/

















































Теги: химия химическая промышленность химические реакции
Год: 1964
Текст
ГОСУДАРСТВЕННЫЙ КОМИТЕТ
ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ ПРИ ГОСПЛАНЕ СССР
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 8
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ
Москва — 196 4
При проведении биологических, микробиологических
и гистохимических исследований (например, выявление
активности ферментов окислительного обмена, функцио-
нальных групп белков, сульфгидрильных групп белко-
вой природы, их локализацию в срезах фиксированной и
нефиксированной ткани и т. д.) наряду с реактивами
общего лабораторного назначения применяется ряд спе-
циальных продуктов, красителей и вспомогательных ве-
ществ для субстратов.
В настоящем сборнике помещены методы получе-
ния реактивов и препаратов указанного назначения, а
также синтез полупродуктов для их изготовления.
СОДЕРЖАНИЕ
Фенилгидразон фенилглиоксалевой кислоты. В. М. Островская,
А. А. Прянишников........................................ ». 5
Феиилгидразои пировиноградной кислоты. В. М. Островская,
А. А. Прянишников............................................ 7
4-Нитрофеиилгидразон фенилглиоксалевой кислоты. В. М. Ост-
ровская, А. А. Прянишников................................... 8
4-Нитрофеиилгилразон бензальдегида.. А. А. Прянишников, В. М. Ос-
тровская, О. Т. Лушина..................................... 10
Диформазан. В. М Островская, А. А. Прянишников.................. 12
Неотетразолий хлористый. В. М. Островская, А. А. Прянишников 14
Йодиитроформазан. В. М. Островская, А. А, Прянишников .... 16
Йоднитротетразолий. В. М. Островская, А. А. Прянишников ... 18
Динитроформазан. В. М Островская, А. А. Прянишников, О. Т. Лу-
шина ........................................................ 21
Нитротетразолиевый синий. В. М. Островская, А. А. Прянишников,
О. Т. Лушина................................................. 24
Тетра-нитрофенил-тетразолий. М. Н. Малев....................... 27
Тетразолий синий. М. П. Малев .... • . -........................ 32
Нитронеотетразолий. М. Н. Малев................................. 36
Метилтиазолилтетразолий. М. И. Малев................• . 39
2,3 - Дифеиил-5 - бензолазо - тетразолий хлористый и 2,3-дифенил-5-
бензолазо-тетразолий бромистый. М. Н. Малев ......... 44
Диазоль сииий С. М. Н. Малев.................................... 48
а-Нафтилацетат. М. И. Малев . . . •............................. 50
|Э-Нафтилацетат. Е. Л. Стряпчева................................ 51
DL-Аланил-^-нафтиламид. М. Н. Малев............................. 53
2-Окси-З-иафтальдегнд. М. Н. Малев, Е. А. Стряпчева............. 56
Фосфат азотола 2,4 МК. М. Н. Малев, Е. А. Стряпчева........... 59
Фосфат азотола А. М. Н. Малев................................... 63
2,2‘-Диокси-6,6‘-динафтилдисульфид. М. И. Малев................. 67
л-Нитро-а-бромацетофенон. В. А. Михалев, А. П. Сколдинов,
М. И. Дорохова, А.П. Арендарук, И. Е. Смолина, Д. Д. Смолин 71
5-Бромацетил-З-нитробеизойиая кислота. И. Б. Краснов.......... 74
У-Этилмалеинимид. И. Б. Краснов............................... 83
АГ-л-Хлорфенил-диамид фосфорной кислоты. М. Н. Малев .... 86
4-Амино-М^диметнлнафтиламин солянокислый. М. Н. Малев ... 88
Ацетилтиохолинйодид. М. Н. Малев ............... 92
Бутирилтиохолинйодид. Af. И. Малев............................ 96
Бриллнаитсульфофлавин ФФ. М. В. Березовская, А. А. Пряниш-
ников . •................................................. 99
Алфавитный перечень соединений, описанных в настоящем выпуске 102
ФЕНИЛГИДРАЗОН ФЕНИЛГЛИОКСАЛЕВОЙ КИСЛОТЫ
В. М. ОСТРОВСКАЯ, А. А. ПРЯНИШНИКОВ
.= 7С00Н
/ с/ ___________
X—Vn—NH—х \
CUHUNSOS М. в. 240,26
Фенилгидразон фенилглиоксалевой кислоты применяют
для получения формазанов [1, 2]. В литературе имеется про-
пись получения фенилгидразона конденсацией фенилгидрази-
на с фенилглиоксалевой кислотой [3] и гидролизом метил-
амидфенилгидразонгидрата фенилглиоксалевой кислоты [4].
Фенилглиоксалевая кислота получается окислением ацетофе-
нона марганцовокислым калием в щелочной водной среде [5].
Для упрощения процесса мы не выделяли фенилглиоксале-
вую кислоту из раствора, а сразу же конденсировали ее с
фениЛгидразином.
СИНТЕЗ ФЕНИЛГИДРАЗОНА ФЕНИЛГЛИОКСАЛЕВОй
КИСЛОТЫ
/-W /—\\ .соон
\__/-NH-NH2‘HC1 + \_/-сч0
^-nh-n. z—
/С-С_/ + Н2О + НС1
СООН
Характеристика основного сырья
Ацетофенон, ч„ ТУ 28—55.
Натр едкий, х.ч., ГОСТ 4328—48.
Калий марганцовокислый, ч.д.а., ГОСТ 4527—48.
О
Фенилгидразин солянокислый, ч., ГОСТ 5834—52.
Кислота соляная, ч„ ГОСТ 3118—46.
Спирт этиловый, ГОСТ 5962—51.
Условия получения
В коническую колбу на 750 мл помещают 25,7 г (0,604 М)
едкого натра, 96 г (0,604 М) марганцовокислого калия и
600 мл дистиллированной воды. Полученную смесь подогре-
вают до 70°. Одновременно, в круглодонной на 2—3 л колбе,
энергично встряхивают смесь из 36 г (0,266 Л1) ацетофенона
и 600 мл воды. К полученной эмульсии приливают горячий
щелочной раствор марганцовокислого калия. Колбу слегка
встряхивают до обесцвечивания фиолетового раствора, после
чего смесь охлаждают на ледяной бане до 20° и фильтруют?
образовавшуюся перекись марганца. К' полученному раст-
вору солей фенилглиоксалевой и бензойной кислот приливают
100 мл концентрированной соляной кислоты. Смесь снова ох-
лаждают на ледяной бане до 0° н выпавшую бензойную
кислоту отфильтровывают. Затем к фильтрату, содержащему
раствор фенилглиоксалевой кислоты, добавляют раствор из
31 г (0,203 М) 95%-ного фенилгидразина солянокислого в
300 мл воды. Через час отфильтровывают выпавший н загу-
стевший желтый осадок, загружают его в колбу с 2 л воды,
тщательно размешивают и снова отфильтровывают. Промыв-
ку водой повторяют два раза. Желтый осадок в количестве
46 г сушат при 65°, затем растворяют в 400 мл этанола, филь-*
труют н добавлением 400 мл дистиллированной воды высажи-
вают из раствора фенилгидразон фенилглиоксалевой кислоты
в виде желтых блестящих игл с т. пл. 152—156° (интервал
плавления 1—2°); после трех перекристаллизаций из спирта
т. пл. 154,5—155°.
Выход продукта 36 г, что составляет 53%. считая на аце-
тофенон, и 74%, считая на фенилгидразин. По литературным
данным, препарат имеет т. пл. 153° [3, 4].
Л итература
1. Е. Wedekind, Liebigs Ann. Chem., 300, 258 —296 (1898).
2. H. Seiler, H. Schmid, Helv. Chim. Acta, 37, 1 (1954).
3^ A. Elbers, Liebigs Ann. Chem., 227, 341 (1885).
4. J. U. Net, Liebigs Ann. Chem., 280, 295 (1894).
5. A. Glaus, W. Neukrauz, J. PraCt. Chem., (2), 44, 80 (1891).
Поступила в январе 1963 г, ИРЕ А
6
ФЕНИЛГИДРАЗОН ПИРОВИНОГРАДНОЙ КИСЛОТЫ
В. М. ОСТРОВСКАЯ, А. А. ПРЯНИШНИКОВ
/СНз
I rNH-N=c<cooH
C^H10O2N8 М. в. 178, 19
уФенилгидразон пировиноградной кислоты применяется
длД синтеза органических соединений и, в том числе, для кра-
сителей группы формазанов [1].
По литературным данным, продукт получают из эквимо-
лекулярных количеств фенилгидразина и пировиноградной
кислоты в эфнрном, водном, уксуснокислом, слабосолянокис-
лом растворах [2, 3], а также щелочным гидролизом фенил-
гидразона нитрила пировиноградной кислоты [4].
Нами проверен способ получения препарата из фенилгид-
разнна солянокислого и пировиноградной кислоты в водном
растворе.
СИНТЕЗ ФЕНИЛГИДРАЗОНА ПИРОВИНОГРАДНОЙ КИСЛОТЫ
< >,-NH-NH2-HCl + CHsCf
7/ х соон
// /СНз
— < NH-N=C<
СООН
Характеристика основного сырья
Фенилгидразин солянокислый, ч., ГОСТ 5834—52.
Кислота пировиноградная, ч.д.а., ВТУ МХП 2838—51.
Спирт этиловый, ГОСТ 5962—51.
Условия получения
Растворяют 43,5 г (0,3 М) солянокислого фенилгидразина
в дистиллированной воде, отфильтровывают нерастворнвший-
ся осадок н, прн размешивании, добавляют к раствору 20,4 р
(0,3 М) пировиноградной кислоты. Образуется светло-желтый
хлопьевидный осадок, который через 1,5 часа отфильтровыва-
ют, высушивают при 60°, затем растворяют в 250 мл спирта и
высаживают водой (250 мл). Выпавший хлопьевидный осадок,
состоящий из блестящих кремовато-белых кристалликов, от-
фильтровывают и высушивают.
7
Выход фенилгидразона пировиноградной кислоты 35 г, что
составляет 66% от теоретического; т. пл. 177-178°. По лите-,
ратурным данным, т. пл. 175° [5], от 178° до 183° в зависимо-
сти от способа нагревания [6], 189° [4], 192° [2, 7], 191° [18],
194° [9].
Л итература
1. A. W. N i n е k a m, Chem. Rev., 55, N3, 355 (1955).
2. Е. Fischer, F. lourdan, Вег., 16, 2241 (1883); E. Fischer,
Ber., 17, 578 (1884). /
IStevens, Ward, J. Amer. Chem. Soc., 125, 1328; Beilst., |5,
11, 124 (1951).
4. J. T h e s i n g, D. W i t z e 1. Ber., 88, 117 (1955).
5. Fianke, Kohn, M„ 20, 885; Beilst., 15. H338 (1932).
6. R. BehrSnd, H. T r v 11 e r, Liebigs Ann. Chem., 283, 227 (1894),
7. Th. Curtins, Ber., 45, 1072 (1912).
8. Subramanian, Sent, Walker, J. Amer. Chem. Soc., 1929,
2490; Beilst., 15, II, 124 (1951).
9. Klein, Fuchs, Biochem. Z., 213, 46; Beilst., 15, II, 124 (1951).
Поступила в январе 1963 г. ИРЕА
4-НИТРОФЕНИЛ ГИДРАЗОН ФЕНИЛГЛИОКСАЛЕВОЙ
КИСЛОТЫ
В. М. ОСТРОВСКАЯ, А. А. ПРЯНИШНИКОВ
^-\\_с/СООН
\=/ 4N_NH_^ ^_no2
СцНиИзО^ ~ М. в. 285, 26
4-Нитрофенилгидразон фенилглиоксалевой кислоты при-
меняют для получения формазанов, содержащих нитрогруп-
пу*. В литературе [1] имеется пропись получения 4-нитро-
фенилгидразона конденсацией 4-нитрофенилгидразина с фе-
нилглиоксалевой кислотой в 5%-ной серной кислоте. Нами
проведена конденсация в реакционном растворе, получаемом
при синтезе фенилглиоксалевой кислоты, как и в случае фе-
нилгидразона фенилглиоксалевой кислоты**.
* См. в наст, сборнике «йодиитроформазан».
** См. в наст, сборнике «Фенилгидразон фенилглиоксалевой кислоты».
8
СИНТЕЗ 4-НИТРОФЕНИЛГИДРАЗОНА ФЕНИЛ ГЛИОКСАЛЕВОЙ
КИСЛОТЫ
,_ч. СООН
O2N-< >-NH-NH2-HCl + < y-Cr;
\ = / \ = /
СООН
OsN-(?__^-NH-N=C-^__^+ Н2О + НС1
Характеристика основного сырья
4-Нитрофенилгидразин, ч., ТУ МХП 387—41.
Кислота соляная, ч., ГОСТ 3118—46.
Спирт этиловый, ГОСТ 5962—51.
Ацетофенон, ч., ТУ 28—55.
Натр едкий, х.ч., ГОСТ 4328—48.
Калий, марганцовокислый, ч.д.а., ГОСТ 4527—48.
Условия получения
Раствор фенилглиоксалевой кислоты получают окислением
36 г (0,266 М) ацетофенона в щелочном растворе марганцо^
вокислого калия, как указано для фепилгидразона фенил-
глиоксалевой кислоты.
Раствор 4-нитрофенилгидразина готовят в конической кол-
бе на 1 л смешением 28 г (0,179 М) этого продукта и 60 мл
(0,73 Л4) концентрированной соляной кислоты в 500 мл ди-
стиллированной воды, нагретой до 60—70°. Оба раствора
сливают вместе. Через час отфильтровывают выпавший оса-
док, заливают его водой (1,5 л) при механическом размеши-
вании и через. 20 минут промывную воду отсасывают обрат-
ным нутчфильтром. Промывку повторяют 2 раза.
Осадок сушат при 70° под инфракрасной лампой. Полу-
ченный продукт в количестве 45 г перекристаллизовывают из
уксусной кислоты (400 мл) при 70° и промывают водой
(400 мл). К маточнику приливают 350 мл воды и высаживают
вторичный осадок, который промывают, сушат и перекристал-
лизовывают, как указано выше. Оба осадка объединяют и пе-
реосаждают из-кипящего этанола (300 мл) дистиллированной
водой (300 мл). Выпавший желтый игольчатый осадок 4-ни-
трофенилгидразона фенилглиоксалевой кислоты отфильтро-
вывают и сушат. Конечный выход продукта 35 г, что состав-
ляет 64,6%, считая на 4-нитрофенилгидразин, и 52,7%, считая
на ацетофенон; т. пл. 158—159°; после перекристаллизации из
9
спирта т. пл. 162,5—163°. По литературным данным, т. пл.
продукта 163—165° [1].
Найдено, И; N —14, 62; 14, 40.
C14H„NSO4. Вычислено, %: N—14, 70.
Литература
1. Н. D. D а к 1 п, Н. W. D a d 1 е у, J. Biol. Chem., 15, 139 (1913).
Поступила в январе 1963 г. ИРЕА
4-НИТРОФЕНИЛГИДРАЗОН БЕНЗАЛЬДЕГИДА
А. А. ПРЯНИШНИКОВ, В. М. ОСТРОВСКАЯ, О. Т. ЛУШИНА
/А
= I! 1
°-n-C_^-nh”n=c\h'!/
C13HiiN8O2 М. в. 241, 25
4-Нитрофенилгидразон бензальдегида применяют для син-
теза формазанов [1, 2, 3].
4-Нитрофенилгидразон бензальдегида получают конденса-
цией 4-нитрофенилгидразина с бензальдегидом [4]. Нами был
уточнен данный способ получения.
СИНТЕЗ 4-НИТРОФЕНИЛ ГИДРАЗОНА БЕНЗАЛЬДЕГИДА
О
Ч I II
O2N-^_^-NH-NH2 + -
н
-> o2n-^_^-nh-n=c-^_J> + н2о
н
Характеристика основного сырья
4-Нитрофенилгидразин, ч., ТУ МХП 387—41.
Бензальдегид, ч., ГОСТ 157—51.
Спирт этиловый, ГОСТ 5962—51.
Условия получения
В трехгорлую колбу емкостью 3 л, снабженную механиче-
ской мешалкой, обратным холодильником, термометром и ка-
пельной воронкой, помещают НО г (0,72 Л4) 4-нитрофенил->
гидразина и 1,26 л спирта. Полученную смесь доводят до ки-
пения и полного растворения 4-нитрофенилгидразина. Затем
подогрев прекращают и при размешивании по каплям добав-
ляют 84 г (0,77 Л4) бензальдегида в течение 15 минут. Смесь
нагревают до кипения в течение 30 минут, затем охлаждают
проточной водой, выпавший объемистый осадок отфильтровы-
вают, загружают в ту же реакционную колбу и снова кипя-
тят с 500 мл этилового спирта в течение 30 минут, после чего
охлаждают проточной водой, отфильтровывают, промывают
на фильтре 170 мл спирта и сушат при 70°.
Выход 4-нитрофенилгидразона бензальдегида 160 г, что
составляет 94%, считая на 4-нитрофенилгидразин. По внеш-
нему виду продукт представляет собой золотисто-оранжевые
иглы с т. пл. 189—190°. По литературным данным, т. пл. 192—
193° [1], 191 — 192° [5], 190° [6].
Примечание. При добавлении бензальдегида к спиртовому раст-
вору 4-нитрофенилгидразина водяную баню удаляют, так как эта реакция
экзотермическая.
Литература
1. Kwan-Chung Tsou, Cha o-S hing Cheng, M. M. N ac h-
l a s, A. M. Seligman, J. Amer. Chem. Soc., 78, 6139-61-44 (1959).
2. S. S. К a r m a r k a r, T. Barrnett, M. M. Nachlas, A. M. Se-
ligman, J. Amer. Chem. Soc., 81, 3771—3775 (1959).
3. S. W. Fox, E. H. Atkinson, J. Amer. Chem. Soc., 72,3629(1950).
4. H. Biltz, F. Si eden. Liebigs Ann. Chem., 324,3’0 321 (1962).
5. Bamberger, Pevnsel, Ber., 36; Beilst., 15, 470 (1932).
6. Ekens t ein, Blankswa, Rec., 22, 439; BellsL, 15, 470 (1932).
Поступила в январе 1963 г.
ИРЕА
ДИФОРМАЗАН
3,6',5,5/-Тетрафенил-1,1/-(4,4/-Дифенилен)-диформазан
В. М. ОСТРОВСКАЯ. А. А. ПРЯНИШНИКОВ
Диформазан применяют для синтеза неотетразолия
[1, 2, 3].
Диформазан может быть получен сочетанием бисдиазоти-
рованного бензидина с фенилгидразоном фенилглиоксалевой
кислоты в содовом водном растворе [2] или в среде метанола
с пиридином [3].
В основу нижеописанной уточненной прописи был положен
метод синтеза в содовом растворе.
СИНТЕЗ ДИФОРМАЗАНА
H2N-^-NHa + 2NaNO24-2HCl —
2С1 +
4- 2 NaCl + HgO
СООН
S\_r/
2 U1 ^N-NH-^\
I II
N = N = N
2CI ->
12
Характеристика основного сырья
Фенилгидразон фенилглиоксалевой кислоты*, ч,, т. пл.
152—156° (см. стр. 5).
Бензидин, ч., ТУ 1320.
Нитрит натрия, ч., ГОСТ 4197—98.
Спирт этиловый, ГОСТ 131—51.
Кислота соляная, ч., ГОСТ 3118—46.
Условия получения
Загружают в колбу 14,4 г (0,078 Л1) бензидина, 300 мл
воды, 36 мл соляной кислоты и подогревают на водяной бане
до растворения бензидина. Раствор фильтруют и охлаждают
до 0°; при этом выпадает дихлоргидрат бензидина. Прибавля-
ют к нему, в течение 30 минут, по каплям, раствор нитрита
натрия (11,4 г; 0,164 Л4) в 35 мл воды. Избыток азотистой
кислоты контролируют по йодкрахмальной бумаге.
Содовый раствор готовят из 36 г (0,15 М) фенилгидразо-
на фенилглиоксалевой кислоты, 12 г соды и 300 мл воды, ох-
лаждают его до 0° и прибавляют к нему тонкой струйкой, при
интенсивном помешивании, тетразораствор бензидина. На
следующий день выпавший мелкокристаллический черно-фио-
летовый осадок отфильтровывают, заливают двумя литрами
горячей воды, интенсивно размешивают и по отстаивании
промывную воду отсасывают обратным нутчфильтром. Очи-1
стку повторяют 4—5 раз до получения бесцветной промывной
воды. Полученный продукт в количестве 45 а сушат при 70°,
промывают при размешивании в 250 мл этанола и отфиль-
тровывают. Операцию повторяют 4 раза. Выход диформазана
35 г, что составляет 74% от теоретического.
По внешнему виду продукт представляет собой вещество
черно-фиолетового цвета с т. пл. в пределах 185—195°
(с разл.).
* Способ приготовления см. на стр. 5.
13
По литературным данным, т. пл. (с разл.) 219—220° [3],
185—190° [2].
Литература
1. Kwan-Chung Tsou, Chao-Shing Cheng, M. M. N а с h-
las, А. М. Seligman, J. Amer. Chem. Soc., 78, 6139 (1956).
2. E. Wedekind, Liebigs Ann. Chem., 300, 258—296 (1898).
3. H. Seiler, H. Schmid, Helv. Chim. Acta, 37, 1 (1954).
Поступила в январе 1963 г. ИРЕА
НЕОТЕТРАЗОЛИЙ ХЛОРИСТЫЙ
2,2/,5,5/-тетрафенил-3,3/-(4,4/-Дифенилен)-дитетразолий
хлористый
В. М. ОСТРОВСКАЯ, А. А. ПРЯНИШНИКОВ
2С1, 2 Н2О
CsgHasNgCl'a • 2Н2О
М, в. 703,65
Неотетразолий хлористый [1] является одним из основных
реактивов в ассортименте красителей для микроскопии [3] и
применяется главным образом в гистохимии [4].
Получают неотетразолий окислением дифорМазана азот-
ной кислотой в присутствии спиртовой хлористоводородной
кислоты [5] или тетраацетатом свинца [2].
Ниже описан разработанный в ИРЕА способ получения
неотетразолия хлористого окислением диформазана изоамил-
нитритом с соляной кислотой; изоамилнитрит применялся ра-
нее для окисления некоторых других формазанов [6].
14
СИНТЕЗ НЕОТЕТРАЗОЛ ИЯ ХЛОРИСТОГО
+ 20 4-2НС1
Характеристика основного сырья
Диформазан, ч„ т. пл. (с разл.) 185—195°.
Кислота соляная, ч., ГОСТ 3118—46.
Изоамилнитрит, ч., ТУ ОРУ 22—55.
Спирт этиловый, ГОСТ 5962—51.
Уголь активированный, марка А, ГОСТ 4453—48.
Условия получения
В трехгорлую колбу емкостью 750 мл, снабженную обрат-
ным холодильником, мешалкой и капельной воронкой, поме-
щают 450 мл спирта, 35 г диформазана, 27 мл изоамилнитри-
та и далее в течение 1 часа по каплям прибавляют 20 мл кон-
центрированной соляной кислоты. Смесь нагревают до 35° и
прикапывают еще 4,5 мл концентрированной соляной кисло-
ты, поддерживая нагревание и размешивание 20—30 минут,
пока окраска реакционной смеси не превратится из фиолето-
вой в коричневую. Затем к реакционной массе, перелитой в
трехлитровую колбу, добавляют 1,5 л воды, нагретой до 50—
60°, при этом выпадает хлопьевидный осадок. После прибавле-
ния к смеси 5 г активированного угля, осадок с углем от-
фильтровывают, а фильтрат упаривают в фарфоровой чашке
15
на водяной бане до объема 1500 мл (для удаления спирта). К
горячему раствору добавляют еще 5 г активированного угля
и оставляют на час при нагревании и помешивании. Отфиль-
тровывают уголь, раствор упаривают до 1/4 объема (~ 370
мл), пока не появятся на поверхности раствора пленки кри-
сталлов. Раствор сливают в стакан и охлаждают. Выпадают
шелковистые иголочки белого цвета со слегка желтоватым от-
тенком. После сушки при 70° выход неотетразолия хлористо-
го 10 г, что составляет 26% от теоретического; т. пл. 225—
230° (с разл.).
По литературным данным, т. пл. 219° (с разл.) [2], 258—
260° (с разл.) [5].
Л итература
1. Kwang — Chung Tso u, Chao-Shing Cheng, M. M.
Nachlas, A. Seligman, J. Amer. Chem, Soc., 78, 6139 (1956).
2. H. Seiler, H. Schmid, Helv. Chim. Acta, 37, 1 (1954).
3. H. J. Conn, Biological stains, Baltimore, U.S.A., 1961.
4. Э. Пирс, Гистохимия, M., ИЛ, 1962,
5. Е. Wedekind, Liebigs Ann. Chem., 300, 258 (1898).
6. A. W. N i n e h a m, Chem. Revs., 55, 457—489 (1955).
Поступила в январе 1963 г. ИРЕА
ЙОДН ИТРОФОРМАЗАН
1-(4-йодфенил)-5-(4-нитрофенил)-3-фенилформазан
В. М. ОСТРОВСКАЯ, А. А. ПРЯНИШНИКОВ
Ч_ \ =
Н С~< \
/ = ч \ //
CieHuN6OaJ М. в. 471, 27 .
йоднитроформазан, по литературным данным, может быть
синтезирован при взаимодействии диазотированного 4-йод-
анилина с 4-нитрофенилгидразоном бензальдегида [1].
Нами йоднитроформазан получен сочетанием 4-йоданили-
на с 4-нитрофенилгидразоном фенилглиоксалевой кислоты.
16
СИНТЕЗ ЙОДНИТРОФОРМАЗАНА
^-NH2 + NaNO3 + 2HC1
J-/_\-N=N
Cl + NaCl + 2H2O
/ = \ + ] - /=\ /СООН
^^-Njc1 + 4_)-c(N_NH4r>_
J4”)-N=\
-» H С-/ ^ + CO, + HC1
Характеристика основного сырья
4-Йоданилин, ч., ТУ РУ 935—53.
Кислота соляная, ч., ГОСТ 3118—46.
Нитрит натрия, х.ч., ГОСТ 4197—48.
4-Нитрофенилгидразон фенилглиоксалевой кислоты, ч,
т. пл. 158—159° (см. стр. 8).
Карбонат натрия, ч., ГОСТ 83—41.
Спирт этиловый, ГОСТ 5962—51.
Пиридин, ч„ ГОСТ 2747—44.
Едкое кали, ч.д.а., ГОСТ 4203—48.
Условия получения
Растворяют 22 г (0,10 М) 4-йоданилина в 450 мл воды и
30 мл концентрированной соляной кислоты при нагревании
до 50°. Раствор фильтруют от механических примесей, охлаж-,
дают до 3—5°, помещают в стакан, снабженный механической
мешалкой, капельной воронкой, термометром, н прибавляют
к сме)си по каплям, в течение 5—10 минут, раствор 7,5 г
(0,108 М) нитрита натрия в 20 мл воды. Конец диазотирова-
ния устанавливают по йодкрахмальной бумажке. После пе-
ремешивания реакционной массы в течение часа диазораст-
вор быстро фильтруют и оставляют на холоду. Затем раство-
ряют 24 г (0,084 М) 4-нитрофенилгидразона фенилглиоксале-
вой кислоты в смеси 20 г (0,188 /И) карбоната натрия и
700 мл воды при 40°. Раствор фильтруют, охлаждают до 10°
и медленно, в течение 30 минут, приливают к нему тонкой
струйкой, при размешивании, диазораствор 4-йоданилина.
Одновременно в реакционную смесь постепенно добавляют
20 г (0,375 М) твердого едкого кали, при этом начинает вы-
падать красно-коричневый осадок формазана.
17
Реакционную смесь выдерживают 3 часа, продолжая ме-
ханическое размешивание. На следующий день из реакцион-
ной смеси отсасывают обратным нутчфйльтром жидкую фа-
зу. Оставшийся на дне стакана осадок промывают водой. Для
этого осадок заливают 2 л нагретой до 70—75° Воды, разме-
шивают 20 минут и промывную воду отсасывают обратным
нутчфйльтром. Операцию повторяют несколько раз до полу-
чения бесцветной промывной воды. Продукт (33 г), высушен-
ный при 65—70° (не выше), загружают в круглодонную кол-
бу, снабженную обратным холодильником, механической ме-
шалкой, заливают 200 мл спирта и нагревают до кипения
спирта.
Через 30 минут осадок отфильтровывают’и эту операцию
проводят еще 2 раза. Осадок (22 г) растворяют в пиридине
(380 мл) при 50° и из профильтрованного раствора высажи-
вают водой (380 мл). Выпавший продукт отфильтровывают,
промывают на фильтре спиртом (200 мл) и снова промывают
кипящим спиртом (200 мл). Выход йоднитроформазана 17 г,
что составляет 43% от теоретического; т. пл. 184—187°
(с разл.). По внешнему виду это бордово-коричневый мелко-
кристаллический, порошок; по литературным данным, т. пл.
препарата 185—186° (с разл.).
Литература
1. S. W. F о х, Е. И. Atkinson, J. Amer. Chem. Soc., 72, 3629
(1950).
Поступила в январе 1963 г. ИРЕА
ЙОДНИТРОТЕТРАЗОЛ ИЙ
3-(4-Йодфенил)-2-(4-нитрофенил)-5-фенилтетразолмй
хлористый
В. М. ОСТРОВСКАЯ, А. А. ПРЯНИШНИКОВ
М. в. 505,72
йоднитротетразолий является одним из основных продук-
тов в ассортименте широко применяемых в гистохимии и мик-
робиологии тетразолиевых. солей [1, 2-, 3, 4; 5].
18
Получают йоднитротетразолий окислением йоднитрофор-
мазана бутилнитритом со спиртовой соляной кислотой. Выход
20% [2]. Ниже дана пропись получения йоднитротетразолия
окислением йоднитроформазана изоамилнитритом с выходом
продукта 75%.
СИНТЕЗ ЙОДНИТРОТЕТРАЗОЛИЯ
4-О + НС1
Характеристика основного сырья
йоднитроформазан *, ч., т. пл. 184—187° (с разл.).
Изоамилнитрит, ч„ ГОСТ 3414—54.
Спирт этиловый, ГОСТ 5962—51.
Аммоний хлористый, ч., ГОСТ 3773—47.
Кислота серная, х.ч., ГОСТ 4204—48.
Эфир серный, ч., ГОСТ 6265—52.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную обрат-
ным холодильником, механической мешалкой и капельной во-
ронкой, загружают 6 г (0,0128 М) формазана, 105 мл спирта,
9 мл (0,0678 М) изоамилнитрита. При механическом размеши-
вании подогревают смесь до температуры 36—39° и прибавляют
по каплям, в течение 40 минут, 10,5 мл (0,0831 М) 33%-ного
спиртового раствора хлористого водорода, разбавленного
15 мл спирта. После прибавления всего спиртового раствора
хлористого водорода реакционную смесь нагревают до темпе-
ратуры 55—60° в течение 75 минут. Затем наносят каплю по-
лученной смеси иа белую фильтровальную бумагу. Если пят-
но желтое и не видно осадка формазана, нагревание прекра-
щают. Если же формазан не полностью прореагировал, под-
нимают температуру до 78° и продолжают нагревание еще в
течение 10—15 минут. После этого реакционную смесь охла-
* См. наст, сборник, стр. 18.
19
ждают, помещают в коническую колбу на 2,5 л и добавляют
2 литра эфира; при этом выпадает белый осадок, превращаю-
щийся на дне колбы в прозрачную пленку. На следующий
день раствор над пленкой сливают, пленку растворяют в
60 мл метилового спирта и высаживают продукт 1 л эфира.
Выпавший белый осадок с желтоватым оттенком отфильтро-
вывают на пористом фильтре № 3 или № 4. Полученный про-
дукт в количестве 5,2 г растворяют в нагретой до 80° дистил-
лированной воде, отфильтровывают от светло-коричневых, не-
растворимых в воде примесей. Водный фильтрат упаривают в
чашке на водяной бане досуха, после чего продукт растворяют
в 60 мл метанола, нагретого до 50—60°, фильтруют и снова
высаживают его 1 л эфира. Выпавший белый кристаллический
осадок, представляющий собой йоднитротетразолий, отфиль-
тровывают и высушивают при 50—60°.
Выход препарата 4,8 г, что составляет 75,8% от теорети-
ческого; т. пл. 226—227° (с разл.).
По литературным данным, т. пл. продукта 229° (с разл.)
[2].
Л итература
1. К w а n-С h u ti g Т s о u, С h а о Shing Cheng, М. М. N а с hi-
la s, А. М. S е 1 i g m a n, J. Amer. Chem. Soc., 78. № 23, 6139 (1956).
2. S. W. Fox, E. H. Atkinson, J. Amer. Chem. Soc., 72, 3629
(1950).
3. Каталог английской фирмы .Light”, 1960.
4. H. J. Conn, Biological stains, Baltimore, U.S.A., 1961.
5. Э. Пирс, Гистохимия, M., ИЛ, 1962.
Поступила в январе 1963 г.
ИРЕА
ДИНИТРОФОРМАЗАН
5,5/-Ди-(4-нитрофенил)-3,3/-дифенил-1,Г-(3,3/-диметокси-
4,4'-дифенилен)-диформазан
В. М. ОСТРОВСКАЯ, А. А. ПРЯНИШНИКОВ, О. Т. ЛУШИНА
ОСН3 Н3СО
NO2 N°2
C40H32N10Oe М. в. 748,77
Динитроформазан применяется для синтеза нитротетразо-
лиевого синего.
В литературе имеется описание синтеза динитроформаза-
на сочетанием бисдиазотированного о-дианизидина с 4-нитро-
фенилгидразоном бензальдегида [1, 2]. Нами уточнен этот
синтез.
СИНТЕЗ ДИНИТРОФОРМАЗАНА
осн3 Н3СО
I. H2N——NH2 + 2NaNO,-Ь 4НС1
осн8 Н3СО
+ = = +
2С1 + 2NaCl + 4Н2О
ОСН3 Н3СО
!= =J
2С1 4-
4-2O2N—=
21
осн3 Н8СО
U д ~
N=N . -ОС.) - О
ЧО“С\\ /н н\ ^с~
N-N N-N
N02 Д и н и т р о ф о р м а з а н N02
ОСН8 Н8СО
/~\ /~N=N
Н\ Х^//
N-N
к/
N02
Моиоформазан (побочный продукт)
Характеристика основного сырья
о-Дианизидин, ч., ГОСТ 4150—54. .
Кислота соляная, ч., ГОСТ 3118—46.
Нитрит натрия, ч., ГОСТ 4197—48.
4-Нитрофенилгидразон бензальдегида, т. пл. 189—190° (см.
стр. 10).
Тетрагидрофуран, т. кип. 65—66°.
Едкое кали, ч., ГОСТ 4203—48.
Диоксан, ч., ГОСТ 3111—52.
Ацетон, техн., ГОСТ 2603—51.
Углекислота твердая (сухой лед).
Спирт метиловый, ч., ГОСТ 6995—54.
Бензол, ч.д.а,, ГОСТ 5955—51.
Условия получения
В трёхгорлую колбу емкостью 500 мл, снабженную термо-
метром, мешалкой, капельной воронкой, помещают 40 г
(0,16 Л4) о-дианизидина, 220 мл дистиллированной воды, на-
гретой до 60°, 86 мл (1,03 М) концентрированной соляной
22
кислоты. Размешивают до получения хорошей суспензии и
охлаждают льдом с солью до —7°. К смеси по каплям, в те-
чение 30 минут, прибавляют раствор 24 а (0,34 Л4) азотисто-
кислого натрия в 60 мл дистиллированной воды и дают 30-
минутную выдержку при 0—5°. Полученный диазораствор
темно-вишневого цвета быстро фильтруют и оставляют в хо-
лодильнике. '
В стакан емкостью 2—3 л, снабженный техническим тер-
мометром, рамной мешалкой и помещенный в баню, загру-
жают 77 г (0,32 М) 4-нитрофенилгидразона, 700 мл тетра-
гидрофурана и размешивают до получения прозрачного жел-
то-оранжевого раствора. К этому раствору, охлажденному до
—25°, прибавляют приготовленный ранее диазораствор (см.
примечание). Полученную смесь охлаждают до —27° и добав-
ляют 40 г (0,57 Л1) едкого кали в 30 мл охлажденной воды;
при этом немедленно образуется черный осадок и температу-
ра реакционной смеси поднимается до —20° Размешивание
реакционной массы продолжают 1 час при минус 25—20° и
30 минут при комнатной температуре. Затем к полученной
смеси добавляют 320 мл диоксана, 700 мл метанола и остав-
ляют до следующего дня.
Выпавший черный осадок отфильтровывают, промывают
на фильтре 300 мл метанола, загружают в стакан, размеши-
вают 1 час с 700 мл метанола и отфильтровывают. Затем оса-
док помещают в 2400 мл кипящей дистиллированной воды,
размешивают 1 час, снова отфильтровывают, промывают на
фильтре метанолом (500 мл), нагретым до 50°, и сушат при
70°. Выход технического продукта равен 80 г.
Половину вещества (40 г) тонко растирают в ступке, по-
мещают в бумажный патрон, который вставляют в экстрак-
тор Сокслета. Примеси экстрагируют из продукта бензолом в
течение 16 дней. Периодически, через каждые 3 дня, экстрак-
цию приостанавливают; патрон вынимают, высушивают, из-
влекают из него вещество и растирают в ступке.
Бензол заменяют свежим и возобновляют экстракцию. Эту
операцию проделывают 4 раза до тех пор, пока экстракцион-
ная жидкость из темно-красной станет светло-розовой. Остав-
шееся в патроне вещество высушивают при 70°.
Выход динитроформазана (из двух аппаратов Сокслета)
60 г, что составляет 50% от теоретического; т. пл. 245—253°
(с разл.), интервал плавления Т.
По литературным данным, т. пл. динитроформазана 252—
253° (с разл.) [1].
Примечания: 1. Охлаждающую смесь готовят постепенным до-
бавлением сухого льда в ацетон.
2. По данным М. Н. Малева, моноформазап экстрагируют в колбе с
обратным холодильником сначала трижды по 12 часов кипящим бензолом
(по 1 л) и затем трижды по 12 часов днметнлформамидом (по 750 мл).
При такой очистке получают диформазан с т. пл. 295—296°.
23
Л итература
1. К wa n-C hung Т sо u, С h a o-S h i ng Cheng, M. M. N ac h 1 a s,
A. M. Seligman, J. Amer. Chem. Soc„ 78, G139 (1956).
2. S. S. К a r m a г к a r, J. В a r r n e 11, M. M. N a c h 1 a s, A. M. Se-
ligman, J. Amer. Chem. Soc., 81, 3771(1959).
Поступила в январе 1963 г,
ИРЕА
- НИТРОТЕТРАЗОЛИЕВЫЙ СИНИЙ (НИТРО-ТС)
2,2/-Ди-(4-нитрофенил)-5,5'-дифенил-3,3,-(3,3/-диметокси-
4,4/-Дифенилен)-дитетразолий хлористый
В. М. ОСТРОВСКАЯ, А. А. ПРЯНИШНИКОВ. О. Т. ЛУШИНА
ОСН3 Н3СО
l= J
// ^N—N N—
I I
/Ч //\
I! | | '!
W Ч/
NO, NO2
C4oHSoN]0OeCl2
2С1
М. в. 817, 67
Нитротетразолиевый синий применяется для определения
в растительных и животных клетках ряда окислительно-вос-
становительных ферментов, а также при исследовании онко-
логических и некоторых других заболеваний [1, 2, 3, 4].
Получают нитро-ТС окислением динитроформ азана изо-
амилнитритом с хлористым водородом [1, 5]. Этот способ на-
ми проверен и уточнен.
24
СИНТЕЗ НИТРО-ТС
Характеристика основного сырья
Динитроформазаи (см. стр. 21).
Изоамилнитрит, ч., ТУ 22—55.
Аммоний хлористый, ч., ГОСТ 3773—47.
Кислота серная, ч., ГОСТ 4204—48.
Тетрагидрофуран, ч., т. кип. 65—66°.
Диоксан, ч., ВТУ МХП 3111—52.
Спирт этиловый, ГОСТ 5962—51.
Спирт метиловый, ч., ГОСТ 6995—54.
Эфир серный, ч., ГОСТ 6265—52.
Условия получения
В стакан на 2 л, снабженный рамной мешалкой, барботе-
ром, термометром, помещают 20 г (0,0267 А1) динитродифор-
мазана, 320 Мл (284 г) тетрагидрофурана и 320 мл (330 г)
Диоксана; по каплям добавляют 24 мл (0,181 7И) изоамил-
нитрита, охлаждают до 0° и пропускают через смесь сухой
хлористый водород, получаемый из 50 г (0,92 М) хлористого
аммония и 10 мл (0,188 М) серной кислоты. Подачу хлори-
25
стого водорода ведут в течение 4 часов с большой осторож-
ностью, регулируя скорость поступления хлористого водорода
в реакционную массу так, чтобы через контрольную склянку
с серной кислотой проходило не более 80—100 пузырьков в
минуту. Такой скорости добиваются, подавая на хлористый
аммоний 10 капель серной кислоты через каждые 15 минут.
Кислотность среды проверяют по универсальной индикатор-
ной бумаге (pH 1—4). Через 1 час после начала пропускания
хлористого водорода добавляют еще 8 мл (0,060 М) изоамил-
нитрита порциями по 2 мл через каждые 20 минут, после че-
го смесь оставляют до следующего дня. Затем реакционную
массу размешивают в течение 2 часов при комнатной темпе-
ратуре, охлаждают до 0°, отфильтровывают зеленовато-жел-
тый осадок и промывают его эфиром (200 мл). Далее полу-
ченный продукт (17 г) растворяют в 300 мл кипящего этило-
вого спирта, фильтруют, охлаждают до 0° и вливают вместе
с частично выкристаллизовавшимся осадком в 1800 мл серно-
го эфира, при этом выпадает светло-желтый нитро-ТС, кото-
рый и отфильтровывают.
Выход продукта равен 15,0 г, что составляет 64% от тео-
ретического; т. пл. 160—162°; при перекристаллизации из ме-
танола т. пл. иитро-ТС 180—182°, после многократного пере-
осаждения из спирта эфиром т. пл. 156°.
По литературным данным, нитро-ТС, переосажденный из
спиртово-эфирной смеси, имеет т. пл. 156° [1], перекристалли-
зованный из метанола — т. пл. 184° [5}.
Литература
1. К w а n-С hung Tsou, Cha o-S h i n g Cheng, M. M. Nach-
las, A. M. Sell gm an, J. Amer. Chem. Soc., 78, 6139 (1956).
2. Э. Пирс, Гистохимия, M., ИЛ, 1962.
3. Н. J. Conn, Biological stains, Baltimore, U. S. A., 1961.
4. H. T. Райхлии, Вопросы онкологии, 7, № 3, 41—47 (1961); там
же, 7, 28—34 (1961); там же, 9, 37—44 (1962).
5. S. S. К а г m а г k а г, J. Barrnett, М. М. N а с h 1 a s, А. М. S е-
ligman, J. Amer. Chem. Soc., 81, 3771 (1959).
Поступила в январе 1963 г.
ИРЕА
ТЕТРА-НИТРОФЕН ИЛ-ТЕТРАЗОЛ ИЙ
2,2/,5,5'-Тетра-(4-нитрофенил)-3,3'-(3,3'-диметокси-4,4г-
дифенилен)-дитетразолий хлористый
М. И. МАЛЕВ '
2С1
C40H28C12Nj2O10 М в. 907,65
Тетра-нитрофенил-тетразолий применяется в гистохимии в
качестве реактива для выявления активности ферментов окис-
лительного обмена [1].
По литературным данным, тетра-нитрофенил-тетразолий
получают окислением соответствующего диформазана изо-
амилнитритом в кислой среде [2].
Тетра-нитрофенил-формазан, в свою очередь, получают
при взаимодействии бисдиазотированного о-дианизидина с
«-нитрофенилгидразоном л-нитробензальдегида в щелочной
среде [2].
Нами был проверен и уточнен указанный метод получения
тетразолия.
СИНТЕЗ ТЕТРА-НИТРОФЕНИЛ-ТЕТРАЗОЛИЯ
н
I. O2N-^“^-NH-NHa + O2N-^—^-С=О -►
н
O2N-("^-C==N-NH-^~^-NO2+H2O
27
ОСНз осн,
п.
+ 2 NaNO2 -*
ОСН8 Н3СО
N = N-^ /“\_/“NN
2Cl 4-
+ 2 NaCl + 4 Н2О
ОСН, ОСНз
|_ _|
4- № N-^_^-^_^-N S n] 2 Cl
ОСНз ОСНз
_ N=N-^
4- O2N-^ ^-Н= =
= N-N
I
NO,
Моноформазан (побочный продукт)
28
IV.
NO,
OCH3 OCH3
N=N-^ ^-N=N
< >" “< )
NO,
N-N
N-N
изоамилнитрит
HC1
Характеристика основного сырья
n-Нитрофенилгидразин, ч., ТУ МХП 381—41, т. пл. 157°.
гг-Нитробензальдегид, ч., ГОСТ 2969—51, т. пл. 105—107°.
о-Дианизидин солянокислый, ч., ВТУ МХП 4163—54.
Изоамилнитрит, ч., ВТУ МХП 3414—54.
Соляная кислота, ч.д.а., ГОСТ ЗГ18—46.
Нитрит натрия, ч., ГОСТ 4197—48.
Хлористый водород (см. синтез л-хлорфенил-диамида фос-
форной кислоты).
Условия получения
Получение п-нитрофенилгидразона п-нитробензальдегида
[3]. В трехгорлой колбе емкостью 500 мл, снабженной мешал-)
29
кой с масляным затвором, капельной воронкой и обратным
холодильником, растворяют 15,3 г (0,1 М) л-нитрофенилгид-
разина в 200 мл этилового спирта при нагревании на водяной
бане. К полученному раствору добавляют в течение 20 минут
раствор 15,1 г (0,1 М) n-нитробензальдегида в 150 мл этило-
вого спирта. Затем реакционную массу нагревают на кипящей
водяной бане 30 минут и, после охлаждения до комнатной
температуры, отфильтровывают выпавший кристаллический
осадок rz-нитрофенилгидразона rz-нитробензальдегида. Выход
25,8 г (90%) продукта в виде красных игл с т. пл. 248—249°,
что соответствует литературным данным [3].
Получение 3,Зг,5,5г-тетра-(4-нитрофенил)-1,Г-(3,3’-диме-
токси-4,4'-дифенилен)-дифор мазана. В фарфоровый стакан
емкостью 200 мл помещают 9,5 г (0,03 М) солянокислого
о-дианизидина (см. примечание 1), 25 мл 5н. (0,125 М) соля-
ной кислоты и около 30 г льда. В полученную смесь при тем-
пературе 0° добавляют по каплям, при размешивании, раст-
вор 4,2 г (0,06 М) нитрита натрия в 20 мл воды. После добав-
ления всего нитрита натрия реакционную смесь выдерживают
30 минут при температуре не выше 5°.
Одновременно с бисдиазотированием о-дианизидина в
фарфоровом стакане емкостью 1 литр, снабженном капельной
воронкой, термометром и мешалкой, растворяют 22,9 г
(0,06 М) л-нитрофенилгидразона rz-нитробензальдегида в сме-
си 200 мл пиридина и 3,9 г (0,07 М) едкого кали в 10 мл во-
ды. Полученный раствор охлаждают в бане со смесью сухого
льда и ацетона до минус 10° и при энергичном размешивании
добавляют по каплям, в течение 30 минут, раствор соли бис-
диазония. Температура реакционной массы не должна превы-
шать минус 5°. Затем реакционную смесь размешивают еще
1 час при температуре минус 5° и оставляют на ночь при тем-
пературе 5°. Выпавший черный осадок, являющийся смесью
моно- и диформазанов, отфильтровывают на воронке Бюхне-
ра, промывают на фильтре последовательно 500 мл холодной
воды1, 1 литром горячей воды и 500 мл ацетона.
Для очистки диформазана от примеси моноформазана по-
лученный черный порошок помещают в круглодонную колбу
емкостью 500 мл, снабженную обратным холодильником, мас-
ляной баней, и экстрагируют моноформазан в течение 1 часа
250 мл кипящего пиридина (см. примечание 2). Эту операцию
повторяют 3 раза. Очищенный от примесей диформазан отса-'
сывают, промывают на фильтре 500 мл ацетона и сушат на
воздухе. Получают .15,6 г (62%) 3,3',5,5'-тетра-(4-нитрофе-
нил)-1,Г-(3,3'-диметокси-4,4'-дифенилен)-диформазана с т. пл.
302—303° (см. примечание 3), слегка растворимого в горячем
пиридине и горячем диметилформамиде, нерастворимого в
других органических растворителях.
30
Получение 2,2',5,5'-четра-(4-ни,трофенил )-3,3'-(3,3'-диме-
токси-4,4'-дифенилен)-дитетразолия хлористого. В фарфоро-
вом стакане емкостью 300 мл суспендируют 8,4 г (0,01 М)
тетранитрофепил-диформазана в 120 мл диоксана и 10 м.г
(0,07 А4) нзоамилнитрита и пропускают сухой хлористый во-
дород (см. примечания к синтезу нитро-неотетразолия) в тече-
ние 30 минут. Затем помещают реакционную смесь в баню
с теплой водой (около 40°), прикапывают еще 5 мл изоамил-
нитрита в течение 1 часа и выдерживают при той же темпера-
туре около 6 часов; ввиду испарения жидкости в реакционной
смеси добавляют диоксан-до постоянного объема. Выпавший
желтый осадок тетразолия отфильтровывают, промывают на
фильтре 10 мл диоксана. Продукт очищают растворением в
50 мл горячего метанола, раствор фильтруют и добавляют
250 мл диоксана. На другой день отфильтровывают выделив-
шиеся светло-желтые кристаллы, промывают на фильтре 50 мл
эфира и сушат на воздухе.
Выход тетра-нитрофенил-тетразолия 7,6 г, что составляет
83% от теоретического; т. пл. 158—159°, что соответствует ли-
тературным данным [2].
Примечания: 1. При работе с о-дианизидином нужно пользовать-
ся резиновыми перчатками и не допускать распыления продукта.
2. В отсутствие примеси моноформазана при встряхивании пробы с
небольшим количеством ацетона последний не должен окрашиваться в ро-
зовый цвет.
3. По литературным данным [2], т. пл. диформазана 300“, выход 12,2%.
Литература
1. С. G. Rosa, Т s о и К w a n-C hung. Nature, 192, 990 (1961).
2. Tsou Kwa n-C hung, Cha o-S h 1 n g Cheng, M. M. N a c h 1 a s,
A. M. Seligman, J. Amer. Chem. Soc., 78, 6139 (1956).
3. E. Hyde, Ber„ 32, 1813 (1899).
Поступала в апреле 1963 г. Институт мозга АМН СССР
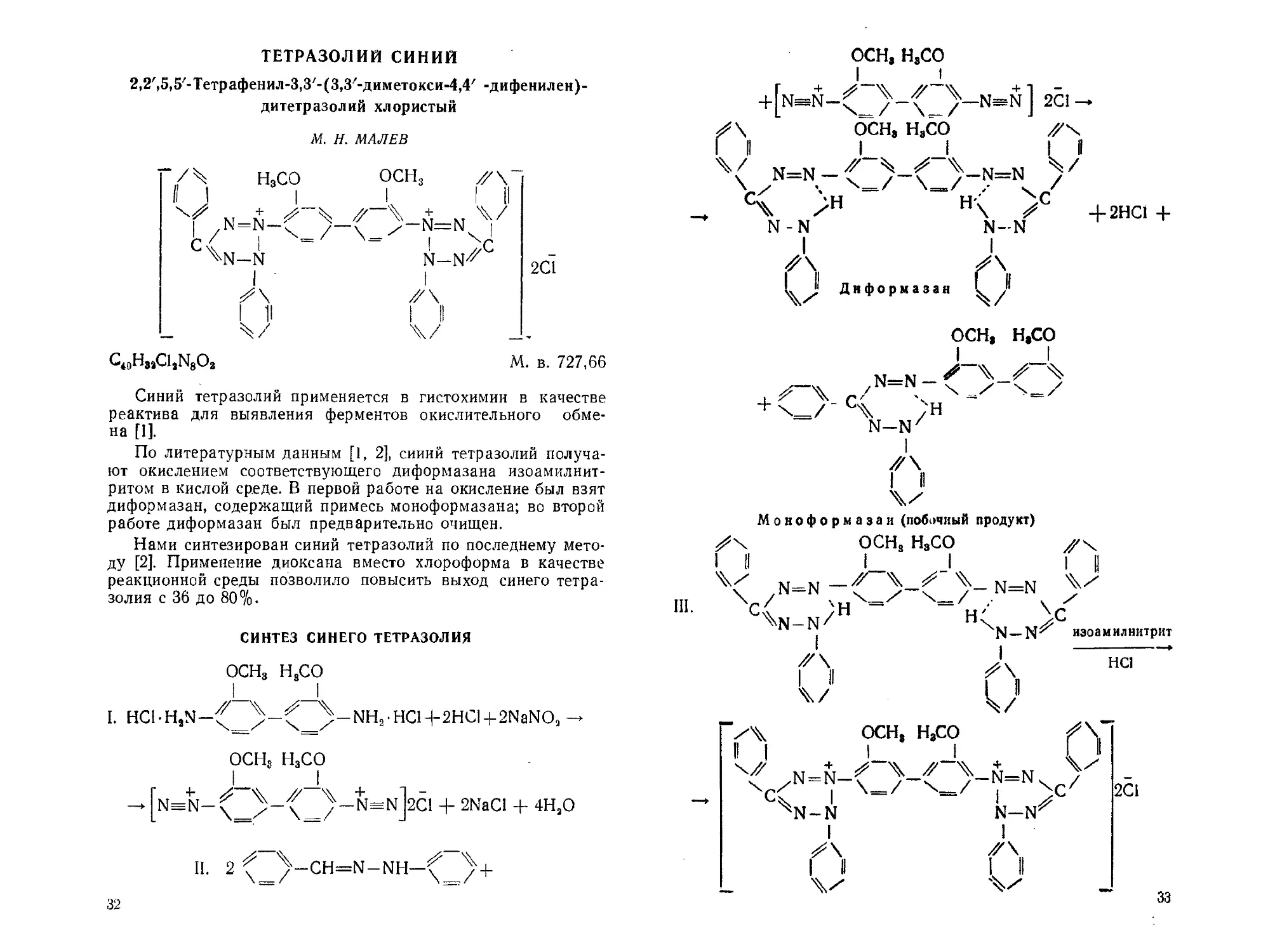
ТЕТРАЗОЛИЙ СИНИЙ
г.г'ДЗ'-Тетрафенил-ЗЛ'-СЗ.З'-диметокоМЛ' -дифенилен)-
дитетразолий хлористый
М. И. МАЛЕВ
ОСН3 //\-
I I II
Z_^-N=N V
N-N-^C
2С1
C^oHajCljNgOa
М. в. 727,66
Синий тетразолий применяется в гистохимии в качестве
реактива для выявления ферментов окислительного обме-
на [1].
По литературным данным [1, 2], синий тетразолий получа-
ют окислением соответствующего диформазана изоамилнит-
ритом в кислой среде. В первой работе на окисление был взят
диформ азан, содержащий примесь моноформазана; во второй
работе диформазан был предварительно очищен.
Нами синтезирован синий тетразолий по последнему мето-
ду [2]. Применение диоксана вместо хлороформа в качестве
реакционной среды позволило повысить выход синего тетра-
золия с 36 до 80%.
СИНТЕЗ СИНЕГО ТЕТРАЗОЛИЯ
ОСНз Н3СО
I. HCl-H,N-^_^-^ ^-NH2.HC14-2HCI-|-2NaNO<1
ОСН3 Н3СО
У~\ /-Nsn]2C1 + 2NaCl + 4НаО
II. 2 \_/-CH=N-NH—
32
осн, н,со
+|n==N-^ У~\ У~N=n] 2С1 —
Диформазан
осн, н,со
zN=N —'
- Ч ;н
N—Nz
Mo но фо рм а за и (побочный продукт)
ОСН, НаСО
N=N
III.
4N-n/H
- N=N
c
иэоамилнитрит
НС1
осн, н3со
,N=N-
cC 1
^N-N
—N=N
N—N'
2С1
XN-IxT
Z\
33
Характеристика основного сырья
Бензальфенилгидразон, т. пл. 154—155° (см. синтез метил-
тиазолилтетразолия).
о-Дианизидин солянокислый, ч., ВТУ МХП 4163—54.
Нитрит натрия, ч., ГОСТ 4197—48.
Соляная кислота, ч.д.а., ГОСТ 3118—46.
Изоамилнитрит, ч., ВТУ МХП 3414—54.
Диоксан, ч„ ВТУ МХП 3111—52.
. Хлористый'водород (см. синтез и-хлорфенил-диамида .фос-
форной кислоты).'
Условия получения
Получение 3,3',5,5'-/етрафёниЛ-1(3,3'-диметокси-4,4'~
дифенилен)-диформазана. В фарфоровом стакане емкостью
1 лигр, снабженном капельной воронкой, мешалкой и термо-
метром, растворяют 39,6 г (0,2 М) бензальфенилгидразона в
150 мл пиридина.н приливают раствор 20 г кристаллического
ацетата натрия в 300 мл этилового спирта.
Одновременно готовят соль бисдиазония, добавляя по кап-
лям, при размешивании, раствор. 14,5 г (0,21 М) нитрита нат-
рия в 50 мл воды к суспензии 31,7 г (0,1 AJ) солянокислого
о-дианизидина в 100 мл 4 н. (0,4 М) соляной кислоты и 100 г
размельченного льда.
Бнсдиазотированный раствор о-дианизидина добавляют
по каплям, в течение 1 часа, п()и интенсивном размешивании,
к охлажденному до-минус 10° раствору бензальфенилгидра-
зона, поддерживая температуру реакционной смеси в интер-
вале минус 10—5°. Размешивание продолжают еще 2 часа,
после чего выпавший черный осадок отфильтровывают на во-
ронке Бюхнера и промывают на фильтре двумя литрами го-
рячей воды. .
Получают около 50 г порошка, являющегося смесью моно-
p. диформазанов. Порошок подвергают экстрагированию в ап-
парате; Сокслета 500 мл ацетона в течение 12 часов для уда-
ления моноформазана; Эту операцию повторяют еще 3 раза
(см. примечание Г). Выход диформазана 20 г (30%) с т. пл.
232—234°; препарат вполне пригоден для дальнейшего син-
теза (см. примечание 2).
Получение 2,2',5,5'-тетрафенил-3,3'- (3,3'-диметокси-4,4'-ди-
фенилен)-дитетразолия хлористого'. В фарфоровом стакане
емкостью 300 мЛ, снабженном капельной воронкой и мешал-
кой, суспендируют 6,6 г (0.01 AI) тонко растертого диформа-
зана в 150 мл диЪксйна, добавляют 7,0 мл (0,05 М) изоамил-
нитрита и в течение 30 минут пропускают сухой хлористый
водород. Газ осушают, пропуская через.склянку Тищенко, на-
полненную на одну треть серной кислотой, со скоростью 40
'34
пузырьков в минуту. Затем Добавляют по каплям еще 3 мл
изоамилнитрита и размешивают реакционную массу еще око-
ло четырех часов. Желтый осадок отсасывают и промывают
на фильтре 20 лм диоксана. Препарат очищают от примеси
диформазана растворением в 40 мл горячего метанола, филь-
трацией раствора и осаждением’ 200 мл диоксана. Выпавшие
светло-желтые кристаллы отфильтровывают на воронке Бюх-
нера. промывают на фильтре 100 мл эфира и сушат на возду-
хе. Выход синего тетразолия 5,7 г, что составляет 80% от тео-
ретического; т. пл. 243—244°, что соответствует литературным
данным. Продукт хранят в герметично закрытой банке из
темного стекла. .
Примечания: Г. Экстракцию моноформазана прекращают тогда,
когда ацетон перестает окрашиваться в розовый цвет.
2. По литературным данным [2], аналитический образец диформазана
получают перекристаллизацией из кипящего пиридина. При охлаждении
юрячсго раствора получают темно-синие игольчатые кристаллы с т. пл.
243—244°.
Л итература
1. A. Rutenburg, А. М. Seligman, Cancer Res., 10. 113 (1950).
2. W. Ried, H. Gick, Liebigs Ann. Chem., 581, 16 (1953)
Поступила в ап, еле 1963 г.. Институт мозга АМН СССР
НИТРОНЕОТЕТРАЗОЛИЙ
2,2/-Ди-(4-нитрофенил)-3,3/-(4,4/-дифенилен)-5,5/-Дифеиил-
дитетразолий хлористый
М. Н. МАЛЕВ
C3gH86Cl2Ni0O4
М. в. 757,60
Нитронеотетразолий может найти применение в гистохи-
мии в качестве реактива при выявлении активности фермен-
тов окислительного обмена.
По литературным данным, нитронеотетразолий получают
окислением диформазана, который, в свою очередь, получают
конденсацией 4-иитрофенилгидразона бензальдегида с бис-
диазотированным бензидином в щелочной среде [1].
Нами проверена и уточнена эта методика.
СИНТЕЗ НИТРОНЕОТЕТРАЗОЛИЯ
I. HCl-HjN—^-NHe-HCl+2HC14-2NaNO1->
№N-^2/-!\ /-N^N
2 Cl + 2NaCl + 4Н2О
H
II. 2 -C=N-NH-^~~^-NO2 +
36
NO, Диформазан NO,
Моноформазан
(побочный продукт)
/ч
N=N--
ш. )н
N-N
_ N= N Ч/
изоамилиитрит
НС(
N—N
Характеристика основного сырья
4-Нитрофенилгидразон бензальдегида (см. стр. 10).
Бензидин солянокислый, ч.д.а., СТ ГОХП 27—1545.
Тетрагидрофуран, ч., т. кип. 65—66°.
Изоамилнитрит, ч., ВТУ МХП 3414—54.
Диоксан, ч., ВТУ МХП 3111—52.
Хлористый водород (см. синтез N'-л-хлОрфенил-диамида
фосфорной кислоты)
Получение 5,5>-ди-(4-нитрофенил)-1,Г-(4,4'-дифенилен)-
3,3'-дифенил-диформазана. В фарфоровом стакане емкостью
300 мл, снабженном мешалкой, суспендируют 25,7 а (0,1 М)
солянокислого бензидина в смеси 80 мл 5 и. (0,4 М) соляной
кислоты и 100 г мелкораздробленного льда. К полученной су-
спензии добавляют по каплям 14,5 г (0,21 М) нитрита натрия,
растворенного в 40 мл воды. Этот раствор добавляют по кап-
лям при интенсивном размешивании в охлажденный до тем-
пературы —10° раствор 48,2 г (0,2 М) п-нитрофенилгидразона
бензальдегида в смеси 400 мл тетрагидрофурана, 22,0 г
(0,55 М) едкого натра и 80 мл воды. Реакцию проводят при
температуре минус 5—10° в фарфоровом стакане емкостью
1 литр, снабженном мешалкой и помещенном в баню с охла-
дительной смесью. Бисдиазотированный раствор бензидина
приливают по каплям в течение 30 минут, при этом наблю-
дается выпадение черного осадка диформазана. Реакционную
смесь размешивают еще 30 минут, полученный осадок отса-
сывают и промывают на фильтре последовательно 1 литром
холодной воды, 2 литрами горячей воды и 500 мл изо-пропи-
лового спирта. Получают 40 г продукта, являющегося смесью
моно- и диформазанов.
Моноформазан отделяют от диформазана экстрагировани-
ем его 500 мл тетрагидрофурана в аппарате Сокслета в тече
ние 12 часов; эту операцию повторяют трижды. Оставшийся
черный осадок отфильтровывают на воронке Бюхнера, про-
мывают на фильтре 100 мл этилового спирта и сушат на воз-
духе. Получают 7,4 г (11%) диформазана с т. пл. 234°, что
соответствует литературным данным [2].
Получение нитронеотетразолия. В фарфоровом стакане
емкостью 300 мл, снабженном мешалкой, суспендируют 6,9 г
(0,01 М) 5,5'-ди-(4-нитрофенил)-1,1'-(4,4'-дифенилен)-3,3'-ди-
фенил-диформазана в 150 мл диоксана, добавляют 7 mi
(0,05 М) изоамилнйтрита и пропускают в. течение 30 минут
сухой хлористый водород (см. примечание 1). Затем при раз-
мешивании добавляют по каплям еще 4 мл изоамилнитрита
в течение 1 часа (см примечание 2). Выпавший зеленый оса-
док соли тетразолия отфильтровывают на воронке Бюхнера,
промывают на фильтре 20 мл диоксана. Препарат очищают
от диформазана, не вошедшего в реакцию, растворением в
38;
30 мл горячего метанола, фильтрацией полученного раство-
ра с последующим осаждением продукта 150 мл диоксана.
Выпавший желтый кристаллический осадок через два часа
отфильтровывают, промывают на фильтре 50 мл эфира и су-
шат на воздухе.
Выход нитронеотетразолия 5,2 г (65% от теоретическо-
го); т. пл. 221—222э,что соответствует литературным данным.
Примечания: 1. Скорость пропускания хлористого водорода в ре-
акционную массу около 40 пузырьков в минуту. Пробулькивание газа ре-
гистрируют в склянке Тищенко, наполненной на одну треть серной кисло-
той. Все работы с хлористым водородом, диоксаном и изоамилнитритом
проводят в вытяжном шкафу при включенной тяге.
2. За ходом реакции удобно следить, отбирая пробы стеклянной па-
лочкой и нанося мазки на фильтровальную-бумагу. Конец реакции опреде-
ляют по образованию светло-желтого пятна и отсутствий) черного осадка
на бумаге.
Литература
1. Т s о и К w a n С h u n g, С h е п g С h a o-S h i n g. M. M. N a c h 1 a s,
A. M, Seligman, J. Amer. Cliem. Soc., 78, 6139 (1956).
2. li 3 11 z, F. S i e d e n, Liebigs Ann. Chem., 324, 321 (19<>2).
Поступила в апреле 19^3 г.
Институт мозга АМН СССР
МЕТИЛТИАЗОЛИЛТЕТРАЗОЛ ИИ
3-(4,'5'-Диметил-тиазолил-2')-2,5-дифенилтетразолий
бромистый
М. Н. МАЛЕВ
N----С-СН3~
II II
+ II II
/N=N—С С-СН8
\=/ 'Sj-N S
вГ
Ci gHjjBrNjS
М. в. 414,34
Метилтиазолилтетразолий применяется в гистохимии в
качестве реактива для выявления ферментов окислительного
обмена [1].
з».
По литературным данным, метилтиазолилтетразолий полу-
чают окислением метилтиазолилформазана бромсукциними-
дом. Формазан, в свою очередь, получают из бензальфенил-
гидразона и диазотированного 2-амиио-4,5-диметилтиазолил-
гидробромида [2].
При проверке указанной методики нами введена перекри-
сталлизация тетразолия бромида, что позволило получить его
в более чистом виде.
СИНТЕЗ МЕТИЛТИАЗОЛ ИЛ ТЕТРАЗОЛ ИЯ
I { Vc€°+h,n-nh-^ —
(2>-ch=n-nh-(_) + н2о
П. СН3СН,СОСН, 4- Br2 — CHgCHBrCOCHs + НВг
Ш. СНзСНВгСОСН, -I- HsN-C-NHa —
II
S
N—С—CHS
II II + нао
- HBr-HjN—С С-СНз
S
N-C-CH,
|| II + НС1 4 NaNO2 ->
IV. HBr-H2N-С С-СНз
S
N-C-CHj
<- II II
№N-C С-СН,
V
С1 4- NaBr 4- 2Н2О
—CH = N-NH —
N —С—СН8~i
N=N-C С- СНз Cl
s
40
N-C-CH,
II II
N=N~C C-CH,
O-<n_n;h у +hc.
N-C-CH,
!l к
.N = N —C C-CH,
v. f W ;h \ / h,c-ch2
S +11
ОС co
\вг
N-C-CH,-
+ II II .
N=N-C C-CH,
к I \Z
x=/ +n-n s
Br
H,C-CH,
ОС CO
\h
Характеристика основного сырья
Бензойный альдегид, ч.д.а., ТУ МХП 157—51.
Фенилгидразии основание, ч.д.а., ТУ МХП 43—47.
Метилэтилкетои, ч., В ТУ МХП 3024—55.
Бром, х.ч., ГОСТ 4109—48.
Тиомочевина, ч., ГОСТ 6344—52.
Бромсукциннмид, ч., В ТУ РУ 1085—54.
Условия получения
Получение бензальфенилгидразона [3]. В фарфоровом ста-
кане емкостью 1,5 л, снабженном мешалкой, растворяют 108 г
(1 М; 96,6 мл) фенилгидразина в 1 литре этилового спирта и
добавляют по каплям, при размешивании, 106 г (1 М; 101 мл)
бензальдегида. По мере прибавления альдегида в реакцион-
ной массе образуются снежно-белые кристаллы. Загустевшую
массу размешивают еще 30 минут, отфильтровывают на во-
41
ронке Бюхнера, промывают на фильтре 50 мл спирта и сушат
на воздухе. Выход бензальфенилгидразона 180 г (91%); т. пл.
154—155°, что соответствует литературным данным.
Бензальфенилгидразон хорошо растворим в пиридине, аце-
тоне, слабо растворим в холодном спирте, нерастворим в воде
(см. примечание 1).
Получение метил-а-бромэтилкетона [4]. В двухлитровый
фарфоровый стакан, снабженный .мешалкой и капельной во-
ронкой, помещают раствор 75 г хлористого калия в 625 мл
воды, 310 г (4,3 М; 388 мл) метилэтилкегона и добавляют по
каплям, при размешивании, 1250 г (7,8 М; 400 мл) брома. В
начале прикапывания брома реакционную массу нагревают
на водяной бане до 50°, затем реакцию ведут при 35—40°.
После добавления всего брома реакционную массу размеши-
вают еще 30 минут и переносят в дёлительную воронку.
Метил-а-бромэтилкетон, содержащийся в нижнем слое, отде-
ляют и встряхивают с 20 г окиси магния и 100 мл воды. Про-
мытый кетон сушат над хлористым кальцием в течение суток
и фракционируют под вакуумом, отбирая фракцию 70770 мм.
Выход метил-а-бромэтилкетона 250 г, что составляет 53% от
теоретического (см. примечание 2).
Получение 2-амино-4,5-диметил-тиазолил гидробромида [5].
В фарфоровом стакане емкостью 500 мл, снабженном ме-
шалкой и капельной воронкой, растворяют 76 г (1 М) тио-
мочевины в 200 мл спирта и при энергичном размешивании
добавляют из воронки 152 г (1 М; 106 мл) метил-а-бромэтил-
кетона в течение 30 минут. Затем реакционную смесь помеща-
ют в круглодонную колбу емкостью 500 мл, снабженную об-
ратным холодильником, и нагревают на кипящей водяной бане
30 минут. Выпавший белый кристаллический осадок 2-амино-
4,5-диметил-тиазолил гидробромида отсасывают, промывают
на фильтре 50 мл спирта и сушат на воздухе. Выход продукта
170 г (81% от теоретического); т. пл. 225°, что соответствует
литературным данным [5].
Получение 3,5-дифенил.-1 -(4‘,5'-диметил-тиазолил-2')-фор-
мазана. В фарфоровом стакане емкостью 500 мл, снабженном
мешалкой, капельной воронкой, термометром, растворяют
4,0 г (0,02 AI) бензальфенилгидразона в 50 мл пиридина и
приливают раствор 10 г кристаллического ацетата натрия в
100 мл метанола. Полученную смесь охлаждают до —10° (см.
примечание 3) и добавляют по каплям, при размешивании,
диазораствор в течение 30 минут.
Диазораствор готовят обычным способом: суспендируют
смесь из 4,2 г (0,02 М) 2-амино-4,5-диметил-тиазолил гидро-
бромида, 50 мл 2 и. (0,1 М) соляной кислоты, 50 г мелко-
раздробленного льда и прикапывают раствор 1,6 г (0,022 М)
нитрита натрия в 10 мл воды.
42
После добавления всего диазораствора реакционную мас-
су размешивают при температуре —5° около 1 часа, прилива-
ют 100 мл воды и оставляют на ночь при 5°. На следующий
день выпавший черный осадок формазана отфильтровывают
на воронке Бюхнера, промывают на фильтре 50 мл спирта н
100 мл горячей воды.
Выход 3,5-дифенил-1- (4/,5'-диметил-тиазолил-2/) -формаза-
на 4,5 г (67%); т. пл. 155—157°. Продукт хорошо растворим в
пиридине, диметилформамиде, нерастворим в холодных спир-
тах, воде (см. примечание 4),
Получение метилтиазолилтетразолия. В конической колбе
емкостью 500 мл смешивают раствор 3,35 г (0,01 М) метил-
тиазолилформазана в 250 мл этилацетата с раствором 3,6 г
(0,02 М) М-бромсукцинимида в 50 мл этилацетата. Через
2 часа отсасывают выпавший желтый кристаллический оса-
док, промывают на фильтре 50 мл этилацетата и сушат на
воздухе. Выход метилтиазолилтетразолия 3,8 г, что составля-
ет 91% от теоретического; т. пл. 163—169°. Продукт очищают
от примеси М-бромсукцинимида растворением в 100 мл горя-
чего ацетона и осаждением 500 мл этилацетата. Получают
желтые игольчатые кристаллы бромистого тетразолия с т. пл.
179—180°, растворимые в воде, диметилформамиде, хлоро-
форме, нерастворимые в эфире (см. примечание 5).
Примечания: 1. Бензальфенилгидразон под воздействием сол-
нечных лучей коаспеет, поэтому его хранят в банке из темного стекла.
2. При длительном хранении жидкость оставляют в закрытой банке
над окисью магния.
3. Охладительную смесь удобно готовить добавлением размельченной
твердой углекислоты в ацетон.
4. Вещество можно перекристаллизовать из ацетона, при этом Полу-
чаются черные блестящие иглы с т. пл. 164°. Однако для дальнейшего син-
теза пригоден и неперекристаллизованный продукт.
5. По литературным данным, метилтиазолилтетразолпй имеет т. пл.
171° [2].
Литература
1 A. G. Е. Pearse, J, Hlstochem. a. Cytochem , 5. 515 (1957).
2. Н. Beyer. Т. Pyl, Chem. Вег, 87, 1511 (1954).
3 Е. Fischer. Вег.. 9, 880 (1876).
4. D. Elliot, D. Ней, J. Chem. Soc.. 1948, 274.
5. R. Kurkjv, E. Brown, J. Ainer. Chem. Soc., 74, 6261 (1952).
Поступила в апреле 1963 г. Институт мозга АМН СССР
2,3-ДИФЕНИЛ-5-БЕНЗОЛАЗО-ТЕТРАЗОЛИЙ ХЛОРИ-
СТЫЙ И 2,3-ДИФЕН ИЛ-5-БЕНЗОЛ АЗО-ТЕТРАЗОЛ Ий
БРОМИСТЫЙ
М. Н. МАЛЕВ
_ N=N-^_/
S '4_N=N_C/ I —
= 4n~n~\_/
c19h16cin.
N=N-^
S Vn=N-C^ I X==
= XN-N-\ /
CjeHigBrNg
Cl
М. в. 362,82
Br
M. в. 407,28
Соли 2,3-дифеиил-5-бензолазо-тетразолия могут найти
применение в гистохимии.
По литературным данным, 2,3-дифенил-5-бензолазо-тетра-
золий хлористый получают при окислении 1,5-дифенил-З-бен-
золазоформазаиа амилиитритом в кислой среде [1] или тетра-
ацетатом свинца с последующим переводом основания тетра-
золия в хлорид [2]. Формазан получают, исходя из малоновой
кислоты [3] или d-галактозы [2].
Формазан и 2,3-дифеиил-5-беизолазо-тетразолий хлори-
стый получены по методике, предложенной Цемплеиом и Ме-
ггером [2]. 2,3-Дифенил-5-бензолазо-тетразолий бромистый на-
ми получен впервые при окислении 1,5-дифенил-З-беизолазо-
формазана бромсукцииимидом в растворе этилацетата.
СИНТЕЗ ХЛОРИСТОГО И БРОМИСТОГО 2,3 ДИФЕНИЛ 5 БЕНЗОЛ-
АЗОТЕТРАЗОЛ ИЯ
I. С^ 1ХН 'N-NH-'f- /-к С€ =
Н-С-ОН + \ NH—NH, -* | хн
I Н-С-ОН
но-с-н 1 но-с-н
но-с-н 1 +н2о но-с-н
н-с-он 1 н-с-он
сн»он сн,он
44
п.
-NHa4-2HCl + NaNOa
V-N = n] Cl + NaCl + 2HjO
xN-NH-^^
C< 4-=x
I XH
H-C-OH
I
HO-C-H
I
HO-C—H
H-C-OH
CH,OH
__ N-N—
^-N=N-C^ \Н= +
= /
OH
H-C-OH
НО-С-Н +HC1
I
HO-C-H
H-C-OH
I
CHaOH
HC1
Pb(OCOCH,)t *
__ N-N
III. ^-N = N-C^ >H 2
= N-N'
IV.
нгс-сн,
I I
ОС CO
NBr
N—N—H2C-CH,
'S-N^N-tff | = Br 4- ОС CO
= N=N-^ X/
+ \_/ J NFi 45
Характеристика основного сырья
d-Галактоза, фарм.
Фенилгидразин основание, ч.д.а., ТУ МХП 43—47.
Анилин, ч.д.а., ГОСТ 5819—51.
Соляная кислота, ч.д.а., ГОСТ 3118—46.
Нитрит натрия, ч.д.а., ГОСТ 4197—48.
Бромсукциннмид, ч., ВТУ РУ 1085—54.
Свинец тетраацетат (см. примечание 1).
Хлористый водород (см. синтез n-хлорфенил-диаМида фос-
форной кислоты).
Условия получения
Получение d-галактозофенилгидразона [4, 5]. В круглодон-
ной колбе емкостью 1 литр, снабженной обратным холодиль-
ником, растворяют 95,0 г (0,55 М) d-галактозы в 250 мл го-
рячей воды и приливают 50 мл (0,5 М) фенилгидразина. Ре-
акционную смесь нагревают на кипящей водяной бане 1 час,
охлаждают до комнатной температуры и отсасывают выде-
лившиеся бесцветные кристаллы d-галактозофенилгидразона.
Осадок промывают на фильтре 200 мл воды и 50 мл этилово-
го спирта, отжимают, сушат на воздухе. Получают 100 Д
(74%) продукта с т. пл. 158—159°, что соответствует литера-
турным: данным {4]. d-Галактозофенилгидразон кристаллизу-
ется из этилового спирта с той же т. плавления; хорошо ра-
створим в пиридине, ацетоне, диоксане, плохо растворим в
воде, эфире.
Получение 1,5-дифенил-З-бензолазо-формазана. В фарфо-
ровом стакане емкостью 200 мл смешивают 10 мл (0,11 М)
анилина, 75 мл этилового спирта и приливают 30 мл (0,33 М)
концентрированной соляной" кислоты. Полученный раствор
солянокислого анилина охлаждают добавлением 50 г льда и
диазотируют раствором 7,6 г (0,11 М) нитрита натрия в
20 мл воды при температуре 0—5°. Реакционную смесь вы-
держивают при той же температуре около 30 минут.
Одновременное проведением диазотирования в фарфоро-
вом стакане емкостью Г литр, снабженном мешалкой, раство-
ряют 27 г (0,1 А4) d;гaлaктoзoфeнилгидpaзoнa в смеси раст-
вора 28 г (0,5 М) едкого кали в. 30 мл воды и 300 мл спирта.
Полученный раствор охлаждают до .температуры минус 5° и
к нему, по’каплям и при размешивании, добавляют диазсгра-
створ анилина в течение 30-минут. Температура реакционной
массы при этом не должна повышаться выше 0°. После добав-
ления всего диазбраствора реакционную массу размешивают
еще 30 минут, приливают 300 мл воды и отсасывают выделив-
шийся красный осадок формазана. Осадок промывают ла
фильтре 500 мл воды- и сушат на воздухе. Получают 20 г
:-К
(61%) 1,5-дифенил-З-бензолазоформазана с т. пл. 139—141°.
После перекристаллизации из 75 мл бутанола получают 10 г
продукта в виде блестящих листочков гранатового цвета с
т. пл. 157—158°, что соответствует литературным данным [2].
Получение 2,3-дифенил-5-бензолазо-тетразолия хлористо-
го. В фарфоровом стакане емкостью 500 мл, снабженном ме-
шалкой, растворяют 3,3 г (0,1 М) 1,5-дифенил-З-бензолазо-
формазана в 200 мл хлороформа и добавляют, в течение 15
минут и при размешивании, 6,6 г (0,015 Л1) тетраацетата
свинца. Реакционную массу размешивают еще 30 минут и в
результате окисления красный раствор формазана превра-
щается в желтый раствор тетразолия, после чего его фильт-
руют и насыщают сухим хлористым водородом. Выпавший
белый кашицеобразный осадок хлористого свинца через 1 час
отфильтровывают на воронке Бюхнера, а фильтрат, содержа-
щий 2,3-дифенил-5-бензолазо-тетразолий хлористый, упарива-
ют наполовину. После охлаждения раствора получают 2,0 г
(55%) желтого кристаллического продукта с т. пл. 250°, что
соответствует литературным данным [2].
Получение 2,3^дифенил-5-бензолаза-тетразолия бромисто-
го. В фарфоровом-стакаire емкостью 200 мл_. растворяют 3,3 г
(0,01 М) 1,5-дифенил-З-бёнзолазоформазана в 50 мл этил-
гцетата и приливают раствор 3,6 г (0,02 М) бромсукциними-
да в 30 мл этилацетата. В результате окисления формазана
получают желтый осадок 2,3-дифе»ил-5-бензолазотетразолия
бромистого. Через 1 час полученный осадок отсасывают и про-
мывают на фильтре 50 мл эфира. Выход поодукта 3.0 г, что
составляет 73% от теоретического; т. пл. 242—243°. 2,3-Дифе-
нил-5-бензолазо-тетразолий бромистый плохо растворим в
этиловом спирте, ацетоне, эфире; растворим в воде, диметил-
формамиде (см примечание 2).
Примечания: 1. Тетраацетат свинца разлагается при хранении,
поэтому его готовят перед употреблением 16].
В колбу Эрлемейера емкостью 1 литр, снабженную мешалкой и. тер-
мометром, помещают 750 г ледяной уксусной кислоты н добавляют-пор-
циями, по 5—10 г, сухого мелкорастертого свинцового сурика (ГОСТ
1787—50).
РЪ,О4 + 8 CHjCOOH —• РЪ(ОСОСНЛ 4- РЬ(ОСОСН,), 4- 4 н,о
Каждую новую порцию сурика добавляют после исчезновения Желтой ок-
раски; температуру реакционной массы поддерживают в интервале 55—
65°. Всего добавляют 300—325 г сурика. Затем полученный раствор охла-
ждают до 10°, отфильтровывают па воронке Бюхнера выпавшие бесцвет-
ные кристаллы тетраацетата свинца. Продукт перекристаллизовывают из
100 мл ледяной уксусной кислоты, нагретой до 50°. По'лучают 150 г пре-
парата, который хранят в эксикаторе над серной кислотой. При разложе-
нии кристаллов поверхность их окрашивается в коричневый цвет:
рь(ососн,т; — рьо, ф 2 (сн,со),о
2. При добавлении формальдегида или аскорбиновой кислоты к Тетра-
золию, растворенному в 0,2 н. водном растворе едкого натра, выпадает
красный осадок формазана.
47
Литература
1. Н. Pechmann, Р. Runge. Вег., 27. 2930 (1894).
2. G. Zemplen, L. Mester, Acta .Chim. Acad, sclent, hung., 2, 9
(1952).
3. H. P 6ch m an n, Ben. 25. 3181 (1892).
4. E. F i s c h e r, Ber , 20. ”21 (1887).
5. H Jacobi, Liebigs Ann. Chem.. 272, ПО (1892).
6. О. D i m г о t h, R. Schweizer, Ben, 56, 1375 (1923).
Поступила в апреле 1963 г.
Институт мозга АМН СССР
ДИАЗОЛЬ СИНИЙ с
3,3'-Диметоксидифенилен-4,4'-бисдиазоний стабилизирован-
ный, соль синего прочного Б
М. Н. МАЛЕВ
ОСН, Н3СО
NsN
2С1,2ZnCl,
Ci4H12CleN4O2Zn3
М. в. 611, 75
Стабилизированные соли диазоиия и бисдиазония приме-
няются в гистохимии в качестве азосоставляющих при выяв-
лении функциональных групп белков [1J.
Диазоль синий С получают бисдиазотироваиием о-диани-
зидииа с последующим осаждением двойной соли бисдиазо-
иия хлористым цинком.
СИНТЕЗ ДИАЗОЛЯ СИНЕГО С
осн3 Н8СО
L J
I. HCl-HjN-^ ^-NH3-HCl4-2HCl4-2NaNO2 —
ОСН, Н,СО
н'й-0-0й“г,_
2С1 + 2NaCl + 4Н3О
осн3 Н8СО
II.
N=N~C_)~C ^-N=N 2С1 + 2ZnCl2 ->
OCHg H3CO
n=i5~G)-C)-,:'-n_
2C1, 2ZnCl2
Характеристика основного сырья
о-Дианизидин солянокислый, ч., ВТУ МХП 4163—54.
Соляная кислота, ч.д.а., ГОСТ 3118—46.
Нитрит натрия, ч.д.а., ГОСТ 4197—48.
Цинк хлористый, ч.д.а., ГОСТ 4529—48.
Условия получения
В фарфоровом стакане емкостью 300 мл, снабженном ме-
шалкой, суспендируют 31,7 г (0,1 М) солянокислого о-диани-
зидина в смеси 80 мл 5 н, (0,4 М) соляной кислоты и 50 г
мелкораздробленного льда. К полученной суспензии, охлаж-
денной до температуры 0°, добавляют по каплям раствор
14,5 г (0,21 М) нитрита натрия в 30 мл воды. После добав-
ления нитрита натрия реакционную смесь размешивают еще
30 минут при температуре 0—5° и полученный раствор соли
бисдиазония профильтровывают на воронке Бюхнера.
К прозрачному фильтрату светло-розового цвета прилива-
ют охлажденный до 5° раствор 30 г (0,22 М) хлористого
цинка в 60 мл воды. Выпавший желтый осадок отфильтровы-
вают на воронке Бюхнера, отжимают на фильтре и сушат в
вакуум-эксикаторе над едким кали в течение двух суток.
Выход диазоля синего С 48 г, что составляет 80% от тео-
ретического. Препарат хранят в герметичной банке из тем-
ного стекла.
Литература
1. Э. Пирс, Гистохимия, 725, 726, 790, 816, 858, М., ИЛ, 1962.
Поступила в апреле 1963 г. Институт мозга АМН СССР
а-НАФТИЛАЦЕТАТ
а-нафтиловый эфир уксусной кислоты
М. Н. МАЛЕВ
CjaH10Oa
ОСОСНз
I II I
4/XZ
М. в. 186, 21
а-Нафтилацетат применяется в гистохимии в качестве суб-
страта при выявлении эстераз [1].
По литературным данным, а-нафтилацетат получают из
а-нафтола при взаимодействии с хлористым ацетилом [2]. На-
ми синтезирован субстрат по этому методу, причем в качестве
среды был использован пиридин, что позволило проводить ре-
акцию в более мягких условиях.
СИНТЕЗ а-НАФТИЛАЦЕТАТА
ОН OCOCHj
| II | + СН3СОС1 - г II | + НС1
х/х/ 4/W
Характеристика основного сырья
а-Нафтол, ч.д.а., ГОСТ 5838—51.
Ацетил хлористый, ч., ГОСТ 5829—51.
Пиридин, ч., ГОСТ 2747—44, высушенный над едким кали
и перегнанный, т. кип. 114—117°.
Условия получения
В трехгорлую колбу емкостью 500 мл, снабженную ка-
пельной воронкой, мешалкой с масляным затвором и обрат-
ным холодильником с хлоркальциевой трубкой, помещают
72 г (0,5 Л1) а-нафтола, 130 мл (1,6 М) сухого пиридина. К
полученному раствору а-нафтола добавляют по каплям, в те-
чение 30 минут, при постоянном размешивании, 44 мл (0,6 М)
хлористого ацетила (см. примечание). Затем реакционную
массу нагревают при температуре водяной бани 80—90° около
30 минут, охлаждают до комнатной температуры и выливают
тонкой струйкой при размешивании в фарфоровый стакан с
50
500 г размельченного льда. Выпавший белый осадок а-наф-
тилацетата отсасывают, промывают на фильтре 500 мл ледя-
ной воды и сушат между листами фильтровальной бумаги.
Продукт перекристаллизовывают из 50 мл этанола и сушат
на воздухе.
Выход а-нафтилацетата 61 г, что составляет 65% от теоре-
тического; т. пл. 45—46°, что соответствует литературным дан-
ным [2};
Примечание. При ацетилировании о -нафтола выделяется хло-
ристый водород, который с пиридином дает белый осадок хлористоводо-
родного пиридина в качестве побочного продукта; а -нафтилацетат хоро-
ню растворяется в пиридине и остается в растворе.
Литература
1. Э. Пирс, Гистохимия, 816, М., ИЛ. 1962.
2. L. Schaeffer, Annalen der Chem. Und Pharm., 152, 287 (1869).
Поступила в апреле 1963 г. Институт мозга АМН СССР
р-НАФТИЛАЦЕТАТ
р-Нафтиловый эфир уксусной кислоты
Е. А. СТРЯПЧЕВА
-ОСОСНз
C^I 1 j jOa
М. в. 186, 21
{З-Нафтилацетат применяется в гистохимии в качестве суб-
страта при выявлении эстераз [1].
По литературным данным, р-нафтилацегат получают при
действии уксусного ангидрида на щелочной раствор р-нафто-
ла (2].
СИНТЕЗ • р-НАФТИЛАЦЕТАТА
I. ^X||/SV0H + NaOH — -I- H2o
51
n. fYY0Na +(CH.CO).O nzVOCOCH, +
-}” CHgCOONa
Характеристика основного сырья
р-Нафтол, ч.д.а., ГОСТ 5835—51.
Натр едкий, ч.д.а., ГОСТ 4328—48.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Условия получения
В фарфоровый стакан емкостью 1 литр, снабженный ме-
шалкой, помещают раствор 29 г (0,2 М) ^-нафтола (см. при-
мечание) в 150 мл 2,5 н. (0,37 М) едкого натра, добавляют
около 300 мл мелкоизмельченного льда и прикапывают, при
размешивании, в течение 20 минут, 32 мл (0,34 М) уксусного
ангидрида. Температура реакционной массы поддерживается
в интервале 0—5°. В результате ацетилирования р-нафтола
образуется белый осадок 0-нафтилацетата, который отсасы-
вают, промывают на фильтре 100 мл воды и сушат в эксика-
торе над едким кали. Выход (З-нафтилацетата равен 25 г, что
составляет 68% от теоретического; т. пл. продукта 68—69°.
После перекристаллизации из петролейного эфира получают
р-нафтилацетат в виде бесцветных игл, растворимых в низ-
ших спиртах, эфире, хлороформе; т. пл. 71°, что соответствует
литературным данным [2].
Примечание. При работе с ₽ -нафтолом нужно пользоваться ре-
зиновыми перчатками и не распылять его в помещении.
Литература
1. Э. Пирс, Гистохимия, М., ИЛ, 1962, 413.
2. Препаративная органическая химия, М., ИЛ, 1959, стр. 377.
Поступила в апреле 1963 г. Институт мозга АМН СССР
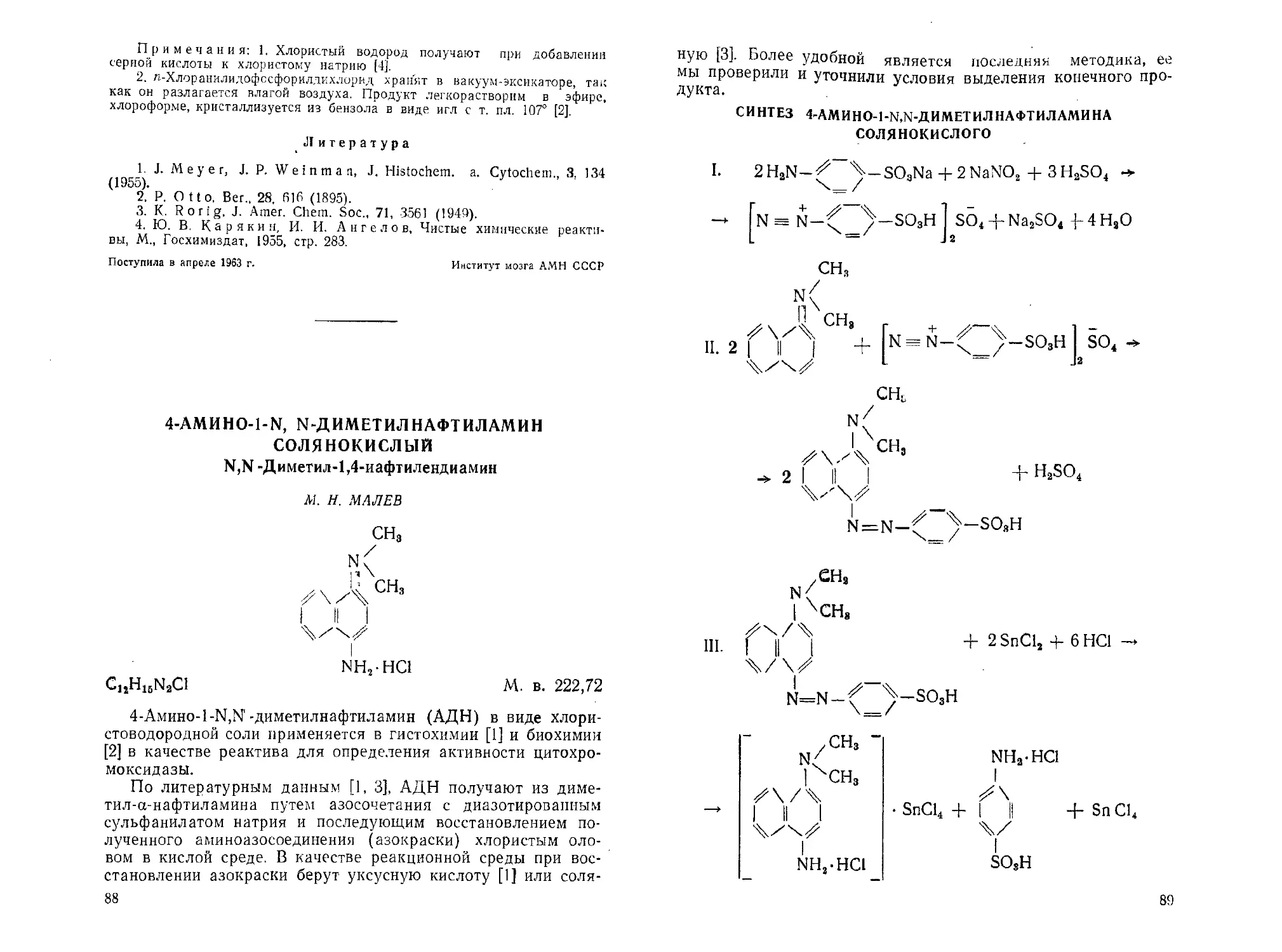
DL-АЛАНИЛ-р-НАФТИЛАМИД
#-(ВЬ-а-Алаиил)-2-иафтиламии
М. Н. МАЛЕВ
CHg-CH-CO-NH-^Y^
nh2 ч/w
Ci8Hi4N3O М. в. 214, 27
DL-Аланил-р-нафтиламид применяется в гистохимии в ка-
честве субстрата при выявлении аминопептидаз [1, 2]. По ли-
тературным данным, его получают при взаимодействии
М-(а-бромпропионил)-2-нафтиламина с аммиаком в закрытом
сосуде под давлением [3]. М-(а-Бромпропионил)-2-нафтиламин
получают при действии бромангидрида а-бромпропионовой
кислоты на р-нафтиламин [3].
Нами проверена и уточнена указанная методика.
СИНТЕЗ DL-АЛАНИЛ- ₽-НАФТИЛАМИДА
I. 8 СН3СН2СООН + 2Р + 5Вга —
-> 8СН3СН2СОВг + 2Н,РО4 + 2НВг
II. 4СН3СНаСОВг + 4Вг2 4СН3СНВгСОВг + 4НВг
Чх/Ч мн
III. | !' 4-CH3CHBrCOBr -
4/XZ
-> CH3CHBrCONH-^ xZ ''| + НВг
Ч/\Ч
IV CH.CHBrCONH-j^ X||/4j.+ NH3 —
ч/w
- CH3CH-CONH-V^I -I- HBr
NH 4/X^
Nils
Характеристика основного сырья
Пропионовая кислота, ВТУ МХП 2492—51.
Бром, ч.д.а., ГОСТ 4109—48.
Фосфор красный, ч., ТУ МХП 1360—46.
53
0-Нафтиламин, ч., ТУ ГКХ ОРУ 130—59,
Аммиак водный, ч.д.а., 25%-ный, ГОСТ 3760—47.
Пиридин, ч., ГОСТ 2747—44.
Условия получения
Получение бромангидрида а-бромпропионовой кислоты
[4]. В трехгорлой колбе емкостью 250 мл, снабженной мешал-
кой с масляным затвором, капельной воронкой и обратным
холодильником с хлоркальциевой трубкой, суспендируют
8,0 г (0,26 М) красного фосфора в 74,6 мл (1 М) пропионовой
кислоты и добавляют по каплям 32 мл (0,625 Л4) брома в те-
чение двух часов. Затем реакционную массу нагревают на
водяной бане (50—55°) и медленно добавляют по каплям еще
54 мл брома (1,05 Af) в течение 12 часов. После прибавления
всего брома постепенно поднимают температуру реакционной
смеси до 90—100° и выдерживают при этой температуре око-
ло двух часов до прекращения выделения бромистого водо-
рода.
Полученную смесь помещают в грушевидную колбу ем-
костью 250 мл, снабженную прямым холодильником с хлор-
кальциевой трубкой, и перегоняют продукт, собирая фракцию
с т. кип. 150—155°. Выход 46,5 мл (96 а), что составляет 45%
от теоретического. По внешнему виду продукт представляет
собой почти бесцветную жидкость.
Получение П-(а-бромпропионил)-2-нафтиламина. В трех-
горлой колбе емкостью 500 мл, снабженной капельной ворон-
кой, термометром и мешалкой с масляным затвором, раство-
ряют 10 г (0,07 М) р-нафтиламина в 300 мл ацетона. Получен-
ный раствор охлаждают в бане с ледяной водой до 5°, добав-
ляют к нему 30 мл (61,8 г; 0,28 М) бромангидрида а-бром-
пропионовой кислоты и прикапывают, при размешивании, в
течение, 30 минут, 50 мл (0,62 Л4) пиридина. Реакцию ведут
при температуре 5—10°. После добавления всего пиридина
реакционную смесь размешивают еще 30 минут, отфильтро-
вывают на воронке Бюхнера выпавший белый осадок броми-
стоводородного пиридина и промывают ею на фильтре 50 мл
ацетона. Фильтрат приливают тонкой струйкой, при разме-
шивании, к 1 литру ледяной воды. При этом выпадает белый
осадок, который отсасывают, промывают на фильтре 100 мл
воды и сушат при температуре 40—50°. Получают 17,5 г
(90%) А-(а-бромпропионил)-2-нафтиламина с т. пл. 168—
169°. После перекристаллизации из 270 мл кипящего метано-
ла выход мелкокристаллического продукта равен 15,5 г, что
составляет 80% от теоретического; т. пл. препарата 172° (см
примечание 1).
Получение DL-аланил-^-нафтиламида. В стеклянную ам
пулу емкостью около 300 мл помещают раствор 2,8 г (0,01 М)
54
А-(а-бромпропионил)-2-нафтиламина в 140 мл метанола и
приливают 140 мл (1,8 М) концентрированного водного ам-
миака. Ампулу запаивают (см. примечание 2), помещают в
водяную баню с терморегулятором и выдерживают при тем-
пературе 60° 24 часа. Затем ампулу осторожно открывают
(см. примечание 3), фильтруют мутный бесцветный раствор,
упаривают под тягой на водяной бане до объема около 25 мл.
К остатку добавляют 50 мл этанола, 0,5 мл 1 н. уксусной кис-
лоты и нагревают смесь на горячей водяной бане. Полученный
раствор обесцвечивают добавлением 0,5 г активированного
угля и фильтруют. К фильтрату добавляют 1 н. раствор ед-
кого натра до слабощелочной реакции (около 40 мл). При
охлаждении раствора выпадают белые пластинчатые кристал-
лы DL-аланил-р-нафтиламида, которые отфильтровывают на
воронке Бюхнера и перекристаллизовывают из 120 мл 25 % -
ного этилового спирта. Выход продукта равен 1,75 г, что со-
ставляет 81% от теоретического; т. пл. 91—92°.
Примечания: 1. По литературным данным, jV-(«-бром пропионил) -
2-иафтиламин представляет собой мелкие иголки с перламутровым оттен-
ком, т. пл. 174° [5].
2. Ампулу перед запаиванием помещают в сосуд Дюара с жидким
азотом. После затвердевания смеси отросток ампулы запаивают.
3. Ампулу перед вскрытием помещают в ледяную воду для конден-
сации паров метанола и понижения давления внутри ампулы.
Литература
1. М. S Burs tone, I. Е. Folk, J. Histochem. a. Cytochem., 4, 217
(1956).
2. M. M. N a c h 1 a s, M. M. F r i e d m a n, A. M. S e 11 g m a n, J. His-
tochem. a. Cytochem., 10, 315 (1962).
3. G. Gomory, Proc. Soc. Exper. Biol. a. Med., 87, 559 (1954).
4. N- Zelynsky, Ber., 20, 2026 (1887).
5. A. T i g e r s t e d t, Ber., 25, 2922 ( 1892).
Поступила в апреле 1963 г. Институт мозга АМН СССР
2-ОКСИ-З-НАФТАЛЬДЕГИД
Альдегид 2-окси-З-нафтойной кислоты
М. Н. МАЛЕВ, Е А. СТРЯПЧЕВА
син8оа
М. в. 172,18
Альдегид 2-окси-З-нафтойной кислоты применяется в ги-
стохимии в качестве реактива при выявлении аминогрупп бел-
ков [1].
В литературе описано два метода получения 2-окси-З-нафт-
альдегида. Синтез с помощью первого метода можно пред-
ставить следующей схемой: 2-окси-З-нафтойная кислота —>
2-ацетокси-З-нафтойная кислота -> хлорангидрид 2-ацет-
окси-3-нафтойной кислоты -> 2-ацетокси-З-нафтальдегид ->
2-окси-З-нафтальдегид [2].
По второму методу [1] альдегид получают из гидразида
2-окси-З-нафтойной кислоты в две стадии. Нами проверен и
уточнен второй метод синтеза реактива, являющийся более
простым и удобным.
СИНТЕЗ 2-ОКСИ-З-НАФТАЛЬДЕГИДА
SO2C1
\ОН ,
I II I + I
^/X^-CONHN^ V
сн3
fYV0H
^>\^-CONHNH
SOa
/\
ч/
сн3
+ НС1
56
1L Ч/W-CONHNH
SO2
NasCO8
OH
V\/-c==o
H
CH3
Характеристика основного сырья
Гидразид 2-окси-З-нафтойной кислоты, ВТУ МГ УХГ1
500—60.
n-Толуолсульфохлорид, ч., ВТУ МХП 3507—52.
Диэтиленгликоль, ч., ВТУ МХП 2784а—51.
Метиловый спирт, х.ч., ГОСТ 6995—54.
Натрий углекислый безводный, ч.д.а., ГОСТ 83—41.
Пиридин, ч., ГОСТ 2747—44, высушенный над едким ка-
ли и перегнанный, т. кип. 113—115°.
Условия получения
Получение п-толуолсульфогидразида 2-окси-З-нафтойной
кислоты. В фарфоровом стакане емкостью 300 мл растворя-
ют 20,2 г (0,1 М) гидразида 2-окси-З-нафтойной кислоты в
200 мл пиридина и добавляют небольшими порциями (при-
мерно по 0,5 г) 20 г (0,11 М) n-толуолсульфохлорида в тече-
ние 20 минут при размешивании. Затем реакционную смесь
выдерживают 3 часа при комнатной температуре и выливают
тонкой струйкой, в течение 30 минут, при размешивании, в
фарфоровый стакан емкостью 2 литра, содержащий смесь
800 а' толченого льда и 250 мл концентрированной соляной
кислоты. Выделившийся белый осадок д-толуолсульфо-
гидразида 2-окси-З-нафтойног! кислоты отфильтровывают на
воронке Бюхнера, промывают на фильтре 500 мл воды и су-
шат в вакуум-эксикаторе над едким кали 1 сутки.
Полученный продукт перекристаллизовывают из смеси
250 мл' метанола с 1 мл уксусной кислоты. Выход 27 г, что
составляет 80% от теоретического; т. пл. продукта 203—204°,
что соответствует литературным данным.
Получение альдегида 2-окси-З-нафтойной кислоты. В круг-
лодонной колбе емкостью 500 мл растворяют при нагревании
на масляной бане 17 г (0,05 М) д-толуолсульфогидразида
2-окси-З-нафтойной кислоты в 370 мл диэтиленгликоля. К по-
лученному раствору светло-желтого цвета при температуре
57
160° добавляют 32 г (0,3 Af) безводного карбоната натрия
небольшими порциями (примерно но 0,5 г). При добавлении
карбоната натрия раствор темнеет и наблюдается выделение
углекислого газа. Затем реакционную смесь выдерживают
при температуре 160° 5—10 минут, охлаждают до 100°, выли-
вают в стакан емкостью 1 литр и приливают, при размешива-
нии, 175 мл горячей воды. Полученную смесь охлаждают до
комнатной температуры, помещают в жидко-жидкостный экс-
трактор и экстрагируют 2-окси-З-нафтилальдегнд эфиром око-
ло 48 часов (см. примечание). Полученный эфирный экстракт
светло-желтого цвета концентрируют, отгоняя эфир, и к остат-
ку добавляют около 300 мл 20%-ной уксусной кислоты. Го-
рячий раствор альдегида фильтруют на воронке Бюхнера и
оставляют на ночь при температуре 5°. Выпавший кристалли-
ческий осадок желтого цвета отсасывают и перекристаллизо-
вывают из 20%-ной уксусной кислоты.
Выход 1,7 г 2-окси-З-нафтилальдегида, что составляет 20%
от теоретического; т. пл. 96—97°, что соответствует литера-
турным данным [1]. Продукт хорошо растворим в эфире, аце-
тоне, хлороформе, плохо растворим в воде, спирте.
Примечание. Альдегид экстрагируют эфиром до обесцвечивания
эфира, находящегося в аппарате над экстрагируемой жидкостью. При не-
прерывной экстракции для нагревания колбы с испаряемым эфиром удоб-
но использовать водяной термостат (баню с терморегулятором), где под-
держивается температура 50—53°.
Литература
1. L. Weiss, K.w а п-С h u п g Т s о и, A. Seligman, J. Histo-
chem. a. Cytochem., 2, 29 (1954).
2. Т. В о с h е m, Е. Р г о f f t, Arch. Pharm., 269, 25 (1931).
Поступила в апреле 1963 г. Институт мозга АМН СССР
ФОСФАТ АЗОТОЛА 2,4 МК
Моно-[3-карбокси-(2'',4''-диметиланилидо)]-2-нафтиловый
эфир ортофосфориой кислоты
М. И. МАЛЕВ, В. А. СТРЯПЧЕВА
I
СНз
Ci9H18NO6P
М. в. 371,33
Фосфат азотола 2,4 МК и его натриевая соль применяют-
ся в. гистохимии в качестве субстрата для выявления фосфо-
таз [1].
По литературным данным, фосфат азотола 2,4 МК полу-
чают при взаимодействии ж-ксилидина 2-окси-З-нафтойной
кислоты [3] с хлорокисью фосфора и последующим разложе-
нием полученного нафтилфосфорилдихлорида водой [2].
Нами проверена и уточнена эта методика. В качестве сре-
ды при взаимодействии ж-ксилидина 2-окси-З-нафтойной кис-
лоты с хлорокисью фосфора вместо тетрагидрофурана был
взят бензол.
СИНТЕЗ ФОСФАТА АЗОТОЛА 2,4-МК
I-1
U\/-C00H
11 1-ОН
+ SOC12 -> +SOa+HCl
J-COCl
NHS
/ \-OH f \-ch3
IL I1 I ‘I + NaHCOj
I
CH,
.59
.^^-он
I I CONH + NaCl + CO2-Ь H.0
4/Wc°NH
fVCHs
4/
I
CH3
/Ч/'Ч OH
I II Г
^/^J-CONH + POCI8 -
4\ rH
I rCH3
4/
I
CH3
^\/\-орос12
!^J4J-conh + hci
fVcH3
'W
I
CH3
^V^-Gpoa,
J^J-CONH + 2HaO
f"rCH>
ch3
A/VoPO(OH)2
L / \ J~conh + 2hci
pl|-CH3
4/
CH3
60
^>/^j-0P0(0H)a
J^-CONH + 2CH3ONa —
|^'"||-СНз
CHS
-* f/4||'/^-0P0(0Na)2
!^JxJ-CONH +2CH3OH
/Л rH
I ||—CHs
w
CH3
Характеристика основного сырья
2-Окси-З-нафтойная кислота, техн., ГОСТ 9251—59.
Тионил хлористый, ч., ВТУ МХП 3591—52, свежеперегнан-
ный
At-Ксилидин, ч., ТУ МХП 197—51, свежеперегнанный.
Фосфора хлорокись, ч„ ВТУ У—166—51.
Пиридин, ч., ГОСТ 2747—44, высушенный над едким кали
и перегнанный, т. кип. 113—115°.
Натрий металлический, ч.д.а., ТУ МХП 1664—50.
Метиловый спирт, х.ч., ГОСТ 6995—54.
Бензол, х. ч., ГОСТ 5955—51.
Диметилформамид, ч., ВТУ РУ 1193—56.
Условия получения
Получение хлорангидрида 2-окси-З-нафтойной кислоты [4].
В круглодонную колбу емкостью 500 мл, снабженную обрат-
ным холодильником с хлоркальциевой трубкой, помещают
94 г (0,5 М) сухой 2-окси-З-нафтойной кислоты и 115 мл
(1,6 М) тионила хлористого. Затем колбу с реакционной мас-
сой помещают в водяную баню и нагревают при 90—95° в те-
чение 4—5 часов до прекращения выделения пузырьков хло-
ристого водорода (см. примечание 1): От полученного в ре-
зультате реакции хлорангидрида 2-окси-З-нафтойной кисло-
ты отгоняют избыток хлористого тионила, меняя обратный
холодильник на прямой и создавая вакуум в конце отгонки.
Получают 103 г продукта, который используют на следующей
стадии синтеза.
61
Получение азотола 2,4 МК. В трехгорлую колбу емкостью
1 литр, снабженную капельной воронкой, масляным затво-
ром с мешалкой и обратным холодильником с хлоркальцие-
вой трубкой, помещают 62 мл (0,5 А1) At-ксилидина, 50 г
(0,6 А1) бикарбоната натрия, 100 мл бензола. Получен-
ную смесь нагревают на водяной бане .(60—70°) и до-
бавляют, при размешивании, в течение 2—3 часов, рас-
твор 103 г (0,5 А1) хлорангидрида 2-окси-З-нафтойной кис-
лоты в 400 мл бензола. Затем реакционную массу размеши-
вают еще 30 минут, отфильтровывают на воронке Бюхнера
выделившийся желтый осадок азотола 2,4 МК, промывают
на фильтре 100 мл бензола. Желтый порошок суспендируют
в 500 мл горячей воды и нейтрализуют избыток бикарбона-
та натрия 1 н. соляной кислотой по бумажке конго. Азотол
2,4 МК фильтруют, промывают 500 мл воды и сушат при
70—80°. Выход сухого-азотола 131 г (90%), т. пл. 216—217°.
После перекристаллизации из 1 литра хлорбензола получают
108 г продукта с т. пл. 220—221°.
Получение фосфата азотола 2,4 МК- В трехгорлую колбу
емкостью 500 мл, снабженную мешалкой с масляным затво-
ром, капельной воронкой и обратным холодильником с хлор-
кальциевой трубкой, помещают 21,9 г (0,1 А4) азотола 2,4 МК,
200 мл бензола й 21 мл (0,23 М) хлорокиси фосфора. Содержи-
мое колбы нагревают на кипящей водяной бане до растворения
нафтола и прикапывают при работающей мешалке 10,6 мл
(0,13 М) пиридина в течение 1 часа. После прикапывания все-
го количества пиридина реакционную массу нагревают еще
2 часа на кипящей водяной бане, охлаждают до комнатной
температуры и полученный раствор нафтилфосфорилдихлори-
да фильтруют. Фильтрат помещают в круглодоиную колбу
емкостью 500 мл и отгоняют бензол на кипящей водяной бане.
Маслянистый остаток, являющийся фосфорилдихлоридом
азотола 2,4 МК, растворяют в 150 мл диметилформамида и
выливают тонкой струйкой, при размешивании, в фарфоровый
стакан, содержащий смесь 300 мл воды и 300 г размельченно-
го льда. Получают светло-желтый аморфный осадок фосфата
азотола 2,4 МК, который отсасывают, промывают на фильтре
500 мл холодной воды и сушат в вакуум-эксикаторе над ед-
ким кали в течение 2 суток. Выход 28 г, что составляет 76%
от теоретического; т. пл. продукта 135° (см. примечание 2).
Получение динатриевой соли фосфата азотола 2,4 МК. В
фарфоровом стакане емкостью 300 мл растворяют 18,6 г
(0,05 Af) азотола 2,4 МК в 75 мл горячего (70—80°) диметил-
формамида и приливают при размешивании раствор 0,1 М
метилата натрия в 50 мл метилового спирта (см. примеча-
ние 3). Полученную смесь охлаждают до температуры 5° и
выделяют натриевую соль фосфата азотола 2,4 МК добавле-
62
нием 400 мл эфира. Желтый кристаллический осадок отфиль-
тровывают, промывают на фильтре 100 мл эфира и сушат на
воздухе. Выход 12,5 г, что составляет 60% от теоретического;
продукт растворим в воде, диметилформамиде, плохо раство-
рим в спиртах, нерастворим в эфире.
Примечания: 1. В начале реакции, при бурном выделении пузырь-
ков газа, временно прекращают внешний обогрев реакционной смеси.
2. В литературе [2] не указывается т. пл. продукта.
3. Метилат натрия получают растворением 2,3 г (0,1 М) металличе-
ского натрия в 50 мл метанола.
Литература
1. М. S. Bu rstone, J. Nat. Cancer Inst., 21, 523 (1958).
2. M. S. Burs tone, J. Nat. Cancer Instit., 20, 601 (1958).
3. Англ. пат. 271146 (1926).
4. H. Meyer, Mh. chem., 22, 791 (1902).
Поступила в апреле 1963 г. Институт мозга АМН СССР
ФОСФАТ АЗОТОЛА А
Моно-(3-карбоксианилидо)-2-нафтиловый эфир
ортофосфорной кислоты
М. Н. МАЛЕВ
^Y^-OPOfOH^
WW~C°NH
I о
w
М. в. 343,28
C17H14NOsP
Фосфат азотола А и его натриевая соль применяются в ги-
стохимии в качестве субстрата для выявления активности
кислой фосфотазы [1].
По литературным данным, фосфат азотола А можно полу-
чить из нафтола АС [2, 3] при взаимодействии его с хлор-
окисью фосфора [1] или пятихлористым фосфором [4J с пос-
63
ледующим разложением полученных продуктов водой. Нами
проверена и уточнена последняя методика [4].
I ! Lсое
nh4
4- | I +NaHCO3 ->
ч/
-CONH
+ NaCl + CO2 + H2O
^A^.-OH
//\
4/
+ PC15 -*
-OPC14
-CONH
+ HC1
/y^-OPCL
L JI J-CONH
^\/^-OPO(OH)2
4 ’ 4-4HC1
L Д J-CONH
^>,/>-opo(OH)2
I + 2CH3ONa ->
L \ J-CONH
64
-CONH
+ 2СН3ОН
Характеристика основного сырья
Хлорангидрид 2-окси-З-нафтойной кислоты (см. синтез
фосфата азотола 2,4 МК).
Анилин, ч.д.а., ГОСТ 5819—51, свежеперегнанный.
Фосфор пятихлористый, ч.д.а., ТУ МХП 386—41.
Диоксан, ч., ВТУ МХП 3111—52, высушенный над метал-
лическим натрием и перегнанный.
Пиридин, ч., ГОСТ 2747—44, высушенный над едким кали
и перегнанный, т. кип. 113—115°.
Диметилформамид, ч., ВТУ РУ 1193—56, т. кип. 152—153°.
Натрий металлический, ч.д.а., ТУ МХП 164—50.
Метиловый спирт, х.ч., ГОСТ 6995—54.
Условия получения
Получение азотола А. В трехгорлую колбу емкостью 1
литр, снабженную мешалкой с масляным затвором, капельной
воронкой и обратным холодильником с хлоркальциевой труб-
кой, помещают 45,6 мл (0,5 А4) анилина, 50 г (0,6 М) безвод-
ного карбоната натрия и 100 мл бензола. Полученную смесь
нагревают на водяной бане (60—70°) и добавляют, при раз-
мешивании, раствор 103 г (0,5 М) хлорангидрида 2-окси-З-
нафтойной кислоты в 400 мл бензола в течение 2—3 часов.
Затем реакционную массу размешивают еще 30 минут при
температуре около 60°, охлаждают до комнатной температу-
ры и отфильтровывают па воронке Бюхнера выпавший жел-
тый кристаллический осадок азотола А. Полученный осадок
суспендируют в 500 мл горячей воды и нейтрализуют избы-
ток бикарбоната натрия 1 н. соляной кислотой по бумажке
конго. Азотол А отсасывают, промывают на фильтре 500 мл
воды и сушат при 70—80°. Получают НО г (90%) продукта
с т. пл. 221—223°.
Для получения препарата, свободного от примесей, азотол
А помещают в круглодонную колбу емкостью 500 мл, прили-
вают 280 мл ледяной уксусной кислоты и нагревают до кипе-
ния. Выпавший светло-желтый кристаллический порошок по
охлаждении раствора отсасывают, промывают на фильтре
500 мл воды и сушат при 60—70°. Выход азотола А 95 г, что
G5
составляет 78% от теоретического; т. пл. вещества 245—246°.
что соответствует литературным данным [2].
Получение фосфата азотола А. В двугорлую колбу ем-
костью 250 мл, снабженную капельной воронкой и обратным
холодильником с хлоркальциевой трубкой, помещают 13,2 г
(0,05 Л4) азотола А, 50 мл диоксана и 26,2 г (0,125 М) йяти-
хлористого фосфора. Полученную смесь нагревают на теплой
водяной бане (около 50°) и прикапывают, в течение 1 часа,
10 мл (0,125 М) пиридина. В результате реакции образуется
раствор хлорида фосфата азотола А. Содержимое колбы ох-
лаждают до комнатной температуры и выливают небольши-
ми порциями, при размешивании, в течение 30 минут, в стакан
с 100 г размельченного льда. Полученный желтый осадок фос-
фата азотола А отфильтровывают на воронке Бюхнера, про-
мывают на фильтре 300 мл ледяной воды и сушат в вакуум-
эксикаторе в течение 2 суток над пятиокисью фосфора. Выход
продукта равен 14 г, что составляет 82% от теоретического;
т. пл. 128°.
Получение динатриевой соли фосфата азотола А. В фар-
форовом стакане емкостью 200 мл растворяют 13,7 г (0,04 А1)
фосфата азотола А в 50 .ил горячего (около 70°) диметилформ-
амида и приливают раствор 0,08 М метилата натрия в 50 мл
метанола (см. примечание). Полученный раствор охлаждают
до температуры 5° и выделяют натриевую соль фосфата азо-
тола А добавлением 300 мл эфира. Выход 7,5 г (50%). Пре-
парат представляет собой желтый кристаллический порошок,
хорошо растворимый в воде, диметилформамиде, плохо ра-
створимый в спиртах и нерастворимый в эфире.
Примечание. Метилат натрия получают растворением 1,9 г
(0,08 М) металлического натрия в 50 мл метилового спирта.
Литература
1. М. Burst one, J. Nat. Cancer Inst., 20, 601 (1958).
2. M. Schopff, Ber., 25, 2740 (1892).
3. Англ. пат. 268877 (1926).
4. G. Jef free, K. Taylor, J. Histochem. a. Cytochem., 9,
93 (1961).
Поступила в апреле 1963 г.
Институт мозга АМН СССР
2,2-ДИОКСИ-6,6-ДИНАФТИЛДИСУЛЬФИД
М. Н. МАЛЕВ
I II I—S— S—। !1 I
CaoH1402S2 М. в. 350,46
2,2'-Диокси-6,6/-динафтилдисульфид (ДДД) применяется
в гистохимии для выявления сульфгидрильных групп (—SH)
белковой природы [1].
По литературным данным, ДДД получают из натриевой
соли 2-нафтол-6-сульфокислоты (соли Шеффера) по методи-
ке, предложенной Цинке [2].
В данной работе нами проверена и уточнена эта методика.
СИНТЕЗ 2,2^ЦИОКСИ-6,6'-ДИНАФТИЛДИСУЛЬФИДА
NaO3S—
/'Х/'Ч
-> I !| I
NaOaS-^J1^^
О
+ С1-С-ОС2НБ + NaOH ->
О
-ОС —ОС2Н6
-h NaCl 4- Н2О
о
^'х/^-ОС-ОС2НБ
IL NaOsS-l 1 J
+ РС15
C10aS
0
/\Z^-OC-OC3HS +
x/\^
POC13 + NaCl
О
/х/'^-ос-ос.,н5
IIL II I
C1O2S-^XX^
Zn
HC1
67
IV.
о
о
^/^-ОС—ОСаН5 FeCl,
2 hsAaJ "
о о
IIBCSO-C-O-^4^^ ^\/Ч_О-С-ОСаН8
WW~S~'S~'A/\/
о о
V. C-O-j^V"4 +
I I II I с С I II I I
Н5С2О A/VA ОС2Н6
НО—/а /Х—он
> I II | о о I II I
Характеристика основного сырья
Соль Шеффера, ч„ ВТУ РУ 882—53.
Этиловый эфир хлоругольной КИСЛОТЫ, Т. КИП. 92°.
Фосфор пятихлористый, ч.д.а., ТУ МХП 386—41.
Уксусная кислота, 98%-ная, ГОСТ 61—51.
Цинковая пыль, 85%-ная, свежеприготовленная.
Спирт этиловый, ГОСТ 5962—51.
Соляная кислота, ч.д.а., ГОСТ 3118—46.
Условия получения
Получение 2-карбэтокси-6-нафталинсульфокислого натрия.
В фарфоровом стакане емкостью 1 литр, снабженном мешал-
кой, растворяют 20 г (0,5 Л!) едкого натра в 500 мл воды и
добавляют 123 г (0,5 Л4) соли Шеффера. К полученному ра-
створу добавляют по каплям, при размешивании, в течение
1 часа, 50 мл (0,51 Л4) этилового эфира хлоругольной кисло-
ты (см. примечание 1). Реакционную массу перемешивают
при комнатной температуре еще 1 час и затем охлаждают до
5°. Выпавший белый осадок отфильтровывают, промывают на
6S
воронке 500 мл холодной воды, сушат в вакуум-эксикаторе
над едким натром 1 сутки, дополнительно подсушивают при
70° и растирают в порошок. Получают 120 г (76%) соли, ко-
торую используют на следующей стадии синтеза без дополни-
тельной очистки. 2-Карбэтокси-6-нафталинсульфокислый нат-
рий легкорастворим в горячей воде, плохо — в холодной воде
и спирте.
Получение 2-карбэтокси-6-нафталинсульфохлорида. В
круглодонную колбу емкостью 500 мл, снабженную обратным
холодильником с хлоркальциевой трубкой, помещают 120 г
(0,38 Л4) 2-карбэтокси-6-нафталинсульфокислого натрия и
120 г (0,58 Л4) пятихлористого фосфора. Полученную смесц
тщательно размешивают, встряхивая колбу в течение 30 ми-
нут, при этом реакционная масса саморазогревается и при-
обретает жидкую консистенцию. Затем колбу с содержимым
помещают в кипящую водяную баню на 2 часа, после чего
полученную смесь выливают тонкой струйкой, при размеши-
вании, в фарфоровый стакан, содержащий 1,5 кг толченого
льда (см. примечание 2). Выпавший белый осадок 2-карбэто-
кси-6-нафталинсульфохлорида отфильтровывают на воронке
Бюхнера, промывают 200 мл холодной воды и кристаллизуют
из 600 мл кипящей уксусной кислоты. Продукт отсасывают,
отжимают на фильтре и, не подвергая сушке, используют на
следующей стадии синтеза. Выход сырого продукта 100 г
(85%); т. пл. 118°. 2-Карбэтокси-6-нафталинсульфохлорид лег-
корастворим в эфире, спирте, горячей уксусной кислоте.
Получение 2-карбэтокси-6-нафтилтиола. В фарфоровый ста-
кан емкостью 1,5 л, снабженный мешалкой, помещают 100 г
цинковой пыли (1,0 М) и 400 мл этилового спирта, нагревают
полученную смесь до 30° и вносят, при размешивании, 100 г
(0,32 Л4) 2-карбэтокси-6-нафталинсульфохлорида, затем при-
капывают 200 мл концентрированной соляной кислоты в те-
чение 30 минут. Полученную кашицеобразную массу перено-
сят в колбу емкостью 1 литр и нагревают с обратным холо-
дильником на кипящей водяной бане около 1,5 часа до полу-
чения желтого прозрачного раствора (см. примечание 3). Го-
рячий раствор полученного 2-карбэтокси-6-нафтилтиола филь-
труют, а осадок на фильтре (цинковая пыль, не вошедшая в
реакцию) промывают 100 мл горячего этилового спирта.
Фильтрат используют на следующей стадии синтеза. В чистом
виде 2-карбэтокси-6-нафтилтиол имеет т. пл. 87°.
Получение 212'-карбэтокси-6,6'-динафтилдисульфида, В
фарфоровый стакан емкостью 2 л, снабженный мешалкой, по-
мещают горячий спиртовой раствор 2-карбэтокси-6-нафтил-
тиола и, при размешивании, приливают теплый (50°) раствор
160 г (1 М) хлорного железа в 150 мл воды. Реакционную
массу перемешивают 30 минут, добавляют к ней 600 мл воды
и охлаждают до 5°. При этом выпадает осадок 2,2'-карбэто-
69
кси-6,б'-динафтилдисульфида; его отфильтровывают, промы-
вают 50 мл воды и кристаллизуют из 600 мл 50%-ной уксус-
ной кислоты. Выход 53 г (70%, считая на 2-карбэтокси-6-наф-
талин-сульфохлорид); т. пл. 127°.
Получение 2,2'-диокси-6,6'-динафтилдисульфида. В кругло-
донную колбу емкостью 1 л помещают 53 г (0,11 М) 2,2'-карб-
этокси-6,б'-динафтилдисульфида, 300 мл воды и раствор 62 г
(1,1 М) едкого кали в 100 мл воды. Реакционную смесь на-
гревают 2 часа на кипящей водяной бане и фильтруют горя-
чий раствор. Полученный щелочной раствор 2,2'-диокси-6,б'-
динафтилдисульфида помещают в фарфоровый стакан ем-
костью 2 л и подкисляют 140 мл концентрированной соляной
кислоты до pH 5. Выпавший белый осадок отсасывают, про-
мывают на фильтре 100 мл воды и кристаллизуют из 500 мл
уксусной кислоты. Выход 2,2'-диокси-6,6'-динафтилдисульфи-
да равен 25 г, что составляет 67% от теоретического; т. пл.
203—205°. После повторной перекристаллизации, из уксусной
кислоты получают реактив с т. пл. 218—220°, что соответст-
вует литературным данным. По внешнему виду вещество
представляет собой светло-желтый мелкокристаллический по-
рошок, хорошо растворимый в ацетоне, спиртах и нераствори-
мый в воде, бензоле.
Пр имечания: 1. Этиловый эфир хлоругольной кислоты представ-
ляет собой бесцветную ядовитую жидкость с резким запахом. При работе
с ним нужно пользоваться резиновыми перчатками и включать вытяжную
вентиляцию.
2. Избыток пятиокиси фосфора, не вошедший в реакцию, и хлорокись
фосфора разлагаются ледяной водой.
3. Через 1 час после начала нагревавия на водяной бане берут пробу
для определения полноты восстановления. Из кислого спиртового раство-
ра при добавлении воды выпадает осадок. Если он является 2-карбэтокси-
6-нафтилтиолом, то он полностью растворяется при добавлении раствора
щелочи. В противном случае (если продукт полностью не восстановился)
в колбу с реакционной массой добавляют 50 г цинковой пыли, 50 мл со-
ляной кислоты н продолжают нагрев.
( Литература
1. R. J. Barrnett, А. М. S е I ig га ari, J. Histochem. a. Cytochem.,
1, 392 (1953).
2. Т. Zinke, R. Dereser, Ber., 51, 352 (1918).
Поступила в апреле 1963 г. Институт мозга АМН СССР
я-Н ИТРО-а-БРОМАЦЕТОФЕНОН
В. А. МИХАЛЕВ, А. П. СКОЛДИНОВ, М. И. ДОРОХОВА,
А. П. АРЕНДАРУК, Н. Е. СМОЛИНА, Д. Д. СМОЛИН
СОСН4Вг
ы
V
NO,
C8HeO3NBr М. в. 244,058
n-Нитро-а-бромацетофенон применяется в гистохимии для
выявления локализации сульфгидрильных групп белков
[1, 2].
n-Нитро-а-бромацетофенон обычно получают бромирова-
нием п-нитроацетофенона. Основными путями синтеза послед-
него являются: взаимодействие n-нитробензоилхлорида и это-
ксимагниймалонового эфира с последующим омылением и
декарбоксилированием образующегося п-ннтробензоилмало-
нового эфира [3]; ацетилирование ацетанилида с дальнейшим
омылением N-ацетильной группы и заменой аминогруппы на
нитрогруппу через диазореакцию [4]; восстановление ацетофе-
нона в фенилметилкарбинол с последующими нитрованием и
взаимодействием образующегося нитроэфира п-нитрофенил-
метилкарбинола с водно-спиртовыми растворами щелочей [5].
Предлагаемый ниже простой и удобный метод получения
n-нитро-а-бромацетофенона состоит в присоединении метило-
вого эфира хлорноватистой кислоты в момент его образова-
ния к стиролу, нитровании полученного метилового эфира
хлоргидрина стирола, дегидрохлорировании изомерных мети-
ловых эфиров нитрохлоргидринов стирола в соответствую-
щие п-нитро-а-метоксистиролы [6] с последующим бромиро-
ванием и гидролизом легковыделяемого п-нитроизомера [7].
СИНТЕЗ П-НИТРО-а-БРОМАЦЕТОФЕНОНА
сн=сн2
/ч
|| | СН,ОН, С1, .
ОСНз
СН~СН2С1
I
/X
|| | HNO„ HgSO4
ОСН3
СН-СН,С1
NaOH
71
осн3
с=сн3
Brs
- Br ОСН3 ~
Н,0
СОСН2Вг
\ J
no2
Характеристика основного сырья
Стирол стабилизированный, ВТУ ЛУ 47—53.
Условия получения
Получение метилового эфира хлоргидрина стирола. В
трехгорлую колбу емкостью 1,5 литра, снабженную мешал-
кой, термометром, трубкой для ввода хлора, достигающей дна
колбы, и трубкой для отвода газа, помещают 750 мл метано-
ла и 83,2 г (0,8 М) стирола. Через раствор, при перемешива-
нии и температуре 20—30°, в течение четырех часов, пропу-
скают около 57 г (0,8 А1) хлора, полученного из 63 г перман-
ганата калия и 400 мл соляной кислоты уд. веса 1,17 (см.
примечание 1). По окончании реакции содержимое колбы
нейтрализуют постепенным прибавлением 55 г кальциниро-
ванной соды, осадок хлористого натрия отфильтровывают и
промывают метанолом. Из собранных вместе метанольных
растворов отгоняют метанол и остаток перегоняют в вакууме,
собирая фракцию с т. кип. 102—103,5° (15 мм), представляю-
щую собой метиловый эфир хлоргидрина стирола. Выход
117 г, что составляет 85,8% от теоретического. По внешнему
виду это бесцветная жидкость со специфическим запахом,
1,1187; «0- 1,5227.
Получение п-нитро-а-метоксистирола. В литровую колбу,
снабженную мешалкой и термометром и погруженную в ба-
ню с водой, охлажденной до 3—5°, помещают 327 г азотной
кислоты уд. веса 1,4 и 440 г серной кислоты уд. веса 1,83. К
полученной смеси, в течение часа, при температуре 26—29°,
прибавляют при энергичном перемешивании 200 г (1,17 М)
метилового эфира хлоргидрина стирола. Реакционную массу
размешивают 3 часа при той же температуре и затем выли-
вают ее при перемешивании в соответствующую емкость, со-
держащую литр ледяной воды. .Выделившееся масло извле-
кают 250 мл дихлорэтана. Дихлорэтановый слой отделяют и
промывают водой до отсутствия кислой реакции на конго.
Из полученного раствора отгоняют на водяной бане дихлор-
72
этан, включая в конце огтонки вакуум. Остаток — желтое
масло — растворяют в 470 мл спирта и прибавляют 140 г
40%-ного водного раствора едкого натра. Реакционную массу
перемешивают при температуре 48—50° в течение часа, затем
охлаждают до 10—15°; выделившийся ;г-нитро-а-метоксисти-
рол отфильтровывают, промывают 100 мл этилового спирта,
охлажденного до 5—10°, несколько раз водой (для удаления
примеси хлористого натрия) и . снова 100 мл охлажденного
спирта. Выход 83 г, что составляет 39,5% от теоретического.
По внешнему виду это желтоватые кристаллы (пластинки) с
т. пл. 80—82°. После перекристаллизации из спирта или меда-
нола n-нитро-а-метоксистирол имеет т. пл. 83,5—84,5° (см.
примечание 2). . .
Получение п-нитро-а-бромацетофенона. Растворяют 20,8 а
(0,115 М) n-нитро-а-метоксистирЬла в 80 мл дихлорэтана, к
раствору прибавляют 0,5 г активированного угля и фильтру-
ют (см. примечание 3). Фильтрат помещают в трехгорлую
колбу емкостью 150 мл, снабженную мешалкой, капельной
воронкой, термометром и отводной трубкой, и в течение 30
минут прибавляют при температуре 20—30° и постоянном пе-
ремешивании 18,4 г (0,114 М) брома. По окончании введения
в реакционную смесь брома размешивание продолжают еще
15 минут и затем выливают ее в трехгорлую колбу емкостью
500 мл, содержащую 250 мл воды и снабженную мешалкой
и обратным холодильником. Содержимое колбы энергично пе-
ремешивают 2 часа при комнатной температуре и оставляют
до разделения слоев; дихлорэтановый слой промывают во-
дой до отсутствия кислой реакции на конго, после чего ди-
хлорэтан отгоняют, включая в конце отгонки вакуум. Полу-
ченный кристаллический n-нитро-а-бромацетофенон размеши-
вают с 50 мл четыреххлористого углерода, отфильтровывают
и высушивают. Выход продукта 25,4 г, что составляет 90% от
теоретического; т. пл. 96—97°.
Примечания: 1. Реакция получения метилового эфира хлоргид-
рина стирола требует строгого соблюдения режима, так как в случае от-
клонений от нормальных условий ведения процесса возможно образова-
ние взрывчатого метилового эфира хлорноватистой кислоты. Это может
произойти в случае взаимодействия метанола с хлором в отсутствие сти-
рола. Поэтому нельзя допускать избытка хлора и к концу реакции следует
делать пробы реакционной массы на присутствие в ней метилового эфира
хлорноватистой кислоты (на активный хлор!). Для этого смешивают 1 мл
реакционного раствора с 5 мл 10%-ного водного раствора йодистого ка-
лия. Появление бурой окраски свободного йода указывает на присутствие
метилового эфира хлорноватистой кислоты. В этом случае прибавляют не-
сколько миллилитров стирола до исчезновения реакции на активный хлор.
2. Гидролиз л-нитро- а -метоксистирола кислотами в водно-спиртовой
среде с выходом 85—90% от теоретического приводит к л-нитроацетофе-
ноиу [6], бромированием которого также может быть получен n-нитро- а-
бромацетофенон [3].
3. Необходимость этой обработки вызвана тем, что присутствие сое-
динений железа в реакционной смеси приводит к ее сильному осмолеиию.
73
Литература
1. В. А. Яковлев, С. Н. Нистратова, Гистохимические методы в
нормальной и патологической морфологии, М„ 1958, стр. 106.
2. Л. М. Герштейн, Гистохимические методы в нормальной и па-
тологической морфологии, М., 1958, стр. 114.
3. L. М. L о ng, Н. D. Troutman, J. Amer. Chem. Soc., 71, 2473
(1949),
4. П. А. Г а н г p с к и й, С. Г. Майрановский, А. П. Яровой,
Авт. свид. СССР 102448; Бюлл. изобр. № 1 (1956).
5. А. М. Г р и г о р о в с к и й, П. М. Кочергин, В. А. Засосов,
Л. С. Блинова, Р. М. Титкова, А. В. Савицкий, Авт. свид. СССР
111768; Бюлл. изобр. № 3 (1958).
- 6. М. И. Д о р о х о в а, А. П. Арен дару к, В. А. Михалев,
А. П. С к о л д и н о в, Н. Е. Смолина, Д. Д. Смолин, Авт. свид. СССР
101280; Бюлл. изобр. № 9 (1955).
7. М. И. Дорохова, А. П. А р е н д а р у к, В. А. Михалев,
А. П. С к о л д и н о в, Н. Е. Смолина, Д. Д. Смолин, Авт. свид. СССР.
100610; Бюлл. изобр. № 5 (1955).
Поступила в апреле 1963 г. ВНИХФИ им. Орджоникидзе, Институт фарма-
кологии и химиотерапии АМН СССР
5-БРОМАЦЕТИЛ-З-НИТРОБЕНЗОЙНАЯ КИСЛОТА
И. Б. КРАСНОВ
СН2Вг
СО
C9HeOsNBr
/\/\
OaN СООН
М. в. 288, 07
5-Бромацетил-З-нитробензойная кислота (БНБ) применя-
ется в гистохимии в качестве реактива для выявления сульф-
гидрильных групп белков в срезах нефиксированной и фикси-
рованной ткани [1, 2J. Предложенный нами синтез 5-бромаце-
тил-3-нитробензойной кислоты проводится по схеме: 3,5-ди-
нитробензойная кислота -> 3,5-динитробензоилхлорид -> 3,5-
динитроацетофенон -> З-амино-5-нитроацетофенон -> нитрил
5-ацетил-З-нитробеизойной кислоты- -* 5-ацетил-З-нитробеи-
зойная кислота -> 5-бромацетил-З-нитробензойная кислота.
74
3,5-Динитробензоилхлорид [3], 3,5-динитроацетофенон [4] и
З-амино-5-нитроацетофенон [3] были синтезированы согласно
литературным данным с уточнением условий проведения ре-
акций.
Нитрил 5-ацетил-З-нитробензойной кислоты, 5-ацетил-З-
нитробензойная кислота и 5-бромацетил-З-нитробензойная
кислота синтезированы впервые.
Нитрил 5-ацетил-З-нитробензойной кислоты получен заме-
щением диазогруппы диазотированного З-амино-5-нитроаце-
тофенона на циангруппу в присутствии комплексной циани-
стомедной соли в качестве катализатора.
5-Ацетил-З-нитробензойную кислоту получали путем омы-
ления ее нитрила 50%-ной серной кислотой.
5-Бромацетил-З-нитробензойную кислоту синтезировали из
5-ацетил-З-нитробензойной кислоты бромированием послед-
ней в ледяной уксусной кислоте.
СИНТЕЗ 5-БРОМАЦЕТИЛ-З-НИТРОБЕНЗОЙНОИ КИСЛОТЫ
СООН СОС1
/А
I. || | + РС15 — || I + НС1 + РОС1,
/\/\
OjN NO2 O2N NO8
C2H5O t
II. 2 C,H6OH 4- Mg ^Mg + H2
C2H5OX
COO-C2H5
C2H5OX I
^Mg + CH2
C2H5O^ I
COO-C2H5
COO-C2H5
- C2H5OH + C2H6OMg- CH
I
COO-C2H6
COO-C2H5 COC1
C2H4OMg-CH + /4 -
COO-C2H6
OjN NO2
75
n°2 COO—C2H6
- o-c°Hh
n^2 COO-C2H6+C2H5OMgCl
2 C2H{OMgCl + H3SO4 -» 2 C2H5OH + MgCl2 + MgSO4
OsNx COO-C2HS
^-CO-CH 4- 2H2O
oZ coo-c2h5
o2n
/=\ t
-C° - CH3 + 2CaHfi0H + 2CO2
o2nZ
с2нБон + CH3COOH ch3-co-oc2h6 -4 h2o
no2
1IL CH3-CO-^-5
XN02
NH3
CH3-CO-(“)
XNOa
CH3
co
IV. /4 + HN^2
s\)\
OaN NH2-HC1
4- 3SnCia 4- 6HC1
4- 3SnCl4 4- 2H2O
ch3
co
Cl + 2Ha0
76
2
Cl + K[Cu(CN)2]
CH3
I
CO .
I t
* 2 Д + KC1 + CuCl + 2N2
AA
o2n cn
CH3 CHa
I I
V co co
I + 2H2O -> | + NH,
/\л /\Л
o2n cn o2n cooh
ch3
VI.
CH2Br
co
o2n cooh
o2n cooh
Характеристика основного сырья
Бром, ч., ГОСТ 4109—48.
3,5-Динитробепзойная кислота, ч., ГОСТ 136—51.
Диэтиловый эфир малоновой кислоты, ч., ВТУ МХП 4162—
51, перегнанный в вакууме при 93—94714 мм рт. ст.
Железо сернистое, ТУ МХП 87—52.
Калий цианистый, ГОСТ 8465—57.
Магний, стружки толщиной 0,25—0,5 мм.
Медь сернокислая, ч.д.а., ГОСТ 4165—48.
Натр едкий, ч.д.а., ГОСТ 4328—48.
Натрий азотистокислый, ч.д.а., ГОСТ 4197—48.
77
Натрий двууглекислый, ч., ГОСТ 4201—48.
Натрий хлористый, ч.д.а., ГОСТ 4233—48.
Олово двухлористое, ч.д.а., ГОСТ 36—40;
Серная кислота, ч., ГОСТ 4204—48, уд. вес. 1,835—1,840.
Соляная кислота, ч., ГОСТ 3118—46, уд. вес 1,174—1,185.
Углерод четыреххлористый, ч., ГОСТ 5827—51, обезвожен-
ный под углекислым калием.
Уксусная кислота ледяная, х. ч„ ГОСТ 61—51.
Фосфор пятихлористый, ч., ТУ МХП 386—41.
Хлороформ, ч.д.а., ГОСТ 3160—51, обезвоженный над уг-
лекислым калием (4:1).
Этиловый спирт, абсолютный (99,98%).
Этиловый эфир, наркозный.
Условия получения
Получение 3,5-динитробензоилхлорида. В круглодонную
колбу емкостью 1 л помещают 212,1 г (1,0 М) 3,5-динитробен-
зойной кислоты и 224 г (1,075 Л4) пятихлористого фосфора,
закрывают пробкой с хлоркальциевой трубкой и реакционную
смесь энергично встряхивают до получения однородной мас-
сы. После этого колбу снабжают обратным холодильником с
хлоркальциевой трубкой и нагревают на водяной бане при
70° до превращения реакционной смеси в однородную жид-
кость и прекращения выделения пузырьков хлористого водо-
рода. Не вошедший в реакцию избыток пятихлористого фос-
фора отфильтровывают на воронке Бюхиера и из прозрачно-
го золотистого цвета фильтрата при вакууме 35 мм и темпе-
ратуре бани 80—84° полностью отгоняют хлорокись фосфора.
Остающийся в колбе 3,5-динитробензоилхлорид застывает в
виде белой кристаллической массы с т. пл. 68—68,5°. По ли-
тературным данным, т. пл. продукта 68—69° [3]. Выход
230,6 г, что составляет 99% от теоретического. Вещество хра-
нят в вакуум-эксикаторе над прокаленным хлористым каль-
цием и без перекристаллизации используют для получения
3,5-динитроацетофенона.
Получение 3,5-динитроацетофенона. В круглодонную колбу
емкостью 1 л помещают 12,77 г (0,525 Af) магния, 116,7 мл
(2,0 А4) абсолютного этанола и 3,1 мл четыреххлористого уг-
лерода и реакционную смесь нагревают с обратным холо-
дильником при температуре бани 88—92° в течение 7 часов.
Образующийся этилат магния имеет белый цвет и сохраняет
очертания магниевых стружек (см. примечание 1).
К полученному этилату магния добавляют 78,95 мл
(83,29 г, 0,52 М) диэтилового эфира малоновой кислоты и
114 мл хлороформа и смесь нагревают 2,5 часа с обратным
холодильником при температуре водяной бани 78—82°. Маг-
ний, не успевший прореагировать с этанолом на предыдущей
7'8
Стадии, за время нагревания полностью превращается в эти-
лат магния, реагирующий затем с диэтилмалоновым эфиром.
Из образовавшейся густой однородной светло-желтой жидко-
сти, при вакууме 150 мм и температуре бани 78—82°, пол-
ностью отгоняют хлороформ, четыреххлористый углерод и
этанол. Оставшийся в колбе этоксимагниймалоновый эфир
медленно кристаллизуется в виде игл светло-желтого цвета.
Полученный этоксимагниймалоновый эфир растворяют в
90 мл хлороформа, раствор переносят в круглодонную колбу
емкостью 1 литр, снабженную мешалкой, обратным холодиль-
ником с хлоркальциевой трубкой и капельной воронкой, и кол-
бу помещают в ледяную баню. Перемешивая раствор и под-
держивая его температуру не выше 30°, в колбу в течение 30
минут приливают раствор 74,43 г (0,325 Л1) 3,5-динитробен-
зоилхлорида в 85 мл хлороформа, при этом первоначальный
светло-желтый цвет реакционной смеси переходит в красный.
При температуре реакционной среды 30° содержимое колбы
выдерживают в течение 1 часа, здтем охлаждают в ледяной
бане до 3—5° и к нему при перемешивании, поддерживая тем-
пературу реакционной массы не выше 20°, в течение 30 минут
приливают 161,5 мл холодной 21,5%-ной серной кислоты. Вы-
падающий белый осадок хлористого магния и сернокислого
магния растворяют добавлением 100 мл воды и смесь разде-
ляют в делительной воронке. Нижний, хлороформный, слой
переносят в круглодониую колбу емкостью 1 литр, откуда при
температуре бани 72—78° и вакууме 170—150 мм полностью
отгоняют хлороформ и воду. В колбе остается прозрачная
светло-желтая жидкость—3,5-динитробензоилмалоновый эфир.
К полученному 3,5-динитробензоилмалоновому эфиру до-
бавляют при размешивании последовательно '65 мл воды,
97,4 мл ледяной уксусной кислоты и 12.1 мл концентрирован-
ной серной кислоты, после чего реакционную смесь кипятят с
обратным холодильником на глицериновой бана при 123—
124° в течение 7,5 часов. По окончании омыления обратный
холодильник заменяют на нисходящий и .при вакууме 170—
150 мм и температуре бани 112—116° отгоняют этилацетат, ук-
сусную кислоту и воду. Оставшийся в колбе 3,5-динитроацето-
фенон еще горячим быстро выливают в колбу, содержащую
400 мл смеси воды со льдом (1:1), где он застывает в виде
светло-желтой кристаллической массы. Надосадочную жид-
кость декантируют. Осадок 3,5-динитроацетофенона сушат при
комнатной температуре, растирают в ступке, суспендируют в
1 литре воды и его водную суспензию нейтрализуют бикар-
бонатом натрия до pH 7,0. После нейтрализации осадок отса-
сывают на воронке Бюхнера, промывают тремя порциями воды
по 50 мл и сушат при комнатной температуре. Выход 55,4 г,
что составляет 81% от теоретического. Температура плавления
продукта 76—-78°. После перекристаллизации из небольшого ко-
79
личества этанола кристаллы имеют вид белых чешуек с т. пл.
80—81°. По литературным данным, т. пл. 3,5-динитроацетофе-
нона 74—76° [4], 82—84° [3].
Получение З-амино-5-нитроацетофенона. В круглодонной
колбе емкостью 1 литр, снабженной мешалкой, термометром,
капельной воронкой и обратным холодильником с хлоркаль-
циевой трубкой, при нагревании на водяной бане растворяют
в 800 мл абсолютного этанола 42,5 г (0,25 Л1) 3,5-динитроаце-
тофенона. Раствор 3,5-динитроацетофенона охлаждают в ледя-
ной бане до температуры 5—8° и к нему, непрерывно переме-
шивая и поддерживая температуру реакционной среды не вы-
ше 10°, в течение 30 минут добавляют холодный раствор 142,2 а
(0,75 М) безводного двухлористого олова (см. примечание 2)
в 200 мл абсолютного этанола, содержащего 60,0 а (1,64 М)
хлористого водорода (см. примечание 3). Затем реакционную
смесь нагревают при температуре бани 73—75° в течение 45
минут, при этом первоначальный желтый цвет смеси изменяет-
ся до темно-красного. По окончании нагревания реакционную
смесь переливают в круглодонную колбу емкостью 1 литр и
большую часть этанола отгоняют при температуре бани 70—
75° и вакууме 170—150 мм. Остаток выливают в колбу, содер-
жащую 2,5 л воды, и раствор освобождают от олова осажде-
нием его в виде сернистого олова, добавляя водный раствор
сероводорода до прекращения выпадения осадка. Одновремен-
но происходит распад двойной оловянной соли З-амино-5-нит-
роацетофенона. Осадок сернистого олова отсасывают на во-
ронке Бюхнера (см. примечание 4), фильтрат концентрируют,
упаривая в чашке на паровой бане до объема 500—600 мл, н
нейтрализуют 10%-ным раствором едкого натра (около
1000 мл) до pH 7,0—7,5 (см. примечание 5). Из охлажденно-
го нейтрализованного раствора в делительной воронке восемью
порциями диэтилового эфира, по 200 мл каждая, экстрагиру-
ют З-амино-5-нитроацетофенон. Эфирный экстракт сушат 3 ча-
са над свежепрокаленным хлористым кальцием и из него, при
температуре бани 50—60°, досуха отгоняют эфир. Остающигы
ся в колбе аморфный желтого цвета'осадок З-амино-5-нитро-
ацетофенона перекристаллизовывают из 100 мл воды. Выход
20,3 г (45% от теоретического). Продукт кристаллизуется в
виде игл лимонно-желтого цвета с т. пл. 155—157°. По лите-
ратурным данным, т. пл. вещества 156—158° [3].
Получение нитрила 5-ацетил-З-нитробензойной кислоты. В
колбе емкостью 1 литр в 540 мл воды суспендируют 18,1 г
(0,1 М) тонкоразмельченного З-амино-5-нитроацетофенона,
добавляют 20,8 мл (0,25 М) концентрированной соляной кис-
лоты, смесь охлаждают в ледяной бане и З-амино-5-нитроаце-
тофенон диазотируют при температуре 1—2°, прибавляя не-
большими порциями раствор 8,63 г (0,125 М) азотистокислого
натрия в 35 мл воды. После этого колбу с ее содержимым
80
энергично встряхивают 5—7 минут. Полученный диазораствор
отфильтровывают на воронке Бюхнера от не вошедшего в ре-
акцию З-амино-5-нитроацетофенона и 'маленькими порциями
добавляют к нагретому до 80° раствору комплексной цианисто-
медной соли (см. примечание 6), непрерывно перемешивая его
и нагревая так, чтобы температура реакционной смеси держа-
лась на уровне 68—73°. Наблюдается энергичное выделение
азота. После добавления раствора диазотата смесь охлажда-
ют, фильтруют и полученный нитрил экстрагируют из филь-
трата в делительной воронке 1,8 л (1,4 кг) эфира. Эфирный
экстракт сушат 3 часа над углекислым калием и эфир отго-
няют досуха при температуре бани 60—70°. Получают 6,0 г
(45%) кристаллического осадка нитрила 5-ацетил-З-нитро-
бензойной кислоты с т. пл. 94—95°. Аналитический образец
после двух последовательных перекристаллизаций из этано-
ла имеет т. пл. 97—98°. Бесцветные кристаллы нитрила раст-
воримы в хлороформе, эфире, ацетоне, этилацетате, этаноле,
горячей воде.
Найдено, %: С 57,14; Н—3,10; N—14,79.
CgHeO3N,. Вычислено, %: С—56,85; Н-3,18; N—14,73.
Получение 5-ацетил-З-нитробензойной кислоты. В кругло-
донную колбу емкостью 50 мл помещают 4,75 г (0,025 М)
нитрила 5-ацетил-З-нитробензойной кислоты и 28 мл 50%-ной
серной кислоты, колбу снабжают обратным холодильником и
реакционную смесь кипятят в течение 4 часов при температу-
ре масляной бани 162—172°. По окончании омыления реак-
ционную смесь охлаждают, выпавший темно-красный кри-
сталлический осадок 5-ацетил-З-нитробензойной кислоты от-
сасывают на воронке Бюхнера, дважды промывают на филь-
тре 20 мл воды (см. примечание 7) и сушат при комнатной
температуре. Полученный продукт перекристаллизовывают из'
250 мл воды. Выход 5-ацетил-З-питробензойной кислоты равен
3,44 г, что составляет 66% от теоретического; т. пл. 171 —172°.
После 3 последовательных перекристаллизаций из ледяной
уксусной кислоты вещество имеет т. пл. 174—174,5° и по вне-
шнему виду это бесцветные кристаллы, растворимые в этил-
ацетате и уксусной кислоте и плохо растворимые в воде, бен-
золе, четыреххлористом углероде, петролейном эфире, хлоро-
форме.
Найдено, %; С-52,08; Н—3,35; N 7,26.
CgH?O5N. Вычислено, %: С —51,68; Н—3,37; N-6,69.
Получение 5-бромацетил-З-нитробензойной кислоты. В
круглодонной колбе емкостью 50 мл растворяют 1,045 г
(0,005 Af) 5-ацстил-З-нитробензойной кислоты в 15 мл ледя-
ной уксусной кислоты. К раствору по каплям, при перемеши-
вании, добавляют 0,799 г (0,005 М) брома (2,56 мл раствора
брома в ледяной уксусной кислоте (1: 10 по объему) и реак-
81
ционную смесь нагревают с обратным холодильником при
температуре бани 65—75° до исчезновения окраски брома.
После этого уксусную кислоту отгоняют в вакууме при 80—
60 леи, температуре бани 66° и температуре паров 55° до пре-
кращения отгонки. Оставшуюся на дне колбы густую светло-
коричневую жидкость выливают на часовое стекло, где она
спустя 20—30 минут начинает кристаллизоваться. Продукт
трижды перекристаллизовывают каждый раз из 130 мл воды.
Выход 5-бромацетил-З-нитробензойной кислоты равен 0,483 г,
что составляет 35% от теоретического. По внешнему виду это
бледно-желтые кристаллы с т. пл. .148,5—149,5°. После допол-
нительной перекристаллизации из воды т. пл. продукта
149,5—150,5°. 5-Бромацетил-З-нитробензойная кислота раство-
рима в ацетоне, этаноле, этилацетате, уксусной кислоте, не-
растворима в воде, хлороформе, четыреххлористом углероде и
бензоле.
Найдено, %: С-37,74; Н—2,11; N— 4,94; Вг—27,72.
CBH,O5NBr. Вычислено, %: С-37,52; Н—2,099; N-4,86; Вг-27,74.
Примечания: 1. В результате реакции между спиртом и магни-
ем образуется водород. За ходом реакции следят по выделению пузырь-
ков водорода через склянку Тищенко, наполненную концентрированной
серной кислотой и соединенную резиновой трубкой через пустую предо-
хранительную склянку с верхним концом холодильника. В процессе реак-
ции не следует вести перемешивание, Реакция между спиртом и магнием
идет в парах.
2. Для получения безводного двухлорнстого олова 300 г кристалличе-
ского двухлористого олова высыпают в фарфоровую чашку и на голом
огне под тягой постепенно нагревают до 1357 При 35° соль становится
жидкой. Температура 135° поддерживается до тех пор, пока вещество при
постоянном помешивании не приобретет консистенцию крутого теста. За-
тем чашка с хлористым оловом переносится для охлаждения в эксикатор
с хлористым кальцием. После охлаждения куски застывшей массы дву-
хлористого олова растирают в ступке и полученный порошок хранят в ва-
куум-экснкаторе над хлористым кальцием. ' \
3. Хлористый водород, полученный при действии концентрированной
серной кислоты на хлористый натрий, сушат, пропуская через колонку с
прокаленным хлористым кальцием, и затем пропускают через абсолютный
этанол, осуществляя контроль за степенью насыщения титрованием ще-
лочью, для чего 0,1 мл анализируемого спирта добавляют к 9,9 мл воды
и полученный раствор тнтруют с фенолфталеином 0,01 н. раствором NaOH.
4. Для ускорения процесса фильтрования осадок сернистого олова от-
сасывают одновременно на 2 воронках Бюхнера диаметром 17 см (двой-
ной фильтр!).
5. Для улучшения выхода З-амиио-5-нитроацетофенона желеобраз-
ный осадок сернистого олова трижды кипятят с 200 мл воды, отсасывают
на воронке Бюхнера н фильтраты присоединяют к основному фильтрату.
6. Вследствие выделения в ходе синтеза отравляющих веществ — ди-
цнана н синильной кислоты — синтез проводят только в вытяжном шкафу
с включенной вытяжной системой и при наличии противогаза. Необходи-
мы резиновые перчатки, клеенчатый фартук.
Для получения раствора комплексной цианистомедной соли в колбе
емкостью 2 литра при нагревании на водяной бане растворяют 90 г
(0,36 Л1) кристаллической сернокислой меди в 340 мл воды и к раствору,
поддерживая его температуру на уровне 75—80°, небольшими порциями при
размешивании приливают раствор 108 г (1,67 М) цианистого калия в
82
170 мл воды. Выпадающий вначале осадок цианистой меди по мере до-
бавления цианистого калия переходит в раствор с образованием комплек-
сной цианистомедной соли.
7. Для улучшения выхода 5-ацетил-З-нитробензойной кислоты маточ-
ный раствор после добавления к нему 20 мл воды оставляют на 12—15 ча-
сов при 2—4°. Выпавший хлопьевидный осадок отсасывают и присоеди-
няют к основному осадку 5-ацетил-З-нитробензойной кислоты.
Литература
1. И. Б. Краснов, Цитология, 3, № 5, 608 (1961).
2. И. Б. К р а с н о в, Ж. общ. химии, 32, № 1, 293 (1962).
3. L. В erend, F. Heymann, J. Pract. Chem., 69, 11, № 10 II, 449
(1904).
4. M. S u z u k 1, B. S h i m i z u, J. Pharmac. Soc. Japan, 73, № 3, 392
(1953).
Поступила в апреле 1963 г, Институт мозга АМН СССР
N-ЭТИЛ МАЛ ЕИ НИМИ Д
И. Б. КРАСНОВ
,с-сн
CH3-CH2-N< II
хс-сн
II
о
CeH,NO2
М. в. 125,13
N-этилмалеинимид применяется в биохимии [1, 2] и гисто-
химии [3] в качестве реактива, специфически реагирующего с
сульфгидрильными группами белков и низкомолекулярных
меркаптанов. По литературным данным, N-этилмалеинимид
подучают из этиламина и малеинового ангидрида с после-
дующей циклизацией получающейся N-этилмалеиновой кис-
лоты [4].
При проверке указанной прописи нами уточнены условия
проведения реакций и выделения продуктов. На стадии цик-
лизации N-этилмалеиновой кислоты в качестве дегидранта
была использована пятиокись фосфора, что позволило полу-
чить N'-этилмалеинимид с более высоким выходом.
S3
СИНТЕЗ N-ЭТИЛМАЛЕИНИМИДА
О
II
,с-сн
I. CH8-CH2-NH2 + O( II
ХС-СН
II
о
н о
CH8-CH2-N-C—сн = сн-соон
н о
I II Р2О5
П. CH9-CH5-N-C-CH^CH-COOH------
о
II
с-сн
ch8-ch2-n; и +2нро8|
хс-сн
Характеристика основного сырья
Диоксан, ч., ВТУ МХП 3111—52.
Кали едкое, ч.д.а., ГОСТ 4203—48.
Малеиновый ангидрид, ч.д.а., ГОСТ 5854—51.
Фосфорный ангидрид, ВТУ 521—41.
Этиловый спирт, абсолютный (99,98%).
Этиламин солянокислый, ч., ВТУ МАП 1006—53.
Условия получения
Получение N-этилмалеиновой кислоты. В трехгорлую круг-
лодонную колбу емкостью 250 мл, снабженную мешалкой,
термометром и капельной воронкой и погруженную в охлаж-
дающую смесь, помещают охлажденный до температуры —5°
раствор 31,86 мл (22,5 а; 0,5 М) этиламина (см. примеча-
ние 1) в 60 мл абсолютного этанола и к нему, при размеши-
вании, поддерживая температуру среды не выше —5°, по кап-
лям? в течение 30 минут, прибавляют раствор 49,0 г (0,5 М)
малеинового- ангидрида в 65 мл абсолютного диоксана (см.
примечание 2). По окончании прибавления раствора малеи-
нового ангидрида начинает выпадать белый кристаллический
осадок N-этилмалеиновой кислоты. Реакционную смесь ос-
81
тавляют на 24 часа при температуре 2—4°, выпавший осадок
отфильтровывают на воронке Бюхнера и сушат при 50—60°.
Получают 44,25 з (61,8%) N'-этилмалеиновой кислоты с т. пл.
121 —123°, которую без последующей очистки используют для
получения У-этилмалеинимида. После двукратной перекри-
сталлизации из этанола т. пл. продукта 124—125°; по литера-
турным данным, —126° [4].
Получение N-этилмалеинимида. В круглодонную с корот-
ким горлом колбу емкостью 50 мл помещают 14,31 г (0,1 ЛГ)
N-этилмалеиновой кислоты и 14,92 г (0,105 М) фосфорного
ангидрида (см. примечание 3). Колбу снабжают нисходящим,
длиной 10 см, воздушным холодильником, подключенным к
вакуумному насосу, и нагревают на масляной бане, быстро
поднимая температуру до 200—210°. С момента начала кон-
денсации паров М-этилмалеинимида в холодильнике включа-
ют вакуумный насос и при вакууме 15—10 мм отгоняют
прозрачную бесцветную жидкость, застывающую при охлаж-
дении в сплошную кристаллическую массу снежно-белого
цвета. Выход N-этилмалеинимида 5,72 а, что составляет
46,74% от теоретического; т. пл. 44°. По литературным дан-
ным, т. пл. продукта 45,5° [4] (см. примечание 4).
Примечания: 1. Для получения этиламина в двугорлую кругло-
донную колбу емкостью 500 мл, снабженную капельной воронкой и газо-
отводной трубкой, помещают 163,1 г (2,0 М) сухого солянокислого этилами-
на и.к нему, по каплям, прибавляют 230 мл (115,02 г, 2,05 М) 50%-ного
раствора едкого кали. Затем колбу с ее содержимым нагревают при тем-
пературе бани 80—90° в течение 5 часов, Газообразный этиламин, выде-
ляющийся из реакционной массы в виде пузырьков газа, сушат, пропуская
через склянку Тищенко, наполненную свежепрокаленным хлористым каль-
цием, и сжижают при температуре минус 15—18° в змеевидном приемни-
ке, погруженном в охлаждающую смесь (спирт + твердая углекислота) в
сосуде Дюара. Сообщающийся с атмосферным воздухом конец змеевико-
вого приемника снабжают хлоркальциевой трубкой, так как этиламин
жадно притягивает влагу из воздуха. Получают 51 мл (36,0 г) прозрач-
ной бесцветной, пахнущей аммиаком жидкости с уд. в. 0,706.
2. Для абсолютизации диоксан кипятят с натрием в течение 12 часов
(обратный холодильник с хлоркальциевой трубкой) и перегоняют над
натрием, собирая фракцию, кипящую при температуре ЮГ.
3. Для поддержания равномерного кипения в вакууме на дно кол-
бы помещают стеклянную вату.
4. N-этилмалеинимид очень гигроскопичен. Полученный продукт хра-
нят в вакуум-эксикаторе над концентрированной серной кислотой.
Литература
1. Е. Roberts, G. Rous er, Analyt, Chem.. 30, № 7, 1921 (1958).
2. F. L e s 1 i e, D. Williams, Q. G о r i n, Analyt. Blochem., 3, № 3,
257 (1962).
3. Э. Пирс, Гистохимия, M., ИЛ, 1962, стр. 718.
4 A. Piutti, Е. G i u s 11 n I a n 1, Gazz. chim. ital., 26, № 5—6,431
(18S6).
Поступила в апреле 1963 г. Институт мозга АМН СССР
85
N-Я-ХЛОРФЕНИЛ-ДИАМИД ФОСФОРНОЙ КИСЛОТЫ
М. Н. МАЛЕВ
О
Cl-\ /-N-P^OH
н nh2
CeH8ClN2OaP
М. б. 206,57
N'-n-Хлорфенил-диамид фосфорной кислоты применяется в
гистохимии в качестве субстрата для выявления активности
фосфоамидаз [1].
По литературным данным, N-n-хлорфенил-диамид фос-
форной кислоты получают-при действии на п-хлоранилидо-
фосфорилдихлорид [2] водного раствора аммиака [3].
Нами был получен продукт по этому методу.
СИНТЕЗ N-Я -ХЛОРФЕНИЛ-ДИАМИДА ФОСФОРНОЙ КИСЛОТЫ
I. С1-(“^-NHS + НС1 -> С1-^~^-NH2 HC1
11. Cl-(“ ^-nh2-hci + РОС13 —
— О
ci-O-NH-p-cl +2hci
\1
о
III. С1-(“ NHP—Cl-{-NHtOH —
Xci
О
— Cl-^—^-NH-P-OH 4- 2НС1
nh2
Характеристика основного сырья
n-Хлоранилин, ч„ СТУ 79-484-х—60.
Натрий хлористый, ч., ГОСТ 4233—48.
86
Серная кислота, ч., ГОСТ 4204—48.
Хлорокись фосфора, ч., ВТУ У 166—51.
Аммиак водный, 25%-ный, ч.д.а,, ГОСТ 3760—47.
Условия получения
Получение солянокислого п-хлоранилина. В двугорлую
круглодонную колбу емкостью 1 литр, снабженную газопод-
водящей и отводной трубками, помещают раствор 64 г
(0,5 М) n-хлоранилина в 500 мл этилового спирта и пропу-
скают струю сухого хлористого водорода (см. примечание 1).
При насыщении раствора хлористым водородом выпадает
желтый кристаллический осадок солянокислой соли п-хлор
анилина. Полученный осадок отсасывают, промывают на
фильтре 50 мл эфира и сушат на воздухе. Выход 74 г, что со-
ставляет 90% от теоретического.
Получение п-хлоранилидофосфорилдихлорида. В кругло-
донную колбу емкостью 500 мл, снабженную обратным холо-
дильником с хлоркальциевой трубкой, помещают 74 г
(0,45 М) хлоргидрата n-хлоранилина и 82 мл (0,9 Л1) хлор-
окиси фосфора. Полученную смесь нагревают на масляной
бане при температуре около 130° в течение 4 часов. В ре-
зультате реакции образуется прозрачная светло-коричневая
жидкость, которую выливают при размешивании в колбу, со-
держащую 400 мл петролейного эфира. Выпавший белый оса-
док п-хлоранилидофосфорилдихлорида отфильтровывают на
воронке Бюхнера, промывают 100 мл петролейного эфира и
помещают в вакуум-эксикатор над смесью едкого кали с
парафином (см. примечание 2).
Выход п-хлоранилидофосфорилдихлорида 102 г, что со-
ставляет 93% от теоретического; т. пл. продукта 102—104°,
что соответствует литературным данным [2].
Получение N-n-хлорфенил-диамида фосфорной кислоты.'В
фарфоровый стакан емкостью 100 мл помещают 50 мл
(0,67 Л1) водного аммиака, охлажденного до 5° и небольши-
ми порциями (по 0,5 г) добавляют 24,5 г (0,1 М) п-хлораии-
лидофосфорилдихлорида. Реакционную массу размешивают
стеклянной палочкой и следят за тем, чтобы температура не
поднималась выше 10°. По добавлении последней пор-
ции п-хлоранилидофосфорилдихлорида полученный раст-
вор фильтруют на воронке Бюхнера, фильтрат нейтрализуют
до pH 3 1 н. соляной кислотой. Выделившийся при нейтрали-
зации белый хлопьевидный осадок N-n-хлорфенил-диамида
фосфорной кислоты отсасывают, промывают па фильтре пос-
ледовательно 200 мл холодной воды, 50 мл горячей воды,
25 мл этилового спирта и, наконец, 50 мл эфира.
Выход продукта 12 г, что составляет 60% от теоретиче-
ского; т. пл. 153—154°, что соответствует литературным дан-
ным [3].
87
Примечания: 1. Хлористый водород получают при добавлении
серной кислоты к хлористому натрию [4].
2. д-Хлоранилидофссфорилдих.торид хранят в вакуум-экснкаторе, так
как он разлагается влагой воздуха. Продукт легкорастворим в эфире,
хлороформе, кристаллизуется из бензола в виде игл с т. пл. 107° [2].
Литература
1. J. Meyer, J. Р. W е i n tn a n, J. Histochem. a. Cytochem., 3, 134
(1955).
2. Р. Otto, Вег., 28, 616 (1895).
3. К. Rorig. J. Amer. Chem. Soc., 71, 3561 (1949).
4. Ю. В. Карякин, И. И. Ангелов, Чистые химические реакти-
вы, М., Госхимиздат, 1955, стр. 283.
Поступила в апреле 1963 г. Институт мозга АМН СССР
4-AMHHO-1-N, N-ДИМЕТИЛНАФТИЛАМИН
СОЛЯНОКИСЛЫЙ
N,N -Диметил-1,4-иафтилендиамин
Al. Н. МАЛЕВ
C11H15N2CI
NH2-HC1
М. в. 222,72
4-Amhho-1-N,N' -диметилнафтиламин (АДН) в виде хлори-
стоводородной соли применяется в гистохимии [1] и биохимии
[2] в качестве реактива для определения активности цитохро-
моксидазы.
По литературным данным [1, 3], АДН получают из диме-
тил-а-нафтиламина путем азосочетания с диазотированным
сульфанилатом натрия и последующим восстановлением по-
лученного аминоазосоединения (азокраски) хлористым оло-
вом в кислой среде. В качестве реакционной среды при вос-
становлении азокраски берут уксусную кислоту [1] или соля-
88
ную [3]. Более удобной является последняя методика, с
мы проверили и уточнили условия выделения конечное р
дукта.
СИНТЕЗ 4-АМИН04-Ы,Ь1-ДИМЕТИЛНАФТИЛАМИНА
СОЛЯНОКИСЛОГО
2HaN-^ ^-SO3Na + 2NaNO2 + 3H2SO4
N = N—^~SO3H SO4-bNa2SO4+-4H3O
= ' Ja
III.
2 SnCl2 4- 6 HC1
,CH3 "
N<
]XCH3
NHj-HCl
NHa-HCl
+ Sn Cl4
89
/CHS
N\
I XCH3
IV.
• SnCl4 + 5NaOH
NHaHCl
,CH8
N(
I XCH3
/Х/Х
I || I +5NaCl 4-Sn(OH)4 + H20
x/w
NHa
,CH3
N\
I XCH3
w Z\/X , Hri
V. | || | ~T HC1
VV
NHS
NH2.HC1
Характеристика основного сырья
Диметил-а-нафтиламин, ч., ВТУ ХСНХ 43—58, т. кип.
272—274°.
Натрий сульфаниловокислый, ч.д.а., ВТУ МХП 2964—51.
Кислота серная, х.ч., ГОСТ 4204—48.
Олово двухлористое, крист., ч.д.а., ГОСТ 36—40.
Натр едкий, х.ч., ГОСТ 4328—48.
Эфир диэтиловый, наркозный.
Уксусная кислота, 98%-ная, х.ч., ГОСТ 61—51.
Хлористый водород (см. синтез N-n-хлорфенил-диамида
фосфорной кислоты).
Условия получения
Получение соли диазония сульфаниловой кислоты. В фар-
форовый стакан емкостью 500 мл, снабженный мешалкой, по-
мещают 120 мл воды, 23 г (0,1 М) кристаллической натрие-
вой соли сульфаниловой кислоты, 7,3 г (0,105 М) нитрита
натрия. Полученный раствор охлаждают до 5—10° и к нему,
при размешивании, добавляют 120 мл 30 %-ной серной кис-
90
лоты. Выпавший белый кристаллический осадок является
солью диазония. Осадок отфильтровывают, промывают, на
фильтре 20 мл воды и используют при сочетании с диметил-а-
нафтиламином.
Получение п-сульфокислоты бензолазодиметилнафтилами-
на. В фарфоровом стакане емкостью 500 мл растворяют 17,1г
Диметил-а-нафтиламина в 100 мл уксусной кислоты и прили-
вают при размешивании суспензию диазосоединения в 150 мл
уксусной кислоты. Реакционную смесь размешивают 1 час
при комнатной температуре и оставляют на ночь при 2—5°.
Затем отсасывают выпавший осадок темно-фиолетового цве-
та, промывают на фильтре 100 мл воды. Полученную азо-
краску кипятят 5—10 минут в 200 мл дистиллированной воды
(для разложения и удаления не вошедшего в реакцию диазо-
соединения), отфильтровывают на воронке Бюхнера и сушат
на воздухе. Выход 25 г (70,5%) п-сульфокислоты бензолазо-
диметилнафтиламина с т. пл. 265—266°, что соответствует
литературным данным.
Получение 4-амино-1-П,П-диметилнаф1иламина. В кониче-
ской колбе емкостью 1 литр помещают 10,7 г (0,03 Л1) п-суль-
фокислоты бензолазодиметилнафтиламина и при размешива-
нии добавляют раствор, содержащий 10,2 г (0,04 М) кри-
сталлического хлористого олова в 50 мл соляной кислоты.
Полученную смесь выдерживают до исчезновения фиолетовой
окраски раствора, па что затрачивается около двух часов (см.
примечание 1). В результате реакции выпадает белый осадок
двойной оловянной соли 4-амино-1-Н,1\-диметилнафтиламина.
Суспензию охлаждают в бане с ледяной водой, добавляют
к ней 150 г толченого льда и 250 мл диэтилового эфира (см.
примечание 2). Затем к полученной смеси добавляют по кап-
лям, при размешивании; около 120 мл 5 н. едкого натра до
получения щелочного раствора с pH 10. При расщеплении
двойной оловянной соли образуется свободное основание
4-амино-1-Ы,М’-диметилнафтиламина, которое экстрагируется
имеющимся в с,меси эфиром.
Эфирный экстракт отделяют на делительной воронке ем-
костью 1 литр, помещают в коническую колбу и сушат све-
жепрокаленным хлористым кальцием в течение 2 часов (см.
примечание 3). Эфирный экстракт амина фильтруют и насы-
щают сухим хлористым водородом до обесцвечивания свет-
ло-желтого раствора. Выпавший белый хлопьевидный осадок
солянокислого 4-амино-1-Н,№-диметилнафтиламииа оставляют
на ночь в холодильнике при температуре около 5°. На сле-
дующий день декантируют эфир с осадка, приливают к осад-
ку еще 200 мл эфира, суспендируют в нем продукт и после
отстаивания снова декантируют эфир. Промытую соль ами-
на помещают в круглодонную колбу емкостью 250 мл и от-
гоняют эфир досуха на водяной бане с терморегулятором при
91
температуре 50—60° (см. примечание 4). Выход солянокисло-
го 4-амино-1-М,М-диметилнафтиламина равен 3 г, что состав-
ляет 45% от теоретического; т. пл. 204—206° (см. примеча-
ние 5). Продукт гигроскопичен, хорошо растворим в спиртах,
воде, нерастворим в эфире.
Примечания: 1. Азокраска должна быть хорошо растерта, иначе
отдельные крупинки ее долго не растворяются. Для ускорения реакции
восстановления колбу можно поместить в горячую водяную баню.
2. Диэтиловый эфир, находящийся в верхнем слое, препятствует
окислению свободного основания амина, образующегося при добавлении
раствора щелочи.
3. Дольше сушить эфирный экстракт не следует, так как амин, на-
ходящийся в растворе, со временем окисляется.
4. При работе с эфиром не следует допускать в помещении приме-
нения открытого огня. При фильтровании эфирной суспензии на воронке
Бюхнера продукт окисляется под воздействием влаги воздуха.
5 Продукт хранят в запарафинированной банке. По литературным
данным, солянокислый 4-амино-1-Ь1,Н-диметилнафтиламнн имеет т. пл.
210° [4].
Л итература
1. М. М. Nachlas и др., J. Histochem. a. Cytochem., 6, 445 (1958).
2. V. V. Portugalov, Е. L. Davedova, V. G. Skrebitsky,
J. Histochem. a. Cylochem, 10, 213 (1962).
3. Y. Yasumasa, Med. J. Osaka Univ., 11, 388 (1961).
Поступила в апреле 1963 г. Институт мозга АМН СССР
АЦЕТИЛТИОХОЛИНЙОДИД
М Н. МАЛЕВ
[CH,-C<-S-CH2-CH2-N(CH8)3] J
CTH16JNOS м. в. 289, 1g
Ацетилтиохолинйодид применяется в гистохимии в качест-
ве субстрата при определении истинной холинэстеразы [1].
11о литературным данным, ацетилтиохолинйодид можно
получить по лногостадийному методу Реншоу и др. [2], а так-
же по более простому методу, предложенному Ивиным [3].
Мы проверили последний метод и уточнили условия неко-
торых его стадий.
92
СИНТЕЗ АЦЕТИЛТИОХОЛИНЙОДИДА
Р
I. (СН8СО)2О + J8-► 2СН3С^
П. СНа—СНа 4- KSCN -> СН2-СН2 + KCNO
с/ S
.0 /О
1П. сн,с^ + сн2-сн2 -> ch3c£s-ch3-ch2j
XJ \ /
S
о
IV. CHgC^S-CHa-CH2J + N(CH3)S ->
.°
-> [CH3(Z —S-CHs-CHa-N(CH8)3]J
Характеристика основного сырья
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52, свежеперег-
нанный, т. кип. 139—14Г.
йод металлический, ч.д.а., ГОСТ 4159—48, мелкорастер-
тый и высушенный над серной кислотой в вакуум-эксикаторе.
Фосфор красный, ч., ТУ МХП 1360—46, высушенный над
серной кислотой в вакуум-эксикаторе.
Окись этилена, ГОСТ 7568—55, т. кип. 13,5°.
Калий роданистый, ч.д.а., ГОСТ 4139—48.
Триметиламин солянокислый, ч., ВТУ МХП 2939—51.
Условия получения
Получение ацетилйодида. В круглодонную колбу емкостью
250 мл, снабженную обратным холодильником с хлоркаль-
циевой трубкой, помещают 55,5 мл (0,58 М) уксусного ангид-
рида и 16 з (0,51 М) красного фосфора. К полученной смеси
прибавляют небольшие порции (по 5—10 г) 152 г (0,6 М)
йода в течение 1 часа. После добавления каждой порции йода
реакционную смесь энергично встряхивают. Затем колбу по-
мещают в масляную баню и нагревают 1 час при температур
ре около 100°. Полученный в результате реакции ацетилйодид
отгоняют, меняя обратный холодильник на прямой и повы-
шая температуру масляной бани до 140°. Продукт собирают
с температурой паров 100—110° (см. примечание 1). Полу-
ченную жидкость, окрашенную в красный цвет арцмесыо
93
йода, перегоняют под вакуумом, собирая фракцию, кипящую
при 34—35755.мм.
Выход ацетилйодида равен 75 г, что составляет 76% от
теоретического. По внешнему виду вещество представляет
собой почти бесцветную жидкость с уд. в. 1,98.
Получение тиоокиси этилена. В плоскодонной колбе ем-
костью 500 мл растворяют 90 г (0,93 М) роданистого калия
в 90 мл воды. Полученный раствор охлаждают до температу-
ры минус 20° и к нему приливают в три приема 90 мл (1,8 ЛГ)
окиси этилена (см. примечание 2). Колбу закрывают корко-
вой пробкой н реакционную массу выдерживают два часа
при минус 10—157 Затем колбу вынимают из охладительной
смеси и оставляют при комнатной температуре на два часа,
причем периодически приоткрывают пробку для удаления не
вошедшей в реакцию окиси этилена (см. примечание 3).
Полученная тиоокись этилена не смешивается с водой и
находится в верхнем слое жидкости. Ее отделяют в делитель-
ной воронке, помещают в плоскодонную колбу емкостью
250 мл, сушат над хлористым кальцием в течение 2—3 часов
и перегоняют, собирая фракцию с т. кип. 46—577 Выход 23 г
(41%) тиоокиси этилена с уд. в. 1,0 (см. примечание 4).
Получение 2-йодэтилтиолацетата. В круглодонную колбу
емкостью 250 мл, снабженную обратным холодильником с
хлоркальциевой трубкой и капельной воронкой, помещают
60 г (0,35 М; 30,3 мл) ацетилйодида и 100 мл четыреххлористо-
го углерода. Колбу с полученным раствором помещают в ба-
ню с ледяной водой и добавляют по каплям, в течение 15 ми-
нут, 22 мл (0,37 М) тиоокиси этилена. Затем реакционную
массу нагревают 1 час на горячей (50—60р) водяной бане,
меняют обратный холодильник на прямой и отгоняют че-
тыреххлористый углерод на кипящей водяной бане. Остав-
шийся в колбе 2-йодтиоэтиловый эфир уксусной кислоты пе-
регоняют при пониженном давлении, собирая фракцию, кипя?
щую в пределах 95—97/10 мм или 79—81/5 мм. Выход про-
дукта 59 г (73%) в виде прозрачной бесцветной жидкости с
уд. в. 1,82.
Получение ацетилтиохолинйодида. В круглодонную кол-
бу емкостью 500 мл, снабженную стеклянной притертой проб-
кой, помещают 56 г (0,29 М\ 30,7 мл) 2-йодэтилтиолацетата и
150 мл сухого диэтилового эфира. Полученный раствор ох-
лаждают до температуры —20° и прибавляют порциями (при-
мерно по 3 мл) 18 г (0,3 М\ 27,5 мл) жидкого триметиламина
(см. примечание 5). После прибавления каждой порции три-
метиламина колбу закрывают притертой пробкой и взбалты-
вают реакционную смесь. Затем колбу герметизируют проб-
кой, которую закрепляют к горлышку колбы изоляционной
лентой, и оставляют при температуре минус 10—15° на одни
сутки. По истечении этого срока начинает выделяться ацетил-.
94
тиохолинйодид в виде белой аморфной массы. Колбу выдер-
живают еще 10—12 дней при комнатной температуре, от-
фильтровывают на воронке Бюхнера полученный белый оса-
док, промывают его па фильтре 200 мл эфира, трижды пере-
кристаллизовывают из 150 мл абсолютного этилового спирта
(см. примечание 6). Выход ацетилтиохолинйодида равен
58 г, что составляет 70% от теоретического. Препарат имеет
вид бесцветных кристаллов с т. пл. 203—204°, что соответст-
вует литературным данным [3].
Примечания: 1. Ацетилйодид на воздухе, взаимодействуя с вла-
гой, сильно дымит; при хранении окрашивается в бурый цвет. От выде-
лившегося йода его очищают встряхиванием с сухой ртутью, а затем от-
деляют от йодистой ртути на стеклянном фильтре № 4.
2. Жидкую окись этилена получают, пропуская газообразную окись
этилена из баллона в змеевиковый приемник, помещенный в сосуд Дюара
с охлаждающей смесью (от —20 до —40е1). Эту смесь готовят растворе-
нием размельченной твердой углекислоты в ацетоне. Окись этилена кон-
денсируется в приемнике в виде бесцветной прозрачной жидкости с уд. в.
0,866.
3. При достижении в реакционной массе температуры 13° начинает
бурно выделяться окись этилена, поэтому колбу на некоторое время по-
мещают в ледяную воду.
4. Все работы, связанные с тиоокисью этилена, необходимо прово-
дить под тягой.
Тноокись этилена легко полимеризуется с образованием белого осад-
ка, поэтому хранить ее более 1 суток не рекомендуется. Посуду осторож-
но дегазируют теплой концентрированной азотной кислотой.
5. Жидкий трнметиламин получают следующим образом: в колбу
Вюрца емкостью 250 мл, снабженную капельной воронкой, помещают 40 г
(0,42 Л4) солянокислого триметиламина и добавляют по каплям 50%-ный
раствор едкого кали. Выделяющийся газообразный триметиламнн сушат,
пропуская через склянку Тищенко с кристаллическим едким натром, и
собирают в змеевидном приемнике, охлаждаемом в сосуде Дюара до
—40°. Получают 18,5 а триметиламина в виде бесцветной прозрачной жид-
кости с уд. в. 0,66 и т. кнп. 3,2’, хорошо растворимой в эфире, спирте,
хлороформе.
6. При. вскрытии колбу помещают в баню с ледяной водой для кон-
денсации газообразного триметиламина, взятого на реакцию в избытке.
Для перекристаллизации ацетилтиохолинйодида можно также ис-
пользовать спирты пропиловый, бутиловый, амиловый.
Литература
1. Q. В. Koelle, J. Pharmacol, a. Exper. Therap., 100, 158 (1950).
2. R. R. R е n s h о w, Р. F. Dreichbach, J. Amer. Chem. Soc., 60,
1765 (1938).
3. С. 3. Ивин, Ж. общ. химии, 22, 267 (1952).
Поступила в апреле 1963 г.
Институт Мозга АМН СССР
БУТИРИЛ ТИ0X0Л И НЙОД ид
М. Н. МАЛЕВ
О
[CH3-CH2-CH2-C^S-CH2-CH2-N(CH3)3]J
CeH20JNOS М. в. 317, 24
Бутирилтиохолинйодид применяется в гистохимии в каче-
стве субстрата при определении псевдохолинэстеразы [1].
Ранее бутирилтиохолинйодид получали по многостадийно-
му методу Реншоу и др. [2]. В 1952 году Ивин разработал но-
вый, более простой метод синтеза тиоацетилхолинйодида [3],
который оказался общим для синтеза тиохолиновых эфиров
карбоновых кислот [4].
Нами получен бутирилтиохолинйодид по методу Ивина.
СИНТЕЗ БУТИРИЛТИОХОЛИНЙОДИДА
I. СН3-СН2-СНа-СООН + NaOH —
СН8—СН2—СНа—COONa + Н20
II. СН8—СН2—СН2—COONa + J2
,0
СНз-СН2-СН2-С^ -ф-NaJ
XJ
О
ш. ch3-ch2-ch2-cAj + СН2-СН2 ->
ch9-ch2-chs-c/-s-ch2-ch2j
IV. СН,-СН2-СНа-C^-S-CH2-CH2J + N(CH3)j -
Z>O +
-> [CH3-CH2-CHs-C^-S-CH2-CH2-N(CH3)3]J
Характеристика основного сырья
н-Масляная кислота, ч„ ТУ МХП 41—47.
Натр едкий, ч.д.а., ГОСТ 4328—48.
Йод металлический, ч.д.а., ГОСТ 4159—48 (см. синтез
ацетилтиохолинйодида).
Фосфор красный, ч., ТУ МХП 1360—46 (см. там же).
96
Т к ь этилена (см. там же).
1риметиламин жидкий т кип 3,2°, уд- в. 0,66 (см. там
же). ’ ‘
Углерод четыреххлористый, ч.д.а., ГОСТ 5827 51.
Эфир диэтиловый, наркозный, высушенный над металли-
ческим натрием и перегнанный.
Условия получения
Получение натриевой соли масляной кислоты. В фарфоро-
вый стакан емкостью 1 литр, содержащий 200 г масляной
кислоты, прибавляют раствор 95 г едкого натра в 150 мл во-
ды. Полученный слабощелочной раствор охлаждают, выпав-
ший белый осадок натриевой соли масляной кислоты отфиль-
тровывают на воронке Бюхнера, промывают на фильтре
20 мл воды, сушат при 80—90° и тщательно растирают в
ступке.
Выход 150 г соли, что составляет 60% от теоретического.
Получение бутирилйодида [5]. В круглодонную короткогор-
лую колбу емкостью 250 мл, снабженную холодильником с
хлоркальциевой трубкой, помещают 55 г (0,5 М) порошко-
образной натриевой соли масляной кислоты, 8 г красного
фосфора и встряхивают до получения однородной массы. За-
тем добавляют небольшими порциями (по 5—10 г) в течение
1 часа 127 г (0,5 Л1) йода. После добавления каждой порции
йода колбу закрывают, а реакционную смесь встряхивают
(см. примечание 1). Полученную смесь нагревают 30 минут
на масляной бане, имеющей температуру около 100°, затем
быстро меняют обратный холодильник на прямой, также
снабженный хлоркальциевой трубкой, повышают температуру
масляной бани до 180° и отгоняют бутирилйодид в виде ко-
ричневой жидкости с т. кип. 146—148°. Полученный' продукт
очищают перегонкой в вакууме, собирая фракцию, кипящую
при 66—67765 мм.
Выход 55 г, что составляет 56% от теоретического. По вне-
шнему виду бутирилйодид — прозрачная жидкость бледно-ро-
зового цвета с уд. в. 1,77 (см. примечание 2).
Получение 2-йодэтилтиолбутирата. В двугорлую колбу ем-
костью 250 мл, снабженную капельной воронкой и обратным
холодильником с хлоркальциевой трубкой, помещают 80 мл
четыреххлористого углерода и 55 г (0,28 М) бутирилйодида.
К полученной смеси добавляют по каплям, при размешивании,
17,5 мл (0,29 Л1| тиоокиси этилена в течение 15 минут. Затем
реакционную смесь нагревают в течение 1 часа на водяной
бане при температуре около 50°, после чего заменяют обрат-
ный холодильник нисходящим и отгоняют четыреххлористый
углерод на кипящей водяной бане. Оставшийся в колбе
2-йодэтилтиобутират перегоняют под вакуумом, собирая
97
фракцию с температурой паров 103—105°/3 мм. Выход 46 г,
что составляет 64% от теоретического. По внешнему виду это
прозрачная бесцветная жидкость с уд. в. 1,654.
Получение бутирилтиохолинйодйда. В круглодонной кол-
бе емкостью 500 мл, снабженной хорошо притертой пробкой,
растворяют 46 г (0,18 М) 2Lfiofl3THflTHo6yTHpaTa в 150 мл ди-
этилового эфира. Полученный раствор охлаждают до минуд
20° и к нему добавляют порциями по 3 мл 12 г (0,20 М;
18,2 мл) жидкого триметиламина. После добавления каждой
порции колбу закрывают пробкой и встряхивают. По оконча-
нии добавления всего триметиламина колбу закрывают стек-
лянной притертой пробкой, которую закрепляют к горлышку
колбы изоляционной лентой.. Реакционную массу оставляют
при температуре минус 10—15° на сутки, а затем при комнат-
ной температуре на 10—12 дней. Выпавший белый аморфный
осадок бутирилтиохолинйодида отфильтровывают на воронке
Бюхнера, промывают на фильтре 200 мл диэтилового эфира
и трижды перекристаллизовывают из 100 мл абсолютного
этилорого спирта в виде бесцветных игл с т. пл. 171—172°.
Выход 35 г, что составляет 60% от теоретического.
Найдено, %: С—33, 95; Н-6, 28; N-4, 37;S—10, 25; J—40, 15.
C,HsoJNOS. Вычислено, %: С-34, 07; Н-6,35; N—4, 41; S—10, 11; J-40, 00.
Примечания: 1. Смесь в колбе постепенно разогревается, приоб-
ретает жидкую консистенцию и окрашивается в коричневый цвет.
2. Свойства бутирилйодида аналогичны свойствам ацетилйодида (см.
синтез ацетилтиохолинйодида). Продукт хранить более двух суток не ре-
комендуется.
Литература
1. G. В. Koelle, J. Pharm. а. Ехрет. Therap., 100, 158 (1950).
2. R. R. R е п s h о w, Р. F. Dreichbach, J. Amer. Chem. Soc,,
60, 1765 (1938).
3. С. 3. Ивин, Ж. общ. химии, 22, 267 (1952).
4. С. 3. Ивин, Ж. общ. химии, 28, 1332 (1958).
5. A. Cohours, Liebigs Ann. Chem., 104, 111 (1857).
Поступила о апреле 1963 г.
Институт мозга АМН СССР
БРИЛЛИАНТСУЛЬФОФЛАВИН ФФ
М. В. БЕРЕЗОВСКАЯ, А. А. ПРЯНИШНИКОВ
СНз
O=C|Z Х|С=О
А А
k/U~so-H
nh2
C19H14O6N2S
М. в. 382,40
Бриллиантсульфофлавин ФФ, впервые полученный Экер-
том в 1927 году [1], является ценным красителем для флюорес-
центной микроскопии и применяется главным образом для
определения жизнеспособности спор бактерий и низших ра-
стений.
В основу разработанной нами методики получения брил-
лиаитсульфофлавииа положен метод конденсации 4-амино-З-
сульфонафталевого ангидрида с я-толуидином [2]. 4-Амино-З-
сульфонафталевый ангидрид получают сульфированием
4-аминонафталевой кислоты.
Бриллиантсульфофлавин ФФ представляет собой мелко-
кристаллический порошок желтого цвета, растворимый в во-
де, спирте, нерастворимый в эфире [3].
СИНТЕЗ БРИЛЛИАНТСУЛЬФОФЛАВИНА ФФ
НООС СООН
20%-ный олеум
О
СНВ
I
I SO8H
NHa
99
Характеристика основного сырья
л-Толуидин, ТУ ОРУ 47—56.
4-Аминонафталевая кислота, ТУ ГАП У 593—54.
Уголь активированный, щелочной марки, ГОСТ 4453—48.
Условия получения
Получение З-сульфо-4-аминонафталевого ангидрида. В
трехгорлую колбу емкостью 300 мл помещают 222 г 100%-ной
серной кислоты и 63 г 20%-ного олеума. К полученной смеси
при помешивании прибавляют 22 г 4-аминонафталевой кисло-
ты, поднимают температуру до 105—110° и поддерживают ее
до тех пор, пока взятая из реакционной массы проба будет
полностью растворяться в воде (~ 1—2 часа). Затем смесь
выливают в стакан, содержащий 900 мл воды, нагревают до
кипения и фильтруют. К фильтрату добавляют насыщенный
раствор поваренной соли (200 мл) для осаждения 3-сульфо-
4-аминонафталевого ангидрида, отсасывают выпавший про-
дукт и сушат при 80—100°. Выход препарата равен 26 г, что
составляет 84% от теоретического.
Получение бриллиантсульфофлавина ФФ. В трехгорлую
крлбу емкостью 400 мл помещают 26 г З-сульфо-4-аминона-
фталевого ангидрида, 29 г л-толуидина и 240 мл воды. Смесь
кипятят 2—3 часа (см. примечание), отгоняют с водяным па-
ром не вошедший в реакцию л-толуидин, остаток кипятят с
активированным углем (3 г) 5—10 минут, фильтруют и из
фильтрата высаживают краситель насыщенным раствором
поваренной соли (^-200 мл). Выпавший осадок отфильтровы-
вают и сушат при температуре 80—90°.
Получают 26 г бриллиантсульфофлавина ФФ, что состав-
ляет 66%. считая на 4-аминонафталевую кислоту. Продукт
соответствует требованиям РТУ № 550—60.
100
Примечание. Критерием конца реакции при п0^®н“ nnLe Взя;
служит оранжевая окраска, появляющаяся при добавлении к р
той из реакционной массы, нескольких капель щелочи.
Литература
1. Color Index, 3, 3466, 1956.
2. Герм. пат. 494446 (1927). „ми Полинг
3. А. А Прянишников, М. В Березовская, Н. В. Донин ,
Уд ост. о регистрации № 19159 (I960).
ИРЕА
Поступила в августе 1963 г.
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ СОЕДИНЕНИЙ, ОПИСАННЫХ В
НАСТОЯЩЕМ ВЫПУСКЕ
Азотол А....................................................... 65
Азотол 2,4 МК.................................................. 62
D L-Аланил-р-нафтиламид........................................ 53
4-Амнно-1-Г4,М-диметилнафтиламин солянокислый.................. 88
2-Амиио-4, 5-диметил-тиазолил гидробромид...................... 42
З-Амино-5-иитроацетофенои...................................... 80
Ацетилтиохолинйодид............................................ 92
Ацетилйодид.................................................... 93
5-Ацетил-З-иитробензойная кислота ....................... 81
Бензальфенилгидразои ........................................
Бриллиантсульфофлавин ФФ.....................................
5-Бромацетил-З-нитробензойиая кислота........................
Бромангидрид а-бромпропионовой кислоты ......................
Г4-(а-Бромпропионил)-2-нафтиламин ...........................
Бутирилйодид ................................................
Бутирилтиохолинйодид . . • ..................................
41
99
74
54
54
97
96
rf-Галактозофенилгидразои......................................... 46
Диазоль синий С.................................................. 48
3,5- Динитробензоил хлористый.................................... 78
Динитроформазан.................................................. 21
2,2'-Днокси-6,6'-динафтилдисульфид............................... 67
3,5-Динитроацетофенон........................................... 78
Динатриевая соль фосфата азотола А............................... 66
Динатриеван соль фосфата азотола 2,4 М К...................... 62
2,3-Днфенил-5-бензолазо-тетразолий хлористый и бромистый ... 44
3,5-Дифенил-1-(4',5'-диметил-тиазолил-2')-формазан............... 42
Диформазан .... ................................................. 12
5,5'-Ди- (4-нитрофеннл)-1,1'-(4,4'-днфенилен) -3,3'-дифенил-диформа-
.................................................•............ 38
1,5- Дифенил-З-бензолазо-формазан................................ 46
Йоднитротетразолий............................................. 18
2-Йодэтилтиолацетат............................................ 94
2-Йодэтилтиолбутират.......................................... 97
Йодиитроформазаи............................................... 16
2-Карбэтокси-6-нафталинсульфокислый натрий..................... 68
2-Карбэтокси-6-иафталннсульфохлорид............................ 69
102
2-Карбэтокси-6-нафгндтиол.....................
Мв-ги.3^?-01***6" Лняафтилдиеульфид.........
Метиловый эфир хлоргидрина стирола............
Метилтиазолмлтетразолий . ....................
Метил-а-бромэтилкетои ? .............
Натриевая'срль масляной кислоты...............
..
₽-Нафтилацетат . . . .......................
Неотетразолий хлористый.......................
п- Нитро^в-бромацетофенон.....................
п-Нитро-а-метоксистирол.......................
Нитрояеотетразолий . . о......................
Нитротетразолиевый синий • • -и...............
Нитрил-5-ацетил-З-нитробеизойной кислоты . . .
4-Нитрофенилгидразон бензальдегида............
, n-Нитрофенилгидразон п-нитробензальдегида . . .
4-Нитрофенилгидразон феиилглиоксалевой кислоты
2-Окси-З-нафтальдегид.........................
69
69
72
39
42
97
50
51
14
71
72
36
24
80
10
29
8
56
Соль диазония сульфаниловой кислоты ............................ 90
га-Сульфокислота бензолазодиметилнафтиламина........................ 91
Тетразолий синий.................................................... 32
Тиоокись этилена ................................................... 94
Тетра-нитрофенил-тетразолий........................................ 27
3,3'5,5'-Тетра-(4-нитрофенил)-1,Г-(3,3'-диметокси-4,4'-дифенилен)-
диформазан...................................................... 30
3,3',5,5'-Тетрафенил-1,Г'-(3,3'-диметокси-4,4'-фифенилен)-диформа-
зан............................................................. 34
2,2', 5,5'-Тетрафенил-3,3'-(3,3'-диметокси-4,4'-дифеиилен)-дитетра-
золий хлористый..................................................... 34
л-Толуолсульфогидразид 2-окси-З-нафтойная киелота............... 57
2.2',5,5'-Тетра-(4-нитрофенил)-3,3'-(3,3'-диметокси-4,4’-дифенилеи)-
дитетразолий хлористый..............•.......................... 30
Фосфат азотола А........................................• . . . . 63
Фосфат азотола 2,4 МК......................................... 59
Фенилгидразон пировиноградной кислоты.......................... 7
Фенилгидразон фенилглиоксалевой кислоты ....................... 5
Хлорангидрид 2-окси-З-нафтойная кислота....................... 61
УУ-л-Хлорфенил-диамид фосфорной кислоты....................... 86
л-Хлораиилни солянокислый..................................... 87
л-Хлоранилидофосфорилдихлорид ................................ 87
W-Этилмалеинимид ............................................. 83
W-Этилмалеиновая кислота...................................... 84
З-Сульфо-4-аминонафталевый ангидрид.......................... 100