/













































































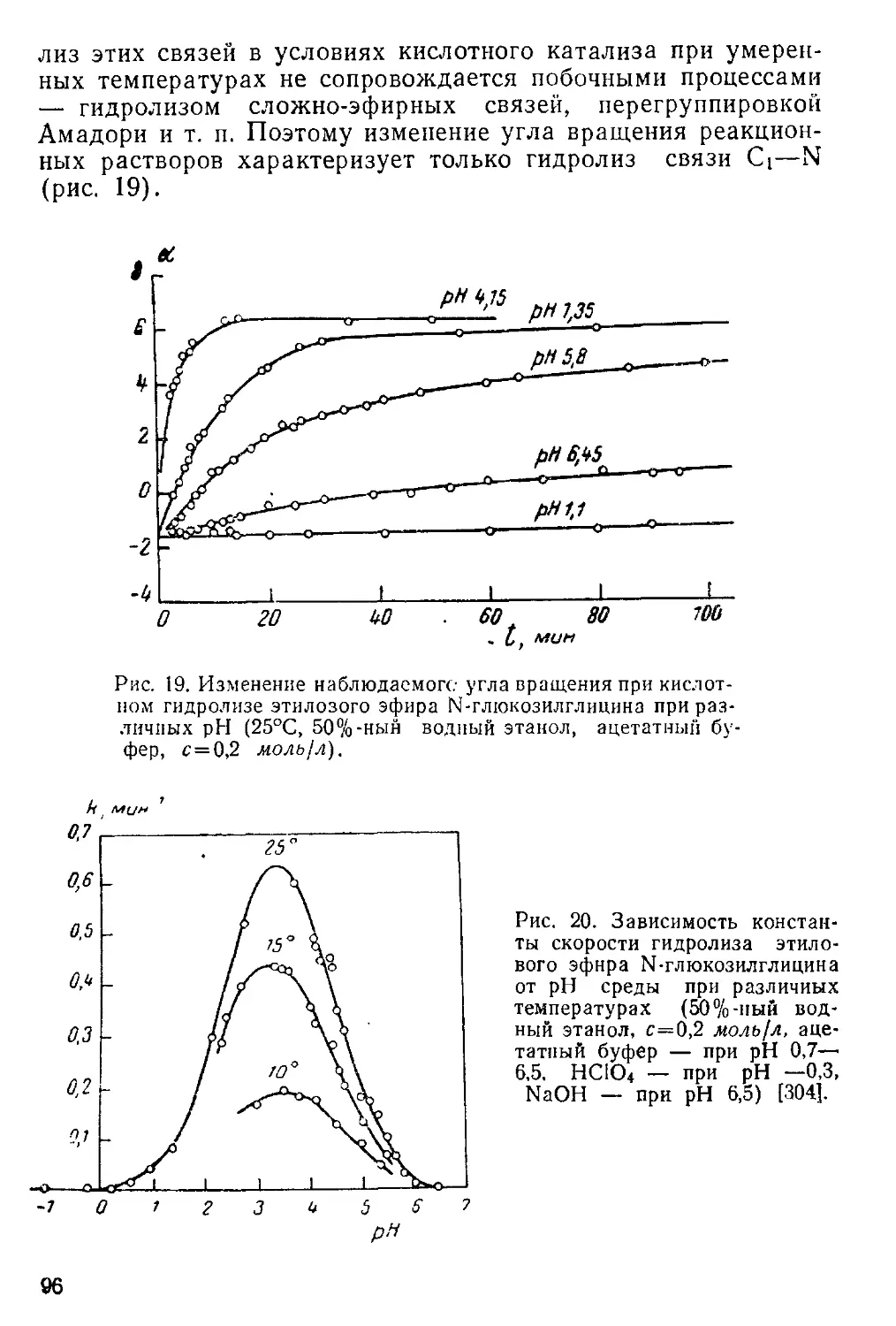






















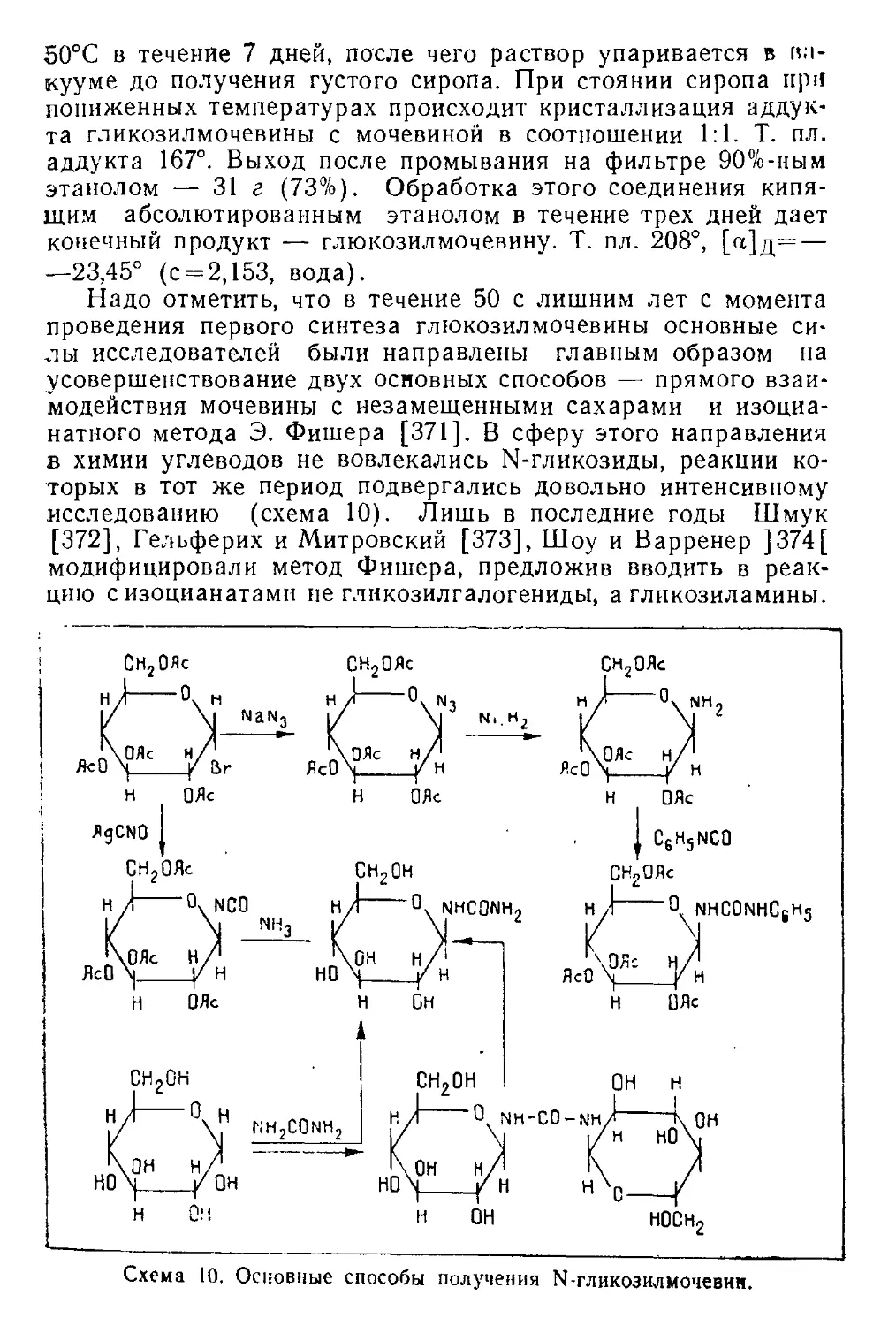




























Текст
Афанасьев В. А., Стревьцова И. Ф., Трушкина Н. И.,
Панарин Ф.В, О.В. Дронов
Строение и реакционная
способность N-гликозидов.
- Ф1- . Ппим 19 3 - 221 с
Глава I
СИНТЕЗ, СТРОЕНИЕ
И ОСНОВНЫЕ СВОЙСТВА N-ГЛИКОЗИДОВ
1.1. Определение
К N-гликозидам относятся углеводсодержащие производ-
ные, в которых гликозидный углеродный атом Ct углеводного
фрагмента связан через атом азота с пеуглеводным компо-
нентом — агликоном. Характерная для этих соединений связь
НС!—N или HC|-N (но не Н2С—N) встречается в самых
разнообразных синтетических и природных продуктах, в част-
ности, в продуктах конденсации сахаров с алифатическими и
ароматическими аминами, аминокислотами, насыщенными и
ненасыщенными азотистыми гетероциклами, мочевиной и ее
алкилпропзводными, гидроксиламином, гидразином и фенил-
тидразипом, семикарбазидом [1—4]. Если использовать
только этот признак, то в класс N-гликозидов следует также
включить ряд биологически важных производных — нуклео-
зидов и нуклеиновых кислот, в которых сахарный компонент
«связан через циклический атом азота с производными пурино-
вых и пиримидиновых оснований [5].
Иногда классифицируют N-гликозиды по типу N-агликона:
а) гликознламины — продукты конденсации сахаров с али-
фатическими и ароматическими аминами; б) гликозилмочеви-
ны (гликозилуреиды) — продукты взаимодействия сахаров с
мочевиной и ее производными; в) нуклеозиды — природные
или синтетические соединения с гетероциклическим N-аглико-
ном пиримидинового или пуринового ряда [4].
Вследствие многоплановости в исследованиях, а также
разнообразия производных для N-гликозидов нет единой об-
щепринятой номенклатуры, как и для их ближайших аналогов
О- и S-гликозидов [2, 4]. В специальной литературе, посвя-
щенной вопросам препаративной химии и биохимии углево-
дов, в названии N-гликозидов указывают характер агликона
и углеводного остатка, тип углеводного кольца и пространст-
венное расположение агликона относительно плоскости угле-
6
водного кольца. Например, циклический продукт присоедине-
ния анилина к С] незамещенной Д-глюкозы, имеющий ше-
стичленный (пиранозный) цикл и N-агликоп в экваториальной
позиции относительно пиранозного цикла, можно назвать
'N-фенил-р-Д-глюкопиранозидом или р-аномером N-глюкозн-
да анилина. В дальнейшем мы будем использовать второе на-
звание, распространенное в литературе по вопросам физико-
химии углеводов. При описании соединений, имеющих струк-
туру шиффовых оснований, таких, как продукты взаимодей-
ствия сахаров с гидроксиламипом и фепилгидразниом, приме-
няют названия «оксим глюкозы», «фсиилгидразон ксилозы»
и т. д.
Преобладающее большинство N-алкил- и N-арплгликози-
,дов представляют собой бесцветные или слегка окрашенные
под цвет соответствующего амина кристаллические соедине-
ния, плавящиеся с разложением, растворимые в воде, водио-
•спиртовых и водио-дноксановых растворах, почти нераствори-
мые в эфире, ацетоне, бензоле, хлороформе, этнлацетате и
многих других органических растворителях. Все они облада-
ют оптической активностью, причем знак угла вращения, как
правило, определяется расположением N-аглнкопа относи-
тельно углеводного кольца.
1.2. Методы получения N-гликозидов
N-глнкознды с простыми алифатическими или ароматиче-
скими агликонами обычно легко получаются при непродолжи-
тельном нагревании смеси амина и моносахарида в неболь-
шом количестве воды или спирта до образования гомогенного
раствора (методы Сорокина [6], Куна и Вейганда [7, 8]). В
качестве катализаторов можно использовать добавки НС1,
СН3СООН, ZnClo, NH4CI [9—12]. Продукт выкристаллизо-
вывается из полученного сиропа при добавлении спирта или
эфира с последующим охлаждением или упариванием. При
этом образуются, как правило, р-аномеры N-глнкозидов. Пря-
мая конденсация моносахаридов с мало основными аминами,
например, с амидами карбоновых кислот, протекает, однако,
очень медленно и требует длительного выдерживания реак-
ционной смеси при повышенных температурах [13—16]. Боль-
шие трудности возникают также и при синтезе гликозидов
а-аминокислот [17—20]. Замещение карбоксила аминокисло-
ты на сложноэфирную группу облегчает образование N-глико-
зидпой связи [21].
N-гликозиды образуются также при взаимодействии аце-
тилированных сахаров с аминами, при этом в ходе N-глико-
7
зилирования возможно отщепление ацетильных групп [22]. В
методах Фишера — Гельфериха [23] и Фрережака [24] син-
тез N-гликозидов осуществляется путем конденсации аминов
(включая третичные амины со стсрически доступным атомом
азота) с О-ацетилированнымн галоген-производными сахаров.
Этим способом можно получить а-аномеры N-гликозидов с
помощью многократной перекристаллизации смеси. Для син-
теза N-гликозидов может быть использована кислотно-ката-
лизируемая реакция N-трансгликозилирования, открытая Ку-
ном с сотр. (см. разд. 3.5). Возможности этого пути еще не
раскрыты в должной мере, однако имеются основания пола-
гать, что трансгликозилирование явится одним из эффектив-
ных и селективных методов получения N-гликозидов, образо-
вание которых в прямых синтезах протекает с большим тру-
дом и не избирательно.
В задачу настоящей работы не входит сколько-нибудь под-
робный анализ деталей синтеза N-гликозидных производных.
Этот вопрос требует специального рассмотрения на уровне
современных обзорных работ по синтетической химии сахаров.
К сожалению, насколько нам известно, подобного обзора по'
методическим разработкам последних лет в литературе не
имеется. Необходимость же в квалифицированном обобщении
новейших данных очевидна, если учесть возрастающий инте-
рес к этому важному классу производных. До недавнего вре-
мени ограничивались изучением преимущественно гликозидов
с простыми арильными и алкильными N-агликонами. Затем в
сферу N-гликозидов были вовлечены производные с разнооб-
разными N-аглнконами, в частности с гетероциклическими
соединениями. Результаты этих исследований опубликованы
в оригинальных сообщениях, список которых приведен в биб-
лиографических сборниках по химии углеводов за пеоиод
1961 — 1968 гг. [25].
1.3. Структура N-гликозидов
По аналогии с моносахаридами и О-гликозидами допу-
скается возможность существования простых N-гликозндов В:
циклических пиранозных (/ и 2) и фуранозных (3 и 4) фор-
мах и в виде ациклических структур (5 и 6) с аминоспирто-
вым и азометиновым фрагментами при Ci [2, 3] (схема 1).
Накопленные к настоящему времени экспериментальные
данные позволяют сделать вывод, что N-гликозиды с просты-
ми алифатическими и ароматическими N-агликонами имеют в
кристаллическом состоянии преимущественно гликопираноз-
8
Схема 1. Структурные формулы N-гликозидов с
простыми агликонами и возможные переходы
между ними.
ную структуру с конформацией С-1 углеводного кольца. Цик-
лические структуры большинства N-гликозидов неустойчивы и.
в водных растворах легко переходят друг в друга с образова-
нием равновесной смеси, в которой преобладают термодина-
мически более устойчивые 0-аномерные формы пираноз. Бог-
нару и Нанаши [26, 27] удалось на нескольких примерах изо-
лировать отдельные аномерные формы N-арилглюкозидов н
показать, что а-апомер с аксиальным N-агликоном имеет по-
ложительный, а 0-аномер с экваториальным N-агликоном —
отрицательный знак угла вращения плоскости поляризован-
ного света.
Для установления преимущественной конформации N-гли-
козидов Соколовский с сотр. [28—31] использовали подход,,
развитый впервые Ривсом применительно к незамещенным
сахарам [32], а именно — поляриметрический и кондуктомет-
рический анализ медноаммиачных комплексов замещенных
N-арилгликозидов (в частности, N-ацетил-тетра-О-ацетил-
глюкопиранозиламинов) в абсолютном метаноле. Проведен-
ные исследования позволили авторам заключить, что N-ариль-
ные производные глюкозы, галактозы, ксилозы и арабинозы
существуют в растворах главным образом в термодинамиче-
ски наиболее устойчивой конформации кресла С-1.
В отличие от О-гликозидов, имеющих стабильную цикли-
ческую структуру, азотистые гликозиды с простыми N-агли-
конами склонны к легкому переходу в ациклические структу-
ры, и, как полагают некоторые исследователи, именно это
обстоятельство позволяет N-гликозидам вступать в реакции,
характерные для соединений в форме азометина — реакции
присоединения HCN по связи C] = N и каталитического гидри-
рования до соответствующих аминопроизводных. Затем ока-
залось, что N-производные вторичных аминов, не способные к
существованию в форме азометипа (если исключить из рас-
смотрения форму «иммониевого иона»), легко присоединяют
HCN и подвергаются каталитическому гидрированию. Таким
образом, обнаружилось, что указанные реакции не могут слу-
жить хорошим тестом для доказательства структуры N-гли-
козидов.
В синтетической химии углеводов широко распространены
методы метилирования и перйодатного окисления для уста-
новления размеров углеводного кольца [4, 33]. Однако при-
менение этих испытанных методов для анализа строения
N-гликозидов не всегда может привести к однозначным вы-
водам (особенно в тех случаях, когда гликозид имеет высоко
основный N-агликон). Так, например, Эллис и Хонемаи [34]
изучили реакцию метилирования N-гликозида анилина и при-
шли к выводу, что этот гликозид реагирует в пиранозной фор-
ме, однако данные опыта не исключают возможности участия
в реакции и других форм. Метод перйодатного окисления, как
известно, основан на способности HJO4 окислять (с разрывом
С—С-связей) концевую группировку СН2ОН до формальде-
гида, а центральные звенья НС—ОН — до муравьиной кис-
лоты. Таким образом, по количеству молей HJO4, НСНО и
НСООН можно судить о структуре углеводного кольца. Ранее
проведенные исследования показали, что N-гликозиды с про-
стыми N-агликонами ведут себя подобно соответствующим
моносахаридам. Однако в N-ацетильных производных N-гли-
козидов не происходит разрыва связи С,—С2 под действием
TTJO4. Вопросы исследования строения N-гликозидов методом
метилирования, и перйодатного окисления достаточно подроб-
но изложены в обзоре Эллиса и Хонемана [1]. Можно пол-
ностью согласиться с мнением авторов работы [35], указав-
ших на то, что «вопрос об истинном строении N-гликозида
нужно решать отдельно в каждом индивидуальном случае,
’так как, по-видимому, в зависимости от структурных особен-
10
яостей амина, а возможно и сахара, речь может идти об от-
крытой или циклической структуре». К этому можно доба-
вить, что лабильность характерной для N-гликозидов группи-
ровки—OUUKJl.— НС[ — NHR варьирует (главным образом, в
зависимости от основности агликона NHR) в очень широких
пределах. Поэтому «классические» методы химии углеводов
могут дать вполне надежную информацию о структуре глико-
зидов с мало основными N-агликонами, такими, например, как
N-гликозилмочевппа, и оказаться ненадежными при анализе
циклической структуры гликозидов с высокой и даже умерен-
ной основностью N-агликонов.
Отметим, что сравнительно недавно Капон и Коннетт [36]
провели доказательство циклической структуры N-гликозида
п-толуидина путем последовательного ацетилирования и де-
зацетилирования правовращающего апомера:
Ас2О в C6H5N
(+) —N-глюкозид *====^' (+) —N-глюкозид (тетрацетат)
NaOH в СН3ОН . trHniI . . _пн
Ас2О в C6H3N * 1 СНЯОП, 4%АсОН
(—)—N-глюкозид — ~ ~ —)—N-глюкозид (тетрацетат)
NaOH в СН3ОН
Образующийся по указанной схеме левовращающий изомер
N-глюкозида п-толуидина имеет заведомо р-пирапозную
структуру, так как он может быть получен независимым пу-
тем — по реакции взаимодействия тетрацетата а-Д-глюкопи-
рапозилбромида с п-толуидином, протекающей с обращением
конфигурации при С|. В пользу пиранозной структуры этого
соединения свидетельствовали также данные ЯМР-спектро-
скопии. Осторожным ацетилированием с помощью уксусного
ангидрида в сухом пиридине он снова может быть переведен
с 98%-ным выходом в левовращающий тетрацетат. Результа-
ты этих исследований надо признать вполне падежными, так
как ацетилированные производные N-гликозидов имеют более
стабильную циклическую структуру по сравнению с незаме-
щенными N-гликозидами.
1.4. Оптическая активность
Угол вращения плоскости поляризованного света является
важной характеристикой N-гликозидов и углеводсодержащих
производных вообще. Способность к вращению плоскости
поляризованного света используется при идентификации гли-
козидов и особенно широко — при изучении кинетики реакций
'(Приложение 2). В большинстве случаев оптическая актив-
11
По аналогии с моносахаридами большинство исследова-
телей принимают, что аномер N-гликозида с положительным
.знаком угла вращения имеет a-конфигурацию с аксиальным
N-агликопом, а левовращающие — 0-конфигурацию с агли-
коном в экваториальной позиции относительно плоскости пи-
ранозного кольца. Соколовский с сотр. [30] предприняли по-
пытку изменить это положение. При этом они исходили из
предположения, что знак оптического вращения зависит от
поляризуемости атомов и атомных групп при С] (схема 26).
Полагая, что атом азота N-гликозидной связи обладает от-
носительно большей поляризуемостью, чем полуацетальные
кислородные атомы в моносахаридах и О-гликозидах, авторы
приходят к выводу, что правовращающим аномерам должна
соответствовать конформация (1) с экваториальным аглико-
ном, а левовращающим — конформация (2) с аксильиым аг-
ликоном. Проведенный анализ представляется нам недоста-
точно убедительным.
Знак оптического вращения N-гликозидов является более
надежной характеристикой при идентификации, чем абсолют-
ное значение наблюдаемого или удельного вращения. Вслед-
ствие высокой основности многих N-гликозидов оптическая
активность их в водных растворах чрезвычайно сильно зави-
сит от pH среды, а также от природы и состава смешанных
растворителей. Поэтому абсолютная величина вращения сама
по себе не является хорошим аналитическим признаком при
установлении структурных особенностей N-гликозидов и при
контроле чистоты продукта. Природа растворителя оказывает
сильное влияние на положение таутомерного (a ii0) равно-
весия. В качестве иллюстрации приведем рис. 1 и табл. 1. В
соответствии со сказанным принимается, что оптическое вра-
щение обусловлено главным образом циклическими формами.
Оптическая деятельность ациклических форм N-гликозидов
так же, как и моносахаридов, неизвестна. В разд. 3.6 будут
рассмотрены поляриметрические данные по кинетике транс-
гликозилирования, позволившие установить зависимость зна-
ка оптического вращения раскрытых диастереоизомерных
форм N-ацеталеп от пространственного расположения N-ar-
ликонов.
1.5. Инфракрасная спектроскопия
В ИК-спектрах простых N-гликозидов так же, как и в
спектрах незамещенных сахаров, различаются четыре группы
полос поглощения, обусловленные колебаниями ряда харак-
терных групп углеводных фрагментов:
1. Интенсивное поглощение в области высоких частот, при-
16
надлежащее валентным колебаниям связей О—Н, включен-
ных в сильные Н-связи (~ 3400 cai-1), и связей С—Н
~2900 саг-1)- Как правило, поглощение ОН-групп характе-
ризуется широким максимумом; в редких случаях можно на-
блюдать отдельные полосы поглощения на фоне широкого
максимума [38]. Полосы поглощения N-—Н, которые для ами-
нов находятся в области 3350—3420 см~', у N-гликозидов пе-
рекрываются интенсивным поглощением ОН-групп.
2. Область деформационных колебаний С—Н и С—О—Н
(1450—1200 см-'). В этой области перекрывается большое
число индивидуальных полос, и она наименее удобна для це-
лей идентификации и решения структурных задач. Тем не ме-
нее поглощение при 1495—1498 см~' считается характерным
признаком ряда N-алкил- и N-арилгликозидов [39—42].
3. Группа наиболее интенсивных полос, которые могут
быть отнесены к валентным колебаниям С—ОН и С—О—С
(1150—1000 см~'). Среди них полосу при 1140—1150 см-',
встречающуюся почти у всех моносахаридов, относят обычно
к асимметричным колебаниям С—ОН. Характер расщепления
этой полосы различен для разных моносахаридов, и при му-
таротации в водных растворах наблюдается монотонное изме-
нение интенсивности отдельных компонент, на основании че-
го в работе [43] сделай вывод, что основной вклад в этой
области вносят колебания гликозидного гидроксила HCi—ОН.
В спектрах N-гликозидов в этой же области также наблюда-
ются различия в положении и относительной интенсивности
полос для а- и 0-аномеров. Например, а-аномер N-ксилозида
п-хлоранилина характеризуется двумя полосами средней
(1080 са<_|) и более высокой (1065 си-1) интенсивности; для
0-аиомера характерны полосы при 1070 см~' и 1040 см~1 (вто-
рая полоса менее интенсивна) [44].
4. Группа низкочастотных полос ниже ~ 1000 см-', среди
которых можно выделить, в соответствии с исследованиями
Баркера с сотр. [45—48], пульсационные колебания углевод-
ного кольца. Указанными авторами были установлены харак-
теристические колебания, принадлежащие аномерным формам
сахаров, примеры которых даны в табл. 2. Эта область (700—
1000 саг-') представляет, пожалуй, наибольший интерес для
структурного анализа простых сахаров и производных. При
съемке твердых образцов наблюдается от 3 до 5—6 интенсив-
ных узких полос, частоты которых различны для разных са-
харов. Воспроизводимость этих полос при изменении условий
съемки (методика прессования с КВч, осаждение на подлож-
ку из AgCI, съемка в масле) вполне удовлетворительная. От-
несение этих полос встречает большие затруднения, и по это-
2 367
17
Таблица 2
Частоты максимумов поглощения в области 700—1000 см- 1
для некоторых сахаров в твердом состоянии*
Соединение V, см—1
Д-глюкоза 774 846 917
Д-галактоза 762, 794 830 960, 974
Д-ксилоза 760 900
L-арабиноза 782 840 890 945
Е-сорбоза 716 814, 880
х-Д-метилглюкозид 745 846 906
«-Д-метил галактозид 784—790 818, 870 922 965
* Спектры сняты на ИКС—14 по методике прессования с КВч.
му вопросу в литературе нет единого мнения. Наиболее веро-
ятным следует признать отнесение их к деформационным ко-
лебаниям С—Н в группах НС—ОН и частично — к пульсаци-
онным колебаниям кольца. Для пиранозных форм N-арилгли-
козидов проявляются полосы при 930—900, 891, 770—740 см~\
Таблица 3
Частоты полос поглощения а—Д-глюкозы (кристаллин.) и их отнесение
по данным ИК-спектроскопии и комбинационного рассеяния света (КРС),
Область 1500—500 см— 1 [51].
V, СМ—1 Отнесение V, сл—1 Отнесение
КРС ик КРС ик
1462 1457 СН2, деф. 1076 1076 Ct—Н + С—О—Н 1442 1054 1047 Ci—Н + С—О—Н 1433 1427 1022 1026 С—О—Н, деф. 1408 1402 С—Н (С2—Н) 998 1011 1375 1378 988 1369 914 911 С|—Н + С—О—Н 1360 С-Н (Ci—Н) 897 890 1346 840 836 Ct—Н 1335 1337 СН2, деф. 779 768 1328 748 1298 1293 721 С-С + С-О 1272 1270 С6—О-Н, Ci-0-H 704 1250 С,—Н 648 645 1224 1219 СН2, деф. 622 1206 1197 601 603 1189 С—О + С— С 581 1153 С—О + С—С 554 555 1142 542 1124 С—Н + С—О—Н 522 1115 1116 495 1104
18
Таблица 4
Частоты полос поглощения N-глюкозида (I), N—галактозида (2)
и Nl-ксилозида анилина (3) и их отнесение. Область 1600—800 см— 1 [43]
V, СМ—1 Отнесение
1 2 3
1601 1594 1600 |
1498 1497 1499 I С=С ароматич., СН2 деф.
1456 1462
1412 1395 1396
1378
1364 1350 1366 С-Н + О-Н, С—О—Н, СН2 деф.
1324
1310 1305 1306
1295 1290 1292 С—N валентные колебания
1274
1242 1236 1232 С—N+N—Н
1198 1209
1173 1174 1174 С—О + С—С, С—О С колебания углеводного
1126 1140 1156 кольца
1104 1114
1082 1086
1072 1072
1046 1054 1054 С—Н+С—О—Н, (С,—Н)
1032 1026 1040
1014 1010
988 996 996
954 949 960
922 924 С—Н + С—О -Н, пульсационные колебания
894 896 896 углеводного кольца
«31 830 830
«18 814 817
Для фуранозных форм характерны полосы при 800, 862 и
875 см-1 [49—50].
В табл. 3 для иллюстрации приведены частоты полос по-
глощения а-аномерной формы глюкозы в твердом состоянии,
измеренные методами ИК-спектроскопии и комбинационное»
рассеяния света [51]. В этой же таблице дано отнесение не-
которых полос к атомным группировкам и химическим свя-
зям. Примеры колебательных спектров N-арилгликозидов да-
ны в табл. 4. В диапазоне частот 1600—500 см-1 перекрывает-
ся большое число полос поглощения; интерпретация колеба-
тельного спектра в этой области исключительно сложна, по-
этому к вопросу о принадлежности отдельных полос к хими-
ческим связям и атомным группам (включая и колебания уг-
леводного кольца в целом) следует подходить с большой ос-
торожностью.
В спектрах N-гликозилмочевип характерной является по-
лоса при 1560 см-1, которая по аналогии с монозамещенными
амидами должна быть отнесена к деформационным колеба-
ниям N—Н (полоса «Амид-2») [52, 53]. Она может служить
надежным аналитическим признаком при идентификации
N-гликозилмочевин [54]. Полосы поглощения самой мочевины
при 1625 см~1 (деформационные колебания N—Н) и при
1680 см~1 (валентные колебания С = О, «Амид-1») суще-
ственным образом не изменяются при образовании
N-гликозидной связи, что позволяет сделать вывод об
отсутствии заметного внутримолекулярного взаимодействия
типа С = О . . . Н—О—С3 (С2), которое можно было бы ожи-
дать по чисто структурным соображениям.
Анализ ИК-спектров некоторых представителей N-арпл- и
N-алкилгликозидов, полученных по методике прессования с
КВг, позволяет сделать следующие основные выводы о строе-
нии и спектральных характеристиках этого класса произ-
водных:
— в кристаллическом состоянии все изученные до сих
пор N-гликозиды находятся в циклической (преимущест-
венно пиранозной, как наиболее устойчивой) форме;
ациклических структур типа оснований Шиффа не най-
дено;
— аномерные формы гликозидов достаточно отчетли-
во различаются по поглощению в низкочастотной обла-
сти за счет деформационных колебаний связи Ci—Н и
пульсационных колебаний углеводного кольца;
— имеется ряд спектральных признаков, позволяю-
щих различать пиранозные и фуранозные структуры, од-
нако опытных данных в этой области пока еще мало;
— для О-замещенных N-гликозидов хорошей харак-
теристикой может служить интенсивное поглощение в
области валентных колебаний связи N—Н, которое ис-
чезает при замещении водорода на алкоксильную группу.
1.6. ЯМР-спектроскопия
Радиоспектроскопия является одним из наиболее эффек-
тивных методов исследования пространственной структуры са-
харов и их производных [55, 56]. При изучении структуры са-
харов до последнего времени использовался только протон-
ный магнитный резонанс (ПМР). Первые исследования ПМР
были выполнены Лемье с сотр. [57] на примере ацетилиро-
ванных сахаров [55, 58]). Спектры ПМР незамещенных мо-
20
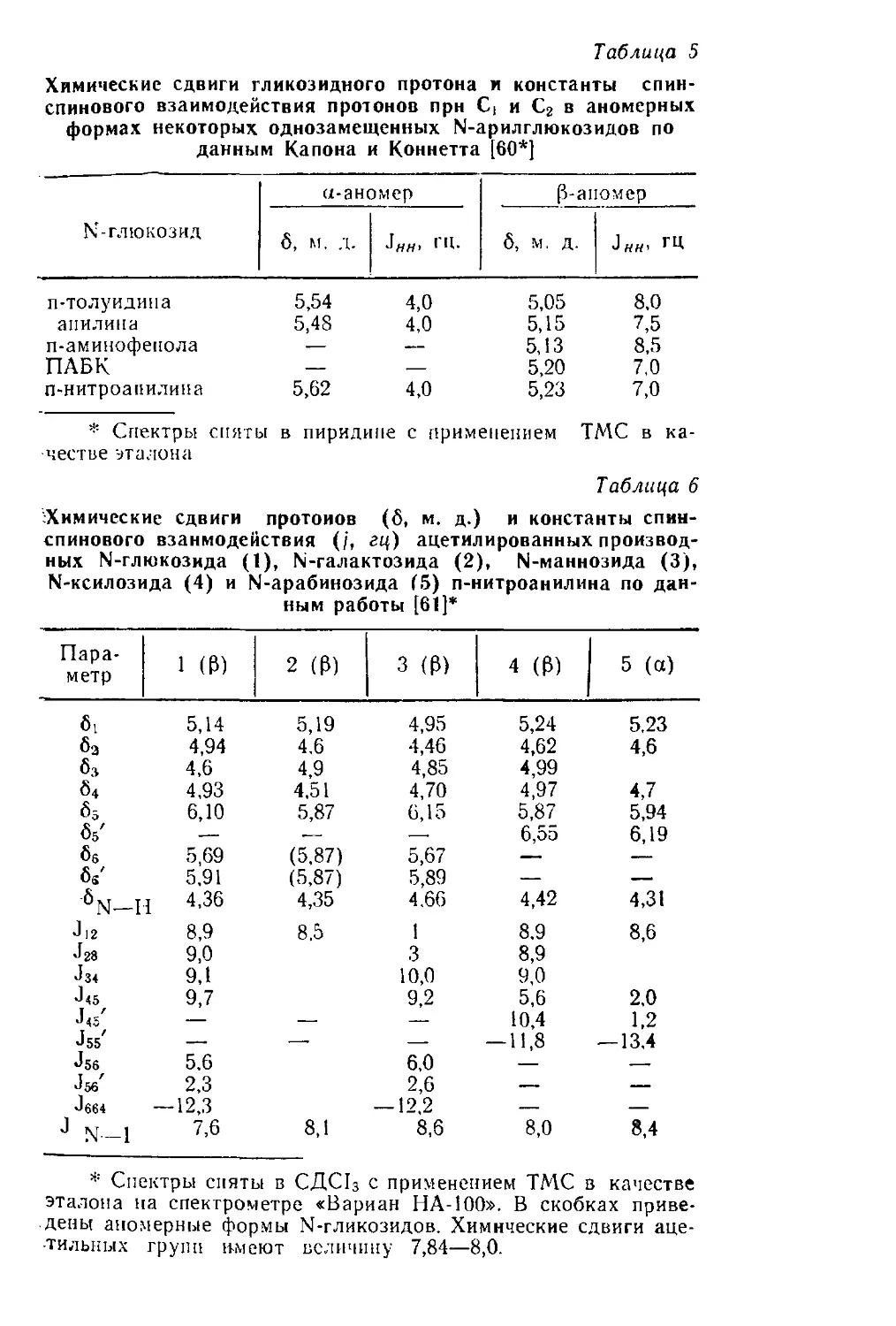
Таблица 5
Химические сдвиги гликозидного протона и константы спин-
спинового взаимодействия протонов при С, и С2 в аномерных
формах некоторых однозамещенных N-арилглюкозидов по
данным Капона и Коннетта [60*]
N-глюкозид а-аномер р-апомер
б, м. д. гц. б, м. д. J««, ru
п-толуидина 5,54 4,0 5,05 8,0
анилина 5,48 4,0 5,15 7,5
п-аминофенола — — 5,13 8,5
ПАБК — — 5,20 7.0
п-нитроапилина 5,62 4,0 5,23 7,0
* Спектры сняты в пиридине с применением ТМС в ка-
честве эталона
Таблица 6
Химические сдвиги протонов (б, м. д.) и константы спин-
спинового взаимодействия (/, гц) ацетилированных производ-
ных N-глюкозида (1), N-галактозида (2), N-маннозида (3),
N-ксилозида (4) и N-арабинозида (5) п-нитроанилина по дан-
ным работы [61]*
Пара- метр 1 (₽) 2 (₽) 3 (₽) 4 (₽) 5 (a)
б. 5,14 5,19 4,95 5,24 5,23
б3 4,94 4.6 4,46 4,62 4,6
6.3 4,6 4,9 4,85 4,99
б4 4,93 4,51 4,70 4,97 4,7
бз 6,10 5,87 6,15 5,87 5,94
б5' — — 6,55 6,19
бб 5,69 (5,87) 5,67 — —
б/ 5,91 (5,87) 5,89 — —
6N—И 4'36 4,35 4.66 4,42 4,31
J12 8,9 8.5 1 8,9 8,6
З38 9,0 3 8,9
J34 9,1 10,0 9,0
J45 9,7 9,2 5,6 2,0
— — — 10,4 1,2
J бб' —. —— — — 11,8 — 13.4
Jss 5,6 6.0 — —.
Ьб' 2,3 2,6 —
J 664 — 12,3 — 12,2 — —
J N—1 7,6 8,1 8,6 8,0 8,4
* Спектры сняты в СДС13 с применением ТМС в качестве
эталона на спектрометре «Вариан НА-100». В скобках приве-
дены аномерные формы N-гликозидов. Химические сдвиги аце-
тильных групп имеют величину 7,84—8,0.
носахаридов в водных растворах рассмотрены в работе Ленца
и Хеешена [59]. Спектры ПМР N-арилгликозидов обследова-
ны Капоном и Коннеттом [60]. Результаты выполненных ис-
следований можно суммировать следующим образом:
— в спектрах простых сахаров достаточно отчетливо
различаются сигналы ПМР трех типов протонов — гли-
козидного Н—Ci, протонов при углеродных атомах
Сг—С4 (Сг>) и метиленовой группы.
— наименее экранированный гликозидный протон
дает сигнал с наибольшим химическим сдвигом б в сто-
рону более слабого Н0-поля (если за нуль шкалы при-
нять сигнал протонов тетраметилсилана ТМС); сигнал
этого протона в а-аномерах наблюдается в более слабом
поле, чем в p-аномере; аналогичная картина наблюдает-
ся и в N-гликозидах (табл. 5);
— сигналы экваториальных протонов располагаются
всегда в более слабом поле, чем сигналы аксиальных
протонов; различие между ними не превышает 0,5 —
0,6 м. д.;
— константа спин-спинового взаимодействия (JHfl
между протонами при Ct и Сг у p-аномера приблизитель-
но вдвое больше, чем у а-аномера; зависимость констан-
ты JHH от «двугранного» угла <р между связями Н—Ci и
Н—Сг в ньюменовской проекции выражается прибли-
женной формулой Карплуса:
/Н!! = 8,5 Cos2<p — 0,28 при 0°<<р<90°
(1)
7НН = 9,5 Cos2<p — 0,28 при 90°<ф<180°
(формулы Карплуса справедливы вообще для вициналь-
ных протонов групп Н—С—С—Н и ’могут быть использо-
ваны для приближенной оценки пространственной струк-
туры молекул [56]);
— замещение ОН-групп в сахарах и N-арилглико-
зидах усложняет спектр ПМР и затрудняет его расшиф-
ровку; тем не менее при достаточно высокой разрешаю-
щей способности спектрометра удается идентифициро-
вать сигналы всех протонов углеводного кольца и оце-
нить константы спин-спинового взаимодействия вици-
нальных и геминальных протонов (табл. 6).
1.7. Хроматография
Бумажная и тонкослойная хроматографии (ТСХ) являют-
ся чрезвычайно распространенными методами исследования
углеводсодержащих систем, в том числе и разнообразных про-
22
изводных по С[. Эти методы широко используются как для
качественной идентификации сахаров, так н для количествен-
ных определений. С помощью хроматографии может быть
также изучена кинетика реакций сахаров и их производных
(как это было сделано, например, Богнаром с сотр. на при-
мере N-трансгликозилировапия; см. разд. 3.5) и динамика на-
копления и расходования промежуточных соединений, обла-
дающих достаточно высокой устойчивостью в условиях хро-
матографирования (см. гл. 6). Наконец, хроматография при-
меняется для препаративного разделения моно-, ди- и олиго-
сахаридов и их производных. Сведения о результатах хрома-
тографических исследований простых сахаров опубликованы
в обзорах и монографиях [3, 62—63]; соответствующего об-
зора по свойствам N-гликозидов и условиях их хроматогра-
фирования в литературе нет, за исключением ряда работ по-
следних лет по хроматографии фенилгидразонов (например,
[64]).
При разделении простых сахаров хорошие результаты по-
казывают слои гипса, кизельгура, силикагеля, их смеси а так-
же порошковая хлопковая целлюлоза, осажденная из водной
суспензии [33]. Те же сорбенты могут быть использованы и
при идентификации N-гликозидов [65]. Как правило, под-
вижность сахаров (мерой которой является величина tff)
уменьшается по мере увеличения длины углеводной цепи:
пентозы>гексозы>дисахариды>трисахариды > олигосахари-
ды. При удачном подборе системы растворителей можно до-
статочно уверенно разделить бинарные и более сложные сме-
Таблица 7
Величины Rf моносахаридов [66—69] и N-гликозидов м-нитро-
аннлина [65], полученных методом ТСХ на слоях различных
сорбентов
Моносахарид Значения /?;
1 2 3 4
Глюкоза 0,17 0,27 (0,77) 1,00
Галактоза 0,18 0,30 0.40 0,90
Манноза 0,23 0,33 0.62 1,09
Ксилоза 0,29 0.58 0,88 1.25
Арабиноза 0.28 0,63 0,75 1,11
Рибоза 0,49 0,79 0,86 : 1,42
1 — кизельгур/гнпс; этнлацетат—изопроианол—вода; 2—
гипс; хлороформ—метанол (19:2); 3 — гипс; хлороформ—ме-
танол (19:3); 4 — целлюлоза, этилацетат—пиридин—вода
(2:1:2); приведены относительные значения подвижностей но
отношению к глюкозе.
.23
Продолжение таблицы 7
N-гликозид м-нитроанълина Значения Rj
1 2 3 4
А. Глюкозид 0,30 0,40 0,60 0,65
Галактозид 0,34 0,54 0,72 0,68
Кснлозид 0,43 — 0,88 0,85
Б, Глюкозид 0,29 0,71 0,30 0,80
Г алактозид 0,35 0,68 0,45 0,83
Ксилозид 0,49 — 0,45 0,72
А — кизельгур/гипс; Б — силикагель КСК/гипс. Системы:
1 — ацетон—вода (4:1); 2 — хлороформ—метанол (19.2);
3 — эфир—толуол (2:1); 4 — ацетон—бутанол—вода (7:2:1).
си незамещенных моносахаридов. Во многих случаях наблю-
дается следующий ряд подвижности моносахаридов: глюко-
за < галактоза < манноза < ксилоза < арабиноза < рибоза,
примером чего могут служить данные, приведенные в табл. 7.
Аналогичная картина часто отмечается и для N-гликозидов с
простыми агликонами.
При работе с N-арилгликозидами удобной является систе-
ма бутанол—пиридин—вода (6:4:3). Однако всегда следует
иметь в виду возможность распада исследуемых гликозидов
(по Ci—N-связи) в процессе длительного разделения метода-
ми бумажной хроматографии. Обычно в этих случаях, наряду
с аномерными формами N-арилгликозидов, на хроматограмме
обнаруживаются интенсивные пятна соответствующих моноса-
харидов. Вообще же надо отметить, что в области хромато-
графирования N-гликозидов предстоит еще большая исследо-
вательская работа по поиску оптимальных условий разделе-
ния и идентификации этих соединений. В частности, следует
обратить внимание на разработку метода газо-жидкостной
хроматографии, который в настоящее время часто исполь-
зуется при анализе смесей моносахаридов и О-гликозидов
[70, 71]. В основе этого метода лежит перевод моносахаридов
в триметилсилильные производные. Этим методом Кадунсу
[72] удалось разделить и идентифицировать смеси некоторых
моносахаридов и N-арилгликозидов.
1.8. Области практического использования
N-гликозидов
Накопленные к настоящему времени сведения о свойствах
N-гликозидов позволяют говорить о возможности использова-
ния их в следующих отраслях [73—78]:
24
Таблица t
Некоторые аспекты практического применения N-гликозндов [73—78]
Соединение Область применения
N-глюкозид 2-диметил—N-диацетил колхицина » N-диацетилколхицнна 2-амино—4,5-диметилаиилипа » Д-(—)трео-1-пара-нит- рофеннл-аминопропандиола—1,3 N-ацетил сульфанил а ми да » сульфадемизина » стрептоцида (а—аномер) » стрептомицина » сульфазола » сульфапнридина » сульфагуанидина N-гликозид ПАСК » аминофеназина » тиосемикарбазида » этил-пара-аминобензоата N-, N-диглюкозид сульфазола » сульгина » сульфидина » стрептоцида N-гликозиды а-нафтиламина—4- сульфокислоты N-глюкоуронид N-ксилозид додециламина N-лактознд додециламина N-глюкозил мочевина N-гликозил мочевины N-глнкозпды А-алкилмочсвин N-рибозид нитроксилидина Аналогичные по действию колхи- цину, но менее токсичны н раст- воримы в воде Промежуточный продукт при син- тезе витамина В|2 Для разделения рацемических сме- сей при синтезе антибиотика «хлорамфеникола» Подавляют жизнедеятельность болезнетворных микроорганизмов; менее токсичны, более растворимы в воде, чем исходные N-агликоны Проявляет противотуберку- лезную активность Промежуточный продукт для синтеза препаратов с туберкулостатическим дей- ствием В ветеринарии против ту- беркулеза животных Компонент кремов против загара Подавляют активность хо- лерных и холероподобных вибрионов При лечении и предупрежде- нии кровотечений Обезвреживающее действие в животных организмах , Защитные оболочки для таблеток Увеличивает прочность бу- маги в мокром состоянии Для синтеза нуклеозидов и их аналогов Стабилизация соков, вита- минных спиртов, джемов Обладают свойствами ПАВ Промежуточный продукт при синтезе рибофлавина
25
Глава 2
ВОПРОСЫ СТРОЕНИЯ
И РЕАКЦИОННОЙ СПОСОБНОСТИ МОНОСАХАРИДОВ
Прежде чем начать обсуждение вопросов строения и ре-
акционной способности N-гликозидов, рассмотрим некоторые
важнейшие свойства моносахаридов. Это целесообразно по
двум причинам. Во-первых, поведение моносахаридов опреде-
ляется некоторыми специфическими особенностями, которые
следовало бы учитывать при анализе механизмов реакций по
гликозидному центру. Они выдвигают перед исследователем
ряд крупных вопросов, решение которых имеет значение для
развития не только химии и физико-химии углеводов, но и ор-
ганической химии в целом. К числу этих вопросов следует от-
нести влияние на кинетику, термодинамику и механизм реак-
ций по Ci таутомерных превращений сахаров, пространствен-
ного расположения заместителей при асимметрических цент-
рах, конформации углеводного кольца и водородных связей в
незамещенных сахарах. Естественно, эти же вопросы могут
быть поставлены и при рассмотрении реакций N-гликозидов.
Отсюда следует и вторая причина — рассмотрение поведения
моносахаридов с точки зрения возможности проведения ана-
логии в свойствах N-гликозидов и их предшественников —
моносахаридов.
2.1. Таутомерия моносахаридов («мутаротация»)
Реакционная способность гликозидного центра в простых
незамещенных сахарах во многом зависит от лабильности
полуацетальной группировки
—НС5—О—НС1—ОН или —НС4—О—НС1—ОН,
которая сочетает в себе одновременно и кислотные, и основ-
ные функции (рис. 2). Поэтому реакции по Ci подвержены как
28
Y—4/ он
-Н^-ОН^* -НС-O’Ка=110'13
-нс-о-нс1он*н+^-нс-6н-нс1он, Кв=8 Ю'17
Рис. 2. а — общая схема мутаротации моносахаридов в вод-
ных растворах;
б — протолитические превращения и соответствующие кон-
станты диссоциации гликозидного гидроксила (Ка) и цикли-
ческого кислорода (Кв) глюкозы. Константы диссоциации рас-
считаны по данным зависимости эффективной константы ско-
рости мутаротации глюкозы от кислотности среды;
в — зависимость константы скорости мутаротации глюкозы от
pH водных безбуферных растворов (поляриметрический метод).
29
'основному, так и кислотному катализу. По той же причине
моносахариды легко взаимодействуют с типичным амфоли-
том — водой, образуя в водных растворах раскрытые альде-
гидные и гидратные формы. С раскрытием полуацетального
кислородного мостика связано таутомерное превращение мо-
носахаридов, играющее важную роль во многих реакциях ну-
клеофильного присоединения по гликозидному центру [4, 85].
С точки зрения конформационных представлений [86—88],
результатом этой перегруппировки является переход глико-
зидного гидроксила из аксиального в экваториальное поло-
жение.
Кроме четырех указанных форм в растворах мутаротнрую-
щих моносахаридов имеются фуранозные структуры, наличие
которых доказано лишь в последние годы с помощью ЯМР-
спектроскопии [89]. В равновесных водных растворах галак-
тозы концентрация фуранозных форм не превышает 1—2%.
В растворах глюкозы, ксилозы, маннозы, арабинозы и ликсо-
зы фуранозные формы радиоспектроскоппчески не обнаружи-
ваются. Поэтому при описании кинетики мутаротации можно
использовать более простые схемы с одним или двумя проме-
жуточными продуктами:
К1 «3
Га-<-=^ Г? (а)
«2 *4
(ср (С2) (С,)
К1 к3
Г« « —— (б)
К2 t| «4
К'5 |<[ К®
Г2
(Г и Г, — аномерные формы моносахарида, Га и Гг —
а р
соответственно альдегидная и гидратная формы).
В рамках формально-кинетической теории [90—92] мута-
ротация может быть описана решением системы линейных
дифференциальных уравнений, например, для схемы (а) —
dC\/dt = K.i,C2—K\Ci ]
dC2ldt = K\C[-\-K3C—(K2-\-Ki)C2\ (I)
dC2ldt = K2C2—к^С-з 1
Эта задача решена в ранних работах по кинетике мутарота-
ции глюкозы [93—96]. Для оценки констант скоростей при-
нимался ряд произвольных допущении (например, равенство
30
•констант К\ = к3 и к2=К4 [96]). Точное решение системы (1)
приводит к громоздким формулам, практическое использова-
ние которых встречает принципиальные затруднения. Для
приближенной оценки констант скоростей можно использовать
метод квазистационарных концентраций [97]. Применение
данного метода к кинетике мутаротации глюкозы [98] пока-
зало, что в нейтральных безбуферных растворах константы
скорости циклизации промежуточного продукта (к2 и к3) при-
близительно на три порядка выше констант скоростей его
образования (к\ и к4). Эта оценка согласуется с данными, по-
лученными в работе [99] полярографическим методом. Таким
образом, малое содержание ациклических форм мутаротирую-
щих моносахаридов является следствием высокой относитель-
ной скорости их циклизации по сравнению со скоростью обра-
зования.
В некоторых случаях образование промежуточных продук-
тов обнаруживается по аномальному изменению оптических
свойств растворов мутаротирующих моносахаридов в началь-
ный период реакции [94—96, 100—102]. В этом отношении
•особенно показательна мутаротация рибозы [94]. Аномалии
усиливаются при исследовании мутаротации в спиртовых рас-
творах, в которых реакция протекает медленнее, чем в вод-
ных [100, 103—107]. Это обстоятельство было использовано в
работе [108] для оценки механизма мутаротации ряда моно-
-сахаридов с помощью регрессионного анализа (см. также
[109—111]).
Наблюдаемая константа скорости может быть представ-
лена в виде суммы констант скоростей каталитических актов:
к=Кнон[Н2О] + кн [Н+] + коп [ОН-], (2)
где кнон, кн, кон — константы каталитического действия во-
ды, Н3О+ и ОН~. Пренебрегая изменением концентрации во-
ды, т. е. полагая к|10Н [Н2О] = к0 и используя «ионное произ-
ведение воды» [Н~] [ОН-]), константу скорости мута-
ротации можно выразить через одну переменную, например
[Н+):
к = Ко +ЛГИ [Н+] + Ко,, /Сг/[Н']> (3)
или
-2.3рН 2,ЗрН
к — Ко = К'н -е + к0!ге (4)
С увеличением pH первый член уравнения (4), описываю-
щий специфический кислотный катализ, уменьшается, а вто-
рой, характеризующий основной катализ, возрастает. Зависи-
мость константы скорости от pH выражается «цепной лнни-
31
ей» с минимумом, положение которого определяется отноше-
нием констант каталитического действия [112—117] (см.
рис. 2):
|Н]% = #х-«он/кн. (5)
Таким образом, по экспериментально найденному минимуму
можно судить об относительных значениях каталитических
констант. В общем случае при кОн>кн Р^опт- <7, при к он<
<кн pH опт- >7, наконец, при кОн = «н рНолгот. =7.
Каталитические константы, входящие в уравнение (2), впер-
вые были определены Осака [112] и Хэдсоном [ИЗ] па приме-
ре мутаротации глюкозы: хн =0,258 и хон=9750 л!моль-мин.
(25°С). В дальнейшем эти величины неоднократно нахо-
дились разными авторами [117—122]. Обращают на себя вни-
мание резкие колебания каталитической константы /сон, на-
пример:
22 300 [112], 14 300 [117], 13 000 [118],
14 600 [119], 4 300 [120], 8 890 [121].
Было отмечено, что константа возрастает с увеличением
концентрации глюкозы и достигает максимума при концент-
рации 0,6 моль!л (25°С). Это дало основание Лосу и Симпсо-
ну [122] предположить, что в щелочных растворах мутарота-
ция глюкозы катализируется не только ионами ОН-, но и от-
рицательно заряженными «глюкозат-ионами», в соответствии
с чем уравнение для наблюдаемой константы скорости сле-
дует представить в следующем виде:
*=*о+кн|Н+]Ч-кон[ОН-]+ка [Г-а ]+^ ], (6^
где /<а и к^ — константы каталитического действия глюкозат-
ионов а- и p-форм глюкозы, [г- ] [Г-р] — концентрации
аномерных ионов. Каталитическая активность ионов глю-
козы на порядок меньше активности ионов ОН-.
Мутаротация моносахаридов как модельная реакция яви-
лась предметом ряда исследований, направленных на отыска-
ние закономерностей гомогенного кислотно-основного катали-
за. Одной из первых следует назвать работу Бренстеда [117]
по исследованию каталитической активности одноосновных
карбонатных кислот при мутаротации глюкозы, в которой
впервые было установлено эмпирическое соотношение между
константами каталитического действия и константой диссо-
циации катализирующих агентов, получившее в литературе
название уравнения Бренстеда.
В соответствии с общими представлениями теории кислот-
но-основного катализа механизм мутаротации можно предста-
32
вить следующим образом. В кислой среде имеет место про-
тонизация циклического кислородного атома (ОццКА.) с по-
следующим переносом протона от гликозидного гидроксила к
молекуле воды и одновременным разрывом связи Ci—Оцикл.
В результате образуется раскрытая альдегидная форма саха-
ра. В щелочной среде происходит образование отрицательного
гликозильного иона в результате отрыва гликозидного прото-
на ионами ОН”. Далее следует перенос протона от молекулы
воды на циклический кислород с одновременным разрывом
связи Ci—Оцикл. В обоих случаях лимитирующей стадией
является раскрытие пиранозного или фуранозного кольца, о
чем свидетельствует соотношение констант скоростей образо-
вания и расходования ациклических форм. Механизм образо-
вания альдегидной формы в нейтральных растворах можно
представить схемой с циклическим переносом протонов от
гликозидного гидроксила к молекуле воды и от последней —
к циклическому кислородному атому.
Мутаротацию по механизму прямого нуклеофильного за-
мещения без раскрытия пиранозного звена
I !
он--г с,н-он но-с,н+ он-
I I
следует признать маловероятной, так как было показано, что
скорость изотопного обмена О18 между молекулами воды и
гликозидным гидроксилом в широкой области pH значитель-
но меньше скорости мутаротации [123].
Можно было предполагать, что циклический перенос про-
тонов, необходимый для раскрытия пиранозного звена, проис-
ходит также при взаимодействии моносахарида с би- и поли-
фуикциональными молекулами, содержащими кислотные и
основные центры. Одной из первых в этом направлении яви-
лась работа Лоури и Фолкнера [124], в которой установлено,
что в смеси крезола и пиридина мутаротация тетраметилглю-
козы протекает значительно быстрее, чем в каждом из этих
растворителей в отдельности. На основании этих наблюдений
сделан вывод о возможности раскрытия пиранозного звена в
результате согласованного (тримолекулярного) взаимодейст-
вия тетраметилглюкозы с кислотными и основными компонен-
тами. Позднее Свэном и Брауном [125] показано, что 2-окси-
пиридин проявляет исключительно высокую каталитическую
активность при мутаротации тетраметилглюкозы, на несколь-
КР порядков превышающую активность смешанных растворов
фенола и пиридина.
3 367 33
Эта работа вызвала большой интерес у ученых, проводив-
ших: исследования в области катализа, строения и реакцион-
ной способности. Во многих монографиях и обзорах по орга-
ническому катализу схема мутаротации под действием 2-окси-
пиридина приводится как иллюстрация исключительно высо-
кой эффективности синхронных механизмов в органической
химии. Однако последующие попытки подбора полифуикцио-
нальиых агентов с целью установления связи между молеку-
лярным строением и их каталитическим действием на мутаро-
тацию, не привели к значительным успехам [126—129]. В
этом отношении 2-оксипиридин остается непревзойденным ка-
талитическим агентом.
Подводя итог краткому анализу явления мутаротации, от-
метим следующие основные моменты. Прежде всего, рассмот-
рение большого литературного материала (а работ по мутаро-
тации уже значительно более трехсот) позволяет высказать
утверждение о том, что на уровне современных эксперимен-
тальных средств механизм этой реакции можно считать до-
казанным. Предпринимавшиеся в последние годы попытки бо-
лее детального изучения [130—138], по существу, не внесли
принципиально нового в установленный ранее механизм. Изу-
чение мутаротации внесло в органическую химию представле-
ние о «циклическом» переносе протонов, что, в свою очередь,
стимулировало развитие идей о полифункциопалыюм взаимо-
действии молекул с ферментными системами [139—142]. Эта
модель представляет интерес и для каталитической химии.
Было бы важно знать, в какой мере распространены эти ме-
ханизмы в органической химии и какими свойствами должны
обладать системы, способные катализировать циклический
перенос протонов. Для химии и физико-химии углеводов зна-
чение таутомерии моносахаридов состоит прежде всего в том,
что в реакциях по Ci могут принимать участие как цикличе-
ские, так и ациклические формы. От этого зависит стереохими-
ческая направленность реакций. Если в реакциях нуклеофиль-
ного присоединения и замещения участвуют преимущественно
ациклические структуры, раскрытие углеводного кольца в
квазиравиовесных стадиях следует рассматривать как про-
цесс, кинетически сопряженный с основной реакцией по Сь
2.2. О конформационной устойчивости сахаров
По данным Исбелла и Пигмана [143], относительная ско-
рость мутаротации в ряду моносахаридов возрастает в поряд-
ке, указанном в табл. 9. Примечательной особенностью моно-
34
сахаридов является то, что реакционная способность их в
различных нуклеофильных реакциях гликозидного центра
возрастает в той же последовательности, как и при мутарота-
ции. Объяснить это явление довольно трудно, и насколько нам
известно, данный вопрос в литературе не решен. Принято свя-
зывать различия в поведении однотипных сахаров с так назы-
ваемой конформационной неустойчивостью углеводного коль-
ца [4, 86—88].
Таблица 9
Относительные скорости мутаротации моносахаридов по Исбеллу и
Питману [143] и конфигурации ОН-групп для двух кресловидных
конформаций пиранозного кольца
Моносахарид Относит, скорость С 1 1 с
с2 С3 С2 С3
Д-глюкоза 1,0 С е е а а а
Д-галактоза 1.3 е е а а а е
Д-манноза 2,75 а е е е а а
Д-гулоза 3,0 е а а а е с
Д-ксилоза 3,2 с е е а а а
Д-талоза 4,1 а е а е а е
L-арабиноза 4,75 е е а а а е
Д-рибоза 7,8 е а е а е а
Д-лнксоза 9,2 а е с е а а
Д-альтроза 12,5 а а е е е а
Для количественной оценки энергетической устойчивости в
литературе введено понятие конформационной энергии [144],
мерой которой является изменение стандартной свободной
энергии при обратимом переходе одной из конформаций в
другую, обладающую минимумом энергии (—Дб^). Для од-
нозамещенных производных циклогексана величина Дб^
может быть определена по равновесию процесса, при кото-
ром имеет место переход заместителя из аксиального поло-
жения в экваториальное без разрыва кольца:
В таблицах Дж. Хирша [144] опубликованы данные но
более чем для сорока производных циклогексана с раз-
35
личными заместителями, из которых можно видеть, что AG#
изменяется в пределах от 0,15 до 3,0 ккал!моль.
Определение конформационной энергии в ряду сахаров
представляет собой несравненно более сложную задачу. До-
статочно вспомнить, что для пираноз возможно существова-
ние восьми конформаций, различающихся положением коль-
цевого кислородного атома — двух кресловидных (С) и ше-
сти ваннообразных (В). Показано, что в растворах конформа-
ционное равновесие сдвинуто в сторону конформаций кресла
[145—148]. Принято считать, что причиной существования ус-
тойчивых и неустойчивых конформаций является взаимное
отталкивание непосредственно не связанных атомов и атом-
ных групп. Так как такое отталкивание наиболее сильно про-
является у аксиальных заместителей, то, как правило, та кон-
формация полагается наиболее устойчивой, которая имеет
наименьшее число аксиальных заместителей в углеводном
кольце.
Ограниченность этого правила становится очевидной при
самом беглом рассмотрении свойств однотипных сахаров и их
производных. Поэтому рядом авторов для количественной
оценки конформационной устойчивости предложены конкрет-
ные схемы расчета, учитывающие разные внутримолекуляр-
ные эффекты отталкивания соседних заместителей (Ривс
[149], Келли [150], Лемье [151]). Эти схемы расчета основа-
ны па использовании некоторых формальных величин (фак-
торов неустойчивости) или свободных энергий различных
стереоизомерных пар заместителей, включая и заместитель
при гликозидном центре. В целом подобного рода расчеты не-
плохо согласуются с экспериментальными оценками конфор-
мационного равновесия в некоторых рядах сахаров.
Для оценки относительной неустойчивости конформаций
типа кресла (С 1 и 1 С) Ривс [149] вводит в рассмотрение
три фактора, от которых зависит увеличение свободной энер-
гии (в скобках указаны относительные безразмерные оценки):
— молекула имеет атомы или атомные группы, от-
личные от атома водорода, находящиеся в аксиальном
положении (1,0);
— одновременно присутствует аксиальный гидроксил
при С[ и аксиальная группа СН2—ОН при С5 (0,5);
— аксиальный заместитель (ОН, OR) при С2 располо-
жен таким образом, что связь С2—О делит угол между
связями Ci—О пополам; в этом случае заместитель при
Ci нахдоится в экваториальной позиции — «Д2-эф-
фект» (2,5).
Предполагая, что указанные факторы являются адднтив-
за
ними и суммируя их для каждой конформации, Ривс показал,
какая из форм должна быть более устойчивой. Оценки Ривса
были несколько улучшены Келлн [150], и расчет конформа-
ций с учетом исправленных факторов приводит к лучшему
соответствию между данными эксперимента и расчета (табл.
10).
Таблица 10
Оценка относительной устойчивости конформаций кресла CI
и IC по Ривсу [149] и относительные свободные энергии этих
конформаций, вычисленные Энжиалом [151] в виде суммы
энергий взаимодействия отдельных групп с учетом аномерного
и Д2—эффектов
Моносахарид Конформация Энергия взаимо- действия ( ккал/моль)
найден- ная предска- занная
С 1 1 1 С
«-глюкоза 1С IC 2.3 7,1
Р-глюкоза IC 1С 1,95 7,9
а-галактоза IC CI 3,2 5,9
Р-галактоза 1С fC 2.85 6,7
«-манноза С1 IC, Cl 2,85 5,1
Р-манноза С1 IC. Cl 3,3 6.7
а-аллоза Cl 4.2 6.65
'Р-аллоза Ci 2,85 6,45
а-альтроза 1С, С1 IC, Cl 4,4 4.3
,Р-альтроза IC, С1 IC, Cl 3,85 4,9
а-гулоза С1 IC. Cl 4.75 5,1
.р-гулоза Cl 3.4 4,9
а-идоза 1С IC 3,75
Р-идоза IC, Cl 5.4 4.35
а-рнбоза С1 Cl 3.85 4.85
^рибоза С1 Cl 2,5 4,4
а-арабиноза 1С IC 4.05 2.5
Р-арабиноза )С !C 3,5 2,85
а-ксилоза С1 Cl 1,95 5,05
р-ксилоза CI Cl 1,6 5,6
а-талоза IC, Cl 4,75 4.9
Р-талоза IC, Cl 5,2 6,5
а-ликсоза 1С, С1 IC, Cl 2.5 3,05
Р-ликсоза 1С, С1 IC, Cl 2,95 4,4
Энжиал и Лемье [151], исходя из данных по равновесию
в реакциях эпимеризации инозитов и их ацетатов, а также
Лемье и Чжу [152] на основании данных по апомеризации
ацетилированных сахаров в смеси уксусная кислота + уксус-
ный ангидрид (1:1) в присутствии НСЮ« предложили схему
расчета с учетом энергий взаимодействия между следующим^
группами (в скобках даны оценки в ккал/моль):
— аксиальная ацетоксигруппа — атом водорода (0,18);
— две син-аксиальные ацетоксигруппы (2,08);
37
— ацетоксигруппы при соседних углеродных атомах (0,55).
Заниженное значение первой оценки по сравнению с вели-
чиной 0,45 ккал/моль для ОН-группы в воде авторы объясня-
ют вкладом «аномерного» эффекта, за счет которого эквато-
риальная позиция при Ci становится менее устойчивой по
сравнению с аксиальной. Сущность «аномерного» эффекта ио
Эдварду заключается в дипольдипольном взаимодействии
между связями Ci—Оцчкл- и С!—OR. Если заместитель нахо-
дится в экваториальной позиции, то имеет место электроста-
тическое отталкивание гетероатомов, располагающих неподе-
ленпыми парами электронов [153].
2.3. Водородная связь и реакционная способность
Углеводы как полигидроксильные соединения склонны к
образованию внутри- и межмолекулярных связей, существова-
ние которых в простых сахарах подтверждено данными опти-
ческой спектроскопии и радиоспектроскопии [154—158]. Для
моно- и дисахаридов расщепление полосы валентных колеба-
ний гидроксильных групп весьма специфично, что позволяет
сделать вывод о наличии индивидуальной системы внутримо-
лекулярных водородных связей у простых углеводов [38]. В
данном случае речь идет о кристаллических состояниях са-
харов. Рентгеноструктурные исследования [159—163] показа-
ли, что в кристаллической a-форме Д-глюкозы все гидрок-
сильные группы вовлечены в межмолекулярпые Н-связи. При
эт.ом молекулы координируются таким образом, что глико-
зидный гидроксил одной молекулы оказывается связанным
Н-связыо с циклическим кислородом другой молекулы. Кста-
ти, такое расположение благоприятно для циклического пе-
реноса протонов, поэтому, как только амплитуда тепловых
колебаний атомов превысит некоторое критическое значение,
может произойти раскрытие пиранозного кольца («термиче-
ская мутаротацня). Эта возможность подтверждается опыта-
ми по мутаротации глюкозы вблизи точки плавления [130].
Показано, что при содержании остаточной воды в образце
0,17% равновесие мутаротации ([Г ]/[Г<] =0,792) достигает-
ся за 15 минут при температуре 151°С.
Используя молекулярные модели, нетрудно установить, что
при благоприятной конформации пиранозного звена водород-
ные связи могут образоваться между всеми гидроксильными
группами с учетом также и циклического кислородного атома.
При рассмотрении молекул в растворенном состоянии необхо-
димо учитывать взаимодействие гидроксильных групп с мо-
лекулами сольватной оболочки,
38
Вопрос о влиянии водородных связей на реакционную спо-
собность органических соединений вообще и углеводов в осо-
бенности сложен. Как показывают экспериментальные дан-
ные [164—167], образование водородных связей может при-
водить как к ускорению, так и замедлению реакции. Однако
до настоящего времени не разработаны основы теории влия-
ния водородных связей на реакционную способность, и на-
блюдаемые эффекты носят в значительной степени иллюстра-
тивный характер.
Из самых общих соображений о механизмах кислотно-ос-
новного катализа следует, что включение реакционных цент-
ров сахаров в водородные связи может привести к двум про-
тивоположным эффектам: а) стабилизации молекулы по отно-
шению к кислотным агентам, б) увеличению реакционной
способности центра за счет потенциально возможного пере-
носа протона под действием внешнего кислотного агента. С
качественной стороны эти эффекты можно иллюстрировать
схемой, учитывающей конкуренцию двух потенциальных про-
тонодоноров за обладание свободной электронной парой, па
реакционном участке:
Случай а представляет собой обычную протонизацию ре-
акционного центра в условиях специфического катализа. Ме-
ханизм блокирующего действия ОН-группы заключается в
смещении электронной плотности от реакционного центра в
сторону протона водородной связи О—Н ... О, вследствие
чего уменьшается вероятность переноса протона от иона гид-
роксония (или другого внешнего кислотного агента) на реак-
ционный центр. В случае б водородный мостик можно рас-
сматривать как канал, по которому возможен перенос прото-
на, ранее принадлежавшего ОН-группе. Переносу, очевидно,
благоприятствует атака кислотного агента на гидроксильный
кислород, участвующий в Н-связи с реакционным центром.
Таким образом, этот механизм, по существу, идентичен меха-
низму миграции протонов в водных растворах [168—170].
Подобного рода эффекты могут играть заметную роль в
реакциях моносахаридов и особенно полисахаридов по Ct и
по гидроксильным группам, и не исключено, что в ряде слу-
чаев [171—174] различия в реакционной способности можно
отнести за счет влияния Н-связей. В этом отношении опреде-
ленный интерес может представить оценка влияния Н-связей
на кинетику и механизм кислотно-катализируемой деструкции
полисахаридов, в частности, целлюлозы. До настоящего вре-
мени роль Н-связей в кинетике деструкции целлюлозы рас-
сматривалась преимущественно с точки зрения стерических
препятствий, создаваемых водородными мостиками в струк-
турно упорядоченных областях целлюлозного волокна. Вме-
сте с тем, учитывая неравномерное расположение Н-связей в
элементарных надмолекулярных образованиях целлюлозы,
можно полагать, что реакционные центры, вовлеченные в
Н-связи, должны быть неравноценными по своим протоноак-
цепторным свойствам. Из этого следует, что и О-гликозидные
связи должны быть неэквивалентными по скорости расщепле-
ния их в условиях кислотного катализа. Этот вопрос рассмот-
рен в работах [175—178].
2.4. Электронная структура сахаров
До последнего времени суждения о распределении элек-
тронной плотности в молекулах углеводов носили главным
образом интуитивный характер. Так, например, полагали, что
первый углеродный атом моносахаридов обладает избыточ-
ным эффективным положительным зарядом, благодаря чему
он способен вступать в типичные реакции нуклеофильного
присоединения. После появления метода индуктивных пара-
40
метров Дель-Ре [179, 180] оказалось возможным довольно
быстро производить оценку cr-электронных зарядов атомов
углеводной молекулы. Этим методом Ю. А. Жданов с сотр.
[181] впервые рассчитали cr-электронную структуру различ-
ных форм пентоз и пятиатомного спирта.
В основе метода Дель-Ре заложена идея о локализаций
сг-электропов на химических связях, в соответствии с которой
рассматривают волновые функции отдельных химических
связей:
<Р12 = С1Ф 1 + с2ф2,
(7)
С21 + С22 = 1
(ci и сг — коэффициенты атомных функций). Число функций
(МО) равно числу химических связей. Вековое (секулярное)
уравнение для каждой из связей имеет предельно простой
где Ни и Н22—кулоновские интегралы атомов (a), Hi2 = H2i—
резонансный интеграл связи (0). Решение уравнения (8) при-
водит к выражению для «орбитальной энергии связи» 1—2:
Е12 = а + 1120. (9)
Специфика метода Дель-Ре заключена в учете взаимного
влияния непосредственно связанных атомов с помощью ин-
дуктивных параметров tif=tJi< численные значения кото-
рых подбираются эмпирически на модельных соединениях.
Для каждого из атомов находятся кулоновские параметры
о/ решением системы неоднородных алгебраических урав-
Лёний:
= (10>
/
Число уравнений (10) равно общему числу атомов рассчиты-
ваемой системы. Далее вычисляются:
параметры Малликена
(11Г
частичный заряд на атоме i, индуцированный соседним ато-
йом /.
41
Тлава 3
МЕХАНИЗМ ОСНОВНЫХ РЕАКЦИЙ
ГЛИКОЗИДНОГО ЦЕНТРА
В любом кинетическом исследовании, направленном на ус-
тановление механизма реакций (как правило, модельных ре-
акций), центральным является вопрос о структуре промежу-
точных соединений или переходных состояний. Если кроме то-
го имеют дело с явлениями гомогенного катализа, в частно-
сти, кислотно-основного, то следует ответить также на вопрос
о механизме каталитического инициирования реакции — ха-
рактере действия каталитических агентов, природы первич-
ных центров реагирующих молекул, подвергающихся дейст-
вию катализаторов, вероятности участия катализаторов на
различных стадиях сложных реакций.
3.1. Общие сведения
о реакциях гликозидного центра
Ряд прямых и косвенных данных указывают па то, что в
процессах взаимодействия гликозидного центра с нуклеофиль-
ными агентами образуются, часто в довольно значительных
количествах, первичные продукты присоединения, имеющие
структуру ацеталей:
ОН ОН OR'
i I I
—НС,—ОН —НС,—OR —НС,—OR'
OH NHR'
—НС,—NHR —НС,—NHR"
Примеры подобного рода соединений и условия, при которых
наблюдается их образование, приведены в табл. 12. Однако
вопрос о степени участия этих соединений в реакциях нуклео-
фильного типа по С,—являются ли они действительно про-
межуточными соединениями или выступают лишь в роли по-
бочных продуктов — требует в каждом отдельном случае спе-
циального рассмотрения и экспериментального доказательст-
ва. Пока такого рода доказательства получены только для
44
Таблица 12
Некоторые продукты присоединения нуклеофильных агентов по гликозид-
ному центру и условия, при которых наблюдается их образование
Структура аддукта Условия получения Литература
1 2 3
ОН 1 —НС—ОН он Может быть выделена одновре- менно с аль-формами пентазаме- щенных моносахаридов. Легко взаимодействует с реагентами на С = О-группу Из иентазамещенных сахаров, в [85] стр. 47
1 частности, при перекристаллиза- [85]
—НС—ОС2П5 ции пентабензоил-аль-формы глю- козы из абсолютного этанола стр. 147, 152
ос2н5 1 —НС—0С2Н5 При взаимодействии незамещенной глюкозы с ортомуравьиным эфи- ром (ОМЭ) в подкисленных НС1 водных растворах. При длитель- ном выдерживании смеси аль- форм сахаров с ОМЭ в присутст- вии NH4NO3; выходы диэтила- цеталей после разделения на А12О3 от 40 до 80% [185]
NH—СО—СНз —НС—NH—СО—СНз Из ацетилированных сахаров пу- тем обработки их метанольным раствором аммиака с последую- щей экстракцией этилацетатом. [186]
nhc6h4ch3 -нс—nhc6h,ch5 При взаимодействии глюкозы с 5-тн кратным избытком п-толу- идина в дноксане [187]
ОС2Н5 —НС—NHC6H„CHS При обработке \-ацеталя абсо- лютным этанолом [187]
NHC6H,R При N-трансглнкознлировании в [188, 189J
1 —нс—nhc6h4r ряду N-арилгликозидов наблюда- ется аномальное изменение угла вращения в начальный период ре- акции замещения (90%-иый вод- ный этанол); продукт присоеди- нения п-ннтроанилина к N-глюко- знду анилина может быть выде- лен обработкой раствора бензо- лом с последующей перекристал- лизацией из смеси спирт—эфир. Возможно также выделение от- дельных диастереоизомерных форм N-ацеталей
NH—СО—CSHS При мягкой обработке пентабензо- ил-аль-глюкозы аммиаком. [190]
—НС—NH— CO-C6HS
45
Продолжение таблицы 12
3
NH—СО—NHj Образуются на первой стадии по- [191]
I лучения гликозилмочевин из N-
—НС—NH—СО—NHj гликозидов ленитроанилина и мо-
чевины, а также при прямом
синтезе гликозилмочевины из мо-
носахаридов и мочевины. В реак-
ционных растворах легко обна-
руживаются бумажной и тонко-
слойной хроматографией
реакции N-трансгликозилирования, связанной с обменом
N-агликонов в N-арилгликозидах (см. разд. 3.5).
Обобщение полученных к настоящему времени экспери-
ментальных данных по реакциям незамещенных моносахари-
дов, N-гликозидов, карбонильных соединений и соединений,
имеющих структуру шиффовых оснований, позволяет устано-
вить некоторые общие черты в их поведении. Это дает осно-
вание рассматривать гликозидный центр моносахаридов как
аналог карбонильной группы, а N-гликозидный центр — как
аналог азометиповой группировки и в соответствии с этой
аналогией конструировать схемы реакций с участием С|. С
этой точки зрения, реакции N-гликозилировапия можно рас-
сматривать как частный случай реакций нуклеофильного при-
соединения азотистых оснований по карбонильной группе, ме-
ханизм которых обстоятельно изучен и изложен в ряде работ
[192—207], из которых следует отметить обзор Дженкса
[203], подводящий итог многолетним исследованиям в этой
области. Экспериментально доказано, что реакции присоеди-
нения аминов по НО-О протекают через стадию образования
промежуточного соединения в форме аминоспирта, при этом
обе стадии реакции катализируются водородными ионами:
—Iic = o+NH2R НС—NHR —HC = NR + Н2О.
I
ОН
Отличительной особенностью многих реакций присоединения
является экстремальный характер зависимости константы ско-
рости от pH растворов — при умеренной кислотности ско-
рость возрастает по мере увеличения концентрации Н3О+, про-
ходит через максимум и затем уменьшается до нуля при вы-
сокой кислотности. Объяснение в общих чертах сводится к
следующему. При умеренной кислотности реакция каталпзи-
46
•руется за счет активации карбонильного соединения, и кон-
станта скорости увеличивается с уменьшением pH. При даль-
нейшем уменьшении pH прогрессивно возрастает концентра-
ция нереакционноспособных протонизованных форм амина и
скорость реакции начинает падать. Оптимальное значение pH
располагается вблизи рКа амина.
Выводы этой теории, однако, не согласуются количествен-
но с результатами кинетических исследований. В связи с этим
Дженксом [197] была разработана схема, предусматриваю-
щая смену лимитирующей стадии реакции при переходе че-
рез максимум на кривой зависимости константы скорости от
кислотности растворов. Согласно этой схеме, в области кис-
лотного катализа (правые ветви зависимости кнабл. от pH
среды) кинетика реакции лимитируется второй стадией —
дегидратацией промежуточного соединения (аминоспирта).
При уменьшении pH ниже оптимального значения (левые
ветви кривых зависимости кНа<>л- от pH среды) определяю-
щей становится стадия присоединения (Ki) вследствие умень-
шения концентрации непротопизоваиных форм нуклеофиль-
ного реагента. Рассмотрение этой схемы в рамках метода
стационарных концентраций позволило получить уравнение,
удовлетворительно описывающее зависимость кна(^л.ог кон-
центрации водородных ионов в реакции оксимирования аце-
тона [197]. Следует заметить, что аналогичные зависимости
кнабл- от [Н+] могут быть получены и при других предполо-
жениях относительно механизма кислотного катализа реакций
карбонильной группы. Поэтому согласие теоретически рассчи-
танных кривых кнабЛ. ([Н+]) с экспериментальными зависи-
мостями не является однозначным доказательством кинетиче-
ской схемы.
Надо отметить, что до середины 60-х годов не проводились
систематические исследования кинетики образования и гидро-
лиза N-гликозидов, за исключением нескольких работ эпизо-
дического характера [208—210]. Одной из первых в этом на-
правлении следует назвать работу Комптона и Вольфрома
[208], в которой изучено взаимодействие ацетата галактозы с
фенилгидразином. На основании кинетических данных эти
авторы пришли к выводу, что суммарная скорость реакции
определяется стадией раскрытия углеводного кольца. Пред-
полагалось, что реакционноспособной является лишь карбо-
нильная форма сахаров. Циклические формы не вступают за-
метным образом в конденсацию с фенилгидразином, поэтому
Скорость раскрытия углеводного кольца определяет скорость
образования фенилгидразона. В свете последних работ по ре-
акциям карбонильной группы [204] этот вывод не представ-
ит
Таблица 13
Аналогия в превращениях незамещенных моносахаридов и N-гликозидов
в условиях кислотно-основного катализа (реакции гликозидного центра)
Моносахариды
N-гликозиды
ЛАутаротация:
Аномеризация:
Перегруппировка Амадори:
Реакция Лобрн де Брюниа —
— А. ван Экенштейна:
—НС!—ОН H2Ci—ОН
НС—ОН С=О
I I
N-глнкозилироваиие:
-HCj-OH N1iR -HCi-NHR
Окисление по С>:
— HCj—ОН-»—С1=О
I
ОН
Вступают в реакции, характерные
для карбонильной группы, нап-
ример:
Н,
—НС1=О -* — H.Cj—ОН
—HCj-NHR
НС—ОН
Н2С,- NHR
I
c=o
N-трансгликозилироваиие:
I NH2R'
—HC,-NHR -» — HCj -NHR'
Окисление по Сц ;j •
—HCi—NHR-»-Cn-O
SlHR
Вступают в реакции, характерные для
азометинозых структур, например:
Н2
—HCi=NR — — II^-NHR
ляется бесспорным. Надо также заметить, что сопоставление
скоростей образования N-гликозидных связей и скоростей му-
таротации соответствующих сахаров не может, строго гово-
ря, привести к однозначному выбору механизма, в частности,
к решению вопроса о лимитирующей стадии реакции.
В упомянутых выше работах по нуклеофильным реакциям
карбонильной группы достаточно надежно установлено, что
при умеренных значениях pH среды превращение промежу-
точных соединений, имеющих структуру ацеталей и полуаце-
талей, протекает медленнее, чем их образование, и скорость
реакции контролируется обычно стадией элиминирования. Ре-
шение этого вопроса для аналогичных реакций гликозидного
48
центра имело бы существенное значение для физико-химии и
химии углеводов и гликозидов. От того, насколько избиратель-
но проходит заключительная стадия при заданной кислотно-
сти среды, зависит аномерный состав продукта реакции. Это-
справедливо, если реакция по Ci идет по пути образования
промежуточных структур. Однако нельзя полностью игнори-
ровать возможность протекания реакций по механизму пря-
мого вытеснения агликона атакующим нуклеофилом с сохра-
нением исходной циклической структуры. В связи с этим воз-
никает задача изменения стереохимической направленности
реакций путем изменения кислотности среды и природы аг-
ликона.
Замещение гликозидного гидроксила на О-агликон резко
снижает реакционную способность Ct по отношению к нуклео-
фильным агентам. Хотя О-гликозидная группировка по хими-
ческой структуре напоминает обычные апетальные системы,
константы скорости кислотного гидролиза алифатических аце-
талей, как правило, на два-три порядка выше констант ско-
ростей гидролиза О-гликозидов [211—228]. Высказывалось
предположение, что особая гидролитическая устойчивость
О-гликозидов связана с отрицательным индуктивным эффек-
том пиранозного кольца [229—232]. Насколько это объясне-
ние соответствует действительности, сказать трудно. Отметим
также, что О-гликозиды в большинстве своем ие мутаротиру-
ют в водных растворах, и следовательно образование О-гли-
козидной связи в свою очередь стабилизирует полуацеталь-
ный кислородный мостик. В отличие от кислородных аналогов
азотистые гликозиды являются весьма реакционноспособными
соединениями и легко вступают в реакции, подобные моноса-
харидам (табл. 13).
3.2. Механизм образования N-гликозидных связей
В литературе описаны данные по кинетике взаимодействия
незамещенных моносахаридов с алифатическими и аромати-
ческими аминами, аминокислотами, а также с гидроксилами-
ном, семикарбазидом и фенилгидразииами. Последние соеди-
нения изучены с кинетической стороны более полно, чем пре-
ДЫдущие [208—210, 233—239]. Скорость реакций образования
N-гликозидных связей очень зависит от кислотности среды.
Как правило, по мере изменения pH скорость реакции вна-
чале возрастает, а затем снижается. Эмпирически установле-
но, что оптимальной для образования N-гликозидной связи
является область pH, не слишком удаленная от соответствую-
4 367 49
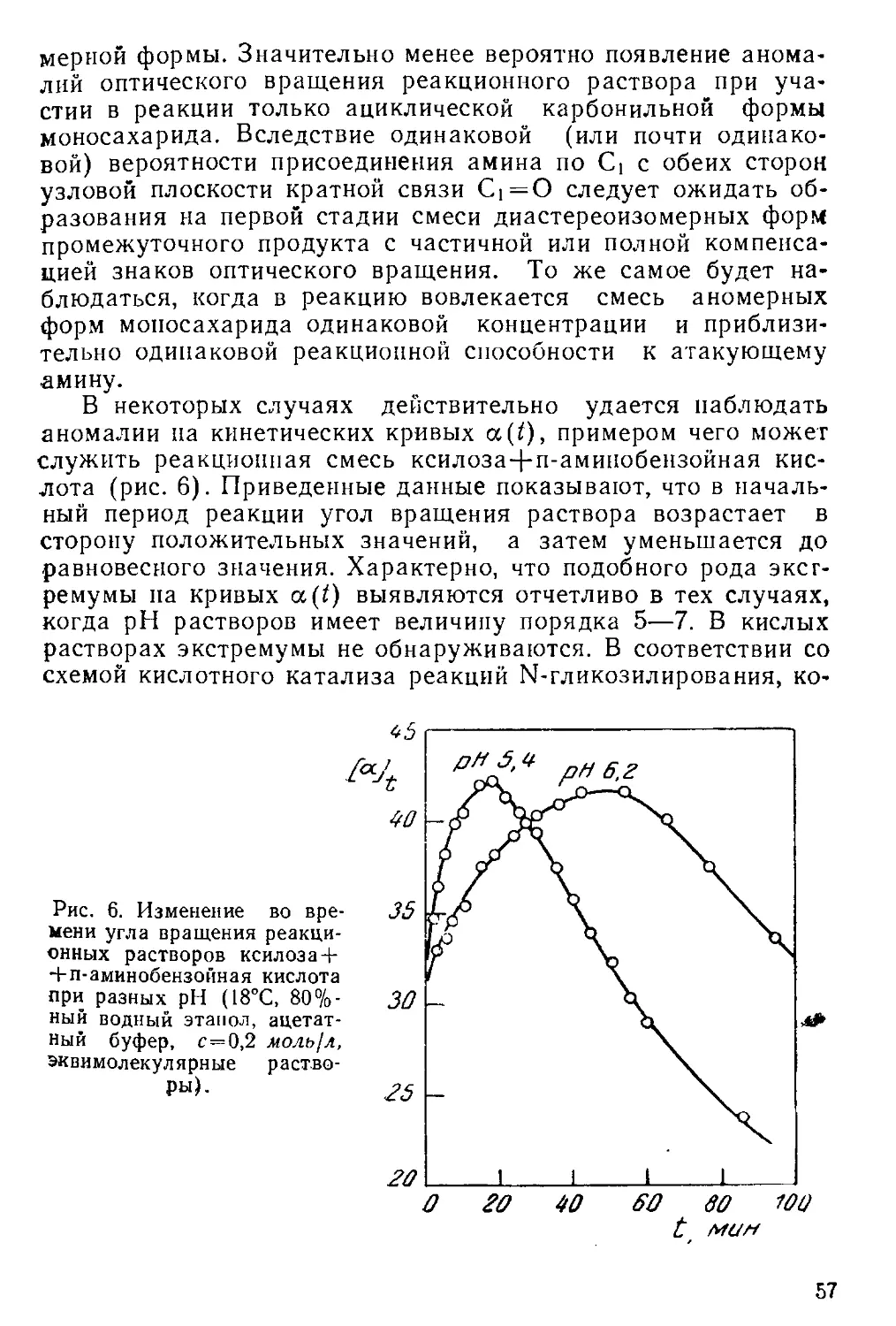
Рис. 3. Характерные зоны основных реакций моносахаридов и N-гликози-
дов с участием гликозидного центра: образования (1) и гидролиза (2)
N-гликозидных связей, аномеризации (3), N-трансгликозилирования в ряду
N-арилгликозидов (4), окислительных превращений no С, моносахаридов
(5) и перегруппировки Амадори для N-арилгликозидов (6).
щего показателя основности рКа атакующего амина (рис. 3).
Опыт показывает, что например, алифатические амины и
а-аминокислоты в умеренно кислых и нейтральных растворах
не вступают заметным образом в реакцию N-гликозилирова-
ния при прямом взаимодействии с незамещенными сахарами.
Оптимальной для образования N-алкилгликозидов является
область pH в окрестности 9—11, однако в ней изучение кине-
тики реакции N-гликозилнровання осложняется рядом побоч-
ных процессов, связанных с превращениями углеводного ком-
понента. В этом отношении лучшим объектом для кинетиче-
ского анализа являются ароматические амины, показатели
основности которых в водно-спиртовых растворах колеблются
в пределах 1—5.
По данным кинетических исследований [240—242], реак-
ционная способность моносахаридов при образовании N-арнл-
гликозидов увеличивается в следующем порядке: глюкоза <
галактоза < ксилоза < арабиноза < рибоза (табл. 14).
В той же последовательности возрастают и скорость тау-
томерных превращений моносахаридов (Исбелл, Пигман
[143]), и равновесная концентрация альдегидных форм (Кан-
тор, Пенистой [243]). Порядок расположения моносахаридов
по реакционной способности сохраняется также в реакциях
образования алкилгликозндов, оксимов, фенилгидразопов и
семикарбазонов сахаров (табл. 15). Примечательно, что ве-
50
Таблица It
Константы скорости образования N-гликозидов
п-толуидина (30°С, pH 5,6—5,8, 80%-ный водный
этанол, ацетатный буфер, С = 0,25 моль/л) и рав-
новесные концентрации альдегидных форм
моносахаридов
Моносахарид Эо к, мин -1 к, л/моль- 1 —пс,=о, мол. %
Глюкоза 0,022 0,088 0,024
Г алактоза 0,074 0,296 0,082
Ксилоза 0,34 1,36 0,17
Арабинозй 0,39 1.56 0,28
Рибоза 0,65 2,60 8,5
Таблица 15
Константы скорости (л/моль-мин) образования оксимов моносахаридов
при 25°С и различных pH [233] и относительные величины, характеризующие
электрофильную реакционную способность Ct
Моносахарид 1 pH 2,5 pH 3.6 pH 4,6 (1g к'/ко ср.
К К К Я/А-'о
Глюкоза 0,021 0 0,0412 0 0,043 0 0
Галактоза 0,088 0,62 0,182 0,65 0,192 0,65 0,64
Манноза 0,118 0,75 0,197 0,68 0,242 0,75 0,73
Ксилоза 0,145 0,84 0.248 0,78 0,259 0,80 0,81
Арабиноза 0,216 1,01 0.439 1,03 0,475 1,04 1,03
Рибоза 0.327 1,19 0,601 1,16 0,679 1,20 1,19
Ликсоза 0,360 1,23 0,700 1,23 0,838 1,29 1,25
личины 1g к/к0, которые можно рассматривать в качестве ме-
ры относительной реакционной способности гликозидного
центра, сохраняют для заданного моносахарида постоянство
при разных значениях pH среды. Если сопоставить получен-
ные относительные скорости оксимирования (ио Хаасу и Ка-
дунсу [233]) и мутаротации моносахаридов (по Исбеллу и
Пигману [143]), то обнаруживается удовлетворительная ли-
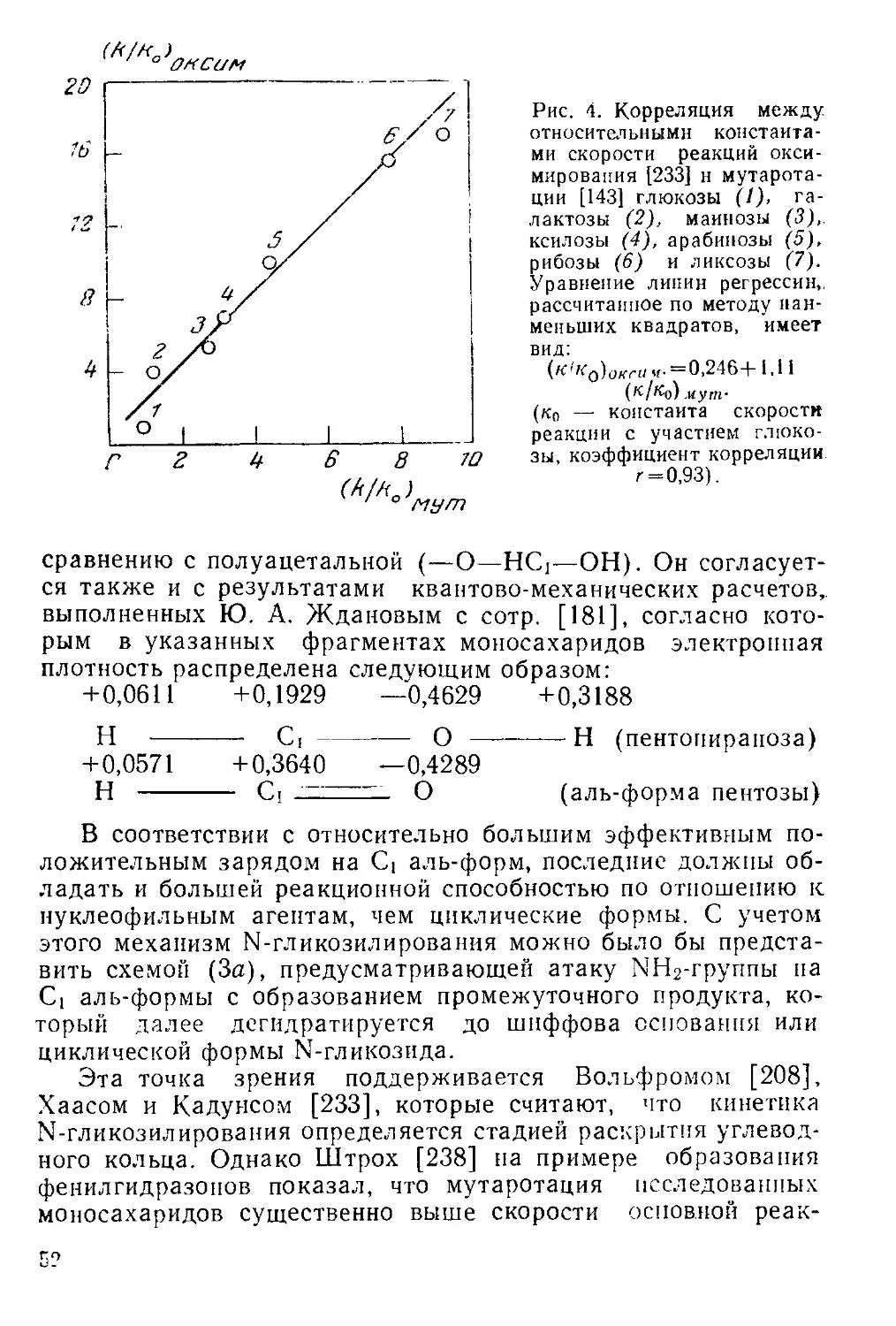
нейная корреляция между этими двумя параметрами (рис. 4)^
Располагая только этими данными, можно было бы сде-
лать вывод, что моносахариды реагируют с арилампнами в
своей альдегидной форме и следовательно N-гликозилирова-
ние представляет собой аналог реакций с участием «ненасы-
щенного углеродного атома» [202, 244]. Этот вывод соответ-
ствует принятой в литературе точке зрения о высокой реак-
ционной способности альдегидной группы (—HCi = O) по
51
Рис. 4. Корреляция между
относительными константа-
ми скорости реакций окси-
мироваиия [233] н мутарота-
ции [143] глюкозы (!)• га‘
лактозы (2), маннозы (3),.
ксилозы (4), арабинозы (5),
рибозы (6) и ликсозы (7).
Уравнение липин регрессии,,
рассчитанное по методу наи-
меньших квадратов, имеет
вид:
(к'К^окси «• = 0,246+ 1.11
(к/Ко) ,иут-
(«о — константа скорости
реакции с участием глюко-
зы, коэффициент корреляции.
г = 0,93).
сравнению с полуацетальной (—О—HQ—ОН). Он согласует-
ся также и с результатами квантово-механических расчетов,
выполненных Ю. А. Ждановым с сотр. [181], согласно кото-
рым в указанных фрагментах моносахаридов электронная
плотность распределена следующим образом:
+0,0611 +0,1929 —0,4629 +0,3188
Н -------- С!----------О ----------Н (пентопираноза)
+0,0571 +0,3640 —0,4289
Н -------- Ci — О (аль-форма пентозы)
В соответствии с относительно большим эффективным по-
ложительным зарядом на Q аль-форм, последние должны об-
ладать и большей реакционной способностью по отношению к
нуклеофильным агентам, чем циклические формы. С учетом
этого механизм N-гликозилирования можно было бы предста-
вить схемой (За), предусматривающей атаку NH2-rpynnbi па
С! аль-формы с образованием промежуточного продукта, ко-
торый далее дегидратируется до шиффова основания или
циклической формы N-гликознда.
Эта точка зрения поддерживается Вольфромом [208],
Хаасом и Кадунсом [233], которые считают, что кинетика
N-гликозилирования определяется стадией раскрытия углевод-
ного кольца. Однако Штрох [238] на примере образования
фенилгидразопов показал, что мутаротация исследованных
моносахаридов существенно выше скорости основной реак-
5?
Схема 3. Возможные механизмы образования N-глнкозидных
связей при прямом взаимодействии аминов с незамещенными
моносахаридами в альдегидной (а) и циклических (б, в) фор-
мах. При участии в реакции N-гликозилнровання циклических
форм сахаров возможно образование диастереоизомерных
форм промежуточного продукта, имеющего структуру амино-
спирта.
ции. Впрочем, в данном случае речь идет о различных реаген-
тах на карбонильную группу и делать какие-либо определен-
ные выводы из этого сопоставления весьма рискованно.
Полученные данные не противоречат также и схеме с у$1-
стием циклических форм сахаров на первой стадии реакции
(схемы бив), если принять во внимание рассмотренные вы-
ше факторы «конформационной неустойчивости» циклических
форм. Мы и в этом случае получим параллелизм между ре-
акционной способностью сахаров и скоростями их мутарота-
Ции. Поэтому корреляция между скоростными параметрами
и содержанием альдегидных групп (табл. 14) не является
53
сильным аргументом в пользу участия в реакции только аль-
форм. Иначе говоря, указанная корреляция вообще не может
служить хорошим тестом для выбора механизма реакции. С
другой стороны, можно полагать, что если бы реакция про-
текала исключительно по механизму то из-за малой кон-
центрации аль-форм (особенно в случае глюкозы) кинетика
реакции лимитировалась стадией раскрытия углеводного
кольца, и константа скорости не зависела бы (или зависела
слабо) от природы атакующего нуклеофила. Однако это не
так. В работе [242] показано, что в ряду однозамещенных
ариламипов константа скорости N-гликозилировапия, отне-
сенная к единице [Н+], зависит от природы заместителя в
бензольном кольце. Наблюдается удовлетворительная корре-
ляция констант скоростей с нуклеофильными о-константами
заместителей по Гаммету; при этом константа р уравнения
Гаммета имеет следующие значения:
р — —3,2 — при образовании N-глюкозидов,
р = —3,0 — при образовании N-арабииозидов. (1)
Кроме того, зависимости констант скоростей мутаротации мо-
носахаридов и реакции N-гликозилирования от кислотности
растворов имеют различный характер. Кривые (pH)
имеют вид «цепной линии» в случае мутаротации и вид «ко-
локола» — при N-гликозилированпп с максимумом в той об-
ласти pH, в которой скорость мутаротации минимальна
(рис. 5). Все это позволяет заключить, что в реакции N-гли-
козилирования могут участвовать одновременно и альдегид-
ные, и циклические формы сахаров, в соответствии с чем ско-
Рис. 5. Типичные кривые зави-
симости констант скоростей му-
таротации моносахаридов (1)и
образования N-гликозидных
связей (2) от pH водных раст-
воров.
рость = (к' [—НС,—ОН] + к" [-НС!=О]) [NH2R], (2)
При этом, хотя реакционная способность аль-форм должна
быть заведомо выше, чем циклических, вклад второго члена
в уравнение (2) может быть относительно малым из-за малой
концентрации аль-форм (см. табл. 14). Важно отметить, что
независимо от вклада каждой из указанных форм в наблю-
даемую скорость их взаимодействие с ариламинами ведет к
одному и тому же промежуточному соединению — амипоспир-
ту, превращение которого в конечный продукт при умеренной
кислотности среды лимитирует скорость реакции N-гликози-
лирования, как это показано в [242].
В циклических формах сахаров атом Ci находится в со-
стоянии 5р3-гибридизации. По этому формальному признаку
такие нуклеофильные реакции следовало бы отнести к реак-
циям «при насыщенном углеродном атоме» [202, 244]. Обыч-
но принимают, что реакции этого типа осуществляются через
переходные состояния в смысле теории абсолютных скоростей
реакций [245]. Возможны два пути движения системы к ко-
нечному или промежуточному соединению: а) одновременный
(согласованный, синхронный) процесс распада старой связи и
образование новой (чистый SN2 -механизм); б) разрыв старой
связи наступает несколько раньше образования повой, и в мо-
мент прохождения переходного состояния имеет место крат-
ковременное возникновение карбониево-ионного состояния
(S 1-механизм). В обоих случаях речь идет о разрыве только
одной заданной связи. В углеводах же имеются две почти эк-
вивалентные связи Ci—О глик-11 Ci—О цикл- и соответственно
две принципиальные возможности для присоединения нуклео-
С2 оч V НВ+ Сх-ОН 1 с2 он V НВ 4- Сх-Оц 1
н С2 Оц V НВ Сх-ОН 1 н ; с2 он V НВ Сх— -Оч &
н С» Оц -+ V НВ - С1+ОН- Н 4 С, ОН 4- V - НВ- Сх Оц-
Если не учитывать эффекта протопизации, механизм
-5^2—Огл. с синхронным вытеснением сильно основного гид-
роксильного иона представляется мало вероятным по энерге-
тическим соображениям [204]. Кроме того, данные по изме-
нению оптической активности реакционных растворов в про-
цессе N-гликозилировапия не дают каких-либо четких ука-
заний на такую возможность. Постулированный Богнаром с
сотр. [246] карбониево-ионный механизм (протонизация гли-
козидного гидроксила с последующим гетеролизом связи
(С,—Огл. ) также следует отвести по следующим соображе-
ниям. При SN1 -механизмах скорость реакции определяется
стадией гетеролиза и поэтому слабо зависит от эффектов за-
местителей у нуклеофильного компонента реакции, что не со-
гласуется с найденными абсолютными значениями констант
Р
Второй случай — механизм 5^2—Оч с синхронным раз-
рывом связи Ci—Оцикл. в момент атаки амина на Ci — пред-
ставляется наиболее вероятным. В соответствии с этим меха-
низмом возможность образования промежуточного продукта
должна зависеть, помимо всего прочего, от лабильности по-
луацетального кислородного мостика, которая определяется
общим запасом свободной энергии пиранозного кольца за-
данной конформации. Чем больше запас свободной энергии,
т. е. чем выше «конформационная неустойчивость» кольца, тем
лабильнее связь С]—Оцикл- и тем выше элементарная ско-
рость образования ациклического промежуточного продукта.
Если учесть, что и при мутаротации моносахаридов различие
в скоростях у разных сахаров может быть отнесено за счет
тех же факторов «конформационной неустойчивости» [149 —
152], то становится понятной отмеченная выше корреляция
между скоростями мутаротации и различных реакций нуклео-
фильного типа по Сь
Заметим, что из предложенной схемы N-гликозилирования
вытекает, что если в реакции участвует только одна из ано-
мерных форм моносахарида, то в результате присоединения
амина по С] на первой стадии возникает соответствующая
диастереоизомерная форма аминоспирта — право- или лево-
вращающая (механизмы б и в). В этом случае при благопри-
ятном соотношении знаков углов вращения всех компонентов
реакционной смеси —• исходного моносахарида, промежуточ-
ного соединения и N-гликозида — можно будет наблюдать
аномальное изменение угла вращения в начальный период ре-
акции, вызванное накоплением промежуточной диастереоизо-
56
мерной формы. Значительно менее вероятно появление анома-
лий оптического вращения реакционного раствора при уча-
стии в реакции только ациклической карбонильной формы
моносахарида. Вследствие одинаковой (или почти одинако-
вой) вероятности присоединения амина по Ci с обеих сторон
узловой плоскости кратной связи Ci=O следует ожидать об-
разования на первой стадии смеси диастереоизомерных форм
промежуточного продукта с частичной или полной компенса-
цией знаков оптического вращения. То же самое будет на-
блюдаться, когда в реакцию вовлекается смесь аномерных
форм моносахарида одинаковой концентрации и приблизи-
тельно одинаковой реакционной способности к атакующему
амину.
В некоторых случаях действительно удается наблюдать
аномалии на кинетических кривых а(0, примером чего может
служить реакционная смесь ксилоза+п-аминобензойная кис-
лота (рис. 6). Приведенные данные показывают, что в началь-
ный период реакции угол вращения раствора возрастает в
сторону положительных значений, а затем уменьшается до
равновесного значения. Характерно, что подобного рода экст-
ремумы па кривых а (0 выявляются отчетливо в тех случаях,
когда pH растворов имеет величину порядка 5—7. В кислых
растворах экстремумы не обнаруживаются. В соответствии со
схемой кислотного катализа реакций N-гликозилирования, ко-
Рис. 6. Изменение во вре-
мени угла вращения реакци-
онных растворов ксилоза+
+п-аминобензойная кислота
при разных pH (18°С, 80%-
ный водный этанол, ацетат-
ный буфер, с=0,2 моль/л,
эквимолекулярные раство-
ры).
57
торая будет рассмотрена в гл. 4, этот результат можно объ-
яснить накоплением промежуточного продукта (предположи-
тельно в форме аминоспирта), превращение которого в конеч-
ный N-гликозид (путем дегидратации с одновременной цик-
лизацией углеводного остатка) затруднено из-за недостаточ-
ной концентрации водородных ионов в растворе.
Поляриметрические измерения согласуются с данными
анализа той же реакционной системы методом бумажной хро-
матографии. Так, при проведении реакции образования
N-ксилозида п-амипобензойной кислоты в 80%-ном водном
этаноле с ацетатным буфером в области pH 5—7 иа хрома-
тограммах проявляется интенсивное пятно вблизи линии стар-
та (/?[~ 0,09), которое обнаруживается на первых же минутах
после сливания растворов ксилозы и п-аминобепзойной кис-
лоты. После элюирования этого пятна горячим метанолом или
водой и последующего хроматографирования полученного
раствора па хроматограмме проявляются характерные пятна
ксилозы, N-ксилознда и п-аминобензойиой кислоты (в поряд-
ке увеличения R,-); каких-либо дополнительных пятен иа хро-
матограмме нет. Эти данные позволяют заключить, что пятно
с R f 0,09 характеризует неустойчивое промежуточное соеди-
нение, которое в условиях элюирования и последующего хро-
матографирования претерпевает распад на исходные компо-
ненты. Таким образом, совокупность экспериментальных дан-
ных и изложенных выше соображений позволяют рассматри-
вать схему 3 образования N-гликозидных связей как наиболее
вероятную.
В дополнение к данным поляриметрии и хроматографии
следует указать на аномалии в температурной зависимости
константы скорости образования некоторых N-арилгликози-
дов, отмеченные в работе [242]. В координатах уравнения
Аррениуса наблюдаются изломы прямых, которые можно
объяснить, предположив, что две последовательные стадии
реакции заметно отличаются энергиями активации (рис. 7).
В этом случае с увеличением температуры относительно
большее ускорение будет приобретать та стадия, для которой
энергия активации больше. При некоторой температуре, зави-
сящей от соотношения энергий активации, наступит выравни-
вание скоростей, после чего скорость реакции будет опреде-
ляться стадией с меньшей энергией активации. Приведенные
в табл. 16 параметры активации показывают, что первая ста-
дия характеризуется низкими значениями энергии активации
и высокими отрицательными значениями энтропии активации,
что свидетельствует о высокой упорядоченности переходного
5?
Рис. 7. Температурная зависимость констан-
ты скорости N-гликозилировапия при взаи-
модействии п-толуидина с глюкозой (1),
ксилозой (2) и арабинозой (3) в координа-
тах уравнения Аррениуса (80%-пый водный
этанол, pH 5,6—5.8, ацетатный буфер,
с = 0,25 моль/л).
состояния первой стадии в смысле теории абсолютных скоро-
стей реакций.
В рамках молекулярно-кинетической теории [247—249]
высокие отрицательные значения энтропии активации первой
стадии можно интерпретировать как переход реакционной си-
стемы из исходного в промежуточное состояние с потерей по-
ступательных степеней свободы, которые вносят наибольший
вклад в энтропию при умеренных температурах. Учитывая
малые энергии активации, можно сделать вывод, что этой
стадией является бимолекулярный процесс, переходное со-
стояние которого отличается высокой степенью синхронности
при разрыве старых и образовании новых химических связей.
Вопрос об интерпретации активационных параметров хими-
ческих реакций требует специального изучения (см. гл. 5). От-
Таблица 16
Энергии и энтропии активации стадий присоедине-
ния (1) и дегидратации (2) реакции N-гликози-
лирования
N-глнкозид анилина Е, ккал!моль AS^. кал!град- моль
1 2 1 2
N-глюкозид 2,4 15,2 —65 —25
N-ксилозид 4,1 12,7 —58 —31
N-арабинозид 2,1 17,3 —62 — 13
59
метим, что указанное выше изменение кинетических парамет-
ров в координатах уравнения Аррениуса служит дополни-
тельным аргументом в пользу рассмотренной схемы N-глико-
зилироваиия, предусматривающей двухстадийное протекание
реакции, скорость которой лимитируется при умеренных тем-
пературах стадией дегидратации.
3.3. Аномеризация N-гликозидов
Как и моносахариды, N-гликозиды испытывают в водных
растворах характерное таутомерное превращение, связанное
с изменением положения N-агликона относительно плоскости
углеводного кольца (явление апомеризации) [250—258]:
Аномеризация N-гликозидов обычно сопровождается в
большей или меньшей степени их гидролизом, что затрудняет
кинетическое исследование с помощью поляриметрического
метода. При измерении оптической активности свежеприго-
товленных подкисленных растворов N-арилгликозидов часто
можно наблюдать быстрое изменение угла вращения в сторо-
ну отрицательных значений, а затем более медленное увели-
чение угла вращения вследствие кислотнокатализируемого
гидролиза N-гликозидных связей в обеих аномерных формах
(рис. 8). Скорость апомеризации даже умеренно основных
N-арилгликозидов значительно выше скорости мутаротации
соответствующих моносахаридов, если речь идет о кислых
средах. С повышением основности N-агликона скорость ано-
меризации возрастает настолько, что в ряде случаев оказы-
вается невозможным зафиксировать ее по изменению оптиче-
ской активности. В щелочных растворах скорость аномериза-
ции, напротив, меньше, чем в кислых средах. Для мутарота-
ции моносахаридов, как мы видели выше (гл. 2), наблюдает-
ся обратная картина. По аналогии с мутаротацией сахаров
константу скорости аномеризации N-гликозидов можно в об-
щем случае выразить уравнением, в котором аддитивно учи-
тываются вклады кислотного и основного катализа:
к набл- = к о + кн[Н+] + К он [ОН ], (3)
где /сни к0|| — константы каталитического действия иоиов
(60
Н50+ и ОН-, ко— констан-
та скорости аномеризации
под действием молекул
воды. По данным работы
[250], каталитические кон-
станты кни к0[[ аиомериза-
ции N-арабинозида ани-
лина равны:
ки=2,6-105 л!моль-мин,
коН = 1,5-10—3 л/моль'мин.
Таким образом, при
аномеризации N-гликози-
дов катализ под действи-
ем основных агентов игра-
ет значительно меньшую
роль, чем при мутарота-
ции моносахаридов. В
умеренно кислых раство-
рах и даже при нейтраль-
ных значениях pH вклад
основного катализа кна/-)Л.
пренебрежимо мал. В
гл. 4 будет рассмотрена
количественная сторона
кислотно-основного ката-
лиза аномерных превра-
щений, здесь же мы оста-
новимся иа основных
Рис. 8. Примеры изменения наблю-
даемого угла вращения свежепри-
готовленных растворов N-глюко-
зида анилина в ходе аномериза-
ции и последующего кислотного
гидролиза (25°С, 70%-пый водный
этанол, с = 2,55, длина трубки —
2 Ли)-
представлениях о механизме этой реакции.
В литературе нет единого мнения относительно механизма
аномеризации. Исбелл и Фраш [251] полагают, что па первой
стадии реакции происходит раскрытие углеводного кольца в
результате протонизации циклического кислородного атома и
образование ациклического иммониевого иона (схема 4). За-
тем следует циклизация с образованием главным образом
термодинамически более устойчивого p-аномера. Кун и Бир-^у
кофер [252], напротив, считают более вероятной протопиза-
цию атома азота N-агликона, поскольку он обладает большей
основностью, чем полуацетальный кислородный атом. Богнар
и Нанаши [254] отстаивают карбониево-ионный механизм,
предусматривающий гетеролиз N-гликозидной связи. Особую
позицию занимают Ходж и Рист [253]. Принимая во внима-
ние тот факт, что N-гликозиды аномеризуются и в неводных
растворителях, например в пиридине, они предложили меха-
61
Схема 4. Механизмы аномеризации N-гликозидов по
Исбеллу и Фрашу (а), Куиу и Биркоферу (б), Богиару
и Напаши (в). В механизме в допускается захват про-
тона от циклического кислорода N-агликоном в момент
его элиминирования.
низм, связанный с внутримолекулярным переносом водород-
ных атомов.
Сравнительное изучение кинетики аномеризации N-арил-
глюкозидов показало, что скорость аномеризации почти ли-
нейно зависит от показателя основности ариламина, входя-
щего в виде N-агликона в N-гликозид [258]. Установлено так-
же, что константы скорости аномеризации одиозаметенных
N-арилгликозидов удовлетворительно коррелируются уравне-
нием Гаммета с константами
р = —2,8 (при 25°С) и р — —2,95 (при 15~С). (4)
Однако эти исследования не внесли ясности в вопрос о меха-
низме аномеризации. В последние годы были проведены ис-
следования кинетики аномеризации N-глюкозида и п-брома-
62
нилина в присутствии бензойной и п-метоксибензойной кис-
лот, трибутиламина и фенолов [259]. Добавки заметно ката-
лизируют реакцию, причем каталитическое действие зависит
от рКа кислот и оснований, что позволяет использовать этот
эффект для оценки констант диссоциации органических сое-
динений.
3.4. Гидролиз N-гликозидных связей
N-гликозиды легко гидролизуются разбавленными мине-
ральными и органическими кислотами [239, 251, 260—267].
В щелочных растворах они. как правило, более устойчивы,
чем в кислых средах. Гликозиды с арильными N-агликонами
в целом более устойчивы, чем соответствующие алкильные
производные. В ряду соединений с одинаковым N-агликоном
скорость гидролиза увеличивается в следующем порядке:
N-глюкозид < N-галактозид < N-ксилозид < N-арабинозид
[266]. В ряду N-арилгликозидов при заданном углеводном
фрагменте скорость гидролиза возрастает с уменьшением
электроноакцепторной способности заместителя в бензольном
кольце. Константа скорости гидролиза N-арилглюкозидов
удовлетворительно коррелирует с а-константами заместителя
в бензольном кольце (по Гаммету) [261, 263]. Установлено,
что знак константы р имеет отрицательное значение (р = —2,3).
Эти наблюдения согласуются с правилом Миттса и Хиксона
[267], согласно которому устойчивость N-гликозидных связей
в условиях кислотного гидролиза возрастает по мере умень-
шения основности N-агликона.
Влияние кислотности растворов на кинетику гидролиза
рассмотрено рядом авторов на примере гидролиза N-арил-
глюкозидов [251, 263, 264]. Определено, что с уменьшением
pH константа скорости гидролиза вначале возрастает, а за-
тем уменьшается по мере дальнейшего увеличения кислотно-
сти растворов. Кривые зависимости кна^л- (pH) имеют коло-
колообразный вид. Положение максимума на шкале pH зави-
сит от основности амина, входящего в состав N-агликона, —
с уменьшением основности максимум смещается в более кис-
лую область.
В вопросе о механизме гидролиза единого мнения нет
(схема 5). Исбелл и Фраш [251] предложили следующий
механизм: а) атака протона на циклический кислородный
атом с последующим разрывом полуацетального мостика п
образованием протонированной формы шиффова основания,
б) присоединение гидроксильного иона к гликозидному цент-
63
Схема 5. Механизмы кислотного гидролиза N-гликозидов по
Исбеллу и Фрашу (а), Капону и Коннетту (б), Богнару и Па-
наши (в).
ру, находящемуся в состоянии карбониевого иона (Ci+), с по-
следующим разрывом связи С!—N и образованием протони-
рованной по кислороду альдегидной формы моносахарида. С
небольшими поправками эта схема принимается Капоном и
Коннетом [263]. Богнару с сотр. [265] представляется более
оправданной, с точки зрения данных эксперимента, схема
гидролиза с образованием карбониево-ионного состояния в
результате атаки протона на азот N-агликона и последующе-
го гетеролиза связи Ci—N. Эта схема, однако, противоречит
отмеченной выше зависимости кна^л (pH). По Богнару сле-
довало бы ожидать, что концентрация карбониевых ионов, а
64
следовательно, и скорость гидролиза, должны возрастать с
увеличением концентрации водородных ионов. В действитель-
ности же наблюдается падение константы скорости гидролиза
ниже некоторого оптимального значения pH.
Симон и Пальм [264] дали подробный анализ возможных
превращений N-гликозидов в водных растворах. Предложен-
ная ими схема гидролиза в основном согласуется с представ-
лениями Исбелла. Следует подчеркнуть, что Симон и Пальм
так же, как и Исбелл, полагают, что протонизация азота в
N-агликоне не активирует, а стабилизирует молекулу N-гли-
козида. Хотя это предположение является несколько неожи-
данным (особенно, если учесть значительные различия в ос-
новностях циклического кислорода и атома азота), оно за-
служивает серьезного внимания.
Известно, например, что в умеренно кислых растворах
скорость гидролиза некоторых ароматических шиффовых ос-
нований снижается по мере уменьшения pH [268—270]. Из
этого наблюдения сделан вывод, что протонированная форма
основания Шиффа более устойчива к гидролитическому рас-
щеплению, чем непротонированиая. Известно также, что в тех
случаях, когда молекула субстрата содержит конкурирующие
протоноакцепторные атомы кислорода и азота (амиды), кис-
лотному гидролизу способствует присоединение протона к
атому кислорода, в то время как протонизация атома азота,
имеющая место в сильно кислых растворах, затрудняет гид-
ролиз С—N [271—278]. Причины этого еще не вполне ясны.
Высказывались предположения, что для гидролитического
расщепления связи С—N необходим одновременно и кислот-
ный, и основной катализ [250, 251]. Поэтому с увеличением
концентрации Н3О+, хотя и увеличивается степень протониза-
ции субстрата (по азоту, если речь идет о гидролизе шиффо-
вых оснований), уменьшается концентрация ионов ОН-, ка-
тализируемых вторую стадию гидролиза, и скорость реакции
начинает уменьшаться ниже некоторого критического значе-
ния pH. Это объяснение не является бесспорным.
Уместно отметить также, что Б. и А. Пюльманами [279]л
предложено полуэмпнрическое правило, согласно которому
легкость гидролиза связей С—N и С—О в амидах, сложных
эфирах, биологически активных полифосфатах и N-гликози-
дах (исключение составляют О-гликозидные связи в олиго- и
полисахаридах) возрастает с увеличением суммарного поло-
жительного заряда на гидролизуемой связи. Это правило ил-
люстрируется примером ферментативного распада N-глико-
зидных связей в некоторых риботидах (см. [279] стр. 538).
Нетрудно заметить, что в действительности скорость гидро-
5 367
65
лиза в ряду риботидов по мере увеличения эффективного по-
ложительного заряда на связи С—N возрастает лишь до оп-
ределенного предела и затем падает. Максимум относитель-
ной скорости гидролиза наблюдается при положительном за-
ряде па атоме азота равном около +0,40 единиц. Причины
этого явления неясны.
3.5. N-трансгликозилирование
При взаимодействии с аминами и моносахаридами N-гли-
козиды вступают в реакцию N-трансгликозилирования, ре-
зультатом которой является замена N-агликона или углевод-
ного остатка на новые фрагменты:
—НС5—R—HCi—NHR' + NH2R"<-
1___О___I
НС5—R—НС!—NHR"+NH2R' (а)
1__О____L
п—NH—R+r2—ОН Г2—NH—R+T1—ОН (б)
(Г1 и Г2 — углеводные фрагменты). Аналогичный обмен
N-агликонов наблюдается при взаимодействии двух разных
N-гликозидов:
Pi—NH—R'+ Г2—NH —R"-> Г1—NH—R" + Г2—NH—R'. (в)
Реакция первого типа (а) открыта Куном и Данси в 1936 г.
[280]. Второй и третий тип N-трансгликозилирования (б), (в)
обнаружены и исследованы Богнаром с сотр. [281—285]. Об-
мен N-агликонов протекает достаточно легко в водно-спирто-
вых растворах при умеренных температурах в присутствии
добавок кислотного характера. Для препаративной химии уг-
леводов N-трансгликозилирование может представить инте-
рес как потенциально возможный метод синтеза новых N-гли-
козидов, образование которых при непосредственном взаимо-
действии моносахаридов с аминами затруднено по кинетиче-
ским или термодинамическим соображениям.
О механизме N-трансгликозилирования имеется ряд пред-
положений. Исбелл и Фраш [251], рассматривая в общем
плане возможные превращения N-гликозидов, полагают, что
обмен N-агликонов происходит по схеме, предусматривающей
протонизацию циклического кислорода и раскрытие пираноз-
ного кольца (схема 6). Далее следует присоединение атакую-
щего амина по С[ шиффова основания с образованием про-
межуточного продукта в форме N-ацеталя. Исходный N-ar-
66
Схема 6. Некоторые из предполагаемых механизмов N транс-
гликозилирования, связанного с обменом N-агликонов при за-
данном углеводном остатке — по Богнару с corp. (а), по Ис-
беллу н Фрашу (б), по Капону и Копнету, а также Симону
с сотр. (в).
।
ликон в момент отщепления захватывает избыточный проток
от входящего N-агликона. Аналогичные схемы предложены
Капоном и Коннеттом [263], а также Симоном и Пальмой
1264].
Критикуя механизм Исбелла, Богцар [281, 282] обращал
внимание на то обстоятельство, что образование N-ацеталь-
ной формы затруднено по стерическим соображениям. По мне-
нию Богнара N-трансгликозилирование протекает через ста-
дию образования карбониевого иона (механизм S •), кото-
67
рый образуется в результате протонизации N-агликона и по-
следующего медленного гетеролиза N-гликозидной связи. Ата-
ка второго амина на С! карбониевого (или оксониевого) иона
приводит к образованию аномерной смеси конечного N-глико-
зида.
Согласно механизму Богнара, реакция должна иметь
первый порядок, поскольку при S N1 -механизмах лимитирую-
щей стадией является, как правило, гетеролитический распад
протонизованной формы субстрата. Однако, как показали
исследования Симона с сотр. [286, 287], трансгликозилирова-
ние первого типа имеет суммарный второй порядок (первый
порядок по исходным компонентам реакции). Сравнивая ско-
рости гидролиза и N-трансгликозилирования в сопоставимых
условиях методом количественной бумажной хроматографии,
Богнар показал, что трансгликозилирование протекает без
предварительного гидролиза исходного N-гликозида. В одной
из последних работ [286] было предложено еще несколько
возможных механизмов: 1) синхронное вытеснение исходного
N-агликона атакующим амином без раскрытия пиранозного
кольца; 2) атака протонизованной формы амина на С1 исход-
ного N-гликозида с одновременным переносом избыточного
протона от аминогруппы на а) циклический кислород с по-
следующим раскрытием пиранозного кольца и образованием
N-ацетальной формы, б) атом азота исходного N-агликона с
последующим отщеплением этого агликона без раскрытия
пиранозного кольца. Однозначных экспериментальных данных
в пользу одного из приведенных выше механизмов в литера-
туре нет. Поэтому до последнего времени вопрос о наиболее
вероятном пути реакции замещения N-агликонов и, тем более,
вопрос о механизме кислотного катализа оставался открытым.
В 1969 г. было установлено, что угол вращения реакцион-
ных смесей N-глюкозид анилина + ариламины аномально
возрастает в первые минуты после начала реакции обмена
[288, 289]. При проведении реакции в обратном направлении,
т. е. при замещении N-агликонов ариламинов на остаток ани-
лина, наблюдается противоположная картина — угол враще-
ния смеси претерпевает аномальное отклонение в сторону от-
рицательных значений. Иллюстрацией этого могут служить
кинетические кривые, показанные на рис. 9, 10. В табл. 17
приведены наблюдаемые углы вращения в максимумах кине-
тических кривых а(/), измеренных в процессе взаимодействия
некоторых N-арилглюкозидов с однозамещенпыми арила-
минами.
Св
Рис. 9. Изменение
наблюдаемого угла
«ращения при N-
1рансглпкозплирова-
мии в системах: N-
глюкозил анилина 4-
4- п-нитроанилин (1)
и N-глюкозид п-иит-
роанилина + анилин
<2) (20°С, 90%-ный
водный этанол, аце-
татный буфер, pH 2,3,
с=0,08 моль/л, экви-
молекулярные кон-
центрации). Кривая 1
описывается уравне-
нием:
az=-4,5 -
-0,85'4
—20,4-е
0,0481
4-24,4-e
Характер изменения угла вращения реакционных раство-
ров свидетельствует о протекании N-трансгликозилирования
через две кинетически различимые стадии с образованием в
заметных количествах промежуточного оптически активного
соединения, обладающего большим положительным углом
вращения в случае вытеснения остатка анилина из исходного
N-глюкозида анилина и отрицательным—при замещении агли-
конов N-арилглюкозидов анилином. Методом количественной
бумажной хроматографии зафиксировать стадийность реак-
ции и образование промежуточного соединения не удается.
Вероятно, это связано с тем, что в условиях длительного хро-
матографического разделения реакционной смеси промежу-
точный продукт претерпевает распад.
Из уравнения (5) видно, что константа скорости первой
стадии более чем на порядок превышает константу скорости
69
Рис. 10. Изменение наблюдаемого угла вращения при N-трансгликозили-
ровании в системе: N-глюкозид л-питроанилина + анилин (20°С, 9О°/о-ный
водный этанол, ацетатный буфер, с=0,08 моль!л). Константы скорости
первой и второй стадий реакции равны:
pH
2,8 I 3,1 : 3,3 | 3.7
к}, мин—' — 0,240 0,147 0,070
кг, мин-' 0,190 0,114 0,047 0,009
(здесь константы скорости выражены в виде произведения константы ско-
рости 2го порядка на начальную концентрацию компонентов).
расходования промежуточного продукта. Этим и объясняется
накопление в заметных количествах промежуточного продук-
та, обладающего положительным углом вращения при вытес-
нении остатка анилина из исходного N-глюкозида и отрица-
тельным — при замещении N-агликонов анилином. Для оцен-
ки относительной концентрации (N2) промежуточного продук-
та в максимуме кинетической кривой можно использовать
соотношение:
ln[N2]= К2 _ к, In («1М2).
(,6>
Например, для рассмотренного выше случая
70
К] = 0,85 мин~\ к2 = 0,048 мин-'.
Подставляя эти величины в уравнение (6), получим N2=0,84.
В той же системе при pH 2,5 (5°С) константы скорости обра-
зования и расходования промежуточного продукта равны:
Ki =0,60 мин-', К2 = 0,005 мин-1.
При указанном соотношении констант скоростей N2 = 0,95.
Таким образом, значительная часть исходных компонентов,
превращается в промежуточный продукт за время от начала
реакции до момента, соответствующего экстремуму кинетиче-
ской кривой. Скорость суммарной реакции определяется ско-
ростью медленной второй стадии.
Наиболее разумным и естественным представляется сле-
дующее объяснение характера изменения угла вращения
в ходе трансгликозилирования. Реакция протекает через две
последовательные стадии — присоединение атакующего амина
к Ci циклической форме гликозида и элиминирование исход-
ного N-агликона из промежуточного продукта в форме N-аце-
таля с последующей (или одновременной) циклизацией угле-
водного фрагмента (схема 7). Рассмотрим экспериментальные
данные, полученные при доказательстве предложенной схемы.
Схема 7. Один из наиболее вероятных механизмов N-транс-
гликознлирования в ряду N-арилгликозидов, предусматриваю-
щий нуклеофильную атаку амина на С-, циклической формы
N-гликозида с «синхронным» раскрытием углеводного кольца
и образованием промежуточных N-ацетальных структур.
Как показывают кинетические кривые, стадии образования
и расходования промежуточного продукта в неодинаковой сте-
пени катализируются водородными ионами. С уменьшением
кислотности среды константа скорости второй стадии падает
значительно быстрее, чем константа скорости первой стадии.
Поэтому выше некоторого критического значения pH N-транс-
сликозилирования фактически «замораживается» на стадии
образования промежуточного продукта. Исходя из этого, ме-
тодика эксперимента заключалась в установлении критиче-
ского значения pH для заданной реакционной системы, вы-
держивании смеси в течение нескольких минут для накопле-
71
пня промежуточного соединения и последующем выделении
этого продукта путем добавления органических растворите-
лей, не смешивающихся с водой. Наиболее достоверные ре-
зультаты получены, в тех случаях, когда в качестве атакую-
щего амина или N-агликона использовался пара- (или мета-)
нитроанилии. В других случаях выделенные продукты быстро
разлагались с выделением соответствующих аминов.
Приведем пример. Растворы N-глюкозида анилина и п-нит-
роанилина (0,08 моль!л, 90%-ный этанол), подкисленные до
pH 1,8, сливались и выдерживались при 20°С в течение одной
минуты. После добавления 25%-ного раствора аммиака до
pH 8—8,5 продукт высаживался бензолом и перекристаллизо-
вывался дважды из смеси этанола и бензола. Выход 21%.
Т. пл. ПО—112°С, [а]д= + 98,2°С (удельный угол враще-
ния, рассчитанный из наблюдаемого угла вращения в макси-
муме кинетической кривой, равен +103°). В предположении
N-ацетальной структуры промежуточного продукта
(C18H23O7N3) вычислено (%): С 55,2; Н 6,4; N 10,5. Найдено
(%): С 54,8; Н 6,5; N 10,1. В спектре поглощения обнаружи-
ваются максимумы поглощения при 285 и 360 ммк, характер-
ные для бензольного кольца анилина и п-нитроанилина. Ко-
ротковолновый максимум при 230 ммк расположен между
максимумами поглощения 238 и 228 ммк, принадлежащими
соответственно N-глюкозиду анилина и п-нитроанилину. Эти
данные свидетельствуют о наличии в промежуточном про-
дукте остатков анилина и п-нитроанилина.
Следовало показать также, что выделенный продукт, имею-
щий строение N-ацеталя, является именно промежуточным
соединением реакции N-трансгликозилирования. С этой целью
была изучена кинетика превращения полученного выше N-аце-
таля в равновесную смесь N-глюкозидов анилина и п-нитро-
анилина в условиях, сопоставимых с условиями реакции
N-трансгликозилирования в той же системе. Пример кинетики
распада промежуточного продукта показан на рис. 11. В ре-
зультате найдено, что величина кг/[Н+] =7,8 л. моль~'мин~}
(20°С) близка константе каталитического действия к2(„) вто-
рой стадии N-трансгликозилирования. Полученный результат
свидетельствует в пользу идентичности выделенного продукта
и промежуточного соединения, фиксируемого в максимуме
кинетических кривых a(t) для системы N-глюкозид анилина +
п-нитроанилин.
В свете предложенного механизма рассмотрим характер
изменения угла вращения реакционных растворов при
N-трансгликозилировании. Приведенные выше примеры кине-
тических кривых показывают, что углы вращения в максиму-
72
Рис. 11. Изменение наблю-
даемого угла вращения при
распаде промежуточного
продукта, выделенного из
реакционной смеси: N-глю-
козид анилина + п-ннтро-
анилин (20°С, 90%-ный вод-
ный этанол, pH 1,3, ацетат-
ный буфер, с = 0,04 моль/л).
Константа скорости распада
Кнабл- =0,39 мин-', кнабл.1
[Н+] = 7,8 моль-мин.
мах кинетических кривых а(/) прямой и обратной реакции
противоположны ио знаку. Из этого можно сделать вывод, что
при N-трансгликозилировании в прямом и обратном направ-
лениях образуются диастереоизомерные формы промежуточ-
ного продукта, различающиеся лишь пространственным рас-
положением заместителей у гликозидного центра:
NH—С6Н4—R'
R—Ci—NH—С6Н4—R"
I
Н
NH-C6H4-R"
R—Ci—NH—С6Н4— R'
н
Эти формы, очевидно, могут возникнуть только в том слу-
чае, если исходный N-гликозид реагирует с амином в своей
циклической форме (см. схему 7). $
В соответствии со схемой 7 нуклеофильная атака амина на
С! направлена со стороны, противоположной циклическому
кислородному атому (0/{цкл). В момент синхронного разрыва
полуацетальной связи С)—Очил-,г.и образования новой связи
Ci—N возникает ациклический продукт (N-ацеталь), в кото-
ром пространственное расположение заместителей при С>
однозначно определяется конфигурацией исходного N-аглико-
на. При протекании реакции в обратном направлении обра-
зуется, очевидно, тот же самый продукт, но с таким располо-
73
жением заместителей при Сг, которое зеркально симметрично
предыдущему случаю (конечно, при условии, что в обоих слу-
чаях исходные N-гликозиды реагируют в одной и той же ано-
мерной форме).
Нетрудно понять, что если бы N-гликозиды реагировали
с аминами в форме шиффовых оснований, как это полагает
Исбелл, то на первой стадии имело бы место образование ра-
цемической смеси промежуточных N-ацетальных форм вслед-
ствие одинаковой вероятности атаки амина на С] с обеих сто-
рон узловой плоскости связи Ci—N. В этом случае на кинети-
ческих кривых не наблюдалось бы экстремумов в начальный
период реакции. По той же причине представляется малове-
роятным карбониево-ионный механизм Богнара с сотр. [283—
285].
Рассмотрим вторую стадию реакции замещения, когда
происходит элиминирование исходного N-агликона с одновре-
менной циклизацией углеводного остатка. В целях общности
обозначим входящий и уходящий агликоны символами Bi и В2
(см. схему а). Эту стадию можно представить в виде после-
довательности двух элементарных актов — атаки гидроксила
при С5 (или С4, если речь идет об образовании фуранозных
форм) па С, и последующего вытеснения исходного агликона
В] в форме НВ]. Реакционный путь, по которому движется
система к переходному состоянию, может быть изображен в
виде а) линии, на которой располагаются последовательно
образующаяся и разрываемая связи С5О—С] п С]—В(. б) цик-
лического контура, в котором, кроме указанных связей, уча-
ствует также связь О—Н гидроксильной группы:
III l'i
-НС.5-0--------В1, -НС,,-о---(?!-
А А
В соответствии с представлениями о реакциях типа 5 N2 пер-
вый путь наиболее вероятен. Посмотрим, к каким последст-
виям приводит этот механизм. Поскольку в данном случае
речь идет о внутримолекулярном процессе, то в переходном
состоянии из-за отсутствия свободного вращения атомных
групп относительно одинарных связей заместители при С] бу-
дут занимать строго фиксированное положение в простран-
стве. При этом входящий агликон В2 может занимать только
две позиции в зависимости от того, какая диастереоизомерная
форма ацеталя претерпевает циклизацию. В свою очередь,
расположение агликонов при Ci в диастереоизомерах одно-
74
значно определяется конфигурацией агликона в исходном'
гликозиде. Поэтому относительное расположение входящего
агликона В2 в переходном состоянии (и в конечном продукте)
целиком зависит от того, в какой аномерной форме участвует
исходный гликозид на стадии присоединения.
Если исходный гликозид реагирует с атакующим нуклео-
филом В2 в виде p-аномера с экваториальным агликоном В(,
то в конечном гликозиде новый агликон займет аксиальную
позицию (схема 8а). При участии па стадии присоединения
Схема 8. Механизм стереохимического контроля реакции нуклеофильного
замещения па завершающей стадии — элиминировании исходного аглико-
на Bi с одновременной циклизацией углеводного фрагмента. Пунктиром
показали линии атаки нуклеофила НВ2 и гидроксила при С5 на гликозид-
ный центр. Случаи а и б соответствуют участию в реакции двух аномер-
ных форм исходного N-гликозида.
а-аномера входящий агликон расположится в продукте реак-
ции в экваториальной позиции (схема 86). Из схемы 86 мож-
но вывести следующее правило «стереохимического контроля»
реакций нуклеофильного замещения на стадии элиминирова-
ния:
— в продуктах реакций нуклеофильного замещения,
протекающих через стадию образования промежуточных
соединений ацетильного типа, входящий агликон зани-
мает конфигурацию, противоположную конфигурации:
уходящего агликона в исходном соединении.
75
'Отклонения от этого правила возможны в тех случаях, ког-
да реакция элиминирования протекает по циклическому ме-
ханизму б. Из этого правила следует также, что на основании
лишь данных по аномерному составу продуктов реакции нель-
зя судить о том, осуществляется ли реакция замещения по
механизму прямого вытеснения агликона из циклической фор-
мы («обращение конфигурации») или по пути раскрытия пи-
ранозного кольца с образованием промежуточных соединении
адетального типа.
3.6. Об оптической активности
диастереоизомерных форм
Как показывают расчеты (6), относительное содержание
промежуточного продукта достигает значительной величины—
от 50 до 90%. Это дает основание отождествить знак угла вра-
щения в экстремумах кинетических кривых a(t) со знаком оп-
тического вращения соответствующего промежуточного про-
дукта. Рассмотрение этих величин в зависимости от природы
уходящего и атакующего N-агликона позволяет установить
определенную закономерность. В табл. 17 суммированы на-
блюдаемые углы вращения для ряда кинетически изученных
систем N-глюкозид ариламнна + арнлампн, при этом N-агли-
коны расположены в порядке возрастания электроноакцеп-
торпых свойств заместителей в бензольном кольце, т. е. по
мере увеличения ст-константы заместителя по Гаммету. Не-
трудно заметить, что положительные значения углов враще-
ния группируются преимущественно (более 80% от общего
числа) в верхней части таблицы — выше «диагональных» си-
стем, в которых объединяются промежуточные соединения с
одинаковыми N-агликонами. Отрицательные углы располага-
ются в нижней левой стороне таблицы. Исключение составля-
ют четыре системы: N-глюкозид п-толуидина-|-лг-толуидин (то
же 4- анилин), N-глюкозид м-толуидина + анилин, N-глюко-
зид м-пнтроанилииа+п-ампнобензойная кислота.
Оказывается, что введение в молекулу N-арилглюкозида
более электроотрицательного N-агликона, чем исходный агли-
кон, приводит к образованию диастереоизомера с положитель-
ным знаком оптического вращения. Противоположный знак
наблюдается при вытеснении исходного N-агликона менее
электроотрицательным заместителем. Используя в качестве
меры электроноакцепторных свойств заместителей гамметов-
ские константы, наблюдаемую зависимость можно сформули-
76
Таблица 17
Углы вращения в экстремумах кинетических кривых реакции N-траисгликозилирования (90%-ный
водный этанол, ацетатный буфер, 20°С, концентрации компонентов 0,08 моль/л, длина трубки—2 <).и)
Заместитель агликоне В Заместитель в атакующем ариламине
1 2 3 1 4 5 6 ! 7 1 8
м-С1!3 (1) — 2,9 — 4,8 + 1.0 4- 6,5 + 3,5 + 14.1
м-СНэ (I) — 3.3 + 3.2 4- 5,0 + 6,8 + 3,8 + 13.7
Н (3) 4- 2,1 + 2,2 + 3,4 + 7,5 + 9,5 + 5,7 + 15.5
м—Вг (5) — 12,7 — 9,0 + 0,8 + 5.6
п-СООС2Н5 (6) - 5,7
м—NOj (7) - 8,8 - 8,7 —5,6 - 9,0 — 7,0 — 7,3 + 0.7
п-СООН (8) - 6.1 — 5.0 + 3.0 + 2.0
г.—NO2 (9) -10,8 —11,1 — 11,0 — 12.0 -13,5 -13,5
Глава 4
КИСЛОТНЫЙ КАТАЛИЗ РЕАКЦИЙ
ГЛИКОЗИДНОГО ЦЕНТРА
4.1. Введение
Гомогенный кислотно-основной катализ чрезвычайно ши-
роко распространен в органической химии [291—295] и в
частности в химии углеводов. Хорошо известна высокая чув-
ствительность реакций моносахаридов и их производных к
добавкам кислот и оснований, однако до последнего времени
в химии углеводов сравнительно мало использовались пред-
ставления и методы кислотно-основного равновесия и катали-
за. Впрочем, в свое время мутаротация глюкозы и гидролиз
сахарозы послужили удобными модельными реакциями, на
примере которых разработан ряд важных положений о тауто-
мерии и специфическом кислотном катализе.
При объяснении явлений кислотно-основного катализа на
электронном уровне обычно исходят из предположения, что
присоединение или отщепление протонов приводит к такому
смещению электронной плотности в реагирующей молекуле,
которое оказывается благоприятным для данного химического
превращения. Иначе говоря, механизм катализа состоит в
поляризации связей. С учетом только этого фактора кислот-
ный катализ нуклеофильных реакций карбонильной группы
(и ее аналогов) можно представить следующей схемой:
Однако эта традиционная точка зрения на катализ как на
процесс, состоящий только из двух основных актов — пере-
80
носа протона и последующего химического превращения мо-
лекулы, — справедлива лишь для простейших одностадийных
реакций, мало вероятных средн органических молекул. Как
правило, органические реакции многостаднйпы, поэтому при
изучении кислотно-катализнруемых реакций надо учитывать
все возможные взаимоотношения между катализирующими
агентами и компонентами сложной реакции. Из этого следу-
ет, что полный анализ реакций, протекающих в условиях кис-
лотно-основного катализа, должен включать в себя два важ-
ных аспекта — установление механизма реакции и изучение
механизма катализа. Обычно для этой цели используют дан-
ные по зависимости констант скоростей реакции от кислотно-
сти среды.
В этой главе мы рассмотрим количественную сторону кис-
лотного катализа с точки зрения методов кинетики стацио-
нарных процессов [296, 297] применительно к реакциям гли-
козидного центра. Наша задача состоит в том, чтобы сфор-
мулировать некоторые достаточно общие правила, дающие
возможность ориентироваться при выборе наиболее вероят-
ного механизма реакции в целом и позволяющие качественно
предвидеть область оптимальной кислотности среды.
4.2. Кислотно-основной катализ
аномеризации N-гликозидов
Аномерные превращения моносахаридов и N-глнкозидов
с простыми алифатическими и ароматическими агликонами
описываются однотипными уравнениями кислотно-основного
катализа следующего вида:
Л к набл- = + кон [ОН ], (1)
где кн и к 01( — константы каталитического действия водо-
родных и гидроксильных ионов. Для установления физическо-
го смысла каталитических констант при аномеризации N-глн-
козидов можно в качестве пробного варианта воспользоваться
аналогией с мутаротацией моносахаридов (разд. 2.1), т. е.^
представить катализ аномеризации в виде схемы, предусмат-
ривающей два основных протолитических превращения N-аг-
ликона под действием кислотных и основных агентов — про-
тонизацию в кислых и депротонизацию агликона в щелочных
средах (речь идет о производных первичных аминов, имею-
щих водородный атом при атоме азота N-аглнкона). Возмож-
ные превращения ионизированных форм N-гликозидов показа-
ны на схеме 8. Для вывода уравнения (1) воспользуемся ме-
6 367
81
тодом стационарных концентраций, расчленив аномеризацию
на два процесса:
Л"а1 аз
NF + НзО+2: Н2О + NTH+ -> Р + Н3О+,
^"д2 ВЗ
Nr + OH-^H2O + Nr-->P + OH-
где NT, NFH+ и NT- — исходный аномер N-гликозида, прото-
низованная и депротонизованная формы N-гликозида, Р —
конечный или промежуточный продукт, образующийся на ста-
дии, лимитирующей реакцию аномеризации в целом, а3 и в3—
константы скорости лимитирующих стадий, Kai и Каг —
константы диссоциации:
[NF] [H+]/[NFH+], [Nr-] [H+]/[NF]. (2)
С учетом уравнений материального баланса по ионизиро-
ванным и неионизированным (исходным) формам выражения
для констант скоростей реакции, протекающей под действием
кислот (Ki) и оснований (к2), имеют вид:
Ki = fl3 [Н+]/(Ка1+[НЧ), «2=в3 Ка»/(Ка» + [Н+]). (3)
Суммарная константа скорости с учетом константы (ано-
меризация под действием молекул воды) равна:
кнабл- =Ko + Ki +К2 =
= к0 + а3 [Н+]/(Ла1+[Н + ])+в3 Ка,/(Ка1+[Н+]). (4)
Если ЛД1>[Н+] и ^Й2<[Н'Ь] (что, вероятно, и имеет место
в большинстве случаев), то уравнение (4) упрощается:
кнабл.=к0+а3 [Н+]/Кв1+в3 КД»/[Н+] (5)
или с учетом «ионного произведения воды» (Кх):
Кнабл.= коЛ-а3 [Н+]/Кй1+в3 KaiKxl[Нт]. (6)
Таким образом, в указанном предположении эмпирические
параметры кни кон уравнения (1) зависят от кислотно-ос-
новных характеристик N-гликозида, т. е. от его протоноакцеп-
торных и протонодонорных свойств, выражаемых количествен-
но константами диссоциации Kai и Кач- По аналогии с мута-
ротацией моносахаридов можно получить выражение для
к наблвключающее константы диссоциации катализирующих
агентов.
82
Уравнение (5) можно переписать следующим образом:
a:1[11+] г+к9Ка1 [Н+] +e9KaiKai m
кнабл.~ Ка1[Н+1 ’ 1 1
Графически уравнение выражается в виде кривой с миниму-
мом при некотором значении [Н+]о, которое легко найти, ре-
шая уравнение (7) на экстремум:
[H+]20 = lg(e3/a3) Ка1Каг, (8)
рНо = О,5 (рКа1+рКа2) — 1g (в3/а3). (9)
В соответствии с уравнением (7) экспериментальные кнадл
должны располагаться на шкале pH в виде кривой «цепной
линии».
Этот вывод в целом согласуется с данными кинетических
измерений аномеризации некоторых N-арилгликозидов. Не-
трудно объяснить и тот факт, что в отличие от моносахаридов
аномеризация N-гликозидов протекает в кислых средах зна-
чительно быстрее, чем в умеренно щелочных. Указанное об-
стоятельство связано с более высокой основностью атома азо-
та N-агликона по сравнению с полуацетальными кислородны-
ми атомами моносахаридов. Поэтому кривые Килбл(рН) ано-
меризации оказываются сильно смещенными в сторону ще-
лочных значений pH по сравнению с кривыми мутаротации.
Если константы tz3 и е3 не очень сильно различаются, то рН0
равен полусумме показателей основности N-гликозида. Та-
ким образом, представляется возможным по эксперименталь-
но найденному минимуму кривой кпа1;л- (pH) оценить прибли-
женное значение одной из констант диссоциации, если извест-
на вторая.
4.3. Кислотный катализ реакций
нуклеофильного присоединения по С,
Рассмотрим реакцию нуклеофильного присоединения аген-
та В по гликозидному центру А, протекающую через стадию
образования промежуточного продукта X, полагая, что в усло-
виях стационарности лимитирующей является стадия превра-
щения промежуточного соединения:
А + В?Х->Р, (и)
кг
83
V«e^.=«3[X]=-^-[A] [В], (10)
^21^3 z
кнабл- =KiK3/(k2+k3). (И)
Если учесть уравнение материального баланса для участвую-
щих в реакции компонентов, то можно получить вместо (10)
выражение, которое трудно использовать при анализе кине-
тических данных. Например, учитывая [В]о = [В] + [Х], полу-
чим для наблюдаемой скорости
V набл = к1кз [А] [В]/(Кг+ Кз + ^i [А]). (12)
В этом случае наблюдаемая скорость реакции не подчиняется
линейной зависимости от концентрации одного из компонентов,
реакции (а) и с увеличением последней стремится к некото-
рому предельному значению. Поэтому понятие KHagA., строго
говоря, теряет ясный физический смысл; т. е. мы не можем ее
даже определить из кинетической кривой. Однако опыт пока-
зывает, что в некотором интервале исходных концентрации А
и В наблюдаемая скорость с хорошим приближением можег
быть представлена линейной функцией от [А]. Поэтому если
относительная концентрация компонента А не очень велика,
можно использовать выражение (10) для количественной
оценки кНа6л. Этот вариант линейного приближения и будем
использовать в дальнейшем.*
Брутто-схема реакции присоединения, записанная в форме
(а), не содержит кислотно-основного катализа. Поэтому ни-
чего нельзя сказать о зависимости эффективных констант
скоростей Klt к2 и к3 от концентрации и свойств каталитиче-
ских агентов (Н3О+ и ОН-). Эту зависимость можно устано-
вить, дополнив схему стационарной кинетики элементарными
стадиями, связанными с переносом протонов. Для простоты
допустим, что в условиях кислотного катализа указанные ста-
дии являются равновесными (часто говорят, что данное об-
стоятельство является условием истинного специфического
катализа [291]. Конечный результат, т. е. зависимость кн.абл-
от [Нч] и величин Ка, определяется тем, какая схема кис-
лотной активации положена в основу.
Рассмотрим типичный случай конкурентной протонизации
исходных реагентов — протонизация молекул А облегчае,
присоединение В, в то же время протопизация молекул В пре-
’ К. Педерсеном и Р. Беллом [291, 29S] показано, что такого род i
упрощения схемы кислотного катализа во многих случаях приводят к урав-
нениям, хорошо согласующимся с экспериментальными да-шымп.
8!
пятствует их присоединению к Л, Исходя из этого, можно за*
писать схему реакции (а) в следующем развернутом виде:
(Zi аз
АН++В^ХН--*Р+Н+
Ла2, *
А ВН+
где Кат_ и К й2 — константы диссоциации активных центров
А и В
Ка1 —[А][Н + ]/[А11 + ], Аа2=[В][Н+1ДВН+]. (13)
Если считать, что стадия аз определяет скорость реакции
в целом, то
УНабл.-а3[ХИ+] (14)
или, так как aJa2~Kp = [ХН+]/ [АН+] [В],
Уцабл- азКр[ АН+] [В]- (15)
Для получения окончательного результата учтем уравне-
ния материального баланса по протонизованным и непротони-
зованиым формам А и В:
[А]о= [А] + [АН+], [В]о= [В] + [ВН+], (16)
•откуда с учетом (13) следует
[АН+] = [A]0[H+I/(Z<al + [H+]), |В] = |В]0Ка2/(Кв2-ИН4-]). (17)
Подставив (17) в (15), получим
^л. = !А0НВ]0а8^[Н+]/(Аа1+[Н+])(Аа2+[Н + ]) (18)
&
и таким образом наблюдаемая константа скорости равна
as/<p[HT]
К аналогичному результату придем, если рассмотрим толь-
ко первую стадию, полагая, что именно она определяет ско-
рость реакции в целом:
АН++В-»ХН+,
Унабл.=а{ [АН+] [В],
85
____________01 [H+J____________
«набл. =И11+1Ч- (Л'й1+Кй8)IН+ ] 4-Ка,Ка2 ‘
(20)
Формула (20) отличается от (19) только тем, что в числителе
вместо множителя а$Кр стоит а,. В обоих случаях зависи-
мость кнаал. от pH выражается колоколообразной кривой с
максимумом при рН0, равном
рНо=0,5 (р/Са14-рАа2).
(21)
Рассмотрим не менее распространенный случай, когда про-
тоноакцепторные свойства А выражены не настолько сильно,
чтобы протонизованные формы могли играть какую-либо за-
метную роль в относительно слабо кислых растворах. Ясно,
что если и в этом случае экспериментальная зависимость
киа(5л.от pH указывает на существование кислотного катализа,
то в рамках принятой схемы (а) единственно возможным ва-
риантом является катализ за счет кислотной активации про-
межуточного продукта:
Каз аз
X + HVXH+->P + H+,
(в)
Хвз=|Х1[Н+]/[ХН+], [Х]0=[Х| + [ХН+], (22)
[ХН+] = |Х0][Н+]/(^+[Н+П. (23)
С учетом (в) схему кислотного катализа следует записать а
следующем виде:
at
А + ВГ
j
ВН+
X
ХН+
V tl«o К„:>
Ка* 4 - а ХЬ
а3
+ Р+Н+ •
(г>
Так по условию протолитические акты совершаются значи-
тельно быстрее химических превращений, то, как и. выше, кон-
станты скорости этих актов не включаются в схему реакции;
учитываются только равновесные концентрации протонизован-
пых и непротонизованных форм В и X. Принимая это во вни-
мание, можно записать для константы равновесия стадии об-
разования промежуточного продукта выражение
Лр=а1/а2=([Х] + [ХН+])/[А] [В] = [Х]0/[А] [В]. (24).
86
По-прежнему полагая
Ц^л.=а3 [ХН+],
получим на основании (22) и (24)
кнабл. |н+тв,Ш1ншл •
(25)
Видно, что и в этом случае зависимость кна^л. от pH вы-
ражается колоколообразной кривой, но теперь максимум кри-
вой располагается при pH, равном
рНо = 0,5(р^24-р^3). (26)
Таким образом, если нет никаких дополнительных сведений
о реакции, оптимум pH является фактически единственным
Рис. 13. Зависимость наблюдаемой
константы скорости N-гликозилирова-
ния от pH среды при взаимодействии
п-толуидина с глюкозой (1), галакто-
зой (2), ксилозой (3) и арабинозой
(4) (ЗО°С, 80%-пый водный этанол,
ацетатный буфер, с=0,25 моль 1л, эк-
вимолекулярные концентрации; здесь
и далее на рнс. 14—16 константы ско-
рости выражены в виде произведе-
ния константы скорости 2-го порядка
на начальную концентрацию компо-
нентов реакционной смеси).
"ST
Рис. 14. Зависимость кон-
станты скорости образова-
ния N-ксилозида анилина от
pH среды (25°С, 80%-ный
водный этанол, ацетатный
буфер, с=0,25 моль/л).
Светлые точки — экспери-
ментально найденные значе-
ния констант, черные —
рассчитанные по уравнению
(33).'
признаком, по которому можно отличить один случай ката-
лиза (активация А) от другого (активация X).
Обратимся к экспериментальным данным. Возьмем в ка-
честве примера зависимость кнавл. от pH, наблюдаемую в ре-
акциях образования N-арилгликозидов. С увеличением кис-
лотности среды кнас)Л- вначале возрастает, а затем выше не-
рН
Рис. 15. Зависимость кон-
станты скорости образова-
ния N-ксилозида п-толуиди-
на от pH среды (30°С. 80 % -
ный водный этанол, ацетат-
ный буфер, с=0,25 лоль/л).
Светлые точки — экспери-
ментально найденные значе-
ния констант, черные — рас-
считанные по уравнению
(35).
S3
Рис. 16. Зависимость константы скорости образо-
вания фенилгидразона глюкозы от pH среды
(30°С, 50%-ный водный этанол, ацетатный буфер,
с=0,1 моль/л). Кривая кнаал. (pH) рассчитана по
\~абл. -6,10~5fH + ]/([H+p+1,13,10-3[Н + Л-
+ 8.10-10).
которой оптимальной кислотности среды монотонно снижает-
ся (рисунки 13—16). Для заданного ариламина положение
максимума кривых на шкале pH практически не зависит от
природы моносахарида, как это можно видеть на примере об-
разования N-глюкозидов п-толуидина (рис. 13). Для задан-
Рис. 17. Корреляция между
оптимальными значениями
pH, при которых константа
скорости образования N
арилглюкозидов максималь-
на, и величинами рК (в
Н2О) ариламинов: п толуи-
дииа (1), л-толуидина (2),
анилина (3), .«-бромапили-
на (4), л-хлорапилииа (5),
л-нитроанилина (6), п-ами-
нобензойпой кислоты (7),
анестезина (S) и п-нитро-
анилина (9). Зависимости
констант скоростей N-глико-
знлирования от pH изучены
В 80%-пом водном этаноле
при 30—45°С 1242].
•О
кого же моносахарида оптимум pH регулярно смещается в
область кислых значений pH с уменьшением основности ата-
кующего амина (рис. 17).
Подобные колоколообразные зависимости кна^л. (pH) на-
блюдались рядом авторов при изучении реакций нуклеофиль-
ного присоединения азотистых оснований по карбонильной
группе [203], однако однозначного объяснения они не полу-
чили. Распространенной среди химиков-органиков точкой зре-
ния на природу экстремалей этого типа является следующая.
Полагают, что протон координируется с кислородом за счет
свободной электронной пары. В результате смещения элек-
тронной плотности увеличивается эффективный положитель-
ный заряд на углероде, тем самым облегчается присоединение
нуклеофильного агента НВ по С:
С = О + Н3О+^Н2О+ (С-ОН 2 С—ОН)
; +НВ
Н++В—С—ОН ч- н+В—с—он
Развивая эту идею, можно полагать далее, что протониза-
ция НВ приведет к образованию нереакционноспособной фор-
мы (НВН+), в которой электронная пара блокирована прото-
ном. Таким образом, имеем пример «конкурентной» протони-
зации участников реакции. Этого достаточно, чтобы понять,
почему с увеличением кислотности среды скорость реакции
сначала возрастает, а затем снижается ниже некоторого оп-
тимального значения pH. Действительно, увеличение скорости
в правой ветви кривой можно связать с ростом концентрации-
протонированных форм субстрата, а снижение ее в левой вет-
ви кривой кнабЛ. (pH) — с уменьшением концентрации сво-
бодной (непротонированной) формы нуклеофильного агента.
Однако, если обратиться к количественной проверке этой
схемы, сразу обнаружится явное несоответствие схемы с ре-
зультатами расчета. Л именно, если рассчитать по формуле
(21) величину рДй1 (характеризующую протоноакцепторные
свойства моносахаридов в соответствии с принятой схемой ка-
тализа), то получим значения, на 5—6 порядков превышаю-
щие показатели основности полуацетальных кислородных ато-
мов (разд. 2.1).
Указанное противоречие можно устранить, если исходить
из предположения, что катализ реакции N-гликозилирования
определяется протонированием промежуточного продукта ре-
акции (аминоспирта). Для этого случая справедливы схема
(г) и уравнения (25) и (26). Из последнего уравнения можно,
сразу оцепить величину рКйз> характеризующую протоноак-
90
цепторные свойства промежуточного соединения. Эта же ве-
личина, а также и другие параметры уравнений типа «коло-
кола», могут быть определены линеаризацией правой и левой
ветвей зависимости кна^л. ([Н+]).
Для правой ветви кривой кна6л. (pH) [Н+]2<Ка2 Kai, по-
этому, пренебрегая квадратичным членом в знаменателе урав-
нения (25) и переходя к обратным величинам, получим
I ' + Кд,
1^набЛ~ а3КрКа2 а3^[Н+] • (27>
В координатах этого уравнения (1/кКйбл. — 1/[Н+]) экспери-
ментальные точки должны располагаться на прямой с угло-
вым коэффициентом а и отрезком в на оси ординат, равными^
(1=Каз/а3Кр, e = (Kai+ Ka3)\a3KpKai- (28)
Для левой ветви кривой, учитывая
[H+]s>/CaaKa3 (29)
и пренебрегая свободным членом в знаменателе уравнения
(25), аналогичным образом получим приближенное соотно-
шение:
Ка9+Ка* , 1Н+]
11Хнабл.--= а3КрКа2 ' а3КрКа3 <30);
В этом случае в координатах 1/Кншуд. — [Н+] эксперименталь-
ные точки должны располагаться на прямой с угловым коэф-
фициентом а'н отрезком в' на оси ординат, которые равны со-т
ответственно
а' == Ha3KpKat, в’ = (Каз+КазУа3КрКа1 (31)
Так как в —в', то имеется три уравнения из четырех, из ко-
торых могут быть вычислены величины Ка1) Ка3 и произведе-
ние а3Кр Если известна величина рКai, то значение рКдз
промежуточного соединения может быть рассчитано по выра-
жению для оптимума pH (26) и по значению константы ско-
рости в максимуме кривой к«аб7г(рН). Приведем два примера.
При образовании ксилозида анилина (рис. 14) максимум
константы скорости наблюдается при рН~-5,0. Используя для
константы диссоциации анилина в 80°/о-ном водном этаноле
[299] величину Ка» = 1,82• 10-4 (25°С, рКа2=3,74), найдем по
(26):
Ка3 =5,5-10~7, рКд3= 6,26. (32)'
9Ь
Уравнение для кна6л-в функции концентрации водородных
ионов для этого случая имеет следующий вид:
1,27-10-5 [Н+]
кнабл- = [НЧ2+ 1,83- 1(Н [Н+Т+ЫО-10’ (33)
Кривая рассчитанная по (33), удовлетворительно
согласуется с экспериментальными данными (рис. 14).
Для реакции образования ксилозида п-толуидина пара-
метры уравнения (25), вычисленные графически с учетом
рК 4,32 для п-толуидина в 80%-ном водном этаноле [300],
равны
а;Л/? = 0,31 лщн-1, Л]гз = 5,3-10-‘, р/у.Д.3, (34)
а уравнение с численными значениями коэффициентов имеет
вид:
1,5-10-5 [Н+]
кнабл = [нД2+4,85- 10--5[Н+] +2,52-10““ ’
Рис. 15 иллюстрирует степень соответствия кривой кна$л,
(pH), рассчитанной по уравнению (35), с экспериментальны-
ми данными. Полученные значения рХ\и свидетельствуют о
более высокой основности промежуточных соединений по
сравнению с исходными аминами. Это согласуется с извест-
ными в литературе фактами увеличения основности арилами-
нов при замещении одного из водородных атомов аминогруп-
пы на алкильный заместитель [299]. Вместе с тем различия
в показателях основности атакующего ариламина (рКй2) и
N-агликопа (р/\ й1) слишком велики, чтобы их можно было
отнести только за счет индуктивного влияния углеводного ос-
татка при С]. Эти различия трудно объяснить также и воз-
можным образованием внутримолекулярных Н-связей (напри-
мер, между гликозидным гидроксилом НС]—ОН и кислород-
ными атомами при С2 и С3). По всей вероятности, увеличение
основности в данном случае обусловлено стерическими препят-
ствиями со стороны объемистого углеводного остатка, ослаб-
ляющими перекрывание п-орбитален атома азота гликозидной
связи С]—N с л-орбиталями бензольного кольца. Известно, что
уменьшение степени сопряжения между п- и л-орбиталями в
ариламинах приводит к существенному увеличению основно-
сти аминогруппы. Впрочем, этот вопрос следует обсуждать
лишь после того, как будут получены оценки основности про-
межуточных соединений реакций присоединения по С| други-
ми (не кинетическими) методами. В настоящее время мы та-
кими сведениями не располагаем. Знание же этих величин
92
имело бы большое значение для решения задач прогнозирова-
ния реакционной способности кислотно-катализируемых реак-
ций присоединения и замещения при С(.
4.4. Катализ реакций гидролиза
N-гликозидных связей
Кислотно-катализируемый гидролиз N-гликозидов и других
продуктов присоединения азотистых оснований по Ci можно
попытаться описать по аналогии с рассмотренной выше схе-
мой катализа прямой реакции. Однако надо заметить, что ко-
личественное истолкование эмпирических зависимостей кна^л.
(pH) для этих реакций встречает большие трудности, чем
объяснение механизма катализа прямых реакций [203, 204].
Выше нами отмечено, что изучение кинетики гидролиза N-гли-
козидных связен в широком диапазоне pH приводит к колоко-
лообразной зависимости кндй7.от pH (разд. 3,4), вполне ана-
логичной «колоколу» для прямой реакции, за исключением
того, что кривые кнарл. (pH) для гидролиза сдвинуты всегда в
более кислую область. При этом нетрудно заметить, что чем
ниже основность N-агликона (азотистого фрагмента при кар-
рн
Рис. 18. Зависимость константы скорости кислот-
ного гидролиза N-глюкозидов п-нитроанилина (1),
п-аминобензойной кислоты (2), анилина (3) и
п-аминофенола (4) от pH среды при 25°С, по дан-
ным работы [263].
93
'бонильном центре), тем в более кислую область смещена кри-
вая кнабЛ. (pH) (рис. 18).
В настоящее время, по-видимому, нет общепринятого объ-
яснения механизма катализа реакций гидролиза. Мы уже от-
мечали (разд. 3.4), что Исбелл и Фраш [251] исходят из то-
го, что для гидролиза N-гликозидной связи необходима одно-
временная атака ионов Н3О+ и ОН-. Аналогичной точки зре-
ния придерживался Вилли [301] при объяснении гидролиза
производных бензилиденанилина. Дженкс с сотр. объясняют
колоколообразный характер кривых кнаал (pH) изменением
лимитирующей стадии в цепи: протонированная форма шиф-
фова основания — аминоспирт — биполярная форма амино-
спирта — конечный продукт распада [197].
При рассмотрении разных вариантов катализа можно по-
казать, что искомое уравнение «колокола» для реакций гид-
ролиза получается, по крайней мере, в трех случаях:
— если исходить из достаточно общего предположения о
том, что эффективность кислотного катализа зависит не толь-
ко от сродства исходной молекулы к кислотному агенту, но
и от способности промежуточного соединения к депротониза-
ции (мерой которой является константа кислотной диссоциа-
ции его) (схема 9);
— если полагать, что в завершающей стадии реакции про-
межуточное соединение участвует в нейтральной форме, для
чего необходимо отщепление протона, присоединившегося к
молекуле на первой стадии;
— наконец, если допустить, что в прямой реакции ката-
лиз осуществляется путем протонизации аминогруппы проме-
жуточного соединения (в форме аминоспирта), а в обратной
реакции за счет протонизации гидроксила при С] той же
формы.
Нетрудно заметить, что во всех трех случаях предусматри-
вается комбинация двух конкурирующих элементарных ак-
тов — протонизация и депротонизация компонентов реакции.
Это — единственное, что объединяет все схемы катализа. Вве-
дение в схему катализа конкурирующих протолитических ак-
тов автоматически приводит к экстремальной зависимости
кнабл- от кислотности среды. Для выбора наиболее вероятной
схемы следовало бы сравнить величину р/<а, найденную ана-
лизом кривых KHaf)q. (pH), с показателем основности промежу-
точных соединений, определенным каким-либо другим незави-
симым методом. Мы не можем пока этого сделать из-за от-
сутствия данных по основности промежуточных соединений.
Поэтому выберем в качестве пробного первый вариант схемы
94
-1
Схема 9. Механизмы катализа аномеризации N-гликозидов под дей-
ствием кислот (а) н оснований (б).
и рассмотрим данные по кислотному гидролизу этилового эфи-
ра N-глюкозилглицина (ЭГГ) [303,304]:
—НС—R—НС—NH—СН2СООС2Н5 + Н2О->
!__о___।
——НС—R—НС—OH + NH2CH2COOC2H5
’________о___1
В гл. 1 отмечено, что замена карбоксила а-аминокислот
на сложно-эфирную группу облегчает образование N-глико-
зидной связи при взаимодействии с незамещенными сахара-
ми. Получающиеся при этом N-гликозиламинокислоты вполне
устойчивы в нейтральных средах в отличие от нестойких про-
дуктов присоединения незамещенных а-аминокислот к моно-
сахаридам. Поэтому сложные эфиры N-гликозиламинокислот
представляют собой удобную модель для изучения свойств
-N-ал кил гликозидных связей в гликозиламинокислотах. Гидро-
95
лиз этих связей в условиях кислотного катализа при умерен-
ных температурах не сопровождается побочными процессами
— гидролизом сложно-эфирных связей, перегруппировкой
Амадори и т. п. Поэтому изменение угла вращения реакцион-
ных растворов характеризует только гидролиз связи С[—N
(рис. 19).
Рис. 19. Изменение наблюдаемого угла вращения при кислот-
ном гидролизе этилозого эфира N-глюкозилглицина при раз-
личных pH (25°С, 50%-нын водный этанол, ацетатный бу-
фер, с = 0,2 моль/л).
Рис. 20. Зависимость констан-
ты скорости гидролиза этило-
вого эфира N-глюкозилглицина
от pH среды при различных
температурах (50%-пый вод-
ный этанол, с=0,2 моль]л, аце-
татный буфер — при pH 0,7—•
6,5, НС1О« — при pH —0,3,
NaOH — при pH 6,5) [304].
86
Кривые зависимости кна^л. от pH показаны на рис. 20.
Форма кривых существенно не изменяется при изменении тем-
пературы реакции. Аналогичные зависимости наблюдаются
также и при гидролизе этилового эфира N-галактозилглицина
[304]. Параметры активации гидролиза ЭГГ мало изменяют-
ся при изменении pH в интервале от 2,5 до 5,5 и в среднем
равны:
7^ ¥-
АН = 4,5ккал/молъ, AS — -54кал]град-моль,
AG =20,4 ккал1моль.
В соответствии со схемой катализа заключительный этап
реакции можно представить последовательностью актов про-
тонизации аминной и депротонизации гидроксильной групп:
ОН о-
I !
—НС!—NHR НС,—N+H2R—>—НС, = 0 + NH2R
В этой цепи наиболее существенным звеном является акт де-
иротинизации гидроксильной группы, инициирующей переход
ее в карбонильную группу (протонизация же сильно основной
аминогруппы осуществляется достаточно легко в кислых сре-
дах). Таким образом, мы имеем два важных в кинетическом
отношении переноса протонов — протонизацию азота в исход-
ном N-гликозиде (или в его раскрытой форме) и депротониза-
цию гидроксила в промежуточном продукте. С учетом этого,
запишем схему стационарной кинетики: *
/С 1
эгг+н3о+ #‘эггн++н,о
Кр Kai
ЭГГН+ + Н2О г ХН+ ХН+-+Н+ (а)
ХН+-—эг+г,
где X, ХН+, ХН +~— промежуточное соединение, его протони-
зованная (по азоту) форма и биполярный ион указанной вы-
ше структуры, и K.ai — константы диссоциации амино-
спирта по азоту и по гидроксильной группе соответственно.
Полагая равновесной стадию присоединения воды к ЭГГН+
и используя выражения для констант диссоциации Kai и К at>
а также уравнения материального баланса по исходному и
7 367
97
промежуточному соединениям, получим уравнение для наблю-
даемой константы скорости:
________^Л1Н+]
к=[Н+р+(К01 + Каа)[Н+]+ад»- (36)
Отметим, что аналогичное по форме уравнение для кНа$л.
в функции [Н+] может быть получено на основании схемы ка-
тализа с участием на первой стадии протонизованного исход-
ного вещества, а на второй — нейтральной формы промежу-
точного продукта:
Н-Н+ +Н2О — Н+ а3
ЭГГ -> ЭГГН+ - ХН+ X—ЭГ+Г.
Хотя и здесь мы имеем дело с двумя конкурирующими прото-
литическими равновесиями на разных стадиях гидролиза, эта
схема, по химическим соображениям, представляется менее
вероятной, чем первая. Подчеркнем еще раз, что для форми-
рования карбонильной группы в переходном состоянии за-
ключительной стадии реакции необходима кислотная диссо-
циация ОН-группы при С[ аминоспирта, и, следовательно, все
факторы (кислотность среды, влияние природы заместителей
при Ci), затрудняющие эту диссоциацию, будут одновременно
подавлять и скорость гидролиза N-гликозидной связи.
В соответствии с (36) зависимость к набл-^рН) выражает-
ся графически колоколообразной кривой гауссовского типа,
симметричной относительно оптимума pH. Выражение для
рНол„г. можно найти, решая задачу на экстремум функции
(36). В результате получим
= /(^2, рЫолот.=0,5(Ма14-/’^а«)- (37)
Входящие в (36) константы К fli и Ка2 характеризуют про-
толитические равновесия по азоту исходного соединения и по
ОН-группе промежуточного аминоспирта. Эти константы мо-
гут быть определены из анализа экспериментальной кривой
««абл-(рН). В отличие от Kat физический смысл константы
Kat менее ясен. Рассмотрим полную схему диссоциации ами-
носпирта (в условиях кислотно-основного равновесия):
ОН ОН
I «1 I +
-HCi-NHR^-HCi-NHaR
«4 t I t I ~«2
o- o-
I «з I
—HCi-NHR?— HC!-N+H2R
98
где «ь к.2, Кз, Kt — индивидуальные константы диссоциации
элементарных равновесий, связанные с макроскопическими
константами (определяемыми, например, потенциометриче-
ским титрованием) уравнениями:
Ki = Ki + K2, «2= 1/(1/«3Ч-1/«4).
Вопрос о том, какой из указанных констант — индивидуаль-
ной («2 или Кз) или макроскопической (К2) — соответствует
определяемая из кинетических измерений константа тре-
бует специального исследования и не может быть решен на
основании только кинетических данных.
Параметры уравнения (36) можно рассчитать линеариза-
цией правой и левой ветвей кривой зависимости к „а(>л. от pH в
координатах: 1/кна6л.— 1/[Н+] и 1/кнабл — [Н 1 ], соответст-
венно (табл. 18). В результате находим
[H+L^. =3,5-10-\ pH Оя/Я.=3,46,
К =6,78-10-3, р/(а1 2,27,
Ка2 = 1,87-10-5, р/Саа4,73,
а3К.р = 240 мин~'.
С учетом полученных данных выражение для кНадл. в функ-
ции концентрации водородных ионов имеет вид:
4,53-Ю-з [Н+]
кноЛ«- — [Н+]2 + 6,8-IO-3 [Н+] + 1,27-10-7' (38)
•Рис. 21. Зависимость констан-
ты скорости гидролиза этило-
вого эфира N-глюкозилглицнна
от pH среды (условия проведе-
ния реакции указаны в подписи
к рис. 20). Кривая кн(,с>л. (pH)
рассчитана по уравнению (38).
’ 99
Кривая КндЛг (рН), рассчитанная по уравнению (38),
удовлетворительно согласуется с экспериментально найден-
ными значениями констант скоростей, как это видно из рис. 21.
Отклонения экспериментальных точек от кривой к набл- (pH)
лежат в пределах ошибок эксперимента.
В исходном соединении наиболее основным центром яв-
ляется атом азота N-гликозидной связи (атомы кислорода
группы СООС2Н5 имеют рКа порядка -2 -О [307]). Одна-
ко полученное значение рКаа 2,3 довольно резко отличается
от рДа NH-группы этилового эфира глицина, который, со-
гласно данным работы Хая и Портера [305], равен 7,69
(25°С). Столь большие отличия нельзя объяснить индуктпв-
Таблица 18
Расчет параметров уравнения (36) применительно к
гидролизу ЭГГ в 50%-ном водном этаноле при 25°С [304]
(а и а' — угловые коэффициенты прямых в координа-
тах 1/к—1/[Н+] и 1/к — [Н । ], в и У — отрезки, отсе-
каемые этими прямыми на оси ординат).
Правая ветвь кривой к.набл- (pH):
pH к, мин—1 [Н+]- 10е (1/IH+H0-4 1/к
4,10 0,488 79 1,3 2,05
4,15 0,472 71 1,4 2,11
4,35 0,450 45 2,2 2,22
4.40 0,453 40 2,5 2,20
4,45 0,442 35 2,8 2,26
4,55 0,350 28 3,6 2,86
5,0 0,179 10 10 5,58
5,3 0,146 5 20 6,85
5,5 0,104 3,2 31 9,6
5,65 0,066 2,2 45 15,1
5,8 0,029 1,6 62 33,4
а= 2,8-10-5 в=/5
Левая ветвь кривой кнабл. (pH):
pH к, мин—' [Н+]-102 1/к
0,20 0,006 63 167
0,55 0,015 28,2 67
0,95 0,044 11,2 22,7
1,35 0,084 4,5 12
2,12 0,295 0,76 3.4
а'=220, в'=1,5
100
ным эффектом углеводного фрагмента при Сь В то же время
шиффовы основания имеют сравнительно низкие показатели
основности. Например, по Вольфендену и Дженксу [306], се-
микарбазон бензальдегида имеет рАа около -1. Оксимы ря-
да алифатических альдегидов обладают рКа близкими к ну-
лю. Таким образом, имеются основания предполагать, что по-
лученное нами pAni 2,3 относится к азоту, включенному в
двойную связь с С[. Иными словами, ЭГГ участвует в реакции
не в циклической форме, а в форме основания Шиффа, т. е.
так, как показано на приведенной выше схеме распада ЭГГ.
В этом случае стадии протонировапия предшествует обрати-
мое раскрытие пиранозного звена с образованием связи
C, = N.
Вторая константа относится, как сказано выше, к
ЮН-группе аминоспирта. Полученное значение этой константы
(р/Саа~4,7) по порядку величины близко к константе диссо-
циации карбоновой кислоты. Значительное усиление кислот-
ной диссоциации данной группы по сравнению с обычными
алифатическими спиртами можно отнести за счет эффекта
влияния основной аминогруппы, непосредственно связанной с
Ci. Механизм такого влияния аналогичен тому, который име-
ет место в обычных «цвиттерионах» [299]: протопизация ами-
ногруппы приводит к сильному смещению электронной плот-
ности от НО—НС[ — в сторону присоединившегося протона и
вследствие этого — к увеличению кислотных свойств гидрок-
сильной группы.
Напомним, что рассмотренный здесь механизм катализа
является лишь наиболее вероятным, но не единственно воз-
можным. Анализ зависимости кнадл. от [Н+], строго говоря,
не дает возможности однозначно выбрать механизм катализа
из нескольких альтернативных схем, так как различные ва-
рианты могут привести к уравнению одного и того же вида:
А-на^-=в1[Н+]/([НН[г+в2[Н+]+бз). (39)
Очевидно, что спрямление ветвей наблюдаемой зависимости
кнабл- от pH само по себе не является достаточным условием
для доказательства механизма катализа. Таким образом, про-
блема выбора наиболее вероятной схемы сводится к «про-
блеме коэффициентов» эмпирической зависимости типа (39),
т. е. к установлению физического смысла постоянных ei, в2 и
б3. Для этого необходимы дополнительные сведения, коаррые
вряд ли можно получить путем измерения кинетики реакций.
В связи с этим возникает вопрос о том, нужно ли вообще
Добиваться детализации механизма катализа? Не достаточно"
101
приведенные выше соотношения получены при самых общих
предположениях относительно природы электрофильного ком-
понента реакции, надо полагать, что сформулированное пра-
вило справедливо для нуклеофильных реакций карбонильной
группы и ее аналогов вообще, а не только для реакций гли-
козидного центра.
В дополнение к описанному выше примеру приведем дан-
ные по равновесию реакции взаимодействия глюкозы с гли-
цином [304]. Зависимость Кр от [Н+] выражается в данном
случае не обычной кривой с «насыщением» при предельных
значениях pH, а в виде «колокола» (рис. 29). Вся кривая
2<р(рН) располагается в щелочной области pH с максимумом
при pH 8,4, что в соответствии с изложенными ранее выво-
дами (разд. 4.3) свидетельствует об участии в реакции по Ci
аминогруппы глицина, имеющей рКа 9,7.
Рассмотрим коротко основы метода, с помощью которого
может'быть произведена оценка величины [303, 315—317].
Рис. 29. Зависимость эффектив-
ной константы равновесия об-
разования продукта присоеди-
нения глицина к глюкозе от pH
среды (35°С, 30%-ный водный
этанол). В нижией части ри-
сунка приведена кривая изме-
нения процентного содержания
анионной формы глицина
(NH2CH2COO—) в зависимости
от pH. Кривая Кр(рН) рассчи-
тана по уравнению (65) [304].
114
В водных растворах существует равновесие между различ-
ными ионными формами а-аминокислоты:
Kai Кн
ЛК АК'-^ АК~.
(г)
где АК+ и АК~ — катионная и анионная формы, АК+~—
цвиттерион, Kat и Ка?— константы диссоциации карбоксиль-
ной и аминной групп. Если ни одна из форм с глюкозой не
реагирует, то при добавлении последней к раствору глицина
pH раствора из-за малой кислотной диссоциации глюкозы за-
метным образом не изменится. При взаимодействии же ани-
онной формы кислотность среды увеличится на величину, про-
порциональную концентрации продукта присоединения, если
его основность ниже основности аминогруппы глицина. Пре
небрегая для нейтральных и слабощелочных растворов кати
онной формой АК+, запишем схемы превращений в чистом
растворе аминокислоты (а) и в присутствии ’моносахарида (б):
Ая2
а)АК'-^АК-+Н\ /<я2 = |АК-][Н -|/[А1(| - 1 (-^)
«а*
б)АК+- АК-+Н+
А К-ГАК- К/,= 1ГЛК-]/[АК-][Г], (58)
где ГАК- — продукт присоединения глицина к глюкозе.
Обозначив через а долю анионных форм аминокислоты,
найдем отношение а/(1—а) для двух рассматриваемых слу-
чаев:
(a) a/(l-a) = [AK-]/[AK+-]=KaS/lH' J (59)
(б) а/(1-а) = ([АК-],+ [ГАК-])/[АК+-] =
-Ля2(Н-Ар[Г])/[Н ' ], (60)
где индексом «штрих» обозначены концентрации ионов в сме-
си моносахарид+ аминокислота. Из (59) и (60) находим
[Н+] = [Н ] (1+ЛДГ]). (61)
После логарифмирования (61) получим окончательно ’**
ДрН = рН - pH' = lg(l+Kp[r]). t (62)
115
весное» распределение электронной плотности валентных
оболочек. От того, в какой последовательности совершаются
акты распада и образования связей В—С и А—В, зависит, в
конечном счете, энергетический баланс переходного процесса.
Нет никаких оснований считать, что во всех случаях химиче-
ское взаимодействие начинается с присоединения атакующего
атома. Равным образом нельзя считать единственно возмож-
ным химический процесс с начальным распадом старой и по-
следующим образованием новой связи. С точки зрения общих
соображений статистической физики, оба варианта следует
признать равновероятными. Возможен также и третий случай,
при котором атакующий и уходящий атомы движутся строго-
согласованно в том смысле, что уменьшению межатомного
расстояния А ... В точно соответствует увеличение расстоя-
ния В ... С.
Посмотрим, какие изменения при этих трех вариантах дви-
жения будут претерпевать энтальпийная и энтропийная со-
ставляющие свободной энергии. Примем, что энтропия есть
мера молекулярного беспорядка системы, а энтальпия — ме-
ра внутренней энергии. При этом нас интересуют, в конечном
счете, разности энтропии и энтальпии системы в переходном
и начальном состояниях.
Случай а — образование связи А—В предшествует раз-
рыву связи В—С. По мере продвижения к переходному со-
стоянию энтропия системы должна, очевидно, уменьшаться за
счет уменьшения числа поступательных и вращательных сте-
пеней свободы, поэтому
— S’ = AS^<0. (25)
С другой стороны, так как энергия, выделяющаяся в момент
формирования новой связи, может частично компенсировать
затраты на разрыв старой связи, энтальпия активации долж-
на иметь в этом случае умеренно низкое значение.
Случай б — разрыв связи В—С предшествует образова-
нию связи А—В. Продвижение системы к переходному состоя-
нию сопровождается увеличением энтропии за счет увеличе-
ния числа степеней свободы в момент разрыва связи В—С,
поэтому
S^-S°=AS^>0. (26)
Энтальпия активации в этом варианте должна быть сущест-
венно больше, чем в случае а, так как в момент распада связи
В—С затраты энергии не восполняются за счет энергии, вы-
деляющейся при формировании связи А—В.
144
Случай в — процессы образования и распада связей про-
текают строго синхронно. Число степеней свободы при этом
остается постоянным на всем пути движения к переходному
состоянию:
5^—5°= AS^ = 0, (27)
а энтальпия активации в соответствии с (4) оказывается рав-
ной свободной энергии активации.
Предложенная модель переходного процесса позволяет
осмысленно подойти к интерпретации фактов линейной зави-
симости активационных параметров. Из нее следует, что в тех
случаях, когда реакция протекает по механизму непрерывного
замещения «старого» атомного остатка на «новый», большим
энтропиям активации должны соответствовать меньшие энер-
гии (энтальпии) активации и наоборот в зависимости от то-
го, в какой последовательности (согласованности) и с какой
степенью одновременности (синхронности) совершаются эле-
ментарные акты распада и образования химических связей по
мере продвижения к активированному (переходному) комп-
лексу. Повторим, что реакционная система сама выбирает
наиболее оптимальный путь движения в соответствии со
«своими возможностями» и в зависимости от окружающей
среды. Поэтому проблему активационных параметров хими-
ческих реакций можно, в принципе, сформулировать в рамках
вариационных принципов механики.
5. 4. О влиянии углеводного фрагмента
на реакционную способность гликозидного центра
Нами неоднократно отмечалось ранее, что в нуклеофиль-
ных реакциях по Ci моносахаридов и простых гликозидов ре-
акционная способность Ci увеличивается, как правило, в сле-
дующем порядке: глюкоза<галактоза<манноза<ксилоза <
<арабиноза<рибоза и т. д. вплоть до ликсозы и альтрозы.
Указанная последовательность углеводных фрагментов в до-
статочной мере постоянна; она соблюдается в разнообразных
реакциях с участием Ci-образования и гидролиза N- и О-гли-
козидных связей, трансгликозилирования, аномеризации
N-гликозидов и мутаротации моносахаридов.
С точки зрения электронных представлений (^реакциях
нуклеофильного присоединения и замещения при насыщенном
углеродном атоме [202, 244], различия в реакционной способ-
ности следовало бы отнести за счет индуктивного влияния уг-
Ю 367
леводного остатка на Сь Трудно однако представить, чтобы
такие углеводные фрагменты, как глюкозил и галактозил, от-
личающиеся друг от друга лишь пространственным располо-
жением ОН-группы при С< (а не числом заместителей), обла-
дали бы заметно различающимися индуктивными эффектами
(табл. 24). Подсчет ст-электронных зарядов по Дель-Ре при-
водит, естественно, к одной и той же величине заряда на Ci в
ряду гексоз или пентоз, вне зависимости от стереохимических
различий у различных углеводных остатков.
Каковы бы ни были причины, обусловливающие различие
в реакционной способности сахаров и их производных, на ос-
новании экспериментальных данных можно приписать каждо-
му фрагменту определенную меру влияния его на состояние
Ci (не уточняя природу и механизм этого влияния). В качест-
ве пробного варианта выберем логарифмы отношения кон-
стант скоростей реакций присоединения азотистых оснований
по Ci (например, данные Хааса и Кадунса, приведенные в
табл. 15). Нами получен следующий ряд относительной реак-
ционной способности Ci для углеводных остатков.
глюкозы 0
галактозы 0,64
маннозы 0,73
ксилозы 0,81
арабинозы 1,03
рибозы 1,19
ликсозы 1,25
Таблица 24
Относительная реакционная способность N-
гликозидов глюкозы и галактозы по данным
кинетических измерений реакций гидролиза,
образования и N-трансгликозилирования
«/х'о К/Ке 1g (к/к0)
3,4 0,53 3,9 0,59
4,4 0,64 4,0 0,60
5,4 0,73 5,2 0,72
4,2 0,62 4,8 0,68
4,1 0,61 6,7 0,83
3,6 0,56 3,36 0,53
3,7 0,57 4,47 0.65
4,0 0,60 2,6 0,41
3,6 0,56 4,1 0,62
3,1 0,49 3,8 0,58
Ср. 1g (к/к0) = 0,61 ±0,06
F46
Располагая этими данными, а также параметрами корре-
ляционных соотношений типа Гаммета-Тафта (разд. 5.2),
можно приближенно оценить вероятность протекания нуклео-
фильных реакций по Ci для неизученных углеводных фраг-
ментов и агликонов. Конечно, в этой области необходимо на-
копление большого числа экспериментальных данных и уточ-
нение параметров относительной реакционной способности.
В связи с этим отметим, что Ю. А. Жданов с сотр. [341]
произвели оценку ст-констант по Гаммету для некоторых уг-
леводов. Однако для этой цели они использовали С-гликози-
ды с п-диметилапилнном в качестве С-агликопа. Поэтому
полученные оценки имеют несколько иной смысл, чем при-
веденные выше величины относительной реакционной спо-
собности.
Г лава 6
ВЛИЯНИЕ N-АГЛИКОНА НА РЕАКЦИОННУЮ СПОСОБНОСТЬ
ГЛИКОЗИДНОГО ЦЕНТРА
6.1. Механизм влияния N-агликона
Для сравнительной оценки реакционной способности неза-
мещенных моносахаридов и N-гликозидов сопоставим скоро-
сти образования одного и того же N-гликозида в реакциях
N-гликозилирования (а) и N-трансгликозилирования (б).
Рассмотрим данные по кинетике образования N-глюкозида
п-толуидина при взаимодействии глюкозы с п-толуидином (а)
и N-глюкозида анилина с п-толуидином (б).
-НС5—R—HCI-OH + NH2C6H4CH3 —'
I О I
—НС5—R—НС1—NHC6H5 + NH2C6H4CH3 ~б
I о_____I
——НС5—R-HC1—NHC6H4CH3
1—0 I
На рис. 41 приведены зависимости константы скорости от
pH для этих двух случаев. Данные показывают, что во всем
изученном интервале pH константы скорости N-трансгликози-
лирования значительно выше констант скоростей образования
N-глюкозида п-толуидина по реакции N-гликозилирования.
Аналогичная картина наблюдается и для других N-арилглико-
зидов и моносахаридов. Напрашивается вывод, что введение
N-агликона приводит к такому изменению электронного со-
стояния гликозидного фрагмента, которое обеспечивает высо-
кую элементарную скорость взаимодействия С1 с нуклеофиль-
ными агентами. Однако, чтобы разобраться в этом вопросе,
надо дать оценку распределения электронной плотности и
рассмотреть разные факторы, изменяющие электронное со-
стояние гликозидного центра.
148
Рис. 41. Зависимость от pH
константы скорости образова-
ния N-глюкозида п-толуидина
с N-глюкозидом анилина (кри-
вая 1, 20°С, 90%-ный водный
этанол, с=0,08 моль!л) и с
глюкозой (кривая 2, 25°С. 80%-
ный водный этанол,
с=0,25 моль[л).
Если исходить из обычных представлений о механизме ре-
акций нуклеофильного присоединения и замещения по карбо-
нильной группе и ее аналогам [244], то увеличение реакцион-
ной способности гликозидного центра Ci у N-гликозидов сле-
довало бы отнести за счет увеличения эффективного положи-
тельного заряда на Ci в результате замены гликозидного гид-
роксила на N-агликон. Однако этому противоречит то обстоя-
тельство, что сродство к электрону атома азота более чем на
порядок меньше сродства к электрону атома кислорода:
азот — 0,05 эв, кислород — 1,47 эв, сера — 2,15 эв
(найдено экстраполяцией по изоэлектронным сериям [342]).
Поэтому поляризация связи С1—N должна быть существенно
меньше поляризации связи С\—О в направлении гетероатома.
В том, что при замене гликозидного гидроксила на N-агли-
кон полярность гликозидной связи должна не увеличиваться,
а снижаться, можно убедиться, если выполнить несложный
подсчет о-электронных зарядов по Дедь-Ре, выбрав в каче-
стве моделей (Ci—С2)-фрагмента ряд простых соединений,
напоминающих аминоспиртовые и ацетальпые формы, обра-
зующиеся в ходе нуклеофильных реакций по Ci (табл. 25).
Действительно, во всех случаях полярность связи С—О за-
метно выше, чем у С—N. Наибольший положительный заряд
наблюдается у Q с геминальными гидроксилами (5), наи-
меньший — у С( этаноламина (1). Можно сделать вывод, что
реакционная способность С, должна была бы снижаться, а не
149
Таблица 2r>
Распределение <т-электронных зарядов в некоторых простых
молекулах (метод «индуктивных параметров»)
Атом Соединение
1 1 2 1 3 1 4 1 5
С| + 0,0012 + 0,047 + 0,1166 + 0,167 + 0,219
о, -0,529 —0,455 -0,5211 —0,462 —0,520 —0,4504
н, 4-0,048 + 0,053 + 0,053 +0,058 + 0,062
С2 + 0,051 + 0,047 + 0,0116 + 0,007 + 0,065
о2 —0,517 —0,455 —0,4469 —0,414 —0.4626
н2 + 0,052 + 0,053 + 0,0604 J- 0,054 + 0,054
Н(О.) + 0,302 + 0,305 + 0,303
Ц(У.) + 0,220 + 0,2207 + 0,223
н (О2) + 0,303 + 0,302 + 0,3033 + 0,306 + 0,302
н н NH,,
1)H —Ct-NHa (2) Н —Ci—OII (3) Н-С—NH»
н-сг-он Н- -С2-ОН н-с2.-он
н н н
II н
(4) H-Ct -NH, (5) II- Ct-OH
н-с2 —ОН н-с2—он
н н
возрастать при введении N-агликона. Противоречие между
данными расчета и результатами эксперимента до некоторой
степени устраняется, если принять во внимание различие в
основности атомов кислорода и азота. Выше говорилось, чго
основность N-агликонов на несколько порядков выше основ-
ности полуацетального кислорода. Следовательно, при одноГг
и той же кислотности растворов концентрация протонизован-
ных форм N-гликозидов должна быть значительно больше
концентрации протонизованных форм соответствующих моно-
сахаридов. В соответствии с этим и скорость реакций нуклео-
фильного присоединения по Ci должна быть выше у N-глико-
зидов. Указанное обстоятельство позволяет рассматривать
N-гликозилирование как своеобразную химическую актива-
цию, сущность которой можно выразить следующим образом:
150
— ведение в молекулу углевода агликона, более основ-
ного, чем гликозидный гидроксил, или какой-либо другой
исходный агликон, увеличивает восприимчивость глико-
зидной связи к кислотным агентам и вследствие этого
повышает скорость кислотнокатализируемых нуклео-
фильных реакций по Ci при заданном значении pH сре-
ды.
Таким образом, имеются основания использовать N-гликози-
лирование в совокупности с N-трансгликозилированием для
развития: нуклеофильного катализа реакций по Сь более эф-
фективных методов синтеза гликозидов, образование которых
при прямом взаимодействии сахаров с нуклеофильными аген-
тами затруднено по тем или иным причинам.
6.2. Нуклеофильный катализ
реакций присоединения
Нуклеофильный катализ в некоторых случаях оказывается
более эффективным, чем кислотно-основной [343—357], одна-
ко количественная сторона его разработана слабее. Этот вид
катализа обычно представляют в виде последовательности
реакций с образованием промежуточных соединений п их рас-
падом, в результате чего катализирующая молекула выделя-
ется в неизменном виде [205, 358]. Применительно к реакци-
ям присоединения по карбонильной группе и ее аналогу —
гликозидному центру, схему нуклеофильного катализа амина-
ми можно записать в следующем виде:
—НС1—OH + NH2R'-^ —НС,—NHR'-*—HGi = NR' + H2O (а)
I
ОН
—HC^NR'+NHzR — — НС,—NHR-— HC, = NR + NH2R/ (б)
I
NHR'
—HC!-OH + NH2R-* —HC,=NR + H2O (в)
NH2R'
Таким образом, катализ аминами можно представить как
совокупность двух последовательных реакций — гликозили-
рования (а) и трансгликозилирования (б), механизмы и кис-
лотно-основной катализ которых рассмотрен нами в преды-
дущих главах. Представляет интерес выяснить, насколько эф-
фективен (и в каких условиях) нуклеофильный катализ реак-
151
ции образования N-гликозидов по сравнению с рассматривае-
мым ранее кислотным катализом. В гл. 4 показано, что выход
реакции N-гликозилирования в условиях кислотного катализа
возрастает с уменьшением кислотности и достигает макси-
мума при относительно больших значениях pH, при которых
скорость реакции становится пренебрежимо малой. Отсюда
следует, что нуклеофильный катализ имеет смысл использо-
вать именно при этих крайних значениях pH, когда кислот-
ный катализ не проявляет себя заметным образом и когда
одновременно равновесие N-гликозилирования смещено в сто-
рону образования продукта.
Что касается выбора нуклеофильного катализатора, то
обычно химики-органики предпочитают использовать высоко-
основные амины. Касаясь требований, необходимых для реа-
лизации нуклеофильного катализа, У. Дженкс пишет [203]:
«Исходное соединение должно быстро реагировать с катали-
зирующим амином, а образующееся при этом нестабильное
промежуточное соединение должно быстро взаимодействовать
со вторым компонентом системы». К этому следует добавить
еще одно немаловажное обстоятельство — катализирующий
агент должен весьма быстро регенерироваться из конечного
промежуточного продукта. Если вспомнить, что из двух N-ar-
ликонов в N-ацетальных формах кинетически легче вытес-
няется более основный, то при выборе катализатора из ряда
однотипных следует отдать предпочтение тому из них, основ-
ность которого выше основности нуклеофильного агента реак-
ции. В этом отношении практический интерес могут представ-
лять, например, алифатические амины и а-амипокислоты, ко-
торые, как показано в работах [359—368], являются весьма
эффективными катализаторами в реакциях по карбонильной
группе и гликозидному центру.
Кинетические исследования, описанные в работах [304,
369], показали также, что а-аминокпслоты заметно ускоряют
и реакцию образования N-арилгликозидов. Примером могут
служить данные, приведенные в табл. 26. Обращает на себя
внимание тот факт, что кажущиеся энергии активации реак-
ций N-гликозилирования, катализируемых глицином, заметно
выше, чем некатализируемых (за исключением образования
N-галактозида анилина). В то же время в присутствии глици-
на наблюдается увеличение энтропии активации в сторону
положительных значений. Таким образом, катализ реакции
обусловлен не снижением энергии активации, а увеличением
энтропии системы в переходном состоянии на стадии, опре-
деляющей кинетику нуклеофильного катализа. Это позволяет
сделать вывод, что в условиях нуклеофильного катализа гли-
152
Таблица 26
1. Константы скорости образования N-гликозидов анилина
(с = 0,2 моль/л) без добавок глицина и в присутствии эквимо-
лекулярных количеств глицина при различных температурах
(30%-ный этанол, фосфатный буфер, pH 8,5)
к - 10э, мин-1
Моносахарид _ в присутствии без глицина „на
19° 40° 50° 19° 40° 50“
Глюкоза 0,80 2,7 4,5 2,1 10,8 17,4
Ксилоза 0.86 3,1 5,7 4,2 28 53
Галактоза 0,93 3,7 18,2 5,4 44 67
Манноза 3,7 10 17 24 97 180
2. Параметры активации реакции N-гликозилировання
(образование N-гликозидов анилина)
Моносахарид
I AS', Дб"
ккал/моль кал/град-моль ккал/моль
а) без глнцина
Глюкоза 10.6 —47,6 24,7
Ксилоза 11,6 —43,4 24,6
Г алактоза 16.8 —26,4 24,4
Манноза 9,3 —45,7 24,0
б) в присутствии глицина
Глюкоза 13,5 —34,8 23,8
Ксилоза 15,8 —25,3 23,1
Галактоза 15,8 —24,4 22,8
Манноза 12,1 —34,5 22,5
цином реакции N-гликозилирования протекают по реакционно-
му пути, отличному от того пути, который имеет место в ус-
ловиях кислотного катализа. Полученные данные не позво-
ляют ответить на вопрос, какая из стадий является лимити-
рующей. Можно лишь предполагать, основываясь на данных
реакции N-трансгликозилирования (гл. 3), что наиболее мед-
ленной является стадия элиминирования исходного N-аглико-
на (в данном случае остатка глицина) из промежуточного
продукта N-ацетальной структуры:
153
(—НС,—OH^—HC, = O)+NH2CH2COO- -
+nh2c6h5
-> НС,—NH—СН2СОО-—HCf = N—СН2СОО--------->
он
—HC1—NH—CH2COO-
I
NH—C6H5
—HC5—R—НС!—NH—CH2COO- ->
I I
OH nhc6h5
HC5—R—HCi—nhc6h5 + nh2ch2coo-
l — O I
При образовании первого промежуточного продукта (амн-
носпирта или шиффова основания) необходимость в кислот-
ном катализе отпадает, так как основность аминогруппы гли-
цина достаточно высока для того, чтобы стадия присоедине-
ния осуществлялась без кислотной активации моносахарида
(не говоря уже о том, что вероятность протонизации гликозид-
ного или полуацетального кислорода в нейтральных и слабо-
щелочных средах исключительно мала). Следующие стадии в
предполагаемом механизме нуклеофильного катализа — при-
соединение ариламина по Ci первого промежуточного продук-
та и элиминирование катализирующей молекулы — имеют
наиболее существенное значение для реализации нуклеофиль-
ного катализа. Именно эти стадии (и особенно стадия элими-
нирования) могут быть наиболее медленными, так как для их
протекания необходима протонизация N-агликонов, а усло-
вия, в которых осуществляется нуклеофильный катализ, не-
благоприятствуют этому акту (достаточно напомнить об ус-
ловиях, при которых N-трансгликозилирование замораживает-
ся на стадии образования промежуточного продукта).
Для обстоятельного выяснения механизма нуклеофильного
катализа требуются дополнительные исследования. Однако
уже сейчас можно отметить, что преимуществом этого вида
катализа является отсутствие некоторых побочных процессов,,
сопровождающих реакцию N-гликозилирования в условиях
обычного кислотного катализа. Полученные этим способом
N-гликозиды обладают достаточно хорошей чистотой и после
перекристаллизации не содержат примесей катализирующего
агента (по данным бумажной хроматографии). Недостатком
способа является его ограниченность. При катализе глицином
154
хорошие результаты получаются только с теми аминами, ко-
торые обладают достаточно высокой основностью. Попытки:
катализировать глицином образование N-гликозидов с мало-
основными агликонами к успеху не привели.
Рассмотрим теперь взаимосвязь между нуклеофильным и-
кислотно-основным катализом. Это может оказаться полезным
как при оценке эффективности нуклеофильного катализа, так
и при выборе каталитических агентов. В том, что такая связь
существует, нетрудно убедиться, рассмотрев схему нуклео-
фильного катализа с точки зрения кислотно-основных свойств
всех компонентов катализируемой реакции. Используя метод
стационарных концентраций, запишем схему нуклеофильного,
катализа в следующем общем виде:
r + Bi^Xf, х, + в2^х,
(г)
X2->P + Bi
где Г — моносахарид, В] — нуклеофильный катализатор, В2—
атакующий амин, Xi, Х2 — промежуточные соединения (амп-
носпирт или шиффово основание и Na-цеталь), Р — конеч-
ный продукт реакции.
Здесь рассматривается случай, когда кинетика лимити-
руется стадией отщепления катализирующего агента Bb Вы-
бор оправдывается тем, что, как показано в гл. 3 и 4, реакции
элиминирования протекают значительно медленнее стадий
присоединения нуклеофильных агентов при сопоставимых зна-
чениях pH. Учитывая далее, что элиминирование Bi осуществ-
ляется в результате его предварительной протонизации
Ках
Х2+Н3О^Х2Н;+НгО , [Xs][H'l
К°*= [Х2НЧ ’
(или Х2+НгО5±ХгН + + ОН-) ('*>
можно записать для наблюдаемой скорости выражение:
V„flft,. = a5|X2H ' ]=а3[Х>]|Н ]/Как, (2).
где as — константа скорости элементарной стадии элиминиро-
вания Bi из протонизованной формы Х2.
Полагая, что в условиях стационарности реакции стадии,
предшествующие лимитирующей, являются равновесными
К1/К2=[Х!]/[Г] [В,] =Kel, (3)f
Кз/к4=[Х2]/[Х,] [В2]=Т<ег
I5S
найдем выражение для [Х2]:
|Х»]=*Л|Г|[В,ЦВ,]. (4)
Из уравнений баланса по протонизованным и непротонизован-
ным формам нуклеофильных агентов В] и В2 и соответствую-
щих констант диссоциации
1В.]0 = [В1]4-[В1Н + ], tfeC=[Bf][H Ч/lBiI-P],
[В11о“1В1] + [В2Н+], ^-[В,][Н+]/[В»Н+] (5)
найдем
1ва]=[в,]0^жав+[н+])- (6)
Подставив (4) и (6) в (2), получим уравнение для скорости,
откуда для наблюдаемой константы скорости будем иметь
го 1 ____a:J^acKae[\\' ]___
>w< -[Bib Ках №c+[H4)№e+[H^]) (7)
или
а»А[Н+]
кхабл--{^ + (Кас+Кав)\П^+КасКав - (8)
где A-[H^KelKeiKacKaeIKaX.
Это уравнение аналогично по форме уравнениям, получен-
ным ранее (гл. 4) для реакции присоединения по С2 в усло-
виях кислотного катализа. Графически, в координатах
pH, оно также выражается колоколообразного вида кривой с
максимумом при
|Н -|2^.= М;в- pHonm.=0,5(p^e+p^e). (9)
Наиболее существенное различие в уравнениях для кис-
лотного и нуклеофильного катализа заключается в том, что в
последнем случае к набл. зависит, кроме всего прочего, от по-
казателя основности нуклеофильного катализатора В|. Уже
из соотношений (9) видно, что в ряду однотипных катализи-
рующих агентов при заданном атакующем амине максимум
кривых к найл. (pH) смещается в сторону щелочных значений
pH с увеличением показателя основности катализатора
(р/Сас). Это положение можно иллюстрировать ранее приве-
денным рис. 22. Иначе говоря, при заданном значении pH
(как правило, нейтральные и слабощелочные растворы) и
прочих равных условиях константа скорости в условиях нук-
156
леофильного катализа
возрастает с увеличени-
ем основности нуклео-
фильного катализато-
ра. При достаточно вы-
сокой основности ката-
лизатора кривые кнавл.
(pH) для кислотного и
нуклеофильного ката-
лиза смещаются отно-
сительно друг друга
так, как это показано
на рис. 42, где кривая 2
характеризует нуклео-
фильный катализ в
«чистом» виде без за-
метного вклада кис-
лотного катализа. С
уменьшением основнос-
ти катализатора кри-
вые кНабл (pH) будут
сближаться до полного
перекрывания. Этот
случай однако не пред-
ставляет практического
интереса, поскольку
нуклеофильный ката-
рН
Рис. 42. Схематическое изображение кри-
вых зависимости константы скорости реак-
ций гликозидного центра в условиях кислот-
ного (1) и нуклеофильного катализа (2) от
pH среды. Относительный вклад нуклео-
фильного катализа при различных pH ил-
люстрируется следующим примером:
pH Q Н pH fl pH
1 90 5 1.2 9 103
2 50 6 2,0 10 10*
3 10 7 11 11 ю5
4 2 8 100 12 10б
Р—отношение функций, описывающих за-
висимость от pH констант скоростей нукле-
офильно- и кислотнокатализируемых реак-
ций. При расчете (S использовались следую-
щие значения констант диссоциации атаку-
ющего амина (Лав ). промежуточного про-
дукта в кислотнокатализируемой реакции
(Кас J и промежуточного продукта в нукле-
офильиокатализируемой реакции (Кар)-'
Ка„«=10-«, Ках=Ю-6. Кас=10-‘°,
Кйр=10-1г.
лиз завуалирован
обычным кислотным, и
его реализация в этих
условиях лишена вся-
кого смысла.
Из уравнения (8)
следует также, что в
ряду однотипных ата-
кующих аминов В2 при
заданном катализато-
ре В( достаточно высокой основности кривые кнабл- (pH) пре
терпевают аналогичное смещение в сторону щелочных значе-
ний pH по мере увеличения основности амина. Иначе говоря,
при заданном значении pH константа скорости при нуклео-
фильном катализе будет тем выше, чем больше основность
атакующего амина. При низкой основности амина нуклео-
фильный катализ может вообще не проявиться достаточно за-
метным образом.
15?
’Из сравнения уравйений для кислотного и нуклеофильного
катализа можно сделать еще один практически важный вы-
вод: скорости кислотно- и нуклеофильнокатализируемых ре-
акций должны известным образом коррелировать друг с дру-
гом. Грубо говоря, чем с большей скоростью протекает кис-
лотнокатализируемая реакция при кислых значениях pH, тем
выше скорость этой же реакции в условиях нуклеофильного
катализа при щелочных значениях pH. Это положение согла-
суется с данными, приведенными в табл. 26, а также с резуль-
татами исследования каталитического действия глицина в ре-
акции метоксиамина с бензальдегидом [347].
6.3. N-трансгликозилирование как метод синтеза
гликозидов с малоосновными агликонами
Выше говорилось, что введение N-агликона облегчает при-
соединение нуклеофильных агентов по Ci, благодаря чему от-
крываются новые возможности для получения гликозидов, в
особенности гликозидов с малоосновными агликонами. Под-
разделение гликозидов по признаку основности до известной
степени условно, так как различие между отдельными пред-
ставителями носит не принципиальный, а количественный ха-
рактер. В этом мы убедились при рассмотрении вопросов кис-
лотного катализа реакций образования и гидролиза N-глико-
зидных связей (гл. 4). Гликозиды с малоосновными агликона-
ми отличаются довольно высокой устойчивостью по отноше-
нию к кислотным и щелочным агентам. Одновременно они со
значительно большим трудом образуются при прямом взаи-
модействии нуклеофильного агента с моносахаридами. Поэто-
му для этой группы производных актуальной является пробле-
ма нахождения простых и вместе с тем эффективных методов
их получения.
Рассмотрим данные по образованию на основе однозаме-
щенных N-арилгликозидов важной группы производных —
гликозилмочевин, которые могут использоваться в частности
для получения синтетических аналогов нуклеозидов [35]. До
последнего времени гликозилмочевины получали главным об-
разом путем прямой конденсации мочевины с моносахарида-
ми в присутствии минеральных кислот как катализаторов
(метод Шоорля [370]). Наиболее трудоемкой частью синтеза
является необходимость длительного выдерживания реакцион-
ной смеси при повышенных температурах [13—15]. Иллюст-
рируем это следующим примером.
Реакционная смесь, состоящая из 27 г глюкозы, 27 г 'моче-
вины, 6 мл H2SO< (27%) и 125 мл воды выдерживается при
158
50°С в течение 7 дней, после чего раствор упаривается в ва-
кууме до получения густого сиропа. При стоянии сиропа при
пониженных температурах происходит кристаллизация аддук-
та гликозилмочевины с мочевиной в соотношении 1:1. Т. пл.
аддукта 167°. Выход после промывания на фильтре 90%-ным
этанолом — 31 г (73%). Обработка этого соединения кипя-
щим абсолютированным этанолом в течение трех дней дает
конечный продукт — глюкозилмочевину. Т. пл. 208°, [а]д=—
—23,45° (с = 2,153, вода).
Надо отметить, что в течение 50 с лишним лет с момента
проведения первого синтеза глюкозилмочевины основные си-
лы исследователей были направлены главным образом на
усовершенствование двух основных способов — прямого взаи-
модействия мочевины с незамещенными сахарами и изоциа-
натного метода Э. Фишера [371]. В сферу этого направления
в химии углеводов не вовлекались N-гликозиды, реакции ко-
торых в тот же период подвергались довольно интенсивному
исследованию (схема 10). Лишь в последние годы Шмук
[372], Гельферих и Митровский [373], Шоу и Варренер ]374[
модифицировали метод Фишера, предложив вводить в реак-
цию с изоцианатами не гликозилгалогениды, а гликозиламины.
Схема 10. Основные способы получения N-гликозилмочевим.
вается возможным объяснить и образование кето-звена при С2
(перегруппировка Амадори), и формирование гидразонных
фрагментов при Ci и С2 (озазонообразование).
7.2. Перегруппировка Амадори
В ледяной уксусной кислоте или в присутствии аминов как
катализаторов реакции N-гликозиды испытывают превраще-
ние в 1-амино-1-дезокси-2-кетозы, известное в литературе под
названием «перегруппировки Амадори» [379—-394]:
О—НС)—NH—R Н2С)—NH—R Н2С)—NH—R
II 1 I
1 нс2—ОН с2=о нс2—о
I I II
Дезоксикетозы могут быть получены при непродолжительном
нагревании (80—120°С) эквимолекулярных количеств моно-
сахарида и сильно основного ариламина (например, толуиди-
на) в сухом состоянии. При конденсации же этих соединений
в спиртовых растворах вначале происходит образование
N-гликозидов (с выходом до 90—95%), и лишь более продол-
жительное нагревание раствора в присутствии кислотного
катализатора приводит к образованию продукта перегруппи-
ровки. Возможность протекания этой реакции у N-глнкозидов
определяется двумя важными факторами — основностью
N-агликона и наличием гидроксильной группы при С2. Напри-
мер, слабо основный N-ацетилированный глюкозиламнн, рав-
но, как и 2-дезокси-производные, превращению в продукты
Амадори не подвергаются.
Перегруппировка катализируется как кислотными, так и
основными агентами. Исследование каталитической активно-
сти ряда органических кислот показало, что наиболее активны
кислоты с показателями рК0, лежащими в интервале 4,2—5,1
[385]. При этих же рКа в наименьшей степени протекают
побочные процессы, приводящие к разложению продукта ре-
акции. Активными катализаторами перегруппировки являют-
ся также третичные амины (триэтиламин. пиридин), комплек-
сы аминов с льюисовскими кислотами (BF3-NR3), меркапта-
ны. Показано, что выход продуктов реакции увеличивается
при добавлении калиевых и натриевых солей бензойной, ук-
сусной и аминобензойпой кислот. Как установили Ходж и Рист
[382], перегруппировка ускоряется также соединениями, со-
держащими активированную метиленовую группу — малоно-
166
вой кислотой, малоновым эфиром, ацетилацетоном, фенилаце-
тоном, дифенилметаном.
Перегруппировка Амадори осложняется протеканием ряда
побочных и еще до конца не изученных реакций, ведущих к
образованию окрашенных продуктов («браун-реакция» [394]),
что затрудняет проведение кинетических исследований. Од-
ним из возможных способов изучения кинетики данной реак-
ции является колориметрический метод с использованием в
качестве титранта натриевой соли 2,6-дихлорфенол-ипдофено-
ла [395]. В щелочных растворах этот краситель легко вос-
станавливается продуктом перегруппировки и не восстанавли-
вается N-арилгликозидами; в умеренно кислых средах часто
наблюдается обратная картина (рис. 44). Перспективен, хотя
и менее точен, метод бумажной и тонкослойной хроматогра-
фии [396]. Из немногочисленных работ по кинетике можно от-
метить [398], в которой установлено, что перегруппировка
4,6—О-бензилиденового производного N-глюкозида п-толу-
идина протекает по уравнению первого порядка в присутствии
ряда кислотных и основных катализаторов. В целом же ко-
личественных сведений пока недостаточно для какого-либо
уверенного суждения о механизме реакции и о природе ката-
литического действия добавок. Поэтому все предложенные до
Рис. 44. Примеры изменения
оптической плотности (Д
при Х = 540 ммк) растворов
2,6-дихлорфенол-индофенол а
в водном метаноле в при-
сутствии N-глюкозида п-то-
лундина (!) и продукта пе-
регруппировки Амадори (2)
(pH 8,13, фосфатный буфер,
25°С). Глюкоза в этих усло-
виях не восстанавливает
краситель.
467
сих пор механизмы Амадорип-перегруппировки являются в
значительной степени умозрительными.
По Куну и Вейганду [379], на первой стадии реакции про-
исходит протонизация атома азота с последующим раскрыти-
ем углеводного кольца N-гликозида и образованием иммоние-
вого иона, который затем в результате взаимодействия с ак-
цептором протона (отщепляющим протон от С2) превращает-
ся в 1,2-энольную форму, а последний изомеризуется в про-
дукт Амадори-перегруппировки (схема 12). Исбелл и Фраш
।--НС—NHR
О 1
1 НС—ОН
а 1 б в
| НА | НА 1 nh2r
р- НС—NH2R |—НС—NHR RHN—НС—NHR 1
О НС—ОН НО+ НС—ОН нс-он
’ ' J - i + —NH2R |
HC=NHR HC=NHR J.
1 1 НС—NHR
НС—ОН НС—он 1
1 1 НС-ОН
1 1 1
НС—NHR НС—NHR I
II II Н2С—NHR
С-ОН С—ОН 1
1 [ г с—он
j 1 1
Н2С—NHR 1 " 1
с=о
Схема 12. Некоторые из предполагаемых механизмов перегруппировки
Амадори: по Куну и Вейганду (а), по Исбеллу и Фрашу (б), по Михел»
и Дийонгу (в).
[251] полагают, что на стадии кислотной активации происхо-
дит протонизация не N-агликона, а циклического кислородно-
го атома; дальнейшие превращения кислотно-активированных
форм в обеих схемах аналогичны друг другу. Эту точку зре-
168
ния поддерживает Готшалк [394]. Такое расхождение во
взглядах на кислотно-основной катализ (прежде всего, на ме-
сто протонизации активных центров углеводных молекул), как
мы видели выше (главы 3 и 4), представляет собой довольно
распространенное явление. Надо сказать, что при оценке ве-
роятности той или иной схемы катализа очень часто не учиты-
ваются показатели основности центров молекул, ответствен-
ных за катализ, например, циклического кислородного атома
и атома азота N-агликона (впрочем, величины рКа для этих
центров остаются пока неизвестными и раздельная оценка их
представляет собой довольно сложную задачу). Конечно, ес-
ли исходить из того, что атом азота обладает заведомо боль-
шей основностью, чем кислородный атом углеводного кольца,
то следует признать более вероятной схему Куна-Вейганда.
Каталитическое действие соединений с метиленовыми груп-
пами Исбелл и Фраш [251] объясняют образованием проме-
жуточного продукта присоединения метиленового соединения
по Ci иммониевого иона, который далее распадается до энолы
ной формы продукта перегруппировки с регенерацией катали-
тического агента.
Аналогичная схема использована для обьяснения катализа
перегруппировки карбоновыми кислотами.
На примере 3, 4, 5, 6-замещенного N-глюкозида и п-толуи-
дина Михель [399] показал, что продукт перегруппировки мо-
жет образоваться из 1,1-бис-Ы-ацеталыюй формы, возникаю-
щей в результате присоединения п-толуидина к N-глюкозиду.
На основе этих данных Михель и Дийонг [400] в 1962 г. при-
шли к выводу о возможности протекания Амадори-перегруп-
пировки по гидридному механизму, который в общих чертах
характеризуется следующей предположительной схемой: а)
образование N-ацетальиой формы из N-гликозида; б) прото-
низация одного из N-агликопов с последующим отщеплением
его и образованием карбониево-ионного состояния сначала
при С|, а затем и при С2; в) образование кето-формы. Гид-
ридный механизм отвергается однако Пальмой и Симоном
[401], которые считают более вероятным протекание реакции
по механизму классической кетоэнольной таутомерии.
7.3. Реакция озазонообразования
Взаимодействие моносахаридов с фенилгидразином, веду-
щее последовательно к образованию фенилгидразонов, а за-
тем и озазонов сахаров впервые изучено Э. Фишером свыше
80 лет назад [402]. Однако вопрос о механизме реакции оза-
169
ПРИЛОЖЕНИЕ I
ОСНОВНЫЕ ПРАВИЛА
(для реакций гликозидного центра в условиях
гомогенного кислотно-основного катализа)
1. Реакции нуклеофильного присоединения и за-
мещения по Ci в ряду моносахаридов и N-гликозп-
дов протекают преимущественно по пути образова-
ния промежуточных ациклических соединений аце-
тильной и полуацетальной структуры (амнноспир-
тов, ампналей и т. п.), возникающих в результате
раскрытия углеводного кольца по связи Ci—ОЧЦАГЛ,
под влиянием атакующего нуклеофила. При уме-
ренной кислотности среды определяющей является,
как правило, стадия элиминирования исходного аг-
ликона, сопровождающаяся одновременной цикли-
зацией углеводного остатка. Путем регулирования
кислотности среды можно добиться такого соотно-
шения констант скоростей присоединения и элими-
нирования, при котором реакция будет фактически
«заморожена» на стадии образования промежуточ-
ного соединения.
2. При участии в реакциях нуклеофильного за-
мещения циклических форм моносахаридов и гли-
козидов образуются дпастероизомерные формы
промежуточного соединения в прямом и обратном
направлениях реакции. Расположение заместителей
при С[ в этих формах однозначно определяется кон-
фигурацией исходного агликона в момент атаки на
С| входящего агликона.
3. Из двух однотипных агликонов в промежу-
точных соединениях ацетальиой структуры кинети-
чески легче вытесняется, как правило, более основ-
ный агликон.
4. В продуктах реакций нуклеофильного заме-
щения, протекающих заведомо через стадию обра-
зования промежуточных соединений ацетального
190
типа, входящий агликон имеет тенденцию занимать
конфигурацию, противоположную той, которую
имел уходящий агликон в исходном соединении
(при условии линейного расположения образую-
щейся и разрываемой связей в переходном состоя-
нии).
5. В ряду N-арилгликозидов знак оптического
вращения диастереоизомерных форм, возникающих
в ходе реакции N-трансгликозилирования, зависит
от электроноакцепторных свойств заместителей в
N-агликонах и относительного расположения N-аг-
ликонов в пространстве. Выбрав в качестве меры
электроноакцепторных свойств параметр <т (по Гам-
мету), эту зависимость можно выразить в виде сле-
дующего приближенного правила — если ориенти-
ровать гликозидный фрагмент N-ацетальных форм
таким образом, чтобы водородный атом Н—Ci был
удален от наблюдателя, то положительному знаку
вращения отвечает расположение N-агликонов по
часовой стрелке в порядке увеличения <т-констант
заместителей в бензольных кольцах агликонов; от-
рицательному знаку соответствует структура с рас-
положением N-агликоиов против часовой стрелки.
6. Характерной особенностью кислотного ката-
лиза нуклеофильных реакций по Ci является экстре-
мальная (обычно «колоколообразная») зависимость
эффективной константы скорости от кислотности
среды. Оптимальное значение pH, при котором кон-
станта скорости имеет максимальное значение, вы-
ражается приближенно в виде полусуммы от пока-
зателей основности компонентов реакции (исходно-
го нуклеофила и промежуточного соединения или
исходного гликозида и промежуточного соедине-
ния). С достаточной для практического использова-
ния степенью приближения справедливо следующее
правило—положение максимума зависимости кна6л.
от pH среды определяется показателем основности
атакующего нуклеофила и с увеличением последне-
го смещается па шкале pH в сторону щелочных
значений.
В принципе, высота максимума колоколообраз-
ной кривой кна$,. (pH) тем больше, чем меньше
разность в показателях основности нуклеофильного
агента и промежуточного соединения при прочих
равных условиях. Из этого следует, что все факто-
191
ры, сближающие указанные показатели основности,
способствуют увеличению скорости нуклеофильной
реакции по Сь
7. Положение равновесия кислотно-катализируе-
мых реакций зависит в общем случае от кислотно-
сти среды. Лишь при равенстве показателей основ-
ности исходных и конечных соединений эксперимен-
тальная константа равновесия представляет собой
постоянную величину, не зависящую от концентра-
ции водородных попов.
Направление смещения равновесия кислотно-
каталпзируемых реакций при изменении кислотно-
сти среды определяется знаком разности показате-
лей основности исходного нуклеофила и продукта
реакции. Иными словами, с увеличением кислотно-
сти среды экспериментальная константа равновесия
возрастает, если продукт реакции обладает боль-
шей основностью, чем исходное соединение (нуклео-
фил или N-гликозид) и, напротив, уменьшается при
относительно меньшей основности продукта реак-
ции. С достаточной для практического использова-
ния степенью точности максимальный выход про-
дуктов реакции определяется отношением констант
диссоциации продукта реакции и исходного соеди-
нения (пли нуклеофила).
8. При корреляциионпом анализе кислотно-ката-
лизпруемых реакций по уравнениям типа Гаммета-
Тафта следует дифференцировать влияние замести-
телей на реакционный центр и па протоноакцептор-
ный участок, ответственный за кислотную актива-
цию реагирующей молекулы. В тех случаях, когда
протоноакцепторный участок расположен на пути
передачи влияния заместителя на реакционный
центр, происходит обращение знака константы р в
уравнениях Раммета-Тафта на противоположный
тому, который имел бы место при прямом воздейст-
вии заместителя на реакционный центр. Отклонение
от этого правила при том же самом расположении
реакционного и активного центров по отношению к
заместителю может свидетельствовать о том, что
заместитель оказывает на реакционный центр су-
щественно большее воздействие, чем на протоноак-
цепторные свойства активного центра (например,
вследствие увеличения электронной проводимости
эффекта влияния).
192
9. Введение в молекулу углевода агликона, бо-
лее основного, чем гликозидный гидроксил (или ка-
кой-либо другой исходный агликон), увеличивает
восприимчивость гликозидного фрагмента к кислот-
ным агентам и вследствие этого — скорость кислот-
но-катализируемых нуклеофильных реакций по С]
при заданном значении кислотности среды.
10. Для получения N-гликозидов с мало основ-
ными агликонами, образование которых при пря-
мом взаимодействии моносахаридов с аминами за-
труднено по кинетическим или термодинамическим
соображениям, может быть использован более эф-
фективный метод N-трансгликозилирования.
При использовании N-арилгликозидов в качест-
ве исходных соединений стереохимическое направ-
ление реакции обменного взаимодействия (N-арил-
гликозид+мало основный амин) существенно зави-
сит от природы заместителя в бензольном кольце
агликона или иначе — от основности исходного
N-аглнкона. В частности, при синтезе N-гликозил-
мочевин по реакции N-трансгликозилирования име-
ет место следующая тенденция: электронодонорные
заместители (типа СНз) в исходном N-арилаглико-
не способствуют преимущественному разрыву
N-гликозидной связи с обращением конфигурации
при Ci, в то время как электроноакцепторные за-
местители (типа NO2) благоприятствуют преиму-
щественному разрыву полуацетального кислородно-
го мостика с образованием промежуточных соеди-
нений в форме N-ацеталей.
11. Для увеличения скорости образования N-гли-
козидной связи в условиях, исключающих кислот-
ный катализ, может быть использован нуклеофиль-
ный катализ алифатическими аминами и особенно
а-аминокислотами. Высокая основность аминогруп-
пы а-аминокислот благоприятствует присоединению
её по Ci моносахарида и одновременно обеспечива-
ет достаточно быстрое элиминирование катализи-
рующей молекулы из промежуточного продукта на
завершающем этапе нуклеофильного катализа ре-
акции N-гликозилирования.
12. Введение N-агликона в молекулу моносаха-
рида способствует возникновению карбониево-ион-
ных состояний гликозидного центра и при благопри-
ятных условиях — дальнейшему развитию реакций
193
карбонпевых ионов. Типичными реакциями этих
ионов в ряду сахаров являются молекулярные пе-
регруппировки ««секстетного» типа, сопровождаю-
щиеся внутримолекулярной миграцией атомных
групп, включая 1,2-гидрндный сдвиг.
В частности, фенилгидразонный фрагмент под
действием карбоновых кислот претерпевает гетеро-
лиз по связи азот—азот, в результате чего образует-
ся ионная пара, время жизни которой сопоставимо
со временем, необходимым для внутримолекуляр-
ной и переггруппировки азониевого и карбониевого
ионов в энергетически более устойчивую кето-пмин-
ную структуру.
ПРИЛОЖЕНИЕ 2
КИНЕТИЧЕСКИЙ АНАЛИЗ ОРГАНИЧЕСКИХ РЕАКЦИЙ
С ПРИМЕНЕНИЕМ ПОЛЯРИМЕТРИЧЕСКОГО МЕТОДА
При изучении кинетики органических реакции очень часто
измеряют не концентрации реагентов, а зависящие от них
физические параметры системы. Одним из таких параметров
для оптически активных соединений является угол вращения
плоскости поляризованного света, линейно связанный с кон-
центрацией компонента реакционной системы:
а = [а] с I (град.), (1)
где [а]—удельный угол вращения, с — концентрация (г!см3),
I — толщина слоя раствора (дм). Идея расчета констант ско-
ростей по данным поляриметрических измерений состоит в
следующем: а) на основании предполагаемой схемы реакции
составляются дифференциальные уравнения, решение которых
приводит к выражению для концентраций (или относительных
концентраций) компонентов в функции времени и констант
скоростей отдельных стадий реакции; б) па основании прави-
ла аддитивности для углов вращения компонентов реакцион-
ной смеси находится выражение для концентраций в функции
углов вращения; в) из полученных соотношений исключаются
концентрации и в результате находится уравнение для угла
вращения в функции времени и констант скоростей.
Для смеси оптически активных веществ наблюдаемый угол
вращения при заданной длине поляриметрической трубки
складывается из углов вращения отдельных компонентов в
соответствии с правилом аддитивности:
а = а1 + аг + аз+ ... (2)
или
а <?o=[ai] С1+[аг] Сг+ [аз] Сз+ (3)
(со—общая концентрация компонентов). Для бинарной смеси
можно установить взаимно однозначное соответствие между
углом вращения и относительным содерхсанием любого из
компонентов в весовых долях. В частности, если в реакции
195
ЛИТЕРАТУРА
1. G. Р. Ellis, J. Honeyman. Adv. Carbohydrate Chem., 10, 95
(1955).
2. P. Богнар. Успехи химии, 21, 734 (1952).
3. W. W. P i g m a n, R. M. Goepp. Chemistry of Carbohydrates.
N—Y., 1948.
4. H. К. Кочетков, А. Ф. Б о ч к о в, Б. А. Дмитриев,
А. И. Усов, О. С. Ч и ж о в, В. Н. Ш и б а е в. Химия углеводов,
М., изд-во «Химия», 1967.
5. R. S. Tipsori. Adv. Carbohydrate Chem., 1, 193 (1945).
6. Б. Сорокин. ЖРФХО, 14, 377 (1877); 18, 129 (1886).
7. R. Kuhn, L. В i г к о f е г. Вег., 71, 621 (1938).
8. F. Weygand. Вег., 72, 1663 (1939); 73, 1259, 1284 (1940).
9. В. В. 3 е л е и к о в а, Б. Н. Степаненко. ДАН СССР, 144,
349 (1962).
10. Б. Н. С т е и а н е н к о, Р. Д. Г р е ш н ы х. ДАН СССР, 170,
121 (1966).
11. Б. Н. С т е п а и е п к о, Э. С. В о л к о в а, М. Г. Ч е н ц о в а. ДА11
СССР, 177, 607 (1967).
12. S. К о 1 k a, J. Sokolowski. Rocz. Chem., 44, 85 (1970).
13. Н. Г. Шкантов а, М. С. Дудкин, С. И. Три н tn п у и,
ЖПХ, 40, 164 (1967).
14. А. Н у п d. Biochem. J., 20, 195, 205 (1926).
15. W. E. Jensen, A. S. Jones, G. W. Ross. J. Chem. Soc., 2463
(1965); M. H. Benn, A. S. J о n e s. J. Chem. Soc., 3877 (1960);
A. S. J о n e s, G. W. Ross. Tetrahedron, 189 (1962); E. A. Ba-
dawi, A. S. J о n e s, M. S t a c e y. Tetrahedron, 281 (1966).
16. R. S c h m i d t, G. Wagner. Pharmazie, 22, 551 (1967).
17. А. В. Степанов, В. В. Мамаева. Биохимия, 9, 10 (1944).
18. К. Н е у п s, Н. Paulsen, Н. Brener. Angew. Chem., 68,
334 (1956); К- Н е у п s, Н. Paulsen. Ann. 622, 160 (1959).
19. А. К I е m е г, F. М i с h е е 1. Вег., 89, 1242 (1956).
20. Р. И. L о w у, Н. В о г s о о k. J. Am. Chem. Soc., 78, 3175 (1956).
21. М. L. W о 1 f г о т, R. D. S с h u 1 z, L. F. С a v а 1 i е г 1. J. Ат.
Chem. Soc., 71, 3518 (1949).
22. В. Н е 1 f е г i с h, A. М i t г о w s ку. Chem. Вег., 85, I (1952).
23. Е. F i s с h е г, В. II е 1 f е г i с h. Вег., 47, 210 (1914).
24. М. Frerejacque С. R., 202, 1190 (1936); 207, 638 (1938).
25. Химия углеводов. Библиографический указатель (1961 —1964),
изд-во «Наука», М., 1966; Химия углеводов. Библиографический
указатель (1965—1968), изд-во «Наука», М., 1971.
206
26. R. В о g n a r, P. N a n a s i. J. Chem. Soc., 323 (1961).
27. R. Bognar, P. N a n a s i. Magyar Kem. folyoirat, 68, 32 (1962).
28. J. Sokolowski, J. Smiatcz, J. Szafranck. Roczn. Chem.,
37, 525 (1963).
29. J. Sokolowski, G. К u p г у szewski, J. Szafranck.
Roczn. Chem., 38, 945 (1964).
30. J. Sokolowski. Roczn. chem., 38, 889 (1964).
31. J. Sokolowski. Zes. Nauk. Wyzszej Szkoly pedagodicznei
Gdansky, 3, 153 (1963).
32. R. E. Reeves. Adv. Carbohydrate Chem., 6, 107 (1951).
33. Методы химии углеводов. Сборник статей под ред. Р. Уистлера и
М. Вольфрома, М., «Мир», 1967.
34. G. Р. Е 11 i s, J. Honeyman. J. Chem. Soc., 2053 (1952).
35. II. К- Кочетков, И. В. Торгов, М. М. Б о т в и н и к. Хи-
мия природных соединений (углеводы, нуклеотиды, стероиды, бел-
ки), гл. 7, М„ Изд-во АН СССР, 1961.
36. В. С а р о п, В. Е. Connett. J. Chem. Soc., 4492 (1965).
37. A. L i р t a k, R. Bognar. Acta. chem. Acad sci. hung., 72, 309
(1972).
38. P. Г. Ж б а и к о в. Инфракрасные спектры и структура углево-
дов, Минск, изд-во «Наука и техника», 1972.
39. В. Д. Щ е р б у х и н, Б. Н. Степ а иеико, О. Г. С е р д ю к.
ЖОХ, 35, 1844 (1965).
40 В Д Щербухии, Р. Д. Грешных, Б. Н. Степаненко.
ДАН СССР, 170, 362 (1966).
41. В. Д. Щербухии. Успехи биол. химии, 9, 198 (1968).
42 Т Н. М а ц к е в и ч, Е. П. Т р а й л и н а, И. А. С а в н ч, В. И. С п и-
цип. ДАН СССР. 188. 601 (1969).
43. В. А. А ф а и а с ь е в, И. Ф. Стрельцова. ЖФХ, 39, 110 (1965).
44 J S m 1 a t с z, J. Sokolowski. Roczn. chem., 44, 757 (1970).
45. S. A. Barker, E. Y. Bourne, M. S t a s e y, D. H. W h i f f e n.
.1. Chem. Soc., 171 (1954).
46. S. А. В a r k e r, E. Y. В о u r n e, R. Stephens D. H. W h i f-
fen.. J. Chem. Soc., 3468, 4211 (1954).
47 S. A. В a r ke r, R. S 1 e p h e n s. J. Chem. Soc., 4550 (1954).
48. N. В a g g e 11, S. А. В a r k e г, Л. В. E о s t e r, R. H. M о о r e,
D. H. W h i f f e n. J. Chem. Soc., 4565 (1960).
49. P. N a n a s i, P. G e г 1 e t t i. Gazz. chim. ital., 92, 576 (1962).
50. P. N an asi, E. N e m e s-N an a si, P. Gerletti. Acta Univ.
Debrecen, ser. phys. et chim., 9, 103 (1963); Gazz. chim. ital., 95,
966, 975 (1965).
51. P. D. V a s k o. J. В 1 a c k w e 1 1, J. L. К о e n i g. Carbohydrate
Res., 23, 407 (1972).
52. Л. Белла м и. Инфракрасные спектры сложных молекул, М.,
ИЛ, 1963.
53. Применение спектроскопии в химии. Сб. под ред. В. Веста, М.—Л.,
ИЛ, 1959.
54. В. А. Афанасьев, Ж. А. Д ж а м а и б а е в. Изв. АН Киргиз.
ССР, № 2. 64 (1973).
55. Дж. П о п л, В. Шнейдер, Г. Бернштейн. Спектры ядер-
ного магнитного резонанса высокого разрешения, М., ИЛ, 1962.
56. Н. Бхакка, Д. Уильямс. Применение ЯМР в органической
химии, М., «Мир», 1966.
57. R. U. Lemieux, R. К. К u 1 1 n i g, Н. J. Bernstein,
W. G. Schneider. J. Am. Chem. Soc., 79, 1005 (1957)- 80
6098 (1958);
207
R. U. L e m i e n x, J. D. S te ven s. Canad. J. Chem., 43, 205$>
(1965).
58. L. D. H all. Adv. in Carbohydrate Chem., 19, 51 (1964).
59. R. W. L e n s, J. P. H e e s c h e n. J. Polimer. Sci., 51, 247 (1961).
60. В. С a p о n, В. E. Connett. J. Chem. Soc., 4492 (1965).
61. J. P. К a m e r 1 i n g, M. J A. de В i e, J. F. G. V 1 i e g e n t h a r t.
Tetrahedron, 28, 3037 (1972).
62. Хроматография на бумаге. Сб. под ред. И. М. Хаиса и К. Мацека,
М., ИЛ, 1962.
63. А. А. А х р е м, А. И. К у з н е ц о в а. Тонкослойная хроматогра-
фия, М., «Наука», 1965, стр. 85.
64. Н. Н. Stroh, Е. Doman, Е. Haschke. Z. Chemie, It,
338 (1962); Н. Н. Stroh, A. Arnold, Н. G. Scharnow.
Chem. Вег., 98, 1404 (1965); Н. Н. S t г о h, Н. G. Scharnow.
Chem. Вег., 98, 1588 (1965).
65. Н. И. С узд алев а, В. В. Зелен ков а. Химия и биохимия
углеводов. Материалы 4-й Всесоюзной конференции по химии и
биохимии углеводов, М., «Наука», 1969, стр. 46.
66. Е. Stahl, U. Kaltenbach. Y. Chromatogr., 5, 351 (1961);
Z. anal. Chem., 181, 303 (1961).
67. G. Pactuska. Z. anal. Chem., 179, 427 (1961).
68. A. S c h w e i g e r. J. Chromatogr., 9, 374 (1962).
69. Ю. А. Жданов, Г. H. Д о p о ф e e н к о, С. В. Зеленская,
ДАН, СССР, 149, 1332 (1963).
70. J. М. R i с h е у, Н. G. Richey, R. S с h г а е г. Anal. Biochem.,
9, 272 (1964).
71. J. Fur и у a. J. Chromatogr., 18, 152 (1965).
72. R. E. Kadun ce. J. Chromatogr., 30, 204 (1967).
73. H. А. Преображенский, Э. И. Генкин. Химия органиче-
ских лекарственных веществ (гетероциклические соединения и их
аналоги). М.—Л., ГНТИ, 1953.
74. С. И. Л у р ь е, М. М. Ш е м я к и н. ЖОХ, 14, 935 (1944).
75. М. М. Шемякин, А. С. Хохлов, М. И. Колосов, Л. Д. Б е р-
гельсон, В. К. Антонов. Химия антибиотиков, М., Изд-во
АН СССР, 1961.
76. J. Goodman. Adv. Carbohydrate Chem., 13, 215 (1958).
77. Б. H. С т e n а н e н к о. Успехи химии, 28, 521 (1959).
78. J. Sykulscki. Wiadom. Chem., 16, 734 (1962).
79. R. Kuhn, G. К r ii g e r. Max-Planck-Inst., Medizinische For-
schug, Heidellerg Inst, fur Chemie Eingegangen, 30, 240 (1959).
80. F. Schneider, H. Geyer. Starke, 16 (10), 309 (1964).
81. К. H ey n s, M. R о I 1 e. Ber., 92, 2439 (1959).
82. К. H e у n s, H. Paulsen. H. Sch roeder. Tetrahedron, 13,
247 (1961).
83. В. И. В e к с л e p, Успехи химии, 33, 951 (1964).
84. E. R e i s t, R. S p e n с e г, В. В a k e r. Y. Am. Chem. Soc., 82,
2025 (1960).
85. Б. H. Степаненко. «Активные формы» простых сахаров и их
отношение к обмену углеводов. М,—Л., «Медгиз», 1945.
86. Э. И л и е л, Н. А л л и н ж е р, С. Э н ж и е л, Г. М о р р и с о н.
Конформационный анализ. М., «Мир», 1969, гл 6.
87. О. Н a s s е 1. Quart. Rews., 7, 221 (1955).
on F; ~ Ree ves- J- Am- Chem. Soc., 72, 1499 (1955).
on m’ R U d Г u m’ D- F- S h 0 w' J- Chem- Soc - 52’ 0965).
90 rnuTi? ? о X b 10 3t Кинетика реакций в растворах, М.—Л.,
208
91. С. Бексол. Основы химической кинетики. М., «Мир», 1964.
92. Р. 3 а г р а д и и к. Успехи химии, 30, 1272 (1961).
93. J. W. В a k е г, С. К. I n g о I d, J. Г. Т h о г р е. J. Chem. Soc.,
125, 28G (1924).
94. С. N. R i i b е г, I. Mi ns a a s. Ber., 59, 2266 (1926).
95. T. M. Lowry. Chem. Rev., 4, 231 (1927).
96. C. !•'. Smith, T. M. L о w r y. J. Chem. Soc., 667 (1928).
97. В. H. Кондратьев. Кинетика химических газовых реакций.
М„ Изд-во ЛН СССР, 1958.
98. В. Л. А ф а и а с ь е в, Н. И. Т р у ш к и н а, Р. И. С а рыб ае в а,
Д. В. Мальковская. И. Ф. Стрельцова. Доклад па 2-й
Всесоюзной конференции по исследованию строения и реакционной
способности физическими методами, Фрунзе, 1966.
99. 1. М. L о s, L. В. S i m р s о п, К. W i е s n е г. J. Am. Chem. Soc.,
78, 1564 (1956).
100. F. Р. W о г 1 е у, J. С. A n d г е w s. J. Phys. Chem., 32, 307 (1928).
101. H. S. I s b e 11, W. W. Pigman. Res. Nat. Stand., 18, 141 (1937).
102. В. С. H e n d r i c s, R. E. R u n d 1 e. J. Am. Chem. Soc., 60, 3007
1938).
103. E. M. R i c h a r d s, J. I. Paul kner, T. M. L о w r y. J. Chem.
Soc, 127, 1733 (1927).
104. H. H. Rowley, S. D. В a i 1 c y. J. Am. Chem. Soc., 62, 2562,
2563 (1940).
105. J. J. G i u 1 i a n o, D. G. H i 11. J. Am. Chem. Soc., 68, 2359
Soc., 74, 1380
(1952).
106.
107.
108.
109.
110.
112.
113.
114.
(1946).
D. G. H i 11, В. A. T h u m. J. Am. Chem.
A. de G r a n d c h a in p-C h a u d u n. Comp, rend., 243, 321 (1956);
258, 6564 (1964); Bull. Soc. chim. biol., 40, 887 (1958).
P. И. Кожахметова. Канд, дисс., Фруизе, 1968.
В. И. Иванов, Н. М. Четвереков, К. Д. Джуидубаев.
ДАН СССР, 160, 112 (1965).
К. Д. Джундубаев, Л. М. Корнева, Р. И. Кожахме-
това. Химия природных соединений, № 3, 168 (1965).
К. Д. Джундубаев. Канд, дисс., Фрунзе, 1965.
Y. О s a k a. Z. phys. Chem., 35, 661
С. S. Hudson. J. Am. Chem.
(1908); 39, 889 (1910).
J. M. N e 1 s о n, F. M. В e e g 1 e.
(1917).
R. Kuhn, P. Jakob. Z. phys.
(1900).
Soc., 29, 1571 (1907); 30, 1569
3. Am. Chem. Soc., 41, 559
Chem., 113, 389 (1924).
Z. anogr. Chem.,
H5.
116. H. V. E u i e г, А. О 1 a n d e r, E. Ru d ber g.
146, 45 (1925); 152, 113, (1928).
J. M. В r 6 n s t e d, E. A. G u g g e n Ti e i m. J. Chem. Soc. 49,
2554 (1927).
T. M. L о w r y, G. L. W i 1 s о n. Trans. Farad. Soc., 24, 683 (1928)
G. F. Smith. J. Chem. Soc., 1824 (1936). ' Л
P - « - Trans. Farad. Soc., 46, 14 (1950)
K- Wynne-Jones. Trans. Farad, Soc, 49,
Simpson. Recuefl, 73, 941 (1954); 75, 267
C.
J.
E.
117.
118.
119.
120.
121.
122.
123.
124.
125.
L. B.
R. P. Bell,
G. К i 1 d e,
243 (1953).
J. M. Los
(1956).
D. R i t t e n b e r g.
T. M. L о w г у, I.
C. G. S w a i n, J.
2538 (1952).
J. E. Prue.
W. F.
Graff. J. Am. Chem. Soc., 80, 3370 (1958)
Faulkner. J. Chem. Soc., 2883 (1925).
Brown. J. Am. Chem. Soc., 74, 2534
367
209
126. A. M. E a s t h a m, E. L. В l а с к a I I, G. A. Latremoni i le
J. Am. Chem. Soc., 77, 2122 (1955).
12/. E. L. В l а с к a 11, A. M. E a s t h a m. J. Am. Chem. Soc., 77,
2184 (1955).
128. C. G. Swai n. J. Am. Chem. Soc., 72, 4578 (1950).
129. C. G. S wai n, A. J. D i m i 1 o, J. P. G a r d n e r. J. Am. Chem.
Soc., 80, 5985 (1958).
130. А. В г о i d a, I. H о u m i n e r, S. P a t a i. J. Chem. Soc., 20, B,
411 (1966).
131. R. E. P in c oc k. Chem. Communs, 864 (1966).
132. H. Schmid. Monatsh. Chem., 94, 1206 (1963); 95,454,1009
(1964).
133. H. Schmid, G. Bauer. Monatsh. Chem., 95, 1781 (1964); 96,
583, 1503, 1508, 1510 (1965); 97, 168, 866 (1966).
134. H. S ch m i d. Monatsh. Chem., 98, 2097 (1967).
135. F. H. De an. J. Colloid. Interface Sci., 24, 280 (1967).
136. A. Kergomaro, M. Penar o. Tetrahedron Letters, 7, 769
(1968).
137. P. R. R о n y. J. Am. Soc., 90, 2824 (1968).
138. W. P i g m a n, H. S. Isbell. Adv. carb, chem., 23, 11 (1968);
24, 14 (1969).
139. M. Д и к с о н, Э. У э б б. Ферменты, At., ИЛ, 1961.
140. T. A. G e i s s m a n. Quart. Rev. Biol., 24, 309 (1949).
141. А. К л ю й в e p, K- Bau-Ниль. Вклад микробов в биологию,
М„ ИЛ, 1959.
142. Л. А. И и к о л а е в. Успехи химии, 33, 580 (1964).
143. Н. S. Isbell, W. W. Р i g m a n. Res. Nat. Bur. Stand., 66A,
233 (1962).
144. Дж. Хирш. Таблица конформационных энергий. В сб. «Избран-
ные проблемы стереохимии», М., «Мир», 1970, стр. 199.
145. Т. Н i 11. J. Chem. Phys., 16, 339, 938 (1948).
146. A. J. Kitaigorodsky. Tetrahedron, 9, 183 (I960); 14,230
(1961); 24, 5917 (1968).
147. J. В. Hendrikson. J. Am. Chem. Soc., 83,4537 (1961); 86,
4854 (1964).
148. В. Г. Дашевский, Ж- структ. химии, 6, 888 (1965); 7, 93
(1966); 9, 289 (1968).
149. R. Е. Reeves. Adv. Carbohydrate Chem., 6, 107 (1951).
150. R. B. Kel 1 y. Canad. J. Chem., 35, 149 (1957).
151. R. U. Lemienx. «Molecular Rearrangement», part 2, Interscience
Pullishers, 1963, p. 735.
152. R. U. Lemieux, N. J. C h ii. Abstracts of Papers Am. Chem.
Soc., 133 (1958).
153. J. T. Ed ward. Chem. Ind (London), 1102 (1955).
Г54. А. А. К о н к и н, Д. H. Ш и г о p и н, Л. И. Н о в и к о в а. ЖФХ,
32, 894 (1958).
155. Н. S р е d d i п g. Methods in Carbohydrate Chem., 1, 539 (1962).
156. A. J. M i c h e 11, H. G. H i g g i n s. Tetrahedron, 21, 1109 (1965).
157. G. G. C a s u. M. R e g g i a n i, G. G. G a l о, A. V i g e v a n i. Tet-
rahedron, 22, 3061 (1966).
158. В. H. H и к и т и и, Б. 3. В о л ч е к. Успехи химии, 37, 504 (1968).
159. С. А. В е е v е г s, W. Cochran. Proc. Roy. Soc., A190, 257
(1947).
160. T. R. R. Me Donald, C. A. Beevers. Acta Cryst., 3, 394 (1950).
161. T. R. R. Me D о n a 1 d. Acta Cryst., 5, 654 (1952).
162. K. J. H a u pt m a n. Acta Cryst., 6, 131 (1953).
210
163. R. С. К i i i e a n, W. G. For r ie;> 1). \V. You n g. Acta Cryst.,
15,911 (1962).
164. Д. Г. Кнорре, H. AY. Эмануэль. Успехи химии, 24,275
(1955).
165. Дж. Пиментел, О. Мак Клелан. Водородная связь. М.,
«Мир», 1964.
166. Э. А м и с. Влияние растворителя на скорость н механизм хими-
ческих реакций. М., «Мир», 1968.
167. И. Д. С а д е к о в, В. И. М и и к и и, А. Е. Л у ц к и й. Успехи
химии, 34, 380 (1970).
168. М. Eigen, L. De М а е у е г. Proc. Rey., А247, 505 (1958).
.169. М. Е i g е n, Z. Е 1 е k t г о с h е m., 64, 115 (I960).
170. Е. Колдин. Быстрые реакции в растворе, М., «Мир», 1966.
171. Р. D. В г a g g, L. Н о u g h. J. Chem. Soc., 4347 (1957).
172. J. F. Mahoney, С. B. Purves. J. Am. Chem. Soc., 64, 15
(1942).
173. В. И. Иванов. Успехи химии, 15, 560 (1946).
174. В. С. Иванова. Канд. дисс. Москва, 1968.
175. В. А. Афанасьев, Р. И. С а р ы б а е в а. Сб. «Развитие хими-
ческих наук в Киргизии». Фрунзе, изд-во «Илим», 1969, стр. 11.
176. В. А. Афанасьев, Р. И. С ары баев а. Изв. АН Киргиз.
ССР, Хе 4, 72 (1969).
177. Р. И. С а р ы б а е в а. Канд. дисс. Фрунзе, 1969.
178. В. И. И в а н о в, В. А. А ф а н а с ь е в, Р. И. С а р ы 6 а е в а.
ДАЧ СССР, 192, 1043, 1970.
179. G. Del Re. J. Chem. Soc., 4031 (1958).
180. G. Del Re, B. Pullman, T. Y on e z a w a. Biochim. et Biop-
hys. Acta, 75, 153 (1963).
181. Ю. А. Ж Д а и о в, В. И. Минкин, Г. H. Д о p о фе e и ко,
Ю. А. Остроумов, Е. Н. Малышева. Химия и биохимия
углеводов. Материалы 4-й Всесоюзной конференции по химии и
биохимии углеводов, 25—31 мая 1967 г. М., «Паука», 1969, стр. 22.
182. Ю. А. Кругляк, Г. Г. Д я дюш а, В. А. Куприевич,
Л. М. Подольская, Г. И. Каган. Методы расчета элек-
тронной структуры и спектров молекул. Киев, изд-во «Паукова
думка», 1969.
183. R. S. М u 1 I i k е п. J. Am. Chem. Soc., 72, 4493 (1950).
184, R. S. М u 1 1 i к е n. J. Chem. Phys., 56, 295 (1952).
185. Ю. А. Ж д а и о в, Г. H. Д о рофеенко, Г. В. Б о г д а и о.в а,
С. М. Лукьянов, В. Г. Алексеева, Ю. Е. А л е к с е е в.• Те-
зисы докладов 5 Всесоюзной конференции по химии и биохимии
углеводов. М., «Наука», 1972, стр. 62.
186. J. О. Diferrari, М. A. Ondetti, U. Deulofeu, J. Org. Chem. 24, 183
(1959).
187. F. M i ch e e 1, S. D e ge n e r, I. D i j о n g. Lieb. Ann. Chem. 701
233 (1967).
188. В. А. А ф а н а с ь e в, Ф. В. П и щ у г и н, Н. И. Т р у ш к и н а,
В. А. Хар мац. Гомогенный катализ. Материалы 1-го Всесоюз-
ного координационного совещания по гомогенному катализу. Фрун-
зе, изд-во «Илим», 1970, стр. 41. ’
189. Ф. В. П ищут ин. Канд, дисс., Фрунзе, 1970.
190. В г i g 1. Miihlschlegel, Schinle. Вег., 64, 2921 (1931).
191. Ж- А. Д ж а м а и б а е в. Каид. дисс., Фрунзе, 1973.
192 1932)^ ° П Я ° Pt В а г 11 е 11. J. Am. Chem. Soc., 54, 2881
21!
193. E. G. А г d a g h, В. К е 1 1 е a in, F. G. Rutherford, Н. Т. W а е-
staff. J. Am. Chem. Soc., 54, 721 (1932).
194. F. H. W e s t h e i m e r. J. Am. Chem. Soc., 56, 1962 (1934).
195. F. P. P r i c e, L. P. Hammett. J. Am. Chem. Soc., 63, 2387
(1941).
196. G. M. S a n ter r e, C. J. H a n s г о t e, T. J. Growell. J. Am.
Chem. Soc, 80, 1254 (1958).
197. W. P. J e n с к s. J. Am. Chem. Soc., 81, 475 (1959).
198. J. С. P о wer s, F. H. Wes the i met. J. Am. Chem. Soc., 82,
5431 (1960).
199. В. M. And erson, W. P. J e n с к s. J. Am. Chem. Soc., 82,
1773 (1960).
200. E. H. С о r d e s, W. P. J e n с к s. J. Am. Chem. Soc., 84, 826,
832, 4319 (1962).
201. Y. Ogata, A. Kawasaki, N. Okamura. J. Org. Chem, 29,
1985 (1964).
202. Г. Беккер. Введение в электронную теорию органических реак-
ций. М, «Мир», 1965.
203. У. Р. Дженкс. Механизм и катализ простых реакций карбо-
нильной группы. Сб. «Современные проблемы физической органи-
ческой химии», М, «Мир», 1967, стр. 343—392.
204. В. Дженкс. Катализ в химии и энзимологии. М, «Мир», 1972.
205. М. Л. Бейдер. Механизмы катализа нуклеофильных реакций
производных карбоновых кислот. М, «Мир», 1964.
206. Т. Брюс, С. Б е н к о в и ч. Механизмы биоорганических реак-
ции. М, «Мир», 1970.
207. Л. Г а м м е т. Основы физической органической химии. М,
«Мир», 1972.
208. J. С о m р t on, М. L. W о 1 f г о m. J. Am. Chem. Soc, 56, 1157
(1934).
209. G. H. Stempel. J. Am. Chem. Soc, 56, 34 (1934).
210. E. G. R. A r d a g h, F. C. R u t h e r f о г d. J. Am. Chem. Soc, 57,
1085 (1935).
211. A. S k r a b a 1 И. A, Z. phys. Chem, 119, 305; 122, 349, 357 (1926).
212. E. A. Moelwyn-Hughes. Trans. Farad. Soc, 25, 81 (1929).
213. R. Leutner. Monatsh. Chem, 60, 317 (1932).
214. L. P. Hammett, M. P a u 1. J. Am. Chem. Soc, 56, 830 (1934).
215. E. Mo e 1 wy n-H u g e s. Z. phys. Chem, B26, 281 (1934).
216. P. M. Leininger, M. К i 1 p a r i c k. J. Am. Chem. Soc, 60,
1268, 2891 (1938).
217. L. P. Ham met, L. Z u c k e r. J. Am. Chem. Soc. 61,2779
2785, 2791 (1939).
218. С. A. В u n t о n, T. A. Lie wel lyn, С. A. V e г n о n. J. Chem.
Soc, 4419 (1955).
219. J. T. E d war d. Chem. and Ind, 1102 (1955).
о»?' Д’ ?• Fo s f e r’ W-G. Overend. Chem. and Ind, 566 (1955).
221. F. A. Long, M. A. Paul. Chem. Rev, 57, 934 (1957).
222- ,s 11 a f i s a d e h. Adv. Carbohydrate Chem, 13, 9 (1958).
223. W. G. О v e r e n d, C. W. R e e s, J. S. S eq ueir a. J. Chem.
Soc, 3429 (1962).
224. F. Shafisadeh. Tappi, 46, 381 (1963).
225- L Er T ' me 1 *• Canad. J. Chem, 42, 1456 (1964).
22"- M.S. Fe at her, J. S. H a r r i s. J. Org. Chem, 30, 153 (1965).
227. В. E. В anks Y. Meinwald, A. J. R h i n d-T u 11, J. S h e 11.
C.A.Vernon. J. Chem. Soc, 3240 (1961).
228. J. A. M 111 s. Adv. Carbodydrate Chem, 10, 1 (1955).
212
229. В. И. Иванов, 3. И. Кузнецова. Изв. АН СССР, ОХН,
646 (1958).
230. В. И. И в а к о в, Н. Я. Ленина а, В. С. И в а и о в а. Изв. АН
СССР, ОХН (1960).
231. V. 1. Ivanov, N. Ja. J е n s h i n a. J. Polymer. Sci., 53, 93
(1961).
232. В. И. Иванов, Г. M. Корнева, Л. А. Сучкова. ДАН
СССР, 156, 1112 (1964).
233. J. W. Haas, R. Е. Kadunce. J. Am. Chem. Soc., 84,4910
(1962).
234. H. H. Stroh, Н. Gamricht. Chem. Ber., 96, 651 (1963).
235. H. H. S t г о h, H. A. R. Hempel, R. A p e 1. Chem. Ber., 98,
2500 (1965).
236. H. H. S t г о h, A. A r n о 1 d. Chem. Ber., 98, 1404 (1965).
237. H. H. Stroh, H.Tengler. Chem. Ber., 101, 751 (1968).
238. H. H. S t г о h, H. Goluke. Z. Chem., 7, 61 (1967).
239. В. C a pon. Chem. Rev., 69, 407 (1969).
240. В. A. X a p м а ц, В. А. А ф а и а с ь e в. ЖФХ, 42, 2078 (1968).
241. В. А. Афанасьев, В. A. X a p м а ц. ЖФХ, 43, 500 (1969).
242. В. А. Харман. Канд. дисс. Фрунзе. 1968.
243. S. М. С a n t о г, L. Р. Р е n i s t о n. J. Chem. Soc., 62, 2113
(1940).
244. Д. Крам, Дж. Хэммонд. Органическая химия. М„ <Мир»,
1964.
245. С. Г л е с с т о и. К- Л е й д е р, Г. Э й р и и г. Теория абсолют-
ных скоростей реакций. М., ИЛ, 1943.
246. R. В о g п а г, Р. N a n a s i. Mag. Kem. Folyoirat, 62, 88 (1956).
247. В. Г. Л с о в и ч. Введение в статистическую физику. М.—Л.,
ТИТТЛ. 1950.
248. Л. Д. Л а н д а у. Е. М. Лифшиц. Статистическая физика.
Гостехнздат, 1958.
249. И. Н. Г о д и е в. Вычисление термодинамических функций по мо-
лекулярным данным. М., ТИТТЛ, 1956.
250. Н. S. Isbell, Н. L. Frush. J. Res. Nat. Bur. Stand., 46, 132
251.
252.
253.
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
(1951); 47, 239 (1951).
H. S. 1 s be 11, ” '
R. Kuhn, L. В irkof er. Ber., 71, 621 (1938).
J. E. Hodge, С. E. R i s t. J. Am. Chem. Soc., 74, 1494 (1952).
P. bl a n a s i. Acta
T. Jasinski
(1966).
T. Jasinski.
Chem.. 39, 827
115. 313 (1968); . ... ____
K. S m i a t a c z о w a. Wiadom. Chem., 25, 343 (1971).
J. W. Baker. J. Chem. Soc.. 1583, 1979 (1928): 1205 (1929).
Z. Pawlak. E. Gorska. Roczn. Chem.. 43, 1237 (1969).
W. Pigman, E. A. С I e ve 1 a n d, D. H. С о u c h. J. H С le-
veland. J. Am. Chem. Soc., 73, 1976 (1951).
J I n о u e, К. О n о d er a. J. Agric. Chem. Soc. (Japan), 22, 70
H948); C. A., 45, 9480 (1951).
В. C ° n n e 11. Tetrahedron Letters, 22, 1395 (1964)
B. Connett. J. Chem. Soc., 8, 4497 (1965).
D. Palm. Chem. Ber., 98 (2), 433 (1965).
* Волкова, M. Г. Ченцова.
H. L. Frush. J. Org. Chem., 23, 1309 (1958).
к.
Univ. Debrecen, 6,
S m i a t a c
z о w a.
107 (1959—1960).
Rocz. Chem., 40, 1273
Smi at ac
K.
(1965); 40, 1273
44, 411 (1970).
z о w a,
(1966);
J. Sokolowski. Rocz.
41, 579 (1967); 42, 107,
B. Capon,
B. Capon,
H. S i m о n.
Б. H. Степаненко, Э. C.
ДАН СССР, 183, 1353, (1968).
213
266. Б. Н. Степаненко, В. А. Игн атюк-Май ст реп ко,
М. Г. Ченцова. Сб. «Химия и обмен углеводов», М., «Наука»,
1965, стр. 100.
267. Е. Mitts, R. М. Hixon. J. Am. Chem. Soc., 66, 483 (1944).
268. R. Л. M о r t о n, G. Л. P i t t. Biochem. J., 59, 128 (1955).
269. T. G. В о n n e r, M. В а г n a r d. J. Chem. Soc, 4176 (1958).
270 E. H. Cordes, W. P. Jencks. J. Am. Chem. Soc., 85, 2843
(1963).
271. T. W. T а у 1 о r. J. Chem. Soc., 2741 (1930).
272 V. К. К r i e b I e, К. A. H о 1 s t. J. Am. Chem. Soc., 60, 2976
(1938).
273. B. S. R a b i n о v i c h, C. A. W i n k 1 e r. Canad. J. Research, 20B,
76 (i942).
274 J. T. Edward, H. S. Chang, K. Yates, R. Stewart.
Canad. J. Chem., 38, 1518 (1960).
275. C. D. Schmulbach, R. S. D r a g o. J. Am. Chem. Soc., 82,.
4484 (1960).
276. R. S. Drago, D. В a f u s. J. Phys. Chem., 65, 1066 (1961).
277 A. Berger. A. Loewenstein, S. M e i b о о m. J. Am.
Chem. Soc.. 81, 62 (1959).
278. M. T a k e d a, E. O. S t e i s k a 1. J. Am. Chem. Soc., 82, 25 (1960).
279. Б. Июль маи. А. П ю л ь м а н. Квантовая биохимия. М„
«Мир», 1965.
R.Kuhn. A. D а п s i. Вег., 69, 1745 (1936).
R. В о g п а г. Р. Nanasi. Nature, 171, 475 (1953); J. Chem.
Soc.. 189 (1955). „ ,
R. Bognar, P. Nanasi. Acta Chim. Hung., 12, 115 (1957).
R. В о g n a r, P. N a n a s i. Tetrahedron, 24, 175 (1961).
R. Bognar. P. Nanasi, E. Neme s-N a n a s i. J. Chem.
Soc., 193 (1955).
R. В о g n a r, P. N a n a s i, J. Chem. Soc., 320 (1961).
G. P h i 1 i p p, H. S i m о n. Carbohydrate Res., 8,424 ((1968).
G. Philipp. Kinetische Untersuchungen zum Mechanismus der
Transglykosidierung von N-Glykosiden, Diss. Dokt. Naturwiss. Fak.
Allgem. Wiss. Techn. Hochschule, Miinchen, 1967.
В. Л. Афанасьев, Ф. В. П ищу г и и. ЖФХ, 44, 1811 (1970),
Ф. В. П и щ у г н н. В. А. А ф а н а с ь е в, ЖФХ, 44, 2085 (1970).
Л. П. Т е р е н т ь е в, В. М. П о т а п о в. Основы стереохимии.
М.—Л., «Химия», 1964.
Р. Бел л. Кислотно-основной катализ п строение молекул. В кн.:
Катализ. Исследование гомогенных процессов, М., ИЛ, 1957, стр. 5.
R. Р. Bell. Acid-Base Catalysis, Oxford, 1941, p. 65.
M. Килпатрик. Кислотный и основной катализ. В кн.: Ката-
лиз. Исследование гомогенных процессов. М., ИЛ., 1957, стр. 67.
Я. К. Сыркин. Проблемы кинетики и катализа, 10, 225 (1960).
Н. М. Чирков. Проблемы кинетики н катализа, 10, 255 (1960).
П. А ш м о р. Катализ и ингибирование химических реакций. М„
«Мир», 1966.
В. Н. Яковлев. Кинетика ферментативного катализа. М., «Hav-
ка». 1965.
280.
281.
282.
283.
284.
285.
286.
287.
288.
289.
290.
291.
292.
293.
294.
295.
296.
297.
298.
299.
300.
301.
302.
К. J. Р е d е г s е n. J. Phys. Chem., 38, 581 (1934).
А. А л ь б е р т, Е. С е р ж а н т. Константы ионизации кислот ж
оснований. М—Л., «Химия», 1964.
N. F. Н а 1 1, М. R. S р г i n k 1 е. J. Am. Chem. Soc, 54, 3469 (1932),
А. V. W i 11 i. Helv. Chim. Acta, 39, 1193 (1956).
И. Ф. Стрельцова. Канд, дисс., Фрунзе, 1971.
214
303. В. Л. Афанасьев, Н. И. Трушкина. ЖФХ, 1836 (1971).
304. Н. И. Трушкин а. Канд, дисс., Фрунзе, 1971.
305. R. W. Hay, J. J. Р о г t е г. J. Chem. Soc., 1261 (1967).
306. R. W о 1 f e n d e n, W. P. J e n c k s. J. Am. Chem. Soc., 83, 2763
(1961).
307. Э. M. Арнетт. Современные проблемы физической органической
химии. М., «Мир», 1967, стр. 195.
308. Е. С. Jackson, С. S. Hudson. J. Am.sChem. Soc., 63, 1229
(1941).
309. R. К- Ness, H. G. F 1 e t c h e r. J. Am. Chem. Soc., 80,2007
(1958).
310. N. J. A n t i a, M. B. Perry. Canad. J. Chem.,-38, 1917 (1960).
311. С. T. В i s h о p, F. P. С о о p e r. Canad. J. Chem., 40, 224 (1962);
41, 2743 (1963).
312. H. А. Халтуринский, Ю. В. Моисеев, Г. А. К о г а и,
В. С. М а р е в ц е в, Г. Е. 3 а н к о в. Изв. АН ССР, серия хим.,
1785 (1970); Н. А. Халтуринский, Ю. В. Моисеев,
Г. Е. Зайков. Изв. АН СССР, серия хим., 2686 (1970).
313 G. D. S t е w а г t, L. Н. D о n n а 1 у. J. Am. Chem. Soc., 54, 3555,
3559 (1932).
314. В. А. Афанасьев, К. А. Д а в л е т к е л ь д и е в а, Н. И. Труш-
кина. Изв. АН Киргиз. ССР, №4, стр. 59 (1973).
315. М. Frankel, A. Katchalsky. Biochem. J., 31, 1595 (1937);
32, 1904 (1938); 35, 1024, 1028, 1034 (1941).
316. S. Lewin. Biochem. J., 63,14 (1955); 65,30 (1957); 76,17
H960).
317. S. Lewin, Z. К о s i n s k i. Trans. Farad. Soc., 54, 22 (1958).
318. M. X. К a p a n e т ь я и ц. Методы сравнительного расчета физи-
ко-химических свойств. М., «Наука», 1965.
319. Ю. А. Жданов, В. И. М и и к и и. Корреляционный анализ в
органической химии. Ростов-на-Дону, Изд-во Ростовского ун-та,
1966.
320. В. А. Пальм. Основы количественной теории органических ре-
акций. Л„ «Химия», 1967.
321. К- Д- Р и ч е, У. Ф. С э д ж е р. Исследование уравнений, связы-
вающих строение и реакционную способность органических соеди-
нений. В кн.: «Современные проблемы физической органической хи-
мии». М., «Мир», 1967, стр. 498.
322. Пространственные эффекты в органической химии. Сб. статей под
ред. М. С. Пьюмеиа. М., ИЛ, 1960, гл. 13.
323. Н. Eyring, М. Р о 1 а п у i. Trans. Farad. Soc., 34, 3 (1938).
324. J. E. L e f f 1 e r. J. Org. Chem., 20, 1202 (1955).
325. F. H. Constable. Proc. Roy. Soc., A108, 355 (1925).
326. G. M. Schwab, E. С r e m e r. Z. Phys. Chem., A144, 243; B5,
406 (1929).
327. Г. M. Шва б. Катализ с точки зрения химической кинетики,
ОНТИ, 1934.
328. Г. М. Шваб. Катализ. Вопросы теории и методы исследования.
М„ ИЛ, стр. 25.
329. Е. Кремер. Компенсационный эффект в гетерогенном катализе.
В кн.; «Катализ. Электронные явления», М., ИЛ, 1958, стр. 86.
330. А. А. Б а л а и д и н. ЖФХ, 4, 3,257 (1933); 31, 4,139 (1957)- ДАН
СССР, 93, 55, 273, 477 (1953).
331. L. G. Hepler, W. F. O’Hara. J. Phys. Chem., 65, 811 (1961).
332. К. J. L a i d 1 e r. Trans. Farad. Soc., 55, 1725 (1959).
215
333. H. S. V e n к a t a r a m a n, C. N. H i n s li e 1 w о о d. J Chem
Soc., 4977, 4986 (1960).
334. R. F. Brown. J. Org. Chem., 27, 3010, 3015 (1962).
335. J. E. L e f f 1 e r. J. Org. Chem., 31, 533 (1966).
336. R. C. Peter s en, J. H. M а г gra 1, S. D. R a s s. J. Am. Chem.,
Soc., 83, 3819 (1961).
337. О. E x n e r. Nature, 201, 488 (1964).
338. Ю. А. Давыдовская, Ю. И. В ай н ш т ей н. Азометины,
Изд-во Ростовского ун-та, 1967, стр. 234.
339. В. И. М и н к и н. К). Л. Ж д а и о в. Азометилы, Изд-во Ростов-
ского ун-та, 1967, стр. 72.
340. В. А. 11 а в л о в. Канд, дисс., Москва, 1969.
341. Ю. А. Жданов, В. А. Поленов. ЖОХ, 35, 589 (1965).
342. В. И. Веденеев, Л. В. Г у р в и ч, В. Н. Кондратьев,
В. Л. Me две де в, Е. Л. Ф р а н к о в н ч. Энергии разрыва хи-
мических связей. Потенциалы ионизации и сродства к электрону,
М., Изд-во АН СССР, 1962.
343. Т. 1. С г о w е 1 1, D. W. Ре с k. J. Am. Chem. Soc, 75, 1075 (1953).
344. В. W i t к о р, Т. W. В е i 1 е г, J. Am. Chem. Soc., 76, 5589 (1954).
345. С. S с h г о е d е г, S. Р г е i s, К. Р- L i п к. Tetrahedron, 13, 23
(1960).
346 R. Cantarel, J. Guenzet. Bull. Soc. Chim. France, 1285
(1961).
347. E. H. G о r d e s, W. P. J e n с к s. J. Am. Chem. Soc., 84, 826
(1962).
348. E. H. G о г d e s, W. P. Jencks. Biochemistry, 1,773 (1962).
349. K. J. Pedersen. J. Am. Chem. Soc., 60, 595 (1938).
350. F. H. We s thei тег, H. С о h e n. J. Am. Chem. Soc., 60, 90
(1938).
351. S. Huni g. Lieb. Ann., 569, 198 (1950).
352. С. A. H a 1 ey, P. M a i t I a n d. J. Chem. Soc., 3155 (1941).
353. F. S. P г о u t. J. Org. Chem., 18, 928 (1953).
354. K. Runge, R. Mayer. Lieb. Ann. Chem., 707, 161 (1967).
355. А. А. Я с н и к о в, К. И. М а т к о в с к и й, Е. М. Г айво рон-
ский. Укр. хим. ж., 22, 88 (1962).
356. И. В. Мельниченко, А. А. Я с н н к о в. Укр. хим. ж., 30,
835 (1964).
357. И. В. М е л ь и и ч е н к о, Е. А. Ш и л о в, А. А. Я с и и к о в. Укр.
хим. ж., 30) 1171 (1964).
358. В. JI а н г ей б е к. Органические катализаторы и их отношение к
ферментам. М., ИЛ, 1961; Л. А. Николаев. Биокатализаторы
и их модели. М., изд-во «Высшая школа», 1964.
359. Н. Euler, Е. Eriksson, Е. Brunius. Lieb. Ann., 467,
213 (1928); 27, 715 (1928).
360. Н. Е u 1 е г, Е. В г u n i u s, К. Joseihson. Z. physiol Chem.
155, 259 (1928).
361- В. Bauminger, Е. L i е b е n. Biochem. Z.. 292, 92 (1937).
Кузин (с сотр.), Биохимия, 3, 481 (1938)- 4, 142 367
4-*9n9)l6, 113 (1941): 13’ 27 <1948)-
n « Биохимия, 6, 146 (1941); 15, 30 (1950).
*’ "•ДАфаиасьев. T. К. Ю н у с а л и е в а. ЖФХ, 45, 1444
365' U957R 27С Е' А' Ши ло в- УкР- хим- ж-. 23, 216» 333
иУЭ'). 47, 639 (1961).
366' Г7 Tio4Mglis’ F' Dukins- ’nd- Eng. Chem. Anal. Ed., 3,
I I (IУоI).
216
367. Н. В. К о з л о в а, И. В. М е л ь н и ч е и к о, А. А. Я с и и к о ь.
кр. хим. ж., 34, 1145 (1968).
368. U. Р. Jeiicks, J. С а г г i и о 1 о. J. Am. Chem. Soc., 82, 4810
(I960).
369. В. Л. Афанасье в, Н. И. Т р у ш к и н а. ЖФХ, 45, 2766 (1971).
370. J. Goo синап. Adv. Carbohydrate Chem., 13, 215 (1955).
371. E. F isher. Ber., 47, 1377 (1914).
3/2. A. A. Schmuck. Zhur. Russ. riz. Khim. Obshchestvo, 61, 1759
(1929); Chem. Zentr., 101, 1, 3173 (1930).
373. B. Helierich, A. Mitrowsky. Chem. Ber., 85, 1 (1952).
374. G. S h a w, R. N. W a r r e n e r. Proc. Chem. Soc., 351 (1957);
81 (1958).
375. В .А. Афанасьев, Ж. А. Джаманбаев. Авт. свидетель-
ство 1647215/23—4 (4.07.72).
376. Д. Бетел, В. Голд. Карбониевые ионы. М., «Мир», 1970.
377. Н. К. Депо. Карбониевые ионы. В кн.: «Современные проблемы
физической органической химии». М., «Мир», 19о7, стр. 393.
378. Г. А. О л а, Ч. Питтмаимл. Спектроскопические свойства
алкилкарбониевых ионов в растворах сильных кислот. В кн.: «Но-
вые проблемы физической органической химии». М., «Мир», 1969,
стр. 338.
379. R. Kuhn, F. Weygand. Ber., 70, 769 (1937).
380. R. К u h и, G. К r ii g e r. Ann., 628, 240 (1959).
381. J. E. Hodge, С. E. Rist. J. Am. Chem. Soc., 75, 316 (1953).
382. J. E. Hodge. Adv. Carbohydrate Chem., 10, 169 (1955).
383. J. G. Ericson. J. Am. Chem. Soc., 77, 2839 (1955).
384. Л. A b r a m s, P. H. L о wy, H. В о r s о о k. J. Am. Chem. Soc.,
77, 4794 (1955).; 78, 3175 (1956).
385. L. Ro sen, J. W. W о о d s, W. P i g m a n. J. Am. Chem. Soc.,
80, 4697 (1958).
386. F. M i c h e e 1, B. S c h 1 e p p i n g h о f f. Chem. Ber., 1702 (1956).
387. F. M i c h e e 1, A. Fr owein. Chem. Ber., 90, 1599 (1957).
388. F. M i c h e e 1, G. H a g e m a n n. Chem. Ber., 92,2836 (1959) ;
93, 2381 (1960).
389. F. Micheel, I. Dijong. Tetrahedron Letters, 21 (1962).
390. F. Micheel, К. H. Heinemann. Tetrahedron Letters, 42,
3769 (1965).
391. H. Paulsen. Tetrahedron Letters, 9,451 (1964).
392. M. S. F e a t h e r, K. R. Russel. J. Org. Chem., 34, 2650 (1969).
393. L. Rosen, J. W. Woods, W. P i g m a n. Chem. Ber., 90, 1038
(1957).
394. А. Готтшалк. Взаимодействие между восстанавливающими са-
харами и аминокислотами в нейтральных и кислых средах. В кн.:
«Гликопротеины», том. 1, М., «Мир», 1969, стр. 105.
•395. S. В а у n е, W. Н. Н о 1 m s. J. Chem. Soc., 3247 (1952).
396. Б. H. Степ а непко. H. И. К а л e т и и а, В. В. 3 е л е н к о в а.
ДАН СССР, 200, 609 (1971).
397. S. К о 1 k a, J. S о к о 1 о w s к i. Rocz. Chem., 46, 147, (1972).
398. J. J о s h i m u r a, M. F u n a b a s h i, H. Simon. Carbohydrate
Res., 11, 276 (1969).
399. F. Micheel. Chem. Ber., 85, 1077 (1952).
400. F. Micheel, 1. Dijong. Antn., 658, 120 (1962).
401. D. P a 1 m, H. S i m о n. Z. Naturforsch., 18b, (5), 419 (1963);
20b, (1). 32 (1963).
402. Л. Физер, M. Ф и з e p. Органическая химия. M., «Химия»,
1970, том 2, стр. 534.
217
403. J. Kenner, E. С. К n i g h t. Ber., 69, 341 (1936).
404. F. W ey g a n d. Ber., 73, 1284 (1940).
405. F. W e у g a n d, H. S i m о n, J. F. К 1 e b e. Chem. Ber., 91,
1567 (1958).
406. M. M. Шемякин, В. И. Май мии д. Изв. АН СССР, 102,
1147 (195а).
407. М. М. S с h е m у a k i n, М. L. М a i m i n d, К- M. E rmol aev,
E. M. В a m d a s. Tetrahedron, 21, 2771 (1965).
408. H. Simon, K. D. К e i 1, F. Weygand. Chem. Ber., 95 17 (1962).
409. H. S i mo n, H. D. D о r r e r,
(1963).
410. H. Simon, G. Hen bach,
3106 (1967).
411. H. Simon, W. Moldenh
A. T r e b s t. Chem. Ber., 96, 1285
H. Wacker. Chem. Ber., 100,
а и e г, А. К г a и s. Chem. Ber.,
102, 2777 (1969).
412. H. Simon, A. Kraus. Fortschr. Chem. Forsch. 14/4, 430 (1969).
413. H. J. Haas, A. S e e 1 i g e r. Chem. Ber., 96, 2427 (1963).
414. V. С. В a rry, P. W. Mitchell. Nature, 175, 220 (1955).
415. G. J. В 1 о i n к, К- H. P a и s а с к e r. J. Chem. Soc., 661 (1952),
416. F. Micheel, I. D i у о n g. Lieb. Ann. Chem., 669, 136 (1963).
417. 1. D i у о n g, F. Michee 1. Lieb. Ann. Chem., 684, 216 (1965).
418. S. К i t а о к a, K. Onodera. J. Org. Chem., 28, 231 (1963).
419. H. EL К h a d e m. Adv. Carbohydrate Chem., 20, 139 (1965).
420. O.LCh apm an, W.J. We 1 s t e ad, T. J.M ur p hy, R. W. К i n g,
J. Am. Cbem. Soc, 86, 732 (1964); 89, 7005 (1967).
421. W. C. Stickler. Hauptversammlung der Gesellschaft Deutscher
Chemicker Bonn, Sept., 1965.
422. L. L. En ge 1. J. Am. Chem. Soc., 57, 2419 (1935).
423. O. Diels, R. Meyer. Lieb. Ann., 519, 157 (1935); E. G. V. Per-
cival. Adv. Carbohydrate Chem., 3, 23 (1948).
424. F. W e у g a n d, H. G r i s e b a c h, K- D. К i r c h n e r, M. Ha-
gel ho r s t. Chem. Ber., 88, 487 (1955).
425. G. H e n s ek e, H. J. В i n t e. Chimia, 12, 103 (1958).
426. G. H e n s e к e, H. К 6 1 e r. Ann. Chem., 105, 614 (1958).
427. O. L. C h a p m a n, R. W. К i n g, W. J. W e I s t e a d, T. J. M и r p-
hy. J. Am. Chem. Soc., 86, 4968 (1964).
428. O. L. Chapman. Tetrahedron Letters, 2599 (1966).
429. H. El Kha dem. Adv. Carbohydrate Chem., 18, 99 (1963).
430. L. M e s t e r. J. Am. Chem. Soc., 77, 4301 (1955).
431. L. M e s t e r, A. Major. J. Am. Chem. Soc., 77, 4297 (1955); 79.
3236 (1957).
432. L. M e s t e r, F. Weygan d. Bull. Soc. Chim. France, 350 (1960).
433. L. M e s t e г, E. M о c z a r, J. P a r e 11 o. J. Am. Chem. Soc.,
87, 596 (1965).
434. L. Mester, A. Stephen, J. P a r e 11 o. Tetrahedron Letters,
4119 (1968).
435. L. Mester, E. M о c z a r, G. Vass, A. S chi tn pl. Carbohydrate
Res., 5, 406 (1967); Tetrahedron Letters, 2943 (1967).
436. L. Mester, G. V a s s. Tetrahedron Letters, 3844 (1969).
437. L. Mester. Chimia, 23, 133 (1969).
438. В. А. Афанасьев, И. Ф. Стрельцова. ЖФХ, 46, 2545
(1972); И. Ф. Стрельцова, В. А. Афанасьев. Изв. АН
Киргиз. ССР № 6, 43 (1971); Труды третьей конференции по химик
и физико-химии полиацеталей, Фрунзе, изд-во «Илим», 1974.
439. F. С. Whi t mo re. J. Am. Chem. Soc., 54, 3274 (1932).
218
440. V. Р г е 1 о g. Angew. Chem., 70, 145 (1958); J. Chem. Soc., 93:
(1963).
441. A. С. Cou p e, M. M. Martin, M. A. Me Kervey. Quart. Rev.,
20, 119, (1966).
442. H. Л. Ч у p с ни а, И. Ф. Стрельцова, В. Л. А ф а и а с ь е в.
Изв. АН Киргиз. ССР, № 3, 64 (1975).
443. Дж. У э й, Ч. Пре тер. Структура и анализ сложных реакцион-
ных систем. В кн.: «Катализ. Полифуикциопальные катализаторы и
сложные реакции», М., «Мир», 1965.
444. Л. Л. Баландин. ДАН СССР, 24,741 (1939); ЖФХ, 15, 615
(1941).
445. N. В. Z w о 1 i n s k 1, Н. Е у г i n g. J. Am. Chem. Soc., 69, 2702
(1947).
446. В. В. Налимов, Н. Л. Чернова. Статистические методы пла-
нирования экстремальных экспериментов. М., «Наука», 1965.
447. М. В. Волькеиштейи, Б. II. Гольдштейн. ДАН СССР,
170, 963 (1966).
448. R. V. Lemieux, \V. Р. S h у 1 u k, G. Huber. Canad. J. Chem.,
33, 148 (1965).
21»