/


























































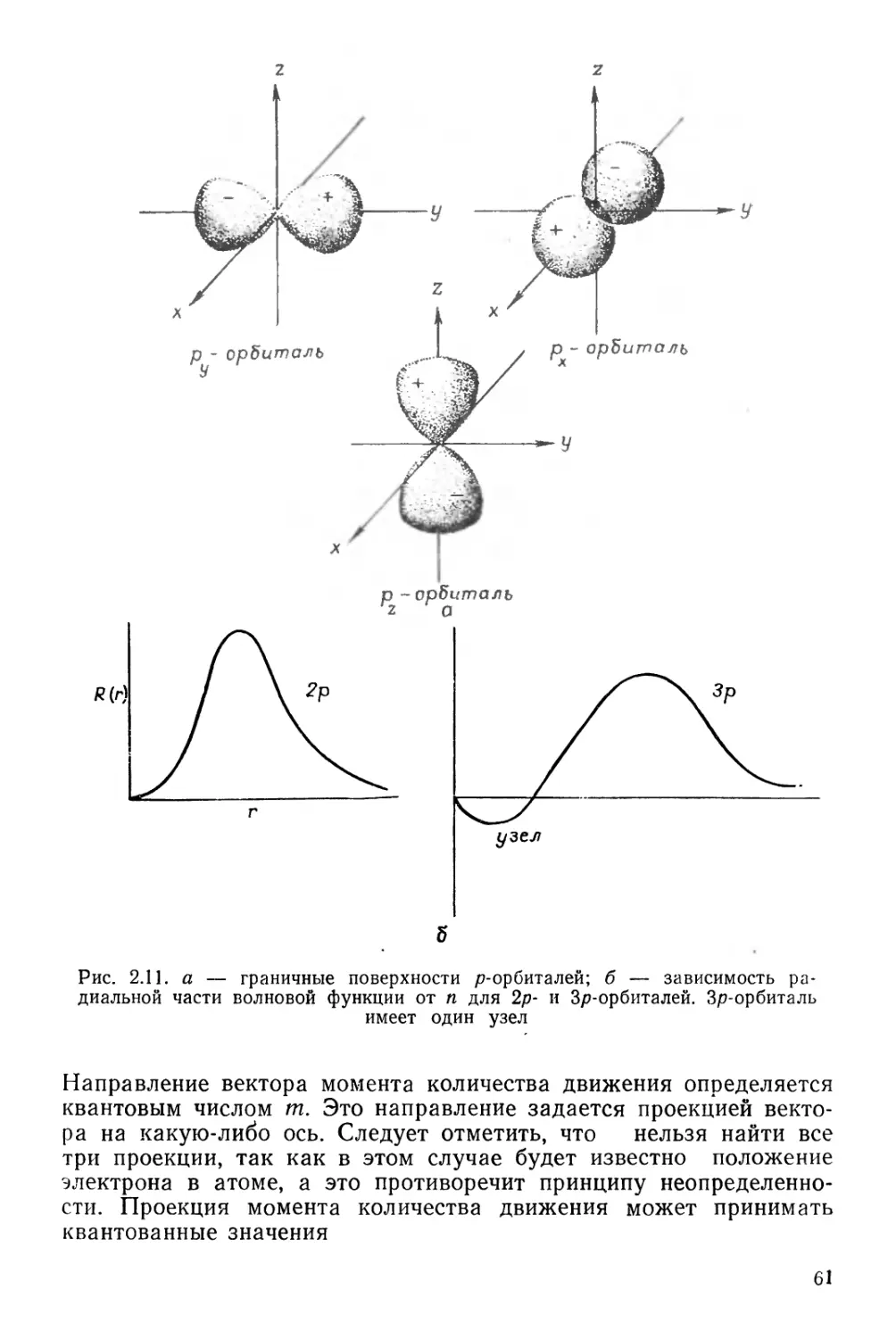
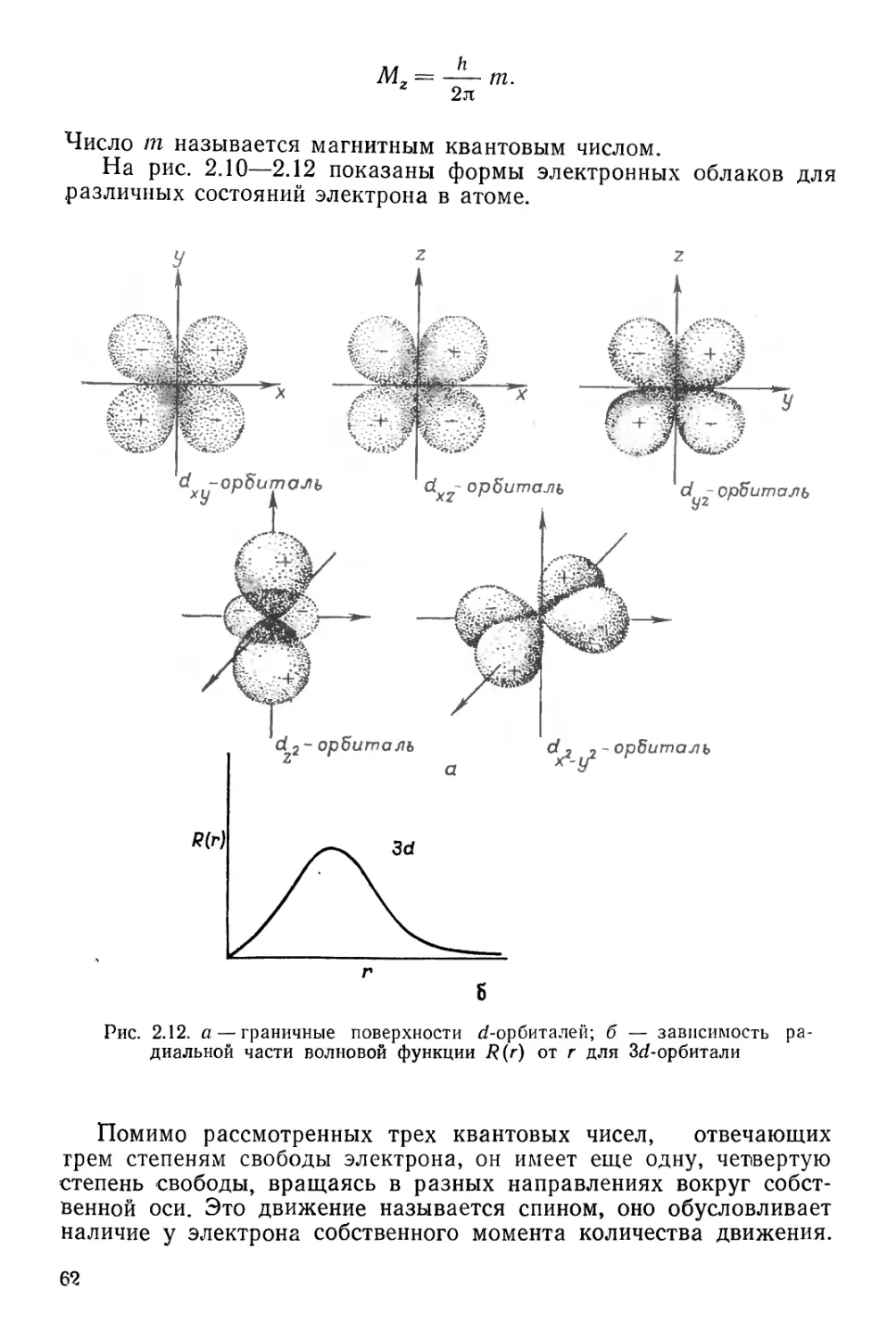













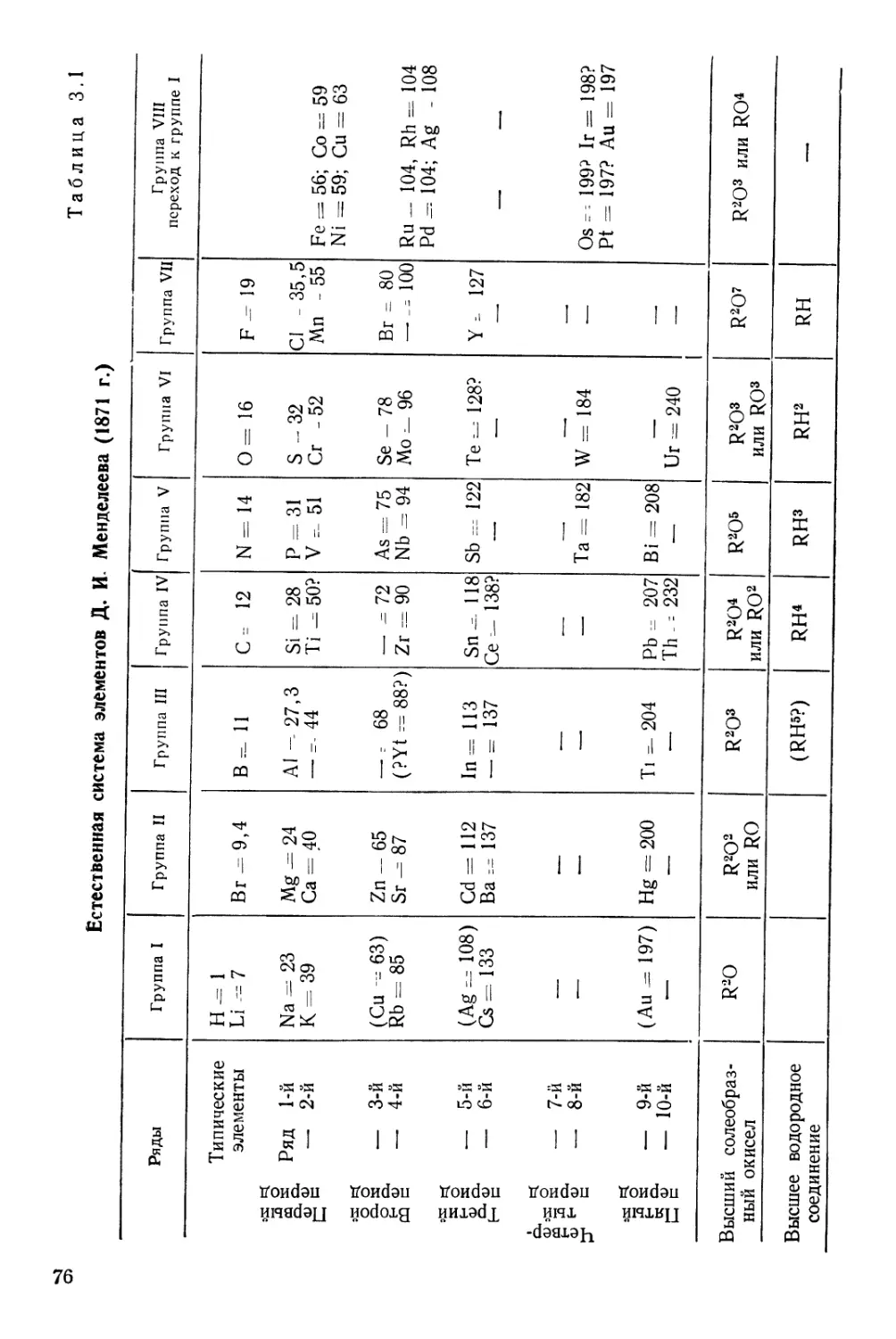























































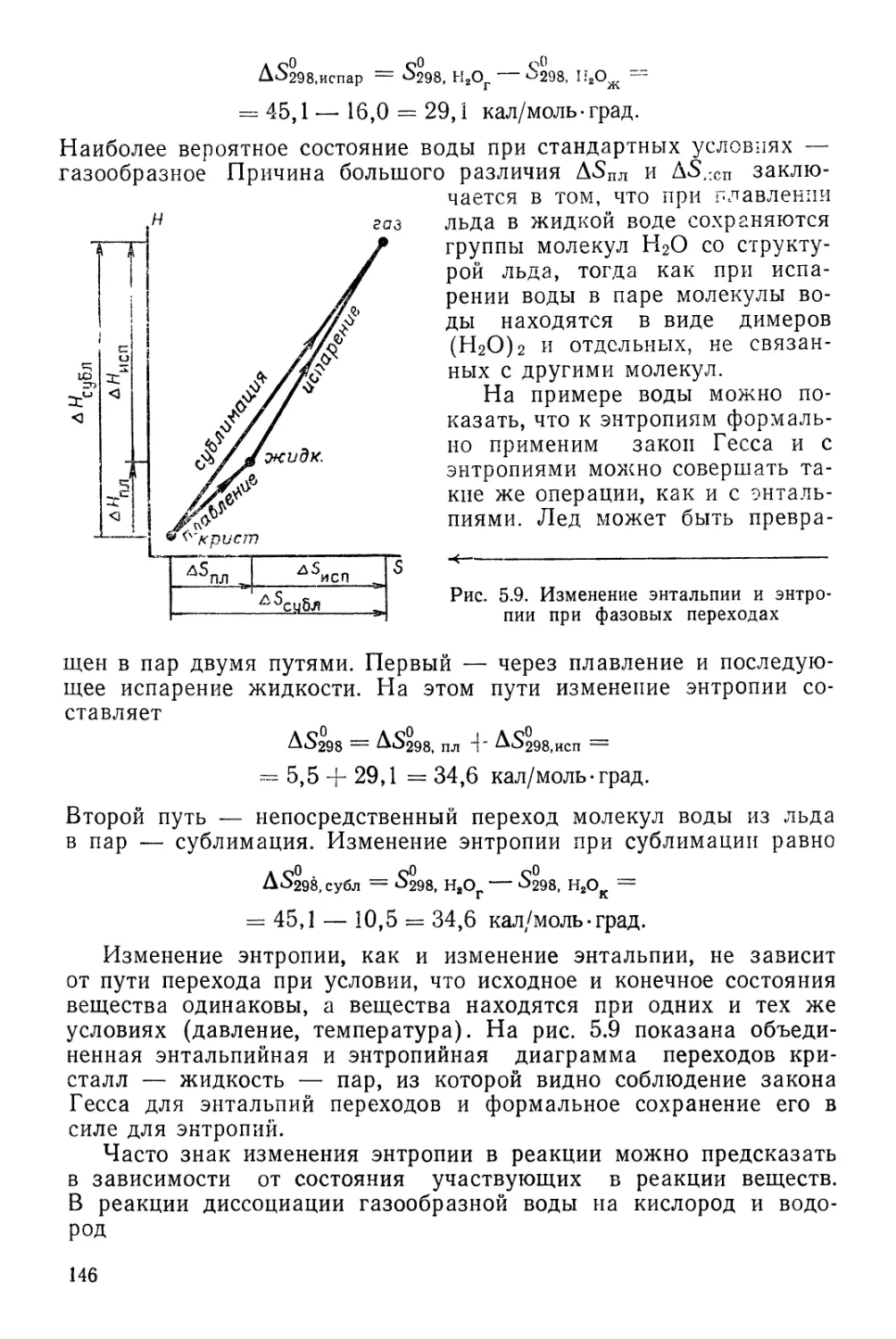













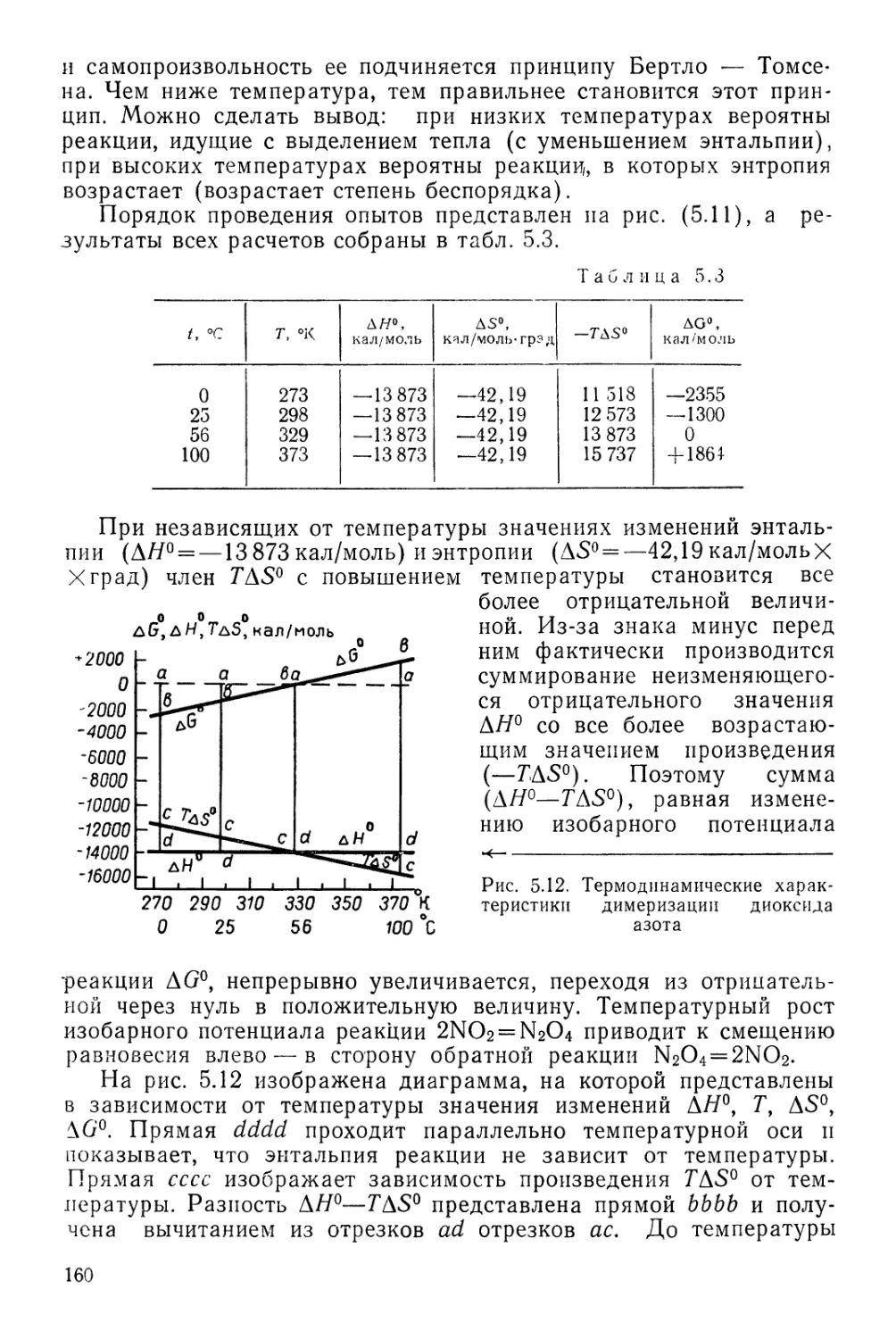











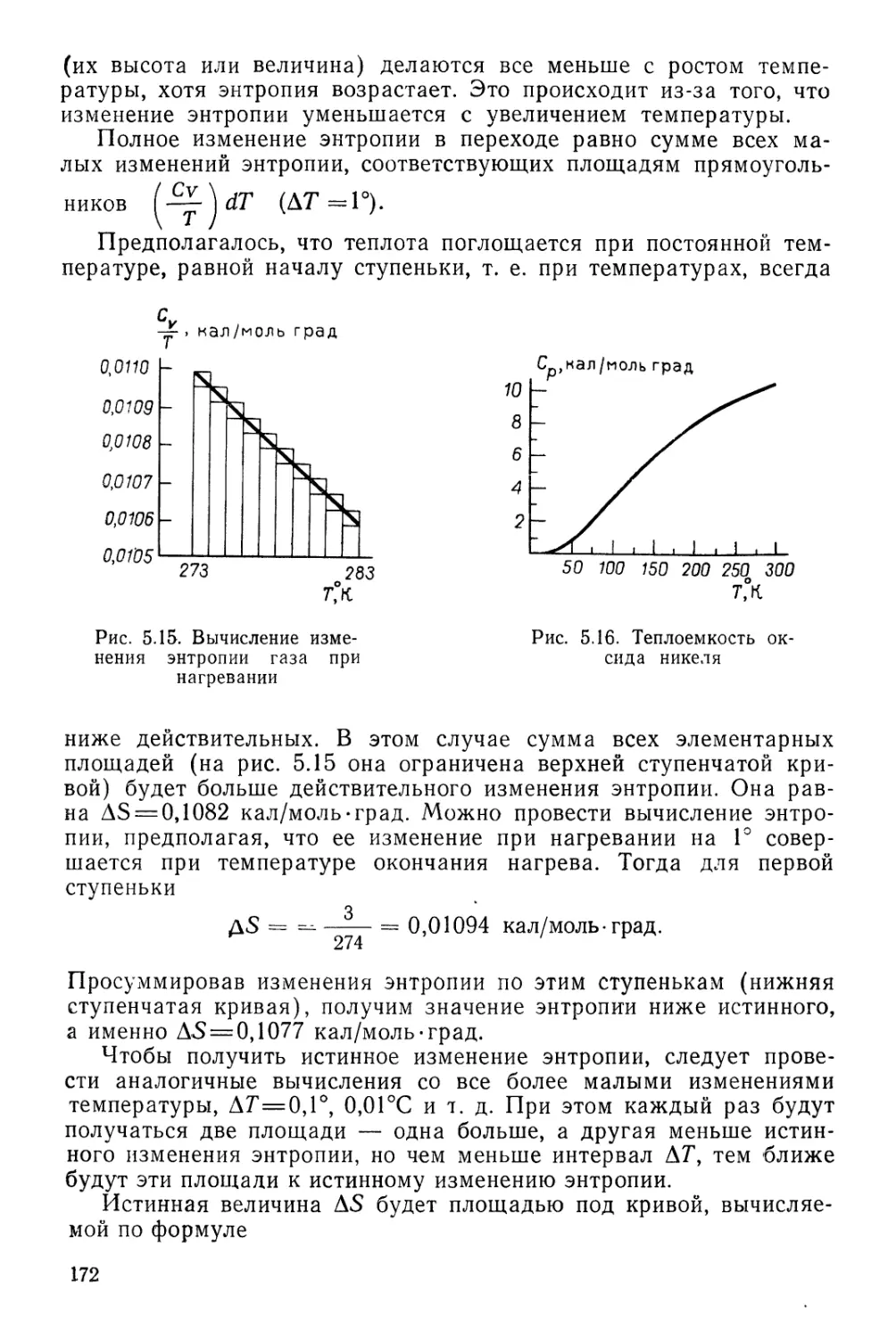






































































































































































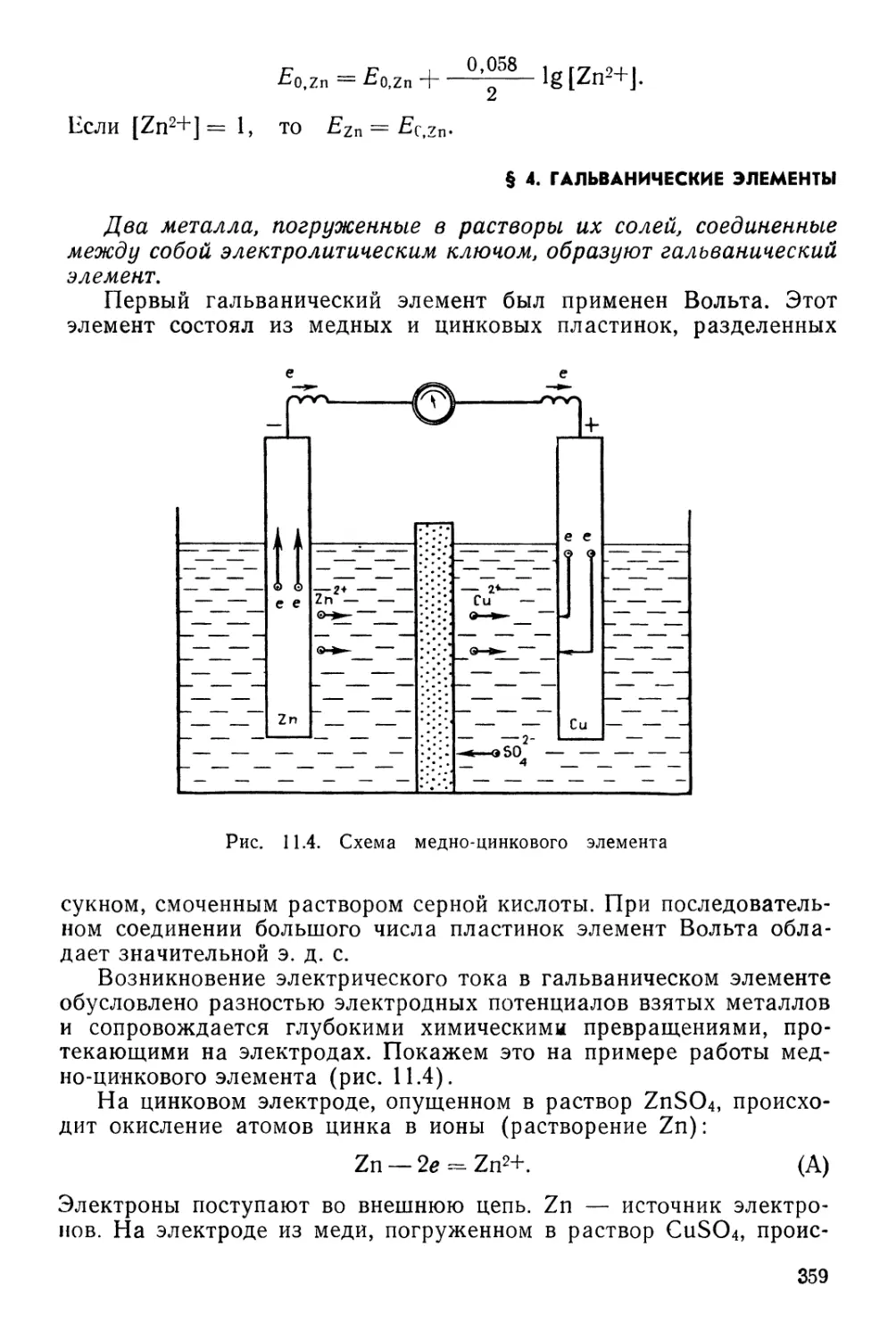















































































































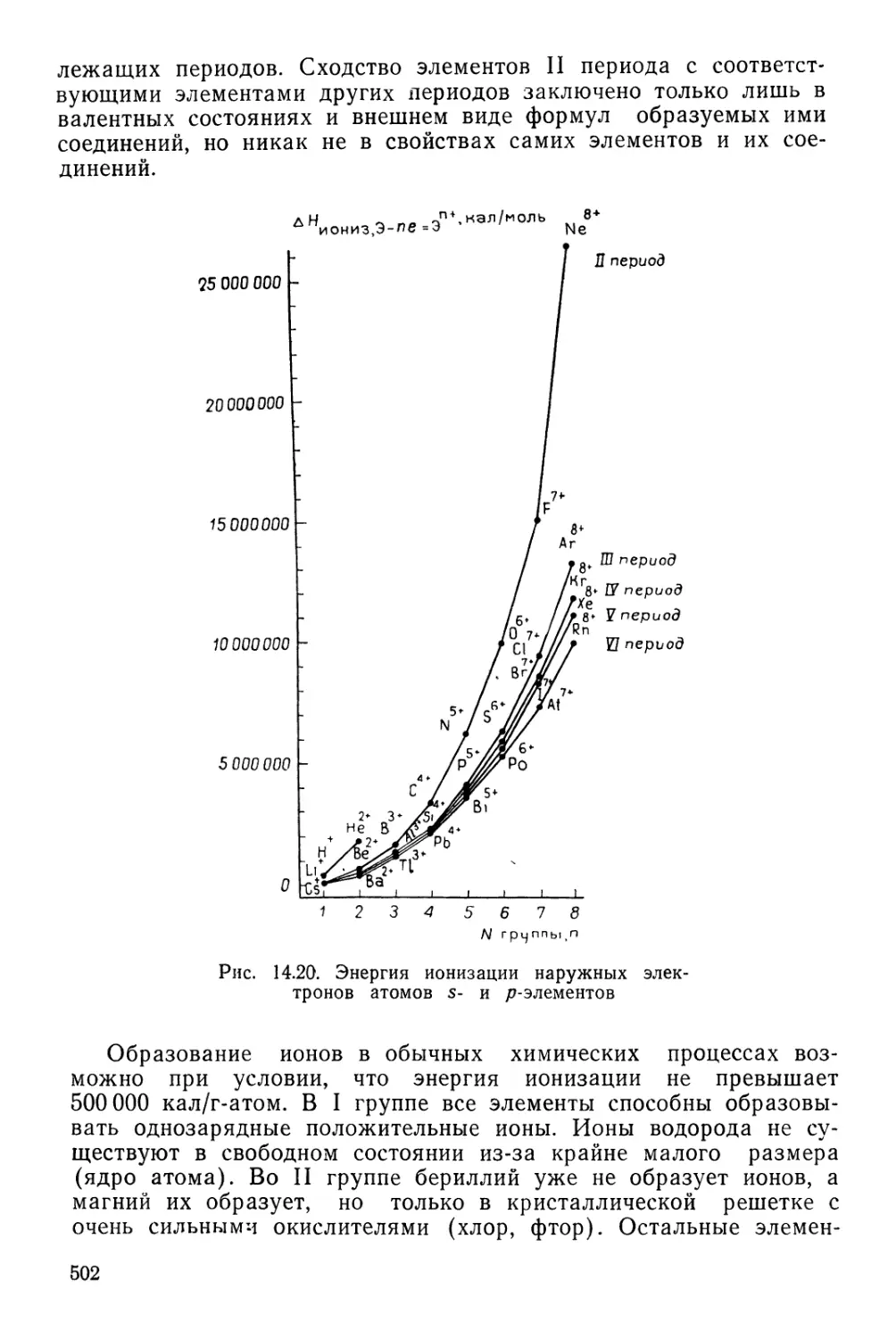
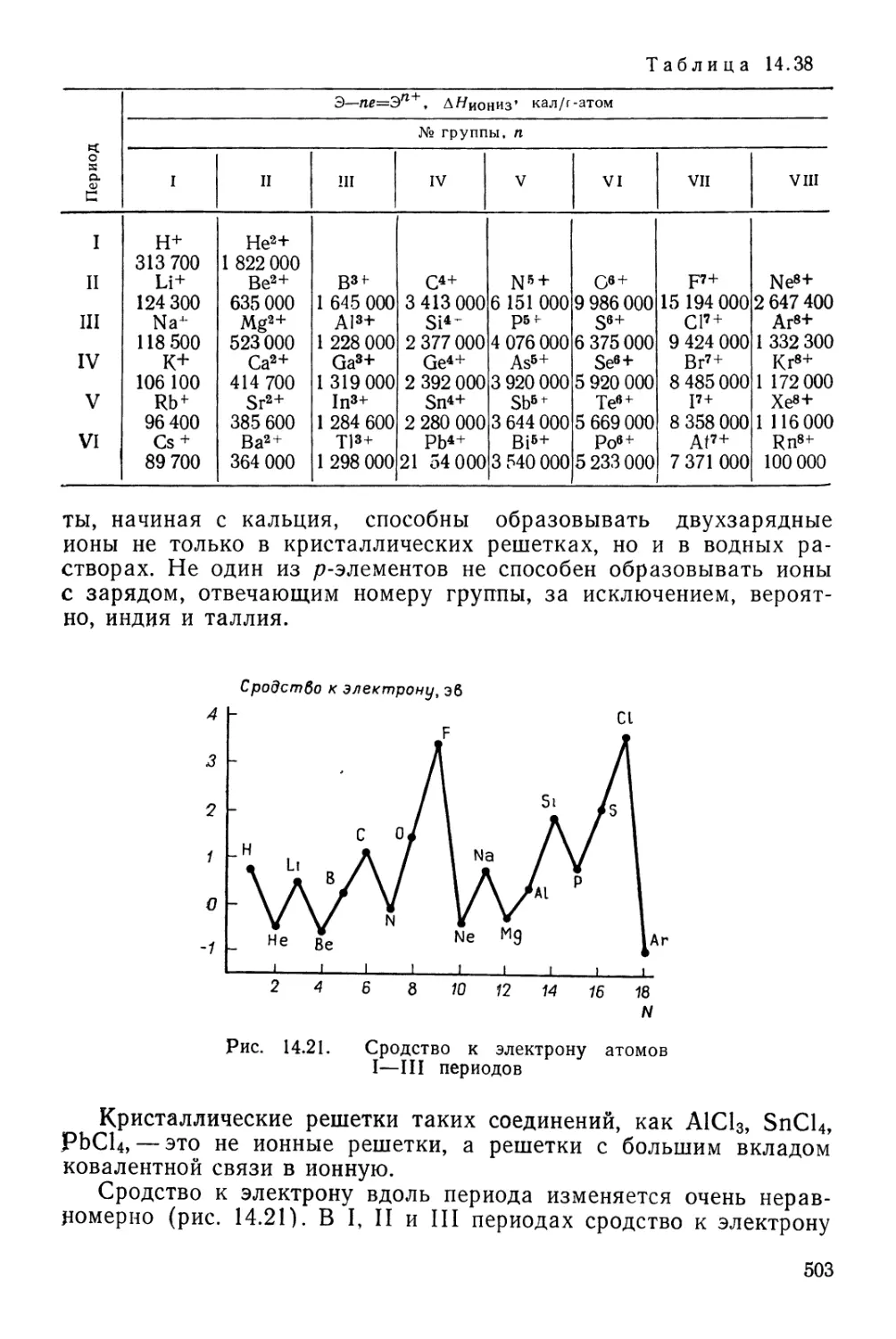
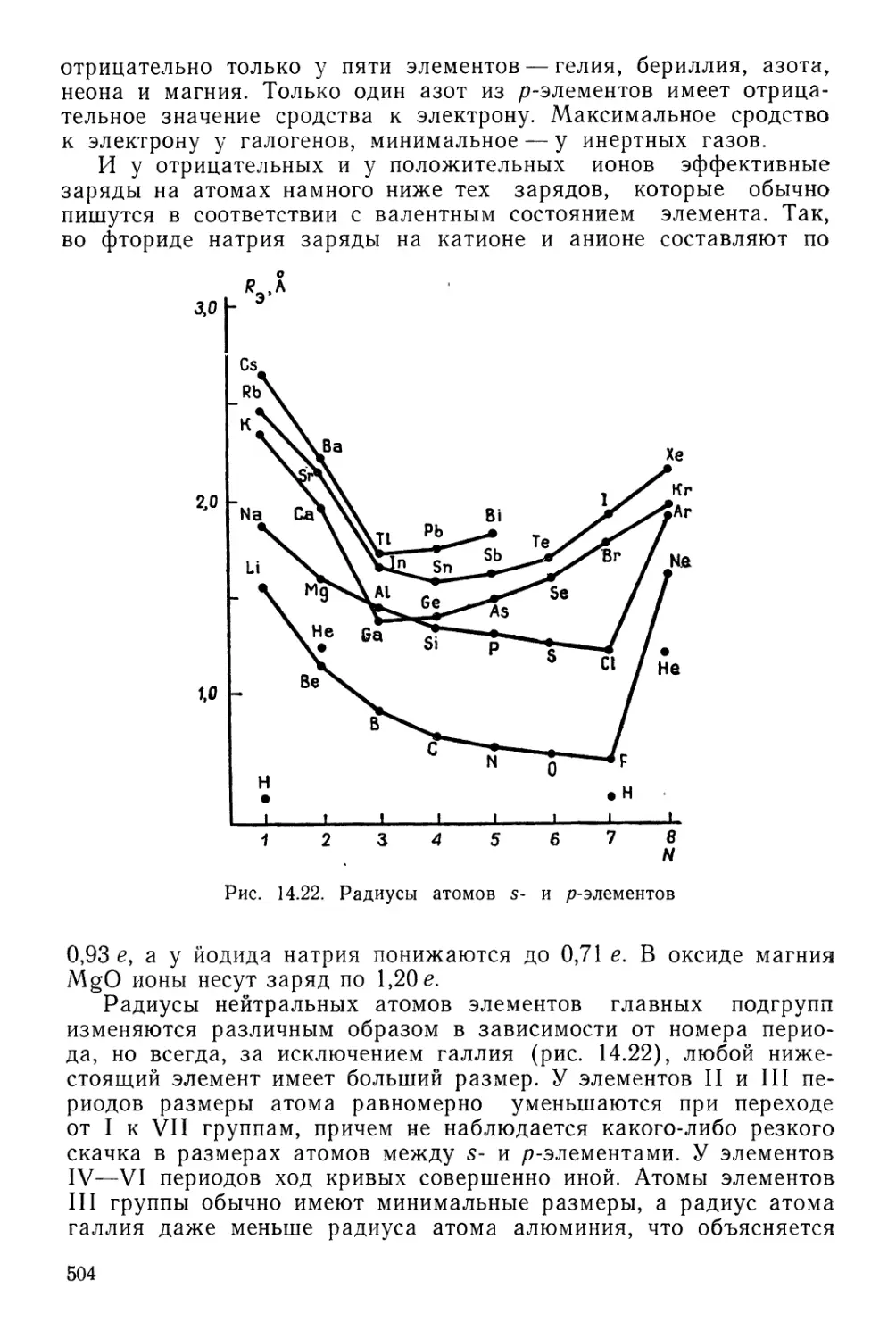
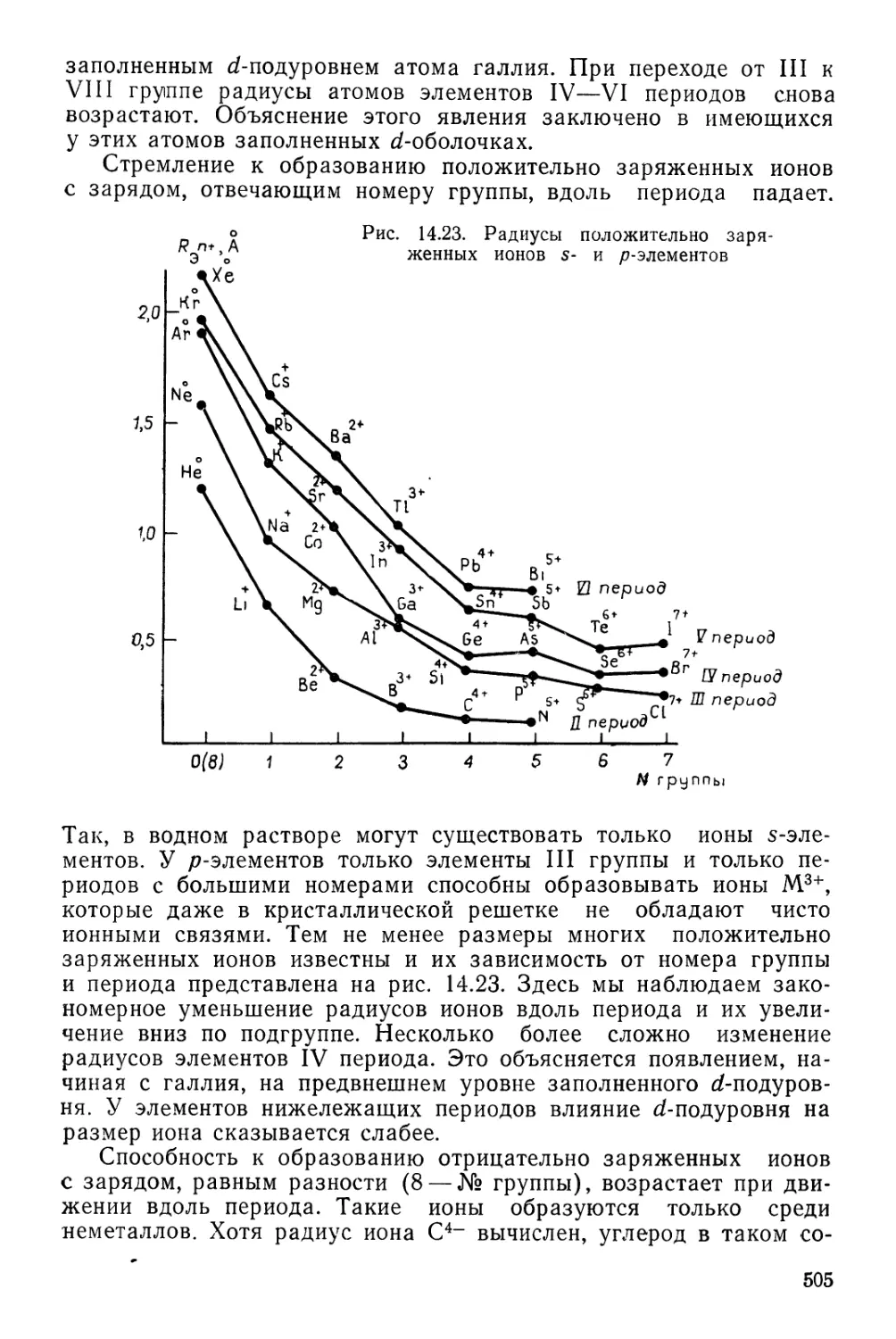

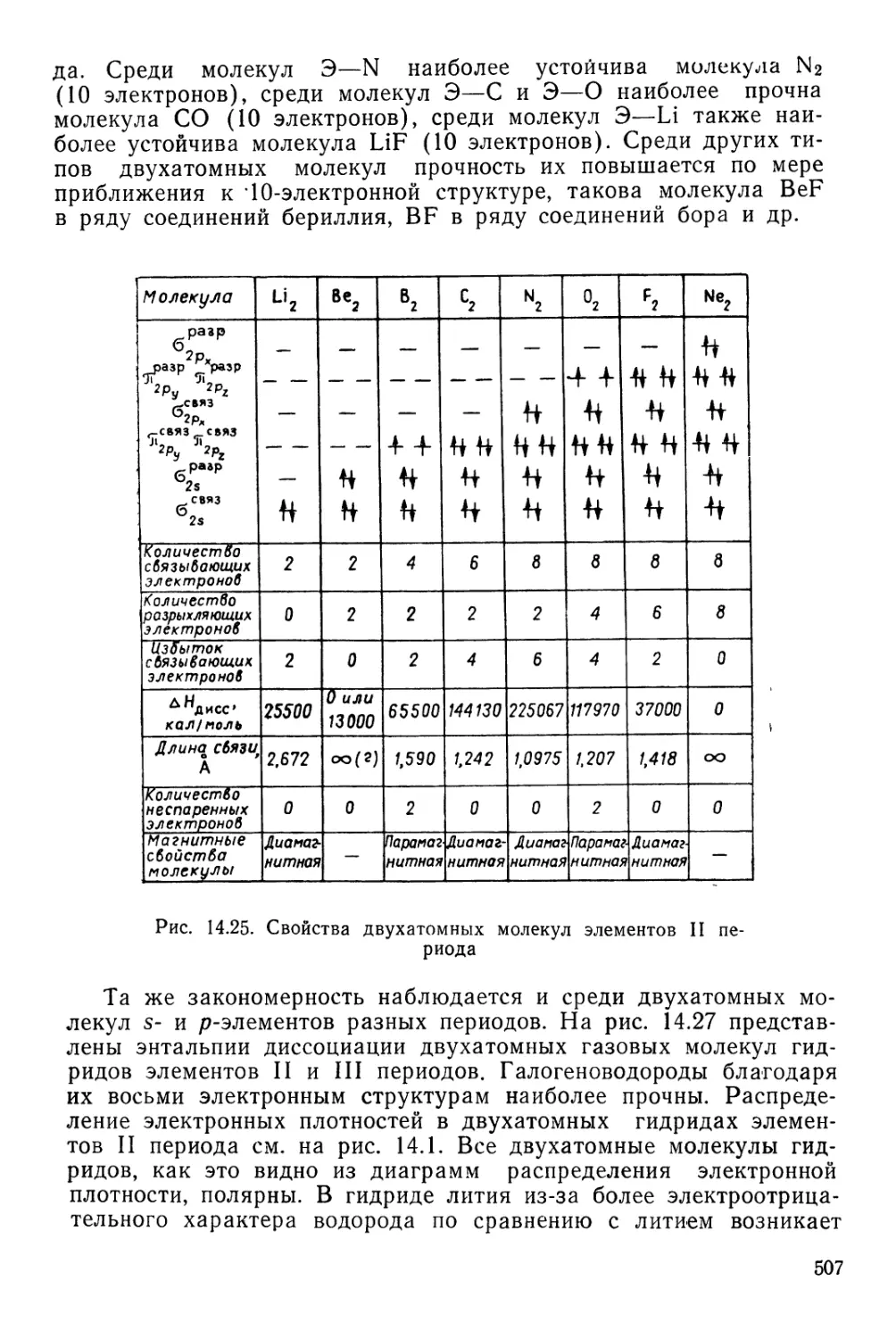


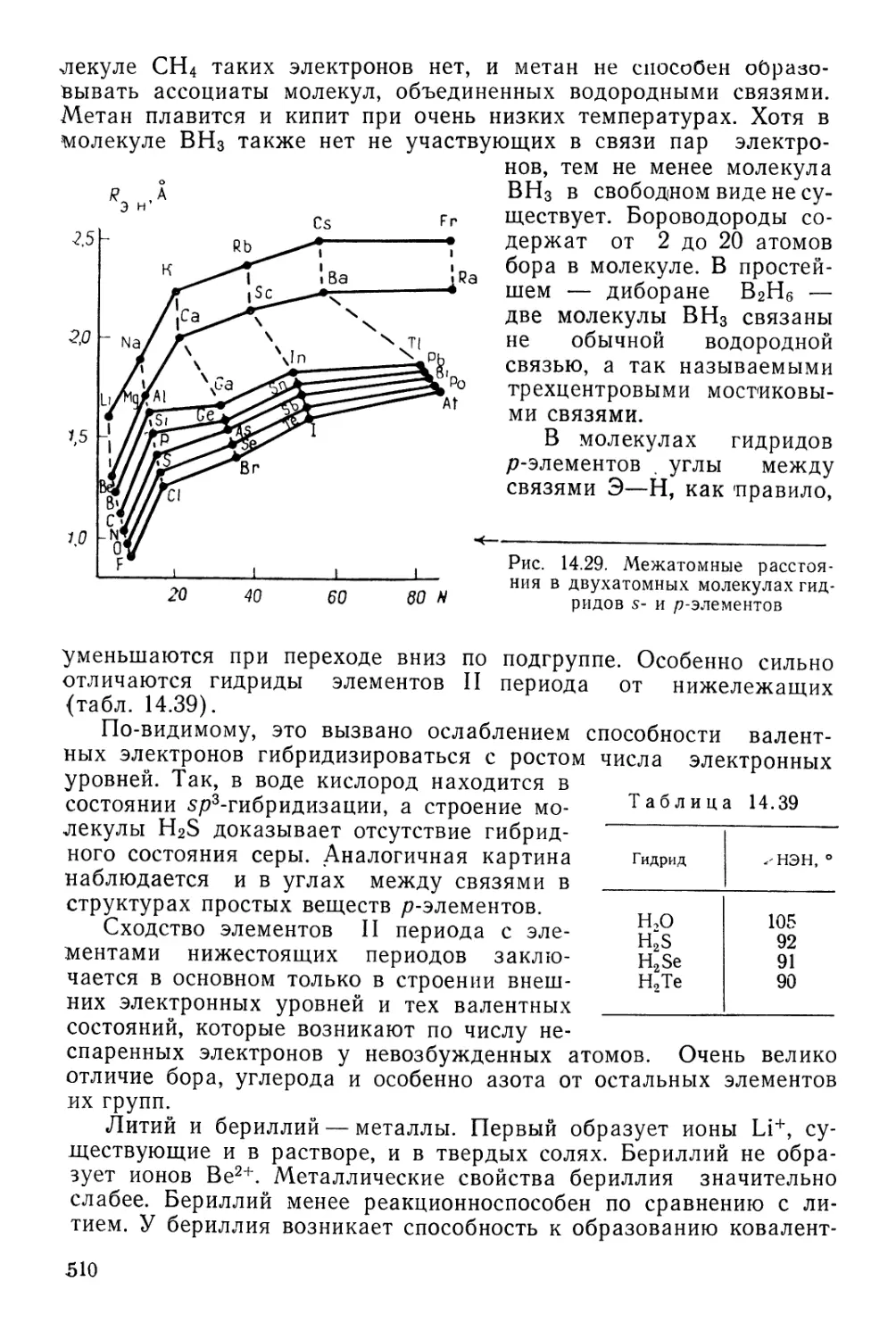


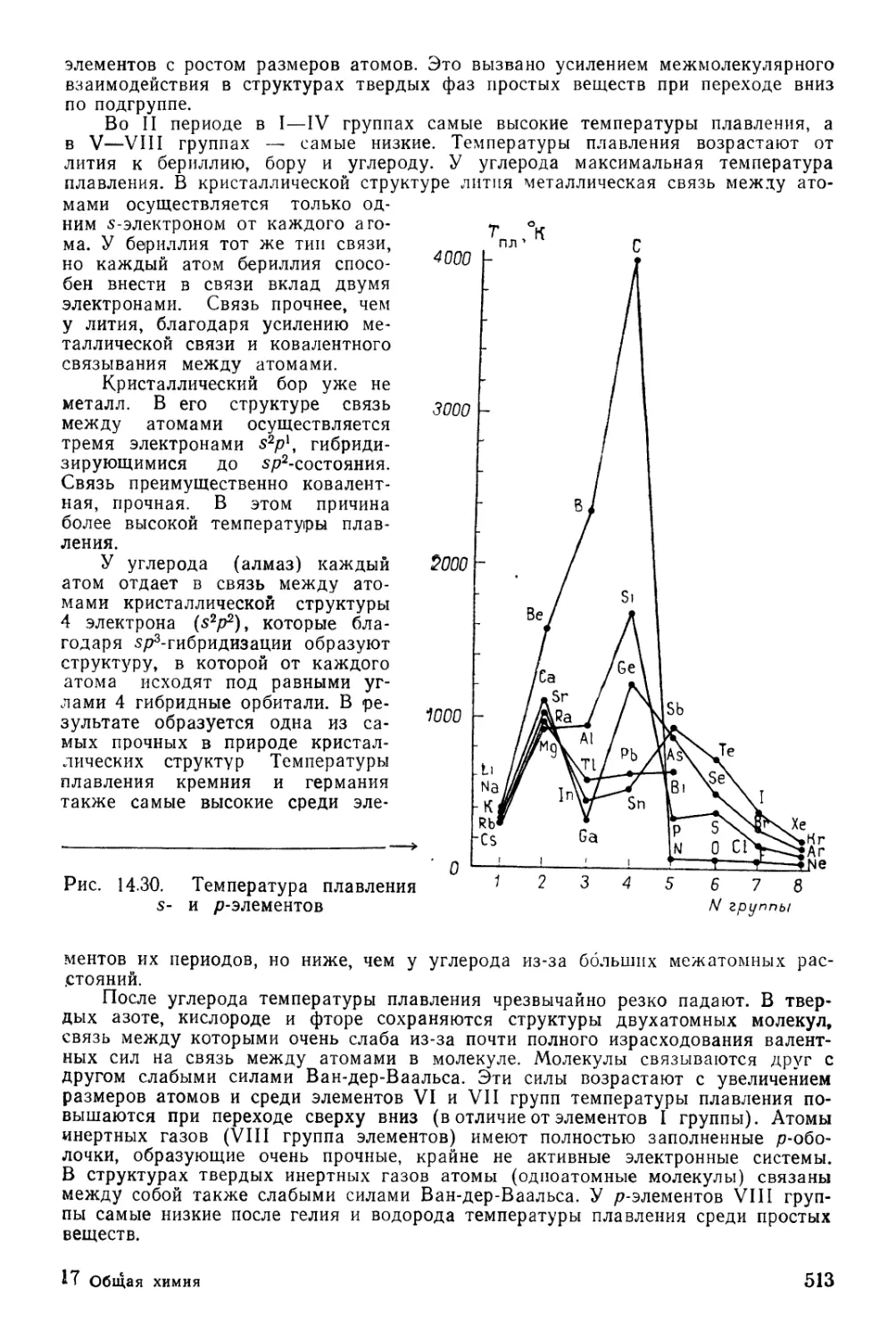
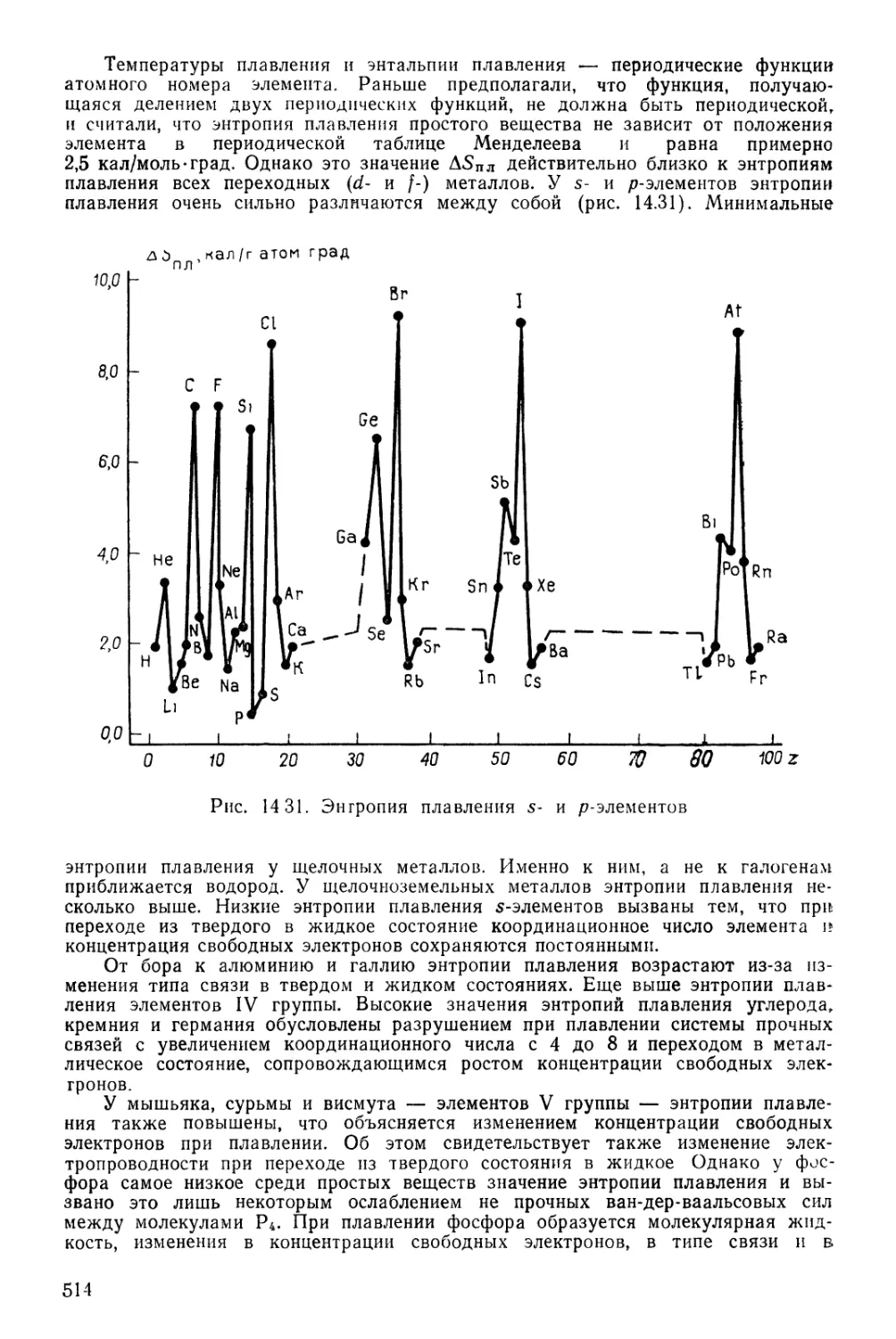





































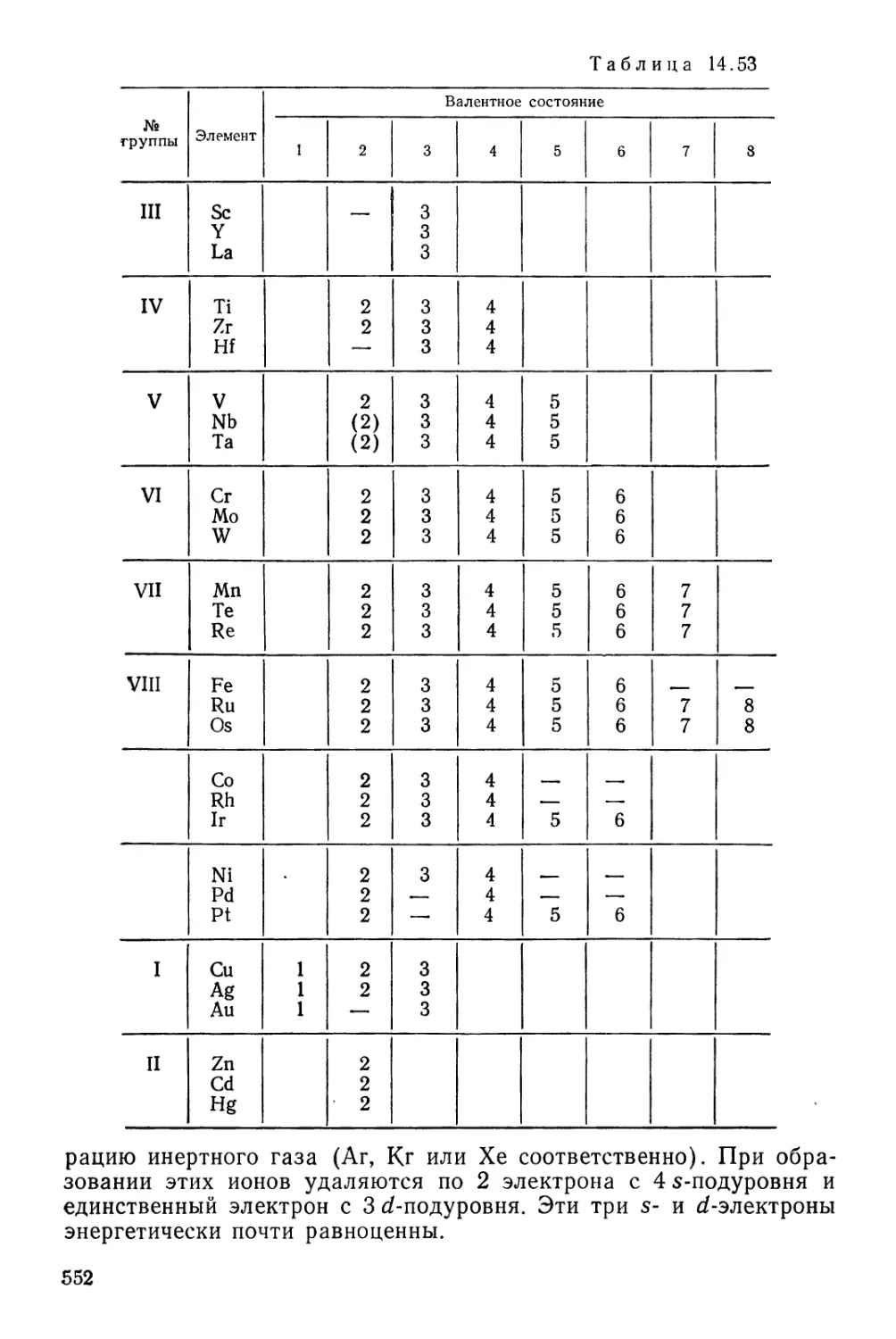


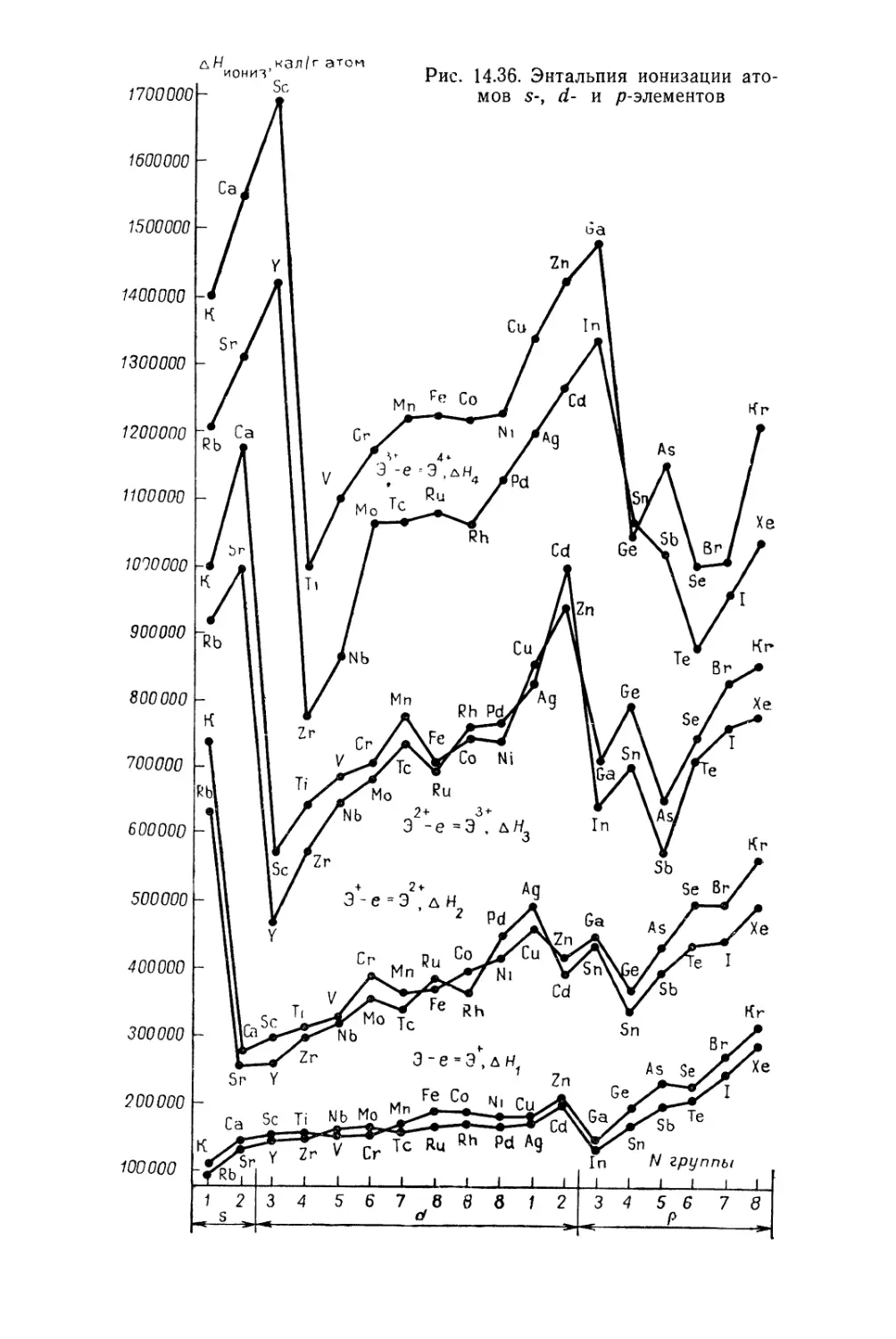
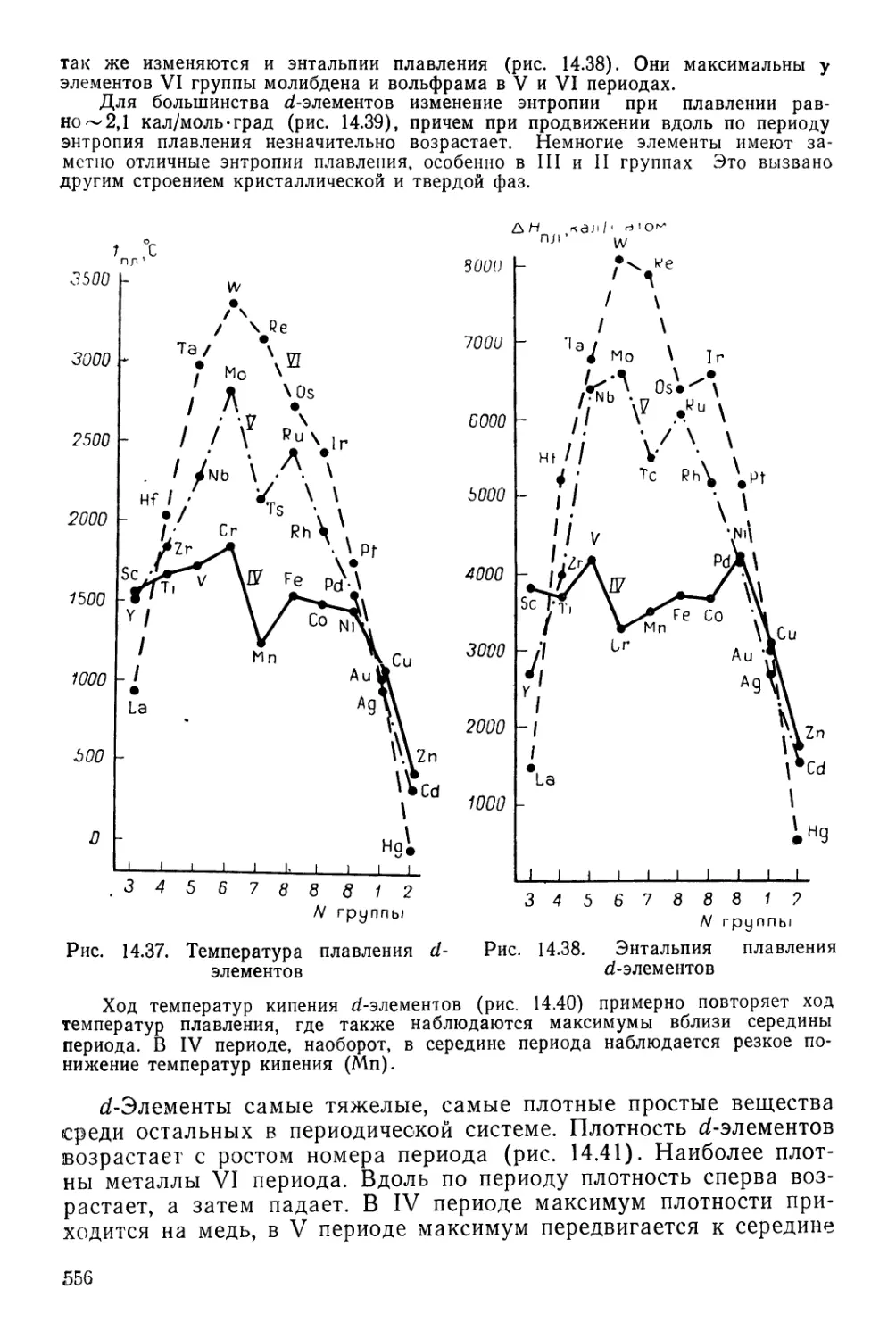
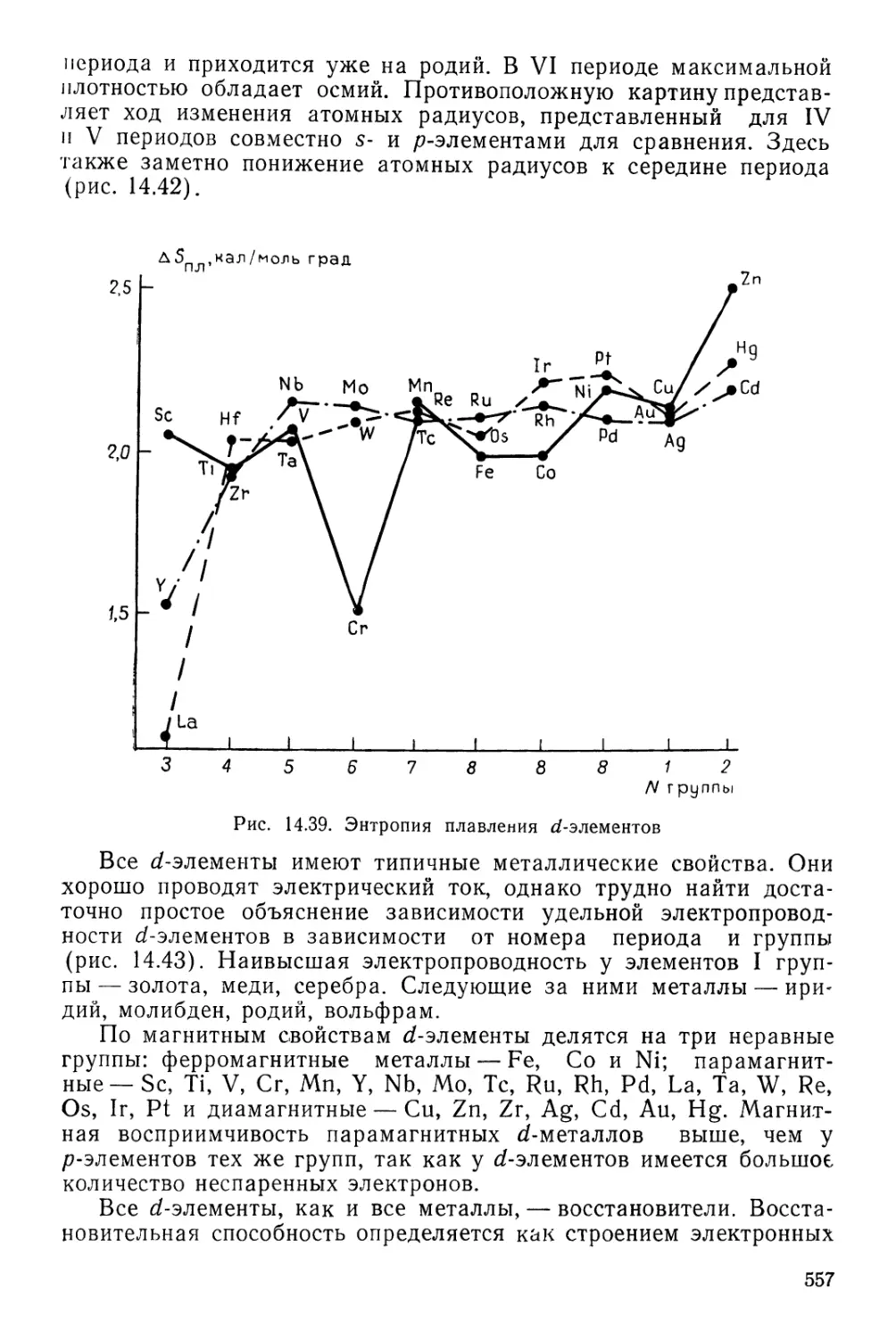










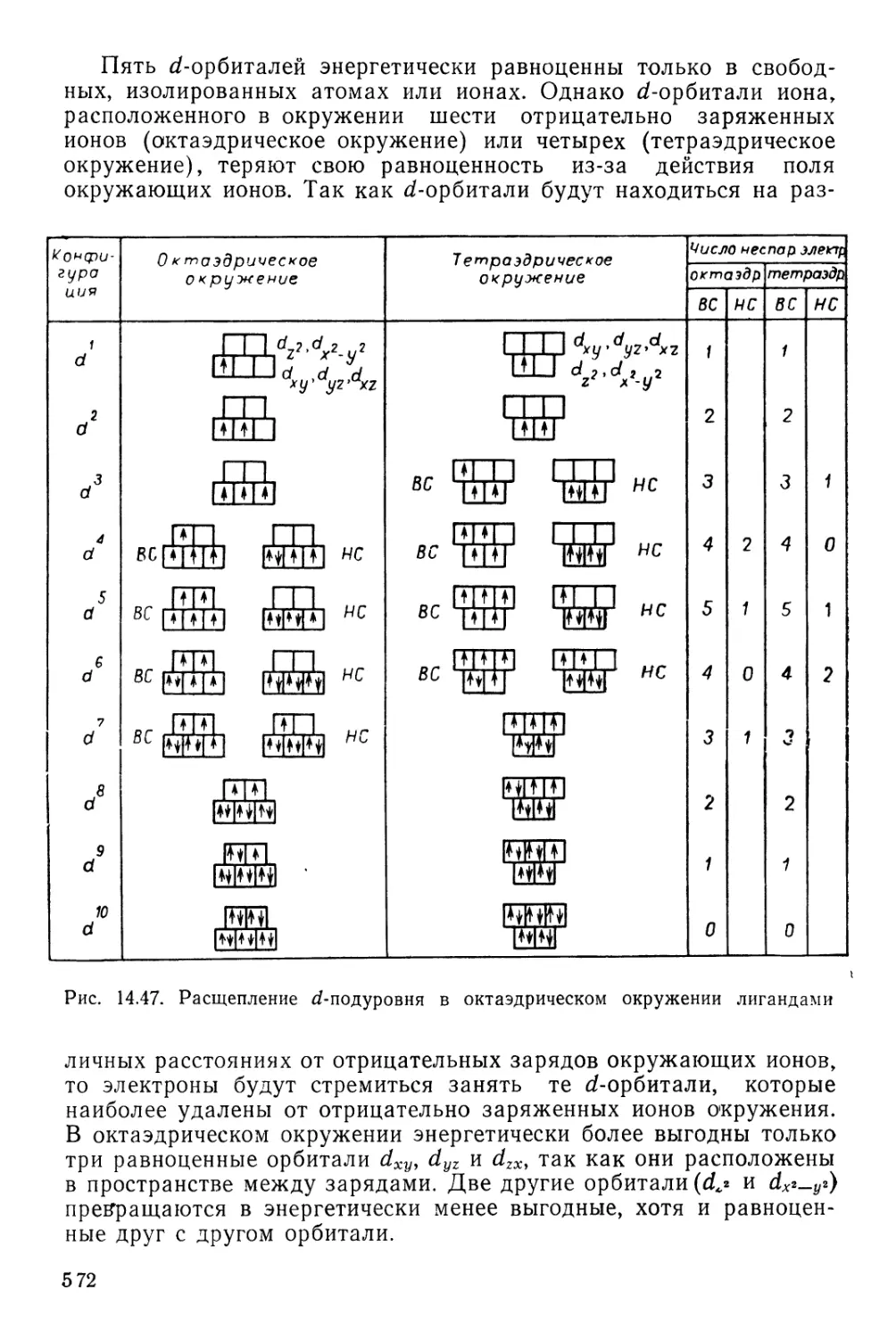



































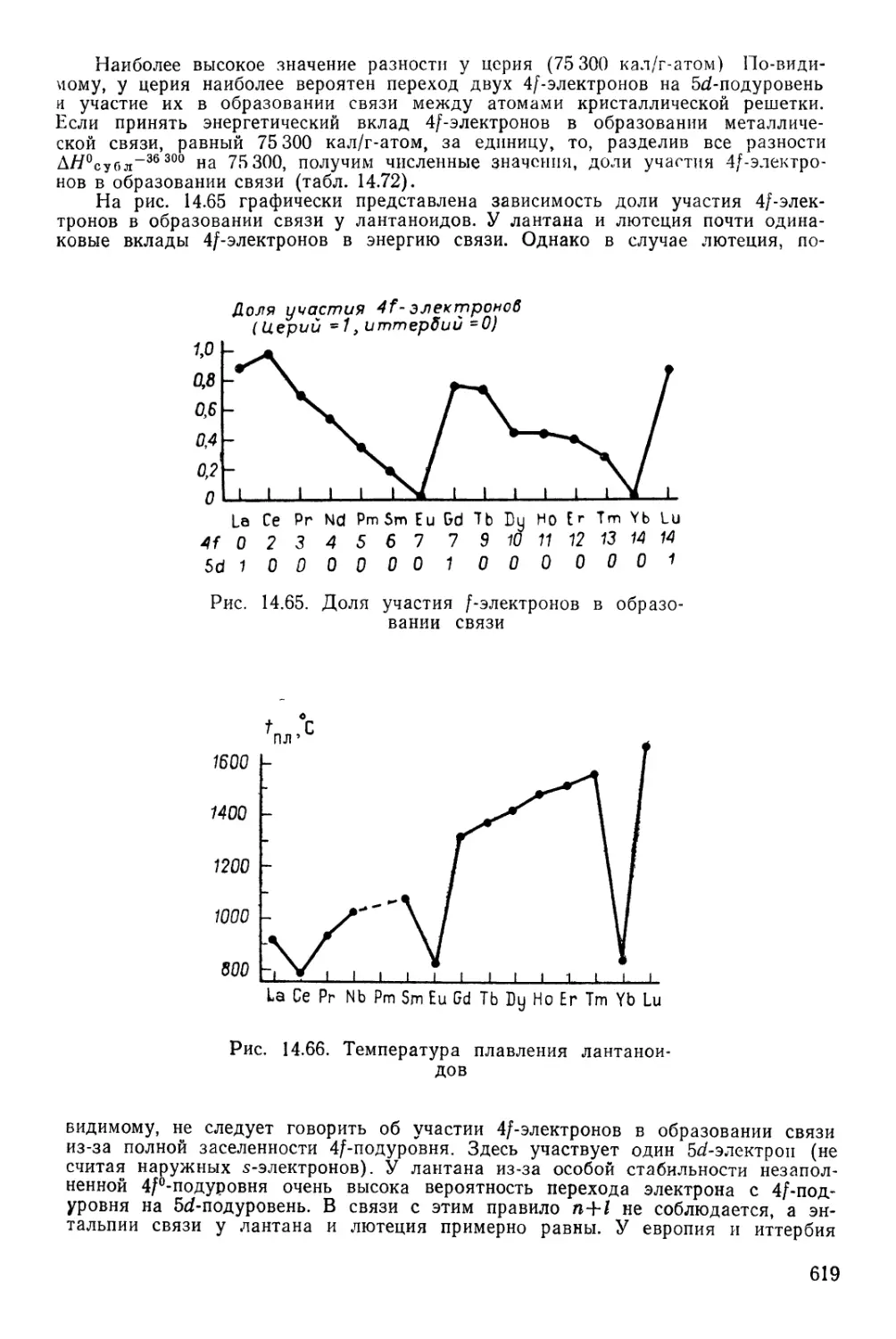
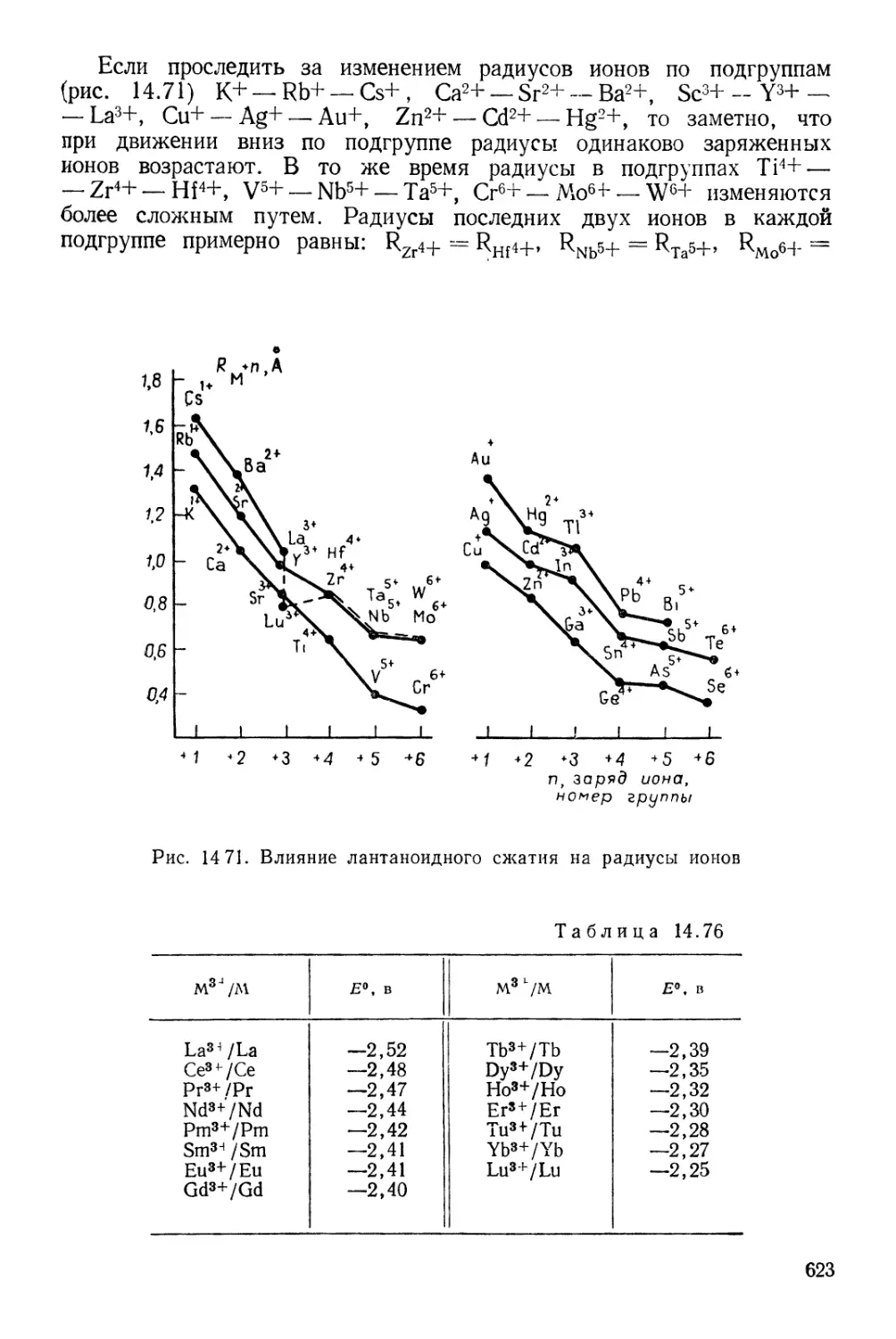
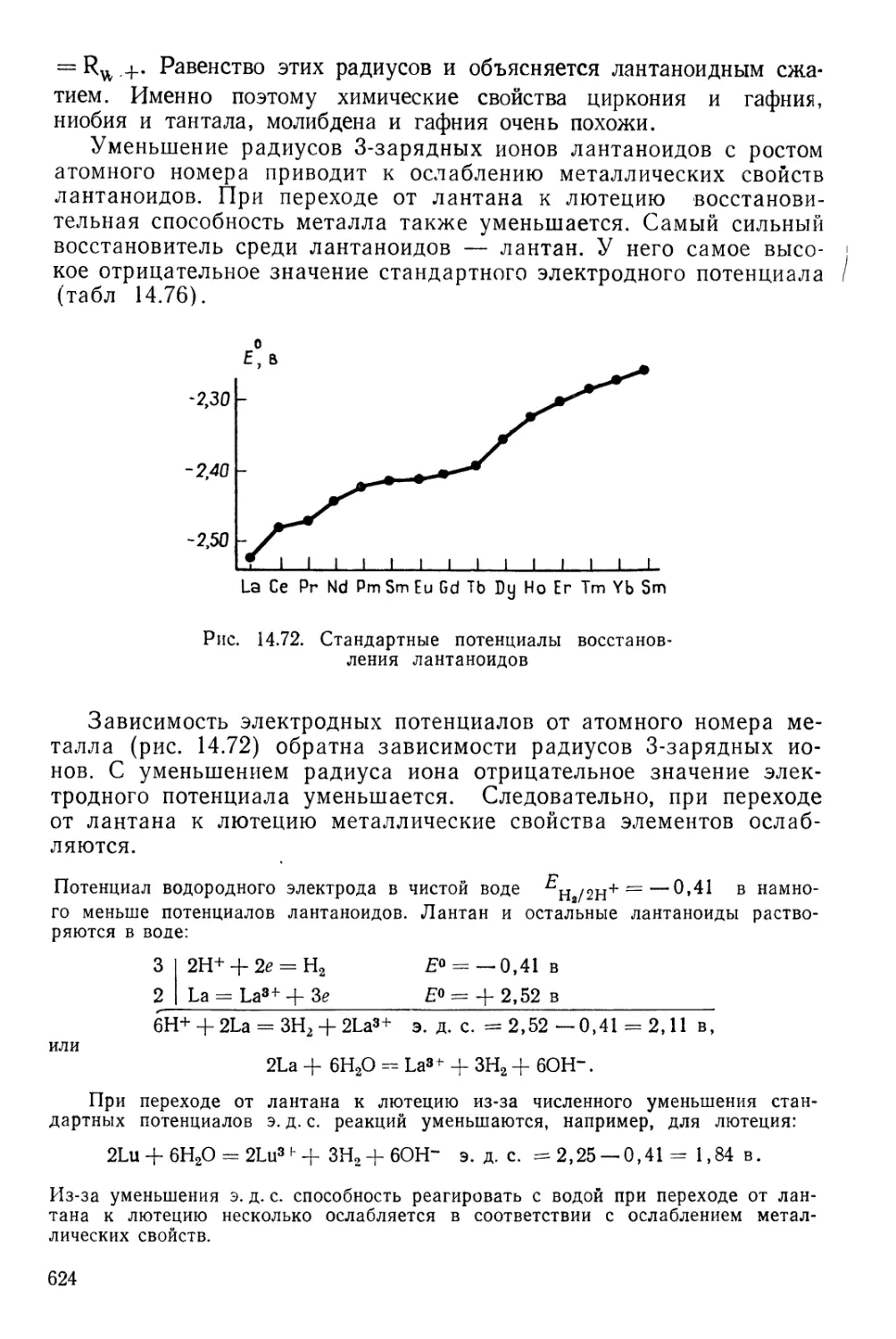


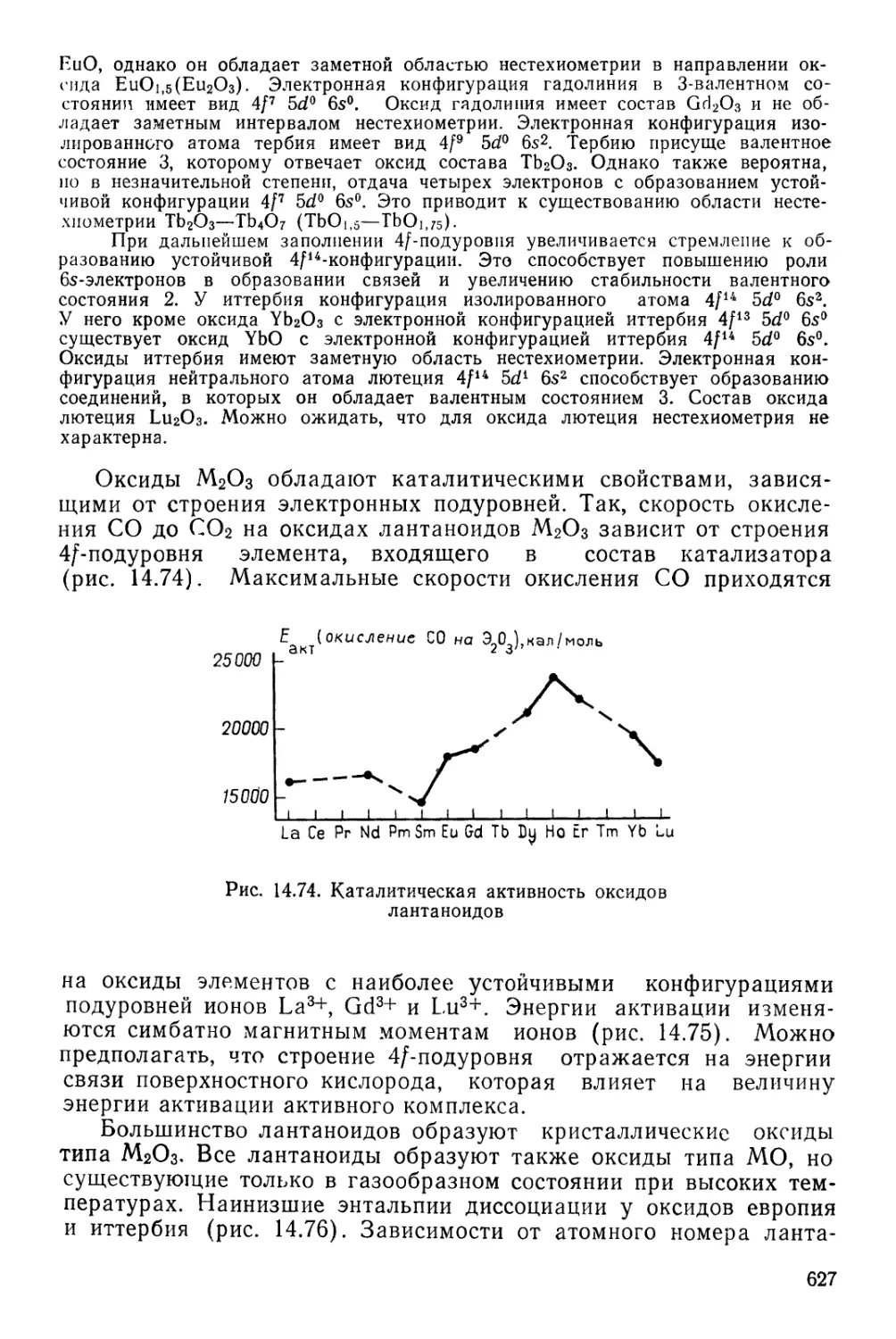
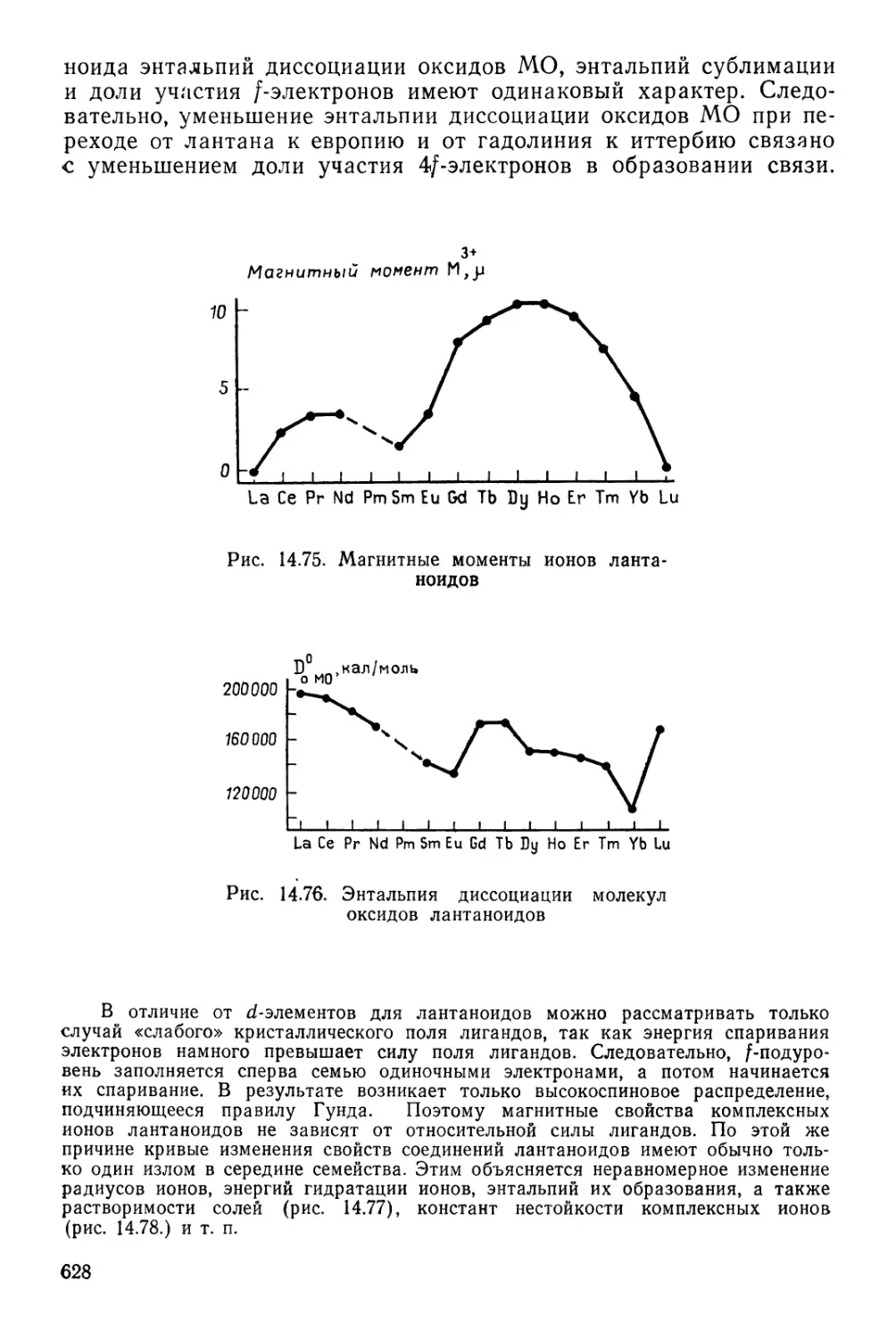

























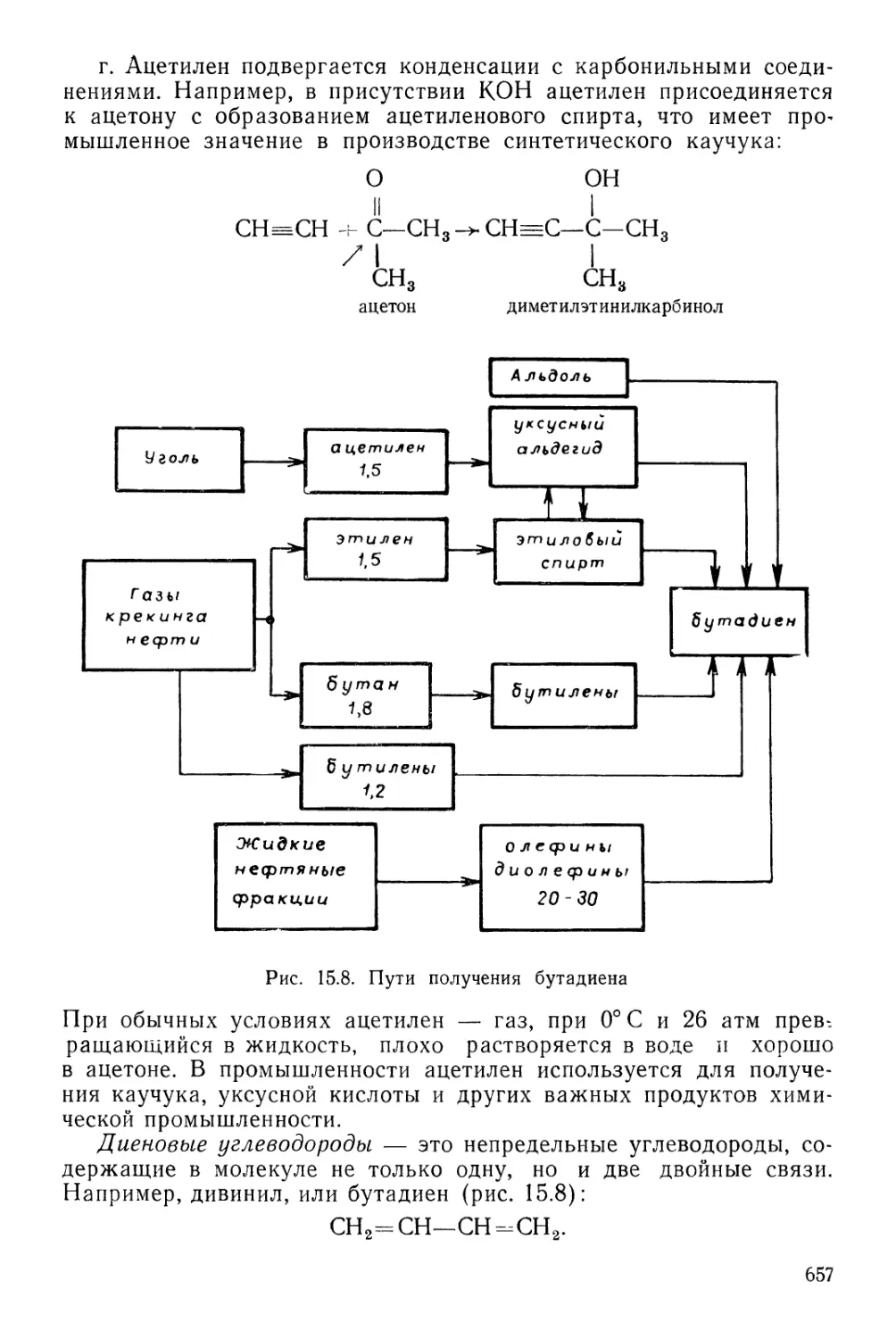















































Текст
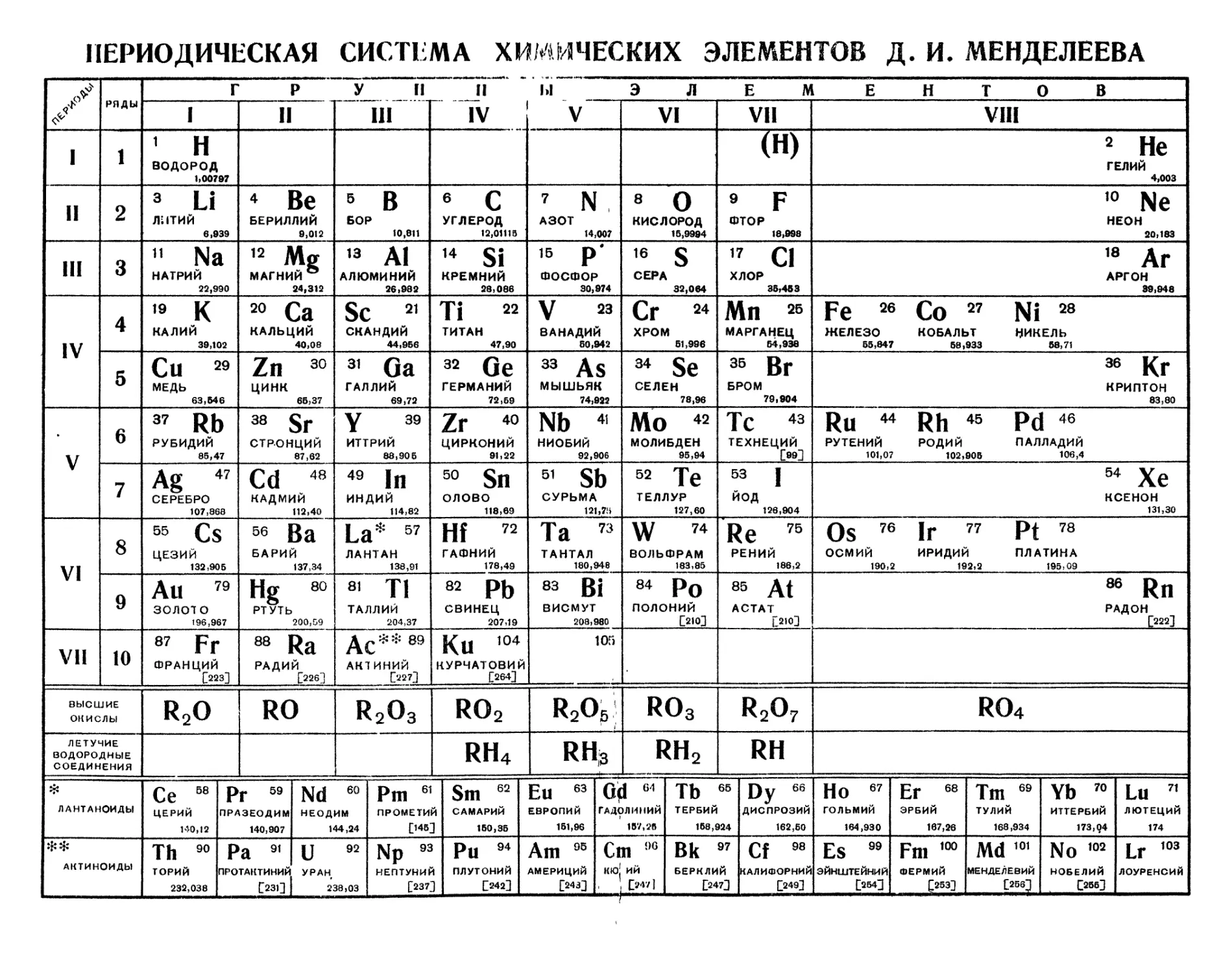
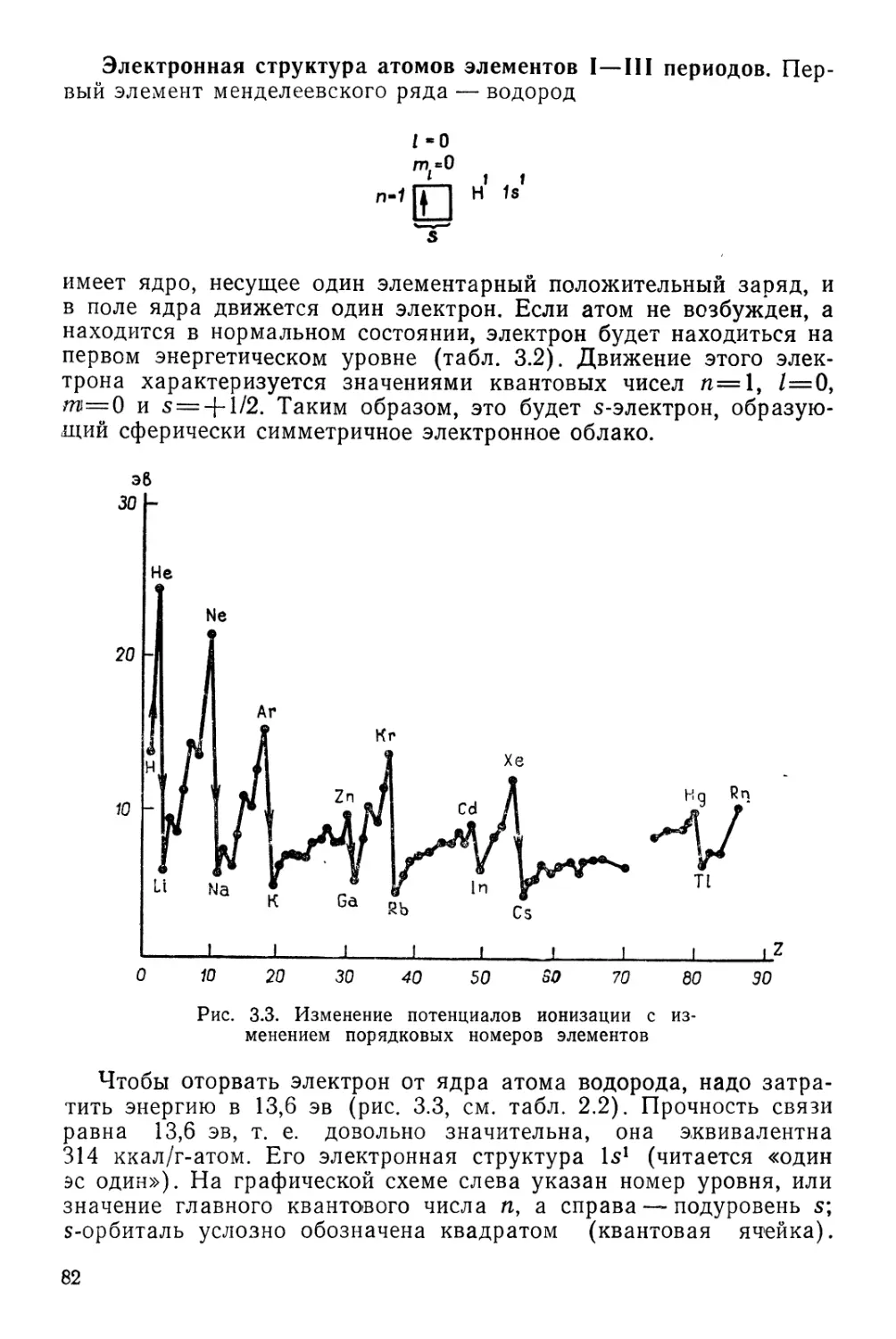
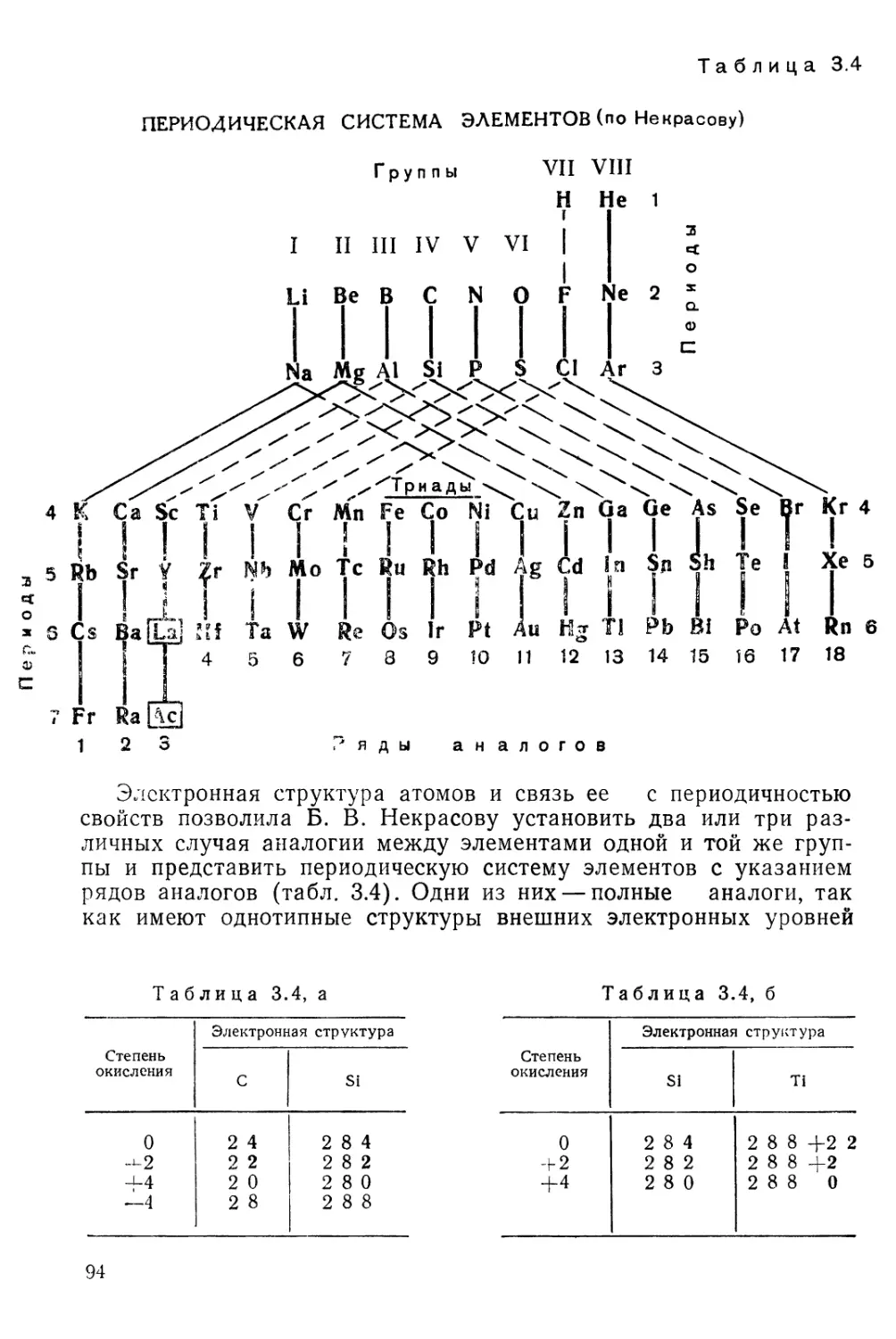
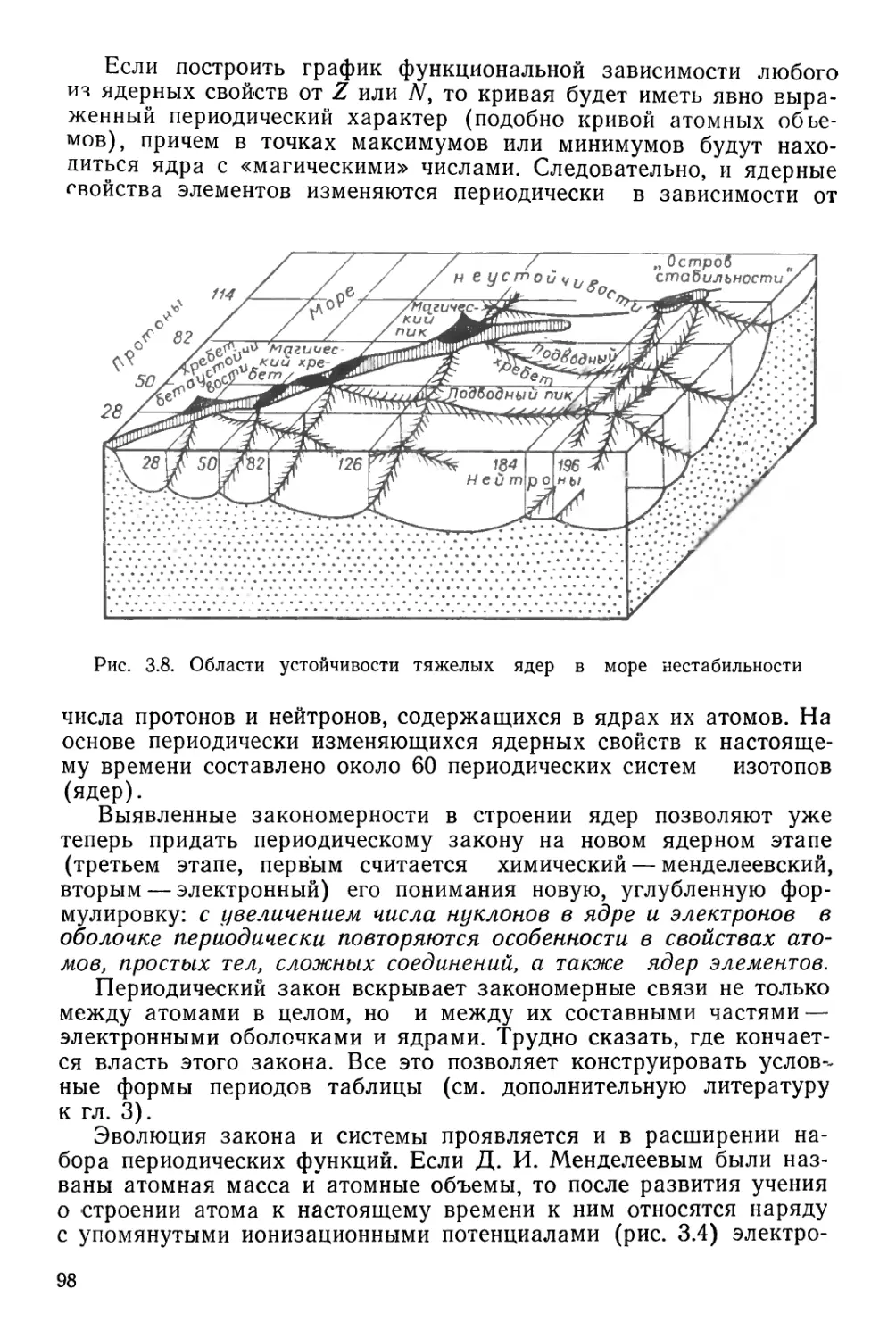
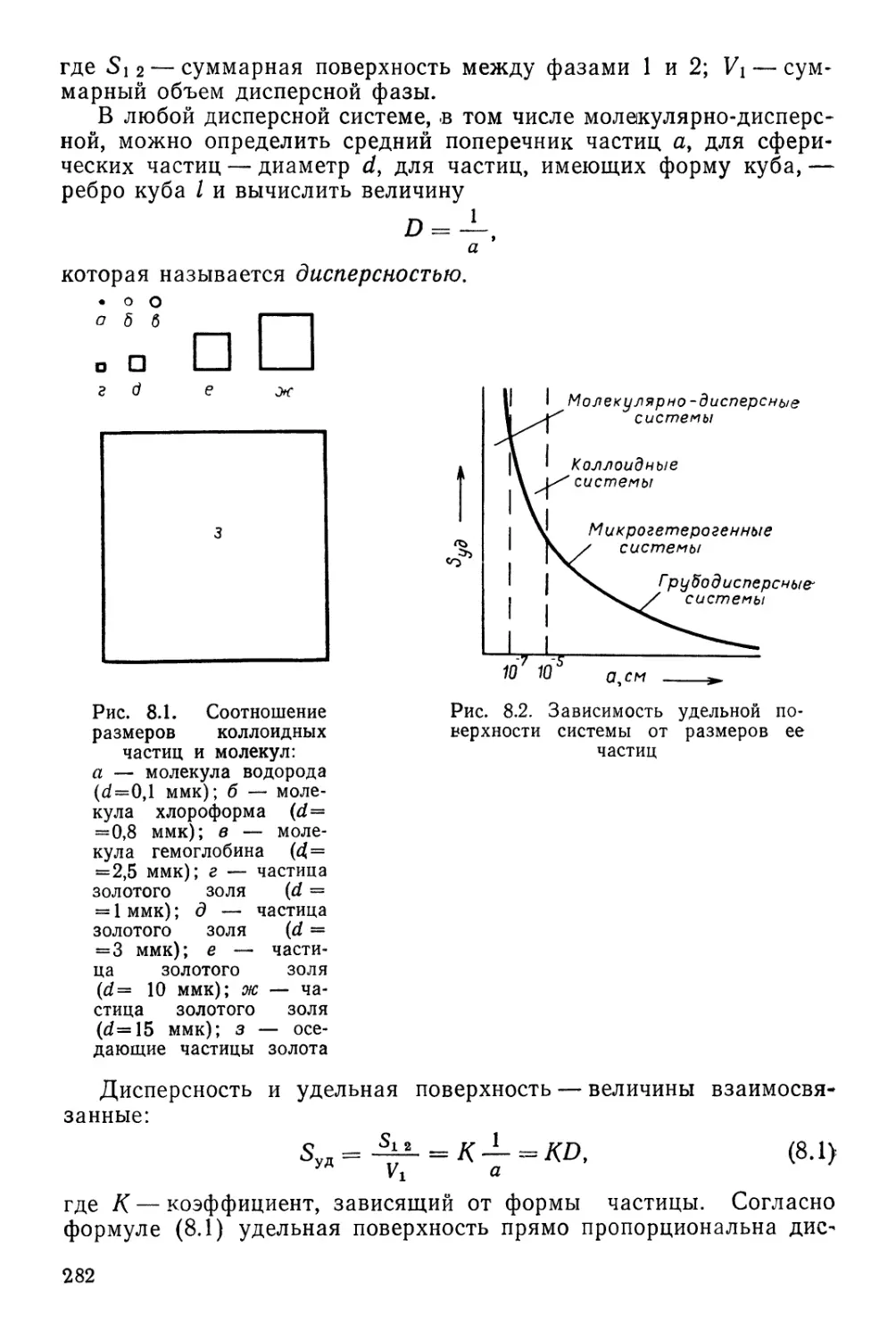
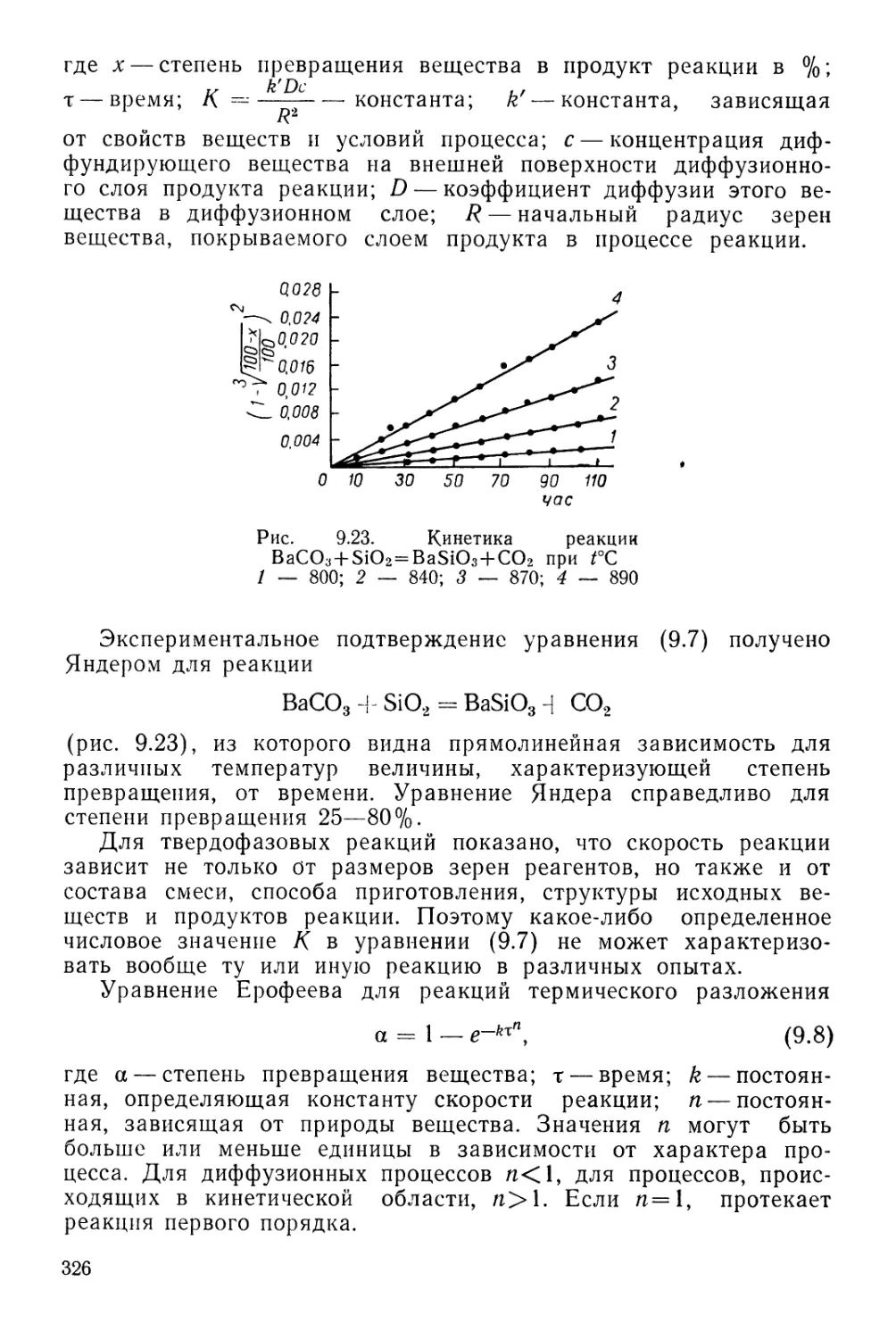
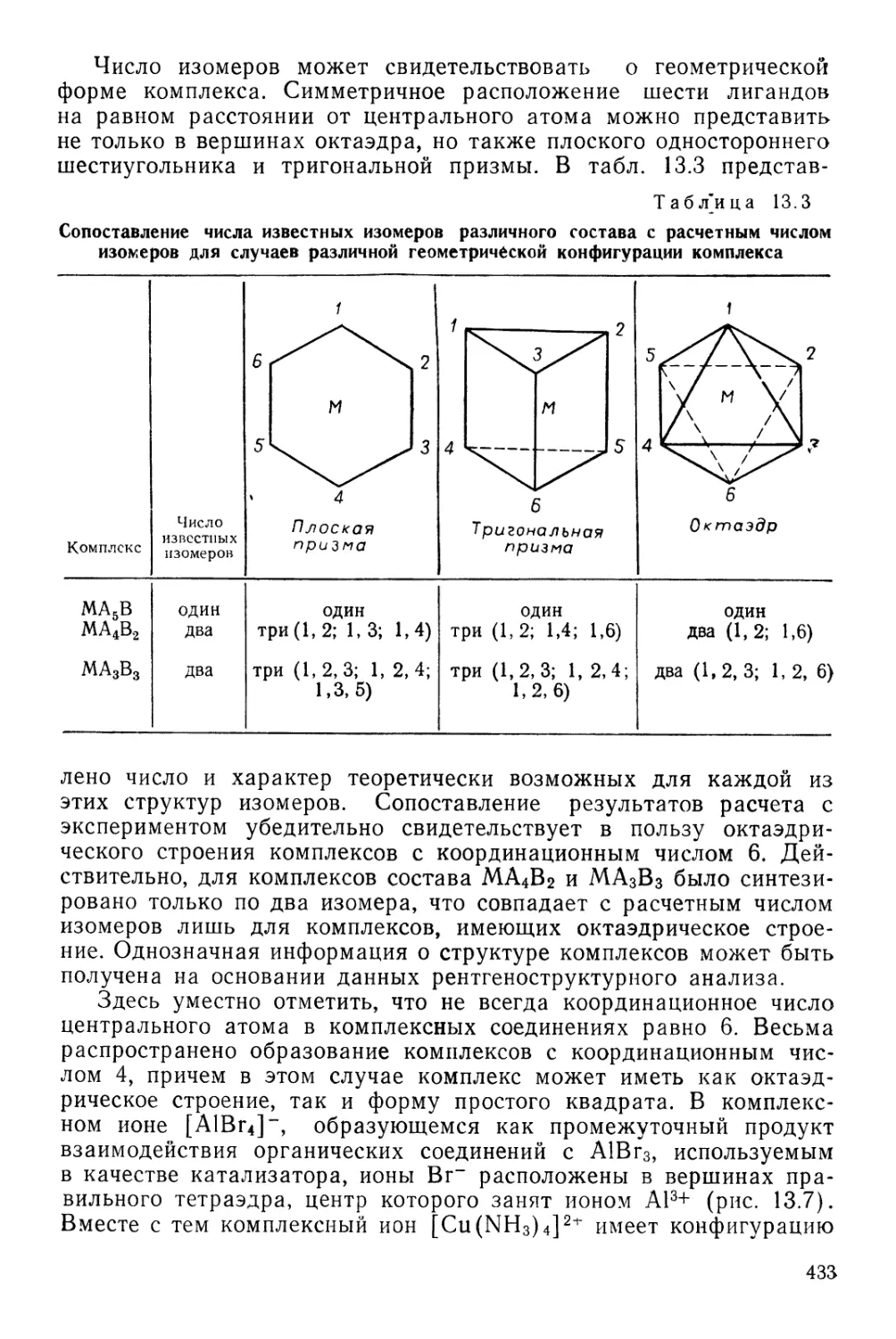
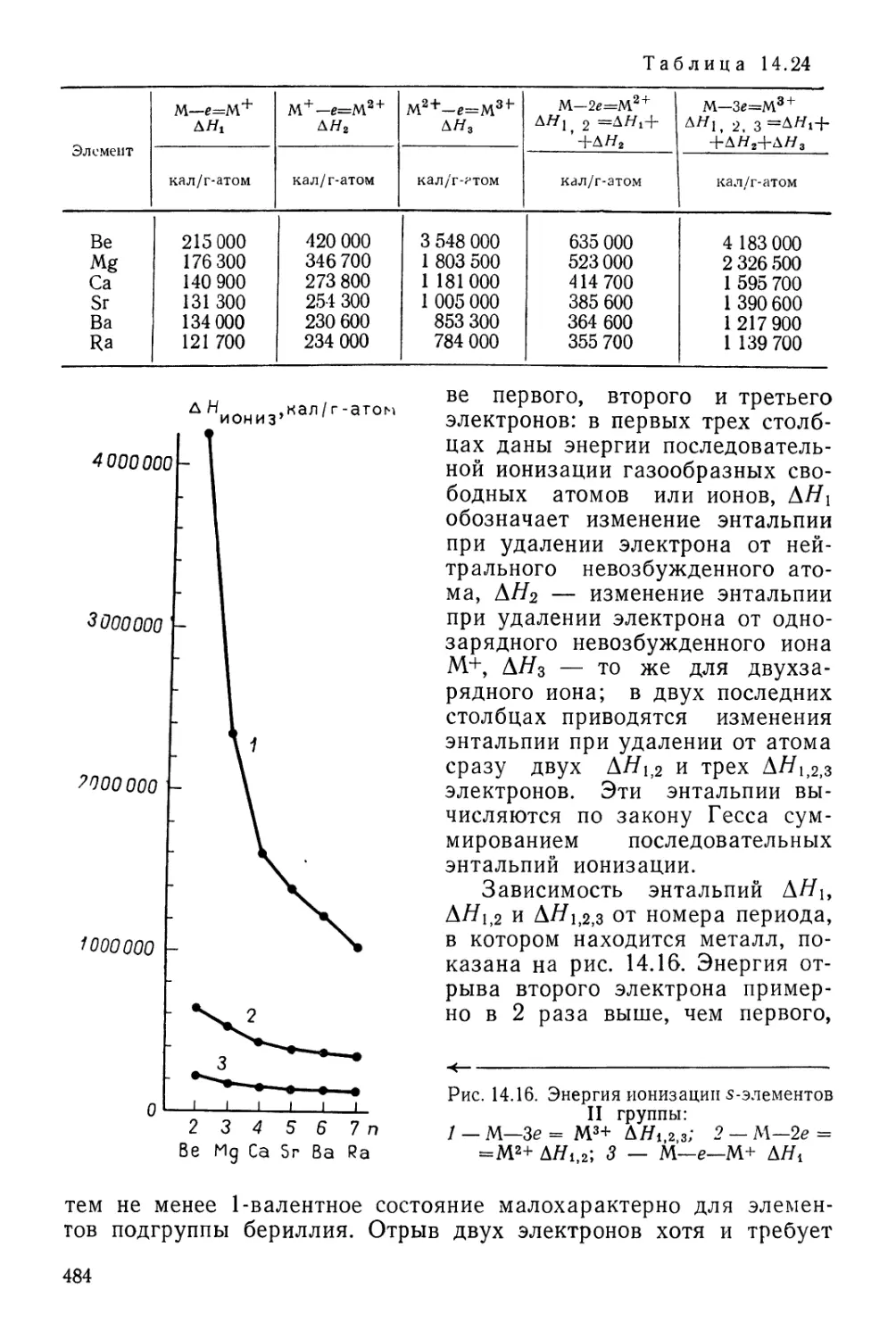
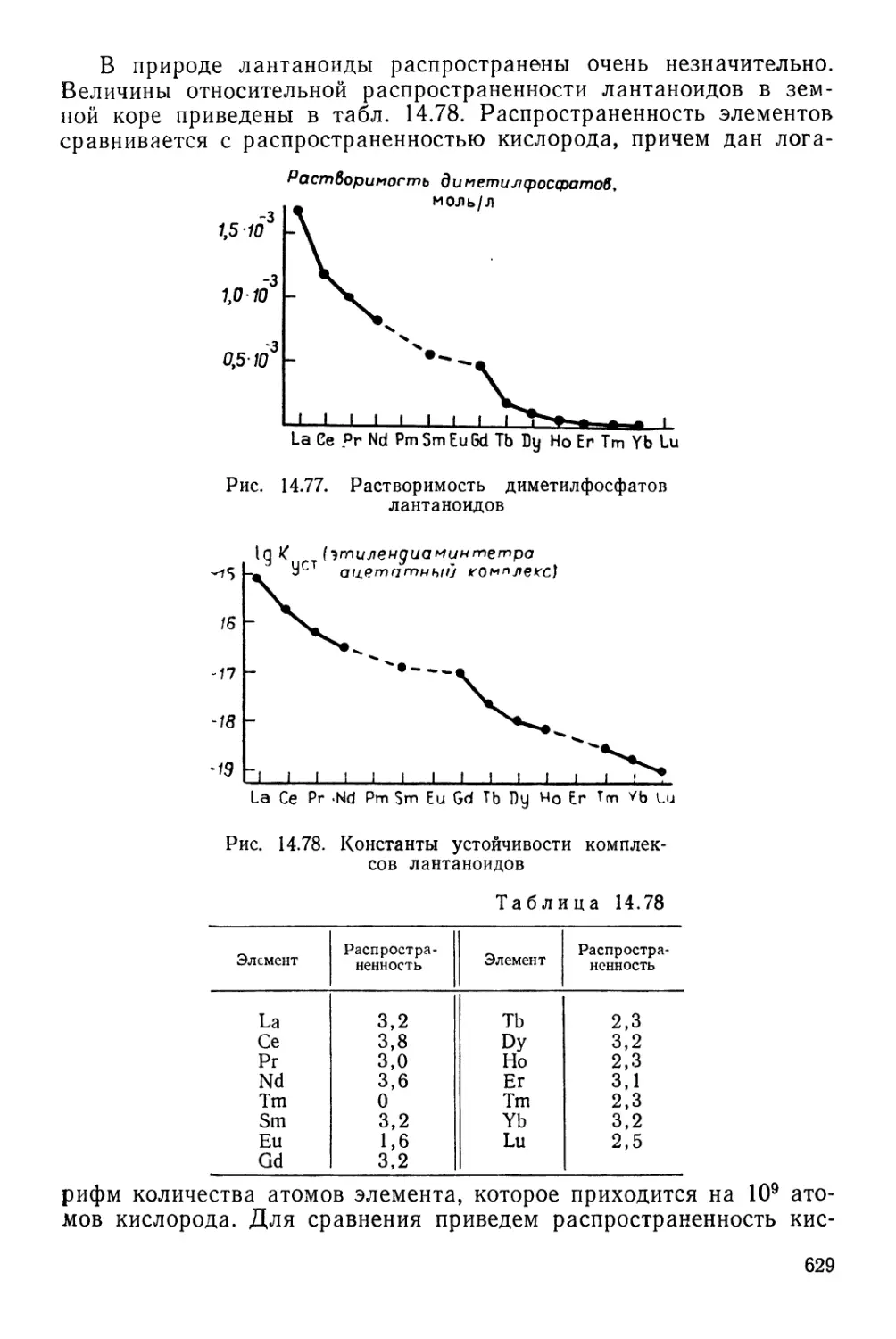
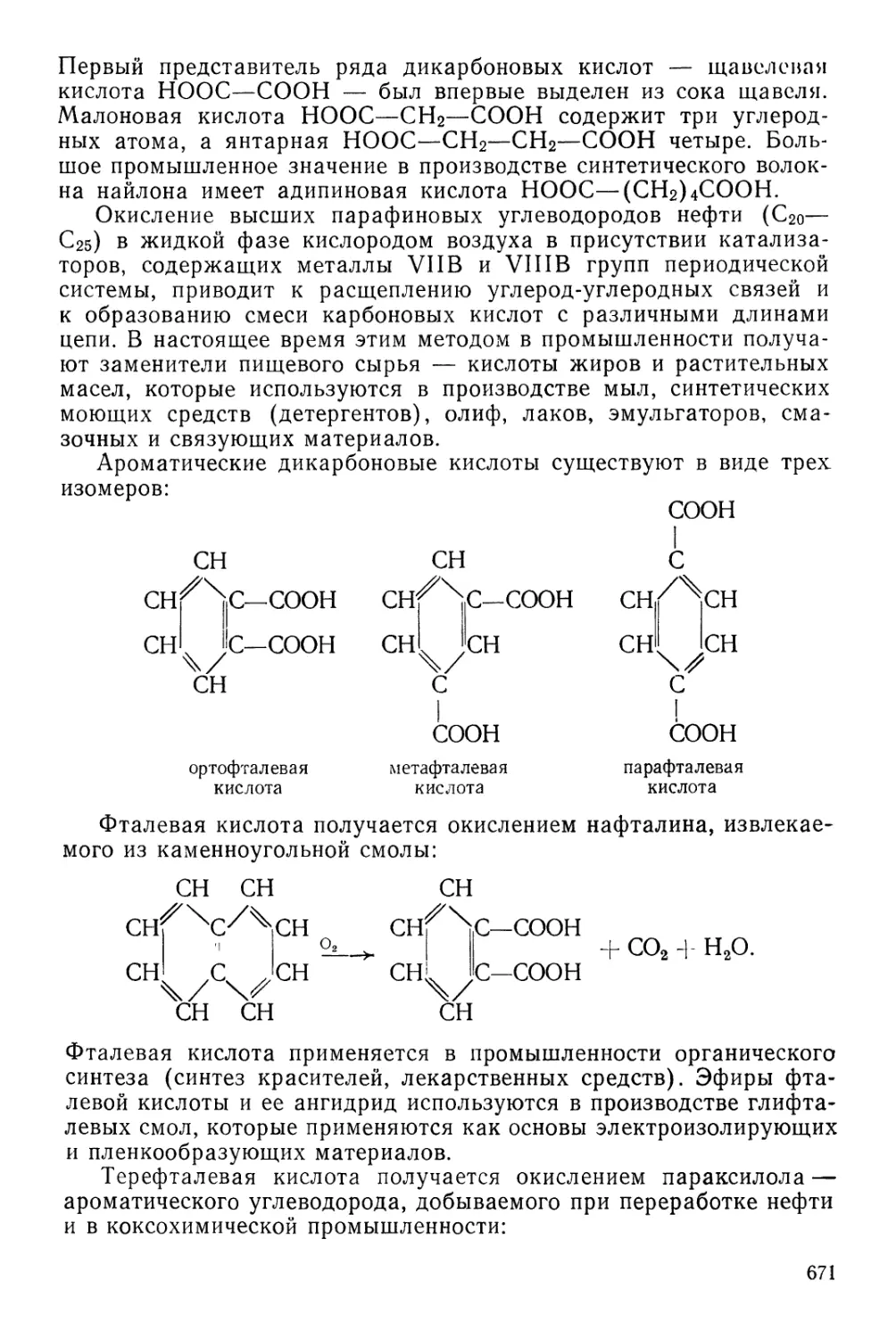
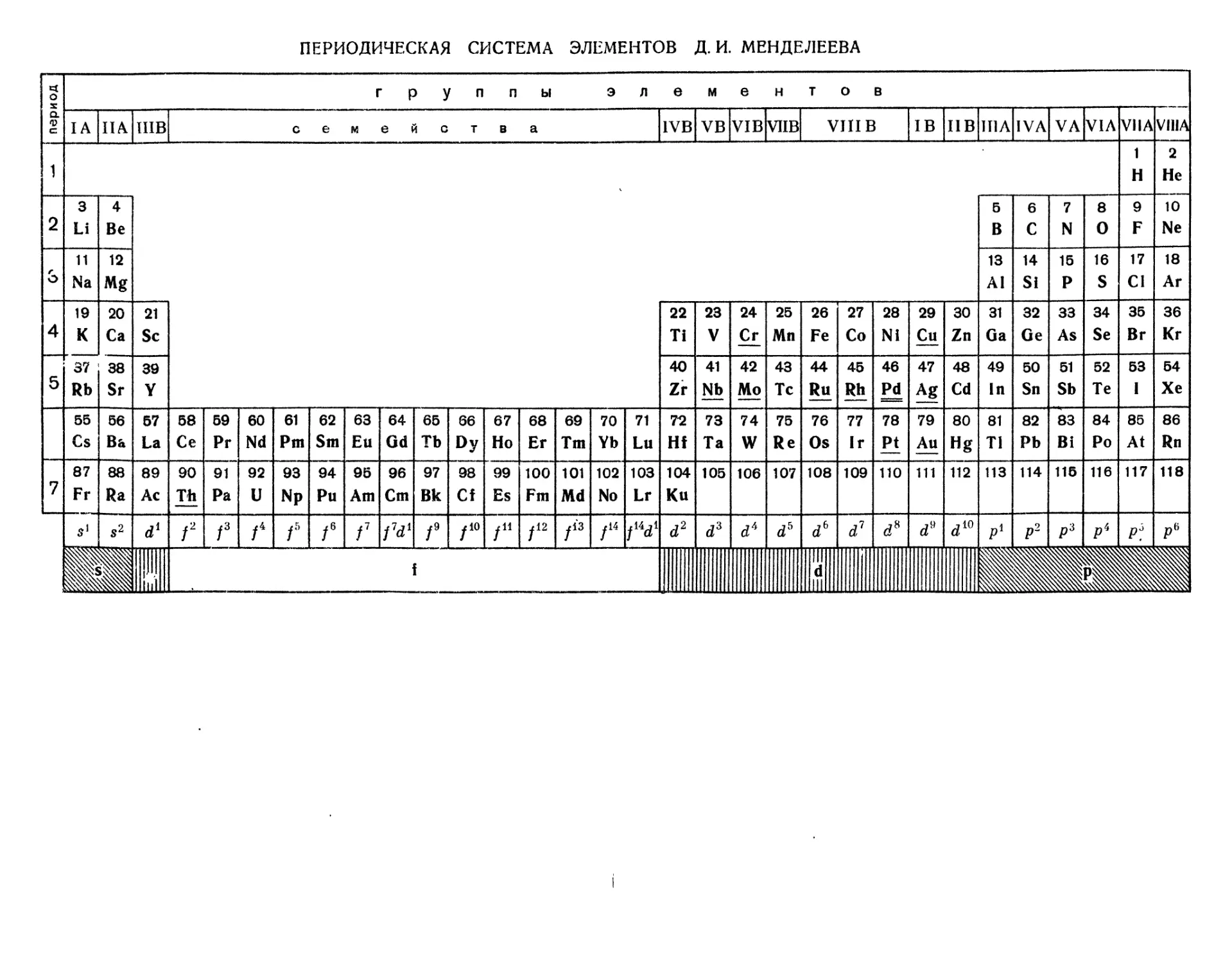
ПЕРИОДИЧЕСКАЯ СИСТЕМА ХИМИЧЕСКИХ ЭЛЕМЕНТОВ Д. И. МЕНДЕЛЕЕВА
1 /
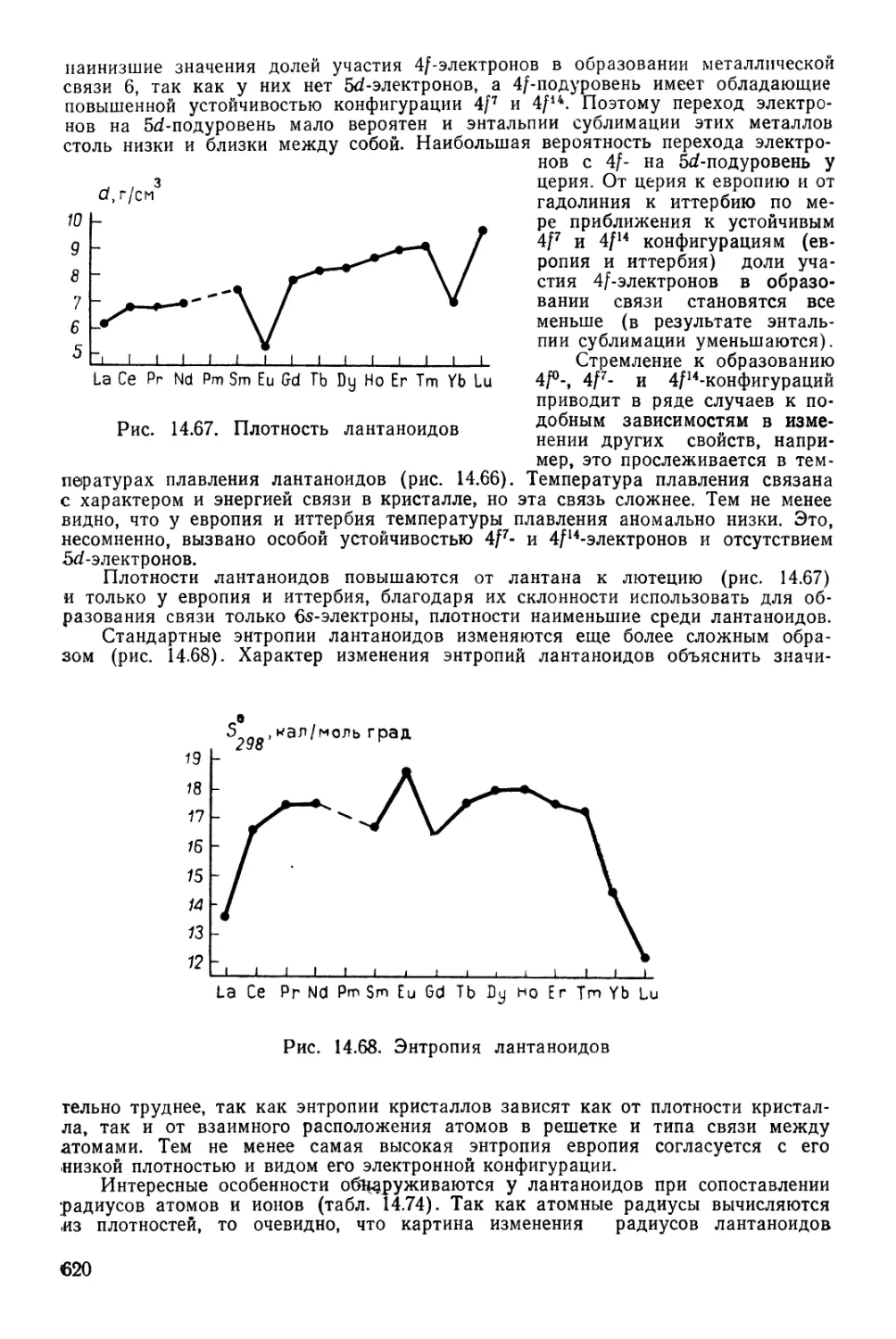
г4
1
II
III
IV
V
VI
VII
РЯДЫ
1
2
3
4
5
6
7
8
9
10
1 ВЫСШИЕ
1 ОКИСЛЫ
1 ЛЕТУЧИЕ
1 ВОДОРОДНЫЕ
[ СОЕДИНЕНИЯ
1 #
1 ЛАНТАНОИДЫ
АКТИНОИДЫ
Г Р У II
1
1 Н
ВОДОРОД
1,00797
3 Li
Л;1ТИЙ
6,939
" Na
НАТРИЙ
22,990
19 К
КАЛИЙ
39,102
Си 29
МЕДЬ
63,646
37 Rb
РУБИДИЙ
85,47
Ag 47
СЕРЕБРО
107,868
65 Cs
ЦЕЗИЙ
132,905
Аи 79
ЗОЛОТО
196,967
87 рг
ФРАНЦИЙ
[223]
R20
Се б8
ЦЕРИЙ
140,12
Th 90
ТОРИЙ
232,038
II
4 Be
БЕРИЛЛИЙ
9,012
12 Mg
МАГНИЙ0
24,312
20 Са
КАЛЬЦИЙ
40,08
Zn 30
ЦИНК
65,37
38 Sr
СТРОНЦИЙ
87,62
Cd 48
КАДМИЙ
112,40
66 Ва
БАРИЙ
137,34
Hg 80
РТУТЬ
200,09
88 Ra
РАДИЙ
[226]
RO~~
Pr S9
ПРАЗЕОДИМ
140,907
Ра 9<
ПРОТАКТИНИЙ
Саз1Д_
III
5 в
БОР
10,811
13 А1
АЛЮМИНИЙ
26,982
Sc 21
СКАНДИЙ
44,956
31 Оа
ГАЛЛИЙ
69,72
Y 39
ИТТРИЙ
88,905
49 In
ИНДИЙ
114,82
La* 57
ЛАНТАН
138,91
8, JI
ТАЛЛИЙ
204,37
Ас**~^
АКТИНИЙ
[227]
R2O3
~Nd 60
НЕОДИМ
144,24
U 92
УРАН
238,03
Pm 61
ПРОМЕТИ»
[1451
Np 93
НЕПТУНИЙ
II
IV
6 С
УГЛЕРОД
12,01115
14 Si
КРЕМНИЙ
28,086
Ti 22
ТИТАН
47,90
32 Ое
ГЕРМАНИЙ
72,59
Zr 40
ЦИРКОНИЙ
91,22
50 Sn
олово
118,69
Hf 72
ГАФНИЙ
178,49
82 рЬ
СВИНЕЦ
207,19
Ки ,04
КУРЧАТОВИИ
[264]
R02
RH4
TSm 62|
Л САМАРИЙ
! | 160,36 1
Ри Э4
1 1 ПЛУТОНИЙ
] 1 [242] 1
Ы ЭЛЕМ Е Н ~Т О ~~В I
1 v
7 N,
АЗОТ
14,007
16 р'
ФОСФОР
30,974
V 23
ВАНАДИЙ
60,942
33 As
МЫШЬЯК
74,922
Nb 41
НИОБИЙ
92,906
61 Sb
СУРЬМА
[ 121,7!i
! Та 73
ТАНТАЛ
180,948
33 Bi
ВИСМУТ
208,980
105
I R2O5;
RH,3
Ей 63
ЕВРОПИЙ
161,96
Am 9Б
АМЕРИЦИЙ
[243]
VI
8 О
КИСЛОРОД
15,9994
16 §
СЕРА
32,064
Сг 24
ХРОМ
51,996
34 Se
СЕЛЕН
78,96
Мо 42
МОЛИБДЕН
95,94
52 Те
ТЕЛЛУР
127,60
W 74
ВОЛЬФРАМ
183,85
84 Ро
ПОЛОНИЙ
[210]
! R03
I RH2
ГАДОЛИНИЙ
187,26
Cm !0
кю{ ий
; О»!
VII
(Н)
9 р
ФТОР
18,998
17 С1
ХЛОР
35,453
Мп 2б
МАРГАНЕЦ
54,936
35 Вг
БРОМ
79.904
Тс 43
ТЕХНЕЦИЙ
[99]
53 |
ЙОД
126,904
Re 75
РЕНИЙ
186,2
85 At
АСТАТ
[2Ю]
R207
RH
Tb 6б
ТЕРБИЙ
168,924
Bk 97
БЕРКЛИЙ
fr<3
Dy 66
ДИСПРОЗИЙ
162,60
Cf 98
КАЛИФОРНИЙ
[249]
VIII 1
2 Не
ГЕЛИЙ 1
4,003 1
10 Ne
НЕОН 1
20,183 |
18 Аг
АРГОН 1
39,948 1
Fe 26 Со 27 Ni 28
ЖЕЛЕЗО КОБАЛЬТ НИКЕЛЬ 1
65,847 58,933 68,71 1
36 Кг
КРИПТОН
83,80 1
Ru " Rh 45 Pd 46
РУТЕНИЙ РОДИЙ ПАЛЛАДИЙ
101,07 102,906 Ю6,4 1
54 Хе
КСЕНОН
131,30 1
Os 76 Гг 77 Pt 78
ОСМИЙ ИРИДИЙ ПЛАТИНА
190,2 192,2 195,09
86 Rn
РАДОН
[222]
RO^ I
Но 67
ГОЛЬМИЙ
164,930
Es "
ЭЙНШТЕЙНИЙ
| [2S4]
Ег 68
ЭРБИЙ
167,26
Fm ,0°
ФЕРМИЙ
£253]
Тт 69
ТУЛИЙ
168,934
Ш101
МЕНДЕЛЕВИЙ
[256^1
Yb 70
ИТТЕРБИЙ
173,Q4
No 102
НОБЕЛИЙ
[266]
Lu 7t 1
ЛЮТЕЦИЙ
174 J
Lr 103
ЛОУРЕНСИЙ
ОБЩАЯ ХИМИЯ
Под редакцией профессоров:
М. СОКОЛОВСКОЙ, Г. Д. ВОВЧЕНКО,
Ю. Д. ТРЕТЬЯКОВА
ДОПУЩЕНО МИНИСТЕРСТВОМ ВЫСШЕГО
И СРЕДНЕГО СПЕЦИАЛЬНОГО ОБРАЗОВАНИЯ СССР
В КАЧЕСТВЕ УЧЕБНОГО ПОСОБИЯ ДЛЯ СТУДЕНТОВ
НЕХИМИЧЕСКИХ СПЕЦИАЛЬНОСТЕЙ УНИВЪРСИ! ЕТОВ"
ИЗДАТЕЛЬСТВО МОСКОВСКОПО УНИВЕРСИТЕТА
1975
УДК 543.06Э
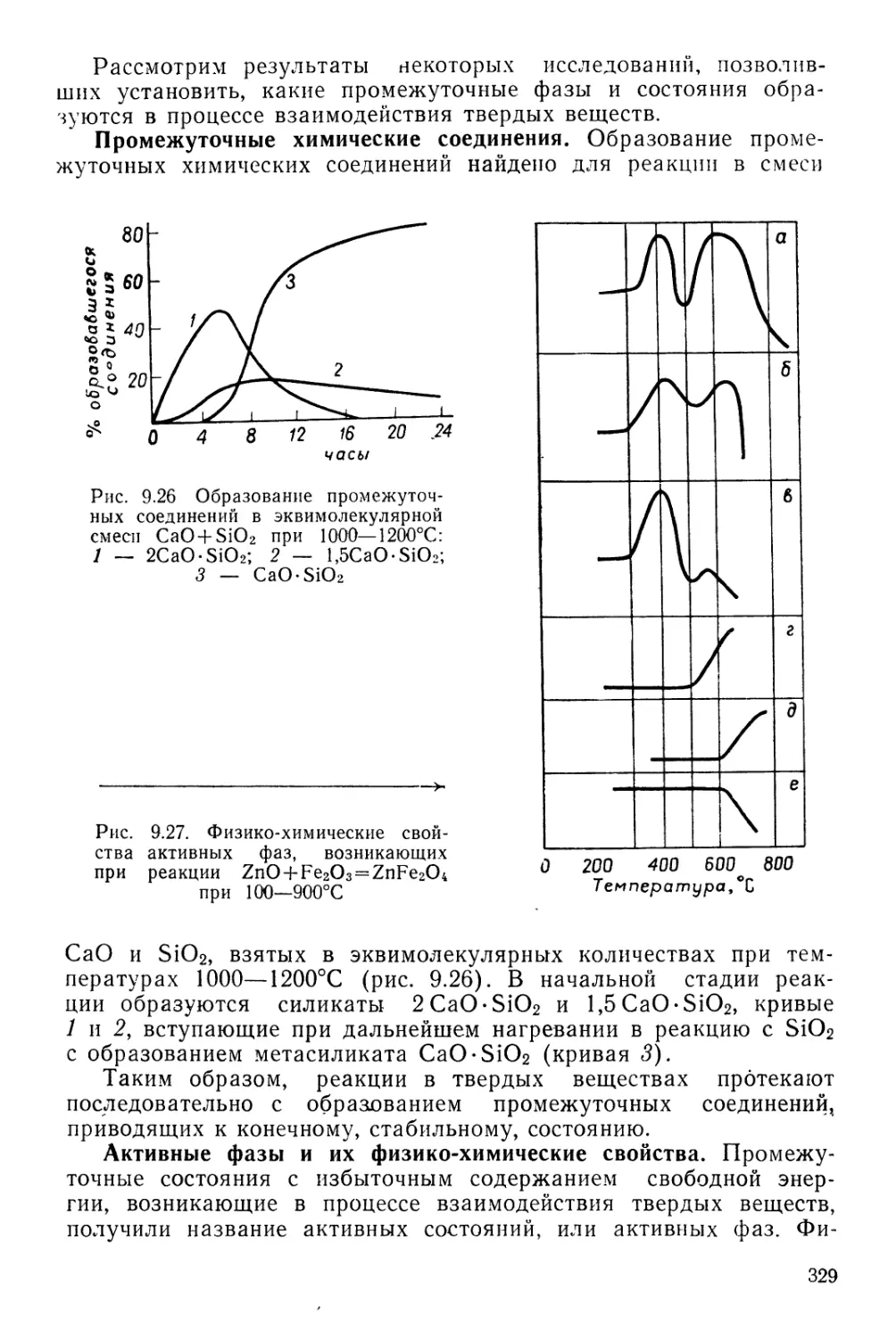
Настоящее учебное пособие заметно огличаеюи о г ныне
принятых учебников по общей химии Это несколько расширенное
и углубленное изложение основных разделов курса общей химии,
который авторы читали в течение ряда лет для студентов
естественных факультетов МГУ не^шмических^пециальностей Большое
внимание уделяется вопросам строения материи, теории строения
атомов и молекул, периодическому закону, химической термодинамике,,
кинетике и механизму химических реакций, катализу, химии
координационных соединений, химии твердого тела и т д. Авторы
широко используют положения квантовой механики к изучению
строения атомов и молекул. Химия элементов различных групп пе.-
теистической систешз!_Д. И. "Менделеева", характерные "особенности
каждой группы, типичные реакции рассматриваются с привлечением
основных положений химической термодинамики и современных
представлений в химии./Изложение ведется на уровне современного
состояния науки, в доступной форме для с1удентов первого курса.
Книга предназначена для студентов нехимических специальностей
университетов, также полезна'Цля желающих составить правильное
и углубленное представление о теоретических основах химической
науки
Рецензенты:
кафедра общей и неорганической химии
Тбилисского государственного университета;
профессор «/7, М. Романцева
о 20502-155 м2_7.
077(02)—75
(5) Издательство Московского университета, 1975 г.
ОГЛАВЛЕНИЕ
Предисловие 7
Введение 9
1. Химия — раздел естествознания .... .... 9
2. Основные этапы развития химии И
3. Химия в СССР. Задачи современной химии 18
Глава 1. ОСНОВНЫЕ ПОНЯТИЯ И ЗАКОНЫ ХИМИИ. АТОМНО-
МОЛЕКУЛЯРНАЯ ТЕОРИЯ 21
§ 1. Материя и энергия .21
§ 2. Закон сохранения материи и движения 23
§ 3. Развитие атомистических представлений в химии . 27
§ 4. Закон постоянства состава 28
§ 5. Закон эквивалентов ф 29
§ 6. Закон кратных отношений. Атомный вес элементов .... 30
§ 7. Закон соединения газов и закон Авогадро 32
§ 8. Уравнение Клапейрона — Менделеева Определение числа
Авогадро 35
§ 9. Современное содержание стехиометрических законов ... 38
Вопросы и упражнения . . 41
Глава 2. СТРОЕНИЕ АТОМА 42
§ 1. Сложность структуры атома 42
§ 2. Модели строения атома. Ядерная модель Э. Резерфорда . . 43
§ 3. Атомные спектры. Спектр атома водорода 43
§ 4. Теория строения атома водорода по Бору 45
§ 5. Развитие теории Бора 48
§ 6. Начала волновой механики. Уравнение Шредингера .... 49
§ 7. Квантовые числа. Спин электрона. Принцип Паули .... 59
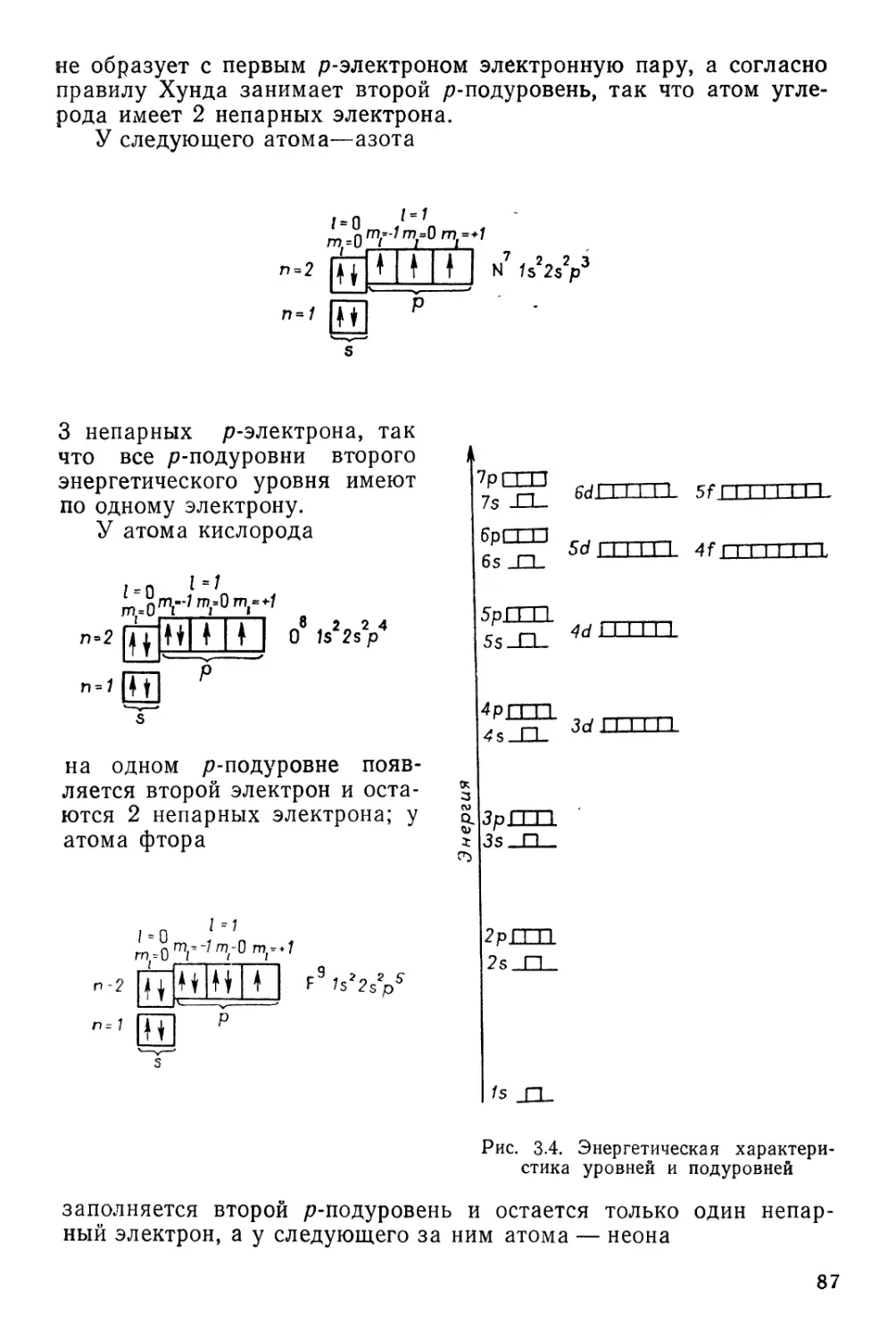
§ 8. Многоэлектронные атомы. Правило Хунда 63
§ 9. Энергия ионизации атома и сродство к электрону .... 65
Вопросы и упражнения 67
Глава 3. ПЕРИОДИЧЕСКИЙ ЗАКОН И ПЕРИОДИЧЕСКАЯ
СИСТЕМА ЭЛЕМЕНТОВ Д. И. МЕНДЕЛЕЕВА 68
§ 1. Понятие о химическом элементе. Классификация элементов до
Менделеева 68
§ 2. Работы Д. И. Менделеева 70
Периодический закон .... 72
Периодическая система элементов Д. И. Менделеева . . 74
^§3. Развитие периодического закона 80
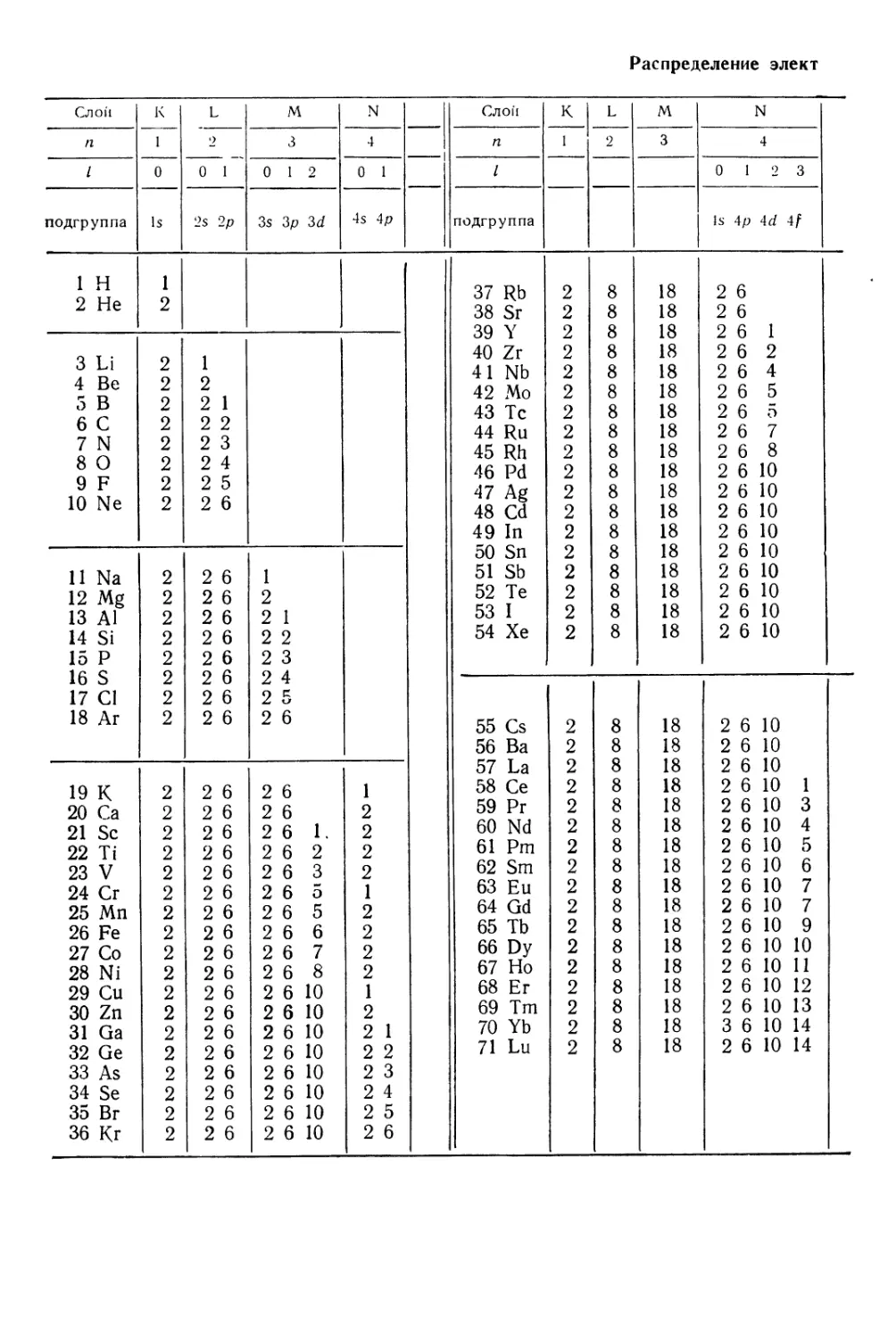
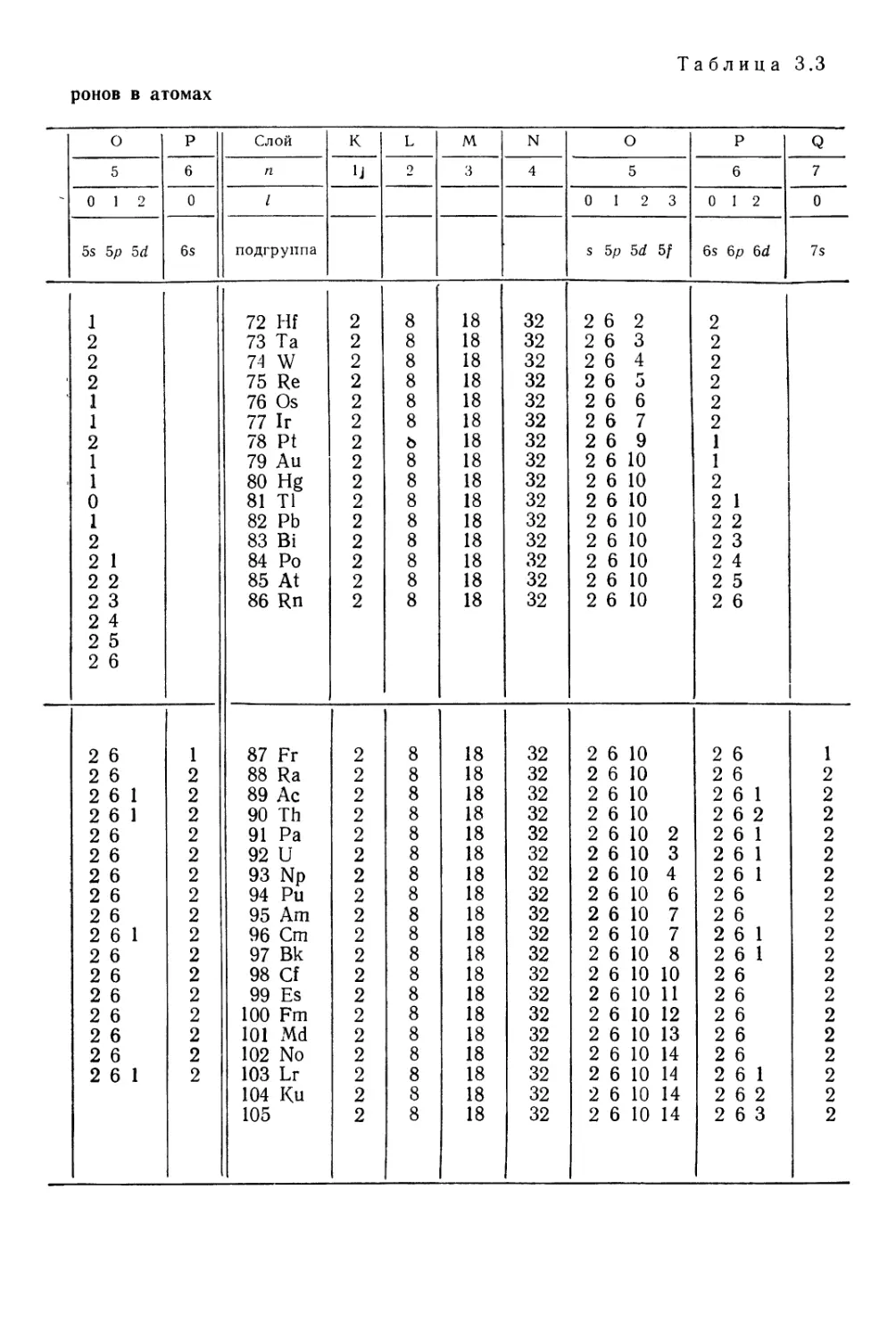
§ 4. Периодическая система и электронные структуры атомов . . 81
Вторичная и внутренняя периодичности 95
§ 5. Периодический закон, периодическая система и современная химия 96
Вопросы и упражнения 101
Глава 4. ХИМИЧЕСКАЯ СВЯЗЬ И СТРОЕНИЕ МОЛЕКУЛ ... 102
§ 1. Валентная связь 102
§ 2. Гибридизация орбиталей 104
§ 3. Метод молекулярных орбиталей 107
§ 4. Делокализаиия электронов. Металлическая связь. Энергетические
зоны 119
§ 5. Ковалентные связи в переходных металлах 122
§ 6. Сравнение метода валентных связей и метода молекулярных
орбиталей 124
Вопросы и упражнения 125
3
Глава 5. ОСНОВНЫЕ ПОНЯТИЯ ХИМИЧЕСКОЙ
ТЕРМОДИНАМИКИ .... 126
§ 1. Изменение внутренней энергии и энтальпии в химической
реакции 126
§ 2. Первый закон термодинамики и закон Гесса 134
§ 3. Изменение энтропии в химическом процессе 141
§ 4. Изобарный потенциал реакции и второй закон термодинамики 148
§ 5. Направление химической реакции 152
§ 6. Третий закон термодинамики и его роль для химии .... 167
Вопросы и упражнения 175
Глава 6. КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИИ. ХИМИЧЕСКОЕ
РАВНОВЕСИЕ. КАТАЛИЗ 178
§ 1. Скорость химических реакций. Основные понятия и определения 178
§ 2. Закон действующих масс 181
§ 3. Реакции в гетерогенных системах 184
§ 4. Влияние температуры на скорость химической реакции. Энергия
активации Jjj*
§ 5. Простые и сложные реакции |°<*
§ 6. Фотохимические реакции ... }~*
§ 7. Молекулярность и порядок реакции ... . J92
§ 8. Теории химической кинетики . . . ... |9j>
§ 9. Химическое равновесие . 198
§ 10. Фазовые равновесия .... . ^06
§ 11. Катализ 208
Гомогенный катализ . 209
Гетерогенный катализ . . . 212
Активность катализаторов ... 215
Избирательность катализатор'ов . 216
Отравление, промотирование, модифицирование
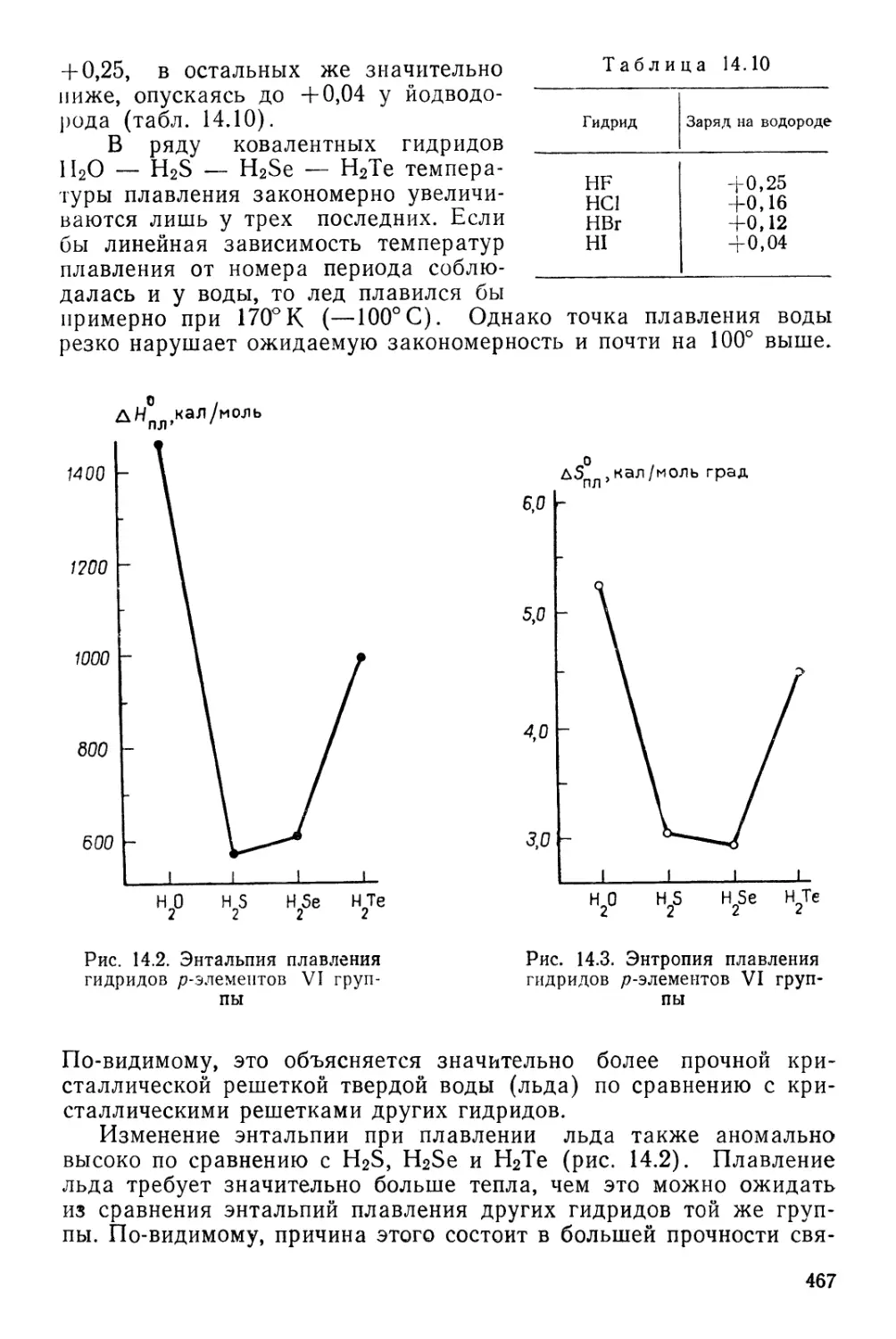
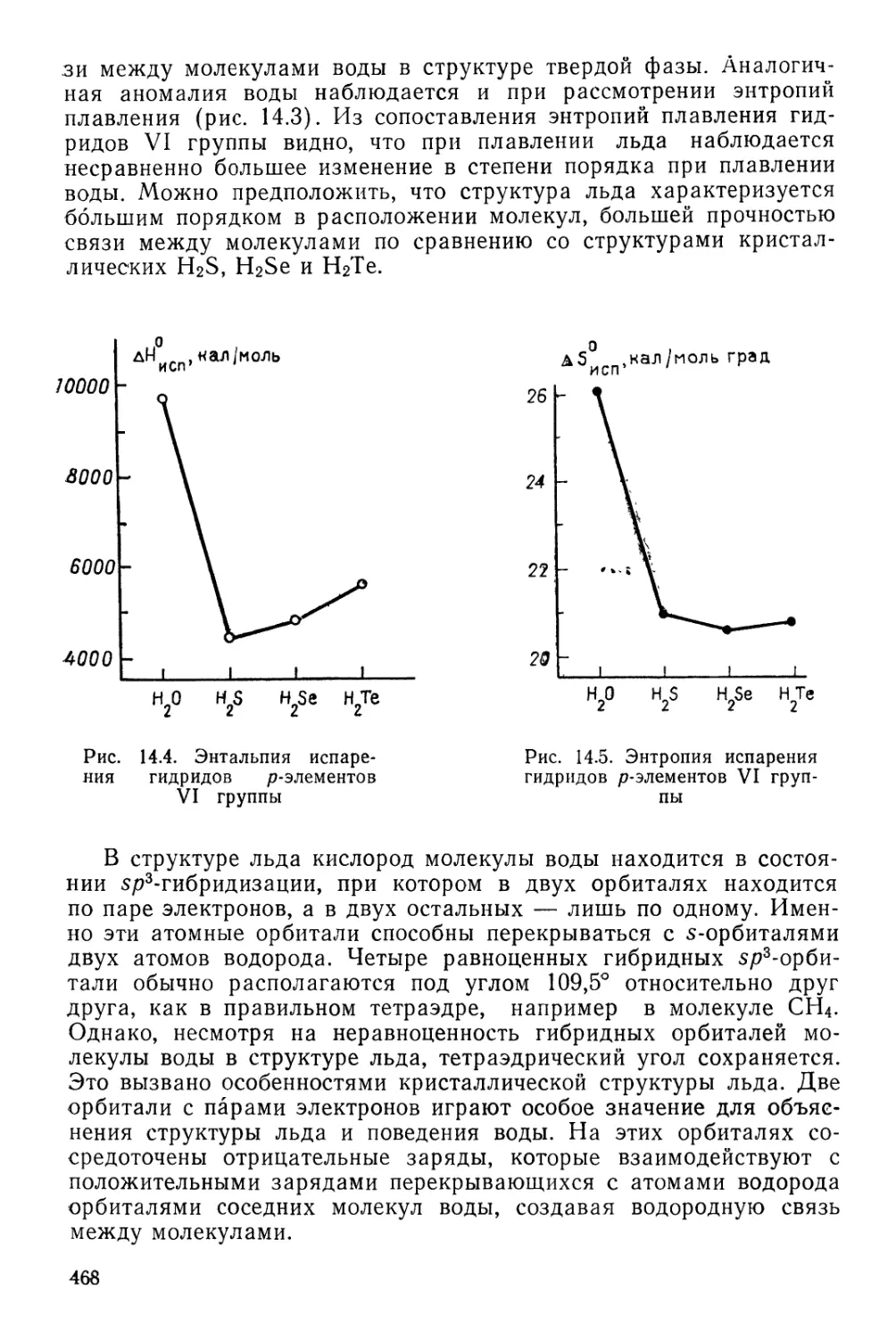
катализаторов 217
Ферментативный катализ 220
Катализ в промышленности .... .... 222
Вопросы и упражнения 223
Глава 7. РАСТВОРЫ 225
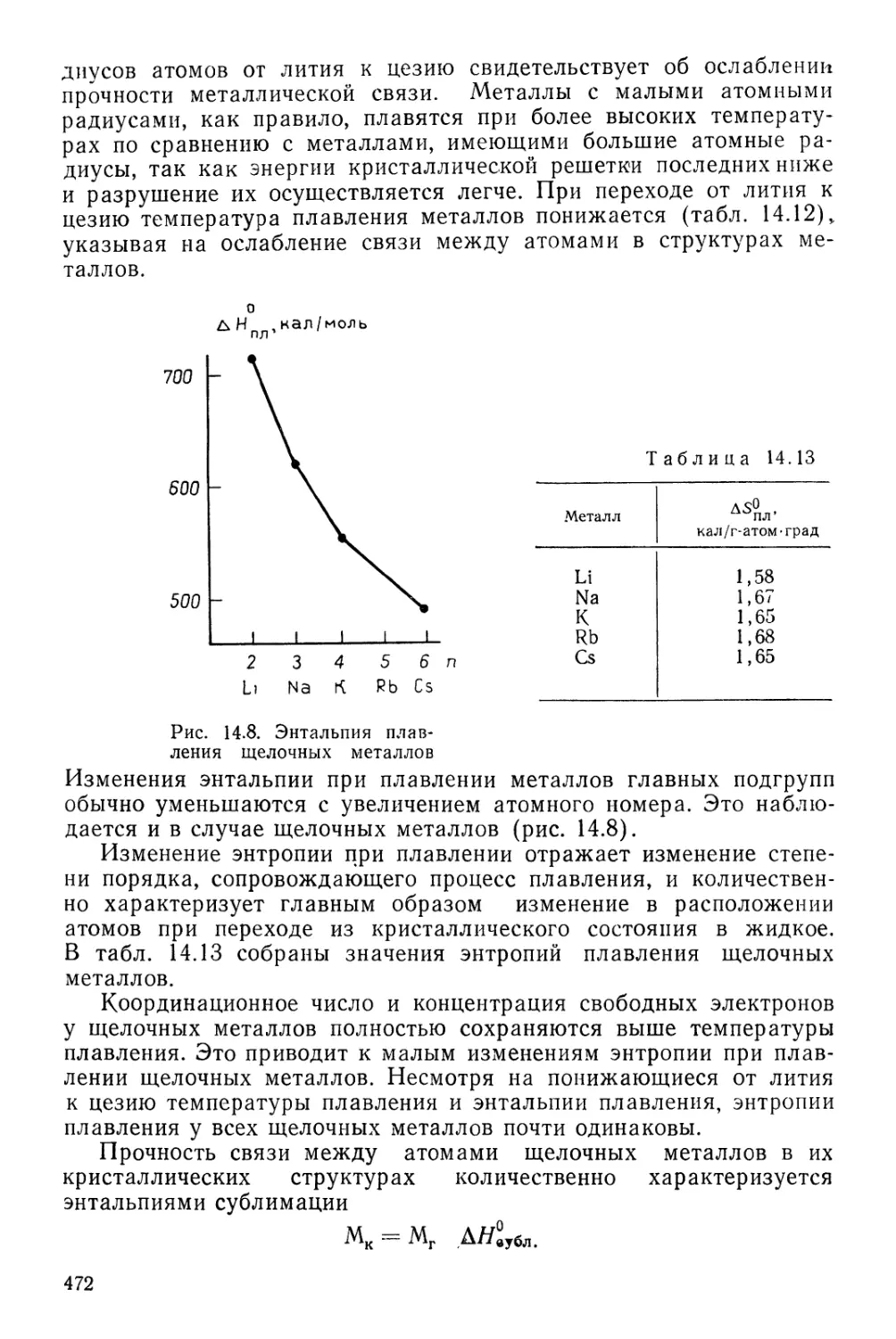
§ 1. Общие положения 225
§ 2. Свойства растворов. Законы Рауля, Генри, Вант-Гоффа . . 229
§ 3. Активность и коэффициент активности компонентов раствора 241
§ 4. Электролитическая диссоциация. Вода 243
Представление о процессе электролитической диссоциации в
водных растворах 251
§ 5. Количественное описание равновесий в растворах электролитов.
Применение ЗДМ к электролитам 257
Гидролиз солей 267
Произведение растворимости .... .... 273
§ 6. Неводные растворители . 276
Вопросы и упражнения 279
Глава 8. КОЛЛОИДНОЕ СОСТОЯНИЕ ВЕЩЕСТВА 281
§ 1. Понятие о коллоидном состоянии вещества и основные виды
коллоидных систем 281
§ 2. Методы получения высокодисперсных и коллоидных систем . . 285
§ 3. Мицеллы коллоидных растворов 287
§ 4. Явление коагуляции и стабилизации коллоидов 29Q
§ 5. Электрические свойства и очистка коллоидов 294
§ 6. Общие представления о микрогетерогенных системах —
суспензиях, эмульсиях, пенах 297
§ 7. Общие представления о полуколлоидах и некоторые свойства
растворов полимеров 299
Вопросы иупра ж нения 302
4
Глава 9. ХИМИЯ ТВЕРДОГО СОСТОЯНИЯ ВЕЩЕСТВА . . . 303
§ 1. Агрегатное состояние вещества ... 303
§ 2. Строение твердых веществ .... 305
§ 3. Реальные кристаллы ... 317
§ 4. Диффузия при химических реакциях в твердых веществах . . 322
§ 5. Кинетика реакций в твердых веществах . . ... 324
§ 6. Механизм реакций в твердых веществах 327
Вопросы и упражнения 330
Глава 10. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПРОЦЕССЫ 332
§ 1. Общие понятия 332
§ 2. Методы составления уравнений окислительно-восстановительных
реакций 338
§ 3. Окислительно-восстановительные потенциалы 341
Вопросы и упражнения 349
Глава 11. ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ. КОРРОЗИЯ . . 351
§ 1. Стандартные электродные потенциалы 351
§ 2. Газовые электроды 353
§ 3. Измерение электродных потенциалов. Ряд напряжений . 354
§ 4. Гальванические элементы 359
§ 5. Поляризационные явления в гальванических элементах . . . 363
§ 6. Электролиз 365
§ 7. Напряжение разложения. Явление перенапряжения. Последова-
тельность разряда ионов ™
§ 8. Законы Фарадея ^
§ 9. Практическое применение электролиза *j™*
§ 10. Аккумуляторы ЗоУ
§ 11. Топливные элементы 371
§ 12. Основные виды коррозии металлов 372
§ 13. Методы защиты от коррозии 376
Вопросы и упражнения 378
Глава 12. ОБЩИЕ СВОЙСТВА МЕТАЛЛОВ. СПЛАВЫ. ОСНОВЫ
ФИЗИКО-ХИМИЧЕСКОГО АНАЛИЗА 380
§ 1. Общая характеристика металлов 380
§ 2. Кристаллическая структура металлов 382
§ 3. Металлическая связь 386
§ 4. Методы получения металлов 388
§ 5. Сплавы металлов. Основы физико-химического анализа . . . 390
§ 6. Химические свойства металлов 408
Вопросы и упражнения 424
Глава 13. ХИМИЯ КООРДИНАЦИОННЫХ СОЕДИНЕНИИ ... 425
§ 1. Основные понятия химии координационных соединений . . . 425
§ 2. Координационная теория Вернера 427
§ 3. Классификация и номенклатура координационных соединений 434
§ 4. Современные представления о природе координационной связи.
Метод валентных связей 436
§ 5. Свойства комплексов в свете теории кристаллического поля и
метода молекулярных орбиталей 438
§ 6. Устойчивость комплексных ионов 450
§ 7. Координационные соединения типа кластеров и клатратов . . 453
Вопросы и упражнения 456
Глава 14. ХИМИЯ ЭЛЕМЕНТОВ И ИХ ВАЖНЕЙШИХ
СОЕДИНЕНИИ 458
§ 1. s-, p-, d- и /-последовательности элементов 458
§ 2. Водород 459
§ 3. Щелочные металлы (s-элементы I группы) . .... 471
§ 4. Бериллий, магний и щелочноземельные металлы (s-элементы
II группы) 483
§ 5. Элементы главных подгрупп III—VIII групп (/7-элементы) . 497
$ 6. Бор, алюминий, галлий, индий и таллий (р-элементы III группы) 519
§ 7. Углерод, кремний, германий, олово, свинец (/7-элементы IV
группы) 525
§ 8. Азот, фосфор, мышьяк, сурьма и висмут (/7-элементы V пруппы) 530
§ 9. Кислород, сера, селен, теллур и полоний (/7-элементы VI лруппы) 536
§ 10. Галогены (р-элементы VII группы) ... . 540
§ 11. Благородные газы (/7-элементы VIII группы) 546
§ 12. Элементы побочных подгрупп (^-элементы) 548
§ 13. Скандий, иттрий, лантан (^-элементы III группы) . . . 581
§ 14. Титан, цирконий, гафний (flf-элементы IV группы) . . . 582
§ 15. Ванадий, ниобий, тантал (d-элементы V группы) . . . 584
§ 16. Хром, молибден, вольфрам (d-элементы VI группы) . . . 588
§ 17. Марганец, технеций, рений (^-элементы VII группы) . . . 591
§ 18. Элементы побочной подгруппы VIII группы (железо, кобальт,
никель и др.) 596
§ 19. Медь, серебро, золото (^-элементы I группы) .... 609
§ 20. Цинк, кадмий, ртуть (^-элементы II группы) .... 614
§ 21. Лантаноиды (4/-элементы) 616
§ 22. Актиноиды (5/-элементы) 630
Вопросы и упражнения 633
Глава 15. ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ 637
§ 1. Строение и свойства органических соединений 637
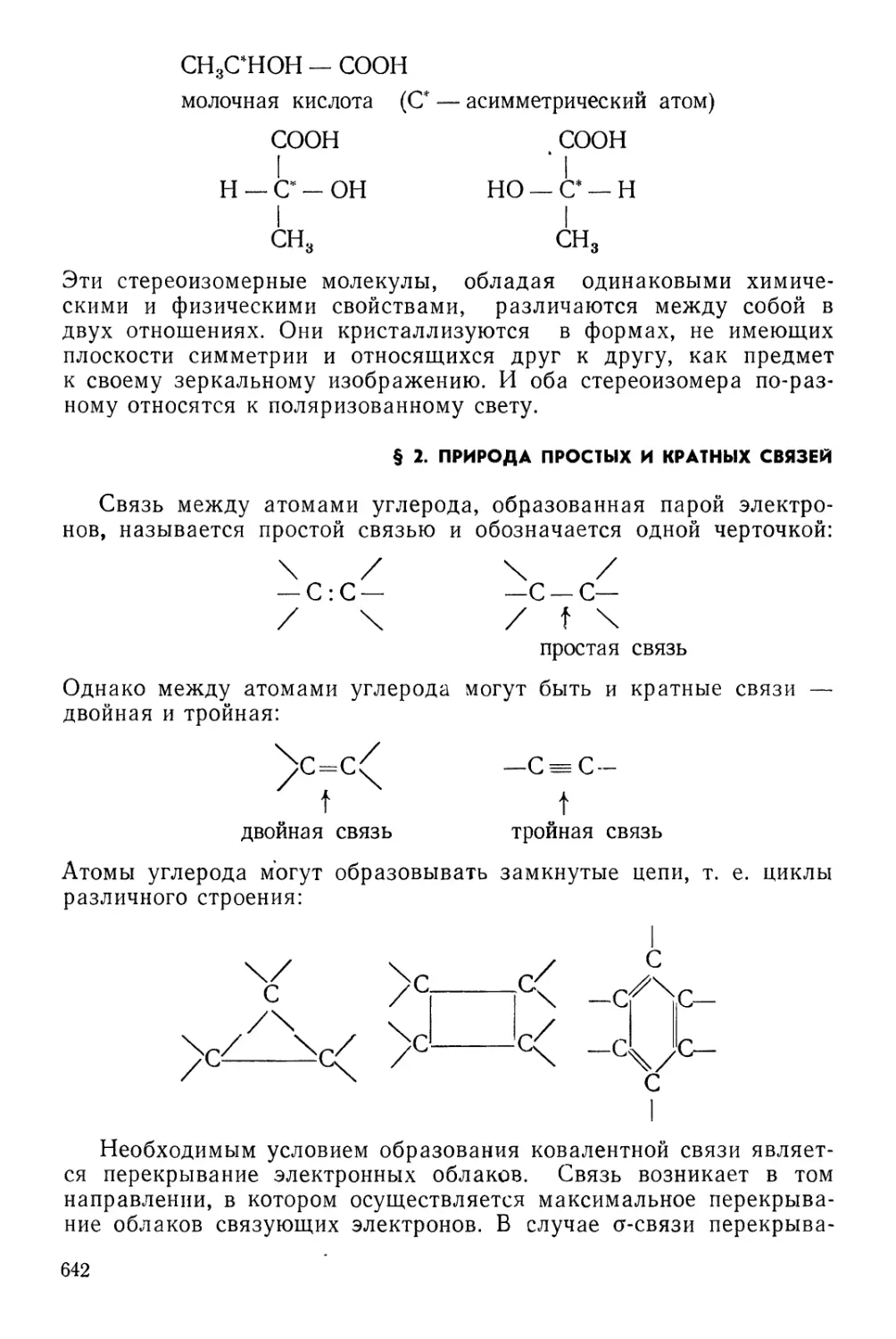
§ 2. Природа простых и кратных связей . . 642
§ 3. Механизмы реакций органических соединений 644
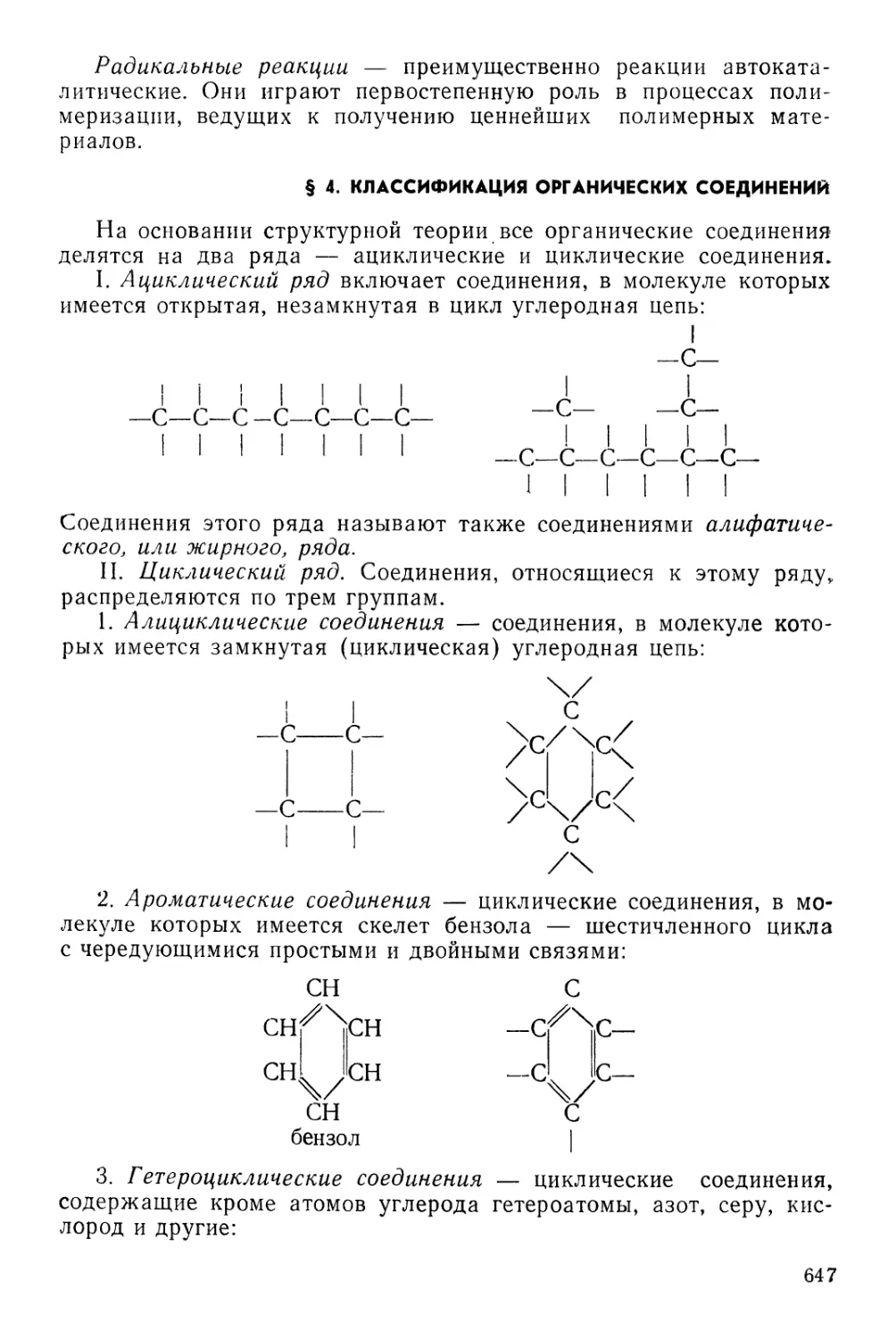
§ 4. Классификация органических соединений 647
§ 5. Углеводороды 648
§ 6. Галоидные производные 658
§ 7. Спирты, или алкоголи 660
§ 8. Альдегиды и кетоны 665
§ 9. Органические, или карбоновые, кислоты 668
§ 10. Амиды кислот 672
§ 11. Аминокислоты 673
' § 12. Белковые вещества 675
§ 13. Амины. Производные, содержащие серу 677
§ 14. Элементоорганические соединения 678
§ 15. Углеводы . 684
Вопросы и упражнения 687
Глава 16. РАДИОАКТИВНОСТЬ И ЯДЕРНЫЕ РЕАКЦИИ ... 689
Вопросы и упражнения 697
Основная литература 699
Дополнительная литература 699
ПРЕДИСЛОВИЕ
В основу настоящего учебного пособия положен курс лекций по
общей химии, который авторы длительное время читают на
химическом факультете Московского государственного университета
им. М. В. Ломоносова для студентов нехимических специальностей.
По своему содержанию книга отвечает программе по общей химии,
утвержденной Министерством высшего и среднего специального
образования СССР для нехимических специальностей естественных
факультетов МГУ.
Авторы предлагаемого учебного пособия использовали
материал, наиболее удачно изложенный в ряде лучших учебников и
учебных пособий по общей, неорганической, физической и органической
химии, изданных как в СССР, так и в зарубежных странах. При
написании ряда разделов широко привлекалось созданное на
кафедре общей химии химического факультета МГУ пособие «Лекции
по общей химии», написанное ныне покойным профессором К. Г.
Хомяковым.
По объему, стилю изложения и логическому построению
настоящее учебное пособие по общей химии — это необходимое
дополнение к учебнику, которое позволяет с большей углубленностью
усвоить основы химической науки. Лекции, читаемые на первом году
обучения в высшей школе, представляются особо ответственными,
поскольку в них доло1спы быть изложены правильные без
упрощенчества представления о развитии современной химической науки в
форме, доступной для студентов первого курса. Нужно также иметь
в виду, что лекторы и преподаватели общей химии обязаны в
достаточной мере подготовить студентов к удовлетворительному усвоению
последующих специальных дисциплин, которые тесным образом
связаны с химией.
В предлагаемом учебном пособии 16 глав, отвечающих
наиболее важным разделам курсов общей и неорганической, физической
и органической химии. В отличие от имеющихся в литературе
пособий здесь значительно шире и глубже представлены разделы,
посвященные энергетике химических реакций, современному содержанию
периодического закона, химической связи, кинетике и катализу,
теории растворов и химии координационных соединений. К каждой
главе даны вопросы и упражнения для самопроверки, приводятся
задачи, а также указан рекомендуемый список учебной и научной
литературы, который может быть использован для более глубокого
самостоятельного изучения предмета. Книга написана коллективом
из 11 авторов — преподавателей кафедры общей химии Московского
университета. В пособии даны не только необходимые сведения по
общей химии, но и показаны те новые направления в науке и
преподавании в высшей школе, которыми так богата эпоха научно-тех-
нической революции Насколько удалось решение птой задачи будут
судить читатели, критические замечания и поо/селанил которых
авторы с благодарностью примут и учтут при дальнейшей работе над
усовершенствованием предлагаемого учебного пособия.
Введение, гл. 6 и 15 написаны Г. Д. В о вч е нко, гл. 1 и 13
Ю. Д. Третьяковым, гл. 2 и 4 Л. А. Р е з ницким, гл. 3 и 12
Е. М. С о коло в с кой, гл. 5 и 14 О. С. Зайце вы м, гл. 7
В. А. Трошкиной, гл. 8 В. Н. Пятницким, гл. 9 Т. И. Б у л-
г а ко в о й, гл. 10 А. И. П лет ю шкиной, гл. 11 Т. М.
Гришиной, гл. 16 Л. С. Г у з ей.
Общее научное редактирование учебного пособия
осуществлено профессорами Е. М. Соколовской, Г. Д. В овченко и
Ю. Д. Третьяковым.
ВВЕДЕНИЕ
1. ХИМИЯ — РАЗДЕЛ ЕСТЕСТВОЗНАНИЯ
Наука возникает на основе практической деятельности людей как
результат обобщения накопленного практического опыта.
Известно, что обобщение опыта приводит к установлению закономерных
связей между явлениями и позволяет проникнуть в их сущность
построением научных гипотез, способных предсказывать
новые факты и явления, подтверждаемые опытом. Результаты
обобщения научных фактов и гипотезы, правильно объясняющие
определенный круг явлений, представляют теоретическую часть науки.
Теоретическая часть науки неотделима от практики, и только в их
единстве возможен прогресс науки, ее развитие, происходящее в
соответствии с развитием материальных производительных сил.
Химия — одна из естественных наук, изучающих материальный
мир во всем многообразии его существования и превращений.
Превращения веществ могут сопровождаться выделением или
поглощением энергии: тепловой, световой, электрической.
Следовательно, химия — это наука о составе, строении и свойствах
веществ, их превращениях и тех явлениях, которыми
сопровождаются превращения одних веществ в другие.
Бесконечное многообразие тел и явлений природы проникнуто
единством, которое заключается в материальности мира. Окру-
жиющий нас мир существует объективно, независимо от
человеческого сознания; он представляет собой различные виды
движущейся материи, различные формы ее проявления.
«...Материя есть то, что, действуя на наши органы чувства, про-
изводит ощущение; материя есть объективная реальность, данная
нам в ощущении...»1.
Материя неразрывно связана с движением. Движение есть
форма существования материи. Материя без движения так же
немыслима, как и движение без материи. Под движением материи
следует понимать все происходящие в природе изменения, начиная
с простого перемещения в пространстве и кончая мышлением.
«...Вся природа, начиная от мельчайших частиц ее до величай-
ших тел, ... находится в вечном возникновении и исчезновении, ...
в неустанном движении и изменении»2.
Каждая наука изучает преимущественно одну из форм
движения материи. Химия изучает ту форму, в результате которой
происходит соединение атомов с образованием определенных веществ.
Поэтому химию можно определить также как науку об атомах и
' И. И Ленин. Поли. собр. соч., т. 18, стр. 149.
2 К. М а р к с и Ф. Э н г е л ь с. Соч., т. 20, стр. 354.
9
их соединениях. При этом областью химии являются такие
превращения, при которых из одних молекул соединением,
разъединением или перегруппировкой входящих в их состав атомов и
изменением связей между атомами образуются другие молекулы, т. е.
новые вещества. Сами же атомы, т. е. химические элементы,
остаются при этом неизменными.
Современное естествознание характеризуется тем, что переход
к познанию тех или иных объектов или явлений природы и
исследование их происходит с позиций и методами двух или нескольких
наук. В результате возникают новые промежуточные пограничные
отрасли науки, например, на стыке физики и химии уже давно
возникла физическая химия, изучающая химические и физические
процессы в их тесной взаимосвязи. Развитие ряда направлений
физической химии привело к выделению из нее других областей
науки — электрохимии, коллоидной химии, фотохимии и др.
Выделившаяся из физической химии — химическая физика,
выросла к настоящему времени в весьма важную отрасль науки. Между
неорганической химией и ядерной физикой возникла новая
наука — радиохимия, между неорганической химией и органической
химией — элементоорганическая химия. Неорганическая химия,
геология и минералогия породили геохимию; широко и весьма
успешно развивается биохимия — синтез биологии, органической
и физической химии.
Характерным для естествознания в наше время является также
его математизация. Математика проникает во все
естественнонаучные дисциплины. Это прогрессивное явление уже привело к
возникновению ряда новых научных дисциплин между математикой
и отдельными отраслями естествознания. Вместе с тем
математизация естественных наук вооружает их новыми методами,
так необходимыми для дальнейшего развития естественных
наук.
Эти примеры показывают, что и естествознание в целом и
входящие в него отдельные науки дифференцируются, широко
развиваются, переплетаются в различных направлениях и порождают
новые ответвления наук. Эта взаимосвязь естественных наук
отражает переход, существующий в самой природе между
соответствующими формами движения материи.
Раньше роль химической науки, как правило, была лишь
вспомогательной в совершенствовании производства, а исследования
с целью развития теоретических представлений или установления
общих закономерностей имели не систематический, а случайный
характер. Современная химическая наука настойчиво стремится
к познанию внутренних скрытых причин, обусловливающих при
химических реакциях изменение или возникновение новых свойств
веществ, что приводит к установлению общих теорий и открывает
широчайшие возможности для практического использования
теоретических знаний. Роль современной науки в производстве
превращается из подсобной в ведущую; наука сама по себе стала
10
непосредственной производительной силой, а производство —
техническим оформлением новых прогрессивных идей теоретической
химии.
2. ОСНОВНЫЕ ЭТАПЫ РАЗВИТИЯ ХИМИИ
Возникновению всякой науки, в том числе и химии, должен
предшествовать более или менее длительный донаучный период
накопления практического опыта и разрозненных наблюдений. Это
накопление стимулируется практической деятельностью людей.
Химические знания возникли в глубокой древности в связи с
практической трудовой деятельностью людей.
История подтверждает, что за тысячелетия до нашей эры люди
научились пользоваться некоторыми химическими превращениями
для получения нужных им веществ. Так, за несколько веков до
нашего летосчисления в Месопотамии из руд добывали железо,
медь, серебро, свинец; химические ремесла существовали в
древности в Египте, Индии, Средней Азии, Китае. Следует при этом
подчеркнуть, что никаких научных представлений о составе
вещества и его превращениях в древнем мире не существовало.
В первые века нашей эры, когда уже были открыты и изучены
многие вещества, например азотная и серная кислоты, ряд солей,
химические знания стали проникать из стран Востока в Грецию и
Рим. Арабы называли химию алхимией. Завоевание арабами
Испании в начале VIII в. способствовало проникновению алхимии
в Западную Европу.
Главной задачей алхимии было при помощи философского
камня превращать неблагородные металлы в золото. Такому
воззрению способствовал ряд явлений, которые были известны уже в то
время (получение амальгам, вытеснение меди железом и др.)-
Указанные производства обслуживали привилегированные слои
населения, а сама химическая наука считалась божественной
наукой и находилась в руках жрецов и тщательно скрывалась от всех
непосвященных. В поисках философского камня алхимики
занимались исследованием различных веществ. Достижения алхимиков
свелись в основном к открытию ряда новых веществ и разработке
некоторых приемов химического исследования.
Медленный темп развития химических знаний в период
алхимии объясняется крайне низким уровнем средневекового
производства, реакционной идеологией правящих классов-41 особенно
господством католической церкви. Энгельс писал: «...существует
очень тесная связь между алхимией и религией. Философский
камень обладает многими богоподобными свойствами...»1.
В Древней Руси алхимия не получила распространения, так
как химические сведения проникли в Русь не с Запада, а с
Востока, через Древнюю Армению. Русская практическая химия вплоть
до XVII в. развивалась почти независимо от западноевропейской
-К. Маркс и Ф. Энгельс. Соч., т. 21, стр. 293.
11
В Западной Европе наряду с алхимией существовала и
развивалась практическая, ремесленная, химия, послужившая
основой для научной химии. Потребность в изучении химических
явлений стала остро ощущаться в XVI в. благодаря начавшемуся
быстрому развитию промышленности. Ф. Энгельс писал: «...уже с
самого начала возникновение и развитие наук обусловлено
производством» 1.
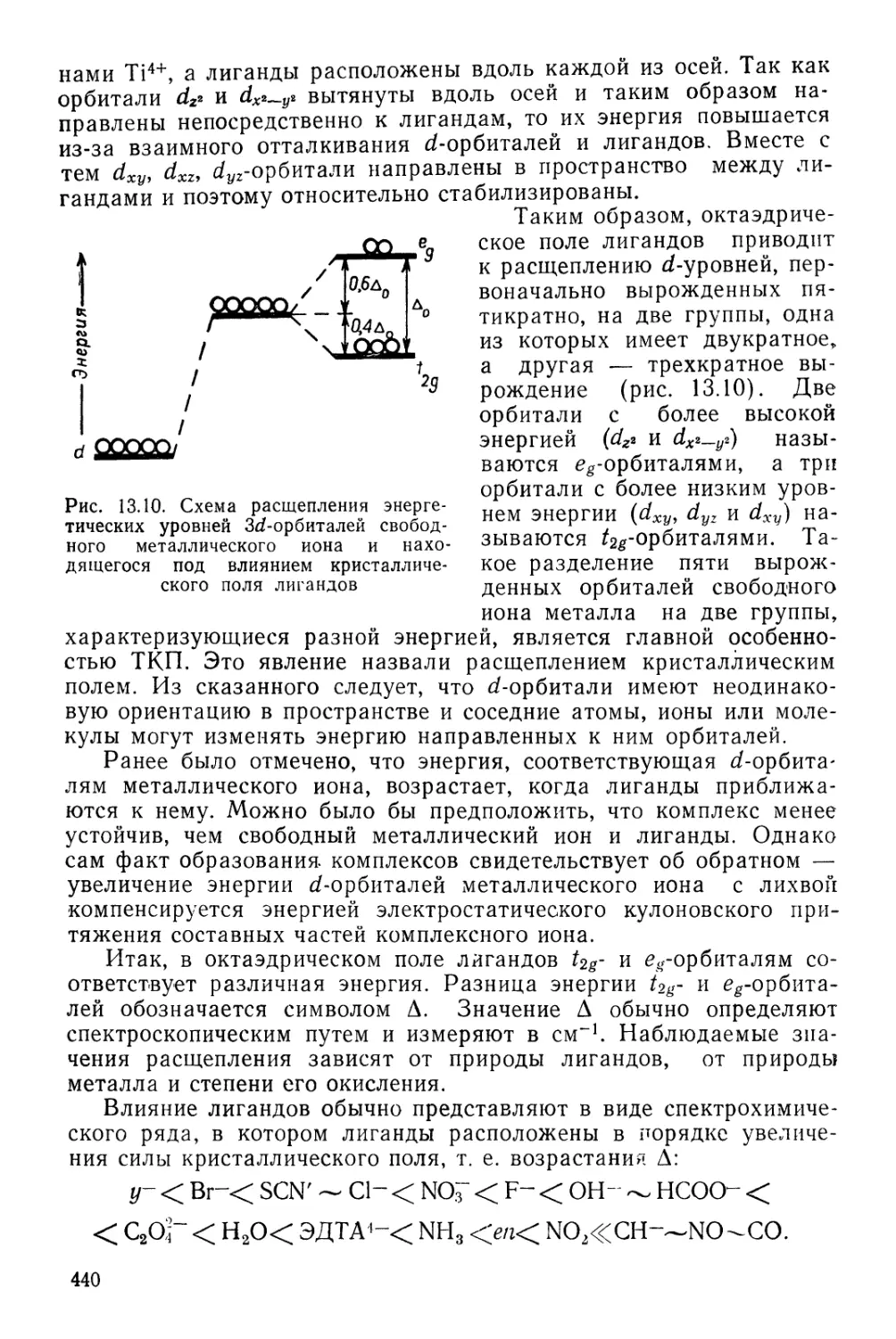
М. В. Ломоносов (1711—1765)
Наиболее крупными реформаторами химии в этот период
выступили Парацельс (1493—1541) и Агрикола (1494—1555).
Парацельс считал, что назначение химии — это изготовление
лекарств (период ятрохимии). По его мнению, все состоит из соли
или тела, ртути или души, серы или духа. Болезнь зависит от
недостатка одного из них. Лечить — это значит вводить в организм
недостающий элемент.
Агрикола работал в области горного дела и металлургии, его
книги служили основными руководствами более 200 лет. Видными
последователями Агриколы были Палисси (1510—1589),
изучавший химические процессы в области керамики и земледелия, и
1 К. Маркс и Ф.Энгельс. Соч., т. 20, стр. 500.
12
Глаубер (1604—1668), работавший в области стеклоделия,
изучения селитр и красящих веществ.
Большое значение в развитии химии имели работы английского
ученого Р. Бойля, который подверг резкой критике алхимические
представления. Он дал определение химического элемента как
предела разложения вещества на составные части.
Экспериментальные исследования Р. Бойля послужили началом химии как
науки. Однако какой-либо теории, обобщающей накопленный
экспериментальный материал, он не выдвинул.
Важнейшим вкладом в развитие химии как науки является
создание корпускулярной теории М. В. Ломоносовым. Ломоносовское
понятие о корпускуле соответствует современному представлению
о молекуле, а об элементе — атому. По теории Ломоносова,
изложенной в книге «Элементы математической химии» (1741 г.),
однородные корпускулы образуют простое вещество, а разнородные
корпускулы — сложное вещество.
Все свойства веществ, их физическую и химическую природу
М. В. Ломоносов объяснял движением и взаимодействием
корпускул и элементов. Поэтому, по мысли Ломоносова, изучение
мельчайших частиц, слагающих вещества, составляет основную
задачу химии.
Свою корпускулярную теорию М. В. Ломоносов дополнил
кинетическими представлениями, указывая, что элементы и корпускулы
обладают движением, наличие которого объясняет многие
свойства простых и сложных веществ.
Следующей ступенью в развитии атомистических представлений
были выдающиеся работы английского физика Дж. Дальтона. Не
зная о работах М. В. Ломоносова и будучи приверженцем
атомистических взглядов И. Ньютона, Дальтон для объяснения
открытого им закона кратных отношений выдвинул атомистическую
теорию, впервые изложенную в книге «Новая система химической
философии» в 1803—1804 гг. В атомистической теории Дальтона
кинетические представления отсутствуют. Таким образом, взгляды
М. В. Ломоносова на природу и свойства веществ были более
совершенны и ближе к современным представлениям, чем взгляды
Дж. Дальтона.
Создатели атомистических представлений М. В. Ломоносов
и Дж. Дальтон сыграли решающую роль в развитии химии как
науки. Ф. Энгельс писал: «Новая эпоха начинается в химии с
атомистики...» 1.
К концу XVII в. химическая практика настолько переросла
теорию, застывшую на уровне алхимических представлений, что
дальше такое положение не могло быть терпимым. Так как химия
была связана главным образом с металлургией, нужно было
объяснить прежде всего процессы горения, окисления и
восстановления.
1 К Маркс и Ф. Энгельс. Соч., т. 20, стр. 608
13
В конце XVII в. немецкий химик Г. Шталь создал
флогистонную теорию. Согласно этой теории все горючие вещества и
металлы содержат в своем составе особое вещество — флогистон; в
процессе сжигания горючих веществ или окисления металлов
флогистон улетучивается. Так, при накаливании металла, например-
железа, происходит выделение флогистона и образование
землистого остатка окалины:
железо -> окалина + флогистон.
Следовательно, флогистонная теория рассматривала процесс
окисления металла как реакцию разложения: металл считался
сложным веществом, а окалина — простым. Считали, что уголь
представляет собой почти чистый флогистон, так как при его сгорании
остается небольшое количество золы.
Хотя теория флогистона была ошибочной, тем не менее в эту
историческую эпоху она сыграла некоторую положительную роль
и способствовала объединению накопленных опытных данных в
определенную систему. Ф. Энгельс считал, что «Химия ...
освободилась от алхимии посредством флогистонной теории» К Со
временем накопилось много наблюдений, которые находились в явном
противоречии с теорией флогистона, и в конце почти векового
господства эта теория стала тормозом в развитии химии. Большое
значение для создания научной теории горения имели открытия
Ломоносова, связанные с опытами по прокаливанию металлов в
запаянных сосудах. Ломоносов установил, что если сосуд с
накаленным железом взвесить, не вскрывая его, то масса остается без
изменения. Если вскрыть сосуд, то в него врывается воздух, в
связи с этим масса увеличивается. На основании этих опытов
М. В. Ломоносов заключил, что при прокаливании металла
происходит соединение его с воздухом. Совершенно очевидно, что
исследования Ломоносова опровергали флогистонную теорию и
намечали новый подход к объяснению процессов горения и
окисления.
Впоследствии французский ученый А. Лавуазье также доказал,
что горение есть реакция соединения вещества с кислородом
воздуха. Лавуазье своими исследованиями также опроверг
флогистонную теорию и создал правильное, строго научное представление о
сущности процессов горения и окисления. «Лавуазье ... поставил
на ноги всю химию, которая в своей флогистонной форме стояла
на голове»2.
Первая половина XIX в. представляет последний этап,
предшествующий периоду современной химии. В конце XIX в.
окончательно были установлены основные понятия молекулярно-ато-
мистической теории. Первоначально неясные представления о раа-
1 К. Маркс и Ф.Энгельс. Соч , т 20, стр. 348.
2 К. Маркси Ф. Энгельс Соч , т. 24, стр. 19—20.
14
личин между атомом как мерой химического элемента и
молекулой как мерой простого и сложного вещества, вступающего в
химическое взаимодействие, получили правильное истолкование.
Этим была устранена одна из причин, мешавших развитию
химической науки. Окончательно было установлено также различие
между атомным и эквивалентным весом и введено понятие о
валентности элементов. Химические формулы и уравнения стали
принимать современный вид. В этот период было открыто много
новых элементов, например: фтор, бром, йод, селен, хром, бор,
алюминий, магний и щелочноземельные металлы и почти все
элементы платиновой группы.
Успехи экспериментальных исследований позволили открыть
существование групп сходственных элементов, внутри которых
свойства правильно и монотонно изменяются с увеличением
атомного веса. Современный период химии начинается с момента
открытия Д. И. Менделеевым периодического закона химических
элементов в 1869 г. и создания А. М. Бутлеровым в 1861 г. теории
химического строения. Открытие периодического закона и
периодической системы элементов оказало огромное влияние на
развитие не только химии, но всего естествознания в целом.
Периодический закон показал, что все элементы образуют
целостную совокупность, связанную общей закономерностью.
Каждый элемент, являясь членом этой совокупности, занимает в ней
строго определенное положение. Основываясь на периодическом
законе, химия впервые получила возможность научного
предвидения и перспективу новых путей исследования свойств элементов.
Значение периодического закона непрерывно увеличивалось по
мере того, как все новые факты подтверждали его справедливость
и выяснялась его руководящая роль во всех последующих
открытиях, касающихся строения вещества. Классики
марксизма-ленинизма всегда подчеркивали выдающееся философское значение
открытия Д. И. Менделеева.
Следует особо подчеркнуть, что вся современная химия и ряд
разделов физики развиваются под воздействием периодического
закона, открытого Д. И. Менделеевым. К началу современного
периода в химии был накоплен большой экспериментальный
материал, касающийся синтеза и свойств углеродных соединений.
Этот материал составил особый раздел химической науки —
органическую химию.
Органические соединения находили многообразное >и все
растущее практическое применение, что вызывало необходимость
усовершенствования методов переработки органического сырья и
разработки методов синтеза новых органических соединений,
наилучшим образом удовлетворяющих запросы практики. Успех
этого дела, в свою очередь, требовал создания руководящей
теории, которая давала бы возможность понять природу органических
соединений, объяснить суть химических реакций, приводящих к их
образованию, и тем самым поднять синтез органических соедине-
15
ний от слепой, наугад проводящейся операции до сознательного,
целеустремленно направленного исследования. Такая теория была
создана А. М. Бутлеровым в 1861 г. Она получила название
теории химического строения.
Сущность теории А. М. Бутлерова заключается в
утверждении, что молекула любого соединения представляет не случайное
произвольное сочетание связывающихся друг с другом атомов, а
частицу, имеющую определенное строение, от которого, так же
как от природы атомов и их числа, зависят химические свойства
этой частицы. Это строение может быть установлено на основании
изучения химических превращений. Знание того, как зависит
реакционная способность, т. е. химические свойства, от внутреннего
строения молекул, открывает необозримые перспективы к
сознательному синтезу новых соединений и к глубокому пониманию
сущности химических превращений.
Теория А. М. Бутлерова, которая первоначально имела в виду
только органические соединения, в настоящее время получила
распространение на все химические соединения. Она определила
главные направления современной химии — изучение строения
химических соединений и выяснение закономерностей между строением,
с одной стороны, и химическими и физическими свойствами — с
другой. В наши дни решение этой грандиозной задачи возможно
при одновременном глубоком изучении строения и свойств атомов
и широкого привлечения к химии физических методов
исследования, т. е. при тесном содружестве с физической наукой.
Химическая наука как раньше, так и в настоящее время всегда опирается
на закон сохранения материи и движения, открытый М. В.
Ломоносовым, на периодический закон химических элементов,
открытый Д. И. Менделеевым, и на теорию химического строения,
созданную А. М. Бутлеровым. М. В. Ломоносова, Д. И. Менделеева
и А. М. Бутлерова законно относят к числу основоположников
современной химической науки.
Современная химия настолько обширна и разнообразна как по
объектам, так и по методам исследования, что представляется
целесообразным и возможным отметить лишь некоторые
важнейшие успехи ее за последнее время.
Открытие законов квантовой механики (1926 г.), управляющих
движением электронов в атомах и молекулах, позволило раскрыть
природу химического взаимодействия. Было окончательно
установлено, что нет особых по своей природе химических сил, которые
существовали бы наряду с электрическими силами, силами
тяготения и ядерными силами. Химическое взаимодействие создается
электрическими силами взаимного притяжения и отталкивания
электронов и атомных ядер, а его особенности вытекают из кван-
товомеханических законов. С этого времени развивается
квантовая химия.
Наряду с изучением строения и свойств веществ все большее
внимание уделяется химическим превращениям как процессам,
16
протекающим во времени, т. е. химической кинетике, что позволяет
в ряде случаев выяснить механизм химических реакций. В связи с
этим интенсивно изучаются короткоживущие активные формы
частиц — свободные радикалы и т. п., являющиеся промежуточными
образованиями в химических реакциях.
Для раскрытия строения сложных химических соединений
наряду с классическими химическими методами весьма широко
используются физические методы — .рентгеноструктурный анализ*
спектроскопия, радиоспектроскопия и др. Физические методы
находят также большое применение и для изучения механизма
реакции.
В послевоенный период неорганическая химия успешно решает
задачи, поставленные новой техникой. В связи с использованием
атомной энергии, освоением космического пространства, широко
развернувшимся строительством и другими научно-техническими
проблемами потребовались материалы с таким ценным сочетанием
физико-химических и механических свойств, которое не
встречается у ранее использовавшихся природных или синтетических
веществ (например, сочетание стойкости к агрессивным средам с
механической прочностью и жаропрочностью) .
Одновременно продолжается широкая разработка методов
получения особо чистых веществ, в первую очередь редких
металлов, крайне необходимых для полупроводниковой и атомной
техники. Вместе с тем продолжают интенсивно развиваться и более
ранние направления неорганической химии. Неорганическая химия
служит научной базой как металлургии, так и основной
химической промышленности (производство кислот, солей и щелочей),
развитие же основной химии крайне необходимо для развития
многих других отраслей промышленности и для химизации
сельского хозяйства.
За последние 25—30 лет больших успехов достигла
органическая химия. Ее основная концепция — о взаимном влиянии
атомов в молекулах — с развитием учения о химической связи
приобрела более точный и глубокий смысл. Оказался возможным
приближенный расчет электронных структур молекул и
коррелирование распределения электронов со свойствами соединений.
К новым теоретическим проблемам, разрешаемым в органической
химии на основе электронных представлений, относятся проблема
«ароматичности», закономерность замещения в ароматическом
ряду, особенности реакций сопряженных систем и др. Достигнуты
большие успехи и в экспериментальной органической химии
последних лет, а именно: разработка обширных областей элементо-
органических соединений, высокомолекулярных соединений
искусственных и природных (например, белков, в том числе синтез,
инсулина); получение витаминов, антибиотиков, гормонов и др.;
изучение процессов жизнедеятельности и поведения веществ в
клетке и ее элементах на молекулярном уровне; синтез
многочисленных фторорганических соединений.
17
Все эти достижения теоретической и экспериментальной
органической химии составили основу развития разнообразных
промышленных отраслей — синтетических каучуков, пластических
масс, искусственных волокон, лекарственных веществ,
инсектицидов, гербицидов, дефолиантов, моющих средств и др.
3. ХИМИЯ В СССР.
ЗАДАЧИ СОВРЕМЕННОЙ ХИМИИ
В жизни современного общества роль химии, как и всей науки,
исключительно велика. Сейчас нет ни одной отрасли народного
хозяйства, которая так или иначе не была бы связана с химией.
Химизация производственных процессов является одним из
основных условий научно-технического прогресса. Природа дает
человечеству, как правило, только сырые материалы, такие, как уголь,
нефть, газы, руды, воздух, воду, соли, дерево и т. д., из которых
человек получает нужные и ценные вещества и изделия. Чтобы
превратить одни вещества в другие, нужно знать и использовать
законы химии и методы, которыми она пользуется для таких
превращений.
Современная промышленность, используя достижения
химической науки, производит высококачественные стали, твердые,
сверхтвердые и жаропрочные сплавы, синтетические каучуки,
пластические массы, искусственные волокна, моторное топливо,
минеральные удобрения, лекарственные препараты, красители, взрывчатые
вещества, соли, кислоты, щелочи и т. д.
Получение многих ценных продуктов из нефти, природных
газов, угля, извести, воды, воздуха стало возможным благодаря
внедрению в химическую технологию принципиально новых
методов.
Достижения в области химии не только умножают число
полезных веществ и расширяют область их применения, но и
позволяют использовать отходы производства, из которых в ряде
случаев получают весьма ценные продукты.
В дореволюционной России химическая промышленность была
весьма отсталой. Это объясняется не отсутствием знаний или
умения русских химиков, а тем, что царское правительство не было
заинтересовано развитием химической промышленности и
подчинялось требованиям иностранного капитала — удовлетворять
минимальный существующий спрос завозом химических продуктов
из-за границы. Несмотря на собственные огромные запасы сырья,
Россия ввозила из-за границы фосфориты, калийные соли, селитру,
лекарственные препараты, красители, цветные металлы, резину и
многие другие виды сырья и химические изделия.
Творческая научная деятельность русских ученых-химиков в
дореволюционное время, среди которых были и выдающиеся
имена, гесьма редко встречала поддержку царского правительства.
Объясняется это политикой царского правительства, направленной
18
на поддержку иностранного капитала в России, не
заинтересованного в высоком техническом уровне русской промышленности.
Отсутствие развитой химической промышленности сильно
сказывалось на состоянии русской химической науки, не имевшей базы
для развития. Несмотря на эти крайне неблагоприятные условия
работы, русские ученые-химики внесли крупнейший вклад в
мировую химическую науку.
Великая Октябрьская социалистическая революция, в корне
устранившая причины, задерживавшие рост русской науки,
создала все условия для ее свободного развития. Уже в первые годы
Советской власти по инициативе В. И. Ленина химической науке
была оказана правительством громадная помощь. Были
организованы первые научно-исследовательские институты и
лаборатории, сыгравшие выдающуюся роль в развитии советской
химической науки и промышленности. В дальнейшем число их стало
быстро возрастать. Во много раз увеличилось число химических
учебных заведений. В университетах и институтах развернулась в
крупных масштабах научно-исследовательская работа как
теоретического, так и прикладного характера, охватившая все отрасли
химии. Советское государство должно было строить и развивать
химическую промышленность почти заново.
Уже в первые годы Советской власти В. И. Ленин указал на
необходимость «...обеспечения материальной основы крупной
индустрии: развития производства топлива, железа, машиностроения,
химической промышленности»1. Выполняя эти указания, советский,
народ под руководством Коммунистической партии уже в годы
первых пятилеток создал современную химическую
промышленность, которая сыграла большую роль в победе советского народа
в Великой Отечественной войне. Война нанесла огромный урон
химической промышленности, но после войны усилиями советского
народа она была быстро восстановлена и получила дальнейшее
развитие.
В послевоенные годы значительно расширились такие отрасли
химической промышленности, как азотная, калийная, пластических
масс, синтетического каучука, органического синтеза, хлора и его
производных. Было создано производство синтетических волокон,
синтетического этилового спирта, органических препаратов для
борьбы с вредителями сельскохозяйственных культур и др.
Особенно возросли темпы развития химической
промышленности после решения пленумов ЦК КПСС, обсуждавших развитие
химии в СССР. Дальнейшее развитие получила химия после
исторических решений XXIV съезда КПСС.
Значение развития химии в период создания
материально-технической базы коммунизма определено в программе КПСС:
«Одна из крупнейших задач — всемерное развитие химической
промышленности, полное использование во всех отраслях народ-
'П.И.Ленин. Поли. собр. соч., т. 36, стр. 188.
19'
ного хозяйства достижений современной химии, в огромной
степени расширяющей возможность роста народного богатства, выпуска
новых, более совершенных средств производства и предметов
народного потребления».
Всемерное и ускоренное развитие химической промышленности
является важнейшей предпосылкой научно-технического
прогресса. Применение химических материалов дает возможность
осуществить качественные преобразования как в промышленности, так
и в сельском хозяйстве. Эти преобразования позволят увеличить
выпуск продукции, повысить ее качество, обеспечить рост
накоплений для расширенного воспроизводства и дальнейшего улучшения
благосостояния народа.
Таким образом, химическая промышленность является одной
из важнейших отраслей народного хозяйства, оказывающей
огромнейшее воздействие на развитие всей экономики СССР.
Обеспечение всесторонней интенсификации общественного производства и
повышение его эффективности, необходимость которых была
подчеркнута на XXIV съезде КПСС, — основная линия
экономического развития нашей страны как на ближайшие годы, так и на
длительную перспективу. «Главная задача сейчас, —сказал
Л. И. Брежнев в докладе «О пятидесятилетии Союза Советских
Социалистических Республик», — это круто изменить ориентацию,
перенести упор на интенсивные методы ведения хозяйства,
обеспечить тем самым серьезное повышение эффективности
экономики».
За счет интенсивных факторов, ускоряющих рост
производительности труда, в девятой пятилетке должно быть получено
-80—85% национального дохода, 87—90% прироста
промышленной продукции, весь прирост продукции сельского хозяйства и т. д.
Благодаря факторам интенсификации, за пять лет будет
достигнута реальная экономия живого труда более 32 миллионов
человек1. Девятая пятилетка — поистине пятилетка интенсификации.
В системе основных факторов интенсификации находится наука,
в которой важнейшее место принадлежит химической науке и
промышленности.
Развитие химической индустрии будет обеспечивать также
всевозрастающие темпы химизации народного хозяйства —
прогрессирующего применения химических материалов и продуктов
промышленности в сельском хозяйстве, а также широкого
использования химических методов во всех отраслях народного хозяйства.
От уровня развития химии в значительной мере зависит развитие
всего народного хозяйства. В наше время полностью
подтвердились слова М. В. Ломоносова, сказанные более 200 лет тому
назад: «Широко распространяет химия руки свои в дела
человеческие».
«Правда», 3 марта 1973 г.
20
ГЛАВА 1
ОСНОВНЫЕ ПОНЯТИЯ И ЗАКОНЫ ХИМИИ.
АТОМНО-МОЛЕКУЛЯРНАЯ ТЕОРИЯ
§ 1. МАТЕРИЯ И ЭНЕРГИЯ
Краеугольным камнем современного научного мировоззрения
является положение о том, что в мире нет ничего, кроме движущейся
материи. Наблюдаемое многообразие явлений в природе и
обществе представляет собой лишь различные формы движущейся
материи. Материя вечна в пространстве и бесконечна во времени.
Материя, существуя независимо от нас, отображается нашими
ощущениями. Эти ощущения зависят от того, какой конкретный
вид материи действует на человека. Вместе с тем материи как
таковой, не связанной с конкретными формами, разумеется, не
существует.
Наиболее исчерпывающее и строго научное определение
материи было дано В. И. Лениным в книге «Материализм и
эмпириокритицизм»: «Материя есть философская категория для
обозначения объективной реальности, которая дана человеку в ощущениях
его, которая копируется, фотографируется, отображается нашими
ощущениями, существуя независимо от них» К Это определение,
направленное против идеализма, играет огромную роль в науке,
позволяя правильно осмыслить любые научные открытия, начиная
от особенностей строения атома и кончая кибернетикой и
молекулярной биологией.
Материя обладает рядом существенных свойств, среди
которых важнейшим является движение. Как уже отмечалось,
материя не мыслима без движения, как и движение без материи. При
этом под движением следует понимать не только простое
механическое перемещение тел, а всякое изменение во всех бесконечно
многообразных его проявлениях.
Формы движения многообразны, но вместе с тем они
взаимосвязаны, так как могут превращаться друг в друга. Мерой
движения материи является энергия. Мы говорим о химической,
тепловой, механической, электрической энергии, подчеркивая тем
самым неразрывную связь меры движения с формой движения
материи, превращающейся или способной превратиться в другие
формы.
С многочисленными примерами таких превращений мы
сталкиваемся на каждом шагу. Многие из них в той или иной мере
связаны с химической формой движения материи. Развитие жизни в
1 В. И. Л енин. Поли. собр. соч., т. 18, стр. 131.
21
любом растительном или животном организме связано с
использованием химической энергии питающих эти организмы веществ.
Превращение химической энергии в электрическую широко
используется в топливных элементах, а обратные превращения являются
основой электролиза. В энергетическом спектре явлений природы
химические превращения
занимают узкую полос\
от 1 до 10 эв (1 эв^
^23 ккал/моль). Для
сравнения в табл. 1.1 приведены
значения энергии некоторых
физических явлений в
пересчете на одну частицу.
Разумеется, что
отдельные формы движения
материи различаются по степени
сложности. Ф. Энгельс в
свое время выделил пять
основных форм движения
материи, перечисляемых
ниже в порядке увеличения
сложности: механическое
перемещение тел в
пространстве, физические
изменения, химические
изменения, органические изменения и социальные процессы1.
Чтобы иметь правильное представление о различных формах
движения и способности их к взаимопревращениям, следует
учитывать, что при возникновении более сложных или высших форм
движения из более простых (например, при превращении тепловой
энергии в химическую или химической энергии в биологическую)
низшие формы не исчезают и высшие формы движения не
возникают вместо них. Новая, более сложная форма движения материи
существует как высшее единство прежних форм, включая их в
себя. Новая форма движения как высшее единство более простых
форм движущейся материи качественно отлична от последних,
так как обладает своими, только ей присущими особенностями,
которых нет у породивших ее простых форм. В обратном процессе
превращения высших форм в низшие последние не рождаются
вновь, а лишь снимается высшее единство, которое обусловливало
качественные особенности сложных форм движения.
В реальных условиях любое тело или явление имеет несколько
форм движения, из которых одна обычно превалирует над
другими. «...Организм,—отмечал Ф. Энгельс,—есть, несомненно,
высшее единство, связывающее в себе в одно целое механику,
физику и химию, так что эту троицу нельзя больше разделить»2. Любой
1 См. К. Маркс и Ф.Энгельс. Соч., т. 20, стр. 564.
2 Там же, стр. 566.
Энергия некоторых физических явлений
в пересчете на одну частицу
Характер явления
Энергия, эв
Движение молекул газа
при комнатной
температуре
Испарение воды
Видимый свет
Ионизация атомов
Жесткое рентгеновское
излучение
Гамма-излучение
Распад радия
Деление урана
Космическое излучение
0,025
1
2
от 5
от 105
от 1-
3
4,
2
от 109
до 25
до 106
10е до
106
8-108
• 108
до Ю10
22
организм может перемещаться в пространстве, в нем непрерывно
протекают физические и химические процессы, однако высшей
формой движения материи, связывающей все остальные, в этом
случае является жизнь — главная форма движения организма.
§ 2. ЗАКОН СОХРАНЕНИЯ МАТЕРИИ
И ДВИЖЕНИЯ
В 1748 г. М. В. Ломоносов сформулировал закон
сохранения материи и движения как всеобщий естественный закон: «Все
перемены в натуре случающиеся такого суть состояния, что
сколько чего у одного тела отнимется, столько присовокупится к
другому; так ежели где убудет несколько материи, то умножится в
другом месте. Сей всеобщий естественный закон простирается и
в самые правила движения» К
Отвергая теорию флогистона, Ломоносов под материей
фактически понимал вещество, а мерой его количества считал вес.
Опыты по прокаливанию металлов в запаянных стеклянных
сосудах, выполненные в 1756 г., привели Ломоносова к выводу о
неизменности веса веществ при химических превращениях, а
следовательно, о справедливости закона сохранения материи. Таким
образом, следствием всеобщего закона сохранения материи в
применении к химическим реакциям является закон сохранения веса
веществ:
Вес всех веществ, вступающих в химическую реакцию, равен
весу всех продуктов реакции.
Этот закон окончательно утвердился в химии после
многочисленных экспериментов А. Лавуазье.
Вторая часть закона Ломоносова касается движения материи.
Мысль о неуничтожаемости движения в применении к простому
перемещению тел высказывалась еще Декартом, но смысл
всеобщего закона природы, охватывающего все виды движения без
исключения, она приобрела лишь благодаря Ломоносову.
Количественная оценка движения была найдена в понятии
энергии, которая определяется теперь как мера движения при
переходе одних его форм в другие. Р. Мейер доказал
эквивалентность различных форм движения материи, выраженных мерой
движения, т. е. энергии, и сформулировал закон, широко известный
ныне как закон сохранения и превращения энергии:
Энергия не творится из ничего и не исчезает бесследно, а
только превращается из одной формы в другую в эквивалентных
количествах.
Важным следствием закона сохранения энергии является
открытый Лавуазье принцип, в соответствии с которым тепловые
эффекты прямого и обратного процессов равны по абсолютной ве-
1 М. В. Ломоносов. Труды по физике и химии, т. II. М., Изд-во АН СССР,
1951, стр. 183—185.
23
личине, но противоположны по направлению. Известно, например,
что взаимодействие водорода с кислородом, ведущее к
образованию воды, сопровождается выделением тепла (58 ккал/моль Н20).
Процесс же разложения воды на простые вещества, имеющий
место при высокой температуре или при электролизе, требует затраты
энергии, эквивалентной теплоте образования Н20, но
противоположной ей по знаку.
А теперь вернемся к закону сохранения веса веществ. Из
элементарного курса физики известно, что вес веществ
пропорционален их массе. Поэтому закон сохранения веса веществ можно
заменить эквивалентным ему законом сохранения массы:
Масса веществ, вступающих в химическую реакцию, равна
массе веществ, образующихся в результате реакции.
Исследование явлений, сопровождающих распад или
образование атомных ядер, привело некоторых ученых к выводу о том,
что закон сохранения массы приближенный. Поскольку масса
веществ неразрывно связана с их энергией (масса вещества
увеличивается при увеличении энергии), то можно было ожидать, что
для химических реакций, сопровождающихся выделением тепла,
масса продуктов реакции должна быть меньше массы реагентов.
Если же химическая реакция происходит с поглощением тепла, то
масса продуктов должна быть больше массы начальных веществ.
При этом предлагалось сравнивать массы начальных веществ и
продуктов реакции при одинаковых условиях, в частности, при
одной и той же температуре.
Чтобы показать несостоятельность выводов о приближенности
закона сохранения массы, следует более подробно рассмотреть
взаимосвязь между массой и энергией. Напомним, что масса и
энергия являются различными свойствами материи. Русский физик
П. Н. Лебедев на основании опытов по измерению давления света
показал, что энергия света Е и его масса m связаны соотношением
Е = шс\ (1.1)
где с — скорость света, равная 3-Ю10 см/сек. Впоследствии
Альберт Эйнштейн доказал, что уравнение, выражающее
взаимосвязь между массой и энергией, универсально в том смысле, что
справедливо для любых форм материи.
Энергия движущегося тела увеличивается при увеличении
скорости движения, и одновременно с этим по мере увеличения
энергии возрастает масса движущегося тела.
Масса движущегося тела
m = m° (1.2)
где т0 — масса покоя; v — скорость движения тела; с — скорость
света в вакууме. Очевидно, что масса покоя, или собственная
масса тела, минимальна и отвечает состоянию тела, в котором v = 0.
24
Различные виды дискретных частиц (протоны, электроны,
нейтроны) имеют различные массы покоя, но существуют и такие
формы движения материи, которые не имеют совсем массы покоя>
иначе говоря, их собственная масса равна нулю. Такие формы ма-
териии называются полями. Наиболее изучено электромагнитное
поле, элементам которого — фотонам — не свойственно состояние
относительного покоя, что и определяет отсутствие у них
собственной массы.
Те виды материи, дискретные частицы которых имеют конечную
массу покоя, или собственную массу, называются веществом. Поля
и вещества находятся в непрерывной связи друг с другом, так как
взаимодействие частиц вещества осуществляется благодаря полям,
через поля. По закону всемирного тяготения все тела
притягиваются друг к другу с силой, пропорциональной их массе, но само
взаимодействие осуществляется через гравитационное поле.
Благодаря электромагнитному полю, возникающему в любой атомной
системе, осуществляется взаимосвязь составляющих ее
элементарных частиц.
Из фундаментального уравнения А. Эйнштейна следует, что
различные состояния тела с одинаковой энергией соответствуют и
одинаковой массе. Предположим, что телу с массой покоя т0
сообщена энергия Д£, в соответствии с уравнением (1.1) масса тела
увеличивается до значения т, причем изменение массы
т — т0 = А т = .
с
При этом не имеет значения форма сообщенной энергии
(тепловая, механическая, химическая). Если тело потеряет энергию Д£,
передав ее другим телам, то его масса уменьшается и снова станет
равной то. Таким образом, фундаментальное соотношение А.
Эйнштейна следует понимать в том смысле, что изменению энергии
всегда отвечает строгое эквивалентное изменение массы и
наоборот. Но тогда следует ожидать, что закон сохранения массы
должен соблюдаться с такой же строгостью, с какой соблюдается
закон сохранения энергии.
Покажем это на конкретных примерах. При определенных
условиях столкновение ядра атома лития с протоном приводит к
образованию двух а-частиц1:
Li + р= 2а. (1.3)
Если сравнить массу покоя частиц, отвечающих начальному и
конечному состоянию системы, то она окажется неодинаковой: сумма
собственной массы ядра атома Li и протона равна 8,0263 атомных
единиц массы (ат. е. м.), а собственная масса двух а-частиц
составляет 8,0078 ат. е. м. Налицо так называемый «дефицит» мас-
1 В радиохимии а-частицами называют ядра атомов гелия, движущиеся с
большой скоростью.
25
сы, который, однако, не следует трактовать как нарушение закона
сохранения массы. Дело в том, что образующиеся а-частицы.
обладают огромной скоростью (порядка 20 тыс. км/сек), а
следовательно, их действительная масса заметно превышает массу покоя.
Пользуясь уравнением (1.2), можно показать, что масса каждой
частицы, движущейся с указанной выше скоростью, составляет
4,01315 ат. е. м., что соответствует строгому соблюдению
сохранения массы.
Итак, а-частица с массой 4,01315 ат. е. м. может существовать
в момент образования по реакции (1.3), но со временем она
постепенно переходит в состояние относительного покоя, передавая
избыточную массу взаимодействующим с ней телам.
Рассмотрим другой, еще более поразительный пример. При
столкновении электрона с позитроном происходит их аннигиляция,
в том смысле, что исчезают частицы вещества, но образуются
фотоны — частицы электромагнитного поля. И здесь закон
сохранения массы соблюдается безукоризненно, ибо собственная масса,
характеризующая пару «электрон + позитрон», равна массе
движения образующегося фотона.
Наконец, при химических реакциях, сопровождающихся
выделением тепла, образующиеся нагретые продукты обладают той же
массой, какую имели реагенты. Если же продукты реакции
охладить до температуры исходных веществ, то их масса уменьшится
за счет изменения состояния окружающей среды, т. е.
одновременного увеличения массы последней.
Разумеется, что фундаментальное соотношение А. Эйнштейна,
выражающее взаимосвязь массы и. энергии, нельзя трактовать,
как закон сохранения массы и энергии, утверждая, что масса
превращается в энергию. Нельзя отождествлять массу с материей,
заявляя о превращении материи в энергию. Масса не материя, а
свойство материи, мера ее инертности, так же как энергия
является свойством материи — мерой ее движения. И масса, и энергия
неотделимы от материи, но не сводятся друг к другу и не
превращаются одно в другое.
Подводя итог, следует снова подчеркнуть, что закон сохранения
материи и движения — это высшее обобщение человеческого
опыта. Выраженный в форме конкретных естественнонаучных
законов — закона сохранения массы и закона сохранения и
превращения энергии, он не теряет своей всеобщности и остается
абсолютным.
Помимо рассмотренных двух форм существования материи
(вещества и поля) следует упомянуть еще две формы —
антивещество (позитрон, антипротон, антинейтрон, предполагаемые
антиэлементы) и вакуум. Некоторые исследователи придают особое
значение последнему. «Большую часть доступного нашему
наблюдению пространства занимает неуловимое непосредственно нашими
органами чувств... материя в особом, мало еще изученном
состоянии, которое характеризуется обычно термином «вакуум». Мысль о
26
том, что вакуум нельзя рассматривать чисто геометрически как
лишенное всякой материи пространство, высказывалась
неоднократно... Луи де Бройль предвидит возможность использования
вакуума для получения энергии, еще более концентрированной,
чем ядерная; вычисляют даже, что энергии вакуума, взятого в
объеме одного литра, могло бы хватить на покрытие нужд всей
земной промышленности и транспорта в течение миллиона лет.
Некоторые ученые предполагают, что вакуум является той пра-
материей, которая способна в неизвестных нам пока условиях
порождать из себя атомы водорода и более простые, элементарные
частицы...»1.
§ 3. РАЗВИТИЕ АТОМИСТИЧЕСКИХ
ПРЕДСТАВЛЕНИИ В ХИМИИ
Пытаясь объяснить причину многообразия окружающего мира,
великие философы в Древней Греции (Левкипп, Демокрит,
Эпикур), по-видимому, впервые в истории человечества создали учение
об атомах как основе строения материи. Они считали, что мир
состоит из мельчайших, неделимых частиц — атомов,
находящихся в непрерывном движении; реальные свойства тел определяются
многообразием формы, размера и порядка расположения атомов,
которые сами по себе неизменны; любые изменения, происходящие
в природе, это результат соединения или разъединения атомов.
Греческая атомистика как выражение стихийного материализма
сыграла большую роль в истории человечества. Отстаивая
реальность существования атомов, она была глубоко чужда любым
идеалистическим представлениям и религиозным догмам. Не
случайно в период средневековья и алхимии атомистические
воззрения рассматривались как одно из наиболее наказуемых
преступлений. Однако атомистика древних греков не оказала сколько-
нибудь заметного влияния на развитие науки о веществе, так как
она носила чисто абстрактный характер и не опиралась на
фактические знания, которые были получены значительно позже.
После крушения алхимии атомистика возродилась, но уже не
как философское учение, а как наука. Важнейший вклад в
развитие новой химической атомистики внес М. В. Ломоносов (см.
введение). Касаясь существа новой атомистики, Ф. Энгельс писал:
«Новая атомистика отличается от всех прежних тем, что она... не
утверждает, будто материя только дискретна, а признает, что
дискретные части различных ступеней (атомы эфира, химические
.тгомы, массы, небесные тела) являются различными узловыми
точками, которые обусловливают различные качественные формы
существования всеобщей материи...»2.
С развитием количественных методов исследования в химии
пыли накоплены экспериментальные факты, обобщение которых
1 г. А. Щ у к а р е в. Лекции по общему курсу химии. Изд-во ЛГУ, 1962, стр. 5.
К Маркс и Ф. Энгельс Соч, т. 20, стр. 608.
27
привело к открытию так называемых стехиометрических
законов — закона постоянства состава, закона эквивалентов и закона
кратных отношений. Именно эти законы способствовали
окончательному утверждению в химии атомно-молекуляриого учения.
§ 4. ЗАКОН ПОСТОЯНСТВА СОСТАВА
Закон постоянства состава, открытый Прустом в 1801 г.,
формулируется следующим образом:
Каждое химически чистое соединение независимо от способа
его получения имеет вполне определенный состав.
Например, двуокись углерода можно получить по любой из
указанных ниже реакций:
с + о2 = со2,
2СО + 02 = 2С02,
СаС03 = СаО + С02,
NaHC03 + НС1 = NaCl + H20 + С02,
но в химически чистом образце С02 всегда содержится)
27,29 вес.% С и 72,71 «вес.% О. Отклонение от указанного состава
свидетельствует о присутствии примесей.
Таким образом, закон постоянства состава утверждает
количественную определенность каждого химического соединения.
Следует отметить, что обратное утверждение — каждому
определенному составу отвечает только одно химическое соединение —
неверно. Действительно, диметиловый эфир и этиловый спирт
имеют одинаковый химический состав — С2Н4О2, но являются
различными химическими соединениями. Различные вещества с
одинаковым химическим составом называются изомерами. Явление
изомерии особенно характерно для органических соединений,
содержащих большое число атомов углерода в молекуле, и
координационным соединениям.
Таким образом, количественный состав еще не определяет
специфику вещества. Вещество изменяется не только при изменении
состава, но и количества присущей ему энергии. «Все качественные
различия в природе, — писал Ф. Энгельс, — основываются либо на
различном химическом составе, либо на различных количествах
или формах движения (энергии), либо, — что имеет место почти
всегда, — на том и другом» К
Итак, в соответствии с законом постоянства состава
химические вещества существуют только в форме соединений постоянного
состава, называемых химическими индивидами. Учение о
химических индивидах сыграло большую роль в химии, определив
надолго направление ее развития (период препаративной химии).
1 К. Маркс и Ф. Энгельс. Соч., т. 20, стр. 385.
28
Вместе с тем применение закона постоянства состава ко всем
соединениям независимо от их состава и агрегатного состояния, как
мы теперь знаем, было неправомерным (см. § 9). Понадобилось
свыше 100 лет, прежде чем Н. С. Курнаков, исследуя характер
взаимодействия в металлических и силикатных системах, доказал
существование соединений переменного состава с широкой
областью гомогенности.
§ 5. ЗАКОН ЭКВИВАЛЕНТОВ
Изучая весовые соотношения, в которых кислоты соединяются
с основаниями при образовании солей, Рихтер в 1792—1800 гг.
пришел к выводу, получившему название закона эквивалентов:
Химические элементы соединяются друг с другом в строго
определенных весовых соотношениях, или эквивалентах.
Для сопоставления соединительной способности различных
элементов в химии введено понятие об эквивалентном весе.
Эквивалентным весом, или эквивалентом, называют весовое количества
элементов, которое соединяется с 1,008 вес. частей водорода или
8 вес. частями кислорода или замещает эти количества в
соединениях.
Из данных химического анализа следует, что в двуокиси
углерода 12 вес. частей углерода соединены с 32 вес. частями
кислорода. Значит, в двуокиси углерода на 8 вес. частей кислорода
приходится 3 вес. части углерода, т. е. эквивалентный вес
углерода равен трем. Кстати, один и тот же элемент может иметь не
один, а несколько эквивалентных весов. В окиси углерода 12 вес.
частей углерода соединены с 16 вес. частями кислорода, откуда
следует, что химический эквивалент углерода равен 6. Заметьте,,
что эквивалентные веса углерода в различных соединениях
относятся между собой, как простые целые числа (3:6=1:2).
Понятие эквивалента можно распространить и на сложные
соединения типа кислот, солей и оснований.
Эквивалентом, или эквивалентным весом, сложного соединения
называют весовое количество этого соединения, содержащее
1,008 вес. частей водорода (кислоты), или эквивалент
металлической составной части (основания, соли).
Рассмотрим конкретный пример. По данным химического
анализа в ортофосфорной кислоте на каждые 98 вес. частей кислоты
приходится 3,024 вес. частей водорода. Следовательно, эквивалент
ортофосфорной кислоты равен
—^—•1,008 = 32,7.
3,024
Весовое количество простого или сложного вещества, выраженное
в граммах и численно равное его эквиваленту, называется грамм-
эквивалентом.
29
Очевидно, что грамм-эквивалент водорода равен 1,008 г,
кислорода — 8,00 г, ортофосфорной кислоты — 32,7 г.
В химии, особенно аналитической и физической, часто
используют водные растворы солей, кислот и оснований.
Число грамм-эквивалентов вещества, содержащихся в 1 л
раствора, называют его нормальностью.
Говорят о нормальном, децинормальном и сантинормальном
растворах, имея в виду, что в 1 л раствора содержится
соответственно 1; 0,1 или 0,01 г-экв растворенного вещества. Например,
раствор едкого натра, в 1 л которого содержится 4,0 г NaOH,
децинормальный (0,1 N), так как 1 г-экв NaOH весит 40 г.
Очевидно, что в самом общем виде закон эквивалентов следует
формулировать следующим образом:
Во всех химических реакциях взаимодействуют друг с другом
эквивалентные количества различных веществ, независимо от того,
являются ли эти вещества простыми или сложными.
Из этого положения, в частности, следует, что если к раствору
основания с нормальностью N\ прибавлять раствор кислоты,
имеющей нормальность /V2, то полная нейтрализация наступит,
•если
N^-NtV» (1.4)
где V\ и V2 — объемы растворов кислоты и основания
соответственно. Соотношение (1.4) широко используется в аналитической
химии при количественном определении кислот и оснований.
§ 6. ЗАКОН КРАТНЫХ ОТНОШЕНИИ.
АТОМНЫЙ ВЕС ЭЛЕМЕНТОВ
Как уже отмечалось выше, некоторые химические элементы
имеют не один, а два или большее число эквивалентных весов,
которые относятся друг к другу как простые целые числа. Это
соответствует возможности образования указанными элементами
нескольких соединений. Наиболее показательны в этом смысле
окислы азота. По данным химического анализа значения
эквивалентного веса азота составляют в азотном ангидриде — 2,8, в
двуокиси азота — 3,5, в азотистом ангидриде — 4,67, в окиси азота—
7,0 и в закиси азота — 14. Легко убедиться, что значения
эквивалентов азота во всех окислах относятся друг к другу как
простые целые числа:
2,8:3,5:4,67:7: 14 = 1 : 2 : 3 : 4 : 5.
Поскольку же различные элементы соединяются друг с другом в
эквивалентных количествах, можно утверждать, что количество
кислорода, приходящееся на 1 вес. часть азота в окислах азота,
тоже относятся друг к другу как простые целые числа. Эта
закономерность впервые была отмечена Дальтоном в 1803 г. и
получила название закона кратных отношений, который формулируется
следующим образом:
30
Атомные веса химических элементов
Таблица 1.2
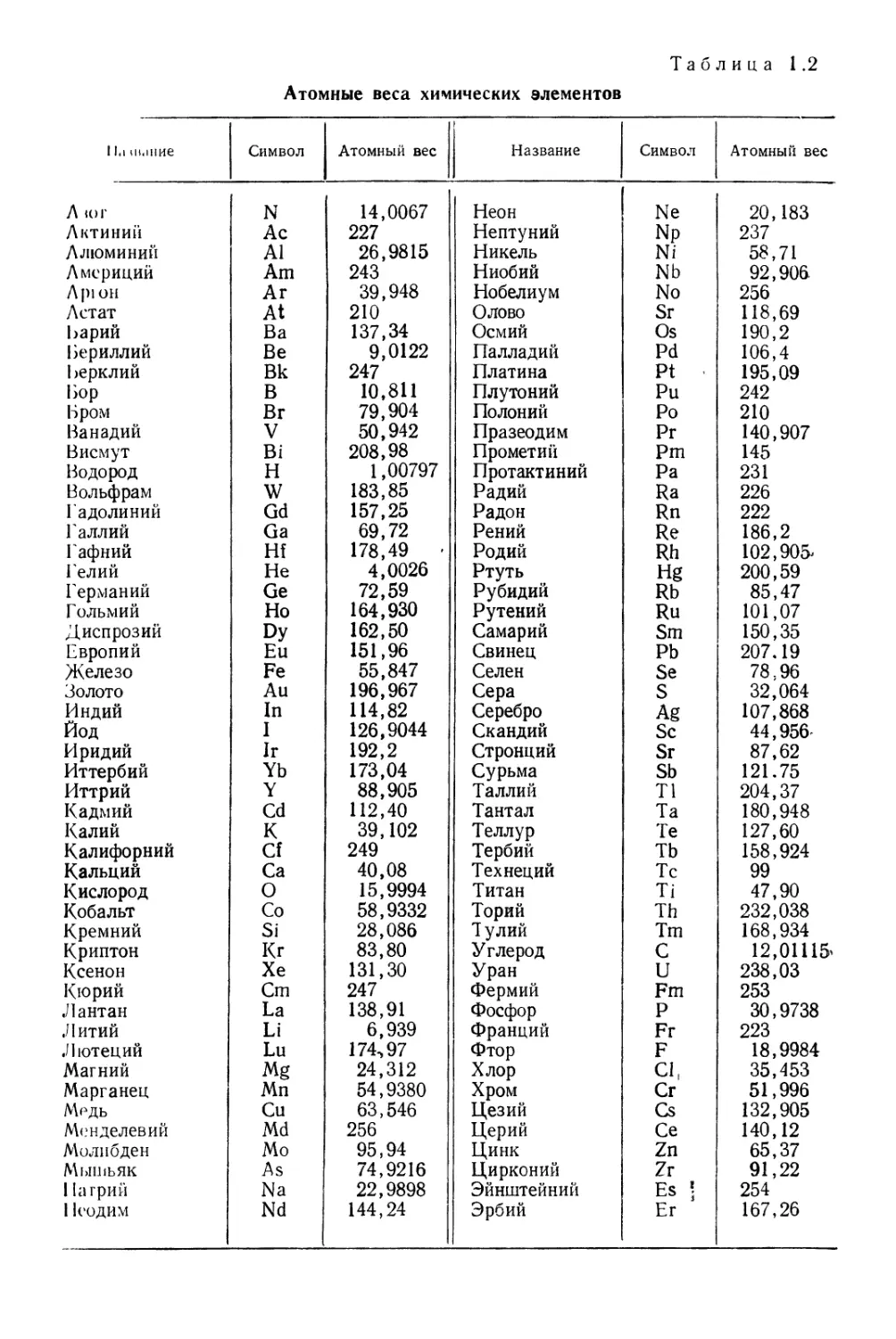
1 la ш.шие
А юг
Актиний
Алюминий
Америций
Артн
Астат
Ьарий
Периллий
Ьерклий
1юр
Ьром
Ванадий
Висмут
Водород
Вольфрам
Гадолиний
Галлий
Гафний
Гелий
Германий
Гольмий
Диспрозий
Европий
Железо
Золото
Индий
Йод
Иридий
Иттербий
Иттрий
Кадмий
Калий
Калифорний
Кальций
Кислород
Кобальт
Кремний
Криптон
Ксенон
Кюрий
Лантан
Литий
Лютеций
Магний
Марганец i
Медь
Менделевий
Молибден
Мышьяк
Пагрий
11 сод им
1
Символ
N
Ас
А1
Am
Аг
At
Ва
Be
Bk
В
Br
V
Bi
H
W
Gd
Ga
Hf
He
Ge
Ho
Dy
Eu
Fe
Au
In
I
Ir
Yb
Y
Cd
К
Cf
Ca
0
Co
Si
Кг
Xe
Cm
La
Li
Lu
Mg
Mn
Cu
Md
Mo
As
Na
Nd
Атомный вес
14,0067
227
1 26,9815
243
39,948
210
137,34
[ 9,0122
247
10,811
79,904
50,942
208,98
1,00797
183,85
157,25
69,72
178,49 •
4,0026
72,59
164,930
162,50
151,96
55,847
196,967
114,82
126,9044
192,2
173,04
88,905
112,40
39,102
249
40,08
15,9994
58,9332
28,086
83,80
131,30
247
138,91
6,939
174,97
24,312
54,9380
63,546
256
95,94
74,9216
22,9898
144,24
Название
Неон
Нептуний
Никель
Ниобий
Нобелиум
Олово
Осмий
Палладий
Платина
Плутоний
Полоний
Празеодим
Прометий
Протактиний
Радий
Радон
Рений
Родий
Ртуть
Рубидий
Рутений
Самарий
Свинец
Селен
Сера
Серебро
Скандий
Стронций
Сурьма
Таллий
Тантал
Теллур
Тербий
Технеций
Титан
Торий
Тулий
[ Углерод
Уран
Фермий
Фосфор
Франций
Фтор
Хлор
Хром
Цезий
Церий
Цинк
Цирконий
Эйнштейний
Эрбий
Символ
Ne
Np
Ni
Nb
No
Sr
Os
Pd
Pt
Pu
Po
Pr
Pm
Pa
Ra
Rn
Re
Rh
Hg
Rb
Ru
Sm
Pb
Se
S
Ag
Sc
Sr
Sb
Tl
Та
Те
Tb
Тс
Ti
Th
Tm
С
и
Fm
P
Fr
F
CI,
Cr
Cs
Ce
Zn
Zr
Es !
Er
Атомный вес
20,183
237
58,71
92,906
256
118,69
190,2
106,4
195,09
242
210
140,907
145
231
226
222
186,2
102,90S
200,59
85,47
101,07
150,35
207.19
78;96
32,064
107,868
44,956-
87,62
121.75
204,37
180,948
127,60
158,924
99
47,90
232,038
168,934
12,01115*
238,03
253
30,9738
223
18,9984
35,453
51,996
132,905
140,12
65,37
91,22
254
167,26
Если два элемента образуют друг с другом несколько
химических соединений, то на одно и то же весовое количество одного из
них приходятся такие количества другого, которые относятся
между собой как простые целые числа.
Дальтон не остановился на открытии закона кратных
отношений, а использовал этот закон и закон постоянства состава для
последовательного обоснования атомистических представлений
химии. Идея дискретности вещества впервые получила
экспериментальное подтверждение. Опираясь на развитые Лавуазье понятия
''химических элементов, Дальтон принял, что все атомы каждого
химического элемента одинаковы и помимо других свойств
обладают строго определенным весом, который он назвал атомным
весом. Считая невозможным определить абсолютный вес атомов,
Дальтон предложил использовать понятие относительного
атомного веса, приняв за единицу вес атома наиболее легкого
элемента — водорода.
Поскольку далеко не все химические элементы непосредственно
соединяются с водородом, то использование водородной единицы
вызывало определенные трудности. Именно этим был продиктован
переход к кислородной единице измерения атомных весов (1850 г.),
численно равной 1/16 веса атома кислорода.
В дальнейшем было обнаружено, что природный кислород
состоит из трех разновидностей, отличающихся друг от друга массой
атомов: 160 (99,759%), 170 (0,037%) и 180 (0,204%). Поэтому
используются две шкалы атомных весов: физическая, в' которой
за единицу была принята 1/16 массы атома 160, и химическая,
в которой за единицу принималась 1/16 средней атомной массы
природного кислорода. Для перехода от одной шкалы к другой
использовалось соотношение
1,000275.
Начиная с 1961 г. в качестве единой была принята углеродная
шкала атомных весов, в которой за единицу принята 1/12 веса
самого легкого изотопа атома углерода 12С. В настоящее время
атомные веса везде даются в углеродных единицах (табл. 1.2).
Итак, атомный вес — это число, которое показывает, во
сколько раз вес данного атома больше 1/12 веса самого легкого изотопа
углерода.
Количество элемента в граммах, численно равное его атомному
весу, называют грамм-атомом.
§ 7. ЗАКОН СОЕДИНЕНИЯ ГАЗОВ
И ЗАКОН АВОГАДРО
Атомное учение Дальтона, несмотря на несомненные
достоинства, имело и серьезные недостатки. Будучи метафизиком, Дальтон
предполагал, что все вещества состоят только из атомов: простые
вещества — из простых атомов, а сложные вещества — из слож-
32
ных атомов, в свою очередь составленных из разнородных
простых. Химические реакции по Дальтону сводились к соединению
простых атомов в сложные или, наоборот, распаду сложных
атомов на простые.
Открытый в 1805 г. Гей-Люссаком закон соединения газов
оказался в противоречии с выводами Дальтона. Согласно этому
закону
Объемы вступающих в реакцию газов относятся друг к другу,
а также к объему получающихся газообразных продуктов как
простые целые числа.
Например, при получении водяного пара из водорода и
кислорода два объема водорода без остатка реагируют с одним
объемом кислорода, образуя два объема водяного пара. Пытаясь
объяснить эти наблюдения, Дальтон предположил, что в равных
объемах различных газов при одинаковых температуре и
давлении содержится одинаковое число атомов (простых или сложных).
Это предположение действительно объясняет, почему два объема
водорода реагируют с одним объемом водорода — по Дальтону
это значит, что один атом водорода соединяется с двумя атомами
водорода. Но при этом должно образоваться столько же сложных
атомов, сколько исчезает простых, т. е. три. Этому должно
соответствовать три объема водяного пара. А в действительности их
образуется только два.
Дли объяснения наблюдавшихся Гей-Люссаком
закономерностей соединения газов оказалось необходимым предположить, что
1) любые газы (в том числе и простые) состоят не из атомов, а
молекул; 2) п равных объемах различных газов при одинаковых
температуре и давлении содержится одинаковое число молекул.
Последнее утверждение, высказанное итальянским ученым
Авогадро в 1811 г., вошло в химию под именем закона Авогадро.
Напомним, что существование молекул как наименьших частиц
вещества, являющихся носителем его свойств, было впервые
предсказано М. В. Ломоносовым. Теперь известно, что число атомов,
составляющих молекулу, может быть различным. Например,
молекулы инертных газов состоят из одного атома, а молекулы
водорода, кислорода, азота, хлора и фтора — из двух атомов.
Кстати, последнее вытекало из экспериментов Гей-Люссака,
согласно которым из одного объема простого газа нельзя получить
более двух объемов газообразных продуктов реакции.
Следовательно, молекула простого газа не может делиться больше, чем на
две части.
Важнейшей количественной характеристикой молекулы
является величина молекулярного веса.
Молекулярный вес — это число, показывающее, во сколько раз
молекула вещества тяжелее 1/12 атома изотопа 12С.
Очевидно, что молекулярный вес химического соединения
равен сумме атомных весов всех атомов, составляющих молекулу
соединения.
£ Общая химия
33
Количество вещества в граммах, численно равное его
молекулярному весу, называется грамм-молекулой, или молем. Важной
физико-химической константой любого вещества, находящегося в
парообразном или газообразном состоянии, является объем грамм-
молекулы при нормальных условиях (0°С и 760 мм рт. ст.).
Из данных эксперимента следует, что в этих условиях 1 л
Н2, 02 и СО весит соответственно 0,09, 1,43 и 1,25 г. Учитывая,
что молекулярный вес Н2, 02 и ССЬ равен 2,016, 32 и 28,01 г
соответственно, легко рассчитать объем 1 г-моль каждого газа:
И„,--i^ll = 22,4 л,
0,09
""—таг ^22'4л-
Аналогичные эксперименты с другими парообразными или
газообразными веществами позволили сделать следующий вывод:
Грамм-молекулярный объем любого вещества, находящегося в
парообразном состоянии, при нормальных условиях равен 22,4 л.
Учитывая же, что в равных объемах различных газов
содержится одинаковое число молекул, можно заключить, что грамм-
молекула любого вещества имеет одинаковое число молекул. Это
число, являющееся фундаментальной константой, равно 6,02-1023
молекул (число Авогадро, обычно обозначаемое символом N).
Число Авогадро одновременно характеризует число атомов в
г-атоме любого элемента.
Число Авогадро настолько велико, что его трудно оценить
обычными мерками. Например, у алюминия атомный вес равен 27,
а удельный вес — 2,7 г/см3. Следовательно, 1 г-атом Ai занимает
объем 10 см3. Учитывая, что предел видимости оптического
микроскопа соответствует долям микрона (1 мк=10-4 см), можно
показать, что минимальная частица А1, которую удается разглядеть
под микроскопом, состоит почти из миллиарда атомов!
Знание числа Авогадро позволяет рассчитать абсолютный вес
атомов и молекул. Например, каждая молекула воды (мол. в. = 18)
1 я
весит — 3-10 23 г. Объем, занимаемый 1 г-молем Н20,
6,02-1023
находящийся в жидком состоянии, легко оценить, зная, что г-моль
Н20 имеет объем 18 см3 (удельный вес воды 1 г/см3). Используя
число Авогадро, находим, что объем 1 молекулы Н20 равен
^—^3-10"23 см3.
6-1023
34
§ 8. УРАВНЕНИЕ КЛАПЕЙРОНА—МЕНДЕЛЕЕВА.
ОПРЕДЕЛЕНИЕ ЧИСЛА АВОГАДРО
Из многочисленных методов определения числа Авогадро
рассмотрим только один, интересный в том отношении, что он убеди-
nvibHo доказывает реальность существования атомов и молекул.
Поскольку при последующих рассуждениях будет использовано
уравнение Клапейрона — Менделеева, начнем с вывода
последнего.
Воспользуемся известными из курса физики законами, описы-
иающими изменения объема газа при изменении температуры или
давления.
1. Согласно закону Бойля — Мариотта произведение объема
газа на его давление есть величина постоянная при неизменных
юмпературе и массе газа, т. е.
P-V = const. (1.5)
2. Согласно закону Гей-Люссака при изменении температуры и
постоянном давлении отношение объема газа к абсолютной
температуре остается постоянным, т. е.
— = const. (1.6)
Напомним, что абсолютной называется температура,
отсчитываемая от абсолютного нуля (0°К), соответствующего — 273,2° С.
Абсолютная температура Т связана с температурой по шкале
Цельсия соотношением
7(°К) = *(°С) + 273,2.
Предположим, что начальное состояние газа, выбранное
произвольно, характеризуется давлением Я, объемом V и температурой
Т. Изменим температуру системы до 0°С, т. е. до Г0 = 273,2°К.
Сели при этом давление газа оставалось постоянным, то его объем
изменится до некоторого значения КоР, которое в соответствии с
уравнением (1.6) выражается соотношением
_L=J^L_, или 1/SP=V.-^-- (1.7)
Изменим теперь давление газа до значения Ро=1атм. Если при
лом температура газа сохранялась постоянной и равной Г0, то его
объем изменился до некоторого значения V0, определяемого
соотношением
Р0У0^РКоТ (1.8)
Исключая из уравнений (1.7) и (1.8) значение VSP, находим
^Л=г, (1.9)
35
где г — некоторая константа. Следовательно, параметры,
характеризующие состояние газа (температура, давление и объем), не
являются независимыми, а связаны соотношением (1.9), которое
называется уравнением Клапейрона. Примечательно, что
значение г является константой лишь для фиксированной массы
данного газа, но изменяется в зависимости от природы газа и его
количества. Если при одинаковых значениях давления и
температуры брать 1 г-моль различных газов, то их объемы тоже
одинаковы, т. е. отношение
-£^- = Д, (1.10)
где Vm — объем г-моля, является константой независимо от
природы газа. Значение R, называемое универсальной газовой
постоянной, легко рассчитать, вспомнив, что при нормальных условиях
(0°С и 760 мм рт. ст.) 1 г-моль любого газа занимает объем 22,4 л:
п_ PV __ 1атм.22,4л = 0)082 атм.л/°К.
Т 273 °К
Уравнение (1.10) можно записать для любого произвольного
количества газа, соответствующего, например, п г-молям. Умножая
обе части уравнения (1.10) на п9 находим
РУщП
nR9 или PVn = nRT, (1.11)
где Vn — объем п г-молей газа. Соотношение (1.11), называемое
обобщенным уравнением Клапейрона — Менделеева, является
основным уравнением атомно-молекулярной теории газов.
Пользуясь этим уравнением, легко показать, что при постоянной
температуре давление газа зависит только от числа частиц в единице
объема.
Пусть п г-молей газа, имеющего молекулярный вес М, весят
Wr и содержат v частиц, каждая из которых весит пг г. Очевидно,
что
n==_W_=jnv_ (1Л2)
мм v '
Так как M = mNy где N — число Авогадро, то из уравнения (1.12)
следует, что
л=—. (1.13)
N
Комбинируя соотношение (1.13) с уравнением Клапейрона —
Менделеева, находим
PVa = -^.RT,
36
II.Ill
P = _^_._*ZL. ' (1.14)
Vn NA
I,ik как при постоянной температуре отношение RT/N является
константой, то можно утверждать, что давление газов
определяется только числом частиц в единице объема. Следовательно,
причину уменьшения атмосферного давления по мере удаления
иверх от земной поверхности следует связывать с уменьшением
числа частиц в единице объема.
Уменьшение числа частиц с высотой выражается выводимым
и курсах физики соотношением
ft= 5'3м107 lgX, (1.15)
м
где h — высота (см), на которой число частиц уменьшается в X
раз при условии, что на всех высотах поддерживается постоянная
1емпература. Из уравнения (1.15) следует, что, например, число
молекул азота уменьшается вдвое на высоте
h= 5'3'10? lg2^0,57-106 см^5,7 км.
28
Нсли представить себе систему, состоящую из огромных молекул
с грамм-молекулярным весом 107, то их число уменьшится вдвое
на высоте всего 1,6 см причем совершенно безразлично,
действительно ли мы имеем совокупность отдельных молекул или их
имитируют частицы, каждая из которых сложена из большого числа
обычных молекул. Как показывает опыт, такие очень малые, но
мпдимые под микроскопом частицы, распределенные в газовой
среде или в жидкости, находятся в непрерывном хаотическом
движении. Это движение открыто английским ботаником Броуном в
1827 г. и получило название броуновского.
Французский физик Ж. Перрен, изучая броуновское движение
микроскопически малых частичек краски гуммигута в воде,
покачал, что в такой эмульсии частички, кажущиеся для
невооруженного глаза плотно осевшими на дно сосуда, в действительности
распределяются по высоте по тому же закону, что и молекулы га-
м\. Прямыми измерениями Перрен обнаружил, что для шариков
гуммигута диаметром 0,5 мк их число уменьшается вдвое на высо-
ю всего 6 мк. Используя эти данные, Перрен по уравнению (1.15)
рассчитал величину М, а, определив экспериментально вес
отдельного шарика, по уравнению (1.15) оценил число Авогадро. Оно
оказалось равным 6,5-1023, что достаточно близко к величине
Г>,02-1023, предсказываемой кинетической теорией и определяемой
экспериментально совершенно независимыми методами. Перрен
писал, что нельзя не удивляться, видя, как согласуются между со-
пой результаты исследований столь различных явлений, и если
шномнить, что одна и та же величина получается в результате
37
варьирования условий и явлений, к которым прилагаются эти
методы, мы придем к заключению, что реальность молекул имеет
вероятность, весьма близкую к достоверности. Независимые
доказательства достоверности атомно-молекулярных представлений
были получены при изучении строения кристаллов с помощью
рентгеновских лучей.
§ 9. СОВРЕМЕННОЕ СОДЕРЖАНИЕ
СТЕХИОМЕТРИЧЕСКИХ ЗАКОНОВ 1
Стехиометрическими законами в химии называют закон
постоянства состава, законы эквивалентов и закон кратных отношений.
Сам термин «стехиометрия» был впервые предложен Рихтером в
1800 г. для выражения соотношения между химическими
элементами, простыми и сложными веществами, вступающими во
взаимодействие друг с другом.
Роль стехиометрических законов в науке исключительно
велика, так как они являются фундаментом, на котором создавалось
современное атомно-молекулярное учение. Примечательно, что в
начале XX в. стехиометрические законы явились предметом
ожесточенных столкновений между идеалистическими и
материалистическими воззрениями в химии. Идеалисты (Оствальд, Вальд и др.)
утверждали что стехиометрические законы лишь умозрительно
:вязываются с атомистической интерпретацией экспериментальных
данных и не являются доказательством реального существования
атомов, что можно придумать и другие объяснения таким фактам,
как целочисленные стехиометрические коэффициенты в химических
формулах и уравнениях. В качестве плацдарма для наступления
на атомистику идеалисты выбрали правило фаз и химическую
термодинамику, которые рассматривают вещество независимо от
его строения и позволяют ограничиться макроскопическими
параметрами системы (температура, давление, концентрация). Однако
идеалисты потерпели поражение. Теперь никто не сомневается в
реальности существования атомов и молекул хотя бы потому, что
современная техника позволяет наблюдать их непосредственно.
Это не означает, что классическая формулировка
стехиометрических законов не нуждается в уточнении. Д. И. Менделеев
обратил внимание на то, что наряду с соединениями, определенными
(к которым «стехиометрические законы применялись до сих пор»)
в природе и в химической практике, имеет место образование
неопределенных соединений (например, сплавов,растворов),«которое
играет столь же существенную роль, что и образование
определенных химических соединений»2.
1 Для глубокого понимания материала, изложенного в § 9, рекомендуется
предварительно познакомиться с элементами термодинамики и химии твердого
тела, изложенными в гл. 5 и 9.
2 Д. И. М е н д е ч е е в. Основы химии, изд. 3. СПб., 1906, стр. 15
38
Исследуя характер химического взаимодействия в
металлических и силикатных системах, Н. С. Курнаков обнаружил
существование многих химических соединений с очень широкой областью
тмогенности, включающей стехиометрический состав или даже
.южащей в стороне от этого состава. Однако авторитет закона по-
сюннства состава был столь велик, что Курнаков объяснял
наблюдаемые явления образованием твердых растворов на основе
с i схиометрических соединений.
Дальнейшее развитие химии твердых тел заставило отказаться
о г такого объяснения и признать существование очень обширного
класса соединений, которые вопреки стехиометрическим законам
имеют переменный состав. К числу соединений с переменным
составом относятся окислы, халькогениды, нитриды, фосфиды, ар-
гппиды, карбиды, силициды и гидриды переходных металлов.
Например, закись титана как индивидуальное соединение
существует не только при соотношении Ti:0=l : 1, но и в широком
интервале составов от ТЮо,7 до TiOi,3. Обратите внимание, что
область гомогенности закиси титана простирается по обе стороны
о г стехиометрического состава, и такие соединения названы
двухсторонними фазами. Рассмотрим другой пример. Закиси железа,
известной в химии и геологии под названием вюстита, формально
приписывают состав FeO. В действительности оказалось, что при
соотношении Fe:0=l : 1 всегда имеют равновесную механическую
смесь железа и вюстита. Однофазный же вюстит существует в
довольно широкой области составов, лежащей в стороне от
стехиометрического, а именно: Fe0,89 О — Feo,95 0.
Для подавляющего большинства кристаллических соединений
отклонения от стехиометрического состава намного меньше, чем
\ закиси титана или железа, однако эти отклонения имеют
принципиальное значение для формирования свойств кристаллов.
Например, прецизионные электрохимические измерения показали, что
закись никеля всегда существует с дефицитом металла, т. е. ее
состав следует выразить формулой Nii_eO. Величина б, зависящая
от условий синтеза, очень мала (6^10~4 при 1000°С), но именно
изменение структуры, обусловленное отклонением от
стехиометрического состава, ответственно за полупроводниковые свойства
закиси никеля.
Образцы сульфида свинца Pbo,999sS и PbSo,9995, являющиеся
однофазными, отличаются не только величиной, но и типом
проводимости, т. е. характером электрических частиц, ответственных за
перенос заряда при наложении внешнего электрического поля.
Таким образом, существует очень обширный класс соединений,
не подчиняющихся стехиометрическим законам. Обратите
внимание, что все эти соединения являются кристаллами, т. е.
нарушения стехиометрических законов связаны с вполне определенным
агрегатным состоянием вещества. Если кристаллы типичного не-
стехиометрического соединения — вюстита — подвергнуть полному
испарению, то в образующейся парообразной фазе обнаруживают
39
строго стехиометрические молекулы FeO и СЬ (соответственно
избытку последнего в вюстите).
Спрашивается, почему химические соединения, находясь в
твердом состоянии, приобретают способность иметь нестехиометриче-
ский состав? Чтобы ответить на этот вопрос, следует напомнить,
что характерной особенностью твердого тела, отличающей его от
жидкости и пара, является наличие кристаллической структуры
с периодическим и строго повторяющимся расположением атомов
или ионов.
Состояние полного структурного порядка отвечает минимуму
внутренней энергии кристалла, тогда как любое нарушение,
связанное с образованием дефектов (несовершенств кристалла),
сопряжено с затратой энергии. Тем не менее дефекты образуются
в кристаллах при любой температуре, отличной от абсолютного
нуля. Это легко понять, если учитывать, что любая система, в том
числе и кристалл, стремится к минимуму свободной энергии, а
последний в соответствии со вторым принципом термодинамики !
может быть достигнут за счет увеличения энтропии, т. е. степени
беспорядка в кристалле. Так как частичная потеря одного из
компонентов кристаллического соединения сопровождается
образованием в решетке точечных дефектов, то нестехиометрическая фаза,
как правило, оказывается термодинамически более устойчивой,
чем стехиометрическая.
Обратите внимание, что нестехиометрия может возникнуть
лишь в немолекулярных кристаллах, т. е. в кристаллах с ионной,
ковалентной или металлической связью. Характерная особенность
таких кристаллов — невозможность выделить отдельные молекулы
в рамках кристаллической структуры. Носителем свойств
соединения в этом случае является не отдельная молекула, а
коллектив атомов или ионов, сильно взаимодействующих друг с другом
и изменяющих взаимно энергетическое состояние. Например, в
кристалле NaCl каждый ион Na+ окружен шестью ионами С\~ и
наоборот, т. е. структурными элементами решетки являются
октаэдры, в центрах которых расположены ионы с одним зарядом, а
в вершинах — ионы с противоположным зарядом. Иногда кристалл
NaCl рассматривают как одну гигантскую молекулу, хотя
подобное сравнение не вполне удачно.
Подводя итог, следует отметить, что современное понимание
стехиометрических законов отличается от традиционного
необходимостью учитывать агрегатное состояние вещества. Так, закон
постоянства состава следует формулировать следующим образом:
Если соединение имеет в данном агрегатном состоянии моле-
кулярную структуру (пар, жидкость или кристалл с
молекулярной связью), то его химический состав (состав молекул) остается
одним и тем же независимо от способа получения.
1 Напомним, что согласно второму закону термодинамики изменение свободной
энергии системы А(7 = АЯ-TAS, где АН — изменение энгалыши, a AS —
изменение энтропии.
40
1:сли же в данном агрегатном состоянии соединение не имеет
молекулярной структуры, то в зависимости от строения атомов,
обусловливающего строение фазы и характер химической связи
и пей, состав соединения и его свойства могут сильно зависеть от
• •иособа синтеза, т. е. могут образовываться соединения
переменного состава.
Закон кратных отношений формулируется следующим образом:
Если два элемента образуют между собой несколько
соединений, имеющих молекулярную структуру, т. е. подчиняющихся за-
1\<>иу постоянства, то весовые количества одного элемента,
соединяющиеся с одним и тем же количеством другого, относятся друг
а ()ругу как небольшие целые числа.
Если же все или только некоторые из этих соединений не
имеют молекулярной структуры, то количества одного элемента,
соединяющиеся с одним и тем же количеством другого, могут
относиться между собой как дробные и изменяющиеся (непостоянные)
числа.
Вопросы и упражнения
1. Пользуясь соотношением (1.2), определите, как изменяется масса тела,
переходящего из состояния движения со скоростью 1000 км/сек в состояние
покоя.
2. Назовите известные вам формы существования материи.
3. Наиболее точное взвешивание можно произвести с точностью до Ю-8 г.
Сколько атомов золота содержится в образце такого веса?
4. Сжатый газообразный кислород находится в стальном баллоне емкостью
'10 л при давлении 130 атм. Сколько весит кислород, заполняющий баллон?
5. Сколько молекул С02 получится при сжигании 1 мг углерода?
6. Какой объем 4,5М H2S04 надо разбавить водой, чтобы получить 100 мл
1,0 М раствора?
7. Рассчитайте объем, занимаемый одним атомом натрия, учитывая, что
удельный вес последнего равен 0,97 г/см3.
8. Предположим, что общее давление в автомобильной шине равно 2,1 атм.
К.чк изменится количество воздуха в шине при увеличении давления до 2,8 атм,
если температура и объем шины остаются постоянными?
9. Почему, характеризуя кристалл NaCl, обычно предпочитают использовать
нмгето термина «молекулярный вес» термин «формульный вес»?
10. Считая молекулярный в€с воздуха равным 29, рассчитайте массу 1 л
шндуха при 0°С на вершине Эвереста (высота 8882 м).
ГЛАВА 2
СТРОЕНИЕ АТОМА
§ 1. СЛОЖНОСТЬ СТРУКТУРЫ АТОМА
В основе атОхМно-молекулярной теории строения вещества лежит
представление о возможности разделения физического тела на
малые части, каждая из которых сохраняет химические свойства
тела. Эти части были названы молекулами.
Предполагалось, что молекулы сложного состава можно
разделить на части, называемые атомами. Атомы в химических
реакциях не изменяются. Если допустить существование движущихся
атомов и молекул, то можно объяснить многие наблюдаемые на
опыте свойства физических тел. При этом считалось, что опытные
факты можно истолковать, считая атомы и молекулы точечными
телами. Однако в конце XIX — начале XX в. были открыты
явления, заставляющие сделать предположение о сложности строения
атомов и молекул.
Изучение прохождения электрического тока через газы и
растворы позволило обнаружить одну из составных частей атома и
молекулы — электрон. Исследование катодных лучей показало, что
они являются потоком частиц с массой 9,1-Ю-28 г и зарядом
4,8-10~10 CQSE. Эти частицы были названы электронами.
Оказалось, что электроны удаляются из любых материалов катода, что
указывает на их содержание в любых атомах.
При изучении прохождения электрического тока через
растворы электролита были открыты законы электролиза и введено
представление о существовании положительно и отрицательно
заряженных атомов или групп атомов — катионов и анионов. При
этом оказалось, что минимальный заряд иона совпадает с зарядом
электрона и равен 4,8-КН0 CGSE.
В 1896 г. А. Беккерель открыл явление радиоактивности урана;
среди частиц, образующихся при распаде урана, также оказались
электроны.
Эти факты привели к мысли о том, что атом можно
представить как сложное образование, состоящее из электронов и
области, несущей положительный заряд. Возник вопрос об
относительном расположении электронов и положительно заряженной
остальной части атома. Для решения этого вопроса были применены
эксперименты по рассеянию электронов и а-частиц при
прохождении их через металлическую фольгу. Опыты показали, что
рассеивание электронов очень невелико. На этом основании В. Томсон
и Д. Д. Томсон в 1904 г. предложили модель строения атома.
Атом представляет положительно заряженный шар, в котором
42
икраплены отрицательно заряженные электроны. При
прохождении пучка электронов через атом искривление их траектории
происходит за счет сил отталкивания, действующих со стороны
атомных электронов.
В 1904 г. японский физик X. Нагаока предложил иную модель
сi роения атома: атом имеет строение, напоминающее строение
планеты Сатурн с ее кольцами спутников. X. Нагаока предположил,
•но основная часть атома — положительно заряженный шар,
вокруг которого по кольцевым орбитам вращаются электроны. X. На-
шока показал, что эта модель механически устойчива, но отметил,
чю согласно электромагнитным явлениям электроны должны бы-
|ц бы излучать электромагнитные волны, теряя кинетическую
энергию и уменьшая свою скорость, пока не упадут на ядро.
§ 2. МОДЕЛИ СТРОЕНИЯ АТОМА.
ЯДЕРНАЯ МОДЕЛЬ Э. РЕЗЕРФОРДА
В 1906—1911 гг. в лаборатории Э. Резерфорда было изучено
прохождение через фольгу дважды ионизированных атомов ге-
лия — а-частиц. Оказалось, что и а-частицы изменяют свою
траекторию, но меньше, чем электроны. Следовательно, область
положительного заряда атома очень невелика по размерам, что
противоречило модели В. Томсона и Д. Д. Томсона. Э. Резерфорд
использовал для объяснения этого явления планетарную модель
атома Нагаоки. Однако он предположил в отличие от Нагаоки,
что масса атома почти целиком сосредоточена в области
положительного заряда. Эта область была названа ядром атома. При
расчетах изменения траектории а-частиц предполагалось, что
заряд ядра сосредоточен в точке. Рассчитанное Резерфордом
распределение частиц по углам отклонения совпадало с опытным.
Ядерная модель атома предложена Э. Резерфордом в 1911 г. Но
модель Резерфорда все же не могла объяснить факта
устойчивости атома.
В дальнейшем изучение строения атома развивалось в двух
им правлениях — исследования атомного ядра и электронной
оболочки атома. Первая количественная теория простейшего атома
иодорода была разработана Н. Бором в 1913 г.
§ 3. АТОМНЫЕ СПЕКТРЫ.
СПЕКТР АТОМА ВОДОРОДА
К началу XX в. физика накопила множество фактов о спект-
рмх химических элементов. В 1885 г. И. И. Бальмер нашел простое
соотношение между длинами волн в эмиссионном спектре атомар-
о водорода. Эмиссионным спектром называется спектр, полу-
чмгмый при пропускании излучения одноатомного водорода через
пи'ктрограф.
43
На рис. 2.1 изображены положения линий спектра водорода.
При переходе от видимой части спектра к ультрафиолетовой
длина волны уменьшается. Если наиболее длинной красной линии в
спектре приписать номер 3, а более коротким линиям номера 4,
Красный Голубой Фиолетовый
Н
I с*
На
1 I 1 I I 1 I I I 1 1 1 I I 1 1 I I I I 1 1 1 I I I I I I I I I I I I i э
7000 6500 6000 5500 5000 4500 4000 3500А
Рис. 2.1. Видимый спектр водорода
5, ..., то длину волны л каждой линии можно рассчитать по
простой формуле Бальмера
X = 3645 —-— (в ангстремах, 1А - 10~8 см), (2. i)
п2 — 4
где п — порядковый номер линии. В 1888 г. К. Рунге переписал
эту формулу, введя величину, обратную длине волны, — волновое
число vn = . В 1890 г. И. Ридберг предложил описывать
эмиссионный спектр водорода формулой
где /г = 3, 4, ..., a R (постоянная Ридберга) = 1Ы0б м-1.
Таким образом, волновое число можно представить в виде раз-
ности двух величин — и —. Это привело к предположению,
что излучение света- происходит при переходе атома из одного
состояния в другое, причем каждое состояние можно обозначить
числом, квадрат которого входит в формулу Ридберга. Но еще в
1900 г. М. Планком было показано, что следует рассматривать
электромагнитные излучения как состоящие из отдельных
прерывистых порций — квантов, пропорциональных частоте колебаний
E—hv, где h — так называемый элементарный квант действия.
Волновое число линии можно связать с энергией,
соответствующей этой линии E = hcv, где с — скорость света (~3108 м/сек),
h = 6,6-Ю-34 дж-сек.
Если умножить левую и правую часть формулы на he,
получим
Е---1^ *£*-. (2.3)
п 22 л3
44
Таким образом, энергия фотона, соответствующая данной линии
спектра, представляет разность двух энергетических состояний
атома Е\ и Е2:
Д£ =Е1 — Е2. (2.4)
Линейчатость атомных спектров связана со скачкообразным
изменением энергетического состояния атомов. Излучение атомов
серия
Рис. 2.2. Схема происхождения Рис 2.3. Строение атома
водородного спектра водорода по Бору
связано с движением электронов. Но по законам классической
электродинамики движущийся электрон должен излучать
энергию непрерывно. При этом спектр должен быть сплошным, в нем
должны присутствовать волны всех длин, отвечающих интервалу
от минимума к максимуму энергии электрона (рис. 2.2). Но
наблюдаемый на опыте спектр оказывается линейчатым, а не
сплошным.
Н. Бор в 1913 г. применил квантовые представления к теории
строения атома водорода.
§ 4. ТЕОРИЯ СТРОЕНИЯ
АТОМА ВОДОРОДА ПО БОРУ
В 1913 г. Н. Бор предложил модель атома водорода, исходя
из гипотезы Резерфорда, дополненной новыми положениями:
1. Электрон может вращаться по определенным круговым
орбитам, не излучая энергии.
2. Ближайшая к ядру орбита отвечает нормальному, наиболее
устойчивому состоянию атома.
3. Поглощение и излучение атомом энергии происходит лишь
при перескоке электрона с одной орбиты на другую.
45
По теории Бора, электрон в атоме водорода движется вокруг
протона по замкнутой круговой орбите (рис. 2.3); т(,— масса
электрона, тр — масса протона, г — радиус орбиты, v — линейная
скорость движения электрона.
Чтобы орбита была устойчивой, центробежная сила —е^—
г
должна быть равна центростремительной силе
электростатического взаимодействия е2/г2:
-^- - —, или mev* - —. (2.5)
г г2 г
Полная энергия электрона равна сумме кинетической и
потенциальной энергии
E=T + V; T= -^L; К = ——. (2.6)
Следовательно,
E^jneVL_I1 (2
2 JK '
Подставляя mev2 = —, получим
Если найти радиус орбиты г, то можно будет вычислить энергию.
е2
В соответствии с уравнением Е = — эта энергия будет
принимать значения от £ = 0 при г-+оо до Е = оо при г->0.
Бор предположил, что момент количества движения mevr
может принимать только дискретные квантованные значения.
.Момент количества движения является величиной, кратной
единичному кванту h/2n:
2jc
где п= 1, 2, 3,... — ряд целых чисел.
Тогда
v = п . (2.9)
2лтег
е2
Подставляя v в условие устойчивости орбиты тр1 = —, найдем
г
mP-n2-h2 P2 n2h2 /0 1/чч
или г = . (2.10)
4n2m2e-r2 r * 4л2тРе2
46
»го уравнение позволяет определить радиусы дозволенных
электронных орбит в атоме водорода. Каждая такая орбита
характеризуется величиной энергии
Е =
2л2тее*
n2h?
(2.11)
300 Е
250
• н, н. н„ н^
200
150
100
50
Л-©о
Л-6
п-5
п«4
п-3
ШХ„,
Представления Бора позволяют установить, что радиусы
дозволенных орбит электронов относятся друг к другу, как I2: 22:
,vl о т-r п2-П2
• З2 : ... : п2. При подстановке в уравнение г = —-—— значения
я=1 и значений А, те, е получается радиус ближайшей к ядру
орбиты г = 0,53 А. Можно вычислить скорость £,ккал
вращения электрона на этой орбите, она
оказывается равной 2,19-108 см/сек.
Спектр атома водорода. Состояние
атома с минимальной энергией называется
основным состоянием. Из уравнения
„ — 2п2тее*
Е = видно, что основное со-
n2h2
стояние отвечает квантовому числу п=\.
Состояния с /2 = 2, 3,... менее устойчивы и
называются возбужденными. При
сообщении электрону энергии он может перейти с
уровня fz=l на уровень с другим значением
п. Наоборот, возвращение электрона с
высшего энергетического уровня в основное
состояние с я=1 будет сопровождаться
выделением энергии. Эта энергия может быть
световой энергией, величина которой
пропорциональна частоте излучения E = h\\
где h — постоянная Планка; v — частота.
Частоту излучения можно регистрировать
при фотоматериалах. Линии, обнаруженные
на излучении, составляют эмиссионный
спектр.
,n-f
Рис 2.4. Уровни энергии атома водорода
(ккал/г-атом)
Если сообщить атому водорода достаточно большую энергию,
то происходит его ионизация с удалением электрона: 313 ккал+
I Н = Н+ + £ (на г-атом Н). Энергия, которую надо затратить для
лого процесса, отвечает д = оо, т. е. полному удалению электрона,
и называется энергией ионизации.
Можно вычислить энергию ионизации, исходя из теории Бора.
II соответствии с уравнением Планка Е = hv = hR
47
В основном состоянии п=\, а т ----- оо. Таким образом,
— " Радиус первой дозволенной орбиты г =
/i2 4л;2т^е2
= я т<е . Тогда £ = е2/2 -- 2,18- К)-11 эрг. Обычно энергию
2г Л2
ионизации выражают в электрон-вольтах, 1 эрг~6,242- 10~и эв.
Тогда энергия ионизации атома водорода £'=13,60 эв.
Экспериментальное значение энергии ионизации составляет 13,595 эв, что
очень хорошо согласуется с вычисленным по теории Бора. На рис.
2.4 приведены уровни энергии водорода в видимой области.
Модель атома водорода Бора количественно описывала спектр
атомного водорода и других водородоподобных атомов, т. е.
атомов, содержащих единственный электрон. Для количественного
расчета водородоподобных атомов, содержащих один электрон,
надо учесть заряд ядра Ze. Тогда центростремительная сила равна
n2h2
Ze2Ir2, а радиус орбиты равен —. Энергия связи элек-
трона с ядром заряда определяется выражением
Е= -2n*mee*Z> (2 12
§ 5. РАЗВИТИЕ ТЕОРИИ БОРА
По мере усовершенствования аппаратуры, применяемой в
спектральном анализе, выяснилось, что единичные линии в действи-
Рис. 2.5. Эллиптическая орбита
тельности состоят из совокупности линий, близко расположенных
друг к другу. Отсюда был сделан вывод, что для каждого
квантового числа существует несколько энергетических уровней.
Возникшее затруднение было преодолено Зоммерфельдом (1916 г.),
рассмотревшим существование эллиптических орбит, по которым
может двигаться электрон. Для круговых орбит единственной
изменяющейся координатой является угол вращения ф (рис. 2.5). Для
эллиптических орбит меняется как радиус-вектор, так и угол ф. Та-
48
кпм образом, электрон получает две степени свободы, которые
обусловливают существование двух квантовых чисел.
В теории Бора квантовое условие имело вид
/2
mvr -= п .
2л
Зоммерфельд обобщил это квантовое условие и для второй
переменной— радиуса-вектора. Величина малой полуоси эллипса
определяется побочным квантовым числом ky для большой полуоси
эллипса действительно соотношение а = п2гу а для малой полуоси
b = n-k-r, где г — радиус орбиты при нормальном состоянии атома
водорода 0,53 А. Побочное квантовое число может принимать
значения последовательных целых чисел, но не может быть
больше главного.
Таким образом, каждому энергетическому уровню с главным
квантовым числом п отвечает несколько подуровней. Наинизшие
подуровни отвечают наиболее вытянутым орбитам. Они
заполняются в первую очередь электронами при построении нового слоя
в многоэлектронных атомах. Электронные слои с одним
значением главного квантового числа обозначаются буквами
К L М N О Р Q
/1 = 1 23 4 567
Существует небольшое различие между энергиями круговой и
эллиптической орбит. Это различие объясняет расщепление главного
уровня энергии на подуровни, близкорасположенные друг к другу.
В дальнейшем было обнаружено, что под действием магнитного
поля спектральные линии расщепляются еще больше. Это —
эффект Зеемана. Он требует (введения еще одного — магнитного —
квантового числа т.
Поскольку для описания поведения электрона в трехмерном
пространстве нужны три кооординаты, то им соответствуют три
степени свободы электрона и три квантовых числа. При наличии
магнитного поля орбитальная плоскость электрона прецессирует
вокруг направления поля. Третье квантовое условие имеет вид
р = т- —. Но даже с этими тремя квантовыми числами невоз-
2л
можно было полностью объяснить спектры многоэлектронных
атомов, например атома гелия. Новый подход к проблеме строения
атомов возник с появлением квантовой волновой механики.
§ 6. НАЧАЛА ВОЛНОВОЙ МЕХАНИКИ.
УРАВНЕНИЕ ШРЕДИНГЕРА
Открытия физики конца XIX — начала XX в. привели к
выводу о том, что свет, который раньше рассматривался как пример
нолнового процесса, можно рассматривать как совокупность ча-
гтиц (фотонов), обладающих механическими характеристиками
49
движения — массой, энергией, импульсом. В 1900 г. М. Планком
было предположено, что процесс излучения атомами световой
энергии происходит не непрерывно, а порциями, квантами, величина
которых зависит от частоты испускаемого света:
E = hv. (2.13)
В 1905 г. А. Эйнштейн показал, что масса тела связана с его
энергией соотношением Е=тс2. Уравнения Планка и Эйнштейна
дают возможность получить
соотношение между массой фотона и длиной
волны света, или частотой световых
колебаний, E = hv = mc2; hv = mc = p
(р — импульс фотона).
В 1924 г. Луи де Бройль
предположил, что корпускулярно-волновая
природа присуща не только свету, но
и любым другим частицам. При этом
длина волны частицы
Ь= — --- —. (2.14)
/пи р
Рис. 2 6. Стоячая электронная волна
с п = Ъ
Уравнение К-= — позволяет определить длину электронной
Р
волны. Для того чтобы электрон, двигаясь по круговой орбите в
атоме водорода, образовал стоячую волну, на длине окружности
2яг должно укладываться целое число волн:
iik = 2nr. (2.15)
Подставляя %=—, получим
Р
п = тог. (2.16)
2т
Это момент количества движения электрона. Таким образом,
волны де Бройля теоретически обосновывают постулат Бора о
квантовании момента количества движения (рис. 2.6).
Длину волны де Бройля можно рассчитать для любой частицы
с известной массой и скоростью. Для электрона с энергией 1,6Х
ХЮ-10 эрг Я= 1,2 А. Эта величина соответствует параметрам
кристаллических решеток.
В 1927 г. Девиссон и Джермер обнаружили, что при
отражении электронов от монокристалла никеля возникает
дифракционная картина, сходная с дифракцией света на кристаллической ре-
50
шетке. Это явление получило название дифракции электронов.
Оно считается экспериментальным доказательством
существования волн де Бройля или корпускулярно-волнового дуализма
электронов. Позже волновые свойства были обнаружены для
нейтронов, атомов водорода и гелия,
В волновых свойствах электрона заложен один из двух
основных принципов волновой механики. Вторым является принцип
неопределенности Гейзенберга (1925 г.).
В механике наблюдение волнового процесса — периодического
колебания сплошной среды — позволяет определить длину волны
и скорость ее распространения. Чем больше область пространства,
it котором распространяется волна, тем точнее можно определить
тш характеристики. Иначе обстоит дело при переходе к объектам
микромира. Гейзенберг (1925 г.) показал, что точное определение
положения электрона на орбите и его скорость не могут быть
зафиксированы. Произведение неопределенностей положения и
скорости никогда не может быть меньше, чем
Ax.&vx>—, (2.16')
где h — постоянная Планка; те — масса электрона.
Представление об атоме с определенными орбитами электронов
заменяется в волновой механике представлением о вероятном
нахождении электрона в той или иной части атома. Оценка этой
вероятности производится при решении уравнения Шредингера,
описывающего движение электрона. Это уравнение может быть
получено следующим образом.
Смещение среды при одномерном волновом движении представляется в ви-
дг уравнения волны
я|з = fl-sin 2я ("7" — vf), (2.17).
|дс а — амплитуда волны; % — длина волны; v — частота; х— положение
/лемента среды; t — момент времени. Смещение яр является функцией х и U
Uiki/кды продифференцировав по х, получим
д2Ц -4я2г|)
■!*=—- (2Л8>
И случае распространения волны в 3-мерном пространстве
а2я|) д2^ а2г|з ' 4я2г|)
"dxT + ~d^+^~dF =~ X2 (2Л9>
к in
4л2г|)
h
i иг у2 — оператор Лапласа. Запишем это уравнение для волн де Бройля Х = .
то
51
Для свободного электрона v = l/ . , где Т — кинетическая энергия. Под-
m
/г2
•ставим v в уравнение к = ; X2 — , тогда
то 2тТ
8л2пгТ
V2^ + " ^ ^=0- (2-21)
Это и есть уравнение Шредингера.
Если электрон движется в потенциальном поле ядра, то получаем
уравнение Шредингера для стационарного состояния
8л2ш (Е — V)
Vя* + ^ ~ =0. (2.22)
Здесь V — потенциал ядра.
При получении этого уравнения использованы выражения, отражающие
корпускулярно-волновой характер электрона.
Физический смысл ф-функции. Физический
смысл ^-функции был предложен М. Борном.
Он предположил, что вероятность нахождения
электрона пропорциональна квадрату
амплитуды волны де Бройля, т. е. величине г|). В том
случае, если волновая функция электрона
выражается комплексным числом i = у/'—1,
то вероятность пребывания электрона в
данной точке пропорциональна квадрату модуля
комплексного числа, т. е. |i|)|2. Далее будем
обозначать вероятность нахождения электрона
без учета возможности комплексного значения
ф-функции, т. е. просто как ф2. Для линейного
у=©о
У=0
■1
Рис. 2.7. Одномерный потен- <*2Ф — 4я2ч|>
циальный ящик Уравнения — = у функция ф пред-
ставляет величину, пропорциональную
амплитуде волны. Из физики известно, что энергия
колебательного движения пропорциональна квадрату амплитуды волны
а2
E = k- —.
2
В случае 3-мерной волны полная энергия тоже пропорциональна квадрату ам-
ллитуды. Теория фотонов связывает пространственную плотность энергии
фотонов с объемной плотностью фотонов как частиц.
Объемная плотность частиц пропорциональна вероятности нахождения
фотонов в данном объеме. Таким образом, вероятность нахождения фотона
пропорциональна квадрату амплитуды волны де Бройля, т. е. величине ф2.
Величина I ф2д(т, где dx— элемент объема dx=dx-dy-dz, является мерой
вероятности нахождения электрона в элементе объема dx, окружающем точку с
координатами (х, у, z), например ядро атома.
Решения уравнения Шредингера могут иметь смысл только для
определенных конечных значений длины волны. На каждом конце г|)(л;)=0, так как в
этих точках система остается фиксированной. Функция ф(я) должна быть
конечной и однозначной.
Квантовомеханическое решение задач в теории атомов и молекул сводится
к нахождению функции я|з(я) и полной энергии Е. В качестве простого
примера приведем решение уравнения Шредингера для электрона в потенциальном
,52
ящике с прямоугольными стенками (рис. 2.7). Положим следующую
зависимого потенциальной энергии от х: при х=0 и х = а У=оо и V=0 при
о л*<а. Это означает, что электрон находится в таком ящике в связанном
иимоянии и не может выйти за его пределы, так как для этого пришлось бы
преодолеть бесконечно большой потенциальный барьер. В этом смысле электрон
п некоторой степени напоминает электрон в атоме, где он также находится
и i вязанном состоянии.
Уравнение Шредингера
d2^ 8л2тЕ
-&-+—»-*-0- <2-23>
Функция
я|)= bs'm l/
8л2тЕ
■*ф (2.24)
h2
удовлетворяет этому уравнению, что проверяется дифференцированием
d2ty 8л2тЕ
dx2 h2
У стенок ящика (0, а) значение г|) = 0. Поэтому
Ч>. (2.25)
, / 8л2тЕ
i|)=6sinl/ — -а = 0, (2.26)
что может быть только при условии, если
/:
8л2тЕ
h2
или
8л2тЕ п2л
-а = п-л,
(2.27)
h2 a2
дде п — целое число. Таким образом получается условие квантования энергии
n2h2
£=——-. (2.28)
8а2т v ;
.! 1рп различных значениях п получим различные значения энергии электрона в
ящике, т. е. набор дискретных уровней энергии. Картина поведения электрона
шизка к той, которая следует из модели Бора, т. е. объясняет необходимость
введения квантовых чисел и дискретных энергетических уровней и согласуется
*• экспериментально наблюдаемыми спектрами атомов.
Уравнение Шредингера может быть решено точно для простых систем
типа атома водорода Для многоэлектронных атомов его решение представляет
очень сложную задачу. Из решения волнового уравнения получаются три кван-
твых числа, которые необходимы для описания поведения электрона.
Обобщая задачу на случай 3-мерного потенциального прямоугольного
ящика, имеем
nl п2
(2.29)
Ь2 с2 Г
«до ах, Ьу и cz ограничивают размеры ящика в пространстве в направлениях
'ii'ofi координат х, у, г, целые числа пХ} пу и nz можно рассматривать как три
53
n2h2
8a*m
Я =
P2
2m
2a
n
h2
2mk2
квантовых числа, которые определяют значения энергии электрона внутри
ящика. При различных п получим набор дискретных уровней энергии электрона
внутри ящика
Если перейти от энергии к длине волны, то оказывается, что электрону в
потенциальном ящике отвечают определенные дискретные длины волн де Бройля
(2.30)
оитп zm zrrtA,-
откуда
2а
(2.31)
Квантовое число п может принимать значения 1, 2, 3... Значение п = 0
исключается, так как при этом Я становится равной бесконечности, а Е равняется
нулю, что не соответствует условию задачи и отвечает отсутствию частицы
в ящике.
Рассмотренный квантовомеханический подход к решению задачи о
возможных значениях энергии электрона в связанном состоянии, например в атоме,
объясняет наличие дискретных энергетических уровней и согласуется с
экспериментально наблюдаемыми линейчатыми спектрами атомов.
Проведем сопоставление представлений классической и волновой механики
для рассмотренного случая. С точки зрения классической механики энергия
частицы может быть равна нулю, с точки зрения волновой квантовой механики
такое состояние запрещено. Классическая механика приводит к выводу о
равной вероятности нахождения частицы в любой точке на оси х в одномерном
потенциальном ящике. Квантовая механика связывает вероятность нахождения
частицы с ее энергией и квантовым числом. Действительно, из уравнения
можно определить
г|)= b-sin( I /
I / ~Ъ~
ф2 = b2 sin2 I I/ —-
h2
/ Sn2mE
х] (2.32)
8л2тЕ
-х\, (2.33)
где ф2 характеризует вероятность обнаружения частицы в ящике. Чем выше
n2h2
квантовое число я, тем выше энергия электрона Е = -——— и тем резче
изменяется вероятность его обнаружения в различных точках ящика. При
значении /г=1 в некоторых точках вероятность обнаружения электрона равна
нулю. На рис. 2.8 приведены 'ф-функция и г|Я-вероятность нахождения
частицы в одномерном потенциальном ящике для разных значений. Видно, например,
а
что при п = 2 для значения л* = — вероятность г|)2 равна нулю.
При больших значениях энергии Е с большим значением массы частицы
картина движения частицы начинает приближаться к классической. Квантовые
уровни энергии сближаются и образуют сплошную полосу, так что их
невозможно различить. Можно считать, что в таком случае частица обладает любой
энергией. Подобное соотношение между явлениями микро- и макромира было
сформулировано Н. Бором в виде принципа соответствия: Переход от квантовой
механики к классической совершается при больших значениях квантового
числа, когда уровни энергии сближаются между собой.
Следует отметить также, что в квантовой механике понятие траектория
частицы теряет смысл вследствие соотношения неопределенностей. Если
зафиксировать положение электрона в какой-то момент времени, то оно становится
совершенно неопределенным в следующий момент времени. Таким образом, не
существует возможности следить в отдельности за каждой из одинаковых час-
54
тиц и тем самым различать их. Поэтому квантовая механика не может делать
•определенных предсказаний относительно будущего поведения электрона.
Задача квантовой механики состоит лишь в определении вероятности результатов
при последующем измерении.
Уравнение Шредингера для основного состояния атома водорода. Чтобы
уяснить принципы решения уравнения Шредингера, рассмотрим решение для
наиболее простой задачи — атома водорода.
Рис. 2.8. г|)-функцня и вероятность |-ф2| нахождения
частицы в потенциальном ящике
Из общих физических соображений можно полагать, что возможной
волновой функцией электрона атома водорода будет экспоненциальная функция
типа
1|з = е
(2.34)
где имеется один параметр г — расстояние от ядра атома Приемлемы
значения с>0. С увеличением г функция стремится к нулю, что отвечает малой
вероятности обнаружения электрона на больших расстояниях от ядра атома.
Составим уравнение Шредингера для атома водорода. Потенциальная энергия
и этом случае
V=-
е*
г
13 сферической системе координат
г = у хг + у2 + z2.
(2.35)
(2.36)
Лайдем лапласиан у2я|э. По правилу дифференцирования сложных функций имеем
^|/. .= г|)^. г^. Отсюда
г'х = [(х*+у2+г*)2Ух = -£(** + У*+г2) 2 *2x^-J- (2-37)
55
Датеэ
Таким образ)м,
Вторая производная
\Ь" = — с
^ = — се
-сг|?.
\рх = — сг|з
(2.38)
(2.39)
**•'—'*■*
•л -г
л* л*
— сф. — • г — — • гЬ
г г
— ctyx2 — — \f> -|- ri|)
: СУ\)
f.l-
* +
ел-2 + — —
(2.40)
Очевидно, что \|) и \|э2 по форме подобны (2.40) и для полного лапласиана можно
записать
V2* = ^ + % + \|?z - —г-
— r+cz2 + —— г
ел-2 + — — г + q/2
2с \
= I са — 1 ф.
11
Используя запись уравнения в виде /?ф= £\|), получим
/г2 / о 2с \ в2
- —— с2- U — * = £*.
8к2т \ г J г
(2.41)
(2.42)
Решение этого уравнения позволит найти разрешенные значения энергии
электрона и плотность электронного облака на различных расстояниях от ядра.
Эту задачу можно решить вариационным методом
Е(с) =
\ ^4%
c2k2
8л2т
— е*с.
Найдем минимум этого выражения и приведем его к нулю:
дЕ ch2
-=. — е2.
дс 4я2/п
Отсюда
4к2те2
h2
f2.43>
с2.44)
(2.45)
Подставив с в уравнение (2.43), найдем минимальное значение энергии для
основного состояния атома водорода
Е -■
16л4/тгУ/г2
8я2т/14
4 л2те*
Л2
• 2л2/7264
h2
(2.46)
56
Выражение для с равно обратному значению первого борорского радиуса с — —,
а0
откуда
4я2тг2
Построим график зависимости энергии от расстояния
Е{с) =
с2/г2,
8я2ш
При с - 0 и £ = 0. В расчеты войдет величина
с= —. В табл. 2.1 приведены числовые значения.
а0
h2
8л2т
-=6,07-i0~24 и
с
и единицах
1/flo
0
0,25
0,50
1,00
1,50
2,00
3,00
Т
(кинетическая
энергия), эрг
0
1,36.10-12
5,42-10-12
21,7.10-i2
49,0.10-12
87-10-12
196-10-12
У
(потенциальная
энергия), эрг
0
—10,9.10-12
-21,7-10-12
-43,4-10-12
—65 • 10-12
—87 • 10-12
—130 -10-12
Таблица 2.1
Общая энергия
T + V = E
Е, эрг
0
—9,54-10-12
-16,3-10-12
—21,7-10-i2
-16,0-10-12
0,000
6,6-10-i2
Е, эв
0
—5,95
—10,2
—13,6
— 10,0
0,000
41
Построенный график Е(с) имеет минимум в точке ——с, которой
отвело
чает максимальная энергия связи электрона с ядром, 13,6 эв. Устойчивым
состояниям атома водорода отвечает область значений с до 2с, где С\ = 2 в
единицах \/а°. Выше этого значения кинетическая энергия превышает по модулю
потенциальную энергию и полная энергия системы окажется положительной.
Это означает, что при c'>2ci электрон не будет находиться в связанном
состоянии.
Отметим, что, когда решалась задача движения электрона в прямоугольном
потенциальном ящике с V=0, получались только положительные значения
кинетической энергии
__ _ п2/г2
Гкин = Епош '~ 8та* '
Теперь можно построить графики функций if — е~г^а° и г|)2 = е~~2г^а°,
которые дадут более полное представление об особенностях плотности
вероятности пребывания электрона в атоме водорода. Но предварительно надо
наложить на волновую функцию еще одно ограничение.
Нормирование волновой функции. Вероятность нахождения электрона во
нсем пространстве, окружающем ядро атома, равна единице. Это условие для
одномерного потенциального ящика записывается в виде
[ ЦЧх = 1,
а для
57
атома водорода I ty2dx — 1, где dx — dx-dy-dz— элемент объема. Это
выражение применяется тогда, когда надо найти полный вид волновой функции.
плх
Постоянная 4 в уравнении гр = л sin называется нормирующим
множителем, его величина должна быть такой, чтобы
г
' урЧх = 1.
г
Можно показать, что нормирующий множитель равен (ла^) " , тогда
оо
наша функция будет удовлетворять условию I 4nr2ty2dr -- 1, что легко про-
6
веряется интегрированием Таким образом, точное выражение волновой фхикции
атома водорода без учета спина
* - (К)
(2.48)
Используем (2 48) для графических построений я|э и г|з2.
Графическое представление волновой
функции и распределения электронной
плотности атома водорода. Вид волновой функции
определен (2.48). Плотность вероятности
г|>2 =- (;иф
-2г/а0
(2.49)
Рис. 2.9. Зависимость
энергии £, волновой функции
г|), плотности вероятности
г|)2 и радиального
распределения плотности D
атома водорода от расстояния
в единицах а0
Представив г в единицах а0, получим
показанные на рис. 2 9 зависимости г|: и г|?2 от
расстояния электрона от ядра. Видно, что при
г>2а0 вероятность нахождения электрона
очень мала.
Особый интерес представляет вычисление
радиальной плотности, т. е. вероятности
нахождения электрона в шаровом слое на
определенном расстоянии от ядра. Радиальную
плотность D можно определить по
соотношению
D = 4яг2а|>2 = 4а^д гЧ~
(2.50)
Здесь 4яг2 — площадь сферы с радиусом г.
Продифференцировав D по г и приравняв производную пулю, легко
убедиться, что расстоянию г=ао = 0,53А отвечает максимум радиального
распределения D. Можно сказать, что вероятность найти электрон на данном
расстоянии от протона имеет максимальное значение, когда это расстояние равно
первому боровскому радиусу 0,53>А.
58
Действительно,
— = 8uq-3 re~2r/a° — 8a30 — e"2r/a° - 0, (2.51)
ОТ CXq
или г = a0 = 0,53A. (2.52)
В то же время, хохя D быстро убывает при больших значениях г, существует
вероятность найти электрон далеко от ядра, в той области, где преобладает
вклад кинетической энергии, всегда положительной по знаку.
Площадь, ограниченная кривой радиальной плотности D и осью абсцисс,
равна полному электронному заряду — единице — при г^оо. Рис. 2.9 наглядно
иллюстрирует вероятностный характер наших знаний, если речь идет о
пространственном распределении электронного заряда в атоме водорода.
Заканчивая на этом описание волновой функции атома водорода,
упомянем, что полная волновая функция содержит еще спиновый множитель, обоз-
h h
начаемый а, если спин s = + ——, и 6, если s=—■ . Основное
2л ' 2я
состояние атома водорода может быть описано одной из двух волновых функций
— _L _ j__
-ф = (яа§) 2 е-г/а°а или я|э = (шф 2 е~г/а°$.
§ 7. КВАНТОВЫЕ ЧИСЛА.
СПИН ЭЛЕКТРОНА. ПРИНЦИП ПАУЛИ
Решение задачи одномерного потенциального ящика приводит
к появлению квантового числа, связанного с энергией электрона.
Поскольку движение электрона происходит в 3-мерном
пространстве, то наличие трех степеней свободы движения приводит в 3-
мерной задаче к появлению трех квантовых чисел. Они
обозначаются буквами п, I и т. Из квантовомеханических представлений
вытекает, что эти числа могут принимать следующие значения:
п = 1, 2, 3, ... , сю
/ =0, 1, 2 (71—1),
т=(0; ±1, ±2, ... , ±/).
Эти квантовые числа характеризуют движение электрона в любом
атоме.
Квантовые числа п и / определяют функцию радиального
распределения вероятности пребывания электрона в атоме. На рис.
2.10 показаны графики этих функций для атома водорода. Видно,
что представление об электронной орбите заменено
представлением о вероятности пребывания электрона в различных областях
пространства. Современным представлениям отвечает понятие об
электронном облаке, плотность которого определяется
вероятностью пребывания электрона. В литературе в настоящее время
пользуются понятием «орбиталь» для обозначения совокупности
положений электрона в атоме.
59
Величина квантового числа п обозначается цифрами, а
значения / буквами по схеме
/012345
5 р d f g h
Запись Is1 читается «один эс один» и означает, что в атоме есть
электрон, у которого п=\ и / = 0. Запись 2р2 обозначает, что
рассматриваются два электрона с /г = 2 и 1=1.
Квантовые числа я, /, т дают
представление о пространственном
распределении электронного облака
и отражают особенности движения
электрона.
Квантовое число п равно числу
узловых поверхностей атомной ор-
битали. Узловой поверхностью
называется геометрическое место
точек, для которых г|) = 0 и г|)2=0.
S - ордиталь
а
Рис. 2 10 а — граничная поверхность s-
орбитали; б — зависимость радиальной
части волновой функции R(r) от /• для
Is-, 2s- и Зя-орбиталей
Узловые поверхности могут быть сферами, центр которых
совпадает с ядром и плоскостями, проходящими через ядро. Число
плоских узловых поверхностей равно I, их число не превышает
(п—1), так как одна из узловых поверхностей лежит бесконечно
далеко от ядра и является сферической. Таким образом,
0^/<(п—1).
Значение главного квантового числа определяет энергию
электрона так же, как и в теории Бора. Квантовое число / определяет
величину орбитального момента количества движения электрона:
м = itVl(l' 1)в
60
z
2
узел
5
Рис. 2.11. а — граничные поверхности /7-орбиталей; б — зависимость
радиальной части волновой функции от п для 2р- и 3/7-орбиталей. Зр-орбиталь
имеет один узел
Направление вектора момента количества движения определяется
квантовым числом га. Это направление задается проекцией
вектора на какую-либо ось. Следует отметить, что нельзя найти все
три проекции, так как в этом случае будет известно положение
электрона в атоме, а это противоречит принципу
неопределенности. Проекция момента количества движения может принимать
квантованные значения
61
м9
2л
т.
Число т называется магнитным квантовым числом.
На рис. 2.10—2.12 показаны формы электронных облаков для
различных состояний электрона в атоме.
,«.. .• ...■.
^&г;*-
^ -ордитоль
d - Ор$италь
d2 2~ор8италь
Рис. 2.12. а —граничные поверхности rf-орбиталей; б — зависимость
радиальной части волновой функции R(r) от г для 3</-орбитали
Помимо рассмотренных трех квантовых чисел, отвечающих
трем степеням свободы электрона, он имеет еще одну, четвертую
степень свободы, вращаясь в разных направлениях вокруг
собственной оси. Это движение называется спином, оно обусловливает
наличие у электрона собственного момента количества движения.
62
Проекция собственного момента количества движения может иметь
, \ h \ h n
только два значения -1 ; ■ . Спиновое квантовое чис-
2 2л 2 2я
, 1 1
ло s может иметь только два значения А и , отвечающие
2 2
различным направлениям вращения электрона.
Квантовые числа п, I, m и s полностью характеризуют
движение электрона.
В многоэлектронном атоме не может быть двух электронов, у
которых все четыре квантовых числа были бы одинаковы. Этот
принцип носит название запрета Паули. По существу принцип
Паули отражает невозможность одновременного пребывания в
одной точке пространства двух электронов.
§ 8. МНОГОЭЛЕКТРОННЫЕ АТОМЫ.
ПРАВИЛО ХУНДА
Уравнение Шредингера точно решается для одноэлектронных
атомов и ионов (например, Не+, Li2+) и молекулярного иона
водорода Нг • В многоэлектронных атомах электрон движется в поле
ядра и в поле других электронов. Состояние электронов в таких
атомах описывается методом одноэлектронного приближения. Суть
метода заключается в следующем. При описании, например,
атома Li рассматривают состояние электрона в ионе Li2+, г. е. в
системе из ядра атома с зарядом 3+ и одного электрона с зарядом
1~. Затем заселяют возможные состояния электрона в атоме Li
последовательно так, чтобы при добавлении каждого очередного
электрона энергия атома оставалась минимальной. Таким
образом, получают представление об электронных структурах атомов.
Значение характера распределения электронов позволяет
объяснить свойства атомов и понять сущность периодического закона.
В многоэлектронных атомах электрон движется в поле других
электронов, это приводит к тому, что энергия электрона зависит не
только от значения главного квантового числа п, но и от
побочного (орбитального) квантового числа L Энергия возрастает с
ростом п и /. Чем больше электронов находится в многоэлектронном
атоме, тем сильнее влияние / на энергию. Для валентного
внешнего электрона атома натрия разность энергий для состояний п=3,
1=0 и л=3, 1=1, т. е. для состояний 3 s и 3 /?, равна 2,1 эв. Эта
разница довольно близка к разности энергий 3 s- и 4 s-состояния,
составляющей 3,1 эв. При фиксированном значении п энергия
электрона растет с увеличением значения /, т. е. по ряду ns, tip, nd.
Энергия состояния в атоме с данным значением п определяет его
энергетический уровень. Энергия состояний в атоме на данном
значении / определяет его энергетический подуровень.
Энергетические подуровни вырождены, т. е. являются энергетически
равноценными в отсутствие электрического или магнитного поля. При
63
наличии поля вырождение энергии снимается за счет
неодинаковых направлений вектора момента импульса.
Последовательность энергетических уровней в порядке
возрастания следующая:
1 s < 2s < 2р < 3s < Зр < 4s ^ 3d <
< 4р< 5s ^ 4d< 5р< 6s ^ Ы ^ 4р< 6р.
Принцип Паули ограничивает число электронов в атоме,
обладающих определенным значением п. Найдем эти числа для
я=1 и л = 2. При п=\ 1--=0 и т = 0. Следовательно, электроны с
л=1 могут различаться только по значениям спиновых квантовых
чисел. На основании этого в атоме может быть только 2
электрона при л=1, обладающих следующими значениями квантовых
чисел:
п I m s
1-й электрон 10 0-1
2-й электрон 10 0
Для п = 2 можно составить 8 комбинаций квантовых чисел:
п I m s
2 0
2 0
2 1
2 1
2 1
2 1
2 1
2 1
Вообще, максимальное число электронов в атоме, которые имеют
главное квантовое число п, равно 2/г2. Величина п определяет
среднее расстояние электронов от ядра, поэтому такую
совокупность электронов называют электронным слоем. Слои
обозначаются буквами
п= 1 2 3 4 5 6 7
К L М N О Р Q
Совокупность электронов с одинаковым значением п называется
электронной оболочкой. Емкость оболочки 2 (2/+1). Говорят об
s-, р- или d-оболочке. Иногда электронный слой именуют
оболочкой, а состояние с определенным значением орбитального
квантового числа /—подоболочкой.
0 + 1/2
0 - 1/2
0 +1/2
0 —1/2
1 +1/2
1 —1/2
-1 -1-1/2
— 1 —Г/2
64
Заполнение s-, p-, d-оболочек происходит таким образом,
чтобы суммарный спин атома был максимальным. Это положение
называется правилом Хунда. Так, для атома азота конфигурации
ls22s22pA отвечают два варианта
n s
2[М_
; \й_
Р s
нчн
2[+1
'ЕЕ
р
м| Ч 1
Анализ спектров и квантовая механика показывают, что
заполнение квантовых ячеек происходит таким образом, что вначале
электроны заполняют ячейки, различающиеся по магнитным
квантовым числам, и лишь затем в ячейках появляются спаренные
электроны. Условно направление спина электрона в ячейке обозначают
стрелкой, направленной вверх или вниз. В одной ячейке в
соответствии с принципом Паули могут находиться только 2
электрона с противоположно направленными спинами.
§ 9. ЭНЕРГИЯ ИОНИЗАЦИИ АТОМА
И СРОДСТВО К ЭЛЕКТРОНУ
Способность атома удерживать электроны является важной
характеристикой его химической активности. Энергией ионизации
/ называется энергия, которую надо затратить, чтобы оторвать
электрон от атома в нормальном состоянии. Энергия ионизации
определяется из спектральных данных.
Для многоэлектронных атомов существует несколько энергий
ионизации, отвечающих отрыву 1-го, 2-го, i-того электронов, при
этом /i</2... </{.
В табл. 2.2 приведены значения
энергии ионизации в электрон-вольтах
для некоторых атомов. Наименьшее
значение энергии ионизации имеют
щелочные металлы, сравнительно
легко отдающие внешний электрон. При
переходе от одного значения Л к
другому /2 для данного элемента
наблюдается резкий скачок энергии
ионизации; это связано с ростом заряда
образующегося положительного иона,
сильно притягивающего электроны.
Следует отметить, что для
понимания энергетических эффектов
химических реакций существенны первые
потенциалы ионизации. Именно они,
Таблица 2.2
Энергия (потенциал) ионизации
для разных ступеней ионизации
Элемент
н -
Не
Li
Be
В
С
N
О
F
Ne
Л. эв
13,6
24,6
5,4
9,3
8,3
11,4
14,5
13,6
17,4
21,6
/2, эв
54
76
18,2
25,1
24,8
29,6
35,2
35,0
40,9
/я. эв
122
154
38
48
47
55
63
64
/4,эв
217
259
65
77
77
87
96
<э Общая химия
65
имеющие порядок величины 5-МО эв, соизмеримы с тепловыми
эффектами реакций.
Важной энергетической характеристикой атома элемента
является способность притягивать свободные электроны. Энергия
притяжения положительным ядром свободного электрона
превышает энергию отталкивания лишнего электрона от электронной
оболочки атома. Таким образом, присоединение электрона
нейтральным атомом приводит к образованию отрицательного иона.
Энергия отрыва Е электрона от такого иона называется сродством
атома к электрону.
Устойчивы лишь однозарядные отрицательные ионы.
Присоединение большего числа электронов к атому термодинамически
невыгодно. Одноатомных многозарядных отрицательных ионов (02~,
N3- и т. п.) в действительности нет ни в свободном состоянии, ни
в молекулах и кристаллах.
Таблица 2.3
Сродство атомов элементов к электрону
Атом
н
Не
Li
Be
В
С
Е, эв
0,75
0,19
0,82 !
—0,19 1
0,33
1,24
Атом
N
О
F
Ne
Е, эв
0,05
1,47 1
3,58
-0,57
Атом
Na
м*
А1
Si
Р
S
Е, эв
0,47
—0,32
0,52
1,46
0,77
2,33
Атом
1
С1
Вг
I
i
Е, эв
3,81
3,56
3,29
В табл. 2.3 приводятся значения электронного сродства для
атомов. Они отвечают тепловому эффекту реакции Э-->Э+е,
выраженному в электрон-вольтах. Обозначения — термодинамические.
Понятия энергии ионизации атома и сродства атома к
электрону могут быть использованы для объяснения одного из типов
химической связи — ионной связи. Пусть в химическую реакцию
вступают атомы А и В, химическая связь осуществляется за счет
перемещения электрона от одного атома к другому. Если электрон
переходит от А к В, то этот процесс связан с выделением энергии
Ев—/а, где Ев — электронное сродство атома В и /а — энергия-
ионизации атома А. При обратном процессе выделялась бы
энергия ЕА — /в. Если электрон перешел от А к В, то образовались
два иона А+ и В". Следовательно,
Ев — /д ^> Еа — /в»
т. е. процесс идет в сторону выделения энергии.
Это неравенство можно записать иначе:
/в + £в>/а-|-£л.
66
Неличина % = называется электроотрицательностью атома.
Электрон смещается к атому того элемента, который имеет
большую величину электроотрицательности. Можно сказать, что элек-
i роотрицательность отражает способность атома к присоединению
электрона при образовании химической связи.
Электроотрицательность атома элемента не постоянная
величина. Она зависит от того, какими атомами в соединении окружен
рассматриваемый атом. Электроотрицательность хлора
неодинакова для свободного атома хлора, для хлора в галогенидах NaCl,
РС13, TiCl4 и т. п. Все же это понятие полезно для трактовки
свойств химической связи во многих соединениях.
Вопросы и упражнения
1. Каким соотношением определяется энергия электронного перехода?
2. Сформулируйте принцип неопределенности Гейзенберга.
3. Вычислите потенциал ионизации атома водорода.
4. Вычислите энергию, поглощаемую атомом водорода при переходе из
состояния с п=\ в состояние п—2.
5. Определите длину волны электрона, имеющего скорость 3-Ю4 м/сек.
6. Объясните резкий скачок величины потенциала ионизации при переходе
эт 4-го к 5-му потенциалу ионизации атома С.
Потенциал ионизации, эв Ix L /3 /4 /5
11,2 22~,4 48 64 392
7. Объясните понижение потенциала ионизации благородных газов с
увеличением порядкового номера.
8. Содержат ли атомы С, Na и Р электроны /С- и АГ-слоев?
9. Какой подуровень заполняется в атомах сначала: 4s или 3d?
10. Определить по формулам высших кислородных соединений номер
группы, в которой находится элемент: ЭОг, Э205, Э203, ЭО4.
3*
ГЛАВА 3
ПЕРИОДИЧЕСКИЙ ЗАКОН
И ПЕРИОДИЧЕСКАЯ СИСТЕМА
ЭЛЕМЕНТОВ Д. И. МЕНДЕЛЕЕВА
§ 1. ПОНЯТИЕ О ХИМИЧЕСКОМ ЭЛЕМЕНТЕ.
КЛАССИФИКАЦИЯ ЭЛЕМЕНТОВ ДО МЕНДЕЛЕЕВА
Одним из основных понятий химии является понятие «химический
элемент». Долгое время не делалось различия между элементом
и простым веществом. Понятие «элемент» в качестве научного
термина впервые использовано Р. Бойлем в 1661 г.
Со времен Бойля элементом считали всякое простое вещество.,
которое можно получить в результате разложения сложных
веществ, но которое не способно к дальнейшему разложению на еще
более простые вещества.
Таких элементов к 1G94 г было ишесгно 14 (Ли; Fe, Sb; P, Ag; Hg, As.
Си; S; Pb; С; Bi; Sn; Zn).
Введение количественных методов исследования великим р>сскпм \ченым
М. В. Ломоносовым позволило определять состав химических соединений, что-
привело к открытию еще ряда элементов.
К концу XVIII в. было известно 25 элементов, а в первой половине XIX в
был открыт еще 31 элемент (Се; Ra; Pd; Os; Ir; Na; Ca; Sr; Ba; Mg; I; Cd;
Se; Li, Si; Th, Al; Zr; La; Be; F; Ti; Y; Та; V; Mo; K; B; Br; Ru; U).
Начало XIX в. ознаменовалось открытием новых
количественных закономерностей. Разработка атомно-молекулярной теории
позволила Дальтону высказать атомную гипотезу и ввести в
химию понятие об относительном атомном весе элементов и
определить атомные веса некоторых элементов. Таким образом,
качественные своеобразия химических элементов начинают связывать с
количественными — относительным атомным весом.
Атомистическая гипотеза в том виде, как она была дана
Дальтоном, не могла внести ничего нового в первое определение
элемента. По Дальтону, элемент можно определить как вид атомов,
характеризующихся определенным значением атомного веса. По
представлению Дальтона, простые вещества всегда состоят из
определенного вида атомов, следовательно, простые вещества и есть
элементы.
Путаница была устранена позже, когда было установлено, что
многие простые вещества образованы из молекул, а не из атомов.
Впервые Менделеев в связи с этим указал на необходимость
ясно различать два понятия: элемент и простое вещество, или
простое тело. «Простое тело, — по Менделееву, — это вещество,
металл или металлоид, с рядом физических признаков и химических
6^
реакций. Ему свойствен частичный вес, содержащий один (как Hg
или Cd, а вероятно, и многие другие простые тела) или несколько
(S6, S2, 02, Н2, С12, Р4 и т. д.) атомов. Оно способно являться в
изомерных и полимерных формах (02 и Оз, S2 и S3) и отличается
от сложных тел только тем, что в простом теле все атомы
однородны. Под именем элементов должно подразумевать те
материальные составные части простых и сложных тел, которые придают
им известную совокупность физических и химических свойств.
Если простому телу соответствует понятие о частице, то элементу
отвечает понятие об атоме. Углерод есть элемент, а уголь, графит и
алмаз суть тела простые» 1.
Следовательно, по Менделееву, химический элемент есть вид
атомов, входящих в состав простых и сложных тел,
характеризующийся определенным значением атомного веса.
Пользуясь понятием о химических элементах, можно сказать,
что важнейшая задача химии состоит в изучении свойств
элементов, в отыскании общих закономерностей в их поведении и в
отношениях между собой.
К середине XIX в. насчитывалось уже 63 элемента и был
накоплен достаточно богатый экспериментальный материал,
касающийся их физических и химических свойств. Был хорошо известен
ряд качественных признаков, характеризующих элементы,
например, их способность образовывать основные или кислотные
окислы.
При этом наряду с индивидуальными свойствами элементов
были установлены и общие групповые свойства. Так, способность
проявлять резко выраженные основные свойства оказалась
присуща группе элементов, которые были названы щелочными
металлами, и в несколько меньшей степени — группе щелочноземельных
металлов, а способность проявления кислотных свойств — группе
элементов, названных галогенами. Наряду с качественными
характеристиками свойств были накоплены сведения о свойствах,
которые в отличие от первых подлежат точному измерению, и среди
них атомный вес элементов и их валентность, т. е. способность
образовывать различные формы соединений.
Итак, открытие новых элементов и изучение свойств
элементов и их соединений, с одной стороны, позволили накопить
большой фактический материал, с другой, — выявили необходимость
его систематизации. Прежде всего нужно было решить основной
вопрос: являются ли химические элементы разрозненными,
независимыми или они закономерно связаны между собой в единую
систему.
Первые попытки решения этой задачи относятся к первой половине XIX в.
Так, Доберейнер (1829 г.) предложил некоторые элементы сгруппировать в
тройки, или триады, сходственных элементов, причем атомный вес среднего члена
1 Д И. Менделеев Основы химии, т 1. М.—Л, Гостехиздат, 1947, стр. 38.
69
триады довольно точно выражается средним из атомных весов крайних членов.
Такие триады сходственных элементов составляют, например, С1; В; I; К; Rb;
Cs; Ca; Sr; Ba; S; Se; Те Делались попытки подойти к решению и общей
задачи. Так, Одлинг (1857 г.) размещал 48 элементов в единую таблицу из
13 групп сходственных элементов; де Шаркунтуа (1863 г.) предложил систему
элементов, где каждые 16 элементов составляли группу; известна произвольная
и примитивная попытка Ньюлендса распределить все известные в то время
(1865 г.) 63 элемента приблизительно в порядке возрастания их атомных весов,
а также таблица элементов, опубликованная немецким химиком Л. Мейером
(1864 г.), где были использованы канницаровские атомные веса с указанием
разности атомных весов двух соседних элементов, причем в таблице
отсутствовали элементы водород, бор и алюминий. Периодичность изменения свойств
элементов из таблицы Л Мейера не вытекала.
Здесь приведены только наиболее известные попытки классификации. Всего
их было не менее пятидесяти. Все они по существу были безуспешными.
При обобщении всех доменделеевских попыток систематизации
элементов возникает вопрос: почему же никому из ученых не
удалось построить естественной системы элементов, отражающей
закон развития материи, хотя многие из них и были близки к
этому? Дело в том, что они не шли далее констатации аналогии и
закономерностей лишь между явно сходными элементами в
отдельных рядах, сами же эти ряды не были между собой связаны в
единое целое —систему. Никому из ученых не приходила
естественная мысль, что должно быть некоторое сходство и между,
казалось бы, несходными элементами (например, между натрием и
хлором) как ступенями развития единой материи. В метафизичном
мышлении этих ученых и заключалась причина их неудач.
Основной причиной успеха труда Д. И. Менделеева по систематизации
элементов является его диалектико-материалистический по
существу подход.
На языке диалектики естественная систематика должна
означать раскрытые взаимосвязи между единичным (элемент) и
всеобщим (система) через особенное (группа). Решение этой задачи
оказалось под силу только такому гению, каким был Менделеев.
Оно состояло в открытии периодического закона и разработке
периодической системы элементов.
§ 2. РАБОТЫ Д. И. МЕНДЕЛЕЕВА
Жизнь и деятельность Д. И Менделеева. Дмитрий Иванович Менделеев
родился 8 февраля 1834 г. в г. Тобольске. Образование Д. И. Менделеев получил
в Тобольской гимназии, а затем в Петербургском педагогическом институте
(1850—1855 гг.). С 1857 г. Д. И. Менделеев доцент, а с 1866 г. — профессор
Петербургского университета, где читал курсы лекций по теоретической и
органической химии
В 1890 г. после 25 лет преподавания Д. И. Менделеев вынужден был
покинуть Петербургский уиивсрешег
В 1898 г'Д И. Менделеев был назначен управляющим Главной палаты
мер и весов, преобразованной после Октябрьской революции в Институт
метрологии.
70
Труды Д. И Менделеева получили широкое признание среди ученых мира,
mi был избран членом ряда академий наук и научных обществ и почетным
профессором ряда университетов.
Литературное наследие Д. И. Менделеева огромно. Оно содержит большое
количество работ в области химии, физикохимии, физики, геофизики, техники,
промышленности и сельского хозяйства, экономических и общественных проблем
■К7' с "*""■"
Д. И. Менделеев (1834—1907)
Следует отметить особо издание Д. И. Менделеевым учебного руководства
«Основы химии», первый выпуск которого вышел в 1868 г. При жизни
Д. И. Менделеева вышло 8 изданий.
Открытие Д. И. Менделеевым периодического закона и создание на его
основе периодической системы химических элементов является главным научным
достижением и на сегодня этот фундаментальный закон естествознания —
основа всех химических открытий.
В 1969 г. наша страна и вся мировая общественность торжественно
отметили 100-летие периодического закона химических элементов Этому событию был
посвящен X юбилейный Менделеевский съезд, который еще раз подчеркнул
значение периодического закона как основного закона природы
71
Периодический закон
Периодический закон был открыт при поисках классификации
химических элементов. Систематикой химических элементов
Д. И. Менделеев стал заниматься с начала 60-х годов XIX
столетия, в связи с его научной и педагогической работой, а также в
связи с подготовкой учебника «Основы химии».
Менделеев избирает за основу систематики элементов двойной
критерий: атомный вес и химические свойства. Атомный вес как
выразитель массы атома, как основной решающий признак
элемента. Атомный вес — рабочее средство систематизации, давшее
ему возможность превратить хаос накопленных химией фактов и
понятий в стройную систему — фундамент всего грандиозного
здания современной химии. Уже в этом выборе ясно сказалась
материалистичность научного мышления Менделеева, ибо масса —
неотъемлемое свойство материи. Выбор этой величины как основы
систематизации Менделеев обосновывал так: «Учитывая, что
масса вещества (атомный вес) по смыслу всех точных сведений о
явлениях природы есть именно такое свойство, от которого должны
зависеть все прочие свойства ... свойство, остающееся постоянным
при всех переменах с телом и при всех его соединениях,
присущее элементу, т. е. ни углю, ни алмазу, ни графиту, а углероду. Я
написал на карточках названия всех известных элементов и их
главные свойства и, расположив их в порядке возрастания
величины атомного веса, подметил повторяемость свойств» 1.
Материалистичность научного мышления Д. И. Менделеева и
на сей раз позволила ему найти правильный подход к анализу
полученного ряда элементов.
Никто до Д. И. Менделеева при попытках классифицировать
элементы не использовал основной диалектической
закономерности— единства противоположностей, т. е. не исследовал
взаимосвязи групп несходных элементов, которая приводит к раскрытию
всеобщего, к естественной систематике элементов.
Кажется, что путь, избранный Менделеевым, невозможно
осуществить. Сопоставить (сличить) несходственные элементы
возможно, имея перед собой непрерывный ряд элементов,
расположенных по величине атомных весов. Но получить необходимый
для решения поставленной задачи полный непрерывный ряд было
невозможно по двум причинам: во-первых, не все элементы этого
ряда были известны к началу работы (1868 г.), поэтому в ряду
должны быть оставлены пустые места для них; во-вторых,
атомные веса многих элементов были определены не точно, что также
могло привести к неправильному расположению элементов в этом
ряду. Только гений Менделеева позволил ему, руководствуясь
величинами атомных весов и химическими свойствами, расположить
элементы в правильный ряд, оставив пустые места, и расположить
некоторые элементы не по возрастанию его, а по близости
химических свойств.
1 Д. И. Менделеев. Периодический закон. М., Изд-во АН СССР, 1958, стр.4.
72
Для осуществления решающей фазы познания периодического
икона Д. И. Менделееву предстояло изучить взаимосвязь между
жтми группами элементов, т. е. раскрыть диалектику перехода от
особенного» (группа элементов) к «всеобщему» (система
элемента). В первую очередь следовало сопоставить группы несходных
•лементов, резко отличающихся своими химическими свойствами,
i. е. группы галогенов и щелочных металлов.
Это сопоставление позволило Менделееву установить, что свой-
ппл элементов, а потому и свойства образуемых ими простых и
«■ложных тел стоят в периодической зависимости от их атомных
иегов. Это и есть формулировка периодического закона, или
закона периодичности, как его называл Менделеев. Оказывается, что
и формы соединений находятся в периодической зависимости от
.1 томных весов элементов. В связи с этим Менделеев дополняет
формулировку закона: свойства простых тел, а также формы и
свойства соединений элементов находятся в периодической
зависимости от величины атомных весов элементов. Следовательно,
исходным пунктом открытия периодического закона явилось
сличение противоположностей, ранее разорванных между собой, а
теперь приведенных к единству, причем основой для этого
послужила близость (сходство) значений их атомного веса.
Уже здесь обнаружилось, что единство полярных
противоположностей (щелочных металлов и галогенов) связано со
своеобразием проявления таких категорий, как качество и количество:
в качественном (химическом) отношении названные элементы
представляют собой полярные противоположности, напротив, в
количественном отношении (по величине их массы) они могут быть
сближены непосредственно друг с другом (аргон, стоящий между
калием и хлором, в то время был неизвестен).
Следовательно, основной закон диалектики проявился здесь и
как прямое сближение противоположностей среди самих
элементов и как установление единства противоположностей двух
сторон у каждого из элементов — качественной (химизм) и
количественной (массы).
Д. И. Менделеев раскрывает содержание периодического
закона следующим образом ]:
1. «Элементы, расположенные по величине их атомного веса,
представляют явственную периодичность свойств».
2. «Величина атомного веса определяет характер элемента, как
величина частицы определяет свойства сложного тела». «Оттого,
например, соединения S и Те и С1 и т. п. при сходстве
представляют и различия весьма ясные».
.'}. «Должно ожидать открытия еще многих неизвестных про-
егых тел, например, сходных с А1 и Si элементов с паем 65—75».
А. «Величина атомного веса элемента иногда может быть
исправлена, зная его аналогии».
1 Д II. Менделеев. Периодический закон, стр. 400.
73
5. «Некоторые аналогии элементов открываются по величине
веса их атома».
6. «Распространенные в природе простые тела имеют малый
атомный вес, а все элементы с малым атомным весом
характеризуются разностью свойств. Они поэтому суть типические
элементы».
Эти шесть пунктов не только характеризуют содержание
закона, но раскрывают его методологическое значение как метода
познания и предвидения.
Ф. Энгельс о значении этого открытия писал: «Менделеев,
применив бессознательно гегелевский закон о переходе
количества в качество, совершил научный подвиг, который смело можно
поставить рядом с открытием Леверье, вычислившего орбиту еще
не известной планеты—Нептуна»1. А. В. Раковский писал об
этом: «... Леверье и Адаме при этом базировались на всеми
признанном законе Ньютона ... Менделеев открывал и предсказывал
свойства, опираясь на пустые клетки в созданной им системе и на
основании открытого им закона, не всеми признанного» 2.
Периодическая система
элементов Д. И. Менделеева
Чтобы подчеркнуть закон периодичности и превратить
непрерывный ряд элементов в периодическую систему, естественно
было разбить весь ряд элементов, расположенных по величине
атомных весов, на отдельные отрезки, внутри которых начинается и
заканчивается периодическое изменение свойств. Менделеев
подписывает каждый такой отрезок ряда один под другим и
получает первый набросок таблицы элементов.
«Днем рождения» системы Д. И. Менделеева является 18
февраля 1869 г. В этот день Менделеев составил первый вариант
таблицы, названной им «Опыт системы элементов, основанной на их
атомном весе и химическом сходстве».
В первом варианте таблицы (рис. 3.1) 63 элемента
расположены в порядке возрастания атомных весов сверху вниз и слева
направо, т. е. по вертикали и горизонтали.
В этом варианте сходные элементы расположены в 19-ти
горизонтальных рядах, которые более или менее соответствуют
подгруппам современной периодической системы. Эти ряды разбиты
на 6 вертикальных столбцов — отрезков единого ряда элементов;
в столбцах проступают черты будущего периода. Они объединяют
несходные элементы самого различного характера (металлы,
неметаллы, элементы с промежуточными свойствами),
характеризующиеся малыми разницами или равномерными скачками
атомных весов. В этом заключалась закономерность. Первый вариант
1 К. М а р к с и Ф Энгельс. Соч., т. 20, стр. 389.
2 «Успехи физических наук», 7, вып. 5, 316, 1927.
74
таблицы Менделеева — это не таблица разрозненных групп, это
система, где все элементы, а не только элементы-аналоги связаны
между собой теми или иными закономерностями, как звенья
развития единой материи. Это единая стройная система, отражающая
собой закон развития материи, всеобщую взаимосвязь элементов.
В первом варианте системы Д. И. Менделеева — таблице
длинной формы (рис. 3.1) —имеются 4 пустых места, которые побуди-
ОЛЫТ СИСТЕМЫ ЭЛЕМЕНТОВ,
ОСНОВАННОЙ НА ИХ АТОМНОМ ВЕСЕ И ХИМИЧЕСКОМ СХОДСТВЕ
Zr = 90
Nb=>94
Mo = 96
Rh =104,4
Ru=104,4
Pt * 106,6
Ag=108
Cd =112
Ur=116
Sn = 118
Sb = 122
Те =128?
J = 127
Cs = 133
Ba = 137
? = 180
Та =182
W =186
Pt =197,4
Ir =198
Os =199
Hg=200
Au=197?
Bi =210?
TI = 204
РЪ = 207
Рис. 3.1. Первый вариант периодической системы элементов Д. И.
Менделеева (1869 г.)
ли Менделеева допустить существование четырех неизвестных
элементов. Используя интерполяцию по вертикалям и горизонталям
•тблицы, Д. И. Менделеев предсказал их атомные веса и свойства.
В табл. 3.1 дан второй вариант таблицы, составленный в
1871 г., представляющий собой повернутое на 90° зеркальное
отражение первого варианта. Это прообраз современной 8-групповой
•тблицы, или таблицы короткой формы. Последняя
конструктивно более сложная, позволила Д. И. Менделееву предсказать уже
11 элементов, а также заурановые.
1
Be =9,4
В -И
С =М2
N =14
0 =16
F =19
7 Na=23
Ni
Mg=24
Al =27,4
Si =28
P =31
S =32
С J =35,5
К =39
Ca=40
? =45
?Er=56
?Yt=60
?ln = 75,6
Ti =50
V =51
Cr =52
Mn=35
Fe =i 56
= Co = 59
Cu =63,4
Zn = 65,2
? =68
? =70
As = 75
Se = 79,4
Br =80
Rb=85,4
Sr =87,6
Ce =92
La =94
D\ =95
Th=H8?
75.
Таблица 3.1
Естественная система элементов Д. И. Менделеева (1871 г.)
Ряды
Типические
элементы
в §[ Ряд 1-й
£* g — 2-й
о о 0 „
а. а — 3-й
н о> — 4-и
PQ С
8£ *
so г „
£§• - 6-И
ш з s — /-и
о; Н си —. 8-И
§§ - 9-й
д & — 10-й
1—. С
Высший солеобраз-
ный окисел
Высшее водородное
соединение
Группа I
Н = 1
Li --■= 7
Na = 23
К = 39
(Си --= 63)
Rb = 85
(Ag=108)
Cs = 133
(Au- 197)
R20
Группа II
Br = 9,4
Mg = 24
Ca = 40
Zn - 65
Sr = 87
Cd = 112
Ba =.= 137
Hg = 200
R202
1 или RO
Группа III Группа IV
B= 11
Al --27,3
— =- 44
— , 68
(?Yt = 88?)
In- 113
— = 137
Ti =- 204
R203
(R№?)
C- 12
Si = 28
Ti = 50?
— - 72
Zr = 90
Sn= 118
Ce -- 138?
Pb - 207
Th - = 232
R20*
или RO2
RH4
Группа V i
N=14
P = 31
V=-_ 51
As = 75
Nb = 94
Sb = 122
Та = 182
Bi = 208
R20&
RH3
Группа VI
0= 16
S -32
Cr -52
Se -78
Mo ^96
Те - 128?
W= 184
Ur = 240
R203
или RO3
RH2
Группа VII
F= 19
CI -35,5
Mn -55
Br - 80
—-- 100
Y=- 127
R20?
RH
Группа VIII
переход к группе I
Fe = 56; Co = 59
Ni =59; Си = 63
Ru - 104, Rh = 104
Pd = 104; Ag - 108
Os= 199? Ir= 198?
Pt = 197? Au = 197
R203 или RO*
—
В этой таблице впервые появляются 8 групп элементов.
Группа-- это совокупность элементов, имеющих, как правило,
валентность по кислороду в их высших солеобразующих окислах, равную
подеру группы. Общие формулы этих окислов приведены
Д. И. Менделеевым внизу под номером группы. Впоследствии
каждая группа разделилась внутри на подгруппы — главную и
побочную. В табл. 3.1 и периоды почти соответствуют по составу
современным, но не отвечают им по номерам.
Периоды — это горизонтальные графы (полосы), включающие
совокупность элементов, расположенных в порядке постепенного
возрастания атомных весов, начинающиеся с типично
металлического элемента (щелочного металла) через амфотерные и
заканчивающиеся типично неметаллическим элементом (галогеном).
Инертные газы, в то время еще неизвестные, в таблице
отсутствуют. Таких периодов было намечено 7.
Если в первом варианте системы Менделеева цепь логического
и конструктивного усложнения представлялась как отдельный
элемент — ряд сходных элементов — совокупность рядов, то в этом
варианте: отдельный элемент — ряд (группа) —вся система.
Современные формы периодической системы химических
элементов Д. И. Менделеева. В связи с открытием все новых
элементов, уточнением их свойств и аналогий между ними появлялись
все новые варианты изображения периодического закона —
таблицы, графики, модели. Общее число всех опубликованных
вариантов превышает 400.
Однако распространение и применение получили лишь
немногие из них. К ним относятся: таблица короткой формы (8-группо-
вой, или 8-клеточный вариант) и таблица длинной формы (32-
клеточный вариант). В основе этих двух форм лежат второй и
первый варианты менделеевских таблиц. Рассмотрим их более
подробно.
В современном 8-групповом варианте короткой формы
таблицы (см. форзац в начале книги) по сравнению с менделеевским
имеются некоторые изменения. Так, водород помещается над
галогенами (это его основное место). Инертные газы в связи с их
выявленными химическими свойствами размещаются в VIII
группе в качестве главной ее подгруппы. А 3 триады—железо, кобальт,
никель и их аналоги — составляют 3 побочных подгруппы VIII
группы, эта группа особая и по своей конструкции и по несовпадению
максимальной валентности большинства ее элементов с номером
группы. Так как каждая из остальных 7 групп состоит из одной
главной и одной побочной подгруппы, то общее число главных
подгрупп— 8, число же побочных— 10.
14 элементов, называемых лантанидами (или лантаноидами),
и 14, называемых актинидами (или актиноидами), размещение
которых по группам встретило затруднения, размещают двумя
рядами под основной таблицей. Тем самым образуются как бы
дополнительные лантаноидно-актиноидные подгруппы. Если учесть,
77
Liv I Be I В
к
Ca
Sr
-44-
ш
M0-* Tc
v-
v-
N
Ее
*Ru
что главные подгруппы делятся на две неравные части: первые 2
и остальные — 6, то получается не 3, а 4 типа подгрупп и число
подгрупп каждого типа — 2—6—10—14. Эти типы называются
электронными семействами: s-семейство (2); р-семейство (6); d-
семейство (10); f-семейство (14) (см. форзац в конце книги).
Уточнились по сравнению с таблицей
1871 г. и периоды. Ныне их 7, и каждый
заканчивается (седьмой, незаконченный,
должен заканчиваться) инертным газом.
Нумерация элементов, соответствующая
менделеевской расстановке их в целом по
возрастанию атомных весов, но с
известными тремя исключениями (аргон — калий;
кобальт — никель; теллур — йод), в
точности совпадает с последовательностью
возрастания зарядов ядер элементов. Очень
важно понятие — место элемента в
системе, занимаемая им клетка,—это
координаты элемента, указывающие на его номер,
на номер периода, номер группы и на тип
подгруппы, или электронное семейство.
Л—I р В периодической системе изменение
w I r* I Osl свойств элементов и их сравнение
прослеживают по трем направлениям:
горизонтальному, вертикальному и диагональному.
По горизонтальному направлению (по
периодам или их рядам) слева направо
усиливается окислительная способность
атомов, т. е. прием электронов, и
ослабевает восстановительная, т. е. отдача
электронов. Если взять простые вещества, соответствующие
элементам, то по этому направлению усиливается их неметалличность
(уменьшается металличность). По вертикальному направлению,
т. е. сверху вниз по подгруппам, картина изменения свойств и
атомов и простых веществ обратная.
Пересечение всех трех направлений получило название «звезд-
ности» системы. Большинство элементов находятся в окружении
восьми других элементов: четырех, расположенных по вертикали
и горизонтали, и четырех — по диагонали. Свойства элемента Тс
будут средними из свойств его 4 главных и 4 дополнительных
соседей (рис. 3.2). Используя это качество системы, Менделеев
сделал свои изумительные по точности предсказания свойств еще
неизвестных элементов. Используя эту «звездность», совершаются
крупные открытия, например открытие рения в минералах.
Следует отметить, что имеется в виду общая тенденция
изменения свойств по этим трем направлениям. На отдельных участках
периодов и подгрупп могут быть отклонения, вызванные
особенностями строения атомов элементов. Причем изменение свойств по
Рис. 3.2. Схема
«звездности»
периодической
системы
78
горизонтальному направлению осуществляется в малых периодах
большими скачками. Это приводит к резкому отличию двух
элементов соседей по периоду. Как мало сходства, например, в
свойствах простых веществ, отвечающих элементам: углерод и азот.
Иначе говоря, элементы малых периодов, включающих наиболее
типичные неметаллы, сильно индивидуализированы.
В больших же периодах изменение происходит более плавно.
На отдельных участках этих периодов наблюдается совсем
незначительное изменение свойств (металличности). В таких случаях
принято говорить о химических семействах. Например, семейство
железа (железо — кобальт — никель), семейство лантана и др.
Рассмотрим таблицу длинной формы, менее компактную, но в
которой нагляднее отражено деление элементов на четыре
категории, или электронных семейства. Это деление чрезвычайно важно.
Оно показывает, на какой подуровень (s, р, d> /) вступает
очередной электрон каждого последующего элемента. У элементов 5- и
р-семейств очередной электрон поступает в s- или р-подуровень
внешнего уровня; у элементов /-семейства — на d-подуровень
предвнешнего уровня; у элементов /-семейства очередной электрон
поступает, как правило, на f-подуровень предпредвнешнего
уровня. s-Элементы образуют две (IA и ПА) подгруппы. р-Эле-
менты образуют шесть (IIIA—VIIIA) подгрупп. Все эти восемь
подгрупп соответствуют главным подгруппам короткой формы.
d-Элементы образуют десять (IB—ХВ) подгрупп, отвечающих
десяти побочным подгруппам короткой формы. /-Элементы
образуют 14 коротких непронумерованных подгрупп, как и в таблице
короткой формы. Общее число подгрупп 2+6+10+14=32. В I
периоде находятся 2 элемента (Н и Не), оба одного s-семейства.
Во II и III периодах элементы уже двух семейств: по 2 s-эле-
мента и по 6 р-элементов, всего по 8 элементов.
В IV и V периодах находятся элементы уже трех семейств: по
2 s-элемента, по 10 d-элементов и по 6 р-элементов, т. е. всего 18
элементов.
В VI периоде представлены элементы четырех электронных
семейств: 2 s-элемента, 14 /-элементов, 10 rf-элементов и 6
р-элементов. Всего в VI периоде — 32 элемента. То же должно
повториться, если он будет закончен, и в VII периоде. Так как самый
длинный период содержит 32 элемента, то он и определяет
конструкцию данной таблицы, состоящей из 32 подгрупп.
Довольно обособленно в этом варианте таблицы распределены
металлы и неметаллы. Все 22 неметалла находятся в первой
верхней части. Как правило, это все р-элементы, за исключением
водорода и гелия (s-элементы). Металлы занимают всю остальную
таблицу. Они охватывают все элементы s-, d- и /-семейств и часть
р-элементов. Как видно, групп в этой таблице нет, их необходимо
воссоздавать сложением IA и IB подгрупп (I группа), ПА и ИВ
(II группа) и т. д.
79
Периодический закон и периодическая система являются
новым методом (инструментом) познания в химии. В ходе открытия
проявилась предсказательная сила периодического закона и
предвидение новых научных открытий.
Д. И. Менделеев, используя метод атом-аналогии, или «звезд-
ность», предсказал 11 еще неизвестных элементов: экацезия, эка-
бария, экабора, экаалюминия, экалантана, экасилиция, экатанта-
ла, экателлура, экамарганца, двимарганца и экайода.
Как сам Д. И. Менделеев, так и химики всех стран с
волнением ожидали открытия этих элементов, предсказанных на основе
периодического закона и периодической системы. Предвидение
(а предвидение — это цель науки) блестяще подтвердилось:
в 1875, 1879 и 1886 гг. были открыты галлий, скандий и
германий— элементы с атомными весами 68, 45 и 70.
После этого периодический закон и система Д. И. Менделеева
получили всеобщее признание, а периодическая система стала в
руках химиков своеобразной картой, которая руководила
поисками новых элементов, контролировала и проверяла открытия
химиков. Эти открытия составили мировую славу и признание
Д. И. Менделееву, его закону и триумф русской науке.
Однако Д. И. Менделеев не смог объяснить ряд вопросов:
причину периодичности свойств элементов; периодичность по рядам
через 2, 8, 18 элементов, связь главных и побочных групп,
количество элементов в периоде, несоответствие в расположении Аг и
К, Те и I, сколько элементов между Н и Не, положение
лантаноидов, положение инертных газов, какова верхняя граница таблицы.
Ответы на эти вопросы были получены только после открытия
строения атома.
§ 3. РАЗВИТИЕ ПЕРИОДИЧЕСКОГО ЗАКОНА
Периодический закон и периодическая система получили свое
полное подтверждение и дальнейшее развитие при установлении
строения атомов элементов.
Мозли в результате исследования рентгеновских спектров
элементов показал, что положительный заряд атома элемента
численно равен порядковому номеру в периодической системе.
Порядковый номер элемента меняется от 1 до 106. Следовательно,
химическая природа элемента определяется не массой (атомным
весом), а новой величиной — зарядом ядра или порядковым
номером.
Периодически изменяющиеся свойства элементов стали
связывать не с величиной атомного веса, а с величиной заряда ядра
или порядкового номера.
Периодический закон претерпел некоторую эволюцию. Теперь
его формулируют так: свойства элементов являются
периодической функцией положительного заряда ядра атомов элементов.
Это позволило установить, что в первом периоде всего 2 элемента
80
Н и Не, между ними нет элементов; подтвердить правильность
расположения аргона и калия, теллура и йода, кобальта и никеля,
ибо расположение их Д. И. Менделеевым отвечало величинам
положительных зарядов ядра; это позволило подтвердить и
установить заряды ядер лантаноидов и их количество.
В связи с этим претерпели эволюцию и некоторые основные
понятия химии, например понятие элемента. Элемент — это вид
атомов с одинаковым положительным, зарядом ядра (или
занимающих одно и то же место в таблице Менделеева).
Очевидно, положение верхней границы периодической системы
определяется тем предельным значением заряда ядра, которого
можно достигнуть при ядерном синтезе.
§ 4. ПЕРИОДИЧЕСКАЯ СИСТЕМА
И ЭЛЕКТРОННЫЕ СТРУКТУРЫ АТОМОВ
Самое замечательное свойство периодической системы то, что
она по существу отображает строение атома любого элемента.
Распределение элементов по периодам и по категориям подгрупп
в точности отвечает распределению очередных электронов этих
элементов по уровням и подуровням электронной оболочки. Как
известно из строения атома, формирование электронной оболочки
атома подчиняется двум принципам: принципу наименьшей
энергии и принципу Паули, уточняемым правилами Хунда и Клечков-
ского. Максимальное число электронов на атомных
энергетических уровнях и подуровнях можно представить табл. 3.2.
Таблица 3.2
Максимальное число электронов на атомных энергетических уровнях и подуровнях
Энергетический уровень
К(п= 1)
L(n = 2)
М (л-3)
N(n = A)
Энергетический
подуровень
s(/=0)
s (/=-0)
р(/- 1)
8 (1 --= 0)
р(Г-1)
d(l ---2)
s(l=-0)
P(/=I)
d(l=-2)
/(/ _-3)
Возможные
значения магнитного
квантового
числа m
0
0
— 1,0, + 1
0
-1,0. +1,
—2, —1,0,
+ 1, +2
0
— 1,0, +1
—2, —1,0,
-И, 4-2
—3, —2, —1,
ft, -fi\ +2~, +3
Число орбиталей
в
подуровне
1
1
3
1
3
5
1
2
5
7
в уровне
1
}'
9
16
Максимальное
число электронов
па
подуровне
2
2
6
2
6
10
2
6
10
14
на уровне
2
1»
18
. 32
81
Электронная структура атомов элементов I—III периодов.
Первый элемент менделеевского ряда — водород
/«О
n-f
он ,s
имеет ядро, несущее один элементарный положительный заряд, и
в поле ядра движется один электрон. Если атом не возбужден, а
находится в нормальном состоянии, электрон будет находиться на
первом энергетическом уровне (табл. 3.2). Движение этого
электрона характеризуется значениями квантовых чисел п=1, /=0,
mi=0 и 5 =+ 1/2. Таким образом, это будет s-электрон,
образующий сферически симметричное электронное облако.
10 20 30 40 50 30 70 ЬО 90
Рис. 3.3. Изменение потенциалов ионизации с
изменением порядковых номеров элементов
Чтобы оторвать электрон от ядра атома водорода, надо
затратить энергию в 13,6 эв (рис. 3.3, см. табл. 2.2). Прочность связи
равна 13,6 эв, т. е. довольно значительна, она эквивалентна
314 ккал/г-атом. Его электронная структура \sl (читается «один
эс один»). На графической схеме слева указан номер уровня, или
значение главного квантового числа пу а справа — подуровень s;
s-орбиталь услозно обозначена квадратом (квантовая ячейка).
82
Второй по порядку элемент непрерывного ряда элементов —
гелий
[=0
т=0
п*1
Н
2 2
Не Is
Заряд его ядра равен двум элементарным положительным
зарядам и, следовательно, в поле ядра движутся 2 электрона.
Минимальной энергии атома, т. е. его нормальному состоянию, будет
отвечать движение обоих электронов на первом энергетическом
уровне, следовательно, оба электрона будут s-электронами, ибо
других орбиталей на первом уровне нет.
Таким образом, оба электрона характеризуются значением
квантовых чисел я=1, /=0 и т!=0, и их состояние отличается
только тем, что для одного электрона четвертое квантовое число
s = + l/2, а для другого s = —1/2. Как видно, принцип Паули,
требующий, чтобы электроны в атоме отличались значением хотя бы
в отношении одного квантового числа, соблюдается. Электронная
формула Не2 Is2. Ионизационный потенциал атома гелия
значительно выше, чем атома водорода, и составляет 24,28 эв, что
эквивалентно 566 ккал. Это больше, чем для водорода, на 252 ккал и
свидетельствует о необычайной прочности связи электронов атома
гелия с ядром. Закончен первый период и закончен первый
уровень.
Третий элемент — литий
лт=0 J. ; '
п=2
п = 1
3 2 1
Lt 1s 2s
h
начинает второй период, он имеет заряд ядра, равный трем
элементарным положительным зарядам, и, следовательно, 3
электрона движутся в поле ядра нейтрального атома; 2 электрона из трех
займут места на первом энергетическом уровне, т. е. окажутся в
тех же состояниях, как и 2 электрона атома гелия. В соответствии
с принципом Паули третьему электрону нет места на первом
энергетическом уровне, так как все возможные значения квантовых
чисел на этом уровне исчерпали первые 2 электрона. Итак,
принцип Паули требует, чтобы третий электрон разместился на втором
энергетическом уровне, т. е. стал 25-электроном.
Поэтому электронная структура лития ls22sl. Заметим, что
электронная формула лития включает в себя электронную
формулу гелия. Прочность связи третьего электрона с ядром невелика
83
Распределение элект
Слой
п
1
подгруппа
1 Н
2 Не
3 Li
4 Be
5 В
6 С
7 N
8 О
9 F
10 Ne
11 Na
12 Mg
13 Al
14 Si
15 P
16 S
17 CI
18 Ar
19 К
20 Ca
21 Sc
22 Ti
23 V
24 Cr
25 Mn
26 Fe
27 Co
28 Ni
29 Cu
30 Zn
31 Qa
32 Ge
33 As
34 Se
35 Br
36 Kr
к
l
0
Is
1
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
L
2
0 1
2s 2/?
1
2
2 1
2 2
2 3
2 4
2 5
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
1 2 6
1 2 6
2 6
2 6
2 6
2 6
2 6
2 6
2 6
M
3
0 12
3s 3p 3d
1
2
2 1
2 2
2 3
2 4
2 5
2 6
2 6
2 6
2 6 1.
2 6 2
2 6 3
2 6 5
2 6 5
2 6 6
2 6 7
2 6 8
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
N
4
0 1
4s Ap
1
2
2
2
2
1
2
2
2
2
1
2
2 1
2 2
2 3
2 4
2 5
2 6
Слой
/г
/
подгруппа
37 Rb
38 Sr
39 Y
40 Zv
41 Nb
42 Mo
43 Tc
44 Ru
| 45 Rh
46 Pd
i V Ag
48 Cd
49 In
50 Sn
51 Sb
52 Те
53 I
54 Xe
55 Cs
56 Ba
57 La
58 Ce
59 Pr
60 Nd
61 Pm
62 Sm
63 Eu
64 Gd
65 Tb
66 Dy
67 Ho
68 Er
69 Tm
70 Yb
71 Lu
К 1
1
2
2 i
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
1 2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
L
2
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
8
и
8
8
8
8
8
8
8
! 8
м 1
3
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
! 18
18
18
18
18
18
18
18
18
18
18
18
N 1
4 1
0 1 2 3
Is Ap Ad Af
2 6
2 6
2 6 1
2 6 2
2 6 4
2 6 5
2 6 5
2 6 7
2 6 8
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
| 2 6 10 1
2 6 10 3
2 6 10 4
2 6 10 5
2 6 10 6
2 6 10 7
2 6 10 7
2 6 10 9
2 6 10 10
2 6 10 11
2 6 10 12
2 6 10 13
3 6 10 14
2 6 10 14
ронов в атомах
Таблица 3.3
-
О |
5
0 1 2
5s Ър bd
1
2
2
2
1
1
2
1
1
0
1
2
2 1
2 2
2 3
2 4
2 5
2 6
2 6
2 6
2 6 1
2 6 1
2 6
2 6
2 6
2 6
2 6
2 6 1
2 6
2 6
2 6
2 6
2 6
2 6
! 2 6 1
Р
6
0
6s
1
2 1
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
Слой 1
п
1
подгруппа
72 Ш
73 Та !
74 W
75 Re
1 76 Os
77 Ir
78 Pt
79 Au
80 Hg
81 Tl
82 Pb
83 Bi
84 Po
85 At
86 Rn
S7 Fr
88 Ra
89 Ac
90 Th
91 Pa
92 U
93 Np
94 Pu
95 Am
96 Cm
97 Bk
98 Cf
99 Es
100 Fm
101 Md
102 No
103 Lr
104 Ku
105
К
1J
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
L i
2
8
8
8
8
8
8
6
8
8
8
8
8
8
8
8
8
8
! 8
1 8
8
8
1 8
i 8
8
8
8
8
8
8
8
8
8
8
8
M
3
18
18
18
18
18 1
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
18
N
4
32
32
32
32 1
32
32
32
32
32
32
32
32
32
32
32
32
32
32
32
32
32
32
32
I 32
32
32
32
32
32
32
32
32
32
32
о 1
5
0 12 3
s Ър 5d 5/
2 6 2
2 6 3
2 6 4
2 6 5
2 6 6
2 6 7
2 6 9
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10
2 6 10 2
2 6 10 3
2 6 10 4
2 6 10 6
2 6 10 7
2 6 10 7
2 6 10 8
2 6 10 10
2 6 10 11
i 2 6 10 12
2 6 10 13
2 6 10 14
1 2 6 10 14
1 2 6 10 14
2 6 10 14
P 1
6
0 1 2 1
6s 6p bd
2
2
2
2
2
2
1
1
2
2 1
2 2
2 3
2 4
2 5
2 6
2 6
2 6
i 2 6 1
12 6 2
2 6 1
2 6 1
2 6 1
2 6
2 6
2 6 1
2 6 1
2 6
2 6
2 6
2 6
2 6
2 6 1
2 6 2
2 6 3
Q
7
0
7s
1
2
2
i 2
i 2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
и составляет всего 5,37 эв. Все 3 электрона атома лития есть s-
электроны, поэтому этот атом имеет сферически симметричное
электронное облако.
Атом четвертого элемента — бериллия
п-~2
п = 1
U
Н
Be4 1s22s7
имеет 4 электрона, заполняющие два уровня: Is и 25 и Be4
ls22s2. Ионизационный потенциал атома бериллия значительно
выше, чем атома лития, и составляет 9,3 эв. Это указывает на то,
что заполнение 25-подуровня двумя электронами, т. е. образование
электронной пары, приводит к увеличению ионизационного
потенциала, поэтому оторвать электрон из электронной пары труднее,
чем непарный электрон.
В следующем атоме — аюме бора
!-- Г
';'u 1
"-'[♦Г
Т~[ | j
о"
5 , ?„ 2 '
В /s 2s p
4 электрона заполняют те же уровни, что и электроны атома
бериллия. Пятый электрон приводит к «открытию» 2/?-подуровня; его
состояние характеризуется л=2, /=1, Щ=\, s = + l/2. В
соответствии со значениями /'на ^-подуровне имеется Зр-орбитали и может
максимально расположиться 6 электронов по 2 в каждой из трех
квантовых состояний рХу ру и pz. Поэтому он постепенно
заполняется в ряду из последующих шести атомов от бора до неона
(табл. 3.3).
Как при этом упрочняются связи электронов, показывают
значения ионизационных потенциалов (рис. 3.3). Следует отметить,
что второй р-электрои атома углерода
п=2
11
Ш_П
С UJ2
2 2
S р
86
не образует с первым р-электроном электронную пару, а согласно
правилу Хунда занимает второй р-подуровень, так что атом
углерода имеет 2 непарных электрона.
У следующего атома—азота
/-о /=;
п-/
н
*♦
♦ ♦ И
2« 2 3
N fs 2s p
3 непарных р-электрона, так
что все р-подуровни второго
энергетического уровня имеют
по одному электрону.
У атома кислорода
/ = 0,
1-1
на одном р-подуровне
появляется второй электрон и
остаются 2 непарных электрона; у
атома фтора
1=1
l;^r-1mrQmr.1
п-2
п=1
Н
И
U НИ
^s р
7рСШ
7s XL-
КрГГП
6s JO-
5рГГП
6dL MILL 5>f м I I I I I I.
CO
3:
5s_D_
4РПП
Sd 11 I 1 1 I 4f г i I 1 l I I I.
4^ i i i i i i
4s П
ЗрГПП
3s П
2рГПП
2s_Q_
3d 1 I 1 I I
fs JDL
Рис. З.4. Энергетическая
характеристика уровней и подуровней
заполняется второй р-подуровень и остается только один
непарный электрон, а у следующего за ним атома — неона
87
/-0 ,=/
п*1
U
п
шщ
10 2 2 fi
Ne is 2s p6
второй энергетический уровень полностью заполнен 8-ю
электронами.
Расположение электронов в атомах от лития до неона
представлено в табл. 3.3. Закончен второй период и заполнен второй
уровень п=2.^Периоды начинаются водородом и литием, у
которых очередной электрон заполняет s-подуровень, заканчивается
период заполнением s2p6.
Следующий за неоном натрий начинает третий период. Он
имеет в атоме 11 электронов. Из них 10 заполняют, как у атома
неона, первый и второй энергетические уровни, а одиннадцатый
должен разместиться на третьем энергетическом уровне п—3 и в
нормальном состоянии атома будет Зз-электроном с п=3 и /==0.
В следующих за натрием атомах Mg, Al, Si, P, S, C1 и Аг
происходит постепенное заполнение s- и р-подуровней третьего
энергетического уровня в том же порядке, как заполнялся второй
энергетический уровень у атомов бериллия — неона.
Обратите внимание на то, что мы исчерпали третий период,
но не использовали третий энергетический уровень.
N2
1 1
m =0
л=3
н
/ 1 1
Н
ч
н
v
и
s
тг-2
т,-/
V
т;-0
т -+/
с/
т^+2
У
Ar,8t/2s22p63sV
п=2
77]
В атоме аргона на третьем уровне всего восемь электронов (3s2
З/?6), а максимально на третьем уровне может быть 2-32= 18
электронов. Таким образом, третий уровень имеет еще 10
незаселенных мест — свободен весь Зй-подуровень с 5-ю орбиталями. И
далее заполнение элементами периодов пойдет быстрее заполнения
соответствующих по номеру уровней. Отметим также, что для
первых 18-ти элементов электронная оболочка заполнялась в полном
соответствии с идеальной последовательностью. Согласно схеме
квантования
88
1 s ->• 2s -»■ 2p -*• 3s -*» 3p
■3d-
• 4s-
-4s-
• 3d-
■4p-
Ap
Ad-
-5s-
-4/-* 5s
• Ad -*■ 5p
(1)
(2)
Затем эта строгая последовательность нарушается и вместо
естественного продолжения по линии (1) наблюдается более сложный
порядок заполнения по линии (2). Это результат экранирования
ядра накапливающимися электронами. Новые электроны
притягиваются к ядру слабее, отходят от ядра на более дальние уровни,
оставляя временно пустые ячейки на более близких уровнях.
Заполнение их будет происходить с «запаз- ,.
дыванием» у последующих элементов.
Принцип наименьшей энергии при этом
не нарушается (рис. 3.4). Именно такая
непоследовательность заполнения
отвечает возрастанию энергии электронов в
соответствии с возрастанием по правилу
Клечковского суммы (п + l) главного и
орбитального квантовых чисел.
Орбитальное к6анто$ое
число I
1
Рис. 3.5. Схема последовательности заполнения
электронных энергетических подуровней в атоме
to
y4py4dy4f y
5s,5pySdy5f
6s,6p 6d 6f
Is 7p nj nr
Id If
Советским ученым В. М. Клечковским 1 установлено правило
фактической последовательности формирования электронной
оболочки по возрастанию суммы главного и побочного квантовых
чисел (рис. 3.5). Если 2 электрона имеют одинаковое значение я+/,
то заполнение идет от меньших значений п к большим (или от
больших значений / к меньшим). Это отвечает принципу
наименьшей энергии (рис. 3.4).
Электронная структура атомов элементов IV и V периодов.
У элементов V периода № 19 (К)
4s
[ШШЕЗ-
3d
К'9 1s22hp63sW
и № 20 (Са)
4s
н
3s
Зр
н
ЩЩ
3d
50 2 г е •> с. ■>
Са 1s 2s 2р 3s Зр 4s
1 См. В. М. Клечковский Распределение атомных электронов и правило
последовательного заполнения (п+l) -групп. М, Атомиздат, 1968, стр. 23.
89
очередные электроны направляются на 45-подуровень,
энергетически более выгодный. Сумма п+1 для Зй-состояния равна 5, а для
45-состояния она составляет 4. Поэтому электронная структура
калия (Ar)4s!; Са—(Ar)4s2.
У последующих десяти элементов побочных подгрупп № 21
(Sc)
4s
Ш
3s
Зр
[п]ШЖ
3d
21 о о с о с 1
Sc 1s 2s 2pb3s Зр 3d 4s
и № 30 (Zn)
4s
Зр
3d
нЮММН
Zn3° yS22s22p63s23p63d'^s2
их десяти очередным электронам энергетически выгоден Зй-под-
уровень, чем 4р. Хотя сумма п-\-1 для обоих подуровней
одинакова и равна 5, преимущество по Клечковскому имеет подуровень
более близкого к ядру уровня с меньшим значением главного
квантового числа п.
Заполнение пяти орбиталей Зй-подуровня и здесь будет
происходить в соответствии с правилом Хунда. Сначала заселение
непарными электронами от № 21 (Sc) до № 26 (Мп) (табл. 3.3), а
затем спаривание от № 26 (Fe) до № 30 (Zn).
Обратите внимание, что имеется два «провала» электронов с
45->3rf у хрома и меди. Следовательно, у цинка
последовательность заполнения электронами следующая: (Ar)4s23d10.
У элементов IV периода 5-го ряда (нечетного) № 31 (Ga)—36
(Кг) очередные 6 электронов займут три орбитали 4р-подуровня
(табл. 3.3). Тогда общая последовательность заполнения
электронами состояний у атома криптона ls22s22/?63s23/?64s23d104p6, что
отвечает полной электронной формуле его ls2|2s22jt?6|3s23p63<i10|4s2-
4/?6 или упрощенной 2|8|18|8.
У 18-ти элементов V периода повторяется та же
последовательность заполнения, что и в IV периоде. Только вместо заполнения
подуровней 4s2-v3d10->4p6 будут заполняться подуровни 5s2->-
->4d10-^5p6 (Табл. 3.3).
Электронная структура атомов элементов VI и VII периодов.
В VI периоде первые два элемента № 55 (Cs) и № 56 (Ва)
очередные электроны направляют в s-подуровень (табл. 3.3). У
элемента III группы лантана № 57 очередной электрон начинает 5d-
подуровень (6s25dl). Однако последующие 14 элементов от Се
(58) до Lu (71) очередные электроны направляют в третий сна-
90
ружи уровень 4/ (табл. 3.3). От Ш (72) до Hg (80) продолжается
заполнение 5й-подуровня. Элементы Т1 (81)—Ru (86) очередные
электроны направят в 6/ыюдуровень. Таким образом, в VI
периоде всего 32 элемента, начинается он с заполнения очередными
электронами s-орбиталей и заканчивается по-прежнему
заполнением трех р-орбиталей (6s26/?6). VII период не окончен, но он
должен быть аналогичен VI периоду.
По тому, какой подуровень заполняется очередными
электронами, элементы разделяются на 4 семейства: s, р, d и f.
При рассматривании электронных структур атомов элементов
связь системы элементов со строением атомов становится
очевидной.
Рассматривая последовательности закономерного заполнения
электронами уровней, можно составить схему конструкции
периодической системы на основе структуры электронных оболочек
атомов
Период
ш
s' s4 р1 р6
s' s2\ р' р1
Вклинивание 3d- и 4d~ подуровней i
s'
'[£-
s2
s2
d' .. d'°
d'...d'°
p'- Pe 1
P1 P6 I
Вклинивание 4f- и 5f~ подуровней.
2s22pe
3s 3p
Число
элементов
2
8
8
s'
»'
s2
s2
d'
d1
f1. f'4
f' f'4
d2. d'°
d\ d'°
p'. VI
p' V1
,4s 3d 4p
5s 4d 5p
2 .10 M 6
6s 5d 4f 6p
7s 6d 5f 7p6
18
/8
32
32
Следовательно, в заполнении электронных оболочек атомов
элементов можно отметить следующие закономерности:
1. Номер периода совпадает со значением главного квантового
числа.
2. Номер периода обычно совпадает с числом заполняемых
электронами уровней у элементов этого периода. Единственное
исключение — палладий (V период, четыре уровня).
3. Максимальное число элементов в периодах и максимальное
число электронов на квантовых уровнях —2, 8, 18, 32 —равно
удвоенному квадрату главного квантового числа 2п2 и определяет
число элементов в периодах. Так, в I периоде 2, во II — 8 и т. д.
91
4. Каждый период начинается с заполнения s-подуровня нового
квантового уровня и заканчивается заполнением р-подуровня
этого же уровня.
5. В каждом большом периоде начиная с IV, с элементов III
группы иттрия, лантана и актиния идет заполнение не внешнего
энергетического уровня, а предвнешнего d-подуровня.
6. У элементов группы лантаноидов и актиноидов идет
заполнение предпредвнешнего подуровня /.
7. Атомы элементов I—III периодов содержат на внешнем
уровне число s- и /^-электронов, равное номеру группы, в которой
находится данный элемент. Это так называемые валентные
электроны.
8. У атомов элементов нечетных рядов 3, 5, 7, 9 больших
периодов содержится также на внешнем уровне число 5- и р-валентных.
электронов, равное номеру группы.
9. Атомы элементов четных рядов (4, 6, 8, 10) больших
периодов содержат не более 2$-электронов (за исключением хрома,
ниобия, молибдена, палладия). Это элементы металлического
характера.
10. Атомы инертных газов имеют на внешнем уровне 8 s2p6-
электронов, что определяет их расположение в конце периодов.
(в VIII группе).
11. У элементов главных или А-подгрупп идет заполнение
электронами s- и р-подуровней внешнего уровня, и валентными могут
быть только эти электроны (например, № 17 CI ls22s22p63s23p5).
12. У элементов побочных или В-подгрупп идет заполнение
электронами d-подуровня предвнешнего уровня, и валентными могут
быть как электроны внешнего уровня, так и d-электроны
предвнешнего (например, № 23 V ls22s22p63s23p*3d4s2).
13. У элементов лантаноидно-актиноидных подгрупп идет
заполнение электронами предпредвнешнего уровня, и валентными
могут быть 5-электроны внешнего уровня, один rf-злектрон
предвнешнего и какое-то число /-электронов из предпредвнешнего
уровня (например, № 92 U —... 5fs6dl7s2).
Итак, деление элементов по электронным семействам имеет
двоякий смысл.
Таким образом, периодическая система элементов предстает
перед нами как структурограмма строения атома любого ее
элемента. Можно устанавливать электронное строение любого атома
не только на основе той фактической последовательности
заполнения подуровней, о которой говорилось, но и на основе самой
таблицы. Это делается последовательным переходом от элемента к
элементу вплоть до заданного и установлением по их положению
в системе, какие уровни и подуровни заполнятся. При этом
необходимо учитывать указанные выше закономерности и особенности
заполнения.
Так, например элемент гафний № 72, находящийся в VI периоде в VIII
(четном) ряду, должен иметь 72 электрона, расположенных на 6 энергетических
92
уровнях; полностью заняты электронами 4 уровня, на внешнем (шестом) два
6s электрона, так как он находится после лантана, следовательно, у него
заполнен второй снаружи уровень, 5<2-подуровень, где имеет 2 электрона; стоит после
лантаноидов, следовательно, 4/-подуровень полностью заполнен и содержит
14 электронов. Поэтому его электронные состояния будут заполняться в
следующей последовательности:
Is21 2s22pe t 3s23p4s22>dl4p* | SsHd^bp* \ 6s25d4f^5d2 \.
Его электронная структура будет следующая:
ls22s22/?e3s23/7e3d104s24/?e4diH/145s25/?65cf26s2.
Это элемент d-семейства, поэтому валентными являются 5<226$2-электроны.
Изложение квантовомеханических представлений по строению
электронных оболочек позволяет установить наличие
периодичности в строении электронных оболочек:
1) по мере увеличения зарядов атомных ядер периодически
начинают заполняться электронами новые энергетические уровни
в атомах и периодически повторяется последовательность
заполнения подуровней;
2) с увеличением зарядов ядер атомов периодически
повторяются электронные структуры атомов;
3) элементы аналогичные, закономерно появляющиеся в
периодической системе, характеризуются одинаковым строением
наружных групп электронов.
Следовательно, причина периодичности свойств элементов
разгадана, она заложена в периодичности изменения строения
электронной оболочки.
Физический смысл периодического закона состоит в том, что
периодическое изменение свойств элементов находится в полном
соответствии с периодически возобновляющимися на все более
высоких энергетических уровнях сходными электронными
структурами атомов. С их закономерным изменением закономерно
изменяются физические и химические свойства. Так образуются
подгруппы системы, т. е. вертикальные ряды элементов-аналогов по их
электронной структуре. Например, в подгруппе углерода
C—ls2'2s22p2
Si— ls22s22pr"3s23p2
Ge— ls22s\2p6 3p63diV';4s24p2
Именно в этой аналогии строения внешних уровней и
заключается причина близости свойств элементов подгруппы.
Следовательно, внутренний (физический) смысл
периодического закона состоит в том, что периодическое изменение свойств
элементов и их соединений является функцией периодически
повторяющихся на все более высоких энергетических уровнях
сходных электронных структур. Но, в свою очередь, периодическое
появление сходных электронных структур является, как это
полагают, функцией периодически повторяющихся на все более
высоких внутриядерных уровнях нуклонных структур, т. е. корни
периодичности следует искать в ядрах атомов.
93
Таблица 3.4
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ (по Некрасову)
Группы
VII VIII
Н Не 1
I II III IV V VI |
Li Be В С N О F Ne 2
з
О
х
О.
<Ю
С
Na Mg Al Si P S CI Ar 3
4 К Ca Sc T\ V" Cr Mn Fe Co Ni "Cu Zn Ga Ge As Se Цт Kr 4
5 Rb Sr Y It ЦЬ Mo fc Ru Rh Pd Ag Cd
В I f T 4 В I S i a
Mi Cu Zn Ga Ge As Se Br Kr 4
i I i I i J i 1 i
?d Ag Cd la $д 5h Те I Xe б
I I i и i I
* S Cs BaO Г» Та W Re 6s Ir Pt Au Hs fl Pb Bi Po At Rn 6
£ I I j 4 5 6 7 3 9 10 11 12 13 14 15 16 17 18
7 Fr RaQc]
1 2 3
,' я д ы
ан алогов
Электронная структура атомов и связь ее с периодичностью
свойств позволила Б. В. Некрасову установить два или три
различных случая аналогии между элементами одной и той же
группы и представить периодическую систему элементов с указанием
рядов аналогов (табл. 3.4). Одни из них — полные аналоги, так
как имеют однотипные структуры внешних электронных уровней
Таблица 3.4, а
Таблица 3.4, б
Степень
окисления
0
•^-2
+4
—4
Электронная структура
С
2 4
2 2
2 0
2 8
Si
2 8 4
2 8 2
2 8 0
2 8 8
Степень
окисления
0
+ 2
+4
Электронная
Si
2 8 4
2 8 2
2 8 0
1 структура
Ti
2 8 8+22
2 8 8+2
2 8 8 0
94
при любой заданной валентности или степени окисления. Это все
подгруппы элементов больших периодов. Таковы, например,
элементы-аналоги (обозначенные в табл. 3.4, сплошной чертой) II
и III периодов углерод и кремний (табл. 3.4, а).
Другие элементы — неполные аналоги: структура их атомов
однотипна лишь при некоторых степенях окисления, исключая
высшую. Например, углерод и кремний, с одной стороны, и германий,
олово и свинец — с другой.
Третьи элементы—также аналоги неполные, но их структура
однотипна только при высшей степени окисления, равной номеру
группы (табл. 3.4, б).
Следовательно, IV группа может быть изображена более уточ-
ненно так:
Si
Ti
Ge
Zr
Sn
Hf
Pb
20
§ 40
1 60
о
'3 '80
Rb
Li Nar
AQ
-oAu
Sc
Вторичная и внутренняя периодичности
Под вторичной периодичностью (ее можно назвать
«вертикальной») подразумевают не монотонное изменение свойств элементов
и их соединений сверху вниз
по подгруппам, особенно по
главным. Впервые это
заметили Л. И. Бирон и С. Д. Щу-
карев. Причина заключается в
сжатии атомов вследствие
заполнения электронами
глубинных cl- и /-подуровней и
экранировании ими ядра атома.
Это ведет к появлению
лантаноидного сжатия —
уменьшению радиуса атома от церия к
лютецию (см. рис. 14, 70).
Вторичная периодичность
хорошо наблюдается в
изменении потенциалов ионизации
элементов (рис. 3.6). Так, в
подгруппе углерода при пере-
М.
7~Зг^Ва
2п Cd La
^^Г"^
7^—^~~~~
Ga
Zr
Ra
"^°Hg
Ac
Tl
Hf
Рис. З.6. Вторичная периодическая
зависимость потенциалов ионизации
95
ходе от углерода к кремнию суммарный потенциал ионизации их
внешних электронов увеличивается, так как rSi>rc. При переходе
же от кремния к германию аналогичный суммарный потенциал
уменьшается, так как rGe<^si. Далее при переходе от Ge к олову
2/ снова увеличивается: rGe>rsn и т. д.
Внутренняя периодичность наблюдается среди лантаноидов.
Это проявляется в повторении валентности (см. рис. 14.73), а
точнее в двухэтапном заполнении электронами f-орбиталсй (сначала
непарными, а затем парными в соответствии с правилом Хунда
(см. табл. 3.3). Внутренняя периодичность характерна для р-эле-
ментов и горизонтальных рядов d-элементов.
§ 5. ПЕРИОДИЧЕСКИЙ ЗАКОН,
ПЕРИОДИЧЕСКАЯ СИСТЕМА И СОВРЕМЕННАЯ ХИМИЯ
Периодический закон и периодическая система оказали
неоценимую услугу для развития теории строения атома. В свою
очередь, познание строения атома привело к эволюции как
периодического закона, так и периодической системы. Наряду с
установлением новой фундаментальной величины — положительного
заряда ядра атома — и совпадением его с порядковым номером
элемента в таблице Д. И. Менделеева, наряду с раскрытием
физического смысла периодического закона, или причин периодичности,
появилась возможность открытия целой плеяды новых элементов
и конструирования периодов таблицы.
Синтез новых элементов. К 1937 г. все природные 88 элементов
были, казалось, открыты. Дальнейшее пополнение системы могло
идти лишь за счет искусственного синтеза их. Возможности этого
синтеза ограничиваются тем предельным значением заряда ядра,
при котором ядро не будет распадаться в момент синтеза.
В 1937 г. бомбардировкой ядер молибдена дейтронами был
получен первый среди искусственных элементов — технеций:
942Mo^D = ^Tc-| In.
Вслед за ним были синтезированы еще 14 элементов от № 91
нептуния до 105 элемента.
В 1964 г. в Дубне в Объединенном институте ядерных
исследований академиком Г. В. Флеровым с сотрудниками был получен
элемент № 104 — курчатовий, в 1970 г.—№ 105, а в 1973 г. — № 106
по следующей ядерной реакции:
2882РЬ-г244Сг^259106^34.
В Объединенном институте ядерных исследований ведутся
работы по синтезу элемента № 114. Однако для трансурановых
элементов характерна неустойчивость (нестабильность) их ядер.
Причем она резко возрастает от плутония—Ti/2 = 50 сек до курчатовия
7V2 = 0,4 сек. Поэтому вначале считали, что синтез элементов с
^6
Z>106 не имеет смысла. Однако развитие теории строения ядра
атома это опровергает.
Согласно наиболее интересной из этих теорий — слоистой, или
оболочечной, предложенной в 1948 г. американским физиком
М. Гсчшерт-Майер и немецким физиком И. Иенсеном, считается,
что нуклоны (протоны и нейтроны) в ядре распределяются так же
по энергетическим уровням и подуровням, как и электроны в
оболочке атома (рис. 3.7); когда уровни и подуровни полностью за-
-3d—D-D-D-D-a ю*1° —3rf-D-D-n-D-D-
-2s П „п 2'2 on 25 Q-
20——20
-«f-D-D-D-D-D-D-a- ,444 ^-D-D-D-D-ChD-n
зр-о-оп 50^6_ 5o—з^-DHD-a
'«MD-D-D-D-D 10*1° -*KHHH>a
*-a 82^L82 ^
6hHD4D4Ir4D4Ir4Ir4Ir-n-D-D-D- 22*22<НЗЧЗ^ПЧЗ<УСНЗ--а-ПЫ
-sf^j-n-o-CHJ-u-a— 14*14 sH^<ya-o-o-u-o
'/>-Q-D-a 6+6 ^P-D-D-D
t
Протонная оболочка Нейтронная оболочка
Число нуклонов
Рис 3 7. Схема заполнения нуклонами ядерных уровней и подуровней
полнены нуклонами, получаются ядра с «магическими» числами
нейтронов и протонов с повышенной устойчивостью. Например:
2Не (2р + 2/г)16 О (8р + 8/г)40 Са (20р + 20п)
являются дважды «магическими» и по числу нейтронов и по
числу протонов. Сюда относятся ядра с «магическими» числами 28,
50, 82. Согласно теоретическим расчетам повышенной
устойчивостью должны обладать и ядра элементов 114, 126, 184 (эка-
свинец, экаплатина, экаэкаовинец), которые не реализованы
сегодня. Следовательно, среди «моря» наступающей нестабильности
элементов возможно наличие «островов» стабильности (рис. 3.8).
Вот почему и поиски в природе и дальнейший синтез элементов
должны продолжаться. Все элементы, как будет видно ниже, в
периодической системе найдут свое «законное» месте.
4 Общая химия
97
Если построить график функциональной зависимости любого
ия ядерных свойств от Z или TV, то кривая будет иметь явно
выраженный периодический характер (подобно кривой атомных обье-
мов), причем в точках максимумов или минимумов будут
находиться ядра с «магическими» числами. Следовательно, и ядерные
свойства элементов изменяются периодически в зависимости от
Рис. 3.8. Области устойчивости тяжелых ядер в море нестабильности
числа протонов и нейтронов, содержащихся в ядрах их атомов. На
основе периодически изменяющихся ядерных свойств к
настоящему времени составлено около 60 периодических систем изотопов
(ядер).
Выявленные закономерности в строении ядер позволяют уже
теперь придать периодическому закону на новом ядерном этапе
(третьем этапе, первым считается химический — менделеевский,
вторым — электронный) его понимания новую, углубленную
формулировку: с увеличением числа нуклонов в ядре и электронов в
оболочке периодически повторяются особенности в свойствах
атомов, простых тел, сложных соединений, а также ядер элементов.
Периодический закон вскрывает закономерные связи не только
между атомами в целом, но и между их составными частями —
электронными оболочками и ядрами. Трудно сказать, где
кончается власть этого закона. Все это позволяет конструировать
условные формы периодов таблицы (см. дополнительную литературу
к гл. 3).
Эволюция закона и системы проявляется и в расширении
набора периодических функций. Если Д. И. Менделеевым были
названы атомная масса и атомные объемы, то после развития учения
о строении атома к настоящему времени к ним относятся наряду
с упомянутыми ионизационными потенциалами (рис. 3.4) электро-
98
отрицательность, температуры плавления и кипения и др. (гл. 12).
Оказалось, что периодичность присуща не только элементам, но
и их соединениям. Для них наряду с указанными выше
величинами периодически меняются и различные термодинамические
характеристики (энтальпии, изобарные потенциалы образования
оксидов, галидов и т. д. (рис. 3.9)).
Рис. 3.9. Зависимость стандартного изобарного потенциала образования AGo6p
хлоридов при 25°С от порядкового номера Z
Электронная классификация элементов периодической
таблицы позволяет объяснять закономерность в комплексообразовании
элементов, распространение элементов в земной коре (см. гл. 14)
и целый ряд других явлений. Знаменательным в этом отношении
является открытие и создание химии соединений инертных газов,
что оказалось возможным при установлении потенциалов
ионизации элементов и сравнении их для кислорода и ксенона.
Все это привело к эволюции представлений об основных
законах и понятиях в химии.
Периодический закон и периодическая система и на сегодня
являются основой химической классификации. Так, дальнейшее
развитие химии привело к появлению целых классов новых
неорганических соединений. Это гидриды, карбиды, нитриды, бориды,
сульфиды, галиды и другие, свойства, условия образования
которых целиком определяются положением элементов в
периодической системе, их такими характеристиками, как величины иони-
4"
99
зационных потенциалов, размеры атомов, тип химической
связи и др.
В качестве примера на рис. 3.10 представлена классификация
гидридов элементов в соответствии с положением их в
периодической системе.
Группа
Период I H
Период II
Период III
Подгруппа
Период IV
Период V
Период VI
Период VII
Li
Na
IA
4
Mgj
IIA
К Ca 1
1 Rb Sr
[ Cs Ba1
Fr Ra
ионные* 1
1 гидр
иды 1
in iv v vj vijo
He
IB С N О F ,Ne
| I
Al Si P S CI JAr
IIIIA IVA VA VIA VIIA VIIIA IB IIВ 'IIIB IVB VB V1B VIIBI О
I л • j j
Sc Ti V Cr Mn Fe Co Ni |Cu Zn' Ga Gc As Se Br |Kr
Y Zr Nb Mo Tc Ru Rh Pd|Ag Cd In' Sn Sb Те I i Xe
|57-7lHi Та W Re Os lr Pi |Au hg TV Pi Bi Po Atl Rn
I промежу- ковалентные
I точные I гидриды
' гидриды I
89-96
гидриды переходных
металлов**
элементы 67-71 (редкоземельные металлы) La, Се, Рг, Nd, Pm,Sm, Eu, Gd, Tb, Dy
Ho, Er, Tm,Yb, Lu
элементы 89-103(актиноиды)Ac,Th, Pa, U, Np, Pu, Am,Cm,Bk, Cf,Es,Fm,Md, No,Zr
*Солеобразные
** Металлоподобные
Рис. 3.10 Классификация гидридов
Следовательно, в настоящее время можно строить
периодическую систему не только атомов, ядер и электронных структур, но
и периодическую систему молекул.
В заключение отметим, что периодическая система по самой
своей сути материалистична. В ней нашли свое яркое выражение
основные положения диалектического материализма. Прежде
всего— это взаимосвязь. Все элементы объединены той или иной
общностью. Характер этой взаимосвязи различен. Так, элементы
одного и того же периода взаимосвязаны одинаковым числом
уровней у их атомов. Элементы одной и той же подгруппы
взаимосвязаны аналогией в строении электронной оболочки их атомов.
Элементы разных подгрупп, но одной группы взаимосвязаны
одинаковой высшей степенью окисления. «Звездность» системы — яркое
выражение этой взаимосвязи. Связаны элементы и единством
материала, из которого они построены (протоны и нейтроны в ядре,
электроны в оболочке).
100
Все элементы — это линия непрерывного развития материи от
простого (атом водорода) к сложному (атомы тяжелых
элементов). Эту особенность подметил Менделеев, когда предсказал
существование элементов, соответствующих пустым местам его
таблицы. Развитие это идет по восходящей линии в сторону
усложнения структуры атомов, в сторону усиления металличности.
Переход количественных изменений (числа протонов и
нейтронов, числа электронов) в изменения качественные совершается
путем «отрицания отрицания»: у фтора отрицаются свойства лития,
у натрия отрицаются свойства фтора, повторяются свойства лития
на «высшем уровне».
И наконец, это единство и борьба противоположностей.
В каждом элементе существуют признаки металличности и неме-
талличности, окислительные свойства и восстановительные. В
широком понимании термина все элементы — амфотерны.
Настоящая история химии началась с открытия периодического
закона и создания периодической системы элементов.
Вопросы и упражнения
1. Чем определяется положение элемента в периоде, группе, подгруппе?
2. Элементами какого типа начинается и заканчивается каждый периоде
3. Что такое вторичная периодичность?
4. Почему на внешнем энергетическом уровне атомов элементов не бывает
больше восьми электронов? (Ответ давать на примере элементов III и VI
периодов.)
5. Какие электронные конфигурации характерны для семейств лантана и
актиния?
6. Представить электронные конфигурации для элементов главной
подгруппы IV группы; побочной подгруппы VI группы; элементов I группы обеих
подгрупп.
7. Написать полные электронные формулы элементов, в атомах которых
содержатся следующие характерные электронные конфигурации: 5s2; Аръ\ 4d9;
б/12. Определить порядковый номер элементов и назвать их.
8 Объяснить следующую электронную формулу:
Is2 2s2 2р6 3s2 З/?6 3d10 4s2 4/?6 4d" 5s2.
По формуле подсчитать порядковый номер элемента; назвать элемент,
пользуясь таблицей Менделеева; ссумировать электроны по квантовым уровням;
определить, к какому периоду относится элемент; указать, к какой группе и
подгруппе он принадлежит.
9. Строение электронной оболочки атома некоторого элемента следующее
(по энергетическим уровням, начиная с низшего): 2 8 18 32 32 10 2. Написать
полную электронную формулу элемента. Указать, к какому периоду, группе,
подгруппе и электронному семейству относится элемент; порядковый номер
элемента, назвать его.
10. Характерная электронная конфигурация элемента 5/137s2. Написать
полную электронную формулу и представить ее графически с учетом правила
Хунда.
11. Напишите электронные формулы элементов N ПО, 122 и 169. К каким
периодам они относятся? Что можно сказать об их свойствах?
ГЛАВА 4
ХИМИЧЕСКАЯ СВЯЗЬ
И СТРОЕНИЕ МОЛЕКУЛ
§ 1. ВАЛЕНТНАЯ СВЯЗЬ
Оптические спектры молекул отличны от атомарных спектров.
Поэтому можно предположить, что электронные оболочки
атомов обладают лишь относительной устойчивостью и могут
перестраиваться при химических реакциях. Льюис (1916 г.) впервые
обратил внимание на наличие четного числа электронов в
большинстве химических соединений. Из этого факта был сделан
вывод об определяющем значении электронной пары при
возникновении связи. На основе этого представления возник метод
валентных связей.
Поскольку электрон обладает спином, то вследствие этого
собственное вращение электрона связано с возникновением
магнитного поля. Электрон можно рассматривать как магнит. Но
два отдельных магнита притягиваются разноименными полюсами,
причем поля замыкаются друг на друга. При сближении двух
атомов, имеющих неспаренные электроны, их магнитные поля
начинают взаимодействовать. Оба атома сближаются и электроны
теперь притягиваются к обоим положительным ядрам. За счет
этих электрических сил стяжения возникает валентная связь.
Таким образом, валентность элемента в соединении определяется
числом неспаренных электронов его атома, участвующих в
образовании связи.
Орбита, на которой расположены эти теперь общие для двух
взаимодействующих атомов электроны, может принадлежать
либо одному атому, либо быть связанной с обоими ядрами.
В первом случае возникает ионная связь. Атом, потерявший
электрон, становится положительно заряженным катионом. Атом,
перетянувший электрон, становится анионом. Противоположно
заряженные ионы притягиваются друг к другу и располагаются
на таком расстоянии, при котором их притяжение
уравновешивается отталкиванием электронных оболочек и ядер.
Если в соединение друг с другом вступают одинаковые атомы,
например два атома водорода, то одностороннего перетягивания
электронной пары не будет. Орбита, на которой располагаются
2 электрона с противоположными спинами, будет принадлежать
обоим атомам. Такая связь называется ковалентной.
Если общая электронная пара лишь частично оттягивается к
одному из атомов, то образуется полярная связь. Для
характеристики сдвигов зарядов в полярной молекуле можно использовать
102
понятие электроотрицательности. По Малликену, за меру
электроотрицательности принимается величина %а = — (/а + £а)>
где /А — потенциал ионизации; Е — энергия сродства к электрону
атома А. Мерой полярного характера связи А—В будет разность
%а—%в- По Поллингу, за величину разности
электроотрицательности принимается выражение %а — %в = 0,208 J/ДЕ, гдеД£ =
= Е(А — В) — 1/£(А — А)£(В — В); здесь £(А—В) — средняя
энергия связи в АВ; Е(А—А) и Е(В—В) —энергия связи в
двухатомных молекулах А2 и В2 в ккал/моль; 0,208 — коэффициент
перехода от ккал к эв. При А£==0 связь А—В часто ковалентная,
большие значения Е указывают на ионный характер связи.
В табл. 4.1 приведены значения Е(А—В) для некоторых
молекул.
Таблица 4.1
Энергии связей
Связь, ккал/моль
Е (А —В)
Е ковалент. среднегеометрич. =
= V Е(А — А) £(В —В)
АЕ
HF
135
73
62
НС1
103
78
25
Молекула
C1F
60
47
13
NaH
48
44
4
NaCl
99
33
66
KCl
102
27
75
Rbri
111
24
87
Использование современных значений потенциалов ионизации
и сродства к электрону приводит к значениям
электроотрицательности по Малликену (табл. 4.2). Электроотрицательность ато-
Таблица 4.2
Электроотрицательности атомов
Н
7,2
Li Be В С N О F
3,1 4,56 4,31 6,18 7,3 7,54 10,45
Na
3,0
К
2,58
Mg
3,66
А1
3,25
Si
4,80
Р
5,62
S
6,25
Se
5,72
CI
8,35
Br
7,69
Те I
5,4 6,91
мов возрастает при переходе от щелочных металлов к галоидам.
Если взаимодействующие атомы сильно различаются по
электроотрицательности, то образующаяся молекула носит ионный
характер. Величину ионного вклада можно оценить по дипольному
моменту молекулы. Так, для молекулы НС1 величина дипольного
103
момента ц = е-/ = 4,8- Ю"10; 0,22-10~8 = 1,06-10~18 ед. GGSE= 1,06D.
В предположении полного переноса заряда от Н к С1 величина
дипольного момента составит 4,8- Ю-10-1,28- 10~8 = 6,11 D.
Таким образом, степень ионности можно оценить отношением
1,06/6,11=0,17, или 17%. В этом расчете 1,28- Ю-8 см —
межатомное расстояние в молекуле НС1, а 0,22-10~8 см — длина диполя.
Оценка ионного вклада в энергию связи в молекуле НС1 по
данным табл. 4.1 приводит к 25/103 = 0,24, или ~25%, что
удовлетворительно совпадает с предыдущим расчетом.
В заключение следует сказать об известной приближенности
оценок ионности связи, выполненных столь примитивными
методами, хотя в некоторых случаях они могут использоваться для
грубой оценки природы связи.
§ 2. ГИБРИДИЗАЦИЯ ОРБИТАЛЕЙ
Подведем некоторые предварительные итоги, вытекающие из
рассмотренной модели образования химической связи. Во-первых,
единичная связь образуется при взаимодействии двух электронов
с противоположными спинами, принадлежащих различным
атомам. При реакции между двумя атомами Н собственные функции
имеют сферическую симметрию, если электроны в 1 s-состоянии.
Поэтому орбитали, на которые переходят электроны
взаимодействующих атомов, носят ненаправленный характер. Картина
образования связи изменится, если будем рассматривать р-электро-
ны; тогда имеют место 3 возможные атомные орбитали (АО),
которые образуют восьмерки, оси которых направлены по осям
х, у, г. Естественно предположить поэтому, что направление
связи определяется тем направлением, при котором происходит
максимальное перекрывание собственных функций
взаимодействующих атомов.
Таким образом, из двух возможных АО та орбиталь будет
образовывать прочную связь с атомом, которая сильнее
перекрывается АО другого атома. Этот второй вывод является
существенным для трактовки многих особенностей химической связи.
Если в молекуле возникают связи с участием /^-электронов, то
угол между ними должен быть 90°. Рассмотрим с этих позиции
образование молекулы Н20.
Теплота образования Н2Ог из Нг и Ог равна 220 ккал, что
лишь на 10% отличается от удвоенного значения теплоты
образования радикалов ОН—101 ккал. Это позволяет сделать вывод о
близком сходстве связей О—Н в обоих частицах. В парафинах
энергии связей С—С и С—Н также примерно постоянны и
составляют 80 и 98 ккал/моль, длины связей также примерно
постоянны, и вычисление теплоты образования парафинов
отличаются от опытных не более чем на 2 ккал/моль. Подобное
постоянство энергетических характеристик можно объяснить тем,
что связывающие электроны локализованы в пределах одной
связи. Такие локализованные орбитали отражают в современной
104
форме основную идею химической связи как связи между двумя
атомами в молекуле.
Пользуясь критерием максимального перекрывания, можно
дать обоснование возникновения локализованных орбиталей. Для
построения орбиталей О—Н исходим из двух АО атомов Н (1 s)
и двух АО атома О (2рх,у).
Максимальное перекрывание Is- и 2р-орби-
галей можно получить, расположив
атомы водорода по осям х, у (рис. 4.1).
Тогда получим две локализованные
между взаимодействующими атомами
О и Н орбитали, угол между
которыми должен быть 90°. В
действительности угол Н—О—Н равен 104,5°.
Увеличение угла вызвано
отталкиванием атомов Н друг от друга
вследствие того, что они несут некоторый
положительный заряд, а также
отталкиванием самих связей О—Н, так как
но сравнению с атомом О на них
Рис. 4.1. Перекрывание 2ps- и 2/?х-орбиталей
атома кислорода с ls-орбиталями атома
водорода
имеется на один электрон больше. Вследствие этого энергия
отталкивания увеличивается. Связь, образованная электронами (точнее
электронными облаками) по линии, соединяющей центры атомов,
называется сигма-связью и обозначается о.
Электронную структуру молекулы воды можно описать так:
О, Is2, 2s2, 2pl[0, 2px, 2ру-±-Н, Is, of.
Из этой формулы видно, что электроны 1 s2, 2 s2 и 2рх атома О
не принимают участия в связях и остаются на атомных орбита-
лях. Электроны, заключенные в квадратные скобки, изменяют
свое энергетическое состояние и переходят на новые —
молекулярные— орбитали (МО), этих электронов 4.
Необходимо обратить внимание на три важных следствия,
вытекающих из модели метода валентных связей (ВС):
1) применение локализованных молекулярных орбиталей,
охватывающих два взаимодействующих атома;
2) использование критерия максимального перекрывания
электронных облаков при образовании связи;
3) использование принципа аддитивности энергий и длин
связи при термохимических и геометрических характеристиках,
молекул.
105
SP,
Отметим, что локализованные МО не применимы при
описании химической связи в металлах и металлоподобных
соединениях, а также для описания связи в ароматических соединениях
типа бензола.
Понятие гибридизации удобно ввести в теорию химической
связи при объяснении факта 4-валентности углерода во многих
органических соединениях. В низшем
энергетическом состоянии атома
С ls22s22pi, 2ply имеются 2 несларенных
электрона, и углерод должен проявлять
валентность, равную двум. Валентный
угол должен быть 90°. В
возбужденном состоянии один из 25-электронов
может перейти на свободную 2р2-ор-
биталь с образованием конфигурации
-<
Рис 4.2. Образование четырех гибридных
sp-орбиталей
ЛШ Ъ/
<-—Щ0Ш0
ls22s12px2py2pz. Теперь можно ожидать, что углерод сможет
образовывать три однотипных связи и одну связь другого типа. Из
Рис. 4.3. Кубическая модель
для определения угла в
тетраэдре
Рис. 4.4. Строение молекулы
СН4 по методу валентных
связей
геометрических соображений ясно, что углы между связями р—р
будут, как и в молекуле Н20, около 90°, а между связями s—p
около 125°. Однако для СН4 физические и химические данные
свидетельствуют об эквивалентности всех четырех связей и
направленности их под углом 109°28' относительно друг друга. Если бы
связи были неэквивалентны, то следовало бы ожидать
существования изомеров типа C(C1)F3 и C(F)C1F2 (выделенный атом
106
галогена помещен на 2$-орбитали атома С). Однако таких
изомеров не обнаружено.
Получить четыре эквивалентных в энергетическом состоянии
АО атома С можно, если смешать («гибридизировать») 2s- и
2р-орбитали. Полученные орбитали называют гибридными. В
случае СН4 такая орбиталь называется s/?3, она содержит 75% р-ха-
рактера. На рис. 4.2 показано образование 5/?3-орбитали (см.
также рис. 4.3 и 4.4). Основное преимущество гибридной АО — ее
высокая степень направленности, которая приводит к более
сильной связи с присоединяющимися атомами. Прочность s- и /?-свя-
зей по сравнению с 5/?3-связью значительно ниже.
Особенно велико значение гибридизации у переходных
элементов, где появляется возможность использования d-орбиталей.
Могут быть получены гибридные АО с энергией связи,
превосходящей энергию чистой s-связи примерно в 3 раза. Это так
называемые 5р3д!2-орбитали, имеющие большое значение при
объяснении строения многих комплексных соединений. Мы вернемся к
вопросу о гибридных орбиталях при обсуждении метода
молекулярных орбиталей.
§ 3. МЕТОД МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
Метод валентных связей — это не единственный
приближенный метод рассмотрения химической связи. Существуют
химические соединения, для которых метод ВС с локализацией
валентных электронов между двумя взаимодействующими атомами не
применим, так как не может объяснить их свойства. Идея о
локализации электронов не может объяснить, например,
электропроводности металлов и интерметаллических соединений. Если
валентные электроны прочно закреплены на связях между
соседними атомами металла, то возникновение проводимости
невозможно. Оказывается, что удовлетворительное объяснение свойств
металлов, ароматических соединений и возбужденных состояний
молекул можно получить в предположении существования
многоцентровых молекулярных орбиталей.
Основные положения метода молекулярных орбиталей
следующие:
1. Каждому электрону в молекуле соответствует волновая
функция г|?, определяющая его «орбиту» в молекуле. Если
молекула состоит из двух атомов (Н2, 02), такая орбиталь будет
двухцентровой. Если молекула состоит из большого числа
атомов, орбиталь называется многоцентровой.
2. Волновая функция характеризуется квантовыми числами,
определяющими форму и энергию уровня, на котором находится
электрон.
3. Электронная структура молекулы устанавливается с
помощью принципа построения, как это делалось при установлении
электронной структуры атома. Сначала определяются дозволен-
107
ные орбитали, а затем в соответствии с принципом Паули на них
размещаются электроны. Применяя некоторое упрощение, можно
сказать, что в двухатомной молекуле валентный электрон
движется по орбите, проходящей вблизи обоих ядер. При этом на
электрон, находящийся вблизи одного из ядер, действуют силы
притяжения со стороны этого ядра и силы отталкивания
находящихся здесь других электронов. Предполагается, что в этом
случае МО приблизительно совпадает с АО. Вблизи другого ядра
- Рис. 4.5. Схема образования свя- Рис. 4.6. Схема образования раз-
зующей МО в ионе Н^" рыхляющей МО в ионе Н^
МО будет напоминать АО этого второго ядра. Исходя из этих
предпосылок, можно полагать, что в целом МО характеризуется
теми же особенностями, что и каждая АО в отдельности. При
конструировании МО пользуются методом линейных комбинаций
атомных орбит. Для гомонуклеарной молекулы Н2 можно
записать MO = \|)(Hi) +г|)(Н2). Если молекула гетеронуклеарна, надо
ввести меру полярности К и записать MO = if)(A) + Ад|)(В). Этот
приближенный метод получил название линейной комбинации
атомных орбиталей — МО ЛКАО.
Какие же орбитали г|)(А) и гр(В) могут комбинировать с
образованием МО? Комбинация бывает эффективной, если:
^энергии, соответствующие яр (А) и г|)(В) атомов А и В, сравнимы по
величине; 2) зарядовые облака гр (А) и ф(В) перекрываются и
3) исходные атомные функции обладают одинаковыми
свойствами симметрии относительно оси молекулы А—А.
Рассмотрим простой пример — молекулярный ион водорода
Hf. Поскольку молекула Hf одноэлектронная, задача расчета
волновой функции и вычисления энергии связи и межъядерного
расстояния в ней имеет такое же значение в теории молекул, как
задача атома водорода в теории атомов.
Линейную комбинацию атомных орбиталей можно получить
сложением или вычитанием АО МО = г|э(А) =tгр (В) (рис. 4.5, 4.6).
Находясь на связывающей орбитали, электрон одновременно
притягивается к двум протонам. При подобном перекрывании
зарядовых облаков сила притяжения достигает большой
величины, электрон «связывает» два ядра в новую стабильную
частицу— молекулу. Связывающая орбиталь обладает цилиндриче-
108
ской симметрией относительно оси, соединяющей ядра. При
вращении вокруг оси ее вид остается без изменений.
При вычитании МО = г|э(А)—г|)(В) получим разрыхляющую
орбиталь с пониженной электронной плотностью между ядрами.
J»3P
Орбиталь
Н
Молекулярные Орбиталь
орбитали Н
разр
Рис 4.7. Изменение электронной плотности if>2 для о*Связ и аразр
Заряд, отвечающий я|)разр, вытолкнут из пространства между
ядрами. На рис. 4.7 показаны изменения электронной плотности г|)2
вдоль оси в случае
притяжения и отталкивания.
Поскольку притяжение единственного
электрона двумя ядрами
сильнее, чем одним в случае атома
Н, то энергия связывающей
орбитали должна возрасти на
некоторую величину р. Если
мы обозначим кулоновский
потенциал ионизации валентного
электрона Is через Е, то
энергия аСВяз будет £ + р, а
энергия разрыхляющей орбитали
будет Е—р.
Потенциальная энергия
рассматриваемой системы выразится
уравнением
1sa /
\ 1su
Ho—
v = -
rAB
Рис. 4.8.
Энергетические
иона Hjj"
уровни МО
Наглядное представление о соотношении энергетических
уровней сТсвяз и <Гразр дает диаграмма энергетических уровней.
Орбиталь Освяз более устойчива, чем исходная АО для JH на величину
энергии связи. Ее уровень лежит ниже уровня АО. Уровень аразр
лежит выше уровня АО на величину р (рис. 4.8).
При изучении основного состояния молекулы следует выбрать
низшие орбитали атома Н. Им соответствуют волновые функции
вида г|) = (яао)-1/2 е~г,а\ с помощью которых вычисляют р для
любого межъядерного расстояния R. Определив электронную
109
энергию Е и складывая ее с энергией кулоновского отталкивания
ядер е2/г, можно найти кривую потенциальной энергии для
молекулы. В точке, где /? = 1,06 А, кривая (Тсвяз имеет минимум.
Потенциальная энергия электрона становится по абсолютной величине
больше, чем в том случае, когда электрон находится у одного
ядра. В случае ара3р возрастания зарядовой плотности между яд-
Н ;^Ш
.*;/.■■'V*v*
'v
ИЛИ
15,
£ Ь.
сбязь/ бающая
пара электронов
Высокая
энергия
I
Низкая
энергия
уа=0 изолированные атомы
_^—^
Увеличение R
Рис. 4.9. Связь в молекуле
водорода за счет пары
электронов
Рис. 4.10. Зависимость энергии
системы, состоящей из двух
атомов водорода, от расстояния
между ядрами
рами нет, и выигрыш в потенциальной энергии недостаточен для
преодоления энергии отталкивания ядер.
Мы нашли МО для системы из двух протонов и двух
электронов. Полученные орбитали пригодны при рассмотрении
Hi" и Н2. В Н2 имеется 2 электрона, которые в соответствии с
принципом Паули могут быть размещены на асвяз- Таким
образом, основное состояние Н2 (аСВяз)2. Основное различие между
Н и Н2 заключается' в появлении второго связывающего
электрона. Потенциальная кривая по форме аналогична кривой для
H<j~, но ее минимум находится в точке # = 0,74 А. Это расстояние
меньше равновесного в Hf (рис. 4.9, 4.10).
Согласно предложению Льюиса можно считать, что
нормальная связь образуется парой электронов. В таком случае число
связей в молекуле равно V2 (число электронов на связывающей
МО минус число электронов на разрыхляющей МО/2). В
соответствии с этим в Н* имеется половина а-связи, а в Н2 — целая
а-связь. Энергия диссоциации соответственно 62 и 104 ккал/моль.
На рис. 4.11 представлена диаграмма энергетических уровней Н2,
при этом связывающие электроны расположены на нижнем
энергетическом уровне. Известно, что атомы или молекулы, имеющие
неспаренные электроны, парамагнитны, а имеющие спаренные
электроны — диамагнитны.
ПО
В полном соответствии с опытными данными из
энергетических диаграмм можно сделать вывод о парамагнитных свойствах
Щ" и диамагнитных свойствах Нг-
Помимо рассмотренных Н2" и Н2 известен также
молекулярный ион водорода Н<Г. Нетрудно видеть, что третий электрон
h
а \
к
Нея
Рис. 4.11. Диаграмма энергетиче- Рис. 4.12. Диаграмма энергетических
ских уровней Н2 уровней Не2 и Hef
может быть размещен только на разрыхляющей орбитали.
Введение третьего электрона приводит к уменьшению энергии
связи— в молекуле HiT £ = 4 ккал, что прямо указывает на ее
неустойчивость. По абсолютной величине такая энергия сравнима
с энергетическими эффектами фазовых превращений, например,
теплота плавления воды 1,4 ккал/моль, а теплота испарения —
9,7 ккал/моль.
Рассмотрев частицы, образуемые атомами водорода, придем
к понятию насыщаемости химической связи. Известно, что
энергия связи в молекуле определяется числом электронов на
связывающих и разрыхляющих орбиталях. Действительно, если
попробуем представить энергетическую диаграмму Не2, то увидим
расположение валентных электронов, указанное на рис. 4.12.
Очевидно, что размещение четырех электронов попарно на
связывающей и разрыхляющей орбиталях приводит к тому, что
число связей в молекуле равно нулю. Это объясняет химическую
инертность Не. Между атомами и молекулами с замкнутыми
оболочками существует отталкивание. В то же время ион Не^"
известен, энергия связи в этом ионе 60 ккал/моль, т. е. близка к
энергии связи в ионе Щ~ с половиной а-связи.
Метод молекулярных орбиталей особенно ценен в том
отношении, что позволяет описывать возбужденные состояния
молекул. Если один из валентных электронов Не2 возбудить на более
высокий уровень, то может получиться устойчивая молекула.
Такие устойчиво возбужденные молекулы обнаружены
спектроскопически. Конфигурация сСВяз, сТра3р встречается во всех
других более тяжелых молекулах.
111
Рассмотрение образования молекулы начинается с
размещения валентных электронов в поле сближающихся ядер. Если этих
ядер несколько, то электроны заполняют многоцентровые орби-
тали в соответствии с принципом Паули. Насыщенность связи —
следствие образования замкнутых оболочек атомов или молекул.
Гомонуклеарные молекулы элементов II периода, а- и я-Орби-
тали. Валентными атомными орбиталями атомов II периода
Е
Е
1 б
/ \ разр
/ \
У v
* \ /8
Рис. 4.13.
Диаграмма
энергетических уровней
Li2
являются 25-, 2рх-, 2ру-, 2/?2-Орбитали. В качестве единой оси для
любой гомонуклеарной молекулы А2 выберем ось г. Будем
конструировать МО при сложении или вычитании АО, способных
перекрываться между собой.
При перекрывании 2я-орбиталей получим аСВяз-орбиталь, как
и в случае молекулы Н2, где перекрывались Is АО. Вследствие
ненаправленного характера s-орбиталей полученная МО имеет
сравнительно малую энергию связи. Такая связь, например,
осуществляется в молекуле Li2. Основное состояние молекулы Li2—
<?2связ> так как оба электрона находятся в соответствии с
принципом Паули на связывающей МО. Такая молекула диамагнитна,
так как нет неспаренных электронов. Межъядерное расстояние
2,67 А, что значительно больше, чем для Н2 с ее ls-связью.
Причиной этого является экранирование валентных электронов
лития электронами на внутренней /(-оболочке. Кроме того,
понижение энергии связи обусловлено отталкиванием двух ls-nap.
Энергия связи Li2 26 ккал.
Молекула Ве2. Валентная конфигурация атома Be в
нормальном состоянии 2s2. Молекула Ве2 должна иметь конфигурацию
<7Связ, ^разр» которая отвечает отсутствию связей.
Действительно, молекула не обнаружена.
На рис. 4.13, 4.14 приведены энергетические диаграммы Li2
и Ве2.
Атомы следующих элементов имеют электроны на р-уровкях.
Система координат показана на рис. 4.15.
/4-bf3P
Be б
ос2 связ
5ис. 4.14. Диаг-
>амма энергети-
[еских уровней
Ве2
2 Z
№ »■ ■*—Mb)-
Рис. 4.15. Система
координат для молекулы типа А2
112
Рассмотрим образование связывающих и разрыхляющих ор-
биталей для р-электронов. На рис. 4.16 показано перекрывание
двух АО /?2-орбиталей с образованием связи. Такая связь будет
а-связью, она симметрична относительно оси молекулы.
При образовании аразр-орбитали плотность зарядового облака
между атомами понижена, между центрами ядер появляется
узел. Орбитали 2ру и 2рх не обладают осевой симметрией
относительно оси х. Вследствие ган-
телеобразной формы этих АО
связывающая и разрыхляющая
МО, получаемые сложением и
вычитанием, имеют разные
знаки от оси г. При повороте
вокруг оси z орбитали изменят
знак на противоположный. Та-
Рис. 4.16. а — перекрытие двух
валентных 25-орбиталей молекулы А2;
б — перекрытие двух валентных 2р-
орбиталей молекулы А2
кая орбиталь имеет узловую плоскость. В отличие от сг2-орбитали„
образованные ру- и /?х-электронами, называются я-орбиталями.
Рис. 4 17. Перекрытие 2/?л--орби-
талей в молекуле А2
Рис. 4 18 Поворот д-МО на 180е
вокруг оси, проходящей через
ядра
узло&ая плоскость
исходная орбиталь
Более устойчивые яСВяз обладают повышенной плотностью
зарядового облака между ядрами, менее устойчивая яразР имеет узловую
плоскость между ядрами (рис. 4.17—4.19).
на
Теперь можно составить полную диаграмму энергетических
уровней таких МО. Известно, что уровень 2s более устойчив,
чем 2р. В атоме Li разность энергий этих уровней 1,9 эв, а в
атоме F — более 20 эв. Поэтому на диаграмме орбиталь 2р должна
размещаться выше 2s. Далее надо отобразить расщепление
исходных атомных уровней с образованием а2,Связ, oZtVa3?, псвя31
Яразр. Относительное расположение oz- и ях, ^-уровней зависит
от гибридизации 5/7-типа. В отсутствие гибридизации орбиталь
Oz, связ лежит ниже, чем лх, у-
При значительном вкладе
s-состояния связь а2,связ
становится менее
устойчивой и ее уровень
располагается выше уровней лХгУ.
Молекулярные орбитали пх
и пу изображены лежащими
на одном уровне, их энергии
равны, орбитали являются
вырожденными (рис. 4.20).
^связ разр л л
-J» и Т\ экбибалентны
_связ _
разр
Рис. 4.19. Граничные поверхности
а- и л-МО, образованных за
счет валентных s- и /7-орбиталей
в гомонуклеарной двухатомной
молекуле
Молекула В2. Валентное состояние атома 2s22pl.
Экспериментальные данные указывают на наличие двух неспаренных
электронов на уровне 2яХ)У)Связ- Таким образом, конфигурация В2
{Ве2) (ях>у,Связ)- Символом Ве2 обозначена сокращенно
конфигурация о^связ, а^разр» которая появляется у Ве2 и затем повторяется
у остальных двухатомных молекул. Энергия связи В2 63 ккал,
Я =1,59 А. Видно, что связь, образованная /7-электронами,
прочнее связи, образованной s-электронами, что прямо связано
с острой направленностью /?-облака в пространстве.
Молекула С2. Валентная конфигурация атома ls22s22p2.
Основное состояние С2 (Ве2) (я*, у)4Связ. Количество неспаренных
электронов равно нулю, £"=150 ккал, /?=1,37 А.
Молекула N2. Валентное состояние атома ls22s22p3. Основное
состояние N2 (Ве2)(ях,у)4
СВЯЗ' Gtf.C
£-226 ккал, Я =1,10 А.
Молекула очень прочна, что согласуется с малой реакционной
способностью газообразного азота. Заполнение всех связывающих МО
закончено на азоте.
Следует ожидать уменьшения энергии связи и возрастания
межъядерного расстояния у молекулы 02. Известно, что 02 пара-
114
магнитен и его парамагнетизм соответствует двум неспареиным
электронам. Можно получить парамагнитную молекулу, разместив
Ордитоли А (а) ОрЬитали А
2
розр
ОрЬитали А(Ь)
ОрЬитали А(о) ОрЬитали А
ОрЬитали А(Ь1
^родр
/ЛЯ^ВЧ^/^РЯ^ч
^^ООЧ^.
* 2
Рис. 4.20. Энергетические уровни МО гомонуклеарных
двухатомных молекул без взаимодействия s—р (а) и
при заметном взаимодействии sp (б)
ЭЛеКТрОНЫ ПО ОДНОМУ На TLx-> 71у, разр" орбиталях. Электронная
конфигурация 02(Ве2)(сг?)СВяз> (%«)связ, (%*)разР. Объяснение
парамагнетизма О2 невозможно, если оставаться в рамках концепции
115
Льюиса. В молекуле 2 связи — одна а и одна я, £=119 ккал,
/? = 1,21 А. При удалении одного разрыхляющего электрона
межатомное расстояние уменьшается до 1,12 А, а энергия связи
возрастает до 154 ккал. Добавление электрона на уровень (яразр)
приводит к образованию иона ОТ с R = 1,26 А, добавление
второго электрона к иону 02~ с R = 1,46 А. На этом примере
отчетливо видно, как зависит прочность связи от соотношения
связывающих и разрыхляющих электронов. По тривиальной схеме
распределения электронов в атоме О по ячейкам ls22s22p4 при
образовании молекулы 02 следовало бы ожидать в соответствии с
принципом Паули размещения валентных электронов второго
атома О в наполовину заполненные р-ячейки с образованием пар
с противоположными спинами. Как следствие этого, молекула 02
должна быть диамагнитной, что противоречит
экспериментальным данным. Объяснение парамагнетизма 02 дает лишь метод
МО ЛКАО.
Молекула F2. Конфигурация атома F ls22s22p5, конфигурация
Р2(а2)связ, (я*,у)связ, (%з)разР, что соответствует одной
результирующей Gz-связи, образованной парой р2-электронов. £ = 36 ккал,
/?= 1,42 А, молекула диамагнитна.
Молекула Ne2 не существует, так как в ней был бы заполнен
уровень (ог)?а^> что отвечает отсутствию связей.
Интересно рассмотреть свойства молекул щелочных металлов
и галогенов как наиболее гетерополярных элементов. В табл. 4.3
представлены межатомные расстояния,
энергия диссоциации и типы связей в
этих молекулах. Видно, что связи,
образуемые 5-электронами, в молекулах
щелочных металлов менее прочны,
чем связи, образуемые р-электронами
в молекулах галогенов. Причина
этого — ненаправленность s-связей.
Молекула Li2 по величине энергии
диссоциации заметно отличается от других
Ме2 вследствие примеси р-характера,
так как разность энергий s- и р-уров-
ней сравнительно невелика (~1,9 эв),
что способствует образованию
яр-гибридной орбитали с большей энергией
связи. Немонотонный характер изменения энергии диссоциации в
молекулах галогенов обусловлен возможностью, начиная с С12,
включения в связь J-орбиталей и уменьшением взаимного
отталкивания электронных пар на р2-орбиталях вследствие увеличения
размеров атомов. Все молекулы диамагнитны.
В заключение подчеркиваем, что в соответствии с принципом
максимального перекрывания я-связь слабее а-связи, т. е. я-связь
более реакционноспособна. Наличие я-связи в молекулах ненасы-
Таблица 4.3
Молекула
R, А
Едисс ккал
Li2
Na2
к2
Rb2
Cs,
F2
CI
Br2
L
2,67
3,08
3,92
—
—
1,42
1,99
2,28
2,67
26
13
13-
10
11
38
58
46
36
116
щенных углеводородов (С2Н4 и др.) обусловливает их
повышенную реакционную способность по сравнению с парафинами.
Гетеронуклеарные молекулы. Одна из простейших гетерону-
клеарных молекул — молекула гидрида лития LiH. Валентные
орбитали лития 2s, 2рх, 2py, 2pz.
Для образования связей необходимо перекрывание сходных
орбнталей Is, 2s, 2pz. Орбитали 2pXi 2py являются л;-орбиталями,
Рис. 4.21. Перекрывание
ls-орбиталей атома
водорода с валентными ор-
биталямн атома лития
О
I
сбяз
<3
Рис. 4.22. Граничная
поверхность связующей
о-МО в молекуле LiH
они обладают отличной от ls-орбитали симметрией. В таком
случае говорят, что эти орбитали ортогональны.
Результирующее перекрывание Is- и я-орбиталей равно нулю, что
соответствует отсутствию перекрывания. На рис. 4.21—4.23 показаны
различные случаи перекрывания АО лития и водорода.
Поскольку ионизационные потенциалы Н и Li сильно
различаются (13,6 и 5,4 эв), то следует ожидать, что электроны сь-свя-
зи будут находиться в основном у ядра Н. Вид связывающего
уровня изображен на рис. 4.24. Уровень АО 2рх-, 2/^-орбиталей
совпадает с уровнем МО — я*-, %-орбиталей, так как в атоме Н
нет орбиталей, способных с ними взаимодействовать.
Составим схему энергетических уровней LiH. В левой части
диаграммы поместим валентные орбитали с учетом их энергии.
В правой части — уровень Is атома Н. В середине диаграммы
находятся уровни (Тсвяз и ^разр МО. Связывающая орбиталь
образуется в основном за счет орбитали Is атома Н, участие в ней
2s- и 2р2-орбиталей атома Li меньше. Уровни я-типа совпадают
с АО атома Li.
Оба электрона на сгСвяз находятся вблизи атома Н, вследствие
чего центры тяжести положительных и отрицательных зарядов в
молекуле не совпадают. Поэтому на Li возникает заряд +, а на
Н — заряд —. Разделение зарядов гетеронуклеарной молекулы
типа LiH вызывает появление электрического дипольного
момента \х = е-1. Дипольный момент (эксперим.) 5,9 D; /? = 1,60 А. Для
117
ерекрыбание
r U-равны, результирующее
перекрыбание рабно нулю
то же для 2py11s
2D результирующее перекрыбание
** между 2р и 1s рабно нулю
чисто ионной связи дипольный момент был бы равен 7,7 D.
Таким образом, частичный заряд равен 0,77 е.
Можно обобщить рассмотрение МО в молекулах типа АхВ^,
указав, что имеется четыре вида МО — несвязывающие орбитали
внутренних оболочек, несвязывающие орбитали валентной
оболочки, связывающие и разрыхляющие. В молекуле Н20 несвязы-
Орбитали Орбитали Орбитали
Рис. 4.23. Граничные по- Рис. 4.24. Энергетические уровни орби-
верхности в2, разр и талей молекулы LiH
o*s, разр-МО в
молекуле LiH
вающими орбиталями будут орбитали iC-оболочки атома О, 2pz-
орбитали О, а связывающими—ls-орбитали атомов Н и 2рХуУ-
орбитали атома О.
Изоэлектронные молекулы. Изоэлектронными называются
молекулы с одинаковым числом внешних электронов,
расположенных на сходных молекулярных орбиталях. В ряде случаев МО
ЛКАО более верно отражает экспериментальные свойства
молекул, чем упрощенная трактовка связи по Льюису. Рассмотрим
диаграмму энергетических уровней молекулы СО, изоэлектрон-
ной с N2 (рис. 4.25). Допустим, что мы превращаем N2 в СО
постепенным оттягиванием положительного заряда от одного ядра
азота и добавлением к другому ядру. Можно представить, что
МО молекулы N2 постепенно превратятся в МО молекулы СО.
Схема МО будет аналогична схеме МО молекулы азота, причем
число связей — три, из них две — я- и одна — а-связь, что
находится в полном соответствии с прочностью молекулы СО
(£ = 255 ккал, /? = 1,13 А).
Если использовать тривиальную схему спаривания электронов
по Льюису, то, приписывая О валентность, равную двум,
получим, что в СО две связи. Однако этот вывод никак не
согласуется с необычной прочностью СО.
118
Молекула BF, которая наблюдается в спектре при высоких
температурах, имеет £"=186 ккал и jR = 1,26 А. Если
предполагать, что валентность F равна единице, то в молекуле должна
быть только одна связь. Это находится в полном противоречии с
высокой энергией связи молекулы, превышающей даже энергию
связи в 02, где имеются две результирующие связи. Однако если
в соответствии с методом МО ЛКАО разместить все валентные
электроны В и F по пяти орбиталям МО, то получим одну а и
две л-связи. Наличие трех связей определяет устойчивость этой
молекулы. Таким образом, понятие валентности расширяется —
в связи могут участвовать не только неспаренные электроны, но
и все валентные электроны с квантовым числом 2.
Рис. 4.26. Строение и валентные
МО я-орбитали молекулы С6Н6
§ 4. ДЕЛОКАЛИЗАЦИЯ ЭЛЕКТРОНОВ.
МЕТАЛЛИЧЕСКАЯ СВЯЗЬ. ЭНЕРГЕТИЧЕСКИЕ ЗОНЫ
В ранее рассмотренных молекулах существуют
локализованные связи. Однако существуют другие классы химических
соединений, к которым идея локализации электронов между двумя
взаимодействующими атомами неприменима. Примером таких
соединений являются ароматические углеводороды типа бензола.
Помимо а-связей С—Н и С—С, на которых располагаются
24 электрона (6 — от Н, 18 — от С), в молекуле бензола имеются
еще три я-связи, на которых располагаются остальные 6
электронов атомов углерода. Таким образом, двойная связь С = С в
бензоле состоит из одной а- и одной (72я)-связи. Если
применить ранее рассмотренный принцип спаривания я-электронов при
образовании связей между соседними атомами С, то сразу
выясняется, что могут быть различные схемы спаривания,
отвечающие различным структурам Кекуле (рис. 4.26).
2
S разр Wi
Рис. 4.25. Схема
энергетических
уровней N2, СО и BF
119
Поэтому нельзя объединять АО попарно, как в случае Н20,
потому что каждая я-орбиталь перекрывается двумя соседними
орбиталями. Молекулярные я-орбитали в С6Нб показаны на
рис. 4.26. Электроны на я-орбиталях движутся свободно по
замкнутому шестиугольнику, они называются подвижными, или
ненасыщенными электронами. Естественно, что если один
электрон движется от атома 1 к атому 2, второй электрон движется
от атома 2 к атому 1, и в нормальном состоянии
результирующий ток равен нулю. Подвижность я-электронов означает, что
молекула С6Нб может иметь свойства, сближающие ее с
металлическими проводниками электричества.
Обычно, излагая сведения о характере металлической связи
в металлах, подчеркивают тот факт, что металл характеризуется
высокой электропроводностью, что связывается с присутствием
свободных электронов. Объясняя строение СбИб, мы видели,
каким образом можно подойти к понятию делокализации
электронов. Для этого надо, чтобы наша модель распределения
валентных электронов учитывала принадлежность электронов
нескольким атомам. Чтобы МО была делокализованной, надо, чтобы
атомная орбиталь одного атома значительно перекрывалась'с АО
нескольких других атомов. При сближении атомов каждый
электрон имеет возможность перейти на АО соседних атомов. Это
уменьшает кинетическую энергию и увеличивает потенциальную
энергию притяжения. Известно, что распространение волновой
функции на соседние атомы ограничено принципом Паули. Когда
несколько электронов занимают один и тот же объем, они
должны находиться в различных энергетических состояних, или иметь
противоположные спины. Если такая возможность существует,
то, как видно на примере молекулы Н2, возможен выигрыш
энергии при переходе электронов на МО. Рассмотрим с точки зрения
метода МО ЛКАО возникновение металлической связи в литии.
По данным рентгеноструктурных исследований, литий имеет
объемноцентрированную решетку, причем каждый атом окружен
14-ю соседними, из которых 8 находятся на ближайшем
расстоянии /?, а 6 — на расстоянии 1,15/?. Один 25-электрон должен
обеспечивать связи по крайней мере с 8-ю соседями на расстоянии
R = 3 А. Иными словами, волновая функция электрона
центрального атома перекрывается с волновыми функциями 8-ми соседних
атомов. С физической точки зрения металлы представляют
бесконечный набор атомов в отличие от молекулы типа бензола, где
число атомов конечно. Число атомов металла в 1 см3 ~ 1023.
Орбитали для такого набора атомов отличаются от
молекулярных орбиталей, с которыми мы встречались раньше.
Если при добавлении к атому А атома В атомная орбиталь
переходит в две двухатомные орбитали г|)(А) ±ф(В), каждой из
которых соответствует одна энергия, то при добавлении третьего
атома получим уже три МО, уровни энергии которых будут
группироваться вблизи первоначальной орбитали. Каждое последую-
120
щее добавление атома приводит к появлению еще одного
энергетического уровня (рис. 4.27). Полное число уровней будет 1023,
каждой из разрешенных орбиталей будет соответствовать одна
полоса, поскольку уровни располагаются очень близко и
сливаются в одну непрерывную полосу. Если АО, например 25 и 2р
К—N
2 V
\ _
3 \
4 число ~-^
amonog сю
2р
2S
Рис. 4 27. а — расположение энергетических уровней
в металлическом литии; б — энергетические зоны
в литии
лития, удалены друг от друга, то перекрытие мало, если же АО
близки, то возможна гибридизация образующейся МО.
Интересно сравнить энергетические характеристики
молекулы Li2 и металлического лития. Теплота атомизации — перехода
в состояние одноатомного газа — металлического лития Д#а =
= 39 ккал/моль, а £,ДИСсЬ12 = 26 ккал/моль. Следовательно, в
расчете на г-атом энергия связи в кристалле лития возрастает
втрое.
Длина связи в кристаллическом литии 3,0 А, что больше
длины связи в Li2 (2,67 А). Электроны проводимости в металле свя-
121
заны сильнее, чем валентные электроны в молекуле, но их
связывающая сила распределена между большим числом атомов.
Если N атомов образуют кристалл, то вместо отдельных
квантовых состояний изолированных атомов образуется зона,
содержащая 2N состояний. В соответствии с принципом Паули в
зоне может находиться 2 N вакантных мест для электронов
щелочного металла. Поскольку на каждый атом лития имеется один
валентный электрон, то зона будет заполнена лишь наполовину.
Возникновение проводимости объясняется тем, что под влиянием
электрического поля электроны могут перераспределиться по
скоростям и сравнительно с небольшой затратой энергии
переходить на свободные уровни в зоне проводимости. Таким образом,
МО молекулы переходят в орбитали проводимости в металле.
При сближении атомов уровни отдельных АО 2р и 2s
расширяются в полосу, при этом возможно перекрытие полос.
Минимум энергии соответствует равновесному расстоянию между
атомами в кристаллической решетке.
В каких веществах будет наблюдаться электронная
проводимость? В тех веществах, где зона проводимости имеет свобрдные
энергетические уровни, на которые могут перейти электроны под
воздействием внешнего поля, следует ожидать металлическую
проводимость. Если в кристалле имеются полностью
заполненные зоны и пустые зоны, то внешнее поле не может
перераспределить электроны заполненной зоны, находящиеся на
молекулярных орбитах. Перевод электронов из состояний одной зоны в
состояние другой зоны с большей энергией возможен лишь в
случае очень большой разности потенциалов (пробой диэлектриков).
В заключение остановимся подробнее на значении
гибридизации валентных электронов для понимания возникновения
механизма проводимости в щелочноземельных металлах. В бериллии
при образовании кристаллов из атомов образуются Is- и 2s-30Hbi,
имеющие по 2N мест, т. е. столько же, сколько имеют
электронов эти N атомов. Следовало бы ожидать, что обе зоны будут
полностью заполнены и Be будет диэлектриком, что
противоречит металлической проводимости Be. Возникновение
проводимости объясняется тем, что возникшая из 2р-уровней Be зона
частично перекрывается с заполненной 2s-30hou. 2/?-зона имеет 6N
вакантных мест для электронов и объединенная 2sp-30Ha имеет
8N мест, из которых заполнены лишь 2N. Существование
свободных уровней в гибридизированной зоне определяет
металлический тип проводимости бериллия.
§ 5. КОВАЛЕНТНЫЕ СВЯЗИ
В ПЕРЕХОДНЫХ МЕТАЛЛАХ
Развитие эксперимента позволило поставить вопрос: какая
часть валентных электронов металла принимает участие в
проводимости? Можно ли представить, например, структуру метал-
122
лического циркония как состоящую из ионов Zr4+ с
концентрацией свободных электронов 4/атом? Целый ряд физических
свойств переходных металлов (температуры плавления, кипения,
теплоты атомизации, твердость, межатомные расстояния)
свидетельствует о том, что нельзя сводить химические связи в
переходных металлах только к типу
металлической связи, характерному
для щелочных и
щелочноземельных металлов.
Физические методы
исследования приводят к заключению,
что в переходных металлах лишь
малая часть валентных
электронов находится в зоне
проводимости. Число свободных электронов
невелико — около 1/атом.
Остальные электроны осуществляют
направленные связи между
соседними атомами. Эти связи в d-ме-
таллах появляются при
достаточном перекрывании d-орбиталей.
Рис 4.28. Зависимость температуры
плавления, температуры кипения,
теплоты испарения АН металлов VI
периода от порядкового номера
Установлено, например, что в Zr имеется 1,4 электрона на атом в
свободном состоянии, а в Nb—1,2 электрона, остальные валентные
электроны находятся в d-зоне и образуют прочные ковалентные
связи. Таким образом, в большинстве элементов с металлическим
типом проводимости в состоянии простого тела имеется
существенная доля межатомной ковалентной связи. Свободные электроны
осуществляют типичную металлическую связь, характерную для
щелочных и щелочноземельных металлов с малой энергией
связи — около 20 ккал/эл.
В переходных rf-металлах, характеризующихся высокими
температурами плавления и кипения, высокими энергиями
атомизации, основной вклад в энергию связи представляют d-электроны.
На рис. 4.28 видно, что металлы V и VI групп наиболее
тугоплавкие и наиболее трудно испаряющиеся. Когда же начинается
заполнение разрыхляющей половины d-зоны, то это приводит к
уменьшению числа связей, что отражается в падении величины
теплоты атомизации Д#а, уменьшении температуры кипения и
зозрастании ковкости переходных металлов к концу периода.
Отметим, что полностью заполненную d-оболочку нельзя
рассматривать как внутреннюю, не участвующую в связях. Изоли-
дН,ккал/г-отом
2001- а «lW
1а 2а 34 5 6 7 W-/ 16 26
8
группы
123
рованный атом Си имеет конфигурацию 3d10 4s1, т. е. вне
замкнутого остова находится один электрон, однако в
противоположность калию, у которого внутренние 2pG электроны не участвуют
в связях, электронные облака d-электронов меди перекрываются.
Это приводит к значительному увеличению энергии связи
Д#аСи = 81 ккал/г-атом; Д#аК = 22 ккал/г-атом.
При объяснении особенностей связи в металлах удобно
пользоваться понятиями метода ВС и метода МО ЛКАО. Метод ВС
применяется при описании энергий связи и межатомных
расстояний, которые зависят от числа связывающих и разрыхляющих
электронов, метод МО ЛКАО — при трактовке явлений
проводимости.
§ 6. СРАВНЕНИЕ МЕТОДА ВАЛЕНТНЫХ СВЯЗЕЙ
И МЕТОДА МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
1. Оба метода приближенны. В методе ВС рассматривается
только одна схема спаривания электронов, остальными можно
пренебречь. Так, в молекуле воды рассматривается спаривание
ls-электронов Н с 2/?х, ^-электронами атома О и не
рассматривается перекрывание атомных орбиталей атома водорода друг с
другом. В методе МО рассматривается поведение электронов у
объединенного ядра молекулы, т. е. предполагается, что одно-
электронные функции, удовлетворительные для атомов, пригодны
и для молекулы. Эти факторы приводят к ошибкам при
определении энергии связи 1—2 эв.
2. В обоих методах вводится понятие о спаривании спинов
электронов.
3. Оба метода указывают на повышение плотности
отрицательного заряда между ядрами.
4. В методе МО вводится понятие о связывающих и
разрыхляющих орбиталях и электронах. Это дает возможность
объяснить, почему в некоторых случаях (02, 0§) энергия связи
возрастает с уменьшением числа электронов, находящихся на связях.
5. В методе ВС валентность атома определяется числом
электронных пар, осуществляющих связь с другими атомами.
Например, валентность азота в NH3 и NH4* равна 3 и 4. В методе
ВС атомы сохраняют свою индивидуальность, а связи
образуются при сближении электронов взаимодействующих атомов. В
методе МО число связей можно определить как полуразность
между числами связывающих и разрыхляющих электронов. Так, в 02>
ОГ, Of и 0\~ число связей между атомами кислорода равно 2;
1,5; 2,5; 1, т. е. возникает возможность дробной валентности.
Молекула в методе МО рассматривается как частица, при
создании которой атомы потеряли свою индивидуальность. В связи с
этим понятие валентности атома в молекуле для метода
молекулярных орбиталей теряет смысл.
124
Таким образом, традиционное понятие валентности атома в
молекуле, вообще говоря, зависит от способа рассмотрения, от
модели химической связи.
Вопросы и упражнения
1. Дайте определение понятиям валентность и степень окисления.
2. Определите степень окисления Fe в FeO, Fe203, Fe304-
3. В сторону какого атома будут смещаться валентные электроны в Na20,
Н20, F20, CaO, С02?
4. Дипольные моменты молекул H2S и S02 равны 0,93Z) и 1,6Ш.
Вычислите длину диполя и укажите более полярную молекулу.
5. Почему молекула С02 не имеет дипольного момента?
6. Нарисуйте энергетические диаграммы молекул N2 и N^j~ и объясните
понижение энергии связи при удалении одного электрона у молекулы
N2(DN — 225 ккал/моль, "D (N^~) =■- 202 ккал/моль).
7. Объясните причину высокой энергии диссоциации молекулы BF
(186 ккал).
8. Какой тип гибридизации атомов углерода в метане, этилене и ацетилене?
9. Вычислите энергию связи в молекуле NH, если теплота образования
NH 83 ккал/моль.
10. Определите понятие «поляризация ионов».
11. Почему температуры плавления галогенидов стронция выше, чем
свинца?
F- CI- Вг- I-
Sr2+ (г — 1 27 А)
' J 1400 870 640 515°С
РЬ2+(г-= 1,32 А) 820 530 370 410°С
12. Почему раствор РЫ2 бесцветен, а твердый йодид — желтого цвета?
13. Какой характер проводимости следует ожидать от боразона BN?
14. Аналогом какого полупроводникового элемента можно считать фосфид
индия InP?
15. Почему металлы V—VII групп имеют высокие температуры кипения?
16. Какие силы объединяются под общим названием сил Ван-дер-Ваальса?
17. Температуры кипения галогенидов бора изменяются следующим образом:
Галогенид BF3 BC13 ВВг3 В13
Гкип, °К 172 286 364 483
Объясните эту зависимость.
18. Как изменяется характер химической связи в твердых веществах по
ряду Ge—As—Se—Вг? Объясните уменьшение теплот атомизации по этом$
ряду (90; 69; 49 и 26 ккал/г-атом).
ГЛАВА 5
ОСНОВНЫЕ ПОНЯТИЯ
ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ
Термодинамика — наука о превращениях различных форм
энергии и законах этих превращений. Превращения энергии (а также
изменения в степени порядка) сопровождают все процессы,
происходящие в природе. Та область термодинамики, которая
занимается изучением энергетических изменений в химических
реакциях, называется химической термодинамикой. Химическая
термодинамика применяет законы термодинамики к поведению
веществ в химических реакциях.
§ 1. ИЗМЕНЕНИЕ ВНУТРЕННЕЙ ЭНЕРГИИ
И ЭНТАЛЬПИИ В ХИМИЧЕСКОЙ РЕАКЦИИ
В каждом теле, в каждом веществе заключена энергия,
количество которой зависит от его количества, состава и состояния.
Эта заключенная в скрытом виде энергия носит название
внутренней энергии U. Количество внутренней энергии системы
характеризует общий запас энергии системы (система — тело или
группа тел, мысленно обособляемых от окружающей среды,
являющейся также системой). Внутренняя энергия системы
включает целый ряд энергий, в том числе энергии поступательного,
вращательного и колебательного движений молекул, энергии
связи атомов в молекулах, энергии размещения электронов на
различных уровнях и подуровнях в атомных и молекулярных
орбиталях, энергии связи ядерных частиц в ядре и ряд других
видов энергий, в том числе и неизвестных нам в настоящее
время. Во внутреннюю энергию системы не входят кинетическая
энергия всей системы в целом и ее потенциальная энергия
положения в поле тяготения.
Абсолютную величину внутренней энергии определить
невозможно, и в этом нет практической необходимости, так как
человечество в своей деятельности имеет дело только с
изменениями энергии при переходе систем из одного состояния в другое.
Можно судить лишь об изменении внутренней энергии системы
при ее переходе в другое состояние по количеству энергии,
отдаваемой системой окружающей среде или же принимаемой
системой от окружающей среды. При этом очень важно одно
условие— объем системы не должен изменяться в ^процессе перехода.
Рассмотрим систему, состоящую из 1 моля'кислорода и 2
молей водорода при комнатной температуре (25°С, 298°К). Пусть
смесь Н2 и 02 характеризуется запасом внутренней энергии U\.
126
Предположим, что прошло взаимодействие и образовалась
газообразная вода по уравнению
2Н2.г+02.г = 2Н2Ог.
Если система не изменила своего объема (реакция проходила в
закрытом сосуде), то, по-видимому, не было совершено никакой
работы — ни самой системой, ни над системой. В то же время в
первый момент после прохождения реакции можно было заме-
Рис. 5.1. Схема калориметра
а
Чг*1/2Ч.Г
1_
о
х04
L
уг
Н2.Г+Нг=НЛ
Ход реакции
Рис. 5.2. Изменение внутренней энергии
при образовании воды
тить повышение температуры в системе из-за теплового эффекта
взаимодействия водорода и кислорода. Затем температура
начала падать и постепенно понизилась до первоначальной, при этом
система выделила в окружающую среду некоторое количество
энергии в виде тепла. Это тепло легко измерить, если опыт
проводить в калориметре (рис. 5.1), представляющем собой
изолированный сосуд с известным количеством воды. Вещества —
реагенты — вводят в реакционную камеру, находящуюся в сосуде и
полностью погруженную в воду. Реакционную смесь
воспламеняют электрическим током и измеряют изменение температуры
воды за счет выделившегося (или поглотившегося) при реакции
тепла. Зная изменение температуры воды, ее теплоемкость и
количество тепла, которое требуется для повышения
температуры калориметра на 1°, можно вычислить количество теплоты
химического процесса для взятых количеств веществ в расчете
на 1 моль продукта или исходного вещества.
Обозначим внутреннюю энергию смеси 2 молей водорода и
1 моля кислорода U\ и ту же энергию 2 молей газообразной
волы U2. Так К1к объемы исходных веществ и продуктов реакции
127
по условию эксперимента не изменились (V = uoct.) и температура
продуктов реакции сравнялась с температурой исходных веществ
(Г = пост.) из-за выделения тепла в окружающее пространство, то
выделение тепла при реакции произошло из-за изменения
внутренней энергии системы.
В термодинамике принято из величины, характеризующей
конечное состояние системы, вычитать величину, характеризующую
ее начальное состояние. Эта разность обозначается знаком Л.
Для процесса
£/2Н2Ог — (U2Ht + Uo2) = L/прод — ^исходи bb = U2 — U1 - AV. (5.1)
Так как в этой реакции тепло выделилось, то U\>U2 и разность
AU— отрицательная величина: U2—£Л<0, AU<0 (рис. 5.2). В то
же время реакция взаимодействия кислорода и водорода —
реакция экзотермическая и сопровождается выделением тепла, т. е. ее
тепловой эффект положителен Qv>0. Так как начальное и
конечное состояние системы характеризуется одним и тем же
объемом и одной и той же температурой (Vi = V2, AV=0, Т\ = Т2,
ДГ=0), то, очевидно, по абсолютной величине изохорный (У=пост.)
тепловой эффект будет равен изменению внутренней энергии, а
отличаться они будут только знаками:
Д£/Г,у=-—Qr.v, (5.2)
где подстрочными индексами Т и V показано, что температура и
объем не изменяются. Ниже, если это не будет специально
оговорено, будем иметь дело с процессами при постоянной
температуре; в понятие же внутренней энергии уже заключено условие
постоянства объема. Поэтому формула (5.2) для краткости
записывается
AU - — Qv. (5.3)
Тепловой эффект Qv, измеренный при постоянном давлении,
называется изохорным тепловым эффектом.
Выделяющаяся при прохождении реакции теплота может
рассматриваться как продукт реакции, и ее записывают вместе с
продуктами реакции с правой стороны уравнения реакции
2Н2,Г + 02.г = 2Н2Ог 4 115000 кал/моль (V = пост.).
Уравнения химических реакций, в которых в левой части
равенства приведены исходные вещества, а в правой — продукты
реакции и тепловой эффект, называют термохимическими
уравнениями. Термохимические уравнения можно переписывать в
обратном направлении, сделав исходные вещества продуктами
реакции, а продукты — исходными, но при этом знак теплового
эффекта должен быть изменен на противоположный:
2Н2Ог =-- 02,г + 2Н2,Г— 115000 кал/моль (V = пост.).
128
Величина теплового эффекта зависит от количеств
участвующих в реакции веществ. В написанных реакциях тепловой
эффект 115000 кал относится к двум молям воды. Уравнение
образования воды можно записать так, чтобы образовалось не 2 моля
воды, а 1 моль. В этом случае величина теплового эффекта
уменьшается в два раза:
Н2(Г + 1/202.г = НаОг + 57500 кал/моль (V = пост.).
Реакция, написанная так, не означает взаимодействия
полмолекулы кислорода с молекулой водорода, а означает, что при
реакции 1/2 г-мол 02 с 1г-мол Н2 образуется 1 г-мол
Н20 и выделяется при этом 57 500 кал тепла р
(при У = пост.).
Величина теплового эффекта зависит от того,
в каких агрегатных состояниях находятся
участвующие в реакции вещества. Чтобы не
возникало никаких сомнений относительно этого,
внизу за формулой вещества указывают его
состояние (газообразное, жидкое или кристалличе- |Zn*HS0-
ское): UnS04+Hj
Рис. 5.3. Вычисление работы расширения реакции
Н2>г + 1/202,г - Н2Ог + 57500 кал/моль(V = пост.),
Н2>г l 1/202,г = Н2Ож -;- 67400 кал/моль (V = пост.),
Н2Ок + 67540 кал/моль (V = пост.).
Н2,г-Ь 1/202.г
Термохимический способ написания реакций в настоящее
время заменяется термодинамическим, в котором справа от
уравнения реакции записывается энергетическое изменение в системе
при реакции. Учитывая равенство (5.3), можно записать
Н2.г+1/202,г = Н2Ог
AU
■ 57500 кал/моль.
Если реакция проводится при постоянном давлении, то ее
тепловой эффект — изобарный тепловой эффект Qv будет, вообще
говоря, отличаться от изохорного теплового эффекта. Система, в
которой при постоянном давлении проходит реакция с
изменением числа молей газообразных веществ, должна для сохранения
постоянства давления изменить свой объем. Это значит, что
система совершает работу или над ней совершается работа.
Предположим, 1 г-атом цинка растворяется в серной кислоте
в цилиндре, поршень которого может перемещаться без трения
(рис. 5.3). В реакции
H2S04,>k + ZnK = ZnS04,p.p + Н2,г + QP
5 Общая химия
129
за счет выделения водорода происходит увеличение объема.
Перед началом реакции поршень находился на отметке h\. После
полного растворения цинка выделился 1 г-моль водорода и для
сохранения р = пост. поршень поднялся до отметки h2, т. е.
переместился на h2—/ii = A/i. Но такое перемещение поршня в
цилиндре совершается против силы F внешнего давления. В
простейшем случае, если направление силы F и направление
движения поршня совпадают, работа, совершаемая поршнем,
вычисляется по формуле
A =F.Aft. (5.4)
Пусть Р есть внешнее давление, которое преодолевает поршень
с поперечным сечением 5. Тогда
F=P-S. (5.5)
Выполненная поршнем работа равна произведению силы на
расстояние ДА, или
A = F.Ah = P.S-Ah. (5.6)
Но S-Ah есть изменение объема AV. Поэтому
А - PAV. (5.7)
Согласно уравнению Менделеева — Клапейрона
р.\' = п%Г. (5.8)
Изменение объема в системе AV при прохождении реакции
связано с изменением числа молей газообразных веществ An
соотношением
PAV = AnRT, (5.9)
где An — изменение числа молей в реакции, т. е. разность чисел
молей газообразных продуктов и исходных веществ:
Atl = /Z2 — U\ ~ #прод — ^исходы . (5.10)
Подставляя формулу (5.9) в формулу (5.7), получаем
выражение для вычисления работы реакции
А = AnRT. (5.11)
Принято считать работу положительной, если она совершается
над окружающей средой. Работа, которая совершается над
рассматриваемой системой, отрицательна.
Очевидно, изобарный тепловой эффект меньше изохорного
теплового эффекта для реакции, идущей с увеличением числа
молей газообразных веществ (п2>П\\ Ап>0), на величину, равную
работе расширения, т. е. работе, которую система совершает над
окружением-
130
Qp = Qv - PAV = Qv— &nRT. (5.12)
Соотношение (5.12) обычно записывают в виде
Qv — Qp = &nRT. (5.13)
В реакциях с уменьшением числа молей газообразных веществ
п2<п{ и Ап = п2—tti<0, но в то же время над реакционной
системой будет совершаться работа, знак которой отрицателен, и для
таких реакций формула (5.13) сохраняет свой прежний вид.
По формуле (5.13) можно вычислять, как различаются
изобарный или изохорный тепловые эффекты при постоянной
температуре, а также величину одного теплового эффекта, если
известен другой.
В уравнения (5.8), (5.9), (5.11) —(5.13) входит газовая
постоянная R. Из последнего уравнения очевидно, что размерность
произведения AnRT должна быть той же, что и у тепловых
эффектов (кал/моль). В этом случае R должна иметь размерность
кал/моль-град.
Чтобы найти значение газовой постоянной, вспомним, что при
Р = 760 мм рт. ст. и Г=273°К (0°С) 1 моль идеального газа
(/г=1) занимает объем 22,4 л. Подставим численное значение Р0,
V0 и Г0 (нормальные условия) в уравнение (5.8) и получим
R = Ро-Уо = 1 атм-22,4 л = g2 ^ьатм g
п-Т0 273°К град-моль ' у
Если давление выразить в динах на см2, а объем в см3, то
Р = 76-13,596-981 дин/см2 (где 13,596 г—вес 1 см3 ртути, а
981 см/сек—ускорение силы тяжести) и
D 76 см-13,593 г/см».981 см/секя-22400 см3
1 моль 1>73,16°К
= 8,314-107 эрг/град-моль. (5.15)
Так как 4,185-107 эрг = 1 кал, то
^ 8,314- 107 1 ПП / /С 1СЧ
£ = —» _ 1 99 кал/моль-град. (5.16)
4,185-К)7 ' F K '
Этим значением газовой постоянной пользуются в химической
термодинамике, часто округляя до 2 кал/моль -град.
Во всех предыдущих рассуждениях объем газа считался
равным грамм-молекулярному объему, т. е. газовая постоянная
относилась к числу Авогадро молекул N=6,02-1023.
Газовая постоянная, отнесенная к 1 молекуле, называется
постоянной Больцмаиа и обозначается символом k:
, _ R _ 8,314-107 эрг/моль»град _
~~ N ~~ 6,02-1023 молекул ~~
= 1,380- Ю-16 эрг/град-молекула. (5.17)
5+
131
Вычислим, как сильно отличается между собой изохорный и
изобарный тепловые эффекты реакции синтеза аммиака при
300°К:
ЗН2.Г + N2,r - 2Ш3,Г,
Ап = 2— (3 + 1)- —2,
Qv — Qp = (_ 2). 2 • 300 - — 1200 кал/моль,
Qp = Qv + 1200 кал/моль.
Рассмотрим другой пример, также для 300 °К:
Ск + 2Н2,г — СН4,Г.
В реакции принимает участие твердый углерод, объем которого
незначителен по сравнению с объемами Н2 и СН4, поэтому
Ап=1 — 2=— 1 и
Qv — Qp = (— 1). 2.300 = — 600 кал/моль.
Заметим, что разница между Qp и Qv равна нулю при 0°К и
возрастает с ростом температуры. Для 1000°К в последнем
процессе
Qv — Qp = — 2.1000 = — 2000 кал/моль.
Знак An влияет на соотношение Qp и Qv. При Ап<0, когда
в реакции происходит уменьшение числа молекул газообразных
веществ, изобарный тепловой эффект больше изохорного:
Qv-Qp<0, или Qp>Qv.
При Дя>0, когда в реакции происходит увеличение числа
молекул газообразных веществ, изобарный тепловой эффект
меньше изохорного:
Qy-Qp>0, или Qp<Qv.
Подставим выражение (5.3) в формулу (5.12):
Qp ---- — AU — РAV = — AU — AnRT
или, изменяя знаки, получаем
— Qp = AU + PAV = AU + AnRT, (5.18)
т. е. изобарный тепловой эффект реакции, взятый с
противоположным знаком, равен изменению внутренней энергии плюс
работа расширения.
Сумму AU+PAV называют изменением энтальпии и
обозначают АН:
AH = AU + pAV. (5.19)
132
Таким образом, изменение энтальпии в химической реакции
равно изменению внутренней энергии плюс работа расширения.
Из сопоставления формул (5.18) и (5.19) следует
ЛЯ = -<Эр, (5.20)
пли же изменение энтальпии в реакции равно изобарному
тепловому эффекту, взятому с противоположным знаком.
Чаще всего химические
эксперименты, технологиче- Н
Н„ +У2НП
ские процессы и химические
превращения в окружающей
нас среде совершаются при
постоянном давлении,
поэтому ниже будем
пользоваться изобарными
тепловыми эффектами и
изменениями энтальпии, особенно
н2т V
дН = -57800нал
Q= 57800 кал
v
Ход реакции
Рис. 5.4. Энтальпийная диаграмма ., +Vnfi =H0
реакции образования воды 2,г 2J г г
последними. В большинстве термодинамических справочников
приводятся именно изменения энтальпии для реакций образования
химических соединений из простых веществ. Пользуясь
формулами (5.3), (5.13), (5.18) — (5.20), по величине АЯ можно вычислить
MJ, QVy Qv.
Изохорный тепловой эффект образования газообразной воды
из простых веществ
Н2.г-Н/2 02,г = НаОг
равен Qv = 57 500 кал/моль при 25 °С. Изобарный тепловой эффект
Qp = 57 500 — (1 — 1 — 1/2).2-298 = 57 500 + 0,5.2-298 =
= 57 800 кал/моль.
Изменение энтальпии в реакции принято записывать справа
от уравнения реакции:
Н2)Г + 1/2 02)Г - Н2Ог Д Я = — 57 800 кал/моль.
На рис. 5.4 изображена энтальпийная диаграмма этой
реакции, аналогичная диаграмме на рис. 5.2, где использовалась
внутренняя энергия.
В газовых реакциях без изменения числа молей газообразных
веществ, а также в реакциях между твердыми веществами и в
реакциях, протекающих в растворах, если при этом не
происходит заметного изменения объема, изменения энтальпии и внут-
133
ренней энергии, так же как и изобарный и изохорный тепловые
эффекты, равны.
Из формул (5.18) и (5.19) видно, что
ДЯ = Д1/ + AnRTf (5.21)
и чем ниже температура, тем ближе АН и AU. При 0° АН = AU.
§ 2. ПЕРВЫЙ ЗАКОН ТЕРМОДИНАМИКИ
И ЗАКОН ГЕССА
Первый закон термодинамики имеет несколько формулировок,
но все они выражают одну идею: неуничтожимость и
эквивалентность различных видов энергии при их взаимных переходах.
Первый закон устанавливает связь между количеством теплоты,
полученной или выделенной в процессе, количеством
произведенной или полученной системой работы и изменением
внутренней энергии системы. В изолированной системе, т. е. в системе,
для которой исключен любой материальный или энергетический
обмен с окружающей средой, сумма всех видов энергии есть
величина постоянная. В любом процессе приращение внутренней
энергии системы AU=U2—U\ равно количеству сообщенной
системе теплоты Q минус количество работы Л, совершаемой
системой
At/^Q —Л. (5.22)
Если в каком-нибудь процессе энергия одного вида исчезает, то
вместо нее появляется энергия другого вида в количестве, строго
эквивалентном первому. Разные виды энергии переходят друг в
друга в строго эквивалентных соотношениях.
В термодинамике подводимое к системе и поглощенное ею
тепло считается положительным, а выделяемое тепло —
отрицательным. В термохимии принята противоположная система
знаков. Учитывая это и подставляя в уравнение (5.22) вместо
теплового эффекта изменение энтальпии из уравнения (5.20),
получаем формулу (5.19)
MJ = -Qp-A,
AU = AH — Л,
АН = bU + Л,
Д# = MJ + р AV - AU + AnRT,
являющуюся математическим выражением первого закона
термодинамики в приложении к химическим реакциям.
Другим очень важным выражением закона сохранения
энергии является закон Гесса, сформулированный русским химиком
Г. И. Гессом в 1840 г. Он гласит: изменение энтальпии (или теп-
134
ловой эффект процесса) зависит только от вида и состояния
исходных веществ и продуктов и не зависит от пути перехода.
Если бы величины тепловых эффектов (или АН и Д£/) при
одинаковых начальных и конечных состояниях на различных путях
были неодинаковы, то, совершая реакцию в прямом направлении
по одному пути, а затем в обратном — по другому, можно было
У2 /f.
см
<
v2f
и х
о о
счЗО
■х. i^
Рис. 5. 5. Энталь-
пийная диаграмма
реакции
образования жидкой и
газообразной воды
JL__,
НЛ
Ход реакции
н + V?o»ho
2,г г 2 ж
бы получать энергию из ничего, т. е. построить вечный
двигатель.
Это можно показать на примере образования воды Н2Ож из
газообразных 02 и Н2. На рис. 5.5 изображена энтальпийная
диаграмма образования воды в двух ее различных состояниях —
жидкой и газообразной:
Н2.г+1/2 02,г = НаОж
Н2.г+1/2 0ог = Н20Р
Д#обр.НьОж =
Д#0бр.Н2Ог = "
-68320 кал/моль.
57800 кал/моль.
Уровень газообразной воды лежит выше уровня жидкой воды,
это и понятно, так как для испарения 1 моля воды
затрачивается 10 520 кал тепла, Qncn=—10 520 кал/моль, АЯИСП=
10 520 кал/моль. В обратном процессе — конденсации — это
количество тепла выделяется, (?конд= +10 520 кал/моль, Д#КОнд=
= —10 520 кал/моль, что можно записать так:
НА
н2ог
= НА
Н2Ож
Д#исп = 10520 кал/моль,
ДЯконд = — 10520 кал/моль.
Следовательно, существуют два пути, по которым можно из
водорода и кислорода получить жидкую воду. 1. Проводить
реакцию так, чтобы сразу получалась жидкая вода. В этом случае
изменение энтальпии составит Д#н,о — — 68320 кал/моль.
2 ж
135
2. Сжигать водород в кислороде так, чтобы получалась
газообразная вода, при этом изменение энтальпии составит Д#н2о =
= —57 800 кал/моль, и далее конденсировать пар в жидкость.
В процессе конденсации изменение энтальпии составит АЯКОнд =
= —10 520 кал/моль.
Если теперь сложить Д#ц2о и ДЯКОнд > то получим изменение
энтальпии образования жидкой воды:
Л//н2ож = _ 57800 + ( — 10520) = — 68320 кал/моль.
Очевидно, что изменение энтальпии (или тепловой эффект) в
реакции образования жидкой воды не зависит от способа ее
получения. В этом и состоит закон Гесса. Таким образом, если из
данных исходных веществ получать различными путями и
способами заданные конечные продукты, то суммарное изменение
энтальпии (тепловой эффект) для всех путей будет одним и тем
же, т. е. не будет зависеть от вида и количества промежуточных
реакций. Закон Гесса точен только для процессов, протекающих
при постоянном объеме или давлении, т. е. суммировать, а также
вычитать или производить другие алгебраические операции
можно только с энергетическими эффектами одного вида — или Д#,
или Д[/, или QP) или Qv. Раньше пользовались понятием
теплового эффекта, а в современной научной литературе и в
большинстве справочников пользуются понятием «изменение энтальпии»,
или просто «энтальпия».
Так как суммарное изменение энтальпии в круговом
процессе, т. е. в процессе, при котором система после прохождения ряда
промежуточных состояний возвращается в исходное состояние,
равно нулю, то из этого вытекают следствия:
1. Энтальпия разложения химического соединения равна и
противоположна по знаку энтальпии его образования из
продуктов разложения
ДЯобр = - Д#разл. (5.23)
2. Разность энтальпий реакций, идущих от одинаковых
начальных состояний к двум различным конечным состояниям
продукта реакции, представляет энтальпию перехода из одного
конечного состояния в другое.
3. Разность энтальпий реакции от различных начальных
состояний исходного вещества к одинаковому конечному
представляет энтальпию перехода от одного начального состояния в
другое.
Пример одной из таких реакций представлен графически на
рис. 5.5. Из энтальпий образования Н2Ог и Н2Ож вычисляется
энтальпия процесса
Н2Ог = Н2Ож,
136
которая называется энтальпией конденсации. Она равна
разности энтальпий образования Н2Ог и Н2Ож (рис. 5.5):
Д#конд = #н2Ог — ^н2°ж = ^^обр.Н2Ор — ^^обр.Н2Ож =
= — 68320 — (— 57800) - — 10520 кал/моль.
Испарение — процесс, противоположный конденсации, — имеет
равную, но противоположную по знаку энтальпию:
АЯисп = #Н2Ог — ^н2°ж ^ Д^обр.Н2Ог — Д^обр.Н80 =
= — (ЛЯобр н2ож — ДЯ0бр.н,ог) = — ДЯконд =• Ю520 кал/моль.
Пользуясь законом Гесса, часто удается вычислить энтальпии
реакций и процессов, которые невозможно определить опытным
н + н
алмаз */
сграфит °2,г
X
О-
о.
о"
Q.
О
ГУ
?J
с +о
^алмаз 2,г
С + 0
^графити2.
— "ВТ
О)
а
ж±
1
-СО
2, Г
=со
Г 2,Г
о
(О
Г
5
о
а.
о
*
_2 j
Ход реакции
Рис. 5.6. Энтальпийная диаграмма
взаимодействия кислорода с алмазом и графитом
путем. Например, пока не удалось непосредственно измерить
энтальпию превращения алмаза в графит, тем не менее она
известна и была найдена по энтальпиям сгорания алмаза и графита:
Сграфит + 02,г =- С02)Г Д#1 = — 94000 кал/моль,
Салмаз + Оо,г = С02,г Д#2 = — 94500 кал/моль.
Энтальпийные уровни алмаза, графита, кислорода и
углекислого газа представлены на диаграмме рис. 5.6. Откуда видно,
что изменение энтальпии при превращении одного грамм-атома
углерода в форме алмаза в графит составляет
А^превр —
■ 94500 — (— 94000) = — 500 кал/г-атом.
137
Таким образом,
Салмаз = Сграфит Д#превр = — 500 Кал/г-аТОМ.
Для нахождения неизвестных энтальпий необязательно
строить диаграммы. Термохимические уравнения можно складывать
и вычитать, как обычные алгебраические уравнения. Например,
чтобы из уравнений
Сграфит + 02 = С02 + 94000 кал/моль,
Салмаз + 02 = С02 + 94500 кал/моль
получить уравнение с неизвестным тепловым эффектом
^алмаз == ^графит?
достаточно из второго уравнения вычесть первое:
_ Салмаз + 02 - С02 + 94500 кал
Сграфит + 02 - С02 + 94000 кал
Салмаз — Сграфит = ~Г 500 Кал/МОЛЬ,
ИЛИ
Салмаз = Сграфит + 500 Кал/МОЛЬ,
Фпревр = 500 кал/моль.
Уравнения, написанные с использованием энтальпий, также
можно складывать и вычитать, но как поступить с несколькими
уравнениями для получения уравнения с неизвестной
энтальпией? Можно посоветовать воспользоваться приемом, который
сбережет много времени при расчетах. Из уравнений с
известными энтальпиями выбираем уравнение с веществом, стоящим в
левой части искомого уравнения, и, если нужно, переписываем
его так, чтобы это вещество находилось в уравнении слева,
изменив знак энтальпии на противоположный. Под этим уравнением
подписываем уравнение, содержащее теперь один из продуктов
этого второго уравнения, и переписываем его так, чтобы эти
продукты находились по другую сторону равенства и так далее,
каждый раз обращая внимание на то, чтобы в нижеследующем
уравнении исходными веществами были продукты вышестоящего
уравнения, и не забывая умножать стехиометрические
коэффициенты веществ и энтальпии на такие коэффициенты, чтобы
количества веществ в уравнениях были одинаковы.
Разберем это на примере превращения алмаза в графит,
энтальпия которого непосредственно не может быть пока
определена из-за огромных экспериментальных трудностей.
Требуется вычислить энтальпию
^алмаз — ^графит Д#ь (I)
138
если известны энтальпии реакций
Салмаз f 02 = С02 Д#2 = — 94500 кал/моль, (II)
Сграфит -т 02 = С03 Д#3 = — 94000 кал/моль. (III)
Достаточно записать эти уравнения так, чтобы после
сложения получилось искомое уравнение, при этом если уравнение
записывается в обратном порядке, то знак энтальпии меняется
на противоположный:
Салмаз + 02 = С02 АЯ2 = — 94500 кал/моль
^графит ' - 02 Д#3 = + 94000 кал/моль
^алмаз ~ ^графит Д/ii = &*~* 2 — Д" 3 ~
= — 94500 + 94000 = — 500 кал/моль.
При расчетах энтальпий на основе закона Гесса особое
значение имеют два вида энтальпий — энтальпия образования и
энтальпия сгорания. Энтальпией образования называется
энтальпия реакции образования химического соединения из простых
веществ, устойчивых при данных условиях. Например, энтальпия
образования S102 равна — 205 000 кал. Это значит, что при
соединении 1 г-атома кремния и 1 моля кислорода происходит
уменьшение энтальпии на 205 000 кал (выделяется 205 000
калорий тепла):
SiK + 02,г = Si02>K A#o6p.sio2tK = — 20500° кал/моль.
Энтальпия образования СаО:
Сак ■-! 1/2 02,г =- СаОк Д#0бР.саок = — 152000 кал/моль.
Энтальпия образования силиката кальция:
Сак + SiK + 3/2 СОо r=CaSi03fK Д/Wcesio^K = — 378000 кал/моль.
Энтальпии образования относят к молю образующегося
соединения и, как правило, приводятся для температуры 25°С (298°К) и
давления 1 атм (стандартные условия). Энтальпии образования
при стандартных условиях называют стандартными энтальпиями
образования из простых веществ (для краткости будем называть
их энтальпиями образования и обозначать Д#0бР, если не будут
оговорены дополнительные условия).
Энтальпия реакции равна разности суммы энтальпий
образования из простых веществ продуктов реакции и суммы энтальпий
образования исходных веществ. Все энтальпии следует брать с
коэффициентами, равными коэффициентам перед формулами
веществ в рассматриваемом уравнении реакции.
Энтальпии образования простых веществ в их наиболее
устойчивых состояниях при стандартных условиях и стандартной тем-
139
пературе принимаются равными нулю. Например, энтальпии
образования Н2,г, 02,г> FeK, CaK равны нулю. Углерод при 298°К
может существовать в виде графита и алмаза. Так как графит
устойчивее алмаза,
в то время как
АЯо0бр.сграфит = 0 кал/г-атом,
Д#обр.салмаз = 500 кал/г-атом.
Вычислим энтальпию реакции окислов кальция и кремния с
образованием силиката кальция:
jO
СаОк + Si02iK = CaSi03,K А/Реакции
СаОк = Сак + 1/2 02,г -ДЯ°бр Саок
" Si02iK = SiK + 02,г -AH°o6p.sio2>K
Сак + SiK -f 3/2 02|Г = CaSi03iK A//o6P.caSio3,K
CaOK + SiOK = CaSi03)K АН°реакщт =
*•*= A^o6p.CaSi03K (Д#обр Са°к + А//обр Si02>K) =
= _ 378000 — (— 152000 — 205000) - — 21000 кал/моль.
Если через 2 АЯ0бр.пРод обозначить сумму энтальпий образования
продуктов реакции, а через 2 ДЯобр.исходн такую же сумму для
исходных веществ, то энтальпия реакции равна
А^реакции ~ ^ Д^обр.ирод ^ ^^обр исходи.
По этому уравнению можно рассчитать любую из содержащихся
в нем энтальпий, если известны значения остальных.
Энтальпией сгорания называется энтальпия реакции
взаимодействия соединения с кислородом с образованием высших
стабильных окислов соответствующих элементов (Н2Ож, С02,г,
Si02,K)- По энтальпиям сгорания всех веществ, участвующих в
реакции, можно рассчитать энтальпию самой реакции. В
примере с комбинированием энтальпий сгорания алмаза и графита
для вычисления энтальпии превращения алмаза в графит
получено
^алмаз = ^графит Д/i превр ~ ^"сгор.Салмаз A" crop Сграфит —
= (А//Сгор.сграфит ДпСГОр.салмаз)-
Следовательно, энтальпия реакции равна разности энтальпий
сгорания продуктов реакции и исходных веществ, взятой с
обратным знаком.
140
§ 3. ИЗМЕНЕНИЕ ЭНТРОПИИ
В ХИМИЧЕСКОМ ПРОЦЕССЕ
Большинство реакций, с которыми мы встречаемся в быту,
которые проходят самопроизвольно в природе или используются
в технологии веществ, сопровождаются выделением тепла, т. е.
уменьшением энтальпии.
Около 95% всех неорганических соединений образуется при
стандартных условиях и температуре с выделением тепла и
уменьшением энтальпии. Таким образом, подавляющее
большинство веществ обладают меньшим запасом энергии по сравнению
с простыми веществами, из которых они образуются.
Это и другие соображения позволили в середине XIX в.
французскому химику Бертло и английскому химику Томсену
сформулировать следующий принцип: «Любой химический процесс
должен сопровождаться выделением тепла».
Принцип Бертло—Томсена возник из-за попытки найти
аналогию в поведении между химическими и механическими системами.
Но химическая форма движения гораздо сложнее механической
и такое сравнение недопустимо; скоро обнаружилось много
исключений из правила.
Каждый знает, что шар, находящийся на верху наклонной
плоскости, стремится скатиться вниз от положения с высокой
потенциальной энергией к положению с более низкой
потенциальной энергией. Обратно шар будет двигаться только при затрате
работы. Бертло и Томсон предполагали, что реакции будут идти,
как и шар катиться, только вниз, в направлении уменьшения
энергосодержания, т. е. с выделением энергии в виде тепла.
Известно много случаев протекания процессов, при которых
тепло поглощается, например растворение. Если растворяемое
вещество не реагирует с растворителем с выделением тепла, то
всегда при растворении наблюдается понижение температуры
раствора. Так, азотнокислый натрий растворяется в воде с
поглощением тепла (энтальпия повышается)
NaN03,K + 400 Н2Ож - NaN03/p-p — 5100 кал/моль
(АЯрастворения =5100 КЭЛ/МОЛЬ).
Важный технологический процесс получения смеси углерода
и водорода
ск + н2ог - сог + н2>г
гакже сопровождается поглощением тепла (Д#<0). Процесс
смешения двух газов протекает самопроизвольно и без
изменения энергии. Обратный же процесс — процесс разделения газовой
смеси на составляющие ее компоненты — сам по себе не пойдет
и требует затраты работы. Движущей силой смешения газов не
является энергия. Следовательно, мы видим, что сведений об
141
энергетических изменениях в системе недостаточно для
предсказания направления реакции.
Рассмотрим систему (система — тело или группа тел,
обособленная мысленно или какой-либо поверхностью раздела от
окружающей среды, являющейся, в свою очередь, также системой
по отношению к своему окружению) из двух газов, не
реагирующих между собой, не склонных к какому-либо
взаимодействию, например аргона и гелия, которые разделены перегородкой
и находятся при одинаковых температуре и давлении (рис 5.7, У).
т 1
Аг
Не
J I
1 2
Рис. 5.7. Процесс смешения
двух газов
Назовем это состояние системы состоянием 1. Удалим
перегородку, не изменив энергетического запаса системы. Несмотря
на одинаковые давления газов справа и слева от места
расположения перегородки, начинается процесс смешения газов, и
через некоторое время молекулы гелия и аргона будут
равномерно распределены по всему объему системы (рис. 5.7, 2). Новое
(конечное) состояние системы назовем состоянием 2.
Газы, разделенные перегородкой (состояние 1),
распределены в системе с большим порядком по сравнению со смесью
газов (состояние 2). По-видимому, движущей силой смешения
газов является их стремление перейти в состояние с меньшим
порядком.
Степень порядка 2<степени порядка 1. Отсюда можно
сделать вывод: самопроизвольный процесс, проходящий без
изменения энергетического запаса системы, совершается только в
направлении, при котором порядок в системе уменьшается. В
химической термодинамике обычно не пользуются выражением
«порядок» или «степень порядка», а применяют выражение,
характеризующее противоположное свойство — «беспорядок» или
«степень беспорядка». Очевидно, что степень порядка и степень
беспорядка находятся в обратной зависимости друг от друга:
чем выше значение одного, тем ниже значение другого. Газы до
смешения в состоянии 1 имели меньшую степень беспорядка,
чем после смешения в состоянии 2. Степень беспорядка 2 >
степени беспорядка 1.
Мы приходим к выводу, что, самопроизвольный процесс,
проходящий без изменения энергетического запаса системы,
совершается только в направлении, при котором беспорядок в
системе возрастает.
Аг
142
Трудно себе представить, чтобы два газа, не разделенные
перегородкой, не смешались. Такое состояние 1 системы
маловероятно. Состояние 2 равномерного распределения молекул
газа по всему объему более вероятно. Каждый человек не может
себе представить, чтобы кусок сахара з стакане с водой остался
неизменным — это невероятно. Состояние равномерного
распределения молекул сахара среди молекул воды — более вероятное
состояние. Движущей силой смешения газов или растворения
сахара является тенденция перейти в более вероятное состояние.
Вероятность состояния 2> вероятности состояния 1.
Мы приходим к новому очень важному выводу:
самопроизвольный процесс, проходящий без изменения энергетического
запаса системы, совершается только в направлении, при
котором система переходит в более вероятное состояние.
Итак, система самопроизвольно и без изменения содержания
энергии может перейти только в состояние, которое имеет
меньший порядок, больший беспорядок и является более вероятным.
В химической термодинамике чаще всего имеют дело именно с
вероятностями. Действительно, вычислить степень порядка и
степень беспорядка при температурах, отличных от 0°К,
невозможно, но вероятность расположения молекул в тОхМ или ином
месте системы (молекулы двух видов разделены, равномерно
распределены в системе, все находятся по одну сторону
перегородки и т. п.) поддается вычислению.
Однако здесь мы сталкиваемся с одним очень существенным
обстоятельством, сильно усложняющим все дальнейшие
рассуждения. Если смешиваются по молю два не реагирующих газа с
разными запасами энергии, то смесь будет обладать суммой
первоначальных запасов энергии. Если объединяются две
системы в одну, то энергетический запас объединенной системы в
простейшем случае равен сумме энергетических запасов
исходных систем (или же просто может быть вычислен по правилу
смешения). Однако аналогичные рассуждения к вероятностям
не применимы: вероятности не складываются, а
перемножаются. Представим себе две системы, имеющие вероятности W\ и
W2- Совместная их вероятность равна не сумме, а произведению:
w=wx.w2.
Требуется найти функцию от вероятности, чтобы вид этой
функции был таков, что когда вероятности перемножаются,
значения функции складываются. Эта зависимость может быть
только логарифмической, а сама функция называется
энтропией S. Таким образом, зависимость S = f(W) имеет вид
S=f(\n W). Подставив коэффициент пропорциональности /?,
получаем
S = R\nW. (5.24)
Энтропия — это логарифмическое выражение вероятности
системы.
143
Пусть две совершенно не связанные системы обладают
вероятностями W\ и W2 и соответственно энтропиями Si и S2. Общая
энтропия двух систем S равна сумме их энтропии:
5 = S, + S2,
(5.25)
а совместная вероятность W равна произведению их
вероятностей:
W^WVW2. (5.26)
Подставив (5.24) в (5.25), получаем
S = S, -,- S2 -=R\nW1 + R In W2 = R In (W^W^ = R\nW.
Таким образом, при сложении энтропии отвечающие им
вероятности перемножаются (рис. 5.8).
1 s>
1 w>
Рис. 5.
и ве]
4-
8. Энтр
юятиос
опия
ть
N.
х
5-S1-S? I
w-w1-w2
С точки зрения химической термодинамики энтропия — это
логарифмическое выражение вероятности существования
веществ или различных их форм.
Возвратимся к системе двух газов (рис. 5.7). Пусть в
состоянии 1, когда газы не смешаны, система характеризуется
роятностыо W\ и энтропией Si:
S1^=R\nW1.
После удаления перегородки и окончания самопроизвольного
процесса смешения газов система переходит в состояние 2 с
вероятностью W2 и энтропией S2:
S2 = R\nW2.
Чтобы оценить изменение энтропии при переходе
(состояние 1)-^-(состояние 2),
нужно, как обычно, из величины какого-либо свойства,
характеризующего конечное состояние, вычесть величину того же
свойства, характеризующего начальное состояние:
AS =-S2 — S, ^RlnW2 — R\nWl =/?ln-
Wx
(5.27)
144
Процесс распределения разнородных газовых молекул по
всему объему системы — процесс самопроизвольный. Обратный
процесс разделения газа по обе стороны воображаемой
перегородки самопроизвольно невозможен (если, конечно, в системе
содержится не слишком малое количество атомов) или
возможен, но с затратой работы. Поэтому вероятность W2>\V\,
S2—Si>0 и AS = S2—Si>0.
Таким образом, самопроизвольный процесс, проходящий без
изменения энергетического запаса системы, совершается только
в направлении, при котором энтропия возрастает (AS>0).
Так как вероятность системы и степени ее беспорядка и
порядка связаны между собой, то энтропия также является и мг-
рой порядка и беспорядка системы. Системы с высокой
степенью порядка и соответственно низкой степенью беспорядка
характеризуются низкими энтропиями и наоборот. В
кристаллах расположение молекул или атомов значительно более
упорядочение, чем в жидкостях, а в
последних более упорядоченно по сравнению с
газовым состоянием того же вещества.
В табл. 5.1 приведены энтропии воды
для различных ее состояний при
стандартных условиях.
Кристаллическая вода (лед) имеет
наинизшую энтропию— 10,5 кал/моль X
Хград (о причине такой размерности
энтропии будет рассказано ниже).
Несколько выше (16,0 кал/моль-град)
энтропия жидкой, а энтропия газообразной
возрастает (45,1 кал/моль. • град).
При стандартных условиях и стандартной температуре 25°С
лед существовать не может, он самопроизвольно (хотя и с
поглощением теплоты) превращается в жидкую воду. При 25° С
существование льда маловероятно. Можно вычислить изменение
энтропии в процессе плавления льда:
Н2Ок = Н2Ож
т
н2о,
состоянье
Газ
Жидкость
Кристалл
а б л и ц а 5.1
S0
^298'
кал/моль град
45,1
16,0
10,5
воды (пара) резко
А5298,пл ~ ^298, Н,0
>298, Н2Ок =
= 16,0 — 10,5 = 5,5 кал/моль-град.
Переход из менее вероятного состояния (лед) в более
вероятное (жидкость) сопровождается возрастанием энтропии (А5>0).
При стандартных условиях и 25° С жидкая вода также не
может долго сохраняться. Ее молекулы самопроизвольно (хотя и с
поглощением теплоты) переходят в газовую фазу и равномерно
распределяются в окружающем пространстве. Вычислим
изменение энтропии в процессе испарения воды:
н2ож = н2ог.
145
газ
л си си си
/А0298,испар = ^298, Н20г — ^298, 118Ож =
= 45,1 — 16,0 = 29,1 кал/моль-град.
Наиболее вероятное состояние воды при стандартных условиях —
газообразное Причина большого различия А5ПЛ и Д5,:сп
заключается в том, что при плавлении
льда в жидкой воде сохраняются
группы молекул Н20 со
структурой льда, тогда как при
испарении воды в паре молекулы
воды находятся в виде димеров
(Н20)2 и отдельных, не
связанных с другими молекул.
На примере воды можно
показать, что к энтропиям
формально применим закон Гесса и с
энтропиями можно совершать
такие же операции, как и с
энтальпиями. Лед может быть превра-
OKvdx.
* Ъ'крист
aS„
aS-
AS,
'си5л
Рис. 5.9. Изменение энтальпии и
энтропии при фазовых переходах
щен в пар двумя путями. Первый — через плавление и
последующее испарение жидкости. На этом пути изменение энтропии
составляет
Д«$298 = ^298, пл 4' Д5298,исп =
= 5,5 + 29,1 = 34,6 кал/моль -град.
Второй путь — непосредственный переход молекул воды из льда
в пар — сублимация. Изменение энтропии при сублимации равно
Д5298,субл = ^298. Н.О^ — So*
298, Н2Ок =
= 45,1 — 10,5 = 34,6 кал/моль-град.
Изменение энтропии, как и изменение энтальпии, не зависит
от пути перехода при условии, что исходное и конечное состояния
вещества одинаковы, а вещества находятся при одних и тех же
условиях (давление, температура). На рис. 5.9 показана
объединенная энтальпийная и энтропийная диаграмма переходов
кристалл — жидкость — пар, из которой видно соблюдение закона
Гесса для энтальпий переходов и формальное сохранение его в
силе для энтропии.
Часто знак изменения энтропии в реакции можно предсказать
в зависимости от состояния участвующих в реакции веществ.
В реакции диссоциации газообразной воды на кислород и
водород
146
НаОг = Н2.г+ 1/2 02,г
объем продуктов реакции больше объема исходных веществ и, по-
видимому, сумма энтропии 1 г-мол Н2 и 1/2 г-мол 02 будет меньше
энтропии 1 г-мол Н2Ог. Действительно,
5н2ог = 45,1; Sh^-31,2; So2r - 49,0 кал/моль-град (при 298 °К).
Изменение энтропии в реакции равно
Д5298 = 5н2>г + 1/2 5о2>г — ^н2ог =
=-31,2 + 24,5 — 45,1 = 10,6 кал/моль-град.
Взаимодействие кристаллической окиси кальция и углекислого
газа с образованием кристаллического карбоната кальция
сопровождается уменьшением объема за счет того, что в реакции
поглощается углекислый газ:
СаОк + С02,г = СаС03,к.
Можно ожидать, что энтропия системы будет уменьшаться
ScaC03fK<(ScaOK+ 5С02>Г).
Энтропии веществ этой реакции равны
5гасо3>к =" 21,2; *$саок = 9,5;
Sco2 г= 51,1 кал/моль- град (при 298 °К).
Изменение энтропии составляет
Д5° = 5саСрз>к — (5СаОк "Г 5с02рГ) =
-21,2 —(9,5 i 51 Л) =21,2 —60,6 = — 39,4 кал/моль-град.
Если же реакция проходит без изменения объема, то
предсказать изменение энтропии таким способом невозможно. В этом
случае нужно пользоваться табличными данными справочников и
проводить вычисления изменения энтропии.
Значения стандартных энтропии веществ при 298° К
определяются по зависимости теплоемкости вещества при его нагревании
от 0 до 298° К. При этом считается, что при нуле градусов по
Кельвину энтропия вещества равна нулю, так как при этой
температуре расположение атомов или молекул в кристаллической
структуре характеризуется максимальным порядком. При нуле
градусов атомы или молекулы в кристалле совершенно
неподвижны. При нагревании кристалла вводимое в кристалл тепло
возбуждает элементы кристаллической решетки и они начинают
колебаться. Чем больше тепла вводится в кристалл, тем сильнее
колебательные движения молекул и колебательные движения ато-
147
мов в молекулах, тем сильнее нарушение порядка в структуре
кристалла, тем выше степень беспорядка, тем выше энтропия.
В то же время, чем больше может быть различных видов
движения у элементов кристаллической структуры и чем интенсивнее
эти движения, тем выше теплоемкость. Специальными методами
по зависимости теплоемкости вещества от температуры
рассчитывается энтропия вещества.
Представление о равенстве нулю энтропии совершенного
кристалла любого вещества при абсолютном нуле температуры
составляет суть третьего закона термодинамики. О нем и методах
вычисления энтропии будет рассказано в § 6.
§ 4. ИЗОБАРНЫЙ ПОТЕНЦИАЛ РЕАКЦИИ
И ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ
Для процессов, происходящих в природе, известны две
движущие силы: первая — стремление перейти в состояние с
наименьшей энергией и выделить тепло при таком переходе, т. е.
понизить энтальпию, и вторая — стремление перейти в наиболее
вероятное состояние с максимально допустимой в данных условиях
степенью беспорядка, максимальной энтропией, т. е. повысить
энтропию.
Если в процессе степень беспорядка не изменяется (AS = 0),
то направление процесса определяется изменением энтальпии и
процесс проходит самопроизвольно в направлении уменьшения
энтальпии (Н2<Ни ДЖО).
Если в процессе не происходит энергетических изменений
(д# = 0), то фактором, определяющим направление реакции,
является энтропия и процесс пойдет самопроизвольно в
направлении, при котором степень беспорядка возрастает, т. е. в сторону
увеличения энтропии (S2>SU AS>0).
В химических процессах одновременно изменяются
энергетический запас системы и степень ее беспорядка. Самопроизвольно
процесс проходит в том направлении, в котором общая
суммарная движущая сила реакции будет уменьшаться.
Общая движущая сила процесса^ (запас энергии в конечном
состоянии — запас энергии в начальном состоянии) — (степень
беспорядка в конечном состоянии — степень беспорядка в
начальном состоянии).
Знак минус перед вторыми скобками ставится для того, чтобы
в самопроизвольном процессе без изменения энергетического
состояния системы сделать изменение общей движущей силы
процесса отрицательной величиной.
Если процесс производится при постоянном давлении, то общая
движущая сила процесса называется изменением изобарного
(р = пост.) потенциала и обозначается AG. Как обычно, изменение
изобарного потенциала в реакции есть разность между суммой
изобарных потенциалов продуктов и исходных веществ:
148
прод ^исходи, в-в»
Запасы энергии продуктов и исходных веществ при постоянном
давлении представляют собой энтальпии, поэтому выражение в
первых скобках можно записать Н2—#i = A#.
Во вторых скобках записана разность степеней беспорядка в
системе в конечном и начальном состояниях. Запас энергии и
соответственно разность запасов энергии выражаются в калориях на
моль вещества. Так как складывать и вычитать можно только
величины одинаковой размерности, то следует найти способ
выражения члена, заключенного во вторые скобки, в тех же единицах,
что и энтальпии. Степень беспорядка и энтропия связаны между
собой пропорциональной зависимостью: чем выше значение одной,
тем выше значение другой.
Если пропорциональную зависимость обозначить знаком ос, то
сказанное можно записать так:
Степень беспорядка ос энтропия.
Такой же зависимостью связаны степень беспорядка й
температура. Чем выше температура, тем больший объем занимают газы,
тем больше растворяется вещества в растворе, тем сильнее
испарение жидкости, тем энергичнее и больше распадается сложных
молекул на более простые молекулы и атомы. Огромное число
других явлений указывает на то, что с повышением температуры
степень беспорядка возрастает. Это записывается следующим
образом:
Степень беспорядка ос температура.
Известно, что если какая-либо величина пропорциональна
нескольким другим, то она пропорциональна и их произведению.
Таким образом, степень беспорядка находится в
пропорциональной зависимости от энтропии и температуры
Степень беспорядка ос Т • S.
Для того чтобы написать знак равенства в последнем выражении,
следует ввести коэффициент пропорциональности к:
Степень беспорядка = к • Т • S.
Так как численное значение степени беспорядка вычислить не
представляется возможным, то можно ввести в эту неизвестную
нам величину коэффициент к. Тогда получим
Степень беспорядка = Г»S.
Окончательно получаем выражение
AG = AH — (T-S2 — Г-Sj),
И9
где T-S2 — величина, связанная со степенью беспорядка в
конечном состоянии (продукты реакции); T-S] — то же в начальном
состоянии (исходные вещества).
Упрощаем последнее выражение:
AG = АН — Т (S2 — Sx) = АН — Г AS.
Таким образом,
AG = AH — TAS. (5.28)
Это одна из наиболее важных термодинамических формул. В этой
формуле:
АО = Сгпрод — ^исходи, в-в — Д00бр. прод Д^обр. исходи. в-в>
All = /7 прод ^"исходи.в-в = Д^обр. прод Д^оСр. исходи.е-в>
Д*Ь = «^прод ^исходи, в-б»
Теперь становится понятной размерность энтропии кал/моль-град.
Произведение энтропии на температуру T-S имеет ту же
размерность, что и энтальпия: (кал/моль-град) -град = кал/моль. Сумма
изменения энтальпии и члена TAS дает изменение изобарного
потенциала той же размерности (кал/моль).
Изменение изобарного потенциала учитывает одновременно
изменение энергетического запаса системы и степени ее
беспорядка. Стремление системы перейти в состояние с минимальным изо-
барно-изотермическим потенциалом теперь можно сравнить с
шаром, двигающимся в сторону положения с минимальной
потенциальной энергией. В любой самопроизвольной реакции происходит
превращение исходных веществ, имеющих некоторую величину
изобарного потенциала Gb в продукты, имеющие меньшую
величину изобарного потенциала G2, G2<Gi. Поэтому все
самопроизвольные реакции характеризуются отрицательным значением
изменения изобарного потенциала
AG=G2 — G!<0.
Если в системе не происходит ни энергетических изменений
АН = 0, ни изменений в степени беспорядка AS = 0, то AG —0 п
система находится в состоянии равновесия.
Если в процессе энтропия системы не изменяется, т. е. AS = 0,
то фактором, определяющим направление реакции, будет
изменение энтальпии. Уменьшение энтальпии (ДЖО) соответствует
уменьшению изобарного потенциала (AG<0), в этом случае
самопроизвольно протекает реакция, идущая с выделением тепла
(Q>0), что согласуется с принципом Бертло — Томсена.
Если в процессе энтальпия реагирующей системы не изменяется,
то самопроизвольно система может перейти только в состояние с
большей энтропией (AS>0), при этом из-за знака минус перед
150
членом TAS изменение изобарного потенциала будет
отрицательной величиной (AG<0).
Знак AG определяет направление процесса. При AG<0 процесс
может протекать только в прямом направлении, т. е. в том
направлении, как написана реакция. При AG>0 реакция может
протекать только в обратном направлении.
Из уравнения
AG = АН —Т AS
следует:
1. Если ДЖО (уменьшение энтальпии, реакция с выделением
тепла) и А5>0 (увеличение энтропии и степени беспорядка), то
в этом случае всегда AG<0. Это значит, что экзотермическая
реакция с увеличением энтропии возможна при всех температурах.
2. Если Д#>0 (увеличение энтальпии, реакция с поглощением
тепла) и AS<0 (уменьшение энтропии, увеличение степени
порядка), то в этом случае всегда AG>0. Это значит, что
эндотермическая реакция с уменьшением энтропии не возможна ни при каких
температурах.
3. Во всех остальных случаях (ДЖО, А5<0, Д#>0, А5>0)
знак AG зависит от соотношения членов АЯ и TAS, и реакция
будет проходить только в том случае, если сопровождается
уменьшением изобарного потенциала.
Для характеристики процессов, идущих при постоянном
объеме, используется изохорно-изотермическии потенциал AF и
изменение внутренней энергии AU:
AF = AU — TAS. (5.29)
Самопроизвольная реакция возможна только при AF<0. Для
случая с AF рассуждения точно те же, что и для AG, поэтому ниже
будем рассматривать только AG ,как движущую силу реакции,
проходящей при постоянном давлении.
Изменение энтальпии и внутренней энергии связаны
формулой (5.19). Легко показать, что изобарно-изотермический
потенциал и изохорно-изотермическии потенциал связаны соотношением
AG = АЯ- TAS = At/ + PAV — TAS =
= AF + PAV = AF + nRT. (5.30)
Для удобства запоминания соотношений между основными
термодинамическими функциями, выраженными уравнениями (5.19),
(5.28) —(5.30), можно воспользоваться диаграммой, приводимой
на рис. 5.10.
AG есть работа химического процесса при постоянном
давлении, но (взятая со знаком минус, a AF — работа химического
процесса при постоянном объеме, также взятая со знаком минус:
AG-— Ар (Р = пост.), (5.31)
AF = — Av (V = пост.). (5.32)
151
Гиббс предложил считать, что свойство, которое определяет
возможность самопроизвольного (спонтанного) протекания
реакции, есть способность системы производить полезную работу при
постоянной температуре и при постоянном объеме или давлении.
Если AG или AF — отрицательные величины, реакция
возможна и она протекает с совершением работы, если AG или А/7 — по-
АН
AU
TaS
Г TaS
рС Э*|
AF
Ав
L РаК '
PaV
Рис. 5.10. Схема для запоминания
важнейших термодинамических
соотношений
ложительные величины, реакция певероягна, невозможна, нужно
затратить работу для проведения реакции. При AG = 0 или AF = 0
реакционная система находится в равновесии. Ниже будем
пользоваться только изобарно-изотермическим потенциалом.
Заключение, что везде и всегда любая самопроизвольная
реакция проходит с уменьшением изобарного потенциала, является
законом химической термодинамики, вытекающим из второго закона
термодинамики. Последний утверждает, что теплота не может
самопроизвольно переходить от холодного тела к более теплому.
Наоборот, в изолированной системе возможен самопроизвольный
переход теплоты только от теплого тела к более холодному. При
этом энергетический запас системы не изменяется, а энтропия
возрастает, достигая максимального значения в момент выравнивания
температуры во всех частях системы, т. е. в момент наступления
термодинамического равновесия.
§ 5. НАПРАВЛЕНИЕ
ХИМИЧЕСКОЙ РЕАКЦИИ
Знак изменения изобарного потенциала указывает на
направление реакции. При AG>0 реакция не проходит в том
направлении, как она написана, возможен лишь обратный процесс (для
него AG будет уже отрицательной величиной). При AG = 0 все
вещества в системе находятся в химическом равновесии, скорости
прямой и обратной реакции равны и внешне не заметно никаких
процессов в системе. При AG<0 реакция протекает в
направлении, как она написана.
Численные величины AG зависят от концентраций
реагирующих веществ. Для сравнения реакций и для выяснения влияния
температуры на потенциал реакции нужно пользоваться
сопоставимыми концентрациями. Принято для характеристики химических
процессов пользоваться стандартным состоянием, при котором
исходные концентрации или парциальные давления всех веществ
152
рассматриваемой системы равны единице (1 моль/л, 1 атм).
В этом случае при протекании химической реакции изобарно-изо-
термический потенциал системы изменяется соответственно на
AF0 и AG0 кал. Обычно в справочниках термодинамические
величины даются для 298° К, и тогда говорят, что процесс протекает
при стандартных условиях и стандартной температуре и величина
AG°298 называется стандартным изменением изобарно-изотермиче-
ского потенцила, или изобарным потенциалом реакции.
В реакции
Н2>г+1/2 02,г = НаОг
изменение изобарио-изотермического потенциала при
взаимодействии 1 моля газообразного водорода и 1/2 моля газообразного
кислорода с образованием 1 моля газообразной воды при 298° К
вычисляется следующим образом.
Из суммы изобарно-изотермических потенциалов образования
продуктов реакции вычитается сумма изобарно-изотермических
потенциалов образования исходных веществ:
Д^реакции = 2AGnpoA— 2ДИСХОдн = AG06p.H2Or—(AG06p.H2fr+ l/2AG06p.02>r)-
Изобарно-изотермические потенциалы простых веществ
принято считать равными нулю. Поэтому
ДС°реакции = AG06p.H2Or.
Откуда
Айреакции = — 54640 кал/моль.
Чем больше отрицательное значение AG0 ,тем сильнее
равновесие смещено вправо — в сторону образования продуктов реакции.
Наоборот, чем меньше отрицательное значение AG0 и чем больше
его положительное значение, тем слабее реакция проходит в
прямом направлении, тем больше в равновесной смеси исходных
веществ и меньше продуктов реакции.
О степени прохождения химической реакции судят по
константе равновесия. Для реакции
A+B=D+F
константа равновесия
к = РИГ]
[А] [В] '
Высокие значения К говорят о сильном взаимодействии
исходных веществ А и В и о большом количестве продуктов реакции D
и F в равновесной смеси. Малые значения К имеют реакции, у
которых А и В слабо взаимодействуют и в равновесной смеси
содержание продуктов реакции D и F мало.
153
Очевидно, что реакции с высокими величинами констант
равновесия характеризуются большими отрицательными значениями
изменений изобарного потенциала (равновесие смещено вправо—
в сторону образования продуктов), а реакции с малыми
величинами констант равновесия характеризуются большими
положительными значениями изменений изобарного потенциала
(равновесие смещено влево — в сторону исходных веществ).
По-видимому, существует связь между константой равновесия и изменением
изобарного потенциала.
Однако константа равновесия не зависит от концентраций
веществ. В каких бы количествах ни брались вещества А, В, D и F,
всегда после установления равновесия в смеси будет такое
соотношение веществ, которое отвечает константе равновесия (при
данной температуре):
„_ [D] [FJ
]А] [В] "
Даже если брать только исходные вещества А и В или только
продукты D и F, при данной температуре образуется смесь всех
четырех веществ с сохранением неизменного значения константы
равновесия.
Однако этого нельзя сказать об изменении изобарного
потенциала, численное значение которого зависит от количеств веществ,
участвующих в рассматриваемой реакции.
Константа равновесия реакции связана со стандартным
изменением изобарного потенциала соотношением
AG°=— RT\nKp, (5.33)
в котором Т — абсолютная температура; R — газовая постоянная,
равная 1,99 кал/моль-град; Кр — константа равновесия,
выраженная через парциальные давления компонентов:
КР= р р •
Для реакции образования воды
н2,г + о2,г = н2ог> *„= в Рн>° .
*Н2'М/202
Для этой реакции AG^s = — 54640 кал/моль.
Это значит, что если при 298° К берется в очень большом
количестве смесь водорода, кислорода и паров воды с равными
1 атм парциальными давлениями
Рщ = Р0г = PHg0 = 1 атм (общее давление = 3 атм),
то в процессе реакции для достижения равновесного состава
произойдет уменьшение изобарно-изотермического потенциала на
154
54640 кал в расчете на 1 моль воды. При этом состав равновесной
газовой смеси может быть найден из формулы (5.33)
1п/Св = ,
или
Откуда
IniC, AG° = 5464° ^40.
р 4,576-Г 4,576-298
к = ^— = 1040.
хр
р pl/2
Смесь Н2, 02 и Н20 с парциальными давлениями, равными
1 атм, после взаимодействия превращается в газовую смесь, в
которой Рн2у Ро2 и Рц2о связаны соотношением
Рн.о-Ш^Ри.-Ро7,2.
При 298° К практически нацело протекает взаимодействие
водорода и кислорода, равновесие смещено полностью вправо.
Большим отрицательным значениям изменения изобарно-изотермиче-
ского потенциала реакции соответствуют высокие константы
равновесия и прохождение процесса вправо — в сторону образования
продуктов реакции.
Из уравнения (5.33) следует, что чем больше константа
равновесия, т. е. чем больше в равновесной смеси содержится веществ,
стоящих в правой части уравнения реакции (продукты), тем
большей отрицательной величиной будет AG0. Если константа
равновесия меньше единицы, то AG0 будет положительной величиной.
Чем меньше /С, т. е. чем меньше в равновесной смеси продуктов
реакции и больше веществ, стояших в левой части уравнения, тем
большей величиной будет AG0. Знак AG0 указывает направление
смещения равновесия, величина AG0 связана со степенью
смещения равновесия.
Процессы, протекающие при постоянном объеме,
характеризуются изменением изохорного потенциала А/70 и константой
равновесия /Сс, выраженной через концентрации:
AFQ=—RT In Кс. (5.34)
Стандартное изменение AF0 достигается только тогда, когда
берется в очень большом количестве смесь всех исходных веществ и
продуктов реакции с концентрацией каждого компонента 1 моль/л
или 1 г-ион/л и система приходит в состояние равновесия с
константой равновесия Кс.
По уравнению
AG0 = Atf° — TAS°
155
можно вычислить AG0 при любой температуре, так как эта
формула выражает линейную зависимость AG0 от температуры, если
считать, что Д#° и А5° от температуры не зависят. Вычислив AG0
при интересующей нас температуре, пользуясь формулой
AG« = — RT \пКр = — 4,576Т lg/Cp, (5.35)
определяем значение константы равновесия при этой температуре.
В качестве примера рассмотрим равновесие между двуокисью
азота и ее димером. О смещении равновесия в ту или иную
сторону можно судить по изменению цвета реакционной смеси:
2N02 +Z N204
красно- бесцветный
бурый
к Уо4
ХР р2
rN02
В табл. 5.2 приведены термодинамические характеристики
рассматриваемых окислов.
Таблица 5.2
Вещество
NO„r
NA.r
ДНобр., 298'
кал/мол1*
8091
2309
кал/моль град
57,46
72,73
Изменение энтальпии в реакции равно:
Д//реакции = АЯ0бр N204,r — 2Д#0бр. N02,r ==
= 2309 —2-8091 = — 13873 кал.
Изменение энтропии в данной реакции:
Аореакции — <JN204(r — ^N02 ==
-72,73 — 2-57,46= —42,19 кал/моль - град.
Энтропия продуктов реакции (N204) меньше энтропии исходных
веществ (2N02). Продукты реакции характеризуются меньшей
степенью беспорядка по сравнению с исходными веществами. Если
в качестве движущей силы реакции рассматривать только
изменение степени беспорядка, не учитывая энергетического вклада, то
реакция самопроизвольно должна была протекать в сторону
образования двуокиси азота как вещества, имеющего большую степень
беспорядка. Если же рассматривать только энергетические
изменения как движущую силу реакции, то реакция должна
самопроизвольно протекать в сторону образования веществ, обладающих
меньшим запасом энергии, т. е. в сторону образования димера.
156
Зависимость изобарно-изотермического потенциала реакции oi
температуры (при р = пост. и при независимости Д#° и AS0 от
температуры) описывается уравнением
AG0 = ДЯ° —TAS° - — 13873 + 42,19т.
Для комнатной температуры 25°С (298°К)
AG298 = — 13873 + 298-42,19 = — 1300 кал/моль.
Отрицательная величина изменения изобарного потенциала
свидетельствует о том, что при комнатной температуре равновесие
смещено в сторону образования продуктов реакции, т. е. четырех-
окиси азота. При этой температуре равновесная смесь двуокиси
и четырехокиси азота имеет строго определенный состав, но
содержание четырехокиси азота в ней выше содержания двуокиси азота.
Равновесная смесь имеет красно-буроватый цвет.
При комнатной температуре смесь N02 и N204, имеющая
равные 1 атм парциальные давления газов, превращается в
равновесную газовую смесь с уменьшением изобарно-изотермического
потенциала на 1300 кал. При 298°К смесь Pno, = ^n2o4 = 1 атм
самопроизвольно превращается в смесь с большим содержанием
димера, характеризующуюся некоторой константой равновесия
Рассчитаем константу равновесия при 298° К:
AG° = —/гПп/Ср = —4,576-71g/Ср = -4,576-298 lg/Cp,
Кр = 100'95 = 8,9.
Итак, из исходной смеси с Pn2o4 = ^no2 = 1 атм
самопроизвольно при 298° К получается смесь со следующим соотношением
компонентов:
^n2o4 = 8,4-Pno2,
т. е. в полученной газовой смеси действительно N2O4 больше, чем
NO2 — равновесие в значительной степени смещено вправо.
Проследим теперь, как будет изменяться цвет газовой смеси при
изменении температуры.
Охладим исходную газовую смесь (Pno2 = ^n2o4 = 1 атм) до
0°С, поместив стеклянный баллончик в тающий лед. Сразу же
заметно ослабление окраски, вызванное смещением равновесия в
сторону образования бесцветного димера N204. Для 0°С (273° К)
AGsts = — 13873 + 42,19 ■ 273 - — 2355 кал/моль.
При понижении температуры изменение
изобарно-изотермического потенциала становится все более отрицательной величиной.
Вероятность самопроизвольного протекания реакции вправо
возрастает.
157
Для 273° К константа равновесия
lg/C„ = ~2355 = + 1,88,
р 4,576-273 ^
/Ср = 101-88 = 75,8.
Из смеси с Pn2o4 = Pno2 = 1 атм получается смесь с соотношением
компонентов
PN2o4-75,8.P9kv
При 0°С выход димера еще более возрос по сравнению с 25° С.
Если бы была поставлена задача получать N204 из NO2, то
следовало бы пользоваться наиболее низкими температурами (если
только скорость реакции не слишком сильно замедляется).
Поместим баллон с исходной реакционной смесью в кипящую
воду (100°С, 373°К). Замечается резкое усиление окраски из-за
смещения равновесия в сторону образования красно-бурого NO2.
Для 373° К
ДОзуз = — 13873 + 42,19-373 = + 1864 кал/моль,
т. е. при этой температуре изобарно-изотермический потенциал
реакции становится уже положительной величиной — член TAS0
превосходит член Д#°. Равновесие смещается в сторону веществ
с большей степенью беспорядка. Положительная величина
изменения изобарно-изотермического потенциала реакции делает
маловероятной протекание реакции вправо. Константа равновесия для
373° К уменьшается:
lgtf,= 1551—= _1,09,
& р 4,576-373
Кр = Ю"1'09-0,081.
Из исходной газовой смеси с Pno2 = ^n2o4 = 1 атм при 100°С
получается смесь с соотношением компонентов
Ры2о4 = 0,081Рк>2.
По сравнению с ранее рассмотренными температурами в
равновесной газовой смеси содержание N204 значительно меньше.
Интересно найти ответ на вопрос, при какой температуре
состав исходной газовой смеси с Pno2 = Pn2o4 = 1 атм не
изменяется, т. е. равен составу равновесной газовой смеси? Этому условию
отвечает константа равновесия, равная единице:
К PN2Q4 1
^N02
158
Тогдз
поэтому
AG0 = Atf° — TAS° = — RT In 1 - 0,
A#° — TAS° = 0,
— 13873+ 42,19-7-0,
13873
T =
42-19
329°K (56°C).
При 56е С исходная газовая смесь является одновременно
равновесной смесью и не замечается процесса взаимодействия
компонентов.
175 С А
F
Ё
Рис. 5.11. Смещение равновесия ди-
меризации диоксида азота при
нагревании
При 56° С движущая сила реакции равна нулю, так как при
этой температуре энтальпийный и энтропийный члены равны:
Д#° = Т.Д5°.
При более высокой температуре TAS0 > Д#°, реакция протекает
только из-за увеличения беспорядка в системе в сторону
образования NO2
N204 = 2N02 — Q.
Этот процесс проходит с поглощением тепла и не может быть
объяснен принципом Бертло — Томсена.
При температуре ниже 56°С A#°>TAS°, реакция протекает в
сторону уменьшения энтальпии
2N02 = N204 + Q
159
и самопроизвольность ее подчиняется принципу Бертло — Томсе-
на. Чем ниже температура, тем правильнее становится этот
принцип. Можно сделать вывод: при низких температурах вероятны
реакции, идущие с выделением тепла (с уменьшением энтальпии),
при высоких температурах вероятны реакции/, в которых энтропия
возрастает (возрастает степень беспорядка).
Порядок проведения опытов представлен па рис. (5.11), а
результаты всех расчетов собраны в табл. 5.3.
t, °с
0
25
56
100
т, °к
273
298
329
373
кал/моль
— 13 873
—13 873
— 13 873
— 13 873
AS0,
кал/моль-грэд
—42,19
—42,19
—42,19
—42,19
Таблица 5.3
—TAS0
11 518
12 573
13 873
15 737
AG0.
кал /моль
—2355
—1300
0
+ 1864
о о о
aG,aH,7aS, кал /моль
При независящих от температуры значениях изменений
энтальпии (ДЯ° = —13 873 кал/моль) и энтропии (Д5° = — 42,19 кал/мольХ
Хград) член TAS0 с повышением температуры становится все
более отрицательной
величиной. Из-за знака минус перед
ним фактически производится
суммирование
неизменяющегося отрицательного значения
Д#° со все более
возрастающим значением произведения
(—TAS0). Поэтому сумма
(ДЯ°—TAS0), равная
изменению изобарного потенциала
-2000
о
^2000
-4000
'6000
-8000
-10000
-12000
-14000
-76000К
270 290 310 330 350 370 К
0 25 56 100 °С
Рис. 5.12. Термодинамические
характеристики димеризации диоксида
азота
реакции AG0, непрерывно увеличивается, переходя из
отрицательной через нуль в положительную величину. Температурный рост
изобарного потенциала реакции 2N02 = N204 приводит к смещению
равновесия влево — в сторону обратной реакции N204 = 2NC>2.
На рис. 5.12 изображена диаграмма, на которой представлены
в зависимости от температуры значения изменений Д#°, Г, AS0,
AG0. Прямая dddd проходит параллельно температурной оси и
показывает, что энтальпия реакции не зависит от температуры.
Прямая сссс изображает зависимость произведения TAS0 от
температуры. Разность АН0—TAS0 представлена прямой ЪЬЪЪ и
получена вычитанием из отрезков ad отрезков ас. До температуры
160
56° С длины отрезков а больше длин отрезков ас и разность —
отрезок ab — отрицательна, AG°<0. При 56°С длины отрезков ad
и ас равны, разность их равна нулю и AG° = 0. Выше 56°С длина
отрезка ас больше длины ad, поэтому разность — отрезок аЬ —
положительна, AG°>0.
Из примера с равновесием 2Ы02 = ^04 можно сделать выводы.
1. Чем ниже температура, тем правильнее становится принцип
Бертло — Томсена. 2. При низких температурах вероятны
реакции, идущие с выделением тепла (с уменьшением энтальпии), при
высоких температурах вероятны реакции, в которых энтропия
возрастает (возрастает степень беспорядка).
Часто требуется знать состав равновесной газовой смеси.
Проведем следующий несложный расчет для равновесия:
2N02^:n2o4-
Пусть общее давление газовой смеси составляет 1 атм, тогда
Pn02 + ^N204 = 1
и
^N02 = 1 —^N204-
Константа равновесия примет вид
К = Pn*°* = pn2o4
rNOg У1 rN204/
Решим уравнение относительно Pn2o4:
M1-Pn2o4)2 = Pn2o4,
#p(1-2PN2o4+Pn2o4)-Pn2o4 =0,
Pn2o4 • Kp -Pn2o4 (2Kp + 1) + /Cp = 0,
p _ (2KP + 1) =fa /2KP + 1 (2-4Kp-KP) _2Kp + l± V*Kp + 1
^°4 WP Wp
Подставим в полученное уравнение значения констант,
вычисленные для четырех интересующих нас температур. Результаты
расчёта сведены в табл. 5.4.
Таблица 5.4 Таблица 5.5
Вещество,
состояние
СаС03, К
СаОк
со2,г
ЛЯ298'
кал/моль
—288 400
—151900
—94 100
0
S298'
кал/моль-град
22,2
9,5
51,1
*, °с
0
25
56
100
AG9,
кал/моль
—2355
—1300
0
+ 1864
«Р
75,8
8,9
1,0
0,081
PN2o4»
атм
0,89
0,71
0,38
0,07
р
N02'
атм
0,11
0,29
0,62
0,93
6 Общая химия
161
Обратите внимание, что при повышении температуры на 100е
константа изменяется почти в тысячу раз, а парциальные дазле*
ния компонентов только в десять раз.
Рассмотрим равновесие с участием твердых фаз, например
диссоциацию карбоната кальция
СаСОэ,к - СаОк + С02,г,
для которого
Кр = Рсо2-
Термодинамические характеристики веществ, участвующих в
равновесии, собраны в табл. 5.5.
Вычислим изменение энтальпии в реакции
Д#298, реакции = (— 151900 — 94100)— (—288400) -42400 кал/моль.
Процесс диссоциации идет с поглощением тепла:
СаСОз.к - СаОк + С02,г — 42400 кал/моль.
Согласно принципу Бертло—Томсена такой процесс невозможен.
Рассчитаем изменение энтропии:
Д<^реакп ии,298 = 51,5 |- 9,5 — 22,2 —- 38,4 кал/моль «град.
Процесс протекает с увеличением энтропии и фактор беспорядка
должен способствовать протеканию процесса вправо.
Зависимость ДО0 от температуры имеет вид
AG°r-42400 — 38,4-7.
Вычислим Кр = РСо2 для комнатной температуры:
AG298 = 42400—38,4-298 = 30957 кал/моль,
р 4.576-298
/Ср=Рсс* = 10-м.7.
При комнатной температуре давление С02 на СаСОз ничтожно
мало. Вероятность протекания реакции в сторону диссоциации
карбоната кальция крайне мала. Наоборот, при комнатной
температуре СаО самопроизвольно реагирует с СОг. При 1500° С =
= 1773°К:
ДС?77з = 42400 — 38,4 -1773 - — 25680 кал/моль.
Отрицательная величина AG0 указывает на большую
вероятность протекания реакции вправо. Для 1500° С
162
Pco2- 103-17 атм.
При 1500° С давление С02 над СаСОз составляет больше 1000 атм,
равновесие смещено вправо — в сторону образования СаО и С02.
Определим температуру, при которой СаСОз, помещенный в
атмосферу с /\xv= 1 атм, не претерпевает никаких изменений, т. е.
находится в равновесии с СОя:
ДС°=— 4,576-74g/Cp-0,
42400 — 38,4. Т = 0,
7* = 424Q0 = 1104°К (831°С).
38,4 '
Итак, в атмосфере С02 (Ясо8 = 1 атм) СаСОз диссоциирует при
температуре выше 831° С. При более низких температурах СаО,
помещенный в углекислый газ, ' взаимодействует с ним и дает
СаС03. На воздухе, где давление С02 значительно ниже,
температура начала распада СаСОз также понижается. Читателю
предлагается вычислить температуру, при которой
РсОг над СаСО, = Рс02 в воздухе =0,03 аТМ.
Интересна и другая, более сложная реакция, идущая с
участием твердых и газообразных фаз:
Fe203fK + ЗН2,г = 2FeK -r ЗН2Ог.
Термодинамические характеристики участников реакции собраны
в табл. 5.6.
Таблица 5.6
Вещество,
состояние
Fe2Os> к
Н-2, г
FeK
Н2Ог
0
A//29S'
кал/моль
—196 300
—57 800
^298'
кал/моль-град
20,9
31,2
6,5
45,1
Для реакции:
ДЯреакщш. 298 = — 3-57800 + 196300 - + 22900 кал,
6*
163
ДЗреакции, 298 = (2-6,5 + 3-45,1) - (20,9 + 3,31) =
^=33,8 кал/моль-град,
AG298- 22900 — 33,8Т.
При комнатной температуре '
ДС298 = 22900 — 33,8-298 =в12828 кал/моль.
Положительная величина AG0 указывает на невозможность вое-
становления Fe203 водородом для получения металлического
железа. Наоборот, противоположный процесс
2Fe + ЗН20 = Fe203 + ЗНа
характеризуется AG° = —12 828 кал, это показывает, что такая
реакция возможна. И, действительно, такая и схожие с ней реакции
самопроизвольно протекают и то, что AG0 этих реакций
отрицательно, есть причина крайне нежелательного процесса —
коррозии.
Рассчитаем температуру, при которой AG° = 0:
п, 22900
33,8
= 678°К.
При этой температуре обе реакции — и восстановления и
окисления железа (коррозия)—равновероятны. При температуре выше
678° К, наоборот, водород восстанавливает Fe203, а процесс
коррозии невозможен.
Иногда температуру, вычисляемую по формуле
^ AG»
AS0
(5.36)
называют температурой начала реакции. Она позволяет судить о
начале развития реакции в желаемом направлении. При этой
температуре окислительные и восстановительные способности веществ
изменяются. В рассмотренном процессе при Г<678°К железо
является более сильным восстановителем по сравнению с
водородом, при Г>678°К восстановительная способность водорода
начинает превышать таковую для железа.
В проведенных вычислениях использовалось только изменение
изобарно-изотермического потенциала при стандартных условиях,
и о направлении реакции мы также судили только при
стандартных условиях, когда парциальные давления всех компонентов
равны 1 атм. Для того чтобы предсказать направление реакции при
любых концентрациях веществ, пользуются другой формулой.
Величина изменения изобарно-изотермического потенциала в ходе
реакции
ак + ЬВ = dD + eE
164
с любыми количествами участвующих веществ вычисляется по
формуле
AG = \nRT PDDa'PJ -RT\nKp, (5.37)
где Pd, Реу Ра, Рв — неравновесные, любые, произвольно
выбранные парциальные давления компонентов; Кр — константа
равновесия процесса с любыми парциальными давлениями компонентов,
в том числе и с начальными Pd = Pe = Pb=^a = 1 атм.
Для реакции, проводимой в условиях постоянства объема, изо-
хорно-изотермический потенциал вычисляют по формуле
AF = RT\n C£C* -ЯТЫКс, (5.38)
где Cd, Се, Са, Св — неравновесные, произвольные концентрации
веществ; Кс — константа равновесия.
Если реакция проводится в стандартных условиях, когда
Рв = Ре = Ра = Рв:= 1 атм или Ст>=Съ = СА = Св=: 1 моль/л, то
получаются известные формулы изменения потенциалов в стандартных
условиях:
Afo = —Д7Чп/е,
с*
Подставляя эти формулы в (5.37) и (5.38), получим их в новой
форме:
AG = AG° + i?71n^^, (5.39)
AF = Д/то + ду In С*'С* . (5.40)
СА'СВ
О возможности реакции не в стандартных условиях, а с
любыми количествами участников реакции судят на основании уже
известных нам правил.
При AG<0 процесс идет в прямом направлении, так как
р« ре
rD<rE
pa pb
<кр.
Это значит, что в системе имеется избыток исходных веществ и
недостаток продуктов, поэтому исходные вещества (стоящие в
левой части уравнения реакции) реагируют между собой. В ходе
реакции увеличиваются количества продуктов (вещества, стоящие
в правой части уравнения). Реакция протекает до тех пор, пока
165
P*.PSF
РаА-РЬВ "'
Изменение изобарно-изотермического потенциала в таком процессе
и есть AG.
При Да = 0 взятая смесь является равновесной и состав ее не
изменяется:
pd ре
па РЬ 'V
^А'ГВ
= К„.
При ДС>0 прямой процесс невозможен. В этом случае
ра рЬ ^АР>
ГЛ*ГВ
в системе имеется избыток продуктов реакции и недостаток
исходных веществ. Реакция может идти только в обратном
направлении, когда продукты реакции, взаимодействуя между собой, дают
исходные вещества. Реакция продолжается до тех пор, пока
парциальные давления всех участников процесса не будут
удовлетворять разепству
D L jr
pa pb
^A*rB
Совершенно аналогичные рассуждения используются и для
реакций, протекающих при постоянном объеме (AFy Kc). Разберем
это на примере. Для реакции
2NO,;tN204
при 25° С /(р = 8,9. Определим направление реакции при общем
давлении 1 атм и при следующих начальных парциальных
давлениях веществ:
а) ^n2o4 = 0,90 атм, Р^о2 = 0,10 атм;
б) PNf0| = 0,71 атм, PNo2 = 0,29 атм;
в) PNto4 = 0,10 атм, PNOf - 0,90 атм.
Решение.
р- р
= 4,576-298 (lg^2i-_lg 8,9),
166
а) AG = 4,576-298 [lg —^ lg 8,9 - 1370 кал/моль.
AG — положительная величина: в начальной газовой смеси был
избыток N204. Процесс превращения N02 и N204 в данной газовой
смеси невозможен. Идет обратный процесс распада молекул
N204.
б) AG - 4,576-298 fig ^ lg 8,9^ - 0 кал/моль.
; \ 0,29-0,29 /
Исходная газовая смесь равновесная.
в) AG = 4,576-298 (lg ^ lg 8,9) - — 2600 кал/моль.
' \ 0,90-0,90 J
В данном случае AG<0, в исходной газовой смеси имеется
избыток двуокиси азота, которая димеризуется в N204, реакция
проходит в прямом направлении.
§ 6. ТРЕТИЙ ЗАКОН ТЕРМОДИНАМИКИ
И ЕГО РОЛЬ ДЛЯ ХИМИИ
Первые два закона термодинамики отличаются от третьего
своей применимостью во всех областях знаний. Третий закон
термодинамики особенно важен для химической термодинамики.
Энтропия есть мера беспорядка системы. При понижении
температуры теплоемкости всех веществ понижаются, так как при
этом замедляется движение атомов в кристаллической решетке.
А это значит, что в системе возрастает порядок и энтропия
системы понижается. Спрашивается, каков же предел понижения
теплоемкости, роста порядка и уменьшения энтропии? При какой
температуре этот предел достигается?
Энтропия связана с вероятностью состояния системы
формулой
S^RlnW.
При приближении температуры системы к 0°К система переходит
в состояние с наименьшей энергией. В этом состоянии
прекращаются все виды движения атомов и молекул. Таких состояний с
наименьшей энергией может быть только одно, поэтому
вероятность существования такого состояния равна единице.
Следовательно,
Sg-tflnW-tflnl -0. (5.41)
Таким образом, абсолютная величина энтропии идеального
кристалла равна нулю при 0°К. Это и есть сущность третьего закона
термодинамики. Следовательно, при 0°К имеет место полный
порядок в расположении молекул. Эти представления о состоянии
167
твердого тела при абсолютном нуле идеализированы, так как
вносимые тепловым движением молекул несовершенства
кристаллической решетки сохраняются, «замораживаются» до самых низких
температур и вносят вклад в энтропию, делая ее не равной нулю
при 0°К. Есть и другая более важная причина неравенства нулю
энтропии при 0°К у некоторых веществ.
В кристаллах простых веществ, например металлов или
кислорода, галогенов и т. п., атомы располагаются в кристаллической
туре кристалла
решетке регулярно и все находятся в окружении одного и того же
числа соседей. При охлаждении до нуля градусов энтропия таких
веществ стремится к нулю.
Кристаллы веществ-, молекулы которых состоят из двух атомов,
могут иметь различное взаимное расположение атомов. Атомы
двух видов могут располагаться совершенно беспорядочно по
отношению друг к другу (рис. 5.13, а) или же взаимно строго
чередуясь (рис. 5.13, б).
Для большинства веществ характерно именно второе, и
происходит это по следующим причинам. Возьмем, например, кристалл
йодистого водорода HI. Ион йода по размерам значительно
превосходит ион водорода, и кристаллическая решетка, по-видимому,
должна состоять из слоев молекул, какие нарисованы на рис. 5.14.
Трудно представить, чтобы в отдельных местах решетки мог
произойти обмен атомами, это неосуществимо из-за различных
размеров атомов. Даже если рассматривать кристалл фтористого
водорода, то и здесь наблюдается строгое чередование атомов
фтора и водорода, хотя размерный фактор делает вероятным
обмен местами между атомами фтора и водорода. Однако этого не
168
происходит, так как атом водорода обладает значительными
валентными силами, оставшимися не использованными полностью
после взаимодействия с атомом фтора, и за счет этих сил (водородная
связь) каждый атом водорода стремится находиться в окружении
атомов фтора. Поэтому у кристаллов веществ гипа галогенидов
при 0° должен быть почти полный порядок в расположении
атомов и So = 0, что и подтверждается экспериментально. Когда же
кристалл состоит из двухатомных молекул, атомы которых близки
по размерам, и при образовании молекулы валентные силы
каждого атома полностью исчерпываются другим атомом, тогда
дополнительное взаимодействие с другими атомами невозможно и
молекулы такого вещества располагаются беспорядочно в
кристаллической решетке. К числу таких веществ относится окись
углерода, молекулы которой образуют кристаллическую структуру
типа
... сосоососсоос...
Если бы молекулы распределялись в кристалле в порядке
чередования атомов углерода и кислорода
... сососососо
то такое состояние могло быть только одним и энтропия при 0°
равнялась бы нулю.
Предположим, кристалл окиси углерода представляет собой
1 г-мол вещества, т. е. состоит из N молекул. По теории
вероятности число возможностей W беспорядочного расположения молекул
равно 2N:
W = 2N,
поэтому
S = kln(2N) = fc.#ln2 = /?ln2 =
= 1,99-2,303-0,301 = 1,379 кал/моль.
Итак, при полном порядке расположения молекул СО
Sq = 0 кал/моль-град,
при полном беспорядке
So = 1,38 кал/моль-град.
Экспериментальное определение дает значение, хорошо
согласующееся с рассчитанным:
So = 1,1 кал/моль- град.
В кристалле окиси углерода большинство молекул
беспорядочно распределено по отношению друг к другу, и то, что энтропия
окиси углерода не достигает значения 1,38 кал/моль -град при 0°К,
свидетельствует о некоторой доле упорядоченности, возникающей
169
из-за небольшого различия в размерах атомов и некоторого
взаимодействия атомов углерода и кислорода соседних молекул.
При несимметричном распределении связей в молекуле число
способов расположения молекул также становится больше
единицы. Так, в молекуле воды связи О—Н находятся под углом
105°, и это приводит к тому, что для воды S0 = 0,81 кал/моль-град,
У водерода была обнаружена значительная величина энтропии.
Затем стало известно, что водород состоит из орто- и
пара-водорода, отличающихся положением ядерных осей в молекуле.
Очень важно, чтобы энтропия отвечала равновесному
состоянию тела при 0°. Из-за «замораживания» добиться такого
состояния практически невозможно. Поэтому избыточная энтрония
особенно велика у твердых веществ, полученных переохлаждением
жидкости и не успевших закристаллизоваться. Так, для
стеклообразного кварца энтропия может равняться I кал/моль-град. Она
еще выше у органических веществ, так, например, у аморфного
глицерина So = 4,6 кал/моль-град.
Так как при 0° энтропия чистого кристалла равна нулю, то мы
имеем нуль отсчета значения энтропии и можем вычислить
абсолютную энтропию при любой температуре. Для этого нужно иметь
данные о теплоемкости вещества от нуля до интересующей нас
температуры.
Нагреем при постоянном объеме 1 моль газа, имеющего
теплоемкость Сг —3 кал/моль-град, на какое-то число градусов,
например на 10°, от 273 до 283° К. При постоянном объеме все
подводимое тепло идет только на увеличение внутренней энергии газа и
дополнительно не расходуется на работу расширения. Поэтому
внутренняя энергия газа увеличивается на величину
Д£/ = Су (Г2 — Тг) = 3-10 = 30 кал/моль.
Предположим, что газ нагревается не непрерывно, а
ступеньками, сперва на 1°, затем еще на 1° и т. д. Из закона сохранения
энергии следует, что независимо от пути нагревания увеличение
внутренней энергии газа составит те же 30 кал/моль. На каждой
ступеньке нагревания газ поглощает по 3 кал тепла и его
температура увеличивается на 1° (ДГ=1°К). Так как на каждой
ступеньке температура повышается очень незначительно, можно
считать, что 3 кал тепла поглощаются системой при постоянной
температуре, например, при температуре начала ступени (начала
элементарного процесса нагревания) или температуре конца ступени
(окончание элементарного процесса нагревания). При поглощении
системой тепла и при повышении температуры энтропия системы
возрастает. Можно считать, что при переходе системы из одного
состояния в другое при нагревании на Г увеличение энтропии
соответствует переходу при постоянной температуре. В таком
переходе Двдерехода ВЫЧИСЛЯеТСЯ ПО формуле
170
AS% = ft*P«»<wa t (5.42)
•'перехода
где (^перехода — количество тепла, поглощенного системой при
температуре Т (Г=пост.).
Формула (5.42) выводится из формулы
AG0-Д#° —TAS0
при помощи следующих простых рассуждений.
Пусть при 0°С смешиваются лед с температурой 0°С и вода с
температурой 0°С. В этом случае изобарный потенциал воды
равен потенциалу льда, в системе не происходит никаких изменений.
Лед и вода находятся в равновесии друг с другом. Поэтому при
температуре плавления изменение изобарного потенциала в
процессе плавления равно нулю:
Откуда
AG°UJl=--&HL-TbSl= 0.
Q0 _ АЯпл
Аналогично (переход совершается при постоянной температуре)
изменение энтропии при испарении при температурах плавления и
кипения определяется по формулам
-гО
0 А^исп
Аоисп —
as!
О АЯисп
В общем случае
AS'
перехода •
ап перехода __ __ ^перехода
* перехода ' перехода
Возвратимся к процессу нагревания газа. При нагревании от 273
до 274° К
Qv Cv з
AS = = = = 0,01099 кал/моль-град.
Т 273 273 iv*
Аналогичные способом вычисляются изменения энтропии для
каждой из последующих одноградусных ступеней, пока не будет
достигнута конечная техмпература 283° К.
Отложим на графике (рис. 5.15) значения Cv/T в зависимости
от Т. Увеличение энтропии в каждой ступени нагревания газа на
Г соответствует площади, прямоугольника с основанием в 1°
(dT=\°) и высотой Cv/T. Заметьте, что площади прямоугольников
171
(их высота или величина) делаются все меньше с ростом
температуры, хотя энтропия возрастает. Это происходит из-за того, что
изменение энтропии уменьшается с увеличением температуры.
Полное изменение энтропии в переходе равно сумме всех
малых изменений энтропии, соответствующих площадям
прямоугольников [Цг)лТ (ДТ=1°).
Предполагалось, что теплота поглощается при постоянной
температуре, равной началу ступеньки, т. е. при температурах, всегда
-%г > кал/моль град
Рис. 5.15. Вычисление
изменения энтропии газа при
нагревании
10
8
6
4
2
С нал/моль град
- ^^
уГ
- /
X
- /
L^T ....I i 1 , I , 1 ,„L
50 100 150 200 250 300
о
7, К
Рис. 5.16. Теплоемкость
оксида никеля
ниже действительных. В этом случае сумма всех элементарных
площадей (на рис. 5.15 она ограничена верхней ступенчатой
кривой) будет больше действительного изменения энтропии. Она
равна AS = 0,1082 кал/моль-град. Можно провести вычисление
энтропии, предполагая, что ее изменение при нагревании на 1°
совершается при температуре окончания нагрева. Тогда для первой
ступеньки
3
aS = — = 0,01094 кал/моль-град.
274 /г
Просуммировав изменения энтропии по этим ступенькам (нижняя
ступенчатая кривая), получим значение энтропии ниже истинного,
а именно Л5 = 0,1077 кал/моль-град.
Чтобы получить истинное изменение энтропии, следует
провести аналогичные вычисления со все более малыми изменениями
температуры, ДГ=0,Г, 0,01°С и т. д. При этом каждый раз будут
получаться две площади — одна больше, а другая меньше
истинного изменения энтропии, но чем меньше интервал ДГ, тем ближе
будут эти площади к истинному изменению энтропии.
Истинная величина А5 будет площадью под кривой,
вычисляемой по формуле
172
283
&S=( -^- dT.
T
273
Предполагалось, что Cv не зависит от температуры. В этом случае
283
CV «„ г, i^ 283
AS = С —^- dT = Су In
273
273
= 3-2,303lg -^ = 0,1081 кал/моль-град.
2*1 о
Экспериментально можно определить теплоемкости от самых
низких, близких к 0°К, температур до необходимых высоких
температур, обычно до стандартной температуры 298,15° К, и
описанным приемом, называемым графическим интегрированием,
вычислять изменение энтропии от 0 до 298,15° К.
Обычно, когда определяется изменение энтропии вещества при
его нагревании от 0 до 298,15° К, результат называют стандартной
энтропией вещества и обозначают не S298,i5—So, a S298,is> считая,
что So = 0.
Таким образом,
298,15
S2Vi5= J ~^rdT. (5.43)
На рис. 5.16 дана кривая зависимости теплоемкости закиси
никеля NiO от температуры. Графическим интегрированием получено
S298,i5 = 9,08 кал/моль-град.
Такое же значение энтропии получается совершенно иным
методом: из констант равновесия реакции закиси никеля с
водородом. Константа равновесия реакции восстановления закиси
никеля:
NiOK + H2,r = NiK + H2Or,
к __ рн2о
рн2
равна 606,7 при 427° С, 574,8 при 527° С и 551,0 при 627° С.
Константа равновесия определена так: при постоянной температуре
над закисью никеля пропускаются смеси водорода и паров воды
такого состава, чтобы одновременно существовали закись никеля
и металлический никель. Их равновесие при данной температуре
характеризуется только одним соотношением парциальных давле-
173
ний паров воды и водорода Рнао/^н2> которое и будет константой
равновесия написанного процесса (концентрация твердых фаз в
константу не входит).
Если состав газовой смеси при 427° С соответствует
н о
2 <С 606,7, то равновесие смещается вправо и идет процесс
восстановления закиси никеля до водорода. Процесс проходит,
пока вся закись никеля не превратится в никель. Если состав
газовой смеси соответствует 606,7, то равновесие смещается влево и
никель окисляется до закиси никеля. Повышение температуры
приводит к уменьшению равновесных значений Рн2о/Рн2. Это
значит, что с повышением температуры равновесие смещается в
сторону восстановления закиси никеля: чем выше температура, тем
меньшее парциальное давление паров воды в смеси с водородом
требуется для перевода NiO в №.
Вычислим по формуле
AG0 = -4,576-74g/Cp
изменение изобарно-изотермического потенциала для каждой
температуры и найдем зависимость его от температуры:
AG0 = 606—И ,87-Т.
В этом уравнении постоянная 606 характеризует изменение
энтальпии в процессе восстановления водородом закиси никеля до
металлического никеля, а коэффициент 11,87 — изменение энтропии в
том же процессе:
дЯ° = 606 кал/моль,
Д5°= 11,87 кал/моль-град.
Пользуясь законом Гесса, найдем изменение энтропии в
реакции как разность между энтропиями продуктов и исходных
веществ:
Л С° С0 1 С° . С0 С0
АОреакции = ^Н2Ог "Г ^NiK — «bNiOR — *^H2, г-
Предположим, что энтропии воды, никеля и водорода известны:
Sh2o =45,13 кал/моль-град,
5н2>г =31,21 кал/моль • град,
S°m =7,12 кал/моль • град.
Следовательно,
С° С0 1С0 С0 А С0
«JNiOK = »^Н2Ог "Г °NiK — °Н2>Г— Д^>реакции
И
5^ю<: = 9,17 кал/моль- град.
174
Из данных по теплоемкости мы получили 9,1 кал/моль-град.
Рассмотренный пример — отличное подтверждение правильности
третьего закона термодинамики. Из третьего закона следует, что
формула
AG°= АЯ° —7AS0
при Т->0 и AS0-*-0 превращается в
AG 7=о = А//г=о.
Только при абсолютном нуле изменение изобарно-изотермиче-
ского потенциала реакции делается равным изменению энтальпии.
На рис. 5.17 изображены изменения
изобарного потенциала и энтальпии реакции
в зависимости от температуры.
Расхождение кривых Л#° и AG0 для
большинства реакций при комнатной
температуре очень мало, и направление
реакции в основном определяется знаком
изменения энтальпии. При высоких
температурах ветви кривых расходятся, и
при температуре Т разница равна TAS.
ко-л/моль
Рис. 5.17. Изменение энтальпии и изобарного
потенциала от температуры
Энтропии всех элементов, простых веществ, кристаллов, газов,
жидкостей и т. и. всегда положительны. Энтропии ионов в
растворе могут быть как положительными, так и отрицательными. Это
объясняется тем, что измерить абсолютную энтропию иона в
растворе невозможно и энтропии ионов измеряются в условной
шкале относительно энтропии иона водорода, которую приняли равной
нулю.
Вопросы и упражнения
1. Сформулируйте первый закон термодинамики и закон Гесса.
2. Чему равно численное значение газовой постоянной R, которое
используется в обычных термодинамических расчетах?
3. Какими соотношениями связаны между собой изохорный и изобарный
тепловые эффекты, изменения энтальпии и внутренней энергии?
4. Приведите примеры реакций, у которых Д#>Д*7 и Д£/>Л#.
5. Зная энтальпию нейтрализации сильных кислот и оснований
Д+р + ОН~р = Н2Ож А /4йтр = - 13340 кал/моль
и энтальпию образования жидкой воды из простых веществ
Н9 _ + — 09 г = Н2Ож АН ^ tj ^ = — 68300 кял/моль
z»1 2 обр.н^о
175
и предполагая, что энтальпия образования иона водорода Н+ в водном
растворе равна нулю:
Т Н2,г = Н£Р А НобР.н+ = ° кал/моль,
I v * р.р
определите энтальпию образования иона гидроксила из простых веществ.
6. Почему при приближении к 0°К энтропии веществ стремятся к
О кал/моль-град?
7. Чем объясняется самопроизвольное прохождение реакций с поглощением
тепла, А# >0?
8. Объясните причину самопроизвольного прохождения реакции,
сопровождающейся поглощением тепла:
СоС12-6Н2Ок + 6SOCI2 ж = СоС12к + 12НС1Г + 6S02r.
9. Что такое стандартные условия и стандартная температура? (Сравните
с нормальными условиями.)
10. Напишите формулу, позволяющую вычислить AG0 процесса по
известным АЯ°, AS0 и Т.
11. В каких случаях изменение изобарно-изотермического потенциала равно
изменению изохорно-изотермического?
12. При комнатных температурах олово может существовать в двух
модификациях: серое олово и белое олово. Термодинамические характеристики
модификаций
Олово
^пбелое
^серое
ЛЯ298, 0бр'кал/моль
0
-500
S2gg, кял/моль-град
12,32
10,55
Какое олово термодинамически устойчиво при стандартной температуре^
13. Можно ли осуществить синтез этилового спирта из углерода, кислорода
и водорода при комнатной температуре, если
Вещество
С2Н5ОНг
^графит
Й2, Г
02, г
АЯ298, обр'кал/моль
—23 000
0
0
0
Sggg, кчл/моль-град
75,43
1,36
31,21
45,01
14. Как связана константа равновесия реакции с изменением изобарно-изо-
гермического потенциала при стандартных условиях?
15. Как определить направление реакции при нестандартных условиях?
16. Произведение растворимости СаСОз равно
прСаСОа = [Са2+] • [СО*"] - 4,8-10*.
Вычислите AG0 растворения карбоната кальция и сделайте вывод о том
процессе, который возможен при стандартных условиях в смеси ионов Сз2+ и СОд"".
17. При 1000°К константа равновесия КР реакции
2Н2Ог = 2Н2 г + 02 г
176
равна 7,62-10~4 атм. Давление кислорода над оксидом FeOK при этой
температуре равно 2,5-Ю-21 атм. Вычислите константу равновесия реакции
FeOK + H2>r=FeK + H2Or
и изменение изобарного потенциала реакции AG?ooO'
18. Константа равновесия реакции
2Н1Г^ Н2г +12)Г
при 360°С равна 0,0162, и при 445°С—0,0240. Вычислите ЛЯ° и AS0 реакциш
диссоциации HI в указанном интервале температур.
19. Константа равновесия диссоциации водорода на атомы при 1 600°К
равна 2,93-10~9 и при 2500° равна 6,26-Ю-4. Вычислите энергию связи молекулы
водорода (в данном температурном интервале).
20. Растворимость углекислого газа в воде при 0°С составляет
7,99-Ю-2 моль/л при Рсо2=1 атм. Вычислите изобарный потенциал
растворения С02 в НгО и сделайте вывод о направлении самопроизвольного процесса
при стандартных условиях.
21. Рассмотрите процесс диссоциации фосфорной кислоты по ступеням..
Термодинамические характеристики участвующих веществ равны:
Вещество (р-р)
н3ро4
Н2Р07
НР02-
ро^-
Н+
ЛЯ298, обр' кал/моль
—308 200
—311300
—310 400
—306 900
0
^29 8'
кал/моль-
42,1
21,3
— 8,6
—52,0
0
град
Вычислите энтальпии, энтропии и изобарные потенциалы процессов
диссоциации при 298°К, а также константы равновесия. Объясните причину
отрицательного значения АН с по первой ступени, а также отрицательных значений
А £дИСС и их рост от первой к третьей стадии.
ГЛАВА 6
КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИЙ.
ХИМИЧЕСКОЕ РАВНОВЕСИЕ. КАТАЛИЗ
§ 1. СКОРОСТЬ ХИМИЧЕСКИХ РЕАКЦИИ.
ОСНОВНЫЕ ПОНЯТИЯ И ОПРЕДЕЛЕНИЯ
Раздел химии, рассматривающий скорости и механизм
химических процессов, называется химической кинетикой.
Представление о том, что химическая реакция протекает во
времени, несомненно, возникло еще у первых химиков.
Сосредоточив свое внимание на конечном этапе реакции, они рассматривали
время jcaK фактор, необходимый для превращения исходного
вещества в конечный продукт.
Однако путь от этих качественных представлений до научного
определения понятия скорости реакции оказался весьма долгим.
Только в начале второй половины XIX в. было дано строгое
количественное определение скорости реакции как изменения
количества реагирующего вещества в единицу времени в единице
реакционного пространства. Это определение справедливо как для
гомогенных, так и для гетерогенных процессов. В первом случае
реакционным пространством является объем реакционного сосуда,
находящегося при температуре опыта, а во втором — поверхность,
на которой протекает реакция.
Основная величина в химической кинетике — скорость
реакции. Химические реакции могут протекать с различной скоростью.
Юдни реакции протекают очень медленно, другие настолько
быстро, что это приводит к взрыву.
Скорости химических реакций' зависят от природы
реагирующих веществ, концентрации, температуры, давления, присутствия
катализаторов, а в случае гетерогенных превращений — и от
ряда других условий, таких, как состояние поверхности раздела фаз,
условий тепло- и массообмена и др. Задача химической кинетики
состоит в выяснении роли этих факторов и в установлении
механизма реакций.
Скорость химических реакций измеряется изменением
концентраций реагирующих веществ в единицу времени. Концентрацию в
этом случае принято выражать числом грамм-молекул вещества
в 1 л раствора, а время — в минутах или секундах. В ходе
химической реакции концентрации вступающих в реакцию веществ
уменьшаются, а концентрации продуктов реакции возрастают, что
приводит к уменьшению скорости реакции (рис. 6.1 и 6.2). Пусть
при неизменном объеме и неизменной температуре концентрация
одного из реагирующих веществ уменьшилась от С{ до С2 за про-
178
межуток времени от xi до Т2. Тогда согласно сказанному скорость
реакции v за данный промежуток времени равна
С<2 "~~ (-«1
V = ^ *-.
т2 —тх
Знак минус в правой части уравнения необходим по следующим
соображениям: скорость реакции всегда представляет
положительную величину, следовательно, и правая часть уравнения в целом
Время
Время
Рис. 6.1. Зависимость
.скорости реакции от времени
Рис. 6.2. Кривые
скоростей с уменьшением и
возрастанием
концентраций
должна быть положительна. Однако числитель правой части
уравнения отрицателен (большая величина Сг вычитается из меньшей
С2), так что правая часть становится положительной величиной
только тогда, когда поставим перед ней отрицательный знак. Чем
меньше промежуток времени, тем меньше и разность
концентраций. Обозначим малый промежуток времени через Дт и
соответствующее ему малое уменьшение концентрации через АС, тогда
v == —
АС
At
Как бы ни был мал промежуток времени Ат, отношение
АС
At
остается постоянной величиной, ибо с уменьшением знаменателя
уменьшается и числитель.
Q Q
Отношение —— — выражает среднюю скорость в интервал
т2 — Ti
ле Т2—ть а отношение —
АС
At
будет тем ближе к истинной
скорости реакции, чем меньше Ат и соответственно АС, так что
отношение предельно малой величины изменения концентрации, которую
обозначим через dC9 к предельно малому промежутку времени d%
179
___ dC
dx
измеряет истинную скорость реакции в момент времени т.
Изучая скорость химической реакции, практически измеряют
ряд концентраций того или другого из реагирующих веществ в
следующие друг за другом моменты времени. Скорость химической
реакции можно определять либо по изменению концентрации ис-
dC л
ходного вещества, тогда v = , либо по изменению концен-
dx
трации получаемого в результате реакции вещества, тогда
i dC ^
0=4- , а в общем виде выражение для истинной скорости
dx
при постоянном объеме системы записывается:
, dC
dx
Безразлично, концентрацию какого из реагирующих веществ брать
для измерения скорости химических реакций, так как
концентрации всех реагентов изменяются в эквивалентных количествах.
Следует при этом подчеркнуть, что измеренные по разным
веществам скорости не равны, а пропорциональны одна другой.
При взаимодействии одновременно двух и более веществ,
например А + В->С, скорость реакции можно выразить через
производную от Са или от Св. Однако следует иметь в виду, если
реагирующие вещества имеют различные стехиометрические
коэффициенты, выбор производной может повлиять на величину
константы скорости. Так, для реакции' А4-2В->С скорость может быть
выражена через концентрации реагирующих веществ двумя
способами:
dCA dCB
V = ИЛИ V =
dx dx
Но из стехиометрического уравнения следует, что на 1 моль
прореагировавшего вещества А приходятся 2 моля веществ В.
Поэтому две скорости, написанные в нашем уравнении, не равны друг
другу. Они будут равны при условии, если написать
— 2
dC* dC]
в
dx dx
Представляет интерес и второй вопрос: каким образом можно из
ряда значений концентрации и отвечающих им моментов времени,
которые дает опыт, вычислить истинную скорость реакции. На эти
вопросы отвечает закон химической кинетики, который называют
законом действующих масс.
180
§ 2. ЗАКОН ДЕЙСТВУЮЩИХ МАСС
Зависимость скорости реакции от концентрации реагирующих
веществ определяется законом действующих масс: скорость
химической реакции при постоянной температуре, протекающей в
гомогенной среде, пропорциональна произведению концентраций
реагирующих веществ, возведенных в степень их стехиометрических
коэффициентов.
Этот закон был сформулирован Гульдбергом и Вааге в 1867 г.
и независимо от них в 1865 г. русским ученым Н. И. Бекетовым.
Справедливость закона действующих масс можно видеть на
примере взаимодействия молекул паров йода и водорода. Пусть
приготовлен 1 л смеси водорода и паров йода при температуре Т.
Молекулы смеси, находясь в беспорядочном движении, будут
соударяться друг с другом, и в момент соударений разнородных
молекул возможно осуществление реакции между ними по
уравнению
Н2 + I, = 2HI.
Очевидно, чем больше будет число соударений разнородных
молекул в единицу времени, тем больше их прореагирует и тем больше
будет скорость реакции. Следовательно, скорость реакции должна
быть пропорциональна числу соударений молекул водорода и йода
в единице объема за единицу времени. Однако число соударений
и число действительно реагирующих молекул не одно и то же.
Число соударений столь огромно, что если бы при каждом из них
происходила реакция, то она закончилась бы мгновенно, т. е.
протекала бы в виде взрыва. Однако известно большое количество
медленно текущих реакций.
Установлено, что только весьма малая доля а общего числа
соударений приводит к химическому взаимодействию, а остальные
соударения оканчиваются безрезультатно. Тогда если общее число
соударений будет равно г, то число реагирующих молекул водо-
рода или иода будет ссг, что составляет грамм-молекул, где
N — число Авогадро.
г az
1ак как представляет число грамм-молекул водорода или
иода, исчезающих в единице объема за единицу времени, то оно
соответствует скорости реакции, т. е.
dC _ az
dx ~ N '
Чтобы подсчитать число соударений z, обозначим концентрации
водорода и йода в смеси, отнесенные к моменту времени и
выраженные в молях на литр, через Сн2 и Ci2. Число отдельных
молекул того и другого газа будет равно NCUz и iVCi2. Подсчитаем
181
сначала число соударении одной молекулы водорода с молекулами
йода за время Ах. Этот промежуток времени будет тем меньше, чем
больше молекул йода в единице объема, т. е. чем больше величина
NCj2. Следовательно, можно написать
Ат
NC}
где k\ — коэффициент пропорциональности. Легко понять, что
величина, обратная Ат, представляет число соударений одной
молекулы водорода с молекулами йода и равна
1 __ Щш
Ат kL
Число соударений не одной, а всех NCh2 молекул водорода со всеми
молекулами йода будет в NCh2 раз больше:
ЛРСн,Сь
Подставив это значение г в уравнение для скорости реакции,
получим
dC az аЛ>2СЫ2 С,
dx N . Nkx
аЛ/
= kCu2C\2\
k^
k'
Так как доля а общего числа соударений зависит только от
температуры и от природы реагирующих молекул и представляет для
данной температуры и данной реакции постоянную величину, так
же как и k\ и N, то и величина k постоянная при тех же условиях.
Поэтому можно сказать, что скорость взаимодействия
газообразных водорода и йода в каждый момент времени
пропорциональна произведению наличных концентраций реагирующих
веществ.
Рассмотрим еще реакцию между газообразным водородом и
кислородом с образованием воды
2Н2 + 02 = 2Н20.
Наши рассуждения в принципе должны быть аналогичны, только
теперь z будет означать число тройных соударений — двух
молекул водорода с одной молекулой кислорода.
Число соударений одной молекулы водорода с молекулами
кислорода, как и раньше, равно
182
где CHjS и Со2 — концентрации водорода и кислорода в молях на
литр и N — число Авогадро.
Нас интересуют такие соударения молекулы водорода с
молекулами кислорода, когда в том же месте окажется еще одна
молекула водорода. Вероятность этого события невелика, и мы
предположим, что это происходит только в небольшой доле х от
общего числа двойных соударений. Эта доля будет тем больше, чем
больше число молекул водорода в смеси, т. е.
х - k"NCHt.
Умножив число двойных соударений на х, получим число тройных
соударений z, которое надо вставить в уравнение (6.6).
Итак,
z = н1_о1_
W
dc а'г <*'b°№C2HiC02 2
~ "5Г == ~Т = ш = *Сн-Со"
k'
где а' — доля общего числа тройных соударений, приводящих
к химическому взаимодействию. Постоянная к и в этом случае
зависит только от температуры и природы реагирующих веществ.
Следовательно, скорость реакции водорода с кислородом
пропорциональна произведению квадрата концентрации водорода на
концентрацию кислорода. Вот почему на основании теоретических и
экспериментальных данных закон действующих масс
формулируется с учетом возведения концентраций в степени их стехиомет-
рических коэффициентов, стоящих перед формулой вещества в
уравнении реакции.
Если обозначить концентрации реагирующих веществ для
реакций
Н2-Ь12=2Н1 и 2NO + Н2 = N20 + Н20
через Сн8, Ci2, Cn0-Ch2, to закон действующих масс можно
записать следующим образом:
o=ftCH,-Cit,
t> = кСко-СИг,
где k — константа скорости реакции, зависящая от природы
реагирующих веществ и температуры, но не зависящая от
концентрации — в этом существенная разница между константой и
скоростью реакции.
183
При концентрации каждого из реагирующих веществ, равных
1 моль/л, k равна скорости химической реакции. Из этого следует,
что константа скорости химической реакции является мерой
реакционной способности молекул и характеризует скорость реакции
при данной температуре. Понятие скорости относится к данной
реакции, а не к отдельным реагирующим веществам. И для
каждой реакции при постоянной температуре ее константа скорости
будет величиной постоянной.
Для реакции, записанной в общем виде
тк + пВ = С,
скорость реакции может быть выражена уравнением
где тип — стехиометрические коэффициенты реагирующих
веществ.
Закон действующих масс применим только к простейшим
реакциям. Если реакция протекает в несколько стадий, закон
действующих масс справедлив для каждой из них, а скорость
сложных химических процессов определяется скоростью наиболее
медленной промежуточной реакции. Для газовых реакций этот закон
является строгим только в пределах применимости к реагирующим
веществам законов идеальных газов. Он также применим и к
реакциям в растворах, но только при больших разбавлениях.
§ 3. РЕАКЦИИ
В ГЕТЕРОГЕННЫХ СИСТЕМАХ
Многие химические процессы протекают в гетерогенных
системах, состоящих из двух или нескольких однородных частей,
называемых фазами, отличающихся своими свойствами и отделенных
друг от друга поверхностями раздела.
Например, в реакциях:
а) С + 02 = С02 — две фазы, твердая в виде угля или кокса и
газообразная — смесь 02 и С02;
б) S + 02 = S03 — две фазы, твердая в виде серы и
газообразная в виде смеси 02 и S03;
в) 3Fe + 4H20^:Fe304 + 4H2 — три фазы, две твердых (Fe и
Fe304) и одна газообразная в виде смеси водорода и водяного
пара.
Если в гомогенных системах реакции происходят во всем
объеме, то в гетерогенных системах химическое взаимодействие
протекает на поверхности. Поэтому течение гетерогенных процессов во-
многом зависит от величины и состояния поверхности.
Как применяется закон действующих масс к гетерогенным
системам? Например, в реакции горения угля
С -I 02 == С02
184
соударения между молекулами кислорода и твердого вещества
могут происходить только на поверхности раздела фаз, а значит
масса твердой фазы не влияет на скорость реакции, тогда
выражение скорости реакции можно записать
v = kCo2,
т. е. скорость реакции пропорциональна только концентрации
кислорода, величина концентрации твердых веществ входит в
значение константы скорости.
Течение гетерогенных процессов в ряде случаев зависит от
характера образующихся продуктов. Если образующиеся продукты
мало растворимы, то, оставаясь на твердой поверхности
реагирующего вещества, замедляют химическую реакцию. Например,
свинец медленно растворяется в серной кислоте, так как на
поверхности образуется труднорастворимый слой сернокислого
свинца. Также известно, что алюминий очень медленно окисляется на
воздухе, так как покрывается малопроницаемой для кислорода
пленкой А120з.
Скорость гетерогенных реакций зависит от вязкости среды,
влияющей на скорость диффузии. Имеет значение и
теплопроводность среды, при малом значении которой может иметь место
разогревание поверхности твердого реагирующего вещества. Поэтому
ожидать совпадения между скоростью по закону действующих
масс и скоростью, полученной в опыте, можно только тогда, когда
скорость всего процесса определяется скоростью химического
взаимодействия, а не диффузионными или другими
сопутствующими явлениями. В химических процессах, протекающих в
несколько стадий, скорость реакции в целом определяется наиболее
медленной стадией.
§ 4. ВЛИЯНИЕ ТЕМПЕРАТУРЫ
НА СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ.
ЭНЕРГИЯ АКТИВАЦИИ
Закон действующих масс остается справедливым при любой
температуре, однако константа скорости, как правило,
увеличивается с ростом температуры, а следовательно, увеличивается и
скорость реакции. Так, вычисленная константа скорости
разложения паров ацетона равна 0,0112 при 503ЭС, при 552°С уже
составляет 0,12, т. е. при увеличении температуры на 49° она
увеличивается в 10,7 раза, или в 2,1 раза при росте температуры на 10°.
Многочисленные опыты показывают, что при повышении
температуры на 10° скорость реакции увеличивается в 2—4 раза
(правило Вант-Гоффа)
v = fy°+10°
185
где kt° — константа скорости при t°\ &/°+ш° —константа
скорости при t° +Ю°; У — температурный коэффициент реакции, т. е.
число, показывающее, во сколько раз увеличивается скорость
реакции при повышении температуры реакции на 10°.
Расчеты показывают, что при повышении температуры в
арифметической прогрессии скорость химической реакции возрастает в
геометрической прогрессии. Влияние
температуры на скорость химической
реакции показана на рис. 6.3.
Как объяснить столь большое
увеличение скорости реакции с ростом
температуры? Казалось бы естественным пред-
Рис. 6.3. Зависимость скорости реакции от тем-
tennepcvypa пературы
положить, что это увеличение скорости — результат возрастания
числа соударений между молекулами.
Однако кинетическая теория показывает, что увеличение
скорости, вызванное увеличением числа столкновений с ростом
температуры, совсем незначительно и не может обеспечить
действительно наблюдающегося возрастания скорости. Число
столкновений частиц можно подсчитать на основе кинетической теории газов.
Рассчитанная на основании этой теории скорость реакции
оказывается значительно больше, чем наблюдаемая на опыте.
Несоответствие теоретически ожидаемой и действительной скорости
привело к выводу, что не каждое соударение эффективно, и
химическое взаимодействие имеет место, только если сталкивающиеся
молекулы обладают определенным избытком энергии по
сравнению со средней энергией данных молекул. Эта избыточная энергия
называется энергией активации, а молекулы, обладающие этой
избыточной энергией,"— активными.
Закон возрастания числа активных молекул с ростом
температуры совсем иной, чем возрастание числа соударений. Если число
соударений при температуре Т{ равно Zx и при температуре Т2
равно Z2, то
При Тг = 273° (0СС) и Т2 = 373° (100°С)
Таким образом, по кинетической теории число соударений с
повышением температуры возрастает незначительно.
186
По теории Аррениуса отношение числа активных молекул NiU<
к общему числу молекул N, т. е. доля активных молекул а равна
е_
а - -^- = е RT,
N
где Е — энергия активации; R — газовая постоянная, равная
1,985 кал/град; е — основание натуральных логарифмов,
равное 2,71828.
Вычислим но уравнению, во сколько раз увеличивается доля
активных молекул, если температура возрастает от 0 до 100°С (при
среднем значении энергии активации £ = 20 000 кал)
2000 100
^И)0_ = е 1,985-273 373 = 20000,
а0
что вполне согласуется с опытом.
Известно, что молекулы одного и того же вещества при
одинаковых условиях движутся с различной скоростью и обладают
поэтому различным запасом кинетической энергии. Молекулы с
большей кинетической энергией будут химически более активны, так
как они легче преодолевают то отталкивание между
электронными оболочками, которое возникает при их сближении и
препятствует реакции. Более активными и способными к химическому
воздействию окажутся и возбужденные молекулы, электроны
которых в момент столкновения находятся на энергетических уровнях
с большим запасом энергии. Но в общем доля активных молекул
очень невелика. По этой причине очень мала и доля химически
эффективных столкновений. При повышении температуры энергия
вещества возрастает и перераспределяется между молекулами
таким образом, что значительно возрастает число активных молекул.
Источником энергии активации может быть энергия в
различных формах: тепловая, лучистая, электрическая, энергия
радиоактивных частиц и т. д.
Обычно реакции между веществами с прочными ковалентпыми
связями, ослабить которые трудно, идут медленно. Это
наблюдается в большинстве реакций между органическими веществами.
Полярные вещества взаимодействуют между собой быстрее, чем
неполярные. Например, реакция между газами — аммиаком и
хлористым водородом
NH3-j-HCl=NH4Cl,
сопровождающаяся почти моментальным появлением белого
тумана — хлористого аммония. Здесь образованию новых химических
связей способствует взаимное притяжение молекул своими
разноименными полюсами, из-за чего продолжительность их
столкновения увеличивается и растет вероятность того, что молекулы
прореагируют.
187
При химическом взаимодействии ионов, заряды которых
противоположны, энергия активации почти не нужна, и такое
взаимодействие отличается очень большой скоростью. Как следует из
сказанного, любое химическое взаимодействие начинается с
преодоления сопротивления (энергетического барьера), возникающего
при сближении атома и молекулы с другой молекулой. Если это
сопротивление преодолено, старые связи исчезают и возникают
новые. Та избыточная энергия молекул, которую называют
энергией активации, необходима, чтобы возникающее сопротивление
было преодолено и соударение между молекулами было
эффективным.
Чем больше необходимая энергия активации, тем меньше та
доля общего числа соударений, которая приводит к химическому
взаимодействию, но одновременно тем больше температурный
коэффициент реакции.
Большую роль в снижении энергии активации играют
катализаторы (см. § 11).
§ 5. ПРОСТЫЕ И СЛОЖНЫЕ РЕАКЦИИ
Простыми реакциями называются такие реакции, которые
протекают в одну стадию в соответствии со стехиометрическим
уравнением. Кинетическое уравнение простых реакций содержит одну
константу скорости.
Сложные реакции представляют совокупность простых реакций.
Поэтому уравнение сложной реакции содержит несколько констант
скоростей. Кинетическое уравнение сложной реакции составляют,
руководствуясь принципом независимости, так как конечное
изменение концентрации данного вещества и скорость сложной
реакции — это результат независимых изменений.
К сложным реакциям относятся обратимые, параллельные,
последовательные, сопряженные, цепные и др.
Обратимые реакции — это реакции, продукты которых
взаимодействуя друг с другом, вновь дают исходные вещества:
А:£В или A+B^C + D,
к2 v2,k2
где k\ и /е2, v\ и v2 — соответственно константы скорости и
скорости прямой и обратной реакций при данной температуре. Общая
суммарная скорость v равна разности скоростей прямой и
обратной реакции (v = V\—и2).
Примеры обратимых реакций:
Н2 + 127^2Н1,
2Н2+02^2Н20,
3H2+N2^2NH3.
188
Параллельные реакции — это реакции, протекающие
одновременно в двух и более направлениях:
А '
г—в
\ С
Примером параллельной реакции является разложение
бертолетовой соли, которое при нагревании идет в двух направлениях;.
2КС1 + ЗОа
6КС103<(
ЗКС104 + КС1
При параллельных реакциях скорость всего процесса будет
определяться в первую очередь скоростью наиболее быстрой стадии.
Последовательные реакции — это реакции с промежуточными,
стадиями:
А-^В->С,
где А — исходное вещество; В — промежуточное вещество; С —
конечный продукт реакции. Здесь к\Фк2. Пример такой
реакции — гидролиз трисахаридов в кислой среде:
Qe^Oie + HaO->-CeHlaOe + С12Н22Оп + Н20 ->ЗС6Н1206.
Сопряженные реакции — это такие сложные реакции, в
которых одна реакция протекает только в присутствии другой:
A + B^D (a),
А + С->Е (б).
Реакция (а) идет самопроизвольно, тогда как (б) идет только в
присутствии первой реакции (а). Так, йодистоводородная -кислота
не окисляется хромовой кислотой непосредственно по реакции
6HI + 2Н2Сг04 -+ 31а + Сг203 + 5Н20.
Однако если в систему ввести FeO, который может непосредствен-
но взаимодействовать с хромовой кислотой по реакции
6FeO + 2H2Cr04 = 3Fe203 + Cr203 Н- 2Н20,
то одновременно с окислением FeO начинается окисление HI.
Цепные реакции. Многие химические процессы представляют
собой цепные реакции. К ним относятся некоторые гомогенные
реакции окисления, галоидирования, расщепления, полимеризации
и другие, обладающие такими особенностям, которые не могут
быть объяснены рассмотренными кинетическими закономерностями.
Часто эти реакции начинаются не сразу, а после индукционного»
189
периода, предшествующего заметному изменению концентрации
реагирующих веществ. Все эти особенности хорошо объясняются
цепным механизмом реакции. Цепной механизм реакций открыт
Н. А. Шиловым (1905 г.), а затем подробно изучен М. Боденштей-
ном, Н. Н. Семеновым и С. Хиншельвудом.
Для цепных реакций характерны три стадии — зарождение
цепи, ее развитие и обрыв. Зарождение цепи может происходить
•о
* + »
+ +
о
20,
/f
У х20о
-0 2
итд,
Рис. 6.4. Схема
разветвляющейся цепной
реакции
Рис. 6.5. Реакция
разложения озона (02 —
возбужденная молекула
кислорода)
под воздействием света, радиоактивного излучения, нагревания
системы и др. Например, образование НВг из Н2 и Вг2 происходит
под воздействием света. В начале процесса молекулы Вг2,
поглотив квант энергии света h\y распадаются на свободные атомы
брома:
Вг2 \-fiv =2Вг.
Затем идет развитие цепи — активные атомы брома
взаимодействуют с молекулами H2:
Вг + Н2 = НВг I- H.
Появившиеся новые активные частицы — атомы водорода
взаимодействуют с молекулами Вг2:
Н + Вг2 = НВг + Вг
и т. д. Обрыв цепи возможен при столкновении двух активных
центров:
Н + Вг=НВг,
190
н + н = н2,
Вг + Вг = Вг2
или при соударении активного центра со стенкой сосуда или
молекулой постороннего вещества. При неограниченном
разветвлении цепей происходит взрыв. Следует различать взрывы цепного
типа, в которых резко увеличивается число активных центров,
и тепловые взрывы, в которых тепло экзотермических реакций не
успевает отводиться.
Мы рассмотрели пример простой цепной реакции, в которой
каждая активная частица дает начало одной цепи, — стационар-
ныеу или, как чаще их называют, неразветвляющиеся цепные
реакции.
Во многих случаях каждая активная молекула или частица
порождает не одну, а две и более активные молекулы или части-
цы, т. е. цепь разветвляется — эти реакции называют
разветвляющимися цепными реакциями (рис. 6.4). Примером
разветвляющейся реакции может служить реакция разложения озона, которая
протекает по схеме, изображенной на рис. 6.5.
Цепные реакции окисления и воспламенения протекают весьма
своеобразно. Н. Н. Семенов и С. Хиншельвуд установили, что такие
реакции возможны, если внешнее давление не превышает
некоторого верхнего предела. Ряд веществ, стойких при атмосферном
давлении, самопроизвольно взрывается при понижении давления,
т. е. существует также и нижний предел давлений, при переходе
которых реакция прекращается.
§ 6. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Реакции, проходящие под действием лучей света, называются
фотохимическими. Поглощение света усиливает вращательное
движение молекул или колебательное движение в них атомов и
атомных групп и может приводить к возбуждению электронов
наружных оболочек атомов. В результате колебаний с большей частотой
можно вызвать образование ионов за счет отрыва наиболее слабо
связанных электронов.
Каждый квант поглощенного света вызывает элементарную
химическую реакцию (закон Эйнштейна). Однако основная
элементарная реакция, протекающая при поглощении одного кванта
света, часто сопровождается побочными реакциями, в которых
участвуют молекулы реагента или другие присутствующие
молекулы. Поэтому число молекул, претерпевающих химические
превращения при поглощении одного кванта света, обычно отличается
от единицы. Квантовым выходом называется величина,
определяемая следующим образом:
число превращенных молекул
квантовый выход = ^-— —.
число поглощенных квантов
191
Закон Эйнштейна является законом эквивалентности между
поглощенной энергией и степенью превращения вещества.
По химическому характеру возбуждаемые реакции могут быть
реакциями синтеза, диссоциации или окисления и др. Процессы
химического разложения под действием света называются фото-
яизом. Различают два вида фотохимических реакций. В одних
реакциях химическое взаимодействие происходит в количествах,
пропорциональных количеству света, поглощенного реагирующими
веществами. Например, реакция разложения галогенида серебра
в фотографическом процессе. В других реакциях поглощение света
играет роль только возбудителя реакции, которая дальше
совершается уже самопроизвольно, независимо от количества
поглощенного света, например, реакция между водородом и хлором при
освещении светом, богатым ультрафиолетовыми лучами. Это
фотокаталитические реакции.
Важнейшими фотохимическими реакциями являются некоторые
природные процессы, использующие энергию солнечного света,
например реакции фотосинтеза, проходящие в растениях. Так,
различные реакции, приводящие к образованию глюкозы, можно
в суммарном виде представить в виде
6СОа + 6НяО->СвН12Ов+60а+2,82 кдж.
Значение поглощения солнечной энергии растениями для про-
дессов фотосинтеза было показано К. А. Тимирязевым, который
впервые установил, что количество поглощенной энергии и
.количество синтезированного вещества находятся в строгом
соответствии между собой и эти соотношения определяются законом
сохранения энергии.
Другим очень важным видом природных фотохимических
реакций является поглощение молекулами кислорода коротковолновой
части солнечной радиации в верхних слоях атмосферы. Процесс
этот происходит с образованием озона, который в свою очередь
способен поглощать некоторую часть ультрафиолетовой радиации.
Благодаря этим процессам, коротковолновая часть солнечного
излучения, являющаяся весьма опасной для живых организмов,
не доходит до земной поверхности. Фотохимические реакции
применяются в фотографическом процессе, в основе которого лежит
способность хлорида, бромида и йодида серебра разлагаться под
действием света.
§ 7. МОЛЕКУЛЯРНОСТЬ
И ПОРЯДОК РЕАКЦИИ
Химические реакции классифицируют по молекулярности и по
порядку реакции.
Число молекул, одновременным взаимодействием между
которыми осуществляется элементарный акт химического превращения,
называется молекулярностью реакции.
192
По этому признаку реакции делят на одно молекулярные, двух
молекулярные и трехмолекулярные. Четырехмолекулярные
реакции практически не встречаются, так как вероятность встречи
одновременно четырех молекул в 108 меньше вероятности
столкновения двух молекул.
а. Одномолекулярные, или мономолекулярные, реакции — эго
реакции, в стехиометрическом уравнении которых в левой части
только 1 моль вещества. В общем виде это можно записать так:
А-*С.
Примеры:
1« = 21,
СН3СОСН3 - С2Н4 + СО + Н2,
N206 = N02 + NO + 02.
Это реакции распада молекулы на более простые части
(термическая диссоциация, реакции изомеризации, самопроизвольный
распад радиоактивных веществ и др.).
б. Двухмолекулярные, или бимолекулярные, реакции — это
реакции, в стехиометрическом уравнении которых в левой части
2 моля вещества. В общем виде это можно записать так:
2А->С или А + В-^С.
Примеры:
Щ + Н1=Н2 + 12,
CHgCOOH + С2Н5ОН - СН3СООС2Н5 + Н20,
СН3СООСН3 + Н20 = CHgCOOH + CH3OH и т. д.
в. Трехмолекулярные, или тримолекулярные, реакции — это
реакции, в стехиометрическом уравнении которых в левой части
3 моля вещества. Эти реакции встречаются весьма редко. В
общем виде это можно записать так:
А + 2В-^С или 2А + В->С.
Примеры:
2NO + 02 - 2N02,
2NO + H2 = N20 + H20.
В случаях, когда в реакциях участвует большое число молекул,
процесс практически проходит в две или большее число стадий.
При определении молекулярности реакции во всех случаях число
молей образующихся продуктов реакции не имеет значения, так
как молекулярность реакции определяется только стехиометриче-
ским числом молей реагирующих веществ.
7 Общая химия
193
Порядок реакции определяется кинетическим уравнением
реакции и равен сумме показателей степеней при /концентрациях в этом
уравнении.
Реакции могут быть нулевого, первого, второго и третьего (не
выше), а также дробного порядка. Дробный порядок характерен
для сложных реакций, протекающих через промежуточные стадии,
т. е. имеющих более одного элементарного акта. В реакциях
нулевого порядка скорость реакции постоянна во времени v = const.
Нулевой порядок наблюдается в таких гетерогенных реакциях,
в которых скорость подвода реагирующего вещества во много раз
больше скорости химического взаимодействия. Порядок и молеку-
лярность реакции совпадают только для простых реакций,
протекающих в одну стадию в соответствии со стехиометрическим
уравнением. В подавляющем большинстве случаев молекулярность
реакции и порядок не совпадают. Объясняется это тем, что стехио-
метрическое уравнение реакции описывает процесс в целом и не
отражает истинного механизма реакции, протекающей, как
правило, через ряд последовательных стадий. Кроме того, можно
встретиться со случаями таких простых реакций, когда
концентрация одного из реагирующих веществ практически остается
постоянной, тогда порядок реакции понижается.
Реакции первого порядка простых гомогенных реакций
описываются кинетическим уравнением первого порядка:
v = -h— = kC1.
— dx
Примеры реакции первого порядка:
1) разложение ацетона
СН3СОСН3 -> СО + СН3СН3,
2) разложение оксида азота-V
2N206-4N02 + 02.
В отношении реакции разложения оксида азота-V отметим
следующее: часто наблюдается несоответствие между действительным
порядком реакции и тем порядком, который вытекает из стехио-
метричеокого уравнения. Разложение оксида азота-V
количественно соответствует реакции
2N205 = 4N02 + 02.
Можно было бы ожидать, что эта реакция будет протекать по
уравнению второго порядка. Однако исследование показывает, что
она подчиняется уравнению первого порядка. Это противоречие
устраняется при рассмотрении истинного механизма реакции,
который определяется двумя последовательными превращениями:
N205-»N203+02,
N205 + N203-4N02.
194
Наиболее медленная первая мономолекулярная стадия, которая
определяет общую скорость и порядок реакции.
Реакции второго порядка простых гомогенных реакций
описываются кинетическим уравнением второго порядка:
ах
или v = kC2 (при С± = С2).
К реакциям второго порядка относятся реакции соединения
А + В->АВ
и реакции обмена
A + B-*C + D.
Пример реакций второго порядка:
разложение оксида азота-IV
2NOa->2NO + Oa.
Реакции третьего порядка простых гомогенных реакций
описываются кинетическим уравнением третьего порядка и встречаются
очень редко:
» = --^- = ЛС1С2С8
dx
или v = kC3 (при Сг — С2 = С3).
Примеры реакций третьего порядка:
2NO + 02->2N02,
2CO-f-02-*2C02.
Среди реакций нулевого порядка, протекающих на поверхности
фаз (гетерогенных), часты случаи, когда скорость практически не
зависит от концентрации веществ, участвующих в реакции:
v = — — = АС°.
dx
Как видно из кинетических уравнений, при одинаковых
концентрациях исходных веществ в правой части уравнений получаются
выражения: kC\ kC2, kC3 и kC°. Следовательно, порядок реакции
по веществу определяется показателем степени концентрации.
§ 8. ТЕОРИИ ХИМИЧЕСКОЙ КИНЕТИКИ
Теории химической кинетики строятся на основе представлений
об активных молекулах. Соответственно этому в химической
кинетике для теоретического расчета констант скоростей используются
два метода: 1) метод активных соударений и 2) метод переходно-
7"
195
го состояния, или активированного комплекса. Оба метода не
противоречат друг другу, а представляют собой лишь разные способы
теоретического обоснования одного и того же явления,
Метод активных соударений Аррениуса. Сущность метода
заключается в том, что соударения между молекулами эффективны
"1
Е1
I7T1
III/ \
гЧИ
Е2
(£_
Ход реакции
Рис. 6.6. Изменение энергии в ходе
экзотермической реакции
Рис. 6.7. Диаграмма уровней энергий молекул
реагирующих веществ, продуктов реакции и
активированных молекул в экзотермических и
эндотермических реакциях
А ктибиробанные
молекулы
А ктибиробаиные
молекулы
реагирующие
бе шест 8 а
А
JL
f
\е
1
1
Jl
т
г
продукты
у реакции
\Е
1
реагирующие
^6emecrnSaji
"Т
продукты
w реакции
*
1
1
Экзотермическая реакция
Эндотермическая реакция
только в том случае, когда соударяющиеся частицы обладают
некоторым избытком энергии по сравнению со средней энергией всех
молекул. Известно, что число столкновений пропорционально
концентрациям веществ, а соответственно росту числа столкновений
растет и скорость реакции.
Однако таких представлений оказалось совершенно
недостаточно для объяснения того, что химическая реакция всегда
протекает значительно медленнее, чем это соответствует теоретически
возможному числу встреч молекул.
Для ряда реакций замечено, что при обыкновенной
температуре некоторые реагенты могут находиться в соприкосновении без
196
и шпмодействия неопределенно долгое время (смесь водорода и
кислорода при обыкновенной температуре, соприкосновение
кислорода с бензином и спиртом и т. д.).
Известно, что число соударений молекул пропорциональна
квадратному корню из температуры, а скорость реакции растет по
экспоненциальному закону, т. е. несравненно быстрее.
Все эти факты находят объяснение в теории активации, в
основе которой лежит утверждение, что реагируют лишь те молекулы,
энергия которых не ниже некоторого предела Е — энергии
активации.
Такое утверждение естественно, так как реакции должно
предшествовать ослабление или разрушение химических связей в
реагирующих молекулах, на что требуется затрата энергии. Величина
энергии активации определяется природой реагирующих веществ.
На рис. 6.6 показано изменение энергии системы в ходе
экзотермической реакции.
Если прямая реакция (переход из состояния / в состояние
//) — экзотермическая, то общий запас энергии продуктов реакции
будет меньше, чем исходных веществ. Система в результате
реакции перешла на более низкий энергетический уровень, разность
уровней I и II равна тепловому эффекту реакции Q. Уровень К
определяет тот наименьший запас, которым должны обладать
молекулы, чтобы их столкновения могли привести к химическому
взаимодействию. Разность между уровнем К и уровнем /
представляет энергию активации прямой реакции Еи а разность между
уровнями К и II — энергию активации обратной реакции Е2. Таким
образом, по пути из исходного состояния в конечное система
должна перейти через энергетический барьер, что также видно из
рис. 6.7.
Только активные молекулы, обладающие в момент
столкновения необходимым избытком энергии, могут преодолеть этот барьер
и вступить в химическое взаимодействие. Число их согласно
закону распределения Максвелла — Больцмана тем больше, чем
меньше величина энергетического барьера. Теория активных
соударений Аррениуса имеет и другой весьма существенный
недостаток — она не учитывает время, необходимое для
перераспределения связей и образования новых молекул при столкновении
активных молекул. Все это не позволяет теоретически рассчитывать
константы скоростей многих реакций. Этот недостаток устраняется
теорией переходного состояния.
Теория переходного состояния. Одним из важнейших
теоретических достижений в развитии химической кинетики было введение
понятия переходного состояния, или активированного комплекса,
предложенного в 1935 г. Эйрингом и Поляньи.
В основе теории переходного состояния лежит закон
действующих масс и необходимость активных соударений молекул, но
последний процесс рассматривается более детально, с точки зрения
197
структурных и энергетических изменений, которые претерпевают
молекулы при их взаимодействии.
Активные соударения согласно теории переходного
состояния — это сложный процесс перераспределения связей в
молекулах, который начинается еще до их столкновения и заканчивается,
только когда молекулы разойдутся на расстояния, превышающие
дальность действия их силовых полей.
Рассмотрим механизм соударения двух частиц в реакции
А + ВС -* АВ + С.
При достаточном сближении атома А с молекулой ВС
ослабляется связь между атомами В и С в молекуле ВС и одновременно
начинает формироваться связь А—В. Образуется так называемый
активный комплекс (переходное состояние) А ... В ... С, в котором
за счет взаимного влияния частиц и ослабления валентных связей
атом В в равной степени принадлежит атомам А и С. По мере
уменьшения расстояния между частицами это влияние возрастает
и в конечном счете приводит к разрыву старых и образованию
новых связей, т. е. АВ и С. Структура переходного комплекса
имеет важное значение для объяснения скорости любой реакции,
так как константа скорости зависит от константы статистического
равновесия Кф процесса образования активного комплекса
Kit к
А + ВС^ А ... В ... С-^ АВ 4- С.
Эта зависимость вытекает из того, что протекание реакции
существенно не нарушает статистически равновесного распределения
молекул по Максвеллу — Больцману. Мерой относительной
статистической устойчивости реагирующих веществ и переходного
состояния является константа Кф. От обычной константы равновесия
она отличается величиной и размерностью. Не следует
отождествлять активный комплекс с промежуточными химическими
соединениями. Активный комплекс — лишь переходное состояние
реагирующих частиц в момент соударения. Время его существования
ничтожно; оно короче времени соударения; конфигурация
переходного комплекса соответствует максимуму потенциальной энергии
{неустойчивость системы). Образование активного комплекса в
каждой реакции требует определенной энергии активации, распад
же его происходит самопроизвольно. Поэтому скорость реакции
равна числу активных комплексов, проходящих в единицу
времени через энергетический барьер в направлении хода реакции, т. е.
она равна так называемой линейной концентрации СФ активного
комплекса вдоль пути реакции, умноженной на среднюю скорость
его движения по этому пути.
§ 9. ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Химические реакции бывают необратимыми и обратимыми.
Необратимые реакции протекают только в одном направлении. Этих
198
реакций не та« много. В качестве необратимых реакций можно
указать на следующие реакции:
2КС103 = 2КС1 + 302 f ,
NaaCO, + H3S04 = Na2S04 + Н20 + C02 f ,
BaCl2 + H2S04 = BaSQ4 + 2НС1.
Строго говоря, полностью необратимых реакций нет> особенно в
закрытых системах.
Большинство реакций являются обратимыми, т. е. они
протекают в противоположных направлениях и не идут до конца, так
как продукты реакции, взаимодействуя между собой, вновь дают
исходные вещества.
Таковы, например, реакции:
3H2 + N2^2NH3,
2Н2+02^2Н20,
Н2 + 12^2Ш.
Обратимость реакции отмечается знаком двух стрелок,
направленным в противоположные стороны =<=£.
В принципе для любого химического процесса можно
подобрать такие условия, при которых он становится обратимым.
Рассмотрим, как протекают обратимые реакции. В качестве примера
возьмем реакцию образования йодистого водорода из водорода
и йода в форме гомогенной газовой реакции:
Н2+12^2Н1. '
Данные анализа смеси показывают, что при температуре около
448°С в закрытом сосуде лишь 78% исходных веществ
превращаются «в йодистый водород.
Если при тех же условиях взять йодистый водород, то 22% его
распадется на водород и йод:
2Ш^:н2 + 12.
Как в первом, так и во втором случае устанавливается такое со
стояние системы, для которого характерно определенное
соотношение исходных и образующихся веществ. В этом случае
устанавливается подвижное химическое равновесие; скорости прямого
процесса (образования йодистого водорода) и обратного (распада
йодистого водорода) становятся равными. Графически этот
процесс хорошо показан на рис. 6.8 и 6.9.
Состояние обратимого процесса, при котором скорости прямой
и обратной реакции равны, называется химическим равновесием.
Концентрации реагирующих веществ, которые устанавливают-
199
ся при химическом равновесии, называются равновесными
концентрациями. Обычно их обозначают при помощи квадратных
скобок, например: [Н2], [Ь], [HI] в отличие от неравновесных
концентраций, обозначаемых Сн2, G2> Снь
п
Л
F 50
*
QJ
^
О
Q.
С:
Ч<*
/ V
( 2
J-
0 20 40 60 80 100
Время, мин
Рис. 6.8. Кривые
образования
(вверху) и разложения .
(внизу) йодоводо-
рода
20 АО 60 80 100
Время,мин
Рис. 6.9. Иллюстрация
двухстороннего подхода к равновесию
При установившемся равновесии
vx - v2.
Следовательно, для обратимого процесса
mk -hnB^lpC 4-9D,
Откуда
[A]m-[B]«
h
K>
где К — константа химического равновесия, заменяющая собой
отношение двух постоянных величин k\ и k2. Константа
химического равновесия не зависит от начальной концентрации реагирую-
200
щих пеществ и изменяется при изменении температуры. Чем
опл мне К, тем больше концентрация конечных продуктов в момент
р.'ншовесия. Она показывает, во сколько раз прямая реакция идет
оыстрее обратной при одинаковой температуре и (концентрациях,
р л иных единице. Это и есть выражение основного закона
химической кинетики — закона действующих масс применительно к
обратимым реакциям. Его формулировка такова:
«При обратимых реакциях равновесие наступает тогда, когда
произведение концентраций образующихся веществ, деленное на
произведение концентраций веществ, вступающих в реакцию,
станет равным некоторой постоянной величине для данной реакции
при данной температуре».
Например, для процессов
равновесия
зн2-
Н2
2NO
будет:
К
К
к =
+ N2:£2NH3,
-г 1,;Г2Н1,
+ 0.2^:2N02.
[NH3p
" [NJ [H2p'
_ [HI]2
[НЛУ '
. [NQ2]a
lNO]« [OJ
Зная равновесные концентрации, легко вычислить константу
равновесия и исходные концентрации реагирующих веществ.
Например, равновесие реакции, протекающей по уравнению
Ha-ria2:2Hi,
установилось при следующих концентрациях участвующих в неЬ
веществ:
[Н2] =0,004 моль/л, [12] -0,025 моль/л,
[HI] -0,08 моль/л.
Необходимо определить константу равновесия и исходные
концентрации водорода и йода. Для решения задачи находим сначала
значение константы равновесия для данной реакции по уравнению
[HI]2
#==■
[Н2] [12]
Подставив значения концентраций реагирующих веществ при
равновесии, находим константу равновесия:
201»
К= °'082 =64.
0,004-0,025
Уравнение реакции показывает, что из одной молекулы Нг и
одной молекулы 12 образуются две молекулы HI. Следовательно,
для образования 0,08 моля HI требуется 0,04 моля Н2 и 0,04 моля
1г. Таким образом, исходные концентрации
[Н2] = 0,04 + 0,04 = 0,08 моль/л,
[12] = 0,025 + 0,04 = 0,065 моль/л.
Пользуясь этим примером, можно решить следующую задачу.
Например, реакция между оксидом азота-П и кислородом
выражается уравнением
2NO + 02^:2N02.
Равновесие установилось при следующих концентрациях веществ:
[NO] -0,02 моль/л; [02] -0,3 моль/л; [N02] =0,06 моль/л.
Определить константу равновесия и исходную концентрацию кис-
+ лорода. (Ответ: /(=30; [02]-= 0,33 моль/л.)
Твердые вещества не входят в выражение константы равновесия
по той же причине, что и при скорости реакции. Например:
3Fe + 4Н20 ^ Fe304 + 4Н2,
к т= [Н2]4
[Н20]4
Зная константу равновесия /С, концентрацию исходных веществ
и уравнение химической реакции, можно рассчитать равновесные
концентрации всех реагирующих веществ.
Для реакций между газами молярные концентрации
пропорциональны парциальным давлениям Р. В этом случае выражение
для константы равновесия можно записать в виде
Рг • ?т> • • •
*, = -£—2 .
гА• *в • • •
Закон действующих масс, строго говоря, применим только
к реакциям между газами при давлениях, близких к
атмосферному, а также к равновесиям в неконцентрированных растворах
неэлектролитов и в сильно разбавленных растворах слабых
электролитов. Для применения его к реакциям всех типов и в большом
интервале условий введено понятие активности. Активность
вещества а пропорциональна его фактической концентрации С:
202
где / — коэффициент пропорциональности, отражающий влияние
окружающей среды.
Таким образом, в общем виде константа равновесия имеет вид
ат . ап
аА ав
При равновесии в каждый момент времени .количество молекул,
образовавшихся в результате взаимодействия исходных веществ,
равно количеству молекул, распавшихся в результате обратной
реакции. Следовательно, с наступлением равновесия реакция не
прекращается; между двумя противоположными процессами
устанавливается подвижное химическое равновесие, вследствие чего
концентрации как исходных, так и образующихся веществ
остаются постоянными. При неизменных условиях химическое равновесие
сохраняется любое время.
Расчеты по константам равновесия находят широкое
применение для количественных характеристик равновесий как в
гомогенных, так и в гетерогенных системах.
Смещение химического равновесия. Химическое равновесие
остается неизменным до тех пор, пока не изменяются условия
(концентрация, температура, а для газов и давление), при
которых оно существует. Изменяя условия, можно перевести систему
из одного равновесного состояния в другое, отвечающее новым
условиям.
Химическое равновесие смещается потому, что изменение
условий неодинаково влияет на скорости прямой и обратной реакций.
Через некоторое время эти скорости вновь сравниваются, и
наступает состояние равновесия, отвечающее новым условиям.
Изменение равновесных концентраций реагирующих веществ, вызванное
изменением какого-либо условия, называется смещением, или
сдвигом, равновесия.
Если при изменении условий увеличилась концентрация
образующихся веществ, т. е. веществ, формулы которых находятся
в правой части уравнения, то говорят о смещении равновесия
вправо. Если изменение условий влечет за собой увеличение
концентраций исходных веществ, формулы которых стоят в левой
части уравнения, то это рассматривают как смещение равновесия
влево.
Смещение химического равновесия с изменением условий
подчиняется правилу, известному под названием принципа Ле Ша-
телье:
«Изменение одного из условий (температуры, давления, кон-
центраций), при которых система находится в состоянии
химического равновесия, вызывает смещение равновесия в направлении
той реакции, которая противодействует произведенному
изменению».
203
Нарастание противодействия, т. е. образование продуктов
реакции, накапливающихся вследствие вызванного смещения
равновесия, продолжается до того момента, пока система не
достигнет нового равновесия, соответствующего новым условиям.
Следует отметить, что принцип Ле Шателье приложим не только
к химическим реакциям, но и ко многим процессам, не имеющим
чисто химического характера: к испарению, конденсации,
плавлению, кристаллизации и др.
Влияние изменения температуры. При повышении
температуры ускоряются оба противоположных процесса, но
эндотермические сильнее, чем экзотермические. При понижении температуры
из двух реакций быстрее протекает экзотермическая.
Следовательно, для суждения о влиянии температуры на химическое
равновесие необходимо знать тепловой эффект процесса. Его можно
найти экспериментально или рассчитать на основе закона Гесса.
Таким образом, направление смещения равновесия при
изменении температуры определяется знаком теплового эффекта. Чем
он больше, тем значительнее влияние температуры. Если же он
близок к нулю, то изменение температуры практически не влияет
на равновесие.
Согласно принципу Ле Шателье при повышении температуры
происходит смещение равновесия в сторону эндотермической
реакции, идущей с поглощением тепла. При понижении
температуры равновесие смещается в направлении экзотермической
реакции, идущей с выделением тепла.
Например, в случае обратимого процесса
N204^2N02~ 56,84 кдж
повышение температуры смещает равновесие .вправо, т. е.
способствует разложению N2O4; понижение температуры смещает
равновесие влево — в сторону образования N204.
Влияние изменения концентрации. Введение в равновесную
систему дополнительных количеств любого из реагирующих
веществ ускоряет ту реакцию, при которой оно расходуется.
Таким образом, увеличение концентрации исходных веществ
смещает равновесие в сторону образования продуктов реакции.
Увеличение концентрации продуктов реакции смещает равновесие в
сторону образования исходных веществ. Степень смещения
равновесия при данном количестве реагента находится в зависимости
от стехиометрических коэффициентов. Так, в случае равновесной
системы
FeCl3 + 3KCNS ^ Fe (CNS)3 + 3KC1
введение FeCl3 или KCNS вызывает смещение равновесия вправо,
что легко обнаруживается по увеличению интенсивности окраски
раствора, зависящего от Fe(CNS)3. Наоборот, введение в раствор
КС1 или Fe(CNS)3 сдвигает равновесие влево — окраска стано-
204
вится менее интенсивной, что указывает на уменьшение в
растворе концентрации Fe(CNS)3.
Константа равновесия реакции
к = [Fe(CNS)3][KCl]3
fFeCl3][KCNS]3 '
Это выражение показывает не только направление, но и степень
смещения равновесия. Изменение концентрации [КС1]
значительнее сдвигает равновесие, чем изменение концентрации
[Fe(CNS)3], поскольку стехиометрический коэффициент при
[КС1] втрое больше коэффициента при [Fe(CNS)3]. To же самое
можно сказать соответственно и о [KCNS] и [FeCl3] с той лишь
разницей, что равновесие при этом сместится в противоположную
сторону. В случае равновесной системы
со + н2опар^со2-ьн2
равновесие может быть смещено вправо увеличением
концентрации СО или Н20 (водяного пара); уменьшение концентрации С02
или Н2 также приводит к смещению равновесия вправо. При
увеличении концентрации С02 или Н2, а также при уменьшении
концентрации СО или Н20 равновесие смещается влево.
Влияние давления. В соответствии с принципом Ле Шателье
увеличение давления смещает равновесие в сторону той реакции,
которая приводит к уменьшению общего числа молекул в газовой
смеси, а следовательно, к уменьшению давления в системе.
Наоборот, при уменьшении давления равновесие смещается в
сторону реакции, сопровождающейся увеличением общего числа
молекул газа, что влечет за собой увеличение давления в системе.
Так, уравнение обратимого процесса
N2 + 3H2^2NH3
показывает, что из одной молекулы азота и трех молекул
водорода образуются две молекулы аммиака. Из-за уменьшения
числа молекул повышение давления вызывает смещение
равновесия реакции вправо — в сторону образования аммиака, что
сопровождается понижением давления в системе. Наоборот, понижение
давления приводит к смещению равновесия влево — в сторону
разложения аммиака, что влечет за собой повышение давления в
системе (рис. 6.10).
В тех случаях, когда в результате реакции число молекул
газообразных веществ остается постоянным, при изменении
давления одинаково изменяются скорости прямой и обратной
реакции, и поэтому равновесие не смещается. К таким реакциям
относятся, например:
N2 + 02^:2NO,
СОа4 Н2£СО гН2Опар.
205
Принцип Ле Шателье имеет большое практическое значение. Он
дает возможность находить такие условия для протекания
химического процесса, которые обеспечивают максимальный выход
желаемого вещества. Технология производства важнейших
химических продуктов основана на применении принципа Ле Шателье
и на расчетах, вытекающих из закона действующих масс. При
решении практических вопросов учитывают влияние температуры,
давления и концентрации не только на равновесие, но и на
скорость реакции.
Р, н/м2 В
Давление
Рис. 6.10. Влияние давления и
температуры (°С) на выход
аммиака
0,01 f,eC
Рис. 6.11. Диаграмма
состояния воды
§ 10. ФАЗОВЫЕ РАВНОВЕСИЯ
При изучении различных химических систем важную роль
играет понятие фазы. Фазой называется совокупность всех
однородных частей системы, обладающих одинаковым химическим
составом и одинаковыми свойствами и отделенных от остальных
частей системы поверхностью раздела. Характерно, что на
границе между двумя фазами всегда существует некоторая
поверхность раздела, при переходе через которую многие свойства
обнаруживают резкое скачкообразное изменение.
Гетерогенные равновесия в процессах перехода вещества из
одной фазы в другую, не сопровождающиеся изменением
химического состояния этого вещества, называются фазовыми
равновесиями. К таким равновесиям можно отнести процессы
испарения, плавления, конденсации различных веществ.
Одним из наиболее общих законов гетерогенных равновесий
(фазовых и химических) является правило фаз, установленное
Гиббсом в 1876—1878 гг. Согласно этому правилу в равновесной
системе число фаз Ф, число степеней свободы С и число
независимых компонентов К связаны простым соотношением
С + Ф-K-f 2 или С = К — Ф + 2.
206
Иными словами, число степеней свободы равно числу
независимых компонентов минус число фаз плюс два.
Компонентом называется такая составная часть системы,
которая является химически однородным веществом, может быть
выделена из системы и может существовать в изолированном
виде в течение длительного времени. Например, в любом водном
растворе, содержащем растворенную соль, компонентами
являются вода и растворенное вещество. При рассмотрении фазовых
равновесий, когда в системе не происходит химических
превращений, понятия «компонент» и «независимый компонент»
совпадают. Однако для системы, в которой протекает химическое
взаимодействие и где, следовательно, относительные количества всех
веществ при равновесии взаимно связаны, эти понятия
различаются. Например, в реакции
2Н2 + 02 - 2Н20
три компонента, но независимы из них только два, так как
содержание любой из этих составных частей определяется
полностью двумя другими.
Известно, что равновесие системы зависит от условий
(температуры, давления, концентрации). Число условий, которые можно
изменить произвольно в известных пределах без изменения числа
и вида фаз системы, называется числом степеней свободы.
Например, жидкая вода и пар могут находиться в равновесии при
различных температурах и давлениях, но при этом каждому
давлению соответствует строго определенная температура и,
наоборот, каждой температуре — строго определенное давление.
Изменение одного из факторов без соответствующего изменения
другого приведет к исчезновению одной из фаз: так, повышение
температуры при постоянном давлении приводит к испарению воды.
Следовательно, для системы вода — пар существует лишь одна
степень свободы, т. е. такая система—одновариантная. Существуют
двухвариантные, или бивариантные, системы (имеющие две
степени свободы), а также системы с большим числом степеней
свободы (поливариантные, или многовариантные, системы).
Системы, не имеющие ни одной степени свободы, называются
инвариантными, или безвариантными. Такие системы могут
существовать только при строго определенных условиях. Например, лед,
жидкая вода и водяной пар при температуре +0,0100°С и
давлении 610,5 н/м2 (4,579 мм рт. ст.).
При изучении фазовых равновесий очень широко применяется
графический метод. На основе опытных данных строят
соответствующие диаграммы, называемые диаграммами состояния.
Диаграмма состояния может быть построена для любого вещества и
позволяет определить, при каких условиях будут устойчивы
данная фаза или равновесие фаз. На рис. 6.11 представлена
диаграмма состояния воды, где на оси абсцисс отложена
температура, а на оси ординат — давление. Каждой фазе отвечает опреде-
207
ленное поле диаграммы, ограниченное от полей других фаз
линией, характеризующей равновесие между данными двумя
фазами. Кривая ОА отвечает равновесию в системе лед — пар, кривая
ОС — равновесию вода — пар, кривая ОВ — равновесию в
системе лед — вода, кривая OD — равновесию в системе
переохлажденная вода — пар. Каждая кривая относится к двухфазной
системе (Ф = 2, С=1, К=1). Все такие системы одновариантны
(С = 1). В точке О пересекаются все три кривые. Эта точка
отвечает равновесию между тремя фазами: лед — вода — пар и
называется тройной точкой Ф = 3, К=1, С = 0. Равновесие в этой
системе возможно, как уже говорилось, только при строго
определенных условиях (давление, температура).
§ 11. КАТАЛИЗ
Одним из наиболее распространенных в химии методов
ускорения химических реакций является катализ.
Катализ — это селективное ускорение химических реакций в
присутствии веществ (катализаторов), которые принимают
участие в промежуточных процессах, но регенерируются в конце
реакции.
В тех случаях, когда вещества замедляют химическую
реакцию, стехиометрически в ней не участвуя, говорят об ингибиро-
вании.
Все проблемы катализа (включая и вопросы подбора
катализаторов) сводятся к основным проблемам химической кинетики,
т. е. к установлению элементарных химических процессов, из
которых складывается весь процесс в целом, и выводу закона, по
которому процесс протекает во времени. Ввиду большой
практической значимости явлений катализа принято выделять этот
раздел из химической кинетики, хотя было бы правильным
рассматривать катализ как раздел химической кинетики.
В дальнейшем будем рассматривать преимущественно
положительный катализ (распространенность и значение которого
несоизмеримо больше, чем отрицательного), называемый просто
катализом. Действие катализатора в процессе изменения
химической системы часто бывает двояким — ускоряющим и
ориентирующим. Ориентирующее действие сказывается в тех случаях, когда
исходная химическая система может в заданных условиях
развиваться в нескольких термодинамически возможных
направлениях.
Каталитическое ускорение реакции достигается в результате
изменения ее механизма. В присутствии катализатора изменяется
путь, по которому проходит суммарная реакция, поэтому
изменяется ее скорость. При этом катализатор входит в состав
промежуточных продуктов, которые образуются и затем переходят в
конечные вещества с большей скоростью, чем при отсутствии
208
катализатора. Понять, как это происходит, и является основной
задачей теории катализа.
Механизм каталитического действия состоит в том, что
катализатор и реагирующее вещество образуют промежуточное
соединение, которое реагирует с другим исходным веществом с
образованием продуктов реакции и регенерации молекул
катализатора по схеме:
А+К^ГАК, (а)
AK-t-B^(AB)*K, (б)
(AB)'K-*C + D + K. (B>
Здесь А и В — реагирующие вещества; К — катализатор; АК —
промежуточное соединение; (АВ)*К — активный комплекс из АК
и В; С и D — продукты реакции.
Природа промежуточных продуктов в катализе чрезвычайно
разнообразна. Чаще всего они представляют лабильные
молекулы или радикалы, существующие ^
весьма короткое время.
Рассматривая каталитический процесс с
энергетической точки зрения,
следует указать, что катализатор
ведет реакцию по иному пути,
чем тот, который отвечает
реакции без катализатора. Энергия
Рис. 6.12. Энергетические кривые
гомогенной некаталитической
(кривая /) и гомогенной каталитической
(кривая 2) реакций
дН реакиии
Продукты
реакции
Ход реакции
активации каталитической реакции значительно ниже обычной
химической некаталитической реакции. Так как энергия
активации входит в показатель степени в уравнении для константы
скорости реакции, то даже весьма небольшое снижение энергии
активации сильно увеличивает скорость реакции. Графически это
наглядно показано на рис. 6.12.
Общим для всех катализаторов является то, что они всегда
изменяют энергию активации, уменьшая ее при положительном
катализе. По фазовому признаку катализ делят на гомогенный и
гетерогенный; отдельно рассматривается ферментативный катализ.
Гомогенный катализ
При гомогенном катализе катализатор и реагирующие
вещества составляют одну фазу — газовую или жидкую. В этом случае
между катализатором и реагирующими веществами отсутствует
поверхность раздела.
209
Газо- и жидкофазные каталитические процессы весьма
многочисленны. Реакции газового катализа протекают как по цепному,
так и по молекулярному механизмам.
Рассмотрим пример катализируемой цепной газовой реакции—
окисления оксида углерода-П в присутствии паров воды. Сухая
смесь СО и 02 взаимодействуют по схеме
СО + 02 ->- С02 + О -|- 35,4 кдж.
Образовавшийся атом кислорода вступает в реакции, ведующие
к обрыву цепи:
0 + СО + Х->С02 + х + 517,2 кдж,
О 4- 02 + Х->-03 + х + 104,5 кдж,
03 + СО-> С02 + 02 + 420,5 кдж.
Здесь X — третье тело (например, стенки сосуда), забирающее на
себя избыточную энергию. Таким образом, в системе СО + 02
вероятность гибели активного центра преобладает над
вероятностью продолжения цепи. Если в эту систему ввести немного
паров воды, то в результате реакции
0 + Н20^20Н + 55,1 кдж
появляются два активных центра, легко дающих разветвленные
цепи:
OH4-CO->C02-f H+ 11,27 кдж,
Н + 02-+ОН + 0 + 22,1 кдж,
0 + Н2-*ОН + Н + 51,5 кдж.
В этих условиях первоначально возникает активный центр —
.атом кислорода, который не исчезает, а дает начало новым цепям.
Обрыв цепей по реакциям
Н+Н + Х-*.На + х,
0 + 0 + Х-*02 + х,
Н-4-ОЩ-Х-*НаО + х
происходит редко из-за малой концентрации в системе частиц
Н, О и ОН. В результате последней реакции регенерируется
катализатор — вода, небольшое количество которой значительно
ускоряет цепной процесс и смещает его пределы взрыва. Это
своеобразный пример катализа, идущего через образование
свободных атомов и радикалов.
Большинство газовых гомогенно-каталитических реакций
протекает по цепному механизму. Приведем пример гомогенного
катализа, протекающего по молекулярному механизму:
210
1. Окисление S02 до S03
N0+ 1/2 02=N02,
N02 4- S02 - S03 f NO.
Здесь катализатор NO, а промежуточное соединение — N02, что
видно из приведенной реакции.
2. Термическое разложение ацетальдегида, катализируемое
парами йода
СН3СНО-*СН4 + СО.
Вычислено, что при температуре 518°С энергия активации этой
реакции в отсутствие катализатора равна 48 ккал/моль, в
присутствии же паров йода энергия активации снижается до
32,6 ккал/моль, а константа скорости реакции возрастает примерно
в 10 000 раз. Это происходит потому, что в присутствии паров
йода реакция протекает в две стадии, энергия активации которых
ниже, чем энергия активации некаталитической реакции:
I СН3СНО + 12 ->■ СН31 4- HI + СО,
II CH3I + HI->CH4 + I2.
Каталитическое разложение ацетальдегида — пример реакции, в:
которой промежуточные вещества относительно устойчивы.
3. В некоторых случаях катализатор не только ускоряет
реакцию, но и изменяет ее направление. Так, в отсутствие катализа*,
тора диэтиловый эфир разлагается по схеме
С2Н5ОС2Н5 -> 2СН4 t 1/2 С2Н4 + СО,
а в присутствии йода эта же реакция идет по схеме
С2Н5ОС2Н5 ->. С2Н6 + СН3СНО.
Очевидно, последняя реакция идет через другой активированный-
комплекс, в котором участвует йод.
4. Разложение перекиси водорода хроматом калия в растворе
K2Cr04 -f 2Н202-* К2СЮб + 2Н20.
пероксохромат
Реакция образования промежуточного соединения в данном
случае сопровождается изменением окраски раствора. Раствор
хромата калия желтого цвета, а при взаимодействии с перекисью,
водорода окраска раствора становится темно-фиолетовой, после
чего снова становится желтой, так как продолжается реакция —
распад:
К2СЮ6-*К2Сг04+02.
В гомогенных каталитических реакциях в растворах во многих
случаях с несомненностью установлено образование в промежу-
2Ц
-точных соединений с участием катализатора, хотя обычно
промежуточные вещества в гомогенно-каталитических процессах
неустойчивы. Изолировать их, как правило, очень трудно, поэтому
их наличие фиксируется различными физико-химическими
методами (спектроскопия, ЭПР, ЯМР и др.)-
Важное место в гомогенном катализе занимает
кислотно-основной катализ, распространенный в растворах, в которых
реакции ускоряются ионами Н30+ в кислой среде и гидроксидионами
ОН" в щелочной среде. К таким реакциям относят омыление эфи-
ров, гидролиз крахмала, амидов, ацеталей, инверсия сахарозы
и др. Подобно ионам Н30+ и ОН", на скорость реакций
оказывают влияние и другие ионы и молекулы, например NH*, НСОГ,
СН3СОО~, NH3, являющиеся обобщенными кислотами и
основаниями. В отличие от специфического кислотно-основного
катализа ионами Н30+ и- ОН", катализ кислотами и основаниями
Бренстада называется общим кислотно-основным катализом.
К общему кислотно-основному катализу относят реакции
изомеризации, алкилирования, конденсации, галоидирования и др.
В кислотном катализе промежуточное соединение возникает
из-за перехода протона от молекулы катализатора (кислоты) к
лтолекуле одного из реагирующих веществ, которые
функционируют как соответствующее сопряженное основание.
При катализе основаниями неустойчивое промежуточное
соединение получается из-за перехода протона от молекулы
реагирующего вещества к основанию. Это видно из схемы
Катализ кислотой Катализ основанием
НХ + НА^НХН+ + А", В + НХ ^ВН+ + X",
В + НХН+^ ВН+ + ХН, Х~ + НА^Г ХН + А",
ВН+ . l а~ ;£ В -f НА. ВН+ + A~^Bf НА.
В этих схемах НХ — субстрат; НА — кислота; В и А" —
основания; НХН" и Х~ — ионизированные формы субстрата; ХН —
продукты реакции.
Недостатком гомогенного катализа в растворах является
ограниченность температурного интервала.
Гетерогенный катализ
В гетерогенном катализе катализатор представляет собой
•самостоятельную фазу. Чаще всего катализатор является
твердым телом, а реагирующее вещество — газ или жидкость.
Промежуточные соединения образуются на поверхности раздела фаз и
во многих случаях представляют собой систему хемосорбционно-
го типа.
,212
Поэтому в гетерогенном катализе особо важную роль играет
•величина поверхности катализатора, ее состояние, характер
активных центров, дефектности структуры катализатора.
В гетерогенных процессах реакции происходят на поверхности
раздела фаз, поэтому многие особенности их кинетики
определяются такими явлениями, как диффузия и адсорбция.
Диффузией называется процесс самопроизвольного
перемещения вещества, приводящий к установлению равномерного
распределения концентраций в объеме.
Роль диффузии в кинетике гетерогенных процессов станет
понятной, если учесть, что для начала процесса необходимо
движение веществ одной фазы к другой. Чтобы процесс протекал с
достаточной скоростью, продукты реакции должны своевременно
удаляться с поверхности раздела фаз, что также происходит
путем диффузии. Движущей силой диффузии является градиент
dC ~
концентрации . Это величина, равная изменению концентра-
dx
ции вещества dC, приходящегося на отрезок пути dx в
направлении диффузии. Но количество переносимого вещества dm
пропорционально также площади поперечного сечения 5 среды, через
которую происходит диффузия, и времени dx.
Таким образом,
dm = — D — Sdx, (a)
dx v 7
яли скорость Диффузии равна
dm ^ о dC
dx dx
Уравнение (а) называется первым законом Фика. Оно описывает
стационарную диффузию, при которой в элементарный объем за
единицу времени входит такое количество вещества, какое
выходит из этого объема. Знак минус указывает на то, что вещество
перемещается в направлении уменьшения концентрации, т. е.
АС*
<0. Коэффициент пропорциональности D называется коэф-
dx
фициентом диффузии. Он имеет смысл постоянной скорости диф-
АС
фузии при 5== 1, = 1 и характеризует влияние среды, в ко-
dx
торой происходит диффузия, на скорость диффузии. Скорость
диффузии в газах D^\ см2/сек, в жидкостях £)=10-5 см2/сек, в
твердых телах £>=10~8 ciyi2/ceK.
Коэффициент диффузии зависит от температуры. Отношение
£ = _!±ul называется температурным коэффициентом (3 диффу-
зии. р в газах и жидкостях равен приблизительно 1,2, в твердых
телах ^1,5—2, т. е. при повышении температуры на 10° скорость
диффузии увеличивается примерно на 20%- Это объясняется
малым значением энергии активации диффузионного процесса.
213
Гетерогенный катализ тесно связан с адсорбцией. Адсорбцией
называется экзотермический процесс самопроизвольного
концентрирования вещества из объема фаз на поверхности раздела
между ними.
Вещества, на поверхности которых происходит адсорбция,
называются адсорбентами. Адсорбируемые вещества называются
Рис. 6.13. Схема поверхностного Рис. 6.14. Изотерма адсорбции
слоя адсорбента
адсорбатами. Частицы поверхностного слоя из-за нескомпенсиро-
ванности молекулярных сил сцепления обладают некоторым
запасом энергии, называемой поверхностной энергией. За счет этой
энергии и происходит притяжение
поверхностью адсорбента молекул газа
или растворенных веществ.
Поверхность адсорбента постепенно
заполняется. При этом может
образовываться несколько слоев адсорбированного
вещества, т. е. имеет место моно- или
полимолекулярная адсорбция.
На рис. 6.13 показана схема
поверхностного слоя адсорбента.
Зависимость количества адсорбируемого
вещества от его концентрации назы-
Рис. 6.15. Адсорбция С02 при различных
давлениях и температурах, °С:
1 __ 80; 2 — 0; 3 76,5
вают изотермой адсорбции. Изотерма адсорбции показана
на рис. 6.14.
Адсорбция происходит с выделением тепла, поэтому с
повышением температуры количество адсорбируемого вещества
уменьшается; понижение температуры ведет к увеличению адсорбции.
Адсорбция также зависит от давления. Зависимость адсорбции
от температуры и давления показана на рис. 6.15. С точки зрения
характера связи, возникающей между адсорбентом и адсорба-
40 60
см рт. ст.
214
том, различают физическую и химическую (или активируемую)
адсорбции.
При физической адсорбции молекулы поглощаемого вещества
(адсорбата) удерживаются поверхностью адсорбента слабыми
межмолекулярными силами притяжения. При постоянной
температуре устанавливается динамическое равновесие
адсорбция ^ десорбция
Хемосорбция — взаимодействие молекул с поверхностью твер
дых тел с помощью химических сил обычного типа. Хемосорбция
происходит лишь на некоторых «избранных» адсорбентах, в то
время как физическая адсорбция возможна на любых
поверхностях. Хемосорбция обычно необратимый процесс. С хемосорбции
реагентов начинается гетерогенный процесс каталитического
превращения, который протекает на поверхности катализатора. Этим
объясняется особая роль хемосорбции в катализе. Поскольку
реагирующие вещества адсорбируются на поверхности
катализатора, на ней возрастает концентрация реагирующих молекул, что
приводит к увеличению скорости. Однако, как показали
исследования, ускоряющее действие адсорбции в большей мере
обусловлено тем, что в процессе адсорбции, особенно при высоких
температурах, молекулы становятся более активными.
Например, молекулы водорода при их адсорбции на
поверхности многих металлов диссоциируют на атомы, а атомарный
водород, как известно, значительно активнее молекулярного.
Активность катализаторов
В ряде случаев достаточно строгой характеристикой
активности катализатора является константа скорости
каталитического процесса, отнесенная к единице поверхности катализатора (так
называемая удельная константа скорости /Суд). Сравнивая /Суддля
разных катализаторов (при одинаковой температуре), можно
сопоставить каталитическую активность веществ.
Если же кинетические уравнения данного процесса для
разных катализаторов неодинаковы, то удельные константы скорости
будут иметь неодинаковую размерность, поэтому в общем случае
мерой каталитической активности принято считать скорость
каталитического процесса, отнесенную к единице поверхности
катализатора при заданном составе реакционной смеси и температуре.
Эта характеристика, называемая удельной 'скоростью, не требует
знания кинетического уравнения.
Если величина поверхности катализатора неизвестна, то
скорость (или константу скорости) относят к единице объема или
единице веса катализатора. Так как каталитическая активность
связана с адсорбцией на поверхности реагирующих веществ, то
наличие сильно развитой поверхности — необходимое условие
активности катализатора.
215
Тейлор впервые показал, что каталитический процесс
обусловлен не всей поверхностью катализатора, а лишь отдельными его
частями — активными центрами. По вопросу о природе активных
центров единого мнения нет. По Тейлору, только активные
центры, представленные гранями кристаллов, выступами, пиками на
поверхности, должны проявлять каталитическую активность. Они
отличаются ненасыщенностью, величиной свободной энергии,
способны активировать молекулу реагирующих веществ,
осуществлять каталитический процесс. Представления Тейлора
подвергались уточнению и детализации, однако сущность этих
представлений одна — физическая неоднородность поверхности.
Теория активных центров правильно объяснила влияние
условий приготовления катализатора на его активность, влияние
контактных ядов, действие промоторов и ряд других эффектов в
катализе. Но эта теория оказалась не в состоянии объяснить
механизм избирательного действия катализаторов.
Сложность и разнообразие процессов, протекающих на
поверхности раздела фаз, в значительной мере определяют собой
множество теорий гетерогенного катализа.
Избирательность катализаторов
В ряде случаев роль катализаторов сводится к ускорению
медленно идущего некаталитического процесса (синтез аммиака,
окисление аммиака, окисление сернистого газа и т. д.). В этих
условиях основные параметры катализатора -- его активность, ста-
Катализаторы
** 2СН+2Н0 А1 О ,Н SOakohu )
2 4 2 2 324
| •* 2СНзСН0+2Н2 Си (босс/т? )
*~ С^Н 0Н+Н20 металлический Na
ими щелочные ка-
. тализаторы
— №Н2+Н2° Zn0Al2°3
—^СНС00СН+2Н медные катализа-
6 2 5 2 т0рЫ с добавками
Рис. 6.16. Получение различных продуктов из этилового
. спирта в зависимости от вида катализатора и условий
проведения реакции
бильность в условиях проведения реакции, устойчивость к
действиям примесей. Однако в катализе главное не просто ускорить
реакцию. В каталитических превращениях органических
соединений, когда число термодинамически допустимых продуктов
реакции оказывается почти необозримым, избирательное действие
катализатора выступает на первый план и счановится не менее
важной его характеристикой, чем активность.
1
С Н ПН
Sn5U
216
Так, в зависимости от вида катализатора и условий
проведения реакции из этилового спирта можно получить до 40 видов
различных продуктов (этилен, диэтиловый эфир, уксусный
альдегид и др.) (рис. 6.16). При взаимодействии оксида углерода-П
и водорода, подбирая соответствующие катализаторы, удается
получить с хорошим выходом самые разнообразные соединения —
алканы, ароматические углеводороды, спирты, альдегиды, кетоны
(рис. 6.17).
К а т ал и з а т оры
-^парафины(преимущественно) Со, Ni, Ru
+олефины (преимущественно) Fe
*Ч:Нз0Н ZnOCuO,2nOCrO
^высшие спирты ZnOCrO^ щелочь
**кислородосодержащис соединения Fe+BeO
Рис. 6.17. Возможные превращения в синтезах из окиси
углерода и водорода в "интервале температур 30О-400°С
Свойство катализатора ускорять только одну из нескольких
возможных реакций называется избирательностью, или
селективностью действия. Всякое определение понятия катализа,
игнорирующее избирательность катализаторов, на современном этапе
развития теории катализа, является шагом назад, возвратом к
определению, данному еще в 1901 г. В. Освальдом, в котором он
учитывал только изменение скорости реакции.
Избирательность, или селективность, катализатора
проявляется в том, что он образует промежуточные продукты только одного
вида. Например, при каталитическом разложении С2Н5ОН
адсорбция молекулы спирта на поверхности катализатора может
происходить в разных положениях, что сопровождается либо
активацией связи — С—С—, приводящей к дегидрогенизации спирта, либо
активации связи —С—О—, приводящей к дегидратации спирта.
Однако такое объяснение селективности применимо не всегда.
Отравление, промотирование,
модифицирование катализаторов
Одним из важнейших отличий гетерогенных катализаторов от
гомогенных является зависимость их свойств от условий
приготовления и высокая чувствительность к действию небольших
примесей инородных веществ. В этой области наблюдается ряд
эффектов, к которым относятся отравление, промотирование и
модифицирование.
Наиболее простой эффект отравления — уменьшение числа
активных мест на поверхности катализатора под действием ядов.
Селективное отравление сложнее. Например, хлороформ на по-
со+н2-
217
верхности Ti02 подавляет процессы дегидратации, но не влияет
на процессы дегидрирования. Различают обратимое отравление,
при котором после прекращения подачи яда активность
катализатора восстанавливается, и необратимое, при котором требуется
специальная обработка по регенерации катализатора для полнот
или частичного восстановления его первоначальной активности.
Действие каталитических ядов 02, Н20, СО и других на
железный катализатор в реакции синтеза аммиака — пример обратимого
отравления:
3H2 + N2^2NH3+Q.
Активность катализатора восстанавливают, добавляя к
реагирующей смеси свежую смесь водорода и азота, которая вытесняет
яды с активных центров.
Примером необратимого отравления того же железного
катализатора в упомянутой выше реакции является добавление H2S,
CS2. Очистить катализатор от ядов в этих случаях возможно
только при добавлении специальных веществ — так называемых
форконтактов — для превращения ядов в неактивную массу. При
гомогенном катализе отравление может вызываться химическим
взаимодействием яда с катализатором, с образованием
каталитически неактивного соединения. Некоторые случаи ингибирования
удалось объяснить отравлением первоначально присутствовавшего
катализатора.
В некоторых случаях одни и те же вещества могут быть
каталитическими ядами по отношению к одним реакциям и
катализатору и не отравлять другие. Например, кислород отравляет железо
при синтезе аммиака, но активирует платиновые катализаторы
при окислении водорода. На полупроводниковые катализаторы
действуют также каталитические яды, но эти катализаторы менее
склонны к отравлению. Поэтому на практике активные
металлические катализаторы часто заменяют полупроводниковыми, менее
активными, но более стойкими к каталитическим ядам. Так, в
реакциях гидрирования и дегидрирования применяют сульфиды
NiS, WS2, CoS, которые не отравляются сернистыми примесями.
Кислотные катализаторы обычно отравляются примесями
оснований, а основные катализаторы — примесями кислот.
Явление активации катализатора под действием примеси
называется промотированием. Промотирование может быть
обусловлено увеличением работающей поверхности катализатора или
изменением природы активных центров. Примером промотора первого
типа является примесь А1203 в железном катализаторе при синтезе
аммиака, а второго типа — примесь КгО для того же катализатора.
В настоящее время отравляющее или промотирующее действие
относят не только к катализатору, как думали раньше, а к
каталитической системе в целом. Например, добавлением Li20 к ZnO
отравляется окисление окиси углерода, но активируется
разложение закиси азота. Механизм промотирования твердых катализа-
218
торов весьма разнообразен. Добавки могут вступать с основаниями
компонентов в химическое взаимодействие, образуя продукты,
обладающие повышенной каталитической активностью,
образовывать твердые растворы, меняющие электронную структуру
каталитически активного компонента в сторону, благоприятную для
повышения каталитической активности, изменять условия
взаимодействия с реагирующими веществами в местах контакта фаз основного
компонента, катализатора и промотора, увеличивать дисперсность
и величину доступной поверхности каталитически активного
компонента, ускорять стадии сложного каталитического процесса,
способствуя направлению реакции в сторону образования
требуемого продукта и т. п.
В современной каталитической химии весьма распространены
методы приготовления катализаторов смешением нескольких
веществ, каталитически активных для данного процесса. Найдено,
что часто такая смесь имеет активность значительно более
высокую, чем сумма активностей компонентов. Такие
многокомпонентные катализаторы называются смешанными, а усиление
каталитической активности—коактивацией. Компоненты смешанного
катализатора могут вступать в химическое взаимодействие между
собой, образуя химические соединения или твердые растворы: из
простых окислов, например из ZnO и Сг203, образуются сложные,
из индивидуальных металлов — их сплавы и т. д. С целью
увеличения доступной поверхности и предохранения катализатора от
рекристаллизации часто применяют носители — инертные материалы
с развитой поверхностью, на которую наносится каталитически
активное вещество.
Носитель вступает в определенное взаимодействие с
катализатором, более или менее глубоко воздействуя на его
каталитические свойства.
Механизм промотирования объясняется современными теориями
катализа. В случае металлических катализаторов часто промоторы-
металлы создают дополнительные дырки в d-зоне основного
металла.
В случае катализаторов-полупроводников роль промоторов,
как правило, заключается в изменении уровня Ферми в промоторе,
благодаря образованию примесных локальных уровней между
валентной зоной и зоной проводимости. Иногда роль промотора
заключается в изменении работы выхода электрона из основного
катализатора. В случае катализаторов кислотного типа повысить
активность главного 'катализатора удается обработкой его
кислотами, при добавлении малых количеств солей меди повышается
каталитическая активность солей железа в разложении перекиси
водорода.
С. 3. Рогинский с сотрудниками нашли еще более сложные
закономерности. Например, при газовом промотировании металлов
под действием кислорода и водорода каталитическая активность
сначала повышается, а затем падает. Одно и то же вещество при
219
различных концентрациях и в разных условиях выступает в роли
промотора и яда. Так как это явление не укладывается в рамки
простых понятий об отравлении и промотировании, Рогинский это
явление назвал модифицированием. Рогинский рассматривает про-
мотирование и отравление катализаторов как частные случаи
модифицирования.
Ферментативный катализ
Ферменты, или биокатализаторы, играют исключительную роль
в биологических процессах и технологии веществ растительного и
животного происхождения, а также в медицине. В настоящее время
известно свыше 2000 ферментов, и их число ежегодно
увеличивается. Сотни ферментов получены в высокоочищенном состоянии
и около 150 — в кристаллическом состоянии.
Все известные до,сих пор ферменты оказались белковыми
веществами. Однако химическое строение белковых молекул —
ферментов — изучено еще недостаточно, чтобы можно было
классифицировать ферменты по их строению.
Известен полный аминокислотный состав для большинства
кристаллических ферментов. Слабее изучена последовательность
чередования аминокислотных остатков, которая установлена пока
лишь для относительно небольшого числа ферментов (рибону-
клеазы, лизоцима, трипсина, химотрипсина и др.). Все известные
ферменты могут быть разделены на два больших класса.
I. Чистые белковые ферменты, каталитические свойства
которых обусловлены только строением самой молекулы (пепсин,
трипсин, химотрипсин, уреаза, папаин и др.).
II. Сложные ферменты, состоящие из белка и небелкового
компонента, в состав которого входят, например, нуклеотиды, гемины,
атомы металлов и т. д. (сюда относятся ферменты окислительно-
восстановительного действия).
Небелковые компоненты сложных ферментов называются про-
стетическими группами, а белковый компонент сложных белков
называется апофермент. Прочность связи между простетической
группой и белком в сложном ферменте различна. Легко
диссоциирующие простетические группы сложных ферментов называются
кооферментами. При потере простетических групп ферменты
теряют свою активность, однако не следует думать, что
каталитическая активность сложных ферментов определяется только
простетической группой, а значение белковой молекулы заключается
лишь в роли пассивного носителя. Наоборот, для гидрогеназ,
аминофераз и других ферментов при одной и той же
простетической группе специфичность действия ферментов определялась
различием белкового компонента. Белковый компонент очень сильно
влияет на эффективность действия сложных ферментов. В простых
ферментах, состоящих из белков, каталитическая активность опре-
220
деляется только химическим строением и пространственной
укладкой самой полипептидной цепи.
В каталитической активности простых и сложных ферментов
принимает участие не вся молекула белка, а лишь определенные
ее участки, получившие название активного центра фермента.
В простых ферментах активный центр непосредственно
образуется определенной группировкой аминокислотных остатков в
спиральной цепи белковой молекулы, а в сложных ферментах он
образуется простетической группой и некоторыми прилегающими к ней
аминокислотными остатками; природа компонентов активного
центра и их взаимное расположение определяют специфичность
действия ферментов.
В среднем один активный центр фермента приходится на
молекулярный вес от 30 000 до 80 000 и на 250—500 аминокислотных
остатков. Данные о структуре активного центра ферментов, его
размерах и пространственном строении сближают ферменты с
гетерогенными катализаторами, а сама реакция происходит на
поверхности молекулы фермента. Вместе с тем почти все
ферментативные реакции имеют аналогии в реакциях гомогенного
катализа, происходящих в растворах. Это позволило некоторым ученым
рассматривать ферментативный катализ как «микрогетерогенный»,
так как многие ферменты «вмонтированы» в среде жестких
компонентов клетки, обладающих микроповерхностью раздела
(митохондрии, рибосомы и др.).
Действие ферментов проходит через стадию образования
промежуточного соединения с молекулой субстрата, которое
происходит при участии различных сил — ковалентных, координационных,,
электростатических, водородных и других связей.
Образование простого адсорбционного комплекса FS между
ферментом и субстратом, как правило, недостаточно для
осуществления реакции, для этого нужно снизить не только энергию
активации, но и изменить энтропийный фактор (что еще важнее).
Ферменты являются бифункциональными и полифункциональными
катализаторами, так как здесь имеет место согласованное
воздействие двух или нескольких групп различной природы в составе
активного центра фермента на поляризацию определенных связей
субстрата. Эта же концепция лежит в основе каталитического
действия фермента и теории кинетики действия ферментов.
Главное отличие ферментов от других катализаторов
заключается в исключительно высокой активности и ярко выраженной
специфичности.
Общие причины превосходства ферментов как катализаторов
заложены в неповторимом, относительно жестком размещении в
молекуле множества функциональных групп. В активном центре
фермента сосредоточены контактирующие функциональные группы,
пространственная и электростатическая топография которых
приближенно, но не идеально комплементарна молекуле субстрата.
Согласованность каталитического действия всех необходимых
221'
групп активного центра фермента достигается благодаря
упорядоченному их распределению в белковой молекуле строго
-комплементарно структуре субстрата.
Катализ в промышленности
Явления катализа очень широко распространены в природе, и
человеку пришлось давно с ними столкнуться, но широкое
промышленное использование катализа началось только в XX
столетии.
Одна из важнейших областей применения катализа в
химической промышленности — контактный способ производства
концентрированной серной кислоты, сыгравшей значительную роль в
развитии других отраслей химической технологии.
В 20-х годах для окисления оксида серы-П были разработаны
новые ванадиевые катализаторы, представляющие оксид вана-
дия-V, нанесенный в смеси с пиросульфатом калия на носители,
содержащие кремнезем. Высокая активность и устойчивость
ванадиевых катализаторов позволили значительно повысить
интенсивность контактного процесса. В настоящее время по этому способу
.вырабатывается свыше 60 млн. тонн серной кислоты.
Очень большое значение имело решение проблемы фиксации
азота воздуха на основе катализа. Только с помощью
катализаторов удалось преодолеть химическую инертность элементарного
азота и осуществить синтез аммиака из азота и водорода. Этот
процесс был реализован в 1918 г. на основе работ Габера, Боша и
Митташа с помощью катализатора, представляющего собой
металлическое железо с добавками окисей калия и алюминия при
температуре 450—550°С и высоком давлении (300—700 атм) для
смещения равновесия в сторону образования аммиака. В настоящее
время таким способом производятся десятки миллионов тонн
аммиака.
Каталитические процессы используются и для производства
водорода, служащего сырьем для синтеза аммиака и ряда других
производств химической технологии. Наиболее дешевым
источником водорода служит природный газ. С помощью катализа была
решена и задача перехода от аммиака к кислородсодержащим
соединениям азота.
Окисление аммиака — пример широко используемой в
промышленности реакции избирательного катализа, при котором
катализатор не только увеличивает скорость химического превращения,
но и направляет его в сторону образования определенного
продукта из ряда возможных. Меняя катализатор, можно изменять и
направление превращения.
Значительно позже каталитические методы нашли
использование при переработке нефти, но их внедрение протекало очень
быстрыми темпами. До 1940 г. при переработке сырой нефти для
изменения состава продуктов изменялись преимущественно терми-
222
•кччшс процессы. В настоящее время свыше 80% нефти перераба-
швается с использованием каталитического крекинга, рифор-
мпнга, гидрогенолиза сернистых соединений, гидрокрекинга и
других каталитических процессов. Это позволило резко увеличить
ныход из нефти ценных продуктов и повысить их качество. Кроме
того, каталитические процессы нефтепереработки являются
источником дешевого сырья для химической промышленности.
Особенно велика роль катализа в осуществлении новых
процессов органического синтеза. Крупнейшим успехом в этом
направлении надо признать получение искусственного
синтетического каучука из этилового спирта, осуществленное в нашей стране
впервые в мире академиком С. В. Лебедевым в 20-х годах.
В 1930 г. был построен опытный завод, а с 1932 г. началось
промышленное производство синтетических каучуков разных
назначений и из различного непищевого сырья.
Приведенные примеры составляют лишь небольшую долю про-
мышленно-каталитических процессов.
Катализ — важнейший метод осуществления в промышленности8
химических превращений. От развития катализа в значительной
степени зависит прогресс химической и других отраслей
промышленности. Реализация многих термодинамически возможных и
экономически выгодных процессов, получение новых продуктов,
осуществление более совершенных технологических схем,
использование более доступных сырьевых ресурсов во многих случаях
лимитируются изысканием достаточно активных катализаторов.
Катализ является основным и наиболее эффективным методом
интенсификации промышленных химических процессов.
Химические производства, использующие катализаторы, перерабатывают
самое дешевое сырье, которое дает природа: природные газы,
воздух, руды, нефть, уголь и другие, преобразуя их в СК и другие
полимеры, высококачественные бензины, аммиак, азотную и
серную кислоты, минеральные удобрения, органические растворители,,
краски и многие другие ценные продукты.
Вопросы и упражнения-
1. Что такое скорость химической реакции, чем она определяется и от
каких факторов зависит?
2. Как формулируется закон действующих масс?
3. Как зависит скорость и константа скорости химической реакции от
температуры?
4. Что такое активные молекулы и энергия активации?
5. Чем отличается молекулярность реакции от порядка реакции? Когда
молекулярность и порядок реакции совпадают и когда не совпадают?
6. Перечислите типы сложных химических реакций.
7. Какие виды цепных реакций Вы знаете?
8. Какие реакции называются фотохимическими? Что такое квантовый
выход фотохимических реакций?
223
9. Что называется смещением равновесия? Какие факторы влияют на
смещение равновесия?
10. В чем заключается принцип Ле Шателье?
11. В какую сторону сместится равновесие при повышении температуры в
«системах
2СО + 02 ^ 2С02 + 567,8 кдж, 2N02 ^ N204 + 56,8 кдж,
N2 + 02->2NO —181,2 кдж, N2 + 3H2^2NH3 + 91,8 кдж?
12. В какую сторону сместится равновесие при повышении давления в
системах
2NO + 02 ^ 2N02, РС15 ^ РС13 + С12,
С02 + С ^± 2СО, Н2 + С02 ^ СО + Н20,
Н2 +12 ^ 2HI, 4НС1 + 02 ^ 2Н20 + 2С12?
13 Как повлияет повышение концентрации С02 на равновесия
СаС03т±СаО+С02; С02+С^±2СО?
14. Что показывает и от каких факторов зависит константа химического
равновесия?
15. Напишите выражения констант равновесия для систем
СО + С12 ^ СОС12; Fe304 + Н2>Г ^ 3FeOTB + Н2Опар;
СН^ г ^ С + 2Н2 г.
16. Равновесие реакции Рез04 + Н2;г=^ЗРеОТв + Н2Опар устанавливается,
когда 85% исходного количества водорода превращается в водяной пар.
Вычислите значение К при этих условиях (ответ: 5,6).
17. Изложите основные понятия и расскажите о значении катализа.
18. В чем сущность гомогенного катализа? Примеры.
19. В чем сущность гетерогенного катализа? Примеры.
20. Расскажите об отравлении, промотировании и модифицировании
катализаторов.
21. Каким энергетическим эффектом сопровождается адсорбция?
22. Какая связь между хемосорбцией и катализом?
ГЛАВ'А 7
РАСТВОРЫ
§ 1. ОБЩИЕ ПОЛОЖЕНИЯ
Растворы — дисперсные системы. В зависимости от агрегатного
состояния распределенного вещества и среды возможны
следующие дисперсные системы (Г — газ, Ж — жидкость, Т — твердое
вещество) :
г—г г—ж г—т
ж-г ж-ж ж-т
т—г т—ж т—т
Устойчивость дисперсных систем в значительной мере зависит
от размеров частиц; чем больше размеры частиц, тем меньшей
устойчивостью характеризуется система. Растворы — дисперсные
системы с молекулярной степенью раздробленности частиц. Это
весьма устойчивые системы, не разделяющиеся ца исходные
составные части при сколь угодно долгом стоянии. Малой
устойчивостью обладают дисперсные системы, получившие название
взвесей. Их частицы значительно превышают размеры молекул
среды. Промежуточную область по устойчивости занимают
коллоидные растворы, в которых размеры распределенных частиц
находятся между размерами частиц взвесей и растворов.
Важнейшие признаки растворов — их однородность в
известных пределах и переменность состава. Растворы образуются при
взаимодействии растворенного вещества и растворителя. При этом
участвуют силы как физической, так и химической природы.
Физическое взаимодействие (физическая сольватация) осуществляется
на расстояниях, значительно превышающих размеры молекул.
Химическое взаимодействие (химическая сольватация)
осуществляется на близких расстояниях при участии молекулярных
(атомных) орбиталей (донорно-акцепторная и водородная связи).
Химическому взаимодействию всегда предшествует физическое.
К продуктам физической сольватации можно отнести гидрати-
рованные ионы цезия [Cs(H20)8] + и хлора [О(Н20)8]~, сольва-
тированные молекулы солей КС1 и KN03 в этиловом спирте
(КСЬС2Н5ОН и KN03-C2H5OH) и т. д. К продуктам химической
сольватации относятся продукты взаимодействия BF3 с
растворителем— жидкой двуокисью серы (BF3-S02), AsCl3 с C5H5N в
серном эфире (AsCU-CsHsN), гидратированные молекулы аммиака
в Н20 (NH3-H20), гидратированные ионы переходных металлов
[СоОН(Н20)5]+, [Сг(ОН)(Н20)5]2+
8 Общая химия
225
В конце XIX в. растворы считались физическими
образованиями, © которых отсутствуют какие-либо взаимодействия между
растворителем и растворенным веществом; образование раствора
объяснялось диспергированием частиц растворенного вещества в
индифферентной среде растворителя. Для правильного понимания
природы растворов важное значение имели работы Д. И.
Менделеева, создавшего химическую (гидратную) теорию растворов.
Менделеев дал следующее определение понятию раствор:
«Растворы представляют жидкие диссоционные системы,
образованные частицами растворителя и растворенного тела и тех
определенных, но экзотермических соединений, которые между
ними происходят, одного или нескольких, смотря по природе
составляющих начал». Это определение имеет силу и в настоящее
время.
Согласно современным представлениям в растворах
осуществляются взаимодействия не только между молекулами исходных
веществ, но между ними и их продуктами. Например, в бинарном
растворе, состоящем из компонентов А и В, возможны такие
взаимодействия:
1. Ассоциация
A f А^:А2; В + В^В2.
2. Диссоциация
А^С ) D; B^lC-!-D.
3. Комплексообразование
А -|- В2 ^ АВ2; С + А2 ^ СА2.
Между динамическими равновесиями 1, 2 и 3 в растворах имеет
место обмен веществом. Рассмотрение порознь каждого из них без
учета возможности наличия других имеет условный характер.
В зависимости от природы сил продукты, возникающие в
растворах, обладают'различной устойчивостью: от короткоживущих
до соединений, которые можно выделить из раствора.
Устойчивость и характер продуктов зависит от природы растворенного
вещества и растворителя, температуры и давления, а также
.концентрации и структуры раствора. Основная задача современной
физико-химической теории растворов заключается в определении
молекулярной и пространственной структуры продуктов раствора,
а также -в изучении элементарных процессов их образования.
В этой области успешно работали и работают советские ученые
Н. А. Измайлов, М. И. Усанович, В. К. Семенченко, К. П.
Мищенко, К. Б. Яцимирский, О. Я. Самойлов и многие другие.
Энергетика растворения. Растворимость. Образование
раствора идет самопроизвольно и необратимо вплоть до состояния
устойчивого равновесия (насыщенного раствора). Движущей силой
процесса растворения является уменьшение свободной энергии
226
(изобарно-изотермического потенциала) раствора AG по
сравнению со свободной энергией исходных составных частей раствора.
При растворении
исходи <0.
Если процесс растворения закончился, система пришла к
состоянию равновесия
AG-0.
Энергетической характеристикой процесса растворения является
теплота (энтальпия) образования раствора Д#, которая при
образовании раствора либо выделяется (Н2—Н{ = —АНР)У либо
поглощается (#2—Н{ = АН). Например, при растворении газов теплота,
как правило, выделяется. Растворение жидких и твердых веществ
может сопровождаться как выделением, так и поглощением тепла.
При образовании растворов значительно изменяется
энтропия AS. Будучи характеристикой неупорядоченности системы, она
возрастает всякий раз, когда растворение сопровождается
увеличением объема по сравнению с исходными компонентами, и падает,
когда объем уменьшается. В частности, при растворении газов в
жидкости энтропия всегда уменьшается, а при растворении
кристаллических тел возрастает.
АН и AS раствора связаны с изобарно-изотермическим
потенциалом соотношением
AG-АЯ-Г AS. (7.1)
Если процесс растворения протекает эндотермически, член TAS в
уравнении (7.1) должен превышать величину АН. При образовании
растворов с выделением тепла возможны две ситуации:
1. AS<0, АЯ<0;
2. AS>0, ДЯ<0.
Чаще реализуется первый случай, что может указывать на
значительную роль в растворах химических взаимодействий.
Энтальпию образования раствора АН следует рассматривать
как сумму эндо- и экзотермических вкладов всех процессов,
сопровождающих растворение. К ним, в частности, относятся эндоэффек-
ты разрушения кристаллической решетки, разрыва связей молекул,
разрушения исходной структуры растворителя; экзоэффекты
образования различных продуктов взаимодействия, в том числе экзо-
эффект химической сольватации.
При растворении твердых тел иногда образуются кажущиеся
равновесные системы — пересыщенные растворы (AG>0). Они
легко переходят в равновесное состояние обычного насыщенного
раствора (AG = 0).
Растворимость веществ зависит от природы растворенного
вещества, природы растворителя, концентрации раствора и темпера-
8*
227
туры. Для газообразных веществ она существенно зависит также
от давления. Абсолютно нерастворимых веществ в природе нет.
Можно говорить о практической нерастворимости данного
вещества в данном растворителе, обусловленной тем, что ЕДЯ и 2А5
сольватации недостаточны для самопроизвольного процесса
растворения.
По растворимости веществ накоплен огромный
экспериментальный материал (см. справочники по растворимости). Общей
теории растворимости пока не создано. Чтобы подчеркнуть
сложность этой проблемы, достаточно указать на то, что даже для
однотипных соединений одной и той же подгруппы периодической
системы Менделеева не наблюдается закономерностей в
растворимости. Так, растворимость Li2C03 в воде при 70° в 100 раз меньше
растворимости К2СО3, a LiN03 и KN03 растворимы
приблизительно одинаково. Большинство экспериментально наблюдаемых
фактов' находят лишь качественное объяснение. Например,
лучшая растворимость йода в бензоле по сравнению с растворимостью
в воде объясняется следующим образом: энергия взаимодействия
между полярными молекулами растворителя Н2О много больше,
чем между ними и неполярными молекулами растворенного
вещества Ь, различия же в межмолекулярном взаимодействии
молекул бензола и йода не столь значительны, так как и те и
другие неполярны.
Важное практическое значение имеют частные закономерности
по растворимости веществ, например: закон Генри, закон
распределения, произведение растворимости и некоторые др.
Растворители. Выбор растворителя определяется в основном
следующими условиями: способностью растворять данное
вещество, доступностью растворителя и простотой работы с ним.
Растворители, характеризующиеся значительной вязкостью или
токсичностью, редко используются на практике (в специальных случаях).
Важной характеристикой растворителя является его
диэлектрическая проницаемость е и структура. 8 — функция полярности и
объема молекулы растворителя. Чем выше полярность и
способность молекулы деформироваться, тем выше диэлектрическая
проницаемость растворителя. Величины е наиболее распространенных
растворителей приведены в табл. 7.1.
Структура растворителя определяет структуру раствора в
целом, и лишь в концентрированных растворах она существенно
изменяется. Долгое время состояние жидкого раствора считалось
аналогом газового состояния. Такая точка зрения находила
подтверждение в физической теории растворов, согласно -которой
растворитель рассматривался как индифферентная среда с
равновероятной во всем объеме плотностью и постоянной величиной
диэлектрической проницаемости. В настоящее время уже не
вызывает сомнения тот факт, что даже в чистой жидкости имеет место
ближний порядок; каждая молекула жидкости стремится
окружить себя определенным числом ближайших соседних молекул,
228
Таблица 7.1
Величины диэлектрической проницаемости некоторых веществ при 25°С
Вещество
Вещество
Аммиак
Ацетон
Бензол
Вода
Гидразин ....
Диэтиловый эфир
Анилин
25
20,
2,
78,
58,
4,
6,
7
3
3
5
1
99
Пиридин
Муравьиная кислота
Уксусная кислота .
Жидкая H2F2 . . .
Синильная кислота
Серная кислота . .
Жидкая двуокись серы
12,3
57,9
6,19
84
96
(0)
15,35
0°С
и эта структура в каждый данный момент времени сохраняется в
объеме всего раствора. Чем выше полярность молекул, способность
образовывать друг с другом дополнительные связи, например
водородные, тем структурированнее растворитель. Установлен
следующий ряд растворителей по структурированности:
Н20 > СН3ОН, С2Н5ОН > СбН5С1 > (СН,)2 СО > СС14 > С6Н6.
Наибольшая структурированность характерна для воды,
наименьшая — для бензола.
Для очень многих явлений, протекающих в растворах, в том
числе для электролитической диссоциации, наряду с
диэлектрической проницаемостью растворителя и его структурой
существенное значение имеет способность растворителя к собственной
ионизации:
2Н20 ^ Н30+ + ОН", 2NH, •£ NHf -j- ОН".
В настоящее время наряду с хорошо известными
растворителями— уксусной кислотой, ацетоном, спиртами, сероуглеродом —
находят большое применение такие растворители, как жидкая
двуокись серы, серная кислота, жидкая фтористоводородная кислота,
хлорокись серы SOCl2, хлорокись фосфора РОС13 и др.
§ 2. СВОЙСТВА РАСТВОРОВ.
ЗАКОНЫ РАУЛЯ, ГЕНРИ, ВАНТ-ГОФФА
Выражение концентрации. Понятие идеального раствора. Для
правильной оценки зависимости свойств раствора от весовой доли
каждого из компонентов небезразлично, в каких единицах
выражать их концентрацию. Чтобы достаточно точно учитывать
количественные соотношения компонентов, удобно выражать
концентрацию растворов в мольных долях Х{ или в единицах моляльности
ni{. Моляльность измеряется числом молей растворенного
вещества в 1000 г растворителя. Например, Зт H2S04 означает, что в
1000 г растворителя растворено 3 моля 100%-ной H2SOt. Под
229
мольной долей Xi компонента понимается отношение числа молей
этого компонента к общему числу молей всех компонентов
раствора. Так, если раствор состоит из компонентов Л, Бу В, то их
мольные доли запишутся соответственно в виде
пА + ПБ + ПВ ^ni
пБ
ХБ = -?-. (7.2)
2 щ
хЕ
S/i/
Сумма мольных долей компонентов раствора равна единице.
Для раствора АБВ
ХЛ+ХБ + ХВ=1.
В разбавленных растворах .концентрацию компонентов можно
выражать в молях в единице объема раствора (в единицах моляр-
ности С моль/л), поскольку мольная доля компонента Х{, его мо-
ляльность rtii и молярность С{ связаны соотношением
Xt = J*-.m,«-^C„ (7.3)
1000 ' 1000 х
где М— молекулярный вес растворителя.
Количественное описание свойств растворов оказалось
возможным с привлечением модели идеального раствора. Идеальным
раствором называется раствор, в котором межмолекулярное поле
сил смешиваемых компонентов одинаково. В таком растворе
безразлично, молекула компонента А находится в окружении
собственных молекул (как, например, в случае чистой жидкости А) или
среди молекул А есть молекулы других компонентов раствора
(Б, 5,...). По этой, причине идеальный раствор будет
образовываться без теплового эффекта Д#; объемы при смешении
компонентов будут суммироваться подобно суммированию объемов при
сливании двух частей одной и той же жидкости. Каждый
компонент будет вести себя независимо от свойств любых других
компонентов раствора. Такие свойства раствора, как понижение
давления пара, температура кипения (замерзания), осмотическое
давление, должны зависеть только от частичной концентрации
растворенного вещества. В частности, будет справедливо соотношение
вида
Ра=--РаХа,
где РА — давление пара растворителя А над раствором; Ра —
давление пара чистого растворителя; ХЛ — мольная доля
растворителя. Вывод этого соотношения приводится ниже.
230
В основе вывода лежит количественная оценка скоростей
испарения и конденсации чистого растворителя — компонента Л, и
скорости испарения и конденсации этого компонента из идеального
раствора (в присутствии компонента Б).
Пусть поверхность раствора составляет 1 см2. Из чистой
жидкости А испарение осуществляется при участии всей поверхности,
в случае же раствора — с той ее части, которая занята
молекулами Л. Остальная часть поверхности, занятая индифферентными
по отношению к А молекулами нелетучего компонента Б, не будет
участвовать в процессе испарения.
Примем, что на долю компонента А с концентрацией ХА
мольных долей приходится 5 частей поверхности, или
51 см2 = S см2.
Тогда на долю компонента Б с концентрацией ХБ мольных долен
будет приходиться (1—5) частей поверхности, или
(1—5)1 см2 = (1— S) см2.
Из-за отсутствия межмолекулярных взаимодействий
компоненты А и Б будут распределяться на поверхности и в объеме
идеального раствора одинаково — пропорционально их концентрации. Это
означает, что в объеме и на поверхности раствора отношение
Ха/Хб — величина постоянная, равная отношению частей
поверхности, приходящихся на долю каждого из компонентов, т. е.
хБ 1-5
и XA=S, ЛБ-(1-5). (а)
Обозначим скорость испарения чистой жидкости с единицы
поверхности Кпар- Тогда скорость испарения компонента А из
раствора от части 5 этой поверхности будет оцениваться соотношением
Vnap^nap'5. (б)
С учетом (a) Vmp = V^XA. (в)
Для конденсации компонента А в отличие от процесса
испарения предоставлена вся поверхность (1 см2) независимо от того,
принадлежит она чистой жидкости А или ее раствору. Скорость
конденсации* будет зависеть от числа молекул пара в единице
объема или от концентрации давления пара над чистым
растворителем и над раствором, т. е.
V°k=KP°ahVk=KPa, (г)
Если раствор идеален, то тем более будет идеальна смесь паров его летучих
компонентов.
231
где VK —скорость конденсации пара над чистой жидкостью Л;
VK — скорость конденсации над ее раствором при данной
температуре УРА и РА —соответственно давление пара чистой жидкости
и давление пара над раствором; К—коэффициент
пропорциональности.
Исключив из (г) коэффициент пропорциональности, получим
V°Pa
tA
Так как чистая жидкость, равно как и ее раствор, в равновесии
с паром представляют динамическую систему, скорость испарения
равна скорости конденсации:
для чистой жидкости ]/паР = УЦ, (е)
для идеального раствора Vnap = VK. (e')
Преобразуем уравнение (е') с учетом уравнений (в) и (е):
Vlp-XA = ^-^. (ж)
Величины Vnap и VK можно сократить в соответствии с
уравнением (е), и тогда
РД=Р°А-ХА. (7.4)
Аналогично для летучего компонента Б получим соотношение
Рб = Р°б • ХБ. (7.4')
Соотношения (7.4) и (7.4') формулируются следующим
образом: при данной температуре давление пара летучего компонента
над идеальным раствором пропорционально его мольной доле
в растворе.
На рис. 7.1 приведено графическое изображение уравнений
(7.4) и (7А'). Отрезок АБ изображает состав бинарной системы
в мольных долях. Вся длина отрезка равна единице. От его левого
конца откладывается мольная доля компонента Б (Хб). В
крайней правой точке отрезка Л"б=1, в крайней левой точке ХА = 1.
Составы промежуточных точек отрезка определяются
соотношением ХА + Хв=\. На перпендикулярах отложены давления пара
чистых растворителей Р°а и Р%. Верхняя прямая изображает
давление пара раствора в зависимости от его состава. Она
описывается уравнением
Р - Ра + Рб - Ра + (Рв - Ра) Ха.
232
Понижение давления пара раствора неэлектролита. В 80-х го-
/i;ix XIX в. Рауль, изучая количественную зависимость свойств
р;пбавленных растворов от их концентрации, пришел к выводу,
Рис. 7.1. Графическое
представление
закона Рауля
х6 V
Рис. 7.2. Кривые
зависимости давления
пара раствора от
состава:
а — с
положительным отклонением от
идеальности; б—с
отрицательным
отклонением от идеальности
•по для разбавленных растворов неэлектролитов относительное
понижение давления пара раствора равно мольной доле нелетучего
растворенного вещества:
р° — Р
Р°
— Хб,
(7.5)
где РА к РА — соответственно давление пара чистого растворителя
(компонента А) и давление этого растворителя над раствором.
И известных пределах равенство (7.5) не зависит от температуры,
i;iк как от нее не зависит Хб- Понижение давления пара раствори-
и'ля над раствором по сравнению с чистым растворителем
обусловлено тем, что часть поверхности раствора занята молекулами
нелетучего растворенного вещества, снижающего скорость
испарения молекул растворителя. Из уравнения (7.5) легко получается
соотношение (7.4). Для этого надо разделить каждый член числи-
к'ли на Р°а и ввести вместо Хб равную ей величину (1—ХА):
233
.-^-.-*д.
или
Ра =-- Р°лХл.
В отличие от соотношения (7.4), соотношение Рауля (7.5)
справедливо не для любых концентраций, как это имеет место в случае
идеальных растворов, а лишь для разбавленных растворов
неэлектролитов, в которых Ха^>Хб.
Только разбавленные растворы неэлектролитов подчиняются
законам идеальности. Чем меньше мольная доля растворенного
вещества, тем ближе реалышй раствор к идеальному. Предельно
разбавленный раствор с бесконечно малой концентрацией
растворенного вещества (Хб-+0) можно рассматривать как раствор
идеальный. В таком растворе молекулы растворенного вещества
в растворителе настолько удалены друг от друга, что их
взаимодействие будет исчезающе мало; соответственно исчезающе малым
будет их влияние на молекулы растворителя. В растворе будет
играть роль лишь межмолекулярное поле сил растворителя как
чистого вещества. Отсюда свойства предельно разбавленного
раствора не будут отличаться от свойств идеального раствора.
В природе идеальных растворов практически не существует К
Свойства реальных растворов, в которых Х{>0,00\, отклоняются
от идеальных. На рис. 7.2 приведены кривые зависимости давления
пара от состава раствора, отражающие поведение реальных
растворов с положительным (7,2, а) и отрицательным (7.2, б)
отклонением от идеальности.
Причиной отклонения принято считать различие в силе
межмолекулярных полей, создаваемых компонентами раствора. В
случае положительного отклонения поле межмолекулярных сил между
одноименными молекулами (А—А) или (Б—Б) больше, чем между
молекулами разной природы (А—Б).
Растворы с положительным отклонением образуются с трудом,
каждый компонент старается вытолкнуть разноименного соседа
из сферы ближайшего окружения и заменить их собственными
молекулами. Процесс смешения компонентов сопровождается
поглощением тепла. Среди продуктов раствора характерны ассоциа-
ты молекул исходных компонентов. Напротив, процесс испарения
компонентов идет с большей легкостью, чем в случае идеального
раствора, что и приводит к положительному отклонению.
Положительное отклонение от законов идеальности
наблюдается в свойствах таких растворов, как четыреххлористый
углерод — бензол, этиловый спирт — этиловый эфир и др.
Отрицательное отклонение от законов идеальности
наблюдается тогда, когда в растворе взаимодействие между молекулами
1 Идеальными можно считать растворы изотопов.
234
разной природы больше, чем между одноименными молекулами.
Смешение таких компонентов сопровождается выделением тепла.
Среди продуктов растворов можно встретить непрочные
химические соединения. Испарение летучих компонентов такого
раствора затруднено; теплоты испарения, как правило, велики.
Отрицательным отклонением характеризуются смеси: СН3С1—(СНзЬСО,
Н20—HN03, H20—HC1 и др.
Как и следовало ожидать, в соответствии с применимостью
закона Рауля к разбавленным растворам неэлектролитов концы
сплошных кривых на рис. 7.2 совпадают с пунктирными прямыми
(линиями Рауля). Из рис. 7.2 следует также, что на участках
сплошных кривых, характеризующих зависимость давления пар?.
растворенного вещества от концентрации, соблюдается прямая
пропорциональность (см. область разбавленных растворов).
Впервые экспериментально это было установлено Генри в 1803 г.
на основании работ по растворимости газов в различных
растворителях.
Прямолинейность участков, отмечаемая на кривых зависимости
давления насыщенного пара растворенного вещества от его
концентрации, приведенных на рис. 7.2, свидетельствует о том, что
для растворенного вещества Б (растворитель А)
Рб = КбХб; (7.6)
для растворенного вещества А (растворитель Б)
Ра = КаХа, (7.7)
где Ка, Кб—коэффициент пропорциональности (константа
Генри). Количественные зависимости (7.7) носят название закона
Генри: в разбавленном растворе при данной температуре давление
пара растворенного вещества прямо пропорционально его мольной
доле.
Закон Генри имеет такое же значение для описания
зависимости давления насыщенного пара растворенного вещества в
реальном растворе от его мольной доли, какое имеет закон Рауля
для растворителя. По форме математические выражения этих
законов отличаются. В законе Рауля коэффициент
пропорциональности численно равен давлению насыщенного пара чистого
компонента (Р°а для Л и Рб для £), о чем свидетельствует совпадение
концов кривых, характеризующих давление пара растворителя над
раствором с пунктирной прямой рис. 7.2. В законе Генри
КлФРаК КбФР1
Различие в поведении растворенного вещества и растворителя,
которое нашло отражение в законах Генри и Рауля, объясняется
неравноценностью взаимного влияния компонентов растворителя
и растворенного вещества. В разбавленном растворе влияние
растворенного вещества на межмолекулярное поле сил раствори-
235
теля ничтожно мало, в то время как влияние межмолекулярного
поля сил растворителя, составляющего практически весь объем
раствора, велико. Чем полнее сходство молекул растворенного
вещества и растворителя, тем ближе значение Кб и Р°б, и тогда,
когда их межмолекулярные силовые поля равны друг другу
(Кб = Рб)> раствор становится идеальным.
Закон Генри — это частный случай закона распределения
с2 ~К«'
(7.8)
Последний формулируется так: «Всякий раз, когда одному и
тому же веществу представляется возможным распределиться
между двумя соприкасающимися индифферентными друг к другу
жидкостями, отношение концентрации этого вещества в обоих
растворителях остается постоянным независимо от его количества
в том и другом растворителях».
Соотношение (7.8) имеет силу при условии, что молекулярный
вес растворенного вещества в обоих фазах сохраняется
постоянным.
Величина Ко называется константой распределения.
Установлено, например, что распределение йода и брома между СС14 и
Н20 характеризуется соотношениями
[ВГз]са* =83,9; [УсС1- =27.
[Вг2]н2С
Ин.о
Понижение температуры замерзания и повышение температуры
кипения раствора неэлектролитов. Растворы неэлектролитов
замерзают при более низкой, а кипят при более высокой
температуре по сравнению с чистым растворителем. Количественная
зависимость понижения температуры замерзания Д*3ам и повышения
температуры кипения Д/кип разбавленного раствора от концентрации
имеет вид
Ызш=-К-т,
(7.9)
Таблица 7.2
Криоскопические и эбулиоскопические
константы некоторых растворителей
Формула
растворителя
н2о
с6н6
QH5NH2
СС14
Е
0,52
2,57
3,69
5,30
К
1,86
5,10
5,87
29,80
где Е и К — соответственно эбу-
лиоскопическая и криоскопиче-
ская константы, зависящие от
природы растворителя (табл. 7.2).
В учебной литературе
соотношения (7.9) часто называют
вторым законом Рауля, а
соотношение (7.4) — первым законом
Рауля. В действительности
соотношения (7.9) являются
следствиями количественной закономер-
236
нос ni (7.4). Раствор кипит при более высокой температуре именно
поюму, что давление пара летучего растворителя над раствором,
содержащим нелетучее растворенное вещество, ниже, чем чистого
рапворителя. По этой же причине раствор не будет замерзать
при температуре замерзания чистого растворителя; его
кристаллизация из раствора будет затруднена присутствием молекул
постороннего вещества и будет происходить
при более низкой температуре.
Графическое изображение соотноше
иия (7.9) дано на рис. 7.3. Наклон
кривых О А и ОВО" определяется
уравненная
dT T(V2 — VJ
(7.10)
Рис. 7.3. Диаграмма повышения температуры
кипения раствора
1 атм
vm где К—мольная теплота испарения или плавления; V2—объем
фазы, характеризующейся большей энергией; V\ — объем фазы с
меньшей энергией. Например, для процесса жидк+±пар: V2—объем
пара, V\ — объем жидкости; для процесса пар^^лед: V2— объем
пара, V\ — объем льда. X процесса лед^пар больше X процесса
жпдк^пар, поэтому кривая ОВО" (кривая сублимации)
поднимается круче кривой О А.
Решение (интегрирование) (7.9) дает уравнение вида
in А.,
_ = _%_ /_1 1_
(7.11)
при условии, что в интервале температур (Т2—Ti) X—величина
постоянная.
Следствие из соотношений (7.9). Эквимолекулярные количества
различных веществ, растворенные в одном и том же весовом
количестве растворителя, понижают его точку замерзания и повышают
точку кипения на одно и то же число градусов. Так, при
растворении 0,1 моля сахара С12Н22О11 в 1000 г воды At= —0,186; такое
же понижение в воде дает и 0,1 моля глюкозы СбН^Об.
Закономерности Рауля (7.9) лежат в основе криоскопического
и эбулиоскопического методов определения молекулярных .весов
нелетучих растворимых веществ, которые нашли широкое
практическое применение. При этом они используются в виде
А, К • g • 1000 , „ ч
Д^зам = (криоскопическии метод),
М • G
. . E-g.1000 . ^ ч
д/ип = —s (эбулиоскопическии метод),
MG
237
где g и G — вес растворенного вещества и растворителя в граммах
соответственно; М — молекулярный вес растворенного вещества.
Важность приведенных методов для современной
физико-химической теории растворов иллюстрируется следующим примером.
Раствор, содержащий 0,32 г серы в 4 г бензола, кипит при 80,9ГС.
Эбулиоскопическая константа £свнв = 2,57; чистый бензол кипит
при 80,1°С. Вычислить, каким будет молекулярный вес серы
в СбН6.
Решение: Д*кип = 80,91 —80,10 =0,81;
Д*кип —
Е-9-1000 __ 0,257.0,32-1000
м а
С.Нв
0,4М
-0,81.
Откуда М серы = 256.
Следовательно, сера в растворителе СбН6 ассоциирована, ее
молекула состоит из
п =
м0
м
теор
256
32
8 атомов.
Осмотическое давление растворов неэлектролитов. Раствор —
однородная система. Это означает, что концентрация
растворенного вещества в любом, мысленно
выделенном объеме остается неизменной во
времени. Неизменность обусловлена тем, что
число выходящих и входящих в этот объем
молекул растворителя и растворенного
вещества одинаково. Выделенный объем
находится в динамическом равновесии с
окружающей средой. Если же взять
растворенное вещество, например CuS04-5H20, и
привести его в соприкосновение с водой, то
на поверхности раздела фаз (твердой —
медного купороса и жидкой — воды)
динамического равновесия не будет. Молекулы
растворенного вещества будут проникать
Рис. 7.4. Простейший прибор для измерения
осмотического давления:
1 — внутренний стакан; 2 — внешний стакан
(диффундировать) в растворитель, а молекулы растворителя в
объем раствора, в направлении от состояния с большей
концентрацией каждого из них к состоянию с меньшей концентрацией.
Результатом такой двусторонней диффузии будет выравнивание
концентрации во всем объеме раствора.
Возьмем сосуд с разбавленным раствором сахара, дно
которого сделано из специального материала, проницаемого для молекул
238
растворителя и непроницаемого для молекул растворенного
вещества, и посмотрим, что произойдет, если этот сосуд опустить в
<такан с водой (рис. 7.4). Молекулы растворителя из объема с их
полыней .концентрацией, т. е. из чистой воды, будут
диффундировать через полунепроницаемую перегородку в водный раствор
сахара, где их концентрация меньше.
Явление односторонней диффузии растворителя через
полупроницаемую перегородку называется осмосом. Сила на единицу
поверхности, заставляющая молекулы растворителей проникать
через полупроницаемую перегородку, получила название
осмотического давления. Вследствие осмоса уровень в сосуде с раствором
<ахара повысится; при этом создается дополнительное
(гидростатическое) давление, препятствующее диффузии. При некоторой
высоте А это дополнительное давление достигает такой величины,
что осмос прекратится совсем.
Гидростатическое давление, равное высоте А, препятствующее
односторонней диффузии растворителя, численно равно
осмотическому давлению Роем-
В 1887 г. Пфефер экспериментально установил две
закономерности, которым подчиняется осмотическое давление неэлектролита.
1. В разбавленных растворах неэлектролитов при данной
температуре осмотическое давление прямо пропорционально
концентрации
Роемое. (7.12)
2. При одной и той же концентрации С (моль/л) осмотическое
давление разбавленного раствора прямо пропорционально
абсолютной температуре
Яос* = /Са7\ (7.13)
Вант-Гофф обратил внимание на аналогию между законами
Пфефера и газовыми законами Гей-Люссака и Бойля — Мариотта.
Анализируя числовые данные Пфефера, полученные при
измерениях осмотического давления, он пришел к выводу, что величина
осмотического давления для разбавленных растворов
неэлектролитов может подчиняться закономерности (закон Вант-Гоффа)
Poc«Y = RT, (7.14)
где V — объем раствора, содержащий одну грамм-молекулу
растворенного вещества; R — постоянная, численно равная величине
газовой постоянной в уравнении состояния Клапейрона —
Менделеева. Уравнению (7.14) можно придать вид
nOCM-^CRT, (7.15)
где Т=С.
239
Формулировка закона Вант-Гоффа: «Осмотическое давление
разбавленного раствора неэлектролита равно тому давлению,
которое производило бы растворенное вещество, если бы оно при
той же температуре находилось в газообразном состоянии и
занимало объем, равный объему раствора». Закономерность,
установленная Вант-Гоффом, так же, как и законы Рауля, используется
для определения молекулярных весов растворенных веществ.
Осмотический метод оказался применимым также для
определения молекулярного веса высокомолекулярных соединений.
Соотношения Рауля для этой цели неприменимы. Зависимость
осмотического давления растворенного высокомолекулярного вещества от
его 'концентрации описывается соотношением
Люсм — - Г Л g *
М
ИЛИ
М
+ Ag9
(7.16)
где g — вес вещества в граммах.
Член (g2-A) учитывает специфику строения и кинетику
полимерных молекул. Экспериментальным путем находят зависимость
величины посм/g от весовой концентрации соединения.
Экспериментальные данные наносят на график (рис. 7.5). Экстраполируя
осмотическое давление n0cJg к нулевой концентрации, определяют
о (1-хА)-хв
Рис. 7.5. Графическое
представление зависимости
RT
М
■Ag
Рис. 7.6. Графический метод
определения коэффициентов
активности
длину отрезка на оси ординат, которая численно равна
величине RT/M.
Из этих данных рассчитывают молекулярный вес
высокомолекулярного соединения.
240
§ 3. АКТИВНОСТЬ И КОЭФФИЦИЕНТ АКТИВНОСТИ
КОМПОНЕНТОВ РАСТВОРА
Известно, что свойства растворов с концентрацией
растворенного вещества более чем 0,001 мольных долей не подчиняются
законам идеальных растворов: Рауля, Генри, Вант-Гоффа. Об этом
свидетельствуют также данные табл. 7.3 и 7.4.
Таблица 7.3
Давление насыщенного пара
над раствором сахара в воде при 0°С
Мольная
доля
растворителя
Давление
пара
растворителя
мм рт. ст.
D0
А
мм рт. ст.)
(Р^=4,6
0,9969
0,9919
0,9827
45,85
45,60
45,09
0,9968
0,9913
0,9802
0,9969
0,9913
0,9919
0,9802
0,9827
Таблица 7.4
Давление пара ацетона
в сероуглеродном растворе при 35,17°С
Мольная
доля
растворенного
вещества
хв
0,900
0,800
0,700
Давление
пара
ацетона Р „,
ь
мм рт. ст.
315
290
275
РБ
КБ
(К=2500)
0,126
0,116
0,110
аБ
Б хв
0,126
0,900
0,116
0,800
0,110
0,700
Однако, если в законе Рауля (Ра = Ра-Ха) вместо мольных
долей растворителя ХА (1-й столбец табл. 7.3; подставить
условные значения мольных долей, равные величинам Ра/Ра (3-й
столбец табл. 7.3), то давление пара растворителя (2-й столбец
табл. 7.3) описывается этим законом.
Условную мольную долю, при подстановке которой в
уравнение закона Рауля вместо концентрации можно получить
величины давлений пара растворителя, совпадающие с наблюдаемыми на
опыте, называют активностью растворителя аА, а степень
отклонения этой величины от мольной доли ХА — коэффициентом
активности растворителя уА (Льюис, 1907 г.).
Согласно Льюису:
Уа
(7.17)
РА=РА-аА=РА(УА.ХЛ).
(7.18)
Коэффициенты активности воды уА в водном растворе даны в
4-м столбце табл. 7.3. Аналогично, если в законе Генри
(РБ =Кб -Хв) вместо мольных долей растворенного вещества Хв
(1-й столбец табл. 7.4) подставить значения мольных долей,
равные величине Рб 1Кб (2-й столбец табл. 7.4), то давление пара
241
ацетона в сероуглероде (2-й столбец табл. 7.4) будет
описываться законом Генри.
Условную мольную долю, при подстановке которой в уравнение
Генри вместо концентрации можно получить величины давления
пара растворенного вещества, совпадающие с наблюдаемыми на
опыте, называют активностью растворенного вещества ав, а
степень отклонения этой величины от мольной доли растворенного
вещества X — коэффициентом активности растворенного
вещества у б
Уб = -~~> (7Л9)
АБ
Рб = КбАб = Кб (Уб ■ ХБ). (7.20)
Коэффициенты активности ацетона в сероуглероде даны в
4-м столбце табл. 7.4.
Коэффициенты активности учитывают суммарный вклад
взаимодействий в реальных растворах, изменяющих (понижающих при
ассоциации частиц типа А + А^А2 или повышающих при
диссоциации части Вг^В + В) концентрацию компонентов раствора.
Коэффициенты активности находят экспериментально, сопоставляя
свойства реального раствора с раствором той же концентрации,
для которого свойства растворителя подчиняются закону Рауля,
а свойства растворенного вещества — закону Генри. На основании
данных рис. 7.6 показан способ определения коэффициентов
активности и активности компонентов раствора с положительным
отклонением от идеальности. Расчет дан для состава точки О;
растворитель и растворенное вещество — компонента А.
Для растворителя (Ха -*• 1)
ра 0б
. *-T£5T~S- <a>
Для растворенного вещества (Ха С 1)
РЛ Об
Ул = — = . (б)
Ка-Хл ос
Так как аА = Уа-Ха* то активность растворителя
*, Y ?A Y Рл
аА =-Уа-Ха = р0 х -Ал = -^-
А
и активность растворенного вещества
а Ра X - Ра
0>А — —Г, 7Г~'ЛЛ —
КА'*А «
А
242
Значение константы Кб определяется тангенсом угла наклона
касательной и кривой давления пара растворенного вещества (пунктирная
линия), т. е.
к ос
*А
Для термодинамики реальных растворов оказалось весьма
плодотворным понятие стандартного состояния раствора, условно
принимаемого за начало отсчета неидеальности. Для растворов
м\ стандартное состояние растворителя принимают состояние
чисти жидкости (Р*а = Ра), за стандартное состояние растворенного
вещества — состояние, в котором его давление пара численно
разно константе Генри (Р^ = /Сгенри).
В стандартном состоянии активность растворителя
Рс7 Р°л
рО рО
^А ^А
активность растворенного вещества
п - Б — К — 1
*б К
§ 4. ЭЛЕКТРОЛИТИЧЕСКАЯ ДИССОЦИАЦИЯ.
ВОДА
Электролиты. Электролитами называются вещества, при
растворении которых раствор может быть проводником электрического
юка. Переносчиками тока являются ионы; последние, как известно,
появляются в растворе либо в результате диссоциации
растворенного вещества, либо за счет его ионизации. Процесс
диссоциации — это процесс разрушения ионной молекулы под влиянием
растворителя. Например (здесь и далее без учета гидратации):
NaCljTNa+ + Cr.
ионный кристалл
Процесс ионизации — это процесс возникновения ионов в
растворе. Простейший случай ионизации — ионизация в воде
полярной молекулы газообразного хлористого водорода
нс1:£Н+ + сг.
Вещества, молекулы которых нацело диссоциируют
(ионизируются) на ионы, принято называть сильными электролитами.
Растворы сильных электролитов не содержат нейтральных
молекул. Вещества, молекулы которых не полностью ионизируются,
принято называть слабыми электролитами. В растворах слабых
243
электролитов наряду с ионами существуют неионизированные
молекулы. Ионизация слабого электролита (АВ) протекает по
уравнению:
АВ^А+ + В~.
Количественной характеристикой способности электролита
распадаться на ионы является степень диссоциации а. Как
известно, она показывает, окакая доля от общего количества
электролита распадается на ионы. В общем случае степень диссоциации —
сложная функция природы электролита, природы растворителя и
концентрации раствора. Так, в концентрированных растворах
органических растворителей, характеризующихся малой
диэлектрической проницаемостью е, сильные в воде электролиты типа КС1
могут оказаться слабыми. Кажущееся понижение способности их
к диссоциации обусловлено в основном процессами ассоциации
ионов с образованием незаряженных частиц — квазимолекул,
например:
К+-,-СГ^:к+сг.
По этой причине деление электролитов на сильные и слабые
нельзя понимать в абсолютном смысле. Такое деление
целесообразно при рассмотрении свойств электролитов в данном
растворителе.
Степень диссоциации обычно определяют из данных
электропроводности. Для разбавленных растворов слабых электролитов
а -= ,
а,
оо
где Хг — эквивалентная электропроводность 1 вещества при данном
разведении; Я,» — электропроводность при бесконечном
разведении (V-*- оо).
При бесконечном разведении а становится равной единице
(100%). Значения степени диссоциации некоторых электролитов,
полученные из электропроводности 0,001 М водных растворов, для
сильных кислот таковы:
соляная НС1 0,993
азотная HN03 0,997
серная H2S04 0,960,
а для слабых кислот (а)
муравьиная НСООН 0,368
уксусная СН3СООН 0,126
цианистоводородная HCN 0,0011.
1 Под эквивалентной электропроводностью %v понимают электропроводность
грамм-эквивалента вещества в объеме V см3, который заключен между
электродами, находящимися друг от друга на расстоянии 1 см.
244
Реакции в растворах электролитов — это реакции между гидра-
гированными (сольватированными) ионами. Так, в реакциях пар
водных электролитов (без учета гидратации)
Na2S04 + ВаС12 = | BaS04 -r 2NaCl,
Li2S04 + Ba (N03)2 = j BaS04 -|- 2LiN03,
H2S04 + Ba (CH,GOO)a = \ BaS04 +2CH3C00H,
Cs2S04 -f BaI2 = | BaS04 -,- 2CsI.
путь взаимодействия выражается ионным уравнением
SOt -f- Ва2+ = | BaS04.
Ионы Na+Cs+Li+NOr и любые другие остаются в растворе в
прежних количествах. Химическая активность приведенных веществ
определяется действующей в растворе концентрацией ионов S04~
С Ва2+.
Коллигативные свойства электролита: давление пара раствора,
понижение температуры замерзания, осмотическое давление и
другие не подчиняются соотношениям Рауля и Вант-Гоффа, поскольку
при диссоциации частичная концентрация раствора возрастает.
Если принять число молей электролита за N, степень
диссоциации за а, число ионов, образующихся при диссоциации одного
моля, за /г, то (N—Na) будет число нераспавшихся молей
электролита, а (Nan) — число ионов, появившихся в результате
диссоциации. Общее число частиц в растворе будет равно [(N—Na) +
+ Лтал].
Отношение
всегда больше единицы. Из уравнения 7.21 следует, что величины
а и i связаны соотношением
a - i=l.; (7.22)
Как и следовало ожидать, чем больше а, тем больше L Опыт
показывает, что для электролита данной концентрации
Д^опыт А^опыт / "опыт
I =■
А*расч А* расч \ * расч
Следовательно, для описания коллигативных свойств электролитов
будут справедливы соотношения
Л/зам = *#т> д^кип = iEm,
Р0А-РЛ = ЬР = 1Р°ХБ, (7.23)
245
хМножитель i получил название изотонического коэффициента
Вант-Гоффа.
Кислоты и основания. Большинство электролитов проявляют
свойства кислоты или основания. В настоящее время не
существует однозначного определения понятий кислоты и основания,
которое в одинаковой мере удовлетворяло бы кислотно-основные
взаимодействия в растворах любых растворителей. По этой причине
находят применение частные определения. Для характеристики
кислотно-основных взаимодействий в водных растворах до сих пор
используется понятие кислоты и основания, данное Аррениусом.
Кислота — вещество, которое диссоциирует с образованием иона
водорода.
Основание — вещество, которое диссоциирует с образованием
иона гидроксила.
Кислотно-основное взаимодействие сводится к реакции
нейтрализации
Н+ + ОН" = Н20.
Ограниченность понятий кислоты и основания, данных
Аррениусом, можно иллюстрировать следующими фактами.
1. Молекула NH3 не содержит иона ОН~, а молекула СОг —
иона Н+, однако в водном растворе первая проявляет свойства
основания, вторая — свойства кислоты.
2. В водоподобных растворителях не обнаружено протона;
вместо него в растворах существуют гидратированные (сольвати-
рованные) ионы: Н30+ в воде, NH4+ в жидком аммиаке
С2Н5ОН2+ в этиловом спирте и т. д.
3. Многие водородсодержащие электролиты диссоциируют в
одном растворителе как кислоты, в другом как основания.
Например, анилин в жидком аммиаке — слабая кислота
C6H5NH2 - NH3 ^ C6H5NH" + NHf,
в воде — слабое основание
C6H5NH2 -r H20 -Z C6HbNHf + ОН".
Широкое распространение (особенно в химии неводных
растворов) получили понятия кислоты и основания, данные Бренстедом
и Лоури (1923 г.).
Кислота — вещество, способное отдавать протон, быть донором
протона.
Основание — вещество, способное принимать протон, быть
акцептором протона.
Процесс ионизации веществ осуществляется в контакте
с растворителем, при этом растворитель выполняет либо функцию
основания, либо функцию кислоты, например,
246
NH8 + H2O^NHf + OH-
l3 "Г А A2
основание кислота
HCl-r NH8^NH^ + Cr,
кислота основание
H20 + NH3 jt Ntif -\- OH",
x2w -]- пн3
кислота основание
CeH5NH2 + NH3^r CeH5NH~ + NH^,
кислота основание
CeH6NH2 + H2O^C6H5NH3+ + OH".
основание кислота
Если сродство к протону у растворителя больше, чем у
растворенного вещества, то растворитель выполняет функцию основания,
а если оно меньше, то — функцию кислоты.
Кислотно-основные реакции Бренстеда между растворителем и
растворенным веществом получили название протолитических
реакций. Подобно окислительно-восстановительной, протолитиче-
окая реакция рассматривается как сумма двух процессов с
участием сопряженных кислотно-основных (к-о) пар, например:
НС1/СГ:НС1:£Н+ + СГ
(к-о пара) I кислх \ сснх
NH4+/NH3 : NH3 ^ Н+:£ NH^
(к-о пара) II осн2 кисл2
HCl + NH3^NH^ + Cr
КИСЛЛ ОСН2 КИСЛ2 OCHi
Чем сильнее .кислота, тем слабее связанное с кислотой основание.
Протолитический процесс идет в направлении переноса протона
от более сильной кислоты НС1, слабо удерживающей протон по
сравнению с кислотой к более сильному основанию 1МНз,
сильнее удерживающему протон по сравнению с основанием С1~.
Концепция Бренстеда более прогрессивна по сравнению с
концепцией Аррениуса, но и она страдает рядом недостатков. Весь
химизм взаимодействия растворителя и растворенного вещества
теория Бренстеда сводит к протолитической реакции — переносу
протона. Как и теория Аррениуса, она не применима к веществам,
проявляющим кислотную функцию, молекулы которых не содержат
водорода. Эта теория исключила важнейший признак кислотно-
основного взаимодействия — реакцию нейтрализации. Любая
реакция сопровождается образованием новой кислотно-основной пары.
С другими современными теориями кислот и оснований можно
ознакомиться в книге А. И. Шатенштейна «Теория кислот и
оснований».
247
Вода как растворитель. Вода занимает исключительное место
среди ионизирующих агентов растворенных веществ. В воде
большинство веществ становится сильными электролитами. Вода как
растворитель характеризуется целым рядом специфических
особенностей, которые не присущи другим растворителям. Эти
особенности определяются строением и свойствами молекул Н20.
Рис. 7.7. Общепринятая модель
расположения зарядов в молекуле воды
Молекула воды имеет четыре полюса зарядов, расположенных
по тетраэдру (рис. 7.7). Дипольный момент воды велик; он
составляет 1,86. Между молекулами воды легко образуются водородные
связи. Энергия каждой водородной связи ~5 ккал. В жидкой воде
за q4£T водородной связи существуют ассоциаты [H30+(H20)^],
где х от 1 до 3. Из-за наличия водородных связей вода кипит при
аномально высокой температуре (100°С), имеет высокую теплоту
испарения (541 ккал/г). Высокая полярность молекул Н20, их
способность образовывать водородные связи обусловливают
высокую структурированность воды как растворителя. Структура
воды характеризуется ближним порядком, свойственным структуре
расплавленного льда.
Согласно рентгеновским данным лед имеет тетраэдрическое
строение; каждая молекула льда окружена четырьмя другими
молекулами. Структура льда характеризуется пустотами, размеры
которых больше размера молекулы Н20. При плавлении пустоты
частично заполняются теми молекулами Н20, которые
освободились от связей, свойственных структуре кристаллического льда.
По этой причине при плавлении плотность воды возрастает.
Пустоты в структуре жидкой воды могут заполняться инородными
молекулами, атомами, ионами.
Жидкая вода характеризуется высокой диэлектрической
проницаемостью е. Это важный фактор ионизирующей активности
растворителя. В соответствии с законом Кулона
р — gl g2
*~ 8 Г2
сила притяжения между двумя противоположными по знаку
зарядами обратно пропорциональна квадрату расстояния между ними
и диэлектрической проницаемости среды. В воде сила притяжения
между ионами в 81 раз слабее, чем в воздухе. Ионизирующая
активность зависит не только от диэлектрической проницаемости
растворителя, но и его сольватирующей способности.
+ н
+н
ЦЗЭА
248
Вода — хороший гидратирующий агент. В воде целый ряд
веществ (NH3, SO2, С02 и т. п.), не являющихся проводниками
электрического тока, становятся электролитами. Высокая гидратирую-
щая активность воды способствует разным по природе
электролитам (например, НС1, H2S04, HN03) быть одинаковыми по силе.
Важное значение для водных растворов имеет гидратация ионов.
Гидратация ионов. В водном растворе каждый ион окружен
оболочкой из молекул воды, создавая ближайшую или первичную
зону гидратации. Вовлеченные в эту зону молекулы воды движутся
вместе с ионом, утрачивая таким образом поступательное движение
молекул растворителя. Число молекул растворителя в первичной
зоне гидратации иона называется координационным числом.
Координационные числа (числа гидратации) некоторых ионов
приведены ниже (по К. П. Мищенко):
Ион. Na+ K+ Rb+ Cs+ F- CI" Br- I-
K. ч. 68 886888
В области первичной зоны гидратации из-за разрушения
структуры растворителя изменяется его диэлектрическая
проницаемость е. Формирование первичной зоны гидратации около
однозарядных ионов с оболочкой инертного газа осуществляется за
счет сил электростатического, ион-дипольного взаимодействия. Для
ионов переходных металлов с недостроенной d-оболочкой в области
первичной зоны заметную роль играют их химические силы (см.
гидролиз).
Как правило, гидратация не заканчивается формированием
первичной зоны гидратации, различают еще две зоны гидратации:
вторичную и зону, называемую границей полной сольватации
(ГПС). Вторичная гидратация состоит из двух-трех слоев,
следующих за первичной зоной гидратации. В этой зоне молекулы
растворителя не теряют способности поступательного движения,
свойственного воде, но из-за влияния заряда иона структурная связь их
с другими молекулами растворителя потеряна; растворитель в зоне
вторичной гидратации деструктурирован.
Под состоянием, определяемым как граница полной
сольватации, подразумевается состояние, когда весь растворитель вовлечен
в зоны гидратации ионов.
За пределами концентрации, отвечающей границе полной
сольватации, раствор утрачивает структуру растворителя, которая
будет определяться структурой сольватированных ионов. В
обычных условиях способность гидратироваться зависит от природы
иона, его заряда, размера, строения электронной оболочки иона и
концентрации раствора. Гидратирующая способность падает в ряду
ионов
А13+ > Сг3+ > Ве2+ > Cd2+ > Zn2+ > Mg2+ > Na+.
249
Анионы и катионы при прочих равных условиях гидратируются
по-разному. В разбавленных растворах этими различиями можно
пренебречь.
Гидратация ионов вносит большой вклад в энергетику
образования раствора. Она сопровождается значительным выделением
тепла. Экспериментально определить теплоту гидратации иона
—АН невозможно. АН ионов рассчитывают из теплот гидратации
солей.
Теплотой гидратации соли принято называть такое количество
тепла Д#, которое выделяется при переходе 1 моля соли из
газообразного состояния в бесконечно разбавленный раствор.
Теплотой гидратации иона принято считать количество тепла,
которое выделилось бы при переходе газообразного нона в
бесконечно разбавленный раствор.
Таблица 7.5
Теплоты гидратации (ккал/г-ион) (по К. П. Мищенко)
Ион
н+
н3о+
Li+
Na+
К+
Rb+
Cs+
Теплота
гидратации
263
110
121
98
80
74
63
Ион
Mg2+
Са2+
Sr2+
Ва2+
Zn2+
Cd2+
Ag+
Теплота
гидратации
470
375
338
312
501
445
109
Ион
F"
СГ
Вг-
I-
он-
N<¥
SO*"
Теплота
гидратации
113
79
72
63
116
72
243
Теплоты гидратации некоторых ионов приведены в табл. 7.5.
При гидратации изменяется энтропия раствора AS. Если
гидратация сопровождается значительным разрушением структуры
растворителя (деструктурированием), энтропия раствора
возрастает. В частности, она возрастает при гидратации объемистых
ионов большого размера, таких, как CNS-, ЫОГ. Если структура
растворителя при гидратации упорядочивается, AS падает.
Гидратация изменяет не только свойства растворителя
(структуру, его концентрацию в концентрированных растворах,
диэлектрическую проницаемость в зоне первичной гидратации иона),
но и свойства ионов. С гидратацией тесно связана
электрохимическая подвижность ионов (направленное перемещение под
действием электрического тока). Она снижает электрохимическую
подвижность ионов. Например, электропроводность расплавленных
ионных кристаллов LiCl выше электропроводности (кристаллов
CsCl. В водных растворах из-за значительно большей гидратирую-
щей способности Li+ по сравнению с Cs+ (табл. 7.5), наоборот,
электропроводность солей CsCl выше, чем LiCl.
250
Под влиянием гидратации деформируются электронные
оболочки ионов, что в большинстве случаев приводит к изменению их
окраски: [Си(Н20)4+ —голубой, [Со(Н20)6]2+ —розовой и т. п.
Гидратированные ионы обладают несравненно большей
термодинамической устойчивостью по сравнению с ионом, лишенным
гидратной оболочки.
Особенно сильно влияет гидратация на ион водорода.
Полученный при гидратации ион гидроксила Н30+ ведет себя совсем иначе,
чем вел бы себя свободный протон.
Гидратация ионов имеет важное значение для всех процессов,
протекающих в растворах, в том числе и для процесса
электролитической диссоциации.
Представления о процессе
электролитической диссоциации
в водных растворах
В теории электролитической диссоциации Аррениуса раствор
рассматривался как механическая смесь из неравноправных
компонентов: растворенного вещества, ответственного за
электропроводность, и растворителя, выполняющего роль физической среды.
Воде приписывалась роль исключительного ионизирующего
агента; ее ионизирующая активность объяснялась высокой
диэлектрической проницаемостью е. Электропроводность раствора в невод-
ных растворителях, характеризующихся, как правило, малой
диэлектрической проницаемостью, не находила объяснения и
относилась к аномальным явлениям.
Большое значение в формировании правильного представления
на природу электролитов имели работы И. А. Каблукова. Будучи
приверженцем гидратной теории Д. И. Менделеева, он утверждал,
что ионы могут вступать во взаимодействие с водой, образуя гид-
раты переменного состава. В своей докторской диссертации в
1891 г. И. А. Каблуков писал: «По-нашему, вода, разлагая частицы
растворенного тела, входит с ионами в непрочные соединения, по
мнению же Аррениуса, ионы свободно двигаются подобно тем
отдельным атомам, которые происходят при диссоциации молекулы
галоидов при высокой температуре». Касаясь механизма
растворения хлористого водорода, он замечал, что молекулы воды
вторгаются в молекулы НС1 настолько, что атомы С1 и Н приобретают
самостоятельность индивидуальных частиц.
Согласно современным представлениям электролитическая
диссоциация невозможна без взаимодействия растворителя и
растворенного вещества. Ионизирующая способность электролита зависит
как от природы растворенного вещества, так от природы и свойств
растворителя. В водных растворах электролитической диссоциации
сопутствуют процессы гидратации.
Каждый ион, находящийся на поверхности ионного кристалла,
окружает себя полярными молекулами воды. При этом в соответ-
251
ствии с законом Кулона силы стяжения между ионами на
поверхности кристалла ослабевают, одновременно увеличивается их
гидратация; процесс завершается образованием гидратированного
иона. Электролитическую диссоциацию ионной молекулы
(типа КО, NaCl) можно представить схемой
Д Л -1- #воды ~*" А воды ~г ^воды.
ионный
кристалл
В концентрированных растворах сильных электролитов на
процесс диссоциации может накладываться процесс ассоциации ионов
с образованием ионных пар
А воды ~г ^воды .<_ А воды ^воды .
При растворении полярных веществ (НС1, HNO3, CuBr2, HgCl2)
процессу диссоциации предшествует ионизация. За счет сил ди-
поль-дипольного взаимодействия полярная молекула растворимого
вещества окружается (гидратируется) полярными молекулами
воды. Под влиянием молекул растворителя она поляризуется,
расстояние между центрами тяжести положительного и
отрицательного электричества увеличивается; в итоге полярная связь
трансформируется в ионную
оео о©
О
©о
или
О О О
ионизация диссоциация .
К А + Яводн > (#-Ь4-)в0ДН >KTow* + Лв
полярная
Так, в частности, ионизируются в воде все галогеноводородьь
На процесс диссоциации водородосодержащих кислот
существенное влияние оказывает гидратация протона:
Н++Н2О^Н80+.
Свободных ионов Н+ в воде не существует. Процесс ионизации
водородосодержащих кислот следует выражать уравнением Брен-
стеда
НС1
t
донор
протона
+ н2о ^н3о+-
t
акцептор
протона
С1-.
252
И ряде случаев ионизируется не растворенное вещество, а продукт
взаимодействия растворенного вещества и растворителя.
Например:
SOa -J- 2H20^ (H2S03-H,0)^H30+ + HSOr,
NH3 + HaO ^ (NH3. H20) ^ NH^ + OH-,
HaO £ (C6H5 NH2. H20) -Z C6H5NH^ + OH-.
C6H5NH
Межионные взаимодействия. Сильные электролиты не
проявляют своей полной диссоциации, если даже их кристаллы состоят
из ионов. Для этих электролитов экспериментально определяемая,
кажущаяся степень диссоциации аКаж меньше 100%, и она
изменяется с разведением. Так, нацело ионизированный в водных
растворах хлористый водород характеризуется следующими
значениями аКаж-
Концентрация
С, моль/л 1 0,1 0,01 0,001
а 0,79 0,92 0,97 0,99
по температуре замерзания
раствора
Полученные из опыта значения изотонических коэффициентов С
сильных электролитов не равны целым числам (табл. 7.6), чего
следовало бы ожидать при их т
100%-ной диссоциации. Эти и другие 1а0лица /ь
факты свидетельствуют О ТОМ, ЧТО дей- Изотонические коэффициенты i
некоторых веществ
ствующая в растворах сильных элек- н ^
тролитов концентрация ионов не
равна их аналитической концентрации С
(г-ион/л).
Наблюдаемые отклонения от
100%-ной диссоциации обусловлены
присутствием в растворах сильных
электролитов ионов — источников
электрических точечных зарядов. Как
известно, разноименные точечные
заряды притягиваются, одноименные —
отталкиваются. По этой причине каждый ион будет стремиться
окружить себя ионами противоположного знака. Результатом
такой межионной упорядоченности будет закономерное
размещение ионов по всему объему раствора. В известной мере эта
закономерность аналогична распределению ионов в расплавленном
кристалле, но отличается от последней тем, что ионы в
растворах гидратированы растворителем и разъединены друг от друга
на значительное расстояние. С повышением зарядности взаимное
электростатическое влияние ионов друг на друга будет
усиливаться.
Электролит
КС1
LiCl
MgS04
Ca(N03)a
Ь онцент-
рация.
моль/л
0,14
0,13
0,38
0,18
i
1,93
1,94
1,20
2,47
253
Важной количественной характеристикой межионных
электростатических взаимодействий в растворах электролитов является
ионная сила раствора
/=т£с^- (7-24)
где Сг, Zi — соответственно молярность и зарядность i-того иона.
В общем случае взаимодействие в растворах нельзя сводить к
межионным взаимодействиям; для концентрированных растворов
является существенным вклад ион-дипольных взаимодействий, для
неводных растворов — диполь-дипольных взаимодействий. Однако
в водных растворах эффект межионных взаимодействий по
сравнению с любыми другими является определяющим. Из-за
взаимных межионных взаимодействий рассчитанная из данных
электропроводности оскаж много меньше ожидаемой, истинной а.
Активность и коэффициент активности электролита. Определяя
активность электролита, как и раньше, исходим из того, что в
предельно разбавленном растворе ионы удалены друг от друга;
между ними электростатические взаимодействия отсутствуют и они,
следовательно, образуют идеальный раствор. Согласно Льюису в
таком растворе активность а электролита численно равна его кон-
дентрации С; коэффициент активности у в соответствии с
соотношением
a =C«y
равен единице.
Если раствор не предельно разбавлен, то на его свойствах,
зависящих от концентрации, будет сказываться наличие межионных
Бзаимодействий. Рассчитанные из эксперимента кажущиеся
концентрации будут меньше истинных концентраций ионов (молекул),
т. е.
афС, уф\.
Как правило, y[<1- Чем больше концентрация ионов в растворе
электролита, тем у значительнее отличается от единицы.
В растворе сильного электролита нет недиссоциированных
молекул, поэтому следует говорить об активности и коэффициенте
.активности ионов, например, для электролита
ЛВ- Л+ + В-,
а+ = Су+,
1 В концентрированных водных растворах сильных электролитов Y иона может
принять значения больше единицы Это обстоятельство обусловлено иными
причинами, главным образом изменением структуры растворителя.
254
и для электролита АпВт = пАт+ + /иЯл""
а+=лС.у+, (7.25)
аг = тС-у^ (7.26)
где С — концентрация электролита; а+у а- — активности катиона
и аниона соответственно; у+> У-— коэффициенты активности
катиона и аниона.
Можно показать, что активность электролита АВ связана с
активностями ионов отношением
a^B--=a+-flL-=Ce-Y--Y+. (7.27)
а в общем случае для электролита АпВт — соотношением
аА в =а']агп==Сп+тпп'тт-уг\'Ут (7.28)
Однако пока не существует методов экспериментального
определения коэффициентов активности ионов, а следовательно, не
представляется возможным рассчитать как активности этих ионов, так
и активности электролитов. В настоящее время возможен только
обратный путь. Сначала экспериментально определяют
коэффициент активности у электролита в целом, рассчитывают его
активность а, а затем ищут пути оценки активности катионов а+ и
анионов а- электролита в отдельности. Для этой цели вводится понятие
среднего коэффициента активности у± и средней активности а±;
у± и а± есть среднее геометрическое из коэффициентов активности
у и активности а катионов и анионов электролита соответственно.
Для электролита АВ
у± = Vy+У-, а± = Va+-a-, (7.29)
для электролита АпВт:
п-\-т/
Y± = УУ1-У1, (7.30)
п-\-т/
а± = Va+.al. (7.31)
После преобразования уравнения (7.28) с учетом соотношений
(7.30), (7.31) получим соотношение, связывающее активность
электролита АпВт с активностями и коэффициентами активности
катионов и анионов и их средними величинами Y+ и а±*
ал в = а\ ат = ai+m = C<l+m)nn mn v'!+m (7.32)
которое используется при расчетах.
Пример1 № 1. Определить активность КО в водном растворе
при моляльности т = 0,02, если известно, что средний коэффициент
активности КО равен 0,894.
1 К. С. Пономарева. Сб. задач по физической химии М., Металлургиздат,
1959.
255
Решение:
а = а+-а-у
а+ = y+ • tn, a— = у- • т,
а = Y2±-m2 = (0,894)2.(0,02)2 = 3,196-Ю-4.
Пример №2. Определить активность и среднюю активность
ZnS04 в 0,1 моляльном растворе, если у± =0,148.
Решение:
1. а ^а+-а_ = у+.т-у--т = у*±-т2 =(0,148)2-(01 )2 = 2,19- 10~4.
2. a±Va+-a„ = 1^2,19-10~4 = 1,48-10~2.
Пример № 3. Определить активность a La (N03)3 в 0,01
молярном растворе, если у± =0,571.
Решение:
La(N03)3^La3+ +ЗГМОГ,
a+ = Y+-C, o- = y-(3C),
я = яц- • aL,
a = y+Cy- (3C)3 = y4±-33-C4 - (0,571)4-33 (0,01>4 - 9,5-10-9.
В 1923 г. Дебай и Хюккель показали, что для разбавленных
водных растворов электролитов с ионной силой раствора /с^0,01
коэффициенты активности ионов могут быть рассчитаны из
выражения (зарядность ионов одинаковая)
■lgY£ = 0,509 z2£ 1/7
(7.33)
Таблица 7.7
Значение коэффициентов активности
ионов в зависимости от ионной силы
раствора
Ионная
сила
0,0001
0,001
0,005
0,01
0,05
0,1
0,2
0,3.
0,5
Коэффициенты активности ионов
однозарядных
0,99
0,96
0,92
0,89
0,81
0,78
0,70
0,66
0,62
двухза-
рядных
0,95
0,86
0,72
0,63
0,44
0,33
0,24
—
—,
трехза-
рмдных
0,90
0,73
0,51
0,39
0,15
0,08
0,04
—
—
где Yf, %i — коэффициенты
активности и заряд /-того катиона
(аниона).
Уравнению (7.33) можно
придать иной вид
lgY±=—0,509z+.z-1/T, (7.34)
где у± — средний коэффициент
активности электролита (ионов).
Закон Дебая и Хюккеля
позволил теоретически обосновать
установленное в 1921 г. правило
ионной силы: «В разбавленных
растворах сильных электролитов
с одной и той же ионной силой
наблюдается равенство
коэффициентов активности катионов и
256
анионов одинаковой зарядности». Коэффициенты активности
некоторых групп ионов в зависимости от ионной силы растворов
приведены в табл. 7.7.
§ 5. КОЛИЧЕСТВЕННОЕ ОПИСАНИЕ РАВНОВЕСИЙ
В РАСТВОРАХ ЭЛЕКТРОЛИТОВ.
ПРИМЕНЕНИЕ ЗДМ К ЭЛЕКТРОЛИТАМ
К любой взаимодействующей системе
пА + тВ^рС (I)
применим закон действующих масс (ЗДМ):
/( = —. (7.35)
ап -а,п
аА аВ
Константа равновесия К—истинная (термодинамическая)
константа, зависит от природы веществ и температуры. Если в
растворе никаких других взаимодействий, кроме предусмотренного
уравнением (I), не происходит, активности а веществ численно равны
молярным концентрациям с (y=1) и
/С = —^—. (7.36)
ЗДМ применим также для (количественного описания процессов
ионизации в растворах электролитов. В том числе для
электролита АВ с концентрацией С (моль/л), ионизирующегося по
уравнению ,
AB^LA+ + Br- (II)
/С= а" '"- . (7.37)
а
С учетом того, что
а+ = С+ • Y-ь #- = С— • Y-> С+ = С - и Снеиониз —С — С+,
К= ^-Y+C-Y- = ^А = Kc-A—t (7.38)
Унеиониз ^неиониз ^леиониз гнеиониз Гнеиониз
где
Кс =
^неиониз С — С+
или в случае малости (С+)
У Общая химия
257
Константа Кс, выраженная через концентрации ионов, в отличие
от термодинамической константы К зависит не только от природы
частиц, температуры, но и ионной силы / раствора. Чем
коэффициент активности частиц ближе к единице, тем ближе значение Кс
к термодинамической константе К.
Соотношение (7.38) можно использовать для количественного
описания любых равновесий, в растворах электролитов таких, как
К+А- ^t/c+-M-,
КЕазимолекула
HSOr^H+ + SC>4~,
[Cu(NH3)4P+^:Cu2+ + 4NH3,
Fe(OH),^[Fe(OH)J+ + OH-
[А1(ОН)2]+^(А10Н)2+ + ОН-,
HgCl2^HgCl++Cl-
СН3СООН ^ СН3СОО- + Н+.
При этом возникают трудности лишь в связи с тем, что не всегда
с достаточной точностью можно определить коэффициенты
активности ионов (средние коэффициенты активности электролитов у±).
Ионизация кислот и оснований. В соответствии с
терминологией Бренстеда кислотой будем называть вещество, которое при
взаимодействии с водой способно образовывать ион гидроксонил
Н30+. К кислотам относятся, например, следующие вещества:
H2S04 + Н20 = HSOr + H3O+, HNO3 + Н20 = NOr + Н30+,
Hsor + н2о -+ so; - + Н3О+, сбн5он + н2о -^ свн5о- + н3о+,
С02 + 2Н20 :£ НСОГ + Н30+, HI + Н20 = I- + Н30+,
НС1 + Н20 = С1- + Н30+, 2HF + Н20 ^ HF^ + Н30+.
Основанием будем называть вещество, которое образует в воде
ион гидроксила (ОН-). Основаниями в растворе будут
А1 (ОН)3 £ [А1 (ОН)а]+ + ОН-, C6H5NH2 + H20^ C6H5NH^+ OH-,
[А1 (ОН)а]+ £Г (АЮН)2+ + ОН-, КОН = К+ + ОН-;
А1 (ОН)2+ •+ А13+ + ОН-, CH3NH2 + Н20 Jt CH3NH^ + ОН-,
NaOH = Na+ + ОН-, LiOH = Li+ + ОН-.
Сильные кислоты (H2S04, HN03, HC1) и сильные основания
(КОН, NaOH) ионизируются нацело. Написание для них
обратимого процесса ионизации с присутствием в растворе неионизиро-
ванных молекул смысла не имеет. Для каждой многоосновной кис-
258
юты (H2S04) или основания типа А1(ОН)3 характерно несколько
ступеней ионизации. Каждая последующая ступень ионизации
протекает слабее предыдущей, так как отнять ион Н+(ОН") от
заряженной кислоты (основания) тем труднее, чем ее заряд больше.
Для описания процессов ионизации слабых кислот и слабых
ч)снований можно использовать более простой вид ЗДМ, чем
уравнение (7.38). Например, примем, что в отсутствие посторонних
ионов сильного электролита межионные взаимодействия не имеют
места, тогда
1, а = С, С+ = С_ = С.а, Сш
с^С
/( =
с+ с
1—а
(l-a)V
(7.39)
Таблица 7.8
Проверка закона разбавления
на растворах уксусной кислоты
при 18°С
где V — разведение (разбавление), или объем моля электролита.
Соотношение (7.39) носит
название закона разбавления
Оствальда.
О применимости закона
разбавления Оствальда к реальным
растворам слабых электролитов
можно судить на основании
табл. 7.8. Соотношение (7.39)
может быть использовано для
количественного описания
процессов ионизации таких слабых
электролитов, как уксусная
кислота. Константа равновесия К
остается практически величиной
постоянной для всех растворов с
концентрацией электролита менее 0,04 моль/л. При более высоких
концентрациях соотношение (7.39) можно использовать лишь для
приближенных расчетов.
Как и следовало ожидать, в присутствии ионов сильного
электролита закон разбавления Оствальда неприменим совсем
(табл. 7.9).
С, моль/л
0,000028
0,000111
0,000218
0,001030
0,05
0,10
a
0,539
0,328
0,248
0,124
0,019
0,0135
К
1,77.10-6
1,78.10-5
1,78.10-5
1,80-10"8
1,84.10-»
1,85-10-5
Таблица 7.9
Проверка закона разбавления на растворах СН3СООН в присутствии КС1 при 25°С
Моляльность
соли
к
0
1,75-10-5
0,01
1,86-10-5
0,05
2,19-10-5
о,ю
2,69-10-5
0,20
2,95- Ю"6
9*
259
Константы ионизации некоторых слабых кислот и оснований
приведены в табл. 7.10.
Таблица 7.10
Константы ионизации некоторых веществ при 25°С
вещество
H2S
HCN
CHgCOOH
H3P04
Н2Сг04
н2с2о4
HNO*
СНС12СООН
H2S03
С6Н5ОН
Кислоты
Кг
8,9-10-8
6,2-10-1°
1,74-10-5
7,6-10-з
5,6-Ю2
5-10-4
5-10-2
1,7-10-2
1,3-10-1°
Кг
1.3-10-13
6,2-10-8
1,8-10-1
5,4-10-5
6,2-10-8
Кг
4,4-10-13
вещество
(NH3-H20)
СН3 (NH2)
C5H5N
Pb (OH)2
Zn(OH)2
Основания
Kt
1,76-10-5
5,25-Ю-з
1,5-10-9
1,96- К)"4
4-10-5
Кг
3,0-10-s
1,5-10-»
Константа ионизации — важнейшая количественная
характеристика слабых кислот и оснований. Она позволяет рассчитать
активность ионов Н30+, (ОН)~, предсказать снижение
(повышение) их активности в присутствии электролита, влияющего на
подвижность данного процесса ионизации.
Рассчитаем, как снизится активность ионов водорода в 0,1 М
растворе СН3СООН, к 1 л которого добавлено 0,005 г-экв
СНзСОСЖа; #сн3соон = 1,74- 10~5.
Решение:
СНоСООН + НаО:£ СН8СОО- + Н30+,
К
асн3соо~'ан3о+
асн3соон'ан2о
СН3 COO"' YCH3COO~ * Н3Р+ ' ^НзО4-
(ссн3соон"~ си 0+) • Ych3cooh
Для растворителя (в растворе слабого электролита) а = 1, для
нейтральных молекул у = 1, поэтому
К-
ch3coo-'ychscoo~
Н8СГ
'нхг
(С{
сн.соон
Сн3о+>
= 1,74.10-5.
В растворе уксусной кислоты, характеризующейся такой малой К>
ионной силой раствора / можно пренебречь и считать у ионов,
равной единице; кроме того, из-за малости величину Сноч =CCHsCOO-
из двучлена в знаменателе можно исключить. Тогда
К = -
а.
С • С
сн3соо~ н.о^
1,74-Ю-5,
260
CHso+ = %,o+ =Kl.74 ■ 10-s. o,i = 1,32-10-3.
В присутствии соли за счет действия избыточных одноименных
ионов СН3СОО~~ ионизация СН3СООН будет подавлена. Пусть в
присутствии соли уксусная кислота диссоциирует с образованием
Сн3о+ = Ссн3соо- = Х (г-ион/л).
При этом концентрация неионизированной части СН3СООН будет
равна (0,1—X) г-моль/л. В растворе кроме СН3СОО~-ионов из
кислоты (X г-моль/л) будут присутствовать также ионы из соли
CH3COONa в количестве 0,005 г-моль/л. Их общая концентрация
равна (0,005+^). Подставим эти величины в выражение
константы ионизации СН3СООН
(0,005 + X). угн гоо- • X 0+ -YH 0+
is __ СН3С00 н8о н3о ;— 1 74 10-5
(0,1 -X)
Величины X из двучленов числителя и знаменателя можно
исключить и, учтя, что Хно+-уио+ = ано+» записать
0,005-v • а +
YCHacoo" н3о+ = j 74.10—5
0,1
Для определенияyCHCOo~ надо рассчитать ионную силу раствора;
практически она будет обусловлена концентрацией ионов соли:
/ = -L (0,005 + 0,005) -.= 0,005.
Для данной силы у = 0,92 (табл. 7.7)1.
С учетом у—
1,75 • 10~5 -0,1
0,005 • 0,92
i,/o . ш ■» ■ u,i qft 1п_4
ан3о+== л ™, г, ^ =3,8.10 4.
Легко видеть, что активность ионов водорода уксусной кислоты
в 0,005 М растворе CH3COONa примерно в 20 раз ниже по
сравнению с ее истинной кислотной активностью.
Ионное произведение воды рН растворов. Процесс ионизации
воды /протекает по уравнению
Н20 + Н20 -Z Н30+ + ОН-. (III)
Вода —типичный амфотерный электролит: одна ее молекула
является кислотой (отдает протон, по Бренстеду),
другая—основанием (принимает протон). Без учета гидратации
н2о:£Н+ + он-.
1 В аналогичных примерах, рассматриваемых ниже, см. табл. 7 7.
261
Константа ионизации воды при 25°С
д_ ан3о+ аон- = g+-a. =1 8.10-i6> (7.40)
а а
Степень диссоциации воды, очень мала; чистая вода практически
не проводит электрический ток. Поэтому можно считать
К = а+ ,а- = нз°ь' он" = 1,8. Ю-16.
а С
Вследствие малой степени ионизации концентрацию чистой воды
можно считать величиной постоянной, равной
1000 -- е „,
= 55,5 молей/л,
18 '
тогда
Кв = аНз0+ • аон- = 55,5.1,8.10~16 = 1 • Ю-14 (при 22 °С).
Соотношение
*в=яНзо+*аон- (7-41)
формулируется следующим образом: Для любого раствора
произведение активностей ионов ^но+иаон" есть величина
постоянная, называемая ионным произведением воды Кв-
Значение Кв значительно изменяется с температурой:
*°с
Кв
0
1,139-10-»
15
5,702-Ю-1»
25
1,008.10-"
50
5,474. Ю-1*
100
5,900-10-13
Постоянство Кв при данной температуре отнюдь не означает
постоянства активностей ионов гидроксония НзО+ и гидро,ксила ОН".
Только для нейтральных растворов аИ 0+ = аон- = V"KB . Для
кислых растворов ан 0+ много выше активности ионов ОН" и,
наоборот, для щелочных растворов аон- выше активности ионоз
гидроксония.
В качестве примера определим активность ионов гидроксония
Н30+ (условно Н+-ионов) для растворов 0,002 MHN03 и 0,001 М
КОН при 25°С.
Ионная сила раствора HN03
/ = — 2 С, ii = — (0,002 + 0,002) = 0,002; у = 0,95;
aHj0+ = C-y+!= 0,0020,95 -= 0,0019 г-ион/л;
1,008-10-u аб4.10_„
он 1,9-10-»
2G2
Ионная сила раствора КОН
7 = 0,001; у =0,92;
аон- =C-y = 0,001-0,92 = 9,2-Ю-3,
1,008-10-и =1
Нз° 9,2 • 10~3
Из этих примеров следует также, что для характеристики
кислотно-основной функции растворов электролитов достаточно знать
активность одного из ионов: Н30+ или ОН~, так как сопряженная
с каждым из них величина легко определяется из соотношения Кв-
Для удобства в практике обычно определяют величину
pH=-lgaH80+>
называемую водородным показателем. Для нейтрального раствора
рН = 7, для кислого рН<7 и для щелочного рН>7.
Шкала кислотности, рН
0,25 0,5 4,5 5 6 7 8 9 10 11 12 13,5
увеличение кислотности среды
увеличение основности среды
Величины аон- и ано+ не могут быть равными нулю. Это
противоречит ионному произведению воды.
рН — важнейшая характеристика раствора. Регулируя рН
среды, можно добиться растворения одних веществ и осаждения
других. Например, известно, что гидроокись магния можно
осадить из раствора, содержащего ионы Mg2+, по уравнению
Mg2+ + 20H-+|Mg(OH)2,
если рН = 10 (рОН = 4); при рН = 9 Mg(OH)2 уже не выпадает
совсем.
Существуют различные методы определения активности ионов
водорода. Одним из простейших является метод с использованием
веществ, называемых индикаторами, которые способны при
определенных условиях изменять окраску. Чаще всего применяются
кислотно-основные индикаторы. Они характеризуются тем, что
изменяют свою окраску ,в зависимости от рН раствора. Всем
известно, например, применение раствора фенолфталеина.
Индикаторы такого типа, как правило, очень слабые кислоты; реже
встречаются индикаторы — слабые основания. Индикаторы кислоты
(основания) при диссоциации образуют кислотно-основную пару,
состоящую из недиссоциированных молекул и ионов индикатора,
различающихся по окраске:
263
H Ind ■£ Ind- + Н+.
(IV)
окраска № 1 окраска № 2
Например, для фенолфталеина (кислота)
Hind ц! Н+ + Ind-. (V)
бесцветная, красная,
кислая форма основная форма
При увеличении концентрации ионов (Н+) равновесие (V)
сместится в сторону увеличения концентрации кислой, бесцветной
формы фенолфталеина; при увеличении концентрации ионов ОН"
(при добавлении щелочи) оно сместится в сторону основной
формы, характеризующейся красным цветом. В любой момент в
системе присутствуют обе формы индикатора. Можно в такой степени
сместить равновесие (V), что концентрация одной из форм будет
подавлена полностью. Кислой формой индикатора называют
форму, которая преобладает в области более кислых растворов, а
основной — в более щелочных растворах. Переход окраски от одной
окрашенной формы к другой происходит в определенном
интервале изменения величины рН, которая носит название интервала
перехода. Обычно этот интервал оценивается двумя единицами рН.
Подвижное равновесие (IV) индикатора как слабого
электролита можно охарактеризовать количественно:
*Hlnd V
Преобразуем это соотношение в вид
^Jnd аш щелочная форма индикатора
= = — . (7.43)
а . aHlnd кислая форма индикатора ч '
aind~
Последнее соотношение указывает на то, что отношение зави-
aHInd
сит от активности ионов водорода.
Быстро и точно рН раствора определяется с помощью
специальных приборов — рН-метров. Широкое применение нашли
рН-метры марки ППМ-ОЗМ.
Буферные растворы. В химической практике часто возникает
необходимость иметь раствор с устойчивой величиной рН, заметно
не изменяющейся при добавлении в раствор небольших
количеств сильной кислоты или сильного основания. Растворы,
обладающие способностью сохранять практически постоянным рН,
получили название буферных смесей. Они, как правило, состоят из
смеси растворов слабой кислоты и соли, образованной этой
кислотой и сильным основанием (CH3COOH + CH3COONa), или из
растворов слабого основания и соли этого основания и сильной
264
кислоты (NH3H2O + NH4CI). Покажем, что буферная смесь,
например (СН3СООН+ CH3COONa), обладает способностью в
определенных пределах поддерживать постоянным рН раствора.
Заметим, что в присутствии соли CH3COONa процесс
сн3соон -z сн3соо- + н+
будет сильно подавлен из-за воздействия на равновесие ионизации
СНзСООН одноименных СН3СОО~-ионов соли. По этой причине
действующая (концентрация СН3СОО~-ионов будет равна
действующей концентрации этих ионов из соли как сильного электролита,
или
аСН3СОО" = ^СН3СОО" ' YcH3COO~ ~ ^снзСОО.Ыа-У<ГН8СОО"".
Константа ионизации СН3СООН запишется в виде
К =
Откуда
ан+ *асн3соо~ _ аи+ ' ас°ли __ ан+'Ссоли <<усн8сосг
аСН3СООН ^кисл ^кисл
ан, ^Скисл . (7.44)
Ссоли'УСНзСОО-
Для приближенных расчетов можно считать y = 1»
ан+ = * ' скисл (745)
^"СОЛИ
И
pH=pK-lg-^- (7-46)
Буферное действие подобных смесей основано на следующем.
Если к буферной смеси, например (CH3COOH + CH3COONa),
прибавить в определенных пределах щелочи, то произойдет реакция
СН3СООН + NaOH -> CH3COONa + Н20,
суть которой заключается в соединении ионов Н+ и ОН~:
Н+ + ОН - = Н20.
Концентрация уксусной кислоты при этом уменьшается, а соли
CH3COONa возрастает. Введение сильной кислоты НС1 в
ацетатную буферную смесь приведет к реакции
НС1 + CH3COONa -> СН3СООН + NaCl
(сильная кислота всегда вытесняет слабую из раствора ее соли),
или в ионном виде
н+ + сн3соо- ->. сн3соон,
265
т. е. будет увеличиваться концентрация уксусной кислоты и
уменьшаться концентрация CH3COONa. При добавлении сильного
основания или сильной кислоты изменение величин концентрации
уксусной кислоты и ее соли может быть заметным, но даже при
изменении отношения СКисл/ССоли в 10 раз логарифм этой
величины в уравнении (7.46) изменится всего лишь на единицу.
Аналогичный процесс буферного действия будет наблюдаться в
случае аммонийной буферной смеси (NH3-H20 + NH4OH). Можно
показать, что в этом случае активность ионов НзО+ в аммонийной
буферной смеси будет определяться соотношением
«*о+ = Чг- = „ , „ .. • <7'47)
#В _ К]
аОН А0сн*Сосн/Ссоли * YNH-|-
Без учета ионной силы раствора
4
рН - 14 —рК + lg-^-. (7.48)
^соли
Рассмотрим буферное действие на следующем примере. В 1 л
буферной смеси, содержащей 0,2 моль/л НСООН и 0,2 моля
HCOONa, прибавили 0,005 г-экв NaOH. Рассчитать рН буферной
смеси. Как изменится рН при добавлении к раствору сильного
электролита (NaOH)? Кнсоон = 2,Ы0~4.
Решение:
1. Ионная сила буферного раствора
/ = -Т(0'2 + 0'2) = 0'2
концентрацией ионов НСООН пренебрегаем); y=0,70.
Согласно (7.45)
/ССКИСЛ = 2,1. 10~*. 0,2
Нз° Ссолиу 0,2-0,70
рН = — lg aHe0+ =4— lg3 = 4 — 0,477 = 3,523.
2. При добавлении к буферному раствору NaOH будет иметь
место реакция нейтрализации
СНСООН + NaOH = HCOOHNa + Н20.
0,005 0,005 0,005
В результате реакций в растворе концентрация кислоты СН3СООН
уменьшится и станет равной 0,2—0,005 = 0,195 г-экв/л (моль/л).
Концентрация соли увеличится и станет равной 0,2 + 0,005 =
= 0,205 (моль/л).
Ионная сила
/ = -L (0,205 + 0,205) =0,205; у = 0,68 (табл. 7.7);
266
2,110-^.0,195 =2>9.ю_4)
Нз° 0,205 . 0,68
рН = 4 — lg 2,9 = 4 — 0,462 = 3,538.
Как и следовало ожидать, из-за буферного действия рН раствора
останется практически постоянным.
Буферным действием могут обладать также смеси солей, из
которых одна вследствие гидролиза (см. ниже) показывает более
щелочную реакцию, другая более кислую, например смесь
(NaH2P04 + Na2HP04).
Буферные смеси различаются по способности оказывать сопро-
тивляющее действие растворам сильных кислот и щелочей. Если
рН одного буферного раствора изменяется при добавлении,
например, сильной кислоты меньше чем другого буфера при
добавлении того же количества кислоты, то говорят, что первый раствор
обладает большей буферной емкостью. Количественно буферную
смесь оценивают числом молей сильного основания или сильной
кислоты, добавление которых к 1 л буферной смеси вызывает
изменение рН на единицу.
Разбавление буферной смеси не сопровождается заметным
изменением рН, так как при разбавлении одновременно происходят
два противоположно действующих процесса. С одной стороны,
разбавление приводит к увеличению концентрации кислоты (или
для щелочного буфера щелочи) из-за увеличения степени
диссоциации; с другой стороны, разбавление влечет за собой
уменьшение концентрации соли.
Наиболее употребительные следующие буферные смеси:
Название Состав смеси рН
Формиатная НСООН + HCOONa 3,7
Бензоатная CeH5COOH + ClC6H4COONa 4,2
Ацетатная СН3СООН + CH3COONa 4,7
Фосфатная NaH2P04 + Na2HP04 6,8
Аммонийная NH3 • Н20 + NH4C1 9,2
Гидролиз солей
Растворение соли, образованной сильной кислотой и сильным
основанием (NaCl, KC1 и т. п.), не изменяет рН чистой воды.
Присутствие соли увеличивает лишь ионную силу раствора.
Растворение соли, образованной сильным основанием и слабой кислотой
(СНзСОСЖа, NaCN), сопровождается заметным изменением рН.
Это обстоятельство обусловлено тем, что ион слабой кислоты
будет взаимодействовать с водой по уравнению
сн3соо- + н2о :£ сн3соон + он ,
CN- + Н20 ^ HCN + ОН-.
267
В общем случае
А- + Н20 :£ НА + ОН-, (VI)
где А — условный анион слабой кислоты. Согласно (VI) часть
ионов Н+(Н30+) из воды связывается в молекулу слабой кислоты,
а ионы ОН~ в соответствии с постоянством величины Кв
накапливаются. Чем больше ионов Н+ свяжется в недиссоциированные
молекулы НА, тем больше будет концентрация ионов ОН"
(больше рН раствора).
Традиционно реакцию взаимодействия ионов соли с водой
называют реакцией гидролиза соли. Реакция (VI) представляет собой
типичный случай гидролиза.
Реакции гидролиза всегда обратимы. Обратимость реакции
(VI) обусловлена тем, что какой бы слабой кислотой НА ни была,
она всегда обладает некоторой ионизацией по уравнению
НА:£Н+ + А-,
характеризующейся определенной величиной
аи+ ' аА~
к = ~——•
*НА
В частности,/Cch3cooh>-/Chcn. Следовательно, раствор NaCN будет
более щелочным по сравнению с раствором CH3COONa той же
концентрации.
К обратимому равновесию (VI) применим ЗДМ:
йхтл • а _
К — °
V ' ан2о '
Так как активность растворителя аНго — 1 (как стандартного
вещества),
Аид • # тт_
Кг = НА он - (7.49)
V
Кг — называется константой гидролиза. Заменив аон- эквивалентной
величиной KJaH+, соотношению (7.49) можно придать удобный для
практических целей вид
Кг= дна • *в ^ Кв (75о)
V ' V *НА
Из соотношения (7.50) легко вывести расчетную формулу
определения активности ионов водорода:
*НА ' аоН~ СнА ' YHA • СОН- * YOH"
Кг
ЯА- ^негидр * YA-
268
В растворах с малой ионной силой коэффициент активности недис-
социированных молекул Yha близок к единице, Сна = Сон- ,
коэффициенты активности y0h~ и Ya~—также можно считать равными.
Следовательно,
К,- с СЬН~ • (7-51)
Ссоли ион~
Решив квадратное уравнение, получим
сон-= ^± V -Г + К,-С.
Если же Сон-<ССОли, то решение (7.51) относительно Сон- приведет
к соотношению
Сон = VKr- С = ]/-£*— • Ссоли • (7.52)
V АКисл
Заменив концентрацию С0^- в соотношении (7.52) на
Соя--
L н3о+ гон
получим
- , -_!_ 1 / ^в - к,
н3о- - v I/ с
кисл
соли
Например, для 0,01 М CH3COONa (ионная сила 7=0,01; у = 0,92):
w = _l_ -I/"1-10"14-1'82'10'8 =4,67-10-»
Нз° 0,92 V 0,01
И
рН- —4,67- Ю-9 =8,33.
Второй типичный случай гидролиза — гидролиз соли, образованной
слабым основанием и сильной кислотой. В этом случае гидролизу
подвергается катион Кат+. Ионное уравнение запишется в виде
Кат+ + Н20 :£ Кат ОН + Н+, (VII)
или
аКатОН ' аи +
а
Кат '
К — -
Аг —
^Кат ОН ' ^в
а _ • а
Кат ОН
Кв
•Косн
(7.53)
269
Концентрация ионов гидроксония
:"»о+ - V*
Сн.о+ = 1/ ~^-С ("л54>
«н.о* =Y+/*,-C -Y+- \/-1Г--С
Г Аосн
(7.55)
Соотношения (7.50) и (7.53) можно представить одним
уравнением
* = -*=-. (7.56)
Апрод
Константа гидролиза растет с температурой, поскольку /Св
возрастает с температурой сильнее, чем константа ионизации
продукта гидролиза /Спрод.
Глубину гидролиза обычно оценивают степенью гидролиза
и — число гидролизованных молей соли _ Сг
общее число молей соли С
Приняв концентрацию соли С моль/л для случая (VI), будем иметь
СР = Сон_=Л:С. (7.57)
С учетом соотношения (7.52)
А=т/—^
V ^кисл
(7.58)
Аналогично для случая гидролиза (VII)
Сг = Сн,о--=/*-С. (7.59)
С учетом соотношения (7.54)
Г
h
-Vitc- (760)
В разобранных выше случаях один из ионов соли подвергался
гидролизу (катион или анион). Если же соль образована слабым
основанием и слабой кислотой, то будут подвергаться гидролизу
иба иона. В частности, для соли CH3COONH4
СН3СОО- + Н2О^СН3СООН + ОН-,
H\lf -г Н20 -Z [NH4OH]! + Н+ -£. NH3 • Н20 + Н+.
После суммирования получим
СН3СОО- + NHt + Н20 ^ СН3СООН -! NH3 • НаО, (VIII)
1 Существование [NH4OH] не установлено.
270
K = ДснзСООН • gNH3-H2Q __ gCH3COOH ' gNH8.H2Q ' ^B
CH3COO— NH+ CH3COO- HsO+ NH+ OH~
a.
Акисл* Aqch
(7.61)
аон-
(7.62)
fl
OH —
a2 .
н,о+ _
KB
^NH3 H20
Kb
4o+ '
^CHgCOOH
^NH3-H20
Образующиеся при гидролизе -количества ионов Н+ и ОН", а
следовательно, их активности будут в прямой зависимости от констант
диссоциации СН3СООН и NH3-H20 соответственно, т. е.
gHaO+ = ^СНзСООН1 ,- gov
^NH3 H20
Заменив в соотношении (7.62)
получим
Откуда QH8o+=l/ Kb'*K™ ■ (7-63)
г Доен
Можно показать, что для солей типа CH3C00NH4 степень
гидролиза h и константа гидролиза Кг связаны уравнением
-rzT" = V%T (7-64)
Плохо растворимые соли, содержащие в растворе над осадком
незначительное количество ионов (Сиона«10~8), практически не
подвергаются гидролизу. Многозарядные ионы (Р03~, СОз"", Fe3+, Al3+
и т. п.) гидролизуются ступенчато. Например, для Na2C03 (без
учета гидратации)
I ступень
СОз~ + Н20 ^ НСОз" + ОН",
II ступень
нсог + н2о •+ н2со3 + он-.
рН растворов солей определяется в основном гидролизом,
идущим по I ступени. В качестве примера оценим гидролиз по I и
II ступени для РЬ(1\ЮзЬ-
^(PboH)-f- ^ * * Ю""7, Ярь(он)2= 3,3- 10~3;
Справедливо в случае близости величин /СДИСс кислот и оснований.
271
I ступень
Pb2+ + H20^ (РЮН)+ + H+; Кг = К» + = |Q~^7 = Ю-
К?ъон+ МО '
II ступень
(РЬОН)+ + Н20 ^ РЬ (0Н)2 + Н+; Кг
Кв
^РЬ(ОН),
10
,-14
3,3.10~3
Кг 10-
= 3,3-Ю~10,
= 200,
*н 3,3-10"
т. е. гидролиз, идущий по I ступени, больше гидролиза, идущего по
II ступени, в 200 раз.
Среди солей можно встретить примеры необратимого
гидролиза. Это соли слабых оснований и слабых кислот, продукты
гидролиза которых, как правило, легко выпадают в осадок или
переходят в газовую фазу (Cr2S3, A12S3, Fe3(C03)3, A12(C03)3 и
некоторые другие). В водных растворах эти вещества превращаются
в продукты гидролиза нацело:
Cr2S3 + 6Н20 = 2Сг (ОН), + 3H2S,
Fe2 (С03)з + 3H20 = 2Fe (ОН)3 + ЗС02.
Согласно Бренстеду реакция гидролиза — протолитическая
реакция; константа гидролиза — константа ионизации гидролизующе-
гося иона. Рассмотрим гидролиз соли NH4C1.
Ионное уравнение гидролиза
NHt + Н20 ;± NH3 • Н20 + Н+. (IX)
Константа гидролиза .
к = aNH3-H2o-flH+ = Кв = Кв (7щ
aNH+ ^NH3-H20 #NH,
В воде имеет место также процесс ионизации аммиака
NH3 + Н20 ^ NH3 • Н20 ^ Ш£ + ОН-; (X)
N
Н+ ОН- NH+ К]
КШз = i = * = -^— . (7.6G)
ИЛИ
272
%на "nh3-"h+
Ki
/с
ч+
NHj- К
NH
Соотношения (7.65) и (7.66) эквивалентны, т. е.
*'—&Г-*•»*■ (7'67>
Гидролиз зависит от ряда факторов. Важнейшим из них
является поляризующее действие иона на молекулы воды в ближайшей
зоне гидратации. Значительному гидролизу подвергаются ионы
переходных металлов. В растворах реальных концентраций солей
переходных металлов под влиянием гидролиза образуются не*
столько ионы состава
[Э(Н20),(ОВД+,
сколько ионы более сложного строения
[(Н,0)х Э - О - Э (Н20),]-+ и др.
Если поляризующее действие катиона заметно превышает
поляризующее действие аниона, то гидролиз протекает по катионно-
му типу, если, наоборот, поляризация молекул воды в зоне
первичной гидратации аниона больше, чем катиона, то гидролиз
протекает по анионному типу. В случае солей, образованных ионами,
характеризующимися сравнимым поляризующим действием,
гидролизу подвергается катион и анион одновременно. Механизм
реакции гидролиза окончательно не установлен.
Сольволиз очень сильно зависит от природы растворителя.
Известно, что в воде сольволиз KN03 не имеет место. В жидкой,
H2SO4 как растворителе он протекает по реакции
KN03 + H2S04^rHN03 + KHS04.
Произведение растворимости
Рассмотрим систему, состоящую из осадка труднорастворимого-'
электролита и насыщенного раствора над ним, например:
AgCl^:Ag+ + Cl-. (XI)
Из-за плохой растворимости концентрации AgCl в растворе очень.
мала, поэтому ионизацию молекулы AgCl можно считать полной.
Между насыщенным раствором и осадком устанавливается
динамическое равновесие: справа налево идет процесс перехода
молекул из насыщенного раствора в осадок, слева направо —
обратный процесс (растворение осадка с образованием ионов в
растворе).
Скорость Vu с которой молекулы AgCl покидают осадок,
пропорциональна величине поверхности осадка S:
t>i=tfi-S,
где коэффициент пропорциональности К\ — константа при
постоянной температуре.
27&
Скорость v2, с которой молекулы AgCl осаждаются из
раствора, пропорциональна как величине поверхности осадка, так и
(в соответствии с основным законом химической кинетики)
произведению активностей ионов Ag+ и С1~:
v2^KrS-aAg+-acl-.
В состоянии динамического равновесия
t>i = t>2,
или
Кг • 5 = К2 • 5 • aAg+ ас1_.
Разделив обе части полученного равенства на K2'S, будем иметь
Постоянную величину слева обозначают ПР и называют
произведением активности (растворимости), так что
nP = aAg+.acl--.
Для труднорастворимого осадка AnBmz2lnAm+ + mBn~
ПР = а^+-я^_. (7.68)
Правило ПР формулируется следующим образом: для
насыщенного раствора труднорастворимого электролита произведение
активности ионов, возведенных в степень их стехиометрических
коэффициентов, есть величина постоянная при данной температуре.
Величины ПР многих веществ определены и сведены ,в таблицы
(см. справочники по аналитической химии).
Выражению (7.68) можно придать другой вид:
ПР = C*m+YV""C^-r-Y- 'mm = [Ат+]п[Вп-]т-Чп±т = ПР'.у±+т>
где ПР' — произведение растворимости, выраженное через
концентрации С (моль/л). В отсутствие сильных электролитов можно
считать y±~ 1,
ПР^=ПР'. (7.69)
Тогда для насыщенного раствора AgCl, характеризующегося
равенством
CAg+ = CCl- (Г'ИОН/Л),
ПР = С2 = S2,
где S — растворимость AgCl (моль/л).
Постоянство величины ПР не означает постоянства
концентрации (активностей) отдельных ионов. Концентрацию каждого из
274
них можно изменить. Можно увеличить концентрацию ионов С1~
в насыщенном растворе AgCl, добавив, например, НС1. Это
сейчас же отразится на состоянии равновесия (XI). Оно нарушится,
сдвинется справа налево, так как избыточное по сравнению с
равновесным количество ионов С1" приведет к увеличению скорости
реакции осаждения (Cl~+Ag+-^AgCl). Вновь установившееся
равновесие будет по-прежнему характеризоваться величиной ПРГ
изменятся лишь равновесные концентрации. Концентрация ионов
Ag+ будет меньше, а ионов О- больше по сравнению с прежним
состоянием равновесия. Сдвиг равновесия справа налево
сопровождается осаждением осадка. Условием осаждения является тот факт,
что произведение активностей (концентрации) оказалось при
добавлении в раствор НС1 больше величин ПР (nPAgci— 1,6-10~10):
«Ag+^Cl->nP- (7'70>
Напротив, если понизить концентрацию, связав, например, ион Ag+
в слабоионизируемый ион [Ag(NH3)2]+, тогда осадок в
соответствии с принципом Ле Шателье начнет растворяться. Условием
растворения осадка является соотношение
aAg+*aci-<np- (7.71)
Правило ПР находит важное применение в химии для разделения
веществ, их очистки и синтеза. По величинам ПР можно найти
растворимость веществ, уменьшить (увеличить) концентрацию
нужного для той или иной цели иона и т. п.
Пример. Вычислить, во сколько раз изменится
растворимость Agl в чистой воде по сравнению с его растворимостью в
0,1 М растворе KI. Произведение растворимости nPAgi=l,5- Ю-16.
В воде
[Ag+] = [I"] - 5AgI = l/? = J- l/ПР.
Из-за малости величины ПР можно принять у± = 1. Тогда SAgi =
= УПР = 1/1,5- Ю-16 = 1,24. Ю-8 (моль/л).
Примем растворимость Agl в растворе за X (моль/л).
Концентрация Ag+ KI также равна X (г-ион/л); концентрация 1~ будет
складываться из величины X и йод-ионов KI, т. е.
[1-]=Х + 0,1;
ПР = Сх_. Yl--CAg+.YAg+ = [I-] [Ag+] Y± = (X + 0,1)X.Y2±.
Ионная сила раствора определяется практически ионами KI:
/ = 1/2(0,1 +0,1) =0,1; Y±=0,78.
Следовательно,
ПР-(Х + 0,1)Х.(0,78)2.
27S
Исключив малую величину X из двучлена (Х + 0,1), будем иметь
ПР = О, IX (0.78)2 = 0,1 -5Agi (0,78)2,
SAgl = 1-5-10"'6 = 1'5-10"'5 = 2,5-10-».
ё 0,1 (0,78)2 (0,78)2
Откуда *&- = 1>24-10"8 = 5.10* раз.
SKi 2,5-Ю-15
§ 6. НЕВОДНЫЕ РАСТВОРИТЕЛИ
По способности участвовать в протолитической реакции типа
НА + *М ^Г НМ, + А- (XII)
растворители разделяются на два класса: протолитические и апро-
тонные.
Протолитические растворители вступают в реакцию (XII).
Апротонные растворители в таком взаимодействии
участвовать не могут. Если в них имеет место кислотно-основное
взаимодействие, то оно осуществляется без участия растворителя. К
таким растворителям относятся бензол, толуол, четыреххлористый
углерод и др.
Разделение на протолитические и апротонные растворители
не следует понимать абсолютно. Известно, например, что бензол
во фтористом водороде в присутствии BF3 проявляет основные
свойства, а в растворе амида натрия NaNFb — кислотные. Прото-
литическая активность растворителя зависит, в свою очередь, от
природы и свойств растворенного вещества, от способности его
сольватироваться. На протолитическую активность растворителя
оказывает влияние собственная ионизация растворителя.
Константы самоионизации получили название констант автопротолиза Ks*
Для некоторых раство.рителей константы Ks приведены в табл. 7.11.
Протолитические растворители могут участвовать во
взаимодействии с растворенным веществом, выполняя либо функцию
кислоты, либо функцию основания, либо одновременно способны
быть и кислотой и основанием. По этой причине их разделяют на
протогенные — (кислые) растворители, протофильные — основные
растворители и амфотерные (амфитропные) растворители.
Указанное деление является также относительным. В зависимости от
природы растворенного вещества протолитическая функция
растворителя может существенно измениться. Вода по отношению к
уксусной кислоте — протофильный растворитель, ибо сродство к
протону у Н20 больше, чем в СН3СООН, по отношению же к жидкому
NH3 вода является протогенным растворителем, поскольку
средство к протону у NH3 больше, чем в воде.
Протолитические растворители обладают способностью
изменять силу кислот относительно их силы в водной среде. По этой
276
Таблица 7.11
Константы автопротолиза (Ks) t° — 20°С
Растворитель
Серная кислота
Муравьиная кислота
Уксусная кислота
Вода
Метиловый спирт
Этиловый спирт
Аммиак (—50°С)
Уравнение самоионизации
2H2S04^H,SO+ + HS07
2НСООН ц! НСООН+ + НСОО
2СН3СООН ^ СН3СООН+ + СНзСОО-
2Н20 ^ Н30+ + ОН-
2СН,ОН ^ СН3ОН+ + СН30-
2С2Н5ОН ^± С2Н5ОН+ + С2НбО~
2NH3^1NH+ + NH-
Ks
ыо-6
5-Ю-7
2,5-10"13
Ю-14
2- Ю-17
8. Ю-20
з. ю~32
способности они разделяются на дифференцирующие и
нивелирующие растворители. Дифференцирующие изменяют
кислотно-основную функцию растворенных веществ, расчленяют их прежний
уровень кислотности, дифференцируя по силе по сравнению с
водной средой, нивелирующие, наоборот, выравнивают различные по
силе растворенные вещества.
Кислые растворители, как правило, дифференцируют силу
кислот и нивелируют силу оснований. Например, в водной среде
все минеральные .кислоты характеризуются одинаковой
способностью к диссоциации, т. е., все являются сильными кислотами.
Более кислый растворитель, чем вода, ледяная уксусная кислота
дифференцирует их по силе.
В среде СН3СООН сила минеральных кислот уменьшается в
ряду
НС104 > HBr > HC1 > HN03.
При этом уксусная кислота выполняет функцию основания. Вместе
с тем, будучи достаточно активным донором протона, СНзСООН
способствует усилению диссоциации веществ, выполняющих
функцию основания. В ее среде многие основания будут нивелированы
по силе: по этой же причине в среде уксусной кислоты круг
веществ с основной функцией будет расширяться, а круг веществ
с кислотной функцией — сужаться.
В среде протофильных растворителей (аммиак, пиридин и т. п.),
в которых акцепторные свойства к протону выражены сильнее, чем
в Н20, будет наблюдаться обратная картина. Они будут
дифференцировать основания и нивелировать по силе кислоты,
увеличивая круг веществ с кислотной функцией и снижая круг веществ
с основной функцией относительно их водных растворов.
277
Амфотерные растворители по своему действию на
кислотно-основную функцию вещества мало отличаются от их отношения в
воде. К этой группе растворителей относятся спирты, фенолы и
некоторые другие.
Дифференцирующее действие в основном зависит от
собственной ионизации растворителя и от способности растворенного
вещества образовать в данной среде ассоциаты, так как последнее
приводит к снижению концентрации ионов.
В случае близкой собственной ионизирующей функции
растворителей дифференцирующее действие будет усиливаться в
растворителе с меньшей диэлектрической проницаемостью. Например,
из двух протогенных растворителей с близкими
кислотно-основными свойствами — муравьиной (е = 57) и уксусной (е = 6), последняя
обладает большей дифференцирующей силой. Дифференцирующее
действие растворителей используется в практике, оно позволяет
подобрать условия разделения веществ, трудно разделимых в
водной среде.
Дифференцирующее действие существенно зависит также от
способности его молекул образовывать водородные связи с
молекулами растворенного вещества, так как характер и прочность
этих связей оказывают влияние на диссоциацию растворенного
вещества.
Нивелирующее действие растворителей объясняется различной
сольватацией молекул растворенного вещества, приводящей к
выравниванию их кислотно-основной функции. В растворителях с
низкой е это выравнивание идет в направлении снижения их
кислотно-основной функции, а в растворителях с большой г — к их
увеличению.
В отличие от протолитичеоких апротонные растворители мало
изучены. Как уже указывалось, они не вступают в протолитиче-
скую реакцию с растворенными веществами. В их средах могут
быть установлены лишь относительные константы диссоциации
растворенных веществ. Все апротонные растворители
характеризуются малыми значениями диэлектрической проницаемости, по
этой причине в них сильно развиты процессы ассоциации.
Способность растворителей вступать в протолитическую
реакцию не исчерпывает всех видов взаимодействий их с
растворенным веществом. Приведенная классификация может служить
свидетельством того, что протолитический тип взаимодействия нашел
в практике широкое распространение.
В неводных растворах из-за малых диэлектрических проницае-
мостей е растворителей процессы сольватации осуществляются
либо за счет сил водородной связи, либо за счет образования
продуктов по донорно-акцепторному механизму. В них чрезвычайно
развиты процессы ассоциации. Сильные в водной среде
электролиты становятся слабыми. Например, LiBr в жидком S02 обладает
/Сдисс = 3-10-5, а в спирте его /Сдисс = 7- Ю-'3. Совсем по-иному
протекают реакции обмена. Например, при взаимодействии AgCl и
278
Ba(N03)2 © жидком аммиаке выпадает осадок ВаС12, в среде
безводной уксусной кислоты можно осадить CuS04 и т. п. Некоторые
реакции в неводных средах приведены ниже:
2AgCl + Ba (N03)2 -^-3-> | ВаС12 + 2AgNOs,
жидкий
H2S04 + Си (N03)2 * \ CuS04 + 2HN03,
изопропил
СНдСООК + НС1 —>. \ КС1 + СНдСООН.
спирт
Последняя реакция лежит в основе количественного определения
ионов калия.
Неводные растворители применяются во всех тех случаях, когда
необходимо: повысить (понизить) растворимость вещества, не
растворимого (растворимого) в воде, усилить или ослабить силу
электролита, или, если это необходимо, перевести вещество из
неонизированного состояния в ионизированное и т. п.
В неводных средах можно осуществить принципиально новые
реакции. Благодаря неводным растворителям значительно
расширилась, например, химия производных фтора, появилась
возможность получить производные таких неустойчивых в воде частиц,
как SOi+> NO 2". Исследования неводных растворителей сыграло
большую роль в формировании современной физико-химической
теории растворов.
Вопросы и упражнения
1. Объясните, что означает термин «идеальный раствор».
2. Раствор KN03 содержит 192,6 г соли в 1 л. Плотность раствора
1,143 г-см~3. Рассчитать концентрации: моляльную, в вес.% и мольных долях.
3. Объясните содержание законов Рауля и Генри и границы их применения.
4. Назовите способы определения молекулярных весов нелетучих
растворенных веществ.
5. 68,4 г глюкозы (М=342) растворено в 1000 г воды. Рассчитать давление
пара, осмотическое давление, точку замерзания. Плотность раствора при
20°С = 1,024 г-см-3. Давление пара воды 17,633 мм рт. ст.
6. Что означает термин активность компонентов?
7. Как можно рассчитать коэффициенты активности ионов и каков их
физический смысл?
8. Вычислите ионную силу раствора, содержащего в 1000 г растворителя
0,005 молей A12(S04)3; 0,005 молей KN03.
9. Назовите границы применимости закона действующих масс в растворах
электролитов и его значение в химии водных растворов.
10. Как вы понимаете явление гидратации (сольватации) в растворах
электролитов?
11. Объясните влияние свойств растворителя на процесс сольватации в
растворах.
12. Что принято понимать под термином «константа автопротолиза»?
13. Объясните термин «произведение растворимости».
279
14. Определите константу ионизации одноосновной кислоты, если степень
ионизации ее 0,2 М раствора равна 0,95%.
Отв.: 1,8Ы0~5.
15. Как изменится степень ионизации 0,2 М СН3СООН при разбавлении ее
водой в 4 раза?
16. Вычислить степень гидролиза 0,1 н. р-ра KCN; /Chcn = 7,2-10_10
Отв.: 1,18%.
17. Определите, во сколько раз растворимость BaS04 в 0,012 М растворе
NaCl больше растворимости в чистой воде. ПРВа$0 = 1,1-Ю-10.
18. Определите растворимость РЬСЬ в чистой воде. ПРрьс1 = 2,4-10~4.
19. Рассчитайте, чему равна активность ионов водорода в растворе,
содержащем в 1 л 0,1 моля HCN и 0,05 моля NaCl. Используйте данные табл. 7.7.
/(hcn = 7,2.10-10.
ГЛАВА 8
КОЛЛОИДНОЕ СОСТОЯНИЕ ВЕЩЕСТВА
§ 1. ПОНЯТИЕ О КОЛЛОИДНОМ
СОСТОЯНИИ ВЕЩЕСТВА
И ОСНОВНЫЕ ВИДЫ КОЛЛОИДНЫХ СИСТЕМ
Впервые термин и понятие «коллоиды» были сформулированы
Грэмом (1861 г.), с появлением трудов которого обычно
связывают возникновение коллоидной химии. В России
основоположником коллоидной химии стал киевский профессор И. Г. Борщев,
работа которого «О свойствах и строении коллоидов,
участвующих в образовании растительных и животных организмов» (1869 г.)
дала направление исследованию сложного состава коллоидных
частиц и роли среды для сцепления частиц. Но еще задолго до
того, как коллоидная химия стала разделом предмета химии,
Берцеллиус (1824—1834), Фарадей (1857 г.), Сельми (1845 г.)
рассматривали коллоидные растворы как псевдорастворы, в
которых «распределение вещества в жидкой среде не сопровождается
разбавлением до молекул».
Монолитное вещество и раздробленное вещество (мелкий
порошок, мелкие пузырьки, мелкие капельки) есть два разных
состояния одного и того же вещества. Монолитное вещество
представляет собой непрерывное, однородное (гомогенное) состояние
вещества. Раздробленное вещество является примером коллоидного
состояния, так как всякое коллоидное состояние представляет
собой дисперсию (раздробление) одного вещества (дисперсная
фаза) в другом (дисперсионная среда). Для порошков
дисперсионной средой могут быть воздух, газы, жидкости, твердые лавы.
Раздробленное вещество называют дисперсной фазой. Каждая
частица этой фазы называется коллоидной частицей. Она состоит
из сотен или тысяч молекул вещества, обладает поверхностью
раздела с окружающей ее средой и находится .в
физико-химическом взаимодействии с нею. Окружающая среда называется
дисперсионной средой.
Таким образом, коллоидное состояние вещества проявляется
в смеси, или в коллоидной системе, когда вещество раздроблено
в какой-либо непрерывной среде. В отличие от однородного
состояния чистых веществ и истинных растворов коллоидная
система характеризуется неоднородностью, или гетерофазностью.
Дисперсная коллоидная фаза может быть охарактеризована
удельной поверхностью 5УД, т. е. межфазной поверхностью,
приходящейся на единицу объема дисперсной фазы:
281
где Si 2 — суммарная поверхность между фазами 1 и 2; V\ — сум-
марный объем дисперсной фазы.
В любой дисперсной системе, в том числе молекулярно-дисперс-
ной, можно определить средний поперечник частиц а, для
сферических частиц — диаметр dy для частиц, имеющих форму куба, —
ребро куба / и вычислить величину
D-i-.
а
которая называется дисперсностью.
• о о
а 5 6
о П
□ □
I Молекулярно -дисперсные
'^ системы
Микрогетерогенные
системы
ГруЬодисперсные-
системы
Рис. 8.2. Зависимость удельной
поверхности системы от размеров ее
частиц
Рис. 8.1. Соотношение
размеров коллоидных
частиц и молекул:
а — молекула водорода
(d=0,l ммк); б —
молекула хлороформа (d=
= 0,8 ммк); в —
молекула гемоглобина (4=
=2,5 ммк); г — частица
золотого золя (d =
= 1 ммк); д — частица
золотого золя (d —
= 3 ммк); е —
частица золотого золя
(<2= 10 ммк); ж —
частица золотого золя
(<2=15 ммк); з —
оседающие частицы золота
Дисперсность и удельная поверхность — величины
взаимосвязанные:
(8Л>
где К — коэффициент, зависящий от формы частицы. Согласно
формуле (8.1) удельная поверхность прямо пропорциональна дис-
282
перенос™ или обратно пропорциональна линейному размеру
частиц.
К коллоидным системам относятся системы, у которых средний
поперечник частиц а находится в пределах от 1 до 100 ммк
(10~7—10~5 см), а дисперсность в пределах 107—105 см-1.
Отдельные молекулы веществ обычно имеют диаметр порядка
10~8 см. Поэтому система с дисперсностью 108 см-1 называется
молекулярно-дисперсной, но не коллоидной. Соотношение размеров
коллоидных частиц и молекул приведено на рис. 8.1. Наименьшая
дисперсность частиц в коллоидных растворах 105 см"1 — это
условная граница, за которой существует большой жласс
микрогетерогенных и грубодисперсных систем (рис. 8.2), имеющих огромное
практическое значение: порошки, суспензии, эмульсии, пены и ряд
других.
Говоря о размерах коллоидных частиц, следует иметь в виду,
что в коллоидных системах частицы редко бывают одного
размера. Монодисперсные системы можно приготовить, пользуясь
специальными приемами. Большинство же коллоидных систем
полидисперсно, т. е. содержат частицы самых разных размеров.
Закономерности взаимодействия между коллоидными частицами
и дисперсной средой относятся к химическим формам движения.
Взаимодействие происходит на поверхности раздела путем
адсорбции и поверхностных химических реакций.
Основные виды коллоидных систем рассматриваются по разным
признакам: по агрегатному состоянию дисперсной фазы и
дисперсионной среды; по степени дисперсности; по интенсивности
химического взаимодействия на поверхности раздела в коллоидной
системе.
Классификация по агрегатному состоянию фаз, представленная
в табл. 8.1, общепризнана, так как она весьма удобна для
рассмотрения всего многообразия коллоидных систем.
Таблица 8.1
Агрегатное
состояние
дисперсионной
среды
Газ
Жидкое
Твердое
Агрегатное состояние дисперсной фазы
газ
коллоидная система
невозможна
пены
включения газов в
твердых телах
(твердые пены)
жидкое
туманы
эмульсии
жидкие включения в
твердых телах
(твердые эмульсии)
твердое
дымы, пыли
суспензии
твердые включения в
твердых телах;
сплавы
Условной является классификация по степени дисперсности,
согласно которой все коллоидные системы можно разделить на
три вида:
283
1. Растворы высокомолекулярных соединений типа гемоглобина
(рис. 8.1), молекулы которого обладают дисперсностью 107 см-1.
Такие растворы находятся на границе между истинными
растворами и коллоидными растворами. Наличие макромолекул в
растворах, несмотря на однофазность (гомогенность) системы, придает
ей некоторые свойства коллоидных систем.
2. Коллоидные растворы с частицами, состоящими из агрегатов
молекул, дисперсностью 107—105 см-1.
3. Микрогетерогенные системы со степенью дисперсности
меньше 105 см-1.
Коллоидные растворы можно подразделить на два вида по
интенсивности взаимодействия дисперсной фазы с жидкой
дисперсионной средой:
1. Необратимые коллоидные растворы — лиофобные
(по-гречески лиос — жидкость, фобо — ненавижу), в которых
дисперсионная фаза неспособна сама по себе растворяться
(взаимодействовать) с дисперсионной средой.
2. Обратимые коллоидные растворы — лиофильные
(по-гречески фило — люблю), в которых дисперсная фаза молекулярно
взаимодействует с молекулами, ионами дисперсионной среды.
Необратимые коллоидные растворы обычно содержат
невысокие концентрации дисперсной фазы, при введении электролитов
дисперсная фаза выпадает в осадок (коагулирует) и почти не
соосаждает с собою частиц дисперсионной среды.
Обратимые коллоидные растворы могут содержать высокие
концентрации дисперсной фазы, мало чувствительны к
электролитам, но способны при достаточно большом количестве
электролита к «высаливанию» (коагуляции) с образованием весьма
объемных, вязких осадков, содержащих большое количество
дисперсионной среды. Обычно таким поведением обладают растворы
высокомолекулярных веществ.
Наряду с обратимыми и необратимыми коллоидными
растворами существуют системы, которые проявляют лиофильно-лиофоб-
ные свойства. Это коллоидные растворы гидроокисей некоторых
металлов (А1(ОН)3, Fe(OH)3 и др.). Частицы гидроокисей в
водной среде гидратированы, и системы можно рассматривать как
лиофильные. Они дают высокие концентрации частиц в растворе.
Раствор мало чувствителен « электролитам, но при определенных
условиях частицы коагулируют без захвата дисперсионной среды,
проявляя поведение лиофобных систем. Когда же эти системы
коагулируют как лиофильные, образуя объемный осадок с
захватом дисперсионной среды (воды), предполагают, что дисперсная
фаза находится в виде макромолекул типа
ОН ОН
НО — А1 — О — А1 — О — А1 — О — А1 — О— ...— А1 — ОН
I ! I
ОН ОН ОН
284
В коллоидной химии все системы, отвечающие коллоидной
степени дисперсности, принято называть золями. В аэрозолях
дисперсионной средой является газ. В лиозолях дисперсионная
среда — жидкость. Гидрозолями или просто золями обычно называют
водные коллоидные растворы.
Гели — это коллоидные системы, образовавшиеся из золей в
результате потери ими устойчивости ('коагуляции). Лиогель, или
просто гель, — это однородная система, в которой частицы
связаны друг с другом за счет молекулярных сил и образуют
пространственные сетки с пустотами, равномерно распределенными по
всему объему, в которых находится жидкая дисперсионная среда.
В отличие от лиогелей различают .каогели, в которых выпадающий
осадок отделяется от дисперсионной среды.
§ 2. МЕТОДЫ ПОЛУЧЕНИЯ ВЫСОКОДИСПЕРСНЫХ
И КОЛЛОИДНЫХ СИСТЕМ
Коллоидные растворы могут быть получены двумя
противоположными путями: 1) раздроблением, или диспергированием,
грубых частиц вещества на более мелкие вплоть до коллоидных:
размеров; 2) агрегацией молекул
или ионов в более крупные
частицы. Первые получили название
методов диспергирования.
Вторые называются
конденсационными.
Диспергирование можно
проводить механическим
измельчением, электрическим
распылением или растворением уже
существующей, но осажденной
дисперсной фазы. Измельчая
вещества дробилками,
работающими по принципу раздавливания
жерновами, шаровыми
мельницами, можно получить высокую
степень дисперсности.
Ультразвуковые волны большой мощности
вызывают эффект
диспергирования вещества. Таким образом
Рис. 8.3 Схема электрического метода
диспергирования металлов
можно получить коллоидные растворы легкоплавких сплавов и
металлов в органических жидкостях, разорвать молекулы
высокополимерных веществ (желатины, каучука, агара и т. д.), получить
устойчивые эмульсии несмешивающихся жидкостей.
285
Электрический способ получения гидрозолей металлов
основан на возникновении электрической дуги от источника
постоянного тока между электродами из диспергируемого металла.
Схема электрического метода диспергирования представлена на
рис. 8.3.
Образование коллоидной системы растворением уже
измельченного вещества называется пептизацией. Пептизация гелей и
рыхлых осадков происходит обычно при действии на них
некоторых веществ, которые способны адсорбироваться на
поверхности коллоидных частиц и таким путем сообщить им сродство к
дисперсионной среде.
Примером образования высокодисперсных растворов
является лиофильное растворение высокомолекулярных веществ:
белков в воде, каучука в бензоле, целлюлозы в эфире и т. д.
Конденсационные методы получения коллоидных растворов
носят как физический, так и химический характер. Физический
метод основан главным образом на явлении конденсации паров.
Конденсационные химические методы получения золей можно
известным образом систематизировать, но всегда следует
помнить, что для приготовления золей недостаточно знания общих
принципов.
Методом замены растворителя получают золи, когда
истинный раствор вещества приливают понемногу к жидкости,
которая смешивается с растворителем раствора, но в которой
растворенное вещество ничтожно мало растворимо и поэтому
выделяется в виде коллоидной фазы. Например, так можно получить
.гидрозоли серы, фосфора, мастики, канифоли, вливая спиртовой
раствор этих веществ в воду.
Окислительно-восстановительный метод заключается в том,
что в истинных растворах в присутствии вещества
стабилизатора происходит окисление или восстановление молекул вещества
€ одновременной конденсацией продукта реакции в коллоидное
состояние вещества. Например, по реакции
2АиС14 + ЗНСНО + 11КОН = 2Аи + ЗНСООК + 8КС1 + 8Н20
происходит образование золя золота. По реакции
2H2S + S02 = 3S + 2Н20
в водной среде можно получить коллоидное состояние серы.
Методом гидролиза чаще всего пользуются для получения
золей гидроокисей металлов. Например, золь гидрата окиси
железа легко возникает, когда раствор хлорного железа приливают
в кипящую воду. Объясняется это следующим образом:
холодный водный раствор хлористого железа содержит слабо
диссоциированные продукты неполного гидролиза трехвалентного
железа Fe(OH)2+, Fe(OH)2~, которые при разбавлении в кипящей
воде гидролизуются полностью с образованием коллоидного
состояния Fe(OH)3.
286
Метод реакции обмена между ионами веществ связан с
образованием малорастворимых соединений, которые в очень
разбавленных растворах выделяются в коллоидном состоянии. Так
можно получить золи AgCl, берлинской лазури и т. п. Размеры
коллоидных частиц, полученных в результате реакций двойного
обмена, зависят от концентрации реагирующих веществ.
При очень высоких и очень низких концентрациях
реагирующих веществ получаются обычно высокодисперсные коллоидные
системы. В первом случае это объясняется возникновением
одновременно очень большого числа центров кристаллизации
(зародышевых центров), что связано с расходом всего реагирующего
вещества. В случае низких концентраций весь возможный
избыток вещества расходуется на возникновение сравнительно
немногочисленных центров кристаллизации, и дальнейший рост
частиц тем самым исчерпывается. При средних концентрациях
реагирующих веществ получаются грубодисперсные частицы,
выпадающие в осадок.
Рассмотрим реакцию двойного обмена с получением
коллоидного состояния берлинской лазури:
4FeCl3 + ЗК4 [Fe (CN)e] = Fe4 [Fe (CN)e]3 + 12 KC1.
1. Если к 5 мл 0,005 н. раствора хлорного железа прилить
5 мл 0,005 н. раствора желтой кровяной соли и затем разбавить
этот раствор 50 мл дистиллированной воды, то получается
прозрачный коллоидный раствор берлинской лазури.
2. Если к 5 мл 0,1 н. раствора FeCl3 прилить 5 мл 0,1 н.
раствора K4[Fe(CN)e], то после разбавления 50 мл воды
наблюдается выпадение осадка берлинской лазури из мутного
раствора. Более сильное разбавление тоже не приводит к
растворению, пептизации осадка.
3. Если к 5 мл насыщенного раствора FeCl3 добавить 5 мл
насыщенного раствора желтой кровяной соли, то образуется
студнеобразный осадок берлинской лазури, который
разбавлением в воде можно перевести в коллоидное состояние, в золь.
В этом случае получается устойчивый золь берлинской лазури.
§ 3. МИЦЕЛЛЫ КОЛЛОИДНЫХ РАСТВОРОВ
Отдельные нейтральные частицы коллоидных систем
называются мицеллами. Мицеллу можно охарактеризовать
размерами (дисперсностью), строением и свойствами, которые зависят
от природы вещества, от метода получения коллоидного
состояния его, от условий окружающей среды.
Рассмотрим общие черты строения мицеллы на примере
йодистого серебра (рис. 8.4), коллоидное состояние которого
получается реакцией обмена в водном растворе добавлением
раствора нитрата серебра к избытку раствора йодида калия:
287
m AgN03 -i- (m + л) KI =■ {m [Agl] nl~ (n — x) K+}*"• xK+ + mKN03,
где m — количество молекул йодида серебра в кристаллике; п—
количество адсорбированных ионов йода на кристаллике; п—х—
количество ионов калия в адсорбированном слое кристаллика;
х — заряд частицы (в данном примере — отрицательный).
Заряженная коллоидная
частица называется гранулой.
Центральная часть ее, состоящая из
нейтральных кристалликов, или
одного кристаллика, называется
ядром. В коллоидном растворе
вокруг гранул находится
диффузионный, подвижный слой
растворителя, в котором сосредото-
Рис. 8.4. Схема строения мицеллы
золя йодида серебра
1 — ядро; 2 — гранула; 3 —
мицелла; 4 — адсорбционный слой; 5 —
диффузионный слой
чиваются ионы противоположного знака, компенсирующие заряд
гранулы.
Электронейтральная коллоидная частица, образованная
гранулой и ее диффузионным слоем, называется мицеллой.
Химический состав мицеллы можно записать так:
{т [Agl] • til- (n — х) К+}*~ • х. К+
ядро •
■адсорбционный-
слой
гранула -
мицелла
• диффузный
слой
В зависимости от концентраций исходных растворов и условий
числовые значения коэффициентов m, n и х могут изменяться в
широких пределах.
Ионы электролита адсорбируются обычно в различной
степени на поверхности ядра мицеллы. Правила избирательной
адсорбции ионов были сформулированы Песковым и Фаянсом.
Согласно первому правилу на всякой твердой поверхности
преимущественно будут адсорбироваться ионы, имеющие общую
с данной поверхностью атомную группировку (или изоморфную
с ней). Поэтому, например, на осадке йодида серебра,
полученном в реакции AgN03 + KI, будут преимущественно адсорбиро-
288
ваться ионы Ag+, либо ионы I" (а также ионы С1+, Вг+), но не
ионы К+ или N07.
В зависимости от размеров ионов и степени их гидратиро-
ванности адсорбционная способность родственных ионов
убывает, например, в такой последовательности:
Cs+ > Rb+ >K+ > Na+ > Li+
Ba2+>Sr2+>Ca2+>Mg2+ (для катионов)
CNs- > I- > Bi~> CI- (для анионов;
Эти ряды называются лиотропными. Они показывают, что
большему атомному весу соответствует большая адсорбционная
способность. Катионы различной валентности по их возрастающей
адсорбционной способности также можно расположить в ряд
К+ < Са2+ < Ag3+ < Th4+,
что подтверждает общее положение: ионы с большей
валентностью адсорбируются лучше.
Гранулы минеральных веществ и высокомолекулярных
органических соединений несут на себе электрический заряд,
который образован в одних случаях в результате избирательной
адсорбции, а в других — за счет диссоциации собственных ионо-
генных групп.
Например, заряд частиц кремниевой кислоты или силиката
натрия в водной среде может быть образован
электрохимической диссоциацией этих веществ по схеме
(Н.О)
[(т+п) Na2Si03] ^{[mNa2Si03nNaSOr]rt--/zNa+}°.
Согласно второму правилу данный ион будет адсорбироваться
твердой поверхностью, если заряд поверхности противоположен
по знаку заряду иона и если образующееся поверхностное
соединение мало растворимо. Правила избирательной адсорбции
позволяют решать вопрос о строении мицелл при ее
образовании и при изменении состава электролита в коллоидном
растворе.
Например, перезарядка отрицательного золя сульфида
мышьяка вызывается многовалентными катионами, в частности
хлорным железом. Схематично эту реакцию между мицеллой и
ионами можно представить уравнением
{т [ As2S3]n S2- • 2 (л— х) Н+р~ • 2х+ Н -+ nFe (ОН) С12 ->
-> п Н20 + я НС1 + {т [As2S3] • rcFeS+ (n — у) С1~}у+ • у С1~.
Этот же пример иллюстрирует и то, что наряду с
избирательной мицеллярной адсорбцией ионов имеет место обменная
адсорбция. В этом случае одни ионы, которые сильнее адсорби-
10 Общая химия
28а
руются, вытесняют в раствор из гранулы другие ионы.
Обменная адсорбция носит специфический характер, который зависит
от природы твердой фазы. Фазы, которые способны к обмену
катионами, называются катионитами, а те, которые способны к
обмену анионами, называются анионитами.
Катиониты и аниониты имеют широкое практическое
применение в извлечении из растворов редких и рассеянных
элементов из сточных вод, электролитических ванн и т. д., в процессе
умягчения воды — очистки ее от ионов кальция. Схематически
умягчение воды с помощью алюмосиликатного поглотителя пер-
мутита Na20-Al203-3Si02-2H20, который выступает в роли ка-
тионита, можно изобразить следующим образом:
{m [H4Al2Si3032]2"*-. 2mNa+}° + т Са2+ -\ m S042
2—
-> {т [H4Al2Si3012]2m-. т Са2+}° + 2mNa+ + m SOf.
Широко известны адсорбционные свойства угля. Химизм
избирательной и обменной адсорбции на угле объяснен в трудах
наших ученых А. Н. Фрумкина и Н. А. Шилова. Согласно их
теории на поверхности угольных частиц адсорбируется
кислород воздуха, который вступает в химическую связь и образует
поверхностные окислы со степенью окисления по кислороду 2
и 3. В водной среде эти окислы гидролизуются и на поверхности
угольных частиц образуются кислотные (—СООН) и гидро-
ксильные ( = С(ОН2)) группы, которые в первом случае
водородными ионами, а во втором случае гидроксильными группами
могут участвовать в ионном обмене:
т(С)х\ —<Z 1 I x¥&
m(C)yl=C/
уон-
§ 4. ЯВЛЕНИЕ КОАГУЛЯЦИИ
И СТАБИЛИЗАЦИИ КОЛЛОИДОВ
Коагуляция — это процесс слипания частиц в коллоидных
системах с образованием более крупных агрегатов. Внешне
коагуляция проявляется в помутнении золя, в изменении окраски
золя, в выпадении твердой фазы в осадок или в образовании
студнеобразной массы. Коагуляция может быть вызвана
самыми разнообразными причинами и прежде всего добавкой
электролита.
290
Минимальное количество любого электролита, выраженное в
миллимолях (1 ммоль = 10~6 моля), которое вызывает
коагуляцию 1 л определенного золя, называется порогом коагуляции /.
Величина / зависит от природы, концентрации золя и от
природы электролита.
Правила коагуляции можно сформулировать следующим
образом:
1. Коагуляцию вызывает ион, заряженный противоположно
грануле золя.
2. Чем больше валентность коагулирующего иона, тем
сильнее его коагулирующее действие (меньше порог коагуляции).
Количественно валентность Z коагулирующего иона и /
связаны соотношением
/ = <х—, (8.2)
где а — множитель. Отсюда соотношение порогов коагуляции
для oaho-(Zi), AByx-(Z2) и трехвалентных (Z3) ионов
соответствует соотношению чисел — сотен, десятков, единиц:
M = /z.:/z.= ^:^:^ = l:^:-^ = 730: 11:1.
Z\ Z% l\ 64 729
Однако надо иметь в виду, что ионы водорода ц ионы
некоторых тяжелых металлов, многие органические катионы
(алкалоиды, красители) проявляют большое коагулирующее
действие, несовместимое с правилом.
Пользуясь табл. 8.2, можно установить все правила
коагуляции нижеперечисленных золей, сопоставив величины порогов
коагуляции золей разными ионами:
{т [Fe (ОН)3] п Fe3+ . 3 (п — х) СГ}3х+ . Зх СГ,
{m[As2S3] • nHS~(n — x)H+Y~ • *Н+,
{т [Agl] . п Г (л — х) К+Г" • х К+.
Явление коагуляции сложно и многообразно. На величину
коагуляции влияет природа иона, сопутствующего
коагулирующему. В табл. 8.2, указано, что коагуляция золя сульфида
мышьяка проводилась в присутствии хлорид иона (хлоридами
металлов), а коагуляция золя йодида серебра — нитратами
металлов.
Пороги коагуляции зависят от скорости прибавления
электролита, от интенсивности перемешивания, а также могут резко
изменяться при совместном действии электролитов.
При смешении двух коллоидных растворов с противоположно
заряженными гранулами наблюдается взаимная мицеллярная
коагуляция. Между гранулами и диффузионными слоями проио
10*
291
Таблица 8.2
Свойство
Радиус иона
Энергия гидратации
Пороги коагуляции:
золь Fe(OH)3
Радиус иона
Энергия гидратации
Пороги коагуляции
As2S3 (анион СГ~)
Agl (анион NOp
^Единица
измерения
о
А
ккал/г-ион
ммоль/л
о
А
ккал/г-ион
ммоль/л
ммоль/л
Ряд анионов по степени коагулирующего действия
(пороги коагуляции)
SO2- > F~ > Cl~ > Вг~ > NO~ > I-
—
243
0,2
1,33
113
—
1,86
79
9,0
1,96
72
12,5
1,89
72
12,0
2,2
63
16
Ряды катионов по степени коагулирующего действия
(пороги коагуляции)
Li+ < Na+ < К+ < Rb+
0,78
121
58
165
0,98
98
51
140
1,33
80
49,5
136
1.49
74
—
126
Cd2+ < Sr2+ < Ba2+
1,06
375
0,65
2,4
1,27
338
0,65
2,38
1,43
312
0,69
2,26
ходят реакции обмена, нейтрализации, комплексообразования
и т. д. Такая коагуляция имеет место при определенных
соотношениях смешиваемых золей.
Большая часть органических веществ, в молекулы которых
одновременно входят и неполярная углеводородная цепь и
полярные группы (например, —ОН, —СООН, —NH2, —СНОН,
—N02, —S03H и др.)» способна к молекулярной адсорбции. Эти
вещества называются поверхностно-активными. Схема молекулы
поверхностно-активного вещества приведена на рис. 8.5.
Полярная группа этих молекул вызывает сродство молекулы
к полярной фазе (например, в воде), так как между полярными
группами более сильное межмолекулярное притяжение, а
неполярная или менее полярная углеводородная цепь ориентируется
к неполярной фазе. Таким образом, поверхностно-активные мо-
292
лекулы обладают двойным сродством, одновременно с полярной
п с неполярной фазами.
Концентрируясь, или адсорбируясь, на поверхности раздела
фаз, поверхностно-активные вещества снижают поверхностное
натяжение, уменьшают значение свободной энергии и делают
поверхность раздела фаз термодинамически более устойчивой.
Отсюда можно сделать вывод:
принцип получения устойчивых коллоидных
систем, т. е. принцип стабилизации
коллоидов, заключается в уменьшении
поверхностной энергии в результате
адсорбции поверхностно-активных веществ
на коллоидных частицах.
В противоположность поверхностно-
активным веществам существуют
поверхностно-неактивные, например
сильные электролиты, которые обычно не
понижают, а несколько повышают
поверхностное натяжение воды. Поверхностное
натяжение — это избыток энергии в
поверхностном слое, отнесенный к 1 см2
поверхности. Поверхностное натяжение
воды при 20° С равно 72,75 эрг/см2. Обычно
гидрофобные вещества (металлы,
электронные полупроводники, парафин и др.)
плохо смачиваются водой, имеют
поверхностное натяжение значительно больше,
чем у жидкостей, но способны
адсорбировать поверхностно-активные вещества
и образовывать относительно устойчивые
коллоидные растворы.
Известно, что если в определенных
отношениях смешать гидрофобную и
гидрофильную коллоидные системы, то мо-
Рис. 8.5. Схема молекулы
поверхностно-активного вещества
жет произойти увеличение устойчивости образовавшегося в смеси
коллоидного раствора, в частности по отношению к действию
электролитов. Это явление назвали защитой гидрофобной системы.
В качестве защитных коллоидов применяют
пленкообразующие вещества: желатин, декстрин, крахмал и др. Эти вещества
образуют защитные пленки на ядрах частиц и значительно
повышают пороги коагуляции. Полярные группы поверхностно-
активных веществ вступают в реакцию ионного обмена с
молекулами воды, и коллоидная система становится гидрофильной.
293
В ряде случаев поверхностно-активные вещества вызывают
пептизацию. В других случаях они препятствуют росту частиц
в процессе образования коллоидной системы.
Однако часто большие добавки полностью насыщают
адсорбционный слой на поверхности частиц, и наблюдается явление
адсорбционного снижения устойчивости системы. Для
количественной характеристики защитных свойств одного и того же
вещества были введены различные относительные числа.
Например, «золотое число» показывает количество миллиграмм
стабилизатора, которое следует добавить к 10 мл золя золота
0,0006-процентной концентрации, чтобы защитить золь от
коагуляции 1 мл 10%-ного раствора NaCl:
желатина .... 0,08 мг
казеин 0,01 мг
крахмал .... 25 мг
Эти числа условны, резко изменяются при переходе от одной
системы к другой, но имеют большое практическое значение в
технологии и анализах в производственных лабораториях.
Физическая теория устойчивости и коагуляции коллоидов
разработана Дерягиным совместно с Ландау. Согласно этой
теории между электрически заряженными гранулами в
дисперсионной среде действуют *ван-дер-ваальсовые силы
молекулярного притяжения и электростатические силы отталкивания,
препятствующие слипанию частиц. Подробно теория изложена в
специальных курсах физической и коллоидной химии.
§ 5. ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА
И ОЧИСТКА КОЛЛОИДОВ
В 1807 г. профессор Московского университета Рейс открыл
новое явление — передвижение коллоидных частиц в
электрическом поле. В кусок мокрой пластичной глины он погрузил на
близком расстоянии друг от друга две стеклянные трубки, в
которые насыпал немного кварцевого песка, налил воды до
одинакового уровня и опустил в воду металлические электроды
(рис. 8.6).
В результате пропускания постоянного тока в анодном
пространстве появилась суспензия глины, понизился уровень воды,
а в катодном пространстве уровень воды повысился. Опыт
показал, что коллоидные частицы были заряжены отрицательно, а
прилегающий к ним слой воды — положительно.
По современной терминологии движение дисперсной фазы в
электрическом поле называется электрофорезом. А явление
переноса дисперсионной среды под влиянием внешней разности
потенциалов называется электроосмосом.
Рассмотрим электрическое поле мицеллы по схеме (рис. 8.4).
Концентрация зарядов на поверхности ядра больше,
294
чем концентрация зарядов в диффузионном слое мицеллы.
Адсорбционный слой гранулы имеет малую, но определенную
толщину 60, которая соизмерима с суммой эффективных диаметров
адсорбированных ионов Г и К+. Разность потенциалов между
поверхностью ядра и поверхностью гранулы имеет
определенную величину фо и называется
потенциалом адсорбционного
слоя гранулы.
Плотность зарядов на
поверхности гранулы выше, чем
плотность зарядов-ионов в
дисперсионной среде, поэтому
существует так называемый
электрокинетический
дзета-потенциал £ между поверхностью
гранулы и условной
поверхностью мицеллы. Этот потенциал
изменчив из-за диффузионной
подвижности ионов в
диффузном слое мицеллы, и он резко
Рис. 8.6. Схема опыта
электрофореза и электроосмоса
суспензия
глины
сырая глина
изменяется, когда к 'коллоидному раствору прикладывается
внешнее электрическое поле и гранулы перемещаются с определенной
скоростью. Адсорбционный и диффузионный слой на ядре
мицеллы называют двойным электрическим слоем мицеллы. А сумма
электрических потенциалов называется потенциалом двойного
электрического слоя.
{m[AgI
пГ(п-х)К+}х~ -*К+
"и0
Фо
-Е-
II
-ф = Фо — С-
-н->
(ф_ общая разность потенциалов между фазами; <р0 — скачок
потенциалов в плотной, адсорбционной части двойного слоя;
£ —скачок потенциала в диффузионной части двойного слоя).
Как измеряют эти потенциалы практически — это большой
самостоятельный раздел предмета электрохимии.
В табл. 8.3 приведены значения потенциала двойного
электрического слоя при скорости движения частиц, близкой к нулю,
и значения электрокинетического потенциала в тех же условиях
на стекле в водных растворах электролитов различной
концентрации.
295
Таблица 8.3
Электролит
KCl
ВаС12
La(N03)3
Th(N03)4
KCl
BaCI2
La (N03)3
Th(N08)4
Концентрация электролитов,
1<Г7
1(Г6
10~б
ф-потенциал, мв
—92
—120
—89
—134
-84
—108
—64
—120
—63
—103
-56
—88
^-потенциал, мв
—95
—29
—11
—3
—38
—26
—7
Н-21
—22
—17
— 1
+ 16
моль/л
кг4
—55
—92
—38
—46
—4
—3
—0,5
Н4
Для лучшего понимания электрических характеристик мицелл
можно самостоятельно сопоставить цифры и представить
строение адсорбционных слоев стекла в различных электролитах.
Приблизительную схему строения мицеллы стекла в водном
растворе можно изобразить следующим образом:
{[т (Na2Si03) п (— SiOa")]2^ 2 (п — x) Na+}2*~ • 2x Na+.
Очистка коллоидных растворов от присутствующих в них
молекулярных и ионных примесей растворенных веществ
называется диализом. Для целей диализа используют мембраны из
животных и растительных перепонок, пленки из нитро- и ацетил-
целлюлозы, в лабораторных условиях — пленки из коллодия и
целлофана. Применяют также пергаментную бумагу и
керамические пористые материалы.
Простейшая схема диализа изображена на рис. 8.7, а.
Коллоидный раствор находится в мешочке А из полупроницаемой
мембраны, который помещен в сосуд с проточной водой. Согласно
явлению осмоса ионы из коллоидного раствора переходят в
проточную воду, а проникающие в мешочек молекулы воды
разбавляют коллоидный раствор. Мицеллы и гранулы задерживаются
мембраной. Если к диализируемому раствору приложить
электрическую разность потенциалов, то в результате
электродиализа очистка коллоидного раствора закончится за несколько часов,
тогда как в обычных условиях диализ может продолжаться
неделями.
Схема электродиализа приведена на рис. 8.7, б, где
соответствующие ионы среды передвигаются к аноду и катоду и
вымываются из коллоидного раствора, а коллоидные частицы, очи-
296
щенные от солей, задерживаются в средней части прибора
между мембранами. Для отделения дисперсионной среды от
дисперсной фазы применяется ультрафильтрация через
специальные полупроницаемые мембраны, которые задерживают кол-
Ф
J5^
iVi'i
i'i'i,'.
SL
"-'-Г
a 5
Рис. 8,7. Схема приборов для диализа и электродиализа
лоидные частицы. Для ускорения ультрафильтрации либо
создают разрежение под фильтром, либо производят давление на
фильтрующийся раствор.
§ 6. ОБЩИЕ ПРЕДСТАВЛЕНИЯ
О МИКРОГЕТЕРОГЕННЫХ СИСТЕМАХ — СУСПЕНЗИЯХ,
ЭМУЛЬСИЯХ, ПЕНАХ
Суспензиями называются микрогетерогенные системы, в
которых дисперсная фаза твердая, а дисперсионная среда жидкая.
Другими словами, суспензии представляют собой взвеси
порошков в жидкостях. В различных суспензиях размеры частиц
колеблются от 10~6 до 10~2 см.
В красках дисперсионной средой является олифа, а
дисперсной фазой твердые пигменты (окись цинка, окись железа
и т. д.). Стабилизацию суспензий можно осуществлять
молекулами мыла или молекулами поверхностно-активного полимера.
Концентрированные суспензии называются пастами.
В эмульсиях дисперсная фаза и дисперсионная среда жидкие.
Степень дисперсности обычно велика: 103—105 см"1. Одна из
фаз чаще всего вода. Другой фазой может быть любая
органическая жидкость, не смешивающаяся с водой. Эту жидкость
независимо от химической природы в коллоидной химии и технике
принято называть маслом. Кроме того, в эмульсии должен
содержаться третий компонент — стабилизатор, или, как его
обычно называют, эмульгатор. В качестве эмульгатора часто приме-
297
няют мыло. Вода и масло могут образовывать эмульсии двух
типов: масла в воде (дисперсная фаза—масло) и воды в масле
(дисперсная фаза — вода).
Эмульгаторы снижают поверхностное натяжение на границе
раздела фаз до 0,1 эрг/см2 и делают коллоидную систему
устойчивой. Без эмульгаторов эмульсии неустойчивы, и в системе
наблюдается явление коалесценции — слипания капель
раздробленной жидкости. Коалесценция продолжается до тех пор, пока
система не распадется на два жидких слоя, на слой масла и
слой воды.
Явление коалесценции можно наблюдать на системе вода —
бензол. Если в эту систему добавить эмульгатор — мыло, то
можно после энергичного встряхивания смесей (1 :100 или
100: 1) получить устойчивые эмульсии.
Коалесценцию стабилизированной эмульсии можно вызвать
центрифугированием (сепарация молока), нагреванием,
воздействием электрического поля высокого напряжения (очистка
добытой нефти от воды), а также с помощью деэмульгаторов,
кислот, некоторых солей и других веществ, которые разрушают
эмульгатор или вытесняют его с поверхности мицелл.
Эмульсии с концентрацией дисперсной фазы более 74%
называют желатинированными. Они подобны гелям и студням.
Примерами таких эмульсий можно назвать консистентные
смазки, сливочное масло, майонез и т. д. Обычными
эмульсиями считаются молоко, сливки, жидкости, применяемые при
обработке металлов.
Важное промышленное значение имеет явление
взаимодействия эмульсий с порошками. На принципе этого
взаимодействия основана флотация, или разделение смесей лиофильного и
лиофобного порошков. Например, для того чтобы разделить
смесь порошков мела и сажи, необходимо залить их водой и
поверх бензолом или другим жидким углеводородом и затем
перемешать. Мел как гидрофильный порошок перейдет в водную
среду, а сажа — гидрофобный порошок — перейдет в
органический растворитель.
Пены—это коллоидные системы, в которых дисперсная фаза—
газ, а дисперсионная среда — жидкость. Чтобы получить
устойчивые пены, необходимы эффективные стабилизаторы —
пенообразователи. Ими могут быть мыла, высокомолекулярные
соединения и другие вещества. Если пленки стабилизаторов способны
отвердевать, то можно получить практически 1 безгранично
устойчивую твердую пену (пенобетоны, пенопласты,
микропористую резину и т. д.). Твердые пены получают, вводя в
пластические массы некоторые вещества (бикарбонат, мочевину, поро-
форы и Др.)» которые разлагаются при 150—180°С, выделяют
газообразные продукты и создают микропористые структуры.
Дым и туман в коллоидной химии называют аэрозолями.
И в том и в другом случае дисперсионная среда — газообразная.
298
В дымах твердые частички являются дисперсной фазой, а в
туманах— жидкие капли.
Изучение устойчивости и свойств аэрозолей представляет
большую самостоятельную главу предмета коллоидной химии.
§ 7. ОБЩИЕ ПРЕДСТАВЛЕНИЯ
О ПОЛУКОЛЛОИДАХ И НЕКОТОРЫЕ СВОЙСТВА
РАСТВОРОВ ПОЛИМЕРОВ
Полуколлоидными системами называют такие растворы, в
которых одно и то же растворенное вещество может
одновременно находиться как в виде молекул, ионов, так и в виде
коллоидных частиц. Это означает, что полуколлоиды —
многокомпонентные системы, в которых вещество дисперсной фазы
находится в динамическом равновесии:
молекулярный раствор ^ коллоидная система
Практически это имеет место в водных растворах мыл,
красителей, танинов (дубящих веществ), алкалоидов. Мыло — это
Рис. 8.8. Схема строения мицеллы масла в водной среде:
а — шарообразной; б — пластинчатой
соли высших жирных кислот. Например, в 1%-ном растворе
олеата натрия наряду с отдельными молекулами уже имеются
мицеллы. В таких растворах мицеллы обычно имеют
шарообразную форму (рис. 8.8, а) и в водной среде молекулы мыла
углеводородными радикалами ориентируются во внутрь.
Гидрофильная полярная группа (—COONa) взаимодействует с водой,
и она ориентирована к растворителю. В 7—8%-ных растворах
мыла образуются сплошные мицеллярные слои и раствор
загустевает. Мицеллярный вес пластинчатых мицелл достигает
20 000 единиц атомного веса.
299
Мицеллы в растворах мыла, подобно мицеллам в
гидрозолях, способны к длительному существованию. Но отличие их
как полуколлоидов состоит в том, что разбавление раствора
вызывает распад мицелл на молекулы мыла, тогда как
разбавление агрегативно устойчивого коллоида просто уменьшает
число мицелл в единице объема. Таким образом, водные растворы
мыл щелочных металлов — лиофильные, термодинамически
устойчивые, обратимые полуколлоидные системы, и они
являются хорошими стабилизаторами почти всех дисперсных систем.
Как известно, углеводороды в воде практически
нерастворимы. Однако если в воде растворено небольшое количество
мыла, то углеводород (например, бензол, гексан и т. д.)
растворится в воде. Это явление называется коллоидной
растворимостью, или солюбилизацией. Частицы углеводорода поглощаются
внутренней частью мицелл мыла. Наиболее важное свойство
растворов мыл — их моющее действие. Представление об этом
развито в работах Ребиндера и его сотрудников. Действие мыла
есть сложный комплекс коллоидно-химических процессов:
смачивания, пептизации, эмульгирования и стабилизации
загрязнений. ;
Чтобы смочить загрязненную поверхность, моющий раствор
должен обладать достаточно низким поверхностным натяжением
(30—40 эрг/см2) по сравнению с чистой водой (73 эрг/см2).
Трение, перемешивание, кипячение способствуют коллоидной
растворимости частиц загрязнений на ткани в пенообразной
коллоидной системе.
Известное моющее средство «Новость» представляет собой
смесь натриевых солей алкилсульфокислот и обладает более
сильным моющим действием, чем растворы олеата натрия.
Почти все каучуки растворяются в бензоле, бензине и
других углеводородах. Белки и полимеры, молекулы которых
содержат много полярных групп, растворяются в полярных
растворителях— воде, ацетоне, этилацетоне и т. д. Многие растворы
высокомолекулярных соединений широко распространены как
клеящие составы.
В разбавленных растворах полимеры находятся в виде
отдельных весьма крупных молекул, называемых
макромолекулами. Линейные размеры этих молекул превосходят размеры
коллоидных частиц, но их поперечное сечение остается в пределах
молекулярных размеров (10~8 см). Такая молекула не имеет
поверхности раздела со средой. И растворы полимеров
являются гомогенными, равновесными, т. е. обладают всеми
признаками истинных растворов. Но по ряду свойств они приближаются
к коллоидным системам.
Молекулы полимеров обладают малой скоростью диффузии,
не способны проникать через полупроницаемые мембраны, и,
кроме того, в присутствии стабилизаторов могут образовываться
300
устойчивые агрегаты макромолекул. Например, присутствие
белковых стабилизаторов делает золь каучука устойчивой, а
размеры частиц (глобул) могут достигать в поперечнике до 10~5 см.
В концентрированных растворах полимеров могут
образовываться структуры, пространственные сетки, сильно загущающие
раствор. Эти структуры создают аномально высокую вязкость.
Застывший раствор обычно со временем претерпевает синере-
зис, разделяясь на две фазы — раствор высокомолекулярного
вещества в растворителе и раствор растворителя в
высокомолекулярном компоненте.
В отличие от низкомолекулярных соединений растворение
высокомолекулярных веществ с линейными молекулами
сопровождается набуханием: растворитель поглощается веществом,
и объем вещества может увеличиться в 10—15 раз.
Устойчивость истинных растворов полимеров можно
нарушить путем добавления нерастворяющей жидкости. Например,
при добавлении воды к раствору нитроцеллюлозы в ацетоне при
определенном содержании воды нитроцеллюлоза выпадает в
осадок.
Выделение белков из водных растворов может быть
осуществлено повышением концентрации солей. Этот процесс
называется высаливанием. Высаливание белков производится
полунасыщенными или насыщенными растворами солей (Na2S04,
(NH4)2S04 и др.) и совершенно отличается от коагуляции лио-
фобных коллоидов слабыми концентрациями электролитов.
Снизить растворимость белков можно добавлением спирта и
действием низкой температуры (до —5°С), а также увеличением
или уменьшением рН раствора относительно «изоточки» белка.
Изоточка белка при определенном значении рН
характеризует такое состояние, когда ион белковой молекулы в растворе
несет на себе одинаковое количество положительных и
отрицательных зарядов. Условно ион белка (аминокислоты) можно
изобразить следующим образом:
ОН~ + {H3N — [—R—] — COO"}0 + Н+,
где R — достаточно длинная углеводородная цепь, соединенная
группами —С—N— Поскольку белок является более сильной
кислотой, то изосостояние белка достигается в кислой среде
при рН меньше 6—5. В зависимости от рН молекула белка
существует то в виде линейной цепочки, то сворачивается в
клубок. Молекула белка представляет пример полиэлектролита,
который обладает свойствами истинных и коллоидных
растворов.
301
Вопросы и упражнения
1. Приведите примеры из окружающих нас предметов, где вещество
находится в коллоидном состоянии.
2. Сопоставьте понятие коллоидного состояния вещества с понятиями
кристаллического, аморфного, жидкого, газообразного состояния вещества.
3. Перечислите характерные признаки коллоидных систем.
4. Какие растворы образуют мыло и вода?
5. Как распознать на любом примере, является ли коллоидная система лио-
фильной, лиофобной или промежуточной лиофильно-лиофобной?
6. Изобразите схему строения мицеллы гидрозоля трехсернистого мышьяка,
если раствор насыщен сероводородом.
7. Начертите мицеллу сернокислого бария в растворе а) при небольшом
избытке серной кислоты; б) при небольшом избытке сульфата натрия.
8. Напишите уравнения реакций, выражающих процессы получения золя
гидроокиси железа методом гидролиза. Изобразите строение мицеллы данного
золя.
9. Вычислите порог коагуляции, если 2 мл золя коагулируют при добавлении
0,2 мл 0,1 н. раствора Na2SC>4.
10. Проанализируйте правила коагуляции по табл. 8.2.
11. Проанализируйте по табл. 8.4 закономерности влияния природы
электролита и концентрации его на величины двойного электрического слоя мицелл.
12. Приведите примеры явления коллоидной растворимости вещества.
ГЛАВА 9
ХИМИЯ ТВЕРДОГО СОСТОЯНИЯ
ВЕЩЕСТВА
§ 1. АГРЕГАТНОЕ СОСТОЯНИЕ ВЕЩЕСТВА
Агрегатным называют состояние вещества, обусловленное
взаимодействиями между его частицами (молекулами, атомами,
ионами), определяющими возможность существования вещества
в твердом, жидком или газовом состояниях. Эти
взаимодействия возрастают при переходе вещества от газового к жидкому
и твердому состояниям.
При низкой температуре частицы вещества обычно
закономерно расположены в пространстве; тепловые колебания не
нарушают этого расположения вследствие того, что энергия
притяжения больше энергии тепловых колебаний. Вещество
существует в твердом состоянии.
При повышении температуры амплитуда колебаний
возрастает и энергия притяжения становится меньше энергии
тепловых колебаний, что приводит к ослаблению связей между
частицами. Теперь частицы совершают колебательные и
вращательные движения, а также могут перемещаться, не разрывая
полностью связи между собой; закономерность в расположении
частиц нарушается. Вещество существует в жидком состоянии.
Дальнейшее повышение температуры увеличивает тепловые
колебания и взаимодействие между частицами практически не
происходит, что дает им возможность перемещаться во всех
направлениях. Вещество существует в газовом состоянии.
При очень высоких температурах (порядка 105—107
градусов) вследствие большой энергии столкновений между
частицами газа молекулы распадаются на атомы, которые теряют
электроны и превращаются в положительные ионы и атомные ядра.
Образуется смесь ионов, электронов и атомных ядер. Вещество
существует в виде плазмы, которую можно назвать четвертым
•состоянием материи. В состоянии плазмы находятся вещества,
образующие Солнце и звезды. На Земле плазма образуется в
молнии и электрической дуге.
Газовое состояние вещества. Газ не имеет ни формы, ни
объема, занимая весь объем сосуда, в котором он находится.
Газовое состояние неупорядоченное. Сжимаемость газов велика,
плотность их мала и изменяется с температурой.
Жидкое состояние вещества. Жидкость не имеет
определенной формы, но имеет определенный объем. Жидкое состояние
называют конденсированным. Сжимаемость жидкостей невели-
303
ка, их плотность близка к плотности твердых тел. но сильнее
изменяется с температурой. При понижении температуры
вещества переходят из газового состояния, в котором отсутствует
связь между частицами и наличие определенной структуры, в
жидкое состояние, в котором частицы взаимосвязаны, но
структура не является определенной, а затем в твердое состояние с
упорядоченной структурой. Однако при понижении температуры
переход жидкого состояния в твердое может происходить и без
упорядочения структуры. Вещество при этом находится в
твердом состоянии, структура которого близка к структуре
жидкости. Частицы совершают колебательные движения, но они не
образуют упорядоченной, правильной, структуры. Такие
вещества называют аморфными. К ним относится, например, стекло.
Рассмотрим строение жидкостей. Переходное состояние
между абсолютным беспорядком (газ) и полной упорядоченностью
(твердое вещество) можно назвать «немного упорядоченным
беспорядком». Жидкость состоит из совокупности
распределенных случайно упорядоченных участков, которые образуются,
распадаются и снова образуются из других частиц.
Совокупность упорядоченных участков остается связанной, но не
образует правильной структуры.
Твердое состояние вещества. В твердом состоянии частицы
вещества не могут перемещаться или вращаться. Они могут
только колебаться около некоторого среднего положения.
Твердое состояние называют конденсированным. Сжимаемость
твердых веществ незначительна, плотность их велика и мало
изменяется с температурой.
Твердое состояние вещества может реализоваться в виде
кристаллической и аморфной структур. Кристаллическая
структура упорядоченная, аморфная — неупорядоченная.
Кристаллическая структура образована частицами
(молекулами, атомами, ионами), закономерно расположенными в
пространстве на определенных расстояниях друг от друга и в
определенных направлениях и совершающими тепловые колебания
около точек равновесия. Совокупность таких частиц образует
пространственную, или кристаллическую, решетку. Идеальный
кристалл характеризуется ближним и дальним порядками в
расположении частиц. Ближним порядком называют
упорядоченное расположение соседних частиц определенного вида, а
дальним порядком — упорядоченное расположение всех частиц
в кристалле.
Свойства кристаллов (механические, оптические,
электрические и магнитные) неодинаковы в различных направлениях. Это
явление, обусловленное внутренним строением кристаллов,
называют анизотропией. Кристаллические тела встречаются
обычно в виде скоплений мелких кристаллов-поликристаллов. Реже
в виде отдельных крупных кристаллов—монокристаллов.
Последние обычно выращивают из жидкой фазы в течение длительного
304
времени. В современной радиоэлектронике монокристаллы
находят большое применение.
В аморфной структуре отсутствует закономерность в
расположении частиц. Свойства аморфных веществ одинаковы во всех
направлениях. Они проявляют изотропию.
§ 2. СТРОЕНИЕ ТВЕРДЫХ ВЕЩЕСТВ
Идеальные кристаллы. Кристаллические решетки. Кристаллы
представляют собой симметричные фигуры. Форму кристаллов
изучает геометрическая кристаллография. Симметричные фигуры
обладают элементами симметрии. В кристаллах различают
следующие элементы симметрии (рис. 9.1). Центр симметрии —
Рис. 9.1. Фигуры, обладающие элементами симметрии:
а — фигура, имеющая центр симметрии С; б — фигура,
имеющая плоскость симметрии АВ\ в — фигура, имеющая
ось симметрии А В (п=4)
точка внутри кристалла, делящая пополам любую проходящую
через нее прямую, проведенную до пересечения с границами
кристалла (рис. 9.1,а, точка С).
Плоскость симметрии — плоскость, делящая кристалл на две
части, каждая из которых является зеркальным изображением
другой (рис. 9.1,6, плоскость АВ).
Ось симметрии — линия, при вращении вокруг которой на
360° кристалл совмещается сам с собой п раз (рис. 9.1,6,
линия АВ, п = 4). Число совмещений кристалла п называют
порядком оси симметрии. Различают оси второго, третьего и более
высоких порядков. Оси первого порядка имеет любой кристалл
вследствие того, что вращение на 360° вокруг любой линии
приводит к совмещению.
В соответствии с геометрической формой кристаллов
существуют следующие кристаллические системы, или сингонии:
кубическая, гексагональная, тетрагональная, ромбическая, моно-
зоа
клинная и триклинная (рис. 9.2). Всего шесть систем, которые
различаются характером расположения координатных осей и
их длиной.
В кубической системе все три оси имеют равную длину и
пересекаются под углом 90°. Основные формы кубической
системы— куб и октаэдр (рис. 9.2,а). В этой системе кристаллизует-
Рис. 9.2. Кристаллические системы
<д — кубическая система; б — гексагональная система; в — тетрагональная
система; г — ромбическая система; д — моноклинная система; е — триклинная
система
'Ся ~8% всех изученных кристаллов, например многие металлы,
алмаз, NaCl, KC1 и др.
В гексагональной системе три оси имеют равную длину и
находятся в одной плоскости под углом 60°, а четвертая
располагается под углом 90° и не равна им. Основные формы
гексагональной системы — гексагональные призма и бипирамида
(рис. 9,2, б). В этой системе кристаллизуется ~7% всех
изученных кристаллов, например многие химические элементы, Н20,
'Si02, NaN03 и др.
В тетрагональной системе все три оси пересекаются под
углом 90°. Две из них имеют равную длину, а третья неравна
им. Основные формы тетрагональной системы — квадратная
призма и бипирамида с квадратным основанием (рис. 9.2,в).
В этой системе кристаллизуется ~5% всех изученных
кристаллов, например Sn, Sn02 и др.
В ромбической системе все три оси пересекаются под углом
90°, но имеют различную длину. Основные формы ромбической
системы — призма и бипирамида (рис. 9.2,г).
В этой системе кристаллизуется ~28% всех изученных
кристаллов, например S, KN03, K2SO4 и др.
306
В моноклинной системе две оси пересекаются под углом,
отличающимся от 90°, а третья ось располагается под углом 90°
к этим двум осям. Все три оси имеют различную длину.
Основные формы моноклинной системы — призма и бипирамида
f^^
У--/У
Рис. 9.3. Комбинации основных форм
кубической системы:
а — комбинация куба и октаэдра; б —
комбинация октаэдра и куба
о^Щ^
оЖ^-^о^Ы
Рис. 9.4. Кристаллическая
решетка йода
(рис. 9.2,(5). В этой системе кристаллизуется ~42% всех
изученных кристаллов, например S, КСЮз, Na2SO4-10H2O и др.
В триклинной системе все три оси имеют различную длину и
располагаются под разными углами, отличающимися от 90°.
О cs
Рис. 9.5. Кристаллическая решетка CsCl
Основные формы триклинной системы — призма и бипирамида
(рис. 9.2, е). В этой системе кристаллизуется ~10% всех
изученных кристаллов, например К2СГ2О7, CuS04-5H20 и др.
Из рис. 9.2 видно, что в различных системах кристаллов
повторяются основные геометрические формы — призмы и
пирамиды. Однако для естественных кристаллов характерны не
только основные формы, но и комбинации этих форм. Так, кристалл,
30?
изображенный на рис. 9.3, а, является комбинацией куба и
октаэдра, а кристалл, изображенный на рис. 9.3,6, — комбинацией
октаэдра и куба.
•ж*
О и*
а
Рис. 9.6. Кристаллическая решетка NaCl
Кристаллические решетки образуются простыми веществами
и химическими соединениями. В зависимости от вида частиц,
образующих кристалл, существуют решетки молекулярные
а
35?
б
в
Рис. 9.7. Кристаллическая
решетка алмаза
Рис. 9.8. Кристаллические решетки
металлов*
а __ Na; б — Си; в — Mg
(рис. 9.4), ионные (рис. 9.5, 9.6), атомные (ковалентные)
(рис. 9.7) и металлические (рис. 9.8).
Молекулярная решетка. В узлах решетки находятся
молекулы вещества, связь между которыми осуществляется силами
308
Ван-дер-Ваальса. Поэтому вещества, имеющие молекулярные
решетки, обычно имеют низкие температуры плавления и
кипения и большую летучесть (сера, йод, С02).
Ионная решетка. В узлах решетки находятся ионы
противоположных зарядов. Связь электростатическая. Кристаллы имеют
относительно высокие температуры плавления и кипения (CsCl,
NaCl).
Атомная решетка. В узлах решетки расположены атомы
элемента, образующего решетку, между которыми осуществляется
ковалентная связь. Вещества имеют высокие температуры
плавления, большую твердость и малолетучи (алмаз).
tf=2,37 A
Рис. 9.9. Истинные и
эффективные радиусы
ионов
Металлическая решетка. В узлах металлической решетки
находятся атомы и ионы металла, а в междоузлиях свободные
электроны, которые перемещаются по всему объему кристалла
и обусловливают характерные свойства металлов: пластичность,
электропроводность, теплопроводность (Na, Cu, Mg).
К наиболее распространенным типам решеток относятся
кубическая объемноцентрированная и кубическая гранецентриро-
ванная. Наименьший объем кристалла, сохраняющий его
форму и содержащий все структурные единицы, образующие
кристалл, называют элементарной ячейкой. Число частиц,
окружающих данную частицу, называют координационным числом.
Ионные радиусы. В кристалле М+Х~ межионное расстояние г
равно сумме эффективных ионных радиусов гм+ и гх-
(рис. 9.9). Поэтому экспериментальное определение г позволяет
еайти величины этих радиусов. Если известны радиусы некото-
Рис. 9.10. Зависимость энергии
кристаллической решетки ионного
кристалла от межионного рас-
309
рых ионов, то можно вычислить остальные, предположив
аддитивность г.
Следует отметить закономерное изменение радиусов ионов в
зависимости от положения элемента в периодической системе
элементов Д. И. Менделеева.
а) У элементов, находящихся в одном периоде, радиусы
катионов меньше, чем анионов. Например, Li+ (0,60); F" (1,36)
(ионные радиусы в А).
б) С увеличением заряда уменьшается радиус в рядах изо-
электронных катионов вследствие сжатия внешних орбиталей.
Например, Na+ (0,98); Mg2+ (0,65); Al3+ (0,50).
в) В рядах изозарядных катионов d-элементов происходит
некоторое уменьшение радиуса с увеличением числа d-электронов.
Например, Ti3+ (0,69); V3+ (0,66); Cr3+ (0,64); Мп3+ (0,62);
Fe3+ (0,60).
г) В ряду лантанидов наблюдается значительное уменьшение
радиуса катионов от La3+ (1,06) к Lu3+ (0,85)—лантанидное
сжатие.
Характер структуры ионных соединений определяется
относительным числом ионов (химической формулой) и их размерами
(отношением ионных радиусов).
Если отношение ионных радиусов находится в пределах
0,414—0,732, образуются структуры типа NaCl с
координационным числом 6. Если отношение радиусов больше 0,732,
образуются структуры типа CsCl с координационным числом 8.
Энергия решетки ионного кристалла. В кристалле каждый
ион находится в окружении других, и поэтому существуют
значительные электростатические взаимодействия.
Энергия кристаллической решетки ионного кристалла —
энергия (теплота) образования решетки кристалла из его ионов в
газообразном состоянии. На рис. 9.10 приведена зависимость
энергии решетки ионного кристалла от расстояния между
ионами; минимум кривой отвечает равновесному состоянию
кристалла с максимальным значением энергии решетки.
Математическое выражение для энергии решетки дается уравнением Бор-
на — Майера
и= NA*z& Л р \
где U — энергия решетки; N— число Авогадро (число ионов в
грамм-ионе, равное 6,023-1023); е — заряд электрона (4,80274Х
Х10~10 эл. ст. ед.); zu z2 — заряды ионов; г — расстояние между
ионами; А — константа Маделунга, характеризующая все
электростатические взаимодействия и зависящая только от типа
структуры (для структуры CsCl—1,76267; для структуры
NaCl—1,7456); р — постоянная величина для данных частиц,
имеющая для большинства кристаллов значение 0,345 А.
310
После подстановки числовых значении констант B = NAe2 и
перевода эргов в килокалории \ принимая, что г равно сумме
ионных радиусов (см), для координационного числа 6 получаем
U = hh— /1 Е | ккал/моль.
Уравнение позволяет вычислить энергию решетки любого
ионного кристалла, предположив, что решетка относится к
структуре NaCl, и подставив приближенные значения ионных
радиусов.
Экспериментальное определение энергии кристаллической
решетки является сложным. Однако можно связать энергию
решетки с другими энергетическими характеристиками
посредством термодинамического цикла Борна — Габера, в котором
кристаллическое соединение MX подвергается следующим
превращениям.
1. Один моль MX распадается на простые вещества в
стандартных (Р=\ атм; Г = 273°К) состояниях. При этом
поглощается количество энергии, равное теплоте образования, ДЯМхт-
2. Атомы простых веществ в их стандартных состояниях
превращаются в газообразные ионы, что связано с изменением
энергии на величины Д# + для катиона и АН _ для аниона.
Соответствующие термохимические уравнения имеют вид
А #м+ = Д#мг - ^м
и
ДЯх_ = Д//х-£х,
Аг^ Г
где Д#м и Д#х —теплоты образования газообразных атомов
из простых веществ в их стандартных состояниях; /м и Ех —
энергия ионизации и сродство к электрону атомов простых
веществ в их стандартных состояниях.
3. Газообразные ионы М+ и Х~ переходят в твердое
состояние и образуют кристалл MX. При этом энергия изменяется на
величину, равную энергии кристаллической решетки, £/мх-
л 1 эрг = 2,389-Ю-11 ккал.
311
Цикл Борна—Габера можно изобразить таким образом:
ЛЯ
MX,
лиц,
*" ^"станд. сост. ~"~ Астанд. сост.
ЛЯ„
/
L
хг
Хт
Применение к этой системе закона Гесса дает возможность
вычислить энергию кристаллической решетки по уравнению
— U мх = АНмхт + ДЯмг + Л#хг + ^м — £"х-
Для соединения NaCl стандартным состоянием натрия
является твердое, и чтобы получить газообразные атомы, необходимо
затратить энергию, равную энергии сублимации SNa, т. е.
A//Na = 5n3. В стандартном состоянии хлор состоит из
газообразных двухатомных молекул. Поэтому энергия, необходимая
для образования газообразных атомов, равна половине энергии
связи С1—С1 и, следовательно, Д#хг =1/2 ^ci-сь где
Lci-ci—энергия связи. Таким образом, для NaCl энергию кристаллической
решетки — f/Naci можно вычислить по уравнению
— ^NaCl = Л#№С1т ~ ^Na + ^ ^Cl-Cl + ^Na — Eq\.
Соответствующие значения энергии приведены в табл. 9.1, из
которой видно, что энергия кристаллической решетки NaCl
составляет —184,7 ккал/моль.
Кристаллические структуры окислов металлов и их
соединений. Окислы. Рассмотрим кристаллические решетки окислов
металлов МО, относящиеся к кубической гранецентрированной
структуре типа NaCl, например MgO, MnO, FeO, CoO, NiCX
На рис. 9.11 изображена кристаллическая решетка МпО. В
узлах решетки находятся ионы Мп2+ и О2". Элементарная ячейка
содержит 4Мп2+ и 402~, всего 8 ионов, или 4 молекулы МпО.
Каждый ион Мп2+ окружен 6 ионами О2- и каждый ион О2- ок-
312
Таблица 9.1
Энергетические величины для NaCl
Теплота распада, равная
теплоте образования NaCIm
Энергия сублимации Na
Половина энергии связи С12
Энергия ионизации Na
Сродство к электрону С1
Формула
A//NaClm
SNa
l/2Lrl_cl
^Na
~^NaCl
ккал/моль
98,2
26,0
29,0
118,0
—86,5
184,7
О Mr ф О
Рис. 9.11. Кристаллическая
решетка МпО
• Fe О О
Рис. 9.12. Кристаллическая
решетка a-Fe203
ружен 6 ионами Мп2+, следовательно, координационное число
равно 6.
В качестве примера кристаллических решеток окислов М203
рассмотрим решетку Fe203 (a-Fe203), относящуюся к триго-
нальной системе типа корунда (рис. 9.12). Ионы О2-,
находящиеся в узлах решетки, образуют гексагональную плотную
шаровую упаковку, в которой имеются октаэдрические междоузлия
(рис. 9.13), расположенные в центре октаэдра, образованного
6 ионами О2-. В этих междоузлиях находятся ионы Fe3+,
занимающие только 2/з общего количества междоузлий.
Элементарная ячейка Fe203 содержит 4Fe3+ и 602~, всего 10 ионов, или
2 молекулы Fe203. Координационное число равно 6.
Соединения окислов. При взаимодействии основных окислов
(МО) с кислотными (М203) образуются соли соответствующих
кислот. Например:
313
MgO + A1203 = MgAl204 — алюминат магния,
FeO + Cr203 = FeCr204 — хромит железа,
MnO + Fe203 = MnFe204 — феррит марганца.
Кристаллическая структура этих твердых веществ относится к
кубической гранецентрированной решетке типа шпинели.
О' 02 о'
Рис. 9.14. Кристаллическая
решетка шпинели iV^+Fe^O^-:
1 — ионы кислорода; 2 —
ионы металла в тетраэдриче-
ских узлах; 3 — ионы металла
в октаэдрических узлах
Рассмотрим кристаллическую решетку шпинели M2+Fe2+C>4~
(рис. 9.14). Относительно большие ионы кислорода,
находящиеся в узлах решетки, образуют кубическую плотную шаровую
упаковку (рис. 9.15). В такой упаковке существуют междоузлия
двух видов: тетраэдрические, находящиеся в центре тетраэдра,
образованного 4 соседними ионами кислорода, и октаэдрические,
находящиеся в центре октаэдра, образованного 6 соседними
ионами кислорода. В междоузлиях находятся относительно
небольшие ионы металлов. Однако они занимают только часть из
всех имеющихся междоузлий, которые в этом случае называют
тетраэдрическими и октаэдрическими, или Л- и В-узлами.
Наличие незанятых междоузлий может вызвать перераспределение
катионов, приводящее к изменениям структуры шпинели.
Элементарная ячейка шпинели состоит из 8 молекул
M2+Fe2+04~~ и содержит 32 О2- в узлах плотной шаровой
упаковки, 96 междоузлий, состоящих из 64 тетраэдрических и 32
октаэдрических, из которых ионами металлов заняты только 8 те-
Рис. 9.13. Плотная упаковка
атомов в гексагональной решетке
314
траэдрических и 16 октаэдрических междоузлий. Число
структурных единиц, входящих в элементарную ячейку шпинели,
приведено в табл. 9.2. Координационные числа ионов металлов,
расположенных в Л- и В- узлах,
соответственно равны 4 О2- и
6 О2".
Распределение катионов по
тетраэдрическим и октаэдриче-
ским узлам. Катионы в шпинели
могут распределяться по
тетраэдрическим и октаэдрическим
узлам (Л- и В-узлам) различным
>.
Рис. 9.15. Плотная упаковка атомов в
кубической гранецентрированной
решетке
образом. Если 8 ионов М2+ занимают 8 тетраэдрических узлов, а
16 ионов Fe3+—16 октаэдрических узлов, то такое распределение
катионов называют нормальным, а шпинель нормальной
шпинелью.
Таблица 9.2
Число структурных единиц в элементарной ячейке шпинели
(M2+Fe3+02~)8
Структурные
единицы
Число
Молекул
M2+Fe3+02-
8
Узлов плотной
упаковки N
32
Междоузлий
тетраэдрических 2N
64
октаэдрических N
32
О
U
О)
о
а
96
Ионов в (M2+Fe^+02 ")e
кислорода в
узлах плотной
упаковки
32
металлов в
тетр
узлах
8
окт
узлах
16
ввего
56
Если 8 ионов М2+ занимают 8 из 16 октаэдрических узлов, а
по оставшимся 8 тетраэдрическим и 8 октаэдрическим узлам
равномерно распределены 16 ионов Fe3+, то такое
распределение называют обращенным, а шпинель обращенной шпинелью.
Если же 8 ионов М2+ и 16 ионов Fe3+ статистически
распределены по тетраэдрическим и октаэдрическим узлам, то такое
распределение называют промежуточным, а шпинель
промежуточной шпинелью.
Рассмотрим распределение катионов в ферритах. Для
промежуточной шпинели распределение катионов можно выразить
формулой
315
M26+Fe?±6[M?±6Fe?|6]0^-,
где ионы в тетраэдрических узлах написаны перед квадратными
скобками, а ионы в октаэдрических узлах помещены в
квадратные скобки. Для нормальной шпинели 6=1, а для обращенной
6 = 0. Для промежуточной шпинели 0<6<1 и при
статистическом распределении катионов 6 = Уз- Величину 6 называют
степенью обращенности шпинели. Она представляет собой долю
М2+, находящуюся в тетраэдрических узлах.
Структуру нормальной шпинели имеют ферриты
Zn2+ [Fe!+]CT и Cd2+[Fe|+]042-;
структуру обращенной шпинели имеют ферриты:
Fe3+ [Fe2+Fe3+] ОГ, Fe3+ [Co2+Fe3+] 0;Г,
Fe3+ [Ni2+Fe3+] ОГ, Fe3+ [Li^Fe^] Ot\
структуру промежуточной шпинели (6 = 0,8) имеет феррит
Mn^Fe3ot[Mn2otFeU]0^-,
(6 — 0,1) имеет феррит
Mg0,iFe3ot[Mg^Fe^]Or.
Изоморфизм и полиморфизм. Твердые вещества, образующие
кристаллические решетки одинакового типа, называют
изоморфными, а явление существования различных веществ в виде
кристаллов одинаковой формы — изоморфизмом. Изоморфные
вещества обладают способностью совместно кристаллизоваться из
растворов в виде кристаллов переменного состава, которые
называют смешанными кристаллами или твердыми растворами
замещения. В узлах решетки смешанного кристалла находятся
частицы изоморфных веществ, например: KA1(S04)2- 12H20 и
KCr(S04)2-12H20 (квасцы), МпС03 и MgC03 (карбонаты),
Ni2C204 и ZnC204 (оксалаты). Совместная кристаллизация
наступает в том случае, когда ионы, входящие в смешанный
кристалл путем замещения друг друга, имеют близкие размеры и
различие составляет ~15%.
Если вещество в зависимости от условий температуры и
давления образует кристаллы различной формы, то такое явление
называют полиморфизмом, а различные кристаллические
структуры— полиморфными модификациями данного вещества.
Например, сера образует две полиморфные модификации:
ромбическую и моноклинную серу. Форма кристаллов зависит от
различного расположения молекул в кристаллах серы. Переход
одной полиморфной модификации в другую связан с изменением
объемов и энергий этих модификаций. Так, превращение низко-
316
температурной модификации в высокотемпературную происходит
с увеличением энтальпии системы (поглощением теплоты).
Между двумя полиморфными модификациями возможно обратимое
превращение, которое получило название энан-
тиотропного превращения и представляет собой
обратимое превращение вещества при
определенных температуре и давлении (95,6° и Р=1 атм
для серы).
Если полиморфное превращение необратимо,
и при охлаждении модификации, полученной
при нагревании, не образуется исходная
модификация, то такое превращение называют моно-
тропным. Например, СаСОз образует две
модификации: арагонит (ромбическая система) и
кальцит (тригональная 1 система):
СаС03-1^-^СаС03.
арагонит кальцит
При охлаждении обратного перехода не происхо,
§ 3. РЕАЛЬНЫЕ КРИСТАЛЛЫ
Дефекты кристаллической решетки. Отметим, что в
идеальной структуре положение ионов в решетке соответствует
минимальному значению энергии кристалла. Такое состояние
отвечает требованию ближнего и дальнего порядков.
Реальные кристаллы характеризуются тем, что в них не все
ионы находятся в положениях, отвечающих минимуму энергии,
вследствие того, что в кристалле появляются нарушения
ближнего и дальнего порядков. Эти отклонения от идеального
расположения ионов в кристалле называют дефектами
(несовершенствами). Все реальные кристаллы содержат дефекты в той или
иной степени.
Дефекты по характеру их природы можно разделить на
следующие группы: точечные дефекты; линейные дефекты, или
дислокации; поверхностные дефекты (границы фаз); объемные
дефекты (пустоты, включения другой фазы).
По энергетическому состоянию дефекты могут быть
равновесными и неравновесными. Равновесными называют дефекты,
находящиеся в термодинамическом равновесии со структурой.
К ним относят точечные дефекты. Неравновесными называют
дефекты, не находящиеся в равновесии со структурой и
вследствие этого имеющие более высокие значения энергии. К ним
относят дислокации, поверхностные и объемные дефекты.
К тригональной системе относятся иногда кристаллы гексагональной системы,
для которых основной формой является ромбоэдр (рис. 9.16).
Рис. 9.16.
Ромбоэдр
317
- + - + -+ + _ + - 4- - +
+ - + _ + _ - + - + - + -
- О - + -+ -»- +._ + _+_ +
+ _ + - + _ - + _ + -. + _
- + - + - + + -+-'+- +
+
+ - + - О - - + - + - + -
-♦-+- + + - + - + -+■
+ - + - + - - + - + _ + _
Равновесные дефекты. Точечные дефекты в ионных
кристаллах. Реальные кристаллы могут содержать два типа дефектов:
дефекты по Френкелю и дефекты по Шоттки.
Дефекты по Френкелю. Катион или анион перемещается из
своего нормального равновесного положения (узла) в
промежуточное положение (междоузлие), образуя ион внедрения и
вакансию, т. е. незанятый узел решетки (рис. 9.17, а). Дефекты в
— +■
- + - + -+-
+ а + - + - *■
- 4- - + - 4-
4- - 4- - Q - 4-
- 4- - 4- - 4-
4- - 4- - 4» - +
- + - + - + -
$
Рис. 9.17. Образование дефектов в кристаллической решетке
— кристалл с дефектами по Френкелю; б — идеальный кристалл; в — кристалл
с дефектами по Шоттки
виде катионов в междоузлиях и катионных вакансий более
распространены, чем дефекты в виде анионов в междоузлиях и
анионных вакансий.
Дефекты по Шоттки. Катион и анион перемещаются из своих
нормальных равновесных положений на поверхность кристалла,
образуя катионную и анионную вакансии в равных количествах
(рис. 9.17, б).
Образование дефектов можно рассматривать как
квазихимическую реакцию. Тогда для дефектов по Френкелю в
соединении MX можно написать уравнение:
где Мм—катион в узле решетки; VJ— междоузлие; Mf — катион в
междоузлии; Ум—катионная вакансия.
Если п\ — число ионов в междоузлиях в единице объема
(концентрация ионов) при равновесии, nv — число вакансий в
решетке в единице объема (концентрация вакансий) при
равновесии, N — общее число ионов в единице объема, равное общему
числу узлов решетки в единице объема (концентрация узлов),
N{— общее число возможных междоузлий в единице объема
(концентрация междоузлий), то к системе можно применить
закон действия масс
318
^ = К. (9.1>
(N-nv)(Ni-ni)
Для дефектов по Френкелю m = nv, и, если кристалл почти
совершенный (неупорядоченность мала), то rti^NNu тогда
= /С и n2.=NNtK. (9.2>
му.
Если £г— энергия, необходимая для перемещения иона из
узла решетки в междоузлие, и можно считать, что процесс
происходит при постоянном объеме, то константа равновесия
/С = e~EilkTy (9.3>
где k — константа Больцмана (1,380-Ю-16 эрг/град); Т —
абсолютная температура.
Тогда из уравнений (9.2) и (9.3)
щ = 1/ЩК - У~Ще~Е1,2кт, (9.4)
т. е. концентрация ионов в междоузлиях, равная концентрации
вакансий, экспоненциально зависит от температуры
образования кристалла.
Количество дефектов по Шоттки можно определить таким же
образом, если предположить, что катион и анион, находящиеся
в узлах решетки, взаимодействуют с вакансиями на
поверхности с образованием двух ионов, адсорбированных на
поверхности кристалла, и двух вакансий в решетке. Процесс
образования дефектов по Шоттки в соединении MX можно выразить
уравнением
М* + Хх + Ум5 + Vts = MUs + Xxs + Vm + V*.
где Мм — катион в узле решетки; Хх — анион в узле решетки; Ум — ка-
тионная вакансия; Ух—анионная вакансия; Ум —поверхностная
катионная вакансия; Ух —поверхностная анионная вакансия; Мм
—катион в поверхностном узле решетки; Хх — анион в поверхностном
узле решетки.
Если nv — число пар вакансий в единице объема при
равновесии, N — число ионных пар в единице объема, Ns — число
ионных пар на единице площади поверхности и Ev — энергия,
необходимая для образования катионной и анионной вакансий,
то для равновесного состояния кристалла можно написать
nvNs
(N-nv)Ns
К и /С = <Г£у/лг. (9.5)
319
Если кристалл почти совершенный (неупорядоченность мала), то
пу «С N, тогда
nv = NK = Ne~EvlkT. (9.6)
Пример Ev= 1 эв, или 1,602-10"12 эрг; k= 1,380- 1СГ16 эрг/град и
Т = 1000 °К. Тогда
п ,lt —1,602-Ю"12 —16,02 11С1
by kl — = = — 11 ,ol
1,380-10-". 108 1,380
И
<Г^//гГ = 2,718 ~и'61, a lg 2,718~п'61 = — 11,6Ь0,434 = — 5,04
получаем nv/N^e~l2~\0~5, и доля вакансий в кристалле будет
-0,001%.
Теперь рассмотрим причины, вызывающие образование
дефектов.
Повышение температуры (тепловые дефекты). Даже в
идеальном при 0°К кристалле с повышением температуры
возникают дефекты. Это происходит потому, что повышение
температуры способствует перемещению ионов кристалла на его
поверхность (дефекты по Шоттки), смещению ионов в промежуточные
положения (дефекты по Френкелю), а также взаимному
замещению ионами друг друга в решетке кристалла.
Примеси (примесные дефекты). В узлах решетки часть
основных ионов заменена ионами примеси — твердые растворы
замещения, или ионы примеси находятся в промежуточных
положениях решетки — твердые растворы внедрения.
Мы рассмотрели образование дефектов в ионных кристаллах.
Теперь напишем равновесные квазихимические реакции,
протекающие в атомных (металлических) кристаллах MX.
Образование дефектов Образование вакансий
по Френкелю по Шоттки
МЙ + V? £М? +V& О^УЙ + V$,
где Мм — атом в узле; М*—атом в междоузлии; V* — междоузлие;
Ум— атомная вакансия; Ух—атомная вакансия.
Равновесие между твердой и газовой фазами (кристалл—
пар) описывается уравнением
мй^мг + у^,
где Мм — атом в узле; Мг — атом в газовой фазе; Ум —атомная
вакансия.
Можно сказать, что наличие тех или иных дефектов в
реальных кристаллах определяется условиями образования этих
кристаллов. Установлено, что дефекты влияют на электрические,
320
магнитные и оптические свойства кристаллов и что химические
реакции в твердых фазах протекают благодаря присутствию
дефектов.
Неравновесные дефекты. Линейные дефекты, или дислокации.
Дислокациями называют линейные нарушения в решетке
кристалла. Дислокации бывают двух видов: краевые и винтовые.
Рис. 9.18. Краевая дислокация в плоскости скольжения
ABCD. R области ARFF ионы смещены более чем на
половину постоянной решетки; в области FECD ионы
смещены менее, чем на половину постоянной решетки
EF — линия дислокации; A'F' — направление
скольжения
Краевые дислокации. Рассмотрим кристалл, в котором
верхний слой ионов сдвинут на одно межионное расстояние в левой
части плоскости скольжения (рис. 9.18). Граница между той
частью кристалла, где произошел сдвиг, и той частью, где сдвиг
не произошел, представляет собой дислокацию, являющуюся,
таким образом, линейным дефектом кристаллической решетки,
наибольшая деформация которой происходит вблизи
дислокации. Краевая дислокация простирается в плоскости скольжения
по всему кристаллу в направлении, перпендикулярном к
направлению скольжения.
Винтовые дислокации. Схематическое изображение винтовой
дислокации дано на рис. 9.19, где указана граница между
смещенной и несмещенной частями кристалла, расположенная
параллельно направлению скольжения, а не перпендикулярно к
нему, как при краевых дислокациях. Образование винтовой
дислокации можно себе представить, если сделать в кристалле
разрез и сдвинуть вниз на одно межионное расстояние часть
кристалла, расположенную справа от разреза. Вертикальная
линия EF—линия дислокации; вблизи этой линии происходит
И Общая химия
321
наибольшая деформация решетки. При винтовой дислокации
атомные плоскости в кристалле расположены по спирали, так
что при каждом обороте вокруг линии дислокации происходит
перемещение на одно межплоскостное расстояние по линии
дислокации.
Краевые и винтовые дислокации относятся к неравновесным
дефектам. Неравновесные дефекты способны взаимодействовать
с равновесными. Так, дислокации
могут поглощать и снова создавать
вакансии.
Дислокации оказывают влияние
на физические свойства кристаллов.
Например, механическая прочность
и пластическая деформация
кристаллов обусловлены наличием
дислокаций. Возникновение дислокаций
происходит путем накопления в
кристалле точечных дефектов
(ионов в промежуточных положениях и
Рис. 9.19. Винтовая дислокация вакансий). Дислокации могут быть
вызваны неравномерным ростом
кристалла, механическими и термическими напряжениями, а
также наличием примесей.
§ 4. ДИФФУЗИЯ ПРИ ХИМИЧЕСКИХ РЕАКЦИЯХ
В ТВЕРДЫХ ВЕЩЕСТВАХ
Реакциями в твердых веществах называют реакции,
протекающие при непосредственном взаимодействии между
частицами кристаллических веществ, а также реакции окисления,
восстановления и термического разложения твердых веществ, при
которых один из продуктов реакции находится в твердом
состоянии.
При химических реакциях в твердых веществах диффузия
происходит через возникающий и увеличивающийся слой
продукта реакции и скорость процесса зависит от скоростей
диффузии и химической реакции. Рассмотрим реакцию
А + В = АВ,
протекающую при нагревании между веществами А и В в виде
таблеток (рис. 9.20). Процесс взаимодействия между
веществами А и В состоит из трех стадий: начало — реакция протекает
на поверхности зерен реагентов (рис. 9.20, а); продолжение —
реакция осуществляется путем массопереноса, сущность
которого состоит в том, что частицы вещества А перемещаются в
зоне реакции к частицам вещества В через разделяющую их
среду, проходя последовательно участки внешней (между зер-
322
нами) и внутренней (в зернах) диффузии (рис. 9.20,6);
окончание реакции — образование вещества АВ (рис. 9.20, в).
Рассмотренная нами реакция относится к области
диффузионно-химических процессов, которые очень сложны вследствие
того, что диффузия сопровождается химической реакцией.
в В
а 6 *
Рис. 9.20. Реакция А+В=АВ, сопровождающаяся
диффузией через слой АВ:
а — начало реакции; б — процесс реакции; в —
окончание реакции
Механизм процесса диффузии. Различают два основных
механизма процесса диффузии в твердых веществах.
1. Диффузия внутри кристаллической решетки или диффузия
по точечным дефектам (рис. 9.21), при которой происходит:
а) смещение атома (иона) в междоузлие с образованием
структуры Френкеля и диффузия по междоузлиям (рис. 9.21, а);
б) смещение атома (иона) 1 в междоузлие с последующим
смещением атома (иона) 2 в вакансию 7, атома (иона) 3 в
вакансию 2 и т. д. — диффузия по вакансиям с начальным
образованием структуры Френкеля (рис. 9.21,6);
в) смещение одновременно двух, трех, четырех атомов
(ионов) в междоузлие с последующим помещением их в
оставленные вакансии—дублетная, триплетная, квадруплетная
круговая диффузия по междоузлиям и вакансиям (рис. 9.21, в);
г) смещение атома (иона) / в имеющуюся вакансию, затем
смещение атома (иона) 2 в вакансию /, атома (иона) 3 в
вакансию 2 и т. д. — диффузия по вакансиям с образованием
структуры Шоттки (рис. 9.21, г).
2. Диффузия вне кристаллической решетки, или диффузия
по протяженным1 дефектам, при которой происходит смещение
атома кристалла из узла на поверхность с образованием
структуры Шоттки и затем:
а) диффузия по поверхности зерен — поверхностная
однофазная диффузия;
б) диффузия по границам различных зерен — поверхностная
межфазная диффузия;
! К протяженным дефектам относятся линейные, поверхностные и объемные
дефекты
11*
323
в) диффузия по внутренним порам, трещинам и
дислокациям— поверхностная внутрифазная диффузия.
Диффузия по протяженным дефектам может сопровождаться
последующей диффузией по точечным дефектам. С повышением
м :•: г>: с*:
г>л £•: с*: с»з
( О / г л
он с*: он он
о: CfTf*:
•: п*з-&«:
с*э-м :•: он
Г# П Атом 6 узле
[_• J^^ Путь атома по узлам
Г П Ьакансия
:•] о: он :•:
/ 2 3
£•: с»: :•: с*з
(•
( j
Путь атома по
междуузлиям
Междуузлие
Рис. 9.21. Возможные механизмы диффузии в кристалле.
а — смещение атома из узла в междоузлие и диффузия по
междоузлиям; б — смещение атома из узла в междоузлие и диффузия по
вакансиям; в — одновременная круговая диффузия по междоузлиям
и вакансиям; г — смешение атома в вакансию и диффузия по
вакансиям
температуры возрастает роль диффузии по точечным дефектам,
с понижением температуры — роль диффузии по протяженным
дефектам.
§ 5. КИНЕТИКА РЕАКЦИЙ
В ТВЕРДЫХ ВЕЩЕСТВАХ
Системы, в которых протекают реакции между твердыми
веществами, реакции окисления, восстановления и термического
разложения твердых веществ, представляют собой гетерогенные
системы, состоящие из нескольких фаз. Поэтому мы будем
рассматривать кинетику гетерогенных химических реакций.
324
При взаимодействии твердых веществ скорость процесса
зависит от скорости химической реакции и скорости диффузии и
определяется скоростью наиболее медленной стадии —
диффузии реагирующих веществ через слой продукта реакции.
Скорость протекающего диффузионно-химического процесса
зависит главным образом от величины поверхности
соприкосновения частиц реагирующих веществ, что определяется степенью
дисперсности реакционной смеси. Величина этой поверхности
обычно составляет ~10~6—10~7 части всей поверхности смеси.
Однако взаимодействие частиц возможно только в начале
процесса, пока поверхность этих частиц еще не покрылась слоем
продукта реакции, после чего процесс может продолжаться
только при наличии массопереноса.
Кинетика образования шпинели. Рассмотрим гетерогенную
химическую реакцию
NiO + Fe203 = NiFe204,
протекающую в смеси окислов NiO и Fe203, взятых в
эквимолекулярных количествах, при температуре 1100°С. Для характе-
-I 1 i i \ ) \ \ \ \
4 8 12 16 20 24 28 32 36 40
час
Рис. 9.22. Кинетика реакции NiO + Fe203 =
= NiFe204 при П00°С
ристики скорости этой реакции определим для различных
значений времени количество образовавшейся шпинели. Построим
кривую зависимости величины, пропорциональной количеству
образовавшейся шпинели (магнитный эффект), от времени
нагревания (рис. 9.22). Ход кривой показывает, что в первые 2 час
нагревания реакция протекает с большой скоростью, после чего
скорость уменьшается. Образование шпинели практически
заканчивается через 4—6 час.
Кинетические уравнения. Рассмотрим кинетические уравнения
для реакций взаимодействия твердых веществ и термического
разложения твердого вещества.
Уравнение Яндера для реакций взаимодействия твердых
веществ, протекающих при постоянной температуре:
325
§Е4О0
%$300
Z§200
*%Ю0
где х — степень превращения вещества в продукт реакции в %;
т — время; /С—- константа; kf — константа, зависящая
от свойств веществ и условий процесса; с — концентрация
диффундирующего вещества на внешней поверхности
диффузионного слоя продукта реакции; D — коэффициент диффузии этого
вещества в диффузионном слое; R — начальный радиус зерен
вещества, покрываемого слоем продукта в процессе реакции.
Q028
Рис. 9.23. Кинетика реакции
BaCO, + Si02=BaSi03 + C02 при ГС
/ — 800; 2 — 840; 3 — 870; 4 — 890
Экспериментальное подтверждение уравнения (9.7) получено
Яндером для реакции
ВаС03 -I Si02 = BaSi03 -\ С02
(рис. 9.23), из которого видна прямолинейная зависимость для
различных температур величины, характеризующей степень
превращения, от времени. Уравнение Яндера справедливо для
степени превращения 25—80%.
Для твердофазовых реакций показано, что скорость реакции
зависит не только от размеров зерен реагентов, но также и от
состава смеси, способа приготовления, структуры исходных
веществ и продуктов реакции. Поэтому какое-либо определенное
числовое значение К в уравнении (9.7) не может
характеризовать вообще ту или иную реакцию в различных опытах.
Уравнение Ерофеева для реакций термического разложения
1 - £Г-*Л
(9.8)
где а — степень превращения вещества; т — время; k —
постоянная, определяющая константу скорости реакции; п —
постоянная, зависящая от природы вещества. Значения п могут быть
больше или меньше единицы в зависимости от характера
процесса. Для диффузионных процессов я<1, для процессов,
происходящих в кинетической области, /г>1. Если п=1, протекает
реакция первого порядка.
326
После математических преобразований уравнение (9.8)
принимает вид
Ig[— lg(l— a)] = lgft + nlgT+lglge.
(9.9)
О 0,2 QU 0,8 0,8 1,0 1,2 1L4 1,6 1,8
Применим уравнение (9.9) к дегидратации CuS04-5H2Or
происходящей при 54°С по реакции
CuS04 . 5Н20 - CuS04 . ЗН20 -\ 2Н20.
Откладывая на оси абсцисс экспериментально полученные
значения lgt, а на оси ординат
значения lg[—lg(l—а)], получаем
прямую линию (рис. 9.24). Для
определения п найдем тангенс
угла наклона прямой к оси
абсцисс. Для определения \gk
продолжим прямую до пересечения
с осью ординат и найдем для
точки пересечения значение
lg[—lg(l—а)], затем подставим
найденное значение в уравнение
(9.9) и вычислим \gk из
уравнения (9.9) при lgT = 0.
Рис 9.24. Кинетика дегидратации
CuS04-5H20 при 54°С
Константа скорости реакции К вычисляется по уравнению
К = nWn.
Следует отметить, что ввиду сложности
диффузионно-химических процессов константа скорости химической реакции не
является константой в обычно принятом смысле слова.
§ 6. МЕХАНИЗМ РЕАКЦИИ
В ТВЕРДЫХ ВЕЩЕСТВАХ
Схематическое изображение механизма реакции А + В = АВ
дано на рис. 9.25, из которого видно, что реакция образования
вещества АВ протекает по стадиям. Исходные вещества
находятся в смеси в виде частиц А и В (рис. 9.25, а). Нагревание
смеси увеличивает процессы диффузии и массопереноса.
Образуется неустойчивый реакционный слой, содержащий
промежуточное соединение А'В' (рис. 9.25, б, в, г). Повышение
температуры смеси вызывает образование мономолекулярного слоя АВ
327
1 п п п п п п
п п п п п п
п п п п п п
■ ■ ■ fl ■ ■
1 а
и п п п п п
п п п п
Нш! в<^ □
0 и ■
О □ D □ И 8
1 *
п п □ п п п
о а ^
■ ■ ■ ■ ■ ■
1 д
□ □ а п п п
п п п п п п
п п п □
И 0 0 ЕЭ
0 fl S 0 fl И
<J
п п п п п п
п п *1
г
п п п п п п
СИСИ СИСИ
шзшз о шзшз а
шзшз шзшзпшзшз
/ч СИСИ СИСИ
w а
и и и и и и
е I
□ Л Ш В g A1 Bf Пи1 /IS
Рис. 9.25. Схематическое изображение механизма реакции
А+В=АВ
(рис. 9.25, в, г). С течением времени вследствие появления
кристаллических зародышей (9.25, д — объединенная частица) слой
АВ принимает характерную для соединения АВ структуру
(рис. 9.25, в, г, д, е).
328
Рассмотрим результаты некоторых исследований,
позволивших установить, какие промежуточные фазы и состояния
образуются в процессе взаимодействия твердых веществ.
Промежуточные химические соединения. Образование
промежуточных химических соединений найдено для реакции в смеси
12 16 20 24
часы
Рис. 9.26 Образование
промежуточных соединений в эквимолекулярной
смеси CaO + Si02 при 1000— 1200°С:
1 — 2CaO-Si02; 2 — l,5CaO-S102;
3 — CaO-Si02
Рис. 9.27. Физико-химические
свойства активных фаз, возникающих
при реакции ZnO + Fe203 = ZnFe204
при 100—900°С
Пт
Ш
L 1 1 1 б\
-п
м
Л
М
тК •
1 1 i_
200 400 600 800
Тем пера ту ра, °С
СаО и Si02, взятых в эквимолекулярных количествах при
температурах 1000—1200°С (рис. 9.26). В начальной стадии
реакции образуются силикаты 2CaO-Si02 и 1,5 CaO-Si02, кривые
1 и 2, вступающие при дальнейшем нагревании в реакцию с Si02
с образованием метасиликата CaO-Si02 (кривая 3).
Таким образом, реакции в твердых веществах протекают
последовательно с образюванием промежуточных соединении,
приводящих к конечному, стабильному, состоянию.
Активные фазы и их физико-химические свойства.
Промежуточные состояния с избыточным содержанием свободной
энергии, возникающие в процессе взаимодействия твердых веществ,
получили название активных состояний, или активных фаз. Фи-
329
зико-химические свойства активных состояний изучены при
исследовании реакции
ZnO + Fe203 =- ZnFe204-
Смесь ZnO и Fe203, взятых в молярном отношении 1:1,
нагревалась в продолжении 6 час при температурах 100—900°С,
после чего подвергалась физико-химическому исследованию.
Результаты даны на рис. 9.27, где на оси абсцисс отложена
температура нагревания, а на оси ординат различные свойства
реакционной смеси:
а) каталитическая активность для реакции
2CQ + 02 - 2С02;
б) каталитическая активность для реакции
2NaO = 2Na + Oi;
в) скорость поглощения паров воды;
г) магнитная восприимчивость;
д) интенсивность рентгенографической интерференционной
линии, характерной для феррита цинка;
е) интенсивность рентгенографической интерференционной
линии, характерной для окиси железа.
В интервале температур 300—400°С на отдельных участках
поверхности раздела возникает химическое взаимодействие
окислов, приводящее к образованию промежуточных фаз переменного
состава, вызывающих рост каталитической активности и
скорости поглощения водяных паров (рис. 9.27, а, б, в). Происходит
активация реакционной смеси. При 400—500°С происходит
дезактивация смеси, вызванная увеличением слоя продукта реакции
у поверхности раздела фаз (рис. 9.27, а, б, в).
При 500—600°С начинается реакция ферритизации между
основными количествами исходных окислов. В области температур
600—800°С скорость ферритизации возрастает. Образование
ZnFe204 сопровождается увеличением магнитной восприимчивости
смеси, ростом интенсивности интерференционной линии феррита
и падением интенсивности интерференционной линии окиси
железа (рис. 9.27, г, д, е).
Установлено, что активное состояние есть общее явление при
протекании реакций в твердых фазах.
Вопросы и упражнения
1. Дайте характеристику газового, жидкого и твердого состояний вещества.
2. Объясните закономерное изменение ионных радиусов в зависимости от
положения элементов в периодической системе элементов Д. И. Менделеева.
3. Чем определяется характер структуры ионных соединений?
330
4. Приведите примеры кристаллических решеток окислов металлов.
5 Объясните структуру кристаллической решетки шпинели.
6. Объясните явления изоморфизма и полиморфизма.
7. Дайте определение идеального и реального кристаллов.
8. Что называют дефектами кристаллической решетки?
9. Укажите, как можно разделить дефекты по их природе и
энергетическому состоянию.
10. Объясните образование дефектов по Френкелю и по Шоттки.
11 Примените закон действия масс к квазихимическим реакциям
образования дефектов.
12 Какие реакции называют реакциями в твердых веществах^
13 Объясните механизм процесса диффузии.
14. От чего зависит скорость процесса взаимодействия твердых веществ?
15. Укажите, по каким стадиям протекает реакция А + В = АВ между
твердыми веществами.
16. Приведите примеры образования промежуточных соединений.
ГЛАВА 10
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ
ПРОЦЕССЫ
§ 1. ОБЩИЕ ПОНЯТИЯ
Окислительно-восстановительными реакциями называют такие ре-
акции, которые сопровождаются передачей всех или части
валентных электронов от одних атомов, молекул или ионов к другим
атомам, молекулам или ионам, что вызывает изменение величины или
знака их электровалентности.
При окислительно-восстановительных реакциях протекают два
взаимосвязанных процесса — процесс окисления и процесс
восстановления. Например, при горении натрия в хлоре
2Na + Cl2-*2NaCl
металлический натрий окисляется до иона натрия, теряя при этом
электрон
Na — е->•№+,
в то же время хлор присоединяет электрон и восстанавливается до
хлор-иона
Cl2 -f &-*2СК
Процесс окисления связан с отдачей электронов отдельными
атомами или группой атомов. Процесс восстановления связан с
присоединением электронов отдельными атомами или группой атомов.
Те атомы, молекулы или ионы, которые отдают электроны,
называются восстановителями. Те атомы, молекулы или ионы,
которые присоединяют электроны, называются окислителями. При
окислительно-восстановительной реакции окислитель
восстанавливается, восстановитель окисляется.
Если рассматривать реакцию горения натрия в хлоре с точки
зрения классических представлений о валентности, то в результате
взаимодействия натрия с хлором не произошло изменение
валентности атомов: до взаимодействия валентность атомов натрия и
хлора равнялась единице, после реакции валентность натрия и
хлора осталась без изменения. В данной реакции один из участников
реакции (натрий) окислился, другой (хлор) восстановился.
Однако встречаются такие окислительно-восстановительные
процессы, которые трудно объяснить в рамках классических
представлений о валентности. Так, например, в присутствии серной
кислоты перманганат калия окисляет щавелевую кислоту до углекис-
332
лого газа, при этом сам восстанавливается до Мп2+. Реакция
протекает по уравнению
2КМп04 + 5Н2С,04 + 3H2S04 = 10СО2 ; K2S04 ] 8H20 \- 2MnS04.
Валентность атома углерода в молекулах щавелевой кислоты и
углекислого газа равна 4. Однако состояние атомов углерода в
этих двух соединениях различное. Состояние атомов
характеризуют степенью окисления.
При изучении окислительно-восстановительных процессов и при
составлении уравнений окислительно-восстановительных реакций
следует различать понятия валентность, или ковалентность, атома
данного элемента и степень окисления, или электрохимическая
валентность этого атома.
Валентность, или ковалентность, атома данного элемента в
соединении определяется числом неспаренных электронов, которые
принимают участие в образовании общих электронных пар с
каким-либо другим атомом; она не учитывает
электроотрицательность атомов соседних с данным и не имеет знака. Но так как
электроны, образующие связь (пара электронов), смещены к
атому, имеющему большую электроотрицательность, то данный атом
в результате перераспределения электронов приобретает
определенный заряд. Заряд атома после такого перераспределения
электронов и называют его степенью окисления или электрохимической
валентностью. Степенью окисления называют электрический заряд
(в единицах заряда электрона), который получил бы данный атом,
если каждая общая пара электронов, связывающая его с другим
атомом полностью переместилась бы к более
электроотрицательному атому.
Степень окисления атома — это кажущийся заряд атома,
возникающий за счет отдачи или присоединения электронов в ионных
соединениях или за счет притягивания или оттягивания электрон-
ных пар от одного атома к другому в полярных соединениях.
Другими словами, степень окисления — это тот условный заряд атома,
который вычисляют, исходя из предположения, что молекула
состоит только из ионов. Степень окисления атома углерода в
молекуле щавелевой кислоты равна +2, а в молекуле углекислого
газа + 4.
Различие между степенью окисления (окислительным числом)
и валентностью видно из следующих примеров:
1. Азот в молекуле азота трехвалентен (N = N), поскольку в
образовании химической связи участвуют по три неспаренных
электрона от каждого атома азота. Степень окисления атомов азота
равна нулю, поскольку в молекуле азота оба атома одинаковы и
электроны распределены равно одинаково между двумя атомами.
2. В молекуле гидразина N2H4 атомы азота тоже
трехвалентны, но степень окисления равна —2.
3. В молекуле этилового спирта оба атома углерода
четырехвалентны, но степень окисления атома углерода, связанного
333
с гидроксил-ионом, равна —1, а у другого атома углерода
равна —3.
Степень окисления (окислительное число) атома в соединении
определяют условно, принимая: 1) степень окисления иона в
веществе, имеющем ионное строение, равна электрическому заряду
этого иона (Na+Cl~); 2) степень окисления атомов в простых
веществах равна нулю (Н2, N2, 02, Zn, Mg и т. п.); 3) в ковалентном
соединении известной структуры степень окисления каждого атома
определяется зарядом, остающимся на данном атоме после того,
как каждая общая пара электронов полностью отнесена к более
электроотрицательному из двух атомов, которым эта пара
принадлежит; 4) степень окисления атома того или иного элемента в
определенном соединении не установленной структуры можно
выяснить на основании окислительных чисел других элементов,
входящих в состав данного соединения; 5) степень окисления
одноатомного иона всегда равна его заряду. Например, для ионов К+, Li+,
Са2+, Fe2+, Cu2+, Cu+ и т. п.; 6) степень окисления кислорода равна
всегда —2, кроме перекисных соединении и О 2 • Степень
окисления водорода, связанного с металлами, равна всегда —1.
Рассмотрим несколько примеров. Валентность (ковалентность)
атома углерода во всех соединениях СН4, С2Н5ОН, СН20, СН3ОН,
НСООН, С02 равна 4, так как четыре его электрона участвуют в
образовании электронных пар:
Н Н Н Н
I II I
Н—С—Н; Н-С-С—О—Н; Н-С-О-Н;
I II I
н н н н
н о
I /уо и
Н—С=0; Ъ—С( ; С
хо—н и
О
Из величин электроотрицательности атомов кислорода, углерода и
водорода видно, что электронные пары оттянуты от углерода к
кислороду и от водорода к углероду:
'♦ и
Н *С* Н ; Н *С— С хОх Н, Н ХС * , Н *С
г- © *•
0**Сх
•х |+
н н
хг. и
•л Н
хх О
Подсчитав суммарный заряд, получим степень окисления атома
углерода в каждом из данных соединений: СН4~ —4; C2HsOH~
334
один атом углерода имеет степень окисления, равную —3, а
другой — 1; СН20- 0; НСООН- + 2 и С02~ +4.
Для определения степени окисления атома не всегда следует
изображать графически молекулу данного соединения. Например,
в соединениях NH3, N2H4, NH2OH, HN02, HN03 степень окисления
атома азота можно определить из условия, что степень окисления
водорода равна +1, а кислорода —2. И тогда степень окисления
атома азота в указанных соединениях соответственно равна ■—3,
—2, —1, +3, +5, так как сумма степеней окисления всех атомов
в молекуле должна равняться нулю. Степень окисления атома в
соединении может равняться и дробному числу. Например, в над-
перекиси калия К02 степень окисления калия равна —1/г-
Понятие степени окисления (окислительного числа) весьма
условно и помогает в основном при уравнивании коэффициентов
окислительно-восстановительных реакций. Однако степень
окисления до некоторой степени дает возможность оценить электрическую
поляризацию атома, а именно: приближение к нему или отдаление
от него его собственных электронов. Окислительное число
показывает, сколько электронов отдалено от атома или притянуто к нему
от другого атома. Мера удаления или приближения электронов к
атому в окислительном числе не отражена.
Для определения знака окислительного числа пользуются
такими физическими характеристиками, как энергия ионизации
атомов / и сродство к электрону Е. Обе эти характеристики являются
периодическими функциями порядковых номеров элементов. При
соединении двух атомов электроны будут перемещаться в большей
или меньшей мере от атома с малым сродством к электрону Е и
малой энергией ионизации / к атому с большими величинами Е и
/. В связи с этим для характеристики способности атомов к потере
и приобретению электронов используют величину
электроотрицательности ЭО:
ЭО=.Е + /.
При соединении двух атомов перемещение электронов происходит
в сторону того из них, у которого величина ЭО больше. Атомы с
большей ЭО могут выполнять роль окислителей, а с меньшей ЭО —
роль восстановителей. В высшей степени окисления атомы
проявляют только окислительные свойства, а в низшей степени
окисления — только восстановительные свойства.
Таким образом, об окислительно-восстановительных свойствах
элементов можно судить по величине их электровалентности. Так,
наиболее сильный восстановитель — франций, а наиболее сильный
окислитель — фтор.
Типичные окислители — кислород, галогены, хлорная и
бромная вода, кислородные кислоты хлора или брома и их соли, дву-
хромовокислый калий, надсернокислый аммоний, бертолетовая
соль, азотная кислота и ее соли, царская водка, соли 3-валентного
железа и все те вещества, которые содержат в своем составе ато-
335
мы, находящиеся в высших степенях окисления, поэтому способные
только понижать свою степень окисления.
Типичные восстановители — металлы (железо, цинк,
алюминий и др.), а также водород, сероводород, йодистоводородная
кислота и ее соли, соляная кислота, соли 2-валентного железа и все
те вещества, которые содержат в своем составе атомы,
находящиеся в низшей степени окисления и, следовательно, способные только
повышать свою степень окисления.
Вещества, которые содержат в своем составе атомы,
находящиеся в промежуточных степенях окисления и, следовательно,
способные как повышать, так и понижать свою степень окисления,
могут выполнять роль как окислителя, так и восстановителя в
зависимости от другого компонента реакции. Так, например, при
взаимодействии сернистой кислоты с сероводородом первая выступает
в роли окислителя:
H2S03 + 2H2S = 3S + 3H20.
В то время как в реакции с перманганатом калия сернистая
кислота — восстановитель.
Соединения азота и серы имеют следующие особенности:
+5 N205
N (HN03), S+6(S03, H2S04) — проявляют только окислительные
свойства;
N~3(NH3); S2~ (H2S) — проявляют только восстановительные
свойства;
N+4(N02),S+4(S02) )
. N+3 (HN02)
N+2(NO)
N+(N20)
№(N2), S°
N~ (NH2OH)
N"2(N2H4)
Если записывают Mn+7, As+5, P+5, S+6, N+5 и т. д., то имеют в виду
не. соответствующие ионы, которых в действительности нет, а ту
степень окисления, которую проявляют атомы данных элементов.
Окислитель и восстановитель реагируют между собой в
отношении их эквивалентов. Эквивалентом окислителя называют такое
количество окислителя, которое отвечает одному присоединенному
электрону в данной окислительно-восстановительной реакции.
Чтобы определить эквивалент окислителя, надо молекулярный вес его
разделить на число электронов, присоединенных одной молекулой
окислителя. Эквивалентом восстановителя называется такое колш
чество восстановителя, которое отвечает одному отданному
электрону в данной реакции. Для определения эквивалента восстано-
проявляют окислительные и восстановительные
свойства
336
вителя надо молекулярный вес его разделить на число электронов,
отданных одной молекулой восстановителя. Например:
2KMn04 + 5K2S03 + 3H2S04 - 2MnS04 + 6K2S04 + 3H20,
эквивалент окислителя Экмпо4 = ,
5
п 158
эквивалент восстановителя 3k2so3 = —.
Все окислительно-восстановительные реакции можно разделить
на три группы: 1) реакции между атомного или
междумолекулярного окисления-восстановления; 2) реакции диспропорционирова-
ния (самоокисления-самовосстановления) и 3) реакции
внутримолекулярного окисления-восстановления.
К первой группе относятся такие реакции, в которых обмен
электронами происходит между различными атомами, молекулами
или ионами.
Например, простейшие реакции соединения и замещения:
2Са + 02 - 2СаО;
Са -* Са2+ + 2е\ 02 + 2е -* 202~;
Fe° -f CuS04 = Cu° +PeS04;
Fe° -* Fe2+ + 2e; Cu2+ + 2e->. Cu°.
Ко второй группе относятся реакции, в которых
восстановителем и окислителем являются атомы элементов с промежуточной
степенью окисления у молекул одного и того же вещества. В
данном случае молекулы или атомы одного, и того же вещества
реагируют друг с другом как восстановитель и окислитель вследствие
того, что содержащиеся в них атомы с промежуточной степенью
окисления способны отдавать и принимать электроны, переходя в
степени окисления — один в низшее, другой в высшее.
Например, реакции диспропорционирования:
3HNOa = НШ3 + 2NO + Н20;
ЗН2Мп04 = 2НМп04 -г Мп02 + 2Н20;
2К2 [Sn (ОН)4] = К2 [Sn (OH)6] + 2Sn° + 2KOH.
К третьей группе относятся реакции, в которых окислитель и
восстановитель находятся в одном и том же веществе. Например,
реакции внутримолекулярного окисления-восстановления:
2HgO = 2Hgo 4- 02°;
337
NH4N02^N2-| 2H20;
(NH4)2 Cr207 = Cr203 I N2 + 4H20.
§ 2. МЕТОДЫ СОСТАВЛЕНИЯ УРАВНЕНИИ
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ РЕАКЦИЙ
При написании уравнений окислительно-восстановительных
реакций применяют метод электронного баланса и электронно-ионный
метод. По существу оба метода базируются на одних и тех же
предпосылках: общее число электронов, отдаваемых
восстановителем, равно общему числу электронов, присоединяемых
окислителем.
Метод электронного баланса. Рассмотрим реакцию
взаимодействия магния с кислородом
Mg + l/202-*.MgO. (10.1)
Исходные вещества (магний и кислород) электронейтральны. Атом
магния, отдавая два электрона атому кислорода, становится
положительно двухзарядным (Mg2+). Atom кислорода, принимая два
электрона, превращается в отрицательно заряженный ион (О2-).
Процесс изменения степени окисления (валентности) этих
элементов может быть изображен электронными уравнениями
2 I Mg — 2е -> Mg2+ — процесс окисления
+
1 I 02 Ч 4е -> 202~ — процесс восстановления
2Mg + 02 = 2MgO
В левой и правой частях уравнений должно соблюдаться
равенство числа атомов каждого элемента и электрических зарядов по
величине и знаку. Слева от написанных уравнений следует поставить
коэффициенты, которые должны уравнять число отданных
восстановителем и присоединенных окислителем электронов, затем
написать суммарное электронное уравнение.
Обычно метод подбора коэффициентов при помощи
электронных уравнений применяется для определения коэффициентов в
реакциях, протекающих в отсутствие воды.
При составлении уравнений окислительно-восстановительных
реакций, протекающих в водных растворах, используют электрон-
но-ионный метод. Чтобы составить уравнение
окислительно-восстановительной реакции электронно-ионным методом, целесообразно
проделать следующее:
1. Написать ионную схему реакции, для этого требуется знание
начальных и конечных продуктов реакции; определить окислитель
и восстановитель.
2. Составить ионную схему реакции, руководствуясь общими
правилами составления ионных уравнений, т. е. сильные электро-
338
литы записать в виде ионов, а слабые электролиты, газы и осадки
и виде молекул. В ионную схему включать только те частицы
(ионы, атомы, молекулы), которые подвергаются изменению, т. е.
выполняют функции окислителя или восстановителя, а также ионы
Н+ и ОН-, характеризующие среду или молекулы воды.
3. Составить электронно-ионные уравнения отдельно для
процесса восстановления и для процесса окисления, руководствуясь
следующим формальным правилом:
а) Если продукт реакции содержит меньше кислорода, чем
исходное вещество МпОГ-^Мп2+, то в кислой среде
освобождающийся кислород связывается с ионами Н+, в результате чего
образуется столько молекул воды, сколько не хватает атомов
кислорода. В нейтральной и щелочной средах освобождающийся кислород
взаимодействует с водой, образуя соответственно удвоенное число
гидроксильных групп (см. ниже пример).
б) На основании закона сохранения массы и энергии должно
быть равенство числа частиц (ионов, молекул, атомов) в левой
и правой частях уравнения, а также суммарное число и знак
электрических зарядов слева и справа от знака равенства должны
быть одинаковы.
Для примера рассмотрим реакцию восстановления пермангана-
та калия в различных средах.
1. В кислой среде реакция идет по схеме
КМп04 -г Na2S03 + H,S04 -*MnS04 -f Na2S04 + H20 + K2S04,
определим окислитель и восстановитель. Для этого надо
установить, какие атомы и каких веществ изменяют свою степень
окисления в процессе реакции. Как видно, в результате реакции степень
окисления у атомов марганца изменилась:
+7 __ +2
Мп04 ->Мп.
У атомов серы тоже изменилась степень окисления:
+4 о +б о
sor->sc>4~.
Следовательно, в данной реакции окислителем является КМп04>
восстановителем — Na2S03. Составляем ионную схему реакции:
МпОГ + SOf" + Н+ -> Мп2+ 4- SO4"" + Н20. (16.2)
2. Запишем раздельно процессы восстановления и окисления в
электронно-ионном виде. Прежде всего из уравнения (10.2) видно,
что МпОГ превратился в Мп2+; четыре атома кислорода иона
МпОГ связываются ионами Н+ в четыре молекулы воды по схеме
МпОГ + 8Н+-^Мп2+ -f 4Н20. (10.3)
процесс восстановления
339
В правой части схемы уравнения (10.3) суммарный заряд равен
2+, а в левой — 7-(-• Чтобы суммы зарядов частиц в обеих частях
схемы уравнения (10.3) стали одинаковыми, надо к левой части
уравнения прибавить 5 электронов:
МпОГ + 8Н" ь 5е - Мп2+ + 4Н20. (10.4)
Процесс окисления S03 сопровождается увеличением числа
атомов кислорода. В водном растворе кислород поступает из воды,
что записывается следующей схемой:
SO3- + H2O^SC>4~ - 2Н+. (10.5)
процесс окисления
В правой части (10.5) суммарный заряд равен нулю, в левой —
2—. Чтобы суммы зарядов частиц в обеих частях схемы были
одинаковыми, надо из левой части отнять 2 электрона:
SOa~ + Н20 — 2е = SO4 " -|- 2Н+. (10.6)
Напишем теперь оба электронно-ионных уравнения друг под
другом и определим коэффициенты для окислителя и восстановителя:
МпОГ f 8Н+ + Ъе = Мп2+ f- 4H20
SOs" + Н20 — 2е •= SO*" + 2Н+.
Так как общее число электронов, принятых окислителем, должно
быть равно общему числу электронов, отданных восстановителем,
умножаем первое уравнение на 2, а второе на 5. Это и есть
коэффициенты в уравнении перед окислителем и восстановителем.
Сложив оба уравнения (с учетом найденных коэффициентов), получим
сокращенное ионное уравнение данной реакции
2МпОГ 4 16Н+ -f 5SO3"" -| 5Н20 = 2Мп2+ + 8Н20 + 5S02i~ -!- ЮН+.
(Ю.7)
3. К полученному сокращенному ионному уравнению (10.7)
слева и справа от знака равенства приписываем противоионы,
причем в одинаковом количестве, не нарушая общего равенства
уравнения, и составляем полное ионное уравнение реакции:
2МпОГ ! 16Н+ -ь бБОз" + 5Н20 = 2Мп2+ -|- 8Н20 -f 5SC>4~ -| 10Н+.
2R+ 8S02~ 10Na+ 2K+ 8S02— 10Na+
4 > 4 >
(10.8)
4. По полному ионному уравнению (10.8) записываем
молекулярное уравнение реакции
2KMn04 + 3H„S04 + 5Na2S03 - 2MnS04 + K2S04 н- 5Na2S04 + 3H20.
(10.9)
340
В нейтральной (и слабощелочной) среде:
KMn04+Na2S03+H20-^Mn02+KOH+Na2S04.
МпОГ 4 2Н20 -| Зе = Мп02 + 40Н~
SOf 4 Н20 — 2е = SO4- + 2Н+
2МпО^ + 3SOj~ 4- 7Н20 = 2Мп02 -\- 80Н~ 4- 3S042~ 4 6НН
2 МпОГ + 2Н20 ч- Зе = Мп02 4- 40H
3 SO^ 4- Н20 — 2е = SO?- -t- 2H+
2Mn04 4 3SOf~ 4 7H20 = 2Mn02 4- 80H~ 4 ЗБО*
к2- l CU +
6Н~
или 2МпОГ + ЗЭОз" + Н20 = 2Мп02 -г 20Н~ + 3S04~;
2К+ 6Na + 2K+, 6Na+
в молекулярном виде:
2КМп04 + 3Na2S03 + Н20 - 2Мп02 + 2КОН + 3Na2S04.
В сильнощелочной среде:
KMn04+Na2S03+KOH~>K2Mn04+Na2S04+H20,
МпОГ t-e-MnO*-
SOf" + 20Н" — 2е =^SOl~ + Н20
2МпОГ + SOt + 20Н~ - 2МпО|~ + S024~ + Н20,
2К+ 2Na+ 2K+ 2K+, 2Na+, 2K+
или 2KMn04 + Na2S03 + 2KOH = 2K2Mn04 + Na2S04 + Н20.
§ 3. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ
Окислительная и восстановительная способность у различных
окислителей и восстановителей выражена различно. Один
окислитель легко окисляет какое-нибудь химическое вещество, другой
провести такое окисление не может. Неодинаковая химическая
активность различных окислителей и восстановителей связана с
неодинаковым их стремлением к присоединению или к отдаче
электронов. Чем больше у данного атома стремление к приобретению
электронов (сродство к электрону), тем ярче выражены его
окислительные свойства. Количественной характеристикой этого
стремления служит стандартный восстановительный потенциал.
Известно, что в медно-цинковом гальваническом элементе
протекает самопроизвольная реакция, которую можно записать
следующей схемой:
Cu2+ + Zn-^Cu-l-Zn2+.
341
Это — реакция окисления-восстановления, а составляющие ее
электродные реакции
Zn — 2e = Zn2+, (a)
Cu2+ 2e - Си0 (б)
показывают, что ион меди, который принимает электроны,
является окислителем, а металлический цинк, отдающий электроны, —
восстановителем.
Рис. 10.1. Установка для определения восстановительно-окислительного
потенциала
Каждая из электродных реакций окисления-восстановления
протекает только на границе раздела электрода и
соответствующего ему раствора соли. Можно построить совершенно аналогичную
окислительно-восстановительную цепь, но с участием только ионов
в каждом из процессов окисления и восстановления, при этом
электроды, например платиновые или графитовые, не изменяются в
данной реакции, но участвуют только лишь в переносе электронов
от восстановителя к окислителю и служат инертными
проводниками электронов в реакции окисления-восстановления. При этом
продукты окисления-восстановления остаются в растворе и не
выделяются на электродах.
Например, если налить в один электродный сосуд раствор
нитрита натрия, а в другой — подкисленный раствор перманганата
калия, опустить в них платиновые электроды и соединить их
электролитическим мостиком (рис. 10.1), то измеритель напряжения,
342
подключенный к электродам, покажет определенную э. д. с. и
переход электронов слева направо — от электрода с NaN02 к электроду
с КМп04.
Переход электронов с электрода в растворе, содержащем ионы
NOV, свидетельствует о том, что, по-видимому, ионы NCJT
претерпевают превращение, в результате которого они отдают
электроны и переходят в ионы с более высокой степенью окисления
азота. Таковыми могут быть только ионы NOi":
Если электроны переходят в сторону электрода, погруженного
в раствор перманганата калия, то, следовательно, на электроде
протекает реакция, при которой ион МпОГ , приобретая электроны,
переходит в ион с более низкой степенью окисления марганца.
В кислой среде это может быть только ион Мп2+:
МпОГ-^Мп2+.
Итак, на левом электроде проходит реакция окисления
ЫОГ г Н20 — 2е - N07 ! 2Н+,
на правом — реакция восстановления:
МпОГ 4- 8Н+ + Ъе = Мп2+ + 4Н20.
Ион МпОГ — окислитель, ион N07 — восстановитель.
Учитывая, что число принятых окислителем электронов должно
равняться числу отданных восстановителем и, суммируя оба
уравнения, получаем уравнение окисления-восстановления:
5NO? + 5Н20 — Юе = 5ЩГ + ЮН+
2МпОГ 16Н+ {- 10* -= 2Мп2+ + 8Н20
5N07 + 2МпОГ Н 6Н+ - 5N07 4- 2Мп2+- + ЗН20,
или в молекулярном виде:
5KN02 И- 2KMn04 -*- 3H2S04 = 5KN03 + 2MnS04 + K2S04 + 3H20.
Величина потенциала каждого электрода может быть
определена его сравнением с водородным электродом (см. гл. 11). Так
как окислительно-восстановительные потенциалы зависят от
концентрации, то такое сравнение принято проводить при
концентрациях окисленной и восстановленной форм, равных по 1 г-ион/л.
Например, NOST/NOr электрод должен содержать все ионы,
участвующие в равновесии, в концентрации, равной 1 г-ион/л:
[N02~] = [N07] = [Н+] - 1 г-ион/л,
343
то же для электрода Мп2+/Мп04 :
[МпОГ] = [Мп2+] = [Н+] = 1 г-ион/л.
Если активности окисленной и восстановленной форм в
растворе равны единице, то возникающий скачок потенциала на границе
электрод—раствор называется стандартным восстановительным
потенциалом.
Стандартный восстановительный потенциал является
количественной характеристикой как процесса окисления, так и обратного
ему процесса восстановления. Он характеризует окислительную и
восстановительную способности окислителя и восстановителя: чем
больше стандартный восстановительный потенциал, тем сильнее
данное вещество как окислитель. И, наоборот, чем меньше
стандартный восстановительный потенциал, тем силь'нее
восстановитель.
Величина равновесного восстановительного потенциала
определяется уравнением Нернста
р /7° "* 1п авосст
^восст/окисл — •f-'восст/окисл А" ,»,
пР аокисл
где £"восст/окисл — восстановительный потенциал системы электрод—
раствор; -Евосст/окисл — стандартный восстановительный потенциал
системы электрод—раствор; R — универсальная газовая
постоянная, равная 8,314 ^ккал/моль; Т — абсолютная температура; F —
число Фарадея; а0кисл — активность окисленной формы
соединения; аВосст — активность восстановленной формы соединения; /г —
число отданных или присоединенных одной молекулой электронов
восстановителя или окислителя.
Если в формуле Нернста подставить значения R, T, F и
перевести натуральные логарифмы в десятичные, то уравнение
принимает вид
F - — Р° 0,058 , Двосст
■С восст/окисл — ^восст/окисл 1ъ
п ^окисл
Как известно, для разбавленных растворов активности с
достаточным приближением могут быть заменены концентрациями, и
уравнение Нернста запишется так:
р _ ро 0,058 , [восст]
■С восст/окисл — -С восст/окисл &г 1 *
п [окисл]
Бели же коэффициенты окисленной и восстановленной форм
данной пары не равны единице, то они должны входить в
уравнение Нернста в качестве показателей степени. В частности, для
пары С12/2С1~, участвующей в процессе С12+2е^2С1, оно будет
иметь вид
[С12
344
Для сложных ионов, претерпевающих изменение в составе,
окислительно-восстановительные потенциалы зависят от рН раствора.
Например, для пары МпОГ -f- 8Н4" /Мп2+ 4- 4Н20, участвующей в
процессе
МпОГ - 8Н+ + Ъе ^ Мп2+ + 4Н20,
уравнение Нернста будет иметь вид
5 [MnO~] [Н+]8
Окислительные свойства молекул или ионов (веществ, стоящих
перед знаком равенства в уравнении реакции) тем сильнее, чем
больше их потенциалы по алгебраической величине.
Например, сравним окислительные способности МпСЬ и КМп04
в кислой среде:
MnOa i- 4H+ 4- 2е = Мп2+ + 2Н20 £° = f 1,23 в;
МпОГ f 8Н+ -f 5е = Мп2+ + 4Н20 £° = -fl ,51 в.
Потенциал второй реакции выше, значит ион МпОГ обладает
более высокой способностью принимать электроны и он будет как
окислитель сильнее и активнее действовать по сравнению с МпОг-
Каждый ион или каждая молекула, находящиеся в уравнении
реакции для восстановительного потенциала перед знаком
равенства и имеющие большую алгебраическую величину его, играют
роль окислителя по отношению к ионам или молекулам, имеющим
меньшую его величину.
В окислительно-восстановительных реакциях окислителем
является то вещество, для которого больше восстановительный
потенциал; восстановителем — то вещество, для которого меньше
восстановительный потенциал.
Абсолютное значение восстановительного потенциала
экспериментально определить нельзя, его можно измерить только
относительно какого-либо стандартного электрода. Чаще всего для этой
цели используют водородный электрод, потенциал которого условно
принят равным нулю. Для экспериментального определения
относительного значения восстановительного потенциала составляют
гальванический элемент, в котором одним полуэлементом служит
инертный электрод, погруженный в исследуемую
окислительно-восстановительную смесь, другим полуэлементом — стандартный
электрод сравнения. Стандартным электродом может быть водородный,
каломельный, хингидронный, хлорсеребряный и др.
В случае окислительно-восстановительных реакций направление
протекания реакции определяется направлением
самопроизвольного перехода электронов с электрода, где их концентрация выше, на
электрод, где их концентрация ниже. Рассмотрим это на примере
реакций
345
МпОГ + 8Н+ -| Ъе - Мп2+ л. 4Н20 Е° = -- 1,51 в;
NOT + 2Н+ 4 2^ N0^ + Н20 £° - + 0,94 в.
Концентрация электронов выше на электроде, находящемся ь
растворе нитрита калия, поэтому на этом электроде при замкнутой
цепи процесс идет в сторону той реакции, которая освобождает
электроны:
N02" + Н20 — 2е = NOJ" + 2Н+ Е° = — 0,94 в.
Следовательно, электродная реакция, характеризующаяся
меньшей величиной потенциала (0,94<1,51), переписывается в
обратном направлении, при этом знак потенциала изменяется на
обратный.
Реакция на электроде Мп2+/МпОГ протекает в том
направлении, в котором она написана, так как этот электрод принимает
электроны. Суммируя одну самопроизвольно протекающую
электродную реакцию и другую, протекающую под действием первой,
получаем общее уравнение реакции, самопроизвольно
протекающей в гальваническом элементе. Суммируя электродные
потенциалы, находим э. д. с. элемента = —0,94+1,51 = +0,57 в.
Положительное значение э. д. с. подтверждает возможность протекания
реакции в целом.
Знак разности потенциалов двух полуреакций, т. е. знак э. д. с,
характеризует направление процесса. Положительный знак э. д. с.
соответствует процессу, который может протекать самопроизвольно
и сопровождается уменьшением изобарно-изотермического
потенциала. Реакция, протекающая самопроизвольно, способна
совершать работу и, как видно в гл. 11, работа реакции численно равна
изменению изобарно-изотермического потенциала.
Самый сильный окислитель — фтор:
£20f-/f2 - + 2,87 в.
Самый сильный восстановитель — литий: Еи/ии = —3,03 в.
Определим, можно ли действием галогена на 2-валентное
железо перевести его в 3-валентное состояние. Для железа
Fe3+ -4- е = Fe2+ £° - Ь 0,77 в.
У фтора, хлора и брома потенциал выше потенциала
£>Fe2+/Fe3f'=0,77 в. В прямом направлении возможна только реакция,
отвечающая большей величине потенциала, например:
Cl2 + 2e-2Qb £°=+ 1,36 в,
3 <
для железа в этих условиях возможна только обратная реакция:
Fe2+ — e = Fe3+ £° = — 0,77 в.
Общая реакция, которая протекает самопроизвольно
С12 -!- 2Fe2+ - 2С1- -|- 2Fe^+,
характеризуется э. д. с. ==1,36—0,77 = 0,59 в.
Положительное значение э. д. с. свидетельствует о возможности
самопроизвольного протекания окислительно-восстановительной
реакции. Таким образом, фтор, хлор и бром могут переводить
2-валентное железо в трехвалентное. Йод же в связи с низким
значением его потенциала не способен осуществить такой переход.
В случае с йодом возможна только противоположная реакция,
в которой ионы Fe3+ будут окислителями для йодид-ионов:
2I- + Fe3+-xI2 + Fe2+
или в молекулярном виде:
2KI Ь FeCl8 - I, -' 2KC1 + FeCl2.
Часто возникает потребность определить, возможно ли взаимодействие
каких-либо двух веществ в водном растворе. Рассмотрим несколько примеров
1. Возможно ли взаимодействие ионов Сг2Оу~ и Fe? Прежде всего
следует установить, можно ли вообще реакцию взаимодействия написать. В
ионе Ct2Oj~ хром находится в своей высшей степени окисления и процесс
может идти только в сторону ее уменьшения, для чего ионы Сг207~~
должны принимать электроны. Ион Fe3+ не может быть превращен в устойчивый
ион с большим зарядом, этот ион также может только принимать электроны.
Таким образом, реакция между этими двумя ионами принципиально
невозможна.
2. Рассмотрим другой более сложный пример. Возможно ли взаимодействие
фторид-иона с брихромат-ионом в кислой среде? Для написания реакций
нужно хорошо знать свойства элементов и их соединений. Бихромат калия в кислой
среде, будучи окислителем, переходит в ион 3-валентного хрома Сг3+. Составим
уравнение реакции
СгХ>2-_+Сг3+,
Сг20?--*2Сг8*\
Cr20~-->2Cr3+ + 7H20,
Cr2Of~ + 14H+ -> 2Сг3+ Н- 7Н20,
Сг20^- |-14Hf+ 6е ^2Сг3+ + 7Н20.
Итак, бихромат-ион может выступать только в качестве окислителя. Для
фторид-иона возможна только реакция
2F- —2^ = F2.
В одной реакции электроны принимаются, в другой отдаются,
следовательно, окислительно-восстановительная реакция может быть написана. Обратимся
к численным значениям для выяснения возможности ее протекания:
347
Cr2Of- -h HH+ + 6j - 2Cr3+ -f 7H20 E -4-1,33 в,
F, j 2e -2F- E -- h 2,87 в.
Потенциал второй реакции выше потенциала первой, следовательно,
самопроизвольно может протекать только вторая реакция, а первая под действием
последней возможна только в обратном направлении. Таким образом,
взаимодействие F~ и СьО2/- ионов невозможно.
Возьмем вместо фторид-иона хлорид-ион. Потенциалы реакций
Cr2Of- -h 14H+ f & 2СгЗ+ -L 7H20 E - \- 1,33 в,
С12 + 2е--2С1- £ = +1,36 b.
Потенциал второй реакции снова выше потенциала первой, поэтому в данных
условиях в прямом направлении протекает только вторая реакция, а первая в
обратном направлении. Тем не менее, если к раствору бихромата калия прилить
соляной кислоты, начинается самопроизвольная реакция вытеснения свободного
хлора и образования ионов Сг3+.
Противоречие теории с опытом объясняется тем, что ^2Сг3+/Сг2о2~~ ~т~1»33
относится к раствору, в котором концентрации всех ионов равны по
1 г-ион/л. Вычислим ^2Сг3+/Гг2о2— для растворов с большей концентрацией
ионов водорода, например для 2н. раствора НС1. Воспользуемся формулой
Нернста
,0 0,058 , ссг3+
-2Cr3f/Cr2o2—~" ^2Гг3+/Сг2о2
^9Гг3+/Сг„о2- : Jg
ССг2о2— СН+
Пусть ССгз+ -- Cq. 02— — 1 г-ион/л. В 2 н. растворе НС1 Сн+ = 2 г-ион/л.
Поэтому
0,058 1 л 0,058 (—14) , ft
£2Cr3Vcr2o^ -1.33-—^ lg — ^i9&-—i—± i-,g2 =
0,058-14.0,301
- 1,33 -f- ■ ■ = 1,33 + 0,04- 1,37 в.
6
Итак, в 2н. растворе кислоты потенциал ^2Сг3+/Сг о2 - > ^2СГ/С1 * в таком
растворе реакция
Ст2027~ + 14H+ + 6е -= 2Сг3+ 4- 7НаО
протекает в написанном направлении, тогда как вторая реакция возможна
только в обратном направлении.
2С1- — 2е--С12.
Умножая стехиометрические коэффициенты второй реакции на 3, чтобы
число принятых и отданных электронов было одинаковым, получаем уравнение
самопроизвольно протекающей окислительно-восстановительной реакции
0,0?," + 6CI- + 14Hf - 2Сг3* -f ЗС12 + 7Н20
348
или в молекулярном виде
К2Сг207 + 14НС1 -- 2СгС13 + ЗС12 + 2KC1 f 7H20.
Практически же потенциал £2^ з+/Сг 02— может быть повышен намного*
2 у
увеличением концентрации ионов Сг2Оу~".
Учет концентрации ионов при предсказании вероятности протекания
реакции следует проводить только з тех случаях, когда потенциалы двух
сравниваемых реакций восстановления лежат очень близко; практически же при
разнице больше, чем в 0,1 в, о возможности протекания
окислительно-восстановительной реакции можно судить по стандартным потенциалам.
В заключение отметим, что использование восстановительного потенциала
представляет частный случай использования изобарно-изотермических
потенциалов. По изобарно-изотермическим потенциалам можно судить о возможности
протекания всех реакций при любых условиях. Восстановительные же
потенциалы используются только при температуре 25°С и давлении 1 атм.
Вопросы и упражнения
1. Какой из ионов Мп04 или Re04 более сильный окислитель?
2. В каком виде 3-валентное железо является более сильным окислителем —
в виде ионов Fe3+ или [Fe(CN)6]3~? Указанные ионы переходят в ионы Fe2+
и [Fe(CN)e]4_ соответственно.
3. В каком виде 2-валентный кобальт проявляет более сильные
восстановительные свойства — в виде ионов Со3+ или [Со(ЫН3)б]2+? В реакции указанные
ионы переходят в Со3+ и [Co(NH3)e]3+ соответственно.
4. Можно ли использовать бихромат калия в качестве окислителя для
осуществления следующих процессов при стандартных условиях:
2F- —2e-F2,
2С1- —2е = С12,
2Вг- — 2е= Вг2,
HN02 + Н20 ~ 2е - NOJ- + 3H+,
Mn2 f 4 4Н20 — Ъе -- МпО^ + 8НЧ ,
2НаО —2е = НД + ?Н+,
H2S — 2е - S + 2Н+?
5. Проанализируйте возможность взаимодействия перхлорат-иона СЮ^" и
двуокиси марганца (в кислой среде).
6. Возможна ли реакция между перманганатом калия и метиловым спиртом
в кислой среде с образованием метана?
7. Возможна ли реакция между перманганатом калия и щавелевой
кислотой Н2С204 в кислой среде с образованием углекислого газа?
8. Вычислите при стандартных условиях э. д. с. элемента, состоящего из
Мп24 /МпО^ и Н202/02 электродов, и напишите уравнение самопроизвольно
протекающей реакции.
349
9. Составьте суммарное уравнение для каждой из следующих реакций:
Спр27- + Fe2+ + Н+ ->- Сг3" ч- Fe34 -\- Н20,
Си + N0^ + Н+ ->■ Си24 + N0 + Н20,
Вг- + SOl~ + Н+ ->- S02 + Вг2 + Н20,
Zn + N0~ + Н+ -> Zn24" + N02 + H20,
h2o2 + i- + h+ + i2,
Cl2 + Br--^Cl"-LBr.,,
NO- + Сг20|~ + H * -> N0^ + Сг3+,
Мп02 + Вг2 + ОН" ->■ МпО2" + Вг-,
Сг О2- + S0~- -г ОН- -*- СЮ7 + SO** -f Н20.
ГЛАВА 11
ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ.
КОРРОЗИЯ
Электрохимия — область химии, изучающая процессы, которые
либо сопровождаются возникновением электрического тока, либо
вызваны электрическим током. Преобразование химической энергии
реакции в электрическую осуществляется в устройствах,
называемых химическими источниками тока, или гальваническими
элементами. Химические же превращения за счет внешней электрической
энергии происходят в электролитических ваннах или
электролизерах.
§ 1. СТАНДАРТНЫЕ ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
Электрохимические процессы относятся к
окислительно-восстановительным. В окислительно-восстановительных реакциях
электроны непосредственно переходят от восстановителя к окислителю
(см. гл. 10). Если же процессы окисления и восстановления
пространственно разделить, а электроны направить по металлическому
проводнику, то такая система будет представлять собой
гальванический элемент.
Причиной возникновения и протекания электрического тока в.
гальваническом элементе является разность электродных
потенциалов.
Поясним, что такое электродный потенциал. Представим себе,
что пластинка какого-нибудь металла опущена в воду. В металле
существует подвижное равновесие, которое можно выразить
уравнением
Ме^М"++ш>, (11.1>
где Me — атом металла; Меп+ — ион металла в электроде; п —
заряд иона и число электронов, е — электрон.
При погружении металла в воду ионы его поверхностного слоя
под действием полярных молекул воды отрываются и гидратиро-
ванными переходят в жидкость. В результате такого перехода
жидкость заряжается положительно, а металл — отрицательно,
поскольку на нем появляется избыток электронов. По мере перехода1
ионов металла в водную среду, увеличивается как отрицательный
заряд металла, так и положительный заряд раствора, при этом
все чаще и чаще ионы металла притягиваются обратно на
металлическую пластинку. Накопление ионов металла в растворе
начинает тормозить дальнейшее растворение металла. В результате
351
устанавливается подвижное равновесие, которое можно
представить в виде
Me -+ тН20 ^ Me (Н20)"гч" + пе. (11.2)
Состояние равновесия зависит как от активности металла, так
и от концентрации его ионов в растворе.
В случае активных металлов (Zn, Fe, Cd, Ni) взаимодействие
с полярными молекулами воды оканчивается отрывом от поверх-
Ч~Утлпнг^-
гстч
Ч^Н«й>*^- Рис. 11.1. Схема
возникновения электродного
потенциала
Рис. 11.2. Водородный
электрод
ности положительных ионов металла и переходом гидратированных
ионов в раствор (рис. 11.1,а). Этот процесс является окислением.
По мере увеличения концентрации катионов у поверхности
становится вероятным обратный процесс — восстановление ионов
металла.
Электростатическое притяжение между катионами в растворе и
избыточными электронами на поверхности образует двойной
электрический слой. Это приводит к возникновению на границе
соприкосновения металла и жидкости определенной разности
потенциалов, или скачка потенциала.
Двойной электрический слой можно уподобить плоскому
конденсатору с определенной разностью потенциалов. Разность
потенциалов, возникающую между металлом и окружающей его
водной средой, называют электродным потенциалом.
При погружении металла не в воду, а в раствор соли этого
металла равновесие (11.2) смещается. Повышение концентрации
ионов данного металла в растворе, очевидно, облегчает процесс пе-
352
рехода ионов из раствора в металл, и следовательно, равновесие
установится при другом потенциале заряда металла. Металлы,
ионы которых обладают значительной способностью к переходу в
раствор, будут заряжаться и в таком растворе отрицательно, но в
меньшей степени, чем в чистой воде.
Для неактивных металлов равновесная концентрация ионов
металла в растворе очень мала. Если такой металл погрузить в
раствор соли с большой концентрацией ионов этого металла, то
в этом случае имеющиеся в растворе положительные ионы
выделяются на металле первоначально с большей скоростью, чем
происходит переход ионов из металла в раствор.
В этом случае поверхность металла получит положительный
заряд, а раствор — отрицательный из-за избытка анионов соли. И в
этом случае на границе металл—раствор возникает двойной
электрический слой и, следовательно, определенная разность
потенциалов (см. рис. 11.1, б).
Таким образом, при погружении металла в воду или в раствор,
содержащий ионы данного металла, на поверхности раздела
металл—раствор образуется двойной электрический слой и
возникает разность потенциалов между металлом и раствором.
Потенциал каждого электрода зависит от природы металла,
концентрации его ионов в растворе и температуры. Если металл
опустить в раствор его соли, содержащий 1 г-ион металла в 1 л
(точнее, активность которого а=1), то электродный потенциал
будет постоянной величиной при данной температуре и давлении.
Такой потенциал называется нормальным, или стандартным
электродным потенциалом.
§ 2. ГАЗОВЫЕ ЭЛЕКТРОДЫ
Благородные металлы Au, Pt и другие благодаря высоким
значениям энергии ионизации не могут создать разность потенциала
за счет выхода положительных ионов в раствор.
В возникновении скачка потенциала на границе благородный
металл—раствор в случае, если последний не содержит катионов
данного металла, важную роль играет избирательная адсорбция
молекул, атомов или ионов среды. Например, платиновый
электрод, покрытый тонким слоем рыхлой платины для увеличения
его поверхности, энергично поглощает атомарный водород. Это
используется в так называемом водородном электроде.
Опытным путем можно определить только относительные
величины так называемых электродных потенциалов, т. е. э. д. с.
цепи, составленной из данного электрода и некоторого
стандартного электрода, потенциал которого условно принимают равным
нулю, таким стандартным электродом, или электродом сравнения,
является обратимый водородный электрод (рис. 11.2).
Он состоит из платиновой пластинки, покрытой платиновой
чернью, т. е. электролитически осажденной платиной. Электрод по-
12 Общая химия
353
гружен в 2 н. водный раствор серной кислоты (точнее с
активностью ионов Н+=1) и омывается струей газообразного водорода
под давлением в 1 атм. Величину потенциала такого электрода
условно принимают за нуль (при всех значениях температур).
При насыщении платины водородом на поверхности металла
устанавливается равновесие Н2^2Н, а на границе электрода с
раствором — равновесие Н^Н++е, а суммарный процесс
выразится уравнением
2Н+ + 2*:£На.
Схематически водородный электрод обозначают Н+|Н2, Pt,
где вертикальная черта обозначает поверхность раздела фаз.
Электродный потенциал такого электрода зависит от
концентрации ионов водорода в растворе, от давления водорода в газовой
фазе и от температуры. Платина здесь играет роль только
инертного проводника. Когда водородный электрод соединяется с
электродом, окисляющимся легче водорода, на нем протекает процесс
разряда ионов водорода:
2Н+ + 2е^Н2.
При соединении водородного электрода с электродом,
окисляющимся труднее водорода, на нем протекает процесс
Н2 — 2е ^ 2Н+.
Подобно водородному может быть создан кислородный электрод
ОН-|02, Pt, на границе раздела фаз которого протекает процесс
02 + 2Н20 + 4е^40Н-.
Потенциал такого электрода зависит от концентрации ОН_-ио-
ноз в растворе и парциального давления кислорода.
На границе инертного электрода, находящегося в растворе,
насыщенном хлором, устанавливается равновесие
С12 + 2е:£2С1-
и возникает потенциал, величина которого зависит от
концентрации ионов хлора в растворе и парциального давления хлора в
растворе. Это хлорный электрод С1~|С12, Pt.
§ 3. ИЗМЕРЕНИЕ ЭЛЕКТРОДНЫХ ПОТЕНЦИАЛОВ.
РЯД НАПРЯЖЕНИЙ
Непосредственно измерить величину потенциала отдельного
электрода нельзя. Ее измеряют относительно величины другого
электрода. Если пластинку металла, погруженную в раствор ее
соли, содержащий 1 г-ион в 1 л (точнее, с активностью ионов,
равной 1), соединить электролитическим ключом со стандартным
водородным электродом, то получится гальванический элемент
354
(рис. 11.3), электродвижущую силу (э. д. с.) которого можно
легко измерить.
Эта э. д. с. будет величиной стандартного электродного
потенциала металла при температуре измерения (так как стандартный
потенциал водородного электрода условно принят равным нулю).
Стандартный электродный потенциал будем обозначать буквой Е0.
Рис. 11.3. Гальваническая цепь для измерения электродного
потенциала:
а — электролитический ключ; б — водородный электрод
Взяв в качестве электрода цинк и соединив его с водородным
электродом, получим гальванический элемент, схема которого
запишется следующим образом:
(-)Zn|Zn2+||H2S04|H2, Pt(+).
В таком элементе осуществляется реакция
Zn + 2Н+ = Zn2+ + Н2
и электроны во внешней цепи протекают от цинкового к
водородному электроду, Е0 такого цинкового электрода —0,76 в.
Взяв в качестве электрода медный электрод при указанных
условиях в сочетании со стандартным водородным электродом,
получим гальванический элемент:
12*
355
(-)Pt, H2||H2SOJCu2+|Cu(+)
и Е0 = + 0,34 в. В этом случае протекает реакция
Си2+ 4- Н2 = Си + 2Н+,
а электроны во внешней цепи будут двигаться от водородного
электрода к медному.
Как видно из приведенных примеров, стандартные потенциалы
электродов, посылающих электроны к водородному электроду,
имеют знак «минус», а принимающих электроны от водородного
электрода, — знак «плюс».
Располагая металлы в порядке возрастания их, получают
электрохимический ряд напряжений металлов, или, точнее, ряд
стандартных электродных потенциалов:
Li, Rb, К, Ва, Sr, Са, Na, Mg, Al, Mn, Zn, Cr,
Fe, Cd, Co, Ni, Sn, Pb, H, Sb, Bi, Cu, Hg, Ag, Pd, Pt, Au.
Числовые значения стандартных потенциалов для ряда
технически важных металлов приведены в табл. 11.1.
Таблица 11.1
Стандартные электродные потенциалы металлов
Электрод
LiMLi
Rb-f | Rb
K+IK
Ва2 + |Ва
Sr2- | Sr
Са2- | Са
Na+ | Na
La3 + | La
Mg2J | Mg
Al3- | Al
Mn2+|Mn
Zn2+ | Zn
Cr3 h | Cr
Fe2^ | Fe
Cd2" | Cd
Co2-|Co
Потенциал,
в
—3,02
—2,99
—2,92
—2,90
—2,89
—2,87
—2,71
—2,37
—2,34
— 1,67 !
— 1,05
—0,76
-0,71
—0,44
—0,40
—0,28
Электрод
Ni2+|Ni
Sn24-|Sn
рь2 ч рь
i
Н4|тНз
sb3 ч sb
Bi3f|Bi
Cu24|Cu
Cu+ | Cu
Hg5+-|2Hg
Ag+ | Ag
Pd2+ | Pd
Hg24Hg
Pt2- | Pt
Au3^ |Au
Потенциал,
в
—0,25
—0,14
—0,13
0,00
+0,20
+ 0,23
+0,34
4 0,52
+0,79
+0,80
-f0,83
-Ь0,86
+ 1,20
+ 1+2
В табл. 11.1 потенциалы расположены по возрастанию их
величин, что соответствует уменьшению восстановительной и
повышению окислительной активности соответствующих систем. Если
составить электрохимическую цепь из двух электродов этого ряда,
то на одном из них, потенциал которого ниже по сравнению с
другим, будет идти процесс окисления, а на другом — процесс
восстановления.
356
Ряд напряжений характеризует химические свойства металлов:
1. Чем левее в ряду напряжений находится металл, тем он
химически активнее, тем легче окисляется и труднее
восстанавливается из своих ионов.
2. Каждый металл этого ряда, не разлагающий воду,
вытесняет (восстанавливает) все следующие за ним металлы из
растворов их солей.
3. Все металлы, стоящие в ряду напряжений левее водорода,
вытесняют (восстанавливают) его из разбавленных кислот (типа
серной или соляной, анионы которых не проявляют окислительных
свойств), а стоящие правее водорода его не вытесняют.
4. Чем дальше расположены друг от друга в ряду напряжений
два данных металла, тем большую э. д. с. будет иметь
построенный из них биметаллический гальванический элемент (т. е.
гальванический элемент, у которого электроды изготовлены из разных
металлов).
Положение некоторых элементов, например Li, Na и других,
в ряду напряжений может показаться неожиданным. Оно прежде
всего свидетельствует о недопустимости отождествления
восстановительной способности металлов в водных растворах с их
восстановительной активностью в твердой фазе. Ряд напряжений
применим только к водным растворам.
Для стандартных электродных потенциалов действительно
выражение
— AG°=/iF£0f] (11.3)
где AG0 — уменьшение свободной энергии для электродной реакции
(см. уравнение 11.2), протекающей в условиях, когда все
реагирующие вещества находятся в стандартном состоянии при
активности, равной единице; п — число электронов; F — число Фарадея.
Так как Е0 для водородного электрода принято равным нулю,
то, следовательно, AG0 реакции, протекающей в водородном
электроде, должно быть также принято равным нулю.
Если пренебречь изменением энтропии, то уравнение (11.3)
можно переписать в следующем виде:
— Atf°~ttF£0. (11.4)
Общая электродная реакция
Мег + aq -ъ MeJ#" + ne (А Я0) (11.5)
состоит из следующих стадий:
Ме"+.aq_>.Me""*" теплота гидратации иона Ме'2+(А//Л) поглощается;
Me"+-f /2£->Мег энергия ионизации п электронов /ме выделяется;
Мег-ьМек энергия сублимации металла SMe выделяется.
357
Таким образом:
ДЯ° = -ДЯЛ —/&е —SMe.
Значения этих величин приведены в табл. 11.2.
Таблица 11.2
Теплоты различных электродных реакций
Электрод
Li
Na
Н
Ag
AHh
—123
—95
—256
—111
in
124
118
312
174
SMe
36
26
52
67
Art9
-37
—49
—108
— 130
Л
+71
+59
—
—22
E0, в
—3,04
—2,71
0,00
+0,80
Разность значений АЯ° соответствующих металлов и водорода,
обозначенная Д, может характеризовать относительную активность
металлов, хотя пропорциональность между А и £0 не вполне
точная. Термохимические величины выражены в ккал/моль.
Из табл. 11.2 видно, что высокоотрицательное значение Eq для
системы Li+/Li определяется большой энергией гидратации иона
лития. Из тех же соображений можно объяснить относительное
положение и других металлов в ряде напряжений.
Величина электродного потенциала металла зависит от свойств
металла, активности его ионов в растворе яМеп+.тН 0 и
температуры Г. Эта зависимость выражается уравнением Нернста
RT
£ме = £о,Ме + —£- 1П aMe«+.mHi0. 0 l 6)
где Еме — потенциал металла в вольтах при данной активности
ионов; Е0> ме — стандартный электродный потенциал металла; R —
универсальная газовая постоянная, равная 9,314 дж/град-моль;
Т — температура в градусах абсолютной шкалы; п — заряд
(валентность) иона металла; F — число Фарадея, равное 96500 к;
амеп+тн2о— активность ионов металла в растворе, в г-ионах/л.
Примем температуру равной 20°С и подставим в уравнение
(11.6) значения R, F и Г, умножив на 2,3 (для перехода от
натуральных логарифмов к десятичным), получим
£ме = £о,Ме + ^ 180Ме*+.тН,О- (ПЛ)
Как известно, для разбавленных растворов активность с
достаточным приближением может быть заменена концентрацией.
Например, для цинкового электрода, помещенного в раствор своей
соли, концентрацию гидратированного иона цинка [Zn2+-mH20]
сокращенно обозначим [Zn2+]:
358
£o,zn = £o,zn + °^58 lg [Zn'+].
Если [Zn2+] = 1, то £zn = £c,zn-
§ 4. ГАЛЬВАНИЧЕСКИЕ ЭЛЕМЕНТЫ
Два металла, погруженные в растворы их солей, соединенные
между собой электролитическим ключом, образуют гальванический
элемент.
Первый гальванический элемент был применен Вольта. Этот
элемент состоял из медных и цинковых пластинок, разделенных
е е
г~ © HL
Рис. 11.4. Схема медно-цинкового элемента
сукном, смоченным раствором серной кислоты. При
последовательном соединении большого числа пластинок элемент Вольта
обладает значительной э. д. с.
Возникновение электрического тока в гальваническом элементе
обусловлено разностью электродных потенциалов взятых металлов
и сопровождается глубокими химическими превращениями,
протекающими на электродах. Покажем это на примере работы
медно-цинкового элемента (рис. 11.4).
На цинковом электроде, опущенном в раствор ZnS04,
происходит окисление атомов цинка в ионы (растворение Zn):
Zn — 2e = Zn2+. (A)
Электроны поступают во внешнюю цепь. Zn — источник
электронов. На электроде из меди, погруженном в раствор C11SO4, проис-
359
ходит восстановление ионов металла в атомы (осаждаются на
электроде):
Cu2+^ 2е-Си. (Б)
Одновременно часть ионов S042~ переходит в растворе через
пористую перегородку в сосуд с раствором ZnS04.
Суммарное уравнение процесса получим, сложив уравнение (А)
и (Б):
Zn — 2e=Zn2+ (анодный процесс),
Си2+ + 2е = Си (катодный процесс)
или в молекулярной форме
Zn + CuS04 = ZnS04 + Cu.
Это — обычная реакция окисления-восстановления. Электрическая
энергия такого гальванического элемента, называемого элементом
Даниэля—Якоби, получается за счет химической энергии последней
реакции.
Цинковый электрод ■— источник электронов, поступающих во
внешнюю цепь, — принято считать отрицательным, а медный
электрод — положительным. Названия электродам дается в
соответствии с процессами, которые на них протекают: электрод, на котором
протекает процесс окисления, называется анодом, а электрод, на
котором протекает процесс восстановления, — катодом.
В рассматриваемом элементе1 цинк—анод (А), медь—катод
(К).
Гальванический элемент можно записать в виде краткой
электрохимической схемы, например:
(-)Zn|Zn2+||Cu2+|Cu(+),
где одна черта означает границу между электродом и раствором,
две черты — границу между растворами, в скобках знаки ионов,
причем анод записывается слева, катод справа.
Необходимое условие работы гальванического элемента —
разность потенциалов его электродов, она называется
электродвижущей силой гальванического элемента — э. д. с.
Э. д. с. всякого работающего элемента — величина
положительная. Э. д. с. любого биметаллического элемента можно
вычислить по разности стандартных электродных потенциалов — для
этого надо из величины более положительного потенциала вычи-
В случае электролиза (см. ниже) название электродов обратное, а именно:
отрицательный электрод называется катодом (соединен с отрицательным
полюсом источника тока), а положительный — анодом (соединен с
положительным полюсом источника тока). Разница обусловлена тем, что условились
называть электрод, уводящий электроны из данной системы, анодом, а электрод,
вводящий электроны, — катодом.
360
гать величину менее положительного. Так, э. д. с. Е медно-цинко-
ного элемента, в котором эти металлы погружены в молярные
растворы их солей, равна разности между стандартными
потенциалами меди (катода) и цинка (анода), т. е.
£=£0,Cd2+|Cu —£0,Zn2+iZn = -г0,34— (—0,7 В) = 1,10 В.
Можно измерить максимальную полезную работу, совершаемую,
например, при выделении 1 г-атома меди в медно-цинковом
гальваническом элементе.
Общие термодинамические условия обратимости применительно
к работе гальванических элементов могут быть сформулированы
следующим образом. Гальванический элемент работает обратимо
при соблюдении двух условий: 1) если его э. д. с. лишь на
бесконечно малую величину превышает приложенную к нему извне и
противоположно направленную э. д. с. и 2) если реакция в
элементе может быть полностью обращена в противоположном
направлении при приложении к нему извне противоположно
направленной э. д. с, которая лишь на бесконечно малую величину
превышает э. д. с. данного элемента.
Как известно, в случае обратимой реакции, протекающей при
постоянных температуре и давлении, получаемая от нее работа
будет максимальной полезной работой реакции ЛмаКс- Работа,
совершаемая элементом в этих условиях, равна э. д. с. элемента Е,
уменьшенной на количество прошедшего электричества:
Л,аМ=-ДОя» = /№ (П.8)
где п — заряд иона; F — число Фарадея.
Таким образом, э. д. с. элемента характеризует максимальную
полезную работу, которая может быть произведена системой при
протекании в ней данной химической реакции.
Так, для медно-цинкового элемента
4iaRC = — AG298 =2.96500-1,1 = 212300 дж = 50,7 ккал.
Это значение AG298 весьма близко к измеренной стандартной
энтальпии для этой реакции, равной —51,85 ккал. Если ту же реакцию
Zn+CuS04=Cu+ZnS04 провести просто, опустив пластинку
цинка в раствор сульфата меди, то практически все изменение
энтальпии системы перейдет в теплоту, а работа будет равна нулю.
Примерно то же произошло бы при коротком замыкании элемента,
так как в этом случае процесс в нем происходил бы в
термодинамически необратимых условиях, когда А<^.АМШС.
Величины стандартных электродных потенциалов и уравнение
Нернста дают возможность вычислять э. д. с. гальванических
элементов при различных концентрациях электролитов. Так, например,
если в том же медно-цинковом элементе
ССи2+ = 1,5 г-ион/л, a CZn2-j- = 0,01 г-ион/л,
361
то потенциал медного электрода
_0
2
£Cu2+/Cu = 0,34 + -Мр- lg 1,5 = 0,345 в;
потенциал цинкового электрода
£Zn2+/zn = -°'76+ °'1|58 IgO.Ql = —0,818 в,
откуда
э. д. с. -£Cu2+/Cu-£Zn2+/Zn== о,345-(-0,818)= 1,163 в.
Необходимую для работы гальванического элемента разность
потенциалов можно создать, используя один и тот же раствор
разной концентрации и одинаковые электроды. В этом случае
гальванический элемент называется концентрационным, а работает он
из-за выравнивания концентраций раствора. Такие элементы
находят применение при измерении концентрации ионов в растворе.
Примером концентрационного элемента может служить элемент
составленный из двух водородных электродов:
Pt, H2|H2S04||H2S04|H2, Pt.
I, П.
Сн+ сн+
Если PUi = 1 атм, а СН4-<СН+, то его э. д. с. при 25°С
определится уравнением
£ = 0,0581g-^— •
Сн+
При Сн+=1 г-ион/л э. д. с. этого элемента будет определяться
концентрацией водородных ионов во втором растворе, т. е. Е=
=0,058 lgCH+=— 0,058 рН.
Определение концентрации ионов Н+ и, следовательно, рН
среды измерением э. д. с. соответствующего гальванического элемента
называется потенциометрическим.
В качестве электрода сравнения при определении э. д. с. элемента вместо
стандартного водородного электрода часто используют другие электроды,
более простые в изготовлении и надежные в работе. Наиболее распространен в
лабораторной практике каломельный электрод. Активная масса его
представляет собой пасту из ртути и каломели Hg2Cl2, в которую погружен электрод
(Pt, Ni, Fe и др.) Паста залита насыщенным раствором хлористого калия.
Схема каломельного электрода: КС1, Hg2Cl2|Hg. Электродная реакция протекает
по уравнению
Hg2Cl2 + 2e^2Hg + 2Cl-
362
Потенциал его определяется соотношением
RT
^калом ~ ^0,калом np 'n \Q~\
и при 25 °С равен +0,2458 в (по нормальному водородному электроду).
§ 5. ПОЛЯРИЗАЦИОННЫЕ ЯВЛЕНИЯ
В ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТАХ
В результате поляризации электродов э. д. с. работающего
элемента всегда меньше той, которая отвечает обратимой
электрохимической реакции. В элементе Даниэля—Якоби растворение
цинкового электрода приводит к накоплению ионов Zn2+ в приэлек-
тродном слое, поэтому потенциал цинка повышается. У медного
электрода концентрация ионов Си2+ уменьшается в результате их
восстановления, а потенциал меди понижается. Смещение
величины потенциала электрода от исходного равновесного значения,
вызванное изменением концентрации потенциал определяющих
ионов в растворе, называется концентрационной поляризацией.
В том случае, когда поляризация вызывается изменением
химической природы электрода, ее называют химической. Так, медный
электрод в элементе Вольта инертный и на границе с раствором
серной кислоты, насыщаясь водородом, образует «водородный»
электрод, потенциал которого ниже потенциала медного электрода.
Поэтому э. д. с. элемента Вольта непрерывно падает.
Процесс устранения поляризации называется деполяризацией.
Концентрационную поляризацию можно в значительной степени
уменьшить перемешиванием раствора. Химическую поляризацию
можно снизить введением специальных веществ
(деполяризаторов), вступающих в реакцию с продуктами, обусловливающими
поляризацию.
Наибольшее значение имеет катодная поляризация, благодаря
которой на катоде накапливается избыток электронов, и он
становится более отрицательным. Для борьбы с ней применяют
вещества, называемые катодными деполяризаторами. Обычно это
окислители, которые принимают электроны от катода, препятствуя
поляризации, например, Си2+ (в медно-цинковом элементе), Мп02
(в марганцово-цинковом элементе) и др. Механизм процесса
деполяризации можно представить уравнениями
Си2+ 4 2е = Си, 2Мп02 + Н20 + 2е = Мп203 + 20H-.
§ 6. ЭЛЕКТРОЛИЗ
Электролизом называется окислительно-восстановительный
процесс, протекающий при прохождении электрического тока через
раствор или расплав электролита.
Химические реакции на электродах осуществляются за счет
электрической энергии. При электролизе катод — восстановитель,
363
так ка,к он отдает электроны катионам, а анод — окислитель, так
как он принимает электроны у атомов. Восстановительное и
окислительное действия электрического тока во много раз сильнее
действия химических восстановителей и окислителей.
На характер и течение электродных процессов при
электролизе большое влияние оказывают состав электролита, растворитель,
материал электродов и режим электролиза (напряжение,
плотность тока, температура и др.)- Прежде всего надо различать
электролиз расплавленных электролитов и их растворов. В последнем
случае в процессах будут участвовать молекулы воды.
Рассмотрим электролиз водного раствора хлорида натрия
(электроды угольные). В этом случае в растворе находятся гидра-
тированные ионы Na+ и О-, а также молекулы воды. При
прохождении тока через раствор катионы Na+ движутся к катоду, а
хлорид-ионы С1~ — к аноду. Для восстановления ионов Na+ к катоду
надо приложить потенциал, равный — 2,71 в, а для восстановления
молекул воды — 0,83 в К Поскольку восстановление молекул воды
требует большей величины потенциала, то на катоде разряжаться
будут молекулы воды, а не ионы натрия:
2Н20 + 2е =На + 20Н-,
а на аноде будут окисляться хлорид-ионы:
2С1- — 2*=С12.
В итоге на катоде выделяется водород, на аноде—хлор, а в
катодном пространстве накапливается NaOH. Сложим уравнения
двух электродных процессов и получим общее уравнение
электролиза водного раствора хлорида натрия:
2Н20 + 2е = Н2 + 20Н-
2С1- — 2е^С12
2Н20 + 2С1- = Н2 + С12 + 20Н-
или в молекулярной форме:
2Н20 + 2NaCl = Н2 + С12 + 2NaOH.
Большое влияние на течение процессов электролиза оказывает
природа анодов. Различают нерастворимые и растворимые аноды.
Нерастворимые аноды изготовляют из угля, графита, платины. При
электролизе нерастворимые электроды сами не посылают
электронов во внешнюю цепь, электроны посылаются в результате
окисления анионов и молекул воды. При этом анионы бескислородных
кислот (S2~, I~, Br~, С1~ и др.) при их достаточной концентрации
окисляются довольно легко. Если же раствор содержит анионы
1 Вода как окислитель реагирует по уравнению 2H20-b2e = H2+20H-. Эта
реакция протекает при катодном потенциале — 0,83 в.
364
кислородных кислот (например, S04~~, N07, CO;f~, POf"), то на
л поде окисляются не эти ионы, а молекулы воды.
В случае растворимого анода электроны во внешнюю цепь
посылает сам анод, а не анионы раствора. Растворимые аноды
изготовляются из меди, серебра, цинка, кадмия, никеля, железа и др.
Однако при определенных условиях нерастворимыми становятся и
аноды из железа, никеля, свинца и др.
В качестве примера можно привести электролиз раствора
хлорида меди (II) при медном аноде. Протекающие процессы можно
изобразить так:
на аноде Си — 2е = Си2+,
на катоде Си2+ j- 2e= Си.
В данном случае процесс сводится к анодному окислению атомов
меди и катодному восстановлению ионов меди, т. е. к переносу
меди с анода на катод. При этом количество ионов Си2+ в растворе
остается неизменным.
Закономерности электролиза водных растворов
распространяются и на расплавы. Однако отсутствие в этом случае воды
сказывается и на характере электродных процессов. Электролиз
расплава, например NaCl, при нерастворимом аноде
сводится к восстановлению катиона (на катоде) и окислению аниона (на
аноде). Но если применять растворимый анод, например медный,
то и в расплавах может происходить анодное растворение металла.
§ 7. НАПРЯЖЕНИЕ РАЗЛОЖЕНИЯ.
ЯВЛЕНИЕ ПЕРЕНАПРЯЖЕНИЯ.
ПОСЛЕДОВАТЕЛЬНОСТЬ РАЗРЯДА ИОНОВ
Процессы электролиза в принципе обратны процессам работы
соответствующих гальванических элементов, и при обратимом
процессе термодинамическая характеристика их должна совпадать.
Однако при практическом проведении электролиза процесс
сопровождается большей частью теми или иными побочными явлениями,
делающими его не вполне обратимым.
Наименьшая разность потенциалов, необходимая для
проведения данного процесса электролиза, называется напряжением
разложения.
При обратимом процессе напряжение разложения равно э. д. с.
гальванического элемента. Однако чаще оно оказывается большим.
Так, при электролизе водных растворов H2S04, HN03, H3PO4,
NaOH, КОН в конечном счете происходит только разложение воды
с выделением на катоде водорода, а на аноде — кислорода.
Напряжение разложения всех этих электролитов близко к 1,70 в.
Казалось бы, оно должно отвечать э. д. с. цепи Pt, H2| кислота IO2,
Pt. Но для этой цепи э. д. с. равно 1,07 в, а найденное напряжение
разложения больше его на 0,63 в. В других случаях при
электролизе нередко нужно применять разность потенциалов, большую,
365
чем э. д. с. обратной цепи. Это явление называется
перенапряжением при электролизе.
Перенапряжение при электролизе равно разности между на-
пряжением (разностью потенциалов), наложенным на электроды,
и э. д. с. гальванического элемента, отвечающего обратной
реакции.
При очень малой плотности тока перенапряжение щ равно
разности между напряжением разложения £разл и э. д. с.
соответствующего гальванического элемента Е, т. е.
т]0 = £разл £•
Перенапряжения на электродах представляют собой
соответствующие составляющие перенапряжения при электролизе.
Катодное перенапряжение — это дополнительное напряжение,
прикладываемое к катоду (при этом потенциал катода смещается
далее в отрицательную сторону), а анодное — к аноду (при этом
потенциал анода смещается далее в положительную сторону).
Можно было бы ожидать, что при разряде положительных
ионов (катионов) на катоде легче всего будут разряжаться те из них.
которым отвечает наибольшее значение положительного
потенциала. Аналогично при переходе с анода в раствор каких-либо
положительных ионов (анодное растворение вещества) легче всего в
раствор будут переходить те из них, которым отвечает наибольшее
значение отрицательного потенциала. Однако указанная
последовательность разряда ионов и их образования на электродах часто
нарушается из-за перенапряжения.
Так, при электролизе кислого раствора сульфата цинка на
катоде в первую очередь должны были разряжаться водородные
ионы, а затем ионы цинка, так как £02Н+/Н|== 0,000 в, а потенциал
£'0Zn2+/Zn-=—0,76 в. Но так как перенапряжение водорода на
цинке очень велико (^0,70 в), то фактически в указанных
условиях будет выделяться и цинк. Выделение таких металлов, как Fe
и РЬ, которые стоят выше водорода в ряду напряжений, может
осуществляться только благодаря тому, что они обладают
перенапряжением, значительно меньшим, чем перенапряжение
водорода на этих металлах, в особенности при высоких плотностях тока.
Поэтому потенциал выделения водорода становится большим, чем
потенциал выделения этих металлов. В других случаях, например
при электролитическом получении водорода, перенапряжение,
наоборот, нежелательно, так как приводит к повышенному расходу
электроэнергии.
§ 8. ЗАКОНЫ ФАРАДЕЯ
Между количеством выделившегося при электролизе вещества
и количеством прошедшего через электролит электричества
существует связь, которая находит отражение в двух законах Фара-
дея.
366
/ закон Фарадея. Массы веществ, выделившихся на электроде
при электролизе, прямо пропорциональны количеству
электричества, прошедшего через раствор электролита.
II закон Фарадея. Одинаковые количества электричества
выделяют при электролизе на электродах эквивалентные массы
различных веществ. Для выделения на электроде одного
грамм-эквивалента любого вещества необходимо затратить одно и то же
количество электричества, а именно 96 493,1, или ~ 96 500 кулона,
называемое числом Фарадея, или фарадеем.
Так, при пропускании 96 500 к электричества через раствор
СиС12 при угольном аноде на катоде выделится 1 г-экв меди
(31,77 г) и одновременно на аноде 1 г-экв хлора (35,46 г).
Из законов Фарадея вытекает уравнение
т = -^-=Э—^-, (11.9)
F 96 500 '
где m — масса окисленного или восстановленного вещества, г;
Э — грамм-эквивалент вещества; Q — количество кулонов
электричества, прошедшего через электролит (Q = I-tt где /—сила тока
в амперах; t — продолжительность электролиза, сек.).
к
Так как Э = » где А — атомный вес элемента; п — его за-
п
рядность в соединении, формулу (11.9) можно представить в виде
r Aft
m = ДУ . (11.10)
п.96 500 '
Если It = 1 к, то вес выделившегося вещества составляет:
+ -—=£, (11.11)
п.96 500
или, что то же
Э
96 500
= Е.
Величина Е называется электрохимическим эквивалентом
вещества и характеризует собой массу вещества, окисляющегося или
восстанавливающегося на электродах при прохождении через
электролит 1 к электричества. Как видно, химический эквивалент
связан с электрохимическим соотношением
3 = E-F. (11.12)
Таким образом, электрохимические эквиваленты веществ
пропорциональны их химическим эквивалентам, а константа Фарадея
F равна произведению числа Авогадро на заряд электрона:
F=Ne.
367
При практическом проведении электролиза действительный
расход тока вследствие протекания тех или иных побочных процессов
(например, взаимодействие образовавшегося вещества с
электродом или электролитом и др.) обычно превышает количество его,
рассчитанное согласно закону Фарадея. Отношение массы
действительно получаемого вещества tri\ к массе, теоретически
вычисленной, называется выходом по току Лт, т. е.
Ат = mi 96500 • 100%. (11.13)
§ 9. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ ЭЛЕКТРОЛИЗА
Процессы электролиза получили широкое и разностороннее
применение в промышленности.
Путем электролиза водного раствора поваренной соли
получают едкий натр (каустическую соду) и хлор.
Получение водорода, потребляемого в больших количествах при
синтезе аммиака, осуществляется во многих случаях
электролитическим разложением воды. Для уменьшения расхода
электроэнергии электролизу подвергают не чистую воду, а раствор такого
электролита, ионы которого, отличные от Н+ и ОН~, разряжаются
много труднее, чем ионы Н+ и ОН". В результате электролит
(КОН, H2SO4) практически полностью сохраняется, а вода
разлагается на водород и кислород.
Электролитическое окисление в случае, если на аноде
образуется атомарный кислород или хлор, часто применяется для
окисления или хлорирования находящихся в растворе неорганических
веществ, которые являются в этом случае анодными
деполяризаторами.
Электролитическое же восстановление в том случае, когда на
катоде образуется атомарный водород, часто применяется для
гидрирования находящихся в растворе как неорганических, так и
органических веществ, являющихся в таких процессах катодными
деполяризаторами.
Металлы, восстанавливающиеся сравнительно легко,
выделяются обычно не путем электролиза, а с помощью наиболее дешевого
в наше время восстановителя — угля, применяемого в виде кокса.
Для металлов, наиболее трудно восстанавливаемых, уголь уже
непригоден, и в этом случае прибегают к катодному
восстановлению, т. е. выделению путем электролиза. Такие металлы могут
окисляться водой, поэтому их соединения подвергаются
электролизу не в водных растворах, а в расплавленном состоянии.
Так, металлический магний получается электролизом
расплавленного MgCb, металлический натрий — электролизом
расплавленного едкого натра, металлический алюминий — электролизом
раствора окиси алюминия в расплавленном криолите 3NaF-AlF3*
368
Все эти процессы проводятся при высокой температуре, для
алюминия, например, при 1000°С.
Другое направление применения электролиза в металлургии —
рафинирование металлов (получение их в чистом виде). В
наибольшем масштабе этот процесс применяется для рафинирования
меди. Электролитом служит C11SO4 и H2S04. Листы сырой
неочищенной (черновой) меди служат анодом. Процесс сводится к
растворению анода и выделению меди на катоде; электролит
регенерируется и сохраняется в растворе.
Электролитические покрытия металлами получили очень
широкое распространение (электролитическое никелирование,
хромирование, серебрение, меднение и др.)-
Хромирование применяется для повышения коррозионной
стойкости черных металлов, а также для увеличения твердости
поверхностного слоя и сопротивления стиранию. Никелирование
применяется для изменения внешнего вида изделия и т. д. Все эти
процессы осуществляются методами, в общем аналогичными
применяемому при рафинировании меди. Покрываемое изделие служит
катодом, покрывающий металл — анодом.
§ 10. АККУМУЛЯТОРЫ
Аккумуляторами называются гальванические элементы
многоразового и обратимого действия. Они способны превращать
накопленную химическую энергию в электрическую (при их разряде), а
электрическую — в химическую, создавая запас ее в процессе их
заряда. Другими словами, после получения от аккумуляторов
электрической энергии (разрядка) их способность снова отдавать
электрическую энергию может быть восстановлена пропусканием через
них электрического тока от внешнего источника (зарядка).
Так как э. д. с. аккумуляторов невелика, их при эксплуатации
обычно соединяют в батареи.
Свинцовый аккумулятор. В наиболее простом варианте
свинцовый аккумулятор состоит из двух перфорированных (с
многочисленными отверстиями) свинцовых пластин, одна из которых
(отрицательная) после зарядки содержит наполнитель пор —
губчатый активный свинец, а другая, положительная — двуокись
свинца (рис. 11.5). Обе пластины погружены в 25—30%-ный
раствор H2S04.
Вначале, перед зарядкой, в поры свинцовых электродов
вмазывается паста, содержащая, помимо органического связующего в.
основном окись свинца РЬО. В результате взаимодействия окиси
свинца с серной кислотой в порах обеих электродных пластин
образуется сернокислый свинец
РЬО + H2S04 - PbS04 + Н20.
Во время разрядки аккумулятора на обоих его электродах
образуется сульфат свинца PbS04, на что расходуется серная кисло-
369>
та. При зарядке сульфат на одном электроде превращается в
губчатый свинец, а на другом — в двуокись РЬ02, причем концентра-
дия H2SO4 в электролите повышается.
ГЙ
D
шшш
Рис. 11.5. Свинцовый аккумулятор:
а — общий вид; б — перфорированная пластина свинцового аккумулятора
Процесс разрядки:
на катоде Pb + SO^ = PbS04 + 2е, или
РЬ — 2е = РЬ2+;
на аноде РЬ03 + 2H2S04 = Pb (S04)2 + 2Н20,
Pb (S04)2 +2e = PbS04 + SOf", или
Pb4+ + 2e=Pb2+.
Суммарно химическую реакцию, протекающую при зарядке
аккумулятора, можно выразить уравнением
разрядка
2PbS04 + 2Н20 £: РЬ + РЬ02 + 2HsS04,
зарядка
т. е. при разрядке равновесие сдвигается слева направо, а при
зарядке — справа налево. Кроме того, при зарядке на
отрицательном электроде возможно восстановление ионов водорода Н+ и
образование газообразного Нг.
370
Если исходить из того, что свинец активнее водорода, то в
первую очередь должен идти именно разряд ионов Н+, но из-за
высокого перенапряжения водорода этот процесс сильно замедлен, и
образование свинца преобладает.
Э. д. с. аккумулятора=£0РЬО1+4Н+_ E0fPb+HfSO4 = 1,68—(-0,36) =
= 2,04 в. На самом деле значение э. д. с. аккумулятора несколько
отличается от 2,04 в, если учесть, что концентрации ионов РЬ2+
п РЬ4+ в растворе не отвечают стандартным условиям. Помимо
свинцового аккумулятора в практике находят применение никеле-
во-кадмиевый, никелево-железный и серебряно-цинковый
аккумуляторы.
§ 11. ТОПЛИВНЫЕ ЭЛЕМЕНТЫ
Проблема преобразования химической энергии
непосредственно в электрическую — одна из актуальных задач науки и техники.
В настоящее время ведутся широкие исследования по
использованию окислительно-восстановительных реакций горения топлива.
В этом случае электрохимические элементы принято называть
топливными элементами. В качестве окисляющихся веществ можно
применять обычное топливо — уголь, кокс, природные и
искусственные горючие газы, в качестве окислителя — кислород или
воздух.
Наиболее реакционноспособный вид топлива — водород. Водо-
родно-кислородные элементы обычно изготовляют с применением
мелкопористых угольных или никелевых электродов, погруженных
в щелочной раствор электролита. Схематически такой элемент
можно представить в виде
(-) (Ni) H21 КОН (30 - 40%) 102 (№) (+).
При работе элемента на отрицательном электроде протекает
электродная реакция
2Н2 + 40Н- - 4Н20 + 4е,
а на положительном
02 + 2Н20 + 4е = 40Н-.
Суммарная реакция
2Н2 + 02 = 2НаО.
Теоретическое значение э. д. с. водородно-кислородного элемента
при 25°С равно 1,229 в и не зависит от состава раствора
электролита.
При разрядке водородно-кислородных элементов напряжение
держится в пределах 0,7—0,9 в в зависимости от плотности тока
на электродах.
37*
Для эффективной работы топливных элементов используют
катализаторы, которые наносят на электрод. Для водородного
электрода катализаторами являются платиновые металлы, а для
кислородного — смешанные катализаторы из Со и А1 или Fe, Mn и
Ag. Высокий коэффициент использования топлива, непрерывность
их действия и другие преимущества открывают перед топливными
элементами перспективы широкого применения. Уже сейчас
используются топливные элементы в спутниках и космических
кораблях. Перспективно применение топливных элементов вместо
двигателей внутреннего сгорания на транспорте и т. д.
§ 12. ОСНОВНЫЕ ВИДЫ КОРРОЗИИ МЕТАЛЛОВ
Коррозией называется процесс самопроизвольного разрушения
металлов под влиянием внешней среды. Ржавление железа на
воздухе, образование окалины при высокой температуре, растворение
металлов в кислотах — обычные примеры коррозии. В
результате коррозии многие ценные свойства металлов ухудшаются:
уменьшается прочность и пластичность, возрастает трение между
движущимися деталями машин, нарушаются размеры деталей.
Вред, причиняемый коррозией в народном хозяйстве, весьма
велик: примерно одна треть добываемого металла выбывает из
технического употребления по причине коррозии.
Коррозия является переходом из металлического состояния в
ионное. Этот процесс сопровождается уменьшением свободной
энергии, поскольку металлическое состояние характеризуется
большим запасом энергии, чем ионное. Подобные процессы протекают
самопроизвольно ввиду стремления вещества переходить в
состояние с наименьшим запасом свободной энергии. Таким образом,
коррозия металлов в атмосферных условиях термодинамически
возможна, что и обусловливает широкое распространение
коррозийных процессов в природе.
Химическая коррозия. Химическая коррозия обусловливается
взаимодействием металлов с сухими газами или жидкостями, не
проводящими электрического тока. К ней относятся образование
окалины на железе при высокой температуре и вообще газовая
коррозия при высокой температуре без участия электролитов.
Механизм реакции сравнительно прост. Продукты реакции
образуются именно на тех участках металлической поверхности,
которые вступили в реакцию. Так, на железе уже при 250—300°С
появляется видимая пленка оксидов. При 600°С и выше
поверхность металла покрывается слоем окалины, состоящей из оксидов
железа различной степени окисления: FeO, Fe304, Fe203. Окалина
не защищает металл от дальнейшего окисления, так как содержит
трещины и поры, которые не могут препятствовать проникновению
кислорода к металлу. Поэтому при нагревании железа выше 800°С
скорость окисления его очень быстро растет с ростом температуры.
372
Образующиеся на металле окисные пленки часто препятствуют
дальнейшему окислению. Образуется так называемая защитная
окисная пленка, которая препятствует проникновению к металлу
как газов, так и жидкостей. Для того чтобы обладать защитными
свойствами, пленка должна покрывать металл сплошь. Расчеты
показывают, что это возможно, если объем окисла металла больше
объема самого металла, пошедшего на образование этого окисла,
т. е. если
V окисла .. ,
V металла
Для щелочных и щелочноземельных металлов это условие не
^ Л7 V окисла ^ л
соблюдается. У них < 1, такие пленки защитными свои-
V металла
ствами не обладают и щелочные металлы ввиду своей химической
Me
МеО
аде
Me
МеО
аде
О
О
О
Me
о о
о о
о о
о о
о о
о о
о о
Условные обозначения -
О Атомы Me е— Электроны Q Атомы кислорода
Q Катионы Me ^% Uohu кислорода
>>Перемещение ионоб ^Перемещение электронов
Рис. 11.6. Схема механизмов диффузии в защитных окислах
активности принадлежат к числу коррозионно-нестойких.
Наоборот, на алюминии и хроме образуются хорошие защитные пленки,
благодаря чему металлы в атмосферных условиях
коррозийно-стойки, несмотря на их химическую активность.
Если металлы, покрытые оксидной пленкой, продолжают
корродировать, это означает, что имеет место диффузия атомов
кислорода сквозь пленку к металлу, и атомов металла в обратном на-
373
правлении. Диффузия металла и кислорода в слое твердого
защитного окисла может осуществляться по одному из двух возможных
механизмов: а) движение ионов в междоузельном пространстве
кристаллической решетки; б) движение ионов по пустым узлам
решетки.
Эти механизмы имеют место при росте защитных пленок:
первый — при образовании пленок ZnO, CdO, ВеО, А1203 (рис. 11.6, а)
и других, второй — при образовании
пленок с пустыми катионными или анионными
узлами в кристаллической решетке,
например: Cu20, FeO, NiO, CoO (рис. 11.6, б),
a = Fe203, Zr02, Ti02 (рис. 11.6, в) и др.
Диффузия катионов в защитной пленке
сопровождается одновременным
перемещением в том же направлении эквивалентного
числа электронов в междоузлиях при
первом механизме и по «электронным
дефектам» (катионам с более высокой
валентностью) при втором механизме.
При повышенных температурах
окисление металлов в воздухе происходит более
Рис. 11.7. Схема действия гальванической пары
интенсивно, так как диффузия через слой оксида протекает с
большей скоростью.
Электрохимическая коррозия. Разрушение металла при
соприкосновении с электролитом с возникновением в системе
электрического тока называется электрохимической коррозией.
В атмосферных условиях роль электролита играет водная
пленка на металлической поверхности, в которой часто растворены
электропроводящие примеси. Электродами являются сам металл
и обычно содержащиеся в нем примеси. В качестве примера
рассмотрим коррозию железа в контакте с медью в растворе соляной
кислоты. При таком контакте возникает гальванический элемент
(—)Fe|HCl|Cu( + ) (рис. 11.7).
Более активный металл — железо — окисляется, посылая
электроны атомам меди, и переходит в раствор © виде ионов Fe2+, а
ионы водорода разряжаются (восстанавливаются) на меди:
2Н+ + 2е= Н2.
Ионы ОН- соединяются с перешедшими в раствор ионами Fe2+:
Fe2+ + 20Н- = Fe (OH)2,
4Fe (OH)2 + 2Н20 + 02 = 4Fe (OH)3.
374
Последний частично отщепляет воду
О
Fe(OH)3-Fe + Н20
\
ОН
и примерно отвечает составу бурой ржавчины.
Согласно теории электрохимической коррозии при
соприкосновении металла с электролитом на его поверхности возникает
множество микрогальванических элементов. При этом анодами
являются частицы металла, катодами — загрязнения, примеси и
вообще участки металла, имеющие более положительный
потенциал. На катодах выделяется .водород, аноды растворяются.
Вместо разряда ионов водорода на катодах может протекать
процесс восстановления кислорода, растворенного в электролите:
02 + 4е + 2Н20 = 40Н-
Это так называемая кислородная деполяризация катода. Какой
процесс будет протекать — это зависит от условий: в кислой среде
обычно выделяется водород, в нейтральной происходит
кислородная деполяризация катода (при коррозии стали, железа) и
водород не выделяется.
Таким образом, при электрохимической коррозии (как в случае
контакта разнородных металлов, так и в случае образования
микрогальванических элементов на поверхности одного металла) поток
электронов направлен от более активного к менее активному
металлу (проводнику) и более активный металл корродирует.
Скорость коррозии тем больше, чем дальше отстоят в ряду
напряжений металлы, из которых образовалась гальваническая пара.
Сильно возрастает коррозия с ростом температуры.
Для образования микропар присутствие примесей
необязательно. Механическая обработка изменяет электродный потенциал,
поэтому соприкосновение двух участков металла —
деформированного и недеформированного — достаточно для появления разности
потенциалов. Корродировать будет деформированный участок
поверхности. Разность потенциалов возникает и там, где
обнаженный участок металла соприкасается с покрытым пленкой.
Было установлено, что коррозия возникает самопроизвольно,
поэтому нельзя связывать ее только с действием локальных или
местных элементов. Наличие этих элементов лишь усиливает
коррозию.
А. Н. Фрумкин развил электрохимическую теорию коррозии
металлов; он показал, что электрохимические реакции,
обусловливающие коррозию (переход ионов металла в раствор и реакция
восстановления водорода или другого окислителя — деполяризатора,
375
например кислорода), могут протекать при одном и том же
потенциале на одном и том же участке поверхности.
Подземная коррозия. Под землей коррозия может развиваться
или под влиянием веществ, растворенных в почве (почвенная
коррозия), или под действием блуждающих токов.
Разрушению подвергаются подземные газо- водо- и нефтетрубо-
проводы, металлические оболочки кабелей, сваи, основания антенн
и другие металлические конструкции.
Коррозия блуждающими токами. Разновидность
электрохимической коррозии — электрическая, или электрокоррозия,
вызываемая блуждающими токами, исходящими от трамваев, метро,
электрических железных дорог и различных электроустановок,
работающих на постоянном токе. Они разрушают подземные
металлические сооружения, трубопроводы, электрокабели.
§ 13. МЕТОДЫ ЗАЩИТЫ
ОТ КОРРОЗИИ
Ввиду больших потерь металла, происходящих в результате
коррозии металлических изделий, издавна принимались различные
меры для ослабления коррозии:
1. Антикоррозионное легирование металла.
2. Электрохимическая защита.
3. Металлические покрытия.
4. Защита путем изменения коррозийной среды.
5. Неметаллические покрытия.
Жаростойкое легирование. Для защиты металлов от наиболее
распространенного и вредного вида химической коррозии —
газовой коррозии используют жаростойкое легирование, т. е. введение
в состав сплава компонентов, повышающих жаростойкость.
Следует указать на три наиболее обоснованные теории
жаростойкого легирования .в зависимости от предполагаемого
механизма действия легирующей добавки:
1) ионы легирующего компонента входят в решетку оксида
основного металла, изменяя в нем скорость диффузии;
2) легирующий компонент образует на поверхности сплава
свой защитный оксид, препятствующий окислению основного
металла;
3) легирующий компонент с основным металлом образует
двойные (смешанные) оксиды типа шпинелей, обладающие
повышенными защитными свойствами.
Согласно первой теории малая добавка легирующего элемента
должна окисляться с образованием ионов определенной
валентности и, растворяясь в оксиде основного металла, уменьшать в нем
число дефектов решетки.
Изменение числа дефектов в решетке оксидов может вызвать
только введение ионов другой валентности. Так, в
полупроводниковых оксидах с избытком металла (например, в ZnO) три иона
376
Ме2+ в узлах решетки оксида заменяются двумя ионами Ме3+
(например, А13+), поскольку должна сохраняться
электронейтральность эти два иона Ме3+ займут места двух ионов Ме2+, а на
хместо Третьего иона Ме2+ перейдет этот же ион из междоузельно-
го пространства, что уменьшит концентрацию междоузельных
катионов и приведет к снижению скорости диффузии катионов и
скорости окисления основного металла, которая контролируется
диффузией катионов.
Согласно третьей теории легирующий компонент может
образовывать с основным металлом двойные (смешанные) оксиды типа
шпинели, например FeCr207, FeAl204, обладающие повышенными
защитными свойствами по сравнению с окислами компонентов
сплава.
По характеру защитного действия против коррозии различают
анодные и катодные покрытия. К анодным относятся такие
покрытия, в которых покрывающий металл обладает в данной среде
более отрицательным электродным потенциалом, чем защищенный,
т. е. стоит выше него в ряду напряжений, например оцинкованное
железо. К катодным относятся покрытия с противоположным
соотношением в свойствах металлов, например железо луженое
или покрытое медью.
Пока слой, покрывающий основной металл, полностью
изолирует его от воздействия окружающей среды, принципиального
различия между этими двумя видами покрытий не возникает. При
нарушении же целостности покрытия создаются совершенно
различные условия. Катодное покрытие (например, олово на железе)
в этом случае перестает защищать и, создавая с основным
металлом гальванический элемент, усилит своим присутствием его
коррозию. Анодное же покрытие (например, цинк на железе) будет
лишь само подвергаться разрушению и, разрушаясь, защищать
основной металл, несмотря на нарушение целостности
покрывающего слоя.
В промышленности часто применяют та(к называемую
протекторную защиту, пригодную в тех случаях, когда защищаемая
конструкция (корпус судна, подземный трубопровод) находится
в среде электролита (морская, почвенная вода). Для
осуществления протекторной защиты используют специальный анод —
протектор (например, старые железные детали, магниевые сплавы и т. д.)
с более отрицательным потенциалом, чем потенциал металла
защищаемой конструкции.
В других методах, называемых катодной защитой, аналогичный
результат достигается присоединением защищаемого металла к
отрицательному полюсу внешнего источника постоянного тока.
Защитное действие осуществляется благодаря повышению
концентрации электронов в поверхностном слое металла, что
затрудняет растворение его.
Скорость процессов кислотной коррозии может быть
значительно уменьшена введением в кислоту тех или иных ингибиторов
377
(замедлителей) коррозии. Это вещества, которые при введении
в незначительном количестве в коррозийную среду заметно
снижают скорость коррозии. К числу ингибиторов относятся
многие вещества, принадлежащие ,к различным классам органических
соединений, например к альдегидам, гетероциклическим
соединениям, белкам и др.
Сложные по составу ингибиторы кислотной коррозии,
употребляемые в большом количестве нашей промышленностью, были
предложены С. А. Балезиным и др. Они вводятся в кислоту в
небольшом количестве, примерно 0,1—0,5%, не мешая растворению
окислов и тормозя коррозию железа в сотни и тысячи раз.
Введение ингибиторов не меняет свойств кислоты. Ингибиро-
ванная кислота действует и на окислы, и на соли, но теряет свою
агрессивность по отношению к металлам. Происходит это
вследствие того, что ингибиторы адсорбируются на поверхности металла,
образуя тонкие пленки. При этом они выводят из строя
коррозийные микропары, препятствуя катодному или анодному процессу
или обоим вместе.
Неметаллические покрытия делятся на неорганические и
органические. Из неорганических покрытий укажем на оксидные и
фосфатные пленки на железе. При кипячении железа в растворах
солей фосфорной кислоты (обычно солей Fe и Мп) получают
фосфатные пленки, хорошо защищающие от коррозии в атмосфере.
Широко распространены органические покрытия, например
лаки. Это растворы веществ, способных при высыхании
образовывать пленки. Если в раствор лаков введены тонко раздробленные
минеральные вещества, то получаются смеси, называемые
красками. Краски употребляют для защиты зданий, железнодорожных
мостов, вагонов, станков и т. п.
Вопросы и упражнения
1. Что называется стандартным электродным потенциалом? От каких
факторов он зависит?
2. Чем вызывается поляризация электродов? Как можно ее устранить^
3. Что вы знаете о явлении перенапряжения?
4. Назовите практические применения электролиза.
5. Какие химические процессы лежат в основе работы свинцовых
аккумуляторов?
6. В чем сущность электрохимической коррозии металлов?
7. Какие методы существуют для борьбы с коррозией металлов?
8. Как опытным путем можно определить величину стандартного
электродного потенциала цинка? Нарисуйте схему гальванического элемента и напишите
уравнения электродных реакций.
9. Какие химические процессы происходят у анода и катода при
электролизе раствора азотнокислого калия, если оба электрода медные?
10. Раствор содержит ионы Fe2+, Hg|"^, Bi3+, Pb2+ в одинаковой
концентрации. В какой последовательности будут разряжаться эти ионы при
электролизе раствора?
378
11. В растворе находилось 0,1 моль HgCl2 и 0,2 моля СиС12. Какие
вещества и в каком количестве выделятся на угольных электродах, если через
раствор пропустить ток в 10 а в течение 1 час?
12. Вычислите э. д. с. и напишите уравнения электродных реакций в
гальваническом элементе (—)Zn|Zn24 | | Fe3+| Fe (—), если
Czn2f = CFe3+== 1 г"ион/л-
13. Вычислите э. д. с. элемента (—)Cd | Cd2+| |Ag+| Ag(+), если
концентрации
CAg+=0,1 и СС(}2+ = 0,001 г-ион/л.
14. Какие процессы происходят на аноде и катоде при электролизе
раствора FeCb, если электроды: а) угольные; б) железные?
15. Определите атомный вес кадмия, если для выделения 1 г кадмия из
раствора его соли потребовалось пропустить через раствор 1717 к электричества?
ГЛАВА 12
ОБЩИЕ СВОЙСТВА МЕТАЛЛОВ.
СПЛАВЫ.
ОСНОВЫ ФИЗИКО-ХИМИЧЕСКОГО АНАЛИЗА
§ 1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТАЛЛОВ
Металлы составляют 4/5 известных элементов (более 80 из 106).
Это определяет их огромное практическое значение.
К металлам в неорганической химии относятся простые
вещества, которые обладают рядом общих химических и физических
свойств.
Атомы металлов способны образовывать элементарные
положительные ионы вследствие легкости удаления или ионизации
валентных электронов. Эти свойства проявляются при
взаимодействии металлов с окислителями, причем металлы при этом
проявляют высокий электроположительный, или основный, характер.
Отрицательных ионов атомы металлов при химическом
взаимодействии никогда не образуют.
Все металлы в компактном состоянии обладают рядом общих
физических свойств. Металлы имеют характерный
«металлический» блеск. Они обладают высокой отражательной способностью
в видимой части спектра. Магний и алюминий сохраняют блеск и
в порошке. Металлы непрозрачны даже в очень тонкой пленке.
Их структура очень плотная и в единице объема содержится
большое число атомов.
Для всех металлов, кроме сурьмы и висмута, характерны
высокая пластичность и ковкость.
Это свойство указывает на то, что кристаллические решетки
металлов не жесткие, плоскости решетки сравнительно легко
сдвигаются одна относительно другой.
Замечательные свойства металлов — высокие тепло- и
электропроводность, причем последняя уменьшается с увеличением
температуры. Это свидетельствует о том, что движение заряженных
частиц через кристаллы металлов происходит легко.
На различии физических свойств металлов основывается их
классификация в технике. Так, по величине плотности металлы
можно разделить на легкие, плотность которых меньше 5 г/см3, и
тяжелые с плотностью более 5 г/см3. Самый легкий — литий,
плотность его составтяет 0,59 г/см3. Самый тяжелый — осьмий, era
плотность равна 22 г/см3.
По температурам плавления (°С) металлы разделяются на
тугоплавкие с температурой плавления выше 1500° и
легкоплавкие— ниже 1000° (табл. 12.1).
380
U со со
< со см
Период
-. 00 е*"
СО ,- см
о
о О
U£2
д
о
S со ц:
_ Чсо
JS8
s
^ о° 8.
0* Ю t^
О О) см
со о
1 ,2 О) —
О см см
п2§
N1 «• 0>
со о
я °о £
w $2
.- ^ 52
5 ■* ^
Z Д сч
■о 2
в СП СО
о ^- ю
QJ Д см
,11
о о
IN СО
с «. *
£ г «
о 2
и. «о *2
г> СО •О
cj i= см
со 2
с* 2
>2°8
о о
СО О
(О *-
н£8
*± 5 *
{/> ^ CN
о
л 5 5
«о
^ со со
* (О N
>
ел «о ~
о
о СМ
см со
с со со
с/) см см
о «Ь
О
^ см со
CJ со t>
о 1>
< О) СМ
-28
2s8
ю о
Sis
о о
о о
- ^ СО
еж см см
U r_ Cft
Н см со
о О
о52
о 2~
00 О
Х> 2 Й
zS?
«8
8S
о
О СО
СЛ N ^-
о
Л ° V—
£ о> о
>
е е
О * W
Ou см Ф
-•8
9 Ift
a. St
О
р5?
о
ьлоо й
3 CD S
< ;г см
0- £ У
о а
см о
ю о
iS5
д с8
О см ю
£8
OS СО Ю
о о
О О
0) Ю
> со со
!> СО Ю
О О
со со
т <Л Ф
я 0) CN
Н ем ю
S CNIO
* о
— ю t»
* t- со
J со со
ев 2 *-
л? ° СО
СО t> iz
г^ оосо
О СМ СО j
>
*& 1
£§1
о
U- см g 1
1—1
Самая низкая температура плавления у ртути (—40°). Она
при комнатной температуре находится в жидком состоянии; галий
имеет температуру плавления 29°, поэтому он плавится в руке.
Самый тугоплавкий — вольфрам (Гпл = 3410°). Для металлов
характерен такой же широкий диапазон изменения прочностных
характеристик от очень мягкого лития, который режется ножом до
очень твердого хрома.
Химическая классификация металлических элементов
основывается на периодической системе Д. И. Менделеева (см. табл. на
форзаце). Металлическими элементами начинается каждый период,
и число их в периоде возрастает с увеличением номера периода.
Резкой границы между металлами и неметаллами провести нельзя.
Можно сказать, что элементы справа от линии, проходящей через
бор, кремний, мышьяк, теллур и астат, — неметаллы, слева —
преимущественно металлы.
Металлические элементы по значениям максимальной
валентности, формам и свойствам главных соединений подразделяются
на группы, соответствующие группам периодической системы.
Элементы, составляющие I А подгруппу периодической системы,
называются щелочными металлами; II А подгруппу (кроме
бериллия) — щелочноземельными металлами. В остальных группах
название дается по первому элементу: группа скандия, титана,
ванадия и т. д.
По особенностям строения атомов элементы металлического
характера разделяются на s-, /?-, d- и /-элементы.
Все физико-химические свойства металлов закономерно
изменяются в зависимости от порядкового номера, например, теплоты,
€ублимации и плавления (рис. 12.1).
Отличительные свойства металлов может объяснить
современная теория металлического состояния. Последнее характеризуется
двумя особенностями — особой кристаллической структурой
металлов и особым типом связи.
§ 2. КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА МЕТАЛЛОВ
Металлы могут быть в трех агрегатных состояниях: твердом,
жидком и газообразном. В зависимости от факторов равновесия
(в данном случае давления и температуры) будет устойчиво то
или иное состояние.
Металлы, за редким исключением, кристаллизуются в решетках
высокой симметрии. Подавляющее большинство металлов
кристаллизуется в одной из следующих трех типов кристаллической
решетки: а) плотная гексагональная решетка; б) плотная
кубическая гранецентрированная; в) объемноцентрированная кубическая
решетка (см. рис. 9.2). В первых двух решетках каждый атом
(ион) на одинаковом расстоянии окружен 12-ю другими атомами.
Такое расположение соответствует самому плотному заполнению
пространства одинаковыми сферами (см. рис. 9.13 и 9.15). В обеих
382
200 r
150
Ю0 h
o 50
[ w
Г м Та Л Re
\ r Mo f\
[■ «
~ /
■ Bf
I
[ I
\M
r
K/He
NbMTc Hff i0s
2rf \Ru 1 \
v 1 \%b 1 r *
с jj Я Oo l\
. fl^Joe t P<A иСе 1 1*
i AI T i 1 ■ 1 1V ^ I Ao 1 в^к I % гпи^^л T
1Л5 / Mn \Obel 1П1/Ьч/ VnJ\fT
L/N?ArVi ... i *«, i., . iXeY , i,.. i..,
>
Tb
tPb
IUb
1 А1Ч
i , ,
Pa
ThM
1 \U
VP/
'/fa Am|
l/Fr
-^750
500»
-1250 «
N /0
20
30
40
50
60
70
ao
90
100
z
не О/о Ar20 30 40 50 60 70 80 90 2
6
Рис. 12.1. Теплоты сублимации (а) и плавления (б) элементов
плотных структурах непосредственно за 12-ю ближайшими
соседями атома следует второй ряд атомов на расстоянии, на 41%
большем, чем у первых. Эти атомы не связаны с первыми. Объемно-
центрированная кубическая решетка несколько более рыхлая
383
Каждый атом имеет 8 равноотстоящих соседей и 6 более
отдаленных. Однако последние удалены от центрального атома всего на
15% больше, чем первые 8 (так, что координационное число цент-
Рольного атома можно считать равным 14). Между шарами плот-
нейшей упаковки имеются пустоты октаэдрические и тетраэдриче-
ские, наличие которых объясняет некоторые особенности в
свойствах металлов и их соединений.
Отметим, что из 52 металлов, структуры которых выяснены
(табл. 12.2), 32 кристаллизуются в плотной кубической или гекса-
Таблица 12.2
Кристаллические структуры и атомные радиусы металлов
Li
а
а,58
Na
в
1,92
К
в
2,38
в
2,53
€s
в
2,72
Fr
Be
б
1,12
Mg
б
1,60
Са
а(б)
1,97
Sr
а
2,15
Ва
в
2,24
Ra
А1
а
1,43
Sc
б(а)
1,66
Y
б
1,82
Л ант.
Ас
Ti
б(в)
1,47
Zr
в(б)
1,60
Hf
б
1,59
Th
а
1,80
V
в
1,35
Nb
в
1,47
Та
в
1,47
Ра
Сг
в(б)
1,29
Мо
в
1,40
W
в(г)
1,41
и
а(г)
Мп
г(г)
1,28
Тс
Re
б
1,37
Fe
в(а)
1,26
Ru
б
1,34
Os
б
1,35
La
а(б)
1,88
Со
б(а)
1,25
Rh
а
1,34
1г
а
1,36
Се
а(б)
1,82
Ni
а(б)
1,25
Pd
а
1,37
Pt
а
1,39
Си
а
1,28
Ag
а
1,44
Аи
а
1,44
Рг
б
1,84
Zn
б
1,37
Cd
б
1,52
Hg
г
1,55
Nd
б
1,81
Ga
г
1,38
In
г
1,67
Т1
а(б)
1,71
Ge
1,39
Sn
1,58
Pb
а
1,75
Ег
б
1,77
Примечания: а — плотная кубическая решетка; б — плотная гексагональная решетка; в —
объемноцентрированная кубическая решетка; г — особые решетки, отличающиеся от трех
предыдущих (для полиморфных металлов в скобках указаны устойчивые при более высоких
температурах решетки).
тональной решетке, а 14 — в объемноцентрированной решетке.
Следовательно, эти структуры наиболее благоприятны. Их
образование легко объяснить на основании природы металлических
связей. Только немногие металлы кристаллизуются в решетках,
отличаются от приведенных выше.
Некоторые металлы кристаллизуются в двух или нескольких
кристаллических формах. Это явление аллотропии. Оно основано
на том же самом едином термодинамическом законе об
устойчивости состояния с наименьшим запасом энергии.
384
Термодинамическая устойчивость той или иной фазы
характеризуется изменением свободной энергии Гиббса или изобарно-
изотермического потенциала AG в зависимости от температуры
и давления. Так, железо существует в двух аллотропных
разновидностях: a-Fe, кристаллизующееся в кубической объемноцент-
рированной структуре, и y-Fe, имеющее гранецентрированную
кубическую решетку. Из схемы (рис. 12.2) видно, что a-Fe
Рис. 12.3. Изменение
свободных энергий а- и
у-модификаций железа
при изменении
температуры
1400 Н
1200 Ч
1000
800 А
600 Н
400
200 А
Рис. 12.2. Кривая
охлаждения и
кристаллизации железа
Время
существует в двух интервалах температур: до 910° и выше 1390°.
Переход одной модификации железа в другую также
объясняется законами химической термодинамики. Каждая
полиморфная разновидность обладает определенным запасом энергии.
Устойчивой при данной температуре является форма с
наименьшим запасом энергии (рис. 12.3). Аллотропические превращения
относятся к фазовым переходам первого рода. Они
сопровождаются скачкообразным изменением свойств при температуре перехода.
Особенности кристаллических структур объясняют и особые
свойства металлов: высокую плотность вследствие плотнейшей
упаковки атомов, а вследствие способности слоев решетки к пере-
13 Общая химия
385
мещению пластичность и ковкость. В кристаллических решетках
металлов могут быть дефекты как точечные, так и линейные, что
отражается на физико-химических свойствах металлов (см.
рис. 9.17).
§ 3. МЕТАЛЛИЧЕСКАЯ СВЯЗЬ
Металлы обладают высокой электропроводностью,
отражательной способностью. Эти свойства металлов ранее объясняли
наличием свободных электронов. Согласно этой теории электроны всех
атомов являются общими. Их называют коллективизированными,
или делокализованными. Они образуют как бы «электронный
Энергия
ионизации 7,76
Низшая энершя 1
возбуждения \Г 1
3,8в y-jr -^
Высший заняу Т 1и
тый уровень
'100
-юоо
-юооо
-3s
.2р
-2s
t Энергия
ионизация
4,36
, Высшая
занятая
полоса
■Зр
■3s
-2р
■ 2s
Энергетические
уровни в атоме
Энергетические
зоны 6 металле
Рис. 12.4. Энергетические уровни атома меди и
металлической меди
газ», который свооодно движется через решетку, состоящую из
положительных ионов металла. Попытка вычислить с помощью
теории электронного газа количественные характеристики
металлов показала несоответствие теоретических расчетов и
экспериментальных данных. Эксперименты по теплоемкости, например, дают
более низкие значения по сравнению с вычисленными. Оказалось,
что не все электроны участвуют в процессе электропроводности, а
только их часть. Это привело к дальнейшей разработке теории
металлического состояния и созданию зонной теории металлов.
Согласно этой теории в сплошном куске металла валентные
электроны располагаются иначе, чем в свободных атомах
металла. Известно, что в свободных атомах электроны находятся на
ограниченном числе энергетических уровней, или орбиталей; в
массе металла подобные уровни занимают только внутренние
электроны (рис. 12.4).
386
Внешние уровни, например все 4s- или З^-уровни атомов меди
в куске металла, сливаются в более широкие энергетические
полосы, или зоны. Каждая энергетическая зона состоит из
множества энергетических уровней, например из всех 4s- или 3^-уров-
ней в случае такого металла, как железо или медь. Эти уровни
принадлежат уже не отдельным атомам, а всему металлу.
Разность энергий уровней одной зоны очень мала. Уровень одной
зоны заполняется электронами в соответствии с принципом
Паули, поскольку на одном уровне могут находиться 2 электрона с
противоположными спинами.
Если рассматриваемая зона содержит N электронов, то для
меди, например, при температуре абсолютного нуля электроны
занимают N/2 низших уровней зоны. При более высокой
температуре некоторые из электронов (электроны проводимости)
переходят на более высокие энергетические уровни. Только это
небольшое число электронов участвует .в переносе тепла и электрического
тока.
Возникновение металлической связи нельзя удовлетворительно
объяснить по методам ВС и МО, ибо в первом случае электроны
все локализованы у отдельных атомов и распределены по их
энергетическим уровням, в МО — все электроны
коллективизированы и располагаются на МО. При образовании металлической
связи происходит нечто среднее: обобществляется,
коллективизируется только часть электронов — валентных электронов внешних
уровней (см. гл. 2 и 4).
Электроны внутренних уровней находятся на орбиталях атомов,
они по-прежнему локализованы у атомов. Эта обобществленная,
коллективизированная часть (валентных) электронов
обеспечивает металлическую связь, взаимодействуя с остовом из
положительных ионов металла, плотноупакованных в кристаллах.
Образование металлической связи происходит также в
результате перекрывания атомных орбиталей соседних атомов. Но, если
в молекуле, например Н2, происходит перекрывание волновых
функций двух атомов, то в кристалле лития каждый атом имеет
8 соседей в первой координационной сфере и 6 во второй и
перекрывание должно происходить с 8 (14) атомами сразу.
Трудно себе представить образование одновременно 8 (14)
связей, так как у атома Li имеется только 1 валентный электрон, а не
8 (14). Но -все 8 (14) атомов обязаны своим существованием этому
единственному валентному электрону. Таким образом,
связывающая сила этого единственного электрона как бы «размазана». Это
следует из сравнения длин связи (2,67 А) в двухатомной молекуле
Li2 с кратчайшим расстоянием (возможным) между атомами в
металле (3,03 А). Увеличение длины «связи» в металле означает,
что она ослаблена, но число связей в металле больше, ибо в 1 см3
металла содержится 1022—1023 атомов. Поэтому полная энергия
связи на один атом возрастает от 13 ккал/моль в молекуле Ыг до
39 ккал/моль в металле (куске) лития.
13
387
В отличие от ковалентной связи, которая насыщена и
направлена, металлическая связь не насыщена, не локализованна и
не имеет определенного направления в пространстве.
На основании представления о расположении электронов в
металлах по энергетическим полосам, или зонам, была разработана
зонная теория твердого тела. Согласно этой теории есть зоны
валентные, зоны проводимости и запрещенные зоны. Их взаимное
расположение объясняет деление твердых тел на диэлектрики,
полупроводники и металлы.
§ 4. МЕТОДЫ ПОЛУЧЕНИЯ МЕТАЛЛОВ
Только немногие металлы в природе встречаются в свободном
состоянии. Та'кие металлы называются самородными. Остальные
металлы в природе встречаются в виде соответствующих
минералов или руд.
Основным принципом получения металлов из руд является
восстановление их до свободных металлов в результате
окислительно-восстановительных процессов. Восстановление может быть
углеродом (карботермия), металлами (металлотермия),
водородом.
Процессы восстановления металлов из водных растворов их
солей относят к области гидрометаллургии.
Процессы катодного восстановления металлов, как из
растворов, так и из расплавов, называют электрометаллургическими.
Типичный пример восстановительного процесса — доменный
процесс, в котором сульфидные руды железа обжигаются,
образующиеся окислы восстанавливаются углем.
Методы получения металлов высокой степени чистоты.
Перечисленные методы не всегда дают металлы высокой степени
чистоты. Однако современная техника предъявляет высокие требования
к чистоте исходных металлов. Десятитысячные доли легкоплавких
примесей влияют на" механические свойства металлов и сплавов
при высоких температурах. Стотысячные доли примесей бора,
лития, гадолиния, кадмия делают «браком» уран для атомных
реакторов. Для металлических материалов термоядерных реакторов
и полупроводниковых приборов содержание вредных примесей не
должно превышать 10~8—10"10%. Поэтому необходима
дополнительная очистка металлов. Используют для этого различные
методы:
1) переплавка в глубоком вакууме для отгона примесей в
металлах;
2) получение чистых металлов термическим разложением их
летучих соединений, например карбонилов, йодидов и др. По
этому методу металл, например титан, нагревают вместе с йодом
до 900°С. При этом образуется летучий тетрайодид титана
Ti + 2I2 = TiI4, который поступает в реактор, где имеется проволока
из чистого титана, нагреваемая электрическим током примерно до
388
1400°С. При этой температуре тетрайодид титана термически
диссоциирует:
TiI4->Ti+2I2
и чистый титан осаждается на проволоке, а йод снова
возвращается в процесс очистки титана.
Этим методом можно получить сверхчистые титан, цирконий,
хром и другие редкие тугоплавкие металлы.
В настоящее время технический уровень той или иной страны
определяется не только количеством произведенного чугуна и
стали, но и редких тугоплавких металлов.
Замечательным методом очистки является так называемая
зонная плавка, или зонная очистка (рис. 12.5).
Рис. 12.5. Схема зонной очистки металлов
Образец кладут в специальную лодочку, которую помещают
в кварцевую трубу, заполненную чистым инертным газом. Вдоль
слитка медленно перемещается нагревательный элемент —
катушка, питаемая высокочастотным генератором. Нагреватель создает
в слитке узкую расплавленную зону, в которой по мере ее
перемещения вдоль слитка накапливается примесь. За нагревателем
остается чистая закристаллизовавшаяся твердая фаза. Таким
образом, примесь, находящаяся в слитке, постепенно оттесняется
в его концевую часть, которая застывает в последнюю очередь.
Для достижения максимальной степени очистки операцию
повторяют несколько раз.
Теория процесса основана на законе распределения вещества
между двумя фазами (в данном случае жидкой с концентрацией
примесей Сх и твердой соответственно С0). Соотношение их
характеризуется коэффициентом распределения К. Чем меньше К,
тем больше степень очистки.
Таким методом впервые удалось получить германий и кремний
с содержанием примесей 10"8%, необходимые для
полупроводниковой промышленности. Сейчас этот метод широко применяется
для очистки всех металлов. Однако идеальными по свойствам
являются не поликристаллические образцы, а монокристаллы, ибо
они содержат меньше и примесей и дефектов кристаллической
структуры. Поэтому ,в настоящее время разработано много
методов получения монокристаллов металлов, в том числе и редких
тугоплавких.
389
Оказалось, что монокристаллы железа, молибдена, вольфрама
теряют хрупкость и приобретают высокую пластичность вплоть до
гелиевых температур. Монокристаллы обладают сверхвысокой
прочностью.
Монокристаллы тугоплавких металлов оказались устойчивыми
при работе в плазме и расплавах других щелочных металлов.
Молибден, хрупкий при обычных условиях, полученный в виде
монокристалла, можно завязать в петлю. Применение
монокристаллов молибдена уже позволило в десятки раз увеличить срок
службы приборов и создать новые.
§ 5. СПЛАВЫ МЕТАЛЛОВ.
ОСНОВЫ ФИЗИКО-ХИМИЧЕСКОГО АНАЛИЗА
Одним из общих свойств металлов является способность их
образовывать спла-вы между собой. Это свойство играет
решающую роль в период научно-технической революции, так как
чистые металлы не могут полностью удовлетворить всем требованиям,
предъявляемым к материалам современной техникой. В технике
необходимы жаропрочные материалы, которые должны работать
в стационарных паровых турбинах при 600°С не менее 100 000 час.
И в то же время для сопел ракет требуются материалы, которые
должны выдерживать температуру ~4000°С в течение 30 мин.
Практически невозможно подобрать металл, который мог бы
удовлетворить всем этим требованиям. И только сочетание двух и
более металлов позволяет получать удовлетворительные по
свойствам композиции. Поэтому и сегодня сплавы металлов — это
ведущие материалы современной техники. В природе существует
более 80 металлов, но из них могут,.быть созданы миллионы
сплавов.
Металлическими сплавами называются продукты химического
взаимодействия металлов (компонентов) между собой. Для
облегчения этого взаимодействия обычно смесь компонентов переводится
нагреванием в жидкое состояние, а затем в результате охлаждения
она кристаллизуется, образуя сплав. В некоторых случаях сплавы
могут быть получены диффузией твердых компонентов при высокой
температуре, или металлокерамическим путем — методом
порошковой металлургии. В основном требования к методам получения
сплавов остаются теми же, что и для металлов, т. е. достижение
особой чистоты и совершенства -кристаллической структуры.
Поэтому нагрев производят обычно в вакууме или инертной
атмосфере (гелия или аргона) в печах, позволяющих получить
соответствующую температуру. В настоящее время наряду с печами
электросопротивления используются и электронно-лучевые и
плазменные печи.
Кристаллическая структура сплавов аналогична чистым
металлам и специфические свойства присущи сплавам в большинстве
своем в кристаллическом состоянии. Свойства сплавов определяют-
390
ся составом, температурой и природой химического
взаимодействия.
Если взять два металла А и В и их расплавить, то при
последующей кристаллизации из расплава можно наблюдать три случая
взаимодействия:
Л + В
исходная смесь (расплав)
4
1
механическая
смесь
кристаллов A -f В
кртл аллиза цим
твердые
растворы
I
химические
(интерметаллические)
соединения
Природа химического взаимодействия металлов между собой
определяется положением исходных металлов в периодической
системе элементов и вытекающими отсюда тремя факторами:
размерным, структурным и электрохимическим, или размерами
атомных радиусов металла; типом кристаллической структуры
исходных компонентов и валентностью и ионизационным
потенциалом.
По мере увеличения различия металлохимичеоких свойств ис-»
ходных .элементов изменяется характер взаимодействия между
ними и в продуктах взаимодействия наблюдается постепенный
переход от металлического типа связи <к ионному. Для установлен
ния природы, или характера, химического взаимодействия обычный
препаративный метод оказался непригодным. Для этого был
разработан новый метод исследования — метод физико-химического
анализа, основанный академиком Н. С. Курнаковым (I860—1941).
Суть метода, по определению Н. С. Курнакова, заключается в
«... определении химической природы одно- и поликомпонентных
систем на основании изучения соотношений между составом и
физико-химическими свойствами» К На осно!вании учения о
гетерогенных равновесиях Гиббса, руководствуясь правилом фаз, Кур-
наков предложил строить диаграммы состояния и диаграммы
состав — свойство и по ним судить о характере химического
взаимодействия компонентов. Разберем это на примере
двухкомпонентных сплавов металлов А и В. Предварительно
готовятся сплавы различных составов от чистого А до чистого
компонента 5. Затем измеряется для каждого сплава одно и то же
физическое свойство. В качестве физико-химических свойств
можно использовать:
1 Н. С. Курнаков. Собрание избранных работ, т. 1, ОНТИ, 1938, стр 217.
391
а) тепловые — термический анализ, плавкость и растворимость;
теплоты образования, теплопроводность, изобарно-изотермический
потенциал, теплоемкость и др.;
б) электрические — электросопротивление, электропроводность,
э. д. с;
в) объемные свойства — удельный вес и удельный объем,
объемное сжатие, коэффициент теплового расширения;
г) оптические свойства—^показатель преломления, спектры
поглощения, двойное лучеиспускание;
д) магнитные свойства — магнитные проницаемость и вос-
приимчимость;
е) давление газов — изотермы упругости паров;
ж) свойства, основанные на сцеплении молекул — внутреннее
трение (вязкость), твердость, давление истечения, модуль упругих
деформаций (сжатие, растяжение, время релаксации,
поверхностное натяжение).
1 А
В J
В А
ш
Рис. 12.6. Диаграммы состав—свойство
I — для непрерывного ряда твердых растворов; II — для
механической смеси; III — для ограниченной растворимости; IV—V —
для металлических соединений
Сюда следует добавить методы исследования макро- и
микроструктуры: нейтронографию, рентгеноструктурный и рентгено-
спектральный анализ, ЯМР, у~РезонанснУю спектроскопию,
электронную микроскопию и др. После изучения одним из указанных
методов всех образцов сплавов заданных составов строится
диаграмма состав — свойство. Для этого на оси абсцисс
откладывается состав сплавов, на оси ординат — изучаемое свойство.
Точки свойств соединяются между собой, и получается изменение
данного свойства в зависимости от состава.
Н. С. Курнаков с сотрудниками установили, что вид этих
кривых бывает разный. Он тесно связан с природой химического
взаимодействия исходных компонентов между собой.
Так, если компоненты А и В взаиморастворимы друг в друге
и образуют непрерывный ряд твердых растворов при кристалли-
392
зации, свойства изменяются по плавным кривым (рис. 12.6, /),
вогнутым или выпуклым к оси состава. Если образуется
механическая смесь, то изменения свойств происходят по прямой,
возрастая или убывая при добавлении одного компонента к другому
(рис. 12.6, II, III). Если образуется химическое соединение
(интерметаллическое), то на кривых изменения свойств от состава
имеется особая точка максимума или минимума, на ординате
химического соединения (рис. 12.6, IV, V). Точка называется
сингулярной. Поэтому подобные диаграммы состав — свойство получили
название химических диаграмм, а закономерности — законами
Курнакова.
На основании полученных результатов Н. С. Курнаковым были
установлены два принципа — непрерывности и соответствия,
которые лежат в основе физико-химического анализа. Принцип
непрерывности гласит, что при непрерывном изменении параметров
(давление, температура и др.), определяющих состояние системы,
непрерывно изменяются и свойства фаз, из которых состоит
система. Принцип соответствия утверждает, что каждой фазе или
комплексу фаз отвечает на диаграмме состав — свойство
определенный геометрический образ, например прямая, кривая.
Диаграммы плавности. Диаграммы состояния металлических
систем. Если в качестве свойств взять температуру плавления
исходных сплавов, то соответственно можно получить диаграмму
плавкости данной двухкомпонентной системы, причем ее вид будет
различен для каждого случая химического взаимодействия
компонентов. Рассмотрим эти случаи.
Для определения температур плавления используют обычно
метод термического анализа. Он заключается в снятии кривых
нагревания и охлаждения сплавов в .координатах температура —
время.
На рис. 12.7, а приведены примеры таких кривых для
некоторых сплавов; вначале температура плавно понижается, в точках
а и с появляется излом, вызванный выделением тепла в
результате кристаллизации, процесс кристаллизации продолжается до
температур, отвечающих точкам bud, при дальнейшем
понижении температуры кривая плавно понижается. Таким путем
можно получить кривые охлаждения ряда сплавов.
Для сравнения на рис. 12.7, а приведены кривые охлаждения
чистых металлов А и В. Они отличаются наличием горизонтальной
площадки, отвечающей температуре кристаллизации. Имея
подобные кривые, можно получить серию точек начала и конца
кристаллизации, соединив которые можно получить две кривые
(рис. 12.7, б). Верхняя кривая ТАС'ТВ описывает зависимость
температур начала кристаллизации сплавов от их состава. Это линия
ликвидуса. Нижняя кривая ТAD'B описывает зависимость
температур конца кристаллизации. Она называется линией солидуса.
Таким образом, можно получить диаграмму плавкости
(рис. 12.7, б) для случая образования металлами кепрерывного
393
Рис. 12.7. Диаграмма
состояния с непрерывным
рядом твердых
растворов:
а — кривые
охлаждения; б — диаграмма
плавности; в —
диаграмма состав—свойства;
г — термодинамический
вывод
состав
% А —
ряда твердых растворов, линии ликвидуса и солидуса разделили
диаграмму на три части, или три поля: выше линии ликвидуса —
однофазное поле жидкого состояния L, ниже линии солидуса —
однофазное поле твердого состояния, или твердых растворов а.
394
Среднее поле представляет собой область сосуществования двух
фаз жидкости и крист 1ллов твердых растворов L + a. Подобные
диаграммы называются диаграммами состояния двухкомпонентных
систем. Следовательно, диаграммы состояния представляют собой
графическое изображение существующих фаз в зависимости от
факторов равновесия. Их строение подчиняется правилу фаз.
Применим правило фаз к диаграмме (рис. 12.7, б). Оно читается так:
число степеней свободы С и число фаз Ф на два превышает число
компонентов К С + Ф = /С + 2. В этом случае 2 — внешние факторы
равновесия — давление и температура.
В случае металлических систем, когда рассматривается
конденсированное состояние, давление пара мало изменяется и им
можно пренебречь. Поэтому на смещение равновесия влияет только
температура. Следовательно, правило фаз в этом случае
упрощается: С + Ф = К+\.
Определим теперь, сколько термодинамических степеней
свободы будет в случае полной взаимной растворимости компонентов
(рис. 12.7, б) в той или иной области диаграммы.
Если сплав состава х будет находиться в однофазном поле
жидкости L или твердого раствора а, то правило фаз запишется
так: С+1=2+1, так как Ф=1; К=2, тогда С = 2, т. е. система
дивариантна и имеет две термодинамические степени свободы.
Если же сплав х находится в поле двухфазного равновесия L + a,
то Ф = 2, правило фаз запишется соответственно С + 2 = 2+1.
Отсюда С=1, тогда система моновариантна и имеет одну степень
свободы.
Рассмотрим особенности процесса кристаллизации сплавов
данной системы, например состава //.
Рассмотрим сначала высокие температуры, например /ь когда
сплав // находится в жидком состоянии. При понижении
температуры до точки С, лежащей на линии ликвидуса, в сплаве никаких
изменений происходить не будет. Как только достигнем
температуры Г2, отвечающей точке С, появляются первые кристаллы
твердого раствора а. Состав их можно найти, если провести прямую,
параллельную оси состава. Пересечение ее с линией солидуса в
точке D' и дает состав твердых кристаллов а, выпадающих из
жидкости состава С
Дальнейшее понижение температуры до ^показывает, что
дальше идет кристаллизация твердого раствора а состава Dx из
расплава состава С.
Линия CD' — линия, концы которой указывают составы фазы
жидкой С, твердой D', находящихся в равновесии при данной
температуре t2, называется конодой.
Следует отметить, что фигуративная точка х делит коноду на
два отрезка С'х и xD\ обратно 'пропорциональные количеству фаз
в сплаве при данной температуре.
Правило рычага, или правило отрезков, позволяет определить
относительные количества жидкой и твердой фаз, находящихся
395
в равновесии при данной температуре. D'x/C'D' — количество
твердой фазы, C'x/C'D' — количество жидкой фазы.
Заканчивается кристаллизация сплава 77 при температуре Г3,
когда фигуративная точка сплава придет в т. F' на линии соли-
дуса. Последние капли жидкости будут иметь состав Е'.
Дальнейшее понижение температуры существенных изменений
вносить не будет. Следует отметить, что в процессе
кристаллизации жидкость меняет овой состав от С до Е\ ее состав движется
по линии ликвидуса от С до Е'. Твердая фаза меняет состав по
линии солидуса от D' до F'.
Подобный тип взаимодействия характерен для металлов,
имеющих близкие металлохимические свойства. Размеры атомов метал-
1500
1400
1300
о 1200
| 1100
о.
с 100
О)
*- о
400
-200
-273
20
40
60
80
^jfl
~—
Жидкость
fi*£
--2
^$г\
Твердый
раствор
Парамагнитный
1
<
У*
4
У
Z_
^е/
«/
ш&
V
'
3681
/
/
\ i
эромагнит-
—__
О
Си
20
40 60
Вес % А
во
100
Ni
/500
1400
1300
400
300
200
100
О
-100
-200
'273
Рис 12.8. Диаграмма состояния системы никель—медь
лов должны отличаться не более чем на 15%. Тип
кристаллической структуры должен быть одинаков, т. е. это должны быть
изоморфные металлы, их электрохимические свойства должны быть
близкими. Следовательно, это металлы, стоящие в одной или
соседних группах таблицы Менделеева. Так, серебро и золото, медь
и никель (рис. 12.8) имеют подобный тип взаимодействия. В
зависимости от механизма образования и кристаллической структуры
различают несколько типов твердых растворов: твердые растворы
замещения, внедрения и вычитания. В системе медь — никель
образуются твердые растворы замещения путем замещения в
кристаллической решетке атомов одного металла-растворителя №
атомами другого металла — Си. Твердые растворы замещения
являются фазами переменного состава с неупорядоченным статисти-
396
ческим расположением атомов в решетке, с металлическим типом
связи и характеризуются плавным изменением свойств с
изменением состава.
Твердые растворы внедрения образуются внедрением атомов
одного элемента в междоузлия кристаллической решетки другого
элемента. Естественно, что внедряться может лишь атом меньших
размеров (водорода, кислорода, азота, углерода и др.)- Примерами
являются твердые растворы внедрения углерода в железо.
Твердые растворы вычитания характерны для полупроводников.
В современной технике сплавы со структурой твердых
растворов находят широкое применение благодаря особым свойствам.
Они коррозионно стойки, пластичны, жаропрочны.
Вывод диаграммы состояния при помощи геометрической
термодинамики. Диаграмму состояния с непрерывным рядом твердых
растворов можно получить при помощи термодинамических
данных. Для этого применяется метод геометрической термодинамики,
или метод изобарно-изотермического потенциала.
Метод геометрической термодинамики — объективный метод,
позволяющий контролировать имеющиеся диаграммы состояния,
а также предсказать возможные типы диаграмм состояния. В
основе метода геометрической термодинамики, использующего
геометрические построения и представления об изобарном
потенциале, лежит первое и второе начало термодинамики (см. гл. 5).
Согласно первому закону
dQ=dU + PdV. (12.1)
Согласно второму-
-f-<dS- (12-2)
Объединяя (12.1) и (12.2), получим
dU + PdV — TdS^O. (12.3)
Изобарный потенциал является функцией переменных Р и Т:
G = f(P,T), (12.4)
или
G = U — TS + PV. (12.5)
Отсюда при Р = пост. иГ = пост, имеем
dG = dU -t- PdV — TdS. (12.6)
Анализируя (12.3) и (12.6), приходим к выводу,что
dG<0. (12.7)
397
Принимая во внимание, что G — функция состояния, изменение
которой не зависит от пути, а только от начального и конечного
состояния (т. е. дифференциал изобарного потенциала является
полным дифференциалом и равняется сумме частных
дифференциалов по независимым переменным), можем записать
дР
dP
dG
дТ
dT.
(12.8)
dG
дТ
-S; [-*-) =V.
1 дР )т
(12.9)
Продифференцируем (12.5) по всем переменным и получим
dG = dU + PdV + VdP — TdS — SdT,
и заменив dU из (12.3), получим
dG = VdP — SdT. (12 Л 0)
Сравнивая (12.8) и (12.10), получим
Следовательно, изменение изобарно-изотермического
потенциала с температурой есть энтропия, т. е. с ростом Т G падает, а с
ростом Р увеличивается.
От состава системы изобарный потенциал изменяется в случае
неограниченной растворимости и образования неограниченных
жидких и твердых растворов плавной непрерывной кривой
(Р = пост. и Г = пост.) (схема а).
Если образуется механическая смесь и компоненты
совершенно нерастворимы во всем интервале концентраций от А до В, то
изобарный потенциал находится по правилу аддитивности, как
это показано на схеме (схема б).
В рассматриваемом случае образования непрерывного ряда
твердых растворов изменение удельного изобарно-изотермического
потенциала для жидкой фазы от состава будет изображаться
плавной кривой, всюду выпуклой к оси состава. Для твердой фазы
имеется аналогичная кривая (BssAs) (рис. 12.7, г).
Если Тпл>Глпл и Твпл, то устойчиво жидкое состояние,
кривая Gi всюду ниже соответствующей кривой для твердого состоя-
398
Рис 12 9 Диаграмма
состояния эвтектического
типа
а — кривые охлаждения;
б — диаграмма
плавкости, в — диаграмма
состав—свойство; г —
термодинамический вывод
состав
ния (рис. 12.7, г, /). Если ^2<Тлпл<ТвПл' то -кривые пересекаются
(рис. 12.7 // и ///), тогда проведение общей касательной к кривым
удельного изобарно-изотермического потенциала жидкой и твердой
фаз даст ib точках касания составы твердой (т. D) и жидкой
(т. С) фаз, находящихся в равновесии при данной температуре.
Проекцией этих касательных на плоскость состав — температура
399
есть не что иное, как коноды, концы которых дают составы
женных фаз (CD').
Соединив точки касания на плоскости температура —
получаем линию ликвидуса (точки касания, указывающие
жидких фаз) и линию соли-
дуса (соответственно точки
касания, указывающие
состав твердых фаз).
Таким образом, можно
по данным изменения изо-
барно-изотермического
потенциала фаз с
температурой построить диаграмму
состояния. В данном случае
она является набором
проекций касательных,
проведенных к кривым
изменениям G для жидкой и твердой
фаз, т. е. набором конод
(рис. 12.7, б).
Эвтектические смеси.
Теперь рассмотрим случай,
когда компоненты А и В в
жидком состоянии
смешиваются неограниченно, а
в твердом состоянии
образуют механическую смесь.
сопря-
состав,
состав
400
Диаграммы состояния этого типа называются
эвтектическими.
Представленное на рис. 12.9, б строение диаграммы данного
типа показывает, что кривая ликвидуса системы состоит из двух
ветвей: ТАЕ —
кристаллизации из расплава
компонента А; ТВЕ — компонента В.
Эти две линии
пересекаются в точке Е — эвтектической
точке, наиболее низкой по
температуре плавления. Солидус
представлен одной
горизонтальной прямой KE\t5. Все
поле диаграммы состояния
разбивается на 4 области: одна
однофазная — жидкость L,
остальные — двухфазные L+A\
L + B и А + В. Построение
подобного типа диаграммы
можно осуществить при помощи
кривых охлаждения (рис,
12.9, а).
Рис. 12.10. Микроструктура сплавов:
а — твердый раствор; б —
эвтектическая смесь; в — смесь А + В
Кристаллизация сплавов заканчивается при эвтектической
температуре образованием двухфазных сплавов, состоящих из смеси
кристаллов компонентов А и В. Соответственно имеется одно бива-
риантное равновесие в поле L с двумя степенями свободы, три
моновариантные равновесия и одно нонвариантное — эвтектическое:
L++A+B. Там сосуществуют одновременно три фазы—одна
жидкая и две твердые А и В. Процессы кристаллизации сплавов
данной системы характеризуются тем, что в них присутствует процесс
кристаллизации эвтектической смеси. Он отмечается горизонтальной
площадкой на кривой охлаждения как нонвариантное равновесие.
Кристаллизация сплавов, за исключением сплавов эвтектического
состава, протекает в интервале температур от U до U. При этом
состав жидкой фазы изменяется по .кривой ликвидуса от D' до
точки £", состав твердой фазы отвечает ординате чистого компонента
А. Количество твердой и жидкой фазы можно определить также
по правилу отрезков.
В дополнение к термическому анализу для построения
диаграммы состояния подобного типа полезно привлечь микроскопический
анализ. Суть этого метода состоит в том, что механическим
шлифованием и полированием готовится зеркальная поверхность
401
-образца и затем, после предварительного действия на нее травите-
.лей, изучается под микроскопом. При этом .выбирается травитель
(например, кислота), который преимущественно растворяет ту или
иную фазу. Выявленная таким образом структура сплавов имеет
определенный вид для каждого случая взаимодействия металлов
между собой (рис. 12.10, а—в).
Чистые компоненты имеют однофазную структуру, только
компонент А под действием травителя имеет светлые полиэдры с
темными границами, компонент В — наоборот. Такова структура
твердых растворов (рис. 12. 10, а). Сплав эвтектического состава
(рис. 12. 10, б) характеризуется одновременной кристаллизацией
А и В, поэтому имеет специфическую структуру эвтектической смеси
мелкую, пластинчатую (может быть и зернистая). Сплав / (до-
эвтектичеокий) имеет две структурные составляющие: первичные
крупные кристаллы компонента А и эвтектическую смесь
(рис. 12.9, в). Несомненно, что можно при помощи геометрической
термодинамики получить подобный тип диаграммы состояния
(рис. 12. 9, г).
Есть аналогия с предыдущим случаем: здесь тоже изотермы
изобарно-изотермического потенциала для жидкой фазы
представлены плавными кривыми. Так как кристаллизуются чистые
компоненты, то состав жидкости, из -которой идет процесс
кристаллизации при данной температуре, находят проведением касательной из
точки изобарно-изотермического потенциала для чистого
компонента к кривой G жидкой фазы. Точки касания Z), Я, G, Е дают
•состав сопряженных фаз при данной температуре. При
эвтектической температуре касательные сливаются в одну общую
касательную (рис. 12.9, г, V). Проекция касательных на плоскость
-состав — температура (рис. 12.9, б) дает диаграмму состояния. Она
представляет собой также набор (конод, концы которых находятся
на линии ликвидуса и ординатах чистых компонентов.
Подобный тип образуют металлы, резко отличные по свойствам,
например висмут — кадмий (рис. 12.11).
Сплавы эвтектических систем вследствие низких температур
плавления используются как флюсы в металлургии,
низкотемпературные охладительные смеси, электроконтакты,
электропрерыватели и др. Многокомпонентная эвтектика, например сплав Вуда,
состоящий из четырех компонентов висмута, кадмия, олова и
свинца, плавится при 68°С. Это почти в 4 раза ниже Гпл самого
низкотемпературного компонента олова (232°С).
Эвтектическая смесь галлия с индием находится в жидком
состоянии при комнатной температуре.
Особой природой металлического состояния определяется и то,
что для образования соединений между металлами так
называемых металлических или интерметаллических характерны иные
закономерности, чем для образования соединений между
неметаллами, а также металлами и неметаллами.
402
f:cC(
400
327
300
200
146
100
Bi (
Cd 11
i
3
W
1
20
80
1
40
60
E
Cd+Bi
i
60
40
1
80
20
В
400
277
200
/00
100%
0
Рис. 12.11. Диаграмма со
стояния системы висмут-
кадмий
Рис. 12.12. Диаграмма состояния системы
с образованием конгруэнтно-плавающего
интерметаллического соединения АтВп\
а — кривые охлаждения сплавов; б —
диаграмма плавности; в — диаграмма
состав—свойства; г — термодинамический
вывод
%А-~
Образование конгруэнтно плавящегося соединения АтВп из
расплава. Пусть соединение АтВп образует с чистыми
компонентами А и В диаграммы простого эвтектического типа. Тогда
строение диаграммы (рис. 12.12) будет иметь следующий вид. На
кривой ликвидуса имеется дополнительный участок ExsE2,
отвечающий процессу выделения из расплава кристаллов химического
соединения АтВп. По ординате химического соединения
диаграмма как бы разделяется на две самостоятельные эвтектического
типа А—АтВп и АтВп-\-В. В левой части нет компонента В как
составляющего, в правой части — компонента А.
Си
Мс
1000
900
'800
£51
500
400 Ь
Си 0 20 40 60 80 100%
Мд 100% 80 60 40 20 0
[—
-
г /~
Г 799° / 1
I м9
к 570° /
\ / о
h v475
' Е1
\ I
ps/ 555
Е2
,_L_ I
ft/ \
V 725°
£з Н
Н
f I
тГс
1063
Рьз^а Вес % Na
3 ♦ 4 5 6
25 30 35 40
Атомн %
Рис. 12.13. Диаграмма состояния
системы магний—медь
Рис 12.14. Свойства сплавов системы
свинец—натрий
Кривая охлаждения сплава состава АтВп аналогична чистым
компонентам (рис. 12.12, а). На кривых свойств оно отличается
особыми сингулярными точками максимума или минимума
(рис. 12.12, б). Подобная точка имеется и на кривой ликвидуса
в случае, если соединение АтВп не склонно к диссоциации в
твердом состоянии.
Подобные соединения Н. С. Курнаков называл дальтонидами
в том случае, если их состав отвечал закону кратных отношений,
если на кривых свойств им отвечали сингулярные точки, а
кристаллическая структура отличалась от исходных компонентов по
своему типу и характеризовалась упорядоченным расположением
атомов и максимумом упорядоченности.
404
Подобные диаграммы можно получить также методом
геометрической термодинамики (рис. 12.12, г). Вывод построен на том, что
химическое соединение является индивидуальным компонентом и
имеет свое значение G для твердого и жидкого состояния,
обозначенного 5. Практически идет построение двух эвтектических
диаграмм: А—5 и 5—В. Было установлено, что соединения имеют
состав, не отвечающий правилам валентности. Например, в
системе Mg—Си (рис. 12.13) образуются два соединения: MgCu,
отвечающее правилам валентности, и Mg2Cu, не отвечающее этому
правилу.
А в А ва В
Рис. 12.15. Схема образования бертоллидных фаз по Н. С. Курнакову
Поэтому впоследствии в отличие от обычных валентных
соединений металлов с неметаллами подобные соединения стали
называться металлическими, или интерметаллическими. Они могут
растворять в себе исходные компоненты и образовывать твердые
растворы на своей основе — тоже фазы переменного состава —
дальтонидные фазы, но с упорядоченным разложением атомов в
решетке и сингулярностью свойств (рис. 12.6, V).
Изучение подобных диаграмм позволило Н. С. Курнакову
установить новые фазы переменного состава в системе натрий —
свинец, таллий — висмут и другие (рис. 12.14). Эти фазы (Р)
характеризовались отсутствием сингулярных точек и были названы
бертоллидами. Они образуются также на основе соединений или
полиморфных модификаций, на так называемых мнимых, не
существующих, ордината которых находится за пределами данной
фазы переменного состава (рис. 12.15).
Исследования последних лет показали, что бертоллидные фазы
самые распространенные в природе, особенно в случае соединений
металлов с неметаллами. Было установлено, что из твердых
растворов в твердом состоянии путем упорядочения могут
образовываться интерметалличеокие соединения.
Классическим примером является система золото — медь, в
которой Н. С. Курнакову впервые удалось обнаружить соединения
CuAu и Cu3Au по сингулярным точкам на кривых свойств
отожженных сплавов. Теплоты образования этих соединений составляют
405
0,37 ккал/моль, рентгенограмма характеризуется наличием
сверхструктуры. Подобные соединения затем были обнаружены во
многих других системах и получили название соединений
Курнакова.
Образуют металлические соединения, как правило, металлы с
резко различными свойствами, природа связи в них может
меняться от чисто металлической до ионной (например, в ряду
Mg2Pb; Mg2Sn; Mg2Ge; Mg2Si; Mg2C).
В настоящее время известно более 4000 соединений. Попытка
их классификации пока не удалась. Однако помимо классификации
Н. С. Курнакова есть классификация их по типам кристаллических
структур (соединения со структурой фаз Лавеса Mg2Zn, Mg2Cu,
Mg2Ni; соединения типа Cr3Si; соединения типа CsCl, NiAs и др.).
Соединения с подобными типами структур обладают целым рядом
специфических свойств (сверхпроводниковых и др.)-
Есть классификация, основанная на электронном строении и
типе химической связи. Фазы Лавеса — соединения с
металлическим типом связи с /с=1,2—1,6, имеющие кубическую и
гексагональную структуру. Остальные соединения имеют примеси кова-
лентной или ионной составляющей.
Есть большая группа так называемых электронных соединений
(табл. 12.3), образование которых происходит при определенных
соотношениях числа валентных электронов к числу атомов.
Типичными примерами существования этих соединений могут быть
промежуточные фазы в системах медь—цинк и медь—олово (латуни
и бронзы).
Таблица 12.3
Формулы некоторых интерметаллических соединений
Электроны/атомы
3/2
Решетка: |3
объемноцент-
рированная
CuBe
CuZn
Cu5Sn
CoAl
3-м n
особая
кубическая
AgsAl
Au3Al
Cu5Si
CoZn3
21/13
V
особая
кубическая
Cu5Zn8
CU9AI4
Fe5Zn21
Ni5Cd2l
Na31Pb8 1
7/4
8
плотная
гексагональная
CuZn3
Cu3Sn
AgZn3
Ag5AI5
Au3Sn
Прочность интерметаллических соединений находится также в
зависимости от типа связи. Например, соединения никеля
Ni3Fe Ni3Ti Ni3Al
Теплота образования, ккал/моль 1,97 0,41 8,4
Температура диссоциации 570 595 1380
Тип соединения соединения дальтонид
Курнакова
406
Переход от чисто металлической связи к ковалентнойвдальто-
нидах магния с элементами четвертой группы ведет к повышению
*0пл,С(Мд2РЬ—355, MgSn—378, Mg2Ge—1070, Mg2Si—1102).
Аналогично с ростом электроотрицательности и потенциала
ионизации идет переход от металлической связи к ковалентной
в этих соединениях.
Класс интерметаллических соединений в настоящее время
имеет большое значение в силу двух обстоятельств: 1) проявление
особых физико-химических свойств и 2) способности к
взаимодействию между собой. Было установлено, что при Г = 0,8 от Гпл
все металлические соединения чрезвычайно пластичны в отличие
от чистых металлов и имеют максимальную прочность.
Среди интерметаллических соединений в настоящее время
открыто большое количество соединений со специфическими
свойствами. Можно прямо сказать, что существование многих областей
современной техники обязано интерметаллическим соединениям.
Это прежде всего соединения с полупроводниковыми свойствами:
Mg2Si, Mg3Cd, CuS, GeS, PbS и жаропрочные соединения Ni3Fe и
Ni3Al. Это большой класс сверхпроводниковых соединений (~900)
с металлическим типом связи и высокими температурами перехода
в сверхпроводящие состояния для Nb 9,7°K, для Nb3Sn 18,5 и
V3Ga 16,5.
Это позволяет использовать их для получения сильных
магнитных полей. 1 кг веса сверхпроводящего магнита создает магнитное
поле, приблизительно равное по силе тока 20-тонному магниту с
железным сердечником. Поэтому сверхпроводниковые соединения
нашли широкое применение в электронике, ядерной энергетике,
космической технике, вычислительной технике и др.
Второе замечательное свойство интерметаллических
соединений — способность их взаимодействовать друг с другом. Причем
типы взаимодействия, факторы, их определяющие, аналогичны
таковым для чистых металлов. Так, для образования твердых
растворов необходим один тип кристаллической решетки, одинаковый
тип химической связи, одинаковый стехиометрический состав,
например Ni3Ti—Ni3Al.
Интересным в практическом отношении оказался случай
образования интерметаллидами эвтектических смесей. Например,
эвтектика Ni3Al + Ni3Nb позволила после направленной кристаллизации
получить новые жаропрочные материалы с высокой рабочей
температурой и с высокими параметрами длительной
прочности.
Для свойств класса интерметаллических соединений имеют
•большое значение вопросы дефектности структуры. Наличие
вакансий в NiAl ведет к отклонению состава от стехиометрического,
что отражается на электросопротивлении и других свойствах.
Диаграммы состояния, отражающие химическую природу
взаимодействия компонентов, служат в современной технике научной основой
выбора сплавов для промышленности. Примером этого может
407
служить система железо — углерод, сплавы которой (стали и чу-
гуны) — основа черной металлургии.
Таким образом, физико-химический анализ и ныне не утратил
своего значения. С помощью диаграмм состояния и на сегодня
решаются основные вопросы смежных наук — геологии,
металлургии и др.
§ 6. ХИМИЧЕСКИЕ СВОЙСТВА МЕТАЛЛОВ
Химические свойства металлов определяются строением их
атомов. Имея на внешнем уровне малое количество валентных
электронов, атомы металлов легко их отдают, превращаясь в
положительные ионы и проявляя при этом восстановительные
свойства.
Схематически этот процесс можно изобразить так:
Ме° — пе-*-Шп+.
Мерой прочности связи электронов в атомах являются величины
энергии ионизации, или ионизационные потенциалы (см. рис. 3.4).
икал/моль
5„ь>
? <>12
2 bCs>
Положит заряд ядра -^*~
Рис. 12.16. Энергия диссоциации двухатомных
молекул
Этим обусловлена способность металлов вступать во
взаимодействие с различными окислителями, в качестве которых могут быть
элементарные вещества, ионы менее активных металлов и др.
При этом происходят типичные процессы
окисления-восстановления. Эти реакции для металлов протекают самопроизвольно, если
408
энергия сродства к электрону окислителя превышает энергию
ионизации металла. Тепловой эффект суммарного процесса сильно
зависит от энергии диссоциации молекулярного окислителя.
На рис. 12.16 приведены энергии диссоциации молекул некоторых
элементарных окислителей. Поэтому окисление металлов
галогенами часто происходит легче, чем кислородом, а окисление азотом
происходит с очень большим трудом, несмотря на сравнительно
большие значения энергии сродства к электрону у кислорода и
азота.
Оксиды и гидроксиды металлов. Почти все металлы
непосредственно взаимодействуют с кислородом. (Только золото, платина
и другие благородные металлы не реагируют с молекулярным
кислородом. Их соединения получаются косвенным путем.)
Образуются при этом соединения общей формулы МехОу — оксиды.
Наряду с величинами ионизационных потенциалов эти процессы
зависят от величин нормальных электродных потенциалов и теплот
образования соответствующих оксидов. Это могут быть собственно
оксиды металлов, т. е. соединения, в молекулах которых все
атомы кислорода непосредственно связаны с атомами металлических
элементов, например СаО. Степень окисления кислорода в них
равна —2. Кроме этих соединений известны пероксиды, или
перекиси, например Na202, Ва02, где атомы кислорода связаны друг с
другом, и супероксиды, в которых атомы металлических элементов
связаны друг с другом (Ti20).
Среди собственных, или нормальных, оксидов можно выделить
простые оксиды, являющиеся соединениями одного металлического
элемента. Состав простых нормальных оксидов определяется
степенью окисления, или валентностью металлического элемента.
Максимальная характеристичная валентность элемента по
кислороду отвечает, как правило, номеру той группы периодической
системы, в которой он расположен. Исключения сравнительно
немногочисленны: сюда относятся медь, серебро, золото, некоторые
лантаноиды (церий, празеодим, тербий) и большинство
актиноидов.
Оксиды, отвечающие характеристичной валентности, Б. В.
Некрасов называет характеристичными. Их теплоты образования
представлены на рис. 12.17.
В подавляющем большинстве характеристичные оксиды
тугоплавки. Исключение представляют немногие (МП2О7 — жидкость,
^Ru04—85°, Os04—4ГС). Наиболее тугоплавкие оксиды
бериллия, магния, кальция, циркония, гафния, тория (2500—3000°С).
Все характеристичные оксиды элементов малых периодов
бесцветны. В больших периодах, напротив, большинство оксидов
окрашено.
Среди низших оксидов доля окрашенных еще значительно
больше. Свойства простых оксидов определяются характером
металлического элемента. Оксиды наиболее активных металлов
характеризуются основными свойствами. По мере уменьшения актив-
409
ности металлов свойства их оксидов непрерывно изменяются от
типично основных через амфотерные к кислотным.
Это, как известно, проявляется при взаимодействии оксидов с
водой. Оксиды активных металлов при этом образуют основания
или щелочи, например К, Са, оксиды мало активных металлов —
Са Sc
13 5 1
135713571357
Валентность
Рис. 12.17. Теплоты образования характеристичных оксидов
кислоты. Но есть оксиды, которые не взаимодействуют с водой.
Отношение оксидов к воде можно представить в следующем виде.
Металл
Отношение к воде
Ru, OS
Щелочные,
щелочноземельные металлы, Cr, Mn, Re
Лантаноиды, актиноиды
Все остальные металлы
растворение без заметного химического
взаимодействия
химическое взаимодействие с образованием
растворимых гидроксидов
химическое взаимодействие с образованием почти
нерастворимых гидроксидов
практически отсутствие химического взаимодействия
и растворения
Типы гидроксидов, образуемых при этом, предста!влены в
табл. 12.4.
Следует отметить, что свойства оксидов и гидроксидов
закономерно изменяются в зависимости от положения металла в
периодической системе элементов
410
Таблица 12.4
Валентность
1
2
3
4
5
6
7
Характерный
тип гидроксидов
эон
Э(ОН)2
Э(ОН)3
нэо2
Э (ОН)4
Н2Э03
н3эо4
н2эо4
НЭ03
Химический характер гидроксидов
основный
Cs, Pb, К
Na, Si, Те
(Си)(Ag)(Au)
Ra, Ba, Si, Ca
Mg, Cr, Mn, Fe
La лантаноиды,
I, Se, Ir, Rh,
Fe, В, Те
Th, Hf, Zr, U
Pa
—
амфотерный
I
Zn, Be, Pb, Sn
Zn, Al, Ca;
Cr, Sb, Au
Ti, Pb, Sr,
Pt, Те
Та, Nb
U, Nb, P
—
кислотный
Br, CI
Bi, As, P
N
Si, C, Ge, Sc,
S
V, Sb, Ag, P,
N, I, Br, CI
W, Mo, Cr
Fe, Sc, C, Mn,
Re, Os, Ru
M, Те, Re, CI, I
Так, в каждой группе сверху вниз в главных подгруппах
усиливается основный характер оксидов и гидроксидов (уменьшаются
теплоты образования оксидов), увеличиваются ионные радиусы и
уменьшаются потенциалы ионизации. В побочных подгруппах
наблюдается обратная зависимость, что хорошо иллюстрируется
изменением изобарно-изотермических потенциалов образования
оксидов элементов подгруппы скандия (рис. 12.18). По периодам,
или горизонтальным рядам, наоборот, идет постепенно рост
кислотных свойств и уменьшение основных свойств. Так, в начале
IV периода в ряду
К20, CaO, Se203, Ti02, V205> Cr03, Mn207
основные амфотерные кислотные
идет постепенно переход от типично основных -к кислотным
свойствам через амфотерные. Это хорошо видно на примере
изменения изобарно-изотермических потенциалов образования оксидов
элементов IV периода (рис. 12.19).
Этому отвечает и повышение степени окисления металлов,
образующих оксиды, от +1 у калия до +7 у марганца. То же
наблюдается при изменении свойств оксидов металлов,
обладающих переменной валентностью. С повышением степени окисления
свойства изменяются от основных .к кислотным, что можно
проследить на известном примере оксидов марганца.
411
В ряду
+ 2 +3 +4 4-5 +6 +7
МпО, Мп203, Мп02, Мп205> Мп03, Мп207
основные амфотерные кислотные
Химические связи в основных оксидах ионные, или сильно
полярные, что сказывается на устойчивости, твердости, температуре
плавления и других свойствах.
Л*2ЭМбрМ203,«'КаЛ/^Ь
-390000 h
-400000
-410000
*Л°зк
XL02°3K
4
Sc
6 Период
La
Рис. 12.18. AG0 ккал/моль
образования оксидов подгруппы скандия
-100000
-200000
-300000
AG ^ .кал/моль
•J 298 оЬр М203 к?
х 2 3,К
У МпА.к
-400000 Ь
/
V0
2 3, К
х Ti О
2 3,К
А\«
JL.
J L
J-
Sc Ti V Cr Mn Fe
Рис. 12.19. AG0 кал/моль
образования оксидов металлов IV
периода
Высшие оксиды малоактивных металлов, например Сг203,
Мп207, обладающие кислотными свойствами, характеризуются
наличием ковалентных связей в молекуле, что обусловливает
непрочность их кристаллических решеток. Соответственно и в
продуктах взаимодействия оксидов с водой металл с кислородом
связан различно.
Если образуется основный гидроксид, то металл с кислородом
связан ионной связью, а водород с кислородом — ковалентной.
В кислотных, наоборот, связь кислорода с металлом — ковалент-
ная, а водорода с кислородом — ионная, или сильно полярная. Это
связано и с ростом степеней окисления металлов, что видно на
примере КОН, Са(ОН)2, Sc(OH)3, Ti(OH)4, Ti(OH)20, V(OH)02,
HV03, H2Cr04, HMn04.
Совершенно аналогичные закономерности проявляются и для
одного и того же металла в разной степени окисления, например
у марганца. В ряду гидроксидов: Мп(ОН)2, Мп(ОН)3, Мп(ОН)4,
Мп(ОН)20, Мп(ОН)30, Н3Мп04, Н2Мп04, НМп04, так же как
412
и оксидов, идет постепенный переход от основного оксида к
кислотному, от основной — гидроксидов марганца II и III к марган-
цеватой и марганцовой кислотам (VI, VII).
Следует отметить, что оксиды металлов имеют состав, не
отвечающий закону простых кратных отношений, отношение атомов
в них не выражается целыми числами. Их состав отличается от
стехиометрического. Это
особенно характерно для оксидов
переходных металлов.
Например, оксид титана TiO может
быть получен любого состава
от TiOo,88 ДО TiOi,2o (при
1200°С). Подобное соединение
относят к фазам, или
соединениям, переменного состава.
Таким образом, в настоящее
время не правильно говорить,
что, например, титан образует
оксид титана TiO, так как
существует оксид титана с
широкой областью гомогенности
0
-20000
'40000
'60000
-80000
'100000
-120000
-140000
^298
PtK
i
-
-
обр
*и
1
,ккал/мбль
мок
Мд 0
1 ~
?.г п+п
Г Pt0H
1 FeOK
'
f * К
Рис. 12.20. AG0 кал/моль
образования МО
исходные
вещества
Продукты
реакции
М +У2 0 = МО
К 2 К
TiO0,88 TiOi,2o. Следовательно, состав TiO отличается от
стехиометрического I : 1 (0,88: 1,2).
Подобные соединения иногда называют нестехиометрическими.
Их образование можно объяснить также на основании
представлений о дефектности .кристаллической структуры. Это хорошо можно
видеть на примере FeO. Аналогично TiO FeO (вюстит)
фактически не существует в виде соединения с такой формулой FeO,
а всегда имеет составы в интервале Fe0,95 до FeOi,2o-
В его решетке не хватает 5—15% катионов Fe2+. Для
сохранения электронейтральности кристалла 5—15% имеющегося железа
находится в состоянии 3-валентного иона Fe^.
Нормальные оксиды металлических элементов получают в
большинстве своем при непосредственном взаимодействии металлов
с кислородом. Этот процесс термодинамически определяется
уравнением AG=AH— TAS. Например, величины изменения изобарно-
изотермических потенциалов образования оксидов платины, железа
к магния из элементов (рис. 12.20) показывают, что при обычных
условиях монооксид платины не образуется, так как он имеет
более отрицательное значение AG.
Величины энтальпий образования оксидов из элементов,
очевидно, в рассмотренном случае имеют те же закономерности.
413
Оксиды высших степеней окисления можно получить окислением
оксидов соответствующих металлических элементов низших
степеней окисления или из-за склонности последних к диспропорцио-
нированию. Например:
2GeO --= Ge02 + Ge„ AG = — 9700 кал/моль,
2SnO = Sn02 + Snrt AG = — 1500 кал/моль,
2PbO = Pb02 + РЬл AG = + 37700 кал/моль.
Этот пример показывает, что легко этот процесс идет для
германия, монооксид свинца устойчивее диоксида его, поэтому
последний процесс практически не идет.
Оксиды металлов и их гидроксиды находятся часто в природе
б виде соответствующих минералов и являются главным
источником для получения металлов. Например: А1203, РЬ02, ТЮ2.
В технике оксиды находят широкое применение главным
образом как кислотоупорные и огнеупорные материалы (А1203, ВеО,
Zr02, Th02). Большое значение имеет покрытие металлов
пленками оксидов для предохранения от дальнейшего окисления (так
называемыми оксидными пленками).
Гидриды металлов. Обычно гидридами называют соединения
металлов с водородом, в которых водород играет роль
электроотрицательного элемента (степень окисления —1).
Гидриды — простейшие соединения. Однако способность
водородных атомов присоединять или отдавать электроны, а также
образовывать многоцентровые орбиты делает группу гидридов
очень своеобразной. Рассмотрим только так называемые
первичные, или бинарные, гидриды, т. е. гидриды, где металл
непосредственно соединен с водородом.
Валентность по водороду химических элементов не
превышает 4 и по группам периодической системы изменяется весьма
закономерно. Формулы водородных соединений даны у каждой
группы внизу. Есть различные классификации гидридов. Согласно
положению в таблице электронных аналогов (см. табл. 3.4)
В. В. Некрасов делит гидриды на 5 групп: 1. Солеобразные
гидриды (I, II группы). 2. Переходные—III группы лантоноидов и
актиноидов. 3. Металлообразные — элементов 6—10 рядов. 4.
Полимерные— 11 —13 рядов, а также Be, Mg, Al. 5. Летучие —
14—17 рядов.
По типу химической связи гидриды делят на ионные, куда
практически относят все солеобразные гидриды щелочных и
щелочноземельных металлов, где присутствует гидрид ион Н"
(см. рис. 3.10).
Ко II группе относятся ковалентные летучие гидриды
кислотного характера. III—VII группы (включают гидриды Be, Al, Ge и
полимерные, электронно-ненасыщенные гидриды — Ge, Sn, Pb, Bi,
Sb, Po), а также промежуточные гидриды II В и III В подгрупп
414
и большая группа гидридов переходных металлов, среди которых
есть металлообразные, или металлические, гидриды внедрения с
металлическим типом связи. Сюда относятся гидриды лантаноидов,
актиноидов и переходных металлов (см. рис. 3.10).
Элементы главных подгрупп II—VII групп образуют
газообразные ковалентные гидриды.
Образование ковалентных летучих гидридов характерно для
электроотрицательных и слабоположительных элементов первой
части и середины таблицы Менделеева, сильно
электроположительные элементы образуют ионные гидриды, гидриды элементов III
группы вследствие недостатка электронов в их молекулах имеют
полимерную структуру.
Ионные гидриды получают нагреванием щелочных и
щелочноземельных металлов, примерно при 350—400°С в атмосфере сухого
водорода; Li, Ca Sr реагируют легко, Na реагирует о трудом.
Все ионные гидриды — бесцветные кристаллические вещества
с температурами плавления 700—500°С. Теплоты их образования-
21—23 ккал/моль.
Ионные гидриды щелочных и щелочноземельных металлов
содержат гидрид-ион Н" наряду с катионами соответствующих
металлов. Это подтверждается электролизом LiH, водород
выделяется на аноде, а не на катоде, как обычно. Следовательно, водород
здесь играет роль С1~ в NaCl. Ионные гидриды щелочных
металлов имеют кубическую гранецентрированную структуру типа
NaCl, радиус гидрид-иона равен 1,54 А (несколько меньше
хлорид-иона 1,8 А).
Ионные гидриды устойчивы .к кислороду при комнатной
температуре, но они энергично взаимодействуют с водой. Процесс-
гидролиза протекает по уравнению
H- + HaO-*H2t + ОН-
и используется как один из методов получения водорода.
Гидролиз является одновременно окислительно-восстановительным
процессом
н- + н+ -> н2.
(из гидрида) (из воды)
Следовательно, гидриды — хорошие восстановители.
Металлообразные гидриды, или гидриды внедрения,
образуются переходными металлами при непосредственном взаимодействии
их с водородом, даже на холоду (палладий, лантан), или чаще
при нагревании.
Гидриды внедрения образуют металл IV В группы (Ti, Zr, Hf),
V В (V, Nb, Та) и VIII В (Fe, Co, Ni, Pd, Pt).
415*
1000
И,бес %
0,25 0,5 1,0 1,5 2
3 4
Количество поглощенного водорода зависит от давления и
температуры. Поэтому гидриды этих металлов имеют нестехио-
метрические составы, например TiHij, ZrHi,g, ТаНо,э, PdHo,3,
VH09, и в отдельных случаях могут рассматриваться ка,к твердые
растворы водорода в металлах (рис. 12.21). Палладий и ванадий
поглощают водород (первый
до 850 объемов) при
комнатной температуре, остальные
металлы только при
нагревании.
При поглощении водорода
решетка металла не
изменяется, но набухает (по данным
рентгеновских и пикнометри-
ческих исследований). Это
подтверждает предположение
о внедрении водорода между
атомами-ионами в решетку
металла.
Природа связи между
атомами водорода и металлом
окончательно еще не выяснена.
н.ат.у»
Рис. 12.21. Часть диаграммы
состояния системы Ti—Н
В гидридах переходных металлов водород содержится в виде
атомов Н (с атомным радиусом 0,37 А).
Предполагается, что электроны водорода занимают свободные
уровни в энергетических зонах атомов металла. По-видимому, при
реакции образования гидридов внедрения потребляется большая
энергия, достаточная для разрыва молекулы Н2 на атомы.
Теплоты образования гидридов внедрения имеют тот же
порядок величины, что и теплоты образования ионных гидридов.
Гидриды элементов побочной подгруппы III группы—
лантаноидов и актиноидов — по своему поведению занимают
промежуточное положение между гидридами внедрения и ионными
гибридами. С первыми они сходны тем, что образуют нестехиометриче-
•ские соединения. Например, LaH2)8, СН2)8 PrH0,o, PuH2, AmH2 —
нестехиометрические и обладают металлической проводимостью.
Со вторыми они сходны своей значительно более высокой
реакционной способностью. Например, UH3 пирофорен, реагирует с
раствором AgN03, образуя нитрат уранила, металлическое серебро
и водород. Гидриды внедрения обладают сильными
восстановительными свойствами, например гидрид палладия восстанавливает
ртуть из ее соединений.
416
Гидриды металлов применяются как ракетное топливо.
Разложение летучих гидридов по нагретой поверхности позволяет
покрывать поверхность металлом. Это имеет значение для
изготовления полупроводников осаждением кремния или германия при
разложении силана SiH4 или германа GeH4. Имеется ряд
возможностей для использования гидридов в технологических
процессах ядерных реакций. Твердые гидриды типа ZrH2 дают при
высоких температурах большую объемную концентрацию
водорода, что может быть использовано для ослабления потока
нейтронов в реакторах как замедлителей. Образование гидридов титана,
циркония и кадмия на поверхности теплообменников может
защитить их от жидких металлов. В порошковой металлургии
гидриды являются наиболее удобным источником быстрого
получения металлических порошков.
Гидриды используют в качестве источника водорода и
восстановителей в химическом синтезе.
Карбиды, нитриды, бориды металлов. Карбиды металлов —
соединения углерода с металлами. Они довольно многочисленны
и многие из них образуются при непосредственном
взаимодействии металлов с углеродом. Факторы, определяющие это
взаимодействие, аналогичны гидридам.
Обычно карбиды подразделяются на 4 группы: 1) ионные, или
карбиды солеобразного характера; 2) ковалентные карбиды;
3) металлического характера, или карбиды внедрения; 4) карбиды
типа цементита Fe3C. Ионные, или солеобразные, карбиды, в
которых углерод играет роль аниона С2-. Они образуются более
электроположительными металлами (Li, Ca, К, Be, A1). Сюда
относятся карбиды, рассматриваемые как продукты замещения
водорода в метане на металл (карбиды алюминия и бериллия).
К этой же группе относятся карбиды, являющиеся
производными ацетилена (карбид кальция), и карбиды, которые при
действии воды и растворов кислот выделяют не только ацетилен,
но и другие углеводороды, а также водород (например, ацетиле-
ниды серебра, меди, кальция и др.). Их кристаллы бесцветны,
ирозрачны и при обыкновенной температуре не проводят
электрический ток.
Поскольку ионы углерода не могут существовать в растворе,
карбиды этой группы разлагаются водой или разбавленными
кислотами, образуя углеводороды.
Различают три наиболее важные группы ионных карбидов:
1. Карбиды, при гидролизе образующие метан (Ве2С и АЦС3).
Они содержат по данным рентгеновского анализа ион С4-
А14С3 + 12Н20 ->ЗСН4 + 4А1 (ОН)3,
С4- + 4Н20 -* СН4 + 40Н~.
2. Карбиды, дающие при гидролизе ацетилен (ацетилениды
Na2C2, K2C2, CaC2, SrC2, BaC2 CuC2, Ag2C2). Они соедржат ацети-
летид-ион Сг2-, который мгновенно реагирует с водой:
14 общая химия
417
СГ~ 2НОН^НС = СН + 20Н-.
3. Карбиды, образующие при гидролизе ацетилен и
ненасыщенные углеводороды (за счет гидрирования водородом
ацетилена): LaC2, CeC2, РгС2, NdC2, GfC2, ThC2.
Ко второй группе относятся карбиды с типично ковалентной
связью. Это очень твердые и жаростойкие соединения кремния,
бора, группы хрома (табл. 12.5), группы марганца. Состав их
весьма различен. Они имеют высокие температуры плавления и
энтальпии образования.
Таблица 12.5
Некоторые свойства карбидов подгруппы хрома
Карбид
Сг4С
Cr3Cs
Сг7С3
Сг2зСб
Мо2С
МоС
W2C
WC
Плотность,
i/CM3
6,68
6,92
6,97
8,9
8,4
17,2
15,6
Температура
плавления, °С
1520
1895
1665
1550
2410
2700 (разл.)
2730
2720 (разл.)
Теплота
образования,
кдж/моль
68,6
87,9
177,8
107,9
17,6
29,7
38,1
Электрические свойства
удельная
электропроводимость,
см/м
1,333-10е
0,918-106
0,788-106
1,41.10е
1,32-106
5,22-105
температура
перехода в
сверхпроводящее
состояние, °К
<1,20
<1,20
<1,20
2,78
9,26
2,74
<1,28
К третьей группе относят металлоподобные карбиды,
представляющие собой фазы внедрения. Карбиды внедрения образуются
слабоэлектроположительными переходными металлами. Они имеют
металлический внешний блеск, их свойства во многом сходны
со свойствами металлов (например, они имеют электронную
проводимость). Часто структура этих карбидов аналогична структуре
тех металлов, из которых они получены, поэтому они могут быть
отнесены к группе интерметаллических соединений.
Цинк, кадмий, ртуть, галий, индий, таллий, германий, олово
и свинец карбидов не образуют. Элементы платиновой группы
растворяют углерод, но индивидуальные соединения не выделены»
4. Карбиды железа, никеля, марганца являются
промежуточными между ионными и металлообразными. Карбид железа —
цементит— играет важную роль в металлургии железа.
Соединения азота и бора с металлами аналогичны карбидам и
по методам получения и по свойствам. Все эти соединения, как и
карбиды, отличаются высокими температурами плавления и
большой твердостью:
418
TiC 7rC HfC NbC TaC WaC WC
*пл, ь
твердость
t °Г
*пл. ^
твердость
3140
8—9
TiN
2950
8—9
3535
8—9
ZrN
2985
8
3890
TaN
3500
TiB
3090 —
3880
ZrB
2995
9
2830
9—10
2720
9
Нитриды — соединения металлов, в которых с атомами
металлов непосредственно связаны атомы азота. Нитриды делят на
ионные, ковалентные и металлоподобные. Образуются они при
непосредственном окислении азотом соответствующих
элементарных металлов. Эти реакции значительно затруднены по сравнению
с кислородными, так как молекула N2 очень прочна и энергия
диссоциации ее очень велика (рис. 12.16).
Аналогично карбидам, щелочные и щелочноземельные
металлы, а также металлы подгруппы меди и цинка дают ионные
соединения. Элементы с внешними р-электронами — ковалентные
нитриды (В, А1, Са), а металлы переходных групп с недостроенными
d-уровнями — металлоподобные.
Соответственно нитриды щелочных металлов Li3N, Na3N
разлагаются водой с выделением аммиака
N3"
3HaO-*NH8t + 30H-
Нитриды титана, циркония, гафния и ванадия (свойства даны
в табл. 12.6) металлоподобны, очень тверды и тугоплавки.
Нитриды малоактивных
Таблица 12.6
Электрическое свойство нитридов,
карбидов, силидов и боридов ванадия
металлов напоминают
интерметаллические
соединения. Нитриды применяют
для получения защитных
пленок азотированием
поверхности металла.
Формулы гидридов,
боридов, карбидов и нитридов
не отвечают обычным
валентностям, часто они не
имеют стехиометрический
состав. Они обладают рядом
специфических свойств,
присущих металлам
(непрозрачные, проводят электрический ток, проявляют
сверхпроводимость и др.). Такие свойства определяются специфической
структурой этих соединений. В их кристаллических решетках атомы
неметаллов занимают пустоты в решетке соответствующего металла.
По этой причине гидриды, карбиды, нитриды, бориды называются
соединениями внедрения. Поскольку они образуются в твердом
Формула
соединения
V2N
VN
VC
VSi2
VB
VB2
Удельная
электропроводность,
см/м
8,14-10»
1,17.10е
1,54-106
1,505.106
2,86—2,5- '10*
5,26-10*
Температура
перехода в
сверхпроводящее
состояние, °К
1,28
7,5—8,2
1,20
1,20
1,28
1,9
14*
419
состоянии диффузией атомов неметаллов в пустоты металлической
решетки, легко понять, почему некоторые пустоты остаются
незанятыми, а состав соединений часто нестехиометрический.
Объясним это подробнее. Как уже отмечалось, большинства
металлов кристаллизуется в плотных гексагональных и
кубических решетках. Между двумя плоскостями атомов плотной
решетки (предполагается, что атомы представляют собой равные
сферы — шары) образуются два вида междоузлий пустот: одни
ограничены четырьмя атомами (тетраэдричеокие пустоты),
другие— шестью атомами (октаэдричеокие пустоты).
Число октаэдрических пустот равно числу сфер. При
заполнении октаэдрических пустот плотной кубической решеткой
образуются соединения MX, например TiC, TiN, VC, TaN, TiB и
другие, которые имеют решетку хлорида натрия. Если заполняется
только половина октаэдрических пустот в правильном порядке, то
образуются соединения М2Х, например W2N, Mo2N и др. Число
тетраэдрических пустот в плотной кубической решетке вдвое
больше, чем число сфер. Эти пустоты меньшего размера могут
занимать только атомы водорода. Если бы все тетраэдрические
пустоты были заняты, то образовалось бы соединение МеХ2 (TiH2).
В гидриде палладия занята, вероятно, 1/4 тетраэдрических пустот,
что приводит к идеальной формуле PdH2.
Металлы с плотными гексагональными решетками образуют
соединения внедрения таким же путем. Например, в V2C, Mo2C
атомы углерода занимают 1/2 октаэдрических пустот, а «в ZrHz
и Ti2H атомы водорода занимают 1/4 тетраэдрических пустот
гексагональной решетки.
Карбиды внедрения образуют только металлы, атомные
радиусы которых больше 1,3 А, так как решетки только этих
металлов имеют октаэдричеокие пустоты, достаточно большие, чтобы
вместить атомы углерода. Этим объясняется, почему некоторые
элементы не образуют карбиды внедрения.
Природа связи между металлом и С, N и В еще не выяснена.
Вполне возможно, что при образовании связи металл — неметалл
в этих соединениях металлическая решетка не может разрушиться,
как при взаимодействии с хлором или кислородом. Вместе с тем
атомы С, N или В в октаэдрической пустоте металлической
решетки не имеют количества электронов, достаточные для образования
ковалентных связей со всеми шестью атомами металла, которыми
они окружены.
Следовательно, необходимо допустить, что эти связи того же
типа, что и в металлических решетках, которые также имеют
дефицит электронов (по этой причине энергетические зоны атомов
металла с электронами, отданными неметаллом, заняты
неполностью). По этой причине карбиды хрома, марганца и железа
имеют следующие особенности: их радиусы атома небольшие,
поэтому внедрение посторонних атомов ведет к деформации решетки.
Эти карбиды обладают кристаллическими решетками, отличными*
420
от исходного металла, и их состав не согласуется с обычными
валентностями (табл. 12.5).
Силициды металлов. Кремний образует ионно-ковалентные
и металлоподобные соединения с металлами — силициды.
Щелочные и щелочноземельные металлы дают
ионно-ковалентные соединения. Таковы, например, силициды лития: Li2Si,
Li4Si, Co2Si. Они бурно реагируют с водой, выделяя силан:
Si4- + 4Н20 - SiH4 + 40Н-.
Если атом металла имеет р-валентные электроны, то
образуются силициды, в которых металлы и кремний связаны кова-
лентными связями.
Переходные металлы с недостроенными оболочками образуют
с кремнием металлоподобные соединения (табл. 12.6). В них доля
ковалентной связи понижена, отчетливо проявляется
металлическая связь.
В ионно-ковалентных соединениях кремний с металлом связан
ионной связью, а атомы кремния друг с другом — ковалентной.
Электронные орбитали кремния гибридизованы (sp3) и атомы
кремния размещаются таким образом, что возникают тетраэдри-
ческие группы из четырех отрицательных ионов кремния, которые
окружены 16 ионами идентичного металла.
Серебро, золото, цинк, кадмий, ртуть, алюминий, галлий, лидий,
талий, сурьма, висмут, германий, олово, свинец, бериллий и бор
не образуют силицидов.
Сульфиды, галиды. К группе бинарных соединений металлов
можно отнести их соединения с галогенами—галиды (табл. 12.7),
с серой — сульфиды, где
Таблица 12.7
Свойства галидов элементов группы IV
В — хрома
атомы галогенов и серы
непосредственно соединены с
металлами. Степень
окисления галогенов в них — 1,
а серы — 2. Они также
разделяются на ионные, кова-
лентные и имеется
постепенный переход от галидов
ионных (солеобразующих) к
галидам ковалентным
(кислотообразующим) .
Среди них много
соединений нестехиометрического
состава. Наиболее
распространенный сульфид
железа также является соедине-
нием переменного состава.
Группу новых бинарных соединений образуют, например,
германий с металлами — германиды — или алюминий с
металлами — алюминиды. Они детально изучены и представляют интерес-
Галид
CrF4
CrF8
CrCl3
СгС12
MoFe
MoCL
WF6
WC18
Теплота
образования,
кдж/моль
1196,6
1112,9
554,4
384,1
1694,5
379,9
1255,2
405,8
о
'пл'С
_
1100
1150
816
17
194
—0,5
284
j° ^
t . с
'кип*
270 (субл
1430
925
1300
36
268
+ 17
337
.)
421
ные классы интерметаллических соединений. Рассмотрим лишь
один из них.
Германиды металлов. Германиды — соединения германия с
металлами, где атомы металла непосредственно связаны с атомами
германия. Германиды имеют много общего с силицидами. По
физико-химическим свойствам и структуре германиды переходных
Пери!
оды
1
2
3
| 4
5
6
\7
1а
fir
N*
К
[ Rb
Cs
|Fr
Подгруппы J
1 Па
lb
lib
IITb
IVb
Vb
VI b
vnblvmjvni vin'
и
Mg
ICa
Sr
Ba
|Ra|
п>
о
L.J»
Cu
Ag
1 Au
Zn\
Cd
H
&
1
Y
La
Ti
Zr
Hf
V
Nb
Та
Cr
Mo
W
Mn
Tc
Re
Fe
Ru
Os
Co
Rh
Ir
Fi
Pdl
•
«j
rib
IVa
Va;
Via
Vna
0
He|
?'
t
1
jGa
jln
LL1.
С
Si
Ge
Sn
Pb
N
p
As
Sb
Bi
0
s
Se
Те
Po
F
CI
Br
1
At
—м
Neil
Ar|
Krj
Xej
Rnj
Ы 1
ГЛантано-
I иды
J Актино-
[Се
J Th
IPr
Pa
Nd
U
[Pm
JNp
Sm
Pu
Eu
Am
Gd
Cm
Tb
Bk
Dy
Cf
Ho
Es
Er
Fm
Tu
Md
Yb
No
lu
1 Ц
I Lrj
Рис. 12.22. Классификация соединений германия:
/ — соединения с ионно-ковалентной связью; 2 — соединения с ковалентно-
металлической связью; 3 — соединения с ковалентно-ионной связью
металлов способны переходить в сверхпроводящее состояние,
имеют довольно высокую электропроводность, металлический блеск
(^ = 2000—2400° С).
Однако в отличие от нитридов и карбидов германиды, как и
силициды, нельзя отнести к соединениям типа фаз внедрения, так
как условие /*х-^а<0,59 не выполняется. Это отношение для Ge с
переходными металлами составляет более 0,84 и увеличивается
до 1,12 для никеля.
Поэтому классифицировать германиды сложнее (рис. 12.22).
Рассмотрим соединения германия с 5-, р- и d-элементами.
Германий имеет 52/?2-конфигурацию. Если совершается s-^p-переход,
получается ^-конфигурация, свойственная алмазу. s-Элек-
троны, переходя к германию, стабилизируют 5/?3-конфигурацию
422
его атомов. Это соответствует образованию ионного типа связей
между щелочными металлами и германием.
Вместе с тем атомы германия способны к взаимной
стабилизации 5/?3-конфигурации, при объединении между собой в ковалент-
ные группы. Поэтому щелочные металлы образуют MeGe2 и поли-
германиды MeGe4. Они обладают полупроводниковыми
свойствами.
Аналогичные соединения следует ожидать также для бериллия,
магния и щелочноземельных металлов. Однако бериллий не
образует германидов. Это связано со способностью конфигурации
бериллия 52 переходить в sp2 и понижать тем самым концентрацию
5-электронов. При переходе к магнию вероятность переходов s-+p-
уменьшается вследствие увеличения стабильности
^-конфигурации. Это ведет к образованию германида также состава Mg2Ge.
При переходе к кальцию появляется вакантный Зй-уровень и
возможны переходы d^^s^^sp и вероятность s-+p уменьшается, а
появляется новая возможность d-функции связи. Поэтому германий
дает германиды различного состава CasGe, CaGe, CaGe2, Ca7Ge
и Ca3Ge.
Для стронция подобные соединения не получены. В подгруппе
меди германиды известны для меди, а серебро и золото не
образуют германидов. Это, вероятно, связано с наличием
^^-конфигурации у меди и переходом s-электрона меди к германию. У
золота и серебра появляются вакантные 4/- и 5/-состояния, d-элек-
троны частично могут переходить на эти уровни и d-состояние
будет незаполненным.
В подгруппе цинка ни один из металлов германиды не
образует. Они имеют й1052-конфигурацию. Германиды s-металлов
имеют ионно-ковалентный тип химической связи, преимущественно
ионный между атомами металла и германием и ковалентный
внутри групп атомов германия и групп атомов металла.
Соединения германия с переходными d-металлами имеют
сложный ковалентно-металлический характер химической связи с
наложением в ряде случаев значительной доли ионного типа связи.
При образовании германидов переходных металлов следует
учесть количество d-электронов ^5 и ^5. Для первых
характерны стабильные конфигурации d° и d5 и, увеличение
статистического веса й5-электронов, а следовательно, и числа
локализованных (участвующих в ковалентной связи) электронов и
понижение относительного числа коллективизированных электронов,
осуществляющих металлическую связь. Так, в металлическом
цирконии локализованы 2,6 d-электрона на атом и 1,4я?-электрона
коллективизировано для ниобия — 3,8 и 1,2 соответственно.
Следовательно, статистический вес ^-конфигурации существенно
возрастает, а ^-конфигурация уменьшается при переходе от циркония
к ниобию.
В связи с этим следует ожидать для элементов IV группы
(Ti, Zr, Hf) перехода нелокализованных электронов к атомам
423
германия и повышения энергетической стабильности их от титана
к гафнию. Поэтому появляются Zr2, Ge, Hf2Ge, а для титана
подобных фаз нет, так как у него статистический вес
^-конфигураций меньше всего.
Подобные закономерности наблюдаются и для элементов W,
Nb, Та, Сг, Мо. Параллельно может идти образование ковалент-
ных группировок из атомов германия, что ведет к образованию
германидов, CruGeig, богатых германием.
Этим объясняется понижение температур плавления
германидов и отсутствие германидов у вольфрама. Электрофизические
свойства германидов металлов d^5 выше, чем силицидов, а
микротвердость ниже. Для них характерны высокие температуры
перехода в сверхпроводящее состояние Гк.
NbsGe имеет ГК=18,6°К, высокие Гк получены при замене
части германия на олово или алюминий, или галлий: Nb3Ge0)5Alo,5
12,6°К; Nb3GeSn 17,6—18°К; Nb3Ge0,5Ga0,5 7,3°K. Если
рассматривать германиды d>5, то следует учитывать стабильные rf5- и к10-
конфигурации. Кобальт образует соединения: Co3Ge, CoGe, Co5Ge8.
У никеля и железа имеются аналогичные соединения. Известны
также RuGe, Ru2Ge3, IrGe, Ir3Ge7, Pd4Ge, Pd2Ge, Th3Ge, U7Ge.
Лантоноиды и актиноды образуют германидные фазы,
аналогичные d-переходным металлам, за счет /-^-переходов.
Неметаллы и полуметаллы дают с германием соединения с ковалентно-ион-
ным типом связи. Получают германиды или синтезом из элементов
(сплавлением или спеканием, или термическим разложением в
вакууме, или электролизом расплавленных сред). Германиды I и II
группы неустойчивы к влаге и дают GeH2. Для германидов
IV группы характерна повышенная устойчивость. Они
разлагаются только смесью азотной и плавиковой кислот и растворяются
концентрированной серной кислотой. Применяются они главным
образом для полупроводниковой и электронной техники.
Вопросы и упражнения
1. Назовите особенности электронной и кристаллической структуры
металлов.
2. Охарактеризуйте особенности металлической связи.
3. Какие вы знаете важнейшие бинарные соединения металлов? Как их
можно классифицировать по типу связи и положению образующего металла
в периодической системе? Какова роль электронной структуры исходного
металла в этой классификации?
4. Что такое сплав? Какова природа химического взаимодействия сплавов?
5. Перечислить основные положения физико-химического анализа.
6. Нарисуйте основные типы диаграмм состояния и состав — свойства.
К какому типу можно отнести диаграммы состояния систем: серебро — золото;
цинк — кадмий; магний — медь.
7. Определить, какие фазы будут находиться в равновесии в системе медь —
никель при 1200°С и каков их состав.
8. Рассчитайте количество жидкой и твердой фазы в сплаве с 20% Bi и
80% Cd системы висмут—кадмий при 20О°С. Какова будет структура этого
сплава после кристаллизации?
ГЛАВА 13
ХИМИЯ КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ
§ 1. ОСНОВНЫЕ ПОНЯТИЯ ХИМИИ
КООРДИНАЦИОННЫХ СОЕДИНЕНИИ
Обширную группу химических соединений составляют так
называемые комплексные, или координационные соединения, В
молекулах этих соединений всегда имеется центральный атом или ион,
окруженный определенным числом других ионов или
молекулярных групп.
Координационные соединения имеют исключительно большое
значение в живой и неживой природе. Достаточно сказать, что
гемоглобин, благодаря которому осуществляется перенос кислорода
из легких к клеткам ткани, является комплексом железа, а
хлорофилл, ответственный за фотосинтез в растениях, — комплексом
магния. Значительную часть природных минералов, в том числе
многие силикаты и полиметаллические руды, также составляют
координационные соединения. Более того, химические методы
извлечения металлов из руд, в частности Al, Cu, W, Au, Pt, Fe связаны
с образованием легкорастворимых, легкоплавких или
.высоколетучих комплексов. Современная химическая промышленность широко
использует координационные соединения как катализаторы (синтез
высокополимерных материалов, производство кислот, химическая
переработка нефти).
Учение о комплексообразовании пронизывает всю современную
химию, так как почти все неорганические и металлоорганические
соединения можно рассматривать как координационные.
Например, образование любых кислородсодержащих кислот и их солей
можно трактовать как результат проявления координационной
связи. Среди хорошо известных химических соединений
комплексными являются многие кристаллогидраты, например медный
купорос CuS04-5H20, железный купорос FeS04-7H20, двойные соли
типа квасцов KA1(S04)2- 12H20 и шенитов K2Mg(S04)2-6H20,
двойные галогениды, комплексные сульфаты, карбонаты, нитраты,
все основные соли.
Как показывает само название, комплексные соединения —
химически сложные и, как правило, состоят из трех или большего
числа разнородных элементов. Однако сложность состава не
единственная специфическая особенность координационных соединений.
Можно назвать немало химически сложных веществ, не
принадлежащих к числу координационных соединений, например
глицерин СН2ОН—СНОН—СН2ОН, а вместе с тем известны комплексы,
сравнительно несложные по химическому составу, например тетра-
425
карбонил никеля Ni(CO)4, широко используемый для экстракции
и очистки никеля.
Что же в таком случае отличает комплексные соединения oi
всех других? В комплексных соединениях помимо обычных типов
связи, уже знакомых нам ранее (см. гл. IV7), действует особая
связь, получившая название координационной. В результате
проявления этой связи возникает возможность образования прочного
соединения из двух других, также устойчивых и, более того,
имеющих насыщенную в обычном понимании валентность. Примером
может служить образование стабильного продукта CoCl3-6NH3 при
взаимодействии хлорида Со3+ и аммиака. Нет сомнений, что оба
реагента термодинамически весьма стабильны и мало склонны
изменять валентное состояние составных частей. Действительно,
в молекуле аммиака NH3 азот (электронная структура
ls22s22px2py2pz) имеет максимальную валентность по водороду,
соответствующую использованию всех неспаренных электронов
2рх, 2ру и 2pz для образования пар с электронами водородных
атомов. Пользуясь диаграммой Льюиса, структуру аммиака
выражают формулой
Н
:Nx H
Н
У хлорида кобальта СоС13 связь осуществляется главным
образом за счет электростатического притяжения ионов Со3+
(\s22s22p63s23p63d6) и хлор-иона, хотя не исключена известная
доля ковалеитности связи, обусловленная частичным перекрыванием
атомных орбиталей электронов, принадлежащих ионам Со3+ и Ch*.
При всех обстоятельствах кобальт не склонен проявлять более
высокую валентность, чем +3.
Проявление координационной связи является также причиной
образования комплексных ионов в растворах и расплавах.
Известно, например, что в водном растворе аммиака концентрация
свободных молекул NH3 ничтожна, тогда как подавляющая часть их
связана за счет образования ионов аммония по реакции
NHg + H+^NH^. (13.1)
Ион аммония — простейший пример комплексного иона. Реакция
(13.1) обратимая, хотя в обычных условиях равновесие сильно
сдвинуто вправо. При 25°С константа равновесия
кр= mti ^i.o.iQ-.
fNH3].[H+]
Используя принцип Ле Шателье, можно указать, как
разрушить комплексный ион. Для этого достаточно удалять один из ре-
426
агентов (нагревание растворов гидроксида аммония приводит к
испарению NH3) или связывать реагенты в еще более прочное
соединение (прибавление щелочи ведет к взаимодействию
н+ + он-^:н2о,
для которого константа равновесия Лр = 5,5-1013 при 25°С).
Следует отметить, что координационная связь,
обусловливающая появление комплексных соединений, в ряде случаев весьма
прочная, это создает известные трудности при осуществлении
химических превращений. Например, безводные хлориды многих
переходных металлов невозможно получить растворением металлов
в соляной кислоте, так как равновесие
М"+ -f /nH20 ^ [M (H20)m]"+
сильно сдвинуто вправо. Более того, попытка удалить
кристаллизационную воду из уже синтезированных хлоридов нагреванием
редко приводит к успеху, так как продуктом нагрева являются не
безводные хлориды, а оксихлориды и другие соединения,
образование которых продиктовано стремлением металлического иона
насытить свою координационную связь и после удаления воды.
§ 2. КООРДИНАЦИОННАЯ ТЕОРИЯ ВЕРНЕРА
Хотя комплексные соединения известны химикам уже более
двух столетий, причина их образования долгое время оставалась
загадкой. Современное понимание природы комплексов стало
возможным благодаря исследованиям швейцарского ученого А. Вер-
нера, который в 1893 г. предложил теорию, известную теперь в
химии под названием координационной теории Вернера.
Основные положения этой теории следующие:
1. Атомы большинства химических элементов проявляют два
типа валентности — главную и побочную. Первая соответствует
степени окисления элемента, а вторая — его координационному
числу, причем последняя может быть истолкована как
максимальное число атомов, ионов или молекул, присоединенных
непосредственно к центральному атому.
2. Атомы каждого элемента стремятся насытить как главную,
так и побочную валентности.
3. Побочная валентность строго фиксирована в пространстве,
окружающем центральный атом.
Смысл первого и основного положения координационной
теории сводится к тому, что соединительная способность данного
элемента, устанавливаемая сравнением с водородом и называемая
валентностью в обычном смысле, не исчерпывает всех сил
сродства, свойственных этому атому. Атом имеет еще и другие силы
сродства, природа которых не была понятна Вернеру, но
проявлялась в форме дополнительной валентности.
42 7
Эффективность координационной теории удобно
иллюстрировать на примере комплексных соединений, образуемых треххло-
ристым хромом с аммиаком. Удалось синтезировать только четыре
таких соединения, состав которых представлен в табл. 13.1. Наи-
Таб л ица 13.1
Физико-химические свойства растворов солей
Состав соли
CrCIs-6NH3
CrCl3-5NH3
CrCIs-4NH3
CrCl3-3NH3
Число
осаждающихся
ионов Ст
3
2
1
0
Мольная
проводимость
1
уменьшается
1
0
Число молей частиц
образующихся при
растворении 1 моля
соли
4
3
2
1
более примечательно сопоставление их физико-химических свойств.
При взаимодействии гексааммиаката CrCl3-6NH3 с нитратом
серебра происходит немедленное осаждение всех ионов С1~ в виде
AgCl. Тот же опыт с солью CrCl3-5NH3 приводит к быстрому
осаждению лишь двух ионов СГ~ из трех, присутствующих в
молекуле. Для комплексов, содержащих 4 и 3 молекулы NH3,
способность к осаждению хлора уменьшается, так что у CrCl3-3NH3
.она полностью отсутствует.
На основании этих наблюдений можно заключить, что в
различных аммиакатах треххлористого хрома характер связи О в
молекуле комплекса не одинаков. Более того, молярная
проводимость растворов в различных аммиакатах резко различна и
уменьшается по мере перехода от CrCl3-6NH3 к CrCl3-3NH3, так что
для последнего комплекса она близка к нулю. Учитывая, что
проводимость водных растворов солей обусловлена их
электролитической диссоциацией, можно ожидать, что у рассматриваемых
аммиакатов способность к диссоциации падает с уменьшением
содержания NH3 и почти отсутствует у CrCl3-3NH3. Дополнительную
полезную информацию о характере связи в молекулах CrCl3-ftNH3
удалось получить на основании криоскопических измерений.
Например, понижение точки замерзания водного раствора
CrCl3-6NH3 указывает на то, что из 1 моля соли в растворе
образуются 4 моля частиц, тогда как из 1 моля соли CrCl3-3NH3
образуется только 1 моль частиц.
Указанные явления превосходно объясняются в рамках
координационной теории Вернера. В соответствии с этой теорией ион
Сг3+, как и любой другой, проявляет два типа валентности —
главную и побочную. Первая из них, судя по степени окисления,
равна трем, а вторая, исходя из состава наиболее богатого
аммиаком комплекса, равна шести.
428
Связанные побочной валентностью молекулы аммиака
непосредственно группируются вокруг иона Сг3+, образуя вместе с ним
координационную сферу, тогда как ионы С1~ находятся дальше
от металлического иона и не связаны с ним столь прочно. По Вер-
неру состав молекулы CrCl3-6NH3 можно представить формулой,
показанной на рис. 13.1, в предположении, что главная связь
изображается сплошной, а побочная — пунктирной линией. Все глав-
мн3
HN 1 /С1
\ /
СГ NH5
/ 1 \>NH»
«? 1 \
нн3 Nci
CI
н>\ I y\
CI Cr NH3
^ 1 \
NH3 Cl
Рис. 13.1. Координационная Рис. 13.2. Координационная
формула CrCl3-6NH3 формула CrCl3-5NH3
ные связи в молекуле CrCl3-6NH3 насыщены хлор-ионами, а
побочные — молекулами NH3. Учитывая относительную прочность
этих связей, можно ожидать, что при растворении в воде из одной
молекулы CrCl3-6NH3 образуются четыре частицы [Cr)NH3)6]3+
и 3 иона С1~. Это полностью согласуется с опытными данными
табл. 13.1.
Что же произойдет при удалении из гексааммиакатов одной
молекулы NH3? При этом должна «обнажаться» одна побочная связь,
но, учитывая, что, по Вернеру, каждый элемент стремится
насытить все свои валентности, резонно допустить, что «обнажившаяся»
валентность будет использована на связь с одним из ионов С1~,
иначе говоря, этот ион должен одновременно насыщать оба типа
валентности. Очевидно, что в этом случае один из ионов С1~
переходит в координационную сферу, как показано на рис. 13.2, и
теряет способность легко отрываться от иона Сг3+ при растворении
аммиакатов в воде. Именно этим объясняется снижение
проводимости при переходе от гекса- к пентааммиакату, уменьшающему
до трех число частиц, образуемых при диссоциации (два иона С1~
и [Cr(NH3)5Cl]2+) и невозможность осаждения всего хлора при
взаимодействии с AgN03.
Свойства тетрааммиаката CrCl3-4NH3 и триаммиаката
CrCl3-3NH3 хорошо объясняются формулами, представленными на
рис. 13.3. У триаммиаката все ионы С1", одновременно
насыщающие главную и побочную валентности, теряют способность
реагировать с AgN03 и отщепляться от молекулы CrCl3-3NH3 при
растворении в воде.
429
Аналогичным образом может быть установлен характер связк
между составными частями любого комплексного соединения.
Очевидно, что в каждом из них можно выделить атом или ион,
являющийся комплексообразователем. Переходные металлы (особенна
d-элементы) настолько часто выступают в роли комплексообразо-
вателей, что химию координационных соединений иногда отожде-
ci Сг ^СРН.
w' Ix" Cl I Cl
a 6
Рис. 13 3. Координационные формулы CrCl3-4NH3 (a) и
CrCl3-3NH3 (б)
ствляют с химией переходных металлов. Однако
комплексообразователем может быть также непереходный металл и даже неметалл.
В качестве примера можно привести А13+ (фторидные комплексы
типа NaAlF4, Na2AlF5, Na3AlF6), Be2+ (фторбериллаты NaBeF3r
Na2BeF4) и уже упоминавшиеся нами N3+ (аммонийные соли).
Атомы или ионы, расположенные непосредственно вокруг комплек-
сообразователя и связанные с ним за счет проявления
дополнительной валентности, называются лигандами. В качестве лигандов
могут выступать различные электроположительные,
электроотрицательные и нейтральные частицы (табл. 13.2).
Лиганды вместе с комплексообразователями составляют
внутреннюю сферу комплекса, которые при написании формулы
координационного соединения помещают в квадратные скобки. Заряд
комплексного иона легко подсчитать, зная валентность его
составных частей. Например, заряд иона [Ti(H20)6] равен +4, если
комплексообразователь находится в четырехвалентном состоянии
(лиганды нейтральны). Заряд иона [Co(NH3bCl] равен + 2, если
кобальт трехвалентен (среди лигандов лишь ион хлора несет
заряд — минус 1). Совершенно аналогичным образом можно
подсчитать заряд иона-комплексообразователя, если известен заряд
комплексного иона и лигандов.
Ионы или нейтральные части комплекса, не являющиеся
лигандами или комплексообразователем, составляют внешнюю сферу
комплексов. Состав рассматривавшихся выше аммиакатов трех-
хлористого хрома следует выразить формулами
гексааммиакат [Cr (NH3)6] Cl3,
430
пентааммиакат [Cr (NH3)5 CI] Cl2,
тетрааммиакат [Cr (NH3)4 Cl2] CI,
триаммиакат [Cr (NH3)3 Cl3].
Таблица 13.2
Наиболее распространенные лиганды
Нейтральные молекулы
Положительные ионы
Отрицательные ионы
Хелатообразующие
лиганды
СО — карбон ил
NH3 — аммин
Н20 — акво
N0 — нитрозил
NH2NH^ — гидразиниум
N0+
F~" — фторо
С1~~— хлоро
0Н~~ — гидроксо
CN~~ — циано
N0^" — нитро
О2- —оксо
СО2- — карбонато
СО2- — оксалато
H2NCH2CH2NH^~ — этилендиамин (еп)
Заметьте, что у последнего соединения внешняя сфера комплекса
отсутствует.
Важнейшей характеристикой любого комплексного соединения
является координационное число, показывающее число лигандов.
Наиболее характерно образование комплексов с координационным
числом, равным 6, причем чаще всего такие комплексы имеют
октаэдрическое строение. Например, в комплексном ионе
[A1F6]3~ (этот анион содержится «в минерале криолите) ион
алюминия можно представить как расположенный в центре октаэдра,
вершины которого заняты ионами F- (рис. 13.4). Другой пример
комплексного иона с октаэдрическим строением — это ион
[Fe^C^h]3-, образующийся в процессе удаления ржавчины с
поверхности железных изделий раствором щавелевой кислоты.
Особенностью этого иона, структура которого изображена на
рис. 13.5, является то, что каждая оксалатная группа занимает
431
два места в координационной сфере, окружающей комплексооб-
разующий ион Fe3+.
Октаэдрическое строение комплексов с координационным
числом, равным 6, не очевидно и нуждается в доказательствах,
которые могут быть получены различными методами. Среди них наи-
Рис. 13.4. Структура
комплексного иона [AlFe]3""
Рис. 13.5. Структура комплексного
иона [Fe(C204)3]3-
большую популярность получил метод, основанный на синтезе
изомеров, т. е. комплексных соединений одинакового состава,
обладающих различным строением. Например, для комплексного иона
H3N
CI
Cr
CI
a
NH3 H3N
NH3 H3N
CI
Cr
NH,
CI
NH,
Рис. 13.6. Изомеры комплексного
иона [Сг(Г\ГНз)4С12]+:
а — трансформа; б — цисформа
Рис. 13.7. Структура тет-
раэдрического комплекса
[AlBrJ-
[Cr(NH3)4Cl2]"L с октаэдрическим расположением лигандов
возможно образование двух изомеров (рис. 13.6). В первом из них
(цисизомер) атомы О расположены по одну сторону от атома Сг,
а во втором (трансизомер) атомы хлора находятся по обе стороны
от центрального атома.
432
Число изомеров может свидетельствовать о геометрической
форме комплекса. Симметричное расположение шести лигандов
на равном расстоянии от центрального атома можно представить
не только в вершинах октаэдра, но также плоского одностороннего
шестиугольника и тригональной призмы. В табл. 13.3 представ-
Табл]ица 13.3
Сопоставление числа известных изомеров различного состава с расчетным числом
изомеров для случаев различной геометрической конфигурации комплекса
Комплекс
Число
известных
изомеров
М
Плоская
призма
Тригональная
призма
\
—^
N
/
/
-i—
/
/
МА5В
МА4В2
МА3В3
один
два
два
один
три (1,2; 1,3; 1,4)
три (1,2,3; 1, 2,4;
1,3,5)
один
три (1,2; 1,4; 1,6)
три (1,2,3; 1, 2,4;
1,2,6)
один
два (1,2; 1,6)
два (1,2,3; 1,2, 6)
лено число и характер теоретически возможных для каждой из
этих структур изомеров. Сопоставление результатов расчета с
экспериментом убедительно свидетельствует в пользу октаэдри-
ческого строения комплексов с координационным числом 6.
Действительно, для комплексов состава МА4В2 и МА3В3 было
синтезировано только по два изомера, что совпадает с расчетным числом
изомеров лишь для комплексов, имеющих октаэдрическое
строение. Однозначная информация о структуре комплексов может быть
получена на основании данных рентгеноструктурного анализа.
Здесь уместно отметить, что не всегда координационное число
центрального атома в комплексных соединениях равно 6. Весьма
распространено образование комплексов с координационным
числом 4, причем в этом случае комплекс может иметь как
октаэдрическое строение, так и форму простого квадрата. В
комплексном ионе [А1Вг4]~, образующемся как промежуточный продукт
взаимодействия органических соединений с А1Вг3, используемым
в качестве катализатора, ионы Вг~ расположены в вершинах
правильного тетраэдра, центр которого занят ионом А13+ (рис. 13.7).
Вместе с тем комплексный ион [Cu(NH3)4]2i~ имеет конфигурацию
433
плоского квадрата, образование которого станет понятным в свете
рассматриваемых позже представлений теорий кристаллического
поля.
В общем случае известны комплексы с координационным
числом от двух до восьми, причем один и тот же комплексообразова-
тель в зависимости от степени окисления и природы лигандов,
составляющих координационную сферу, может проявлять различные
значения координационного числа. В качестве общей следует
рассматривать тенденцию к снижению значения координационного
числа при переходе к комплексообразователям и лигандам,
обладающим более высокой поляризующей способностью и
поляризуемостью электронных оболочек.
§ Э. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ
Исключительное многообразие комплексных соединений
затрудняет любую попытку их классификации. Как уже отмечалось
выше, в роли комплексообразователя может выступать любой
химический элемент, однако наиболее распространены металлические
комплексы. Последние иногда делят на две группы:
1) вернеровские комплексы,
2) карбонилы металлов и металлоорганические соединения
(например, K[Pt(C2H5)Cl3]).
Различие между ними сводится к тому, что у соединений
второй группы в отличие от первой молекулы содержат, по крайней
мере, одну связь Me—С и, как следствие, химическая связь
является в значительной мере ковалентной. Соединения первой
группы имеют солеобразный характер с более выраженной ионной
структурой.
По составу координационной сферы можно выделить несколько
наиболее распространенных классов вернеровских комплексов:
1. Аквокомплексы типа [Сг(Н20)б]С13 (лиганды —
нейтральные молекулы воды).
2. Аммиакаты типа [Ag(NH3)2]Cl (лиганды — молекулы
аммиака).
3. Ацидокомплексы типа Кз[А1Рб] или K[Al(S04h] (лиганды—
кислотные остатки кислородных или бескислородных кислот).
4 Гидрооксисоли типа Na[Zn(OH)3] (лиганды — гидроксиль-
.ные группы).
5. Полигалогениды типа Те [13] или Sb[ICl4].
6. Изо- и гетерополикислоты и их производные.
Формально изополикислоты можно рассматривать как продукт
"взаимодействия кислоты с ее же ангидридом, например:
Н2СЮ4- Сг03 = Н2Сг207 = Н2[Сг04 (СЮ3)] — двухромовая кислота,
Н2СЮ4 • 2СЮ3 = Н2Сг3О10 = Н2 [СЮ4 (Сг03)2] — трихромовая кислота,
434
I I2Cr04 • 3Cr03 = H2Cr04013 = H3 [Cr04 (Cr03)2] — тетрахромовая
кислота.
Образование изополикислот характерно для хрома, вольфрама,
молибдена, ванадия, ниобия и тантала.
Гетерополикислоты представляют собой комплексные
производные кислородных кислот, в которых О2" полностью или частично
заменен кислотными остатками других кислот. Например,
гидрату Н7[РОб] отвечают следующие типы гетерополикислот:
Н7[Р(Мо207)6], Н7{РО(Мо207)5], H7[P(V206)6], Н7[Р(Мо207)3],
H7[P(Mo207)3(V206)3] и т. д. Образование гетерополикислот
характерно для фосфорной, кремниевой, борной и мышьяковой кислот
с ангидридами вольфрамовой, молибденовой и ванадиевой кислот.
Разумеется, что указанная классификация комплексных
соединений чисто условная, ибо во многих случаях координационная
сфера комплекса содержит два или больше типов разнородных
лигандов. Более того, как будет показано в § 7, комплексы со
смешанными лигандами иногда термодинамически более устойчивы за
счет благоприятного энтропийного фактора.
Особую группу составляют так называемые внутрикомплексные
соединения, в которых центральный атом связан с молекулой
лиганда одновременно главной и побочной валентностью,
примером может служить гликолят меди
ОС О О СО
I X I '
Н2С HaN NH2 СН,
в молекуле которого кислородные остатки NH2CH2COO~
захватывают «клешнями» центральный ион Си2+ (клешнеобразная
структура).
Принятая ЮПАК (Международный союз по теоретической и
прикладной химии) номенклатура комплексных соединений
предусматривает следующие правила:
1. Сначала называют катион, а затем анион.
2. Называя лиганды, начинают с отрицательно заряженных
частиц, затем переходят к нейтральным и, наконец, к положительно
заряженным. При этом внутри каждой из указанных групп
лиганды перечисляются в порядке увеличения их сложности.
Например, перечисляя лиганды в комплексе [Co(NH3)4(NCS)Cl]3+,
сначала называют хлор, затем кислотный остаток кислоты HCNS и,
наконец, нейтральные частицы NH3.
3. Названия анионных комплексов оканчиваются на «ат», тогда
как в катионных или нейтральных комплексах металл не имеет
специфического окончания. Например, комплекс Кз[А1(С204)з]
называют калия триоксалатоалюминат (III).
4. Нейтральные лиганды называют так же, как
соответствующие молекулы, к лигандам-анионам прибавляют «о», а к лиган-
435
дам-катионам — «иум». Исключение составляют молекулы воды
и аммиака, которые называют акво- и аммин- соответственно.
5. Степень окисления центрального атома обозначают римской
цифрой в круглых скобках в конце названия комплекса.
Названия наиболее распространенных лигандов приведены в
табл. 13.2, Руководствуясь этими правилами, составим названия
некоторых комплексов:
[СО (NH3)e] Cl3 — гексааминкобальт (III) хлорид
K2[PtCle] — калия гексаплатинат (IV),
[Pt (en) Cl4] — тетрахлороэтилендиаминплатина,
[Fe (H20)e] S04 — гексааквожелезо (II) сульфат.
§ 4. СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯ
О ПРИРОДЕ КООРДИНАЦИОННОЙ СВЯЗИ.
МЕТОД ВАЛЕНТНЫХ СВЯЗЕЙ
Координационная теория Вернера, сыгравшая огромную роль
в развитии химии комплексных соединений, возникла значительно
раньше современных представлений об электронном строении
атома и естественно, что введенная Вернером концепция побочной
валентности, или, как ее теперь называют, координационной
связи, нуждалась в более глубокой интерпретации. Для описания
природы связи в координационных соединениях в настоящее время
применяются три метода: метод валентных связей (МВС), теория
кристаллического поля (ТКП) и теория молекулярных орбиталей
(ТМО).
В основу метода валентных связей положено предположение,
что связь между комплексообразователем и лигандами ковалентна.
Отличительной же чертой ковалентной связи является наличие
одной или нескольких электронных пар, в одинаковой мере
принадлежащих соединяющимся атомам. Например, в молекуле хло-
ра :С1Х Ох связь осуществляется за счет электронной пары,
XX
образующейся из первоначально неспаренных электронов
изолированных атомов. Итак, обычная ковалентная связь возникает в
результате спаривания электронов, первоначально
принадлежавших различным атомам.
В соответствии с МВС связь в комплексах также
осуществляется электронной парой, но оба составляющих ее электрона
поставляются одним и тем же атомом. В качестве примера
рассмотрим образование аммиачных комплексов серебра
н
Ag+ + :N:H ->
Н
" Н -
Ag:N:H
. Н -
" Н Н
Н :N: AgH :N: H
. Н Н .
+
436
Г NH8 "12+
и меди Cu2+ + 4 :NH3 -> H3N:Cu: NH3 .
L NH, J
В обоих случаях свободная электронная пара молекулы
аммиака используется для связи с ионом-комплексообразователем.
Такую связь обычно называют донорно-акцепториой, или
координационной ковалентной связью. Роль донора (поставщика
электронной пары) играет лиганд, а акцептором, принимающим
электроны, является ион-комплексообразователь.
Очевидно, что общее число электронов вокруг атома или иона-
комплексообразователя складывается из электронов, ранее
принадлежавших этому атому, и электронов, передаваемых лигандом.
Н. Сиджвик назвал это число эффективным атомным номером
(ЭАН) и придавал ему важное значение при определении состава
комплексов. Он считал, что комплексообразователь координирует
вокруг себя такое число лигандов, чтобы его эффективный
атомный номер равнялся числу электронов в атоме инертного газа, т. е.
соответствовал наиболее стабильной электронной конфигурации.
Это правило позволяет предсказать состав простейших кар-
'бонилов. Например, атом железа имеет 8 валентных электронов
(3d6 4s2) и способен присоединять еще 10 электронов от пяти
карбонильных групп (по одной электронной паре от каждой молекулы
СО). Тогда общее число электронов становится равным 18, что
соответствует внешней электронной оболочке атома криптона.
Аналогичным образом можно предсказать состав других карбонилов
Ni(CO)4, Mn(CO)5, Сг(СО)6 и т. д.
Чтобы понять некоторые особенности донорно-акцепторной
связи, уместно вспомнить, что каждый электрон обладает собственной
атомной орбиталью, т. е. областью пространства, в которой,
вероятно, нахождение этого электрона достаточно велико. Связь между
атомами является результатом перекрывания их атомных орбита-
лей с образованием молекулярных, принадлежащих обоим
соединяющимся атомам. Донорно-акцепторная связь возникает как
результат перекрывания валентных вакантных орбиталей комплек-
сообразователя с заполненными орбиталями донора.
Чем больше степень перекрывания орбиталей центрального
атома и лигандов, тем прочнее связь между ними. Как показал
Полинг, все орбитали комплексообразователя, участвующие в
связи с лигандами, независимо от исходного состояния одинаковы по
форме и энергии, т. е. вырождены. Строение и прочность
комплекса зависят от характера орбиталей, участвующих в
гибридизации. Если в образовании связи участвуют две d-, одна 5- и три
/7-орбитали (^/^-гибридизация), то комплекс имеет октаэдриче-
ское строение; гибридные 5р3-орбитали приводят к образованию
структуры плоского квадрата и т. д.
Свойства комплексного иона существеннейшим образом
зависят от интенсивности перекрывания орбиталей комплексообразо-
437
вателя и лиганда. Покажем это на примере двух комплексов
[FeFe]4- и [Ре(СЫ)б]з4_, имеющих одинаковый центральный атом
Fe2+. Образование первого из них происходит по схеме
2+
Fe
3d
\4з
МИ * * *
4Р
4d j
+ 6F
*♦ Mtt**
ИМ
$р d -гибридизация
Ввиду того что F~ — сравнительно слабый лиганд, то
поставляемые им электронные пары занимают совершенно вакантные орби-
тали центрального атома, тогда как З^-орбитали, содержащие по
одному электрону, остаются без изменений. Образуемый в этом
случае комплекс, называемый внешнеорбитальным, обладает
магнитным моментом, величина которого определяется числом неспа-
ренных электронов.
Совсем иная картина наблюдается при образовании цианидного
комплекса: CN~-hoh (сильный лиганд) использует для
гибридизации частично застроенные Зй?-орбитали центрального атома, как
показано ниже
J i
3d 1 4s 4p I
еЛнНМИн] И ihihihi
JnNCN CU CN CN CNi
ч
CN CN CN CN CN CN |
• 2 3 '
L-d sp - гибридизация -J
В этом случае гибридизации d2sp3-mna предшествует
спаривание двух Зй-электронов с образованием свободной Зб(-орбитали.
Очевидно, что возникающий комплекс, называемый внутриорби-
тальным, немагнитен, -так как не содержит неспаренных
электронных спинов.
МВС является весьма наглядным методом, но, к сожалению,
не позволяет производить количественные расчеты, направленные
на объяснение оптических свойств комплексов и их относительной
прочности. Более эффективной в этом смысле оказалась теория
кристаллического поля, объясняющая многие специфические
особенности комплексных соединений.
§ 5. СВОЙСТВА КОМПЛЕКСОВ
В СВЕТЕ ТЕОРИИ КРИСТАЛЛИЧЕСКОГО ПОЛЯ
И МЕТОДА МОЛЕКУЛЯРНЫХ ОРБИТАЛЕИ
В отличие от МВС теория кристаллического поля
предполагает, что связь между комплексообразователем и лигандами
чисто ионная, поэтому энергию координационной связи UCB можно
438
Рассчитать, используя кулоновское соотношение
t/CB=-tt, (13.2)
где q\ и <72—истинные или наведенные в результате поляризации
наряды взаимодействующих частиц; г — расстояние между ними.
а ооооо
I I
I
I
А /
Щ I
Ц I
Ц I
'ооооо/
СвоЬодныи ион (Ti4*)
Рис. 13.8. Схема энергетических
уровней 3^-орбиталей свободного
иона Ti4+ и того же иона,
имеющего «равномерное» окружение
отрицательно заряженных ионов
Значения энергии связи, полученные по уравнению (13.2),
хорошо согласуются с экспериментальными данными для
комплексов непереходных металлов, но оказываются заниженными для
комплексов переходных металлов. Это несоответствие между
простейшей электростатической моделью комплексного иона и его
реальным поведением было устранено благодаря теории
кристаллического поля (ТКП), получившей в настоящее время широкое
распространение в неорганической химии.
Основная идея ТКП состоит в признании энергетической
неравноценности орбиталей комплексообразователя, возникающей
под влиянием лигандов. Для иллюстрации рассмотрим
образование комплекса [TiF6]"2 из ионов Ti4+ и F", первоначально
бесконечно удаленных друг от друга. Для свободного иона Ti4+,
изолированного в пространстве, электронная конфигурация выражается
формулой ls22s22p63s23p63d° и все d-орбитали свободны. Они
вырожденные, т. е. энергетически равноценные. При сближении
ионов Ti4+ и F" (за счет отталкивающего действия последних)
энергия rf-орбиталей увеличивается. Если бы при этом ионы F~
оказывали одинаковый эффект на все d-орбитали, то они оставались бы
вырожденными, хотя и соответствовали более высокой энергии,
чем та, которая свойственна свободному иону Ti4+ (рис. 13.8).
Однако такое состояние чисто гипотетическое.
Комплекс [TiF6]2" имеет октаэдрическую структуру, которую
можно изобразить (рис. 13,9), полагая, что начало ее занято ио-
Tv
,^н
Г
Рис. 13.9. Строение октаэдриче-
ского комплекса [TiF6]2~
439
1
/
/
0МЛ
/
/
25
w ООООО/
Рис. 13.10. Схема расщепления
энергетических уровней 3£/-орбиталей
свободного металлического иона и
находящегося под влиянием
кристаллического поля лигандов
нами Ti4+, а лиганды расположены вдоль каждой из осей. Так как
орбитали d2* и с1Х2^.У2 вытянуты вдоль осей и таким образом
направлены непосредственно к лигандам, то их энергия повышается
из-за взаимного отталкивания d-орбиталей и лигандов. Вместе с
тем dxyy dxzy с/у2-орбитали направлены в пространство между ли-
гандами и поэтому относительно стабилизированы.
Таким образом, октаэдриче-
ское поле лигандов приводит
к расщеплению d-уровней,
первоначально вырожденных
пятикратно, на две группы, одна
из которых имеет двукратное,
а другая — трехкратное
вырождение (рис. 13.10). Две
орбитали с более высокой
энергией (dz% и dx*—y2)
называются е^-орбиталями, а три
орбитали с более низким
уровнем энергии (dxy, dyz и dxy)
называются ^g-орбиталями.
Такое разделение пяти
вырожденных орбиталей свободного
иона металла на две группы,
характеризующиеся разной энергией, является главной
особенностью ТКП. Это явление назвали расщеплением кристаллическим
полем. Из сказанного следует, что d-орбитали имеют
неодинаковую ориентацию в пространстве и соседние атомы, ионы или
молекулы могут изменять энергию направленных к ним орбиталей.
Ранее было отмечено, что энергия, соответствующая ^-орбита-
лям металлического иона, возрастает, когда лиганды
приближаются к нему. Можно было бы предположить, что комплекс менее
устойчив, чем свободный металлический ион и лиганды. Однако
сам факт образования комплексов свидетельствует об обратном —
увеличение энергии d-орбиталей металлического иона с лихвой
компенсируется энергией электростатического кулоновского
притяжения составных частей комплексного иона.
Итак, в октаэдрическом поле лигандов t2g- и е^-орбиталям
соответствует различная энергия. Разница энергии i2g- и ^-орбита-
лей обозначается символом А. Значение А обычно определяют
спектроскопическим путем и измеряют в см"1. Наблюдаемые
значения расщепления зависят от природы лигандов, от природы
металла и степени его окисления.
Влияние лигандов обычно представляют в виде спектрохимиче-
ского ряда, в котором лиганды расположены в порядке
увеличения силы кристаллического поля, т. е. возрастания А:
у- < Br-< SCN' ~ С1- < ЫОГ < F- < ОН- ~ НСОО- <
< C20;i~ < H20< ЭДТА*-< NH3 <e/z< N02«CH-~NO-CO.
440
Напомним, что энергетическое расщепление d-уровней в
кристаллическом поле является результатом повышения устойчивости
/2£-орбиталей (т. е. уменьшение их энергии) с одновременным
понижением устойчивости е^-орбиталей (т. е. увеличение их энергии)
под влиянием лигандов. С точки зрения стабильности комплекса
большой интерес представляют разности E(t2g)—Ed и E(eg)—£<*,
где Ed — энергия вырожденного уровня. Их можно рассчитать как
функцию величины А, учитывая, что по определению
E{eg)-E(t2g) = b, (13.3)
2E(eg) + 3E(t2g) = bEd. (13.4)
Последнее уравнение вытекает из так называемого принципа
сохранения центра тяжести, согласно которому сумма
произведения энергии подуровней после расщепления на кратность их
вырождения равна произведению энергии подуровня до расщепления
на кратность его вырождения.
Из уравнений (13.3) и (13.4) следует, что
E(eg) — Ed = 0№, (13.5)
E(tu)-Ed = -094A. (13.6)
Если в октаэдрическом комплексе металлический ион имеет
только один электрон (например, в комплексе [Ti(H20)6]3+
электронная конфигурация Ti3+ соответствует ls22s22p63s23<il), то этот
d-электрон должен занять орбиталь с наинизшей энергией. Такой
орбиталью является t2g, и попадание на нее электрона
сопровождается энергетическим выигрышем ОДДо1. Для комплекса
[Т1(Н20)б]3+ величина 0,4До называется энергией стабилизации
кристаллическим полем (ЭСКП). Учитывая соотношение (13.5) и
(13.6), энергию стабилизации поля лигандов для любого октаэд-
рического комплекса можно рассчитать по уравнению
AHL = —{0,4n(t2g) — 0,6/1 (*,)}• А,
где n(t2g) и n(eg) —число электронов, занимающих Ug- и е#-орби-
тали соответственно.
Порядок заполнения орбиталей электронами определяется
двумя конкурирующими фактами: во-первых, в кристаллическом
поле d-электроны стремятся занять орбитали с низким значением
энергии и таким образом избежать насколько возможно
отталкивающего действия лигандов. Во-вторых, в соответствии с правилом
Хунда электроны стремятся остаться неспаренными. Спаривание
электронов сопряжено с затратами энергии и будет происходить
лишь в том случае, если энергия, выигрываемая за счет стабили-
Подстрочный индекс указывает, что речь идет об октаэдрическом комплексе.
441
Таблица 13.4
Число d-элек-1
тронов 6
ионах металлал
i \
г
г
к
5
6
7
8
9
10
Высок оспи-
новый
комплекс 1
1гд
®оо
ФфО
®ф®
ффф
фф®
®ф®
®®ф
®®®
®@®
®®®
е'\
оо
оо
оо
ФО
©ф
фф
фф
фф
®ф
ф®
эксп\
о,ч
0,8
\ 0,6
0,0
0,4
0,8
0,6
0,0
Низко спи но-1
Выи
комплекс
{г*
|®Ф®
®®ф
®®®
®®®
е9
[ © О О О
| о о о о
эщ
щ
2А
U
зации полем лигандов, достаточно велика, чтобы превзойти
энергию спаривания электрона. В табл. 13.4 приведены значения
ЭСКП в октаэдрических комплексах как низкоспиновых (слабое
поле лигандов), так и высокоспиновых (сильное поле лигандов).
Низкоспиновыми называют комплексы с максимально возможным
числом неспаренных электронов, определяемым стремлением
системы следовать правилу Хунда (в слабом поле лигандов).
Высокоспиновыми называют комплексы, в которых вопреки правилу
Хунда происходит спаривание электронов благодаря
значительному энергетическому выигрышу, достигаемому в сильном поле
лигандов.
ТКП позволяет объяснить многие свойства соединений
переходных металлов. Известно, например, что экспериментально
найденная энергия кристаллической решетки соединений переходных
металлов (£/Эксп) выше рассчитанной по циклу Борна — Габера
(^расч). Это повышение легко связать с закономерностью
изменения энергии стабилизации поля лигандов у различных переходных
металлов. Например, у фторидов Fe2+ и Ni2+ £/ЭКсп>£/расч> т. е.
A#L>0; у фторидов Мп2+, напротив, £/ЭКсп мало отличается от
*/расч, так как Д#г„ = 0 (ион Мп2+ во фториде имеет конфигурацию
Ы*(е8)2).
442
Другие термохимические величины, например, теплота гидра-
кщпи двух трехзарядных ионов З^-элементов, следуют анало-
1 ичной закономерности, т. е. у двухзарядных ионов они
максимальны для Ni2+ (AHL) и минимальны для Мп2+ (АЯ2==0), а у
трехзарядных ионов максимум приходится на Со3+, минимум —
на Fe3+.
Стереохимия соединений переходных элементов также хорошо
объясняется теорией кристаллического поля. Исходя из ТКП,
можно ожидать, что правильное октаэдрическое строение имеют
такие комплексы, у которых d-орбитали иона-комплексообразова-
теля симметрично заполнены по отношению к октаэдрическому
полю лигандов, т. е. все 6 лигандов испытывают одинаковое
отталкивание. Симметричными конфигурациями являются
(t2g)°(eg)«, например Ti*+ в ТЮ2 и [TiF6]2~;
(tu)*(eg)*, например Сг3+ в [Сг (С204)з]3~;
(t2g)3 (е^)2(спин-свобэдная конфигурация), например Мп2+ в [MnFe]4+
и Fe3+ в [FeF6]3~;
(hg)6(^g) (спин-спаренная конфигурация), например в [Fe(CN)6]4_ и
Со3+ в [Co(NH3)]3+.
У металлических ионов с несимметричным заполнением d-орби-
талей по отношению к полю лигандов можно ожидать различной
степени отталкивания лигандов и, как следствие, искажения
правильной октаэдрической структуры. Этот эффект должен
проявляться в следующих конфигурациях:
(t2g)s(eg)1 (спин-свободная), например Мп3+, Сг2+;
(t2g)G (eg)x (спин-спаренная), например Со2+;
(t2g) (eg)3, например Cu2+.
Действительно, эксперимент подтвердил, что соединения CoF2,
MnFe3, CuF2 имеют искаженную структуру типа рутила. Искажение
симметричных структур, являющееся следствием частичного
заполнения электронных энергетических уровней, называется
внутренней асимметрией или эффектом Яна — Теллера.
Ионы Ni2+, Pd2+, Pt2+ имеют конфигурацию типа (^)6 (%)2>
которая в принципе может быть симметричной по отношению к
октаэдрическому полю лигандов, если один из d-электронов
расположен на dz>, а другой на dx*-y* -орбитали. Однако если поле
лигандов достаточно велико, то более стабильной будет
конфигурация, когда оба ^-электрона переходят на dz* -орбиталь,
которая находится в области более низкой электронной плотности, чем
dx*-.y* -орбиталь. Образуется конфигурация, которую называют
44
спин-спаренной ^-конфигурацией. У нее 2 лиганда, расположен*
ные вдоль оси z9 претерпевают сильное отталкивание от
электронов, находящихся на ^-орбитали, тогда как 4 лиганда,
расположенные в плоскости ху, не испытывают отталкивания. В
результате образуется сильно искаженная тетраэдрическая структура, а
во многих случаях происходит полная потеря пары лиган-
дов с образованием конфигурации плоского квадрата.
Примеры: CuO, [Cu(NH3)4]2+, [Ni(CN)4]2" большинство комплексов
Pt2+ и Pd2+
1 оо .**" а*1-у2
куЦ^-»^ о
Н
QQQi
л
.'Л*Ч'
\ UWO i
■"Ж
Тетрагональ- ахгЛуг
интаздр пая Ьипира- Плоении
мида нЬадрат
Рис. 13.11. Схема
расщепления
кристаллическим полем лигандов
d-орбиталей в
комплексах с различной
симметрией
Рис. 13.12. Схема
расположения комплексо-
образователя и
лигандов в тетраэдрическом
комплексе
Схема расщепления кристаллическим полем d-орбиталей
центрального атома в комплексах с различной симметрией
представлена на рис. 13.11. При переходе от правильного октаэдра к
плоским квадратным структурам изменение сводится к удалению из
октаэдра каких-либо двух лигандов, находящихся в
трансположении, например, расположенных по оси г. При некотором
смещении лигандов расстояние металл—лиганд вдоль оси z станет
больше расстояния металл — лиганд в плоскости ху, в результате чего
возникает тетрагональная бипирамида. Это позволит лигандам
плоскости ху приблизиться к центральному атому и оказывать на
d-орбитали, расположенные в этой плоскости, большее
отталкивающее действие, чем в правильной октаэдрической структуре.
В результате увеличивается энергия dxy и с/^-^-орбиталей.
В то же время d-орбитали, ориентированные по оси z или в
плоскостях xz и yz, испытывают меньшее отталкивание от
лигандов, удаленных по оси z на некоторое расстояние. Это приводит
к значительному уменьшению энергии я^-орбитали и
небольшому уменьшению энергии dxz- и <^2-орбитали по сравнению с окта-
444
эдрической конфигурацией. Полное удаление лигандов по оси гу
приводящее к образованию конфигурации плоского квадрата,
сопровождается дальнейшим увеличением энергии (1хг-уг и dxy-
орб'италей и уменьшением энергии dz2-, dxz- и rf^-орбиталей.
Чтобы установить, как d-орбитали центрального атома
расщепляются полем лигандов, образующих тетраэдрическую
структуру, мысленно представим тетраэдр, вписанный в куб, так, чтобы
четыре вершины тетраэдра находились в вершинах куба
(рис. 13.12). Если провести оси х9 у и z так, что они пройдут через
центр куба и центры всех его граней, то можно представить
положение четырех лигандов относительно d-орбиталей центрального
атома. Орбитали, простирающиеся вдоль осей координат
(dx*-.y2 и dz*), больше удалены от лигандов, чем орбитали,
расположенные между осями (dxyy dxz и dyz). Поэтому %-орбиталям
(dx2_y2, d2*) в тетраэдрических комплексах соответствует меньшая
энергия, чем ^-°рбиталями (dxy, dyz и dxz). Было обнаружено,
что расщепление кристаллическим полем лигандов, образующих
тетраэдрическое А* и октаэдрическое А0 окружение, связано соот-
ношением А, = —Д0. Следовательно, эффект кристаллического
поля способствует образованию октаэдрических комплексов в
большей степени, чем в тетраэдр ических.
Одной из распространенных кристаллических структур, в
которой кристаллизуются огромное число минералов и синтетических
соединений, является шпинель. Структура шпинели образуется в
результате кубической почти плотной упаковки ионов кислорода,
октаэдрические и тетраэдрические пустоты которой частично
заняты металлическими ионами. Простейшая формула шпинели
А2+ВЛ+04. Если все 2-валентные ионы находятся в тетраэдриче-
ских узлах, а все 3-валентные ионы — в октаэдрических, то
говорят, что шпинель нормальная.
Если же все валентные деоны находятся в октаэдрических
узлах, а 3-валентные ионы поровну распределены между тетра-
и октаэдрическими узлами, то шпинель обращенная. Возможен и
такой случай, когда разнородные ионы
статистически-беспорядочно распределены между тетра- и октаэдрическими узлами
(смешанная шпинель).
На основе ТКП легко определить, какую шпинельную
структуру имеет то или другое соединение. Возьмем, к примеру, хромит
цинка ZnCr^O^ Судя по данным табл. 13.5, ион Zn2+ не имеет
предпочтения к определенным узлам решетки, тогда как для иона
Сг3+ октаэдрическому окружению соответствует больший
энергетический выигрыш, чем тетраэдрическому. Следовательно, ион.
Сг3+ займет октаэдрические узлы и ZnCr204 имеет структуру
нормальной шпинели.
В алюминате цинка ZnAl204 ни один из ионов, входящих в
состав соединения, не имеет предпочтения к определенному типу
узлов, поэтому структура должна быть смешанной.
445^
Таблица 13.5
Число 3<i-
^лектронов !
0
1
2
3
4
5
6
7
8
9
10
Ионы
Са2 Sc3'
Ti4r V5 +
Ti3
V3
Cr3
Mn*4
Mn3i Cr2f
Mn2"
Fe3-
Fe2 4-
Co3+
Co2+
Ni2+
Cu2*
Zn2" Ga3
Се*'
!
Окта-узел
(hg)° (eg)0
('«)l (eg)ft
C2g)2 (««)•
('*)» (eg)0
C.g)3 (eg)0
('«)» (4Y
(hg)3 (eg)*
(<2g)3 (eg)2
('«)* (^)2
(<2g)4 (eg)2
Сад)» (eg)2
(hgf (eg)2
('«)• (^)3
(<«)• (eg)4
Тетра-узел
(eg)° (<„)•
(*«)J (/«)-
(eg)2 (<«)•'
(eg)2 C.g)1
(eg)2 (^g)1
(eg)2 (t2g)2
(eg)2 (<2g)3
(eg)2 (hg)3
(eg)3 C2g)3
(eg)3 (^g)3
(eg)4 (/2g)3
(eg)*(t,g)3
(eg)4 (M5
(eg)4 (<2g)e
Предпочтение
к окта узлу в Д
0
0,1
0,2
0,8
0,8
0,4
0
0
0,1
0,1
0,2
0,8
0,4
0
В феррите никеля NiFe204 (см. табл. 13.5) ион Fe3+ не имеет
предпочтения к определенным узлам решетки, тогда как для иона
Ni2+ октаэдрическому окружению соответствует больший
энергетический выигрыш, чем тетраэдрическому. Можно ожидать, что
ион Ni2+ займет тетраэдрические узлы и, следовательно, NiFe204
имеет обращенную структуру.
Большим успехом TKJI является возможность объяснить
магнитные свойства комплексов переходных металлов. Известно, что
вещества, содержащие атомы с неспаренными электронами,
парамагнитны (притягиваются магнитом), а вещества, содержащие
только электронные пары, диамагнитны (магнитом не
притягиваются).
В ионе [CoF6]3~ поле лигандов относительно невелико, из двух
возможных конфигураций (низкоспиновая и высокоспиновая) в
соответствии с правилом Хунда образуется первая, т. е. (^)4(ея)2-
Опыт показывает, что [CoFe]3~ парамагнитен, причем магнитный
момент ji близок к пяти, что соответствует присутствию четырех
неспаренных электронов.
Диамагнитность комплекса [СоЫН3)б]3+ объясняется тем, что
в нем поле лигандов велико по сравнению с фторидным
комплексом и вопреки правилу Хунда образуется низкоспиновая
конфигурация (t2g)6(eg)°j не имеющая неспаренных спинов.
Самым большим достижением ТКП является успех в
объяснении цвета соединений переходных металлов. Следствием
сравнительно небольшой разности энергии А между неэквивалентными
(t2g и eg) орбиталями переход электрона с низкого на более
высокий уровень энергии может осуществляться за счет поглощения
видимого света, что и приводит к окраске комплекса. Например,
446
водный раствор Ti3+ имеет фиолетовый цвет, что объясняется в
рамках схемы, изображенной на рис. 13.13.
Энергия перехода электрона Е связана с длиной волны К
поглощаемого света уравнением Планка
Е =
he
где с — скорость света (3-Ю10 см/сек); h — постоянная Планка
(6,62-10"27 эрг-сек). Подставляя значение констант и выражая
энергию в ккал/моль, а X в ангстремах, находим
Л--
2,84.105
57
Поскольку для комплекса [Ti(H20)6]3+ £ = 57 ккал/моль, то
Я = 5000 А, что действительно соответствует максимуму
поглощения в спектре комплекса [Ti(H20)6]3+.
g9
'[TUH2o)6f
Свет
©О
ооо.
Чтимы*
Рис. 13.13. Схема возбуждения
3^-электрона в комплексном
ионе [Ti(H20)e]3+ под
действием света
Ccf Ti2* О2* Fe2* Nr* In2
Sc2* V2* Mn2* Co2+ Cu2'
Рис. 13.14. Зависимость
констант устойчивости
комплексов [ML6]2+ от природы ком-
плексообразователя
(пояснения в тексте)
Металлические ионы, не испытывающие расщепления уровней
в кристаллическом поле лигандов, должны образовывать
бесцветные комплексы, что подтверждается экспериментом. Например,
акво-комплексы К+, Са2+, Sc3+Cu+, Zn2+, In3+ действительно
бесцветны, а высокоспиновые комплексы Сг3+ и V2+, орбитали
которых испытывают наибольшее расщепление кристаллическим
полем лигандов, имеют фиолетовую окраску.
ТКП позволяет объяснить и относительную термическую
стабильность комплексных соединений переходных металлов.
Покажем это на примере высокоспиновых октаэдрических комплексов
типа [ML6]2+, где М — двухвалентные ионы элементов
периодической таблицы с порядковым номером от 20 до 31. Учитывая
одинаковый заряд иона-комплексообразователя и монотонное умень-
447
шение ионного радиуса при переходе от Са2+ к Zn2+ без учета
энергии стабилизации кристаллическим полем лигандов, можно
было ожидать монотонного увеличения константы стойкости
(показано пунктиром на рис. 13.14). Однако найденная
экспериментально кривая зависимости констант стойкости от порядкового
номера указанных элементов показывает два максимума, которые
обусловлены различием энергии стабилизации кристаллическим
лолем лигандов у различных ионов. Электронная конфигурация
интересующих нас ионов в октаэдрическом поле лигандов такова*
Са2+-(^)0(^)о; Sc^-(^)0(^)i; W-(tu)*(eg)*i
Учитывая, что энергия стабилизации кристаллическим полем
выражается уравнением
M*L = {0,4n(t%g)~09bn(eg)}.b,
легко установить, что величина AHL равна нулю для ионов
<-а , Mn , Zn2+ и принимает максимальное значение для ионов
Рис. 13.15. Схема относительного
расположения ^g-орбиталей ком-
плексообразователя и орбиталей
лигандов в октаэдрическом комплексе
осгакзво
dx'-u*
Рис. 13.16. Схема относительного
расположения eg-орбиталей комплек-
сообразователя и орбиталей
лигандов в октаэдрическом комплексе
V2 , Ni2+. Это полностью объясняет относительное
значение-констант устойчивости комплексов [ML6]2+, изображенных на
рис. 13.14 кружками.
Несмотря на успешное применение ТКП к объяснению свойств
координационных соединений, не следует забывать, что эта теория
весьма 'приближенная, поскольку полностью игнорирует ковалент-
ный вклад в образование координационной связи. В этом смысле
несомненным шагом вперед является применение теории молеку-
448
лярных орбиталей (ТМО), позволяющей учесть одновременно и
ионный и ковалентный вклады в образование связи между комп-
лексообразователем и лигандами.
Основная идея ТМО состоит в признании того, что образование
молекул из атомов приводит к возникновению новых, так называе-
Рис. 13.17 Схема образования о- и
я-связей между комплексообразова-
телем и лигандами иона [Fe(CN6)]4_
мых молекулярных орбиталей. Форма и энергия этих орбиталей,
каждая из которых может содержать максимум 2 электрона,
зависит от характера взаимодействующих орбиталей.
Электроны комплексообразователя, которые в кристаллическом
поле лигандов распределены между t2g- и %-орбиталями, в
принципе могут участвовать в образовании 0- и я-связей. Поскольку
в октаэдрическом комплексе ^g-орбитали (dxy, dxz и dyz)
расположены между орбиталями лигандов (рис. 13.15), то они не
перекрываются, а следовательно, ^-электроны не могут участвовать
в образовании а-связи.
В том же октаэдрическом комплексе е#-орбитали (dx*-y* и d2*),
направленные на орбитали лигандов (рис. 13.16), могут
перекрываться с ними, образуя а-связь.
Из геометрических соображений следует, что ^-орбитали в
октаэдрическом комплексе могут перекрываться с орбиталями ли-
ганда, образуя я-связь.
Необычно высокая устойчивость ионов типа [Fe(CN)6]4-
обусловлена одновременным образованием а- и я-связей между комп-
лексообразователем и лигандами, как показано на рис. 13.17.
Очевидно, что у иона-комплексообразователя Fe2+ (электронная
конфигурация отвечает формуле ls22s22p63s23p63d6) в низкоспиновом
октаэдрическом комплексе все ^-орбитали заняты, тогда как
е^-орбитали свободны. Образование а-связи в ионе [Fe(CN)6]4~
может происходить только за счет электронной пары, переносимой
на пустую 4^-орбиталь металлического иона (рис. 11.17, а).
Образование же я-связи в комплексе [Fe(CN)6]4" может
осуществляться за счет электронной пары, переносимой с заполненной
^-(например, dxy) орбитали металлического иона к лигандам.
Аналогичная ситуация имеет место в молекулах карбонилов, у
которых связь Me—С образуется в результате перекрывания
заполненных ^-орбиталей металлического атома со свободными
я-орбиталями молекул СО. В этом случае в отличие от а-связи
Fe-CN е-связь
Fe-CN л-сбязь
15 Общая химия
449
донором является металлический атом, поставляющий свои d-элек-
троны на свободные орбитали лиганда.
§ 6. УСТОЙЧИВОСТЬ КОМПЛЕКСНЫХ ИОНОВ
Как правило, комплексные соединения хорошо растворимы в
воде, и это обстоятельство широко используется для перевода в
раствор трудно растворимых или нерастворимых соединений.
Применение тиосульфата Na2S203 для фиксирования в
фотопроцессе эффективно потому, что галогенид серебра, входящий в
состав эмульсии пленки, растворяется в результате образования
устойчивого и растворимого в воде комплекса
Ag+ + 2S.O*- -*[Ag (S203)2]3-.
Добавление комплексующих агентов к жесткой воде приводит
к образованию устойчивых и растворимых комплексов тех
металлических ионов, например Са2+, которые определяют жесткость
воды, благодаря чему предотвращается образование
нерастворимых солей с обычным мылом.
Однако следует иметь в виду, что образование комплекса, даже
самого устойчивого, — это обратимый процесс; он, как и любая
другая обратимая реакция, характеризуется определенной
константой равновесия. Если образование комплекса выразить
уравнением реакции
М"+ + xL ^ [MLJ"+, (13.7)
то константа равновесия, называемая в этом случае константой
устойчивости комплекса, выражается соотношением
[мя+нц*
в котором в квадратных скобках фигурируют равновесные
концентрации комплексного иона, иона-комплексообразователя и ли-
гандов. Очевидно, что чем больше величина Кусту тем более
устойчив комплексный ион. Учитывая, что для любого обратимого
химического процесса изменение свободной энергии
AG = — RT In Kp = ДЯ — TAS,
легко убедиться, что значение константы устойчивости
определяется изменением энтальпии АН и энтропии AS реакции (13.7).
В соответствии с предыдущим уравнением
in д VCT = ,
уст т RT •
т. е. комплексный ион тем более устойчив, чем больше тепла
выделяется при его образовании (Д#<0) и чем значительней увели-
450
чище энтропии (AS>0). Учитывая, что энтропия системы
является мерой беспорядка, можно утверждать, что образованию
устойчивого комплекса способствует наибольший беспорядок в
структуре продуктов относительно исходных веществ.
Простая электростатическая модель неплохо объясняет
влияние энтальпийного фактора. Можно предполагать, что наиболее
усюйчивы те комплексы, которые состоят из противоположно
заряженных частиц, причем, чем больше заряд и меньше размер
попа, тем устойчивее комплекс.
Увеличение устойчивости комплексов с увеличением заряда
комплексообразующего иона хорошо иллюстрируется рядом гидро-
ксо-комплексов, для которых значения константы устойчивости
равны
Кист — 2, i<Mg(CH)8 = Ю2, /CYOH2+ = 107, #ThOH3+ = 10го-
Эффект уменьшения ионного радиуса комплексообразователя
виден из сопоставления константы устойчивости ионов МОН4 для
щелочноземельных металлов
К
ВеОН+
= 4, К
СаОН-
30. ^меон+ = 120, /СВеОН+ = Ю'.
Таблица 13.6
Влияние заряда и радиуса катиона
на устойчивость гидроксо-комплексов
мож*-1)*
мл-* + он- т^ монс*-1)-*-
Наиболее удобным критерием
для оценки комплексообразую-
щей способности металлических
попов является величина
отношений заряда к ионному радиусу.
Как видно из данных табл. 13.6,
величина константы
устойчивости гидроксо-комплексов
увеличивается по мере увеличения
отношений заряд/ионный радиус,
причем влияние заряда более
существенно, чем влияние ионного
радиуса.
Влияние энтропийного
фактора на относительную
устойчивость комплексных ионов можно
иллюстрировать на примере реакции
[М (Н20)в]«+ Ь У L = [М (Н20)6_, L,]»+ + уН20,
для которой АЯ^О, когда L — нейтральный лиганд.
Самопроизвольное протекание процесса слева направо обусловлено тем, что
исходный комплексный ион имеет совершенно упорядоченную
сольватную оболочку, которая разупорядочивается при частичном
замещении лигандами L. Как следствие, энтропия системы
возрастает и ее состояние становится термодинамически более
выгодным, чем исходная.
л*П *
Л\
Li +
Са2+
Ni2+
уз^
Th4+
А1з+
Ве2+
Ионный
радиус
0,60
0,99
0,69
0,93
1,02
0,50
0,31
Заряд
радиус
1,7
2,0
2,9
3,2
4,0
6,0
6,5
кмон(^-1)ь
2
3101
3.103
МО7
Ы010
МО9
1-107
15*
451
По тем же самым причинам (роль энтропийного фактора) при
образовании комплекса [ML6]n+ из [М(Н20)6]п замена каждой
последующей молекулы Н20 лигандом затруднена по сравнению с
предыдущей. Так, например, для реакции
[Ni (H20)6]2+ + 6NH3^ [Ni (NH3)6]2+ + 6Н20
ступенчатые константы равновесия равны
Кх = 5-102, /С2= 1,3- 102, УС3= 4-101, 7(4 = 1,2-101,
/С5 = 4, ^6 = 0,8.
Очевидно, что чем меньше молекул Н2О остается в
комплексном ионе, тем меньше вероятность замены их молекулами NH3 и
больше вероятность обратного процесса. Примечательно, что
/Сс<1, т. е. комплексный ион [Ni(NH3)5H20]2+ более стабилен,
чем [Ni(NH3)6]2+. Этого следовало ожидать, учитывая, что ион
[Ni(NH3)5H20]2+ имеет более разупорядоченную структуру по
сравнению с ионом [Ni(NH3)6]2+.
Проявлением энтропийного фактора объясняют высокую
прочность металлических комплексов этилендиамина по сравнению с
аналогичными комплексами аммиака. Молекула этилендиамина
(еп), состав которой выражается формулой H2NCH2CH2NH2,
занимает в координационной сфере иона-комплексообразователя два
места и ведет себя как две связанные между собой молекулы
аммиака. Поэтому структуру комплексного иона [Ni(en)3]2+, по Вер-
неру, можно выразить формулой
&г
/
NK
-NH
*^*ь
тогда как аналогичный аммиачный комплекс [Ni(NH3)6]2+ имеет
структуру
HN,
Шя
\ /
H3N— Ni—NH3
/ \
,H3N NH3.
2+
452
Очевидно, что замена одной молекулы Н20 в комплексе
| Ni (Н20)б]2+ молекулой NH3 и еп равновероятна. Однако замена
и горой молекулы Н20 аминной группой уже координированной
молекулы этилендиамина более вероятно, чем замена ее
свободной молекулой аммиака из раствора, так как молекула еп уже
< низана с ионом металла и свободный ее конец находится в
непосредственной близости от молекулы Н20, которую он заменяет.
Поэтому образование [Ni(en) (H20)4]2+ более вероятно, чем
образование комплексного иона fNi(NH3)2(H20)4]2b.
Явление увеличения прочности комплексного иона при
перехоле к лигандам, молекулы которых занимают два или более
координационных мест, называется хелатным эффектом К По
энтропийным соображениям можно ожидать, что хелатный эффект
выражен тем сильнее, чем больше мест во внутренней сфере
комплекса занимает одна молекула лиганда. Самый активный лиганд
в этом смысле — ЭДТА (этилендиаминтетраацетат)
-:ооссн2 сн2соо:~
\nch2ch2n/
-:ооссн2 сн2соо:-
который может занимать вплоть до шести координационных мест.
Способность ЭДТА давать весьма устойчивые и хорошо
растворимые комплексы с Са2+ широко используется для устранения
жесткости воды. Прочность комплексов, образованных ЭДТА с
некоторыми двухвалентными металлами по реакции
ЭДТА4~ + [М (Н20)6]2+-> [МЭДТА]-- + 6Н20,
положена в основу количественного определения этих металлов
комплексометрическим методом (комплексон — товарное
название ЭДТА и его производных).
§ 7. КООРДИНАЦИОННЫЕ СОЕДИНЕНИЯ
ТИПА КЛАСТЕРОВ И КЛАТРАТО»
В последнее время интерес исследователей привлекли
необычайные координационные соединения типа кластеров и клатратов.
Название кластеры, происходящее от английского слова «cluster»,
что означает, «рой, скопление», получили соединения, в которых
существуют ковалентные связи металл — металл. Известно, что
такие связи характерны металлической подрешетке переходных
металлов. Однако способность сохранять хотя бы частично, связи
Me—Me при переходе от металлической решетки к решетке
соединений — довольно необычное явление, которое теперь неоспо-
1 Хелатами называют лиганды, которые способны присоединяться к
центральному атому двумя или более атомами.
453
римо доказано благодаря применению рентгеноструктурного и
магнитного анализов.
Например, высокая температура кипения металлического
молибдена (~4800°С) обусловлена прочностью связи Me—Me,
которая в свою очередь определяется сильным перекрыванием атомных
орбиталей атомов Мо, находящихся на расстоянии 2,725 А друг
от друга. Так как в решетке Мо02 расстояние между ближайшими
атомами еще меньше (2,50 А), трудно ожидать, что в двуокиси
молибдена связи Мо—Мо отсутствуют или непрочны.
Другим доказательством существования в окислах и галогени-
дах (особенно низших) ковалентных связей Me—Me являются
результаты измерения магнитного момента. Известно, что
последний определяется числом неспаренных электронов по формуле
Ив= |Ai(/z-j-2).(iB, (13.8)
где jwr— магнетон Бора (т. е. элементарный магнитный момент).
Возьмем к примеру йодид рения Rel2. Если считать, что .в этом
Рис. 13.18. Структура кластера
[Nb6Ie]3+ (а) и кластера
[Nb6Cli2]2+ (б)
соединении рений 2-валентен, т. е. каждый атом рения использует
на образование связи с I два электрона, то на ионе Re должно
остаться три 5й-электронов, т. е. магнитный момент, рассчитанный
по уравнению (13.8), должен быть равен 3,87 |ыв, тогда как в
действительности магнитный момент намного меньше. Это означает,
что часть неспаренных электронов «расходуются» на образование
связи Me—Me.
Классическим примером соединения, в котором существует
связь Me—Me, является каломель — соединение Hg2Cl2. При
формальном подсчете (произведение числа атомов О на их
валентность делят на число атомов Hg) валентность ртути получают
равной 1. Однако в действительности валентность атомов ртути
в каломели равна 2, но одна из этих валентностей расходуется на
связь металлических атомов друг с другом. Поэтому структура
каломели выражается формулой О—Hg—Hg—CI.
Существуют и еще более странные соединения типа кластеров,
например Nbli)83. Исходя из закона простых кратных отношений,
можно было ожидать, что Nbl^ss является механической смесью
монойодида Nbl и дийодида Nbb, взятых в определенных
соотношениях. Однако в действительности Nbli,83 — индивидуальное
соединение, структура которого изображена на рис. 13.18. В ее
454
основе лежит октаэдр из шести атомов Nb, находящихся па
расстоянии 2,85 А (в металлическом Nb расстояние между
ближайшими атомами равно 2,856 А). Поэтому можно полагать, что в
рассматриваемом кластере атомы Nb прочно связаны между собой.
Восемь атомов I расположены в вершинах куба, описанного вокруг
октаэдра. Поэтому состав кластера выражается формулой
| Nbels]3+. Положительный заряд кластера компенсируется тремя
ионами 1~, т. е. соединение с валовым составом Nbli,s3 можно
записать как [Nb6l8]b.
Соединение состава NbCl2,33 также содержит кластеры
[Nb6Cli2]2+, которые вместе с дополнительными ионами С1~,
обеспечивающими электронейтральность решетки, составляют
молекулу [Nb6Cli2] СЬ. Очевидно, что в качестве комплексообразова-
теля в обоих случаях надо рассматривать октаэдр атомов Nb,
соединенных друг с другом ковалентной связью.
Комплексные ионы типа кластеров довольно устойчивы и
склонны оставаться неизменными при химическом и термическом
воздействии. Например, при растворении в соляной кислоте
хлористого молибдена, имеющего кластерную структуру типа
[Мо6С1з]С14, в растворе обнаруживают комплексный катион
[Мо6С18]4_, а при испарении треххлористого рения, состоящего из
кластеров Re3Cl9, в парах обнаруживают не мономолекулу ReCl3,
а кластеры.
Перейдем теперь к комплексным соединениям типа клатратов,
которые образуются, если молекулы растворителя, кристаллизуясь,
образуют ажурные структуры с пустотами, заполненными
посторонними молекулами.
Например, молекулы Н20 во многих клатратах образуют
сложную кристаллическую решетку, отличающуюся от обычного льда
большой рыхлостью. В ней на 46 молекул Н20 приходится 6
пустот размером 5,9 А и 2 пустоты размером 5,2 А. Если
внедряющиеся молекулы сравнительно невелики (Аг, Кг, СН4, СН3С1, С02,
SO2), то возможно заполнение всех 8 пустот. Очевидно, что
предельное отношение числа внедренных молекул к числу молекул
Н20 равно 8:46=1:5,75. И действительно, состав гидрата аргона
выражается формулой Аг-5,75 Н20.
Более крупные молекулы (С12, Вг2СН3Вг) могут внедряться
только в большие пустоты, а их только 6 на 46 молекул Н20.
Значит, предельное отношение числа внедренных молекул к числу
молекул воды составит 6:46=1:7,67. И действительно, хлор
образует кристаллогидрат приближенного состава С12-8Н20.
Многие органические соединения (например, фенол и
гидрохинон) также могут служить матрицей для образования
клатратов. У гидрохинона С6Н4(ОН)2, как и у воды, кристаллическая
структура в клатрате иная, нежели в свободном состоянии. Если
в обычных условиях устойчив так называемый а-гидрохиион, то в
клатрате гидрохинон образует более рыхлую решетку р-гидрохи-
нона. В ней молекулы гидрохинона, соединенные водородными свя-
455
зями, расположены так, что между ними остаются пустоты. Эти-то
пустоты и заполняются молекулами «посторонних веществ»,
например S02, H2S, HC1, СН3ОН и др. Во всех клатратах на 3
молекулы гидрохинона приходится одна пустота. Поэтому предельный
состав клатратов гидрохинона — ЗС6Н4(ОН)2)-X (X —
внедренная молекула). В клатратах между матрицей и внедренными
молекулами имеет место сильное взаимодействие, связанное, в
частности, с перекрыванием электронных оболочек их атомов.
С образованием клатратов приходится иметь дело на практике.
Например, в газопроводах при низкой температуре (—29° С) и
высоком давлении (26 атм) возможно возникновение гидрата
метана.
Ученые нашей страны внесли огромный вклад в развитие химии
координационных соединений. Начатые по существу Д. И.
Менделеевым и Н. С. Курнаковым систематические исследования
комплексов, получили блестящее развитие в работах Л. А. Чугае-
ва и его учеников И. И. Черняева, А. А. Гринберга, О. Е.
Звягинцева, а также академика В. И. Спицина и др.
Вопросы и упражнения
1. Определите степень окисления комплексообразователя в следующих
соединениях Na2[PtCle], K2[ReHg], K3[Co(NH2)6], Саз[Со(Н20)б]2.
2. Почему NH3 легко образует комплексы, а NH+ — нет?
3 Назовите известные вам комплексные соединения, в которых в качестве
центрального атома выступают Ag+, Fe2+, Al3+, N2+, Cu2+, Zn2+.
4. Изобразите возможные структуры октаэдрического комплекса Ni с
четырьмя молекулами аммиака и двумя роданид-ионами SCN-.
5. Изобразите координационные формулы следующих соединений:
PtCl4-6NH3, PtCl4-4NH3, PtCl4-2NH3. Во всех случаях степень окисления хрома
равна 3 + , а координационное число — шести
Предскажите относительную проводимость растворов этих соединений,
степень понижения температуры замерзания эквимолярных растворов и
возможность быстрого осаждения хлора при взаимодействии с AgN03.
6. Назовите каждое из следующих соединений
K3[Fe(CN)6j, [Cu(H20)(N03)2], [Co (NH3)4(CNS) CI] NO.,
NH4 [Cr (NH3)> (CNS)4], [Pt (NH3)4 S04] Br2,
[Cr(H20)3(NH3)3]Cl, lPt(*/i)]Cl4, Ыа2[Ш(ЭДТА)].
7. Напишите формулы следующих соединений: калия пентахлороамминпла-
теат, дибромотетраамминрубений (III) нитрат, кальция трисульфитокобальтиат,
сульфопентаамминкобальтихлорид, цезия фторотрихлородат (III), калия тера-
нитроднамминкобальтиат.
8. Исходя из спектрохимического рода лигандов, определите, какое из
комплексных соединений трехвалентного железа K3[Fe(CN)6] или K3[FeF6]
является намного более магнитным, чем другое.
9. Константа устойчивости иона [Ag(CN)2]~ равна ЫО21. Рассчитайте
концентрацию ионов серебра в 0,Ш растворе Na[Ag(CN2)], содержащем также
0,01М NaCN в 1 л раствора.
456
10. Какова величина энергии стабилизации кристаллическим полем лиган-
дов в октаэдрических комплексах V2+, Cr3+, Мп4+?
11. Какова структура октаэдрических комплексов Си2 •", если учесть
проявление эффекта Яна—Теллера?
12. Укажите, какие из перечисленных ниже соединений имеют структуру
нормальной и обращенной шпинели: NiFe204, С0СГ2О4, NiGa204, Fe304r
CdMn204, CuAl204, Mn304.
13. Чем объясняется диамагнитность комплексов, образованных ионами
Sc3+, Ti4+, V5+ и Сгб+?
14. Почему акво-комплексы Cu+, Zn2+, Ga3+ бесцветны, а акво-комплекс
Сг3+ окрашен?
15. Объясните, почему карбонилы железа Fe(CO)5 и хрома Сг(СО)6
содержат различное, но вполне определенное для каждого из них число
карбонильных групп?
ГЛАВА 14
ХИМИЯ ЭЛЕМЕНТОВ
И ИХ ВАЖНЕЙШИХ СОЕДИНЕНИЙ
§ 1. S-, р-, rf- и /-ПОСЛЕДОВАТЕЛЬНОСТИ
ЭЛЕМЕНТОВ
Несмотря на большое разнообразие соединений — несколько сотен
тысяч неорганических, а органических свыше двух миллионов,
благодаря развитию техники синтеза и анализа вещества
открываются все новые и новые соединения. Использование основной идеи
периодического закона Менделеева дает возможность объединить
элементы по общности их свойств и изучать их по группам и
периодам периодической системы. Состав и свойства соединения
определяются типом химической связи, которая зависит от
строения внешнего или близких к нему электронных уровней и
подуровней атомов. Периодичность изменения свойств соединений
обусловлена периодичностью строения этих уровней и подуровней.
По мере роста атомных масс элементов увеличивается число
уровней, участвующих в образовании химической связи. Появившийся
новый подуровень (s-, /?-, d-, /-) затем повторяется во всех
последующих уровнях: 5-подуровень существует во всех атомах; р-под-
уровень появляется во втором периоде и повторяется у всех
последующих; ^-подуровень появляется у элементов четвертого и
последующих периодов; /-подуровень имеется у элементов шестого
и седьмого периодов.
Появление в непрерывно возрастающей по атомным массам
последовательности элементов каждого нового s-, p-9 d- и
/-подуровня приводит к. резкому изменению химических и физических
свойств элементов. Все элементы на внешнем уровне имеют s-элек-
троны и большинство элементов — р-электроны. Они оказывают
наибольшее влияние на свойства элементов. Заполнение предвнеш-
него d-иодуровня электронами несколько слабее сказывается на
свойствах. Еще глубже расположен /-подуровень, он еще слабее
влияет на свойства. Общность и различие в структурах s-, p-9 d- и
/-подуровней атомов элементов лежат в основе конструкции
периодической системы.
Последовательность элементов, в которой каждый
последующий элемент отличается от предыдущего на один добавочный
электрон на 5-подуровне, называют s-элементами. Ту же
последовательность элементов, в которой элементы отличаются количеством
электронов на р-подуровне, называют р-элементами. Аналогично
выделяются последовательности d- и /-элементов. Таким образом,
все 8 групп элементов в короткопериодном варианте таблицы
458
Менделеева и все 16 подгрупп в длиннопериодном варианте
объединяются более крупными совокупностями s-9 р-, d- и /-элементов;
/-элементы обычно помещают отдельно внизу периодической
системы.
Ниже изучение элементов будет проходить по частям, объединяющим s-, p->
d- и /-элементы. В то же время появление каждого нового электрона на s- и
/?-подуровнях сильно сказывается на свойствах элементов, поэтому
соответствующие элементы удобно рассматривать по группам периодической системы,
обращая внимание на характер изменения свойств при продвижении вниз по
мере увеличения номера периода, т. е. в направлении увеличения числа
электронных уровней. Интересно изучать ^-элементы как вдоль длинного периода,
когда происходит увеличение числа ^-электронов, так и по подгруппам,
сравнивая свойства элементов побочных подгрупп со свойствами элементов главных
подгрупп. В периодической системе /-элементы стоят обособленно, число s-, p-
и ^-электронов у них остается неизменным (за редкими исключениями). Эти
элементы интересны тем, что иллюстрируют слабую зависимость свойств от
количества электронов на третьем снаружи электронном уровне.
Благодаря периодическому закону уменьшаются трудности в
ознакомлении со свойствами, составом, названиями огромного
числа химических соединений: классификация элементов на S-, р->
d- и /-элементы поможет лучше понять закономерности в
химических и физических свойствах элементов, облегчит ориентировку
в «необъятном разнообразии неорганических соединений.
§ 2. ВОДОРОД
5-Элементы — это элементы» главных подгрупп первой (Li, Na,
К, Rb, Cs, Fr) и второй (Be, Mg, Ca, Sr, Ba, Ra) групп
периодической системы. Элементы первого периода—водород и гелий также
могут быть причислены к s-элементам, но обычно из-за специфики
свойств их рассматривают вместе с элементами других групп:
водород с галогенами, гелий — с инертными газами.
Все щелочные металлы
и водород имеют на внеш- Таблица 14.1
нем энергетическом уровне
по одному 5-электрону.
У водорода — это
единственный электрон,
непосредственно связанный с ядром,
и для его удаления
требуется значительное количество
энергии. У щелочных
металлов удаление s-электрона
тем легче, чем больше
номер периода элемента
(табл. 14.1).
Отрыв единственного электрона от атома водорода требует
затраты почти в 3 раза больше энергии по сравнению с атомами
щелочных металлов. Образование ионов Э+ хотя и свойственно
Элемент
н
Li
Na
К
Rb
Cs
Потенциал
ионизации,
эв
13,60
5,39
5,14
4,34
4,18
3,89
Эн
?ргия ионизации,
ДЯиониз' кал/моль
О-
-е-
=Э^"иониз>
313 700
124 300
118 500
106 100
96400
89 700
459
рассматриваемым элементам, но энергетически очень затруднено
у водорода. Обычно говорят о ионах водорода Н+ в водных
растворах, однако ион водорода — это протон, который может
существовать лишь в вакууме и при любом контакте с другим ионом
или молекулой он проникает в их электронные оболочки,
оттягивая на себя значительную долю электронной плотности. В водном
растворе ионы водорода существуют лишь в гидратированном
виде: Н+-пН20. Простейший гидрат протона — ион гидроксония
Н30+. Энергия гидратации иона водорода очень высокая и это в
значительной мере компенсирует затраты энергии на ионизацию.
Ионы щелочных металлов также в растворе гидратированы:
М^.дН20.
Все рассматриваемые элементы в газовой фазе образуют
двухатомные молекулы Э2. Для водорода его состояние в виде
молекул Н2 — обычное устойчивое состояние при стандартных
условиях. При тех же условиях щелочные металлы существуют в виде
металлических кристаллов. Устойчивость молекул водорода
объясняется очень высокой энергией диссоциации, высокой энергией
связи по сравнению с молекулами М2 щелочных металлов
(табл. 14.2).
Таблица 14.2 Таблица 14.3
Молекула
На
Li2
Na2
к2
Rb2
Cs.,
ДЯдисс.М2>г = 2Мг'
кал/моль
103 300
25 500
17 300
11800
10 800
10 000
Молекула
нГ
Li+
Na+
к2+
Rb+
Cs+
АЯдисС кал/моль
61000
35 700
23 800
17 300
16 800
16 000
Все молекулы Э2 образуются благодаря двум связывающим
электронам а-связи. Однако между молекулами водорода и
молекулами щелочных металлов существует значительное различие.
Энергия диссоциации молекулы Н2 в 4 раза больше энергии
диссоциации молекулы Li2. Расстояния между атомами в молекулах
Н2 и Li2 также различаются очень сильно (0,74 А в Н2 и 2,67 А в
Li2). Однако самое главное различие молекул Н2 и М2 состоит в
том, что при удалении связывающего электрона связь в молекуле
Н2 ослабляется, а в молекулах щелочных металлов, наоборот,
упрочняется (табл. 14.3).
При удалении связывающего электрона из молекулы Н2 связь
становится длиннее, а при удалении того же электрона из
молекулы М2 связь укорачивается.
460
Причина этих различий лежит в том, что хотя в молекуле Li2
перекрываются электроны 25-подуровня, между Is- и 25-орбита-
лимп нет никаких других подуровней (s, р, или d), поэтому
I s-орбиталь как бы выталкивает 2 5-орбиталь, ослабляя ее.
Удаление электрона из молекулы Li2 приводит к уменьшению
ослабляющего действия ls-молекулярной орбитали. Из-за
возросшего притяжения двумя ядрами одного электрона происходит
сгущение электронной плотности между ядрами и в конечном счете
усиление связи, в результате чего энергия связи возрастает, а
межъядерное расстояние уменьшается. У остальных щелочных
металлов переход от молекулы М2 к молекуле М2 также приводит
к увеличению энергии связи. В этом отношении молекула Н2 по
своему поведению отличается от молекул галогенов Г2, у которых
переход в молекулу Г2 также приводит к усилению энергии свя-
ш: в молекуле F2 энергия связи 37 000 кал/моль и межъядерное
расстояние 1,42 А, а в молекуле FJ" энергия связи 76 500 кал/моль
при межъядерном расстоянии 1,33 А. Но здесь отличие
закономерное — из молекулы галогена удаляется разрыхляющий электрон.
Изучение водорода вместе с элементами главной подгруппы
первой группы оправдывается тем, что все эти элементы являются
^-элементами, имеют на внешнем уровне по одному электрону и
образуют ионы Э+. Спектры этих элементов очень близки.
Водород и щелочные металлы связаны реакциями взаимного
вытеснения. Все они — типичнейшие восстановители.
С галогенами и прежде всего фтором водород объединяет
следующее. Недостаток одного электрона до полного заполнения
подуровня — у водорода 5-подуровня, у галогенов р-подуровня —
приводит к образованию соединений водорода и галогенов с
металлами (NaF, NaH и др.). Двухатомные молекулы водорода и
галогенов по энергетическим характеристикам стоят значительно
ближе друг к другу по сравнению с двухатомными молекулами
щелочных металлов и водорода. Водород и галогены образуют ряд
однотипных комплексных соединений, например MA1F4 и МА1Н4,
MBF4 и МВН4 (М — металл).
Ко всем перечисленным примерам можно было бы добавить
много других, доказывающих аналогию водорода и с щелочными
металлами и с галогенами. Но прежде всего нужно помнить, что
водород — это единственный и особый элемент среди всех других
элементов, поэтому он стоит особняком, и его следует
рассматривать отдельно от других элементов.
99,985% всего водорода на Земле — это водород, ядро которого
состоит из одного протона iH. Иногда этот водород называют
протаем. Другой встречающийся в природе изотоп — дейтерий —
содержится на Земле в количестве 0,015%. Его ядро состоит из
одного протона и одного нейтрона (iH, или D). В связи с
присутствием дейтерия атомная масса обычного водорода равна не
единице, а несколько больше — 1,00797.
461
Изотоп водорода с ядром, состоящим из протона и двух
нейтронов, называется тритием (i H, или Т). Его ядро неустойчиво, и
тритий радиоактивен с периодом полураспада 12,26 года.
Известно еще два искусственно приготовленных неустойчивых
изотопа водорода: i H и i H.
Хотя изотопы водорода все имеют один ls-электрон, тем не
менее химические и физические свойства двухатомных молекул и
соединений с другими элементами заметно различаются.
Стандартные энтальпии всех водородов как простых веществ
равны нулю, однако стандартные энтропии их различны
(табл. 14.4). Поэтому равновесия реакций, отличающихся
изотопным составом водорода, различны и константы равновесия их не
одинаковы.
Таблица 14.4 Таблица 14.5
Водород
н2
D2
т2
S298 г' кал/моль-град
31,21
34,63
36,63
Водород
н2
D2
т2
ДНсвязи' кал/моль
Н2|Г==2НГ
103 300
105 100
105 900
Связи между атомами в молекулах Н2, D2 и Т2 также не
равносильны по прочности, причем с увеличением массы атома связь
становится прочнее (табл. 14.5). Это объясняется тем, что чем
больше массы атомов, тем слабее колебательные движения атомов
в молекуле и тем ниже колебательная энергия связей между
атомами. Ослабление колебательной энергии в ряду Н2—D2—Т2
сопровождается усилением связи, на разрыв которой требуется
большее количество энергии.
Даже такое незначительное изменение в энергии связи сильно
сказывается на скоростях реакций, отличающихся изотопным
составом водорода. Это вызвано
тем, что энергия активации
реакций тем выше, чем
сильнее связи в молекуле
реагента. Поэтому реакции с
дейтерием и тритием протекают
заметно медленнее по
сравнению с водородом. В табл. 14.6
приведены примеры
нескольких реакций с участием
водорода и дейтерия. Во всех
случаях обнаруживается повышенное значение энергии активации
реакций с дейтерием..
Тяжелая вода не пригодна для жизненных процессов, так как
более прочные О—D связи изменяют скорости биологических
процессов и смещают равновесия.
Таблица 14.6
Реакция
н + н2 - н2 + н
Н + D2 = HD + D
С1 + Н2 = НС1 + Н
C1+D2-DCI + D
сн3 + н2 = сн4 + н
CH3 + D2-CH3D + D
ЕакГ кал/моль
6 860
7 300
5 260
7 030
9 900
12 300
462
Стандартный электродный потенциал водорода принят равным
пулю. Электродный потенциал дейтерия в растворе с
|Ь'] = 1 г-ион/л и Яо2 = 1 атм при 298°С равен E°DJ2D+ =
— 0,0034 в.
В гальванической цепи, состоящей из двух газовых электродов — стан-
длртного водородного и дейтериевого, находящегося также при стандартных
условиях, протекают реакции
2Н+ + 2е = Н2
Р2 = 2D+ + 2е
£0-0,0000 в;
£=+0,0034 в
2Н+ + D2 = Н2 + 2D+ э. д. с. = + 0,0034 в.
Таким образом, газообразный дейтерий вытесняет обычный водород из
раствора и является более сильным восстановителем, чем Н2.
При электролизе смеси тяжелой D20 и обычной воды Н20 из-за
различия электродных потенциалов в первую очередь происходит
разложение обычной воды и выделение Н2, тяжелая же вода
накапливается в электролизерном сосуде. На этом явлении основан
один из способов получения тяжелой воды и дейтерия. Из-за
сравнительно близких свойств Н и D получение тяжелой воды является
крайне трудоемким и дорогостоящим процессом.
Обычная вода состоит из смеси молекул воды различного
изотопного состава. При постоянном изотопном составе кислорода
можно выделить, синтезировать и изучить воду шести составов:
Н20, D20, T20, HDO, НТО, DTO.
Свойства воды из дейте- ~ . лл _
- Таблица 14.7
рия, трития и обычного во-
дорода заметно
различаются (табл. 14.7).
Наибольшей энтальпией
образования обладает тритиевая
вода, так как связь Т—О
наиболее прочна среди связей
Н—О и D—О. В то же
время наиболее высокой
энтропией характеризуется вода
состава DTO, как состоящая
из двух различных изотопов
водорода самой большой
массы.
При температуре 20,4°К (—253°С) водород переходит в жидкое состояние
и при 14°К затвердевает. В твердом состоянии водород образует молекулярные
кристаллы, в которых решетка образована молекулами Н2. В твердом состоянии
водород — диэлектрик. Однако нахождение водорода в первой группе
периодической системы позволяет предположить, что твердый водород при высоких
давлениях может перейти в металлическое состояние.
Широко распространенное мнение о высокой химической
активности водорода (молекулярного) неоправдано и связано только с
Вода (газ)
н2о
D40
т2о
HDO
НТО
DTO
АП298,обр'
кал/моль
—57800
—59 561
—60 308
—58 630
—58 980
—59 200
^298'
кал/моль-град
45,13
47,40
48,80
47,71
48,42
49,47
463
представлениями о взрывоподобных опытах с кислородом
(гремучий газ) или хлором. Нельзя обсуждать активность элемента, не
обращая внимания на свойства другого участника реакции. Кроме
того, высокие скорости взаимодействия водорода с кислородом или
хлором вызваны особым механизмом прохождения этих реакций—
цепным. На самом же деле благодаря очень прочной связи
атомов в молекуле водород довольно инертен.
Совершенно чистые атомы водорода (без примеси каких бы то
ни было молекул), не соприкасающиеся со стенками сосуда,
теоретически могут сохраняться, не превращаясь в молекулы Н2
неограниченно долгое время. Соударение двух атомов приводит к
образованию не стабильной молекулы Н2, а молекулы Н2,
имеющей большой избыток энергии, делающий связь между атомами
непрочной. Для удаления этого избытка энергии в месте
соударения необходимо присутствие третьего тела — или молекулы Н2,
или любой другой молекулы, или наличие стенки сосуда.
Атомарный водород очень реакционноспособен и участвует во
многих реакциях, в которых молекулярный водород не участвует.
Многие гидриды могут быть получены с использованием
атомарного водорода:
Hg + H = HgH,
Ge + 4Н = GeH4,
As + ЗН = AsH3,
Те Н- 2Н = Н2Те.
Молекулярный водород не реагирует при комнатной температуре
с кислородом. Атомарный водород реагирует с кислородом и
образует пероксид водорода. Молекулярный водород при тех же
условиях очень медленно взаимодействует с хлором в темноте и
практически не взаимодействует с бромом и йодом. С участием
атомарного водорода эти реакции проходят быстро.
Невысокая реакционная способность молекулярного водорода
по сравнению с атомарным объясняется тем, что на разрыв связи
Н—Н требуется затрата большого количества энергии, поэтому
энергии активации реакций с участием Н2 высоки и сами реакции
проходят медленно.
Энергия ионизации водорода по сравнению с щелочными
металлами очень высока. При отрыве электрона от газообразного
атома водорода образуется чрезвычайно малый по размерам ион
Н+ (протон), который существует лишь в вакууме. Не известно ни
одного вещества, содержащего в своем составе водород в
состоянии чистого иона Н+ (хотя щелочные металлы существуют в виде
ионов в солях LiF, KC1, NaN03). Очень часто пользуются
выражением «ион водорода», когда говорят о водных растворах, тем не
менее водород не является элементом, склонным к образованию
464
ионных связей. Ковалентность связи в соединениях с водородом
имеет преимущественный характер.
В соединениях с щелочными и щелочноземельными металлами
атом водорода способен отнимать у них и присоединять к себе
второй электрон, образуя гидрид-ион. Связь в соединениях
МЩ и МПН2 носит преимущественно ионный характер, и эти
гидриды обладают свойствами ионных солеобразных соединений.
Наличие гидрид-иона Н- подтверждается электролизом
расплавленных гидридов: на аноде выделяется водород. Однако даже в
гидриде LiH можно говорить только о частично ионном характере
молекулы Li+H~. По мере продвижения от Li к F вдоль периода
(рис. 14.1) увеличение электроотрицательности атомов изменяет
направление смещения заряда от водорода к атому другого
элемента. В молекуле HF протон проникает в электронные оболочки
иона фтора, поэтому распределения электронной плотности в ионе
F~ и в молекуле HF очень похожи. В том же направлении (от LiH
к HF) увеличивается энергия связи и уменьшается межъядерное
расстояние (табл. 14.8).
Таблица 14.8 Таблица 14.9
Гидрид
LiH
NaH
кн
RbH
CsH
Заряд на водороде
—0,50
—0,50
—0,58
—0,58
—0,59
Молекула
LiH
ВеН
ВН
СН
NH
ОН
HF
АН
""связи'
кал/моль
56 000
53 000
69 000
80 000
83000
101 360
134 320
Межъядерное
о
расстояние, А
1,59
1,30
1,24
1,12
1,03
0,97
0,92
При образовании гидридов с щелочными и щелочноземельными
металлами водород выступает в качестве окислителя. Хотя эти
гидриды образуют ионные кристаллические решетки, заряд на
водороде даже у гидрида цезия не достигает —0,59, понижаясь при
переходе к натрию и литию (табл. 14.9).
Благодаря ионным решеткам эти гидриды — твердые вещества,
летучие только при высоких температурах и разлагающиеся
вблизи температуры плавления.
Гидриды других менее металлических элементов имеют связи
со значительно меньшей долей ионности и обладают свойством
образовывать полимерные твердые кристаллы (В2Нб, 1пН3 и др.)-
Отрицательный заряд на водороде незначителен: В2Н6 (—0,05),
1пН3 (—0,05), SiH4 (—0,05), РН3 (—0,01). В метане водороднесет
очень слабый положительный заряд ( + 0,01).
В соединениях с галогенами водород — восстановитель, однако
тояько в одной молекуле HF его положительный заряд достигает
465
LiH
BeH
NH
Рис. 14.1. Распределение электронной плотности в молекулах гидридов
элементов II периода
Гидрид
Заряд на водороде
+ 0,25, в остальных же значительно Таблица 14.10
ниже, опускаясь до +0,04 у йодводо-
рода (табл. 14.10).
В ряду ковалентных гидридов
ц20 — H2S — H2Se — Н2Те
температуры плавления закономерно
увеличиваются лишь у трех последних. Если
бы линейная зависимость температур
плавления от номера периода
соблюдалась и у воды, то лед плавился бы
примерно при 170°К (—100°С). Однако точка плавления воды
резко нарушает ожидаемую закономерность и почти на 100° выше.
HF
НС1
НВг
HI
+0,25
+0,16
+0,12
+0,04
&Н ,кал/моль
И 00 \-
1700
6,0
5,0
4,0
3,0
л5пл,кал/моль град
- \
\
\
i I I I
->
L_
н2о
h2s
Н Se H Те
2 2
Рис. 14.2. Энтальпия плавления
гидридов р-элемеитов VI
группы
Рис. 14.3. Энтропия плавления
гидридов /?-элементов VI
группы
По-видимому, это объясняется значительно более прочной
кристаллической решеткой твердой воды (льда) по сравнению с
кристаллическими решетками других гидридов.
Изменение энтальпии при плавлении льда также аномально
высоко по сравнению с H2S, H2Se и Н2Те (рис. 14.2). Плавление
льда требует значительно больше тепла, чем это можно ожидать
из сравнения энтальпий плавления других гидридов той же
группы. По-видимому, причина этого состоит в большей прочности свя-
467
зи между молекулами воды в структуре твердой фазы.
Аналогичная аномалия воды наблюдается и при рассмотрении энтропии
плавления (рис. 14.3). Из сопоставления энтропии плавления
гидридов VI группы видно, что при плавлении льда наблюдается
несравненно большее изменение в степени порядка при плавлении
воды. Можно предположить, что структура льда характеризуется
большим порядком в расположении молекул, большей прочностью
связи между молекулами по сравнению со структурами
кристаллических H2S, H^Se и Н2Те.
дН . , нал /моль
И СП
70000
S000
6000
4000 h
Н2°
H2S
26
24
22
2Q
а5
-
-
"
~
чкал/моль град
\
\
\
...,\
J 1 1 L
H2Se H2Te
¥
H2S
H2Se H2Te
Рис. 14.4. Энтальпия
испарения гидридов /7-элементов
VI группы
Рис. 14.5. Энтропия испарения
гидридов /7-элементов VI
группы
В структуре льда кислород молекулы воды находится в
состоянии 5/?3-гибридизации, при котором в двух орбиталях находится
по паре электронов, а в двух остальных — лишь по одному.
Именно эти атомные орбитали способны перекрываться с 5-орбиталями
двух атомов водорода. Четыре равноценных гибридных 5/?3-орби-
тали обычно располагаются под углом 109,5° относительно друг
друга, как в правильном тетраэдре, например в молекуле СН4.
Однако, несмотря на неравноценность гибридных орбиталей
молекулы воды в структуре льда, тетраэдрический угол сохраняется.
Это вызвано особенностями кристаллической структуры льда. Две
орбитали с парами электронов играют особое значение для
объяснения структуры льда и поведения воды. На этих орбиталях
сосредоточены отрицательные заряды, которые взаимодействуют с
положительными зарядами перекрывающихся с атомами водорода
орбиталями соседних молекул воды, создавая водородную связь
между молекулами.
468
Существование в молекуле воды двух 5/73-орбиталей, в которых
каждая имеет по паре электронов, приводит к возникновению за
их счет двух водородных связей. Еще две связи обеспечивают два
водородных атома. Таким образом, только одна молекула воды
в состоянии образовать четыре водородные связи. Поэтому в
структуре льда каждый атом кислорода окружен четырьмя
другими атомами кислорода,
связанными через водород водо- , ^
родными связями. ? б
Исследование процесса пе- II J\ xpa5p АО
рехода молекул воды из жид- // \чбр
кой фазы в газообразную до- //j \\z и н
казывает существование водо- АО j/. \-4 t— is
родных связей и высокий по- ® // ' /f I ]
рядок расположения молекул 2р\ \ \ д / / \ \ /j% несвя*
воды в структуре жидкости.
— - ..„. —- *№&£г/'к
Температуры кипения H2S, \\J 7 I \ "\{/
H2Se и Н2Те линейно возра- w\/ fj6
стают с номером пеоиода, и, ,» А / /
связ
стают с номером периода, и,
если бы подобная закономер- '/ \ t 1 ^
ность сохранялась у воды, она /Л ' * ' вр
кипела бы примерно при _ // * \ г
180°К (^—90°С). Истинная ±X-t \ I
U
СВЯЗ
Рис. 14.6. Энергетическая диаграмма
молекулярных орбиталей Н20
связ
^ Т 1 ,.' s
н
же темп. кип. воды +100°С, т. е. почти на 200° выше
ожидаемой, что объясняется наличием у воды в отличие от H2S, H2Se и
Н2Те водородных связей, препятствующих разрушению льдоподоб-
ной жидкой структуры. Разрыв водородных связей требует
дополнительного количества энергии при испарении, что сказывается на
аномально высокой энтальпии испарения жидкой воды (рис. 14.4).
Изменение энтропии при испарении в точке кипения вычисляют по формуле
А^исп — т
1 кип
У большинства жидкостей изменение энтропии при испарении в точке
кипения равно 20—22 кал/моль-град (правило Трутона). Это означает, что
изменение степени порядка при переходе жидкого состояния в газообразное у
большинства веществ примерно одинаковое. Правило Трутона соблюдается
только тогда, когда состав жидкости одинаков с составом пара. У жидкостей,
у которых молекулы ассоциированы или объединены в группы, величина Д5Исп
выше, чем требуется по правилу Трутона, так как в теплоту испарения
включается теплота, требующаяся на разрушение группировок молекул. Так, для
воды
г0
9717
А5ИСП = 373 =26,04 кал/моль-град.
469
Это значение энтропии испарения сильно отличается от энтропии испарения
H2S, H2Se и Н2Те (рис. 14.5), которые из-за отсутствия водородных связей
подчиняются правилу Трутона.
Согласно методу молекулярных орбиталей б валентных
электронов кислорода и 2 электрона двух атомов водорода
распределяются по молекулярным орбиталям молекулам воды (рис. 14.6).
Две разрыхляющие орбитали не заполнены. Это приводит к
высокой энергии связи в молекуле воды и ее высокой стабильности.
Рис. 14.7. Распределение электронной плотности в молекуле Н20
Расчет распределения электронной плотности связывающих
электронов в молекуле воды (рис. 14.7) в плоскости Н—О—Н
показал, что молекула воды действительно имеет треугольную
форму, а атомы водорода не «погружены» в электронные оболочки
атомов кислорода. Такая картина строения молекулы воды сильно
отличается от ранее общепринятого представления о ней как
шарообразного тела, образуемого анионом О2", в котором находятся
протоны Н+.
Многие реакции с участием воды, протекающие в водных
растворах, имеют порядок на единицу ниже, чем следовало бы
ожидать по уравнению реакции. Например, каталитическое (в
присутствии ионов водорода) превращение сахара в глюкозу и
фруктозу:
С12Н22Оп + Н20 = СбН12Об + С6Н1206
сахароза глюкоза фруктоза
470
в разбавленных растворах протекает как реакция первого
порядка, так как в ходе реакции концентрация воды сохраняется
постоянной.
Вода играет исключительную роль во многих реакциях,
оказывая каталитическое действие. Так, в отсутствие воды фосфор не
окисляется кислородом, то же касается и натрия; фтороводород
не реагирует с Si02 и не разъедает стекло. Реакции между
водородом и кислородом при отсутствии следов воды не идет даже при
1000° С.
Практически все вещества, с которыми мы имеем дело, содержат ничтожные
количества зоды, и физические постоянные вещества относятся именно к такому
ях состоянию. Удаление ничтожных количеств воды резко изменяет свойства
вещества. Например, этиловый спирт после многолетней осушки над Рг05 кипит
при 138°С, хотя во всех справочниках приводится температура его кипения —
+ 78,5°С. Введение следов воды возвращает спирту его обычные свойства.
§ 3. ЩЕЛОЧНЫЕ МЕТАЛЛЫ ( S-ЭЛЕМЕНТЫ
I ГРУППЫ)
Элементы главной подгруппы I группы периодической системы
названы щелочными, так как при взаимодействии с водой сами
металлы и их оксиды образуют растворы сильных оснований —
щелочи.
К главной подгруппе I группы периодической системы
относятся следующие элементы: литий, натрий, калий, рубидий, цезий
и франций. Атомы щелочных металлов имеют на внешнем
электронном уровне один s-электрон. На предвнешнем электронном
уровне только у лития 2 электрона, а у остальных щелочных
металлов — 8 электронов. Различие в строении электронных уровней
является причиной заметного отличия лития от остальных
щелочных металлов: некоторыми свойствами литий напоминает
щелочноземельные металлы и даже алюминий.
При переходе от лития к цезию размеры атомов щелочных
металлов равномерно возрастают (табл. 14.11).
Таблица 14.11 Таблица 14.12
Металл
Li
Na
К
Rb
Cs
w °c
181
98
63
39
28
Металл
Li
Na
К
Rb
Cs
о
R, A
1,65
1,89
2,36
2,48
2,68
"Связь между атомами в кристалле щелочных металлов однотипна,
так как все они кристаллизуются по типу центрированного куба.
Однотипные связи тем прочнее, чем они короче, и увеличение ра-
471
диусов атомов от лития к цезию свидетельствует об ослаблении
прочности металлической связи. Металлы с малыми атомными
радиусами, как правило, плавятся при более высоких
температурах по сравнению с металлами, имеющими большие атомные
радиусы, так как энергии кристаллической решетки последних ниже
и разрушение их осуществляется легче. При переходе от лития к
цезию температура плавления металлов понижается (табл. 14.12),
указывая на ослабление связи между атомами в структурах
металлов.
о
дН кал/моль
Таблица 14.13
Металл
Li
Na
К
Rb
Cs
AS°n*.
кал/г-атом-град
1,58
1,67
1,65
1,68
1,65
Рис. 14.8. Энтальпия
плавления щелочных металлов
Изменения энтальпии при плавлении металлов главных подгрупп
обычно уменьшаются с увеличением атомного номера. Это
наблюдается и в случае щелочных металлов (рис. 14.8).
Изменение энтропии при плавлении отражает изменение
степени порядка, сопровождающего процесс плавления, и
количественно характеризует главным образом изменение в расположении
атомов при переходе из кристаллического состояния в жидкое.
В табл. 14.13 собраны значения энтропии плавления щелочных
металлов.
Координационное число и концентрация свободных электронов
у щелочных металлов полностью сохраняются выше температуры
плавления. Это приводит к малым изменениям энтропии при
плавлении щелочных металлов. Несмотря на понижающиеся от лития
к цезию температуры плавления и энтальпии плавления, энтропии
плавления у всех щелочных металлов почти одинаковы.
Прочность связи между атомами щелочных металлов в их
кристаллических структурах количественно характеризуется
энтальпиями сублимации
МК^МГ Д#°убл.
700
600
500
472
Таблица 14.14
«QQOO h
30 000
20000
Л Нс flll.,<aя^r'drOt,
Яо Сь
Рис. 14.9. Энтальпия
сублимации щелочных металлов
Металл
Li
Na
К
Rb
Cs
Относительная удельная
электропроводность
(цсзий=1)
2,2
4,2
2,8
1,6
1
В ряду щелочных металлов литий занимает особое положение,
выделяясь очень высокой энтальпией сублимации (рис. 14.9). Это
вызвано малыми размерами атомов лития и поэтому относительно
большим перекрыванием s-орбиталей атомов лития в
кристаллической решетке металла. У лития наиболее прочная
металлическая связь. Остальные щелочные металлы обладают монотонно
уменьшающимися энтальпиями сублимации. Атомы цезия в
структуре металла связаны между собой наименее прочно —
металлическая связь из-за больших размеров атомов и малой электронной
плотности в зонах перекрывания s-орбиталей наиболее слаба.
Все металлы — хорошие проводники. Усиление металлических
свойств от лития к цезию, казалось бы, должно сопровождаться
увеличением электропроводности в том же направлении.
В табл. 14.14 приведены относительные удельные
электропроводности щелочных металлов. За единицу принята
электропроводность цезия.
Электропроводность металлов сложным образом зависит от
энергии ионизации, типа, энергии и длины связи между атомами
в кристаллической решетке, координационного числа атома,
концентрации дефектов, температуры и других факторов. Однако
цезий, являясь самым типичным металлом, тем не менее обладает
наименьшей удельной электропроводностью среди щелочных
металлов. Это объясняется не тем, что в металлическом цезии
электроны прочнее связаны с атомами, наоборот, в цезии электроны
наиболее свободны, делокализованы, а тем, что при определении
удельной электропроводности последняя вычисляется на единицу
веса металла. При переходе к цезию атомные веса металлов
возрастают, в единице веса содержится все меньшее количество
атомов металла и соответственно все меньшее количество электронов,
способных принимать участие в переносе электрического тока.
473
Аномально низкая электропроводность лития (можно было бы
ожидать, что она будет выше, чем у натрия) объясняется высокой
прочностью металлической связи между атомами и значительно
меньшей способностью электронов к свободному перемещению в
металле. Энергии ионизации атомов щелочных металлов малы и
уменьшаются при переходе от лития к цезию (рис. 14.10). Способность
дН кал/г-атом
иониз'
130000
120000
110000
100000
90000
150
W0
0,50
Na
РЬ CS
2
Li
3 4
Na К
5 б п
Rb Cs
Рис. 14.10. Первая энергия
ионизации атомов щелочных
металлов
Рис. 14.11. Радиусы ионов
щелочных металлов
атома элемента терять электрон характеризует его металличность.
Чем легче атом элемента теряет электрон, тем ярче у него
выражены металлические свойства. Цезий — элемент с самыми
сильными металлическими свойствами как среди щелочных металлов,
так и среди элементов периодической системы.
Радиусы однозарядных ионов в том же направлении
равномерно возрастают (табл. 14.15, рис. 14.11). Литий и в меньшей сте-
Таблица 14.15 Таблица 14.16
Ион
Li+
Na+
К+
Rb+
Cs+
о
R, A
0,68
0,98
1,33
1,49
1,65
Ион
Li ь
Na^
К+
Rb+
Cs+
Относительное
число гидратации п
(цезий = 1)
5,5
3,3
1,8
1,3
1
474
пени натрий по химическим свойствам значительно отличаются от
остальных щелочных металлов. Натрий в состоянии иона Na+
ближе стоит к калию, чем к литию. Ион лития способен значительно
сильнее притягивать к себе противоположно заряженные ионы
(гидроксид лития — самое слабое основание в ряду оснований
МОЙ) и взаимодействовать с молекулами растворителя.
Энергия гидратации иона Li+ максимальна в ряду ионов щелочных
металлов.
В водном растворе ионы щелочных металлов гидратируются,
т. е. окружаются молекулами воды с образованием сравнительно
прочных комплексов М+-пН20. В табл. 14.16 даны относительные
числа гидратации ионов щелочных металлов, причем число
гидратации иона цезия принято за единицу. Приведенные числа
гидратации — не целые, хотя, разумеется, ион может быть окружен
только целым числом молекулы воды. Несмотря на то что
радиусы ионов возрастают от лития к цезию, тем не менее в том же
направлении числа гидратации уменьшаются, хотя, казалось бы,
больший ион должен окружаться большим количеством молекул
воды. Ион лития, обладая малым радиусом, удерживает вокруг
себя наибольшее число молекул. Точнее было бы сказать, что
связь иона лития с окружающими его молекулами воды наиболее
прочная. Это находит свое выражение и в энтальпиях гидратации,
количестве тепла, выделяющегося в процессе взаимодействия иона
с молекулами воды:
М+ + п Н20 - М+-л НаО А #г°идр.
Энтальпии гидратации ионов щелочных металлов представлены
графически на рис. 14.12. Ион лития обладает аномально
высокой энтальпией гидратации из-за своих малых размеров. Это
приводит к различного рода аномальным свойствам и явному отличию
от остальных щелочных металлов. Уменьшение энтальпии
гидратации от лития к цезию объясняется тем, что с ростом размера
иона плотность заряда на последнем s-подуровне уменьшается и
связь с гидратирующими молекулами воды ослабляется. Гидраты
типа М+-Н20 по свойствам и строению сильно отличаются от
гидрата иона водорода Н+-Н20 (Н30+), иона гидроксония. Это видно
из табл. 14.17.
Таблица 14.17
Гидрат
Н+-Н,0 (Н,0+)
Li+-H20
Na+-HoO
Угол Н—О—Н п
молекуле Н20
120
106
104,5
Энтальпия сиязи
М+—Н20, кал/моль
173 240
36 020
25 160
В то время как ион водорода, внедряясь в электронные
оболочки молекулы воды, связывается с молекулой воды очень прочной
475
связью и изменяет угол между связями Н—О—Н, возможно даже
изменяя тип гибридизации кислорода, ионы щелочных металлов
не проникают в электронные оболочки молекулы воды, связь их
с молекулой воды значительно слабее, а угол Н—О—Н
сохраняется как в нейтральной молекуле воды.
-60000
-ьоооо
-юоооо
-120000
ЛН ,кал/г-ион
гидр' ;
Li Na К Rb Cs
Рис. 14.12. Энтальпия гидратации
ионов щелочных металлов
Подбижногп.с М
(ус/>с6нь*е еси^иць
Рис. 14.13. Подвижность
ионов щелочных металлов
Энтальпия гидратации ионов щелочных металлов в растворе
много выше энергии водородной связи между молекулами воды в
структуре жидкой воды. Это значит, что добавляемые в воду ионы
будут разрушать водородные связи и в растворе будут окружаться
более плотными гидратными оболочками. Поэтому процесс
растворения иона в воде сопровождается выделением значительного
количества тепла (Д#<0) и одновременно увеличением порядка
в системе (AS<0).
Сравнение подвижностей ионов щелочных металлов в водных
растворах (скорости передвижения ионов в электрическом поле)
подтверждают высокую степень гидратации ионов лития
(рис. 14. 13). Ион лития, несмотря на свой самый малый радиус,
обладает наименьшей подвижностью из-за прочной гидратной
оболочки.
476
Стандартные восстановительные потенциалы щелочных
металлов
изменяются при переходе от лития к цезию (табл. 14.18) без
какой-либо закономерности, не следуя правилу усиления
металлических свойств при переходе сверху вниз по группе.
Таблица 14.18 Таблица 14.19
Металл
Li
Na
К
Rb
Cs
F R
расплав'
—2,1
—2,4
—2,6
—2,7
—2,9
Реакция
Li++e-
Na++e
K++e=
Rb++<?=
Cs++e=
=Li
-Na
=K
=Rb
=Cs
E\ в
—3,03
—2,71
—2,93
—2,93
—2,91
У лития наибольший отрицательный потенциал не только среди
щелочных металлов, но и среди всех металлов. У натрия —
наименьший среди щелочных металлов. У калия, рубидия и цезия
восстановительные потенциалы примерно равны.
Несмотря на свое положение в периодической системе, литий
согласно стандартным потенциалам металлов — самый сильный
восстановитель в водном растворе как среди щелочных металлов,
так и среди всех других известных металлов. Такое поведение
лития объясняется исключительно высокой энтальпией гидратации
его иона.
Электродные потенциалы щелочных металлов в расплавленных солях
изменяются так, как это можно ожидать по их положению в группе — их
отрицательные значения возрастают от лития к цезию (табл. 14.19). Цезий обладает
наиболее сильными восстановительными свойствами среди щелочных металлов.
В расплаве один щелочной металл вытесняет другой согласно их электродным
потенциалам. Любой щелочной металл, имеющий большее отрицательное
значение потенциала, вытесняет из расплава другой, имеющий меньшее
отрицательное значение потенциала. Например:
LiCl+Cs=CsCl + Li,
или в ионном виде
Li+-F Cs = Li + Cs+.
В твердом состоянии в отсутствие воздуха и влаги щелочные
металлы имеют металлический блеск. Для предохранения от
окисления Li, Na, К и Rb хранят в керосине. Цезий настолько легко
и энергично окисляется воздухом, что его хранят в откачанных
стеклянных ампулах.
Оксид лития Li20 — единственный оксид такой формулы,
получающийся прямым взаимодействием простых веществ:
2Li-f l/2 02 = Li20.
477
Таблица 14.20
Оксиды остальных щелочных металлов получают косвенным
путем. Энтальпии образования оксидов формулы М20 (табл. 14.20)
хорошо показывают особое положение лития в подгруппе и
заметное отличие натрия от нижестоящих
металлов.
При горении металлического лития
в кислороде кроме оксида лития в
небольшом количестве образуется перок-
сид лития
2Li + 02 =■ Li202.
При нагревании натрия на воздухе и в
кислороде также образуется пероксид
натрия
Оксид
Li20
Na,0
к2о
Rb20
Cs2Q
А^°298, обр,
кал/мол ь
— 142 300
— 99 450
— 86 200
— 82 900
— 82 100
2Na + 02 = Na202.
Пероксид натрия — сильный окислитель. Реакцией
Na202 + С02 - Na2C03 + 1/20.
пользуются для одновременного поглощения углекислого газа и
получения кислорода. Пероксиды щелочных металлов содержат
ион 022". Кроме оксида и пероксида калия известно другое его
кислородсодержащее соединение — надпероксид калия КОг (или
К2О4), получающееся при сжигании металлического калия в
избытке кислорода:
К + 02 = К02,
а также при электролизе на холоду концентрированного раствора
гидроксида калия. Надпероксид калия очень сильный окислитель:
2Кр2 + С = КаС03+1/2 02.
Кислородные соединения рубидия и цезия аналогичны по составам
Y\ свойствам кислородным соединениям калия.
Гидроксиды щелочных металлов — сильные основания. Их
основные свойства в согласии с ростом металлического характера
элементов при увеличении атомного веса возрастают при переходе
от лития к цезию. Электролитическая диссоциация гидроксидов
МОН у щелочных металлов совершается только по основному типу
мои = м+ + он-.
Теоретически возможна диссоциация по кислотному типу
мон = н+н мо-,
но расчеты показывают, что константа этой диссоциации
значительно меньше константы диссоциации воды. Диссоциация по тому
478
или иному типу зависит от прочности связи металл — кислород.
Кислород и водород во всех гидроксидах несут постоянный заряд,
поэтому прочность связи М—О будет обусловливаться радиусом
иона М+ (и зарядом иона металла в гидроксидах М(ОН)п). Чем
меньше радиус (и больше заряд) иона металла, тем сильнее
взаимодействие иона металла с кислородом, тем больше вероятность
диссоциации тю кислотному типу с образованием ионов МО- и Н+.
При переходе от лития к цезию происходит усиление диссоциации
по основному типу и ослабление диссоциации по кислотному типу.
Таблица 14.21
Металл
Li
Na
К
Rb
Cs
Характер взаимодействия с водой
Реакция протекает относительно спокойно без плавления металла
на поверхности воды.
Реакция протекает значительно энергичнее, металл плавится на
поверхности воды.
Реакцию трудно наблюдать, калий воспламеняется и с треском
разлетается.
Металл при соприкосновении с водой мгновенно воспламеняется,
реакция крайне энергична.
Взрывоподобная реакция.
Реакция взаимодействия щелочных металлов с водой протекает
тем энергичнее, чем ниже расположен металл (табл. 14.21). Столь
различный характер прохождения реакций вызван не различными
количествами тепла, выделяющимися при взаимодействии металла
с водой (табл. 14.22) по реакции
Мк + Н2Ож = МОНр.р 4" 1 /2 H2il Д/^еакции,
а другими причинами.
Литий, натрий и калий в воде не тонут и процесс взаимодействия с водой
совершается на поверхности воды. Натрий, калий, рубидий и цезий плавятся
при температуре ниже +100°С, их можно расплавить в воде. Натрий плавится,
при этом резко возрастает поверхность соприкосновения металла с водой, в
результате реакция натрия с водой проходит намного энергичнее, чем с литием,,
хотя тепла выделяется меньше. Калий в воде не тонет и плавится при
температуре более низкой, чем натрий — реакция проходит с воспламенением калия
и выделяющегося водорода. Рубидий и цезий тонут в воде, где расплавляясь,,
дробятся на мелкие капли, которые пузырьками водорода поднимаются на
поверхность воды. Реакция принимает взрывоподобный характер. Следовательно,,
объяснение различного поведения щелочных металлов в реакции с водой
кроется не в химических и термодинамических явлениях, а в физических процессах,
сопровождавших эту реакцию.
Часто наблюдается, что наиболее реакционноспособным среди
щелочных металлов является цезий — это объясняется самым
малым значением его энтальпии сублимации, которая входит в
одну из начальных стадий реакции. Самые низкие энтальпии
479
Таблица 14.22
Таблица 14.23
Гидрид
LiH
NaH
кн
RbH
CsH
Д^0298, обр,
кал/моль
—21 590
— 14 400
— 14 240
—13000
—13 500
м
Li
Na
К
Rb
Cs
AW°298. реакции,
кал/моль
—53 200
—44 100
—47 100
—44 900
—45 300
(энергии) кристаллических решеток соединений цезия в ряду
однотипных соединений щелочных металлов приводят к высокой
реакционной способности соединений
цезия и к их сравнительной
неустойчивости.
Другой важный класс
соединений щелочных металлов — их
гидриды. Щелочные металлы
настолько сильные восстановители, что в
атмосфере водорода, даже при не
сильном нагревании, превращаются
в гидриды МН. Самый устойчивый
из них — гидрид лития, у него же
наивысшее отрицательное значение
энтальпии образования (табл. 14.23).
Рис 14.14. Давление диссоциации гидридов
щелочных металлов
При высоких температурах гидриды щелочных металлов
диссоциируют на металл и водород. У гидридов, кроме LiH, при
температурах до 600° С давление диссоциации повышается при
переходе от натрия к цезию. При более высоких температурах (выше
900° С) наблюдается обратная последовательность — давление
диссоциации возрастает от цезия к натрию (рис. 14.14). Этот
пример хорошо иллюстрирует то, как характер изменения свойства
зависит от внешних условий и даже изменяется на обратный.
Гидрид лития — самый устойчивый гидрид. В отсутствие влаги
устойчив на воздухе, но водой, как и остальные гидриды,
разлагается по уравнению
LiH -|- НОН - Н2 + L1+ + ОН-.
Этой реакцией пользуются для получения водорода. Чем ниже в
группе находится гидрид, тем энергичнее проходит реакция с
водой, напоминая ту же последовательность взаимодействия щелоч-
lg P (мм рт ст
800 900 1000
480
пых металлов с водой. Если вместо воды взять этиловый спирт,
то образуется алкоголят лития (натрия, калия):
С2Н5ОН + LiH = LiOC2H5 + Н2.
Водород в гидриде лития вытесняется хлором (энергия
кристаллической решетки хлорида лития выше энергии решетки гидрида
лития). Гидрид натрия еще более реакционен — он
воспламеняется в атмосфере хлора
NaH + С12 = NaCl f HC1.
Гидрид натрия взаимодействует с водой значительно энергичнее,
чем гидрид лития, и за счет теплоты реакции водород
воспламеняется. Гидрид натрия — сильный восстановитель; он
восстанавливает четыреххлористый титан до металлического титана:
TiCl4 + 4NaH = Ti + 4NaCl +■ 2H2.
Гидрид калия также очень реакционен и взаимодействует с хлор-
водородом:
КН + НС1 = КС1 + Н2.
Гидриды рубидия и цезия
изучены значительно хуже из-за их
неустойчивости и высокой
реакционной способности. Они не
представляют практического
интереса.
Гидриды щелочных металлов
являются ионными, солеобраз-
ными соединениями. Лишь
гидрид лития имеет черты ковалент-
Рнс. 14.15. Энергия кристаллических
решеток галогенидов щелочных
металлов
ного соединения, поэтому он в значительной степени растворим в
органических растворителях.
При нагревании гидрида лития в атмосфере азота в
зависимости от условий опыта образуется амид лития LiNH2, имид лития
Li2NH или нитрид лития Li^N. Это говорит о близости свойств
лития и водорода. Можно считать, что эти соединения — продукты
замены водорода на литий в аммиаке.
Некоторые соединения в расплавленном гидриде лития
диссоциируют на ионы. Например, карбид кальция диссоциирует на
ионы Са2+ и Сг2-, и при электролизе в расплавленном гидриде
лития на электродах выделяются металлический кальций и
углерод.
АН , кал/моль
298 крист реш
16 общая химия
481
Галогениды щелочных металлов получаются непосредственной
реакцией между простыми веществами, реакциями между водным
раствором галогеноводородов и соответствующим раствором гид-
роксида или карбоната металла. Фторид лития — вещество с
самой высокой энергией кристаллической решетки. Энергии решеток
последовательно уменьшаются при переходе от лития к цезию и
от фтора к хлору (рис. 14.15). Наиболее высокая энергия
кристаллической решетки у фторида лития объясняется малыми
размерами как катиона, так и аниона. Кристаллов, достаточно
близких к ионным, немного. К чисто ионным близки только галогениды
щелочных металлов, хотя и у них степень ионности не превышает
90 о/0.
Литий по некоторым свойствам похож на водород. Учитывая их близость
в периодической системе, можно предположить, что литий, аналогично водороду,,
может образовывать связь, похожую на водородную — литиевую связь. В
газовой фазе обнаружены циклические димеры (LiF)2 и тримеры (LiF)3.
Литиевая связь отличается от водородной тем, что в ней могут принимать участие
незаполненные р-орбитали лития. Литиевая связь прочнее водородной, но
водородная связь хорошо изучена и известны многочисленные ее примеры (вода,
фторводород, спирты, аммиак, органические азотсодержащие вещества, белки и
др.). Примеров литиевой связи значительно меньше, она не встречается в
органических системах и изучена слабо.
Щелочные металлы непосредственно соединяются с
большинством неметаллических элементов и с азотом в том числе, при
этом образуются нитриды состава M3N. Формально нитриды
щелочных металлов можно рассматривать как производное
аммиака, водород которого замещен на атомы металлов. Так, при
пропускании азота над гидридом лития образуется амид лития LiNH2
и имид лития LJ2NH:
3LiH + N2 = LiNH2 -f Li2NH.
При взаимодействии нитрида, имида лития и амида лития с водой
выделяется аммиак
Li3N + 3H20 = NH3 + 3LiOH,
Li2NH f 2H20 = NH3 + 2L10H,
LiNH2 + H20 = NH3 + LiOH.
Сульфиды щелочных металлов типа M2S также получаются
непосредственно прямым синтезом. Известен целый ряд сульфидов
другого состава M2S2, M2S3, M2S'4, M2Ss, M2S6 и др. Только карбид
лития получается непосредственно из элементов при нагревании
2Li + 2С = Li2C2.
Карбиды остальных щелочных металлов получаются
взаимодействием металла с ацетиленом. При низкой температуре
образуются кислые карбиды
2М + 2Н2С2 = 2МНС2 + Н2.
482
При высокой температуре образуются обычные карбиды
м. + н2ся = маса + н2.
При высокой температуре кислые карбиды распадаются по схеме
2МНС2 = М2С2 + Н2С2.
Карбиды щелочных металлов — бесцветные кристаллические
вещества с чрезвычайно высокой химической активностью, даже в
углекислом газе они самовоспламеняются, с водой взрывают.
§ 4. БЕРИЛЛИЙ, МАГНИЙ
И ЩЕЛОЧНОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ
|s-ЭЛЕМЕНТЫ II ГРУППЫ)
Элементами, которыми заканчивается заполнение s-подуровня,
являются гелий, бериллий, магний, кальций, стронций, барий и
радий. Обычно гелий изучают вместе с инертными газами,
бериллий и магний отдельно, а кальций, стронций, барий и радий
объединяют в семейство щелочноземельных металлов. Отнесение гелия
к группе инертных газов (введение его в 8-ю группу периодической
системы) оправдывается исключительно только инертностью гелия,
но не связано ни с его электронным строением, ни с его
свойствами, ни с некоторыми чертами его химического поведения. Гелий
следует считать таким же особым элементом периодической
системы, как и водород.
В главной подгруппе II группы находятся бериллий, магний,
кальций, стронций, барий и радий. Кальций, стронций и барий
раньше назывались щелочноземельными металлами из-за
сходства их оксидов с оксидами щелочных металлов, сообщающих
водному раствору щелочную реакцию, и с оксидом алюминия, из-за
тугоплавкости и труднорастворимости именовавшимся «землей».
Такое разделение во многом оправдано из-за очень сильного
отличия бериллия и отчасти магния от нижестоящих элементов.
Атомы элементов имеют в основном состоянии два s-электрона
на внешнем уровне, а на предвнешнем — по 8 электронов, за
исключением бериллия. У беррилия на предпоследнем уровне
только 2 электрона и вследствие этого наблюдается заметное
отличие его от остальных элементов, во многом аналогичное отличию
лития от нижестоящих элементов. Однако бериллий отличается
от остальных элементов II группы несравненно сильнее, чем литий
от остальных щелочных металлов.
Многие физические и химические свойства элементов главной
подгруппы II группы изменяются не монотонно. Только у
свободных атомов наблюдается последовательное изменение свойств.
Типичным периодическим свойством является энергия ионизации.
В табл. 14.24 приведены данные об энергиях ионизации при отры-
16*
483
Таблица 14.24
Элемент
Be
Mg
Са
Sr
Ва
Ra
М—е-М+
A#i
кал/г-атом
215 000
176 300
140 900
131 300
134 000
121 700
М+— е=М2 +
АЯ2
кал/г-атом
420 000
346 700
273 800
254 300
230 600
234 000
М2+—e=M3f
дя3
кал/г-^том
3 548 000
1803 500
1 181000
1 005 000
853 300
784 000
М—2е=М.2+ \
Чая,
кал/г-атом
635 000
523 000
414 700
385 600
364 600
355 700
М—3<?=М3 +
ДЯ1, 2, 3=Д^1+
+ДЯ2+ДЯ3
кал/г-атом
4 183 000
2 326 500
1 595 700
1 390 600
1217 900
1 139 700
ве первого, второго и третьего
электронов: в первых трех
столбцах даны энергии
последовательной ионизации газообразных
свободных атомов или ионов, A#i
обозначает изменение энтальпии
при удалении электрона от
нейтрального невозбужденного
атома, Д#2 — изменение энтальпии
при удалении электрона от
однозарядного невозбужденного иона
М+, Д#3 — то же для двухза-
рядного иона; в двух последних
столбцах приводятся изменения
энтальпии при удалении от атома
сразу двух A#i)2 и трех Д#1)2,з
электронов. Эти энтальпии
вычисляются по закону Гесса
суммированием последовательных
энтальпий ионизации.
Зависимость энтальпий Д#ь
Д#1,2 и Д#1,2,з от номера периода,
в котором находится металл,
показана на рис. 14.16. Энергия
отрыва второго электрона
примерно в 2 раза выше, чем первого,
ч
Рис. 14.16. Энергия ионизации s-элементов
II группы:
/ _ м—Зе = М3+ Д#1,2)з; 2 - М-2е =
=М2+ A#i,2; 3 — М—е—М+ АН,
тем не менее 1-валентное состояние малохарактерно для
элементов подгруппы бериллия. Отрыв двух электронов хотя и требует
дНиониз>кал/г-атом
4 000000
3000000
У 000 000
юооооо ь
0« 1 1 1 1 ' '
2 3 4 5 6 7 п
Be Mg Са 5г Ва Ra
484
большей затраты энергии, но окупается общим уменьшением
энергии из-за выделения тепла при образовании двух связей за счет
энергии кристаллической решетки или энергии гидратации. Отрыв
трех электронов не может перекрыться энергиями химических
процессов, и рассматриваемые элементы в 3-валентном состоянии
неизвестны. Трех- и более зарядные ионы могут быть получены
только посредством мощных физических воздействий. Обращает
внимание резкое падение энергии ионизации до 3-зарядных ионов
с ростом номера периода.
Многие физические свойства элементов в металлическом
состоянии изменяются немонотонно, это связано с тем, что эти
элементы кристаллизуются в различных кристаллических решетках.
Радиусы атомов металлов подгруппы бериллия (табл. 14.25)
значительно меньше радиусов щелочных металлов.
Таблица 14.25 Таблица 14.26
Металл
Be
Mg
Са
Сг
Ва
Ra
пл' ч
1556
923
1123
1043
987
973
'кип'
2744
1376
1762
1630
1907
1809
Металл
Be
Mg
Са
Sr
Ва
Ra
Ям, А
1,13
1,60
1,97
2,15
2,24
2,35
Плотность щелочноземельных металлов значительно выше, чем
щелочных, так как 2 валентных электрона образуют более
прочную металлическую связь.
Температуры плавления и кипения элементов подгруппы
бериллия (табл. 14.26) намного выше, чем у щелочных металлов и в
отличие от последних изменяются не монотонно, а скачками от
элемента к элементу. Ход изменения температур плавления
обычно объясняют, ссылаясь на различные кристаллические структуры
металлов и вытекающие отсюда различия в энергиях
кристаллических решеток.
Гелий и элементы подгруппы бериллия объединяет
заполненность 5-подуровня парой электронов, и таким образом,
равенство числа связывающих и разрыхляющих электронов в
двухатомных молекулах, что приводит к их нестабильности.
Невозбужденная молекула Нег действительно не обнаружена. Единого мнения
о возможности существования молекулы Вег нет. По разным
источникам энтальпия диссоциации Вег или равна нулю (молекула
не существует), или равна 14 000 кал/моль. Для двухатомных
молекул магния и кальция известны энтальпии диссоциации
(табл. 14.27), уменьшающиеся в сторону кальция.
485
Молекула
Ве2
Mg3
Са2
Sr2
Ва2
д связи•
кал/мол
0 или 14000
7200
5520
0
0
Таблица 14.27 для объяснения причин
образования молекул Эя следует вспомнить, что
атом гелия не имеет /^-электронного
подуровня, а у атома бериллия и у
всех атомов нижестоящих элементов
он имеется. Поэтому возможен
переход части электронной плотности на
р-подуровень, причем если такой
переход совершается в молекуле, то
электроны попадают на связывающие ор-
битали как энергетически более
выгодные. Разумеется, связывающее действие электронов р-подуров-
ня у бериллия будет наиболее сильное и ослабляется с ростом
размеров атомов при переходе к барию.
Рассмотрение энергий ионизации атомов изучаемых элементов доказывает
невозможность образования ими трехзарядных ионов и вытекающего отсюда
3-валентного состояния. В подавляющем большинстве своих соединений
элементы подгруппы бериллия 2-валентны. Тем не менее простой расчет не исключает
возможности существования соединений с одновалентными элементами.
Суммированием двух реакций ионизации и их энергий получаем реакцию
взаимодействия нейтрального атома с двухзарядным ионом, в которой образуется
однозарядный ион:
Мг — е = М+
М+ — е-
■Ж2+
ДЯХ
АЯ9
МГ + М2+ = 2М+ ДЯ^ДЯх — ДЯ2
Результаты вычислений энтальпий образования однозарядных ионов
представлены в табл. 14.28.
Таблица 14.28
Реакция
Мг-е=М+
МГ+М2+=2М+
АН
ДЯХ
дя2
ДЯ^ДЯ!—
-АН2
АН, кал/моль
Be
215 000
420 000
—205000
Mg
176 300
346 700
—170 400
Са
140 900
273 800
—132 900
Sr
131 300
254 300
—123 000
Ва
134 000
230 600
—96 600
Реакция образования однозарядного иона из атома и двухза-
рядного иона термодинамически не запрещена, причем вероятность
существования однозарядных ионов падает от бериллия к барию.
При высокой температуре в газовой фазе, содержащей пары
металлов и двухзарядные катионы, действительно были обнаружены
двухатомные молекулы СаН, CaF, CaCl, Cal, BaCl и др. Делались
486
попытки сохранить эти молекулы быстрым охлаждением газовой
смеси, но из-за конденсации металла равновесие
Mr -f М?+ - 2М+
смещается влево, и однозарядные катионы в газовой фазе при
низких температурах перестают существовать.
При быстром охлаждении расплава галогенида кальция с
металлическим кальцием удалось выделить в виде кристаллов СаС1.
Моногалогениды распадаются по схеме
2МГК = Мк -f МГ2.
Из-за низких температур скорость распада очень мала, тем не менее следует
дать ответ, почему однозарядный ион термодинамически не запрещен, а
соединения с одновалентными металлами неустойчивы? Термодинамические
характеристики кристаллических моногалогенидов известны лишь для нескольких
веществ (табл. 14.29. Здесь так же приведены
энтальпии соответствующих дигалогенидов). Таблица 14.29
Энтальпии образования из простых
веществ и моногалогенидов и дигалогенидов
АН298, обр>
кал/моль
MgClK
MgCl2,K
СаВгк
СаВга,к
Са1к
Са12|К
— 55 000
—153 400
— 54 000
—161 300
, — 43 500
—127 800
отрицательны. Это значит, что и те и другие Вещество
термодинамически предпочтительны по отно-
шению к простым веществам три стандартных
условиях, но энтальпии образования
дигалогенидов примерно в 3 раза ниже по сравнению
с энтальпиями моногалогенидов, и это
означает, что при стандартных условиях и
стандартной температуре (и при других не
высоких температурах) будут образовываться
дигалогениды, а не моногалогениды.
Чтобы не возвращаться больше к моногалогенидам элементов
подгруппы бериллия, заметим, что они энергично взаимодействуют
с водой, выделяя водород:
2СаС1к + 2Н2Ож = СаС12,к + Са(ОН)2,к + Н2,г.
Увеличение заряда ядер атомов элементов подгруппы бериллия
по сравнению со стоящими рядом атомами щелочных металлов
приводит к значительно большим энергиям ионизации и меньшим
металлическим радиусам. Это делает элементы главной подгруппы
II группы обладателями значительно более слабых металлических
свойств по сравнению с соответствующими щелочными металлами.
Наиболее слабыми металлическими свойствами среди металлов
s-элементов обладает бериллий. Радиус двухзарядного иона
бериллия в два с лишним раза меньше, чем у магния (табл. 14.30),
и почти в 1,5 раза меньше, чем у лития. У бериллия значительно
слабее металлические свойства по сравнению с литием и магнием.
Ион Ве2+ неизвестен ни в растворе, ни в кристаллическом
состоянии. В таких, казалось бы, ионных соединениях, как BeF2 или
ВеО, связи между элементами носят очень сильный ковалентный
характер. ,
Таблица 14.30
Таблица 14.31
Иои
Ве«+
Mg2+
Са2+
Sr2+
Ва2+
Л//°гидр'
кал/моль
—606 000
—474 000
—395 000
—354 000
—326 000
Ион
Ве2+
Mg2+
Са2"*-
Sr2+
Ва2+
Ra2+
*М2+' А
0,34
0,74
1,04
1,20
1,38
1,44
При переходе к магнию ковалентный характер связи в
соединениях ослабляется, а соединения кальция и остальных
щелочноземельных металлов перимущественно ионные. Во всех главных
подгруппах периодической таблицы первый элемент сильно
отличается от нижестоящих.
Энергия гидратации иона бериллия (табл. 14.31) почти в 5 раз
выше энергии гидратации иона лития и она настолько велика, что
процесс гидратации — это по существу химическая реакция иона
с молекулами воды. Прочность образующихся связей настолько
велика, что свободный ион Ве2+ не обнаружен даже в очень
кислых растворах.
Изучение щелочных и щелочноземельных металлов приводит к
важнейшему выводу о так называемом диагональном сходстве
элементов. Оно проявляется в том, что вышестоящий s-элемент
первой группы напоминает s-элемент, расположенный на период
ниже в соседней группе. Это объясняется характером изменения
размеров ионов вдоль по периоду (уменьшение радиуса) и вниз
по подгруппе (увеличение радиуса). Действительно, в этом
отношении ион лития близок к иону магния, ион натрия к иону кальция,
калия к стронцию и т. д. Соли лития (фторид, фосфат и карбонат)
в отличие от таких же солей щелочных металлов плохо
растворяются в воде, напоминая соли магния (и кальция). Основные
свойства гидроксидов лития и магния также близки. Однако не
следует думать, что таким способом можно сопоставлять все
свойства элементов и их соединений. Диагональное сходство элементов
главных подгрупп проявляется не только у s-элементов, но и у
р-элементов и между \s- и р-элементами.
Все элементы главной подгруппы II группы образуют гидриды
типа МН2. Все гидриды, кроме гидрида бериллия, можно
синтезировать из простых веществ:
Мк + Н2,г =-- МН2,Г.
Гидрид магния можно получить по этой реакции из простых
веществ, но реакция проходит с трудом.
Гидрид бериллия легко получается взаимодействием хлорида
бериллия с гидридом лития в эфире:
ВеС12 + 21ЛН = ВеН2 + 2L1C1.
488
Гидриды бериллия и магния по своим свойствам занимают
промежуточное положение между ионными и ковалентными гидридами.
Последнее свойство особенно присуще бериллию. Гидрид
существует в виде полимера благодаря водородным мостикам:
Н Н
\3е;' ^Ве^' ")Ве^
н н
В этом отношении он напоминает гидриды бора и алюминия и в
еще большей степени гидрид лития, имеющий более ковалентныи
характер, чем гидриды остальных щелочных металлов. Однако
гидриды бериллия и магния значительно более реакционны.
Гибриды Be и Mg термически менее устойчивы по сравнению с
гидридом лития, но в то же время гидрид
магния устойчивее гидрида алюминия Таблица
Стандартные энтальпии образова-
ния гидридов элементов II группы гидрид
(табл. 14.32) ясно показывают отли-
А//298. обр,
кал/моль
— 1000
—19 000
—45 100
—42 300
—40 960
чие гидридов бериллия и магния как в „
друг от друга, так и от гидридов ще- MgH^K
лочноземельных металлов. СаН2,к
Некоторое уменьшение численных SrH2)K
значений энтальпий гидридов от каль- ВаН2,к
ция к барию вполне понятно — в этом
же направлении уменьшаются энергии
решеток ионных кристаллов (размеры катионов возрастают).
Ионность всех гидридов подгруппы проявляется в очень
энергичной реакции с водой — в этой реакции гидриды являются
восстановителями:
МН2,К + Н2Ож = М (OH)2iK + н2.г.
Гидриды щелочных и щелочноземельных металлов имеют со-
леобразный характер (у гидрида бериллия он выражен очень
слабо, у гидрида лития уже заметно). Водород в них —
отрицательно заряженный ион. Гидрид кальция — сильный
восстановитель, но он не так активен, как гидриды щелочных металлов.
При горении металлы подгруппы бериллия образуют
оксиды МО:
Мк+1/2 02 = МОк.
Все оксиды имеют высокие отрицательные значения энтальпий
образования (рис. 14.17). У оксидов бериллия, магния и
стронция АЯ298, обр примерно одинаковы, у оксида кальция
наибольшее отрицательное значение, у оксида бария — наименьшее.
489
Можно обратить внимание, как и в ряде, других случаев, на
равномерное уменьшение значения Л #298, обр оксидов
щелочноземельных металлов (Са—Sr—Ва).
Несмотря на самое низкое значение энтальпии образования
ВаО, барий очень энергично реагирует с кислородом и даже
воспламеняется от удара. Объяснение этому явлению заключено в
меньшей устойчивости
кристаллической решетки бария. Магний
на воздухе сам не
воспламеняется, но, будучи подожжен, горит
ярким пламенем.
Кристаллические оксиды
магния и кальция обладают ионной
проводимостью (1000°С), что
доказывает присутствие ионов М2+
и О2".
Энергии кристаллических
решеток оксидов щелочноземель-
-130000
-140000
-150000
0
ЛНооо к мл ♦ «ал/моль
298,обр М0К
"BeSqQ A0
YCaO
1 , .J I I I
б п
Рис. 14.17. Энтальпия образования
оксидов 5-элементов II группы
ных металлов изменяются параллельно энтальпиям образования,
уменьшаясь по абсолютной величине от СаО к SrO и ВаО. На
примере этих оксидов очень хорошо прослеживается зависимость
реакционной способности оксидов от энергии их решеток.
Сравнение температур начала заметного взаимодействия с оксидами
щелочноземельных металлов ясно показывает, что с увеличением
величины энергии решетки эти температуры возрастают
(табл. 14.33).
Оксид
СаО
SrO
ВаО
решетки»
кал/моль
831 000
760000
727000
Та
блица 14.33
Температура (°С) начала
взаимодействия с
MgCO,
525
455
345
ZnS04
520
425
340
Все оксиды элементов главной подгруппы II группы
реагируют только с кислотными оксидами, проявляя основную
природу:
СаОк 4 Si02,K = CaSi03,K.
490
Оксид бериллия, будучи амфотерным, играя роль или основного
или кислотного оксида, взаимодействует и с кислотными и с
основными оксидами:
2ВеОк + Si02,K - Be2Si04,K,
ВеОк + Na20K = Na2Be02,K.
Реакция между оксидом металла и водой с образованием гид-
роксида проходит только с оксидами кальция и нижестоящих
металлов:
МОк + НяОж = М(ОН)2,к.
Свежеприготовленный оксид магния реагирует с водой очень
медленно, а кристаллический MgO совсем не реагирует. Оксид
бериллия не реагирует с водой
ни при каких условиях. Гидрок- ы 7 180*
сиды мало растворимы в воде. &ь^У~^\
С ростом температуры раствори- М)—*~Ве~г~9о
мость их очень сильно возраста- 1>4А %Р&«
ет, за исключением кальция, pa- ° и
створимость которого из-за
положительного теплового эффекта •
растворения уменьшается с тем- ^\^\ S^~^\
пературой. ^Д/^д-^-0о
Бериллий и магний разлагают 1.9А vjfcg»
воду очень медленно, даже при Ъщ v*
нагревании вследствие малой
растворимости гидроксидов.
Остальные металлы реагируют с
водой при обычной температуре,
причем барий взаимодействует с
Рис. 14 18. Строение молекул дигидро-
ксидов s-элементов И группы
водой легче всех благодаря лучшей растворимости его гидроксида.
Гидроксид бериллия — амфотерен, у остальных гидроксидов
основной характер возрастает от Mg(OH)2 к Ва(ОН)2. Гидроксид
бария настолько сильное основание, что приближается к гидро-
ксидам щелочных металлов. ,
Гидроксиды щелочноземельных металлов — ионные
кристаллы, имеющие ионы М2+. По сравнению с гидроксидами щелочных
металлов у них заряды ионов металлов выше, а радиусы ионов
меньше, поэтому взаимодействие между ионами М2+ и ОН~
сильнее по сравнению с соответствующими ионами щелочных
металлов и энергии кристаллических решеток гидроксидов
щелочноземельных металлов выше. Следовательно, их растворимость и
491
основность ниже. С ростом радиуса иона М2+ эти свойства
усиливаются, поэтому гидроксид бария — самое сильное основание
в этой подгруппе.
Изучение строения молекул гидроксидов показало их интересные особенности.
Все гидроксиды М(ОН)2 не линейны. Угол О—М—О равен 180° только у
гидроксидов бериллия и магния. У гидроксидов щелочноземельных металлов он
составляет 120°. Угол Н—О—М у всех гидроксидов равен 120°, за исключением
гидроксида магния, у которого он равен 110° (рис. 14.18). Все расстояния О—Н
равны 0,96 А, катион на них не влияет. Расстояния О—М возрастают от
бериллия к барию. Молекулы гидроксидов бериллия и магния, по всей вероятности,
плоские (все атомы лежат на одной плоскости), но молекулы гидроксидов
щелочноземельных металлов — не плоские; протоны располагаются выше и ниже
плоскости атомов О—М—О. Столь различное строение молекул гидроксидов
объясняется возможностью участия в связях d-орбиталей атомов магния и
нижележащих элементов.
При высокой температуре в газовой фазе существуют
молекулы оксидов и гидроксидов одновалентных металлов МгО и
МОН. При высоких температурах равновесие
МОг + Мг = М2Ог
отчасти смещено в сторону образования М20, но при понижении
температуры равновесие резко смещается влево, и одновалентные
кристаллические оксиды получить не удается.
При введении паров воды в атмосферу, содержащую одновалентные оксиды,
проходит газовая реакция
М2Ог + Н2Ог = 2МОНг
в результате которой образуются гидроксиды одновалентных элементов.
Строение их молекул изучено недостаточно. Известно, что у гидроксидов
щелочноземельных металлов угол М—О—Н равен 105°, а расстояние М—О несколько
меньше, чем у дигидроксидов (Са—О 2,00 A, Sr—О 2Д2А, Ва—О 2,15 А).
При действии на раствор соли бериллия щелочью получается
гидроксид бериллия в виде геля Be(OH)2-ftH20. Это вещество
растворяется и в кислотах
Be (ОН)2 + 2Н+ = Ве2+ +2Н20
и в щелочах. При растворении в щелочи продукты различны в
зависимости от условий. При комнатной температуре и не
слишком высокой концентрации щелочи образуется соль состава
NaBe(OH)3:
Be (OH)2 + NaOH = NaBe (OH)3,
или
Be (OH)2 + ОН- = Be (ОН)Г.
При большом избытке щелочи реакция проходит по уравнению
Be (OH)2 + 2NaOH = Na2Be (OH)4,
или
Ве(ОН)2 + 20Н- = Ве(ОН)^.
492
Бериллаты натрия и калия получаются сплавлением гидроксида
бериллия с щелочью:
Be (ОН)2.к + 2№ОНж - Na2Be02,K + 2Н2Ог.
Водой бериллаты гидролизуются по реакции
К2Ве02,к + 2Н2Ож = Be (ОН)2,к + 2K+ + 20Н-.
Несмотря на то что бериллат-ион в водном растворе вообще
не существует, уравнения с участием бериллат-иона ВеО|~
писать можно исключительно только для удобства и простоты.
Например, растворение бериллия в растворе щелочей описывается
простейшим уравнением
Век + 20Н7-р = ВеО^-р + Н2,г.
Это уравнение не отражает истинного процесса и служит только
для указания на амфотерность бериллия. Все соли бериллия
имеют вследствие гидролиза кислую реакцию
Ве2+ + Н20 - Be (ОН)+ + Н+.
В концентрированных кислых растворах бериллий находится в
виде ионов Ве(Н20)4+. Структура этого иона из-за очень прочной
связи с молекулами воды может сохраняться и в твердом
состоянии, например, хлорид бериллия — это настоящая комплексная
соль, в которой бериллий имеет координационное число 4:
[Ве(Н20)4]С12. Растворы солей бериллия способны растворять
оксид и гидроксид бериллия, что объясняется способностью
бериллия образовывать комплексы с цепочками Be—О—Be и
Be—ОН—Be. Исключительно малая величина радиуса иона
бериллия объясняет склонность бериллия к образованию ковалент-
ных связей, не свойственных остальным s-элементам.
Амфотерность бериллия проявляется и в свойствах его
сульфида. Сплавлением могут быть осуществлены реакции
BeS + Na2S = Na2BeS2,
сульфидобериллат натрия
BeS + SiS2 = BeSiS3.
бериллиевая соль сернокремниевой
кислоты
Как и у щелочных металлов, при переходе от бериллия вниз
по подгруппе реакция взаимодействия с водой протекает все
легче. Однако отличие в характере этой реакции у I и II группы
элементов огромное. Все металлы подгруппы бериллия тонут в
воде, плавятся при температурах, намного больших температуры
кипения воды, а энтальпии образования гидроксидов не слишком
сильно различаются. Характер взаимодействия этих металлов с
493
Таблица 14.34
Металл
Be
Mg
Ca
Sr
Ba
Ra
£°298, В
—1,85
—2,37
—2,87
—2,89
—2,90
—2,92
водой определяется ходом изменения стандартных
восстановительных потенциалов, которые при переходе от бериллия к радию
приобретают все более отрицательные значения (табл. 14.34).
Ход изменения Е° от номера
периода металла еще раз подтверждает
обособленное положение бериллия и
магния среди элементов их подгруппы
и большое сходство всех
щелочноземельных металлов.
Несмотря на невысокий
восстановительный электродный потенциал
(£ве/Ве24 =— 1,85 В), ГОВОРЯЩИЙ О
возможности растворения бериллия в
1 н. растворах сильных кислот,
бериллий в них не растворяется вследствие
образования непроницаемой окисной пленки. Соли остальных
металлов могут быть получены растворением их в кислотах.
Хлорид бериллия ВеС12, единственный среди хлоридов
изучаемых элементов, является не солеобразным соединением.
Безводный хлорид бериллия — это полимер, в котором молекулы ВеСЬ
связаны донорно-акцепторными связями (хлор — донор,
бериллий— акцептор):
С1 С1 С1 С1
к ^Ве/ ^Ве/ ^Ве/ ^Ве/ ^
С1 С1 С1 С1
Энергий химических процессов (гидратации или энергии образования
кристаллической решетки) из-за высоких значений энергии ионизации бериллия
недостаточно для удаления электронов из атома с образованием иона Ве2+. В то
же время при образовании ковалентных связей необходимы распаривание двух
25-электронов и sp-гибридизация. В газовой фазе образуются линейные
молекулы типа ВеГг, имеющие' смешанные ковалентно-ионные связи.
Координационное число бериллия в этих молекулах только 2 и у него остается значительная
способность к координации, проявляющаяся при конденсации молекул в твердую
фазу. Кристаллический ВеС12 — по существу полимер, в котором связь между
атомами бериллия осуществляется через мостики атомов хлора, при этом
каждый атом хлора играет роль донора электронной пары. Координационное число
бериллия в твердом хлориде бериллия равно четырем. Угол С1—Be—С1 равен
98°, а не 109°. Это значит, что четырехугольники Be—CI—Be—C1 вытянуты вдоль
цепи атомов бериллия. Газообразный хлорид бериллия при высоких
температурах состоит из молекул ВеС12, но при низких температурах пар хлорида
бериллия содержит значительное количество молекул димера ВегСЦ.
Координационное число бериллия в димере равно трем.
Амфотерными свойствами могут обладать не только оксиды,
но и другие соединения, например галогениды. Так, фторид
бериллия с фторидом калия дает фторобериллат калия
BeF2 + 2KF=K2BeF4.
494
В этой реакции фторид бериллия проявляет кислотные свойства.
В реакции с тетрафторидом кремния фторид бериллия
характеризуется основными свойствами:
BeF2 + SiF4 = BeSiFe,
так как образуется соль бериллия.
Известны фторбериллаты кальция, стронция и бария, МВеГ4,
устойчивость которых возрастает с усилением различия между
бериллием и металлом, т. е. в направлении CaBeF4—SrBeF4—
BaBeF4, в котором металлические свойства катиона усиливаются.
Расплавленный фторид бериллия содержит катионы и анионы,
в состав которых входит бериллий. Реакцию образования ионов
можно записать так:
2BeF2 = BeF+ + BeFT.
В этой реакции одна молекула фторида бериллия проявляет
кислотные свойства, а другая — основные.
Галогениды бериллия в расплавленном состоянии плохие
проводники электрического тока. Хлорид бериллия в водном
растворе гидролизуется, и раствор имеет кислую реакцию
ВеС12,к + Н2Ож = ВеОН+р + 2С1~Р + Н+р.
Ион магния Mg2+ не гидролизуется, раствор хлорида магния
нейтрален. Ионы нижестоящих металлов также не гидролизуются.
Все молекулы дигалогенидов подгруппы бериллия должны
согласно теории валентных связей быть линейными благодаря
-sp-гибридизации. Между тем среди фторидов линеен только
фторид бериллия, а остальные фториды представляют собой
угловые молекулы. Например, в молекуле MgF2 угол F—Mg—F
равен 150°. При переходе к йоду
способность образовывать
линейные молекулы возрастает, и
только йодид бария имеет угловую
структуру. Примерно половина
всех дигалогенидов нелинейна
(табл. 14.35).
Дипольные моменты
дигалогенидов увеличиваются при
переходе от бериллия к барию и от
йода к фтору, что подтверждает
расположение нелинейных
молекул в нижнем левом углу
табл. 14.35.
Причина этого явления состоит в том, что при переходе от
бериллия к барию возрастает способность к изменению sp-гибриди-
зации на sd-гибридизацию, так как чем выше число уровней я,
тем энергетически ближе наружные подуровни. В то же время
Металл
Be
Mg
Са
Sr
Ва
Таблица 14.35
Галоген в МГ2
F
Л
У
1 у
У
У
С1
л
л
л
У
У
Вг
Л
л
л
л
У
I
л
л
л
л
У
495
при переходе от фтора к йоду возрастает способность этих
атомов изменять гибридизацию центрального атома металла,
способствуя увеличению степени ионной связи.
Щелочноземельным металлам не свойственно комплексообра-
зование, но известны некоторые соли, в которых
координационные числа чаще всего 4 и 6, а у кальция даже 8: [Be(NH3)4]Cl2,
[Mg(NH3)6]Cl2, [Ca(H20)6]Cl2, [Ca(NH3)8]Cl2. Склонность к
образованию комплексных соединений падает с увеличением
размера атома-комплексообразователя, т. е. от бериллия к барию.
Среди многочисленных солей элементов подгруппы бериллия
особая роль принадлежит карбонатам и сульфатам, в
особенности карбонату и сульфату кальция.
Карбонаты рассматриваемых металлов плохо растворимы в
воде. Их термическая устойчивость возрастает при переходе от
бериллия к барию. Избыток углекислого газа переводит
нерастворимые карбонаты Са, Sr и Ва в легкорастворимые кислые
бикарбонаты:
СаСОз.к + С02>г + Н2Ож = Са2р+ + 2НС032^Р.
Карбонаты бериллия и магния могут существовать в
присутствии воды только при наличии атмосферы углекислого газа.
В воде, не насыщенной углекислым газом, они гидролизуются с
образованием основных карбонатов различного состава.
Например, основной карбонат магния имеет состав 4MgC03-Mg(OH)2»
•5Н20. Ему приписывают строение
Н20 - MgC034 yC03Mg - ОН2
>Mg<
Н20 - MgCCV XC03Mg - ОН2
(ОН), . Н20.
При осторожном нагревании одна молекула воды может быть
удалена и образуется состав 4MgC03-Mg(OH)2-4H20.
Вода, содержащая ионы НС03, образовавшиеся шд
действием углекислого газа и воды на карбонаты, называется временно
жесткой водой и может быть смягчена кипячением:
Са*+ + 2НСОзТР-р - СаС03,к + С02,Г + Н2Ож,
Mgp$ + 2НСОзТР.р = MgC03,K + С02,Г + Н2Ож.
При этом карбонат кальция выделяется в виде накипи.
Аналогичные реакции проходят и с ионами железа, поэтому корка
накипи имеет грязно-коричневый цвет. Та же реакция совершается
и при испарении воды из концентрированного раствора
бикарбоната кальция. Так в природе образуются сталактиты и
сталагмиты.
В постоянно жесткой воде содержание бикарбонат-иона
недостаточно для связывания всех катионов (обычно избыточное
относительно бикарбонат-иона количество катионов приходится на
496
сульфат-ион), и для смягчения воды применяют ионно-обменные-
смолы или соду:
Са2+ (+ SOf- + 2Na+) + СОГ = СаС03,к + (Sdt + 2Na+).
В быту смягчение воды обычно происходит при пользовании
мылом (смесь натриевых солей стеариновой и пальмитиновой-
кислот). Часть мыла вступает во взаимодействие с катионами
магния и -кальция и выпадает в осадок (магниевые и кальциевые
соли стеариновой и пальмитиновой кислот в воде не
растворимы), а оставшаяся часть мыла, гидролизуясь на щелочь и
жирную кислоту, действует как моющее средство.
При нагревании оксида кальция с углем образуется карбид
кальция
СаОк + ЗСК = СаС2 + СОг.
Его реакция с водой (гидролиз) проходит энергично и является;
одним из способов получения ацетилена:
[Са2+ С!~]к + 2Н2Ож = С2Н2,Г + Са (ОН)2,к.
Бериллий образует карбид состава Ве2С прямой реакцией иа
простых веществ (при 1900°С). При действии воды выделяется
метан
Ве2Ск f 4Н2Ож = СН4,Г Ь 2Ве (ОН)2|К.
Карбиды типа МС2 образуют все элементы, но получаются
они различными путями: береллия и магния нагреванием в
ацетилене, остальные металлы реагируют с углеродом при высоких
температурах. При 400°С магний в атмосфере ацетилена
переходит в карбид MgC2, но при 600°С получается карбид другого
состава Mg2C3. Предполагают, что он содержит ионы С3"~~, так как
при гидролизе выделяется метилацетилен СН3—С = СН:
Mg2C3,K + 4Н2Ож --= 2iMg(OH)2,K + С3Н4.Г.
При красном калении карбид кальция реагирует с азотом
воздуха, образуя цианамид кальция Ca(N = C = N), содержащий
цианамид-ион CN2~~. Цианамид кальция — широко используемое
азотное удобрение.
§ 5. ЭЛЕМЕНТЫ ГЛАВНЫХ ПОДГРУПП III—VIII ГРУПП
(р-ЭЛЕМЕНТЫ)
К р-элементам относятся элементы главных подгрупп III—
VIII групп периодической системы. У элементов III группы
появляется первый р-электрон и начинается последовательное
заполнение /?-подуровня, заканчивающееся у элементов VIII группы —
инертных газов.
497
Свойства р-элементов, если их рассматривать как вдоль
периода, так и вниз по подгруппе, изменяются часто не равномерно.
Это вызвано строением электронных уровней и подуровней. При
переходе вдоль по периоду заметно стремление /7-подуровня или
не быть незаполненным, или заполненным наполовину, или же
заполненным полностью. Так, у элементов III группы обращает
внимание склонность к образованию соединений с 1-валентным
состоянием элемента. Хорошо изучены различные соединения
алюминия, существующие в газовой фазе, а у таллия
I-валентное состояние известно даже и для его ионов в водных растворах.
Наполовину заполненный /^-подуровень у азота и нижестоящих
элементов приводит к заметному различию в свойствах по
сравнению с кислородом и нижестоящими элементами, имеющими
уже одну пару спаренных электронов на р-подуровне. У инертных
газов — элементов главной подгруппы VIII группы — р-подуро-
вень заполнен; это согласуется с его чрезвычайно высокой
устойчивостью.
При переходе вниз по подгруппе на свойства элементов и их
•соединений оказывает влияние как появление новых подуровней
на одном уровне, так и заполнение внутренних подуровней.
Элементы II периода резко отличаются от нижележащих элементов.
Причина этого заключена в имеющемся у элементов III периода
свободном d-подуровне, способном принимать электроны с р-под-
уровня при их распаривании. У элементов IV периода на пред-
внешнем третьем уровне появляется заполненный Зй-подуровень,
что приводит к их заметному отличию от элементов III периода.
Точно так же сказывается наличие заполненного 4 /-подуровня
на третьем снаружи уровне у элементов VI периода. Разумеется,
все эти факторы влияют не только на р-элементы, но и на уже
изученные s-элементы.
Элементы II периода характеризуются невысокими
валентными состояниями—не более 4. Способность к проявлению
нескольких валентных состояний для них не характерна. Только бор и
углерод проявляют два различных валентных состояния: 1 и 3
у бора, 2 и 4 у углерода. Это объясняется пустыми электронными
ячейками р-подуровня, способными при возбуждении атома
принимать электроны с 5-подуровня. Валентные состояния азота,
кислорода, фтора и неона определяются числом неспаренных
электронов и равны соответственно 3, 2, 1 и 0. Отсутствие d-под-
уровня у атомов элементов II периода исключает возможность
распаривания электронов. Такая возможность появляется у
элементов III периода, у которых при переходе от натрия к хлору
возрастает как значение максимального валентного состояния
(7 у хлора), так и набор различных валентных состояний
(4 у хлора).
Благодаря чрезвычайной стабильности полностью
заполненного р-подуровня аргон химически инертен, но теоретически он
может образовывать 8-валентное состояние и целый набор дру-
498
гих меньших четных валентных состояний. У элементов III и
нижестоящих периодов номер групп совпадает с максимальны^
валентным состоянием, т. е. с максимально достижимым числом
неспаренных электронов. Такой механизм возникновения
валентных состояний подтверждается формулами оксидов, гидроокси--
дов, кислот, гидридов и других соединений (табл. 14.36).
Таблица 14.36
Соединение
Оксиды
Гидроксиды
Гидриды
I
Э20
ЭОН
ЭН
п
эо
Э(ОН)2
эн2
III
Э303
Э (ОН)3
Н3Э03
нэо2
эн3
№ группы
IV
эо2
Э(ОН)4
Н4Э04
Н2Э03
ЭН4
V
э2о5
н3эо4
НЭ03
ЭН3
VI
Э03
н2эо4
эн2
VII
э2о7
нэо4
ЭН
VIII.
ЭО*.
н,эо&
Первые энергии ионизации атомов элементов второго периода
возрастают, но не равномерно, от лития к неону (табл. 14.37),
Таблица 14.37
Период
II
III
IV
V
VI
I
Li
124 300
Na
118 500
К
106 100
Rb
96400
Cs
89 700
Энергии ионизации Э— е=Э~*~, Л#иониз, I, кал/г-атом
II
Be
215 000
Mg
176 300
Ca
140 900
Sr
131 800
Ba
134 000
№ групп
Ш
В
191 400
Al
137 900
Ga
138 400
In
133 500
Tl
140 900
IV
С
259 700
Si
188 000
Ge
181 700
Sn
169 100
Pb
171 100
V
N
335 300
P
243 300
As
226 200
Sb
199 300
Bi
168 100
VI
О
313 900
S
238 900
Se
224 900
Те
207 800
Po
189 100
VII
F
401 800
CI
300 000
Br
273 100
I
240 800
At
212 200
VIII
Ne
497 200
Ar
363 500
Kr
322 90G
Xe
279 800
Rn
247 900
Самая низкая энергия ионизации у лития, что объясняется
легкостью потери одного-единственного наружного электрона с
образованием иона Li+. У бериллия энергия ионизации значительно
выше и этот элемент не образует ни ионов Ве+, ни ионов Ве2+,
а его 2-валентное состояние в соединениях в большей степени ко-
валентиое с частично ионным характером. Все /7-элементы уже
49а
не способны образовывать положительно заряженные ионы.
Первая энергия ионизации бора ниже энергии ионизации атома Be.
Это вызвано отрывом первого р-электрона, который связан с
ядром заметно слабее s-электронов. От бора к азоту первые
энергии ионизации равномерно возрастают, но у кислорода
наблюдается новый разрыв монотонности изменения энергий
ионизации. У нейтрального атома азота все три р-ячейки заполнены по
одному электрону.
Наполовину заполненный любой подуровень обладает
повышенной устойчивостью. Введение одного электрона в наполовину
заполненный р-подуровень у кислорода приводит к ослаблению
связи электронов с ядром, и первая энергия ионизации
кислорода ниже, чем у азота. От кислорода к неону первые энергии
ионизации снова равномерно возрастают.
Точно такая же картина в характере изменения первых
энергий ионизации наблюдается у атомов элементов III периода и
других нижележащих, за исключением VI периода. Следовало бы
ожидать, что первые энергии ионизации будут падать при
переводе по группе элементов сверху вниз. Но такое поведение часто
не наблюдается. Во II группе энергия ионизации бария выше,
чем у стронция. В III группе первые энергии ионизации
алюминия, галлия, индия и таллия очень близки друг другу и намного
меньше энергии ионизации бора. Они располагаются в
следующем порядке: Т1—Ga—А1—In. У элементов IV группы свинец
нарушает ожидаемый ход, располагаясь над оловом. Внешние
электроны очень чувствительны к строению предвнешних
электронных уровней, особенно это касается первого р-электрона.
р-Электрон бора заметно слабее связан с ядром по сравнению
«с s-электронами бериллия. р-Электрон алюминия еще слабее
связан с ядром по сравнению с электронами магния. У галлия
появляется заполненный ^-подуровень, и d-электроны занимают
пространство вокруг ядра значительно менее плотно и не столь
равномерно, как 5-электроны или р-электроны. В результате
наружный р-электрон галлия притягивается ядром сильнее, и энергия
ионизации галлия больше. У таллия на третьем снаружи уровне
имеется заполненный четырнадцатью электронами 4/-подуровень,
еще слабее экранирующий притяжение ядра, а также
заполненный 5 d-подуровень. Это обусловливает заметное увеличение сил
притяжения наружного р-электрона таллия к ядру. В результате
первая энергия ионизации таллия больше, чем у алюминия и
галлия.
При введении второго р-электрона в наружный уровень
влияние строения предвнешних оболочек сказывается уже слабее.
Первая энергия ионизации свинца выше, чем у олова. Наружный
р-электрон свинца отличается от наружного р-электрона олова
тем, что у него имеются заполненные 4f- и 5 d-подуровни,
характеризующиеся меньшей экранирующей способностью к силам
притяжения ядра. У элементов V и остальных групп все ниже-
500
стоящие элементы обладают, как и следует ожидать, меньшей
энергией ионизации.
Отличие в стабильности незаполненного, наполовину
заполненного и полностью заполненного р-подуровня от других его
состояний постепенно сглаживается при возрастании номера
периода. Это заметно на уменьшении скачка в энергиях ионизации при
рассмотрении элементов V и VI групп (рис. 14.19). У элементов
И период
500000
400000
300000
200000
100000
иониз
-
-Li
-Na3
Rb;
Be
/Mg
//ta\
f// Ba
Cs i
j, нал/г атом
Ne
С
s/Sl
Згьа
Al
In
J I
r
/ Ar
N / A
Л44* Cl//
/ ° /л
1 lfoTf
fiij//
Sn
1 1 1 L
Ш период
Ш период
F период
Ш период
1 2 3
5 6 7 8
N группы
Рис. 14.19. Первая энергия ионизации атомов
S-, р-элементов
V периода скачок исчезает, рассматриваемая особенность
сказывается только на наклоне линий, соединяющих элементы Sb—
Те—I. В VI периоде влияние наполовину заполненного /?-подуров-
ня на энтальпию ионизации полностью исчезает.
Перейдем к энергиям ионизации всех внешних электронов.
Энергии ионизации атомов s-элементов уже обсуждались, и они
приведены в табл. 14.38 для сравнения. Энергии ионизации при
отрыве всех внешних электронов с образованием положительного
иона с зарядом, отвечающим номеру группы. Эп+ увеличивается
чрезвычайно быстро от лития к неону (рис. 14.20). Эти энергии
ионизации возрастают равномерно в отличие от первых энергий
ионизации. Резко отличаются элементы I периода — водород и
гелий — от остальных и элементы II периода от элементов ниже-
501
лежащих периодов. Сходство элементов II периода с
соответствующими элементами других периодов заключено только лишь в
валентных состояниях и внешнем виде формул образуемых ими
соединений, но никак не в свойствах самих элементов и их
соединений.
ЛН ~ г,а -зп + -нал/моль
25 000 000
20 000000
15 000 000 Ь
10 000000
5 000 000 Ь
Я период
gv Ш период
8* Ш период
• в* У период
Rn
Шпериод
Рис. 14.20. Энергия ионизации наружных
электронов атомов s- и /?-элементов
Образование ионов в обычных химических процессах
возможно при условии, что энергия ионизации не превышает
500 000 кал/г-атом. В I группе все элементы способны
образовывать однозарядные положительные ионы. Ионы водорода не
существуют в свободном состоянии из-за крайне малого размера
(ядро атома). Во II группе бериллий уже не образует ионов, а
магний их образует, но только в кристаллической решетке с
очень сильными окислителями (хлор, фтор). Остальные элемен-
502
Таблица 14.38
Перио;
I
II
III
IV
V
VI
Э—пе—Эп+, А^иониз' кал/г-атом
№ группы, п
I
313 700
Li+
124 300
Na4-
118 500
К+
106 100
Rb +
96 400
Cs +
89 700
н
Не2+
1 822 000
Ве2+
635 000
Mg2 +
523 000
Са2+
414 700
Sr2+
385 600
Ва2+
364 000
ш
ВЗ+-
1 645 000
А13+
1228 000
Ga3+
I 319 000
In3+
1 284 600
Т]3 +
1 298 000
IV
С4+
3 413 000
2 377 000
Ge4+
2 392 000
Sn*+
2 280 000
Pb4+
21 54 000
V
, N5 +
6 151 000
P6f
4 076 000
As5+
3 920 000
Sb6 +
3 644 000
Bi5+
3 540 000
VI
06+
9 986 000
S6+
6 375 000
Se6+
5 920 000
Te«+
5 669 000
p06+
5 233 000
VII
p+
15 194 000
Cl7 +
9 424 000
Br7+
8 485 000
I?+
8 358 000
At7+
7 371 000
VIII
Ne8+
2 647 400
Ar8+
1 332 300
Kr8+
1 172 000
Xe*+
1 116 000
Rn84-
100 000
ты, начиная с кальция, способны образовывать двухзарядные
ионы не только в кристаллических решетках, но и в водных
растворах. Не один из /^-элементов не способен образовывать ионы
с зарядом, отвечающим номеру группы, за исключением,
вероятно, индия и таллия.
Сродство к электрону, эб
J 1 1 1 1 1 i '
2 4 б 8 10 12 14 16 18
N
Рис. 14.21. Сродство к электрону атомов
I—III периодов
Кристаллические решетки таких соединений, как А1С13, SnCl4,
РЬСЦ, — это не ионные решетки, а решетки с большим вкладом
ковалентной связи в ионную.
Сродство к электрону вдоль периода изменяется очень
неравномерно (рис. 14.21). В I, II и III периодах сродство к электрону
503
отрицательно только у пяти элементов — гелия, бериллия, азота,
неона и магния. Только один азот из р-элементов имеет
отрицательное значение сродства к электрону. Максимальное сродство
к электрону у галогенов, минимальное — у инертных газов.
И у отрицательных и у положительных ионов эффективные
заряды на атомах намного ниже тех зарядов, которые обычно
пишутся в соответствии с валентным состоянием элемента. Так,
во фториде натрия заряды на катионе и анионе составляют по
| Cs
Л 1 I I I 1 1 L
1 2 3 4 5 6 7 8
N
Рис. 14.22. Радиусы атомов s- и /?-элементов
0,93 е, а у йодида натрия понижаются до 0,71 е. В оксиде магния
MgO ионы несут заряд по 1,20 е.
Радиусы нейтральных атомов элементов главных подгрупп
изменяются различным образом в зависимости от номера
периода, но всегда, за исключением галлия (рис. 14.22), любой
нижестоящий элемент имеет больший размер. У элементов II и III
периодов размеры атома равномерно уменьшаются при переходе
от I к VII группам, причем не наблюдается какого-либо резкого
скачка в размерах атомов между s- и /^-элементами. У элементов
IV—VI периодов ход кривых совершенно иной. Атомы элементов
III группы обычно имеют минимальные размеры, а радиус атома
галлия даже меньше радиуса атома алюминия, что объясняется
504
заполненным d-подуровнем атома галлия. При переходе от III к
VIII группе радиусы атомов элементов IV—VI периодов снова
возрастают. Объяснение этого явления заключено в имеющихся
у этих атомов заполненных d-оболочках.
Стремление к образованию положительно заряженных ионов
с зарядом, отвечающим номеру группы, вдоль периода падает.
Рис. 14.23. Радиусы положительно
заряженных ионов s- и /7-элементов
V период
№ период
'7+ Ш период
6 7
N группы
Так, в водном растворе могут существовать только ионы s-эле-
ментов. У р-элементов только элементы III группы и только
периодов с большими номерами способны образовывать ионы М3+,
которые даже в кристаллической решетке не обладают чисто
ионными связями. Тем не менее размеры многих положительно
заряженных ионов известны и их зависимость от номера группы
и периода представлена на рис. 14.23. Здесь мы наблюдаем
закономерное уменьшение радиусов ионов вдоль периода и их
увеличение вниз по подгруппе. Несколько более сложно изменение
радиусов элементов IV периода. Это объясняется появлением,
начиная с галлия, на предвнешнем уровне заполненного d-подуров-
ня. У элементов нижележащих периодов влияние ^-подуровня на
размер иона сказывается слабее.
Способность к образованию отрицательно заряженных ионов
с зарядом, равным разности (8 — № группы), возрастает при
движении вдоль периода. Такие ионы образуются только среди
неметаллов. Хотя радиус иона С4- вычислен, углерод в таком со-
505
стоянии не существует, а образует лишь ковалентные связи. В
нитридах связь между металлом и азотом только частично ионная.
В оксидах доля ионности связи возрастает, но сохраняется
преимущественно ее ковалентный характер. Только элементы
VII группы — галогены — способны образовывать ионы Г" как в
водных растворах, так и в кристаллах.
Зависимость радиуса иона при движении вдоль периода
сложна (рис. 14.24). В II и III периодах в рядах С4" — N3"—О2-— F~
и Р3~ — S2" — С1~ размеры ионов падают, а в IV и V периодах в
том же направлении
размеры ионов изменяются очень
незначительно или же
возрастают. И в этом случае
различный ход
зависимостей размеров ионов в
периодах может быть
объяснен появлением у ионов IV
и V периодов заполненного
d-подуровня.
Xе V период
ТГг
ГУ период
д7 Ш период
и период
7 в
N группы
Рис. 14 24. Радиусы отрицательно
заряженных ионов некоторых
р-элементов
Все /?-элементы способны образовывать двухатомные
молекулы Э2, различающиеся по устойчивости. Наиболее устойчивые
молекулы Э2 образуют элементы И периода — N2, 02 и F2.
Устойчивость молекул возрастает при переходе от III к IV и V
группам, а затем падает к VIII группе. У большинства элементов при
движении вниз по подгруппе прочнос1ь молекул падает, за
исключением подгруппы галогенов (аномальные свойства фтора).
Характер изменения свойств молекул Э2 вдоль периода
(энтальпии диссоциации, длины связей, магнитные характеристики)
объясняется наилучшим способом методом молекулярных орбита-
лей. На рис. 14.25 представлены энергетические диаграммы
молекул Э2 II периода (s-элементы приведены для сравнения).
Прочность химической связи возрастает с увеличением числа
связывающих электронов и падает с увеличением числа
разрыхляющих электронов. Молекула N2 характеризуется общим
количеством электронов, равным 10, что соответствует шести
связывающим электронам на трех 2р-орбиталях и четырем взаимно
компенсирующим друг друга связывающим и разрыхляющим
электронам на двух 25-молекулярных орбиталях. Десятиэлектронные
молекулы двух различных элементов одного и того же периода
также характеризуются наибольшей прочностью.
На рис. 14.26 представлены энтальпии диссоциации всех
возможных сочетаний двух атомов между собой элементов II перио-
506
да. Среди молекул Э—N наиболее устойчива молекула N2
(10 электронов), среди молекул Э—С и Э—О наиболее прочна
молекула СО (10 электронов), среди молекул Э—Li также
наиболее устойчива молекула LiF (10 электронов). Среди других
типов двухатомных молекул прочность их повышается по мере
приближения к 10-электронной структуре, такова молекула BeF
в ряду соединений бериллия, BF в ряду соединений бора и др.
Молекула
разр
разр *разр
«С
<-связ ,_связ
.СВЯЗ
2s
количество
с 6я 36/ б ающих
\злектронов
количество
{разрыхляющих
\злектронов
избыток
\с вязы ваюилих
электронов
дНдисс»
I кал/поль
Длина связи
А
{количество
шеспаренных
Ьлектронов
Магнитные
\сбойства
молекулы
U2 I
н
2
0
■ 2
\?5500
2.672
0
Щиамаг-
шитная
**2
н
2
2
0
(7 или
13000
оо(2)
0
-
~*Г]
^
2
2
65500
/,590
2
транаг
\нитная
"VI
н
б
2
4
И4Ш
1,242
0
Шиамаг-
шитная
~*А
5
2
б
I225067
/.0975
0
| Дианаг
шитная
02 [
4 +
4+
Н !
Ц
8
4
4
\117970
1,207
2
Шрамаг
\нитная
1
F2
4+Н
в
б
2
37000
1,418
0
\Лиамаг
шитная
"ёЛ
4+4+I
4+
в
8
0
0
ОО
0
-
Рис. 14.25. Свойства двухатомных молекул элементов II
периода
Та же закономерность наблюдается и среди двухатомных
молекул s- и р-элементов разных периодов. На рис. 14.27
представлены энтальпии диссоциации двухатомных газовых молекул
гидридов элементов II и III периодов. Галогеноводороды благодаря
их восьми электронным структурам наиболее прочны.
Распределение электронных плотностей в двухатомных гидридах
элементов II периода см. на рис. 14.1. Все двухатомные молекулы
гидридов, как это видно из диаграмм распределения электронной
плотности, полярны. В гидриде лития из-за более
электроотрицательного характера водорода по сравнению с литием возникает
507
с электронами, смещенными в сто-
д И ,кал/моль Лп
дисс IU
СО
частично ионная связь LifH
рону протона.
В гидриде бериллия электронные плотности валентных
электронов распределены примерно одинаково между двумя атомами.
В гидриде бора явно чувствуется смещение электронной
плотности в сторону бора. Однако по
мере продвижения к фтору не
наблюдается дальнейшего
смещения электронной плотности
в сторону элемента, и ионная
связь не возникает. Это
объясняется увеличением размера
атома элемента и количества
электронов на наружном
уровне. Поэтому взаимодействие с
протоном все слабее изменяет
электронную плотность атома
элемента, и в молекуле HF
протон так слабо влияет на
электроны фтора, что
молекула HF по существу сохраняет
форму атома фтора.
Это подтверждается и ди-
польными моментами гидридов
(рис. 14.28). Гидрид лития
имеет наибольший дипольный
момент среди гидридов II
периода. На диаграмме
электронной плотности молекулы
отчетливо замечается деление
молекулы на два атома —
750000
200000
150000
100000
50000\-
-
-
-
-
hLiF Bef
I BeOJ
LLiO /
[ /Вей
Tun/
!L'fl
/ \ N
bf/ / \
ВОД / W
//MA/
// r\
2 / 1
AboWf
:n\/\1
Ano 11
VN0 \ 11
\ V,fU
\ \ япВеР
/bcY Be0JACF\\l
' L ЛВе^л\ \
BeN/6b / \ / /\\ 111
/B2/Be/\
if /NF\
/ / LiH
' /Вев/
e&/ Уис
/Xlib
ЪВе
J NFU 11
/\of\\ \j
2 Wl
i -i ■ V
5" 6 7
N группь
Ne3 рис. 14.26. Энтальпия диссоциации
двухатомных молекул s- и р-эле-
ментов
связь между атомами преимущественно ионная. Дипольный
момент молекулы гидрида бериллия почти равен нулю. Молекулы
гидридов р-элементов имеют примерно одинаковые дипольные
моменты, и в газовых молекулах связь между атомами элемента и
водородом носит в основном ковалентный характер. У гидридов
р-элементов дипольные моменты меньше и незначительно
возрастают от А1Н к НС1. В то же время гидриды s-элементов III
периода имеют большие дипольные моменты по сравнению с
элементами II периода.
При переходе вниз по подгруппе среди s-элементов
межатомные расстояния Э—Н в гидридах возрастают (рис. 14.29).
Особое
бенно силен рост межатомного расстояния в II—IV периодах.
У р-элементов характер изменения межатомных расстояний
сильно отличается. От ВеН к А1Н межатомные расстояния
увеличиваются, но у GaH оно падает, затем несколько возрастает,
сохраняясь постоянным у InH и Т1Н. Заполненный d-подуровень у
галлия слабо защищает наружные электроны от действия ядра,.
ЛНДиСС'КаЛ/МаПЬ
ЮО000 h
50000Ь
1 2 3 4 5 6 7 8
N группы
Рис. 14.27. Энтальпия диссоциации
двухатомных молекул гидридов
элементов II и III периодов
<-2,0
О
-2.0
-4,0
-6,0
-8,0
^р.дебай
вн СН NH OH^f
на
/ biH _^f~
Me
J I i i i i i
1 2 3 4 5 6 7
N группы
Рис. 14.28. Дипольные
моменты двухатомных
молекул гидридов
элементов II и III
периодов
и увеличение числа протонов в ядре на 10 не уравновешивается
увеличением числа электронов на то же число. В дальнейшем
при переходе вправо по периодической таблице в подгруппах
влияние d-электронов ослабляется, и у галогенов межатомные
расстояния Н—F возрастают от HF к HAt.
Наиболее ярко выраженной способностью участвовать в
образовании водородных связей обладают р-элементы II периода:
азот, кислород и фтор. Элементы III и нижестоящих периодов
утрачивают такую способность. Температуры плавления воды,
аммиака и фторводорода аномально высоки по сравнению с
температурами плавления аналогичных гидридов нижележащих
элементов. То же самое касается и температур кипения. В
молекулах NH3, H20 и HF азот, кислород и фтор имеют соответственно'
одну, две и три орбитали, заполненные парами электронов.
Именно они и ответственны за образование водородных связей. В мо-
509
лекуле СН4 таких электронов нет, и метан не способен
образовывать ассоциаты молекул, объединенных водородными связями.
Метан плавится и кипит при очень низких температурах. Хотя в
молекуле ВН3 также нет не участвующих в связи пар
электронов, тем не менее молекула
ВН3 в свободном виде не
существует. Бороводороды
содержат от 2 до 20 атомов
бора в молекуле. В
простейшем — диборане В2Нб —
две молекулы ВН3 связаны
не обычной водородной
связью, а так называемыми
трехцентровыми мосгиковы-
ми связями.
В молекулах гидридов
/7-элементов . углы между
связями Э—Н, как -правило,
Рис. 14.29. Межатомные
расстояния в двухатомных молекулах
гидридов 5- и /7-элементов
Таблица 14.39
уменьшаются при переходе вниз по подгруппе. Особенно сильно
отличаются гидриды элементов II периода от нижележащих
(табл. 14.39).
По-видимому, это вызвано ослаблением способности
валентных электронов гибридизироваться с ростом числа электронных
уровней. Так, в воде кислород находится в
состоянии 5/?3-гибридизации, а строение
молекулы H2S доказывает отсутствие
гибридного состояния серы. Диалогичная картина
наблюдается и в углах между связями в
структурах простых веществ /7-элементов.
Сходство элементов II периода с
элементами нижестоящих периодов
заключается в основном только в строении
внешних электронных уровней и тех валентных
состояний, которые возникают по числу не-
спаренных электронов у невозбужденных атомов. Очень велико
отличие бора, углерода и особенно азота от остальных элементов
их групп.
Литий и бериллий — металлы. Первый образует ионы Li+,
существующие и в растворе, и в твердых солях. Бериллий не
образует ионов Ве2+. Металлические свойства бериллия значительно
слабее. Бериллий менее реакционноспособен по сравнению с
литием. У бериллия возникает способность к образованию ковалент-
Гидрид
НХ>
H2S
H2Se
НоТе
.'НЭН, °
105
92
91
90
510
ных связей. Металлические свойства бора настолько слабы, что
его обычно уже считают неметаллом. Ион В3+ не существует.
В своих соединениях бор трехковалентен. Углерод —
неметалл, во всех своих соединениях углерод образует ковалентные
связи, причем в большинстве соединений углерод 4-ковалентен.
Азот как простое вещество — очень инертный газ вследствие
высокой энергии тройной связи в молекуле N2, но в атомарном
состоянии и при высоких температурах азот вступает в реакции
с очень многими элементами. В своих соединениях азот 3-кова-
лентен благодаря трем неспаренным электронам и
невозможности распаривания наружных s-электронов. Соединения с
металлами — нитриды — в основном ковалентные соединения. Вопрос
о существовании нитрид-иона N3~ окончательно не разрешен, иу
по-видимому, все нитриды ковалентны с некоторым вкладом
ионной связи, зависящим от природы 'катиона.
Существование иона кислорода О2- в кристаллах оксидов
металлов также часто оспаривается, но доля ионности в оксидах
значительно выше, чем в нитридах, и зависит в сильной степени
от природы катиона. Фтор значительно реакционнее кислорода.
В соединениях фтор образует как ковалентные, так и ионные
связи, ион F" существует, а в соединениях с ковалентными
связями фтор вносит значительный ионный вклад в связь. Неон не
обнаруживает никакой склонности вступать в химическое
взаимодействие даже с самыми реакционноспособными веществами.
Хотя простые положительные или отрицательные ионы у
большинства р-элементов в чистом виде не существуют, тем не менее
часто пишут реакции с их участием. Например, пишут реакции,
проходящие в водных растворах с ионами Al3+, Sn4+, Pb4+ и т. п.
Однако написание реакций с участием ионов Э+п чисто условное,
так как все положительно заряженные ионы р-элементов в
водном растворе сильно гидратированы. Реакции равновесий
диссоциации, гидролиза, перехода иона из осадка в раствор и другие
записываются с участием не гидратированных ионов с одной-
единственной целью — экономии времени при расчетах
концентраций ионов водорода или гидроксила (рН раствора) или
вычисления концентрации элемента в растворе. Ионы С4", N3~,02_ не
могут существовать в водных растворах, только один фторид-ион
F~ устойчив в воде, хотя и подвергается гидролизу.
При переходе от верхнего периода к нижним сохраняются все
типы химических связей, характерные для соединений верхнего
периода, и появляются новые типы химической связи, не
наблюдающиеся у соединений верхних периодов. В том же направлении
возрастает как склонность элементов образовывать комплексные
соединения (их прочность), так и разнообразие их типов
(количество различных координационных частиц). В I периоде, в
соединениях водорода, реализуется только а-связь и координационное
число 2, например в [F—Н—F]~. В соединениях элементов II
периода участвуют а- и л-связи, по отдельности или совместно.
511
Координационные числа элементов II периода равны 2,3 и 4.
В соединениях элементов более низких периодов кроме а- и
л;-связей появляются б-связи и другие еще более сложные типы
связей, а координационные числа элементов, сохраняя прежние
значения, повышаются до 5, 7, 8 и даже до 12.
У р-элементов при переходе сверху вниз устойчивость
максимальной положительной степени окисления уменьшается и
возрастает устойчивость низших положительных степеней окисления
(углерод +4, свинец +2, или алюминий +3, таллий +/).
Физические свойства простых веществ р-элементов
чрезвычайно различны. Среди них есть вещества, плавящиеся и кипящие
при очень низких температурах (неон, аргон) и при очень
высоких (бор, углерод). Характер изменения свойства по периоду или
по подгруппе у р-элементов часто бывает сложным,
сопровождается скачками и не всегда может быть объяснен строением
электронных уровней атомов, типом химической связи и
координационным числом атома.
В структурах простых веществ при продвижении вниз по
подгруппе обычно наблюдается уменьшение углов между связями.
Это явление было отмечено ранее среди гидридов VI группы
Н20—H2S—H2Se—Н2Те. В табл. 14.40 приведены углы между
связями в структурах простых веществ элементов V и VI групп.
Таблица 14.40
V группа элементов II VI групп* элементов
Вещество
Фосфор
Мышьяк
Сурьма
Висмут
угол между
связями
104°30'
97°
95°35'
95°29'
вещество
Кислород (в 03)
Сера (в S8)
Селен
Теллур
угол между
связями
117°
108°
102°50'
101°46'
Описываемая закономерность, по-видимому, может быть
объяснена или ослаблением стремления к гибридизации валентных
электронов при увеличении размеров атомов или же
уменьшением жесткости связи с увеличением размера атома, что
позволяет связям менять взаимное положение и направление при
сохранении одного и того же валентного состояния атома.
Рассмотрим изменение температур плавления и кипения элементов вдоль
периода совместно с s-элементами для сравнения (рис. 14.30). У элементов
I группы температуры плавления при переходе от лития к цезию закономерно
ладают, указывая на ослабление связи между атомами в кристаллической
решетке одного и того же типа. У элементов II группы такая закономерность
нарушается в связи с различными структурами. В III группе элементов
температуры плавления изменяются также не закономерно, падая от бора к
алюминию, затем следует таллий, индий и галлий. Похожая картина наблюдается в
IV и V группах, что вызвано различными структурами простых веществ.
Однако с VI группы наблюдается равномерное повышение температур плавления
512
4000
3000
элементов с ростом размеров атомов. Это вызвано усилением межмолекулярного
взаимодействия в структурах твердых фаз простых веществ при переходе вниз
по подгруппе.
Во II периоде в I—IV группах самые высокие температуры плавления, а
в V—VIII группах — самые низкие. Температуры плавления возрастают от
лития к бериллию, бору и углероду. У углерода максимальная температура
плавления. В кристаллической структуре лития металлическая связь между
атомами осуществляется только
одним 5-электроном от каждого а
тома. У бериллия тот же тин связи,
но каждый атом бериллия
способен внести в связи вклад двумя
электронами. Связь прочнее, чем
у лития, благодаря усилению
металлической связи и ковалентного
связывания между атомами.
Кристаллический бор уже не
металл. В его структуре связь
между атомами осуществляется
тремя электронами s2p\ гибриди-
зирующимися до я/Асостояния.
Связь преимущественно ковалент-
ная, прочная. В этом причина
более высокой температуры
плавления.
У углерода (алмаз) каждый
атом отдает в связь между
атомами кристаллической структуры
4 электрона (s2p2), которые
благодаря sp3-гибридизации образуют
структуру, в которой от каждого
атома исходят под равными
углами 4 гибридные орбита ли. В
результате образуется одна из
самых прочных в природе
кристаллических структур Температуры
плавления кремния и германия
также самые высокие среди эле-
2000
1000
Рис. 14.30. Температура плавления
s- и р-элементов
6 7 8
/V группы
ментов их периодов, но ниже, чем у углерода из-за больших межатомных
расстояний.
После углерода температуры плавления чрезвычайно резко падают. В
твердых азоте, кислороде и фторе сохраняются структуры двухатомных молекул,
связь между которыми очень слаба из-за почти полного израсходования
валентных сил на связь между атомами в молекуле. Молекулы связываются друг с
другом слабыми силами Ван-дер-Ваальса. Эти силы возрастают с увеличением
размеров атомов и среди элементов VI и VII групп температуры плавления
повышаются при переходе сверху вниз (в отличие от элементов I группы). Атомы
инертных газов (VIII группа элементов) имеют полностью заполненные р-обо-
лочки, образующие очень прочные, крайне не активные электронные системы.
В структурах твердых инертных газов атомы (одиоатомные молекулы) связаны
между собой также слабыми силами Ван-дер-Ваальса. У р-элементов VIII
группы самые низкие после гелия и водорода температуры плавления среди простых
веществ.
IT Общая химия
513
Температуры плавления и энтальпии плавления — периодические функции
атомного номера элемента. Раньше предполагали, что функция,
получающаяся делением двух периодических функций, не должна быть периодической,
и считали, что энтропия плавления простого вещества не зависит от положения
элемента в периодической таблице Менделеева и равна примерно
2,5 кал/моль-град. Однако это значение ASnn действительно близко к энтропиям
плавления всех переходных (d- и /-) металлов. У 5- и ^-элементов энтропии
плавления очень сильно различаются между собой (рис. 14.31). Минимальные
Щ
80 Ь
w ь
4,0
2,0
0.0
л а ,кал/г атом град
пл
J L
Кг Sn<
Хе
Rb In Cs
J 1 , 1 L
Af
Rn
i
[
Ra
nJPb "F
JL
0 10 20 30 40 50 60 70 SO WO z
Рис. 14 31. Энтропия плавления 5- и ^-элементов
энтропии плавления у щелочных металлов. Именно к ним, а не к галогенам
приближается водород. У щелочноземельных металлов энтропии плавления
несколько выше. Низкие энтропии плавления s-элементов вызваны тем, что при
переходе из твердого в жидкое состояние координационное число элемента i?
концентрация свободных электронов сохраняются постоянными.
От бора к алюминию и галлию энтропии плавления возрастают из-за
изменения типа связи в твердом и жидком состояниях. Еще выше энтропии
плавления элементов IV группы. Высокие значения энтропии плавления углерода,
кремния и германия обусловлены разрушением при плавлении системы прочных
связей с увеличением координационного числа с 4 до 8 и переходом в
металлическое состояние, сопровождающимся ростом концентрации свободных
электронов.
У мышьяка, сурьмы и висмута — элементов V группы — энтропии
плавления также повышены, что объясняется изменением концентрации свободных
электронов при плавлении. Об этом свидетельствует также изменение
электропроводности при переходе из твердого состояния в жидкое Однако у
фосфора самое низкое среди простых веществ значение энтропии плавления и
вызвано это лишь некоторым ослаблением не прочных ван-дер-ваальсовых сил
между молекулами Р4. При плавлении фосфора образуется молекулярная
жидкость, изменения в концентрации свободных электронов, в типе связи и в
514
координационном числе не наблюдается. Точно теми же причинами
объясняется очень низкое значение энтропии плавления у серы, так как несмотря на
плавление в основном сохраняется молекулярная структура колец S8. У
кислорода энтропия плавления выше, что объясняют переходом кристалла в
жидкость, состоящую из молекул О2 и 03.
У селена энтропия плавления еще выше Хотя при плавлении селена тип
связи не меняется, но образующаяся жидкость содержит не только
кольцеобразные, но и цепеобразные молекулы Se8 и более длинные. При плавлении
теллура образуется большой набор различной длины цепей. Это, а также
диссоциация молекулы Те2 на атомы приводит к сильному изменению в составе
вещества при плавлении и к высокому значению энтропии плавления теллура.
Причина максимальных значений энтропии плавления галогенов (VII
группа) из-за отсутствия данных о составе и строении твердой и жидкой фаз пока
не объяснена. Энтропии плавления инертных газов (VIII группа) ниже по
сравнению с галогенами, но достаточно значительны, и объяснение этого,
по-видимому, лежит в разрушении очень слабого частичного перекрывания электронных
облаков соседних атомов кристалла инертного газа.
При изучении s- и р-элементов обнаруживается интересная и
важная особенность: диагональное сходство элементов. Энергии
ионизации увеличиваются по периоду слева направо, а в группе
снизу вверх. Это приводит к тому, что в диагональном
направлении в периодической системе будут находиться атомы с примерно
близкими энергиями ионизации, что, в свою очередь,
обусловливает сходство элементов и их соединений в отношении
физических и химических свойств (рис. 14.32). Если обратиться к
радиусам положительных ионов с зарядами, равными номеру груп-
лы (табл. 14.41), то по размерам ионов можно обнаружить, что
Таблица 14.41
о
я
с7
II
III
IV
V
VI
I
Li+
0,68
Na1
0,98
К+
1,33
Rb+
1,49
Cs+
1,65
II
Ве2+
0,34
Mg2+
0,74
Са2+
1,04
Sr2+
1,20
Ва2+
1,38
Яэп+, ?
№ группы
III
В3"
0,20
А18+-
0,57
Ga3+
0,62
In3+
0,92
Т1з+
1,05
IV
с44-
0,17
Si4^
0,39
Ge4^
0,41
Sn4+
0,67
Pb4+
0,76
1
n
V
N*f
0,15
p5~
0,35
As*+
0,47
Sb5+
0,62
Bi&+
0,74
VI
Se-
0,29
Se«^
0,35
Te°b
0,56
VII
Cl7f
0,26
Br7'
0,33
v+
0,50
ион Li+ ближе всего стоит к HOHyMg24" или ион Mg2+ к иону Ga3+
и т. д.; замечается сходство элементов по диагонали в ряду:
Li+—Mg2+—Ga3+—Sn4+—Bi5+. Радиусы ионов Р5+ и Se6+ равны.
Ион натрия ближе к иону кальция, а не к своим
непосредственным соседям по группе и периоду.
17*
515
Сходство элементов из соседних групп проявляется и в их
химическом поведении. Так, литий схож с магнием. Это видно в
реакциях образования нитридов и карбидов, в близких раствори-
мостях солей, способности реагировать с рядом органических
соединений и образовывать литийорганические и магнийоргани-
ческие соединения и т. п.
Первые энергии ионизации
э - е « э+, дН, эв
Г Р У
ы
1 Л Ш IF Y Ш Ш
\\> ЧВ \Vn \) V
K5.3S\&32 8,30 11,2&\]4,54 Щ11Щ
^Na >1g4NAl 4Si V^OS ЧС1
.5,14 7,64\&8 $15 10,55\Ю36 Щ
К Са Ч6а\Ье 4As 4SeV4Br
4.34 б, 11 £,0ГО5б 9,8? 9,
"Rb Sr In SnVSb Че
4,18 5,69^ $7{ 7,3$\Ш 9,01 11,4
Рис. 14 32. Диагональное сходство среди
s- и /?-элементов
Средняя энергия
ионизации
эв
17,4
13,3
12,2
Щ7
8,9
8,0
6,7
6 J
5,0
З.Э
кал/г атом]
401000
307000
281000
247000
205000
185000
155000
141000
125000
115000
90000
Свойства элементов
Неметаллы
Окислите ли.
Оксиды кислотного
характера
Промежуточное
положение между
металлами и
неметаллами.
Восстановители.
Металлы
Восстановители
Оксиды основного
характера.
Сходство бериллия и алюминия проявляется в способности
обоих металлов растворяться в щелочах с выделением водорода
и образованием амфотерных гидроксидов. Гидроксиды бериллия
и алюминия, растворяясь в щелочах, дают соответственно берил-
латы и алюминаты. И бериллий и алюминий не растворяются в
азотной кислоте, покрываясь защитной окисной пленкой. Хотя
энтальпии образования оксидов ВеО и А1203 имеют высокие
отрицательные значения, металлы на воздухе не горят, покрываясь
защитной пленкой оксидов. Безводные галогениды бериллия и
алюминия склонны к димеризации посредством образования мо-
516
стиковых связей М—Г—М. Безводные галогениды бериллия и
алюминия используются в качестве катализаторов при
органических синтезах. Термическая устойчивость солей бериллия и
алюминия в ряде случаев близка.
Бор наиболее близок кремнию, а не углероду или алюминию.
Это проявляется хотя бы в свойствах оксидов Si02 и В203,
способных, в частности, образовывать стекла, в свойствах борной и
кремниевой кислот (эти кислоты очень слабые и в высшей
степени склонны к образованию поликислот). Точно так же можно
предсказать и доказать сходство фосфора с селеном, мышьяка с
теллуром.
Обращает внимание характерная особенность s- и р-элемен-
тов, особенно последних, заключающаяся в очень сильном
отличии элементов II периода от элементов нижестоящих периодов, в
заметном сходстве элементов III и IV периодов и вновь
проявляющемся отличии элементов IV периода от V. Известно, что
явление, о котором идет речь, называется вторичной
периодичностью. Она не имеет еще окончательного и строгого объяснения,
а примеры ее выявлены не все. Вторичная периодичность в
свойствах соединений элементов является следствием вторичной
периодичности в свойствах свободных атомов, наиболее ярко
выражающейся в периодическом изменении энергий ионизации при
движении по подгруппе сверху вниз. Вторичная периодичность
сказывается и на характере изменения размера атома в группе
элементов.
При переходе от III периода к IV вследствие появления
первого заполненного d-подуровня и при переходе от V периода к VI
в результате появления второго заполненного d-подуровня и
заполненного jT-подуровня наблюдается замедление в увеличении
атомных радиусов в направлении сверху вниз. Это приводит к
периодическому изменению размеров атомов в группах. Наиболее
четко вторичная периодичность проявляется в соединениях
элемента, имеющего валентное состояние, отвечающее номеру
группы. Вторичная периодичность почти не обнаруживается у s-эле-
ментов (I и II группы), незначительно проявляется в III группе
элементов, но хорошо заметна в более высоких группах.
Часто обнаруживается, что элементы нечетных периодов в
какой-то мере более похожи друг на друга, как и элементы
четных периодов. Так, фосфор и сурьма схожи между собой и
отличаются от мышьяка и висмута, сходных, в свою очередь, между
собой. Энтальпии образования оксидов фосфора и сурьмы
отличаются в большей степени от энтальпий оксидов мышьяка и
висмута, чем между собой.
Подобное можно сказать и о сере и теллуре, кислороде и
селене, фторе и броме, хлоре и йоде. Сера и теллур образуют
несколько устойчивых оксидов: S02, S03, ТеО, Те02, Те307, Те03
и др. У селена только один оксид устойчив — Se02 (оксид Se03
очень неустойчив). Оксиды фтора и брома могут быть получены
517
с трудом, они малочисленны и неустойчивы, но хлор и йод
образуют целый ряд оксидов, значительно более устойчивых.
Влияние d-электронов (и отчасти f-электронов) на свойства
элементов главных подгрупп (s- и р-элементов) очень
разносторонне. Элементы II периода имеют только s- и р-подуровни и
отличаются от элементов III периода тем, что у последних
появляется незаполненный Зй-подуровень, способный принимать
электроны при их возбуждении с 3s- и 3/?-подуровней. В этом
отношении элементы III и IV периодов похожи, но у элементов IV
периода, начиная с галлия, предвнешний З^-подуровень полностью
заполнен и имеется пустой 4/-подуровень. Сходство
элементов IV—VI периодов было бы весьма полным, если бы в VI
периоде, начиная с таллия, не появлялся заполненный
^-подуровень.
Энергии связей в двухатомных молекулах щелочных металлов
малы и убывают вниз по подгруппе закономерно, у двухатомных
молекул галогенов энергии связи велики и изменяются не
закономерно. Такие же аномалии наблюда-
Таблица 14.42 ются и в энергиях одинарных связей
Э—Э в V и VI группах элементов
(табл. 14.42). Энергии одинарных
связей элементов II периода меньше, чем
у элементов нижележащих периодов.
Одинарные связи Э—Э у всех
щелочных металлов, а также у кислорода,
азота и фтора обладают невысокой
прочностью, в то время как
аналогичные связи у фосфора, серы, хлора и
находящихся под ними элементов
значительно более прочны.
Объяснение этого заключено в электронной структуре
рассматриваемых атомов, а именно в d-подуровне. У фосфора, серы,
хлора и соответствующих нижележащих элементов помимо
наружных s- и р-подуровней имеются свободные d-подуровни,
которых нет у элементов II периода (кислород, азот, фтор). У
элементов III и нижестоящих периодов помимо связей валентными
электронами происходит дополнительное взаимодействие
электронов одного атома с пустыми d-орбиталями другого атома. При
этом в одинарных связях может появиться значительная
дополнительная кратность связи, приводящая к упрочению связи в
целом. Поэтому истинные одинарные связи Э—Э осуществляются у
щелочных металлов, азота, кислорода и фтора, у которых нет
возможности дополнительного взаимодействия за счет d-орбита-
лей. Эти настоящие одинарные связи из-за 5-5- и р-/?-электронов
обладают невысокой прочностью. Обычно рассматриваемые как
одинарные связи Р—Р, As—As, Sb—Sb, S—S, Se—Se, Те—Те,
CI—CI, Br—Br, I—I на самом деле не одинарные, а кратные
из-за участия в них d-орбиталей.
Связь
N—N
jP—P
As—As
Sb—Sb
О—О
s—s
Se—Se
Энергии
одинарных связей Э—Э.
кал/моль
21000
48 000
35 000
29 000
35 000
64 000
41000
518
При изучении s-элементов обращалось внимание как на
различие элементов главных подгрупп I и II групп, так и на
различие элементов внутри подгруппы. У р-элементов эти особенности
выражены значительно резче и различие в свойствах соседних
элементов по периоду или по подгруппе может проявляться столь
сильно, что объединение этих элементов в одно р-семейство часто
оказывается формальным.
Все р-элементы и особенно р-элементы II периода образуют
многочисленные соединения между собой и с s-, d-, /-элементами.
Подавляющее большинство известных соединений — это
соединения с участием р-элементов. Поэтому изучение р-элементов имеет
особое значение в химии.
§ 6. БОР, АЛЮМИНИЙ, ГАЛЛИЙ,
ИНДИИ И ТАЛЛИИ | р-ЭЛЕМЕНТЫ III ГРУППЫ)
Заполнение каждого нового р-подуровня начинается с
элементов, расположенных в главной подгруппе III группы. Все s-эле-
менты как I, так и II групп проявляют значительно большее
сходство между собой как внутри подгруппы, так и между
подгруппами по сравнению с р-элементами III группы. Хотя р-эле-
менты III группы обнаруживают сходство между собой, заметны
глубокие различия в их свойствах. При переходе от бора к
таллию неметаллические свойства изменяются на металлические,
кислотные через амфотерные на основные, связь в соединениях с
ковалентной изменяется на преимущественно ионную.
Аналогичная картина наблюдается и у р-элементов других подгрупп.
Целый ряд свойств как простых веществ, так и соединений
изменяется при переходе вниз по подгруппе неравномерно. Так,
первые энергии ионизации атомов всех элементов, за
исключением бора, очень близки друг к другу. Атомные радиусы в том
же направлении возрастают, причем очень сильно от бора к
алюминию и от галлия к индию. Можно было бы думать, что
меньшему атомному радиусу должна соответствовать более прочная
химическая связь в структуре простого вещества и более высокая
температура плавления. Действительно, у бора максимальная в
подгруппе температура плавления (2300°К), но затем она падает
к алюминию (932°К) и галлию (312°К) и снова возрастает к
индию (429°К) и таллию (577°К). В то же время температуры
кипения от бора к таллию понижаются (4000, 2700, 2500, 2300,
1740°К). Обычно температуры плавления дают указания на
сравнительную прочность кристаллических решеток. Энтальпии
сублимации также характеризуют прочность кристаллических решеток.
Однако если температуры плавления в подгруппе изменяются
неравномерно, то энтальпии сублимации падают от бора к
таллию (135 000 кал/г-атом у В, 77 500 у AI, 65 300 v Ga, 58 000 v In
и 43000 у Т1).
519
Ионный радиус бора неизвестен. Ионные радиусы элементов в
трехвалентном состоянии возрастают от алюминия к таллию
(А13+ 0,45, Ga3+ 0,60, In3+ 0,81, Т13+ 0,95А). Обычно в ряду одина-
ковозарядных ионов энтальпии гидратации с увеличением
размера иона уменьшаются. Однако у Al, Ga, In и Т1 это правило не
соблюдается: энтальпии гидратации их ионов в трехвалентном
состоянии равны соответственно 1121000, 1124 000, 994 000 и
984 000 кал/г-ион.
Энтальпии сублимации и энтальпии гидратации входят в
качестве слагаемых в энтальпию перехода металла в раствор в
виде гидратированного иона и поэтому связаны со стандартными
потенциалами металлов. Несмотря на неравномерность
изменений ДЯСубл и АЯгидр в ряду изучаемых элементов, стандартные
потенциалы их изменяются, так что от алюминия к таллию и
индию отрицательное значение Е° уменьшается (—1,67, -0,52,
—0,34 в), становясь положительным у таллия (+0,72 в). Таллий
не растворяется в кислотах-неокислителях с образованием трех-
зарядных ионов.
Бор — неметалл. Его электропроводность мала и
увеличивается с ростом температуры, что не свойственно металлам. Хотя бор
формально проявляет окислительное состояние +3 в его оксидах
и галогенидах, ион В3+ неизвестен. Бор связывается ковалентно
с неметаллами, а в соединениях с металлами — боридах — он
является акцептором электронов, например в MgB2, А1В2 и др.
Триоксид бора В203—кислотный оксид. При действии на него
воды образуется борная кислота, простейшая формула которой
Н3В03. Это очень слабая, одноосновная кислота. Одноосновность
кислоты раньше объяснялась тем, что в растворе она существует
в виде НВ02, однако ни ионы ВОГ, ни молекулы НВ02 не
обнаружены. Одноосновность борной кислоты лучше всего
объясняется дополнительной гидратацией Н3В03 до состава H5BO4. Эта
кислота диссоциирует как комплексное соединение, в котором
бор как комплексообразователь имеет координационное число 4
и окружен четырьмя лигандами — ионами гидроксила:
Н6В04-Н+ + В(ОН)7.
Поэтому процесс растворения триоксида бора в воде и кислый
характер раствора объясняются процессом
В203,к + 5Н20 = 2B (ОН)47Р-р + 2Н+р.
Кислотное действие борной кислоты иногда приписывают не ее
диссоциации, а гидролизу
В (ОН)3 + Н20 = В (ОН)Г + Н+.
В этой реакции бор проявляет себя как акцептор электронов,
принимая их от ОН".
Анион В03~ в водном растворе также неизвестен. Соли
борной кислоты образуют сложные ионы, например В406(ОН)4~.
520
Этот анион соответствует соли Na2B407 — буре, т. е. анион
В4От~ гидратируется двумя молекулами воды: (В407-2Н20)2~,
Строение тетраборат-иона, как предполагают, таково:
Н
О
/°-В-°\
НО—В
/
\
о
I
-О—В—О
/
>в—он
о
н
С водородом бор образует серию летучих соединений,
названных боранами. Простейший представитель — диборан В2Н6 —
может быть получен реакцией гидрида лития с трифторидом бора:
6L1H -t- 8BF3 - 6LiBF4 + В2Н6.
Нагревание диборана до 300°С приводит к образованию целого
ряда других боранов, объединяемых двумя гомологическими
рядами: BnHn+4 и ВпНп+б, например В4Ню, В5Н9, В5Ни, В6НШ,
Рис. 14.33. Структура молекулы
В2Нб по методу валентных связей
Рис. 14.34. Связывающие
молекулярные орбитали молекулы В2Н6
В9Н15, ВюНм, В10Н16. Соединения ВпНп+4 устойчивее ВпН„+6.
Низшие бораны самозагораются на воздухе и легко гидролизуют-
ся водой до борной кислоты.
Связь между группами ВН3 в диборане — это новый тип связи,
хотя и напоминает водородную связь, но из-за высокой
прочности связи имеет другую природу. Структура В2Н6 по методу
валентных связей изображена на рис. 14.33. Бор находится в
состоянии 5/?3-гибридизации, причем одна из гибридных орбиталей
пуста. Три остальные орбитали перекрываются с атомами
водорода. Пустая орбиталь бора оттягивает на себя часть
электронной плотности от одного атома водорода, связанного с другим
атомом бора. В то же время этот последний атом бора, находясь
521
также в состоянии 5р3-гибридизации, оттягивает часть
электродной плотности от одного из атомов водорода, связанного с
первым атомом бора. Таким образом, между двумя атомами бора
располагаются два атома водорода, которые и связывают атомы
бора. Электронная плотность двух связей В—Н—В
выравнивается и они в молекуле В2Н6 равноценны. Связь типа В—Н—В
называется мостиковой. У мостиковых В—Н—В связей длина
связей больше, а энергии связи ниже, чем у соседних, не
мостиковых В—Н связей во фрагментах ВН2.
На рис. 14.34 молекула В2Н6 представлена так, что св«и
В—Н—В изображены в виде молекулярных орбиталей. От ка;ж.-
дого атома бора к другому исходят две молекулярные орбитали,
по форме напоминающие бананы. В каждой мостиковой связм
слились воедино s-орбиталь атома водорода и по две sp3-op6HTai~
ли атома бора. Каждая мостиковая связь — трехцентровая связь..
Бор очень сильный комплексообразователь. Так как у бора;
есть свободные р-орбитали, то бор в реакциях комплексообразо-
вания ведет себя как акцептор электронов. Например:
BF3 + NH3 - F3BNH3 (F3B +- NH3),
BF, + F- = BFr (F3B+-F-).
В этих реакциях фторид бора принимает пару электронов или!
от NH3 или от F". Свойство галогенидов бора быть акцепторами,
электронов обусловливает их широкое применение как
катализаторов в реакциях синтеза органических соединений.
Алюминий по своим свойствам резко отличается от бора.
Больший размер атома и возможность переходов электронов на
пустой d-подуровень увеличивают свободу электронов, степень их
делокализации, а следовательно, придают элементу большую
электро- и теплопроводность. Алюминий обладает ярко
выраженными металлическими свойствами.
Обычное валентное состояние алюминия 3. Несмотря на
довольно высокое отрицательное значение электродного потенциала
А13р+-Зв-А1к £° = -!,66в,
алюминий в воде не растворяется из-за устойчивой оксидной
пленки.
Координационное число алюминия 6, когда лигандами
являются молекулы воды. В водном растворе, даже в очень кислом,
свободные ионы А13+ не существуют из-за гидратации. Кислая среда
растворов солей алюминия, образованных сильными кислотами,
объясняется диссоциацией иона А1(Н20)е+ по типу
диссоциации слабого электролита:
А1(Н20)36+ р= А1 (Н20)5ОН2р+р +Н+Р.
522
При прибавлении щелочи из-за связывания ионов водорода
равновесие смещается вправо, и диссоциация по второй ступени
усиливается:
А1 (Н20)5 ОН* + 4- ОН-р = А1 (Н20)4 (ОН)£р.р + Н20.
При диссоциации по третьей ступени образуется осадок гидрати-
рованного тригидроксида алюминия.
А1 (Н20)4 (OH)2tp.p -i- ОН-р - Al (H20)3- (ОН)з.к + Н20.
Осадок, имеющий гелеобразный вид, постепенно дегидратируется
и переходит в А1(ОН)3. Процесс дегидратации ускоряется
нагреванием. При сильном нагревании тригидроксид алюминия
отщепляет воду и переходит в оксид А1203:
2А1(ОН)з,к = А1А.к + ЗН2Ог.
При приливании кислоты А1(Н20)3(ОН)з и свежеосажденпый
А1(ОН)3 быстро растворяются:
А1 (Н,0)3 (ОН)3,к L ЗН+Р = А1 (Н20)2+ р,
А1 (ОН)3,к + 3Hptp + ЗН20 = Al (HaO)g+р.
Оксид А1203 в кислотах не растворяется.
При приливании щелочи к осадкам А1(Н20)3(0Н)3 и А1(0Н)3
также происходит растворение их с образованием анионов,
содержащих алюминий в трехвалентном состоянии:
А1 (НаО), (ОН)3.к + ОН-р = А1 (ОН)^р.р + ЗН20,
А1 (ОН)3,к Ч- ОН~Р = А1 (ОН)-4.р.Р.
А1203, особенно не свежеприготовленный, растворяется только в
концентрированных и нагретых растворах щелочей и к тому же
очень медленно:
А120з,к + 20Н~Р Ь ЗНаО = 2А1 (ОН^р.р.
Простой алюминат-ион состава А107 не обнаружен.
Координационное число алюминия в кислой среде 6 (лиганды—вода) и
в щелочной 4 (лиганды — ионы ОН"). Ионы легко взаимно
превращаются при изменении рН раствора:
А1 (ОН)^р.р + 4Н+р - 2Н20 = А1 (Н20)£р\р,
Al (H20)j?+p + 40Н~Р = А1 (ОН)47Р-р 4- 6Н20.
Галогениды алюминия А1Г3 в газовой фазе существуют в виде
смеси мономерных молекул А1Г3 и димерных АЬГв, причем при
низких температурах преимущественно в виде последних. Связь
523
между двумя атомами алюминия осуществляется двумя мостико-
выми связями, как это имеет место в структуре молекулы дибо-
рана ВгНб-
Галлий во многом похож на алюминий. Ему присуще то же
валентное состояние. В растворе ионы галлия ведут себя так же,
как и ионы алюминия. В кислой среде — это ионы Ga(H20)6 ,
в щелочной —Ga(OH)7. В растворах солей галлия,
образованных сильными кислотами, среда кислая. Из-за больших размеров
иона Ga3+ по сравнению с ионом А13+ диссоциация иона
Ga(H20)6+ проходит сильнее:
А1 (Н20)бь - А1 (Н20)бОН2+ + H+ /Сж = 1.1- Ю-5,
Ga(H20)H+ = Ga(H20)5OH2+ -f Н+ Ki - 2,5-10~3.
Сравнение электродных потенциалов алюминия, галлия и индия
А13+ + Зе = А1к Е°= —1,66 в,
Ga3+ + Зе - GaK Е° = — 0,52 в,
1п3+ + Зе = 1пк Е° - — 0,34 в
творит об ослаблении восстановительных свойств металлов при
переходе вниз по подгруппе. Несмотря на большие
отрицательные значения потенциалов галлия и алюминия по сравнению с
потенциалом разложения воды (£° = 0,41 в), оба металла с водой
не реагируют из-за того, что их поверхность покрыта оксидной
пленкой. Индий вообще не способен реагировать с водой (при
комнатной температуре). Все три металла растворяются в
растворах с [Н+] = 1 г-ион/л (стандартное условие).
У индия есть устойчивые при низких температурах
соединения, в которых он одновалентен. У алюминия и галлия такие
соединения обнаружены только при высоких температурах.
Таллий, так как его поверхность не защищена оксидной
пленкой, окисляется на воздухе с образованием Т120з- Однако
1-валентное состояние таллия для него обычно:
Т\1+ + 2е = Т1+р £0 = 1,25 в.
В 1-валентном состоянии таллий и его соли напоминают
соответствующие соединения щелочных металлов.
При переходе вдоль периода ясно замечается следующая
особенность групп: в I и II группах у s-элементов различие в
свойствах элементов внутри подгруппы у щелочных металлов
значительно слабее, чем у щелочноземельных. Различие в свойствах
р-элементов III группы заметно резче по сравнению с
элементами двух предыдущих групп и еще более усиливается в IV группе.
За,тем в V и VI группах степень различия в свойствах /7-элемен-
тов каждой подгруппы почти одна и та же. р-Элементы VII груп-
ЪП
пы уже не столь сильно различаются между собой по сравнению
с /^-элементами VI группы. Наконец р-элементы VIII группы —
инертные газы—единственная подгруппа элементов со столь
близкими свойствами.
§ 7. УГЛЕРОД, КРЕМНИЙ, ГЕРМАНИИ,
ОЛОВО, СВИНЕЦ \р -ЭЛЕМЕНТЫ IV ГРУППЫ^
Хотя в подгруппе элементов С—Si—Ge—Sn—Pb явно
заметна постепенная смена неметаллических свойств на
металлические, свойства элементов изменяются в этом же направлении
неравномерно. Энергии ионизации от углерода к олову
уменьшаются, но у свинца энергия ионизации несколько возрастает.
В то же время атомные радиусы возрастают. Температура
плавления максимальна у углерода (4100°К), затем уменьшается
более чем в 2 раза у кремния (1685СК), далее понижается у
германия (1210°К) и олова (505°К) и несколько возрастает у свинца
(60ГК). Энтальпии сублимации в том же направлении
понижаются, указывая на ослабление связей между атомами в
кристаллических решетках. Температуры кипения также понижаются.
Все эти элементы проявляют только два различных
валентных состояния — 2 и 4. Последнее наиболее характерно для
углерода и кремния и роль его снижается при переходе к свинцу.
Значение валентного состояния 2 в том же направлении
возрастает и у свинца оно становится наиболее устойчивым.
Углерод — совершенно особый элемент, так как способен
образовывать бесконечно большое количество органических
соединений. Причина заключается в целом ряде свойств, отличающих
углерод от остальных элементов. Связь С—С значительно
прочнее связей между двумя атомами других элементов той же
подгруппы (табл. 14.43).
Таблица 14.43 Таблица 14.44
Связь
С—С
Si—Si
Ge—Ge
Sn—Sn
ЛЯсвязи' кал/моль
82 000
53 000
45000
37 000
Связь
С—F
Si—F
С—CI
Si—CI
ЛЯСВЯЗИ' К1Л/М0ЛЬ
110 000
135000
80 000
91000
У углерода велика способность к гибридизации. В состоянии
5р3-гибридизации атом углерода образует 4а-связи с углами
между связями по 109° (метан, этан и весь гомологический ряд
CnH2n+2, все соединения углерода с одинарными связями). В
состоянии $/?2-гибридизации атом углерода образует две одинарные
<г-связи и одну двойную связь 0 + я (этилен, гомологический ряд
СпН2п, все другие соединения с двойными связями). В состоянии
525
s/7-гибридизации от атома углерода исходит одна одинарная
сг-связь и тройная связь а + 2я (ацетилен, гомологический ряд
С„Н2п-2, все вещества, содержащие тройные связи).
Благодаря малым размерам атома углерода все связи с его
участием довольно прочны. я-Связи, возникающие при боковом
перекрывании р-орбиталей, у углерода обладают повышенной
прочностью из-за малых размеров атома углерода и благодаря
этому сильно перекрываются. У других элементов я-связи
намного слабее и даже вообще не образуются. Связь с атомами
водорода у углерода также наиболее прочна (С—Н, 98 000 кал/моль,
Si—H, 75 000 кал/моль).
Интересно, что связи с атомами галогенов у углерода слабее,
чем у кремния (табл. 14.44). Меньшая прочность связей в CF4 по
сравнению с SiF4, вызвана меньшими размерами атома углерода и
возникающему благодаря этому более сильному отталкиванию
атомов фтора друг от друга.
С кислородом углерод образует три оксида: СО, С02 и Сз02-
Монооксид углерода СО — самая прочная из известных
двуатомных молекул благодаря тройной связи С^О, состоящей из одной
0-связи (гибридная sp-орбиталь) и двух я-связей (рх и ру
негибридные орбитали).
Несмотря на высокую прочность связи С = 0, монооксид
углерода легко сгорает, превращаясь в диоксид:
2СОг + 02 = 2С02,Г А Н° = — 125 200 кал/моль.
Реакция проходит самопроизвольно и с выделением большого
количества тепла, потому что сумма энергий всех связей в двух
молекулах СО и одной 02 меньше, чем сумма энергий всех
связей в двух молекулах С02.
С02 плохо растворим в воде (0,034%). При растворении С02
в воде незначительная часть молекул С02 гидратируется с
образованием угольной кислоты (0,4% молекул С02 от всего
количества растворенного С02):
СО_)>р.р -j- Н2Ож = Н2СОз,р-р.
Большая часть молекул Н2С03 находится в состоянии
диссоциации
Н2СОз,р-Р ^ Нр.р -|- НС03,р-р,
и в этом отношении угольная кислота — не слабый электролит»
Однако ее обычно считают слабым электролитом. Такое мнение
вызвано тем, что только очень малая часть растворенного в воде
углекислого газа присутствует в форме Н2С03.
Константу диссоциации Н2СОз
[H+p]fHC03-p.pl
526
правильнее было бы писать так:
со2,р.р + н2о - н+р f нсо^;р.р,
Lcu2, p-pJ
С металлами углерод образует многочисленные соединения —
карбиды. При замене в структуре алмаза половины атомов
углерода на атомы кремния структура получающегося карбида—•
карборунда SiC — сохраняется той же, что и у алмаза.
Карборунд— одно из самых твердых веществ, уступает по твердости
лишь алмазу. Карбид кремния — ковалентный карбид.
Некоторые карбиды углерода могут рассматриваться как
производные метана или ацетилена. Например, карбид алюминия
как производное метана. При гидролизе карбида алюминия
образуется метан и гидроксид алюминия (пример необратимого
гидролиза):
А14С3>К + 12Н2Ож - ЗСН4.Г 4- 4А1 (ОН)3.к.
В А14С3 связь носит частично ионный характер. Карбид кальция
СаС2 имеет еще более ионный характер и в кристаллическом
состоянии обнаружены ионы С2~. Карбид кальция — производное
ацетилена (ацетиленид кальция)—при гидролизе выделяет
ацетилен
СаС2>к ! - 2Н2Ож - Сар^ + 20Н~Р + С2Н2.Г.
При высоких концентрациях иона Са2+ образуется осадок гидро-
ксида кальция Са(ОН)2.
Кристаллическая структура кремния та же, что и у алмаза
(5/?3-гибридизация), однако из-за больших размеров атомов и
соответственно р-орбиталей зоны перекрывания гибридных sp3-op-
биталей не столь плотны, как у алмаза. Связи Si—Si в
кристаллической структуре кремния поэтому слабее, и кремний не
обладает твердостью алмаза. Кроме того, больший размер гибридных
5р3-орбиталей приводит к значительно меньшей связи электронов
с ядрами атомов. Поэтому некоторая часть электронов не
локализуется у атомов кремния. Последнее обстоятельство приводит
к металлическому блеску кремния и его электропроводности
(несравненно более низкой, чем у металлов).
Кристаллический кремний в отличие от углерода растворяется
в концентрированных растворах щелочей:
SiK /- 20Н~Р + Н20 = SiOaVp + 2Н2,Г.
При комнатной температуре кремний с кислородом не реагирует,
но при высокой сгорает с образованием кристаллического
диоксида Si02 — кварца. Это одно из самых устойчивых веществ.
527
В кварце атомы кремния образуют подрешетку, сходную с
кристаллической решеткой алмаза, атомы же кислорода
располагаются между атомами кремния.
Известны гидриды кремния общей формулы SinH2n+2, по
структуре и по характеру связей похожие на предельные
углеводороды. От последних они отличаются меньшим значением п
и меньшей прочностью связей Si—Si. На воздухе они сгорают с
образованием Si02 и воды и выделением большого количества
тепла.
Гидриды типа SinH2n и SinH2n_2, аналогичные таким же у
углерода, не известны из-за неспособности кремния в гибридных
sp2- и sp-состояниях образовывать я-связи.
Диоксид кремния Si02 обладает только слабокислотными
свойствами, медленно растворяясь в концентрированных
растворах щелочей:
Si02fK +- 20Нр-р = Si032^-p + Н20.
Кремниевая кислота — слабая кислота. В растворе она находится
в виде целого ряда кислот и ионов различного состава в
зависимости от концентрации. Молекула H2Si03 способна присоединять
дополнительные количества молекул Si02 и Н20, и в общем
случае состав кремниевой кислоты отвечает формуле ttSiCVmH^O.
Соли кремниевой кислоты — силикаты также имеют различный
состав.
Монооксид кремния SiO известен только в газовой фазе при
высоких температурах. С галогенами кремний реагирует
непосредственно. Все галогениды SiT4 не являются ионными
соединениями.
Германий похож на кремний, но сильно отличается от
углерода. Кристаллический Ge имеет структуру алмаза с атомами в
состоянии ^-гибридизации. Электропроводность германия
значительно выше, чем у предыдущих элементов — при комнатной
температуре германий полупроводник.
В растворах щелочей германий растворяется:
GeK -г 20Н~Р -г Н20 = GeOlp-p + 2Н2)Г.
Диоксид германия, получающийся непосредственно из простых
веществ при нагревании, также растворяется в щелочах:
Ge02iK + 20Нр-_р - GeO^-p + Н20.
Соли германиевой кислоты H2GeC>3 — германаты — в растворе
сильно гидролизуются. Из-за того что германиевая кислота в
свободном состоянии не существует, выпадает осадок диоксида:
GeO^.p -г Н20 =- Ge02,K + ЮН^-р.
Гидриды германия GenH2„+2 еще более неустойчивы и известны
только для п не более 8.
528
В 2-валентном состоянии германий известен в виде некоторых
галогенидов и сульфида GeS. Их синтезируют обычно
восстановлением металлическим германием соединений с
четырехвалентным германием:
Ge + GeF4 = 2GeF2,
Ge + GeS2 = 2GeS,
Ge + Ge02 - 2GeO.
Все эти соединения — сильные восстановители. Известны также
соли, отвечающие кислоте H2Ge02— германиты.
Олово и свинец — металлы. Валентное состояние 2
приобретает у них большее значение, а для 4-валентного, наоборот,
понижается. Из гидридов известны только два: SnH4 и РЬН4. Тетра-
галогениды олова мало устойчивы, а у свинца PbF4 и РЬСЦ
вообще неустойчивы и распадаются:
РЬСЦ.к = РЬС12,к -I- С12рГ.
У олова двухвалентное и 4-валентное состояния примерно
одинаковы по устойчивости. Оксиды SnO и S11O2 при действии на
них щелочами растворяются с образованием станнит- и станнат-
ионов соответственно
SnOK + 20Н~Р - SnO^p'.p -г Н20,
Sn02,K + 20Н7-Р = SnO^-p + H20.
Координационное число олова равно 6, и правильнее
представлять формулы станнит- и станнат-ионов как Sn(0H)6~ и
Sn(OH)6~, а процесс растворения записывать в виде
SnOK + 40Н~Р + Н20 - Sn (OH)^.p,
Sn02,K + 20Н~р + 2Н20 - Sn (ОН)^-р.
В кислой среде станнит и станнат-ионы превращаются в
катионы, содержащие олово в тех же валентных состояниях. Состав
катионов точно неизвестен. При прибавлении к раствору
Sn(OH)6~ соляной кислоты образуется ряд катионов и
анионов Sn2+, SnCl+, SnCl2, SnCir и SnClj" в зависимости от
концентрации хлорид ионов. В солянокислой среде станнат-ион
Sn (OH)6~, по-видимому, сразу переходит в ион ShCIg":
Sn (OH)^.p + 6С1~р -!- 6Н+р - SnCl^.p + 6Н20.
Состав ионов, образующихся при действии других кислот,
неизвестен. Диоксид олова — амфотерен, в щелочной среде он
растворяется с образованием ионов Sn (ОН)б", в солянокислой — ионов
SnCle".
529
Для свинца наиболее характерно 2-валентное состояние.
Азотнокислый свинец Pb(N03)2 в водном растворе сильно
гидролизу ется: J
РЬ*+ -г Н20 = РЬОН+р -г Н+р.
При прибавлении щелочи к раствору соли свинца образуется
осадок РЬ(ОН)2, растворяющийся в избытке щелочи с
образованием плюмбит-иона РЬ (ОН)7:
РЬ (ОН)2.к + ОНрТр = РЬ (ОН)^р.р.
Плюмбит-иону отвечает не существующая в свободном состоянии
кислота Н2РЬ02 и плюмбит-ион простейшей формулы РЮ2"~,
также, по-видимому, не существующий.
Галогениды свинца РЬГ2 плохо растворимы в воде, но
растворимы в растворах, содержащих соответствующие галогенид-
ионы, в связи с образованием комплексных ионов РЬГУ:
РЬГ2,К + Г~р - РЬГз~р.Р.
В растворах галогенидов обнаружены ионы РЬГ+, РЬ2+, а
также нейтральные молекулы РЬГ2 в концентрациях, зависящих как
-от концентрации свинца, так и галогенид-иона.
О поведении в растворе ионов 4-валентного свинца известно
очень мало, так как такое состояние для него не характерно.
Диоксид свинца очень сильный окислитель:
Pb02 + 4H+ -f 2е = РЬ2+ + 2Н20 Е° = 1,46 в.
Отвечающая 4-валентному состоянию свинца, свинцовая кислота
Н2РЬ03 в свободном состоянии неизвестна, плюмбат-ионы
состава РЬ03~ также в растворе неизвестны, однако кристаллические
соли получены. Например, оксид свинца состава РЬ203 можно
рассматривать как соль свинцовой кислоты свинца в
двухвалентном состоянии: PbnPbIV03. Оксид состава РЬ304 аналогично
можно считать солью свинцовой кислоты свинца в 4-валентном
состоянии: PbIV(Pbn02)2 = Pb304.
§ 8. АЗОТ, ФОСФОР, МЫШЬЯК,
СУРЬМА И ВИСМУТ (/-ЭЛЕМЕНТЫ У ГРУППЫ)
Азот, фосфор, мышьяк, сурьма и висмут считаются не
металлическими элементами. Конфигурации валентных уровней: s2p3.
Энергии ионизации при переходе вниз по подгруппе равномерно
уменьшаются. Атомные радиусы в том же направлении
возрастают. В то же время температуры плавления от азота к
мышьяку возрастают, а затем падают. Это вызвано различием
кристаллических решеток простых веществ. В твердом азоте
молекулы N2 связаны слабыми силами Ван-дер-Ваальса, а в структуре
.530
фосфора — ковалентными связями. В структуре мышьяка кова-
лентные связи наиболее прочны. У сурьмы и висмута с
появлением нелокализованных электронов (металлическая связь)
структура становится менее прочной. Температуры кипения простых
веществ повышаются при переходе вниз по подгруппе. Азот и
фосфор — неметаллы. У мышьяка и сурьмы появляются
металлические свойства, висмут — металл, но с невысокой
электропроводностью.
Наиболее характерны валентные состояния элементов 3 и 5.
Устойчивость валентного состояния 3 в гидридах при переходе
вниз по подгруппе быстро уменьшается. Только один NH3
устойчив, остальные гидриды неустойчивы, а гидрид висмута BiH3
обнаружен только в виде следов. Основный характер гидрида
азота NH3 изменяется на слабо основный у РН3, а у остальных
гидридов основные свойства отсутствуют. Оксиды азота и фосфора
N203 и Р203— кислотные оксиды, As203 и Sb203— слабо
кислотные оксиды, a Bi203 — основный оксид. В том же направлении
изменяются свойства оксидов с элементами в 5-валентном
состоянии Э2О5: от сильнокислотных к слабокислотным.
Молекула азота — самая прочная из двухатомных молекул,
состоящих из атомов одного сорта. В молекуле N2 одна а- и две
зт-связи. При невысоких температурах, когда недостаточно
энергии на разрыв прочной связи N = N, азот — инертный газ и он не
реагирует с большинством элементов и их соединений. Однако
уже при 300°С азот очень энергично взаимодействует с литием с
образованием нитрида лития, при более высоких температурах
проходят реакции с магнием, алюминием и другими элементами.
Азот в гидридах щелочных и щелочноземельных металлов
связан с атомами металлов не чисто ионными связями, а только*
частично ионными со значительной долей ковалентной связи.
В воде нитриды s-элементов гидролизуются с выделением
аммиака:
Li3NK + ЗН2Ож = ЗЦГр f ЗОН~р -\- NH3,r.
Нитриды неметаллических элементов — ковалентные нитриды
с небольшим вкладом ионной связи. Нитриды s- и /?-элементов
чаще всего по составу отвечают обычным валентным
соотношениям в отличие от нитридов переходных элементов, например
W2N, TiN и др.
С кислородом азот образует несколько оксидов: N20, NO,
N203, N02, N205 и N03. Оксид N03 обнаружен только в газовой
фазе спектроскопически. Оксид N02 способен димеризоваться в
оксид N204.
Оксид N20 изоэлектронен диоксиду углерода. Его молекула,
как и молекула С02, линейна: N—N—О с расстояниями N—N
1Д2А и N—О 1,19А. При нагревании N20 распадается на
кислород, азот и N0.
531:
В молекуле оксида N0 на один электрон больше, чем в N2
или СО, и этот лишний электрон занимает разрыхляющую орби-
таль. Поэтому при его удалении образовавшийся газовый ион
N0+ обладает более высоким значением энергии связи и
меньшим межатомным расстоянием (в NO R = 1,15 А; А# связи=
= 150000 кал/моль; в N0+ #=1,06 А; Д#связи = 244 000 кал/моль).
Ион NO+ хорошо изучен не только в газовой фазе, а в растворах
и кристаллических веществах. Если азотистый ангидрид ^03или
смесь NO и N204 обработать концентрированной серной
кислотой, в растворе образуется ион NO+:
N203 - 3H2S04,>K - 2NO+p -| H20 + ЗЫБО^.р + Н+р.
При охлаждении раствора образуются кристаллы NOHS04,
содержащие в кристаллической решетке ионы NO+ и HSOlf.
Газообразный NO окисляется непосредственно кислородом с
образованием диоксида азота N02:
2NOr -f 02(г = 2N02,r.
N02 — газ красно-бурого цвета. Цвет диоксида обусловлен
одним неспаренным электроном азота. Благодаря этому
электрону молекула N02 димеризуется в бесцветный N204. При
димеризации угол О—N—О не изменяется (134°), а расстояние N02
незначительно уменьшается (в N02 i?NO=l,19A; в N204 Rno =
= 1,18А). Молекула N204 — плоская, расстояние N—N довольно
велико (1,75°). Из-за того, что при димеризации и при обратном
распаде N204 структура N02 не искажается, скорости процессов
димеризации и распада велики и равновесие быстро смещается в
ту или иную сторону при изменении температуры и давления.
Триоксид N203 во многих реакциях ведет себя как смесь N02
и NO, и только в газовой фазе подтверждено существование
молекул N203. N2O3 или N02 и NO в растворе щелочи переходят в
состояние ионов N07:
N203>r + 20Н~Р = 2N02:P-P + Н20,
NOr + N02 + 20Н-Р = 2N02:P.P ~ Н20.
Диоксид азота N02 с холодной водой диспропорционирует на
азотную и азотистую кислоты:
2N02,r + Н2Ож = HN02,P.p + Н+р + NOaTp.p.
Азотистая кислота существует только в холодных растворах. При
нагревании она диспропорционирует по реакции
3HN02,p.p - NOaTp.p + 2NOr + Н20 -f- н£р.
.532
Таблица 14.45
Следовательно, если диоксид азота N02 растворять в горячей
воде, то образуется азотная кисдота и монооксид азота N0:
3N02,r + Н20 = 2Н+Р + 2N037p-P + NOr.
В ряду NO2", N02 и N07 угол 0N0 постепенно уменьшается
(табл. 14.45).
При обезвоживании азотной кислоты сильным осушителем,
например пентоксидом фосфора Р2О5, образуется пентоксид азота
N205, являющийся ангидридом азотной кислоты. В газовой фазе
молекула N20s действительно обнаружена, но в кристаллическом
состоянии решетка состоит из ионов NO2 и N07-
Нитрат-ион N07 — плоский с углом
между связями 120°. Расстояния О—N в
нем равны по 1,218 А. При присоединении
иона Н+ в образовавшейся молекуле HNO3
углы между связями 0N0 изменяются:
\Р
/"J130°
о
Две связи ON очень незначительно возрастают, а третья связь с
кислородом, связанным с водородом, значительно удлиняется.
Азотная кислота очень сильный окислитель. Ее стандартный
восстановительный потенциал изменяется в очень широких пределах
в зависимости от среды раствора:
N07 -г 12Н+ -f Ше - N2 + 6На0 j£° = + 1,24 в,
Ион, молекула
^ONO
N0+
N02
NO7
180
134
116
N03
f H20 + e = N02 -f- 20Н- £° - — 0,86 в.
Вид образующихся веществ и их относительные количества
зависят от силы восстановителей, концентрации азотной кислоты (от
концентрации ионов N07 и рН раствора).
По ряду свойств фосфор напоминает азот, но и не менее
сильно от него отличается. Отличие связано с появлением Зй-под-
уровня, который может принимать электроны с 35-подуровня при
их распаривании. Но в это же время способность образовать
я-связи у фосфора значительно слабее. Поэтому фосфор не
образует в заметных количествах в газовой фазе молекул Р2,
аналогичных N2. В газовой фазе фосфор, как и сера, находится в
состоянии нескольких различных по составу молекул. Эти
молекулы частично сохраняются и в твердой фазе, обусловливая
различные модификации фосфора. В газовой фазе в наибольшем
количестве фосфор содержится з виде молекул Р4,
равносторонних пирамид с углами по 60°. Устойчивая при стандартных усло-
533
виях кристаллическая модификация фосфора — красный
фосфор— представляет молекулярную структуру, состоящую иа
тетраэдров Р4. Поэтому во всех реакциях с участием фосфора,
как простого вещества, обычно пишут его формулу как Р4.
При нагревании фосфора с оксидом кальция образуется
фосфид кальция и оксид фосфора в трехвалентном его состоянии:
ЗСаОк + 2Р4,ж,г = 2Са3Р2,к + Р4Об,г.
Фосфид кальция напоминает те нитриды, которые гидролизуются
с образованием аммиака. При гидролизе Са3Р2 выделяется фос-
фин:
Са3Р2,к + 6Н20 - 2РН3,г -г ЗСа (ОН)2,р.р.
На воздухе фосфор сгорает до триоксида Р4О6, а в чистом
кислороде до пентоксида Р40ю- В молекулах оксидов
сохраняется структура молекулы Р4. В молекуле Р4О6 между каждой
парой атомов расположен атом кислорода. Молекула Р4О10
сохраняет структуры Р4 и Р4О6, но к каждому атому фосфора,
находящемуся в вершинах пирамиды, присоединено по атому кислорода.
Триоксид фосфора Р4О6 при растворении в воде дает кислоту
состава НР02. Формула фосфористой кислоты точно не
установлена, но известно, что она является двухосновной, причем
диссоциацию ее пишут, исходя из формулы ортофосфористой кислоты
НзР03:
н3ро3 = н+ -г н2РОз~ к = 1,6 • кг2,
Н2РОз" - Н+ + НР05"" К2 = 7,0 . 1(Г7.
Р205 является ангидридом фосфорной кислоты:
Р205,к + ЗН2Ож - 2Н3Р04,р.р.
Фосфорная кислота — Кислота средней силы, дающая соли
различной степени замещенности, например Na3P04, Na2HP04 и
NaH2P04.
Непосредственно фосфор соединяется со всеми галогенами с
образованием РГ3 и РГ5 в зависимости от количества галогена.
В газовой фазе молекулы РГ5 действительно отвечают этому
составу и связи носят ковалентный характер, однако в
кристаллическом состоянии решетка их построена различно. Решетка PCU
состоит из ионов PC\t и PC1JT (суммарный состав Р2С1ю).
Кристаллический РВг5 состоит из ионов PBrf и Вг~~ (суммарный
состав РВг5). Различие двух структур вызвано большими
размерами брома и невозможностью плотного расположения шести его
атомов вокруг атома фосфора, как в PC1JT.
При действии на галогениды фосфора водой образуются
соответствующие фосфорные и галогеноводородные кислоты:
534
РС13>к f ЗН2Ож - НаРОз,р-р + ЗН+р + ЗС1~Р,
РВг5,к 4 4Н2Ож = H8POifP.p + 5Нрь.р + 5Вгр.р .
Мышьяк во многих отношениях похож на фосфор. В газовой
фазе он существует в виде молекул As4. На воздухе и в
кислороде сгорает до As406 и As4Oi0, имеющих то же строение, что и у
фосфора.
Оксид As406 имеет амфотерный характер (в отличие от РгОб).
Предполагается, что в растворе мышьяковистая кислота
находится в виде молекул HAs02, а мышьяковая — H3As04. Последняя в
отличие от фосфорной проявляет окислительные свойства в
кислых растворах, переходя или в ион AsO~ или даже в AsH3.
Соли мышьяковистой кислоты называются арсенитами. Арсе-
ниты в зависимости от среды раствора получаются в виде
производных от HAs02 и H3As03, смотря от того, в какую сторону
смещены равновесия:
AsO^'.p 4- 2Н+Р = AsO^p.p + Н20,
As0^p.p -f 20H~p = AsC^.p -f H20.
В щелочных растворах преобладают арсенит-ионы As03~~, в
кислых— метаарсенит-ионы AsOJ"- Соли мышьяковой кислоты
называются арсенатами. И арсениты и арсенаты получают
растворением оксидов в щелочах:
As406,k + 120Нр:р = 4As0^.p + 6Н20,
As4Oio,k4- 120Н~р - 4As034^p -j- 6H20.
Арсенит-ион окисляется даже йодом в слабощелочной среде
до арсенат-иона:
AsO," p + I2 f Н20 = As04~ + 21" + 2Н+.
При изменении среды на кислую реакция изменяет направление
на обратное. В сильнокислых средах арсенит-ион переходит в
гидратированный ион As3+:
AsO^.p + 6Н+р = As3p+ + 3H20.
Оксид сурьмы Sb203 обладает амфотерными свойствами:
Sb203,K -4- 6Н+р = 2Sbj£ + 3H20,
Sb203,K + 60H" = 2Sb03~ 4 3H20.
Соли сурьмянистой кислоты H3Sb03 называются антимонитами.
Соли сурьмы Sb3+ в водных растворах сильно гидролизуются с
образованием ионов антимонила (или стибила):
535
Sb3ptp + H20 = SbO+ p -f 2Hptp.
Ион SbO+ входит в состав некоторых кристаллических солей,
например хлорида антимонила SbOCl.
Оксид Sb502 обладает кислотными свойствами:
Sb205(K + 60Н~р = 2SbO^" Н- ЗН20.
Соли сурьмяной кислоты называются антимонатами. Оксид Sb20s
в кислых растворах нерастворим, но в присутствии
восстановителей переходит в раствор в виде ионов SbO+, проявляя свойства
окислителя:
Sb205,K + 6Н£р + 4е = 2SbOptp -f ЗНаО Е° - 0,58 в.
Висмут окисляется кислородом только до оксида Bi203,
растворимого в кислотах с образованием висмутил-иона:
Bi203,K -г 2Н+Р - 2ВЮ+Р + Н20.
Ионы Bi3+ в растворе не обнаружены, так как даже в сильно
кислых средах гидролизуются до BiO+.
При действии очень сильных окислителей (хлор) на Bi203 в
присутствии щелочи образуется вещество предполагаемого
состава NaBi03, являющееся очень сильным окислителем.
§ 9. КИСЛОРОД, СЕРА, СЕЛЕНГ
ТЕЛЛУР И ПОЛОНИЙ (р-ЭЛЕМЕНТЫ VI ГРУППЫ}
Кислород, сера, селен, теллур и полоний имеют строение
наружного электронного уровня s2p4, что позволяет им проявить
валентные состояния 2, а также, за исключением кислорода, 4 и 6.
Энергии ионизации по подгруппе падают, радиусы атомов в
том же направлении возрастают. Температуры плавления,
однако, изменяются неравномерно (О — 54°, S — 392, Se — 490,
Те — 723, Ро — 527°К). Параллельно им изменяются и
температуры кипения (0 — 90°, S —718, Se—960, Те—1260, Ро—1235°К).
В подгруппе происходит постепенный переход от
неметаллических элементов к металлическим, от окислителей к
восстановителям, от элементов кислотного характера к элементам основного
характера.
Кислород в свободном состоянии устойчив только в виде
двухатомной молекулы 02, обладающей парамагнитными свойствами.
Парамагнетизм молекулы 02 может быть объяснен с
использованием метода молекулярных орбиталей, так как пара электронов
вступает <на разрыхляющие орбитали лрх и лру.
Озон 03 — аллотропное видоизменение кислорода. Молекула
озона имеет нелинейное строение (,/1000= 117°). Расстояние
О—О в молекуле 03 то же, что и в ионе 07. Это наводит на
536
мысль, что связи в озоне промежуточные между одиночными и
двойными. Озон — чрезвычайно сильный окислитель:
03.г + 2Н+р + 2е = 02,г + Н2Ож £° = 2,07 в.
Только инертные газы (не все) не образуют с кислородом
соединений. Свойства оксидов рассматриваются при изучении
соответствующих элементов. Свойства оксидов зависят от
положения элементов в периодической системе. Среди s- и ^-элементов
основные оксиды расположены в нижнем левом углу, а
кислотные в верхнем правом. Оксиды d-элементов в их низких
валентных состояниях имеют преимущественно основный характер, с
повышением валентного состояния кислотные свойства оксидов,
как правило, усиливаются.
Известно несколько соединений кислорода с водородом,
устойчивость которых резко падает при увеличении количества атомов
кислорода в молекуле. Вода при стандартных условиях вполне
устойчивое вещество, но пероксид водорода Н202
самопроизвольно распадается с выделением кислорода:
2НаО2.ж=2НаОя + 02,г,
хотя по отношению к простым веществам пероксид водорода
вполне устойчив:
Н2,г -(- 02>г = Н202,ж ДЯ = — 44700 кал/моль.
Пероксид водорода проявляет как окислительные, так и
восстановительные свойства:
Н202 + 2Н+р + 2е = 2Н20 Е° = + 1,77 в,
02tr + 2H+р + 2е = На02 Е° = + 0,68 в.
Если у кислорода известно только две модификации Оз и 03,
то у серы их несколько в кристаллическом состоянии, а в парах
сера — смесь молекул различного состава S, S2, S4, Se, Sg. Чем
выше температура, тем меньше в смеси больших молекул.
Интересно отметить, что в устойчивых молекулах серы всегда
содержится четное число атомов.
При непосредственном соединении серы с водородом
образуется сероводород H2S. Раствор сероводорода в воде — слабая
кислота:
h2sp.p = н+р + hs~p /с = 1,1 • ю-7,
HSp.p = Нр.р -)- Sp7p /C2 = 1-10
Соли H2S — сульфиды, могут быть синтезированы из простых
веществ. Сульфиды 5-элементов растворимы в воде и очень сильно
гидролизованы:
537
Sp.p + H20 = SHp.p + OHp.p,
к _ /Св _ Ю-14 _ 1
Л ГИДР — -г? - — — L .
Адисс SH Ю~14
Сульфиды d-элементов плохо растворяются в воде. Их
кристаллические решетки не чисто ионные, а со значительным
вкладом ковалентной связи.
Кроме простых сульфидов существуют многочисленные соединения,
содержащие большее количество атомов серы. Если считать, что в простых сульфидах
содержится ион S2-, то в сложных содержатся ионы 5^~" > где п=2—6. Связь
группировки S„~~ с катионами щелочных и щелочноземельных металлов в основном
ионная, а в соединениях переходных металлов имеет большую долю ковалент-
ности. Вещества, содержащие группировки S~~~ , называются полисульфидами.
Они могут быть синтезированы непосредственной реакцией элементов с
избытком серы или же обработкой раствора сульфида мелкораздробленной серой.
При подкислении раствора полисульфида выделяются сульфаны общей
формулы H2Sn (n=2—6).
При сгорании серы на воздухе образуется диоксид серы, хотя
термодинамически более выгодно образование триоксида серы
SK + 02,г = S02,r Д#° = — 70 660 кал/моль,
S02,r -J- V202.r = S03,r АЯ° = — 23490 кал/моль.
Вторая реакция на воздухе и в кислороде практически не
проходит из-за ее медленности, но катализаторы (Pt, V2O5 и др.)
значительно ускоряют ее. В молекуле S02 и S03 все углы между
связями равны по 120°, расстояния S—О также равны по 1,43 А,
несмотря на различие молекул. Растворение S02 в воде
приводит к образованию слабой сернистой кислоты H2S03.
Растворение S03 в воде сопровождается выделением большого
количества тепла и образованием раствора серной кислоты.
Безводная серная кислота H2S04 — слабый электролит.
Небольшая ее электропроводность объясняется диссоциацией
2Н2804,Ж = H3S04,p.p + HSO^p.p.
По мере разбавления водой серной кислоты ее свойства
изменяются и из слабого электролита в концентрированных
растворах она превращается в сильный электролит:
H2S04,p.p = Нр.р -|- HS04,p.p,
HS04,p.p = Нр.р -f S04,p-p.
В концентрированных растворах серная кислота ведет себя-
как сильный окислитель и способна растворять металлы, не
растворимые в кислотах-неокислителях:
CuK + Haso4tP.p = Cu2+ ; so?,, + н2о.
538
Свободная H2S03 не выделена, как не выделена свободная
угольная кислота. Равновесие в растворе, содержащем
растворенный S02, описывается уравнениями
S02,p-p -г Н20 = HS03fP.p 4- Нр-р,
НБОз.р.р = Н + БОз.р.р.
Диоксид серы в водных растворах проявляет слабые
окислительные и восстановительные свойства:
H2S03,p_p + 2H2Sp.p = 3SK -f 3H20,
5H2S03,p-p + 2Мп0^р.р - 5SO?" р + 2Mnptp + 4Н+Р + 3H20.
При прибавлении мелкораздробленной серы к раствору S02 в
воде растворимость последнего резко увеличивается. Это
объясняется реакцией
ЬОз,р-р "~г Ьк = Ь2Оз,рр.
В этой реакции кристаллическая сера ведет себя, как
кислород в реакции окисления S02 в S03, т. е. является окислителем.
Образовавшийся тиосульфат-ион S205~ по структуре аналогичен
иону S04~\ в котором один атом кислорода заменен на атом
серы. Оба иона имеют тетраэдрическое строение.
В кислых растворах ион S205~ разрушается с образованием
серы и диоксида серы:
S20^.p i 2H+p = S02.r + SK i-H20.
В щелочах, нейтральных и слабо кислых растворах тиосульфат-
ион ведет себя как слабый восстановитель. Это его свойство
используется в методе количественного анализа — йодометрии,
так как тиосульфат-ион переводит йод в йодид-ион:
S4Og~~ + 2е = 2S20^~ £° = 4- 0,09 в
12 Ч- 2в = 21- £°= 4-0,54 в
2S20^.p + 12.к = S40^-p -> 21~р э. д. с. - 0,54 — 0,09 = f- 0,45 в.
Химия селена и теллура отличается от химии серы тем, что у
этих двух элементов появляются металлические свойства. Селен
и теллур — полупроводники. С металлами селен и теллур
образуют соединения, похожие на сульфиды. Связь в селенидах и тел-
луридах носит более ковалентный характер по сравнению с
сульфидами.
Устойчивость всех гидридов /7-элементов VI группы сильно
понижается при переходе вниз по подгруппе:
539
н2
н2,
н2,
+ V А
r + Sr =
,г + Ser =
= н2ог
: H2Sr
= H2Ser
А #298 ==
Д#298 =
А#298 =
-57800 кал/моль,
■4800 кал/моль,
20500 кал/моль,
Н2,г + Тег = Н2Тег ДЯгэв = + 36900 кал/моль.
В том же направлении возрастает их диссоциация в водных
растворах:
Н20 = Н+ + ОН- /С = 1,8- Ю-16,
H2S = H+ + HS- К = 1.1 -10—7.
H2Se - Н+ + HSe- К = 2,0.10~4,
Н2Те = Н+ Н-!НТе- К = 2,3-10~3.
Диоксиды селена и теллура Se02 и Те02 — твердые
кристаллические вещества. При переходе от S02 к Se02 и ТеОг ковалент-
ность связи Э—О постепенно понижается и у Те02 доля ионной
связи уже превышает долю ковалентной. Диоксид селена,
растворяясь в воде, образует слабую селенистую кислоту H2Se03.
Диоксид теллура в воде не растворим и кислота, отвечающая
такому валентному состоянию теллура, неизвестна. Однако теллу-
риты, содержащие ион ТеОз~~, могут быть получены действием
щелочей на Те02.
В шестивалентном состоянии селен образует селеновую
кислоту H2Se04, аналогичную серной кислоте как по структуре
молекулы и характеру связей, так и по силе. Селеновая кислота —
более сильный окислитель по сравнению с серной:
SOfcp + 4Н+р + 2е = H2S03,p-p + Н20 £° - 0,17 в,
SeOfcp + 4Н+Р + 2е - H2Se03,p-p + Н20 £° - 1,15 в.
Поэтому сернистая кислота (или сульфит-ионы) может быть
переведена в серную кислоту, или сульфат-ионы, действием
селеновой кислоты:
H2S03,p.p 4- SeO^p.p = SO^p.p + H2Se03,p.p
E° = 1,15 — 0,17 = +0,98 в.
Теллур не образует соединения, аналогичного серной или
селеновой кислоте. В 6-валентном состоянии для него известно
соединение Те (ОН) б, являющееся очень слабой кислотой.
§ 10. ГАЛОГЕНЫ (/-ЭЛЕМЕНТЫ VII ГРУППЫ)
/^-Элементы VII группы — фтор, хлор, бром и йод
объединяются одним общепринятым названием — галогены. Все они ярко
выраженные неметаллы. Только йод в очень редких случаях об-
540
наруживает некоторые свойства, указывающие на его отдаленное
сходство с металлами.
В невозбужденном состоянии у атомов галогенов
конфигурации наружных электров — s2p5. Из-за отсутствия ^-подуровня,
способного принимать распаренные электроны, фтор —
единственный из галогенов, имеющий в соединениях только одно
валентное состояние 1. Энергии ионизации вниз по подгруппе
равномерно уменьшаются. Все галогены в свободном состоянии —
окислители. Сила их как окислителей уменьшается при переходе от
фтора к йоду. В кристаллическом, жидком и газообразном (при
не очень высоких температурах) состояниях все галогены
существуют в виде отдельных молекул. Атомные радиусы в том же
направлении возрастают, что приводит к повышению температур
плавления и кипения.
Уже при 1000°К в газовой фазе среди двухатомных молекул
обнаруживаются атомы галогенов. Фтор диссоциирует на атомы
даже лучше йода. В табл. 14.46 даны константы диссоциации
галогенов. Константа диссоциации водорода приводится для
сравнения:
Гг.,
Кр
Таблица 14.46
Молекула
н2
F2
С1,
Вго
L
кр
5,1-10-1°
9,2-10-3
1,6.10-*
3,3-10-5
3,0-10"8
Молекула водорода несравненно прочнее молекул галогенов.
Прочность молекул от хлора к йоду уменьшается (табл. 14.47),
Невысокая по сравнению с другими галогенами прочность и
соответственно энтальпия связи фтора объясняются отсутствием у
фтора d-орбиталей. Предполагается, что у хлора и других
нижележащих галогенов при образовании связи между двумя
атомами часть электронной плотности с наружных s- и р-орбиталей
переходит на d-орбитали, которые, участвуя в связывании
атомов, упрочняют молекулу.
Электродные потенциалы галогенов определяются изменением
изобарного потенциала в процессе
Г2,г + 2е = 217р.
. = 2Г
— ^1 г,
Р2
_ г
1 2
Таблица 14.47
Молекула
н2
F2
С12
Вго
I*
АЯсвязи' ^ал/моль
103 260
37 000
57 000
45 400
35 600
541
Так как при Г = 298°К AG°^A#°, то электродные потенциалы
могут быть вычислены из энтальпий отдельных стадий перехода
галогена из газообразного состояния в состояние однозарядного
отрицательного иона:
Г2г = 2ГГ ДЛГ
Гг -f е - ГГ Л#<
ДИССС)
сродства к электоону»
rr(-MHaoj = r^p
Д#,
гидр •
Энтальпии этих процессов и стандартные электродные
потенциалы галогенов приведены в табл. 14.48.
Таблица 14.48
Галоген
F2
Вь
1*~
АЯдисс'
кал/г-атом
18 500
28 500
22 700
17 800
^сродства'
кал/г-атом
—90 900
—85 300
—84 000
—76 100
А^иДР-
кал/г-ион
-113000
—79000
—72 000
—63000
Е°, в
2,87
1,36
1,09
0,54
При переходе вниз по подгруппе электродные потенциалы
галогенов понижаются. У фтора самый высокий электродный
потенциал, фтор самый сильный окислитель,
хотя энтальпия связи в молекуле фтора
меньше энтальпии связи в молекуле хлора,
а сродство к электрону понижается от
фтора к йоду. Столь высокий потенциал
фтора объясняется крайне высокой энтальпией
гидратации ионов фтора. Энтальпия
гидратации зависит от радиусов ионов. Радиус
иона фтора F~ резко отличается от
остальных галогенов своей аномально малой
величиной (табл. 14.49).
Любой вышестоящий свободный галоген вытесняет любой
нижестоящий, находящийся в состоянии отрицательного
однозарядного иона в растворе, например:
Таблица
г
F"
CI-
Вг"
I-
14.49
о
R, А
1,33
1,81
1,96
2,19
CUr-u2e = 2Cl
Вг2ж ;-2е = 2Вг~
р-р
р-р
£» = 1,36 в
£°= 1,09 в
С12.г f 2Вг~р =-2С1~Р -'-Вг2.ж э. д. с. = 1,36—1,09 = 0,27 в.
Электропроводность безводного HF очень низка и вызвана
диссоциацией
3HF = H„F+ -f HFj
■542
Электропроводность других безводных галогеноводородов еще
ниже и указывает на значительное ослабление процесса
диссоциации.
В водных растворах галогеноводороды, за исключением HF,
хорошо проводят электрический ток, являясь сильными
электролитами. Раствор HF проводит ток плохо, так как благодаря
системе водородных связей молекулы HF объединяются в
устойчивые агрегаты. Фтористоводородная кислота лишь на порядок
сильнее уксусной:
HF + Н20 - Н30+ + F~ К = 7 • 1(Г4.
Остальные галогеноводороды в водных растворах
слоты и почти нацело
диссоциируют на ионы. Их сила
возрастает в направлении НС1—НВг—
—HI, причем оилы НВг и HI
близки.
Интересно определить, какими
факторами определяется сила
галогеноводородных кислот.
Можно предположить, что сила
кислот связана с энтальпией
образования галогеноводородов из
простых веществ или же с энтальпиями связей (табл. 14.50).
Из этих данных видно, что энтальпии образования
галогеноводородов изменяются параллельно силе их как кислот.
Наименьшая энтальпия у HF, несомненно, характеризует HF как
очень слабую кислоту. Различие в АЯ0бр между соседними гало-
геноводородами отвечает различию их как кислот. По энтальпиям
связи галогеноводороды располагаются в том же направлении.
Таблица 14.51
злых раст
нг
HFr
НС1Г
НВгг
Н1г
ворах — сильные ки-
Га блица 14.50
ЛЯ298.обр'
кал/моль
—64 000
—22 240
—8 600
—3 000
А//связи'
кал/моль
134 320
102 200
86 090
70 410
нг
HF
НС1
НВг
HI
ЛЯ298,дисС
кал/моль
—1000
—10 000
— 11000
—9 000
АС0
А,ь298,дисс'
кал/моль-град
—21
-13
—10
—3
TAS9,
кал/моль
—6300
—3900
—3000
—900
ЛГ0
ДС/298,дисс-
кал/моль
5300
—6100
—8000
—8100
Однако эти данные не дают правильного представления о силе
галогеноводородных кислот. Так, энтальпии диссоциации этих кислот примерно одина-
7 29 8, дн ее
ковы, за исключением HF, имеющей аномально малое значение Л#2<
(табл. 14.51).
Изменение энтропии при диссоциации галогеноводородных кислот
отрицательно, что указывает на уменьшение степени беспорядка или увеличение
степени порядка. Степень порядка при диссоциации возрастает, так как ноны во-
543
дорода Н+, а также и анионы сильно гидратируются, что приводит к
образованию более упорядоченных структур. У фтористоводородной кислоты
наибольшая отрицательная величина энтропии диссоциации из-за очень высокого
стремления малого по размеру иона фтора быть окруженным гидратной
оболочкой. Численные значения энтропии диссоциации падают по мере перехода к
HI, у ионов I- стремление к гидратации сильно ослаблено. Изменение
изобарного потенциала при диссоциации только у HF положительно, HF — слабая
кислота. У всех остальных кислот AG отрицательно и численно возрастает к
HI — самой сильной в этом ряду кислоте.
По количеству оксидов различного состава хлор занимает
среди галогенов первое место, за ним следует бром, затем фтор
и, наконец, йод. Некоторым оксидам отвечают соответствующие
валентному состоянию галогена кислоты и в то же время часто
у галогенов имеется кислота, но нет соответствующего ей оксида
(табл. 14.52).
Таблица 14.52
Валентное
состояние
1
2
3
4
5
6
7
Фтор
оксид
F?0
FA
—.
кислота
HFO
—
.—
—.
—
—
—~"
Хлор
оксид
С120
—
—
сю2
С1206
С12о7
кислота
НОС1
—
нсю2
—
НСЮ3
—
нсю4
Бр
оксид
Вг20
—
—
Вг02
Вг03
—
ом
кислота
НОВг
—
—
—
НВгОо
—
—
Йод
оксид
,
—
—
—
IA
—
'—
кислота
HOI
—
—
—
НЮ3
—
НЮ4,
Н5Юв
Все галогены, за исключением фтора, гидролизуются водой по
уравнению
Гг.г т- Н20 = Нр.р + Гр.р г НОГр.р.
Константа равновесия реакции гидролиза
v _ [НЧ [Г-] [НОГ]
резко уменьшается при переходе от хлора к йоду (4-10~4, 7-Ю"9
и 2• 10~13 для С12, Вг2 и 12 соответственно). Следовательно,
измеримые количества кислоты НОГ могут быть получены
гидролизом только у хлора (хлорноватистая кислота).
Хлорноватистая, бромноватистая и йодноватистая кислоты —
очень слабые кислоты. Их константы диссоциации понижаются
при переходе вниз по подгруппе (/(ДИСс НОС1 2-10~8, НОВг
2-Ю-9, HOI МО"11). Все кислоты неустойчивы и не выделены в
свободном состоянии, но анионы устойчивы в составе
кристаллических солей (гипохлориты и т. п.). Хлористая кислота НСЮ2 —
544
кислота средней силы. В водных растворах неустойчива, но
соли— хлориты — устойчивы. Аналогичных кислот у других
галогенов не известно. Кислоты типа НГ03 (хлорноватая, бромноватая,
йодноватая) получаются диспропорционированием анионов ГО~
в щелочной среде, например, из гипохлорит-иона образуется
хлорат-ион:
ЗС10- = 2С1- f СЮГ.
Кислоты НГ03 —сильные кислоты. Только кислота НЮ3
получена в чистом виде; НС103 и НВг03 известны только в виде
водных растворов. Хлорноватокислый калий, так называемая
бертолетова соль, — сильный окислитель, как и соли остальных
галогенов.
Кислоты типа НГ04 известны только для хлора и йода.
Хлорная кислота НСЮ4 — самая сильная из всех кислот.
Концентрированные растворы хлорной кислоты взрывоопасны. Перхлорат-
ион отличается от всех остальных ионов исключительной
неспособностью выступать в качестве лиганда и образовывать
комплексные ионы. Именно поэтому хлорная кислота наиболее удобна
для изучения свойств ионов в кислых растворах в зависимости
от рН, а перхлорат калия для создания растворов с требуемой
ионной силой. Неспособность перхлорат-иона быть лигандом
вызвана его большим размером, малым зарядом и высокой
симметричностью.
Хлорная кислота — очень сильный окислитель в
концентрированных растворах. Это свойство относится в основном к
органическим соединениям, так как с неорганическими
восстановителями реакция проходит очень медленно.
Кислородные кислоты галогенов — хороший пример изменения
кислотных и окислительных свойств в зависимости от валентного
состояния элемента. В ряду кислот НС10 — НСЮ2 — НС103 —
НС104 ясно заметно усиление кислотных свойств и ослабление
окислительных свойств (в водных разбавленных растворах).
Поведение йодной кислоты в водных растворах сильно
отличается от хлорной. В сильнокислых растворах она существует в
виде слабой кислоты состава HsI06, диссоциирующей по
уравнению
2НбЮ6,р.р = НЮб^.р +Ю47р-р + ЗН+р + 2Н20.
Все галогены непосредственно реагируют между собой с образованием
различных соединений типа ГТП. Обычно, если галоген Г' расположен ниже Г в
подгруппе, значение п бывает нечетным и не превышает 7, например C1F,
IBr, C1F3, IF3, CIF5, IFe, IF?. Молекулы этих соединений часто имеют
необычное строение. Так, молекула C1F3 имеет Т-образный вид:
F—CI— F
18 Общая химия
В молекуле C1F3 углы между связями равны по 87,5°. Два
противоположных атома фтора удалены от центрального атома хлора на 1,70 А, а третий
только на 1,60 А, т. е. этот атом фтора энергетически не равноценен двум
другим — он связан с хлором несколько прочнее.
В молекуле IF5 атомы фтора располагаются по вершинам пирамиды,
имеющей основанием квадрат, а атом I находится в центре. Молекула IFV еще
более сложна. Пять атомов фтора образуют правильный пятиугольник, в центре
которого располагается атом йода, остальные два атома фтора находятся над
атомом йода и под ним, т. е. атомы фтора образуют пентагональную бипира-
миду.
В водных растворах галогенидов растворимость свободных галогенов сильно
возрастает, что объясняется комплексообразованием:
Чк + Vp = з.р-р»
Вг2,ж + ^р = 1Вг<ГР-р'
С12(Г + 1-р = 1С12-р.р.
Ионы I^~\ IClJT, IBrJ"— линейные.
§ 11. БЛАГОРОДНЫЕ ГАЗЫ (/-ЭЛЕМЕНТЫ VIII ГРУППЫ)
Гелий, неон, аргон, криптон и ксенон (а также радон) обычно
помещают или в нулевую или в восьмую группу периодической
системы. Среди них только гелий является 5-элементом, все
остальные — р-элементы, поэтому логичнее их размещать
в VIII группе.
Двухатомные молекулы благородных газов в
невозбужденном состоянии неизвестны, так как число связывающих электронов
равно числу разрыхляющих. В возбужденном же состоянии с
разрыхляющих орбиталей часть электронов переходит на
энергетически более высокие связывающие орбитали, и в этом случае
были зарегистрированы короткоживущие двухатомные молекулы.
В то же время положительно заряженные двухатомные молекулы
типа Не^, Ne^, Ait, Ы или Xef известны, причем наибольшая
энергия связи у молекулы Не2 и она уменьшается в ряду
перечисленных молекул. Существование молекул типаЭз
обусловлено отнятием разрыхляющего электрона и превышением числа
связывающих электронов над разрыхляющими. Описаны также и
другие более сложные положительно заряженные молекулы,
например НеН+, АгН+, NeHe+ и даже СН3Хе+. Все эти
положительно заряженные молекулы могут существовать только в таких
газовых атмосферах, в которых отсутствуют вещества, способные
отдавать электроны. Как только положительно заряженная
молекула приобретает электрон, она сразу распадается на атомы,
например:
Не£г + Nar = 2Нег + Na^,
или
Не2+Г + е = 2Нег.
546
Таким образом, He2 и аналогичные другие молекулы ведут себя
и газовом состоянии как очень сильные окислители.
В дуговом разряде гелий реагирует со фтором и образуются
наряженные молекулы HeF+, HeF2+ и HeF2+. Самая устойчивая
из них — первая, энтальпия связи ее составляет примерно
50000 кал/моль, что меньше энтальпии связи иона Het равной
60000 кал/моль.
Образование соединений гелия возможно только при условии
возбуждения нейтрального атома и распаривания двух 1 s-элект-
ронов и перевода по крайней мере одного из них на 2 s-под-
уровень. Энергия для такого возбуждения чрезвычайно велика
Is2 - Is1 2s1 A H = 460 000 кал/моль
и недостижима никакими известными химическими реакциями.
Поэтому такое возбуждение возможно лишь в электрическом
разряде при низких давлениях гелия и в этих условиях масс-спек-
троскопически обнаружен целый ряд двухатомных молекул
различных элементов с гелием, например, HgHe, FeHe, Hel, HeS,
He2S. При повышении температуры или давления из-за
столкновений молекул они распадаются, и выделить эти соединения не
удается. Энтальпии связи в этих молекулах очень низки, например
у молекулы HgHe она составляет всего 70 кал/моль.
Известно соединение переменного состава PtHex, полученное
в твердом виде и отщепляющее гелий уже при нагревании выше
комнатной температуры. Структура и характер связи в этом
соединении не определены.
У гелия самая низкая Гкип (4,18°К) и самая низкая ЛЯИСП
(22 кал/моль). Это вызвано невозможностью возникновения
связей между атомами посредством пары электронов. Связь между
атомами гелия и других инертных газов — это слабая ван-дер-
ваальсова связь, сила которой прямо пропорциональна размерам
атомов и обратно пропорциональна энтальпии их ионизации.
Энергия ионизации ксенона несколько выше, чем у молекулы
кислорода, который с гексафторидом платины образует
соединение O^PtFe" (оксид гексафторида платины). Гексафторид
платины — сильнейший акцептор электронов, энтальпия сродства его
электрону несколько превышает энергию ионизации ксенона и
образование соединения Xe+PtFe" — гексафтороплатината
ксенона — оказывается энергетически выгодным. После открытия этого
соединения были обнаружены и исследованы многие другие
соединения ксенона со фтором XeF2, XeF4, XeF6, с кислородом ХеОз и
оксифториды XeOF2, XeOF4. Энтальпии образования соединений
ксенона довольно низки и отрицательны:
Xer + F2>r = XeF2 А #обр.,298 = — 30000 кал/моль,
18*
547
Хег + 2F2,r = XeF4 Д#обр.,298 =■ — 69000 кал/моль,
Хег -j- 3F2,r = Хе F6 Д#обР ,298 = — 96000 кал/моль,
Хег+3/202|Г= ХеОо Л#обР.,298 = — 96000 кал/моль.
Несмотря на отрицательные значения Д#°бр 298в обычных
условиях (нестандартные условия), все эти вещества распадаются на
составляющие их элементы. ХеОз в сухом виде — чрезвычайно
взрывчатое вещество. В одном растворе Хе03 — крайне сильный
окислитель.
Из щелочного раствора ХеОз выделена соль состава Ыа4ХеОб
(перксенонат натрия), производное ксеноновой кислоты H4Xe(V
И соль и кислота — окислители, более сильные, чем перманганат-
ион.
§ 12. ЭЛЕМЕНТЫ ПОБОЧНЫХ ПОДГРУПП
[d -ЭЛЕМЕНТЫ}
Изучение химии элементов начиналось с s-элементов. Обзор s-элементов
проводился как путем сравнения свойств элементов главных подгрупп I и
II групп элементов при увеличении числа s-электронов от одного до двух, так
и обсуждением влияния количества электронных уровней на физические и
химические свойства элементов и их соединений. Свойства /7-элементов нельзя
обсуждать изолированно, не возвращаясь к s-элементам, поэтому, когда шла
речь о ^-элементах, было обращено внимание на сравнении свойств элементов
всех главных подгрупп периодической системы, на закономерностях их
изменений в малых периодах и на влиянии ^-подуровня на свойства р-элементов
главных подргупп больших периодов.
Заполнение d-электронного подуровня происходит у элементов
больших периодов. d-Элементы расположены в больших периодах
непосредственно после s-элементов, а за ними следуют /7-элементы,
например в IV периоде: за калием и кальцием (s-элементы) идет
последовательность d-элементов: Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn,
затем период заканчивается /^-элементами: Ga, Ge, As, Se, Br, Kr.
Поэтому при обсуждении свойств d-элементов следует сравнивать
их свойства с 5- и р-элементами, т. е. рассматривать длинные
периоды целиком. Такое непрерывное усложнение материала курса
неорганической химии связано со структурой периодической
системы и непрерывным усложнением строения атома элемента с
ростом его атомного номера.
d-Элементами, или переходными, считают те элементы, у
которых в нейтральных, свободных атомах, d-подуровни частично
заполнены электронами. Однако к переходным элементам также
относят и другие элементы, у которых d-подуровни частично
заполнены электронами не у нейтрального атома, а у атома или
иона в каком-либо валентном состоянии. Так, медь, серебро и
золото считают переходными металлами, так как Си2+ имеет
конфигурацию d-подуровня 3 d9, Ag2+ —4 d9, Au3+ —5 ds. Эти
548
Са
Sc
Mn
Fe
Co
Ni
Cu
элементы считают переходными и потому, что их химические
свойства близки к свойствам обычных переходных элементов.
Первым d-элементом
является скандий с
конфигурацией 4s2 3d1 (рис. 14.35).
У следующих за скандием
восьми элементов (Ti, V, Cr,
Mn, Fe, Co, Ni, Cu) З^-под-
уровень заполнен частично
либо у свободного атома,
либо у ионов. С иттрия,
имеющего конфигурацию
f)S2 4d\ вновь начинаются Ti
переходные элементы: Zr,
Nb, Mo, Tc, Ru, Rh, Pd и Ag. v
У элементов, следующих за
лантаном, с конфигурацией о
Gs2 5d\ подуровень 4/
несколько более устойчив, чем
Ы. Поэтому у 14 элементов
электроны заполняют 4/-
подуровень, пока он не
заполнится у атома лютеция.
Лютеций имеет
конфигурацию 4/14 Ых 6s2. Поскольку
у La и Lu d-подуровень
заполнен лишь частично, а все
остальные подуровни
полностью, оба эти элемента
можно было бы отнести к
^-элементам. Однако, судя
по химическим и
физическим свойствам, все 15
элементов от La до Lu ведут
себя аналогично. Все эти
элементы называют
лантаноидами и относят к
отдельному семейству /-элементов.
Следует отметить, что
иттрий и скандий по своим
свойствам заметно
отличаются от обычных d-элемен-
тов. Иттрий в
значительной мере, а скандий во
многом напоминают
лантаноиды.
Третий ряд переходных Рис. 14.35. Заполнение подуровней
элементов начинается с гаф- мов IV периода
7 4
Zn з
Ga
Ge
As
Se
Br
Кг
1 S1 Pi C*
Ml
:*lhwl 1 II 1
IHI 1 | 1 | | | |
1UIHIW»! 1 1 1 1
N 1 1 1 || | 1
NblHlHHI I I I
N 1 1 1 1 1 1 1
IhimIhIhUM 1 1
1**1 1 1 1 II 1 1
hmtMHltmtl 1
ill i 1 1 1 1 ||
HkiMnUUUUh
hi 1 1 1 1 1 1 1
МшМшМФ
M | 1 1 1 I 1 1
1нИМнЫмш1'
И 1 1 1 i 1 1 1
кшиМнМшм
M 1 | 1 1 | | |
1и1н1м1н!ШММ;
1
1
\\
III 1 1 1 1 1 1 1
МнМшнЫик
\щ И 1 1 1 1 1
Ун1Н1Ш1нУнк
|Ш| 1 1 | 1 1 I
MhIhImhIhihIw
|МШ| MM
к1м1и1и1п1шМ*
J
L
n
\\
|А||ШШ | 1 I 1
1шЫмш*1икм
t
|*#t|*|H 1 1 1 1
1иКМнм1Ш|Нк
|4MN^| || 1 |
кмМНММиМ*
i
J
1МЦМАМ | 1 1 1
IHM^^tylHM*
iL
4s
1
2
2
2
2
1
2
2
2
2
1
2
2
2
2
2
2
2
3d
1
2
3
5
T
6
7
8
10
10
10
10
10
10
10
10
4p.
1
2
3
4
5
6
ч
T
i }
cu 1
I
T
3 1
i ^
О 1
a
T
ния, имеющего электронную конфигурацию 6s2 Ы2. Помимо
гафния в этот ряд входят элементы Та, W, Re, Os, Ir, Pt, Au, Hg.
В обычных состояниях окисления и в нейтральных атомах (за
исключением Au) у этих элементов 5^-подуровень заполнен частично.
После ртути, следующей за золотом, расположено еще
несколько элементов и, наконец, актиний с конфигурацией
электронов 7 s2 6 dl. Здесь снова возникает ситуация, аналогичная той,
которая наблюдалась у лантаноидов. Следует ожидать, что после
актиния начнется заполнение 5 /-подуровня и образуется новый ряд
из 15 элементов, подобный ряду лантаноидов. Однако в случае
лантаноидов электронная конфигурация определялась тем, что
4 f-орбитали энергетически заметно выгоднее 5 of-орбиталей.
У элементов непосредственно за актинием различие в энергии
5f- и бй-орбиталей не столь велико. Поэтому они и нейтральные
атомы таких элементов могут иметь электроны либо на 5 /-, либо
на 6 d-подуровнях, либо на тех и других одновременно. Лишь
после того как к конфигурации Ас добавится 4 или 5 электронов,
5/-орбитали становятся более устойчивыми. Аналогия в
химических свойствах заметна лишь у элементов, следующих за
америцием. Тем не менее сейчас принято считать, что группа из 15
элементов начинается с актиния и называется группой актиноидов.
При заполнении d-подуровня часто наблюдается характерная
особенность—повышенная устойчивость незаполненного,
наполовину заполненного и полностью заполненного d-подуровня. Эта
особенность отмечалась на р-подуровне, но там она проявлялась
значительно менее ярко. Наиболее устойчивое состояние у d-элемен-
тов соответствует наполовину заполненному d-подуровню
предпоследнего уровня, как это имеет место у металлов VII
группы— Mn, Tc, Re и полностью заполненному d-подуровню. Это
имеет место у металлов II группы — Zn, Cd, Hg.
Стремление к заполнению d-подуровня наполовину проявляется
в переходе наружного s-электрона на предвнешний d-подуровень
у Сг, Мо и Nb. Стремление к полностью завершенному d-подуров-
ню также приводит к переходу с s-подуровня на ^-подуровень
одного электрона у Си, Ru, Ro, Ag, Pt и Au. У палладия же на
^-подуровень переселяется сразу два s-электрона. Все это
налагает заметный отпечаток и на устойчивости тех или иных
валентных состояний.
Посмотрим, в какой последовательности заполняются орбитали
у атомов элементов IV периода (см. рис. 14.35). Аргон имеет
электронную конфигурацию 1 s2 2 s2 2 р3 3 рг 3 s2 3 р6. У следующих
за ним калия и кальция появляются 4 s-электроны. Со скандия
начинается заполнение З^-подуровня. Энергии 45- и 3^-орбиталей
близки между собой. Поэтому возможны переходы 4 s-электрона
на 3 d-подуровень при образовании более энергетически выгодных
йъ и d10 конфигураций.
У скандия, титана и ванадия последовательно увеличивается
число 3 d-электронов. Для хрома можно было бы ожидать конфи-
550
|урации 3 dA 4 s2, но более устойчивой оказывается другая
конфигурация 3d5 4s1. У меди можно было бы ожидать 3d9 4s2, но
осуществляется конфигурация 3d10 4s1. Это связано с особой
устойчивостью наполовину и целиком заполненных подуровней.
Высокая устойчивость 3 ^-конфигурации сказывается на
химических свойствах соответствующих элементов. Этим объясняется
малая устойчивость соединений 2-валентиого хрома и 3-валентно-
ю марганца. Например, в IV периоде все элементы образуют
устойчивые оксиды типа МО, за исключением хрома (СгО не
получен). Хотя известно кислородное соединение марганца состава
Мп203, однако предполагают, что это вещество является
соединением одновременно 2- и 4-валентного марганца (МпО-Мп02).
В главных подгруппах все атомы в невозбужденном состоянии
имеют аналогичные электронные конфигурации. У d-элементов эта
закономерность наблюдается не всегда. В ряде случаев переходные
элементы, стоящие в одной подгруппе, имеют отличающиеся
электронные конфигурации, например в V группе: V 3 d3 4 s2, Nb 4 d4
5 s1, Та 5d3 6 s2.
Такие же отклонения наблюдаются и у ^-элементов VIII
группы. Палладий — единственный элемент, у атома которого в
невозбужденном состоянии на внешнем уровне нет s-электронов. Однако
поскольку энергетические уровни nd и (n-\-\)s сравнительно
близки, разница в электронном строении атомов d-элементов,
находящихся в одной подгруппе, мало сказывается на их химических
свойствах.
У свободных атомов электроны d-подуровня никогда не
являются внешними электронами. Они заполняются электронами лишь
после заполнения s-подуровней оболочек с большим квантовым
числом, и, следовательно, их энергия .несколько выше последних.
Электроны d-подуровней участвуют в образовании связей лишь
после того, как s-электроны внешнего уровня окажутся
связанными.
d-Элементы отличаются от остальных элементов (в том числе
и от /-элементов) многообразием валентных состояний
(табл. 14.53). Из-за того, что по мере продвижения вниз по
подгруппе удаление электрона с ^-подуровня становится все более
легким, обнаруживается повышение * максимальных значений
валентных состояний. Наивысшие валентные состояния 8
обнаружены у двух элементов периодической таблицы — рутения и
осмия.
d-Элементы не образуют в кристаллическом состоянии и в
растворах одноатомных отрицательных ионов, так как прием
электронов для них не характерен и не сопровождается
уменьшением энергии. d-Элементы сравнительно легко теряют электроны
с образованием положительных ионов, поэтому все они — типичные
металлы.
d-Элементы III группы Sc, Yu и La образуют лишь ионы с
максимальной степенью окисления +3, которые имеют конфигу-
551
Таблица 14.53
№
группы
III
IV
V
VI
VII
VIII
I
II
Элемент
Sc
Y
La
Ti
Zr
Hf
V
Nb
Ta
Cr
Mo
W
Mn
Te
Re
Fe
Ru
Os
Co
Rh
Ir
Ni
Pd
Pt
Cu
Ag
Au
Zn
Cd
Hg
Валентное состояние
1
1
1
1
2
—
2
2
2
(2)
(2)
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
3
3
3
3
3
3
3
3
3
3
3
з
3
3
3
3
3
3
3
3
3
3
3
3
3
3
4
4
4
4
4
4
4
4
4
4
4
4
4
4
4
4
4
4
4
4
4
4
5
5
5
5
5
5
5
5
5
5
5
5
5
5
5
6
6
6
6
6
6
6
6
6
6
6
6
7
i 7
7
7
7
7
8
8
8
рацию инертного газа (Аг, Кг или Хе соответственно). При
образовании этих ионов удаляются по 2 электрона с 4 s-подуровня и
единственный электрон с 3 d-подуровня. Эти три s- и d-электроны
энергетически почти равноценны.
552
Остальные d-элементы (элементы побочных подгрупп IV—
VII групп) помимо соединений, в которых они обладают
максимальной валентностью, равной номеру группы, образуют
соединения с низкими валентными состояниями. Так, титан образует
соединения, в которых он 2-, 3-, 4-валентен, а ванадий 2-, 3-, 4- и
5-валентен. При образовании этих соединений используются
электроны внешнего s-подуровня и частично или все электроны d-под-
уровня.
В VIII группе максимальная валентность наблюдается лишь у
двух элементов — рутения (Ru04) и осмия (Os04, OsF8). У
остальных элементов этой группы валентность не превышает 6, а у
Со и Ni даже 3.
Элементы первой подгруппы Си, Ag и Аи являются
единственными элементами, которые имеют валентность, большую, чем
максимальная валентность группы. Так, атом меди может
использовать в химической связи 2 электрона, а атомы Ag и Аи даже
по 3.
Внешние подуровни атомов 5- и d-элементов побочной
подгруппы II группы (Zn, Cd, Mg) полностью заняты (соответственно
двумя и десятью электронами). Валентными могут быть только
лишь внешние s-электроны, и эти элементы всегда 2-валентньь
Обычно в низших валентных состояниях (1, 2 и 3) d-элементы
образуют соединения со значительным вкладом в химическую
связь ионной связи. В соединениях с более высокими валентными
состояниями возникают связи преимущественно ковалентного
характера.
Соединения с низшими валентными состояниями чаще всего
обладают основными, а с более высокими состояниями —
кислотными свойствами. Примерно так же делятся они на восстановители
и окислители.
Часто валентные состояния элементов не могут быть объяснены
каким-либо простым способом. Кристаллический TiCb вполне
устойчив в присутствии кислорода, но в водных растворах ион
Ti2+ не существуют. TiCl3 в кристаллическом состоянии также
устойчив, но в расплаве титан переходит в состояние Ti2+. Ионы
Ti3+ в водном растворе неустойчивы и переходят под действием
кислорода воздуха в Ti4+. В водных растворах большинство ионов
переходных элементов в свободном состоянии неизвестны. Они
энергично гидратируются, и их следует рассматривать как
комплексные ионы, например Ti(H20)62f и т. п.
Сравним свойства переходных элементов со свойствами
соответствующих элементов главной подгруппы той же группы.
Различие в свойствах элементов главной и дополнительной подгрупп
одной и той же группы тем сильнее, чем ниже валентность
элемента. Чем выше валентные состояния, тем ближе свойства элементов
и соответствующих элементов главных подгрупп.
Например, сравним свойства соединений марганца и хлора.
Атом Мп может использовать для образования связи все 7 элект-
553
ронов (2 s-электрона и 5 d-электронов). При валентности
марганца и хлора, равной 7, конфигурации атомов таковы;
1 s2 2 s2 2 р6 3 s2 3 /?6 и 1 s2 2 52 2 р6. Подобие электронных
конфигураций приводит к подобию свойств соединений 7-валентных Мп
и С1. Например, Мп207 и С1207 в обычных условиях — жидкости.
Они малоустойчивы, разлагаются от удара и нагревания. Оба
оксида — ангидриды сильных кислот формулы НЭ04. Оба оксида,
как и соответствующие им кислоты и соли, — окислители. Когда
марганец проявляет не высшую валентность, для образования
связи используются в первую очередь s-электроны и остается то
или иное число d-электронов. У хлора в его невысших валентных
состояниях имеются р-электроны. Это приводит к большому
различию в свойствах соединений Мп и С1, отвечающих невысшим
валентностям. Например, низший оксид хлора С120 представляет
собой газообразное вещество, являющееся ангидридом
хлорноватистой кислоты, тогда как низший оксид марганца МпО — твердое
кристаллическое вещество, характеризующееся основными
свойствами.
На рис. 14.36 представлены значения первой, второй, третьей
и четвертой энергий ионизации атомов элементов IV и V периодов.
У s-элементов первые энергии ионизации АЯЬ возрастают от
элементов I группы (К и Rb) к элементам III группы — кальцию и
стронцию. У элементов IV периода при переходе от скандия к
хрому энергии ионизации почти не изменяются, затем возрастают
к марганцу и железу, далее незначительно понижаются к меди и
резко возрастают к цинку. У цинка самая высокая первая энергия
ионизации среди d-элементов IV периода. Причина этого — в
заполнении всех предвнешних подуровней 18 электронами и
внешнего 5-подуровня двумя электронами — полностью заполненные
подуровни обладают повышенной устойчивостью.
Элементы, расположенные ниже в подгруппе и имеющие
больший размер, имеют меньшие энергии ионизации. Это правило
верно только для '5- и р-элементов. Но у d-элементов немало
исключений: так, у ниобия и молибдена энергии ионизации
меньше, чем у ванадия и хрома соответственно, хотя первые два
элемента из V периода, а последние два из IV. Имеются исключения
и в изменениях второй и третьей энергий ионизации ^-элементов.
Это вызвано очень большими значениями энергий
последовательной ионизации (АНи АЯ2, Д#3) ^-элементов IV и V периодов (и VI,
не показанного на рис. 14.36).
Температуры плавления d-элементов (рис. 14.37) выше температур
плавления соседних с ними 5- и р-элементов. Обычно при перемещении вниз по
подгруппе температуры плавления повышаются. Это относится к средним частям
периодов (IV—VIII группам). У s-элементов наблюдается противоположное
явление: при переходе вниз по подгруппе температура плавления понижается.
d-Элементы, находящиеся в тех же группах, что и s-элементы, обнаруживают
ту же закономерность. Наиболее тугоплавкие металлы в каждом периоде —
это элементы VI группы — хром, молибден и вольфрам. Наиболее высокая
температура плавления из всех известных простых веществ у вольфрама. Примерно
554
1700000
&Н кал/г атои
ИОНИЗ'
Sc
Рис. 14.36. Энтальпия ионизации
атомов s-, d- и р-элементов
300000
200000
100000 1
так же изменяются и энтальпии плавления (рис. 14.38). Они максимальны у
элементов VI группы молибдена и вольфрама в V и VI периодах.
Для большинства ^-элементов изменение энтропии при плавлении
равно ~2,1 кал/моль-град (рис. 14.39), причем при продвижении вдоль по периоду
энтропия плавления незначительно возрастает. Немногие элементы имеют
заметно отличные энтропии плавления, особенно в III и II группах Это вызвано
другим строением кристаллической и твердой фаз.
3500
3000 Ь
2500
2000
1500
1000
ЪОО
5 б
7 8 8 в 1 2
N группы
3001)
700U
0000
ЬООО
4000
3000 \- /I
2000
1000
'la
ал/
/
ч
к
1
Mo
Л
о^
\
\
\
0s#
1г
Л
// V7 Уи \
Ht / / V \v
pt
J I L
3 4 5 6 7
Рис. 14.37. Температура плавления d-
элементов
Рис. 14.38.
8 8 8 1?
N группы
Энтальпия плавления
d-элементов
Ход температур кипения ^-элементов (рис. 14.40) примерно повторяет ход
температур плавления, где также наблюдаются максимумы вблизи середины
периода. В IV периоде, наоборот, в середине периода наблюдается резкое
понижение температур кипения (Мп).
d-Элементы самые тяжелые, самые плотные простые вещества
среди остальных в периодической системе. Плотность d-элементов
возрастает с ростом номера периода (рис. 14.41). Наиболее
плотны металлы VI периода. Вдоль по периоду плотность сперва
возрастает, а затем падает. В IV периоде максимум плотности
приходится на медь, в V периоде максимум передвигается к середине
556
периода и приходится уже на родий. В VI периоде максимальной
плотностью обладает осмий. Противоположную картину
представляет ход изменения атомных радиусов, представленный для IV
и V периодов совместно s- и р-элементами для сравнения. Здесь
также заметно понижение атомных радиусов к середине периода
(рис. 14.42).
AS кал/моль град
/
/
3 4 5 6 7 8 8 8 12
N группы
Рис. 14.39. Энтропия плавления ^-элементов
Все ^-элементы имеют типичные металлические свойства. Они
хорошо проводят электрический ток, однако трудно найти
достаточно простое объяснение зависимости удельной
электропроводности d-элементов в зависимости от номера периода и группы
(рис. 14.43). Наивысшая электропроводность у элементов I
группы — золота, меди, серебра. Следующие за ними металлы —
иридий, молибден, родий, вольфрам.
По магнитным свойствам d-элементы делятся на три неравные
группы: ферромагнитные металлы — Fe, Co и Ni;
парамагнитные— Sc, Ti, V, Сг, Mn, Y, Nb, Mo, Tc, Ru, Rh, Pd, La, Та, W, Re,
Os, Ir, Pt и диамагнитные — Cu, Zn, Zr, Ag, Cd, Au, Hg.
Магнитная восприимчивость парамагнитных d-металлов выше, чем у
/^-элементов тех же групп, так как у d-элементов имеется большое
количество неспаренных электронов.
Все d-элементы, как и все металлы, — восстановители.
Восстановительная способность определяется как строением электронных
557
конфигураций, так и размерами ионов. Стандартный
восстановительный потенциал зависит главным образом от энергии
ионизации и энтальпии гидратации иона, которая зависит от размера
иона. Энергии ионизации щелочноземельных металлов намного
Gf,r/CM
6000
5000
4000
3000
2000
1000
кип
—
Та/
Hf У*
f Nb
w
^ в
' У
Мо ,
20
15
10
I
I
I
Os
Г
/
/
/w
1
1
1
Ir
^
\
\Аи
\
\
1
\
\
Ru
Rh
I
Н9
Mo,
/
/
/ Nb/
>•'
Pd
У
yg
Co Ni Cu X
3 4 5 6 7 8-881 2
N группы
Рис. 14.40. Температура кипения
^/-элементов
. ~.г.->
Рис. 14.41. Плотность ri-элементов
меньше, чем у d-элементов, поэтому, казалось бы, переходные
металлы должны находиться в ряду напряжений за водородом.
Однако цинк, например, стоит в ряду напряжений перед
водородом. Это связано с тем, что из-за малого размера иона цинка
энтальпия гидратации его довольно высока. Поэтому высокие
энергии ионизации ^-элементов компенсируются высокими значениями
энтальпий гидратации соответствующих ионов и в результате
этого их электродные потенциалы отрицательны, а
восстановительная способность достаточно высока.
558
Таблица 14.54
Процесс
Sc3 г + Ъе = Sc
Ti2 ' + 2е = Ti
V2+ + 2е .- V
Сг3' + 3e-=Cr
Mn2+ + 2e = Mn
Fe2+ + 2e ^= Fe
Fe3 ь + 3e = Fe
Co2+ + 2e = Co
Co3+ + Зг = Со
Ni2 *■ + 2e = Ni
Cu+ + e -= Cu
Cu24 + 2e = Cu
Zna+ + 2e = Zn
^298' B
—2,08
—0,37
—1,20
—0,74
—1,19
—0,44
—0,04
—0,28
+0,33
—0,23
+0,52
+0,34
—0,76
Рйс. 14.42. Радиусы атомов ^/-элементов
В табл. 14.54 собраны стандартные восстановительные
потенциалы ^-элементов IV периода. Только медь не способна
вытеснять водород из воды. Практически и остальные переходные
элементы также не растворяются в воде, но это объясняется
образованием различного типа нерастворимых гидроокисных пленок.
Из-за отрицательных значений стандартных восстановительных
потенциалов все переходные
Ъдельная^ тлектропроводность
|JOM См'
Ад
Си
8 8 1?
N группы
металлы, кроме меди,
должны растворяться в 1 н.
растворах кислот с
выделением водорода, если этот
процесс не осложняется
другими причинами.
С увеличением атомного
номера восстановительные
свойства металлов
постепенно уменьшаются, достигают
минимума у переходных
элементов I группы (медь и
др.) и возрастают к
переходным элементам II
группы (цинк и др.)- Тяжелые
элементы VIII и I групп за
свою инертность получили
название благородных
металлов.
Прочность однотипных
соединений rf-элементов
Рис 14 43. Удельная
электропроводность rf-элементов
уменьшается с ростом порядкового номера, достигая минимума у
элементов I и VIII групп и несколько возрастая у элементов
II группы. У элементов главных подгрупп устойчивость
соединений, в которых элемент проявляет высшее валентное состояние,
имеет тенденцию уменьшаться при движении вниз по подгруппе.
У d-элементов, наоборот, при движении вниз по подгруппе
устойчивость соединений, отвечающих высшим валентностям, растет.
Так, железо в обычных условиях имеет максимальное валентное
состояние 3 (6-валентные соединения не устойчивы), а его
аналоги — рутений и особенно осмий, — проявляют валентность 8.
Среди переходных элементов V группы производные низших
валентностей характерны лишь для ванадия и мало характерны для
ниобия и тантала.
С ростом порядковых номеров d-элементов в периоде
максимальное валентное состояние, отвечающее устойчивым соедине-
560
ниям, сначала увеличивается (за счет увеличения числа d-элект-
ронов), затем уменьшается. Например, у Sc, Ti, V, Cr, Mn макси*
мальное устойчивое валентное состояние равно номеру группы, в
то время как у железа в обычных условиях оно равно 3, а у меди
и цинка — 2. Соответствующим образом изменяется и устойчивость
низших валентных состояний, например 2-валентного: если TiO
и VO — сильные восстановители, то NiO, ZnO и CuO не обладают
такой особенностью.
У элементов IV периода сравнительная легкость отрыва
4 5-электронов (по сравнению с d-электронами) приводит к тому,
что для этих элементов низшее валентное состояние чаще всего
равно 2.
Рассматривая элементы главных подгрупп периодической
системы, наблюдаем большую аналогию свойств элементов по
вертикали. Для переходных элементов характерно также
горизонтальное сходство. В ряде случаев сходство между соседними
переходными элементами по горизонтали больше, чем между соседями
по вертикали. Например, Fe, Co, Ni имеют больше общего в
свойствах, чем Fe, Ru и Os.
В химических реакциях с участием d-элементов образование
связи обусловлено как s-электронами, так и всеми или частью
d-электронов. При этом возникают соединения с различными
валентными состояниями d-элементов. Например, у кремния гало-
гениды имеют только один состав Sir4, хлоридов же титана
известно три: TiCb, TiCl3 и TiCl4.
То же самое касается и гидроксидов металлов, которых у
каждого переходного элемента может существовать по нескольку, в
зависимости от валентного состояния. Оно же резко сказывается
на кислотно-основных свойствах гидроксидов. Гидроксиды,
отвечающие низшим валентным состояниям, обычно проявляют
основные свойства, а отвечающие высшим валентным состояниям —
кислотные. Граница между теми и другими чаще всего
соответствует степеням окисления 3 и 4 и смещается к более низким
степеням окисления с ростом атомного номера. Гидроксиды,
отвечающие этим степеням окисления, часто амфотерны.
Гидроксиды Sc(OH)3, Y(OH)3 и La(OH)3 — типичные
основания. Гидроксид Ti(OH)4 амфотерен. Как титановая кислота он
растворим в щелочах с образованием титанатов. Гидроксид
Zr(OH)4 также амфотерен и растворим в щелочах (образуются
соли циркониевой кислоты — цирконаты). Ванадий в 5-валентном
состоянии обладает кислотными свойствами. Пентоксид ванадия
V205 растворим в воде и раствор имеет кислую реакцию. Такому
состоянию ванадия соответствуют только соли — ванадаты,
например NaV03. Ванадий в 4-валентном состоянии амфотерен.
Оксид V02 растворим в щелочах с образованием ванадитов, в то
же время в кислых растворах существует гидратированный ион
V4+. Гидроксид хрома Сг(ОН)3 типично амфотерен. Ему
соответствуют соли типа Cr(N03)3 и NaCr02 (хромиты). Хрому в 6-ва-
561.
лентном состоянии соответствуют две кислоты — хромовая Н2Сг04
(соли — хроматы, Na2Cr04) и двухромовая Н2Сг207 (соли — би-
лроматы, К2Сг207).
На соединениях марганца особенно легко проследить
изменение кислотно-основных свойств гидроксидов при изменении
валентного состояния металла. Так, Мп(ОН)2— слабое основание,
растворимое в кислотах с образованием иона Мп2+. Мп(ОН)4 —
типично амфотерное основание, которое может растворяться в
кислотах (ион Мп4+ не устойчив) и в щелочах с образованием
манганит-иона МпОз-, которому соответствует марганцеватистая
кислота Н6Мп03. Марганец в 6- и 7-валентном состояниях имеет
только кислотные свойства и существует в виде манганат-ионов
(Мп04~) и перманганат-ионов (МпОГ), которым соответствуют
марганцеватая кислота Н2Мп04 (соли манганаты, K2MnOi) и
марганцевая кислота НМ04 (соли перманганаты, КМп04).
Гидроксид железа Fe(OH)2 — слабое основание. Гидроксид
Fe(OH)3 амфотерен с преобладанием основных свойств. Fe(OH)3
и оксид Fe203 соответственно растворяются в щелочных растворах
или сплавляются с щелочью с образованием ферритов (KFe62).
Соединения с 6-валентным железом гораздо менее устойчивы,
обладают только кислотными свойствами, соли их, например
K2Fe04 — ферраты, получаются сплавлением железа с
азотнокислым калием.
Си(ОН)2 обладает слабокислотными свойствами, он растворим
в концентрированной щелочи с образованием солей —купратов,
например Na2Cu02. Гидроксид цинка типично амфотерен, при
растворении в кислоте получается раствор Zn2+, при растворении
в щелочи раствор соответствующего цинката (K2Z11O2).
Оксиды тех d-металлов, у которых начинается заполнение
.d-подуровня, имеют высокие отрицательные изобарные потенциалы
и энтальпии образования. Металлы легко окисляются, а оксиды
трудно восстанавливаются (Sc, Ti и V). По мере увеличения
порядкового номера ^-элемента изобарные потенциалы оксидов
уменьшаются и возрастает легкость их восстановления. Оксиды
железа, кобальта, никеля и меди легко восстанавливаются
водородом при сравнительно низких температурах.
В табл. 14.55 приведены термодинамические характеристики
реакций образования оксидов
Мк+1/202.1 =М0К.
Оксиды типа МО для скандия и хрома неизвестны. При
стандартных условиях и температуре все изобарные потенциалы
отрицательны (рис. 14.44), что говорит о возможности взаимодействия
металлов с кислородом. Металлы устойчивы в атмосфере
кислорода только благодаря крайне медленному процессу окисления
при комнатных температурах.
Даже при температурах 2000°К все оксиды металлов
сохраняют отрицательные значения потенциалов образования, за
,562
Таблица 14.55
Оксид
ScO
ТЮ
VO
СЮ
MnO
FeO
CoO
NiO
CuO
ZnO
ЛЯ298,обр'
кал/моль
— 123 900
—100 000
—
—92 000
—63 700
—57 200
—58 000
—57 100
—33 200
AS298,o6p'
кал/моль -град
—23,52
—22,25
—
—17,82
—18,09
—20,18
—22,42
—22,07
—24,05
AG298,o6p'
кал/моль
—116 900
—93 400
—
—86 700
—58 300
—51 200
—51 300
—30 500
—76 000
AG2000,o6p
кал/моль
_
—76 900
—55 500
—
—56 400
—27 500
— 16 800
—13 200
+7 000
—25 100
исключением меди. Это
означает, что оксид меди,
помещенный при 2000° К в
атмосферу кислорода с
Ро2=1 атм (стандартные
условия), становится
неустойчивым и диссоциирует.
Все остальные оксиды при
тех же условиях не
диссоциируют. Оксид меди —
наименее устойчив в
рассматриваемом ряду оксидов.
Рис. 14.44 Изобарные
потенциалы образования оксидов
^'-элементов IV периода
1 — М02ооо°к; 2 — М0298°к; 3 ~
М2О3)2000о К; 4 — М20з,2980К
Элементы от скандия до железа образуют устойчивые при
стандартных условиях оксиды типа М2О3. Термодинамические
характеристики реакций образования
2МК | 3/2 02рГ = М208.к
приведены в табл. 14.55,а и представлены на рис. 14.44.
Хотя и известны составы, отвечающие формуле М203 у
кобальта, никеля и даже у меди, но свойства их не определены. При
переходе от скандия к железу отрицательные значения изобарных
потенциалов оксидов М203 резко уменьшаются, указывая на
различную зависимость устойчивости 3-валентного состояния по
сравнению с 2-валентным при заполнении d-подуровня.
AG с ,кал/моль
оор
'100000
-200000
-300000
-400000
J—I—I I I |_] 111
Sc Ti V Сг MnFeCo N1 Си In
Металл
5ба
Таблица 14.55а
Оксид
Sc203
Ti203
V203
Craos
МП9О3
Fe203
ЛЯ298,обр'
кал/моль
—411000
—362 900
—296 100
—270 000
—232 100
—196 500
Л,ь298,обр'
кал/моль- град
—68,98
—69,33
—64,02
—65,46
—62,28
—64,98
ЛСг298,обр-
кал/мо ль
—390 400
—342 200
—277 000
—250 500
—213 500
—177 100
Л/~0
А(/2000,обр'
кал/моль
—273 000
—224 200
—168 100
—130 800
—107 500
—66 500
Влияние температуры на устойчивость оксидов МО и М203 очень различно и
-сильнее сказывается на оксидах М2О3. Последнее является следствием того,
что в реакции образования М203 в расчете на один атом металла участвует
в 1,5 раза больший объем кислорода. Это приводит к большему изменению
энтропии в реакции и соответственно к большему возрастанию члена TAS при
повышении температуры.
Изобарные потенциалы образования оксидов М203 имеют большие
отрицательные значения по сравнению с потенциалами оксидов МО. Однако это не
означает, что оксиды М203 устойчивее оксидов МО. Пользоваться изобарными
лотенциалами для сравнения устойчивости соединений можно только в случае
однотипных соединений (т. е. соединений, имеющих одинаковые формулы и
образующихся из веществ в соответственно одинаковых агрегатных состояниях).
Судить об устойчивости соединений, образующихся из веществ с участием
газовой фазы, можно по парциальному давлению газообразного вещества,
возникающего при диссоциации.
Изобарные потенциалы реакций диссоциации численно равны изобарным
потенциалам реакций образования, но противоположны по знаку.
МОк = Мк + 1/202.г AG&0 = -AG°6pMO ,
м2о3,к = 2МК + з/2 о2г да^0з = - Д0°6р.МгОз.
Давления диссоциации, являющиеся константами равновесия этих реакций,
определяются из уравнений
•ДОобр.мо--4-^^.
А<о-
jm2o,
>Ы2
ДО°м n = - Да°йо.МЛ = - 4.576Г Ig р%,
__ АЧзбр,м2о3
да
обр. МО
над МО к
Ы\
2надМ203)К
4,576-Г -°-и2надт2и3)К 4,576- Т
Давления диссоциации при стандартной температуре настолько малы, что
не имеют физического смысла, однако ими можно пользоваться для сравнения
устойчивости соединений (табл. 14.56). Давления диссоциации оксидов М203
Таблица 14.56
564
Оксид
ScO
TiO
VO
СЮ
MnO
FeO
CoO
NiO
lgPQ
298 °K
— 171,4
— 137,0
—
— 127,2
—85,5
—75,1
—75,2
, атм
2000 °K
— 16,8
—12,1
—
—12,3 J
—6,0
—3,7
—29 |
Оксид
CuO
ZnO
Sc.,03
TiA
VA
Cr203
MnA
1 Fe203
igP0
KJ2
298 °K
—44,7
— 111,4
—190,9
— 167,3
—135,4
— 122,4
—104,4
—26,6
, атм
2000 °K
-1,5
-7,7
—19,9
— 16,3
— 12,2
—9,5
-7,8
—4,8
пыше, чем у оксидов МО, так как соединение, имеющее большее количество
кислорода (или другого элемента, способного переходить в газовую фазу) на
один атом другого элемента, характеризуется большим давлением диссоциации.
При 2000°К оксиды FeO, CoO, NiO, CuO, ZnO, Cr203, Mn203 и Fe203 имеют
измеримые давления кислорода. Следует только помнить, что в
действительности диссоциация большинства оксидов М203 не протекает сразу до металла и
кислорода, а проходит через ряд промежуточных ступеней, благодаря которым
15 действительности давление кислорода много выше. Так, в системе Fe—О
последовательное отщепление кислорода совершается через стадии
Fe203 -^.Fe304 -♦ FeO -> Fe
3Fe203)K -= 2Fe304 K + 1/2 02>г К - P#,
Fe304 K = 3FeOK + 1/2 02 r К - Р#я,
FeOK-FeK+l/2 02r К--P^.
Известно много кристаллических соединений, особенно оксидов
и сульфидов переходных элементов, в состав -которых атомы
входят не в соотношениях целых чисел, как требуется по закону
простых кратных отношений. Это вызвано особенностями
кристаллических решеток, их несовершенствами, когда место катиона или
аниона может остаться свободным, не занятым (дефекты Шоттки)
или же дополнительные количества атомов внедряются между
ионами (дефекты Френкеля), при этом электрическая
нейтральность кристалла поддерживается изменением валентности другого
иона.
Основное отличие оксидов и сульфидов d-элементов от оксидов
и сульфидов элементов главных подгрупп заключается в том, что
у d-элементов они часто являются соединениями переменного
состава, например оксид титана может быть получен любого
состава— от TiOo,88 До TiOi,2o (при 1200°С). Таким образом,
неправильно говорить, что, например, титан образует оксид TiO, так
как он существует в области гомогенности TiOo.88-i.2o (1200 °С).
Все d-элементы способны образовывать соединения, в которых
ятомы металла связаны между собой непосредственно. Такие связи
обнаружены как в структурах твердых тел, так и в отдельных
молекулах. Связи Э—Э не являются редкостью — они встречаются
и среди /^-элементов. Это — связь О—О в пероксиде водорода,
аналогичные связи S—S, связи С—С в органических соединениях,
связь N—N в N204, но у rf-элементов это явление очень широко
распространено, хотя его изучение началось совсем недавно. В
молекулах карбонилов типа Мп2(СО)ю атомы металла
непосредственно связаны друг с другом. В ряде оксидов также обнаружено
существование связей М—М, например в диоксиде молибдена
Мо02. В структуре этого оксида атомы молибдена образуют цепи,
состоящие из чередующихся атомов молибдена и двух атомов
кислорода, причем расстояния Мо—Мо различны. Короткое рас-
565
стояние приписывают образованию связи Мо—Мо. Доказано
существование связей Fe—Fe в оксиде железа FeCb (вюстите). С
увеличением нестехиометрии (^bFe0.x) количество связей Fe—Fe
возрастает.
s29eMK- «ал/моль- град
ь 5,ae.MCl, „.кал/моль град
28 ^
11000
10000
9000
2. К
Рис. 14.45. Термодинамические
характеристики дихлоридов ^-элементов IV
периода
* __
Рис. 14.46. Изобарные потенциалы
различных хлоридов ^-элементов IV
периода:
1 — МС1; 2 — МСЬ; 3 — МС13
-50 000
-100000 Ь
0^00000
-700000 h
Sc Ti V Cr Mn Fe Co Ni Cu Zn
Металл
V Cr Mn Fe Co Ni Си In
Максимальная валентность металла в хлоридах d-элементов
часто значительно ниже, чем максимальная валентность того же
металла в оксиде. Это вызвано тем, что геометрически в
пространстве легко расположить в 2 раза меньшее количество атомов
кислорода по сравнению с атомами хлора при одной и той же
валентности металла. Поэтому несмотря на то, что могут
существовать такие устойчивые оксиды, как V2O5, Сг03, МпОг, ИегОу, Ru04,
Os04, этим металлам отвечают устойчивые хлориды с
меньшей валентностью металла: VCl4, CrC!3, MnCl3, ReCl6, R11CI3,
OsCl4.
При движении по подгруппе вниз устойчивость высших гало-
генидов возрастает. Так, СгСЦ, крайне неустойчив, МоС15 и WC16
вполне устойчивы. Аналогично, VC14 и ТаС15 устойчивы примерно
в одинаковой степени. Такое поведение может быть объяснена
566
тем, что при переходе вниз по подгруппе размер катиона
возрастает. (Вспомните, что у р-элементов при переходе вниз по
подгруппе возрастает устойчивость низших валентных состояний.)
Термодинамические характеристики галогенидов состава МСЬ
в зависимости от положения d-элемента в периоде изменяются
сложным образом (рис. 14.45). Кривая зависимости AG^s.oep
повторяет ход зависимости Д#298,обР хлоридов и отличается ог
кривой AG298.o6P оксидов большей устойчивостью хлорида
марганца. Хотя энтропии металлов и хлоридов, а также изменение
энтропии при образовании хлоридов из простых веществ
изменяются очень сложным образом в зависимости от структур
веществ, изменение изобарного потенциала в основном определяется
изменением энтальпии образования хлорида: член T&S при
стандартной температуре вносит незначительное влияние на
Д#298,обр МС12.
Изобарные потенциалы трихлоридов МС13 различаются между
собой значительно сильнее, чем дихлоридов. Трихлорид марганца
МпСЦ не нарушает равномерного уменьшения устойчивости
трихлоридов при переходе от ScCl3 к FeCl3 (рис. 14.46).
В общем случае тенденцию к образованию соединения с тем или иным
валентным состоянием металла можно оценить, исходя из термодинамических
данных. В табл. 14.57 приведены стандартные энтальпии и изобарные
потенциалы образования хлоридов некоторых ^-элементов.
Таблица 14.57
Хлорид
FeCla
FeCl3
CrCl2
CrCI,
TiCl,
Tici;
TiCl4
VCIa
VC13
VC14
АЯ298,обр'
кал/моль
—81 900
—26 400
—93 000
—132 500
—123 000
—170 000
—192 100
—115 000
—163 000
—165 000
ло298,обр'
кал/моль
—72 700
—79 200
i -83 600
—116 000
—111900
—153 200
— 176 200
—104 000
—146 400
—145 200
кал/экв
—36 350
—26 400
—41800
—38 700
—55 950
—51 060
—44 050
—52 000
—48 800
—36 350
Интересно, что чем выше валентное состояние металла, тем больше
различие между Д#£бр и AGo6p из простых веществ, например:
FeK + С12г = FeCl2>K Д#£98 =- — 81 900 кал/моль,
AGSqs ~ ~~ 72700 кал/моль,
567
AW — AGO = __ 9200 кал/моль,
FeK -4- 1/2 C!2 r - FeCI3>K АЯ^98 -= — 96400 кал/моль,
AG298 - = — 79200 кал/моль,
АЯ« — AG0 -= — 17200 кал/моль.
Это вызвано тем, что с ростом валентного состояния металла в реакции
участвует большее число молей газообразного вещества и в связи с этим на
численное значение AG0 в большей степени сказывается влияние члена TAS0.
По величинам изобарных потенциалов образования галогенидов, оксидов*
сульфидов и других соединений одного и того же элемента в его различных
валентных состояних нельзя судить об относительной устойчивости соединений.
Действительно, для хлоридов железа*
FeK + С12 г -= FeCl2 K AG°98 = — 72 700 кал/моль,
FeK -|- 3/2 С12 г -= FeCl3,K AG°98 = — 79 200 кал/моль.
Образование трихлорида железа сопровождается большим изменением AG0 и„
казалось бы, FeCl3,K должен быть устойчивее FeCl2,K.
К решению вопроса об устойчивости соединения можно подойти двумя
путями. 1. Определить температуры, при которых давления диссоциации разно-
валенгных соединений одинаковы. Вещество, достигающее требуемого значения
давления диссоциации при более высокой температуре, более устойчиво. 2.
Определить давления диссоциации тех же соединений при одной и той же
температуре. Вещество, обладающее меньшим давлением диссоциации, более
устойчиво.
В первом случае для определения температур одинаковых давлений
диссоциации необходимо знание значений АН0 и AS0 реакций. Во втором случае
достаточно знать AG298 реакции и определить давление диссоциации при
298°К. Найдем давление диссоциации дихлорида железа при 298СК:
FeCI2fK -= FeK + Cl2r AG°q8 - - AG2°9 ,обр FeCIs = - 4,576-298 \g Pcl>
72700
lgPr1 -=— з—53,4 (атм).
с1г 4,576-298 ;
Для трихлорида железа: •
FeC13K = FeK -г 3/2 С12г AG2°98 = - AG2098>o6p Рес1л ■= - 4,576-298. Ig P^
79 200-2
lg Prl = — = — 38,2 (атм).
6 С1г 4,576-298-3 v '
Столь низкие давления диссоциации не имеют физического смысла и,
разумеется, не являются давлениями диссоциации. Они лишь характеризуют
устойчивость вещества по отношению к распаду на простые вещества. Дихлорид
железа устойчивее трихлорида.
Чтобы не проводить каждый раз расчеюв давления, диссоциации,
достаточно рассчитать AG^ обр на 1 г-экв металла, т. е. в случае хлоридов
разделить AGq6p (кал/моль) на число атомов хлора в молекуле. Так, для FeCl*
и FeCl3
п — 72700
AG°98 = = — 36 350 кал/г-экв,
568
AG298 =
— 79200
3
— 26 400 кал/г-экв.
Более устойчиво то вещество, у которого меньшее положительное и большее
отрицательное значение эквивалентного изобарного потенциала образования.
Энтальпии образования галогенидов падают от фторидов к
йодидам. Способность d-элементов проявлять максимальные
валентности также падает от фторидов к йодидам. Если переходный
элемент образует несколько галогенидов, то высшие галогениды
проявляют окислительные свойства, а низшие —
восстановительные.
Из-за возможности существования переходного элемента в
различных валентных состояниях соединения переходных элементов
очень склонны к диспропорционированию:
2ТЮ13,к = TiCl2(K + TiCl4,r,
2Т1С12,к - TiK + TiCl4.r,
4FeOK = Fe304 -f- FeK.
Большинство элементов периодической системы может
выступать в качестве комплексообразователей, однако по способности
к комплексообразованию они сильно отличаются. Под
способностью к комплексообразованию понимается одновременно
способность давать как прочные, так и разнообразные комплексные
ионы и соли.
/^-Элементы могут быть как комплексообразователями, так и
лигандами, т. е. элементами, строящими непосредственное
окружение центрального атома — комплексообразователя. Следует
особо подчеркнуть, что последнее свойство присуще только р-эле-
ментам. Многие /^-элементы могут быть комплексообразователями,
но значительно более слабыми по сравнению с d-элементами.
У р-элементов II периода (от бора до фтора) координация
возникает благодаря участию 2 5- и 2 р-орбиталей центрального атома,
поэтому координационное число не больше 4. У элементов III и
нижестоящих периодов в образовании координационных связей
могут участвовать кроме наружних s- и р-орбиталей также ^ d-op-
битали, при этом координационные числа повышаются до 5 (PF5),
6(PF-6), 7(IF7) и даже до 8(XeF8).
Все rf-элементы — активные комплексообразователи.
Способность к образованию комплексных соединений в сильной степени
зависит как от количества d-электронов (т. е. от места элемента
в большом периоде), так и от количества электронных уровней
(т. е. от номера периода). Наименее способные к
комплексообразованию элементы находятся в главных подгруппах I (щелочные
металлы), VII (галогены) и VIII (благородные газы) групп.
Максимум способности к комплексообразованию приходится на d-эле-
менты и особенно на те d-элементы, которые расположены в по-
569
бочной подгруппе VIII группы (Fe, Co, Ni, Pt и др.). К ним
примыкают элементы побочных подгрупп I и II групп (Си, Ag, Zn
и др.) и побочной подгруппы VII группы (Мп). Это —элементы
с незаполненными d-подуровнями. При переходе вдоль большого
периода отчетливо наблюдается -возрастание способности к
комплексообразованию примерно в обоих направлениях к центру
периода. Хотя элементы малых периодов (s- и р-элементы)
значительно худшие комплексообразователи, но и у них наибольшая
склонность к комплексообразованию также приходится примерно
на середину периода.
При переходе вниз по подгруппе способность к
комплексообразованию изменяется сложным путем. Она связана с зарядом иона
и его радиусом. Невысокие заряды ионов и их большие радиусы
приводят к уменьшению прочности комплексных ионов, но при
этом часто наблюдается большее разнообразие комплексных
соединений. Наоборот, высокозарядные ионы и их малые радиусы
способствуют увеличению прочности комплексов, но при этом
снижается число возможных комплексных соединений. У s-элементов
наблюдается уменьшение способности к комплексообразованию
при переходе сверху вниз, но в побочных подгруппах часто
замечается противоположное явление.
Количество комплексных соединений, образуемых элементом,
проявляется в многообразии координационных чисел. У
s-элементов I периода известны лишь координационные числа 1 и 2,
причем число 2 встречается очень редко. У s-элементов II и
нижестоящих периодов всегда сохраняются координационные числа
предшествующих периодов и наблюдается увеличение разнообразия
координационных чисел, но это связано не с участием р- или
d-орбиталей, а с увеличением размера иона-комплексообразова-
теля и вытекающей отсюда большей геометрической способности
координировать ионы.
По мере заполнения d-подуровня максимальные
координационные числа d-элементов постепенно уменьшаются (табл. 14.58).
Таблица 14.58
Период
IV
V
VI
III
8(Sc)
8(Y)
9(La)
IV
8(Ti)
8(Zr)
8(Hf)
V
8(V)
8(Nb)
8(Ta)
VI
8(Cr)
8(Mo)
8(W)
Группы
VII
8(Mn)
7(Tc)
9(Re)
7(Fe)
8(Ru)
6(Os)
VIII
6(Co)
6(Rh)
6(Ir)
i
6(Ni)
6(Pd)
j 6(Pt)
IX
6(Cu)
6(Ag)
6(Au)
У ^-элементов наблюдается отчетливая тенденция к
достижению максимума устойчивости (минимум в значении константы
нестойкости) комплексного иона по мере приближения -к середине
^-последовательности элементов. В табл. 14.59 приведены констан-
570
Таблица 14.59
Число
(/-электронов
0
1
2
3
4
5
6
7
8
9
10
Ион
Са2+
Sc2+
Ti2+
V2^
Cr2 +
Mn2+
Fe2^
Co2+
Ni2+
Cu2+-
Zn2+
Анест
10-io,6
Ю-12.7
10-13.б
Ю-14.4
10-I6.1
Ю-18,5
,0-18.9
,0-16.6
Ион
Sc3f
Ti3 +
уз^
Cr3^
Mn3 +
Fe3
Co3-
Ni3^
Cu3f
Ga3'
•^нест
,0-23.1
,0-21,3
,0-25,9
,0-25,1
Ю-26
Ю-20.3
Таблица 14.60
ты нестойкости комплексов d-элементов (с этилендиаминтетрааце-
татом) в зависимости от числа d-электронов у иона.
Вторичная периодичность замечена не только у р-элементов
при переходе по подгруппе сверху вниз, но и у d-элементов при
передвижении вдоль периода. При этом наблюдаются повышенные
стабильности заполненных наполовину (d5) и полностью (d10)
^-подуровней. Наблюдается также аналогия во многих свойствах
между парами комплексных соединений ионов с числами d-элект-
ронов, различающихся на 5 электронов, т. е. между ионами с
электронной структурой dm и d5+m. Например, комплексы со
структурой центрального иона dz и ds имеют одинаковые
координационные числа, одинаковые скорости реакций с их
участием, близкие спектры поглощения
и т. п.
В табл. 14.60 даны константы
скоростей замещения молекул
воды в аквакомплексах
некоторых d-элементов в зависимости
от числа d-электронов на d-под-
уровне.
Молекулы воды быстрее
замещаются в акваионах со
структурой d4 и d9 (разница 5
электронов). Наинизшие скорости
замещения воды наблюдаются в
комплексах с d? и d8 (разница
5 электронов).
Ион
V2 +
Сг2+
Мп2 +
Fe2+
Со2-
Ni2-
Cu2+
Zn2+
Число
^-электронов
3
4
5
6
7
8
9
10
k
102'0
И8,4
106'7
106'1
105'4
104'2
108'4
107'4
571
Пять d-орбиталей энергетически равноценны только в
свободных, изолированных атомах или ионах. Однако d-орбитали иона,
расположенного в окружении шести отрицательно заряженных
ионов (октаэдрическое окружение) или четырех (тетраэдрическое
окружение), теряют свою равноценность из-за действия поля
окружающих ионов. Так как d-орбитали будут находиться на раз-
гура
иия
О к тоэдрическое
ок ружение
7етраэдрическое
окружение
\Число неспор элегтй
октаэдр
ВС \НС
тетраэдр]
ВС \НС
ш
ПИП
газа
BCQjjjf) ЩЩ] НС
ГТТ*1 ГП
ИГЛ ГП
вс gift] цЩ нс
ПТП
ЙМН|
mm ■
|И|ЩЩ
шг
ВС ЧШ
8С тшг
тжгнс
гтттп ггп
бС ШЯ ШГ нс
бСРДШ] РХП
гтттт |t|ti i
таг
15ШГ
шшг
Рис. 14.47. Расщепление rf-подуровня в октаэдрическом окружении лигандаыи
личных расстояниях от отрицательных зарядов окружающих ионов,
то электроны будут стремиться занять те d-орбитали, которые
наиболее удалены от отрицательно заряженных ионов окружения.
В октаэдрическом окружении энергетически более выгодны только
три равноценные орбитали dxy, dyz и dzx, так как они расположены
в пространстве между зарядами. Две другие орбитали (d^ и dx*-y')
превращаются в энергетически менее выгодные, хотя и
равноценные друг с другом орбитали.
572
Количество
неспаренных
электронов
5
4
3
2
1
0
S\
: jcX\
-А X
s xXcX
i i i t i i i i i Т
/ 234 56789 10
Количество d-электроноб
Рис. 14.48. Число неспаренных
электронов у ионов d-элементов в
зависимости от силы поля лигандов
d-Орбитали в окружении четырех отрицательных зарядов
также становятся не равноценными. Однако в этом случае
энергетически более выгодны (т. е. имеют меньший запас энергии) две
орбитали dZ2 и dx*-y*i а три оставшиеся dxy, dyz и dXZ9 будучи
равноценными между собой, располагаясь ближе к
отрицательным ионам окружения, превращаются в энергетически менее
выгодные (т. е. имеют больший запас энергии).
Магнитные свойства
комплексных ионов d-элементов
находятся в прямой зависимости
от числа неспаренных
электронов иона-комплексообразова-^
теля. В октаэдрическом
окружении (рис. 14.47) первыми
заполняются согласно правилу
Гунда три нижележащие d-op-
битали dXY, dyz, dxz.
Следующий, четвертый, d-электрон
имеет две различные
возможности вступления на d-орбита-
ли, или на более низкую
орбиталь, образуя пару электронов,
или на более высокую
орбиталь, в неспаренное состояние.
В первом случае из четырех электронов только два сохраняют
неспаренное состояние. Такая конфигурация носит название
низкоспиновой (НС). Во втором случае все 4 электрона неспарены и
конфигурация называется высокоспиновой (ВС).
Выбор той или иной конфигурации определяется плотностью
электронных зарядов окружающих ионов. Если ионы окружения
обладают сильным электрическим полем, достаточным для
выталкивания электронов с d22 - и dx*—y* - орбиталей, и энергия этого поля
превышает энергию спаривания электронов, электрон вступает на
нижележащую орбиталь. В этом случае образуется низкоспиновое
состояние. Если электрическое поле окружающих ионов
недостаточно для преодоления сил отталкивания между двумя
электронами в одной орбитали (слабое кристаллическое поле), электрон
заполняет dZ2 - или d^-y2 - орбиталь и возникает высокоспиновое
состояние. Заполнение электронами d-подуровня с образованием
высокоспиновых состояний согласуется с правилом Гунда, и
магнитные свойства ионов должны изменяться в соответствии с
числом неспаренных электронов (рис. 14.48). Пятый, шестой и
седьмой d-электроны имеют две возможности заполнения d-подуров-
ня — с образованием высокоспинового или низкоспинового
состояния. Остальные 3 электрона заполняют оставшиеся места вне
зависимости от силы поля окружающих ионов.
Точно таким же способом можно рассмотреть порядок
заполнения d-подуровня иона, находящегося в тетраэдрическом окруже-
57а
иии. Однако низкоспиновое состояние здесь мало выгодно, так как
энергия электростатического отталкивания двух электронов, на*
ходящихся в одной ячейке, значительно выше, чем энергия
выталкивания электронов с орбиталей dxy, dyz, dxz в поле близлежащих
ионов тетраэдрического окружения. Поэтому в тетраэдрическом
окружении заполнение d-подуровня происходит в согласии с
правилом Гунда и соответствует максимальному количеству неспа-
ренных электронов. Низкоспиновые тетраэдрические комплексы
пока не обнаружены. На рис. 14.48 зависимость числа неспарен-
ных электронов от общего числа электронов на ^-подуровне для
низкоспиновых тетраэдрических комплексов представлена
пунктирной линией.
Лиганды — Н2О, F" — относятся к числу лигандов,
оказывающих слабое воздействие на ион-комплексообразователь и в их
окружении ион-комплексообразователь находится в состоянии,
когда электроны стремятся занять максимальное число ячеек, т. е.
образуется высокоспиновое состояние. NH3, CN~, NO<T —
лиганды, обладающие сильным действием на
ион-комплексообразователь. В их присутствии электроны заполняют d-подуровень не по
правилу Гунда, образуются пары электронов на нижележащих
dxy, dyz и dxz ячейках при пустых dz* и dxz_yz ячейках. В
результате ожидаемое суммарное спиновое число не достигается и
возникает низкоспиновое состояние.
В комплексах Ti (H20)e+ (d1), Ti (Ш2)б~(#), V (H20)i+ (d2),
О (H20)e+(d3), Cr (Ш3)б+ (d3), Cr(CN)6~~(d3), несмотря на
различную силу лигандов, спиновое состояние одно и то же, т. е.
соответствует числу электронов на d-подуровне. Ионы парамагнитны
и магнитный момент комплексного иона примерно совпадает с
числом (неспаренных) электронов. Однако при конфигурациях
d4, d5, dQ и d7 ион-комплексообразователь в октаэдрическом
окружении при одинаковом числе электронов на d-подуровне может
давать два типа ионов, отличающихся магнитным числом, т. е.
количеством неспаренных электронов. Ионы Мп(С1М)б~ и MnF6~"*,
несмотря на одинаковое количество электронов на d-подуровне,
одинаковые заряд иона и координационное число, различаются
магнитными свойствами. Магнитный момент Мп3+ в ионе
Мп (СЫ)б~" примерно в 2 раза ниже магнитного момента в ионе
MnFl-. Такое понижение магнитного момента связано с
уменьшением числа неспаренных электронов от четырех в
высокоспиновом комплексе MnFe"" до двух в низкоспиновом Mn(CN)l~.
У комплексов Fe (С1М)б~~ и Fe (Н20)б+ различие в величинах
магнитного момента Fe3+ еще значительнее. В первом комплексе
ион Fe3+ (d5) находится в окружении сильнодействующих
лигандов и возникает низкоспиновое состояние с одним неспаренным
О I
электроном. В комплексе Fe (Н20)б молекулы воды создают
слабое кристаллическое поле и все 5 d-электронов неспарены. В ре-
574
зультате высокоспиновое состояние с магнитным моментом
примерно в 5 раз выше.
Октаэдрические комплексы с шестью d-электронами бывают
двух типов — парамагнитные и диамагнитные. Так, в комплексе-
CoF6~" ион-комплексообразователь Со3+ (d6), будучи окруженным
слабодействующими лигандами, находится в высокоспиновой
конфигурации с четырьмя неспаренными электронами, но в комплексе
Со(ЫН3)б+ слабое кристаллическое поле заменяется на сильное
и конфигурация d6 превращается ,в низкоспиновую с заполненными
парами электронов dxy-9 dyz- и с?х?у-орбиталями, поэтому комплекс
диамагнитен.
Кобальт в 2-валентном состоянии Со2+ (d7) также образует
два типа комплексов, различающихся по магнитным свойствам.
В низкоспиновом комплексе Co(N02)<3~ неспаренный электрон, а
в высокоспиновом Со(Н20)б 3 неспаренных электрона. Ионы-
комплексообразователи, имеющие на d-подуровне по 8 и 9
электронов, обладают одинаковыми магнитными свойствами независимо
от силы кристаллического поля. Таковы ионы
№(Ш3)б+ и №(Н20)б+ (Ni2+, d8), Cu(CN)46"~ и Си(Н20)е+ (Cu2+,d9).
Устойчивость комплексных ионов d-элементов чрезвычайно
сильно зависит как от количества электронов на d-подуровне, так
и от воздействия на них поля окружающих лигандов.
Восстановительные потенциалы ионов-комплексообразователей в окружении-
сильнодействующих или слабодействующих лигандов весьма
различны (табл. 14.61).
Таблица 14.61
Реакция
Fe(H20)3+ + e = Fe(H20)2+
Fe(CN)36- + ^Fe(CN)46-
Со (Н20)3+ + e = Со (Н20)2+
Co(NH3)3+ + e = Co(NH3)^
£°, в
0,77
0,36
1,84
0,10
При замене слабодействующих лигандов (Н20, F") на
сильнодействующие (CN-, NQT, NH3) стремление к окислению ионов-
комплексообразователей возрастает (Fe2+ ->Fe3+, Со2+ ->Co3+
и т. п.).
Лиганды CN~ или NH3 образуют с ионами М3+ значительно
более прочные комплексы по сравнению с ионами М2+, и это
способствует окислению. Комплексный ион Со(Н20)б в водном
57S
растворе довольно неустойчив и окисляет воду до кислорода, но в
водных растворах Со2+ в присутствии N0^, CN~~ или аммиака
легко переходит в Со3+ с образованием октаэдрического комплекса.
В окислительно-восстановительном процессе
Со (НаО)Г + 6NH3 — е = Со (Ш8)Г + 6Н20
одновременно с удалением электрона от комплексообразователя
и заменой слабодействующих лигандов на сильнодействующие
происходит переход высокоспинового состояния d7 в Со2+ в низко-
спиновое dQ в Со2+ с высокой его стабилизацией кристаллическим
полем.
К 2*
нест МЛ
1 ш
\Г)
*Ч
V
,ислоЬиые
2+
Mn
2+ ш
Сгу/Ч
1 1 1
единицы
2 +
Iе 1+ п*<
J 1 Т*1 х-
Л 1 2 3 4 5 6 7 8 9 10
Число d-электроноб
10
09
08
01
-I 1 1 1 1 I I I < i i
0 723456789/0
Число d-электронов
Рис. 14.49. Константы нестойкости Рис. 14.50. Радиусы ионов ^-элементов
^-элементов IV периода IV периода
Хотя на магнитных свойствах ионов оказывается только
различие в конфигурациях <24, d5, d6 и d7, разделение октаэдрическим
окружением ^-подуровня на два подподуровня с двумя и тремя
d-орбиталями обнаруживается в ряде других свойств. Аналогично
тому, что каждый подуровень обладает повышенной устойчивостью
в наполовину и полностью заполненном состоянии, наблюдается
повышенная устойчивость подподуровня из трех орбиталей dxy,
dyzy dxz, когда он наполовину или полностью заполнен. Такие
состояния в октаэдрическом окружении сильных лигандов
возможны, когда на d-подуровне находится 3 или 8 электронов.
На рис. 14.49 показано изменение констант нестойкости окта-
эдрических ионов:
МЛё+ = М2+ + 6Л,
АнрСТ
[М2 ] [Л]6
[МЛ|+]
576
Таблица 14.62
где М — металл-комплексообразователь, Л — лиганд. Устойчивость
октаэдрических комплексов со слабодействующими лигандами
(Л==Н20 и др.) возрастает по мере заполнения dxy, dyz- и dxz- op-
биталей, достигая первого максимума у иона VJI2+6, затем
понижается к марганцу, снова начинает возрастать, становясь
максимальной у иона №Лб и затем понижается к цинку. Если бы
расщепления (разделения) d-подуровня не происходило, следовало
ожидать, наоборот, особых свойств у иона Мп2+, имеющего 5 d-эле-
ктронов, которые заполняют d-подуровень (в целом) наполовину.
По-видимому, в октаэдрическом окружении
сильнодействующих лигандов, создающих низкоспиновые конфигурации, также
должно наблюдаться два максимума устойчивости комплексных
ионов, но при трех (d3) и шести (d6) электронах, заполняющих
соответственно наполовину и полностью подподуровень из dxy~9 dyz-
н dxz- орбиталей.
Наблюдаемый ход изменения констант нестойкости
комплексных ионов при переходе от Са2+ <к Zn2+ связан с изменением
радиусов ионов М2+ (рис. 14.50, табл. 14.62).
От иона Са2+ к иону V2+ радиусы
уменьшаются. Это означает, что
окружающие молекулы или ионы подходят
на более близкие расстояния к комп-
лексообразователям. У иона Са2+
d-электроны отсутствуют. Двухзаряд-
ный ион скандия неизвестен. Ион Ti2+
имеет конфигурацию d2, и 2 электрона
находятся на двух орбиталях из трех
dxy, dyz или dxz. В октаэдрическом
окружении орбитали располагаются не
на линиях лиганд — комплексообра-
зователь, а в областях пространства,
наиболее удаленных от лигандов.
Поэтому лиганды могут подойти ближе к
центральному иону, а это означает
уменьшение размера иона. То же
имеет место и у иона V2+. Однако хотя заряд ядра ванадия возрос
на единицу, а количество d-электронов также возросло на
единицу, но d-электроны, распределяясь в пространстве не равномерно,
не сферически, плохо экранируют воздействие сил протяжения
ядра и не компенсируют его. В результате лиганды подходят еще
ближе к комплексообразователю и радиус иона сокращается.
У ионов хрома и марганца конфигурации rf4 и d5. В окружении
слабодействующих лигандов (вода) d-подуровень заполняется в
соответствии с правилом Гунда непарными электронами.
Появление электронов на орбиталях dzt и dx*—y* (высокоспиновое
состояние) приводит к значительно более полному использованию
окружающего пространства, лиганды выталкиваются из него и
радиусы ионов Сг2+ и Мп2+ возрастают. Начиная с иона Fe2+,
Ион
Са2'
(Sc2+)
Ti2 -
V2+-
Cr2f-
Mn2'
Fe2+
Co2r
Ni2J
Cu2f
Zn2J
Конфиг y-
рация
0
1
2
3
4
5
6
7
8
9
10
о
M +
1,04
—
0,78
0,72
0,83
0,91
0,80
0,78
0,74
0,80
0,83
19 общая химия
577
происходит спаривание электронов на орбиталях dxy, dyz и dXZy
т. е. на тех орбиталях, которые в меньшей степени препятствуют
приближению лигандов. Заполнение этих орбиталей не
компенсирует возрастающего заряда ядра и при переходе к иону Ni2+
радиусы ионов уменьшаются. Ион Ni2* имеет конфигурацию d8y
при которой орбитали dxy, dxz и dyz заполнены парами электронов.
У ионов Си2+ и Zn2+ спаривание электронов происходит на
орбиталях dZ2 и dxt-y*. Степень заполнения пространства электронами
и экранирование от воздействия ядра возрастают, лиганды сильнее
выталкиваются из межорбитального пространства и радиусы
ионов Си2+ и Zn2+ возрастают.
Энтальпии гидратации двухзарядных ионов, т. е. энтальпии
реакций
м;
6Н2Ож (+ оо Н2Ож) = М (Н80)|+р А Я,
1 гидр >
зависят от размера иона М2+. Чем меньше ион, тем больше
тепловой эффект гидратации и тем больше отрицательное значение
энтальпии гидратации.
Изменение энтальпии
гидратации двухзарядных ионов
d-элементов IV периода
повторяет ход изменения
радиусов (рис. 14.51).
Влияние заполнения d-подуровня
и возникновение неравно-
- 600000
-бьоооо
700000
АН
298
f"V*
U U_
гидр о1* 2
М +6H20=M(HJ))
^ г*""*
>*J>>4 2+
V**-«er\Fe ?4
%, ^° 2+ , '
-1—i—1—1—1—L_i i
0 1 2 3 А 5 6 7 8 9 Ю
Число d элекгг>[,с,ч(,(1
Рис. 14.51. Энтальпия гидратации
ионов d-элементов IV периода
ценности d-орбиталей в окружении поля лигандов или
кристаллического поля обнаруживается в ряде других тепловых эффектов,
например в энергиях кристаллических решеток хлоридов МГ?
(рис. 14.52) или оксидов (рис. 14.53).
d-Элементы обладают удивительной и чрезвычайно важной в
практическом отношении особенностью — все они активные
катализаторы. Даже весьма различные по своему составу катализаторы
обладают одним общим свойством — они обязательно содержат
хотя бы один из d-элементов. В большинстве случаев активность
катализатора при ее рассмотрении вдоль периода (по мере
заполнения d-оболочки) возрастает, проходит через максимум и затем
снова падает. Однако активность катализатора зависит от
реакции. Так, в газовой реакции синтеза аммиака наиболее высокая
каталитическая активность у железа (рис. 14.54), а в реакции
окисления водорода — у никеля. В реакции рекомбинации атомов
водорода максимум каталитической активности приходится на
кобальт. На рис. 14.55 представлено изменение каталитической
активности d-металлов IV, V и VI периодов в реакции разложения
578
аммиака. Периодичность в изменении каталитических свойств
очевидна. Максимум активности лежит у железа и его аналогов
рутения и осмия.
дН кал/моль
реш,
NiCL
CuCl
650000 h-
600000 h
550000 h
ZnCL
0 1 23456789 Ю
Число d-электроноб
Рис. 14.52. Энергия кристаллических решеток ди-
хлоридов ^-элементов IV периода
Д ^L~ ,кэл/моль
реш'
1000000
ZnO
S 50 000 \-
900000 \-
850000
0123456789 10
Число d-электронов
Рис. 14.53. Энергия кристаллических решеток
оксидов d-элементов IV периода
d-Металлы катализируют многие
окислительно-восстановительные реакции, проходящие в растворе. В этом случае катализатор,
.^-элемент, участвуя в процессе гомогенного катализа, является
комплексообразователем, а реагирующие вещества входят в виде
лигандов в состав комплексного иона. Реакция ускоряется благо-
19*
579
даря тому, что при ооразовании промежуточного соединения
улучшается необходимая для взаимодействия ориентация
(увеличивается энтропия активации), понижаются энергии связей, которые
2NH =N *3H^
К N +ЗН =-2NH
б I 2 2 3
Ю Н
Ю
ю
Л L
J L.
Сг Mn Fe Co Nj Cu
Рис. 14 54.
Каталитическая активность
некоторых d-эчементов
IV периода в реакции
синтеза аммиака
от и
Ю Ь
Ti V Сг Mn Fe Co Ni Cu
А цегпилацетонатные
комплексы
Рис. 14.55. Каталитическая
активность ^-элементов в
реакции разложения аммиака
разрываются в реакции
(уменьшается энергия активации),
облегчаются электронные переходы
благодаря участию в них центрального
атома.
Воздействие всех этих факторов
существенно зависит от
электронной структуры комплексообразова-
теля d-элемента. Чаще
максимальная активность приходится на
элементы, содержащие 7—9
электронов на ^-подуровне. На рис. 14.56
представлена каталитическая ак-
Рис. 14 56. Каталитическая активность
ацетилацетонатных комплексов d-эле-
ментов IV периода в реакции
гидрирования бензола
тавность сложных катализаторов, содержащих комплексы (аце-
тилацетонатные) d-элементов для реакции гидрирования бензола.
580
Го же изменение активности наблюдается и для металлов V и VI
периодов, при этом каталитическая активность возрастает при
переходе вниз по подгруппе.
§ 13. СКАНДИИ, ИТТРИИ, ЛАНТАН
[d -ЭЛЕМЕНТЫ III ГРУППЫ)
В третью группу кроме скандия, иттрия и лантана входят
также 14 элементов, непосредственно следующих за лантаном.
У них происходит заполнение третьего снаружи /-подуровня.
Поэтому эти элементы очень похожи друг на друга и обычно
выделяются в отдельное семейство /-элементов. Точно так же 14
элементов, следующих за актинием, выделяются во второе семейство
/-элементов.
Хотя скандий — rf-элемент, а алюмииий — р-элемент, их
свойства довольно похожи. Скандий энергично взаимодействует с водой
с образованием водорода (точно так же как освобожденный от
окисной пленки алюминий). Оксид скандия Sc203 не растворим в
воде, как и оксид А1203. Из-за большего размера иона Sc3+
скандий более основен по сравнению с алюминием. У скандия и
алюминия при обычных условиях устойчивы только соединения, в
которых они находятся в валентном состоянии 3. Иттрий
проявляет в своих устойчивых соединениях то же валентное состояние.
Оксид иттрия Y203 не растворим в воде, но в отличие от А1203
растворим в кислотах. Иттрий проявляет еще более основные
свойства. Лантан, находящийся в своих соединениях в 3-валентном
состоянии, также обнаруживает ярко выраженные основные
свойства.
Стандартные электродные потенциалы /?- и d-элементов
III группы приведены в табл. 14.63,
Таблица 14.63
Период
III
IV
V
VI
Реакция
AI31 -гЗе ^ Ai
Sc3 •■ + Зе =- Sc
Ga3 j -f Зг = Ga
Y3 ь + Зе = Y
In3- + 3?-In
La3 ь + Зг = La
£«
р-элемент
— 1,66
—
—0,56
—
—0,33
—■
, в
d элемент
—2,08
—2,37
-2,52
Все металлы III группы имеют отрицательные значения
электродного потенциала, т. е. все они растворяются в растворах
кислот с концентрацией ионов водорода 1 г-ион/л. Однако у р-элемен-
тов с переходом сверху вниз по группе (А1—Ga—In—Т1)
численное значение потенциала уменьшается. Это указывает как на
ослабление стремления реагировать с водой, так и на возможность
581
вытеснения нижестоящего /^-металла из раствора его соли другим
вышестоящим, например:
А1 + 1п3+-А13+Ч In э. д. с. = — 0,56 —(1,66) = + 1,10 в.
Наоборот, у d-элементов III группы при переходе сверху вниз чис-
леные значения потенциала возрастают. Лантан реагирует с
растворами кислот (и с водой) значительно более энергично по
сравнению с иттрием или скандием. В то же время вышестоящий
металл способен вытеснить нижестоящий из раствора его соли,
например:
Y + Sc3+ = Y3+ i- Sc э. д. с. = — 2,08 — (— 2,37) = + 0,29 в.
§ 14. ТИТАН, ЦИРКОНИЙ, ГАФНИЙ
({/-ЭЛЕМЕНТЫ IV ГРУППЫ)
d-Элементы IV группы (Ti, Zr и Hf) имеют на предвнешнем
d-подуровне и внешнем 5глюдуровне по 2 электрона. Все эти
элементы проявляют валентное состояние 4, а титан и цирконий — 2
и 3. Устойчивость низких валентных состояний при переходе вниз
по группе у rf-элементов уменьшается, в то время как среди
/^-элементов той же группы (Ge, Sn и Pb) при переходе вниз
устойчивость низких валентных состояний возрастает. Диоксид
свинца РЬ02 — сильный окислитель, переходящий в ион РЬ2+,
однако гафний таким свойством не обладает.
Хотя изобарные потенциалы диоксидов титана, циркония и
гафния — большие отрицательные величины, тем не менее
металлы на воздухе вполне устойчивы. Они также довольно устойчивы
к действию различных химических веществ. Это объясняется
образованием на поверхности металлов прозрачных и очень устойчивых
пленок диоксидов.
Таблица 14.64
Период
IV
V
VI
Оксид
тю2
Ge02
Zr02
Sn02
НЮ2
PbOo
ЛЯ298,обр
р-элемент
— 138 700
—138 800
—66 100
, кал/моль
^-элемент
—225 500
—260 500
—266 000
Оксид
ТЮ
GeO
ZrO
SnO
НЮ
PbO
Л//298,обр
р-элемент
—61 000
—68 400
—52 400
кал/моль
rf-элемент
—123 900
— 151000
—
В табл. 14.64 приведены энтальпии образования оксидов и
диоксидов р- и d-элементов IV группы. У диоксидов р-элементов
(Ge02, Sn02 и Pb02) энтальпии образования численно понижаются
при переходе вниз по группе, а у d-элементов (ТЮ2, Zr02 и НЮ2),
наоборот, возрастают. В ряду Ge02, Sn02, РЬ02 у первых двух
582
устойчивость 4-валентного состояния примерно одинакова, затем
понижается. В ряду ТЮ2, Zn02, Hf02, наоборот, устойчивость
4-валентного состояния возрастает. Что касается монооксидов р- и
^/-элементов III группы, то ясно видна значительно большая
устойчивость монооксидов d-элементов (так же как и их диоксидов).
У тетрахлоридов металлов IV группы также наблюдается
значительно большая устойчивость тетрахлоридов d-элементов
(табл. 14.65). В ряду хлоридов GeCl4, SnCl4, РЬСЦ устойчивость
Таблица 14.65
11 ериод
IV
V
VI
Хлорид
Т*С14,Ж
ОеС14,ж
ZrCl4,K
SnCI4>)K
HfCI4,K
РЬС14ж
ЛНобр,298-
р-элемент
—129 000
—126400
—78 500
кал/моль
d-элемент
—191 500
—234 700
-250 000
Хлорид
Tid2,K
GeCI2,r
ZrCl2K
SnCI, K
Hfci2,K
PbCI2.K
ЛЯобр,298
p- элемент
—42 000
—79 100
—86 000
кал/моль
^-элемент
—120 600
—145 000
— 148000
4-валентного состояния падает (отрицательное значение Д#0бР
уменьшается). В ряду хлоридов d-элементов той же группы TiCl4,
ZrCl4, HfCl4, наоборот, устойчивость 4-валентного состояния
возрастает (отрицательные значения Д//0бР возрастают). В ряду
дихлоридов р-элементов GeCl2, SnCb, РЬСЬ, наоборот,
устойчивость 2-валентного состояния возрастает (отрицательные значения
АЯобр возрастают), а в ряду аналогичных соединений d-элемен-
тов TiCU, ZrCb, HfCl2 устойчивость 2-валентного состояния также
возрастает. Таким образом, сходство между двумя подгруппами
IV группы во многих отношениях не слишком близкое.
Важнейшее валентное состояние олова и особенно свинца — 2, а не 4, у
d-элементов, наоборот, 4, а не 2. Среди валентных состояний
титана 2, 3 и 4 последнее наиболее распространено и эти соединения
с 4-валентным титаном наиболее устойчивы.
Соединения титана валентности 2 могут быть получены
восстановлением металлическим титаном соединений титана в
4-валентном состоянии:
Ti02 + Ti = 2 TiO,
TiCl4 + Ti = 2TiCl2.
Монооксид титана TiO в некотором отношении похож на оксиды
щелочноземельных металлов: он обладает основными свойствами,
связь носит преимущественно ионный характер. Титан в 2-ва-
583
лентном состоянии — сильный восстановитель. Ионы Ti2+
восстанавливают воду с образованием водорода, поэтому растворы солей
2-валентного титана не существуют. Даже кристаллический TiO
разлагает воду.
Титан в 3-валентном состоянии также может быть получен
восстановлением соединений 4-валентного титана:
2ТЮ2.К + Н2,г - Т12Оз,к + Н2Ог-
Оксид Ti203 обладает основными свойствами. Ионы Ti3+ в водных
растворах устойчивы, но проявляют восстановительные свойства
с такими окислителями, как MnO^", Fe3+ или кислород воздуха.
Диоксид титана Ti02 в воде не растворим, однако растворим в
сильных щелочах и кислотах. В растворах щелочей образуется
титанат-ион. Его простейшая формула ТЮ3~ отвечает титановой
кислоте H2Ti03, однако предполагается, что в растворе
4-валентный титан находится в виде ионов [Ti02 (OH)2f~ или ТЮз~~-Н20.
Гидратированный титанат-ион - - комплексный ион, в котором
комплексообразователь Ti4+ обладает координационным числом 4.
В сильнокислых растворах ион Ti4+ не существует. Вместо него
образуются в зависимости от концентрации ионов водорода ионы
типа Ti (ОН)з~, Ti (OH)2+, Ti (Н20)б+ и др. Существование
последнего иона наиболее вероятно. Свободный ион Ti4+ не обнаружен
даже в ничтожно малой «концентрации.
С увеличением валентного состояния тип связи в галогенидах
существенно изменяется. Так, TiCl2 и TiCl3 — кристаллы. В
'кристаллической решетке TiCl2 связь носит заметно ионный характер.
В трихлориде TiCl3 доля ионности понижается. Тетрахлорид
титана — легко испаряющаяся жидкость. Связи в молекуле TiCU уже
не несут ионного характера и близки к ковалентным.
Цирконий и гафний но физическим и химическим свойствам
очень похожи друг на друга. Между лантаном и гафнием (№ 57 и
№ 72) располагаются 14 элементов, у которых заполняется третий
снаружи /-подуровень. В ряду /-элементов размеры атомов и ионов
понижаются. В связи с этим гафний и близок к цирконию.
Оксиды Zr02 и Ш02 более основны по сравнению с Ti02 из-за
больших размеров ионов Zr4+ и Hf4+. Диоксиды циркония и гафния
поэтому хуже растворимы в щелочах и лучше — в кислотах. Соли
циркония и гафния в водных растворах менее гидролизованы по
сравнению с солями титана (IV), однако и у этих элементов
свободные ионы Zr4+ и Ш4+ в водных растворах также не
обнаружены.
§ 15. ВАНАДИЙ, НИОБИЙ, ТАНТАЛ
Id-ЭЛЕМЕНТЫ V ГРУППЫ)
^-Элементам V группы — ванадию, ниобию и танталу —
доступен большой выбор валентных состояний: 2, 3, 4 и 5. У ванадия
все эти валентные состояния более или менее устойчивы, ниобию
584
и танталу присуще валентное состояние 5 и несколько менее
присуще 3. Соотношение между свойствами ^-элементов V группы
(побочная подгруппа) и р-элементами той же группы (главная
подгруппа) примерно то же, что и в предыдущей IV группе.
При обычных условиях металлы V, Nb и Та устойчивы к
действию кислорода и других химических реагентов благодаря
плотной защитной пленке оксида. При высоких же температурах они
энергично реагируют с окислителями (Ог, СЬ и др.), так как
изобарные потенциалы (и энтальпии образования) оксидов и
галогенидов имеют большие отрицательные значения.
Обладая способностью к переменной валентности, ванадий
образует несколько оксидов, рассмотрение которых является
прекрасной иллюстрацией зависимости свойств от валентного
состояния.
Монооксид ванадия VO окрашен в черный цвет. Имеет
широкую область нестехиометрии по кислороду: от VOo,85 до VOi,25-
В воде не растворим, но, обладая основным характером,
растворяется в разбавленных кислотах (неокислителях) с образованием
катиона V2+, окрашенного в фиолетовый цвет. V2O3 окрашен в
черный цвет. Имеет основный характер. Плохо растворим в кислотах.
Ион V3+ окрашен в зеленый цвет.
V02 окрашен в черно-синий цвет. Обладает амфотерными
свойствами, но основные свойства преобладают над кислотными.
При растворении в щелочах образуются соли — ванадиты:
V02)K ^ 20Н"Р - VOsVp + Н2Ож.
При растворении в кислотах возникают соли катиона ванадила
V02+, окрашенные в синий цвет:
У02.к-|-2Н+р-УО^+ + НаОж.
V2O5 имеет цвет от оранжево-желтого до кирпично-красного.
Кристаллическая структура оксида представляет собой длинные
цепи V07", которые связаны между собой кислородными
мостиками в сетчатую плоскость.
V2O5 может быть получен непосредственно из элементов. V2O5
проявляет амфотерные свойства, растворяясь и в кислотах, и в
щелочах. В кислых средах образуется ион VO2":
V2Os,k + 2Н+р - 2VO£p.p -f- H20.
При растворении V2O5 в щелочах образуются ванадаты различного
состава, зависящего от рН среды. Из-за слабо основных свойств
V2O5 растворим в кислотах с образованием кислородсодержащих
катионов, например простейшего — VO2 . Пентоксид ванадия
растворим в воде.
Химия ионных равновесий с участием ванадия очень сложна. В различных
средах в зависимости от концентрации соли ванадия образуются многочислен-
585
ные ионы, часто способные полимеризоваться и при изменении условий
медленно переходящие в равновесное состояние.
Состав ванадиевых ионов чрезвычайно сильно зависит от среды раствора
и концентрации ванадия. Чем больше концентрация ванадия, тем больше
склонность ионов объединяться в поливанадиевые комплексы. В табл. 14.66
представлены те ионы, которые преимущественно существуют при данном рН в
0,1 М растворе ванадия.
Таблица 14.66
рН
<1
1
1—1,5
«1,6
«1,7
2—7
7
7—9
9
9—10,5
10,5—10
>12
Равновесие или формула преобладающего иона
VO+
10 (V02]+ + 80Н- == [HsV10O28] + 2Н+
[H«Vie028]
5VA + 3H20 = [H.V, Ad
[HeV10O28l = [V:e028]«- + 6H+
[V10O28]«-
2 [V10O28]*- +40H- = 5 [V4012]*- + 4H+
[V4o12]«-
rvAJ*- + 2H2o = 2 [V2o7]4- + 4H+
[VAJ*-
[V207]*" + OH- = 2 [V04]s" + H+
[V04J8-
Ванадил-ион VO^" способен существовать только в самых кислых, а ва-
надат-ион VO^~~ — только в самых щелочных растворах. В растворах с
интервалом рН=1—12 5-валентный ванадий находится в состоянии
поливанадиевых ионов, причем в декаванадат-ионы охватывают наибольший интервал рН.
В табл. 14.66 приведены только те ионы, которые встречаются в растворах при
данном значении рН в преимущественных концентрациях. Следует помнить, что
границы существования тех или иных ионов значительно шире и в растворе
содержатся в меньших количествах ионы, которым присуще другое значение рН.
Интересно, что при рН=1,6 из раствора выпадает пентоксид ванадия V2O5.
Все соли, соответствующие описанным ионам, могут быть выделены в
кристаллическом состоянии: V02C1, КеУюОгв, K4V4O12, K4V2O7, K3VO4. Получены
некоторые кислые соли:' КвНУюОгв, ^НгУюОга и другие, а также смешанные
соли: К^ГУСЬЫУюОгв], КзГ^ОгЫНУюОгз] и др. Примеры двух последних солей
доказывают, что ионы существуют не в узком интервале значений рН, а область
их существования простирается на области соседних ионов.
В слабокислых растворах ион VO^~ гидролизуется и одновременно поли-
меризуется, например:
10VO+ р.р + 8Н20 = H2V10O*- p.p+ 14Н+р .
В сильнощелочных растворах V2O5 переходит в раствор в виде
отрицательно заряженных ионов, например VO|~~# Этот ион можно рассматривать как
происходящий от кислоты H3V04, аналогичной фосфорной кислоте Н3Р04. Ион
VOJ аналогичный иону NO^~, не обнаружен.
При добавлении кислоты к щелочному раствору ионов начинается процесс
•амфотерного превращения:
2VO^.p + зн+р = hva3-p + на
586
Образующийся ион HVgOy с новыми порциями ионов водорода и с оставшимися
3—
ионами V04 способен к дальнейшему превращению:
VOj- p +HV20 з-р +ЗН+р = V303-.p + 2Н20.]
В конечном итоге при подкислении раствора происходит полимеризация иона
3V04,7-P +6Н£р = V3° 9^-р + ЗНА
При действии восстановителей (металлический цинк) на кислый
раствор соли ванадия в 5-валентном состоянии возникают
различные по составу ионы с ванадием в 4-валентном состоянии,
например V02+, ванадил-ион, придающий растворам желтый цвет.
Этот ион обнаружен рентгенографически в кристаллических солях
VOS04 или VOCl2. Ванадий в 4-валентном состоянии в некотором
отношении похож на титан в том же валентном состоянии. В этом
состоянии ванадий амфотерен. При действии щелочи на раствор
V02+ образуется осадок диоксида ванадия:
V02p+ + 20Н~р - V02,K + Н20.
Диоксид ванадия при дальнейшем увеличении концентрации ионов
гидроксила снова растворяется:
V02,K + 40Н~р = VOl"p + 2Н20.
Образующийся ион VOt~~, однако, может существовать в
незначительных концентрациях, так как подвергается полимеризации.
Хлорид ванадия VC14.— соединение со связями значительно
ковалентного, а не ионного характера. Это — легколетучая
жидкость, напоминающая тетрахлорид титана.
При восстановлении или ванадия (5) или ванадия (4)
образуется ванадий в 3-валентном состоянии в виде ионов V(H20)+3
зеленого цвета. При подщелачивании образуется осадок трио-кси-
да ванадия
2V3+ + 60Н~Р = V203.k + ЗН20.
Все соли, содержащие ванадий в 3-валентном состоянии, имеют
связи преимущественно ионного характера. Ион V^ обнаружен
рентгенографически. При дальнейшем восстановлении образуется
ион V2+ в виде иона V(H20)e+ фиолетового цвета. Этому
валентному состоянию ванадия отвечает устойчивый оксид VO,
основный по своему характеру. Соединения 2-валентного ванадия
во многом похожи на соединения титана в том же состоянии.
Ниобий и титан чрезвычайно близки друг к другу по свойствам
из-за равенства атомных и ионных радиусов благодаря
лантаноидному сжатию при заполнении 4 /-подуровня. Их типичное
валентное состояние 5. Их пенiоксиды Nb2Os и Та2Об растворяются
587
в расплавленных щелочах, давая ниобаты и танталаты простейших'
формул NaNb03 или NaTa03. При изменении концентраций обра-,
зуются другие ионы. В водных растворах ниобий и тантал в 5-ва-1
лентном состоянии образуют ряд полимерных ионов, состав
которых не установлен, но, по-видимому, близок к ионам в растворах
ванадия того же валентного состояния. Nb205 и Ta2Os в кислотах
не растворимы. Галогениды NbT5 и ТаГ5 — низкоплавкие,
легколетучие соединения преимущественно ковалентного характера.
Диоксиды ниобия и тантала Nb02 и Та02, тетрахлориды NbCl4
и ТаС14 могут быть получены, но они менее устойчивы по
сравнению с соединениями 5-валентных элементов. В валентном
состоянии 3 известны только галогениды NbT3 и ТаГ3, валентное
состояние 2 становится еще более редким: известно только два
вещества ТаС12 и NbBr2.
При изучении этих элементов еще раз обнаруживается
особенность, что у d-элементов при переходе сверху вниз по подгруппе
устойчивость соединений с низкими валентными состояниями
надает.
§ 16. ХРОМ, МОЛИБДЕН, ВОЛЬФРАМ
(^-ЭЛЕМЕНТЫ VI ГРУППЫ)
В VI группу периодической системы из d-элементов входят
хром, молибден и вольфрам. Все элементы способны проявлять
несколько валентных состояний, однако у хрома наиболее
устойчивыми являются 3 и 6, а у молибдена и вольфрама только 6.
Для хрома наиболее типично 3-валентное состояние. Триоксид
хрома растворяется и в кислотах и щелочах:
Сг203,к -[- 6Н+р = 2Cr*+ + 3HaO,
Сг203,к + 20Н"р - 2 СгО^р + Н20.
Хромит-ион СгОГ существующий,в щелочных растворах,
обнаружен лишь в незначительных концентрациях из-за сильной
гидратации:
СгО^р -Ь 2Н20 = Сг (ОН)^р.р.
Ион Сг(ОН)Г —комплексный ион, в котором
координационное число хрома 4. Поэтому правильнее писать процесс
растворения триоксида хрома Сг203 в виде
Сг20з,к + 20Нр:р + ЗН20 = 2Сг (ОН^р-р.
Ион Сг3+ в растворах даже сильнокислых в свободном
состоянии не существует из-за чрезвычайно сильной гидратации,
приводящей к образованию гекс-аакваиона Сг (ОН2)б :
Сгр:+ + 6Н20=Сг(ОН2)^.р.
588
В комплексном ионе Сг(ОН2)б+ координационное число хрома 6 и
ион Сг3+ находится в октаэдрическом окружении.
Ион Сг(Н20)б+ благодаря высокой прочности связи с атомами
кислорода воды проявляет слабые кислотные свойства:
Сг(Н20)^+ - Сг(Н20)5ОН2+ + Н+,
к _ [Сг(НдО)8ОН»г]-[НЧ _ , о 1П_4
АДИСС „,, 1,0'Ш
[Сг(Н20)б+1
Обычно гексааквакомплексы переходных элементов в
3-валентном состоянии легко теряют молекулы воды и быстро
переходят в комплексы с другими лигандами. В этом отношении хром
отличается от остальных d-элементов. Его комплекс Сг(Н20)б+
более устойчив, поэтому реакции 3-валентного хрома в водных
растворах при комнатной температуре протекают очень медленно.
В ряду ионов Ti3+, V3+, Cr3+, Mn3+, Fe3+, Co3+ их
восстановительные свойства в кислых растворах постепенно сменяются
на окислительные. Так, ионы Ti3+ и V3+ — восстановители, ионы
Мп3+, Fe3+ и Со31" — окислители. Только ион Сг3+ не обнаруживает
окислительно-восстановительных свойств. Однако в щелочных
растворах, когда разрушаются прочные гидратные оболочки,
3-валентный хром легко окисляется до 6-валентного:
Cr(OH)i:p.p-;-40H-p — 3* = СгО;-р I 4 Н20,
Сг (ОН)3,к г 50Н~Р — Зе = СЮ^.Р + 4Н20,
Сг02;р.р + 40Н- — Зе = СЮ^.р + 2Н20.
Хромат-ион CrOl~ в кислом растворе переходит в бихромат-ион:
2CrOfcp + 2Н+Р = Сг20^.р + Н20.
Обратный процесс совершается в щелочной среде и описывается
уравнением
Сг20?~ + 20Н~Р - 2Сг0^.р + Н20.
Бихромат-ион чрезвычайно сильный окислитель:
Сг20?" + 7H+ + 6е = 2Сг3+ + Н20 £° = 1,33 в.
Стандартный потенциал воды равен
4Н+ + 02 + Ае = 2Н20 £° = 1,23 в.
Следовательно, возможна реакция
2Сг202Г + 16Н+ = 4Сг3+ + 8Н20 + 302 э. д. с. - 1,33 —
— 1,23=0,10 в.
589
Однако скорость этой реакции настолько мала, что растворы би-
хромата калия могут храниться годами без заметного
образования кислорода.
Бихромат калия ускоряет распад пероксида водорода. Скорость распада
Н202 под влиянием бихромата калия сильно зависит от концентрации
пероксида водорода. При невысоких концентрациях Н2О2 (ниже 0,5 моль/л) распад
проходит как реакция первого порядка. При увеличении исходной концентрации
пероксида водорода возникают отклонения от первого порядка и при
Сн о ~ ^ моль/л порядок реакции переходит в нулевой. При этом скорость
реакции перестает зависеть от исходной концентрации пероксида водорода.
Независимость скорости реакции от концентрации может быть объяснена
связыванием практически всего количества катализатора (ионов Сг2С>7~~) в
промежуточный комплекс состава Сг209~:
Сг2°7,7-Р + 2Н202 = Сг20* р.рЧ- 2НаО. (1)
В этом случае скорость распада Н202 определяется скоростью распада
промежуточного продукта Cr2Og :
Cr»Ofo.p = Cr,0?-p + 02iI. (2>
Регенерируемые ионы С^О^"" снова связываются имеющимися в большом
избытке молекулами пероксида водорода. Скорость процесса (1) больше
скорости процесса (2). Таким образом, концентрация иона Сг209"~ определяется
только количеством исходного катализатора Сг2о7~" и не изменяется при
прохождении реакции. Следовательно, хотя процесс (2) мономолекулярен,
скорость его постоянна во времени (пока концентрация не упадет до 4 моль/л),
и реакция является реакцией нулевого порядка.
При действии на кристаллический бихромат калия
концентрированной серной кислоты выпадает осадок триоксида хрома СгОз:
К2Сг207,к + 2H2S04,*< = 2К£Р + 2HSO^p.p + Н20 + 2Cr03(K.
Триоксид хрома хорошо растворим в воде:
2СЮ3,к + Н2Ож = 2Н+р + Cr2072 V.p
и в щелочных растворах:
СЮз,к + 20Н~р = СгО^р + Н2Ож.
Хром без доступа воздуха растворяется в разбавленных
кислотах с образованием ионов 2-валентного хрома Сг2+, которые гидра-
тированы шестью молекулами воды [Сг(Н20)6]2+. Кислородом
воздуха 2-валентный хром легко окисляется ,в 3-валентный Сг3+ с
сохранением шести молекул воды: [Сг(Н20)6]3+. При действии
на растворы, содержащие хром в 3-валентном состоянии,
металлическим цинком также образуются ионы хрома в 2-валентном
состоянии:
Сг3+ + е = Сг2+ Е° = — 0,41 в.
590
Раствор ионов Сг2+ в воде — один из самых сильных
восстановителей. Кислородом воздуха ионы Сг2+ чрезвычайно энергично
окисляются до ионов Сг3+.
Для молибдена и вольфрама наиболее типично 6-валентное
состояние, хотя известны соединения с их валентными
состояниями 4 и 5.
МоОз и W03 — кислотные оксиды, растворимые в щелочах с
образованием многочисленных сложных и плохоизученных ионов,
например: кМо209Г, Мо70§Г, W2Or~, А^ОгГ-
§ 17. МАРГАНЕЦ, ТЕХНЕЦИЙ, РЕНИЙ
(d-ЭЛЕМЕНТЫ VII ГРУППЫ)
Из ^-элементов VII группы наиболее распространен марганец.
Химия его соединений играет наибольшее значение. Технеций в
природе не найден, а все его искусственно полученные изотопы
имеют очень короткие периоды полураспада. Рений — очень
редкий элемент, нашедший применение лишь в последние
десятилетия (катализ, техника высоких температур).
Марганец — элемент, обладающий наибольшим набором
различных по устойчивости валентных состояний: 2, 3, 4, 5, 6 и 7.
При растворении металлического марганца в кислоте образуются
ионы Мп2+:
Мп2+ + 2е = Мп Е° = — 1,18 в.
Несмотря на гидратацию, ионы Мп2+ обнаружены в растворе в
свободном состоянии. При действии щелочи на раствор выпадает
осадок, переходящий при нагревании без доступа кислорода в
оксид марганца МпО (основый оксид). В противоположность
ионам титана, ванадия и хрома в их 2-валентном состоянии ион
Мп2+ не обладает восстановительными свойствами:
Мп3+ + е = Мп2+ £° = 1,51 в.
Из-за высокого положительного значения потенциала процесс
перевода ионов Мп2+ в Мп3+ в водном растворе очень затруднен
(AG>0). Ионы Мп3+ -в водном растворе неустойчивы из-за их
взаимодействия с водой:
2Мп*+ + 2Н20 = Мпр+ + Мп02,к + 4Н+р э. д. с. = 0,56 в.
В 3-валентном состоянии марганец может существовать только
в неводных средах или в кристаллах.
Триоксид марганца может быть получен окислением дигидрок-
сида марганца:
2Мп(ОН)2 + 1/202 = Мп203 + 2Н20.
Мп203 — основный оксид.
591
Известно только одно устойчивое соединение марганца, в
котором он 4-валентен — диоксид марганца Мп02. Действием соляной
кислоты на Мп02 получить тетрахлорид марганца невозможно:
Мп02,к -t- 4НС1р.р - МпС12,р.р - С12.г + 2Н20.
Это объясняется тем, что в кислых средах Мп02 сильный
окислитель:
Мп02,к -- 4Н+Р + 2е = Mrip+ -г 2Н20 Е° = 1,23 в.
Хотя в щелочном растворе в процессе участвует то же самое
количество электронов, потенциал имеет другой знак:
Мп02,к + 2Н2Ор.р + 2е = Мп (ОН)2 + 20Н~ Е° = — 0,50 в.
У соединений марганца, как и во всех других случаях, оксиды
и кислородсодержащие анионы — более сильные окислители
только в кислых, а не в щелочных или нейтральных растворах.
Другими словами, синтез кислородных соединений d-элементов в их
высших валентных состояниях удобнее проводить в щелочных
растворах или пользоваться методом плавления с окислителем и
щелочью. Так, манганат калия КгМп04 получается при
нагревании Мп02 с кристаллическими KN03 и КОН.
В манганат-ионе Мп04~ марганец находится в 6-валентном
состоянии. Ион Мп04~~ устойчив только в щелочных растворах.
В кислых растворах он переходит в более высокое и более низкое
валентные состояния одновременно (диспропорционирование):
ЗМ11О4" + 4Н+ - 2Мп07 -г Мп02 + 2Н20 э.д.с. - 1,70 в.
Ион МпОГ — перманганат-ион содержит марганец в его
высшем валентном состоянии. Перманганат калия — очень сильный
окислитель как в кислой, так и в нейтральной и щелочной средах:
МпОГ + 8Н+'-|- Ъг = Мп2+ + 4Н20 £° = - - 1,51 в,
МпОГ + 2Н20 + Зе - Мп02 + 40Н~ Е° = + 0,60 в,
МпОГ + е = MnOf Е° = + 0,56 в.
Несмотря на то что потенциал первой реакции достаточен для
окисления воды до кислорода, растворы перманганата калия
довольно устойчивы и могут храниться продолжительное время.
Черезвычайно интересен тот факт, что КМп04 изоморфен BaS04>
хоти по химическим свойствам обе соли не имеют ничего общего.
Заряды и ионные радиусы катионов и анионов также весьма различны
(Ява,, = 1,43, RK+ = 1,33, R^ = 2,30; *Mno_= 2,40 А).
592
Изоморфизм вызван исключительно тем, что расстояния катион —
анион у обоих солей одинаковы:
Ва2+ — БОГ 1,43 + 2,30 = 3,73 А,
К+_МпОГ 1,33 + 2,40 = 3,73А.
При действии на перманганат калия концентрированной
серной кислоты образуется крайне неустойчивый оксид марганца
Мп207:
2КМп04,к + 2H2SOv* - 2Kptp + 2H2S04, p.p + Мп207,ж + Н20.
Оксид Мп207 носит ярко выраженный кислотный характер.
На примере оксидов марганца интересно
обсудить вопрос о стабильности соединений с
различными валентными состояниями
элемента.
Изобарные потенциалы образования
кристаллических оксидов марганца приведены в
табл. 14.67.
Судить о сравнительной устойчивости этих
оксидов невозможно, так как изобарные
потенциалы вычислены из констант равновесия,
в которых давление кислорода возводится в
различные степени. Например:
1
Мпк + 1/2 02г - МпОк, К =-- —т~г ,
дсо = _ 4,576Г \g Pq0'5 = — 86800 кал/моль,
3MnK + 202 г - Mn304 К. /С = ——,
Рк
AG» ^ — 4,576 Г Ig Pq22 = — 306 000 кал/моль.
Вопрос об устойчивости соединения содержит в себе два совершенно
разных смысла. Во-первых, можно говорить о термической устойчивости и
располагать соединения как в порядке температур, при которых достигается одно
и то же давление диссоциации, так и в порядке давлений диссоциации при
постоянной температуре. Во-вторых, можно говорить об устойчивости
соединения к воздействию атмосферы, в которой он находится: МпО окисляется
кислородом воздуха, а Мп02 устойчив к действию кислорода и не превращается
в Мп207.
Судить о термической стабильности оксидов можно по давлениям
диссоциации при некоторой температуре или по величинам изобарных потенциалов
реакций диссоциации, отнесенных к 1 г-молю кислорода:
Мп02к = Мпк + 02>Г AGO = _ AGo6peMn0f - + 114 000 кал/моль,
Таблица 14.67
Оксид
МпО
Мп304
Мп203
Мп02
А^0
АС/298, обр-
кал/моль
—86 800
—306 000
—212 000
—114 000
593
AG0
3/2 Mn203 K - 4/3 MnK + 02 r AG* - — o6pm"*°» - 141 000 кал/моль,
3/2
1/2 Mn304#K = 3/2 MnK + 02r AGO _ _.—обР.Мп,о4 - 153 000 кал/моль,
2MnOK = 2MnK + 02>r AGO = __ AG°6p MnQ • 2 - 17 300 кал/моль.
Из изобарных потенциалов диссоциации, отнесенных к 1 г-молю кислорода,
давления вычисляются по соотношению
AGO = _4,576Г \gK^~- 4,576Г Jg Р^.
Однако вычисленные давления диссоциации (за исключением МпО) будут
относиться к воображаемым процессам диссоциации непосредственно до
кристаллического марганца. Реально же все оксиды, содержащие металл не в самом
низком валентном состоянии, диссоциируют по ступеням, переходя все стадии
от более сложных оксидов к простым и, наконец, к металлу:
Мп02, Мп208, Мп304> МпО, Мп.
Поэтому, строго говоря, давления диссоциации следует вычислять для
каждого процесса в отдельности:
4Мп02>к=2Мп2О3,к + °2,г
AGO =4-21 600 кал/моль,
6Мп203,к = 4Мпз04,к + 02(Г
AGO = +48 000 кал/моль,
2Мп304к=6МпО + 02г
AGO = _j_ 91 200 кал/моль,
2МпОк = 2Мпк + 02 г
AGO — 4- 173 600 кал/моль.
Оксиды марганца располагаются в порядке увеличения положительного
значения изобарного потенциала диссоциации следующим образом: МпОг,
Мп20з, Мп204, МпО.
В том же порядке уменьшаются давления кислорода для реакции
последовательного перехода из одного оксида в другой:
AGQ
g °'~ 4,576-Г *
Давления кислорода над оксидами марганца представлены графиком на
рис. 14.57. Наименьшим давлением обладает монооксид МпО. Процесс
диссоциации его сопровождается наибольшим положительным изменением изобарного
потенциала. Наименьшее давление кислорода у диоксида марганца МпОг,
процесс диссоциации которого до МпгОз сопровождается наименьшим изменением
AG Св расчете на 1 г-моль кислорода).
Следовательно, для определения относительной термической стабильности
оксидов достаточно расположить их по изменению изобарного потенциала
диссоциации (в расчете на 1 г-моль кислорода). Несмотря на очень высокую тер-'
594
In P мм рт ст
3 o2' r
мическую стабильность монооксида марганца, подверженный невысокому
нагреванию на воздухе, он окисляется до МпОг- Остальные оксиды также способны
поглощать некоторое количество кислорода и превращаться в более сложный
оксид, вплоть до МпОг-
Это вызвано тем, что для каждого оксида марганца, кроме МпОг, давление
кислорода в воздухе выше равновесного давления диссоциации и равновесие
смещается в сторону взаимодействия
с кислородом, т. е. окисления.
Следовательно, при стандартной
температуре (298° К) и стандартных
условиях (Pq2 = * атм) устойчивым будет
только тот оксид, у которого
давление диссоциации равно 1 атм.
Оксиды с другими давлениями
диссоциации или будут поглощать кислород
(окисляться) или будут
диссоциировать. Практически эти процессы не
замечаются из-за очень низких
скоростей реакции. Такое же поведение
у оксидов па воздухе (Р0 =
0,21 атм).
Химия технеция изучена
очень слабо. Синтезированы
немногочисленные и
неустойчивые соединения технеция с
валентными состояниями 3, 5
и б и более устойчивые
соединения с валентными
состояниями 4 и 7. При нагревании
технеция на воздухе
образуется оксид ТС2О7 —
кристаллическое вещество, плавящееся
при 119° С. Оксид имеет
кислотный характер и при
растворении в воде дает ионы Тс04~:
Тс207,к + Н20 - 2Тс0^р.р +
+ 2Н+Р.
Рис. 14.57. Давление диссоциации
оксидов марганца
500
W00
1500
/;с
В 7-валентном состоянии технеций (ионы ТсОГ) в отличие от
марганца обнаруживает лишь слабые окислительные свойства.
Оксиды Тс03 и Тс02 могут быть получены восстановлением Тс207,
например:
ЗТс207(К + Тск - 7Тс03к.
В противоположность марганцу технеций образует летучие гало-
гениды, например ТсСЦ, ТсС16 и TcF7, имеющие сильно
выраженный ковалентный характер.
595
Химия рения очень похожа на химию технеция. Валентные
состояния рения 3, 4 и 7 устойчивее валентных состояний 5 и 6.
Поэтому соединения, отвечающие последним валентным
состояниям, синтезировать труднее. На воздухе рений при нагревании
окисляется до Re207—кислотного оксида, дающего при
растворении в воде перренат-ионы ReOr (слабый окислитель). Известны
оксиды Re03 и Re02. Галогенидов рения известно больше, чем у
технеция. Синтезированы все галогемиды Rer4, а также ReF5,
ReCl5, ReBr5, ReF6, ReCl6, ReF7.
§ 18. ЭЛЕМЕНТЫ ПОБОЧНОЙ ПОДГРУППЫ VIII ГРУППЫ
(ЖЕЛЕЗО, КОБАЛЬТ, НИКЕЛЬ И ДР.)
В VIII группу периодической системы включено 9 d-элементов,
которые обычно изучаются семействами по 3 элемента в
зависимости от номера периода. В IV периоде в VIII группу входит
железо, кобальт и никель. Сходство вдоль по периоду у элементов
VIII группы значительно заметнее, чем по подгруппе (Fe, Ru, Os
и др.). Электронные конфигурации наружного и предвнешнего
уровней железа, кобальта и никеля 3 d6 4 s2, 3 d7 4 s2 и 3 d8 4 s2
соответственно. Энергии ионизации, атомные радиусы,
температуры плавления и кипения у всех трех металлов очень близки
(табл. 14.68). Все они ферромагниты.
Таблица 14.68
Элемент
Fe
Со
Ni
Д иониз'
кал/г-атом
182 000
181 000
176 000
о
R, А
1,16
1,16
1,15
пл х
1812
1768
1728
кип * х
3150
3150
3160
По химическим свойствам элементы также очень похожи друг
на друга. Наиболее типичные валентные состояния 2 и 3.
Известны и более высокие, но они неустойчивы и соединения с этими
валентными состояниями проявляют сильные окислительные
свойства.
При действии газов-окислителей на d-металлы образуются
соединения с различными валентными состояниями элементов,
например FeO, Fe203, Fe304 или FeCl2, FeCl3(Fe2Cl6).
При действии хлора на металлическое железо возможно
образование как 2-валентного, так и 3-валентного железа:
FeK + С12,г = FeCl2,r Кг = -^-.
ИС\2
2FeK + ЗС12,Г = Fe2Cl6, К, = Р%с1' .
рс\2
596
После преобразования констант равновесия получаем
^FeCl2 = Ki'Pc\2j
Р?е2С\« = К2'Рс\2-
Из уравнений следует, что повышение парциального давления
хлора способствует повышению выхода Fe2Cl6 в большей степени, чем
FeCl2. Следовательно, при хлорировании металла, способного
иметь несколько валентных состояний, повышение парциального
давления хлора способствует образованию хлорида металла с
более высокими валентными состояниями. Повышение общего
давления в системе оказывает тот же эффект, т. е. равновесие
смещается в сторону реакции, протекающей с наибольшим изменением
объема. В общем случае можно заметить, что повышение давления
реагирующего газа или общего давления способствует вхождению
вещества из газовой фазы в твердую фазу и повышению
валентного состояния металлического элемента.
С кислородом железо образует три оксида: вюстит FeO,
магнетит Fe304 и гематит Fe203. Наибольшее количество кислорода
содержится в Fe203. При нагревании триоксид железа Fe203
переходит в оксид Fe304, а затем в моноксид FeO, который при
дальнейшем нагревании разлагается с образованием железа:
6Fe203,K --= 4Fe304,K + 02,г /С, - P0f,
2Fe304,K = 6FeOK + 02,r Kt = ^o2,
2FeOK = 2FeK-;-02fr Ks = Poz.
Все эти реакции проходят с Д#>0 (поглощение тепла), поэтому
повышение температуры способствует смещению равновесия
вправо. Повышение давления кислорода смещает равновесия влево.
Таким образом, низкие температуры или высокие давления
кислорода содействуют получению оксидов с высоким содержанием
кислорода, и наоборот. Получение оксидов с высоким содержанием
кислорода из металла или других оксидов с меньшим содержанием
кислорода можно проводить, обрабатывая последние в атмосферах
газов-окислителей (02, С02, Н20) при возможно более низких
температурах. Наоборот, чтобы уменьшить содержание кислорода
в твердой оксидной фазе, следует богатые кислородом оксиды
обрабатывать газами-восстановителями (Н2, СО) при высоких
температурах:
3Fe203.K + COf - 2Fe304,K + C02r \
Fe304,K + COr = 3FeOK + C02r 1К = -^-,
FeOK + COr - FeK -f C02,r ) co
597
или
3Fe203 + Н2,г = 2Fe304 + Н20г
Fe304 + H2,r = 3FeOK + H2Or
FeOK + H2,r = FeK + H20r
Все эти равновесия имеют большое практическое значение при
производстве железа из природных руд.
Каждый из оксидов железа легко переходит в другой с
большим или меньшим содержанием кислорода при изменении
температуры, парциального давления кислорода или в случае смеси
газов отношения парциального давления газа-окислителя к
парциальному давлению газа восстановителя (Рияо/Рнл\ PcqJPcq)*
Восстановление оксида железа Fe203 до металла проходит
различно в зависимости от температуры. При температуре ниже
572°С восстановление проходит только в две стадии, через
образование промежуточного состава Fe304:
Fe203 — Fe304 — Fe (t < 572°C).
При температуре выше 572°С восстановление проходит в три
стадии, через последовательное образование магнетита и вюстита:
Fe203 — Fe304 — FeO — Fe (t > 572°C).
Это объясняется тем, что при температуре ниже 572°С вюстит не
устойчив и диспропорционирует по уравнению
4FeOK-Fe304,K + FeK.
На диаграмме состояния железо — кислород (рис. 14.58)
видно, что поле вюстита простирается только до температуры 572°С.
На графике давления кислорода над оксидами железа (рис. 14.59)
видно, что кривые давления кислорода над оксидами FeO и Fe304
выше 572°С располагаются отдельно, а при этой температуре
сливаются в одну ветвь. '
Каждый из оксидов железа обладает способностью содержать
переменное .количество кислорода в твердой фазе в зависимости от
условий его синтеза или обработки в той или иной окислительно-
восстановительной атмосфере.
Легкость перехода одного оксида в другой и их переменный
состав объясняются как различными валентными состояниями
железа, так и особенностью кристаллических структур оксидов.
Структуру любого оксида железа можно представить «как плотно
упакованную кубическую решетку из ионов кислорода. Между
ионами кислорода располагаются октаэдрические и тетраэдриче-
ские пустоты. Если все октаэдрические пустоты заполнить ионами
Fe2+, то на один атом кислорода будет приходиться один атом
железа и состав оксида будет соответствовать идеальному FeO.
Если небольшое количество ионов Fe2+ окислить до Fe3+ — для
сохранения электронейтральности кристалла такая операция соот-
К
/\
н2о
н2
598
ветствует замене некоторого количества ионов Fe2+ двумя третями
ионов Fe3+ — образуется вюстит переменного состава FenO,
например Fe0,95O (при 572°С, рис. 14.58). В зависимости от условий
состав вюстита может изменяться в интервале FeOi,o2—FeOi,og.
При высоких давлениях кислорода был синтезирован вюстит
стехиометрического состава FeO.
пп I J 1 1 Li 1|_ 1 i . . . ■ i i i—i—i—L
22' 24 2S ]2в 3G1 500 WOO o 1500
F«0 FeO FeO, A,C
3 4 2 S
Рис. 14.58. Часть диаграммы состояния желе- Рис. 14.59. Давление диссоциации
зо—кислород оксидов железа
Нестехиометрия вюстита хорошо показана на диаграмме
состояния железо — кислород (рис. 14.58), где видно, что с
повышением температуры область вюстита расширяется, достигая
максимальной ширины при температуре плавления. При повышении
температуры низкокислородная граница поля вюстита (Fe—FeO)
изменяется незначительно, а высококислородная (FeO—Fe304)
смещается в сторону увеличения количества атомов кислорода.
Температура плавления вюстита непостоянна, она повышается
с увеличением количества кислорода в твердой фазе.
Последующее окисление вюстита, или, что то же, замена двух
третей всего количества ионов Fe2+ на ионы Fe3+ приводит к
составу Fe304, в котором на каждый ион Fe2+ приходится два
иона Fe3-*-. В оксиде Fe304 все ионы Fe2+ расположены в октаэдри-
ческих пустотах, а ионы Fe3+ поровну в октаэдрических и тетра-
599
эдрических. Дальнейшее окисление оставшихся ионов Fe2+ — или
замена их на ионы Fe3+ — даст состав Fe203, содержащий только
железо в 3-валентном состоянии. Таким образом, все взаимные
переходы между оксидами железа происходят без заметного
нарушения кислородной решетки и кристаллической структуры оксида
в целом, что и объясняет легкость превращения одного оксида
в другой.
Поле магнетита значительно уже поля вюстита.
Низкокислородная граница магнетита почти не изменяется с температурой,
а высококислородная с ростом температуры смещается в сторону
большего содержания кислорода.
н0о
Рис. 14.60. Изотерма
восстановления магнетита и вюстита
15
10
0,5
ТгЧ
х в FeO.
На рис. 14.60 приведена изотерма восстановления водородом машешта
и вюстита (1250°К), где изображена зависимость равновесны к отношений
РН2о1РИ2 для магнетита, вюстита и процессов их восстановления. Пунктиром
показан вид изотермы в предположении, что магнетит (линия 1) и вюстит
(линия 3) имеют составы, отвечающие формулам Fe304 и FeO соответственно.
В этом случае восстановление магнетита сопровождалось бы переходом вдоль
оси состава из точки a (Fe304) до точки б (FeO) и такому переходу отвечала
бы при данной температуре одна константа равновесия
Fe3OiiK+H2ir
-= 3FeOK -!- Н2Ог К ■
НоО
Однако из-за того, что восстановление магнетита начинается от его
низкокислородной границы (Л) и заканчивается получением вюстита с высоким
содержанием кислорода (Б), процесс восстановления заключается в переходе от
точки Л в точку В:
FeO
! 29;к + 0,18Н2 г = FeOj и -f- 0,18Н2Ог К ■
D0,18
H2Q
эС,18
Н2
600
Изменение изобарного потенциала в этом процессе вычисляется по формуле
Do,is
н2о
AG°- — 4,576-74g-
р0,8
Участок изотермы БВ соответствует непрерывному уходу кислорода из
вещества твердой фазы и переходу состава Fed,и в состав FcOi)02
FeOliI1.K + 0,9H2.r=0,9FeOli02.K + H2Or.
Константу равновесия этого процесса написать невозможно, но изменение
изобарного потенциала вычисляется при помощи специального приема —
графического интегрирования. Для этого предполагают, что восстановление проходит
ступенчато, например через 0,01 состава по кислороду, по изотерме находят
состав газовой фазы (^н2о/"н2)' отвечающей уходу такого количества
кислорода. Это отношение ^Hjo/^h, принимают за константу равновесия и
обычным приемом вычисляют изменение изобарного потенциала на данной ступени
восстановления. Затем, суммируя изменение изобарных потенциалов всех
ступенек, находят общее изменение AG.
Кривая 4 соответствует восстановлению вюстита состава FeOj,o2 до
металлического железа.
реО1,02;к f~ ]'02 H2,r = Fe« "!" 102 Н*°г
к
р1.02
гн2о
р1.02 •
Н2,г
Из-за растворимости водорода в железе часть изотермы ВГ имеет вид не
прямой, а кривой, проходящей через точку Г, что следует учитывать при
вычислении AG этого процесса.
Железо в 2-валентном состоянии в виде ионов образуется при
растворении металла в разбавленных сильных кислотах:
Fe2+ -f 2e = Fe E° = — 0,44 в.
Растворы, содержащие Fe2+, — зеленоватого цвета, а безводные
соли, например FeCl2, бесцветны. Это объясняется гидратацией
ионов Fe2+ и превращением их в октаэдрические комплексы
Fe(OH2)6+- На воздухе растворы Fe2+ постепенно изменяют цвет
на красновато-коричневый из-за окисления ионов Fe2+
кислородом:
2Fe2+ + ± 02,г + 2Н+р = 2Fe3+ + Н20 э.д.с. = 0,46 в..
Хлорид железа FeCb в растворе на воздухе гидролизуется и
одновременно окисляется:
2FeCl2,K + —02,г -1- 2Н20 - Fe203,K + 4НС1р.Р,
или
2Fe;+ + — 02.г + 2Н2Ож - FeА.к - 4Н+Р,
2
601
js 1 РР]
i^ep-pJ Н02
При действии раствора щелочи на раствор Fe2+ образуется
белый осадок Fe(OH)2, обладающий амфотерными свойствами: в
горячем концентрированном растворе щелочи осадок растворяется
из-за образования ионов Fe(OH)6~, которые входят в состав
комплексной плохорастворимой соли Ыа4|Те(ОН)б]. Однако
основные свойства Fe(OH)2 (растворение в кислотах)
преобладают над кислотными.
Дигидроксид железа на воздухе очень быстро темнеет из-за
окисления и перехода в тригидроксид железа. Fe(OH)3 — амфо-
терное соединение. При растворении в концентрированных растворах
щелочей образуются феррит-ионы простейшей формулы FeO<T-
Fe (ОН)з,к + ОН^р =, Fe02-" Р + 2Н20.
Отвечающая феррит-иону железистая кислота HFe02 неизвестна,
но соли ее — ферриты — известны и могут быть получены
нагреванием смесей оксидов, гидроксидов, карбонатов, нитратов и других
солей:
Fe203,K + 2ЫаОНж = 2NaFe02,K + Н2Ог,
2Fe (ОН)з,к + СаОк = Са (Fe02)2,K + ЗН2Ог,
2Fe (ОН)з,к + NiC03,K - Ni (Fe02)2.K + С02,г f ЗН2Ог,
Fe203,K + MnOK = Мп (Fe02)2)K.
Обычно формулу ферритов пишут в виде MnFe204, например
MnFe204, CoFe204 и т. п.
Оксид железа Fe304, который может быть синтезирован из
двух оксидов или солей железа, содержащих железо в различных
валентных состояниял, правильнее считать солью железистой
кислоты:
2Fe* (0Н)з,к + Fe"0K = Fe" (Fe"i 02)2,к + 3H20,
следовательно, магнетит Fe304 — это феррит железа Fe1IFeI2II04.
В водных растворах ион Fe3+ в свободном виде не существует,
так как подвергается сильной гидратации шестью молекулами
воды Fe(OH2)63+. Гексаакваион диссоциирует как слабая кислота:
Fe (ОН2)36+р = Fe (ОН2)5 ОНр+ -f Н+р Кг = 9 • 10~4,
Fe(OH2)5OH2p+=Fe(OH2)4(OH)2+p + Hp+p /Си = 5,510~4 и т. д.
Диссоциация объясняет явление гидролиза и кислую среду
растворов солей Fe3+. Хотя реакцию гидролиза обычно пишут в виде
процесса
602
Fej% + H20 = Fe(0H)2P+ +H+P (I),
FeOH2p+ + H20 - Fe (OH)tp.p + Hp+P (II) и т. д.,
правильнее ее писать в виде вышенаписанных уравнений кислотной
диссоциации гексаакваионов и говорить не о гидролизе ионов, а о
диссоциации гидратированных ионов.
При нагревании гидролиз проходит по следующим ступеням:
Fe (ОН) (Н20)25+ р = Fe (ОН)2 (Н,0)£р.р + Н £р,
Fe (ОН), (Н20)4+р-р - Fe ЮН), (Н20)3>к + Н+р.
Молекулы гидратированного тригидроксида железа из-за
отсутствия заряда объединяются, образуется гель и осадок
тригидроксида железа.
Кроме ионов, содержащих по одному атому железа, в водном
растворе хлорида железа образуются в зависимости от
концентрации железа и температуры различные полиядерные комплексы и
молекулы. Например:
2Fe (H,0)&p - Fe, (ОН), (H,0)ft.p ^ 2НР!:Р + 2H20.
Повышение температуры способствует полимеризации и
объединению ионов. Связь между атомами (ионами) железа в
полиядерных -комплексах не непосредственная, а через молекулы воды,
по природе близкая к водородной связи.
При нагревании раствора хлорида железа возрастают как
скорость реакции гидролиза, так и степень гидролиза. Поведение
продуктов гидролиза зависит от начальной концентрации соли.
Если раствор, содержащий 5—30% FeCl3, сначала нагреть до
кипения, а затем охладить, то осадок вновь растворяется с
образованием исходного продукта (обратимый гидролиз). У растворов
с концентрацией FeCl3 1—4% обратная реакция протекает очень
медленно, а в растворах с концентрацией FeCl3, меньшей 1%,
реакция гидролиза необратима.
С ростом валентного состояния d-элемента характер связи в
галогенидах становится все более ковалентным. При этом гидролиз
галогенидов усиливается. Это правило полностью применимо к
хлоридам железа. В отличие от FeCl3 дихлорид железа FeCU
гидролизуется очень слабо.
Ионы Fe(OH2)6+бесцветны. Цвет водных растворов трихлорида
железа объясняется цветом образующихся при диссоциации
ионов Fe(OH2)5OH2+ и Fe(OH2)4(OH)+. Равновесие диссоциации
гексаакваиона смещается влево при прибавлении сильной кислоты
(Н+), и раствор обесцвечивается благодаря переходу гидроксо-
акваионов в акваионы Fe(OH2)6 .
Ион Fe(OH2)5OH2+ очень устойчив и сохраняется даже в
сильнокислых растворах. Так, в растворе хлорной кислоты (ион С10Г не
603
склонен образовывать комплексы) при рН = 2 98% ионов Fe-]+
содержится в виде Fe(OH2)6+ и 2% в виде Fe (ОН2)5 ОН2+.
Поведение иона Fe3+ существенно меняется в ^растворах кислот, анионы
которых образуют комплексные ионы. В растворе серной кислоты при
рН = 3 только 5% ионов Fe3+ содержатся в виде иона Fe(OH2)e+,
35% в виде иона Fe(OH2)5OH2+ и 60% в виде комплексного иона
(ионной пары) FeSO^- В том же растворе серной кислоты при рН=2
в отличие от раствора хлорной кислоты того же значения рН ионы
Fe(OH2)5OH2+ исчезают, но ионов Fe(OH2)e+ содержится только
50%, остальные ионы Fe3+ связаны в комплексы FeSOf.
При действии хлора на осадок Fe203 в растворе щелочи
железо переходит в 6-валентное состояние и образуются феррат-ионы
Fe04~. Им отвечает железная кислота H2Fe04, не известная в
свободном виде.
Феррат-ионы устойчивы только в щелочных растворах. В
кислых или нейтральных средах феррат-ионы переходят в ионы
Fe(OH2)g+:
4Fe04VP + 20Н+р + 14Н20 - 4Fe (ОН2)^.р + 302,г.
Феррат-ион — сильный окислитель.
Сплавы железа с углеродом — основа важнейших технических
сплавов — сталей и чугунов. Поведение сплавов зависит как от
количества углерода, так и от термической обработки сплава.
Последняя обусловлена модификациями железа.
Железо существует в зависимости от температуры в виде трех
модификаций: а, у и б; а- и б-железо кристаллизуется в
объемноцентрированной кубической решетке, а ^-железо — в
гранецентрированной кубической. Ниже 910°С устойчиво а-желе-
зо, между 910 и 1400°С устойчива ^-модификация, а выше 1400°С
и до температуры плавления б-железо. Низкотемпературная а- и
высокотемпературная б-формы железа во всех отношениях
похожи, за исключением постоянной решетки: ниже 910°С aa_Fe —
о о
=2,867 А, а выше 1400°С Яб-Fe^ 2,94 А. (Постоянная решетки YFe =
= 3,64 А.) Поэтому часто а- и б-модификации отождествляют.
Если нагревать вещество, то его температура будет равномерно
повышаться по мере подвода тепла. На кривой «температура —
время» при этом не должно регистрироваться каких-либо пере*
гибов, скачков, изломов или горизонтальных площадок. Но если
при нагревании в кристаллическом веществе произойдет при
некоторой температуре превращение, то поглощаемая превращением
теплота нарушит равномерность повышения температуры вещества
и на кривой «температура — время» будет отмечен или излом или
остановка. Последняя характерна для чистых веществ. На
604
парамагнитное
°*- ферропо * ни ти ое
8ре^я
Рис. 14 61. Кривая
нагревания железа
рис. 14.61 схематично представлена кривая нагревания чистого
железа. Первая площадка соответствует точке Кюри —
температуре, при которой железо из ферромагнитного состояния переходит
в парамагнитное (769°С). Две последующие остановки при 910 и
1400°С отвечают переходом а^-у и У~*& (а) соответственно.
Последняя остановка имеет место при
1538° С и железо из кристаллического
состояния переходит в жидкое.
При добавлении к железу углерода
температуры плавления и
полиморфных превращений изменяются в
зависимости от количества углерода.
Кривые зависимостей этих температур от
концентрации углерода ограничивают
поля на диаграмме состояния системы
железо — углерод (рис. 14.62).
Кривые, расположенные вблизи чистого
железа, начинаются от точек
температур полиморфных превращений и
плавления. При добавлении углерода
к железу температура плавления
железа сначала снижается (от Л до С), а затем вновь повышается
(от С kD).
Углерод образует с железом твердые растворы внедрения, &
которых атомы С, имеющие небольшие размеры (i?c = 0,77A),
размещаются в междоузлиях кристаллической решетки металла,
состоящей из более крупных атомов (/?Fe=l,26 А). Твердый раствор
углерода в низкотемпературном а-железе называется а-ферри-
том (не путать с ферритами!), в высокотемпературном б-железе —
б-ферритом, а в ужелезе— аустенитом.
Расплавы, содержащие до 1,75% углерода, после быстрого
охлаждения до 1150°С представляют собой твердый раствор —
аустенит. Обычно из него готовят сталь. При содержании углерода
более 1,75% после охлаждения до 1150°С образуется аустенит и
кристаллическая эвтектика, которая мелкими кристаллами
располагается между крупными кристаллами аустенита. Такие составы
называются чугунами. Эвтектика может кристаллизоваться двумя
способами. При быстром охлаждении эвтектическая смесь состоит
из кристаллов аустенита и кристаллов карбида железа состава
Fe3C, называемого цементитом. Карбид Fe3C неустойчив, поэтому
при медленном охлаждении он распадается на железо и углерод
(графит). Железо переходит в аустенит, а графит остается в виде
отдельной фазы. Таким образом, при медленном охлаждении
образуется смесь кристаллов аустенита и графита. Чугун,
содержащий цементит (быстрое охлаждение), называется белым, а
содержащий графит (медленное охлаждение) — серым. При
некоторых средних скоростях охлаждения образуются чугуны,
содержащие и графит и цементит.
605
Полиморфные превращения в железоуглеродных сплавах
связаны с перестройками кристаллических решеток на основе а-, б-,
«у-железа. В зависимости от условий охлаждения и нагревания
полиморфные превращения совершаются с различными скоростями
и этим путем удается изменять структуру сплавов, получая
материалы с разнообразными требуемыми свойствами. Наиболее часто
16001-
1538
Uoov
A S+m
1200 h
1000
ж
ж + :
гж + цементит
(Аустенит)
Аустенит -*• цементит
600
Феррит + цементит
_J I I I I I L
JL
-L
4
I
5"
j I
6
1
Стали
Чугунь/
%С
О 10 20 ' 30 40 50 50 70 80 90 100 %Fe С
Рис. 14.62. Часть диаграммы состояния железо—углерод
пользуются так называемой закалкой, заключающейся в нагреве
сплава и последующем быстром охлаждении для сохранения
исходной кристаллической структуры и предотвращения
низкотемпературных превращений. Закалка возможна только тогда, когда
скорость охлаждения выше скорости нежелательных
низкотемпературных превращений. Поэтому закаленные структуры
термодинамически не устойчивы, при нагреве совершаются
соответствующие превращения и сплав переходит в более устойчивое состояние.
Как видно из рис. 14.62, термодинамически устойчивым
состоянием при температурах выше кривой GSE является
аустенит — раствор углерода в у-железе. При медленном охлаждении
606
аустенита от температур ниже этой кривой он распадается на
феррит и цементит. Для получения твердой, прочной и
малопластичной стали необходимо избежать распада аустенита. Закалка, т. е.
быстрое охлаждение от температуры существования аустенита,.
предохраняет его от распада. Закаленная сталь отличается
хрупкостью, поэтому после закалки ее часто подвергают отпуску, т. е.
нагреву закаленной стали до температуры 260—300°С. При этом
хрупкость устраняется, но твердость сохраняется прежней.
Регулируя содержание углерода в составе исходного расплава,,
скорость охлаждения, продолжительность выдержки при
некоторых определенных температурах, можно получать самые
различные сорта сталей и чугуна.
Если сплав охлаждать по заданному режиму до некоторой
требуемой температуры, а затем закалить, т. е. резко охладить от
этой температуры, то все дальнейшие превращения, которые
должны произойти при более низких температурах, сильно
замедлятся и сохраняется та структура, которая образовалась к моменту
закалки.
Обычный серый чугун содержит углерод в виде графита, а
белый чугун — в виде цементита. Из-за большей твердости и
хрупкости по сравнению с серым чугуном белый чугун пригоден для
отливок и идет на приготовление ковкого железа.
Для превращения белого чугуна в ковкий чугун его
подвергают отжигу — длительной выдержке при 900—1000°С. При этом
происходит разложение карбида железа, образующиеся
включения графита повышают пластичность металла и делают его
ковким.
Кобальту, как и железу, присущи два валентных состояния —
2 и 3, однако кобальт в 3-валентном состоянии значительно более
сильный окислитель по сравнению с железом:
Рез+ + е = ре2+ Е° - 0,771 в,
Со3+ + е = Со2+ Е0 - 1,84 в.
Кобальт в 3-валентном состоянии быстро переходит в водных
растворах в 2-валентное состояние, окисляя воду до кислорода.
В водных растворах кобальт в 3-валентном состоянии устойчив
только в составе комплексных ионов, так как при этом
электродный потенциал кобальта значительно понижается:
[Со (NH3)6]3+ + е= [Со (NH3)6]2+ £° = 0,1 в.
Химия кобальта и железа во многих отношениях схожа. Ди-
гидроксид кобальта Со(ОН)2 амфотерен: легко растворяется в
кислотах, но в щелочах только концентрированных с образованием
ионов Со(ОН)42~. Оксиды СоО и Со304 имеют те же
кристаллические решетки, что и соответствующие оксиды железа. Однако»
оксид С02О3 не существует (известен только его гидрат
Со203-Н20).
607
При переходе от железа к кобальту и никелю электродные
потенциалы металлов численно понижаются:
Fe2+ + 2е = Fe Е° = — 0,44 в,
Со2+ 4- 2е = Со Е° = — 0,28 в,
Ni2+ + 2е = Ni £° = — 0,23 в.
Кобальт и никель обладают меньшей восстановительной
способностью по сравнению с железом, но все же растворяются в
разбавленных кислотах. В этом отношении между кобальтом и
никелем большое сходство — никель слегка слабее как восстановитель
по сравнению с кобальтом.
Дигидроксид никеля Ni(OH)2 в отличие от дигидроксидов
железа и кобальта обладает только основными свойствами.
Монооксид никеля NiO имеет ту же структуру, что FeO и СоО. При
нагревании смеси NiO и Fe203 образуется феррит никеля:
NiOK + Fe203,K - NiFe204)K.
Существование оксида Ni203 не установлено. Предполагается, что
он существует, но только в виде гидрата Ni203-H20. Известно и
другое соединение никеля с кислородом — оксид, по составу
приближающийся к Ni02. При нагревании Ni02 он постепенно теряет
кислород, проходя через фазы переменного состава до оксида NiO.
У железа также известно соединение Fe02, но это пероксид
2-валентного железа.
В концентрированных растворах (0,1 —1,5 моль/л) гидролиз
иона никеля протекает с образованием сложных многоядерных
гидрооксокомплексов Ni4(OH)]+ и Ni2OH3+:
4Ni2+ + 4Н20 - Ni4 (ОН)4+ -ь 4Н1+,
2№2+ 4- Н20 - Ni2OH3+ + Н+.
Остальные 6^-элементов VIII группы являются элементами V
и VI периодов. Между собой элементы каждого периода похожи
в той же мере, как и элементы IV периода (Fe, Ni, Co). Энергии
ионизации у элементов каждого периода Ru—Rh—Pd и Os—Ir—Pt
несколько возрастают с увеличением номера элемента
(табл. 14.69). Атомные размеры изменяются в том же
направлении.
Несмотря на близость энергий ионизации и особенно атомных
радиусов, температуры плавления, температуры кипения и
энтальпии сублимации у элементов различаются довольно сильно
(табл. 14.70).
Рутений и осмий — единственные элементы, способные
проявлять валентное состояние 8: Ru04, Os04. Кроме этих оксидов они
дают диоксиды Ru02 и Os02. Диоксиды известны также у иридия
и платины: 1г02 и Pt02. У родия известен только один оксид
608
Таблица 14.69
Таблица 14.70
Элемент
Ru
Rh
Pd
Os
Ir
Pt
А^иониз, I,
кал/г-атом
170000
172 000
192 000
200 000
200 000
210 000
о
R, A
1,24
1,25
1,25
1,26
1,26
1,29
Элемент
Ru
Rh
Pd
Os
Ir
Pt
*W °*
2523
2236
1827
3300
2720
2045 1
кип' ч
4450
4000
2937
5300 1
4650
4100
Д"субл'
кал/г-атом
157 000
130000
86000
187 000
157 000
132 000
Rh203. Моноксиды образуют платина и палладий PtO и PdO.
Химия галогенидов также очень своеобразная: часто даже соседние
металлы проявляют различные валентные состояния и образуют
один тип соединений с одним галогеном и не образуют его с
другим.
В валентном состоянии 6 осмий и рутений образуют соли
осматы и рутенаты: Na20s04 и Na4Ru04. Хотя платина и
считается одним из самых устойчивых к химическому воздействию
металлов, тем не менее платина легко реагирует с фосфором, серой,
мышьяком при невысоких температурах, а при высоких
температурах заметно проходят реакции (мало изученные) с различными
оксидами, сульфидами и др. Палладий и в меньшей мере платина
легко растворяют водород, давая металлоподобные гидриды но
составу, отвечающие некоторым валентным состояниям, а также
и гидриды переменного состава.
§ 19. МЕДЬ, СЕРЕБРО, ЗОЛОТО
((/-ЭЛЕМЕНТЫ I ГРУППЫ]
Медь, серебро и золото расположены в 1 группе, но их сходство
с щелочными металлами заключено лишь в металлических
свойствах и в численном значении валентного состояния 1, самого
важного для d-металлов I группы. Валентное состояние 1 присуще
серебру, но менее характерно для меди и золота. Валентное
состояние 2 типично для меди. У серебра известно только несколько
соединений в этом валентном состоянии, а у золота такие
соединения не описаны. Валентное состояние 3 характерно для золота,
и только незначительное число соединений известно для меди и
серебра.
Элементы I группы Li, Na, К, Rb и Cs по целому ряду
характеристик похожи на находящиеся с ними в одной группе медь,
серебро и золото. Так, первая энергия ионизации серебра не
намного больше, чем у калия. У всех этих элементов есть s-элект-
роны, все элементы — типичные металлы. Однако d-элементы
I группы очень сильно отличаются от s-элементов той же группы.
Сродство к электрону у меди, серебра и золота значительно
превышает сродство щелочных металлов (s-элементов I группы).
Медь, серебро и золото значительно тверже, более тугоплавки,
20 Общая химия
609
электропроводны, тяжелы, значительно менее реакционноспособны,
не реагируют с водой. По многим свойствам оказывается, что они
более металличны. Причина этого явления — наличие заполненного
предвнешнего ^-подуровня. d-Электроны меди, серебра и золота
участвуют в образовании металлической связи наряду с s-электро-
нами, что приводит к большей прочности кристаллической решетки
этих металлов по сравнению с щелочными металлами.
Первые энергии ионизации меди и серебра очень близки
(178 000 и 175 000 кал/г-атом соответственно), а энергия ионизации
золота значительно выше (213 000 кал/г-атом). У меди вторая
энергия ионизации более высокая, поэтому одновалентное
состояние меди более устойчиво, чем у других элементов. В то же время
атомные радиусы серебра и золота одинаковы — по 1,44 А
(атомный радиус меди 1,28 А). Ионные радиусы металлов в их
1-валентном состоянии равномерно возрастают: Си+ — 0,98, Ag+— 1,13
и Аи+—1,37 А. Только строго определенные соотношения между
энергиями ионизации, атомными и ионными радиусами приводят к
устойчивости тех или иных валентных состояний и объясняют
столь большое различие в химии d-элементов I группы.
Стандартные электродные потенциалы металлов:
Cu+-re=Cu £0=0,52 в,
Си2+4-2е- Си £°-0>34 в
Ag+ -f e = Kg £°-0,80 в,
Ag2+ + 2e-Ag £°-2,80 в,
Аи+ + *=Аи £° = 1,7 в,
Аи3+ + Зе=:Аи £0-1,5 в.
Значения электродных потенциалов показывают, что медь в
растворе устойчива только в виде ионов Си2+, серебро — Ag+, а
золото — Аи3+.
Комбинируя две полуреакции
Си+ + *?- Си £°^о,52 в>
Си2+ + 2г =. Си Е° = 0,34 в,
получаем 2Си+ + 2е - 2Си £° = 0,52 в,
Си — 2;^ С\гг Е°-= — 0,31 в
2Си+ = Си + Си2+ э. д. с. = 0,18 в.
В растворе ионы 1-валентной меди диспропорционируют на металлическую
медь и ионы 2-валентной меди. Константа равновесия этого процесса
„ [Си2+]
^ __ ^ вычисленная из соотношения
[Си+]2
nEF-^RT \n /С,
610
равна
, [Cu2+]
/f=- -^--= 1,2-10».
[Cu+]3
Следовательно, в водном растворе ионы Си+ не могут существовать даже в
достаточных для измерения концентрациях. Пусть металлическая медь
находится в 0,01 М растворе медного купороса. Тогда
0,01
' 0 = 1,2.10е,
[Си+]2
^УЖ='■■»-•
Хотя 1-валентная медь в виде ионов Си1" в водном растворе не
существует, она может существовать в виде комплексных ионов.
Так, в растворе с высокой концентрацией хлорид-ионов образуются
ионы CuClJ", отвечающие комплексной соли КСиС12:
Си2+ + 4С1~ [ Си=2СиС1Г.
Ионы СиС1<Г устойчивы только при высоких концентрациях.
При разбавлении раствора они подвергаются реакции гидролиза,
сопровождающейся выпадением осадка монохлорида меди CuCl.
Аналогичными способами получаются CuBr и Cul. При переходе
от хлорида к йодиду меди растворимости солей сильно
понижаются: ПРСис1 = 3,2.10-7, ПРсиВг = 5,9-10-9; ПРСи1=1,Ы0-12.
Кристаллический хлорид меди CuCl, испаряясь, переходит в газовую фазу
в виде полимерных молекул и ионов: Си3С13, СщСЦ и Си5С15, причем Си3С13
содержится в наибольшем количестве. Предполагают, что Си3С13 имеет
кольцевое строение, структура остальных хлоридов пока не изучена. Кроме молекул
в газовой фазе зарегистрированы различные положительные ионы, например:
Cu5Cl^, Cu5Cl+,Cu4Clf, Cu4Cl^, Cu3Cl^, Cu3Cl^, Cu+, CuXl^, Cu+,Cu2Cl',
CuCl4 и Cu+. Это прекрасный пример того, что процесс испарения есть
не только физическое явление, а в ряде случаев главным образом химическое
явление, сопровождающееся многочисленными химическими реакциями.
Ранее не раз отмечалось, что d-элементы в валентном
состоянии, отвечающем номеру группы, сходны с /^-элементами главных
подгрупп в том же валентном состоянии. Однако это не относится
к s-элементам. Галогениды 1-валентной меди совершенно не
похожи на галогениды калия: у них совершенно различные
кристаллические структуры, различные энергии кристаллических решеток,
энтальпии образования, устойчивость, растворимость, различный
тип связи. У галогенидов щелочных металлов связь
преимущественно ионная, а у галогенидов меди — преимущественно кова-
лентная.
20*
611
То же самое относится и к Cu20 и C112S, не растворимым в
воде, обладающим областями нестехиометрии и имеющим
преимущественно ковалентный характер.
Медь в 2-валентном состоянии похожа на остальные d-элемен-
ты. В водном растворе ионы Си2+ гидратированы четырьмя и
шестью молекулами воды, а образующиеся акваионы
диссоциируют как слабые кислоты:
Си (Н20);+ - Си (Н20)3 ОН+ + Н+,
Си (Н20)е+ = Си (Н20)5 OH+ + Н+.
Написанные реакции — это по существу реакции гидролиза,
которые другим способом пишутся в виде
Си2+ 4- Н20 •= Си (ОН)+ + Н+.
Среда в растворе медного купороса и любой другой соли,
образованной сильной кислотой, — кислая.
Сульфат меди кристаллизуется с пятью молекулами воды:
CuS04-5H20. При нагревании легко удаляется только одна
молекула воды, что находит объяснение в связи четырех молекул воды
непосредственно с ионом меди, а пятой молекулы воды с
кислотным остатком. Четыре молекулы воды образуют с медью четыре
донорно-акценторные связи, а кислотный остаток связан с
внутренней сферой водородными связями через пятую молекулу воды:
Н
I
О—Н Н—О О
н2о\ / \ / \ /
Си2- О S
н2о/ \ / \ / \
о—н н~о о
I
н
В разбавленном водном растворе ионы 2-валентной меди
находятся в виде октаэдрических гексааквакомплексов Си(Н20)б *
которые придают раствору голубовато-синий цвет. При добавлении
в раствор анионов происходит замещение ими молекул воды и
заметное изменение цвета. Так, хлорид-ионы последовательно
замещают молекулы воды, вплоть до образования тетраэдрического
комплекса CuCl*"" желто-зеленого цвета. Цвет растворов бромида
меди другой: молекулы СиВг2 придают ему коричневый цвет, а
ионы CuBrij" окрашивают раствор в вишневый цвет.
При прибавлении к раствору Си2"'" щелочи выпадает осадок
Си (ОН) 2, который растворяется в избытке щелочи и кислоте,
указывая на амфотерность меди:
Си (ОН)2.к -]- Н+р1 V- 4Н20 = Си (Н20)|£-р,
Си (ОН)2,к + 20Нр~р = Си (ОН)24^-р.
612
По последней реакции образуются купрат-ионы Cu(OH)i •
Безводные купраты имеют состав Na2Cu02.
У серебра наиболее устойчиво 1-валентное состояние. При при-
ливании щелочи к раствору азотнокислого серебра выпадает не
гидроксид, а оксид серебра:
2Ag+p + 20Н-Р - Ag20K !- Н20.
Оксид серебра обладает слабыми кислотными свойствами,
растворяясь в концентрированных щелочах:
Ag20K + 20Н^р + Н20 = 2Ag (ОРЩ
р-р.
Хлорид серебра плохо растворим в воде, но в присутствии веществ,
имеющих в своем составе ионы или молекулы, способные быть лигандами, серебро
переходит в раствор. Рассмотрим это на примере растворения AgCl в растворе
тиосульфата натрия. Комбинирование двух равновесий
AgClK -= Ag+p + С1-р ПР = 2,8- 10-ю,
Ag (S2Q0i-р - Ag+p + 2Sp2- p Л:нест - 9,9• 10""
приводит к третьему
AgCIK + 2StOg- p = Ag (S A)i^-p "I" CI^p
с величиной константы равновесия, равной
[Ag(SA)3+][Cl-] 2,8.10-1"
к = 1 = —- = 4,8 • 103.
[Sp2-]« 9,9 • 10-"
Это говорит о сильном смещении равновесия вправо и высокой растворимости
хлорида серебра в тиосульфате натрия, гипосульфите, веществе, используемом
в фотографии для удаления незасвеченных частиц галогенида серебра. Еще
ниже константа нестойкости цианидного комплекса
Ag (CN)2-p.p = Ag+p + 2CN-p /(„ест -1,8 10-1»,
однако в фотографии этот процесс использовать нельзя из-за ядовитости
цианистых солей.
Золото — одно из наиболее устойчивых к химическим
воздействиям веществ. В раствор золото может быть переведено только в
присутствии как сильных окислителей, так и в присутствии ионов,
могущих служить лигандами. Так, растворение золота в царской
водке:
Аик + ЗЫОзТр-р + 4С1рТр + 6Н+р = AuCUTp-p -u ЗЫО^.г i- 3H20
объясняется образованием прочного комплексного иона АиС1Г-
613
Еще более прочен цианидный комплекс золота:
Au (CN)7 = Au+ + 2CN- KttQCT = 5-10~39.
Низкое значение константы нестойки приводит <к изменению знака
электродного потенциала золота и его растворению:
AujS-b3e==AuK E«= 1,5 в,
Au (CN)^p.p + e = AuK n 2CN~ £° = — 0,6 в.
Золото с цезием образуют соединение с преимущественно
ионной связью Cs+Au-. Образование этого соединения возможно
только благодаря тому, что у золота очень высокое сродство к
электрону (большее, чем у кислорода).
§ 20. ЦИНК, КАДМИЙ, РТУТЬ
(/-ЭЛЕМЕНТЫ II ГРУППЫ)
Zn, Cd, Hg — d-элементы, входящие во II группу
периодической системы, заметно отличаются от остальных d-элементов и
очень сильно различаются между собой. Несмотря на то что у всех
этих элементов элекронная конфигурация внешних подуровней
имеет вид dl0s2, свойства но подгруппе изменяются неравномерно.
Так, энергии ионизации Zn, Cd и Hg равны соответственно 217 000,
207 000 и 240 000 кал/г-атом. В то же время в том же направлении
атомные размеры возрастают (1,25, 1,41 и 1,44 А), температуры
плавления понижаются (1131°, 1040 и 630° К).
Сравним свойства галогенидов главной и побочной подгрупп.
В табл. 14.71 приведено сопоставление некоторых свойств
галогенидов кальция и цинка (IV период, II группа).
Таблица 14.71
Вещество
CaF2
СаС12
СаВг2
Са12
ZnF,
ZnClo
ZnBr>
Заряд на
голо! сне, доли
электрона
—0,47
—0,40
—0,36
—0,30
—0,24
—0,18
—0,15
Л\сжатомное
расстояние,
А
2,10
2,51
2,67
2,88
1,81
2,05
2,21
W °с
1418
782
760
740
872
275
394
л«°
Л//обр'
кал/моль
—293 600
—189 800
—163 300
—128 500
—182 700
— 88 300
— 78 800
У одинаковых галогенидов кальция и цинка заряды на атоме
галогена различаются почти точно в 2 раза. Из-за меньших
зарядов на атоме галогенов у d-злементов температуры плавления их
ниже. Энтальпии образования также ниже. Однако межатомные
расстояния у дигалогепидов d-элементов короче, чем у
однотипен
пых дигалогенидов s-элементов. Таково влияние заполнения пред-
внешнего d-подуровня.
Если цинк и отчасти кадмий в некоторых отношениях похожи
па s-элементы той же группы, то ртуть ближе к серебру или меди.
Кадмий и цинк растворяются в кислотах с выделением водорода и
образованием ионов, в которых они находятся в 2-валентном
состоянии. Ртуть не растворяется в кислотах — неокислителях.
Кроме ионов типа Hg2^ ртуть образует ионы Hg22+, в которых она
формально находится в 1-валентном состоянии.
Оксиды всех трех элементов плохо растворимы в воде, но
растворимы в кислотах:
МОк^-2Н+р = М*+ г Н20.
Только оксид цинка растворим в сильных щелочах:
ZnOK ; 20Н~Р I- H20 - Zn (OH)fcp.
Цинкат-ион Zn(OH)4~ имеет простейшую формулу Zn02~~,
являясь производным от несуществующей в свободном состоянии
цинковой кислоты H2Zn02. Взаимодействием кристаллических
NaOH и ZnO при повышенных температурах получается цинкат
натрия Na2Zn02. Из водных растворов цинкат натрия выпадает в
виде кристаллогидрата Na2Zn02-4H20, причем координационное
число цинка 4, а молекулы воды играют роль лигандов.
Ионные радиусы Zn2+, Cd2+ и Hg2+ равны соответственно 0,74,
0,97 и 1,10 А. Из-за больших ионных размеров кадмий и особенно
ртуть не амфотерны и имеют основные свойства.
Галогениды цинка ZnT2 в расплавленном состоянии проводят
электрический ток, указывая на ионную природу солей; только
фторид ртути HgF2 ведет себя также. Остальные галогениды ртути
носят ковалентный характер, и в кристаллическом состоянии в их
структурах подтверждено наличие молекул типа HgCl2.
Хлорид ртути относится к немногочисленному классу солей,
диссоциирующих как слабые электролиты:
HgCl2>p.p = HgClp+p -;- ClpTp Ki - 3,2- Ю-7,
HgCl+p = Hg2p+ + ClpTp Kn = 1,8- Ю-7.
Ионы Hg2^" в водном растворе сравнительно неустойчивы. Стандартные
электродные потенциалы ртути и ионов Hg2+ и Hg2 составляют:
Hg2+ + 2е = Hg» £° = 0,854 в,
2Hg2p+ + 2е - Hg2+_p £о , 0,921 в.
Комбинированием реакций получаем
Щж + Hg*+ = Hg2+р э. д. с. --- 0,921 - 0,854> 0,067 r .
615
Положительное значение э д с. говорит о том, что ионы Hg2+ в присутствии
жидкой ртуги способны превращаться в ионы g2 . Однако из-за
небольшого значения стандартной э. д. с. она изменяет знак в соответствии с
уравнением Нернста при увеличении концентрации Hg2+ или уменьшении
концентрации Hgi^ и становится возможным обратный процесс. В этом процессе
происходит днепропорционирование ионов Hg2 •
Hg22+р = Hg2p+ + Н§ж.
Если в раствор Hg2Cl2 пропускать сероводород, то из-за
образования осадка HgS процесс диспропорционирования смещается
вправо и образуется жидкая ртуть:
Hg22+р + H2Sr = HgSK + Нёж + 2НР+Р.
При действии раствором щелочи на раствор соли по тем же
причинам образуется оксид ртути и ртуть:
НЙ+р ^ 20Н"р = HgOK + Н§ж + Н20.
§ 21. ЛАНТАНОИДЫ (4/-ЭЛЕМЕНТЫ)
В ечнову построения периодической системы элементов Д. И. Менделеева
заложен принцип у каждого следующего элемента появляется один
добавочный электрон. Порядок заполнения электронами энергетических уровней и
подуровней диктуется стремлением системы «атомное ядро — электроны» к
состоянию с минимумом энергии. Энергетическое состояние электрона
определяется в основном значениями главного и орбитального квантового чисел.
Поэтому сначала заполняются те подуровни, для которых сумма квантовых чисел
п+1 наименьшая. Если ж суммы п+1 для некоторых подуровней совпадают,
то сначала заполняется подуровень с меньшим значением п. Подуровни б/?,
5d и 4/ все имеют п + 1 = 7у поэтому заполнение должно происходить в
последовательности 4/—bd—6р.
Барий имеет такую электронную конфигурацию, в которой нет
незаполненных полностью подуровней (рис. 14.63). Следующий электрон согласно правилу
п+1 должен находиться на 4/-подуровне. Однако вопреки этому из-за очень
близких энергий 4/- и 5^-подуровней и из-за особой устойчивости
незаполненного 4/-подуровня электрон вступает на 5^-подуровень. Возникающая
электронная структура отвечает невозбужденному атому лантана. У церия,
следующего за лантаном, распределение электронов подчинено правилу п+1,
поэтому на 4/-подуровне у него находится 2 электрона—один новый, а другой
перешедший с 5с?-подуровня.
В дальнейшем заполнение 4/-подуровня происходит согласно правилу Гунда
так, что в энергетических ячейках электроны располагаются сперва по одному,
и, заполнив 4/-подуровень наполовину (у европия) неспаренными электронами,
далее вступают на 4/-подуровень, образуя пары электронов. Наполовину
заполненный 4/-подуровень обладает повышенной устойчивостью, поэтому у
гадолиния, следующего за европием, на 4/-подуровне сохраняется прежнее
количество электронов (7), а новый электрон входит в 5^-подуровень, нарушая
правило п + 1 У тербия на 4/-подуровень вступает новый электрон и происходит
переход электрона с 5^-подуровня. Таким образом, тербий отличается от
гадолиния двумя добавочными электронами на 4/-подуровне. Иттербием
заканчивается заполнение 4/-подуровня и уровня с п = 4. Новый электрон входит
на 5^-подуровень, создавая электронную конфигурацию атома лютеция,
отличающегося от конфигурации лантана только полностью заполненным 4/-под-
уровнем.
616
Элементы, у которых происходит последовательное заполнение ^-подуровня,
называются ^-элементами. Стремление к образованию устойчивых незаполненой
4/°, наполовину заполненной 4/7 и полностью заполненной 4f14 электронных
конфигураций, сказывается не только на распределении электронов, но и на
Ба
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
»У
Ho
Er
Tm
Yb
Lu
Hf
4s
4p
4*4+4+
4*4+4+
4*4+4*
4+4+4+
4+4+4+
t*4+4+
4*4+4+
t*4*4+
4*4+4+
t+r+4+
4+4+4+
♦♦♦♦♦♦
4+4+4+
t*4+4*|
4*4+4+
4d
4+4+4+4+4+
4+4+4+4+*+
4+4+4+4+4+
4+4*4+4+4+
4+4+4+4+4+
4+4+4+4+4+
4+4+4+4+4+
4+4+4+4*4*
1*4*4*4*4+
4+4+4+4+4+
4+4+4+4+4+
4+4+4+4+4*
t+4+4*4+4+
4+4*4+4+4+
4+4+4*4+4+
4+4*4+4+4+
4f
t 4
* 4 *
till
4 4 4 4 4
t 4 4 4
4
t 4
4 4
4
4 4 4
4+4+ ♦ 4 4
4+4*4*4 4 4 4
4*4*4*4* 4 4 4
4*4*4*4*4* 4 4
4*4*4+4+4+4+4
4*4*4*4*4*4*4*
4*4*4*4*4*4*4*
4*4*4*4+4*4*т-*
№
Sp
4+7*77
4*4*4*
4+4*4+
4*4*4*
4+4*4*
4*4*4+
4*4*4*
4*4*4*
4*4*4+
4*4*4*
4+4Й*
4t4+4*
4*4*4*
4*4*4*
4t4*4*
4*4*4+
4+ilil
Sd
4
4 *
fisl
Щ
4*
4+1
4*!
4*
4+
4+
4+
4+
H
4+
4+
Ц
4+
Щ
Щ
Ш
Рис. 14.63. Электронные структуры атомов лантаноидов
Элемент
La
С1
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
Количество
электронов
4f
0
2
3
4
5
6
7
7
9
10
11
12
13
14
14
Ы
1
0
0
0
0
0
0
1
0
0
0
0
0
0
1
AW298'
кал/г-атом
103 000
111600
89 100
78 300
63 000
49 500
42 100
95 700
93 000
71400
70 000
66400
59 100
36 300
102 200
Таблица 14.72
Д^субл-зезоо,
кал/г-атом
66 700
75 300
52 800
42 000
26 700
13 200
5 800
59 400
56 700
34 900
33 700
30 100
22 800
0
65 900
Доля
участия 4/-
электро-
нов
0,89
1,00
0,70
0,56
0,35
0,18
0,08
0,79
0,75
0,46
0,45
0,40
0,30
0
0,88
617
ряде физических и химических свойств. Энтальпии сублимации металлических
/-элементов приведены в табл. 14.72 и представлены графически на рис. 14.64
Энтальпии сублимации лантана и лютеция примерно одинаковы. В то же
время у них конфигурации /-подуровня 4/° и 4/14 соответственно, а на
^-подуровне находится по 1 электрону (3d1). Европий и иттербий имеют электронные
110000
90000
70000
50000
-
~l
суЪл,298'
1 1 1 1
кал/г этом
1 1 1 1 1 1 1 1 ? 1
La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Vb Lu
Рис. 14.64. Энтальпии сублимации лантаноидов
Таблица 14.73
Элемент
Ва
Sr
Eu
Yb
ЛЯсубл., 298,
кал/г-атом
46 700
39 100
42 100
36 300
конфигурации 4/7 и 4/14 соответственно, в то же время их энтальпии
сублимации близки к энтальпиям сублимации бария и стронция (табл. 14.73).
У металлических стронция и бария связи
между атомами в кристаллической решетке
возникают из-за перекрывания s-орбиталей.
Близость энтальпий сублимации европия и
иттербия к энтальпиям сублимации бария и
стронция позволяет предположить, что у
европия и иттербия связи между атомами
образуются при перекрывании s-орбиталей.
Трудно представить,чтобы внутренние 4/-электроны
могли участвовать в образовании связей
между атомами. В то же время у лантаноидов
очень различные энтальпии сублимации
Видимо, имеется некоторая вероятность перехода
одного или нескольких 'электронов с 4/-подуровня на 5^-или 6/7-подуровни,
находясь на которых электроны принимают участие в образовании связи.
Маловероятен переход с 4/-подуровня на бр-подуровень, так как энергетически два
этих подуровня заметно различаются. Но переход с 4/-подуровня на 5^-под-
уровень энергетически не вызывает затруднения и для простоты будем считать,
что именно такой переход и совершается.
У каждого лантаноида на бя-подуровне имеется по 2 электрона и их вклад
в энергию связи у всех лантаноидов, по-видимому, один и тот же.
Следовательно, различия в энтальпиях сублимации лантаноидов и в энергиях связи между
атомами в решетках кристаллов вызваны различным количеством 5^-элементов,
перешедших с 4/-подуровня. Наименьшая энтальпия сублимации у иттербия.
У него полностью заполнен 4/-подуровень, 5^-электроны отсутствуют, и
металлическая связь возникает только благодаря перекрыванию электронов Gs-под-
уровня. Энергия металлической связи атомов иттербия в его кристаллической
решетке равна энтальпии сублимации, т. е. составляет 36 300 кал/г-атом.
Предполагая, что и у всех остальных лантаноидов бя-электроны дают одинаковый
вклад в энергию связи, т. е. 36 300 кал/г-атом,вычитанием из энтальпий
сублимации лантаноидов энергетического вклада бя-электронов, равного 36 300 кал/г-
атом, получаем вклад в энергию связи, приходящийся на 4/-электроны,
перешедшие на 5^-подуровень.
618
Наиболее высокое значение разности у церия (75 300 кал/г-атом)
По-видимому, у церия наиболее вероятен переход двух 4/-электронов на 5^-подуровень
и участие их в образовании связи между атомами кристаллической решетки.
Если принять энергетический вклад 4/-электронов в образовании
металлической связи, равный 75 300 кал/г-атом, за единицу, то, разделив все разности
АЯ°суГ)л_3630° на 75 300, получим численные значения, доли участия 4/-электро-
нов в образовании связи (табл. 14.72).
На рис. 14.65 графически представлена зависимость доли участия 4/-элек-
тронов в образовании связи у лантаноидов. У лантана и лютеция почти
одинаковые вклады 4/-электронов в энергию связи. Однако в случае лютеция, по-
Доля участия 4f-электронов
(Церий =1, иттербий -0)
La Се Pr Nd Pm Sm Eu Gd Tb Du И0 Er Tm Yb Lu
4f 0 2 3 4 5 6 7 7 9 10 11 12 13 14 14
Sd 1 0 0 0 0 0 0 1
DU ПО
10 11
0 0 0 0 0 0
Рис. 14.65. Доля участия f-электронов в
образовании связи
1600
ыпп
1200
1000
800
VC
*
-V
L-1—I 1 1 I I I I I I I I L L
La Се Рг Nb Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
Рис. 14.66. Температура плавления
лантаноидов
видимому, не следует говорить об участии 4/-электронов в образовании связи
из-за полной заселенности 4/-подуровня. Здесь участвует один 5^-электроп (не
считая наружных s-электронов). У лантана из-за особой стабильности
незаполненной 4/°-подуровня очень высока вероятность перехода электрона с 4/-под-
уровня на 5б/-подуровень. В связи с этим правило п+1 не соблюдается, а
энтальпии связи у лантана и лютеция примерно равны. У европия и иттербия
619
наинизшие значения долей участия 4/-электронов в образовании металлической
связи 6, так как у них нет 5^-электронов, а 4/-подуровень имеет обладающие
повышенной устойчивостью конфигурации 4/7 и 4/14. Поэтому переход
электронов на 5^-подуровень мало вероятен и энтальпии сублимации этих металлов
столь низки и близки между собой. Наибольшая вероятность перехода
электронов с 4/- на ^/-подуровень у
d, г/см
J I I I l I L
J I L
церия. От церия к европию и от
гадолиния к иттербию по
мере приближения к устойчивым
4f7 и 4/14 конфигурациям
(европия и иттербия) доли
участия 4/-электронов в
образовании связи становятся все
меньше (в результате
энтальпии сублимации уменьшаются).
Стремление к образованию
4/°-, 4/7- и 4/14-конфигураций
приводит в ряде случаев к
подобным зависимостям в
изменении других свойств,
например, это прослеживается в
температурах плавления лантаноидов (рис. 14.66). Температура плавления связана
с характером и энергией связи в кристалле, но эта связь сложнее. Тем не менее
видно, что у европия и иттербия температуры плавления аномально низки. Это,
несомненно, вызвано особой устойчивостью 4f7- и 4/14-электронов и отсутствием
5^-электронов.
Плотности лантаноидов повышаются от лантана к лютецию (рис. 14.67)
и только у европия и иттербия, благодаря их склонности использовать для
образования связи только бз-электроны, плотности наименьшие среди лантаноидов.
Стандартные энтропии лантаноидов изменяются еще более сложным
образом (рис. 14.68). Характер изменения энтропии лантаноидов объяснить значи-
La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
Рис. 14.67. Плотность лантаноидов
5 ,кал/моль град
298
La Се Рг Nd Pm Sm Eu Gd Tb Бу ho Er Tm Yb Lu
Рис. 14.63. Энтропия лантаноидов
тельно труднее, так как энтропии кристаллов зависят как от плотности
кристалла, так и от взаимного расположения атомов в решетке и типа связи между
атомами. Тем не менее самая высокая энтропия европия согласуется с его
низкой плотностью и видом его электронной конфигурации.
Интересные особенности об"Наруживаются у лантаноидов при сопоставлении
радиусов атомов и ионов (табл. 14.74). Так как атомные радиусы вычисляются
,из плотностей, то очевидно, что картина изменения радиусов лантаноидов
620
(рис. 14.69) будет противоположной изменению плотностей Низким плотностям
отвечают высокие радиусы. Радиусы атомов европия и иттербия максимальны,
так как они обладают устойчивыми 4/7- и 4/14-конфигурациямк. 4/-элементы
этих атомов не участвуют в образовании связи, что приводит к большим
расстояниям между атомами и их большим радиусом.
La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb lu
Рис. 14.69. Радиусы атомов лантаноидов
Химические свойства элемен- Таблица 14.74
тов главным образом связаны со
структурой внешних электронных
уровней. Изменение количества
электронов на третьем снаружи
4/-подуровне слабо отражается
на химических свойствах
элементов. Поэтому все 4/-элементы
очень похожи по свойствам. Все
лантаноиды обладают валентным
состоянием 3 и образуют ионы
типа М3+. Наиболее характерно
3-валентное состояние для
лантана, гадолиния и лютеция, так
как в этом валентном состоянии
возникают конфигурации 4/°, 4f7
4f14 соответственно.
Радиусы 3-зарядных ионов
заметно отличаются от радиусов нейтральных атомов (табл. 14.74).
Радиусы трехзарядных ионов лантаноидов постепенно
уменьшаются с ростом атомного номера (рис. 14.70), как это имеет место
и у радиусов атомов, однако в данной зависимости нет
максимумов у европия и иттербия. Равномерность изменения радиусов
ионов вызвана одинаковыми конфигурациями 6s и 5й-подуровней
(они пустые) и равномерным увеличением числа 4/-электронов.
Явл-ение уменьшения радиусов трехзарядных ионов лантаноидов с
ростом их атомного номера известно под названием лантаноид-
Металл
1
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
ЛМ» А
1,87
1,83
1,82
1,82
—
1,81
2,02
1,79
1,77
1 1J7
1,76
1,75
1,74
1,93
1,74
Кмз;* А
1,04
1,02
1,00
0,99
0,98
0,97
0,97
0,94
0,89
0,88
0,86
0,85
0,85
0,81
0,80
621
ного сжатия. Оно объясняется тем, что электроны 4/-подуровня
находятся внутри атома и ожидаемое увеличение радиусов
благодаря увеличению числа электронов на этом подуровне
компенсируется все возрастающим притяжением электронов к ядру, заряд
которого непрерывно возрастает.
Лантоноидное сжатие заметно сказывается на свойствах
элементов, следующих за лантаноидами. В табл. 14.75 приведены
размеры ионов некоторых элементов IV, V и VI периодов, причем
положительный заряд иона равен номеру группы.
1,00
0,90
0,80
* а*,А
м
Ij—i i i i— i _i
f
i i i i i i i
La Се Рг Nd Pm Sm Eu Gd Tb Dq Ho Er Tm Yb Lu
Рис. 1470. Радиусы ионов лантаноидов
Таблица 14.75
Ион
к+
Rb+
Cs+
Cu+
Ag+
Au+
Ca2+
Sr2+
Ba2+
in
Ga3+
In3+
Tl3+
Sc3+
уз+
La3+
Lu3+
*. К
1,33
1,49
1,65
0,98
1,13
0,37
1,04
1,20
1,38
0,83
0,98
1,12
0,62
0,92
1,05
0,83
0,97
1,04
0,80
Тип
элемента
S %
d
s
d
P
d
f
Группа
I
II
III
Ион
Ge*+
Sn*+
Pb*+
Ti4+
Zr*+
Hf*+
As^b
SbB+
Bi*+
V»+
Nb6+
Ta6+
Se6+
Tee+
Po8+
Cr6+
R,A
0,44
0,67
0,76
0,64
0,82
0,82
0,47
0,62
0,74
0,40
0,66
0,66
0,35
0,56
0,35
0,65
0,65
Тип
элемента
P
d
P
d
P
d
Группа
IV
V
VI
622
Если проследить за изменением радиусов ионов по подгруппам
(рис. 14.71) К+ — Rb+ — Cs+ , Са2+ — Sr2+ — Ва2+, Sc3+ - Y^-H _
— La3+, Cu+— Ag+— Au+, Zn2+— Cd2+— Hg2+, то заметно, что
при движении вниз по подгруппе радиусы одинаково заряженных
ионов возрастают. В то же время радиусы в подгруппах Ti4+ —
— Zr4+ — Hi4+, V5+ —Nb5+ —Ta5+, Cr6+— Мо6-Ь — W6+ изменяются
более сложным путем. Радиусы последних двух ионов в каждой
подгруппе примерно равны: Rzr4+= RHf4+, RNb5+ = RTa5+> Rm0<H- =
_J I I I 1 L l I t I i I
41 + 2 +3 +4 ■» 5 +6 +1 + 2 +3 + 4 + 5 + 6
п, заряд иона,
номер группы
Рис. 14 71. Влияние лантаноидного сжатия на радиусы ионов
Таблица 14.76
m8J/m
La»4/La
Ce3VCe
Ргз+ /рг
Nd3+/Nd
Pm3+/Pm
Sm34 /Sm
Eu8+/Eu
Gd3+/Gd
E°, в
—2,52
—2,48
—2,47
—2,44
—2,42
—2,41
—2,41
—2,40 |
M3 U/M
Tb3+/Tb
1 Dy3+/Dy
Ho3+/Ho
Er8+/Er
Tu3+/Tu
Yb3+/Yb
Lu3,-/Lu
E°, в
—2,39
—2,35
—2,32
—2,30
—2,28
—2,27
—2,25
623
= R^ .4-. Равенство этих радиусов и объясняется лантаноидным
сжатием. Именно поэтому химические свойства циркония и гафния,
ниобия и тантала, молибдена и гафния очень похожи.
Уменьшение радиусов 3-зарядных ионов лантаноидов с ростом
атомного номера приводит к ослаблению металлических свойств
лантаноидов. При переходе от лантана к лютецию
восстановительная способность металла также уменьшается. Самый сильный
восстановитель среди лантаноидов — лантан. У него самое
высокое отрицательное значение стандартного электродного потенциала
(табл 14.76).
-2,30 Ь
'2,40 h
-2,50
La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Sm
Рис. 14.72. Стандартные потенциалы
восстановления лантаноидов
Зависимость электродных потенциалов от атомного номера
металла (рис. 14.72) обратна зависимости радиусов 3-зарядных
ионов. С уменьшением радиуса иона отрицательное значение
электродного потенциала уменьшается. Следовательно, при переходе
от лантана к лютецию металлические свойства элементов
ослабляются.
Потенциал водородного электрода в чистой воде н2/2Н+~—^,41 в
намного меньше потенциалов лантаноидов. Лантан и остальные лантаноиды
растворяются в воде:
3
2
2Н+ + 2е = Н2
La=La3++3e
£"° = —0,41 в
£<>:= + 2,52 в
6Н+ + 2La = ЗН2 + 2La3+ э. д. с. =2,52—0,41 = 2,11 в,
или
2La + 6Н20 - La3+ + ЗН2 + 60Н~.
При переходе от лантана к лютецию из-за численного уменьшения
стандартных потенциалов э. д. с. реакций уменьшаются, например, для лютеция:
2Lu + 6Н20 - 2Lu3 ь + ЗН2 + 60Н- э. д. с. = 2,25 — 0,41 = 1,84 в.
Из-за уменьшения э. д. с. способность реагировать с водой при переходе от
лантана к лютецию несколько ослабляется в соответствии с ослаблением
металлических свойств.
624
Трехзарядные ионы лантаноидов имеют различные шкча
(табл. 14.77). Последовательность изменения окраски у мерных
семи ионов повторяется в
обратном порядке у последних семи.
Различие в цвете ионов
лантаноидов обусловлено различным
количеством электронов на 4/-
подуровне и различной
вероятностью их переходов на наружные
уровни. Одинаковую окраску
имеют трехзарядные ионы с
одинаковым числом неспаренных
электронов. Ионы с устойчивыми
4/°-, 4/7- и 4/14-конфигурациями
бесцветны.
Металлические лантаноиды на
воздухе быстро тускнеют, легко
горят, превращаясь в оксиды
типа М203, за исключением церия,
который образует оксид СеОг.
Для всех лантаноидов
характерно 3-валентное состояние. Оксиды М20з поглощают из воздуха
диоксид углерода и воду:
М203,к f 3C02 = М2 (СОз)з.к,
М203+ЗНаО = 2М(ОН)8.
Гидроксиды М(ОН)3 можно получить действием раствора
аммиака или щелочи на раствор соли. Они не амфотерны и не
растворяются в избытке щелочи. Чем меньше радиус положительно
заряженного иона, тем прочнее должны связываться с ним
отрицательно заряженные ионы гидроксила. Поэтому в ряду гидроксидов
La(OH)3—Lu(OH)3 связь М3+—ОН усиливается. Следовательно,
с увеличением атомного номера в соответствии с уменьшением
ионного радиуса основной характер гидроксидов в ряду
La(OH)3—Lu(OH)3 понижается. В том же направлении
понижаются и их растворимости.
Ионы М3+ частично гидролизуются в водном растворе.
Растворы хлорида лантана и хлоридов других лантаноидов имеют
кислую среду:
La3+ - Н,0 = М (ОН)2+ -,- Н+.
Кислотность растворов этих солей можно объяснить тем, что
гидратированные шестью молекулами воды ионы М(Н20)63+ ведут
себя как слабые кислоты:
М (Н20)36+р = М (Н20)5ОН*+ + Н+р.
Число
неспаренных
электронов
Цвет
La3+
Се3+
ргз+
Nd3+
Pm3(-
Sm3~
Eu3 +
Gd3+
ТЬз+
Dy3+
Ho3+
Er3f
Tm3+
Yb3t
Lu3+
0
1
2
3
4
5
6
7
6
5
4
3
2
1
0
бесцветный
бесцветный
зеленый
красный
желтый
желтый
розовый
бесцветный
розовый
желтый
желтый
красный
зеленый
бесцветный
бесцветный
625
Благодаря уменьшению ионных радиусов 3-зарядных
ионов склонность к гидролизу в ряду La3+ — Lu3+ увеличивается.
Поэтому растворы солей одинаковой концентрации при переходе
от лантана к лютецию будут все более кислыми.
У лантаноидов наиболее устойчиво валентное состояние 3.
Состояние +1 для лантаноидов неизвестно. Валентные состояния
2 и 4 мало характерны, но их наличие можно связать с
особой устойчивостью незаполненного 4/°, наполовину заполненного в
4//7- и полностью заполненного 4/14-подуровня. Валентное состояние
2 могут иметь европий и иттербий, так как ион Ей24 имеет
конфигурацию 4/75d°6s5, a Yb2+ конфигурацию 4/145d° 6s°. Валентное
состояние + 4 могут иметь церий и иттербий, так как Ce4f имеет
конфигурацию 4/05d06s°, a Tb4+ конфигурацию 4/75d°6s°. Возможные
валентные состояния лантаноидов изображены на рис. 14.73.
Различный диаметр кружков на схеме соответствует сравнительной
устойчивости того или иного валентного состояния.
валентное состояние
" О ° О о
ооооооооооооооо
о О о О
-i_J—I—I—l_J—I I I 1 t I I I I
La Ce Pr № Pm Sm Eu Gd Tb Dy Ho Er Tm Vb Lu
Рис. 14.73. Валентные состояния лантаноидов
Существование у лантаноидов различных валентных состояний приводит к
часто наблюдаемой нестехиометрии кристаллических соединений с кислородом,
азотом, углеродом и др. Для лантана, атомы которого в изолированном
состоянии имеют конфигурацию 4/° Ъй 6s2, наиболее вероятна отдача трех электронов
с приобретением устойчивой конфигурации 4/° 5d° 6s°. Этой конфигурации
соответствует оксид Ьа^Оз (LaOi,s). Из-за постоянства валентного состояния
лантана можно ожидать, что оксид будет обладать узкой областью нестехно-
метрии.
У церия, имеющего в состоянии невозбужденного изолированного атома
электронную конфигурацию 4/2 5d° 6s2, вероятна отдача четырех электронов и
приобретение конфигурации 4/° 5d° 6s°. Это приводит к преимущественному
валентному состоянию церия 4 и к образованию оксида состава Се02. Однако
у церия имеется определенная вероятность образования конфигурации 4/1. При
этом валентное состояние церия понижается до 3, что обусловливает
существование широкой области нестехиометрии в интервале составов
Ce02—CeOi)5(Ce203).
Празеодим наряду с основным валентным состоянием 3 способен
образовывать соединения с более высоким валентным состоянием. Это приводит к
существованию оксида состава Рг6Оц или PrOi,83 и широкой области
нестехиометрии оксида PrOi,5 (Рг20з). Оксиды неодима и самария имеют формулы
Nd203 и Sm203, но у самария уже возможно образование соединений с
валентным состоянием 2, что отвечает оксиду SmO. Поэтому оксиды самария
обладают заметными интервалами иестехиометрии У европия при образовании
химических соединений возникает устойчивая 4/7 5<2° ^-конфигурация и
валентное состояние его равно преимущественно двум. Оксид европия имеет состав
4
3
2
626
FuO, однако он обладает заметной областью нестехиометрии в направлении
оксида Еи01)5(Еи20з). Электронная конфигурация гадолиния в 3-валентном
состоянии имеет вид 4/7 Sd° 6s°. Оксид гадолиния имеет состав Grl203 и не
обладает заметным интервалом нестехиометрии. Электронная конфигурация
изолированного атома тербия имеет вид 4f9 Sd° 6s2. Тербию присуще валентное
состояние 3, которому отвечает оксид состава ТЬ203. Однако также вероятна,
но в незначительной степени, отдача четырех электронов с образованием
устойчивой конфигурации 4/7 5d° 6s°. Это приводит к существованию области
нестехиометрии Tb203—TD4O7 (TbOi,5—TbOi,75).
При дальнейшем заполнении 4/-подуровпя увеличивается стремление к
образованию устойчивой 4/14-конфигурации. Это способствует повышению роли
бя-электронов в образовании связей и увеличению стабильности валентного
состояния 2. У иттербия конфигурация изолированного атома 4/14 5d° 6s2.
У него кроме оксида Yb203 с электронной конфигурацией иттербия 4/13 5d° 6s°
существует оксид YbO с электронной конфигурацией иттербия 4/14 5d° 6s°.
Оксиды иттербия имеют заметную область нестехиометрии. Электронная
конфигурация нейтрального атома лютеция 4/14 5d{ 6s2 способствует образованию
соединений, в которых он обладает валентным состоянием 3. Состав оксида
лютеция LU2O3. Можно ожидать, что для оксида лютеция нестехиометрия не
характерна.
Оксиды М203 обладают каталитическими свойствами,
зависящими от строения электронных подуровней. Так, скорость
окисления СО до СОг на оксидах лантаноидов М203 зависит от строения
4/-подуровня элемента, входящего в состав катализатора
(рис. 14.74). Максимальные скорости окисления СО приходятся
Ед ^{окисление СО на Э 0 ), кал /моль
25000
20000
150U0
La Се Рг Nd Pm Sm Eu Gd Tb Dy Но Ег Tm Yb Lu
Рис. 14.74. Каталитическая активность оксидов
лантаноидов
на оксиды элементов с наиболее устойчивыми конфигурациями
подуровней ионов La34-, Gd3+ и Lu3+. Энергии активации
изменяются симбатно магнитным моментам ионов (рис. 14.75). Можно
предполагать, что строение 4/-подуровня отражается на энергии
связи поверхностного кислорода, которая влияет на величину
энергии активации активного комплекса.
Большинство лантаноидов образуют кристаллические оксиды
типа М20з. Все лантаноиды образуют также оксиды типа МО, но
существующие только в газообразном состоянии при высоких
температурах. Наинизшие энтальпии диссоциации у оксидов европия
и иттербия (рис. 14.76). Зависимости от атомного номера ланта-
1 1 1 1 1 I I I _.]_.!
1 1 1 1
\
1 1
627
ноида энтальпий диссоциации оксидов МО, энтальпий сублимации
и доли участия /-электронов имеют одинаковый характер.
Следовательно, уменьшение энтальпии диссоциации оксидов МО при
переходе от лантана к европию и от гадолиния к иттербию связано
с уменьшением доли участия 4/-электронов в образовании связи.
Магнитный момент M,}i
10
5
О
La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Vb Lu
Рис. 14.75. Магнитные моменты ионов
лантаноидов
D нал/моль
J I > ' ' » I ' ' » '
La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
Рис. 14.76. Энтальпия диссоциации молекул
оксидов лантаноидов
В^ отличие от ^-элементов для лантаноидов можно рассматривать только
случай «слабого» кристаллического поля лигандов, так как энергия спаривания
электронов намного превышает силу поля лигандов. Следовательно, /-подуро-
вень заполняется сперва семью одиночными электронами, а потом начинается
их спаривание. В результате возникает только высокоспиновое распределение,
подчиняющееся правилу Гунда. Поэтому магнитные свойства комплексных
ионов лантаноидов не зависят от относительной силы лигандов. По этой же
причине кривые изменения свойств соединений лантаноидов имеют обычно
только один излом в середине семейства. Этим объясняется неравномерное изменение
радиусов ионов, энергий гидратации ионов, энтальпий их образования, а также
растворимости солей (рис. 14.77), констант нестойкости комплексных ионов
(рис. 14.78.) и т. п.
628
200000
160000
120000
J I I l
В природе лантаноиды распространены очень незначительно.
Величины относительной распространенности лантаноидов в
земной коре приведены в табл. 14.78. Распространенность элементов
сравнивается с распространенностью кислорода, причем дан лога-
-з
1,5 10
-3
10-10
0,5-10 h
Растворимость ди метил фосцютоб,
моль/л
1111 I « » ' '
La Се ?r Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
Рис. 14.77. Растворимость диметилфосфатов
лантаноидов
^5
/6
~18 t
-19
lg К (ътилендиаминтетра
Уст ацетптныи комплекс)
J I L
J I L
I I 1 1 L_J 11)1111 i !
La Ce Pr «Nd Pm $m Eu Gd Tb Dy w0 Er Tm vb Lu
Рис. 14.78. Константы устойчивости
комплексов лантаноидов
Таблица 14.78
Элемент
La
Се
Рг
Nd
Tm
Sm
Eu
Gd
Распространенность
3,2
3,8
3,0
3,6
0
3,2
1,6
3,2 1
Элемент
Tb
Dy
Ho
Er
Tm
Yb
Lu
Распространенность
2,3
3,2
2,3
3,1
2,3
3,2
2,5
рифм количества атомов элемента, которое приходится на 109
атомов кислорода. Для сравнения приведем распространенность кис-
629
лорода 9,0, водорода 8,5, углерода 7,0, кремния 8,5, железа 7,4,
натрия 7,5, магния 7,5 и алюминия 8,0.
Из рис. 14.79 видно, что элементы с четными порядковыми
номерами распространены значительно больше, чем с нечетными.
Следовательно, ядра с четным числом протонов более устойчивы.
Такая закономерность справедлива не только для лантаноидов, но
и для всех элементов.
Распространенность, q
j гЛогарисрп числа атомов элемента на Ю атомов
' г" ^< а кислорода
L i i 1 V i i ' i ' i 1 i i l.
La Се Рг Nd Pm Sm Eu &d Tb Dy Ho Er Tm Yb Lu
N 57 58 59 SO 61 62 63 64 65 66 67 68 69 W 11
Рис. 14.79. Распространенность лантаноидов
В природе лантаноиды очень рассеяны и всегда встречаются
все вместе. Из-за общности химических свойств лантаноиды не
разделяются в природных процессах. Методы искусственного
разделения лантаноидов могут основываться на различии их ионных
радиусов и вытекающих отсюда различных стандартных
потенциалов, различий в рН осаждения гидроксидов, растворимостей
гидроксидов и солей, различной устойчивости комплексных
соединений, возможности образования различных валентных
состояний и т. п.
§ 22. АКТИНОИДЫ (5/-ЭЛЕМЕНТЫ)
Семейство актиноидов состоит из 14 элементов с атомными
номерами 90—103. Актиноиды располагаются в VII периоде
системы Менделеева за актинием. Они, как и лантаноиды, относятся к
III группе элементов. Все актиноиды не имеют стабильных
изотопов. Только торий, протактиний и уран обнаружены в природе.
Положение трансурановых элементов в периодической системе
связано со строением их атомов и проявляемой валентностью.
Теория строения атома предсказывает, что в начале VII периода
системы Менделеева должно происходить заполнение электронами
третьего снаружи 5/-подуровня, аналогичного подуровню 4/ у
лантаноидов. Предполагается, что заполнение 5/-подуровня
начинается после актиния. В результате возникает семейство элементов с
номерами 90—103. По химическим свойствам актиноиды очень
похожи как друг на друга, так и на лантаноиды, что вызвано
одинаковым (в большинстве случаев) строением двух наружных
630
уровней. Наиболее распространено среди актиноидов валентное
состояние 3. Однако существуют и значительные различия между
актиноидами и лантаноидами, которые заключаются в том, что у
актиноидов значительно более распространены высокие валентные
состояния (табл. 14.79). Совершенно четко заметно деление
семейства актиноидов на две половины. Б первой половине семейства,
когда происходит заполнение 5/-подуровня неспаренными электро-
Таблица 14.79
Состояние
1
2
3
4
5
6
7
Актиноид
Th
3
4
Ра
3
4
5
и
3
4
5
6
Np
3
4
5
6
7
Pu
3
4
5
6
7
Am
3
4
5
6
Cm
3
4
Bk
3
4
Cf
2
3
Es
3
Fm
3
Md
2
3
No102
2
3
нами, ясно видна тенденция к увеличению числа валентных
состояний и к росту устойчивых высших валентных состояний у
элементов урана, нептуния и плутония. Во второй половине семейства
наиболее устойчиво 3-валентное состояние, и у последних
элементов семейства актиноидов появляется 2-валентное состояние.
Именно поэтому из-за столь необычных валентных состояний
раньше некоторые из актиноидов помещались в основных
подгруппах. Для тория, протактиния и урана высшие валентности
(4, 5 и 6 соответственно) совпадают с номерами групп, в которых
они могут быть последовательно расположены за актинием.
Однако спектральные исследования показывают, что первые
б/электроны, появляются, начиная с протактиния. Кристаллографические
же исследования свидетельствуют о том, что торий, протактиний
и уран не имеют 5/-электронов, и первый 5/-электрон появляется
только у урана. Изучение же магнитных свойств указывает на
появление 5/-электронов начиная с урана.
Протактиний (№ 91) раньше занимал место под танталом, однако сейчас
его размещают среди актиноидов, хотя он во многих отношениях на них не
похож. Оксид протактиния имеет формулу Ра205, нерастворим в HN03, но
хорошо растворим в HF. Известны соединения 4-валентного протактиния.
Соединения протактиния в наиболее распространенном среди актиноидов
3-валентном состоянии вообще неизвестны. Благодаря энергетической близости 5f- и
6с/-подуровней протактиний ведет себя то как типичный d-элемент (сходство с
ниобием и танталом), то как актиноид.
Уран раньше также считался аналогом молибдена и вольфрама и его
размещали в VI группе. Однако, несмотря на проявление 6-валентного состояния,
уран отличается от Мо и W.
Молибден и вольфрам способны образовывать соединения с галогенами в
2-валентном состоянии. Для урана такие соединения неизвестны. Соединения
631
3-валентного урана больше похожи на соединения лантаноидов и значительно»
отличаются от соединений 3-валентного молибдена и вольфрама. Триоксиды
молибдена и вольфрама Мо03 и WCb легко растворимы в щелочах и не
растворимы в кислотах. Триоксид урана U03, наоборот, легко растворим в кислотах,
но не растворим в щелочах.
Молибден и вольфрам, а также хром образуют гетерополисоединения, но
уран не обладает такой склонностью. Молибден и вольфрам образуют гексакар-
бонилы Э(СО)6, тогда как уран их не образует. Диоксид урана имеет иную
кристаллическую структуру, чем диоксиды молибдена и вольфрама. В природе
минералы вольфрама и молибдена никогда не сопутствуют урановым. Таким
образом, размещение урана в VI группе не соответствует его химическому
поведению. Выделение элементов, следующих за актинием, в одно семейство
оправдано как их свойствами, так и закономерностями распределения электронов*
по уровням и подуровням в свободных атомах.
Нептуний и плутоний проявляют переменную валентность от 3
до 7, причем для нептуния характерна валентность 5, а для
плутония 4. Если водные суспензии нептунатов (VI) и плутонатов (VI)
K2Np04 и К2Р11О4 окислять озоном в сильнощелочной среде,
осадки растворяются с образованием нептунат- и плутонат-ионов (VII)
2KaNp04iK -'- Оз.г + 20Н-р - 4К£Р + 2ЫрО|£Р + Н2Ож -J- 02,г.
Элемент № 101 — менделевий — первый элемент среди
актиноидов, полученный в 1-валентном состоянии, хотя типичное
валентное состояние его 3 и 2. В водных растворах Md3+ легко
восстанавливается до Md2+, проявляя сходство со своим аналогом из
семейства лантаноидов — тулием. Возможность существования
1-валентных тулия и менделевия вытекает из правила повышенной
устойчивости полностью заполненного подуровня. Однако
1-валентный тулий до сих пор не получен, а 1-валентный менделевий
получен в спиртовом солянокислом растворе, причем в этой среде
1-валентное состояние менделевия наиболее устойчивое.
Подтверждением 1-валентности менделевия явилось то, что он
кристаллизовался совместно с труднорастворимыми солями щелочных
металлов. Элемент № 102 проявляет почти всегда валентность 2, а для
перевода его в 3-валентное состояние необходимы сильные
восстановители. Здесь ярко выраженное отличие от свойств
иттербия — аналога из 4/-семейства.
Лоуренсий заканчивает семейство актиноидов. Его устойчивая
3-валентность делает его похожим на аналогичный 4/-элемент —
лютеций, для которого также характерна 3-валентность. Элемент
№ 104 — курчатовий — уже не входит в семейство актиноидов,
а является d-элементов, находящимся в одной подгруппе с
гафнием, цирконием и титаном. Эксперименты указывают, что,
по-видимому, для курчатовия характерна валентность 4, так как его
валентное состояние не похоже на валентное состояние лантаноидов
и тяжелых актиноидов.
Формально элемент № 105 должен быть аналогом тантала,
элемент № 106 — вольфрама и т. д. Но в связи с очень близкими
632
энергиями 5/- и 6 d-подуровней можно ожидать проявления
больших валентных состояний.
Трехзарядные ионы актиноидов по поведению в водных
растворах очень похожи между собой. При переходе тория к лоуренсию
число электронов на 6- и 7-м уровнях почти не изменяется. Из-за
роста положительного заряда ядра притяжение внешних
электронов к ядру возрастает и размеры нейтральных атомов и ионов
уменьшаются при увеличении номера актиноида. Это явление
носит название актиноидного сжатия. Поэтому радиусы трехзаряд-
ных ионов уменьшаются с ростом атомного номера:
Th3+1,08; Ра3+1,06; U3+l,04; Np3+1,02; Pu3+1,01; Am3+1,00A
и т. д. Благодаря актиноидному сжатию характер изменения
свойств актиноидов в их 3-валентном состоянии такой же, как и у
лантаноидов. При переходе вдоль семейства металлические
свойства актиноидов незначительно ослабляются.
Из-за близости химических свойств актиноидов, особенно элементов второй
половины семейства, проявляющих только низкие валентные состояния, отделить
их друг от друга крайне сложно. Практически единственный метод разделения
актиноидов — это метод ионнообменной хроматографии. Раствор смеси
элементов в 3-валентном состоянии пропускают через заполненную органической
смолой трубку (колонку). Ионы реагируют с материалом смолы, замещая в них
положительно заряженные ионы (Na+, K+, Са2+ и др.) или же связываясь с
полимером химически. Сила удерживания полимером ионов актиноидов зависит
от радиуса иона. Когда через колонку, содержащую актиноиды, пропускают
чистую воду или раствор солей-комплексообразователей, происходит вымывание
ионов. Можно так подобрать условия, чтобы колонку покидали ионы в
зависимое ги от их радиусов, например, чтобы в первую очередь уходили ионы с
наименьшими радиусами. Так как радиусы ионов от тория к лауренсию
уменьшаются, то ионы будут выходить из колонки в последовательности от лоурен-
сия к торию, т. е. в последовательности уменьшения атомных весов актиноидов.
Вопросы и упражнения
1. Какой принцип положен в основу построения периодической системы
элементов Д. И. Менделеева?
2. Укажите расположение s-, p-t d- и /-элементов в периодической системе.
3. Где место водорода в периодической системе?
4. Каково отличие молекулярных орбиталей двухатомных молекул щелочных
металлов от молекулы Н2 и других двухатомных молекул?
5. Объясните причины различий в свойствах молекул, содержащих обычный
водород и дейтерий.
6. Как следует относиться к выражению «ион водорода»?
7. Как изменяются свойства двухатомных гидридов при переходе вдоль
по периоду?
8. В чем причина аномальных свойств воды?
9. Какие сведения о структуре жидкости можно получить по численному
значению энтропии плавления?
10. Как изменяются свойства щелочных металлов (размер атома,
температура плавления, энтропия плавления, энтальпия сублимации, электропроводность,
633
энергия ионизации, радиус иона, число шдратации, энтальпия гидратации,
подвижность иона, стандартный восстановительный потенциал)?
11. Как изменяются электродные потенциалы щелочных металлов в
расплавленных солях?
12. Какие кислородные соединения образуют щелочные металлы?
13. В чем причина различного характера реакций взаимодействия щелочных
металлов с водой?
14. Почему литий сильно отличается от остальных 5-элементов I группы?
15. Какие валентные состояния способны проявлять s-элементы II группы?
16. Как изменяются свойства щелочноземельных элементов по подгруппе"?
17. В чем состоит отличие бериллия от нижестоящих элементов?
18. Приведите примеры диагонального сходства элементов.
19. Как изменяются свойства гидридов, хлоридов, оксидов, гидроксидов
при переходе вниз по подгруппе?
20. Дайте общую картину изменения свойств р-элементов.
21. Как изменяется характер связи (ионность, ковалентность) у соединений
с участием р-элементов?
22. Каковы закономерности в изменении свойств 2-атомных молекул при
переходе вдоль по периоду?
23. Какие элементы проявляют склонность к участию в образовании
водородных связей?
24. В чем причина различного строения однотипных молекул одной и той
же подгруппы?
25. Как изменяются энтальпии и энтропии плавления среди р-элементов?
Объясните.
26. В чем причина сходства бериллия и алюминия?
27. Дайте примеры вторичной периодичности.
28. В чем состоит влияние d- и f-электронов на свойства элементов
главных подгрупп?
29. Какова основность борной кислоты?
30. Опишите структуру молекулы диборана.
31. Напишите реакции, показывающие амфотерность алюминия.
32. Как объясняется кислая среда растворов А1С13, A12(S04)3, или А1(Ы03)з?
33. В чем главное отличие таллия от вышестоящих элементов?
34. Каковы валентные состояния р-элементов IV группы?
35. Почему только углерод способен образовывать бесконечно большое
количество различных соединений (органические соединения)?
36. Почему связь С—F слабее связи Si—F?
37. Как следует правильно писать константу диссоциации угольной кислоты
и почему?
38. Составьте формулы германат-, 1ерманит-, станнат-, станнит-, плюмбат- и
плюмбит-ионов. Напишите реакции амфотерных переходов с участием
перечисленных ионов. Какова устойчивость' этих ионов в водных растворах? Дайте
примеры окислительно-восстановительных реакций с участием этих ионов.
39. Какие ионы содержатся в солянокислом растворе хлорного олова?
40. Опишите состав и свойства оксидов азота.
41. Какова структура оксидов фосфора?
42. В чем проявляется сходство мышьяка и сурьмы с вышестоящими
элементами?
43. Каковы магнитные свойства молекул 02, Oj~ и 02' и в чем
заключена причина?
44. Составьте примеры реакций, в которых пероксид водорода проявляет
окислительные или восстановительные свойства.
45. Почему растворимость сернистого газа в воде при прибавлении
порошка серы возрастает?
46. В чем состоит суть йодометрии?
47. Дайте краткое описание химии селена и теллура.
48. Как определяются энергии связи в 2-атомных молекулах?
49. Каковы особенности фтора? В чем причина аномальных свойств фтора
среди галогенов?
634
50. Как изменяются электродные потенциалы галогенов?
51. Какие кислородные соединения образуют галогены?
52. Дайте примеры реакций диспропорционирования.
53. В чем состоят особенности межгалоидных соединений^
54. Приведите примеры реакций с участием инертных газов.
55. Как сказывается повышение устойчивости незаполненного, наполовину
заполненного и полностью заполненного электронами подуровня на свойствах
атомов и соединений?
56. Каковы закономерности в проявлении различных валентных состояний
среди ^-элементов?
57. Опишите характер изменения свойств элементов по мере заполнения
^/-подуровня.
58. Как изменяются кислотно-основные свойства гидроксидов в различных
валентных состояниях элементов?
59. Дайте представление о химии оксидов ^-элементов.
60. Дайте представление о химии хлоридов ^/-элементов.
61. Каковы закономерности в химии комплексных соединений d-элементов?
62. Как влияют лиганды на магнитные свойства комплексных ионов?
63. Покажите плодотворность применения теории поля лигандов для
объяснения свойств соединений ^-элементов.
64. Какова каталитическая активность ^-элементов и некоторых их
соединений?
65. На примере элементов III группы покажите сходство и различие в
свойствах р- и ^-элементов.
66. Как изменяется устойчивость оксидов в рядах ТЮ2—ZrCh—Hf02 и
Ge02—Sn02—Pb02?
67. Как изменяется устойчивость хлоридов в рядах TiCU—ZrCl4—HfCI4,
GeCl4—SnCU—РЬСЦ, TiCl2—ZrCl2—HfCl2, GeCl2—SnCl2—PbCl2?
68. Какова формула титанат-иона?
69. Опишите кислородные соединения ванадия
70. Как изменяется состав ванадиевых ионов в зависимости от рН раствора?
71. Как изменяются окислительно-восстановительные свойства в ряду ионов
Ti3-f __ уз — Сг3 — Мп3' — Fe3 ь — Со3^ ?
72. Чем объясняется устойчивость бихромат-иона в водном растворе?
73. Дайте описание кислородных соединений марганца.
74. Почему BaS04 и КМп04 способны давать между собой твердые
растворы?
75. На основании каких признаков можно судить о сравнительной
устойчивости оксидов марганца?
76. Как можно регулировать изменением условий выход дихлорида и три-
хлорида железа в реакции хлорирования железа?
77. Как влияют внешние условия (Г, Р) на образование различных оксидов
железа?
78. Каковы особенности нестехиометрических оксидов железа?
79. Напишите примеры формул ферритов и ферратов.
80. Каково валентное состояние железа в магнетите?
81. Составьте уравнения реакций гидролиза солей железа.
82. В каких полиморфных модификациях может существовать железо?
83. В чем заключены теоретические предпосылки технологии стали и чугуна?
84. Как изменяются свойства элементов и их соединений в ряду Fe—Со—Ni?
85. Какие валентные состояния характерны для меди, серебра и золота?
86. Как можно осуществить взаимные переходы ионов Hg ~*~ и Ь^-""?
87. Расскажите о последовательности расчета доли участия f-электронов в
образовании химической связи.
88. В чем причина немонотонного изменения некоторых свойств у f-элемен-
тов по мере заполнения /-подуровня?
89. Каковы валентные состояния лантаноидов?
635
90. В чем состоит влияние лантаноидного сжатия на свойства следующих
за лантаноидами элементов?
91. Можно ли применять теорию поля лигандов к соединениям лантаноидов?
92. Как изменяется распространенность лантаноидов по мере роста
атомного номера?
93. Каковы валентные состояния актиноидов?
94. Почему уран не аналог молибдена и вольфрама?
95. Дайте примеры соединений нептуния и плутония в их семивалентном
состоянии.
96. В чем проявляется актиноидное сжатие?
97. Как разделяются актиноиды друг от друга?
ГЛАВА 15
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
§ 1. СТРОЕНИЕ И СВОЙСТВА
ОРГАНИЧЕСКИХ СОЕДИНЕНИИ
Органическими соединениями называют вещества, в состав
которых входит углерод. По своей способности соединяться друг с
другом углерод занимает особое положение в периодической системе.
В настоящее время известно свыше ста тысяч соединений,
образуемых всеми элементами периодической системы, за исключением
углерода. Соединений же только одного углерода изучено и
описано свыше двух миллионов.
Атомы углерода, как никакого другого элемента, способны ко-
валентно связываться друг с другом, образуя молекулярные цепи
различной длины и различной конфигурации. В отличие от других
элементов в углеродных соединениях бывают цепи, содержащие
десятки тысяч связанных друг с другом атомов. Способность
атомов углерода соединяться друг с другом, по-видимому, совершенно
неограничена. Это одна из причин многообразия углеродсодержа-
щих соединений.
Молекулярные углеродные цепи могут быть не только
линейными с незамкнутыми концами, но могут также создавать
замкнутые циклы. Кроме того, отдельный атом углерода, входя в
молекулярную углеродную цепь, может на связь с соседним атомом
углерода затрачивать не одну валентность, а две С—С —С и три
С—С = . Это вторая причина большого разнообразия органических
соединений. Углерод может также соединяться почти со всеми
химическими элементами с образованием молекул цепной и
циклической структуры самых различных размеров. В состав цепей и
циклов молекул органических соединений кроме атомов углерода
могут входить кислород, сера, азот, фосфор, мышьяк, кремний,
германий, олово, свинец, бор, титан, железо и другие элементы.
Если цепи атомов кислорода, азота, фосфора, серы, бора и
других элементов мало устойчивы, то длинные цепи атомов углерода
в силу большой энергии углерод-углеродных связей (347,3 кдж)
весьма устойчивы.
В развитии химии органических соединений огромное значение
имела теория химического строения, разработанная в 1861 г
А. М. Бутлеровым. Он открыл закон зависимости свойств веществ
от их химического строения, лежащий в основе современной
теории строения химических соединений: «Химическая натура
сложной частицы определяется натурой элементарных составных частей,
количеством их и строением» 1.
1 А. М Бутлеров Соч , т. I. M., Изд-во АН СССР, 1953, стр. 70.
S37
Теория химического строения — это учение о взаимной связи,
расположении и взаимном влиянии атомов в молекулах
органических веществ.
В дальнейшем теория химического строения А. М. Бутлерова
была дополнена трудами многих ученых и особенно Ф. Кекуле,
А. Купера, В. В. Марковникова и др.
А М. Бутлеров (1828—1886)
Основные положения теории химического строения заключаются
в следующем:
а) каждое вещество имеет определенное, свойственное ему
химическое строение, т. е. в молекулах веществ атомы соединены
друг с другом в строго определенной последовательности;
б) атомы, находящиеся в молекуле, как связанные
непосредственно, так и связанные через другие атомы, оказывают друг на
друга взаимное влияние;
в) свойства веществ зависят от порядка соединения атомов в
молекулах и от влияния их друг на друга;
631
г) химическое строение сложного вещества может быть
установлено в результате изучения его химических превращений, и,
наоборот, по строению данного вещества можно судить о его
свойствах.
В основе этой теории лежат такие представления:
1) каждый атом углерода, имея четыре единицы валентности,
может присоединять к себе атомы или группы атомов других
элементов;
2) все валентности углерода равнозначны;
3) атомы углерода способны соединяться друг с другом,
образуя «цепи» атомов или «углеродный скелет» молекулы. Например:
! I ! I I I
_с—с—с—с—с—с—
I ! I ! I I
Органические соединения, которые обладают одинаковым
составом и одинаковой молекулярной массой, но различным
расположением атомов в молекуле, называют изомерами. Например,
формуле С4Ню отвечают два углеводорода, которые обладают
разными свойствами. Объясняется это различным расположением
атомов в молекулах:
СНЯ
CHS — СН2 — СН2 — СН3,
нормальный бутан
СН3 ■— СН — СН3
изобутан
Изомерия — также одна из причин многообразия органических
соединений, так как с увеличением числа атомов в молекуле резко
возрастает число возможных изомеров. С увеличением числа
атомов углерода в молекуле растет число структурных изомеров,
(табл. 15.1).
Таблица 15.1
Формула
сн4
С2Н6
СзН8
С4Н10
с5н12
с6н14
с7н1в
CgH18
QH20
Нлзваиис
метан
этан
пропан
бутан
пентан
гексан
гептан
октан
нонан
Число изомеров в ряду алканов
Число
изомеров
1
1
1
2
3
5 1
9 ;
18
35
Формула
с„н24
СцЛе
Q3H28
Q4H30
Ci5HM
С20Н42
~с25н52
СзоНб:
Q(}H82
Название
ундекан
додекан
тридекан
тетрадекан
пентадекан
эйкозан
пентакозан
триаконтан
тетраконтан
Число изомеров
159'
355
802
1858
4 347
366 319
36 797 57&
4 111846 76а
62 491 178 805 831
639
Примером взаимного влияния атомов может служить
различная реакционная способность водорода, связанного с атомом
кислорода (связь —О—Н), и водорода, связанного с атомом углерода
(связь С—Н). В первом случае атом водорода при действии
металлического натрия относительно легко замещается металлом, в
то время как атом водорода, связанный с углеродом, замещается
с большим трудом и не всегда. Причина различной реакционной
способности водорода заключается в различной полярности связей
О—Н и С—Н. Первая более полярна, так как электронная пара
смещена к более электроотрицательному атому кислорода.
Следствием взаимного влияния атомов, непосредственно не
связанных друг с другом, является увеличение диссоциации карбо-
новых кислот с введением в их молекулу галогенов. Так, уксусная
О
кислота СН3— С^ значительно слабее диссоциирует, чем мо-
ОН
О
нохлоруксусная СН2С1—С^ . Это объясняется влиянием ато-
ОН
ма хлора: чем ближе расположен атом хлора к карбоксильной
группе, тем сильнее сказывается его влияние. Введение атома
хлора в молекулу уксусной кислоты увеличивает ее дипольный
момент, так как к атому хлора оттягивается электронная пара
среднего атома углерода, а это, в свою очередь, вызывает смещение
других электронных пар, и в частности, от атома водорода. Такое
взаимное влияние атомов можно изобразить следующим образом:
Н
I II
н о
Главная заслуга в установлении законов и правил взаимного
влияния атомов в молекуле принадлежит В. В. Марковникову
(1837—1904).
Теория органического строения получила объяснение с позиций
квантовой химии, которая позволила понять природу химической
связи и тем самым способствовала дальнейшему развитию
представлений о строении вещества. А. М. Бутлеров рассматривал
химическую связь как особый вид движения атомов. Он также
полагал, что молекулы находятся в постоянном движении и
превращении, подвергаясь многочисленным внутренним перестройкам.
По теории Бутлерова представление об устойчивых и
неустойчивых изомерных формах молекул является чисто формальным, так
64Ф
как в ряде случаев изомерные молекулы настолько легко переходят
друг от друга, что оба изомера находятся в состоянии подвижного
равновесия:
СН,
С — СН9 — СООСоН. ZL СН, — С = СН — GOOCH,
О
кетонная форма
ОН
енольная форма
ацетоуксусный эфир
В других случаях, наоборот, равновесие между изомерами сильно
или почти полностью смещено в сторону одного из них:
СН3 СН3 СН3 СН3
\/ \/
СН —it CBr
СНЗг
сня
Взаимные превращения изомеров могут осуществляться без
какого-либо внешнего воздействия, и, наоборот, иногда требуются
интенсивные внешние воздействия для того,
чтобы вызвать обратимые переходы
изомеров друг в друга.
Распространенное в органической
химии явление, заключающееся в
существовании двух или нескольких изомерных
форм молекул, находящихся в
состоянии динамического равновесия,
называется таутомерией. Исследования
показали, что при одних внешних условиях
вещества могут быть вполне
устойчивыми изомерами, тогда как при других —
таутомерами.
Вопрос о пространственном
расположении атомов получил развитие в
работах Ле-Беля и Вант-Гоффа, которые
независимо друг от друга установили тетраэдрическое строение
атома углерода в органических молекулах. Это означает, что четыре
атома водорода, связанные с атомом углерода, расположены не в
одной плоскости, а в вершинах правильного тетраэдра, в центре
которого, в свою очередь, находится углеродный атом (рис. 15.1).
При одинаковой последовательности связей атомов в молекулах
возможна изомерия, обусловленная различным расположением
атомов в пространстве. Этот вид изомерии называется
пространственной или стереоизомерией.
Органические вещества, содержащие асимметрический
углеродный атом, существуют в виде двух изомерных молекул с
различным пространственным расположением атомов.
Рис. 15.1.
Тетраэдрическое строение атома
углерода
^1 Общая химия
641
СН8СНОН — СООН
молочная кислота (С
СООН
н
он
■ асимметрический атом)
СООН
'I
но — с* — н
СН3 СН3
Эти стереоизомерные молекулы, обладая одинаковыми
химическими и физическими свойствами, различаются между собой в
двух отношениях. Они кристаллизуются в формах, не имеющих
плоскости симметрии и относящихся друг к другу, как предмет
к своему зеркальному изображению. И оба стереоизомера
по-разному относятся к поляризованному свету.
§ 2. ПРИРОДА ПРОСТЫХ И КРАТНЫХ СВЯЗЕЙ
Связь между атомами углерода, образованная парой
электронов, называется простой связью и обозначается одной черточкой:
\ / \ /
— С:С— —С —С—
/ \ / f \
простая связь
Однако между атомами углерода могут быть и кратные связи —
двойная и тройная:
чс-с7
/С7С\
—с = с-
t ч t
двойная связь тройная связь
Атомы углерода могут образовывать замкнутые цепи, т. е. циклы
различного строения:
с
/\
\г/ \г/
/Я
/с
с/
ч
с/
Ч
-сЛ,с-
-с
с
Необходимым условием образования ковалентной связи
является перекрывание электронных облаков. Связь возникает в том
направлении, в котором осуществляется максимальное
перекрывание облаков связующих электронов. В случае а-связи перекрыва-
642
ние облаков осуществляется по прямой, соединяющей два атома.
Если перекрывание электронных облаков происходит в плоскости,
перпендикулярной а-связи, то возникает я-связь. Перекрывание
облаков я-электронов менее полно, чем о-электронов. Этим и
объясняется, что я-связи между атомами углерода менее прочны, чем
простые, поэтому легче разрываются при реакциях
присоединения (рис. 15.2).
Рис. 15 2. Схема расположения я- и а-связей в
молекулах*
а — этилена; б — ацетилена
В молекулах с двойными связями электроны, образующие
я-связь, всегда смещены в сторону более электроотрицательного
атома ^С^о * ^ результате такой поляризации атом
углерода приобретает частичный положительный заряд, а атом
кислорода — частичный отрицательный ^>С6+ = Об~~- Эти заряды во
многом определяют течение различных реакций.
В молекуле могут быть две или несколько чередующихся
двойных связей, называемых сопряженными:
Рис. 15.3. Схематическое изображение молекулы бутадиена (а);
вид сверху (б)
На рис. 15.3 показано, что все а-связи бутадиена лежат в одной
плоскости, а я-связи — в плоскости, перпендикулярной к ней, и
21*
643
образуют единую сопряженную систему, охватывающую все
четыре атома углерода. Теория сопряжения в значительной мере
обязана своим возникновением учению о взаимном влиянии атомов.
Эффекты сопряжения проявляются в молекулах, имеющих л-элек-
троны, которые значительно более подвижны, чем а-электроны.
Рис. 15.4. Облака Jt-электронов в бензоле, перекрываясь друг
с другом, образуют одно общее облако я-электронов (а);
максимальные плотности этого облака находятся выше и
ниже плотности бензольного кольца
К системам с сопряженными связями относится бензол. Все
атомы углерода бензола расположены в вершинах правильного
шестиугольника и связаны друг с другом и с атомами водорода
а-связями. Облака л-электронов, направленные параллельно друг
к другу и перпендикулярно плоскости связей, образуют единое
молекулярное облако с максимальной электронной плотностью по
обе стороны плоскости молекулы (рис. 15.4). В результате этого
все атомы углерода в бензольном кольце равноценны, что и
обусловливает его особую устойчивость.
§ 3. МЕХАНИЗМЫ РЕАКЦИИ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Изучение реакционной способности органических веществ
показало, что разрыв ковалентной связи между атомами может
осуществляться так, что электронная пара остается у одного из двух
ранее связанных атомов:
А|:В-*А+ + :В".
! !
Такой тип разрыва ковалентной связи, приводящий к
возникновению свободных ионов, называется гетеролитическим расщеплением.
Образование ковалентных связей в ходе гетеролитических реакций
происходит путем передачи электронных пар свободными
органическими анионами:
A|:B + C|:D-*A:D + C:D.
I I
644
Большинство реакций между органическими соединениями — это
гетеролитические реакции. Если ионные реакции неорганических
молекул обычно протекают мгновенно, то ионные реакции
органических молекул — медленно, по стадиям. Так, например,
взаимодействие нейтральной или слабополярной молекулы А—В с
ионами С и D состоит из трех стадий:
А^В-^А** 8 U),
д""+У--^а - а (2),
в": * С*—~& - С (3)
Полярные реагенты разделяются па нуклеофильные и
электрофильные. Нуклеофильные, или электронодонорные, реагенты отдают
свои электроны углеродному атому в органической молекуле,
образуя с ним химическую связь. Электрофильные, или электро-
ноакцепторные, реагенты приобретают электроны от углерода
органической молекулы, образуя с ним химическую связь.
Нуклеофильные реагенты отдают электроны, потому их можно
рассматривать как восстановители. Электрофильные реагенты
оттягивают электроны, тем самым проявляют окислительные
свойства. Подобно процессам окисления и восстановления,
нуклеофильные и электрофильные реакции взаимно связаны. Например,
молекула акролеина СН2 = СН—СН = 0 может по связи С = С
присоединять как нуклеофильный реагент—гидросульфит натрия,
так и электрофильный реагент — молекулу хлора:
НоС=СН—Gf \- NaHS03->CH9—CH2—Cf ,
ХН | NH
S03Na
HX=CH-Cf + С12 ->. CHoCl—CHC1—cf .
ХН " NH
В первом случае молекула акролеина проявляет электрофильные,
а во втором — нуклеофильные свойства. Такие реагенты, как
аммиак и вода, в зависимости от условий реакции и природы
взаимодействующего с ним вещества проявляют как
нуклеофильные, так и электрофильные свойства.
Г о молит иче с кое расщепление ковалентной связи происходит
вследствие разрыва электронной пары
А -т- В->.А' + В.
Гомолитическая диссоциация молекул обычно вызывается
термическим или фотохимическим разложением органических веществ.
645
Фотоны с достаточной энергией превращают молекулы в
свободные радикалы. Поэтому действие света, равно как и облучение
УФ-лучами, инициирует процесс гомолитической диссоциации
молекул. В результате гомолитических реакций образуются
свободные радикалы. Установлено, что многие органические реакции
(окисление, галогенирование, нитрование и др.) протекают с
образованием свободных радикалов.
Радикальные реакции обычно цепные, так как взаимодействие
свободного радикала с молекулой приводит к образованию нового
свободного радкала или атома с развитием цепи химических прев-
вращений. Примером цепной радикальной реакции является
реакция металепсии, заключающаяся во взаимодействии галогенов с
углеводородами при освещении:
CI: CI -f- hv ->- 2C1 инициирование,
CH4 + ci->ch; + hci
развитие или рост цепи.
СНз + С12 ^СН3С1 + CI' "
Обрыв цепи происходит вследствие исчезновения из газовой фазы
свободных радикалов в результате их рекомбинации:
С1- + С1 = С12 ]
1 обрыв цепи.
СНз + СНз = С2Н6 ]
Причиной обрыва цепи является также столкновение радикалов
со стенками сосуда и с молекулами примесей. При взаимодействии
свободных радикалов или атомов с молекулами с двойными
связями происходит гомолитический разрыв я-связи и образование
нового свободного радикала:
Br24-ftv->Br -f Br
вг + /C=c/ -*.\c-c-/,
Br
\c-с/ i вг ->. Ъс —c(.
/| \ /| |\
Br Br Br
Энергия, необходимая для разрыва я-связи (пары электронов),
тем меньше, чем больше устойчивость образовавшегося при этом
свободного радикала.
646
Радикальные реакции — преимущественно реакции
автокаталитические. Они играют первостепенную роль в процессах
полимеризации, ведущих к получению ценнейших полимерных
материалов.
§ 4. КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
На основании структурной теории все органические соединения
делятся на два ряда — ациклические и циклические соединения.
I. Ациклический ряд включает соединения, в молекуле которых
имеется открытая, незамкнутая в цикл углеродная цепь:
-С—С-С -С—С—С—С-
-С-
I
—с-
I
—С-
-С—С—С—С—С-С-
Соединения этого ряда называют также соединениями
алифатического, или жирного, ряда.
II. Циклический ряд. Соединения, относящиеся к этому ряду,
распределяются по трем группам.
1. Алициклические соединения — соединения, в молекуле
которых имеется замкнутая (циклическая) углеродная цепь:
С
—С-
—С-
-С-
/с\/с\
I I с
/\
2. Ароматические соединения — циклические соединения, в
молекуле которых имеется скелет бензола — шестичленного цикла
с чередующимися простыми и двойными связями:
сн с
сн/^сн
CHl IICH
сн
бензол
-сАс-
С
3. Гетероциклические соединения — циклические соединения,
содержащие кроме атомов углерода гетероатомы, азот, серу,
кислород и другие:
647
>п<
0
>с/\с<
\г L/
/с\/с\
N
7 i 1 Ч
/С\/<
S
В каждом ряду органические вещества распределяются по
классам. Принадлежность соединения к тому или иному классу
(спирты, альдегиды, кетоны, кислоты, амины, эфиры и т. д.)
определяются характером функциональной группы, которая содержит^
ся в его молекуле.
§ 5. УГЛЕВОДОРОДЫ
Простейшими органическими соединениями являются
углеводороды, которые образуют, как и другие органические соединения,
гомологические ряды. Каждый последующий член такого
гомологического ряда насыщенных углеводородов отличается от
предыдущего на —СН2-группу, называемую гомологической разностью.
Гомологические ряды объединяют органические соединения с
общими химическими свойствами и сходным химическим строением.
Другие классы органических соединений можно рассматривать
как производные углеводородов, образовавшиеся замещением в
молекулах последних одного или нескольких атомов водорода
другими атомами или группами атомов. Все производные
углеводородов можно рассматривать как вещества, молекулы которых
состоят из двух частей: углеводородных радикалов и групп атомов,
обладающих характерной для нее реакционной способностью.
Именно эти группы" в большинстве химических реакций
подвергаются изменению, остальная же часть молекулы — радикал —
целиком переходит во вновь образующееся соединение.
Подвергающиеся изменению группы атомов получили название
функциональных групп; они обусловливают химический характер молекул и
их принадлежность к определенному классу соединений.
Если в молекулах углеводородов имеются только простые
связи, то такие углеводороды называются насыщенными. Их также
называют предельными углеводородами, парафинами, или алка-
нами. Если в молекулах углеводородов имеются кратные связи —
двойные и тройные, то их называют непредельными
углеводородами.
Предельные, или насыщенные, углеводороды имеют общую
формулу С?гН2п+2 и составляют гомологический ряд, первым
членом которого является метан (табл. 15.2).
648
Таблица 15.2
Гомологический ряд насыщенных, углеводородов
Название
Метан
Этан
Пропан
Бутан
Пентан
Гексан
Гептан
Молекулярная
формула
сн4
с2н6
с3н8
С4Н10
с5н12
С6Н14
С7Н16
Состояние
газ
»
»
»
жидкость
»
»
Название
Октан
Нонан"
Декан
Гексадекан
Пентаэйкосан
Триаконтан
1 Гептаконтан
Молекулярная
формула
QH18
СэН20
СюН22
CieH34
Q5H52
Сзо"в2
^70^! 42
Состояние
жидкость
»
»
твердое
вещества
»
»
»
В молекулах этих углеводородов атомы углерода соединены
между собой а-связью, прочность которой обусловливает малую
химическую активность этих соединений.
Химические реакции насыщенных углеводородов
осуществляются в результате разрыва а-связи С—Н и замены атома
водорода на какой-либо другой атом или группу атомов. Окисление
высших насыщенных углеводородов сопровождается также
разрывом связи, но разрыв происходит в этом случае между атомами
углерода С—С. Реакции окисления имеют важное промышленное
значение для получения мыл, поверхностно-активных веществ,
смазочных материалов, синтетических смол, лаков, эмалей и т. д.
Первые четыре представителя предельных углеводородов
нормального строения называются так: метан СН4, этан С2Нб, пропан
СзН8, бутан С4Н1С. Далее названия углеводородов образуются от
греческих и латинских числительных с добавлением окончания
«ан»: пентан C5Hi2, гексан С6Нн, гептан С7Н16, октан С Н}8, нонан
С9Н2% декан СПН22 и т. д.
Чтобы дать название изомерам с разветвленной цепью,
необходимо знать наименования простейших одновалентных
органических радикалов, т. е. остатков, образующихся в результате
отрыва атомов водорода от предельных углеводородов. Названия
радикалов с заменой окончания «ан» на «ил», например:
метан СН4 —СНЭ метил,
этан С2Н6 —С2Н5 этил,
пропан С3Н8 —С3Н7 пропил и т. л.
Названия некоторых радикалов образуются от названия
соответствующих спиртов. Так, от амилового спирта С5НцОН берет
название радикал амил —С5Н1Ь от винилового спирта СН2 = СНОН—
радикал винил СН2=СН—.
649
Источниками насыщенных углеводородов являются нефть и
природные газы. Если нефть подвергнуть перегонке, можно
получить несколько фракций, отличающихся друг от друга по
температуре кипения. Фракции с более низкой температурой кипения
содержат углеводороды меньшего молекулярного веса, а высоко-
кипящие фракции — углеводороды большего молекулярного веса.
Природные газы более чем на 90% состоят из метана СН4.
Насыщенные углеводороды в химическом отношении вещества
относительно инертные. Только при высоких температурах под
влиянием катализаторов или других инициаторов они вступают в
химические реакции. Насыщенные углеводороды применяются в
качестве топлива (газообразное, жидкое) и сырья для химической
промышленности при производстве различных химических
продуктов.
Рис. 15.5. Изображение а- и .тт-связей в этилене
Природный газ как химическое сырье используют для
получения сажи, водорода, ацетилена, окиси углерода, метилового спирта
и других важных продуктов, которые находят широкое
применение в различных отраслях промышленности.
Для насыщенных углеводородов характерны реакции
замещения.
Непредельные, или ненасыщенные, углеводороды имеют в
молекулах кратные связи — двойные и тройные. Ненасыщенные
углеводороды, содержащие двойную связь, называются олефино-
выми углеводородами. Их также называют этиленовыми
углеводородами, или алкенами. Ненасыщенные углеводороды с тройной
связью называются ацетиленовыми углеводородами, или алкинами.
Олефиновые, или этиленовые, углеводороды (алкены, или оле-
фины) с общей формулой CnH2n имеют двойную связь между
атомами углерода (рис. 15.5). Непрочность я-связи 5/?2-гибриди-
зации придает этим соединениям большую химическую
активность. Для них характерны реакции присоединения и
полимеризации', причем последние могут протекать как по радикальному, так
и по ионному механизмам:
650
/С—G^ -v /С—Сч радикальный,
^С=С^-»^С- СС ионный .
Присоединение брома является качественной реакцией на
двойную связь С = С:
Н ,С=СН2 I- Вг2 ->■ СН2Вг—СН2Вг.
При этом происходит обесцвечивание водного раствора брома.
Реакция алкенов со фтором сопровождается взрывом. Труднее
всего к алкенам присоединяется йод. Продукты присоединения
к алкенам минеральных кислот и воды применяются в
производстве пластмасс, химических волокон, синтетических каучукову
моющих средств, растворителей. Алкены легко окисляются
нейтральным и щелочным раствором перманганата калия. Эта
реакция, связанная с присоединением по двойной связи двух гидро-
ксильных групп, приводит к гликолям:
ЗСН2 = СН2 - 2КМп04 -!- 4Н20 -> ЗСН2 — СН2 -г 2Mn02 + 2KOH.
I I
ОН ОН
этиленгликоль
Изомерия этиленовых углеводородов зависит не только от
последовательности соединения атомов углерода, но и от положения
двойной связи в цепи. Этилен и пропилен (этен и пропен)
изомеров не имеют:
Н2С -- СН2
этилен (этен)
бутену отвечают три изомера:
СН3—СН2—СН=СН2 СН3—СН—СН-
бутен-1 бутен-2
СН3 — СН = СН2,
пропилен (пропен)
-СН3 СН,—С-СН2
1
СНз
2-метилпропенг
(изобутилен).
Характерной группой химических превращений этиленовых
углеводородов являются реакции полимеризации. Полимеризацией
называется процесс соединения нескольких молекул олефиновых
углеводородов друг с другом. Индивидуальный олефиновый
углеводород называется мономером, а вещество, образующееся в
результате соединения нескольких молекул мономера, называется
полимером.
При образовании из мономеров полимера элементарный состав
вещества не изменяется, но при этом происходит изменение
характера связей между атомами углерода.
651
Различают следующие типы полимеризации этиленовых
углеводородов.
1. Ступенчатая полимеризация. При ступенчатой
полимеризации соединяется ограниченное число молекул мономера. Так, под
влиянием серной или фосфорной кислот происходит соединение
двух молекул изобутилена, при этом может быть получен димер
изобутилена следующего строения:
СН2 СН3
СН,—С-ЬСН8=С—СН8
5 4| 3 2 1
• СН3—С—СН—С—СН3,
СН3
изобутилен
сня
сн,
СН,
2, 4, 4-триметилпентен-2
2,4,4-триметилпентен-2 при гидрировании превращается в 2,2,4-три-
метилпентан (изооктан), который является ценным компонентом
жидкого топлива:
СНо СНо
5
СН,
4
-с-
3
-сн=
2
=с-
-сня
Но ->■ СНо
2|
-С-
А3
-СН9
4
-СН-
5
-СН,.
СН3 СН3 СН3 СН3
2, 4, 4-триметилпентен-2 2, 2, 4-триметилпентан (изооктан)
2. Цепная полимеризация. При цепной полимеризации
происходит соединение многих молекул мономера. В результате
образуются высокомолекулярные соединения. Из этилена при цепной
полимеризации могут быть получены как жидкие, так и твердые
полимеры:
СН2=СН2 Ьл(СН2=СНа) + СН2-СН2->.
этилен
—*• —СН2—СН2 [СН2—СН2],г—2—СН2—СН2—.
полимер этилена
Вязкие полимеры этилена применяются в качестве смазывающих
веществ. Твердый полимер этилена называется полиэтилен, или
политен. Он обладает высокой прочностью, эластичностью,
устойчивостью и высокими электроизоляционными свойствами.
Политен — один из цепных полимерных материалов в настоящее время.
При полимеризации изобутилена получают каучукоподобный
материал, который называется полиизобутиленом (оппонол):
сн3'
СН2=С '-л
СН,)
СН.2=С
СН,
СН,
сня
-j- CH2=C—►•—СН2—С— СН2—С
сн3
I
СН,—С-
СН3 v CH3j
изобутилен
CHS
СН
3-
СН
3_!п~2
сня
полиизобутилен
652
Из этиленовых углеводородов большое практическое значение
ицеют этилен и пропилен. При обычной температуре — это
газообразные вещества. В воде они практически не растворяются,
горя? светящимся пламенем. В промышленности из этилена
получает этиловый спирт, этиленгликоль, хлористый винил, дихлор-
этай, иприт, полиэтилены различной степени полимеризации и др.
Под влиянием этилена
ускоряется вызревание лимонов.
Пропилен используют в
качестве исходного вещества
при получении ацетона,
глицерина и полипропилена
(рис. 15.6).
си
сн
сн.
гн сйд
№.
Ир Н р
|СН3 in
СИ.
СН,
4
CHL
Рис. 15 6. Макромолекула
полипропилена*
а — стереорегулярный полимер,
б — нерегулярный полимер
Иг Н У
'Сн/
ч—
-4-
W
/НС
Нр
Исходным продуктом при получении пропилена служит газ
пропилен СН2=СН—СНз, который получают из газов
нефтепереработки. Полимеризацию пропилена можно выразить схемой
СН3 снз
I I
лСНа=СН -*• [—СНа—СН—]я.
Катализаторами в этой реакции служат триэтилалюминий и три-
хлорид титана. В результате этой схемы получается полипропилен
со строгим закономерным расположением отдельных групп атомов
в его макромолекуле. Такой полимер называется стереорегуляр-
ным. Стереорегулярные полимеры в настоящее время
синтезируются также из других мономеров.
Полипропилен применяется для изготовления труб, химической
аппаратуры, деталей радио и электротехнической аппаратуры, для
изоляции кабелей, различных предметов широкого потребления.
Синтетическое волокно, полученное из пропилена, по своей
прочности превосходит капрон и нейлон, находит применение в
технике.
В молекулах ацетиленовых углеводородов имеется тройная
связь. Общая формула гомологического ряда ацетиленовых
углеводородов — СпН27г-2.
Первым членом этого ряда является ацетилен (рис. 15.7).
Н—Gs=C—Н, или СН^СН
653
Заменяя один из водородов в молекуле ацетилена на
метальную группу, получаем следующий член этого ряда — метилаиети-
лен: /
СН3 ... /
\
Н—С=С—Н, СН3—CsCH.
ацетилен метилацетилен
Рис. 15.7. Изображение о- и я-связей в ацетилене
Все ацетиленовые углеводороды называются алкинами.
Ацетилен — наиболее практически важный углеводород из алкинов.
В технике его получают взаимодействием карбида кальция с
водой:
СаС2 ~ 2Н20 -> С2Н2 + Са (ОН),,
Сч НОН СН
или ||i ; Са -J- -> HI + Са (ОН)2.
сх но!н сн
В промышленности ацетилен получают также путем пиролиза
метана и других углеводородов:
2СН4-+СН = СН~т ЗН2.
Ацетилен является химически активным соединением. При
образовании ацетилена из элементов происходит не выделение, а
поглощение тепла:
2С |-Н2-^СН = СН —54,9 кал/моль.
Следовательно, ацетилен сильно эндотермическое вещество, т.е. оно
обладает большой потенциальной энергией. Этим объясняется
взрывоопасность ацетилена, а также высокая температура
(2700° С) пламени при сгорании его в кислороде. Ацетилен —
соединение еще менее насыщенное, чем этилен. Поэтому
характерными реакциями для ацетилена и его гомологов являются реакции
присоединения. Например:
а) при гидрировании ацетилена в присутствии катализаторов
можно получить этилен и даже этан:
654
СН ^ СН 4~ Н2 —^ СН2 = СН2,
СН2 = СН2 ~г Н-2 —>* СН3 — СН3;
\б) также ступенчато к ацетилену присоединяются галогены:
СН = СН + Вг2 -►• СНВг - СНВг,
1 сим. дибромэтилен
СНВг - СНВг -f Вг2 -+ СНВг2 — СНВг2;
сим. дибромэтилен сим. тетрабромэтан
в) из ацетилена и хлористого водорода получают хлористый
винил, который является исходным мономером при получении
полихлорвинила:
i
Н=СН + Н С1 ->. СН2 = СН — С1;
t
хлористый винил
г) реакция гидратации ацетилена, т. е. присоединение воды к
ацетилену (реакция Кучерова), важная промышленная реакция,
протекающая в присутствии катализатора HgS04:
СН = СН - Н20 -►. СН3 — СОН
ацетальдегид (альдегид уксусной кислоты)
(уксусный альдегид — исходный продукт для промышленного
получения этилового спирта, уксусной кислоты, этилацетата);
д) из спиртов и ацетилена получают простые виниловые эфи-
ры, которые легко полимеризуются:
t
СН = СН + Н ОС2Н5 -*СН2 - СН — О - С2Н5
этиловый спирт винилэт иловый эфир
(группа СН2 = СН — называется винильным радикалом);
е) из кислот и ацетилена образуются сложные виниловые
эфиры:
О
СН3—Cf | НС = СН->СН2=СН8—С—О—СН=СНа.
хОН f
уксусная кислота винилацетат, виниловый
эфир уксусной кислоты
Кроме реакций присоединения для ацетилена характерны и
другие реакции.
655
а. Образование металлических производных ацетилена
тилепидов замещением его водородов на металлы:
2СН^СН 4- 2Na ^2HC=CNa + Н2,
ацетиленид натрия
2HC=CNa 4- 2Na ^-2NaC=CNa -+- Н9.
/
/
I
ацетиленид натрия
диацетиленид натрия
Легко образуются ацетилениды и с некоторыми другими
металлами. Все эти реакции — качественные реакции на ацетиленовые
углеводороды.
б. Ацетилен подвергается полимеризации:
I
♦СН9=СН—С=СН
СН=СН + СН=СН
t
винилацетилен
в. При повышенной температуре из трех молекул ацетилена
образуется циклический углеводород — бензол:
СН СН
СН^СН
сн7// >сн
СНо
СН
СН
сн' усн
СН
Углеводороды, в молекулах которых содержится бензольное
кольцо, называются ароматическими углеводородами с общей
формулой СпН2?г-б. Ароматические соединения характеризуются
устойчивостью бензольного кольца и при химических превращениях ведут
себя преимущественно как предельные соединения. Приведенная
формула бензола не отражает этих свойств ароматических
соединений и обозначенные двойные связи в настоящее время считают
чисто условными, показывающими присутствие л-связей.
Гомологами бензола являются толуол (метилбензол), ксилолы ^(диметил-
бензолы) и др. Радикал С0Н5 называют фенилом. Простейший
ароматический углеводород с двойной связью в боковой цепи —
стирол:
с—сн=сн2
HCf^CH
НС>ч /СН
Присутствие дополнительной двойной связи в молекуле стирола
объясняет его повышенную по сравнению с бензолом реакционную
способность. Стирол легко полимеризуется.
656
г. Ацетилен подвергается конденсации с карбонильными
соединениями. Например, в присутствии КОН ацетилен присоединяется
к ацетону с образованием ацетиленового спирта, что имеет
промышленное значение в производстве синтетического каучука:
О ОН
СН=СН -ь С—СН3-
/I
СН3
ацетон
• СН=С—С-СН3
I
сня
диметилзтинилкароинол
Уголь L
Газы
крекинга
н еаэт и
ацетилен
1 1'5
Этилен
1,5
* i Л
6 утан
1,8
6 у тилены
1,2
Жидкие
нефтяньге
Ф
рс
IKU,
ии
Альдоль L
уксусний
альдегид
П~~^
I этиловый
К спирт
J бугпиленьг
♦ 1 ■
Г
I
бутадиен
Т 1
0 Я С фи НЫ
диолефиньг
20-30
1 Л
Рис. 15.8. Пути получения бутадиена
При обычных условиях ацетилен — газ, при 0°С и 26 атм прев-.
ращающиися в жидкость, плохо растворяется в воде и хорошо
в ацетоне. В промышленности ацетилен используется для
получения каучука, уксусной кислоты и других важных продуктов
химической промышленности.
Диеновые углеводороды — это непредельные углеводороды,
содержащие в молекуле не только одну, но и две двойные связи.
Например, дивинил, или бутадиен (рис. 15.8):
СН2~ СН—СН—СН2.
657
Две двойные связи, разделенные одной одинарной, как в
молекуле дивинила, называются сопряженными. Чрезвычайно важной
особенностью соединений с сопряженными двойными связями
является их способность присоединять одновалентные атомы или
радикалы не к двум соседним атомам углерода, связанным
двойной связью (в положении 1 и 2), а к двум крайним углеродным
атомам системы сопряженных связей (1 и 4), причем образуется
двойная связь между двумя соседними атомами (2 и 3):
СН2-СН—СН=СН2 -f Вг2-ъВгСНа—СН-СН—СН2Вг.
Для диеновых углеводородов с сопряженными двойными
связями особенно характерна способность полимеризоваться, т. е.
соединяться друг с другом в длинную цепь. На этом основано
получение каучуков.
Рассмотрим классы производных углеводородов.
§ 6. ГАЛОИДНЫЕ ПРОИЗВОДНЫЕ
Это вещества — производные углеводородов, в которых один
или несколько атомов водорода замещены галоидом. Различают
фтористые, хлористые, бромистые и йодистые углеводороды. Из
метана могут быть получены четыре типа галогенопроизводных:
СН4 СН3Х СН2Х2 СНХ3 СХ4
моно- ди- три- тетра-
галогенопроизводные метана (X — галоген)
Н2С—СН2, Н2С—СНС1,
этен хлорэтен (хлористый^ винил)
.СбН6, С6Н6С1.
бензол хлорбензол
Первые члены галогеналканов — газы, остальные — жидкости.
Реакция хлора с метаном протекает под действием света или при
высокой температуре (300—600° С) и дает смесь
галогенопроизводных:
1. СН4 -г С12 -»СН3С1 + НС1; 2. СН3С1 + С12 -*-СНаС12 + НС1;
хлористый хлористый
метил метилен
3. СН2С12 + С12 ^СНС13 + НС1; 4. СНС13 |- С12->СС14 -у НС1.
хлороформ четырех-
хлористый
углерод
,658
Хлористый этил C2H5CI применяется в производстве тетраэтил-
свинца РЬ(С2Н5)4, являющегося антидетонатором для моторного
топлива. Хлористый метилен — негорючий и легко летучий
растворитель. Трихлорметан, или хлороформ, СНС13 применяется как
растворитель жиров и других органических веществ, а также как
наркотическое средство. Четыреххлористый углерод ССЦ —
негорючий растворитель для смол, жиров, масел. Из смешанных поли-
галогенопроизводных метана практическое значение имеет дифтор-
хлорметан CF2C12 — представитель групп фреонов, используемых
в холодильной промышленности как хладоагенты. CF2CI2 —
бесцветный газ, без запаха и вкуса, легко сгущается в жидкость с
температурой кипения — 30°С, токсичный.
Из галогенопроизводных алкенов широкое применение имеет
хлористый винил. При комнатной температуре и атмосферном
давлении хлористый винил — бесцветный газ со слабым запахом,
очень мало растворим в воде, но хорошо растворим в
углеводородах нефти, дихлорэтане, хлороформе. Промышленный синтез ви-
нилхлорида основан на взаимодействии ацетилена с хлороводоро-
дом в газовой или жидкой фазе:
НС^СН f HC1-^H2C=CH—С1.
Взаимодействие л-электронов с неподеленными парами электронов'
атома хлора приводит к упорядочению связи углерод—хлор и
поляризации двойной связи в молекуле:
н c-ch-cj-
По этой причине атом хлора при углероде с двойной связью
малоподвижен и в обычных условиях не способен к реакциям
замещения.
Из фторзамещенных алкенов особенно широкое применение в
технике нашел политетрафторэтилен (F2C = CF2)2, известный как
фторопласт-4, или тефлон, сочетающий высокую химическую и
термическую стойкость с низкой водопоглощаемостью, значительной
плотностью и высокими и диэлектрическими показателями.
Реакции галогенирования ароматических углеводородов
осуществляются на свету или в присутствии катализаторов. При
фотохимическом хлорировании бензол присоединяет три молекулы
хлора с образованием гексахлорциклогексана:
СН
НС^^СН
HCk iCH
СН
659-
Гексахлорциклогексан под названием гексахлоран применяется
как инсектицид. Хлорирование и бромирование бензола в
присутствии катализаторов (железа, хлорного железа, йода) приводит к
последовательному замещению водорода ароматического ядра на
галоген:
С6Н6 + С12 -+СбН5С1 + НС1, С6Н6 + 6С12 -> С6С16 -t- 6HC1.
хлорбензол гексахлорбензол
§ 7. СПИРТЫ, ИЛИ АЛКОГОЛИ
Спирты, или алкоголи, содержат в молекуле гидроксильную
группу — ОН, связанную с углеводородным радикалом; их можно
рассматривать как гидроксильные производные углеводородов.
Молекулы спиртов в твердом и жидком состоянии, так же как
и молекулы воды, ассоциированы, при этом существенно
увеличивается молекулярный вес и, следовательно, уменьшается летучесть
вещества. При испарении спиртов ассоциация нарушается. Явление
ассоциации объясняют возникновением водородных связей между
молекулами. Водородная связь — это особый вид
межмолекулярной связи. Она осуществляется при участии водорода гидроксиль-
ной или аминогруппы одной молекулы и атома, имеющего
электроотрицательный характер (О, N, F, C1), другой молекулы. Чем
большим частичным положительным зарядом обладает атом
водорода и чем больше способность другого атома отдавать свои непо-
деленные электронные пары, тем легче происходит образование
водородной связи и тем она прочнее.
В гидроксильной группе ковалентная связь кислород—водород
поляризована, что обусловливает межмолекулярное взаимодействие
водорода, несущего частичный положительный заряд, с
кислородом, несущим частичный отрицательный заряд:
. .. Н—О ... Н—О ... Н—О . ..
. I I I
Н Н Н
ассоциация молекул роды
н—о ... н—о ... н—о ...
I I I
R R R
ассоциация молекул спирта
'(водородная связь изображена точками). Энергия образования
водородной связи обычно не превышает 5—7 ккал/моль, в то
время как средняя энергия ковалентной связи колеблется в пределах
80—110 ккал/моль. При растворении спиртов в воде у них
возникают новые водородные связи с молекулами воды. Чем меньше
разветвлена углеродная цепь спирта и чем меньше алкильных
групп связано с атомом углерода, при котором стоит гидроксил,
660
тем легче осуществляется ассоциация. Поэтому первичные
спирты и спирты нормального строения имеют более высокую
температуру кипения. При замещении гидроксильного водорода в
спиртах другими атомами или группами водородная связь исчезает и
это сразу сказывается на температуре кипения
СН3СН,ОН СН3СН2С1 СН3СН2—0-СН2СН3
78,3 °С 13 °С 34,6 °С
Эти примеры водородной связи относятся к межмолекулярным
связям. Водородная связь может возникать и внутри одной и той
же молекулы.
Прочные водородные связи образуют также карбоновые
кислоты и такие биологически важные вещества, как белки, ферменты,
целлюлоза и др.
Простейший представитель класса спиртов — метиловый спирт,
или метанол, получают в промышленности каталитической
гидрогенизацией окиси углерода в присутствии катализатора смеси
оксидов хрома и цинка:
СО - 2Н2^СН3ОН.
Этиловый спирт, или этанол, получают в промышленности прямым
присоединением воды к этилену:
Н2С - СИ, 4- Н20 -^ СН3СН2ОН.
Гидратация этилена осуществляется в присутствии фосфорной (на
носителе из силикагеля или пористого алюмосиликата) или серной
кислот.
Этиловый спирт, или винный, с древнейших времен получают
брожением сахаристых веществ
С6Н1206 = 2С2Н5ОН Г 2С02.
Большое количество спирта в настоящее время получают
гидролизом древесины (гидролизный спирт).
В спиртах предельного ряда, или алкоголях, углеводородный
радикал соединен о одной гидроксильной группой. Поэтому
названия алкоголей выводят из названия радикала: метиловый,
этиловый, пропиловый, бутиловый и т. д.
Атомность спиртов определяется числом гидроксильных групп в
молекуле:
одноатомный спирт СН3СН2ОН этиловый спирт,
двухатомный СН2—ОН этиленгликоль,
I
сн2—он
трехатомный спирт СН2ОН—СНОН—СН2ОН глицерин.
661
В зависимости от положения функциональной группы в молекуле,
спирты разделяются на первичные, вторичные и третичные, так,
например:
СН3—СН2—СН2—СН2ОН СН8—СН8—СНаОН
первичный бутиловый спирт i
сн3
первичный изобутиловый спирт
В первичных спиртах группа —ОН связана с атомом углерода,
который соединен только с одним атомом углерода:
СН3
1
СН3—СН—СН2—СН.> СН3—С—СН3
I ■ I
он он
вторичный бутиловый спирт третичный изобутиловый спирт
Во вторичных спиртах группа —ОН связана с атомом углерода,
который соединен с двумя атомами углерода. В третичных спиртах
группа —ОН стоит при атоме углерода, связанном с тремя
атомами углерода.
В группе —ОН электронная пара смещена в сторону более
электроотрицательного атома кислорода, поэтому водород этой
группы подвижен и замещается атомами щелочных металлов. При
взаимодействии спиртов с металлическим натрием образуются ал-
коголяты натрия, например:
2СН3ОН -г 2Na -> H2 f + CH3ONa
метиловый спирт алкоголят Na
(метилат натрия)
При действии сильных окислителей спирты окисляются. При
окислении первичных спиртов образуются альдегиды, например:
2СН8—СН2—СН2—СН2—ОН -Ь 02->
первичный бутиловый спирт
-*2СН3—СН9-СН2—cf -- 2Н90.
ХН
масляный альдегид
Из спиртов и кислот (органических и неорганических) образуются
сложные эфиры:
ноч о
СН3—СН,—ОН -ь ^>S^ ■£.
этиловый спирт НО' и
серная кислота
662
сн3-сн2-оч Х/о
S? + Н20,
но7 ^о
этиловый эфир серной кислоты
о
«о
сн3сн2он - но— с-сн3^гсн3—с/
\q-qh,.
этиловый спирт
уксусная
кислота
2*V
этиловый эфир
уксусной кислоты
Эти реакции называются реакциями этерификации. Они обратимые,
так как сложные эфиры под действием выделяющейся при их
образовании воды подвергаются гидролизу.
Фенолы — гидроксильные производные ароматических
углеводородов. Их можно рассматривать как продукт замещения одного
или нескольких атомов водорода в бензольном ядре гидроксиль-
ной группой. Простейший фенол (карболовая кислота) является
производным бензола:
С—ОН
не/ \сн
но
сн
сн
или С6Н5ОН.
Реакция гидролиза хлорбензола лежит в основе непрерывного
способа получения фенола:
С1
i
с
нс,^\сн
НС
сн
сн
-!- Н,0
Up,
л1,о,
НС
не!
он
I
с
V
сн
СН + HC1.
сн
Гидролиз осуществляется водяным паром над катализатором АЬОз
или 6—8%-ным раствором NaOH при 300°С под давлением 150—
200 атм.
Фенолы проявляют слабые кислотные свойства. В водных
растворах они диссоциируют с отщеплением иона водорода от
группы ОН:
СвНвОН:
:С6Н80-
н+.
663
Поэтому замещение водорода гидроксильной группы происходит
не только при взаимодействии фенола с металлами, но и при
взаимодействии со щелочами:
2CeH5OH -J- 2Na-»- 2C6H5ONa + H2 f ,
CeH5OH — КОН-* СвН5ОК + Н20,
образующиеся при этом вещества называются фенолятами.
Ближайший гомолог фенола — метилфенол, или крезол,
существует в трех изомерных формах:
сн сн
сн/^сн—он
НЧС—СН^СН—ОН
сн' ;сн—сн3
сн
ортокрезол,
СН' ]'СН
\/
сн
метакрезол,
сн
сн
СН—ОН
НЯС-
-CHL JCH
сн
паракрезол.
Орто-, мета- и паракрезолы являются составной частью
каменноугольного, торфяного и сланцевого дегтя. Они применяются в
производстве пластмасс и взрывчатых веществ, для пропитки дерева
с целью предохранения его от гниения и т. п.
При замещении водородного атома гидроксилом в боковой
цепи ароматического углеводорода получаются ароматические
спирты:
С6Н5—СН3 С6Н5—СН2—ОН С6Н5—СН2—
толуол, бензиновый спирт, радикал бензил.
Фенол и особенно крезолы обладают сильными бактерицидными
свойствами и используются для дезинфекции.
Простые эфиры содержат кислород, связанный с двумя
углеводородными радикалами. Простые зфиры образуются путем
замены водорода гидроксильной группы спирта на алкильный радикал,
например:
R'
R-O—H
спирт,
R—О—R'
простой эфир,
664
СН3—О—СН3 СН3СН2—О—СН2—СН3
демитиловый эфир, диэтиловый эфир.
Их можно получить отщеплением воды от двух молекул спирта:
СН3СН210Н+Н|ОСН2СН3-^^-^СН3СН2— О—СН2—СН3.
этиловый спирт диэтиловый эфир
Диэтиловый, или серный, эфир — прекрасный растворитель для
многих органических веществ. В химическом отношении простые
эфиры довольно устойчивые соединения.
§ 8. АЛЬДЕГИДЫ И КЕТОНЫ
Альдегиды и кетоны содержат в молекуле двухатомную группу,
называемую карбонильной группой, или радикалом карбонилом.
В альдегидах карбонил соединен с атомом водорода и
углеводородным радикалом, так что получается одноатомная группа
//°
—С—Г , называемая альдегидной группой. В кетонах карбонил
NH
соединен с двумя углеводородными радикалами.
Альдегиды получают названия от тех кислот, в которые они
превращаются при окислении. Например:
^°
Н—С' альдегид муравьиной кислоты — формальдегид,
//°
СН3—С\ альдегид уксусной кислоты — ацетальдегид,
NH
СН3—СН2—С\ альдегид пропионовой кислоты.
NH
Кетоны называют по радикалам, с которыми связана
карбонильная группа. Например:
О
II
СН3—С—СН3 диметилкетон,
О
II
СН3—С—СН2—СН3 метилэтилкетон,
О
II
СН3—С—СН2—СН2—СН3 метилпропилкетон.
12 3 4 5
665
Получают альдегиды и кетоны, например, так:
а) альдегиды образуются при окислении первичных спиртов:
Н
СН3—С—ОН -±°__*
н
этиловый спирг
он
СНз-С—ОН
н
^-* сн3-с/
о
SH
альдегид
уксусной кислоты
б) кетоны образуются при окислении вторичных спиртов:
ОН
I +0
СН3—С—СН3
I
н
вторичный пропило-
вый спирт
он
I
СН3—С—СН3
I
он .
-н.о
о
I!
->* СНо—С—СНо
ацетон
Из реакций, характеризующих свойства альдегидов и кетонов,
важнейшими являются следующие:
1. Восстановление
СНЗЧ
уксусный альдегид
)С=0 -f Н2 ->-СН3СН2ОН.
этиловый спирт
2. Окисление
2СН3—CH2--Cf ^02-^2СН3—СН2—С(
ХН ХОН
пропионовыи альдегид
пропионовая кислота
3. Конденсация. Реакция образования больших молекул из
молекул с меньшим числом углеродных атомов, сопровождающаяся
выделением молекул воды, аммиака, галогеноводородов и т. п.,
называется реакцией конденсации. Например:
CHS—Cf + СН3—cf
молекула
уксусного
альдегида
молекула
уксусного
альдегида
сн3—сн-сн—с{ | н.о.
хн
молекула
кротонового
альдегида
666
\/ лдом^ттл. атомами С и
Р
Следует подчеркнуть, что в группе —Cf между
ХН
О одна связь является а-связью, а другая — я-связью. Поэтому
альдегиды — химически активные вещества и при определенных
условиях способны присоединять водород (за счет разрыва я-свя-
зи) с образованием первичных спиртов, например:
/Р
сн3—с< +н2-*сн3—сн.3он.
уксусный альдегид эаиловый спирг
Простейшим альдегидом ароматического ряда является
бензойный альдегид, в котором карбонильная группа непосредственно
связана с бензольным ядром:
СН^ V-C—Н
ХСН-СН/ ||
О
Бензойный альдегид применяется в кондитерском производстве,
парфюмерной промышленности, в производстве красителей и
лекарственных средств.
Альдегиды легко окисляются, превращаясь в органические или
карбоновые кислоты:
2R—Cf +02^2R-Cf .
NH NOH
Таким образом, спирты, альдегиды и органические кислоты
генетически связаны между собой:
окисление
спирт восстановление альдегид-жис лота.
восстановление
Важнейшим представителем альдегидов является муравьиный
альдегид, или формальдегид. В промышленности его получают
каталитическим дегидрированием' метанола при 650—700°С:
сн3он - сн2о + н2
и прямым окислением метана кислородом воздуха при 400 — 600 °С:
СН4 г-02-^СН20 f Н20.
40%-ный водный раствор формальдегида, называемый
формалином, применяют для синтеза различных органических соединений
и как дезинфицирующее средство. Формалин также применяется в
667
производстве пластических масс, искусственной шерсти, лаков,
клеев и т. д.
Ацетальдегид, или уксусный альдегид — важный представитель
альдегидов. Новейший промышленный синтез уксусного альдегида
состоит в окислении этилена, добываемого при переработке
нефтяных газов, в присутствии солей палладия и меди:
/°
Н,С=СН, f Q2 PdCL+cuci,^ CH3-Cf .
хОН
Ацетальдегид, или уксусный альдегид, — важнейший продукт
тяжелого органического синтеза. Он легко окисляется в уксусную
кислоту:
2СН3-СС 02-*2СН8—Of
NH хОН
Простейший кетон — ацетон СН3СОСН3 смешивается с водой в
любых отношениях. Применяется в различных отраслях
промышленности как растворитель. Его широко используют в
производстве бездымного пороха, лаков, пленок и смазочных масел.
§ 9. ОРГАНИЧЕСКИЕ, ИЛИ КАРБОНОВЫЕ, КИСЛОТЫ
Органические кислоты содержат в молекуле карбоксильную
//°
группу —Cq , представляющую собой сочетание карбониль-
хОН
ной и гидроксильной групп. Поэтому карбоновые кислоты
рассматривают как производные углеводородов, в которых один или
несколько атомов водорода замещены на карбоксильную группу:
-сУ°
R-(/
ЮН
Кислоты бывают одноосновные (монокарбоновые),
двухосновные (дикарбоновые) и многоосновные (поликарбоновые). По
характеру радикала, связанного с карбоксильной группой, кислоты
могут быть предельными, непредельными, ароматическими и др.
Для карбоновых кислот применяют эмпирические названия:
/° /° ^°
Н—Cv СН3—С. СН3—СН2—С^
хОН хОН хОН
муравьиная кислота уксусная кислота прэпионовая кислота
(метановая кислота) (этановая кислота) (пропановая кислота)
CH,—CHS—CH8—C-/ СН3—СН-С^
хн I хон
сн3
масляная кислота изомасляная кислота
(бутановая кислога)
Низшие карбоновые кислоты (Ci—С3) — подвижные жидкости,
они смешиваются с водой во всех отношениях. Кислоты (С4—С9) —
маслянистые жидкости, малорастворимы в воде. Высшие
кислоты — твердые вещества. Карбоновые кислоты устойчивы к
нагреванию, разлагаются лишь при высокой температуре, сравнительно
устойчивы к действию окислителей и восстановителей. Карбоновые
кислоты — слабые кислоты, так как протолитическое равновесие
в растворах этих кислот сильно смещено влево:
R—C< ^_ R—СОО- + Н+
хон
В карбоксильной группе имеет место сопряжение я-электронов
одного из атомов кислорода с я-электронами карбонильной группы:
Это сопряжение уменьшает электронную плотность атома
кислорода гидроксильной группы и тем самым облегчает отщепление
протона. Поэтому карбоновые кислоты проявляют значительно более
сильные кислотные свойства, чем фенолы.
Ненасыщенные органические кислоты — карбоксилпроизводные
ненасыщенных углеводородов широко применяются как мономеры.
Простейшие ненасыщенные одноосновные кислоты с двойной
связью в молекуле — акриловая
Н2С=СН—Gf и метакриловая Н2С=С—С?
хОН | ^ОН
СН3
кислоты. Это — бесцветные жидкности с резким запахом,
смешиваются с водой во всех отношениях. Акриловая и особенно
метакриловая кислоты очень легко полимеризуются и применяются в
производстве пластических масс.
669
Метиловый эфир метакриловой кислоты
СН„—С—Cf
I \о-сн3
сн3
мономер в производстве органического стекла (плексиглас).
В ароматических карбоновых кислотах карбоксильная группа
непосредственно связана с бензольным ядром. Простейшая
одноосновная ароматическая кислота — бензойная кислота:
с с/°
снГ^сн Хон
сн
XN/CH
сн
Радикал бензойной кислоты — бензоил CeH=Cf . Бензойная кис-
/-0
лота — белые кристаллы, трудно растворимые в воде. Константа
диссоциации бензойной кислоты выше, чем у алифатических
кислот. Бензойная кислота широко используется как антисептик в
медицине и в консервной промышленности, в синтезе красителей,
фармацевтических препаратов, в производстве сахарина и
парфюмерии. Перекись бензоила — продукт замещения атомов водорода
в перекиси водорода остатками бензойной кислоты:
О
II
Н-0 С6Н5С-0
Н—О С6Н5С—О
II
о
Перекись бензоила — бесцветные кристаллы с температурой
плавления 106—108°С, разлагается со вспышкой, в результате чего
образуются свободные радикалы:
(С6Н5СОО)3-> С6Н5СОО Ь ОД + С02.
По этой причине перекись бензоила применяется как катализатор
реакции полимеризации ненасыщенных соединений в
промышленном производстве многих полимерных материалов.
Двухосновные, или дикарбоновые, кислоты — производные
углеводородов, содержащие в молекуле две карбоксильные группы.
670
Первый представитель ряда дикарбоновых кислот — щавелевая
кислота НООС—СООН — был впервые выделен из сока щавеля.
Малоновая кислота НООС—СН2—СООН содержит три
углеродных атома, а янтарная НООС—СН2—СН2—СООН четыре.
Большое промышленное значение в производстве синтетического
волокна найлона имеет адипиновая кислота НООС—(СН2)4СООН.
Окисление высших парафиновых углеводородов нефти (С20—
С2б) в жидкой фазе кислородом воздуха в присутствии
катализаторов, содержащих металлы VIIB и VIIIB групп периодической
системы, приводит к расщеплению углерод-углеродных связей и
к образованию смеси карбоновых кислот с различными длинами
цепи. В настоящее время этим методом в промышленности
получают заменители пищевого сырья — кислоты жиров и растительных
масел, которые используются в производстве мыл, синтетических
моющих средств (детергентов), олиф, лаков, эмульгаторов,
смазочных и связующих материалов.
Ароматические дикарбоновые кислоты существуют в виде трех
изомеров:
СООН
сн
сн
СЕ
/\
СН
[
|С—СООН
!с—соон
[
ортофталевая
к
ислота
сн
сн
СН
/\
Ч/
с
С—СООН
сн
СООН
метафталевая
(
<ислс
та
сн
сн
с
/Ч
с
1
сн
сн
СООН
парафталевая
кислота
Фталевая кислота получается окислением нафталина,
извлекаемого из каменноугольной смолы:
СН СН СН
сн^^с/Чсн сн^\с—соон
°5—>. I +С02-1-Н20.
>сн сн^ к
сн
\/V
С—СООН
сн сн
сн
Фталевая кислота применяется в промышленности органического
синтеза (синтез красителей, лекарственных средств). Эфиры фта-
левой кислоты и ее ангидрид используются в производстве глифта-
левых смол, которые применяются как основы электроизолирующих
и пленкообразующих материалов.
Терефталевая кислота получается окислением параксилола—
ароматического углеводорода, добываемого при переработке нефти
и в коксохимической промышленности:
671
сн,
соон
с
сн/^сн
сн
сн/^сн
с
CHI"
сн
с
,сн
н,о.
сн3
параксилол
соон
терефталевая кислота
Терефталевая кислота — мономер в производстве полиэфирных
волокон (лавсан, или терилен). Изофталевая кислота применяется
в производстве модифицированных полиэфирных смол.
§ 10. АМИДЫ КИСЛОТ
В амидах карбоновых кислот гидроксил карбоксильной группы
замещен на аминогруппу:
сн3с^
о
снч—с
//
о
он
4NH,
уксусная
кислота
амид уксусной кислоты
(ацетамид)
Амид угольной кислоты — карбаминовая кислота:
НО—С—ОН
II
о
угольная кислота
H2N-C—ОН
О
карбаминовая кислота
В радиоэлектронной промышленности применяются полимеры на
основе сложных эфиров карбаминовой кислоты — уретанов:
H2N-
-С-0-С2Н5
О
Полиуретаны сочетают повышенную влагостойкость,
вибростойкость, антикоррозийную стойкость с высокими и стабильными
диэлектрическими показателями.
Полный амид угольной кислоты — карбамид, или мочевина:
NHa—С—NHa
О
672
Мочевина — высококонцентрированное азотистое удобрение. Она
используется на всех почвах и под все культуры. В условиях
орошаемого земледелия особенно важны мочевинно-альдегидные
удобрения. Мочевина имеет существенное значение в химизации
животноводства, в повышении питательности кормов.
Карбамид-исходное вещество при получении карбамидных смол, мочевинно-
формальдегидных смол, синтетического волокна «урилон»,
лекарственных средств (люминал, веронал, бромурал).
§ 11. АМИНОКИСЛОТЫ
В молекулах некоторых органических веществ имеются две или
несколько различных функциональных групп. Такие вещества
называются соединениями со смешанными функциями, например
аминокислоты, простейшим представителем которых является ами-
ноуксусная кислота NH2—СН2—СООН. Так как аминокислоты
содержат в молекулах и основную (NH2) и кислотную (СООН)
группы, то они амфотерны и образуют соли как с основаниями, так
и с кислотами. Взаимное влияние амино- и карбоксильной групп в
молекулах аминокислот проявляется в их амфотерных свойствах:
H2N—CH2-Cf ^ H3N+—CH2—(X
NOH X)-
В кислой среде они реагируют как основания:
H3N+—СН2—СОО- + Н+ ^ H3N+—СН2—СООН,
а в щелочной — как кислоты:
H3N+—СН2—СОО- + ОН- ^ H2N—СН2—СОО- + Н20.
Аминокислоты можно рассматривать как продукт замещения
одного или нескольких атомов водорода в углеводородном
радикале карбоновой кислоты аминогруппой. Например:
СН3СООН NH2CH2COOH
уксусная кислота аминоуксусная кислота
(гликоколь, или глицин),
СН3—СН2—СООН CH3CHNH2—СООН CHaNHa—СН2—СООН,
пропионовая а-аминопропионовая р-аминопропионовая
кислота кислота кислота
СООН СООН
I !
СН2 СН2
I I
СН2 CHNH2
I I
СООН СООН
янтарная кислота амнноянтарная кислота (аспарагиновая)
22 Общая химия
673
Аминокислоты как бифункциональные мономеры, содержащие
две реакционноспособные группы, являются исходными
веществами в производстве синтетических волокон. Полиамидные волокна—
капрон и энант — синтезируются путем поликонденсации е-амино-
капроновой кислоты с со-аминоэнантовой кислотой:
^°
H2N—СН,—СН,-СН,—СН,—СН,—С? ,
NOH
е-аминокапроновая кислота
H2N—СН2—СН2-СН2—СН,—СН,—СН,—С^
Ч)Н
со-аминоэнантовая кислота
В производстве капрона (или перлона) используется не сама е-
аминокапроновая кислота, а ее внутренний циклический имид —
капролактам:
СН,—СН. Of
сн,—сна—сн /
^NH
12 ^llj
Поликонденсация высших аминокислот приводит к
образованию линейных полимеров с пептидными связями в цепи, подобно
натуральным белковым волокнам.
Следует указать на специфические свойства аминокислот — их
превращения при нагревании. Приведем примеры:
а) При нагревании а-аминокислот от двух молекул
аминокислоты отщепляются две молекулы воды и образуются циклические
соединения, которые называются дикетопиперазинами:
рсн3
I
аСН
O^C^^NH
СНЧ
СН
о=с./\ш
ОН ОН z!lbo^
NH
с=о
H.N C=0 \£
«сн с'н3
рсн3
а-аминопрошюновая кислота диметилдикетопиперазин
674
б) а-Аминокислоты при нагревании превращаются м iH'iiptvir.'ii.iiMi'
кислоты:
NH2
СН3—СН—СООН -=™i-> СНа=СН—СООН.
0 а
сх-аминопропионовая акриловая кислота
кислота
в) От у- и 6-аминокислот вода отщепляется внутримолекуляр-
но, которая образуется из гидроксильной группы карбоксила и
водорода аминогруппы; получаемые при этом вещества называются
лактамами:
СН9—СН.>—СН2—С< .z±£_>. СН2-СН0-СН2-С-0
ji * Х°Н I NH I
н н
Y-аминомасляная лактам у-аминомасляной
кислота кислоты
§ 12. БЕЛКОВЫЕ ВЕЩЕСТВА
Белки образуются путем поликонденсации а-аминокислот с
образованием пептидных связей:
NH2—СН—СООН + HNH—СН—СООН j-
I I
Ri R2
+ HNH—СН—СООН -f ... -=ML+
R3
Н Г NH—CH—C—NH—CH-C—NH—CH—C
I II I il I II
Ri О R2 O R3 O
ОН|вНД
Пептидное звено —CH—С—NH— является структурным эле-
I II
R О
ментом белков — линейных биополимеров, связанных пептидной
22* 675
связью —С—NH—. В построении белков участвуют 20 необхо-
II
О
димых организму аминокислот, отличающихся друг от друга
составом и строением бокового радикала R. Последовательность
расположения остатков аминокислот в полипептидной цепи называют
первичной структурой этого биополимера. Из 20 аминокислот 8 не*
заменимы и обязательно должны входить в состав пищи, являясь
самой ценной и дефицитной ее частью.
Белки представляют собой наиболее сложные по составу и
строению высокомолекулярные соединения; при их гидролизе
образуются аминокислоты. В состав белков входят углерод, водород,
кислород и азот. Многие белки содержат серу, известны белки,
содержащие фосфор и другие элементы.
Белки играют исключительную роль в природе. Они
синтезируются растениями из минеральных веществ. Животные организмы
такой функцией не обладают, поэтому нуждаются в готовой
белковой пище. Белки вследствие этого в основном используются в
качестве питательных веществ. Всевозрастающее значение белки
приобретают в текстильной, кожевенной промышленности, бытовой
химии и т. п.
Белковые вещества имеют огромный молекулярный вес. При
растворении белков в воде образуются коллоидные растворы. Если
к растворам белков добавлять спирт, ацетон или растворы
минеральных солей, то белки выделяются из раствора в осадок. Белки
очень чувствительны к нагреванию, а также к действию некоторых
сильных реагентов (азотная кислота, трихлоруксусная кислота
и др.). Под влиянием этих реагентов, а также нагревания белок
подвергается сложным необратимым процессам, которые приводят
к денатурации белка.
Расщепление молекул белковых веществ осуществляется
методом гидролиза. При полном гидролизе из белков получают а-ами-
нокислоты, которые являются структурными элементами молекул
белков, построенных по типу амидов. При неполном гидролизе
белков образуются полипептиды, молекулы которых построены из
нескольких аминокислот. В составе сложных белков (протеидов)
белок связан с небелковым компонентом: нуклеиновой кислотой,
гемоглобином, фосфорной кислотой, углеводами.
Еще в 50-х годах был осуществлен синтез двух белковых
гормонов — окситоцина и вазопрессина. Это сравнительно простые
полипептиды — в состав каждого из них входят остатки 9
аминокислот. Установлено строение белкового гормона инсулина,
регулирующего сахарный обмен в организме, а также строение рибонук-
леазы —■ фермента, катализирующего гидролизующее расщепление
рибонуклеиновых кислот на простые нуклеотидные остатки.
Молекула рибонуклеазы имеет цепь из 124 аминокислотных остатков,
которая удерживается четырьмя дисульфидными мостиками.
Упомянутыми примерами далеко не исчерпываются достигнутые в на-
676
стоящее время успехи в расшифровке строения белков. Особое
внимание уделяется изучению белков, обладающих каталитическим
действием, а именно ферментов и гормонов. Изучение строения
белков, выполняющих важные физиологические функции,
позволило в ряде случаев вскрыть химическую первопричину некоторых
болезней.
Ученые давно уже задумываются над проблемами синтеза
продуктов питания, и в первую очередь, наиболее ценной части
пищи — белков. В настоящее время микробиологический синтез
белков из углеводородов нефти ведется в промышленном масштабе.
При этом из 1 г углеводородов получается 0,7 г полноценных
белковых веществ. В образовавшейся массе содержатся также
витамины группы В. Сырьем служат тяжелые фракции нефти с
добавками обычных калийных, азотных и фосфорных удобрений, а
также микроэлементов. Эту смесь микроорганизмы перерабатывают
в белковую массу, которую используют в животноводстве,
добавляя к ней наиболее «дефицитные» аминокислоты, получаемые
синтетическим путем.
На стыке биологии и химии возникли новые науки — биохимия
и в самое последнее время биоорганическая химия. Белковые
вещества, нуклеиновые кислоты, ферменты, витамины, антибиотики,
терпены и стероиды — это далеко не полный перечень веществ,
которые стоят в центре внимания исследований в области бурно
развивающейся биоорганической химии.
§ 13. АМИНЫ. ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ СЕРУ
Органические производные аммиака называются аминами. Они
образуются замещением атомов водорода в молекуле аммиака
углеводородными радикалами. Например:
CH3I + 2NH3-*CH3NH2 + NH4I.
Если в молекуле аммиака один атом водорода замещен
углеводородным радикалом, то амин называется первичным, если два —
вторичным, если все три — третичным. Например:
Н СН3 СН3
I I I
N—CH3 N—СН3 N—СН3
i I I
н н сн3
метиламин диметиламин триметиламин
(первичный амин) (вторичный амин) (третичный амин)
Амины, подобно аммиаку, реагируют с водой, образуя основания:
CH3NH2 + Н20 ->• [СН3 NH3]+ ОН.
677
При взаимодействии аминов с кислотами образуются соли, сходные
с солями аммония:
CH8NHa + HCly= [CH3NH3]C1 (или CH3NH2HC1).
метиламин солянокислый
метиламин
Эти реакции показывают, что аминогруппа является
функциональной группой, придающей органическому соединению свойства
реагировать с водой и в водном растворе диссоциировать с
отщеплением ионов гидроксила.
Большое практическое значение имеет простейший
ароматический амин — фениламин C6H5NH2, или анилин. Он получается
восстановлением нитробензола:
* C6H5N02 + ЗН2 -> C6H5NH2 л. 2Н20.
Анилин растворяется в спирте, эфире, бензоле. Он является
исходным продуктом в производстве разнообразных красителей,
пластмасс, фотопрепаратов и т. д.
Органические соединения, в молекулах которых содержится две
и более групп NH2, называются соответственно диаминами, триа-
минами и т. д.
Важное значение имеет гексаметилендиамин NH2—(СН2)6—
NH2. Из него и двухосновной адипиновой кислоты НООС —
(СН2)4 — СООН образуется полимер, из которого получают
волокно нейлон. Поликонденсацию можмо выразить схемой
хоон
/*NHa — (CHa)e —NH9 f-/i(CH2)/ -+
ХЮОН
^ [_ NH — (СН2)6 - NH - СО — (СН2)4 — СО]й - 2п Н20.
Фосфины, арсины, стибины и висмутины — соединения,
построенные аналогично аминам.
Из производных, содержащих серу, можно отметить:
а) тиоспирты, или меркаптаны, содержащие группу —S—Н;
Ч
б) тиоэфиры общей формулы )S (R — углеводородный радикал);
r/
в) сульфокислоты, содержащие одноатомный радикал сульфо-
ксил —S02~OH.
§ 14. ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Это соединения, которые содержат атомы элементов,
непосредственно связанные с атомами углерода. Различают «полные» эле-
ментоорганические соединения, в которых атом элемента связан
СН3.
только с углеводородным радикалом, например /Hg,
СН
■з
678
и «смешанные» элементоорганическис соединения, где атом
элемента связан еще также с неуглеродным атомом, например
СН
34)Hg, CHa=CH-CHa-HgON02,
V
и т. д.
Органические соединения, в которых имеется непосредственная
связь элемент—углерод, принято объединять под общим
названием элементоорганические соединения, которые составляют
важнейший раздел современной химической науки.
Классифицируют эти соединения прежде всего по
содержащемуся в них элементу. Органические производные элементов,
принадлежащих к одной группе периодической
системы, имеют сходные свойства, поэтому их можно
объединить и рассматривать совместно. Так
возникла естественная классификация элементоорга-
нических соединений по группам периодической
системы.
Среди элементоорганических соединений мож- Fe
но выделить более узкую область металлооргани-
ческих соединений. Наличие частичного
отрицательного заряда на углероде и определяет своеоб-
Рис. 15.9. Ферроцен
разные свойства металлоорганических соединений—их способность
вступать в электрофильные, а не нуклеофильные реакции.
В настоящее время получены и всесторонне изучены
органические соединения лития, натрия, калия, рубидия, цезия, бериллия,
магния, кальция, стронция, бария, цинка, кадмия, ртути, бора,
алюминия, галлия, индия, таллия, кремния, германия, олова,
свинца, фосфора, мышьяка, сурьмы, висмута, хрома, марганца,
железа, никеля, которые нашли разнообразное применение в народном
хозяйстве. Это кремнийорганические каучуки, смолы и
диэлектрики, свинецорганические антидетонаторы, фосфорорганические
инсектициды и лекарственные средства, ртутноорганические
бактерицидные вещества, мышьяк и сурьмяноорганические химиотерапев-
тические средства, фторорганические хладоагенты и пластические
массы, кобальторганические катализаторы в нефтехимическом
синтезе и т. п.
Начиная с 50-х годов наибольшее внимание исследователей
сосредоточено на изучении органических соединений переходных
металлов. Многие из них образуют органические соединения
необычного строения. Одним из примеров такого рода соединений может
служить дициклопентадиенилжелезо, или ферроцен Ci0Hi0Fe. Это
соединение представляет собой кристаллы цвета ржавчины, с
температурой плавления 174°С. Ферроцен кипит без разложения при
679
250°С, растворяется в органических растворителях. Необычные для
металлорганического соединения свойства ферроцена — следствие
его своеобразного строения (рис. 15.9). Структуры подобного
типа получили название «сендвичеобразных». В них атом железа
прикрыт сверху и снизу циклопентадиенильными циклами. Такое
строение ферроцена подтверждено рентгеноструктурным анализом,
на него указывают и спектральные данные. В изучении ферроцена
и его производных большой вклад внесен А. Н. Несмеяновым и
его учениками.
Кремнийорганические соединения. Химия кремнийорганических
соединений приобрела в наше время исключительно важное
теоретическое и практическое значение. Эти соединения нашли
применение в строительстве, ракетной технике,
электромашиностроении, радиоэлектронной технике, металлургии, оптикомеханической
промышленности.
Органические соединения кремния впервые были получены при
взаимодействии четыреххлористого кремния с этиловым спиртом
SiCl4 + 4С2Н5ОН ->Si (ОС2Н5)4 + 4НС1.
Кремнийорганические соединения, в цепь которых входят
атомы кремния и кислорода, называются органосилоксанами:
— Si —О —Si —
I I
Соединения, в цепи которых содержатся атомы кремния и
углерода, образуют группу карбосиланов:
I I I
— Si —С— Si —
I I I
Сравнительное изучение свойств и реакций органических и
кремнийорганических соединений показало также и довольно сильное
различие между ними. Причина этого кроется в различной
прочности химических связей кремний—кислород и кремний—углерод:
i i ii
— Si —О —Si—, — С —Si—
II II
I I
Силаксановые связи —Si — О —Si— обладают очень высокой
I I
прочностью и придают веществу твердость и особую стойкость. Эти
связи характерны для кварца, песка и ряда минералов и
обусловливает их исключительную термостойкость.
680
Карбосилановые связи —С — Si— обусловливают эластич-
i i
ность и растворимость вещества. Кремнийорганические
соединения, содержащие в молекуле силаксановые и карбосилановые
связи, обладают свойствами, присущими как силикатам, так и
органическим соединениям. Это ценное сочетание столь различных
физических и химических свойств позволяет использовать кремний-
органические соединения и их полимеры в самых различных
отраслях народного хозяйства.
Атомы кремния, подобно атомам углерода, обладают
способностью связываться друг с другом в цепи или кольца. Примером
могут служить кремневодороды, или силаны:
н н н н н н н
III I I I I
Н — Si — Si — Si — Н Н — Si — Si — Si — Si — Н
III i I I I
H H H H H H H
Si3H8 Si4H10
Однако кремневодороды менее прочны, чем углеводороды; они
самовоспламеняются на воздухе, сгорая до двуокиси кремния и
воды.
Продукты замещения атомов водорода в молекулах силановна
органические радикалы представляют собой кремнийорганические
соединения, проявляющие значительную устойчивость. Большой
прочностью обладают цепи из атомов кремния, связанных друг с
другом через атомы кислорода. Устойчивую силоксановую
группировку — кремний кислородный скелет — можно изобразить
следующим образом:
I ! I I I I
— Si — О — Si — О — Si — О — Si— О — Si — О —
Sill ! I I 1
К. А. Андриянов разработал метод получения высокомолекулярных
кремнииорганических соединений, обладающих весьма ценными в
практическом отношении свойствами. Если в ортокремниевой
кислоте атомы водорода заместить углеводородными радикалами, то
образуются эфиры ортокремниевой кислоты
С2Нб —О ОН С2Н5 —О О — С2Н5
\ / \ /
Si Si
/ \ / \
С2Н6-0 ОН С2Нб-0 0-С2Н5
кислый этиловый эфир полный этиловый эфир
ортокремниевой кислоты ортокремниевой кислоты
681
При поликонденсации кислых эфиров ортокремниевой кислоты
образуются силоксановые полимеры
OR OR OR OR
i I i I
„O — Si — O — Si — O— Si —O — Si —
I i I I
OR OR OR OR
Среди кремнийорганических полимеров известны жидкие продукты
различной консистенции, смолы и синтетические каучуки. Эти
жидкости применяют в качестве смазочных масел и консистентных
смазок. Кремнийорганические смолы сочетают характерную для
силикатов теплостойкость с легкой перерабатываемостью,
эластичностью, водостойкостью и высокими электроизоляционными
свойствами, присущими органическим полимерам. Их применяют в
производстве пластмасс, которые широко используются в
электроизоляционной технике. Кремнийорганический каучук, обладающий
высокой морозо- и теплостойкостью, который сохраняет эластичность
при температурах от —60 до +250°С, применяется для
изготовления теплостойкой изоляции проводов, кабелей, высоковакуумных
прокладок. Эти полимеры в сочетании со стеклянной тканью,
слюдой и асбестом используются как изоляция.
Введение в главную полимерную цепь наряду с атомами
кремния и кислорода других химических элементов (В, Р, Al, Ti, Sn,
Rb) сильно расширяет возможности синтеза новых полимерных
материалов.
Фосфорорганические соединения. Фосфор как элемент пятой
группы периодической системы аналог азота. Между
органическими производными этих элементов наблюдается определенное
сходство, например:
R —NH2 R — РН2
первичные амины первичные фосфины
Подобно соответствующим соединениям азота известны вторичные
и третичные фосфины, а также аналоги четвертичных аммониевых
соединений — соли фосфония. Так же как и амины, фосфины
являются основаниями, способными давать с кислотами соли:
СН3РНо + НС1 -^[СН3РН3]++С1-.
метилфосфин хлористый метилфосфоний
Однако основные свойства фосфинов значительно слабее, чем у
аминов. Отличия соединений фосфора от соединений азота
связаны главным образом со способностью фосфинов легко реагировать
с элементами, обладающими большим сродством к электронам
(галоиды, кислород). Объясняется это тем, что свободная элек-
682
тронная пара фосфинов и других соединений трехвалентного
фосфора находится дальше от ядра, чем в азоте. Чем дальше
расположен электрон от ядра, тем меньше он притягивается ядром, тем
легче он может перейти к другому атому. В частности, фосфины
легко окисляются с образованием соответствующих окисей (на
воздухе простейшие фосфины самовоспламеняются). При
окислении первичных и вторичных фосфинов образуются соответственно
алкилфосфоновые и диалкилфосфицовые кислоты, из третичных
фосфинов — окиси:
RPH^R — P(OH)„
алкилфосфоновая кислота
(г>)
R9PH->-R2 — P(OH),
II
О
диалкилфосфиновая кислота
(О)
R3p-R3-p = o.
окись третичного фосфина
Эти же соединения образуются из хлорфосфинов. В результате
присоединения к последним хлора получаются хлорфосфораны, гидро-
лизующиеся при действии воды:
сл2 н2о
RPCU -* RPCI3 ->R - Р (ОН),,
II
О
r2PC1 i R2PC18 *%2 - P (OH).
Фосфор в сочетании с углеродом образует многочисленные и
р-азнообразные соединения. Первую группу фосфорорганических
соединений составляют производные фосфина:
РН3 PH2R PHR2 PR8
фосфин первичный вторичный третичный
фосфин фосфин фосфин
683
Фосфины алифатического ряда токсичны, самопроизвольно
окисляются и воспламеняются на воздухе. Ко второй группе фосфор-
органических соединений относятся производные кислот фосфора:
ОН ОН Н3С
/ / \
НО — Р Н8С — Р Р —ОН
II \ II \ /I!
О ОН О ОН Н3С О
ортофосфорная метилфосфоновая диметилфосфиновая
кислота кислота кислота
Соединения пятивалентного фосфора типа алкил- и арилфосфорных
кислот образуются вследствие участия d-орбиталей фосфора.
Среди фосфорорганических соединений имеются мощные
средства защиты растений
С2Н50 О С2НВ О
р р
/ \ / \
С2НбО F C2H5 S-(CH2)SC2H5
диэтилфторфосфат меркаптофос
отравляющие вещества со сверхвысокой токсичностью
С2Н50 О (СН3)2СНО О
Р Р
/ \ / \
(CH3)2N CN CH3 F
табун зарин
и эффективные лекарственные средства.
Эфиры алкил- и арилфосфиновых кислот — мономеры в
производстве полимерных материалов с повышенной огнестойкостью и
теплостойкостью.
§ 15. УГЛЕВОДЫ
К углеводам относятся различные сахара, крахмал, клетчатка
и др. Все известные в настоящее время углеводы можно разделить
на следующие три группы: моносахариды, дисахариды,
полисахариды. Последние две группы характеризуются тем, что в водных
растворах они гидролизуются до моносахаридов.
Моносахариды — соединения со смешанными функциями; они
представляют собой альдегидоспирты и кетоноспирты. Это белые
кристаллические вещества со сладким вкусом, легко растворяются
684
в воде. Они встречаются в растительных и животных организмах.
Важнейший представитель моносахаридов — глюкоза С6Н1206,
представляющая собой альдегидоспирт, придает виноградным
ягодам, грушам, яблокам сладкий вкус.
Строение молекулы глюкозы можно представить так:
н н н н н о
I i I I I /
Н —С-С — С — С— С—С ^
I I I I I \
он он он он он н
I
н н н н н н
I I ! I I I
1ГН —С—С — С— С— С— С—ОН
он
он он он
о
II
Между I и II состояниями устанавливается в водных растворах
равновесие.
Другой моносахарид — фруктоза — изомер глюкозы имеет
строение
Н Н Н Н Н
I I I I I
н_с—с—с—с—с—с—н
I I I I II I
ОН ОН ОН ОН О ОН
К группе дисахаридов относятся углеводы, которые
представляют собой продукты уплотнения моносахаридов; из двух молекул
моносахарида образуется одна молекула дисахарида, при этом
выделяется молекула воды:
—н2о
2СсН12Об -^С^Н^Оц.
Из дисахаридов широко распространены и имеют наибольшее
значение тростниковый и свекловичный сахар. В Советском Союзе
он получается из сахарной свеклы, содержащей 16—20% сахара.
При кипячении водного раствора сахара в присутствии кислоты он
гидролизуется, т. с. расщепляется водой, при этом образуются
моносахариды — глюкоза и фруктоза:
+н8о
Q2H22O11 -*" С6Н12Об-|- С6Н12Об.
глюкоза фруктоза
моносахариды
685
Молекулы полисахаридов представляют собой связанные в
цепи остатки глюкозы или других моносахаридов числом от 100 до
20 000 и более. Молекулярная масса их точно неизвестна, поэтому
их состав выражается формулой (СбНюОбК, где п — среднее число
остатков моносахаридов. Этой формуле соответствуют крахмал и
клетчатка.
Клетчатка или целлюлоза — это полисахарид с массой порядка
200 000 и более. В природе клетчатка — главная составная часть
оболочки растительных клеток. Целлюлоза — это многоатомный
спирт. При обработке клетчатки едкими щелочами образуются ал-
коголята (как в трехатомном спирте)
[СбН702 (ОН)3]я + rcNaOH -> лН20 ~- [C6H702 (OH)2 ONa]n.
С кислотами клетчатка образует сложные эфиры. Наибольшее
значение имеют ее эфиры с азотной и уксусной кислотами и кис^
лый эфир днтиоугольной кислоты H2COS2. Азотнокислый эфир
клетчатки, или пироксилин, содержит до 14% азота и имеет
большое значение в качестве взрывчатого вещества. Эфиры,
содержащие 10,7—12,5% азота, образуют с пластификаторами и другими
добавками пластическую массу, называемую целлюлойдом.
Последний имеет широкое применение (лаки, пленки, искусственная
кожа, киноленты и др.). При действии на целлюлозу смеси
концентрированных азотной и серной кислот (последняя водоотнимаю-
щее средство) образуется сложный эфир азотной кислоты и
целлюлозы, называемый нитроцеллюлозой, или нитроклетчаткой:
[С6Н702 (ОН)81я + 3/iHON02 = [С6Н702 (ОШ2)3] + ЗяН20.
Нитроцеллюлозу применяют для получения пластмассы (целлю-
лойда), искусственной кожи, нитролаков и бездымного пероксили-
нового пороха. Огромным недостатком таких эфиров клетчатки
является их исключительная горючесть. Более ценным продуктом
считается негорючая ацетилцеллюлоза, которая получается при
действии на клетчатку ангидрида уксусной кислоты. Раствор
ее в ацетоне служит материалом для получения ацетатного
искусственного шелка.
При взаимодействии алкоголята клетчатки и сероуглерода
образуется эфир днтиоугольной кислоты и целлюлозы — ксантогенат
целлюлозы:
ОСвН702(ОН)2-
[C6H702(OH)2ONa]„ + rtCS2
/
C = S
\
SNa
Ксантогенат целлюлозы — вязкая масса. Продавливая ее через
тонкие отверстия, получают нити, которые в растворе кислоты
превращаются в нити вискозного шелка. При действии кислоты на
ксантогенат целлюлозы происходит его разложение на целлюлозу
686
и сероуглерод (одновременно происходит образование натриевой
соли добавленной кислоты). Из вискозной массы подобным же
образом готовят пленки и ленты различной ширины, известные под
названием целлофана. Обработка древесины в производстве
бумаги сводится к отделению клетчатки от других веществ. Это
достигается варкой измельченной древесины пли с едким натром, или с
бисульфатом кальция.
В зеленых растениях на солнечном свету из углекислого газа
и воды образуются моносахариды, которые путем сложных
процессов полимеризации превращаются в первичные зерна крахмала
(С6Ню05)п, где п — того же порядка, что и в целлюлозе. Крахмал
добывают из клубней картофеля, из зерен риса, пшеницы,
кукурузы и других растений. Гигантские молекулы крахмала
подвергаются расщеплению при действии кислот, щелочей, ферментов и др.
Полному гидролизу с образованием глюкозы крахмал
подвергается при кипячении его в воде с примесью минеральных кислот —
серной или соляной. В технике этот процесс используют для
изготовления патоки.
Главный потребитель крахмала — пищевая, текстильная и
бумажная промышленности. В последнее время крахмал стали
использовать при изготовлении пластических масс.
Вопросы и упражнения
1. Чго понимают под названием органические соединения в настоящее
время?
2. В чем сущность теории химического строения, разработанной А. М.
Бутлеровым0
3. Что такое изомерия и таутомерия? Напишите формулы всех возможных
изомеров гексана.
4. Что такое простые и кратные связи? Приведите примеры соединений,
имеющих простые и кратные связи
5. Что мы называем сопряженными связями0
6. Что называется гетеролитическим и гемолитическим расщеплением?
Приведите примеры.
7. Какова классификация органических соединений0
8. Как можно объяснить, что атом углерода, имеющий два неспаренных
электрона, проявляет валентность 2 и 4°
9. Что такое гомологический ряд0 Что называется гомологической
разностью0
10. Какие углеводороды называют предельными, или насыщенными, какая
их общая формула0 Какая связь между атомами углерода в предельных
углеводородах и как это влияет на их химические свойства0
11. Какие углеводороды называются непредельными, или ненасыщенными,
какие у них общие формулы0
12. Какие химические свойства наиболее характерны для непредельных
углеводородов0 Объясните почему.
13. Что такое полимеризация? Полимером какого углеводорода является
натуральный каучук0
14. Назвать простейший углеводород ароматического ряда, привести его
формулу и указать его химические свойства.
15. Что такое галоидные производные углеводородов? Назовите их
важнейшие представители.
687
16. Какие наиболее характерные свойства спиртов?
17. Какие соединения называют простыми эфирами? Приведите примеры.
18. Чем отличаются альдегиды от кетонов? Назовите важнейшие свойства
альдегидов и кетонов.
19. Что представляет собой процесс поликонденсации? Чем он отличается
от процесса полимеризации?
20. Какая функциональная группа характеризует органические, или карбо-
новые кислоты?
21. Приведите примеры получения сложных эфиров.
22. Какие соединения называют амидами? Амидом какой кислоты является
мочевина?
23. Какие соединения называют аминокислотами? Каковы их свойства0
24. Какие соединения получаются в результате гидролиза белков?
25. На какие группы делятся углеводы? Дайте их краткую характеристику.
26. Назовите важнейшие кремнийорганические соединения. Где они
применяются?
27. Какие соединения мы называем элементоорганическими? Приведите
примеры.
ГЛАВА 16.
РАДИОАКТИВНОСТЬ
И ЯДЕРНЫЕ РЕАКЦИИ
До конца XIX в. атомы рассматривали как неделимые и
неизменные частицы, поскольку не было известно ни одного случая
превращения элементов друг в друга. Допускалось, что каждый
элемент состоит из идентичных атомов, обладающих одинаковой
массой и имеющих одинаковые физические и химические свойства.
Атомы, из которых состоят все элементы, считали простейшей
формой материи.
В 1896 г. Беккерель обнаружил, что соли урана испускают
какие-то лучи, которые проходят через черную бумагу и
засвечивают фотопластинку, подобно известным уже в то время лучам
Рентгена. Эти же лучи вызывают флуоресценцию некоторых
веществ, а также появление электропроводности в воздухе.
Открытое явление было названо радиоактивностью. Беккерель установил,
что радиоактивность — это свойство элемента урана,
независящее от его агрегатного состояния или формы химических
соединений, в состав которых он входит.
Вскоре были открыты другие радиоактивные элементы. В 1898 г.
Пьер и Мария Кюри открыли радиоактивные полоний и радий, а
Шмидт обнаружил радиоактивность тория. В 1899 г. Дебьерн
открыл актиний. В настоящее время известно около 50 различных
радиоактивных атомов, которые существуют в природе.
В 1903 г. Резерфорд и Содди показали, что радиоактивное
излучение появляется при распаде атомов радиоактивных элементов,
с превращением их в атомы других элементов. После того как
было установлено строение атома, стало ясным, что
радиоактивность — это свойство ядер атомов.
Как известно, атомное ядро состоит из нуклонов, т. е. из про-
тонов и нейтронов. Число протонов в ядре совпадает с его
порядковым номером Z в периодической системе элементов; число
нейтронов N=A—Z, где А — массовое число. Ядра с одинаковым
числом протонов Z, но различным числом нейтронов называются
изотопами. Ядра с одинаковым числом нуклонов А называются
изобарами. Ядра с одинаковым числом нейтронов JV, но различным
числом протонов называются изотопами.
Атомное ядро имеет приблизительно сферическую форму. Его»
радиус
R = г0Л|/3, гАе го ^ 1>2-10~13 см — радиус нуклона.
Нуклоны в ядре связаны ядерными силами. Поэтому энергия
ядра всегда меньше, чем энергия всех (разрозненных) нуклонов,
68&
из которых состоит ядро. В соответствии с соотношением
Эйнштейна между массой и энергией (Е=тс2) это означает, что масса
ядра меньше суммы масс образующих его нуклонов — так
называемый дефект массы. Чем он больше, тем устойчивее ядро.
Радиус действия ядерных сил очень мал. Каждый нуклон
может взаимодействовать только с ближайшими соседями, т. е. центр
притяжения в ядре отсутствует. Это дало возможность Н. Бору
построить капельную модель ядра, согласно которой ядро
представляет сферическую каплю из заряженной несжимаемой
сверхплотной (р^Ю14 г/см3) ядерной жидкости. Эта теория позволила
объяснить многие ядерные явления (например, деление ядер) и
рассчитать с хорошей точностью массу и энергию связи ядер,
образованных заданным числом нуклонов.
Однако более точные подсчеты энергии связи обнаруживают
небольшие, но вполне заметные отклонения от капельной модели:
устойчивость ядра зависит от числа и соотношения содержащихся
в нем нуклонов. Оказалось, что большей устойчивостью обладают
ядра, имеющие четное число протонов или особенно нейтронов.
Табл. 16.1 (Б. Некрасов) иллюстрирует этот факт. Видно, что наи-
Таблица 16.1
Распрастраненность атомов в земной коре
Число нейтронов
Содержание атомов, ат. % .
четное
четное
73,17
нечетное
четное
25,77
четное
нечетное
1,03
нечетное
нечетное
0,03
более распространены в природе и, значит, наиболее устойчивы
атомы, содержащие четное количество нуклонов.
Самыми стабильными, обладающими наибольшей энергией
связи, оказываются ядра, содержащие «магическое» число протонов
или нейтронов (2, 8, 20, 50, 82, 126 и, по некоторым данным, 138,
146). Это свойство ядер объясняется в другой модели атомного
ядра — модели ядерных оболочек. Последняя строится по аналогии
с моделью электронных уровней. Полагают, что и в ядре
существуют энергетические уровни, или оболочки, заполняемые парами
нуклонов (два протона или два нейтрона), спины которых антн-
параллельны. Ядра с полностью заполненными оболочками
становятся «магическими».
Капельная модель ядра и модель ядерных оболочек, дополняя
друг друга, позволяют описывать широкий круг ядерных явлений.
Однако все свойства ядер ими не исчерпываются. Существуют и
другие модели.
690
Атомное ядро может находиться в различных энергетических
состояниях. Энергетический уровень с минимальной энергией
называется основным. Все остальные энергетические состояния ядра
называются возбужденными.
Ядра в возбужденных состояниях образуются как в природных
условиях (в результате а- или р-распада), так и искусственно (при
i i i I L
J I L
30 40 50 60 10 80 90 100 110 120 130140 1Ь0160170180 190 200 210 220 230
Массовое число
10 20
Рис. 16.1. Зависимость дефекта массы, приходящегося в среднем на каждый
нуклон в ядре, от массового числа
бомбардировке ядер ускоренными частицами). Из возбужденного
состояния ядро самопроизвольно переходит в менее возбужденное,
или основное состояние, изучением у-кванта. у-Излучателями
являются практически все дочерние ядра — продукты а- и р-радио-
активных ядер, так как они образуются не только в основном, но
и в возбужденном состояниях. Энергия у-квантов лежит в пределах
от 0,5 до 2,5 Мэв.
Обычно время жизни возбужденного состояния ядра бывает
очень небольшим (т^10~13 сек). Однако в некоторых случаях
возбужденное состояние может существовать очень долго: секунды,
часы, годы и даже тысячелетия. Такие состояния (уровни)
называются метастабильными, а ядра, имеющие такие состояния,
называются изомерами. Изомеры могут обладать различными типами
радиоактивности. Например, одна треть атомов s3~Th ее-, а две
трети — р-радиоактивны.
Как уже указывалось, количественной характеристикой
стабильности ядра может служить дефект массы. На рис. 16.1
показана его зависимость от массового числа. Максимальной
стабильностью обладают ядра со средними значениями масс. Это
означает, что энергия должна выделяться как при соединении самых
легких ядер (процесс осуществленный при термоядерном синтезе и в
водородных бомбах), так и при распаде самых тяжелых ядер
(процессы в атомных реакторах и ядерных бомбах). В природных
691
условиях процессы первого типа наблюдаются на звездах, в
частности на Солнце, а второго типа — на Земле. Радиоактивный
распад, таким образом, является экзотермическим процессом.
Например, 1 г радия (вместе с продуктами своего распада) дает 136 кал
в час. Если учесть общее количество радиоактивных элементов в
земной коре, то окажется, что каждая ее тонна получает за их счет
в среднем 8 кал ежегодно, а энергия, получаемая всей корой,
составляет ~ 1 % энергии, получаемой Землей от Солнца, т. е. имеет
важное значение для теплового баланса земного шара.
Существуют различные типы радиоактивного распада.
Некоторые из них будут рассмотрены ниже.
1. В процессе а-распада из радиоактивного ядра испускается
-а-частица, т. е. ядро гелия Не, состоящее из двух протонов и двух
нейтронов (дважды магическое ядро). Таким образом, дочернее
ядро имеет на 2 протона и 2 нейтрона меньше, чем исходное.
Например, реакция
291U^He + 2^Th.
Кинетическая энергия а-частиц у большинства ядер заключена
в небольших пределах 4—9 Мэв, и а-частицы имеют скорости
порядка 1,4-109—2-Ю9 см/сек, т. е. 1/15—1/20 скорости света.
Каждое ядро имеет строго определенную энергию испускаемых
а-частиц. Эта энергия может быть охарактеризована длиной пробега
в воздухе.
В табл. 16.2 приведены некоторые примеры.
Таблица 16 2
Ядро
Длина пробега, см
238и
92U
2,67
234и
92и
3,23
*>
4,08
214Ро
6,95
Наряду с а-частицами, характеризующимися нормальными для
данного радиоактивного атома длинами пробега, изредка
наблюдается испускание атомом более длиннопробежных а-частиц. На-
218
пример, у 84 Ро на каждый миллион случаев нормального пробега
(в воздухе) 8,53 см приходится 34 случая пробега по 9,69 см и 190
случаев пробега по 11,54 см. Возникновение подобных
длиннопробежных частиц обусловлено какими-то особо возбужденными
состояниями ядра в момент их испускания. Из того обстоятельства,
что промежуточных длин пробега не наблюдается, вытекает
характерность для ядра только некоторых определенных энергетических
уровней.
2. В процессе р-распада из радиоактивного ядра
самопроизвольно испускается электрон (электронный, или р~-распад) или
позитрон (позитронный, или р+-распад), которые возникают в са-
692
мый момент р-распада. (Их нет в ядре.) Электроны образуются в
результате превращения нейтрона в протон по реакции
Jn~>i/7 + е~" + v.
Позитроны образуются в результате превращения протона в
нейтрон:
jp-*ofl + e+ +v.
Первая реакция может проходить как в атомном ядре, так и
со свободным нейтроном, так как масса последнего больше массы
протона и электрона. А вторая реакция возможна только внутри
ядра и за счет его энергии, так как масса протона меньше массы
нейтрона и позитрона. Третьим видом р-распада является захват
ядром электрона из электронной оболочки своего атома (£-захват,
или /(-захват). Во всех трех случаях р-распад сопровождается
испусканием нейтрино (v) или антинейтрино (v). В результате
Р"-распада количество протонов в ядре возрастает и его заряд
повышается на единицу. Например:
28эАС -42о Th + ег + v (Р~-распад).
В результате р+-распада и £-захвата заряд ядра понижается
на единицу (исчезает один протон). Например:
293sNp -*Л342и + е+ + v (Р+-распад),
294бАт->294?Ри (£-захват).
Массовое число ядра (т. е. количество нуклонов) при всех
видах р-распада остается неизменным: ядро превращается в изобар
(один нейтрон превращается в протон или наоборот).
В отличие от строго определенной энергии а-частиц,
излучаемых данным сортом ядра, р-частицы могут иметь любую энергию
от практически нулевой до максимальной, характерной для
данного атома. Это объясняется тем, что часть энергии уносят
образующиеся при р-распаде нейтрино или антинейтрино.
3. В процессе спонтанного деления происходит
самопроизвольное расщепление ядер с Z^90 (торий, протактиний, уран и
трансурановые элементы) на два ядра-осколка с примерно равными
массами (Mi : М2=2 : 3). Например:
2932U^13586Ba+f6Kr + 8i/z.
Следует отметить, что при таком распаде образуются
разнообразнейшие осколки, которые могут испытывать дальнейшие
радиоактивные превращения, сопровождающиеся а-, р- и 7_из«71учениями.
Так что вышеприведенное уравнение весьма условно.
Радиоактивный распад является результатом внутриядерных
процессов и не связан с взаимодействием между различными яд-
693
рами. Поэтому ракпия радиоактивного распада описывается
кинетическим уравнением первого порядка:
_^__XiV. (16.1)
dx
Таким образом, скорость распада, или количество распадов в
единицу времени, пропорциональна количеству имеющихся
радиоактивных ядер N. Коэффициент пропорциональности (имеющий
смысл константы скорости для реакций первого порядка) %
называется постоянной распада и имеет для каждого радиоактивного
элемента вполне определенное значение. Интегрирование
уравнения (16.1) дает
N = Nffir-te. (16.2)
Здесь N — количество еще не распавшихся атомов к моменту
времени т из общего первоначального количества атомов No. Выбрав
время т=Г, за которое распалась половина первоначального
количества, так что N=X/2N0, и подставив эти значения в уравнение
(16.2), получим
— - е~кт
2
и
1п2-Х7\ (16.3)
Таким образом, постоянная распада однозначно связана с так
называемым периодом полураспада Т. Зная одну величину, легко
рассчитать вторую.
Именно величиной периода полураспада чаще всего
характеризуется устойчивость тех или иных ядер атомов. Эта величина
может меняться в очень,широких пределах. Например, 84Р0
испытывает а-распад с Г=3-10~7 сек, a goTh — спонтанное деление с
Г=1,4-1018 лет. (Указанные ядра имеют изомеры, распадающиеся
и по другому механизму с другими величинами Т.)
В качестве примера на рис. 16.2 показана последовательность
радиоактивных превращений природного урана. Над стрелками
указан тип распада и в скобках для а-частиц указана длина их
пробега (в см) в воздухе при 15°С и 760 мм рт. ст. Под стрелкой
приведен период полураспада. Разветвления указывают на наличие
изомерных ядер; в этом случае указана доля атомов,
распадающихся по данному типу.
Из закона радиоактивного распада вытекает важное следствие,
касающееся количественных соотношений между отдельными
членами радиоактивного ряда. В самом деле, допустим, что имеется
некоторое количество чистого радия. При его распаде образуется
радон, претерпевающий, в свою очередь, дальнейший распад
694
(см. рис. 16.2). Так как скорость распада и радия, и радона
зависит от их наличных количеств, то вначале, когда атомов радона
еще мало, его будет образовываться из радия больше, чем
распадаться. Однако по мере накопления радона его распад станет
ускоряться, и наконец наступит равновесие, при котором будет
распадаться столько же атомов радона, сколько их образуется за это
23* ы(2,бЗ) 234Tk fi^ 234
4,5-10 лет ",'а»
0,15% у 234 Д у
%,Ра v. 234 *(3,22) 230
^> U »- 1
99,85% А у / 92 2,3 10 мет 90
6,7час
Ы(З.И),у 225Ра d (3,30),у 222 <*К05) 2»8
8,3/0 лет °° 1590лет 86 3,8 дн 84
0,04%oL (4,1) 2Ю J3, f
9т%^(4,64) г* м а «у /97ми; a/l7iF7
3,05 мы* 62 26,8mUW 83, ' """[21
99,9%7(5,53)/ \&9,ЭбХДу . в2РЬ~""
0,03 %j* 2'8At / \ Po-^-J,
^85T\ar/0JJ 218 4I61) 4 84 1,45 Ю сек
\ 1—^ Rn ' 1
86 0,02 сек
-5
5 10 % с* 206 p
M , ™. / «П ^23^X206Pb
22,3 года g3 \ fry 21° ?q *(3,85) / 82
4,95Эн 84 /39 дн
Рис. 16.2. Радиоактивный ряд урана
же время. (Это равновесие устанавливается приблизительно за
2 месяца.) Но число образующихся атомов радона равно числу
распадающихся атомов радия. Отсюда следует, что при
равновесии за одно и то же время распадается одинаковое число атомов
радия и радона, т. е. скорости их распада равны.
Очевидно, аналогичное рассуждение полностью применимо и к
любой другой паре последовательных членов данного радиоактив-
695
ного ряда. Поэтому между всеми ними должно в конце концов
установиться радиоактивное равновесие, характеризующееся тем, что
скорости распада всех членов ряда одинаковы, т. е. в соответствии
с уравнением (16.1)
Или, учитывая уравнение (16.3),
Nt _ N2 = Л^3 ^
7\ ' Т2 Т3
Так как периоды полураспада Т различны, то должны быть и
различны относительные количества находящихся в
радиоактивном равновесии атомов. В табл. 16.3 в качестве примера
приведены количества различных элементов, находящиеся в
радиоактивном равновесии в природном уране (расчет сделан на 1 т урана).
Таблица 16.3
Изотоп
т
Количество
вещества, г
238тт
92и
4,5-10» лет
9,93-10б
90Th
24,1 дн.
1,4.10-»
234и
92и
2,3-105 лет
50,1
226Ra
88ка
1590 лет
0,33
86Кп
0,02 сек
ью-"
Таким образом, зная период полураспада хотя бы одного
члена ряда и количественные соотношения их при радиоактивном
равновесии, можно рассчитывать периоды полураспада всех
остальных членов. Именно таким образом по периоду полураспада
радия был определен период полураспада урана, который
невозможно измерить непосредственно из-за слишком медленного распада.
Как уже указывалось, в настоящее время известно около 50
природных радиоактивных изотопов различных элементов. Еще
больше радиоактивных атомов получено искусственно в результате
облучения стабильных изотопов в атомных реакторах (например,
по реакции
27Со 4- сп =2?Со,
в которой образуется 27С0, обладающий |3~-, у-РаДиоактивностыо с
периодом полураспада 5,3 года и нашедший широчайшее
применение в науке, технике, медицине как источник у-излучения) или в
ускорителях заряженных частиц (например, по реакции
123CU , 4тт« 126т - 1
5ibb -f 2He = 5з1 + оя,
в которой образуется р~-радиоактивный йод).
669
Вопросы и упражнения
1. Перечислите изотопы водорода, кислорода, урана.
2. Пользуясь периодической системой Д. И. Менделеева, приведите
примеры атомов-изобаров и изотонов.
3. Вычислите радиусы ядер атомов кислорода, урана.
4. Назовите элементы с «магическими» ядрами атомов.
5. Приведите схемы радиоактивного распада изомеров атома 822^*
6. В 1919 г. Резерфорд при облучении азота ()4N) а-частицами
обнаружил появление протонов. Напишите уравнение соответствующей ядерной
реакции.
ОСНОВНАЯ ЛИТЕРАТУРА
1. Некрасов Б. В. Основы общей химии, т. 1, 2 М., «Химия», 1973.
2. Хомяков К. Г. Лекции по общей химии. Изд-во МГУ, 1966.
3. Г л и н к а Н. Л. Общая химия. Л , «Химия», 1973
4. П о л и н г Л. Общая химия. М., «Мир», 1974.
5. Неницеску К. Общая химия. М, «Мир», 1969.
6 Ахметов Н. С. Неорганическая химия. М., «Высшая школа», 1975.
7. Барнард А. Теоретические основы неорганической химии. М., «Мир», 1968.
8. Коттон Ф.Уилкинсон Дж. Современная неорганическая химия, ч. I —III.
М., «Мир», 1967.
9 Рем и Г. Курс неорганической химии. М., «Мир», 1972
10. Третьяков Ю. Д, Зайцев О. С. Программированное пособие по
общей химии. Изд-во МГУ, 1971.
П. Третьяков Ю. Д., Зайцев О. С. Программированное пособие по
общей и неорганической химии. Изд-во МГУ, 1975.
12 Kvpc химии Под ред Г. А. Дмитриева, Г. П Лучинского и В. И. Ссмишина.
М , «Высшая школа», 1967.
К е м и б е 1 Д. Современная общая химия, т 1—3 М , «Мир», 1975.
Дополнительная литература
Глава 1
1 Энгельс Ф. Диалектика природы. М., Госполитиздат, 1969.
2. Ленин В. И Материализм и эмпириокритицизм Поли. собр. соч., т. 18.
3 Щукарев С. А. Лекции по общему курсу химии. Изд-во ЛГУ, 1962.
4 Кузнецов В. И. Эволюция представлений об основных законах химии.
М., «Наука», 1967.
Глава 2
1. ПисаржевскийЛ В. Электрон в химии. Избранные труды. Киев, Изд-во
АН УССР, 1956.
2. С и е н к о М., П л е й н Р., X е с т е р Р. Структурная неорганическая химия.
М., «Мир», 1968.
3. Барнард А. Теоретические основы неорганической химии. М., «Мир», 1968.
Глава 3
1. Сб. «100 лет Периодического закона химических элементов» (доклады на
пленарных заседаниях X юбилейного Менделеевского съезда). М., «Наука»,
1971.
2. Менделеев Д И. Основы химии, изд 4. 1908.
3 Менделеев Д И. Избранные лекции по химии. М, «Высшая школа»,
1968.
4. М е н д е л е е в Д. И. Собр. соч М , Изд-во АН СССР, 1954.
699
5. Менделеев Д. И. Научный Архив, Периодический закон. М, Изд-во АН
СССР, 1953; Растворы, 1961.
6. М а к а р е н я А. А. Д. И. Менделеев и физико-химическая наука. М., Атом-
издат, 1972.
7. Кедров Б. М. День одного открытия. М., Соцэкиздат, 1958.
8. С е м и ш и н В. И. Периодическая система химических элементов. Д. И.
Менделеев. М., «Химия», 1972.
9. Астахов К. В. Современное состояние периодической системы Д. И.
Менделеева. М., «Знание», 1969.
10. А г а ф о ш и н Н. П. Избранные главы общей химии. М., «Просвещение»,
1959.
П.Трифонов Н. Д. Структура и границы периодической системы. М,
Атомиздат, 1970.
Глава 4
1. Карапетьянц М. X., Дракин С. И. Строение вещества. М., «Высшая
школа», 1967.
2. Грей Г. Электроны и химическая связь. М., «Мир», 1967.
3. Дей К. и Селбин Д. Теоретическая неорганическая химия. М., «Химия»,
1969.
Глава 5
1. Карапетьянц М. X. Введение в теорию химических процессов. М,
«Высшая школа», 1970.
2. 3 а й ц е в О. С. Химическая термодинамика к курсу общей химии. Изд-во
МГУ, 1973.
3. Льюис и Рендалл. Химическая термодинамика. М., ОНТИ, 1936.
4. Кричевский И. Р. Понятия и основы термодинамики, изд. 2. М., «Химия»,
1970.
5. Термические константы веществ Справочник в десяти выпусках под ред
В. П Глушко. М., Изд-во АН СССР, 1965-1975
Глава 6
1. Ройтер В. А., Голод ец Г. И. Введение в теорию кинетики и катализа.
Киев, «Наукова думка», 1972.
2. П о л т о р а к О. М. Лекции по теории гетерогенного катализа Изд-во МГУ,
1968
3. Сокольский А. В, Друзь В. А. Теория гетерогенного катализа. Алма-
Ата, «Наука», 1968.
4. Же р мен Ж. Гетерогенный катализ. М., ИЛ, 1961.
5. Пасы некий А. Г. Биофизическая химия. М., «Высшая школа», 1968.
6. Боресков Г. К. Катализ (курс лекций), ч. 1 и 2. Новосибирск, «Наука»,
1971.
7. Диксон М., У э б б Э. Ферменты. М., «Мир», 1966.
8. Краткий курс физической химии, под ред. С. М. Кочергина и С. Н.
Кондратьева. М, «Высшая школа», 1968.
Глава 7
1. Кир ее в В А. Курс физической химии М., «Химия», 1975.
2. Крешков А. П. Основы аналитической химии, т. II. М., «Химия», 1971.
3. Карапетьянц М. X. Введение в теорию химических процессов М.,
«Высшая школа», 1970.
700
4 Самойлов О Я. Структура водных растворов и гидратация ионов М,
Изд-во АН СССР, 1957.
5 НадеинскийБ. П. Теоретические обоснования и расчеты в аналитической
химии. М, «Наука», 1956.
6 Даниэльс Ф., Альберти Р. Физическая химия. М., «Высшая школа»,
1967.
7. Л и л и ч С. С. Некоторые аспекты современных представлений в растворах
электролитов. Изд-во ЛГУ, 1967.
Глава 8
1. Волоцкий С. С. Курс коллоидной химии. М., «Химия», 1964.
2. Жуков И. И. Коллоидная химия. Изд-во ЛГУ, 1949.
3. Путилова И. Н. Руководство к практическим занятиям по коллоидной
химии. М., «Высшая школа», 1961.
Глава 9
1. Булгакова Т. И. Реакции в твердых фазах. Изд-во МГУ, 1972.
2. Хенней Н Химия твердого тела М, «Мир», 1971
3 КрёгерФ Химия несовершенных кристаллов. М, «Мир», 1969
Глава 10
1. Кудрявцев А. А. Составление химических уравнений. М., 1969.
2. Л а м и т е р Б. М. Окислительные состояния элементов и их потенциалы в
водных растворах. М., 1954.
Глава 11
1. Фрумкин А. Н. и др. Кинетика электродных процессов. Изд-во МГУ, 1952
2. Дамаскин Б. Б, Петрий О. А. Современная электрохимия. М., «Наука»,
1965.
3. Акимов Г. В. Теория и методы исследования коррозии металлов. М., Изд-во
АН СССР, 1945.
4. Шаталов А. Я. Электрохимические основы теории коррозии металлов
Изд-во Воронежского ун-та, 1971.
5. Тодт Ф. Коррозия и защита от коррозии. Л., «Химия», 1967.
6. Антропов Л. И. Теоретическая электрохимия, изд. 2. М., «Высшая
школа», 1969.
7. Жук И. П Курс коррозии и защиты металлов. М., «Металлургия», 1968.
Глава 12
1 НемиловВ. А. Общая металлография. Изд-во МГУ, 1974.
2. Курнаков Н. С. Введение в физико-химический анализ. Л, ОНТИ-хим-
теорет., 1936.
3. МихееваВ И. Метод физико-химического анализа в неорганическом
синтезе. М, «Наука», 1975
4. Агеев Н В. Химия металлических сплавов. М., Изд-во АН СССР, 1947.
5 Аносов В Я, Погодин С А. Основные начала физико-химического
анализа М, Изд-во АН СССР, 1949
Глава 13
1. Гринберг А. А Введение в химию комплексных соединений. М.-Л,
«Химия», 1966.
701
2. Басоло Ф, Джонсон Р Химия координационных соединений. М, «Мир»,
1966.
3 Карапетьянц М X, Драк и н С. И. Строение вещества. М, «Высшая
школа», 1970.
Глава 14
1 Б о к и й Г. Б. Кристаллохимия. М, «Наука», 1971.
2 К о т т о н Ф., У и л к и н с о н Дж. Современная неорганическая химия,
т. 1—3. М., «Мир», 1969.
3. Ахметов Н. С. Неорганическая химия, изд. 2. М, «Высшая школа», 1975.
4. Н и к о л а е в Л А. Современная химия. М, «Просвещение», 1970.
Глава 15
1. Несмеянов А. М., Несмеянов М. А. Начала органической химии,
ч. I и II М, «Химия», 1969.
2. Чичибабин А. Е. Основные начала органической химии, ч. I и II. М.,
Госхимиздат, 1953, 1959.
3. П р о с т а к о в Н. С. Лекции по органической химии. М., Изд-во УДН
им. П Лумумбы, 1967.
4 П о т а п о в В М , Т а т а р и н ч и к С. II. Органическая химия. М., «Химия»,
1972.
5 Курс химии, ч I, под ред Г. А. Дмитриева, Г. П. Лучинского и В И. Семи-
шина. М, «Высшая школа», 1965
:1АМ1-:ч1-:нные опечатки
. !|)()!ч,1
Напечатано
Следует читать
0 сверху
II сверху
18 снизу
тлбл. 7.3 2-й
столбец
Габл. 7.3 4-й
столбец
8 сверху
13 сверху
15 сверху
6 сверху
Табл. 12.4 4-й
столбец 3 строка
Табл. 12.4 5-й
столбец 7 строка
Табл. 12.4 5-й
столбец 10 строка
Табл. 12.4 5-й
столбец 12 строка
11 снизу
2 сверху
3 снизу
5 сверху
6 сверху
7 сверху
10 снизу
8 снизу
4 сверху
Табл. 12.5 5-й
столбец
Табл. 12.6 2-й
столбец
22 сверху
7 сверху
Табл. 12.7
11 сверху
2 сверху
5 сверху
1 снизу
5 сверху
6 сверху
1На]=0,04-Ю,04 =
-^0,08 моль/л
ОБО"
ОБО"
45,85
45,60
45,09
0,9969
иуть
С Ва+2
(541 ккал/г)
Еу/кТ
Zn, Al, Ca;
V, Sb, Ag, P
Fe, Sc, С, Mn
М, Те, Re, CI, I
Se203
от основной
более
Qen
Sn„
РЬп
гибри—-
СН2,8
Gf
см/м
см/м
Р(1Н2
Co2Si
группы IV В
sp2
Zr2,Ge
W
->A:D+C:D
-н3о
>
0,9969
Суть
и Ва+2
(541 кал/г)
~~£V/kT
Zn, Al
V, Sb, As, P
Fe, Mn
Mn, Tc, Re, CI,
Sc2o3
от оснований
менее
Ge
Sn
Pb
гидри—
СеН2>8
Ht
см/ом
см/ом
Pd,H
Ca2Si
группы VI В
sp
Zr2Ge
V
^A:D4C:B
—H20
[Ha] = 0,0014-0,04=
— 0,044 моль/л
00"
OO"
4,585
4,560
4,509
0,9968
*ак 179
ОБЩАЯ ХИМИЯ
Редактор Л Н Лукиных
Переплет художника Е Л Михельсона,
Технический редактор 3 С Кондрашова
Корректоры Н П. Стерина, Н. И. Коновалова,
С Ф. Будаева, А. А. Алексеева
Тематический план 1975 г. № 142
Сдано в набор 25/V 1975 г. Подписано к печати 13/XI
1975 г Л-23008 Формат 60X90Vi« Бумага тип. № 1
Физ печ л 44,0 Уч -изд л. 43,95 Изд. № 2293
Зак. 179 Тираж 10 000 экз Цена 1 р 34 к
Издательство Московского университета
Москва, К-9, ул Герцена, 5/7
Типография Изд-ва МГУ. Москва, Ленинские горы
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ Д. И. МЕНДЕЛЕЕВА
период 1
1
|2
г
о
4
|5
7
IA
группы эле
ha|iiib
3
Li
11
Na
19
К
37
Rb
56
Cs
87
Fr
■
4
Be
12
Mg
20
Ca
38
Sr
56
Ba
88
Ra
s2
1 семейства
м е
H
ivb|vb|vib|viib
21
Sc
39
Y
67
La
89
Ac
d1
Я
JML
58
Се
90
Th
л
59
Pr
91
Pa
Л
60
Nd
92
U
JL
61
Pm
93
Np
f
62
Stn
94
Pu
JL
63
Eu
95
Am
_f_
64
Gd
96
Cm
am.
65
Tb
97
Bk
JL
66
Dy
98
Cf
JL
67
Ho
99
Es
JL
68
Er
100
Fra
JL.
69
Tm
101
Md
JL
70
Yb
102
No
JL
71
Lu
103
Lr
fdx
22
Ti
40
Zr
72
Hf
104
Ku
d2
23
V
41
Nb
73
Та
105
d*
24
Cr
42
Mo
74
w
106
d"
25
Mn
43
Tc
75
Re
107
d*
T
О
в
VIIIВ
26
Fe
44
Ru
76
Os
108
db
27
Co
45
Rh
77
lr
109
d1
28
Ni
46
Pd
78
Pt
110
d«\
IB
29
Cu
47
79
Au
111
d9
iib|iiia|iva|va|via
30
Zn
48
Cd
80
Hg
112
di0
6
В
13
Al
31
Ga
49
in
81
Tl
113
6
С
14
Si
32
Ge
50
Sn
82
Pb
114
P2\
7
N
16
P
33
As
51
Sb
83
Bi
115
P3\
f 8
0
16
s
34
Se
52
Те
84
Po
116;
P*\
ж
VIIA
1
H
9
F
17
CI
35
Br
53
I
85
At
117
VI1IA
2
He
; io
Ne
18
Ar
36
Kr
54
Xe
86
Rn
118
p* \ рь \