
4) обладает ли фосфатное покрытие защитными свойствами?
IX
основы
ЗАНЯТИЙ
ПРАКТИЧЕСКИХ
Правила поведения
и порядок работы
в химической
лаборатории
Реактивы, посуда
и оборудование
учебной химической
лаборатории
Техника эксперимента
Лабораторные работы наряду с лекциями и
практическими занятиями являются неотъем-
неотъемлемой частью любого курса химии. Благодаря
этой форме занятий при изучении общей хи-
химии в техническом университете студенты на
практике знакомятся с лабораторным обору-
оборудованием, приборами и установками, а также
осваивают основные несложные методы про-
проведения эксперимента и изучают химические
свойства веществ.
При домашней подготовке к лабораторной
работе студент должен хорошо изучить тео-
теорию по теме, используя учебники, методиче-
методические разработки и конспекты лекций. При вы-
выполнении лабораторной работы необходимо
четко представлять себе ее цель и этапы, при
возникновении вопросов следует обращаться к
преподавателю.
20 ¦ Правила поведения и порядок
ццг] работы в химической лаборатории
В химической лаборатории всегда присутствуют источники повы-
повышенной опасности, к которым относятся сами химикаты, часто яв-
являющиеся агрессивными, ядовитыми, пожаро- или взрывоопасными,
а также электро- и газовые приборы. В связи с этим все работающие
должны строго выполнять правила техники безопасности. Эти пра-
правила преподаватель специально излагает на вводном занятии.
20.1. Общие требования
При выполнении опытов в химической лаборатории нужно
соблюдать следующие правила.
1. Содержать рабочее место в чистоте и порядке, проводить
эксперименты в чистой посуде.
2. Сухие вещества отбирать чистой ложечкой или шпателем.
При отливании жидкостей из склянок этикетка должна находиться
на верхней стороне емкости (под рукой) во избежание загрязнения
стекающими каплями.
3. Избыток твердых и жидких реактивов нельзя возвращать в
склянки, где они хранятся.
4. Пробки и крышки от банок с реактивами следует класть на
стол в перевернутом виде, т.е. поверхностью, не соприкасающейся
с веществом. При этом важно не перепутать пробки от разных ем-
емкостей.
5. Реактивы общего пользования (в том числе стоящие в вы-
вытяжном шкафу) нельзя уносить на свое рабочее место.
6. Если не конкретизируется количество вещества, необхо-
необходимое для проведения опыта, то твердое вещество следует брать в
количестве, покрывающем дно пробирки, а жидкости наливать
15.. .20 % от объема пробирки.
7. Остатки металлов после проведения опытов нельзя выбра-
выбрасывать в раковину во избежание засорения гидрозатвора, их нужно
собирать в специальную банку. Остатки концентрированных ки-
кислот, щелочей, горючих веществ (например, органических раство-
растворителей) сливать в склянки, стоящие в вытяжном шкафу. Дорогие
реактивы (например, соли драгоценных металлов) не выбрасывать,
а собирать в специальную емкость.
8. Отходы (битую посуду, использованные фильтры, индика-
индикаторную бумагу, спички и др.) выбрасывать в урны для мусора.
20.2. Техника безопасности и противопожарная безопасность 699
9. Запрещается выполнять опыты, не описанные в руководстве к лабо-
лабораторной работе.
10. Во избежание загрязнения и порчи одежды реактивами в лаборато-
лаборатории следует находиться в халате. При работе с концентрированными кисло-
кислотами, щелочами, щелочными металлами и другими веществами, обладающи-
обладающими повышенной химической активностью, применять защитные очки.
11. Для ополаскивания вымытой посуды и приготовления водных рас-
растворов нужно использовать дистиллированную воду.
20.2. Техника безопасности и противопожарная безопасность
Основные правила техники безопасности и противопожарной безо-
безопасности при работе в химической лаборатории сводятся к следующим по-
положениям.
1. Нельзя загромождать проходы между столами и рабочие места
лишними предметами.
2. Опыты с ядовитыми, неприятно пахнущими веществами, а также с
концентрированными растворами кислот и щелочей нужно проводить в вы-
вытяжном шкафу; с легковоспламеняющимися жидкостями — на безопасном
расстоянии от открытого огня и нагревательных приборов.
3. При переливании реактивов, нагреве жидкостей или сплавлении
веществ запрещается наклоняться над отверстиями сосудов во избежание
попадания брызг в глаза, на кожу или на одежду.
4. При нагреве раствора в пробирке ее отверстие нужно направлять в
противоположную от себя и работающих вблизи соседей сторону, так как
при местном перегреве жидкости возможен ее выброс.
5. При идентификации веществ по запаху нельзя близко наклоняться
над склянкой и сильно вдыхать пары; сосуд нужно держать на расстоянии, а
пары вещества вместе с воздухом направить к себе легким взмахом ладони и
осторожно понюхать.
6. В лаборатории запрещается принимать пищу, курить, громко разгова-
разговаривать, брать вещества или дотрагиваться до них руками и пробовать на вкус.
7. Разбавляя концентрированные растворы кислот, особенно серной,
следует приливать всегда небольшими порциями при перемешивании ки-
кислоту в воду, но не наоборот.
8. При работе со щелочными металлами необходимо избегать их кон-
контакта с водой (возможность самовоспламенения!). Остатки этих металлов
нельзя выбрасывать в урну, их следует сдать лаборанту.
9. Все работы с агрессивными веществами, например, с твердыми ще-
щелочами, фосфорным ангидридом, а также с концентрированными раствора-
700 20. Правила поведения и порядок работы в химической лаборатории
ми кислот и щелочей необходимо проводить с использованием инди-
индивидуальных средств защиты (защитных очков и резиновых перчаток). Рабо-
Рабочее место следует тщательно очищать от остатков этих веществ.
10. При включении электроприбора в сеть соединительный шнур сна-
сначала подключить к прибору, а затем к розетке. При отключении прибора
операции со шнуром проводить в обратном порядке.
11. Следует осторожно обращаться с открытым пламенем и нагрева-
нагревательными приборами. При зажигании газовой горелки сначала необходимо
поднести горящую спичку или зажигалку к горелке, а затем открыть кран.
12. Для тушения пожара в лаборатории использовать углекислотные
огнетушители, песок, воду, листовой асбест, войлок (кошму).
13. При возникновении очага возгорания необходимо обесточить ра-
рабочее место, отключить газовый стояк и гасить огонь имеющимися средст-
средствами пожаротушения в зависимости от природы очага:
воспламенившиеся горючие жидкости нельзя гасить водой, для туше-
тушения применять твердую углекислоту, песок или лист асбеста;
горящие щелочные металлы нельзя гасить углекислотой или водой,
для ликвидации возгорания применять сухой песок;
горящий фосфор гасят водой или мокрым песком.
14. При невозможности ликвидации очага возгорания собственными
силами следует без промедления вызывать пожарную команду.
20*3. Оказание помощи при несчастных случаях
В любой химической лаборатории по правилам техники безопасно-
безопасности должна находиться медицинская аптечка с утвержденным минималь-
минимальным набором лекарств, необходимых для оказания первой помощи при
несчастных случаях. Большинство травм в лаборатории происходит не от
незнания правил техники безопасности, а от пренебрежения ими по раз-
различным причинам. Если повреждение (чаще всего это порезы стеклом,
химические или термические ожоги) легкое, пострадавшему оказывается
первая медицинская помощь силами сотрудников лаборатории. В серьез-
серьезных случаях травмирования необходимо после оказания первой помощи
срочно вызвать врача.
При порезах стеклом из раны удаляют осколки, промывают ее рас-
раствором перманганата калия, пероксида водорода или спиртом, края обраба-
обрабатывают спиртовым раствором йода. Затем накладывают повязку из бинта.
При ожоге кожи концентрированными растворами кислот место ожо-
ожога промывают большим количеством воды или 2%-ным раствором гидро-
гидрокарбоната натрия. Затем накладывают повязку из ваты, смоченной 3%-ным
20.4. Рабочий журнал 701
раствором перманганата калия или раствором танина. Если на кожу попала
концентрированная щелочь, место ожога промывают большим количеством
воды или 2%-ным раствором уксусной кислоты, после чего накладывают
такую же повязку, как при ожоге концентрированными растворами кислот.
При промывке мест химических ожогов следует использовать холодную или
имеющую комнатную температуру воду. Применение горячей воды может
усугубить ожог.
При попадании капель кислоты или щелочи на слизистую оболочку
глаза необходимо сразу же промыть поврежденный глаз большим количест-
количеством воды комнатной температуры, после чего закапать 1-2 капли касторо-
касторового масла и обратиться к окулисту.
При термическом ожоге горячими жидкостями или твердыми предметами
(металлы, стекло и др.) пострадавший участок кожи смачивают 3%-ным рас-
раствором перманганата калия, а затем накладывают повязку с мазью от ожогов.
В случае отравления ядовитыми газами (хлор, пары брома, сероводо-
сероводород, угарный газ и др.) пострадавшего следует немедленно вывести на све-
свежий воздух и обратиться к врачу.
20.4. Рабочий журнал
Все проводимые опыты, описание действий и наблюдений эксперимен-
экспериментатора, расчеты, графики и выводы по выполняемой лабораторной работе
должны быть отражены в рабочем журнале. Для рабочего лабораторного
журнала удобно использовать общую тетрадь в клетку. В подписи журнала
указывают фамилию и инициалы студента, номер группы и название практи-
практикума. Записи следует вести только чернилами кратко, четко и аккуратно.
Полученные результаты нужно заносить только в лабораторный жур-
журнал. Использование разного рода черновиков не допускается. Если резуль-
результат изображен ошибочно, его аккуратно зачеркивают и сверху записывают
правильные цифры. Нельзя исправлять одни цифры на другие, так как это
ведет к небрежному оформлению отчета и нечеткости записи.
При оформлении отчета по лабораторной работе необходимо придер-
придерживаться следующего порядка изложения сведений:
1) дата выполнения и название лабораторной работы;
2) краткая теоретическая часть;
3) экспериментальная часть:
последовательность операций и условия проведения опытов;
схема прибора или установки;
качественные характеристики эксперимента (изменение окраски,
реагентов, описание выделившихся газов и осадков);
702 20. Правила поведения и порядок работы в химической лаборатории
количественные характеристики опыта (объемы и концентрации исполь-
использованных растворов, объем выделившегося газа, изменение температуры, элек-
электрической проводимости или других свойств реакционной среды и т.д.);
результаты измерений удобно представлять в табличной форме;
уравнения реакций со стехиометрическими коэффициентами;
подробные расчеты с выводом размерностей величин;
графики экспериментальных зависимостей, которые лучше всего вы-
выполнять карандашом на бледной миллиметровой бумаге, а в случае ее от-
отсутствия — на тетрадном листе в клетку;
выводы;
4) ответы на контрольные вопросы, имеющиеся в руководстве или
выданные преподавателем.
Подробнее остановимся на количественных характеристиках экспе-
эксперимента. Их выражают в виде чисел, показывающих, как изменяется неко-
некоторое свойство системы (функция) при изменении какого-либо параметра
(аргумента). Например, скорость химической реакции возрастает при увели-
увеличении температуры системы и концентрации взаи-
взаимодействующих веществ; ионное произведение
воды увеличивается с ростом температуры и т.д.
При проведении опыта математическую
связь указанных функции и аргумента сначала вы-
выражают в табличной форме в виде трех столбцов:
номер столбца, значение аргумента, значение
функции. Однако табличные данные при всей пол-
полноте количественного описания эксперимента не
обладают наглядностью, поэтому таблицы в опы-
опытах всегда дополняются графиками. По графику
можно судить о характере изменения функции. На
нем четко видны так называемые характерные
точки (экстремумы функции, точки перегиба, точ-
точки изменения наклона линий) и ошибки измерений
(выпадающие точки).
По оси абсцисс откладывают значения аргу-
аргумента, а по оси ординат — значения функции.
Возле осей указывают обозначения величин и их
размерности. Масштабы по осям выбираются та-
таким образом, чтобы углы наклона линий графика
были близки к 45°. Для удобства определения коор-
координат любой точки на расстоянии между двумя
соседними делениями оси должны размещаться
одна, две, пять или десять единиц размерности из-
о
о
Рис. 20.1. Построение гра-
графика по эксперименталь-
экспериментальным точкам:
а — неправильно; б — пра-
правильно; А — выпавшая
(ошибочная) точка
20.4. Рабочий журнал
703
где п — целое
меряемой величины (либо то же число, умноженное на 10*"
число).
Для построения графика экспериментальные точки нужно соединить
между собой. Наиболее простой способ соединения всех точек ломаной лини-
линией (рис. 20.1, а) обычно не применяется, так как это означает скачкообразный
характер соотношения между функцией и аргументом. Гораздо более распро-
распространенной в природе является плавная зависимость свойства системы
(функции) от ее параметра (аргумента), поэтому через экспериментальные
точки проводят некоторую усредненную плавную кривую, не обязательно
включающую все точки (рис. 20.1, б). Точки, удаленные от кривой, указыва-
указывают на ошибку в эксперименте или в расчете. Этот экспериментальный график
может быть с определенной степенью точности описан (аппроксимирован)
математической функцией у=/(х), выраженной в виде формулы (уравнения).
При сравнении на одном графике экспериментальных данных с близ-
близко расположенными расчетными экспериментальную кривую обычно не
строят, чтобы не загромождать рисунок. Для разных условий эксперимента
(давления, температуры) или нескольких объектов изучения используют
различные обозначения экспериментальных точек (обычно крестики, зачер-
зачерненные и светлые геометрические фигуры).
Графики можно использовать для операций графической интерполя-
интерполяции или экстраполяции. Интерполяцией называется нахождение неизвестно-
неизвестного значения функции у = f(x), лежащего между двумя экспериментальными
значениями^! -у2, т.е. внутри некоторого интервала. Если .неизвестное зна-
значение функции находится вне интервала
расположения экспериментальных точек,
нахождение этого значения называется
экстраполяцией. При проведении экстра-
экстраполяции делается предположение о со-
сохранении функциональной зависимости
У = Лх) вне интервала проведения
эксперимента.
Для проведения графической ин-
интерполяции по большому числу экспе-
экспериментальных точек, показывающих
форму графика, строят усредненную о
кривую зависимости у = f(x) (рис. 20.2), рис Ж1 Графическая ИНтерполя-
после чего определяют значение функ- ция (СШЮшные линии со стрелка-
ции у при любом заданном значении ар- ми) и экстраполяция (пунктирные
гумента х (или наоборот) внутри интер- линии со стрелками) по экспери-
вала расположения экспериментальных ментальным точкам; х\-х2 — ин-
точек. тервал проведения эксперимента
¦
- — —т
7
**
у'
/
1
1
1
i
1
1
1
704 20. Правила поведения и порядок работы в химической лаборатории
Для проведения графической экстраполяции плавную усредненную
кривую зависимости у = f(x) продолжают влево и вправо за пределы интер-
интервала проведения эксперимента с сохранением характера зависимости, после
чего снимают значение у при заданном х (или наоборот). Точность снимае-
снимаемых данных можно увеличить, если аппроксимировать зависимость у = f(x)
математическим уравнением и по нему производить вычисления.
Иногда для удобства вычислений функциональную зависимость у = f(x)
(кривая) на некотором малом участке АВ аппроксимируют отрезком прямой.
В этом случае интерполяцию называют линейной и ее результат у' будет
отличаться от у. Линейную интерполяцию удобно применять при выраже-
выражении экспериментальных данных в табличной форме как предварительный
этап расчета.
Реактивы, посуда и оборудование
учебной химической лаборатории
21.1. Реактивы
Все вещества и их растворы в лабораториях хранятся в бутылях,
склянках и других емкостях с этикетками, на которых написаны фор-
формулы или названия реагентов и при необходимости указана концен-
концентрация раствора. Емкости закрываются резьбовыми крышками или
хорошо подогнанными стеклянными пробками. Большинство реаген-
реагентов в лаборатории хранится на полках. Исключение составляют кон-
концентрированные кислоты, щелочи, неприятно пахнущие и сильно ле-
летучие вещества, которые обычно располагают в вытяжном шкафу.
По степени очистки реактивы подразделяют на шесть классов
(квалификаций):
1) технические (техн.);
2) очищенные (оч.);
3) чистые (ч.);
4) чистые для анализа (ч.д.а.);
5) химически чистые (х.ч.);
6) особо чистые (о.ч.).
По мере увеличения номера квалификации возрастает степень
очистки вещества. В особо чистых реактивах содержится ничтожно
мало примесей A0~10-10~5 %). Для лабораторных опытов чаще всего
используются реактивы квалификации «ч.», «ч.д.а.» и «х.ч.».
21.2. Посуда
При проведении химического превращения в лабораторных ус-
условиях в качестве «реактора» используется какая-либо химическая
посуда. Чаще всего ее изготавливают из специальных сортов стекла.
Основные требования, предъявляемые к стеклу, — устойчивость к
воздействию химических реагентов и способность противостоять рез-
резким колебаниям температуры. Выполнение первого требования обу-
обусловлено химическим составом, а второго — малым значением тем-
температурного коэффициента объемного расширения. Повышенной
термической устойчивостью отличается посуда, изготовленная из
стекла марки «Пирекс» (температура размягчения около 800 °С) или
из кварца (температура размягчения около 1600 °С).
23 - 9795
706 21. Реактивы, посуда и оборудование учебной химической лаборатории
а б
Рис. 21.1. Пробирки:
а — цилиндрической;
б — конической фор-
формы (для центрифуги-
центрифугирования)
а б в г
Рис. 21.2. Колбы:
а — коническая (Эрленмейера); б — плоскодонная; в -
круглодонная; г — с боковым отводом (Вюрца)
Существенным недостатком стекла является хрупкость. Если стеклян-
стеклянная посуда не нагревается во время опытов, то ее иногда для увеличения
устойчивости к удару покрывают снаружи пленками органических полиме-
полимеров, например поливинилхлорида.
В предлагаемых лабораторных работах многие реакции удобно прово-
проводить в пробирках, имеющих цилиндрическую (рис. 21.1, а) или коническую
(рис. 21.1, б) формы и размещающихся в пластмассовых, металлических или
деревянных штативах. Объем цилиндрических пробирок составляет обычно
10... 15 мл, конических 5...7 мл. На конические пробирки заводским спосо-
способом может быть нанесена градуировочная шкала. Они могут применяться
как мерная посуда.
Для проведения опытов используют различные колбы (рис. 21.2) —
конические (колбы Эрленмейера), плоскодонные, круглодонные, с боковым
отводом (колбы Вюрца) и химические стаканы (рис. 21.3), часто имеющие
для удобства слива жидкостей оттянутый носик. Колбы изготавливают из
химического стекла разных сортов, а химические стаканы — из стекла или
фарфора. В конических и плоскодонных колбах, а также в химических ста-
стаканах допускается нагревать реакционные смеси до
температуры 100 °С или несколько выше (кипение
водных растворов). При нагреве до существенно бо-
более высокой температуры применяют круглодонные
колбы.
Небольшие количества жидкостей, например
растворы индикаторов, хранят в капельницах (рис. 21.4)
и отливают из них. Наиболее часто встречаются две,
конструкции капельниц: с пипеткой (рис. 21.4, а) и с
носиком (рис. 21.4, б). Из первой капельницы нужное
а б
Рис. 21.3. Химические
стаканы:
а — без носика; б — с
носиком
21.2. Посуда
707
число капель раствора переносят с
помощью пипетки, снабженной ре-
резиновым колпачком, из второй —
откапывают из носика, перевернув
сосуд и обхватив его ладонью. Жид-
Жидкость выливается вследствие расши-
расширения воздуха в колбочке при нагре-
нагревании от руки.
Мерная химическая посуда
предназначена для измерения объе-
объемов жидкостей. К ней относятся мер-
мерные цилиндры (рис. 21.5, а), мензурки
(рис. 21.5, б), пипетки (рис. 21.5, в, г),
бюретки (рис. 21.5, д, ё) и мерные
колбы (рис. 21.5, ж).
Рис. 21.4. Капельницы:
а — с пипеткой; б — с носиком
_J_00
±90
S70
_|40
ф
Г
Г
= 10
20
мл
20°С
И
О
а б в г д е ж
Рис. 21.5. Мерная химическая посуда:
а — цилиндр; б — мензурка; в, г — пипетки (простая и градуированная);
д, е — бюретки (со стеклянным и шариковым кранами); ж — мерная колба
23*
708 21. Реактивы, посуда и оборудование учебной химической лаборатории
Мерные цилиндры и мензурки изготавливают из толстостенного стек-
стекла (нельзя нагревать), градуируются в миллилитрах A мл = 1 см3; 1 л = 1 дм3).
Они применяются в тех случаях, когда не требуется высокая точность изме-
измерения объема. Емкость цилиндров составляет от 5... 10 мл до 1 л и более.
Пипетки предназначены для точного отмеривания и дозировки опре-
определенных объемов жидкости. Обычные пипетки для жидкости (или пипетки
Мора) (см. рис. 21.5, в) представляют собой стеклянные трубки малого диа-
диаметра с расширением в средней части и с оттянутым нижним концом, диа-
диаметр которого составляет примерно 1 мм. Пипетки имеют кольцевую гра-
дуировочную метку в верхней части, показывающую, до какого уровня сле-
следует заполнить пипетку, чтобы объем вылитой из нее жидкости был равен
величине, указанной на корпусе. Емкость пипеток составляет от 1 до 100 мл.
Существуют градуированные пипетки (см. рис. 21.5, г), обычно не имеющие
вздутия посередине, снабженные шкалой с ценой деления 0,1 мл.
Чтобы отобрать пипеткой определенный объем жидкости, на ее
верхний конец надевают резиновую грушу, а нижний конец опускают в
емкость с жидкостью. Затем, сжав и отпустив грушу, набирают жидкость в
пипетку таким образом, чтобы ее уровень был на 2...4 см выше градуиро-
вочной отметки, после чего быстро снимают грушу и указательным паль-
пальцем правой руки зажимают верхнее отверстие пипетки, держа ее средним
и большим пальцами. Излишек жидкости медленно выпускают из пипетки,
регулируя скорость истечения величиной зазора между указательным
пальцем и зажимаемым краем. Когда мениск жидкости опустится до гра-
дуировочной отметки, пипетку плотно зажимают ука-
указательным пальцем.
Для смачивающих стекло жидкостей мениск
имеет вогнутую форму (рис. 21.6, я), для несмачи-
вающих — выпуклую (рис. 21.6, б). В первом случае
отсчет производят по нижнему краю мениска, во вто-
втором — по верхнему краю. Оставшуюся на конце пи-
пипетки каплю жидкости удаляют прикосновением к
стенке сосуда. Жидкость переносят в колбу и, открыв
верхний конец пипетки, дают ей стечь, касаясь концом
пипетки стенки сосуда. Жидкость, оставшуюся в носи-
носике, не вытряхивают и не выдувают, так как пипетка
отградуирована с учетом объема этой жидкости. Неаг-
Неагрессивные жидкости, обычно разбавленные растворы
допускается осторожно втягивать в пипетку ртом при
соблюдении правил гигиены.
Бюретки для работы с жидкостями представ-
представляют собой градуированные стеклянные трубки, за-
а б
Рис. 21.6. Форма ме-
мениска жидкости в
стеклянной трубке:
а — смачивающая
жидкость; б — несма-
чивающая жидкость
21.2. Посуда
709
крепленные в штативе и снабженные в нижней части
запорным устройством. Химики-профессионалы исполь-
используют бюретки с притертыми стеклянными кранами
(см. рис. 21.5, д\ но в учебных лабораториях чаще при-
применяются бюретки с капилляром (рис. 21.5, е), подсоеди-
подсоединяемым к корпусу посредством резиновой трубки. Труб-
Трубка перекрывается с помощью зажима или находящегося
внутри нее твердого тела (стеклянного шарика или ци-
цилиндрика с закругленными краями). Удобство примене-
применения бюреток второго типа связано с «залипанием» стек-
стеклянных кранов при использовании щелочных растворов.
Для сливания жидкости из бюретки нащупывают
стеклянный шарик в трубке и слегка сдавливают резинку
большим и указательным пальцами в месте ее соприкос-
соприкосновения с шариком. При этом образуется зазор между
шариком и внутренней стенкой трубки, через который
жидкость стекает в капилляр и далее в колбу. Скорость
Рис. 21.7. Удале-
Удаление пузыря воз-
воздуха из капилляра
бюретки
истечения жидкости зависит от величины зазора, а последний — от силы
сдавливания резинки. Скорость вытекания жидкости обычно составляет
10.. .20 мл/мин, а при необходимости снятия показаний (например, поиск точ-
точки эквивалентности при титровании) — 1.. .2 капля/с.
Бюретка заполняется раствором известной концентрации с помощью
воронки. При этом обычно жидкость немного не доливают до нулевой от-
отметки, а в случае перелива жидкости ее избыток сливают через запорное
устройство до нулевой отметки или чуть ниже ее. Точное совпадение на-
начального уровня жидкости в бюретке с нулевой отметкой не является обяза-
обязательным, поскольку расчет объема жидкости, понадобившегося для реак-
реакции, ведется по разности конечного и начального уровней.
При заполнении бюретки следует обращать внимание и на заполнение
капилляра. В случае наличия в нем пузырька воздуха последний вытесняет-
вытесняется жидкостью. Для этого резиновую трубку ?/-образно изгибают так, чтобы
капилляр и корпус бюретки при нажатии на шарик образовали сообщаю-
сообщающиеся сосуды (рис. 21.7).
Показания уровней жидкости в бюретке следует снимать с возможно
большей точностью. Самые распространенные жидкостные бюретки имеют
объем 50 и 100 мл. Цена большого деления составляет 1 мл, а малого 0,1 и
0,2 мл соответственно. Даже неопытный экспериментатор может снять по-
показания уровня жидкости с точностью 0,5 цены деления (т.е. 0,05 и 0,1 мл), а
при наличии некоторого опыта — с точностью 0,25 цены деления. При от-
отсчете уровня жидкости необходимо следить за тем, чтобы глаз наблюдателя
находился в одной горизонтальной плоскости с краем мениска жидкости.
710 21. Реактивы, посуда и оборудование учебной химической лаборатории
Если прозрачная жидкость смачивает стекло, то на фронтальной проекции
бюретки будет четко виден вогнутый нижний край мениска, по которому
ведут отсчет уровня жидкости. Для непрозрачной смачивающей жидкости
на фронтальной проекции будет виден лишь верхний край мениска в виде
прямой линии, по нему отсчитывают уровень. Для несмачивающей стекло
жидкости прозрачность не имеет никакого значения, так как на фронтальной
проекции в любом случае мы увидим выпуклый верхний край мениска, по
которому сделаем отсчет.
Мерные колбы предназначены для приготовления растворов строго
заданной концентрации. Они представляют собой плоскодонные колбы с
узким длинным горлышком, на которое наносится кольцевая градуировоч-
ная метка, соответствующая объему жидкости (обычно при Т = 20 °С), ука-
указанному на колбе.
Для приготовления раствора в колбу наливают дистиллированную воду
(приблизительно 25...50 % ее объема) и вносят навеску вещества, взвешенную
на аналитических весах с точностью 0,1 мг. Затем растворяют вещество и доли-
доливают воду, не доводя до метки приблизительно 1 см. Последние капли воды
добавляют с помощью пипетки, после чего колбу закрывают притертой стек-
стеклянной или резиновой пробкой и переворачивают 10-15 раз горлышком вниз и
обратно для полного перемешивания раствора. Зная массу, молярную массу
вещества и объем раствора, можно вычислить массовую концентрацию раство-
растворенного вещества (г/л) или его молярную концентрацию (моль/л).
21.3. Оборудование
К оборудованию учебной химической лаборатории относятся различ-
различные вспомогательные инструменты и устройства: штативы, держатели для
пробирок, стеклянные палочки для перемешивания жидкостей, часовые и
предметные стекла, шпатели (рис. 21.8), капельные (медицинские) и капил-
капиллярные пипетки (рис. 21.9, 21.10), фарфоровые и полимерные пластинки с
углублениями, лупы для рассматривания осадков, промывалки объемом
100...150 мл (рис. 21.11), фарфоровые чашки и чашки Петри (рис. 21.12,
21.13), ступки с пестиками (рис. 21.14), тигли и тигельные щипцы (рис.
21.15), лодочки (рис. 21.16), асбестированные сетки (рис. 21.17), а также
специальное оборудование, используемое в технике эксперимента и рас-
рассматриваемое далее в гл. 22.
Лабораторные штативы (рис. 21.18, а) используют как основу для креп-
крепления составных частей любой лабораторной установки. Часовые и предмет-
предметные стекла, а также пластинки с углублениями применяются при проведении
опытов полумикрометодом, который иначе называют капельным.
21.3. Оборудование
711
\ /
/ \
а б
Рис. 21.8. Шпатели (а) и стеклян-
стеклянная палочка (б)
\1
Рис. 21.9. Капельная
(медицинская) пипетка
Рис. 21.10. Капил-
лярная пипетка
а б
Рис. 21.12. Фарфоро- Рис. 21.13. Чашка Петри (а) и ча-
вая чашка совое стекло (б)
_
_
У
Рис. 21.11. Промы-
валка, изготовленная
из полиэтиленового
сосуда
v_y
Рис. 21.14. Фарфоровая ступ-
ступка (а) с пестиком (б)
а б
Рис. 21.15. Тигель (а) и
тигельные щипцы (б)
Рис. 21.16. Лодочка для прокаливания
712 21. Реактивы, посуда и оборудование учебной химической лаборатории
а б
Рис. 21.17. Асбестированная сетка (а) и тренога (б)
1
Рис. 21.18. Лабораторный штатив (а) и под-
подставка для пробирок (б)
С помощью капиллярных пипеток отделяют осадки от надосадочных
жидкостей. В фарфоровых чашках обычно осуществляют выпаривание рас-
растворов (удаление воды при нагревании или кипячении), в фарфоровых или
агатовых ступках с помощью пестиков измельчают твердые вещества, в
тиглях и лодочках их прокаливают.
Асбестированные сетки используют при нагревании стеклянной посу-
посуды для большей равномерности обогрева и предотвращения разрушения
вследствие теплового удара при соприкосновении с голым пламенем.
Техника эксперимента
При выполнении опытов в учебной химической лаборатории ис-
используют общие экспериментальные операции и приемы, рассмот-
рассмотренные ниже.
22.1. Мытье и сушка посуды
Вся применяемая в лабораторном практикуме посуда должна
быть чистой настолько, чтобы на ее стенках при тщательном осмотре
нельзя было обнаружить каких-либо загрязнений. Вода должна сво-
свободно стекать по стенкам, не оставляя капель. В зависимости от сте-
степени загрязнения для мытья посуды применяют различные средства:
а) сравнительно чистую посуду достаточно заполнить горя-
горячей водой и оттереть загрязнения ершиком;
б) жировые загрязнения хорошо удаляются теплым мыльным
раствором или теплым насыщенным раствором технической соды
(карбоната натрия Na2CO3);
в) грязную посуду моют щелочным раствором перманганата
калия КМпО4 или хромовой смесью (хромпиком).
Для получения щелочного раствора перманганата калия не-
необходимо 5 г твердого КМ11О4 растворить в 100 мл горячего 10 %-
ного раствора едкого натра NaOH или едкого кали КОН. Хромовая
смесь представляет собой дихромат калия К2Сг207, растворенный в
концентрированной серной кислоте H2SO4 (конц)- Обычно для приго-
приготовления хромпика в H2SO4(kohu) (p = 1,84 г/мл) добавляют около
5 % (от массы H2SO4) тонкоизмельченного дихромата калия и осто-
осторожно нагревают на водяной бане в фарфоровой чашке до его пол-
полного растворения.
Согласно другому способу, 15 г дихромата калия растворяют
в 100 мл горячей воды. После охлаждения раствора к нему по кап-
каплям при постоянном перемешивании добавляют 100 мл H2SO4 (КОнц)-
При попадании хромпика на кожу ее промывают большим количе-
количеством холодной воды, а затем раствором гидрокарбоната натрия
NaHCO3. Вследствие агрессивности (сильное окисляющее дейст-
действие) щелочного раствора КМпО4 и хромовой смеси ершики при мы-
мытье посуды использовать нельзя. Обычно хромпик на 5-10 мин зали-
заливается в отмываемый сосуд, после чего его сливают обратно в бу-
бутыль, где он хранится, а посуда несколько раз ополаскивается теп-
теплой водопроводной и потом дистиллированной водой.
714
22. Техника эксперимента
Вымытую химическую посуду нельзя вытирать. Обычно ее высуши-
высушивают в сушильных шкафах или с помощью специальных электросушилок, в
которых сжатый под небольшим избыточным давлением воздух нагревается
и проходит через узкие выходные трубки, на которые надевается высуши-
высушиваемая посуда.
22.2. Нагревание и прокаливание. Нагревательные приборы
В лабораторном практикуме часто приходится проводить реакции при
повышенной температуре. Для нагрева реагентов используют различные
устройства: газовые горелки, бани, электрические плитки, печи и др. Наибо-
Наиболее распространены в лабораториях газовые горелки Бунзена и Теклю
(рис. 22.1), которые конструктивно различаются способом подачи воздуха.
Горелка Бунзена (рис. 22.1, а) устроена проще. Она представляет со-
собой стальную трубку, в нижней части которой перпендикулярно оси про-
просверлено сквозное цилиндрическое отверстие 7, служащее для подачи воз-
воздуха. Эта трубка на резьбе ввинчена в корпус горелки, снабженный боковым
штуцером для подвода газа. Смешение горючего (природного газа, в кото-
котором примерно 95 % составляет метан) и окислителя (кислорода воздуха)
происходит в трубке, а количество поступающего воздуха регулируется с
помощью кольцевой муфты 2.
В горелку Теклю (рис. 22.1, б) воздух подается через зазор между
вращающимся по резьбе на корпусе горелки диском 1 и нижней частью
расширяющейся на конус трубки 2.
Количество поступающего воздуха
определяется шириной этого зазора
и регулируется точнее, чем в горелке
Бунзена. Предусмотрена также ре-
регулировка притока газа с помощью
винта 3, расположенного на корпусе
горелки.
В зависимости от количеств
горючего и окислителя процесс го-
горения происходит различным обра-
образом. При существенном недостатке
воздуха природный газ сгорает све-
светящимся коптящим пламенем (не-
а б полное сгорание, восстановительное
Рис. 22.1. Газовые горелки Бунзена (а) и пламя). При стехиометрическом со-
Теклю (б) отношении горючего и окислителя
22.2. Нагревание и прокаливание. Нагревательные приборы
715
или при небольшом недостатке одного из компо-
компонентов газ сгорает практически несветящимся пла-
пламенем без копоти (полное сгорание). При большом
избытке воздуха может произойти опасное явление,
называемое «проскоком» пламени, при котором газ
загорается внутри горелки с характерным свистя-
свистящим звуком. Горелка сильно накаляется, что может
вызвать пожар в лаборатории. При возникновении
«проскока» следует срочно закрыть газовый кран.
Пользоваться горелкой можно только после ее ох-
охлаждения и уменьшения расхода воздуха.
Распределение температуры в пламени нор-
нормально работающей газовой горелки приведено на
рис. 22.2. Здесь можно различить три конические
зоны. В нижней (ближней к отверстию горелки)
зоне 1 горючее и окислитель смешиваются и по-
подогреваются от 300 до 520 °С. Температура увели-
увеличивается по мере удаления от выходного отвер-
отверстия. В зоне 2 имеется небольшой недостаток ки-
кислорода, поэтому пламя является восстановитель-
восстановительным. Средний конус восстановительного пламени
окружен зоной 3 окислительного пламени, в кото-
Рис. 22.2. Распределе-
Распределение температуры в пла-
пламени нормально рабо-
работающей газовой горелки
рои происходит полное сгорание газа с участием кислорода окружающих
слоев воздуха.
При соотношении горючее—окислитель, близком к стехиометри-
ческому, значения температуры в зонах 2 и 3 различаются мало. При ис-
использовании газовой горелки в качестве нагревателя нагреваемый предмет
помещают в верхнюю часть пламени на расстоянии B/3)Л от выходного от-
отверстия (h — полная высота пламени). Пламя газовой горелки имеет макси-
максимальную температуру 1550... 1570 °С.
Бани применяют при нагреве веществ в интервале температур от
комнатной до 400 °С. В зависимости от нагревающей среды бани могут
быть водяными, воздушными, глицериновыми, масляными, песчаными, си-
силиконовыми и др.
Простая водяная баня (рис. 22.3, а) представляет собой алюминиевую
кастрюлю, крышка которой является наборной и состоит из концентриче-
концентрических налегающих друг на друга колец. Вынимая кольца, можно подобрать
подходящий диаметр отверстия для нагреваемого сосуда (стакана или кол-
колбы). Баня нагревается на газовой горелке, причем по достижении кипения
размер пламени уменьшают. Бани могут иметь электрический нагреватель и
указатель уровня воды (рис. 22.3, б). На водяных банях реагенты нагревают
716
22. Техника эксперимента
а б в
Рис. 22.3. Бани:
а — водяная простая; б — водяная с электрическим подогревом и указанием
уровня воды; в — песчаная
до температуры, не превышающей 100 °С (температура кипения воды), на
глицериновых и масляных — приблизительно до 200 °С, на песчаных — до
300 °С, на силиконовых — до 400 °С.
Электрические плитки, применяемые в лабораториях, обычно имеют
закрытый нагревательный элемент и регулятор потребляемой мощности.
Электрические печи используются для нагрева веществ до высокой
температуры. Нагрев кристаллических веществ до температуры более 400 °С
называют прокаливанием. Наиболее распространены следующие конструк-
конструкции печей: муфельные, тигельные и трубчатые. В муфельные и тигельные
печи прокаливаемые субстанции вносят в специальных сосудах - тиглях
(см. рис. 21.15, я), а в трубчатые печи — в лодочках (см. рис. 21.16). Тигли
и лодочки изготавливают из устойчивых к нагреву материалов: фарфора,
платины, кварца. Во всех упомянутых печах можно достичь температуры
1000... 1200 °С. Печи включаются в сеть через лабораторный автотрансфор-
автотрансформатор (ЛАТР). При разогреве печей нельзя включать их сразу на полную
мощность (это может вызвать перегорание нагревательных элементов).
Мощность следует увеличивать постепенно по мере повышения температуры.
22.3. Измерение температуры
При изменении температуры среды варьируются такие свойства сис-
системы, как объем, давление, электродвижущая сила (ЭДС), электрическое
сопротивление и др.
Наиболее часто температуру среды в лабораторном практикуме изме-
измеряют с помощью жидкостных дилатометрических термометров. При работе
в интервале температур -30...+360 °С применяют ртутные химические тер-
термометры со шкалой в 100, 150, 200, 250, 300 и 360 °С. При температуре
22.3. Измерение температуры 1\1
-38,9 °С ртуть замерзает, поэтому низкотемпературные термометры запол-
заполняются рабочими жидкостями, имеющими в шкале Цельсия большие отри-
отрицательные температуры кристаллизации (этанолом, толуолом или пента-
ном). Эти жидкости бесцветны, для удобства наблюдения за столбиком в
них добавляют красители синего или красного цвета.
При измерении температуры жидкости термометр вводят в сосуд та-
таким образом, чтобы его рабочий резервуар (нижняя часть) не касался стенок
или дна емкости и возможно большая часть термометра находилась в среде
с измеряемой температурой. Необходимо помнить о том, что термометр из-
изготовлен из стекла, являющегося хрупким материалом, и аккуратно обра-
обращаться с ним. Категорически запрещается нагревать термометр выше мак-
максимальной температуры, на которую он отградуирован. Перегрев может
привести к разрыву жидкостного резервуара, что имеет особо нежелатель-
нежелательные последствия в случае ртутных термометров, поскольку пары ртути ядо-
ядовиты. Если разбился ртутный термометр и произошла утечка рабочей жид-
жидкости, следует аккуратно бумажкой собрать капли ртути, перекатывая и по-
постепенно укрупняя их, а затем перенести в склянку. Место, где была разлита
ртуть, посыпают порошком серы или заливают концентрированным раство-
раствором хлорида железа (III).
Другой способ измерения температуры, часто применяемый в химиче-
химических лабораториях, заключается в использовании термометров сопротивления
(болометров) и термоэлектрических термометров (термопар). Работа термомет-
термометра сопротивления основана на хорошо воспроизводимой зависимости сопро-
сопротивления чистого металла R от температуры Т — с увеличением температуры
сопротивление металла возрастает. Болометр представляет собой металличе-
металлическую спираль (чаще всего из платины или меди), намотанную на каркас из изо-
изолятора (слюда, кварц) и помещенную в защитную кварцевую трубку.
Сопротивление болометра измеряется с помощью мостика Уитстона, а
температуру находят по градуированному графику R = f(T) или рассчиты-
рассчитывают по формуле. На практике обычно градуируют гальванометр, включен-
включенный в мостик Уитстона, по температурам плавления чистых металлов или
солей. Платиновые термометры сопротивления позволяют измерять темпе-
температуру от -190 до +600 °С, медные — от -55 до +200 °С.
Термопара состоит из двух проводников, представляющих собой раз-
различные металлы или сплавы. На одном конце (так называемый горячий спай)
проводники соединены между собой, на другом конце (холодный спай) — с
двумя медными проводами, ведущими к гальванометру. При равенстве тем-
температур горячего и холодного спаев термоэлектродвижущая сила Е в цепи
равна нулю. При нагревании горячего спая в цепи возникает ЭДС, величина
которой зависит от природы металлов (сплавов) и разности температур го-
горячего и холодного спаев.
718
22. Техника эксперимента
Градуируют термопару, помещая оба спая в тающий лед (О °С) и ус-
устанавливая гальванометр на нуль. Затем холодный спай оставляют в сосу-
сосуде Дьюара со льдом, а горячий поочередно помещают в различные чистые
металлы или соли с известной температурой плавления, снимают показа-
показания Е и строят градуировочный график Е =/(Г). Точность метода достига-
достигает сотых долей градуса. В лабораториях наиболее широко применяются
термопары платинородий A0 %) — платина (+250...+1450 °С), хромель—
алюмель (-200...+1200 °С), медь—константан (-185...+500 °С), железо—
константан (-200...+750 °С).
22.4. Весы и взвешивание
Химия является точной наукой. Все реагенты взаимодействуют друг с
другом в строго определенных соотношениях по массе и количеству веще-
вещества, поэтому одной из основных операций, проводимых в химической ла-
лаборатории, является взвешивание. Взвешиванием называется сравнение
массы данного тела (или навески) с известной массой гирь. Прибор для
взвешивания называется весами и может иметь различную конструкцию.
По точности взвешивания все весы подразделяются на следующие виды:
1) для грубого взвешивания
(точность до 1 г);
2) для точного взвешивания
(точность до 0,01.. .0,001 г);
3) аналитические (точность
обычных весов 10 г, микро-
микрохимических 1(Г* г);
4) специальные (пробирные,
торзионные, квадрантные
и др.).
В учебных химиче-
химических лабораториях наибо-
наиболее часто встречаются тех-
технические весы для точного
взвешивания (технохими-
ческие) и аналитические
весы. Технохимические
Рис. 22.4. Технохимические весы: весы ( 22.4) просты
1 — коромысло; 2 — балансировочные грузы; 3 —
опорная колонка; 4 - серьга; 5 - чашка; б- стрелка; П0 конструкции, надежны
7 — шкала; 8 — отвес; 9 — установочный винт; 10 — И имеют высокую ТОЧ-
ручка арретира НОСТЬ.
22.4. Весы и взвешивание 719
Основной частью весов является коромысло 7, представляющее собой
равноплечий рычаг, снабженный тремя треугольными призмами из высоко-
высокосортной стали и балансировочными грузами 2. Центральная призма зафик-
зафиксирована посередине коромысла ребром вниз. Это ребро опирается на
стальную подушку, укрепленную на стержне, проходящем внутри опорной
колонки 3. Две боковые призмы расположены симметрично на концах ко-
коромысла ребрами вверх. На эти ребра опираются подушки специальных
устройств — серег 4, к которым на дужках и крестовинах подвешиваются
чашки 5 весов. Стрелка 6 закреплена верхней частью в центре коромысла,
нижняя ее часть при колебаниях чашек перемещается по шкале 7, располо-
расположенной у основания опорной колонки. По стрелке определяется уравнове-
уравновешенность весов. Отвес 8 служит для установки корпуса весов в горизон-
горизонтальное положение с помощью установочных винтов 9. Ручка арретира 10
предназначена для подъема и опускания коромысла и призмы. При опуска-
опускании коромысло опирается на два стерженька, весы находятся в нерабочем
состоянии, исключается контакт призмы с подушкой (говорят, что весы ар-
ретированы или арретир включен). Таким образом, центральная призма
дольше сохраняет заостренное состояние, от которого напрямую зависит
чувствительность весов. При подъеме коромысло вывешивается на цен-
центральной призме, которая приходит в контакт с подушкой, что соответству-
соответствует рабочему состоянию весов.
При взвешивании следует соблюдать следующие правила:
1) произвести правильную установку весов по отвесу и уравновесить
их. Арретир необходимо плавно отключить. При этом стрелка исправных
весов будет колебаться вправо-влево от середины шкалы. Если разность чи-
чисел делений, на которые отклоняется стрелка, не превышает трех, значит,
нулевая точка расположена не далее полутора делений от центра шкалы и
весы готовы к работе. При разности, большей трех, лаборант должен отре-
отрегулировать весы осторожным вращением балансировочных грузов;
2) взвешиваемый предмет всегда помещается на левую чашку весов, а
разновес — на правую. Разновес — это набор гирь (массой 1, 2, 5, 10, 20, 50,
100 г) и алюминиевых пластинок (разновесок) (массой 10, 20, 50, 100, 200,
500 мг), помещенный в коробку с пинцетом. Гири и разновески разрешается
брать только пинцетом во избежание их загрязнения. Разновес должен нахо-
находиться либо на чашке весов, либо в коробке, его нельзя ставить на рабочие
столы;
3) взвешиваемый образец и разновес следует класть на чашки весов
только при включенном арретире и размещать в центре чашки;
4) нельзя помещать вещества непосредственно на чашку весов. Для
взвешивания применяют листки гладкой бумаги или кальки, часовые стекла,
тигли, химические стаканы и др. Очень удобно взвешивать вещества в бюксах
720 22. Техника эксперимента
(рис. 22.5) — небольших стеклянных стаканчиках с при-
притертой крышкой. Масса взвешиваемого вещества опре-
определяется как разность между массами заполненной и
пустой тары;
5) взвешиваемое вещество должно иметь комнат-
комнатную температуру; нельзя взвешивать горячие и теплые
предметы, так как конвективные воздушные потоки ис-
искажают результат;
Рис. 22.5. Бюкс 6) гири и разновески помещают на чашку весов в
стаканчик с при- порядке убывания их массы, не пропуская ни одного
тертой крышкой значения. Избыточность или недостаточность массы
для взвешивания разновеса проверяют кратковременным отключением
арретира;
7) по окончании взвешивания массу предмета удобно подсчитывать
по пустым гнездам в коробке разновеса;
8) при необходимости проведения нескольких взвешиваний в одной
работе следует использовать постоянные весы и разновес.
Аналитические весы в отличие от технохимических устанавливают в
специальном застекленном корпусе с открывающимися боковыми дверками,
чтобы исключить влияние потоков воздуха на результат взвешивания. Для
повышения точности весов призмы изготовлены из агата. Весы снабжены
так называемым демпфером — устройством, позволяющим уменьшить чис-
число и время колебаний чашек до наступления равновесия, а также специаль-
специальным устройством для малой загрузки коромысла и шкалой с подсветкой.
22.5. Перемешивание, осаждение и фильтрование
Перемешивание жидкостей и гетерогенных смесей проводят с помо-
помощью стеклянной палочки (см. рис. 21.8, б), механической (с электроприводом)
или магнитной мешалки. Осаждение твердых частиц на дно реакционного
сосуда происходит под действием силы тяжести или центробежной силы в
центрифуге.
Фильтрованием называют метод разделения осадка и надосадочной
жидкости в гетерогенной системе. Эта операция является эффективной
только в случае достаточно большой разности плотностей твердых частиц и
жидкости. Разделение происходит вследствие проникновения молекул жид-
жидкости сквозь поры фильтра под действием силы тяжести (обычное фильтро-
фильтрование), в то время как имеющие больший размер твердые частицы остаются
на фильтре. Для ускорения процесса при близких значениях плотностей
твердой и жидкой фаз, малом размере частиц или рыхлости осадков создают
22.5. Перемешивание, осаждение и фильтрование
721
\
/ *¦
-0,5
/ — длина образующей
воронки
Рис. 22.6. Изготовление простого (а) и складчатого (б) фильтров
избыточное давление над фильтром (в промышленных установках) или ва-
вакуум под фильтром (в лабораториях и промышленности). Если фильтрование
под вакуумом оказывается неэффективным, используют центрифугирова-
центрифугирование, при котором разделение фаз осуществляется под действием центро-
центробежной силы, создаваемой при вращении барабана центрифуги, где нахо-
находятся пробирки с разделяемым содержимым.
По окончании процесса фильтрования дальнейшие исследования могут
проводиться с твердым осадком или надосадочной жидкостью (фильтратом).
В случае востребования осадка применяют простой фильтр с неразвитой
фильтрующей поверхностью, от которой осадок легко отделяется. Процесс
изготовления такого фильтра изображен на рис. 22.6, а.
Кусок фильтровальной бумаги со стороной квадрата 2/ складывается
на четыре равные части. Сторона получившегося квадрата / должна быть
равна или несколько больше образующей конической воронки (рис. 22.7), в
которую вставляется фильтр. Затем фильтр обрезается ножницами по дуге
24 - 9795
722
22. Техника эксперимента
Рис. 22.7. Кониче-
Коническая воронка
так, чтобы радиус получившегося сектора был прибли-
приблизительно на 0,5 см меньше образующей воронки. Фильтр
расправляется, вставляется в воронку и смачивается
дистиллированной водой для более плотного прилегания
к стенкам воронки.
Осадку дают отстояться в стакане или колбе, за-
затем осторожно сливают слой жидкости на фильтр. Эта
операция называется декантацией. Оставшийся в стака-
стакане осадок заливают порцией дистиллированной воды и
тщательно перемешивают (промывание осадка), после
чего жидкость декантируют. Промывание повторяют 2-3
раза, затем содержимое стакана переносится на фильтр.
При востребовании фильтрата для ускорения
процесса используют так называемый складчатый фильтр, имеющий разви-
развитую поверхность, изготовление которого изображено на рис. 21.7, б. Сло-
Сложенный вчетверо фильтр расправляется на две равные части, которые скла-
складываются гармошкой от краев к центру. Готовый фильтр расправляется по
секторам и вставляется в воронку. Осадок взбалтывают в стакане, и взвесь
быстро переливают на фильтр.
В лабораторном практикуме часто применяют метод очистки веществ,
называемый перекристаллизацией. Для этого загрязненное примесями ве-
вещество растворяют в горячем растворителе, в котором очищаемое соедине-
соединение растворяется гораздо лучше, чем примеси и имеет сильную зависимость
коэффициента растворимости от температуры.
Полученную гетерогенную систему необходимо
фильтровать на воронке с подогревом (горячее фильтро-
фильтрование), ибо при охлаждении раствора вещество будет
выпадать в осадок, забивая фильтр и сливное отверстие
воронки. Для нагрева стенок воронки ее помещают в
электрический обогреватель, имеющий выемку соответ-
соответствующей формы, или в конический змеевик, через ко-
который пропускают водяной пар.
Хвостовая часть воронки для горячего фильтрова-
фильтрования максимально укорочена, так как она мало подверга-
подвергается нагреву. По окончании процесса механические не-
нерастворимые примеси остаются на фильтре, а очищенное
вещество переходит в фильтрат, при охлаждении которо-
которого оно выпадает в осадок и отделяется вторичным (хо-
(холодным) фильтрованием.
Для ускоренного фильтрования под вакуумом рИс. 22.8. Колба
применяют толстостенные колбы Бунзена и фарфоровые Бунзена A) и во-
воронки Бюхнера специальной формы (рис. 22.8), имею- ронка Бюхнера B)
22.6. Приборы для получения газов 723
щие перегородку из того же материала с отверстиями, на которой распола-
располагается фильтр, как правило, двойной, состоящий из двух слоев фильтро-
фильтровальной бумаги.
Нижняя часть фильтра по размеру совпадает с перегородкой или изго-
изготавливается чуть меньшего диаметра, так чтобы закрывались все отверстия
перегородки. Верхний фильтр вырезается несколько большего диаметра, что-
чтобы при помещении в воронку он имел бортик высотой примерно 0,5 см.
Боковой отросток колбы Бунзена соединяется с вакуумным водо-
водоструйным насосом посредством резинового толстостенного вакуумного
шланга. Воронка Бюхнера и колба Бунзена соединены с помощью резино-
резиновой пробки или шлифов. Перед началом фильтрования смачивают фильтр
дистиллированной водой, включают вакуум и переливают взвесь (суспен-
(суспензию) на фильтр. Далее отсасывают фильтрат в колбу и потоком воздуха под-
подсушивают осадок в течение 5... 10 мин.
По окончании процесса фильтрования воду в водоструйном насосе
отключают осторожно, постепенно уменьшая поток, чтобы не было резкого
перепада давления, иначе водопроводная вода попадет в колбу с фильтром.
Для предотвращения этого явления иногда в магистраль включают двугор-
двугорлую склянку из толстого стекла. Колбы и склянки, находящиеся под ваку-
вакуумом, из соображений техники безопасности помещают в мешки, изготов-
изготовленные из мелкой металлической сетки или плотной ткани, а за неимением
их обертывают полотенцем.
22.6. Приборы для получения газов
Многие газы в лаборатории получают взаимодействием жидкости, напри-
например водного раствора кислоты, с твердыми веществами (металлами, солями).
При взаимодействии металлов, стоящих в ряду стандартных элек-
электродных потенциалов до водорода, с кислотами-неокислителями (НС1,
H2SO4 разб.) получают водород:
Zn + 2HC1 -> ZnCl2 + H2t
При реакции карбоната кальция или сульфида железа (II) с хлорово-
хлороводородной (соляной) кислотой образуются соответственно оксид углерода (IV)
или сероводород:
СаСОз + 2НС1 -> СаС12 + Н2О + СО2Т
FeS + 2HC1 -> FeCl2 + H2St
Эти три распространенных газа получают в лаборатории при проведе-
проведении реакций в аппарате Киппа (рис. 22.9), который представляет собой два
24*
724
22. Техника эксперимента
Рис. 22.9. Аппарат Киппа:
1 — верхний сосуд; 2 — воронка; 3 —
средний резервуар; 4 — перетяжка; 5 —
нижняя полусферическая часть; 6 — ту-
тубус для загрузки твердого реагента и от-
отвода газа; 7 — кран; 8 — пластмассовое
кольцо с отверстиями; 9 — тубус для
замены отработанной кислоты
соединенных друг с другом сосуда.
Верхний сферический сосуд 1 заканчи-
заканчивается длинной воронкой 2, вставляе-
вставляемой на шлифе в средний сферический
резервуар 3. Последний через перетяж-
перетяжку 4 переходит в нижнюю полусфери-
полусферическую часть резервуара 5, образуя
единый сосуд фигурной формы.
Жидкость из верхней емкости 1
попадает в среднюю часть 3 не непо-
непосредственно, а только после заполнения
нижнего резервуара 5.
Перед началом опыта твердый
реагент загружают в среднюю емкость
3 через тубус б, который затем закры-
закрывают пробкой. В пробку вставлена га-
газоотводная трубка с краном 7. Щель
между воронкой и перетяжкой закры-
закрывают при помощи пластмассового
кольца 8 с отверстиями, чтобы куски
реагента (металла или соли) не проско-
проскочили на дно сосуда.
Верхнюю емкость заполняют со-
соляной кислотой. При открытом кране 7
кислота через воронку 2 проходит в нижнюю часть 5, заполняет ее и через
щель между воронкой и перетяжкой поднимается в среднюю часть, где про-
происходит химическая реакция. Посредством крана 7 можно регулировать
скорость выделения газа. При закрытии крана 7 в среднем резервуаре созда-
создается избыточное давление, кислота полностью вытесняется в нижнюю
часть, реакция прекращается. После открытия крана 7 процесс получения
газа возобновляется.
Через тубус 9, закрытый в рабочем состоянии пробкой, осуществ-
осуществляется замена отработанной кислоты, когда ее концентрация вследствие
реакции значительно снижается. Сверху аппарат Киппа закрывается пре-
предохранительной воронкой, заполненной поглотителем. Газ, выходящий
из аппарата, обычно загрязнен примесями паров воды, хлороводорода, а
также твердыми частицами сульфида или карбоната. Для очистки его
пропускают через промывные склянки, заполненные нужными поглоти-
поглотителями.
В ряде случаев, когда требуется собрать небольшой объем газа, мож-
можно применять простейший прибор, состоящий из колбы или пробирки,
22.7. Определение плотности и реакции растворов. Отбор проб 725
Рис. 22.11. Прибор для получения и сбора газа
методом вытеснения воды
Рис. 22.10. Простей-
Простейший прибор для по-
получения газа:
/ — пробирка; 2 —
штатив
закрепленной в штативе и закрытой пробкой с газоот-
газоотводной трубкой (рис. 22.10). В таком приборе можно
получать газ нагреванием твердого вещества (получе-
(получение кислорода из перманганата или хлората калия),
нагреванием жидкости (получение аммиака при сли-
сливании растворов солей аммония со щелочами), взаи-
взаимодействием твердого вещества с кислотой или щелочью (см. реакции, опи-
описанные выше).
Выделяющийся газ тяжелее воздуха собирают методом вытеснения
воздуха в пробирку, отверстие которой направленно вверх, газ легче воздуха —
в пробирку, перевернутую вверх дном. Если плотность газа близка к плот-
плотности воздуха и этот газ плохо растворяется в воде, его обычно собирают
методом вытеснения воды (рис. 22.11). В этом случае газ не будет содержать
примесей составных частей воздуха (кислорода и азота).
22.7. Определение плотности и реакции растворов. Отбор проб
Плотность растворов в лаборатории наиболее просто определяется с
помощью специального прибора, который называют ареометром (см рис.
Л.3.2). Ареометр представляет собой нагруженный металлическими шари-
шариками и запаянный стеклянный поплавок с верхней узкой частью, на которой
нанесена шкала плотности. В лабораториях имеются наборы ареометров,
предназначенных для измерения плотности жидкостей в различных преде-
пределах. Зная плотность раствора, можно по таблице найти его концентрацию.
Реакцию раствора определяют с помощью кислотно-основных инди-
индикаторов. Наиболее распространенными являются фенолфталеин (в кислот-
кислотной и нейтральной средах бесцветный, в щелочной при рН > 9 — малино-
726 22. Техника эксперимента
вый), метиловый оранжевый (при рН < 3,1 — розовый, при рН > 4,4 — жел-
желтый, внутри интервала перехода при 3,1 < рН < 4,3 — оранжевый) и лакмус
(при рН < 5 — красный, при рН > 8 — синий, внутри интервала перехода
при 5 < рН < 8 — фиолетовый). Существуют также наборы универсальной
индикаторной бумаги с приложением шкалы цветов ее окрашивания в зави-
зависимости от водородного показателя рН среды.
Отбор проб в лабораторных экспериментах проводят с помощью мик-
микрошпателей (твердые вещества) или пипеток и мерных колбочек (жидко-
(жидкости), представленных на рис. 21.8, 21.9 и 21.5, ж соответственно.
Приложения
Фундаментальные физические постоянные
Универсальная газовая постоянная
Постоянная Авогадро
Постоянная Больцмана
Постоянная Фарадея
Молярный объем идеального газа при нормаль-
нормальных условиях (Т= 273,15 К,/? = 101325 Па)
Электрическая постоянная, диэлектрическая
проницаемость вакуума
Постоянная Планка
Атомная единица массы
Масса покоя протона
Масса покоя нейтрона
Масса покоя электрона
Отношение массы протона к массе электрона
Элементарный заряд
8,31441 Дж/(моль-К)
6,022045 • 1023 моль
1,380662- 103Дж/К
9,648456 • 104 Кл/моль
22,41383- 10м3/моль
8,85418782- 102Ф/м
6,626176- 10-34Дж/Гц
1,6605655- 107кг
1,6726485- 107кг
1,6749543- 107кг
9,109534-101 кг
1836,15152
1,6021892- 109Кл
Соотношения между единицами некоторых
физических величин
Единицы силы:
1 Н= 105 дин = 0,102 кгс
Единицы работы, энергии и количества теплоты:
1 Дж [Н • м] = 107 эрг = 0,239 кал = 2,78 • 10 Вт • ч = 0,102 кгс • м =
= 6,24 • 1018 эВ
Единицы мощности:
1 Вт [Дж/с] = 107 зрг/с = 0,239 кал/с
Единицы давления и механического напряжения:
1 Па [Н/м2] = 10"э бар = 1,02 • 10 кгс/м2 = 7,5 • 10"' мм рт. ст. =
= 7,5 • 10~3 торр
Единицы температуры:
Г, К (градусы Кельвина) = 273,15 + Т °С (градусы Цельсия) = 255,37 +
+ Т °F (градусы Фаренгейта)
Единицы удельной теплоемкости:
1 Дж/(кг • К) = 107 эрг/(г • °С) = 2,39 • КГ* ккал/(кг • °С)
Единицы теплопроводности:
1 Вт/(м • К) = 105 эрг/(с • см • °С) = 0,860 ккал/(ч • м • С)
Единицы количества электричества:
1 Кл [А • с] = 2,78 • 10 А • ч
728
Приложения
П.1. Наименования неорганических веществ
Таблица ПЛ Л
Систематические и традиционные названия некоторых неорганических
Формула
В(ОНK
(Н3ВОз)
Н2СО3*
H2Si03*
H4Si04*
HNO2
HNO3
НРО3
Н3РО4
Н4Р2О7
H3AsO4*
Н2О2
H2S
H2SO3*
H2SO4
H2SO3S
HF
HC1
нею*
HclO2*
HclO3*
HC1O4
HBr
HBrO*
HBrO3*
HBrO4
HI
HIO*
HIO3
н5ю6
H2Cr04*
H2Cr207*
HMnO4*
кислот и солей
Кислоты
Систематическое название
Тригидроксид бора
Триоксокарбонат (IV) водорода
Триоксосиликат (IV) водорода
Третраокососиликат (IV) водорода
Диоксонитрат (III) водорода
Триоксонитрат (V) водорода
Триоксофосфат (V) водорода
Тетраоксофосфат (V) водорода
Гептаоксодифосфат (V) водорода
Тетраоксоарсенат (V) водорода
Диоксид диводорода
Сульфид водорода
Триоксосульфат (IV) водорода
Тетраоксосульфат (VI) водорода
Триоксодисульфат (II) водорода
Фторид водорода
Хлорид водорода
Оксохлорат (I) водорода
Диоксохлорат (III) водорода
Триоксохлорат (V) водорода
Тетраоксохлорат (VII) водорода
Бромид водорода
Оксобромат (I) водорода
Триоксобромат (V) водорода
Тетраоксобромат (VII) водорода
Иодид водорода
Оксоиодат (I) водорода
Триоксоиодат (V) водорода
Гексаоксоиодат (VII) водорода
Тетраоксохромат (VI) водорода
Гептаоксодихромат (VI) водорода
Тетраоксоманганат (VII) водорода
Традиционное
название
Борная
(ортоборная)
Угольная
Метакремниевая
Ортокремниевая
Азотистая
Азотная
Метафосфорная
Ортофосфорная
Пирофосфорная
Мышьяковая
Пероксид
водорода
Сероводородная
Сернистая
Серная
Тиосерная
Фтороводородная
Хлороводородная
Хлорноватистая
Хлористая
Хлорноватая
Хлорная
Бромоводородная
Бромноватистая
Бромноватя
Бромная
Иодоводородная
Иодноватистая
Йодноватая
Ортопериодная
Хромовая
Дихромовая
Марганцовая
Соли
Традиционное
название
Бораты
(ортобораты)
Карбонаты
Метасиликаты
Ортосиликаты
Нитриты
Нитраты
Метафосфаты
Ортофосфаты
Дифосфаты
Арсенаты
Пероксиды
Сульфиды
Сульфиты
Сульфаты
Тиосульфаты
Фториды
Хлориды
Гипохлориты
Хлориты
Хлораты
Перхлораты
Бромиды
Гипобромиты
Броматы
Перброматы
Иодиды
Гипоиодиты
Иодаты
Ортопериодаты
Хроматы
Дихроматы
Перманганаты
В свободном виде не выделена.
Приложения
729
Таблица П. 1.2
Тривиальные названия и формулы некоторых неорганических веществ
Тривиальное название
Аммонийная селитра
Алюмокалиевые квасцы
Английская соль
Баритовые белила
Белая сажа
Белый графит
Берлинская лазурь
Бертолетова соль
Болотный (рудничный) газ
Боразон
Бура ювелирная
Веселящий газ
Гашеная (едкая) известь, пушонка
Гипс (другие формы: алебастр, мариенглас)
Глауберова соль
Глинозем (другие формы: корунд, алунд)
Двойной суперфосфат
Едкий барит
Едкий натр
Едкое кали, калиевый щелок
Железный купорос
Железокалиевые квасцы
Желтая кровяная соль
Жженая (негашеная) известь, известковая земля, кипелка
Жженая магнезия
Жженый (строительный) гипс
Известковая (норвежская) селитра
Индийская (калийная) селитра
Каломель
Кальцинированная сода
Каменная (поваренная) соль
Карборунд
Каустик, каустическая сода
Формула
NH4NO3
KA1(SO4J12H2O
MgSO4-7H2O
BaSO4
SiO2«H2O
BN(reKC)
KFe[Fe(CNN]
KCIO3
CH4
BN(Ky6)
Na2B4O710H2O
Na2B4O7-5H2O
N2O
Ca(OHJ
CaSO4-2H2O
Na2SO410H2O
A12O3
Ca(H2PO4J
Ba(OHJ
NaOH
KOH
FeSO4-7H2O
KFe(SO4J12H2O
K4[Fe(CNN]-3H2O
CaO
MgO
2CaSO4H2O
Ca(NO3JH2O
KNO3
Hg2Cl2
Na2CO3
NaCl
SiC
NaOH
730
Приложения
Окончание табл. П. 1.2
Тривиальное название
Киноварь
Красная кровяная соль
Красный сурик
Кремнезем
Кристаллическая сода
Медный купорос
Медная лазурь
Мел (другие формы: мрамор, известняк)
Натронная (чилийская) селитра
Нашатырный спирт
Нашатырь
Питьевая сода
Поташ
Пруссеновская лазурь
Свинцовые белила
Свинцовый купорос
Сернистый газ
Силикагель
Сулема
Сурик
Сусальное золото
Сухой лед
Турнбулева синь
Угарный газ
Углекислый газ
Фреон-12
Фосген
Хлорное железо
Хромовый купорос
Хромокалиевые квасцы
Хромпик
Цементит
Цинковый купорос
Формула
HgS
K3[Fe(CNN]
Pb3O4
SiO2
Na2CO310H2O
CuSO4-7H2O
Cu(OH)r2CuCO3
CaCOj
NaNO3
NH4OH
NH4CI
NaHCO3
K2CO3
Fe4[Fe(CNN]3
Pb(OH)r2PbCO3
PbSO4
SO2
SiO2«H2O (w<6)
HgCl2
Pb3O4
SnS2 (тонкие пластинки)
CO2(T)
+2
KFe[Fe(CNN]
CO
co2
CF2C12
COC12
FeClr6H2O
CrSO4-7H2O
KCr(SO4J12H2O
K2Cr207
Fe3C
ZnSO4-7H2O
Приложения
731
Таблица П.1.3
Технические названия некоторых дисперсных систем (смесей, растворов)
Техническое название
Аммиачная вода
Баритовая вода
Белильная(хлорная)
известь
Бордосская жидкость
Бромная вода
Водяной газ
Генераторный газ
Гидравлический гипс
Гипсовая вода
Гремучий газ
Жидкое стекло
Известка
Известковая вода
Известковое молоко
Известь венская
Йодная настойка
Купоросное масло
Ляпис
Олеум
Пергидроль
Плавиковая кислота
Свинцовый уксус
Серая известь
Сероводородная вода
Синильная кислота
Соляная кислота
Термит
Уксус
Уксусная эссенция
Формалин
Хлорная вода
Хромовая смесь
Царская водка
Состав
Водный раствор NH3
Насыщенный водный раствор Ва(ОНJ
Смесь Са(ОС1J, СаС12 и Са(ОНJ
Раствор C11SO4 в известковом молоке
Водный раствор Вг2 (содержит НВЮ и НВг)
Смесь СО и Н2
Смесь СО B5 % об.), N2 G0 % об.) и СО2 D % об.)
Смесь CaSO4 и СаО
Насыщенный раствор CaSO4
Смесь Н2 B/3 объема) и О2 A/3 объема)
Щелочной водный раствор Na2Si03 и K2Si03
Смесь Са(ОНJ, песка и воды
Насыщенный водный раствор Са(ОНJ
Суспензия Са(ОНJ в известковой воде
Смесь СаО и MgO
Раствор, содержащий 5 г 12, 2 г KI, 50 мл 96%-ного
раствора этилового спирта на каждые 50 мл воды
Концентрированный раствор H2SO4
Смесь AgNO3 и KNO3
Раствор SO3 в H2SO4 (содержит H2S2O7)
30 %-ный водный раствор Н2О2
Концентрированный водный раствор HF
Водный раствор РЬ(СН3СООJ
Неочищенный Са(СН3СООJ
Насыщенный водный раствор H2S
Водный раствор HCN
35-36 %-ный водный раствор НС1
Смесь порошкообразных А1 и Fe3O4
3-7 %-ный водный раствор уксусной кислоты
СН3СООН
80 %-ный водный раствор СН3СООН
37 %-ный водный раствор формальдегида НСНО
Водный раствор С12 (содержит НС1О и НС1)
Смесь концентрированной H2SO4 A/2 объема) и насы-
насыщенного водного раствора К2Сг207 A/2 объема)
Смесь концентрированной HNO3 A/4 объема) и соля-
соляной кислоты C/4 объема)
732
Приложения
П.2. Основные характеристики атомов и молекул
Таблица U2 А
Основные свойства атомов s-9 р- и а1- элементов: заряд ядра Z, молярная масса М,
атомный радиус R, первая энергия ионизации Е/ ь сродство к электрону ЕА,
электроотрицательность %
Название
Азот
Алюминий
Аргон
Барий
Бериллий
Бор
Бром
Ванадий
Висмут
Водород
Вольфрам
Галлий
Гафний
Гелий
Германий
Железо
Золото
Индий
Иод
Иридий
Иттрий
Кадмий
Калий
Кальций
Кислород
Кобальт
Кремний
Криптон
Ксенон
Лантан
Литий
Магний
Марганец
Медь
Сим-
Символ
N
А1
Аг
Ва
Be
В
Вг
V
Bi
Н
W
Ga
Hf
Не
Ge
Fe
Аи
In
I
Ir
Y
Cd
К
Ca
0
Co
Si
Kr
Xe
La
Li
Mg
Mn
Си
Z
7
13
18
56
4
5
35
23
83
1
74
31
72
2
32
26
79
49
53
77
39
48
19
20
8
27
14
36
54
57
3
12
25
29
M,
г/моль
14,007
26,981
39,948
137,327
9,012
10,81
79,904
50,941
208,980
1,0079
183,85
69,723
178,49
4,0026
72,61
55,847
196,966
114,82
126,904
192,22
88,905
112,411
39,098
40,078
15,999
58,933
28,085
83,80
131,29
138,905
6,941
24,305
54,938
63,546
R,
ПМ
71
143
174
217
113
88*
114*
132
155
78
137
122
156
128
122
124
144
163
133*
136
181
149
227
197
66*
125
117
189*
218
188
152
160
124
128
кДж/
/моль
1402,3
577,4
1520,4
502,8
899,4
800,6
1140
650
703,2
1312
770
578,8
642
2372,3
762,1
759,3
890,1
558,3
1008,4
880
616
867,6
418,8
589,7
1313,9
760,0
786,5
1350,7
1170,4
538,1
513,3
737,7
717,4
745,4
ЕЛ,
кДж/
/моль
-7
44
-35**
-46
-18
27
324,7
50,7
91,3
72,8
78,6
30
0
0
116
15,7
222,8
30
259,2
151
29,6
-26
48,4
-186
141
63,8
133,6
-39**
-41**
50
59,6
-21
<0
118,5
Электроотрицательность %
поПолингу
3,04
1,61
-
0,89
1,57
2,04
2,96
1,63
2,02
2,20
2,36
1,81
1,3
-
2,01
1,83
2,54
1,78
2,66
2,20
1,22
1,69
0,82
1,00
3,44
1,88
1,90
-
2,6
1,10
0,98
1,31
1,55
1,90
по Малликену, эВ
7,30
3,23
7,7***
2,40
4,9
4,29
7,59
3,6
4,69
7,18
4,40
3,2
3,8
12,3***
4,6
4,06
5,77
3,1
6,76
5,4
3,19
4,33
2,42
2,2
7,54
4,3
4,77
6,8***
5,85
3,1
3,01
3,75
3,72
4,48
Приложения
733
Окончание табл. П.2.1
Название
Молибден
Мышьяк
Натрий
Неон
Никель
Ниобий
Олово
Осмий
Палладий
Платина
Радий
Рений
Родий
Ртуть
Рубидий
Рутений
Свинец
Селен
Сера
Серебро
Скандий
Стронций
Сурьма
Таллий
Тантал
Теллур
Технеций
Титан
Углерод
Фосфор
Фтор
Хлор
Хром
Цезий
Цинк
Цирконий
Сим-
Символ
Мо
As
Na
Ne
Ni
Nb
Sn
Os
Pd
Pt
Ra
Re
Rh
Hg
Rb
Ru
Pb
Se
S
Ag
Sc
Sr
Sb
Tl
Та
Те
Тс
Ti
С
P
F
Cl
Cr
Cs
Zn
Zr
Z
42
33
11
10
28
41
50
76
46
78
88
75
45
80
37
44
82
34
16
47
21
28
51
81
73
52
43
22
6
15
9
17
24
55
30
40
A/,
г/моль
95,94
74,921
22,989
20,179
58,69
92,906
118,710
190,2
106,42
195,08
226,025
186,207
102,905
200,59
85,467
101,07
207,2
78,96
32,066
107,868
44,9559
87,62
121,75
204,383
180,947
127,60
98,906
47,88
12,011
30,973
18,998
35,453
51,996
132,905
65,39
91,224
R,
ПМ
136
125
154
—
125
143
140
135
137
138
223
137
134
160
247
134
175
215
104
144
160
215
182
170
143
143
136
145
77*
110*
58*
99*
125
265
133
160
Ei и
кДж/
/моль
685,0
947,0
495,8
2080,6
736,7
664
708,6
840
805
870
509,3
760
720
1007,0
403,0
711
715,5
940,9
999,6
731,0
631
549,5
833,7
589,3
761
869,2
702
658
1086,2
1011,7
1681
125,1
652,7
375,7
906,4
660
Ел,
кДж/
/моль
72,0
78
52,9
_29**
156
86,2
116
106
53,7
205,3
—
14
109,7
-18
46,9
101
35,1
195,0
200,4
125,7
18,1
-146
101
20
14
190,2
96
7,6
121,9
72,0
328
349
64,3
45,5
9
41,1
Электроотрицательность %
по Полингу
2,16
2,18
0,93
—
1,91
1,6
1,96
2,2
2,20
2,28
0,89
1,9
2,28
2,00
0,82
2,2
2,33
2,55
2,59
1,93
1,36
0,95
2,05
1,62
1,5
2,1
1,9
1,54
2,56
2,19
3,98
3,16
1,66
0,79
1,65
1,33
по Малликену, эВ
3,9
5,3
2,85
10,6***
4,40
4,0
4,30
4,9
4,45
5,6
—
4,02
4,30
4,91
2,34
4,5
3,90
5,89
6,22
4,44
3,34
2,0
4,85
3,2
4,11
5,49
3,91
3,45
6,27
5,62
10,41
8,30
3,72
2,18
4,45
3,64
* Ковалентныи радиус.
** Расчетные значения.
*** Приближенное значение.
734
Приложения
Таблица П. 2.2
Длины связей и валентные углы молекул и ионов с одним центральным атомом
Частица
А1С13
[А1СЦГ
АН3
Asl3
ВВгз
BF3
BeF2
Bel2
[BeF4]2"
BiBr3
BrF3
[BrF4]"
CC14
CC12O
co2
cs2
[C1F2]+
CIF5
GaCl3
GeCl2
GeO2
HCN
HOF
[ici2r
[IC12]+
NCS"
NH4+
NH2"
Тип гибри-
гибридизации
валентных
орбиталей
центрально-
центрального атома
sp2
sp3
sp2
sp2
sp2
sp2
sp
sp
sp3
sp3
sp3d
sp'd2
sp3
sp2
sp
sp
sp3
sp'd2
sp2
sp2
sp
sp
sp3
sp*d
sp3
sp
sp3
sp3
Длина связи, пм
Al-Cl 206
Al-Cl 209
Al-I 244
As-I 256
B-Brl89
B-F 131
Be-F 140
Be-I210
Be-F 155
Bi-Br 263
Br-F172;Br-F* 181
Br-F 188
C-Cl 177
C-C1175;C-O117
C-0 117
C-S 155
Cl-F 156
Cl-F 167; Cl-F* 158
Ga-Cl 209
Ge-C1211
Ge-0 163
C-H 107,C-N115
O-H97,O-F144
I-C1*255
I-C1230
C-N 117, C-S 160
N-H 104
N-H 103
Валентный угол
C1-A1-C1 120°
C1-A1-C1 109,5°
I-Al-I 120°
I-As-I 100°
Br-B-Br 120°
F-B-F 120°
F-Be-F 180°
I-Be-I 180°
F-Be-F 109,5°
Br-Bi-Br 100°
F-Br-F* 86°; F*-Br-F* 188°
F-Br-F 90°
Cl-C-Cl 109,5°
Cl-C-Cl 111°; Cl-C-0 124°
O-C-0 180°
S-C-S 180°
F-Cl-F 100°
F-Cl-F* 86°
Cl-Ga-Cl 120°
Cl-Ga-Cl 107°
O-Ga-0 180°
H-C-N 180°
H-O-F 97°
C1*-I-C1* 180°
Cl-I-Cl 95°
N-C-S 180°
H-N-H 109,5°
H-N-H 104°
Приложения
735
Окончание табл. П.2.2
Частица
N02
NOF
NOF3
0F2
PCI3
РС15
РС130
PF5
[PF6]"
РЬС12
РЬСЦ
РЫ2
S02
S03
SbCl5
SbF3
SeF4
SiF4
[SiF6]2"
SnCl2
SnCl4
SnO2
TeCl4
XeF2
[XeF3]+
XeF4
[XeF5]+
XeOF4
XeO2F2
Тип гибри-
гибридизации
валентных
орбиталей
центрально-
центрального атома
sp2
sp2
sp3
sp3
sp3
sp3d
sp3
sp3d
sP4
sp2
sp3
sp2
sp2
sp2
sp3d
sp3
sp3d
sp3
sp'd2
sp2
sp3
sp
sp3d
sp2d
sp3d
sP4
sp'd2
spV
sp'd2
Длина связи, пм
N-0 120
N-O114;N-F151
N-O116;N-F143
O-F 141
P-Cl 204
P-Cl 202; P-Cl* 212
P-Cl 204; P-O 146
P-F 153; P-F* 158
P-F 159
Pb-Cl 246
Pb-Cl 243
Pb-I279
S-0 143
S-0 142
P-Cl 229; P-Cl* 234
Sb-F 188
Se-F 168; Se-F* 177
Si-F 156
Si-F 170
Sn-Cl 234
Sn-Cl 228
Sn-Cl 181
Te-Cl 233; Te-Cl*>233
Xe-F* 198
Xe-F 183; Xe-F* 191
Xe-F 194
Xe-F 190; Xe-F* 177
Xe-O* 171; Xe-F 190
Xe-0 171; Xe-F* 190
Валентный угол
O-N-0 134°
O-N-F1100
O-N-F112°;F-N-F101°
F-O-F 103°
Cl-P-Cl 100°
Cl-P-Cl 120°; Cl-P-Cl* 90°
Cl-P-Cl 104°; Cl-P-O 115°
F-P-F 120°; F-P-F* 90°
F-P-F 90°
Cl-Pb-Cl 96°
Cl-Pb-Cl 109,5°
I-Pb-I 95°
O-S-O1190
O-S-0 120°
Cl-P-Cl 120°; Cl-P-Cl* 90°
F-Sb-F 95°
F-Se-F 100°; F*-Se-F* 169°
F-Si-F 109,5°
F-Si-F 90°
Cl-Sn-Cl 100°
Cl-Sn-Cl 109,5°
O-Sn-0 180°
Cl-Te-Cl 120°; Cl-Te-Cl* 120°
F*-Xe-F* 180°
F-Xe-F*80-82°; F*-Xe-F* 162°
F-Xe-F 90°
F-Xe-F...; F-Xe-F* 79-83°
F-Xe-F...;F-Xe-O* 91°
O-Xe-0 106°; O-Xe-F* 92°
Концевые атомы, находящиеся в аксиальных позициях.
736
Приложения
Таблица П.2.3
Постоянные дипольные моменты \i, длины связи / и энергии связи ?А-в
некоторых двухатомных молекул
Вещество
HF
НС1
НВг
HI
KF
КС1
КВг
KI
LiH
LiF
LiBr
Lil
NaCl
Nal
RbF
RbBr
CsF
CsCl
Csl
BrF
ц,Д
1,91
1,08
0,79
0,42
7,33
11,05
10,41
9,20
5,88
6,60
6,20
6,25
10,00
8,50
8,80
10,00
7,88
10,42
12,10
1,29
/, пм
92
128
141
161
217
267
282
305
160
156
217
239
236
271
227
295
235
291
332
176
?А-в> кДж/моль
566
432
366
298
497
425
382
325
236
577
423
353
411
290
500
387
514
443
336
233
Вещество
C1F
IC1
IBr
СО
CS
NO
н2
F2
Cl2
Вг2
h
Li2
Na2
к2
Rb2
Cs2
c2
N2
o2
s2
0,65
0,65
1,21
0,11
1,97
0,16
0
0
0
0
0
0
0
0
0
0
0
0
0
0
/, пм
163
232
247
113
154
115
74
141
199
228
267
267
308
392
410
430
124
110
121
189
?А_в, кДж/моль
251
212
179
1076
714
632
436
159
243
194
153
102
73
74
49
42
605
945
498
426
П.З. Физические свойства неорганических веществ
Таблица П. 3.1
Температуры плавления Тпл и окраска некоторых солей
Соль
LiF
LiCl
NaF
NaCl
NaBr
Nal
KF
KC1
KBr
KI
BeF2
BeCl2
CaF2
CaCl2
PbCl2
Tm,°C
845,1
610
997
800,8
755
661
857
770
734
681
803
550
1419
782
501
Окраска
Белая
Белая
Белая
Белая
Белая
Белая
Белая
Белая
Белая
Белая
Белая
Светло-зеленая
Белая
Белая
Белая
Соль
РЫ2
SbF3
SbCl3
AgCl
AgBr
CoF2
CoCl2
NiF2
NiCl2
A1C13
GaCl3
CdCl2
HgCl2
ScCl3
YC13
Tnn,°C
402
287
72,3
455
432
1127
740
1160
1009
192,6
77,8
568,5
280
967
721
Окраска
Желтая
Белая
Белая
Белая
Светло-желтая
Розово-красная
Голубая
Желто-зеленая
Желто-коричневая
Белая
Белая
Белая
Белая
Белая
Белая
Приложения
Таблица П. 3.2
Тип кристаллической решетки и плотность р некоторых металлов при Т= 293 К
Металл
Алюминий А1
Барий Ва
Бериллий а-Ве
Ванадий V
Вольфрам W
Гадолиний a-Gd
Гафний a-Hf
Гольмий а-Но
Германий Ge
Европий Ей
Железо a-Fe
Золото Аи
Иридийн*
Иттрий a-Y
Кадмий a-Cd
Калий К
Кальций а-Са
Кобальт а-Со
Лантан a-La
Литий E-Li
Магний Mg
Марганец а-Мп
Медь Си
Тип кристал-
кристаллической
решетки
гцк
оцк
ГПУ
оцк
оцк
ГПУ
ГПУ
ГПУ
ПК
оцк
оцк
гцк
гцк
ГПУ
ГПУ
оцк
гцк
гцк
ГПУ
гцк
ГПУ
оцк
гцк
р>
кг/м
2698
3594
1848
6110
19300
7900
13310
8795
5323
5243
7874
19320
22560
4465
8650
862
1550
8900
6145
534
1738
7440
8960
Металл
Молибден Мо
Натрий a-Na
Никель Ni
Ниобий Nb
Олово a-Sn
Осмий Os
Палладий Pd
Платина Pt
Рений Re
Родий Rh
Рубидий Rb
Свинец РЬ
Серебро Ag
Скандий a-Sc
Стронций a-Sr
Таллий а-Т1
Тантал Та
Титан a-Ti
Хром Сг
Цезий Cs
Цинк2п
Цирконий a-Zr
Эрбий a-Er
Тип кристал-
кристаллической
решетки
ОЦК
ГПУ
гцк
оцк
ПК
ГПУ
гцк
гцк
ГПУ
гцк
оцк
гцк
гцк
ГПУ
гцк
ГПУ
оцк
ГПУ
оцк
оцк
ГПУ
ГПУ
ГПУ
р>
кг/м
10220
971
8902
8570
5750
22590
12020
21450
21020
12410
1532
11350
10500
2989
2540
11850
16654
4540
7190
1873
7133
6506
9066
Таблица П. 3.3
Плотность р некоторых оксидов металлов при Т= 293 К
Оксид
А12О3
ВаО
СаО
Си2О
FeO
К2О
MgO
МоО3
р-10,кг/м3
3,97
5,72
3,37
6,10
5J
2,32
3,58
4,5^,7
Оксид
Na2O
Nb2O5
NiO
Pb3O4
SnO
Та2О5
WO3
ZnO
р-10~3,кг/м3
2,36
4,47-5,29
6,67
9,1
6,446
8,235
7,2-7,4
5,7
738
Приложения
IL4. Свойства воды и водных растворов
Таблица П.4.1
Давление насыщенного водяного пара рн 0 при различных температурах
г,°с
0
1
2
3
4
5
6
7
8
9
10
И
12
13
14
15
16
17
18
19
20
Рн2о
Па
610,4
657,2
705,2
757,3
813,3
871,9
934,6
1001,2
1073,2
1147,8
1227,9
1311,9
1402,5
1497,2
1598,5
1705,1
1817,1
1947,1
2063,8
2197,1
2337,8
мм рт. ст.
4,58
4,93
5,29
5,69
6,10
6,54
7,01
7,51
8,05
8,51
9,21
9,84
10,52
11,23
11,99
12,79
13,63
14,53
15,48
16,48
17,53
Т,°С
21
22
23
24
25
26
27
28
29
30
32
34
36
38
40
50
60
70
80
90
100
Ри2о
Па
2486,4
2643,7
2809,0
2983,7
3167,2
3361,0
3564,9
3779,6
4004,9
4242,2
4753,0
5318,0
5940,0
6623,0
7374,0
12334,0
19920,0
31160,0
43360,0
70100,0
101325,0
мм рт. ст.
18,65
19,83
21,07
22,38
23,76
25,21
26,74
28,35
30,04
31,82
35,66
39,90
44,56
49,69
55,32
92,51
149,38
233,70
.355,10
525,76
760,00
Плотность р воды при различных температурах
Таблица П. 4.2
г,°с
0
1
2
4
6
8
10
12
14
16
18
20
22
р, г/л
999,841
999,900
999,941
999,973
999,941
999,849
999,700
999,498
999,244
998,943
998,595
998,203
997,770
Г/С
24
25
26
28
30
32
34
36
38
40
42
44
46
р, г/л
997,296
997,044
996,783
996,232
995,646
995,02
994,37
993,68
992,96
992,21
991,44
990,63
989,79
Г,°С
48
50
52
55
60
65
70
75
80
85
90
95
100
р, г/л
988,93
988,04
987,12
985,70
983,21
980,56
977,78
974,86
971,80
968,62
965,31
961,89
958,35
Приложения
739
Таблица П. 4.3
Ионное произведение воды Кно при различных температурах
г,°с
0
10
15
20
25
30
V10'4
0,11
0,29
0,45
0,68
1,00
1,47
Г,°С
35
40
50
60
80
-100
*Hi0-ioM
2,09
2,92
5,47
9,61
25,1
59,0
Таблица П. 4.4
Плотность р водных растворов некоторых веществ при Т= 293 К
NaCl
со
0,01
0,02
0,04
0,06
0,08
0,10
0,12
0,14
0,16
0,18
0,20
0,22
0,24
0,26
р, г/л
1005,3
1012,5
1026,8
1041,3
1055,9
1070,7
1085,7
1100,9
1116,2
1131,9
1147,8
1164,0
1180,4
1197,2
HNO3
со
0,040
0,110
0,176
0,240
0,300
0,359
0,421
0,484
0,551
0,627
0,716
0,824
0,967
0,998
р, г/л
1020,0
1060,0
1100,0
1140,0
1180,0
1220,0
1260,0
1300,0
1340,0
1380,0
1420,0
1460,0
1500,0
1512,0
H2SO4
со
0,003
0,032
0,062
0,091
0,120
0,147
0,174
0,201
0,227
0,252
0,277
0,302
0,326
0,350
р,г/л
1000,0
1020,0
1040,0
1060,0
1080,0
1100,0
1120,0
1140,0
1160,0
1180,0
1200,0
1220,0
1240,0
1260,0
NH3
со
0,019
0,038
0,058
0,078
0,099
0,120
0,143
0,167
0,191
0,215
0,240
0,267
0,293
0,344
р,г/л
0,990
0,982
0,974
0,966
0,958
0,950
0,942
0,934
0,926
0,918
0,910
0,902
0,894
0,880
НС1
со
0,004
0,024
0,044
0,064
0,085
0,105
0,125
0,145
0,165
0,184
0,204
0,223
0,243
0,262
р, г/л
1000,0
1010,0
1020,0
1030,0
1040,0
1050,0
1060,0
1070,0
1080,0
1090,0
1100,0
1110,0
1120,0
1130,0
NaOH
со
0,019
0,056
0,092
0,128
0,164
0,201
0,237
0,274
0,311
0,350
0,390
0,431
0,473
0,505
р, г/л
1020,0
1060,0
1100,0
1140,0
1180,0
1220,0
1260,0
1300,0
1340,0
1380,0
1420,0
1460,0
1500,0
1530,0
СНзСООН
со
0,04
0,08
0,12
0,16
0,20
0,24
0,28
0,32
0,36
0,40
0,44
0,48
0,52
0,56
р, г/л
1004,1
1009,8
1015,4
1020,8
1026,1
1031,2
1036,0
1040,5
1044,8
1048,8
1052,5
1055,9
1059,0
1061,8
КОН
со
0,046
0,089
0,131
0,173
0,194
0,214
0,254
0,293
0,330
0,367
0,404
0,439
0,474
0,521
р, г/л
1040,0
1080,0
1120,0
1160,0
1180,0
1200,0
1240,0
1280,0
1320,0
1360,0
1400,0
1440,0
1480,0
1535,0
740
Приложения
Таблица П.4.5
Растворимость неорганических веществ в воде при Т = 298 К
Ионы
А<
А13+
Ва2+
Ве2+
Са2+
Cd2+
Со2+
Сг3+
Cs+
Cu2+
Fe2+
Fe3+
H+
Hg2+
к+
Li+
Mg2+
Mn2+
NH4+
Na+
Ni2+
Pb2+
Zn2+
CH3COO"
M
M
p
M
p
p
p
p
p
p
-
p
p
p
p
p
p
p
p
p
p
p
CN"
T
-
P
-
P
M
T
T
p
T
T
T
p
p
p
p
T
T
p
p
T
T
T
CO32"
T
-
H
-
T
-
T
-
p
T
T
-
M
-
p
p
M
T
p
p
T
T
T
СГ
T
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
M
p
F"
p
M
M
p
T
p
p
M
p
p
M
T
p
-
p
T
M
p
p
p
p
M
M
NO3-
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
OH"
-
T
p
T
M
T
T
T
p
T
T
T
p
-
p
p
T
T
p
p
T
T
T
PO32"
T
T
T
T
T
T
T
T
p
T
T
T
p
T
p
M
T
T
p
p
T
T
T
s2-
T
-
p
-
p
T
T
-
p
T
T
-
M
T
p
p
T
T
-
p
T
T
T
SO42"
M
p
T
p
M
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
p
T
p
Примечание, р - растворимые (>10 г/л Н2О); м - малорастворимые A0...0,01 г/л
H2O); т - труднорастворимые (<0,01 г/л Н2О); прочерк - полностью гидролизуются
или не существуют.
Таблица П.4.6
Эбулиоскопическая Е и криоскопическая К постоянные некоторых растворителей
Растворитель
Вода
Бензол
Анилин
Этанол
Ацетон
Хлороформ
Диэтиловый эфир
Е, К • кг/моль
0,52
2,57
3,69
1,16
1,50
3,89
2,02
К, К • кг/моль
1,86
5,10
5,87
-
2,40
4,90
1,73
Приложения
1А\
Таблица ПА. 7
Давление насыщенных паров рр и плотность р водных растворов некоторых солей
с массовой долей соли со при температуре Т = 293 К
СиС12
со
0,10
0,15
0,20
0,25
РР • Ю2, Па
22,2
21,4
20,7
19,7
р, г/л
1096,0
1149,0
1205,0
1265,5
KI
со
0,10
0,20
0,30
0,40
РР' Ю2, Па
23,3
22,7
21,9
20,4
р, г/л
1076,1
1166,0
1272,2
1395,9
NaBr
0)
0,10
0,20
0,30
0,40
РР • Ю2, Па
23,2
21,1
18,9
17,0
р, г/л
1076,1
1174,5
1284,1
1413,8
CuSO4
со
0,05
0,10
0,15
0,20
РР • Ю2, Па
23,3
22,8
22,0
21,1
р, г/л
1051,0
1107,0
1167,5
1219,0
Mg(NO3J
со
0,10
0,15
0,20
0,25
Рр • Ю2, Па
22,9
21,6
20,0
18,5
р, г/л
1076,2
1118,5
1163,0
1210,1
ZnSO4
со
0,05
0,15
0,25
0,30
РР • 102, Па
20,0
21,1
21,1
20,9
р, г/л
1051,2
1168,0
1304,0
1378,0
Таблица П.4.8
Предельная молярная электропроводность Х°° и удельная электропроводность х на-
насыщенных растворов некоторых солей при Т = 298 К
Соль
Т1С1
SrSO4
MgF2
CaF2
BaF2
РЬС12
PbBr2
Pbl2
AgCl
AT, Cm • м2/моль
1,51 • 10
2,79 • 10'2
2,17 • 10
2,30 • 10
2,38 • 10
2,60 • 10
2,64 • 10
2,65 • 10
1,38 • 10
x, Cm/m
2,40 • 10
1,28- 10
2,54 • 10~2
4,99 • 10
1,78 • 10
4,42 • 10
3,46 • 10
3,31 • 10~2
1,50 10
742
Приложения
Таблица П. 4.9
Предельная молярная электропроводность Xе0 некоторых ионов при Т= 298 К
Ионы
Н+
NH4+
ОН"
А,00 • 104,
См • м2/моль
349,8
73,55
198,3
Ионы
НСОО"
СН3СОО~
с2н5соо-
г • ю4,
См • м2/моль
56,7
40,9
3,8
Таблица П. 4.10
Константы диссоциации некоторых кислот К& и оснований Къ при Т= 298 К
Кислота
Формула
СНзСООН
НВЮ
HCN
Н2СО3
Н2С2О4
нсоон
нею
Н2СЮ4
Н2Сг207
HF
НЮ3
HNCS
HNO2
Н3РО4
H2S
H2SO3
H2SO4
H2Se
КЛ
1,74 10
2,06 • 10"9
4,93 • Ю0
(I) 4,27 • 10
(II) 4,68-10-"
(I) 6,46 • 10
(II) 6,17 • 10
1,78 10
2,82 • 10"8
(II) 3,16 • 10
(II) 2,29 • 10
6,67 • 10
1,69- 10"'
1,41 • 10
5,13 • 10
(I) 7,24 • 10
(II) 6,17 • 10"8
(III) 4,57 • 10"'3
(I) 1,05 • 10
(II) 1,23 • 10~13
(I) 1,40 10
(II) 6,31 • 10"8
(II) 1,12 • 10
(I) 1,55 • 10
(II) 1,0 • 10""
4,76
8,69
9,31
6,37
10,33
1,19
4,21
3,75
7,55
6,50
1,64
3,18
0,77
0,85
3,29
2,14
7,21
12,34
6,98
12,91
1,85
7,20
1,95
3,81
11,00
Основание
Формула
AgOH
A1(OHK
Ba(OHJ
Ca(OHJ
Cd(OHJ
Со(ОНJ
Cr(OHJ
Cu(OHJ
Fe(OHJ
Fe(OHK
LiOH
Mg(OHJ
Mn(OHJ
NH4OH
Ni(OHJ
Pb(OHJ
Sr(OHJ
Zn(OHJ
5,0 • 10
(I) 7,41 • 10"9
(II) 2,14 • 10"9
(III) 1,05 • 10"9
(II) 2,3 • 10
(II) 5,8 • 10~2
(I) 8,13 • 10
(II) 4,17 • 10
(I) 7,9 • 10
(II) 7,9 • 10
(II) 3,98 • 10""8
(I) 6,61 • 10"8
(I) 1,2 • 10-2
(II) 1,3 • 10
(II) 1,82-10""
4,37 • 10
,(H) 2,63 lO
(II) 3,89 • 10
1,74- 10
(II) 8,32 • 10
(I) 5,01 • 10
(II) 1,41 • 10"8
(II) 1,48-10"'
(I) 1,32 • 10
(II) 2,00 • 10"9
P*b
2,30
8,13
8,67
8,98
0,64
1,23
3,09
6,38
4,10
5,10
7,40
7,18
1,92
3,89
10,74
0,36
2,58
3,41
4,76
3,08
3,30
7,85
0,83
4,88
8,70
Приложения
743
Таблица ПАЛ I
Произведение растворимости ПР малорастворимых электролитов при Т= 298К
Электролит
AgBr
Ag2Cr04
Ag2Cr207
Agl
AgOH
ВаСЮ4
BaSO4
Ва(ОНJ
СаСО3
СаС2О4
СаНРО4
Са(Н2РО4J
Са3(РО4J
Са(ОНJ
CaSO4
CdCO3
ПР
5,0 • 103
1,2-102
2,0- 10
8,5 • 107
1,6- 10~8
1,1 • 100
1,8-100
5,0- 10
4,4-10'9
2,3 • 10~9
2,2 • 10
1,0- 10
1,0- 10~25
6,3 • 10
3,7 • 10
2,5 • 104
Электролит
Cd(OHJ
СоСОз
Со(ОНJ
Сг(ОНJ
CuCN
CuOH
Cu(OHJ
CuS
FeCO3
Fe(OHJ
FeO(OH)
Fe(OHK
FePO4
HgS
LiOH
MgCO3
ПР
4,3 - 105
1,5 • 10-10
1,6- 105
1,0-10'17
3,2 • 100
1,0-10'14
5,6-100
1,4 • 106
2,9-lO1
7,9 • 10~16
2,2-lO2
6,3 • 10'38
1,3 • 102
1,4-105
4,0 • 10
7,9-10^
Электролит
MgF2
Mg(OHJ
Mg2(OHJCO3
Mg3(PO4J
MnCO3
Mn(OHJ
NiCO3
Ni(OHJ
PbCl2
РЬСЮ4
Pb(OHJ
PbSO4
Snl2
Sr(OHJ
ZnCO3
Zn(OHJ
ПР
6,4 • 10"9
6,8 • 10-'2
<6,8- 10~12
3,9 • 106
4,9- 10""
2,3 • 10~13
1,3 • 10-7
1,6 • 10"'4
1,7 • 10-5
2,8 • 103
5,5 • 10-'6
1,7 • 10-8
8,3 • lO
3,2 • 10
5,3 lO""
3,1 • 10-'5
Таблица П. 4.12
Константы нестойкости Кн некоторых комплексных ионов при Т= 298 К
Комплексный ион
[Ag(NH3J]+
[Ag(OHK]2;
[Cd(NH3N]2+
[Co(C5H5NJ]2+
К„
6,17- 10"8
6,31 • 10
7,24 • 10
2,88 • 10
Комплексный ион
[Co(NH3N]2+
[Cu(NH3N]2+
[Hg(NH3L]2+
[Ni(NH3N]2+
K.
4,07 • 10
1,26- 10"9
3,39 • 10"8
4,90 • 10"9
Комплексный ион
[Pb(OHK]"
[Zn(NH3L]2+
[Fe(CNN]3"
[FeCCN^]4-
KH
1,20- 10"'4
2,40 • 10-9
1,26 -lO4
1,26- 107
Таблица И4.13
Средние ионные коэффициенты активности у± некоторых сильных электролитов
в водных растворах различной моляльной концентрации Ст при Т= 298 К
С„, моль/(кг Н2О)
0,001
0,005
0,01
0,02
0,05
0,1
0,2
0,5
1,0
Средний ионный
AgNO3
-
0,925
0,897
0,860
0,793
0,734
0,657
0,536
0,429
CuSO4
0,74
0,573
0,438
0,317
0,217
0,154
0,104
0,062
0,043
коэффициент активности у+ электролита
Pb(NO3J
0,88
0,76
0,69
0,60
0,46
0,37
0,27
0,17
0,11
NiSO4
-
-
-
-
-
0,150
0,105
0,063
0,042
CdSO4
0,726
0,505
0,399
0,307
0,206
0,150
0,102
0,061
0,041
ZnSO4
0,700
0,477
0,387
0,298
0,202
0,150
0,104
0,063
0,043
744
Приложения
П.5. Термодинамические характеристики некоторых веществ
Таблица П. 5.1
Стандартные энтальпии образования А///® , энтропии SQ2n
и энергии Гиббса образования
некоторых веществ
Вещество
Ag(T)
AgCl(T)
AgNO3(T)
Ag2O(T)
A1(T)
A1C13(T)
А12О3(Т)
А1(ОНK(Т)
A12(SO4K (Т)
Аи(т)
Аи(ОНK(т)
Au2O3(T)
Ва(т)
ВаСО3(т)
Ba(NO3J(T)
ВаО(т)
BaS(T)
BaSO4(T)
ВаТЮ3(т)
Вг2(ж)
С (т, графит)
СС14(Ж)
СН4(Г)
С2Н2 (Г)
С2Н4(Г)
С2Н6 (г)
С4Н6 A,2-бутадиен) (г)
С4Н8A-бутен)(г)
С4НШ (бутан) (г)
С6Н6(Ж)
C6Hi2(r)
СН3ОН (ж)
СН3СООН (ж)
С2Н5ОН (ж)
С3Н6О(Ж)
3 П 298 '
кДж/моль
0
-126,78
-124,52
-30,54
0
-704,17
-1676,0
-1315,0
-3441,80
0
-477,80
-13,0
0
-1210,85
-992,07
-553,54
-460,5
-1465,0
-1663,6
0
0
-132,84
-74,85
+226,80
+52,30
-84,67
+162,21
-0,13
-126,15
+49,03
-123,14
-201,00
-484,09
-277,60
-248,11
?2°98, Дж/(моль ¦ К)
+42,55
+96,23
+140,92
+121,75
+28,33
+109,29
+50,92
+70,10
+239,20
+47,40
+121,0
+134,30
+60,67
+112,13
+213,80
+70,29
+78,3
+132,0
+108,03
+152,21
+5,74
+216,19
+186,27
+200,80
+219,45
+229,46
+293,01
+305,60
+310,12
+173,26
+298,24
+239,76
+159,83
+160,67
+200,41
A/G° j кДж/моль
0
-109,54
-33,60
-10,90
0
-628,58
-1582,0
-1157,0
-3100,87
0
-349,80
+78,70
0
-1132,77
-797,23
-525,84
-456,0
-1353,0
-1574,90
0
0
-62,66
-50,85
+209,20
+68,14
-32,93
+198,44
+71,26
-17,19
+124,38
+31,70
-162,38
-389,36
-174,15
-155,42
Приложения
745
Продолэюение табл. П.5.1
Вещество
CN-(p)
со(г)
со2(г)
СОС12(Г)
CS2 (Г)
Са(т)
СаСО3(т)
СаС12(т)
СаНРО4(Т)
СаО(т)
Са3(РО4J(т)
CaSO4(T)
Cd(T)
CdCl2(T)
Cd(NO3J-4H2O(T)
ci2(r,
Co(T)
CoO(T)
Cr(I)
Cr2O3(T)
Cr(OHK(T)
Cr2(SO4K(T)
Cu(T)
Cu2CO3(OHJ(T)
Cu(NO3J(T)
CuO(T)
Cu(OHJ(T)
CuS(T)
F2(r)
Fe(T)
Fe2+(P,
Fe3+(P)
[FeCCNy4-^
[FeCCNM3-^
Fe(COM()lc)
FeO(T)
Fe2O3(T)
Fe3O4(T)
Fe(OHK(T)
FeSO4(T)
1сДж/моль
+150,62
-110,52
-393,51
-220,30
+116,70
0
-1206,90
-795,0
-1808,56
-635,50
-4120,82
-1436,28
0
-390,79
-1653,20
0
0
-239,30
0
-1140,56
-999,98
-3308,0
0
-1051,0
-305,30
-162,0
-444,30
-53,14
0
0
-87,86
-47,70
+530,11
+635,13
-764,0
-264,85
-822,16
-1117,13
-826,60
-927,59
52°98, Дж/(моль • К)
+96,45
+197,54
+213,68
+283,90
+237,77
+41,63
+92,90
+113,60
+111,38
+39,70
+235,98
+106,68
+51,76
+115,27
+393,0
+222,98
+30,04
+43,90
+23,64
+81,17
+80,33
+288,0
+33,14
+211,60
+192,50
+42,60
+84,0
+66,53
+202,67
+27,15
-113,39
-293,30
+92,05
+267,78
338,0
+60,75
+87,45
146,19
+105,0
+107,53
A/G° »кДж/моль
+171,58
-137,14
-394,38
-266,90
+66,55
0
-1128,80
-750,20
-1675,38
-604,20
-3887,90
-1323,90
0
-343,24
-1236,50
0
0
-213,40
0
-1058,97
-849,02
-2984,0
0
-900,90
-117,15
-129,40
-359,40
-53,58
0
0
-84,88
-10,53
+769,44
+803,75
-695,20
-244,30
-740,34
-1017,14
-699,6
-819,77
746
Приложения
Продолэюение табл. П. 5.1
Вещество
FeSO4-7H2O(T)
Fe2(SO4K(T)
H(r)
Н2(Г)
HBr(r)
HCOH(r)
нсоон(ж)
HCl(r)
нсю4(ж)
HF(r,
HNO3W
H2O(r)
н2о(ж)
н2о2(ж)
H2S(r)
H2SO4()i()
Ь(г)
K(T)
K2CO3 (T)
KC1(T)
KHCO3(T)
KNO2(T)
KNO3(T)
K2O(T)
K2SO4(T)
Mg(T)
MgCO3(T)
MgCl2(T)
MgO(T)
Mg(OHJ(T)
MgSO4(T)
Mn(Tja)
MnCO3(T)
MnCl2(T)
MnO(T)
MnO2(T)
N(r)
N2(r)
A/>C>
кДж/моль
-3016,0
-2584,0
+217,98
0
-36,38
-115,90
-424,76
-92,31
-34,50
-273,30
+26,36
-173,00
-241,82
-285,83
-187,86
-21,0
-813,99
+62,43
0
0
-1150,18
-436,68
-959,30
-370,30
^92,46
-363,20
-1433,69
0
-1095,85
-644,80
-601,49
-924,66
-1287,42
0
-881,66
-481,16
-385,10
-521,49
+472,78
0
S298, Дж/(моль • К)
+409,10
+282,80
+114,60
+130,52
+198,58
+218,78
+128,95
+186,79
+188,0
+173,67
+206,48
+156,16
+188,72
+70,08
+109,60
+205,72
+156,90
+260,60
+116,14
+71,45
+155,52
+82,55
+128,70
+117,0
+132,88
+94,10
+175,56
+32,68
+665,10
+89,54
+27,07
+63,18
+91,55
+32,01
+109,54
+118,24
+61,50
+53,14
+153,20
+191,50
A fG° , кДж/моль
J 298
-2512,0
-2253,0
+203,28
0
-53,43
-109,94
-361,74
-95,30
+84,31
-275,41
+1,58
-79,90
-228,61
-237,24
-120,52
-33,80
-690,14
+19,39
0
0
-1064,87
-408,93
-860,60
-218,60
-392,75
-322,10
-1316,04
0
-1012,15
-595,30
-569,27
-833,75
-1173,25
0
-811,40
-440,41
-363,34
-466,68
+455,50
0
Приложения
747
Продолжение табл. П. 5.1
Вещество
NH3(r)
NH4C1(T)
NH4C1O4(T)
(NH4JCrO4(T)
(NH4JCr2O7(T)
NH4NO3(T)
NO(r)
NO2(r)
N2O(r)
N2O3(r)
N2O4(r)
N2O5(r)
N2O5(T)
Na(T)
Na2CO3(T)
Na2CO3- 10H2O(T)
NaCl (T)
NaC103(T)
NaClO4(T)
NaNO3(T)
NaOH(T)
Na2SO3(T)
Na2SO4(T)
Na2Si03(T)
Ni{T)
NiC!2(T)
NiO(T)
NiSO4(T)
O(r)
O2(r)
Оз(г)
" (т, красный)
* (т, черный/
?4 (т, белый)
РСЬ(г)
РС13(ж)
РС15(г)
PCI5 (ж)
Р4Ою(г)
Р4Ою(т)
Pbm
кДж/моль
-465,94
-314,22
-295,90
-1182,40
-1799,12
-365,43
+91,26
+34,19
+82,01
+83,30
+11,11
+13,30
-42,70
0
-1130,80
-4083,50
-411,12
-365,40
-382,80
-466,70
-426,35
-1089,43
-1384,60
-1561,43
0
-304,18
-239,74
-873,49
+249,17
0
+142,26
-17,60
-38,90
0
-287,02
-320,91
-374,89
-445,89
-2897,49
-2984,03
0
52°98, Дж/(моль • К)
+192,66
+95,81
+184,30
+167,78
-
+151,04
+210,64
+240,06
+219,83
+307,30
+304,35
+356,65
+178,40
+51,57
+138,80
+564,70
+72,13
+122,70
+140,0
+116,50
+64,43
+146,02
+149,50
+113,76
+29,87
+98,07
+37,99
+103,85
+160,95
205,04
+238,82
+22,80
+22,70
+41,10
+311,79
+218,49
+364,47
+170,80
+394,55
+228,86
+64,81
A/G° 9 кДж/моль
-16,48
-203,22
-88,80
-995,80
-
-183,93
+87,58
+52,29
+104,12
+140,60
+98,29
+117,14
+114,20
0
-1048,20
-3424,30
-384,13
-275,0
-282,0
-365,97
-380,29
-1001,21
-1266,80
-1467,50
0
-258,03
-211,60
-463,76
+231,75
0
+162,76
-11,90
-33,47
0
-267,58
-274,08
-305,10
-318,36
-2657,46
-2697,60
0
748
Приложения
Окончание табл. П.5.1
Вещество
РЬС12(Т)
PbCO3(T)
Pb(NO3J(T)
PbO(T)
PbO2(T)
PbS(T)
PbSO4(T)
Pdw
PdO(T)
Pt(T)
PtO2(T)
^ (т, монокл)
S (т, ромб)
so2(r)
so3(r)
so3W
SiH4(r)
SlO2 (т, аморф)
olvJ2 (т, а-кварц)
ЬП (Т> серое)
^П (т> белое)
SnCl2(T)
SnO(T)
SnO2(T)
SrCO3(T)
StO(t)
SrSO4(T)
Ti(T)
TiCl4(T)
ТЮ2 (x, рутил)
V(T)
v2o3(T)
W(x)
wo3(T)
Zn(T)
ZnO(T)
Zn(OHJ(T)
ZnCO3(T)
ZnS(T)
Zr(T)
ZrCl4(T)
Af»l'
кДж/моль
-359,82
-699,56
-451,70
-217,61
-276,56
-100,42
-920,48
0
-115,50
0
-134,0
+0,38
0
-296,90
-395,85
-439,0
+34,73
-903,49
-910,84
-2,10
0
-330,95
-285,98
-580,74
-1218,4
-592,04
-1444,74
0
-804,16
-944,75
0
-1219,10
0
-842,91
0
-348,11
-645,43
-812,53
-205,18
0
-979,77
52°98,Дж/(мольК)
+135,98
+130,96
+217,90
+68,70
+71,92
+91,21
+148,57
+37,70
+38,90
+41,55
+69,10
+32,55
+31,92
+248,10
+256,69
+122,05
+204,56
+46,86
+41,84
+44,20
+51,55
+131,80
+56,48
+52,30
+97,10
+54,39
+117,57
+30,63
+252,40
+50,33
+28,90
+98,30
+32,64
+75,90
+41,63
+43,51
+76,99
+80,33
+57,66
+38,99
+181,42
Д fG° , кДж/моль
J 298
-314,56
-625,87
-256,90
-188,20
-217,55
-98,77
-813,67
0
-85,30
0
-84,0
+0,19
0
-300,20
-371,17
-368,04
+57,18
-850,71
-856,67
-0,13
0
-288,40
-256,88
-519,83
-1137,60
-562,10
-1332,42
0
-737,32
-889,49
0
-1139,40
0
-764,11
0
-318,10
-555,92
-730,66
-200,44
0
-889,27
Приложения
749
Таблица П. 5.2
Стандартные энтальпии сгорания Ac#°98 некоторых веществ
Формула, название
-а л,
кДж/моль
Углеводороды
СН4(Г)Метан
С2Н2 (Г) ацетилен
С2Н4 (г) этилен
С2Н6(Г) этан
С3Н8 (г) пропан
С4Н,0(г)бутан
С4Ню(Г) изобутан
QHi2(r) пентан
С6Н6(Ж) бензол
С6Н6(Г) бензол
СбН12(ж) циклогенксан
СбНщг) циклогексан
С6Н14(ж) гексан
C6Hi4(r) гексан
С7Н8(Ж) толуол
С7Н8 (г) толуол
С8Н1О(Ж) Л1-КСИЛОЛ
С8Ню(ж)И-, 0-КСИЛОЛ
C8H18(«) октан
СюН8(т) нафталин
890,31
1299,63
1410,97
1559,88
2220,00
2877,13
2868,76
3536,15
3267,58
3301,51
3919,91
3953,00
4163,05
4194,75
3910,28
3947,94
4551,81
4552,80
5470,58
5156,78
Азотсодержащие соединения
CH3O2N (Ж) нитрометан
CH4ON2 (т) карбамид
CH2N (г) метиламин
C2H7N (r) диметиламин
C3H5O9N3 (Ж) нитроглицерин
C3H8N (Г) триметиламин
C5H5N (ж) пиридин
C6H5O2N (Ж) нитробензол
C6H5O2N (т) «-нитробензол
C6H7N (Ж) анилин
708,77
632,20
1085,08
1768,59
1541,40
2442,92
2755,16
3091,20
2884,00
3396,20
Формула, название
-ая;8,
кДж/моль
Кислородсодержащие соединения
СО(Г) оксид углерода
СН2О (Г) формальдегид
СН2О2(Ж) муравьиная кислота
СН4О (Ж) метанол
С2Н4О (Г) ацетальдегид
С2Н4О2 (Ж) уксусная кислота
С2Н6О (Ж) этанол
С3Н6О (Ж) ацетон
С3Н8О (Ж) 1-пропанол
С3Н8О (Ж) 2-пропанол
С3Н8О3 (ж) глицерин
С4Н8О2 (Ж) этилацетат
С4Н10О (Ж) бутанол
ОНюО (ж) диэтиловый эфир
C5Hi20 (Ж) амиловый спирт
С6Н6О(Т) фенол
C6Hi206 (т) а-глюкоза
С6Н12О6 (т) C-глюкоза
С7Н6О2 (Т) бензойная кислота
СШН16О(Т) камфора
Ci2H22On (т) сахароза
Ci8H36O2(T) стеариновая к-та
Хлорсодержащие соединения
СС14 (Ж) тетрахорметан
СНС13 (ж) хлороформ
СН3С1 (Ж) хлорметан
С6Н5С1 (Ж) хлорбензол
282,92
561,07
254,58
726,60
1193,07
874,58
1370,68
1785,73
2010,41
1986,56
1661,05
2246,39
2671,90
2726,71
3320,84
3063,52
2802,04
2808,04
3226,70
5924,84
5646,73
11274,60
260,65*
428,06**
759,64***
3110,30***
Серосодержащие соединения
COS (Г) серооксид углерода
CS2 (Ж) сероуглерод
H2S (Г) сероводрод
553,12
• 1075,29
578,98
Примечание. Продукты сгорания — СОад, Н2О(Ж), а также для азотсодержа-
азотсодержащих соединений — N2(r), для серосодержащих соединений — SO2(r), для хлорсодер-
жащих соединений — С12(Г); С12(г) и НС1(Р); НС1(Р).
750
Приложения
П.6. Электрохимические системы
Таблица П. 6.1
Ряд стандартных электродных потенциалов (ф °) окислительно-восстановительных
(О-В) систем в водных растворах при Т= 298 К
(по отношению к стандартному водородному электроду)
О-В система
(сопряженная
пара): Оф/Вф
Схема, или условная
запись электрода
Электроды первого рода,
Li+/Li
К+/К
Ва2+ / Ва
Са2+ / Са
Na+/Na
Mg2+ / Mg
А13+/А1
Ti2+/Ti
Ti3+ / Ti
Mn2+/Mn
0*7 Cr
Zn2+/Zn
Cr3+/Cr
Fe2+ / Fe
Cd2+ / Cd
Co2+/Co
Ni2+/Ni
Sn2+/ Sn
Pb2+/Pb
Fe3+ / Fe
H+/H2
Cu2+ / Cu
Cu+ / Cu
HgT/Hg
Ag+/Ag
Hg2+/Hg
Pd2+/Pd
Pt^/Pt
Au3+/Au
Au+/Au
Уравнение электродного процесса
обратимые относительно катиона
(электрохимический ряд напряжений металлов)
Li+|Li
К+|К
Ва2+ | Ва
Са2+ | Са
Na+ | Na
Mg2+ | Mg
Al3+ | Al
Ti2+ | Ti
Ti3+ | Ti
Mn2+|Mn
Cr2" | Cr
Zn2+ | Zn
Cr3+ | Cr
Fe2+ | Fe
Cd2+ | Cd
Co2+ | Co
Ni2+ | Ni
Sn2+ | Sn
Pb2+ | Pb
Fe3+ | Fe
H+|H2,Pt
Cu2+ | Cu
Cu+ | Cu
Hg22+ | Hg
Ag+| Ag
Hg2+ | Hg
Pd2+ | Pd
Pt2+| Pt
Au3+ | Au
Au+ | Au
Li+ + e <=> Li
К+ + е <=> К
Ва2+ + 2е <z> Ва
Са2+ + 2е <± Са
Na+ + e <=i Na
Mg2+ + 2е <=± Mg
Al3+ + Ъе <=> А1
Ti2+ + 2е <=> Ti
Ti3+ + 3e ^ Ti
Mn2+ + 2е <± Мп
Сг^+ + 2е <=± Сг
Zn2+ + 2е <± Zn
Сг3+ + Ъе <=± Сг
Fe2+ + 2е <± Fe
Cd2+ + 2е <=± Cd
Со2+ + 2е ^± Со
Ni2+ + 2е <=± Ni
Sn2+ + 2е <± Sn
Pb2+ + 2е <=± Pb
Fe3+ + Ъе <=± Fe
2Н++2е ±± Н2
Си2+ +2е ^± Си
Си+ +е <=± Си
Hg^+ +2e ^± 2Hg
Ag+ +e ?=> Ag
Hg2+ +2e <=i Hg
Pd2+ + 2e <=> Pd
Pt2+ +2e <=> Pt
Au3+ + Ъе <=> Аи
Au+ +e fi Аи
ф\в
-3,045
-2,924
-2,906
-2,866
-2,714
-2,363
-1,662
-1,628
-1,208
-1,180
-0,913
-0,763
-0,744
-0,440
-0,403
-0,277
-0,250
-0,136
-0,126
-0,036
0,000
+0,337
+0,521
+0,798
+0,799
+0,854
+0,987
+1,190
+1,498
+1,691
Приложения
751
Продолжение табл. П. 6.1
О-В система
(сопряженная
пара): Оф/Вф
Схема, или условная
запись электрода
Уравнение электродного
процесса
Ф°,В
Электроды первого рода, обратимые относительно аниона
Se/Se2"
S/S2"
12/Г
Вг2 / ВГ
Se2" | Se
S2"|S
r|I2,Pt
Br|Br2I,Pt
Se + 2e <± Se'
S + 2e <± S~
I2(T) +2e <=> 2Г
Вг2(ж) +2е ?=± 2Br"
Газовые электроды первого рода, обратимые относительно катиона либс
Н2О / Н2
Н+/Н2
S/H2S
О2 / ОН"
NO~/NO
О2 / Н2О
С12 / СГ
F2/F"
H2O,ObT|H2,Pt
H+|H2;Pt
S,H+|H2S,Pt
H2O,OH-|O2,Pt
NO3", H+, H2O | NO, Pt
H2O,H+|O2,Pt
СГ| Cl2, P t
F|F2,Pt
2H2O +2e ^± H2 + 2OH"
2H+ +2e <=± H2
S +2H+ +2e <± H2S
O2 +2H2O+4e ?± 4OH"
NO3 + 4РГ+ 3e <± NO + 2H2O
O2 +4H++4e <=> 2H2O
Cl2 +2e ?± 2СГ
F2 + 2e ^± 2F"
-0,920
-0,447
+0,536
+1,065
»аниона
-0,828
0,000
+0,142
+0,401
+0,960
+1,229
+1,360
+2,871
Электроды второго рода, обратимые относительно катиона и аниона
Mg(OHJ/Mg
MgCO3 / Mg
Zn(OHJ/Zn
ZnCO3 / Zn
Fe(OHJ / Fe
Cd(OHJ / Cd
Ni(OHJ/Ni
PbO / Pb
PbCO3/Pb
PbI2/Pb
PbSO4 / Pb
PbCl2/Pb
Agl/Ag
AgBr/Ag
HgO/Hg
AgCl/Ag
Hg2Cl2/Hg
малорастворимого соединения
OH" | Mg(OHJ , Mg
CO32-|MgCO3,Mg
OH" | Zn(OHJ, Zn
CO2"|ZnCO3,Zn
OH" | Fe(OHJ, Fe
OH" | Cd(OHJ, Cd
OH" | Ni(OHJ, Ni
H2O ,OH" | PbO , Pb
СОз'|РЬСО3,РЬ
Г | Pbl2, Pb
SO32"|PbSO4,Pb
СГ|РЬС12,РЬ
Г | Agl, Ag
ВГ | AgBr, Ag
H2O,OH" | HgO , Hg
Cr|AgCl,Ag
СГ|Н&С12,Н8
Mg(OHJ +2e<dMg + 2OH~
MgCO3 + 2e <=± Mg + CO32-
Zn(OHJ + 2e ^± Zn + 2OH"
ZnCO3 + 2e ^=> Zn + СОз"
Fe(OHJ+2^ ^± Fe+2OH"
Cd(OHJ + 2e ±± Cd + 2OH"
Ni(OHJ + 2e <=> Ni +2OH"
PbO + H2O + 2e <=> Pb +2OH"
PbCO3 + 2e <=> Pb + COj"
Pbl2 + 2e +± Pb + 2Г
PbSO4 + 2e ^± Pb + SO'"
PbCl2 + 2e <=> Pb + 2СГ
Agl + e <=± Ag + Г
AgBr + e <=i Ag + Br"
HgO + H2O + 2e <=> Hg +2OH"
AgCl + e +± Ag + СГ
Hg2Cl2+2e 4=^ 2Hg + 2Cr
-2,694
-1,500
-1,245
-1,060
-0,877
-0,809
-0,729
-0,578
-0,506
-0,365
-0,359
-0,268
-0,152
+0,071
+0,098
+0,222
+0,268
752
Приложения
Окончание табл. П.6.1
О-В система
(сопряженная
пара): Оф/Вф
Ag2O / Ag
Ag2Cr04 / Ag
Ag2SO4/Ag
Схема, или условная
запись электрода
Н2О,ОН-1 Ag2O , Ag
CrO*IAg2CrO4,Ag
SO*IAg2SO4,Ag
Уравнение электродного процесса
Ag2O + H2O + 2e ±± 2Ag+2OH"
Ag2CЮ4 + 2e ^± 2kg + CvO\'
Ag2SO4 + 2e +± 2Ag + SOf
Ф°,В
+0,345
+0,464
+0,654
Электроды третьего рода или окислительно-восстановительные электроды
(инертный металлический электрод — платина)
SO*"/SO2"
Cr3+/Cr2+
Н3РО4/ Н3РО3
Sn4+/Sn2+
Cu2+/Cu+
сю;/сю;
12/Г
ClOj/CIO"
Fe3+/Fe2+
СЮ / CI-
CINQ"/HNO2
Вг2/ВГ
MnO2/Mn2+
Сг2О72"/Сг3+
PbO2/Pb2+
MnO~/Mn2+
Се4+/Се3+
Н2О2/Н2О
Со3+/Со2+
s2o82/sor
SO,,SO2~,ObriPt
Cr3+, Cr2+ 1 Pt
H+,H3PO4,H3PO3IPt
Sn4*, Sn2+1 Pt
Cu2+, Cu+1 Pt
cio4~, cio;,OH-iPt
I2,riPt
ClO-,ClO",OHIPt
Fe3+,Fe2+IPt
CIO", СГ, OH" 1 Pt
H+,NO;,HNO2IPt
Br2,Br~IPt
H+, MnO2, Mn2+ 1 Pt
H+,Cr2O*~,Cr3+IPt
IT, PbO2, Pb2+ 1 Pt
H+,MnO~,Mn2+IPt
Ce4+, Ce3+1 Pt
H+,H2O2IPt
Co3+, Co2+ 1 Pt
s2o82",sor|pt
SO2; + H2O+ 2e ^± SO 3" +2OH"
Cr3+ + e +± Cr2+
H3PO4 + 2H++e ±± HjPO3+H2O
Sn4+ + 2e ^± Sn2+
Cu2+ + e +± Cu+
CIO; + H2O + 2e & СЮ" +2ОН"
12(водн) + 2е ^ 2 Г
ClOj + H2O + 2e+± C1O+2OH-
Fe3+ + 2e *± Fe2+
ClO"+H2O + 2e +± СГ+2ОН-
N0^+3^+26 <=> HNO2 + H2O
Вг2(вода) + 2е ^± 2Br"
MnO2 + 41Г+ 2e ±± Mn2++ 2H2O
Cr2O'"+ 14H++ 6e *z* Cr3+ +
+ 7H2O
PbO2+4H++2e ±± Pb2++2H2O
МПО4 + 8H+ + be ?± Mn2+ +
+ 4H2O
Ce^ + e +± Ce3+
H2O2 + 2H++2e?± 2H2O
Co3+ + e *± Co2+
S2Oj"+2«?±2SOj"
-0,930
-0,408
-0,276
+0,151
+0,153
+0,360
+0,621
+0,661
+0,771
+0,890
+0,940
+1,087
+1,230
+1,330
+1,455
+1,511
+1,610
+1,776
+1,818
+2,010
Примечание. Запись сопряженных пар (О-В) систем упрощена: приведены
только определяющие данную пару частицы стехиометрические коэффициенты,
указания на агрегатные состояния опущены.
Приложения
753
Таблица П. 6.2
Характеристики некоторых типов гальванических элементов
Тип элемента
Свинцово-
кадмиевый
Марганцево-
магниевый
Серебряно-
цинковый
Марганцево-
цинковый
Окисно-
ртутный
Медно-
окисный
Система
PbO2|H2SO4|Cd
MnO2|MgBr2|Mg
Ag2O|KOH|Zn
MnO2|NH4Cl|Zn
HgO|KOH|Zn
CuO|NaOH|Zn
Уравнение токообразующеи реакции
Cd + 2H2SO4+PbO2 =
= CdSO4+PbSO4+2H2O
Mg + H2O + 2MnO2 =
= Mn2O3+Mg(OHJ
Zn + 2KOH + Ag2O =
= 2Ag + K2ZnO2+H2O
Zn + 2NH4Cl + Mn02 =
= [Zn(NH3J]Cl2+Mn203
Zn + 2KOH + HgO =
= Hg + K2ZnO2+H2O
Zn + 2NaOH + CuO =
= Cu + Na2ZnO2+H2O
ЭДС,В
2,2
2,0
1,85
1,5-1,8
1,34
0,85
Таблица П. 6.3
Характеристики некоторых типов аккумуляторов
Тип аккумулятора
Свинцовый
Серебряно-
цинковый
Серебряно-
кадмиевый
Никель-
цинковый
Железо-
никелевый
Никель-
кадмиевый
Система
PbO2|H2SO4|Pb
Ag2O|KOH|Zn
Ag2O|KOH|Cd
NiOOH|KOH|Zn
NiOOH|KOH|Fe
NiOOH|KOH|Cd
Уравнение токообразующеи
реакции
Pb+PbO2+2H2SO4 =
= 2PbSO4+2H2O
Zn+2KOH+Ag2O =
= 2Ag+K2ZnO2+H2O
2Cd+2Ag2O+H2O =
= 4Ag+CdO+Cd(OHJ
Zn+NiOOH+2H2O =
= 2Ni(OHJ+Zn(OHJ
Fe+NiOOH+2H2O =
= 2Ni(OHJ+Fe(OHJ
Cd+NiOOH+2H2O =
= 2Ni(OHJ+Cd(OHJ
ЭДС,В
2,10
1,85
1,50
1,70
1,40
1,36
25 - 9795
754
Приложения
Таблица П.6.4
Минимальные перенапряжения водорода и кислорода
на некоторых электродах при наименьшей плотности тока
Материал
электрода
Pt (чернь)
Pd
Аи
Pt
Ag
Ni
С (графит)
Перенапряжение, В
водорода
0
0
-0,02
-0,08
-0,10
-0,14
-0,14
кислорода
+0,25
+0,42
+0,52
+0,44
+0,40
+0,12
Материал
электрода
Fe
Си
Cd
Pb
Zn
Hg
Co
Перенапряжение, В
водорода
-0,17
-0,19
. -0,39
-0,40
-0,48
-0,57
кислорода
+0,24
+0,25
+0,42
+0,30
+0,13
Таблица П.6.5
Значения констант аи Ьв уравнении Тафеля для реакции катодного выделения
водорода на некоторых металлах при 298 К и плотности тока i = 1 А/см2
Металл
Ag
А1
Аи
Cd
Со
Си
Fe
Hg
Mn
Mo
Ni
Pb
Pd
Pt
Sn
Ti
Zn
a
0,95
1,00
0,40
1,40
0,62
0,86
0,70
1,41
0,80
0,66
0,63
1,56
0,24
0,10
1,20
1,55
1,24
Значения
кислотного
b
0,10
0,10
0,12
0,18
0,14
0,12
0,12
0,11
0,10
0,08
0,11
0,11
0,03
0,03
0,13
0,14
0,12
констант для раствора
щелочного
а
0,73
0,64
-
1,05
0,60
0,96
0,76
1,54
0,90
0,67
0,65
1,36
0,53
0,31
1,28
-
1,20
Ь
0,12
0,14
-
0,16
0,14
0,12
0,11
0,11
0,12
0,14
0,10
0,25
0,13
0,10
0,23
-
0,12
Приложения
755
Таблица 17.6.6
Соответствия между значениями глубинного показателя скорости коррозии,
баллами шкалы и группами коррозионной стойкости металлов
Глубинный показатель
скорости коррозии гглуб, мм/год
< 0,001
0,001-0,005
0,005-0,01
0,01-0,05
0,05-0,1
0,1-0,5
0,5-1,0
1,0-5,0
5,0-10,0
>10
Коррозионная стойкость металлов
Балл
1
2
3
4
5
6
7
8
9
10
Группа
Совершенно стойкие
Весьма стойкие
Стойкие
Пониженно-стойкие
Малостойкие
Нестойкие
Примечание. Коррозионная стойкость металлов оценивается при гглуб
0,5 мм/год по группам, при гглуб < 0,5 мм/год — по баллам.
Список рекомендуемой литературы
Учебники и учебные пособия
Ахметов НС. Общая и неорганическая химия. М.: Высш. шк., 1998.
Васильев В.П. Термодинамические свойства растворов электролитов. М.:
Высш. шк., 1982.
Теоретические основы общей химии: Учеб. для студентов технических уни-
университетов и вузов/ А.И. Горбунов, А.А. Гуров, Г.Г. Филиппов и др.; Под ред.
А.И. Горбунова. М.: Изд-во МГТУ им. Н.Э. Баумана, 2001.
Минкин В.И., Симкин Б.Я., Миняев P.M. Теория строения молекул. Ростов-на-
Дону: Феникс, 1997.
Степин Б.Д., Цветков А.А. Неорганическая химия. М.: Высш. шк., 1994.
Суворов А.В., Никольский А.Б. Общая химия: Учебник для вузов. СПб.:
Химия, 1997.
Физическая химия / Под ред. К.С. Краснова. Т. 1, 2, 4. М.: Высш. шк., 1995.
Фримантл М. Химия в действии. В 2 ч. Ч. 1,2/ Пер. с англ. М.: Мир, 1998.
Хаускрофт К., Констебл Э. Современный курс общей химии. В 2 т. Пер. с
англ. М.: Мир, 2002.
ХъюиДж. Неорганическая химия. М.: Мир, 1987.
Практикумы и задачники
Ахметов Н.С., Азизова М.К., Бадыгина Л.И. Лабораторные и семинарские за-
занятия по общей и неорганической химии. М.: Высш. шк., 1994.
Гольбрапх З.Е., Маслов Е.И. Сборник задач и упражнений по химии: Учеб.
пособие для хим.-технол. спец. вузов. М.: Высш. шк., 1997.
Гольбрапх З.Е. Практикум по неорганической химии: Учеб. пособие для
хим.-технол. спец. вузов. 3-е изд., перераб. и доп. М.: Высш. шк., 1986.
Зайцев О.С. Исследовательский практикум по общей химии. М.: Изд-во
МГУ, 1994.
Коровин КВ., Мингулина Э.И., Рыжова Н.Г. Лабораторные работы по химии:
Учеб. пособие для техн. направ. и спец. вузов / Под ред. Н.В. Коровина. 2-е изд.,
перераб. и доп. М.: Высш. шк., 1998.
Практикум по общей и неорганической химии: Учеб. пособие для студентов
нехимических специальностей вузов / Л.С. Гузей, В.Н. Кузнецов, Г.П. Жмурко и
др.; Под ред. С.Ф. Дунаева. М.: Изд-во МГУ, 2000.
Суворов А.В., Никольский А.Б. Вопросы и задачи по общей химии. СПб.:
Химиздат, 2002.
Список рекомендуемой литературы 757
Справочники
Артеменко АЖ, Тикунова КВ., Малеванный В А. Справочное руководство по
химии. М.: Высш. шк., 2002.
Лидин Р. А,, Андреева Л. Л., Молочко В А. Справочник по неорганической химии.
М.: Химия, 1987.
Свойства неорганических соединений: Справочник / А.И. Ефимов, Л.П. Бе-
лорукова, И.В. Василькова, В.П. Чечев. Л.: Химия, 1983.
Химическая энциклопедия. В 5 т. М.: «Большая российская энциклопедия»,
1988-1998.
Химия: Справ, изд. / В. Шретер, К.-Х. Лаутеншлегер, X. Бибрак и др.: Пер. с
нем. 2-е изд., стереотип. М.: Химия, 2000.
Предметный указатель
Автокатализ 283
Адсорбат 284
Адсорбент 285
Адсорбция 284
- физическая (физисорбция) 285
- химическая (хемосорбция) 287, 289
- активированная 289
- неактивированная 289
Агрегатное состояние 149
- аморфное твердое 151
-газообразное 149
-жидкое 151
- кристаллическое 151
Аккумулятор(ы) 563
-, зарядка 563
-, разрядка 563
Акт химический элементарный 240
Активированный комплекс (переход-
(переходное состояние) 261
Активность 330, 441
- средняя ионная 441
Актиноиды 99
Акцептор 116
Аллотропия 11
Аллотропная модификация 11
Анализ количественный 43
Ангидриды 15
Анизотропия (векторность свойств) 151
Анионы 422
Анод 526
Ассоциаты 146
Атмосферы защитные 633
Атом 10
-, возбужденное состояние 95
-, орбитальный радиус 102
-, основное состояние 95
-, потенциал ионизации 103
-, сродство к электрону 103
-, электронная конфигурация 95
-, электроотрицательность 104
-, энергия ионизации 102
Аэрация неравномерная 623
Бертоллиды
Бронзы 630
38
Валентность 111
Валентный угол 107
Вероятность термодинамическая 193
Вещество простое 10
- растворенное 40, 420
- сложное 11
-, стандартное состояние 184
Взаимодействие(я) межмолекулярное
143
- дисперсионное 144
- индукционное 143
- ориентационное 143
- диполь-дипольные 155
- ион-дипольные 146
- ион-молекулярные 146
Вода 148
-, декарбонизация 470
-, жесткость 465
-, -, градусы 466
-, -, единицы 466
-, - карбонатная (временная, устрани-
устранимая) 465
-, — остаточная 465
-, - некарбонатная 465
-, - общая 465
-, известкование 470
- нестабильная 464
-, обессоливание (деминерализация) 471
-, область электрохимической устой-
устойчивости 546
-природная 461
-, термическая обработка 469
-, термохимическое умягчение 470
- техническая 461
-, умягчение 469
-,-, известково-содовый метод 471
-, химическая обработка 470
Предметный указатель
759
Восстановитель 19, 26
Восстановление 19
Время полупревращения 244
- защитного действия 473
Вязкость 150
Галогенид-ионы 26
Галогениды 12
Галогены 21,27
Гальванический(е) элемент(ы)
- Даниэля-Якоби 551, 576
- концентрационные 556
-макро- 611
- микро- 608
- короткозамкнутый коррозионный
608
-, напряжение 552, 555
- сухие 562
-химические 551
- без переноса 556
- с переносом 556
Гальванопластика 571
Гальваностегия 572
Герметичность покрытия 638
Гибридизация валентных орбиталей
118
Гидратация 421
Гидраты 13, 17
Гидриды 12, 26
Гидроксид-ионы 13
Гидроксиды 12
- амфотерные 13,15
- кислотные 13
-основные 13
Гидролиз
- сложных эфиров 279
- соли 447
- по аниону 448
- по катиону 448
- по катиону и аниону 449
Граница устойчивости 629
Группы активные функциональные
(или ионогенные) 471
- комплексообразующие 471
Давление(я) диссоциации 376
- насыщенного пара 428
- осмотическое 433
- парциальные 37
- равновесные 329
Дальтониды 38
Двойной электрический слой 528
Деаэрация термическая 635
- химическая 634
Денсиметрия 43
Деполяризация 577
-, смешанный вид 610
Деполяризаторы 577, 609
Десорбция 285
Десублимация 178
Диаграмма Пурбе 545
- реакции энергетическая 190
Диполь мгновенный 145
- электрический 110
—, длина 110
Дисперсионная среда 418
Диффузия 267
Дихромат калия 24
Диэлектрики (изоляторы) 142
Длина волны де Бройля 77
Доля массовая 40
- молярная (мольная) 41
- равновесная 328
Донор 116
Жаропрочность 629
Жаростойкость 629
Жесть луженая 641
- электролитическая белая 641
Жидкость(и) 150
Закон(ы) Авогадро 36
- Бойля—Мариотта 36
- Гей-Люссака 36
-Генри 427
-Гесса187
—, следствие второе 188
—, первое 188
- действующих масс 245, 331, 343
—, кинетический вывод 334
760
Предметный указатель
—, термодинамический вывод 340
- действующих поверхностей 286
- кратных отношений 35
- независимого движения ионов
(Кольрауша) 459
- объединенный газовый 36
- объемных отношений 35
-Ома 456
- парциальных давлений (Дальтона) 36
- периодический Д.И. Менделеева 100
- постоянства состава 35
- предельный Дебая—Хюккеля 442
- разбавления (разведения) Оствальда
439
-Рауля 429
- роста оксидных пленок 605
- линейный 605
- логарифмический 605, 606
- параболический 605
- сохранения массы 35
- стехиометрические 35
- термодинамики второй 191
- первый 179
--третий 198
- Фарадея второй 526
- первый 526
—, обобщенное выражение 527, 572
- Фика первый 267
- эквивалентов 35, 37
Защита электрохимическая 635
- анодная 635
- катодная 635
- наложенным (внешним) потенциалом
635
- протекторная 636
Золи 418
Зона валентная 141
- проводимости 141
- энергетическая 141
Изомер(ы) 111
Изоморфизм 154
Изоморфные вещества 154
Изотропность 150
Ингибиторы 631
-анодные 631
- катодные 632
-летучие (парофазные) 633
- нелетучие (контактные) 633
-, критическая концентрация 632
- органические катодные замедлители
(травления) 632
- экранирующие 632
Индикатор 43
Индикаторная ошибка титрования 45
Интерметаллиды 12, 140
Иониты 471
-, емкость 472
-, - динамическая 472
-, - полная 472
-, - рабочая 472
-, - статическая 472
-, избирательность (селективность) 473
Ионная атмосфера 436
Ионная пара 436
Ионное произведение воды 44,444
Ионный обмен 471
Ионы 422
- водорода 14, 25
- металлов 25
-, поляризация 138
-, поляризующее действие 138
-, электрическая подвижность 456
Испарение 178
Истинная точка эквивалентности 44
Катализ 274
-гетерогенный 274,289
- гомогенный 274, 279
- кислотно-основный 279
- специфический 280
- окислительно-восстановительный
281
-, схема слитная 276
-, - стадийная 277
Катализатор(ы) 274, 360
-, активность 277
-, отравление 278
-, селективность 275
-, - дифференциальная 275
Предметный указатель
761
-, - интегральная 275
-, специфичность 276
-, число оборотов 277
Каталитический (активный) центр 276
Катионы 422
- металлов 27
Катод 526
Квантовая механика 76
Квантовое(ые) число(а) 82
- главное 82
- магнитное 85
- орбитальное 83
- спиновое 87
Кинетика гетерогенных каталитиче-
каталитических реакций 291
- химическая 175, 240
- электрохимических процессов 572
Кислород 21
Кислота(ы) 14,279
- азотистая 28
-азотная 21
- бескислородные 14
- кислородсодержащие (оксокислоты) 14
- галогенов 24, 27
-комплексные 17
- серная 23
Кислотность оснований 14
Клатраты 154
Комплексы 17
-аква- 17
-аммино- 17
-анионные 17
-ацидо- 17
-, внешняя сфера 16
-, внутренняя сфера 16
-гидроксо- 17
- катионные 17
-нейтральные 17
- смешанного типа 17
Комплексные соединения 13,16,122,446
Комплексообразователь 16
-, координационное число 16
Конвекция 266
Конденсация 178
Константа Больцмана 193
- гидролиза 449
- диссоциации (ионизации) 437
- концентрационная (аналитическая)
442
- термодинамическая 442
- нестойкости 446
- скорости химической реакции 245
- равновесия 330
—, выражение 336
- стандартная 343
- термодинамическая 331
- эмпирическая (концентрационная)
332
Концентрация(и) исходные (началь-
(начальные) 329
- неравновесные (текущие) 329
-массовая 41
- моляльная (моляльность) 41
- молярная (молярность) 41
- равновесная 329
- химического эквивалента (нор-
(нормальность) 41
-процентная 41
- равновесные 327, 329
- молярные 329
Координата реакции 261
Корпускулярно-волновой дуализм 76
Коррозия 593
-, активаторы 621
- атмосферная 622
- влажная 622
- мокрая 623
- сухая 622
- в естественных условиях 622
- в морской воде 626
- водородная 599
- высокотемпературная газовая 598
—, кинетика 600
—, термодинамика 600
-, деполяризаторы 609
- кавитационная 595
- контактная 608, 611
- локальная (местная) 594
- межкристаллитная 595
- неравномерная 594
762
Предметный указатель
-, определение 594
- под напряжением 612
-, продукты 594
-,-вторичные 610
-, - первичные 610
- равномерная 594
- с анодным контролем 615,617
- с водородной деполяризацией 610
- с катодным контролем 615,616
- с кислородной деполяризацией 610
- под действием блуждающих токов
626
- подземная 625
- селективная (избирательная) 595
-, влияние факторов 620
-,-давление 621
-, - кислотность раствора 620
-,-скорость движения электролита 621
-,-состав электролитной среды 621
-, - температура 620
- сплошная (общая) 594
- транскристаллитная 595
- фреттинг- 595
- химическая 595, 597
-, экологический аспект 593
-, экономический аспект 593
- электрохимическая (гальваническая)
595, 608
—, кинетика 615
—, термодинамика 613
Коэффициент(ы) активности 441
- средний ионный 441
- диссоциации (ионизации) 430
- диффузии 267
- изотонический 434
- растворимости 421
- стехиометрические 12
- температурный скорости реакции
Вант-Гоффа 256
--ЭДС 561
- трансмиссионный 262
- электрической проводимости 459
Краска 642
Кривая(ые) кинетические 243
- поляризационные 573
- сорбции выходная 472
Кристаллизация 178
Кристаллиты (зерна) 595
Кристаллическая решетка 151
- атомно-ковалентная 152
-ионная 152
- металлическая 152
-молекулярная 152
-, узлы 152
-, элементарная ячейка 153
-, - координационное число 153
-, - кратчайшее расстояние 153
-, - число частиц, необходимое для
построения 153
Кристаллогидраты 13, 17
Кристаллография 153
Кристаллохимия 420
Кристаллы жидкие 155
- нематические 155
- смектические 155
- холестерические 155
-смешанные 154
Лак 642
Лантаноиды 99
Легирование объемное 629
- поверхностное 630
Лиганды 16
-, дентатность 16
Мезоморфная фаза 155
Мембраны жидкие 550
Металл(ы) 10, 26, 141
- высокой термодинамической ста-
стабильности 615
- катодного контакта 612
- низкой термодинамической стабиль-
стабильности 614
- пассивный 603
- практически полной термодинамиче-
термодинамической стабильности 615
- промежуточной термодинамической
стабильности 615
-термодинамическинестабильные 615
Метод(ы) активностей Льюиса 330, 441
Предметный указатель
763
- валентных связей 115
- кислотно-основного титрования 44
- молекулярных орбиталей 128
- определения порядка реакции 253
- -, Вант-Гоффа 254
—, подстановки 254
—, по периоду полупревращения 255
- термохимических схем 189
- электронного баланса 32
- электронно-ионных полуреакций 29
Многоэлементные соединения 12
Молекула(ы) 10
-, геометрическая форма 117
- гетероатомные 107, 134
- гомоатомные 107, 131
- двухатомная 131, 135
-, дипольный момент 110
- изоэлектронные 118
-, электронная конфигурация 129
Моль 34
Молярная масса 34
- химического эквивалента вещества
35, 527
Молярный объем 34
- химического эквивалента вещества
35, 528
Неметаллы 10, 26
Неэлектролиты 423
Нитраты 22
Нитриты 28
Обескислороживание десорбционное
635
- сорбционное 635
Окисление 19
- высокотемпературное 600
Окислитель 19, 21
Окислительно-восстановительная
двойственность 20, 27
Оксидирование термическое 642
- химическое 642
- электрохимическое 642
Оксиды 12, 15
- амфотерные 16
-кислотные 15
— несолеобразующие 15
— основные 15
— солеобразующие 15
Орбиталь(и) атомная(ые) 82, 83
— валентные 115
— водорода 89
— вырожденные 89
— гибридные 118
— диагональные 119
— неэквивалентные 121
— тетрагональные 119
— тригональные 119
— эквивалентные 120
-молекулярная 129
— делокализованная 129, 135
— локализованная 135
— многоцентровая 129
— несвязывающая 130
— разрыхляющая 129
— связывающая 129
Осмос 433
— обратный 434
Осмотическое давление 433
Основание(я) 13, 279
— комплексные 17
— сопряженное 279
Основность кислот 14
Параметры состояния 176
— интенсивные 177
— экстенсивные 177
Пассиваторы 618, 631
Пассивация 566, 607, 618
— высокотемпературная 607
Переменная химическая 207, 241
Перенапряжение 575
— диффузионное 575, 576
— электрохимическое 575, 576
—, теория замедленного разряда
Перепассивация 619
Период индукционный 283
Периодическая система элементов 100
-, период 100
-, группа 101
764
Предметный указатель
Перманганат калия 24
Пероксид(ы) 16
- водорода 27
Пероксокислоты 14
Питтинг 594
Плавление 178
Пленка защитная 603
- оксидная 602
Плотность тока 572
- коррозионного 597
-обмена 573
- предельная 574
Поверхностное натяжение 151
Погрешность систематическая 44
Показатель водородный 44,445
- гидроксидный 445
- скорости коррозии глубинный 596
- массовый 596
- объемный 596
- токовый (плотность коррозионного
тока) 596,597
- титрования 45
Покрытия защитные 638
- анодные (протекторные) 638
- катодные (коррозионно-стойкие) 638
-конверсионные 641
- лакокрасочные 642
-, методы нанесения 640
Полимерные материалы 642
Полиморфизм 154
Полиморфные модификации 154
Полупроводники 142
Полуреакция анодная 552
- катодная 552
Поляризуемость 138
- деформационная 144
Порядок ближний 150
-дальний 151
Постоянная Авогадро 34
-Генри 427
- криоскопическая 432
- универсальная газовая 36
-Фарадея 457,527
- эбулиоскопическая 432
- электролитической ячейки 456
Постулат Клаузиуса 192
- Планка (третье начало термодина-
термодинамики) 199
- химической кинетики основной 245
Потенциал(ы) диффузионный 529
- контактный 529
- равновесный 529
- Леннарда—Джонса 145
- мембранный 550
- окислительно-восстановительный
равновесный 548
- стандартный 549
- разложения 566
- химический 206
- электродный 530, 537
- равновесный 532
- стандартный 535
Правило
-Вант-Гоффа 256
- Клечковского (п + /) 93
-фазГиббса 351
-Хунда 94
Принцип
- Бертло—Томсена 204
- Ле Шателье—Брауна 361,454
- максимального перекрывания 123
- микроскопической обратимости 338
- наименьшей энергии 92
-неопределенности 78
-Паули 93
- титрования 43
Проба 43
Проводимость электрическая (электро-
(электропроводность) 456
- молярная 457
предельная 459
- удельная 456
Произведение растворимости 453
Промотирование 115
Промоторы 278
Протектор 637
-, защитное действие 637
Процесс(ы) 177
-изобарный 181
- изотермический 180
Предметный указатель
765
-изохорный 180
- коррозии вторичные 610
- первичные 610
-, критерии направленности протека-
протекания 201
- неравновесный (нестатический) 177
- обратимый 177
- равновесный (квазистатический) 177
- самопроизвольный 177
- электродные (потенциалопределяю-
щие) 524
-, последовательность 566
Работа 179
Равновесие 365
-, влияние изменения давления 365
-,- концентраций 360
-,- температуры 364
-, - присутствия инертного газа 365
-,- катализатора 360
- устойчивое 326
- фазовое 325
- химическое 321, 326
—, виды 325
-, возможность достижения 326
- гетерогенное 325
- гомогенное 324
—, динамический характер 326, 335
- заторможенное 326, 327
- истинное 326, 338
—, кинетический критерий (условие)
335
—, кинетическое описание 334
—, количественные характеристики
328
—, особенности (признаки) 325, 326
—, подвижность 326
—, положение 325
—, смещение (сдвиг) 326
—, состояние 325
—, термодинамические критерии (ус-
(условия) 340
—, термодинамическое описание 340
—, термодинамическая устойчивость
326
Равновесный выход продукта 330
Разбавление 369,425
Разрушения коррозионные эвтектиче-
эвтектического сплава 611
Раствор(ы) 40,417
- газовые 420
- жидкие 420
- идеальные 426
-, ионная сила 436,442
- коллоидные 418
- молекулярные (истинные) 418
- насыщенный 421
- твердые 420
- внедрения 154
- замещения 154
- титрованные 43
Растворимость 421
Растворитель 40, 420
Рафинирование электролитическое 571
Рациональное конструирование 628
Реагенты 11
Реакция(и) анодные 609
- бимолекулярная 241
- второго порядка 250
- в условиях псевдопервого порядка 253
- гетерогенные 18, 265, 324
-, диффузионный режим 266
-, кинетический режим 267
-, переходный 267
- гетерофазные 325
- гомогенные 18, 241, 324
- гомофазные 325
- каталитические 18
- ферментативные 274
—, цикличность 277
- катодные 609
- кинетически необратимые 322
- кинетически обратимые 322, 324
-, компоненты (участники) 322
-, механизм 240
-, молекулярность 240
- мономолекулярная 240
- некаталитические 18
- нулевого порядка 246
-обменные 18,38
766
Предметный указатель
- окислительно-восстановительные
18, 19, 38
- внутримолекулярные 28
- диспропорционирования (дисмута-
ции) 28
- контрпропорционирования (конму-
тации) 29
- межмолекулярные 28
- первого порядка 247
-, порядок (или частный порядок реак-
реакции) 245
- практически необратимые 323
-, признаки необратимости 322
-пробег 333
-, продукты 12
-, -, равновесный выход 330
- совершенно (истинно) необратимые
323
- сопряженные 609
- твердофазные 270
—, топохимический характер 271
- токообразующая 552
- транспортные 375
- тримолекулярные 241
-, энтальпийный фактор 203
-, энтропийный 203
- фотохимические 18
-химическая 10, И
—, тепловой эффект 182
-экзотермические 18, 179
- элементарные 240
- электрохимические 18
- эндотермические 18, 179
Ржавление 594
Ряд стандартных электродных потен-
потенциалов 536
Самодиффузия 150
Свойства диамагнитные 132
-магнитные 132
- парамагнитные 132
Связь водородная 146
-, дипольный момент 110
-, длина 107
-ионная 137
- ковалентная 112
-кратная 117,121
- металлическая 140
-, механизм образования
-,-дативный 116
-, - донорно-акцепторный 116
-,-обменный 112
-, направленность 107
-, насыщаемость 117
-, поляризация 137
-, полярность 107
-, порядок 132
-, - частный 245
-, энергия 106
- химическая 106
Силициды 12
Силы близкодействующие 145
- Ван-дер-Ваальса 143
- дальнодействующие 145
Система(ы) дисперсные 418
-, состояние равновесное 177
-, - стационарное 177
- термодинамическая 176
—, внешние факторы 357
— гетерогенная 178
— гомогенная 178
— закрытая 176
— изолированная 176
—, критерий равновесия 342
- -, независимые компоненты 353,352
— открытая 176
—, составляющие вещества (компо-
(компоненты) 352
—, число сосуществующих фаз 356
—, число степеней свободы (вариант-
(вариантность) 352
- термомеханическая 180
- химически нереагирующая 373
- электрохимическая 524
— правильно разомкнутая 528
Скорость реакции 542
— гетерогенной 268
— гомогенной 242
— мгновенная 243
— по компоненту 242
Предметный указатель
161
- при постоянном объеме 243
- средняя 243
- электродного процесса 573
Слитность сечения 628
Смазки ингибированные 633
Солевой мостик 530
Соли 13, 15
-двойные 15
- кислые (гидросоли) 15
-комплексные 17
- основные (гидроксосоли) 15
- смешанные 15
- средние (нормальные) 15
Сольватация 420
- ближняя 420
- дальняя 420
-, координационное число 420
Сольватная оболочка 420
Сольваты 146
Сорбенты ионообменные (или ионооб-
менники) 471
Состав равновесный 328
Спин(ы) 87
- антипараллельные 113
- параллельные 113
Среда коррозионная 594, 597
-, изменение состава и свойств 631
Сродство к электрону 103, 107
Стадия лимитирующая 246
Стали 630
Степень гидролиза 449
- диссоциации 423
- кажущаяся 440
-окисления 19
- превращения вещества 269
- равновесная 330
Стехиометрия 34
Сублимация (возгонка) 178
Температура перехода 155
- разложения 376
Теория активированного комплекса 260
- активных столкновений 258
- зонная 140
- ионной ассоциации 436
- кислот и оснований
- протонная Брёнстеда и Лоури 423
- электронная Льюиса 423
- координационная Вернера 16
- отталкивания электронных пар ва-
валентных орбиталей 124
- парно-электронной связи Льюиса 111
- адсорбционная 619
- пленочная 619
- сильных электролитов электростати-
электростатическая 436
- химического строения Бутлерова 110
- электролитической диссоциации 423
Теплоемкость 196
Теплота 179
- приведенная 192
Термодинамика химическая 175
Термохимия 182
Тиокислоты 14
Титранты 43
Титриметрия 43
Токи блуждающие 626
Топливные элементы 565
Торможение релаксационное 459
- электрофоретическое 459
Углекислота свободная 463
-, гидрокарбонатная форма 463
-, карбонатная форма 463
- связанная 463
Узловая поверхность 83
Уравнение Аррениуса 256, 460
- Больцмана 193
- Ван-дер-Ваальса 149
-ВанЛаара 434
-Вант-Гоффа 433
- Гиббса—Гельмгольца 203, 348
- де Бройля 77
- Дэвис 442
- изобары химической реакции 348
- изотермы 345
--Ленгмюра 285,287
--Генри 287
- химической реакции 12, 345
, в общем виде 346
768
Предметный указатель
стандартное 347
- изохоры химической реакции 350
- кинетическое 245
-Кирхгофа 208,209
- Клапейрона—Менделеева 36
- Нернста 533, 554, 561, 576
- Планка—Ван Лаара 368
- состояния идеального газа 36
- стандартного сродства 347
- стехиометрическое 12
- термохимическое 182
- Тафеля 577
- токообразующего процесса 552
-химическое 12
-Шрёдингера 79,81,112
- Эйринга 263
- электродного процесса 532
Условие(я) ориентационного соответ-
соответствия 604
- сплошности Пиллинга—Бедворса
603
- стандартные термодинамические 183
Фаза 178
-дисперсная 418
Фазовый переход 178
Фактор сплошности Пиллинга—Бед-
вордса 604
- эквивалентности 35
Ферменты 274
Фиксаналы 42
Формула графическая 11
- молекулярная 11
-Стокса 456
- химическая 10
- эмпирическая 11
Формульная единица вещества 34
Фугитивность 330
Функция(и) волновая 79
- антисимметричная 113
- симметричная 113
- радиального распределения вероят-
вероятности 91
-собственная 81
-состояния 176
Халькогениды 12
Химическая номенклатура ИЮПАК 12
Химические соединения 11
- бинарные 12
- многоэлементные 12
Химический источник тока 525, 551
-, применение 562
Химический эквивалент 34
Химический(е) элемент(ы) 10
-5- 95
-/?- 95,96
-d- 95,98
-/- 95,99
Химия 9
- коллоидная 420
Хромат калия 25
Царская водка 23
Цеолиты 471
Цикл 177
-Гесса 189
Число эквивалентности (эквивалентное
число) 34,527
Электрическая проводимость (элек-
(электропроводность) 456
- молярная 457
- молярная предельная иона 459
- удельная 456
Электрод(ы) водородный стандартный
537
-второгорода 546
- газовые 543
- ионообменные (ионоселективные) 549
- кислородный стандартный 543
-, классификация 542
- необратимые 542
- обратимые 542
-окислительно-восстановительные 548
- первого рода 542
-, поляризация 573
-, - концентрационная 576
- сравнения 537
Электродвижущая сила 537, 552, 554
Предметный указатель
769
Электролиз 551, 566
-, применение 570
Электролизер 526, 551
Электролит(ы) 422
- бинарный 438
- сильные 423, 435
- слабые 423
Электролитическая диссоциация (ио-
(ионизация) 423
Электроны проводимости 141
- спаренные 94
Электронная пара несвязывающая 121
- общая 111
- связывающая 121
Электроотрицательность 104, 107
- абсолютная (по Малликену) 107,108
- по Полингу 107, 108
Электрохимическая эквивалентная
масса 526
Эмали силикатные 641
Энергетический подуровень 88
Энергетический профиль пути реакции
262
Энергетический уровень 88
Энергия активации 257
- истинная 259
- опытная (аррениусовская) 256
-внутренняя 178
- стандартная 183
- Гельмгольца 201
-Гиббса 201
—, температурная зависимость 208
- ионизации 102, 107
- образования вещества стандартная
Гиббса 204
Энтальпия 181
- гидратации интегральная 424
- образования вещества 185
- стандартная 185
- растворения 424
- дифференциальная 425
- последняя 425
- интегральная 424
первая 425
полная 425
промежуточная 425
-стандартная реакции 183
—, температурная зависимость 208
- сгорания вещества стандартная 186
Энтропия 191
-вещества стандартная 199
-, изменения в процессах 195
- растворения 424
- реакции стандартная 208
—, температурная зависимость 208
Эрозия 595
- коррозионная 595
Эффект перезащиты 637
Яды каталитические 278
Именной указатель
Авогадро Амедео (Avogadro A.) A776-1856) — итал. физик и химик 34, 36
Акимов Георгий Владимирович A901-1953) — сов. физикохимик 619
Аррениус Сванте (Arrhenius S.) A859-1927) — швед, физикохимик 256,423, 460
Баландин Алексей Александрович A898-1967) — сов. физикохимик 290
Бертло Марселей (Berthelot M.) A827-1907) — франц. химик 204, 533
Берцелиус Йене (Berzelius J.) A779-1848) — швед, химик 273
Бойль Роберт (Boyle R.) A627-1691) — англ. физик и химик 36, 180, 197
Больцман Людвиг (Boltzmann L.) A844-1906) — австр. физик 193
Бор Нильс (Bohr N.) A885-1962) — дат. физик 76
Борн Макс (Born M.) A882-1970) — нем. физик-теоретик 78
Браун Карл (Braun К.) A850-1918) — нем. физик 361, 439, 454
Брёнстед Йоханнес (Bronstedt J.) A879-1947) — дат. физикохимик 279, 423
Бройль Луи де (de Broglie L.) A892-1987) — франц. физик 77
Бунзен Роберт (Bunsen R.) A811-1899) — нем. химик 714
Бутлеров Александр Михайлович A828-1886) — рус. химик-органик 110
Бьёррум Нильс (Bjerrum N.) A879-1958) — дат. химик 436
Вааге Петер (Waage P.) A833-1900) — норв. химик 245, 331, 340
Ван-дер-Ваальс Йоханнес (van der Waals J.) A837-1923) — нидерл. физик 143, 149,
285, 340
Ван Лаар Иоганнес (van Laar J.) A860-1938) — нидерл. физикохимик 434
Вант-Гофф Якоб (Van't Hoff J.) A852-1911) — нидерл. физикохимик 254,256,345,433
Вернер Альфред (Werner A.) A866-1919) — швейц. химик 16
Гейзенберг Вернер (Heisenberg W.) A901-1976) — нем. физик 78
Гей-Люссак Жозеф (Gay-Lussac J.) A778-1850) — франц. физик и химик 35, 36
Гейтлер Вальтер (Heitler W.) (р. 1904) — нем. физик и химик-теоретик 111
Гельмгольц Герман (Helmholtz H.) A821-1894) — нем. физик и физиолог 175,201,203
Генри Уильям (Henry W.) A774-1836) — англ. химик 287,427
Гесс Герман Иванович A802-1850) — рус. химик 187
Гиббс Джозайя (Gibbs J.) A839-1903) — амер. физик-теоретик 201, 203, 358
Гиллеспи Рональд (Gillespie R.) (р. 1924) — канад. физикохимик 124
Гульдберг Като (Guldberg С.) A836-1902) — норв. физикохимик и математик
245,331,340
Дальтон Джон (Dalton J.) A766-1844) — англ. химик и физик 35, 36, 329
Даниэль Джон (Daniel J.) A790-1845) — англ. физик и химик 551, 576
Дебай Петер (Debye P.) A884-1966) — нидерл. физик 143, 436, 442
Джоуль Джеймс (Joule J.) A818-1889) — англ. физик 175
Карно Никола (Carnot N.) A796-1832) — франц. физик и инженер 175
Кирхгоф Густав (Kirchhoff G.) A824-1887) — нем. физик 208, 209
Кистяковский Владимир Александрович A865-1952) — сов. физикохимик 619
Клапейрон Бенуа (Clapeyron В.) A799-1864) — франц. физик и инженер 36, 177
Клаузиус Рудольф (Clausius R.) A822-1888) — нем. физик 191
Клечковский Всеволод Маврикиевич A900-1972) — сов. агрохимик 93
Именной указатель 771
Кобозев Николай Иванович A902-1974) — сов. физикохимик 290
Колотыркин Яков МихайловичA910-1995) — рос. физикохимик 619
Кольрауш Фридрих (Kohlrausch F.) A840-1910) — нем. физик и физикохимик 459
Лавуазье Антуан (Lavoisier A.) A743-1794) — франц. химик 35
Ленгмюр Ирвинг (Langmuir I.) A881-1957) — амер. физик и физикохимик 285, 287
Леннард-Джонс Джон (Lennard-Jones J.) A894-1954) — англ. химик-теоретик 145
Ле Шателье Анри (Le Chatelier H.) A850-1936) — франц. физикохимик и металло-
металловед 361,439,454
Ломоносов Михаил Васильевич A711-1765) — первый рус. ученый-естествоиспы-
ученый-естествоиспытатель 35
Лондон Фриц (London F.) A900-1954) — нем. физик-теоретик 111, 144
Лоури Томас (Lowry Т.) A874-1936) — англ. химик 279,423
Льюис Гилберт (Lewis G.) A875-1946) — амер. физикохимик 111, 330,423
Майер Юлиус (Mayer J.) A814-1878) — нем. врач и естествоиспытатель 175
Малликен Роберт (Mullikan R.) A896-1986) — амер. физикохимик 107
Мариотт Эдмунд (Mariotte E.) A620-1684) — франц. физик 36, 180, 197
Менделеев Дмитрий Иванович A834-1907) — рус. ученый-энциклопедист, химик
34, 36, 100, 108, 177
Нернст Вальтер (Nernst W.) A864-1941) — нем. физикохимик 533, 554, 561, 576
Ом Георг (Ohm G.) A787-1854) — нем. физик 456
Онзагер Ларе (Onsager L.) A903-1976) — амер. физик и физикохимик 436,442
Оствальд Вильгельм (Ostwald W.) A853-1932) — нем. физикохимик 255,439
Паули Вольфганг (Pauli W.) A900-1958) — швейц. физик 93, 145
Пауэлл Джордж (Powell G.) (р. 1910) — амер. химик 124
Планк Макс (Planck M.) A858-1947) — нем. физик 76, 199
Полинг Лайнус (Paulihg L.) A901-1994) — амер. физик и химик 104, 107, 115
Пруст Жозеф (Proust J.) A754-1826) — франц. химик 35
Рауль Франсуа (Raoult F.) A830-1901) — франц. физик и химик 429
Рейнитцер Фридрих (Reinitzer F.) A857-1927) — нем. химик 155
Рихтер Иеремия (Richter J.) A762-1807) — нем. химик 35
Сиджвик Невил (Sidgwick N.) A873-1952) — англ. химик 124
Слейтер Джон (Slater J.) A900-1976) — амер. физик-теоретик 115
Стоке Джордж (Stokes G.) A819-1903) — англ. физик и математик 456
Тафель Юлиус (Tafel J.) A862-1918) — нем. физикохимик 577
Томсен Ханс (Thomsen H.) A826-1909) — дат. химик 204, 533
Фарадей Майкл (Faraday М.) A791-1867) — англ. физик и химик 457, 526
Фрумкин Александр Наумович A895-1976) — сов. электрохимик 576, 619
Хунд Фридрих (Hund F.) (р. 1896) — нем. физик-теоретик 94
Хюккель Эрих (Hiickel E.) A896-1980) — нем. физик и химик-теоретик 436,442
Шрёдингер Эрвин (Schrodinger Е.) A887-1961) — австр. физик 79, 81, 112
Эванс Мередит (Evans М.) A904-1952) — англ. физикохимик 619
Эйнштейн Альберт (Einstein A.) A879-1955) — нем. физик-теоретик 40
Эйринг Генри (Eyring H.) A901-1981) — амер. физикохимик 263
Якоби Борис Семенович A801-1874) — рус. физик и электротехник 551, 576
Оглавление
Предисловие 5
I. Ведение в химию • 9
1. Понятия вещества и химической реакции 10
1.1. Основные определения 10
1.2. Классификация и номенклатура неорганических соединений 12
1.3. Окислительно-восстановительные реакции 18
2. Стехиометрия. Закономерности изменения и способы определения
количества вещества 34
2.1. Основные определения 34
2.2. Количественные законы протекания химических реакций 35
2.3. Растворы. Общие понятия 40
Практические занятия 46
Фундаментальные понятия и законы химии 46
Примеры решения задач 46
Задачи для самостоятельного решения 52
ЛР 1. Окислительно-восстановительные реакции 55
ЛР 2. Химический эквивалент 65
ЛР 3. Приготовление раствора и определение его концентрации 71
II. Строение вещества 75
3. Строение атомов 76
3.1. Основные положения квантовой механики 76
3.2. Стационарное уравнение Шрёдингера 79
3.3. Уравнение Шрёдингера для атома водорода 81
3.4. Квантовые числа 82
3.5. Атомные орбитали водорода 89
3.6. Принципы построения электронной структуры атомов элементов 92
3.7. Электронные конфигурации атомов элементов Периодической
системы 95
3.8. Периодический закон и Периодическая система химических
элементов Д.И. Менделеева 100
3.9. Периодическое изменение некоторых свойств атомов химических
элементов 102
4. Химическая связь и строение вещества 106
4.1. Основные характеристики химической связи 106
4.2. Квантово-механические представления о природе химической связи.
Основные методы описания ковалентной связи 111
4.3. Метод валентных связей 115
4.4. Метод молекулярных орбиталей 128
Оглавление 773
4.5. Невалентные типы связей 137
4.6. Межмолекулярное взаимодействие 143
4.7. Строение вещества в конденсированном состоянии 149
Практические занятия 156
Строение атомов и ионов. Периодический закон и периодическое
изменение свойств элементов 156
Примеры решения задач 156
Задачи для самостоятельного решения 160
Химическая связь и строение вещества 163
Примеры решения задач 163
Задачи для самостоятельного решения 172
III. Закономерности протекания химических процессов 175
5. Тепловой эффект химической реакции 176
5.1. Основные понятия и определения химической термодинамики 176
5.2. Первый закон термодинамики и его приложение к процессам в
идеальном газе 179
5.3. Понятие теплового эффекта химической реакции 181
5.4. Стандартные энтальпии образования и сгорания веществ 184
5.5. Закон Гесса и следствия из него 187
6. Направление химической реакции 191
6.1. Второй закон термодинамики. Энтропия как функция состояния
системы 191
6.2. Изменение энтропии в некоторых процессах 195
6.3. Третий закон термодинамики. Абсолютные значения стандартных
энтропии веществ 198
6.4. Критерии направленности самопроизвольного процесса в закрытой
системе 201
6.5. Химический потенциал 205
6.6. Температурная зависимость стандартных энергии Гиббса, энтальпии
и энтропии химической реакции 208
Практические занятия 212
Энергетика химических реакций и фазовых переходов 212
Примеры решения задач 212
Задачи для самостоятельного решения 217
Направление самопроизвольного протекания химических реакций 221
Примеры решения задач 221
Задачи для самостоятельного решения 228
ЛР 4. Определение энтальпий химических реакций и процессов
растворения солей 232
IV. Кинетика химических процессов 239
7. Кинетика гомогенных химических реакций 240
7.1. Основные понятия и определения 240
7.2. Основной постулат химической кинетики 244
7.3. Кинетические уравнения односторонних реакций 246
7.4. Методы определения порядка химических реакций 253
774 Оглавление
7.5. Влияние температуры на скорость химических реакций 256
7.6. Теоретические представления о скоростях элементарных реакций 258
8. Особенности кинетики гетерогенных реакций 265
8.1. Реакции на границе раздела твердое тело — газ и твердое тело —
жидкость 265
8.2. Скорость гетерогенных реакций 268
8.3. Твердофазные реакции 270
9. Основы катализа 273
9.1. Основные понятия и определения 273
9.2. Механизм протекания каталитических реакций 276
9.3. Гомогенный катализ 279
9.4. Адсорбция 284
9.5. Гетерогенный катализ 289
Практические занятия 294
Кинетика химических реакций 294
Примеры решения задач 294
Задачи для самостоятельного решения 297
ЛР 5. Кинетика гомогенных химических реакций 305
ЛР 6. Гетерогенные химические реакции 309
ЛР7. Каталитические реакции 313
V. Химическое равновесие 321
10. Виды, особенности и характеристики химического равновесия 322
10.1. Обратимые и необратимые химические реакции 322
10.2. Виды и особенности химического равновесия 325
10.3. Количественные характеристики химического равновесия 328
10.4. Кинетический вывод закона Гульдберга—Вааге 334
11. Термодинамическое описание химического равновесия 340
11.1. Термодинамические критерии. Термодинамический вывод закона
Гульдберга—Вааге 340
11.2. Уравнения изотермы, изобары и изохоры химической реакции 345
11.3. Правило фаз Гиббса 351
12. Влияние различных факторов на химическое равновесие. Особенности
равновесий в гетерогенных системах 360
12.1. Введение катализатора и изменение концентрации компонентов 360
12.2. Изменение температуры 364
12.3. Изменение давления и разбавление системы инертным газом 365
12.4. Особенности описания равновесия в гетерогенных химических
системах 373
Практические занятия 378
Химическое равновесие в гомогенных и гетерогенных системах 378
Примеры решения задач 378
Задачи для самостоятельного решения 390
ЛР 8. Химическое равновесие 401
Оглавление 775
VI. Растворы 417
13. Общие свойства растворов 418
13.1. Основные понятия и определения 418
13.2. Термодинамические характеристики процесса образования
растворов 423
13.3. Коллигативные свойства растворов 428
14. Растворы электролитов 435
14.1. Влияние различных факторов на свойства растворов электролитов 435
14.2. Диссоциация слабых электролитов 437
14.3. Растворы сильных электролитов. Ионная сила и активность 440
14.4. Ионные равновесия в водных растворах электролитов 444
14.5. Гидролиз солей 447
14.6. Произведение растворимости 453
14.7. Электропроводность растворов электролитов 455
15. Свойства природной воды 461
15.1. Состав природной воды 461
15.2. Жесткость воды: виды, единицы измерения 465
15.3. Требования, предъявляемые к питьевой и технической воде 467
15.4. Способы умягчения воды 469
Практические занятия 475
Общие свойства водных растворов 475
Примеры решения задач 475
Задачи для самостоятельного решения 483
Свойства водных растворов электролитов 487
Примеры решения задач 487
Задачи для самостоятельного решения 494
ЛР 9. Растворы электролитов 498
ЛР 10. Жесткость и умягчение воды 515
VII. Электрохимические явления и процессы 523
16. Электродные процессы 524
16.1. Основные определения. Законы Фарадея 524
16.2. Потенциалы электрохимической системы. Двойной электрический
слой 528
16.3. Уравнение Нернста 533
16.4. Стандартный водородный электрод. Ряд стандартных электродных
потенциалов металлов 536
16.5. Классификация электродов 542
17. Химические источники тока и электролиз 551
17.1. Химические и концентрационные гальванические элементы. Элемент
Даниэля—Якоби 551
17.2. Термодинамика окислительно-восстановительных реакций,
протекающих в гальванических элементах 557
17.3. Практическое применение химических источников тока 562
17.4. Электролиз. Потенциал разложения, последовательность процессов
на электродах 566
776 Оглавление
17.5. Практическое применение электролиза 570
17.6. Кинетика электрохимических процессов 572
Практические занятия 578
Электрохимические процессы 578
Примеры решения задач 578
Задачи для самостоятельного решения 583
ЛР 11. Химические источники тока 587
VIII. Коррозия и борьба с ней 593
18. Коррозия металлов и сплавов 594
18.1. Классификация коррозионных сред, разрушений и процессов.
Показатели скорости коррозии 594
18.2. Химическая коррозия: виды и разновидности 597
18.3. Термодинамика и кинетика газовой коррозии 600
18.4. Законы роста толщины оксидных пленок 605
18.5. Электрохимическая коррозия: причины и механизм возникновения 608
18.6. Термодинамика электрохимической коррозии 613
18.7. Кинетика электрохимической коррозии 615
18.8. Влияние различных факторов на скорость электрохимической
коррозии 620
18.9. Коррозия в естественных условиях 622
19. Защита от коррозии 628
19.1. Основные факторы рационального конструирования. Легирование
металлических материалов 628
19.2. Изменение состава и свойств коррозионной среды 631
19.3. Электрохимическая защита: виды и механизм действия 635
19.4. Защитные покрытия: виды, методы нанесения и области применения ..638
Практические занятия 643
Коррозия и защита металлов и сплавов 643
Примеры решения задач 643
Задачи для самостоятельного решения 653
ЛР 12. Коррозия металлов и сплавов 660
ЛР 13. Защита от коррозии 682
IX. Основы практических занятий 697
20. Правила поведения и порядок работы в химической лаборатории 698
20.1. Общие требования 698
20.2. Техника безопасности и противопожарная безопасность 699
20.3. Оказание помощи при несчастных случаях 700
20.4. Рабочий журнал 701
21. Реактивы, посуда и оборудование учебной химической лаборатории 705
21.1. Реактивы 705
21.2. Посуда 705
21.3. Оборудование 710
22. Техника эксперимента 713
22.1. Мытье и сушка посуды 713
22.2. Нагревание и прокаливание. Нагревательные приборы 714
Оглавление 111
22.3. Измерение температуры 716
22.4. Весы и взвешивание 718
22.5. Перемешивание, осаждение и фильтрование 720
22.6. Приборы для получения газов 723
22.7. Определение плотности и реакции растворов. Отбор проб 725
Приложения 727
П.1. Наименования неорганических веществ 728
П.2. Основные характеристики атомов и молекул 732
П.З. Физические свойства неорганических веществ 736
П.4. Свойства воды и водных растворов 738
П.5. Термодинамические характеристики некоторых веществ 744
П.6. Электрохимические системы 750
Список рекомендованной литературы 756
Предметный указатель 758
Именной указатель 770
Учебное издание
Александр Алексеевич Гуров
Фатих Захарович Бадаев
Людмила Петровна Овчаренко
Валентин Николаевич Шаповал
Химия
Редактор ГА. Нилова
Художник НТ. Столярова
Корректор Г. С. Беляева
Компьютерная верстка С. Ч. Соколовского
Подписано в печать 02.03.2004. Формат 70x100 1/16.
Печать офсетная. Бумага офсетная. Гарнитура «Тайме».
Печ. л. 49. Усл. печ. л. 63,7. Уч.-изд. л. 49,5.
Тираж 2000 экз. Заказ № 9795
Издательство МГТУ им. Н.Э. Баумана,
105005, Москва, 2-я Бауманская, 5.
Отпечатано в полном соответствии
с качеством предоставленных диапозитивов
в ППП «Типография «Наука»
121099, Москва, Шубинский пер., 6