/






















































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































Автор: Гиллебранд В.Ф. Лэндель Г.Э. Брайт Г.А. Гофман Д.И.
Теги: химия неорганическая химия
Год: 1966




Текст
рактическое руководство
’ ' - 'If
по
НЕОРГАНИ ЧЕТКОМУ АНАЛИЗУ
Dr. W. F. HILLEBRAND, Dr. G. E. F. LUNDELL
APPLIED INORGANIC ANALYSIS WITH SPECIAL REFERENCE TO THE ANALYSIS
OF METALS, MINERALS AND ROCKS.
Revised by SECOND EDITION
Late Chief, Chemistry Division Dr. G. E. F. LUNDELL
Chief, Analytical Chemistry Section H. A. BRIGHT, M. S.
Assistant Chief, Chemistry Division Dr. J. I. HOFFMAN
NATIONAL BUREAU OF STANDARDS.
JOHN WILEY & SONS, INC., NEW YORK CHAPMAN & HALL, LIMITED, LONDON 19 5 3
В. Ф. ГИЛЛЕБРАНД, Г. Э. ЛЕНДЕЛЬ.
Г. А. БРАЙТ, Д. И. ГОФМАН
ПРАКТИЧЕСКОЕ
РУКОВОДСТВО
ПО НЕОРГАНИЧЕСКОМУ АНАЛИЗУ
Перевод с английского Е. II. ГУЛЬДИНОЙ и Ю. Ю. ЛУРЬЕ
Под редакцией проф. Ю. Ю. ЛУРЬЕ
ИЗДАНИЕ ТРЕТЬЕ СТЕРЕОТИПНОЕ, ИСПРАВЛЕННОЕ
Читальный Зал
ИЗДАТЕЛЬСТВО «X И М II Я» МОСКВА 19 6 6
543-7 Г-47
В книге изложены проверенные методы отделения и определения большинства химических элементов. Подробно рассмотрены методы определения редких элементов. Описаны также методы анализа силикатных и карбонатных горных пород.
Книга предназначена для работников химико-аналитических лабораторий научно-исследовательских учреждений и промышленных предприятий, ведущих химический анализ горных пород, руд, минералов и т. п. Она может служить также пособием для студентов химических факультетов вузов при изучении ими курсов специального анализа.
Настоящее издание полностью соответствует изданию 1957 г.
СОДЕРЖАНИЕ
Предисловие редактора................................................ 19
Из предисловия авторов к первому изданию............................... 21
ЧАСТЬ ПЕРВАЯ. ОБЩИЕ УКАЗАНИЯ
Глава I. Введение ..................................................... 25
Точность анализа.................................................. 27
Факторы пересчета и представление результатов..................... 30
Глава II. Весы и разновески ........................................... 31
Весы ............................................................. 31
Качество весов ............................................. 31
Уход за весами.............................................. 31
Аналитический разновес............................................ 32
Разновески класса М ........................................ 32
Разновески класса S ........................................ 33
Проверка разновесок и уход за ними.......................... 35
Взвешивание....................................................... 36
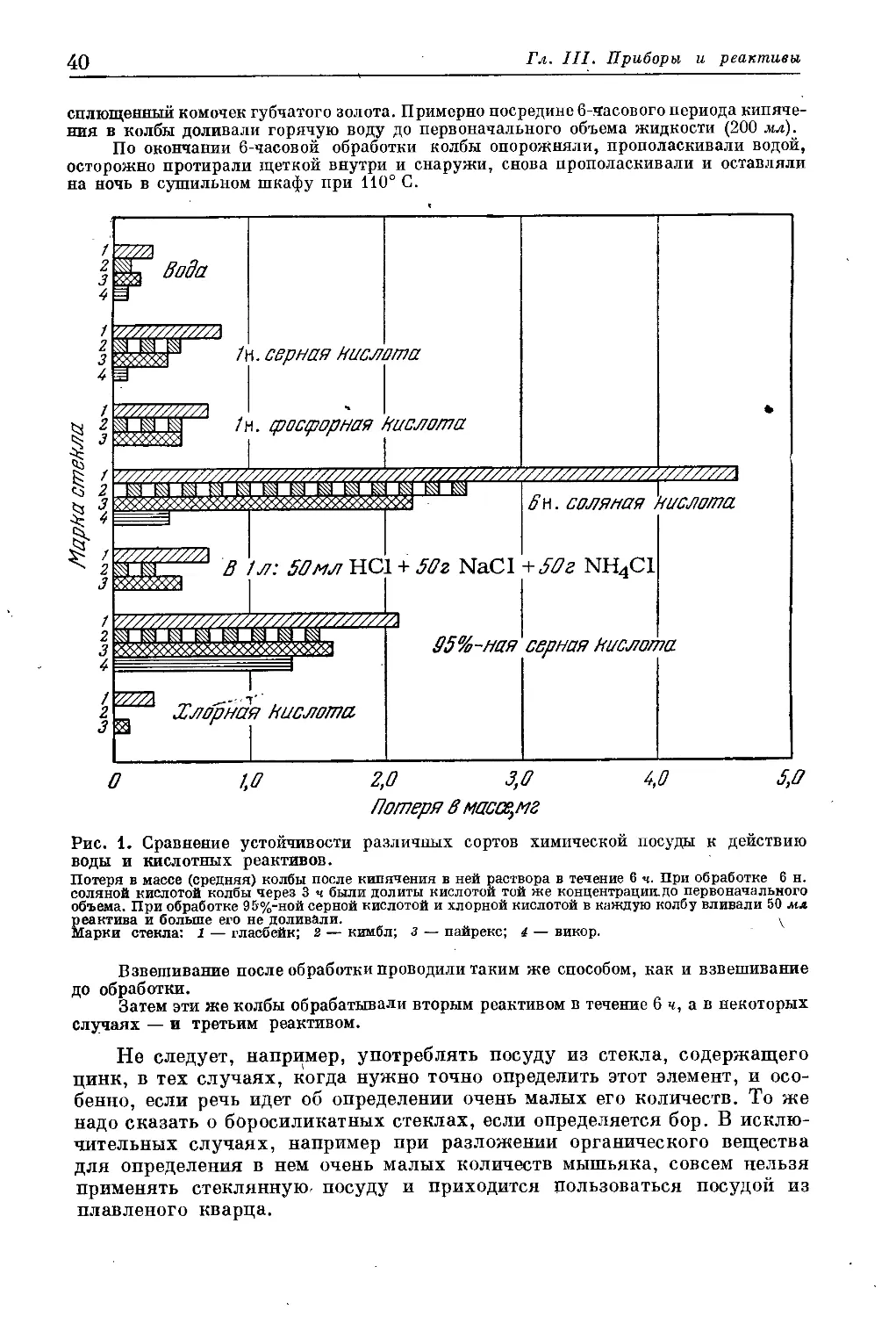
Глава III. Приборы и реактивы.......................................... 39
Приборы.......................................................... 39
Стеклянная и фарфоровая посуда.............................. 39
Платина и заменяющие ее сплавы............................. 44
Необходимые инструменты и приборы........................... 47
Реактивы.......................................................... 58
Общие замечания ............................................ 58
Специальные реактивы........................................ 61
Растворы реактивов.......................................... 66
Газообразные реактивы ...................................... 68
Поглотители двуокиси углерода .............................. 68
Высушивающие вещества ...................................... 71
Глава IV. Общие методы и операции химического анализа.................. 77
Отбор пробы ...................................................... 77
Приготовление пробы для взвешивания............................... 79
Взвешивание пробы................................................. 79
Выбор метода анализа ............................................. 80
Приготовление раствора для анализа ............................... 82
Осаждение посредством сульфид-ионов............................... 83
Сильнокислые растворы (рН<1)................................ 83
Слабокислые растворы (pH от 2 до 3)......................... 85
Почти нейтральные растворы (pH от 5 до 6)................... 86
Щелочные растворы (рН>7).................................... 87
6
С одержание
Осаждение в растворах, содержащих комплексные анионы.............. 88
Осаждение сульфидом аммония ...................................... 90
Разделение элементов в осадке от сероводорода, полученном в сильно-кислом растворе................................................... 92
Разделение групп меди и мышьяка ............................ 93
Разделения в группе меди.................................... 94
Разделения в группе мышьяка................................. 95
Осаждение посредством изменения концентрации ионов водорода в растворе ...................................................... 100
Осаждение аммиаком ...................................... 102
. Осаждение в виде основных ацетатов......................... 103
Осаждение сукцинатом натрия (янтарнокислым натрием) . . . 106
Осаждение бензоатом аммония.............................. 107
Осаждение суспензиями карбонатов и окисей................ 108
Осаждение добавлением йодатов пли броматов............... 109
Осаждение едким натром .................................. 109
Осаждение слабыми органическими основаниями.............. 111
Разделение элементов в осадке от аммиака, ацетата натрия или сукцината натрия ...................................................... ИЗ
Обычные методы разделения.................................. 114
Другие методы разделения................................... 117
Подробный анализ осадка от аммиака......................... 118
Фильтрование..................................................... 123
Воронки.................................................... 123
Фильтровальная бумага...................................... 123
Бумажная масса............................................. 124
Асбест..................................................... 125
Вата из кремпекислоты...................................... 127
Платина................................................... 127
Алунд, фарфор и стекло..................................... 128
Промывание осадков............................................... 128
Высушивание и прокаливание осадков.............................. 131
Глава. V. Специальные операции........................................ 135
Редуктор Джонса................................................. 135
Восстановление в редукторе с серебром........................... 139
Определение железа......................................... 140
Определение молибдена..................................... 140
Восстановление жидкими амальгамами............................... 141
Другие методы восстановления..................................... 142
Осаждение купфероном ........................................... 143
Определение железа, циркония, галлпя, титана и ванадия .... 145
Определение урана ......................................... 146
Определение олова.......................................... 146
Определение галлия......................................... 146
Определение ниобия и тантала .............................. 147
Определение других элементов............................... 147
Экстрагирование купфератов................................. 147
Осаждение оксихинолином.......................................... 148
Осаждение алюминия из уксуснокислого раствора.............. 150
Осаждение магния из аммиачного раствора.................... 150
Титрование оксихинолятов .................................. 150
Осаждение таннином............................................. 151
Осаждение фенилгидразином...................................... 154
Осаждение хлоридом тетрафениларсопия........................... 155
Осаждение и-оксифениларсоновой кислотой........................ 156
Методы определения с дифенилтиокарбазоном (дитизоном)............ 156
Методы определения с комплексоном................................ 157
Взаимное превращение хлоридов, нитратов и сульфатов щелочных металлов ........................................................ 159
Превращение сульфатов в хлориды ........................... 159
Превращение хлоридов и нитратов в карбонаты обработкой щавелевой кислотой ....................................... 160
Превращение сульфатов в карбонаты......................... 160
Содержание
7
Превращение сульфатов в тартраты, оксалаты и др............ 160
Превращение хлоридов в окислы.............................. 160
Удаление солей аммония.......................................... 161
Хлорид аммония ............................................ 161
Нитрат аммония............................................. 161
Сульфат аммония ........................................... 161
Извлечение эфиром .............................................. 161
Электролитические методы ....................................... 164
Электролиз с контролируемым электродным потенциалом . . . 165
Электролиз с ртутным катодом............................... 166
Внутренний электролиз ..................................... 167
Полярографический анализ........................................ 171
Фотометрические методы.......................................... 173
Флуоресцентный анализ........................................... 176
Спектрохимический анализ........................................ 177
Фотометрия пламени ............................................. 180
Рентгеновский метод химического анализа ........................ 181
Хроматографический анализ....................................... 183
Масс-спектрометрия ............................................. 184
Капельный анализ................................................ 184
Микрохимия и химическая микроскопия............................. 185
Глава VI. Объемный анализ............................................. 188
Приборы для измерения объема.................................... 188
Общие требования........................................... 188
Специальные требования..................................... 190
Правила обращения с измерительными приборами............... 192
Весовые бюретки ........................................... 193
Ацидиметрия и алкалиметрия..................................... 193
Индикаторы................................................. 193
Установка титра кислот..................................... 202
Установка титра оснований ................................. 207
Методы окисления и восстановления............................... 212
Окислительно-восстановительные индикаторы.................. 212
Установка титра растворов перманганата калия............... 214
Установка титра растворов бихромата калия.................. 218
Цериметрия................................................. 219
Иодометрия................................................. 220
Потенциометрическое титрование ............................ 229
ЧАСТЬ ВТОРАЯ. ОПРЕДЕЛЕНИЕ ЭЛЕМЕНТОВ
ГРУППА СЕРОВОДОРОДА
Серебро, ртуть, свинец, висмут, медь, кадмий, мышьяк, сурьма, олово, германий, молибден, селен, теллур, золото, рутений, родий, палладий, осмий, иридий, платина (таллий)
Элементы, сульфиды которых нерастворимы в кислотах и в растворах сульфидов щелочных металлов
Серебро, ртуть, свинец, висмут, медь, кадмий
(и полностью или частично рутений, родий, палладий и осмий)
Глава VII. Серебро...................................................... 235
Общие замечания................................................... 235
Разложение минералов, содержащих серебро......................... 236
Методы разделения ................................................ 236
Методы определения................................................ 237
Весовое определение в виде хлорида серебра.................. 237
Определение титрованием роданидом калия или аммония . . . 238
Другие методы .............................................. 240
Г лава VIII. Ртуть...................................................... 242
Общие замечания................................................... 242
Разложение соединений ртути....................................... 242
8
Содержали е
Методы отделения.................................................... 245
Методы определения.................................................. 248
Объемное определение титрованием роданидом аммония .... 248
Весовые определения .......................................... 249
Объемное или весовое определение в виде перйодата ртути . . . 251
Определение малых количеств ртути............................. 253
Глава IX. Свинец '....................................................... 257
Общие замечания................................................... 257
Разложение минералов, содержащих свинец............................. 258
Методы отделения.................................................... 258
Методы определения.................................................. 262
Весовые определения........................................... 262
Электролитическое определение в виде двуокиси свинца .... 264
Объемное определение хроматным методом........... 265
Другие методы................................................. 267
Г лава X. Висмут......................................................... 268
Общие замечания .................................................... 268
Разложение минералов, содержащих висмут........................... 268
Методы отделения.................................................... 269
Методы определения.................................................. 274
Весовые определения........................................... 274
Колориметрические определения................................. 277
Другие методы ................................................ 279
Глава XI. Медь......................................................... 281
Общие замечания .................................................... 281
Разложение минералов, содержащих медь............................ 282
Методы отделения.................................................... 282
Методы определения.................................................. 285
Электролитический метод....................................... 286
Йодометрический метод......................................... 287
Роданидный метод.............................................. 290
Сульфидный метод.............................................. 292
Колориметрические методы...................................... 293
Другие методы................................................. 293
Глава XII. Кадмий........................................................ 296
Общие замечания..................................................... 296
Разложение минералов, содержащих кадмий........................... 296
Методы отделения.................................................... 296
Методы определения.................................................. 299
Определение взвешиванием в виде сульфата кадмия....... 299
Электролитическое определение................................. 299
Другие методы................................................ 300»
Элементы., сульфиды которых нерастворимы в кислотах, но растворимы' в растворах сульфидов щелочных металлов’
Мышьяк, сурьма, олово, германий, молибден, селен, теллур (и полностью или частично золото, платина и иридий)
Глава XIII. Мышьяк..................................................... 302
Общие замечания .................................................. 302
Разложение минералов, содержащих мышьяк........................... 303
Методы отделения.................................................. 303
Методы определения . ............................................. 309
Весовое определение в виде сульфидов мышьяка................ 309
Объемные определения........................................ 310
Определение отгонкой в виде мышьяковистого водорода......... 312
Колориметрический метод..................................... 315
Другие методы............................................... 316
Содержание
9
Глава XIV. Сурьма...................................................... 317
Общие замечания.................................................. 317
Разложение минералов, содержащих сурьму.......................... 317
Методы отделения................................................ 321
Методы определения............................................... 324
Определение титрованием перманганатом...................... 324
Весовые определения........................................ 326
Определение отгонкой в виде сурьмянистого водорода (стибипа) 327
Другие методы ............................................. 328
Глава XV. Олово........................................................ 332
Общие замечания.................................................. 332
Разложение минералов, содержащих олово........................... 332
Методы отделения................................................. 334
Методы определения............................................... 337
Йодометрический метод...................................... 338
Весовое определение в виде двуокиси олова.................. 341
Другие методы ............................................. 343
Глава XVI. Германий.................................................... 345
Общие замечания.................................................. 345
Разложение германиевых минералов п окпсп цинка, содержащей германий 346
Методы отделения................................................. 347
Методы определения............................................... 348
Осаждение в виде сульфида германия и взвешивание в виде окиси германия .................................................. 348
Взвешивание в виде ортогермапата магния.................... 349
Объемные определения....................................... 351
Колориметрические методы................................... 352
Глава XVII. Молибден................................................... 356
Общие замечания ................................................. 356
Разложение молибденовых минералов................................ 356
Методы отделения................................................. 357
Методы определения................................................. 361
Титрование перманганатом................................... 361
Осаждение а-бензоиноксимом................................. 364
Определение в виде молибдата свинца........................ 366-
Осаждение в виде сульфида молибдена п прокаливание до окпсп 367
Колориметрический метод.................................... 368
Другие методы.............................................. 370
Глава XVIII. Рений ...................................................... 372
Общие замечания.................................................... 372
Разложение минералов, содержащих рений........................... 373
Методы отделения................................................... 373
Методы определения......................................-........ 375
Весовые методы............................................... 375
Объемные методы............................................ 377
Колориметрический метод.................................... 378
Электролитические методы..................................... 380
Глава XIX. Селен и теллур................................................ 382
Общие замечания ................................................. 382
Разложение минералов, содержащих селен и теллур.................. 383
Методы отделения................................................. 385
Методы определения................................................. 389
Селен......................................................... 389
Восстановление сернистой кислотой в солянокислом растворе 389
Восстановление солянокислым гпдроксиламппом................ 390
Объемные методы............................................ 391
10
С одержание
Теллур......................................................... 392
Восстановление сернистой кислотой и гидразином............. 392
Объемные-методы............................................. 393
Колориметрический метод определения теллура и селена . . . 394
Благородные металлы
Золото, осмий, рутений, платина, палладий, родий и иридий
Глава XX. Платиновые металлы........................................... 395
Общие замечания................................................. 39(5
Обычные содержащие платину материалы........................ 396
Поведение платины в ходе анализа............................ 397
Разложение минералов и сплавов................................... 399
Методы разделения................................................ 406
Разделение платиновых металлов и золота..................... 406
Отделение платиновых металлов и золота от других металлов 412
Методы определения............................................... 416
Общие замечания............................................. 416
Ход анализа................................................. 419
Новые методы анализа ....................................... 422
Систематический ход разделения и определения платиновых металлов 423
Рекомендуемый ход анализа................................... 424
ГРУППА СУЛЬФИДА АММОНИЯ
Железо, никель, кобальт, цинк, марганец, ванадий, уран, таллий, индий, галлий, алюминий, бериллий, хром, торий, скандий, редкоземельные металлы, цирконий, титан, ниобий и тантал
' Элементы, образующие при действии (NH^S растворимые в кислотах сульфиды
Железо, никель, кобальт, цинк, марганец, ванадий, уратг, таллий, индий, галлий
Глава XXI. Железо...................'.................................. 434
Общие замечания.................................................. 434
Разложение минералов, содержащих железо.......................... 435
Методы отделения ................................................ 437
Методы определения............................................... 439
Весовой метод............................................... 439
Объемные методы............................................. 440
Колориметрические методы.................................... 152
Глава XXII. Никель ................................................... 456
Общие замечания.................................................. 456
Разложение минералов, содержащих никель.......................... 457
Методы отделения................................................. 457
Методы определения............................................... 460
Осаждение диметилглиоксимом................................. 460
Электролитический метод..................................... 463
Цианидный метод............................................. 466
Объемный метод (титрование диметилглиоксимом)............... 467
Другие методы .............................................. 467
Глава XXIII. Кобальт................................................... 469
Общие замечания ................................................. 469
Разложение минералов, содержащих кобальт......................... 469
Методы отделения................................................. 470
Методы определения............................................... 471
Электролитический метод .................................... 471
Калиево-питритный метод .................................... 471
а-Нитрозо-|3-нафтоловый метод............................... 473
Колориметрические методы ................................... 475
Другие методы .............................................. 476
С одержание
И
- Глава XX/V. Цинк....................................................... 478
Общие замечания ................................................... 478
Разложение минералов, содержащих цинк.............................. 479
Методы отделения................................................... 479
Методы определения................................................. 481
Осаждение в виде сульфида цинка.............................. 481
Осаждение в вйде фосфата цинка............................... 485
Осаждение в виде роданомеркуриата цинка...................... 486
Гексацианоферратный метод ................................... 489
Другие 'методы .............................................. 491
Определение малых количеств цинка............................ 492
'Глава XXV. Марганец ................................................... 493
Общие замечания .................................................. 493
Разложение минералов, содержащих марганец.......................... 493
Методы отделения ................................................. 494
Методы определения ............................................... 497
Объемные методы ............................................ 497
Весовые методы............................................., 503
Колориметрические методы ................................... 505
.Глава XXVI. Ванадий ................................................... 507
Общие замечания .................................................. 507
Разложение минералов, содержащих ванадий.......................... 508
Методы отделения.................................................. 509
Методы определения ............................................... 513
Восстановление сернистым ангидридом ........................ 513
Метод с применением сульфата железа (II) п персульфата аммония 514
Осаждение купфероном ...................................... 515
Потенциометрическое определение ............................. 515
Колориметрический метод ................................... 516
Другие методы ............................................... 518
Определение ванадия в шлаках............................... 520
Определение ванадия в специальных сталях и чугунах........... 520
..Глава XXVII. Уран..................................................... 522
Общие замечания.................................................. 523
Разложение минералов, содержащих уран.............................. 523
Методы отделения................................................. 524
Методы определения................................................ 526
Весовое определение после осаждения аммиаком............... 527
Объемное определение восстановлением цинком и титрованием перманганатом ........................................... 529
Весовое определение после осаждения купфероном............... 531
Другие методы ............................................. 531
Глава XXVIII. Таллий...................................................... 536
Общие замечания .................................................. 536
Разложение минералов, содержащих таллий.......................... 537
Методы отделения.................................................. 538
Методы определения................................................ 540
Определение в виде хромата таллия........................... 540
Определение в виде окиси таллия (III)....................... 540
Определение в виде йодида таллия (I)........................ 541
Другие методы ............................................. 541
Глава XXIX. Индий........................................................ 544
Общие замечания ................................................. 544
Разложение минералов, содержащих индий........................... 545
Методы отделения................................................. 545
Методы определения............................................... 547
Осаждение аммиаком ........................................ 547
Колориметрический метод .................................... 548
Содержание
Глава XXX. Галлий ................................................... 549
Общие замечания ................................................. 549
Разложение минералов, содержащих галлий.......................... 550
Методы отделения ................................................ 550
Методы определения .............................................. 554
Осаждение аммиаком ........................................ 554
Осаждение купфероном ...................................... 555
Осаждение бисульфитом аммония ............................. 556
Колориметрические методы .................................. 556
Элементы, образующие при действии сульфида аммония гидроокиси или основные соли
Алюминий, бериллий, хром, торий, скандий, редкоземельные металлы, цирконий, титан, ниобий и тантал
Глава XXXI. Алюминий . . ............................................. 559
Общие замечания ................................................. 559
Разложение минералов, содержащих алюминий........................ 560
Методы отделения................................................. 561
Методы определения .............................................. 565
Осаждение аммиаком ........................................ 565
Осаждение в виде фосфата алюминия.......................... 569
Осаждение оксихинолином ................................... 571
Определение малых количеств алюминия «0,1 лг).............. 575
Другие методы ............................................. 578
Глава XXXII. Бериллий ................................................ 580
Общие замечания ................................................. 580
Разложение минералов, содержащих бериллий........................ 582
Методы отделения ................................................ 582
Методы определения .............................................. 586
Осаждение аммиаком ........................................ 586
Другие методы ............................................. 588
Глава XXXIII. Хром.................................................... 589
Общие замечания ................................................. 589
Разложение минералов, содержащих хром ........................... 589
Методы отделения................................................. 590
Методы определения .............................................. 592
Титрование сульфатом железа (II) и перманганатом........... 592
Колориметрические методы.................................. 595
Другие методы ............................................. 596
Глава XXXIV. Торий ................................................... 598
Общие замечания ................................................. 598
Разложение минералов, содержащих торий........................... 599
Методы отделения ................................................ 600
Методы определения .............................................. 607
Осаждение в виде оксалата тория ........................... 607
Осаждение в виде йодата тория.............................. 607
Колориметрические методы .................................. 609
Комплексометрические методы ............................... 611
Глава XXXV. Скандий .................................................. 613
Общие замечания ................................................. 613
Разложение минералов, содержащих скандий......................... 613
Методы отделения ................................................ 614
Методы определения .............................................. 616
Глава XXXVI. Редкоземельные металлы................................... 617
Общие замечания ................................................ 618-
Разложение редкоземельных минералов............................. 619'
Содержание
13
Методы отделения ................................................ 620
Методы определения .............................................. 628
Колориметрические методы определения церия................. 633
Глава XXXVII. Цирконий (гафний)........................................ 635
Общие замечания ................................................. 636
Разложение минералов, содержащих цирконий........................ 636
Методы отделения ................................................ 638
Методы определения .............................................. 640
Осаждение в виде фосфата циркония.......................... 640
Купфероновый метод ........................................ 643
Осаждение в виде основного селенита циркония............... 644
Другие методы ............................................. 646
Колориметрические методы .................................. 648
Глава XXXVIII. Титан................................................... 650
Общие замечания.................................................. 650
Разложение минералов, содержащих титан........................... 651
Методы отделения................................................. 652
Методы определения .............................................. 654
Колориметрический метод ................................... 655
Объемный метод ............................................ 659
Осаждение купфероном ...................................... 661
Гидролитическое осаждение из уксуснокислых или солянокислых растворов...............'................................... 661
Другие методы ............................................. 662
Глава XXXIX. Ниобий и тантал........................................... 663
Общие замечания.................................................. 663
Разложение минералов, содержащих ниобий и тантал.............. 666
Разложение фтористоводородной кислотой и последующая обработка ................................................ 666
Разложение сплавлением с едким кали, карбонатом калия или перекисью натрия................................................ 668
Разложение сплавлением с пиросульфатом и дальнейшая обработка 669
Разложение хлористой серой и последующая обработка......... 672
Методы отделения................................................. 673
Отделение малых количеств ниобия и тантала в присутствии больших количеств циркония ................................... 674
Отделение больших количеств ниобия и тантала в присутствии любых количеств циркония................................ 674
Методы определения .............................................. 680
Отделение тантала от ниобия осаждением таннином в щавелевокислом растворе ...................................... 681
Отделение ниобия от тантала обработкой окислов оксихлоридом селена...................................................... 682
Разделение ниобия и тантала по методу Мариньяка............ 683
Объемное определение ниобия ............................... 686
Непосредственное колориметрическое определение ниобия . . . 688
Фотометрическое определение тантала ................. 691
Протактиний...................................................... 692
ЩЕЛОЧНОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ И МАГНИЙ
Кальций, стронций, барий, магний (радий)
Глава XL. Щелочноземельные металлы — кальций, стронций, барий .... 693
Общие замечания ................................................. 693
Разложение минералов, содержащих щелочноземельные металлы . . . 694
Методы отделения ................................................ 694
Отделение кальция от магния............................... 694
Отделение кальция, стронция и бария от других элементов . . . 696
14
Содержание
Отделение кальция, стронция и бария друг от друга........ 697
Отделение кальция от стронция и бария.................... 697
Отделение бария и стронция друг от друга................. 700
Методы определения............................................. 701
Кальций ................................................. 701
Стронций................................................. 712
Барий ................................................... 713
Глава XLI. Магний................................................... 715
Общие замечания ............................................... 715
Разложение минералов, содержащих магний ....................... 715
Методы отделения .............................................. 715
Методы определения ............................................ 719
Определение в виде пирофосфата магния.................... 719
Осаждение оксихинолпном ................................. 725
Объемный метод........................................... 727
ЩЕЛОЧНЫЕ МЕТАЛЛЫ
Литий, натрий, калий, рубидий, цезий
7 лава XLII. Щелочные металлы....................................... 729
Общие замечания ............................................... 730
Методы определения ............................................ 731
Хлороплатинатный метод .................................. 731
Определение натрия с и-бутиловым спиртом и этилацетатом . . . 734
Определение лития ....................................... 737
Определение рубидия и цезия ............................. 740
Определение одного калия................................. 744
Отделение малых количеств калия от больших количеств натрия 747
Определение одного натрия................................ 748
КИСЛОТООБРАЗУЮЩИЕ ЭЛЕМЕНТЫ
Кремний, вольфрам, фосфор, сера, хлор, бром, иод, фтор, бор, углерод, водород и азот
Глава XLIII. Кремний................................................ 752
Общие замечания .............................................. 752
Разложение пород и минералов, содержащих кремний.............. 752
Методы отделения.............................................. 753
Методы определения............................................ 756
Глава XLIV. Вольфрам ............................................... 764
Общие замечания............................................... 764
Разложение минералов, содержащих вольфрам..................... 765
Методы отделения . . . ....................................... 765
Методы определения............................................ 769
Определение обработкой кислотами с добавлением цинхонина . . 770
Колориметрический метод ................................ 774
Глава XLV. Фосфор................................................... 777
Общие замечания............................................... 777
Разложение соединений, содержащих фосфор...................... 778
Методы отделения.............................................. 779
Методы определения............................................ 781
Осаждение фосфора в виде фосфоромолибдата аммония и обработка полученного осадка ............................ 782
Непосредственное осаждение фосфора в виде фосфата магния и аммония.......................................... . . . 791
Колориметрические методы ............................... 792-
Содержание
15
Глава XLVI. Сера :......................................................... 793
Общие замечания...................................................... 793
Разложение минералов, содержащих серу .............................. 794
Методы отделения..................................................... 796
Методы определения ............................................ 797
Определение в растворах, содержащих только серную кислоту 801
Определение в растворах, содержащих умеренные количества сульфатов в присутствии солей щелочных металлов или аммонийных солей..................................................... 802
Определение в растворах, содержащих большие количества сульфатов в присутствии железа и подобных ему элементов .... 803
Объемные методы ............................................... 804
Турбодиметрический и колориметрический методы ................. 805
Глава XLVII. Хлор, бром и иод............................................ 806
Общие замечания.................................................... 806
Разложение соединений галогенов ..................................... 807
Методы отделения.................................................... 808
Методы определения ................................................. 811
Хлор.............................................................. 811
Взвешивание в виде хлорида серебра............................. 811
Осаждение нитратом серебра и титрование роданидом.............. 813
Метод Мора .................................................... 814
Бром.............................................................. 815
Осаждение в виде бромида серебра............................... 815
Осаждение нитратом серебра и титрование роданидом.............. 816
Иод............................................................... 816
Взвешивание в виде иодида серебра ............................. 816
Осаждение в виде иодида палладия (II).......................... 816
Окисление галогенидов серебра серной и хромовой кислотами 817
Метод Фольгарда................................................ 817
Другие методы ................................................. 818
Глава XLVIII. Фтор ........................................................ 819
Общие замечания .................................................... 819
Разложение соединений фтора ........................................ 821
Методы отделения.................................................... 823
Методы определения.................................................. 824
Метод отгонки в виде H2SiFe и титрования нитратом тория . . . 824
Осаждение в виде фторохлорпда свинца.......................... 826
Колориметрические методы...................................... 828
Другие методы................................................. 829
Глава XLIX. Бор............................................................ 830
Общие замечания .................................................... 830
Разложение минералов, содержащих бор................................ 832
Методы отделения................................................... 833
Методы определения ................................................. 834
Титрование едким натром ...................................... 834
Титрование едким натром после экстракции ..................... 840
Взвешивание в виде бората кальция............................. 841
Другие методы ................................................ 843
Глава L. Углерод и водород................................................. 846
Общие замечания..................................................... 846
Разложение веществ, содержащих углерод ............................. 847
Методы отделения.................................................... 848
Методы определения ................................................ 848
Определение углерода, присутствующего в виде карбонатов . . . 848
Определение общего содержания углерода и водорода прямым сжиганием...........................................,,.... 850-
16
Содержание
Определение общего содержания углерода «мокрым сжиганием» 856
Другие методы определения углерода ....................... 857
Определение водорода в горных породах и минералах............ 858
Глава LI. Азот........................................................... 859
Методы определения................................................. 859
Метод Дюма................................................... 859
Метод Кьельдаля ............................................. 862
Осаждение в виде азотнокислого нитрона....................... 867
Другие методы ............................................... 868
Определение очень малых количеств азота, находящегося в виде аммиака или нитрата ..................................... 869
ЧАСТЬ ТРЕТЬЯ. АНАЛИЗ СИЛИКАТНЫХ ПОРОД
Глава LII. Введение в анализ силикатных пород............................ 875
Значение полных анализов .......................................... 875
Составные части силикатных горных пород............................ 881
Предварительный качественный анализ................................ 883
Суммирование результатов анализа и пределы допустимых ошибок . . . 883
Представление результатов анализа ................................. 887
Продолжительность выполнения анализа .............................. 888
^Глава LIII. Методы анализа силикатных пород............................. 889
Предварительные замечания . 890
Приготовление пробы для анализа.................................... 891
Величина средней пробы, поступающей в лабораторию............ 891
Измельчение.................................................. 891
Растирание .................................................. 893
Истирание ступки и пестика................................... 895
Масса пробы ................................................. 895
Вода. Общие замечания.............................................. 896
Роль водорода в минералах.................................... 896
Изменение содержания воды при измельчении.................... 900
Применение для анализа воздушно-сухой пробы............... 902
Включение гигроскопической влаги в таблицы результатов анализа 903
Вода. Методы определения........................................... 903
Косвенные методы............................................. 904
Прямые методы................................................ 908
Методы разложения горных пород..................................... 915
Разложение при помощи плавней................................ 915
Разложение кислотами......................................... 930
Кремний ........................................................... 931
Специальные случаи .......................................... 931
Анализ силикатов............................................. 936
Ход анализа после выделения кремнекислоты.......................... 946
Металлы, осаждаемые сероводородом............................ 948
Совместное осаждение железа, алюминия, титана, циркония, хрома, редкоземельных металлов, фосфора и ванадия вместе с марганцем и без него........................................... 949
Марганец, никель, кобальт, медь и цинк............................. 959
Затруднения, встречающиеся при точном определении марганца весовым способом........................................... 959
Осаждение всей группы и разделение ее составных частей .... 960
Колориметрическое определение марганца................ 962
Кальций, стронций (барий)......................................... 963
Магний............................................................. 964
'Титан............................................................. 964
Общие замечания ............................................. 964
Колориметрическое определение с перекисью водорода........... 966
Весовые методы............................................... 967
Объемные методы ............................................ 968
Содержание
17
Барий (цирконий, редкоземельные металлы, общее содержание серы, хром)...........................................................
Общий метод ...............................................
Цирконий........................................................
Метод Гиллебранда .........................................
Другие методы .............................................
Редкоземельные металлы..........................................
Первый метод...............................................
Второй метод...............................................
Фосфор..........................................................
Предварительные замечания .................................
Метод, применяемый при большом количестве материала для анализа ......................................................
Метод, применяемый при малом количестве материала для анализа
Хром............................................................
Весовые методы.............................................
Колориметрический метод . .................................
Сравнительные данные ......................................
Ванадий (хром) и молибден...............,.......................
Распространение ванадия и молибдена........................
Валентность ванадия в породах..............................
Метод Гиллебранда .........................................
Железо..........................................................
Железо (II)................................................
Железо (III)...............................................
Щелочные металлы ...............................................
Общие замечания ...........................................
Метод Лоуренса Смита ......................................
Метод Берцелиуса ..........................................
Двуокись углерода; углерод......................................
Качественное испытание.....................................
Количественное определение ................................
Определение углерода.......................................
Хлор............................................................
Состояние хлора в горных породах...........................
Определение растворимого в воде хлора......................
Определение хлора, переходящего в раствор при обработке кислотами ......................................................
Определение хлора сплавлением со щелочами..................
Фтор (кремнекислота в присутствии фтора)........................
Общие замечания ...........................................
Прямое определение фтора...................................
Непрямое определение малых количеств фтора.................
Сера ...........................................................
Определение различных форм, в которых сера находится в горных породах ...................................................
Определение серы в горных породах..........................
Бор.............................................................
Общие замечания ...........................................
Качественное испытание.....................................
Компоненты, встречающиеся в породах в виде следов...............
Газы и пары, выделяющиеся при нагревании........................
Особые исследования.............................................
Открытие нефелина в присутствии оливина....................
Определение «растворимой» кремнекислоты....................
968 969
971
971
973
974
974
975
975
975
975 977
978
978
979
980
980 980 981
982
986
986
1004
1004 1004
1006
1011
1014
1014
1015
1016
1017
1017
1017
1017
1018 1018 1018
1019
1025
1028
1028
1029
1031
1031
1032
1033
1034
1036
1036
1037
ЧАСТЬ ЧЕТВЕРТАЯ. АНАЛИЗ КАРБОНАТНЫХ ПОРОД
Глава LIV. Введение в анализ карбонатных пород........................1041
Качественное сравнение карбонатных пород с силикатными..........1041
Минеральный состав карбонатных пород............................1043
Распознавание различных карбонатов по их отношению к разным реактивам ............................................................1045
2 Заказ 522.
4729Ь
18
Содержание
Глава LV. Точные методы анализа карбонатных пород...................1048
К ремнекисл ота, отделение ее от окиси алюминия и других окисей . . . 1048
Методы разложения породы.................................1048
Отделение кремнекислоты .................................1051
Алюминий, общее содержание железа, тктан (кремнекислота, марганец); осаждение вместе с фосфором............................. 1052
Осаждение алюминия, железа и аналогичных элементов .... 1052
Обработка фильтратов от гидроокисей алюминия и других элементов 1054
Растворение и разделение окислов ....................... 1054
Марганец . . ..................................................1055
Определение в фильтратах после осаждения.................1055
Отдельное определение .................................. 1050
Медь, пикель, кобальт, свинец, редкоземельные металлы, хром, ванадий и молибден.....................................................1056
Кальций, стронции, барий, магний (марганец)....................1057
Фосфор..................................-......................1057
Железо (II) , .................................................1058
Определение в отсутствие углистых веществ................1058
Определение в присутствии нерастворимых углистых веществ . . 1059
Щелочные металлы ..............................................1000
Двуокись углерода, углерод (вода)..............................1060
Определение углерода и водорода углистых веществ.1060
Одновременное определение воды и общего содержания углерода 1061
Хлор...........................................................1061
Фтор...........................................................1062
Сера ..........................................................1062
Установление формы соединений серы. Определение сульфатной серы...............................................1062
Определение общего содержания серы.......................1062
Вода ..........................................................1063
Гигроскопическая вода ...................................1063
Связанная вода ..........................................1064
Глава LVI. Сокращенный анализ карбонатных пород.....................1065
Разложение и растворение.................................1065
Кремнекислота..............’.............................1065
Алюминий, железо и другие элементы.......................1066
Марганец.................................................1067
Кальций..................................................1068
Магний...................................................1069
Определение других компонентов...........................1070
Потеря в массе при прокаливании..........................1070
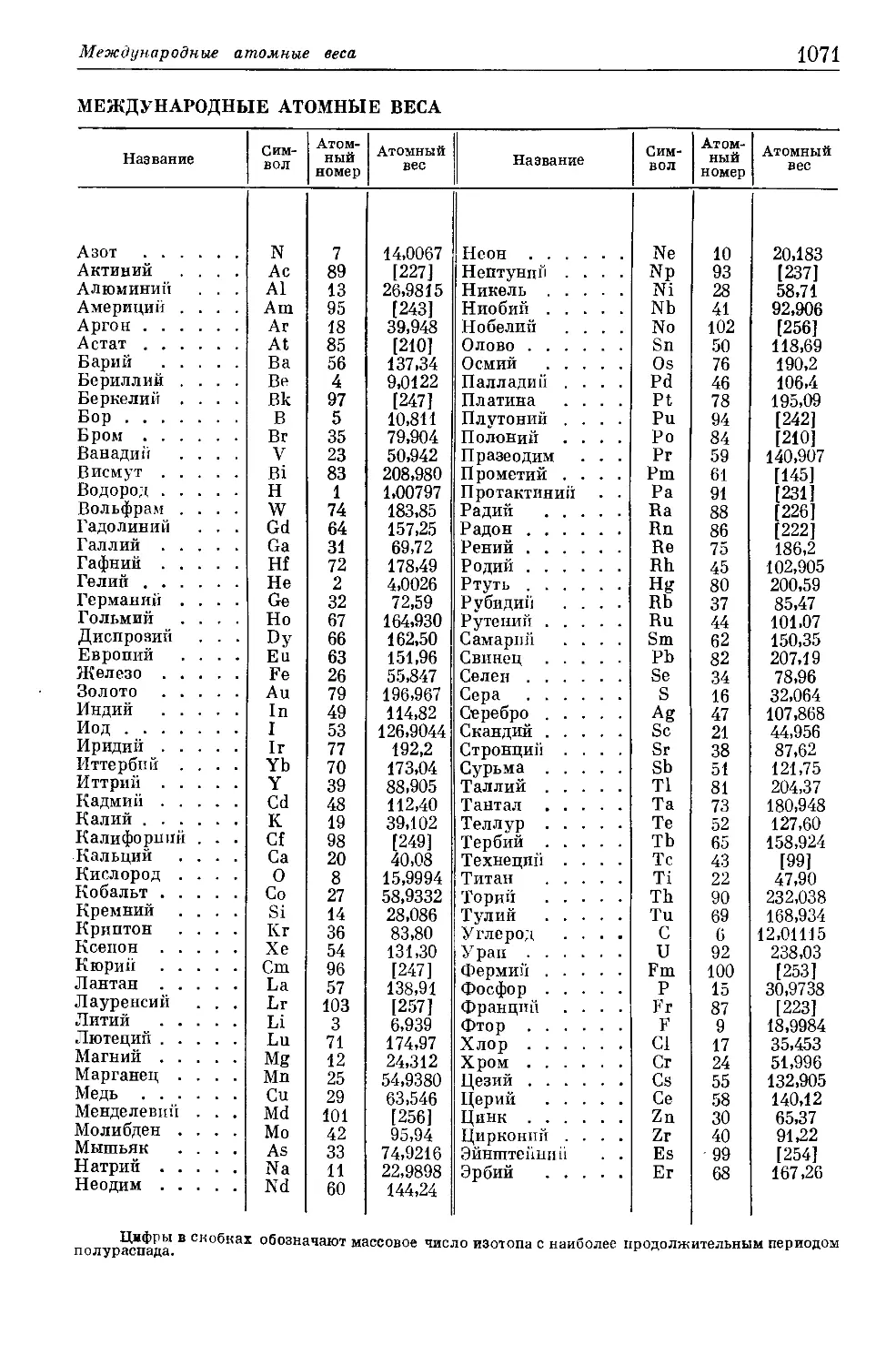
Международные атомные веса..........................................1071
Именной указатель ..................................................1072
Предметный указатель ...............................................1088
ПРЕДИСЛОВИЕ РЕДАКТОРА.
Книга В. Ф. Гиллебранда и Г. Э. Ленделя «Практическое руководство по неорганическому анализу» хорошо известна советскому читателю по изданному у нас в 1935 г. переводу на русский язык первого американского издания (1929 г.).
Эта книга является настольным руководством для всех химиков-аналитиков, как проводящих научные исследования в области аналитической химии, так и выполняющих рядовые анализы горных пород, минералов, продуктов металлургических производств, сплавов и т. п.
Старший из авторов книги — В. Ф. Гиллебранд скончался в 1925 г., еще до выпуска первого американского издания книги; в 1950 г. умер второй автор — Г. Э. Лендель. Их ученики и сотрудники по работе Дж. И. Гофман и Г. А. Брайт выпустили в 1953 г. второе издание книги. При подготовке книги к переизданию Гофман и Брайт пополнили ее новыми методами, вошедшими в практику анализа за 24 года, отделяющие второе издание от первого, но сохранили построение книги и ее основные характерные черты нетронутыми. Их работа выразилась в небольших по размерам, но многочисленных вставках и исправлениях, помещенных почти во всех разделах книги. При этом в книге сохранен тот принцип, который был принят Гиллебрандом и Ленделем в их первоначальном труде: помещать в руководстве лишь наиболее проверенные методы, особенно те, которые были испытаны в Бюро Стандартов США, и исключать методы, применимость которых ограничена анализом чистых солей и несложных по составу смесей.
Более значительной переработке подверглись главы, посвященные анализу редкоземельных элементов, металлов платиновой группы, олова, ниобия и тантала, урана, а также физико-химическим методам анализа. Заново написана глава о рении.
Сравнительно небольшим изменениям подверглась III часть книги — «Анализ силикатных пород», где Гофман и Брайт ограничились небольшими вставками, исправлениями и ссылками на оригинальную литературу. 2*
20
Предисловие редактора
Однако, как они указывают, этими методами были проведены с величайшей точностью десятки тысяч полных анализов различных горных пород, включая и наиболее сложные по составу. И если при выполнении отдельных определений иногда отступают от описанных методов, то в основном ход полного анализа силикатов остается и в настоящее время неизмененным.
Следует все же указать, что в книге имеется и ряд существенных пробелов. Это более всего следует отнести к ссылкам на исследования советских ученых, работы которых вообще слабо освещены в этом руководстве (за исключением, быть может, работ по анализу металлов платиновой группы). Не упомянуты и многие интересные исследования американских ученых (особенно за последние годы), возможно, потому, что эти работы считаются еще недостаточно проверенными практикой.
Обращая основное внимание на методы разделения элементов и на те методы их определения, в которых отделение и определение сливаются в одну операцию, авторы сравнительно мало места уделили специфическим методам определения отдельных элементов, особенно колориметрическим методам.
В настоящем издании перевода этой книги на русский язык редактором и переводчиком сделана попытка заполнить указанные выше пробелы путем внесения представляющихся необходимыми дополнений, отмеченных в тексте звездочкой (*). Из огромной литературы по аналитической химии, опубликованной в течение последних двадцати лет, в качестве дополнений дано сравнительно немногое, причем отдано предпочтение методам, пригодным для анализа сложных смесей и проверенным практикой работы в наших аналитических лабораториях.
Перевод глав I—IV, VI—XV, XXI-XXV, XL, XLI и LII—LVI сделан Ю. Ю. Лурье, а глав V, XVI—XX, XXVI—XXXIX и XLII — LI — Е. И. Гульдиной.
Ю. Ю. Лурье
ИЗ ПРЕДИСЛОВИЯ АВТОРОВ К ПЕРВОМУ ИЗДАНИЮ
За последние пятьдесят лет в области развития методов определения элементов и выяснения теоретических основ аналитических методов достигнуты крупные успехи. Многие элементы как сами по себе, так и в. простейших соединениях могут быть определены с большой точностью. Однако приходится сознаться, что для анализа более или менее сложных смесей, в которых обычно встречаются элементы, до сих пор еще не выработано методов, на точность и правильность которых можно было бы вполне положиться, т. е., иными словами, методы разделения элементов в основном остаются до сих пор неизменными, а новейшие методы их определения в отношении «селективных возможностей» мало чем отличаются от старых. Так, например, известны точные условия для определения алюминия путем осаждения его аммиаком, но перед аналитиком все же стоит задача отделить сначала алюминий от тех разнообразных элементов, в смеси с которыми он обычно встречается и которые также осаждаются аммиаком.
Поэтому в конечном счете химик-аналитик прежде всего должен уметь количественно получать чистые химические препараты. Самые тщательные предосторожности при взвешивании, отмеривании и конечном определении элементов не приводят к желаемым результатам, если аналитик не сумеет количественно приготовить соединение, на основании которого производят вычисления, и количественно отделить его от всех других соединений, могущих повлиять на правильность вычислений. Насколько большие трудности может представлять эта задача, можно судить по тому, какие делались ошибки, несмотря на огромные затраты времени и усилий, при определениях атомных весов. Так, например, в 1921 г. общепринятые атомные веса для алюминия, кремния и сурьмы были равны соответственно: 27,1, 28,3 и 120,2. В 1925 г. атомные веса этих элементов были изменены на 26,97; 28,06 и 121,77, причем разница здесь, выраженная pro mille, равна 4,8; 8,6 и 12,9.
22
Из предисловия авторов к первому изданию
В добавление к затруднениям общего порядка анализ минералов усложняется тем, что идеально чистые минералы встречаются лишь очень редко. Зависит это не только от того, что вследствие изоморфизма в минерал часто входят в качестве составных частей родственные члены данной изоморфной группы, но также и от того, что часто встречаются твердые растворы не родственных Между собой минералов. Кроме того, мине-ралы часто загрязнены механическими примесями, совершенно посторонними веществами, которые не удается удалить полностью. Поэтому анализ минералов превращается, собственно говоря, в анализ смеси минералов, аналогичный анализу горных пород, и эта сравнительно простая задача может стать чрезвычайно трудной не только по выполнению, но также и в отношении правильного толкования полученных результатов.
При беспристрастном разборе большинства имеющихся в печати трудов, посвященных методам определения элементов, видно, что большая часть этих методов разработана на основании опытов, проведенных с чистыми растворами, и очень мало или совсем не говорится о том, каким образом можно применять эти методы и какие результаты получаются при анализе более или менее сложных продуктов, в состав которых эти элементы обычно входят. Вообще, как правило, не имеется недостатка в удовлетворительных методах для анализа чистых соединений, но, с другой стороны, чувствуется большая потребность в развитии количественных способов разделения и определения соединений, входящих в состав сложных смесей. Так, например, точное определение ниобия, которое не представляет трудности в случае его чистых соединений, делается совершенно невозможным, когда этот элемент встречается вместе с танталом, Поэтому в настоящей книге основное внимание уделяется не описанию способов, которые с уверенностью могут быть применены только в специальных случаях, а приготовлению растворов, в которых потом будет произведено то или иное определение. Вполне очевидно, что если бы было возможно разработать метод анализа, пригодный для любых случаев, то он был бы слишком громоздким.
Применяемый в книге в параграфах под заголовком «Общие замечания» термин «обычный метод анализа» относится к общему способу проведения анализа, при котором пробу разлагают, упаривают полученный раствор с соляной кислотой для выделения кремнекислоты и последовательно осаждают затем аммиаком, оксалатом аммония и двузамещенным фосфатом аммония — для выделения группы В2О3, окиси кальция и окиси магния.
Часть первая
ОБЩИЕ УКАЗАНИЯ
Глава I
ВВЕДЕНИЕ
Аналитическая химия подразделяется на два больших раздела — качественный и количественный анализы. Как показывают названия, задачей первого является установление качественного состава вещества, задачей второго — определение относительного количественного содержания в нем каждой составной части. Иногда количественный анализ определяют, как приложение качественного анализа на практике. Это можно признать справедливым только условно, так как цели качественного и количественного анализа совершенно различны. В одном случае целью аналитика является выделение той или иной составной части в количестве, достаточном для ее идентификации, тогда как в другом случае он должен полностью отделить всю эту составную часть от сопровождающих ее веществ. Кроме того, в качественном анализе не имеет значения, вступают ли вещества в реакцию в стехиометрических отношениях или нет и имеет ли конечный продукт реакции определенный состав. По этой причине многие реакции, применяющиеся в качественном анализе, совершенно непригодны для целей количественного анализа или требуют значительного уточнения условий их проведения.
Если какую-нибудь составную часть надо определить в веществе, состав которого неизвестен, то в соответствующих местах хода анализа надо путем качественных испытаний установить, не присутствуют ли элементы, которые могут помешать определению. Эти качественные испытания в одних случаях должны предшествовать определению (например, перед определением марганца висмутатным методом надо проверить, нет ли в растворе кобальта), в других — они следуют за определением (например, после прокаливания и взвешивания осадка, полученного добавлением аммиака, исследуют, нет ли в этом прокаленном осадке иных веществ, помимо предполагавшихся). Проводить полный качественный анализ перед выполнением полного количественного анализа редко имеет смысл. Обычно выбирают метод анализа, который дает возможность выделить естественные группы элементов, а затем пли проводят дальнейшие разделения и определения внутри каждой группы (так поступают, например, с группой элементов, выделенных сероводородом из кислого раствора), или же всю выделенную группу взвешивают и уже после этого определяют ее составные части, как, например, при выделении щелочных металлов методом Смита.
Применяемые в количественном химическом анализе методы обычно основаны на вполне определенных химических реакциях. В весовых методах продукт реакции отделяется и взвешивается. В объемных методах конечная реакция определения проводится добавлением отмериваемого
26
Гл. I. Введение
объема раствора с известным содержанием в нем реагирующего вещества. Некоторые аналитические методы основаны на измерении количественных характеристик определенных физических свойств, например отношения растворов к свету: интенсивности их окраски, опалесценции, флуоресценции, преломляющей способности, вращения плоскости поляризации; в других случаях измеряют объем, занимаемый веществом в газообразном состоянии при определенных условиях температуры и давления. Применяют также спектрографические, спектрофотометрические, потенциометрические, полярографические, хроматографические и микроаналитические методы.
Следует различать анализ чистых веществ и анализ смесей. Определение элемента в его чистых соединениях объемным или весовым методом является простой технической операцией, которая к анализу смесей относится так же, как упражнения для развития пальцев относятся к игре на рояле. Такого рода упражнения необходимы, но надо понимать, что это только начало. Слишком часто обучение студентов заканчивается на этой ступени. Если не продолжить обучения дальше, то они унесут с собой из аналитической лаборатории совершенно ложное представление о задачах, возникающих у аналитика, и о точности, к которой он должен стремиться. Об искусстве аналитика можно судить по его способности выбрать правильный путь анализа каждого попадающего в его руки для исследования вещества, по его способности изменять существующие методы анализа или придумывать и разрабатывать новые методы анализа, если это необходимо, и, наконец, по способности доводить анализ до конца и правильно истолковывать получаемые результаты.
Первыми операциями в прикладном неорганическом анализе являются: 1) отбор пробы из поступившего для анализа образца, 2) приготовление лабораторной пробы, 3) приготовление пробы для взвешивания, 4) выбор подходящего метода анализа или разработка нового метода, 5) взятие навески пробы и С) получение раствора для определения. Если определение проводится дальше весовым методом, то следующими операциями будут: осаждение, фильтрование, промывание осадка, высушивание его, прокаливание, охлаждение и взвешивание. Если определение заканчивается объемным методом, то дальше следуют: приготовление раствора для титрования, установка титра этого раствора и само титрование.
Иногда условия таковы, что весь анализ можно сделать по стандартной методике. В этом случае выполнение анализа становится механическим, и он может быть сделан лицом, не имеющим специальной химической подготовки. Нет ничего особенно трудного или непонятного в таких операциях, как взвешивание, измерение, титрование, осаждение, фильтрование или промывание и прокаливание осадков. Несомненно, что в такой работе высокая точность достигается большой практической сноровкой, а последняя не требует специального химического образования, нужны лишь способности и навык.
Механические детали количественного анализа разработаны вполне удовлетворительно, чего нельзя сказать о химической стороне дела. Другими словами, весы, разновесы, объемно-аналитические приборы, реактивы, стеклянные, фарфоровые и платиновые изделия, приборы для фильтрования, горелки, сушильные шкафы, печи и даже методы определения отдельных элементов — все это для обычных целей мало нуждается
Точность анализа
27
в улучшении. Но методы количественного разделения элементов далеки от совершенства. Большая часть плохих определений получается из-за применения неверных или неточных методов анализа, а не из-за ошибок, допущенных при взвешивании, отмеривании и тому подобных операциях. Мы не хотим умалить значения тщательности в выполнении анализа, но следует более обращать внимание на то, что взвешивают, чем на то, как проводится взвешивание х. -
ТОЧНОСТЬ АНАЛИЗА
При обсуждении результатов химического анализа пользуются терминами точность и воспроизводимость 2, часто заменяя одно слово другим, что недопустимо, так как воспроизводимость является мерой того, как повторяются результаты при многократном проведении определения одним способом, а точность говорит о правильности полученных результатов, т. е. об их соответствии действительному содержанию. Аналитик заинтересован и в том, и в другом, ибо его результаты должны быть достаточно точными для той цели, для какой анализ проводится, и он не может достигнуть этой точности, если нет воспроизводимости, тем более что ему часто приходится давать результат на основании лишь одного определения и редко на основании более чем трех параллельных определений. Работник, получающий результаты анализа, заинтересован только в их точности и только в той точности, которая достаточна для его целей. Точность может быть равна лишь 10% относительным при определении фосфора или серы в некоторых сталях или доходить до 0,01 % относительного при определении основного металла" в чистых металлах или при определении атомных весов. Если достаточна малая точность, принимать •особые меры для достижения высокой точности — пустая трата времени.
Найти воспроизводимость того или иного метода легко, независимо от того, применяется ли он для анализа данного вещества одним аналитиком или группой квалифицированных аналитиков; надо только собрать результаты и обработать их подходящими статистическими методами 3. Если речь идет об оценке этих результатов, то величина воспроизводимости лишь указывает, чего можно ожидать при применении данного метода к данному материалу. Если этот метод подходит для анализа данного материала, то полученная средняя цифра будет близка к истинному значению, и найденную воспроизводимость метода можно принять за точность его. Но в том случае, когда метод не подходит для анализа данного материала, получение идентичных или близких результатов указывает лишь на высокую воспроизводимость метода, зря потраченные усилия и неверную информацию того лица, для которого этот анализ произво
ди. G. Е. F. L u п dell, Ind. Eng. Chem., Anal. Ed., 15, 221 (1933).
a* Термины «accuracy» п «precision» иногда переводят словами «правильность» и «точность», но так как под словом «точность» у нас чаще понимают то, что авторы называют термином «accuracy», и выражение «анализ сделан с правильностью до ±0,1%» звучало бы уж очень непривычно, мы предпочли оставить слово «точность» за термином «accuracy». «Воспроизводимость» достаточно хорошо передает понятие, выражаемое термином «precision». Прим, ред*
3 См., например, W. J. Y о u d е п. Statistical Methods for Chemists. John Wiley and Sons, 1951; R. F. M о r a n, Ind. Eng. Chem., Anal. Ed., 15, 361 (1942); ASTM Manual on Prcentation of Data, American Society for Testing Materials, 1916, Race St., Phila-delphis. Pa 1945; G. Wernimonl. Ind. Eng. Chem., Anal. Ed., 18. 587 (1946); там же, 19, 943 (1947); Anal. Chem., 20. 1132 (1948).
28
Гл. I. Введение
дился (если только аналитик не имеет возможности найти поправку, на которую надо помножить полученные результаты, чтобы увеличить их точность). Другими словами, если мы переходим от воспроизводимости к точности, то должны иметь в виду не только полученные результаты, но и то, какой материал анализировался и какой применялся метод анализа.
Точность химического определения, если оно заканчивается весовым способом, зависит прежде всего от того, насколько количественно выделен определяемый элемент в виде соединения, которое будут взвешивать, и насколько состав взвешиваемого соединения отвечает формуле, по которой будут проводить последующий расчет. Если другие элементы, присутствовавшие в анализируемом материале, не могут быть отделены от этого конечного соединения, то они также должны быть переведены в соединения определенного состава и их количества должны быть известны.
Так, например, при определении железа взвешиванием его в виде Fe.,O3 прокаленный осадок не должен содержать ни Fe3O4, ни тем более SiO2, А12О3 или Р2О5. Если смесь, полученная осаждением аммиаком, предполагается состоящей из А12О3, Сг2О3 и TiO2 и содержание А12О3 находят пр разности после определения Ст2О3 и TiO2, то в прокаленном осадке не должно быть ни СгО3, ни неоткрытых V2O5 или ZrO2.
То же можно сказать и в отношении объемных определений. Если объемное определение молибдена основано на восстановлении его цинком и титровании стандартным раствором перманганата, то такие вещества, как нитраты, мышьяк, вольфрам, ниобий, которые не восстанавливаются до определенной степени окисления, должны быть предварительно удалены. Такие элементы, как железо, хром, титан, ванадий, надо или удалить или точно определить, чтобы можно было внести на их присутствие соответствующую поправку.
В большинстве анализов само определение элемента является конечной ступенью после ряда проведенных операций. К ошибке определения прибавляются поэтому все ошибки, накопленные с начала анализа. Кроме того, имеются еще ошибки от недостаточно полного удаления веществ, мешающих конечному определению. Накопляющиеся с начала анализа ошибки могут быть случайными ошибками из-за плохо выполненных отдельных операций или же ошибками, свойственными процессам, применявшимся при предварительной обработке. Первых ошибок можно избежать, вторых — нельзя. Понятно, что ошибка в конечном результате анализа зависит также и от состава исследуемого материала. Фосфор в ферровольфраме нельзя определить с такой же точностью, как в ферросилиции.
Следует помнить, что литературные данные о точности аналитических методов часто показывают только, чего можно ожидать при использовании этих методов для анализа чистых солей. Но определить тот или иной компонент в смесях, в каких он обычно встречается, так же точно, как его определяют в чистых солях, в большинстве случаев невозможно. При анализе чистых солей аналитик имеет дело с реакциями одного вещества, при анализе смесей он сталкивается с реакциями ряда веществ. Приближение к предельной точности, которое может быть достигнуто опытным аналитиком, в значительной мере зависит от того, сколько времени последний может уделить анализу и насколько полно может быть отде
Точность анализа
29
лена определяемая составная часть от веществ, мешающих ее определению данным методом, или от того, насколько точно может быть учтено влияние на результат анализа тех примесей, которые не были или не могли быть отделены. Дополнительная работа, которую приходится выполнять при проведении таких анализов, в действительности представляет собой отчасти работу по приготовлению чистой соли, по которой устанавливалась вероятная точность метода. Но в этом отношении следует помнить, что производство чистых солей, при котором не имеет значения потеря небольшого количества вещества, значительно быстрее и проще, чем превращение всего определяемого компонента в чистую соль, как это необходимо в количественном анализе.
Трудности, которые возникают при попытке вывести наиболее вероятное содержание определяемого компонента, находя средние числа из результатов анализа, полученных разными аналитиками, можно иллюстрировать на следующих примерах. При определении окиси свинца в' свинцово-бариевом стекле результаты, полученные восемью аналитиками, работавшими параллельно, дали в среднем 17,25%, а среднее отклонение от этого среднего числа было равно 0,20%. В число результатов, из которых выводилось это среднее, входило и наиболее вероятное значение 1 17,50 %, которое оказалось, таким образом, за пределами среднего отклонения. При определении окиси магния в том же стекле среднее из данных, полученных восемью аналитиками, составило 0,047%, а среднее отклонение от этого среднего 0,017%. И здесь среди полученных результатов было наиболее вероятное значение содержания окиси магния 0,029%, но эта величина также выходила за пределы среднего отклонения. При определении содержания кремнекислоты в железной руде было получено 28 результатов анализа, проведенного 23 аналитиками. Среднее из всех определений составило 1,02%, а среднее отклонение 0,02%. Однако наиболее вероятная величина оказалась равной 1,05%.
В общем можно сказать, что наиболее вероятное значение содержания какого-нибудь компонента можно установить только после очень тщательного его определения методами, возможно более отличающимися один от другого 2.
При оценке результатов следует учитывать не только полученные цифры, но и то, какими методами эти цифры получены, какие другие компоненты входили в состав анализированного вещества и какова сумма результатов определения всех компонентов, если был выполнен полный анализ. Если анализ проводился двумя или несколькими аналитиками, надо учитывать и личные их особенности, если они известны. Здесь снова встречаются затруднения, ибо ни в коей мере нельзя быть уверенным, что аналитик, имеющий большой опыт в одном типе анализа, окажется
1 Оно было найдено в результате наиболее тщательных анализов, проведенных различными методами, и затем подтверждено суммированием результатов полного анализа и повторными анализами, сделанными со специальными предосторожностями другими аналитиками.
2 Например, при определении кремнекислоты в свинцово-барпевом стекле, содержавшем 17,5% РЬО и 1,41% ВаО, Г. Б. Ноульз получил 65,35; 65,3(5 и 65,35%, проводя выпаривание с соляной кислотой, и 65,38; 65,38 и 65,36%, выпаривая с хлорной кислотой. Если сделан полный анализ, иногда бывает желательным проверить наиболее вероятный результат определения какого-нибудь компонента, определяя его в искусственно приготовленной смеси, имеющей тот же состав, что и анализируемый материал, и содержащей известную добавку этого компонента.
30
Гл. I. Введение-
так же искусен, впервые выполняя совершенно новый для него вид определения или исследуя новый, незнакомый ему материал. В этом заключается слабая сторона многих так называемых арбитражных анализов. Такой анализ будет хорош только в том случае, если арбитр так же хорошо знаком с методом анализа и так же опытен в применении его именно к данному объекту анализа, как и тот аналитик, который дал вначале более правильный результат. Арбитр должен быть также очень осторожным и вдумчивым, потому что часто необходимость в арбитражном анализе возникает вследствие необычных условий, которые были обнаружены одним из аналитиков, но остались незамеченными другими.
ФАКТОРЫ ПЕРЕСЧЕТА И ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ
В весовых методах анализа фактором пересчета называется десятичная дробь, выражающая отношение массы определяемого компонента к эквивалентному весу некоторого другого вещества. В большинстве обычных анализов вряд ли имеются основания для применения факторов, рассчитанных с точностью, превышающей 1 на 2000. В некоторых руководствах, например, фактор для пересчета окиси железа на железо (Fe = = 55,847) дается в виде 0,699436, в других руководствах 0,6994. Применение первого фактора пересчета означало бы точность 1 на 699436, что бессмысленно; применение второго фактора предполагает точность 1 на 6994, такая точность возможна, хотя и трудно достижима в анализе. В действительности очень мало весовых определений железа пострадало бы при применении фактора 0,7. Точно так же фактор для пересчета сульфата бария (Ва = 137,34; S = 32,064; Н = 1,00797) на серную кислоту иногда дается в виде 0,42017, чаще в виде 0,4202. Но если не применяются совершенно исключительные меры для получения особенно точных результатов, аналитик имеет очень мало оснований применять более точный фактор, чем 0,42. Подобным же образом редко оправдывается применение фактора 0,7403 вместо 0,74 для пересчета окиси циркония на цирконий (Zr = 91,22) или 0,7930 вместо 0,793 для пересчета окиси вольфрама на вольфрам (W = 183,85). Наконец, фактор для пересчета хлорида серебра (Ag = 107,868, Cl = 35,453, Н = 1,00797) на соляную кислоту следует принимать равным 0,2544, а не 0,254 или 0,25441 или 0,254405, так как точность определения здесь выше, чем 1 на 600, но не выше, чем 1 на 25 000.
Аналитики обычно бывают довольно небрежными при представлении результатов анализов, и это приводит к тому, что возникают сомнения не только в отношении точности результатов, но и в отношении того, можно ли вообще им доверять.
Каждый полученный результат следует округлить до первой сомнительной для аналитика цифры. Например, полученный при определении кремнекислоты в стекле результат 75,15% следует округлить до 75,2%, если аналитик не проделал исключительно большой дополнительной работы, которая дает ему право утверждать, что точность анализа превышает 1 на 1500. Подобным же образом результат 3,513, полученный при определении углерода в чугуне, надо округлить до 3,51, а результат 0,0567 при определении серы в стали надо округлить до 0,057, потому что второй десятичный знак в первом результате и третий десятичный знак во втором результате всего являются сомнительными.
Глава II
ВЕСЫ И РАЗНОВЕСКИ
Точность аналитической работы в значительной мере зависит от точ-. ноети применяемых весов и разновесок, и поэтому на выбор тех и других должно быть обращено самое серьезное внимание.
ВЕСЫ
Качество весов
Хорошие весы должны удовлетворять следующим требованиям: а) Они должны быть точными и должны показывать одну и ту же массу при последовательных взвешиваниях одного и того же предмета.
Для этого плечи коромысла весов должны быть равной длины, коромысло не должно гнуться при нагрузке и ребра всех трех призм должны лежать в одной плоскости и быть параллельными одно другому.
б) Они должны реагировать на самые малые изменения в массе. Для обычных анализов достаточно, чтобы при нагрузке в 10 г изменение на 0,1 мг вызывало смещение нулевой точки на одно деление шкалы.
Чувствительность весов прямо пропорциональна длине плеч коромысла и обратно пропорциональна его массе и расстоянию между центром тяжести и осью колебания коромысла.
в) Они должны быть устойчивыми, т. е. коромысло должно возвращаться к своему горизонтальному положению после качания.
Ото зависит от правильного положения центра тяжести.
г) Коромысло должно качаться быстро, чтобы взвешивание могло быть сделано скоро. Период колебания должен быть не более 10 сек.
Это зависит от правильного положения центра тяжести, а также от длины и массы коромысла.
Уход за весами
Весы должны находиться в помещении, воздух которого должен быть свободен от разъедающих газов, и должны быть установлены на таком месте, чтобы оба плеча коромысла всегда имели одинаковую температуру. Весы нельзя, следовательно, ставить одной стороной близко к окну или к радиатору или на прямом или отраженном солнечном свете. Весы должны покоиться на прочном основании и должны быть возможно лучше защищены от сотрясений и колебаний. Идеальной подставкой является каменный или бетонный столб, погруженный основанием в грунт и
32
Гл. II. Весы и разновески
введенный в комнату без соприкосновения со зданием. Но это обычно бывает неосуществимо, и весы приходится ставить на прочный стол или полку и защищать от сотрясения поглощающим толчки материалом, например подставлять под стол толстую пробковую пластину и плотные резиновые кружки под уравнительные винты весов. Если пользуются искусственным освещением, источник его должен находиться вверху и позади головы взвешивающего и в таком положении, чтобы тепло от источника света нагревало оба плеча коромысла одинаково. Можно пользоваться также дневным рассеянным светом, если окно находится не слишком близко.
Помещать в шкафчик весов вещества, поглощающие влагу или двуокись углерода, обычно не имеет смысла, разве лишь в тех случаях, когда атмосферные условия, в которых находятся весы, могли бы вызвать их сильную коррозию. Эти вещества почти не предохраняют во время взвешивания, потому что небольшое количество применяемого поглотителя (к тому же обычно частично насыщенного) не может быстро очистить воздух, входящий в шкафчик весов при открывании дверец. В тех случаях, когда эти реактивы все же применяются, следует пользоваться натронной известью или плавленым хлоридом кальция, но никогда не надо применять серную кислоту.
Аналитический разновес
При выборе аналитического разновеса надо проявлять очень большую осмотрительность; при дальнейшей работе требуется осторожное •обращение и тщательный уход за разновесом. Хороший набор должен содержать разновески от 100 г до 5 мг. Бюро Стандартов США допускает два класса аналитических разновесов — М и S. К классу М относятся разновесы, пригодные для проверки стандартов, для работ очень высокой точности и для применения в исследованиях, требующих особенно точного определения массы. В класс S включаются разновесы, обычно применяемые в физических и химических лабораториях для большинства точных взвешиваний. Следующие ниже описания и таблицы допустимых отклонений взяты из циркуляра Бюро Стандартов *.
Разновески класса М
Материал, из которого сделаны разновески класса М, должен быть твердым, не магнитным и не должен корродировать на воздухе. Он не должен иметь ни малейшей пористости. Можно применять позолоченные разновески, хотя золото — мягкий металл.
Платина является наиболее подходящим материалом для мелких разновесок массой даже меньше 1 мг, но для разновесок массой от 20 мг и меньше можно применять алюминий. Все разновески, имеющие вид плоских пластинок, должны быть целиком сделаны из материала, поверхность которого не нуждается в защите от окисления или коррозии.
Все разновески должны быть из одного материала; допускаются только золотые и платиновые покрытия на разновесках из бронзы и т. и. На разновесках не должно быть глубоких впадин, острых углов и других
1 Circular 3, The Design and Test of Standards of Mass, 3d ed., 1918.* Допустимые погрешности для разновесов, выпускаемых в СССР, см. Н. Рудо, Точное взвешивание. Лениздат, 1949.*
Аналитический разновес
33
особенностей, которые могут повлиять на их неизменяемость. Разновески из латуни,-бронзы или других металлов, тускнеющих на воздухе, должны быть покрыты золотом, платиной или другим подходящим металлом. Поверхность разновесок не должна темнеть, и на ней не должно появляться пятен, если разновески положить на некоторое время в кипящую воду и затем высушить при 110° С, как это делается перед их поверкой.
Вся поверхность разновески должна быть гладкой и хорошо отполированной. Если на разновеске указана ее масса, то-линии пометки должны быть неглубокими и не должны иметь выступов и острых углов.
ТАБЛИЦА 1
Точность поправок и допустимые отклонения, класс М
(Лабораторные стандартные разновески высшей точности)
Колонка, озаглавленная «допустимые отклонения», показывает максимальные отклонения от помеченной массы, допустимые для каждой разновески, а колонка, озаглавленная «точность поправок», содержит данные о степени точности, до которой разновески обычно испытываются в Бюро Стандартов США.
Масса Точность поправон Допустимые отклонения Масса Точность поправок . Допустимые отклонения
г мг мг
кг г мг 2 0,01 0,10
20 0,01 100 1 0,01 0,10
10 0,01 50 мг
мг 500 0,001 0,05
5 1 30 200 0,001 0,05
2 1 10 100 0,001 0,05
1 1 5 50 0,001 0,03
г 20 0,001 0,03
500 1 3 10 0,001 0,02
200 0,1 1 5 0,001 0,02
100 0,1 0,5 2 0,001 0,01
50 0,1 0,3 1 0,001 0,01
20 0,01 0,2 0,5 0,001 0,01
10 0,01 0,15 0,2 0,001 0,01
5 0,01 0,15 0,1 0,001 0,01
Массы разновесок этого класса должны быть в пределах допустимых отклонений, указанных в табл. 1. Они могут быть выверены или соот-
ветственно их истинным массам, или по их кажущимся массам, определенным сравнением с латунными стандартными разновесками в воздухе; это соответствует «весу в воздухе по сравнению с латунью». Последнее вообще предпочтительнее, так как позволяет применять мелкие платиновые или алюминиевые разновески одновременно с латунными.
Разновески класса S
Разновески класса S должны быть сделаны из материала, не корродирующего быстро на воздухе. Материал этот должен быть твердым, не пористым и не магнитным. (Никель, хотя он и магнитен, может быть использован для покрытия им других металлов.) Большие разновески до 1 г включительно должны быть из латуни или бронзы или из другого материала, имеющего приблизительно такую же плотность, как эти металлы. Для разновесок, меньших 1 г, можно применять платину, а для разновесок, меньших 50 мг, — алюминий. Все разновески, имеющие форму пластинок, должны быть сделаны из материала, поверхность которого не
3 Заказ 522.
34
Гл. II. Весы, и разновески
нуждается в защите от коррозии. Разновески могут иметь любую из обычных форм, если в них нет особенностей, которые могли бы отразиться на их надежности. У разновесок не должно быть углублений с нижней стороны. В этом классе допускаются разновески с привинченными головками.
Поверхность разновесок должна быть гладкой и полированной. Если металл, из которого сделаны разновески, не обладает такой устойчивостью к коррозии, как никель или алюминий, то его следует покрыть платиной, золотом или никелем или же полакировать. Применяемый лак должен быть твердым, слой его должен быть умеренной толщины и гладкий. Нельзя покрывать лаком плоские разновески. В зависимости от влажности воздуха лак поглощает различные количества влаги, вследствие чего происходят такие колебания в массе разновесок, которые могут иметь значение. Так как свойства лака изменяются в широких пределах, нельзя дать определенных цифр, но для средних значений влажности, например от 40 до 70%, . можно ожидать изменения массы разновески в 100 г на 0,1 мг и несколько большего изменения в такой же суммарной массе меньших разновесок.
На каждой разновеске, за исключением рейтера и других разновесок, сделанных из проволоки, должна быть отчетливо помечена ее номинальная масса. На самых больших разновесках набора и на разновесках-пластинках до 100 мг включительно эти пометки должны иметь также и название единицы массы (г, мг). На прочих разновесках достаточно одной цифры. Линии, образующие цифры, должны быть неглубокими, широкими и не должны иметь засечек и острых углов. В каждом наборе имеется по две и по три разновески одинаковой массы. На них должны быть пометки, чтобы их легко было отличать одну от другой. Лучше всего поставить рядом с цифрой на одной разновеске одну звездочку, на другой — две звездочки, а если имеются три разновески одинаковой
ТАБЛИЦА 2
Точность поправок и допустимые отклонения, класс S (Обычные аналитические разновески) Колонка, озаглавленная «допустимые отклонения», показывает максимальные отклонения от помеченной массы, допустимые для каждой разновески, а колонка, озаглавленная, «точность ч поправок», содержит данные о степени точности, до которой разновески обычно испытываются в Бюро Стандартов США.
Масса Точность поправок Допустимые отклонения Масса Точность поправок Допустимые отклонения
г мг мг
кг г мг 2 0,05 0,10
20 0,1 100 1 0,05 0,10
10 0,01 50 мг
5 0,01 30 500 0,01 0,05
2 0,01 10 200 0,01 0,05
мг 100 0,01 0,05
1 5 5 50 0,01 0,03
г 20 0,01 0,03
500 1 3 10 0,01 0,02
200 1 1 5 0,01 0,02
100 0,5 0,5 2 0,01 0,01
50 0,1 0,3 1 0,01 0,01
20 0,1 0,2 0,5 0,01 0,01
10 0,05 0,15 0,2 0,01 0,01
5 0,05 0,15 0,1 0,01 0,01
Аналитический разновес
35
массы, то на третьей — три звездочки. Часто в таких случаях одну из разновесок оставляют совсем без звездочки, но это менее удобно.
Рекомендуется иметь для разновесок непроницаемый для пыли ящичек или коробку, в которой гнезда для цилиндрических разйовесок должны быть выложены каким-нибудь мягким материалом, например бархатом. Для каждой разновески должно быть отдельное гнездо, и гнезда должны быть достаточно широкими, чтобы вынимание и вкладывание разновесок не требовало усилий. В наборе должен быть специальный пинцет. Части пинцета, приходящие в соприкосновение с разновесками, могут быть из дерева или слоновой кости или могут быть оклеены бархатом или другой мягкой материей; они должны быть гладкими и не иметь острых ребер.
Разновески этого класса должны быть выверены в соответствии с допусками, приведенными в табл. 2.
Разновески этого класса можно выверить так же, как и разновески класса М, по их истинной массе или по их кажущейся массе, определяя последнюю сравнением с латунными стандартными разновесками в воздухе, т. е. по «весу в воздухе в сравнении с латунью». Последний способ предпочтительнее, так как позволяет применять мелкие платиновые и алюминиевые разновески одновременно с латунными.
Проверка разновесок и уход за ними
Новые разновески всегда следует проверить. Еще более необходимо проверить их снова через несколько месяцев, даже если ими не пользовались.
Разновески бывают самого различного качества, и только проверив каждый отдельный набор разновесок, можно сделать вывод об их точности и постоянстве.
Неточность разновесок часто остается необнаруженной не только из-за неуменья или отсутствия возможности их проверить, но часто из-за опасения, что калибровка разновесок дело слишком трудное и продолжительное для проведения в обыкновенной лаборатории. Грубая проверка на отсутствие больших погрешностей может быть сДелана очень небольшим числом взвешиваний. Нахождение поправки для каждой разновески, но без сравнительных взвешиваний, требует меньшей затраты времени, чем два прямых взвешивания каждой разновески или чем одно взвешивание методом тарирования х.
К разновескам никогда не следует прикасаться пальцами. Переносить их из ящика в шкафчик весов надо с помощью пинцета (имеющего кончики из слоновой кости), возможно меньше нажимая на разновеску. Разновески высшей точности никогда не следует оставлять открытыми в шкафчике весов. Разновески, применяемые в обычной работе, можно
1 Описания методов быстрого калибрования даны в NBS Sci. Paper, 527 (1925) и в статьеТ. W. R i с ha г d s [J. Am. Chem. Soc., 22,144 (1900)]. Более точные методы приведены в Циркуляре № 3 Бюро Стандартов США и в трудах: J. R. В е п о i t, Travaux et memoires du Bureau International des Poids et Mesures, 1907, p. 13 и В. Weinstein, Handbuch der Physikalischen Maasbestimmungen, 1888.* Проверка разновесок методами замещения и двойного взвешивания подробно изложена, в руководстве И. М. Кольтгофи Е. Б. Сендэл, Количественный анализ, Госхим-издат, 1948, стр. 233.*
3*
36
Гл. II. Весы и разновески
'Оставлять в весах на соответствующей подставке при условии, что шкафчик достаточно плотно закрывается и разновески Проверяются по крайней мере один раз в полгода Ч
ВЗВЕШИВАНИЕ
Весы должны быть обязательно арретированы, когда на них не работают или когда к их чашкам прикасаются. Опускать арретир надо очень осторожно, и приводить в движение коромысло легким взмахом руки или незначительном поворотом винта арретира.
Разновески и взвешенный предмет следует всегда помещать в центре чашек. Прежде чем приступить к взвешиванию, надо подождать, чтобы взвешиваемый предмет принял температуру весов. Окончательное взвешивание проводится при закрытом шкафчике весов. Предметы, которые электризуются при трении, не следует обтирать непосредственно перед взвешиванием; кварцевые разновески иногда электризуются, когда их вынимают из покрытых плюшем гнезд футляра 1 2.
Вещества гигроскопичные, летучие и вообще изменяющиеся на воздухе, надо взвешивать в закрытых сосудах, которые приоткрывают на мгновение перед взвешиванием. Пористые или порошкообразные вещества, высушенные нагреванием или в эксикаторе, также нужно взвешивать в закрытых сосудах, так как во время взвешивания они могут поглотить значительные количества влаги.
Некоторые гигроскопичные вещества, как, например, прокаленную окись алюминия или известь, можно взвешивать в тех тиглях, где их прокаливали, при условии, что тигли плотно закрыты крышками и что второе взвешивание после вторичного прокаливания проводится быстро <с разновесками, заранее установленными на чашке весов.
В обычных аналитических работах вполне удовлетворительную точность дают взвешивания методом коротких качаний 3 или методом одного «отклонения 4. Первый метод основан на том, что при состоянии, близком к равновесию, качания становятся настолько короткими, что при однократном колебании стрелки-указателя нельзя заметить их замедления, и потому средняя точка одного качания соответствует точке равновесия. Колебания стрелки могут меняться от четырех делений обычной шкалы до самых коротких колебаний, которые еще можно заметить. Колебания
1 J. J. Manley [Phil. Mag., 16 (VII), 489 (1933)], разбирая происхождение и природу тех пленок, которые образуются на разновесках, отмечает, что пленки могут появиться, если при изготовлении внутренней отделки ящичка применялся клей, содержащий свободные кислоты, или если бархат, находящийся в ящичке, был плохо промыт. Существуют весы, дающие возможность совсем избежать прикосновения к разновескам; в этих весах разновески переносятся с особых площадок, где они стоят внутри весов, на весы и обратно при помощи стержня, конец которого выведен из шкафчика наружу.
2 F. W. Van Straten и W. F. Ehret [Ind. Eng. Chem., Anal. Ed., 9, 443 (1937)] предлагают для удаления электростатических зарядов свой «высокочастотный вакуумный» прибор. Если воздух' очень сухой, заряд может быть снят действием ультрафиолетовых лучей. Продолжительность облучения зависит от интенсивности света и от расстояния между источником этих лучей и объектом. Так, в опыте, проведенном Родденом, был снят заряд с конической колбы из стекла пайрекс в течение 8 мин. Колба стояла на расстоянии 60 см от электрической дуги; температура в комнате была 23° С, относительная влажность 30%. Масса колбы в результате изменилась на 8 мг.
3 Н. L. Wells, J. Am. Chem. Soc., 42, 411 (1920).
4 P. H. В r i n t о n, J. Am. Chem. Soc., 41, 1151 (1919).
В звешивание
37
от 1/г до 2 делений шкалы достаточны, и обычно бывает удобным при определении равновесия считать центр шкалы средней точкой колебания. Обычная установка аналитических весов дает колебания в 5 делений на 1 мг. При такой чувствительности, если стрелка отклоняется от центра колебания в каждую сторону меньше чем на деления, можно считать,, что масса определена с точностью до 0,1 мг.
Если шкафчик весов снабжен отдельным арретиром для коромысла и отдельным для чашек \ то взвешивание можно сделать быстро и точно методом одного отклонения.
Тело, которое взвешивается в воздухе, теряет в своем весе столько,, сколько весит вытесненный им воздух. Поэтому два тела, уравновешивающие друг друга при взвешивании, имеют один и тот же вес только* в том случае, если объемы их равны или если взвешивание происходит в безвоздушном пространстве. Потерю веса при взвешивании в воздухе надо учитывать в работах, требующих высочайшей точности, например при определении атомных весов различных элементов или при сравнении методов установки титра кислот. Взвешивание небольших осадков (0,5 г и меньше), получаемых в обычных анализах, не требует введения поправки. Например, исправленная масса 0,5 г А12О3, взвешенной с латунными разновесками в воздухе при нормальных условиях, равна 0,50008 г; разность между этими величинами явно не выходит за пределы вероятной точности определения. Значения таких поправок на взвешивание в воздухе различных веществ приведены в табл. 3.
• ТАБЛИЦА 3
Истинные массы веществ, которые при взвешивании их в воздухе
(/> = 760 мм pm. ст', £ = 25° С) платино-иридиевыми или латунными разновесками показали массу точно 1 г
Вещество Плотность г/см* Применялись разновески Вещество Плотность г/см* Применялись разновески
платино-иридие-вые (<2 = 21,5) латунные (d=8,4) платино-иридие-вые (d = 21,5) латунные (d = 8,4)
Be 1,85 1,00060 1,00051 Na2CsO4 2,34 1,00046 1,00038
РЬ 11,34 1,00005 0,99997 КНС8Н4О4 1,64 1,00067 1,00059
А12О3 3,5 1,00029 1,0002 СвН6СООН 1,32 1,00086 1,00079
Fe3O3 5,24 1,00017 1,00009 AS2O3 3,86 1,00025 1,00017
СаО 3,40 1,0003 1,00021 К2СГ2О7 2,69 1,00039 1,00031
При взвешивании больших предметов следует ставить в качестве тары на вторую чашку весов предмет такого же размера и из того же материала. Если нет особых указаний, то всегда предполагается, что данные анализа основаны на взвешиваниях, проведенных с латунными разновесками в воздухе.
Другие приемы особо,точной работы, как, например, применение очень чувствительных весов, взвешивание замещением, двойное взвешивание, внесение поправок на очень малые ошибки в массе разновесок, входящих в хороший их набор, и т. п., являются лишь напрасной потерей
1* Такие весы выпускаются некоторыми иностранными фирмами. У нас они мало известны, поэтому подробности взвешивания методом одного отклонения мы опускаем. Прим, ред.*
38
Гл. it. Весы, и разновески
' ТАБЛИЦА 4
Приведение веса в воздухе к весу в пустоте 1 * * * * * * *
Вес тела в пустоте । находят добавлением к кажущемуся его весу в дрозду хе поправки, равной разности между весом воздуха, вытесненного этим телом, и весом воздуха, вытесненного разновесками:
М — вес в пустоте; W—кажущийся вес в воздухе;
р —плотность воздуха; dt— плотность взвешенного тела;
«1—плотность разновесок.
Таблица вычислена для р = 0,0012
Плотность взвешенного тела г/саи* Поправочный коэффициент k Плотность взвешенного тела , г/см9 Поправочный коэффициент k
применялись разновески применялись разновески
платино-иридие-вые (d—21,5) латунные (d = 8,4) кварцевые или алюминиевые (d — 2,65) платин о-иридие-вые (d = 21,5) латунные (<М,4) кварцевые или алюминиевые (с?= 2,6 5)
0,5 +2,35 +2,26 +1,95 5,0 +0,18 +0,10 —0,21
0,6 +1,95 +1,86 +1,55 6,0 +0,15 +0,06 —0,25
0,7 +1,66 +1,57 +1,26 7,0 +0,12 +0,03 —0,28
0,8 +1,45 +1,36 +1,05 8,0 +0,10 +0,01 —0,30
0,9 +1,28 +1,19 +CL88 9,0 +0,08 —0,01 —0,32
1,0 +1,14 +1,06 +0,75 10,0 +0,06 —0,02 -0,33
1,1 +1,04 +0,95 +0,64 11,0 +0,05 —0,03 —0,34
1,2 +0,94 +0,86 +0,55 12,0 +0,04 —0,04 —0,35
1,3 +0,87 +0,78 +0,47 13,0 +0,04 —0,05 -0,36
1,4 +0,72 +0,40 14,0 +0,03 —0,06 —0,37
1,5 +0,74 +0,66 +0,35 15,0 +0,02 —0,06 —0,37
1,6 +0,69 +0,61 +0,30 16,0 +0,02 —0,07 -0,38
1,7 +0,65 +0,56 +0,25 17,0 +0,01 —0,07 —0,38
1,8 +0,61 +0,52 +0,21 18,0 +0,01 -0,08 —0,39
1,9 +0,49 +0,18 19,0 +0,01 -0,08 —0,39
2,0 +0,54 +0,46 +0,15 20,0 0,00 —0,08 —0,39
2,5 +0,42 +0,34 +0,03 21,0 0,00 —0,09 —0,40
3,0 +0,34 . +0,26 —0,05 22,0 0,00 -0,09 —0,40
3,5 +0,29 +0,20 —0,11 23,0 0,00 -0,09 -0,40
4,0 +0,24 +0,16 -0,15 24,0 -0,01 -0,09 —0,40
* Циркуляр №19 Бюро Стандартов США (1924).
времени и денег, если их применяют в обычной аналитической работе, потому что точность других операций в такой работе настолько относительно невелика, что все эти предосторожности теряют всякий смысл. Несоблюдение их приводит к ошибкам х, не превышающим 0,1—0,2 мг. Если необходима большая точность, то, конечно, все эти усложнения становятся неизбежными. Вес в воздухе может быть приведен к весу в пустоте, как это показано в табл. 4.
1 Следует помнить, что такого рода ошибки не обязательно связаны с большими
осадками. Они возникают при взвешивании и небольших осадков в больших сосудах,
например в чашках, масса которых, может измениться между взвешиваниями в резуль-
тате изменения барометрического давления, температуры или влажности воздуха.
Влияние значения силы тяготения на величину поправки при приведении веса в воз-
духе к весу в пустоте следует учитывать лишь в самых точнейших анализах. О методе
взвешивания, дающем точность в 0,001 мг при нагрузке в 100 г путем очень незначительных конструктивных изменений в обычных весах, см. в статьях: А. Е. С о п-
г a d у, Proc. Roy. Soc., London, 101, (A), 211 (1922) и W. H. J. V er n on, J. Soc.
Chem. Ind., 53, 211 (1934).
Глава III
ПРИБОРЫ И РЕАКТИВЫ
Качество приборов и реактивов значительно улучшилось в последние годы, и аналитик имеет теперь гораздо меньше причин для жалоб, чем раньше. Несмотря на это, выбор прибора для каждой данной операции требует осторожности, а реактивы надо проверять на отсутствие в них примесей, которые могли бы повлиять на результаты анализа.
ПРИБОРЫ
Стеклянная и фарфоровая посуда
Все стеклянные изделия, приходящие в соприкосновение с растворами, применяемыми в анализе, должны быть сделаны из химически стойких сортов стекла. Насколько продуманным должен быть выбор стекла для применения его в том или ином анализе, видно из приведенных в табл. 5 результатов анализа четырех распространенных в США сортов химического стелла 1 (анализ проводился в 1941 г.) и из данных по исследованию их химической стойкости, графически представленных 2 на рис. 1, 2 и 3.
В рассматриваемом исследовании все испытания химической стойкости проводили на одних и тех же партиях конических колб (емкостью 250 мл) из стекла каждого сорта. Перед испытаниями колбы осторожно промывали внутри и снаружи пемзовым мылом, тщательно прополаскивали дистиллированной водой, высушивали в электрическом сушиль'ном шкафу при 110° С (их оставляли в шкафу на ночь) и взвешивали. Одну из колб каждого сорта стекла, обработанную таким же способом, применяли в качестве тары. Колбы, служившие тарой, прополаскивали и высушивали перед каждой серией взвешиваний, а мыли их мылом только между обработками различными реактивами.
Обработку реактивами проводили следующим образом. Брали по три колбы из стекла каждого сорта, йаливали в них по 200 мл того или иного реактива и одновременно ставили их на электрическую плитку. Нагревали до слабого кипения раствора и поддерживали его 6 ч. Во время этой обработки каждая колба была прикрыта воронкой, сделанной из стекла того же сорта; воронка частично служила холодильником. Для предупреждения толчков кипящей жидкости в каждую колбу помещали
1 Е. W i с h е г s, A. N. F i n n, W. S. С 1 a b a u g h, J. Research NBS, 26, 537 (1941), Ind. Eng. Chem., Anal. Ed., 13, 419 (1941); См. также P. H. Walker, F. W. Smither, J. Ind. Eng. Chem., 9, 1090 (1917); NBS Tech. Paper, 107 (1918). Последние авторы находили в некоторых сортах стекла до 10,9% ZnO, а также такие компоненты, как PbO, MnO, Р2О6, SO3 и Li2O. В старых стеклах содержится обычно больше мышьяка, чем в современных.
2 Е. W i с h е г s, A. N. F i n n, W. S. С 1 a b a u g h, цит. выше.
40
Гл. III. Приборы, и реактивы
сплющенный комочек губчатого золота. Примерно посредине 6-часового периода кипячения в колбы доливали горячую воду до первоначального объема жидкости (200 мл).
По окончании 6-часовой обработки колбы опорожняли, прополаскивали водой, осторожно протирали щеткой внутри и снаружи, снова прополаскивали и оставляли на ночь в сушильном шкафу при 110° С.
Рис. 1. Сравнение устойчивости различных сортов химической посуды к действию воды и кислотных реактивов.
Потеря в массе (средняя) колбы после кипячения в ней раствора в течение 6 ч. При обработке 6 н. соляной кислотой колбы через 3 ч были долиты кислотой той же концентрации.до первоначального объема. При обработке 95%-ной серной кислотой и хлорной кислотой в каждую колбу вливали 50 мл реактива и больше его не доливали. >
Марки стекла: 1 — гласбейк; 2 — кимбл; з — пайрекс; 4 — викор.
Взвешивание после обработки проводили таким же способом, как и взвешивание до обработки.
Затем эти же колбы обрабатывали вторым реактивом в течение 6 ч, а в некоторых случаях — и третьим реактивом.
Не следует, например, употреблять посуду из стекла, содержащего цинк, в тех случаях, когда нужно точно определить этот элемент, и особенно, если речь идет об определении очень малых его количеств. То же надо сказать о боросиликатных стеклах, если определяется бор. В исключительных случаях, например при разложении органического вещества для определения в нем очень малых количеств мышьяка, совсем нельзя применять стеклянную, посуду и приходится пользоваться посудой из плавленого кварца.
Приборы
41
На химическое стекло действуют все растворы. Действие кислотных растворов в общем слабее, чем действие чистой воды и щелочных растворов. Поэтому щелочные растворы при необходимости хранить их продолжительное время в стеклянной посуде надо, когда только это допустимо, предварительно подкислять.
1 ZZZZZZZZZZZZZZ^ZZZZZZZZZZZZZ^ZZZZZZZZ
з Хлорид натрия (без буферного раствора)
4 э
z Хлорид натрия + 0,00'/н соляная кислота
2
3
Хлорид натрия + буферный раствор (pH -6,2)
з Хлорид^натрия + буферный раствор (р Н = 7,0)
4 ' ~1 ..... I I ।
раствор ( р Н = 7,7)
2
3
/Vz^T^^Z^^ZZZZZZ^ZZZZ^ZZZZZZZZZZZZZ^ZZZZZZZZZZZZ^ZZZZZ
з Хлорид натрия + буферный раствор
у I ) р н =8,6)
2 Фосфатный буферный раствор ( pH =6,8)
2 з
20
Потеря в массе, мг
Рис. 2. Сравнение устойчивости различных сортов химической посуды к действию нейтральных реактивов.
Потеря в массе (средняя) колбы после кипячения в ней раствора в течение 6 ч. Применяли 5%-ный раствор хлорида натрия и 7%-ный раствор хлорида калия. В качестве буферных растворов применяли смеси KHfPO4 и Na2HPO4 соответствующих значений pH.
Марки стекла: 1 — гласбейк; 2 — кимбл; 3 — пайрекс; 4 — викор.
Следует также обратить внимание на часовые стекла, особенно потому, что они делаются обычно из обыкновенного стекла. Количества вещества, переходящего в раствор из часового стекла, могут быть очень значительными. Так, например, при кипячении воды в течение 60 мин в литровой платиновой чашке, покрытой часовым стеклом, вода извлекла из последнего 1 мг кремнекислоты. При кипячении в тех же условиях раствора аммиака в течение 30 мин в него перешло из часового стекла 1,4 мг кремнекислоты.
42
Гл. III. Приборы, и реактивы
*В дополнение к данным, о составе американских стекол (табл. 5) приводим опубликованные в справочной литературе данные о составе стекол для химической посуды, выпускаемых в СССР (табл. 6). В этой же таблице приведен и состав йенского стекла, производимого в Германии.
/ 2
3
4
£ 3
/
3
1
2
3
О 50 " ЮО >50 200 250 ЗОО
Потеря вмассе,мг
Рис. 3. Сравнение устойчивости различных сортов химической посуды к действию щелочных реактивов.
Потеря в массе (средняя) колбы после кипячения в ней раствора в течение 6 ч. Потеря в массе после обработки 0,5 н. раствором едкого кали в спирте (95%-ном) рассчитана умножением в 6 раз средней величины потери после кипячения с обратным холодильником в течение 1 ч.
Марки стекла: I — гласбейк; 2 — кимбл; 3 — пайрекс; 4 — викор.
' ТАБЛИЦА 5
Анализы химического стекла (в %)
Химический состав Марка стекла
Гласбейк Кимбл Пайрекс Викор 1
SiO2 4 78,4 74,7 81,0 96,3
В„О3 14,0 9,6 13,0 2,9
В2О32 2,5 5,6 2,2 0,4
ZnO Не обнар. 0,1 Не обнар. Не обнар.
СаО 0.1 0,9 Следы Следы
ВаО Не обнар. 2,2 Не обнар. Не обнар.
MgO Следы Следы Не обнар. Не обнар.
Na2O 5,0 4 к 6,4 3,6 <0,02
као Следы 0,5 0,2 <0,02
AS2O3 0,037 0,027 0,002 0,005
Sb2O3 0,038 0,009 Не обнар. Не обнар.
1 Веществ, не определенных в анализе, 0,3%.
8 Главным образом AltOs.
Химический состав распространенных лабораторных стекол 1 (в % весов.)
ТАБЛИЦА 6
Приборы
Химический состав, %
Сорт стекла SiO, BjOs Al,О, Na,0 CaO K,0 ZnO BaO MgO MnO PbO P,O, As2Oa Fe2O« NO,
№ 23 (химическое) . . . 68,39 2,66 3,89 9,42 8,51 7,14
ВБ (Безборное) .... 69,06 — 3,88 9,42 8,50 7,14 — — 2,0 — — — — — —
№ 2, то же 72,60 — 4,00 14,5 8,70 — — — — — — — — — —
№ 846 (химическое) . . 74,0 3,0 3,0 10,0 6,0 — — — 4,0 — — — — — —
Белое 72,0 — 1,5 13,50 10,0 0,5 — — 2,5 — — — — — —
Нейтральное № 59 (термометриче- 72,5 6,0 4,0 8,5 7,0 2,0 — — — — — — — — —
ское) 72,0 12,0 5,0 11,0 — — — — — — — — — —
VI-B (свинцовое) . . . 66,0 — 1,0 9,0 2,5 7,0 — — — — 14,5 — — — —
№ 12, то же 55,0 — — 3,8 0,26 9,2 — — 0,04 — 3,0 1,7 — — —-
№ 112 (баритовое) . . . 71,5 — — 10,0 4,5 5,5 — 7,0 1,5 — — — — — —
БД-1, то же 69,5 — — 1275 5,5 4,0 — 5,0 3,5 — — — — —
№71 (доломитовое) . . 73,62 — — 17,0 5,46 — — — 3,91 — — — — — —
№ 46 (молибденовое) 68,25 16,50 4,00 6,00 — — 5,00 — — — — — 0,25 — —
ЗС-5, то же ..... . 68,0 20,0 3,0 4,0 — 5,0
ЗС-8, » 66,5 23,0 3,0 3,7 — 3,8 — — — — — .
Жаростойкое . . \ . . 63,0 24,9 1,5 9,1 — 1,5
№35 80,5 12,0 2,0 4,0 0,5 1Ю — — — — —
Пайрекс I 80,5 12,0 1,5 5,0 — — — — 0,5. — — — 0,5 —
Йенское 64,70 10,9 4,2 7,5 0,63 0,37 ' 10,9 —— 0,21 0,01 — 0,14 0,25 .
Дюробакс 65,7 7,8 6,0 2,0 7,8 2,9 — 6,8 1,0 — —
Мазда 57,6 — 25,0 — 7,4 2,0 — — 8,0 —
Сверхпайрекс 85,4 8,32 1,99 3,54 — — — — 0,34 — — — 0,35 1
Кварцевое 99,95 — 0,01 0,04 — 0,028 — — 0,012 — — — — 0,04 —
16Ш 67,5 2,0 2,5 14,0 7,0 — 7,0 — — — — — — —
1 По С. Ф. Веселовскому, Стеклодувное дело, Изд. АН СССР, 1952.
44
Гл. III. Приборы и реактивы
Стекло № 23 идет на производство стаканов, колб и т. и. Из стекла 16ш вырабатываются ареометры, термометры и мерная посуда. Стекло № 59 служит только для производства термометров, показывающих температуру выше 360° С. Предельная разность температур, выдерживаемая стеклом № 23, равна 135° С, стеклом № 846 — 160° С, «жаростойким» стеклом — 250° С. «Жаростойкое» стекло по составу и свойствам близко к американскому стеклу пайрекс.
Приводим также значения линейного коэффициента расширения различных стекол, от величины которого зависит сопротивление стекла резким колебаниям температуры: обыкновенное стекло — 9 • 10-6, жаростойкое стекло и пайреке — 3,2 • 10~6, стекло викор — 8 • 10-7, прозрачное кварцевое стекло — 5 • 10“7.
•Исследования химической стойкости стекла в СССР проводятся методами, значительно отличающимися от метода, который описан выше авторами (обрабатывают, например, стекло, измельченное до определенной степени величины зерна, или подвергают стекло действию иодэозина и т. п.). Результаты этих исследований несравнимы с теми, которые даны на рис. 1, 2 и 3, и поэтому мы их не приводим. Доп. ред*
На фарфор растворы действуют вообще слабее, чем на стекло. Для сравнения с действием растворов едкого натра на стеклянную посуду (см. рис. 3) можно привести результаты, полученные 1 при обработке этим реактивом фарфоровых чашек: при нагревании в течение 6 ч на паровой бане чашек, наполненных 2,5 н. раствором едкого натра, каждая из них потеряла в массе только 2,5 мг. Если какой-нибудь раствор надо оставить на продолжительное время, лучше поместить его в фарфоровую посуду, а не в стеклянную. Нужно помнить, однако, что химический состав стекла и фарфора совершенно различен и что при употреблении стекла вместо фарфора иногда может произойти меньшее загрязнение каким-нибудь элементом, например алюминием.
Линейный коэффициент расширения фарфора равен в среднем 3,56 • 10~в при температурах от 20 до 200° С и постепенно возрастает до 4,69-Ю-6 при 1000° С. Верхней температурной границей применимости глазурованного фарфора является температура 1250° С, неглазурованного фарфора 1400° С.
Для внесения поправок в результаты анализа на ошибки, вызванные извлечением различных веществ из стеклянной или фарфоровой посуды, обычно ставят «холостой» опыт с одними реактивами (без анализируемого йещества), проводя его через все стадии анализа. Это, однако, не приводит к желаемым результатам, если присутствующие в растворе вещества реагируют в «холостом» опыте иначе, чем при проведении самого анализа; так, например, получающийся в анализе большой осадок гидроокисей алюминия и железа увлекает с собой всю кремне кислоту, мышьяк и сурьму, извлеченные из стекла, а очень маленький осадок гидроокисей алюминия и железа, получаемый в «холостом» опыте, может и не захватить полностью этих веществ.
Платина и заменяющие ее сплавы
Почти все платиновые изделия содержат иридий, родий, медь или медь вместе с родием, которые прибавляются к платине для увеличения ее твердости. Потеря в массе от улетучивания, происходящая при сильном
1 С. Е. Waters, NBS Tech. Papers, 105 (1917).
Приборы
45
прокаливании (выше 1100° С) некоторых платиновых изделий, имеет в большинстве случаев своей причиной присутствие в них иридия. Поэтому чем меньше платиновая посуда содержит иридия, тем лучше. Однако некоторое оптимальное содержание иридия в платине необходимо для придания изделию требуемых механических свойств.
В Бюро Стандартов США для тиглей применяют сплав, содержащий 0,6—1 % иридия; для чашек содержание иридия в платине доводят до 2-2,25%.
Имеются указания 1 на то, что родий повышает прочность платиновых тиглей и понижает потерю, происходящую от прокаливания и от действия кислот. По данным авторов \ наиболее высокими качествами обладает сплав платины с 3—5% родия. Для подтверждения этого следует, однако, произвести дальнейшие исследования таких сплавов. При более высоком содержании родия на тиглях при их использовании могут появиться трещины.
Прежде платиновые изделия всегда содержали железо, и теперь еще надо опасаться его присутствия. Если железо присутствует в платине в значительных количествах, его легко обнаружить, нагревая металл до темно-красного каления, лучше всего в муфельной печи. Тогда поверхность платинового предмета покрывается темным или даже красноватым налетом окиси железа. Для доказательства присутствия железа прокаленное изделие из платины обрабатывают затем соляной кислотой — получается окрашенный в желтый цвет раствор, в котором можно открыть железо обычными реакциями качественного анализа. Этим способом обнаруживают следы железа даже тогда, когда исходный металл, служивший для изготовления платиновой посуды, железа не содержал, но железо попало в платину из применявшихся матриц или вальцев. В таких случаях железо находится на поверхности платины. Поэтому рекомендуется перед нагреванием все новые тигли подвергать обработке горячей соляной кислотой.
Иногда после прокаливания можно открыть присутствие кальция по щелочной реакции, которую обнаруживают, прикладывая к поверхности прокаленной платины смоченную водой красную лакмусовую бумажку. Эта реакция наблюдается лишь в отдельных местах 2.
Не следует прокаливать платиновые сосуды в соприкосновении с какими-либо другими металлами; обычной ошибкой является употребление нихромовых треугольников. Также надо избегать соприкосновения платины с соединениями легко восстанавливающихся металлов и с фосфидами, арсенидами, сульфидами. Сильно разрушают платину окиси, гидроокиси, нитраты, нитриты и цианиды щелочных металлов и бария. Карбонаты щелочных металлов только слегка разрушают платину, когда тигель прокаливают на голом пламени, и несколько сильнее при прокаливании в муфеле. Пиросульфаты щелочных металлов действуют в большей мере: обычно при сплавлении с этими веществами в раствор переходит от 1 до 3 мг платины. Нельзя, конечно, помещать в платиновую посуду вещества, которые реагируют друг с другом с выделением хлора,
1 G. К. Burgess, Р. D. Sale, J. Ind. Eng. Chem., 7, 561 (1915).
2 Бюро Стандартов США опубликовало несколько циркуляров и статей о качестве обычной платиновой посуды, обращении с нею, исследовании ее свойств. См. G. К. Burgess, Р. D. S а 1 е, J. Ind. Eng. Chem., 6, 452 (1914); 7, 561 (1915); NBS Sei. Paper, 254 (1915); G. K. Burgess, R. G. Waltenberg, ibid., 280 (1916).
46 Гл. III.. Приборы и реактивы
например царскую водку, смеси соляной кислоты с пиролюзитом или некоторыми окислами редкоземельных металлов.
Растворяющее действие на платину кипящей серной кислоты может быть почти полностью устранено, если нагревание вести в присутствии избытка сернистого ангидрида \ который или образуется в самом растворе действием горячей серной кислоты на уголь или с-еру, или вводится в раствор извне по трубке. Было найдено, что после кипячения в течение 6 ч 50 мл серной кислоты с 2 г серы в покрытой платиновой чашке емкостью 200 мл кислота совсем не содержала платины, а серы осталось только 0,2 мг. Но когда в покрытой чашке кипятили 20 мл одной только серной кислоты в течение 1 ч, в раствор перешло 7,7 мг платины. При кипячении хлорной кислоты в платиновой посуде платина или совсем не -переходит в раствор, или переходит в ничтожно малых количествах.
Прямое соприкосновение платиновых изделий с несгоревшим газом, коптящим пламенем, тлеющим углем и т. п. приводит к образованию карбида платины, который делает тигель хрупким и легко дающим трещины.
Платиновые тигли должны быть всегда чистыми, полированными и иметь правильную форму. Очищать тигель можно, производя в нем осторожное плавление пиросульфата в течение нескольких минут, после чего сплав выливают на сухой металл или камень, а тигель и крышку опускают ненадолго в горячую разбавленную (1:1) соляную кислоту. Для очистки от некоторых загрязнений могут применяться другие плавни и кислоты, например карбонат натрия Или фтористоводородная кислота. Когда тигли начинают тускнеть, их надо осторожно полировать песком. Для этой цели следует применять песок с округленными зернами, предпочтительно очень тонкий, например в 75—100 меш (d —0,2—0,15 мм). Песок смачивают водой и полируют им тигель, растирая песок пальцем или мягкой тканью. В настоящее время платиновые тигли и чашки продаются вместе с деревянными формами, которые незаменимы для разглаживания всяких впадин и бугров, образующихся на этих сосудах.
Для выполнения многих операций платиновые тигли можно заменить тиглями из других материалов, а в некоторых случаях такая замена обязательна. Так, например, кварцевые тигли * 2 вполне могут применяться для прокаливания и взвешивания в них многих веществ, а также для сплавления с пиросульфатом, если не требуется последующего определения кремне кис лоты. Сплавление с перекисью натрия надо проводить в железных, никелевых или фарфоровых тиглях, в зависимости от цели этой операции. Сплавление с едкими щелочами лучше всего проводить в золотых или серебряных тиглях.
Было показано 3; что сплавление, производимое для определения щелочных металлов методом Смйта (стр. 1006), можно вполне удовлетворительно проводить в железных и никелевых тиглях; тигли эти должны иметь такую же форму, как и платиновые тигли, применяемые в методе Смита, и должны быть снабжены плотно закрывающимися крышками.
Ч.Т. Conroy, J. Soc. Chem. Ind., 22, 465 (1903); L. W. M с С a у, 8th Intern. Congr. Appl. Chem., 1—2, Sect. I—II, 1912, p. 351.
2 Кварцевые тигли легко электризуются и слегка гигроскопичны. Например, тигли, весившие 13,4608 и 11,1330 г, простоявшие 1 ч при 25° С в атмосфере с относительной влажностью 45%, увеличились в массе соответственно на 0,8 и 0,7 мг.
3 A. W. Е р р е г s о n, R. В. R u.d у, Ind. Eng. Chem., 17, 35 (1925).
Приборы
Если учитывать ограничения, связанные с низкой температурой плавления, то для многих операций вместо платиновой посуды можно применять золотые или золотые, покрытые платиной изделия (например, содержащие приблизительно 25% платины, наплавленной на золотую основу), особенно чашки, которые редко подвергаются нагреванию до температур, близких к температуре плавления золота. Азотная и хлорная кислоты на золото действуют сильнее, чем на платину.
Необходимые инструменты и приборы
Дать полный список оборудования, которым должна быть снабжена современная аналитическая лаборатория, было бы трудно. Мы остановимся здесь лишь на некоторых предметах, которые^почти незаменимы.
Рис. 4. Тигельные щипцы с платиновыми наконечниками.
Среди них нет ни одного принципиально нового, но многие из них пока еще мало распространены, гораздо меньше, чем они того заслуживают, поэтому мы их здесь и опишем.
Тигельные щипцы. На рис. 4 показаны тигельные щипцы с платиновыми наконечниками особой формы, предложенные много лет тому назад. Части АВ, сделанные из платины, имеют внутри гнезда, в которые вставляются концы ручек, изготовляемых из более дешевого металла. Такими щипцами можно крепко захватывать горячий тигель и придавать ему любое положение, а при плавлении на паяльной горелке сообщать содержимому тигля вращательное движение, не сдвигая при зтом крышки, как это приходится делать, когда применяют тигельные щипцы обычной формы.
Воздушные бани (радиаторы) для выпаривания жидких и удаления летучих твердых веществ. На рис. 5 показано очень полезное приспособление, с помощью которого можно на своем рабочем столе и особенно под тягой быстро выпаривать жидкости почти при любой желаемой температуре; можно также обезвоживать твердые вещества, причем значительно безопаснее, чем на железной плитке или песочной бане. Мы не можем указать, кто первый предложил эту форму воздушной бани, но йриме-няется она уже давно и в принципе идентична «никелевому стакану» Яннаша. Никель имеет перед железом известное преимущество в том, что он не ржавеет, но зато радиатор, имеющий форму, показанную на рис. 5, легко сделать из куска листового железа. Дно прикрепляется своим зубчатым краем к краю конической верхней части. Тигель, помещенный внутри радиатора на платиновом треугольнике, равномерно нагревается горячим воздухом, и таким образом можно быстро выпарить, без кипения и разбрызгивания, большое количество жидкости, даже серной кислоты. Сделав дно радиатора из платинового листа, можно этим увеличить эффективность и срок службы радиатора; с другой стороны, и железное дно можно много раз сменять, пока не износятся бока
48
Гл. III. Приборы и реактивы
7
Рис. 5. Радиатор (воздушная баня) для быстрого и безопасного выпаривания:
1 — коническая часть; 2 — основание.
радиатора. Последние можно сделать более стойкими к действию высокой температуры, покрыв их «серебряной» краской — порошком алюминия. В настоящее время можно приобрести радиаторы из кварца с вплавленными в них кварцевыми треугольниками.
Радиатор для быстрого выпаривания показан на рис. 5. Его коническая часть 1 сделана из листового железа или алюминия (или из никеля, по Яннашу). Высота ее около 7 см, диаметр вверху 7 см, у дна 5 см. Основание 2 может быть из железа, никеля или платины, но не из алюминия, который не выдерживает прямого нагревания пламенем газовой горелки. Лучше всего применять платину вследствие продолжительного срока ее службы и большой излучающей способности. Для прикрепления дна к конической части зубцы его загибают кверху. При желании можно значительно ускорить выпаривание, вставляя часть 1 в чугунное кольцо несколько большего внутреннего диаметра, чем отверстия радиатора.
Удобную воздушную баню на не-.сколько тиглей можно сделать, покрыв железную сковороду листом асбестового картона, имеющим отверстия, подходящие для тиглей разных размеров; отверстия должны быть таких размеров, чтобы донышки тиглей не доходили до дна сковороды по крайней мере на 1 см. Неиспользуемые отверстия закрывают тигельными крышками.
Вместо сковороды можно согнуть в кольцо узкую полоску железа и поставить на железную пластинку.
Такого же рода установки применяются для выпаривания жидкостей, высушивания осадков и даже для обугливания фильтров. В них происходит нагревание небольшого пространства с помощью отраженных инфракрасных лучей; нагревание это производится сверху и с боков, что предупреждает часто наблюдаемое в обычных условиях «выползание» солей ^верх по стенкам сосудов.
Таким же способом можно удалять из платиновых чашек значительные количества аммонийных солей и других летучих твердых веществ, не опасаясь разбрызгивания или перегрева. Для этого чашку с сухим или почти сухим веществом вставляют в другую чашку, большего размера, так, чтобы дно внутренней чашки было на некотором расстоянии от дна внешней чашки. Внешняя чашка может быть железной, но большая теплопроводность и излучающая способность платины заставляют предпочитать ее всякому другому металлу. Алюминий нельзя применять, так как он не выдерживает сильного нагревания.
Непосредственное соприкосновение обеих чашек друг с другом устраняется путем наложения полосок асбестового картона на края внешней чашки. Если содержимое внутренней чашки вначале влажно, то его высушивают, нагревая чашки небольшим пламенем; затем пламя можно уси-
Приборы
49
железа или при опреде-
Рис. 6. Установка для изолирования содержимого тигля от газов пламени при прокалива нии:
1 — асбестовый картон;
2 — платиновый нружок.
лить и дальше вести нагревание уже без наблюдения. Вместо радиатора можно иногда применять песочную баню Ч
Дырчатая пластинка для тиглей. На рис. 6 показано устройство, при помощи которого можно во время прокаливания изолировать содержимое тигля от соприкосновения с продуктами горения газа, как это необходимо, например, при прокаливании окиси лении серы сплавлением с карбонатами.
Для этой цели сначала были предложены1 2 дырчатые глиняные пластинки для определения зольности углей. Позднее Лунге и другие рекомендовали асбестовый картон с проделанным в нем отверстием для тигля. Однако асбестовый картон имеет тот недостаток, что он быстро разрушается и волокна его пристают к тиглю.
Лучше всего — вырезанная кружком пластинка из толстого листа платины: она может служить очень долго, чиста, хорошо проводит тепло. В этой пластинке надо проделать отверстие такого диаметра, чтобы тигель вошел в него на две трети своей высоты. Желательно иметь набор таких пластинок для тиглей разного размера, хотя зто, конечно, несколько затруднительно вследствие высокой цены платины.
Такой платиновый кружок можно приме-. нять отдельно, помещая на кольцо обыкновенного штатива в горизонтальном положении, или, лучше, вместе с асбестовым картоном, в котором надо вырезать отверстие большего диаметра, чем диаметр тигля. Картон укрепляют в наклонном положении, как показано на рис. 6, чтобы продукты горения газа отводились в сторону.
Если нет платинового кружка, можно пользоваться и одним асбестовым картоном с отверстием.
При прокаливании на паяльной горелке
тогда также получаются хорошие результаты, но при применении обыкновенной горелки хорошие результаты получаются не всегда.
Лодочка или совочек для взвешивания. На рис. 7 изображен очень удобный совочек для отвешивания пробы вещества. Совочек изготовляют из платинового листа. Длина совочка около 7,5 см, ширина в развернутом виде 5 см. На весах совочек уравновешивают свинцовой гирькой. Преимуществом такого совочка по сравнению с часовым стеклом является то, что из него очень удобно пересыпать навеску в узкогорлые сосуды.
Стеклянный футляр для взвешивания тиглей. Для взвешивания тиглей часто бывают необходимы стеклянные футляры разных размеров.
1 В статье J. A. Scherrer [Ind. Eng. Chem., Anal. Ed., 5, 22 (1933)] описано применение особых резиновых колец для поддерживания стаканов в паровых банях с целью ускорения процесса выпаривания.
2 J. Lowe, Z. anal. Chem., 20, 224 (1881)s
4 Заказ 522.
50
Гл. III. Приборы и реактивы
На рис. 8 показан футляр из стекла пайрекс, применяемый тогда, когда горячий тигель, содержащий гигроскопическое вещество, надо перенести в эксикатор, охладить и затем взвесить при минимальном соприкосновении его с воздухом. Нижняя часть футляра должна быть невысокой, чтобы горячий тигель можно было поставить в него щипцами, показанными на рис. 4 (стр. 47), не задевая крышки тигля. Верхнюю часть футляра надевают так, чтобы маленькое отверстие в ней пришлось почти против такого же отверстия в нижней части, оставляя совсем ничтожный просвет для уравнивания давления внутри и вне футляра. Тигель с фут-
ляром помещают в эксикатор и по - охлаждении взвешивают, поместив
другой такой же футляр на вторую чашку весов
в качестве тары.
Ступка и пестик. В каждой лаборатории должна быть агатовая ступка с таким же пестиком. Если можно приобрести только одну ступку, рекомендуется взять ступку с внешним диаметром 9 см; если
имеется возможность купить две ступки, желательно, чтобы одна из них имела диаметр 7,5 см, вторая — 12,5 см. Для дробления
Рис. 7. Платиновый совочек для взвешивания.
очень твердых веществ необходима так называемая «алмазная» ступка (см. рис. 37, стр. 891), чсделан-ная из закаленной стали. В настоящее время можно приобрести ступки, в которых чаша из карбида бора
Рис. 8. Футляр для взвешивания тиглей:
1 — тигель с крышкой;
2 — крышка футляра; з — шлиф.
вставлена в оправу ий нержавеющей стали, а пестик также сделан из карбида бора и вставлен в ручку
из нержавеющей стали. Такие ступки с пестиками
оказывают чрезвы-
чайно большое сопротивление трению (их твердость уступает только твердости алмаза), и их применение не приводит обычно к загрязнению анали-
зируемого вещеотва.
Колориметры и фотометры., В хорошо оборудованной лаборатории должен быть визуальный колориметр типа Дюбоска. Необходим также фотоколориметр.- Если в лаборатории проводятся научные исследования, большую помощь в них может оказать и спектрофотометр. Здесь нет возможности подробно разобрать преимущества и недостатки всех имеющихся в продаже приборов этого типа, но на стр. 173 мы остановимся немного на общих принципах проведения фотометрических определений.
Нефелометры. Нефелометры 1 применяются для определения следов тончайших осадков, не оседающих на дно сосуда, проходящих сквозь обычные фильтры, но отражающих или рассеивающих лучи света. Они
1 Т. W. R i с h а г d s, Z. anorg. Chem., 8, 269 (1895); Т. W. R i с h а г d s, R. C. W e 11 s, Am. Chem. J., 31, 235 (1904); T. W.-Ri chards, ibid., 35, /510 (1906); 8th Intern. Congr. Appl. Chem., 1—2, Sect. I—II, 1912, p. 423; P. А. К о b e r, J. Biochem., 13, 485 (1913). Теория нефелометрии в простой форме дана в статье Р. V. W е 11 з, J. Am. Chem. Soc., 44, 267 (1922).* Нефелометрические методы анализа описаны в книге Д. Г, Й оу, Фотометрический химический анализ, т, II, Нефелометрия, ОНТИ, 1936.* ’
Приборы
51
не могут применяться в тех случаях, когда осадок оседает на дно в течейие 2—3 ч, необходимых для проведения определения. Главным источником ошибок при использовании нефелометра является различное состояние осадков, полученных в разных условиях. Поэтому условия осаждения должны соблюдаться очень точро и должны быть совершенно одинаковыми и при приготовлении стандарта, и при выполнении анализа. Разбавление раствора до определенного объема после того, когда осаждение проведено, недопустимо. Для полного образования осадка необходимо некоторое время, обычно часа два или больше. Нужна,, большая осторожность, чтобы избежать загрязнений, которые могут повлиять на результат анализа, например наличие взвешенных частиц или присутствие хлоридов в применяемой дистиллированной воде отразится на определении следов хлоридов.
К большинству выпускаемых в настоящее время колориметров прилагаются дополнительные приспособления для выполнения нефелометрических определений.
В указанной выше работе Ричардс описывает конструкцию и работу простого нефелометра. Две пробирки поставлены близко и слегка наклонно одна к другой, так что их можно закрывать от яркого источника света скользящей заслонкой. Пробирки рассматриваются сверху, через две тонкие призмы, которые дают их изображения рядом, как в обычном полутеневом поляриметре. В одной из пробирок осаждают определяемое вещество в виде тонкой опалесцирующей мути добавлением соответствующего реактива; в другой пробирке таким же способом обрабатывают раствор, содержащий известное количество того же вещества. Оба осадка отражают свет, и пробирки кажутся слабо светящимися. Если при одинаковом положении заслонок обе половины поля зрения кажутся освещенными одинаково, то можно принять, ято количества обоих осадков равны. Если обе половины поля зрения кажутся неравно освещенными, изменяют положение заслонок, пока не получится одинаковое освещение, и по их положению рассчитывают относительные количества осадков в двух пробирках. Пробирки освещаются горизонтальными лучами сильного источника света, находящегося на расстоянии по крайней мере полуметра. Надо защищать пробирки от исходящего от источника света тепла каким-нибудь прозрачным экраном. Рисунок этого нефелометра и более подробное его описание приведены в первых двух статьях, указанных выше (в сноске1).
Потенциометр. Если в лаборатории часто проводят объемные определения таких элементов, как хром или ванадий, рекомендуется приобрести потенциометр. Он также необходим в тех случаях, когда надо очень точно определять концентрацию водородных ионов (см. «Потенциометрическое титрование», стр. 229).
Спектроскоп. Маленький визуальный спектроскоп бывает очень полезным при исследовании осадков, получаемых в различных стадиях анализа, например для обнаружения стронция в остатке после прокаливания оксалатов или лития — в остатке хлорида натрия.
Лупы. При исследовании анализируемых проб и получающихся осадков часто приходится пользоваться лупами; рекомендуются небольшие лупы, дающие 10—20-кратное увеличение.
Центрифуга. Нет сомнения в том, что многие аналитические операции можно значительно упростить и ускорить, применяя центрифугу. Хорошо известно, например, ее применение при определении фосфора в сталях или в фосфористой бронзе. При применении центрифуги особенно большое значение имеет физическое состояние осадка, поэтому ход анализа должен быть очень тщательно разработан и условия его проведения надо строго соблюдать. Центрифуга может быть очень полезной 4*
52
Гл. III. Приборы и реактивы
при выполнении таких операций, как промывание осадков декантацией х.
Электрооборудование лаборатории. Желательно следующее оборудование: 1) сушильный шкаф с терморегулятором, который можно установить на любую температуру до 250° С; 2) электрическая муфельная печь, которую можно нагревать до температуры 1200° С, с термопарой и пирометром, если температуру надо точно регулировать, как, например, при прокаливании вольфрамовой кислоты; 3) печь для сжигания длиной 25—30 см, которую можно нагревать до 1200° С; 4) печь для сжигания, применяемая при выполнении элементарных анализов органических веществ; она должна быть разборной (из трех частей) и давать температуру До 1000° С; 5) электрические плитки с регуляторами на три температуры; 6) специальные нагревательные приборы, например маленькая печь для определения содержания летучих веществ в углях.
Не следует думать, что применение электрической печи устраняет все источники загрязнения при прокаливании осадков или сплавлении анализируемой пробы с различными плавнями; это особенно справедливо в тех случаях, когда в одной и той же печи проводят различные операции.
Например, при сплавлении с карбонатом натрия для определения серы при проведении «холостых» опытов в одном случае было получено 0,6 мг и потом снова 0,6 мг BaSC4, когда сплавление проводилось в электрической печи, специально защищенной от возможности загрязнения плава серой. При сплавлении в обычной лабораторной печи, в которой проводились и все другие операции, «холостые» опыты дали осадки BaSO4 в 3,5 и 3,0 мг. Аналогично этому при приготовлении А12О3 с определенным содержанием В2О3, применяющейся в качестве спектрохимического стандарта, было найдено, что в «холостых» опытах, где В2О3 не прибавлялся, и в стандарте, в который было прибавлено 0,05% В2О3, после прокаливания в муфельной печи обнаруживаются одинаковые количества В2О3.
Тогда «холостые» опыты были повторены следующим образом. Три порции чистого алюминия поместили в платиновые чашки, растворили в соляной и серной кислотах, растворы выпарили и остатки прокалили при 1000—1100° С. Одну порцию прокаливали в муфеле, в котором однажды была просыпана бура; вторую порцию — в муфеле общего применения, но в котором ни буру, ни борную кислоту никогда не рассыпали; третью порцию — на горелке Мекера. В результате было найдено в этих порциях соответственно: 0,01%, 0,001% и 0,0000% борного ангидрида (ВгО3).
Газовое оборудование. Обычно применяют газовые горелки двух типов: горелки типа Терриля (*или Теклю*), дающие температуру до 1000° С, и горелки типа Мекера или паяльные, с помощью которых можно нагревать до 1200° С и выше. Если в лаборатории нет электрического оборудования, то приходится применять газовые сушильные шкафы, печи для сжигания и плитки. Последние желательно иметь в форме сковородок с толщиной дна по крайней мере 6 мм и с утолщенными краями, чтобы избежать их коробления.
Паровые бани. В лабораториях, в которых должны проводиться многочисленные выпаривания, работа очень облегчается, если имеются паровые бани, присоединённые к паровой сети и действующие круглые сутки.
Паровые бани необходимы, например, в лабораториях, где постоянно определяют кремнекислоту выпариванием солянокислых растворов досуха. Бани могут иметь форму ящиков, сделанных из некорродирующего
1 О применении центрифуги при определении серы в стали, кальция в виде оксалата, алюминия в виде гидроокиси, никеля в виде диметилглиоксимата и нитратов в виде азотнокислого нитрона см. в статье: Н. S. G г е е n, J. Am. Chem. Soc., 53, 3275 (1931).
Приборы
53
материала, с отверстиями наверху, снабженными концентрическими кольцами разных размеров для установки нагреваемых сосудов.
Выпаривание на таких банях можно значительно ускорить, если над поверхностью жидкости пропускать горячий воздух, а нагреваемый сосуд поместить в чашку, имеющую в дне отверстие, а на наружной стороне фланец, посредством которого чашка держится на бане, будучи погружена в нее до желаемой глубины. Если нет возможности устроить в лаборатории паровую баню, то надо иметь две или более водяных бань с кольцами и устройством для поддержания постоянного уровня.
Аппараты для перегонки воды. В продаже имеются аппараты любых размеров и любой вместимости для получения дистиллированной воды. Аппараты, дающие до 8 л дистиллированной воды в 1 ч, обычно нагреваются газом или электричеством. Аппараты больших размеров чаще всего нагреваются паром, но устанавливать их имеет смысл только в том случае, если расход дистиллированной воды в лаборатории превышает 100 л в день. В продаже также имеются перегонные аппараты с особым устройством для удаления .из воды аммиака и других газов. Небольшие перегонные аппараты надо чистить по крайней мере раз в 3 месяца.
Водоструйные насосы. Если в лаборатории нет специальной вакуумной проводки, надо приобрести латунные насосы, действующие как инжекторы; они облегчают фильтрование растворов и высушивание осадков. О применении колокола для фильтрования под вакуумом см. стр. 126.
Промывалки. Для выполнения анализов, в которых приходится учитывать количество применяемой промывной жидкости, были рекомендованы 1 простые градуированные промывалки: градуированный цилиндр, снабженный пробкой и трубками для холодных растворов, и градуированная пробирка в деревянном футляре (для горячих растворов).
Большие бутыли. Большие бутыли из химически устойчивого стекла всегда находят в лаборатории широкое применение. Лаборатория должна иметь в большом количестве 1-литровые склянки для хранения растворов, которые употребляются не часто, а также 2-литровые и даже 6- и 10-литровые бутыли для хранения растворов, которые постоянно расходуются в больших количествах.
Приспособление для титрования. Если в лаборатории часто проводятся титрования каким-нибудь раствором, надо, чтобы сосуд, в котором хранится этот раствор, был постоянно соединен с бюреткой или пипеткой, из которой раствор приливается при титровании.
Платиновая посуда. Абсолютно необходимо, чтобы каждая аналитическая лаборатория имела хотя бы минимальное количество платиновых тиглей с хорошо подходящими к ним крышками. Если лаборатория может приобрести только два тигля, следует выбрать тигли емкостью 25 мл. Маленькие тигли емкостью 10 мл можно применять при прокаливании осадков, но для проведения сплавления они обычно непригодны. В большинстве случаев применяются тигли емкостью 20 и 25 мл, изредка оказываются полезными и большие тигли — емкостью 50 и 100 мл.
В'лабораториях Бюро Стандартов США было отмечено, что платиновые тигли, содержащие от 0,3 до 0,5% ирйдия, и тигли, содержащие 3,5% родия, примерно одинаково хороши. Первые лучше сохраняют блестящую полировку, вторые лучше сохраняют свою форму и несколько
1 Е. R. С а 1 е у, Ind. Eng. Chem., Anal. Ed., 1, 162 (1929).
54
Гл. III. Приборы, и реактивы
меньше теряют в массе после прокаливания или проведения в ник сплавления с щелочными или кислыми плавнями х. Спекание для определения щелочных металлов , можно проводить в тиглях емкостью 25 мл, но лучше применять для этой цели тигли специальной формы (см. рис. 45, стр. 1007).
Платиновые чашки чаще применяются емкостью 100 и 300 мл. Последние можно с успехом заменить чашками емкостью 500 и 1000 мл. Маленькие чашки применяются для прокаливания и взвешивания в них небольших остатков-, например солей щелочных металлов при определении их в горных породах; большие чашки нужны при точном определении крем-некислоты. Для первых крышки не нужны, для вторых желательно иметь крышки, сделанные из тонкой листовой платины и укрепленные по краям платиновой проволокой.
Большое применение, без сомнения, получат предложенные 2 сравнительно недавно специальные чашки, в которых можно сплавлять силикаты с карбонатами и, не отделяя полученного сплава, тут же обрабатывать его кислотой и выпаривать для отделения кремнекислоты.
Из других платиновых изделий следует упомянуть платиновые дырчатые конусы (диаметр 2,2 см) и диски (для тиглей Гуча). И те и другие применяются для фильтрования с отсасыванием. Необходимы также платиновые палочки (толщина —2,5 мм, длина 12—15 см) для перемешивания растворов, содержащих плавиковую кислоту, и около 30 см толстой платиновой проволоки (толщина — 0,8 мм), которую нарезают на короткие куски и применяют, например, для перемешивания вязких сплавов с борным ангидридом.
Кварцевая посуда. Многие из перечисленных выше платиновых изделий вполне можно заменить соответствующими изделиями из кварца. Например, кварцевыми тиглями можно пользоваться для сплавления с пиросульфатом, кварцевыми чашками и стаканами — при определении щелочных металлов. Совершенно незаменимы треугольники из плавленого кварца или из нихромовой проволоки с трубочками из плавленого кварца. Кварцевая посуда обладает исключительной стойкостью к резким переменам температуры (кварцевые тигли можно, например, накалить до 1000° С и затем погрузить в холодную воду) и к действию кислот, за исключением плавиковой кислоты, а при высоких температурах — и фосфорной кислоты.
Кварцевое стекло обладает следующими важными свойствами: 1) коэффициент расширения его равен 5,4-10~7; 2) температура размягчения его лежит около 1500° С; 3) его летучесть при 1200° С меньше, чем летучесть платины; 4) при обычных давлениях и температуре 1200° С прозрачное кварцевое стекло не пропускает газов, за исключением гелия; это свойство оно теряет при расстекловывании (при переходе и кристаллическую форму); 5) расстекловывание начинается тогда, когда кварцевое стекло подвергается многократным резким изменениям температуры; в восстановительной атмосфере расстекловывание сильнее, чем в окислительной; 6) посуда из прозрачного кварцевого стекла прочнее н более устойчива к расстекловыванию, чем из непрозрачного стекла; 7) если кварцевые трубки надо нагревать до температур, превышающих 1000° С, рекомендуется пользоваться трубками, которым не- давали охлаждаться ниже 300° С, — тогда расстекловывание будет происходить медленнее.
1 О поведении их при прокаливании см. J. I. Н о f f m a n, G. Е. F. L u n d е 11, J. Research NBS, 5, сноска на стр. 290 (1930) и там же, 15, 415 (1935).
* См. I. I. Н о f f m a n, J. Research NBS, 25, 379 (1940).
Приборы. 55
В настоящее время можно приобрести различные изделия (тигли, стаканы, колбы, чашки, пробирки, трубки для сжиганий) из стекла викор, содержащего 96% кремнекислоты (см. табл. 5, стр. 42). Очень низкий коэффициент расширения этого стекла (8 • 10"7, в пределах температур от 19 до 350° С) дает возможность очень быстро его нагревать и охлаждать. Наивысшая температура, до которой можно это стекло нагревать, около 900° С.
Золотая и серебряная посуда. Для сплавления с едким натром и едким кали нужны золотые или серебряные тигли емкостью 25 или 35 мл. Золотые тигли лучше, так как они обладают большей химической стойкостью. Недостатком, препятствующим широкому применению тех и других тиглей, является то, что золото и серебро имеют низкие температуры плавления — первое 1050° С, второе 960° С.
’ Во многих выпариваниях и прокаливаниях, где применяются невысокие температуры, можно пользоваться золотыми чашками взамен платиновых. Серебряные чашки применяются главным образом для выпаривания в них щелочных растворов.
Электроды. Наиболее удобными электродами для применения во всех случаях анализа являются платиновые цилиндры, сделанные из тонкой сетки приблизительно на 50 меш \ обработанной песком при помощи пескоструйного аппарата (толщина проволоки около 0,21 мм); чтобы придать цилиндрам постоянную форму, края сетки загибают в верхней части и у основания цилиндра приблизительно на 3 мм и прикрепляют сетку к плоскому платиновому стержню, припаянному по всей длине образующей цилиндра.
Цилиндры обычно делают высотой 5 см, диаметром 5 см для анода и 4 см для катода; выступающая часть стержня имеет 1,5 мм в диаметре и 8 см в длину. Стержень может быть сделан из платино-иридиевого, платино-родиевого или платино-рутениевого сплава.
Если электрод должен вращаться, применяют более прочные цилиндры из платиновой сетки или дырчатой листовой платины с прочным стержнем в середине. Конструкция второго электрода зависит от его назначения: если на электроде должен выделяться осадок, электрод лучше всего сделать из платиновой сетки плоским или цилиндрическим, но таких размеров, чтобы между ним и первым электродом оставалось достаточное пространство. 1
Для специальных целей могут оказаться более подходящими электроды другой формы, например один электрод — платиновая чашка, второй — плоская спираль 2.
1 * Диаметр отверстия 0,29 мм. Прим, ред.*
2 Насколько можно судить по пока еще ограниченному числу опытов, платину можно заменить танталом для осаждения золота, серебра, никеля и меди, но не для выделения свинца или цинка и не для применения в качестве анода. L. W. S t г о с к и Н. S. Luk е ns [Trans. Am. Elektrochem. Soc., 56, 409 (1929)] рекомендуют тантал как удовлетворительный заменитель платины для электроосаждения меди, если поверхность катода сохранять свободной от окиси. В. М ear s и Р. R. Pine [Ind. Eng. Chem., Anal. Ed., 2, 298 (1930)] утверждают, что медь можно удалять с танталовых катодов разбавленным (1 : 1) раствором аммиака, к которому добавлена трихлоруксусная кислота до 10%-ной концентрации.
* О применении тантала (и ниобия) вместо платины см. также D. F. Calhaue, С М. А 1 Ь е г, Trans. Am. Electrochem. Soc., 63, 173 (1933) и В. Fethenheuer, Е. Creme г, Wiss. Veroffentlichungen Siemens — Konzem, 12, 168 (1932).*
56
Гл. III. Приборы и реактивы
Отложившиеся на электроде осадки меди, серебра, кадмия и ртути можно удалить погружением электрода в разбавленную (1:1) азотную кислоту, ополаскиванием водой и затем кипячением со свежей порцией разбавленной азотной кислоты в течение 5—10 мин. Для удаления осадка никеля лучше первую обработку проводить неразбавленной азотной кислотой. Осадки двуокиси свинца лучше всего удалять погружением анода в азотную кислоту одновременно с катодом, с которого надо удалить медь; перед кипячением с разбавленной азотной кислотой в этом случае надо прибавить 1—2 капли спирта. Иногда после такой обработки на анодах остаются темные пятна. Обычно их можно полностью удалить, прокаливая анод 10—30 мин приблизительно при 1000° С на одной или двух больших горелках Мекера.
*В тех случаях, когда надо только удалить двуокись свинца с анода, I
а на катоде одновременно медь не выделяется А. М. Шаврин 1 предлагает i
обрабатывать анод разбавленной азотной кислотой с добавлением медной или латунной стружки. Можно к азотной кислоте добавлять и нитрит калия (в этом случае также выделяются окислы азота, служащие восстановителем, как и при добавлении меди) или другие восстановители — перекись водорода, щавелевую кислоту и т. п. i
Замене платиновых электродов электродами из более дешевых металлов посвящено большое число работ. Кроме упомянутого авторами тантала, предлагались в качестве катодов сетки из вольфрама 2, серебра 3, '
хромово-никелевых сплавов4, нержавеющей стали 5, никеля, латуни, ;
покрытой медью 6, и меди, покрытой серебром 7. В качестве анодов предлагали «пассивированное железо» 8, хромированную сталь 9, свинец 10 и графит 11. Эти материалы не могут заменить платину во всех электро- J
аналитических осаждениях, но в отдельных случаях для осаждения того или иного металла они применялись с успехом. Доп. ред*
Посуда из бакелита. Если в лаборатории часто применяется плавиковая кислота, работа с ней значительно облегчается при наличии стаканов, мерных цилиндров, пипеток и палочек, сделанных из баке-лита. !
Тигли Розе. Главным отличием тиглей Розе является просверленная крышка с трубкой для введения в тигель газа, в атмосфере которого должно происходить прокаливание. Мы считаем, что для обычных работ лучше применять кварцевые или глазурованные фарфоровые тигли, снабженные кварцевой крышкой и трубкой, а не те тигли из неглазуро-ванного фарфора, которые обычно имеются в продаже.
1 А. М. Ш а в р и н, Зав. лаб., 9, 476 (1940).
2 А. Фишер, А. Шлейхер, Электроанализ, ОГИЗ, 1931.
3 J. Gewecke, Chem. Ztg., 41, 297 (1917); В. М. Ш а лф еев, А. П. Б е з-заботнова, Зав. лаб., 5, 1311 (1936).
4 М. В leesen, Z. anl. Chem., 63, 209 (1923).
5 A. Schleicher, Z. angew. Chem., 39, 822 (1926); А. П. К p e ш к о в, С. С. О лен ии, Зав. лаб., 20, 404 (1954).
6 Л. Фаворский Г. Зотов, ЖПХ, 10, 1700 (1937).
7 О. Cal hap е, F. Wheaton, J. Soc. Chem. Ind., 33, 279 (1914).
8 О. В a r u e b y, J. Am. Chem. Soc., 36, 1444 (1914).
9 J. Guzman и сотрудники (1929—1934 гг.), цит. по A. Schleiche Elektroanalysische Schnellmethodes, Stuttgart, 1947.
” А. П. Крешков, С. С. О л e н и н, цит. выше.
11 В, М. Шалфеев, А. П. Беззаботнова, цит. выше.
Приборы,
57
Никелевые и железные тигли. Если часто приходится проводить сплавления с перекисью натрия, надо иметь запас никелевых или железных тиглей. Они должны быть емкостью по крайней мере в 25 мл, но лучше, если емкость их составляет 30—35 мл. Тигли должны быть штампованы из возможно более чистого металла. Мы предпочитаем тигли, штампованные из листового чистого железа толщиной 0,8 мм. Большего размера тигли, емкостью приблизительно 60 мл, можно применять в качестве радиаторов (см. рис. 5, стр. 48).
Эксикаторы. Обычно применяемые эксикаторы (типа Шайблера) наиболее удобны при аналитической работе. Эксикаторы с тубусом сбоку или в крышке, для высушивания в вакууме, необходимы, когда вещества настолько гигроскопичны, что нужно возможно быстро удалить влагу, попавшую при открывании эксикатора, или если нужно ускорить высушивание очень влажных веществ. Внутренняя пластинка или полочка, на которую ставят тигли, чашки и т. п., должна соответствовать исполняемой работе, но не должна препятствовать свободной циркуляции паров и не должна разрушаться под действием высушиваемого вещества или помещенных на ней предметов. Высушивающее вещество надо помещать внизу, а не над высушиваемым предметом. Выбор того или иного высушивающего вещества надо делать продуманно. Например, такое сильно гигроскопичное вещество, как прокаленная окись алюминия, нельзя охлаждать над хлоридом кальция, а возогнанный иод нельзя сушить над серной кислотой. За высушивающим веществом в эксикаторе надо наблюдать и сменять его, как только появится подозрение, что оно перестало поглощать влагу 1 или загрязнилось (например, серная кислота потемнела от попавшей в нее пыли).
Наилучшим высушивающим веществом в эксикаторе при обычной работе является концентрированная серная кислота, налитая на пемзу, битое стекло или куски фарфора так, чтобы она полностью их не покрывала. Если применяется хлорид кальция, надо брать гранулированный или плавленый; кристаллический гидратированный хлорид кальция бесполезен. Фосфорный ангидрид надо применять только тогда, когда это действительно нужно, и сменять его, как только появится корка на его поверхности. Надо помнить, что влажность воздуха внутри эксикатора через очень короткое время после того, как он был открыт, почти не отличается от влажности окружающего воздуха. Поэтому очень гигроскопичные вещества, например прокаленную окись кальция, надо на это время защищать, применяя тигли с плотно прилегающими крышками 2.
1 В связи с этим интересно отметить наблюдение G. Boehm [Chem. Ztg., 53, 323 (1929)], что в серной кислоте, содержащей 18 г/л растворенного сульфата бария, осадок не появляется, пока серная кислота не будет разбавлена поглощенными парами воды до 93%-ной концентрации. При концентрации ее от 93 до 84% выделяются игольчатые кристаллы1 BaSO4 • 2H2SO4 • Н2О. Дальнейшее разбавление серной кислоты, поглощаемой водой, приводит к превращению игольчатых кристаллов в мелкокристаллический порошковидный BaSO4, и тогда серная кислота теряет свои высушивающие свойства.
2 б том, как поразительно медленно происходит высушивание воздуха в эксикаторе, содержащем окись бария, фосфорный ангидрид, хлорид кальция или серную кислоту, сообщают Н. S. Booth и L. Н. McIntyre [Ind. Eng. Chem., Anal. Ed., 8, 148 (1936)].
58
Гл. III. Приборы и реактивы
РЕАКТИВЫ
Общие замечания
В количественном анализе следует применять наиболее чистые реактивы, какие только можно получить в открытой продаже или по специальным заказам, или, наконец, очистив реактивы в самой лаборатории А Проверка чистоты применяемых в лаборатории реактивов должна проводиться постоянно и очень тщательно 1 2.
За последние 20 лет качество выпускаемых в продажу реактивов значительно улучшилось, и тем анализам их, которые печатаются на этикетках, можно гораздо больше доверять, чем это было в прошлые годы. Тем не менее каждый новый реактив, приобретенный лабораторией, должен быть проверен на его чистоту 3. Поэтому надо приобретать реактивы’ большими партиями, чтобы не приходилось проводить проверку их слишком часто. Из каждой такой партии надо тщательно отобрать для анализа среднюю пробу,, избегая смешивать различные партии, и результаты анализа (или номер, под которым анализ записан в журнале) надо' поместить на приклеенной к сосуду этикетке.
Особое внимание должно быть обращено на качество дистиллированной воды, применяемой в анализе. Через короткие промежутки времени дистиллированную воду надо исследовать, или определяя ее электропроводность, или выпаривая по крайней мере 1 л воды в платиновой чашке; при этом надо соблюдать все необходимые меры предосторожности, чтобы в воду не попала пыль или пары каких-либо веществ. Вода, применяемая в Бюро Стандартов США, обычно имеет электропроводность в 1-10-® обратных омов. 1 л лучшей дистиллированной воды однократной перегонки, выпаренной в надлежащих условиях, дает остаток, весящий около 1 мг. После осторожного прокаливания этого остатка, т. е. удаления из него всех летучих солей, масса его уменьшается до 0,5 мг. Хранить дистиллированную воду лучше всего в сосудах, покрытых изнутри оловом, и разливать ее через трубку из чистого олова. Если дистиллированную воду приходится держать в стеклянных бутылях, то последние
1 Очистка реактивов в лаборатории часто бывает очень сложной, и ее следует избегать, если купленный реактив не настолько загрязнен, что могут возникнуть сомнения в правильности получаемых результатов анализа'. Так, например, при проведении осаждения аммиаком и персульфатом аммония лучше ставить «холостые» опыты, чем пытаться очистить персульфат.
2 Мы находили барии в перекиси водорода, органические вещества — в фтористоводородной кислоте, свинец — в винной кислоте, двуокись серы — в серной кислоте.
8 Е. Wichers, A. Isaacs и I. С. Schoonover в статье о 200 химических реактивах, хороших и плохих [Ind. Eng. Chem., Anal. Ed., 3, 227 (1931)], утверждают, что из 236 партий химических реактивов, исследованных ими в течение 1928—1930 гг., только 65%. отвечали стандартам Ам. Хим. О-ва или тем анализам, которые были даны фабрикантами на этикетках.
Все чистые для анализа химические реактивы должны удовлетворять техническим условиям, ’разработанным реактивным комитетом Ам. Хим. О-ва (ACS Analytical Reagents, март 1941). См. также Ind. Eng. Chem., Anal. Ed., 16, 281 (1944) и книгу Reagent Chemicals, ACS Specifications, изД. Am. Хим. О-ва 1950. Об испытании реактивов, не вошедших в число исследованных реактивным комитетом, см. J. Rosin, Reagents Chbmicals and Standards, 2d ed., Van Nogtrand Co, 1946.* Методы испытания химических реактивов, применяемые в СССР, см. Реактивы неорганические, сборник стандартов, ч. I—III, Стандартгиз, 1949; Реактивы органические, сборник стандартов, Стандартгиз, 1951; Реактивы органические, сборник технических условий, Стандартгиз, 1951.*
Реактивы
59
должны быть из химически стойкого стекла, и применять такую воду надо возможно более свежей, так как ее действие даже на химически стойкое стекло довольно велико; это особенно относится к горячей воде, которую держат в промывалках. Следует помнить, что дистиллированная вода может содержать двуокись углерода, кислород, азот, аммиак; если присутствие этих веществ недопустимо, то при перегонке воды и охлаждении дистиллята надо принимать особые меры.
Кислоты можно получить большой чистоты, и обычно они не нуждаются в перегонке. Когда открывают новую бутыль с кислотой, надо тщательно удалить всю замазку, прежде чем вынимать пробку; затем, следует внимательно осмотреть пробку и горло бутыли, чтобы убедиться в том, что в кислоту не попали посторонние вещества при ее запечатывании.
Раствор аммиака постепенно разъедает химическую посуду и загрязняется извлеченными из нее веществами, растворимыми и нерастворимыми. Если раствор аммиака время от времени приходит в соприкосновение с воздухом, то он- загрязняется также карбонатом аммония. Применение такого загрязненного реактива в анализе, например при определении алюминия, приводит к ошибке, даже если вводить поправку на «холостой» опыт, проведенный с одними реактивами (см. гл. «Алюминий», стр. 568). Если расдвор аммиака содержит карбонат аммония, а в анализируемом растворе, кроме алюминия, присутствуют такие элементы, как кальций или магний, положение еще ухудшается, так как в «холостом» опыте с одними реактивами карбонат аммония улетучивается и не оказывает влияния, а при введении его в анализируемый раствор он вызывает соосаждение щелочноземельных металлов. Растворы аммиака следует поэтому, периодически, через короткие промежутки времени перегонять, предварительно взбалтывая их с гашеной известью для разложения карбоната аммония.
Для некоторых специальных целей надо хранить растворы аммиака в сосудах из церезина, полиэтилена, золота или платины. Защитные покрытия из парафина или церезина на внутренней поверхности стеклянных сосудов обычно оказываются неудовлетворительными, так как эти покрытия имеют тендеццию' со временем отставать от стекла, особенно в жаркую погоду.
Если в лаборатории имеется баллон с жидким аммиаком, можно легко приготовлять растворы любой концентрации, пропуская газообразный аммиак сначала через промывную склянку с водой, а затем в бутыль, на две трети наполненную водой'и охлаждаемую льдом или током холодной воды. Поглощение аммиака обычно прекращают, когда плотность полученного раствора достигает величины 0,926 г!см3 (20% NHe), после чего раствор переливают в бутыль из церезина, если предполагают сохранять его в течение нескольких месяцев. Если раствор аммиака не должен совершенно содержать карбоната аммония, то при его приготовлении газообразный аммиак надо пропускать через раствор гидроокиси бария. Применяемую для поглощения аммиака воду надо сначала прокипятить' и охладить, пропуская через нее воздух, освобожденный от двуокиси углерода, а во время поглощения аммиака раствор надо защищать от СО 2 соответствующей предохранительной трубкой.
В табл. 7 приведены данные о концентрациях продажных кислот и раствора аммиака.
60
Гл. III. Приборы и реактивы
Концентрация продажных кислот и раствора аммиака
ТАБЛИЦА 7
Реактив Содержание % по массе (приблизительно) Плотность, г/см* (приблизительно) Нормальная концентрация (приблизительно)
Соляная Кислота 36 1,18-1,19 12
Азотная кислота 68—70 1,41 15,5
Серная кислота 96 1,84 36,0
Хлорная кислота 70 1,67 11,6
Фтористоводородная кислота . . . 46 1,15 26,5
Фосфорная кислота 85 1,69 14,7—44
Иодистоводородная кислота .... 45 1,48 5,2
Бромистоводородная кислота . . . 40 1,38 6,8
Уксусная кислота 99,5 ’ 1,05 17,4
Раствор аммиака 27 (NH3) 0,90 14,3
В настоящем руководстве всюду, где нет указаний на концентрацию, подразумеваются концентрированные кислоты и концентрированный раствор аммиака. Разбавленные реактивы обозначаются отношением, указывающим степень разбавления.
В выражениях (1 : 2), (1 : 19) и т. п. первая цифра означает объем взятого реактива, вторая — объем прибавленной к нему воды. Например, выражение «разбавленная соляная кислота (1 : 2)» означает, что реактив был приготовлен смещением 1 объема продажной концентрированной соляной кислоты (пл. 1,18 г/см3) с 2 объемами воды. Выражение «разбавленный аммиак (1 : 19)» означает, что 1 объем продажного концентрированного раствора аммиака (пл. 0,90 г/см3) был разбавлен 19 объемами воды. Если никакого разбавления не указано, подразумевается продажный концентрированный реактив.
Карбонат натрия — один из важнейших реактивов в анализе силикатов — можно иногда достать очень чистым, с содержанием примесей, не превышающим 2,5 мг на 20 г (0,012%). Такой реактив надо сохранять для применения его в тех анализах, где требуется определение кремне-кислоты, алюминия и т. п. При определении других компонентов, например фосфорной кислоты, фтора, серы, можно пользоваться менее чистым карбонатом натрия, если он не содержит веществ, мешающих данному определению, и или совсем не содержит определяемого элемента, или содержит его в таком количестве, которое может быть точно определено и вычтено из результата анализа. Даже в очень чистых препаратах карбоната натрия содержатся небольшие количества фосфата натрияг
Определение содержания кремнекислоты и веществ, осаждаемых аммиаком, в карбонате натрия проводят следующим способом. Растворяют 10 г карбоната натрия в 50 мл воды, осторожно прибавляют 30 мл разбавленной (1 : 1) серной кислоты, выпаривают и нагревают до обильного выделения паров серной кислоты. Затем охлаждают, растворяют в 100 мл воды, прибавляют несколько капель раствора метилового красного, нагревают до кипения и приливают разбавленный (1 : 1) аммиак до перехода красной окраски в желтую. Кипятят 2 мин, фильтруют и промывают осадок горячим нейтральным 2%-ным раствором хлорида или нитрата аммония. Осадок прокаливают и взвешивают. Масса его не должна превышать 0,001 г. Если масса превышает 0,0005 г, его обрабатывают несколькими каплями серной и плавиковой кислот, выпаривают, прокаливают и взешивают. Потеря в массе (кремнекислота) не должна превышать 0,0005 г.
Реактивы.
61
Висмутат натрия и персульфат аммония — реактивы, которые могут применяться только тогда, когда они обладают извёстной минимальной концентрацией активного вещества, поэтому надо тщательно проверять их перед употреблением; только при этом условии можно иметь уверенность, что анализы, проводимые с их применением, дадут правильные результаты. Мы встречали препараты персульфата аммония, которые содержали менее 25% этого вещества.
Проверка качества органических реактивов: диметилглиоксима, а-нитрозо-Р-нафтола, купферона и др., так же как и органических индикаторов, — дело очень трудное. Можно лишь рекомендовать приобретать такие реактивы непосредственно у фирм, специализировавшихся на их производстве, и проверять каждую партию того или иного реактива, проводя с ним анализ известного вещества. Нет никаких сомнений, что в продажу иногда поступают органические реактивы очень низкого качества, и это приводит к затруднениям в анализе и ошибочным результатам даже тогда, когда применяются вполне удовлетворительные методы.
Специальные реактивы
Пиросульфаты щелочных металлов. При сплавлении с бисульфатами (кислыми сульфатами) щелочных металлов последние сначала превращаются в пиросульфаты и лишь потом взаимодействуют с анализируемым веществом. Но значительно лучше сразу пользоваться готовыми пиросульфатами, тогда при сплавлении не образуется пена, сплавление происходит без разбрызгивания и значительно быстрее.
Для превращения реактивного бисульфата в пиросульфат расплавляют в платиновой чашке некоторое количество соли и держат в расплавленном состоянии при возможно более низкой температуре, пока не прекратится вспенивание и не начнут выделяться белые пары. Затем плав наливают тонким слоем в другие чашки, охлаждают, разбивают на куски и хранят в банках. Если в лаборатории нет хорошего бисульфата, его можно приготовить, смешав эквивалентные количества сульфата калия и серной кислоты. Но в этом случае надо предварительно проверить качество сульфата калия, так как он иногда содержит значительные количества свинца, кальция и кремнекислоты. В некоторых случаях лучше применять пиросульфат натрия, а не пиросульфат калия, так как он действует быстрее и с такими прокаленными осадками, как окись алюминия, образует более растворимые соли. Небольшим недостатком пиросульфата натрия является то, что последующее разложение прокаленной массы замедляется из-за склонности натриевой соли образовывать на поверхности корку.
Обе соли должны быть совершенно свободными от кремнекислоты и от элементов, подлежащих определению после сплавления. Для определения кремнекислоты в этих солях растворяют 5 г соли в возможно малом количестве воды, осторожно прибавляют 10 л1л серной кислоты и выпаривают до появления белых паров. Затем охлаждают, приливают 100 мл воды, нагревают, пока не растворятся все растворимые соли, немедленно фильтруют и продолжают анализ, как описано на стр. 956.
Персульфат аммония или персульфат калия. Для получения правильных результатов при определении марганца персульфатным методом необходимо, чтобы реактив этот был высокого качества, т. е. содержал
62 Гл. III. Приборы и реактивы
большое количество основного вещества. Кроме того, если персульфат применяется в анализе горных пород для окисления марганца с целью переведения его в осадок аммиаком вместе с алюминием, железом и др., то этот реактив должен быть‘свободным от осаждаемых аммиаком элементов, особенно от алюминия. Хороший персульфат должен содержать не меньше 85% этой соли и не больше 0,0001 % марганца, 0,001 % хлорид-ионов и 0,03% окислов тяжелых металлов.
Содержание персульфата в реактиве определяют следующим способом. Точно отвешивают 0,3 г соли и переносят навеску в склянку емкостью 200 мл, снабженную притертой пробкой. В эту склянку предварительно наливают раствор 2 г иодида калия в 50 мл воды. Прибавив 10 мл 10%-ной серной кислоты, закрывают склянку пробкой, осторожно перемешивают ее содержимое, вращая склянку, и дают постоять 30 мин, в темноте, время от времени взбалтывая. Затем титруют выделившийся иод 0,1 н. раствором тиосульфата. 1 мл точно 0,1 н. раствора последнего соответствует 0,01141 е (NH^jSjOg или 0,01352 г KaSaOg. В результат вносят поправку, полученную проведением «холостого» опыта с одними реактивами.
Методы очистки персульфата, предназначаемого для применения в осаждениях аммиаком (стр. 949), требуют настолько большого труда и дают в результате продукт с таким малым содержанием персульфата, что лучше не проводить этой работы; надо приобрести возможно более чистый персульфат и в получаемые результаты вносить поправки на «холостые» опыты, проведенные с этим реактивом.
Персульфаты быстро разлагаются в растворе или при соприкосновении с влажным воздухом. Поэтому сухой реактив надо хранить в плотно закрытой пробкой банке и применять только свежеприготовленные его растворы.
Висмутат натрия. Висмутат натрия не является истинной солью висмутовой кислоты. Он должен, однако, содержать не менее 4% активного кислорода, что соответствует'по крайней мере 70% NaBiOe. Содержание в нем марганца и хлорид-ионов не должно превышать соответственно 0,0005 и 0,001%.
Неудачи при определении марганца висмутатным методом часто объясняются применением реактива, не удовлетворяющего указанным выше условиям, особенно по содержанию активного кислорода.
Определение содержания активного кислорода в висмутате натрия проводят следующим способом. Навеску в 0,700 г висмутат натрия помещают в коническую колбу и прибавляют 25 мл стандартного кислого раствора сульфата железа (II) приблизительно 0,2 н. концентрации [для его приготовления 7 г сульфата железа (II) растворяют в 90 мл свежепрокипяченной и охлажденной дистиллированной воды и добавляют серную Кислоту до объема 100 мл', раствор этот приготовляют и устанавливают его титр непосредственно перед употреблением]. Закрыв колбу пробкой, дают постоять 30 мин, временами взбалтывая, и титруют избыток соли железа (II) стандартным раствором перманганата.
Для проверки на содержание марганца 2 г висмутата натрия обрабатывают 35 мл разбавленной (2 : 5) азотной кислоты и кипятят раствор. Затем прибавляют 0,5 мл сернистой кислоты (раствор должен стать прозрачным) и кипятят для удаления окислов азота. После этого охлаждают до 15® С, всыпают 0,5 г того же висмутата и оставляют на 5 л»вн, изредка перемешивая. Потом приливают 25 мл воды и фильтруют через асбест. Цвет фильтрата не должен быть интенсивнее, чем.цвет раствора, полученного обработкой 0,01 мг марганца тем же способом и теми же количествами реактивов.
Для испытания на хлорид-ионы 2 г висмутата натрия всыпают в 25 мл воды, нагревают до кипения и держат при температуре кипения 10 мин. Затем разбавляют водой до 50 мл, фильтруют и к фильтрату прибавляют 1 мл азотной кислоты и 1 мл 0,1 н. раствора нитрата-серебра. Образующееся помутнение не должно быть сильнее, чем помутнение, производимое 0,01 мг хлорид-ионов в таком же объеме раствора, со
Реактивы
63
держащего те же количества азотной кислоты и нитрата серебра, какие были добавлены при испытании висмутата.
Перекись водорода. Для обычных аналитических целей достаточно иметь 3%-ный раствор перекиси водорода, если он свободен от содержания фтора. Реактив разлагается при хранении, особенно в жаркую погоду, или если он стоит в теплом месте. Поэтому перекись водорода следует приобретать небольшими партиями. Если сосуд, в котором она была приобретена, открыли, его не следует закрывать плотно, и время от времени надо проверять концентрацию раствора.
Для определения концентрации перекиси водорода отбирают 10 мл ее, разбавляют до 100 мл дистиллированной водой и хорошо перемешивают. Затем пипеткой берут аликвотную часть в 10 мл, разбавляют до 300 мл, прибавляют 10 мл разбавленной (1 : 1) серной кислоты и титруют 0,1 н. раствором перманганата: 1 мл точно 0,.1 н. раствора перманганата соответствует 0,0017 г Н2О2; на титрование должно быть израсходовано не менее 17,6 мл этого раствора.
Для испытания на содержание фтора 50 мл перекиси водорода обрабатывают карбонатом натрия, прибавляя последний в небольшом избытке, и нагревают раствор. Фильтруют, если выпал осадок, и к кипящему раствору прибавляют в избытке хлорид кальция. Осадок отфильтровывают и прокаливают. После этого обрабатывают осадок разбавленной уксусной кислотой, прибавляя ее по каплям, пока весь карбонат кальция не растворится, фильтруют и остаток умеренна промывают. Затем его осторожно прокаливают и испытывают на содержание фтора обычным способом с серной кислотой.
Перекись водорода обычно содержит ацетанилид, что может быть причиной затруднений в анализе, например при восстановлении цинком с последующим титрованием перманганатом; при этом ацетанилид также восстанавливается и титруется. В таких случаях, и вообще при выполнении очень точных работ, следует предпочесть 30%-ный раствор перекиси водорода. Последний хотя и реже, но также иногда содержит примеси: в одной из партий, поступивших в нашу лабораторию, было значительное количество бария. '
Для приготовления 20%-ной перекиси водорода и для концентрирования ее до 90%-ной рекомендуют1 прибавлять небольшими порциями, медленно и при непрерывном перемешивании х. ч. перекись натрия к сильцо охлаждаемой (ниже 20° С) 20%-ной серной кислоте, пока 95% последней не будет нейтрализовано. Раствор затем фильтруют я фильтрат перегоняют в вакууме при 60—65° С. Полученную таким способом 20%-ную перекись водорода освобождают от хлоридов добавлением сульфата серебра в избытке и вторичной перегонкой в вакууме. Затем дистиллят оставляют на 3 дня в вакуум-эксикаторе над концентрированной серной кислотой при комнатной температуре и так получают 90%-ную перекись водорода.
Для некоторых целей рекомендуется 2 так называемая «твердая перекись водорода», или «гиперол», CO(NH2)2-НгОг, которая содержит 35% НгОг- Это белый кристаллический порошок, устойчивый при обычных температурах, но начинающий разлагаться при 60° С. С водой он дает раствор, содержащий Н2О2 и мочевину, а с эфиром — раствор свободной Н2О2. 1
фтористоводородная кислота. С появлением сосудов из церезина и полиэтилена отпала необходимость в повторных перегонках плавиковой кислоты, за исключением, может быть, некоторых особых случаев. Обычными примесями в плавиковой кислоте являются железо, соляная кислота и органические вещества. Влияние первых двух может быть учтено тщательно проведенными контрольными опытами, а примесь органических, веществ вообще влияния не оказывает, за исключением двух случаев анализа:
1 М. L. К i 1 р a t г i с к, О. М. R е i f f, F. О. R i с е, J. Am. Chem. Soc., 48, 3019 (1926).
2 С. T а н а т а Рл ЖРФХО, 40. 376 (1909).
64
Гл. III. Приборы, и реактивы,
определения двухвалентного железа в горных породах и определения пятивалентного мышьяка в стеклах. Все примеси можно удалить перегонкой плавиковой кислоты из платиновой посуды с добавлением сульфата серебра, если имеется примесь соляной кислоты, и перманганата калия, если присутствуют органические вещества или двуокись серы (которая изредка также встречается в плавиковой кислоте). При выпаривании 30 мл плавиковой кислоты и прокаливании полученного остатка масса последнего не должна превышать 1 1 мг. При хранении плавиковой кислоты в сосудах из эбонита она окрашивается и загрязняется заметными количествами извлеченного из сосуда вещества. То же наблюдается и при хранении плавиковой кислоты в бакелитовых сосудах, но пипетки и градуированные цилиндры из бакелита незаменимы при работе с плавиковой кислотой.
Хлорная кислота, 70—72%-ная (пл. 1,67—1,70 г!см 3). Хлорная кислота такой концентрации имеется в продаже, и ее также можно приготовить в лаборатории из перхлората аммония 2. О применении ее было сказано следующее 2: «Следует обратить внимание на некоторые ценные свойства этой кислоты. Она не ядовита, не взрывчата и вполне устойчива (в отличие от безводной хлорной кислоты). Свое окислительное действие она, как правило, проявляет только при температурах, близких к температуре кипения (203° С), а так как последняя очень высока, то хлорная кислота может вытеснить соляную, плавиковую, азотную и другие летучие кислоты из их солей. Большая часть солей хлорной кислоты 'легко растворима не только в воде, но и в таких органических растворителях, как спирт или ацетон. Соли хлорной кислоты очень хороши для различного рода электрохимических работ, так как они не восстанавливаются при электролизе 3. Хлорная кислота — прекрасный реактив для применения в ацидиметрии; ее титрованные растворы очень устойчивы и особенно удобны в тех случаях, когда требуется нелетучая кислота, а серная кислота почему-либо неприменима. Ее можно применять взамен серной кислоты при выполнении перманганатометрических определений. Выпаривание раствора хлорида железа (III) с хлорной кислотой до полного удаления хлорид-ионов не сопровождается образованием малорастворимых основных солей, как это имеет место при использовании серной кислоты, и остаток легко растворяется в небольшом количестве воды».
При высоких температурах хлорная кислота является энергичным окислителем. Имеются указания 4 на то, что наиболее эффективной является смесь хлорной и серной кислот, например при обработке по Кьельдалю (стр. 862) или при окислении хрома и церия. Смесь равных частей 96%-ной серной кислоты и 72%-ной хлорной кислоты легко окисляет сульфат церия (III) до сульфата церия (IV) примерно при 140° С, а окисление перхлората церия (III) одной хлорной кислотой не происходит и при 168—200° С.
1 О фотоколориметрическом определении малых количеств (0,005—0,3%) кремнефтористоводородной кислоты в плавиковой кислоте, основанном на образовании окрашенной в желтый /цвет кремнемолибденовой кислоты в присутствии борной кислоты, см. G. N. С a d е, Ind. Eng. Chem., Anal. Ed., 17, 372 (1945).
2 H. H. Willard, J. Am. Chem. Soc., 34, 1480 (1912).
3 О 'Применении хлорной кислоты в электроанализе см. Н. S.Carhart, Н. Н. Willard, W. D. Henderson, Trans. Am. Electrochem. Soc., 9, 375 (1906); см. также W. S. H e n d r i x s о n, J. Am. Chem. Soc., 34, 389 (1912).
4 G. F. Smith, Ind. Eng. Chem., Anal. Ed.,'6, 229, 252 (1934).
Реактивы
65
Хлорная кислота имеет особую ценность, когда надо определять кремнекислоту в присутствии веществ, которые с соляной, серной или азотной кислотами образуют нерастворимые соединения.
В то время как 70%-ную хлорную кислоту можно безопасно кипятить приблизительно при 200° С, с неразбавленной хлорной кислотой надо обращаться с очень большой осторожностью. Соприкосновение кипящей неразбавленной хлорной кислоты или ее горячих паров с органическими веществами или даже с легко окисляемыми неорганическими веществами, как, например, с соединениями трехвалентной сурьмы, приводит к сильному взрыву.
Мы наблюдали, например, в одном случае взрыв, когда горячие пары кипящей хлорной кислоты в перегонной колбе пришли в соприкосновение с резиновой пробкой, соединяющей боковой отросток колбы с холодильником, а в другом случае, когда хлорная кислота нагревалась в чашке на горячей плитке, после того как разбавленный раствор хлорной кислоты и спирта был выпарен насколько возможно на паровой бане, снова разбавлен водой и снова выпарен. Имеется сообщение \ что висмут и его сплавы при обработке их концентрированной хлорной кислотой (70%-ной) образуют взрывчатые вещества. Соли висмута, полученные обработкой металла другими кислотами, не обнаруживают этого свойства. А. Нойес и В. Брэй утверждают 2, что ЗЬзОз может взорваться, когда ее нагревают с одной хлорной кислотой, но что взрыва не происходит, если нагревание проводить сначала с азотной кислотой, а потом заканчивать его с хлорной кислотой.
Насколько мы знаем, нет оснований опасаться взрыва, когда при анализе в растворы вводят небольшие количества хлорной кислоты, например прибавляют 1—5 мл хлорной кислоты для разрушения следов продуктов обугливания в конце обработки серной кислотой по Кьельдалю (см. «Азот», стр. 862), или если предварительная обработка проводится смесью азотной и хлорной кислот при температуре ниже 100° С.
Согласно Нойесу и Брэю, «единственным возражением против применения хлорной кислоты (для разрушения органических веществ) является тот факт, что при неправильном ее применении может произойти опасный взрыв, но опыт показывает, что взрьрв никогда не происходит, если органические вещества нагревать сначала со смесью концентрированных азотной и хлорной кислот при температуре паровой бани»3. Смеси хлорной и азотной кислот так же стойки, как и каждая из этих кислот в отдельности. Поэтому, если такая смесь применяется часто, лучше заготовить ее заранее в больших количествах. Таким образом будет сэкономлено время на добавление каждой кислоты отдельно, а главное, будет гарантия, что как-нибудь по рассеянности не прибавят только одну хлорную кислоту, забыв о необходимости прилить еще и азотную кислоту. Следует заметить, что в лабораториях, где выпаривание с хлорной кислотой проводится часто и в больших количествах, имеется опасность конденсации хлорной кислоты на деревянных частях вытяжного шкафа и органических веществ в трубах тяги. Это может привести к воспламенению вытяжного шкафа или к взрывам в дымоходах. В таких случаях надо все деревянные части и дымоходы промывать еженедельно. Применяющиеся для этого щетки и т. п. должны быть сделаны из невоспламеняющегося материала, и каждый раз перед употреблением их надо хорошо вымыть. Если вытяжной шкаф будет использоваться для работы с хлорной кислотой, то при его изготовлении не слег дует цементировать керамические части с помощью содержащих органические вещества цементов, например смесью глета с глицерином.
1 D. G. N i с h о 1 s о n, J. Н. R е е d у, J. Am. Chem. Soc., 57, 817 (1935).
2 А. Н о й е с, В. Б р э й, Качественный анализ редких элементов, ОНТИ, 1936.
3 A. A. N о у е s, W. С. В г а у, Qualitative Analysis, Macmillan Со, 1927.
5 Заказ 522.
66
Гл. III. ПриЬоры и реактивы
Растворы реактивов
Растворы твердых реактивов, вследствие растворяющего их действия на стекло, следует приготовлять или непосредственно перед их применением или лишь в таких количествах, которые будут израсходованы в самый короткий срок. Что это не излишняя предосторожность (при выполнении действительно точных анализов), мы не раз имели возможность убедиться; особенно это относится к таким реактивам, как оксалат аммония и двузамещенный фосфат аммония.
Описываемые ниже растворы реактивов применяют так часто, что их надо иметь заготовленными в достаточном количестве.
Раствор молибдата. Обычно применяемый раствор молибдата содержит приблизительно 5% молибденовой кислоты, от 5 до 10% нитрата аммония и от 20 до 25 % (по объему) азотной кислоты. Его можно приготовить следующим образом. Смешивают 100 г чистого молибденового ангидрида или 118 г 85%-ной молибденовой кислоты с 400 мл воды, медленно и при сильном перемешивании прибавляют 80 мл раствора аммиака и, когда все растворится, фильтруют. Отдельно приготовляют второй раствор: 400 мл азотной кислоты смешивают с 600 мл воды. Этот второй раствор сильно перемешивают током воздуха и очень медленно через трубку, погруженную в жидкость, вливают в него первый раствор. Когда все будет прибавлено, продолжают еще пропускать воздух 1—2 ч. Затем дают постоять, фильтруют, если нужно, и сохраняют в склянке с притертой стеклянной пробкой. Если раствором приходится пользоваться часто, склянку снабжают сифоном.
Рекомендуется также1 применение раствора молибдата аммония, приготовленного следующим способом. В два стакана емкостью по 1 л помещают в каждый по 91 г кристаллического х. ч. молибдата аммония, по 85 г нитрата аммония и по 70 мл раствора аммиака. Затем нагревают, пока соли не растворятся, добавляя при этом в каждый стакан по 700 мл воды. Когда растворение будет закончено, фильтруют содержимое обоих стаканов, собирая фильтрат в одну склянку емкостью 3 л, разбавляют водой до 3 л и хорошо перемешивают. Этот раствор, по данным автора, может сохраняться очень долгое время. При использовании этого раствора молибдата надо в анализируемый раствор, содержащий фосфор, вводить больше азотной кислоты, чем при применении кислого реактива, описанного в тексте2 3. КМ fc '
Магнезиальная смесь. Обычно рекомендуемые аммиачные магнезиальные смеси разъедают стеклянные сосуды и, как было доказано ®, совершенно не нужны, так как вполне можно пользоваться нейтральными растворами. В Бюро Стандартов США было найдено 4 полезным добавление очень небольшого количества соляной кислоты, и в настоящее время в США этот реактив приготовляется следующим образом. Растворяют 50 г MgCh • 6Н2О и 100 0 NH4CI в 500 мл воды, прибавляют раствор аммиака в небольшом избытке, оставляют на ночь и, если за это время выпал осадок, его отфильтровывают. Затем раствор доводят до кислой реакции добавлением соляной кислоты, разбавляют водой до 1 л и сохраняют в склянке с притертой пробкой.
1 С. М. J о h n s о п, Chemical Analysis of Special Steels, 4th ed., John Wiley and Sons, 1930, p. 555>.
2 См. также Chemical Analysis of Metals, ASTM, 1950, p. 35.
3 О. К u h n t, Chem. Ztg., 44, 586 (1920).
4 G. E. F. L u n d e 11, J. I. H о f f m a n, Assoc. Offic. Agr. Chemists, 8, 194 (1924).
Реактивы
67
Хлороплатиновая кислота. Обычно применяемый для определения щелочных металлов раствор хлороплатиновой кислоты содержит 1 г платины в 10 мл. Для осаждения калия из 0,1 г хлорида калия требуется 1,31 мл этого раствора, для определения натрия из 0,1 г хлорида натрия— 1,68 мл. Лучшим способом приготовления раствора хлороплатиновой кислоты является анодное растворение чистой губчатой платины с применением в качестве электролита концентрированной соляной кислоты. Таким образом избегают образования нитрозосоединений платины. Подробности этого метода описаны в оригинальной статье Ч
При растворении платины в царской водке также получается хлороплатиновая кислота, но, кроме того, образуются нитрозосоединения платины. Поскольку эти нитрозосоединения не легко разлагаются, а их соли с щелочными металлами отличаются по своей растворимости от хлороплатинатов щелочных металлов, следует предпочесть метод приготовления реактива анодным растворением металлической платины. Преимуществом растворения в царской водке является то, что в этом случае не требуется никакого специального прибора, но полученный раствор надо несколько раз выпаривать с соляной кислотой до сиропообразного состояния, разбавлять водой и снова выпаривать, чтобы удалить таким образом нитрозогруппу.
Если для приготовления реактива применяют платиновый лом или металл, выделенный из остатков от определения калия хлороплатинат-ным методом, платину надо очистить, как описано ниже.
а) Извлечение платины из остатков от определения калия. Осадки хлороплатината калия и спиртовые фильтраты в лабораториях обычно собирают, чтобы извлечь из них платину для приготовления реактива — хлороплатиновой кислоты. Для извлечения платины применяют метод, в основном разработанный Прехтом1 2. По этому методу сначала выпариванием удаляют избыток этилового спирта и альдегид. При этом надо опасаться образования воспламеняющегося этиленхлорида платины, который в сухом виде имеет склонность взрываться. Поэтому выпаривание нельзя доводить совершенно досуха. Затем выделяют платиновую чернь, подщелачивая раствор и прибавляя к нему восстановитель: глицерин, муравьиную кислоту или декстрозу. Прехт рекомендует добавление раствора едкого натра (пл. 1,2 г/см3), содержащего 8% глицерина. Восстановление начинают при умеренной температуре и заканчивают при кратковременном кипячении. Поскольку платиновая чернь имеет склонность удерживать-соли щелочных металлов, рекомендуется растворить ее, затем осадить платину хлоридом аммония и прокалить осадок до получения губчатой платины, как описано ниже (п. б.). Одного цикла операций достаточно.
б) Извлечение платины из платинового лома. Источником платины для приготовления хлороплатиновой кислоты может также служить различный платиновый лом, собираемый в лаборатории. Этот лом, помимо платины, может содержать и другие металлы платиновой группы, особенно иридий. Поэтому его надо очищать. Лом растворяют в царской водке, полученный раствор выпаривают на паровой бане до сиропообразного состояния, приливают немного воды, нагревают на паровой бане, прибавляют равный объем соляной кислоты и снова выпаривают до сиропообразного состояния. Затем растворяют остаток в воде, отфильтровывают то, что не растворилось, приводят фильтрат к концентрации 5 г платины в каждых 100 мл, нагревают до кипения и прибавляют по 50 мл 20%-ного раствора хлорида аммония на каждые 100 мл раствора. По охлаждении раствор фильтруют через уплотненную фильтровальную бумагу с применением отсасывания. Осадок переносят на фильтр струей раствора хлорида аммония и промывают сначала тем же раствором хлорида аммония, а под конец 95%-ным этиловым спиртом или водой, охлажденной льдом. После высушивания осадка его помещают в фарфоровый тигель и прокаливают, постепенно повышая температуру,
1 Н. С. Р. W е Ь е г, NBS Sci. Paper, 82; NBS Bull., 4, 365 (1907); J. Am. Chem. Soc., 30, 29 (1908).
2 H. Precht, Z. anal. Chem., 18, 509 (1879).
' 5*
68
Гл. III. Приборы, и реактивы
пока осадок не превратится в металлическую губку. Весь описанный цикл операций надо повторить 2 или 3 раза в зависимости от содержания других платиновых металлов в первоначальном ломе.
Раствор диметилглиоксима. Диметилглиоксим растворяют в спирте или в растворе щелочи. Если имеется чистый этиловый спирт, следует предпочесть спиртовый раствор; его приготовляют растворением 1 г диметилглиоксима в 100 мл 95%-ного этилового спирта и, если надо, филь- ' труют. 10 мл этого раствора применяют для осаждения 0,025 г никеля. Если нет чистого спирта, реактив приготовляют растворением 1 г диметилглиоксима в 65 мл раствора аммиака и разбавлением водой до 100 мл. Можно также пользоваться свежеприготовленным раствором 3 г диметилглиоксима в 100 мл 3 %-ного раствора едкого натра.
Газообразные реактивы
Такие газообразные реактивы очень большой чистоты, как двуокись углерода, кислород, азот, аммиак, сернистый ангидрид и сероводород, можно приобрести в баллонах. Применение газов из баллонов значительно удобнее и лучше, чем добывание их в небольших количествах в аппаратах Киппа и т, п. Там, где газ приходится применять часто и он необходим в больших количествах*, надо всегда приобретать его в сталь- t
ных баллонах Ч В настоящее время можно достать в баллонах и менее часто применяемые газы, например аргон, трехфтористый бор, фтористый водород, хлористый водород, шестифтористую серу.
Вр избежание несчастных случаев баллоны с газообразными реактивами должны быть правильно маркированы.
Поглотители двуокиси углерода
Наиболее употребительными поглотителями двуокиси углерода ?
являются раствор едкого кали, натронная известь, натронный асбест и *
едкий натр. Для количественного определения двуокиси углерода эти реактивы помещают в сосуды особой формы: раствор едкого кали — 1
в кали-аппараты Ванье, Либиха и т. п., а твердые поглотители — в стеклянные колонки небольшой массы, простой2 или сложной 3 формы. Все перечисленные поглотители при поглощении двуокиси углерода выделяют воду, и поэтому, когда они применяются для количественного опре- '
деления СО г, за ними в колонке должно быть помещено высушивающее вещество, задерживающее пары воды. Различные реактивы в приборах, л в каких они обычно применяются, могут поглотить приблизительно еле- '
1 Хотя и мало вероятно, чтобы баллоны с такими газами, как сернистый ангидрид или сероводород, могли неожиданно дать серьезную течь, все же возможные случайности должны быть предупреждены, особенно, если баллонами пользуются в местах, где быстрое проветривание помещения или быстрый выход из него невозможны.’ Химики склонны пренебрегать ядовитыми свойствами сероводорода. В высоких концентрациях он приводит к смерти; сероводород не следует допускать в атмосфере лаборатории в каких бы то ни было концентрациях. Все операции, при которых выделяется «сероводород, надо проводить под хорошей тягой в шкафу.
2 Типа: Midvale [G. L. Kelley, Ind. Eng. Chem., 8, 1038 (1916)], Stetser и Norton [J. B. S t e t s e r, R. H. Norton, Iron Age, 102, 443 (1918),], Kelley л Evers [G. L. К e 11 e у, E. W. E ver s, Ind. Eng. Chem., 13, 1052 (1921)], Nesbitt.
3 W. B.Fleming, Iron Age, 93, 64 (1914).
Реактивы
69
дующие количества двуокиси углерода: раствор едкого кали — 1г; натронная известь — от 1 до 4 г; натронный асбест — от 6 до 15 г; твердый едкий натр — от 9 до 20 г.
Раствор едкого кали (пл. 1,4 г/см3), приготовленный растворением двух частей едкого кали (в палочках) в трех частях воды, прежде часто применялся для поглощения двуокиси углерода. Раствор едкого кали более удобен, чем раствор едкого натра, потому что карбонат калия значительно более растворим, чем карбонат натрия, и потому не так скоро выделяется в виде кристаллов, закупоривающих прибор. Однако раствор едкого кали имеет ряд недостатков: он сильно пенится, что не дает возможности пропускать через него быстрый ток газа, и отдает значительно больше воды, чем твердые поглотители. По этим причинам применение раствора едкого кали не рекомендуется.
Натронная известь является смесью двух частей окиси кальция и одной части едкого натра; она приготовляется обработкой раствора едкого натра негашеной известью, выпариванием раствора досуха, прокаливанием до красного каления и измельчением охлажденной массы до частиц, в большинстве своем проходящих через сито в 10 меш х. Полученный продукт должен быть пористым, а не таким твердым и малопогло-щающим, какой употребляется иногда при сжигании азотистых органических веществ. Для наиболее эффективного поглощения двуокиси углерода он должен содержать 2% влаги. Натронную известь не следует набивать в трубки слишком плотно, потому что, поглощая воду, она увеличивается в объеме. При прохождении сухого воздуха через натронную известь она отдает ему влагу, особенно если при этом происходит поглощение СО2. Натронный асбест и твердый едкий натр настолько лучше натронной извести, что от применения последней следовало бы отказаться.
Натронный асбест приготовляют следующим образом 1 2. Растворяют 1 кг едкого натра в 1 л воды. На каждые 500 мл полученного раствбра прибавляют еще по 1 кг едкого натра, измельченного в порошок, и перемешивают. Затем прибавляют небольшими порциями, при перемешивании, разделенный на волокна асбест до тех пор, пока смесь не утратит способности его смачивать. Тогда нагревают всю массу на воздушной бане при-150—180° С в течение 4 ч. В начале нагреванця время от времени прибавляют еще асбест, пока масса не примет такой же вид, какой она имела до нагревания, после чего ее охлаждают и измельчают настолько, чтобы большая часть ее проходила через,сито в 10 меш (с? = 1,65 мм). Если натронный асбест предполагают применять при уменьшенных давлениях, то его надо измельчить сильнее — до 30 (d = 0,54 мм) или 40 меш (d = = 0,37 мм), чтобы увеличить поглощающую поверхность и ввести в него больше влаги.
Поглотитель, приготовленный таким способом, действует в 4 раза сильнее натронной извести и может поглотить двуокись углерода в количестве около 20% от своей массы. Полное поглощение происходит даже тогда, когда газы проходят через поглотитель со скоростью 500 мл в минуту. Натронный асбест меняет свою окраску при насыщении двуокисью углерода, являясь таким образом индикатором своего истощения. Это самый
1 * Диаметр отверстия 1,65 мм. Прим, ред.*
2 G. L. К е 11 е у, цит. выше.
70
Гл. III. Прибора и реактива
лучший поглотитель двуокиси углерода для обычной аналитической работы, несмотря на то что он дороже твердого едкого натра и поглощает меньше двуокиси углерода на единицу своей массы г.
Твердый едкий натр был рекомендован1 2 как вещество, превосходящее по своим качествам и натронную известь и натронный асбест. Для приготовления поглотителя едкий натр измельчают так, чтобы частицы его проходили через сито в 5 меш (d = 3,96 мм) и оставались на сите в 20 меш (d = 0,83 мм); при таком измельчении он поглощает двуокись углерода в количестве, равном почти одной трети своей массы. Измельчение и просеивание едкого натра проводится с трудом, особенно когда его влажность велика. Еще труднее приготовить таким способом едкое кали вследствие его большей гигроскопичности, к тому же поглотительная способность на единицу массы у едкого кали меньше, чем у едкого натра. Указанные авторы нашли, что «хозяйственный щелок», в котором частички едкого натра несколько меньше рисовых зерен, дает прекрасные результаты. Пригоден также для поглощения двуокиси углерода и едкий натр в виде лепешек. Во всех случаях за едким натром надо поместить высушивающее вещество,'потому что едкий натр отдает влагу сухому воздуху, особенно при поглощении больших количеств двуокиси углерода.
Пемза, пропитанная безводным сульфатом меди. Безводный сульфат меди не применяется как поглотитель двуокиси углерода, но мы рассматриваем его здесь, потому что пемзу, пропитанную безводным сульфатом меди, часто включают в поглотительную цепь для удаления из очищаемого газа малых количеств хлористого водорода и сероводорода.
Препарат приготовляют следующим образом. Измельчают пемзу до кусков размером примерно 0,5 см, отсеиващт мелочь и 60 г полученного продукта переносят в фарфоровую чашку или кастрюлю. Залив кусочки пемзы концентрированным раствором 30—35 г сульфата меди, выпаривают досуха при непрерывном перемешивании, после чего нагревают еще 3—4 ч при 150—160° С на воздушной бане. Охлаждают в эксикаторе и хранят в банке с притертой пробкой. При помещении этого препарата в поглотительную цепь надо перед ним и после него ставить сосуд с высушивающим веществом.
Из других веществ, предложенных для той же цели, следует отметить: раскаленную серебряную сетку3, горячую двуокись свинца, не содержащую окиси свинца4 *, раствор сульфата серебра (0,6 г Ag2SO4 в 100 мл воды, содержащей 3—4 капли H2SO4) и раствор арсенита серебра в разбавленной серной кислоте6. Последний приготовляют следующим образом. Растворяют 2 г As2O3 в возможно меньшем количестве раствора КОН, разбавляют до 200 мл и нейтрализуют разбавленной серной кислотой. Прибавляют раствор AgNO3, пока происходит образование осадка, сохраняя при этом раствор нейтральным по лакмусу добавлением разбавленного раствора КОН. Перемешивают до коагуляции осадка, дают ему осесть и промывают декантацией водой. Затем рас
1StetsernNorton (цит. выше) приготовили несколько иной вид натронного асбеста, который теперь имеется в продаже под названием «аскарит»; они утверждают, что этот препарат лучше, чем описанный выше, и не выделяет влаги при работе. Последнее не совсем правильно, особенно в тех случаях, когда поглощается много двуокиси углерода или когда реактив близок к истощению. Для поглощения двуокиси углерода имеются еще и другие патентованные препараты гранулированного едкого натра; эти препараты называются «микобайт» и «кароксайт».
2 G. L. К е 11 е у, Е. W. Е vers, Ind. Eng. Chem., 13, 1052 (1921).
3 F. К о p p e г, Z. anal. Chem., 17, 23 (1878).
4 M. Dennstedt, F. Hassler, Z. anal. Chem., 42, 417 (1903).
6 Lord, Demorest, Metallurgical Analysis, 5th ed. McGraw-Hill Co., 1924, p.23.
Pea ктивы
71
творяют осадок в 10%-ной H2SO4, прибавляя ее в очень небольшом избытке, разбавляют водой до 150 мл и фильтруют. Этот раствор поглощает H2S, НС1 й С12 и не поглощает заметных количеств СО2.
Высушивающие вещества
Выбирать подходящее высушивающее вещество для каждого отдельного случая его применения следует с гораздо большим вниманием, чем это обычно делается аналитиками. Нужно обращать внимание и на состояние, в котором это высушивающее вещество находится. Сказанное относится не только к тем случаям, когда высушивающее вещество применяется в поглотительной цепи при определении воды или двуокиси углерода, но и к обычному применению его в эксикаторах. Непродуманное использование различных высушивающих веществ в поглотительных цепях не может не привести к затруднениям,-так как величина поглощающей способности и скорость поглощения влаги у них различны. Воздух, прошедший через хлорид кальция, будет отдавать влагу серной кислоте, а воздух, прошедший через фосфорный ангидрид, будет отнимать влагу у хлорида кальция; два различных высушивающих вещества могут иметь одинаковую поглощающую способность при медленном токе газа и совершенно различную — при быстром токе. В поглотительную цепь можно поместить два и несколько высушивающих веществ при условии, что каждое последующее имеет большую поглотительную способность, чем предыдущее, и что в конце помещено то же вещество, какое находится во взвешиваемом сосуде. Само собой разумеется, что высушивающие веще-' ства надо возобновлять достаточно часто, чтобы обеспечить максимальное их действие. Для этой цели нужно вести запись массы поглощенной воды, если это возможно, или хотя бы записывать даты наполнения сосудов свежим высушивающим веществом.
Обычными высушивающими веществами являются: хлорид кальция, серная кислота и фосфорный ангидрид. Менее часто применяются безводный перхлорат магния, трехводный перхлорат магния, окись бария, окись алюминия, безводный сульфат кальция, окись кальция и плавленое едкое кали.
Хлорид кальция имеется в продаже в нескольких видах. Безводный хлорид кальция, применяемый для высушивания, по техническим условиям Ам. Хим. О-ва должен содержать не менее 96% СаСН. Его можно приобрести в виде кусочков (гранул) различного размера: 4, 8, 12, 20 и 40 меш (d = 4,7; 2,4; 1,4; 0,83 и 0,37 мм). Этот реактив показывает щелочную реакцию вследствие частичного разложения во время его производства; в нем по техническим условиям Ам. Хим. О-ва допускается содержание до 0,020% Са(ОН)2. Поэтому в тех случаях, когда хлорид кальция применяют в поглотительной цепи при определении двуокиси углерода (для высушивания газа, содержащего СОг, перед поглощением последнего щелочью), надо предварительно обработать его двуокисью углерода. Но и такая обработка не вполне достигает цели, потому что нейтрализация гидроокиси кальция двуокисью углерода происходит только на поверхности гранул, а по мере поглощения воды поверхность снова становится щелочной. Хлорид кальция не является сильно высушивающим веществом, и его поглотительная способность зависит от температуры.
72
Гл. III. Приборы и реактивы
Показано1, что воздух, насыщенный водяными парами и затем высушенный на- ' сколько возможно лучше над хлоридом кальция, содержит в 1 л еще 0,3 мг влаги при 0 °C; 1 мг — при 15 °C; 3,3 мг — при 30 °C, что соответствует 6, 8 и 11% от всего количества влаги, первоначально присутствовавшей в воздухе. Если хлорид кальция применяется в различных местах поглотительной цепи, все наполненные им трубки должны иметь приблизительно одинаковую температуру.
В качестве высушивающего вещества предложен2 безводный перхлорат бария Ва(С1О4)2, который поглощает примерно столько же влаги (15%), сколько и хлорид кальция, но эффективнее последнего при комнатной и более низших температурах и его можно легко регенерировать, не вызывая разложения, нагреванием при 140— 400 °C.
Серная кислота — значительно лучшее высушивающее вещество, чем хлорид кальция. Ее действие одинаково эффективно при всех температурах от —30 до +25° С; при температурах выше -|-30о С оно становится слабее. Разность между поглотительными способностями серной кислоты и хлорида кальция видна по приводимым в таблицах (см., например, табл. 8, стр. 76) значениям поглотительной способности последнего, так как остаточная влажность в каждом случае означает то, что было поглощено серной кислотой после возможно более полного высушивания над хлоридом кальция. Было показано 3, что летучесть серной кислоты очень незначительна и что 1 л воздуха, пропущенного через чистую серную кислоту при комнатной температуре, может содержать не более 0,003 мг серного ангидрида. Серная кислота иногда содержит двуокись серы и очень редко — пиросерную кислоту. Первую можно удалить, пропуская через серную кислоту сухой воздух, вторую — добавлением 5 % воды. СО2 легко удаляется из серной кислоты пропусканием воздуха.
Фосфорный ангидрид — наиболее энергичное из обычно применяемых высушивающих веществ; его следует предпочесть в тех случаях, когда необходимо очень полное высушивание, особенно если газ проходит с очень большой скоростью. 1 л воздуха, высушенного насколько возможно полно при 25° С серной кислотой, содержащей не более 8,4 % воды, отдает еще около 0,002 мг влаги фосфорному ангидриду при пропускании через него со скоростью около 2 л в час 3>4 5 * *.
Поглотительная способность фосфорного ангидрида почти не изменяется 8 при 90° С. Применение его в обычной работе мало удобно, потому что, поглощая воду, он образует вязкую жидкость, которая или закупоривает трубку, прекращая ток газа, или приводит к образованию каналов. Это затруднение может быть отчасти преодолено чередованием рыхлых слоев фосфорного ангидрида со слоями стеклянной ваты и предварительным пропусканием осушаемого газа через другие высушивающие вещества, например через серную кислоту или трехводный перхлорат магния. Фосфорный ангидрид в некоторых случаях содержит более лету
1 Н. С. D 1 Ь Ь i t s, Z. anal. Chem., 15, 121 (1876).
2 G. F. Smi t h, Chemist Analyst, 17, 21 (1928).
8 E. W. Morley, Am. J. Sei., [3], 30, 140 (1885).
* H. C. D i b b i t s, цит. выше.
5 В более позднем сообщении [J. Am. Chem. Soc., 26, 1171 (1904)] Morley -
утверждает: «воздух, проходящий co скоростью 2 л в час через 25 см3 фосфорного анги-
дрида, правильно положенного в трубку для высушивания, содержит значительно меньше 1 мг водяного пара в 40 000 л; сколько именно, половину этого количества или десятую часть, никто не может сказать», и далее «ни один из весовых экспериментов,
которые проводятся в настоящее время в научном мире, не потребовал бы учета той влаги, которая остается в газе после прохождения его через фосфорный ангидрид».
Реактивы
73
чий фосфористый ангидрид, который можно удалить возгонкой в токе сухого воздуха.
Имеются указания 1 на то, что поглотительная способность пористой окиси бария (ее приготовляют прокаливанием смеси карбоната бария с углем при умеренно высоких температурах) равна поглотительной способности фосфорного ангидрида или даже слегка ее превышает; Это утверждение основано, однако, на опытах, в которых газ проходил через поглотитель с очень малой скоростью, около 250 мл в час. Плавленая окись бария, получаемая в электрической печи, менее активна и содержит карбиды, которые при соприкосновении с влагой выделяют ацетилен.
По данным авторов \ предложенная, ими пористая окись бария имеет следующие преимущества: 1) ее поглотительная способность на единицу массы примерно такая же, как у фосфорного ангидрида; 2) она может применяться для высушивания газов, являющихся в химическом отношении основаниями, например для аммиака; 3) гидроокись бария при комнатных температурах имеет неизмеримо малое давление пара; 4) ее поглотительная способность не уменьшается при повышении температуры до 1000° С. Недостатки у пористой гидроокиси бария следующие: 1) приходится применять большие поглотительные трубки, потому что высушивающее вещество изготовляется в гранулах, имеющих меньшую общую поверхность, чем тонкие порошки фосфорного ангидрида; 2) при поглощении влаги окись бария расплывается, и ее нужно поэтому перекладывать стеклянной ватой; 3) реактив должен быть свежим, а не частично погашенным; 4) отработанная окись бария не может быть регенерирована.
Безводный перхлорат магния и тригидрат перхлората магния были предложены2 в качестве высушивающих веществ в 1922 г. По данным авторов, этот реактив при 25° С является таким же эффективным высушивающим веществом, как фосфорный ангидрид, и поглощает влагу в количестве до 60% от своей массы, т. е. в 3—5 раз больше, чем фосфорный ангидрид, при условии, что газ проходит через безводный перхлорат магния со скоростью, не превышающей 5 л в час. Трехводная соль мало отличается по своему действию от безводной соли при 0° С и равных скоростях тока газа, но при более высоких температурах она менее эффективна. Безводная соль имеет следующие преимущества: 1) масса поглощенной воды, приходящейся на единицу массы высушивающего вещества, в несколько раз больше, чем при использовании фосфорного ангидрида; 2) частички этой соли, поглощая влагу, не слипаются, не образуют каналов, и объем их при поглощении воды уменьшается; 3) высушивающее вещество можно многократно регенерировать; 4) реактив является нейтральным веществом, которое можно применять для высушивания таких газов, для которых применение фосфорного ангидрида и серной кислоты недопустимо 3.
* Безводный перхлорат магния можно приготовйть следующим способом 4. В стакан, содержащий 30 %-ную хлорную кислоту, прибавляют
1 Н. S. В о о t h, L. Н. М с I n t yre, Ind. Eng. Chem., Anal. Ed., 2, 12(1930). 2 H. H. W i 11 a r d, G. F. S ih i t h, J. Am. Chem. Soc., 44, 2255 (1922).
3 В позднее опубликованной статье [Ind. Eng. Chem., 19, 411 (1927)] Smith рекомендует употреблять смесь 26,5% безводного перхлората магния и 73,5% безводного перхлората бария и утверждает, что этот реактив поглощает 40% (по массе) воды и может быть регенерирован постепенным нагреванием до 250° С при давлении 102 мм рт. ст.
4 И. П. А л и м а р и н, Зав. лаб., 9, 915 (1940).
Ilk
Гл. III. Приборы и реактивы
небольшими порциями в избытке химически чистую окись магния, отфильтровывают то, что не перешло в раствор, через стеклянную воронку с пористым дном и фильтрат, имеющий щелочную реакцию, нейтральной хлорной кислотой до слабокислой реакции по бумаге конго. Раствор после этого упаривают на электрической плитке и охлаждают холодной водой. Выделившуюся кристаллическую массу переносят на фарфоровую воронку с дырчатым дном (отверстия должны быть маленькими, так как фильтровальную бумагу применять нельзя). Отсасывают маточный раствор в течение 10—15 мин, растворяют кристаллы в небольшом количестве горячей воды; полученный раствор снова упаривают до тех пор, пока не начнется выделение кристаллов на его поверхности, и охлаждают. Кристаллы отделяют, как в первый раз. Оба маточных раствора соединяют и из них выделяют дополнительное количество соли. Полученная шестиводная соль Mg(C104)a • 6Н2О имеет вид белых игольчатых кристаллов, которые при растворении должны давать прозрачный раствор, имеющий нейтральную реакцию. Вследствие большой разности в растворимости Mg(C104)2- 6Н2О в горячей и холодной воде выход кристаллического продукта составляет около 90%.
Кристаллы шестиводной соли помещают в фарфоровую чашку и нагревают на электрической плитке; при 145—147° С они сначала плавятся в своей кристаллизационной воде., потом по мере удаления воды жидкость затвердевает, и получается пористый продукт, состоящий главным образом из трехводной соли.
Для предупреждения образования сплошной малопористой массы операцию обезвоживания следует вести при энергичном перемешивании. Затем температуру повышают до 170—200° С, причем соль снова плавится, образуя прозрачную бесцветную жидкость, с одновременным выделением некоторого количества воды; в таком состоянии массу выдерживают 1—2 ч. Нагревать выше 230—240° С ни в коем случае не следует, так как при такой температуре трехводная соль разлагается с образованием хлора, хлорида магния и окиси магния. Поэтому температуру все время контролируют термометром, погруженным в жидкость.
После охлаждения затвердевший перхлорат магния измельчают до зерен размером 3—4 мм и помещают в круглодонную колбу, которую присоединяют при помощи толстостенной резиновой трубки к колонке с хлоридом кальция, а последнюю — к масляному вакуум-насосу, дающему разрежение до 0,1 мм рт. ст.
Колбу помещают в сушильный шкаф и нагревают при непрерывно действующем насосе и температуре около 170° С в продолжение 2—3 ч, потом температуру повышают до 220—240° С и нагревание продолжают еще 3 ч. Удаление воды при этом происходит без. плавления соли. Полученный безводный перхлорат магния представляет собой белую пористую массу, содержащую около 0,1 % воды. Водный раствор его должен быть прозрачным и нейтральным и не должен давать реакции на хлорид-ионы. Хранить полученный продукт надо в плотно закрывающихся банках. Регенерацию бывшей в употреблении соли проводят нагреванием в вакууме, как описано выше. Доп. ред. *
Окись алюминия, приготовленная при низкой температуре 1, является таким же энергичным высушивающим веществом, как фосфорный анги
1 F. М. G. J ohnson, J. Am. Chem. Soc., 34, 911 (1912).
Реактивы
75
дрид, если применяется при температуре 18° С, и газ через нее проходит со скоростью 2—3 пузырька в секунду. Когда она утеряет свою высушивающую способность, что наступает при увеличении ее массы приблизительно на 18%, ее можно регенерировать нагреванием при 400° С при одновременном, пропускании воздуха, высушенного серной кислотой Е
Окись кальция вдвое менее эффективное высушивающее вещество, чем хлорид кальция, но ее иногда применяют для предварительного высушивания газов, способных реагировать с другими высушивающими веществами. В таких случаях применяют также плавленое едкое кали, которое по своей высушивающей способности (в отсутствие двуокиси углерода) примерно равноценно хлориду кальция.
Борный ангидрид 1 2 (для его приготовления расплавленную борную кислоту нагревают приблизительно до 800° С, выливают в четыреххлористый углерод, охлажденный до 0° С, и измельчают в порошок) может поглотить воду в количестве 25 % от своей массы и является лучшим высушивающим веществом, чем хлорид кальция. Следует учитывать, что эффективное его действие начинается лишь после того, как он поглотил некоторое количество влаги, и что скорость тока газа при его применении не должна превышать 2 л в час.
<. В табл. 8 приведены результаты испытания 3 различных высушивающих веществ, выполненного в Бюро Стандартов США. Метод испытания состоял в пропускании соответственно кондиционированного воздуха со скоростью от 1 до 5 л в час через цепь U-образных трубок, содержащих два или несколько высушивающих веществ, расположенных в порядке возрастания их поглотительной способности. После того как отмеренное количество воздуха было пропущено через цепь, в которой поддерживалась температура 30,5 ± 0,5° С, было определено увеличение в массе каждой из U-образных трубок. Сумма увеличений в массе всех трубок, которые следуют за каждой данной трубкой, показывает, сколько влаги прошло через эту трубку непоглощенной. Последняя трубка содержала фосфорный ангидрид; все результаты таким образом даны по отношению к поглотительной способности фосфорного ангидрида. Исследовались продажные продукты в той форме, в какой они выпускаются на рынок как высушивающие вещества.
Если высушивающее вещество нужно взвешивать, то его помещают в легкую U-образную трубку с притертыми пробками. В некоторых приборах, например в приборе Флеминга для поглощения двуокиси углерода, высушивающее вещество помещается в специально отведенном для него месте. Хлорид кальция накладывают в трубку рыхлым слоем и покрывают сверху асбестовыми тампонами, чтобы предупредить его распыление. При применении серной кислоты трубку наполняют на две трети ее емкости стеклянными бусами и серную кислоту наливают в таком количестве, чтобы она смочила бусы и заполнила колено трубки. Фосфорный ангидрид накладывают рыхлыми слоями вперемежку со слоями стеклянной ваты или асбеста. Если нужно поглотить много воды, то перед фосфорным ангидридом помещают трубку с другим высушивающим
1 Поглощение влаги окисью алюминия объясняется скорее адсорбцией, чем ее гидратацией. Об адорбции паров окисью алюминия см. L. A. Mu нго, F. М. G. J о h п-s о н, Ind. Eng. Chem., 17, 88 (1925).
2 J. H. W a 1 t о n, С. К. R os enbau m, J. Am. Chem. Soc., 50, 1648 (1928).
8 J. H. Bower, J. Research NBS, 12, 241 (1934); 33, 199 (1944).
76
Гл. Ill Приборы и реактивы
ТАБЛИЦА .8
Поглотительная способность некоторых высушивающих веществ
Вещество Объем воздуха в мл, прошедшего в 1 ч через 1 МЛ поглотителя Общий объем воздуха в л, прошедшего через 1 мл поглотителя Влага, оставшаяся в 1 л воздуха, мг
данные Бюро Стандартов США литературные данные
CuSO4 (безв.) . . 36—50 0,45-0,7 2,8 (2,7—2,9) 1,7 (30° С)1
СаС12 (гран.) . . 66—165 6,1-24,2 1,5 (1,4-1,6)
СаС12 (техн, безв.) 115—150 4,0-5,8 1,25 (1,23|—1,27)
ZnCl2 (палоч.) . . 120-335 0,8-2,1 0,98(0,94-1,02) 0,95 (30° С) 2 *
Ва(СЮ4)2 (безв.) 26-36 2,3-3,7 0,82 (0,78-0,83) 0,29 (27° С)8
NaOH (палоч.) 75—170 2,3—8,9 0,80 (0,78—0,83) 0,16 (25° С) *
СаС12 (безв.) . . 75-240 1,2-7,8 0,36 (0,33—0,38) 0,36 (25° С) 5
Mg(C104)2-3H20 65-160 4,0-7,2 0,031 (0,028—0,033)
КОН (палоч.) . . 55—65 3,2—7,2 0,014 (0,010—0,017) 0,002 (25° С) 4
Силикагель . . . 43-59 2,1-5,2 0,006 (0,002-0,01)
CaSO4 (безв.) . . 75—150 1,2—18,5 0,005 (0,004—0,006) 0,005 (25° С) •
СаО 60-90 7,6—10,1 0,003 (0,003-0,004) 0,2-0,3 (25° С)1
Mg(ClO4)2 (безв.) 43-53 2,8-5,9 0,002 (0,0016-0,0024)
AI2O3 •36-63 5,6-6,2 0,001 (0,0008—0,0012) 0,003 (25° С)1
ВаО ....... 64-66 10,6—25 0,0007 (0,0006—0,0008) Меньше 0,0003 при «обычной температуре» 7
1 М. V. Dover, J. W. Harden, J. Am. Chem. Soc., 39, 1613 (1917).
2 G. P. Baxter, R. D. Warren, J. Am. Chem. Soc., 33, 344 (1911).
3 G. F. Smith, Ind. Eng. Chem., 19, 411 (1927).
* G. P. Baxter, H. W. Starkweather, J. Am. Chem Soc., 38, 2038 (1916). <
5 G. P. Baxter, H. W. Starkweather, J. Am. Chem. Soc., 38, 2041 (1916).
6 W. A. Hammond, J. R. Withrow, Ing. End. Chem., 25, 653 (1933).
’ H. S. В о о t h, H. Me Intyre, Ind. Eng. Chem., Anal. Ed., 2, 12 (1930).
О поглотительной способности высушивающих веществ, не включенных в табл. 8, см. в следующих статьях; '
ZnBr2, CaBr2: G. Р. Baxter, R. D. Warren, цит. выше.
В2О3: J. Н. W а 11 о n, С. К. Rosenbaum, J. Am. Chem. Soc., 50, 1648 (1928)-
MgO: M. V. Dover. J. W- M a r d e n, цит. выше.
H2SO4, P2O5: E. W. Morley, Am. J. Sci., 30, 141 (1885); J. Am. Chem. Soc., 26, 1171 (1904).
веществом, например с трехводным перхлоратом магния. Последний уменьшается в объеме при поглощении воды, не слипается и его надо накладывать так, как указано для хлорида кальция т.
1 О приготовлении свинцово-натриевого сплава (90 частей свинца, 10,5 части
натрия), применяемого для высушивания легко воспламеняющихся жидкостей, на-
пример эфира, см. в статье Н. S о г о о s, Ind. Eng. Chem., Anal. Ed., 11, 657 (1939).
Глава IV
ОБЩИЕ МЕТОДЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
•ОТБОР ПРОБЫ
Отбор пробы вещества, подлежащего анализу, всегда имеет большое лндчение, а иногда является даже более важной операцией, чем выполнение самого анализа. Целью отбора пробы во всех случаях является получение сравнительно небольшого количества вещества, в котором определяемый компонент был бы в таком7 же относительном количестве, в каком он содержится во всей массе анализируемого материала. Методы отбора пробы значительно различаются для разных материалов и могут быть как простыми, так и очень сложными. Для определения влаги в исходном материале обычно отбирается отдельная проба *.
Отбор проб горных пород рассматривается в третьей части этой книги (стр. 890). Подробные описания методов отбора проб других веществ приводятся в трудах, посвященных анализу этих веществ 2.
При анализе руд и других подобных веществ в лабораторию обычно поступает большая проба подлежащего анализу материала, из которой должна быть отобрана средняя лабораторная проба. Этот отбор проводится различными методами, но в общем они сводятся к следующему. Всю присланную в лабораторию большую пробу раздробляют до кусков подходящей небольшой величины, затем квартованием или каким-либо другим аналогичным приемом отбирают часть этой пробы, измельчают до кусков несколько меньшей величины и снова делят. Эту операцию повторяют до тех пор, пока не получится проба, по величине достаточная для выполнения всего анализа, но не слишком большая во избежание излишней работы при ее окончательной подготовке. Всю эту пробу 3
\ См., например, Standard Methods'of Laboratory Sampling and Analysis of Coal, ASTM Standards, Part 5, 1950, p. 583 и T. A. W r i g h t, Sampling and Evaluating Secondary Nonferrous Metals, AIME Tech. Pub., 81. •
2 См., например, W. J. Sharwood, M. von Bern ewitz, Bibliography of Literature of Sampling, U. S. Bur. Mines Pub. 2336; Chemists U. S. Steel Corp., Sampling and Analysis of Carbon and Alloy Steels, Reinhold Publishing Corp., 1938; Sampling and Analysis of Pig Iron, 3d ed., 1934; Sampling and Analysis of Iron and Manganese Ores, Carnegie Steel Co., Pittsburgh, Pa, 1926; ASTM Standards, Part. 1—6, 1949; W. F. Hi lie bra nd, J. Ind. Eng. Chem., 8, 466 (1916).* Г. A. X a H, E. А. А н ф и м о в а, Опробование сырья и продуктов промышленности, Госхимиз-дат, 1953.*
3 См. стр. 890—893.
78
Гл. IV. Общие методы и операции химического анализа
полностью измельчают до такого размера частиц, чтобы свести к минимуму ошибки при взятии навесок для анализа и чтобы степень измельчения была достаточной для последующей химической обработки *.
Измельчение следует проводить так, чтобы при этой операции не было химичесйих изменений 1 2 или селективного распыления отдельных компонентов.
Отбор средней про^ы металла или сплава часто оказывается таким же сложным делом, как отбор средней пробы руды. В большей или меньшей степени, но практически всегда имеет место неоднородность (сегрегация) металла. Кроме того, окончательно отобранную среднюю пробу нельзя, как правило, измельчить до такой степени, чтобы устранить ее неоднородность. Это хорошо иллюстрируется теми трудностями при отборе проб, которые вызываются присутствием графита в чугунах и хрупких компонентов в подшипниковых сплавах.
Если для получения пробы чугуна обрабатывать его на станке, то проба не только будет содержать свободный графит, но в более мелких ее частичках будет меньше углерода, чем в более крупных. Например, после удаления из пробы свободного графита и просеивания всей остальной части через ряд сит, в порции, состоявшей из частиц в 30—40 меш (d = 0,54—0,137 мм), найдено 2,04% углерода, в порции из частиц в 20—30 меш (d = 0,83—0,54 мм) найдено 2,27%, а в порции частиц в 14—20 меш (d = 1,17—0,83 мм) найдено 2,45%.
О затруднениях, возникающих вследствие присутствия хрупких компонентов, дают представление следующие результаты анализа измельченной пробы обыкновенного подшипникового сплава3:
Составные части, % Величина частиц
20 меш (d = 0,83 «’<) 20—3 0 меш (d= 0,83— 0,54 л’..м) 30—80 меш (d= 0,54— 0,18 мм) от 80 меш (d — 0,18 мм и до пыли)
Медь . . . 70,0 70,2 67,6 63,2
Свинец . . 24,4 24,1 27,9 31,2
Олово ... 4,9 4,9 4,7 4,4
Пробы от стальных изделий отбирают следующим образом. От болванок, брусьев и других больших предметов пробу отбирают в средней части изделия между наружной его поверхностью и центром; от пустотелых стальных изделий пробу берут в середине стенок между внутренней и внешней их поверхностями; от тонких стальных изделий пробу отбирают со всего поперечного сечения 4.
1 Согласно G. F. Smith, L. V. Hardy и Е. L. G а г d [Ind. Eng. Chem.], Anal. Ed., 1, 228 (1929)], смеси веществ различной плотности не могут сегрегировать вследствие сотрясения или под влиянием вибрационных колебаний помещения, где они хранятся, независимо от того, как сильно отличаются друг от друга по плотности отдельные частицы смеси, если смесь измельчена так, что она проходит через сито в 200 меш (d = 0,074 мм) и хорошо перемешана. Отбор средней лабораторной пробы материалов, содержащих металлические частицы, например «чешуйки» в рудах золота, надо проводить по указаниям, которые даются в специальных руководствах.
2 См. стр. 898, 900 и 987.
3 См. также J. A. S с h е г г е г, G. Е. F. L u n d е 11, J. Research NBS, 5, 891 (1930).
4 ASTM Methods of Chemical Analysis of Metals, 1950, стр. 58 — стали и чугуны, стр. 63 — ферросплавы.
Взвешивание пробы
79
Если имеется вероятность сильного различия в составе у разных по величине частиц одной и той же пробы, рекомендуется поступать следующим образом.
При необходимости провести особо точный анализ всю большую пробу надо просеять через сита разных размеров (избегая потерь от распыления), взвесить каждую из полученных таким образом фракций и приготовить пробу для анализа, отбирая от каждой фракции количество, пропорциональное массе этой фракции.
При выполнении обычных рядовых анализов можно всю пробу измельчить достаточно тонко, чтобы, взяв из нее после перемешивания некоторое число мелких порций (как при отборе проб) и смешав их, получить навеску для анализа, состав которой соответствовал бы среднему составу присланного образца.
ПРИГОТОВЛЕНИЕ ПРОБЫ ДЛЯ ВЗВЕШИВАНИЯ
Общее правило приготовления пробы для взвешивания заключается в том, что из пробы должны быть удалены все посторонние вещества, попавшие в нее при отборе или из упаковки, например щепки, волокна, металлические частицы или масло, примененное при сверлении или строгании стали. Общих правил высушивания пробы не существует. В ряде случаев, например при анализе некоторых горных пород (стр, 900), лучше анализировать пробу, как. она есть, не высушивая ее предварительно. В других случаях принято высушивать при 105—110° С. Если достигнуть постоянной массы пробы при этих температурах очень трудно и для этого требуется много времени, приходится применять более высокие температуры; например, при анализе пиролюзита его высушивают при 125° С, при анализе боксита — при 140° С. Некоторые вещества при нагревании медленно окисляются, и поэтому их надо высушивать в неокисляющей атмосфере.
Результаты анализа, показывающие содержание компонентов «в расчете на сухое вещество», означают, что для анализа была взята проба, высушенная при определенной температуре (обычно при 105—110° С), или же что для анализа была взята воздушно-сухая проба, но полученные результаты были пересчитаны на сухое вещество на основании определения влажности этого материала, проведенного в отдельной его пробе, взятой в то же самое время, что и проба для анализа.
«Анализ сухим путем» есть нечто совершенно другое — это анализ проведенный с применением сухих твердых реактивов при высоких температурах, как, например, определение золота методом купелирования
ВЗВЕШИВАНИЕ ПРОБЫ
Если пробу надо отвешивать в воздушно-сухом состоянии, то особых предосторожностей принимать не приходится; не требуются они и при взвешивании предварительно высушенных негигроскопичных веществ. Слабо гигроскопичные вещества можно высушивать и взвешивать в тиглях с плотно закрывающимися крышками или в стаканчиках для взвешивания. Последние следует предпочесть при выполнении точных анализов, и их всегда надо применять при взвешивании заметно гигроскопичных веществ.
80
Гл. IV. Общие методы, и операции химического анализа
Нужно иметь в виду, что некоторые вещества, будучи только умеренно гигроскопичными, поглощают в короткий промежуток времени большую часть того количества влаги, которое они вообще могут поглотить из воздуха. Навески'таких веществ нельзя отбирать последовательно из одной высушенной пробы, находящейся в стаканчике для взвешивания; нельзя также брать навеску, отсыпая порцию высушенной пробы и затем снова взвешивая стаканчик, из которого она была отсыпана (см. «Марганец», стр. 497). В этих случаях надо высушить только такое количество вещества, какое необходимо для одного определения, закрыть стаканчик крышкой, охладить в эксикаторе и перед взвешиванием открыть крышку на мгновение, чтобы наполнить стаканчик воздухом. При взвешивании надо поставить на другую чашку весов подобный же стаканчик, прошедший вместе с первым через все операции. После взвешивания высыпают из стаканчика по возможности всю пробу, стараясь, чтобы не было потери от распыления. Не очищая кисточкой внутренних стенок стаканчика, его закрывают крышкой и снова взвешивают, поставив ту же тару на вторую чашку весов. (О методах взвешивания см. «Взвешивание», стр. 36.)
ВЫБОР МЕТОДА АНАЛИЗА
Нужно быть .осмотрительным, приступая к анализу неизвестного вещества или даже хорошо известного материала, если неизвестно его происхождение.
Такая осмотрительность необходима не только для того, чтобы получить правильные результаты при определении известных компонентов этого вещества, но и для того, чтобы не упустить каких-либо важных его составных частей;
Если какой-либо метод дает вполне удовлетворительные результаты при определении того или иного элемента в растворе чистой его соли или даже в некоторых смесях, это вовсе не является гарантией, что правильные результаты будут получены этим методом и в других условиях. Нет возможности испытать и дать указания о применимости метода в каждой из тысяч возможных комбинаций, которые могут встретиться аналитику. И совершенно невозможно разработать такой метод, который был бы применим во всех без исключения случаях. Трудно даже быть уверенным, что он всегда будет давать вполне удовлетворительные результаты при применении в ограниченной области. Например, совершенно удовлетворительный метод определения углерода в простой углеродистой стали, содержащей 0,1 % углерода и 0,04% серы, прямым ее сжиганием может дать слишком высокие результаты, если его применить без изменения к стали, содержащей то же количество углерода, но 0,1 % серы.
Примеры несостоятельности хорошо известных методов при некоторых условиях весьма многочисленны. Метод определения кремневой кислоты выпариванием досуха солянокислого раствора и обезвоживанием сухого остатка дает хорошие результаты в обычном случае, но его нельзя применять в присутствии таких элементов’, как бор, фтор, сурьма или висмут. Осаждением смесью едкого йатра и карбоната натрия можно очень хорошо отделить алюминий от железа и кальция, но не от железа
Выбор метода анализа
81
и магния. Оксалатный метод, который применяется обычно для определения кальция в присутствии магния, неприменим, если кальция очень мало, а магния много. Фосфатный метод определения магния не дает хороших результатов в присутствии большого количества оксалатов. Висмутатный метод определения марганца не оставляет желать лучшего, если раствор не содержит кобальта или хрома. Определение свинца в виде его сульфата дает вполне удовлетворительные результаты, если это определение не, пытаются проводить, когда присутствуют барий, кальций, серебро или сурьма.
Таким образом, прежде чем выбрать тот или иной метод анализа, аналитик должен рассмотреть Все известные его ограничения и решить, можно ли его применить к данному материалу. Во всех случаях надо иметь уверенность, что анализируемый материал не содержит веществ, мешающих определению, в отношении которых не приняты меры предосторожности. Если присутствуют вещества, влияние которых находится под сомнением, то нужно сначала удостовериться в их поведении, проведя анализ приготовленной в лаборатории смеси, близкой по составу к анализируемому материалу и содержащей известное количество определяемой составной части. Ничего нельзя принимать на веру.
Всегда полезно составить схему анализа материала, качественный состав которого известен; изображенная в виде диаграммы схема помогает сразу обнаружить ошибки и слабые места, которые иначе могли бы остаться незамеченными.
Если, например, анализируется минерал турмалин и аналитик намерен выполнить анализ по обычной схеме, то предварительное изображение ее в виде диаграммы будет иметь следующий вид:
Турмалин
Сплавление с Na2CO3
I
Выпаривание с НС1
SiO2 NH4OH
I I___________________________________I
«R2O3», осадок от аммиака ’ (NH4)2C2O4
I
CaO (СаС2О4) (NH4)2PO4
_______________________I
MgO (Mg]\fH4PO4) Раствор
отбрасывается
Если теперь рассмотреть, какие составные части могут содержаться в турмалине (Si, Al, Fe, Мп, Са, Mg, К, Na, Li, F и В) и расставить их по местам в предположенной схеме анализа, то ясно видно, что схема эта будет^непригбдной в присутствии фтора, бора и марганца, потому что: 1) при выпаривании с соляной кислотой будет происходить улетучивание кремния; 2) бор будет частично захвачен остатком кремнекислоты и улетучится вместе с последней при выпаривании с плавиковой кислотой; 3) некоторая часть имевшегося вначале фтора вероятно останется в растворе и будет препятствовать полному осаждению алюминия аммиаком,
6 Заказ 522.
82
Гл. IV. Общие методы и операции химического анализа
а также вызовет присоединение к этому осадку некоторого количества кальция в виде фторида; 4) некоторая часть бора будет захвачена осадком от аммиака; 5) некоторая часть марганца выпадет при осаждении оксалатом аммония вместе с неосажденными фтором и алюминием и 6) большая часть марганца выпадет при осаждении фосфатом. Если начертить также диаграмму для определения в турмалине щелочных металлов методом Лоуренса Смита, то придется подумать, что произойдет в ходе анализа с необычными элементами — фтором, бором и литием, и не следует ли в обычный метод анализа ввести какие-либо изменения.
Данные, полученные при исследовании простых соединений, не могут быть с уверенностью применены к анализу смесей. Так, например, титан, ниобий и тантал могут быть количественно осаждены гидролизом их солей из слабокислых растворов, но если присутствует цирконий, осаждение будет неполное, а может и вовсе не произойти. Точно так же, галлий совсем не осаждается сероводородом в слабокислом растворе, но в присутствии цинка, серебра, меди и мышьяка его осаждение может быть полным. Наконец, растворимость хлороплатината калия в 80%-ном спирте хорошо известна, но она совершенно отличается от его растворимости в 80 %-ном спиртовом растворе, содержащем хлороплатинат натрия и" свободную платинохлористоводородную кислоту, как это обычно имеет место в ходе анализа.
Много затруднений вызывают вещества, которые препятствуют осаждению или замедляют его, если они не удалены предварительно или не обезврежены, как, например, ванадий (V) при осаждении фосфора в виде фосфоромолибдата аммония и фтор или органические вещества при осаждении алюминия аммиаком. Равным образом должно быть исключено присутствие веществ, вызывающих нежелательные реакции, как, например, нитратов или аммонийных солей при осаждении серы в виде сульфата бария, оксалата аммония при осаждении тория в виде оксалата, карбонатов при осаждении урана аммиаком, мышьяка при титровании сурьмы перманганатом калия и хлоридов при титровании ртути роданидом калия.
Наконец, наибольшим источником затруднений в прикладном анализе является то, что многие из применяемых реакций не приводят к точным разделениям. Так, кремнекислота не может быть полностью отделена от бора выпариванием кислых растворов досуха и обезвоживанием; цинк при осаждении сероводородом осаждается частично вместе с медью и кобальт — вместе с оловом (IV); осадок гидроокиси алюминия удерживает медь и цинк даже после повторного переосаждения аммиаком; кальций осаждается оксалатом аммония не полностью; магний склонен увлекать с собой щелочные^металлы при его осаждении в виде фосфата.
ПРИГОТОВЛЕНИЕ РАСТВОРА ДЛЯ АНАЛИЗА
Приготовление раствора чистого соединения, предшествующее определению его весовым или объемным методом, является сравнительно простым делом. Совсем иные условия наблюдаются в случае анализа веществ, с которыми обычно приходится сталкиваться. Здесь успех анализа в значительной степени зависит от той.тщательности, с какой был подготовлен раствор к определению, а подготовка эта не может быть выполнена путем слепого следования определенным предписаниям. Каждый случай должен быть рассмотрен особо. Метод приготовления раствора
Осаждение посредством сулъфид-ионов
83
для определения фосфора в простой углеродистой стали совершенно отличен от метода, применяемого в случае анализа быстрорежущей инструментальной стали; метод, применяемый при приготовлении раствора для электролитического определения меди в латуни, совершенно не похож на метод, применяемый при определении меди в молибденовых рудах.
Одним из больших затруднений при приготовлении растворов для анализа является то, что некоторые компоненты анализируемой пробы могут улетучиться во время предварительного прокаливания, сплавления или выпаривания. Например, мышьяк может быть потерян при прокаливании пробы, если он сопровождается органическими веществами; фтор — при сплавлении с карбонатами материалов, содержащих сульфиды; сурьма — при выпаривании растворов, содержащих соляную кислоту; бор и мышьяк (III) — при обработке пробы плавиковой кислотой \
ОСАЖДЕНИЕ ПОСРЕДСТВОМ СУЛЪФИД-ИОНОВ
Осаждения добавлением сульфид-ионов имеют очень важное значение в количественном анализе не только для выделения отдельных элементов, но и для отделения групп элементов друг от друга. Осаждения могут быть проведены при самых различных условиях как в отношении концентрации ионов водорода, так и в отношении других особенностей раствора, в зависимости от преследуемых целей. Например, изменяя концентрацию ионов водорода, можно мышьяк (V) отделить от свинца, свинец от цинка, цинк от никеля, никель от марганца и марганец от магния. В щелочных растворах некоторые сульфиды образуют растворимые соединения, что может быть использовано для разделения элементов внутри группы, например для отделения свинца от молибдена. Разделения внутри группы возможны также путем превращения одного или нескольких ее членов в комплексные анионы, которые не реагируют с сульфид-ионами, например отделение кадмия от меди в растворе цианида, меди или сурьмы (III) от олова (IV) в растворе фтористоводородной кислоты, и сурьмы от олова в растворе, содержащем щавелевую кислоту и оксалат.
Осаждения сероводородом могут быть приблизительно разделены на пять классов в зависимости от того, в каком растворе проводится осаждение.
Применяются: 1) сильнокислые растворы (pH <1); 2) слабокислые растворы (pH от 2 до 3); 3) очень слабокислые, почти нейтральные растворы (pH от 5 до 6); 4) щелочные' растворы (рН> 7); 5) растворы, содержащие комплексные анионы.
Сильнокислые растворы (pH <С 1)
Под осаждением сероводородом в сильнокислом растворе понимается осаждение, проводимое при любом значении pH, соответствующем концентрациям от 0,25 до 13 н. соляной кислоты. Осаждение при более низких концентрациях кислоты представляет обычный случай и является
1 См. G. Е. F. L u n d е 11, J. I. Н оИшал, Outlines of Methods of Chemical Analysis, 1938, p. 28. Теми же авторами описывается улетучивание соединений металлов при 200—230° С во время выпаривания их хлорнокислых илп серпокислых растворов [J. Research NBS, 22, 465 (1939)].
6*
84
Гл. IV. Общие методы и операции химического анализа
общепринятым способом осаждения элементов так называемых групп меди и мышьяка. К группе меди относятся: медь, серебро, ртуть, свинец, висмут, кадмий, рутений, родий, палладий и осмий. В группу мышьяка входят: мышьяк, золото, платина, олово, сурьма, иридий, германий, селен, теллур и молибден. Таллий, индий и галлий также осаждаются полностью или частично в присутствии некоторых членов сероводородной группы; точно так же ведут себя ванадий и вольфрам в отсутствие винной кислоты. Последние два элемента образуют сульфосоли и присоединяются к группе мышьяка, а первые три не дают сульфосолей и осаждаются с группой меди.
Осаждение групп меди и мышьяка может быть осуществлено количественно и представляет собой превосходный способ отделения этих элементов от многих других. Нужно отметить, что элементы других групп могут также осаждаться сероводородом в сильнокислом растворе вследствие образования смешанных сульфидов. .
Так, цинк 1 увлекается в осадок медью, кадмием, ртутью; таллий 2 3— медью, мышьяком и сурьмой, а никель и кобальт’8 * * — оловом (IV). Последнее имеет также склонность увлекать в осадок железо, если осаждение проводится из солянокислого раствора, содержащего железо в виде Fe2+-HOHOB.
Некоторые элементы сероводородной группы также образуют друг с другом смешанные сульфиды, например ртуть с кадмием, олово с медью.
Разделения внутри группы путем тщательного подбора pH раствора при осаждении сероводородом, как правило, не дают удовлетворительных результатов.
Растворимость некоторых сульфидов возрастает приблизительно в следующем порядке:
Asv, As111, MoVI, Hg11, Cu11, Sbra, Bi111, SnIV, Cdn, Pbn и Sn11
Из разделений внутри группы наиболее удовлетворительно протекает выделение мышьяка (V) осаждением сероводородом из холодного раствора, концентрированного по содержанию НС1 (стр. 305) 4 * * *. Менее
1 I. М. К olthof f, J.’C. V а в D i j k, Chem. Weekblad, 59, 1351 (1922); Chem. Zentr., 94, II, 440 (1923); F. F e i g 1, Z. anal. Chem., 65, 25 (1924—1925).
Согласно G. Luff [Z. anal. Chem., 65, 97 (1924)]', кадмий может быть отделен от цинка предварительным осаждением сероводородом в горячем растворе, сильно подкисленном НС1 или H2SO4 и содержащем 15% NH4C1 или (NH4)2SO4, с последующим постепенным разбавлением раствора во время его остывания.
2 L. Bruner, J. Zawadski, Z. anorg. Chem., 65, 136 (1910).
3 V. Auger, L. О d i n о t, C. r., 178, 710 (1914); Chem. Zentr., 95, I, 1836 (1924); F. F e i g 1. цит. выше. Адсорбция (или последующее осаждение) ионов кобальта при осаждении сульфида олова уменьшается с повышением температуры и с повышением концентрации ионов водорода, и ее можно сделать совсем ничтожной, проводя осаждение в присутствии акролеина (стр. 470).
4 См. также G. Panajotow, Z. anal. Chem., 49,118 (1910); G. Luff, Chem.
Ztg., 45, 229 (1921); Z. anal. Chem., 65, 97, 151 (1924-1925); W. D. Treadwell,
K. S. Guiterman, там же, 52, 459 (1913); J. Lang, Ber., 18, 2714 (1885); W.
Manchot, G. Grass 1, A. Schneeberger, Z. anal. Chem., 67, 177 (1925—
1926). Последние авторы, например, показали, что осаждение кадмия сероводородом
при 20° С и давлении приблизительно в 2 атм происходит полностью в растворах,
содержащих в 100 мл меньше 11 мл НС1, и что осаждение не происходит совсем в растворе, содержащем в 100 мл более 23 мл НС1. Подобным же образом висмут полностью
осаждается в присутствии 31 мл НС1 в 100 мл и совершенно не осаждается в растворе,
Осаждение посредством сулъфид-ионов
85
удовлетвориуельно происходит отделение меди от кадмия (стр. 95), при котором, однако, оба элемента осаждают вместе и сульфиды затем кипятят с разбавленной (1 : 5) серной кислотой. Разделения внутри группы при помощи фтористоводородной кислоты (стр. 89), основанные на образовании комплексных анионов, не следует смешивать с разделениями путем измерения pH раствора.
Осаждения сероводородом обычно проводят в солянокислых растворах. Некоторые элементы, например платина и молибден х, осаждаются легче в сернокислых растворах. В концентрированных азотнокислых растворах осаждение сероводородом, конечно, невозможно; более разбавленные растворы азотной кислоты, содержащие 4% ее (по объему), могут применяться даже горячими.
Как общее правило, при выделении этой группы хорошо начинать осаждение сероводородом в растворе более кислом, чем это необходимо в конце осаждения, и затем продолжать осаждение после разбавления до соответствующего объема. Например, для качественного анализа рекомендуется 2 добавлять 2,4 мл соляной кислоты или 1,5 мл серной кислоты на объем в 40 мл, нагревать до кипения в конической колбе, пропускать сероводород до насыщения им раствора и затем нагревать 10— 15 мин при 70—90° С. После этого раствор надо охладить, разбавить до 60 мл водой, снова насытить сероводородом, закрыть колбу пробкой, встряхнуть и оставить стоять 15 мин.
Значение осаждения сероводородом под давлением часто весьма переоценивают. Например, применение его, при осаждении молибдена совершенно не является необходимым, если молибден во все время осажденйя остается шестивалентным (стр. 357), и имеет сомнительную ценность, если последнее условие не соблюдается. Почти во всех случаях осаждения сероводородом желательно, чтобы все промывание осадка на фильтре или по крайней мере большая его часть проводилась водой, насыщен-, ной сероводородом и подкисленной той кислотой, какая имеется в растворе.
Слабокислые растворы (pH от 2 до 3)
Осаждение сероводородом в растворах, имеющих кислотность, соответствующую pH от 2 до 3, предполагает предварительное осаждение членов предшествующей группы и отсутствие элементов, соли которых
содержащем в 100 мл 37 мл этой кислоты. Указывается также, что ртуть может быть отделена от кадмия осаждением в растворе, содержащем в 100 мл 45 мл НС1, и промыванием осадка сульфида кислотой той же концентрации; что руть может быть ртделена от висмута или свинца осаждением в растворе, содержащем в 100 мл от 40 до 45 мл НС1, и промыванием осадка кислотой той же концентрации; что сурьма может быть отделена от кадмия осаждением в кипящем растворе, содержащем в 100 мл 18 мл НС1, разбавлением 100 мл кипящей воды, вторичной обработкой сероводородом, фильтрованием и промыванием осадка разбавленной (1 : 10) НС1. Согласно G. V о г t-шаппи А. М е t ? 1 [Z. anal. Chem., 44, 555 (1905)], полное осаждение сурьмы невозможно в,кипящих растворах, содержащих в 100 мл более 24 мл НС1'
1 Некоторые элементы можно осадить полностью, обрабатывая сероводородом концентрированные по содержанию серной кислоты растворы их сульфатов (обработка ведется 30—60 мин при 25° С), например: Ag1, Hgn, Си11, Asra, Asv, Sb111, Sbv, SeIV> TeIV, Au111 и Pdiv.
2 A. A. N о у e s, W. С. В г a y, Technot., Quart., 19, 235 (1906); А. Нойес, В. Брэй, Качественный анализ редких- элементов, ОНТИ, 1936.
86
Гл. IV. Общие методы и операции химического анализа
могут гидролизоваться или реагировать с образованием осадков в такой разбавленной кислоте. Отделение от членов следующей далее группы происходит не четко, и при выполнении точных анализов или в тех случаях, когда члены этой группы присутствуют в значительных количествах приходится делать повторные переосаждения. Наиболее успешно из элементов этой группы отделяется цинк, и это происходит потому, что другие элементы, которые могли бы вызвать затруднения вследствие своей склонности частично осаждаться в этих условиях, как таллий, индий и галлий, встречаются редко. Наиболее подходящая кислотность для осаждения элементов этой группы соответствует 0,01 н. серной кислоте; подробности осаждения см. гл. «Цинк» (стр. 481).
Почти нейтральные растворы (pH от 5 до 6)
В практике только одно отделение проводят действием сероводорода в почти нейтральном растворе — это отделение кобальта и никеля от марганца в отсутствие железа. Таллий и индий также осаждаются полностью, железо осаждается не вполне, если присутствует большое количество уксусной кислоты, а галлий в присутствии некоторых элементов осаждается частично. Конечно, этот метод следует применять после отделения предшествующей группы элементов и элементов, соли которых гидролизуются или образуют в этих условиях осадки других соединений. Подходящую кислотность получают прибавлением органических кислот вместе с их солями; обычно применяется уксусная кислота вместе с ацетатом натрия или аммония, главным образом по причине предварительного применения их при отделении железа. В этой группе труднее получить полное осаждение, чем в предыдущих группах, и потому приходится дополнительно извлекать осаждаемые элементы из фильтрата. Для этого фильтрат обычно доводят почти до нейтральной реакции и осаждают последние следы вместе с большим или меньшим количеством элементов следующей группы, например марганца, а затем проводят повторное разделение, повышая кислотность, чтобы растворить сульфид марганца и оставить никель или кобальт в осадке. Осаждение сероводородом обычно проводят в растворе, нагретом до 70—80° С и содержащем уксусную кислоту и по крайней мере 5 г ацетата натрия на 1 г осаждаемого никеля или кабальта.
* Как показали исследования \ отделение никеля и кобальта от марганца осаждением их сероводородом лучше всего проводить из растворов, содержащих пиридин и его солянокислую соль и имеющих pH около 4,5. В этих условиях осаждение происходит медленно, вследствие чего получается кристаллический, легко отделяемый фильтрованием осадок. Марганец при такой относительно высокой кислотности раствора не выделяется.
К 200 мл анализируемого нейтрального или слабокислого раствора прибавляют раствор солянокислого пиридина (5 мл соляной кислоты, пл. 1,19 г)см\ разбавляют 20—25 мл воды и нейтрализуют чистым пиридином по индикатору метиловому красному до появления оранжевой
1Э. А. Остроумов, Зав. лаб., 7, 20 (1938); Ind. Eng. Chem., Anal. Ed., 10, 693 (1938); Э. А. Остроумов, Г. С. Масленникова, Зав. лаб., 7, 267 (1938); Ind. Eng. Chem., Anal. Ed., 10, 965 (1938).
Осаждение посредством сулъфид-ионов
87
окраски). Затем раствор нагревают до кипения, прибавляют 5—10 мл 20%-ного раствора пиридина и через горячий раствор при взбалтывании пропускают 10—15 мин сероводород. Выделившиеся в кристаллической форме сульфиды никеля и кобальта отфильтровывают и промывают сероводородной водой, содержащей несколько капель пиридина. Из фильтрата, не удаляя пиридина, можно выделить марганец в виде фосфата марганца и аммония (если отсутствуют кальций и магний). Доп. ред*
Щелочные растворы (pH 7)
Сульфиды щелочных металлов осаждают из раствора многие элементы. В обычном случае осаждение сульфид-ионами в щелочных растворах следует после предварительного отделения сероводородом в сильнокислых растворах, т. е. после удаления элементов, осаждающихся в сильнокислых растворах. Осложнений, вызываемых такими элементами, как алюминий, титан, хром, уран и редкоземельные металлы, легко можно избежать добавлением тартратов, но тогда можно осаждать только марганец (в присутствии тартратов он осаждается не полностью), железо и элементы, осаждающиеся в слабокислых или почти нейтральных растворах. Иногда элементы данной группы осаждают вместе с группой меди, отделяя их все таким способом от группы мышьяка, как зто описано ниже (см. «Осаждение сульфидом аммония», стр. 90).
В противоположность общепринятому мнению, осаждение этой группы требует большей тщательности, чем осаждение предыдущих групп, как это будет видно из дальнейшего. Осаждение может быть выполнено: 1) пропусканием сероводорода в щелочной раствор; 2) пропусканием сероводорода в кислый раствор с последующим подщелачиванием раствора и 3) прибавлением сульфида, бисульфида или полисульфида щелочного металла к слабокислому или щелочному раствору. Все эти способы находят применение. Сульфиды щелочных металлов ведут себя в общем одинаково по отношению ко всем элементам, способным образовать сульфиды; исключение составляет ртуть. На элементы, не образующие сульфидов, они действуют как растворы соответствующих гидроокисей; например, алюминий и бериллий осаждаются сульфидом аммония, но растворимы в растворе сульфида натрия. Сульфид аммония, как правило, следует предпочесть сульфидам натрия и калия. Последние применяют главным образом в металлургическом анализе при отделении меди, свинца, железа и цинка от олова или алюминия. Сульфид калия употребляют редко; он применяется только тогда, когда есть к тому достаточное основание, например в присутствии значительного количества сурьмы. Сульфиды натрия и калия лучше применять вместе с соответствующими гидроокисями. То же справедливо и в отношении сульфида аммония, хотя в небольшом избытке NH4HS или (NH4)2S' можно добавлять и без аммиака. Как общее правило, применения полисульфидов следует избегать, потому что их присутствие ведет к неполному осаждению марганца, а также меди, никеля и кобальта; в то же время полисульфиды осаждают щелочноземельные металлы, так как содержат сульфаты. Чрезмерного количества аммонийных солей цужно также избегать, потому что это ведет к неполному осаждению марганца. Осаждение в холодном растворе дает вполне удовлетворительные результаты и часто лучшие, чем в горячем. Никель лучше всего осаждать в охлажденном льдом растворе, защищенном
88
Гл. IV. Общие метода и операции химического анализа
от действия воздуха. Осадок промывают холодным бесцветным раствором сульфида аммония, содержащим хлорид аммония, а в некоторых случаях тартрат аммония и сульфит аммония.
* Ввиду того что различные катионы третьей аналитической группы выделяют осадки сульфидов в наиболее плотной форме при несколько различных значениях pH, Э. А. Остроумов и Р. И. Бомштейн 1 предложили осаждать их сероводородом из горячего слабокислого раствора при добавлении 7—8 г гексаметилентетрамина (уротропина), который в слабокислой .среде разлагается:
(CH2)eN4+4НС1 + 6Н2О = 4NH4C1 + 6НСНО
Таким образом связывается как свободная кислота, первоначально присутствовавшая в растворе, так и кислота, образующаяся в результате выделения сульфидов. pH раствора медленно и постепенно возрастает, и сульфиды всех металлов выделяются в наиболее для них подходящих условиях. '
Образующийся формальдегид реагирует с •, сероводородом, выделяя белый осадок циклического соединения (CH2)3S3. Подробности осаждения см. в оригинальной статье. Доп. ред*
ОСАЖДЕНИЕ В РАСТВОРАХ, СОДЕРЖАЩИХ КОМПЛЕКСНЫЕ АНИОНЫ
Некоторые элементы предыдущих групп можно связать в комплексные анионы, в результате чего они теряют способность осаждаться суль-фид-ионами. Это их свойство позволяет проводить разделения внутри групп, как описано ниже.
Разделение посредством образования сульфо-анионов. Элементы группы мышьяка, в противоположность большинству элементов группы меди, образуют сульфо-анионы и растворяются поэтому в растворах сульфидов и полисульфидов щелочных металлов. Из группы меди только ртуть, медь и висмут ведут себя отчасти аналогично элементам группы мышьяка. Сульфид ртути практически нерастворим в растворах сульфида аммония, мало растворим в растворах полисульфида аммония и растворим в смеси растворов сульфида натрия и едкого натра или едкого кали. Сульфид меди нерастворим в растворах сульфидов щелочных металлов, свободных от полисульфидов, но несколько растворим в присутствии последних. Сульфид висмута нерастворим в растворах сульфида и полисульфида аммония и в растворах бисульфидов калия и натрия (NaHS и KHS), но заметно растворим в растворах K2S и Na2S, в смесях их с едкими щелочами и в растворах полисульфидов натрия и калия.
Различия в поведении сульфидов этих элементов позволяют проводить их разделения. Выбор между R2S и R2SX зависит главным образом от валентности присутствующих в растворе элементов; в количественном анализе предпочитают обычно работать с R2S и элементами группы мышьяка в их высшей валентности. Лучше проводить разделение, осаждая элементы группы меди в щелочном растворе, чем осаждать всю группу в кислом растворе и после обрабатывать смесь сульфидов раствором сульфида щелочного металла. В зависимости от обстоятельств, обра-
1 Э. А. Остроумов, Р. И. Бомштейн, Зав. лаб., 9, 139 (1940).
Осаждение в растворах, содержащих комплексные анион»
89
, ---------1----
батывают щелочной раствор, сероводородом или сульфидом щелочного металла иди же вливают слабокислый анализируемый раствор в раствор сульфида щелочного меТаллД, взяв последний в избытке; все эти способы находят применение.
Во всех случаях осажденные сульфиды отделяют фильтрованием и промывают разбавленным раствором, соответствующим среде, в которой было сделано осаждение. Если осадок очень объемист или анализ требует очень большой точности, полезно растворить осадок и повторить осаждение. Если из фильтрата надо выделить в осадок группу мышьяка, то фильтрат подкисляют соляной или серной кислотой, стараясь ввести столько кислоты, чтобы кислотность раствора соответствовала требуемой при осаждении этой группы сероводородом.
Разделение посредством образования фторо-анионов. Осаждение сероводородом в растворах, содержащих фтористоводородную кислоту, не пользуется тем вниманием, какого оно заслуживает, и без сомнения, это объясняется отсутствием навыков работы с такими растворами и необходимостью употребления особых сосудов вместо обычных стеклянных и фарфоровых. Тем не менее этот способ осаждения заслуживает серьезного внимания, например для отделения мышьяка, сурьмы, свинца, меди и др. от германия и олова, для отделения двухвалентного олова от четырехвалентного, а также трехвалентных мышьяка и сурьмы от соединений, содержащих их в пятивалентной форме. Руководящим указанием здесь является то, что в этих условиях не осаждаются элементы, образующие комплексные фторо-анионы, и что pH раствора должен быть приблизительно таким же, как и при обычных осаждениях этих элементов. Для отделения Sbin от SnIV было рекомендовано 1 проводить осаждение в растворе, содержащем 5 мл соляной кислоты, 5 мл 48%-ной фтористоводородной кислоты и по 0,15 г сурьмы и олова при общем объеме раствора 300 мл.
Разделение посредством образования комплексных анионов с оксалат-или тартрат-ионами. Единственное разделение, основанное на образовании комплексных анионов с оксалат-ионами, — это отделение олова (IV), остающегося в растворе, от выпадающих в осадок сульфидов мышьяка, сурьмы и др. 2 Возможно, что германий при этом осаждении ведет себя подобно олову. Разделения, основанные на образовании комплексных анионов с тартрат-ионами, распадаются на две группы в зависимости от того, в какой среде проводится осаждение, в кислом или в щелочном растворе. В первом случае винная кислота прибавляется для предупреждения частичного осаждения вольфрама и ванадия; во втором случае ею пользуются иногда для той же цели, подкисляя потом щелочной раствор, но главное, ее назначение здесь — предупредить осаждение элементов, способных выделяться в щелочной среде в виде гидроокисей.
Разделение посредством образования комплексных анионов с цианид-ионами. Разделения, основанные на образовании комплексных анионов с цианид-ионами, мало применяются в количественном анализе. Осаждение в растворе KCN применяется только для отделения меди и серебра (образующих в щелочном растворе не разлагаемые сероводородом комплексные цианиды) от других элементов группы меди.
1 L. W. М с С а у, J. Am. Chem. Soc., 31, 373 (1909).
2 F. W. С 1 а г k е, Chem. News, 21, 124 (1870).
90
Гл. IV. Общие метода и операции химического анализа
ОСАЖДЕНИЕ СУЛЬФИДОМ АММОНИЯ
После удаления сероводородной группы иногда бывает нужно предварительно осадить сульфидом аммония (обычно из раствора хлоридов) железо — одно или совместно с другими элементами. Здесь будут рассмотрены два случая: а) железо, одно или вместе с цинком, марганцем, никелем, кобальтом, ураном, индием и таллием, а иногда с металлами, осаждаемыми сульфидом аммония в виде гидроокисей, — отделяется от щелочных и щелочноземельных металлов и магния; б) железо, одно или вместе с цинком, никелем, кобальтом, медью, — отделяется не только от щелочных, щелочноземельных металлов и магния, но и от металлов, осаждаемых сульфидом аммония в виде гидроокисей.
Ход анализа, а) Перед осаждением необходимо убедиться в отсутствии свободной серы, двуокиси углерода и окислителей, например хлора, и в том, что раствор содержит достаточно хлорида аммония или соляной кислоты, чтобы предупредить осаждение магния, когда в растворе появится избыток аммиака. Анализируемый раствор помещают в колбу подходящей величины (емкостью от 150 до 250 мл), нейтрализуют аммиаком, прибавляют еще 2 мл избытка аммиака, разбавляют до 100 мл водой и охлаждают. Насыщают раствор сероводородом, прибавляют еще 2 мл аммиака, доливают свежепрокипяченной и охлажденной водой до шейки колбы, закрывают пробкой и оставляют стоять в течение нескольких часов, лучше всего на ночь. Фильтруют (не прерывая фильтрования) через бумажный фильтр и промывают осадок холодным 2%-ным раствором хлорида аммония, содержащим немного сульфида аммония. Зеленое окрашивание промывных вод указывает на недостаточное количество осадителя или электролита в промывном растворе. Во время фильтрования и промывания осадка воронку по возможности все время следует держать покрытой часовым стеклом. Наконец, дают промывной воде стечь, оставляя осадок почти сухим.
Если щелочноземельные металлы отсутствуют или их частичный переход в осадок не имеет значения, то перед окончательным разбавлением прибавляют достаточное количество сульфита аммония, чтобы предотвратить образование полисульфидов. Сульфит аммония удобно готовить насыщением раствора аммиака сернистым ангидридом; перед употреблением отбирают некоторое количество этого раствора и слегка подщелачивают по лакмусу; обычно достаточно прибавить 5 мл такого раствора. Его применение устраняет необходимость фильтрования для удаления серы, если последняя образовалась в предыдущих операциях в не слишком большом количестве, позволяет заменить сероводород заранее приготовленным раствором сульфида аммония, который иначе мог бы вызвать затруднения вследствие содержания в нем полисульфидов, и улучшает осаждение меди, кобальта и особенно никеля.
При указанных выше условиях осаждение железа, кобальта, меди и цинка будет полным; полностью осаждается также уран, если отсутствуют ванадий и карбонаты. Осаждение марганца и никеля почти полное, в растворе обычно остается не более 0,1 мг. Алюминий осаждается не полностью, но количество его, остающееся в растворе, не превышает 1 мг. Некоторое количество ванадия осаждается вместе с ураном; если ванадий значительно преобладает, он может помешать полному осаждению урана. Железо не может быть отделено от ванадия даже при повтор
Осаждение сульфидом аммония
91
ном осаждении сульфидом аммония. Осаждение ванадия может быть полным в присутствии алюминия и титана.
Если последние элементы преобладают или присутствуют щелочноземельные металлы, осаждение фосфора может быть также полным. Фосфор не выпадает, если присутствует только одно железо.
б) К 100 мл раствора прибавляют винную кислоту в количестве, превышающем по крайней мере в 3 или 4 раза содержание растворенных металлов в расчете на их окислы; подщелачивают аммиаком (если винной кислоты было прибавлено достаточное количество, раствор должен оставаться прозрачным), подкисляют снова соответствующей кислотой, разбавленной (1 : 1), и прибавляют избыток последней в 2 мл. Раствор насыщают сероводородом, чтобы восстановить железо и предупредить осаждение вместе с сульфидом железа небольшого количества титана, если он присутствует. Другим основанием для проведения операции предварительного восстановления железа является то, что сульфид железа (II) легче отделяется фильтрованием, чем сульфид железа (III).
Фильтруют, если выпал осадок сульфидов или выделилось много серы, затем сильно подщелачивают аммиаком, пропускают еще сероводород, если необходимо, и дальше поступают, как в п. «а», но прибавляют к промывочной жидкости немного тартрата аммония. При желании осаждение можно в большинстве случаев провести и в горячем растворе, после чего надо дать раствору постоять на водяной бане. Если осадок велик или нужна очень большая точность определения, надо осадок растворить и снова осадить его таким же способом, а фильтраты от обоих осаждений соединить вместе.
Этим методом нельзя достигнуть отделения осажденных сульфидов от фосфора, если в первоначальном растворе присутствовал магний или какой-нибудь щелочноземельный металл. Кроме того, этот метод не дает полного отделения марганца. Уран не осаждается вовсе. Кобальт, медь и цинк осаждаются полностью, а никель — почти полностью. Этот метод обычно применяется не для осаждения всей группы сульфидов, а для отделения железа от одного или от всех следующих ниже элементов: фосфора, алюминия, ванадия 1, хрома, титана, циркония, бериллия, ниобия и тантала. Как и в методе, описанном в п. «а», осаждение никеля и кобальта идет лучше на холоду после прибавления к аммиачному анализируемому раствору нейтрального сульфита аммония и затем сульфида аммония.
Обработка промытого осадка. После выделения осадка методом, описанным в п. «а» или «б», осадок обрабатывают для разделения и определения отдельных ее составных частей. В зависимости от состава осадка применяется тот или иной метод обработки. При этом важно, чтобы при растворении осадка фильтровальная бумага по возможности не подвергалась действию горячей кислоты, потому что из нее
1,В противоположность осаждению в растворах/содержащих один сульфид аммония (п. «а»), осаждение в присутствии тартрата дает хорошее отделение железа от ванадия. Например, никаких следов ванадия не было обнаружено в осадке сульфида железа после двухкратного осаждения его из смеси, первоначально содержавшей 0,1 г железа и 0,05 г ванадия. Добавление тартрата служит также для отделения урана и, вероятно, помогает отделению других элементов, например вольфрама и сурьмы.
92
Гл. IV. Общие методы и операции химического анализа
может быть извлечено органическое вещество, которое будет препятствовать полному осаждению аммиаком и некоторыми другими осадителями. Особенно это относился к осаждению гидроокиси алюминия (стр. 124).
Обработка фильтрата. Фильтрат, полученный после осаждения по п. «а», может быть сразу применен для определения кальция и магния. Фильтрат, полученный после обработки по п. «б», может содержать некоторые металлы, которые должны быть предварительно выделены. Для этого нужно сначала разрушить тартраты. Раствор выпаривают в большой платиновой чашке с 10—12 мл серной кислоты и осторожно нагревают до тех пор, пока не начнется ясное обугливание. Слегка охлаждают, покрывают часовым стеклом и осторожно приливают 5 мл азотной кислоты (лучше дымящей); когда бурная реакция прекратится, постепенно нагревают до тех пор, пока органические вещества полностью не окислятся; обработку азотной кислотой, если нужно, повторяют х. Чашку охлаждают, растворяют остаток в воде и прибавляют раствор аммиака, чтобы осадить алюминий, титан, цирконий, бериллий, ниобий, тантал и уран, а также фосфор и ванадий, если количество этих двух элементов не превышает того, которое может соединиться с основаниями в виде фосфатов и ванадатов. В присутствии алюминия избытка аммиака надо избегать. Если фосфор и ванадий присутствуют в количестве большем, чем то, какое может быть, связано алюминием, титаном и др., то в осадке можно ожидать присутствия щелочноземельных металлов. После растворения осадка 1 2 в горячей разбавленной (1 : 1) соляной кислоте дальнейшее разделение идет обычным путем.
Фильтрат, содержащий тартрат аммония, можно также выпаривать досуха в большой платиновой чашке без прибавления серной кислоты. В этом случае остаток обугливают и сплавляют с карбонатом натрия и небольшим количеством селитры. Плав выщелачивают водой и раствор фильтруют. Титан и цирконий остаются на фильтре, а хром и ванадий вместе с алюминием и фосфором переходят в фильтрат в виде хромата и ванадата. О дальнейшем отделении последних двух элементов от хрома и ванадия при анализе горных пород см. стр. 978.
РАЗДЕЛЕНИЕ ЭЛЕМЕНТОВ В ОСАДКЕ ОТ СЕРОВОДОРОДА, ПОЛУЧЕННОМ В СИЛЬНОКИСЛОМ РАСТВОРЕ
Разделение элементов сероводородной группы в осадке при анализе горных пород проводится редко, потому что в пробе массой 1 г, взятой для анализа, редко присутствуют определимые количества каких-либо членов этой группы. Но при анализе минералов или руд и металлургических продуктов дело обстоит иначе — здесь можно ожидать наиболее сложных смесей. Мы приводим дальше описание разделений тех смесей, которые могут встретиться в анализе, когда не имеется в виду выделение только одной какой-либо составной части. Термин «осадок элементов серо
1 Винную кислоту можно разрушить нагреванием приблизительно при 60° С с концентрированной серной кислотой, прибавлением небольших порций персульфата калия и окончательным нагреванием до появления паров серной кислоты. Полного разложения 1 г винной кислоты в 10 мл серной кислоты можно легко достичь добавлением 10 г персульфата. Персульфат калия вызывает меньшее вспенивание жидкости, чем персульфат аммония.
2 Иногда бывает трудно удалить весь осадок из пор фильтра. В сомнительных случаях бумагу фильтра лучше сжечь и полученный остаток исследовать.
Разделение элементов в осадке от, сероводорода
93
водородной группы» означает осадок, полученный в результате обработки сероводородом растворов, имеющих pH <1 (стр. 83).
Золото и металлы платиновой группы здесь це рассматриваются; их присутствие не является обычным, и методы их разделения приведены в гл. «Платиновые металлы» (стр. 395).
Разделение групп меди и мышьяка
Разделение обработкой сульфидом щелочного металла. Отделить элементы сероводородной группы, не образующие растворимых сульфоанионов, можно либо обработкой всей осажденной группы раствором сульфида щелочного металла, либо осаждением сульфид-ионами в щелочном растворе. Последнее значительно лучше, потому что полное растворение многих осажденных сульфидов (например, сульфидов селена, теллура и молибдена) обработкой раствором сульфида щелочного металла происходит с трудом и часто даже невозможно. Способ, каким проводят осаждение сульфид-ионами в щелочном растворе, зависит от растворимости осадка в таком растворе. Если практически все растворяется, как, например, составные части нечистого молибдена в аммиаке или продажного олова в растворе едкого натра, то осаждение лучше всего проводить, обрабатывая щелочной анализируемый раствор сероводородом или сульфидом щелочного металла. Если же большая часть сульфидов не растворяется, как, например, компоненты бронзы при обработке едким натром, то тогда лучше прилить слабокислый анализируемый раствор к раствору сульфида щелочного металла, взятому в избытке. Употребления растворов полисульфидов следует избегать, кроме тех случаев, когда нет лучшего способа разделения (например, для выделения сульфида ртути приходится пользоваться нолисульфидом аммония). Применение полисульфидов не является необходимым, если элементы, образующие сульфосоли, находятся в состоянии их высшей валентности.
Выбор сульфида щелочного металла зависит от того, какое надо провести разделение. Например, нужно взять сульфид аммония или бисульфид щелочного металла (NaHS или KHS), если висмут должен остаться вместе с группой меди; сульфид натрия или сульфид калия вместе с соответствующей едкой щелочью, когда нужно, чтобы ртуть осталась с группой мышьяка. Сульфид натрия следует также предпочесть, когда в осадке должен остаться сульфид меди, тогда как сульфид калия более желателен для отделения сурьмы. (Описание метода см. «Осаждение посредством образования сульфо-анионов», стр. 88.)
Разделение электролизом в растворе, содержащем смесь азотной и фтористоводородной кислот. По имеющимся в литературе данным х, серебро, ртуть, медь и свинец можно отделить от олова, сурьмы, молибдена и вольфрама в их высшей валентности, проводя электролиз раствора в смеси азотной и фтористоводородной кислот. Осадки ртути и меди, полученные таким способом, содержат немного платины, а двуокись свинца содержит фтор. Двуокись свинца можно очистить погружением электродов в азотную кислоту, проведением электролиза с обратным направлением тока, пока не растворится РЬО2, и повторным электролизом с первоначальным направлением тока или же переведением РЬО2 в сульфат.
1 L. W.McCay, J. Am. Chem. Soc., 36, 2375 (1914); см. также L. W- M с С а у. N. Н. Furman, там же, 38, 640 (1916). >
94
Гл. IV. Общие методы и операции химического анализа
Разделения в группе меди
Отделение ртути.. Хороший метод отделения ртути от серебра, висмута и свинца, основанный на нерастворимости сульфида ртути в кипящей разбавленной азотной кислоте (пл. 1,2—1,3 г/см3), был предложен очень давно х. Если присутствует свинец, часть его может превратиться в нерастворимый сульфат свинца, который затем надо удалять из остатка сульфида ртути. Медь, олово и кадмий образуют с ртутью смешанные сульфиды, что препятствует их полному отделению.
Отделение ртути обработкой осадка сульфидов смесью сульфида натрия и едкого натра не дает вполне удовлетворительных результатов в присутствии кадмия, меди или висмута. Образования смешанных сульфидов можно, вероятно, избежать, приливая медленно при перемешивании слабокислый анализируемый раствор к взятому в избытке раствору смеси сульфида щелочного металла и едкой щелочи. Висмут отделяется не полностью, потому что сульфид его заметно растворим в указанной смеси сульфида щелочного металла и едкой щелочи.
Отделение серебра. Отделение серебра от свинца, висмута, меди и кадмия осаждением его в виде хлорида в азотнокислом растворе, как описано в гл. «Серебро» (стр. 236), проходит удовлетворительно. Если присутствует свинец, осадок хлорида серебра надо переосадить, а если имеется висмут, то в раствор надо ввести достаточное количество азотной кислоты, чтобы предупредить осаждение висмута в результате гидролиза.
-Отделение висмута. Отделение висмута должно предшествовать отделению меди электролизом и желательно также — выделению свинца в виде сульфата, потому что висмут частично осаждается электролизом вместе с медью и при некоторых условиях может загрязнить осадок сульфата свинца. Осаждение висмута в виде оксибромида и оксихлорида висмута является лучшим методом отделения небольших количеств висмута от свинца и кадмия. Большие количества висмута могут быть отделены в виде оксибромида или в виде основного нитрата. Каждый из этих методов может также служить для отделения висмута от меди, но для этой цели обычно применяется осаждение карбонатом аммония. (Описание этих методов-см. в гл. «Висмут», стр. 269.)
Отделение свинца. Свинец может быть отделен от меди, кадмия и ртути осаждением его в виде сульфата. Отделение от висмута этим способом считается не вполне надежным, но мы при использовании этого метода получили хорошие результаты. Неудачные разделения объясняются или гидролизом соли висмута, вызванным излишним разбавлением раствора, или же уменьшением растворимости этой соли вследствие применения спирта. При осаждении сульфата свинца в присутствии висмута спирт прибавлять не следует. Сульфат серебра настолько плохо растворим, что он частично остается со свинцом, поэтому серебро надо предварительно отделить; это же можно сказать и о сурьме.
Применение электролиза в азотнокислом растворе для разделения металлов группы меди на практике ограничивается отделением свинца от меди, хотя он также с успехом может быть применен и для отделения свинца от кадмия и ртути. Отделение свинца электролизом идет неудовлетворительно в присутствии серебра или висмута, которые загрязняют
1 G. von Rath, Pogg. Ann., 96, 322 (1855),
Разделение элементов в осадке от сероводорода
95
осадок двуокиси свинца на аноде. Электролизом не следует выделять более 0,1—0,2 г свинца, потому что большие количества осажденной двуокиси свинца не держатся на аноде. (Описание метода см. стр. 264.)
Отделение меди. Электролитический метод редко применим непосредственно для отделения меди, потому что практически все другие элементы сероводородной группы в небольшом количестве осаждаются вместе с медью на катоде; кадмий и свинец составляют исключение. Поэтому электролитический метод анализа должен применяться после предварительного отделения группы мышьяка, а из группы меди — ртути, серебра и висмута.
При осаждении меди в виде роданида меди после восстановления сернистой кислотой в разбавленном сернокислом или солянокислом растворе получается удовлетворительное отделение меди от висмута и кадмия (из группы меди) и от сурьмы, олова и мышьяка (из группы мышьяка). Если присутствуют элементы, соли которых легко гидролизуются, например висмут, сурьма и олово, полезно прибавить винную кислоту. Метод этот часто применяется, когда нужно определить одну медь. Если другие элементы также должны быть определены, то лучше последовательно отделять все мешающие элементы, как описано выше (см. «Отделение ртути, серебра, висмута»).
Менее надежны способы отделения меди от кадмия, основанные на разной растворимости их сульфидов в разбавленной серной кислоте и на осаждении меди тиосульфатом натрия. (Описание этих методов см. в гл. «Медь», стр. 282).
Разделения в группе мышьяка
Способ, который применяется для растворения сульфидов группы мышьяка, имеет весьма существенное значение, если присутствуют германий, мышьяк (III), олово (IV), сурьма (III) или селен, потому что при выпаривании солянокислых растворов могут произойти значительные потери этих элементов. В сомнительных случаях сульфиды лучше растворить в горячем разбавленном растворе едкого натра с добавлением хлора, перекиси водорода или перкарбоната калия, затем раствор охладить и подкислить кислотой, требующейся при предполагаемых отделениях. Если после предшествующих разделений ртуть осталась вместе с группой мышьяка, ее обычно выделяют до подкисления или окисления щелочного раствора, например обработкой нитратом аммония, как описано в гл. «Ртуть» (стр. 245).
Отделение германия. Отделять германий, мышьяк, сурьму и олово от других элементов сероводородной группы и один от другого удобно перегонкой. Германий может быть сначала отделен от мышьяка (V) перегонкой из солянокислого раствора, а мышьяк затем отделяют от других членов группы повторной перегонкой после восстановления его до трехвалентного. При этом нужно обращать внимание на температуру, при которой происходит дистилляция. Главная трудность заключается в том, чтобы предупредить одновременное улетучивание олова, сурьмы и отчасти селена. Очевидно, что необходима большая осторожность для предупреждения потери этих элементов во время таких предварительных операций, как кипячение солянокислых растворов германия, селена и трехвалентного мышьяка. (Описание метода см. в гл. «Германий», стр. 345.)
96
Гл. IV. Общие методы и операции химического анализа
Отделение мышьяка, сурьмы и олова. Очень удобным методом отделения этих.трех элементов является перегонка в приборе, показанном на рис. 9, в котором все части сделаны из стекла (без соединений посредством резиновых трубок и пробок). Емкость колбы 1 — 200 мл\ емкость воронки 2 — 65 мл. Расстояние от дна колбы до отводной трубки приблизительно 17 см.
Рис. 9. Прибор для перегонки:
1 — колба; 2 — воронка.
Если относительные количества мышьяка, сурьмы и олова сильно различаются, то однократной перегонкой нельзя достичь полного разделения. Когда требуется очень большая точность, всегда приходится проводить вторичную перегонку. Поэтому практически часто бывает неудобным определять все три элемента из одной навески. Так, например, при анализе сплава на оловянной основе, содержащего очень мало мышьяка (0,05%), значительное количество сурьмы (8%) и очень много олова (88%), для каждого определения надо брать отдельную навеску.
Перегонку проводят следующим образом х. Анализируемый раствор, содержащий мышьяк (III), сурьму (III) и олово (IV), занимающий объем 10 мл и содержащий серную кислоту в концентрации1 2 1:1, переносят в перегонную колбу прибора с помощью 100 мл соляной кислоты, вставляют термометр в предназначенную для него трубку прибора и подставляют под холодильник приемник, содержащий 50—100 мл дистиллированной воды, следя за тем, чтобы конец трубки холодильника был погружен в воду. Затем пропускают через прибор ток двуокиси углерода, нагревают раствор до кипения и слабо кипятят, пока объем раствора в колбе не уменьшится до 50 мл. Скорость прохождения через прибор двуокиси углерода. регулируют показывал 111—112° С. Не прекращая тока СОа
так, чтобы термометр
и нагревания, опускают вниз приемник, обмывают конец трубки хо-
1 J. A. S с h е г г е г, J. Research NBS, 16, 253 (1936); 21, 95(1938). Первая статья содержит общие указания, во второй статье описано применение этого метода для определения указанных трех элементов в сплавах на свинцовой, оловянной и медной основах.
2 Мышьяк и сурьма могут быть легко восстановлены до трехвалентных кипячением концентрированного сернокислого раствора этих элементов с серным цветом или с сульфатом гидразина. При такой обработке олово остается в четырехвалентном состоянии. Сульфат гидразина следует предпочесть сере, потому что при этой обработке он полностью разрушается, а непрореагировавшую серу надо потом отделять фильтрованием. Простое кипячение разбавленных кислых растворов с сернистой кислотой или сульфатрй гидразина не всегда эффективно. Сульфат железа (II) и хлорид меди (I) — реактивы, часто применяемые для восстановления мышьяка и вполне пригодные для этой цели здесь не рекомендуются, потому что они образуют осадки в перегонной колбе. Эти осадки вызывают толчки жидкости, когда температуру поднимают для последующей отгонки сурьмы и олова. Вообще присутствие в дистилляционной колбе нерастворимого осадка или образование непрозрачной сиропообразной жидкости, каково бы ни было их происхождение, нежелательно, так как осадок вызывает толчки в колбе и препятствует отгонке при надлежащей температуре. Из твердых веществ, которые могут образоваться в колбе при анализе цветных металлов, чаще всего встречаются сульфат свинца и фосфат четырехвалентного олова. Толчков жидкости в колбе можно в значительной степени избежать, вводя в колбу 0,1—0,2 г хлопьевидного графита.
Разделение элементов в осадке от сероводорода
97
лодильника водой и полученный дистиллят сохраняют для определения мышьяка 1 2.'//Затем подставляют второй приемник, содержащий 50— 100 мл воды, вливают в перегонную колбу 7 мл 85%-ной фосфорной кислоты и, продолжая пропускать двуокись углерода, постепенно нагревают, пока температура в колбе не достигнет 155° С. Тогда через воронку 2 вливают в колбу соляную кислоту со скоростью 30—40 капель в минуту, следя за тем, чтобы температура была в границах 155—165° С. Когда таким образом будет прибавлено 75 мл соляной кислоты, гасят горелку, заменяют приемник новым, а полученный дистиллят сохраняют для определения сурьмы а. Когда раствор в колбе 1 остынет до 140° С, снова зажигают горелку под прибором и в воронку 2 наливают смесь, состоящую из трех частей соляной кислоты и одной части бромистоводородной кислоты. Затем поворачивают кран воронки, чтобы жидкость из нее выливалась в колбу 1 со скоростью 30—40 капель в минуту (как при перегонке сурьмы), и поддерживают температуру в 140° С. После добавления 50 мл смеси кислот нагревание прекращают, удаляют прием-цик с дистиллятом, в котором определяют олово, и останавливают ток двуокиси углерода 3 * * * 7.
Описанным методом можно разделить по 10 мг каждого из этих трех элементов. Для разделения больших количеств каждую отгонку надо продолжать, добавляя в колбу еще кислоты. Количество последней определяют по имеющемуся опыту или жр ее добавляют до тех пор, пока проба дистиллята на отгоняемый элемент не станет давать отрицательный результат.
1 Перегонка мышьяка не представляет затруднений и происходит полностью, если мышьяк был восстановлен до трехвалентного и анализируемый раствор не содержит такого большого количества суспендированных веществ, чтобы это могло помешать отгонке. Результаты определения мышьяка (в присутствии сурьмы) получаются несколько повышенными, особенно если определение заканчивается объемным методом. Источниками ошибок являются: сернистый ангидрид, не полностью удаленный из раствора перед началом перегонки, и перешедшая в небольших количествах в дистиллят сурьма. Присутствие сернистого ангидрида нежелательно и тогда, когда определение заканчивается весовым методом (осаждением мышьяка в виде AsaS8), так как SOa реагирует с сероводородом, образуя серу, которую надо затем удалять. Затруднений, вызываемых сернистым ангидридом, можно избежать кипячением разбавленного анализируемого раствора после восстановления, но до прибавления соляной кислоты. Сурьма всегда переходит в дистиллят, если ее количество значительно превышает содержание мышьяка и если не применяются приспособления для фракционированной перегонки. Если пользуются обычным перегонным аппаратом, сурьму отделяют вторичной перегонкой. «
2 После удаления мышьяка добавляют обычно фосфорную кислоту для связывания олова и поднимают температуру до 160 ± 5° С для отгонки сурьмы. При этой температуре перегонка хлорида сурьмы происходит без затруднений, если фосфат олова (IV) не находится в растворе в таком большом количестве, что образуется тяжелый осадок его. В последнем случае рекомендуется отгонять сурьму без добавления фосфорной кислоты; при этом вместе с сурьмой отгоняется значительное количество олова. Для определения олова берут тогда другую, меньшую по величине навеску. Если анализируемая проба содержит большое количество сурьмы, вследствие чего время перегонки значительно удлиняется, то в дистиллят вместе с сурьмой, перейдет и некоторое количество олова, даже если была прибавлена фосфорная кислота. В этом случае дистиллят, содержащий сурьму, перегоняют вторично и остаток в колбе присоединяют к первому остатку для определения олова.
3 Олово можно также отгонять, добавляя по каплям разбавленную (1 : 2) бро-
мистов од сродную кислоту, содержащую немного соляной кислоты. Отгонку ведут при
180° С; отгонка тогда происходит очень быстро и бромистоводородной кислоты требуется
минимальное количество.
7 Заказ 522.
98 Гл. IV. Общие методы и операции химического анализа
Этому разделению мешают некоторые элементы группы мышьяка: германий количественно перегоняется вместе с мышьяком; модибден, рений-, селен и, возможно, теллур частично переходят в дистиллят с сурьмой, частично, — с оловом. Если присутствует ртуть, она перегоняется с сурьмой и оловом и, возможно, также с мышьяком х.
Отделение селена и теллура. Избежать улетучивания селена вместе с трехвалентным мышьяком при перегонке последнего очень трудно, а потому селен, так же как и теллур, лучше предварительно отделять. Это может быть сделано после отгонки германия следующим образом. Раствор кипятят с обратным холодильником до полного удаления хлора.и восстановления селена и теллура до четырехвалентного состояния. Затем раствор разбавляют так, чтобы он содержал 30—40% (по объему) содя-ной кислоты, и. осаждают селен и теллур, как описано в гл. «Теллур» (стр. 392), прибавляя сернистой кислоты и солянокислого гидразина и. нагревая с обратным холодильником, пока не закончится восстановление. Затем раствор охлаждают и осадок отфильтровывают.
Отделение мышьяка. Перегонка трехвалентного мышьяка из солянокислого раствора является наиболее удовлетворительным методом отделения мышьяка от других членов этой группы, за исключением германия и селена. Последние встречаются редко и отделяются, как указано в разделах «Отделение германия», стр. 95 и «Отделение селена и теллура» (см. выше). В обычных случаях в предварительных отделениях необходимости нет, и мышьяк восстанавливают и перегоняют, как описано в гл. «Мышьяк» (стр. 303). Если в оставшемся растворе должны быть проведены другие разделения, следует выбрать такой восстанавливающий реактив, чтобы он при этих операциях не мешал.'
Мышьяк (V) можно отделить от сурьмы (V) и (III), осаждая его быстрым током сероводорода на холоду (0° С) в 10 н. солянокислом растворе 1 2, как описано в гл. «Мышьяк» (стр. 310). Хотя эта реакция обычно применяется для отделения мышьяка-от сурьмы, она должна приводить к удовлетворительным результатам и при отделении мышьяка от некоторых других элементов сероводородной группы, например от кадмия, висмута, свинца и олова. Что касается отделения мышьяка от германия, молибдена, селена, теллура и ртути, то здесь применимость этой реакции находится под сомнением.
Мышьяк (V) осаждается магнезиальной смесью в аммиачном растворе, содержащем тартраты; при этом сурьма, олово, молибден и, вероятно, также германий, селен и теллур не выпадают. Реакция эта пригодна больше для отделения мышьяка, чем для его определения. Нужно избегать большого избытка аммонийных солей, а также чрезмерного промывания осадка; лучше даже совсем не промывать осадок, а очищать его переосаждением. (Дальнейшие подробности см. в гл. «Мышьяк», стр. 306).
Отделение олова. Осаждение сероводором в щавелевокислом растворе 3 дает удовлетворительное отделение сурьмы (III) и мышьяка (III)
1 О выполнении в том же приборе отгонок со смесями кислот: НС1—HC1O4S НВг—НС1О4, НС1—1Г3РО4—НС1О4 и НВг—Н3РО4—НС1О4 Для отделения хрома, ртути, осмия и рутения см. в статье J, I. Н o f f m a n, G. E. F. L u n d e 11, J. Research ‘NBS,’ 22, 465 (1939).
. 2 F. Neber, Z. anal. Chem., 32, 45 (1893).
3 F. W. C 1 a r k e, Chem. News, 21, 124 (1870).
Разделение элементов в осадке от сероводорода 99
от олова (IV). Поведение при этом других членов группы нам неизвестно, но можно думать, что молибден, теллур и селен также перейдут в осадок, а германий — нет. '
Отделение может быть выполнено следующим образом. Приготовляют .раствор, содержащий не более 0,3 г металлов в виде их сульфосолей, , переводят в стакан емкостью 500 лм, накрывают часовым стеклом и прибавляют понемному перекись натрия до обесцвечивания раствора. Раствор кипятят 15 мин и прибавляют к нему твердую щавелевую кислоту до кислой реакции на лакмус и сверх того избыток в 10 г. Если появится, осадок, то раствор кипятят, пока он не растворится, прибавляя, если нужно, еще щавелевой кислоты. Разбавляют до 100 мл горячей водой, нагревают до кипения и пропускают в кипящий раствор быструю струю сероводорода в течение 15 мин, считая с момента появления осадка, окрашенного в оранжевый цвет. Через 15 мин кипячение прекращают, но продолжают пропускать газ еще 15 мин. Раствор с выпавшим осадком оставляют стоять приблизительно при 50° G в течение 30 мин, после’ чего его фильтруют теплым и промывают осадок сульфида сурьмы горячим 1 %-ным раствором щавелевой кислоты, насыщенным сероводородом, фильтрат сохраняют. Чтобы извлечь олово, увлеченное осадком сульфида сурьмы, смывают осадок обратно в колбу, растворяют остаток на фильтре в разбавленном растворе едкого калц, содержащем немного перекиси водорода, и повторяют обработку сероводородом в щавелевокислом растворе. К соединенным фильтратам от обоих осаждений прибавляют столько серной кислоты, сколько нужно, чтобы связать все основания и чтобы остался избыток ее в 5—10 мл", выпаривают До выделения па^ов серной кислоты, охлаждают, прибавляют воды и затем перманганата калия до неисчезающей розовой окраски. Наконец, разбавляют раствор так, чтобы он содержал приблизительно 1 мл серной кислоты в 100 мл, и осаждают сероводородом, как описано в гл. «Олово», стр. 342.
Более быстрый, но несколько менее надежный метод осаждения олова в соединенных щавелевокислых фильтратах следующий; раствор слабо подщелачивают амйиаком, затем прибавляют сульфид аммония, пока образовавшийся сначала осадок снова не растворится, потом добавляют уксусной кислоты в небольшом избытке, чтобы разложить сульфосоль, и, наконец, оставляют раствбр стоять при 30—40° С, цока осадок не осядет.
Описаны 1 методы отделения олова, основанные на образовании комплексных его анионов с фторид-ионами. На том же принципе основан метод отделения мышьяка от германия 2. Эти методы разделения дают удовлетворительные результаты, и главным препятствием к их широкому распространению являются неприятные свойства фтористоводородной кислоты и необходимость иметь сосуды, устойчивые к ее действию. Олово можно- также полностью отогнать пропусканием сухого хлористого водорода через раствор соли олова (IV) в серной кислоте, нагретый приблизительно до 200° С.
Отделение сурьмы. Если сурьму предполагают определить титрованием перманганатом, как описано в гл. «Сурьма» (стр. 324), а олово — титрованием иодом, как описано в гл. «Олово» (стр. 338), то нет необходимости отделять эти элементы один от другого. Мышьяк, однако, должен
1 L. W. М с С а у, J. Am, Chem. Soc., 31, 373 (1909),
2 J. Н. М ii 11 е г, J. Am, Chem. Soc., 43г 2549 (1921),
7*
100
Гл. IV. Общие методы и операции химического анализа
быть удален, лучше всего перегонкой, как описано в гл. «Мышьяк» (стр. 303). Сурьму можно также полностью удалить в виде летучего хлорида пропусканием сухого или влажного хлористого водорода через раствор соли сурьмы (III) в серной кислоте, нагретый приблизительно до 200° С. Если хотят выделить сурьму для ее идентификации, то сначала удаляют германий и мышьяк перегонкой из солянокислого раствора, а потом теллур и селен — восстановлением сернистым ангидридом в разбавленном солянокислом растворе, содержащем винную кислоту. Сурьма может быть-затем отделена от олова осаждением сероводородом в растворе, содержащем щавелевую рли фтористоводородную кислоты, а от молибдена — восстановлением свинцом в кислом растворе, как описано в гл. «Молибден» (стр. 360).
Отделение молибдена. Наилучшим методом отделения малых количеств других элементов группы мышьяка от молибдена, по-видимому, является введение в раствор достаточного количества соли железа и осаждение этих элементов вместе с железом добавлением аммиака, как описано в гл. «Молибден», стр. 359. Метод этот оказался весьма удовлетворительным для отделения молибдена от мышьяка и сурьмы, и нет оснований предполагать, что отделение олова, германия, селена и теллура не будет проходить так же хорошо. Для отделения от молибдена больших количеств этих элементов могут служить следующие методы: перегонка с соляной кислотой — для удаления мышьяка и германия; восстановление сернистым ангидридом — для удаления теллура и селена; восстановление свинцом — для удаления сурьмы и осаждение сероводородом в присутствии щавелевой или фтористоводородной кислоты — для отделения олова.
ОСАЖДЕНИЕ ПОСРЕДСТВОМ ИЗМЕНЕНИЯ КОНЦЕНТРАЦИИ ИОНОВ ВОДОРОДА В РАСТВОРЕ
Осаждение посредством изменения концентрации ионов водорода в растворе является одной из самых обычных операций, принятых в аналитической химии. Осаждение гидроокиси того или иного металла происходит при достижении определенной концентрации ионов водорода. На рис. 10 представлен ряд металлов в порядке тех концентраций ионов водорода, при которых осаждаются соответствующие гидроокиси из разбавленных растворов солей этих металлов 1>а. Хотя нельзя сказать с уверенностью, что этот порядок отражает действительную величину силы разных гидроокисей металлов как оснований, но на нем построены многие реакции, применяемые в анализе.
Приведенное на рис, 10 расположение металлов показывает, например, что чистое отделение марганца от алюминия и железа (III) может быть достигнуто, если тщательно установить требуемую концентрацию иойов водорода в растворе, и что медь и цинк будут загрязнять осадок в большей степени, чем никель и кобальт. Все это было подтверждено экспериментально одним из нас 3. Другие интересные подтверждения мы . J И. Т. Sr В г i 11 о n, J. Chem. Soc., 1272, 2157 (1925).
а В статье В. G i 1 с h г i s t [J. Research NBS, 30, 89 (1943)] приводятся результаты испытаний на полноту осаждения 40 химических элементов в условиях pH от 1 до 10 и обсуждаются возможности аналитических разделений путем контролируемого гидролитического осаждения.
* G. Е. F. L u n d е 11, Н. В. К п о w 1 е s, J. Am, Chem. Soc., 45, 676 (1923),
Осаждение посредством изменения, концентрации ионов водорода
101
находим: 1) в наблюдениях Джилеса \ который показал, что суспензия чистого карбоната свинца осаждает торий, цирконий, церий (IV) и
Z7/H.HCI__=-1
Z7./H H2SO4--
-Os
#<7/h.HCI----2 n
/Н. CH3 COOH---—Fe"1
Zr
IV
т.сн^оонада/1| НС . 3^ee,g.
<7,Z7h.CH3COOH—= -------------------Th
^н.СНзСООН W/H.HC1
7нП _ —5 \гк(при0лиз.')
Рис. 10. Концентрации СиО — РЬСО3 — CdCO3 — 0,12М NaHCO3, насыщ. СО2 ВаСО., ,0,25 М NaHCO3,A77£W. СО2_ J HgO СаСОз 0,1 И NaHCO3 ^Z7Z7h.NH4OH— MgO — ^/h.NH4OH — Cr1'1 —Fe" — Be -6-Pb—Ru”1 p^ili или iVp^j H1 uNi—Cd,„ 7 n C°xrVt Sm — 7—Pr—Nd —rCe— Hg" -8 —Mn La —9—Ag- -Ю' — Mg
ионов водорода, при которых осаждаются гидроокиси металлов. W/n.NaOH— <7,/h.NH4OH — /h.NH4OH WNaOH —= CaO — -11 -12 — Ca
4/H.NaOH —-- -13
/H.NaOH—=-И
железо (III) полностью; уран, хром (III) и алюминий — не вполне; церий (III), лантан, неодим, празеодим, иттрий, самарий и иттриевую группу (поскольку она была исследована) не осаждает совсем; 2) в одной из
1 W. В. Giles, Chem. News, 92, 1, 30 (1905),
102 1'л- IV- Общие методы и операции химического анализа
старых работ х, где было показано, что в холодных растворах хлоридов окись ртути полностью осаждает гидроокиси железа (III), алюминия и хрома, частично — гидроокиси цинка, кобальта, никеля, бериллия, церия (III) и лантана и не осаждает гидроокиси марганца (II).
Большинство приведенных на рис. 10 данных взято из книги Бриттона 1 2 3. Значения pH осаждения гидроокисей платиновых металлов взяты из работы R.’Gilchrist (Бюро стандартов США). Значение pH осаждения платины на графике не приведено, но известно, что платина (IV) начинает осаждаться при pH более высоком, чем pH осаждения других металлов платиновой группы. ,
Разделения можно было бы проводить гораздо лучше, если бы удавалось доводить рас'гвор до желаемой концентрации ионов водорода, избегая местных снижений этой концентрации в отдельных частях раствора. Этого никогда не удается избежать при Добавлении щелочных реактивов, например раствора аммиака, хотя довольно удовлетворительные результаты можно получить, если разбавленный реактив прибавлять очень осторожно и если имеется значительная разница между концентрациями ионов водорода, при которых осаждаются разделяемые металлы. Лучшие результаты получаются, если к анализируемому раствору, содержащему минеральную кислоту, прибавлять такую соль, как ацетат натрия. Но даже и в этом случае желаемая концентрация ионов водорода местами может быть на мгновение перейдена.
Более точных разделений можно ожидать при использовании в качестве осаждающих реактивов некоторых чистых окислов и карбонатов, хотя надо учитывать затруднения в получении этих веществ в чистом виде, свободными от,примесей. Наилучшие условия создаются, когда осаждение происходит одновременно во всем растворе, что имеет, например, место при осаждении гидроокиси алюминия мочевиной х.
Наиболее часто эти осаждения проводятся добавлением раствора аммиака. Ход работы при использовании этого реактива, а также и некоторых других описан ниже.
Осаждение аммиаком
Осаждение аммиаком — одна из самых обычных операций, применяемых в анализе. Она проводится либо для определения осажденного соединения весовым путем, либо для совместного отделения двух или нескольких металлов от других металлов. Если эта операция выполняется для количественного весового определения, то ей должно предшествовать выделение кремнекислоты и отделение элементов группы сероводорода; некоторые из этих элементов также более или менее полно осаждаются аммиаком. Вследствие того, что предварительно удалить всю кремнекислоту обычным методом невозможно, оставшееся .небольшое количество ее увлекается осадком гидроокисей, и эту кремнекислоту следует выделить и. определить, как указано в разделе «Кремний» (стр. 955). Число металлов, осаждаемых аммиаком, очень велико. Сюда входят; алюминий, железо (III), хром, таллий, галлий, индий, редкозе
1 Е. S’. Smith, Р. R. H eyl, Z.anorg. Chem., 7, 87 (1892).
* X. Г. С. Бриттон, Водородные ионы, ОНТИ, 1936.
3 L. Gordon, Anal. Chem., 24, 459 (1952).
. Осаждение посредством изменения концентрации ионов водорода
103
мельные металлы, уран, титан, цирконий, бериллий, ниобий и тантал (стр. 112). К ним надо прибавить пятивалентные фосфор, мышьяк и ванадий, которые осаждаются в виде фосфатов, арсенатов и ванадатов одного или нескольких из перечисленных металлов. При большом содержании этих трех элементов осаждение их не будет полным; фосфор и мышьяк в большем или меньшем количестве осаждаются в виде фосфатов и арсенатов щелочноземельных металлов и магния, если последние присутствуют х. Поэтому в таких случаях осаждение аммиаком недопустимо. Неудовлетворительные результаты получаются также, когда раствор содержит большое количество цинка, особенно в присутствии хрома; плохо удается разделение и в присутствии кобальта или меди. Бор мешает осаждению,' и поэтому должен быть предварительно удален методом, описанным на стр. 833.
Способ осаждения аммиаком зависит от элементов, которые содержатся в растворе. При осаждении алюминия, а также фосфора вместе с железом или алюминием и, вероятно, таких менее обычных элементов, как бериллий, скандий и галлий, требуется тщательное соблюдение определенной концентрации ионов водорода в растворе, и в этом случае может быть допущен только очень небольшой избыток аммиака (см. гл. «Алюминий», стр. 565). Так как алюминий присутствует почти всегда, то такой способ осаждения применяется наиболее часто. Нужно, однако, иметь в виду, что в столь тщательной нейтрализации нет необходимости при осаждении элементов, крторые количественно осаждаются и при более высокой концентрации ионов водорода (железо, титан и цирконий). Тщательная нейтрализация не требуется и во всех тех случаях, когда полнота выделения алюминия не имеет значения, например при предваритель-ном отделении железа для его определения объемным методом.
Если для промывания осадка применяют раствор хлорида аммония и осадок содержит большое количество железа, то при последующем прокаливании осадка может произойти небольшая потеря железа вследствие улетучивания FeCl3. Этого можно избежать, промывая осадок раствором нитрата аммония (если только при последующих операциях, проводимых с фильтратом, допустимо присутствие в нем NH4NO3), или же если после обычного промывания раствором NH4C1 промыть Аего 1 рай 5—10 мл горячей воды.
Осаждение в виде основных ацетатов
Общие замечания. Метод осаждения в виде основных ацетатов раньше очень широко применялся при анализе горных пород, но в настоящее время он в значительной степени вытеснен методом осаждения аммиаком, условия проведения которого теперь известны лучше (см. «Алюминий», стр. 565). Этот старинный стандартный метод, который и в настоящее время может оказаться полезным, не следовало бы предавать забвению. В настоящее время его применяют главным образом для: 1) отделения железа (III) от кобальта, меди и'цинка; 2) отделения железа (III) от никеля, когда оба элемента присутствуют в больших количествах, и 3) отделения больших количеств железа (III) от марганца. Положительной
1 В осадках, полученных двукратным осаждением аммиаком, как описано в гл« «Алюминий» (стр. 565), из растворов, первоначально содержавших 0,05 г СаО, 0,005 г Р2О6 и в 10 раз большее количество железа или алюминия, чем требовалось для образования нормальных фосфатов этих металлов, мы не обнаружили кальция.
104
Гл. IV. Общие методы и операции химического анализа
стороной этого метода является то, что осаждение здесь выполняется без заметных местных понижений концентрации ионов водорода в растворе, которая должна поддерживаться постоянно на одной высоте, чтобы предотвратить одновременное осаждение перечисленных выше двухвалентных металлов.
Окончательная кислотность раствора зависит от количества свободной минеральной кислоты, оставшейся в растворе после предварительной его нейтрализации, и от количества прибавленного ацетата А
Алюминий не осаждается ацетатом так полно, как железо, но чем больше преобладает последнее, тем полнее вместе с ним осаждается алюминий. t Поэтому метод редко применяется для отделения алюминия в отсутствие железа и он совершенно не применяется для отделения хрома, урана и большинства редкоземельных металлов 1 2. Фосфор осаждается полностью, если он не содержится в избытке по сравнению с тем количеством, которое необходимо для образования нерастворимых фосфатов с осаждаемым металлом или металлами. Если фосфор находится в избытке, его осаждение можно сделать полным, вводя предварительно в раствор известное количество чистого железа в виде Fede. Такое предварительное удаление фосфора значительно облегчает определение щелочноземельных металлов и магния при анализе некоторых фосфатов.
Большая часть авторов предлагает или предпочитает применять ацетат натрия вместо ацетата аммония за исключением тех случаев, когда введение в раствор соли натрия могло бы помешать в последующих операциях. При работе с ацетатом натрия применяется следующий ,ход анализа. ,
Ход анализа. 1.'Обычный способ. Для осаждения применяют окисленный, предпочтительно солцнокислый, раствор хлоридов. В обычном случае анализа горных пород этот раствор разбавляют 3 до 100 мл. Медленно и при постоянном перемешивании к нему приливают разбавленный (1 : 3) раствор аммиака до тех пор, пока не появится заметное красное окрашивание без образования неизчезающего осадка. После этого прибавляют таким же способом раствор карбоната аммония. Когда окраска раствора начнет темнеть, карбонат аммония прибавляют еще медленнее и с более продолжительным перемешиванием между следующими друг за другом каплями. Когда осадок, вызываемый каждой каплей, начнет растворяться с трудом, реактив прибавляют только тогда, когда раствор станет вполне прозрачным после добавления предыдущей капли. При некотором опыте это не трудно заметить, даже когда жидкость становится очень темной. Интенсивность цвета зависит от количества присутствующего железа и от разбавления. Поэтому при малых количествах' железа существует большая опасность перейти за конечную точку. Если, несмотря на сильное перемешивание, муть после прибавления последней капли реактива скорей увеличивается от перемешивания, чем уменьшается, то добавляют 1 или 2 капли соляной кислоты. Если от этого
1 Например, 400 мл воды, содержащие 0,5 мл НС1, имеют pH = 5,4 после прибавления 5 г и CH3COONa и pH = 5,75 после прибавления 10 г ацетата.
2 Осаждение CeIV, Th и Zr, по-видимому, происходит удовлетворительно в кипящем растворе. ’
3 Большее разбавление в начале и в конце хода анализа необходимо, если присутствие более 0,2 г осаждаемых веществ. Например, при осаждении 1 г железа начальное разбавление должно быть доведено до 200 мл, а окончательное — до 700 мл.
Осаждение посредством изменения концентрации ионов водорода
105
жидкость не проясняется, то лучше прилить небольшой избыток кислоты и повторить процесс нейтрализации.
Затем раствор разбавляют кипящей водой приблизительно до 400 мл и нагревают до кипения. При правильно проведенной нейтрализации осаждение не должно начаться прежде, чем температура достигнет 70° С. Когда будет достигнута температура кипения, постепенно прибавляют раствор 3 г ацетата натрия в 10—25 мл воды и продолжают кипячение 3 мин. Фильтруют, как только осадок соберется на дне. При этом вначале лучше обходиться без отсасывания и применять фильтр такой величины, чтобы весь осйдок мог в нем поместиться, не наполняя его доверху. Осадок умеренно промывают горячей водой, к которой прибавляют 1 г ацетата на 100 мл, так как иначе фильтрат неизбежно будет мутным. Наконец, отсасывают осадок по возможности досуха, растворяют его в соляной кислоте и осаждают — на этот раз аммиаком, как описано в гл. «Алюминий» (стр. 565). Об обработке фильтрата, полученного при анализе горных пород, с целью выделения из него не выпавшего в осадок алюминия, перед осаждением сульфидов см. стр. 952. Марганец, цинк, никель и кобальт осаждают в соединенных фильтратах, как описано в разделе «Осаждение сульфидом аммония» (стр. 90).
2. Видоизменение Брунка — Ф у н к а. Способ Брунка х, немного измененный и подробнее разработанный Функом 1 2, по данным авторов приводит к полному отделению железа (и, вероятно, титана и циркония) от марганца, цинка, никеля и кобальта однократным осаждением. Метод неприменим к анализу материалов, содержащих алюминий, вследствие слишком большой кислотности конечного раствора. В действительности при использовании этого способа может остаться не-осажденным даже небольшое количество железа (до 0,5 мг Fe2O3). Этот метод отличается от предыдущего тем, что здесь не применяют осторожной нейтрализации, а прибавлением хлорида калия достигают образования двойной соли с хлоридом железа, которая мешает появлению нерастворимого основного хлорида железа во время высушивания. Эти изменения, по-видимому, улучшают первоначальный метод, если алюминий отсутствует.
Раствор хлоридов, содержащий не больше 0,3 г железа, переносят в большую чашку, прибавляют по 0,35 г хлорида калия на каждую 0,1 г железа и выпаривают на паровой бане до кажущегося сухого состояния, но не уделяя всей свободной кислоты. Перемешивают остаток с поверхности и снова ставят чашку на 5—10 мин на баню. Затем приливают 10—20 мл холодной воды; после перемешивания должен получиться прозрачный раствор (если не присутствуют фосфор или титан или оба эти элемента вместе). Необходимо, чтобы раствор содержал небольшое количество свободной минеральной кислоты для выделения ею уксусной кислоты из ацетата при последующих операциях. Затем прибавляют ацетат натрия в количестве, в 1,5—2 раза превышающем теоретически требуемое (3 молекулы CH3COONa • ЗН2О на 1 атом железа, или в круглых цифрах 1,5 г ацетата на каждые 0,1 г железа). Ацетат натрия вводят в виде его раствора, слабо подкисленного уксусной кислотой. Разбавляют холодной водой до 400—500 мл на каждые 0,2 г железа, нагревают
1 О. Brunck, Chem. Ztg., 28, 514 (1908).
2 W. Funk, Z. anal. Chem., 45, 181 (1906).
106 ^л. IV- Общие методы и операции химического анализа
постепенно, при перемешивании, до температуры осаждения (60—70°jC), дают осадку осесть, декантируют раствор через бумажный фильтр, переносят осадок на фильтр, промывают его горячей водой и обрабатывают осадок и фильтрат, как описано выше (см. «Обычный способ», стр. 104).
3. Способ Митташа1. Следующий метод, в котором применяется ацетат аммония вместо ацетата натрия, претендует на отделение однократным осаждением больших и малых количеств марганца от железа. Отделение никеля, кобальта и цинка происходит, также почти количественно, а осаждение железа полное. Метод, однако, неприменим к анализу Материалов,; содержащих алюминий, вследствие несколько высокой кислотности раствора (солянокислого вначале), который должен быть холодным, не превышать 100 мл по объему и содержать не более 1 0,3 г железа.
Анализируемый раствор нейтрализуют сначала раствором карбоната аммония (200 г/л) до тех пор, пока образующийся осадок не станет растворяться медленно, затем продолжают нейтрализацию более разбавленным раствором карбоната аммоний (10 г/л), пока, несмотря на перемешивание, небольшой осадок не будет оставаться, в течение 1—2 мин. Тогда прибавляют, соответственно количеству этого осадка, от 3 до 5 мл 2 н. уксусной кислоты, если будут дальше применять продажный ацетат аммония (приблизительно отвечающий формуле GH3COONHi'• С2Н4О2), й 10 мл этой кислоты, если применяют нейтральный раствор ацетата аммония, который получают нейтрализацией уксусной кислоты аммиаком. Затем раствор разбавляют водой приблизительно до 400 мл и нагревают почти до кипения. Не обращая внимания на осадок, который может появиться, прибавляют 20 мл раствора кислого ацетата (полумолярного по концентрации) или смесь 5 мл уксусной кислоты и 10 мл раствора нормального ацетата (по концентрации одномолярного) и продолжают кипячение 1 мин.
Осаждение происходит полностью при 60° С, но фильтрование идет легче, если была достигнута температура кипения. Фильтруют, как только весь осадок соберется на дне, и промывают его без перерыва сначала одной горячей водой, потом горячей водой, содержащей немного ацетата и немного уксусной кислоты. Приставшие к стенкам стакана частицы осадка растворяют в НС1 ц снова осаждают, на этот раз аммиаком, как описано выше (см. «Обычный способ», стр. 104). Этот небольшой осадок не следует собирать на один фильтр с главным осадком, если последний не был промыт до конца, ввиду опасности осаждения аммиаком еще не отмытого марганца. Осйдок и фильтрат обрабатывают, как описано на стр. 105.
Осаждение сукцинатом натрия (янтарнокислым натрием)
Метод осаждения сукцинатом натрия является • также старинным методом отделения железа (и алюминия) от марганца, цинка, никеля и кобальта. Этот метод никогда не применялсй так широко, Как ацетатный метод, однако он заслуживает более тщательного изучения, чем это было до сих пор, потому что опубликованные данные в отношении его преимуществ и недочетов недостаточны и не согласуются между собой. Сук-
1 А. М i 11 a s с h, Z. anal. Chem., 42, 492 (1903),
Осаждение посредством изменения концентрации ионов водорода 107
цинатный метод не уступает ацетатному в отношении отделения никеля, но хуже ацетатного при отделении кобальта, марганца и цинка. Он цроще ацетатного, потому что при нем не требуется таких предосторожностей при предварительной нейтрализации. Его преимущество заключается еще и в том, что алюминий осаждается полнее, чем при использовании ацетатного метода.
Специально в применении к отделению железа метод заключается в следующем. Свободную соляную кислоту нейтрализуют аммиаком до тех пор, пока раствор не станет красно-коричневым, затем прибавляют ацетат йатрйя или аммония до интенсивно красного цвета. Разбавляют водой до 200—600 мл соответственно количеству присутствующего железа и прибавляют 3 г нейтрального сукцината натрия (янтарнокислого натрия), растворенного в небольшом количестве воды. Нагревают почти до кипения, фильтруют и промывают осадок сначала горячим 2%-ным раствором хлорида аммония, затем горячим разбавленным раствором аммиака и отсасывают по возможности досуха. При действии аммиака светло-коричневый осадок сукцината железа переходит в коричневую гидроокись железа. Затем растворяют осадок в соляной кислоте и снова осаждают аммиаком, как описано в гл. «Алюминий» (стр. 565). Двухвалентные металлы: цинк, марганец, никель, , кобальт, остающиеся в фильтрате, лучше всего осаждать сульфидом аммония, как описано на етр. 90.
* Осаждение бензоатом аммония
Из всех методов, в которых железо, алюминий и Другие элементы осаждают добавлением буферной смеси слабой органической кислоты и ее соли, по-видимому, наилучшие результаты дает бензоатный метод х. Однако в этом методе также требуется переосаждение в случае необходимости отделить железо, алюминий, хром и другие элементы от больших количеств марганца, цинка или никеля и, кроме того, очень небольшое количество алюминия может остаться в фильтрате.
К анализируемому раствору, содержащему от 1 до 2,5 миллимолей алюминия, железа (1П) и т. п., разбавленному до 100 мл, приливают 2 капли раствора метилового оранжевого и затем по каплям и при непрерывном перемешивании — 6 н. раствор аммиака, пока окраска раствора не перейдет из красной в оранжевую. Тогда прибавляют 3 мл ,6 н. уксусной кислоты и медленно при перемешивании вливают 20 мл 0,7 М раствора бензоата аммония NH4C7H5O2. Не прекращая перемешивания, нагревают раствор до кипения и осторожно кипятят в течение 5 мин. Затем фильтруют через фильтр диаметром 11 см и промывают 10 раз горячим промывным раствором (0,07 'М по содержанию бензоата аммония и 0,35 М по содержанию уксусной кислоты). При,большом содержании цинка, марганца,, никеля и других двухвалентных металлов осадок растворяют в соляной кислоте и осаждение повторяют.
При осаждении этим методом хрома кипячение раствора надо Продлить до 20 мин; соосаждение других металлов в этом случае будет больше, чем при осаждении алюминия и железа. Доп. ред.*
1 I. К о 11 h о f f, V. Stenger, В. M о s t о V i t z, J. Am, Chem. Soc. 56, 812 (1934),
108
Гл. IV. Общие методы и операции химического анализа
Осаждение суспензиями карбонатов и окисей
Карбонат бария. Взбалтывание анализируемого раствора со взятым в избытке взвешенным в воде карбонатом бария и настаивание смеси на холоду или при нагревании дает возможность очень хорошо отделять железо (III), алюминий, титан, цирконий, хром и уран (а также фосфор и ванадий в присутствии перечисленных элементов) от марганца, цинка, никеля, кобальта и железа (II). Отделение от цинка и марганца происходит более полно, чем от никеля и кобальта. Осаждение бериллия будет полным, если его проводить в горячем растворе, и неполным — в холодном растворе. Элементы цериевой группы осаждаются на холоду, хотя некоторые из них выпадают медленно и осаждение их лучше удается в горячем растворе; элементы иттриевой группы осаждаются очень медленно или вовсе не осаждаются в холодном растворе и не полностью — в горячем растворе.
Суспензия свежеосажденного карбоната бария (а также карбоната кальция и карбоната кадмия) значительно более активна, чем старая или приготовленная из сухого препарата. Свежую суспензию удобно приготовлять, смешивая равные объемы растворов хлорида бария и карбоната натрия такой концентрации, чтобы первый остался в небольшом избытке. Рекомендуются растворы, содержащие 36 ? карбоната натрия КагСОз и 90 г хлорида бария BaCh • 2НгО в 1 л; при смешении они образуют суспензию, pH которой приблизительно равен 1 7,25.
Карбонат кальция. Суспензия карбоната кальция имеет pH, приблизительно равный 1 7,4, но эта суспензия не так хороша для осаждения, как суспензия карбоната бария, вследствие гораздо большей величины взвешенных частиц.
Карбонат кадмия. Суспензия карбоната кадмия имеет pH, приблизительно равный 6,5; она применяется 2 для отделения хрома и ванадия от железа (П). Главное преимущество в ее применении заключается в легкости, с которой осадитель может быть удален из осадка и фильтрата сероводородом, и в том, что сульфаты этому осаждению не . мешают.
Карбонат свинца. Суспензия карбоната свинца 3 имеет pH, приблизительно равный 6,2; она приготовляется и применяется, как описано в гл. «Торий» (стр. 605).
Окись цинка. Суспензия окиси цинка 4 имеет pH, приблизительно равный 5,5; она особенно удобна для осаждения в растворах, содержащих серную кислоту и сульфаты.
Суспензия окиси цинка не должна показывать щелочной реакции по фенолфталеину. В присутствии большого количества железа (ГП), что имеет место, например, при анализе стали, после окисления раствора пробы, осадок от окиси цинка будет содержать все железо, вольфрам, ванадий, хром, уран, цирконий, титан, алюминий, фосфор, мышьяк, олово и почти полностью медь, молибден и кремний. Железо (II), вольфрам (если они не полностью окислены) и малые количества кремния, меди, молибдена, сурьмы и свинца могут оказаться в фильтрате, если они присутствовали в первоначальном растворе в значительных количествах. Фильтрат содержит марганец и кобальт почти полностью; если осадок переосадить и соединить фильтраты, то отделение марганца и кобальта можно считать полным. Отделение никеля не так удовлетвори
1 Е. Wichers, J. Am. Chem. Soc.f 46, 1826 (1924).
2 J. R. Cain, J: Ind. Eng. Chem., 3, 478 (1911).
3 W. B. G i 1 e s, Chem. News, 92, I, 30 (1905).
* W. E. Bradt, R. Ё. L у о n s, J. Am. Chem. Soc., 48, 2644 (1926)s
Осаждение посредством изменения концентрации ионов водорода
109
тельно, особенно если его было вначале много. Сами по себе олово (IV), ванадий (IV) и уран (IV) осаждаются полностью, а олово (II), ванадий (V), уран (VI), вольфрам (VI) и сурьма (V) не осаждаются1.
Окись магния. Суспензия окиси магния имеет pH, приблизительно равный 10,5, и применяется в тех случаях, когда желательна довольно высокая щелочность.
Окись ртути. Суспензия окиси ртути имеет pH, приблизительно равный 7,4. Для некоторых целей ее применение особенно удобно потому, что избыток осадителя может быть полностью удален из осадка прокаливанием, а ртуть, перешедшая в раствор, может быть осаждена сероводородом. Осаждение лучше всего удается из растворов хлоридов. Железо, хром и алюминий осаждаются количественно в холодном растворе, но осадки могут быть загрязнены щелочноземельными металлами, если последние присутствовали в растворе. Цинк, кобальт, никель, бериллий, церий и лантан осаждаются до некоторой степени на холоду и в большей степени из горячих растворов; марганец осаждается на холоду только при долгом стоянии 2.
Суспензия приготовляется следующим образом. Сублимируют чистый хлорид ртути (II) из смеси, содержащей ГО частей (по массе) HgCh и 1 часть HgO. Растворйют его'в небольшом количестве воды до получения насыщенного раствора, который нагревают до кипения и медленно при перемешивании вливают в горячий 10%-ный, не содержащий железа, взятый в избытке раствор едкого натра. Осадку дают осесть на дно, после чего основательно промывают его декантацией горячей водой, переносят на фильтр и продолжают промывание до удаления хлоридов. Затем взбалтывают осадок с водой и сохраняют в сосуде из церезина или в парафинированной склянке. После осторожного прокаливания 5 г полученной окиси ртути в фарфоровом тигле (под хорошей тягой) не должно оставаться видимого или весомого остатка.
Осаждение добавлением йодатов или броматов
Смеси йодата и иодида калия или бромата и бромида калия применяются для осаждения таких элементов, как алюминий, хром, железо, кобальт, никель и олово 3 и для разделения некоторых элементов, например для отделения висмута от свинца, меди, кадмия и цинка 4.
Осаждение едким натром
Осаждение едким натром широко не используется аналитиками вследствие не всегда высокого качества реактива и слизистого характера получаемого осадка. Первое возражение не может считаться достаточно основательным, потому что в настоящее время легко приобрести очень
1 Подробности этих разделений см. стр. 470 и в статье J. I. Hoffman. J. Research NBS, 7, 883 (1931).
2 J. Volhard, Ann. 198, 331 (1879); E. F. S m i t h, P. R. H e у 1, Z. anorg. Chem., 7, 82 (1894).
s S. E. M o о d y, Am. J. Sci., [4], 20,181 (1905); 22, 176, 379, 483 (1906). Раствор, приготовленный смешением равных объемов 7%-ного раствора КЮ3 и 10%-ного раствора KI, имеет pH около 7,5.
4 L. Moser, W. Maxymowicz, Z. anal. Chem., 67, 248 (1925—1926).
110
Гл. IV. Общие методы и операции химического анализа
хороший едкий натр. Второе возражение имеет некоторое значение, особенно когда приходится фильтровать концентрированные растворы; однако если применяют горячие 5 %-ные растворы едкого натра, операция фильтрования и промывания осадков не труднее, чем При работе с аммиачными растворами. *
В наших опытах этот метод оказался весьма удовлетворительным, особенно когда для последующего анализа можно было взять аликвотные порции фильтрата.
Осаждение едким натром служит для отделения железа, титана, циркония, редкоземельных элементов и хрома от алюминия, фосфора и ванадия.
Если едкий натр применяют вместе с карбонатом натрия, то в осадок переходят также и щелочноземельные металлы. Полнота осаждения титана зависит от присутствия железа. Хром осаждается вполне удовлетворительно х. В растворах, содержащих магний, никель (III или Й), отделение алюминия не полно. Если количество никеля не превышает .содержание алюминия, а железа (III) по крайней мере в 5 раз больше, чем никеля, то захват алюминия осадком ничтожен 2. В присутствии карбонатов или ванадия уран осаждается частично или совсем не осаждается, а осадок, полученный в присутствии ванадия, всегда им заррязнен. Нечеткость разделения в отношении хрома, ванадия и урана легко устраняется путем осаждения в присутствии окислителей, например перекиси натрия или пергидроля, с добавлением карбоната натрия, если присутствует уран. При такой обработке уцомянутые три элемента переходят в фильтрат.
В случае отделения никеля или кобальта желательно прибавление брома или подобного ему окислителя, потому что это приводит к образованию компактных осадков гидроокисей трехвалентных никеля и кобальта, которые гораздо легче отфильтровываются и промываются, чем осадки гидроокисей двухвалентных никеля и кобальта.
Лучшие результаты получаются при медленном прибавлении горячего слабокислого анализируемого раствора солей этих элементов к горячему раствору едкого натра, взятому в избытке. Если применяется перекись натрия, то ее прибавляют к раствору едкого натра, который в этом случае заранее не нагревают; после смешения, однако, раствор нужно хорошо прокипятить, чтобы осадить титан, ниобий и др,, и обеспечить полное осаждение железа.
Ход анализа. Для осаждения берут 100 мл кислого раствора, пред-, почтительно не содержащего солей аммония. Прибавляют почти до нейтральной реакции едкий натр,'нагревают до кипения и вливают раствор медленно и при постоянном перемешивании в 100 мл горячего свежеприготовленного 10%-ного раствора едкого натра. Кипятят смесь 2— 3 мин, слегка перемешивая, дают осесть и фильтруют через бумажный фильтр средней плотности (фильтр следует предварительно промыть горячим 5%-ным раствором едкого натра; содержащим немного сульфата натрия, пока не получится 50 мл совершенно бесцветного фильтрата). Осадок промывают этим же промывным раствором и фильтрат сохраняют.
1 Например, 0,4 мг Сг было найденб в фильтрате после осаждения 0,06 г Сг из 200 мл кипящего 2,5%-иого раствора NaOH и 0,5 мг Сг осталось после осаждения из такого же объема 5%-ного раствора NaOH.
’ Н. А. В г i g h t, R, M. F о w 1 e г, J. Research NBS, 10, 327 (1933),
Осаждение посредством изменения концентрации ионое водорода Щ
При очень точных анализах или если полученный осадок очень велик, а количество подлежащих отделению неосаждаемых элементов также значительно, осадок растворяют в горячей разбавленной серной кислоте и переос4ждают. Фильтраты соединяют и проводят анализ объединенных фильтратов и осадка.
*Ораждение слабыми органическими основаниями
Э. А. Остроумовым 1 разработан ряд методов гидролитического осаждения, в которых требуемая концентрация ионов водорода создается добавлением слабых органических оснований или смесей этих оснований с их солйми. Для анализов применяют: пиридин, смесь пиридина с азотнокислым пиридином и а-пиколин. Чистота разделений, проводимых этими методами, связана с тем, что многие металлы образуют с указанными органическими основаниями растворимые комплексные соединения.
При. добавлении пиридина к слабокислому анализируемому раствору в нем создается pH, приблизительно равный 6,5. В этих условиях осаждаются железо (III), алюминий, хром, уран, индий, галлий, титан, цирконий, торий и скандий. В то же время марганец, кобальт, никель и цинк (а также и металлы сероводородной группы — медь и кадмий) образуют с пиридином комплексные ионы состава Me(C6H5N)2+, остающиеся в растворе. Для создания в растворе указанного- значения pH при определений металлов, присутствующих в обычных аналитических концентрациях, требуется добавление пиридина в избытке около 8 эквивалентов.
• По данным автора осаждение пиридином имеет следующие преимущества: . .
1. Вследствие того что пиридин является слабым основанием,/выделение водных гидроокисей происходит сравнительно медленно. Это является причйной быстрой дегидратации гидроокисей и перехода их в малодисперсное состояние, в котором они обладают значительно меньшей поверхностью и, следовательно, меньшей адсорбционной способностью. Этр приводит к получению более чистых осадков, к лучшему отделению от ионов, остающихся в растворе.
2. Низкая величина pH раствора, создаваемая пиридином, устраг няет явление пептизации осадков гидроокисей, что облегчает их отфильтровывание и промывание.
Эти преимущества особенно сказываются -при выделении гидроокисей, имеющих амфотерный характер, как, например, гидроокисей алюминия, галлия и хрома.
Осаждение надо проводить из растворов хлоридов и нитратов при температуре, близкой к кипению. Сульфаты мешают разделению, образуя
1 Э. Д. Остроумов. Новые методы химического анализа с применением органических оснований, Госгеолиздат, 1952. Осаждение пиридином: Э. А. О с т р о-у м о в, Зав. лаб., 4, 1317 (1935); 6, 16 (1937); Z. anal. Chem., 106, 170, 243 (1936); Ann. chim. analyt., 19, № 4 (1937); 20, № 1, 9 (1937); Э. А. О с т p о у м о в, Р. И. Бомштейн, Зав лаб., 11,146 (1945); Э. А. Остроумов, Б. Н.Иванов-Эмин, там же, 11, 1034 (1945); Б. Н. Иванов-Эмин, Э. А. Остроумов, тем же, 12, 674 (1946); Э. А. Остроумов, С. Беручьян, там же, 12, 802 (1946). Осаждение смесью пиридина и его азотнокислой соли: Э. А. Остроумов, Зав. лаб., 8, 1226 (1939); 13, 404 (1947); ЖАХ, 2, 111 (1947); 3, 153 (1948). Осаждение а-пиколином: Э, А. О с т р о у м о в, Б. Н. Иванов-Эмин, Зав. лаб., И, 386 (1945),
112
Гл. IV, Общие методы и операции химического анализа
комплексные анионы, не осаждаемые пиридином, например с ураном, торием и железом (III). Мешающее влияние сульфатов, если количество их не слишком велико (не более 2—3 г) устраняется добавлением 10— 20 г хлорида аммония. Кроме того, прибавление хлорида аммония способствует удержанию в растворе марганца, никеля, кобальта и др. в виде их комплексных ионов с пиридином.
Ход анализа. Кислый анализируемый раствор, содержащий не более 0,1 г осаждаемых металлов, нейтрализуют аммиаком до появления слабой мути и растворяют последнюю добавлением 1—2 капель разбавленной соляной кислоты. Прибавляют 5 г хлорида аммония (10—20 а, если присутствуют сульфаты) и нагревают до кипения. Сняв стакан с огня, осторожно прибавляют по каплям при перемешивании 20%-ный раствор пиридина до изменения окраски метилового красного и сверх того еще 10—15 мл избытка. Снова нагревают жидкость до начала кипения, переносят на водяную баню и оставляют на 30 мин. Затем фильтруют и осадок промывают' горячим 3 %-ным раствором хлорида аммония, содержащим несколько капель пиридина. На фильтрование и промывание осадка требуется около 15 мин. Если из фильтрата желают выделить никель диме-тилглиоксимом, то для удаления пиридина, который мешает этому осаждению, прибавляют немного карбоната натрия и кипятят. После удаления пиридина раствор подкисляют и осаждают никель обычным способом.
Применяя смесь пиридина и его азотнокислой соли, имеющую pH «=« 4,1—4,2, можно отделить висмут от свинца (а также от меди и кадмия) (стр. 272) и торий от церия (III) и редкоземельных металлов. Пользуясь другой буферной смесью, содержащей 1 эквивалент азотной кислоты на 4 эквивалента пиридина и имеющей pH 5,4, можно отделить скандий от церия (III) и других редкоземельных металлов.
а-Пиколин, являющийся несколько более сильным основанием, чем пиридин, создает в растворе pH, равный примерно 7,0. Это дает возможность количественно осаждать этим реактивом бериллий, который пиридином осаждается не полностью. Марганец, цинк, кобальт и никель образуют с а-пиколином прочные комплексные соединения, остающиеся в растворе (стр. 583). Доп. ред*
РАЗДЕЛЕНИЕ ЭЛЕМЕНТОВ В ОСАДКЕ ОТ АММИАКА, АЦЕТАТА НАТРИЯ ИЛИ СУКЦИНАТА НАТРИД
Если принять во внимание, что почти половина всех катионов осаждается аммиаком, а большая часть их также и другими реактивами, указанными в заголовке, то становится ясным, что осадки, полученные таким способом при анализе горных пород, минералов, руд и мегаллурических продуктов, должны иметь очень сложный состав. Например, исключая те элементы группы сероводорода, которые также осаждаются названными реактивами и которые, как предполагается, 0ыли удалены раньше, получим следующий перечень элементов, которые могут встретиться в весомых и легко открываемых количествах в сложных случаях анализа изверженных, метаморфических или осадочных горных пород: кремний, титан, цирконий, алюминий, железо, хром, ванадий, фосфор (изредка — уран), бериллий, тантал, ниобий и редкоземельные металлы. В этот список не вошли те элементы, которые могут попасть в осадок при непра
Разделение элементов в осадке от аммиака
113
вильно проведенном осаждении, как, например, марганец, затем такие элементы, как магний и щелочноземельные металлы, которые могут оказаться в осадке при некоторых особых обстоятельствах, и, наконец, бор, который будет всегда находиться в осадке, если он присутствовал в заметных количествах в исходном материале. Несомненно, что многие из перечисленных элементов встречаются редко, некоторые могут быть удалены до осаждения специальной обработкой, другие могут быть количественно определены из отдельных навесок пробы. Все же остается так много элементов, что надо очень тщательно исследовать взвешенный осадок, прежде чем вычислять содержание алюминия по разности, как это обычно делается.
Может, показаться, что анализ такого сложного осадка очень труден и ненадежен. В действительности это не так. Достаточно полный анализ осадка сделать не так уж трудно, и он даже не очень сложен, если схема анализа правильно продумана и указанные элементы присутствуют в количествах, допускающих их определение (см. «Подробный анализ осадка от аммиака», стр. 118).
По нелетучему остатку, получающемуся после обработки нечистой кремнекислоты смесью серной и фтористоводородной кислот, можно установить, какие компоненты нужно искать в сложном осадке от аммиака. Если этот первый остаток весит не более 2—3 мг и после сплавления с небольшим количеством карбоната натрия легко растворяется (обычный случай) в горячей разбавленной соляной кислоте, то можно с полной уверенностью считать, что тантал и ниобий не будут найдены в последующем осадке от аммиака и что исходный анализируемый материал не содержит заметных количеств фосфора, циркония или титана. Большой нелетучий остаток после обработки HF + H2SO4 или остаток, не дающий после сплавления с содой прозрачного раствора при растворении в соляной кислоте, ясно указывают на присутствие необычных составных частей. Так, остаток может содержать сульфат бария, сульфат свинца, окислы ниобия, тантала или сурьмы; или титан, цирконий и олово, одни или вместе с фосфором. В таких случаях даже лучше исследовать раствор нелетучего остатка отдельно, прежде чем присоединять его (целиком или аликвотную чать) к фильтрату, полученному после отделения кремнекислоты, если только в результате тщательно проведенного предварительного качественного анализа это не стало излишним х.
Хотя обычно принято вычислять содержание алюминия по разности, но в особых случаях могут потребоваться другие методы его определения. Так, может случиться (хотя и не при обычном анализе горных пород), что анализируемая проба содержит очень мало алюминия и что необходимо или желательно сделать прямое определение этого элемента, чтобы определить его содержание с большей точностью, чем это возможно
1 При анализе ряда горных пород из Leucite Hills Wyoming одним из нас был получен после удаления кремнекислоты большой остаток, единственный в своем роде. К концу операции отгонки, когда начали выделяться в твердом виде сульфаты, остаток имел молочный или эмалеподобный вид и вел себя подобно смеси окислов ниобия и тантала, но после исследования оказался в основном фосфатом титана (может быть, с небольшой примесью фосфата циркония). В дальнейшем ходе анализа этой горной породы осадок, полученный при осаждении аммиаком, сплавленный с пиросульфатом и выщелоченный холодной водой, дал белый, более или менее хлопьевидный осадок такого же или подобного состава.
8 Заказ 522.
114 Гл. IV. Общие методы и операции химического анализа'
косвенным методом, по разности, а в некоторых случаях, — чтобы даже просто установить, действительно ли алюминий присутствует. Если анализируемая проба содержит много титана, то может оказаться желательным определение этого элемента с большей точностью, чем это возможно' колориметрическим методом; в таком случае необходимо иметь методы отделения титана от алюминия и железа.
В дальнейшем мы не будем рассматривать различные методы разделения ниобия и тантала, так как они встречаются редко и ид присутствие серьезно нарушает ход анализа и требует применения специальных методов анализа (см. «Ниобий и тантал», стр. 666).
Ванадий встречается часто, а уран редко; когда оба эти элемента содержатся вместе, они взаимно мешают определению один другого; так, ванадйй, в зависимости от его количества, мешает осаждению большего-или меньшего количества урана сульфидом аммония. Бериллий не принадлежит к числу обычных составных частей горных пород; в большинстве разделений он сопровождает алюминий.
При использовании наиболее обычных методов разделения железа и алюминия последний остается вместе с фосфором. Методы разделения других элементов различны. Например, при применении фенилгидрази-нового метода практически удаляется только один Элемент -- железо (II); при использовании купферонового метода железо, титан, цирконий и ванадий отделяются от всех прочих металлов, за Исключением редкоземельных, которые частью осаждаются, частью остаются в растворе; при сплавлении с карбонатом натрия и извлечении водой отделяются железо, титан, цирконий и редкоземельные металлы от алюминия, хрома (VI), фосфора и.ванадия (уран разделяется: частью остается в остатке, частью переходит в раствор); осаждение, раствором едкого натра отличается от описанного выше тем, что хром (III) и уран осаждаются, если ванадий отсутствует, в противном случае уран и ванадий разделяются между осадком и раствором; при отделении сульфидом аммония в присутствии тартрата аммония железо отделяется от всех других элементов. При применении большей части этих методов небольшое количество кремнекис-лоты, содержащееся в осадке от аммиака, может быть до начала разделения' выделено выпариванием сернокислого раствора пиросульфатного плава (стр. 956). Все изложенные ниже методы применимы к осадкам, полученным добавлением ацетата натрия, сукцината натрия или других реактивов, осаждающих рассматриваемые элементы.
Обычные методы разделения
Осаждение фенилгидразином. Когда железо находится в трехвалентном состоянии, главное различие между осаждением фенилгидразином и осаждением аммиаком заключается в том, что первый не вполне осаждает некоторые редкоземельные металлы, а уран не осаждает совсем. Но если железо находится в двухвалентном состоянии, то в слабокислом (по лакмусу) растворе оно совсем не осаждается фенилгидразином, и этот реактив дает возможность полностью отделять некоторые компоненты осадка от аммиака (в частности, алюминий) от железа.
Метод осаждения фенилгидразином»обычно применяется для анализа смесей железа и алюминия, особенно когда желательно иметь видимое-подтверждение присутствия последнего или когда количество алюминия:
Разделение элементов в осадке от аммиака
115
так мало, что при использовании других методов его присутствие осталось бы под сомнением.
Ход анализа. Прокаленный и взвешенный осадок от аммиака спла-. вляют с пиросульфатом калия в платиновом тигле (стр. 955), растворяют плав в воде, раствор нейтрализуют, восстанавливают железо и осаждают, как описано ниже (см. «Осаждение фенилгидразином», стр. 154).
Если осадок содержал много железа или желательна очень большая точность, растворяют осадок в кислоте и повторяют нейтрализацию, восстановление и т. д.
Осаждение купфероном! Осаждение купфероном [аммонийная соль нитрозофенилгидроксиламина CeH8N(NO)ONHd отличается от осаждения аммиаком тем, что первый совершенно не осаждает алюминия, хрома фосфора и урана (VI), а редкоземельные элементы, осаждает не полностью. Купферон количественно осаждает уран (IV). Этим реактивом можцо прекрасно отделить железо, титан, цирконий и ванадий от алюминия, хрома и фосфора (когда последний присутствует в малых количествах; он дает также наиболее эффективное отделений ванадия от урана (VI). ' Ход анализа. Взвешенный осадок от аммиака'сплавляют с пиросульфатом калия в платиновом тигле (стр. 955), плав растворяют в разбавленной (1 : 9) серной кислоте и далее проводят осаждение купфероном в холодном растворе (охлаждение льдом), как описано ниже (см. «Осаждение купфероном», стр. 143)х. Промытый осадок прокаливают и взвешивают, а затем обрабатывают соответственно его составу. В фильтрате разрушают все органические вещества (стр. 144), и проводят нужные определения.
Осаждение сульфидом аммония в растворе, содержащем тартрат аммония. Если сернокислый раствор осадка от аммиака обработать винной кислотой, затем сероводородом и, наконец аммиаком или сульфидом аммония, то железо осаждается, а другие обычные элементы остаются в растворе. Цинк, кобальт, никель и большая часть марганца (если о пи раньше были увлечены в осадок при осаждении аммиаком) также осаждаются. Этот метод иногда применяют для выделения и определения железа. Однако главное применение он находит для отделения железа, особенно перед осаждением титана купфероном, и ’г. п.
Осадок сульфида железа никогда не прокаливают непосредственно, но растворяют его в соляной и азотной кислотах и снова осаждают аммиаком.
Ход анализа. Взвешенный осадок от аммиака сплавляют, как описано выше (см. «Осаждение фенилгидразином»), и растворяют плав в воде или . разбавленной (5 : 95) серной кислрте, последнее — в тех случаях, когда присутствую^ элементы, образующие легко гидролизующиеся соли, например титан или цирконий. Прибавляют достаточное количество прозрачного насыщенного раствора чистой винной кислоты, чтобы впоследствии удержать алюминий и др. в растворе; винная кислота не должна содержать желёза и других мешающих злементов; большого избытка ее следует избегать, особенно в тех случаях, когда в фильтрате надо будет ее разрушать; достаточно 3—4-кратного ее количества по отношению к массе осадка от аммиака.
1 При выполнении очень точных анализов перед осаждением купфероном надо удалить платину.
8*
116 Гл. Общие методы и операции химического анализа
Осаждают, как указано в разделе «Осаждение сульфидом аммония» (стр. 90), не забывая о сохранении фильтрата; осадок обрабатывают, как указано в гл. «Железо» (стр. 440), а в аммиачном фильтрате после отделения гидроокиси железа определяют марганец, цинк, никель и кобальт, если они присутствовали в первоначальном растворе.
Обработка фильтрата, полученного после осаждения сульфидом аммония, зависит от дальнейшего хода анализа. Если последует осаждение купфероном, то нейтрализуют раствор серной кислотой, прибавляют избыток ее в.10% (по объему) и кипятят, чтобы удалить сероводород. Если предполагают применить другие методы, прибавляют избыток серной кислоты в 10 мл, выпаривают и разрушают винную кислоту, как описано на стр. 92.
Сплавление с карбонатом натрия с последующим выщелачиванием водой. Сплавление взвешенного осадка от аммиака с карбонатом натрия и последующее выщелачивание плава водой (возможно повторенное 1 или 2 раза, в зависимости от массы и состава осадка) дает вполне удовлетворительное отделение железа, титана, циркония, бериллия и редко- < земельных металлов от алюминия, хрома, ванадия и фосфора (уран разделяется и находится частью в остатке, частью в растворе). Отделение хрома является результатом его окисления до хромата во время сплавления; это окисление идет обычно до конца, и прибавления окисляющих реактивов вроде селитры не требуется (разве только для ускорения окисления). Сплавление и выщелачивание являются предварительной ступенью, за которой должны последовать анализы соответствующими методами водной вытяжки и остатка.
Ход анализа. Взвешенный осадок от аммиака смешивают с 20-крат-ным (по массе) количеством Na2CO3, сплавляют при умеренной температуре, пока сплав не перестанет пениться, и затем нагревают в течение 30 мин при 1100—1200° С на пламени паяльной лампы (не в муфеле, см. стр. 924). Охлажденный плав выщелачивают холодной водой и, когда все растворимые соли перейдут в раствор, фильтруют. Остаток на фильтре промывают холодной водой, пока не будут извлечены соли натрия. При выполнении очень точных анализов или в случае'большого остатка его прокаливают и повторяют сплавление. Водную вытяжку сохраняют; при заметных количествах хрома или урана она имеет желтую окраску.
Нерастворимый остаток прокаливают, сплавляют с пиросульфатом и продолжают анализ, применяя для осаждения фенилгидразин (стр. 114), купферон (стр. 115), сульфид аммония (стр. 115) и другие реактивы.
Стакан, содержащий водную вытяжку плава, покрывают часовым стеклом и раствор осторожно подкисляют минеральной кислотой, выбор которой зависит от предполагаемой дальнейшей обработки. Нужно иметь в виду, что в этом растворе аммиак может не осадить количественно ванадия и фосфора по причине удаления некоторых оснований, содержавшихся в первоначальном растворе.
Осаждение едким натром. При осаждении едким натром железо, титан, цирконий, редкоземельные металлы, хром и уран отделяются от алюминия, фосфора и ванадия. Полнота осаждения титана зависит от количества присутствующего железа. Осаждение урана полное, если отсутствуют ванадий и карбонаты; в том случае, когда ванадий и уран присутствуют вместе, последний осаждается не полностью (осадок содержит ванадий) или совсем не осаждается. Осаждение хрома не вполне количе
Разделение элементов в осадке от аммиака
117
ственное: 0,1—0,2 мг Сг2О3 остаются в растворе. Неопределенность в отношении хрома, ванадия и урана легко устраняется проведением осаждения в присутствии окислителей, например перекиси натрия или пергидроля, и добавлением карбоната натрия, если присутствует уран. В этом случае все три указанных элемента переходят в фильтрат.
Ход анализа. Взвешенный осадок от аммиака сплавляют с пиросульфатом в платиновом тигле (стр,- 955) и растворяют плав в 100 мл воды. Нагревают раствор до кипения и осаждают, как описано выше (см. «Осаждение едким натром», стр. 109). Полученный осадок растворяют в минеральной кислотё, выбранной соответственно дальнейшему х'оду анализа. Щелочной фильтрат подкисляют минеральной кислотой, также выбранной в зависимости от последующего хода анализа, ндпример азотной кислотой’ если будут определяться фосфаты, или серной кислотой, если ванадий будет восстанавливаться сернистым ангидридом.
Осаждение алюминия в виде хлорида. Выделение алюминия в виде гидрата хлорида алюминия обеспечивает при однократном осаждении достаточно полное отделение алюминия от бериллия. Этим методом можно также достигнуть хорошего отделения алюминия от железа, цинка, меди, ртути и висмута и от некоторых других элементов.
Ход анализа. Взвешенный осадок от аммиака сплавляют с карбонатом натрия (стр. 116) и растворяют охлажденный плав в разбавленной соляной кислоте, прибавляя последнюю сначала осторожно, а затем нагревая, если нужно до полного растворения. Разбавляют до 200 мл, осаждают аммиаком (стр. 102), фильтруют и промывают осадок 2 или 3 раза водой. Затем растворяют осадок в горячей разбавленной (1:1) соляной кислоте и продолжают анализ, как описано в гл. «Алюминий» (стр. 564).
Другие методы разделения
Существует много других методов разделения, которые могут быть применены к раствору осадка от аммиака; они мало чем отличаются от тех, которые были описаны выше. Из них отметим следующие..
Осаждение ацетатом натрия. Титан, цирконий, и торий можно отделить от железа осаждением ацетатом натрия после восстановления железа сероводородом 4; образование сульфида железа (II) помогает при предварительной нейтрализации раствора. Этим методом алюминий, фосфор и, вероятно, некоторые другие элементы осаждаются полностью или частично. Этот метод осаждения сходен с методом осаждения фенилгидразином.
Добавление гипосульфита натрия. Гипосульфит натрия (Na2S2O4, не тиосульфат2) был применен 3 для одновременного восстановления железа и осаждения алюминия и бериллия. Цирконий, титан и некоторые другие элементы, вероятно, также при этом осаждаются.
Гидролиз после восстановления железа. При использовании гидролитического метода 4 титан и цирконий отделяются от железа и алюминия
1 М. Dittrich, S. Freund, Z. anorg. Chem., 56, 337 (1907).
2 * По старой номенклатуре соль Na2S2O4 называлась гидросульфитом натрия. Прим. ред. *
3 Р. Barbier, Bull. Soc. chim., (4), 7, 1027 (1910).
4 C. Baskerville, J. Am. Chem. Soc., 16, 427, 475 (1894).
118 Гл. IV. Общие методы и операции химического анализа
кипячением в течение нескольких минут почти полностью нейтрализованного солянокислого раствора, содержащего эти элементы, после предварительного восстановления железа сернистой кислотой. При этом методе для растворения первоначального осадка нельзя применять серную кислоту. Нейтрализацию надо проводить очень осторожно,' инАче осаждение не будет полным или осадок будет содержать примеси. Осадок редко бывает чист и, несомненно, содержит другие элементы, например торий и фосфор, если они присутствовали в осадке от аммиака.
Отгонка с соляной кислотой и хлором. Железо может быть полностью удалено и отделено от алюминия, циркония, бериллия и хрома нагреванием осадка от аммиака при 200—300° С в токе сухого хлористого водорода, смешанного с небольшим количеством хлора х. Имеются также указания 1 2 и на то, что железо можно удалить нагреванием прокаленного осадка от аммиака в токе смеси паров соляной кислоты и воздуха» Оба метода могут найти применение, когда железо мешает и его надо удалить, но они не так удобны в тех случаях, когда железо необходимо определить количественно.
Осаждение в растворе, содержащем едкое кали и перекись водорода. Рядом авторов3 было показано, что железо можно, отделить от титана, если холодный кислый раствор солей обоих элементов обработать сначала 30%-ной перекисью водорода, ..взятой в значительном избытке, затем едким кали и выделившийся осадок отфильтровать и промыть холодной водой, содержащей перекись водорода. Некоторые другие элементы также останутся полностью или частично в растворе вместе с титаном, например алюминий/ цирконий, ниобий, тантал, хром, ванадий и фосфор. При нагревании раствора титан частично осаждается, а при кипячении осаждение титана становится полным.
Осаждение салициловой кислотой и салицилатом аммония. Отделение циркония, тория, ниобия и тантала от титана и алюминия может быть достигнуто их осаждением в растворе, содержащем салициловую кислоту и салицилат аммония 4. Холодный, сдабокислый анализируемый раствор вливают медленно и при перемешивании в кипящий раствор 10 г салицилата аммония в. 50 мл воды; кипячение продолжают 30 мин, затем фильтруют и осадок промывают горячим 5%-ным раствором салицилата аммония.
Подробный анализ осадка от аммиака
При исследовании горных пород, которое может быть приведено в качестве примера, обычный ход анализа требует определения в осадке от аммиака кремнекислоты (оставшейся после выделения главной ее массы в начале анализа), железа и титана. Дальнейшее вычитание из суммы окислов (см. стр. 113) проводят на основании результатов анализов отдельных навесок анализируемого материала. Например, цирконий и редкоземельные металлы определяют в одной навеске, фосфор — в другой, хром и ванадий — в третьей. Такой метод хорош в тех .случаях,
1 F. A. Gooch, F. S. Н avens, Am. J. Sci., [4], 7, 37.0 (1899).
2 H. В о г c k, Z. angew. Chem., 25, 719 (1912).
3 А. С 1 a S s е п, Вег., 21, 370 (1888); К. Borneman n. Н. Schirmeis-t е г, Metallurgie, 7, 726' (1910).
4 М. Dittrich, S. Freund, Z. anorg. Chem., 56, 344 (1907).
Разделение элементов в осадке от аммиака
119
когда в распоряжении имеется достаточно большое количество анализируемой пробы. Но если имеющаяся для анализа проба так мала, что большая часть компонентов или все они должны быть определены в одном осадке, полученном после действия аммиака, ацетата натрия или сук-цината натрия, то отдельные определения можно провести или попытаться •йрйвести по приводимой ниже схеме. (Случай, когда все рассматриваемые ниже элементы присутствуют одновременно, встречается очень редко).
Эта схема предусматривает прежде всего выделение остаточной кремнекислоты. Затем отделяют железо, титан и редкоземельные металлы, осаждая их едким натром в присутствии окислителя и карбоната натрия. В. фильтрате остаются алюминий, фосфор, ванадий, хром и бериллий. Из осажденных элементов железо выделяют в виде сульфида осаждением сульфидом аммония в присутствии тартрата аммония; титан определяют в фильтрате колориметрически, после разрушения винной кислоты; цирконий осаждают в растворе, содержащем перекись водорода, употребленном для определения титана, и, наконец, редкоземельные металлы осаждают вместе с Гидроокисью титана в фильтрате от осаждения циркония и отделяют от титана в виде фторидов. Окраска фильтрата после осаждения едким шатром указывает на присутствие хрома или ;урана, если последние содержатся в количествах, достаточных, чтобы окрасить раствор. Дальше ведут анализ следующие путем. Сначала определяют ванадий объемным методом, затем выделяют, фосфор в виде фосфоромолибдата аммония и, наконец, осадок, полученный осаждением аммиаком фильтрата от фосформолибдата, испытывают на алюминий, бериллий и другие элементы. :
При выполнении большей части описанных выше операций растворы .должны иметь возможно меньший объем, потому что количества различных элементов, за исключением немногих самых главных (железо и алюминий в случае анализа горных пород), вероятно, будут незначительны. Так как заметное количество осадка может остаться в порах фильтровальной бумаги даже после тщательной обработай растворителем фильтра с осадком, промытый фильтр следует сжечь, сплавить золу с щепоткой соды или пиросульфата и растворить пл*ав.
Нужно твердо помнить, что приведенная схема не может быть применена ко всем смесям, даже содержащим одни и те же компоненты. В зависимости от отношений, в которых эти‘компоненты входят в состав смеси окислов, эта схема в одном случае может быть применена, в другом — ее применять нельзя. Опытный аналитик во многих случаях может счесть необходимым изменить указанный ход анализа или разработать новый.
Ход анализа. Взвешенный осадок от аммиака сплавляют с пиросуль--фатом (стр. 955) и растворяют плав в разбавленной (1 : 20) серной кислоте. Полученный раствор делят на две приблизительно равные части, обрабатывают одну часть несколькими каплями перекиси водорода (30%-ной) и сравнивают окраски обеих частей. Это испытание обнаруживает присутствие заметных количеств титана или ванадия; появление чисто желтой или оранжево-желтой окраски раствора указывает на при-сутствие титана, красно-коричневый оттенок окраски — на присутствие ванадия (который может маскировать титан), а отсутствие изменения •ркраски — на отсутствие обоих элементов. Выполненная в такой форме,
120 Гл. IV. Общие методы и операции химического анализа:
эта проба более чувствительна, чем обработка перекисью водорода всего раствора.
Кремний. После пробы перекисью водорода обе части раствора соединяют, выпаривают до появления паров серной кислоты, отделяют и отгоняют кремне кис лоту, как указано на стр. 956, а фильтрат сохраняют. Нелетучий остаток (если таковой остался после отгонки кремнекислоты) сплавляют с возможно меньшим количеством пиросульфата и охлажденный плав растворяют в полученном ранее фильтрате. Если нелетучего остатка очень мало, как это обычно бывает, то можно считать доказанным отсутствие заметных количеств сульфата бария, а также фосфора в соединении с титаном, цирконием, ниобием или танталом, за исключением того случая, когда значительное количество титана может удержать последние два элемента в растворе. Большое количество остатка — явление необычное и потому остаток надо тщательно исследовать и только после этого прибавлять раствор его к фильтрату.
Фильтрат от кремнекислоты выпаривают дЗ такого малого объема, чтобы лишь удержать в растворе содержащиеся в нем соли, и осаждают (под конец при кипячении) раствором едкого натра, содержащим перекись водорода, а при наличии урана — также и соду (см. «Осаждение едким натром», стр. 109). Осадок переосаждают и щелочные фильтраты соединяют. Затем осадок растворяют в. горячей разбавленной серной кислоте; полученный раствор сохраняют.
Анализ щелочного фильтрата. Хром и уран. Если соединенные щелочные фильтраты бесцветны, то это указывает на отсутствие заметных количеств хрома и урана. Однако при содержании этих элементов в количестве нескольких сотых процента в таком разбавленном растворе они могут остаться незамеченными. Если нужно более точное исследование, то подкисляют раствор серной кислотой, кипятят с 1—2 каплями сернистой кислоты, осаждают аммиаком, добавляя его в небольшом избытке, фильтруют и сохраняют фильтрат. Осадок растворяют в малом количестве разбавленной серрой кислоты, полученный раствор выпаривают до 10—20 мл, охлаждают, прибавляют 1—3 капли перекиси водорода (30%-ной) и затем едкий натр, пока осадок не растворится. Если в этом растворе не появится окрашивания, то содержание хрома и урана не превышает «следов» и раствор нужно соединить с сохраненным аммиачным фильтратом: объединенный раствор служит для определения ванадия.
Если первоначальный щелочной фильтрат окрашен в желтый цвет, то можно считать вероятным присутствие хрома или урана, особенно при исследовании горных пород, и вести анализ дальше, исходя из предположения, что окрашивание вызвано хромом. С возможностью того, что желтое окрашивание происходит от органических веществ, извлеченных из фильтра, считаться не приходится, если фильтр был перед фильтрованием хорошо промыт горячим раствором щелочи. Если окрашивание слабое, то хром определяют колориметрически (стр. 595) и сохраняют раствор для определения ванадия. Действительное присутствие хрома или урана может быть впоследствии проверено после отделения фосфора, но лучше это делать непосредственно, определяя эти элементы из отдельной большой навески (стр. 596). Если окраска раствора слишком интенсивна для колориметрического определения хрома, то кипятят щелочной раствор, пока вся перекись водорода не разложится, подкисляют серной
Разделение элементов в осадке от аммиака
121
кислотой, определяют хром титрованием сульфатом железа (II) и перманганатом (стр. 592). Раствор сохраняют для определения ванадия. Этот метод определения хрома применим в присутствии урана. Маленькая ошибка происходит, если раствор был профильтрован через бумажный фильтр, потому что извлеченное при этом небольшое количество органических веществ восстанавливает при титровании перманганат, вследствие чего определение дает несколько пониженное по сравнению с истин-* ным содержание Сг2О3. Эта потеря не превосходит 0,2—0,5 .мг Сг2О3, и ее можно избежать, если фильтровать щелочной раствор через очищенный асбест.
Ванадий. Раствор, служивший для определения хрома, подкисляют серной кислотой и определяют содержание ванадия восстановлением сернистым ангидридом и титрованием перманганатом (стр. 513), если раствор не содержит железа, или восстановлением сульфатом железа (II), окислением избытка последнего персульфатом и титрованием перманганатом (стр. 514), если железо присутствует. Раствор после определения ванадия сохраняют.
Фосфор. Раствор после определения ванадия -подщелачивают аммиаком, прибавляют избыток его в 10 мл и подкисляют азотной кислотой, вводя избыток этой кислоты в 5 мл. Если предыдущее определение показало присутствие ванадия, прибавляют сульфат железа (II) в достаточном количестве, чтобы восстановить ванадий до четырехвалентного, и осаждают фосфор при комнатной температуре молибденовым реактивом (стр. 785). При отсутствии ванадия сульфат железа (II) не прибавляют, а нагревают раствор до 50° С и осаждают молибденовым реактивом. В обоих случаях раствор оставляют стоять в течение ночи, после чего-его фильтруют и определяют фосфор в виде пирофосфата магния Mg2P2O7 (стр. 785). Фильтрат и промывные воды после промывания фосфоромолиб-дата сохраняют.
Алюминий. Определение алюминия имеет смысл только при одновременном проведении через все стадии анализа двух или более «холостых» опытов. Если это условие выполнено, то алюминий, выделенный в следующей операции, переводят в А12О3 и в массу последнего вводят поправку на среднюю величину результатов, полученных при проведении «холостых» опытов. Фильтрат от фосфоромолибдата нагревают до кипения, осторожно прибавляют в небольшом избытке разбавленный аммиак, кипятят 2 мин, фильтруют и промывают осадок 2 или 3 раза водой. Затем растворяют осадок в горячей разбавленной соляной кислоте и осаждение повторяют. Переосажденный осадок снова растворяют и осаждают алюминий в виде хлорида из зфирно-солянокислого раствора (стр. 117); если нужно, определяют в осадке А12О3. Фильтрат и промывные воды сохраняют.
Бериллий. Эфирно-солянокислый фильтрат осторожно нагревают на краю паровой бани, пока не улетучится эфир, прибавляют несколько капель азотной кислоты и отделяют бериллий (и галлий) от небольших количеств железа, хрома и следов марганца осаждением последних едким натром (стр. 109). Фильтруют, фильтрат подкисляют, кипятят и прибавляют аммиак в небольшом избытке. Если появится осадок, его отфильтровывают и исследуют на названные выше элементы по методам, указанным в соответствующих главах. Нужно помнить, что осадок вместе-с бериллием будет содержать и ванадий, если этот элемент присутствовал
122
Гл. IV. .Общие методы, и операции химического анализа
и остался в растворе при осаждении фосфора, и что в осадке может также оказаться уран, если он присутствовал в исследуемом материале вместе с ванадием.
Анализ осадка от едкого натра. Осадок от едкого натра обрабатывают так, чтобы получить 100 мл сернокислого раствора, содержащего 4 мл-свободной серной кислоты. Осаждают сероводородом, фильтруют и осадок промывают (стр. 85). Фильтрат сохраняют.
Осадок обычно состоит из сульфида платины и серй, появившихся в результате проведения сплавления в платиновом тигле и восстановления железа (III) сероводородом. Исследование осадка на содержание в нем титана, ниобия или тантала, которые могли осесть вследствие гидролиза, выполнить нетрудно, и это следует сделать, если вид осадка вызывает сомнение.
Элементы сероводородной группы здесь не рассматриваются, так как предполагается, что все они были удалены раньше.
Железо. К сохраненному фильтрату прибавляют винную кислоту (свободную от железа) в количестве, достаточном для того, чтобы удержать все основания в растворе (четырехкратное количество от массы основных окислов), затем аммиак,.добавляя избыток его в 5 мл, и в течение 10 мин пропускают сероводород. Осадку дают осесть, поставив стакан на край паровой бани, после .чего его отфильтровывают и промывают холодной водой, содержащей хлорид аммония, бесцветный сульфид аммония и немного тартрата аммония, не давая жидкости стекать полностью с осадка, чтобы не окислилось железо. Фильтрат, который при стоянии в течение нескольких часов не должен выделять нового осадка сульфидов, сохраняют. Обработку осадка сульфидов и определение железа продолжают, как описано на стр. 115.
В обычном случае осадок содержит все железо в виде, сульфида и очень мало других веществ. Если осаждение сделано правильно, то осадок будет содержать также весь цинк и кобальт, практически весь никель и большее или меньшее количество марганца, увлеченного в осадок при осаждении аммиаком.
Титан. Фильтрат от сульфидов подкисляют соляной кислотой в стакане емкостью 400 мл, выпаривают до небольшого объема, накрывают стакан стеклом и разрушают аммонийные соли нагреванием с царской водкой. Затем разрушают всю винную кислоту и другие органические вещества, для чего прибавляют серную кислоту (если после разложения сульфата аммония ее не осталось от 5 до 10 мл), нагревают до выделения паров серной кислоты и осторожно приливают азотную кислоту (лучше дымящую), вводя ее время от времени маленькими порциями через носик стакана. Если органическое вещество устойчиво, последние следы его могут быть разрушены осторожным прибавлением нескольких капель концентрированного раствора перманганата. Наконец выпариванием удаляют всю азотную кислоту. Охлаждают, разбавляют до определенного 1 объема в мерной колбе емкостью 50 мл или больше, соответственно количеству присутствующего титана, и определяют его колориметрически (стр. 655).'Раствор сохраняют. '
Цирконий. Раствор от предыдущего определения выпаривают .до 50 мл, охлаждают, прибавляют 1—2 мл перекиси водорода (30%-ной), доводят концентрацию серной кислоты До 10% (по объему), прибавляют 1 мл сиропообразной фосфорной кислоты, оставляют раствор стоять.
•Фильтрование * 123
в течение нескольких часов при 40° С и определяют цирконий в виде ZrP2O7 (стр. 642). Фильтрат сохраняют.
Редкоземельные металлы. К фильтрату от циркония прибавляют приблизительно 0,05 г Fe2O3 в виде сульфата железа (III), нагревают раствор до кипения и прибавляют едкий натр, вводя его избыток в 1—2%. Кипятят, чтобы разложить перекись водорода, фильтруют и промывают осадок горячей водой. Затем растворяют осадок в фтористоводородной кислоте, отделяют нечистые фториды редкоземельных металлов и определяют их (стр. 621).
ФИЛЬТРОВАНИЕ
Фильтрование играет настолько важную роль\в анализе, что продуктивность и качество работы аналитика в значительной степени зависят от избранных им способов фильтрования и возможностей для их осуществления. Чаще всего фильтрование проводится через бумажный фильтр при атмосферном давлении. При фильтровании может быть потеряно много времени, если воронка сделана неправильно, фильтры плохого качества или вложены неверно; кроме того, последующее промывание осадка тогда будет неполным, а затянувшееся проведение операции может привести к изменениям в составе осадка, например к окислению сульфида меди до растворимого сульфата меди.
Воронки
Хорошие воронки должны иметь правильный угол в 60° ±1°, не должны иметь сужений или расширений у шейки, трубка воронки должна •быть длиной не более 15 см. Фильтрование идет много быстрее, если воронки сделаны ребристыми; для этого внутреннюю поверхность воронки покрывают парафинбм, прорезывают в нем 10—12 линий длиной в две трети расстояния от вершины конуса воронки до края и затем травят на небольшую глубину плавиковой кислотой?
\
Фильтровальная бумага
Фильтровальную бумагу следует выбирать, согласуясь с характером осадка и предполагаемой дальнейшей обработкой осадка и фильтрата. Создавать большой запас фильтровальной бумаги с неизвестным содержанием золы не имеет смысла, если только в лаборатории не проводится такое большое число массовых фильтрований, при которых запас найдет себе скорое применение. При работе нужно иметь в виду возможное загрязнение анализируемых растворов, например извлечение железа из фильтровальной бумаги в фильтрат, в котором затем надо будет определять очень малые количества железа, как это требуется, например, при анализе белого песка, идущего на производство стекла.
Фильтровальная бумага, обработанная соляной и плавиковой кислотами, прочна, содержит небольшое количество золы и имеет однородную структуру во всей массе. Необходимо иметь под рукой фильтровальную бумагу по крайней мере трех степеней плотности: 1) для очень тонких осадков^ как сульфат бария или метаоловянная кислота; 2) для жела
124 >Гл. IV. Общие методы и операции химического анализа:
тинозных осадков, вроде гидроокиси алюминия, и 3) для обычных осадков, к которым относится, например, большая часть сульфидов металлов.
Диаметр фильтров может колебаться от 5,5 до 15 см; чаще всего применяются фильтры диаметром 9 см. Если прокаленный осадок вместе с золой фильтра будет взвешиваться, то размер фильтра следует сообразовать с величиной осадка, а не с объемом жидкости. Весь осадок не должен занимать более чем одну треть вместимости фильтра. Край фильтра, вложенного в воронку, должен быть на 5—15 мм ниже края последней.
Правильно вложенный фильтр должен плотно прилегать вверху и не должен касаться стекла во всей нижней половине. Это не может быть достигнуто складыванием фильтра в форме четверти круга, как это обычно предписывается. Лучше поступать следующим образом. Осторожно складывают бумагу вдоль диаметра и затем сгибают полученный полукруг таким образом,_ чтобы сложенные края образовали между собой угол в 3—4°, а края полукруга совпадали, образуя непрерывную ду?у. Осторожно прижимают кончик и затем от сектора, который находится сверху, отрывают кусочек приблизительно на одну треть, так чтобы край фильтра у места обрыва был скошен. Раскрывают фильтр, придерживая три сектора вместе между указательным и большим пальцами у оборванного места, прижимают бумагу к воронке осторожно указательным пальцем и смотрят, правильно ли входит фильтр в воронку, касаясь ее только по краям, но не у дна. Если фильтр оказывается подогнанным неудовлетворительно, его исправляют, изменяя угол внутренней складки. Наконец, тщательно смачивают водой внутреннюю часть воронки и быстро прижимают край фильтра к стеклу так, чтобы не оставалось пузырьков воздуха и каналов. Если фильтр подогнан правильно, то вода будет быстро проходить через него и трубка воронки будет всегда заполнена х.
Бумажная масса
Применение массы из фильтровальной бумаги 1 2 очень помогает фильтрованию таких студенистых осадков, как гидроокись алюминия, или очень тонких слизистых оиадков, как сульфид олова (IV). Кроме того, применение бумажной массы имеет еще и то преимущество, что осадок после прокаливания оказывается тонко разрыхленным, вместо того чтобы превратиться в сплошной комок, в результате чего окисление происходит быстрее, а время, требующееся для последующего растворения, значительно сокращается.
Бумажная масса должна оставлять после сжигания очень мало золы, если осадок подлежит взвешиванию, и прибавленное ее количество должно быть известно, если масса золы может оказать влияние на результат анализа. Массу можно приготовить сильным встряхиванием фильтровальной бумаги в закрытой пробкой конической колбе с достаточным количеством воды до получения однородной массы консистенции сметаны. Для приготовления бумажной массы можно употреблять пульте фильтры, обрезки фильтров или таблетки фильтровальной бумаги, которые продаются специально для этой цели. Бумажную массу следует прибавлять после
1 В литературе описаны некоторые специальные методы складывания фильтровальной бумаги для быстрого фильтрования; см., например, Р. Е. С 1 а г k, Chemist Analyst, 28, (i), 18 (1939).
2 М. Dittrich, Anleitung zur Gesteinsanalyse, 1905, s. 10, 11, 14; Ber., 37, 1840 (1904).
•Фильтрование
125
образования осадка и затем хорошо перемешивать с ним. Если бумажную массу прибавлять до осаждения, то некоторое количество осадка образуется в самих волокнах бумаги и это может сделать их несгораемыми. Если нежелательно разбавлять раствор водой, прибавляя к нему бумажную массу, то ее можно перед прибавлением отжать или дать воде стечь с нее на фильтр, затем приступить к фильтрованию анализируемого раствора и смешивать бумажную массу с осадком в воронке в процессе фильтрования.
Применяя бумажную массу, следует иметь в виду, что из нее может быть извлечено некоторое количество органических веществ, которые могут повлиять на результаты анализа. Влияние этих веществ, вероятно, будет меньше после того, как осадок образовался. Это является второй причиной, почему бумажную массу следует прибавлять всегда после осаждения. Если присутствие в растворе органических веществ имеет значение, как например, при осаждении гидроокиси алюминия, то бумажную массу не следует прибавлять в тех случаях, когда осадок будет растворяться в сильной кислоте для последующего переосаждения.
Асбест
Асбест при выполнении аналитических разделений применяется очень часто. Поэтому выбор соответствующей разновидности его имее?*важное значение. К сожалению, не существует стандартного сорта асбеста, и аналитик должен Выбирать его по собственному усмотрению и затем судить, является ли купленный материал подходящим для его целей. Гуч 1 подробно описывает «белый, шелковистый, безводный асбест». Применяемый для аналитический целей асбест должен быть кальциево-маг-незиальным силикатом амфиболовой разновидности, не содержащим мелких включений магнезита и железа (II), которое может замещать кальций или магний в силикате. Такой силикат безводен и нерастворим в кислотах. Хризотиловая или серпентиновая разновидности асбеста не годятся, потому что они заметно растворимы в кислотах и при прокаливании отдают воду, теряя свое волокнистое строение.
Некоторые аналитики предпочитают приготовлять асбест для фильтрования самостоятельно. Для этого совершенно белый волокнистый асбест скоблят ножом или растирают на латунном сите до получения тонкого короткого пуха и затем промывают декантацией до тех пор, пока не будут удалены пыль и слишком мелкие частицы, которые могли бы забить приготовленный фильтр. Затем асбест обрабатывают горячей разбавленной (1 : 5) соляной кислотой; раствор декантируют и операцию повторяют, пока не будет удалено растворимое железо; наконец волокна промывают водой до полного удаления хлоридов. При использовании асбеста для некоторых специальных целей его надо подвергнуть еще дальнейшей обработке. Например, если предполагают применять его при определении марганца висмутатным методом, то его нужно нагревать 1—2 ч с горячей разбавленной (1 : 2) азотной кислотой после удаления хлоридов, чтобы окислить железо (II) и иметь уверенность в полноте удаления хлоридов. Если асбест будет применяться при сожжении углерода, его следует высушить, осторожно прокалить в токе кислорода и снова смешать с водой 2.
Если асбестовый фильтр должен взвешиваться, то его обычно приготовляют на донышке фарфорового или платинового тигля Гуча. Реже
1 F. A. Gooch, Proc. Am. Acad. Arts and Sci., 13, 342 (1878). В другой статье [там же, 12, 390 (1884—1885)] Гуч рекомендует в качестве материала для приготовления фильтрующего слоя антрацен и фенантрен.
2 В статье В. L. К a s р i n [Ind. Eng. Chem., Anal. Ed., 12, 517 (1940)] описывается метод разделения асбеста на волокна током сжатого воздуха.
Гл. IV. Общие методы и операции химического анализа
Рис. 11. Колокол для фильтрования под вакуумом.
126 • » :—-
употребляют специальный тигель \ у которого дно вырезано, а на оставленных закраинах удерживается дырчатый фарфоровый или платиновый диск. После использования такого тигля очень легко вытолкнуть-диск вместе с его асбестовой прокладкой и осадком.
Все эти.тйгли применяются для фильтрования под давлением (с отсасыванием), и к ним требуются особые держатели для тиглей и Приемные склянки, достаточно прочные, чтобы выдержать частичное разреже-вие внутри. Такими, держателями могут быть стеклянные трубки или воронки с резиновым кольцом или пробкой; они должны удовлетворять следующим условием: 1) обеспечивать очень.
• плотное соединение посредством резины во время отсасывания; 2) удерживать тигель на достаточной глубине, чтобы стекающая жидкость не касалась резины; 3) отводить 4иЛьтРат достаточно глубоко в приемный сосуд, чтобы он не мог быть- , втянут в насос. Приемные склянки должны быть снабжены отверстием для держателя тигля и отводом для соединения с насосом.
Очень удобно применение колокола, установленного на пришлифованной стеклянной пластинке вместо дна и имеющего вверху и сбоку тубусы; применяя его, можно собирать фильтрат непосредственно в тот сосуд, в котором он будет дальше обрабатываться (рис. 11). Хорошо иметь у насоса двухходовой кран, чтобы можно было легко прекращать отсасывание. Между приемной склянкой- и насосом следует поместить предохранительную склянку для защиты фильтрата от обратного тока воды при работе с водоструйным предотвращения случайных закупорок линии или
насосом или же для порчи масляного насоса, если прибор соединен с центральной вакуумной сетью.
Для приготовления фильтрующего слоя Лендель пользовался водными суспензиями асбестового волокна трех размеров: крупного, среднего и мелкого. Сначала в тигель вливают суспензию крупного волокна и, не отсасывая, ждут, пока не стечет вся вода; затем йри слабом отсасывании вливают суспензию среднего волокна и придавливают, особенно по краям, расплющенным концом стеклянной палочки. Йаконец, при , более сильном отсасывании кладут верхний слой из тонкого асбеста. Затем фильтр промывают при самом полном отсасывании дистиллированной водой или тем промывным раствором, который'будет применяться в анализе, если установлено, что, таким способом можно- удалить вещества, не удаленные при приготовлении асбеста. Промывание продолжают до тех пор, пока волокна не перестанут проходить в фильтрат. Тигель, приготовленный таким способом, прокаливают при той же температуре, какая будет , применена при анализе, охлаждают в эксикаторе и взвешивают 2.
1 G. С. Gal dw ell, J. Am. Chem. Soc,, 13, 105 (1891).
2 Тигель co сдоем асбеста должен иметь в вовдушно-сухом состоянии практически-, ту же самую массу, , какую он имеет после прокаливания при темно-красном калении-Например, тигель со слоем асбеста, весивший в-воздушно-сухом состоянии 15,2160 после прокаливания его в радиаторе приблизительно при 700° С весил 15,2158 г~
Фильтрование 127
Правильно приготовленный слой должен быть не толще 2—3 мм-и должен весить не более'50—100 мг. Чтобы предупредить разрыхление слоя, тигель следует всегда вставлять в держатель при слабом отсасывании и фильтруемый раствор нужно всегда лить по палочке, держа последнюю у внутренней стенки тигля. Некоторые аналитики предпочитают помещать поверх асбеста дырчатый диск, чтобы предупредить взрыхле-' ние слоя, Платиновые тигли Гуча снабжены колпачками, надеваемыми на дно тигля, так что их можно прокаливать на голом пламени, не опасаясь смещения .частиц асбеста или осадка газами горелки, которые иначе проходили бы через фильтрующий слой. Тигли с незащищенным, дном следует прокаливать в муфеле или радиаторе (стр. 47) Ч
Вата из кремнекислоты
Имеются указания 1 2 3 на то, что замена асбеста в тиглях Гуча ватой из кремнекислоты (диаметр волокон порядка 3 • 10-3 мм) дает преимущества не только перед обычными тиглями Гуча с асбестом, но и перед стеклянными фильтрующими тиглями с пластинками из плавленого пористого стекла.
Вата из кремнекислоты устойчива.к действию химических реагентов, не гигроскопична, и ее можно нагревать до 600° С. Для получения слоя такой ваты; хорошо задерживающего частицы осадка, в тигле емкостью* 20 мл достаточно поместить 50 мг ее.
Платина
Тигли Гуча с губчатой платиной вместо асбеста называются тиглями1 Мунро ®. Главное преимущество таких тиглей в их стойкости по отношению к химическим воздействиям. Правильно приготовленный слой платины допускает быстрое фильтрование, удерживает тончайший осадки и служит неопределенно долгое время, потому что осадки можно удалять любой обработкой, которая не приводит к растворению платины. Чтобы получить фильтрующий слой,-обрабатывают концентрированный раствор платинохлористоводородной кислоты хлоридом аммония, добавляя его в небольшом избытке, и промывают осадок несколько раз водой, а потом 80%-ным спиртом. Затем помещают платиновый тигель Гуча на несколько-листов фильтровальной бумаги й переносят в него такое количество полученного осадка хлороплатината аммония, чтобы после поглощения спирта бумагой образовался ровный слой толщиной 5 .юи. Тигель осторожно нагревают, пока спирт не испарится, надевают на дно тигля колпачок, постепенно повышают температуру до разложения хлороплатината и потом прокаливают при темно-красном калении. Тигель охлаждают, слегка придавливают слой платины расплющенным концом стеклянной палочди и повторяют весь процесс снова, если окажется, что слой слиш-
1 В статье W. М. Т horn t он и J. G. Smith, [Ind. Eng. Chem., Anal. Ed., 9, 95 (1937)1 приводится описание регулируемого держателя для тиглей Гуча, который; поддерживает во время прокаливания и тигель и колпачок на нем.
2 W. W. R u s s е 11, J. Н. А. Н а г 1 е у, Ind. Eng. Chem., Anal. Ed., 11, 168-(1939).
3 С. E. M u n г о e, J. Anal. Appl. Chem., 2, 241 (1888); Chem. News, 58, 101 (1888); H. N e u h a u e r, Z. anal. Chem., 39, 501 (1900); T. W. R i c h a r d s, A, Staebler. J. Am. Chem. Soc., 29, 633 (1907); W. O. S n e 11 i n g, там же, 31, 456 (1909).
128 Гл- IV- Общие методы и операции химического анализа
ком тонок или местами ненадежен. Наконец тщательно протирают палочкой всю поверхность слоя, пока она не отполируется. Перед фильтрованием растворов, содержащих соляную кислоту, тигель надо сначала промыть кипящей водой для удаления кислорода, иначе некоторое количество цлатины может раствориться.
Алунд, фарфор и стекло
Такие фильтрующие материалы, как алунд и неглазурованный фарфор, применяются преимущественно в тех случаях, когда нужен только фильтрат х.
В продаже имеются фильтрующие тигли, бюхнеровские воронки и экстракционные аппараты из стекла, в которые впаяны стеклянные фильтрующие пластинки 1 2 из измельченного и потом сплавленного стеклянного порошка. Эти пластинки имеют различную пористость (100—120, 40—50, 20—30 и 5—10 мк, *что отвечает соответственно № 1, 2, 3 и 4*). Если такие тигли подлежат взвешиванию вместе с осадком, аналитик должен сначала продумать, не произойдет ли како-либо изменение в массе фильтрующей пластинки в результате той обработки, которой она должна подвергнуться. Стеклянные фильтрующие тигли следует нагревать только в муфеле, постепенно повышая температуру, но не выше 600° С. Охлаждать их надо также постепенно, не вынимая из муфеля, чтобы избежать деформации стекла. Такого же рода кварцевые тигли с кварцевыми фильтрующими пластинками можно нагревать сильнее, и они не боятся резких изменений температуры.
Следует сказать несколько слов о перенесении на фильтр последних частиц осадка из сосуда, в котором происходило осаждение. Обычно это делают при помощи стеклянной палочки с кусочком резиновой трубки на конце. Это допустимо при выполнении рядовых анализов, если применяемая резиновая трубка сделана из чистого каучука или не содержит веществ, которые могли бы помешать анализу (например, серы и сурьмы в тех случаях, когда определяют содержание этих элементов, или неорганических наполнителей, которые будут взвешены с осадком после прокаливания, если частицы резины оторвутся и попадут в осадок). При выполнении очень точной работы вполне удовлетворительные результаты дает применение птичьего пера с подстриженной бородкой.
ПРОМЫВАНИЕ ОСАДКОВ
• Существует только два общих правила, которыми следует руководствоваться при промывании осадков: во-первых, промывная жидкость, там где это возможно, должна содержать одноименные с осадком ионы и, во-йторых, объем промывной жидкости не должен превышать некото-
1 Некоторые кристаллические овадки можно переносить в фильтрующие тигли на слой стеклянной ваты или асбеста и затем взвешивать их без предварительного прокаливания. В этих случаях вполне удовлетворительных результатов можно достичь, заканчивая промывание осадка пропусканием через него подходящей летучей жидкости, например спирта или эфира, и взвешиванием тигля после высушивания его под уменьшенным давлением или пропусканием через него сухого воздуха при комнатной температуре в течение 5—15 мин. Пустые тигли перед их взвешиванием надо, ронечно, обрабатывать ' подобным же способом.
2 L. М-о set, W. Маху mowic z, Chem. Ztg., 48, 693 (1924); G. F. Hii t-t i g, M. Nette. Z. anal. Chem., 65, 385 (1925).
Промывание осадков
129
рого наименьшего количества, необходимого для удаления посторонних веществ, так как ни один осадок нельзя считать абсолютно нерастворимым.
Совершенно справедливо также и то, особенно если осадок несколько растворим, что многократное промывание маленькими порциями промывного раствора, которому дают каждый раз полностью стекать, лучше, чем промывание одной или двумя большими порциями или же прибавлением свежей промывной жидкости, пока предыдущая порция еще имеется на фильтре. Во всех случаях никогда не следует наполнять фильтр осадком и даже раствором до верху, если осадок имеет тенденцию всползать на стенки воронки. Осадок в фильтре должен занимать не более */з—его объема, а уровень раствора при наполненном фильтре должен быть на 5—10 мм ниже его края. В последней стадии промывания струю из промывалки направляют вдоль края фильтра и затем спирально вниз, пока осадок не соберется в глубине фильтра. При отфильтровывании выползающих осадков фильтр должен быть пригнан особенно тщательно.
Количество промывного раствора, необходимое для промывания, зависит от природы осадка. Студенистые осадки, вроде гидроокиси алюминия, требуют более длительного промывания, чем кристаллические, как оксалат кальция; мелкокристаллические осадки приходится промывать более тщательно, чем крупнокристаллические.
Часто имеет значение выбор промывной жидкости. Как общее правило, применять чистую воду не следует, если нет уверенности, что она не растворит заметного количества осадка, не разложит его и не переведет в коллоидный раствор. Если осадок заметно растворим в воде, то обычно прибавляют соль, содержащую одноименные ионы, по крайней мере к тем порциям промывной жидкости, которые применяются для переноса осадка на фильтр, и в начале промывания. Например, если отфильтровывают оксалат кальция, который должен быть потом прокален, промывание может быть до конца сделано разбавленным раствором оксалата аммония; если же оксалат кальция предполагают титровать перманганатом, то сначала отмывают посторонние вещества тем же раствором оксалата аммония, а затем промывают осадок таким количеством холодной воды, которое достаточно для удаления оксалата аммония.
Если осадок имеет тенденцию переходить в коллоидный раствор по отмывании большей части примесей, то нужно применять промывную жидкость, содержащую электролиты. В этом случае обычно выбирают летучий электролит, удаление которого путем прокаливания не вызывает затруднений. Так, раствор хлорида аммония вполне пригоден для промывания осадка гидроокисей алюминия и железа, но когда он применяется для промывания гидроокисей олова или германия, возникают затруднения вследствие образования летучих хлоридов этих элементов. Некоторые осадки склонны окисляться во время промывания. В таких случаях нельзя давать жидкости стекать полностью с осадка, и нужно подобрать специальный корректирующий промывной раствор, который возвращал бы окислившееся соединение в первоначальное состояние. Так, например, при промывании сульфида меди применяют воду, насыщенную сероводородом.
При выполнении различных разделений хорошим правилом является: проводить промывание раствором такого же состава, каким было сделано осаждение. Таким образом, если железо было отделено от титана,
9 Заказ 522.
130
Гл. IV. Общие методы и операции химического анализа
алюминия, циркония, тантала и т, п. осаждением сульфидом аммония в присутствии хлорида аммония и тартрата аммония, желательно, чтобы промывание осадка проводилось раствором, содержащим все эти три соли, по крайней мере пока не пройдет надобность в тартрате; затем,, если нужно, его можно исключить из промывной жидкости и окончить промывание раствором, содержащим только хлорид и сульфид. Подобным же образом, если разделение зависит от присутствия в растворе избытка едкого натра, как в случае отделения железа от алюминия, промывной раствор должен содержать некоторое количество едкого натра с добавлением какого-нибудь электролита, например сульфата натрия.
Наилучший способ промывания осадка и количество промывного-раствора, необходимое для удаления мешающих веществ, должны быть установлены в каждом отдельном случае. Если осадок быстро закупоривает поры фильтра или если его нужно держать покрытым раствором (так как иначе он или окисляется или образуются каналы при полном стекании с него жидкости), то промывать надо декантацией, насколько только это возможно. При замедлении фильтрования или невозможности давать жидкости стечь перед наливанием новой порции промывного раствора отмывание примесей происходит медленно. Иногда нет необходимости переводить осадок на фильтр, например можно оставлять в стакане остаток после сплавления цо Лоуренсу Смиту или фосфоромолибдат аммония, если его потом растворяют для алкалиметрического определения фосфора. В таких случаях чем меньше осадка попало на фильтр, тем лучше.' При всяком декантировании следует учитывать возможность применения центрифуги.
Испытать на полноту промывания нужно тогда, когда имеются основания полагать, что большая часть примесей отмыта. Для этого собирают маленькие порции вытекающей из воронки жидкости и испытывают их на присутствие нежелательных примесей. Некоторые осадки имеют склонность разлагаться по удалении избытка осадителя. Если ионы осадителя при этом освобождаются и снова переходят в раствор, то нельзя делать испытание на эти ионы для проверки полноты промывания, а надо проверять полноту удаления других ионов, которые присутствовали с самого начала. Так, при промывании осадка фосфата алюминия или фосфата циркония, осажденных в присутствии хлоридов, можно считать, что избыток фосфат-ионов отмыт, если проба на хлориды дает отрицательный результат.
Данными о растворимости осадков можно пользоваться при их промывании лишь очень ограниченно, потому что во время первой части промывания остается достаточное количество первоначально присутствовавших веществ, чтобы они могли повлиять на растворяющую способность промывного раствора; кроме того, раствор никогда не остается в соприкосновении с осадком достаточно долго, чтобы могло наступить равновесие х.
1 Заканчивая обсуждение вопроса о промывании, следует отметить, что даже такая кажущаяся очень простой операция, как промывание стеклянной посуды, может оказаться более сложной, чем это предполагают. Е. Р. L a u g [Ind. Eng. Chem., Anal. Ed., 6, 111 (1934)] утверждает, например, что около 3-икг хромат-ионов может все еще оставаться на внутренней поверхности стеклянного стакана, который для очистки обработали смесью хромовой и серной кислот и затем тщательно прополоскали 10 раз отдельными порциями воды. ,
При промывании бумажных фильтров вымыть из них ионы хрома (VI) труднее,, яем ионы хрома (III). -
Высушивание и прокаливание осадков
131
ВЫСУШИВАНИЕ И ПРОКАЛИВАНИЕ ОСАДКОВ
После того как осадок собран на фильтре и промыт, он должен быть переведен в соединение постоянного состава, в виде которого его взвешивают. Не существует метода обработки, одинаково применимого ко всем осадкам. Так, способы обработки могут изменяться от осторожного высу-простого прокаливания на
шивания до прокаливания при 12UU I и от воздухе до прокаливания в атмосфере водорода или азота. Осадки, подлежащие высушиванию при низких температурах, следует отфильтровывать через тигли Гуча, Мунро или тигли с пористой стеклянной пластинкой (стр. 128), но ни в коем случае не через бумажные фильтры. Высушивание и прокаливание осадков вообще лучше проводить в электрических шкафах или муфельных печах; снабженных термометрами или пирометрами и терморегуляторами.
Осадки, которые восстанавливаются при соприкосновении с обугленной бумагой, меняют свою массу не только вследствие восстановления (как, например, сульфат свинца, переходящий в сульфид свинца), но и вследствие улетучивания (например, восстановление арсенатов до летучего мышьяка). Дальнейшая опасность состоит в том, что продукт восстановления может соединиться с материалом сосуда, в котором проводится прокаливание (мышьяк, фосфор, свинец и т. п. элементы соединяются с платиной), вызывая таким образом не только потерю вещества,
Рис. 12. Высушивание влажных фильтров с осадками.
но и повреждение или даже разрушение тигля. Никакого вреда- не будет, если продукт восстановления нелетуч, не действует на тигель и снова окисляется после выгорания бумаги при длительном прокалицании на воздухе или в атмосфере кислорода (например, тонко измельченная окись железа, какой она получается при прокаливании гидроокиси железа, смешанной с бумажной массой). Восстанавливающиеся осадки, которые не относятся к.этой категории, лучше всего собирать в тиглях Гуча и т. п.
Если осадок может быть безопасно сожжен в соприкосновении с бумагой, его лучше всего не сушить в воронке, а сложить влажный фильтр вместе с его содержимым и высушить в тигле (рис. 12).
Если небольшое пламя, хорошо защищенное от движения воздуха, помещено, как показано на рис. 12, высушивание осадка происходит быстро и без риска потери вещества. Когда высушивание закончится, пламя немного увеличивают, так чтобы шла медленная сухая перегонка бумаги, пока от последней не останется лишь черный уголь. Не следует допускать воспламенения бумаги, так как это приводит к механическому распадению тонких частиц осадка. Если случайно-бумага воспламенится, то пламя гасят, быстро закрывая тигель крышкой, и затем продолжают обугливание. Когда пары перестанут выделяться, • придвигают пламя ко дну тигля и дают углю медленно выгореть, увеличивая пламя по мере 9*
132
Гл. IV. Общие методы и операции химического анализа
надобности. Только после того как уголь полностью сгорит, можно закрыть тигель и увеличить пламя до максимума, но никогда не следует помещать тигель так низко, чтобы он касался внутреннего темного конуса пламени горелки Бунзена. Если это произойдет, тигель испортится вследствие образования карбида платины. В пламени горелки Мекера тигель не должен касаться зеленой зоны, находящейся непосредственно над отверстием горелки Е При работе с фарфоровыми тиглями приемы в общем остаются теми же, что и при работе с платиновыми.
С тиглями типа Гуча, как платиновыми, так и фарфоровыми, работа идет значительно быстрее. Высушивание может быть сделано, как и при использовании обыкновенного тигля, или же каким-либо другим удобным способом, не сопряженным с риском потери, но при этом даже по окончании высушивания нагревание для прокаливания осадка должно быть постепенным. При работе с этими тиглями, в особенности при прокаливании восстанавливающихся веществ, следует избегать проникновения в тигель газов пламени через отверстия в дне тигля. До некоторой степени это достигается надеванием колпачка на дно тигля, но лучше помещать тигель внутри радиатора (стр. 47), а самое лучшее — прокаливать его в муфеле. В общем фильтрующие тигли не так удобны для прокаливания при высоких температурах на горелках, как тигли обычного типа, потому что колпачок понижает температуру у дна тигля.
Платиновые тигли значительно лучше для прокаливания при высоких температурах на пламени, чем фарфоровые, потому что в них можно получить более высокую температуру. При нагревании в муфеле это преимущество исчезает, и поэтому прокаливание при высоких температурах фарфоровых и кварцевых тиглей следует проводить в муфеле.
Нужно всегда иметь в виду, что при нагревании платины выше 1000° С может произойти постепенная потеря ее массы. Потеря эта возрастает с повышением температуры и происходит в результате испарения металла, в частности иридия, входящего в состав' сплава, применяемого для изготовления тиглей. Содержание иридия различно в разных изделиях, а потому каждый аналитик должен сам определить потерю в массе своих тиглей при прокаливании. При необходимости длительного прокаливания на пламени паяльной горелки веществ, масса которых должна быть определена, скорость уменьшения массы самого тигля должна быть заранее определена, чтобы можно было ввести поправку 1 2. Если это не было сделано, то следует определить массу тигля после осторожного удаления из него прокаленного осадка.
При прокаливании на пламени горелки должно быть принято во внимание еще то обстоятельство, что когда пламя окружает тигель полностью, атмосфера внутри тигля представляет собой не воздух, но обычно
1 J. D. М. S m i t h [Trans. J. Soc. Chem. Ind., 44, 539 (1925)] советует применять особое приспособление — зубчатую крышку, которая целиком входит внутрь тигля; край крышки имеет зубцы, вследствие чего крышка позволяет свободно выходить выделяющимся парам, но предупреждает всползание осадка, а также потери от разбрызгивания или растрескивания. Тигель лучше всего применять низкий и широкий, а крышку — из прозрачного кварца. Для прокаливания гигроскопичных осадков можно изготовить вторую крышку, плотно прилегающую снаружи; ее надевают в конце прокаливания, с нею тигель охлаждают и взвешивают.
2 Доказательства этого см. в статье W. F. Hillebrand, J. Am. Chem. Soc., 24, 373 (1902).
Высушивание .и прокаливание осадков
133
смесь азота и продуктов сгорания газа, среди которых главной составной частью является водяной пар. Поэтому в некоторых случаях приходится этого избегать, прокаливая на наклонном пламени паяльной горелки, направленном только на нижнюю часть тигля, или вставляя тигель в отверстие, проделанное в асбестовой или платиновой пластинке. Основное действие водяного пара заключается в том,4 что в его присутствии необходима более высокая температура для полного обезвоживания некоторых минералов, и полное обезвоживание, вероятно, не достигается. Этим можно объяснить, почему в некоторых руководствах рекомендуется очень длительное прокаливание для превращения оксалата кальция или карбоната кальция в окись кальция, между тем как в надлежащих условиях это превращение может быть выполнено в несколько минут. Другая ошибка заключается в том, что допускают пламя целиком охватывать закрытый тигель при прокаливании восстанавливающихся веществ, вроде SnO2. Ошибка в анализе из-за присутствия в пламени не вполне сгоревших газов получается даже в том случае, когда вид пламени указывает на полное'сгорание.
Наконец, обязанностью каждого аналитика является испытание эффективности действия применяемых им горелок и муфелей. Расхождение многих результатов исследований может быть приписано разнице в температуре прокаливания, примененной различными авторами.
Указать точные температуры нагрева тиглей в пламени разных горелок невозможно вследствие различного типа этих горелок и местных различий в составе применяемого газа и его давлении. Тем не менее желательно, чтобы аналитик имел хотя бы некоторое представление о получаемых температурах. Приведенные ниже температуры прокаливания были получены при использовании карбюрированного водяного газа, имевшего теплотворную способность 600 британских тепловых единиц на 1 куб. фут (*5340 ккал на 1 л3*). Во всех случаях термоэлектрическая пара помещалась внутри накаливаемого тигля у его дна и испытанию подвергалось несколько горелок каждого типа. Под термином «красное каление» обычно понимается температура, близкая к 600 °C.
Горелки Тигли Температура °C
Бунзена Закрытый платиновый 900-1050
Тирриля То же . 1050—1150
Мекера » 1150—1250
Паяльная .1100—1300
Тирриля Закрытый фарфоровый в радиаторе 700
Мекера То же *“ 725-800
Паяльная » 900
♦Распространенные у нас горелки Теклю дают приблизительно ту же температуру, что и горелки Тирриля*.
Хорошее пламя паяльной горелки должно легко расплавлять несколько сотых грамма порошка ортоклаза, помещенного на дно 15-граммового платинового тигля; это указывает на температуру, равную приблизительно 1200° С. Если прокаливание должно быть проведено при тщательно регулируемой температуре, как, например, в случае окиси вольфрама или окиси молибдена, то следует пользоваться муфелем, снабженным пирометром.
Обычно прокаленный осадок надо защищать от влаги, пока он охлаждается до температуры, при которой его можно взвесить. Для этого
134 Гл. Общие методы и операции химического анализа
тигель с прокаленным осадком помещают в эксикатор, содержащий соответствующее высушивающее вещество (см. раздел «Высушивающие вещества», стр. 71). В обычном случае для охлаждения достаточно 30 мин, чрезмерно продолжительный промежуток времени между прокаливанием и взвешиванием нежелателен. Если осадок гигроскопичен, как, например, окись кальция или окись алюминия, то его следует прокаливать и взвешивать в тигле, снабженном хорошо прилегающей крышкой. При взвешивании после второго прокаливания надо поставить разновески на чашку весов заранее, чтобы массу тигля можно было определить быстро. Изредка случается, что осадок настолько гигроскопичен, что с ним нельзя работать описанными выше способами. В таких случаях осадок надо охладить в эксикаторе, из которого насколько возможно быстро выкачивают воздух, затем тигель быстро помещают в стаканчик для взвешивания и взвешивают, помещая подобный же стаканчик на другую чашку весов для уравновешивания тары.
Глава V
СПЕЦИАЛЬНЫЕ ОПЕРАЦИИ
I
I
РЕДУКТОР ДЖОНСА
Редуктор Джонса 1 состоит из стеклянной трубки, внизу которой находится фильтрующий слой, а над ним — столб из кусочков амальгамированного цинка. Если через трубку пропустить солянокислый или сернокислый раствор солей таких элементов, как железо, молибден и т. п., то они весьма быстро восстанавливаются. Содержание восстановленных, элементов можно определить титрованием соответствующим окислителем. Раствор может быть использован также и для других целей, для которых требуется предварительное восстановление находящихся в нем элементов. Восстановление в редукторе сопряжено с введением в раствор солей цинка; загрязнение ртутью настолько незначительно, что в рядовой работе им можно пренебречь.
Удобная форма-редуктора приведена на рис. 13. Как видно из рисунка, столб цинка помещен на фильтрующем слое, а конец трубки редуктора доходит до дна приемной колбы. Имеется приспособление, которое дает возможность легко отделять колбу от редуктора, а также поддерживать в колбе давление на уровне атмосферного или создавать в ней вакуум. Внутренний диаметр редуктора обычно равен 19 мм. Трубку делают такой длины, чтобы в ней помещался столб цинка высотой 25— 40 см. Фильтрующий слой должен быть достаточно плотным, чтобы на нем задерживались все кусочки цинка. Его обычно делают из асбеста или, лучше, из стеклянной ваты, которую помещают на платиновую сетку или дырчатую пластинку. Цинк не должен содержать ни восстанавливающихся элементов вроде железа, ни элементов, способных окисляться, как, например, углерод. Цинк измельчают до 20—30 меш (d = 0,5— 0,8 мм) и амальгамируют 0,1—5% ртути2.
Имеется указание3, что скорость реакции между окислителем и амальгамированным цинком зависит главным образом от природы окислителя и концентрации цинка на поверхности амальгамы.
*С. Jones, Trans. AIME, 17, 411 (1888—1889).
2 Вместо цинка для восстановления в редукторе можно пользоваться и другими металлами, найример кадмием, губчатым свинцом [см. W. D. Treadwell, Helv. Chim. Acta, 5, 732, 801 (1922); Analyst, 46, 342 (1921) и 47, 533 (1922)] и серебром (стр. 139). Восстановление всеми этими металлами не обязательно идет до одной и той же валентности, так как это зависит от потенциала применяемого металла.
3 Н. W. S t о n е, D. N. Hu me, Ind. Eng. Chem., Anal. Ed., 11, 598 (1939).
136
Гл. V. Специальные операции
Для восстановления таких окислителей, как Fein и GeIV, который реагируют и с цинком и с ртутью, можно пользоваться 1 %-ными, а в случае необходимости даже 5 %-ными по содержанию ртути амальгамами (в частности, при высокой кислотности раствора). Восстановление таких элементов протекает быстро и без изменения состава амальгамы, так как
Рис. 13. Редуктор Джонса:
1 — двухходовой кран; 2 — дырчатая пластинка; 3 — слой асбеста| 4 — амальгамированный цинк.
в процессе реакции из нее извлекается как цинк, так и ртуть. Для восстановления же элементов, которые со ртутью не реагируют, как, например, Сгш и TiIvr следует пользоваться амальгамами с минимальным содержанием ртути (0,1—1%), при котором удовлетворительно регулируется выделение водорода, так как при введении больших количеств ртути в амальгаму процесс восстановления замедляется. Такие элементы восстанавливаются медленнее, и продолжительное употребление амальгамы приводит к значительному понижению скорости реакции, так как с поверхности амальгамы удаляется один только цинк, который через ртуть диффундирует медленно.
Наиболее совершенная амальгама должна обеспечивать быстрое и полное восстановление соответствующих элементов при минимальном выделении водорода. Количество ртути, которое следует вводить в цинк, зависит главным образом от кислотности раствора, природы окислителя, размера редуктора и скорости прохождения раствора через редуктор.
Амальгамирование проводят энергичным встряхиванием в течение 3 мин измельченного до 20 меш (d = 0,83 мм) цинка с соответствующим количеством 0,25 М раствора нитрата или хлорида ртути (II). Цинк предварительно промывают 1 мин 1 н. соляной кислотой, взятой в количестве достаточном, чтобы покрыть весь металл. По окончании амальгамирования раствор -сливают, амальгаму промывают водой, а затем сохраняют под водой, подкисленной несколькими каплями соляной кислоты. Амальгамы с незначительным содержанием ртути при хранении под водой, не защищенной от доступа воздуха, покрываются белым налетом, что снижает активность их действия.
Когда редуктор не находится в работе, он должен быть наполнен водой, чтобы избежать образования основных солей цинка, которые могут его закупорить. Через редуктор нельзя пропускать аммиачные
растворы, так как аммиак реагирует со ртутью, вследствие чего прекращается ее действие. Медь, никель и другие элементы, восстанавливающиеся в редукторе до металла, естественно, должны быть предварительно удалены.
Восстановление можно осуществлять в растворах, содержащих 3— 15% (по объему) соляной кислоты или 1-*-10% (по объему) серной кислоты. Если присутствие в растворе соляной кислоты затрудняет проведение дальнейших операций, лучше пользоваться сернокислыми растворами. Процесс восстановления в редукторе с высотой столба амальгамированного цинка 40 см в большинстве случаев проходит вполне удовле
Редуктор Джонса
137
творительно при комнатной температуре *. В случае же восстановления таких элементов, как ниобий и титан, вероятно, следует применять растворы, нагретые до 40—60° С, особенно при меньшей высоте столба амальгамированного цинка. Это надо установить опытным путем. Если редуктор находился некоторое время без употребления, его обычно предварительно промывают 100—200 мл разбавленной кислоты, а затем таким же количеством воды. Промывную жидкость отбрасывают. По обычно принятому способу восстановления растворы пропускают через редуктор со скоростью 100 мл в минуту в следующем порядке: сначала 25— 50 мл разбавленной кислоты, затем 100—200 мл анализируемого раствора, после этого снова 25—50 мл разбавленной кислоты и, наконец, 100— 200 мл воды, чтобы вымыть восстановленный раствор и кислоту, которая в противном случае без надобности будет растворять цинк. Каждый раствор сливают лишь настолько, чтобы в редукторе оставалось его примерно на 1 см выше уровня столба амальгамированного цинка, после чего в воронку наливают следующий раствор. Цинк ни в коем случае не следует оставлять обнаженным. Последнюю порцию промывной воды прекращают сливать, когда воронка еще до половины ею заполнена. По окончании промывания закрывают двухходовой кран в трубке, соединяющей прибор с вакуумной линией, чтобы предотвратить доступ воздуха в колбу. Редуктор отделяют от колбы и поднимают его настолько, чтобы конец его был выше уровня раствора. Кран в редукторе открывают и дают промывной воде стечь в колбу через соединительную трубку, после чего эту трубку снаружи ополаскивают струей воды из промы-валки. Кран в редукторе снова закрывают, когда цинк еще полностью покрыт промывной водой. Редуктор вынимают из колбы и, закрыв воронку стаканом, отставляют. В определенных условиях растворы, вероятно, можно пропускать через редуктор с большей скоростью, но это надлежит устанавливать экспериментальным путем. При выполнении рядовых анализов нет необходимости пропускать через редуктор кислоту до испытуемого раствора и после него. Можно лишь несколько повысить кислотность анализируемого раствора, а после восстановления промыть редуктор достаточным количеством воды, чтобы этот раствор полностью извлечь.
До определенной валентности в редукторе восстанавливаются железо, титан, европий, хром, молибден и ванадий. Уран частично восстанавливается ниже четырехвалентного, но этого можно избежать, если проводить восстановление в охлажденных растворах и после восстановления дать им некоторое время постоять на воздухе i 2, как описано в гл. «Уран» (стр. 530). Восстановление вольфрама, ниобия и рения также идет не до определенной валентности, но способ устранения этого, как для урана, неизвестен.
Растворы восстановленных веществ, за исключением растворов солей железа (II) и урана (IV), крайне неустойчивы и от соприкосновения с воздухом должны быть предохранены каким-нибудь инертным газом, например двуокисью углерода или азотом, или же их следует сливать из редуктора непосредственно в раствор сульфата железа (III). Восстановленные элементы при этом окисляются либо до устойчивых соединений более
i G. Е. F. Lun dell, Н. В. К п о w 1 е s, Ind. Eng. Chem., 16, 723 (1924).'
2 G. E. F. L u n d e 11, H. В. К n о w 1 e s, J. Am. Chem. Soc., 47, 2637 (1925)..
138
Гл. V. Специальные операции
высокой промежуточной валентности, как ванадий и молибден, либо до состояния высшей валентности, как титан, с образованием эквивалентного количества железа (II). Для окисления в большинстве случаев достаточно пятикратного количества железа \ Если окраска железа (III) вызывает затруднения при последующем титровании, то прибавляют к раствору 2—5 мл фосфорной кислоты, за исключением тех случаев, когда в растворе присутствует титан, который при этом образует малорастворимый фосфат. Некоторые восстановленные соединения, как, например, рения, настолько неустойчивы, что из всех пропускаемых через редуктор растворов необходимо предварительно полностью удалить воздух продолжительным кипячением. Охлаждать эти растворы следует в атмосфере, свободной от кислорода, как, например, в атмосфере очищенного азота или двуокиси углерода.
Объемному определению каждого из элементов после восстановления в редукторе, само собой разумеется, мешают все прочие восстанавливающиеся наряду с ним элементы, а именно: железо, титан, европий, хром, молибден, ванадий, уран, ниобий, вольфрам и рений. Помимо того, следует упомянуть азотную кислоту, органические вещества, олово, мышьяк, сурьму и политионаты. Наиболее часто приходится сталкиваться с азотной кислотой, которая восстанавливается до гидроксиламина и других соединений, на которые при титровании расходуется окислитель. Например, при определении железа в белой глине можно получить неверные результаты вследствие содержания нитрата аммония в осадке от аммиака, даже тщательно промытом. Для полного удаления азотной кислоты обычно требуется двукратное, даже лучше трехкратное, выпаривание раствора с серной кислотой до появления ее паров, причем стенки сосуда необходимо каждый раз тщательно обмывать. Иногда, как, например, в присутствии урана 1 2 или при разрушении фильтровальной бумаги обработкой азотной и серной кислотами, азотная кислота 3 4 удерживается настолько прочно, что для ее удаления двукратного выпаривания с серной кислотой недостаточно. При разрушении фильтровальной бумаги можно избежать введения азотной кислоты, Для чего к раствору, выпаренному в закрытом стакане до появления паров серной кислоты, прибавляют осторожно по каплям насыщенный раствор перманганата калия до появления неисчезающей розовой окраски, а затем продолжают нагревание в течение нескольких минут.
О наличии в растворе органических веществ обычно бывает заведомо известно, но в некоторых случаях они могут быть введены с реактивами, как, например, ацетанилид с перекисью водорода. Не всякие органиче
1 L. D. Randall [Am. J. Sci., [4], 24, 313 (1907) и некоторые другие авторы рекомендуют пользоваться железо-аммонийными квасцами. G. Е. F. Lundell и Н. В. К п о w 1 е s [J. Am. Chem. Soc., 45, 2621 (1923)] предпочитает сернокислый раствор сульфата железа (III), для приготовления которого растворяют мягкое железо или арестую углеродистую сталь в царской водке. Для удаления хлора к раствору прибавляют серную кислоту и выпаривают до начала выделения ее паров. После этого раствор разбавляют до соответствующего объема. Подходящей концентрацией является й,02 г жедаэа в 1 мл 8%-ной (по объему) серной кислоты. Такой раствор по сравнению с раствором железных квасцов имеет бойее светлую окраску, он дешевле, и отсутствие в нем железа (II) и хлоридов обеспечено.
2 J. A. Holladay, Т. R. Cunningham, Trans. Am. Electrochem. Soc.,
43, ' 329 (1923)'.
4 Опыты Лендедя.
Восстановление в редукторе с серебром
139
ские вещества удается разрушить одним только введением в раствор перманганата до появления неисчезающей окрйски. Некоторые из них, в частности ацетанилид, в этих условиях устойчивы, восстанавливаются затем в редукторе и при последующем титровании раствора снова окисляются. Поэтому в сомнительных случаях лучше поступать так, как это рекомендуется для разрушения фильтровальной бумаги.. Олово, мышьяк и сурьма легко удаляются осаждением сероводородом. При этом, однако, образуются политионаты, на которые расходуется окислитель. Эти соединения не разлагаются в процессе восстановления в редукторе или при кипячении раствора с разбавленной кислотой Ч Их влияние можно устранить обработкой раствора, перед пропусканием его через редуктор, перманганатом, взятым в небольшом избытке. Платина восстанавливается .до металла в верхней части редуктора и поэтому не мешает титрованию раствора, прошедшего через редуктор.
Следует указать еще на два возможных источника ошибок, а именно: недостаточно тщательная промывка редуктора, остающегося продолжительное время без употребления, и проникновение воздуха в редуктор во время восстановления. В первом случае получаются повышенные результаты, что подтверждается данными титрования серной кислоты, пропущенной через редуктор без предварительной промывки его кислотой. Часто утверждают, что проникновение воздуха в редуктор во время восстановления приводит к образованию озона или перекиси водорода. Мы этого не наблюдали 1 2, но можно считать бесспорным, что наличие воздуха в редукторе в процессе восстановления является причиной получения пониженных результатов.
ВОССТАНОВЛЕНИЕ В РЕДУКТОРЕ С СЕРЕБРОМ
Редукторы с серебром, имеющие внутренний диаметр 2 см и длину колонки приблизительно 12 см, с шариком емкостью 50—75 мл вверху (короткая трубка Джонса), могут быть использованы для определения железа 3, молибдена 4, урана 5 и меди5. Колонку редуктора наполняют приблизительно 18 г серебра, для приготовления которого растворяют 29 г нитрата серебра в 400 мл воды, подкисленной несколькими каплями азотной кислоты. Раствор энергично перемешивают пластинкой листовой электролитной меди, Имеющей поверхность 10 см2, до полного осаждения серебра. После промывки осадка разбавленной серной кислотой серебро при помощи струи воды переносят в редуктор, встряхивают для уничто
1 G. Е. F. L u n d е 11, Н. В. Kn owles, J. Am. Chem. Soc., 43, 1560 (1921).
2 G. E(. F. Lundell, H. В. К n owl es, Ind. Eng. Chem., 16, 724 (1924).
В действительности перекись водорода полностью разлагается в сернокислом растворе при прохождении его через редуктор.”
3 G. Н. Walden Jr., L. Р. Н a m m е t t, S. M. E d m о n d s, J. Am. Chem. Soc., 56,' 350 (1934); C. F. F г у 1 i n g, F. V. T о о 1 e у, там же, 58, 826 (1936). При испытании этого метода для микроанализа последние авторы наблюдали нарушение процесса, вызванное образованием перекиси водорода, которое было уменьшено насыщением раствора водородом и проведением восстановления в атмосфере водорода в специальном аппарате.
4 N. Birnbaum, G. Н. Walden Jr., J. Am. Chem. Soc., 60, 64 (1938); см. также C. F. H i s k e у, V. F. S p r i n g e г, V. W. M e 1 о c h e, там же, 61, 3125 (1939).
5N. Birnbaum, S. M. Edmonds, Ind. Eng. Chem., Anal. Ed., 12, 155 (1940).
140
Гл. V. Специальные операции
жения воздушных пробок и снова промывают разбавленной серной кислотой до удаления меди. Редуктор хранят наполненным 1 М соляной кислотой. При продолжительном употреблении серебро постепенно покрывается слоем хлорида серебра. Когда этот слой достигнет трех четвертей высоты столба серебра, хлорид восстанавливают, для чего редуктор наполняют 1 М серной кислотой и вводят в нее цинковый стержень, который соединяют с серебром.
Пропускаемые через серебряный редуктор растворы должны быть свободны от солей платины, так как последние восстанавливаются до металла, в результате чего образуется пара: платина — серебро с более высоким восстановительным потенциалом. Это приводит к нежелательному восстановлению и других металлов, например титана.
При восстановлении некоторых элементов серебром следует более тщательно регулировать температуру и кислотность раствора, чем при восстановлении их в цинковом редукторе. Так, в серебряном редукторе в 2 М соляной кислоте восстановление молибдена идет до пятивалентного, а в 4 М кислоте — до трехвалентного, тогда как в цинковом редукторе молибден восстанавливается до трехвалентного при колебании в растворе концентрации кислоты в довольно широких пределах.
Разработаны условия восстановления следующих металлов до указанной валентности: Fe11, Мо1П, Mov, Viv, UIV и Си1. Вполне вероятно, что и другие элементы, как, например, WVI, восстанавливаются до более низкой валентности, а Ап111 и SeIV — до элементарного состояния. К числу металлов, не восстанавливающихся серебром, относятся несколько таких, которые восстанавливаются цинком, например ReVI1, Сгш и TIV. При анализе материалов, содержащих эти металлы, как, например, для определения железа в присутствии титана, применение серебряного редуктора весьма целесообразно.
Приводимые ниже методы определения железа и молибдена являются примерами использования серебряного редуктора.
Определение железа
Ход анализа. Холодный 1 М по концентрации соляной кислоты раствор, содержащий 0,15—0,2 г железа и свободный от других элементов, восстанавливающихся серебром, пропускают через редуктор со скоростью примерно 30 мл в минуту. Ополаскивают стакан, в котором находился анализируемый раствор, и промывают редуктор примерно 150 мл 1 М соляной кислоты, вводимой небольшими порциями, так, чтобы серебро не обнажалось. К восстановленному раствору прибавляют 200 мл 10 М серной кислоты, охлаждают его, прибавляют 1 киплю 0,025 М раствора ферроина и титруют 0,01 М раствором сульфата церия (IV). Вводят поправку на количество сульфата церия, расходуемого на титрование одних реактивов.
Титан, хром, марганец, а также умеренные количества ванадия и азотной кислоты (0,2 а) не влияют на определение.
Определение молибдена
Ход анализа. К 2 М по концентрации соляной кислоты раствору, содержащему не более 0,48 г молибдена в 50 мл и свободному от нитратов, железа, меди, урана'и других восстанавливающихся серебром элементов,
Восстановление жидкими амальгамами
141
добавляют 10 мл соляной кислоты (пл. 1,19 г/см1 2 3), 3 мл фосфорной кислоты (пл. 1,69г/см3) и нагревают до 60—80° С. Раствор пропускают со скоростью 10 мл в минуту через редуктор, предварительно нагретый обработкой горячей 2 М соляной кислотой. Ополаскивают стакан и редуктор 150 мл нагретой 2 М соляной кислоты, повысив скорость прохождения последних 100 мл жидкости. В охлажденный раствор вводят каплю 0,025 М раствора ферроина и титруют пятивалентный молибден 0,1 М раствором сульфата церия (IV), вводя последние несколько десятых миллилитра по каплям при сильном перемешивании раствора.
ВОССТАНОВЛЕНИЕ ЖИДКИМИ АМАЛЬГАМАМИ
Объемные методы, основанные на восстановлении амальгамами цинка 1, кадмия 2, висмута 3 или свинца 4, с последующим титрованием соответствующим окислителем, обычно перманганатом калия, вполне надежны для определения таких элементов, как железо, титан, молибден, уран и ванадий. Восстановление осуществляется в приборе, показанном на рис. 14.
Для удаления воздуха всю нижнюю часть прибора до крана заполняют свежепрокипяченной водой и закрывают кран 4. В расширенную часть 5 прибора вводят 100—200 г амальгамы5, а затем — анализируемый раствор. Если восстановленные элементы легко окисляются, вытесняют воздух из ч^сти 5 током двуокиси углерода, впуская его через трубку 6 и выпуская через кран 8. Закрывают краны 7 и 8 и встряхивают прибор до полного восстановления раствора, о чем судят на основании опыта или по окраске раствора. Открывают кран 4 и сжимают резиновую трубку 2, чтобы амальгама прошла в нижнюю часть 1 прибора, где она промывается вытесняемой ею водой. Закрывают кран 4 и окисляют восстановленные элементы титрованным раствором окислителя, вводимым через кран 8.
Если раствор проходит недостаточно легко, повышают давление
1 Т. Nakazono, J. Japan Chem. Soc., 42, 526, 761 (1921); S. Kikuchi, там же, 43, 173, 544 (1922); S. Hakomori, там же, 734; I. W a d a, T. N a к a-z о n o, Sci. Papers Inst. Phys. Chem. Research, 1, 139 (1923); N. К a n o, J. Japan. Chem. Soc., 43, 330, 550 (1922); 44, 37 (1923); Kin’ichi Someya, Sci. Rep., Toboku Imp. Univ., [1], 14, 47, 571 (1925).
2 N. Kano, цит. выше, 333.
3 К in' i c hi Someya, цит. выше, 14, 60, 225, 244 (1925); 15, 399 (1962); 16, 515, 521 (1927); 17, 131 (1928).
* К i n’i c hi Someya, цит. выше, 14, 574 (1925).
5 Для приготовления амальгамы смешивают 3 г чистого гранулированного или в палочках цинка примерно со 100 г ртути и небольшим количеством разбавленной серной кислоты. Смесь нагревают на водяной бане. По охлаждении тщательно промывают амальгаму водой и для отделения от твердого вещества пропускают ее через делительную воронку. Твердое вещество можно сохранить и затем добавлять к амальгаме по мере ее израсходования. Подобным же образом готовится кадмиевая, а также и висмутовая амальгамы. В последнем случае вместо серной кислоты применяют разбавленную соляную кислоту.
Для приготовления свинцовой амальгамы чистый свинец промывают соляной кислотой и смешивают со ртутью. Смесь нагревают до получения однородной жидкой массы и затем пропускают через делительную воронку.
142
Гл. V. Специальные операции
в воронке 9, что проще всего сделать, надавливая ладонью руки на верхушку воронки \
Согласно указаниям цитируемых авторов, процесс восстановления
может пройти успешно до валентностей, указанных в табл. 9, при усло-
Рис. 14. Прибор для восстановления жидкими амальгамами:
1 — нижняя часть прибора; 2—резиновая трубка; 3— зажим; 4,7, 8 — краны;
5 — расширение; 6 — отводная трубка; 9—воронка. Емкости частей 1, 5 и 9 равны соответственно 15— 20, 150—200 и 10 мл.
вии, если соблюдаются надлежащие температура и кислотность раствора и обеспечивается соответствующая атмосфера над ним. В данном случае, так же как и при использовании редуктора Джонса, все посторонние элементы, восстанавливающиеся амальгамой, должны быть предварительно отделены, если влияние их на результаты титрования восстановленного раствора нельзя точно учесть.
'Таблица э Восстановление амальгамами
Элемент
Амальгама
цинка висмута свинца
Валентность элемента после восстановления
Fe II II II
Ti III III III
v- II IV II
U III IV IV
w III III III
Mo III V или III * III
Cr II — —
Sn — II . —
Cu - I —
* В зависимости от концентрации кислоты в растворе.
ДРУГИЕ МЕТОДЫ ВОССТАНОВЛЕНИЯ
Для определения молибдена, железа, ванадия и сурьмы предложен 1 2 метод, основанный на восстановлении их ртутью в слабосолянокислом растворе с последующим оксидиметрическим титрованием. Для восстановления железа в присутствии титана и ванадия рекомендуют 3 пользоваться сплавом Вуда.
1 Восстановление титана цинковой амальгамой в видоизмененном приборе см. Н. В. Н о р е, R. F. М о г а п, А. О. Р 1 о е t z, Ind. Eng. Chem., Ana], Ed., 8, 48 (1936). См. также стр. 686; G. F. S m i t h, L. T. К u г t z [там же, 14, 854 (1942)] указывают, что метод может быть упрощен проведением, процесса восстановления в конической колбе. По окончании восстановления в раствор нужно ввести достаточное количество четыреххлористого углерода, чтобы закрыть всю амальгаму, а затем титровать, перемешивая раствор настолько осторожно, чтобы слой четыреххлористого углерода оставался в покое.
2 N. Н. F и г m a n, W. М. М и г г а у Jr., J. Am. Chem. Soc., 58, 428, 1689, 1843 (1936).
3 G. F.S mith, C. S. Wilcox, Ind. Eng. Chem., Anal. Ed., 9, 419 (1937).
Осаждение купфероном
143:
В гл. «Сурьма» (стр. 323) описаны методы восстановления некоторых металлов до элементарного состояния гипосульфитом натрия (Na2S2O4y' в щелочной среде.
Описаны 1 методы разделения, открытия и определения легко восстанавливающихся элементов, таких, как мышьяк, серебро, золото, платина, палладий, селен, теллур и иод, основанные на обработке солянокислых растворов избыточным количеством хлорида ртути (I).
ОСАЖДЕНИЕ КУПФЕРОНОМ
Купферон 2 CeH5N(NO)ONH4, аммонийная соль нитрозофенилгидроксиламина, является весьма ценным реактивом в химическом анализе. Осаждением купфероном в сильнокислых растворах можно осуществить, полное отделение железа, ванадия, циркония, титана, олова, ниобия и тантала от алюминия, бериллия, фосфора, бора, марганца^ никеля и урана (VI). Соединения, образуемые купфероном, являются солями, в которых аммоний замещен на металл. Осаждение проводится в охлажденных , льдом растворах, содержащих свободную минеральную или органическую кислоту. Применяется холодный 6%-ный раствор купферона, который вводят в анализируемый раствор медленно при перемешивании до тех пор, пока избыток его не будет отмечен по появлению быстро исчезающего тонкого белого осадка, отличного от хлопьевидного нерастворимого соединения купферона с металлами. Осаждение обычно происходит сейчас же по добавлении реактива. Если осадок надо будет прокалить и взвесить, то фильтровать лучше при осторожном отсасывании через бумажный фильтр, вложенный в конус. Если же предполагается провести только разделение элементов и для дальнейшей работы нужен будет фильтрат, то лучше всего перед осаждением вводить мацерированную бумагу и фильтровать через бюхнеровскую воронку. 1
Для промывания осадков в приводимых методах применяется холодная разбавленная соляная или серная кислота, содержащая небольшое
1 G. G. Р i е г s о n, Ind. Eng. Chem., Anal. Ed., 6, 437 (1934); 11, 86 (1939).
2 О. Baudisch, Chem. Ztg., 33, 1298 (1909). См. также G. E. F. Lunael 1, H. В. К n о w 1 e s, Ind. Eng. Chem., 12, 344 (1920); V. A u g e r, C. r., 170, 995 (1920). Позднее О. Baudisch и S. Holmes [Z. anal. Chem., 119, 241 (1940)] предложили применять неокупферон
N=O I n-o-nh4
образующий еще менее растворимое соединение с железом (и, возможно, с другими элементами). Об использовании органических реагентов в неорганическом анализе и о факторах, которые должны быть приняты во внимание при систематических исследованиях по применению новых реактивов или по их усовершенствованию, см. F. F е i g 1, Ind. Eng. Chem., Anal. Ed., 8, 401 (1936). См. также F. F e i g 1, Chemistry of Specific, Selective and Sensitive Reactions, Academic Press, New York, 1949; F. J. Welcher, Organic Analytical Reagents, Vol. I—IV, D. Van Nostrand Co., 1947— 1948; J. F. F1 a g g, Organic Reagents, Intersciens Publishers, New York, 1948; * Л. M. Кульберг, Органические реактивы в аналитической химии, Госхимиздат, 1950; В. И. Кузнецов, ЖАХ, 2, 67 (1947); Химические реактивы и препараты (Справочник), под ред. В. И. Кузнецова, Госхимиздат, 1953.*
144
Гл. V. Специальные операции
количество купферона. Осадки от купферона нельзя непосредственно взвешивать, их необходимо прокаливать до окисей металлов. Вначале осадок прокаливают крайне осторожно при невысокой температуре, чтобы избежать механических потерь, так как влажный осадок разжижается и вскипает, а из сухого выделяется значительное количество летучих веществ.
Купфероновый метод вполне надежен для определения железа, титана, циркония, ванадия и в отдельных случаях — олова, ниобия, тантала, урана (IV), галлия и, вероятно, гафния. Этим методом можно определять также медь и торий, но осаждать их следует из слабокислых растворов; результаты определения этих элементов менее удовлетворительны, чем при обычно принятых методах. Из числа элементов, мешающих применению купферонового метода, следует упомянуть таллий (III), сурьму (III), палладий, ниобий, тантал, молибден, висмут, церий, торий, вольфрам и большие количества кремния, фосфора, щелочноземельных и щелочных металлов х. Торий и церий частично выделяются купфероном даже из растворов, содержащих 40% (по объему) серной кислоты. Уран (VI) не влияет на осаждение купфероном. Число элементов, мешающих определению купфероном, может показаться очень значительным, но нужно принять во внимание, что часть из них относится к группе сероводорода и может быть легко отделена перед осаждением купфероном, а некоторые элементы встречаются редко. Здесь следует указать на представляющие интерес разделения, которые можно осуществить этим методом, а именно: 1) отделение железа, титана, циркония, галлия и ванадия при анализе чистых алюминия, никеля, цинка и т. и.; 2) отделение осаждающихся купфероном элементов от алюминия, хрома, магния и фосфора при анализе различных руд и горных пород; 3) отделение ванадия (V) от урана (VI), разделение урана (IV) и урана (VI) и отделение ванадия от фосфора. Осаждение купфероном может быть осуществлено в присутствии винной кислоты, что дает возможность предварительно отделять железо в виде сульфида. Для этого в раствор вводят достаточное количество винной кислоты, чтобы он оставался прозрачным при последующем добавлении аммиака. В кислом растворе восстанавливают железо сероводородом и затем подщелачивают аммиаком. Выделившийся осадок сульфида железа отфильтровывают, как описано при осаждении сульфидом аммония (стр. 115), фильтрат подкисляют серной кислотой, удаляют сероводород кипячением и после этого проводят осаждение купфероном.
Если фильтрат должен быть использован для определения алюминия и др. элементов, винную кислоту и купферон можно разрушить выпариванием сернокислого раствора до 20—50 мл, добавлением 20—25 мл азотной кислоты и медленным выпариванием до появления паров серной кислоты. Если при этом органические вещества полностью не разрушаются, снова добавляют азотную кислоту.
1 Согласно А. Р i n k u s и F. Marlin [Chem. et Ind., 17, 182 (1927)], мышьяк (III и V), а также сурьма (V) не осаждаются купфероном, тогда как сурьма (III) осаждается.
Вольфрам частично осаждается, независимо от концентрации серной и винной кислот в растворе. S. G. С 1 а г k е [Analyst, 52, 466. 527 (1927)] установил, что вольфрам не выделяется купфероном в присутствии HF и может быть таким образом отделен от ванадия.
Осаждение купфероном
145
Купферон можно разрушить также следующим способом. Раствор кипятят, пока объем его не уменьшится приблизительно до 50 мл, затем прибавляют 25 мл азотной кислоты и продолжают кипячение до тех пор, пока объем раствора снова не станет равным 50 мл. После этого вводят еще 10 мл азотной кислоты, 15 мл хлорной кислоты и выпаривают до появления паров хлорной кислоты.
Установлено *, что купферон не влияет на осаждение мышьяка (V) магнезиальной смесью, никеля — диметилглиоксимом, цинка в виде фосфата, серебра в виде хлорида, а также на электролитическое осаждение кадмия из цианидных растворов и никеля из холодных аммиачных растворов.
Определение железа, циркония, галлия, титана и ванадия
Анализируемый раствор не должен содержать кремния, вольфрама и металлов сероводородной группы. Следует избегать также содержания в растворе значительных количеств фосфора, щелочноземельных и щелочных металлов, в противном случае осадок будет несколько загрязнен ими. Редкоземельные металлы частично осаждаются купфероном и поэтому должны быть предварительно отделены в виде фторидов или оксалатов, илк же содержание их должно быть определено позже во взвешенном осадке. Анализируемый раствор разбавляют до 200 мл. и приливают такое количество серной кислоты, чтобы общее содержание ее составило 20—25 мл. Если отсутствует винная кислота, вводят раствор перманганата до появления слабо-розового окрашивания, а в присутствии винной кислоты — до полной уверенности, что все содержащиеся в растворе элементы окислились до состояния, высшей валентности. Раствор охлаждают до 10° С и затем медленно при перемешивании добавляют холодный 6%-ный раствор купферона 2 до прекращения образования нераство-ряющегося осадка. После этого вводят немного мацерированной бумаги, дают раствору отстояться 2—3 мин и затем при слабом отсасывании фильтруют через бумажный фильтр, вложенный в конус, собирая фильтрат в приемник (в котором находится немного прозрачного раствора купферона) для проверки полноты осаждения. Осадок промывают холодной 10%-ной (по объему) серной или соляной кислотой, содержащей 1,5 г/л купферона. Жидкость возможно полнее отсасывают, а осадок переносят в достаточно большой фарфоровый или платиновый тигель, осторожно высушивают и затем нагревают до тех пор, пока бумага не начнет обугливаться. Нагревать следует осторожно, так как осадок обладает свойством разжижаться и вскипать. Постепенно повышают температуру до полного сгорания углерода, а под конец прокаливают при температуре, необходимой для получения окисла определяемого элемента.
1 A. Pinkus, J. Dernies, Bull. Soc. chim. Belg., 37, 267 (1928).
2 Раствор нужно готовить по мере надобности. Купферон можно изготовлять согласно указаниям С. S. М а г i е 1 и О. К a m m [J. Am. Chem. Soc., 41, 276 (1919)]. Приобретать же его следует непосредственно на предприятиях, специализировавшихся на его производстве. Сухой реактив необходимо хранить в холодном темном месте, лучше под карбонатом аммония, подвешенным в мешочке к пробке' склянки.
Согласно F. G. Germuth [Chemist Analyst, 17 , 3 (1928)], устойчивость раствора купферона может быть повышена добавлением 0,05 г ацетфенетидина (C2H5OCeH4NHCOCH3) на 100 мл. Приготовленный таким образом раствор устойчив в продолжение 20 дней и очень мало изменяется в течение 30 дней, даже на солнечном свету.
10 Заказ 522. '
146
Гл. V. Специальные операции
Определение урана
При осаждении купфероном уран следует предварительно восстановить до четырехвалентного. Концентрация кислоты в растворе при этом должна быть несколько ниже, чем в описанном выше случае Если для определения урана используется фильтрат, полученный после отделения некоторых элементов купфероном по предыдущему методу, прозрачный раствор выпаривают приблизительно до 50 мл, прибавляют 20 мл азотной кислоты и продолжают выпаривание до появления паров серной кислоты. Если при этом органические вещества полностью не разрушаются, снова прибавляют азотную кислоту, тщательно обмывают стенки сосуда водой и повторяют выпаривание для удаления азотной кислоты. После охлаждения раствор разбавляют водой с таким расчетом, чтобы в 100 мл содержалось 6 мл серной кислоты, снова охлаждают и пропускают его через редуктор Джонса (стр. 135). Редуктор промывают 896-ной серной кислотой. Охлаждают раствор до 5—10° С и прибавляют холодный 6%-ный раствор купферона (в небольшом избытке). После этого вводят мацерированную бумагу и дают раствору отстояться. Осадок отфильтровывают, промывают холодной 4%-ной серной кислотой, содержащей 1,5 г/л купферона, и затем осторожно прокаливают сначала, как описано выше, а под конец при 1000—1050° С. По охлаждении взвешивают в виде U3O8. Состав окисла не совсем точно отвечает этой формуле, что указано в гл. «Уран» (стр. 528).
Определение олова
Имеются указания1 2-3, что олойо количественно осаждается купфероном из раствора, содержащего фториды и бораты. Это интересно тем, что медь, свинец, мышьяк (III) и сурьму (III) можно отделить от олова (IV) осаждением сероводородом в присутствии фтористоводородной кислоты (стр. 89), а затем, удалив из фильтрата сероводород кипячением и прибавив борную кислоту, можно выделить олово купфероном. Если при кипячении раствора выделяется сульфид, то для его растворения вводят перекись водорода, избыток которой разрушают кипячением. Один из авторов3 проводил осаждение олова добавлением в избытке 10%-ного раствора купферона к раствору, содержащему в 200—500 мл 0,1—0,3 г олова, 5 мл 48%-ной фтористоводородной кислоты, 4 г борной кислоты, 2—5 мл серной кислоты и 5—10 мл соляной кислоты. Раствор перемешивали до тех пор, пока осадок не становился компактным и зернистым. После фильтрования осадок промывали холодной водой и осторожно прокаливали до окиси SnO2.
Определение галлия
Галлий количественно осаждается купфероном из разбавленного (7 : 93) сернокислого раствора и отделяется таким образом от индия и алюминия (подробно см. стр. 555).
1 J. A. Holladay, Т. R. Cunningham, Trans. Am. Electrochem. Soc., 43, 329 (1923).
2 А. К 1 i n g, A. L a s s i e u г, C. r., 170, 1112 (1920).
3 N. H. F u г m a n, Ind. Eng. Chem., 15, 1071 (1923); см. также A. P i n k u s, J. Claessens, Bull. Soc. chim. Belg., 31, 413 (1927).
Осаждение купфероном
147
Определение ниобия и тантала
Тантал и ниобий количественно осаждаются купфероном из сильносернокислого раствора, содержащего щавелевую или винную кислоту \ что дает возможность отделять эти элементы от алюминия, хрома и урана (VI). Осаждение купфероном может быть осуществлено после отделения редкоземельных металлов в виде оксалатов, олова — сероводородом из сернокислого раствора, содержащего винную кислоту, и железа — сульфидом аммония из аммиачного раствора в присутствии тартратов 1 2.
Определение других элементов
’ В нейтральных или слабокислых растворах купферон образует нерастворимые соединения и с некоторыми другими элементами, кроме тех, которые упоминались выше. При pH = 4,6 (синее окрашивание бромфенолового синего) количественно осаждаются алюминий (даже в присутствии винной кислоты), бериллий и некоторые редкоземельные металлы (иттрий, церий, галлий и эрбий). Частично осаждаются хром (III), таллий (III), индий, торий и уран (VI). Число элементов, которые полностью или частично осаждаются купфероном, значительно возрастает, когда значение pH раствора приближается к 7. Насколько известно, купферон не образует осадков в аммиачных растворах с такими элементами, как медь или ванадий, которые не осаждаются аммиаком, или с такими, как железо и титан, которые не выделяются из аммиачных растворов в присутствии тартрата.
Экстрагирование купфератов
Растворимость некоторых купфератов в органических растворителях, в частности в хлороформе и диэтиловом эфире, дает возможность осуществлять разделение ряда элементов.
Имеется указание3 на то, что купфераты меди, железа (III), олова (IV), титана, урана (IV) и ванадия (V) количественно экстрагируются из разбавленной (.1 : 9) соляной кислоты. Молибден (IV) также практически полностью переходит в слой органического растворителя. Из разбавленной (1 : 9) серной кислоты медь извлекается значительно хуже, но другие элементы, из перечисленных выше, в этих' условиях также отделяются количественно, за исключением молибдена, микрограммы
1 Н. Р i е d, С. г., 179, 897 (1924).
2 Н. В. Knowles наблюдал незначительные потери тантала и ниобия, хотя в фильтрате после осаждения купфероном он эти элементы не обнаружил. Так, например, при осаждении купфероном из растворов, содержавших 5% винной кислоты и 5% серной кислоты (по объему), было определено 0,1930 г Nb2O5 вместо взятых 0,1936 а и 0,1961 а Та2О6 вместо 0,1962 а. В растворах, содержавших 5% винной кислоты и 10% H2SO4, вместо 0,1947 г Nb2O5 и 0,1955 а Та2О6 было найдено 0,1942 a Nb2O5 и 0,1946 а Та2О6. Осадки промывались холодной разбавленной (1 : 9) HCI. Потери можно отнести за счет примесей, содержавшихся в исходном материале, несмотря на предварительную тщательную его обработку, а также за счет механических потерь при прокаливании осадков купфератов и некоторой летучести хлоридов.
3 N. H.Furman, W. В. М a s о n, J. S. Р е к о 1 a, Anal. Chem., 21, 1327 (1949).
10*
148
Гл. V. Специальные операции
которого остаются в водном слое после трехкратного экстрагирования. Извлечение купфератов органическими растворителями является эффективным способом отделения железа, титаца, молибдена и ванадия при определении других элементов х. Экстрагирование целесообразно использовать также для концентрирования малых количеств (0,001—1 лег) железа, титана и ванадия, особенно при определении их фотометрическими методами.
ОСАЖДЕНИЕ ОКСИХИНОЛИНОМ f
Некоторые из реакций оксихинолина 2 были впервые использованы для идентификации 3 уже давно. '
Позднее применение оксихинолина в количественном анализе было описано многими авторами 4-5.
Оксихинблин реагирует с солями металлов с образованием соединений, в-которых водород гидроксильной группы замещен на металл, как, например, Mg(C9HeNO)2 или Al(C9HeNO)3. Осаждение проводится из слабокислых или щелочных растворов, в зависимости от преследуемой цели. Количественные методы осаждения разработаны для меди, висмута, кадмия, ванадия (V), алюминия и цинка, которые выделяются из уксуснокислых растворов, содержащих ацетат, и для магния, осаждающегося из аммиачного раствора в. ,Ряд других элементов также осаждается оксихинолином более или менее количественно. Так, например, молибден, серебро, ртуть (II), свинец, сурьма (III) и сурьма (V), ванадий (IV) и ванадий (V), уран, железо (II) и железо (III), титан, цирконий, тантал, ниобий, марганец, никель и кобальт выделяются из уксуснокислых рас-
1 Например, при определении урана в рудах титрованием бихроматом железо, титан и др. отделяются экстрагированием купфератов хлороформом из разбавленного (1 : 9) сернокислого раствора. См. С. J.R odden, Analytical Chemistry of the Manhattan Project, McGraw-Hill Book, Co., 1950, p. 139.
OH
A /N4
2 Структурная формула соединения | | ; оно называется также 8-окси-
хинолином, о-оксйхинолином, 8-гидроксихинолнном, а некоторыми авторами — оксином.
3 Z. Н. Skraup, Monatsh., 2, 139, 518 (1881); 3, 381, 531 (1882).
4 R. В е г g, J. prakt. Chem., 115, 178 (1927); Z. anal. Chem., 70, 341 (1927); 71, 23, 171, 321, 369 (1927); 72, 177 (1927); 76, 191 (1929); F. L. H a h n, К. V i e w e g, Z. anal. Chem., 71, 122 (1927); J. R о b i t s c h e k, J. Am. Ceram. Soc., 11, 587 (1928); I. M. К о 1 t h о f f, E. B. S a n e 11, J. Am. Chem. Soc., 50, 1900 (1928); G. E. F. Lu nd ell. H.B. Knowles, J. Research NBS, 3, 91 (1929).
s О применении оксихинолина в химическом анализе см. Р. Берг, Применение zj-оксихинолина в аналитической химии, ОНТИ, 1937.
Результаты систематического изучения азопроизводных оксихинолина в качестве аналитических реактивов представлены в статье Т. Boyd, Е. F.Degering, R, N.Shreve, Ind. Fing- Chem., Anal. Ed., 10, 606 (1938).
• H. V. M о у e г и W. J. R e m i n g t о n [Ind. Eng. Chem., Anal. Ed., 10, 21241^38)] установили, что при отделении умеренных количеств цинка от магния и железа от алюминия pH раствора должен быть соответственно 4,6—5,5 и 3,5—4,0. R. O.S со tt н R. L. Mitchell [J. Soc. Chem. Ind., 62, 4 (1943)] рекомендуют определять кобальт, никель, молибден, медь и цинк в растительных продуктах и почвенных вытяжках спектрографическим методом, после концентрирования этих металлов выделением из* раствора оксихинолином при нагревании и pH около 5,1. Осадок оксихииолята прокаливают й смешивают с кварцем.
Осаждение оксихинолином
149
творов. Все перечисленные элементы, кроме серебра, но включая бериллий, кальций, стронций, барий, магний, олово (II) и олово (IV), осаждаются из аммиачных растворов.
Из растворов, содержащих едкий натр, оксихинолином в основном осаждаются те же элементы, что из аммиачных растворов, за исключением олова, алюминия, бериллия и щелочноземельных металлов. В присутствии минеральных кислот осаждение оксихинолином не происходит.
* Некоторые элементы, в частности “медь, ванадий, молибден, сурьма, висмут, палладий, осаждаются в виде оксихинолятов из минеральнокислых растворов.
Условия осаждения оксихинолином ряда элементов приведены в литературе Доп. перев*
Для количественного определения осадок можно высушить и взвесить непосредственно в виде оксихинолята или растворить в соляной кислоте и оттитровать раствором бромат-бромида калия. Менее удовлетворительные результаты получаются при переводе осадка в окисел прокаливанием под слоем щавелевой кислоты иди при использовании обычно принятых методов определения (после разложения осадка обработкой азотной и серной кислотами). Поскольку оксихинолином осаждается такое большое число элементов, на первый взгляд может показаться, что применение его должно быть 'ограничено испытанием чистых солей. Известно, однако, несколько весьма интересных возможностей применения этого реагента, из которых следует упомянуть: 1) отделение магния от щелочных металлов: 2) отделение ряда других элементов от щелочных металлов; 3) отделение алюминия от некоторых элементов и 4) отделение некоторых элементов от алюминия.
Магний можно количественно отделить от щелочных металлов осаждением оксихинолином из аммиачного раствора даже в присутствии оксалата аммония. Благодаря применению для разделения этих элементов летучего реагента можно в фильтрате после отделения магния определять также и щелочные металлы, если они не были введены в процессе анализа. Что касается второго случая применения оксихинолина, то он Основан на том, что оксихинолином осаждаются многие элементы, и поэтому в фильтрате вместе с щелочными металлами нередко остается один лишь кальций.
Возможность отделения алюминия от других элементов обусловлена тем, что он осаждается оксихинолином из растворов, содержащих: а) уксусную кислоту и ацетат аммония, б) аммиак, в) аммиак и перекись водорода и г) карбонат аммония. В первом случае алюминий отделяемся от таких элементов, как магний и бериллий; во втором — от фосфатов, арсенатов, бора и фтора; в третьем — от молибдена, ванадия, титана, ниобия и тантала и, наконец, в четвертом — от урана. Отделение ряда элементов от алюминия может быть выполнено благодаря тому, что алюминий не осаждается оксихинолином из растворов, содержащих тартрат натрия и умеренные количества едкого натра, тогда как медь, кадмий, цинк й магний в этих условиях образуют нерастворимые оксихиноляты 1 2.
Ниже приводится описание методов осаждения оксихинолином из кислых и щелочных растворов, а также способы обработки осадков.
1 Е. С.Тииовская, Зав. лаб., 17, 387 (1951).
2 R. Berg, Z. anal. Chem., 71, 370 (1927).
150
Гл. V. Специальные операции
Осаждение алюминия из уксуснокислого раствора
Раствор, содержащий не более 0,1 г окиси алюминия и 1—2 капли свободной минеральной кислоты на 100 л»л, нагревают до 50—60° С. Прибавляют избыток (от 10% в обычных случаях и до 50% при содержании значительных количеств бериллия) уксуснокислого раствора реагента г, а затем медленно вводят 2 н. раствор ацетата аммония до появления неисчезающего осадка и сверх этого еще 20—25 мл, чтобы обеспечить полноту осаждения. Раствору дают отстояться, после чего его фильтруют через асбест, пористое стекло или пористый фильтр. Осадок промывают холодной водой, сушат при 120—140° С и взвешивают в виде A1(C9H6ON)3, содержание в котором А12О3 составляет 11,10%. Осадок склонен окклюдировать некоторое количество оксихинолина, что/приводит к получению несколько повышенных результатов при взвешивании его после высушивания или титровании после растворения.
«
Осаждение магния из аммиачного раствора
Слабокислый раствор, содержащий не более 0,1 г окиси магния и достаточное количество аммонийных солей, чтобы в дальнейшем воспрепятствовать выделению гидроокиси магния, нагревают до 60—70° С, добавляют избыточное количества (обычно около 10%) реактива и затем при перемешивании медленно вводят аммиак до явно щелочной реакции. При наличии избытка осадителя раствор приобретает желтую окраску. Раствору дают отстояться, затем осадок отфильтровывают и промывают горячим разбавленным (1 : 40) аммиаком. Осадок сушат при 130—140° С и взвешивают в виде Mg(C9HeON)2, который содержит 12,91% MgO.
Более чистые осадки получаются при добавлении оксихинолина к горячим аммиачным растворам соли магния1 2. Оксалат аммония замедляет осаждение; введения значительных количеств оксалата поэтому в некоторых случаях следует избегать, а при определении малых количеств магния оксалат должен быть разрушен выпариванием с концентрированной азотной кислотой. Рекомендуется следующий метод: к 100 мл раствора, содержащего 10—50 мг окиси магния и 2 г хлорида аммония, добавляют 0,5 мл о-крезолфталеина (0,02%-ный спиртовый раствор), вводят 6 н. раствор аммиака до появления фиолетовой окраски (pH = 9,5); а затем избыток аммиака в 2 мл. Нагревают до 60—80° Сив соответствии с содержанием магния медленно при постоянном перемешивании вводят 5%-ный или 1%-ный уксуснокислый раствор оксихинолина (2 н. и 0,4 н. соответственно) до появления небольшого избытка, что определяется по глубокой желтой окраске верхнего слоя жидкости. Затем раствор нагревают 10 мин, на водяной бане и горячим фильтруют через стеклянный тигель с пористым дном, пользуясь фильтратом для перенесения осадка на фильтр. Промывают осадок 50 мл горячей воды. При употреблении для промывания 100 мл горячей воды растворяется 0,09 мг осадка, а при применении 1 н. раствора аммиака — 0,07 мг.
Титрование оксихинолятов
При титровании броматом калия каждая молекула оксихинолина связывает 4 атома брома. Таким образом, одной молекуле А12О3 соответствует 24 атома брома, а одной молекуле MgO — 8 атомов брома.
1 Приготовляют растворением 5 г тонко измельченного оксихинолина в 100 мл
2 н. уксусной кислоты при нагревании до 60° С и перемешивании. Раствор фильтруют и сохраняют в склянке из оранжевого стекла. Приготовленный таким образом раствор устойчив в течение нескольких недель. 1 мл раствора достаточно для осаждения 0,0031 г алюминия или 0,0058 г окиси алюминия. Спиртовыми растворами оксихинолина пользоваться не рекомендуется, так как некоторые оксихиноляты, особенно оксихинолят алюминия, несколько растворимы в спирте.
2 С. С. Miller, I. С. McLennan, J. Chem. Soc., 1940, 656.
Осаждение таннином
151
Титрование проводится следующим способом.
Осадок отмывают от избытка осадителя, а затем растворяют его в 200—300 мл 2—3 н. раствора соляной кислоты. Раствор охлаждают и вводят из бюретки титрованный раствор смеси бромата и бромида калия в небольшом избытке (1—2 мл) %
Смесь осторожно, перемешивают и оставляют стоять 30—60 сек для того, чтобы бромирование прошло полностью. Затем добавляют 15 мл 20%-ного раствора иодида калия и снова тщательно перемешивают. Выделившийся иод' титруют раствором тиосульфата натрия, пока большая часть иода не будет оттитрована, а'затем добавляют раствор крахмала и заканчивают титрование, как обычно.
Для вычисления результата определения из введенного объема бромата вычитают количество его, эквивалентное тиосульфату, израсходованному на титрование иода, выделенного избытком бромата.
Раствор КВг + КВгО3 может быть 0,05, 0,1 или 0,2 и. в зависимости от содержания определяемого элемента и должен содержать немного больше КВг, чем требуется по соотношению КВгО3 : 5КВг. Если применяется, чистый бромат, можно пользоваться теоретическим титром. Для практической работы, однако, лучше устанавливать титр раствора, как описано в гл. «Алюминий» (стр. 574) и «Магний» (стр. 726). Соотношение между приблизительно 0,1 н. раствором бромид-бромата и раствором тиосульфата можно устанавливать следующим образом. 25 мл раствора бромид-бромата вводят в 300 мл холодной разбавленной (1 : 9) соляной кислоты и затем добавляют 10 мл 10%-ного раствора иодида калия. Острожно перемешивают и сразу же титруют раствором тиосульфата почти до обесцвечивания. После этого вводят несколько капель раствора крахмала и продолжают титрование до исчезновения синей окраски раствора. Когда приходится проводить большое число титрований, концентрации титрованных растворов лучше устанавливать так, чтобы они были эквивалентными. Так как раствор бромата более устойчив, чем раствор бромид-бромата, бромид можно вводить в анализируемый раствор непосредственно перед титрованием. Предложен метод непосредственного потенциометрического титрования броматом калия нагретого до 50 °C солянокислого раствора оксихинолята, в который введен бромид 2.
ОСАЖДЕНИЕ ТАННИНОМ
Таннин применяется для осаждения ряда элементов 1 2 3.
G осаждаемыми элементами таннин не образует соединений определенного состава, и часто его применение основано на .способности усиливать действие других реагентов. В некоторых случаях он придает осадкам требуемые физические свойства, иногда способствует осаждению, коагулируя коллоидные соединения, и, наконец, по причинам, пока еще не
1 Когда бромирование закончено, освобождается бром, что можно установить различными способами. Один из них заключается в следующем. Каплю анализируемого раствора прибавляют к 1 капле раствора иодида калия и 1 капле крахмала, нанесенным на фарфоровую пластинку. В случае появления синего окрашивания перемешивают раствор 1 мин и опять проводят испытание, чтобы удостовериться в том, что реакция прошла полностью. Если на этот раз синее окрашивание не появляется, вводят в раствор еще некоторое количество бромата и повторяют реакцию с иодидом и крахмалом. Появление в растворе свободного брома можно установить также следующим образом. Вводят в раствор 2—3 капли 0,2%-ного спиртового раствора метилового красного и титруют до исчезновения красной окраски или же в процессе титрования время от времени испытывают раствор добавлением капли индикатора. На окисление метилового красного расходуется бром, что должно быть учтено при вычислениях, если в раствор введено более 3 капель индикатора.
2 G. F. S m i t h, R. L. M a y, J. Am. Ceram. Soc., 22, 31 (1939).
3 Подробно о применении таннина в весовом анализе см. W. R.Schoeller. The Analytical Chemistry of Tantalum and Niobium, Chapman and Hall, London, 1937,
152
Гл. V. Специальные операции.
выясненным, вызывает выделение осадков в тех случаях, когда в его отсутствие они не образуются. Таннин применяется в присутствии таких различных по характеру реагентов, как аммиак, соляная кислота и некоторые органические соединения, например уксусная, винная, салициловая и щавелевая кислоты или их соли. При осаждении танинном обычно требуется более или менее тщательное регулирование концентрации ионов водорода в растворе. Это осаждение всегда проводят из содержащих электролит горячих растворов. При действии таннина выделяются объемистые хлопьевидные осадки, отфильтровывание которых не вызывает затруднений, особенно если вводить мацерированную бумагу и применять умеренное отсасывание. Если для дальнейшей работы используют фильтрат, таннин в нем можно легко разрушить обработкой дымящей азотной кислотой.
Осаждению из аммиачных растворов таннин может способствовать вследствие образования комплексных танниновых соединений вместо гидроокисей, которые осаждает аммиак (как, например, при осаждении алюминия х, бериллия 2 и редкоземельных металлов 3), или же вследствие образования осадков в растворах, содержащих органические ком-плексообразователи, препятствующие осаждению одним аммиаком (что наблюдается в случае выделения таких элементов, как тантал, ниобий, титан, цирконий, уран, редкоземельные металлы, бериллий и марганец)4.
В растворах, содержащих винную кислоту, могут быть созданы условия, при которых происходит осаждение таннином тантала, ниобия, титана, циркония, тория, урана, хрома, алюминия, .бериллия, ванадия и, вероятно, других элементов. Это имеет существенное значение, так как исключается необходимость разрушения винной кислоты, которую иногда приходится вводить в предыдущих операциях, как, например, при осаждении сероводородом металлов сероводородной групщи и группы сульфида аммония, которое проводится в присутствии винной кислоты сначала из кислого, а затем из аммиачного раствора. Осаждение таннином может быть осуществлено в сероводородном фильтрате после его подкисления, удаления сероводорода кипячением и последующей нейтрализации раствора.
Ниже приводится метод осаждения алюминия (также и галлия) таннином после предварительного отделения железа.
Ход анализа. К раствору алюминия и железа в 0,2—0,5 и. кислоте прибавляют 2—3 г винной кислоты и 10 г хлорида аммония и пропускают сероводород до полного восстановления железа. Если при этом выпадает осадок, не похожий на серу, его отфильтровывают и промывают подкисленной сероводородной водой. К фильтрату при перемешивании прибавляют разбавленный (1:1) аммиак в небольшом избытке, снова пропускают сероводород и оставляют стоять в течение нескольких часов, лучше на ночь. Осадок отфильтровывают и возможно быстрей промывают ^раствором, содержащим аммонийную соль, тартрат и сульфид. Для. полного разделения может потребоваться переосаждение осадка. Если нужно определить железо, осадок сохраняют. I
----г-—'
1 R. Ё. Divine, J. Soc. Chem. Ind., 24, И (1905).
2 L. Moser, J. Singer, Monatsh., 48, 673 (1927).
s G; F. Smith,.R. L. May, J. Am. Ceram. Soc., 22 , 31 (1939).
4 W. R. Schoeller, The Analytical Chemistry of Tantalum and Niobium, Chapman and Hall, London, 1937.
Осаждение таннином
153
Фильтрат, который должен иметь бледно-желтую окраску без зеленоватого оттенка, подкисляют соляной кислотой и кипятят для удаления сероводорода. К горячему раствору добавляют разбавленный (1:1) аммиак до слабощелочной реакции, затем 10%-ный раствор таннина в таком количестве, чтобы соотношение между таннином и содержащейся в растворе А12О3 составляло 40 : 1, и, наконец, 5 г ацетата аммония. После этого по каплям вводят разбавленный (1:1) аммиак до тех пор, пока 1—2 капли отстоявшейся жидкости не окрасят в яркий пурпурный цвет 1 каплю 0,04%-ного раствора бромкрезолового пурпурового, нанесенную на фарфоровую пластинку. После этого к раствору прибавляют еще 2 капли разбавленного аммиака, кипятят 5 мин и затем нагревают 30 мин при 80—100° С. При наличии одного алюминия осадок имеет белый цвет, слегка измененный реагентом. Танниновые осадки отдельных элементов обладают характерными окрасками. Так, например, осадок титана окрашен в оранжево-желтый цвет, циркония — в белый, железа — в черный, а ванадия — в темно-синий.
Раствор сливают декантацией через фильтр средней плотности, на котором находится небольшое количество беззольной мацерированной бумаги. К оставшемуся в стакане осадку прибавляют мацерированную бумагу, затем переносят его на фильтр и дают жидкости стечь. В рядовой работе осадок вместе с бумажной массой смывают обратно в стакан при помощи ,100 мл 2 %-ного раствора хлорида аммония, содержащего 1,0 г/л таннина и нейтрализованного по бромкрезоловому пурпуровому. Перемешивают и на этот раз полностью переносят осадок из стакана на фильтр и тщательно промывают. Для лучшего отделения алюминия от других элементов, таких, как кальций и магний, вместо промывания осадок лучше растворить в разбавленной соляной кислоте и повторить осаждение, а затем уже количественно перенести осадок на фильтр. После того как жидкость стечет, фильтр с осадком переносят во взвешенный тигель, лучше кварцевый, если окись алюминия должна быть затем сплавлена с пиросульфатом. Осторожно обугливают бумагу и осадок прокаливают до А12О3, как обычно.
Объемистые танниновые осадки можно отфильтровывать под слабым вакуумом через неплотный фильтр, вложенный в платиновый конус или в конус из плотной бумаги. В этом случае фильтрование следует приостанавливать прежде, чем осадок становится слишком сухим, так как он может растрескаться и отделиться от фильтра до того, как будет, окончательно промыт.
Осаждение титана, циркония, ванадия и, возможно, также тантала, ниобия, тория, урана, железа и хрома из растворов, содержащих винную кислоту, лучше проводить при pH = 6—7.
Из методов, основанных на осаждении таннином из ацетатных растворов, можно указать: 1) отделение бериллия от алюминия х, от железа,
1 L. Moser, М. Niessner, Monatsh., 48, 113 (1927); Митчел А. Д., У орд А. М., Новые методы количественного химического анализа, Госхимтехиздат, 1933. Согласно М. L. Nichols и J. М. Shempf [Ind. Eng. Chem., Anal. Ed., 11, 278 (1939)], алюминий можно отделить от бериллия, таннином при условии, если pH раствора приведен точно к 4,6 и для промывания осадка применяют 5%-ный раствор ацетата аммония, приведенный к тому же pH и содержащий немного таннина. 1 Бериллий можно осаждать в фильтрате добавлением избыточного количества таннина и аммиака до достижения pH раствора порядка 7,5.
154
Гл. V. Специальные операции
хрома, тория, ванадия, титана и циркония 1 и от олова 2; 2) определение урана 3 4 *, галлия * и меди 6.
Другие случаи применения таннина приводятся при оксалатном методе отделения тантала от ниобия (стр. 681) и при таннино-цинхонино-вом методе 'осаждения вольфрама (стр. 766).
ОСАЖДЕНИЕ ФЕНИЛГИДРАЗИНОМ
Некоторые органические основания (фенилгидразин, анилин, пиридин и др.) применяются в качестве реагентов для отделения алюминия и других,’гидролитически легко осаждающихся металлов от элементов, соли которых мало гидролизуются 6. Из названных соединений фенилгидразин в настоящее время находит довольно широкое применение в техническом анализе для отделения алюминия от железа (II). Осадок гидроокиси алюминия прокаливают и взвешивают в виде А12О3. Фенилгидразин можно использовать также и в других случаях, особенно тогда, когда надо доказать присутствие очень малых количеств алюминия.
Фенилгидразин количественно осаждает также титан, цирконий, торий и хром лучше всего из растворов их сульфатов. Фосфор и ванадий осаждаются, если содержание их в растворе не превышает таких количеств, какие могут быть увлечен!! выделяющимися металлами. Бериллий выделяется не количественно. Церий (IV) и железо (III) частично восстанавливаются и полностью не осаждаются. Цинк, кадмий, ртуть, кобальт ?
и никель при достаточной концентрации образуют с фенилгидразином
малорастворимые продукты присоединения. Щелочноземельные металлы (из растворов хлоридов или нитратов), магний., марганец и железо (II) не осаждаются.
Первоначально предложенный метод осаждения фенилгидразином 7 -
был впоследствии несколько видоизменен. Ниже приводится этот видоизмененный метод для определения алюминия 8.
Анализируемый раствор, лучше всего сернокислый, в котором все ) металлы находятся в виде сульфатов (кроме тех случаев, когда присут- ) ствуют щелочноземельные металлы), свободный от кремнекислоты, метал- j лов сероводородной группы и значительных количеств цинка, кобальта j
и никеля, слабо подщелачивают аммиаком по метиловому оранжевому. :
Затем подкисляют его лишь настолько, чтобы удержать металлы в рас- 1
творе, и разбавляют до 100—200 мл, в зависимости от содержания алюми- j
ния. Раствор нагревают и восстанавливают железо насыщенным раство- 1
ром бисульфита аммония (5—20 капель, в зависимости от содержания железа в растворе). Если раствор окрашивается сульфитом железа (III) в темно-красный цвет, что указывает на его низкую кислотность, прибавляют несколько капель соляной кислоты, так как при недостаточной
1 L. М о s е г, J. Singer, Monatsh., 48, 673 (1927).
2 L. М о s е г, F. L i s t, Monatsh., 51, 1133 (1929).
3P. N. Das-Gupta, J. Indian. Chem. Soc., 6, 763 (1929); L. M о s e r, K. Neumayer, K. W i n t e r, Monatsh., 55, 85 (1930).
4 L. M о s e г, А. В r u k 1, Monatsh., 50, 567 (1928).
6 M. B. D a r b i n i a n, A. G. К a n k a n i a n, Z. anal. Chem., 99, 29 (1934).
6 См. мелкий шрифт на стр. 303.
7 W. Н. Н е s s, Е. D. С a m р b е 11, J. Am. Chem. Soc., 21, 776 (1899).
8 Е. Т. Allen, J. Am. Chem. Soc., 25, 421 (1903). i
I
.=i
Осаждение хлоридом тетрафениларсония
155
концентрации кислоты восстановление железа сульфитом идет -слишком медленно. Затем раствор быстро нейтрализуют аммиаком и добавляют 6—7 капель разбавленной (1:1) соляной кислоты. Если эта операция проводится недостаточно быстро, то за счет окисления кислородом воздуха может образоваться небольшое количество гидроокиси железа (III), которая не всегда достаточно легко растворяется в разбавленной кислоте. К раствору прибавляют, в зависимости от содержания алюминия, от 1 до 3 мл фенилгидразина х, немного мацерированной беззольной бумаги, перемешивают, пока осадок не станет хлопьевидным, и дают отстояться. Проверяют полноту осаждения прибавлением нескольких капель реактива к отстоявшемуся раствору или к фильтрату. Отстоявшаяся жидкость должна иметь щелочную реакцию по метиловому оранжевому и явно кислую по лакмусу. Коричневая окраска осадка не должна вызывать сомнений, так как она обусловлена не гидроокисью железа, а окрашенным веществом, которое всегда содержится в препарате фенилгидразина, если он не был перегнан незадолго до его npHMeHeHHrf. При продолжительном стоянии раствора количество этого продукта окисления под действием воздуха увеличивается и на поверхности раствора и стенках стакана образуется коричневая нерастворимая пленка, которая в дальнейшем может вызвать затруднения. Однако этого можно избежать, так как равновесие устанавливается быстро. Осадок отфильтровывают и промывают раствором фенилгидразинсульфита 1 2.
При точных анализах, а также в случае содержания значительных количеств железа, осадок растворяют в горячей разбавленной серной или соляной кислоте и осаждение повторяют. Осадок прокаливают и взвешивают в виде А12О3 совместно с окислами других перечисленных выше металлов, если они присутствовали в растворе.
ОСАЖДЕНИЕ ХЛОРИДОМ ТЕТРАФЕНИЛАРСОНИЯ
Хлорид тетрафениларсония (C6H5)4AsC1 может быть использован в качестве реагента в весовом и объемном анализе для определения ртути, олова, золота, платины, кадмия, цинка, перхлоратов, перйодатов, перманганатов и перренатов 3. В растворах, содержащих хлорид натрия (1,0—2,5 М) и разбавленную кислоту (0,2—1,0 М), исключая азотную, тетрафениларсоний реагирует с последними четырьмя соединениями с образованием нерастворимых солей, которые могут быть взвешены. Остальные элементы не образуют осадков, пригодных для взвешивания. Они осаждаются при добавлении избыточного количества реактива, которое можно затем определить потенциометрическим титрованием иодом.
1 Реагент должен быть свободен от неорганических примесей, присутствие котовых может исказить результаты анализа. Получив при анализе повышенные результаты, Allen обнаружил в осадке олово.
2 Приготовляется прибавлением холодного насыщенного раствора сернистой кислоты к нескольким миллилитрам фенилгидразина до тех пор, пока образующийся вначале кристаллический сульфит не растворится в избытке сернистой кислоты. Затем при сильном перемешивании прибавляют раствор фенилгидразина до исчезновения запаха сернистого ангидрида. К 100 мл горячей воды прибавляют 5—10 мл этого раствора.
3 Н. Lamphrey, диссертация, Univ. Michigan; Н. Н. W i 1 1 а г d, G. М. Smith, Ind. Eng. Chem., Anal. Ed., 11, 186, 269 (1939).
e
156 Гл. V. Специальные операции
Эти методы имеют ограниченное применение, так как многие ионы препятствуют определению. Помимо перечисленных, весовому определению мешают железо, висмут, сурьма (III), мышьЯк (III), фториды, бромиды, иодиды, оксалаты, ацетаты, цитраты, роданиды, фосфаты, молибдаты, хроматы, вольфраматы и большие количества нитратов. На результаты объемного определения влияют все ионы, которые окисляют иодид или восстанавливают иод.
ОСАЖДЕНИЕ п-ОКСИФЕНИЛАРСОНОВОЙ КИСЛОТОЙ
n-Оксифениларсоновая кислота НО—СвН4—AsO(OH)2 была рекомендована для определения титана, циркония, а также циркония в присутствии титана х. Титан осаждается из растворов приблизительно 0,6 н. по "концентрации соляной или 1,8 н. по концентрации серной кислоты. Осаждение циркония может быть осуществлено при концентрации кислоты, равной 3,0 н. Для отделения титана в раствоф вводят перекись водорода. При наличии в растворе железа прибавляют 1—2 г роданида аммоний на каждые 100 мл раствора. Осадок промывают раствором, соответствующим по составу фильтрату, переносят в фарфоровый тигель и прокаливают под хорошей тягой до окиси.
Титан можно осаждать в присутствии железа (II и III), алюминия, цинка, кобальта, никеля, бериллия, хрома (III), марганца (II), кальция, магния, таллия, церия (III), тория, натрия, калия, аммония, а также фосфатов, молибдатов, хроматов, ванадатов, перманганатов, уранила и ванадила. Мешают определению ионы циркония, церия (IV) и олова. Перекись водорода также должна отсутствовать. На осаждение циркония влияют церий (IV), олово, большие количества фосфата, а также титан при отсутствии в растворе перекиси водорода.
МЕТОДЫ ОПРЕДЕЛЕНИЯ С ДИФЕНИЛТИО КАРБАЗОНОМ (ДИТИЗОНОМ)
Дитизон образует ярко окрашенные внутрикомплексные соединения со многими элементами и имеет широкое .применение для отделения и определения малых количеств некоторых тяжелых металлов, например свинца, цинка и кадмия. Для выполнения реакции к водному раствору соли ' тяжелого металла, имеющему соответствующий pH, добавляют раствор дитизона в не смешивающемся с водой органическом растворителе, например в хлороформе или четыреххлористом углероде. Образующаяся при этом окрашенная комплексная соль дитизона с металлом растворяется в органическом растворителе. Специфичность реакции может быть повышена тщательным регулированием pH водного раствора, введением таких комплексообразователей, как цитраты, тартраты, цианиды, роданиды, или в некоторых случаях (например, при орределении олова) переведением элемента в соответствующее валентное состояние.
Содержание тяжелого металла в органическом растворителе, в котором наряду с дитизонатом этого металла находится избыток дитизона,
1 С. Т. Simpson, G. С. С h a n d 1 е е, Ind. Eng. Chem., Anal. Ed., 10, 642 (1938),
Методы определения с комплексоном , 157
может быть установлено спектрофотометрическим методом или же визуально сравнением окраски раствора с серией стандартов, содержащих одинаковые с испытуемым раствором количества дитизона х.
♦ МЕТОДЫ ОПРЕДЕЛЕНИЯ С КОМПЛЕКСОНОМ 2
К числу заслуживающих внимания органических реагентов относится также двунатриевая соль этилендиаминтетрауксусной кислоты
NaOOCCH24 zCH2COONa
)N-CH2-CH2-N< HOOCCH2Z XCH2COOH
которая относительно недавно получила применение в аналитической химии. Этот реагент, известный под названием3 комплексон III (трилон Б), в последнее время приобретает все большее значение, и, судя по литературным данным, может быть использован в самых разнообразных областях неорганического анализа. В аналитической практике комплексон III впервые был применен в качестве титранТа для определения жесткости воды (т. е. кальция и магния) 4. Первоначально предложенный метод сводился к алкалиметрическому титрованию кислоты, освобождающейся в результате взаимодействия металла с комплексоном III:
Ме2+ + Н2Х2" —> МеХ2“ + 2Н+
Впоследствии был предложен 5 и получил широкое распространение прямой метод тцтрования кальция и магния комплексоном III в присутствии индикатора, взаимодействующего с этими- металлами с образованием яркоокрашенного соединения. При этом в точке эквивалентности, когда все ионы металла связаны в прочный комплекс с комплексоном III, в растворе появляется окраска, присущая свободному индикатору.
Помимо кальция и магния, многие другие двухвалентные, а также и трехвалентные металлы с комплексоном III образуют растворимые в воде прочные внутрикомплексные соединения, что дало возможность значительно расширить область применения j, комплексометрического метода титрования. В настоящее время описаны условия титрования комплексоном III меди, никеля ®, свинца, цинка, таллия, кадмия, кобальта,
1 Подробности и библиографию см. Е. Б. С е н д э л, Колориметрическое определение следов металлов, Госхимиздат, 1949. Из числа органических реагентов, нашедших применение, хотя и менее обширное, чем дитизон, следует упомянуть бен-зоицоксим (см. «Молибден», стр. 364), а-нитрозо-(3-нафтол (см. «Кобальт», стр. 473), нитрон (см. «Азот», стр. 867 и «Рений», стр. 373), фениларсоновую кислоту (см. «Цир-, копий», стр, 638, «Торий», стр. 606 и «Олово», стр. 334), н-пропиларсоновую кислоту (см. «Цирконий», стр. 638), солянокислый бензидин (см. «Сера, стр. 804) и таннин (стр. 151).
2 См. Р. П р ш и б и л, Комплексоны в химическом анализе, Издатинлит, 1955.
3 Этилендиамин*гетрауксусная кислота известна под названием комплексон II. В английской литературе эту кислоту называют версен, сокращенно обозначаемый иногда ЭДТА, а ее натриевую соль — версенат.
1 G. S с h w-a rzenbach, W. Biedermann, F. Banger ter, Helv. Chim. Acta, 29, 811 (1946). Эти авторы впервые назвали реагент комплексоном, откуда предложенный ими метод титрования впоследствии получил название комплексометрического.
5 G. Schwa г z е nba ch, W. Biedermann, Helv. Chim. Acta, 31, 678 (1948).
• H. Flaschka, Mikrochim. Acta, 39, 38 (1952); W. Harris, T. Sweet, Anal. Chem., 23, 1032 (1952).
158
Гл. V. Специальные операции
марганца, ртути, индия \ железа 1 2, тория 3 4 и многих других элементов с применением индикаторов или с потенциометрическим контролем точки эквивалентности *. '
‘Известный интерес представляет возможность выполнения титрования комплексоном III в присутствии осадков, что используется, например, для определения кальция и магния в растворах, содержащих цинк и медь, влияние которых устраняется осаждением их в виде сульфидов 5. На этом же принципе основано комплексометрическое определение магния в присутствии кальция, который осаждают в виде оксалата 6 7.
Используются также окрашенные внутрик&мплексные соединения некоторых элементов с комплексоном III для их колориметрического определения. Такие методы описаны, например, для хрома (III) марганца (III) 8 9, никеля и меди ®.
Комплексообразующее действие комплексона III успешно используется в аналитической практике для устранения влияния посторонних элементов. Так, например, способность двух- и трехвалентных металлов образовывать прочные комплексные соединения с комплексоном III дает возможность осаждать уран 10 и титан и, а также и бериллий 12 (который в отличие от большинства двухвалентных металлов не образует комплексных соединений с комплексоном III) аммиаком в присутствии многих элементов, в том числе алюминия и железа, что имеет весьма важное практическое значение. Описано также, применение комплексона III при определении вольфрама и молибдена осаждением оксихинолином в ацетатной среде. Установлено, что в этих условиях осаждаются только молибден, вольфрам, уран и ванадий (V) 13.
Комплексон III в качестве комплексообразователя применяется также при колориметрическом определении некоторых элементов. Так, при определении бериллия в меднобериллиевых бронзах по реакции с алюминоном влияние посторонних элементов, в том числе и меди, устраняется 14 введением в раствор комплексона III. Медь связывается в комплекс с комплексоном III также и при колориметрическом определении ртути с дитизоном 15. введением в раствор комплексона III устраняется
1 Н. Flaschka, Mikrochim. Acta, 39, 315 (1952); 40, 42 (1952); J. К i n n u-nen, B. Mericanto, Chemist Analyst, 41, 76 (1952); N. Strafford, Analyst, 78, 733 (1953); H. Flaschka, Chemist Analyst, 42, 56 (1953); H. Flaschka, A. A m i n, Z. anal. Chem., 140, 6 (1953).
2 D. L у d e r s e n, O. G jems, Z. anal. Chem., 138, 249 (1953).
3 K. ter Haar, J. В a z e n, Anal. Chim. Acta, 9, 235 (1953); J. Fritz, J. Ford, Anal. Chem., 25, 1640 (1953).
4 R. Prihil, Z. К ou del а, В. B. Matyska, Coll. Czech. Chem. Comm., 16, 80 (1951).
5 T. Б. Стюнкел ь, E. M. Якимец, Д. А. Савиновский, ЖАХ, 8, 163 (1953).
6 3. В. Архангельская, Зав. лаб., 20, 44 (1954).
7 R. Prihil, J. Kluhalova, Coll. Czech. Chem. Comm., 15, 42 (1950).
8 R. Prihil, E. Hornichova, Coll. Czech. Chem. Comm., 15, 456 (1950).
9 P. Sweetser, C. Bricker, Anal. Chem., 25, 253 (1953).
10 R. Prihil, J. Uorlichek, Chem. Listy, 46, 216 (1952).
11 R. Prihil, P. Schneider, Coll. Czech. Chem. Comm., 15, 886 (1950).
12 R. Prihil, J. Kucharsky, Coll. Czech. Chem. Comm., 15, 132 (1950).
13 R. P r i b i 1, M. M a 1 a t, Coll. Czech. Chem. Comm., 15, 120 (1950); R. P r i-bil, V. S e d 1 a r, Coll. Czech. Chem. Comm., 16, 69 (1951).
14C. Luke, M. C a m p b e 11, Anal. Chem., 24, 1056 (1952).
16 A. V a s”a k, V. S e d i v e c, Coll. Czech. Chem. Comm., 15, 1076 (1950).
Взаимное превращение хлоридов, нитратов и сульфатов
159
влияние даже больших количеств железа, никеля, кобальта и марганца на колориметрическое определение меди по реакции с диэтилдитиокарбаматом натрия в аммиачной среде. В этих условиях с диэтилдитиокарбаматом реагируют только медь, серебро, ртуть, свинец и висмут х. Комплексообразующее действие комплексона III, кроме того, используется для сдвига окислительно-восстановительного потенциала системы при потенциометрическом 1 2 и амперометрическом3 титровании в полярографии 4. Доп. перев.*
ВЗАИМНЫЕ ПРЕВРАЩЕНИЯ ХЛОРИДОВ, НИТРАТОВ
И СУЛЬФАТОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Превращение хлоридов, нитратов и других солей щелочных металлов в сульфаты выпариванием с серной кислотой осуществляется достаточно просто, и эта операция не нуждается в пояснении;
То же можно сказать и о превращении хлоридов в нитраты повторным выпариванием с азотной кислотой. Количественное превращение нитратов в хлориды более затруднительно и не обеспечивается выпариванием с соляной кислотой. В таких случаях лучше сначала перевести нитрат в карбонат, как описано ниже. Последующее превращение карбоната в хлорид не вызывает особых затруднений. Рассмотрим некоторые представляющие интерес методы превращения одних солей' в другие.
\
Превращение сульфатов в хлориды
Превращение сульфатов в хлориды обычно проводится осаждением сульфат-иона эквивалентным или избыточным количеством хлорида бария. Избыток осадителя затем удаляют прибавлением карбоната аммония. Оба метода страдают известными недостатками, и вместо них предлагается следующий способ 5.
Водный раствор сульфата обрабатывают избыточным количеством ацетата свинца, слегка нагревают, фильтруют и умеренно промывают осадок горячим разбавленным раствором ацетата свинца. Прозрачный горячий раствор насыщают сероводородом. Осадку сульфида свинца дают отстояться, затем его отфильтровывают и промывают разбавленной насыщенной сероводородом уксусной кислотой.
Прозрачный фильтрат выпаривают почти досуха, прибавляют в избытке соляную кислоту, снова выпаривают и сухой остаток прокаливают приблизительно при 250° С. Вместо обычно применяемого ацетата свинца можно пользоваться хлоридом свинца. В этом случае прибавление соляной кислоты излишне. При отсутствии элементов, восстанавливающихся сероводородом, можно не опасаться его окисления до серной кислоты, если раствор не оставлять слишком долгое время стоять перед фильтрованием.
1 V. V a s a k, V. S е d i v е с, Coll. Czech. Chem. Comm., 15, 260 (1950).
v 2 R. P r i b i 1, L. S v e s t к a, Coll. Czech. Chem. Comm., 15, 31 (1950); J. D o-1 e z a 1, V. H e n c i, V. Simon, Chem. Listy, 46, 267 (1952); V. S i m о n, J. S e-kerka, J. Dolezal, Chem. Listy, 46, 613 (1952):
3 V. Simon, J. Sekerka, J. Dolezal, Chem. Listy, 46, 613 (1952).
‘ С. И. Синякова, ЖАХ, 8, 333 (1953).
6 J. L. S m i t h, Am. J. Sci., 15, 234 (1853); 16, 53 (1953); Erdm. J. Pract. Chem., 59, 159 (1853); 60, 244 (1853).
160 Гл. V. Специальные операции
f
Превращение хлоридов и нитратов в карбонаты обработкой щавелевой кислотой
Этот способ основан на замещении летучих соляной и азотной кислот более устойчивой щавелевой кислотой х.
Раствор хлоридов или Нитратов щелочных металлов обрабатывают в колбе избытком щавелевой кислоты при нагревании на водяной бане Вначале реакция идет довольно медленно, но когда температура дость гает примерно 100° С, она становится бурной и в случае необходимости должна быть замедлена охлаждением раствора. Раствор выпаривают досуха, прибавляют немного воды и, если требуется, повторяют выпаривание, следя за тем, чтобы в смеси всегда находился избыток щавелевой кислоты. Под конец остается смесь щавелевой кислоты и оксалатов, которые могут быть осторожным прокаливанием переведены в карбонаты.
Превращение сульфатов в карбонаты
Превращение сульфатов в карбонаты основано на реакции двойного обмена с карбонатом бария 1 2.
Сульфаты помещают в колбу и растворяют в воде (20—30 мл воды на 1 г соли). Воду насыщают двуокисью углерода до или после прибавления ее к сульфату. Затем вводят свежеприготовленный карбонат бария в виде жидкой пасты р количестве, приблизительно на 50% превышающем теоретически необходимое для образования сульфата бария. Закрывают колбу и оставляют на несколько часов, часто взбалтывая. После Этого нагревают до кипения, фильтруют и осадок промывают.
Превращение сульфатов В тартраты, оксалаты и др.
Несмотря на то что и тартрат, и оксалат бария мало растворимы в воде, превращение может быть выполнено следующим образом 2. К раствору сульфата, находящемуся в чашке, прибавляют карбонат бария, растертый с водой до густой пасты, в количестве, соответствующем приблизительно 7 частям карбоната на 5 частей сульфата. Массу нагревают _ и вводят понемногу требуемое количество щавелевой или винной кислоты. Растворение карбоната бария и осаждение сульфата бария происходит быстро, и разложение вскоре заканчивается. Фильтруют и промывают осадок горячей водой. Если в исходном сульфате содержится магний, то он при этом отделяется от щелочных металлов.
Из оксалатов и тартратов щелочных металлов можно, затем получить карбонаты выпариванием раствора досуха и прокаливанием остатка.
Превращение хлоридов в окислы
Хлориды многих металлов могут быть легко и быстро перевбдены в окислы, если смешать их с водной суспензией окиси ртути, приготовлен- -ной, как описано на стр. 109, смесь выпарить досуха и затем осторожно прокалить под хорошей тягой до удаления хлорида ртути и избытка окиси ртути 3.
1 J. L. Smith, Am. J. Sci., 16, 373 (1853).
2,J. L. S m i t h, Am. Chemist, 3, 341 (1873); Chem. News, 27, 316 (1873).
3 J. Volhard, Ann., 198, 331 (1879); E. F. S m i t h, P. H e у 1, Z. anorg. Chem., 7, 82 (1894).
Удаление солей аммония 161
УДАЛЕНИЕ СОЛЕЙ АММОНИЯ
Хлорид аммония
Иногда в процессе анализа в растворе концентрируются такие значительные количества солей аммония, что они затрудняют проведение дальнейших операций, и их необходимо удалить (например, при определении следов кальция и магния). Обычно для этой цели раствор упаривают досуха и хлорид аммония осторожно возгоняют. Такая операция довольно продолжительна, и требуется большая аккуратность, чтобы избежать механических потерь, а также потерь, связанных с летучестью определяемых компонентов. Значительно надежнее и более удрбен следующий метод х.
Раствор слабо подкисляют соляной кислотой, выпаривают в стакане или большой чашке и затем добавляют на каждый грамм хлорида аммония примерно 3 г азотной кислоты (избыток не мешает). Стакан закрывают часовым стеклом, осторожно нагревают до прекращения бурного выделения газов и затем раствор выпаривают досуха. Этим способом можно легко, не опасаясь потерь, отделить до 100 г хлорида аммония от нескольких миллиграммов магния, щелочных или щелочноземельных металлов.
Нитрат аммония
При аналитических операциях редко приходится сталкиваться с содержанием в растворе больших количеств нитрата аммония, но в тех случаях, когда это необходимо, нитрат аммония также может быть удален выпариванием раствора досуха и последующим прокаливанием. В отличие от хлорида аммбния, нитрат аммония плавится при температуре около 165° С й начинает разлагаться на воду и окислы азота при температуре около 185° С. При 240° С разложение идет настолько бурно, что смесь может взорваться. Более удобным является способ, основанный на принципе, описанном выше (см. «Хлорид аммония»), с той разницей, что к выпаренному до небольшого объема раствору следует добавить не азотную, а соляную кислоту. Разложение нитрата аммония идет не так легко, как хлорида аммония, и выпаривание иногда приходится повторять несколько раз.
Сульфат аммония
Сульфат аммония, так же как нитрат и хлорид аммония, можно удалить возгонкой;. Разложение сульфата аммония осуществляется обработкой раствора царской водкой. При- этом конечным продуктом реакции является серная кислота.
ИЗВЛЕЧЕНИЕ ЭФИРОМ
Извлечение хлорида железа (III) эфиром из солянокислого раствораf является удобным методам отделения больших количеств железа от меньших количеств других элементов, например никеля
1 J. L. Smith, Am. J. Sci., 15, 94 (1853); Am. Chemist, 3, 201 (1873); A. C. Langmuir, J. Am. Chem. Soc., 22, 104 (1900).
s J. W. Rothe, Stahl u. Eisen, 12, 1052 (1892); A. L e d e b u г, там же, 13, 333 (1893); A. C. L a n g m u i r, J. Am. Chem. Soc., 22, 102 (1900); E. F. К e r n, там же, 23, 689 (1901); F. N. S p ell er, Chem. News 83, 124 (1901).
И Заказ 522.
162
Гл. V. Специальные операции
ТАБЛИЦА 1 О
Извлечение хлоридов некоторых элементов эфиром
(Значения приблизительные и были получены в разных условиях)
Элементы Извлечение % Литературный источник Элементы Извлечение % Литературный источник
Al ’ 0 1 Mo (МоО3) 80-90 6
Sb(SbCl3) ' 6 2 Ni 0 7
Sb(SbCl5) 81 2 Os 0 3
As(AsCl3) 68 2 P (P2O5) Заметные 4
количества
As(AsC15) 2—4 3 Pd (PdCl2) 0 ' 2
Be 0 3 Pt (PtCl4) Следы 2
Bi 0 3 РЗЭ 0 6
Ca 0 4 Rh 0 3
Cd 0 3 Se Следы 3
Cr 0 1 Ag 0 2
Co 0 1 Те (TeCl4) 34 2
Cu 0,05 2 Th (ThCl4) 0 6
Ga 97 3 T1 (T1C13) 90-95 . 8
Ge 40-60 3 Sn (SnCl4) 17 2
Au(AuCl3) 95 2 Sn (SnCl2) 15—30 3
In Следы 5 Ti 0 8
It (IrCl4) 5 2 W (c PO4) 0 3
Fe (FeCl3) 99 2 ' u О 9
Fe(FeCL) 0 3 V (V2O5) Следы 3
Pb 0 • 2 V (V2O4) » 4
Mn 0 1 Zn 0,2 2
Hg(HgCl2) 0,2 2 Zr 0 8
1 F. N. Speller. Chem. News, 83, 124 (1901).
2 F. Mylius, C. Huttne, Ber., 44, 1315 (1911).
3 California Institute of Technology (не опубликовано).
4 National Bureau oj Standards (не опубликовано).
& I. W a d a, S. A to, Sci. Papers Inst. Phys. Chem. Research, Tokyo, 1, 70 (1 922).
6 Massachusetts Institute of Technology (не опубликовано).
1 A. C. Langmuir, J. Am. Chem. Soc., 22, 102 (1900).
S A. A. Noyes, W. C. Bray, E. B, 'Spear, J. Am. Chem. Soc., 30, 51 5, 559 (1908).
9 E F. Kern, J. Am. Chem. Soc., 23, 689 (1901).
и алюминия !. Экстрагирование эфиром может быть с успехом применено также и для отделения некоторых других элементов, как, например, молибдена, золота, галлия и таллия (III).
Данные, приведенные 1 2 в табл. 10, показывают поведение ряда элементов при однократном извлечении эфиром, не содержащим спирта.
1 Помимо никеля и алюминия, в водном слое остаются также железо (II), кобальт, марганец, хром, титан и уран. Небольшие количества меди и ванадия переходят совместно с железом в эфирный слой, но для большинства целей разделение достаточно удовлетворительно. То же может быть сказано об отделении железа от серной и фосфорной кислот. Изучая поведение «следов» (<0,005%) различных элементов при извлечении хлорида железа из разбавленного солянокислого раствора изопропиловым эфиром, Р. Е. Eighty и J. D. Moulton нашли, что значительная часть As, Mo, Se и Те переходит совместно с железом в эфирный слой, потери В и V умеренны (около 20%), a Al, Sb, Ba, Bi, Cd, Са, Сг, Со, Си, Pb, Mg, Мп, Ni, Si, Ag, Sn, Ti, W и Zn полностью остаются в водном слое. Опыты проводились следующим образом. Солянокислый раствор (с несколько более высокой концентрацией соляной кислоты, чем 1 : 1), содержащий около 3 мг примесей и 30 г железа (III) в виде хлорида, экстрагировался шестью порциями эфира (в общей сложности 900 мл) и затем, после переведения хлоридов в сульфаты, исследовался спектроскопически.
2 Е. Н. Swift, J. Am. Chem. Soc., 46, 2378 (1924).
Извлечение эфиром
163
Железо лучше всего экстрагировать из солянокислых растворов (пл. 1,1—1,115), содержащих около 1 г железа в 20 мл. Для извлечения такого количества железа требуется 50 мл эфира, свободного от спирта. Экстрагируемый раствор не должен содержать таких веществ, как свободный хлор или азотная кислота (разлагающих эфир), солей, нерастворимых в соляной кислоте, насыщенной эфиром, как, например, хлориды
щелочных металлов, а также серную кислоту, которая снижает степень
извлечения железа в эфирный слой. Железо никогда сразу не извлекается
количественно, 1—2 мг его обычно остается в водном слое. Небольшие количества хлоридов таких элементов, как никель и медь, могут раствориться в эфире в случае большой концентрации их в растворе. Поэтому солянокислый раствор для более полного отделения железа сначала экстрагируют эфиром по меньшей мере дважды, а затем эфирный экстракт встряхивают с соляной кислотой для удаления никеля и других элементов. Экстрагирование может быть выполнено в обычных делительных воронках или в специальном приборе Роте (рис. 15), более удобном для смешивания и сливания растворов. Для отделения 5 г железа пригодна воронка емкостью 110 мл; при более высоком содержании железа емкость воронки должна быть увеличена до 250 мл. Необходимо иметь в виду, что экстрагирование нельзя проводить близко от огня, так как пары эфира могут воспламениться.
Ход анализа. Приготовляют растворы: А) Соляная кислота пл. 1,10 г/см3; Б) Соляная кислота пл. 1,19 г/см3, насыщенная эфиром; В) Соляная кислота пл. 1,10 г/см3, насыщенная эфиром. Для получения растворов Б и В соляную кислоту соответствующей концентрации охлаждают, встряхивают с небольшой порцией эфира и затем снова охлаждают до 10° С. Эту
«
.4
Рис. 15. Прибор Роте для экстрагирования эфиром.
операцию повторяют до тех пор, пока над кислотой не останется слой эфира. В 100 мл концентрированной соляной кислоты растворяется 150 мл эфира, а в 100 мл кислоты пл. 1,10 г/см3 растворяется 30 мл.
Анализируемый солянокислый раствор, содержащий железо в виде FeCl3 и свободный от перечисленных выше мешающих экстрагированию
элементов, выпаривают в чашке до тех пор, пока он не станет вязким, що кристаллизация еще не начнется. Переносят раствор в делительную воронку 3 и несколько раз ополаскивают чашку и воронку 4 соляной кислотой, порциями по 1—2 мл, употребив для этого возможно меньшее количество кислоты.
Раствор перемешивают, охлаждают и на каждый грамм содержащегося в растворе железа добавляют по 6 мл раствора Б- Осторожно встряхивают и затем снова охлаждают. После этого на каждый грамм железа вводят по 30 мл эфира. Закрывают воронку пробкой, охлаждают до 10° G под струей холодной воды и встряхивают. Дают отстояться, после чего водный слой сливают в воронку 1. Ополаскивают воронку 3 и кран 5 мл раствора В, осторожно вращая воронку.
Дают отстояться и сливают кислоту в воронку 1. Для извлечения из эфирного слоя никеля, меди и т. и. элементов вводят в воронку 3 через
11*
164
Гл. V. Специальные операции
воронку 4 10 мл раствора В. Для повторного извлечения железа из кислого раствора в воронку 1 через воронку 2 вводят 50 мл, или более, эфира. Охлаждают, хорошо встряхивают, дают отстояться и затем сливают водный слой из воронки 1 в стакан. После этого через воронку 2 вводят 5 мл раствора В, осторожно вращают воронку, а затем дают жидкости отстояться и кислоту сливают в тот же стакан.
Для извлечения посторонних хлоридов из' эфирных экстрактов, находящихся в воронках, кислый раствор из воронки 3 сливают в воронку 1, после чего через воронку 4 снова вводят 10 мл раствора В. Воронку 1 встряхивают, дают раствору отстояться и затем водный слой сливают в стакан. Отстоявшийся кислый раствор из воронки 3 спускают в воронку 1, встряхивают и после расслоения этот водный слой также сливают в стакан. Экстрагирование таким путем повторяют сообразно с концентрацией нелетучих хлоридов в растворе, а также с требуемой четкостью разделения. Эфир из кислого раствора удаляют осторожным нагреванием закрытого часовым стеклом стакана на краю водяной бани. Из эфирного экстракта извлекают железо встряхиванием с водой \
Имеется указание 1 2, что для полного извлечения железа многократным экстрагированием эфиром процесс следует проводить при слабом искусственном освещении или в темноте, так как железо в эфирном растворе фотохимически восстанавливается до двухвалентного 3. Для многократного экстрагирования эфиром весьма удобен видоизмененный жидкий экстрактор Фридриха 4.
Экстрагирование эфиром растворов см. стр. 161, а экстрагирование купфератов — стр. 147.
ЭЛЕКТРОЛИТИЧЕСКИЕ МЕТОДЫ
Методы, основанные на электролитическом осаждении металлов или их гидроокисей, нашли широкое применение в аналитической практике *. Эти методы особенно удобны тем, что обычно они не требуют введения в раствор посторонних веществ; сводят к минимуму затруднения, связан
1 Согласно J. А х е 1 г о d и Е. Н. S w i f t [J. Am. Chem. Soc., 62, 33 (1940)], Р,Р'-дихлорэтиловый эфир обладает тем преимуществом перед диэтиловым и изопропиловым эфирами, что он может быть применен для экстрагирования сильнокислых растворов (>7 н.) и повторное извлечение можно проводить в той же воронке, так как эфир переходит в нижний слой. Неудобством является более медленное разделение водной и эфирной фаз.
2 S. Е. Q. A s h 1 е у, W. М. М и г г а у, J. Ind. Eng. Chem., Anal. Ed., 10, 367 (1938).
3 При наличии в эфире перекиси в процессе экстрагирования может произойти взрыв [М. S. К harasch, М. Gladstone, J. Chem. Education, 16, 498 (1939)]. Поэтому перед употреблением эфир всегда следует проверять на содержание перекиси и в случае положительной реакции разрушать ее обработкой бисульфитом натрия или сульфатом железа (II). Для испытания эфир встряхивают с подкисленным раствором • иодида калия. Появление через 5 мин по окончании встряхивания коричневой окраски указывает на наличие перекиси. Можно также, взболтав эфир с сульфатом железа (II) без доступа воздуха, провести пробу с роданидом калия на содержание железа (III) в растворе. Методы.испытания приведены в статье G. М i d d 1 е t о n, F. С. H у m a s, Analyst, 53, 201 (1928).
* f. В. Heberling, Anal. Chem., 21, 331 (1949).
3 S. E. Q; A sh 1 ey, Anal. Chem., 21, 70 (1949); A. Schleicher, Elektroanaly-tische Schnellmethoden, 3-te AufL, F. Enke, Stuttgart, 1947.
Электролитические методы.
165
ные с процессом соосаждения элементов, и, кроме того, экономят время работника.
Единственным недостатком электролитических методов является то, что количественное осаждение достигается лишь в редких случаях вследствие теоретической зависимости между концентрацией ионов в растворе и электродным потенциалом. Однако в большинстве случаев имеется возможность так подобрать соответствующий потенциал электрода, чтобы в растворе оставались лишь ничтожные количества осаждаемого элемента.
Поскольку для осаждения обычно пользуются током постоянного напряжения, потенциал электрода в процессе электролиза может колебаться в широких пределах.
Электролитические методы, как правило, не отличаются большой специфичностью.
Однако различными способами электролитическое осаждение можно сделать избирательным, например: 1) контролем электродного потенциала; 2) применение ртутного катода и 3) внутренним электролизом.
Электролиз с контролируемым электродным потенциалом
Для каждого элемента, который осаждается 1 из водных растворов, имеется определенное напряжение разложения, ниже которого электролитическое осаждение не наступает. Соответствующим подбором и регулированием электродного потенциала можно достигнуть разделения многих элементов 2. Некоторые металлы или их окислы при электролитическом выделении плотно пристают к электроду и настолько химически чисты, что их можно непосредственно взвешивать. Нередко электролитическое осаждение используется для отделения посторонних элементов от определяемых, остающихся в растворе.
Одной из причин, препятствующих применению электролиза с контролируемым электродным потенциалом, является недостаток в соответствующем оборудовании, необходимом для установления и поддержания требуемого напряжения. Нет нужды подчеркивать, что регулирование напряжения от руки является трудоемкой и утомительной операцией. Появившиеся в последнее время автоматические приборы для поддержания постоянного потенциала 3 значительно улучшили это положение.' Опубликовано несколько способов с применением этих приборов в связи с полярографией 4 s *, а также и в других- областях анализа 8.
1 G. Е. F. Lundell, J. I.Hoffman, Outlines of Methods of Chemical Analysis, John Wiley and Sons, 1938, p. 168.
2 H. J. S. S a n d, Electrochemistry and Electrochemical Analysis, Vol. II, Blakie. and Son, London and Glasgow, 1940.
3 C. W. Cal dwell, R. С. P arker, H. D i e h 1, Ind. Eng. Chem., Anal. Ed., 16, 532 (1944); J. J. L i n g a n e, там же, 17, 332 (1945); R. W. Lamphere, Anal. Chem., 23, 258 (1951); M. L. G r e e n о u g h, W. E. Williams, J. К. T a y-1 о r, Regulated Low Voltage Supply for Electrolysis and Other Uses, Rev. Sci. Instruments, 22, 484 (1951).
4 J. J. Lingane, Ind. Eng. Chem., Anal. Ed., 16, 147 (1944); 18, 429 (1946).
s H. Diehl, Electrochemical Analysis with Graded Cathode Potential Control,
G. F. Smith Chemical Co., McKinley Ave., Columbus, Ohio, 1948, p. 867.
166
Гл. V. Специальные операции
Электролиз с ртутным катодом
Рис. 16. Прибор Мелавена для электролиза с ртутным катодом.
Одним из заслуживающих внимания методов разделения элементов является электролиз с ртутным катодом в слабосернокислых растворах. В этих условиях алюминий, титан, цирконий, фосфор, ванадий, уран и другие элементы количественно отделяются от хрома, железа, кобальта, никеля, меди, цинка, галлия, германия, молибдена, родия, палладия, серебра, кадмия, индия, олова, рения, иридия, платины, золота, ртути, таллия и висмута, осаждающихся на ртутном катоде1. Электролиз может быть проведен в специальном приборе2 (рис. 16) следующим образом. Анализируемый раствор выпаривают до появления паров серной кислоты и затем разбавляют водой с таким расчетом, чтобы в 50—100 мл содержалось 0,15— 0,30 мл серной кислоты.
При анализе цветных металлов в приготовленном для электролиза растворе часто появляется' осадок. Если это гидроокись олова или сурьмы или же сульфат свинца, то его можно перенести в прибор для электролиза, так как под действием тока эти соединения разлагаются. Сульфат свинца легко разлагается при силе тока в 2—3 а, если применять анод из проволоки, погруженной приблизительно на 5 см в раствор, и перемешивать электролит. При высокой плотности тока осадок перекиси свинца сходит с анода и свинец постепенно переходит на ртутный катод. На фосфат олова электрический ток заметно не действует, и перед электролизом его, так же как и обезвоженную кремнекислоту, лучше отделить фильтрованием спльнокислого раствора.
С самого начала в раствор следует вводить
возможно меньше серной кислоты, чтобы при ее последующей нейтрализации до требуемой концентрации не образовалось чрезмерно большого количества солей. В прибор вливают нужное количество ртути (около 200 г), переносят туда раствор и ведут электролиз при плотности тока около 0,15 а/сл?, установив анод возможно ближе к поверхности раствора. Ртуть слабо перемешивают мешалкой (рис. 17) и электролиз продолжают до тех пор, пока проба раствора не даст отрицательную реакцию на элементы, которые отделяют. По окончании электролиза раствор сливают и промывают ртуть водой, не прерывая тока, после чего проводят анализ электролита, к которому присоединяют промывные воды. Извлечение из ртути осажденных элементов очень затруднительно, и проводить его не рекомендуется за исключением тех случаев, когда
1 J. R. С a i п, J. Ind. Eng. Chem., 3, 476 (1911); G. E. F. L u n d e 11, J. I. Hoffman, H. A. Bright, там же, 15, 1064, 1923); D. H. В г о p h у, там же, 16, 963 (1924); J. A. Maxwell, R.P. Graham, Chem. Rev., 46, 471 (1950).
На ртутном катоде можно осадить 10' г меди или никеля или же 5 г железа. Изучая электролитическое осаждение хрома, R. Chirnside'n L. Н. Dauncey [Analyst, 68, 175 (1943)] нашли, что высокая плотность тока, повышение температуры и концентрации, а также понижение кислотности раствора увеличивают скорость осаждения хрома, в то время как восстановители, сульфат аммония и вращение анода препятствуют его выделению. Влияние сульфата аммония можно устранить добавлением к электролиту небольших количеств никеля или очень малых количеств (но не менее некоторого необходимого минимума) серебра.
2 A. D. Mel a ven, Ind. Eng. Chem., Anal. Ed., 2, 180 (1930).
Электролитические методы
167
Рис. 17. Измененный прибор для электролиза с ртутным катодом:
1 — мешалка; 2 — часовое стекло; 3 — резиновая трубка.
отделяют «следы» какого-либо элемента от основного компонента, не осаждающегося на ртути.
При необходимости осажденные в ртути металлы можно отделить от нее после растворения амальгамы в соответствующей кислоте или же отгонкой ртути, как это делают, например, при определении приме сей в уране *.
Ртуть можно в достаточной степени очистить сильным перемешиванием с 10%-ной (по объему) азотной или соляной кислотой. Для этого ртуть и кислоту помещают в колбу из прочного стекла, снабженную пробкой с двумя отверстиями, в которые вставлены трубки. Одна из этих трубок доходит до дна колбы, а другую, заканчивающуюся выше уровня кислоты, присоединяют к вакуумному насосу. Значительно более эффективная очистка достигается обработкой нитратом закиси ртути 1 2. Была предложена также установка для полной очистки ртути 3. Если ртуть сохраняет блестящую чистую поверхность 4 5 или же при встряхивании с водой образуется пена, которая исчезает в течение 5—15 сек 8, то это означает, что содержание менее благородных металлов в ртути не достигает 0,0001 %. Более благородные, чем ртуть, металлы — серебро, золото и элементы платиновой группы — таким способом не обнаруживаются.
Внутренний электролиз
Внутренний электролиз в качестве аналитического метода с успехом использовался еще
в 1866 г. 6, но даже в 1927 г. 7 он все еще расценивался как процесс любопытный, но не имеющий практического значения, и только в последние годы этот метод стал широко применяться.
Термин «внутренний электролиз» впервые был предложен 8 в 1930 г. для таких процессов электроосаждения, которые происходят без подведения тока извне, а за счет электродвижущей силы, возникающей в результате замыкания цепи непосредственным соединением анода и катода.
Внутренний электролиз целесообразно применять для отделения примесей от основного компонента при анализе металлов, руд и солей,
1 N. Н. Furman, С. Е. Bricker, В. McDuffie, J. Wasch. Acad. Sci., 38, 159 (1948).
2 J. H. Hildebrand, J. Am. Chem. Soc., 31, 933 (1909); см. также H. F. E a s 1 y, Ind. Eng. Chem., Anal. Ed., 9, 82 (1937).
3 W. A. Carlson, L. F. Borchardt, Ind. Eng. Chem., Anal. Ed., 10, 94 (1938).
4 E. Wi c h e r s, Chem. Eng. News, 20, 1111 (1942).
5 О. H. M ii 11 e r, Chem. Eng. News, 20, 1528 (1942).
6 C. Ullgren, Ofversigt kgl. Vetenskaps — Akademiens Forhandlingar, 9, 277 (1866).
7 A. Classen, Quantitative Analyse durch Electrolyse, 7-te Anfl., 1927.
8 H, J. S. Sand, Analyst, 55, 309 (1930).
168 Гл. у. Специальные операции
как, например, при определении висмута, меди и серебра в свинце и припоях1; висмута — в свинцовых рудах2; кадмия, меди и никеля — в цинковых рудах и цинке3; свинца — в рвотном камне4 C4H4K(SbO)O6; меди — в железе, стали s или кадмии 6; меди и олова — в алюминиевых сплавах 7 8 и, наконец, для отделения ртути от других металлов при. анализе латуни и бронзы ®. В некоторых случаях определение может быть произведено непосредственным взвешиванием электрода, но обычно после электролиза анализ заканчивают, пользуясь методами, соответствующими техническим условиям.
Большим удобством метода внутреннего электролиза является быстрота, с которой происходит осаждение и разделение элементов. В большинстве случаев ’этот процесс длится около 30 Мин, Для достижения этого требуется тщательное соблюдение соответствующих условий температуры и кислотности раствора, а также сильное перемешивание электролита.
Известны систематические исследования по применению внутреннего электролиза для разделения и определения некоторых металлов 9.
В усовершенствованном приборе для внутреннего электролиза 10, показанном на рис. 18, анод и катод разделены алундовыми диафрагмами, в которые заключены аноды. Катодом обычно служит платиновый сетча-’ тый электрод, а анодом — соответствующий неблагородный металл, специально подбираемый для каждого случая. Для ускорения осаждения обычно применяют два анода, которые присоединяют к одной клемме.
Примером применения внутреннего электролиза является приводимый ниже метод определения серебра в товарном свинце. Согласно имеющимся указаниям, для анализа следует брать навеску свинца в 100 а, в которой содержится не более 10 мг серебра. Можно пользоваться большими или меньшими навесками, в зависимости от содержания серебра, но изменив сообразно с этим и количество применяемых реактивов. В качестве анода применяют проволоку из меди высокой чистоты (не содержащей серебра и не корродированной). Катодом служит обычный платиновый сетчатый электрод, приспособленный так, чтобы его легко было отделять от прибора. Катод предварительно взвешивают. Концентрация азотной кислоты в растворе должна быть достаточно высокой, чтобы предотвратить соосаждение висмута. Сурьма, мышьяк и олово в тех коли
1 Е. М. Collin, Analyst, 55, 312 (1930); В. L. С 1 а г к е, L. A. Wooten, С. L. Luke, Ind. Eng. Chem., Anal. Ed., 8, 411 (1936); ASTM, Methods of Chemical Analysis of Metals, 1950, p. 371.
2 E. M. С о 11 i n, Analyst, 55, 680 (1930).
3 E. M. С о 11 i n, Analyst, 55, 495 (1930); J. G. Fife, там же, 61, 681 (1936); В. L. Cl arke, L. A. W о о t en, Trans. Electrochem. Soc., 76, 63 (1939).
4 E. M. С о 11 i n, H. J. S. S a n d, Analyst, 56, 90 (1931).
.s J. G. F i f e, S. T о г r a n c e, Analyst, 62, 29 (1937).
8 J. G. F i f e, Analyst, 65 , 562 (1940).
7 B. L. C 1 а г k e, L. A. W о о t e n, цит. выше; H. А. Суворовская,
Зав. лаб., 11, 474 (1945).
8 J, G. F i f е, Analyst, 63, .650 (1938).'
9 A. Schleicher, Т. Todoroff, Z. anal. Chem., 126, 412 (1944); Z. Elektrochem., 50, 2 (1944); Chem. Ztg., 68, 48 (1944).
10 B. L. C1 a r k e, L. A. W о о t e n C. L. Luk e, Ind. Eng. Chem., Anal. Ed., 8, 411 (1936).
Электролитические методы.
169
чествах, которые обычно содержатся в чистом товарном свинце, не влияют
на осаждение, если они окислены до высшей валентности. Если содержание этих элементов достаточно велико, чтобы образовался осадок, то при растворении пробы вводят минимальное ко-
личество фтористоводородной кислоты, какое необходимо для получения прозрачного раствора.
Ход анализа. 100 г свинцовой стружки или опилок помещают в стакан емкостью 800 мл и обрабатывают 5 г винной кислоты и 320 мл разбавленной (1 : 3) азотной кислоты. Осторожно нагревают до полного растворения материала и затем кипятят до удаления окислов азота. Если остается небольшой темный остаток, его отфильтровывают и растворяют в 5 мл разбавленной (1:1) азотной кислоты. По удалении окислов азота кипячением этот раствор присоединяют к основному раствору, разбавляют холодной водой приблизительно до 600 мл, охлаждают и затем при помешивании прибавляют по каплям 1%-ный раствор КМпО4 до появления неисчезающей розовой окраски. В окисленный раствор вводят 5 г винной кислоты и нагревают его до 80—90° С. Анодную ячейку заполняют промывной жидкостью, содержащей нитрат меди 1 и свободной от ионов хлора. После этого ведут электролиз в течение 15—30 мин при энергичном перемешивании, подогревая электролит, чтобы сохранить его первоначальную температуру.
Через 5 мин заменяют раствор в анодной ячейке свежим промывным раствором нитрата меди и так же поступают еще раз спустя 10 мин. Замена одного раствора другим производится через отверстие 11, показанное на рис. 18.
По окончании осаждения серебра, что контролируют погружением новой части электрода в электролит, не прекращая пере-
Рис. 18. Схема прибора для внутреннего электролиза:
1 — электрическая плитка; 2 — свинцовый анод; 3 — сосуд из алунда; 4 — резиновая пробка; 5 — пластинка для укрепления электродов; в — стеклянный тройник; 7 — воронка; 8 — резиновая трубка-; 9 — зажим; 1 о — стеклянная трубка) 11— отверстие для промывания; 12— алундовый сосуд с анодом; 13 — мешалка; 14 — катод.
мешивания и не прерывая при этом тока, сливают раствор соли свинца из стакана сифо-нированием, для чего постепенно добавляют воды, в общей сложности около 1 л. После этого осторожно опускают стакан и быстро заменяют его другим, содержащим воду. Электрод промывают водой, затем спиртом, сушат при 110° С, охлаждают и взвешивают. Как Правило, катодный осадок серебра имеет блестящую поверхность и достаточно чист для непосредственного взвешивания. Если осадок' загрязнен, что определяется
1 8 г меди высокой чистоты растворяют в 35 мл азотной кислоты (пл. 1,42), раствор кипятят до удаления окислов азота и разбавляют до 2 л. '
170
Гл. V. Специальные операции
по его цвету, его растворяют в 5 мл разбавленной (1 : 1) азотной кислоты и повторяют осаждение внутренним электролизом или же заканчивают определение в виде AgCl.
*Внутренний электролиз известен в двух основных вариантах. По первому варианту, приведенному выше, католит отделяется от анолита пористой перегородкой. Другой вариант, разработанный Ю. Ю. Лурье с сотрудниками \ отличается тем, что электролиз проводят без диафрагмы и без перемешивания электролита, благодаря чему можно пользоваться крайне простой аппаратурой. В этом случае катод и анод соединяют друг с другом проволокой или Муфтой и погружают непосредственно в анализируемый раствор. Применение этого способа ограничивается определением малых количеств металла, не более 20 мг, так как при повышенной концентрации осаждаемого элемента в исходном растворе возникают явления цементации на аноде. Примером использования метода без диафрагмы может служить определение меди.
Прибор для определения меди состоит из платиновой сетки, служащей катодом, й алюминиевой пластинки, являющейся анодом (длина около 130 мм, ширина 6—7 мм, толщина 1—2 мм). Контакт между электродами осуществляется при помощи клеммы или любым другим надежным способом. Алюминиевая пластинка должна быть предварительно тщательно очищена наждачной бумагой.
, Ход анализа. Анализируемый раствор, содержащий не более 20 мг меди, лучше всего в виде сульфата, помещают в стакан емкостью 400 мл, подкисляют 4 мл разбавленной (1 : 1) H2SO4 или 5—10 мл концентрированной уксусной кислоты и разбавляют водой до 200 мл. В нагретый до 85—95° С раствор опускают соединенную пару электродов. Платиновый катод предварительно высушивают при 100—110° С и взвешивают. Электролиз ведут 40 мин, нагревая раствор на электрической плитке или на небольшом пламени горелки, чтобы сохранить его первоначальную температуру. По окончании электролиза вынимают электроды и обмывают их водой. Отделяют катод и промывают его сначала водой, затем спиртом, высушивают при 100—110° С и взвешивают. Увеличение в массе катода показывает содержание меди. Если привес катода не превышает 2 мг, определение лучше закончить колориметрическим методом, посл!е растворения осадка меди в азотной кислоте.
Совместно с медью осаждаются висмут, серебро и ртуть. В случае содержания этих элементов в исходном растворе выделенные металлы можно растворить в азотной кислоте и в полученном растворе определить медь колориметрическим или объемным методом. Помимо этих металлов, наряду с медью выделяются также золото и платина. Влияние больших количеств железа устраняется восстановлением его перед электролизом, для чего в раствор вводится маленькими порциями 1 г сульфата гидразина.
, Предложен 1 2 также и третий вариант метода внутреннего электролиза, который основан на непосредственном покрытии анода коллодиевой
1 В. И. Колосов, Ю. Ю. Лурье, Промстандарт СССР, Цветные металлы, вып. 45; Ю. Ю. Л у р ь е, М. И. Троицкая, Зав. лаб., 5, 1420 (1936); 6, 33 (1937); Ю. Ю. Л у р ь е, там же, 6, 1040 (1937); Ю.Ю. Л урье, Л. Б.Гинзбург, там же, 7, 1 (1938); 7, 535 (1938); Ю. Ю. Л у р ь е, М. И. Т р о и ц к а я, там же, 7, 675 (1938).
2 J. Guzman, S. С е 1 s i, С. А., 33, 87 (1930); Anales farm, bioquim., 9, 43 (1938).
Полярографический анализ
171
пленкой, что, не усложняя прибора, дает возможность практически полностью выделять металл при относительно высокой концентрации его в исходном растворе. Методом внутреннего электролиза, с применением защитной пленки на аноде, возможно практически полностью выделить относительно большие количества (порядка 100 мг) ртути, серебра, меди, висмута, свинца, олова, индия и молибдена \ Осаждение олова проводилось из солянокислого раствора хлорида олова (II) при комнатной температуре с применением кадмиевого или цинкового анода. Анализируемый раствор содержал в 250—300 мл 2 мл разбавленной (1 ; 2) соляной кислоты, 5 г хлорида аммония и 1 г солянокислого гидроксиламина. Кобальт, нике(ль и железо (II) не мешают выделению олова.
Прибор, которым пользовались авторы, состоит из широкого стакана емкостью 500 мл, платинового сетчатого электрода, служащего катодом, и металлического анода, имеющего форму палочки диаметром 10 мм. Оба электрода замкнуты накоротко медной проволочкой.
Коллодиевую пленку наносят непосредственно на анод, для чего, тщательно очистив поверхность металла напильником и наждачной бумагой, погружают его на несколько минут в коллодий. Затем анод высушивают на воздухе до тех пор, пока коллодий не перестанет прилипать к пальцам, и снова опускают в коллодий для нанесения второго слоя. Обычно двукратного покрытия бывает достаточно. Аноды, приготовленные таким способом, весьма устойчивы и могут служить для многократных определений, если сохранять их в дистиллированной воде.
Электролит перемешивают током двуокиси углерода или воздуха, который в данном случае окисляющего действия не оказывает.
Перед началом электролиза контакты всех металлических частей следует тщательно очистить напильником и наждачной бумагой, иначе металл может выделиться не полностью.
В последнее время внутренний электролиз с анодами, покрытыми коллодийными пленками, применен для микроопределения золота и серебра 1 2, висмута, меди и сурьмы 3. Доп. перев*
ПОЛЯРОГРАФИЧЕСКИЙ АНАЛИЗ
Когда постепенно возрастающая электродвижущая -сила налагается на электроды, погруженные в ^раствор, то прохождение тока начнется лишь после того, как разность потенциалов достигнет величины, необходимой для возникновения электрохимических реакций на электродах. Как только этот потенциал разложения будет достигнут, возникает ток, сила которого возрастает с повышением напряжения. Если при этом один из электродов очень мал, то вблизи него вскоре устанавливается состояние концентрационной поляризации и сила тока достигает определенной величины, которая зависит от скорости притока электрохимически реагирующих веществ к этому электроду. При полярографических измерениях таким малым электродом являются капли ртути, вытекающие из очень тонкого отверстия капиллярной трубки. Второй электрод должен иметь достаточно большую поверхность, чтобы избежать поляризации.
1 Ю. А. Ч ернихов, Г. А. Б о л ь ш а к о в а, Зав. лаб., 14, 3 (1948).
2 L. S о m m е г, Coll. Czech. Chem. Comm., 20, 1, 46 (1955).
3 А. О k a c, L. S о m m e г, Coll. Czech. Chem. Comm., 20, 95 (1955).
172
Гл. V. Специальные операции
Полярограф— это прибор, посредством которого регистрируются, автоматически или вручную, кривые: ток — напряжение. Эти кривые, или полярограммы, показывают внезапное резкое увеличение силы тока при напряжениях, характерных для каждого отдельного вещества, реагирующего у капельного ртутного электрода. Резкое изменение силы тока, называемое волной или ступенью на кривой, является мерилом концентрации вещества. )
В основе качественного полярографического анализа лежит определение потенциала, при котором сила тока равна половине своего предельного значения.
Этот так называемый потенциал полуволны не зависит от концентрации вещества и является, таким образом, характерной особенностью реакции, происходящей у электрода. На величину потенциала полуволны могут влиять комплексообразователи, pH и ионная сила раствора. Кроме того, потенциалы полуволны нескольких подвергающихся электролизу веществ могут оказаться столь близкими, что волны их сольются. Это в известной мере ограничивает применение полярографического метода для качественной идентификации отдельных веществ. Однако во многих случаях путем подбора состава электролита можно изменить потенциал полуволны мешающих ионов.
Количественный полярографический анализ основан на измерении Диффузионного тока (высоты волны), который при сохранении постоянства остальных факторов пропорционален концентрации электрохимически разлагающихся веществ. Сила тока зависит также от размеров капель ртути, скорости их вытекания из капилляра и температуры раствора. Установлены отклонения, которые допустимы при регулировании этих факторов J. Полярографические волны часто имеют неправильную форму или максимумы, которые для получения точных результатов должны быть устранены. Отсюда следует, чтс полярографические определения лучше проводить путем сравнения результатов измерения на одном и том же приборе исследуемого и стандартного растворов приблизительно одинакового состава 1 2.
Зависимость между силой тока и характеристикой электрода была выведена Ильковичем 3. Хотя по поводу уравнения, определяющего эту зависимость, в последнее время и были выражены некоторые сомнения, но для внесения поправок на неточности эксперимента, с которыми приходится сталкиваться в повседневной практике, оно достаточно удовлетворительно.
Предельная точность, которая может быть достигнута при рядовых полярографических определениях, лежит в пределах ±1 % (относительного), хотя некоторые исследователи считают полярографический метод более точным. Следовательно, полярография лучше всего применима для определения малых количеств веществ. Измерению обычно поддаются концентрации, лежащие в пределах от 0,01 до 10 миллимолярной. При концентрациях, выходящих за эти пределы,' растворы концентрируют или разбавляют. При концентрациях ниже 0,2 миллимолярной точность определения значительно уменьшается.
1 F. В п с k 1 е у,' J. К. Т а у 1 о г, Trans, Am. Electrochem. Soc., 87, 197 (1945).
2 J. К. T а у 1 о r, Anal. Chem., 19, 368 (1947).
3 D. Ilkovic, Coll. Czech. Chem. Comm., 6, 498 (1934).
Фотометрические методы
173
Хотя и описаны микроэлектролизеры для долей миллилитра раствора, но обычно берут не менее 5—10 мл раствора, особенно когда требуется' большая степень точности. Полярографическим методом измеряется концентрация раствора, поэтому точность определения общего содержания вещества не может превышать точности измерения объема анализируемого раствора.
Полярографический метод с успехом применяется в неорганическом анализе. В тех случаях, когда обеспечиваемая этим методом точность удовлетворяет предъявляемым требованиям, он может быть использован для определения основных компонентов, как, например, при текущем контроле гальванических ванн. Наиболее широкое применение метод получил для определения компонентов, содержащихся в небольших количествах, например легирующих элементов в черных и цветных металлах, а также для определения примесей. Метод особенно привлекателен в тех случаях, когда раствор для полярографии может быть приготовлен при минимальном числе операций, причем один или несколько элементов могут быть определены без предварительного разделения. Можно ожидать, что полярография найдет все возрастающее применение в рядовых анализах, но о возможности ее применения не следует забывать и при исследовательских работах. Благодаря своей чувствительности и многосторонности она будет находить особенно широкое применение в специальных областях неорганического анализа \
ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
Фотометрические методы в химическом анализе сводятся к измерениям, в основу которых положено свойство растворов поглощать или рассеивать лучистую энергию. К ним относятся колориметрия, спектрофотометрия, турбидиметрия и нефелометрия. Во всех этих методах используются химические реакции, при которых определяемые элементы, вступая в соединения, образуют соответствующие светопоглощающие или светорассеивающие системы. Фотометрические методы наиболее применимы для определения малых количеств веществ. При рядовой работе погрешность определения лежит около 2% (относительных). Применение спектрофотометров позволяет распространить эти методы и на определение элементов при относительно высоком процентном содержании их с точностью, приближающейся к точности обычного весового и объемного анализа, если химизм процесса такую точность гарантирует 1 2 * * s. Многие элементы могут вступать в реакции, в результате которых образуются окрашенные соединения, являющиеся основой колориметрических методов. Для определения интенсивности получаемых окрасок используется источник света, дающий сплошной спектр в видимой части, а светопо-
1 Подробные сведения можно найти в следующих работах: I. М. Kolthoff,
J. J. Lingane, Chem. Rev., 24, 1 (1939); И. M. К о л ь т г о ф, Дж. Дж. Лин-
гейм, Полярография, Госхимиздат, 1948; J. Heyr-ovsky, Polarographie; Sprin-ger-Verlag, Vienna (1941); Bibliography of Polarographie Literature, E.—90 (1), Leeds and Northrup Co., Philadelphia, 1950; J. J. Lingane, Anal. Chem., 21, 45 (1949); 23, 86 (1951).* Я. Г e й p о в с к и й, Полярографический метод, теория и практическое применение, ОНТИ, Химтеорет, 1937; Я. Гейровский, Техника полярографического исследования. Сборник статей, Издатинлит, 1951.*
1 J. В. С u 1 b е г t s о n, R. М. F о w 1 е г, Steel, 122, № 21, 108 (1948); С. F. Н i-
s к е у, J. Rabinowitz, I. G. Young, Anal. Chem., 22, 1464 (1950).
174
Гл. V. Специальные операции
глощением анализируемого раствора сравнивается с светопоглогцением стандартного раствора. Сравнение можно осуществлять различными способами, например: 1) введением титрованного раствора определяемого вещества в раствор для сравнения до получения одинаковой с анализируемым раствором окраски; 2) сравнением окраски анализируемого раствора с серией стандартов, в которых содержатся известные количества определяемого элемента (если раствор окрашенного соединения подчиняется закону Бера); 3) сопоставлением высот столбов анализируемого раствора и стандарта, при которых интенсивность их окрасок уравнивается Ч Визуальная колориметрия все еще широко применяется в тех случаях, когда обычная точность простых, быстрых методов удовлетворяет предъявляемым требованиям 1 2, но такие методы неприменимы в присутствии посторонних окрашенных соединений. Это затруднение устраняется при использовании фильтрофотометров.
Фильтрофотометр является относительно недорогим прибором, посредством которого устраняется влияние посторонних примесей, когда максимумы светопоглощения этих примесей и окрашенного соединения определяемого элемента не слишком близки между собой. Действие таких приборов заключается в выделении посредством светофильтров нужного 3 участка спектра. Для практической работы в большинстве случаев достаточно комплекта из 10—12 фильтров. Имеются визуальные приборы, но более распространены приборы, снабженные фотоэлементами и электроизмерительными устройствами для измерения фототока 4. *(Такие приборы называют фотоколориметрами).* При надлежащем выборе источника света, светофильтра и фотоэлемента может быть выполнено огромное большинство аналитических определений невысоких содержаний искомого вещества (до нескольких процентов), если разработаны достаточно надежные химические методы. Применение этих приборов не достигает цели, если максимум светопоглощения определяемого вещества лежит в неблагоприятной для измерения области спектра. В некоторых случаях неудовлетворительная работа прибора связана с шириной полосы спектра световых лучей, пропускаемых светофильтром. Тогда вместо фотокэлориметра применяют спектрофотометр.
Спектрофотометр дает возможность выделить волны света одной длины или очень узкого участка спектра (50 А или менее). Этот прибор обычно более совершенен, но и дороже, чем фотоколориметр. Большинство спектрофотометров снабжено диффракционными решетками или призмами, соответствующими источниками света и имеет щели для выделения узких участков в ультрафиолетовой, видимой и инфракрасной областях спектра. В некоторых приборах используется прерывистый пучок света, как, например, ртутная дуга, и светофильтры для выделения
1 М. G. Mellon, Editor, Analytical Absorption Spectroscopy, Chapter 3, by W. B. Fortune, John Wiley and Sons, 1950.* А. К. Б a.6 к о,. A. T. Пилипенко, Колориметрический анализ, Госхимиздат, 1951 *.
2 Примеры использования этих методов в повседневной работе см. Reagent Chemicals, ACS Specifications, American Chemical Society, 1950; Standard Methods for the Examination of Water and Sewage, 9th ed., American Public Health Association, 1947.
3 M. G. M e 11 о n, цит. выше, p. 369.
4 M. G. Mellon, Editor, Analytical Absorption Spectroscopy, Chapter 4, by R. H. Miiller, John Wiley and Sons, 1950; ASTM, Methods of Chemical Analysis of Metals, 1950, p. 43.
Фотометрические методы
175
нескольких узких участках спектра. Преимущества таких приборов особенно выявляются в тех случаях, когда светопоглощение раствора определяемых веществ резко изменяется от небольшого изменения длины волны. В качестве примера можно привести анализ смеси редкоземельных элементов. Пропуская через раствор световые лучи различных длин волн, можно выполнить раздельное определение некоторых элементов этой группы (стр. 630). Другое преимущество спектрофотометра вытекает из того факта, что при измерении интенсивности окраски многих систем, подчиняющихся закону Бера в пределах, требуемых для аналитических целей, в большинстве фотоколориметров получаются значительные расхождения вследствие несовершенства оптики и механизма этих приборов. Эти колебания иногда серьезно ограничивают точность «дифференциальных» 1 методов 2, вводимых в практику для устранения необходимости химических разделений. Наконец, возможность применения некоторых спектрофотометров для анализа растворов, обладающих относительно большим светопоглощением, позволяет проводить «дифференциальные» измерения и определять отдельные компоненты при высоком их содержании с погрешностью порядка нескольких тысячных долей. Преимуществом спектрофотометрических методов является также возможность использования ультрафиолетовой и ближайшей инфракрасной областей спектра.
В турбидиметрии и нефелометрии 3 искомое вещество переводят в нерастворимое соединение, которое должно быть равномерно диспергировано в растворе. Это требование очень часто ограничивает применение таких методов, так как приготовление неорганических суспензий, имеющих воспроизводимые оптические свойства, не всегда легко выполнимо. . "
В турбидиметрии свет, поглощенный раствором или прошедший через него, обычно измеряют таким же способом, как и ₽ случае окрашенных растворов. Применяемый иногда визуальный способ основан на определении высоты столба суспензии, при которой нить накаливания лампы перестает быть видимой. Такой способ более всего пригоден для анализа растворов, содержащих определяемое вещество в значительной степени в виде взвеси, и его можно рекомендовать для массовых анализов однотипных продуктов, как, например, для определения серы в цементе (стр. 804).
В нефелометрии свет, отраженный или рассеянный частицами испытуемой освещенной суспензии, сравнивают со светом, отраженным или рассеянным стандартной суспензией, приготовленной аналогичным
1 * Речь идет Об определении двух окрашенных соединений, одновременно присутствующих в растворе, путем измерения светопоглощения этого раствора при пропускании через него света с различными длинами волн. Прим, ред.*
I 2 Примеры этого типа методов см. F. W. Haywood, A. R. Wood, Metallurgical Analysis, Adam Hilger Ltd., London, 1944. Дальнейшее обсуждение см. E. Б. С e н д э л, Колориметрическое определение следов металлов, Госхимиздат, 1949; Е. В. S a n d е 11, Colorimetric Determination of Traces of Metals, Interscience Publishers, New York, 1950, p. 74; ASTM, Symposium on Analytical Colorimetry and Photometry, 1944, p. 716; M. G. Mellon, Editor, Analytical Absorption Spectroscopy, Chapter 7, John Wiley and Sons, 1950; * F. D. S n e 11, С. T. S n e 11, Colorimetric Methods of Analysis, vol. 1—4, New York, 1949—1954* .
3 Дж. Г. Йоу, Фотометрический химический анализ, т. II, Нефелометрия, 0НТИ, 1936; Р. V. Wells, Chem. Rev., 3, 331 (1927).
176
Гл. V. Специальные операции
/
образом и одинаково освещенной. Этот способ очень чувствителен и дает возможность определять относительно малые количества вещества, находящегося в растворе в виде взвеси
ФЛУОРЕСЦЕНТНЫЙ АНАЛИЗ
Методы, основанные на флуоресценции веществ, большей частью разработаны в области органического анализа. В неорганическом анализе они нашли применение сравнительно недавно 1 2. Флуоресцентный анализ заключается в исследовании света, испускаемого веществом, облучаемым ультрафиолетовыми лучами (обычно длиной волны между 3000 и 4000 А). Используются также и другие средства возбуждения флуоресценции, как, например, рентгеновские и катодные лучи. При использовании этого метода для количественного анализа необходимо тщательно продумать выбор источника возбуждения, светофильтров, условий подготовки образца и способа измерения интенсивности флуоресценции.
Флуоресцентные методы мало приемлемы для идентификации минералов, но могут служить для открытия и определения в них следов различных элементов, например хрома в сапфирах и рубинах, урана в рудах и молибдена в шеелите.
Необходимо при этом иметь в виду, что под влиянием одних примесей флуоресценция может ослабляться, а при наличии других, наоборот, усиливаться. Влияние измерений pH на флуоресценцию настолько велико, что метод этот может быть применен для определения конечной точки титрования. Этот вопрос должен рассматриваться в связи с определениями, основанными на реакциях органических реагентов с неорганическими соединениями. Например, установлено *, что 3—4 капли нафтионовой кислоты, введенные в 10 мл воды, не дают флуоресценции при pH = 3, но становятся фиолетовыми при pH = 14, синими при pH = = 6, яркосиними при pH = 7, зёленовато-синими при pH = 10 и желтовато-зелеными при pH =12.
Вполне возможно, что флуоресценция будет причислена к специфическим методам открытия элементов, особенно при изучении свойств новых реагентов. Например, установлено, что чувствительность оранжево-красной флуоресценции (соответствующей красной полосе, лежащей между длинами волн 6365 А и 6975 А), которая вызвана облучением ультрафиолетовыми лучами уксуснокислых растворов алюминия, обработанных красителем пантохром черно-синий R, достигает 1 части на 5 000 000 частей воды. На эту флуоресценцию не влияет бериллий и
1 О приборах для нефелометрии см. R. В. В а г n е s, С. R. S toe k, Anal. Chem., 21, 183 (1949).
2 Обзор флуоресцентных методов см. С. Е. White, Ind. Eng. Chem., Anal. Ed., 11, 63 (1939); Anal. Chem., 21, 104 (1949); 22, 69 (1950); см. также П. В. Д а н к-в о р т, Люминесцентный анализ в фильтрованном ульрафиолетовом свете, ГНТИ, Ленхимсектор, 1931; J. W. Edwards, Ann. Arbor., Mich. (1944); F. Feigl, Spot Tests, 3d ed., Elsevier Publishing Co., New York, 1946; J. A. Radley, J. Grant, Fluorescence Analysis in Ultraviolet Light, 3d ed., D. Van Nostrand Co., 1939; *M. А.. К о н с т а н т и н о в а-Ш л е з и н г е р, Люминесцентный анализ, Изд.
АН СССР, 1948.*
«М. Deribere, Ann. chim. anal. chim. appl., 18, 173 (1946).
Спектрохимический анализ
177
другие элементы, совместно с которыми алюминий обычно встречается х.
Флуоресцентный метод применен для определения бора, причем в качестве реагента как для качественного, так и для количественного определения используют бензоин. Определение проводят в спиртовом растворе, и, по данным авторов, чувствительность метода составляет 0,2 мкг бора 2.
Другие примеры применения флуоресцентного анализа приведены в гл. «Галлий» (стр. 557), «Алюминий» (стр. 579), «Бериллий» (стр. 588), «Цинк» (стр. 492) и «Уран» (стр. 531).
СПЕКТРОХИМИЧЕСКИЙ АНАЛИЗ
Метод анализа, основанный на исследовании эмиссионных спектров, приобретает все большее значение для определения металлов в металлических и неметаллических продуктах. Помимо использования его как обычного лабораторного способа в качественном и количественном анализе, он нашел широкое применение для экспрессного аналитического контроля металлургических процессов. Особыми преимуществами метода являются: 1) возможность проведения анализа с небольшим количеством анализируемого материала; 2) возможность с помощью только нескольких операций провести качественное исследование материала на 70 элементов; 3) возможность открывать очень малые концентрации многих элементов и 4) возможность быстро выполнять количественные определения после того, как ход анализа установлен.
Анализ с погрешностью, в 10% (относительных) осуществим сравнительно легко. Точность 1—2 % достигается в результате тщательного изучения метода, применения элемента сравнения и использования для сравнения стандартных образцов, близких по химическим и физическим свойствам к анализируемому материалу.
Применение метода ограничивается: 1) трудностью наблюдения спектральных линий большинства неметаллов: 2) сложностью оборудования и 3) недостаточной точностью определения компонентов, содержащихся в количествах свыше 5—10% (кроме нескольких благоприятных случаев). В результате усовершенствования аппаратуры и изучения новых областей применения спектрохимического анализа появилась обширная литература, по которой имеются соответствующая библиография и обзорные статьи А
1 С. Е, W h i t е, С. S. Lowe, Ind. Eng. Chem., Anal. Ed., 9, 430 (1937). Эти же авторы [там же, 12, 229 (1940)] приводят определение алюминия посредством фотометрического измерения интенсивности флуоресценции растворов, в которых происходит реакция между алюминием н морином. Многие элементы, в том числе и бериллий, мешают этому определению.
1 С. Е. W h i t е, A. W е i s s 1 е г, D. Busker, Anal. Chem., 19, 802 (1947).
• W- F. M e gg e r s, В. F. Scribner, Index to the Literature on Spectrochemi-ca| Analysis, pt. I, 2d ed., ASTM, 1941, p. 1920; B. F. Seri h ner, W. F. M egger s, тай Же, pt. II, 1947, p. 1940—1945. Последние обзорные статьи опубликованы W. Ff Meggers, Anal? Chem., 21, 29 (1949); 22, 18 (1950). См. также W. R. В г о d e, Chemical Spectroscopy, 2d ed., John Wiley and Sons, 1943; G. R. Harrison, R. C. Lord; J. R. L о о f b о u г о w, Practical Spectroscopy, Prentice — Hall, 1948; N. H. N achtrieb, Principles and Practice of Spectrochemical Analysis, McGraw-Hill Book Co.,. 1950. * В. В. Герлях, Спектрохимический эмиссионный анализ, » ОНТИ, Химтеорет, 1936; Г. Ш е й б е, Химический спектральный анализ, ОНТИ, Химтеорет, 1938; А. К. Русанов, Спектральный анализ руд и минералов, Гос-геолиздат, 1948 ♦.
12 Заказ 522. "
178
Гл. V. Специальные операции
Для рядовой аналитической работы применяются спектрографы с призмами или с диффракционными решетками, имеющие дополнительное устройство для возбуждения спектра, фотографирования спектров, фотометрического измерения интенсивности линий и (для быстрого контроля) графических расчетов. Спектрограф, имеющий линейную дисперсию 5 А на миллиметр в области 3000 А, служит для наблюдения простых и сложных спектров. Наблюдению подвергаются спектры, возбужденные при испарении вещества в пламени, электрической дуге или в искре. Пламя обычной бунзеновской горелки может возбудить только спектры щелочных и щелочноземельных металлов. Более высокотемпературное пламя, кислоррдно-ацетиленовое или кисЛородно-водородное возбуждает спектры 35 элементов, в том числе и обычно определяемых в сельскохозяйственных продуктах А Приборы для фотометрии пламени, обеспечивающие быстрые определения, особенно щелочных элементов, вполне доступны. Дуга постоянного тока является обычно принятым источником возбуждения в качественном анализе и для количественного определения следов элементов 2. Высоковольтная дуга может служить для более точного контроля и применяется при определении более высоких концентраций элементов, главным образом для рядовых анализов металлов и сплавов 3. Другие источники возбуждения, как, например, разрядные трубки, находят более ограниченное применение.
Визуальное наблюдение спектра служит иногда для идентификации образцов. Грубые количественные испытания, имеющие целью сортировку металлов, также можно проводить визуально, но более часто как в качественном, так и в количественном анализе спектры фотографируются, а спектрограмма является документом анализа.
Исследования последнего времени по усовершенствованию фотоэлектрического оборудования привели к разработке конструкции приборов с непосредственной фотоэлектрической регистрацией спектра; эти приборы имеют исключительно важное значение для текущих анализов на крупных металлургических производствах 4. При помощи таких приборов, в которых объединены источники возбуждения, спектрометр, комплект фотоэлементов и самопишущий аппарат, можно провести полный анализ металла в течение 1—2 мин. Приборы этого типа хорошо приспособлены для массовых анализов ограниченной группы металлов и сплавов. Для разнообразной аналитической работы наиболее приемлемым остается метод фотографирования.
Спектрохимический анализ нашел обширное и разнообразное применение в контроле производства металлов 5. Этим методом качественный анализ практически любого продукта может быть проведен без специальной его обработки. Полуколичественные методы используются для оценки концентраций компонентов в разнообразных продуктах с точностью
1 Н. G. Lundegardh, Die Quantitative Spektralanalyse der Elemente, T. 2, Gustav Fischer, Jena, 1934.
2 Подробное описание методов анализа с применением дуги постоянного тока, см. L. Н. A h г е n s, Spectrochemical Analysis, Addison-Wesley Press, Cambridge
H. В. Vincent, R. A. Sawyer, J. Appl. Phys., 8, 163 (1937).
4 M.F.Hasler, J. W.Kemp, H. W.D ietert, ASTM Bull., 139, 22 (1946); M. F. Hasler, Iron Age, 164, 96 (1949); R. O’b. Carpenter, E. Dubois, J. Sterner, J. Optical Soc. Am., 37, 707 (1947).
5 J. R. Churchill, Ind. Eng. Chem., Anal. Ed., 17, 66 (1945).
Спектр о химический анализ
179
до 50%, а иногда и более высокой.1. Чувствительность определения элементов колеблется в широких пределах. Для большинства металлов она достигает 0,001 %, а в некоторых случаях и меньшей величины. Для элементов, имеющих сложные спектры (уран, торий, редкоземельные элементы и платиновые металлы), чувствительность метода более низка, и чтобы открыть их в сложных смесях, может потребоваться концентрация не ниже нескольких сотых долей процента.
Предел чувствительности определения для каждого элемента не является постоянной величиной и зависит от сложности спектра, источника возбуждения и дисперсии спектрографа. Для повьцпения чувствительности определения Особо важных элементов имеется возможность подобрать соответствующие условия. Одним из способов, не изменяя существа метода, применить его для определения очень малых количеств веществ являются предварительные химические отделения. Так, например, можно в 500 раз повысить концентрацию следов некоторых элементов в золе растений, отделив их от основных компонентов, таких, как щелочные и щелочноземельные металлы и фосфор, осаждением оксихинолином 2. Фракционная дистилляция в источнике возбуждения спектра также может быть использована как средство концентрирования искомого элемента с целью повышения чувствительности метода. При анализе урановых продуктов на содержание следов примесей анализируемую пробу переводят в окись, прибавляют окись галлия в качестве коллектора и отгоняют 33 летучих элемента прокаливанием в вольтовой дуге 3. В результате этого чувствительность определения повышается, достигая от нескольких миллионных частей до 0,1 %. Этот процесс в достаточной мере поддается контролю, чтобы его можно было использовать для количественного анализа.
В большинстве случаев спектрографический анализ проводят непосредственно после незначительной подготовки анализируемого материала. Если анализируют металлы, то из них могут быть отлиты стержни, которые используют в качестве электродов в дуге или искре. Стекла, породы, минералы, руды измельчают в ступке, навеску в 1—10 мг помещают в отверстие угольного электрода и затем возбуждают спектр в дуге.
Спектральный анализ имеет особенно важное значение для тех материалов, которые обычными химическими методами анализировать трудно или невозможно, как, например, смеси редкоземельных металлов 4. В тех случаях, когда стандарты определенного состава недоступны, образец переводят в раствор и непосредственно наблюдают искоровой спектр, применяя пористый графитовый электрод, в который помещают исследуемую жидкость 5. Как и в химическом анализе, растворение является прекрасным средством превращения анализируемого материала в однородный продукт, и этим же способом можно легко приготовить стандарты из чистых элементов.
Помимо рядовой аналитической работы, спектроскопические методы используются для разрешения некоторых специальных проблем, как,
1 С. Е. Н а г v е у, A Method of semi-quantitative Spectrografic Analysis, Applied Research Laboratories, Glendale, California, 1947.
2 R. L-Mitchel 1, R.O.Scott, Spectrochim. Acta, 3, 367 (1948).
3 B. F. S с r i b n e r, H. R. M u 11 i n, J. Research NBS, 37, 379 (1946).
4 V. A. F a s s e 1, H. A. W i 1 h e 1 m, J. Optical Soc. Am., 38, 518—26 (1948).
5 C. Feldman, Anal. Chem., 21, 1041 (1949).
12*
180 Гл> V. Специальные операции
например, непосредственный анализ различных включений в стали определение незначительных количеств свинца в воздухе 2 и анализ остаточного газа в электронных лампах 3.
ФОТОМЕТРИЯ ПЛАМЕНИ
Фотометрия пламени представляет собой аналитический метод, заключающийся в распылении анализируемого раствора в пламени, выделении характерной для определяемого элемента длины световой волны и измерении интенсивности излучения 4. Этим методом можно быстро и надежно определять щелочные и щелочноземельные металлы! Имеются указания, что точность определения этих элементов лежит в пределах 1—3% (относительных).
Прибор для фотометрии пламени состоит из горелки, приспособления для распыления раствора в пламени, 'светофильтра или рассеивающей системы для выделения света определенной длины волны, интенсивность которого надлежит измерить, фотоэлемента и гальванометра для измерения тока, пропорционального интенсивности излучения. Простейший из известных — прибор со светофильтром — успешно служит для определения щелочных и щелочноземельных металлов, спектральные линии которых отличаются высокой природной интенсивностью и находятся в видимой области спектра, где спектр относительно несложен.. Широко применяются фильтрофотометры (стр. 174), например, для определения натрия и калия в портландском цементе 5; эти методы заменяют в значительной степени в массовых анализах классический метод 6 Лоуренса Смита.
По имеющимся наблюдениям, источником ошибок в этом методе являются различия в концентрации кислоты или других компонентов в анализируемом и стандартном растворах, что приводит к различной скорости их испарения.
С целью сведения до минимума действия этих факторов фотометр со светофильтрами видоизменили так,'чтобы при определении были обеспечены условия одинакового и одновременного наблюдения двух линий: одной от определяемого элемента в анализируемой пробе, другой — от внутреннего стандарта, например лития, который вводится и в пробу, и в стандартные растворы.
При использовании этого видоизмененного способа, сложность которого не всегда оправдывается, достигают более точных результатов в случаях, когда состав анализируемой пробы сильно колеблется, что имеет
J. Convey, J. Н. О 1 if i е 1 d, Iron and Steel, London, 18, 580 (1945).
2 H. Aughey, J. Optical Soc. Am., 39, 292 (1949); 0. G. К о p p i u s, там же, 39, 294 (1949).
’ R. H. Zachariason, Anal. Chem., 21, 1285 (1949).
4 С фотометрией пламени можно ознакомиться в работе V. W. Meloche, A Review of Flame Photometry, и в статьях, помещенных в Symposium on Flame Photometry, 1951, ASTM, Philadelphia, Feb., 1952, и в Flame Photometry, Anal. Chem., 22, 1202 (1950).
8 Sodium Oxide and Potassium Oxide in Portland Cement by Flame Photometry, ASTM Standard C 228—49T, pt. 3, 1949, p. 133.
8 W. R. E u b a n k, R. H. В о g u e, J. Research NBS, 43, 173 (1949); J. J. Dia-m ond, L. Bean, Use of the Beckman and Perkin-Elmer Flame Photometers for the Determination erf Alkalies in Portland Cement, ASTM Preprint, 1951, p. 128.
Рентгеновский метод химического анализа 181
место, например, в клинических анализах. Исследования, направленные на повышение чувствительности определения щелочных металлов и на распространение этого метода на определение других элементов, привели к разработке конструкции приборов, обладающих более широкой разрешающей способностью. В одном из этих приборов интерференционные светофильтры служат для более резкого выделения спектральных линий, а усилители фотоэлектрического тока — для повышения чувствительности. В другом имеющемся в продаже приборе приспособлением для получения пламени снабжен обычный спектрофотометр.
Исцользование в более новых приборах высокотемпературного пламени, например оксиацетиленового, дает возможность определять значительно большее число элементов.
Однако по сравнению со спектрографами, которыми обычно пользуются в спектрохимическом анализе, приборы для фотометрии пламени обладают значительно меньшей разрешающей способностью.
Применение фотометрии пламени для определения многих элементов ограничивается неблагоприятным соотношением между интенсивностями спектральных линий фона и определяемого элемента и наложением их линий спектра. В таких случаях успеху фотометрического определения может способствовать предварительное отделение мешающих элементов обычными химическими методами.
РЕНТГЕНОВСКИЙ МЕТОД ХИМИЧЕСКОГО АНАЛИЗА
Заслуживают внимания три вида рентгеновского метода анализа: рентгеновский эмиссионный спектральный анализ, включая рентгено-флуоресценцию; абсорбционный рентгеновский анализ и рентгеновский диффракционный порошковый анализ.
Первый ид этих методов первоначально сводился к прикреплению ' исследуемого образца к антикатоду рентгеновской трубки и сопоставлению его рентгеновского линейного спектра со стандартными таблицами. Этот метод применим для открытия элементов с атомными номерами выше 10. Преимуществом его перед спектральным методой является значительно большая простота спектра и возможность точно предсказывать длины волн. В опытных руках метод дает однозначные результаты, так ч как они зависят только от атомного номера искомого элемента; этот метод можно применять как для качественного, так и для количественного анализа. Недостаток метода заключается в сложности и трудоемкости про-.цессов, что ранее в известной мере и ограничивало его использование. Важрую роль Метод сыграл в открытии новых элементов, таких, как ‘техшщий (43), прометий (61), гафний (72) и рений (75).
В последнее время эмиссионный рентгеновский анализ значительно упрощен благодаря использованию рентгено-флуоресценций. В этом случае образец помещают вне рентгеновской трубки и облучают интенсивным пучком коротковолновых рентгеновских лучей. Получающийся в результате этого спектр вторичных рентгеновских лучей наблюдается в счетчике Гейгера, причем регистрация может быть произведена в очень короткий промежуток времени. Оборудование, необходимое для использования этого метода, доступно. Описано применение рентгено-флуорес-х центного метода для определения гафния в цирконии и тантала
Ig2 Гл. V. Специальные операции
в ниобйи \ а также для анализа легированных сталей, жаростойких и высокотемпературных сплавов типа хром — никель — кобальт 1 2.
Рентгеновский абсорбционный анализ, основанный на поглощении рентгеновских лучей, может быть использован для идентификации элементов в несложных смесях. В этом случае слой жидкого или твердого образца помещают перед рентгеновской трубкой и наблюдаемое селективное поглощение рентгеновских лучей, имеющих различные длины волн, сопоставляют с результатами, полученными для стандартов. Метод находит все возрастающее применение для определения свинца и серы в нефтяных продуктах, урана в растворах, гафния в цирконии и др. 3.
Рентгеновский порошковый диффракционный анализ дает возможность определить характер химического соединения каждого из присутствующих элементов и применим к твердым продуктам, имеющим кристаллическую структуру. Для этого типа анализа не требуется спектрометра. Метод заключается в следующем. Измельченный материал помещают в трубку, через которую пропускают пучок монохроматических рентгеновских лучей. Спектр фотографируется или, что еще лучше, регистрируется электронным самопишущим аппаратом. Полученную рентгенограмму затем сравнивают с типовыми рентгенограммами, предварительно полученными для многих веществ. Каждое кристаллическое вещество дает свойственную ему, всегда одинаковую, рентгенограмму, независимо от присутствия других веществ. Таким образом, наблюдаемая рентгенограмма смеси представляет собой сумму наложенных друг на друга рентгенограмм, которые дает каждое вещество в отдельности при действии на него рентгеновских лучей в продолжение такого же промежутка времени. Этот закон распространяется не только на положение диффракцион-ных линий, но и на их интенсивности при условии, если абсорбция каждого компонента ничтожна; благодаря этому метод может быть использован для количественного анализа. При работе по этому методу можно не обнаружить всех компонентов, входящих в смесь, например при анализе аморфных продуктов, таких, как уголь или стекло. Не удается обнаружить вещества, находящиеся в виде твердого раствора в кристаллах, а также компоненты, содержащиеся в малых количествах. Диффракционный метод обычно применим для определения компонентов, концентрации которых составляют около 5%; ни в отдельных случаях он может быть применен и для определения веществ при более низких концентрациях — порядка 1—2%.
Метод (особенно удобен для предварительных исследований, результаты которых при необходимости могут быть дополнены спектрохими-ческим или микрохимическим анализом.
К преимуществам диффракционного метода можно отнести следующие: 1) образцы анализируются непосредственно в том виде, в каком они получены, без разрушения; 2) процесс подготовки образца и получения рентгенограммы прост, непродолжителен и экономичен; 3) вещества определяются в виде их истинных химических соединений; 4) можно наблюдать различные кристаллические фазы, определять степень гидратации и'окисления, а также физическое состояние компонентов; 5) резуль-
1 L. S. В i г k s, Е. J. В г о о k s, Anal. Chem., 22, 1017 (1950).
2 J. L. A b b о t t, Iron Age, 162, 58, 121 (1948).
3 Обзор, касающийся этого вопроса, см. Н. A. Liebhafsky, Anal. Chem., 21, 17 (1949); 22, 15 (1950); 23, 14 (1951).
Хроматографический анализ
183
тэты анализа достоверны даже в том случае, если аналитик располагает ничтожно малым количеством исследуемого материала; 6) результаты определения всегда сохраняются в виде рентгенограмм *.
ХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ
Хроматографические разделения основаны на селективной адсорбции и дифференциальной диффузии. Во многих случаях успешное разделение достигается обменом ионов, как, например, замена Са2+ на 2Na+ при умягчении воды. В других случаях искомые вещества адсорбируются из раствора и удерживаются адсорбентом, так что раствор непрерывно от них освобождается. Это имеет место, например, при удалении окрашенных веществ из растворов сахара твердыми адсорбентами. До настоящего времени хроматографический метод остается в значительной степени эмпирическим, т. е. нет определенных принципов, на которых можно основываться при выборе адсорбента, растворителя и элюента 1 2, когда требуется провести разделение какой-либо смеси. Выбор этот следует делать, основываясь на накопленном опыте. В последнее время, однако, эмпирические сведения накапливаются быстро, и уже сделано достаточно обобщений, чтобы дать некоторые теоретические основы, которыми можно руководствоваться при выборе методов для осуществления требуемых разделений.
Способность к обмену различных ионов качественно можно представить следующим образом: 1) при низких концентрациях в водных растворах и обыкновенной температуре способность к обмену возрастает с повышением заряда иона (Na* <Са2+ <^А13+ <Th4+); 2) при низких концентрациях в водных растворах, обыкновенной температуре и постоянном заряде способность к обмену возрастает с повышением атомного номера иона (Li < Na < К < Rb < Cs; Mg Са < Sr <£ Ва) 3.
Отличительной чертой хроматографических методов является возможность их широкого применения. Хроматография может быть использована для разделения как больших, так и малых количеств элементов. Она может быть с одинаковым успехом применена к оргайическим и неорганическим веществам, для больших и малых молекул, для анионов и катионов. Кроме того, имеется возможность примерять разнообразные растворители и элюенты. В области аналитической химии хроматография открывает большие возможности для разделения редкоземельных металлов, для отделения ниобия от тантала, гафния от циркония и т. д. Она может приобрести также большое значение для упрощения некоторых продолжительных методов анализа. Так, например, при определении пятиокиси фосфора в апатите сначала из раствора Са3(РО4)2 извлекают хроматографически ионы Са2+, а затем титруют освобожденную фосфорную кислоту.4 Техника хроматографии разнообразна, но для аналитических
1 Обсуждение этих методов см. W. Р. D a v е у, J. Appl. Phys., 10, 820 (1939), а также обзор Н. S. Kaufman, I. Fankuchen, Anal. Chem., 21, 24 (1949); 22, 16 (1950). Компилирование данных порошкового диффракционного анализа см. J. D. Hanawalt, Н. W. R i n n, L. К. F г е v е 1, Ind. Eng. Chem., Anal. Ed., 10, 457 (1938); L. К. F г e v e 1, там же, 16, 209 (1944).
2 * Элюент — раствор для извлечения поглощенного вещества (иона) пз адсорбента. Прим, перев. *
3 R. К u n i п, Anal. Chem., 21, 87 (1949).
'‘К. Н е г 1 i с h, W. R i е m a n, Ind. Eng. Chem., Anal. Ed., 19, 651 (1947).
184
Гл. V. Специальные операции
целей наиболее удобна колонка адсорбента, подобная редуктору Джонса. Метод с применением этого приема описан в гл. «Редкоземельные металлы» (стр. 628). За ходом процесса или степенью разделения можно проследить, отбирая небольшие количества пропущенной через колонку жидкости (элюата) и наблюдая изменение ее окраски или испытывая флуоресцентным методом' или методом радиоактивных индикаторов \ В последнее время значительное развитие получила хроматография на бумаге. Она заключается в разделении£смесей по различной адсорбции на полосках бумаги 1 2. '
МАСС-СПЕКТРОМЕТРИЯ
Масс-спектрометр в аналитической химии используется главным образом для анализа газов. Этим сложным и довольно дорогим прибором можно идентифицировать и определять компоненты в простых и сложных смесях углеводородов, в горючих, отработанных и редких газах. Мало внимания уделяется анализу твердых продуктов, хотя последние работы показывают, что масс-спектрометрия имеет некоторое применение и в этой области 3. Удовлетворительные масс-спектрограммы' были получены для паров многих жидкостей.
Описание приемов масс-сцектрометрии слишком длинно, чтобы его помещать здесь. В литературе имеется обзор последних работ, касающихся этого вопроса 4
КАПЕЛЬНЫЙ АНАЛИЗ
Качественный и полуколичественный анализ органических и неорганических продуктов можно выполнять посредством реакций, которые осуществляются в одной капле раствора на пластинке, фильтровальной бумаге, в специальных приборах или даже непосредственно на самом образце 5. Такие методы можно рассматривать как особую разновидность микроанализа.
Подобно микроанализу, капельный анализ особенно полезен, когда имеющееся в Наличии количество материала очень мало или когда тре
1 Применив соответствующую смолу и радиоактивные носители, L. Schubert (Бюро стандартов США) отделил 1 .мкг цезия от 43 г нитрата бария.
2 Другие области применения, приемы и общие обзоры по вопросам, касающимся хроматографического анализа, читатель может найти в следующих работах: Some Applications of Selective Adsorption and Differential Diffusion in Chemical Analysis by Beverly L. Clarke, in Colloid Chemistry Theoretical and Applied, vol. V, Jerome Alexander, Reinhold Publishing Corp., 1944; H. H. Strain, Anal. Chem., 21, 75 (1949); 22, 41 (1950); D. Clegg, там же, 48; R. К u n i n, там же, 21, 87 (1949); 22, 64 (1950). * О. Самуэльсон, Применение ионного обмена в аналитической химии.’Издатинлит, 1955; Ф. М. Шемякин, Э. С. Мицеловский, Д. В. Романов, Хроматографический анализ, Госхимиздат, 1955 *.
3 Си. J. G. Gorman, Е. J. J a m е s, J. A. Hippie, Anal. Chem., 23, 438 (1951).
4 J. А.' Н i р р 1 е, М. S h е р h е г d, Anal. Chem., 21, 32 (1949); 22, 23 (1950).
8 В. S. Evans, D. G. Higgs, Spot-Tests for the Identification of Certain Metalic Coatings and of Certain Metals in Bulk, W. Heffer and Sons, Cambridge, 1943. См. также Symposium on Rapid Methods for the Identification of Metals, ASTM, 1950.
Микрохимия и химическая микроскопия
185
буется избежать разрушения анализируемого образца. Использование-этого способа, однако, этим не ограничивается \ Описаны усовершенствования'в технике капельного анализа и метод с применением реактивной бумаги для анализа больших объемов раствора 1 2.
В последние годы методы капельного анализа были значительно усовершенствованы, и область их использования весьма расширилась-благодаря открытию и применению новых органических реагентов, а также маскирующих и демаскирующих реакций3. Использование флуоресценции , еще больше увеличило возможности капельного анализа. Так, например, испытание на натрий по реакции образования тройного-ацетата значительно более эффективно в ультрафиолетовом свете. Интересным примером капельных колориметрических методов анализа без разрушения образца является электролитический метод, в котором образец металла или сплава используется в качестве анода в соответствующей среде. Растворяющиеся при этом незначительные количества искомого-компонента электролитически переносятся к катоду на бумагу или другой материал, пропитанный соответствующим реактивом 4.
МИКРОХИМИЯ И ХИМИЧЕСКАЯ МИКРОСКОПИЯ
В области микрохимии за -последние годы достигнуты большие успехи, благодаря чему микроскопия и микроанализ стали самостоятельными разделами в аналитической химии.
Микроанализ служит главным образом для количественных определений в техническом анализе. В органическом анализе он почти совершенно вытеснил более старые макрометоды, особенно в рядовых анализах, где для определения микрометодами таких компонентов, как углерод, водород и азот, требуется почти вдвое меньше времени, чем при использовании макрометодов. Использование и возможное применение микрометодов в анализе неорганических продуктов весьма многообразно, но в настоящее время в этой области микроанализ чаще служит в качестве специального, а не обычно применяемого способа. В этом случае, так же как и в органическом анализе, микрометоды удобны вследствие
1 Ф. Ф а й г л ь, Капельный анализ, ОНТИ, 1937; F. F е i g 1, Spot tests, v. I, II, Elsevier Publishing Co, 1954; H. А. Тананаев, Капельный метод,. Госхимиздат, 1954; Н. А. Тананаев, Бесстружковый метод анализа черных, цветных и драгоценных сплавов, Металлургиздат, Свердловск — Москва, 1948.
2 В. L. Clarke, Н. W. Н е г m а n с е, Ind. Eng. Chem., Anal. Ed., 9, 292 * (1937); 10, 591 (1938); См. также H. Ya g о d а, там же, 9, 79 (1937); Н. Y a g о d а,. Mikrochemie, 24, 117 (1938); R. Е. Stevens, Н. W. Lakin, U. S. Geol. Survey Circ. № 1, 63 (1949).
8 F. F e i g 1, Chemistry of Specific, Selective and Sensitive Reactions, Academic Press, New. York, 1949, p. 67. Экспериментальная часть капельного анализа органических и неорганических продуктов полностью обработана F е i g 1 в труде Qualitative Analysis by Spot Test, 3d English ed.-, Elsevier Publishing Co., New York, 1946.
4 H. Fritz, Z. Anal. Chem., 78, 418 (1929); R. J irkovsky, Miftochemie, 15, 331 (1934); Ed. Arnold, C., 1933, I, 2846; R.J irkovsky, Chem. Listy, 25, 254 (1931); A. Glasunov, J.Krivohlavy, Z. phys. Chem., 161, 373 (1932); B. L. Cl a rke, . H. W. H e r m a n c e, Ind. Eng. Chem., Anal. Ed., 9, 292 (1937); H. Y a g о d а, там же, 12, 698 (1940); 15, ' 135 (1943). * Л. M. К у л ь б е р г, Г. С. А л ь т е р з о н, Р.. П. Вельтйан, Капельный анализ, Госхимиздат, 1951, стр. 116. *
186
Гл. V. Специальные операции
экономии времени, оборудования и реактивов, когда требуется провести количественное исследование продуктов, достаточно однородных, чтобы можно было пользоваться маленькими навесками. Там, где строгая однородность материала не существенна, как, например, в качественном анализе, значение микро- и полумикрометодов возрастает.
В противоположность микроанализу, химическая микроскопия нашла применение главным образом в качественном анализе, где она обладает особыми преимуществами перед макроанализом. Так, например, групповые реагенты в этом методе отличаются большей специфичностью, лучше наблюдается соотношение между фазами в анализируемом материале, которые перед их определением часто можно разделять механическими способами, что дает лучшую идентификацию, чем.валовый анализ. В таких разделениях часто полезно применение тяжелых жидкостей \
Приблизительные величины навесок, требуемые при применении различной техники работы 1 2, приведены ниже (в мг):
Макрометоды...............100 и более
Полумикрометоды............ 10—30
Микрометоды................. 2—10
Ультрамикрометоды........... 0.001 .
Преимущество, которым обладают микрохимия и химическая микроскопия, заключается в возможности применения для анализа маленьких навесок (10 мг). Это очень важно, если в распоряжении имеется мало материала или когда материал очень ценен, или желательно избежать заметного разрушения образца. Микротехника особенно удобна при анализе некоторых минералов и руд 3. Неспециалистами часто упускается из виду тот факт, что химическая микроскопия и микроанализ распространяются на определение главным образом основных, а не второстепенных компонентов. Другими словами, эти методы не применяются непосредственно для определения компонентов, содержащихся в таких малых количествах, как 0,1 % или менее. В этих случаях удобно пользоваться способом, который состоит в следующем. Берут, насколько позволяют условия, большую навеску, концентрируют определяемый компонент макрометодом, а затем, собрав его в небольшом объеме, заканчивают определение микрометодами.
В основе макро- и микрохимических методов лежат одинаковые принципы. Основная разница заключается в приемах работы, что обусловлено маленькими масштабами операций в микрометодах. Этими приемами не так трудно овладеть, как может показаться.
1 См. J. D. S ul 1 i v a n, Heavy Liquids for Mineralogical Analysis, U. S. Bur. Mines Techn. Paper, 1927, p. 381. * E. В. Копчено в а, Минералогический анализ шлихов, ГоЬгеолиздат, 1941. *
2 См. также П. Кир к, Количественный ультрамикроаналпз, Издатин-лит, 1952.
3 См. Е. S. Larsen, Н. Berman, The Microscopic Determination of the Nonopaque Minerals, 2d ed., U. S. Geol. Survey Bull., 848 (1934); M. N. Short, Microscopic Determination of the Ore Minerals, 2d ed., U. S. Geol. Survey Bull., 914 (1940).
Микрохимия и химическая микроскопия
187
Подробное описание методов и специальной аппаратуры, такой, как микроскопы или микрохимические весы, выходит за пределы круга вопросов, которые подлежат освещению в этой книге
1 Подробное описание аппаратуры, методов, приемов п применения приводят Е. М. Chamot, С. W. Mason, Handbook of Chemical Microscopy, vol. I, Principles and Use of Microscopes and Accessories, Physical Methods for the study of Chemical Problems, 1938; vol. II, Chemical Methods and Inorganic Qualitative Analysis, John Wiley and Sons, 1940; F. E m ich, Lehrbuch der Mikrochemie, 2d ed., J. F. Bergmann, Munich, Germany, 1926; F. P r e g 1, H. Roth, Quantitative Organic Microanalysis, 3d ed., translated by E. B. Daw, P. Blakiston’s Son and Co., Philadelphia, 1937; F. Hecht, Die Quantitative Mikro-Mineralanalyse und ihre Epgebnisse, Micro-chim. Acta, 2, 120 (1937); Micro-Silicatanalyse, там же, 188. См. также F. Hecht, J. D о n a n, Anorganische Mikrogewichtsanalyse, Julius Springer, Vienna, 1940; photolithoprint reproduction by Edwards Brothers, Ann. Arbor., Mich, 1943; N. H. H a r t s-horne, A. Stuart, Crystals and the Polarising Microscope, 2d ed., Edward Arnold and Co., London, 1950; /* И. M. К о p e н м а н, Микрокристаллоскопия, Госхимиздат, 1955. *
Глава VI
ОБЪЕМНЫЙ АНАЛИЗ
ПРИБОРЫ ДЛЯ ИЗМЕРЕНИЯ ОБЪЕМА
Приводимые ниже описания приборов для измерения объема, взятые нами из1 циркуляра Бюро Стандартов США С434 (1941), имеют большое значение в объемном анализе.
Единицы емкости. Литр определяется как объем, занимаемый при 4° С количеством чистой воды, имеющей массу 1 кг, а одна тысячная часть литра, называемая миллилитром \ принимается за Единицу емкости.
Стандартная температура. Бюро Стандартов США принимает 20° С за стандартную температуру при работе со стеклянными приборами, предназначенными для измерения объема2., (*Эта же температура принята и в СССР*). В табл. 11 приведены массы различнйх объемов воды при температурах от 15 до 30° С при взвешивании на воздухр латунными разновесками.
Общие требования
Применяемое стекло и его отжиг. Приборы для измерения объема жидкостей должны изготовляться из стекла лучшего качества, прозрач-( ного, свободного от Полос и устойчивого к воздействию химических реактивов.
Все приборы перед их градуированием следует основательно отжечь при 400° С в течение 24 ч и медленно охладить.
Форма приборов й их отделка. Поперечное сечение всех приборов должно быть круглым, а форма каждого сосуда должна допускать полное его опоражнивание и высушивание.
Приборы, имеющие основание или ножку, должны стоять устойчиво на плоской поверхности; основания должны быть такой величины, чтобы приборы не падали, если их поставить на плоскость, наклоненную на 15° к горизонтальной.
1 Миллилитр часто обозначают буквами см3. Но кубический сантиметр не равен точно одной тысячной части литра; 1 мл = 1,000028 «№ (La Creation du Bureau International ’des Poids et Mesures et sori Oeuvre, Ch. Ed. Guillaume, 1927, стр. 258). Хотя эта разница не имеет значения в обычной работе, мы ее отвечаем, чтобы избежать путаницы в применении обеих единиц.
' 2 М. G. М ell an [Ind. Eng. Chem., Anal. Ed., 2, 260 (1930)] описывает метод
учета изменения температуры при выполнении объемного анализа.
Приборы, для измерения объема
189
Пробки и краны должны быть дршЛлифованы так, чтобы они легко действовали и не допускали утечки.
Части, на которых нанесены деления, должны быть цилиндрическими по крайней мере на расстоянии 1 см по обе стороны от каждой метки, но в других местах они могут быть расширены, чтобы обеспечить требуемую вместимость при удобной длине.
' ' ТАБЛИЦА 11 Масса воды, взвешенной на воздухе латунными разновесками
Б табл. 11 приведены наблюдаемые массы при взвешивании различных количеств воды, имеющей температуру от 1 5 до 30° Сь латунными разновесками. Влажность воздуха принимается равной 50%, барометрическое давление 760 мм рт. ст. Таблица удобна для калибрования мерной посуды. Принимается, что воздух имеет одинаковую с водой температуру.
Температура °C Емкость сосуда, мл
2000 1000 500 400 300 250 150
Масса воды, г
15 1996,11 998,05 499,03 399,22 299,42 249,51 149,71
16 1995,80 997,90 498,95 399,16 299,37 249,48 149,68
17 1995,48 997,74 498,87 399,10 299,32 249,43 149,66
18 1995,13 997,56 498,78 399,03 299,27 249,39 149,63
19 1994,76 997,38 498,69 398,95 299,21 249,34 149,61
20 1994,36 997,18 498,59 398,87 299,15 249,30 149,58
21 1993,95 9Й6.97 498,49 398,79 299,09 249,24 149,55
22 . 1993,51 996,76 498,38 398,70 299,03 249,19 149,51
23 1993,06 996,53 498,26 398,61 298,96 249,13 149,48
24 1992,58 996129 498,15 398,52 298,89 249,07 149,44
25 1992,09 996,04 498,02 398,42 298,§1 249,01 149,41
26 1991,57 995,79 497,89 398,31 298,74 248,95 149,37
27 * 1991,04 995,52 497,76 398,21 298,66 248,88 149,33
28 1990,49 995,24 497,62 398,10 298,57 248,81 149,29
29 1989,92 994,96 497,48 397,98 298,49 248,74 149,24
30 ' 1989,33 994,66 497,33 397,87 298,40 248,67 149,20
Деления должны быть одинаковой ширины, непрерывны и тонко, но отчетливо начерчены и должны быть перпендикулярны к оси и параллельны основанию прибора.
Все деления должны охватывать по крайней мере половину окружности. Кроме того, на тех приборах, где имеются мелкие подразделения отметок, каждое десятое деление, а при отсутствии их все метки должны охватывать полностью всю окружность прибора.
Интервалы между смежными метками должны быть не меньше 1 мм. Промежутки между метками на приборах, где имеются подразделения, не должны иметь видимых неправильностей, и достаточное число делений Должно быть пронумеровано, чтобы ясно можно было определить емкость, -отвечающую каждому делению. }
Надписи. На каждом приборе должны быть обозначены четким шрифтом: его емкость, температура в градусах Цельсия, при которой его надо применять, и метод его употребления, т. е. предназначен ли он «на вли-. вание» или «на выливание»; на приборах, из которых жидкость выпускается через Оттянутый кончик, должно быть также указано время, требуемое для опоражнивания всей номинальной емкости прибора при полностью открытом выходном отверстии.
190
Гл. VI. Объемный анализ:
На каждом приборе должна быть обозначена фирма или фабричная марка. Каждый прибор должен иметь номер; отдельные вынимающиеся его части, например пробки, краны и т. п., должны быть помечены тем же номером.
Специальные требования
Мерные колбы. Внутренний диаметр шейки колбы около метки (или меток) ее вместимости может колебаться в следующих пределах:
Емкость колбы, мл 25
Диаметр шейки, мм
максимум ... 8
минимум .... 6
50 100 200 250 500 1000
10 12 14 15 18 20
6 8 9 10 .12 14
2000 3000 4000 5000 6000
25 30 35 40 45
18 20 22 25 30
Шейка колбы не должна выше метки сужаться. Метка емкости колбы должна находиться от конца цилиндрической шейки на расстоянии не меньшем, чем указано ниже:
Емкость, мл...............................
Расстояние от верхнего конца шейки, см . .
Расстояние от нижнего конца шейки, см . .
100 или менее
3
1
Более 100 6
2
Мерные колбы могут быть градуированы одновременно и «на вливание» и «на выливание» при условии, что значения обеих меток отчетливо указаны и что расстояние между ними не меньшем 1 мм. В колбах, градуированных более чем на одну емкость, ошибка в объеме между двумя соседними метками не должна превышать половины; допустимой ошибки в объеме, показанном первой меткой. Приводим допустимые отклонения Ъ
Емкость, мл . . . . 25 50 100 200 300 500
Предел погрешности
на вливание . . . . 0,03 0,05 0,08 0,10 0,12 0,15
на выливание ..... . . . . 0,05 0,10 0,15 0,20 0,25 0,30
Емкость, мл . . . . 1000 2000 3000 4000 5000 6000
Предел погрешности
на вливание . . . . 0,30 0,50 0,75 1,00 1,2 1,5
на выливание 0,50 1,00 1,50 2,0 2,4 3,0
Обыкновенные пипетки. Обыкновенные пипетки — это пипетки, градуированные на выливание с одной меткой на верхней трубке, через которую всасывают жидкость. Их еще называют «пипетками для переноса».
Трубка для всасывания жидкости каждой такой пипетки должна иметь в длину по крайней мере 16 см, а трубка для выливания — не менее и не более 25 см. Внутренний диаметр обыкновенной пипетки около метки ее емкости должен быть не менее 2 мм и в зависимости от объема не должен превосходить следующих предельных значений:
Емкость пипетки, мл, включительно до . . 25 50 200
Диаметр, мм....................... 4 5 6
1 О методах проверки объемно-аналитических приборов см.: NBS Reprint 92; NBS Bull. 4, № 4 (1907—1908); H. N. M о г s е, Т. L. В 1 а 1 о с k, Am. Chem. J.r 16, 479 (1894); W. Schloesser, Z. angew. Chem., 16, 953, 977, 1004, 1061 (1903); W. Schloesser, C. Grimm, Chem. Ztg., 30, 1071 (1906): W. Schloesser, Z. anal. Chem., 46,- 392 (1907).
Приборы для измерения объема
191
Внешний диаметр верхней и нижней трубок обыкновенной пипетки,, не считая кончика, должен быть не менее 5 мм.
Метка емкости пипетки должна находиться не далее 6 см от рнсши- z ренной ее части.
Выпускное отверстие кончика обыкновенной пипетки должно быть, такой величины, чтобы свободное вытекание жидкости продолжалось, не более 1 мин и, в зависимости от емкости пипетки, не менее указанного ниже времени: /
Емкость пипетки, мл
Время вытекания, сек
5 10 50 100 200
15 20 30 40 50
Допустимые отклонения 1 приведены ниже:
Емкость, мл, включительно до..........
Предел погрешности, мл................
2 5 10 30 50 100 200’
0,006 0,01 0,02 0,03 0,05 0,08 0,10
Бюретки и измерительные пипетки. Следует применять только бюретки, опоражнивающиеся через кончик, неподвижно прикрепленный внизу. Боковые трубки, если они не снабжены кранами, у бюреток не допускаются. (Так называемые бюретки Шельбаха, имеющие заднюю стенку из молочного стекла с окрашенной центральной линией, Бюро Стандартов США для испытания не принимаются.)
Расстояние между крайними делениями не должно превосходить 70 см у бюреток и 35 см у измерительных пипеток.
Скорость истечения жидкости из бюреток и измерительных пипеток должна определяться диаметром отверстия кончика, и для каждого градуированного интервала время свободного вытекания должно быть не более 3 мин и не менее приведенного ниже, в зависимости от длины градуировки:
Длина градуировки, см.............. 15 20 25 30 35 40 45 50 55 60 65 70
Время вытекания, сек, не менее . . 30 35 40 50 60 70 80 90 105 120 140 160
Деления измерительной пипетки должны начинаться на расстоянии не менее чем 10 см от верхнего конца ее и кончаться на расстоянии не менее 4 см от нижнего конца.
На бюретках вместимостью 50 и 100 мл самое верхнее деление должно находиться на расстоянии не менее 4 см и не более 10 см от верхнего конца бюретки. На бюретках, имеющих вместимость 25 мл или меньше, это расстояние должно быть не менее 3 см и не более 6 см.
Кончики бюреток и пипеток должны иметь равномерную конусообразную заостренность на протяжении от 2 до 3 см.
Сильное сужение у конца не допускается; кончик должен быть хорошо отшлифован.
Для облегчения удаления капель и для избежания разбрызгивания, когда прибор находится в вертикальном положении, кончик может быть слегка загнутым.
1.Если пипетка удовлетворяет приводимым требованиям, то при ее применении в обычном анализе никаких поправок вводить не надо. Следует, однако, помнить, что . это может привести к ошибке порядка 1 : 500 (в случае применения пипеток на 5 и 10 мл) и что при неразборчивом применении разных пипеток нельзя ожидать получения совпадающих результатов.
192
Гл. VI. Объемный анализ
Допустимые отклонения:
Емкость градуированной части, мл . . . 2 5 10 30 50 100
Предел погрешности всей или частичной
емкости, мл:
бюретки — • 0,01 0,02 0,03 0,05 0,10
измерительной пипетки 0,01 0,02 0,03 0,05 0,08 0,15
Правила обращения с измерительными приборами
Приводим основные правила обращения с объемно-аналитическими приборами. Только при соблюдении этих правил осуществляются условия, необходимые для получения точных измерений.
Жидкость для калибровки. Калибровка прибора проводится водой, и емкость его определяется, следовательно, объемом воды, содержащейся в приборе или вылитой из него при стандартной температуре.
Способ отсчета. Во всех приборах, где объем жидкости отграничивается мениском, отсчет делается по нижнему краю мениска. Чтобы можно было легко наблюдать нижний край, за бюреткой непосредственно под мениском нужно поместить экран-из какого-нибудь темного материала; при этом профиль мениска становится темным и ясно видимым на светлом фоне. Удобным приспособлением для этой цели является кусок толстой черной резиновой трубки, сделанный в виде воротничка, с вырезом с одной стороны, и такой величины, чтобы он плотно охватывал заднюю половину трубки прибора.
Чистота приборов. Приборы должны быть настолько чистыми, чтобы они смачивались водой равномерно по всей поверхности.
Мерные колбы и цилиндры. При наполнении мерных колб и цилиндров смачивается вся внутренняя поверхность сосудов; поэтому перед отсчетом нужно подождать 2 мин, чтобы дать жидкости стечь со стенок.
Мерные колбы и цилиндры, если они употребляются «на выливание», следует опоражнивать постепенным наклонением их до тех пор, пока не прекратится непрерывное вытекание жидкости и они не примут почти вертикальное положение. Продержав их полминуты, в этом положении, приводят горлышко в соприкосновение с влажной поверхностью приемного сосуда, чтобы удалить последнюю каплю.
Пипетки и бюретки. При наполнении пипеток и бюреток избыток жидкости, приставший к кончику, должен быть по окончании наполнения удален.
При опоражнивании бюреток их нужно держать в вертикальном положении, и после прекращения непрерывного выливания кончик должен быть приведен в соприкосновение с влажной поверхностью приемного сосуда, чтобы докончить выливание. При выливании жидкости из обыкновенных Пипеток их нужно держать в вертикальном положении и вытекание должно быть непрерывным, пока поверхность жидкости не дойдет до верхнего отверстия нижней трубки; тогда прикасаются кончиком к влажной поверхности приемного сосуда и держат так, пока выливание не закончится. Не следует выдувать остающуюся в кончике жидкость.
При пользовании кранами последние должны быть полностью открыты во время выливания.
Ацидиметрия и алкалиметрия
193
Бюретки и пипетки нужно наполнять приблизительно на 2 см выше нуля, и установку на нуль нужно проводить медленным выливанием жидкости от этой точки. Хотя обычно измерения делаются от нулевой точки, но при использовании бюреток стандартной формы можно начинать выливание жидкости и от других точек.
Весовые бюретки
При выполнении анализов самой высокой точности нужно применять весовые бюретки, потому что при работе с ними исключается
влияние температуры, а количество раствора может быть опреде-
лено отвешиванием с большей точностью, чем отмериванием. Так, например, израсходовав при титровании 50 мл раствора, легко найти массу этого раствора с точностью до 1 мг, т. е. с точностью 1 на 50 000, тогда как сделать отсчет на бюретке с точностью до 0,01 мл, т. е. 1 на 5000, было бы уже трудно. Весовые бюретки имеют, кроме того, следующие преимущества *.
1) Так как эти бюретки не градуированы, то не приходится тратить время на их калибрование и исключаются связанные с этим ошибки.
2) Отпадает необходимость в частой чистке, бюреток, так как капли, приставшие к стенкам бюретки, не влияют на получаемый результат титрования 1 2.
3) Результат титрования не зависит от температуры раствора.
4) Не приходится ждать, пока жидкость стечет со стенок бюретки. За время, обычно затрачиваемое на это ожидание, бюретку можно взвесить.
На рис. 19 показана одна из применяемых форм весовой бюретки.
Рис. 19. Весовая бюретка.
АЦИДИМЕТРИЯ И АЛКАЛИМЕТРИЯ
Индикаторы
Общие замечания. При титровании кислотами и щелочами достигаемое в конце титрования состояние определенной степени нейтрализации устанавливается обычно при помощи индикатора. Процесс нейтрализации разбавленных растворов кислот и щелочей можно также проконтролировать потенциометрическим методом (стр. 229).
1 Е. W. W ashb urn, J. Am, Chem. Soc., 30, 31 (1908). J. C. Hostetter и H. S. Roberts [там же, 41, 1350 (1919)] описывают весовую бюретку, имеющую дополнительный маленький резервуар, градуированный на интервалы в 0,01 мл, служащий для добавления очень малых объемов титрующего раствора вблизи конечной точки титрования.
2 Это относится к стеканию жидкости. Капли, образующиеся на стенках бюретки в результате испарения жидкости при ее длительном пребывании в бюретке, следует снимать перемешиванием жидкости в бюретке перед ее употреблением.
13 Заказ 522.
194
Гл. VI. Объемный анализ-
Индикатору, применяемые при титровании, перечислены в табл. 12 и 13. Необходимо помнить, что концентрация ионов водорода х, при которой один индикатор меняет свою окраску, может быть совершенно отлична от концентрации, требуемой для изменения окраски другого индикатора^ Поэтому концентрация раствора, определенная при титровании с одним индикатором, может» отличаться от найденной при титровании с другим индикатором. Например, при титровании одного и того же количества соляной кислоты будет израсходовано больше едкого, натра для достижения конечной точки по фенолфталеину, чем по метиловому оранжевому.
Поэтому устанавливать титр того или иного раствора и затем приме- -пять этот раствор следует по возможности с одним и тем же индикатором, при одной и той же концентрации индикатора и титруя до одного и того же оттенка при переходе окраски.
Индикаторы, перечисленные в табл. 12, могут быть классифицированы следующим образом.
Нейтральные индикаторы (интервал перехода — около pH =7) — феноловый красный и нейтральный красный.
Индикаторы, чувствительные к кислотам (интервал перехода — pH больше 7), — фенолфталеин, тимоловый синий и тимолфталеин.
Индикаторы, чувствительные к основаниям (интервал перехода — pH меньше 7), — метиловый желтый, бромфеноловый синий, метиловый оранжевый и метиловый красный.
С водой самой высокой степени чистоты первые индикаторы дают переходную окраску, вторые — кислотную, а третьи — щелочную. Угольная кислота и карбонаты должны быть исключены при титровании с индикаторами, чувствительными к кислотам, а также, желательно, и при титровании с метиловым красным. В табл. 13 приведен список кислот и оснований и те индикаторы, которые наиболее подходят для их титрования.
При титровании сильных кислот сильными основаниями можна применять любой индикатор, но конечная точка не будет соответствовать одному и тому же pH. Например, к 100 лм чистой воды нужно прибавить 0,08 лм 0,1 н. раствора кислоты, чтобы получить оранжево-желтый оттенок с метиловым оранжевым, и 0,2 лм 0,1 н. раствора щелочи для получения красной окраски с фенолфталеином. Интервал между конечными точками при таком разбавлении равняется, следовательно, 0,1 мл 0,1 н. раствора. Нужно также отметить, что при титровании
1 1 Показатель ионов водорода, изображаемый символом pH, является удобным^
способом выражения концентрации этих ионов. Первоначально величина pH определялась как отрицательный логарифм концентрации ионов водорода, а позднее — как отрицательный логарифм активности ионов водорода. За исключением простых разбавленных растворов, таких, например, как 0,01 н. соляная кислота, точная концентрация ионов водорода не может быть определена. Современная шкала значений pH основывается на ряде значений pH, приписываемых определенным растворам, принятым в качестве стандартов. Однако на практике величину pH все еще обычно понимают так, как она объяснялась первоначальным определением. Например, pH = 2 означает, что концентрация ионов водорода равна 10“а, pH = 7 означает концентрацию ионов-водорода, равную 10”’, т. е. концентрацию этих ионов в чистой воде. Более полные сведения приводятся в статье: Standardization of pH Measurements Made with the Glass Electrode, NBS Letter Circ. LC 933; см. также R. G. В a t e s, Chem. Rev., 42, 1 (1948).
ТАБЛИЦА 12
Индикаторы, применяемые в объемном анализе
Индикатор Интервал перехода окраски. pH Изменение окраски от кислоты к щелочи Концентрация приготовленного раствора индикатора, % Количество раствора индикатора в каплях на 100 мл титруемого раствора Объем раствора реактива, необходимый для изменения окраски *, мл
1 н. 0,1 н. 0,01 н.
Метиловый желтый (диметиламиноазобензол) 2,9-4,0 От красного к желтому 0,1 1-2 0,01 0,1 1,0
Бромфеноловый синий (тетрабромфе-нолсульфофталеин) 3.0—4.6 От желтого к синему 0,04 3-5 0,01 0,1 1,0
Метиловый оранжевый (диметиламино-азобензолсульфонат натрия) 3.1-4,4 От красного к оранжево-желтому -1 0,02 3-5 0,008 0,08 0,8
Метиловый красный (диметиламино-азобензол-о-карбоновая кислота) 4,2-6,3 От красного к желтому 0,02 .3—5 0,000 0,01 0,1
Вромкрезоловый пурпурный (дибром-о-крезол сульфофталеин) 5,2—6,8 От желтого к пурпурному 0,04 3-5 0,000 0,01 0,1
Нейтральный красный (диметилдиаминотолуол феназин) 6,8-8,0 От красного к желтому 0,1 1-2 0,000 0,00 0,0
Феноловый красный (фенолсульфофталеин) , 6,8-8,4 От желтого к красному 0,02 3-5 0,000 0,00 0,0
Фенолфталеин 8,2-10,0 От бесцветного к красному 1,0 1-3 0,002 0,02 0,2
Тимоловый синий (тимолсульфофталеин) 8,0-9,6 От желтого к синему 0,04 3-5 0,002 0,02 0,2
Тимолфталеин 9,3—10,5 Олг бесцветного к синему 0,04 3-5 0,01 0,1 1,0
* Число миллилитров реактива, которое необходимо прибавить к 100 мл воды, содержащей требуемое количество индикатора, чтобы вызвать ясное изменение его «нейтральной» окраски.
алкалиметрия
ь*. со СЛ
196
Гл. VI. Объемный анализ
ТАБЛИЦА 13
Индикаторы, подходящие для титрования различных кислот и оснований
Титруемые вещества
Индикаторы
Кислоты
Сильные кислоты Любой индикатор (см. текст)
Фосфорная и мышьяковая как одноосновная как двухосновная как трехосновная Метиловый желтый; метиловый оранжевый; бромфеноловый синий Тимолфталеин; фенолфталеин; тимоловый синий (раствор насыщают NaCl) Фенолфталеин; тимоловый синий (в концентрированном растворе СаС12)
Пирофосфорная как двухосновная как четырехосновная Метиловый желтый; метиловый оранжевый; бромфеноловый синий (до pH = 4) Фенолфталеин; тимолфталеин; тимоловый синий (в присутствии ВаС12)
Фтористоводородная Фенолфталеин; тимоловый синий; феноловый красный; нейтральный красный
Сернистая как одноосновная как двухосновная Метиловый желтый; метиловый оранжевый; бромфеноловый синий Фенолфталеин; тимоловый синий (в присутствии ВаС12)
Угольная как одноосновная как двухосновная Фенолфталеин; тимоловый синий (в присутствии NaCl или глицерина) Фенолфталеин; тимоловый синий (в- присутствии ВаС12)
Хромовая Фенолфталеин; тимоловый синий
Борная при добавлении многоатомного спирта без многоатомного спирта Фенолфталеин; тимоловый синий Нитрамин; тропеолин 0
Синильная Нитрамин; тропеолнн 0
Мышьяковистая как одноосновная Нитрамин; тропеолин 0
Муравьиная и ее гомологи Фенолфталеин; тимоловый синий; феноловый красный; нейтральный красный
Щавелевая и ее гомологи фенолфталеин; тимоловый синий; феноловый красный; нейтральный красный
Бензойная и ее гомологи Фенолфталеин; тимоловый синий
Фталевая Фенолфталеин; тимоловый синий
Ацидиметрия и алкалиметрия
197
Продолжение табл. 13
Титруемые вещества Индикаторы
Основания •
Сильные основания Любой индикатор
Аммиак г 1 Метиловый красный; бромкрезоловый пурпурный; метиловый желтый; бромфеноловый синий
Соли щелочных металлов и борной кислоты Метиловый оранжевый; метиловый желтый; бромфеноловый синий; метиловый красный; бромкрезоловый пурпурный
угольной кислоты . Метиловый оранжевый; метиловый жёлтый;
сероводородной кислоты бромфеноловый синий Метиловый оранжевый; метиловый желтый; бромфеноловый синий; метиловый красный;
фосфорной кислоты (до однозамещенной соли) бромкрезоловый пурпурный Метиловый оранжевый; метиловый желтый; бромфеноловый синий]
очень слабых растворов, например 0,01 н., лучше применять метиловый красный или фенолфталеин и титровать до цвета индикатора в чистой воде.
Для титрования слабых кислот сильными основаниями требуются индикаторы, чувствительные к кислотам; для титрования слабых оснований сильными кислотами нужны индикаторы, чувствительные к основаниям.
Выбор индикатора для титрования многоосновных кислот или многокислотных оснований. Выбор индикатора при таких титрованиях зависит от констант диссоциации этих веществ. Когда константы всех ступеней диссоциации велики, как, например, у серной кислоты или гидроокиси бария, многоосновные кислоты и многокислотные основания ведут себя подобно сильным одноосновным кислотам или основаниям, и конец титрования соответствует образованию нормальной соли. Если вторая или третья константы диссоциаций значительно меньше, чем первая, то можно выбрать индикатор, который будет показывать образование однозамещенной, двузамещенной и даже трехзамещенной соли. Например, при титровании фосфорной кислоты метиловый оранжевый меняет окраску, когда закончится образование однозамещенной соли; фенолфталеин (в растворе, насыщенном хлоридом натрия) — когда произойдет превращение в двузамещенную соль, а тимоловый синий (в концентрированном растворе хлорида кальция) — когда образуется средняя, трехзамещенная соль. Подобным же образом угольная кислота реагирует как одноосновная кислота при титровании ее с фенолфталеином в присутствии хлорида натрия, а сернистая кислота ведет себя как одноосновная кислота при титровании с метиловым оранжевым, и как двухосновная
198
Гл. VI. Объемный анализ
кислота — при титровании с фенолфталеином в присутствии солей бария х.
В табл. 14 приведен набор индикаторов, предложенный Кларком и Лабсом 1 2, а на рис. 20 показаны кривые диссоциации этих индикаторов. Во второй из приведенных в ссылке 2 статей отмечается, что если взять только два индикатора для общего употребления — метиловый красный и тимоловый синий, то их будет достаточно для выполнения большей части титрований в тех рядовых анализах, которые приходится про-
Рис. 20. Кривые диссоциации индикаторов, перечисленных в табл. 14.
Индикаторы здесь рассматриваются как простые одноосновные кислоты. В таблице показано изменение окраски (в процентах) для каждого значения pH. Жирные части кривых — области наиболее полезного применения индикаторов: 1 — тимоловый синий (первый переход); 2 — бромфеноловый синий; 3 — метиловый красный; 4 — бромкрезоловый пурпурный; 3 — бромтимоловый синий; в — феноловый красный; 7 — крезоловый красный; з — тимоловый синий (второй переход).
водить среднему химику. Тимоловый синий дает яркие изменения окраски при двух совершенно различных концентрациях ионов водорода1,2’3-При концентрации ионов водорода, отвечающей значениям pH между 1 и .3, окраска этого индикатора из темно-красной переходит в желтую; последняя остается, пока показатель водородных ионов pH не достигнет 8, когда начинается переход в темно-синюю окраску. Поэтому тимоловый синий можно применять для различных дифференциальных ацидиметрических й алкалиметрических титрований, как, например, для раздельного определения бензойной и соляной кислот или уксусной и серной кислот при их совместном присутствии 8.
Смешанные индикаторы. Для получения более отчетливых изменений окраски были предложены различные смешанные индикаторы. Так, вместо метилового оранжевого предложено 4 * * * применять смесь метилового оранжевого и индигокармина: 1 г метилового оранжевого и 2,5 г индигокармина растворяют в 1000 лм воды. Этот индикатор имеет в щелочном растворе желтовато-зеленую окраску, которая отчетливо изменяется из
1 Более полный обзор индикаторов дается в книгах W. М. С 1 а г k, The Determination of Hydrogen Ions, 3d ed., Williams and Wilkins Co., Baltimore, 1928 и I. M. К о 11-h о f f, N. H. Furman, Indicators, John Wiley and Sons, 1926; * см. также И. M. К о л ь т го ф, В. А. Стенгер, Объемный анализ, т. I, Госхимиздат, 1950, стр. 114—171, т.,П, 1952, стр. 61 и Ю. В. Карякин, Кислотно-основные индикаторы, Госхимиздат, 1951 *.
2 W. М. С 1 а г k, Н. A. L u b s, J. Bact., 2, 1, 109, 191 (1917); Н. A. L u b s, Ind. Eng. Chem., 12, 273 (1920).
8 A. В. Clark, H. A. Lubs, J. Am. Chem. Soc., 40, 1443 (1918).
4 R. Luther, Chem. Ztg., 31, 1172 (1907); F. X. Moerk, Am. J. Pharm.,
93, 675 (1921); J. Am. Pharm. Assoc. 10, 743 (1921); I. M. К о 11 h о f f, N. H. F и r-
m a n, цит. выше; I. M. К о 1 t h о f f, Biochem. Z., 189, 26 (1927). * См. также цитиро-
ванную выше литературу на русском языке. *
Ацидиметрия и алкалиметрия
199
Набор индикаторов Кларка и Лабса
ТАБЛИЦА 14
Химическое название Обычное название Концентрация натриевой соли 8 % Изменение окраски Интервал pH
Тимолсульфофталеин (кислотная область) Тимоловый синий 0,04 Красная—желтая 1,2-2,8
Тетрабромфенолсуль-фо фталеин Бромфеноловый синий 0,04 Желтая— синяя 3,0—4,6
Ортокарбоксибензол-азодиметиланилин Метиловый красный 0,02 Красная—желтая 4,4—6,0
Дибромортокрезол-сульфо фталеин Бромкрезоловый пурпурный 0,04 Желтая—пурпурная 5,2-6,8
Дибромтимолсульфо-фталеин Бромтимоловый синий 0,04 Желтая—синяя 6,0—7,6
Фенолсульфофталеин феноловый красный 0,02 Желтая—красная 6,8-8,4
Ортокрезолсульфофталеин Крезоловый красный 0,02 Желтая—красная2 7,2-8,8
Тимолсульфофталеин (щелочная область) Тимоловый синий 0,04 Желтая—синяя 8,0-9,6
Ортокрезолфталеин Крезолфталеин 0,02 Бесцветная—красная 8,2-9,8
’ Натриевые соли приготовляются растиранием 0,1 а сухого количествами 0,05 в. раствора NaOH и разбавлением до 250 мл после
красителя со следующими полного растворения:
Индикатор
Объем 0,05 н. ,, „ раствора NaOH
Молекулярный на 0 1 г 8ео красителя
мл
Феноловый красный..................
Бромфеноловый синий................
Крезоловый красный.................
Бромкрезоловый пурпурный...........
Тимолсвый синий....................
Бромтимоловый синий ...............
Метиловый красный..................
354 669
381
540
466
624
269
5,7
3,0
5,3
3,7
4,3
3,2
7,4
, Применяется 0,04%-ный спиртовый раствор ортокрезолфталеина. Метиловый красный иногда приготовляется растворением красителя в 100 мл спирта и разбавлением водой до 250 мл. Для колориметрических определений 5 капель приготовленного индикатора прибавляют к 10 мл исследуемого раствора; для титрования достаточно 5 капель на 100 мл раствора.
Приготовленные указанным способом растворы индикаторов вполне пригодны для применения их в обычных целях, но если они должны служить для определения концентрации ионов водорода в растворах с малой буферной емкостью, их надо предварительно привести к концентрациям ионов водорода, соответствующим pH серЬдины интервала перехода их окрасок (например, бромфеноловый синий—3,80; бромкрезоловый пурпурный—5,80; бромтимоловый синий— 6,80; феноловый красный—7,60) [W. Н Р 1 е г г е, J. F. Pudge, J. Am. Chem. Soc., 50, 1254 (1928)]. Те же авторы отмечают, что концентрация ионов водорода в растворах индикаторов а заметно изменяется при хранении этих растворов в стеклянной посуде; в сосудах из обыкновенного стекла она понижается, в сосудах из стекла пайреск и в парафинированных внутри колбах она повышается.
• Следует отметить, что крезоловый красный изменяет окраску от красной к желтой при переходе pH раствора от 0,2 до 1,8 [Р. R. McCrumb, W. Е. К enny, J. Am. Chem. Soc., 51, 1458 (1929)].
зеленой через серую в фиолетовую, причем изменение это легко заметно даже при искусственном свете.
Смес!ь 1 г метилового оранжевого и 1,4 г ксиленолцианола красного в 500 мл 50%-ного спирта 1 имеет зеленую окраску в щелочном растворе, красную окраску1 в кислом растворе и принимает стально-серый оттенок в точке перехода, соответствующей pH = 3,8,
1 К. С. D. Hickman, R. Р. L i n s t е a d, J. Chem. Soc., 121, 2502 (1922).
200
Гл. VI. Объемный анализ
Некоторыми исследователями 1 были предложены смеси сульфофталеинов. Например, смесь бромкрезолового пурпурного и бромтимолового синего отчетливо меняет окраску от зеленовато-желтой при pH = = 6,0 до чисто синей при pH = 6,8; смесь бромкрезолового пурпурного и бромфенолового синего имеет интенсивно желтый цвет при pH от 3,4 до 3,6, желто-зеленый при pH от 3,8 до 4,2 и пурпурный при pH от 5 до 6,2, а смесь бромфенолового синего и крезолового красного имеет зеленовато-синюю окраску при pH от 4,6 до 6,8, сине-пурпурную — при pH от 6,8 до 7,6 и пурпурно-фиолетовую 2 при pH от 7,6 до 8,8.
"На переходную окраску индикаторов влияют: концентрация индикатора 3 4, температура раствора *, количество спирта в растворе 5 и концентрация нейтральных солей 6 (а также других соединений, например белковых веществ).
Влияние нейтральных солей, образующихся при титровании разбавленных растворов, например децинормальных, не имеет значения при применении индикаторов, перечисленных в табл. 12, за исключением бромфенолового синего, который не следует применять в очень разбавленных растворах электролитов. Влиянием спирта можно пренебречь, если концентрация его низка, как это и бывает обычно. Концентрация индикатора, однако, имеет большое значение, в особенности при титровании многоосновных кислот. Большое значение имеет и температура, при которой ведется титрование. Нужно заметить попутно, что тот или иной инди
1 J. L. L i z i u s, Analyst, 46, 355 (1921); A. Cohen, J. Am. Chem. Soc., 44, 1851 (1922); F. H. С а г г, Analyst, 47, 196 (1922).
2 Описание «универсальных» индикаторов см. в следующих статьях (pH от 3 до 11,5)—Н. W. van U г k, Pharm. Weekblad, 65, 1246 (1928); (pH от 2 до 12) — там же, 66, 157 (1929); (pH от 1 до 10) — I. М. К о 1 t h б f f, там же, 66, 67 (1929).
3 Различные концентрации индикаторов не дают одного и того же изменения окраски при данном значении pH, особенно при использовании одноцветных индикаторов. Так, разбавленный раствор фенолфталеина при pH = 9 может показать менее интенсивную окраску, чем более концентрированный раствор фенолфталеина при pH = 8. Поэтому важно всегда применять индикатор одинаковой концентрации. Обычно применяемые концентрации индикаторов приведены в табл. 12 и 14. Для некоторых целей необходимы значительно более высокие концентрации индикаторов.
4 При 100° С окраска чувствительных к щелочам индикаторов смещается в щелочную сторону (например, в случае метилового оранжевого от pH = 3,1 при 18° С до pH = 3,66 при 100° С), а чувствительных к кислотам индикаторов — в кислотную сторону (например, окраска фенолфталеина смещается от pH = 8,2 при 18° С до pH = = 8,1 при 100° С).
5 Добавление спирта сильно уменьшает константы диссоциации индикаторов, и потому в его присутствии индикаторы, которые ведут себя как кислоты, становятся более чувствительными к ионам водорода, независимо от того, является ли индикатор чувствительным к кислотам или щелочам. Напротив, индикаторы, являющиеся слабыми основаниями, становятся менее чувствительными к ионам водорода. Кроме того, повышение температуры в спиртовых растворах действует в противоположном направлении по сравнению с водными растворами. Например, если прибавить спирт к слабо окрашенному водному раствору фенолфталеина, окраска исчезнет, а при нагревании она снова появится.' Метиловый спирт вообще действует слабее, чем этиловый спирт. См. также A. Thiel, W. Springeman n, Z. anorg. allgem. Chem., 176, 64, 112 (1928).
6 Нейтральные соли имеют тенденцию изменять окраску кислотных индикаторов в щелочную сторону, а окраску основных индикаторов — в кислотную сторону; например, поправка величины pH в 0,5 н. растворе.поваренной соли равна +0,1 для метилового красного и —0,17 для фенолфталеина. Кроме того, соли могут влиять и на общую интенсивность окраски. См., также I. М. Kolthoff, J. Phys. Chem., 32, 1820 (1928). * О влиянии нейтральных солей, так называемом «солевом эффекте», см. в книге: И. М. К о л ь т г о ф, Г. А. Л а й т и н е н, Определение концентрации водородных ионов и электротитрование, Издатпнлпт, 1947, стр. 68. *
Ацидиметрия и алкалиметрия
201
катор может оказаться непригодным для определения pH в некоторых специальных условиях, например ализарин в присутствии алюминия (с которым он образует лак) или метиловый красный в присутствии окислителей.
Буферные растворы. Буферными растворами называются растворьц почти не изменяющие pH при прибавлении к ним кислоты или щелочи. Смеси, которые проявляют такие свойства, называются буферными или регулирующими смесями, и они обычно состоят из какой-либо слабой кислоты и ее соли со щелочным металлом + Смешивая эти вещества в определенных отношениях, легко можно приготовить раствор с известным значением pH, который дает возможность стабилизировать концентрацию ионов водорода в известных пределах. Поэтому буферные растворы представляют значительную ценность в аналитической работе; например, при осаждении висмута в виде бромокиси, прибавляют буферную смесь бромата калия и бромида калия (стр. 269); буферные смеси вводятся также в раствор при титровании арсенатов иодом (стр. 224) и при исследовании изменений окраски индикаторов.
Буферное действие указанных растворов зависит от природы и концентрации их составных частей, от характера прибавляемой кислоты или щелочи и от охватываемой раствором области значений pH.,
Приготовление растворов индикаторов. Метиловый желтый. Перекристаллизовывают продажный краситель из разбавленного спирта и приготовляют 0,1 %-ный раствор его в 90%-ном спирте.
Метиловый оранжевый. Растворяют 0,02 г этой сульфокислоты в 100 мл горячей воды, охлаждают раствор и, если выделился осадок (метасульфокислоты), фильтруют. Если индикатор приготовляется из натриевой соли, то растворяют 0,022 г реактива в 100 мл горячей воды, прибавляют 0,67 мл 0,1 н. соляной кислоты и обрабатывают, как указано выше.
Метиловый красный. Растворяют 0,02 г свободной кислоты1 2 в 100 мл горячей воды, охлаждают раствор и фильтруют.
1 W. М. С 1 а г к и Н. A. Lubs [J. Biol. Chem., 25, 479 (1916)] рекомендуют 0,05 М растворы, приготовленные из 0,2 М растворов хлорида калия и соляной кислоты,бифталата калия и едкого натра, гидрофосфата калия и едкого натра, борной кислоты, хлорида калия и едкого натра. О приготовлении этих растворов см. в перечисленных выше руководствах.
Следующая таблица показывает, как приготовляются некоторые буферные растворы:
pH Смесь
2 . . . 50 лсл0,2 М КС1 +10,6 мл 0,2 М НС1
4 . . . 50 » 0,2 М бифталат калия+ 0,4 мл 0,2 М NaOH
6 . . . 50 » 0,2 М бифталат калия+ 45,45 мл 0,2 М NaOH
8 . . . 50 » 0,2 М КН2РО4+46,80 мл 0,2 М NaOH
10 . . . 50 » 0,2 М Н3ВО3, 0,2 М KCl + 43,90 мл 0,2 М NaOH
Все растворы затем разбавляют до 200 мл. '
См. также стандарты 185, 186, 187 и 188 Национального Бюро Стандартов США о приготовлении растворов, имеющих pH около 3,6; 4,0; 6,8 и 9,2.
2 Согласно S. Palitzsch [С. г. trav. lab. Carlsberg, 10, 162 (1911)], этот индикатор можно очистить таким способом. Нагревают 4 г его с 30 мл ледяной уксусной кислоты, фильтруют, прибавляют воду, пока раствор не начнет мутнеть, нагревают до просветления и выделяют кристаллы быстрым охлаждением. Его можно также очистить перекристаллизацией пз горячего толуола.
202
Гл. VI. Объемный анализ
Нейтральный красный. Растворяют 0,5 г реактива в 300 мл спирта и разбавляют водой до 500 мл.
Фенолфталеин. Перекристаллизовывают реактив из метилового или этилового спирта, растворяют 5 г очищенного реактива в 400 мл спирта и разбавляют водой до 500 мл.
Тимолфталеин. Растворяют 0,2 г реактива в 300 мл спирта и разбавляют водой до 500 мл.
Лакмус. Этот индикатор представляет собой естественный краситель, который в настоящее время употребляется главным образом для изготовления индикаторной бумаги. Чувствительность различных марок этой бумаги сильно различается, и интервал перехода' окраски у нее очень велик (pH от 4,8 до 8,0), потому что лакмус является смесью индикаторов, состав которой меняется в зависимости от способа приготовления. Было предложено 1 заменить лакмус дибромкрезолсульфофта-леином или дибромтимолсульфофталеином. Первый изменяет свой цвет от желтого (pH = 5,2) до ярко-пурпурного (pH = 6,8), а второй — от желтого (pH = 6) до ярко-синего (pH = 7,6). Оба эти индикатора могут быть получены в очень чистом виде и, следовательно, совершенно •однородными.
Тропеолин 0 и нитрамин. Изредка находят применение тропеолин 0 (натриевая соль азорезорцинсульфаниловой кислоты) и нитрамин (2,4,6-тринитрофенйлметилнитрамин). Первый применяется в 0,1 %-ном водном растворе; он изменяет окраску от желтой при pH = = 11,0 до оранжево-коричневой при pH =13,0. Второй индикатор дает • более удовлетворительные результаты; он применяется в 0,1 %-ном растворе в разбавленном спирте (3 : 2). Раствор нужно держать в темноте. Изменение окраски — при pH 10,8—13 от бесцветной до красно-коричневой 2.
Установка титра кислот
Титрованные растворы соляной кислоты находят более широкое применение, чем растворы других кислот, главным образом потому, что хлориды хорошо растворимы. Растворы других кислот применяются в некоторых специальных методах, например серная или хлорная кислоты применяются в известном методе Кьельдаля, а азотная кислота — в- алкалиметрическом методе определения фосфора. Серная или хлорная кислоты особенно необходимы в тех случаях, когда раствор приходится нагревать или кипятить. Титрованные растворы щавелевой кислоты несколько непостоянны, особенно децинормальные или еще более разбавленные 3.
Титрованные растворы соляной кислоты можно приготовлять простым разбавлением — методом 4 * *, основанным на неизменяемости (при
1 См. сноску 2 на стр. 216.
2 В статье I. М. Kolthoff [Ind. Eng. Chem., Anal. Ed., 8, 237 (1936)] разбираются кислотно-основные индикаторы, окислительно-восстановительные индикаторы, адсорбционные индикаторы и специфические индикаторы и указываются пути дальнейшего развития йх применения. * См.7также И. М. К о л ь т г о ф, В. А. Стенгер,
Объемный анализ, т. I, гл. V, Госхимиздат, 1950 *.
® 8. Ishimaru [Bull. Chem. Soc. Japan, 2, 139 (1927)] указывает, однако, что
в его опытах 0,1 н. раствор щавелевой кислоты практически не изменился в течение
149 дней, когда его хранили в склянке, обернутой в черную бумагу.
* G. А. Н u 1 е 11, W. D. В о n n e г, J. Am. Chem. Soc., 31, 390 (1909).
Ацидиметрия и алкалиметрия
203
определенном атмосферном давлении) состава соляной кислоты, имеющей постоянную температуру кипения. Но обычно титрованные растворы соляной кислоты, а также и других кислот приготовляются разбавлением водой рассчитанного количества реактива и установкой титра полученного раствора. Если совершенно необходимо иметь раствор точно определенного титра (например, точно 0,1 н.), то его приготовляют сначала несколько более концентрированным, затем после установки титра точно разбавляют водой и снова устанавливают его титр. Для обычных работ можно применять объемные бюретки. Весовые бюретки следует применять в работах, требующих самой высокой точности, и во всех тех случаях, когда небольшие различия между температурой, при которой был установлен титр раствора, и температурой, при которой этот раствор применяют, могут иметь значение. Во всех случаях вода, конденсирующаяся на стенках сосуда над поверхностью титрованного раствора, в склянке, где он хранится, или в весовой бюретке, должна быть смешана с остальным раствором до его употребления.
Титр растворов соляной и серной кислот может быть установлен весовыми или объемными способами. Первые требуют значительной затраты труда и времени, если их проводить надлежащим образом. Последние более быстры, вполне точны и их следует предпочесть, потому что устанавливать титр можно так, чтобы применяемый индикатор и направление титрования (прямое или обратное) были такими же, как и при последующем пользовании титрованным раствором. -В табл. 15 дана сводка результатов, полученных 1 при установке титра приблизительно 0,1 н. раствора соляной кислоты различными способами.
ТАБЛИЦА 15
Установка титра раствора соляной кислоты
Метод
Полученная поправка для приведения концентрации раствора к точно 0,1 н.
Разбавлением кислоты с постоянной точкой кипения............................
По AgCl................... ........
По H2SO4-BaSO4—NaOH-HCl............
По H2SO4 —Na2C2O4 —NaOH — HC1......
По C6H6COOH — Ba(OH)2 — НС1........
По CeH5COOH —NaOH — НС1............
0,9980
0,9984
0,9984
0,9982
0,9984
0,9981
Очевидно, что при соблюдении- надлежащих мер предосторожности все методы приводят к одинаковым результатам. Такие же удовлетворительные результаты, без сомнения, будут получены при замене оксалата натрия чистым карбонатом натрия или бензойной кислоты — бифталатом натрия или калия 2. При выполнении подобных сравнительных исследований различных способов установки титра (а также при проведении
1 G. W. М о г е у, J. Am. Chem. Soc. 34, 1032 (1912).
’ W. S. Hendrixson, J. Am. Chem. Soc., 37, 2352 (1915),
204
Гл. VI. Объемный анализ
особо точных его установок) результаты всех взвешиваний надо приводить к пустоте Ч
Установка титра методами весового анализа. Для установки титра азотной кислоты удовлетворительного весового метода не существует. Весовые способы установки титра соляной и серной кислот основаны на предположении, что устанавливаемый раствор не содержит других кислот, кроме той, титр которой устанавливается, и что другие вещества, осаждаемые применяемым реактивом, отсутствуют. Если это предположение не отвечает действительности, то вычисленный титр не будет соответствовать истинной нейтрализующей способности титрованного раствора кислоты: так, титр, найденный для серной кислоты, будет слишком низким, если раствор содержит сернистую кислоту, и слишком высоким, если в растворе содержатся сульфаты.
Поэтому установке титра весовыми методами должна предшествовать специальная подготовка раствора или тщательное испытание его на возможное содержание нежелательных примесей. Кроме того, при точной установке титра соляной кислоты взвешиванием хлорида серебра (стр. 811) необходимо, чтобы не осевший хлорид серебра, перешедший в фильтрат и промывные воды, был определен в них нефелометрическим способом. При весовой установке титра серной кислоты нужно определить сульфат бария в фильтрате и промывных водах их выпариванием, а взвешенный осадок BaSOa должен быть исследован на присутствие в нем хлоридов. При соблюдении таких предосторожностей можно получить очень хорошие результаты 1 2.
Установка титра объемными методами. Установка титра по титрованному раствору щелочи. Установка титра раствора кислоты по титрованному раствору щелочи является быстрым и точным методом при условии, что титрование проводится до той же самой концентрации ионов водорода, какая достигалась при установке титра применяемого раствора щелочи. Это ограничивает применение метода практически лишь теми случаями, когда при установке титра может быть исключено влияние угольной кислоты, так как титры растворов щелочи обычно устанавливают при помощи органических кислот.
Установка титра по карбонату натрия.. Установка титра раствора кислоты по карбонату цатрия дает вполне удовлетворительные результаты, если препарат карбоната натрия тщательно приготовлен самим химиком. Это лучший метод установки титра кислот, если эти кислоты предполагают применять при выполнении титрований в присутствии
1 Чтобы можно было непосредственно сравнивать результаты, полученные при установке титра какого-нибудь раствора по различным исходным веществам, например при установке титра кислоты по карбонату натрия и по тетраборату натрия, I. М. Kolthoff [J. Am. Chem. Soc., 48, 1449 (1926)] рекомендует применение «рациональных эквивалентных весов». * Подробнее о них см. в руководстве И. М. К о л ь т г о ф, В. А. С т е н г е р, Объемный анализ, т. II, Госхимиздат, 1952, стр. 44 *.
2 С. W. Foulk и L. A. Pappenhagen [Ind. Eng. Chem., Anal. Ed., 6, 430 (1934)] описывают метод установки титра растворов соляной кислоты по чистому металлическому серебру. D. N. Craig и J. I. Hoffman нашли, что листовое серебро практически всегда содержит в себе кислород (до 0,01%). Этот кислород можно удалить нагреванием листа серебра в течение 25 ч или более, в зависимости от его толщины, в атмосфере водорода при температуре около 800° С и затем охлаждением его в атмосфере гелия.
Ацидиметрия и алкалиметрия
205
угольной кислоты. Карбонат натрия можно приготовить из очищенного бикарбоната или из оксалата натрия. Первое представляет меньше затруднений.
Установка по карбонату натрия, полученному из бикарбоната. Растворяют 35 г чистого бикарбоната натрия в 350 мл воды, фильтруют и выпаривают фильтрат в платиновой или фарфоровой чашке при температуре не выше 40° С, пока не появятся кристаллы соли. Охлаждают раствор, защитив его от пыли, и оставляют стоять, пока не выделится около трех четвертей всей растворенной соли. Декантируют прозрачную жидкость, промывают кристаллы один раз холодной водой и высушивают, насколько возможно, отжимая их между листами фильтровальной бумаги. Наконец сушат при 120° С, охлаждают и сохраняют в банке с притертой пробкой.
Для установки титра 0,1 н. раствора кислоты помещают около 0,25 г очищенного бикарбоната натрия в платиновый тигель, вставлякй' тигель в песочную баню так, чтобы песок снаружи и бикарбонат натрия внутри тигля были на одном уровне, и нагревают, пока температура внутри тигля не достигнет 260—265° С. Тигель выдерживают при этой температуре 30 мин, перемешивая время от времени и не давая температуре подняться выше 270° С, чтобы не произошло разложения карбоната натрия х. Помещают тигель вместе с его содержимым в стаканчик для взвешивания, охлаждают в эксикаторе, взвешивают и повторяют нагревание, пока не будет достигнута постоянная масса 1 2. Получение спекшейся или сплавленной массы указывает на частичное разложение карбоната, и тогда этот карбонат натрия надо отбросить, потому что нейтрализация его потребует повышенного количества кислоты на единицу массы и в результате получйтся пониженный титр этой кислоты. В обычных работах можно брать большее количество бикарбоната (около 8 г), прокаленный карбонат охлаждать прямо в эксикаторе, потом переносить в стакан для взвешивания и для анализа отвешивать отдельные порции, отбирая их из этого стакана3.
Отвешенный карбонат натрия растворяют в 100 мл воды, прибавляют 3 капли 0,02%-ного раствора метилового оранжевого и титруют до первого неисчезающего красноватого оттенка, который легко можно заметить, сравнивая его со «свидетелем» 4. Если в качестве индикатора
1 W. С. F е г g u s о n, J. Soc. Chem. Ind. London, 24, 781 (1905). См. также G. F. S m i t h, V. R. Н а г d у [J. Chem. Educ., 10, 507 (1933)] и G. F. S m i t h, G. F. С г о a d [Ind. Eng. Chem., Anal. Ed., 9, 141 (1937)], которые рекомендуют проводить разложение при 290 —300° С и давлении 1—4 мм рт. ст.
* Приготовленный таким способом препарат может содержать до 0,05% влаги. Если это имеет значение, его надо прокалить в токе чистой двуокиси углерода до начала плавления соли и затем охладить при постепенной замене двуокиси углерода воздухом [Т. W. Richards, С. R. Hoover, J. Am. Chem. Soc., 37, 99 (1915)].
* I. M. К о 11 h о f f [J. Am. Chem. Soc., 48, 1448 (1926)] утверждает, что результаты, полученные таким образом, могут давать ошибку приблизительно в 0,1% вследствие поглощения влаги при открывании стаканчика, когда берут навески.
4 Конечная точка титрования не является истинной точкой эквивалентности, потому что растворенная в воде двуокись углерода окрашивает метилевый оранжевый до оттенка, который углубляется присутствием соли, образовавшейся во время титрования. Это не имеет значения в обычных работах. В работах высокой точности перед концом титрования нужно удалить СО2, а конечный оттенок сравнить с оттенком раствора, имеющего тот же самый объем и те же концентрации индикатора и соли, и вычесть количество кислоты, требуемое для доведения этого последнего раствора до того же конечного оттенка.
206
Гл. VI. Объемный анализ
применяют фенолфталеин, то СО2 следует удалить раньше достижения конечной точки, и потому применение летучей кислоты нежелательно, даже если прибавлен небольшой ее избыток и кипячение проводится осторожно.
При установке титра серной кислоты получаются очень хорошие результаты следующим образом. Растворяют взвешенный карбонат в 200 мл воды и прибавляют устанавливаемый раствор серной кислоты в избытке от 0,1 до 0,5 лм. Кипятят, пока не будет удалена двуокись углерода и объем раствора не уменьшится до 100 лм. Тогда охлаждают, пропуская одновременно через раствор ток воздуха, свободного от двуокиси углерода, и, продолжая пропускать воздух, титруют, как описано на стр. 209, раствором едкого натра, йе содержащим карбоната. Из общего количества прибавленной серной кислоты вычитают кислотный эквивалент раствора едкого натра, определенный таким же способом
Установка по карбонату натрия, полученному из оксалата. Превращением оксалата натрия в карбонат 1 2 нельзя получить абсолютно чистый карбонат, хотя продукт превращения и имеет щелочность, соответствующую взятому оксалату; поэтому надо вести расчет на массу оксалата натрия, а не на массу продукта превращения. Это является слабой стороной метода, потому что существует опасность механических потерь во время разложения. Недостатком метода является также необходимость защиты карбоната во время прокаливания от соприкосновения с продуктами горения газа, например с SO 2.
Точно отвешивают от 0,2 до 0,3 г чистого оксалата натрия во взвешенном платиновом тигле, закрывают тигель крышкой и ставят его в Отверстие асбестовой пластинки. Нагревают постепенно на спиртовой горелке, пока не разложится соль, и затем более сильно, полуоткрыв крышку, пока не сгорит уголь. До сплавления не доводят. Охлаждают, смывают содержимое тигля в колбу емкостью 300 лм и дальше поступают, как при установке титра по карбонату натрия, полученному из бикарбоната.
Установка титра по тетраборату натрия. Установка титра растворов кислот по тетраборату натрия находит много приверженцев, среди которых можно назвать И. М. Кольтгофа 3, горячо рекомендующего этот метод.
Надлежащим образом приготовленный препарат изменяет свой состав меньше .чем на 0,1 %, если его хранить в плотно закупоренной банке в течение даже 1 года. Рекомендуется следующая обработка препарата. Продажный химически чистый тетраборат натрия очищают перекристаллизацией, применяя по 50 мл воды на растворение каждых 15 г буры. Такая концентрация предупреждает выделение кристаллов при температуре выше 55° С и, следовательно, образование кристаллов Na2B4O7-5H2O вместо кристаллов Na2B40,-10H20. Выделившиеся кристаллы буры отфильтровывают
1 Как и с метиловым оранжевым, здесь также наблюдается некоторый солевой эффект, и потому в результаты титрования следует ввести поправку на «холостой» опыт с раствором, имеющим тот же объем и те же концентрации индикатора и соли.
’ S. Р. L. S б г е n s е n, Z. anal. Chem., 36, 639 (1897); 42, 333, 512 (1903); 44, 141 (1905); Sorensen, А. С. A n d е г s о п, там же, 44, 156 (1905); 45, 217 (1906); J. Sebelien, Chem. Ztg., 29, 638 (1905); G. Lunge, Z. angew. Chem., 17, 195 (1904); 18, 1520 (1905).
3 I. M. Koi t h о f f, J. Afii. Chem. Soc., 48, 1453 (1926); T. Milobedzki, H. К a m i n s k a, Bull. Soc. chim., 41, 957 (1927).
Ацидиметриями алкалиметрия
207
с применением отсасывания и дважды промывают водой. Затем промывают их двумя порциями 95%-ного спирта и двумя порциями эфира, каждый раз расходуя по 5 мл промывной жидкости на 10 г выделенных кристаллов и давая этой жидкости полностью стечь с кристаллов; применяют отсасывание. Когда кристаллы буры высохнут,, распределяют их тонким слоем на часовом стекле, оставляют на некоторое время, пока весь эфир не испарится, и переносят в' банку, снабженную плотно входящей пробкой.
Имеются указания на то, что тетраборат натрия можно сохранять неопределенно' долгое время без каких-либо изменений в его составе, если держать его над раствором, насыщенным сахарозой и поваренной солью.
Ход установки титра следующий. Чистую буру перекристаллизовывают из воды и высушивают в эксикаторе, имеющем относительную влажность 60% (на дно эксикатора помещают насыщенный раствор NaBr • 2Н2О). Отвешивают 0,4—0,6 г полученной соли Na2B4O7 • ЮН20, растворяют в 200 мл воды, прибавляют 8 капель 0,2%-ного раствора метилового красного и титруют раствором кислоты, титр которого хотят установить (лучше всего приблизительно 0,1 н.). Концом титрования считают момент, когда окраска титруемой пробы сравняется с окраской 200 мл раствора, содержащего то же самое количество индикатора, борную кислоту — в количестве, отвечающем 0,1 М, и хлорид натрия — в количестве, отвечающем 0,05 М. В указанной конечной точке титрования pH раствора около 5.
Установка титра основании
Для титрования кислот чаще всего применяются растворы едкого натра. Применение для этой цели растворов аммиака мало удобно потому, что они меняют титр вследствие улетучивания NH3, особенно если эти растворы концентрированнее полунормаль-ных. Растворы едкого барита также очень неудобны, так как они загрязняют приборы, если доступ двуокиси углерода полностью не устранен.
Растворы едкого натра, достаточно полно освобожденные от карбоната и пригодные для вс^х обычных титрований, при использовании такйх индикаторов, как фенолфталеин, можно легко приготовить следующим способом т. Растворяют 100 г едкого натра в 100 мл дистиллированной воды и переводят раствор в цилиндр из химически стойкого стекла емкостью 150 лм, стараясь не смочить верхней его части. Плотно
закрывают цилиндр корковой пробкой, обернутой станиолем, и оставляют его стоять, пока верхний слой жидкости не станет прозрачным. Прозрачный раствор можно получить быстрее, фильтруя жидкость через стеклянный фильтрующий тигель в приборе, где устранен доступ двуокиси углерода (рис. 21). Осторожно отбирают пипеткой нужное количество едкого натра
Рис. 21. Прибор для фильтрования концентрированных растворов едкого натра:
1 —карбонат натрия; 2 — раствор едкого натра; з — аскарит; 4 — тигель с фильтрующей стеклянной пластинкой.
1 F. Н. Н url е у, Ind. Eng. Chem., Anal. Ed., 8, 220 (1936); 9, 237 (1937).
208
Гл. VI.. Объемный анализ
(8—8,5 г или 6,5 мл для приготовления 1 л 0,1 н. раствора NaOH), переносят в парафинированную склянку 1 и быстро разбавляют свежепроки-пяченной дистиллированной водой. Склянка должна быть снабжена пробкой с двумя отверстиями, через одно из которых проходит стеклянная сифонная трубка с краном, а через другое —.предохранительная трубка с натронной известью или натронным асбестом.
Для приготовления раствора едкого натра, свободного от карбоната, предлагают 2 получать сначала амальгаму натрия следующим образом. Перегнанную ртуть наливают толстым слоем в коническую делительную воронку и вводят в ртуть катод из платиновой проволочки, вставленной в предохранительную стеклянную трубку. На ртуть наливают концентрированный раствор перекристаллизованного хлорида натрия, а в «тот раствор погружают платиновый анод. Проводят электролиз при напряжении 4—6 в и время от времени осторожно встряхивают воронку, чтобы разбить кристаллы амальгамы, образующейся на поверхности ртути. Одновременно кипятят 1 л (или больше) дистиллированной воды, пока не будет удалена вся СО2, и затем, когда кипение еще продолжается, закрывают колбу пробкой с тремя отверстиями, через которые проходят сифон и предохранительная трубка с натронной известью, а третье, закрытое пробкой, предназначается для делительной воронки. Затем после охлаждения воды в отверстие пробки вводят трубку делительной воронки и выливают амальгаму в воду. Реакции дают идти, пока переведенная сифоном в бюретку и оттитрованная порция раствора не покажет, что образовалось достаточное количество щелочи. Тогда спускают сифоном приблизительно требуемое количество раствора в определенный объем прокипяченной и защищенной от- СО 2 воды, тщательно перемешивают и устанавливают титр полученного раствора.
Титр растворов щелочей, предназначенных для титрования с такими индикаторами, как фенолфталеин, лучше всего устанавливать по органическим кислотам, а растворов, которые предполагают применять для титрования в присутствии СО2, — по растворам кислот, установленным в свою очередь по карбонату натрия, или по соляной кислоте с постоянной температурой кипения. Для установки титра растворов щелочей, не содержащих карбонатов, могут быть применены различные органические, кислоты. В этих случаях можно посоветовать применять гидрофталат калия или бензойную кислоту. Из них следует предпочесть гидрофталат калия вследствие его большей растворимости в воде и более высокого молекулярного веса 3.
Установка титра по гидрофталату Калия. Исследования показали, что в условиях, существующих в большинстве химических лабораторий, стандартные водные растворы гидрофталата калия не изменяют своей концентрации 4. Однако приготовление таких растворов в больших объемах мало оправдывается, потому что операции взвешивания небольшой навески гидрофталата, растворения ее в воде и немедленного титрования полеченного раствора устанавливаемым раствором щелочи сравнительно просты.
1 Склянку сначала тщательно моют и затем сушат. После этого ее слегка нагревают и вливают в нее такое количество расплавленного парафина, чтобы он мог образовать толстый слой на стенках склянки. Затем вращают осторожно склянку, пока не получится ровный толстый слой на ее стенках и парафин не будет близок к затвердеванию. Тогда ставят склянку прямо, так что на дне образуется очень толстый слой парафина. О применении каучуковых лаков для защиты стеклянных склянок см. В. A. S о u 1 е, Ind. Eng. Chem., Anal. Ed., 1, 109 (1929).
2 W. M. Clark, см. сноску 1 на стр. 198.
8 H. В. KellognA. М. Kellog (Ind. Eng. Chem., Anal. Ed., 6, 25 (1934)] рекомендуют в качестве основного вещества для установки титра растворов щелочей — пирослизевую кислоту С4Н3ОСООН.
« J, I. Н о f f m a n, J, Research NBS, 15, 583 (1935),
Ацидиметрия и алкалиметрия
209
Ход работы.^ Прежде всего измельчают несколько граммов гидрофталата калия примерно до 100 меш (d — 0,147 льм), высушивают 1—2 ч при 120° С, помещают в стаканчик, снабженный притертой крышкой, и охлаждают в эксикаторе. Навеску около 1 г высушенного таким способом препарата переносят в колбу емкостью 300 лгл, совершенно освобо
жденную от двуокиси углерода продуванием (рис. 22), прибавляют 50 зел воды (25—28° С), не содержащей СО2, закрывают колбу пробкой и осторожно взбалтывают, пока навеска полностью не рас-
творится. Затем полученный раствор титруют устанавливаемым свободным от карбоната натрия приблизительно 0,1 н. раствором едкого натра до pH = =8,6. При титровании принимают меры для устранения двуокиси углерода. В качестве индикатора применяют или pH-метр со стеклянным электродом или добавляют 3 капли 1%-ного раствора фенолфталеина. В последнем случае конец титрования определяется сравнением с цветом буферного раствора, имеющего pH — 8 и приготовленного смешением 25 мл раствора 0,2 М по содержанию Н3ВОз< и 0,2 М по содержанию КС1 с 6 мл 0,2 М раствора NaOH и 3 каплями 1%-ного раствора фенолфталеина и разбавленного до 100 мл водой, свободной от двуокиси углерода.
Для внесения поправки определяют количество едкого натра, затрачиваемое в конце титрования
для возникновения окраски индикатора. С этой целью в другую колбу наливают воду, свободную от двуокиси углерода, в объеме, равном объему оттитрованного раствора, прибавляют к ней индикатор и титруют раствором едкого натра до той же окраски. Это последнее количество NaOH вычитают из затраченного при первом тйтровании и подсчитывают нормальность раствора щелочи на основании уравнения
Рис. 22. Колпак для титрования в отсутствие двуокиси углерода:
1 — вводная трубка для воздуха, очищенного от двуокиси углерода;
2 — отверстие для кончика бюретки; з — боковое отверстие, снабженное пробкой, через которое обмывают стенки колбы.
HKCgHjOj-j-NaOH = NaKC8H4O4+H2O
В ацидиметрических титрованиях 204,216 г гидрофталата калия эквивалентны 1,008 г водорода и 1,0211 г КНС8Н4О4 эквивалентны 50 мл 0,1 н. раствора.
Установка титра по бензойной кислоте. Было найдено \ что бензойная кислота слегка гигроскопична: навеска, сохранявшаяся больше года в стаканчике с притертой пробкой, поглотила около 0,07 % влаги. Поглощение влаги происходит не быстро, поэтому при оставлении навески бензойной кислоты на короткое время открытой на воздухе никаких изменений в ее массе обнаружить нельзя.
Если имеет значение ошибка порядка 1 на 2000, то можно предварительно высушить бензойную кислоту осторожным плавлением ее при температуре ниже 130° С. Это может быть сделано в покрытой часовым стеклом и нагреваемой на воздушной бане платиновой чашке. Нагревание
1 Е. R, Weaver, J. Am, Chem, Soc,, 35, 1309 (1913), 14^3аказ 522.
210
Гл. VI. Объемный анализ-
не должно продолжаться дольше, чем это абсолютно необходимо. Слишком высокая температура или длительное плавление приводят к образованию продуктов разложения, которые окрашивают расплавленную массу в заметно желтый цвет.
Ход работы. Навеску около 1,0000 г (отвешенную точно) сухой бензойной кислоты помещают в колбу емкостью 300 мл, освобожденную от двуокиси углерода. Приливают 20 мл спирта (95%-ного), закрывают колбу пробкой и оставляют стоять, пока навеска не растворится. К полученному раствору добавляют 3 капли 1 %-ного раствора фенолфталеина и титруют устанавливаемым приблизительно 0,1 н. раствором щелочи при пропускании через раствор тока воздуха, свободного от двуокиси углерода.
Затем для внесения поправки определяют объем титрованного раствора едкого натра, который вызывает возникновение такой же окраски раствора, содержащего те же количества спирта, воды и индикатора, какие были в оттитрованном растворе бензойной кислоты. Израсходованный в этом титровании объем раствора NaOH вычитают из объема того же раствора, израсходованного в основном титровании, и вычисляют нормальность раствора едкого натра на основе уравнения
С6Н5СООН + NaOH = C6H5COONa + Н2О
Установка титра по титрованному раствору кислоты. Титрованные растворы щелочей, предназначенные для применения в присутствии СО,, лучше всего устанавливать или по растворам кислот, титр которых установлен по карбонату натрия, или по соляной кислоте с постоянной температурой кипения.
• Применение соляной кислоты с постоянной температурой кипения для установки титра растворов щелочей было исследовано многими авторами 1’2 . В табл. 16 приводятся концентрации растворов соляной кислоты, имеющих постоянную температуру кипения при обычно встречающихся атмосферных давлениях. Концентрация такой кислоты не меняется годами, по крайней мере в пределах точности, требуемой в обыч-. ных определениях, при условии ее хранения в темном прохладном месте 2 в склянке хорошего химического стекла, плотно закрытой притертой пробкой.
Ниже описан способ приготовления такой кислоты, пригодный для общего применения. При установке титра, основанной на употреблении этой кислоты, достигается точность 2, превышающая 1 на 1000.
Ход работы. Собирают перегонный аппарат, несколько сходный с применяемым при определении азота по Кьельдалю, только колба должна быть емкостью 1—2 л, шарик насадки не должен иметь изогнутой внутренней трубки, а внутренний диаметр соединительных трубок равен 6—10 мм. Колбу прибора помещают в отверстие асбестовой пластинки и ее верхнюю часть во избежание теплоизлучения закрывают асбестовой бумагой. Соединяют колбу новой резиновой пробкой со стеклянным холодильником, имеющим водяную муфту длиной 75 см и внутреннюю
1 G. А. Н u 1 е t t, W. D. В о n n e r, J. Am. Chem. Soc., 31, 390 (1909); G. W. M o-г e у, там же, 34, 1032 (1912); W. S. Hendrixson, там же, 37, 2352 (1915); C. W. F о u 1 k, M. Hollingsworth, там же, 45, 1220 (1923).
2 J. A. Shaw, Ind. Eng. Chem., 18, 1065 (1926).
Ацидиметрия и алкалиметрия
211
ТАБЛИЦА 16
Концентрация растворов соляной кислоты, кипящих при постоянной температуре
Давление, мм pm. cm. Содержание HC1 no массе в пустоте, % Масса в воздухе дистиллята кислоты, кипящей при постоянной температуре, соответствующая 1 моль хлористого водорода, взвешенного в пустоте, г
770 20,197 180,407
760 20,221 180,193
750 20,245 179,979
740 20,269 179,766
730 20,293 179,555
трубку диаметром 6—10 мм, и конец шариковой насадки вдвигают в холодильник по крайней мере на 2 см дальше пробки. Свободный конец холодильника вставляют в шейку конической приемной колбы так, чтобы он не был погружен в дистиллят. Если воздух лаборатории загрязнен аммиаком или пылью, приемная колба должна быть помещена в сосуд с воздухом, очищенным промывкой.
Разбавляют не менее чем 500 мл соляной кислоты водой до плотности, приблизительно равной 1,10 г/см3, и вливают в перегонную колбу. Перегонку ведут со скоростью 8—10 мл в минуту. Собирают отдельно и оставляют для обычного употребления дистиллят в количестве 75% первоначального объема разбавленной кислоты и затем собирают 10—15%, уже как стандартную кислоту с постоянной температурой кипения. Остаток в перегонной колбе, объем которого ни в коем случае не должен быть меньше 100 мл, отбрасывают. Показания барометра с точностью до миллиметра отмечают 3 раза: в начале перегонки, после отгонки 75% жидкости и в конце перегонки. Берут среднее из последних двух отсчетов и вычисляют концентрацию кислоты по данным табл. 16.
При установке титра отбирают при помощи весовой бюретки такое количество кислоты, которое требуется для нейтрализации приблизительно 40 мл устанавливаемого раствора щелочи; кислоту вливают прямо из бюретки в 100—150 мл дистиллированной воды, держа кончик бюретки на 1—2 см выше поверхности воды.
Простой перегонкой можно получить стандартный раствор хлорной кислоты 1 (содержащий 73,60 ± 0,03% НС1О4). Для приготовления 1 н. ее раствора отвешивают 136,4201 г этой стандартной кислоты (предполагается применение обыкновенных разновесок и средние значения влажности и температуры воздуха) и разбавляют до 1000 мл. При взвешивании стандартной кислоты следует принимать меры предосторожности, связанные с тем, что кислота эта слегка гигроскопична и очень слабо дымит на воздухе.
Утверждают 2 3, что очень хорошим основным веществом для установки титров щелочей является чистый бииодат калия КН(Ю8)2, потому что эквивалентный вес этой соли велик, и йодноватая кислота является сильной кислотой. Чистую соль можно получить двукратной или трехкратной перекристаллизацией хорошего продажного препарата из водного раствора (нагревать не выше 60° С) и высушиванием при 100° С.
Рекомендуется 3 применение сульфаминовой кислоты NH2SO8H, потому что ее легко можно очистить и обезводить; это кристаллическое и негигроскопичное вещество,
1 G. F. Smi t h, W. W. К och, Ind. Eng. Chem., Anal. Ed., 3, 52 (1931).
2 I. M. Koi t hof f и L. H. van В erk, J. Am. Chem. Soc., 48, 2800 (1926).
3 С. M. I. Butler, G. F. S m i t h и L. F. A u d r i e t h, Ind. Eng. Chem., Anal. Ed., 10, 690 (1938).
14*
212
Гл. VI. Объемный анализ
являющееся сильной кислотой; в водных растворах она может быть оттитрована щелочью с применением индикаторов, имеющих интервалы перехода в границах pH от 4 до 9.
В качестве основного вещества можно применить1 соль 3CdSO4-8H2O. В этом . методе точно отвешивают определенное количество указанной соли, растворяют в воде, подвергают раствор электролизу со ртутным катодом, пока весь кадмий не выделится, и получают таким образом в эквивалентном количестве свободную серную кислоту.
В табл. 17 приведены плотности растворов соляной кислоты, имеющих постоянную температуру кипения при различных давлениях 2.
ТАБЛИЦА 1 7
Плотность и составы растворов соляной кислоты, имеющих постоянную температуру кипения при различных давлениях
Давление .iWJH pm. cm. Плотность при 25±0,02° C г/см3 Содержание HC1 % Давление, awh pm. cm. ПЛОТНОСТЬ при 25±0,02° C s/cjw* Содержание HCI %
50 1,1118 23,42 600 1,0980 20,638
150 1,1073 22,520 640 1,0973 20,507
250 1,1042 21,883 700 1,0966 20,360
350 1,1019 21,437 760 1,0959 20,222
400 1,1010 21,235 • 800 1,0955 20,155
450 1,1002 21,075. 1000 1,0933' 19,734
500 1,0993 20,916 1220 1,0915 19,358
540 1,0987 , 20,777
Если на оси абсцисс отложить процентное содержание хлористого водорода в растворах НС1, а на оси ординат — соответствующие плотности этих растворов, то зависимость между ними выразится прямой линией, уравнение которой следующее:
Г-0,9966
Л 0,004912
где У — плотность раствора при 25° С.
В границах обычных барометрических давлений это уравнение точно до 0,05%. Таким способом можно приготовить раствор соляной кислоты с постоянной точкой кипения при любом удобном давлении атмосферы, определить его плотность при 25° С и сразу вычислить содержание в нем хлористого водорода с точностью, достаточной для обычной объемноаналитической работы. Барометрическое давление, конечно, не должно значительно изменяться во время перегонки кислоты.
МЕТОДЫ ОКИСЛЕНИЯ И ВОССТАНОВЛЕНИЯ
Окислительно-восстановительные индикаторы
Некоторые органические соединения в своих окисленных формах имеют окраски, настолько отличающиеся от окрасок восстановленных форм, что их можно использовать в качестве индикаторов, указывающих на полноту протекания различных окислительно-восстановительных реак
1 S. Е. Q. A s h 1 е у, G. А. Н u 1 е 11, J. Am. Chem. Soc., 56, 1275 (1934).
2 W. D. В о n n e г, A. С. T i t и s, J. Am. Chem. Soc., 52, 634 (1930).
Методы, окисления и восстановления
213
ций. Изменение состава и цвета каждого из этих индикаторов происходит при определенном окислительном потенциале, и у большинства индикаторов такое изменение цвета обратимо х. В табл. 18 приведен список некоторых типичных окислительно-восстановительных индикаторов.
ТАБЛИЦА 18
Окислительно-восстановительные индикаторы
1 Индикатор Потенцией перехода окраски (нормальный водородный электрод равен 0), в Изменение окраски при переходе от восстановленной формы к окисленной
Метиленовая синяя 0,53 х Бесцветная—синяя
Дифениламин 0,76 Бесцветная — фиолетовая 1 2
Дифенилбензидин 0,76 Бесцветная — фиолетовая 2
Дифениламиносульфоновая кислота (Ва- или Na-соль) 0,83 3 Бесцветная — красно-фиолетовая 2
Дифенилбензидинсульфоновая кислота (Na-соль) 0,87 Бесцветная — фиолетовая
н-Фенилантраниловая кислота 1,08 Бесцветная— розовая
Дипиридил-сульфат железа (II)2 1,06 4 Красная — бледно-голубая
о-Фенантролин-сульфат железа (II)2 1,06 4 Красная — бледно-голубая
Нитро-о-фенантролин-сульфат железа (II) (нитроферроин) 1,25 Красная — бледно-голубая
1 При рН = 2,86.
* Не стойкая в присутствии окислителей.
• В 0,5—2,0 н. растворе H,SO4.
4 В 1 М растворе НС1 или HjSOi.
Об окислительном потенциале ионов фенантролина и ионов нитро-, бромо-, хлоро-, метия-и нитрометилфенантролина в 1—8 М растворах серной кислоты см. О. F. S m i t h, F. P. Richter, Ind. Eng. Chem., Anal. Ed., 16, 580 (1944). См. также брошюру: Phenanthroline and Substituted Phenanthroline Indicators, изданную в 1944 г. G. F. Smith. Chemical C°, и статью W. W. BrandtnG. F. Smith [Anal. Chem. 21, 1313 (1949)] о многозамещенных фенантролинах и бипиридинах как окислительно-восстановительных индикаторах.
* Индикатор н-фенилантраниловая кислота (о-дифениламинкарбоновая кислота) был предложен В. С. Сырокомским и В. В. Степиным [Зав. лаб. 5, 144 (1936)]. А. В. Кирсановым и В. П. Черкасовым [(там же, 5, 143 (1936)] были предложены в качестве индикаторов о.л'-дифениламиндикарбоновая кислота и о,о'-дифениламиндикарбоновая кислота. Потенциал перехода окраски первого индикатора—1,12 в, второго—1,26 в. При окислении обоих индикаторов окраска их переходит из бесцветной в сине-фиолетовую. Применять их надо в сильнокислых растворах (15 — 20 н. серная кислота). *
На изменение окраски всех индикаторов расходуется некоторое количество окислителя или восстановителя, но им можно пренебречь, если только определяются не очень уже малые количества титруемого вещества и требуется не слишком высокая точность определения.
Некоторые окислители сами действуют как индикаторы, например перманганат калия и иод. Цвет иода обычно усиливают, экстрагируя его малым объемом подходящего растворителя, например четыреххлористого углерода, или добавляя крахмал к концу титрования.
Очень неудобные и отнимающие много времени у аналитика внешние индикаторы теперь почти полностью вытеснены более удобными и не требующими продолжительного времени титрования внутренними окислительно-восстановительными индикаторами: например, при титровании железа (II) внешний индикатор гексацианоферрат (III) калия заменен дифениламинсульфонатом.
1 Небольшое число индикаторов этого типа необратимо, например метиловый
оранжевый в реакциях титрования броматом калия в кислых растворах.
214
Гл. VI. Объемный анализ
Установка титра растворов перманганата калия
Для приготовления нормального раствора перманганата калия можно растворить навеску перекристаллизованной чистой соли в воде, подвергнутой предварительно специальной очистке перегонкой после обработки щелочным раствором перманганата. Полученный раствор затем разбавляют до определенного объема При этом способе требуется значительная затрата труда, причем столь же хорошие результаты могут быть получены гораздо легче, если растворить требуемое количество чистого перманганата калия в обыкновенной дистиллированной воде, раствор оставить стоять в течение нескольких недель, профильтровать через очищенный асбест (стр. 125), собирая фильтрат в чистую склянку, и затем установить его титр по оксалату натрия 1 2. Децинормальные и более концентрированные растворы, освобожденные таким образом от двуокиси марганца, не подвергаются заметному изменению (не более 1 на 2000) при стоянии на рассеянном дневном свете в течение нескольких месяцев, при условии, что они защищены от пыли и других восстанавливающих веществ и что в склянку проникает только очищенный воздух. Титр растворов более разбавленных, чем 0,1 н., следует часто проверять. Обычай разбавлять профильтрованные растворы перманганата дистиллированной водой не имеет обоснования, если только вода не была предварительно перегнана после обработки ее щелочным раствором перманганата. При наличии условий, при которых происходит быстрое восстановление нейтральных растворов перманганата, например в присутствии пыли, восстанавливающих газов или осевшей двуокиси марганца, разложение может быть замедлено прибавлением 1 % едкого кали.
Титрование перманганатом калия обычно проводится в растворе, содержащем серную кислоту. Можно также применять разбавленную азотную кислоту, как это делается, например, в висмутатном методе определения марганца (стр. 497); разбавленная соляная кислота также допустима, если она необходима по ходу анализа, но при условии применения смеси Рейнгардта, как описано в гл. «Железо» (стр. 446). Можно применять и хлорную кислоту. Титрование нельзя проводить в присутствии фтористоводородной кислоты, так как в этих условиях перманганат окисляет марганец (II) (стр. 993). В таком случае можно получить совершенно удовлетворительные результаты, если к титруемому раствору прибавить в достаточном избытке борную кислоту, чтобы связать весь фтор во фтороборную кислоту. Титрование в нейтральном или слабощелочном растворе, как, например, при определении марганца по методу Фольгарда, проводится редко; в этих условиях восстановление перманганата происходит не до двухвалентного, а до четырехвалентного марганца.
Установка по оксалату натрия. Лучшим веществом для определения активной концентрации перманганата является чистый оксалат натрия. Сухой оксалат натрия не претерпевает заметного изменения в составе и свойствах при хранении его в закрытых пробками склянках даже на
1 W. Blum, J. Am. Chem. Soc., 34, 1379 (1912).
2,Согласно T. Kato (J. Chem. Soc. Japan, 48, 408 (1927)], устойчивый раствор перманганата можно получить скорее, если свежеприготовленный раствор его прокипятить, оставить стоять одни сутки и профильтровать через асбест в обработанную паром склянку, обернутую черной бумагой.
Методы, окисления и восстановления
215
•свету. Перекристаллизованный из воды препарат (в очень мелких кристаллах) поглощает максимум около 0,01 % воды при относительной влажности воздуха 70%, а препараты, осажденные спиртом, поглощают в тех же условиях 0,04% воды, что, несомненно, является следствием большей поверхности кристаллов. Если банка с оксалатом натрия многократно открывалась и подозревают присутствие влаги в кристаллах, то их можно снова высушить нагреванием при 105° С. Если навеску для анализа берут на открытом часовом стекле, то заметной ошибки от поглощения влаги при этом, по-видимому, не происходит.
Оксалат натрия не подвергается заметному разложению при нагревании даже до 240° С, но в этом случае следует применять электрический сушильный шкаф, потому что в шкафу, обогреваемом газом, может происходить поглощение сернистых соединений из продуктов сгорания газа. Обычно в нагревании до таких высоких температур нет необходимости, если нет подозрения, что оксалат содержит бикарбонат натрия или окклюдированную воду'.
Растворы оксалата натрия заметно не разлагаются при кипячении, но сильно разъедают стекло. В небольшой степени разъедающее действие растворов оксалата натрия наблюдается и в том случае, когда их хранят в стеклянной посуде при обыкновенной температуре, причем происходит образование оксалата кальция, что затрудняет точное отмеривание аликвотных частей раствора. По этой причине, а также ввиду возможного разложения оксалата натрия на солнечном свету не следует делать запасных его растворов для проверки титра растворов перманганата.
В основе установки титра перманганата лежит титрование щавелевой кислоты, освобождаемой из отвешенного количества оксалата натрия действием серной кислоты. Для окисления 5 молекул оксалата требуется 2 молекулы перманганата. Отсюда легко вычислить концентрацию раствора перманганата, а также и его эквиваленты в других реакциях, имеющих отчетливый конец и протекающих количественно, например, с солями железа (II), иодистоводородной кислотой или оксалатом кальция.
Титр раствора перманганата, получаемый при использовании описываемого ниже хода работы х, хорошо совпадает в границах допустимых ошибок с титрами, установленными при применении других основных веществ. При использовании весовых бюреток легко можно получить совпадение между параллельными определениями титра с отклонениями, ‘не превышающими1 2! : 5000. Точность при применении объемных бюреток несколько меньше, так как зависит от точности их калибрования, тщательности в отсчете объема и от постоянства температуры раствора. Поэтому ввиду возможного повышения температуры раствора перманганата во время титрования употребление весовых бюреток рекомендуется во всех случаях, когда желательна точность большая, чем 1 : 1000.
Ход работы. Точно отвешивают около 0,3 г химически чистого оксалата натрия (высушенного при 105° С) и количественно переносят в стакан емкостью 600 мл. Приливают 250 мл разбавленной (5 : 95) серной кислоты, предварительно прокипяченной 10—15 мин и потом охлажденной
1 R. М. Fowler, Н. A. Bright, J. Research NBS, 15, 493 (1935).
2 Например, при выполнении пяти установок титра 0,11850 н. раствора перманганата среднее отклонение полученных результатов от их среднего значения составило 0,01%, а максимальное отклонение — 0,02%.
216
Гл. VI. Объемный анализ
до 27 ± 3° С, и перемешивают до растворения оксалата натрия. Затем добавляют 1 39—40 мл устанавливаемого 0,1 н. раствора КМпО4 со скоростью 25—35 мл в минуту при медленном перемешивании раствора и дают постоять до исчезновения розовой окраски (около 45 сек) 2. Нагревают до 55—60° С и заканчивают титрование добавлением перманганата до тех пор, пока 'слабо-розовая окраска раствора не будет сохраняться 30 сек. Последние 0,5—1 лм раствора перманганата прибавляют по каплям, тщательно следя за тем, чтобы каждая последующая капля попадала в раствор только после исчезновения окраски от предыдущей капли.
Отдельно определяют, какое количество перманганата необходимо для того, чтобы сообщить розовую- окраску раствору. Это может быть сделано добавлением перманганата к взятой в таком же объеме разбавленной серной кислоте, предварительно прокипяченной и охлажденной до 55—60° С, до тех пор, пока окраска кислоты не станет одинаковой с окраской оттитрованного раствора. Полученная поправка обычно составляет 3 0,03—0,05 мл. При потенциометрических титрованиях поправкой можно пренебречь в том случае, если к конечноц точке титрования приближаться медленно.
Установка по мышьяковистому ангидриду. Непосредственное титрование мышьяка (III) перманганатом в слабокислых растворах не дает удовлетворительных результатов, потому что реакция не проходит в стехиометрических отношениях. Но если прибавить в небольшом количестве иодид калия или иодат калия, реакция идет нормально с образованием мышьяка (V) и марганца (И)4 *-6. Следующий ход работы® приводит к получению очень точных результатов, совпадающих с получаемыми при применении оксалата натрия (отклонения не превышают 1 на 3000).
Ход работы. Высушивают химически чистый мышьяковистый ангидрид при 105° С и точно отвешивают около 0,25 г высушенного препарата. Переносят навеску в стакан емкостью 400 мл, прибавляют 10 мл холодного 20%-ного раствора едкого натра (свободного от присутствия в нем окислителей и восстановителей) и дают постоять 8—10 мин, изредка перемешивая. Когда растворение полностью закончится, приливают 100 мл воды, 10 мл соляной кислоты (пл. 1,18 г/см3) и 1 каплю 0,0025 М раствора иодида калия или йодата калия, после чего титрование может быть выполнено потенциометрическим или визуальным способом.
1 На титрование 0,3000 г оксалата натрия требуется 44,73 мл.0,1 н. раствора перманганата.
- 2 Если розовая окраска не исчезнет из-за того, что устанавливаемый раствор перманганата имеет слишком высокую концентрацию, то раствор надо отбросить и начать все сначала, приливая на несколько миллилитров меньше раствора перманганата, чем было прибавлено раньше.
3 При выполнении особо точных работ эту поправку лучше всего находить иодо-метрическим методом (W. С. В г а у, J. Am. Chem. Soc., 32, 12Q4 (1910)] следующим образом. Охлаждают оттитрованный раствор до 25° С, прибавляют 0,5 г иодида калия.
2 мл раствора крахмала и титруют выделившийся иод 0,02 н. раствором тиосульфата.
Для нахождения соотношения между растворами тиосульфата и перманганата наливают в другую колбу 300 мл разбавленной (5 : 95) серной кислоты, прибавляют 1 мл раствора перманганата, перемешивают, добавляют 0,5 г иодида калия и титруют раствором тиосульфата, вводя раствор крахмала перед самым концом титрования.
4 R. L a n g, Z. anal. Chem., 152, 197 (1926); I. М. К о 11 h о f f, Н. A. L a i t -
n ей, J. J. L ingane, J. Am. Chem. Soc., 59, 429 (1937).
6 H. A. В r i g h t, J. Research NBS, 19, 691 (1937).
Методы окисления и восстановления
217
Потенциометрическое титрование. Титруют раствором перманганата до максимального значения изменения потенциала на единицу объема прибавленного раствора перманганата &EI&V. Последние 1—1,5 мл прибавляют по каплям, ожидая установления равновесия, прежде чем произвести отсчет ДЕ1. При выполнении потенциометри- / ческого титрования поправкой на «холостой» опыт можно пренебречь при условии, что применяемые реактивы не содержат мешающих загряз-.нений.
Визуальное титрование. Титруют раствором перманганата до появления слабо-розовой окраски раствора, сохраняющейся 30 сек. Последние 1—1,5 мл добавляют по каплям, следя за тем, чтобы каждая последующая капля попадала в раствор только после полного исчезновения окраски от предыдущей капли. Для внесения поправки определяют объем раствора перманганата, вызывающий появление окраски в конечной точке титрования. Для этого в другой стакан'наливают те же количества едкой щелочи, кислоты и катализатора, какие вводились в первый раствор, и прибавляют раствор перманганата до получения такой же окраски. Эта поправка не должна превышать 0,03 мл.
Конец титрования можно определять и с помощью индикатора фенан-тролин-сульфата железа (II) (ферроина). В этом случае при приближении к концу титрования прибавляют 1 каплю 0,025 М раствора указанного индикатора и потом добавляют по каплям перманганат, пока розовая окраска индикатора не перейдет в бледно-голубую. При использовании индикатора средняя поправка на «холостой» опыт составляет 1 0,02 мл.
Установка по стандартным образцам (нормалям). Если раствор перманганата служит для определения вещества, которое перед титрованием должно пройти через более или менее сложную химическую обработку, то его титр лучше всего устанавливать по соответствующему стандартному образцу (нормали). При этом навеска стандартного образца должна быть проведена через все стадии анализа. Если, например, раствор перманганата служит для определения железа в рудах после восстановления его хлоридом олова (II), как описано на стр. 441, то титр раствора перманганата лучше всего устанавливать по стандартному образцу железной руды, выпускаемому Бюро Стандартов США (* в СССР — Свердловским институтом металлов *).
Установка другими методами. Титр растворов перманганата можно удовлетворительно устанавливать потенциометрическим титрованием чистого иодида калия в разбавленном растворе серной кислоты 2 (не слабее 0,13 н.).
Были предложены также различные другие методы установки титра растворов перманганата, например по кристаллической щавелевой кислоте НаСаО4 • 2H2Q, соли Мора FeSO4 • (NH4)2SO« • 7Н2О и по железной проволоке. От этих способов лучше отказаться. Трудно приготовить щавелевую кислоту, которая содержала бы точно 2 молекулы воды. Против соли Мора можно возразить то, что эту соль трудно получить ________ )
1 О применении монохлорида иода в качестве катализатора и комплекса о-фе-нантролин-сульфат железа (II) в качестве индикатора при установке титра растворов перманганата по мышьяковистому ангидриду см. D. Е. Metzler, R. J. Myers, Е. Н. S w i f t, Ind. Eng. Chem., Anal. Ed., 16, 625 (1944).
2 W.S. Hendrixson, J. Am. Chem. Soc., 43, 858 (1921); I. M. Kolthoff, H. A. L a i t i n e n, J. J. L i n g a n e, там же, 59, 429 (1937).
218
Гл. VI. Объемный анализ
в чистом виде и предупредить слабое окисление ее и потерю воды при хранении. Железная проволока непригодна, если она не была предварительно точно проанализирована и если установка титра не проводится таким методом, при котором на результаты титрования не влияют примеси, содержащиеся в проволоке, например связанный углерод.
Установка титра раствора бихромата калия
Хромовая кислота не так легко, как марганцевая, восстанавливается органическими веществами и соляной кислотой — одной или в присутствии железа (III). Поэтому ее применение в некоторых титрованиях очень удобно, особенно в тех случаях, когда проводят титрование пробы предварительно обработанной соляной кислотой. Если титрование проводится потенциометрическим методом или с дифениламином или комплексом 1 о-фенантролин-сульфат железа (II) в качестве внутренних индикаторов, то такой метод не оставляет желать лучшего. Употребление свежего раствора гексацианоферрата (III) калия в качестве внешнего индикатора, например при определении железа, менее надежно, и от него следует отказаться.
При отсутствии уверенности в достаточной чистоте бихромата калия его надо очистить следующим способом. Самый чистый продажный препарат перекристаллизовывают по крайней мере 3 раза, полученные кристаллы высушивают при 150° С, после чего растирают их в тонкий порошок и снова высушивают при 150° С до постоянной массы. Очищенный таким способом препарат сохраняют в банке с притертой пробкой. Если нужен раствор бихромата калия точно определенного титра, то лучше точно взвесить соответствующее количество очищенного препарата, растворить в воде и разбавить раствор до точного объема или массы, чем полагаться на установку титра его титрованием. Если, как это часто бывает, титр раствора неизвестен, то его лучше всего устанавливать по стандартному образцу материала, проводя его через все операции предполагаемого к применению метода 2. Растворы бихромата калия очень стойки 3.
1 G. Н. Walden, L. Р. Hammett, R. Р. Chapman, J. Am. Chem. Soc., 53, 3908 (1931). См. также брошюру: Ortho-Phenanthroline, A High Potential, Reversible, Oxidimetric Indicator, G. F. Smith Chemical C°, Columbus, Ohio. -
2 H. H. Willard и P. Young (Ind. Eng. Chem., Anal. Ed., 7, 57 (1935)] утверждают, что окислительный эквивалент твердого бихромата калия или титр его растворов можно определить с очень большой точностью, применяя в качестве исходного вещества мышьяковистый ангидрид. Для этого сернокислый раствор мышьяковистой кислоты, полученной из навески мышьяковистого ангидрида, обрабатывают бихроматом калия, взятым в количестве несколько меньшем эквивалентного. Затем избыток восстановителя титруют или сульфатом церия (IV) или перманганатом калия в присутствии четырехокиси осмия в качестве катализатора и комплекса о-фенантро-лин-сульф&т железа (II) в качестве индикатора. Последнее титрование можно также проводить потенциометрическим методом броматом калия в солянокислом растворе.
3 W. М. С а г е у (J. Am. Pharm. Assoc., 16, 115 (1927)] отмечает, что титр приготовленного им 0,1 н. раствора бихромата калия заметно не изменился в течение 24 лет.
Методы, окисления и восстановления
219
Цериметрия
Титрование сульфатом церия (IV) было всесторонне изучено многими исследователями V Пользуясь этим реактивом, можно выполнить почти все те титрования, которые проводятся перманганатом калия. Преимуществами этого метода являются: 1) изменение валентности церия в результате титрования только на одну единицу, CeIV—Се111; 2) устойчивость как холодных, так и горячих растворов сульфата церия (IV); 3) возможность проводить титрование в присутствии соляной кислоты. Недостатком является отсутствие заметного изменения окраски реактива при титровании, что требует или выполнения титрования потенциометрическим методом или применения окислительно-восстановительного индикатора, например ферроина [комплекса о-фенантролин-сульфат железа (II)]. Некоторые реакции титрования требуют также добавления катализатора — четырехокиси осмия.
Было показано 1 2, что механизм цериметрических титрований основан не на отношении концентраций простых ионов (Се4+) : (Се3+), а на отношениях концентрации комплексных ионов:
[Ce(S04)|~] и [Се(С1О4)3~] [Се3+] [SO|~]3 [Се3+] [С1О;]в
Это объясняет те различия в значениях нормальных окислительных потенциалов по отношению к водородному электроду, которые были получены в солянокислых (1,28 в), сернокислых (1,44 в), азотнокислых (1,61 в) и хлорнокислых (1,70 в) растворах солей церия (IV).
Для приготовления растворов указанных солей сначала получают гексанитроцерат (IV) аммония (NH4)2Ce(NO3)6 из окиси церия (IV) 40%-ной чистоты 3, не содержащей тория. Полученную соль затем превращают в хлорид церия (III) нагреванием с соляной кислотой и, по желанию, в нитрат, сульфат или перхлорат церия (III) выпариванием с соответствующей кислотой, взятой в избытке. Соли церия (III) можно затем превратить в соли церия (IV) анодным окислением их растворов в приборе для электролиза с платиновыми электродами и диафрагмой. Безводный сульфат церия (IV), сульфат церия (IV) и аммония, гексанитроцерат аммония и перхлоратоцериевая кислота имеются в продаже 4.
Титр раствора сульфатоцерата можно установить потенциометрически, титруя этим раствором горячий (выше 70° С) раствор навески оксалата натрия в разбавленной (2 : 98) серной кислоте. Можно также отмеренный объем раствора сульфатоцерата, взятый в избытке, добавить к сернокислому раствору оксалата натрия, нагревать до 50° С в течение
1 См., например, Н. Н. W i 1' 1 а г d с сотрудниками и N. Н. Furmanc сотрудниками, статьи в J. Am. СЬепц Soc., 50—53 (1928—1931) и Ind. Eng. Chem. 20, 972 <1928).
2 G. F. S m i t h, C. A. G e t z, Ind. Eng. Chem., Anal. Ed., 10, 191, 304 (1938).
3 G. F. S m i t h, V. R. S u 1 1 i v a n, G. F r a n k, Ind. Eng. Chem., Anal. Ed., 8, 449 (1936).
4 О методах приготовления различных комплексных цератов см. G. F. S m i t h, G. F r a n k, A. E. Kott, Ind. Eng. Chem., Anal. Ed., 12, 268 (1940). Извлечение церия из остатков после титрования описано F. R. D u к е и К. G. Stone (там же, 16, 721 (1944)].
220
Гл. VI. Объемный анализ
5 мин, охладить, добавить индикатор ферроин и оттитровать раствором сульфата железа (II).
Растворы нитрато- и перхлоратосолей церия несколько нестойки, даже при хранении их на рассеянном свету. Наиболее стойки растворы нитратоцерата (IV) аммония в хлорной (3 М) или азотной (1 М) кислотах. Титр растворов перхлоратоцерата (IV) в хлорной кислоте требуется периодически проверять. Умеренные количества других элементов цериевой группы не отражаются заметно на устойчивости растворов \
Титры перхлоратоцератных и нитроцератных растворов можно устанавливать, титруя ими при комнатной температуре растворы оксалата натрия или мышьяковистого ангидрида в 2 н. хлорной кислоте (мышьяковистый ангидрид растворяют сначала в растворе едкого натра, потом подкисляют и добавляют четырехокись осмия, служащую катализатором). Конец титрования определяют потенциометрическим методом или визуально с применением нитроферроина (нитро-о-фенантролина — Fe2+) в качестве индикатора.
Цериметрия в растворах хлорной кислоты дает' возможность проводить титрования окислителем с таким высоким окислительным потенциалом, какой не имеет ни один из ранее известных стойких в растворе окислителей. Этот потенциал равен 1,7 в в 1 н. растворе кислоты и 1,87 в в 8 н. растворе кислоты (по отношению к нормальному водородному электроду). Титрование при таких высоких окислительных потенциалах можно проводить потенциометрическим методом или с применением обратимых окислительно-восстановительных йндикаторов, таких, как ферроин или нитроферроин, у которых потенциалы перехода равны соответственно 1,06 и 1,25 в.
Иодометрия'
Раствор крахмала. В литературе описано много способов приготовления раствора крахмала, применяемого в йодометрических титрованиях. Хорошую стойкость и чувствительность при использовании в титрованиях, проводимых в кислых растворах, например при определении серы методом отгонки сероводорода1 2, показывает раствор крахмала, приготовляемый в Бюро Стандартов США следующим способом. Растирают в пасту 5 г растворимого крахмала, обрабатывают 400 мл кипящей воды и охлаждают приблизительно до 20° С. Затем прибавляют 50 мл 25%-ного раствора иодида калия и 50 мл холодного 10%-ного раствора едкого натра. Длй употребления в «нейтральных» растворах насыщают двуокисью углерода.,
Приводим еще два метода приготовления стабилизированных растворов крахмала, описанных в литературе.
1 G. F. Smith, С. A. Getz, Ind. Eng. Chem., Anal. Ed., 12, 339 (1940).
2 В Бюро Стандартов США для определения серы сжиганием применяют раствор крахмала, приготовляемый следующим способом. К 4,5 г растворимого крахмала прибавляют 5—10 мл воды и перемешивают до получения однородной пасты. Затем эту смесь медленно вливают в 250 мл кипящей воды, охлаждают, добавляют 7,5 г KI и перемешивают до растворения этой соли, потом разбавляют до 500 мл. Полученный раствор нестоек и его надо готовить перед употреблением. При хранении в холодильнике он сохраняет свои свойства 1—2 недели.
Методы, окисления и восстановления
221
Первый метод 1 следующий. Тонко измельчают 5 г аррорутного крахмала в стеклянной или агатовой ступке, затем растирают в пасту с небольшим количеством холодной воды и вливают медленно при перемешивании в 1 л кипящей воды. Кипятят раствор 2 ч. К концу кипячения прибавляют 30 г борной кислоты. Борная кислота защищает от развития плесени, но не от действия амилазы слюны; если последняя не попадает в раствор, то он даже по истечении нескольких месяцев не отличается от свежеприготовленного раствора, если хранится в колбе, покрытой только перевернутым вверх дном стаканчиком2.
По второму методу3 растирают 2,5 г картофельного крахмала с 10 лм холодной воды в тонкую пасту и вводят ее небольшими порциями при непрерывном перемешивании в 1 л кипящей воды. Кипятят 15 мин, не прекращая перемешивания. Затем несколько охлаждают, прибавляют 1,25 г салициловой кислоты и перемешивают до ее растворения. Полученный раствор крахмала очень чувствителен и сохраняется почти безграничное время даже в открытом сосуде. При титровании добавляют примерно 1 мл этого раствора на 100 мл титруемой жидкости и к концу титрования вводят 0,1—0,2 г иодида калия, если его в растворе не было.
Приводим также два метода приготовления раствора крахмала без добавления стабилизаторов.
Первый из них 4 состоит в следующем. Тонко измельчают 5 г аррорутного крахмала в стеклянной или агатовой ступке, затем растирают его в пасту, добавляя небольшое количество холодной воды, и медленно при перемешивании вводят в 1 л кипящей воды. Кипятят 2 мин, охлаждают, помещая стакан в холодную воду, оставляют стоять на ночь и фильтруют в небольшие, емкостью 50 лл, аптекарские склянки. Погружают склянки до шейки.в водяную бдню, нагревают 2 ч и закрывают мягкими пробками, стерилизованными медленным проведением через огонь. Растворы крахмала, стерилизованные таким способом и сохраняемые в закрытых склянках, не плесневеют неопределенно долгое время. При испытании такой раствор оставался совершенно чистым в течение 18 мес. и был так же чувствителен, как и свежеприготовленный. При открывании склянки плесень в ней появляется через неделю.
По второму способу 8 сухой пшеничный крахмал перемалывают в обыкновенный жерновой мельнице в течение 122 ч, и достаточное количество измолотого крахмала, чтобы получить 2%-ный его раствор, медленно просевают в дистиллированную воду, которую перемешивают электрической мешалкой. Через час раствор центрифугируют со скоростью около 2000 об/мин. Центрифугирование продолжают полчаса,
1 R. М. Chapin, J. Am. Chem. Soc., 41, 354 (1919).
2 N. К a n о [Sci, Rep. Tohoku Imp. Univ., 16, 861 (1927)] рекомендует прибавлять 6,5 мл 2 н. раствора НС1 на 50 мл раствора крахмала, если последний будет применяться в кислом растворе, и несколько капель CS2, если крахмал будет применяться в щелочном растворе. Для защиты растворов крахмала (и таннина) от разложения рекомендуют также прибавление 0,1% пирослизевой кислоты или ее натриевой соли [А. М. Р 1 a t о w, Chemist Analyst, 28, 30 (1939)].
3 М. S. N i с h о 1 s, Ind. Eng. Chem., Anal. Ed., -1, 215 (1929). См. также G. J. Hough, J. hid. Eng. Chem., 11, 767 (1919).
4 H. N. S t о k*e s, см. Ф. П. T p e д в e л л, В. Т. Г о л л, Курс аналитической химии, т. II, ч. 2, ОНТИ, 1935, стр. 142.
6 С. L. А 1 s Ь е г g, Е. Р. G г i f f i n g, J. Field, J. Am. Chem. Soc., 48, 1299 (1926).
/
L.
222
Гл. VI. Объемный анализ
декантируют прозрачную жидкость с осадка и хранят ее под слоем толуола в закрытой склянке. Приготовленный таким способом раствор содержит от 0,1 до 0,5% крахмала и остается прозрачным в течение месяцев. При замене пшеничного крахмала картофельным получаются опалесцирующие растворы, которые надо очищать повторным фильтрованием под давлением через инфузорную землю.
Первое окрашивание, получающееся при прибавлении иода к раствору крахмала, — розовое х. В обычной аналитической работе эту точку немного переходят, и конечной точкой титрования считают появление хорошо известного синего окрашивания. Для получения настоящей окраски раствор должен содержать приблизительно 1 г иодида калия в 100 мл и температура раствора не должна быть выше 25° С. Чувствительность конечной точки при 30° С ниже, и окрашивание полностью исчезает при слабом нагревании. В очень холодных растворах проводить титрование также не следует вследствие того, что замедляется течение реакций при низких температурах 1 2.
Титрованные растворы иода. Давление паров иода в его 0,05 н. и 0,1 н. растворах очень велико 3. Раствор иода, содержащий 4% иодида калия, не теряет заметных количеств иода при приливании его из бюретки по каплям жидкость, уровень которой находится от бюретки на расстоянии нескольких сантиметров, но его нельзя взбалтывать в колбе или оставлять в открытом стакане 4 *’6. В обычной рядовой работе можно тркже применять растворы иода, содержащие 1 % иодида калия, если титрование проводится быстро и в условиях, которые сводят к минимуму опасность улетучивания иода.
Присутствие йодата калия в растворе иода нежелательно, потому что это приводит к различному значению титра, в зависимости от того, проводится ли титрование в нейтральной или в кислой среде. Если титрованный раствор иода предполагают применять в различных средах, то нужно обязательно вводить иодид калия, свободный от йодата калия, или же разрушать последний прибавлением соответствующего количества серной кислоты.
Главными причинами изменения титра растворов иода являются: улетучивание иода, попадание в раствор пыли или примесей, содержащихся в иоде или в воде, взятой для приготовления раствора. Ни в каком случае нельзя употреблять корковые или резиновые пробки. Для переливания раствора нужно применять чистую пипетку или стеклянный сифон (целиком из стекла), но не выливать через горлышко. При взбалтывании раствора не нужно давать ему смачивать пробку. Устойчивость правильно
1 Е. W. Washburn, J. Am. Chem. Soc., 30, 42 (1908).
2 Некоторой неопределенности, связанной с применением крахмала, можно избежать, проводя титрования потенциометрическим методом. Пример такого титрования см. S. Р о р о f f, J. L. Whitman, J. Am. Chem. Soc., 47, 2261 (1925).
3 Вместо титрованных растворов иода можно применять титрованные растворы смеси йодата калия и иодида калия. Их преимуществом является то, что выделение из них иода происходит только в процессе титрования (* при введении раствора в титруемую жидкость, содержащую свободную кислоту *)• Еще более стойкими являются растворы, содержащие только один иодат калия, но при их применении иодид калия приходится добавлять в титруемый раствор. *
4 R. М. С h а р i n, J. Am. Chem. Soc., 41, 354 (1919).
8 F. О. Rice, М. Kilpatrick, W. Lemkin, J. Am. Chem. Soc., 45,
1361 (1923).
Методы- окисления и восстановления
223
приготовленного и правильно сохраняемого раствора иода иллюстрируется следующим наблюдением х. Титр раствора иода, постоянно употреблявшегося в течение двух месяцев, изменился за этот период от 0,0048504 до 0,0048505 г по As2O3.
Для работ высшей точности титрованный раствор иода следует готовить из иода, очищенного особым методом 1>2. Для обычных целей иод можно очищать следующим способом. Растирают 5 г иода с 2 а иодида калия, переносят смесь в сухугй глубокую фарфоровую чашку или кастрюлю, ставят ее на. радиатор (стр. 48) и закрый^ют круглодонной колбой, наполненной водой и закрытой пробкой с двумя трубками, через которые можно в дальнейшем пропустить ток холодной воды. Чашку осторожно нагревают, пока на нижней поверхности колбы не осядет достаточное количество возогнанного иода; дают чашке остыть. Колбу снимают и пропускают через нее ток холодной воды, чтобы вызвать сжатие стекла, затем переносят корку возогнавшегося иода на часовое стекло, осторожно соскабливая его стеклянной палочкой. Большие куски иода измельчают и снова его возгоняют, на этот раз без предварительного прибавления иодида калия. Второй возгон снимают со стекла колбы тем же способом и измельчают в стеклянной или агатовой ступке. Высушивают в эксикаторе над хлоридом кальция, следя за тем, чтобы на внутреннюю поверхность эксикатора не попала смазка со шлифа.
Титрованные растворы иода обычно получают установкой титра приготовленного раствора, а не растворением точно взятой навески иода и доведением раствора до точной массы или объема.
Приготовление приблизительно 0,1 н. раствора. Отвешивают в маленьком стаканчике 12,7 г чистого иода, прибавляют 40 г чистого иодида калия, не содержащего йодата калия, и заливают дважды перегнанной водой. Оставляют стоять, помешивая время от времени, пока не закончится растворение, фильтруют через асбест в чистую литровую склянку и разбавляют дважды перегнанной водой приблизительно до 1 л. Прежде чем устанавливать титр, раствор оставляют стоять 2—3 дня.
Установка титра по оксалату натрия. Наиболее удобный метод установки титра раствора иода основан на применении раствора тиосульфата натрия, установленного в свою очередь по титрованному раствору перманганата.
Такой метод установки титра, который сводится в конце концов к применению в качестве исходного вещества оксалата натрия, может вызвать возражения, так как точность окончательного результата зависит от чистоты взятого оксалата натрия и от случайных ошибок трех титрований. Если применяют оксалат натрия высокой чистоты, то получаемый титр должен совпадать с титром, установленным прямым титрованием по очищенному мышьяковистому ангидриду в пределах 0,05%.
Ход работы. Растворяют 0,5 г иодида калия в 120 мл воды и в полученный раствор вливают определенное количество, например 40 мл, раствора иода, держа кончик бюретки возможно ближе к поверхности раствора иодида калия. Титруют установленным раствором тиосульфата, пока окраска раствора не станет слабо-желтой; прибавляют 2 мл 0,5 %-ного раствора крахмала и продолжают титрование до обесцвечивания
1 См. М. Guicjiard, Ann. chim., 7, 27 (1917).
2 R. M. Chapin, J. Am. Chem. Soc., 41, 354 (1919).
224 Гл. VI. Объемный анализ
раствора. Затем снова прибавляют раствор иода до появления неисчезающего синего окрашивания. Записывают объемы израсходованных растворов иода и тиосульфата. Отдельно растворяют 2 г иодида калия в 200 мл воды, прибавляют 2 мл раствора крахмала и титруют раствором иода до того же оттенка, какой получился в первом растворе. . Объем израсходованного на зто титрование раствора иода вычитают из объема раствора иода, пошедшего на первое титрование, и вычисляют титр раствора иода по титру раствора тиосульфата.
Установка титра по мышьяковистому ангидриду. Установка титра растворов иода по мышьяковистому ангидриду имеет то преимущество, что это — Прямой способ установки титра х. Мышьяковистый ангидрид надо очищать, ёсли чистота его вызывает сомнения, и концентрацию ионов водорода надо сохранять в узких границах (pH от 5 до 9) в течение всего титрования. Для установки титра обычно берут отмеренные или отвешенные порции раствора арсенита, приготовленного, как описано ниже (стр. 227), а не отдельные навески сухого мышьяковистого ангидрида.
В описываемом способе pH раствора сохраняется достаточно близко к требуемому благодаря бикарбонату натрия, содержащемуся в растворе арсенита, и СО2, освобождающемуся В результате происходящей реакции. При выполнении особо точных исследований существенно, чтобы раствор был все время насыщен двуокисью углерода. Так как это довольно трудно выполнимо, рекомендуется 1 2 проводить титрование в растворах буферных смесей Na2HPO4 4- NaH2PO4 или Н3ВО3 + + Na3BO3, лучше в первом. Удовлетворительные результаты получались также при использовании смеси буры и борной кислоты 3>4. Имеются указания 4 на то, что в фосфатной смеси реакция идет слишком медленно. Во всех случаях следует вычислить или определить экспериментально отношение смешиваемых солей, чтобы сохранялась нейтральность раствора.
Ход работы. Точно отвешивают около 0,2 г мышьяковистого ангидрида в чистом, предварительно взвешенном бюксе и переносят последний вместе с его содержимым в круглодонную колбу емкостью 200 мл из стекла пайрекс. Приливают 10 мл 1 н. раствора едкого натра, вставляют в горло колбы воронку с короткой трубкой и осторожно перемешивают до полного растворения навески. Затем приливают 15 мл 1 н. серной кислоты, тщательно перемешивают и осторожно прибавляют 50 мл раствора, содержащего 40 г!л бикарбоната натрия. Обмывают воронку водой и вынимают ее. Затем медленно титруют жидкость устанавливаемым приблизительно 0,1 н. раствором иода (12,7 г очищенного иода и 60 г чистого иодида калия растворяют в 1 л дистиллированной воды) при непрерывном перемешивании, пока бблыпая часть иода не будет таким образом прибавлена (на 0,2 г As2O3 требуется приблизительно 40,4 мл 0,1 н. раствора иода). Затем, прилив 5 мл 0,5%-ного раствора крахмала, продолжают титрование, пока первоначально розовое окрашивание не перейдет в чисто синее.
1 Быстрый и простой метод получения мышьяковистого ангидрида, свободного от сурьмы, см. С. W. F о u 1 k, Р. G. Horton, J. Am. Chem. Soc., 15, 2416 (1929).
2 E. W. Washburn, J. Am. Chem. Soc., 30, 42 (1908).
3 O. L. В a г n e b e y, J. Am. Chem. Soc., 38, 330 (1916)»
4 R. M. Chapin, цит. выше,
Методы окисления и восстановления
225
Из израсходованного объема раствора иода вычитают то его количество, которое требуется для получения такой же окраски в растворе, содержащем все прибавленные в первый раствор реактивы и 40—50 мл свежепрокипяченной и охлажденной воды, в которой предварительно растворяют 5 г иодида калия. Титр раствора иода вычисляют на основе реакции
HAsO2 +12 + 2Н2О = H2AsO; + 21" + зн+
Титрованный раствор тиосульфата натрия. Существует много исследований, посвященных вопросу о хранении растворов тиосульфата натрия. Часто повторяемое утверждение о вредном влиянии двуокиси углерода в действительности имеет мало оснований К Кольтгоф утверждает, что на солнечном свету разложение растворов тиосульфата ускоряется, что 0,01 н. растворы разлагаются быстрее, чем 0,1 н., что осадок серы, образующийся при разложении раствора тиосульфата, ускоряет дальнейшее разложение и что небольшие количества иодида ртути (II) (10 мг на 1 л) или некоторых щелочных веществ, сильно замедляют его. Он высказал предположение, что разложение может быть вызвано некоторыми потребляющими серу микроорганизмами. Это предположение было затем подтверждено 2: разложение происходит под действием бактерий Thiobacillus thioparus, превращающих тиосульфат в сульфат и элементарную серу.
Титрованные растворы тиосульфата следует поэтому стерилизовать. Если это сделано, то можно устанавливать титр раствора и применять его немедленно при приготовлении. Такой раствор не показывает увеличения титра при стоянии, концентрация его уменьшается, но чрезвычайно медленно. Было найдено 2, например, что раствор, концентрация которого вначале была равна 0,014085 н., после 8 мес. пользования им изменил свою концентрацию только до 0,014016 н. Двуокись углерода, кислород и малые количества едкого натра влияют очень мало. Если в чистоте препарата тиосульфата натрия имеются сомнения, то его нужно перекристаллизовать, для чего приготовляют его насыщенный при 30— 35° С раствор в свежепрокипяченной и охлажденной дистиллированной воде, затем охлаждают раствор при сильном перемешивании и выделившуюся соль отсасывают.
Приготовление приблизительно 0,1 н. раствора. Отвешивают около 25 г кристаллического тиосульфата натрия Na2S2O3 • 5Н2О и растворяют приблизительно в 1 л свежей дважды перегнанной и охлажденной воды 3.
Для сохранения тиосульфата рекомендуется растворять его или в 0,05 М растворе буры, содержащем Na2HPO4B 0,1 н. концентрации, или в воде, насыщенной сероуглеродом 4. Растворы тиосульфата натрия, приготовленные растворением обычной чистой соли в дистиллированной воде, не изменяют своего титра, если к ним прибавлять 0,1% натриевой соли пирослизевой кислоту5. Рекомендуют к растворам тиосульфата, имеющим pH от 6,2 до 6,8, прибавлять по 3 капли хлороформа СНС13 на 1 л
1 G. Т о р f, Z. anal. Chem., 26, 137 (1887); I. М. К о 1 t h о f f, Pharm. Weekblad, 56, 878 (1919); Z. anal. Chem., 60, 344 (1921); M. K. Kilpatrick, Jr., M. L. Kilpatrick, J. Am. Chem. Soc., 45, 2132 (1923); F. O. R ice, M. Kilpatrick, W. Lemkin, там же, 45, 1361 (1923).
2 M. Kilpatrick, M. L. Kilpatrick, цит. выше.
3 I. Y ос hi da, J. Chem. Soc. Japan, 48, 26 (1927); C. A., 21, 3030 (1927).
* A. M. Plato w, Chemist Analyst, 28, 30 (1939).
5 J. L. К a s s n e r, E. E. К a s s n e r, Ind. Eng. Chem., Anal. Ed., 12, 655 (1940).
15 Заказ 52?.
226
Гл. VI. Объемный анализ
и хранить их в склянках с резиновыми пробками; если концентрации растворов ниже 0,05 н., склянки для их хранения должны быть оранжевого стекла.* Количество добавляемого хлороформа лучше повысить до 1 мл на 1 л раствора. Можно также взамен хлороформа добавлять амиловый спирт (несколько миллитров на 1 л раствора).*
Установкатитрапо перманганату калия. Титр раствора тиосульфата натрия удобнее всего устанавливать по титрованному раствору перманганата калия. Если соблюдать простые меры предосторожности, то титр, полученный таким способом, не будет отличаться более чем на 0,05% от полученного путем установки по очищенному иоду, проведенной в самых лучших условиях х. Раствор перманганата должен быть установлен по оксалату натрия, как описано выше (стр. 214), и его реакцию с иодидом калия нужно проводить на рассеянном свету. Под действием прямого солнечного света кислород воздуха реагирует с иодистоводородной кислотой, выделяя заметные количества свободного иода.
Ход работы. В каждой из двух конических колб, снабженных стеклянными притертыми пробками, растворяют по 10 г иодида калия в 100 мл свежепрокипяченной и охлажденной дистиллированной воды и прибавляют по 2 мл соляной кислоты. В одну из колб затем медленно приливают из бюретки отмеренное количество, например 40 мл, титрованного приблизительно 0,1 н. раствора перманганата при очень осторожном вращении колбы. Во вторую колбу приливают 40 мл воды. Закрывают колбы пробками и оставляют стоять в темноте 10 мин. Выделившийся иод титруют устанавливаемым раствором тиосульфата, пока раствор не примет слабую соломенно-желтую окраску, прибавляют 2 мл 0,5%-ного раствора крахмала и продолжают титрование раствором тиосульфата, пока синий цвет не исчезнет а. Объем раствора тиосульфата, израсходованный в контрольном оТтыте, вычитают из объема раствора тиосульфата, израсходованного в основном титровании, и титр раствора тиосульфата по иоду вычисляют на основании отношения КМпО4 : 51.
Установка титра по чистому иоду. Применение чистого иода для установки титра раствора тиосульфата имеет преимущество в том, что непосредственно дает титр, выраженный по иоду. Главный недостаток способа заключается в необходимости приготовления чистого сухого иода и в трудности нахождения точной массы иода, употребленного для установки титра. Очистка иода проводится, как описано на стр. 223. Иод обычно взвешивают в того или иного вида стаканчике для взвешивания, содержащем концентрированный раствор иодида калия 3.
Ход работы. Осторожно отвешивают приблизительно 0,5 г иода в стаканчик для взвешивания или другой сосуд, предохраняющий от потери иода из-за улетучивания во время взвешивания или во время переноса в сосуд для титрования и не изменяющий своей массы во в^емя взвешивания. После перенесения иода в колбу и разбавления водой должен получиться раствор, содержащий 2 г иодида калия и занимающий объем около 160 мл. Не следует оставлять раствор открытым или пере-
1 S. Popoff, J. L. Whilman, J. Am. Chem. Soc., 47, 2259 (1925); S. Popoff, A. H. Kunz, там же, 51, 1307 (1929).
2 Конечную точку можно также определять потенциометрическим титрованием; см., например, Popoff и Whitman, цит. выше.
3 PopoffnWhitman (цит. выше) утверждают, что при каждом снимании крышки со стаканчика для взвешивания, содержащего концентрированный раствор иодида калия, теряется около 0,2 мг.
Методы окисления и восстановления
227
мешивать его дольше, чем это нужно. Раствор иода титруют устанавливаемым раствором тиосульфата, пока почти весь иод не будет оттитрован. Затем прибавляют 2 мл 0,5%-ного раствора крахмала и продолжают титрование до исчезновения синего окрашивания.
Установка титра по титрованному раствору иода. Если раствор иода был установлен по чистому мышьяковистому ангидриду (стр. 224), то его можно в свою очередь использовать для установки титра раствора тиосульфата. В этом случае поступают точно так же, как указано на стр. 226, и вычисляют титр раствора тиосульфата на основе имеющегося титра раствора иода.
Установка титра другими методами. Установка титра раствора тиосульфата по бииодату калия или бихромату калия дает такие же точные-результаты, как и установка указанными выше способами, но эти методы применяются мало х. Если пользуются бииодатом калия, то необходимо сначала установить его чистоту. При использовании бихромата калия некоторым затруднением является окраска восстановленной соли, если не применяется потенциометрическое титрование.
Следующий ход установки титра растворов тиосульфата натрия по бихромату калия рекомендован 1 2 в особенности для работы в жаркое время года (30—40° С). Растворяют 0,2—0,22 г К2Сг2О7 в 125 мл воды в конической колбе емкостью 500 мл, прибавляют 5 г чистого иодида калия, перемешивают до растворения и, не прекращая перемешивания, вливают 5 мл 6 н. раствора соляной кислоты. Затем обмывают стенки колбы водой, не перемешивая раствор, так, чтобы образовался наверху бесцветный слой, закрывают пробкой и оставляют в темноте на 6 мин. Потом прибавляют 165 .ил воды и титруют устанавливаемым 0,1 н. раствором тиосульфата натрия при постоянном взбалтывании, пока раствор не станет зеленовато-желтым, и заканчивают титрование после добавления 0,6%-ного раствора крахмала (2 мл при 20° С, 3 мл при 40° С). Вводят поправку на «холостой» опыт. Вместо твердого бихромата калия можно применять его титрованный раствор. В этом случае вливают в коническую колбу 45 мл 0,1 и. раствора бихромата калия и продолжают, как описано выше.
Титрованные растворы арсенита натрия. Для работ высшей точности титрованные растворы арсенита натрия следует приготовлять из мышьяковистого ангидрида известной чистоты или из препарата, очищенного специальными методами 3. Достаточно чистый для применения в обычных работах мышьяковистый ангидрид можно приготовить следующим способом. Около 10 г тонко измельченного продажного мышьяковистого ангидрида вносят в фарфоровую чашку, покрывают часовым стеклом и осторожно нагревают, пока не соберется достаточное количество возгона. Если возгон имеет желтоватый оттенок, то, вероятно, он содержит сульфид мышьяка. В этом случае растворяют возгон в горячей разбавленной (1 : 2) соляной кислоте, фильтруют и охлаждают фильтрат. Сливают с кристаллов прозрачную жидкость, промывают их водой, высушивают и снова возгоняют. Охлаждают и сушат 12 ч в эксикаторе над
1 Описание метода установки титра по бииодату калия (а также различные методы иодатометрии) см. в книге: G. S. Jamieson, Volumetric Jodate Methods, Chemical Catalog Co., New York, 1926. * См. также И. M. К о ль т г о ф, Е. Б. С е н-д э л, Количественный анализ, изд. 3-е, Госхимиздат, 1948, стр. 644—646, где подробно излагаются методы установки титра растворов тиосульфата по йодату калия, бромату калия, бихромату калия и гексацианоферрату (III) калия*.
2 S. О. Rue, Ind. Eng. Chem., Anal. Ed., 14, 802 (1942).
3 Например, методом, описанным в статье R. М. С h а р i n [J. Ind. Eng. Chem., 10, 522 (1918)].
15*
228 Гл. VI. Объемный анализ
серной кислотой. Для приготовления растворов, титр которых будет устанавливаться и которые будут применять в обычной рядовой работе, например при определении марганца (стр. 502), можно пользоваться чистым продажным арсенитом натрия.
Приготовление приблизительно 0,1 н. раствора. Для приготовления титрованного, приблизительно 0,1 н. раствора арсенита натрия, предназначаемого для применения в работах высшей точности, поступают следующим образом. 4,95 г чистого мышьяковистого ангидрида вносят в маленькую трубочку для взвешивания, высушивают в вакууме над серной кислотой 1 и взвешивают. Затем высыпают навеску без потерь в чистую литровую мерную колбу, снабженную пробкой, и снова взвешивают трубочку 2. Навеску в колбе смачивают небольшим количеством воды. Отдельно растворяют 10—12 г чистого едкого натра (не содержащего железа) в 30 мл воды, свежеперегнанной из щелочного раствора перманганата, чтобы обеспечить отсутствие органических веществ и растворенного кислорода. Раствор едкого натра фильтруют через асбест, фильтрат приливают к As2O3 в мерной колбе, закрывают колбу пробкой и оставляют стоять или осторожно вращают, пока растворение не закончится 3. Прибавляют 200 мл дистиллированной воды и насыщают раствор чистой двуокисью углерода. Удаляют и обмывают вводную трубку, разбавляют точно до 1 л дистиллированной водой и тщательно перемешивают. Раствор, приготовленный таким образом и сохраняемый при комнатной температуре, не изменяет свой титр очень долгое время.
Для обычных работ раствор может быть приготовлен следующим способом. Навеску мышьяковистого ангидрида переносят в литровую мерную колбу, смачивают водой и приливают'раствор 15 г едкого натра в 100 мл воды. Содержимое колбы осторожно перемешивают, пока мышьяковистый ангидрид полностью не растворится, разбавляют до 250 мл, насыщают раствор двуокисью углерода и разбавляют водой до 1 л.
Раствор мышьяковистого ангидрида в едком натре можно нейтрализовать фосфорной кислотой, если предполагают применять буферную фосфатную смесь Na2HPO4 + NaH2PO4 для поддержания нужной концентрации ионов водорода во время титрования 4. Для применения с Н3ВО3 + Na3BO3H некоторыми другими буферными смесями 5 6>G можно
1 Чистый мышьяковистый ангидрид почти не гигроскопичен и нет необходимости его высушивать перед употреблением, если не требуется точность, превышающая 1 : 2000. Например, препарат мышьяковистого ангидрида Бюро Стандартов США не потерял в массе при высушивании в течение 1 чпри 105° С, но увеличил свою массу на 0,014%, когда его оставили на две недели в атмосфере, имеющей относительную влажность 90%. Если мышьяковистый ангидрид надо высушить, то нагревания в течение 1 ч при 105° С достаточно. Взвешивание в открытом сосуде не вводит значительной ошибки.
2 Величину навески лучше определять по разности, как указано выше, потому что мышьяковистый ангидрид трудно без потерь счистить кисточкой с металлической или стеклянной поверхности.
3 Реактивный едкий натр может содержать загрязнения, которые приводят к слабому, но измеримому окислению мышьяковистого ангидрида во время его растворения. В работах особо высокой точности или когда чистота едкого натра неизвестна рекомендуется растворять мышьяковистый ангидрид при слабом нагревании в концентрированном растворе чистого карбоната натрия, затем разбавлять полученный раствор и насыщать его двуокисью углерода.
4 Е. W. Washburn, J. Am. Chem. Soc., 30, 42 11908).
s R. M. Chapin, J. Am. Chem. Soc., 41, 354 (1919).
6 R. C. R о ark, С. С. M c D onnell, J. Ind. Eng. Chem., 8, 327 (1916).
Методы окисления и восстановления
229
мышьяковистый ангидрид растворять в серной кислоте следующим образом-. Помещают 5 г чистого мышьяковистого ангидрида в колбу емкостью 1000 мл с длинной шейкой и растворяют кипячением с 500мл воды, содержащей 10 мл серной кислоты. Не следует кипятить дольше, чем это необходимо. Когда все растворится, раствор охлаждают и доливают водой до 1 л.
Установка титр а по иоду или по титрованному раствору иода. Обычно титр растворов арсенита натрия устанавливают точным отвешиванием чистого мышьяковистого ангидрида, растворением его без потерь и разбавлением до точного объема или массы, как уже описано. Если необходима установка титра полученного раствора, То ее лучше всего проводить титрованием отвешенных порций иода или отмеренных порций титрованного раствора иода тем же общим методом', который описан выше (см. «Установка титра раствора иода по мышьяковистому ангидриду», стр. 224).
Потенциометрическое титрование
Во многих случаях конец реакции в ацидиметрии, алкалиметрии, оксидиметрии, иодометрии или в-методах осаждения можно легко и точно определить потенциометрически, т. е. отмечая изменения потенциала подходящего электрода-во время титрования1. Такие титрования легко выполнимы как в бесцветных растворах, которые можно титровать с применением внутренних или внешних индикаторов, так и в растворах, окрашенных настолько, что применение индикаторов становится невозможным. Кроме того, потенциометрическим титрованием во многих случаях можно определять последовательные ступени реакций, например при нейтрализации фосфорной кислоты или при окислении смеси хлорида олова (II) и хлорида железа (II).
Потенциометрическое титрование основано на тех разностях потенциалов, которые возникают, когда неметаллический или металлический электрод погружен в раствор, содержащий его ион, или когда химически стойкий металл, например платйна, находится в контакте с раствором, содержащим такие ионы, которые способны отдавать электроны платине или же отнимать их от нее. Потенциал электрода является функцией концентрации ионов в растворе при остающихся постоянными прочих условиях и претерпевает резкое изменение в конце большинства титрований вследствие значительного изменения концентрации одного из реагирующих веществ в конечной точке. Резкий скачок потенциала является удобным показателем момента окончания реакции 2.
1 Для выполнения обычных, рядовых работ наличие в лаборатории потенциометрической установки может не оправдать себя, если не приходится часто определять какой-нибудь один элемент, например ежедневно производить массовые определения железа в песках для производства стекла или хрома и ванадия в сталях.
2 Обзор работ по потенциометрическому, кондуктометрическому и полярографическому титрованиям за период 1931—1942 гг. дан в статье N. Н. Furman, Ind. Eng. Chem., Anal. Ed., 14, 367 (1942); см. также обзоры в Anal. Chem., 21, 1—167 (1949) и 22, 1—126 (1950).
230
Гл. VI. Объемный анализ
Кислоты и щелочи можно титровать потенциометрическим методом, измеряя электродный потенциал между водородом на поверхности водородного электрода и ионами водорода в окружающем растворе \ но хорошего метода измерения этого потенциала нет. Можно, однако, определять разность потенциалов между этим водородным электродом и другим водородным электродом, погруженным в раствор, содержащий ионы водорода и известной концентрации, или, что более удобно, сравнивать первый электрод с любым другим, имеющим постоянный потенциал, например с каломельным полуэлементом. При многих таких титрованиях нет необходимости измерять действительные значения потенциалов, а достаточно, чтобы применяемый прибор давал возможность отмечать указанные резкие изменения потенциала.
Если, например, в раствор окислителя погрузить платиновый электрод и раствор соединить с каломельным полуэлементом, то эта система покажет определенную разность потенциалов, которая может быть уравновешена падением потенциала на проволоке, включенной последовательно с сопротивлением в цепь аккумулятора. Если теперь к этому раствору постепенно прибавлять раствор восстановителя, то заметного изменения разности потенциалов нашей системы наблюдаться не будет, пока не израсходуется весь окислитель и в растворе не появится первая капля избытка восстановителя. Если изменение потенциала в конечной точке титрования достаточно велико, то оно может быть замечено по резкому отклонению стрелки чувствительного гальванометра. Если изменения потенциала мало заметны, то электродвижущую силу системы определяют после добавления каждой порции титрующего раствора, а затем строят кривую изменения электродвижущей силы, откладывая ее значения по оси ординат, а объемы титрующего раствора — по оси абсцисс. В этом случае надо правильно определять относительные значения потенциалов; абсолютные же их значения не нужны.
Во многих случаях при построении кривой вместо нанесения на график значений электродвижущей силы Е и объема прибавленного реактива V гораздо удобнее откладывать по оси ординат значения отношения ДЕ7Д V, а по оси абсцисс объем 1 2 V. Практически то же самое может быть достигнуто, если в течение титрования образовывать концентрационные элементы временным изолированием порции раствора вокруг одного из электродов, каждый раз пока не будет прилита следующая порция титрующего раствора 3.
Применяются также 4 биметаллические системы, в которых нормальный каломельный электрод заменен вольфрамом или каким-нибудь метал
1 О водородном электроде и других электродах, например кислородном и воздушном, высших окислов различных элементов, хингидронном, стеклянном, вольфрамовом или других металлических электродах, см. J. Н. Hildebrand, J. Am. Chem. Soc., 35, 847 (1913); W. M. Clark (сноска 3 на стр. 198); И. К о л ъ т г о ф, Н. Ф у р-м а н, Потенциометрическое титрование, Химтеорет, 1935.
2 J. С. Hostetter, Н. С. R о b е г t s, J. Am. Chem. Soc., 41, 1341 (1919); D. A. MacI nnes, P. T. J ones, там же, 48, 2831 (1926), W. A. R о t h, Z. Elektro-chem., 33, 127 (1927).
3 D. A. M a с I n n e s, P. T. J о n e s, цит. выше; N. F. H a 11, M. A. J e n s e n, S. А. В a e c k s t г о m, J. Am. Chem. Soc., 50, 2217 (1928); D. A. M a с I n n e s, M. D ole, там же, 51, 1119 (1929).
1 J. C. Hostetter, H. S. Roberts, цит. выше; H. H. W i 11 а г d, F. Fenwick, J. Am. Chem. Soc., 44, 2504, 2516 (1922).
Методы окисления и восстановления
231
лом или сплавом платиновой группы, кроме чистой платины или чистого палладия. При использовании таких систем труднее определить конечную точку титрования, но если используется поляризационный ток, потери активности электродов не происходит и не надо прерывать титрование для той или иной обработки электрода.
Описание применения «электрометрических индикаторов» в установке, где стрелка останавливается при достижении конца титрования (в методах нейтрализации, окисления-восстановления или осаждения), приводится в литературе х. Одним из преимуществ таких установок является применение простых электродов из платиновой проволоки и отсутствие электродов сравнения.
Аппараты для потенциометрического титрования можно собирать в лаборатории 2 или приобретать их готовыми. Для определений ионов водорода, не требующих большой точности, или для титрования кислот и щелочей вполне пригоден аппарат, собранный по схеме, показанной на рис. 23.
По применению потенциометрического титрования было проведено большое число исследований, и теперь этот метод рекомендуется для
1 См. Н. II. W i 11 а г d, F. F е n w i с k, J. Am. Chem. Soc., 45, 715 (1923); С. W. Foulk,A. Т. Bawden, там же, 48, 2045 (1926); А. Н. W г i g h t, F. H.-G i fa-son, Ind. Eng. Chem., 19, 749 (1927); D. R. С 1 i p p i n g e r, C. W. F о u 1 k, Ind. Eng. Chem., Anal. Ed., 11, 216 (1939); G. W e г n i m о n t, F. J. Hopkinson, там же, 15, 272 (1943).
2 Подробности сборки аппаратов для определения концентрации ионов водорода см. в руководстве И. К о л ь т г о ф, Н. Ф у р м а н, Потенциометрическое титрование, цит. выше. Специальные методы описаны в следующих статьях: водородный электрод: J. Н. Н i 1 1 е Ь г a n d, цит. выше; биметаллические электроды: Н. Н. Willard, F. F е n w i с к, цит. выше; J. Am. Chem. Soc., 45, 715 (1923); N. Н. F u г т а п, там же, 50, 268, 273, 277 (1928); кислородный и воздушный электроды; N. Н. Furman, там же, 44, 2685 (1922); Р. A. Van der Meulen, F. Wilcoxon, Ind. Eng. Chem., 15, 62 (1923); электроды из высших окислов разных элементов: Н. С. Parker, там же, 17, 737 (1925); хингидронный электрод: Е. В u 1 m a n n, Ann. chim., (9), 15, 109 <1921); 16, 321 (1921); 19, 137 (1923); Е. В й 1 m a n n, J. К г а г и р, J. Chem. Soc., 125, 1954 (1924); I. М. К о 1 t h о f f, Rec. trav. chim., 42, 186 (1923); А. И. P а б п н o-в и ч, В. А. К a p г и h, Z. Elektrochem., 33, 11 (1927); сурьмяный электрод: A. U h 1, V. Kestranek, Monatsh., 44, 29 (1923); I. M. К о 1 t h о f f, B. D. H a r t о n g, Rec. trav. chim., 44, 113 (1925); вольфрамовый электрод: J. R. В а у 1 i s, Ind. Eng. Chem., 15, 852 (1923); стеклянный электрод: F. H a b e r, Z. Klemen-siewicz, Z. phys. Chem., 67, 385 (1909); W. S. Hughes, J. Am. Chem. Soc., 44, 2860 (1922); D. A. Maclnnes, M. Doi e, Ind. Eng. Chem., Anal. Ed., 1, 57 (1929).
Подробности конструкции аппаратов для титрования методами осаждения, окисления или восстановления см. в книге И. К о л ь т г о ф, Н. Фурман, Потенциометрическое титрование, цит. выше, а специальные методы в следующих статьях: каломельно-платиновые электроды: G. L. К е 11 е у, J. R. A d a m s, J. A. Wiley, J. Ind. Eng. Chem., 9, 780 (1917); H. S. R о b er t s, J. Am. Chem. Soc., 41, 1358 (1919); биметаллические системы: H. H. Willard, F. Fenwick, там же, 44, 2504, 2516 (1922); D. С. С о х, там же, 47, 2138 (1925); N. Н. Furman, там же, 50, 268, 273, 277 (1928); поляризованные биметаллические системы: Н. Н. Willard, F. Fenwick, там же, 44, 2516 (1922).
* Кроме приведенной выше литературы по потенциометрическому титрованию, см. Ю. С. Л я л и к о в, Физико-химические методы анализа, изд. 3-е, Госхимиздат, 1960, стр. 316—370; В. Гильтнер, Практика потенциометрических анализов, ОНТИ, 1936; И. М. К о л ь т г о ф, Г. А. Л а й т и н е н, Определение концентраций водородных ионов и электротитрование, Госиноиздат, 1947, Н. Я. X л о и и н, Потенциометрическое некомпенсационное титрование в заводской лаборатории, Свердловск — Москва, ОНТИ, 1937*.
232
Гл. VI. Объемный анализ
определения очень многих веществ. Среди его применений можно упомянуть следующие: ацидиметрия и алкалиметрия, иодометрия, определение галогенных солей, ванадия, урана, молибдена, мышьяка и сурьмы, меди,
Рис. 23. Схема прибора для потенциометрического титрования методом Гиллебранда:
1 — сухой гальванический элемент;
2 — мостик Кольрауша; 3>— милливольтметр (от 0 до ' 1,2 в);
4 — переносный гальванометр д'Ар-сонваля; 5 — входное отверстие для водорода; 6 — водородный электрод Гиллебранда; 7 — сосуд для титрования; S — солевой мостик; О — сосуд с насыщенным раствором хлорида калия; 10 — каломельный электрод; 11 — ключ.
серы в растворимых сульфидах, селена, гексацианоферратов (II) и гексацианоферратов (III), железа, марганца, никеля, кобальта, цинка и хрома1.
1 См. G. Е. F. Lu nd е 11, J. I. Н о f f m a n, Outlines of Methods of Chemical Analysis, J. Wiley and Sons, 1938, p. 165.
Часть вторая
ОПРЕДЕЛЕНИЕ
ЭЛЕМЕНТОВ
ГРУППА СЕРОВОДОРОДА
СЕРЕБРО, РТУТЬ, СВИНЕЦ, ВИСМУТ, МЕДЬ, КАДМИЙ, МЫШЬЯК, СУРЬМА, ОЛОВО, ГЕРМАНИЙ, МОЛИБДЕН, СЕЛЕН, ТЕЛЛУР, ЗОЛОТО, РУТЕНИЙ, РОДИЙ, ПАЛЛАДИЙ, ОСМИЙ, ИРИДИЙ, ПЛАТИНА (ТАЛЛИЙ)
ЭЛЕМЕНТЫ, СУЛЬФИДЫ КОТОРЫХ НЕРАСТВОРИМЫ В. КИСЛОТАХ И В РАСТВОРАХ СУЛЬФИДОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
СЕРЕБРО, РТУТЬ, СВИНЕЦ, ВИСМУТ, МЕДЬ, КАДМИЙ (И ПОЛНОСТЬЮ ИЛИ ЧАСТИЧНО РУТЕНИЙ, РОДИЙ, ПАЛЛАДИЙ И ОСМИЙ)
Глава VII
СЕРЕБРО
Серебро встречается в самородном состоянии, в виде сульфида, теллурида, арсенида, антимонида, хлорида, бромида, иодида и в виде многочисленных сульфосолей. Оно обычно содержится в самородном золоте и встречается также в самородной меди.
Из окисленных соединений серебра в природе известен только а р-гентоярозит AgFe3(OH)6(SO4)2, представляющий собой сульфат серебра и железа. Незначительные следы серебра содержатся в морской воде х.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород практически все серебро выпадает в виде хлорида серебра вместе с кремнекислотой. Присутствие значительных количеств серебра узнается по характеру остатка кремне-кислоты и изменению его окраски при выпаривании. При сжигании бумажного фильтра с остатком кремнекислоты, содержащей хлорид серебра, и прокаливании остатка большая часть хлорида серебра, а иногда и весь хлорид серебра восстанавливается и получающееся металлическое серебро сплавляется с тиглем.
Небольшое количество серебра, не увлеченное кремнекислотой и оставшееся в фильтрате, пройдет через весь анализ незамеченным, если после осаждения аммиаком не проводилось еще и осаждение сульфидом аммония. В присутствии серебра кремнекислоту лучше обезвоживать серной или азотной кислотами и удалять серебро сероводородом перед осаждением железа и алюминия аммиаком.
1 F. W. С 1 а г k е, U. S. Geol. Survey Bull., 770, 20.
236
Гл. VII. Серебро
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ СЕРЕБРО
Сульфиды, арсениды и теллуриды серебра лучше всего разлагать обработкой азотной кислотой и сплавлением нерастворимого остатка с карбонатом натрия. Галогениды серебра сплавляют с карбонатом натрия и плав выщелачивают водой для отделения серебра от галогенида натрия, после чего нерастворимый в воде остаток растворяют в азотной кислоте.
МЕТОДЫ РАЗДЕЛЕНИЯ
Отделение серебра основано на осаждении его в виде хлорида в разбавленном азотнокислом растворе (стр. 237). Этому отделению мешают: свинец, палладий, ртуть (I), медь (I) и таллий (I), также образующие нерастворимые хлориды, цианиды и тиосульфаты, растворяющие хлорид серебра и соединения сурьмы и висмута, гидролизующиеся в слабокислом растворе, который необходим для полного осаждения хлорида серебра. Мешают также органические вещества, иногда препятствующие надлежащей коагуляции хлорида серебра.
Помехи, вызываемые органическими веществами, цианидами, тиосульфатами, ртутью, медью и таллием, можно устранить предварительным кипячением с царской водкой при наличии таллия и с азотной кислотой в присутствии остальных веществ.
Влияние свинца можно обезвредить повторным осаждением серебра в виде хлорида из разбавленного раствора или предварительным удалением свинца двукратным осаждением его в виде сульфата х. Палладий, сообщающий розовую окраску осадку, удаляется двукратным растворением смеси хлоридов в аммиаке и осаждением кислотой. Такая же обработка достаточна для отделения солей висмута или сурьмы, которые, гидролизуясь, загрязняют хлорид серебра. При наличии остальных мешающих веществ применяемая обработка зависит от состава смеси.
Серебро количественно осаждается сероводородом в кислом или щелочном растворах, причем сульфид серебра нерастворим в растворах сульфидов или полисульфидов щелочных металлов. Эти реакции позволяют успешно отделять серебро от элементов группы мышьяка и последующих групп.
Малые количества серебра (20 мг) можно отделить от большого количества меди (8 г), проводя электролиз холодного раствора их сернокислых солей, содержащего 1 % (по объему) свободной серной кислоты и 2—3% (по массе) персульфата калия. Электролиз проводится 15—30 мин, при напряжении тока 2,0—2,5 в и плотности тока 0,2—0,7 0,1дм? поверхности катода.
Избыток персульфата в растворе должен сохраняться во все время электролиза.
1 Согласно G. V о г t man п и О. Hecht [Z. anal. Chem., 67, 276 (1925)], свинец может быть отделен от серебра осаждением либо фосфатом аммония, либо иодидом калия в слабоаммиачном растворе, содержащем тартрат аммония. Первый реактив количественно осаждает свинец в виде фосфата, второй осаждает серебро в виде иодида.
Методы, определения
237
МЕТОДЫ ОПРЕДЕЛЕНИЯ '
Наиболее точными методами количественного определения серебра (за исключением метода анализа сухим путем) являются: весовой метод, основанный на осаждении серебра в виде хлорида, и объемный метод, при котором серебро осаждают титрованным раствором роданида калия или аммония в присутствии железа (III) в качестве индикатора.
Весовое определение в виде хлорида серебра
Наиболее точным методом определения серебра является осаждение его в виде хлорида. Полученный осадок отделяют, промывают и взвешивают. Специальные операции, применяемые при определении атомных весов или в пробирном анализе серебряных слитков, здесь приводиться не будут А
Растворимость хлорида серебра в разбавленном растворе азотной кислоты, содержащем небольшой избыток хлорид-ионов, очень мала и в обычных анализах ею можно пренебречь. При 25° С хлорид серебра менее всего растворим 1 2 в воде, содержащей хлорид-ионы в 0,01 н. концентрации, растворимость эта равна 0,01 мг AgCl в 5 л. Однако растворимость хлорида серебра значительна в горячей или холодной чистой воде 3, в концентрированных растворах-соляной и азотной кислот и в растворах, содержащих большие количества хлоридов или нитратов щелочных и щелочноземельных металлов. Хлорид серебра легко растворяется в растворах цианидов, тиосульфата натрия и гидроокиси аммония.
При проведении. очень точных работ количество неосажденного хлорида серебра должно быть вычислено по данным растворимости, а то количество AgCl, которое вновь перешло в раствор при промывании осадка разбавленной кислотой или водой 4 и оставшееся на стенках сосуда, должно быть определено нефелометрическим методом — сравнением со стандартным раствором, как описано в гл. III (стр. 50).
Хлорид серебра имеет склонность окклюдировать как хлорид натрия, так и нитрат серебра 5 6. По этой причине осаждение должно проводиться медленно, постепенным прибавлением разбавленной соляной кислоты к разбавленному раствору соли серебра, свободному, насколько возможно, от других растворенных в нем веществ.
1 Описания специальных методов, применяемый при определении атомных весов, см. в статьях: Т. W. R i с h а г d s, R. С. W е 11 s, Pub. Carnegie Inst, of Wash., № 28 (1905); T. W. Richards, F. O. A n d e r e g g, J. Am. Chem. Soc., 37, 13 (1915); G. P. В a x t e r, F. A. H i 1 t о n, Jr., там же, 45, 695 (1923). Метод Гей-Люссака, применяемый в пробирном анализе серебряных слитков, описывается в специальных руководствах по пробирному анализу.
2 G. S. Forbes, J. Am. Chem. Soc., 33., 1937 (1911).
3 G. S. W h i t b у [Z. anorg. Chem., 67, 108 (1910)] показал, что при 100 С растворимость хлорида серебра в воде равна 21,7 мг!л, а при 21° С растворимость падает до 1,54 мг!л. В 1%-ной соляной кислоте она равна 0,2 мг/л, в,5%-ной — 33 мг!л, а в 10%-ной — 74,0 мг!л. х \ .
4 Количество хлорида серебра, переходящее в раствор при промывании его осадка
водой или разбавленной азотной кислотой, зависит от физической характеристики осадка. Точное количество хлорида серебра должйо быть определено в каждом отдельном случае [Т. W. Richards, R. С. Wells, Pub. Carnegie Inst, of Wash., № 28 (1905)].
6 J. S. S t a s, Oeuvres completes Bruxelles, 1, 337 (1894); T. W. Richards, R. C. W e 11 s, цит. выше.
238
Гл. VII. Серебро
О веществах, мешающих осаждению серебра в виде хлорида, и о тех мерах, которые следует предпринять в случае их присутствия, сказано выше (стр. 236).
Хлорид серебра плавится при температуре около 455° С и энергично при этом реагирует с платиной. Для обычных целей высушивания при 130—150°С достаточно. При выполнении работ высокой точности хлорид серебра нагревают приблизительно до 280° С; взвешивают и остающуюся в нем влагу (приблизительно до 0,01 %) определяют, перенося остаток в фарфоровый тигель и нагревая до полного его плавления. Продолжительного нагревания при такой высокой температуре надо избегать из-за возможных потерь вследствие улетучивания.
Ход определения. Во всех последующих операциях хлорид серебра должен быть как можно лучше защищен от света. Приготовляют раствор, содержащий 0,1—0,2 г серебра в 200—400 мл разбавленной (1 : 99) азотной кислоты; он не должен содержать значительных количеств других солей, а также мешающих примесей, о которых упоминалось выше. Раствор нагревают до 70° С и медленно прибавляют приблизительно 0,2 н. соляную кислоту при постоянном перемешивании до прекращения выделения осадка. Прибавления большого избытка кислоты следует избегать. Нагревают, пока осадок не осядет на дно, дают остыть до 25—30° С и осторожно приливают к прозрачной, жидкости над осадком 1 или 2 капли соляной кислоты, для того чтобы убедиться в полноте осаждения. Раствор рекомендуется оставлять в темном месте на несколько часов, лучше всего на ночь. Жидкость с осадка затем сливают через взвешенный тигель Гуча или Мунро (стр. 127), промывают осадок декантацией примерно 0,01 н. соляной кислотой и переносят его в тигель. Тигель с содержимым высушивают сначала при 100° С, затем при 130—150° С, охлаждают в эксикаторе и взвешивают осадок в виде AgCl. Высушивание повторяют до постоянной массы.
Определение титрованием роданидом калия иди аммония
Метод1 основан на большом различии в растворимости роданида железа (III) и. роданида серебра. Растворимый роданид железа не образуется до полного осаждения серебра в виде белого роданида серебра AgCNS. Титрование проводят в разбавленном азотнокислом растворе (от 1 до 10% азотной кислоты по объему) при температуре не выше 25° С. Концентрированная азотная кислота замедляет образование роданида железа, более высокая температура вызывает уничтожение окраски.
Осадок роданида серебра адсорбирует и нитрат серебра, и роданид калия или аммония 2. В обыкновенных анализах это не ведет к большой ошибке, если раствор роданида применяется в тех же условиях, в которых проводилась установка его’титра. Если установку титра раствора KCNS проводить методом, описанным ниже, а затем приливать его в анализируемому раствору до полного осаждения серебра (т. е. поступать аналогично методу Гей-Люссака, но применяя KCNS вместо NaCl), то результаты получаются повышенными.
1 Р. Charpentier, Bull. Soc. ing. civ. France, 135, 325 (1870); J. Vol-hard, J. prakt. Chem., (2), 9, 217 (1874); Ann., 190, 1 (1878).
a С. H о i t s e m a, Z. angew. Chem., 17, 647 (1904).
Методы определения
239
Анализируемый раствор должен быть свободен от азотистой кислоты, хлорида серебра и сульфатов. Первая образует окрашенное в красный цвет соединение с роданистоводородной кислотой; хлорид серебра до-некоторой степени реагирует с роданидом, а в присутствии сульфатов образуется смешанный осадок роданида и сульфата серебра х. Кроме-концентрированных растворов солей, мешающих определению своей окраской, вредны главным образом соединения ртути (II) и палладия. Медь (I), образующая также нерастворимый роданид (стр. 290), обычно не присутствует, так как предварительной обработкой вся медь окисляется до двухвалентной. Медь (II) не мешает определению, если отношение меди к серебру не превышает 7 : 10. Не мешают также мышьяк, сурьма, свинец, висмут, кадмий, железо, марганец, цинк, никель и кобальт.
Для проведения определения требуются три раствора. Первые два из них — растворы роданида аммония или калия и нитрата серебра должны быть титрованными и желательно эквивалентными. Третьим является насыщенный раствор железных квасцов. Роданид аммония следует предпочесть роданиду калия, так как он реже бывает загрязнен хлоридами.
Для приготовления раствора роданида растворяют 9 г роданида аммония или 10 г роданида калия в 1 л воды и устанавливают титр по стандартному образцу (нормали) серебра или по титрованному раствору цитрата серебра методом, описанным ниже, после чего раствор разбавляют до точно децинормального.
Для приготовления раствора нитрата серебра помещают 10,788 г химически чистого серебра в коническую колбу, вставляют в ее горло воронку для предотвращения механических потерь и обрабатывают серебро разбавленной азотной кислотой, не содержащей хлоридов. По окончании растворения кипятят для удаления окислов азота, разбавляют водой и, наконец, доводят объем раствора точно до 1000 мл водой при температуре, при которой будут с этим раствором работать. Если имеется нитрат серебра известной чистоты, то растворяют 16,989 г этой соли (измельченной и высушенной при 130° С в течение 1 ч) в воде и раствор разбавляют до нужного объема. Если же чистота взятой соли неизвестна, то титр раствора может быть установлен весовым способом (см. стр. 237), а затем раствор разбавлен точно до децинормального.
Раствор железных квасцов приготовляют, растворяя достаточное-количество соли для получения насыщенного раствора и прибавляя затем чистой бесцветной азотной кислоты до исчезновения бурой окраски. Все описанные растворы стойкие.
Ход определения. Приготовляют 200 мл разбавленного (приблизительно 5 : 95) азотнокислого раствора, содержащего около 0,3 г серебра и не содержащего перечисленных выше мешающих определению веществ. 'Прибавляют 4 мл раствора железных квасцов и затем по каплям приливают чистую азотную кислоту, пока раствор не станет практически бесцветным, после чего титруют раствором роданида до не исчезающей при сильном взбалтывании слабо-розовой окраски. Приливать раствор роданида надо осторожно, так как к концу титрования окраска исчезает медленно. В сомнительных случаях к оттитрованному раствору следует
1 L. W. Andrews, J. Am. Chem. Soc., 29, 275 (1907).
240
Гл. VII. Серебро
прибавить отмеренное количество титрованного раствора нитрата серебра и снова оттитровать раствором роданида х.
Другие методы
Метод Мора чаще применяется для определения хлоридов (стр. 814), чем для определения серебра. Серебро этим методом определяют следующим способом. К нейтральному раствору нитрата серебра прибавляют при перемешивании отмеренный объем титрованного раствора хлорида натрия так, чтобы ввести избыток его. Затем прибавляют раствор хромата калия в качестве индикатора и в заключение — титрованный раствор нитрата серебра, так же как и при определении хлоридов (стр. 814), до появления устойчивой красноватой окраски хромата серебра, не исчезающей при сильном перемешивании. Метод Мора менее точен и удобен, чем метод Фольгарда.
Серебро можно вполне удовлетворительно выделить электролизом из азотнокислого 1 2 аммиачного 3 и цианидного 4 раствора. При электролизе выделяются целиком или частично и многие другие элементы, и потому электролитический метод мало пригоден для обычных анализов, так как перед электролизом надо провести большое число предварительных разделений.
Очень малые количества серебра (1—2 мг/л) удобно определять нефе-„лометрическим (стр. 175) или колориметрическими методами 5.
*Из колориметрических методов определения серебра, по-видимому, наилучшим является дитизоновый 6, подробно описанный в его нескольких вариантах в руководстве Е. Б. Сендэла 7. Определению серебра этим методом мешают только палладий, золото, ртуть и большие количества меди.
1 О титровании серебра иодидом калия в сернокислом растворе, содержащем сульфат церия (IV) и аммония, а также крахмал, см. А. В I о о m, W. М. Me Nab b, Ind. Eng. Chem., Anal. Ed., 8, 167 (1936); H. R obinson иН. Hugg (там же, 9, 565 (1937)] рекомендуют потенциометрическое титрование иодидом калия с применением пары электродов (одного из золота, второго — угольного, насыщенного азотной кислотой), присоединенных к микроамперметру. Авторы утверждают, что за исключением палладия ни один из элементов, обычно присутствующих в серебряных сплавах, не мешает.
2 С. Luckow, Z. anal. Chem., 19, 15 (1880); F. W. К ii S t e г, H. Stein-w e h r, Z. Elektrochem., 4, 451 (1898); L. G. К о 11 о c k, E. F. S m i t h, J. Am. Chem. Soc., 27, 1536 (1905).
3 C. L u с k о w, цит. выше; J. К г u t w i g, Ber., 15, 1267 (1882); F. A. G о о c h, J. P. F e i s e r, Am. J. Sci., [4], 31, 109 (1911).
4 C. Luckow, Dinglers Polytech. J., 178, 42 (1865); Z. anal. Chem., 19, 15 (1880); Chem. News, 41, 213 (1880); F. A. Gooch, H. E. M e d w a v, Am. J. Sci., 4, 15, 320 (1903).
5 G. S. Whitby, Z. anog. Chem., 67, 62 (1910); W. В 6 t t g e r, Z. angew. Chem., 25, 1992 (1912). Об объемных и колориметрических методах определения малых количеств серебра «0,1 мг) в присутствии свинца, цинка, кадмия, мышьяка, сурьмы, висмута, меди, ртути и золота с помощью дитизона (дифенилтиокарбазона) см. в статье: Н. Fischer, G. L е о р о 1 d i, Н. von U s 1 а г, Z. anal. Chem., 101, 1 (1935).;
6H. Fischer, G. Leopold i, H. von U s 1 a r, Z. anal. Chem., 101. 1 (1935).
7 E. Б. Сендэл, Колориметрическое определение следов металлов, Госхим-изат, 1949. стр. 452. См. также Р. И. Алексеев, Зав. лаб., 7, 415 (1938).
Методы определения .
241
Предложен также колориметрический метод определения серебра с применением о-толидина 1 (1 % -ный раствор в этиловом спирте) в ацетатном буферном растворе (pH = 4). Окрашивание голубое.
В качестве специфического реактива для открытия серебра рекомендуется 2 формазилкарбоновая кислота, которая с серебром в уксуснокислых растворах образует окрашенное в бордово-красный цвет соединение. Открытию серебра не мешают барий, кальций, магний, бериллий, алюминий, железо (II и Ш), марганец, никель, кобальт, торий, таллий (I и III), индий, цинк, кадмий, свинец, ртуть (I и II), медь (I и II), висмут, уранил, олово (II) и сурьма (III). Доп. ред*
1Л. М. Кульберг и С. Б. Серебряный, Материалы Института химической технологии АН УССР, № 4, 37 (1937).
2 Л. М. К ульберг, А. М. Леднева, ЖАХ, 2, 131 (1947). В статье приводится метод синтеза этого реактива.
16 Заказ 522.
Глава VIII
РТУТЬ
В больших количествах в, природе ртуть не встречается, и общее распространение ее в горных породах, по-видимому, также невелико. Однако при выполнении анализа горных пород ничтожные следы ртути легко можно пропустить, и поэтому возможно, что распространение ее в земной коре больше, чем думают. Ртуть встречается изредка в самородном состоянии, но обычно ее находят в месторождениях в виде сульфида ртути (II) — к и н о в а р л (HgS). Менее часто она встречается в виде окиси, хлоридов, селенида и теллурида.
ОБЩИЕ ЗАМЕЧАНИЯ /
Присутствие ртути не вносит затруднений в обычный ход анализа горных пород. Ojia может быть не обнаружена вследствие летучести ее соединений. ‘
РАЗЛОЖЕНИЕ СОЕДИНЕНИЙ РТУТИ
Следует помнить, что ртуть полностью улетучивается при прокаливании и сплавлении ее соединений с содой и частично улетучивается, когда растворы солей двухвалентной ртути, особенно хлорида ртути (II), выпаривают на водяной бане или кипятят. Величина потери от выпаривания досуха на водяной бане зависит больше от формы сосуда, чем от концентрации ртути в растворе х. Было найдено, например, что при выпаривании одинаковых объемов раствора хлорида ртути (II) одной и той же концентрации в большой чашке улетучилось 40% ртути, в маленькой чашке улетучилось 15,8%, а при выпаривании в стеклянной колбе — только 5,3%. Потеря ртути при кипячении растворов менее велика. В одном из опытов 1 2 было найдено, что при отгонке 150 мл из 300 мл раствора хлорида ртути (II) в течение 40—45 мин потеря равнялась 0,25 мг HgCh, когда первоначальная концентрация раствора была равна 0,1 %, и 2,2 мг, если был взят 1 %-ный раствор.
При анализе минералов ртуть обычно определяют отгонкой ее из отдельной навески пробы. Для определения остальных составных частей берут вторую навеску, при анализе которой уже не обращают внимания
1 Е. Esteve, Chem. Ztg., 35, 1152 (1911).
а A. Minozzi, Bull. chim. farm., 43, 745 (1904).
Разложение соединений ртути
243
на присутствие ртути. При отгонке ртути ее поглощают металлическим серебром или золотом или же собирают под водой. В первом случае увеличение в массе серебра или золота очень точно показывает содержание ртути в анализируемом минерале Ч Во втором случае ’сконденсировавшуюся под водой ртуть надо отделить, высушить и взвесить или, лучше, растворить в азотной кислоте и оттитровать, как описано на стр. 248-Если ртуть находится в виде хлорида, иодида или сульфида, то для предотвращения частичного ее улетучивания р виде этих солей без разложения должны быть приняты специальные меры.
Общий метод заключается в следующем. Берут трубку для сжигания длиной приблизительно 60 см и внутренним диаметром 1,5 см, один ее конец вытягивают и сгибают под прямым углом так, чтобы образовался отвод с узким выходным отверстием и длина оставшейся горизонтальной части была около 55 см. В горизонтальную часть трубки на расстоянии около 5 см от места изгиба помещают слой асбеста в 2 см и вводят затем последовательно туго скатанную медную сетку длиной 2 см, еще слой асбеста длиной 2 см, слой свежепрокаленной и измельченной в порошок окиси кальция длиной 15 см, слой тщательно перемешанной смеси 1 г анализируемого минерала, 1 г окиси меди и 0,5 г окиси кальция (всея виде тонкого порошка); последний слой должен занять около 0,5 см по длине трубки. Далее следует слой в 5 см окиси кальция, которой проводили «сухую промывку» ступки, где измельчалась навеска анализируемого минерала, затем еще слой окиси кальция в 5 см, слой асбеста в 1 см и скатанная медная сетка длиной 1 см. Наполненную таким способом трубку ставят в горизонтальное положение, слегка постукивают, чтобы обеспечить свободный проход газа через нее, и вводят в печв так, чтобы обе медные сетки лежали в нагреваемой области. Соединяют конец для ввода газа с баллоном, содержащим СО2, а конец для выхода газов соединяют с двумя последовательно стоящими маленькими колбами, содержащими: первая — воду, а вторая — разбавленную (1:1) азотную кислоту, налитые в таком количестве, чтобы отводные трубки были только чуть-чуть погружены в жидкость. Пропускают через прибор ток сухой двуокиси углерода со скоростью около 3 пузырьков в секунду до удаления большей части.воздуха и затем, продолжая пропускать газ, постепенно нагревают трубку для сжигания, начиная от выводного конца и затем распространяя нагрев на заднюю часть трубки, пока вся трубка не прогреетсй равномерно приблизительно до 700° С. В заключение осторожно нагревают изогнутый выводной конец голым пламенем, для того чтобы окончательно отогнать всю ртуть. При этом надо следить за тем, чтобы не опалить пробку. По окончании отгонки трубку охлаждают, удаляют вторую колбу, отрезают согнутую часть трубки для сжигания и смывают приставшую к ее стенкам ртуть в колбу. Удаляют пробку и трубки из второй склянки, нагревают азотную кислоту до 70—80° С и раствор, переливают через согнутую трубку в первую колбу, ополаскивая трубки, после чего их удаляют. Раствор нагревают, прибавляя, если необходимо для растворения ртути, еще азотной кислоты, и дальше поступают, как указано ниже (стр. 248).
1 Об этих методах см. А. Е s с h k a, Dinglers Polytech. J., 204, 47 (1872);
(1906) С h s Trans. AIME* 28$ 444 (1898); G. T. Holloway, Analyst, 31, 66
16*
244
Гл. VIII. Ртутъ
Содержание хлорида ртути (I) и общее содержание ртути в рудах, в состав которых входят каломель и оксихлориды ртути, можно определить 1 в одной навеске пробы отгонкой в стеклянной трубке, в которой выделяющиеся пары сначала соприкасаются с карбонатом натрия, а потом — с предварительно взвешенной золотой фольгой. Выделяющийся при отгонке хлор задерживается карбонатом натрия в виде хлорида натрия, который зат^м превращают в хлорид серебра и пересчитывают на хлорид ртути (I), а пари металлической ртути задерживаются золотой фольгой, которую потом взвешивают.
Природный или свежеосажденный сульфид ртути (II) можно разложить, не вводя в раствор хлора, следующим способом 2. Навеску сульфида ртути помещают в маленькую кьельдалевскую или коническую колбу и вставляют в колбу воронку с коротко, обрезанной трубкой, затем вливают 5 мл серной кислоты и перемешивают. После этого добавляют маленькими порциями при сильном перемешивании от 0,5 до 1 г кристаллического перманганата калия, вливают еще 5 мл серной кислоты, обмывая ею стенки колбы, и дают постоять до видимого прекращения реакции, время от времени взбалтывая содержимое колбы. Приблизительно через 30 мин начинают постепенно нагревать колбу, все время сильно взбалтывая ее содержимое. Наконец нагревают до кипения. Если внешний вид смеси в колбе будет указывать на неполное разложение сульфида ртути (II), колбу охлаждают, прибавляют еще перманганата калия, дают постоять и снова нагревают. Затем снимают с огня, и не охлаждая, добавляют маленькими порциями щавелевую кислоту, пока вся двуокись марганца не будет восстановлена и не перейдет в раствор. Затем снова нагревают до появления паров серной кислоты, чтобы разрушить таким образом избыток 'щавелевой кислоты, охлаждают, разбавлют водой до 100 мл и продолжают, как описано на стр. 248.
Руды и органические вещества, содержащие ртуть, можно разложить следующим образом. Навеску пробы смешивают с 2—4-кратным количеством по массе йодноватой кислоты (частично обезвоженной при 95° С), переносят в трубку для сжигания, снабженную охлаждаемым водой конденсатором и перегородками у выходного конца, замедляющими прохождение воздуха, и осторожно нагревают, медленно пропуская воздух, пока не Прекратится реакция. Образовавшийся красный иодид ртути (II) вместе с избытком иода затем растворяют в растворе тиосульфата натрия и осаждают ртуть сероводородом 3.
Органические вещества также разложить нагреванием до 270° С с азотной кислотой по методу Кариуса 4 или с серной кислотой при добавлении перманганата или дымящей азотной кислоты в колбе Кьель-даля 5.
1 J. J. Fahey, Ind. Eng. Chem., Anal. Ed., 9, 477 (1937).
2 A. H. Low, Chemist Analyst, 29, 13 (1920); О. T о m i ce k, Chem. Ztg., 49., 281 (1925).
3 J. Sandilands, Analyst, 65, 13 (1940).
4 O. Tomicek, цит. выше. He следует вводить более 4 г азотв:ой кислоты в трубку емкостью 50 мл во избежание взрыва [С. R. F г е s е n i u s, А. I. С о h n, Quantitative Chemical Analysis, vol. II, J. Wiley and Sons, 1904, p. 118; A. W. В e s fa-get о о r, Ind. Eng. Chem., Anal. Ed., 1, 92 (1929)].
5 E. R и p p, Chem. Ztg., 32, 1077 (1908); LA. К о t e n, R. A d a m s, J. Am, hem. Soc., 46, 2769 (1924).
Методы отделения
245 -
О II
Разложение соединений ртути типа R—С—S—HgR удобно проводить в конической колбе с коротко обрезанной воронкой, вставленной в ее горлышко. Сначала пробу нагревают с 10 мл дымящей серной кислоты (7 % SO3) и 5 мл дымящей азотной кислоты, а затем, продолжая нагревание, добавляют маленькими порциями 10 мл воды х, после чего содержимое колбы слегка взбалтывают; должен получиться прозрачный раствор. Затем прибавляет в избытке насыщенный раствор перманганата для окисления азотистой кислоты и ртути (I) и некоторое количество сульфита железа (II) для восстановления избытка перманганата. В заключение раствор титруют, как указано на стр. 248.
Такая обработка не годится, однако, для соединений типа RHgX (где X — галоген, a R — органический радикал) вследствие летучести галогенидов ртути. В этом случае рекомендуется следующий ход анализа. Навеску в 0,2—0,3 г пробы помещают в коническую колбу емкостью 300 мл, прибавляют 40 мл ледяной уксусной кислоты, обрабатывают 2 мл брома и дают постоять 20 мин. Вставляют в горло колбы маленькую воронку с обрезанной трубкой, прибавляют 3 мл соляной кислоты и восстанавливают ртуть, прибавляя небольшими порциями цинковую пыль при температуре ниже 50° С. По-исчезновении окраски брома прибавляют избыток цинковой пыли и дают раствору стоять 3 ч или, лучше, оставляют его на ночь. Затем прибавляют 0,5 г тонко измельченного силикагеля для удаления коллоидной ртути и декантируют при слабом отсасывании через тигель Гуча, в который предварительно укладывают довольно толстый слой асбеста, покрытый слоем тонко измельченного силикагеля. Осадок несколько раз промывают декантацией и затем переносят обратно в колбу или весь тигель или же только слой асбеста с находящимся на нем осадком и обмывают тигель над колбой 20 мл разбавленной (Г: 1) азотной кислоты. По окончании реакции прибавляют 10 мл азотной кислоты, слабо нагревают до растворения всей ртути и приливают концентрированный раствор перманганата до появления розовой окраски, не исчезающей в течение 5 мин. Удаляют избыток перманганата, прибавляя по каплям свежеприготовленный раствор сульфата железа (II), и далее ведут анализ, как описано на стр. 248.
МЕТОДЫ ОТДЕЛЕНИЯ
Как уже было сказано, при* определении ртути в минералах ее отгоняют; если же хотят определить остальные элементы, помимо ртути, то анализ проводят так, как если бы ртути не было. Следовательно, методы отделения в количественном анализе не имеют большого значения.
Осаждение сероводородом (стр. 83) служит для отделения ртути от элементов последующих групп, за исключенйем цинка и таллия, которые частично выпадают вместе с ртутью в виде смешанных сульфидов 1 2. Ртуть
1 I. А. К о t е n, Chemist Analyst, 46, 8 (1937).
2 Согласно F. F е i g 1 [Z. anal. Chem., 65, 37 (1924)], при пропускании сероводорода в раствор HgCl2 и ZnCl2 в сильной минеральной кислоте может выпасть до 3% присутствующего цинка в виде смешанного сульфида ртути и цинка.
246
Гл, VIII. Ртуть
образует смешанные сульфиды также с.кадмием1, медью 2 и оловом3, осложняя, таким образом, методы анализа, основанные на предположении о выделении чистого сульфида ртути (II).
В отсутствие олова и меди ртуть можно удовлетворительно отделить от мышьяка и сурьмы обработкой сульфидов горячим полисульфидом аммония (желтым). Придакой обработке в раствор переходят только следы ртути. Растворимость сульфида ртути >(11) увеличивается, если обработку проводить холодным раствором или пользоваться бесцветным сульфидом аммония вместо желтого полисульфида. В присутствии олова часть его остается в осадке вместе со ртутью, а часть ртути переходит в раствор вместе с оловом 4. Сульфид меди также растворяется до некоторой степени, и весьма вероятно, что он увеличивает растворимость сульфида ртути (II).
Ртуть может быть успешно 4 отделена от серебра и свинца 5 6 (но не от кадмия и смешанного сульфида ртути и цинка) нагреванием сульфидов® со смесью 15%-ных растворов сульфида калия и едкого кали, взятых в равных количествах. Для этого сульфиды осаждаю? сероводородом из горячего солянокислого раствора хлоридов, содержащего свободную-НС1 в большой концентрации, и постепенно разбавляют горячей водой, пока раствор не будет содержать 1% свободной соляной кислоты по объему. Фильтруют, осадок промывают подкисленной сероводородной водой и смывают его в стакан горячей водбй, содержащей равные количества едкого кали и сульфида калия. Прибавляют по меньшей мере по 10 мл указанных выше растворов едкого кали и сульфида калия на каждую 0,1 г ртути, кипятят 1—2 мин, разбавляют горячей водой и фильтруют. Остаток промывают водой, содержащей несколько капель раствора K2S + КОН. Фильтрат, содержащий ртуть, обрабатывают 20%-ным раствором нитрата аммония 7 до прекращения выделения осадка, нагревают на водяной бане 30 мин и фильтруют. Осадок промывают водой, содержащей немного сульфида аммония, и далее поступают, как указано на стр. 248 или на стр. 249.
Как ранее было сказано, разделение этим способом не приводит к удовлетворительным результатам, если присутствуют кадмий, цинк или олово. При отсутствии олова ртуть полностью отделяют от мышьяка и сурьмы вторичным осаждением.
1 Ф. Файгль утверждает, что смешанный сульфид кадмия и ртути образуется при такой кислотности, при которой один кадмий не выпадает.
2 F. F е i g 1, цит. выше.
3 Т. Вильм, ЖРФХО, 1, 60 (1887); Вег., 20, 232 R (1887).
4 К. Bulow, Z. anal. Chem., 31, 697 (1892); Chem. News, 67, 174 (1893).
5 К. Вюлов нашел, что отделение ртути от висмута и меди протекает также удовлетворительно. Едва ли это можно считать правильным, так как известно, что ртуть образует смешанный сульфид с медью, а сульфид висмута значительно растворим в смеси Na2S (или K2S) и NaOH (или КОН).
6 Для приготовления раствора сульфида калия берут требуемое количество 15%-ного раствора едкого кали, насыщают сероводородом, разбавляют равным количеством указанного раствора едкого кали, слегка нагревают, дают отстояться, сифонируют и сохраняют прозрачный раствор в закрытой пробкой колбе. Для приготовления раствора едкого кали несколько капель раствора сульфида калия прибавляют к другой порции 15%-ного раствора едкого кали,- и раствор после слабого нагревания и отстаивания сифонируют в другую колбу.
7 Раствор нитрата аммония приготовляют, растворяя 20 г сухой соли в 100 мл воды, прибавляя затем несколько капель раствора сульфида аммония и фильтруя через 12 ч. '
• Методы отделения 247
Для отделения ртути от большинства элементов группы сероводорода часто применяется метод, основанный на нерастворимости сульфида ртути в кипящей разбавленной азотной кислоте (пл. 1,2—1,3 г/см3). Отделение это не удается, если сульфид ртути был осажден в растворе, содержащем медь, кадмий или цинк, или в присутствии соляной кислоты и хлоридов. В последнем случае надо принять меры К превращению двойного' соединения состава 2HgS • HgCl2 в сульфид ртути; кроме того, перед разделением надо отмыть осадок от соляной кислоты и хлоридов.
Для выполнения этого разделения сначала осаждают сульфид ртути из сернокислого или солянокислого раствора, в котором вся ртуть должна 'быть в двухвалентной форме, затем фильтруют, промывают осадок подкисленной сероводородной водой до полного удаления хлоридов, переносят его в фарфоровую чашку, прибавляют 50 мл разбавленной азотной кислоты (пл. 1,2—1,3 г/см3), покрывают часовым стеклом и слабо кипятят около 30 мин. Затем приливают 100 мл воды, фильтруют и промывают остаток горячей разбавленной (5 : 100) азотной кислотой. Кроме сульфида ртути и серы, остаток содержит также И сульфат свинца, если свинец присутствовал в значительном количестве. Для отделения свинца этот остаток растворяют при слабом нагревании в царской водке в покрытом часовым стеклом сосуде, разбавляют водой, фильтруют и промывают остаток, содержащий большую часть сульфата, свинца. Перешедший в раствор свинец отделяют от ртути осаждением сульфидов и нагреванием осадка со смесью едкого кали и сульфида калия, как было описано выше.
Серебро можно отделить от ртути (II) осаждением его соляной кисло-/той (стр. 236), декантацией раствора через фильтр, нагреванием осадка хлорида серебра с небольшим количеством разбавленной (1 : 2) азотной кислоты и втрричным осаждением серебра добавлением воды и нескольких капель соляной кислоты. Ртуть затем определяют в соединенных фильтратах.
Восстановление ртути (II) фосфористой кислотой в солянокислом растворе, как указано на стр. 251, является хорошим способом отделения ртути от, кадмия, меди, цинка и, с некоторыми видоизменениями, от висмута, сурьмы, селена и теллура. Отделение это не является совершенным вследствие некоторой растворимости хлорида ртути (I). При отделении висмута для предупреждения гидролиза необходимо прибавление достаточного количества соляной кислоты. Для предупреждения гидролиза соединений сурьмы рекомендуется прибавить 3—5 г винной кислоты. В присутствии же селена и теллура лучше всего осадить все сульфиды, растворить их при слабом нагревании в царской водке, затем прибавить хлорную воду, разбавить до 1000 мл и добавить фосфористую кислоту; после этого следует дать постоять по меньшей мере 24 ч и отфильтровать хлорид ртути (I).
Галлий не осаждается вместе со ртутью при обработке сероводородом, если эта обработка вначале проводится в достаточно кислом растворе и заканчивается постепенным разбавлением водой.
Ртуть от таллия лучше всего отделять осаждением последнего иодидом калия, добавляемым в избытке, как описано в гл. «Таллий» (стр. 541). Таллий частично увлекается ртутью при обработке солей обоих элементов сероводородом в кислом растворе.
248
Гл. VIII. Ртуть
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Определение ртути титрованием ее роданидом аммония является наилучшим во всех случаях, когда ртуть может быть переведена в азотнокислый или сернокислый раствор без введения хлора и без потери ее от улетучивания. При анализе руд очень удобным методом является возгонка ртути с последующим растворением ее в азотной кислоте, как описано на стр. 243. Сульфидный метод точен, но в отличие от предыдущего его не всегда можно применять. Определение ртути в виде хлорида ртути (I) дает пониженные результаты, но для выполнения рядовых анализов метод удобен.
Объемное определение титрованием роданидом аммония
Нитрат ртути (II), подобно нитрату серебра, образует при добавлении роданида аммония нерастворимый роданид ртути (II) и потому может быть оттитрован способом, аналогичным описанному для серебра (стр. 238). В отличие от нитрата хлорид ртути (И) не реагирует с роданидом, следовательно, метод неприменим в присутствии соляной кислоты или хлоридов. Это составляет главное препятствие его широкому применению, так как при обычных методах разложения руд требуется применение соляной кислоты, которую потом нельзя удалить выпариванием вследствие летучести хлорида ртути (II). Соли ртути (I) также должны отсутствовать \ так как они реагируют с растворимыми роданидами, образуя металлическую ртуть и роданид ртути (II).
Роданидный метод точен и особенно интересен тем, что может применяться в присутствии элементов, трудно отделяемых обычными методами: свинца, меди, висмута, кадмия, олова, мышьяка, сурьмы, таллия, железа, цинка, марганца, никеля и кобальта. Меры предосторожности, указанные при описании титрования серебра (стр. 239), следует применять и при определении ртути.
Ход определения. Приготовляют раствор солей ртути (II), содержащий приблизительно 10 мл азотной кислоты и 5 мл верной кислоты в 100 мл и не содержащий серебра, палладия и хлора. Чтобы быть уверенным в отсутствии азотистой кислоты и ртути (I), прибавляют по каплям и при перемешивании 5%-ный раствор перманганата до появления розовой окраски или осадка двуокиси марганца, не исчезающих в течение 5 мин. Избыток перманганата разрушают осторожным прибавлением мелких кристаллов сульфата железа (II), раствора этой соли или разбавленной перекиси водорода. Все эти реактивы должны быть свободными от хлора. Затем прибавляют 2 мл насыщенного раствора железных квасцов на каждые 100 мл анализируемого раствора и титруют 0,1—0,01 н. раствором роданида аммония, в зависимости от, содержания ртути, до не исчезающей при сильном взбалтывании заметной розовой окраски. Следует остерегаться ошибки от недотитрования, так как окраска к концу титрования исчезает медленно 1 2.
1 Е. Rupp, Chem. Ztg., 32, 1077 (1908).
2 Точность метода может быть иллюстрирована следующими результатами, полученными Н. В. Knowles путем растворения навески чистой сухой ртути и анализа полученного раствора приведенным выше методом:
Взято ртути, г ... 0,0517 0,1033 0.1550 Найдено ртути, г . . 0,0518 0,1032 0,1552
Методы, определения 249
Раствор железных квасцов и титрованный раствор роданида аммония приготовляют, как описано при определении серебра (стр. 239). Титр роданида аммония должен быть установлен прямым титрованием по навеске чистой сухой ртути, которую растворяют в горячей разбавленной (1 : 1) азотной кислоте, и полученный раствор анализируют описанным выше методом х.
Весовые определения
Определение в виде сульфида ртути (II). Весовое определение ртути в виде сульфида ртути (II) является точным методом, но метод этот менее пригоден, чем предыдущий, так как ртуть должна быть предварительно отделена от всех остальных элементов группы сероводорода, и если применяется метод Фольгарда, то и от элементов, осаждающихся сульфидом аммония. К этому надо добавить, что осадок.сульфида ртути (II) увлекает с собой серу, которая должна быть удалена перед взвешиванием. Осаждение сульфида ртути (II) обработкой сульфосоли нитратом аммония 1 2 протекает быстрее, чем прямое осаждение сероводородом в кислом растворе, и имеет те преимущества, что может проводиться в присутствии окислителей, например азотной кислоты, и дает возможность отделить от ртути серебро, свинец, висмут, Мышьяк и сурьму. Метод этот не удается при анализе растворов, содержащих цинк, кадмий или медь, как указано выше (стр. 246).
Ход определения. Приготовляют кислый раствор соли ртути (II), содержащий не более 0,1 г ртути в 100 мл и свободный от кадмия, цинка, олова и от элементов, подобных алюминию, образующих соли, растворимые в растворе сульфида натрия и осаждаемые сульфидом аммония. Анализируемый раствор нейтрализуют почти полностью раствором чистого карбоната натрия, обрабатывают свежеприготовленным сульфидом аммония 3, добавляя его в небольшом избытке, и затем приливают при сильном перемешивании 10%-ный раствор едкого натра 4 до начала осветления раствора. Тогда нагревают до кипения и добавляют еще раствор едкого натра, пока анализируемый раствор не станет совсем светлым, что указывает на переход всей ртути в сульфосоль. Если раствор не совсем прозрачен, его фильтруют и промывают остаток горячей водой, содержащей по 10 мл указанных выше растйоров едкого кали и сульфида калия на 1 л. К фильтрату постепенно прибавляют достаточное количество 25%-ного раствора нитрата аммония 5 6 для превращения едкого натра в нитрат натрия и для разложения сульфосоли ртути. (Объем прибавленного раствора нитрата аммония должен быть равен объему ранее
1 I. М. KolthoffnJ. J. Lingane [J. Am. Chem. Soc., 57, 2377 (1935) J указывают на то, что небольшие отклонения от теоретического значения титра происходят в результате побочных реакций.
2 J. Volhard, Ann., 255, 255 (1889).
3 Получается насыщением трех объемов аммиака (пл. ~0,9 г/см3) сероводородом и прибавлением двух объемов аммиака той же концентрации. Раствор оставляют стоять в закрытой пробкой колбе и в случае выпадения осадка сифонируют.
4 Свободного от-элементов, осаждаемых сульфидом аммония (железо, алюминий,
кремний).
6 Освобожденного от элементов, осаждаемых сульфидом аммония, для чего раствор нитрата аммония обрабатывают несколькими каплями этого реактива и фильтруют после отстаивания в'.течение ночи.
250
Гл. VIII. Ртуть
прибавленного раствора едкого натра). Раствор кипятят для удаления большей части аммиака и дают осадку отстояться. Фильтруют через взвешенный тигель Гуча и промывают осадок последовательно сероводородной водой, горячей чистой водой, спиртом, сероуглеродом, снова спиртом и, наконец, эфиром.
Эта обработка имеет целью удаление свободной серы, находящейся в осадке. Для получения осадка, не содержащего серы или содержащего ее лишь немного, надо перед обработкой нитратом аммония прибавить несколько миллилитров раствора бисульфита аммония. Из таких сульфидных осадков свободную серу можно также удалять экстрагированием сероуглеродом следующим способом. После промывания осадка сульфидов спиртом помещают тигель на треугольной стеклянной подставке в стакан, содержащий 25—50 мл сероуглерода. Накрывают стакан круглодонной колбой, наполненной холодной водой, и ставят на горячую водяную баню. В течение часа осадок сульфидов обрабатывается сероуглеродом, конденсирующимся на стенках и дне круглодонной колбы и падающим каплями. Затем тигель удаляют, извлекают сероуглерод из осадка в тигле, промывая его небольшим количеством спирта, и высушивают.
Промытый осадок высушивают при 105—110° С и взвешивают в виде HgS.
Вместо растворения серы, находящейся в осадке сульфида ртути (II) (или присутствующей в продажной киновари, если анализируется последняя), можно 1 поступать в обратйом порядке: растворять сульфид ртути (II) и оставлять нерастворенйой элементарную серу. В этом случае осадок сульфида ртути (II) собирают во взвешенном стеклянном или фарфоровом фильтрующем тигле, промывают холодной водой, тщательно высушивают при 110° С и взвешивают. Затем снова вставляют тигель в его держатель, не приводя в действие насоса для отсасывания, и приливают холодную стабилизированную (добавлением фосфорноватистой кислоты НаРО2) иодистоводородную кислоту с постоянной температурой кипения в количестве 5 мл на 1 г осадка. Смесь перемешивают стеклянной палочкой, пока не исчезнут все черные частички сульфида ртути (II), включают отсасывание и пропускают раствор через тигель, рстаток элементарной серы в тигле промывают сначала тремя-четырьмя последовательными порциями по 5 мл разбавленной (5—10%-ной) иодистоводородной кислоты, а затем холодной водой. Тигель с его содержимым после высушивания в течение 2 ч в вакуум-эксикаторе взвешивают. Разность между результатами обоих взвешиваний показывает содержание чистого сульфида ртути (II) в осадке.
Определение в виде хлорида ртути (1). Результаты, получаемые взвешиванием ртути в виде хлорида ртути (I), обычно понижены вследствие некоторой растворимости этого ооединения. Растворимость Hg2CI2 в растворах, содержащих хлор-ионы, больше, чем в чистой воде; в умеренных концентрациях она изменяется в прямой зависимости от количества прибавленного избытка хлорида. Так, например, в 100 мл чистой боды растворимость хлорида ртути (I) равна 2 0,28 мг при 24,6° С; в таком же объеме воды, содержащей 0,585 г хлорида натрия, растворимость при 25° С равна 3 0,41 мг, а в 100 мл воды, содержащей 3,169 г (приблизительно 7 мл) соляной кислоты, растворимость при 25° С равна 3,4 мг.
1 Е. R. С а 1 е у, М. G. В u г f о г d, Ind. Eng. Chem., Anal. Ed., 8, 43 (1936).
2 F. Kohlrausch, Z. phys. Chem., 64, 150 (1908).
3 T. W. flic lia rds, E. H. Archibald, Z. phys. Chem., 40, 385 (1902).
Методы определения
251
Если в начале анализа ртуть находится в виде ионов Hg|+, то при описанном ниже ходе определения нет необходимости прибавлять фосфористую кислоту. Если же последняя прибавлена, то осаждение должно проводиться на холоду, иначе ртуть восстановится до металла. При наличии в растворе азотной кислоты большая часть ее должна быть нейтрализована карбонатом натрия перед прибавлением соляной кислоты, и осаждение следует проводить в растворе, в 3—4 раза более разбавленном, чем указано в ходе определения.
Ход определения. Приготовляют солянокислый раствор солей ртути (II), свободный от соединений, образующих осадок с фосфорной кислотой. Раствор разбавляют так, чтобы он содержал приблизительно по 1 мг ртути и соляной кислоты в 1 мл, причем он должен остаться прозрачным. Раствор охлаждают до 20—30° С, прибавляют по 1 мл 50 %-ного раствора фосфористой кислоты на каждые 100 мл анализируемого раствора и оставляют стоять на 12 ч или более. Фильтруют через взвешенный тигель Гуча осадок, умеренно промывают водой, Высушивают при 100—105° С и взвешивают в виде Hg2Cl2. '
Определение в виде металлической ртути. В описываемом ниже методе ртуть взвешивают в виде металла после восстановления ее хлоридом олова (II) в солянокислом растворе. По данным авторов х, железо, кадмий, висмут, медь, свинец, сурьма, нитраты и сульфаты определению не мешают.
Ход определения. К 50 мл холодного нейтрального анализируемого раствора прибавляют 25 мл соляной кислоты и 5 мл свежепрофильтрован-ного раствора хлорида олова (II) [содержащего 1,125 г SnCl2 • 2Н2О в 1 мл НС1 (пл. 1,09 г/см3) и сохраняемого над металлическим оловом]. Перемешивают и дают осадку осесть в течение 1 ч или центрифугируют. Затем осадок отфильтровывают через взвешенный фарфоровый фильтрующий тигель, предварительно высушенный пропусканием через него ацетона и сухого воздуха, умеренно промывают его разбавленной (1 : 1) соляной кислотой, затем водой и, наконец, ацетоном, после чего высушивают в течение 5 мин просасыванием через тигель сухого воздуха и взвешивают. Увеличение в массе тигля показывает содержание ртути в пробе. К этой величине прибавляют еще 1 мг — поправку на потерю ртути вследствие улетучивания (см. стр. 242).
• Объемное или весовое определение в виде перйодата ртути 1 2
В этом методе ртуть (II) осаждают в виде перйодата ртути Hg5(IOe)2 из 0,15 н. азотнокислого или 0,1 н. сернокислого раствора и затем или взвешивают в виде этого соединения или титруют иодометрическими методами. Умеренные количества алюминия, кадмия, цинка, меди, никеля, кальция и магния не мешают. Железо мешает, так как оно осаждается в виде перйодата железа (III). Хлориды и другие галогениды должны отсутствовать, потому что они препятствуют полному осаждению ртути.
Осаждение ртути. Раствор, содержащий не более 0,5 г ртути в 150 мл 0,15 н. азотной кислоты или 0,1 н. серной кислоты, нагревают до кипения и добавляют медленно и при постоянном перемешивании
1 Н. Н. Willard, A. W. В о 1 d у г е f f, J. Am. Chem. Soc., 52, 573 (1930).
2 H. H. Willard, J. J. Thompson, Ind. Eng. Chem., Anal. Ed 3, 398 <1931).
252
Гл. VIII. Ртуть-
раствор 2 г перйодата калия в 50 мл воды. Затем охлаждают, осадок отфильтровывают через фильтрующий тигель с пористой стеклянной пластинкой или с фарфоровым пористым дном и промывают горячей водой.
Весовой метод. Промытый осадок высушивают при 100° С в течение 2—3 ч и взвешивают в виде Hg5(IO6)2.
Объемные методы. Титрование тиосульфатом натрия. Промытый осадок в фильтрующем тигле обрабатывают 2—3 г твердого иодида калия и 10—15 мл воды и перемешивают до растворения всего перйодата ртути (II), собирая раствор в колбу для отсасывания емкостью 150 мл. Раствор подкисляют 10 мл 2 н. соляной кислоты и титруют 0,1 н. раствором тиосульфата натрия с применением крахмала в качестве индикатора. Протекающие реакции выражаются следующими уравнениями:
Н£6(1Об)2 + 34I-+24Н+= 5HgPf + 8!а-Н2И2°
8I2 + 16S8O|- = 8S4O®-+16J-
Титрование арсенитом натрия и йодатом калия. Промытый осадок в фильтрующем тигле обрабатывают отмеренным количеством 0,1 н. раствора арсенита натрия, прибавляя его в избытке, и добавляют' 35 мл соляной кислоты, собирая раствор в колбу для отсасывания емкостью 150 мл. Промывают тигель водой, раствор вместе с промывными водами переносят в коническую колбу емкостью 150 мл, снабженную притертой пробкой, разбавляют до 100 мл и титруют 0,1 н. раствором йодата калия, пока цвет раствора не станет светло-коричневым. Затем прибавляют 4—5 мл хлороформа и продолжают титрование, сильно взбалтывая после каждого прибавления реактива, пока пурпурное окрашивание раствора иода в хлороформе не исчезнет. В конце титрования раствор должен содержать, от 28 до 45 мл соляной кислоты в общем объеме 100 мл. Растворение осадка перйодата ртути (II) в растворе мышьяковистой кислоты проходит по уравнению
Hg6(IO6)2 + 6H3AsO3 + 12НС1 = 5HgCl2 + 2IC1 + 6H3AsO4 + 6H2O
Образующийся в результате реакции однохлористый иод — красная жидкость, растворимая в концентрированной соляной кислоте, но нерастворимая в хлороформе, который в присутствии IG1 остается бесцветным. Однохлористый иод при разбавлении раствора гидролизуется с образованием элементарного иода
51С1-ЬЗН2О = 5НС1 + 212 + Н1О3 окрашивающего хлороформ; поэтому концентрация соляной кислоты в растворе во все время титрования не должна падать ниже 12%.
При титровании йодатом калия избытка мышьяковистой кислоты сначала происходит окисление последней, сопровождаемое выделением иода:
5H3AsO3+2HIO3 = 5H3AsO4+Н2О +12
Выделяющийся иод окрашивает хлороформ в пурйурный цвет, который .к концу приведенной реакции достигает максимальной интенсив ности. При дальнейшем титровании иодистоводородная кислота вступает в реакцию с иодом:
212 + НЮ3 + 5НС1 = 51С1 + ЗН2О и окраска хлороформа постепенно исчезает.
Методы определения
253
Складывая уравнения обеих реакций, получаем:
, 10H3AsO3 + 4НЮ3 = 10H3AsO4 + 2Н2О + 212
' 212 + НЮз+5НС1=51С1+ЗН20
IOH3ASO3 + 5Н.10з+5НС1 = 10H3AsO4 + 51 Cl + 5Н2О, или после сокращения:
' 2НзАзОз + НЮз + НС1 = 2НзА5О4+1С1 + Н2О
Эти уравнения показывают, что на 2 молекулы H3AsO3 всего расходуется 1 молекула КЮ3 и что максимальная окраска хлороформа от выделившегося иода достигается тогда, когда расходуется четыре пятых всего требуемого количества йодата калия. Это дает возможность заранее приблизительно рассчитать, когда должен наступить конец титрования, и последние капли титрованного раствора йодата калия «прибавлять с большой осторожностью, основательно встряхивая титруемую жидкость после каждого добавления этого раствора х. Доп. ред*
Определение малых количеств ртути
Концентрирование и открытие малых количеств ртути в некоторых растворах может быть проведено восстановлением порошкообразной медью и последующим амальгамированием на золотой фольге 2. Подкисляют 500 мл анализируемого раствора 10—20 мл соляной кислоты, переливают в большую колбу и обрабатывают 1—2 г медной пыли. Плотно закрыв колбу пробкой, раствор встряхивают*- несколько минут и оставляют стоять на ночь. Затем декантируют прозрачную жидкость, переносят всю медь на фильтр и промывают ее водой, потом спиртом и, наконец, эфиром. Высушив медную пыль на воздухе, вводят ее через воронку с тонкой трубкой в маленькую колбу, следя за тем, чтобы она пё пристала к стенкам колбы. Затем прибавляют немного измельченного в порошок магнезита, вставляют в шейку колбы небольшое количество асбеста и осторожно сужают трубку на 2 см выше шаровидной части колбы. В суженную часть трубки помещают маленькие кусочки листового зубоврачебного золота и постепенно нагревают колбу до красного каления, охлаждая в то же время суженную часть трубки обвертыванием ее влажной бумагой. По данным автора этого метода, амальгамирование, вызываемое даже таким малым количеством, как 0,001 мг ртути, легко видно невооруженным глазом. Меньшие количества ртути могут быть обнаружены при рассматривании точечного амальгамирования через ручную лупу.
При .разложении органических веществ разбавленной (1 : 10) серной кислотой или разбавленной (1:1) азотной кислотой и перманганатом в колбе с обратным холодильником некоторое количество ртути выделяется в коллоидном состоянии и не осаждается сероводородом. Поэтому рекомендуется 3 после разложения обрабатывать раствор сероводородом, затем удалять избыток последнего током воздуха и улавливать коллоидную ртуть обработкой 50%-ным раствором едкого натра до тех пор, пока
1 Подробности различных определений титрованием йодатом калия в сильно -солянокислой среде в присутствии хлороформа см. Ю. Ю. Лурье, Минеральное -сырье, № 7 (1931).
8 J. А. Е 11 i о 11, J. Am. Med. Assoc., 68, 1693 (1917).
3 H. 8, В о о t h, N.; E. Schreiber, K. G, Zwik, J, Am. Chem, Soc., 48, 1815 (1926).
254 Гл. VIII. Ртуть
не образуется слабый, но неисчезающий осадок гидроокиси марганца. Раствор затем оставляют на ночь, после чего осадок отфильтровывают, высушивают, прокаливают в смеси с хроматом свинца в маленькой трубке и выделяющуюся ртуть определяют, собирая ее в капиллярной трубочке (с одинаковым внутренним диаметром по всей длине) и измеряя ее объем х.
Для открытия следов ртути в органических жидкостях рекомендуется также 1 2 применять электролиз с золотым катодом в холодном разбавленном азотнокислом растворе с последующим спектроскопическим исследованием катода в трубке Дюпре. Золотой катод перед употреблением нагревают в электрической печи при 900° С до тех пор, пока спектроскопическое испытание не покажет отсутствия ртути. Таким образом, электролизом 10 мл раствора при силе тока 0,2 а в течение 10—15 мин, может быть.открыта одна часть хлорида ртути (II) в 100 миллионах частей раствора. Взяв для анализа большие количества раствора и сильно перемешивая его при электролизе, можно увеличить чувствительность метода еще в 100 раз. С сильно разбавленными (1 : 10 миллионам) растворами солей ртути, даже подкисленными азотной кислотой, нельзя работать в стеклянной или кварцевой посуде, поскольку ртуть адсорбируется стенками сосуда. Кроме того, заметная потеря ртути наблюдается и при выпаривании в кварцевой чашке, растворов, содержащих ртуть, несмотря на прибавление к ним азотной кислоты, соляной кислоты с перхлоратом калия или серной кислоты с перманганатом калия 3.
Рекомендуется следующий электромикрокачественный метод открытия ртути в отсутствие других веществ, осаждающихся на катоде при электролизе. Анодом служит маленькая платиновая проволока, катодом — заостренный конец маленькой медной проволоки. Каплю раствора помещают на предметное стекло и пропускают через нее ток силой 1,3—1,5 ма при напряжении 1,5—2 в в течение 2—3 мин. Затем медный катод удаляют, промывают водой и его кончик исследуют на присутствие ртути (амальгамирование) или невооруженным глазом, или под микроскопом. Чувствительность метода равна 1 части ртути на 2 миллиона частей раствора (это соответствует одной капле раствора, содержащего 0,5 мг ртути в 1 л); чувствительность может быть повышена до 1 части на миллиард изотермическим концентрированием раствора при комнатной температуре.
При взаимодействии солей ртути (II) с тетрароданодиамминохро-миатом аммония (солью Рейнеке) в 0,1 н. солянокислом растворе образуется бледно-красный объемистый осадок 4 Hg[GNS)4Cr(NH3)2]2. Реакцией можно обнаружить 2,5 мкг ртути в 5 мл раствора, если после добавления реактива дать постоять 2 мин. Мешают золото, серебфо и таллий.
*Полученный осадок можно отфильтровать через асбест или стеклянную пористую пластинку, промыть и растворить-в растворе тиомочевины в метилэтилкетоне (с добавлением, при желании, этанола или аце
1 Н. S. В о о t h, N. Е. S с h г е i b е г, К. G. Z w i k, J. Am. Chem. Soc., 50, 1620 (1928).
8 К. С. В г о wni ng, J. Chem. Soc., Ill, 236 (1917). *Ряд работ по микроэлек-тролитическому выделению следов ртути на золотой или медной проволоке с последующей отгонкой ртути в капиллярной трубке был проведен A. Stock, Вег., 71, 550 (1938); 72, 1844 (1939); Z. angew. Chem., 39, 466, 791 (1926); 41, 429 (1919); 44, 200 (1931); 46, 62, 187 (1938); 47, 641 (1934). См. также А. А. С а у к о в, Н. X. А й д и-н ь я и, ДАН СССР, 32/358 (1941)*.
s Н. S. Booth, N. Е. Schreiber, J. Am. Chem. Soc., 47, 2628 (1925).
4 C. Mahr, Z. anal. Chem., 104, 241 (1936).
Методы, определения
255
тона) х. По окраске полученного раствора содержание ртути можно затем определить колориметрически. Доп. ред*
Рядом авторов 1 2 описаны фотометрические методы определения ртути, основанные на поглощении резонансной волны длиной 2537 А, излучаемой парами ртути.
*Из колориметрических методов определения очень малых количеств ртути, по-видимому, наилучшим методом является дитизоновый, подробно описанный в руководстве Е. Б. Сендэла 3. Перед определением этим методом часто извлекают следы ртути из раствора сероводородом, применяя в качестве коллектора соли мышьяка (V) или кадмия. Выпадающий осадок сульфида мышьяка (V) или сульфида кадмия количественно увлекает с собой сульфид ртути (II). Таким способом можно извлечь 1 мкг ртути из 100 мл раствора. Выбор мышьяка и кадмия из всех других металлов, осаждаемых сероводородом в кислой среде, объясняется тем, что эти металлы не мешают последующему определению ртути дитизоновым методом.
Выделить ртуть можно также, медленно пропуская анализируемый раствО/р (pH которого предварительно доводится до 5—7) через асбест, импрегнированный сульфидом кадмия. Таким способом можно извлечь даже 0,5 мкг ртути из 200 мл раствора. Определению ртути дитизоновым методом мешают медь, золото, палладий, платина (II) и большое количестве серебра. Следует отметить, что при введении в анализируемый раствор комплексона III (этилендиаминтетраацетата натрия) реакция на ртуть с дитизоном становится специфичной — мешает только серебро, которое можно также маскировать добавлением роданида 4 *.
Из других методов колориметрического определения ртути интересен метод с применением реактива, аналогичного дитизону, — ди-Р-наф-тилтиокарбазона 8. Этот реактив более чувствителен, чем дитизон. При его взаимодействии со ртутью получается соединение, окрашивающее слой хлороформа в пурпурно-красный цвет (при использовании дитизона слой хлороформа окрашивается в значительно менее яркий желтый цвет).
Для колориметрического определения ртути применяются также дифенилкарбазид и дифенилкарбазон 6 *. В обоих случаях получается окрашенное в синий или пурпурный цвет производное дифенилкарбавона, переходящее в коллоидный раствор. Определению мешают цинк, свинец, медь, железо, хром, никель и кобальт, от которых ртуть надо предвари-
1 С. М a h г, Z. angew. Chem., 53, .257 (1944).
2 Об определении 0,02—0,60 мкг ртути с точностью ±0,02 мкг в 150—400 раствора см. А. Е. В а 11 а г d, С. D. W. Thornton, Ind. Eng. Chem., Anal. Ed., 13, 893 (1941). В. статьях T. T. W о о dson, Rew. Sci. Instruments, 10, 308 (1939). * V. F. Hanson [Ind. Eng. Chem., Anal. Ed., 13, 119 (1941)]* иМ. Shepherd, S. S c h u h m a n n [J. Research NBS, 26, 358 (1941)] описываются методы открытия очень малых количеств (до 5 мкг) ртути в 1 м3 воздуха.*
3Е. Б. Сендэл, Колориметрическое определение следов металлов, Госхим-издат, 1949, стр. 410. См. также: Р. И. А л е к с е е в, Зав. лаб., 7, 415 (1938); Н. С. К у-з я ♦ и н а, там же, 8, 174 (1939); J. F. Reith, К. W. Gerri tsma, Rec. trav. chim., 64, 41 (19j45); H.I rving, G. Andrew, E. J. Risdon, J. Chem. Soc., 1949, 541.
4 V-. V a s a k, V. S e d i vec, Chem. Listy, 45, 10 (1951); P. П p ш и б и л,
Комплексоны в химическом анализе, Издатинлит,' 1955, стр. 119.
6 А. Е.Ballard, С. D. W. Thornton, Ind. Eng. Chem., Anal. Ed., 13, 893
(1941). И. Б. Супрунович, ЖОХ, 8, 839 (1938). См. также J. С h о 1 a k, D. М. Hubbard, Ind. Eng. Chem., Anal. Ed., 18, 149 (1946).
’ M. P. C a s e n e u v e, C. r., 130, 1478 (1900).
256 Гл. VIII. Ртуть
тельно отделить. Мешают также большие количества электролитов в растворе, так как они вызывают коагуляцию окрашенного соединения.
Для определения ртути в воздухе широкое применение получил метод Н. Г. Полежаева в котором ртуть выделяется в виде комплексного соединения GuHgI3 вместе с белым иодидом меди (I) Gul, образуя суспензию, имеющую окраску от желтовато-розовой до оранжевой. Применяемый реактив содержит хлорид меди (II), сульфит натрия, иодид калия и бикарбонат натрия. Одновременно с анализом пробы приготовляют серию стандартных растворов, с окрасками которых сравнивают окраску анализируемого раствора. Доп. ред*
1 Н. Г. Полежаев, Гигиена и безопасность труда, № 5—6 (1934); Гигиена труда и техника безопасности, 14, № 6, 86 (1936); Гигиена и санитария, № 5, 34 (1946).
Глава IX
СВИНЕЦ
Свинец часто находят в горных породах, если берут для анализа достаточйо большие навески исследуемого материала. Он входит, например, в состав горных пород Ледвилля, Колорадо и Британской Гвианы.
Сульфид свинца — свинцовый блеск, галенит (PbS), нередко встречается в метаморфических известняках, особенно в таких, которые подверглись изменению благодаря вторжению в них изверженных пород. Сульфид свинца вместе с некоторыми другими сульфидами образует руды, имеющие большое промышленное значение. В результате окисления из этих руд образуются в природе столь же или еще более ценные руды, например различные окислы, сульфаты и карбонаты. Самородный свинец встречается редко. Известны, также многие суЛьфороли свинца, силикаты свинца, фосфат свинца, арсенат свинца и несколько ванадатов свинца. Важнейшая руда — галенит — часто встречается вместе с пиритом, марказитом и сфалеритом. Область применения свинца в промышленности очень велика, и методы его определения имеют поэтому большое значение.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализе горных пород присутствие больших количеств свинца узнается по выделению хлорида свинца в солянокислом растворе. При прокаливании с кремнекислотой часть хлорида свинца образует силикат свинца, но большая часть его теряется и при этом возможно разрушение им платины. Свинец, оставшийся в фильтрате после ^осаждения кремнекислоты, будет количественно осажден аммиаком, если имеется значительное количество железа или алюминия. Большая часть этого свинца останется в виде окиси свинца в остатке «полуторных» окислов после прокаливания и будет принята за окись алюминия. Отсюда ясно, что присутствие свинца должно учитываться в начале анализа. При его наличии кремнекислоту и свинец выделяют совместно путем обезвоживания серной кислотой, как указано на стр. 258. Можно также кремнекислоту выделить отдельно обезвоживанием хлорной кислотой (стр. 258), а затем удалить свинец сероводородом. Реже применяют обезвоживание кремнекислоты азотной кислотой, после чего осаждают свинец электролизом в виде его двуокиси. Если кремнекислоту определять не надо,
17 Зака» 52?.
258
Гл. IX. Свинец
ее можно удалить в самом начале анализа обработкой пробы фтористоводородной и азотной кислотами и повторным выпариванием с последней для удаления фтористоводородной кислоты (стр. 823).
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ СВИНЕЦ
' Разложение сульфидных руд свинца лучше всего начинать с осторожного нагревания их с соляной кислотой.’Если при этой обработке остаются нерастворимые сульфиды, то их разлагают прибавлением азотной кислоты и новой порции соляной кислоты в количестве, достаточном для растворения всего хлорида свинца. Под конец прибавляют в избытке серную кислоту и раствор выпаривают до появления паров серной кислоты, как описано ниже.(стр. 262). Силикатные горные породы сплавляют с содой. Если для удаления большей части кремнекислоты плав выщелачивают водой, то надо помнить, что некоторое количество свинца при этом переходит в раствор и должно быть затем определено в фильтрате *.
МЕТОДЫ ОТДЕЛЕНИЯ
Главный метод отделения свинца основан на нерастворимости его сульфата. Описанное на стр. 262 выпаривание с серной кислотой служит для отделения свинца от многочисленных элементов, образующих растворимые сульфаты. При необходимости точного определения свинца в растворах, содержащих соляную или азотную кислоту, их следует выпаривать до появления паров серной кислоты два или три раза, после каждого выпаривания обмывая стенки сосуда, чтобы быть уверенным в полном удалении соляной или азотной кислоты, так как эти кислоты частично растворяют PbS04. Следует также избегать добавления хлорной кислоты, так как она растворяет небольшое, но все же заметное количество сульфата свинца, даже и в тех случаях, когда в растворе имеется избыток свободной серной кислоты. Сульфат свинца слегка растворим также и в разбавленной серной кислоте, поэтому в точных работах его надо затем извлекать из фильтрата. При выполнении рядовых анализов, когда определяют только один свинец, сульфат свинца достаточно промывать разбавленным раствором серной кислоты, насыщенным сульфатом свинца при той же температуре, при которой применяется раствор. Часто рекомендуемое прибавление спирта уменьшает растворимость сульфата свинца, но одновременно вызывает осложнения вследствие загрязнения осадка сульфата свинца сульфатами кальция и висмута, и поэтому в тех случаях, когда фильтрат надо подвергнуть дальнейшему анализу, спирт добавлять не следует. Вместе с сульфатом свинца выделяется кремнекислота, а также и вольфрам, ниобий, тантал, барий и менее полно стронций и кальций. Висмут, сурьма, серебро, медь, а также, без сомнения, и некоторые другие элементы отчасти загрязняют сульфат свинца. Никель и хром иногда создают затруднения, если серная кислота нагревалась выше температуры появления ее паров или почти полностью была выпарена.
Если присутствует олово, то полученный сернокислый раствор после его разбавления надо немедленно профильтровать, так как сульфат олова
1 О выделении малых количеств свинца (например, при анализе силикатных горных пород) соосаждением вместе с введенным в раствор стронцием путем добавления разбавленной серной кислоты см. I.T. R osenquist, Am. J. Sci., 240, 356 (1942).
Методы отделения
259
(IV) имеет склонность гидролизоваться с образованием нерастворимого осадка.
Малые количества кремнекислоты могут быть отделены от сульфата свинца растворением последнего в горячем растворе, содержащем 20 г ацетата аммония и 3 мл 80%-ной уксусной кислоты в 100 мл. Большие количества кремнекислоты лучше удалять обработкой фтористоводородной и серной кислотами с последующим выпариванием до появления паров последней.
Сульфат бария слегка растворим в растворе ацетата аммония 1 и, кроме того, препятствует полному растворению сульфата свинца. В присутствии бария следует предварительно отделить свинец (как будет описано ниже) перед осаждением бария в виде сульфата или же взвесить сульфаты свинца и бария вместе и ввести поправку после определения одного из них.
* При наличии в анализируемой пробе тяжелого шпата (сульфата бария) добавление серной кислоты при разложении пробы с последующим выпариванием до появления паров серной кислоты недопустимо х. Нельзя также выпаривать досуха и с одной азотной кислотой или с царской водкой, так как при разложении сульфидных руд этими кислотами образуется серная кислота. В обоих случаях, когда при выпаривании раствора концентрация серной кислоты сильно повышается, начинает разрушаться кристаллическая решетка тяжелого шпата с переходом части ионов бария в раствор. При последующем разбавлении водой кристаллы тяжелого шпата покрываются снова слоями BaSO4 с неизбежной окклюзией свинца, ионы которого оказываются внутри кристаллов тяжелого шпата. Окклюдированный таким образом свинец не может быть потом извлечен никаким растворителем.
Если после разложения руды царской водкой ввести в раствор достаточное количество хлорида натрия, то при последующем выпаривании досуха вся образовавшаяся в результате окисления сульфидов серная кислота превращается в сульфат натрия, который не разрушает кристаллической решетки тяжелого шпата, и окклюзия свинца, таким образом, исключается.
В указанной статье 2 приводятся два метода анализа свинцовых руд, содержащих тяжелый шпат. Доп. ред.*
Если присутствует большое количество вольфрама, то его лучше отделить нагреванием с азотной кислотой (стр. 765). Для определения свинца, захваченного осадком вольфрамовой кислоты, осадок растворяют в аммиаке и ацетате аммония и осаждают свинец сульфидом аммония. Свинец может быть отделен от ниобия и тантала осаждением сероводородом в кислом растворе, содержащем винную кислоту, или в щелочном растворе, содержащем тартраты (стр. 89).
Отделение свинца от' щелочноземельных металлов проводится осаждением сероводородом в разбавленном солянокислом растворе, не содержащем окислителей, или электролизом в азотнокислом растворе, как указано ниже (стр. 264).
* Малые количества свинца нельзя осадить полностью сероводородом из разбавленных минеральнокислых растворов даже при использовании
1 Е. Т. А 11 е n, Е. G. Z i е s, J. Am. Ceram. Soc., 1, 777 (1918).
2 Ю. Ю. Л у р ь е, Г. А. Т а р а т о р и н, Зав. лаб., 9, 522 (1940).
17*
260
Гл. IX. Свинец
коллекторов х. Для полноты извлечения малых количеств свинца осаждение следует проводить при pH около 3 (а по мнению некоторых авторов, даже при pH 4—5) и в качестве коллектора добавлять медь или ртуть 1 2. Если присутствует железо (III), особенно вместе с фосфат-ионами, следует добавить лимонную кислоту или ее соли. Железо и фосфаты, если они присутствуют в больших количествах, мешают также полноте выделения свинца в виде его двуокиси электролизом. Доп. ред*
Серебро и висмут лучше всего отделять перед осаждением свинца, первое — повторным осаждением в' виде хлорида (стр. 237), второй — в виде хлорида (стр. 274), бромида или нитрата висмутила (стр. 269—270).
Загрязнение сульфата свинца сурьмой уменьшается при добавлении винной кислоты.
Например, для рядовых анализов сплавов, содержащих свинец и сурьму, J. A. Scherrer рекомендует следующий метод. Сплав растворяют в смеси 60 мл воды, 2—4 г винной кислоты и 15 мл азотной кислоты. По окончании растворения прибавляют 15 мл серной кислоты и выпаривают на паровой бане до прекращения выделения красных паров и начала выделения белых паров. Разбавляют раствор до 200 мл, фильтруют, осадок сульфата свинца умеренно промывают и сохраняют фильтрат, содержащий теперь большую часть сурЬмы и немного свинца. Осадок сульфата свинца смывают в чашку, обрабатывают 10 мл разбавленной серной кислоты и выпаривают до появления ее густых паров. Разбавляют, отфильтровывают сульфат свинца, умеренно промывают его разбавленной серной кислотой и прокаливают, как обычно. Фильтрат соединяют с прежде полученным растворбм, подщелачивают едким кали, обрабатывают 25 мл 20%-ного раствора сульфида натрия и фильтруют после стояния в течение ночи. Осадок сульфидов растворяют в азотной кислоте и подвергают электролизу для определения меди и остаточного свинца.
Очень большое значение имеют методы отделения свинца, основанные на нерастворимости его сульфида в кислых и щелочных растворах. Этими способами свинец легко отделяется от элементов, не принадлежащих к группе сероводорода, и от элементов подгруппы мышьяка (см. «Осаждение сульфидов», стр. 83 и 85).
Осаждение свинца в виде его гидроокиси и в виде хлорида свинца в обычных условиях не бывает полным. Если, однако, количество свинца невелико и в растворе преобладают такие элементы, как железо, то вместе с ними свинец осаждается аммиаком полностью. Осаждение свинца в виде хлорида иногда применяется для удаления большей части свинца, из раствора, в котором надо провести определение других элементов 3.
Электролиз с платиновым матированным электродом в азотнокислом или азотно-сернокислом растворе является очень хорошим методом отделения небольших количеств свинца в отсутствие веществ, препятствующих его осаждению или загрязняющих осадок (см. стр. 264). Свинец, так же как и медь, серебро и ртуть, можно отделить от оловянной, сурьмяной, вольфрамовой и молибденовой кислот электролизом в растворе, содержащем азотную и фтористоводородную кислоты 4. Полученная таким способом двуокись свинца будет загрязнена фтором, и ее надо или пере-
1 I. Т. Rosenquist, Am. J. Sci., 240, 356 (1942).
8 E. C. D a ws о n, A. R e e s, Analyst, 71, 417 (1946); F. F e i g 1, N. В r a i 1 e, там же, 69, 147 (1944).
3 В. L. Clarke, L. A. Wooten и J. D. Struthers [(Ind. Eng. Chem., Anal. Ed., 9, 349 (1937)] описывают интересный прибор, в котором происходит растворение пробы свинца в соляной кислоте и одновременно отделение свинца от некоторых загрязняющих его элементов, например от олова.
4 L. W. McC ay, N. Н. Furman, J. Am. Chem. Soc., 38, 640 (1916).
Методы отделения
261
осадить йосле промывания и растворения обратным током в азотной кислоте или превратить в сульфат.
При осаждении свинца в виде хромата, как описано на стр. 265, свинец хорошо отделяется от меди, цинка и от остальных элементов, хроматы которых растворимы в уксусной кислоте *. Если осаждение проводится при 70° С в растворе, содержащем, 1 мл 60%-ной хлорной кислоты в общем объеме 200 мл, то свинец выделяется количественно и отделяется также и от бария 1 2.
Если свинец находится в виде хромата, сульфата, арсената, селената или теллурата, то его можно отделить от связанных с ним анионов, превратив его в гидрат двуокиси свинца нейтрализацией раствора едким натром до pH 7—10, добавлением бромата натрия и кипячением 3.
Свинец можно отделить купфероном от железа и, возможно, от некоторых других элементов, осаждаемых этим реактивом в солянокислом растворе (стр. 143). Для этого добавляют к кислому анализируемому раствору, содержащему осадок, выпавший от добавления купферона, какой-нибудь не смешивающийся с водой органический растворитель, например хлороформ или эфир, и отделяют после взбалтывания водный слой 4.
* Свинец можно количественно отделить от щелочноземельных металлов в виде оксихлорида свинца, осаждая его добавлением аммиака и хлорида аммония 5. Выпадающий при этом осадок гидроокиси железа способствует коагуляции оксихлорида свинца. От щелочноземельных металлов свинец можно также отделить восстановлением его металлическим алюминием. При этом, конечно, вместе со свинцом выделятся такие металлы, как медь, серебро и др.
При осаждении тиомочевиной CS(NH2)2 из охлажденного до 0° С азотнокислого раствора в виде 2Pb(NO3)2 • 11CS(NH2)2 свинец отделяется от бария, алюминия, хрома, железа, марганца, цинка, никеля, кобальта, меди и висмута. Осадок промывают насыщенным и охлажденным до 0° С раствором тиомочевины, растворяют в горячей воде и осаждают свинец в виде хромата 6.
Малые количества свинца (0,5—2,5 мг) можно извлечь из разбавленных растворов его солей (например, из природных или промышленных сточных вод) добавлением 0,2 г карбоната кальция и встряхиванием в закрытом сосуде в течение получаса 7. Доп. ред.*
1 Согласно Н. Н. WillardnJ.L. К assner [J. Am. Chem. Soc., 52, 2402 (1930)], хромат свинца практически нерастворим в разбавленных растворах хлорной кислоты, содержащих хромат-ионы в избытке. Свинец не был, например, совершенно обнаружен в растворе после того, как хромат свинца находился 15 ч при 25° С в 0,5 М растворе НС1О4, содержащем Na2Cr2O7 в 0,03 М концентрации.
2 D. J. Brown, J. A. Moss, J. В. Willi а-m s, Ind. Eng. Chem., Anal. - Ed., 3, 134 (1931).
3 R. G i 1 c h r i s t, J. Research NBS, 30, 89 (1943).
4 L. Panouse-Digeaud, H. Cheftel, Ann. fals. et fraudes, 32, 296
(1939).
6 В. П. Ш в e д о в, ЖОХ, 17, № 1, 33 (1947); ЖАХ, 3, 113 (1948).
’ С. M a h r, Z. anorg. Chem., 234, 224 (1937), цит. по руководству С.Ю.Файн-z б e p г, Анализ руд цветных металлов, Металлургиздат, 1953,.
7 Ю. Ю. Л у р ье, А. И. Рыбникова, Методы химического анализа производственных сточных вод, Госхимиздат, 1958, стр. 108.
262
Гл. IX. Свинец
МЕТОДЫ ОПРЕДЕЛЕНИЯ
По-видимому, наиболее удовлетворительным методом определения свинца следует считать весовой метод, по которому свинец взвешивают в виде сульфата свинца, несмотря на то что при выполнении точных анализов приходится извлекать свинец дополнительно из фильтрата, куда он переходит вследствие растворимости PbSO4. Это извлечение, равно как и определение малых количеств свинца (1—2 мг), лучше всего проводить электролизом в азотнокислом или азотно-сернокислом растворе с взвешиванием осажденной на аноде двуокиси свинца РЬО2.
Электролитический метод рекомендуется также при рядовых определениях средних количеств свинца.
Взвешивание свинца в виде молибдата равноценно его определению в виде сульфата за исключением того, что этот метод может применяться реже и что введение в раствор молибдена может вызвать затруднения в тех случаях, когда фильтрат после отделения свинца подлежит дальнейшему анализу. Объемное определение свинца в виде хромата является очень хорошим методом для рядовых определений свинца в рудах, сурике и т. п. . *
Весовые определения
Определение в виде сульфата свинца. Как уже было указано, отделение свинца в виде его сульфата в разбавленном сернокислом растворе не является точно количественным. Прибавление спирта понижает растворимость сульфата свинца, но одновременно загрязняет осадок такими элементами, как серебро, висмут или кальций. Растворимость сульфата свинца увеличивается в присутствии соляной, азотной или хлорной кислот. Кроме того, следует помнить, что ацетаты, как и вообще органические вещества, а также и некоторые катионы (стр. 258), мешают осаждению. Соли щелочных металлов, особенно калия, склонны образовывать с сульфатом свинца двойные соли аналогично тому, как это наблюдается для сульфата бария (стр. 798). При выполнении точных анализов лучше, освободить растворы от мешающих определению элементов перед осаждением сульфата свинца, чем пытаться очистить осадок PbS04.
Ход определения. Приготовляют раствор, содержащий до 0,5 г свинца и такое количество серной кислоты, чтобы избыток последней при дальнейшем разбавлении равнялся 5 мл на 100 мл раствора. Раствор, который не должен содержать мешающих осаждению упомянутых выше веществ, за исключением летучих кислот, выпаривают до появления паров серной кислоты и, если присутствовали другие кислоты, помимо серной, охлаждают, обмывают стенки сосуда и снова выпаривают до появления паров серной кислоты. Органические вещества разрушают повторным добавлением азотной кислоты. Раствор охлаждают и прибавляют 100 мл воды на каждую 0,1 г присутствующего свинца. Раствор тщательно перемешивают и оставляют стоять по меньшей мере на 1 ч. Затем фильтруют через фарфоровый «тигель с пористым дном (имеющим минимальную пористость) или через слой губчатой платины в тигле Гуча и промывают
Методы определения
263
осадок разбавленной (1,: 99) серной кислотой Ч Помещают тигель в радиатор или муфельную пень и постепенно нагревают до 500—600° С и при этой температуре прокаливают до постоянной массы. Охлаждают в эксикаторе и взвешивают в виде PbSOi.
Определение в виде молибдата свинца. Осаждение свинца в виде его молибдата РЬМоО4 является хорошим методом определения свинца в растворах, не содержащих щелочноземельных металлов, также образующих нерастворимые молибдаты, и таких веществ, как хроматы, арсенаты, фосфаты, образующих нерастворимые соединения со свинцом, и веществ, легко гидролизующихся, подобно солям титана или олова.
Осадок РЬМоО4 менее растворим, чем сульфат свинца, и не изменяется при прокаливании. Преимуществом его является большой молекулярный вес.
Ход определения 1 2 3 *. Нейтральный раствор, содержащий, около 0,5 г свинца, разбавляют до 200 мл, прибавляют 8 капель азотной кислоты ®, нагревают до кипения и, поддерживая температуру близкой к кипению, медленно прибавляют из бюретки при перемешивании 2—2,5%-ный раствор молибдата аммония. По окончании осаждения кипятят 1 мин, дают осадку осесть, и чтобы убедиться в полноте осаждения, прибавляют еще каплю осадителя к жидкости над осадком. Большого избытка осадителя следует избегать. Когда осаждение закончится, прибавляют из бюретки по каплям разбавленный (1 : 2) раствор аммиака до нейтральной или слабощелочной реакции на лакмус. Затем подкисляют несколькими каплями уксусной кислоты и оставляют стоять несколько минут. Отстоявшуюся жидкость декантируют череЗ беззольный фильтр или через тигель Гуча с асбестовым или платиновым слоем и промывают осадок горячим 2—3%-ным раствором нитрата аммония. Для промывания декантацией достаточно 3—4 порций по 75 мл раствора нитрата аммония. Затем переносят осадок на фильтр и промывают его до тех пор, пока проба не покажет полноту удаления молибдата. Если применялся бумажный фильтр, то его сушат при 100° С, отделяют осадок от фильтра, сжигают последний в фарфоровом тигле, всыпают осадок и прокаливают при темно-красном калении. Если был применен тигель Гуча, то его помещают в радиатор (стр. 48) или муфельную печь и постепенно повышают температуру до темно-красного каления, после чего охлаждают в эксикаторе и взвешивают осадок в виде РЬМоО4.
1 Согласно D. N. CraignG. W. Vinal [J.,Research NBS, 22, 55 (1939)], растворимость сульфата свинца в разбавленных растворах серной кислоты при 25° С с повышением концентрации последней сначала постепенно падает, достигая величины 4,55 мг/л в 0,3%-ной (по массе) кислоте, затем повышается до 6,68 мг/л в 10%-ной кислоте и снова падает до 1,08 мг/л в 50%-ной кислоте. Некоторое количество сульфата свинца остается поэтому в растворе во всех случаях, и при выполнении очень точных анализов надо принять соответствующие меры для внесения поправки на растворимость PbS04. При выполнении рядовых анализов поправка вносится автоматически применением для осаждения PbSO4 и для промывания осадка разбавленной серной кислоты, насыщенной сульфатом свинца при тех же концентрации и температуре, при которых она применяется. В арбитражном анализе пользуются серной кислотой различных концентраций, не насыщая ее сульфатом свинца, но извлекают потом свинец из всех растворов, например электролизом одновременно с выделением меди.
2 Н. В. W е i s е г, J. Phys. Chem., 20, 659 (1916). 5
3 Согласно Н. Н. Willard, J. L. К a s s n е г [J. Am. Chem. Soc., 52, 2402
(1930)], молибдат свинца менее растворим в хлорной кислоте (в присутствии или
в отсутствие одноименных ионов), чем в азотной кислоте той же концентрацйи. Растворимость хромата свинца при 25° С в 0,5 М растворе хлорной кислоты, содержащем Na2MoO4, в 0,05 М концентрации, равна 0,27 мг в 100 мл.
264
Гл. IX. Свинец
Электролитическое определение в виде двуокиси «винца
Электролитическое осаждение двуокиси свинца 1 лучше всего проводить на сетчатом платиновом электроде с матированной поверхностью >(стр. 55). Прочный, не осыпающийся с электрода осадок можно получить, осаждая на таком аноде самое большее 0,1 г свинца 2. Осаждение двуокиси свинца происходит особенно хорошо в присутствии меди. Свинец и медь могут быть определены одновременно. Лучше всего сначала осадить свинец так, как указано ниже, а затем прибавить небольшое количество серной кислоты и осадить медь. Можно также начинать осаждение из раствора, содержащего приблизительно 10 мл азотной кислоты и 2 мл серной кислоты в 100 л1л электролита 3.
При проведении электролиза в горячих растворах, содержащих только одну азотную кислоту (*при отсутствии меди *), имеется опасность отложения свинца на катоде, если концентрация азотной кислоты не поддерживается достаточно высокой, нйпример, 20%-ной. При очень низком концентрации азотной кислоты свинец может отлагаться на катоде и в холодном растворе.
Если свинец был выделен в виде сульфата, то для осаждения его электролизом осадок надо обработать раствором аммиака, введенным в избытке, довести до кипения, и влить эту смесь при непрерывном перемешивании в раствор азотной кислоты, взятой с таким расчетом, чтобы после смешения получился избыток ее в 15 лл на 100 мл раствора. ‘
Электролитическому осаждению свинца мешает присутствие хлоридов, ртути, мышьяка, теллура, селена и фосфора, которые препятствуют полному его выделению, и присутствие висмута, олова, сурьмы, серебра, кобальта и марганца (если только раствор не сильно кислый), загрязняющих осадок.
Ход определения. Приготовляют 100—150 мл раствора, содержащего не более 0,1 г свинца в виде нитрата свинца и 15—25 мл азотйой кислоты и не содержащего упомянутых веществ, мешающих осаждению. Электролиз ведут в течение ночи с платиновыми сетчатыми матированными электродами при силе тока около 0,5 а и напряжении 2 в. Раствор испытывают на полноту осаждения, прибавляя воду и наблюдая при продолжающемся электролизе 10—15 мин, не появляется ли новый осадок на свежепокрытой жидкостью поверхности анода.
Когда полнота осаждения будет достигнута, не прерывая ток, приподнимают анод и обмывают его водой. Затем анод высушивают 30 мин при 125° С, охлаждают в эксикаторе, взвешивают и вычисляют содержание свинца, умножая массу осадка на коэффициент 0,866.
1 С. Luckow, Dinglers Polytech. J., 178, 42 (1865); А. Классен, Электроанализ, Госхимтехиздат, 1934.
2 Плотно прилегающие к аноду осадки больших количеств двуокиси свинца можно получить даже на нематированных анодах, если проводить электролиз в горячем растворе и сильно перемешивать раствор вращающимся анодом или стеклянной мешалкой. «
3 Для рядовых определений свинца (7%) в рудах W.T. SchrenknP. Н. Delano [Ind. Eng. Chem., Anal. Ed., 3, 27 (1931)] рекомендуют, чтобы раствор содержал 20—30% свободной азотной кислоты и 0,25 мл серной кислоты и чтобы электролиз начинался при температуре раствора 90° С и продолжался 1,5—2 ч при силе тока 3 а и постепенном охлаждении раствора.
Методы определения
265
* При выделении малых количеств свинца рекомендуется заканчивать определение колориметрическим методом 1 следующим способом. Платиновый анод с выделившейся на нем двуокисью свинца погружают на 1 сек в кипящую воду, затем стряхивают с него, капли воды и опускают в стакан, содержащий 25 мл свежеприготовленного 1 %-ного раствора тетраметилдиаминодифенилметана в ледяной уксусной кислоте (этот раствор устойчив в течение 24 ч). Двуокись свинца, растворяясь, окисляет указанный реактив, образуя окрашенное в синий цвет соединение. Окраску полученного раствора сравнивают с серией стандартных растворов, приготовленных таким же способом из известных количеств двуокиси свинца, или же определяют светопоглощение полученного раствора в фотоколориметре, применяя светофильтр с максимумом пропускания при 600 ммк после чего рассчитывают содержание свинца по калибровочной кривой. Доп. ред*
Для удаления осадка двуокиси свинца с анода последний помещают в стакан, содержащий горячую разбавленную (1 : 3) азотную кислоту, и прибавляют небольшое количество какого-либо восстановителя, например спирта или перекиси водорода 2 3.
Объемное определение хроматным методом
В хроматном методе определения свинца, вероятно, наилучшим из объемных методов определения этого элемента, свинец осаждают в виде РЬСгО4 в уксуснокислом растворе, осадок растворяют в соляной кислоте и полученный раствор обрабатывают иодидом калия. Выделившийся иод оттитровывают раствором тиосульфата натрия ®. Обычно свинец сперва выделяют в виде сульфата свинца. Сурьма, висмут, серебро, барий и гель кремнекислоты нежелательны и должны быть отделены или перед осаждением свинца в виде сульфата или последующей специальной обработкой.
* Осадок хромата свинца можно растворить в кислоте и закончить определение колориметрическим методом или по желтой окраске хромата или по красно-фиолетовой окраске, получаемой при добавлении к полученному раствору 10 мл 0,02 %-ного раствора дифенилкарбазида. Чувствительность метода можно значительно повысить, осаждая свинец в виде двойной соли 4 К2РЬ(СгО4)2 в почти нейтральном растворе (pH 6,6—7,4). Необходимые условия создаются добавлением сначала раствора аммиака до перехода окраски фенолового красного в розовую, а затем — разбавленной уксусной кислоты до перехода' окраски этого индикатора в оранжево-желтую, после чего прибавляют раствор хромата калия и оставляют на ночь. Доп. ред*
1 А. Д. Петров, ЖРФХО, 60, 311 (1928); М. В. Неустроева, Труды Государственного института радия, 4, 304 (1938); Н. Muller, Z. anal. Chem., 113, 161 (1938).
2 Операция превращения осадка двуокиси свинца в РЬО осторожным его прокали-ванием, как рекомендовал W.-C. May [Am, J. Sci. (3), 6, 255 (1873))], при исследовании нами не показала никакого преимущества по сравнению с обычным методом высу-пшвания.
3 Н. A. G u е s s, Trans AIME, 35, 359 (1904); J. W a d d е 11, J. Ind. Eng. Chem., 3, 638 (1911); A. H. Low, A. J. W e i n i g, W. P. S c h о d e r, Technical Methods of Ore Analysis, 11th. ed., 1939, p. 153.
4T. V. Letonoff, J. G. Reinhold, Ind. Eng. Chem., Anal. Ed., 12, 280 (1940); там же, 13, 631 (1941).
266
Гл. IX. Свинец
Ход определения. Осаждают свинец (желательно в количестве, не пре-' вышающем 0,1 з) в виде сульфата, осадок отфильтровывают и промывают, как описано на стр. 262. Фильтр с осадком осторожно развертывают к смывают возможно большее количество сульфата свинца в колбу емкостью 250 мл минимальным количеством горячей воды х. Обрабатывают фильтр 25 мл горячего экстрагирующего раствора 1 2 3 в небольшом стакане и фильтруют в колбу, содержащую главную массу осадка. Стакан, фильтр и воронку промывают горячим экстрагирующим раствором и нагревают колбу до полного растворения сульфата свинца. Разбавив, если это необходимо, до объема 150 мл, нагревают до кипения и прибавляют из пипетки 10 мл насыщенного раствора бихромата калия. Осторожно кипятят 5—10 мин, фильтруют и промывают колбу, фильтр и осадок десять раз порциями по 10 мл горячего разбавленного раствора ацетата натрия ®. В присутствии висмута перед фильтрованием прибавляют 2 г лимонной кислоты, растворенной в небольшом количестве горячей воды. Помещают промытую колбу под воронкой, растворяют осадок на фильтре солянокислой смесью 4 и тщательно промывают ею же фильтр, употребляя на все не менее 50 мл. Затем промывают фильтр 50 мл холодной воды. К полученному раствору прибавляют 1—2 г иодида калия, растворенного в небольшом количестве воды, и осторожно перемешивают. Затем тотчас же титруют раствором тиосульфата натрия почти до полного исчезновения иода, прибавляют 5 мл раствора крахмала (стр. 220) и продолжают титровать, пока цвет раствора не перейдет в зеленый без малейшего оттенка синего. Титр тиосульфата натрия устанавливают по чистому свинцу, растворяя его в азотной кислоте и проводя раствор через все стадии анализа.
Свинец можно количественно определить, и при этом очень точно отделить его от меди, серебра, кадмия, никеля, марганца, цинка, железа (III), алюминия, бария, стронция и кальция следующим способом5. Помещают нитраты указанных металлов в 250 мл раствора, содержащего 5—15 мл (5 мл в присутствии меди и никеля, 10 мл — когда надо отделять от кальция и бария, 15 мл — при отделении от серебра) 0,3 н. азотной кислоты, нагревают до кипения и при сильном перемешивании раствора прибавляют 3—5-кратное количество 1 н. раствора хромата аммония. Реактив добавляют
1 В присутствии бария сульфат свинца смывают обратно в колбу струей горячей воды, употребляя возможно меньшее ее количество. Споласкивают также под складкой фильтра. Прибавляют 10 мл соляной кислоты и выпаривают раствор почти досуха. Затем приливают 25 мл экстрагирующего раствора (см. ниже, сноска 2), кипятят и продолжают прибавлять этот раствор до растворения сульфата свинца, употребляя на это не более 75 мл. Затем фильтруют через тот же фильтр и промывают остаток сульфата бария горячей водой. *Как было указано на стр. 258, если при получении сульфата свинца в присутствии сульфата бария проводили выпаривание до паров серной кислоты (что приводит к частичному растворению обоих сульфатов), то при последующем разбавлении водой выделяются кристаллы BaSO4, окклюдирующие ионы свинца, и тогда предлагаемая авторами обработка не достигает цели: часть свинца остается в нерастворимом остатке.*
2 Для его приготовления насыщенный на холоду раствор ацетата натрия фильтруют, прибавляют двукратный объем воды и затем ледяную уксусную кислоту в количестве 25 мл на 1 л раствора.
3 Приготовляется разбавлением 50 мл насыщенного на холоду раствора ацетата натрия водой до 1 л.
4 Получается добавлением к 1 л прозрачного насыщенного раствора хлорида натрия 300 мл разбавленной (1 : 1) соляной кислоты.
5 Z. Karaoglanov, М. Michov, Z. anal. Chem., 103, 113 (1935).
Методы определения
267
по каплям так, чтобы добавление продолжалось 10 мин. Затем дают постоять несколько часов, фильтруют через стеклянный фильтрующий тигель, умеренно промывают осадок горячей водой, высушивают при 140° С и взвешивают1 РЬСгО4.
Другие методы
Из других методов определения свинца могут быть упомянуты: 1) молибдатный метод2 3, по которому уксуснокислый раствор ацетата свинца титруют титрованным раствором молибдата аммония, применяя таннин в качестве внешнего индикатора; 2) гексацианоферратный метод ®, по которому уксуснокислый раствор соли свинца титруют титрованным раствором гексацианоферрата (II) калия с применением ацетата уранила в качестве внешнего индикатора; 3) перманганатный метод 4, в котором свинец осаждают в уксуснокислом растворе в виде оксалата, затем осадок растворяют в серной кислоте и титруют раствором перманганата.
Очень малые количества свинца обычно определяют колориметрическими методами: сравнением коричневой окраски сульфида свинца, получаемого в разбавленном уксуснокислом или в щелочном растворе, с окрасками стандартных растворов 5, или же дитизоновым методом 6.
От дитизонового метода нельзя ожидать специфичности, потому что по крайней мере 17, а возможно, 21 элемент образуют соединения с дитизоном. Тем не менее, регулируя- pH раствора и прибавляя комплексо-образователи, можно точно определить малые количества свинца и других элементов. В общем случае дитизон прибавляют к слабощелочному раствору (pH 8—10), содержащему свинец. Образующееся вишневокрасное соединение извлекается хлороформом или четыреххлористым углеродом. Содержание свинца определяется сравнением полученной окраски с окрасками - стандартных растворов, содержащих известные количества свинца и подвергнутых такой же обработка, как и испытуемый образец. Более подробно об условиях определения см. в оригинальных статьях 6.
1 * Этот метод получил дальнейшее развитие в работе С. Ю. Файнберга, Л. Б. ЗайчиковойиС. М. Фрайберг, Зав. лаб., 16, 771 (1950)*.
- 2 G. Schindler, Z. anal. Chem., 27, 137 (1888); Н. Alexander, Eng. Mining. J., 55, 1 (1893); J. Chem. Soc., (11), 64, 599 (1893); A. H. L о w, A. J. Wei-n i g, W. P. S ch о der, Technical Methods of Ore Analysis, 11th ed., 1939, p. 158; R. C. W i 1 e у [Ind. Eng. Chem., Anal. Ed., 2, 124 (1930] рекомендует титровать почти нейтральный раствор с применением смеси хлорида олова (II) и роданида калия в качестве внешнего индикатора.
3 А. Н. Low, J. Am. Chem. Soc., 15, 550 (1893); А. С. В e e b e, Chem. News, 73, 18 (1896); I. С. В u 11, Z. anal. Chem., 41, 653 (1902).
4 W. Hempel, Jahresber., 627 (1853); A. H. Low, J. Am. Chem. Soc., 15, 550 (1893); A. H. L о w, A. J. W e i n i g, W. P. S c h о d e г, цит. выше, стр. 160.
5 J. Pelouze, Ann. chim. phys., (3), 79, 108 (1841); J. M. W i 1 k i e, J. Soc., Chem. Ind., 28, 636 (1909); там же, 29, 7 (1910); L. W. W i n k 1 e r, Z. angew. Chem., 26, 38 (1913).
6 H. J. W i c h m a n n, C. W. Murray, M. Harris, P. A. Clifford, J. H. Loughrey, F. A. V orhes, J. Assoc. Offic. Agr. Chemists, 17, 108 (1934); P. A. С 1 i f f о r d, там же, 26, 26 (1943); * E. Б.Сен д э л, Колориметрическое определение следов металлов, Госхимиздат, 1949, стр. 427—438.*
Глава X
ВИСМУТ
По нахождению в природе висмут подобен сурьме, но встречается реже. Главные его руды — самородный висмут и сульфид висмута — висмутинит Bi2S3. Относительно редкими минералами являются: два силиката висмута, некоторые сульфовисмутиды, теллурид висмута, окись висмута^ карбонат висмута, молибдат висмута, ванадат висмута и арсенат висмута.
ОБЩИЕ ЗАМЕЧАНИЯ
При анализе горных пород можно считать, что висмут в них не содержится вовсе, а в минералах встречается редко. Он, однако, часто находится в некоторых рудах свинца, олова, серебра, меди, никеля, кобальта и в легкоплавких сплавах. В обычном ходе анализа большее или меньшее количество висмута сопровождает кремний в виде оксисоединений висмута, если обезвоживание кремнекислоты проводилось выпариванием солянокислого или азотнокислого растворов. Это, без сомнения, приводит к ошибке при определении кремния вследствие изменения состава этого соединения висмута при обработке нечистой кремнекислоты фторо-ристоводородной и серной кислотами и прокаливании оставшихся в тигле примесей. При обычном ходе анализа, если не принять специальных мер предосторожности, висмут попадет в осадок от аммиака и будет принят за алюминий. Осаждение висмута аммиаком происходит практически полностью, если в анализируемом растворе имеются хлорид-ионы, но тогда в выделяющемся осадке будет в большей или меньшей мере присутствовать хлорид висмутила, и потому осадок нельзя прокаливать, рассчитывая взвесить потом Bi2O3. Из растворов, содержащих сульфат-и нитрат-ионы (но не содержащих хлорид-ионов), висмут осаждается аммиаком не полностью, и осадок также содержит оксисоединения висмута. При анализе металлургических продуктов присутствие висмута создает затруднения главным образом при выполнении электроаналити-ческих определений, потому что висмут осаждается и на катоде й на аноде, большей частью на первом.
life
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ВИСМУТ '
Большинство минералов, содержащих висмут, легко растворяется при следующей обработке. Разложение ведут сначала азотной кислотой, затем прибавляют небольшое количество соляной кислоты и в заключе
Методы отделения
269
ние выпаривают с серной кислотой, прибавленной н избытке. Остаток, получающийся после разбавления водой и нагревания, может содержать висмут, а в присутствии сульфата свинца всегда содержит висмут. Поэтому этот остаток обрабатывают следующим образом. Его отфильтровывают, промывают разбавленной серной кислотой, затем водой и высушивают.' Высушенный остаток отделяют от фильтра насколько возможно полнее, фильтр сжигают отдельно в фарфоровом тигле при возможно низкой температуре, при которой еще можно окислить углерод, и предупреждают таким образом сплавление золы с глазурью тигля. Затем золу соединяют с главной массой остатка и смешивают с содой в платиновом тигле. После сплавления плав тщательно выщелачивают водой для удаления сернокислых солей и остаток растворяют в азотной кислоте, после чего из раствора выделяют висмут, осаждая его в виде BiOCl, как описано на. стр. 274.
Свинец и кремнекислоту удобнее определять в отдельной навеске минерала после обработки его, как описано выше, и выделения кремнекислоты и загрязненного сульфата свинца. Из этого осадка сульфат свинца сначала извлекают уксуснокислым раствором ацетата аммония (стр. 259) и в остатке определяют кремнекислоту, как обычно. В ацетатном растворе свинец осаждают в виде сульфида, который обрабатывают для определения элементов группы мышьяка,. затем растворяют в азотной кислоте и после отделения висмута осаждают в виде сульфата свинца.
Серу удобно определять, как описано в гл. «Сурьма» (стр. 321),
МЕТОДЫ ОТДЕЛЕНИЯ «
Описываемые методы отделения висмута основаны на осаждении его в виде хлорокиси висмута BiOCl и бромокиси висмута BiOBr. Первая образуется при гидролизе хлорида висмута в очень разбавленном солянокислом растворе. Висмут этим способом обычно выделяют после осаждения его вместе со всей сероводородной группой и после отделения от подгруппы мышьяка, так как соли ряда других элементов — циркония, титана, сурьмы, олова — также легко гидролизуются. Если присутствует серебро или ртуть (I), то, конечно, висмут должен быть отделен также и от них. Отделение висмута в виде хлорокиси может быть количественно проведено, как описано на стр. 274, причем этому отделению не мешает присутствие большого количества свинца 2, меди, кадмия и, по всей вероятности, присутствие ртути (II).
Осаждение висмута в виде его бромокиси 3 является лучшим способом отделения висмута от свинца, меди, цинка, кадмия, чем осаждение его в ниде хлорокиси или основного нитрата. Осаждение это лучше проводить в отсутствие хлоридов и аммонийных солей. Если присутствуют хлориды, следует прибавить большее количество бромида и бромата и провести двукратное осаждение. Аммонийные соли замедляют осаждение;
1 * * Подробное изложение описанных в литературе методов отделения и определения висмута дадо в монографии А. И. Б у с е в а, Аналитическая химия висмута, Изд. АН СССР, 1953. Прим. ред. *
’ Для определения очень малых количеств висмута в свинце можно брать боль-шие навески последнего и доводить их до подходящих размеров трейбованием.
* L. М о s е г, W. М а х У m о w i с z, Z. anal. Chem.,' 67, 248 (1925—1926).
270
Гл. X. Висмут
в их присутствии требуется более продолжительное кипячение и возможно большее разбавление.
Ход анализа в присутствии свинца следующий. Приготовляют азотнокислый раствор пробы и медленно прибавляют к нему раствор карбоната натрия до тех пор, пока осадок не станет растворяться с большим трудом, после чего разбавляют до 200—300 мл, прибавляют 2 г твердого бромата калия или бромата натрия и нагревают до кипения. Если образовавшаяся муть не исчезнет при кипении, то добавляют по каплям азотную кислоту до полной прозрачности раствора. Затем к кипящему раствору из пипетки по каплям прибавляют 10%-ный раствор бромида калия или бромида натрия, пока муть снова не появится и раствор не окрасится в коричневый цвет. Cocyft покрывают часовым стеклом, кипятят, пока раствор не станет прозрачно-желтым, прибавляют еще немного бромида и так продолжают до тех пор, пока не перестанет выделяться осадок как при дальнейшем прибавлении бромида, так и при добавлении нескольких капель раствора бромата. Раствор кипятят до удаления всего брома, дают осадку осесть, после чего его отфильтровывают и промывают горячей водой. Для того чтобы быть уверенным в полноте отделения свинца, особенно если присутствовали хлорид-ионы, растворяют осадок в горячей разбавленной азотной кислоте и повторяют операцию. Под конец растворяют осадок в горячей азотной кислоте и определяют висмут, как указано на стр. 278. Однократного осаждения достаточно для отделения висмута от меди, цинка и кадмия, если только последний не присутствует в больших количествах. Как и при отделении от свинца, лучше, если аммонийные соли и хлориды будут отсутствовать. Но все же отделение висмута от меди, цинка и кадмия можно проводить в растворах, содержащих хлориды и сульфаты этих металлов.
Менее удовлетворительно проходит отделение висмута в виде основного нитрата висмута. При этом отделении азотнокислый раствор выпаривают до сиропообразной консистенции, обрабатывают водой и выпаривают досуха. Обработку водой и выпаривание повторяют, пока прибавление воды уже не будет вызывать помутнения раствора. Наконец, растворимые соли извлекают холодным 0,25 %-ным раствором нитрата аммония, а нерастворимый остаток прокаливают до окиси свинца или подвергают дальнейшей обработке. Хлориды и сульфаты, образующие также основные соли висмута, должны, очевидно, отсутствовать. Также должны отсутствовать арсенаты и хроматы, образующие с висмутом нерастворимые осадки и элементы, соли которых способны гидролизоваться, как, например, олово и сурьма.
Выделение висмута в виде сульфида часто применяется как групповое отделение и для отделения висмута от хлорид- и сульфат-ионов, когда наличие последних недопустимо в последующем ходе анализа. Если сульфид висмута надо отделить от сульфидов элементов, входящих в подгруппу мышьяка, то следует помнить, что сульфид висмута значительно растворим в растворах нормальных сульфидов M2S и дисульфидов M2S2 натрия и калия, а также в растворах смесей сульфидов и гидроокисей щелочных металлов. С другой стороны, сульфид висмута нерастворим в растворе сульфида аммония и в растворах гидроокисей или гидросульфидов MHS натрия и калия Ч
1 G. С. S t о n е, J. Am. Chem. Soc., 18, 1091 (1896); Т. В. S t i 11 т а п, там же, 683; I. К по х, J. Chem. Soc., 95, 1760 (1909).
Методы отделения
271
Висмут можно полностью осадить сероводородом в виде сульфида висмута при концентрации серной кислоты в растворе, доходящей до 18 н. Удовлетворительное отделение висмута от кадмия 1 может быть получено пропусканием сероводорода через горячий анализируемый раствор, содержащий серную кислоту в отношении 1 : 3, фильтрованием и промыванием осадка сульфида висмута разбавленной (1 : 3) серной кислотой, насыщенной сероводородом.
Висмут количественно не осаждается из растворов его солей едким натром или едким кали. Так как при электролизе в кислом растворе висмут отлагается и на аноде и на катоде, то перед определением меди или свинца электролитическим методом его надо предварительно отделить.
Висмут легко отделяется от меди прибавлением к азотнокислому раствору обоих элементов аммиака почти до нейтральной реакции, разбавлением до 200—300 мл, добавлением карбоната аммония в небольшом избытке и нагреванием до кипения. В этом случае висмут выделяется в виде основного карбоната неопределенного состава, который отфильтровывают и промывают горячей водой. Так как осадок несколько растворим в избытке аммиака или карбоната аммония, то эти реактивы надо прибавлять осторожно. Если этим методом проводится отделение больших количеств висмута или присутствует большое количество меди, то лучше осадок растворить и снова осадить.
Приблизительно такой же метод 2 обработки может служить и для отделения висмута от ртути. Ход анализа следующий: приготовляют раствор солей обоих металлов, содержащий 5 мл свободной азотной кислоты и, желательно, не более 0,1 з ртути в 100 мл раствора. Нагревают до 60° С и медленно прибавляют раствор, содержащий 10% карбоната аммония и 1% аммиака, до щелочной реакции. Затем повышают температуру Д° 80° С и оставляют стоять, часто перемешивая, пока не прекратится выделение СОг из раствора. Дают осадку осесть, отфильтровывают его и промывают 1 %-ным раствором аммиака до удаления ртути. Если в растворе присутствовали только нитраты, то осадок прокаливают до Bi2O3. Если присутствовали хлориды, растворяют осадок в азотной кислоте и выпаривают раствор несколько раз, добавляя новые порции азотной кислоты.
Висмут можно количественно осадить купфероном 3 в холодном, 1 н. по содержанию соляной или азотной кислоты, растворе и таким способом отделить его от щелочных металлов, серебра, ртути (II), свинца, кадмия, мышьяка, сурьмы (V), цинка, марганца, никеля, кобальта, алюминия и хрома. Лучше проводить осаждение из солянокислого раствора. Раствор купферона прибавляют по каплям при непрерывном перемешивании. Надо прибавить приблизительно по 5 мл 6%-ного раствора купферона на каждую 0,1 г висмута. Отфильтрованный осадок промывают холодным 0,1 %-ным раствором купферона, слегка подкисленным кислотой (до 0,1—1 н. концентрации). Если осаждение проводили из азотнокислого раствора, то фильтровать и промывать осадок надо как можно скорее. Полученный осадок можно превратить в Bi2O3 осторожным прокаливанием до 700° С, обработкой остатка несколькими каплями азотной
1 W. Н. Keefe, I. L. Newell, Chemist Analyst, 21, (2), 8 (1932).
2 A. P. Castanares, 8th Intern. Congr. Appl. Chem., 25 (I—V), 39 (1912).
3 A. Pinkus, J.D ernies, Bull. Soc. Chim. Belg., 37, 267 (1928).* См. также Э. А. О с т p о у м о в, Зав. лаб., 4, 1016 (1935); Z. anal. Chem., 106, 36 (1936).*
272
Гл. X. Висмут
кислоты, выпариванием и вторичным прокаливанием до .700° С. Железо и некоторые другие элементы (стр. 143) осаждаются вместе с висмутом. Если эти элементы присутствовали, можно фильтр с осадком обработать до полного разложения смесью азотной и серной кислот (стр. 91) и провести в полученном растворе дальнейшие разделения. Хорошим методом отделения малых количеств висмута от больших количеств свинца является метод внутреннего ‘электролиза (стр. 167).
Висмут можно отделить * 1 от свинца, кадмия, меди, цинка, алюминия, хрома, железа, никеля, бария, кальция, натрия и калия, осаждая его галловой кцслотой в 3%-ном (по объему) растворе азотной кислоты при 70° С. Для хорошего отделения требуется переосаждение. Полученный осадок превращают сначала в основной карбонат, потом прокаливают до окиси висмута. Сурьма, ртуть, олово и серебро мешают определению.
При отделении кремнекислоты от висмута необходимо подобрать такие условия, чтобы не происходило ни образования оксисоединений, ни гидролиза. По этим причинам в присутствии висмута обезвоживание кремнекислоты лучше проводить в сернокислом или, возможно, хлорнокислом растворе.
Отделение свинца от висмута выпариванием с серной кислотой не является количественным, поскольку висмут захватывается осадком сульфата свинца.
Для отделения малых количеств висмута «0,02 г) от больших количеств свинца, например при анализе металлического свинца, поступают следующим образом 2. Анализируемые металлы получают в виде их нитратов в растворе объемом 300 мл, прибавляют 3—5 капель 1,5%-ного раствора конго красного, приливают раствор едкого натра до перехода окраски в синевато-красную, что соответствует pH 3—5,2, перемешивают и добавляют 20 мл 0,7%-ного раствора солянокислого цинхонина. Дают постоять 30 мин, фильтруют и проверяют фильтрат на полноту осаждения висмута, прибавив к нескольким его каплям иодид калия (в присутствии висмута получается коричневое окрашивание, при отсутствии висмута выделяется только желтый иодид свинца). Если висмут будет обнаружен в фильтрате, последний снова нейтрализуют щелочью и повторяют осаждение солянокислым цинхонином. Отфильтрованный осадок отмывают от свинца холодной водой, содержащей 10 мл указанного выше раствора цинхонина в 1 л, и обработанной азотной кислотой до перехода окраски конго красного в синевато-красную. Промытый осадок растворяют в горячей азотной кислоте, раствор выпаривают с серной кислотой до появления паров последней, разбавляют водой, отфильтровывают сульфат свинца, если он выделился, и определяют висмут колориметрическим иодидным способом. ч
Малые количества висмута, так же как и малые количества мышьяка и сурьмы, могут быть осаждены аммиаком вместе с гидроокисью железа. Для этого в раствор предварительно вводят небольшое количество соли железа (1П), если последнее не находилось уже в растворе в достаточном количестве (стр. 360). В связи с этим надо отметить, что осаждение висмута аммиаком или едкими щелочами протекает не полно, если в растворе отсутствуют другие осаждаемые этими реактивами элементы. Осаждение аммиаком протекает, однако, почти количественно в присутствии избытка хлорида аммония 3.
* Тиосульфат натрия при кипячении осаждает висмут вместе с медью, серебром и частично с мышьяком и сурьмой. Кадмий и катионы первых трех аналитических гр'уЦп остаются в растворе.
1 L. К i е f t, G. С. Chandie е, Ind. Eng. Chem., Anal. Ed., 8, 392 (1936).
1 C. Frick, H. Engemann, Chem. Ztg, 53, 601 (1929).
* Об отделении висмута, основанном на восстановлении его до элементарного
состояния гипосульфитом натрия Na2S2O4, см. В. S. Evans, Analyst, 54, 395 (1929).
Метод» отделения
273
Отделение висмута от свинца методом внутреннего электролиза 1 может быть проведено точно и быстро, если применять прибор без диафрагмы (см. стр. 170).
Условия выделения висмута внутренним электролизом в приборе без диафрагмы в присутствии многих других элементов были подробно исследованы Ю. Ю. Лурье и Л. Б. Гинзбург 2, разработавшими метод выделения висмута из свинцово-цинковых, медных и медно-цинковых руд.
Ход отделения. Навеску 0,5—2,0 г руды (в зависимости от содержания меди и висмута, суммарное количество которых не должно превышать 20 мг) помещают в коническую колбу емкостью 250 мл, разлагают добавлением смеси азотной и соляной кислоты (3:1) и выпаривают почти досуха.
При анализе окисленных свинцово-цинковых руд и концентратов разложение их лучше проводить одной соляной кислотой. К остатку после выпаривания прибавляют 20—30 мл разбавленной (1 : 1) азотной кислоты и кипятят несколько минут. Затем прибавляют 15—20 мл горячей воды, нагревают до кипения и фильтруют через фильтр средней плотности. Полученный азотнокислый раствор осторожно нейтрализуют аммиаком до появления осадка гидроокиси железа, растворяют этот осадок добавлением концентрированной уксусной кислоты (80%-ной) и приливают еще 10 мл ее избытка. Разбавляют водой до 200 мл, нагревают до 90° С и восстанавливают железо добавлением гидразина до обесцвечивания раствора (можно применять сернокислый гидразин, но лучше пользоваться основанием гидразина, чтобы не вводить в раствор слишком большого количества сульфат-ионов). Опускают в раствор соединенные электроды (см. стр. 170) и оставляют их в растворе на 40 мин, поддерживая температуру раствора 85—90° С.
Затем вынимают электроды, разъединяют их, промывают платиновый катод, опуская его на несколько минут в, стакан с водой, подкисленной уксусной кислотой, переносят в другой стакан, приливают 40 мл разбавленной (1 : 4) азотной кислоты и нагревают до растворения висмута и меди. Платиновый электрод обмывают после этого над стаканом водой.
Если определение висмута заканчивают колориметрическим методом с применением тиомочевины, то никаких дальнейших разделений проводить не надо. Если предполагают закончить определение висмута колориметрическим методом с применением иодида калия, то надо отделить висмут от меди. Для этого к раствору добавляют 0,2 г алюминиевых квасцов, аммиак до слабощелочной реакции и 0,1 г карбоната аммония, после чего нагревают до тех пор, пока осадок не станет хлопьевидным. Отфильтровав его и промыв 2%-ным раствором карбоната аммония,' растворяют в горячей разбавленной соляной, кислоте и проводят определение.
Следует отметить, что при определении висмута колориметрическим методом с тиомочевиной (см. стр. 278) в большинстве случаев его можно не отделять от других элементов.
Предложен 3 метод отделения висмута от свинца, меди и кадмия с помощью буферной смеси, состоящей из пиридина и нитрата пиридина,
1 В. И. К о л о с о в, Ю. Ю. Л у р ь е, см. сноску 1 на стр. 170.
2 Ю. Ю. Лурье, Л. Б. Гинзбург, Зав. лао., 7, 11 (1938).
3 Э. А. Остроумов, Зав. лаб., 8; 1226 (1939).
18 Заказ 522.
274
Гл. X. Висмут
имеющей pH около 4,2. Если содержание свинца в растворе превышает 0,1 г, требуется переосаждение, в других случаях достаточно одного-осаждения. По этому методу азотнокислый анализируемый раствор объемом около 100 мл нагревают примерно до 80° С и нейтрализуют 10 %-ным раствором аммиака, прибавляя его осторожно по каплям при сильном перемешивании до появления неисчезающей мути. Последнюю растворяют, приливая 5—6 капель азотной кислоты, к прозрачному раствору добавляют 6—8 г нитрата аммония, нагревают до начала кипения и, сняв с горелки, прибавляют при непрерывном перемешивании по каплям из капельной воронки 20 мл осаждающего реактива х. Затем раствор нагревают до кипения и дают постоять 30—40 мин на электроплитке (не доводя до кипения), покрыв часовым стеклом и периодически перемешивая. Выделившийся осадок отфильтровывают (во время фильтрования не следует давать раствору охлаждаться), промывают горячим 3 %-ным раствором нитрата аммония, к 100 мл которого прибавлен 1 мл осаждающего реактива, и растворяют на фильтре в горячей разбавленной (1 : 1) азотной кислоте. Раствор собирают во взвешенную ^платиновую чашку и упаривают на водяной бане досуха, сухой остаток смачивают 1—2 мл воды, прибавляют 5—10 капель 10%-ного раствора аммиака, снова выпаривают досуха, осторожно прокаливают для удаления аммонийных солей, затем повышают температуру и прокаливают до образования окиси висмута. В фильтрате можно определить свинец, медь и кадмий обычными методами. Отделение висмута от свинца и других двухвалентных металлов может быть также успешно проведено при помощи комплексона III (стр. 157). Для этого к кислому анализируемому раствору приливают в небольшом избытке 0,1 М раствор комплексона, равный объем 0,1 М раствора нитрата кальция и аммиака до запаха последнего. Выпадает осадок гидроокиси висмута.
При осаждении малых количеств висмута (меньше 5 мг), после добавления осаждающего реактива осадок сразу не выпадает. В этом случае прибавляют немного бумажной массы, снова нагревают до кипения, переносят на электроплитку и дают постоять при частом перемешивании 2—3 ч. Доп. ред.*
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Висмут обычно определяют весовыми методами (*болыпие его количества, малые — обычно определяют колориметрическими методами*). Наиболее удобным является конечное взвешивание в виде Bi2O3, особенно когда определяют большие количества висмута. Взвешивание в виде ВЮС1 возможно только в тех случаях, когда содержание висмута не, превышает нескольких миллиграммов.
Весовые определения
Определение в виде BiOCl (количество висмута не превышает 5 мг). Хлорокись висмута обычно осаждают нейтрализацией азотнокислого раствора соли .этого элемента аммиаком, прибавлением нескольких капель
1 К 20 мл перегнанной азотной кислоты пл. 1,32—1,33 (или 15 мл азотной кислоты пл. 1,4), отмеренным точным цилиндром, прибавляют примерно равный объем воды, затем при перемешивании приливают 34 мл пиридина 'и разбавляют водой до 100 лм; 20 мл этого реактива достаточно для осаждения 0,2 г висмута. '
- Методы определения
275
соляной кислоты, сильным разбавлением водой и нагреванием на паровой бане. При этом должны отсутствовать: серебро, ртуть (I), таллий (I), образующие нерастворимые хлориды, элементы, соли которых легко гидролизуются, как, например, сурьма, олово и цирконий, а также суль-фат-ионы, фосфат-ионы и арсенат-ионы, которые образуют с висмутом нерастворимые осадки. При выделении больших количеств висмута состав полученного таким способом осадка может полностью не соответствовать формуле BiOCl, и поэтому его нельзя непосредственно взвешивать.
Ход определения. Приготовляют азотнокислый раствор, свободный от мешающих определению перечисленных выше веществ, разбавляют до 100 мл и затем при постоянном перемешивании приливают из бюретки по каплям разбавленный (1 : 2) раствор аммиака до появления слабой опалесценции. Если выпадает осадок, то его растворяют добавлением разбавленной (1,: 4) азотной кислоты и раствор снова тщательно нейтрализуют аммиаком. Прибавляют 5 мл разбавленной (1 : 9) соляной кйс-лоты, разбавляют раствор горячей водой до 400 мл, нагревают до кипения, накрывают часовым стеклом и оставляют стоять 2 ч или, лучше, на ночь на краю паровой бани.
Фильтруют через бумажный фильтр и промывают стакан, фильтр и осадок двумя-тремя маленькимц порциями горячей воды. Осадок растворяют в 4 мл нагретой до кипения разбавленной (1 : 9) соляной кислоты, приливая кислоту из пипетки на края фильтра и собирая раствор в стакан, где происходило осаждение. Промывают фильтр горячей водой, затем 1 мл горячей разбавленной соляной кислоты и снова горячей водой. Полученный раствор разбавляют горячей водой до 400 мл и снова проводят осаждение, как описано выше. Если количество висмута мало, то фильтруют через взвешенный тигель Гуча, промывают осадок горячей водой, затем спиртом, сушат при 100° С, охлаждают в эксикаторе и взвешивают в виде BiOCl.
Определение в виде Bi2O3 (количество висмута выше 5 мг). Когда количество висмута превышает 5 мг, его лучше определять, взвешивая в виде Bi2O3 после осаждения в виде основного карбоната висмута раствором карбоната аммония. При этом осаждении должны отсутствовать хлориды и сульфаты, которые могут образовать нерастворимые основные соли висмута, а также и другие элементы и соединения, осаждающиеся сами или вместе с висмутом при прибавлении к раствору карбоната аммония. Теми малыми количествами хлорид- и сульфат-ионов, которые могут быть введены в раствор, если висмут был предварительно отделен в виде хлорокиси или сульфида, а затем растворен в азотной кислоте, можно пренебречь, и поэтому осаждение висмута в виде основного карбоната может следовать за этими предварительными отделениями. Если присутствуют только медь и висмут, то отделение висмута может быть проведено непосредственно аммиаком, но осадок в этом лучше переосадить. Основной карбонат висмута несколько растворим в растворе карбоната аммония, поэтому при осаждении надо избегать избытка последнего.
Ход определения. Если присутствует свинец, висмут предварительно отделяют повторным осаждением в виде хлорокиси, как указано выше, или осаждением в виде бромокиси или основного нитрата (стр. 269—270). Осадок растворяют в горячей разбавленной (1 : 3) азотной кислоте и раствор дважды выпаривают до сиропообразной консистенции с добавлением
18*
276
Гл. X. Висмут
азотной кислоты в тех случаях, когда предварительное выделение проводилось в виде хлорокиси или бромокиси висмута. Раствор нейтрализуют почти до конца аммиаком, разбавляют до 200—300 мл и прибавляют насыщенный раствор карбоната аммония до небольшого его избытка. Нагревают до кипения и дают постоять на водяной бане 2 ч. Фильтруют через бумажный фильтр и умеренно промывают осадок горячей водой. Если осадок мал, его растворяют в горячей разбавленной азотной кислоте, собирая раствор во взвешенный платиновый тигель или чашку, и выпаривают досуха. Если осадок велик, фильтр с его содержимым высушивают, переносят бблыпую часть осадка на часовое стекло и растворяют оставшийся на фильтре осадок, как указано выше. Раствор выпаривают досуха, прибавляют главную массу осадка и прокаливают сначала осторожно, потом на полном пламени бунзеновской горелки (около 1000° С) при хорошем доступе воздуха. Охлаждают и взвешивают в виде В12О3.
Определение в виде BiPO4 (количество висмута до 250 мг). Фосфат висмута BiPO4 — белый, тяжелый кристаллический усадок, практически нерастворимый в воде и разбавленной азотной кислоте и вполне устойчивый при прокаливании. Сульфат- и хлорид-ионы в небольших количествах соосаждаются и их надо предварительно отделить, так же как ионы свинца, циркония и других элементов, образующих нерастворимые фосфаты в разбавленных растворах кислоты. Кадмий мешает в незначительной степени, натрий, калий, магний, кальций, цинк и медь не мешают.
Ход определения х. К 100 мл холодного разбавленного азотнокислого раствора, содержащего не более 250 мг висмута в виде его нитрата и свободного от сульфат- и хлорид-ионов, а также от элементов, образующих нерастворимые фосфаты в разбавленных растворах азотной кислоты, медленно и при непрерывном перемешивании приливают разбавленный (1 : 1) раствор аммиака до появления мути, которую растворяют добавлением 5 мл азотной кислоты. Нагревают до кипения и прибавляют 30 мл 10%-ного раствора (NH4)2HPO4 в разбавленной (1 : 9) азотной кислоте. Реактив прибавляют из бюретки, сначала очень медленно по каплям при непрерывном перемешивании раствора. После прибавления всего указанного количества осаждающего реактива разбавляют до 300 мл кипящей водой и дают осадку осесть в течение получаса при 80° С. Затем фильтруют через предварительно взвешенный фарфоровый тигель с пористым дном, переносят в тигель осадок и промывают его горячим 2 %-ным раствором нитрата аммония, содержащим несколько капель азотной кислоты в 1 л. Промытый осадотГПрокаливают сначала очень осторожно, потом при 800° С.
Можно также перенести осадок на бумажный фильтр, затем фильтр с осадком после его промывания осторожно обуглить, прокалить при возможно более низкой температуре до сгорания всего угля и, наконец, довести температуру прокаливания до 800° С. В том и другом случае тигель с прокаленным осадком охлаждают в эксикаторе и взвешивают BiPO4.
1 Подробности см. в статьях: W. R. Schoeller, Е. F. Waterhouse, Analyst, 45, 435 (1920); W. С. В 1 a s d a 1 e, W. G. Parle, Ind. Eng. Chem., Anal. Ed., 8, 352 (1936); W. R.Schoeller, D. A. Lambie, Analyst, 62, 533 (1937). В последней статье приводится метод определения висмута в богатых этим металлом рудах.
Методы определения
277
Колориметрические определения
Определение с иодидом калия. Малые количества висмута, от 0,05 до 0,5 мг, лучше всего определять колориметрическим методом, сравнивая желтую или коричневую окраску, полученную в результате обработки разбавленного азотнокислого раствора соли висмута иодидом калия, с окраской стандартного раствора. Определению мешают медь и железо (III), которые реагируют с иодидом калия, выделяя иод, некоторые члены мышьяковой группы, также дающие окрашенные растворы с иодидом калия, и, наконец, соли, которые сами сильно окрашены (как, например, нитрат никеля), если они присутствуют в достаточном количестве. Эти вещества должны быть удалены общими, или специальными способами отделения соответственно каждому отдельному случаю х. Свинец не создает затруднений, если не присутствует в очень больших количествах, потому что желтый иодид 'свинца можно отфильтровать перед определением висмута. Большие же количества иодида свинца могут увлечь в осадок висмут.
Ход определения. Приготовляют 20—30 мл бесцветного раствора, содержащего 2 мл свободной азотной кислоты и не содержащего железа, меди и членов группы мышьяка. Раствор переводят в цилиндр Несслера, прибавляют 1 мл насыщенного раствора SO2, 5 мл 30%-ного раствора иодида калия и разбавляют до 50 мл. В другой цилиндр наливают 40 мл воды, те же количества иодида калия, азотной кислоты и насыщенного раствора SO2 и добавляют по каплям стандартный раствор соли висмута 1 2 до тех пор, пока жидкости в обоих цилиндрах не окажутся совершенно одинаково окрашенными. Сернистая кислота придает раствору слабое окрашивание, поэтому при очень малом содержании висмута ее надо добавлять в умеренном количестве, всего 1—3 капли. В этом случае после того, когда сравнение окрасок было проведено, надо прибавить еще немного сернистой кислоты, чтобы быть уверенным, что окраска не была частично вызвана иодом. Для этой же цели после определения прибавляют к обоим растворам немного раствора крахмала 3.
1 Например, при анализе медной проволоки рекомендуется следующий метод: 100 г пробы помещают в стакан емкостью 2500 мл и растворяют в 700 мл разбавленной (1 : 1) азотной кислоты. Прибавляют 0,5 г карбоната кальция, разбавляют до 1500 мл, кипятят и осторожно прибавляют разбавленный (1:1) раствор аммиака до растворения всех основных солей. Прибавляют 2—3 г карбоната аммония н оставляют стоять по меньшей мере на 30 мин. Фильтруют через пластинку Витта при отсасывании и промывают осадок разбавленным (1 : 20) раствором аммиака, содержащим 1% карбоната аммония, до удаления растворимых солей меди. Остаток растворяют в разбавленной (1 : 1) азотной кислоте, разбавляют до 150 мл, подщелачивают аммиаком, кипятят, снова осаждают, как описано выше, осадок отфильтровывают и промывают. Промытый осадок растворяют в растворе 2 мл азотной кислоты в 10 мл воды и промывают фильтр так, чтобы объем полученного раствора не превысил 30 мл. Для выделения малых количеств висмута из растворов, получаемых при анализе металлической меди, рекомендуется добавление хлорида железа (III) (в умеренном количестве) и осаждение аммиаком.
2 Приготовляется растворением 1 г висмута в 10 мл азотной кислоты и разбавлением до 1 л.
3L. С. Nickolls [Analyst, 58, 684 (1933)] рекомендует проводить осаждение добавлением 0,05—5 мл раствора сульфата олова (II) [10 г хлорида олова (II) растворяют в 100 мл 6 н. серной кислоты и дают осадку осесть, пока жидкость над ним не станет прозрачной]; в этом случае раствор в «холостом» опыте остается бесцветным.
278
Гл. X. Висмут
Определение с тиомочевиной. Несколько большие количества висмута (от 0,1 до 4 мг) могут быть определены фотометрически * 1 в разбавленном азотнокислом растворе добавлением тиомочевины и измерением свето-поглощения образовавшегося окрашенного в желтый цвет комплексного соединения при длине волны света 425 ммк. Сурьма, палладий, осмий и рутений также образуют с тиомочевиной в кислом растворе окрашенные комплексные соединения2. Добавление фтористоводородной кислоты предупреждает образование окрашенного соединения сурьмы; серебро, ртуть, свинец, медь, кадмий и цинк образуют белые осадки, когда присутствуют в значительных количествах; если же содержание этих элементов невелико, то ни осадков, ни окрашивания раствора не получается. Железо, при содержании его, превышающем 0,1 мг в 50 мл, должно быть удалено или восстановлено до двухвалентного состояния 3. Селен и теллур мешают определению 4 *.
* Колориметрический метод определения висмута с тиомочевиной имеет то преимущество, что он значительно меньше подвержен мешающему влиянию других присутствующих в растворе элементов. Висмут в металлическом свинце может быть определен без предварительных отделений, если проводить это определение так, как указано ниже. Больше других металлов мешают сурьма и железо (III). Сурьму можно связать в бесцветное комплексное соединение добавлением фторидов, но в присутствии свинца лучше для этой цели применять винную кислоту. Мешающее влияние железа объясняется присутствием в продажных препаратах тиомочевины примеси роданида аммония, образующего с железом (III) окрашенное в красный цвет комплексное соединение. Для предотвращения этого железо (III) может быть предварительно восстановлено (частично оно восстанавливается и самой тиомочевиной, но при этом розовое окрашивание все же может появиться). Поэтому рекомендуется вводить в стандартный раствор при колориметрировании такое же количество нитрата железа, какое имеется в анализируемом растворе.
Исследования 8 показали, что 1) концентрация азотной кислоты в растворе должна быть в пределах 0,4—1,2 н.; 2) интенсивность окрашивания повышается с увеличением концентрации тиомочевины в растворе, но при любой ее величине пропорциональность между концентрацией висмута и величиной светопоглощения раствора сохраняется. В присутствии свинца не рекомендуется все же увеличивать концентрацию тиомочевины выше 1 %; 3) подчинение закону Ламберта — Бера наблюдается при концентрации висмута в растворе в пределах 2—16 мг!л (при толщине кюветы I — 13 мм)', относительная ошибка определения не превосходит тогда 1%; 4) окрашивание устойчиво в течение 1,5 ч, а если применяемая тиомочевина не содержит примесей, то и значительно дольше.
Для определения висмута в металлическом свинце рекомендуется8 следующий ход анализа. Навеску в 1 г анализируемого свинца раство-
' 1 ASTM, Methods of Chemical Analysis of Metals, 195P, p. 374, 384; В. В. В e n-
digo, R. K. Bell, H. A. Bright, J. Resarch NBS, 47, 252 (1951).
1 J. H. Yoe, L. G. О vernolser, Ind. Eng. Chem., Anal. Ed., 14, 435 ;
(4942).
» C. Mahr, Z. anal. Chem., 94, 161 (1933); 97, 96 (1934).
4 P. W. West, J. V. Tokos, Ind. Eng. Chem., Anal. Ed., 16, 761 (1944). (
6 Ю. Ю. Лурье, Л. Б. Гинзбург, Зав. лаб., 15, № 1, 21 (1949).
Методы определения
279
ряют при нагревании в 15 мл разбавленной (1 : 9) азотной кислоты, прибавляют 20 мл воды, 1 г винной кислоты, 10 мл 5 %-ного раствора тиомочевины, разбавляют водой точно до 50 мл и перемешивают. Затем переносят часть растворд в кювету фотоколориМетра и измеряют его свето-поглощение, применяя фиолетовый светофильтр с максимальным пропусканием при 400 ммк. В качестве нулевого раствора следует применять раствор такой же навески свинца со всеми реактивами, кроме тиомочевины. Содержание висмута находят по калибровочной кривой, для построения которой приготовляют серию растворов, содержащих такое же количество свинца в виде его нитрата, какое содержится во взятой навеске, но различные количества висмута, и вводят такие же количества азотной кислоты и тиомочевины. .
Для определения очень малых количеств висмута в металлическом свинце 1 (десятитысячные доли процента) висмут предварительно отделяют от большей части свинца гидролизом. Навеску свинца в 10 г растворяют в разбавленной (1 : 4) азотной кислоте, выпаривают до малого объема для удаления большей части свободной кислоты, остаток растворяют в небольшом-количестве воды, нейтрализуют 2 н. раствором карбоната натрия до появления мути, прибавляют сверх того еще 1—2 мл 2 н. раствора карбоната натрия и кипятят 1—2 мин. Выделившийся осадок, содержащий весь висмут и некоторое количество свинца, растворяют в разбавленной (1 : 4) азотной кислоте и определяют висмут колориметрическим методом с тиомочевиной. Доп. ред.* *
Другие методы
Другие методы определения висмута — весовые, электролитические и объемные — менее удовлетворительны. Среди весовых методов имеются такие, в которых висмут определяется в виде сульфида Bi2S3 и в виде металла после восстановления окиси висмута или сульфида висмута сплавлением с цианидом калия 2. Но ни один из этих методов не может считаться точным. То же самое можно сказать и об электролизе в разбавленном сернокислом или азотнокислом растворе 3 и об объемном методе определения висмута осаждением его в виде основного оксалата с последующим титрованием перманганатом А
Висмут можно осадить 8 в виде комплексного роданохромиата висмута Bi[Gr(CNS)eI добавлением роданохромиата калия K3[Cr(CNS)e] к холодному раствору соли висмута 0,3—1 н. по содержанию азотной или серной кислоты. Так, висмут отделяется от молибдена, железа, алюминия, хрома, цинка, марганца, никеля, кобальта и магния. Осадок переносят в стеклянный фильтрующий тигель, промывают холодной водой и высушивают при 120—130° С. Можно также разложить осадок добавлением азотной кислоты, в полученном растворе определить содержание хрома объемным способом и пересчитать на содержание висмута.
1 А. И. Б у с е в, Н. П. Корец, Зав. лаб., 15, 30 (1949).
* 2 Н. Rose, Pogg. Ann., 110, 425 (1860).
8 О. Bru nek, Вег., 35, 1871 (1912).
* М. М. Pattison, Muir, J. Chem. Soc., 33, 70 (1878). Критический обзор объемных методов определения висмута, оксалатного, хроматного, молибдатного и др. дан в статье L. Moser, Z. anal. Chem., 46, 231 (1907)..
8 С. Mahr, Z. anorg. und allgem. Chem., 208, 313 (1932).
280
Гл. X. Висмут
Предложен также 1 способ осаждения висмута в виде соли иодовисмутовой кислоты с хинальдином C8HeNGH3 • HBiI4. Осадок можно затем растворить и определить содержание иода титрованием раствором йодата калия. Этим способом в разбавленном (1 : 9) сернокислом растворе можно осадить даже такое малое количество висмута, как 0,3 мг, в присутствии свинца, меди, кадмия, сурьмы, олова, мышьяка, марганца, никеля, кобальта, цинка, железа, хрома, урана, алюминия, бериллия и фосфора. Определению мешают ртуть, серебро и большие количества хлорид-ионов.
*Из других методов определения висмута следует' отметить также: 1) осаждение его пирогаллолом в виде BiO3G6H3; определение можно заканчивать взвешиванием этого осадка 2 или колориметрическим методом 3, основанным на растворении осадка в соляной кислоте, прибавлении фосфорномолибденововольфрамовой кислоты (реактива Фолина и Дениса) и соды до щелочной реакции, в результате чего возникает голубое окрашивание; 2) купфероновый метод (см. выше, стр. 271); 3) дитизоновый колориметрический метод 4 5; 4) осаждение висмута оксихинолином и иодидом калия в виде C8H7ON • HBiI4, после чего осадок отфильтровывают и заканчивают определение объемным йодометрическим методом 8. Полученное соединение можно также экстрагировать смесью ацетона и амилового спирта или циклогексанола 6 * или же одним циклбгексанолом и заканчивать определение колориметрированием полученного красного экстракта.
Большое число других методов отделения и определения висмута описано в указанной выше (стр. 269, сноски 1) монографии А. И. Бу-сева. Доп. ред*
1 J. R. Hayes, G. С. Chandlee, Ind. Eng. Chem., Anal. Ed., 11, 531 (1939).
2 F. F e i g 1, H. О г d e 1 t, Z. anal. Chem., 62, 372 (1923); см. также Э. A. О c-т p о у м о в, Зав. лаб., 4, 1016 (1935); Z. anal. Chem., 106, 36 (1936).
3 M. Teitelbaum, Z. anal. Chem., 82, 366 (1930).
4 D. M. Hubbard, Ind. Eng. Chem., Anal. Ed., 11, 343 (1939); Anal. Chem., 20, 363 (1948); см. также E. Б. Сендэл, Колориметрическое определение следов металлов, Госхимиздат, 1949, стр. 172.
5 R. Berg, О. Wurm, Вег., 60, 1668 (1927); I. М. Kolthoff,
F. S. Griffith, Mikrochim. Acta, 3, 46 (1938).
eR. Sazerac, J. Pouzergues, C. r. Soc. biol., 109, 370 (1932).
Глава XI
МЕДЬ
В изверженных горных породах медь редко определяют количественно, хотя небольшие ее количества встречаются в них часто. Главными медными рудами являются самородная медь, сульфиды, окиси и карбонаты меди. Менее важны арсениды, арсенаты, антимониды, фосфаты, сульфаты и силикаты меди.
Определение меди в минералах, рудах и продуктах металлургического производства имеет очень важное значение особенно потому, что медь в этих материалах определяется непосредственно, а не рассчитывается по разности, как это имеет место при определении железа, алюминия и цинка в сплавах, где эти элементы преобладают.
ОБЩИЕ ЗАМЕЧАНИЯ
При определении малых количеств меди в горных породах надо особенно внимательно следить за тем, чтобы не произошло загрязнения анализируемой пробы медью из латунных или бронзовых сит, реактивов или из других источников. В обычном ходе анализа материалов, содержащих умеренные количества меди, большая часть меди совершенно не осаждается, если обработка сероводородом опускается и, как правило, применяется двукратное осаждение последующими реактивами. Незначительная часть меди увлекается, однако, осадком от аммиака и принимается за алюминий. Другая незначительная часть меди осаждается вместе с магнием в виде фосфата и принимается за магний. В осадок оксалата вместе с кальцием медь не переходит, если перед осаждением оксалатом аммония было прибавлено достаточное количество аммиака, чтобы удержать медь в растворе. Малые количества меди, встречающиеся в горных породах, обычно выделяют вместе с марганцем, никелем и т. п. осаждением бесцветным сульфидом аммония после отделения кремнекислоты и осадка от аммиака. Такой метод допустим только при наличии малого количества меди, потому что в том слабощелочном растворе, какой необходим для полного осаждения алюминия (стр. 565) \ осадок от аммиака склонен увлекать с собой медь, несмотря на двукратное осаждение.
1 G. Е. F. L u n d е 11, Н. В. Knowles, J. Am. Chem. Soc., 45, 680 (1923). Например, прп анализе смеси 0,1 г алюминия и 0,05 г меди в осадке от аммиака оказалось 0,0211 г меди при первом его осаждении и 0,0077 г меди при втором осаждении. Осаждение аммиаком проводилось в условиях, благоприятных для полного выделения алюминия (стр. 568). Лучшего отделения меди можно достичь, прибавляя NH4OH и NH4C1 в избытке, но тогда такие элементы, как алюминий, осаждаются не полностью.
282
Гл. XI. Медь
При наличии значительного количества меди или в случае необходимости особо точного ее определения медь должна быть выделена из кислого раствора сероводородом или электролизом перед осаждением «полуторных окислов» аммиаком х.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ МЕДЬ
Разложение' медных руд и минералов не вызывает затруднений и может быть проведено обработкой минеральными, кислотами. Лучше обрабатывать сперва соляной кислотой для растворения окисленных минералов и затем прибавлять азотную рислоту для растворения сульфидных минералов. При наличии в пробе силикатов и других нерастворимых веществ может потребоваться обработка остатка фтористоводородной кислотой или сплавление его с карбонатом натрия или пиросульфатом калия.
Для разложения труднорастворимых сульфидных медных руд рекомендуется 1 2 0,15—3,0 г тонко измельченной руды обработать смесью 40 мл 15 н. азотной кислоты и 3 мл 12 н. соляной кислоты, прокипятить примерно 30 мин,, прибавить 20 мл 36 н. серной кислоты, выпарить до появления густых паров последней (при этом получается сульфат меди), охладить и разбавить водой.
МЕТОДЫ ОТДЕЛЕНИЯ
Наиболее удовлетворительные методы отделения небольших количеств меди от большого числа других элементов основаны на свойствах сульфида меди.
1) Медь может быть отделена от элементов, не принадлежащих к группе сероводорода, и даже от кадмия (являющегося членом этой группы) осаждением сероводородом в разбавленном солянокислом или сернокислом растворе (стр. 84) 3.
2) От элементов группы мышьяка медь может быть отделена осаждением едким натром и сульфидом натрия (стр. 87).
3) От кадмия, висмута и др. медь отделяется осаждением этих элементов в виде их сульфидов в щелочном цианидном растворе (стр. 89).
4) От олова и германия медь отделяется осаждением сероводородом в растворе, содержащем фтористоводородную или щавелевую кислоты (стр. 89).
Некоторые из перечисленных выше элементов удобно, однако, отделять индивидуальными реакциями, например серебро — в виде хлорида
1 Об отделении закиси меди от металлической меди и от окиси меди при анализе смеси всех трех и об определении каждой из них в отдельности см. L. С. Ни г d, A. R. Clark, Ind. Eng. Chem., Anal. Ed., 8, 380 (1936).
2 T. H. Whitehead, Ind. Eng. Chem., Anal. Ed., 6, 298 (1934).
3 Об отделении медп от цинка см. стр. 479. В опытах Н. В. Knowles полное отделение 0,5—0,05 г меди от 0,05—0,5 г кадмия достигалось при соблюдении следующих условий: 1) оба эти элемента в виде их сульфатов находились в 100 мл раствора, в котором концентрация свободной серной кислоты была в границах от 9 до 18 н.; 2) температура раствора была равна 95—100° С, и сероводород пропускался через него 30 мин\ 3) фильтрование после осаждения проводилось немедленно и 4) осадок сульфида меди промывали горячей, предварительно насыщенной сероводородом серной кислотой той же концентрации, какая была в анализируемом растворе; всего на промывание расходовалось 100 мл этой кислоты.
' I
)
Методы, отделения
283
>(стр. 236), свинец — в виде сульфата (стр. 262) или двуокиси (стр. 264), висмут — осаждением карбонатом аммония (стр. 270), ртуть — обработкой сульфидов азотной кислотой (стр. 247), мышьяк и германий — перегонкой их хлоридов или фторидов (стр. 303 и 346).
Предварительное отделение очень малых количеств висмута, ' мышьяка, сурьмы, селена и теллура от больших количеств меди проводится лучше всего прибавлением 0,1—0,2 г железа (III) или алюминия в виде их солей и осаждением аммиаком (стр. 359). Большие количества селена и теллура лучше всего отделять осаждением двуокисью серы в солянокислом растворе (стр. 386).
Осаждение меди едким натром (стр. 109) служит для отделения ее от таких элементов, как молибден, ванадий, мышьяк и вольфрам, при условии, что органические соединения отсутствуют. Конечная концентрация едкого натра в растворе не должна превышать 1 %, осаждение надо проводить в горячем растворе и затем кипятить 2—3 мин, после чего дать постоять 30 мин и фильтровать. Отделение меди от олова и сурьмы этим •способом неудовлетворительно, потому что при добавлении едкого натра в количестве, достаточном для удержания олова и сурьмы в растворе, медь осаждается не полностью. Медь может быть количественно отделена от мышьяка, селена и теллура осаждением едким натром в кипящем растворе, конечная щелочность которого тщательно приводится к pH = 10. При pH = 1,5 медь не осаждается, и поэтому ее можно отделить от элементов, количественно осаждающихся при этой концентрации ионов водорода х, например от олова (IV) в солянокислом растворе.
Прекрасным методом отделения меди от кобальта, никеля, марганца, цинка, мышьяка, олова, висмута и сурьмы является осаждение ее в виде роданида меди (I). Ход анализа 1 2 следующий. Приготовляют раствор, содержащий 0,1 а меди в виде ее сульфата в 5 мл серной кислоты, прибавляют 30 мл 10%-ного раствора винной кислоты и нагревают до растворения растворимых солей. Немного охлаждают, приливают раствор аммиака .до щелочной реакции, затем серную кислоту точно до кислой реакции и сверх того еще 1 мл избытка. К раствору, который должен быть теперь горячим, прибавляют 2 мл сульфита натрия, размешивают до растворения соли и затем вливают раствор 1 г роданида калия в небольшом количестве воды. Сильно перемешивают, нагревают до кипения и дают отстояться несколько минут. Фильтруют через плотный бумажный фильтр . и промывают осадой раствором, содержащим 1% роданида калия и такое же количество винной кислотй. Фильтр с осадком помещают обратно в сосуд, где происходило осаждение, и обрабатывают его 20 мл разбавленной (1 : 2) азотной кислоты. Покрыв стакан часовым стеклом, нагревают , до кипения, прибавляют 2Q мл воды, фильтруют, промывают фильтр вместе с бумажной массой, сжигают их при низкой температуре в фарфоровом тигле; растворяют золу в разбавленной азотной кислоте и полученный раствор прибавляют к главному раствору. Затем кипятят для разру-шения роданистоводородной кислбты и определяют медь электролизом, как описано далее (стр. 286).
1 R. Gilchrist, J. Research NBS, 30, 89 (1943).
2 G. J. Demorest, J. Ind. Eng. Chem.', 5, 216 (1913). По данным этого автора, остается неосажденным небольшое количество меди, но величина эта не превышает 0,5 мг, а в большинстве случаев она не больше 0,2 мг.
284
Гл. XI. Медь
Восстановлением солей меди до металла с помощью алюминиевой или цинковой пластинки в кислом, предпочтительно сернокислом, растворе можно удовлетворительно отделить медь от железа и молибдена. Для этого необходимо, чтобы все железо было предварительно восстановлено, кислотность раствора к концу восстановления была низка и чтобы некоторое количество восстановителя было помещено на фильтр во время фильтрования. Кадмий, сурьма и олово при этом сопроврждают медь.
При прибавлении сероводородной воды, обычно практикуемом для осаждения невосстановленной меди, увеличивается количество веществ, загрязняющих осадок меди. Если для выделения меди применяют алюминий, то содержание меди в нем должно быть известно, что дает возможность внести поправку, взвешивая пластинку алюминия до и после восстановления.
Электролитическое осаждение меди с целью только ее отделения от других элементов применяется редко, за исключением отделения меди от кадмия (стр. 299) х. Это объясняется отсутствием уверенности в полноте осаждения меди и тем, что большое количество других элементов может частично или даже полностью отложиться на катоде вместе с медью. Отделять медь от кадмия этим способом лучше всего из азотнокислого раствора. Малые количества кадмия можно отделить от меди электролизом в серно-азотнокислом растворе. Никель, кобальт, цинк и умеренные количества железа не мешают. В присутствии первых трех элементов электролиз лучше проводить в азотнокислом или в азотно-сернокйслом растворе. Электролиз в аммиачном растворе или в таком же растворе, но с добавлением фторидов дает хорошее отделение меди от солей мышьяка и сурьмы 1 2.
Осаждение купфероном, хотя оно и было первоначально рекомендовано для определения меди, не дает полного ее отделения, так как многие другие элементы также осаждаются этим реактивом (стр. 143).
,Медь может быть количественно отделена от кадмия, свинца, ртути, марганца, цинка, никеля и магния осаждением нитрозо-Р-нафтолом в сильно разбавленном солянокислом растворе хлоридов или сульфатов 3.
♦Большое распространение в заводских лабораториях имеет метод выделения меди тиосульфатом натрия из сернокислого или солянокислого раствора. Реактив прибавляют небольшими порциями к кипящему раствору до его обесцвечивания (восстановление железа), а затем вводят небольшой избыток. Медь при этом выделяется в виде сульфида меди (I) вместе с серой. Кроме меди, тиосульфатом натрия полностью осаждаются серебро и висмут и частично мышьяк и сурьма. Катионы первых трех аналитических групп, а также и кадмий тиосульфатом не осаждаются (железо, если оно присутствует в больших количествах, частично сорбируется выпадающим осадком).
1 Согласно L. W. М с С а у и N. Н. Furm an [J. Am. Chem. Soc., 38, 640 (1916)], медь может быть отделена от вольфрамовой, молибденовой, оловянной и сурьмяной кислот электролизом в растворе, содержащем азотную и плавиковую кислоты (см. стр. 165, где указана литература, содержащая описания оборудования для проведения электролитических разделений при контролируемом потенциале).
2 L. W. McCay, Chem. Ztg., 14, 509 (1890); N. Н. Furman, Ind. Eng. Chem., Anal. Ed., 3, 217 (1931).
3 E. Hintz, Z. anal. Chem., 28. 234 (1889).
Методы определения
285
Внутренним электролизом медь выделяется в тех же условиях, в каких выделяется висмут (см. стр. 170 и 273).
О выделении меди солью Рейнеке, бензоиноксимом и салицилальдок-симом см. стр. 293. Доп. ред* I
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Из всех методов определения меди наиболее точным является электролитический метод 1 при условии, что удалены все мешающие электролизу элементы, как, например, металлы, стоящие в ряду напряжений ниже меди, и если проведено извлечение последних следов меди из электролита. Преимуществом электролитического метода является и то, что этим методом можно выделить до 5 а меди, в то время как прщдругих методах определения меди допускается ее содержание в растворе не выше 0,2 г. Вслед за электролизом по распространению и точности стоит йодометрический метод 2, обладающий тем преимуществом, что при работе им присутствие других элементов влияет меньше. Наряду с йодометрическим методом, хотя он и не является широко распространенным, стоит роданидный метод в его весовой и объемной модификациях 3.
Определение меди осаждением в виде сульфида и прокаливанием осадка до окиси меди требует предварительного удаления большого числа элементов, причем метод этот неприменим для определения меди в количествах, превышающих 0,01 з, так как трудно разрушить сульфат меди, образующийся при прокаливании. Колориметрические методы удобны для быстрых массовых определений меди или для определения малых ее количеств. Цианидный метод 4 неточен и пригоден только для быстрых контрольных определений. Взвешивание меди в виде окиси меди после осаждения ее щелочью и прокаливания осадка дает совершенно неудовлетворительные результаты вследствие невозможности избежать загрязнения окиси меди щелочью. Представляют интерес, несмотря на их редкое применение, методы, в которых медь осаждается купфероном 5, ацетиленом 6 и сероводородом с превращением полученного осадка 7 сульфида меди (II) в сульфид меди (I).
1S. Skowronski, ASTM Bull., 174, 6 (May 1951); А. Классен, Электроанализ, ОНТИ, 1934, стр. 133; Е. F. Smith, Electro-Analysis, 6th ed., 1918, p. 73.
2 E. de H а ё n, Ann., 91, 237 (1854); A. H. Low, J. Am. Chem. Soc., 18, 458 (1896); 24, 1082 (1902); F. A. Gooch, F. H. Heath, Am. J. Sci., [4], 24, 68 (1907); F. H. Heath, там же, [4], 25, 513 (1908).
3 L. E. Rivot, C. r., 38, 868 (1854); S. W. Parr, J. Am. Chem. Soc., 22, 685 (1900); R. G. Van Name, Z. anorg. Chem., 26, 230 (1901);, D. J. Demorest, J. Ind. Eng. Chem., 5, 215 (1913); W. H. Swanger, E. Wishers,!. Am. Chem. Soc., 46, 1814 (1924).
4 Dr. Steinbeck, Z. anal. Chem., 8, 1 (1869); Chem. News, 19, 207 (1869);
R. S. D u 1 i n, J. Am. Chem. Soc., 17, 346 (1895).
6 O. Baudisch, Chem. Ztg., 33, 1298 (1909); О. В a u di s ch, V. L. К i n g, J. Ind. Eng. Chem., 3, 629 (1911).
3 H. Erdmann, O. Makowka, Z. anal. Chem., 46, 128 (1907); A. W a e g-n e г, там же, 44, 565 (1905); H. G. S о d e r b a u m, там же, 44, 564 (1905).
’ W. Натре, Chem. Ztg., 9, 1441 (1885); Z. anal. Chem., 38, 465 (1894); R. Wegscheider, там же, 38, 466 (1894).
286
Гл. XI. Медь
Электролитический метод
При электролитическом методе определения меди требуется получение прозрачного раствора, свободного от мышьяка, сурьмы, олова, молибдена, золота, платиновых металлов, серебра, ртути, висмута, селена (IV/ и теллура (IV), загрязняющих осадок выделяющейся меди. Кроме того, должны отсутствовать: роданистоводородная кислота, присутствие которой делает осадок меди губчатым, и соляная кислота, действующая аналогично и, кроме того, вызывающая растворение платины на аноде и переход ее на катод. Затем должны отсутствовать окислители, как, например, окислы азота, большие количества нитрата железа (III) или азотной кислоты, которые вначале препятствуют осаждению меди, а потом служат причиной получения высоких результатов, если в конце концов удалось добиться полноты осаждения медиЭлектролиз может быть проведен в азотнокислом или сернокислом растворе,“и обычно его проводят в смеси обеих кислот. Если применяется одна азотная кислота, имеется опасность замедленного или неполного осаждения. Этого можно избежать, прибавляя 1 каплю 0,1 н. раствора соляной кислоты перед началом электролиза 1 2. Катод и анод желательно иметь в виде открытых сетчатых платиновых цилиндров с матированной поверхностью, полученной при помощи пескоструйного-аппарата (стр. 55).
Наиболее точные результаты получаются при медленном осаждении меди на холоду, без перемешивания раствора. Быстрое осаждение при перемешивании электролита воздухом, вращающимися электродами или магнитным вращением обычно дает повышенные результаты, независимо от того, проводится ли это осаждение на холоду или при нагревании. Стакан для электролиза должен быть покрыт двумя половинками часового стекла с вырезанными в них отверстиями для электродов, чтобы избежать потерь от разбрызгивания или загрязнения извне. В полноте осаждения меди можно убедиться, опуская в раствор верхнюю непогруженную часть катода и продолжая электролиз иди отбирая пробу электролита и обрабатывая ее сероводородной водой. Во всех анализах, требующих особой точности, электролит и промывные воды после электролиза надо сконцентрировать выпариванием, обработать сероводородом и определить содержание меди в выпавшем осадке. Отложенная на катоде медь должна иметь шелковистую структуру, оранжево-розовый цвет (цвет семги) и плотно держаться на катоде. Тусклый цвет меди указывает на ее окисление или на присутствие посторонних элементов. Крупнокристаллические или губчатые осадки дают повышенные результаты. При электролизе
1 Мешающее влияние мышьяка не имеет серьезного значения, если осаждение меди проводится из азотнокислого раствора. Его можно преодолеть, проводя пере-осаждение выделившегося осадка меди, для чего платиновую сетку с осадком меди делают анодом. Молибден, наоборот, создает меньше затруднений при применении сернокислого раствора электролита, и влияние его также преодолевается вторичным осаждением меди. Железо мало мешает, если его сначала восстановить до двухвалентного и проводить электролиз в холодном растворе. Хорошего отделения меди от солей мышьяковой кислоты можно также достичь, выделяя ее электролизом из аммиачного раствора (см. стр. 308). Соединения селена и теллура, в которых эти элементы шестивалентны, не мешают, если присутствуют в умеренных количествах и раствор электро-' лита азотнокислый.
2 J. A. Scherrer, R. К. Bell, W. D. Mogerman, J. Research NBS, 22, 697 (1939).
Методы определения
287
растворов чистой меди следует опасаться неполноты осаждения (что приводит к получению низких результатов) и окклюзии газов или окисления (что может быть причиной высоких результатов). Потери меди можно избежать, обрабатывая весь электролит после осаждения меди сероводородом, как сказано выше. Окклюзию газов можно сделать минимальной, осаждая медь медленно, на холоду, и удаляя катод, как только достигнута полнота осаждения. Таким же образом избегают и окисления. Кроме того, высушивание надо проводить, нагревая катод с выделенной на нем медью очень непродолжительное время.
Ход определения.. Приготовляют раствор, содержащий от 0,5 мг до 5 г меди, 5 мл серной кислоты, 3 мл азотной кислоты и не содержащий мешающих определению упомянутых выше веществ. Если первоначально присутствует большее количество кислоты, чем указано, то, если этот избыток невелик, его нейтрализуют аммиаком, если же он значителен, его удаляют выпариванием. Переносят раствор в стакан емкостью 250 мл, разбавляют до 200 мл водой и помещают в него взвешенный цилиндрический катод так, чтобы поверхность катода отстояла от поверхности анода по меньшей мере на 5 мм. Покрывают стакан двумя половинками часового стекла с вырезанными в них отверстиями для электродов и ведут электролиз лучше всего в течение ночи (без перемешивания) током в 0,5 а при напряжении 2 в. Обмывают часовые стекла и испытывают раствор на полноту осаждения. Если медь выделилась полностью, то, не прекращая тока, постепенно вынимают катод и тщательно обмывают его водой. Затем прекращают ток, вынимают электроды, ополаскивают катод спиртом, высушивают 1 мин при 100° С, охлаждают в эксикаторе и взвешивают. При особо точных анализах электролит следует сконцентрировать и после этого проверить на содержание меди сероводородом. Если выделится осадок, следует определить содержание в нем меди. Отложившуюся же на катоде медь проверяют на присутствие в ней тех примесей, каких можно было ожидать, исходя из состава анализируемого материала и той обработки, которой он подвергался х.
Йодометрический метод
Йодометрический метод определения меди основан на том, что при обработке подкисленных растворов солей меди (II) иодидом калия образуется иодид меди (I) и выделяется иод. По точности этот метод очень близок к электролитическому методу и обладает тем преимуществом, что при работе мало отражается присутствие посторонних веществ; это преимущество имеет особенно большое значение при анализе материалов сложного состава; например медных руд. Йодометрическому определению меди мешают: окислы азота, соединения мышьяка (III) и сурьмы (III), реагирующие с иодом; соединения железа (III), молибдена (VI) и селена^!), выделяющие иод из иодида калия; минеральные кислоты в присутствии мышьяка (V) и сурьмы (V), а если последних нет, то помехи возникают, когда концентрация кислот превышает 3% (по объему), и, наконец, избыточные количества ацетата аммония, если из кислот
1 При желании переосадить выделившуюся на катоде медь надо очень осторожно обработать катод разбавленной азотной кислотой в высоком стакане, так как вследствие выделения газов при растворении меди имеется большая опасность механических потерь.
288
Гл. XI. Медь
присутствует только уксусная кислота. Определению не мешают: цинк, мышьяк (V) и сурьма (V), висмут, свинец и серебро. Три последних элемента вступают, однако, в реакцию с иодидом калия, выделяя осадок, и требуют поэтому прибавления добавочного количества этого реактива.
Реакция обыкновенно проводится в уксуснокислом растворе, причем концентрация уксусной кислоты может доходить до 50% по объему. Необходим избыток иодида калия; обычно прибавляют 5 г K.I на 100 мл раствора. Выделившийся иод титруют раствором тиосульфата натрия, титр которого устанавливают по навеске меди, которую растворяют и проводят через все стадии анализа. При титровании крахмал прибавляют лишь тогда, когда большая часть иода будет оттитрована, чтобы воспрепятствовать образованию труднорастворимого соединения иода с крахмалом х.
Следующий ниже метод, применяемый при анализе руд, пригоден вообще для анализа веществ сложного состава. При анализе простых смесей и чистых солей этот метод можно упростить.
Ход определения. Навеску анализируемого вещества, содержащую от 0,05 до 0,25 г меди, обрабатывают 7 мл соляной кислоты в стакане емкостью 250 мл и нагревают. Прибавляют 10 мл азотной кислоты и снова осторожно нагревают до разложения руды. Затем приливают 7 мл серной кислоты, выпаривают до появления ее паров, охлаждают и прибавляют 30 мл воды. Нагревают до растворения ее растворимых солей, охлаждают и прибавляют 4 г гранулированного цинка. Взбалтывают 5—10 мин, нагревают до раств.орения цинка и прибавляют 25 мл сероводородной воды. Отфильтровав осадок, промывают его слегка подкисленной серойо-дородной водой до удаления железа и переводят осадок обратно в стакан возможно меньшим количеством воды. Прибавляют 7 мл азотной кислоты, нагревают до растворения меди и кипятят для удаления оцислов азота. Горячий раствор сливают через фильтр, промывают фильтр несколькими миллилитрами бромной воды и, наконец, промывают фильтр и стакан горячей водой. Фильтрат кипятят для удаления брома, охлаждают и прибавляют аммиак, пока раствор не станет синим. Избытка аммиака следует избегать. Раствор подкисляют уксусной кислотой, приливают избыток в 3—4 мл 80%-ной уксусной кислоты и охлаждают до комнатной температуры. Затем прибавляют 3—5 г иодида калия, растворенного в небольшом количестве воды, хорошо перемешивают и сейчас же титруют раствором тиосульфата натрия. Желательно, чтобы 1 мл последнего соответ-
1 Согласно А. Н. L о w, A. J. 'WeinignW. Р. Schoder ITechnical Methods of Ores Analysis, 11th ed., p. 116 (1939)], конец титрования в присутствии большого количества иодида меди (I) можно сделать более отчетливым, прибавляя приблизительно 1 мг нитрата серебра. Желтоватый цвет иодида серебра маскирует розоватую окраску иодида меди (I). Н. W. Foote и J. Е. Vance [J. Am. Chem. Soc., 57, 845 (1935)] рекомендуют прибавление 2 г роданида аммония перед самым введением в раствор крахмала. Это приводит к превращению окрашенного в цвет буйволовой кожи иодида меди (I) в менее растворимый белый роданид меди (I); адсорбированный иод таким образом освобождается, и конец титрования становится более резким. J. R. Cald-w е 11 [J. Am. Chm. Soc., 57, 96 (1935)] утверждает, что конец титрования можно значительно улучшить, медленно добавляя при непрерывном перемешивании 0,5— 1,0 мл 4%-ного спиртового раствора белого шеллака, когда большая часть иода будет оттитрована. Перед самым окончанием титрования надо дать осадку осесть в течение 20—30 сек. Шеллак выполняет здесь двойную функцию: он вызывает быструю флоккуляцию осажденного иодида меди (I) насаждение его в виде тяжелых хлопьев, и, кроме того, шеллак деактивирует поверхность осадка, в результате чего дальнейшая адсорбция синего иодокрахмального соединения уменьшается.
Методы определения
289
ствовал 0,005 г меди. Когда коричневая окраска Почти исчезнет, прибавляют 5 мл раствора крахмала (стр. 220) и продолжают титрование до тех пор, пока прибавление одной капли тиосульфата не изменит цвет раствора от голубого до желтовато-белого.
Мешающее влияние железа может быть преодолено следующим способом (при котором также обеспечивается полнота окисления мышьяка, когда анализируются руды, содержащие такие минералы, как арсенопирит): навеску в 1 г пробы обрабатывают 15 мл азотной кислоты и выпаривают до объема 5 мл. Затем прибавляют 10 мл соляной кислоты, 10 лм. серной кислоты и выпаривают до появления густых белых паров. Чтобы быть уверенным в полноте окисления мышьяка, добавляют еще 10 мл азотной кислоты и 10 мл соляной кислоты, снова выпаривают до появления густых белых паров для удаления всей азотной кислоты и разбавляют 20 мл воды. (Взамен последней обработки можно прибавить 20 мл воды, 10 мл насыщенной бромом воды и прокипятить до удаления брома.) Затем приливают раствор аммиака до начала появления синего комплексного соединения меди и потом осторожно по каплям вливают 6 н. раствор аммиака до слабого его запаха. После этого вводят в раствор 2 г кислого фторида аммония (1 г NH4HFa на каждую 0,1 г железа), перемешивают до растворения, приливают 10 мл 3 М раствора иодида калия и титруют тиосульфатом натрия. В этом методе1 железо переходит в стойкие комплексные фторидные ионы, а восстановление мышьяковой кислоты (также сурьмяной и селеновой кислот) исключается поддерживанием pH раствора, близким к 3,3.
* С целью удешевления йодометрического метода определения меди было предложено 2 заменить иодид калия смесью иодида калия и роданида аммония 3: 50 г роданида аммония и 6 г иодида калия растворяют в воде и разбавляют до 1 л. При добавлении этой смеси к раствору, содержащему ионы меди (I), протекают реакции
2Gu2+ + 4I-=Cu2I2 + Ia (1)
Cu2I2 + 2CNS- = Cu2(CNS)2 + 2I- (2)
в результате которых медь переходит в очень малорастворимый роданид меди (I) и на каждый атом меди выделяется атом свободного иода, который и оттитровывают тиосульфатом натрия:
i2+2s2o;--=2i-+s4o’- (3)
Если в растворе частично образовался роданид меди (II) Си (CNS)2, то он будет также титроваться Na2S2O3. И в этом случае на каждый атом меди будет расходоваться 1 молекула тиосульфата натрия:
2Cu(CNS)2 + 2S2O2-=Cu2(CNS)2 + 2CNS-+S4O2- (4)
Таким образом, расход тиосульфата натрия будет одинаковым независимо от того, идет ли реакция по уравнениям (1), (2), (3) или по уравнению (4).
1 W. R. С г о w е 11, Т. Е. Hillis, S. R. R i 1t e n b e г g, R. F. Even-son, Ind. Eng. Chem., Anal. Ed., 8, 9 (1936). Дальнейшие подробности этого метода см. в статьях: В. Р а г к, там же, 3, 77 (1931); Н. W. Foote, J. Е. Vance, там же, 8, 119 (1936); 9, 205 (1937); W. R. Crowell, S. Н. Silver, А. Т. S р i-h е г, т!ам же, 10, 80 (1938); W. R. Crowell, там же, И, 159 (1939); W. R. С г о-w е 11, А. Т. S р i h е г, там же, 12, 147 (1940). * См. также: Г. А. Т а р а т о р и н, Зав. лаб., 8, 723 (1939); М. А. П о п о в, там же, 9, 227 (1940); М. А. Попов, Поле-; вые методы химического анализа, Госгеолиздат, 1953; С. Ю. Файнберг, Анализ
- руд цветных металлов, Металлургиздат, 1953; Ю. Н. У с а т е н к о, О. В. Д а-
' ценно, Зав. лаб., 13, 116 (1947). *
Г 2 G. Bruhns, Chem. Ztg., 42, 301 (1918); Z. anal. Chem., 59, 337 (1920);
f. W. О r 1 i k, W. T i e t z e, Chem. Ztg., 54, 174 (1930).
I 3 Роданид калия стоит дороже иодида калия.
19’ заказ 522.
t
290
Гл. XI. Медь
Титрование меди надо проводить в сернокислой или солянокислой среде, но не в уксуснокислой, и титровать надо сейчас же после прибавления раствора смеси солей. Доп. ред. *
Роданидный метод ,
Роданидный метод определения меди основан на осаждении нерастворимого роданида меди (I) после ее восстановления сернистой кислотой в слабокислом растворе. Определение может быть закончено взвешиванием этого осадка, но обычно его растворяют и титруют раствором йодата калия или перманганата калия. Осаждение может быть проведено в присутствии 1% (по объему) серной или соляной кислот, предпочтительно последней, если раствор содержит большое количество мышьяка. Прибавление 2—3 г винной кислоты, как указано на стр. 283, желательно для предотвращения гидролиза солей висмута, сурьмы или олова, если они присутствуют. Раствор должен содержать не более 0,2 г меди в 100 мл\ осаждающий реактив нужно прибавлять в 3—5-кратном количестве. Большой избыток этого реактива нежелателен, так как осадок несколько растворим в концентрированных растворах роданидов х. Осадок также более растворим в горячих, чем в холодных растворах. При выполнении особо точных анализов осаждение можйо проводить в горячем растворе, который затем перед фильтрованием следует охладить. Рядовые анализы лучше проводить в горячих растворах для более быстрого осаждения и фильтрования.
Фильтровать можно через бумажный фильтр, асбест и губчатую платину и осадок следует промывать холодным 1 %-ным раствором сульфата аммония и затем 20 %-ным спиртом, если определение будет заканчиваться взвешиванием этого осадка.
Осаждению препятствуют: окислители, высокая кислотность раствора, избыток в нем аммонийных солей или избыток осаждающего реактива — роданида аммония.
Загрязняют осадок: свинец, ртуть, благородные металлы 1 2, теллур и селен, если последний присутствует в значительном количестве, особенно в солянокислом растворе. Соосаждение некоторых из этих элементов не всегда обязательно приносит вред: свинец, например, не мешает в том случае, когда определение заканчивается электролитическим методом. Если после осаждения роданида меди (I) определение заканчивают йодометрическим методом, то захват осадком свинца, ртути или серебра не мешает.
Не мешают определению: висмут, сурьма, олово, мышьяк, железо, никель, кобальт, марганец, цинк и кадмий.
Ход определения. К 150—300 мл сернокислого или солянокислого раствора, содержащего не более 0,1 г меди и не содержащего указанных выше мешающих примесей, прибавляют насыщенный раствор сернистой кислоты порциями по 20 мл, пока после стояния в течение получаса на водяной бане раствор не будет иметь явственного запаха SO2 и не станет
1 Согласно I. М. Kolthoff и G. Н. Р. v. d.Meene [Z. anal. Chem., 72, 337 (1927)], концентрация роданида аммония не должна превышать 0,05 н.
2 С известными предосторожностями медь этим методом может быть отделена от платины, палладия, иридия и родия (W. Н. Swanger, Е. W i chers, см. сноску 3 на стр. 285).
Методы определения
291
бледно-желтым. Охлаждают и нейтрализуют большую часть свободной кислоты, прибавляя раствор едкого натра до начала образования неисчезающего осадка. Раствор осветляют, прибавляя 8—10 капель соляной кислоты, и затем приливают по каплям из бюретки при постоянном перемешивании по 10 мл 2%-ного раствора роданида аммония в насыщенном растворе сернистой кислоты на каждые 50 мг присутствующей меди. "Дают постоять несколько часов, лучше в течение ночи, фильтруют через плотный бумажный фильтр, в который помещают предварительно немного бумажной массы, и промывают холодным 1%-ным раствором сульфата аммония.
Об осаждении в присутствии висмута, сурьмы и др. см. стр. 283.
Определение может быть закончено весовыми или объемными методами.
Весовые методы. 1) Фильтр с осадком помещают в фарфоровый тигель и высушивают при 110° С. Затем накрывают тигель крышкой Розе и вытесняют воздух водородом. Не прекращая тока водорода, нагревают тигель слабым пламенем до разложения роданида меди (I), удаляют крышку, прокаливают на воздухе до выгорания всего углерода, охлаждают, снова накрывают крышкой, вытесняют воздух водородом и, нако-. нец, прокаливают в токе водорода до восстановления окиси меди. Охлаждают, пропуская непрерывно водород, и взвешивают осадок в виде металлической меди.
2) Если осадок роданида меди (I) был собран на слое асбеста или тонко измельченной платины, то конечное его промывание можно проводить 20%-ным спиртом и осадок после высушивания при 105—120° С взвешивать в виде CuCNS.
Объемные методы. 1. Йодатный метод1. При прибавлении йодата калия к концентрированному солянокислому раствору роданида меди (I) последний окисляется и выделяется иод. По окончании окисления дальнейшее прибавление йодата ведет к превращению иода в бесцветный хлористый иод 1С1.
Фильтр с осадком CuCNS помещают в склянку с притерной пробкой емкостыо'около 250 мл. Приставшие к стенкам стакана и палочке частицы осадка снимают кусочком влажной фильтровальной бумаги и переносят в ту же склянку. Прибавляют около 5 мл хлороформа, 20 мл воды и 30 мл соляной кислоты.
Титруют 0,2 н. раствором йодата калия, прибавляя его небольшими порциями. После каждого добавления йодата калия склянку закрывают пробкой и сильно взбалтывают. Окраска слоя хлороформа сначала усиливается, потом ослабевает. Когда начнется ослабление окраски, раствор йодата калия начинают прибавлять все более малыми порциями, пока при добавлении последней порции реактива окраска сразу не исчезнет.
1 G. S. J ami es on, L. Н. Lev у, Н. L. W ells, J. Am. Ghem. Soc., 30, 760 (1908). * Более подробно от определении меди титрованием йодатом калия см. дополнение редактора в книге В. Т. Г о л л, Учебник количественного анализа, Гос-химтехиздат, 1933, стр. 118—119; вообще о титрованиях йодатом и о титровании им мышьяка, сурьмы, олова и цинка см. Ю. Ю. Лурье, Минеральное сырье, № 7, 731 (1931). * О применении этого метода для потенциометрического определения меди в плавах см. Н. В. Hope, М. Ross, Ind. Eng. Chem., Anal. Ed., 6,316 (1934).
19*
292
Гл. XI. Медь
* В первой части процесса титрования (усиление окраски слоя хлороформа) протекает реакция
lOCuCNS +1410“+ 14Н+ = lOCu2 + + 10SO2“ + 10HCN + 712 + 2Н2О
Во второй части (ослабление окраски) идет реакция
212 + 1О“ + 6Н+ + 5СГ=^51С1 + ЗН2О
Доп. ред.*
2. Перманганатный метод х. Осадок, который в этом Случае лучше собирать в тигле Гуча, а не на бумажном фильтре, растворяют в 30 мл горячего 10%-ного раствора едкого натра, хорошо промывают асбест водой и нагревают раствор до 50° С. Медленно титруют раствором перманганата калия, применяя сильно подкисленный раствор хлорида железа (III) в качестве внешнего индикатора.
Когда капли раствора на фарфоровой пластинке будут окрашиваться индикатором лишь в бледно-розовый цвет, прибавляют 30 мл разбавленной (1 : 1) серной кислоты, перемешивают до полного растворения двуокиси марганца и продолжают титрование перманганатом до появления обычной розовой окраски.
Сульфидный метод
Осадок, получаемый сульфидным методом, редко бывает чистым, если только предварительные отделения не были проведены особенно тщательно. Он легко окисляется и образует коллоидный раствор. При фильтровании и промывании осадка сульфида меди (II) фильтр надо держать все время наполненным на две трети жидкостью и никогда не следует промывать осадок горячей водой. При промывании сульфида меди умеренным количеством холодной воды или воды, насыщенной сероводородом, происходят лишь очень небольшие потери, который можно еще уменьшить, если осадок промывать подкисленной сероводородной водой.
Сульфид меди (II) обычно прокаливают до окиси меди при 700—900° С в хороших для окисления условиях. Прокаленная окись меди немного гигроскопична, но может быть взвешена в хорошо закрытом тигле. Так как при прокаливании трудно удалить всю серу из большого осадка, то этим методом не рекомендуется пользоваться при определении более 0,01 г меди.
Превращение сульфида меди (II) в сульфид меди (I) смешением его с серой и нагреванием до темно-красного каления в атмосфере водорода в тигле Розе представляет утомительную операцию и не может быть рекомендовано.
Ход определения. Приготовленный сернокислый или солянокислый, содержащий медь раствор, свободный от остальных членов сероводородной группы и, желательно, от азотной кислоты, подкисляют или разбавляют так, чтобы он содержал 5% кислоты по объему, нагревают до кипения и пропускают сероводород, пока раствор не охладится до комнатной температуры. По осаждении прекращают ток газа, дают осадку осесть и фильтруют, следя за тем, чтобы фильтр всегда был наполнен раствором. Осадок на фильтре промывают 1 %-ным раствором кислоты, насыщенным сероводородом. Во время промывания фильтр по-прежнему
1 D. J. Demorest, см. сноску 3 на стр. 285.
Методы определения
293
постоянно должен быть наполнен раствором, чтобы, сульфид меди (II) не мог окислиться кислородом воздуха. Под конец дают раствору стечь насколько возможно, помещают фильтр с осадком в фарфоровый тигель, высушивают над небольшим пламененем горелки, обугливают фильтр, не допуская его воспламенения, и затем сжигают уголь. Прокаливают при температуре около 1100° С, охлаждают в эксикаторе и взвешивают в виде СнО. Прокаливание повторяют до постоянной массы. При желании, можно конечное прокаливание провести в атмосфере водорода (стр. 432) и взвесить полученную металлическую медь.
Колориметрические методы
Из колориметрических методов определения меди чаще других применяют два метода: аммиачный 1 и диэтилдитиокарбаматный 2. Первый из них относительно мало чувствителен и пригоден больше всего для определения меди в количествах нескольких миллиграммов. При его применении должны отсутствовать органические вещества и.,элементы, образующие осадки или окрашенные растворы при добавлении избытка аммиака. Полученный аммиачный раствор нельзя фильтровать через бумажный фильтр, потому что медь в таких растворах вступает в соединение с примесями, присутствующими в целлюлозе, или восстанавливается ими.
Диэтилдитиокарбаматный метод очень чувствителен; его можно применять лишь для определения меди в количестве нескольких микрограммов (мкг). Применяемый реактив реагирует со многими элементами 3, но большинство из них можно связать в прочные комплексные соединения добавлением лимонной или винной кислот, диметилглиоксима и этилендиаминтетраацетата натрия 4, тогда метод становится специфичным в отношении меди. Комплексное соединение меди с диэтилдитиокарбаматом можно экстрагировать органическими растворителями, например бутилацетатом, и окраску полученного раствора сравнивать с окрасками стандартных растворов в обыкновенном колориметре или же измерять свето-поглощение полученного раствора в фотоколориметре.
* Другие методы
Из других методов определения меди заслуживают внимания следующие:
1) Выделение меди сернокислым гидразином 5 в виде очень мало растворимой в разбавленной кислоте соли CuSO4 -(N2H4)2- H2SO4. Осадок или
1 G. Bischof, Dingier’s Polytech. J., 184, 433 (1867); J. M i 1 b a u e r, V. Stanek, Z. anal. Chem., 46, 644 (1907); A. H. L о w, A. J. W e i n i g, W. P. Scho der, Technical Methods of Ore Analysis, 11th ed., John Wiley and Sons, 1939, p. 129; О. I. Milner, Anal. Chem., 18, 94 (1946). О применении триэтаноламина см. J. Н. Y о е, С. J. В а г t о n, Ind. Eng. Chem., Anal. Ed-, 12, 456 (1940); J. P. M e hl i g, D. Durst, Chemist Analyst, 37, (3), 52 (1948).*
2 T. Callan, J. A. R. Henderson, Analyst, 54, 650 (1929).
3 D. L. D r a b k i n, J. Assoc. Offic. Agr. Chemists, 22, 320 (1939); A. H. L о w, A. J. Weinig, W. P. S cho der, цит. выше, О. I. Milner, цит. выше. * См. также Ю. А. Чернихов, В. М. Добкина, Применение диэтилдитиокарбамата в аналитической химии, Зав. лаб., 15, 1143 (1949). * ,
* L. A. Haddock, N. Evers, Analyst, 57, 495 (1932); L. I. Butter, H. O. Allen,!. Assoc. Offic. Agr. Chemists, 25, 569 (1942); V. S e d i v e с, V. V a-
s a k, Coll. Czech. Chem. Comm., 15 , 260 (1950); J. L. H a g u e, E. R. Brown, H. A. Bright,!. Research NBS, 47, 380 (1951).
’ Ц. Г. Райхинш те й н, Зав. лаб., lf>, 1407 (1949).
294
Гл. XI. Медь
взвешивают после высушивания при 100—110° С или титруют раствором бромата калия:
3CuSO4 • (N2H4)2 • H2SO4 + 4BrO; = 3Cu2+ + 6H+ + 6S O1 2’ + 6N2 + 4Br- + 12H2O
В качестве индикатора применяют тропеолин 00, фуксин или тимоловый синий. 1 мл 0,1 н. раствора бромата калия соответствует 0,000795 г меди.
2) Осаждение меди а-бензоиноксимом С6Н5—СН(ОН)—C(NOH)—СвН5, образующим 1 с медью нерастворимое внутрикомплексное соединение, которому Файгль приписывает строение:
Н5Св-СН-С-С6Н5
: I II : : О NO j
!..Си.....:
Осаждение проводится в аммиачном растворе; для предупреждения мешающего влияния осаждающихся при действии аммиака элементов прибавляют тартрат натрия или калия. Определение заканчивается взвешиванием осадка или колориметрированием коллоидного раствора, обра-, зующегося в присутствии желатина 2.
3) Осаждение меди салицилальдоксимом C6H4OHCHNOH в виде внутрикомплексного 3 соединения
CeH4—CH = NOH HON=CH—СвН4
\>---------Си-----o'
Осаждение проводят в кислой среде. Этим способом медь можно отделить от всех элементов, кроме палладия, золота и серебра.
4) Осаждение меди солью Рейнеке 4 — диаминтетрароданохромиатом аммония NH4[Ct(NH3)2(CNS)4]-2Н2О. Осаждение проводят из кислой среды, концентрация кислоты не должна быть выше 3,0 н. Реактив этот обладает очень большой избирательностью: осаждению не мешает олово, сурьма, мышьяк, висмут, свинец, кадмий, молибден, никель, кобальт, железо, алюминий, хром, титан, марганец, цинк, бериллий, магний, барий, кальций, стронций, щелочные металлы, тартрат-, оксалат- и суль-фат-ионы. Мешают ртуть, таллий и серебро. Выпадающий осадок имеет состав: Си [Cr(NH3)2(CNS)J. Его высушивают при 110° С и взвешивают. Теоретический коэффициент пересчета массы этого осадка на медь равен 0,1666, но С. Ю. Файнберг рекомендует применять эмпирический коэффициент 0,1636, так как осадок, высушенный при 110° С, удерживает небольшое количество влаги. Подробности осаждения меди и метод приготовления осаждающего реактива (соли Рейнеке) приведены в цитирован
1 F. F е i g I, Вег., 56, 2083 (1923); Mikrochemie, 1, 76 (1923); И. Р. Ши к, Зав. лаб., 9, 542 (1940); В. Evans, D. Н ig g s, Analyst, 70, 75 (1945); E. И. Никитина, Зав. лаб., И, 231 (1945).
2 С. Ю. Файнберг, Анализ руд цветных металлов, Металлургиздат, 1953, стр. 127.
3 Е. Е р h a i ш, Вег., 64, 1210, 1215 (1931); W. R е i f, Z. anal. Chem., 88, 38 (1932); И. P. Ши к, Зав. лаб., 10, 261 (1941).
4 С. М a h г, Z. anorg. Chem., 225, 386 (1936); С. Ю. Файнберг, Анализ руд цветных металлов, Металлургиздат, 1953.
Методы определения
295
ном руководстве С. Ю. Файнберга. Осадок можно также отфильтровать, растворить в тиомочевине и метилэтилкетоне и колориметрировать полученный раствор х.
5) Колориметрическое определение меди пиридинродановым методом 1 2. Ионы меди (II) образуют с пиридином и роданид-ионами комплексное соединение [Cu(C5HaN)2(CNS)2], нерастворимое в воде, но растворяющееся в хлороформе или четыреххлористом углероде с образованием окрашенного в зеленый цвет раствора. Колориметрическому определению меди этим методом мешают только ртуть (I), никель, кобальт, серебро и железо (II). Железо (III) надо связать в комплекс лимонной кислотой.
Ход определения 3. В стакан емкостью 50 мл переносят аликвотную порцию анализируемого раствора, содержащую не более 0,2 мг меди, прибавляют 3—5 мл 50%-ного раствора лимонной кислоты и аммиака до щелочной реакции. Затем подкисляют раствором лимонной кислоты той же концентрации и прибавляют 1 мл ее 'избытка. Полученный раствор пере-. носят в цилиндр для колориметрирования, прибавляют 1,5 мл 30%-ного раствора роданида калия или роданида аммония, 0,5 мл пиридина, 5 мл хлороформа или четыреххлористого углерода и сильно встряхивают. В другой цилиндр вносят все реактивы, какие были прибавлены в анализируемый раствор, и титруют типовым раствором соли меди (1 мл которого содержит 0,020 мг меди), прибавляя его из микробюретки при сильном взбалтывании раствора до тех пор, пока окраски слоев органического растворителя в обоих цилиндрах не сравняются по интенсивности. Доп. ред*
1 С. Mahr, Angew. Chem., 53, 257 (1944).
2 G. S p a c u, Bui. soc. stiinte Cluj, 1, 352 (1922); H. T. Gebhardt, H. H. Sommer, Ind. Eng. Chem., Anal. Ed., 3, 24 (1931); Ю. Ю. Лурье, Л. Б. Гинзбург, Зав. лаб., 8, 271 (1939).
3 Ю. Ю. Лурье, Л. Б. Гинзбург, цит. выше.
Глава XII
КАДМИЙ
Кадмий — металл относительно редкий \ Он встречается вместе с цинком, на который походит по своим свойствам. Основным минералом, в виде которого находят кадмий, является его сульфид, г р е е и о-к и т CbS. Хотя кадмий и не встречается в значительных количествах в металлургических продуктах, все же он присутствует в них настолько часто, что методы его определения заслуживают внимания.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород кадмий, если он не присутствует в очень больших количествах, проходит через весь ход анализа незамеченным, так как он не осаждается ни аммиаком, ни оксалатом аммония в присутствии хлорида аммония, а осаждение его в виде фосфата в сильноаммиачных растворах происходит очень медленно и неполно.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ КАДМИИ
Разложение минералов, содержащих кадмий, проводится так же, как и разложение минералов, содержащих цинк (стр. 479).
МЕТОДЫ ОТДЕЛЕНИЯ
Методы отделения кадмия особенно важны потому, что для его определения необходимо, чтобы в анализируемом растворе отсутствовало большинство других элементов. Главные методы отделения кадмия основаны на свойствах его сульфида. Кадмий может быть отделен от элемен-'тов, не входящих в группу сероводорода, осаждением сероводородом в кислом растворе (стр. 83). От мышьяковой группы кадмий может быть отделен осаждением сульфидом натрия 2, от меди — осаждением сероводородом в щелочном цианидном растворе и от некоторых других элементов сероводородной группы — установлением соответствующей кислотности раствора перед пропусканием сероводорода. Сульфид кадмия лучше всего осаждать из сернокислого раствора. Присутствие больших коли
1 F. W. Clarke, U. S. Geol. Survey Bull., 770, p. 14.
* Согласно В. S. Evans [Analyst, 54, 395 (1929)], олово препятствует осаждению кадмия «сульфидом аммония» (который имеет обычно состав NH4HS); осаждение раствором (NH* *)2S проходит вполне удовлетворительно.
Методы отделения
297
честв хлоридов щелочных металлов препятствует осаждению кадмия даже из очень слабых солянокислых растворов х.
Об отделении кадмия от меди см. стр. 286. В опытах, проведенных Ноульзом, раствор, содержавший в 300 мл по 0,05 г кадмия, меди, висмута, мышьяка, сурьмы и олова и серную кислоту в концентрации 9 н., нагревали до 95—100° С и через него 30 мин пропускали сероводород. Выделившийся осадок был немедленно отфильтрован и промыт 9 н. серной кислотой, насыщенной сероводородом. После этого в полученном осадке было обнаружено 1,5—2,0 мг кадмия. Растворение осадка и вторичное его осаждение привело к полному отделению от кадмия.
Полнота отделения кадмия от цинка достигается с трудом, особенно если содержание цинка значительно превосходит содержание кадмия. Наилучшие результаты получаются при пропускании сероводорода в горячий 3 н. по содержанию свободной серной кислоты раствор, в котором кадмий и цинк присутствуют в виде их сульфатов. Пропускание сероводорода продолжают 1—1,5 ч, в течение которых раствор постепенно остывает. Если цинк преобладает, то выделившийся осадок надо растворить в горячей разбавленной (1 : 1) соляной кислоте, прибавить серную кислоту в незначительном избытке, выпарить до появления паров серной кислоты и после соответствующего разбавления водой снова осадить сероводородом.
Вполне удовлетворительные разделения проводятся путем электро лиза с неподвижными электродами в горячих щавелевокислых растворах, содержащих оксалаты, и с вращающимся катодом — в очень слабых сернокислых растворах 1 2 3 * * *.
Кадмий можно количественно осадить, прибавляя к горячему раствору его соли едкий натр до 5 %-ной концентрации последнего, но это не приводит к хорошему отделению от цинка.
Хорошим способом8 предварительного отделения кадмия от больших количеств цинка (как, например,' при анализе листового цинка) является следующий: 20 г нарезанного на кусочки листового цинка обрабатывают 150 мл воды и 30 мл серной кислоты. Когда действие кислоты почти прекратится, фильтруют, промывают остаток «металлов» небольшим количеством воды и сохраняют. К фильтрату, объем которого не должен превышать 200 мл, прибавляют несколько капель сернистой кислоты и 2 г иодида калия. Затем охлаждают до 20° С или более низкой температуры и приливают раствор 0,1 г р-нафтохинолина в 10 мл 2 н. серной кислоты. Если образуется осадок, его отфильтровывают и дают фильтрату стечь с осадка, не промывая последнего. Фильтрат, содержащий большую часть цинка, отбрасывают. Осадок на фильтре растворяют в горячей воде и к полученному раствору присоединяют сохраненный ранее остаток
1 A. S. Cushman, Z. anal. Chem., 34, 368 (1895).
2 F. E. Townsend и G. N. Cade [Ind. Eng. Chem., Anal. Ed., 12, 163 (1940)] описан метод, в котором кадмий восстанавливается до металла кипячением разбавленного (0,5 : 99,5) сернокислого его раствора с гранулированным « 40 меш; d < 0,37 Л1л) алюминием и таким способом отделяется от цинка и других не восстанавливаемых алюминием элементов.
3 A. Pass, А. М. Ward, Analyst, 58, 667 (1933); R. В е г g, О. W u г т,
Вег., 60, 1664 (1927). Последние утверждают, что осаждение кадмия р-нафтохинолином.
может служить также для отделения его от кобальта, никеля, марганца, железа (II),
а в присутствии винной кислоты — и от олова и сурьмы.
.298
Гл. XII. Кадмий
«металлов», содержащий большую часть кадмия. Смесь обрабатывают азотной и серной кислотами и выпаривают до появления густых паров последней. Остаток охлаждают, разбавляют водой и отделяют кадмий от оставшегося цинка двукратным осаждением его сероводородом. В указанных условиях 0,2 мг кадмия образуют с 0-нафтбхинолином муть, а 0,5 мг — легко отделяемый фильтрованием осадок.
Кадмий может быть отделен от цинка электролитическим осаждением из щелочного цианидного раствора при контролируемом катодном потенциале, равном —1,3 в по отношению к насыщенному каломельному электроду 1 2. С кадмием соосаждается менее 0,3 мг цинка, но результаты определения кадмия этим методом оказываются слегка повышенными. Для получения точных результатов надо осадок кадмия растворить и затем определить кадмий весовым способом, взвешивая его в виде сульфата или пирофосфата.
Кадмий от меди легко отделяется электролитическим осаждением последней из раствора, сильно подкисленного азотной кислотой (стр. 286), или же из азотно-сернокислого раствора, если содержание кадмия невелико. Электролиз в сернокислом растворе не дает удовлетворительных результатов. Медь можно также отделить от кадмия: 1) осаждением в виде роданида меди (I) (стр. 283); 2) кипячением с тиосульфатом натрия (5— 10 г) в разбавленном сернокислом растворе (3—5 мл H2SO4 на 100 мл раствора).
Свинец отделяют от кадмия в виде сульфата, (стр. 258) или, если он присутствует в небольших количествах, электролизом в виде двуокиси свинца (стр. 260).
Висмут отделяют осаждением его в виде хлорокиси висмута, бромокиси висмута, основного нитрата висмута или сульфида висмута (стр. 269—270).
Отделение ртути от кадмия .основано на нерастворимости сульфида ртути в азотной кислрте (стр. 247).
* Отделение кадмия от больших количеств цинка и одновременное его определение может быть проведено с большой точностью методом внутреннего электролиза 2. Для этой цели можно применять простейший прибор без диафрагмы (стр. 170). Кадмий выделяют из раствора, содержащего в объеме 250 мл 1,65 мл 80%-ной уксусной кислоты и 5,9 г ацетата натрия. pH такого раствора равен 5,2 (колебания в величине pH допустимы в пределах 4,6—5,6). Анодом служит пластинка цинка. Электролиз ведут 30—40 мин при 70—80° С. Выделившийся осадок промывают водой, подкисленной уксусной кислотой и содержащей небольшое количество электролита — сульфата аммония. В промывной воде указанного состава электроды, соединенные друг с другом, оставляют на 20—30 мин при 70— 80° С (если в момент погружения электродов некоторое количество кадмия перейдет в раствор, то в течение этого времени оно снова выделится на катоде). Потом промывают 95%-ным этиловым спиртом (но не разбавленным спиртом). Вместе с кадмием выделяется медь, содержание которой можно потом определить колориметрическим методом после раство
1 Н. Diehl, Electrochemical Analysis with Graded Cathode Potential Control, •G. F. Smith Chemical Co, Columbus, Ohio, 1948, p. 52.
2 Ю. Ю. Лурье, M. И. Трои цй а я, Зав. лаб., 5, 1425 (1936); 6, 507 <1937); 7, 675 (1938); Ю. Ю. Лурье, там же, 6, 1040 (1937).
Методы определения
299
рения выделившегося на катоде осадка. Определению кадмия мешают свинец, сурьма, мышьяк, висмут, которые можно легко отделить 1 предварительно. Доп. ред.*
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наиболее удовлетворительным методом определения кадмия является взвешивание его в виде сульфата кадмия после отделения всех веществ, дающих остаток после выпаривания с серной кислотой. Из других методов заслуживает внимания только электролитический.
Определение взвешиванием в виде сульфата кадмия
Главным затруднением при использовании этого метода (как и аналогичных методов определения цинка и марганца) является то, что полное удаление серной кислоты из остатка сульфата кадмия происходит с большим трудом. Прокаленный сульфат кадмия следует поэтому растворить в воде, раствор снова выпарить, сухой остаток прокалить, взвесить и операции эти повторять до получения постоянной массы.
Ход определения. Отделяют кадмий от всех веществ, дающих остаток после выпаривания с серной кислотой. Если кадмий связан с какой-либо летучей кислотой, то раствор его переносят во взвешенный платиновый тигель, обрабатывают разбавленной (1 : 1) серной кислотой и выпаривают, насколько это возможно, на водяной бане. Затем помещают тигель в радиатор (стр. 48), осторожно нагревают до прекращения выделения паров серной кислоты и затем усиливают нагрев приблизительно до 500° С. Охлаждают, растворяют остаток в небольшом количестве воды, осторожно выпаривают и снова нагревают. Вновь охлаждают, взвешивают и повторяют эти операции до получения постоянной массы.
Если кадмий был предварительно выделен в виде сульфида, то переносят, насколько возможно полно, осадок в маленький стакан и фильтр с оставшимися на нем частицами осадка сохраняют. Стакан покрывают часовым стеклом, обрабатывают осадок разбавленной (1 : 3) соляной кислотой, осторожно нагревают до его растворения и прекращения выделения сероводорода, после чего часовое стекло снимают и обмывают над стаканом. Подставив стакан под воронку, растворяют приставший к фильтру остаток сульфида кадмия обработкой горячей разбавленной (1 : 3) соляной кислотой. Полученный раствор выпаривают до малого объема, переносят его во взвешенный платиновый тигель и поступают, как описано выше.
Сульфат кадмия должен быть белым, и раствор его в воде должен быть прозрачным. Желтый или коричневый цвет прокаленного остатка указывает на частичное разложение сульфата. В этом случае остаток надо смочить разбавленной серной кислотой и операцию повторить при несколько более низкой температуре.
Электролитическое определение
Точное электролитическое определение кадмия лучше всего проводить с неподвижными электродами в слабощелочном растворе, содержащем только такое количество цианида калия, какое необходимо для удер
1 См. С. Ю. Файнберг, Анализ руд цветных металлов, Металлургиздат, •1954, стр. 534.
300
Гл. XII. Кадмий
жания кадмия в растворе х. Менее точное, но более быстрое определение-при выполнении рядовых анализов может быть проведено при большей плотности тока, с вращающимся катодом — в сильно разбавленном сернокислом растворе 1 2, ' или с вращающимся анодом — в сернокислом, муравьинокислом или уксуснокислом растворах 3. Осаждению кадмия из сернокислого раствора препятствуют те же самые элементы, какие были указаны при описании электролитического метода определения меди (стр. 286). Ряд элементов, как, например, цинк и серебро, препятствуют осаждению кадмия и из цианидного раствора, поэтому кадмий обычно предварительно отделяют практически от всех остальных катионов.
Ход определения. Приготовляют сернокислый раствор, свободный от всех мешающих определению примесей, выпаривают до появления паров серной кислоты, охлаждают, разбавляют и, если окажется нерастворимый остаток, фильтруют. Прибавляют каплю фенолфталеина, затем едкий натр или едкое кали до неисчезающей розовой окраски и, наконец, приливают по каплям 10%-ный раствор цианида калия или цианида натрия при непрерывном перемешивании до растворения гидроокиси кадмия. Избытка цианида тщательно избегают. Раствор разбавляют до 100— 15Q мл и проводят электролиз с взвешенным сетчатым платиновым катодом при силе тока 0,5—0,7 а и напряжении 4,8—5,0 в. После 5—6 ч электролиза увеличивают силу тока до 1—1,2 а и продолжают электролиз еще 1 ч. Обмывают часовое стекло и стенки стакана и ведут электролиз еще 15 мин. Если вновь погруженная в раствор поверхность катода останется чистой, то, не прекращая тока, опускают стакан с электролитом и обмывают катод водой. Затем электроды вынимают, ополаскивают катод спиртом, потом эфиром, высушивают, его при 100° С, охлаждают и взвешивают. Электролит проверяют на присутствие в нем кадмия, насыщая его сероводородом.
Другие методы
Весовое определение кадмия в виде сульфида не заслуживает внимания вследствие того, что состав сульфида кадмия, получаемого при осаждении как из кислого, так й из щелочного растворов, неопределенный -и осадок точного состава нельзя получить даже прокаливанием с серой в токе водорода или сероводорода. Титрование сульфида кадмия иодом в кислом растворе также не дает удовлетворительных результатов 4.
Довольно хорошие результаты можно получить осаждением кадмия в виде фосфата в холодном слабокислом растворе (см. «Цинк», стр. 485), свободном от большого избытка аммонийных солей, и прокаливанием этого осадка до пирофосфата 5 при 800—900° С. Этот метод, однако, не так точен, как сульфатный метод, и требует удаления многих элементов, как, например, цинка, висмута и железа.
1 F. Beilstein, L. J awein, Ber., 12, 759 (1879).
2 H. E. Medway, Am. J. Sci., [4], 18, 56 (1904); С. P. Flora, там же, [4], 20, 268, 392, 454 (1905).
3 E. F. Smith, Eletro-Analysis, 6th ed., 1918, p. 94.
4 Согласно H. B. W e i s e г и E. J. Durham [J. Phys. Chem., 32, 1061 (1928)], сульфид кадмия, полученный в кислом растворе, не является двойной солью, но представляет собой сульфид кадмия, загрязненный адсорбированной им солью кадмия.
3 М. Austin, Am. J. Sci., [4], 8, 214 (1899); 14, 156 (1902); E. H. Miller, R. W. Page, Z. anorg. Chem., 28, 233 (1901).
Методы определения
301
Применению весового метода определения кадмия осаждением его в видё молибдата и взвешиванием полученного осадка (а также объемного метода его определения титронанием раствором молибдата) мешает такое большое число элементов, что эти методы могут применяться только после полного отделения кадмия х.
* Из колориметрических методов определения малых количеств кадмия наибольшее значение имеют методы определения его дитизоном 1 2 и динафтилтиокарбазоном 3. Можно также осаждать кадмий солью Рей-неке (см. «Медь», стр. 294), растворять осадок в тиомочевине и метил-этилкетоне и полученный раствор колориметрировать4 5 *. Заслуживает внимания весовой метод определения 8 Кадмия, в котором его осаждают бруцином в сернокислой среде в присутствии бромида калия. Состав осадка — (C23H2eO4N2)'2-CdBr2 ^НВг. Об определении кадмия осаждением fJ-нафтохинолином и внутренним электролизом сказано выше (стр. 298). Доп. ред.*
1 R. С. Wiley, Ind. Eng. Chem., Anal. Ed., 3, 14 (1931).
2H. Fischer, G. Leopold i, Mikrochim. Acta, 1, 30 (1937);.E. Б. С e h-д э л, Колориметрическое определение следов металлов, Госхимиздат, 1949, стр. 249.
3J. Cholak, D. М. Hubbard, Ind. Eng. Chem., Anal. Ed., 16, 333 (1944).
* M. Mahr, Z. angew. Chem., 53, 257 (1944).
5 E. И. Никитина, Зав. лаб., 7, 409 (1938); И. М. К о р е н м а н, там же,
-6, 1461 (1937).
ЭЛЕМЕНТЫ, СУЛЬФИДЫ КОТОРЫХ НЕРАСТВОРИМЫ ж В КИСЛОТАХ, НО РАСТВОРИМЫ В РАСТВОРАХ СУЛЬФИДОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
МЫШЬЯК, СУРЬМА, ОЛОВО, ГЕРМАНИЙ, МОЛИБДЕН, СЕЛЕН, ТЕЛЛУР (И ПОЛНОСТЬЮ ИЛИ ЧАСТИЧНО ЗОЛОТО, ПЛАТИНА И ИРИДИЙ)
Глава XIII
МЫШЬЯК
Мышьяк очень широко распространен в природе, и следы его обычно присутствуют даже в живых организмах. Он встречается в самородном состоянии в виде двух сульфидов As2S2 и As2S3 в различных арсенидах и сульфоарсенидах тяжелых металлов, в виде окиси мышьяка и большого числа арсенатов. Наиболее распространенным мышьяковым минералом является арсенопирит FeAsS. Мышьяк часто встречается в воде минеральных источников, особенно горячих х.
В продуктах металлургического производства мышьяк чаще всего считается загрязнением, и обычное содержание в них мышьяка не превосходит 0,1%.
ОБЩИЕ ЗАМЕЧАНИЯ
В анализе горных пород малые количества мышьяка не создают затруднений, так как мышьяк (III), остающийся в растворе после разложения образца горной породы, улетучивается во время выпаривания с соляной кислотой при обезвоживании кремнекислоты. Мышьяк (V) осаждается в виде основного арсенита железа или алюминия вместе с осадком от аммиака и, вероятно, целиком восстанавливается и улетучивается при последующем сожжении фильтра с осадком и прокаливании. Иное дело при анализе продуктов металлургического производства, навеску пробы которых обычно обрабатывают окисляющими растворами. Например, при анализе черных металлов присутствие мышьяка затрудняет определение в них фосфора; при анализе сплавов цветных металлов присутствие мышьяка может помешать определению олова, сурьмы и меди.
1 F. W. С 1 а г k е, U. S. Geol. Survey Bull., 770, р. 14.
Методы отделения
303-
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ МЫШЬЯК
При всякой обработке мышьяковых минералов необходимо избегать улетучивания мышьяка (III) в виде его хлорида, для чего следует пользоваться окисляющими реактивами.
Для растворения арсенопирита тонко измельченную навеску пробы сперва обрабатывают при комнатной температуре водным раствором брома и бромида натрия, затем осторожно малыми порциями добавляют азотную кислоту и выпаривают досуха. Нитраты, если желательно, могут быть после этого превращены в хлориды или сульфаты. Металлический мышьяк, сульфид мышьяка (III) и большинство арсенидов тяжелых металлов могут быть растворены в дымящей азотной кислоте или в царской водке. Арсениды, не растворяющиеся при такой обработке, сплавляют со смесью соды и селитры или суспендируют в растворе едкого кали и обрабатывают хлором. Для разложения некоторых природных арсенатов требуется сплавление с содой. Если минерал полностью при этом не окисляется, прибавляют селитру, в присутствии же сурьмы соду заменяют поташом. При сплавлении с содой и селитрой мышьяк не теряется, но заметные количества мышьяка улетучиваются, если соединения мышьяка (III) сплавлять с одной содой х. Рекомендуют также сплавление с обезвоженным и истолченным в порошок тиосульфатом натрия 1 2.
При обработке соединений мышьяка (III) и (V) фтористоводородной и серной кислотами улетучивается только мышьяк (III) в виде фторида мышьяка 3. В этих условиях сульфат железа (II) не восстанавливает мышьяка (V), а сульфат железа (III) не окисляет мышьяка (III). При обработке одной серной кислотой мышьяк (III) не улетучивается при температуре ниже 200° С.
МЕТОДЫ ОТДЕЛЕНИЯ
При выполнении всех операций отделения мышьяка следует помнить, что соли мышьяка (III) в солянокислом растворе летучи. Надо избегать продолжительного кипячения соединений мышьяка (V) в солянокислом растворе, не содержащем окислителей, так как и в этом случае вследствие медленного восстановления мышьяка может произойти его улетучивание.
Наилучшим методом отделения мышьяка от остальных элементов является перегонка его из солянокислого раствора, содержащего весь мышьяк в трехвалентном состоянии. При этом отделении, насколько известно, только германий количественно отгоняется вместе с мышьяком. Другие элементы, как сурьма, олово (IV), ртуть (II), могут улетучиваться
1 Е. Т. Allen, Е. G. Zies, J. Am. Ceram. Soc., 1, 741 (1918). В статье F. Р. Carey, G. В 1 о d g е 11 и H. S. Satterlee [Ind. Eng. Chem., Anal. Ed., 6, 327 (1934)] описывается метод разложения материалов, содержащих малые количества мышьяка, сжиганием их в бомбе, в которую нагнетают кислород под давлением.
2 Е. D о n a t h, Z. anal. Chem., 19, 23 (1880).
8 Фтористоводородная кислота иногда содержит вещества, извлеченные из сосуда, в котором она хранится. Этих веществ, а также и растворенных органических веществ следует избегать. Если нет возможности быстро очистить кислоту (см. «Реактивы», стр. 63), надо пользоваться вместо HF концентрированным раствором чистого прокаленного NaF.
304
Гл. XIII. Мышьяк
лишь в незначительных количествах, если отгонка проводится при соблюдении требуемых условий. Этим загрязнением можно пренебречь, если перегонять из разбавленного раствора солей этих элементов и температуру пара 1 держать ниже 108° С. В "редком случае, когда присутствует германий, лучше всего его сперва отделить от мышьяка (V) отгонкой из солянокислого раствора в токе хлора (стр. 347), а затем отогнать мышьяк после удаления хлора и восстановления мышьяка до трехвалентного. Умеренные количества серной кислоты не мешают. Азотная кислота и нитрозосоединения должны быть удалены добавлением серной кислоты и выпариванием до густых паров. Эта- операция, однако, не должна быть продолжительной. Так же надо избегать перегрева, в противном случае мышьяк будет медленно улетучиваться. Так как при обычно применяемых методах разложения руд мышьяк переводится в пятивалентное состояние, то перед перегонкой его надо восстановить кипячением с серой в концентрированном сернокислом растворе (стр. 314) или добавлением восстановителей: хлорида меди (I), сульфата железа (II), сернистой, иодистоводородной или бромисто- • водородной кислот.
Прибор для перегонки (рис. 24) состоит из колбы емкостью 500—1000 мл. снабженной пробкой с тремя отверстиями, В одно из них вставляют термометр, нижний конец которого не доходит до уровня жидкости на 4—5 см, во второе отверстие помещают делительную воронку и в третье — выводную трубку с каплеуловителем. Эта трубка соединена с вертикально поставленным холодильником, как показано на рисунке. Каплеуловитель облегчает стекание механически увлеченного паром раствора
обратно в колбу, непрерывный же ход пара обеспечивается с помощью отверстия в трубке на расстоянии 2 см от нижнего ее конца, находящегося в колбе. Если мышьяк в дистилляте определяют Йодометрическим методом, то резиновые пробки и трубки не должны содержать серы. Если определяют небольшое количество мышьяка, то при перегонке нет необходимости пропускать ток хлориЬтого водорода через раствор. При большом количестве мышьяка это желательно. Хлористый водород можно получить в отдельной колбе, обрабатывая приливаемой по каплям серной кислотой соляную кислоту, насыщенную хлоридом аммония. При малом содержании мышьяка дистиллят собирают под водой в стакане, охлаждаемом снаружи льдом или холодной водой. Если же количество мышьяка велико или если пропускается хлористый водород, то возникает опасность неполного поглощения. В этом случае поглощение надо проводить в более сложном приборе, например в колбе Мейера или в закрытой колбе, снабженной предохранительными трубками. Дистиллят следует охлаждать. При обыч
1 Теория отгонки мышьяка разбирается в статье L. Moser, J. Ehrlich, Вег., 55, 437 (1922).
Методы отделения
305
ной перегонке к содержащему мышьяк раствору, находящемуся в дистилляционной колбе, прибавляют 150—300 мл соляной кислоты, затем 15— 30 г хлорида меди (I) или сульфата железа (II) и перегоняют, пока не соберется 100—200 мл дистиллята или температура пара на начнет подниматься выше 108° С. Если перегоняют более 0,01 г мышьяка, то к дистилляту прибавляют 10 мл фосфорноватистой кислоты и 50 мл соляной кислоты и перегонку повторяют х.
Имеются указания1 2’3 на то, что вольфрамовая и мышьяковая кислоты образуют друг с другом комплексные соединения, из которых обычными методами отогнать мышьяк невозможно. В таких случаях рекомендуется проводить перегонку после выпаривания анализируемого раствора до малого объёма и обработки его 20 мл ледяной уксусной кислоты, 120— 150 мл соляной кислоты и 30 мл чистого метилового спирта. Перегонка продолжается около 1 ч. Если содержание мышьяка велико, прибавляют еще 15 мл соляной кислоты и 15 мл метилового спирта и продолжают перегонку в течение получаса.
Для отделения очень малых количеств мышьяка (1 мг и менее) от больших количеств вольфрамового ангидрида (10 г) был предложен 3 следующий способ. Вольфрамовый ангидрид растворяют в 20%-ном растворе едкого натра, подкисляют фосфорной кислотой, нагревают 1 ч на водяной бане и перегоняют в неокисляющей атмосфере после добавления разбавленной (5 : 1) соляной кислоты, 10 г хлорида меди (I) и 1 г бромида калия. Для перегонки применяют прибор, в котором все стеклянные части притерты друг к другу.
Следующим по значению является метод отделения мышьяка осаждением его сероводородом в сильно солянокислом растворе. Обычно это отделение проводят после предварительного выделения всей группы мышьяка. Отделение это хорошо проходит в присутствии олова и сурьмы — элементов, обычно сопровождающих мышьяк, но оно не удается в присутствии германия, молибдена, ртути и меди, также образующих малорастворимые сульфиды. Осаждение сульфида мышьяка (V) должно проводиться при пропускании быстрого тока сероводорода через охлаждаемый льдом, 10 н. по содержанию соляной кислоты, раствор (см. стр. 310). В менее кислых растворйх осаждение идет медленно, в теплых растворах получается смесь As2S5 и As2S3. Чаще применяется осаждение сульфида мышьяка (III), так как оно может проводиться при кислотности нитке 9 и. и при обыкновенной температуре (стр. 310).
Как мышьяк (III), так и мышьяк (V) можно отделить от элементов, не входящих в сероводородную группу, осаждением сероводородом в кислом растворе (стр. 83); от элементов группы меди — осаждением в виде сульфида в щелочном растворе (стр. 87) или, менее удовлетворительно, — обработкой смеси сульфидов раствором сульфида щелочного металла (стр. 87).
Мышьяк (III) можно отделить от олова (IV) и от германия осаждением сероводородом из раствора, содержащего соляную и фтористоводородную кислоты (стр. 89).
1 Дистилляционный метод, в котором последовательно отгоняются мыщьяк, сурьма и олово, описан на стр. 96.
2 L. Moser, J. Е h г 1 i с h, Вег., 55, 430 (1922).
• Т. Millner, F. К u п о s, Z. anal. Chem., 107, 96 (1936).
20 Заказ 522.
306
Гл. XIII. Мышьяк
От германия1 мышьяк (III) можно количественно отделить осаждением сероводородом под давлением из раствора, 0,004—0,09 н. по содержанию свободной серной кислоты и содержащего 1—2% сульфата аммония. Серную кислоту и сульфат аммония можно заменить соляной кислотой и хлоридом аммония в тех же концентрациях, если осаждение прово- ' дится только с целью выделения мышьяка. Но такая замена недопустима, когда надо также определять и германий, так как последний может частично улетучиться в виде хлорида германия при последующих операциях. После осаждения сероводородом осадок сульфида мышьяка (III) отфильтровывают и промывают сероводородной водой, содержащей суль-фат аммония. Фильтрат подкисляют серной кислотой до 6 н. ее концентрации и осаждают германий также в виде сульфида. Если в осадке сульфида германия будет более 0,2% мышьяка, он будет заметно окрашен в желтый цвет.
Сульфид мышьяка (V) можно растворить в аммиаке и затем окислить до арсената перекисью водорода, лучше всего 30%-ным ее раствором.
Мышьяк (V) можно отделить от сурьмы и олова осаждением магнезиальной смесью 2 в холодном прозрачном аммиачном растворе, содержащем тартрат или цитрат аммония. Осадок будет, конечно, содержать и фосфор, если последний присутствовал в анализируемом растворе. Этому осаждению мешают те же вещества, какие мешают и осаждению фосфора (стр.'785). Арсенат магния и аммония более растворим в аммиаке и в растворах аммонийных солей, чем фосфат магния и аммония, и его нельзя прокаливать в присутствии органических веществ.
Относительно лучших условий осаждения арсенат-ионов магнезиальной смесью мнения расходятся. По нашим опытам, осаждение надо проводить двукратно, избыточных количеств аммонийных солей следует избегать, магнезиальную смесь надо добавлять в относительно большом избытке и промывать осадок умеренным количеством разбавленного (5 : 95) раствора аммиака, но ни в коем случае не раствором, содержащим хлорид \ или нитрат аммония. Добавлением тартрата или цитрата аммония можно почти полностью избежать загрязнения осадка оловом, сурьмой и, вероятно, германием. Малые количества мышьяка, медленно осаждающиеся или совсем не осаждающиеся, могут быть выделены замораживанием раствора, после чего выделившемуся льду дают расплавиться при комнатной температуре 3.
В обычных случаях ход отделения следующий. Приготовляют кислый, содержащий мышьяк (V) раствор, свободный от указанных выше мешающих элементов. Если мышьяк был отогнан в виде хлорида мышьяка (III), то сперва осаждают его в виде сульфида (стр. 309), растворяют осадок в малом количестве раствора едкого натра при помощи окислителя, например перекиси водорода, подкисляют раствор азотной кислотой и кипятят до малого объема. Затем разбавляют до 100 мл водой, если присутствует не более 0,1 г мышьяка, и прибавляют 25 мл магнезиальной смеси (стр. 66). В присутствии олова, германия или сурьмы прибавляют 3 г лимонной или винной кислоты и 50 мл магнезиальной смеси. Затем приливают раствор аммиака по каплям и при постоянном перемешивании
11 Н. J. Abrahams, J. Н. Muller, J. Am. Chem. Soc. 54 , 86 (1932). a A. L e v о 1, Ann. chim., (3), 16, 501 (1846).
’ F. A. Gooch, M. A. Phelps, Am. J. Sci (4), 22, 492 (1906).
Методы отделения
307
до тех пор, пока анализируемый раствор не станет щелочным, а после этого еще по 5 мл аммиака в избытке на каждые 100 мл раствора.
Оставляют стоять на ночь, фильтруют через бумажный фильтр и промывают осадок двумя порциями по 20 мл разбавленного (5 : 95) раствора аммиака. Фильтрат сохраняют, если надо определять олово, сурьму и пр. Осадок растворяют в возможно малом количестве разбавленной (1 : 2) соляной кислоты, прибавляют 10 мл магнезиальной смеси, разбавляют до 25—100 мл, в зависимости от количества мышьяка, и снова осаждают медленным прибавлением раствора аммиака, которого под конец приливают избыток, равный 5% по объему. Оставляют стоять на несколько часов, лучше на ночь, фильтруют, осадок очень умеренно промывают, как прежде, разбавленным (5 : 95) раствором аммиака и соединяют фильтрат с прежде полученным фильтратом. Затем растворяют осадок в горячей соляной или азотной кислоте и ведут определение мышьяка х, как указано на стр. 309.
Для отделения мышьяка от сурьмы рекомендуют 2 осаждать его в виде арсената серебра из слабощелочного раствора, содержащего фториды. Это разделение обычно следует за отделением от олова и проводится следующим образом. Сульфиды мышьяка, сурьмы и олова (IV) переносят в платиновую или кварцевую чашку, обрабатывают серной кислотой и 1—2 г чистой серы и кипятят до их полного растворения. Затем разбавляют раствор до 200—400 мл, нейтрализуют, если это необходимо, так, чтобы осталось от 2 до 4 мл свободной кислоты, прибавляют 3—6 мл фтористоводородной кислоты (45%-ной) и фильтруют, собирая раствор в платиновую чашку. Осаждают сероводородом, фильтруют и промывают осадок сульфидов мышьяка и сурьмы подкисленной сероводородной водой. Фильтрат выпаривают в платиновой чашке для удаления фтористоводородной кислоты и определяют олово осаждением его аммиаком или же, не выпаривая фильтрата, удаляют сероводород током двуокиси углерода, прибавляют 4—8 г борной кислоты и осаждают олово сероводородом или купфероном.
Осадок сульфидов мышьяка и сурьмы переносят в платиновую чашку, обрабатывают дымящей азотной кислотой и удаляют большую часть последней нагреванием. Затем приливают 2 мл фтористоводородной кислоты и неболыпое количество воды, нагревают, пока раствор не станет прозрачным, и разбавляют до. 100 мл водой. Накрывают кварцевым стеклом, нагревают до слабого кипения и прибавляют небольшими порциями 5 г персульфата калия. Охлаждают, прибавляют каплю метилового оранжевого и затем аммиака до пожелтения раствора. Нагревают до кипения, прибавляют в небольшом избытке нитрат серебра, сильно перемешивают, приливают аммиак точно до щелочной реакции по лакмусу (если это необходимо) и испытывают на полноту осаждения. Затем охлаждают, фильтруют, промывают осадок водой, содержащей 5 г нитрата аммония и 0,25 г нитрата серебра в 1 л, и потом небольшим количеством спирта. Осадок растворяют в азотной кислоте, в полученном растворе определяют содержание серебра, как указано на стр. 310, и вычисляют содержание мышьяка. Избыток серебра в фильтрате удаляют добавлением минимального
> Применение магнезиальной смеси для открытия или количественного определения мышьяка (V) в присутствии мышьяка (III) не дает удовлетворительных результатов {О. L u t г, R. S winne, Z. anorg. Chem., 64, 298 (1909)].
* L. W. M с С a у, J. Am. Chem. Soc., 50, 368 (1928).
20*
308
Гл. XIII. Мышьяк
количества соляной кислоты, фильтруют, собирая фильтрат в кварцевую чашку, и прибавляют 10—15 мл серной кислоты. Выпаривают, насколько возможно, на водяной бане, а затем на голом пламени до появления паров серной кислоты, прибавляют немного серы, кипятят 20 мин, охлаждают и определяют сурьму, как описано на стр. 324.
Мышьяк (V) не осаждается электролизом из аммиачного раствора и может быть таким способом количественно отделен от серебра, меди, кадмия и никеля.
Был предложен метод \ в котором трех- или пятивалентный мышьяк осаждают в виде элементарного мышьяка восстановлением фосфорнова-тистой кислотой Н(Н2РО2). Для этого анализируемый раствор, 33%-ный по содержанию соляной кислоты, после прибавления указанного реактива кипятят с обратным холодильником. Этим способом мышьяк отделяют от железа, меди, свинца, хрома, ванадия, молибдена, никеля, кобальта, олова и сурьмы.
Прекрасным методом предварительного отдел* .±ия мышьяка, встречающегося в малых количествах во многих материалах, является осаждение его аммиаком в виде основного арсената железа. Этот метод применяется при анализе медных и молибденовых руд. В этих случаях разложение исходного материала ведут так, чтобы весь мышьяк получился в пятивалентной форме, затем црибавляют 0,1—0,2 г соли железа (III) (если последнее не присутствует уже в растворе в достаточном количестве) на каждые 10 мг мышьяка и осаждают, как указано в гл. «Молибден» (стр. 360). Р-яд других элементов: селен, теллур, фосфор, вольфрам, ванадий, олово и сурьма — также осаждается этим методом. Применение соли алюминия вместо соли железа (III), не дает таких удовлетворительных результатов 1 2.
Малые количества мышьяка (<0,1 мг} могут быть извлечены из раствора соосаждением их с фосфатом магния и аммония, с которым аналогичная соль мышьяка образует смешанные кристаллы. Для этого мышьяк надо сначала перевести в пятивалентное состояние, прибавить двузамещенный фосфат аммония в таком количестве, чтобы в 500 мл раствора содержалось 0,5 г Р2О5, осадить магнезиальной смесью, отфильтровать а промыть осадок, как это делается при определении фосфат-ионов (стр. 784). Мешающего влияния железа, сурьмы, олова, алюминия и цинка можно избежать, прибавляя винную кислоту в количестве, достаточном для удержания этих элементов в растворе. Промытый осадок затем растворяют и в полученном растворе определяют содержание мышьяка любым способом.
Для отделения следов (менее 0,1 мг) мышьяка от большинства других элементов простейшим методом является его отгонка в виде арсина AsHs действием цинка и серной или соляной кислоты на раствор, содержащий мышьяк в трехвалентном состоянии (стр. 313).
1 В. S. Evans, Analyst, 54, 523 (1929).
2 Проводились опыты, в которых алюминий вводили в раствор в 60-кратном количестве по сравнению с отвечающим отношению 1А1: lAs, и затем однократно осаждали гидроокись алюминия, как описано на стр. 567 (метод Блюма); при этом соосаждение мышьяка как трехвалентного, так и пятивалентного, было далеко не полным. В противоположность распространенному мнению, тот мышьяк, который был увлечен выделенным осадком гидроокиси алюминия, количественно не улетучивался, когда отфильтрованный осадок прокаливали при температуре 1200° С.
Методы, определения
309
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наиболее удовлетворительными методами определения мышьяка, если содержание его превышает 1 мг, являются следующие три метода: 1) весовое определение его в виде сульфида мышьяка (III) As2S3; 2) осаждение его в виде арсената серебра с последующим определением серебра в осадке объемным методом Фольгарда; 3) объемный метод йодометрического титрования солей мышьяка (III). По точности первые два метода примерно равны. Второй метод менее зависит от присутствия других элементов и особенно удобен, когда в растворе присутствуют сурьма, олово %ли германий. В идеальных условиях третий метод, по всей вероятности, наиболее точен из всех трех. При использовании этого метода чаще других элементов мешает сурьма в трехвалентном состоянии. Определение мышьяка, основанное на осаждении его в виде дрсената магния и аммония и прокаливания осадка до пироарсената магния, может дать хорошие результаты только при соблюдении очень точно установленных условий, и вследствие этого, будучи ненадежным для широкого применения, оно может быть рекомендовано только для отделения мышьяка, как указано на стр. 305.
Мышьяк в количествах, меньших 0,1 мг, лучше всего определять выделением в виде арсина, как описано далее (стр. 312).
Предварительное отделение мышьяка восстановлением его до трехвалентного с последующей отгонкой его с соляной кислотой является очень удобной операцией, облегчающей дальнейшее определение мышьяка любым методом. Это отделение приводит к получению мышьяка в сильнокислом солянокислом растворе, из которого он может быть выделен разбавлением раствора и осаждением сероводородом. Если сульфид мышьяка (III) не будет взвешиваться как таковой, то его можно перевести в раствор различными способами. Наиболее распространена обработка осадка едкой щелочью вместе с каким-либо окислителем, например перекисью водорода или хлором. Все применяющиеся реактивы должны быть предварительно тщательно испытаны на содержание в них мышьяка, так как в реактивах он редко совершенно отсутствует.
Весовое определение в виде сульфидов мышьяка
Мышьяк может быть осажден и взвешен в виде сульфида мышьяка (III) As2Ss и сульфида мышьяка (V) As2S5. И тот и другой осадок могут содержать в виде примесей ртуть, молибден и германий, сульфиды которых очень мало растворимы в сильных кислотах, а также серу, образующуюся в результате окисления сероводорода такими веществами, как соли железа (III) или ванадиевой кислоты. As2S3 более растворим в кислотах, чем As2S5. Осаждение мышьяка в виде сульфида мышьяка (III) особенно удобно после отгонки его с соляной кислотой.
Осаждение в виде As2S3. Приготовляют раствор, 9 н. по содержанию соляной кислоты, содержащий весь мышьяк в трехвалентном состоянии и не содержащий других металлов группы сероводорода, осаждающихся при такой кислотности раствора. Осаждение проводят при 15—20° С быстрым током сероводорода и оставляют стоять на 1 ч или долее, если мышьяка выделилось немного. Фильтруют через взвешенный тигель Гуча или через стеклянный фильтрующий тигель. Если пленка сульфида мышьяка
310
Гл. XIII. Мышьяк
плотно пристала к сосуду, где проводилось.осаждение, то ее растворяют в аммиаке и вновь осаждают сульфид мышьяка (III), прибавляя некоторое количество кислой промывной жидкости. Осадок в тигле умеренно промывают концентрированным (8 н.) раствором соляной кислоты, насыщенным сероводородом, затем спиртом, потом сероуглеродом до удаления свободной серы и под конец снова спиртом. Высушивают при 105° С до постоянной массы и взвешивают в виде As2S3.
Если чистота осадка вызывает сомнения, то растворяют его в горячем разбавленном растворе аммиака при слабом отсасывании, промывают тигель и асбестовый слой водой, снова высушивают и взвешивают.
Осаждение в виде As2S5. Приготовляют 10 н. по содержаниюделяной кислоты раствор, в котором мышьяк находится в пятивалентной форме. Раствор не должен содержать других элементов сероводородной группы, осаждающихся при этой кислотности. Такие элементы, как олово, сурьма и кадмий, сульфиды которых растворимы в кислоте указанной концентрации, осаждению не мешают. В начале определения мышьяк должен быть полностью в пятивалентной форме, и соляную кислоту следует прибавлять медленно, при постоянном перемешивании, в пределах температуры от 0 до 10° С, так как при повышении температуры раствора выше 10° С произойдет частичное восстановление мышьяка. Раствор охлаждают льдом до 0° С и пропускают сильную струю сероводорода 60 мин. После этого закрывают колбу пробкой и, охлаждая льдом, оставляют стоять !—2 ч. Затем фильтруют через тигель Гуча или стеклянный фильтрующий тигель и промывают осадок 4 н. раствором соляной кислоты, пока промывные воды не перестанут выделять осадок при разбавлении их и пропускании сероводорода. Потом промывают водой и горячим спиртом для ускорения высушивания, сушат при 105—110° С и взвешивают в виде 1 As2S5.
Объемные определения
Определение превращением мышьяка в арсенат серебра и титрованием методом Фольгарда. Осаждение мышьяка (V) в виде арсената серебра, растворение последнего в азотной кислоте и титрование серебра в полученном растворе методом Фольгарда является очень хорошим способом определения мышьяка, особенно пригодным для применения после отгонки мышьяка с соляной кислотой и отделения его в виде сульфида. Германий и те малые количества сурьмы и олова, которые могут в этом случае сопровождать мышьяк, определению не мешают. Этот метод не может применяться для анализа веществ неизвестного качественного состава, так как имеется большое число анионов, также осаждающихся в виде солей серебра, например фосфат-, ванадат-, молибдат- и хромат-ионы. Следует избегать большого избытка аммонийных и натриевых солей,
1 Согласно L. W. М с С а у [Am. Chem. J., 9, 174 (1887); Chem. News, 56, 262 (1887)], осадок сульфида мышьяка (V) можно получить быстро и без образования As.2S3 или серы следующим образом. Раствор, содержащий от 0,1 до 0,3 г мышьяка (V), помещают в специальную склянку (емкостью 200 мл) для осаждения под давлением, сильно подкисляют концентрированной соляной кислотой, наполняют склянку почти доверху свежепрокипяченной и охлажденной водой и насыщают сероводородом. Затем склянку плотно закупоривают и нагревают 1 ч в кипящей водяной бане, после чего охлаждают и сильно взбалтывают. Сульфид мышьяка отфильтровывают, промывают сначала водой до удаления хлоридов, потом небольшим количеством спирта и высушивают при 110° С.
Методы, определения
311
так как первые несколько растворяют арсенат серебра, а цоследние захватываются выделяющимся осадком.
Ход определения,. Мышьяк, полученный в солянокислом дистилляте (стр. 304), осаждают сероводородом (стр. 309) и растворяют осадок сульфида мышьяка (III) в 2—3 мл аммиака или 10%-ного раствора едкого натра. В редких случаях, когда присутствуют значительные количества сурьмы, сульфиды растворяют в дымящей азотной кислоте, так как растворение в аммиаке в этом случае затруднительно. Промывают асбестовый слой небольшим, но достаточным количеством воды, выпаривают раствор почти досуха и прибавляют 10 мл концентрированной азотной кислоты. Снова выпаривают почти досуха, прибавляют 100—150 мл воды и такое количество нитрата серебра, чтобы связать весь мышьяк и чтобы оказался избыток его приблизительно в 10 мл 0,1 н. раствора. Затем прибавляют По каплям 10%-ный раствор едкого натра до выпадения осадка или пока раствор не станет щелочным по лакмусу. При наличии очень малого количества мышьяка осадок не всегда образуется, и тогда нейтрализацию следует проводить по лакмусу. Прибавляют по каплям разбавленную (1 : 1) азотную кислоту до растворения осадка или до покраснения лакмусовой бумаги и затем приливают 10 мл насыщенного раствора ацетата натрия. Нагревают до кипения, чтобы осадок арсената серебра скоагулировал, охлаждают, фильтруют через асбест и умеренно промывают осадок малыми порциями холодной воды до удаления избытка ни- ч трата серебра. Осадок арсената серебра растворяют в 30 мл разбавленной (1 : 1) азотной кислоты, разбавляют, если мышьяка много, до 100— 150 мл, прибавляют в качестве индикатора раствор железных квасцов и титруют 0,01 н. или 0,1 н. раствором роданида кадия, как описано в гл. «Серебро» (стр. 238). Реакция протекает согласно теоретическому уравнению, содержание мышьяка вычисляют из соотношения 3 атома серебра на 1 атом мышьяка. Титр раствора роданида калия должен быть установлен по чистому серебру в присутствии тех же количеств азотной кислоты и индикатора, какие применялись при анализе х.
Определение титрованием иодом. Процесс окисления мышьяка (III) до мышьяка (V) титрованием его раствором иода протекает полностью при условии применения чистых растворов солей мышьяка. При титровании образуется иодистоводородная кислота, которая, если ее не удалить, будет препятствовать полному окислению мышьяка в конце титрования. Поэтому обычно титрование проводят в присутствии бикарбоната натрия, который нейтрализует иодистоводородную кислоту и не реагирует с иодом, в отличие от едких щелочей и средних карбонатов. Конечную точку титрования можно сделать более резкой, прибавляя иодид калия и насыщая раствор двуокисью углерода (стр. 220).
При использовании этого метода к растворам, получаемым в ходе анализа, возникают затруднения, так как в анализируемом растворе
1 Проверочные опыты, проведенные J. I. Hoffman этим методом, дали следующие
результаты:
Присут- еТвовало Найдено Присутствовало Найдено Присутствовало Найдено
A.S2O3, <2 As2Os, г As2O3, г As2O3, г A.S2O3, г As2O3, г
0,0500 0,0502 0,2000 0,1998 0,1000 * 0,1003
0,1000 0,0999 0,1500 0,1498 0,0500 * 0,0503
0,1500 0,1503 0,0500 0,0500 ' 0,1000* 0,0998
Звездочкой отмечены опыты, в которых прибавлена 0,1 г GeO2.
312
Гл. XIII. Мышьяк
должны отсутствовать как вещества, на окисление которых расходуется иод, например сурьма (III), сульфиды, сульфиты, тиосульфаты, так и вещества, выделяющие иод, как, например, железо (III). Большинство затруднений устраняется применением этого метода после отгонки мышьяка с соляной кислотой. В этом случае сурьма остается единственным мешающим элементом. Сероводород и сернистую кислоту обычно удаляют предварительной обработкой перед перегонкой. Если мышьяк определяют не в солянокислом дистилляте, то весь мышьяк должен безусловно находиться в трехвалентном состоянии и раствор должен быть свободен от мешающих определению примесей (включая аммонийные соли). Раствор, содержащий мышьяк, сначала делают кислым, а потом подщелачивают бикарбонатом натрия, прибавляя его в избытке.
Ход определения,. Если определение проводится в солянокислом дистилляте, полученном, как указано на стр. 304, то дистиллят обрабатывают едким натром до щелочной реакции и затем соляной кислотой до слабокислой реакции. Прибавляют 15—25 мл холодного насыщенного раствора бикарбоната натрия, несколько миллилитров раствора крахмала (стр. 220) и 1 г иодида калия. Титруют 0,0! н. или 0,1 н. раствором иода до первого появления синей окраски. Вычитают объем израсходованного раствора иода, потребовавшийся на титрование в «холостом» опыте, проведенном через все стадии анализа, включая и дистилляцию. Содержание мышьяка вычисляют, исходя из расчета lAs : 21.
Определение отгонкой в виде мышьяковистого водорода
Очень мальйэ количества мышьяка (0,1 мг и менее) лучше всего могут быть определены отгонкой мышьяка в виде мышьяковистого водорода AsH3, обработкой последнего тем или иным реактивом и сравнением продукта этой реакции с полученным при обработке в тех же условиях стандартным раствором, содержащим известные количества мышьяка. Для этого сравнения были предложены различные реакции \ как, например, образование мышьякового зеркала в приборе Марша, получение окраски с нитратом серебра или, что лучше всего, получение окраски на сухой полоске бумаги, пропитанной хлоридом ртути (II). Последний метод имеет точность порядка 5—10% от истинной величины.. Минимальное количество мышьяка (в расчете на As2O3), которое может быть открыто этим методом, равно 0,00008 мг, но практически открывают не меньше 0,001 мг.
1 С. R. Sanger, О. F. Black, Proc. Am. Acad. Arts. Sci., 43 , 297 (1907); J. Snr Chem. Ind. London, 26, 1115 (1907); W. S. Allen, R. M. Palmer, 8th Intern. Congr. Appl. Chem., 1—2 (I—II), 9 (1912). См. также J. R. Nelle r, J. Assoc? Offic. Agr. Chemists, 12, 332 (1929); J. W. Barnes, C. W. Murray, Ind. Eng. Chem., Anal. Ed., 2, 29 (1930); H. E. Crossley, J. Soc. Chem. Ind. London, 55, 272 T (1936) и С. С. C a s s i 1, J. Assoc. Offic. Agr. Chemists, 20, 171 (1937). Согласно Cassil, до 10 г органического вещества, например табака, креветок, рыбьего жира, можно полностью разрушить (не теряя при этом мышьяка) в течение 30—60 мин следующим способом. Переносят пробу в колбу Кьельдаля, смачивают водой, если она сухая, и прибавляют смесь 20 мл серной кислоты и 15 мл азотной кислоты. Нагревают, когда бурная вначале реакция ослабеет, и прибавляют время от времени по 10 мл азотной кислоты, если вещество в колбе становится коричневым или черным. После прибавления 20 мл азотной кислоты приливают 10 мл 60 %-ной хлорной кислоты и продолжают нагревание, прибавляя небольшими порциями азотную кислоту, если происходит обугливание смеси. Наконец, кипятят для удаления большей части хлорной кислоты.
Метода определения
313
Для успешного определения мышьяка этим методом 1 (называемым часто методом Гутцайта) надо принимать во внимание следующее: 1) скорость образования мышьяковистого водорода’ имеет очень большое
значение, и эта скорость не всегда пропорциональна скорости выделения водорода (например, хлорид платины ускоряет выделение водорода, но препятствует восстановлению соединений мышьяка до мышьяковистого водорода)2; 2) процесс восстановления и образования мышьяковистого водорода успешнее протекает в присутствии железа (II), хлорида олова (II), цинка и кислоты, чем при наличии только двух последних веществ; 3) железо (II) всегда должно присутствовать в одинаковом количестве, лучше всего от 0,05 до 0,1 г в расчете на FeO; 4) выделение водорода 2 в опытах со стандартным и анализируемым веществами должно протекать с одинаковой скоростью.
Во всех методах, основанных на образовании мышьяковистого водорода, мышьяк должен находиться сначала в трехвалентном состоянии. Должны отсутствовать азотная кислота, хлор, бром, иод и соединения, образующие в этих условиях сероводород, сернистый ангидрид и фосфины. Эти вещества легко могут быть удалены кипячением с азотной кислотой; последняя же в свою. очередь может быть удалена выпариванием с серной кислотой до появления густых паров. При такой обработке мышьяк переходит в пятивалентный и должен быть перед определением восстановлен, лучше всего сульфатом железа (II). Присутствие ртути, платины, серебра, палладия, никеля, кобальта и их солей нежелательно. Нежелательно также присутствие большого количества меди в виде сульфата 3. Все эти вещества перед определением должны быть удалены. Присутствие сурьмы в количестве менее 0,1 мг не оказывает вредного влияния; большие же количества сурьмы должны быть отделены предварительно отгонкой мышьяка (III) в токе хлористого водорода, как это описано на стр. 304, но в приборе меньших размеров.
Ход определения. Приготовление бумаги, пропитанной хлоридом ртути (II). Из чистой рисовальной холодного прессования бумаги, 1 м2 которой весит около 160 г, нарезают полоски одинаковой ширины (4 мм). Полоски тщательно вымачивают, проводя их через 5%-ный раствор чистого перекристаллизованного хлорида ртути (II), высушивают, подвешивая их на
Рис. 25. Прибор для определения малых количеств мышьяка:
1 —склянка; 2, 5, 6 — стеклянные трубки; 3—фильтровальная бумага, пропитанная раствором ацетата свинца и высушенная; 4 — стеклянная вата, смоченная раствором ацетата свинца; 7 — полоска фильтровальной бумаги (7 cjhX-х 3 мм), пропитанной раствором хлорида ртути (И).
стеклянных палочках, укрепленных горизонтально, и
по высыхании разрезают на куски длиной 7 см, удаляя концы, за которые держали бумагу при погружении в раствор. Полоски
1 W. S. Allen, R. М. Palmer, цит. выше.
2 W. D. Harkins, J. Am. Chem. Soc., 32, 518 (1910).
3 W. P. H e a d d e n, B. Sadler, Am. Chem. J., 7, 342 (1885—1886).
314 Гл. XIII. Мышьяк
помещают в закрытый сосуд над хлоридом кальция и хранят в темноте х.
Прибор для восстановления. Прибор для восстановления показан на рис. 25. Этот прибор состоит из широкогорлой склянки 1 емкостью примерно 60 мл, в пробку (№ 4) которой вставлена стеклянная трубка 2 длиной 7. см и диаметром 1,25 см, оттянутая в нижней своей части так, чтобы она легко могла входить в пробку. В отверстии резиновой пробки (№ 00), находящейся в верхней части трубки 2, вставлена трубка 5 такого же диаметра, но более короткая, длиной 4 см. Наконец, в отверстие второй резиновой пробки (№ 00) в верхней части трубки 5 помещена стеклянная трубка 6, имеющая внутренний диаметр 4 мм и длину 10 см; трубка 6 сужена на расстоянии 6 см от одного конца, как показано на рисунке. В трубке 2 находится собранная в складки бумага, пропитанная раствором ацетата свинца, служащая для удержания сероводорода. Бумагу эту после каждого опыта надо менять. Трубка 5 неплотно набита стеклянной ватой, смоченной раствором ацетата свинца, для удержания последних следов сероводорода и увлажнения мышьяковистого водорода. Полоска 7 сенсибилизированной бумаги помещается точно в середине верхней части трубки 1 2 6,
Специальные реактивы. 1. Раствор железных квасцов. Растворяют 8,4 г железо-аМмонийных квасцов в воде, содержащей 1 г хлорида натрия и 2 ли серной кислоты, и разбавляют до 100 мл. 2 лил этого раствора содержат приблизительно 0,1г железа в пересчете на Fe2O3.
2. Раствор ацетата свинца. Растворяют 1 г ацетата свинца в воде, прибавляют такое количество уксусной кислоты, чтобы раствор стал прозрачным, и разбавляют до 100 мл.
3. Цинк. Кусочки не содержащего мышьяка цинка диаметром от 8 до 4 мм обрабатывают соляной кислотой для очистки поверхности, промывают водой и сохраняют под водой 3.
4. Хлорид олова (II). Растворяют 8 г хлорида олова (II) в 9,5 мл воды, содержащей 0,5 мл соляной кислоты.
1 Н. Е. Crossley (цит. выше) рекомендует полоски размером 2,5 х 115,0 лип для определения мышьяка в количестве, меньшем 18 мкг, и 5,0 X 115,0 мм для определения от 18 до 45 мкг мышьяка (в последнем случае применяют соответственно более широкую трубку). Полоски эти вырезают ид фильтровальной бумаги, применяемой для весовых количественных определений. Их погружают на несколько секунд в 1,5% -ный раствор бромида ртути (II) в 95%-ном спирте, высушивают 5 мин на стеклянной подставке, временами их переворачивая, и переносят в эксикатор.
2Н. Е. Crossley (цит. выше) утверждает, что при надлежащей температуре и влажности можно получить более равномерную окраску пропитанной раствором соли ртути бумаги, если верхнюю часть трубки 7, в которой эта бумага находится, окружить рубашкой, через которую пропустить холодную воду (20° С). Для придания большей устойчивости всему прибору склянка внизу и трубка 7 прикреплены зажимами к латунному стержню, вделанному в металлическое основание, где устроено специальное углубление для склянки прибора.
3 * Вместо цинка, часто загрязенного мышьяком, можно применять алюминиевую проволоку, которая обычно мышьяка не содержит (Ю. Ю. Лурье, Труды VI Менделеевского съезда, т. II, вып. 2, 1933). Отвешивают 1,5 г алюминиевой проволоки диаметром 1 мм и свертывают ее в спираль вокруг трубки диаметром 20 мм. Вместо серной кислоты применяют х. ч. соляную кислоту, концентрация которой в растворе перед определением должна соответствовать разбавлению 1 : 4. После прибавления SnCl2 и железных квасцов раствор разбавляют вдвое горячей (70—80° С) водой, опускают спираль, закрывают пробкой и продолжают, как описано выше. Прим. ред. *
Метода определения
315
5. Ацетатно-свинцовая бумага. Фильтровальную бумагу хорошо пропитывают раствором ацетата свинца, высушивают и нарезают полосками размером 7x5 см.
Шкала стандартных окрасок. Приготовляют стандартную шкалу окрашенных полосок бумаги, полученных при отгонке известных количеств мышьяка методом, описанным ниже. Отдельные полоски шкалы должны отвечать различным количествам мышьяка, начиная с 0,001 мг As2O3 и кончая 0,Q5 мг As2O3. От действия света и отчасти от действия водяных паров окраски бледнеют. Если полоски должны служить продолжительное время, можно добиться относительного постоянства окрасок, погружая бумажки в расплавленный парафин и сохраняя затем их над пятиокисью фосфора в пробирке, которая должна постоянно быть в темноте.
Ход определения. В склянку прибора помещают 50 мл анализируемого раствора, содержащего мышьяк, 2,5—3,5 мл серной кислоты, 2 мл раствора железных квасцов и 0,5 мл раствора хлорида олова (II). Нагревают приблизительно до 25° С. Помещают в нижние трубки полоски ацетатно-свин-цовой бумаги и стеклянную вату,' смоченную насыщенным раствором ацетата свинца. Соединяют трубку 5 с трубкой 6, содержащей полоску бумаги, пропитанной хлоридом ртути (II) (см. стр. 313), всыпают в раствор 35 г зерненного цинка, свободного от мышьяка, и закрывают сосуд пробкой, держащей верхнюю часть прибора. Осторожно взбалтывают и дают постоять на бане при 25° С в течение 1 ч. Вынимают полоску сулемовой бумаги и сравнивают ее со шкалой стандартов, которую приготовляют одновременно с анализом пробы, или же погружают полоску в парафин и сравнивают ее с ранее приготовленной стандартной шкалой г.
♦Лучшие результаты получаются 1 2 при использовании прибора, в котором выделяющиеся газы проходят сквозь кружок фильтровальной бумаги, пропитанной раствором хлорида или бромида ртути (II). Бумагу помещают перпендикулярно движению газов. В этом случае не происходит потеря части AsH3 и повышается чувствительность метода. Доп. ред. *
Колориметрический метод
При отсутствии фосфора и германия малые количества мышьяка в пределах 0,005—0,3 мг As2O5 можно определить превращением мышьяка в синее комплексное соединение молибдена и сравнением полученной окраски со стандартом в обычном визуальном колориметре или измерением светопоглощения раствора в фотоэлектрическом колориметре 3. Метод этот особенно пригоден для определения мышьяка в дистилляте.
1 В статье С. Е. Lachele [Ind. Eng. Chem., Anal. Ed., 6, 256 (1934)] описан метод, в котором вместо полоски бумаги применяется бумажная диафрагма, пропи-
танная бромидом ртути (II).
3 См. Ю. Ю. Лурье, А. И. Рыбникова, Методы химического-анализа производственных сточных вод, Госхимиздат, 1958.
3 Ch. Zinzadze, Ind. Eng. Chem., Anal. Ed., 7, 230 (1935); R. B. D e e m e r, J. A. Schricker, J. Assoc. Offic. Agr. Chemists, 16, 226 (1933). Основы метода изложены в статье J. A. Schricker и Р. R. Dawson {там же, 22, 167 (1929)], приложение его к определению мышьяка в металлах — в статье С. J. Rodden [J. Research NBS, 24, 7 (1940)] и к определению мышьяка в^еди и медных сплавах — в статье О. Р. Case [Anal. Chem., 20, 902 (1948)].
316
Гл. XIII. Мышьяк
♦Другие методы
Для определения малых количеств мышьяка широко применяется гипофосфитный метод 1 в его двух вариантах: объемном и колориметрическом. В обоих случаях мышьяк выделяют в виде элементарного мышьяка добавлегу?ем гипофосфита натрия или гипофосфита кальция и нагреванием:
2As3++ЗН2РО;+ЗН2О = 2 As+ЗН2РО^ + 6Н+
При выполнении анализа объемным вариантом метода раствор после добавления гипофосфита кипятят, отфильтровывают осадок мышьяка, промывают его сначала разбавленной (1 : 3) соляной кислотой, содержащей немного гипофосфита, потом 5%-ным раствором хлорида аммония, растворяют в отмеренном количестве 0,01 н. или 0,1 н. раствора иода и после добавления в избытке раствора бикарбоната натрия оттитровывают /избыток иода титрованным раствором мышьяковистой кислоты. Можно также растворять мышьяк в отмеренном количестве 0,01 н. или 0,1 н. раствора бихромата калия и оттитровывать избыток последнего титрованным раствором соли Мора в присутствии дифениламина или фенилантраниловой кислоты в качестве индикатора. В колориметрическом варианте метода 2 анализируемый раствор после добавления гипофосфита не кипятят, а нагревают определенное время на водяной бане. Выделяющийся в коллоидной форме элементарный мышьяк окрашивают раствор, который потом колориметрируют.
Из объемных методов определения мышьяка большое применение имеет также титрование раствора, содержащих мышьяк (III), раствором бромата калия или бромата натрия. При этом протекает реакция
3As3++BrO; + 6H+ —> 3As5+ + Вг” +ЗН2О
Индикатором служит метиловый оранжевый, который обесцвечивается в конце титрования.бромом, образующимся после окисления всего мышьяка. Можно также применять3 в качестве индикатора 0,01%-ный водный раствор флуоресцеина, содержащий несколько капель раствора NaOH (прибавляют 10 капель индикатора на 400 мл раствора). Этот индикатор не разрушается при действии брома, а изменяет свою окраску в точке эквивалентности из зеленовато-желтой в красно-коричневую. Доп. ред. *
1 В. Evans, Analyst, 54, 523 (1929); 57, 492 (1932); С. Ю. Файнберг, Л. Б. Гинзбург, Зав. лаб., 1, 733 (1932); Е. Фогельсон, Н. В. К а л-м ы к о в а, там же, 5, 584 (1936).
2 Б. С. Цивина, Б. М. Добкина, Зав. лаб., 7, 115 (1938).
3 F. L. Hah n, Ind. Eng. Chem., Anal. Ed., 14, 571 (1942).
<
Глава XIV
СУРЬМА
Хотя сурьма является обычным, не редким элементом, однако содержание ее в земной коре меньше, чем мышьяка, и она не так широко рассеяна в горных породах, как мышьяк. Сурьма встречается в самородном виде, но чаще в виде сульфида, стибнита Sb2O3, а также в различных антимонидах и сульфоантимонидах тяжелых металлов и в окисях вторичного происхождения. Сурьма в отличие от мышьяка имеет большое применение в металлургии и часто входит в состав сплавов цветных металлов.
ОБЩИЕ ЗАМЕЧАНИЯ
Сурьма редко, почти никогда не встречается в анализе горных пород и лишь изредка — в анализе минералов. В обычном ходе анализа сурьма частью улетучивается при выпаривании с соляной кислотой во время определения кремнекислоты; некоторое количество ее захватывается осадком кремнекислоты в виде хлорокиси сурьмы и частью улетучивается при прокаливании кремнекислоты, частью попадает в осадок от аммиака и принимается за алюминий.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ СУРЬМУ
Чистый хлорид сурьмы (V) кипит при 140° С при атмосферном давлении и при этой температуре медленно распадается на хлорид сурьмы (III) и хлор. Хлорид сурьмы (III) кипит при 223° С ji в значительной степени улетучивается из солянокислых растворов при температуре в 110° С и выше. Следовательно, при выпаривании солянокислых растворов, содержащих сурьму, теряется некоторое количество последней и величина потери зависит от характера обработки. Поэтому при разложении минералов, содержащих сурьму, следует избегать обработки кипящей соляной кислотой. Если же этой операции нельзя избежать, то ее надо проводить в закрытом сосуде или, лучше, с обратным холодильником. А если холодильник не применяется, то обработку надо проводить при возможно более низкой температуре и в течение как можно более короткого промежутка времени. Обработку никогда не следует проводить в открытых чашках для выпаривания, на стенках которых может образоваться корка.
Величина потери сурьмы при кипячении или выпаривании солянокислых ее растворов иллюстрируется опытами Н. A. Buchheit. В трех порциях по 50 мл
318
Гл. XIV. Сурьма
разбавленного (1 : 1) солянокислого раствора SbCl3 найдено при титровании методом, описанным ниже (стр. 324), 0,2728; 0,2730 и 0,2728 г Sb. В других трех порциях того же раствора найдено 0,0628; 0,1016 и 0,1204 г Sb после выпаривания до сиропообразного состояния (в чашке на паровой бане), обработки 100 мл разбавленной (1 : 1) соляной кислоты и вторичного выпаривания до сиропообразного состояния. Еще в трех порциях раствора того же состава найдено 0,2675; 0,2678 и 0,2674 г Sb после добавления 100 мл НС1, кипячения в стакане емкостью 600 мл (пока объем не уменьшился до 10— 15 мл), добавления еще 100 мл НС1 и вторичного кипячения до объема в 8—10 мл. В новых трех порциях того же раствора найдено 0,1564; 0,1447 и 0,0804 г Sb; эти порции обрабатывали 40 мл НС1 и 10 мл HNO3, нагревали несколько минут в покрытой часовым стеклом чашке, выпаривали до сиропообразного состояния на паровой бане, обрабатывали остаток 100 мл разбавленной (1 : 1) НС1; вторично выпаривали до сиропообразного состояния, разбавляли, нагревали до появления паров H3SO4 и титровали после восстановления Нг8О3, как описано на стр. 324. Наконец, последние три порции того же раствора показали содержание в них 0,2288; 0,2304 и 0,2307 г Sb после обработки их 10 мл H2SO4 и 100 мл разбавленной (3 : 2) НС1, выпаривания в чашке на паровой бане, нагревания до появления паров HaSO4 и титрования после восстановления, как указано на стр. 324.
В опытах, проведенных в несколько иных условиях, J. A. Scherrer получил следующие результаты. В двух порциях по 50 мл разбавленного (1:1) солянокислого раствора SbCl3 найдено 0,2467 и 0,2467 г Sb после осаждения сурьмы в виде сульфида, растворения в азотной кислоте, нагревания с H2SO4 + K2SO4 и обработки в дальнейшем, как описано на стр. 324. В двух других порциях того же раствора найдено содержание 0,2461 и 0,2461 г Sb после обработки 200 мл разбавленной (1 : 1) НС1 и уменьшения объема до 100 мл кипячением в стакане емкостью 400 мл, частично покрытом часовым стеклом. Еще в двух порциях того же раствора найдено содержание 0,246,6 и 0,2467 г Sb при аналогичной обработке после окисления КС1О3. В двух новых порциях того же раствора найдено 0,2148 и 0,2223 г Sb после обработки 200 мл разбавленной (1 : 1) НС1 и 12 л1л H2S04 и выпаривания в чашке емкостью 350 мл, насколько это £ыло возможно, на паровой бане. Наконец, в последних двух порциях этого раствора нашли 0,2268 и 0,2215 г Sb после такой же обработки, как в предыдущем случае, но с последующим выпариванием до появления паров H2SO4. \
Надо также иметь в виду, что соли сурьмы легко гидролизуются, образуя малорастворимые основные соли, если кислотность раствора уменьшается в результате его нейтрализации, разбавления или выпаривания досуха. Добавление винной кислоты в большинстве случаев предупреждает это осаждение.
Методы разложения сурьмяных минералов различны в зависимости , от того, какие элементы будут затем определяться. Сурьму и другие элементы, образующие летучие хлориды, лучше всего определять после растворения минерала в серной кислоте или сплавления его с едкими щелочами. Серу определяют из отдельной навески после специальной ее обработки. Для определения остальных элементов пробу можно растворять в соляной, азотной и серной кислотах, а нерастворимый в этих кислотах остаток сплавлять с карбонатами щелочных металлов или с едкими щелочами.
Металлическая сурьма практически нерастворима в холодных разбавленных минеральных кислотах, но легко растворяется в горячей серной кислоте. Переведение в раствор металлической сурьмы и свинцово-сурьмяных сплавов может быть достигнуто осторожным сплавлением с бисульфатом калия в фарфоровом или кварцевом тигле и растворением охлажденного плава в 200 мл воды, содержащей 10 мл соляной кислоты и 30 мл серной кислоты Ч
Влажные сульфиды сурьмы растворяются при нагревании с серной кислотой и сульфатом калия или сульфатом аммония. Для определения
1 К. Stanford, D. С. М. Adamson, Analyst, 62, 23 (1937).
, Ризложение минералов, содержащих сурьму
319
сурьмы и других элементов, образующих летучие хлориды, поступают следующим образом. Переносят 1 г тонко измельченной пробы в колбу Кьедьдаля, прибавляют 5 г сульфата аммония, 1 г сульфата калия, 15 мл серной кислоты и около 0,2 г серного цвета для облегчения восстановления сурьмы и мышьяка до трехвалентного состояйия. Смесь нагревают на голом пламени сначала осторожно, потом более сильно, пока не исчезнет свободная сера и раствор не станет бесцветным. Колбу охлаждают, давая плаву растекаться по стенкам сосуда, и прибавляют 25 мл воды и 2—3 г винной кислоты. Нагревают до полного растворения растворимых солей, фильтруют, если это необходимо, умеренно промывают остаток разбавленной (1:9) серной кислотой и затем водой для удаления кислоты.
Нерастворимый остаток, в особенности если это сульфат* свинца, может содержать сурьму. Поэтому при наличии остатка поступают следующим образом. Фильтр с остатком высушивают при 100° С, снимают с фильтра возможно большее количество остатка, обугливают фильтр в фарфоровом тигле и прокаливают при низкой температуре до выгорания всего углерода. Прибавляют снятый раньше с фильтра остаток, затем всыпают достаточное количество смеси, состоящей из равных количеств карбоната натрия и серы, хорошо перемешивают и сплавляют при низкой температуре. Плав охлаждают, растворяют в воде и раствор фильтруют. Фильтрат подкисляют серной кислотой, прибавляя ее в небольшом избытке, и после того как выделившиеся сульфиды осядут на дно сосуда, фильтруют. Осадок растворяют в нескольких миллилитрах горячего 10%тного раствора едкого кали, прибавляя, если нужно, перекись водорода. Полученный раствор подкисляют серной кислотой, прибавляя избыток последней в 3—5 мл, выпаривают до появления паров серной кислоты, разбавляют до 10—15 мл водой и прибавляют полученный раствор к первоначальному фильтрату. Соединенные растворы выпаривают до объема 25—30 мл, охлаждают и, если присутствует мышьяк, отделяют его, прибавляя 50 мл концентрированной соляной кислоты и пропуская быстрый ток сероводорода (стр. 304). Фильтруют с отсасыванием через поддерживаемый конусом двойной фильтр, предварительно смоченйый разбавленной (2 : 1) соляной кислотой, и промывают осадок соляной кислотой той же концентрации.
Осадок содержит весь мышьяк и большее или меньшее количество-меди, молибдена, германия и других элементов, если эти элементы присутствовали в руде. Фильтрат разбавляют 3—4-кратным (по объему) количеством горячей воды, насыщают раствор сероводородом, дают осадку осесть и фильтруют. Осадок промывают разбавленной (1 : 99) серной кислотой, насыщенной сероводородом, до удаления хлоридов. Фильтр с осадком переносят в колбу Кьельдаля и нагревают с 8 мл серной кислоты и 2 г сульфата калия для удаления свободной серы и обесцвечивания раствора. Охлаждают, разбавляют 50 мл воды и кипятят несколько минут, чтобы быть уверенным в удалении сернистой кислоты. Раствор теперь готов для определения сурьмы перманганатным методом (стр. 324).
Кроме этого метода обработки, в тех случаях, когда надо определить только одну сурьму, особенно в окисленных материалах, не легко растворяющихся при нагревании с серной кислотой, применяют метод сплавления, как, например, сплавление с едким .натром в железном тигле или
320
Гл. XIV. Сурьма
с обезвоженным тиосульфатом натрия 1 или со смесью карбоната натрия и серы в фарфоровом тигле.
Разложение сурьмяных руд для определения в них серы — очень сложная операция. Изложенный ниже метод выполнения этого разложения является видоизменением метода, предложенного 2 для анализа пиритов. Этот метод дал при анализе стибнитов хорошие результаты. Переносят 1,373 г (факторная масса) тонко измельченной пробы в чашку, покрывают ее часовым стеклом и обрабатывают 10 мл 10%-ного раствора брома в четыреххлористом углероде, вводя его осторожно через носик чашки. Затем медленно прибавляют 5 мл брома и оставляют стоять 1 ч, время от времени перемешивания. Охлаждают чашку в ледяной воде, прибавляют 15 мл азотной кислоты и оставляют стоять еще 30 мин, изредка* перемешивая. Прибавляют 15 мл концентрированной соляной кислоты, оставляют стоять при комнатной температуре около 30 мин, затем медленно нагревают, чтобы удалить четыреххлористый углерод, и выпаривают до сиропообразной консистенции (не перегревать и не выпаривать досуха!). Прибавляют 10 мл соляной кислоты и снова выпаривают до сиропообразной консистенции. Затем приливают 20 л»л соляной кислоты, нагревают до растворения растворимых веществ и переносят в коническую колбу емкостью 500 .ил. Объем полученного раствора не должен превышать 100 л»л. Затем всыпают 5 г железных стружек и оставляют стоять около 1 ч, чтобы практически вся сурьма была выделена. Фильтруют и тщательно промывают осадок водой. Фильтрат разбавляют до 1600 мл и осаждают сульфат бария, прибавляя 125 л»л 6%-ного раствора ВаС12 *2^0 из капельной воронки со скоростью 5 мл в минуту. Оставляют стоять на ночь, фильтруют через тигель Гуча, умеренно промывают осадок холодной водой, высушивают и прокаливают 3.
Для определения других элементов, кроме серы, кремния и тех, которые, подобно сурьме, образуют летучие хлориды, раствор пробы может быть приготовлен обычным методом: обработкой соляной кислотой, затем азотной кислотой и сплавлением нерастворимого остатка.
Обработка тонко измельченного анализируемого материала медленным током хлора с нагреванием в конце при низкой температуре служит для отделения породы и нелетучих хлоридов серебра, свинца и меди от летучих хлоридов ртути, сурьмы и мышьяка.
Сурьмяная кислота, получающаяся вместе с метаоловянной кислотой при выпаривании с азотной кислотой, может быть легко растворена нагреванием осадка с серной кислотой, содержащей небольшое количество сульфата натрия. Количественное окисление сурьмы (а также и мышьяка) персульфатом аммония требует 2—3-минутного кипячения, если проводить его в слабокислом растворе 4.
1 Е. Donath, Z. anal. Chem., 19, 23 (1880).
2 W. S. Allen, H. B. Bishop, Ind. Eng. Chem., 11, 46 (1919).
3 Об определении сТибнитной серы в рудах и минералах см. J. А. Т s с h е г n i-h о w, Т. A. Uspenskaya, Ind. Eng. Chem., Anal. Ed., 7, 309 (1935) [* см. также Ю. А. Ч e p н и x о в, T. А. Успенская, Зав. лаб., 4, 387 (1936) *]; в статье Н. С. Haller [Chemist Analyst, 29 (I), И (1940)] приведен метод быстрого экстрагирования свободной серы из сульфида сурьмы.
4 N. Н. Furman, Ind. Eng. Chem., Anal. Ed., 3, 217 (1931).
Методы отделения
321,
МЕТОДЫ ОТДЕЛЕНИЯ
Из известных методов отделения сурьмы важнейшие основаны на' свойствах ее сульфида. Так, сурьма отделяется от элементов, не входящих в группу сероводорода, осаждением сероводородом в кислом растворе (стр. 83) и от элементов группы меди — растворением сульфида сурьмы в щелочном растворе (стр. 87). Далее, сурьму можно отделить: от мышьяка — осаждением очень мало растворимого сульфида последнего в сильно солянокислом растворе (стр. 305); от олова и германия — осаждением сероводородом в растворе, содержащем фтористоводородную кислоту (стр. 89), и от олова — осаждением сероводородом в щавелевокислом или виннокислом растворе (стр. 89). Из всех этих методов отделения наиболее важным является отделение мышьяка в сильно солянокислом растворе, так как мышьяк во всех методах мешает определению сурьмы. Мышьяк можно отделить как в виде сульфида мышьяка (III), так и в виде сульфида мышьяка (V) (стр. 309), и отделение может быть проведено прямо в кислом растворе анализируемого вещества или после совместного осаждения сурьмы и мышьяка в виде сульфидов и растворения их в кислоте.
Отделение мышьяка от сурьмы отгонкой его из солянокислого раствора (стр. 303) проходит вполне удовлетворительно, если только температура пара удерживается ниже. 108° С. Прекрасно проходит отделение германия от сурьмы кипячением с соляной кислотой и хлором (стр. 304). Хлорид сурьмы (III) может быть отогнан и полностью отделен от меди, свинца, молибдена и других элементов в сернокислом растворе, если прибавить немного серы, нагреть приблизительно до 200° С и пропустить через раствор ток пара, получаемого при кипячении разбавленной (1 : 1) соляной кислоты или обработкой соляной кислоты концентрированной серной кислотой, добавляемой по каплям. Для предотвращения образования мало летучего соединения сурьмы (V) необходимо присутствие восстановителя, например серы. (Метод отгонки, в котором последовательно отгоняются мышьяк, сурьма и олово, описан на стр. 96.)
При осаждении мышьяка (V) магнезиальной смесью в аммиачном растворе, содержащем тартраты (стр. 306), мышьяк полностью не отделяется от сурьмы вследствие того, что арсенат магния и аммония немного растворим и, кроме того, удерживает небольшие количества сурьмы.
Так как сурьма редко определяется взвешиванием в виде сульфида, то ее отделение (вместе с мышьяком) от олова (IV) осаждением сероводородом в щавелевокислом растворе 1 теперь применяется редко, за исключением того случая, когда необходимо предварительное отделение сурьмы в виде сульфида, а содержание олова настолько велико, что оно причинит много неудобств при дальнейшем определении, если осядет вместе с сурьмой. Даже при наилучших условиях полное отделение сурьмы от олова осаждением сероводородом является трудной операцией, и осадок сурьмы, кроме того, загрязняется мышьяком, молибденом и, вероятно, многими другими элементами.
Отделение это обычно проводится после получения олова и сурьмы в виде растворимых сульфосолей в растворе щелочного металла. К раствору, содержащему не более 0,3 г обоих металлов в виде растворимых
1 F. W. Clarke, Chem. News, 21, 124 (1870); Z. anal. Chem., 9, 487 (1870).
21 Заказ 522 '
322
Гл. XIV. Сурьма
сульфосолей, приливают раствор 6 г едкого кали и 3 г винной кислоты х. Прибавляют 30%-нук) перекись водорода до обесцвечивания раствора и затем еще 1 мл избытка, и кипятят несколько минут для окисления сульфосолей в сульфаты и разрушения избытка перекиси водорода. По прекращении выделения кислорода раствор немного охлаждают, покрывают стакан часовым стеклом и осторожно приливают горячий раствор 15 г-перекристаллизованной щавелевой кислоты. По прекращении бурной реакции нагревают до кипения и кипятят для разрушения избытка перекиси водорода. Разбавляют до 100 мл, пропускают быстрый ток сероводорода, нагревая в то же время раствор, и продолжают пропускание сероводорода ровно 15 мин (точно по часам) после первого появления оранжевого осадка. Разбавляют раствор горячей водой до 250 мл, продолжая пропускать сероводород при нагревании, и по истечении 15 мин удаляют пламя. Пропускание сероводорода продолжают еще 10 мин, после чего фильтруют через тигель Гуча, дважды промывают осадок декантацией 1 %-ным раствором щавелевой кислоты и дважды — разбавленной (1 : 99) уксусной кислотой; оба эти раствора должны быть горячими и насыщенными сероводородом. Осадок сульфида сурьмы можно затем растворить и обработать, как указано в разделе «Методы определения», стр. 324, а олово может быть выделено из фильтрата электролизом или осаждением в виде сульфида после разрушения щавелевой кислоты выпариванием с серной кислотой.
Было предложено 2 заменить щавелевую кислоту фосфорной кислотой; по данным авторов этого предложения хорошие результаты получаются при однократном осаждении следующим методом.
Растворяют смесь сульфидов олОва и сурьмы в разбавленной (1 : 1) соляной кислоте в конической колбе, прибавляют несколько капель фенолфталеина и нейтрализуют концентрированным раствором карбоната натрия или едкого натра. Приливают равный объем фосфорной кислоты (пл. 1,3 г/см3)-п затем на каждые 100 мл раствора добавляют по 20 мл соляной кислоты. Нагревают до начала кипения, погружают колбу в кипящую воду и осаждают сурьму быстрым током сероводорода. По окончании осаждения сурьмы, о чем судят по изменению окраски от желтой . к красной и осветлению раствора, снижают скорость тока сероводорода до 2—3 пузырьков в минуту, чтобы предупредить осаждение олова. Время от времени, осторожно вращая колбу, перемешивают ее содержимое, пока осадок не станет кристаллическим и черным. Тогда колбу вынимают из бани и разбавляют содержимое ее равным объемом горячей воды так, чтобы сульфид сурьмы, приставший к стенкам сосуда, был полностью смыт. Снова помещают колбу в горячую водяную баню и продолжают пропускать медленный ток сероводорода, пока раствор не станет прозрачным. Несколько охлаждают и фильтруют через тигель Гуча или бумажный фильтр, в зависимости от последующей обработки осадка. Авторы не ука- ' зывают подробно состава промывного раствора, но, вероятно, наиболее подходящим будет раствор, приближающийся по составу к анализируемому раствору и насыщенный сероводородом.
1 1 Если винную кислоту добавлять перед окислением сурьмы и олова перекисью водорода и последующим прибавлением щавелевой кислоты, то, как указывает F. Н е и к [Z. anorg. Chem., 37, 18 (1903)], получаются лучшие результаты.
а G. Vortmann, А. .М е t z 1, Z. anal. ,Chem., 44, 532 (1905).'
Методы отделения
323
Выделение малых количеств сурьмы из растворов таких веществ, как медные и молибденовые руды, осаждением их аммиаком совместно с гидроокисью железа (III) является вполне удовлетворительным методом, так же как и аналогичное отделение малых количеств мышьяка (стр. 308). Соэса-ждение сурьмы с гидроокисью алюминия вместо гидроокиси железа протекает менее удовлетворительно х.
Отделение больших количеств сурьмы от молибдена может быть проведено восстановлением свинцом в солянокислом растворе, как описано в гл. «Молибден» (стр. 360).
Нагреваний или выпаривание досуха с азотной кислотой не дает полного отделения сурьмы, за исключением того случая, когда одновременно в значительном избытке находится олово. Осадок при этом осаждении редко получается чистым 1 2.
Отделение свинца от сурьмы осаждением первого в виде сульфата никогда не должно применяться lip'll точных анализах, потому что сульфат свинца удерживает значительное количество сурьмы. Это количество, однако, сильно уменьшается, если раствор содержит 1 % винной кислоты, но тогда раствор нельзя выпаривать до появления паров серной кислоты, и часть свинца остается в растворе.
Серебро, медь и кадмий можно выделить электролизом из аммиачного- раствора и таким способом.количественно отделить от сурьмы (V) (см. «Медь», с?р. 285).
Кремний в присутствии сурьмы следует отделять в сернокислом растворе выпариванием до появления тяжелых паров серной кислоты, охлаждением, быстрым прибавлением воды при перемешивании, пока содержание кислоты в растворе не станет равным 10% по объему, и фильтрованием. Возможно, что обезвоживание хлорной кислотой также дает хорошие результаты. При использовании хлорной кислоты сурьма должна быть в пятивалентном состоянии (см. стр. 65). х
Было отмечено 3 *, что гипосульфит натрия (* по старой номенклатуре — гидросульфит натрия Na2S2O4 *) восстанавливает медь, серебро, ртуть,
1 Например, 1 мг сурьмы (V) не был полностью увлечен в осадок при осаждении 100 мг алюминия, тогда как такое же количество сурьмы (III) и сурьмы (V) и даже 10 мг сурьмы (V) количественно осаждаются вместе с 100 мг железа (III).
Согласно Н. Blumenthal [Z-. anl. Chem., 74, 33 (1928)], железо не полностью захватывает с собой сурьму, если последняя присутствует в количестве 50 мг. Лучшим методом выделения сурьмы, по его мнению, является растворение меди (содержащей Сурьму) в азотной кислоте, нейтрализация почти всей свободной кислоты и осаждение сурьмы вместе с двуокисью марганца, для чего прибавляют сульфат марганца и кипятят, добавляя небольшими порциями 1 н. раствор перманганата калия.
2 М. В. R а п е, К. К on dai ah и М. К. R a tnam [J. Indian Chem-Soc., 13, 544 (1936)] указывают на то, что растворы азотной кислоты, более концентрированные, чем 8 н., не оказывают растворяющего действия на безводные Sb2O4 и Sb2O5. При действии азотной кислоты на содержащие сурьму сплавы образуются водные окислы сурьмы, степень гидратации которых и растворимости в азотной кислоте зависит от характера обработки. Например, при выпаривании раствора и высушивании остатка при 120° С образуется соединение Sh20e • 2Н2О, а если высушивать при 160° С, то получается 38Ь2Об-2Н2О. Первое из этих соединений нерастворимо в растворах азотной кислоты, концентрация которых превышает 10 н., а второе — в растворах азотиой кислбты более концентрированных, чем 8 н. Эти растворимости, однако, имеют место лишь в идеальных условиях и изменяются в присутствии других элементов (например, олова). ‘
3 В. S. Evans, Analyst, 54, 395 (1929); см. также J. М е у е г, Z. anorg. Chem.,
34,43(1903). . S
- 21*
324
Гл. XIV. Сурьма
свинец, висмут, мышьяк, сурьму и селен в щелочных растворах их солей до элементарного состояния. В этом случае такие окислители, как азотная кислота, не мешают осаждению указанных элементов, что имело бы место, если бы осаждение проводилось в кислой среде. Добавление цианида калия предупреждает выделение меди, но способствует выделению сурьмы.
Автор описывает ряд практических случаев применения этого метода, например отделение сурьмы от меди и отделение висмута от олова и цинка.
* Небольшие количества сурьмы перед их колориметрическим определением часто выделяют на медной фольге. Последнюю опускают в анализируемый раствор, содержащий соляную кислоту в соотношении 1 : 4, и нагревают 2 ч при температуре, близкой к кипению. Если в растворе присутствует железо (III) или медь (II), их предварительно восстанавливают гипофосфитом натрия, солянокислым гидроксиламином или другим восстановителем. Вместе с сурьмой выделяются висмут и мышьяк. Последний обычно отделяют гипофосфитом натрия перед осаждением сурьмы. Выделенную на медной фольге сурьму растворяют, обрабатывая фольгу водой с перекисью натрия или щелочным раствором перекиси водорода. Доп. ред. *
МЕТОДЫ ОПРЕДЕЛЕНИЯ
По-видимому, наиболее точным методом определения сурьмы, когда количество ее превышает несколько миллиграммов, является объемный метод, в котором сурьма (III) титруется до сурьмы (V) в серно-солянокислом растворе раствором перманганата. Положительной стороной этого метода является то, что после титрования сурьмы тот же раствор может быть использован для йодометрического определения олова (стр. 338). Весовые методы определения сурьмы в виде сульфида сурьмы (III) Sb2S3 или четырехокиси сурьмы Sb2O4 менее удовлетворительны; их целесообразно применять лишь в тех случаях, когда содержание сурьмы настолько мало, что ошибки титрования становятся ощутимыми. Очень малые количества сурьмы (0,1 мг и менее) лучше всего определять выделением сурьмы в виде сурьмянистого водорода (стибина) и сравнением окраски, полученной при действии этого газа на полоску бумаги, пропитанную хлоридом ртути (II), со стандартной шкалой окрасок1.
Определение титрованием перманганатом
Присутствие соляной кислоты имеет существенное значение для быстрого и полного окисления сурьмы перманганатом. Содержание ее должно быть не менее 10% и не более 25% по объему. Желательно также присутствие серной кислоты в количестве, приблизительно равном 10% по объему.
Титрование должно проводиться при 5—10° С. Происходящая реакция настолько соответствует теоретическому ее уравнению, что можно пользоваться теоретическим титром перманганата (при отсутствии в лаборатории чистой сурьмы или сурьмянистых солей с известным содержанием
1 * Малые количества сурьмы значительно лучше определяются колориметрическими методами, изложенными ниже (стр. 329—331). Прим. ред. *
Методы определения
325
сурьмы для установки титра). Однако если такая возможность имеется, титр перманганата лучше устанавливать по сурьме, так как теоретический титр все же несколько отличается от эмпирического: он обычно на 0,001 часть ниже.
Главным элементом, мешающим определению, является мышьяк, который поэтому должен быть предварительно удален. Если процентное содержание мышьяка известно, и оно не превосходит нескольких миллиграммов, то определение можно провести титрованием в присутствии мышьяка с вычетом 1,86-кратного содержания мышьяка из найденного процентного содержания сурьмы.
Присутствие ванадия не мешает, если сурьма перед определением была предварительно выделена в виде сульфида. Однако наличие ванадия отразится на результате титрования, если анализируемое вещество было прямо растворено в серной кислоте и затем обработано сернистой кислотой для перевода сурьмы в трехвалентное состояние. Желательно отсутствие железа, хотя малые его количества не подвергаются значительному восстановлению при непродолжительной обработке сернистой кислотой в горячем растворе концентрированной серной кислоты. Если при предварительной обработке выделилось большое количество сульфата свинца, то результат определения сурьмы обычно - получается пониженным. Этого можно избежать, так же ка-к и ошибок, связанных с присутствием мышьяка и других элементов, проводя отгонку сурьмы, как описано на стр. 321. Несколько менее эффективной обработкой является добавление соляной кислоты, осторожное нагревание до растворения сульфата свинца и разбавление водой. Последующее осаждение хлорида свинца значения не имеет.
Ход определения,. Приготовляют 1 раствор, содержащий сурьму (III) и свободный от мышьяка (III), ванадия (IV), железа (II), сернистой кислоты, органически'х веществ и других вещестг, титруемых перманганатом. Раствор должен содержать приблизительно 10 мл серной кислоты и 10 мл соляной кислоты в 100 мл.
Охлаждают до 5—10° С и титруют 0,03—0,1 н. раствором перманганата калия до появления слаборозовой окраски, не исчезающей при перемешивании в течение 5 сек 2. Содержание сурьмы вычисляют по уравнению реакции [окисление сурьмы (III) до сурьмы (V)] или, пользуясь эмпирическим титром раствора КМпО4, полученным при установке его по навеске сурьмы, растворенной в серной кислоте и обработанной, как описано выше.
Если имеется сомнение в том, находится ли сурьма в начале определения полностью в трехвалентном состоянии, то перед прибавлением соляной кислоты горячий сернокислый (1 : 2) раствор сурьмы обрабатывают сернистой кислотой и кипятят до полного удаления сернистого ангидрида. Затем раствор охлаждают, разбавляют водой и обрабатывают соляной кислотой. Эта предосторожность особенно необходима в тех случаях,
1 Например, помещая бумажный фильтр, содержащий сульфид сурьмы (III) или метаоловянную и сурьмяную кислоты, в колбу Кьельдаля и нагревая с H2S04 + K2S04 до разрушения бумаги и полного растворения осадка.
2 * Рекомендуется перед титрованием добавить раствор соли марганца, иначе, несмотря на низкую температуру, возможен переход за конечную точку титрования, сопровождающийся выделением хлора. Если добавить соль марганца, можно титровать и при обычной комнатной температуре. Прим, ред.*
326
Гл. XIV. Сурьма
когда сурьму переводят в раствор продолжительной обработкой горячей концентрированной серной кислотой, при которой она может несколько окислиться.
Можно также выпарить сернокислый раствор до выделения густых паров серной кислоты, прибавить приблизительно 0,2 г серного цвета и прокипятить 10 мин. Для удаления сернистого ангидрида и избытка серы дают раствору охладиться, разбавляют его 25 мл воды, кипятят, пока объем раствора не уменьшится вдвое, и фильтруют через быстро фильтрующий фильтр. Затем сосуд и фильтр с остатком серы промывают водой.
Весовые определения
Определение в виде сульфида сурьмы (III). Из весовых методов определения сурьмы наиболее удовлетворительным является ее взвешивание в виде Sb2S3 после осаждения1 и высушивания осадка 2. Осажденный сульфид может содержать как трехвалентную, так и пятивалентную сурьму; присутствие серы не мешает. Но применимость метода ограничена тем, что недопустимо присутствие других элементов, осаждаемых сероводородом в кислом растворе, и тем, что сульфид сурьмы приходится высушивать и затем прокаливать при 280—300° С в атмосфере СО2.
Ход определения. Приготовляют раствор соли сурьмы в разбавленной (1 : 4) соляной кислоте. Быстро нагревают до кипения и немедленно осаждают сурьму быстрым током сероводорода при температуре раствора около 90—100° С. После того как сульфид сурьмы окрасится в красный цвет, содержцмое колбы время от времени осторожно перемешивают, избегая, однако, размазывания осадка по стенкам. Когда -осадок потемнеет, ток сероводорода можно ослабить. Реакция обычно требует 30— 35 мин и заканчивается, когда весь осадок станет кристаллическим и почернеет. Разбавляют раствор равным объемом воды, перемешивают и снова нагревают, пропуская через раствор сероводород. Когда раствор станет прозрачным, охлаждают его и фильтруют через тигель Гуча, предварительно высушенный при 280—300° С, охлажденный и взвешенный. Осадок промывают несколько раз водой для удаления кислоты, а потом спиртом. Затем тигель с содержимым высушивают, нагревая его 2 ч при 100—130° С в атмосфере двуокиси углерода, свободного от воздуха, после этого поднимают- температуру до 280—300° С и прокаливают еще 2 ч при этой температуре для превращения всей сурьмы в Sb2S3 и удаления серы. Охлаждают в атмосфере двуокиси углерода, помещают в эксикатор на 15—30 мин и взвешивают в виде Sb2S3.
Определение в виде четырехокиси сурьмы. Вместо взвешиваний сурьмы в виде сульфида сурьмы (III) методом, описанным выше, влажный сульфид сурьмы можно растворить и после выпаривания раствора прокалить и взвесить в виде четырехокиси сурьмы Sb2O4. Этот метод проще в выполнении, чем первый, но при недостаточно тщательной работе он может привести к большим ошибкам. Четырехокись сурьмы очень легко
• l V or t ma nn, Metzl, Z. anal. Chem., 44, 525 (1905).
2 F. Henz, Z. anorg. Chem., 37, 18 (1903).
Методы определения
327
восстанавливается и при прокаливании ее следует тщательно защищать ют восстанавливающих газов. Обычно сульфид сурьмы обрабатывают азотной кислотой. Эта операция требует большой осторожности, и, вероятно, обработка аммиаком и перекисью водорода, рекомендуемая для сульфида германия (стр. 348), может дать более удовлетворительные результаты.
Ход определения. Полученный сульфид сурьмы (III) или сульфид сурьмы (V) высушивают на бумажном фильтре при 100° С. Затем переносят большую часть осадка на часовое стекло, помещают фильтр в небольшую чашку и извлекают из него остатки сульфида сурьмы кипячением с 5 мл свежеприготовленного раствора сульфида аммония. Фильтруют раствор через маленький фильтр, собирая фильтрат в фарфоровый тигель емкостью 30 мл, и снова обрабатывают фильтр сульфидом аммония, пока не будет уверенности, что вся сурьма из него извлечена. Раствор в тигле выпаривают досуха и присоединяют главную массу осадка.
Непосредственная обработка сульфида сурьмы азотной кислотой в тигле может привести к механическим потерям, поэтому лучше оставить тигель с содержимым на ночь под колоколом рядом с чашкой, наполненной дымящей азотной кислотой. После такой предварительной обработки к содержимому тигля прибавляют азотную кислоту и нагревают на водяной бане, пока осадок не побелеет и большая часть кислоты не будет удалена( Прибавляют немного воды и затем аммиака при перемешивании до щелочной реакции. Выпаривают досуха на водяной бане, помещают тигель на асбестовом щите в радиатор (стр. 48) или лучше в электрическую муфельную печь, и нагревают сначала до удаления па-• ров серной кислоты, а затем полчаса 1 при 800—850° С. Охлаждают в эксикаторе, быстро переносят в стаканчик для взвешивания со стеклянной притертой пробкой и взвешивают, поставив такой же стаканчик на ДРУГУЮ чашку весов. Перед взвешиванием оба стаканчика должны несколько минут находиться в футляре весов. Прокаливание повторяют до постоянной массы.
Определение отгонкой в виде сурьмянистого водорода (стибина)
При содержании сурьмы менее 0,1 мг ее лучше всего определять отгонкой в виде стибина 2. Окраску, полученную при действии стибина на полоску бумаги, пропитанной хлоридом ртути (II), сравнивают со стандартными окрасками,, полученными при такой же обработке растворов, содержащих известные малые количества сурьмы. Мышьяк, сульфиды и фосфиды, также окрашивающие бумагу, пропитанную хлоридом ртути (II), должны отсутствовать. Мышьяк может быть отделен отгонкой -его с соляной кислотой; присутствия сульфидов и фосфидов легко избежать соответственной предварительной обработкой.
1 Пятиокись сурьмы Sb20s (полученную, например, растворением сурьмы в азотной кислоте, выпариванием и нагреванием остатка) можно количественно превратить в четырехокись сурьмы Sb2O4, прокаливая ее при 800° С. Примерно при 900° С четырехокись сурьмы медленно разлагается с образованием трехокиси сурьмы Sb2O3. Трех--окись сурьмы затем возгоняется, медленно при 900° С и быстро при температурах выше 1000° С. Смеси соединений олова и сурьмы, Получаемые растворением сплавов в азотной «слоте, нагреванием, фильтрованием и промыванием осадка, нельзя количественно ( превратить прокаливанием в SnO2 4- Sb2O4 или в SnO2 • Sb2O6 • Н2О, как это утверждает A. G. Dunbar-Poole [Analyst, 65, 453 (1940)].
а С. R. Sanger, J. A. Gibson, J. Soc. Chem. Ind., 26, 535 (1907),
328 Гл. XIV. Сурьма
Ход определения. Поступают, как описано в гл. «Мышьяк» (стр. 313), пока на полоске бумаги, пропитанной хлоридом ртути (II), не появится окраска. Тогда погружают эту полоску в разбавленный (1 : 4) раствор аммиака на 3 мин, пятикратно промывают водой, погружают на 5 мин в 1%-ный раствор АиС13, еще раз пятикратно промывают водой и сравнивают окраску со шкалой стандартных окрасок, приготовленной таким же способом и хранящейся в темноте в банке с притертой пробкой, содержащей несколько миллилитров воды.
Другие методы
Из остальных методов определения сурьмы можно указать на: электролитический метод 1 и объемные — титрованием иодом, броматом 2 и тиосульфатом 3.
Электролитический метод определения сурьмы обычно дает повышенные результаты. Выделение сурьмы проводится из раствора сульфида сурьмы в сульфиде натрия, освобожденном от полисульфидов прибавлением цианида натрия.
При титровании иодом [методом, только тем отличающимся от описанного при определении мышьяка (стр. 311), что для удержания сурьмы в растворе необходимо добавление тартрат-ионов], так же как и при титрованйи броматом 4, сурьма окисляется от трехвалентной до пятивалентной. Оба метода дают хорошие результаты в растворах, содержа
1 G. Parrod i, A. Mascazzini, Z. anal. Chem., 18, 587 (1879); A. С 1 a s-s e n, Ber., 27, 2074 (1894); F. H e n z, Z. anorg. Chem., 37, 31 (1903).
2 S. G у о г у, Z. anal. Chem., 32, 415 (1893); * А. Васильев, В. Каргин, Труды Института им. Карпова, № 5 (1928)*; G. F. Smith, J. Am. Chem. Soc., 53, 2091 (1931); ASTM Methods for Chemical Analysis of Metals, 1950, p. 394.
3 A. Weller, Ann., 213, 364 (1882); G. S. Jamieson, J. Ind. Eng. Chem., 3, 250 (1911).
4 Это титрование проводится в сильно солянокислом растворе (15—35% по объему) медленным прибавлением при сильном перемешивании 0,1 н. раствора КВгО3. За 1—2 мл до конечной точки титрования прибавляют 3—4 капли метилового оранжевого п продолжают при сильном перемешивании очень медленно титровать до исчезновения розовой окраски. Согласно G. F. Smith титрование лучше проводить при комнатной температуре. * Обычно титрование проводят при 60—70° С, так как при комнатной температуре реакция проходит слишком медленно. *
О применении в этом титровании а-нафтофлавона в качестве обратимого внутреннего индикатора см. F. S с h u 1 е k, Z. anal. Chem., 102, 111 (1935). Титрование тогда проводят в разбавленном (1 : 5) сернокислом растворе с добавлением достаточного количества вииной кислоты для удержания сурьмы в растворе. (* О применении с той же целью флуоресцеина см. «Мышьяк», стр. 316. *)
Свинец, циик, серебро, олово, хром и сульфат-ионы не мешают определению сурьмы броматометрическим методом. Мышьяк (III) титруется подобно сурьме, азотная кислота, окислители, железо и медь мешают, так же как и большие количества кальция, магния и аммония. *Ю. А. ЧерниховиП. А. К о л од у б [Зав. лаб., 9, 467 (1940)] разработали метод броматометрического определения сурьмы в присутствии железа. В присутствии большого количества,сульфата свинца получаются пониженные результаты как при титровании броматом, так и при титровании перманганатом. Это вызвано тем, что осадок сульфата свинца захватывает с собой небольшое количество сурьмы. По окончании титрования следует слить раствор с осадка PbSO4, прилить концентрированную соляную кислоту, несколько капель метилового оранжевого и дотитровать ту сурьму, которая была захвачена осадком PbSO4. См4 А, В а-сильев, В. Каргин, цит. выше. *
Методы определения -
329
щих только сурьму, но титрованию мешает мышьяк и, как правило,, эти методы менее точны, чем перманганатометрический методх.
Описан также метод, основанный на титровании тиосульфатом натрия, который заключается в том, что сурьма (V) восстанавливается до трехвалентной подкисленным раствором иодида калия и выделившийся иод титруется раствором тиосульфата натрия. Этот метод менее точен, чем оба предыдущих, и для точных анализов не может быть рекомендован.
*Для определения малых количеств сурьмы в настоящее время применяются следующие колориметрические методы.
Пиридин-иодидный метод1 2. Сурьму (III) определяют по желтому окрашиванию ее комплексного соединения с пиридином и иодид-ионами, Py-HI-SbI3, образующегося в кислых растворах. Это соединение удерживается в коллоидном состоянии добавлением гуммиарабика или желатины. Максимальная по интенсивности окраска получается в растворе, 6—8 н. по содержанию серной кислоты. Концентрация иодида калия после добавления всех реактивов должна быть равна 1 %. Хлорид-ионы ослабляют окраску, а в больших количествах ее разрушают. Слишком большие количества пиридина также несколько . ослабляют окраску. Мышьяк и олово в малых количествах (десятые доли миллиграмма) не мешают определению сурьмы (большие количества мышьяка надо предварительно удалить гипофосфитом натрия). Висмут, никель, кобальт и цинк мешают, образуя осадки. Сурьму обычно предварительно выделяют на медной фольге (см. выше, стр. 324) или содсаждением с двуокисью марганца 3. От висмута сурьму отделяют сульфидом аммония.
Для колориметрического определения сурьмы в цилиндр с притертой пробкой вводят: 10 мл 1%-ного раствора гуммиарабика или 0,2%-ного раствора желатины, 5 мл 20%-ного раствора иодида калия, 1 мл 10%-ного водного раствора пиридина, 1 мл раствора сернистого ангидрида (насыщенный раствор разбавляют водой в 10 раз) или, что лучше, 3—5 мл 10%-ного раствора тиомочевины, 60 мл разбавленной (1 : 3) серной кислоты и 20 мл анализируемого раствора (в котором должно содержаться не более 1 мг сурьмы). Указанный порядок введения реактивов следует строго соблюдать. В другой такой же цилиндр наливают все реактивы в таком же порядке, но только серной кислоты (1 : 3) добавляют 75 мл и титруют типовым раствором сурьмы, содержащим 0,1 мг ее в 1 мл, до получения одинаковых окрасок в обоих цилиндрах (объемы растворов в обоих цилиндрах при этом уравнивают добавлением серной кислоты той же концентрации).
Если в том или ином растворе выделится иод (проба с крахмалом на фарфоровой пластинке), то прибавляют еще несколько капель раствора тиомочевины или сернистой кислоты. При определении очень малых
1 * Трудно согласиться с авторами. Мышьяк мешает определению сурьмы перманганатометрическим методом в такой же мере, как и определению броматометрическим методом, но титрование броматом дает очень резкую конечную точку титрования, и ири ием не наблюдается переход за эту точку, сопровождающийся выделением хлора, как это иногда случается при титровании перманганатом, если не была добавлена соль марганца. Прим. ред. *
2 S. G. С 1 а г k е, Analyst, 53, 373 (1928); С. Ю. Файнберг, Зав. лаб., 6, г . 36 (1937); А. А. Васильев, М. Е. Ш у б, ЖПХ, 6, 560 (1933); М. Я. Ш а-I ж р о, Зав. лаб., 8, 968 (1939).
а’ . 3 А. X у д ы н ц е в, Цветные металлы, № 7, 103 (1930).
-330
Гл. XIV. Сурьма
количеств сурьмы предложено 1 экстрагировать полученное окрашенное соединение добавлением 10—15 мл амилового спирта и после взбалтывания сравнивать окраски слоев экстракта.
Иодидныи метод2. При добавлении к раствору соли сурьмы (III) большого количества иодида калия получается желтое комплексное соединение KSbI4, достаточно интенсивно окрашенное. В присутствии железа (III) выделяется иод, но его восстанавливают добавлением раствора тйомочевины или аскорбиновой кислоты. Умеренные количества меди и мышьяка не мешают определению. Висмут мешает, образуя также окрашенное соединение. Определение выполняют следующим способом. К анализируемому раствору, содержащему 0,005—0,5 мг сурьмы, прибавляют разбавленную (1 : 1) серную цислоту до получения концентрации 1:5, прибавляют равный объем реактива, содержащего 11,2% иодида калия и 2% аскорбиновой кислоты, перемешивают и через 5 мин определяют светопоглощение раствора в фотоколориметре с применением синих светофильтров. В присутствии висмута получают суммарное содержание сурьмы и висмута. Тогда к другой пробе прибавляют реактив, содержащий только 0,16% иодида калия и 2% аскорбиновой кислоты, и, умножая результат измеренного светопоглощения на поправку 1,11, получают содержание только одного висмута. По разности находят содержание сурьмы.
Метод с кристаллическим фиолетовым или метиловым фиолетовым 3. Лучшие результаты получаются с гексаметилпарарозанилином, называемым также кристаллическим фиолетовым.
Сурьма должна находиться в растворе в виде анионов SbCle, которые и входят в состав образующегося комплексного соединения4. Некоторое затруднение создает то, что эти анионы легко гидролизуются даже в сильно солянокислых растворах, а другие ионы сурьмы (V) не образуют комплексного соединения с применяемым реактивом. Требуемые ионы SbCle можно получить только окислением сурьмы (III) до сурьмы (V) в сильно солянокислом растворе, содержащем большое количество хло-рид-ионов. Если сурьма была раньше частично или полностью в пятивалентном состоянии, ее надо сначала восстановить до трехвалентной хлоридом олова (II), затем окислить до SbCle в указанных выше условиях нитритом натрия и разрушить избыток нитрита добавлением мочевины. Образующееся с кристаллическим фиолетовым комплексное соединение растворяется в толуоле, окрашивая его в фиолетовый цвет. \
Ход определения. К анализируемому раствору, содержащему 0,005— 0,06 мг сурьмы, прибавляют при перемешивании 10%-ный раствор хлорида олова (II) до обесцвечивания раствора (восстановление железа), избегая его избытка. Затем приливают 1—2 мл 10%-ного раствора нитрита натрия и оставляют стоять на 5 мин. Разбавив раствор равным объемом воды, приливают 1 мл насыщенного раствора мочевины, перемешивают, переносят в делительную воронку, разбавляют до 50 мл водой, прибав-
1 А. А. Васильев, М. Е. Шуб, цит. выше.
а М. L. Fa nc h о n, J. pharm. chim., (8), 25, 537 (1937); Е. W. McChesney, Ind. Eng. Chem., Anal. Ed., 18, 146 (1946).
3 Ю. Ю. Лурье, H. А. Филиппова, Зав. лаб., 18, 30 (1952); 19, 771 <1953).
* В. И. Кузнецов, ЖАХ, 2, 179 (1947).
-Методы определения
331
ляют 20 капель 0,2%-ного раствора кристаллического фиолетового, 1 10 мл толуола и взбалтывают 30 сек. Водный слой сливают в другую делительную воронку и повторяют извлечение толуолом до получения бесцветного слоя толуола. Для полного извлечения 0,06 мг сурьмы достаточно 30 мл толуола. Все толуольные вытяжки соединяют, разбавляют, •если надо, толуолом до 30 мл и измеряют светопоглощение этого раствора .в фотоколориметре с зеленым светофильтром (максимальное светопро-пускание при 620—630 ммк).
Для построения калибровочной кривой растворы с известным содержанием сурьмы обрабатывают таким же способом.
Метод образования молибденовой сини Ч Для йриготовления применяемого в этом методе фосфоромолибденового реактива растворяют 20,6 г безводного молибдата натрия в 100 мл воды, к раствору прибавляют 3 г Na2HPO4-12H2O, растворенного при нагревании в 25 мл воды, и добав-, ляют по каплям разбавленную (1 : 1) азотную кислоту, пока раствор не станет золотисто-желтым, что примерно соответствует pH = 3,0. Анализируемый раствор, содержащий 0,05—0,5 мг сурьмы, нейтрализуют, разбавляют до 25 мл, прибавляют 3 мл разбавленной (1 : 4) серной кислоты, 3 мл насыщенного раствора сернистого ангидрида и кипятят до удаления SO2. Снова разбавляют до 30 мл, приливают 1 мл указанного фосфоромолибденового реактива и нагревают 10 мин на кипящей водяной бане. Затем охлаждают до комнатной температуры, приливают 8 мл разбавленной (1:4) серной кислоты для разложения избытка реактива, оставляют на 5 мин, периодически взбалтывая, и разбавляют в мерной колбе до 50 лм. Светопоглощение полученного синего раствора измеряют в фотоколориметре.-Висмут определению сурьмы этим методом не мешает. Мешает присутствие даже малых количеств железа, поэтому сурьму рекомендуется предварительно выделять на медной фольге.
Родаминовый метод 1 2. Этот метод основан на реакции сурьмы (V) с родамином В в минеральнокислом растворе в присутствии хлорид-ионов. Применению его мешают: мышьяк (больше 1 мг), железо (больше 10 мг) и медь (больше 10 мг). Не мешают: висмут, цинк, олово, свинец, ртуть, молибден, вольфрам и кобальт. Доп. ред*
1 А. И. Кокорин, Зав. лаб., 12, 64 (1946).
2 F. Е egriwe, Z. anal. Chem., 70, 400 (1927)-; W. G. Frederick, Ind. Eng. Chem. Anal. Ed., 13, 922 (1941); T. H. M a r e n, Anal. Chem., 19, 487 (1947); L. D. Freedman, там же, 19, 502 (1947); В. И. Кузнецов, Цветные металлы, 20, 59 (1947); Л. Е. Сабинина, А. П. Золотухина, Зав. лаб., 15, 398 (1949).
Глава XV
ОЛОВО
Олово в виде двуокиси олова — касситерита SnO2 — встречается в высококремнистых горных породах, гранитах и пегматитах, где его присутствие вызвано главным образом пневматолитическими процессами. Следы олова могут также встретиться в ильмените, слюдах, полевых шпатах, ниобатах и танталатах. Самородное олово встречается. очень редко. В рудах олово находится преимущественно в виде касситерита SnO2 и менее часто в виде станнита SnS2.
ОБЩИЕ ЗАМЕЧАНИЯ
При анализе горных пород и минералов олово определяют не часто. Тем не менее методы его определения важны, так как этот металл широко применяется в промышленности.
Если не были приняты специальные меры, то при обычном ходе анализа горных пород часть олова улетучится, а часть выпадет в осадок вследствие гидролиза и выделится вместе с кремнекислотой. Продукты гидролиза солей олова, присоединяясь к осадку кремнекислоты, могут быть причиной ошибки в определении кремния, так как они изменяют свой состав, когда нечистая кремнекислота обрабатывается фтористоводородной и серной кислотами и остаток примесей прокаливается. В обычном ходе анализа горных пород та часть олова, которая не улетучится при выпаривании с соляной кислотой, попадет в осадок от аммиака и будет принята за алюминий, потому что большинство реактивов, применяемых для восстановления железа, не восстанавливает олова. Цинк, являющийся исключением, обычно восстанавливает все олово в редукторе Джонса до металла. Таким образом и при использовании цинка для восстановления железа олово не оказывает влияния на титрование железа, если только оно не перейдет в раствор до конца титрования.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ОЛОВО
При разложении оловянных руд растворы, содержащие хлорид, олова (IV) в соляной кислоте или царской водке, не должны выпариваться досуха даже на водяной бане, так как хлорид олова (IV) заметно летуч. Улетучивание также происходит, если раствор концентрируется в открытых выпарных чашках, когда над раствором образуется кольцо сухих
Разложение минералов, содержащих олово
333
солей. Улетучивания, однако, не происходит при кипячении разбавленных солянокислых растворов, содержащих олово (IV), в покрытых часовым стеклом сосудах или при выпаривании соляно-сернокислых растворов солей этого элемента до появления паров серной кислоты.
Например, в опытах, проведенных с аликвотными порциями титрованного разбавленного раствора хлорида олова (IV), Н. В. Knowles получил 0,0036 г БпОг при действительном содержании его 0,1998 г, после того как 50 мг раствора были обработаны 50 мл разбавленной (3 : 2) соляной кислоты, выпарены досуха на паровой бане и эта операция была повторена 5 раз. Во втором опыте было получено 0,2002 г БпОг после разбавления 50 мл того же раствора 50 мл воды и 100 мл соляной кислоты и кипячения в течение 3 ч в покрытом часовым стеклом стакане емкостью 400 мл, причем объем раствора не менялся благодаря прибавлению новых порций кипящей при постоянной температуре соляной кислоты. В третьем опыте было получено 0,1999 г SnO2 после обработки 50 мл того же раствора 10 мл разбавленной (1 : 1) серной кислоты и 50 мл разбавленной (3 : 2) соляной кислоты, выпаривания до появления паров серной кислоты, вторичной обработки 50 мл разбавленной (3 : 2) соляной кислоты и вторичного выпаривания.
Оловянные руды, не содержащие серы, могут быть разложены сплавлением с перекисью натрия 1 или со смесью этого реактива с 25% карбоната натрия. Для выполнения этого разложения помещают 0,5—1,0 г сухой, тщательно измельченной пробы в тигель из никеля или чистого железа емкостью 50 мл и прибавляют 6—10 г перекиси натрия. Тщательно перемешав содержимое тигля, покрывают его крышкой и слабо нагревают для удаления влаги. Затем осторожно повышают температуру, пока смесь не расплавится, доводят до темного вишнево-красного каления, берут тигель щипцами и для полного перемешивания содержимому тигля осторожно придают вращательное движение, продолжая нагревание еще,3—5 мин. Затем охлаждают, растворяют плав в воде и подкисляют соляной кислотой. Для сплавления некоторых руд с перекисью натрия может оказаться пригодным метод взрыва (стр. 919).
Оловянные руды, содержащие олово в виде его двуокиси, можно •также легко разложить нагреванием в токе водорода. Для этого навеску в 0,5 г руды помещают в фарфоровую лодочку (тщательно перемешивают с 0,3—0,4 г чистой окиси кальция и нагревают 2 ч при 750° С в токе чистого сухого водорода 2. По охлаждении переносят лодочку с ее содержимым в коническую колбу и растворяют образовавшееся металлическое олово обработкой разбавленной соляной кислотой при нагревании. Если анализируемая проба содержит сульфиды, то навеску ее перед восстановлением водородом надо или обработать разбавленной азотной кислотой и выпарить досуха, или смешать с полуторакратным по массе количеством окиси кальция, покрыть еще слоем окиси кальция и прокалить.
Руды, содержащие более чем несколько процентов сульфидной серы, надо перед сплавлением обработать азотной кислотой. В этом случае навеску пробы обрабатывают разбавленной азотной кислотой и выпаривают почти досуха. Затем разбавляют горячей водой, фильтруют, нерастворимый остаток промывают и сохраняют. К фильтрату приливают раствор аммиака до щелочной реакции, осадок отфильтровывают, промывают и присоединяют его к сохраненному ранее нерастворимому
1 При хранении перекиси натрия в жестяных коробках она может быть загрязнена оловом, особенно если коробка несколько раз открывалась.
а Вместо водорода можно применять аммиак (’’’или светильный газ*).
334
Гл. XV. Олово-
остатку. Прокаливают их при невысокой температуре в условиях хорошего окисления для сжигания всех углистых частиц и сплавляют, как было описано выше.
Менее часто применяют следующие методы разложения оловянных руд: 1) сплавление с едким кали или с едким натром1; 2) сплавление с бурой или длительное сплавление со смесью равных частей карбоната, натрия (или карбоната калия) и серы 2 или с безводным тиосульфатом, натрия в тигле, из которого возможно тщательнее удаляют воздух 3;. 3) спекание с окисью кальция или окисью цинка или прокаливание^ цинковой пылью 4. Сплавление с карбонатами щелочных металлов дает удовлетворительные результаты при разложении большинства нерастворимых силикатов, но не шпинели, двуокиси олова и сульфидов 5.
МЕТОДЫ ОТДЕЛЕНИЯ
Важнейшие методы отделения олова основаны на свойствах его сульфидов. Так, например, олово может быть отделено: от элементов, не входящих в группу сероводорода, осаждением сероводородом в умеренно-кислом растворе (стр. 85); от сульфидов элементов группы меди осаждением последних в растворах сульфидов щелочных металлов (стр. 87); от мышьяка — осаждением этого элемента сероводородом в сильно солянокислом растворе (стр. 83) и от мышьяка (III) и сурьмы (III) — осаждением последних сероводородом в растворе, содержащем олово в четырехвалентном состоянии и либо щавелевую, либо фтористоводородную-кислому (стр. 89).
Осаждение олова раствором аммиака в присутствии аммонийных солей успешно применяется для предварительного отделения олова от-больших количеств меди, никеля или кобальта перед определением его-йодометрическим методом. Это отделение, совершаемое в присутствий других осаждаемых аммиаком элементов, например железа (III), служит также для извлечения и концентрирования малых количеств олова6.
1 А. Н. Low, A. J. W е i n i g, W. Р. S с h о d е г, Technical Methods of Ore Analysis, 11th ed., J. Wiley and Sons, 1939.
2 H. 0. Hoffman, Chem. News, 62, 57 (1890); 0. Beck, H. .Fischer, School Mines Quart., 20, 372 (1899).
3A. Froehde, Pogg. Ann., 119, 317 (1863); E. D о n a t h, Z. anal. Chem., 19,. 23 (1880).
4 W. van Tongeren, Ind. Nederland Indie, 7 (5), 51 (1941).
5 H. N. Warren [Chem. News, 67, 16 (1893)] утверждает, что «недавно было, доказано», что окиси сурьмы и олова улетучиваются при сплавлении с щелочами при-высоких температурах. Температура и другие условия, а также величина потери им не приводятся.
• J. S. К п а р р е г, К. A. Craig и G. С. С h a n d 1 е е [J. Am. Chem. Soc., 55, 3945 (1933)] утверждают, что олово можно количественно осадить фениларсоновой кислотой C6HBAsO(OH)2 в горячем разбавленном (5 : 95) солянокислом растворе-и таким образом отделить от цсех элементов, встречающихся в бронзах и латунях, за исключением железа. В присутствии железа осадок олова надо переосадить. Полученный осадок можно превратить в SnO2 прокаливанием, которое надо проводить сначала-осторожно до сжигания всего углерода, а затем при 1100° С до постоянной массы. Согласно В. Tougarinoff [Bull. Soc. chim. Belg/, 45, 542 (1936)], очень малые количества олова (например, 0,1 мг в 30 мл) можно обнаружить, осаждая его из 15— 20%-ного (по объему) раствора соляной кислоты, добавлением нитрофениларсоновой кислоты C6HgOHNOaAsO(OH)2. Медь, цинк, кобальт, никель, железо и те малые количества висмута и сурьмы, какие могут остаться в растворе после проведения предварительных операций, не мешают.
Методы отделения
335
Оловянные руды и концентраты часто содержат минералы, в состав-которых входят элементы, которые необходимо отделять перед восстановлением олова и йодометрическим его определением. При выполнении быстрых рядовых анализов мешающее влияние малых количеств меди, мышьяка, сурьмы, висмута и германия устраняется или в значительной мере преодолевается обработкой растворов олова в соляной кислоте-чистым железом (Ferrum Reductum). Осажденные железом металлы захватывают небольшие количества олова, которые можно извлечь растворением этих металлов и вторичным осаждением.
Имеются указания 1 на то, что олово может быть полностью осаждено-при pH = 1,5 добавлением едкого натра к кипящему раствору, содержащему хлорооловянную кислоту, которая получается при растворении сплавов в разбавленной царской водке. Если олово получено в сернокислом растворе (как, например, при растворении осадков, содержащих олово, вместе с фильтром в серной и азотной кислотах, выпаривании и нагревании при температуре выделения густых паров серной кислоты для окисления органических веществ и удаления окислов азота), его-можно полностью осадить разбавлением раствора до 1,8 н. концентрации серной кислоты (5 мл H2SO4 в 100 мл) и нагреванием на паровой бане в течение 1 ч. Для полного отделения таким способом олова от железа (III) и индия, гидролизующихся при низких значениях pH, требуется двукратное или многократное осаждение.
Олово может быть отделено от малых количеств вольфрама осаждением сероводородом в разбавленном солянокислом растворе, содержащем, винную кйслоту (стр. 89). В присутствии значительного количества вольфрама отделение лучше всего проводить повторным осаждением аммиаком в присутствии аммонийных солей, растворением полученного под конец осадка в соляной и винной кислотах и осаждением олова сероводородом 2. Каждый раз, когда удаляют гидроокись олова с фильтровальной бумаги, необходимо принимать меры предосторожности для уверенности в полном ее растворении. Олово легко потерять вследствие захвата осадка бумажными волокнами. Единственный способ избежать потерь 'состоит в разрушении фильтра с остатком олова обработкой азотной и серной кислотами (стр. 438) до полного растворения или в сжигании фильтра при низкой температуре в фарфоровом тигле. ^Золу переносят в платиновый тигель, сплавляют с небольшим количеством карбоната натрия и растворяют плав в кислоте.
Если для растворения гидроокиси олова была применена соляная кислота, то необходимо принять дополнительные меры предосторожности, а именно: хорошо отмыть фильтр от хлоридов, чтобы не было потери, олова вследствие улетучивания.
При определении олова в горных породах и минералах его редко-отделяют в виде метаоловянной кислоты нагреванием с азотной кислотой, как это делается при анализе цветных металлов, содержащих небольшие количества железа или совсем не содержащих железа. Это объясняется тем, что горные породы содержат так много железа, что полное осаждение олова этим способом делается невозможным и выделяемый осадок метаоловянной кислоты никогда не бывает чистым. Если анализируемое-
1 R. Gilchrist,!. Research NBS, 20, 745 (1938).
* См. также гл. «Вольфрам» стр. 765.
336
Гл. XV. Олаво
вещество растворимо в азотной кислоте и содержит не более 1—2 мг железа, то практически полное отделение олова может быть достигнуто растворением в разбавленной (1 : 1) азотной кислоте, разбавлением до концентрации азотной кислоты 1 : 5, нагреванием при 80—100° С в течение 3 ч или более, фильтрованием горячего раствора через плотный фильтр и промыванием осадка горячей водой или горячей разбавленной (1 : 20) азотной кислотой. Умеренные количества серной кислоты не мешают этому осаждению. Полученный осадок обычно бывает загрязненным большим числом примесей: кремнекислотбй, ниобием, танталом, вольфрамом, •сурьмой, мышьяком, фосфором и даже такими элементами, как железо, медь и цинк х.
Очистка осадка сплавлением с содой и серой требует большого труда, ненадежна и не дает удовлетворительных результатов, потому что: 1) необходимо повторное сплавление; 2) полное осаждение олова сомнительно и 3) после сплавления олово остается все же связанным с сурьмой и мышьяком. По этим причинам осаждение олова обработкой азотной кислотой должно быть рассматриваемо как предварительное его выделение, но не как количественное отделение 1 2.
Отделение олова от сурьмы описано на стр. 96, от мышьяка — на стр. 303 и от свинца — на стр. 260. Олово можно отделить от многих элементов восстановлением его до металла цинком, алюминием или кадмием в солянокислом растворе, а затем освободить от избытка примененного для осаждения металла растворением в азотной кислоте, нагреванием на водяной бане и отфильтровыванием образовавшейся метаоловян-ной кислоты.
Олово может быть полностью отогнано и отделено от меди и молибдена пропусканием тока сухого хлористого водорода через нагретый приблизительно до 200° С сернокислый раствор, содержащий все олово в четырехвалентном состоянии. Метод отгонки, в котором мышьяк, сурьма и олово последовательно отгоняются, описан на стр. 96.
Интересное отделение олова от некоторых элементов основано на применении купферона 3. Пользуясь этим реактивом, как описано на стр. 143, можно олово количественно осадить и отделить от алюминия, хрома, марганца, цинка, кобальта, никеля и, вероятно, также от сурьмы (V). Поскольку отделение от некоторых металлов группы сероводорода этим методом неудовлетворительно (стр. 144), то его, по-видимому, следует применять лишь для предварительного выделения малых количеств
1 Например, при определении олова в бронзе (88,33% меди, 7,9% олова, 1,89% цинка, 1,52% марганца и 0,12% железа) растворением 1 г пробы в азотной кислоте, нагреванием в течение нескольких часов или выпариванием досуха и прокаливанием осталось в растворе в обоих случаях около 0,1 мг олова, а метаоловянная кислота содержала приблизительно 1,5 мг железа, 0,4 мг меди, 0,2 мг цинка, но не содержала свинца. При анализе навесок чистого олова величиной 0,2 г в среднем в растворе оставалось 0,4 мг олова. Следует отметить, что алюминий не мешает осаждению метаоло-вянной кислоты и не загрязняет осадка.
2 Согласно Е. Stelling [Ind. Eng. Chem., 16, 346 (1924)], метаоловянная кислота, даже после прокаливания ее до темно-красного каления, может быть растворена обработкой 50 мл концентрированного раствора сернистой кислоты, нагреванием при 60—70° С в течение нескольких минут, прибавлением 10 мл соляной кислоты и кипячением.
3 A. Kling, A. L a s s i е и г, С. г., 170, 1112 (1920); N. Н. Furman, Ind. Eng. Chem., 15, 1071 (1923); A. Pinkus, J. Claessens, Bull. Soc. chim, Belg., 31, 413 (1927).
Методы определения
337
олова, содержащихся в продажных сортах алюминия, цинка и кобальта. Кроме того, этим путем осаждают олово из фильтратов, получаемых после выделения сурьмы и мышьяка осаждением их сероводородом в присутствии фтористоводородной кислоты. В этом случае надо удалить сероводород и перед осаждением прибавить борную кислоту.
Для отделения примесей от больших количеств олова (а также мышьяка и сурьмы) с целью их определения, как, например, при анализе олова высокого сорта в чушках, в дистилляционную колбу помещают 25 г полученных сверлением стружек анализируемого металла (или целый кусок его). В колбу должны быть вставлены: трубка для ввода в нее двуокиси углерода, капельная воронка, термометр и трубка, присоединяющая колбу к холодильнику. Пустив слабый ток двуокиси углерода, приливают (сначала небольшими порциями) смесь (5 : 1) 48 %-ной бромистоводородной кислоты и брома. Когда бурная реакция прекратится, приливают ту же смесь так, чтобы она покрыла остаток, нагревают до 130—140° С и поддерживают эту температуру, пока все олово не будет отогнано. Затем охлаждают, разбавляют водой и переливают раствор из колбы в подходящий сосуд. Для уверенности в полноте удаления олова можно еще обработать раствор едким натром и сульфидом натрия (стр. 87). В меньшей степени может быть рекомендована обработка пробы той же смесью в открытом сосуде, выпаривание досуха, обработка остатка смесью кислот и вторичное выпаривание.
Такие сплавы, как бронза, «никелевое серебро» и т. и., можно рас-. творять, отгоняя олово, мышьяк и сурьму, следующим способом, 5 г тонко измельченной пробы помещают в стакан емкостью 600 мл, приливают 35 мл 48 %-ной бромистоводородной кислоты и 5—10 мл брома, р покрывают часовым стеклом и нагревают при 70—80° С до разложения ц, пробы. Затем часовое стекло снимают,7обмывают над стаканом и удаляют,
к- Раствор выпаривают досуха, пользуясь излучающим теплом от источника
[". его, находящегося вверху над раствором. Стакан покрывают сухим часо- вым стеклом, поместив его на стеклянных крючках, и нагревают 10— р’1 15 мин. на умеренно горячей цлитце. Затем охлаждают, приливают 15 мл
, .. азотной кислоты и 25 мл 70%-ной хщорной кислоты, выпаривают до по-
v явления паров последней и осторожно кипятят 10—15 мин. Снова охла-ждают, приливают 100 лм горячей воды, перемешивают до растворения солей и, если окажется нерастворимый остаток кремнекислоты, его отфиль-tv тровывают и промывают.
В- '
К > МЕТОДЫ ОПРЕДЕЛЕНИЯ
&-.. Наилучший метод определения олова основан на окислении его до К четырехвалентного состояния титрованным раствором иода после того, ж ; > как оно было восстановлено до двухвалентного в сернокислом или соляно-BfeX кислом растворе гранулированным свинцом х, железными стружками 2 pj или никелем 3.
1 A. R. Р о w el 1, J. Soc. Chem. Ind., 37, 287T (1918); G. E. F. L u n d e 1 1,
J. A. S c h e г г e г, J. Ind. Eng. Chem., 14, 426 (1922).
a A. H. L ow( A. J. W e i n i g, W. P. S c h о d e r, Technical Methods of
Ote Analysis, 11th ed., 1939, p. 239.
t * R. L. H a 11 e 11, J. Soc. Chem. Ind. London, 35, 1087 (1916).
ftp . 22 Закаа 522.
338
Гл. XV. Олово
Определение олова окислением его до четырехвалентного и осаждением аммиаком с последующим прокаливанием до SnOa вполне удовлетворительно, если в растворе содержится только одно олово. Однако применять этот метод можно редко, так как наряду с оловом осаждаются и другие элементы. Отделение олова в виде сульфида олова с последующим прокаливанием до SnOa требует тщательного отделения всех остальных металлов группы сероводорода, применение этого метода должно быть ограничено определением малых количеств олова, так как сульфид олова с трудом отфильтровывается и промывается и его трудно количественно превратить в SnOa.
Йодометрический метод
Йодометрический метод определения олова часто применяют без предварительного отделения олова от других элементов. Однако в присутствии элементов, которые также восстанавливаются применяемым восстановителем и затем окисляются иодом, полное отделение олова необходимо.
При выполнении точных определений олова йодометрическим методом основными требованиями являются: количественное восстановление всего олова до двухвалентного состояния и предупреждение возможного окисления его кислородом воздуха. Первое требование не вызывает особых затруднений и может быть вьйюлнено с помощью многих металлов и соединений. Для удовлетворения второго требования приходится поддерживать неокислительную атмосферу в продолжение всей операции, чего невозможно достичь применением клапана Бунзена или введением в раствор нескольких граммов карбоната натрия.
Следует отметить, что различные металлы и соединения, применяемые для восстановления олова в кислых растворах до двухвалентного состояния, ведут себя различно по отношению к изменениям температуры и кислотности раствора. Например, восстановление свинцом может быть замедлено до ничтожной скорости одним лишь охлаждением кислого раствора, а восстановление фосфорноватистой кислотой — охлаждением раствора и разбавлением его водой, освобожденной от воздуха. Но такая обработка оказывается недостаточной при применении в качестве восстановителя металлического железа, алюминия или цинка, избыток которых перед титрованием иодом надо удалить.
Влияния мешающих элементов, если они присутствуют, нельзя с уверенностью предвидеть, потому что они могут или восстановиться до губчатого металла и при этом окклюдировать небольшие количества олова1 или же восстановиться в большей или меньшей степени до низших степеней валентности, в которых они титруются иодом. Например, германий большей частью, если не полностью, восстайавливается, образуя нерастворимое соединение, и не мешает определению олова, если для восстановления последнего применяется свинец. Но если восстановление олова проводится
1 Если для восстановления олова применяются свинец, железо, цинк или алюминий, то одновременно осаждается сурьма, которая захватывает с собой заметные количества олова, и, кроме того, выделившаяся сурьма может в некоторой мере восстановить четырехвалентное олово снова до двухвалентного во время титрования последнего иодом. При использовании порошковидной сурьмы в качестве восстановителя результаты определения олова могут оказаться как повышенными, так и пониженными, в зависимости от степени измельчения применяемой сурьмы [S. G. Clarke, Analyst, 56, 82 (1931); В. S. Evans, D. G. Higgs, там же, 69, 201, 291 (1944)].
Методы определения
339
с применением железа, то германий частично восстанавливается, образуя при этом растворимое соединение, титруемое иодом. Малые количества германия (менее 10 мг) не оказывают заметного влияния.
Для восстановления олова до двухвалентного состояния или до металла применяют;.; свинец, никель, железо, алюминий и цинк. Из этих металлов следует отдать предпочтение свинцу и никелю, так как их восстанавливающее действие легко останавливается охлаждением раствора после восстановления, и потому удаление избытка этих металлов не обязательно.
При использовании свинца в качестве восстановителя определению олова мешают азотная кислота, вольфрам, молибден, хром и ванадий. Азотная кислота реагирует с иодистоводородной кислотой, выделяя иод, отчего получаются для олова пониженные результаты. Вольфрам восстанавливается с образованием соединения, окрашенного в синий цвет, и присутствие большого его количества маскирует конечную точку титрования (окрашивание крахмала иодом). Если же вольфрама мало и синяя окраска получаемых после его восстановления продуктов слаба и не мешает обнаружить конечную точку титрования с крахмалом, то результаты получаются точные, так как соединения восстановленного вольфрама не титруются иодом.
Молибден, хром и ванадий восстанавливаются свинцом, и так как продукты их восстановления титруются иодом, то для олова получаются повышенные результаты. Присутствие этих элементов обнаруживается по изменению окраски раствора при восстановлении олова. Молибден, например, после восстановления окрашивает раствор в коричневый цвет, а ванадий — в пурпуровый. Малые количества мышьяка не мешают определению х. Из остальных веществ, не мешающих титрованию, можно отметить сульфаты, фосфаты, иодиды, бромиды, фториды, железо, никель, кобальт, цинк, марганец, уран, алюминий, свинец, висмут, магний и щелочноземельные металлы.
Титан, независимо от его содержания в исследуемом растворе, приводит к получению нескольких повышенных результатов, если для титрования олова (II) применяется раствор иода, содержащий растворенный в нем воздух 1 2.
Некоторые элементы, как, например, никель, присутствуя в большом количестве, мешают окраской своих солей. Небольшие количества меди (II) (0,005 г и менее) восстанавливаются до металла или до не титруемых иодом ионов меди (I). Восстановление больших количеств меди (II) в этих условиях может иногда вызывать сомнения, а при неполноте их восстановления получаются пониженные результаты определения олова, так как вследствие реакции между солями меди (II) и образующейся при титрова
1 Например, Н. A. Buchheit получил 0,2000 г олова при анализе раствора, содержавшего 0,2000 г олова и 1 мг мышьяка; 0,2018 г олова — при анализе раствора, содержавшего 0,2009 г олова и 100 мг мышьяка.
2 F. L. О k е 11 и J. Lumsden [Analyst, 60, 803 (1935)] утверждают, что при использовании растворов иода, не содержащих воздуха, получаются точные результаты. W. van Tongeren [Ind. Nederland Indie, 7 (5), 57 (1941)] рекомендует для устранения влияния титана применять растворы иода, содержащие не менее 90 г иодида калия в 1 л W. Н. R. Allen [Mining Mag., 40, 25 (1929)] также останавливает внимание на помехах, вызываемых титаном, и для устранения их применяет раствор, содержащий 17,5 г иода и 318 г иодида калия в 1 л воды.
22*
340
Гл. XV. Олово
нии иодиСтоводородной кислотой выделяется иод. Кроме того, в присутствии большого количества меди небольшая часть олова может восстановиться до металла, особенно при низкой кислотности раствора и продолжительном его кипячении. Малые количества сурьмы (меньше 15 мг) не оказывают заметного влияния; большие количества сурьмы приводят к ошибкам, которыми нельзя пренебречь.
Ход определения. Восстановление свинцом. Приготовляют раствор, содержащий 0,2 г или менее олова, 10—15 мл серной кислоты, 100 мл соляной кислоты и не содержащий нитратов, вольфрама, молибдена, хрома и ванадия Ч Переносят раствор в коническую колбу емкостью 500 мл, разбавляют до 300 мл водой и прибавляют 2—3 г реактивного свинца. Закрывают колбу пробкой с тремя отверстиями. Через одно отверстие проходит трубка для ввода двуокиси углерода, доходящая почти до дна колбы; через второе отверстие проходит отводная трубка длиной 15—20 см, служащая холодильником; третье отверстие закрыто маленькой пробкрй. f
Пропускают в колбу медленный ток двуокиси углерода, нагревают до кипения и осторожно кипятят 30—40 мин. Затем охлаждают льдом до 10° С, усилив предварительно ток двуокиси углерода во избежание снижения давления. Последнее можно обнаружить, присоединяя шариковую трубку («гусек») к трубке для выхода газа. По охлаждении раствора продолжают пропускать газ, вынимают пробку из третьего отверстия, приливают через него при помощи пипетки 5 мл прозрачного раствора крахмала и затем вставляют в это отверстие кончик бюретки с титрованным раствором иода. Титруют до неисчезающей синей окраски. В результат титрования вводят поправку на «холостую» пробу, проведенную через все стадии анализа. Титр раствора иода должен быть установлен по чистому олову, растворенному и обработанному так же,как и при выполнении определения. Полученный таким способом титр несколько выше теоретического, что объясняется небольшим окислением олова (II) воздухом, содержащимся в применяемом растворе иода 1 2.
Восстановление никелем. Раствор, содержащий в 300 мл не более 0,25 г олова, 65 мл соляной кислоты и не содержащий нитратов, вольфрама, молибдена, хрома, ванадия, меди и сурьмы, переносят в коническую колбу емкостью 750 мл и цводят 2—3 полоски чистого никеля (размером 2 X 28 см), свернутых в спирали или согнутых так, чтобы они стояли на ребре. Затем закрывают колбу резиновой пробкой, в которую вставлена отводная сифонная трубка диаметром 7 мм, суженная на обоих концах. Внутренний конец этой трубки должен быть у самой пробки, внешний — на расстоянии около 1,2 см от дна колбы. Раствор нагревают до кипения и слабо кипятят 60 мин, избегая большого уменьшения его
1 Во многих случаях анализа сернокислый раствор олова приготовляют, помещая фильтр с метаоловянной кислотой или смесью сульфидов в колбу Кьельдаля и разлагая кипячением с серной кислотой и сульфатом натрия, так же как и при определении азота методом Кьельдаля (стр. 862).
2 F. L. О k е 11 и J. Lumsden (цит. выше) утверждают, что при применении раствора иода, освобожденного от растворенного воздуха, можно пользоваться теоретическим титром. См. также В. S. Е vans, D. G. Higgs, Analyst, 69 , 201 (1944). При желании можно проводить титрование раствором сульфата церия (IV) в присутствии иодида калия и крахмала [L. G. Bassett, L. F. Stumpf, Ind-Eng. Chem., Anal. Ed., 6, 477 (1934)].
Методы определения
341
объема. Не прекращая кипячения, погружают внешний конец сифонной трубки в насыщенный раствор бикарбоната натрия, приготовленного на \ не содержащей воздуха воде. Затем колбу вместе с запирающим выходное отверстие раствором бикарбоната натрия удаляют от источника нагрева и дают остыть. Когда раствор охладится до 10—15° С, осторожно вынимают пробку, приливают 5 мл раствора крахмала и титруют титрованным раствором иода так, чтобы струя его шла по внутренней стенке колбы. Анализируемый раствор не перемешивают и не взбалтывают, пока не будет прибавлено почти все требуемое количество иода (останется добавить около 1 мл раствора). Затем заканчивают титрование при перемешивании раствора, титруя до синего окрашивания.
Восстановление алюминием или цинком. Восстановление олова (IV) металлическим алюминием или цинком в солянокислом растворе проходит успешно,' если анализируемый раствор не содержит других осаждаемых этими металлами элементов, которые могут окклюдировать олово, и не содержит также веществ, восстанавливающихся в этих условиях и потом титруемых иодом. При проведении такого восстановления надо следить, чтобы выделившаяся губка металлического олова полностью растворилась, превращаясь в хлорид олова (II), и лишь потом приступать к титрованию.
Восстановление фосфорноватистой. кислотой1. Восстановление проводят в горячих разбавленных (1:1) солянокислых растворах с добавлением тонко раздробленной ртути в качестве катализатора. В этих условиях олово восстанавливается до двухвалентного состояния, сурьма — до трехвалентного, мышьяк — до металла. Восстанавливающее действие фосфорноватистой кислоты Н3РО2 прекращают разбавлением и охлаждением раствора в атмосфере инертного газа. . Выделившийся мышьяк перед титрованием удаляют; сурьма не мешает, если добавлять иодид калия и оксалат аммония и проводить титрование в достаточно сильнокислом растворе. Мешают железо, медь и нитрат-ионы.
Весовое определение в виде двуокиси олова
Как уже было сказано, весовые методы, в которых олово определяется взвешиванием в виде двуокиси олова, обычно требуют более или менее полного предварительного отделения элементов, могущих загрязнить осадок.
При применении этого метода нужно следить за тем, чтобы прокаливание осадка происходило в хороших для окисления условиях, так как двуокись олова легко восстанавливается. После прокаливания двуокиси олова в закрытом тигле на полном пламени горелки Теклю нередко находят в тигле корольки металлического олова. Метаоловянную кислоту, полученную растворением олова в азотной кислоте и выпариванием досуха, можно количественно превратить в двуокись олова йпОз даже при 700° С. Чистая двуокись олова после охлаждения имеет белый цвет.
1 В. S. Evans, Analyst, 56, 171 (1931); 69, 201 (1944).* См. также С. Ю. Ф а й н-1 . б ер г [Анализ руд цветных металлов, Металлургиздат, 1953, стр. 233], по данным которого фосфорноватистая кислота не выделяет ни мышьяка, ни меди, а восстанавливает их до низших форм валентности. Допустимо содержание мышьяка до 40 мг, меди — до 20 мг,
342
Гл. X V. Олово
Голубоватый ее оттенок указывает на загрязнения, чаще всего сурьмой х.
Ход определения. Осаждение аммиаком. Приготовляют содержащий олово кислый раствор, свободный от органических веществ и от всех веществ, которые Выпали бы в осадок вместе с оловом при осаждении аммиаком. Осаждают, как описано в гл. «Алюминий» (стр. 554), и промывают осадок горячим 2 %-ным раствором нитрата аммония. Надо убедиться в том, что хлориды, если они присутствовали, удалены из осадка полностью. Бумажный фильтр с содержимым осторожно высушивают во взвешенном фарфоровом тигле, обугливают бумагу и прокаливают до выгорания всего угля в условиях хорошего окисления. Желательно, чтобы тигель был защищен от восстанавливающих газов пламени с помощью щита и чтобы прокаливание проводилось при возможно более низкой температуре. Под конец прокаливают при 1100—1200° С в муфеле или помещая тигель над горелкой так, чтобы пламя целиком его не охватывало. Охлаждают в эксикаторе, взвешивают и повторяют прокаливание до постоянной массы. При проведении особо точных анализов следует перенести прокаленную двуокись олова в платиновый тигель и ввести поправку на содержание в ней кремнекислоты обработкой фтористоводородной и серной кислотами.
Осаждение сероводородом. Помимо необходимости отделения других элементов, осаждаемых сероводородом, определение олова этим методом представляет еще то неудобство, что слизистый характер выделяющегося осадка затрудняет фильтрование и промывание. Кроме того, нельзя быть уверенным в полноте превращения сульфида олова в двуокись олова при прокаливании. По этим причинам метод не должен применяться, если содержание олова превышает несколько миллиграммов 1 2.
В разбавленном кислом растворе, содержащем олово и не содержащем других металлов группы сероводорода, осаждают олово так, как описано на стр. 84. Оставляют стоять 2—3 часа или лучше на ночь. Затем фильтруют через взвешенный тигель Гуча, снабженный колпачком для дна, и промывают осадок 2 %-ным раствором нитрата аммония до удаления посторонних солей, особенно хлоридов. Надевают колпачок на дно тигля, придают тиглю наклонное положение и нагревают сперва очень осторожно во избежание разбрызгивания. С этой целью горелку помещают впереди тигля (см. рис. 12, стр. 131) и постепенно подвигают ее ко дну тигля. Когда опас
1 Поправку на присутствие некоторых загрязнений в прокаленном осадке двуокиси олова можно ввести следующим образом. Прибавляют иодид аммония, прокаливают при 425—475° С, охлаждают, обрабатывают остаток азотной кислотой, выпаривают досуха, осторожно прокаливают и снова взвешивают. При такой обработке олово улетучивается в виде иодида олова (IV), а примеси железа, меди, свинца, кремния и вольфрама остаются в виде их окисей. Сурьма также полностью улетучивается, цинк улетучивается частично, фосфор — в зависимости от условий (* количества железа и других примесей*) ]Е. R. С а 1 е у, М. G. Burford, Ind. Eng. Chem., Anal. Ed., 8, 114 (1936)].
2 D. A. L a mbi e и W. R. Schoeller [Analyst, 65, 283 (1940)] утверждают, что можно получить плотный зернистый осадок сульфида олова (IV), легко переходящий в двуокись олова при светло-красном калении, следующим образом. Олово перед осаждением должно быть в виде сульфата олова (IV) в растворе, имеющем объем 200—400 мл и содержащем 5—15 мл серной кислоты, 20—50 мл 20%-ного раствора хлорида аммония и немного бумажной массы. Раствор насыщают сероводородом, дают осадку осесть, фильтруют и промывают осадок разбавленной (1 : 99) серной кислотой, содержащей сероводород.
Методы, определения
343
ность разбрызгивания исчезнет, нагревание ведут при 600—700° С, пока не прекратится выделение сернистого ангидрида. Охлаждают и прибавляют к осадку кусочек карбоната аммония. Снова осторожно нагревают до удаления аммонийных солей, затем прокаливают при 1000—1200° С в благоприятных для окисления условиях. Охлаждают, взвешивают в виде SnOa и повторяют прокаливание до получения постоянной массы. Двуокись олова не гигроскопична.
Осаждение купфероном. Заслуживающим внимания методом определения олова в дистилляте, получаемом при разделении мышьяка, сурьмы И олова (стр. 96), является осаждение его купфероном (стр. 146) с последующим прокаливанием осадка до SnOa. При выполнении этого определения нужно соблюдать следующие условия: 1) избегать больших концентраций хлорида аммония или бромида аммония, которые вызывают разбрызгивание во время высушивания и прокаливания осадка; 2) применять купферон в 6—7-кратном избытке; 3) полностью удалять галогенид-ионы при промывании осадка, потому что их присутствие в осадке приводит к улетучиванию олова во время прокаливания; 4) высушивать осадок очень медленно, постепенно, чтобы избежать его разбрызгивания. Для соблюдения этих условий олово перегоняют из смеси 1—2 частей воды и 1 части бромистоводородной кислоты, содержащей 5—10 мл соляной кислоты в 100 мл, и разбавляют дистиллят так, чтобы кислоты в нем стало 5—10% по объему, но не нейтрализуют кислоту аммиаком. Общий объем дистиллята можно довести до 1000 мл. При высушивании фильтра с осадком его следует сначала нагревать 4—5 ч приблизительно при 45° С, а затем постепенно довести температуру до 1000° С, начиная нагревание в холодном муфеле Ч
Другие методы
Из остальных методов определения олова важнейшим является электролитическое его осаждение на сетчатом платиновом катоде из растворов, содержащих щавелевую кислоту и оксалат калия или оксалат аммония 1 2. Здесь не приводится описания этого метода, потому что он менее точен, чем йодометрический метод, и требует большого числа предварительных отделений, а потому более длителен.
* Очень хорошим методом определения олова является весовой метод, в котором олово выделяется на катоде внутренним электролизом (см. стр. 171) с применением цинкового анода, покрытого защитной пленкой коллодия3. Перемешивание во время электролиза проводится путем пропускания через раствор тока воздуха или двуокиси углерода. В течение 1—3 ч можно выделить от 25 до 200 мг олова. Железо надо предварительно восстановить до двухвалентного гидроксиламином. Для устранения мешаю
1 Подробности см. в статье W. D. Mogerman, J. Research NBS, 33, 307 (1944). Отделение олова и определение его в дистилляте можно также произвести гидролизом его соли при pH не ниже 1,5, как это рекомендует R. Gilchrist (цит. выше, стр. 335, сноска 1), фильтрованием, промыванием осадка и прокаливанием его до двуокиси олова. Как отмечалось выше, при промывании осадка необходимо полное удаление галогенид-ионов.
2 A. Classen, М. A. Reis, Вег., 14, 1622 (1881); F. Н е n z, Z. anorg. Chem., 37, 40 (1903).
3 Ю. А. Ч е р н и х о в, Г. А. Большакова, Зав. лаб., 14, 1 (1948); Ю. А. Ч е р н и х о в, Р. Н. Рощина, там же, 14, 383 (1948).
344
Гл. XV. Олово
щего влияния вольфрама вводят 100 мг щавелевой кислоты. Медь^мышьяк, свинец, висмут и другие элементы мешают, и их надо предварительно отделить. Доп. ред.*
Описан метод 1 определения олова в органических веществах, не содержащих других нелетучих составных частей.
Колориметрическое определение олова может быть проведено 2 отгонкой 0,02—1,0 мг олова в виде хлорида олова (IV), восстановлением до двухвалентного и получением синей окраски с силикомолибденовой кислотой.
*Наилучшим колориметрическим методом определения малых количеств олова, по-видимому, является метод, основанный на реакции его с дитиолом3 (1-метил-3,4-димеркаптобензолом). Этот реактив образует с оловом (II) розово-красный осадок, а при малых количествах олова — коллоидный раствор, для стабилизации которого прибавляют агар-агар. Мешают висмут, медь, серебро, ртуть, молибден, ванадий, теллур, мышьяк, сурьма, германий, большие количества хрома, никеля и кобальта. Доп. ред.*
1 Н. Gilman, W. В. King, J. Am, Chem. Soc., 51, 1213 (1929).
2 I. Baker, M. Miller, R. S. G i b.b s, Ind. Eng. Chem., Anal. Ed., 16, 269 (1944). *См. также N. Strafford, Mikrochim. Acta, 2, 306 (1937), в этой работе в качестве реактива предлагается фосфоромолибденовая кислота*.
3 R. Е. D. Clark, Analyst, 61, 242 (1936); 62, 661 (1937); В. А. Назаренко, Л. Е. Шварцбуд, И. А. Сойферман, Зав. лаб., 15 , 387 (1949).
Глава XVI
ГЕРМАНИИ
Германий является очень редким металлом, сходным с оловом. Его содержание в изверженных породах земной коры составляет1 1-10~10%. Германий встречается главным образом в .некоторых цинковых обманках Северной Америки. К важнейшим содержащим германий минералам относятся аргир'одит - сульфид германия и серебра (4AgaS-GeSnS2), канфильдит — сульфид серебра и олова (4Ag2S-(Ge, Sn)S2), в котором олово частично заменено германием, ифранкит — сульфидный минерал, содержащий наряду с германием свинец, олово и сурьму. Кроме того, германий встречается в энергите2 Cu2AsS4, в.оловянных 2, танталовых и ниобиевых минералах, а также в некоторых углях и коксе. В ничтожных количествах он был обнаружен в некоторых минеральных водах.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород германий, по-видимому, полностью улетучивается в виде хлорида в процессе выделения кремнекислоты упариванием с соляной кислотой, так как хлорид германия (IV). кипит при 86° С.
Насколько известно, в металлургических продуктах германий содержится лишь в виде примеси. В тех случаях, когда его следует определять, необходимо принимать специальные меры, чтобы не произошла потеря его вследствие улетучивания.
1 F. W. Clarke, Н. S. Washington, Proc. Natl. Acad: Sci., 8, 114 (1922). *По данным А. П. Виноградова (1950), содержание германия в литосфере равно 7 • 10-4%*.
2 J. Р а р i s h, F. М. Brewer, D. A. Holt, J. Am. Chem. Soc., 49, 3028 (1927). Эти авторы описывают дуговой спектрографический метод открытия и определения германия. J. Р а р i s h [Econ. Geol., 23, 660 (1928)] указывает на содержание германия в станните (следы), в боливийском пираргирите Sb2S3 • 3Ag2S (ниже 3%), в самородной меди (0,001%) и в каламине Н27п28Ю6(следы).
Для определения 0,019 мг или более германия в присутствии больших количеств мышьяка S. А. С о a s е [Analyst, 59, 462 (1934)] рекомендует электролитическое восстановление в щелочном растворе до германистого водорода GeH4 с последующим термическим разложением и сравнением образующегося при этом зеркала со стандартами, приготовленными аналогичным образом.
346
Гл. XVI. Германий
РАЗЛОЖЕНИЕ ГЕРМАНИЕВЫХ МИНЕРАЛОВ И ОКИСИ ЦИНКА, СОДЕРЖАЩЕЙ ГЕРМАНИЙ
При определении германия обычные методы разложения материала соляной кислотой, царской водкой, смесью серной или хлорной кислоты с соляной или бромистоводородной кислотой и даже выпаривание с одной хлорной кислотой неприемлемы вследствие летучести хлорида (а также бромида) германия (IV). Из растворов, содержащих фтористоводородную кислоту, германий, по-видимому, заметно не улетучивается, если выпаривание и последующее прокаливание проводить медленно Ч
Аргиродит или канфильдит могут быть разложены сплавлением с 5—6-кратным количеством (по массе) смеси равных частей карбоната натрия и серы. Плав выщелачивают водой, раствор фильтруют и осадок промывают водой. Так как извлечение германия в раствор при этом редко бывает количественным, сплавление и выщелачивание повторяют. Фильтраты объединяют и германий совместно с мышьяком и некоторыми другими элементами группы мышьяка осаждают сероводородом из 6 н. по концентрации серной кислоты раствора. Колбу закрывают пробкой и оставляют на 24—48 ч. После этого сульфиды отфильтровывают, промывают 6 н. серной кислотой, насыщенной сероводородом, и растворяют в едком натре или аммиаке с перекисью водорода или хлором.
Для определения германия в окиси цинка наиболее удобна непосредственная отгонка его в виде хлорида 1 2, которая проводится следующим образом. 20—110 г окиси цинка, высушенной при 110° С, растирают в ступке с водой и затем сливают в колбу, содержащую раствор едкого натра. Соотношения должны быть следующие: 1 часть едкого натра, 2 части сухой окиси цинка и 5 частей воды. Колбу закрывают пробкой с тремя отверстиями, в которые вставлены трубка, достигающая дна колбы, маленькая делительная воронка и маленькая дистилляционная колонка. К боковому тубусу этой колонки присоединяют либиховский холодильник. Другой конец холодильника соединяют с трубкой, проходящей через пробку с двумя отверстиями, вставленную в коническую колбу, служащую приемником. Эта трубка доходит почти до дна колбы. Приемную колбу соединяют с другой (предохранительной) колбой посредством трубки, достигающей дна' второй колбы. В обе колбы вводят достаточное количество воды, чтобы закрыть концы вводных трубок, и затем колбы погружают в ледяную воду. Дистилляционную колбу погружают в толченый лед, прибор заполняют хлором и затем, не прекращая слабой струи газа, в колбу из делительной воронки медленно вводят соляную кислоту в количестве, достаточном для нейтрализации едкого натра и образования избытка, равного удвоенной массе окиси цинка. Лед удаляют и колбу слабо нагревают до тех пор, пока половина жидкости не перейдет в приемник. В дистилляционную колбу прибавляют соляную кислоту до первона
1 Так, например, если смесь 1 мл НС104, 2 мл H2SO4, 5 мл НС1, 5 мл HF и 1 мл Н2О, содержащую 0,003 г GeO2 в виде Ge(SO4)2, выпарить па водяной бане, затем нагреть на воздушной бане и после этого осадок прокалить при 900° С в течение 15 мин, то потерь германия не наблюдается. См. также W. С. A i t k е n h e a d, A. R. Middle t о n, Ind. Eng. Chem., Anal. Ed., 10, 633 (1938).
2 L. M. Dennis, J. P a p i s h, J. Am. Chem. Soc., 43, 2140 (1921). Cm. также C. James, H. C., Fogg, там же, 51, 1459 (1929).
Методы отделения
347
чального объема раствора и продолжают перегонку до тех пор, пока в колбе опять не останется половина жидкости. После этого снова добавляют соляную кислоту и снова перегоняют. По окончании дистилляции растворы, находящиеся в приемных колбах, объединяют, охлаждают во льду и прибавляют серную кислоту в количестве, необходимом для получения 6 н. концентрации H2SO4. Раствор насыщают сероводородом, закрывают колбу и оставляют стоять 24—48 ч. Осажденный сульфид германия обрабатывают, как указано в предшествующем способе.
МЕТОДЫ ОТДЕЛЕНИЯ
Основной метод отделения германия заключается в отгонке его в виде хлорида германия (IV). Применяя соответствующую дистилляционную колонку, германий можно отделить не только от элементов, которые не отгоняются из солянокислых растворов, но и от мышьяка, олова, сурьмы, селена и теллура, хлориды которых летучи. Для этой цели может служить колонка \ состоящая из трубки длиной 680 см и диаметром 20 мм, наполненной стеклянными бусами диаметром 7—9 мм. Трубка заключена в стеклянный кожухи снабжена специальной насадкой для перегонки. Дистилляция проводится, как описано выше (стр. 346). Хлорид германия (IV), заключенный в пузырьках воздуха, конденсируется с трудом, поэтому следует применять соответствующую поглотительную установку или, еще лучше, заменить концентрированную соляную кислоту газообразным хлористым водородом 1 2.
Для отделения германия от других элементов используются также методы, основанные на применении сероводорода и сульфидов щелочных металлов. Так как германий относится к подгруппе мышьяка сероводородной группы металлов, он может быть осажден сероводородом из кислых растворов, а затем отделен от элементов подгруппы меди обработкой сульфидного осадка сульфидами или полисульфидами щелочных металлов. Кроме того, в кислых растворах, содержащих фтористоводородную кислоту, германий ведет себя подобно олову, благодаря чему его можно отделять от мышьяка (III) и сурьмы (III), которые в этих условиях оса-' ждаются сероводородом (стр. 88). Количественное осаждение германия в виде сульфида происходит значительно труднее, чем осаждение большинства других элементов сероводородной группы. Выделять его лучше всего, насыщая сероводородом холодный раствор, 6 н. по концентрации серной кислоты. Образующемуся при этом почти коллоидному осадку дают отстаиваться в течение 48 ч, закрыв колбу пробкой. Осадок сульфида германия следует промывать 6 н. серной кислотой, насыщенной сероводородом.
Таннином германий осаждают из кислых растворов следующим образом 3. Нейтральный раствор, лучше свободный от хлоридов, содержащий в 150—250 мл 50—60 мг двуокиси германия, обрабатывают 5—15 мл 2 н.
1 L. М. Dennis, Е. В. Johnson, J. Am. Chem. Soc., 45, 1380 (1923).
2 Такие незначительные количества германия, как 0,0003%, могут быть определены в силикатных породах отгонкой хлорида германия (IV) и последующим колориметрированием дистиллята, обработанного сульфатом железа (II) и молибдатом аммония [A. G. Hybbinette, Е. В. S a n d е 11, Ind. Eng. Chem., Anal. Ed., ' 14, 715 (1942)].
3 G. R. Davies, G. Morgan, Analyst. 63, 388 (1938).
348
Гл. XVI. Германий
серной кислоты, 8—10 г сульфата аммония и нагревают почти до кипения. Сильно перемешивают и медленно вводят 10—30 мл свежеприготовленного 5%-ного раствора таннина. Осадку дают отстояться, для чего ставят стакан на край водяной бани, затем отфильтровывают его через беззольный фильтр при слабом отсасывании. Фильтр с осадком промывают 5 %-ным раствором нитрата аммония, содержащим немного таннина и 5 мл 2 н. азотной кислоты на 100 мл. Для лучшей промывки, особенно если присутствуют хлориды, осадок переносят обратно в стакан, перемешивают с 50 мл промывной жидкости, затем снова его отфильтровывают и продолжают промывать на фильтре. Промытый осадок с фильтром переносят во взвешенный тигель и прокаливают, сначала осторожно при температуре ниже 700° С, пока не сгорят органические вещества, а затем при 900—1000° С. Осадок и бумагу можно разложить также обработкой смесью азотной и серной кислот, выпариванием раствора досуха и осторожным прокаливанием. Согласно указаниям авторов, этот метод дает возможность отделять германий от мышьяка, галлия, цинка, меди, железа, магния, ванадия, титана и циркония, но не от молибдена.
*При осаждении таннином из сернокислых растворов 1 происходят заметные потери германия; величина потери зависит от количества добавляемого таннина, в избытке которого осадок германия частично растворим. Наилучшие результаты получаются при добавлении 0,5—2 мл 5%-ного раствора таннина на 1 мг GeO2. Несмотря на известную специфичность таннинового метода, применять его непосредственно к анализу растворов, содержащих посторонние компоненты, неудобно, так как таннин образует растворимые комплексные соединения со многими элементами, что затрудняет расчет его количества, необходимого для осаждения германия.
На получение не вполне удовлетворительных результатов при применении таннинового метода осаждения германия указано и в некоторых других работах2. Доп. перев.*
Хлорид германия (IV) очень мало растворим в концентрированной соляной кислоте (0,3 мг в 1 г кислоты при 0° С) и может быть отделен от растворимых хлоридов мышьяка (III), сурьмы (V), олова (IV) и титана (IV) многократным встряхиванием с концентрированной соляной кислотой в делительной воронке 3. Германий не мешает при использовании эфирнохлористоводородного метода (стр. 564), так как хлорид герма- •• ния (IV) раствором в соляной кислоте, насыщенной хлористым водородом, и не осаждается при разбавлении раствора эфиром.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Осаждение в виде сульфида германия и взвешивание в виде *
окиси германия |
Лучшим методом определения германия можно считать осаждение а
сероводородом с последующим переведением сульфида в окись, GeO2. |
Как указано выше (стр. 347), осаждение германия в виде сульфида сопря-жено с некоторыми трудностями. Последующее окисление сульфида гер- ;
1 И. П. Алимарин, О. А. Алексеевская, ЖПХ, 13, 1393 (1940).
2 Н. J. С 1 u 1 е у, Analyst, 76, 517 (1951); Н. Н о 1 n е s s, Anal. Chim. Acta , 2, 254 (1948).
3 E. R. Allison, J. H. Muller, J. Am. Chem. Soc., 54 , 2833 (1932).
Методы определения
349
мания не вызывает особых затруднений, если осуществлять его обработкой аммиаком и перекисью водорода х. Менее удовлетворительные результаты получаются при окислении азотной кислотой, так как реакция протекает очень бурно и трудно отделить выделяющуюся при этом свободную серу, избежав восстановления и улетучивания некоторых количеств германия.
Более или менее значительные количества сульфида германия можно перевести в окись кипячением с водой до разложения сульфида и удаления сероводорода 1 2. После этого раствор можно выпарить досуха во взвешенном тигле, обработать несколькими каплями азотной кислоты, снова выпарить досуха и затем прокалить при 900° С.
Ход определения. Сульфид германия, полученный осаждением сероводородом (см. стр. 347), растворяют в очищенном перегонкой.10 н. растворе аммиака, который вводят небольшими порциями. Раствор собирают во взвешенную платиновую чашку или большой тигель. На растворение осадка расходуется незначительное количество аммиака, так как.сульфид германия хорошо в нем растворим. Иногда может остаться нерастворен-ным небольшое количество серы, выделившейся совместно с сульфидом германия. Раствор фильтруют и промывают фильтр небольшими порциями воды до обесцвечивания промывной жидкости. К раствору прибавляют 20 мл 3 %-ной перекиси водорода, и дают пройти окислению на холоду. Весьма существенно, чтобы в перекиси водорода не содержалось нелетучего остатка. Выпаривают раствор досуха при 105° С, смачивают сухой остаток серной кислотой и осторожно нагревают содержимое чашки на пламени бунзеновской горелки до удаления сульфата аммония и серной кислоты. Прокаливают в условиях, обеспечивающих хорошее окисление, под конец при 900° С, до постоянной массы. Окись германия плавится 3 примерно при 1100° С.
Взвешивание в виде ортогерманата магния
Германий (IV) можно количественно осадить в виде ортогерманата магния Mg2GeO4 из аммиачного раствора 4. Этот метод подобен методу определения фосфора и мышьяка. Основное отличие его заключается в том, что германий осаждают смесью сульфатов магния и аммония вместо обычно применяемой магнезиальной смеси. Это делается для того, чтобы осадок ортогерманата магния был свободен от хлоридов, в присутствии которых германий при прокаливании может частично улетучиться в виде хлорида германия (IV). В присутствии тартрата аммония ортогерманат магния не осаждается, что дает возможность отделять фосфор и мышьяк от германия.
Ход определения. Сульфид германия растворяют в аммиаке, взято в небольшом избытке, с добавлением перекиси водорода, свободной от фосфорной кислоты. Раствор кипятят до полного разложения перекиси водорода, после чего слабо подкисляют серной кислотой и разбавляют до
1 Е. В. Johnson, L. М. D е n n i s, J. Am. Chem. Soc., 47, 790 (1925).
8 J. H. Muller, A. Eisner, Ind. Eng. Chem., Anal. Ed., 4, 134 (1932).
‘J. H. Muller, H. R. Blank, J. Am. Chem. Soc., 46, 2358 (1924);
A. W. Laubengayerr, D. S. Morton, там же, 54, 2310 (1932).
4 J. H. Muller, J. Am. Chem. Soc., 44, 2493 (1922).
350
Гл. XVI. Германий
100 мл. В раствор вводят 20—25 мл 2 н. раствора сульфата аммония и 15— 20 мл 1 н. раствора сульфата магния. Затем осторожно при перемешивании прибавляют аммиак в таком количестве, чтобы получился избыток его в 15—20 мл на 100 мл раствора. Нагревают раствор до кипения, охлаждают и дают отстояться 10—12 ч. Осадок отфильтровывают, промывают небольшими порциями разбавленного (1 : 9) раствора аммиака, употребив его не более 50 мл, прокаливают в фарфоровом тигле так же, как пирофосфат магния (стр. 722), и взвешивают в виде Mg2GeO4.
*На основании аналогии свойств поликислот кремния и германия в качестве осадителя для определения германия был предложен оксихинолин1. Оксихинолин с германат-ионами не реагирует, а с германомолиб-деновой кислотой образует мало растворимый в минеральных кислотах мелкокристаллический осадок желтого цвета. В этом осадке на 1 атом германия приходится 4 молекулы оксихинолина, и состав его может быть представлен формулой [C9H7ON]4-H4 [Ge (Мо12О40) ]. В соответствии с этим содержание германия в осадке составляет 2,965%. Благодаря высокой чувствительности реакции (предельная концентрация равна 1 : 1 000 000) метод может быть применен для определения малых количеств германия.
Определению мешают кремневая и фосфорная кислоты. Селенистая и теллуристая кислоты в кислой среде образуют осадки с молибдатом аммония и поэтому должны отсутствовать. Небольшие количества селеновой и теллуровой кислот, а также олова и сурьмы не мешают осаждению германия. Определение можно осуществить в присутствии небольших количеств мышьяка, если указанное выше соединение осаждать на холоду.
Метод сводится к следующему: в 50 мл нейтрального или слабокислого раствора германата, содержащего не более 0,02—0,03 мг GeO2, на каждый миллиграмм двуокиси германия вводят по 2 мл 5 %-ного водного свежеприготовленного раствора молибдата аммония, полученного перекристаллизацией из спирта. Затем прибавляют 3 мл 10%-ной серной кислоты. Появление лимонно-желтого окрашивания указывает на образование гетерополикислоты. Раствор разбавляют водой до 100 мл, через 5 мин прибавляют 9 мл соляной кислоты и тотчас же медленно при энергичном перемешивании вводят 20 мл 2%-ного уксуснокислого раствора оксихинолина (20 г оксихинолина растворяют в 120 мл концентрированной уксусной кислоты и раствор разбавляют водой до 1 л). Оставляют стоять при комнатной температуре около 3 ч, а при малом содержании германия — на ночь. Отфильтрованный осадок промывают жидкостью, приготовленной добавлением к 1 л воды 7 мл соляной кислоты и 25 мл 2 %-ного уксуснокислого раствора оксихинолина.
Определение можно закончить непосредственным взвешиванием высушенного при 110° С осадка. Для вычисления содержания GeO2 авторы рекомендуют пользоваться эмпирическим фактором 0,0448 (или 0,0311 для пересчета на Ge).
Определение можно закончить также объемным путем, бромированием связанного оксихинолина и последующим йодометрическим титрованием. Для этого осадок растворяют в горячей смеси 10 мл соляной кислоты и 10 мл этилового спирта, не содержащего примесей посторонних органических соединений, способных окисляться бромом.*
1 И. П. Алимарин, О. А. Алексеевская, ЖПХ, 12, 1900 (1939).
Методы определения
351
Объемные определения
Объемные методы обычно применяются после выделения германия в виде сульфида, который переводят в раствор обработкой едкой щелочью с перекисью водорода. Избыток перекиси разрушают кипячением.
Можно указать на два объемных метода определения германия. Один из них 1 основан на свойстве двуокиси германия реагировать в водных растворах с маннитом (и другими многоатомными спиртами) с образованием сильной одноосновной комплексной кислоты, которая титруется раствором едкого натра по фенолфталеину. Маннитогерманиевая кислота реагирует также с иодат-иодидной смесью с выделением свободного иода, который можно оттитровать тиосульфатом.
Алкалиметрическое титрование маннитогерманиевой кислоты было проверено Н. С. Полуэктовым на растворах германата натрия, содержащих 1—50 мг германия 2. Испытуемый раствор сначала слабо подкисляли серной кислотой, а затем, так же как при определении бора (стр. 840), нейтрализовали по паранитрофенолу и после введения маннита титровали по фенолфталеину.
По аналогии с методом, применяемым для титрования бора 3, при определении германия также рекомендуют 4 нейтрализовать раствор, до введения маннита, и последующее титрование маннитогерманиевой кислоты проводить до одинаковой величины pH. Отмечено, что в этих условиях менее сказывается влияние посторонних ионов и на «холостой» опыт расходуется меньшее количество едкой щелочи, что особенно важно при определении малых количеств германия. Титрование проводят следующим образом. К 80 мл слабокислого раствора соли германия прибавляют 7 капель бромкрезолового пурпурного и нейтрализуют приблизительно 0,02 н. свободным от карбоната раствором едкого натра до pH = 6,2, что определяется сравнением окраски анализируемого раствора с окраской буферного раствора (33,9 мл 0,1 М лимонной кислоты и 66,1 мл 0,2 М раствора двузамещенного фосфата натрия), содержащего такое же количество индикатора. Затем прибавляют 10 г маннита и титруют раствором едкого натра до pH = 6,2.
Титр раствора едкого натра устанавливают по чистой двуокиси германия.
Мышьяк (III) в количестве до 100 мг не мешает определению. Мышьяк (V) предварительно восстанавливают сернистым ангидридом.
Германий, подобно олову (стр. 341), можно определять йодометрическим титрованием после его восстановления гипофосфитом натрия ®. В условиях восстановления германия мышьяк восстанавливается до эле-' ментарного состояния и удаляется фильтрованием. Метод заключается в следующем. Анализируемый раствор, объем которого должен быть около 50 мл, а концентрация соляной кислоты 1:1, переносят в прибор для восстановления. Этот прибор представляет собой круглодонную колбу емкостью 250—350 мл с резиновой пробкой, в отверстия которой вставлены
1 A. Tchakirian, С. г., 187, 229 (1928).
2Н. С. Полуэктов, Зав. лаб., 5, 27 (1936).
3 F. J. Foote, Ind. Eng. Chem., Anal. Ed., 4, 39 (1932); M. Hollander, W. R. R i e m a n, там же, 18, 788 (1946).
4 H. J. С 1 u 1 e у, Analyst, 76, 517 (1951).
6 Б. H. Иванов-Эмин, Зав. лаб., 13, 161 (1947).
352
Гл. XVI. Германий
обратный холодильник, капельная воронка и трубка для подвода двуокиси углерода, доходящая почти до дна .колбы.
Перед введением раствора в колбу пробку и холодильник ополаскивают разбавленной (3 : 2) соляной кислотой во избежание гидролиза хлот рида германия (IV). К раствору прибавляют 2,5—3 г гипофосфита натрия, закрывают колбу пробкой и 5 мин пропускают ток двуокиси углерода. Затем, прекратив ток газа, кипятят раствор 40—50 мин. Выделяющийся при этом осадок элементарного мышьяка отфильтровывают через плотный фильтр с небольшим количеством бумажной массы, промывают 5—6 раз разбавленной (3 : 2) соляной кислотой, употребляя каждый раз возможно меньшее количество кислоты (—40 мл).
Находящийся в фильтрате хлорид германия (II) в процессе фильтрования частично окисляется, поэтому восстановление следует повторить. Для этого к раствору прибавляют еще 2—3 г гипофосфита натрия и кипятят 15—20 мин, пропуская слабый ток двуокиси углерода (1 пузырек в секунду) и, не прекращая тока газа, охлаждают жидкость до 20° С или ниже.
К охлажденному раствору через капельную воронку прибавляют 250 мл свежеприготовленного раствора, содержащего 10 г лимонной кислоты, 0,4 г иодида калия и 5—6 мл свежеприготовленного раствора крахмала. Холодильник удаляют и вместо него в пробку вставляют кончик бюретки, содержащей 0,05 н. раствор иода. Быстро вводят из бюретки в колбу избыточное количество раствора иода, вынимают из колбы пробку и оттитровывают избыток иода раствором тиосульфата натрия. 1 мл 0,05 н. раствора иода соответствует 0,002675 г двуокиси германия.
Колориметрические методы
Для колориметрического определения германия используют реакцию образования растворимой в воде германомолибденовой кислоты и реакцию взаимодействия германия (IV) с 2,3,7-триокси-9-фенил-6-флуороном (фе-нилфлуороном) в солянокислой среде.
Определение по реакции с молибдатом аммония. Колориметрический метод определения германия, основанный на его реакции с молибдатом аммония, впервые был разработан И. П. Алимариным и Б. Н. Ивано-вым-Эминым 1, хотя на возможность использования германомолибденовой кислоты для определения германия указывалось и раньше 2. Реакция эта может быть выполнена в азотнокислой, сернокислой или уксуснокислой среде. Чувствительность реакции в азотнокислой среде выше, чем в уксуснокислой. Интенсивность окраски практически не зависит от изменения концентрации молибдата аммония, если содержание его в растворе превышает 0,1 %, но избыток реагента благоприятно отражается на устойчивости окраски. В продолжение 15—20 мин после введения реагентов окраска остается постоянной, а затем постепенно ослабевает. Закон Бера распространяется на концентрации до 40 мг GeO2 на 1 л.
Минимальная поддающаяся определению концентрация равна 1 : 1 000 000.
Окраску молибденовой кислоты ослабляют или полностью разрушают винная, щавелевая, лимонная кислоты и ионы фтора. Влияние фтора
1 И. П. Алима р и н, Б. Н. И в а н о в - Э м и н, ЖПХ, 9, 1334 (1936).
2 Grosscup, J. Am. Chem. Soc., 52, 5154 (1930).
Методы определения
353
можно устранить введением в раствор солей алюминия или циркония. Селен (LV) и (VI) и теллур (IV) и (VI) также несколько снижают интенсивность окраски. Усиливают окраску такие элементы, как железо, хром, кобальт и др., ионы которых образуют окрашенные растворы, а также кремневая, фосфорная и мышьяковая кислоты, образующие с молибдатом окрашенные гетерополикислоты.
Влияние примеси мышьяка устраняется проведением реакции на холоду.
Рекомендуемый ход колориметрического определения в азотнокислой среде заключается в следующем. На каждые 100 мл холодного анализируемого раствора германата прибавляют по 2—4 мл 5 %-ного свежеприготовленного раствора молибдата аммония, а затем — азотную кислоту в таком количестве, чтобы кислотность раствора была 0,15—0,35 н. Спустя 2—3 мин окраску раствора сравнивают в колориметре с окраской стандартного раствора, приготовленного одновременно и таким же способом. В качестве стандарта можно также пользоваться растворами хромата или пикриновой кислоты. Авторы указывают, что окраска раствора, полученного растворением 10 мг х. ч. пикриновой кислоты в 1000 мл воды, соответствует окраске раствора, содержащего 74,8 мг СеОг в том же объеме.
Позднее реакция образования германомолибденовой кислоты изучалась спектрофотометрическим методом, в результате чего были разработаны условия колориметрирования германия в уксуснокислой среде Ч Наилучшие результаты получаются при 5 н. концентрации уксусной кислоты и содержании в растворе 0,25% молибдата аммония. Окраска более интенсивна и устойчива при введении в раствор сначала уксусной кислоты, а затем молибдата аммония.
Известно, что молибден, входящий в состав гетерополикйслот, относительно легко восстанавливается с образованием продуктов, окрашенных в синий цвет. Основываясь на этом свойстве, Н. С. Полуэктов 1 2 разработал колориметрический метод, согласно которому содержание германия определяется по интенсивности окраски молибденовой сини, образующейся при действии восстановителя на раствор германомолибденовой кислоты. В качестве восстановителя автор применил сульфат железа (II), который вводится в подкисленный несколькими каплями серной кислоты анализируемый раствор вместе с молибдатом аммония в виде реактива, содержащего в 100 мл 4 мл 15%-ного раствора молибдата аммония, 4 мл концентрированной азотной кислоты, 4 мл 5%-ного раствора соли Мора и 20 мл насыщенного раствора ацетата натрия.
В 50 мл конечного раствора должно содержаться 25 мл этого реактива.
Колориметрическое определение германия по желтой окраске германомолибденовой кислоты и по синей окраске продуктов восстановления молибдена проводится после предварительной дистилляции германия и выделения его в виде сульфида, который переводят в раствор обработкой едким натром с перекисью водорода. Метод Полуэктова был несколько видоизменен и применен 3 непосредственно к дистиллятам хлорида германия (IV).
1 R. Е. К i t s о n, М. G. Mellon, Ind. Eng. Chem., Anal. Ed., 16, 128 (1944).
2 H. С. Полуэктов, Зав. лаб., 5, 27 (1936).
3A. G. Hybbinette, E. B. Sandell, Ind. Eng. Chem., Anal. Ed., 14, 715 (1942).
/ 23 Заказ 522.
354
,Гл. XVI. Германий
Определение по реакции с фенилфлуороном1. Германий реагирует с фенилфлуороном в кислой среде с образованием комплексного соединения розового цвета. Благодаря желтой окраске самого реагента раствор в присутствии германия приобретает оранжевый цвет. С течением времени германий выпадает в осадок, поэтому для стабилизации раствора необходимо вводить защитный коллоид. Определению германия препятствуют галлий, титан, олово, мышьяк (III) и (V), висмут, молибден (IV), железо (II) и сурьма (III). Установлено, что влияние мышьяка весьма незначительно, а галлия, олбва, сурьмы и молибдена наиболее ощутимо. Сильные окислители, такие, как бихромат и перманганат, также мешают определению, так как они разлагают реагент. По утверждению автора, этот метод почти в 4 раза чувствительнее, чем метод колориметрирования по молибденовой сини. Для отделения германия от мешающих элементов используется дистилляция. Колориметрическое определение проводится непосредственно в дистилляте.
Сравнительно недавно для отделения германия от посторонних элементов при колориметрическом его определении по реакции с фенилфлуороном предложено 2 вместо дистилляции применять экстракцию четыреххлористым углеродом. Для аналитических целей наилучшие результаты получаются при экстракции германия из 8—9 н. соляной кислоты. Коэффициент распределения не зависит от концентрации германия в исходном растворе. При низких концентрациях Германия равновесие наступает очень быстро после встряхивания раствора с четыреххлористым углеродом в продолжение 1—2 мин.
Помимо перечисленных выше элементов, окрашенные продукты с фенилфлуороном дают также ниобий, тантал, цирконий и вольфрам (VI). Однако при экстракции четыреххлористым углеродом все элементы, реагирующие с фенилфлуороном, практически полностью отделяются от германия. Исключение составляет мышьяк, но в предлагаемых авторами условиях колориметрирования он не оказывает заметного влияния на результаты определения германия.
Ниже приводится рекомендуемый авторами ход анализа.
Ход определения. Анализируемый раствор, объем которого должен быть 20—25 мл, может содержать 0,5—10 мкг германия. Концентрация соляной кислоты в растворе должна быть 8 М (лучше 9 М). Окислители, выделяющие хлор в солянокислой среде, должны отсутствовать или влияние их следует устранить введением избыточного количества сульфата железа (II).
Охлажденный ниже 20° С раствор [чтобы избежать потерь хлорида германия (IV) вследствие улетучивания] энергично встряхивают в делительной воронке в течение 2 мин с 10—15 мл реактивного четыреххлористого углерода. Слой четыреххлористого углерода сливают в другую делительную воронку и повторяют экстракцию со свежей порцией экстрагента. Затем для удаления оставшихся капелек водного раствора экстракт встряхивают с 3—5 мл 9 М или более концентрированной соляной кислоты, которую отбрасывают.
Слой четыреххлористого углерода энергично встряхивают 2 мин с точно отмеренными 6 мл воды (или соответственно большим количеством,
1 Н. J. С 1 u 1 е у, Analyst, 76, 523 (1951).
2 W. Schneider Jr., Е. В. S a n d в 11, Mikrochim. Acta, № 2, 263 (1954).
Метода определения
355-
если содержание германия превышает 5 мкг.' Дают жидкости расслоиться и аликвотную часть' в 5 мл водного раствора переносят в мерную колбу емкостью 10 мл. Прибавляют 1 мл раствора гуммиарабика 1 и точно 3 мл раствора фенилфлуорона 2, разбавляют до.метки и перемешивают. Оставляют стоять При комнатной температуре 1 ч, если температура не выше 25° С, в противном случае на 30 мин помещают колбу в баню, имеющую температуру 20° С, а затем держат при комнатной температуре еще 30 мин. После этого измеряют светопоглощение раствора при 510 ммк, применяя для сравнения раствор всех реактивов. Содержание германия вычисляют по калибровочной кривой.
*Для определения германия в силикатных породах поступают следующим образом. 0,5 г тонко измельченной породы обрабатывают в платиновой чашке 3 мл разбавленной (1 : 1) серной кислоты, 0,5 мл концентрированной азотной кислоты и 5 мл фтористоводородной кислоты. Перемешивают и выпаривают до появления паров серной кислоты. По охлаждении прибавляют 2—3 мл воды, перемешивают и выпаривание повторяют, избегая продолжительного и обильного выделения паров кислоты. Операцию эту повторяют еще раз, после чего прибавляют 5 мл воды и нагревают почти до кипения в течение нескольких минут для разложения остатка. Охлаждают и возможно полно переводят массу в делительную воронку при помощи 5 мл соляной кислоты. Воронку закрывают пробкой и оставляют стоять, время от времени встряхивая, до растворения солей или в продолжение 30 мин, если осадок полностью не растворяется. Остающийся нерастворенным сульфат кальция в дальнейшем не вызывает затруднений. Прибавляют 10 мл соляной кислоты для повышения кислотности до порядка 9 М, перемешивают, охлаждают до температуры ниже 25° С и проводят экстракцию и колориметрирование германия так, как это указано выше. Через все эти операции проводят также «холостую» пробу со всеми применявшимися реактивами. Доп. перев*
1 Растворяют 1 г гуммиарабика в 200 мл горячей воды и фильтруют. При появлении плесени раствор становится негодным.
2 Растворяют 50 мл чистого фенилфлуорона в 40 мл соляной кислоты и разбавляют чистым спиртом до 250 мл. Раствор устойчив в продолжение нескольких недель.
* Колориметрический метод с применением фенилфлуорона использован также для определения германия в пылях свинцового и цинкового производств (Л. Б. Г и н з-бург, С. Д. Гурьев, А. П. Шебаренкова, Сборник трудов Гинцветмета № 10, 1955, стр. 378). Прим, перев*
23*
Глава XVII
МОЛИБДЕН
Распространение молибдена в силикатных породах земной коры (в таких количествах, какие возможно открыть в нескольких граммах анализируемого вещества), по-видимому, ограничивается некоторыми более кислыми силикатными породами. Наиболее часто молибден встречается в граните в виде сульфида, м о л и б д е ни т a, MoS2. Известны также минералы, представляющие собой соли молибденовой кислоты, например молибдат железа — молибдит Fe2 (МоО4)3-7Н4О, молибдат кальция — повеллит Са(Мо, W)O4 й молибдат свинца — вульфенит РЬМоО4. С анализом пород на молибден приходится сталкиваться сравнительно редко, тогда как определение молибдена в минералах, рудах и металлургических продуктах представляет собой довольно обычное явление. Принимая во внимание высокую стоимость молибдена, а также его влияние на свойства сплавов, в которые он вводится, анализ руд и металлургических продуктов на содержание в них молибдена должен проводиться с достаточной точностью.
ОБЩИЕ ЗАМЕЧАНИЯ '
Чистые молибденовые минералы имеют относительно простой состав, и анализ их не относится к сложным задачам аналитической химии. Но довольно часто эти минералы настолько тесно связаны с другими минералами, что количественно разделить их не удается. Это свойство, особенно характерное для руд с нерезко выраженной кристаллизацией, нередко приводит к различным недоразумениям. Так, например, смесь двух минера- ; лов — молибденита и висмутового блеска была описана как молибденит с высоким содержанием висмута. 1
В обычном ходе анализа горных пород, когда не проводится осаждение сероводородом в кислом растворе, те небольшие количества молиб- > дена, которые могут содержаться в анализируемом материале, проходят , через все стадии анализа незамеченными и остаются в фильтрате после отделения магния. При полном анализе молибденовых минералов молиб- ? ден, совместно с другими металлами сероводородной группы, осаждают J сероводородом после отделения кремнекислоты.
РАЗЛОЖЕНИЕ МОЛИБДЕНОВЫХ МИНЕРАЛОВ J
Наиболее распространенными рудными минералами молибдена яв- 1 ляются молибденит MoS2 и вульфенит РЬМоО4. Молибденит нередко со-провождается продуктами его окйсления, из которых наиболее часто |
Методы отделения
357
встречается молибдит или молибденовая охра — гидратированный молибдат железа, ранее принимавшийся за окись молибдена. Молибдит количественно извлекается обработкой пробы разбавленной соляной или серной кислотой, которые на молибденит практически не действуют. Молибдит разлагается также щелочами.
Молибденит легко разлагается концентрированной азотной кислотой и царской водкой, причем молибден и сера окисляются до молибденовой и серной кислот. Если нет необходимости определять серу, вводят еще и серную кислоту, а затем выпаривают раствор до появления ее паров. Сульфаты обрабатывают водой и нерастворимый остаток, представляющий собой кварц и другие неразложившиеся минералы, отфильтровывают.
После прокаливания и взвешивания остаток можно анализировать соответствующим способом. Свинец, находящийся в нерастворимом остатке в виде сульфата, извлекают до прокаливания обработкой на фильтре горячим раствором ацетата аммония, содержащим немного уксусной кислоты.
Молибдаты разлагаются также кислотами, некоторые даже одной азотной или одной соляной, но на практике обычно вводят и серную кислоту, прибавляя иногда еще 1—2 капли фтористоводородной кислоты, чтобы обеспечить более полное растворение материала. При анализе вульфенита серная кислота служит одновременно для осаждения свинца, который можно определить обычным путем в виде сульфата после отделения его от неразложенной части материала, как указано .выше. Для разложения повёллита (CaMoCU, содержащий иногда вольфрамат кальция) серную кислоту вводить не следует.
Методы сплавления, как, например, сплавление с едким натром или с перекисью натрия, применяются иногда для разложения окисленных или сульфидных минералов (см. стр. 915).
В некоторых случаях эти методы могут одновременно обеспечить отделение молибдена и серы от ряда других элементов и тем самым упростить дальнейший ход анализа.
МЕТОДЫ ОТДЕЛЕНИЯ
Применение того или иного метода отделения молибдена от других элементов, естественно, зависит от характера анализируемого материала и от принятого хода анализа. Обычно в одной из стадий анализа молибден выделяют из раствора в виде сульфида M0S3. Количественное осаждение M0S3, хотя он и нерастворим в неокисляющих кислотах, сйязано с большими затруднениями, особенно осаждение из солянокислых растворов, или из растворов, содержащих такие примеси, как ванадий. Неполнота осаждения сульфида вызывается частичным восстановлением молибдена сероводородом. Восстановленное соединение осаждается крайне медленно, даже под давлением, и единственный способ, который дает возможность .полностью выделить молибден из раствора, заключается в следующем.
Выделенный сульфид молибдена отфильтровывают и промывают. Фильтрат кипятят до удаления сероводорода, восстановленный молибден окисляют персульфатом и затем раствор снова насыщают сероводородом. В присутствии солей железа (III) восстановление молибдена замедляется, вследствие чего достигается более полное его осаждение.
Сульфид молибдена осаждают сероводородом из кислых, лучше сернокислых, растворов его солей, или же подкисляя щелочные растворы,
358
Гл. XVII. Молибден
содержащие сульфиды. В первом случае в раствор, содержащий 5% по объему серной кислоты или 15% по объему соляной кислоты, пропускают сильную струю сероводорода в течение 15 мин, после чего раствор разбавляют равным объемом горячей воды. Снова 10 мин пропускают сероводород, кипятят 3 мин, затем опять пропускают сероводород 10 мин и оставляют при комнатной температуре на 1 ч.
Из. щелочного раствора сульфидов молибден осаждают следующим образом. Раствор нагревают до кипения, насыщают сероводородом в течение 10 мин, подкисляют разбавленной серной кислотой так, чтобы содержание свободной кислоты составляло'2 % по объему, после чего снова 10 мин пропускают сероводород и нагревают на водяной бане 2 ч. Вместо сероводорода можно пользоваться свежеприготовленным раствором щелочного сульфида, свободного от тиосульфатов. При наличии в растворе вольфрама или ванадия более четкое разделение достигается в присутствии винной кислоты, с которой эти элементы образуют устойчивые комплексные соединения. Но если по ходу анализа какие-нибудь элементы должны быть осаждены из фильтрата аммиаком, винную кислоту следует вводить лишь в случае крайней необходимости, так как перед добавлением аммиака она должна быть разрушена х. Для промывания осадка сульфида молибдена, если вольфрам и ванадйй отсутствуют, пользуются разбавленной (1 : 99) серной кислотой, насыщенной сероводородом. В присутствии же этих элементов к промывной жидкости добавляют еще 20 г винной кислоты на 1 л.
При осаждении вольфрама из раствора, содержащего молибден, некоторая часть последнего захватывается осадком 1 2. В связи с этим осадок вольфрамовой кислоты целесообразно растворять в аммиаке, содержащем тартрат аммония, и затем из раствора выделять сульфид молибдена сероводородом, как было описано выше. При наличии больших количеств вольфрама осадок сульфида молибдена рекомендуется переосаждать. Извлечение молибдена серной кислотой из непрокаленного осадка вольфрамовой кислоты не дает хороших результатов. Этот метод 3 в несколько видоизмененном виде 4 заключается в следующем. Свежеосажденную вольфрамовую кислоту 30 мин нагревают с концентрированной серной кислотой в фарфоровой чашке на голом пламени, прибавляя время от времени
1 Отделение молибдена от вольфрама после добавления винной кислоты лучше всего достигается при пропускании сероводорода в кислый раствор, который затем делают щелочным, снова пропускают сероводород и после этого опять подкисляют. Наличие в растворе других элементов сероводородной группы, например меди, способствует осаждению молибдена. I. Koppel [Chem. Ztg., 48, 801 (1924)] указывает, что при осаждении сульфида молибдена в муравьинокислом растворе’, как рекомендуют J. S t е г Ь а, В о h m и J. V о s t г е b а 1 [Z. anorg. Chem., 110, 81 (1920)], достигается достаточно удовлетворительное отделение от вольфрама, причем муравьиная кислота имеет то преимущество по сравнению с винной, что она может быть легко удалена. Согласно этому методу щелочной раствор, содержащий оба элемента, нейтрализуют 80 %-ной муравьиной кислотой, прибавляют избыточное количество свежеприготовленного сульфида аммония и затем опять муравьиную кислоту в таком количестве, чтобы в 100 мл раствора содержалось 5 мл свободной кислоты. Нагревают на водяной бапе, фильтруют и промывают осадок сульфида 5%-ным раствором муравьиной кислоты.
2 Например, при анализе 1 г ферровольфрама, содержащего 78% вольфрама и 0,2% молибдена, более 75% молибдена захватывается осадком вольфрама, независимо от того, выделяют ли его обезвоживанием с H2SO4 и HNO3 пли осаждают цинхонином.
3 М. J. Ruegen berg, Е. F. S m i t h, J. Am. Chem. Soc., 22, 772 (1900).
4 W. H о m m e 1, Inaugural Dissertation, Giessen, 1902.
Методы отделения
359
1—2 капли разбавленной азотной кислоты, если осадок приобретает зеленоватый оттенок. По окончании обработки разбавляют раствор трехкратным по объему количеством воды, фильтруют и осадок вольфрамовой кислоты промывают холодной разбавленной серной кислотой х.
Для отделения от молибдена умеренных количеств многих элементов целесообразно пользоваться осаждением аммиаком с переосаждением осадка, если он велик, и последующей обработкой фильтрата сульфидом аммония. Осаждение аммиаком, при наличии в растворе достаточного количества железа (III), позволяет отделять от молибдена железо, фосфор, мышьяк, сурьму и, возможно, другие элементы, например висмут, олово, ' германий и редкоземельные металлы 1 2. Свинец при этом должен отсутствовать, иначе выделяется молибдат свинца. Обработкой фильтрата сульфидом аммония полностью удаляют кадмий, серебро и большую часть, а возможно, и всю медь. В тех случаях, когда не требуется 'определять железо и щелочноземельные металлы, осаждение аммиаком целесообразно проводить, как описано на стр. 363. Необходимо указать, что при медленном введении аммиака в слабокислый раствор некоторое количество молибдена захватывается осадком 3, поэтому рекомендуется прозрачный анализируемый раствор вливать при сильном перемешивании в избыточное количество аммиака. В некоторых случаях, как, например, для лучшего отделения меди, аммиак можно заменить едким натром и сульфидом натрия. Сплавление породы или окисленных минералов с карбонатом натрия и последующее извлечение молибдена в раствор обработкой плава водой также может служить для отделения умеренных количеств молибдена от целого ряда элементов. Следует иметь в виду, что все эти методы отделения молибдена от других элементов не равноценны и заменить друг друга не могут. Так, при осаждении аммиаком мышьяк совместно с другими элементами выделяется в осадок, тогда как при применении едкого натра или при выщелачивании карбонатного плава водой он практически полностью переходит с молибденом в раствор. Медь же, наоборот, переходит вместе с молибденом в аммиачный фильтрат, а при обработке раствора
1 Отделение малых количеств молибдена (5 мг) от больших количеств вольфрама (1 г) экстрагированием ксантогената молибдена хлороформом из разбавленного сернокислого раствора см. D. Hall,!. Am. Chem. Soc., 44, 1462 (1922). Возгонку молибдена и вольфрама в токе СС14 и С02 см. Р. J annash, Z. prakt. Chem., 97, 93, 141, 154 (1918). *Р. Б. Голубцова и Ф. М. Шемякин [ЖАХ, 4, 232 (1949)] рекомендуют отделять вольфрам от молибдена осаждением fj-нафтохинолином в сильнокислой среде. Нейтрализовав затем фильтрат до слабокислой реакции, осаждают молибден тем же реагентом.*
2 Согласно A. Schoep hW. Steinkuhler [Bull. Soc. chim. Belg., 31, 156 (1922)], значительно более удовлетворительное разделение урана (VI) и молибдена достигается обработкой аммиачного раствора сульфидом аммония, чем при осаждении одним аммиаком. Это относится также и к кальцию, осадок которого при осаждении оксалатом аммония из аммиачного раствора, содержащего значительные количества обоих этих элементов, увлекает некоторую часть молибдена.
3 D. A. Lambie и W. R. Sen о е 11 е г [Analyst, 65, 281 (1940)] указывают, что удовлетворительное разделение олова и молибдена может быть достигнуто следующим образом. В 10 мл солянокислого раствора, содержащего эти элементы, вводят хлорид железа (III) в таком количестве, чтобы содержание железа в нем равнялось количеству находящегося в растворе'олова. Раствор разбавляют до 100 мл, нагревают почти до кипения и затем при сильном перемешивании вливают в 100 мл горячего разбавленного (1:4) раствора аммиака. Кипятят 30 сек и после этого фильтруют. Осадок умеренно промывают 3%-ным раствором нитрата аммоция, затем растворяют в соляной кислоте и переосаждают. Аммиачные фильтры объединяют.
360
Гл. XVII. Молибден
едким натром выделяется в осадок. Цинк и щелочноземельные металлы не отделяются от молибдена в случае осаждения аммиаком или едкий натром, но полностью остаются в нерастворимом остатке после выщелачивания карбонатного плава, водой.
Для отделения больших количеств мышьяка от малых количеств молибдена наиболее целесообразно восстанавливать мышьяк до трехвалентного состояния и затем удалять его отгонкой из солянокислого раствора (стр. 303). Худшие результаты получаются при осаждении мышьяка (V) из аммиачного раствора магнезиальной смесью (стр. 306).
Разделение сурьмы и молибдена рекомендуется осуществлять следующим образом1. Солянокислый раствор, свободный от нитратов и сульфатов и содержащий 20—25 мл соляной кислоты в объеме 100 мл, слабо ки-пятят 20—30 мин с 5 г чистого гранулированного или тонкого прокатанного свинца. Затем раствор разбавляют кипящей водой, быстро фильтруют и осадок промывают горячей водой. В фильтрате молибден окисляют азотной кислотой и осаждают его в виде молибдата свинца. Осадок сурьмы и непрореагировавшего свинца растворяют в серной кислоте и определяют сурьму титрованием перманганатом, как описано на стр. 324. '
Установлено 2, что молибден можно количественно отделить от хрома (III) и ванадия (IV) осаждением перхлоратом свинца в кипящем растворе, содержащем 2% хлорной кислоты. Железо окклюдируется осадком и должно быть предварительно отделено.
Разделение молибдена и рения см. стр. 379.
Большой интерес представляет метод отделения молибдена от целого ряда элементов, основанный на экстракции его эфиром из холодного солянокислого раствора 3 (пл. 1,1 г/см3), содержащего большой избыток хлорида железа (III) 4 (стр. 375). Этот метод применяется главным образом при анализе стали и обеспечивает практически полное 'отделение молибдена от меди, .марганца, никеля, кобальта, хрома и алюминия. Молибден можно также удалить возгонкой из многих соединений нагреванием при 250— 300° С в токе сухого хлористого водорода 5.
1W. R. Schoeller, A. R. Powell, The Analysis of Minerals and Ores-of the Rarer Elements, 2d ed., J. B.'Lippincott Co., Philadelphia, 1941, p. 192.
2 H. H. Willard, F. F e n w i c k, J. Am. Chem. Soc., 45, 932 (1923). ,
3 *И. П. Алимарин и В. Н. Полянский [ЖАХ, 8, 266 (1953)], исследуя экстракцию эфиром хлоридного комплекса молибдена, установили, что молибден наиболее полно экстрагируется из 5—6 и. соляной кислоты. Авторы объясняют это тем, что при меньших концентрациях соляной кислоты в растворе не происходит достаточно полного образования комплексной кислоты Н[МоОС16] или Н[Мо02С13], а при более высоких концентрациях соляной кислоты увеличивается растворимость в ней эфира и большие количества хлоридного комплекса молибдена задерживаются в водном растворе. Показано, что присутствие в растворе хлоридов аммония, кальция и алюминия не влияет на степень извлечения молибдена, хотя это противоречит утверждениям G. Morrison [Anal. Chem., 22, 1388 (1950)].
В этой же статье описан простой экстракционный аппарат неперывного действия, применение которого значительно сокращает время отделения молибдена при одновременном увеличении процента экстракции и снижения потерь экстрагируемого вещества. Прим, перев.*
4 A. A. Blair, The Chemical Analysis of Iron, 8th ed., J. B. Lippincott Co., 1918, p. 205.
5 P. J. К о s k e y, Chemist Analyst, 29, 53 (1940). См. также «Вольфрам» (стр. 767).
Методы определения
361
Молибден количественно отделяется от кальция двукратным осаждением последнего оксалатом аммония из аммиачного (стр. 705) или щавелевокислого (стр. 708) раствора.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Для количественного определения молибдена в растворах его чистых солей с одинаковым успехом можно применять объемный метод (после восстановления молибдена в редукторе Джонса) и весовые методы, основанные на осаждении молибдена а-бензоиноксимом (с последующим прокаливанием осадка и взвешиванием в виде МоОз) или йа осаждении и взвешивании в виде молибдата свинца. Определению молибдена бензоиноксимо-вым методом мешает Меньшее число элементов, и поэтому он более широко применим в аналитическо!! практике.
Определение молибдена весовым путем в виде МоОз после осаждения его в виде сульфида не достаточно надежно, так как прокаливание сульфида молибдена до окиси связано с некоторыми потерями молибдена за счет улетучивания. Этот метод применим лишь для определения малых количеств молибдена, порядка нескольких миллиграммов. При определении еще меньших количеств молибдена (порядка 1 мг и менее) объемным и весовыми методами трудно получить даже приближенно точные результаты. В таких случаях целесообразно пользоваться колориметрическим методом, основанным на реакции с роданидом и хлоридом олова (II), лучше после предварительного выделения молибдена в виде сульфида 1..
Титрование перманганатом
Метод определения молибдена, основанный на том, что пропущенный через редуктор Джонса раствор вводят в раствор сульфата железа (III) и образующееся при этом железо (II) титруют раствором перманганата калия, вполне приемлем как по точности результатов, так и по продолжительности анализа. Прежнее представление о том, что в редукторе Джонса молибден не полностью восстанавливается до трехвалентного состояния, было опровергнуто2, причем показано, что пониженные результаты определения молибдена вызваны частичным окислением его кислородом воздуха. Этого можно легко избежать, если анализируемый раствор непосредственно из редуктора вливать в раствор сульфата железа (III). При этом молибден окисляется, но не до шестивалентного состояния, а до устойчивого соединения промежуточной валентности, с образованием эквивалентного количества железа (II). Количественное восстановление молибдена в редукторе проходит быстро крк в сернокислом, так и в солянокислом растворе, даже на холоду 3. При использовании солянокислых растворов перед титрованием перманганатом необходимо вводить смесь
1 * В техническом анализе осаждение молибдена в виде сульфида обычно опускают и колориметрирование осуществляют непосредственно в растворе, полученном после разложения пробы кислотами.
О. Т. Боберкова [Зав. лаб., 8, 324 (1939)] предлагает разлагать молибденовые руды сплавлением с едкими щелочами и затем определять молибден колориметрически реакцией с роданидом непосредственно после выщелачивания плава водой И подкисления раствора серной кислотой. Прим, перев. *
а D. L. Randall, Am. J. Sci., [4], 24, 313 (1907).
3 G. E. F. L u n d e 1 1, H. B. Knowles,- Ind. Eng, Chem., 16, 723 (1924).
362
Гл. XVIJ. Молибден
Рейнгарда (стр. 446). Прошедший через редуктор раствор сливают в раствор сульфата железа (III), взятый в пятикратном количестве по отношению к теоретически необходимому для окисления Мо2О3 до 2МоО3. Если при титровании сернокислых растворов окраска сульфата железа (III) затрудняет установление конечной точки титрования, в раствор вводят фосфорную кислоту. Для расчета результата определения можно пользоваться теоретическим титром перманганата, считая, что молибден количественно окисляется до МоО3.
*3десь следует понять, что хотя молибден и не окисляется сульфатом железа (III) до шестивалентного, но в дальнейшем при титровании перманганатом происходит окисление как образовавшейся соли железа (II), так и этого промежуточного соединения молибдена полностью до молибдена (VI).
Если условно принять это промежуточное соединение за НМоО3 (точный состав его неизвестен), то происходящие реакции можно представить уравнениями:
ЮН2МоО4+ 30H2SO4+ 15Zn = 5Mo2(SO4)3 + 15ZnSO4 + 40H2O 5Mo2(SO4)3 + l(We2(SO4)3 + 30H2O = 10HMoO3 + 20FeSO4 + 25H2SO4 . 10HMo03-|-20FeS04-|-19H2S04-|-9KMn04 = = 10H2Mo04+10Fe2(S04)3 + 3K2S044-6MnS04+14H20
Таким образом, на 10 атомов молибдена идет 6 молекул КМпОа, т. е. столько, сколько пошло бы при непосредственном восстановлении MoVI до Мош. Доп. ред.'*
Посторонние вещества, восстанавливающиеся в редукторе с образованием растворимых соединений, должны отсутствовать. К этим веществам относятся азотная кислота, органические соединения, политионовые кислоты, соли железа, хрома, титана, мышьяка, сурьмы, ванадия, урана, вольфрама и ниобия. Применяемые методы отделения, естественно, зависят от характера присутствующих посторонних элементов и должны соответствовать методам, приведенным в разделе «Методы отделения». Так, разрушение органических веществ обычно достигается обработкой горячего концентрированного сернокислого раствора азотной кислотой. Последующим повторным выпариванием раствора до появления паров серной кислоты удаляют азотную кислоту Двукратное осаждение аммиаком, при наличии в растворе избытка Железа, служит для отделения железа, хрома, титана, мышьяка, сурьмы, ванадия, урана и ниобия. Для отделения молибдена от вольфрама и политионовых кислот аммиачный фильтрат обрабатывают винной кислотой и сероводородом, фильтруют, фильтрат подкисляют и затем снова фильтруют.
Предосторожности, которые нужно соблюдать при подготовке анализируемого раствора, указаны в следующем ходе анализа1 2. Навеску в 0,5—5 г измельченной до
1 При разрушении органических веществ обработкой серной и азотной кислотами соединения азота могут остаться в растворе даже после нагревания до выделения обильных паров серной кислоты. Некоторые из этих соединений восстанавливаются цинком и затем окисляются, но не разрушаются перманганатом в разбавленном сернокислом растворе. В таких случаях в концентрированный сернокислый раствор осторожно вводят 10%-ный раствор перманганата калия в небольшом избытке.
2 Анализ разработан совместно J. A. Holladay (of the Electro Metallurgical Co) и A. M. Smoot (of Ledoux and Co).
Методы, определения
363
100 меш (d = 0,147 мм) пробы помещают в закрытый стакан емкостью 250 мл и обрабатывают при нагревании 10—35 мл HNO3 и 7—10 мл H2SO4. По окончании разложения раствор выпаривают до появления паров H2SO4. Охлаждают, прибавляют 40— 50 мл воды, сильно кипятят и снова охлаждают. Остаток отфильтровывают и промывают несколько раз горячей водой, затем 6—8 раз разбавленным (1 : 3) раствором аммиака и после этого снова горячей водой. Фильтрат сохраняют. При анализе вульфенита необходимо удалять PbSO4. Для этого остаток промывают кипящим раствором CH3COONH4 [к разбавленному (1 : 1) раствору аммиака прибавляют СН3СООН (99,5%-ную) до кислой реакции]. Затем его прокаливают при низкой температуре в платиновом тигле, обрабатывают HF и H2SO4 и выпаривают досуха.
Остаток сплавляют с K2S2O7, плав растворяют в воде, прибавляют винную кислоту и испытывают на содержание молибдена сероводородом. При анализе молибденита остаток прокаливают в фарфоровом тигле, охлаждают, затем переносят в платиновый тигель, обрабатывают HF и H2SO4 и выпаривают досуха. Сплавляют с Na3CO3, ллав выщелачивают водой и водную вытяжку присоединяют к основному фильтрату или подкисляют и отдельно обрабатывают винной кислотой и сероводородом. К основному раствору прибавляют Fe2(SO4)3 в таком количестве, чтобы содержание в нем железа в 10 раз превышало содержание мышьяка в растворе (обычно достаточно 0,3— 0,4 г). После этого вводят раствор аммиака почти до нейтральной реакции, но так, чтобы раствор оставался светло-желтым, а не становился коричневым. Нагревают почти до кипения и медленно при сильном перемешивании вливают в 75 мл почти кипящего разбавленного (1 : 6) раствора аммиака. Ополаскивают стакан горячей водой, а затем небольшим количеством горячего раствора аммиака. Прибавляют немного мацерированной бумаги, фильтруют в стакан емкостью 500 мл и тщательно промывают осадок горячей водой. Фильтрат сохраняют. Осадок растворяют в небольшом количестве разбавленной (1 : 1) серной кислоты и повторяют осаждение. Аммиачные фильтраты объединяют.
При наличии значительных количеств мышьяка основную массу его следует удалить перед осаждением аммиаком. Для этого после разложения материала и удаления HNO3 добавляют 20 мл H2SO3, накрывают стакан, упаривают раствор до 10 мл и затем снова вводят 10 мл H2SO3 и 50 мл НС1. Сильно кипятят, пока объем раствора не уменьшится до 10—20 мл, прибавляют 10 мл HNO3 и выпаривают до появления паров H2SO4.
К объединенным фильтратам прибавляют 3 г винной кислоты, нагревают и пропускают сильную струю H2S, пока большая часть NH4OH не превратится в (NH4)2S. Осадок отфильтровывают, промывают разбавленным раствором (NH4)2S и отбрасывают. Фильтрат нагревают, стакан накрывают часовым стеклом и осторожно прибавляют разбавленную (1 : 1) H2SO4 в таком количестве, чтобы избыток ее составлял 1 мл на 100 мл раствора. Оставляют не менее чем на 15 мин в теплом месте, дают отстояться, фильтруют и тщательно промывают осадок насыщенной сероводородом разбавленной (1 : 99) H2SO4, содержащей в 1 л 20 г винной кислоты. Осадок сохраняют для определения молибдена.
К фильтрату прибавляют 15 мл HNO3 и выпаривают до появления паров H2SO4, добавив, если требуется, еще HNO3. Охлаждают, прибавляют 5 мл HNO3 и снова выпаривают. Эту операцию повторяют до полного разрушения органических веществ. По охлаждении сульфаты растворяют в воде, затем прибавляют 2 г винной кислоты и раствор аммиака в таком количестве, чтобы получилось 5 мл избытка. Раствор нагревают, насыщают сероводородом, затем подкисляют и оставляют стоять не менее 1 ч при 40— <60° С. Осадок отфильтровывают, промывают, как описано раньше, и присоединяют к основному осадку сульфидов или же анализируют отдельно.
Осадок MoS3 вместе с фильтром переносят в стакан емкостью 250 мл, накрывают часовым стеклом и прибавляют 10 мл HNO3. Перемешивают до разложения бумаги, обрабатывают 10 мл H2SO4 и осторожно нагревают до появления ее паров. По охлаждении прибавляют еще 5 мл HNO3 и снова выпаривают. Эту операцию повторяют до полного исчезновения желтой окраски раствора. После этого осторожно вводят несколько капель насыщенного раствора КМпО4 для большей уверенности в полном разрушении органических соединений азота. Охлаждают, ополаскивают часовое стекло и стенки стакана небольшим количеством воды и для полного удаления HNO3 снова выпаривают до появления паров H2SO4. По охлаждении прибавляют 75 мл воды, раствор кипятят несколько минут, прибавляют 2 г чистого гранулированного цинка (содержащего не более 0,002% Fe) и продолжают кипятить до полного восстановления молибдена и осаждения меди. Фильтруют через асбест и промывают осадок разбавленной (1 : 99) H2SO4.
364
Гл. XVII. Молибден
Ход определения. К раствору соединения молибдена в 3—10%-ной по объему серной кислоте, свободному от посторонних восстанавливающихся в редукторе соединений, прибавляют раствор перманганата калия до появления слабо-розовой окраски. Холодный раствор восстанавливают в редукторе и из редуктора вливают его в колбу, содержащую раствор сульфата железа (III), как описано на стр. 136. Затем вводят несколько миллилитров фосфорной кислоты и титруют раствором перманганата калия, титр которого установлен по химически чистому оксалату натрия (стр. 214). В результаты титрования вносят поправку, вычитая количество перманганата, израсходованное на титрование всех применяющихся реактивов, проведенных через все стадии анализа. Содержание молибдена вычисляют, исходя из реакции окисления Мо2О3 до МоО3.
Точность метода характеризуется данными, приведенными в табл. 19.
ТАБЛИЦА 19'
Результаты определения молибдена методом титрования перманганатом после отделения мешающих элементов
Элемент Введено примесей г Содержание молибдена г Определено молибдена г Найдено молибдена в фильтрате после вторичного осажденид его H2S, г
As 0,002 0,0947 0,0946 0,0001
и 0,022 0,1261 0,1258 0,0007
V 0,0041 0,1262 0,1263 0,0006
V 0,004 2 0,1262 0,1282 0,0011
W 0,002 0,1179 0,1182 0,0104
Си ..... . 0,005 0,1179 0,1177 0,0004
РЬ 0,05 0,1195 0,1193 0,0004
Bi 0,011 0,1261 0,1264 0,0038
Sn 0,011 0,1261 0,1263 0,0038
Р ...... . 0,002 0,1195 0,1196 0,0001
Сг 0,07 0,1261 0,1262 0,0005
1 В промывную жидкость (разбавленная HaSO4, насыщенная HjS) введена винная кислота.
2 В промывную жидкость винная кислота не вводилась.
Осаждение а-бензоиноксимом
Лучшим весовым методом определения молибдена является метод, основанный на осаждении его а-бензоиноксимом
С6Н6-СН-С-СвН6 к
I II
ОН N-ОН
из холодных растворов, содержащих 5% по объему уксусной, фосфорной, ^азотной, соляной или серной кислоты. При отсутствии элементов, образующих малорастворимые сульфаты, лучше всего пользоваться серной кислотой, концентрация которой может быть увеличена до 20%. Фтористоводородная и винная кислоты препятствуют полноте выделения молибдена. Для осаждения требуется 2—5-кратное количество реагента по сравнению с теоретически необходимым по соотношению
1Мо : ЗС6Н5—СНОН-С = NOH-C6H5
Методы, определения
365
В растворе должен находиться небольшой избыток брома для предотвращения частичного восстановления молибдена, которое приводит к неполноте осаждения. После добавления осадителя раствор оставляют стоять в течение 10 мин, время от времени перемешивая, и затем фильтруют. Осадок промывают холодной разбавленной серной кислотой, содержащей небольшое количество реагента. Продолжительное стояние приводит к некоторому разложению осадка. Осадок не может быть непосредственно взвешен, а должен быть осторожно прокален до окиси МоО з.
Серебро, свинец, ртуть, висмут, медь, кадмий, мышьяк, сурьма, олово, алюминий, железо, титан, цирконий, хром (III), ванадий (IV), церий, уран, никель, кобальт, марганец и цинк не осаждаются а-бензоиноксимом. Селен, теллур, рений, рутений, родий, осмий, иридий и платина не осаждаются, когда они находятся одни в растворе, и, возможно, не выделяются в осадок также и совместно с молибденом.
Ниобий, кремний, палладий, вольфрам и тантал загрязняют осадок и должны быть отделены перед осаждением молибдена или же следует ввести соответствующую поправку после определения их во взвешенном осадке. Ванадий (V) и 1ром (VI) мешают определению, но это влияние может быть устранено восстановлением их сернистой кислотой соответственно до четырех- и трехвалентного состояния перед введением осадителя.
Ход определения. К 200 .мл раствора, содержащего не более 0,15 г молибдена (VI) в 5%-ной по объему серной кислоте, в случае наличия ванадатов и хроматов прибавляют свежеприготовленную сернистую кислоту в количестве, достаточном для восстановления. этих соединений. Затем раствор кипятят до исчезновения запаха сернистого ангидрида, охлаждают до 5—10° С и медленно при перемешивании приливают 10 мл 2 %-ного спиртового раствора а-бензоиноксима и сверх того еще по 5 мл на каждую 0,01 г молибдена. Перемешивание прекращают и прибавляют бромную воду до появления бледно-желтой окраски, а затем вводят еще несколько миллилитров осадителя. Стакан помещают в охлаждающую смесь на 10— 15 мин, временами перемешивая его содержимое, прибавляют немного мацерированной бумаги и фильтруют через плотный фильтр (синяя лента). Фильтрование можно значительно ускорить, если пользоваться неплотным фильтром (черная или красная лента), однако в этом случае следует •очень тщательно проверить фильтрат, и если он окажется не вполне прозрачным, первую порцию профильтровать вторично. Осадок промывают 200 мл холодного свежеприготовленного раствора, содержащего в 1 л 25— 50 мл реактива и 10 мл серной кислоты. Если в анализируемый раствор введено достаточное количество реактива, в фильтрате при стоянии выделяются игольчатые кристаллы.
Промытый осадок переносят во взвешенный платиновый тигель, осторожно высушивают, фильтр обугливают на небольшом пламени газовой горелки, не давая ему воспламениться, затем прокаливают в электрической муфельной печи при 500—550° С до постоянной массы и взвешивают в виде * 1 МоО з.
1 Более детально см. Н. В. Knowles, J. Research NBS, 9, 1 (1932). C.Ster-
1 i n g и W. P. S p u h r [Ind. Eng. Chem., Anal. Ed., 12, 33 (1940)] предлагают видоизмененный метод, согласно которому осадок оксимина растворяют в аммиаке с перекисью водорода, раствор кипятят до разрушения перекиси, затем осаждают молибден ацетатом свинца в кипящем ацетатном растворе и взвешивают в виде молибдата свинца.
366
Гл. XVЛ. Молибден
Определение в виде молибдата свинца 2
Осаждение молибдена в виде молибдата свинца обычно осуществляется медленным добавлением раствора ацетата свинца к горячему разбавленному раствору соли молибдена, содержащему ацетат аммония.
Этот метод применим в присутствии меди, кобальта, никеля, марганца, цинка, магния и ртути. Хорошие результаты получаются также в присутствии щелочноземельных металлов, алюминия, урана и кадмия, если осаждение проводить медленным добавлением ацетата аммония к горячему солянокислому раствору молибдена, содержащему небольшой избыток свинца. Соли щелочных металлов не препятствуют определению, за исключением сульфатов, которые должны быть удалены в случае наличия в растворе щелочноземельных металлов. В отсутствие последних небольшие количества сульфатов, такие, какие могут образоваться при растворении сульфида молибдена, не оказывают влияния на осаждение. При наличии в растворе сульфатов и хлоридов следует избегать введения в раствор большого избытка свинца. Свободные минеральные кислоты и винная кислота препятствуют количественному осаждению молибдена, а железо, хром (III), алюминий, ванадий, вольфрам и кремний, если присутствуют в значительных количествах, загрязняют осадок. Фосфор, хроматы и арсенаты должны отсутствовать.- К элементам, мешающим определению, относятся также олово, титан и другие элементы, соли которых легко гидролизуются.
Если нет уверенности в том, что раствор свободен от мешающих определению элементов, следует предварительно отделить молибден способами, описанными на стр. 357—360. При выполнении полных анализов метод, как правило, применяется после переведения в раствор предварительно выделенного чистого осадка сульфида молибдена. Обычно ход определения молибдена зависит от состава анализируемой npo6ti. Так, например, умеренные количества железа (0,1-т-0,2 е) можно отделить осаждением аммиаком, а большие количества — едким натром.
Ход определения. Анализируемый раствор, если он имеет щелочную реакцию, слегка подкисляют уксусной кислотой или, если он кислый, добавляют аммиак в небольшом избытке, а затем — уксусную кислоту. После этого прибавляют 25 мл 50%-ного раствора ацетата аммония и разбавляют раствор в соответствии с содержанием в нем молибдена и посторонних солей. Наиболее подходящей концентрацией является примерно 0,1 г Мо в 200 мл раствора.
Раствор нагревают до кипения и, не прекращая нагревания, в почти кипящую жидкость вводят по каплям из бюретки при. непрерывном перемешивании раствор ацетата свинца (содержащий 40 г соли и 10 мл уксусной кислоты в 1 л). Раствор, вначале молочно-белый, при введении небольшого избытка осадителя становится заметно прозрачным. После этого его кипятят 2—3 мин при перемешивании, дают отстояться и для проверки полноты осаждения прибавляют еще несколько капель реактива.
Введения большого избытка осадителя следует избегать, так как это затрудняет промывку осадка и, кроме того, избыток осадителя нежелателен в случае наличия в растворе значительных количеств хлоридов и сульфатов. В присутствии этих соединений испытание на полноту оса- 1
1 Т. М. С h a t а г d, Am. J. Sci., [3], 1, 416 (1871).
Методы определения
367
ждения лучше проводить в отобранной капле раствора с 5%-ным свежеприготовленным раствором таннина. Когда эта проба дает положительный результат, прибавляют 2—5 мл избытка раствора ацетата свинца. Осадку дают отстояться 10—60 мин, оставляя стакан на краю водяной бани, после • чего раствор декантируют через бумажный фильтр или через взвешенный тигель Гуча. Осадок 3—4 раза промывают декантацией горячим 2—3%-ным раствором нитрата аммония, порциями по 75 'мл, затем переносят на фильтр и продолжают промывать до удаления растворимых солей.
Если предполагают, что осадок загрязнен, например, солями щелочных металлов или сульфатом свинца, его растворяют в горячей соляной кислоте, раствор нагревают до кипения, прибавляют разбавленный аммиак до тех пор, пока образующаяся муть не начнет растворяться с трудом, а затем по каплям при сильном перемешивании вводят ацетат аммония в количестве несколько большем, чем требуется для связывания соляной кислоты. После этого прибавляют’ несколько капель раствора ацетата свинца, чтобы обеспечить количественное осаждение молибдена.
Тигель Гуча с отфильтрованным осадком высушивают и прокаливают при темно-красном калении в муфеле или радиаторе (стр. 48). Если фильтруют через бумажный фильтр, осадок вместе с фильтром переносят во взвешенный фарфоровый тигель, фильтр озоляют при возможно более низкой температуре, затем осадок прокаливают в окислительном пламени при темно-красном калении и взвешивают в виде PbMoOi.
При отсутствии в растворе сульфатов ацетат аммония можно не вводить и осаждение проводить следующим образом Г Раствор слегка подкисляют 0,25 мл азотной кислоты и прибавляют 4%-нып раствор нитраад свинца, а затем аммиак до слабощелочной реакции по лакмусу. Прежде чем молибдат свинца начнет оседать, слегка подкисляют раствор уксусной кислотой 1 2.
Осаждение в виде сульфида молибдена и прокаливание до окиси
Метод, основанный на осаждении молибдена в виде сульфида и последующем прокаливании до окиси МоОз, менее удовлетворителен, чем предыдущий метод; его применяют преимущественно для определения относительно небольших количеств молибдена (2—10 мг). Это обусловлено главным образом трудностью количественного переведения MoS з в МоОз прокаливанием. Возгонка окиси молибдена начинается при 500° С, И при 500—600° С потеря молибдена не превышает 0,1 мг в час 3.
Ход определения. Из сернокислого раствора или из раствора в сульфиде щелочного металла, свободного от других элементов сероводородной группы, выделяют молибден сероводородом, как указано на стр. 358. Осадок отфильтровывают и промывают сероводородной водой, подкисленной серной кислотой. Фильтрат кипятят до удаления' сероводорода,
1 Н. В. W е i s е г, J. Phys. Chem., 20, 640 (1916).
2 Молибден можно взвешивать также в виде молибдата серебра после осаждения нитратом серебра в слабокислом растворе и последующего прокаливания осадка^при 250° С [L. W. М с С а у, J. Am. Chem. Soc., 56, 2548 (1934)]. Этот метод не более специфичен, чем метод определения в виде молибдата свинца.
3 Р. Н. В г i n t о п, A. F. S t о р р е 1, J. Am. Chem. Soc., 46, 2454 (1924).
368
Гл. XVII. Молибден
окисляют оставшиеся в растворе небольшие количества молибдена несколькими кристаллами персульфата или другим соответствующим окислителем и снова насыщают раствор сероводородом. Если выделяется осадок, его отфильтровывают, промывают и присоединяют к основному осадку сульфида ‘ молибдена. Объединенные осадки прокаливают до постоянной массы, как указано на стр. 365.
При правильном проведении операции выделения сульфида молибдена из сернокислого раствора в фильтрате редко остается более 1 мг молибдена после первого и более 0,1 мг после второго осаждения. В некоторых случаях количественное осаждение молибдена достигается и в один прием, однако это недостаточно надежно, даже когда анализируемый раствор насыщают сероводородом под давлением.
Колориметрический метод
Приведенные выше методы недостаточно надежны для определения малых количеств молибдена (менее 1 мг). В таких случаях целесообразно выделить молибден в виде сульфида, осадок прокалить при температуре не выше 500° С -и взвесить. Затем, для проверки содержания молибдена, осадок растворяют в аммиаке, раствор подкисляют соляной кислотой, прибавляют хлорид олова (II) и роданид калия; после этого интенсивность появляющейся окраски сравнивают со стандартом Е Применение этой реакции для определения больших количеств молибдена не дает достаточно точных результатов и приемлемо лишь для рядовых анализов. Рений мешает колориметрическому определению молибдена с роданидом 1 2. Платина оказывает значительное влияние на реакцию, и поэтому в процессе подготовки раствора для колориметрирования не следует пользоваться платиновой посудой. Азотная кислота должна быть удалена, так как она образует с роданидом окрашенное соединение, которое экстрагируется эфиром. В тех случаях, когда при подготовке раствора для колориметрирования вводят азотную и серную кислоты, выпаривание до появления густых паров серной кислоты следует повторить по меньшей мере 2 раза, ополаскивая каждый раз стенки стакана водой. Интенсивность и’ устойчивость окраски соединения молибдена с роданидом в солянокислом растворе зависят от кислотности этого раствора и концентрации в нем солей 3.
Как правило, комплексное соединение молибдена с роданидом более устойчиво в органических растворителях, чем в водных растворах. В случае экстрагирования окрашенного соединения эфиром медь, никель,
1 Согласно F. С. Krauskopf и С. Е. Swartz [J. Am. Chem. Soc., 48, 3021 (1926)], состав окрашенного соединения соответствует формуле Mo(SCN)3. Это, однако, сомнительно, так как молибден (III), полученный в результате восстановления цинком, не дает окраски с роданидом. Это соединение является, вероятно, солью молибдена (V). См. J. I. Hoffman, G. Е. F. L u n d е 11, J. Research NBS, 23, 498 (1939). *Рядом исследований было доказано, что окрашенный комплекс с роданидом образует молибден (V) [В. Ф. Мальцев, Зав. лаб., 5, 264 (1939); Н. А. Т а и а-наев, А. П. Лохвицкай, там же, 11, 6 (1945); А. К. Бабко, Л. Давыдов, ЖОХ, 17, 642 (1947).]*
2 J. I. Hoffman, G. Е. L u n d е 11, J. Research NBS, 23, 497 (1939).
3 Н. Сохи А. А. Р oil i tt (J. Soc. Chem. Ind., 63, 375 (1944)] установили, что окраска соединения молибдена с роданидом устойчива в течение 15 мин в растворе, содержащем 2% НС1О4 и 10—15% H2SO4 (по объему). Светопоглощение окрашенного соединения измерялось при 525 ммк.
Методы определения 369
кобальт, хром, ванадий и уран на определение молибдена заметно не влияют.
При определении малых количеств молибдена в вольфраме рекомендуется1 экстрагировать роданид молибдена эфиром, в котором это соединение более устойчиво. Кроме того, в этом случае устраняется влияние окрашенных соединений никеля и хрома, которые эфиром заметно не извлекаются.
Установлено 2, что для приготовления раствора для колориметрирования можно с успехом применять хлорную кислоту. Для экстрагирования окрашенного соединения автор рекомендует пользоваться бутилацетатом вместо эфира, так как при этом не происходит разогревания раствора и не повышается давление в делительной воронке. Экстрагирующий раствор приготовляют следующим образом. 250 мл бутилацетата (темп. кип. 118—127° С) встряхивают в делительной воронке с 5 мл 5%-ного раствора роданида натрия и 25 мл 25'%-ного раствора хлорида олова (II). Жидкости дают отстояться, водный слой сливают и отбрасывают. Экстрагирующий раствор не устойчив и должен приготовляться каждые 24 ч.
Рекомендуется3 пользоваться для экстрагирования циклогексанолом, насыщенным хлоридом олова (II), роданидом калия и соляной кислотой, так как он является лучшим растворителем и менее летуч, чем эфир. Кроме того, он достаточно устойчив при комнатной температуре, и продукты его гидролиза не вызывают ослабления интенсивности окраски.
Для стабилизации комплексного соединения молибдена с роданидом рекомендуют 4 вводить в раствор некоторые водорастворимые органические жидкости, как, например, гликолевый эфир, бутилкарбитол (монобутиловый эфир диэтиленгликоля) и бутилцеллозольв (монобутиловый эфир этиленгликоля). В присутствии этих жидкостей интенсивность окраски роданида молибдена нарастает в продолжение 10 мин и затем остается постоянной 24 ч. Способ применяют главным образом в массовых анализах стали, так как он исключает необходимость экстрагирования, но в этом случае следует устранять влияние хрома, который дает окрашенные ионы, и меди, которая образует нерастворимые соединения с роданидом.
*Причина относительно быстрого ослабления окраски роданидного комплекса молибдена заключается в большой разности потенциалов систем SnIV/Sn11 и Movi/Mov, вследствие чего возможно дальнейшее восстановление молибдена до более низких степеней окисления 5 6 *. Основываясь на этом, для колориметрирования в водной среде, без экстракции, рекомендуется пользоваться в качестве восстановителя иодидом калия, который имеет более положительный потенциал, чем хлорид олова (II). Авторы нашли, что в этих условиях окраска роданида калия не только |более устойчива, но и быстрее достигает максимальной интенсивности.
При колориметрировании в водной среде для восстановления молибдена предложено также применять тиомочевину ®. Окраска растворов роданида молибдена (в этом случае устойчива в течение нескольких часо^. Небольшие количества меди не мешают колориметрированию, так как медь образует с тиомочевиной растворимые бесцветные комплексные соединения. Доп. перев*
1 W. J. King, Ind. Eng. Chem., 15, 375 (1923).
2 L. H. James, Ind. Eng. Chem., Anal. Ed., 4, 89 (1932).
3 L. C. Hurd, F. Reynolds, Ind. Eng. Chem., Anal. Ed., 6, 477 (1934).
4 M. Kapron, P. L. H e h m a n, Ind. Eng. Chem., Anal. Ed., 17, 573 (1945),
5 Л. Б. Гинзбург, Ю. Ю. Лурье, Зав. лаб., 14, 538 (1948).
6 Л. Б. Зайчикова, Зав. лаб., 15, 1025 (1949).
24 заказ 522.
370
Гл. XVII. Молибден
Ниже приводится метод определения малых количеств молибдена в стали с применением бутилацетата для экстрагирования роданида молибдена.
Ход определения. Навеску анализируемого материала величиной около 0,1 г (0,02—0,4% молибдена) растворяют при нагревании в конической колбе емкостью 150 мл в 10 мл разбавленной (1 : 1) хлорной кислоты. Раствор нагревают до кипения, закрывают колбу часовым стеклом и продолжают кипятить до полного разложения карбидов. После этого слегка охлаждают, прибавляют 25 мл воды и затем кипятят несколько минут для удаления свободного хлора. К раствору прибавляют 10 мл раствора лимонной кислоты [250 г лимонной кислоты на 1 л разбавленной (1 : 99) серной кислоты] и 30 мл раствора едкого натра (200 г/л). Нагревают примерно до 80° С в течение 5 мин и затем охлаждают. Вводят 25 мл разбавленной (1:1) серной кислоты и охлаждают до комнатной температуры. Раствор переносят в делительную воронку емкостью 250 мл и дважды ополаскивают колбу разбавленной (1 : 9) серной кислотой порциями по 5 мл.
Приливают 10 мл раствора NaCNS (50 г/л), встряхивают 20—30 сек, прибавляют 10 мл раствора хлорида олова (II) [350 г SnCl2 • 2Н3О в 1л разбавленной (1 : 9) соляной кислоты] и сильно встряхивают точно 1 мин. После этого вводят пипеткой 20 мл бутилацетата, закрывают воронку пробкой, встряхивают 20—30 сек, охлаждают, лучше в проточной воде, и снова встряхивают. Дают отстояться, водный слой сливают и отбрасывают. Затем вводят 50 мл разбавленной (1 : 9) серной кйслоты, 10 мл раствора NaCNS и 5 мл раствора SnCl2, хорошо встряхивают, охлаждают в проточной воде и снова встряхивают. После этого дают отстояться, водный слой сливают и отбрасывают, а бутилацетатный экстракт переносят в пробирку и закрывают пробкой. Помещают 5—10 мл прозрачного экстракта в кювету фотоколориметра и измеряют светопоглощение 1 при длине волны примерно 540 ммк. Содержание молибдена вычисляют по калибровочной кривой.
Другие методы
Помимо приведенных выше, известен еще ряд методов определения молибдена, но они большого интереса не представляют, хотя некоторые из них, как, например, метод осаждения нитратом ртути (I) из почти нейтрального карбонатного раствора, дают весьма точные результаты при анализе чистых растворов молибдена. Нитратом ртути (I) осаждаются также хром, ванадий, молибден, вольфрам, фосфор и мышьяк, и эта реакция в отдельных случаях применяется лишь для предварительного выделения молибдена из карбонатных растворов, получаемых в результате выщелачивания водой плава породы с карбонатами щелочных металлов 2.
Метод йодометрического восстановления молибдена 3 является эмпи
1 Максимум светопоглощения комплексного соединения молибдена с роданидом соответствует приблизительно 470 ммк, но измерение проводят при большей длине волны, чтобы избежать влияния вольфрама.
2 W. F. Н i 11 е Ь г a n d, J. Am. Chem. Soc., 20, 461 (1898); U. S. Geol. Survey Bull., 700, 185. См. также «Ванадий» (стр. 510).
3 C. Friedhe im, H. Euler, Ber., 28, 2066 (1895); F. A. Gooch, C, Fairbanks, Am. J. Sci., [4], 2, 160 (1896); F. A. Gooch, O. S. Pulman, там же, [4], 12, 449 (1901).
Методы определения
371
рическим, и при его применении возможны многочисленные ошибки. Метод титрования молибдена раствором ацетата свинца с использованием таннина в качестве индикатора применим лишь при массовых определениях ‘больших количеств молибдена. Этот метод не пригоден для определения малых количеств молибдена и никогда не применяется в точных анализах. Известный интерес представляют метод, основанный на восстановлении молибдена (VI) до молибдена (V) потенциометрическим титрованием сульфатом титана (III) \ и метод восстановления в серебряном редукторе 1 2.
* Описан также 3 редуктометрический метод титрования молибдена солью хрома (II) в сильнокислой среде (концентрация соляной кислоты 1:1) с потенциометрическим контролем эквивалентной точки. Метод позволяет определять молибден при одновременном присутствии вольфрама и железа. Большие количества вольфрама удерживаются в растворе щавелевой кислотой в случае титрования солянокислых растворов, если же титрование проводится в сернокислых растворах, можно пользоваться фосфорной кислотой. Доп. перев*
Прокаливание сульфида с серой в атмосфере водорода с последующим взвешиванием в виде MoS2 требует выделения вначале чистого сульфида молибдена и не имеет никаких преимуществ перед осторожным прокаливанием MoS3 до МоО3.
1 Н. Н. Willard, F. Fenwick, J. Am. Chem. Soc., 45, 928 (1923).
2 N. Birnbaum, G. H. Walden Jr., J. Am. Chem. Soc., 60, 64 (1938).
’В. Г. Горюшина, T. В. Черкашина, Зав. лаб., 14 (1948).
24*
Глава XVIII
РЕНИЙ
Рений был открыт в 1924 г. Г Он является самым новым из встречающихся в природных условиях устойчивых элементов, которые были выделены в чистом виде и свойства которых были изучены.
Хотя в самых богатых минералах содержится менее 0,001 % рения, известно, что из остатков после извлечения меди из мансфельдских (Германия) сланцев ежегодно в качестве побочного продукта получали около 250 кг рения2.
Использование рения в промышленности настолько незначительно, что основная задача аналитической химии в настоящее время сводится к качественному открытию этого металла в природных продуктах.
ОБЩИЕ ЗАМЕЧАНИЯ
В систематическом ходе качественного анализа рений обычно обнаруживается только в осадке сульфидов, выделенных сероводородом из кислых или щелочных растворов.
Рений (VII) не осаждается аммиаком (лишь в случае высокой концентрации ReO” выделяется NH4ReO4), оксалатом аммония и двузамещенным фосфатом аммония.
Наиболее надежным методом испытания, в случае содержания незначительных количеств рения, является метод, основанный на реакции образования желтовато-коричневого соединения при обработке солянокислого раствора рения роданидом калия и хлоридом олова (II).
Непосредственное спектрохимическое исследование минералов на содержание рения обычно не проводится вследствие низкой концентрации в них репиц, многообразия спектров сопровождающих его элементов, таких, как железо, молибден, а также вследствие подавления интенсивности спектра рения некоторыми другими содержащимися в значительных количествах элементами. Поэтому спектральный, а также и другие методы испытания на рений применяются лишь после предварительного обогащения исследуемого материала.
1 W. N о d d а с k, I. Т а с к е, О. Berg, Natunyiss., 13, 567 (1925).
4 W. Feit, Z. angew. Chem., 43, 459 (1930); Minerals Yearbook U. S. Bur. Mines. 1937, p. 772 и 1948, p. 1338.
й
Методы, отделения
373
В соответствующих, растворах рений может быть открыт также и микроскопическим методом по реакциям образования специфических перренатов рубидия, цезия, калия, серебра и таллия (I). В виде рубидиевой и цезиевой солей в определенных условиях можно открыть до 0,25 мкг ReO“. Этому испытанию, естественно, мешают перхлораты, хлоростаннаты и хлороплатинаты.
Имеются указания на то, что рений проявляет в своих соединениях все степени валентности: от —I до +VII. Точно установлены соединения Re J, Re+I, Re+IV, Re+VI и Re+vn. Заслуживает внимания то, что некоторые соединения рения могут самоокисляться \ Так, например, при сплавлении с едким натром без доступа кислорода из ReOz образуются Re2O7 и Re, a ReO3 переходит в Re2O7 и ReO2. Интересным является также тот факт, что рений образует более или менее устойчивые соединения, в которых он находится в низших степенях окисления. Так, хлоро-рениевая кислота H2ReCle вполне устойчива против окисления, и из ее растворов рений медленно осаждается лишь сероводородом.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ РЕНИЙ
Необходимо иметь в виду, что при выпаривании сернокислых и хлорнокислых растворов, содержащих соли рения, до появления паров этих кислот имеют место потери рения, независимо от присутствия в таких растворах соляной или азотной кислоты. В процессе кипячения солянокислых, азотнокислых или соляно-азотнокислых растворов потери рения не наблюдаются; но тем не менее такие раствбры не следует выпаривать досуха и нельзя прокаливать остаток.
Минералы, содержащие рений, растворяют в азотной кислоте или, если это требуется, в смеси азотной и соляной кислот. Нерастворимые в этих кислотах минералы сплавляют с карбонатом натрия или со смесью карбоната и нитрата натрия. Рений придает карбонатному плаву желтую окраску. В тех случаях, когда рений находится в виде металла, применяют окислительную обработку, в результа!е которой он переходит в семивалентное состояние.
МЕТОДЫ ОТДЕЛЕНИЯ
Единственной реакцией рения с неорганическим реагентом, применимой для отделения его от других элементов, является реакция с сероводородом в кислом или щелочном растворе. В первом случае происходит отделение рения от элементов, не образующих сульфидов, а во втором случае — от элементов, сульфиды которых растворимы в растворах сульфидов щелочных металлов (главным образом от молибдена).
При обработке сероводородом кислого или щелочного раствора образуется сульфид рения (VII) Re2O7. Для осаждения сульфида рения из кислых растворов требуется продолжительная обработка сероводородом, причем осаждение более надежно в случае высокой концентрации кислоты в растворе: 4 н. (1 : 2) для соляной кислоты или 6 н. (1 : 5) для серной кислоты. При более низкой кислотности количественное осаждение
1 W. В i 1 t z, Z. anorg. Chem., 214, 225 (1933).
374
Гл. XVIII. Рений
рения достигается лишь при насыщении горячих растворов сероводородом под давлением Ч
Из щелочных растворов сульфид рения выделяется медленно и количественное осаждение достигается лишь в том случае, если насыщенный сероводородом раствор оставляют стоять в течение нескольких часов, лучше в продолжение ночи. Независимо от способа получения, сульфид рения Re2S? мало растворим в полисульфиде аммония.
Рений не образует осадков с целым рядом органических реагентов, которые применяют для количественного осаждения различных элементов. Так, рений не осаждается диметилглиоксимом и оксихинолином ни в кислом, ни в щелочном растворах, а также а-нитрозо-|3-нафтолом, купфероном и а-бензоиноксимом. Изучалось отделение молибдена от рения осаждением бензоиноксимом1 2 и оксихинолином в уксуснокислой среде 3 4. Другие способы разделения, как, например, отделение молибдена от рения осаждением купфероном в кислом растворе, не дали положительных результатов.
Перспективны методы отделения рения, основанные на летучести некоторых его соединений. Так, рений отгоняется при введении по каплям азотной кислоты в горячий (около 200° С) сернокислый раствор перрениевой кислоты. Количественная отгонка рения достигается при 200—220 ° С добавлением по каплям соляной или бромистоводородной кислоты к хлорнокислому раствору или бромистоводородной кислоты к сернокислому раствору2. Применению этого метода препятствуют элементы, образующие в этих условиях летучие соединения, как, например, селен, германий и мышьяк.
В некоторых случаях влияние летучих элементов монжо устранить соблюдением особых предосторожностей. Так, в присутствии больших количеств молибдена в раствор вводят фосфорную кислоту, а при малых его количествах проводят повторную отгонку.
При разделении мышьяка, олова и сурьмы дистилляцией по обычно принятому способу (стр. 96) рений в процессе дистилляции мышьяка с соляной кислотой при 1Q5—110° С не отгоняется, но при дистилляции сурьмы из серно-фосфорнокислого раствора при 155—165° С и введения по каплям соляной кислоты может улетучиться до 50% рения. Большая часть оставшегося рения, но обычно не весь, отгоняется вместе с оловом при добавлении по каплям соляной и бромистоводородной кислот к сернофосфорнокислому раствору, нагреваемому до 140° С.
Рений количественно осаждается электролизом с ртутным катодом в слабосернокислом растворе, что применяется для его отделения и может быть использовано также и для его определения.
Из разбавленного (1:1) солянокислого раствора, при отсутствии элементов, извлекаемых эфиром, рений эфиром не экстрагируется. Этот
1 W. G е i 1 m a n n, G. Lange, Z. anal. Chem., 126, 321 (1944).
2 Н. В. Knowles, J. Research. NBS, 9, 1 (1932).
3 W. Geilmann и F. Weibke [Z. anorg. Chem., 199, 347 (1931)]; L. C. Hurd [Ind. Eng. Chem., Anal. Ed., 8, 12 (1936)] установил, что способ отделения больших количеств молибдена от малых количеств рения непосредственным осаждением оксихинолином неприменим вследствие адсорбции перрениевой кислоты объемистым осадком соединения молибдена с оксихинолином.
4 J. I. Hoffmann G. Е. F. Lundell[J. Research NBS, 22, 465 (1939)], а также С. F. Н i s к е у и V. W. М е 1 о с h е [Ind. Eng. Chem., Anal. Ed., 12, 503 (1940)] отгоняли малые количества рения, пропуская смесь воздуха и водяного пара через сернокислый раствор, нагретый до 260—270° С.
Методы определения
375
метод может служить только для частичного отделения рения от молибдена, особенно в присутствии хлорида железа (III). Суспензией окиси цинка рений осаждается также только вместе с другими элементами.
* Четкое разделение рения и молибдена может быть достигнуто экстракцией рения хлороформом в виде перрената тетрафениларсония Ч Реагент также экстрагируется хлороформом в виде хлорида, поэтому при увеличении концентрации хлорид-ионов в водной фазе большая часть тетрафениларсония переходит в слой хлороформа в виде хлорида и соответственно меньшая часть — в виде перрената. При концентрации тетрафениларсония, равной 0,01 М (растворимость реагента в воде равна 0,072 М), и 0,1 М концентрации хлорида коэффициент экстракции рения равен 1000. Хлорид тетрафениларсония и перренат тетрафениларсония являются солями сильных кислот и сильных оснований, поэтому экстракция рения не зависит от кислотности раствора. По данным автора, коэффициент экстракции не изменяется при колебании кислотности раствора от 6 н. по соляной кислоте до pH = 13. При более высокой концентрации соляной кислоты в растворе извлечение рения затрудняется.
При pH от 7 до 13 молибден не осаждается тетрафениларсонием и не экстрагируется хлороформом. Известно, что хлоро-комплексы других металлов [висмута, кадмия, железа (III), олова (IV) и цинка] осаждаются хлоридом фениларсония.
Элементы, дающие в водных растворах устойчивые сульфиды, образуют мало растворимые в водной среде карбаматы [соли диэтилдитио-карбаминовой кислоты (C2H5)2NCSSMe], извлекающиеся органическими растворителями 1 2. К числу таких элементов относится также и рений, карбамат которого экстрагируется хлороформом из концентрированной соляной кислоты. Это свойство может быть использовано для отделения рения от ряда элементов. Доп. перев*
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Рений можно определять весовым, объемным, колориметрическим и электролитическим методами. Однако ни один из этих методов не дает вполне надежных результатов при определении рения даже в чистых его соединениях, как, например, в КВеО4,'и во всех случаях требуется более или менее сложная предварительная обработка.
Весовые методы
Заслуживают внимания лишь три весовых метода определения рения, а именно — взвешивание: 1) в виде металла, 2) в виде «перрената нитрона» и 3) в виде перрената тетрафениларсония (СвН5)4А8ВеО4. Все эти методы применимы для определения нескольких миллиграммов и больших количеств рения.
Определение в виде металла. В этом случае рений отделяют от сопровождающих его нелетучих элементов, затем переводят в соответствующее соединение (например, Re2S7) и прокаливают в атмосфере водорода при 900° С до металла. При взвешивании рения в виде сульфида нам не удалось получить удовлетворительных результатов.
1 S. Tribalat, Anal. Chim. Acta, 3, ИЗ (1949).
a Ю. А. Ч e p н и x о в, Б. М. Добкина, Зав, лаб., 15, 906, 1143 (1949).
37$
Гл. XVIII. Рений
Определение в виде перрената нитрона. Рений осаждают нитроном — 1,4-дифенил-3,5-(эндоанил)-дигидро-1,2,4-триазолом, имеющим формулу
C6H5-N-----N
При этом одна молекула осадителя реагирует с одной молекулой перрениевой кислоты с образованием соединения C20HieN4 • HReO4; фактор пересчета на рений равен 0,3306.
Определению рения с нитроном мешают вольфрам, молибден, палладий, платина и золото. Марганец в высшей степени валентности, нитраты и хроматы также должны быть удалены. Если рений предварительно выделяют в виде сульфида Re2S7, последний' лучше растворять в едком натре, содержащем перекись водорода. При использовании аммиака и перекиси водорода получаются повышенные результаты.
Ход определения *. Нейтральный раствор, содержащий не более 0,1 г рения в виде перрената, разбавляют до 50 лиг, прибавляют 1 мл 2 и. серной кислоты и нагревают приблизительно до 80° С. Вводят 5 %-ный раствор ацетата нитройа 1 2 в таком количестве, чтобы анализируемый раствор содержал 0,3—0,4% свободного нитрона. Охлаждают до комнатной температуры, помещают в ледяную воду и перемешивают 2 ч. Фильтруют через высушенный при 110° С и взвешенный тигель с плотным пористым дном, перенося осадок в тигель охлажденным до температуры льда фильтратом. В зависимости от количества осадка промывают его 10—20 мл ледяного 0,3 %-ного водного раствора ацетата нитрона, порциями по 3—5 мл, давая каждый раз жидкости полностью стечь. Для удаления задержанного осадком нитрона промывают еще 2—3 раза ледяной водой, насыщенной перренатом нитрона, приливая ее порциями по 2—3 мл. Сушат 2—3 ч при 110° С и взвешивают. Фактор пересчета на рений равен 0,3306.
Определение в виде перрената тетрафениларсония. Рений осаждают хлоридом тетрафениларсония (стр. 155) из растворов, варьирующих от сильноаммиачных (6 М) до умеренйо кислых (5 М НС1) 3. Мешают определению перманганат-, перхлорат-, перйодат-, иодид-, бромид-, фторид-и роданид-ионы, а также ртуть, висмут, свинец, серебро, олово и ванадил. Нцтраты могут присутствовать лишь в очень незначительных концентрациях. Вольфрам и ванадаты не мешают определению. Молибден не влияет, если осаждение проводят из аммиачного раствора (6 М) или в присутствии винной кислоты (0,6 М).
Ход определения. К 5—25 мл горячего раствора, свободного от перечисленных мешающих элементов и содержащего 0,5—100 мг рения в виде
1 W. G е i 1 m a n n, A. Voigt, V. anorg. Chem., 193, 312 (1930).
2 Смешивают 5 г нитрона с 3 мл уксусной кислоты, смесь растворяют в 100 мл воды и перед употреблением фильтруют через тигель с пористым дном.
3 Н. Н. Willard, G. М. Smith, Ind. Eng. Chem., Anal. Ed., 11, 305 (1939).
Методы определения
377
перрената и хлорид натрия в таком количестве, чтобы в конечном растворе концентрация его'была 0,5 н., прибавляют при перемешивании избыточное количество 1%-ного раствора хлорида тетрафениларсония. Конечный объем раствора не должен превышать 25—60 мл. Перемешивают, охлаждают и оставляют в течение нескольких часов, лучше в продолжение ночи. Осадок отфильтровывают через тигель Гуча и промывают несколько раз ледяной водой. Сушат при 110° С, охлаждают в эксикаторе и взвешивают в виде перрената тетрафениларсония (CeH5)4AsReO4.
Объемные методы
Известны два объемных метода определения рения, которые могут давать удовлетворительные результаты при строгом соблюдении соответствующих условий.
1) Согласно первому из этих методов рений переводят в перрениевую кислоту, которую титруют едкой щелочью. Этот метод дает весьма точные результаты, так как перрениевая кислота является сильной кислотой. При титровании можно пользоваться индикаторами с переходом окраски в интервале pH от 4 до 9. Максимальные количества рения, которые можно определять таким способом, не ограничены. Титрование необходимо проводить весьма тщательно, пользуясь весовыми бюретками, так как титр раствора в пересчете на рений относительно высок — 1 мл 0,1 н. раствора соответствует 0,02513 г HReCU. Необходимость переведения рения в перрениевую кислоту без введения других кислот ограничивает применение этого метода особыми случаями. Он может быть использован, например, для титрования водных растворов Re2O,, при анализе' окислов рения, обработанных перекисью водорода, или при анализе электролитически осажденного рения, подвергавшегося воздействию влажного воздуха или кислорода. Металлический рений превращается в Re2Or при прокаливании в атмосфере кислорода, но это связано с опасностью потери рения вследствие улетучивания.
2) Вторым, представляющим интерес методом (хотя он и не имеет большого практического применения) является метод, основанный на восстановлении рения цинком и последующем титровании его растворами окислителя. Очевидно, что такие элементы, как молибден, вольфрам, ванадий, ниобий и железо, восстанавливающиеся и окисляющиеся в тех же условиях, должны быть предварительно удалены. Метод имеет следующие недостатки: а) он может быть применен для определения только малых количеств рения (<30 мг); б) раствор, пропускаемый через редуктор, должен быть свободен от растворенного кислорода и в) пропущенный через редуктор раствор следует сразу же вливать в раствор сульфата железа (III), или же должны быть приняты меры, предохраняющие его от соприкосновения с воздухом. При восстановлении валентность рения изменяется 1 от -J-VII до —1 и при последующем титровании перманганатом от —I до +VII.
Ход определения. Перед восстановлением анализируемого раствора редуктор промывают сначала горячей разбавленной (5 : 95) серной кислотой, затем холодной водой (свободными от воздуха). После этого для
1 G. Е. F. Lund ell, Н. В. Knowles, J. Research NBS, 18, 629 (1937);
S' О. Т о m i с е k, F. Т о m i с е k, Coll. Czech. Chem. Comm., 11, 626 (1939).
В78
Гл. XVIII. Рений
удаления поглощенного цинком воздуха наливают в приемник воду, плотно закупоривают заполненный водой редуктор и затем попеременно включают и выключают вакуум, пока не прекратится выделение газа. Выливают из приемника воду, вводят в него 20 мл раствора сульфата железа (III), содержащего 0,02 г железа в 1 мл (стр. 136), и снова присоединяют к редуктору. Из всех растворов, применяемых в последующих операциях, воздух должен быть удален кипячением. Охлаждают растворы в атмосфере двуокиси углерода, свободной от кислорода.
Через редуктор пропускают сначала 50 мл холодной разбавленной (5 : 95) серной кислоты, а затем 500 мл разбавленного сернокислого анализируемого раствора, содержащего не более 30 мг рения и свободного от посторонних восстанавливающих веществ (а также от воздуха). Промывают редуктор 50 мл холодной разбавленной (5 : 95) серной кислоты, которую затем отмывают 100 мл холодной воды. Титруют раствором перманганата калия. Вводят поправку на количество перманганата, израсходованного на титрование одних реактивов. 1 мл 0,1 н. раствора КМиСЧ соответствует1 0,002329 г Re.
Колориметрический метод
Колориметрический метод определения рения основан на образовании коричневато-желтого соединения рения с роданидом, которому приписывают состав ReO(CNS)4. Это соединение образуется при обработке солянокислого раствора, содержащего рений, смесью SnCl2 и KCNS. Окраску раствора можно или непосредственно сравнивать со стандартами, приготовленными аналогичным образом, или же после извлечения органическими растворителями, такими, как эфир и бутилацетат. Определению мешают главным образом молибден и платина. Влияние молибдена можно устранить, восстанавливая его в кислом растворе ртутью в присутствии роданида с последующим извлечением эфиром 2. Молибден и рений можно разделить, также экстрагируя хлороформом и бензолом металлорганическое соединение молибдена с этилксантатом. Рений при этом остается в водном слое 3. Отделить рений от молибдена можно также дистилляцией из смеси хлорной и фосфорной кислот, в которую медленно вводят бромистоводородную кислоту. (Подробности см. в сноске 4, стр. 374).
Колориметрический метод обычно применяют для определения 0,0005—0,5 мг рения.
Ход определения рения в растворах, свободных от мешающих элементов 4. Почти нейтральный раствор, содержащий не более 500 мкг рения в виде перрената в 15—20 мл, переносят в несслеровскую пробирку соответствующего размера. Смешивают в стакане соляную кислоту, хлорид олова (II), роданид калия и воду в таких количествах, чтобы после добавления к анализируемому раствору перрената общий объем составлял 50 мл, а концентрация соляной кислоты была 2%, роданида калия 0,4% и хлорида олова (II) 0,2%. Эту смесь прибавляют к раствору перрената, находящемуся в пробирке, и перемешивают не долее, чем требуется для
1 Подробно см. G. Е. F. L u n d е 1 1, Н. В. Knowles, цит. выше.
2 J. I. Hoffman, G. Е. F. L u n d е 11, J. Research NBS, 23, 497 (1939).
3 F. Е. М а 1 о и f, М. G. White, Anal. Chem., 23, 497 (1951).
4 L. C. Hurd, В. J. В a b 1 e r, Ind. Eng. Chem., Anal. Ed., 8, 112 (1936),
Методы, определения
379
получения однородного раствора. Оставляют на 7 мин и затем окрашенное соединение рения экстрагируют соответствующим количеством эфира, лучше предварительно обработанного смесью, содержащей одинаковые с анализируемым раствором количества соляной кислоты, роданида калия и хлорида олова (II). Окраску сравнивают со стандартными растворами, содержащими определенные количества рения и приготовленными таким же способом, как и анализируемый раствор.
Ход определения малых количеств рения в присутствии относительно больших количеств молибдена *. Этот метод применим для анализа растворов, содержащих 10 мг молибдена и не более 1 мг рения.
Разделение рения и молибдена. К почти сухому остатку, содержащему молибден и рений, прибавляют 10 мг железа в виде хлорида железа (III) (если это количество железа не содержится в анализируемой пробе) и 1—2 капли насыщенного раствора перманганата калия. Затем остаток обрабатывают небольшим избытком аммиака и нагревают несколько минут на водяной бане, чтобы обеспечить переход рения и молибдена в перренат и молибдат. Осадок растворяют добавлением 25 мл холодной разбавленной (1 : 4) соляной кислоты и прибавляют 2 мл 20%-ного раствора роданида калия а. Раствор переносят в делительную воронцу, содержащую около 25 г ртути. Немедленно вводят 20 мл эфира, плотно закрывают воронку пробкой и сильно встряхивают до обесцвечивания раствора 3. Обычно достаточно сильного встряхивания в течение 1 мин. После отслаивания эфирного слоя ртуть и кислый раствор сливают в другую делительную воронку. Эфирный экстракт, который содержит основную массу молибдена, сохраняют.
В делительную воронку, содержащую кислый раствор и ртуть, вводят 1 мл 20%-ного раствора роданида калия и 15 мл эфира. Сильно встряхивают 1 мин и после отстаивания снова сливают кислый раствор и ртуть. Эту операцию повторяют еще один раз 4, после чего сливают ртуть в делительную воронку и сохраняют ее для следующего определения, а в кислом растворе (раствор А) определяют рений 5. Эфирные экстракты объединяют (раствор Б).
Определение рения. Для определения рения к раствору А прибавляют 1 мл 20%-ного раствора роданида калия и 1 мл раствора хлорида олова 6 (II), слегка встряхивают и оставляют стоять 5 мин. Вводят 20 мл эфира, снова сильно встряхивают и дают отделиться эфир
1 J. I. Hoffman, G. Е. F. L undell, J. Research NBS, 23, 497 (1939).
2 Осадок, который иногда растворяется с большим трудом, очень быстро переходит в раствор после добавления роданида.
3 В присутствии таких элементов, как хром, ванадий, кобальт и никель, раствор имеет окраску, свойственную этим элементам, и встряхивание следует продолжать .лишь до тех пор, пока не исчезнет окраска роданида железа.
4 Если в этот момент эфир имеет еще отчетливую окраску, указывающую на заметное содержание молибдена, экстрагирование следует повторить.
5 От 1 до 5% присутствующего рения переходит при этом в эфирный экстракт вместе с молибденом. Лучше, однако, потерять такие количества рения, чем оставить в растворе следы молибдена, заметно отражающиеся на результатах определения рения. Фактические потери рения не имеют практического значения, так как общее количество рения, находящееся в экстрагируемом растворе, редко превышает 0,5 мг. Опыты показали, что потери рения цропорциональны его содержанию в растворе и не выходят за пределы неопределимых количеств.
9 Растворяют 350 г SnCl • 2Н2О в 200 мл разбавленной (1 : 1) соляной кислоты. Охлажденный раствор разбавляют до 1 л свежепрскипяченной водой.
380
Гл. XVIII. Рений
ному слою. Кислый раствор сливают в другую делительную воронку и затем дважды повторяют экстракцию эфиром порциями по 15 мл. Кислый раствор отбрасывают. К объединенным эфирным экстрактам прибавляют 10 мл разбавленной. (1 : 4) соляной кислоты, Сильно встряхивают и дают раствору расслоиться. Кислый раствор сливают и отбрасывают. Промывка кислотой имеет целью извлечение железа, которое постепенно окисляется и дает розовую окраску. Эфирный экстракт, содержащий рений, переносят в мерную колбу емкостью 50 мл, разбавляют до метки специально приготовленным эфиром \ и плотно закрывают колбу пробкой.
Содержание рения определяют сравнением интенсивности окраски полученного раствора с окраской стандартного раствора или с серией стандартов, приготовленных следующим образом. Соответствующее количество типового раствора рения 1 2 переносят в делительную воронку, содержащую 10 мг железа в виде хлорида железа (III) и 25 мл разбавленной (1 : 4) соляной кислоты. Затем прибавляют 2 мл 20%-ного раствора роданида калия и 1 мл раствора хлорида олова (II). Слегка встряхивают, оставляют стоять 5 мин, после чего экстрагируют эфиром, объединяют эфирные экстракты и разбавляют зфирный раствор точно так, как при приготовлении анализируемого раствора.
Аналогичный метод применяют для отделения относительно больших количеств рения от малых количеств молибдена. Только в этом случае при восстановлении ртутью и экстракции эфиром применяют более разбавленную солянук) кислоту (3 : 200 вместо 1:4). Более подробные указания можно найти в оригинальной работе.
*Н. С. Полуэктов 3 предложил косвенный каталитический метод определения рения, который основан на свойстве рениевой кислоты и ее солей каталитически ускорять восстановление теллурата натрия хлоридом олова (II) до элементарного теллура. При прочих равных условиях количество восстановленного теллура пропорционально концентрации рения, которую можно определить, измерив светопоглощение коллоидного раствора теллура, после'введения в него защитного коллоида. Этим методом можно определять от 0,001 до 0,1 мкг рения с точностью 10—20%. Молибден мешает определению. Азотная кислота подавляет реакцию. Другие кислоты также влияют на интенсивность окраски. Доп. перев.*
Электролитические методы
Электролитические методы определения рения недостаточно надежны, так как определению мешают многие элементы. Количества рения, которые могут быть выделены электролизом, относительно невелики (<Д5 мг), осадок рения загрязнен кислородом, кроме того, не удается достигнуть количественного осаждения. Ошибки, обусловленные-
1 Эфир для разбавления эфирных растворов молибдена и рения приготовляют следующим образом. В большую делительную воронку наливают 25 мл разбавленной (1 : 1) соляной кислоты, 2 мл 20%-ного раствора роданида калия и около 10 г ртути. Вводят 100—200 мл эфира и встряхивают 1 мин. После отделения эфирного слоя сливают ртуть и кислоту.
2 Приготовляют слабокислый раствор, содержащий 0,1 мг рения в 1 мл. Для этого 0,155 г чистого перрената КВеО4 растворяют в 1 л разбавленной (1 : 99) серной кислоты. Для определения менее чем 0,1 мг рения этот раствор разбавляют в 10 раз.
3 Н. С. Полуэктов, ЖПХ, 14, 695 (1941).
Методы, определения
381
содержанием кислорода в осадке и неполнотой осаждения, могут взаимно компенсироваться. При тщательной работе, когда осаждение проводится из слабосернокислых растворов (5 : 95), ни одна,из этих ошибок не превышает 0,5 мг.
Наиболее удовлетворительные результаты получаются при: 1) применении неподвйжнбго обработанного песком для получения матовой поверхности платинового сетчатого катода и спирального анода, заключенного в неглазурованный керамический цилиндр; 2) использовании разбавленной (5 : 95) серной кислоты в качестве электролита и 3) проведении электролиза в течение ночи при плотности тока 0,25 а на 1 дм? и напряжении 2,34 в. После окончания электролиза осадок быстро промывают водой, затем спиртом и эфиром. Сушат 10 сек при 105° С. Охлаждают в эксикаторе, содержащем хороший поглотитель влаги, и через 15 мин взвешивают.
Выделенные электролизом осадки количественно переходят в перре-ниевую кислоту HReO4 под действием влажного кислорода или воздуха.
Глава XIX
СЕЛ^Н И ТЕЛЛУР
В природе селен и теллур встречаются совместно и в соединении? с серой. Селен, являющийся неметаллическим элементом, встречается в самородном состоянии и в виде селенидов меди, серебра, ртути, свинца, висмута и таллия. Некоторые селениты образуют вторичные минералы. Селен находится в небольших количествах во многих пиритах и содержится поэтому в серной и соляной кислотах.
Теллур — полуметаллический элемент — является наименее распространенным элементом группы серы. Он встречается в самородном состоянии и в виде теллуридов золота, серебра, свинца, висмута, ртути, никеля и меди. Двуокись теллура и некоторые редко встречающиеся теллураты и теллуриты известны как продукты выветривания пород. Теллур ассоциируется главным образом с золотыми, серебряными и висмутовыми рудами. Он содержится также в мышьяково-железных пиритах и в незначительных концентрациях в медных рудах \
ОБЩИЕ ЗАМЕЧАНИЯ
Селен и теллур, подобно сере, образуют двуокиси и трехокиси. Они вступают в реакции с различными металлами, образуя соединения, аналогичные сульфидам. Селеновая кислота H2SeO4 очень сходна с серной кислотой и образует нерастворимые соли со свинцом, барием и подобными им элементами. Существенным отличием является то, что селеновая кислота и ее соли легко восстанавливаются соляной кислотой до селенистой кислоты H2SeO3. Селенистая кислота, так же как и ее соли, легко растворима в воде и ни при каких условиях не является восстановителем. Реакции теллуровой и теллуристой кислот в основном сходны с реакциями соответствующих кислот селена. Теллуристая кислота, однако, мало растворима и при сильном разбавлении водой солянокислого раствора
1 Нахождение в природе, физические свойства, качественное и количественное определение селена и теллура описаны в статье V. L е n h е г, Trans. AIME, 69, 1035 (1923) и Proc. Am. Phil. Soc., 65, 33 (1926). В первой статье изложены методы анализа различных продуктов, содержащих селен и теллур. О распространении селена в природе см. L. W. Str ос k, Am. J. Pharm., 107, 144 (1935). О нахождении селена в пиритах см. К. Т. Williams, Н. G. Byers, Ind. Eng. Chem., Anal. Ed., 6, 296 (1934).
Разложение минералов, содержащих селен и теллур
383
соли теллура она выделяется в осадок, который легко может быть принят за оксихлорид сурьмы. Теллуриты и селениты не окисляются азотной кислотой, но их можно окислить хлором, хроматом, перманганатом или окислительным сплавлением. Соединения селена (IV) и теллура (IV) окисляются также, если их слабокислый или аммиачный раствор обработать 1—2 г кристаллического персульфата калия при кипячении в течение 5—10 мин, затем добавить еще 1 г соли и продолжать кипятить 5 мин V
В обычном ходе анализа горных пород селен и теллур, оставшиеся в растворе после предварительной обработки анализируемой пробы, переходят в осадок от аммиака, если имеется достаточный избыток железа, алюминия или щелочноземельных металлов. В тех случаях, когда присутствие селена и теллура не учитывается, их принимают за алюминий. Содержание значительных количеств селена и теллура обнаруживается при определении железа в процессе восстановления.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ СЕЛЕН И ТЕЛЛУР
При разложении анализируемой пробы необходимо всегда иметь, в виду, что селен легко улетучивается из кипящих солянокислых растворов в виде SeO2 • 2НС1, а из сильно солянокислых растворов — даже при нагревании на водяной байе. Заметных потерь селена не наблюдается 1 2 в случае нагревания слабо солянокислых растворов (<<6 н.) при температуре ниже 100° С.
Теллур улетучивается из кипящих растворов в концентрированной соляной кислоте, но не отгоняется из разбавленной соляной кислоты, а также из растворов в концентрированной соляной кислоте, нагреваемых ниже 100° С. Соли щелочных металлов не препятствуют улетучиванию селена и теллура. Следует учесть также, что летучий монохлорид селена Se2Cl2 легко образуется в сильно солянокислых растворах при действии таких восстановителей,, как сероводород, сернистый ангидрид и даже волокна фильтровальной бумаги. Значительные потери селена могут происходить в горячих растворах, когда восстановление протекает медленно или когда в них находится недостаточное количество восстановителя, чтобы обеспечить восстановление селена до элементарного состояния. В процессе выпаривания сернокислых растворов селенистой кислоты (до появления густых паров серного ангидрида потерь селена не наблюдается 3, но различные количества сёлена улетучиваются при выпаривании (до появления белых паров) сернокислых или хлорнокислых растворов, содержащих хлориды или бромиды4.
1 N. Н. Furman, R. С. Newton (частное сообщение).
2 Так)' например, в одном из опытов кипятили в высоком стакане емкостью 1,3 л солянокислый раствор селенистой кислоты, содержавший 0,2069 г селена, пока объем раствора не уменьшился от 200 до 50 мл\ раствор повторно упаривали до 50 мл после добавления 150 мл соляной кислоты. Потеря селена составляла 0,0173 г. При такой же обработке раствора, но в присутствии 10 г хлорида натрия было потеряно 0,022 г селена. В растворе, содержавшем 0,02025 г селена, после трехкратного выпаривания на водяной бане досуха с последовательно добавляемыми порциями соляной кислоты было определено 0,1009 г селена. В другой порции раствора, в которую было введено 10 г хлорида натрия, после аналогичной обработки осталось 0,1238 г селена. И, наконец, после упаривания на водяной бане 50 мл раствора 0,0776 г селена в разбавленной (1 : 1) соляной кислоте до 25 мл было определено 0,0777 г селена.
2 Н. Н. Willard, F. Fenwick, J. Am. Chem. Soc., 45, 936 (1923).
4 J. I. Hoffman, G. E. If. L u n d e 11, J. Research NBS, 22, 467 (1939).
384
Гл. XIX. Селен и теллур
При сплавлении минералов, содержащих селен и теллур, тигель подбирают в зависимости от характера минерала и компонентов, которые должны быть определены. Для сплавления селенидов, действующих на платину, можно пользоваться фарфоровыми тиглями, если требуется определить только селен и теллур. В большинстве случаев сплавление с перекисью натрия или со смесью карбоната натрия и селитры можно проводить в никелевых тиглях. Холодный плав выщелачивают водой, фильтруют, раствор подкисляют соляной кислотой и напревают при температуре ниже 100° С до удаления хлора и полного восстановления соединений селена и теллура до селенитов и теллуритов.
При определении незначительных количеств селена и теллура, когда приходится пользоваться большими навесками исходного материала, разложение осуществляют обработкой царской водкой или соляной кислотой с хлоратом калия при нагревании. В обоих случаях свободный хлор удаляют и затем восстанавливают селенаты и теллураты до четырехвалентного состояния, обработкой разбавленной соляной кислотой при нагревании на водяной бане *. При обработке персульфатом калия сернокислых или азотнокислых растворов или же хлором, бромом или перекисью водорода щелочных растворов образуются соединения шестивалентных селена и теллура.
В результате обработки азотной или серной кислотой, а также царской водкой образуются соединения, в которых эти элементы четырехвалентны.
Руды, содержащие селениды и теллуриды, разлагают нагреванием при темно-красном калении в токе хлора. Образующиеся при этом летучие хлориды селена и теллура улавливают разбавленной (1:1) соляной кислотой.
Мышьяк, сурьма, ванадий, висмут, железо и сера также полностью или частично в этих условиях отгоняются. Солянокислый раствор, содержащий селен и теллур, обрабатывают, как было указано выше.
При сплавлении пробы с пяти-, шестикратным количеством цианида калия, которое часто рекомендуется, образуются растворимые соли — теллурид калия и селеноцианат калия, но при этом имеют место потери вследствие улетучивания.
Имеются указания 1 2 на то, что. селен, входящий в состав органических соединений, можно перевести в селенистую кислоту нагреванием пробы с азотной кислотой в запаянной трубке, как в методе Кариуса. В нейтрализованном по метиловому оранжевому растворе потом определяют селен титрованием нитратом серебра, применяя хромат в качестве внешнего индикатора.
Разложение содержащих селен органических соединений рекомендуют 3 также проводить в стеклянной колбе Кьельдаля с дистилляционной насадкой, «смазанной» серной кислотой и снабженной капельной воронкой и отводной трубкой, которая.проходит в содержащий воду шарик Фрезениуса для определения азота. Обработку осуществляют следующим образом.
В колбу помещают анализируемую пробу, обрабатывают ее 5 мл серной кислоты и затем в продолжение 20 мин при осторожном нагре
1 О разложении пиритов азотной и серной кислотами и об источниках ошибок, • которые имеют место при определении содержания в них селена, см. К. Bruckner, Z. anal. Chem., 94, 305 (1933).
2 W. E. Brad t, Б. E. Lyons, J. Am. Chem. Soc., 48', 2642 (1926).
2 A. F r e d g a, J. prakt. Chem., 121 (New series), 57 (1929).
Методы отделения
385
вании вводят по каплям азотную кислоту с такой скоростью, чтобы колба все время была заполнена окислами азота. Под конец температуру повышают, пока серная кислота не закипит. При разложении ароматических углеводородов кипячение продолжают несколько минут, после чего охлаждают, прибавляют еще 1—2 мл азотной кислоты и снова нагревают. Переносят раствор в фарфоровую чашку, нагревают на водяной бане до удаления основной массы азотной кислоты и, если есть необходимость, фильтруют. Раствор нейтрализуют аммиаком, слегка подкисляют серной кислотой и осаждают селен сернокислым гидразином. Нагревают, пока осадок не осядет на дно, затем фильтруют через стеклянный фильтр. Осадок промывают сначала водой, затем спиртом и сушат при 110° С.
При анализе пиритов, глинистых, сланцев, почв, природных вод, зерна и животных тканей, содержащих малые количества селена, наиболее целесообразно предварительно 'отделять селен, совместно с мышьяком и германием, отгонкой с бромистоводородной кислотой в стеклянном аппарате \ В цитируемой статье имеются указания, как надо производить разложение этих материалов. Дистилляция может быть успешно выполнена в дистилляционной колбе (см. рис. 9, стр. 96).
Когда содержание селена превышает 0,5 мг, его взвешивают в элементарном состоянии после восстановления ангидридом, газом и солянокислым гидроксиламином. В случае определения меньших количеств селена осадки диспергируют гуммиарабиком и содержание селена устанавливают колориметрическим методом путем сравнения со стандартами. Германий и мышьяк в процессе восстановления не осаждаются и после отделения селена фильтрованием могут быть извлечены из раствора дистилляцией.
МЕТОДЕ! ОТДЕЛЕНИЯ
Основные методы отделения селена и теллура сводятся к выделению их из кислых растворов в элементарном состоянии различными восстановителями. Большей частью восстановление проводят сернистым ангидридом 1 2 в солянокислой среде, причем селен и теллур должны находиться в четырехвалентном состоянии. Азотная кислота должна отсутствовать. Селен (VI) и теллур (VI) можно легко восстановить до четырехвалентного состояния нагреванием соединений этих элементов с разбавленной (<6 н.) соляной кислотой при температуре ниже 100° С до удаления хлора. Азотную кислоту можно разложить выпариванием с разбавленной (< 6 н.) соляной кислотой при температуре ниже 100° С или кипячением с обратным холодильником.
Серная кислота не препятствует восстановлению, если присутствует соляная кислота. Из солянокислых растворов селен осаждается количественно, если концентрация соляной кислоты выше 3,4 н. (около 28%
1 W. О. Robin'son, Н. С. Dudley, К. Т. Williams, Н. G. Byers, Ind. Eng. Chem., Anal. Ed., 6, 274 (1934); H. C. Dudley, H. G. Byers, там же, 7, 3 (1935); К. T. Williams, Н. W. L a k i п, там же, 7, 409 (1935).
2 Многие другие вещества, как, например, иодид калия, хлорид олова (II), хлорид титана (III), металлические цинк и алюминий, фосфористая и фосфорноватистая кислоты, восстанавливают селениды и теллуриды до металла в холодных кислых растворах. Для количественных определений эти восстановители, однако, не пригодны вследствие окклюзии осадком продуктов реакции. О применении фосфорноватистой Кислоты для открытия и определения селена и теллура в меди см. Н. J. G. С h а 1-1 i s, Analyst, 67, 186 (1942).
25 Заказ 522.
• 386
Гл. XIX. Селен и теллур
по объему), а теллур при концентрациях соляной кислоты от 1,2 до 5 н. (10—42% по объему). Из растворов выше 8,8 и. по концентрации соляной кислоты (около 73% по объему) теллур не осаждается, если находится один. Из холодной концентрированной соляной кислоты он не выделяется даже и совместно с селеном. Обычно селен и теллур сначала осаждают вместе восстановлением их сернистым ангидридом в солянокислых растворах, имеющих 3,7—4,8 и. концентрацию соляной кдслоты (приблизительно 30—40% по объему). Осадок, в котором могут находиться также золото, палладий и небольшие количества сурьмы, висмута, меди и других элементов, промывакгг водой. Эти примеси отделяют от селена и теллура следующим способом.
Золото осаждается совместно с селеном и теллуром при использовании большинства восстановителей. Для его отделения промытой водой осадок обрабатывают при нагревании разбавленной азотной кислотой (пл. 1,25 г/см3), раствор разбавляют и остаток, содержащий золото, отфильтровывают. Фильтрат выпаривают почти досуха, прибавляют едкий натр и затем отделяют селен от теллура осаждением сернистой кислотой или гидроксиламином, как указано на стр. 389 или 390. Золото можно количественно осадить и отделить от теллура восстановлением кипящих растворов хлорида золота (III) и теллуристой кислоты в 0,3— 0,6 М соляной кислоте сульфатом железа (II). Осадок отфильтровывают через плотный фильтр или пористую пластинку и промывают разбавленной (1 : 99) соляной кислотой \
Четырехвалентные селен и теллур осаждаются сероводородом из кислых растворов и могут быть отделены таким образом от элементов последующих групп. Соединения селена (VI) и теллура (VI) выделяются сероводородом из слабокислых растворов медленно или вовсе не осаждаются 1 2 3.
Осадок соединения селена, выделенный сероводородом из холодного раствора, имеет лимонно-желтую окраску, а из горячего — оранжевожелтую. Осадок соединения теллура окрашен в красновато-коричневый цвет. Соединения селена и теллура, образующиеся при осаждении сероводородом, быстро разлагаются на серу и соответствующий элемент в свободном состоянии. Эти соединения, если осаждение сероводородом проводили из холодных растворов, не содержащих элементов группы меди, растворяются в растворах сульфидов щелочных металлов. Осадки же, полученные из горячих растворов, значительно менее растворимы, и если они выделены совместно с группой меди, то селен из них извлекается не : количественно, а теллур — с трудом. Теллур после необходимой предварительной обработки 3 можно отделить от свинца, висмута, ртути, J меди, золота и селена осаждением сульфитом натрия из раствора его в растворе сульфида щелочного металла.
1 V. Lenher, G. В. L. Smith, D. С. Knowles Jr., Ind. Eng. Chem., Anal. Ed., 6, 43 (1934). *
2 Согласно А. НойесиВ. Брэй [Качественный анализ редких элементов, ОНТИ, Гл. ред. хим. лит., 1936, стр. 238 и 308], из растворов селеновой кислоты сероводород полностью осаждает селен при 12 н. концентрации соляной кислоты, выделяет ,< незначительную часть селена при 6 н. концентрации кислоты и вовсе не образует осадка, 1 если концентрация соляной кислоты соответствует 0,3 н. Из растворов селенистой j кислоты в 3 н. фтористоводородной кислоте селен количественно осаждается сероводо- I родом при комнатной температуре. 1
3А. Brukl, W. Maxymowicz, Z. anal. Chem., 68, 14 (1926). |
Методы отделения
387
Метод осаждения теллура заключается в следующем. Раствор слегка подщелачивают аммиаком, слабо нагревают и прибавляют по каплям полисульфид аммония (желтый) до полного растворения осадка. Затем вводят 20—25 мл насыщенного раствора сульфита натрия, кипятят 10— 20 мин, разбавляют до 200—300 мл, прибавляют еще несколько миллилитров раствора сульфита натрия, снова кипятят и проверяют полноту осаждения. Раствору дают отстояться (при содержании малых количеств теллура, в продолжение нескольких часов), после чего осадок отфильтровывают через тигель Гуча и промывают последовательно горячей водой, спиртом и эфиром, а затем сушат при 100° С в атмосфере азота или воздуха. К фильтрату прибавляют еще сульфита натрия и кипятят, чтобы удостовериться в полноте осаждения.
Свинец отделяют в виде сульфата, как описано на стр. 262. Фильтрат обрабатывают, как указано выше, предварительно отфильтровав сульфид свинца, который может' образоваться при добавлении сульфида аммония. Для отделения висмута кислый раствор обрабатывают карбонатом натрия до появления слабой мути, прибавляют по 2 г бромида и бромата калия и затем кипятят до удаления брома. Полноту осаждения проверяют добавлением небольших количеств бромида и бромата калия, после чего снова кипятят. Дают отстояться, фильтруют и в фильтрате осаждают теллур, как было указано выше. Оксибромид висмута растворяют в горячей разбавленной азотной кислоте и определяют висмут, как указано в гл. «Висмут» (стр. 274). Для отделения ртути (II) раствор нейтрализуют едким натром до’ слабощелочной реакции, затем прибавляют сульфид натрия до растворения осадка, кипятят и прибавляют кристаллический хлорид аммония до полного осаждения ртути и появления запаха сульфида аммония. Кипятят, дают отстояться, осадок отфильтровывают через тигель Гуча и промывают, как указано на стр. 250. Теллур определяют в фильтрате, как описано ранее. Медь (а также и золото) отделяют следующим образом. Раствор, содержащий медь и теллур, делают аммиачным и прибавляют полисульфид аммония (желтый) до полного осаждения меди и пожелтения анализируемого раствора. Если медь и теллур предварительно выделены в виде сульфидов, то их обрабатывают полисульфидом аммония, а затем разбавляют до 100 мл водой. Прибавляют цианид калия до растворения осадка, нагревают до кипения и вводят 10—15 мл насыщенного раствора сульфита натрия. Слабо кипятят 10—20 мин, разбавляют до 200—300 мл, прибавляют небольшое количество цианида и дают отстояться в течение нескольких часов, если содержание теллура незначительно. Осадок отфильтровывают и промывают разбавленным раствором цианида. Из фильтрата медь можно осадить в виде сульфида, для чего фильтрат подкисляют и кипятят под хорошей тягой. Золото сопровождает медь.
Селен при всех описанных операциях будет частично сопровождать теллур. Для разделения этих элементов их растворяют в растворе сульфида щелочного металла, раствор обрабатывают цианидом калия до обесцвечивания и затем нагревают до кипения. Вводят 10—20 мл насыщенного раствора сульфита натрия, кипятят, пока осадок не скоагули-рует и жидкость не станет прозрачной, затем дают отстояться и фильтруют, как указано выше. Определять селен в фильтрате нецелесообразно, так как в процессе анализа могут иметь место потери его вследствие улетучивания.
25*
388
Гл. XIX. Селен и теллур
Селениды и теллуриды можно отделить от металлов, хлориды которых нелетучи, нагреванием в токе хлора. Летучие хлориды селена (IV) и теллура (IV) улавливают водой и затем восстанавливают сернистым ангидридом или солянокислым гидроксиламином. Для разложения четырех-или шестивалентных соединений селена и теллура вместо хлора можно применять газообразный хлористый водород.
Селен можно отделить от теллура дистилляцией солянокислого раствора следующим образом \ Анализируемую пробу помещают в колбу емкостью 150 мл, прибавляют серную кислоту и нагревают до 300— 330° С, пропуская через раствор струю хлористого водорода. Отгон собирают в холодную воду и осаждают селен сернистым ангидридом, как указано на стр. 389. Раствор в колбе разбавляют так, чтобы концентрация серной кислоты в нем стала 4—5%-ной по объему, и осаждают теллур сернистым ангидридом и солянокислым гидразином, как указано на стр. 392. Количественное отделение как шестивалентного, так и четырехвалентного селена от теллура можно осуществить также в растворе, содержащем бромистоводородную, фосфорную и селенистую кислоты. Разделение проводят таким путем 1 2. Смесь окислов обоих элементов помещают в соответствующую колбу и растворяют в едком кали. Раствор нейтрализуют фосфорной кислотой (пл. 1,70 г!см3) и затем добавляют 20 мл избытка этой кислоты. Прибавляют 1 г бромида калия и разбавляют до 50 мл. Соединяют с колбой, наполненной водой, пропускают через прибор СО2 и кипятят, пока объем раствора не уменьшится до 15 мл. Вместо фосфорной кислоты и бромида калия можно пользоваться бромистоводородной кислотой или смесью бромистоводородной кислоты с бромом. Мышьяк, германий, олово и сурьма частично перегоняются совместно с селеном.
Очень небольшие количества селена и теллура, как, например, в рафинированной меди, можно осадить совместно с гидроокисью железа, как описано в гл. «Молибден» (стр. 359). Для этого в растворе должно содержаться не менее 0,1—0,2 г железа (III), и для осаждения следует прибавить избыточное количество аммиака.
Определение селена в серной кислоте лучше всего проводить следующим образом s. Растворяют 0,5—1,0 г КВт в 3—4 мл бромной воды, переносят в реторту емкостью 200 мл и затем вводят 100 г анализируемой кислоты. В цилиндр емкостью 25 мл, охлаждаемый льдом, наливают 3—4 мл соляной кислоты, насыщенной SO2. Конец реторты вводят в цилиндр и погружают в соляную кислоту, а затем перегоняют селен до обесцвечивания серной кислоты. Дистиллят переносят в маленькую колбу, насыщают SO2, закрывают и оставляют стоять на 24 ч до полного осаждения красного селена4.
1 V. L е n h е г, D. Р. Smith, Ind. Eng. Chem., 16, 837 (1924).
2 F. A. Gooch, A. W. Pierce, Am. J. Sci., [4]., 1, 181 (1896); A. H ойес, В. Брэй, цит. выше.
3 R. С. Wells, J. Wash. Acad. Sci., 18, 127 (1928).
1 Определение малых количеств селена в сере см. G. G. Marvin, W. С. Schumb, Ind. Eng. Chem., Anal. Ed., 7, 423 (1935). В органических соединениях, не содержащих ванадия, можно открыть 0,0001% селена следующим способом. Анализируемую пробу обрабатывают по Кьельдалю. Полученный раствор охлаждают, затем прибавляют 2 капли водного 3%-ного раствора сернокислого кодеина, охлаждая
и встряхивая после добавления каждой капли реагента. Появление зеленой окраски, быстро переходящей в синюю, указывает на присутствие селена [М. J. Horn, Ind. Eng. Chem., Anal. Ed., 6, 34 (1934)], К. W. Franke, R. BurrisnR. S. Hut-t о n [там же, 8, 435 (1936)] описывают колориметрический метод определения 0,005— 0,15 мг селена, основанный на сравнении окраски слоя свежеосажденного сульфата бария (получаемой в результате фильтрования через сульфат бария коллоидной суспензии селена) с окрасками стандартов, приготовленных таким же способом.
Методы определения
389
Для определения малых количеств селена и теллура в различных рудах, металлах и побочных продуктах металлургии рекомендуется1 следующий ход анализа.
Материал обрабатывают кислотой (предпочтительно азотной) или сплавляют с перекисью натрия. Плав растворяют в соляной кислоте, в раствор вводят нитрат железа и осаждают селен и теллур; совместно с железом, аммиаком для отделения от азотной кислоты и меди. Осадок растворяют в насыщенной бромом соляной кислоте и осаждают оба элемента восстановлением хлоридом олова (II). После этого проводят разделение селена и теллура осаждением сернистым ангидридом в две стадии* как описано ниже.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
СЕЛЕН
Селен обычно взвешивают в элементарном виде после осаждения сернистым ангидридом2 или гидроксиламином3 в сильнокислом растворе. Наиболее часто применяют сернистый ангидрид. Восстановление гидроксиламином особенно удобно при анализе растворов, содержащих только селен и теллур, преимущественно в их высшей валентности, а также в присутствии азотной или серной кислоты. Излагаемые ниже два метода служат для отделения селена от теллура.
Восстановление сернистой кислотой в солянокислом растворе
При определении селена восстановлением сернистой кислотой в солянокислом растворе селен должен находиться в четырехвалентном состоянии. Концентрация кислоты, в растворе должна быть не менее 3,4 н. (около 28 % по объему) для количественного осаждения селена и не менее 8,8н. (около 73 % по объему) для обеспечения полного отделения от теллура. Кроме того, концентрация Селена или теллура в растворе не должна превышать 0,25 а в 150 мл. Селен выделяется при значительно более высокбй кислотности, если в процессе осаждения раствор охлаждают. Осаждение предлагается4 осуществлять довольно быстрым введением значительного избытка сернистого ангидрида в холодный (15—20° С) раствор, причем аморфный осадок красного селена не рекомендуется переводить нагреванием в кристаллическую серо-черную модификацию. При медленном насыщении концентрированного солянокислого раствора сернистым ангидридом образуется монохлорид селена, в связи с летучестью которого могут иметь место некоторые потери селена. При нагревании раствора может улетучиться весь селен. Переведение красного селена в черную модификацию приводит к получению повышенных результатов вследствие окклюзии веществ, находящихся в растворе, в частности
1 W. R. S с h о е 11 е г, Analyst, 64, 318 (1939).
2 М. Shimose, Chem. News, 49, 26 (1884); Е. Divers, М. Shimose, там же, 51, 199 (1885); Е. Keller, J. Am. Chem. Soc., 19, 771 (1897); 22, 241 (1900); V. Lenher, С. H. К а о, там же, 47, 769 (1925).
s P. Jannash, M. Muller, Ber., 31, 2389 (1898).
4 V. Lenher, С. H. Kao, J. Am. Chem. Soc., 47, 2454 (1925).
390
Гл. XIX. Селен и теллур
теллура и воды. Наличие воды в осадке может вызвать окисление 1 селена в процессе высушивания, а удалить ее полностью при температуре ниже начала возгонки селена не удается. Осадок может быть загрязнен серебром, золотом, медью, висмутом, сурьмой и ртутью2. Серебро можно удалить в начальной стадии анализа в виде хлорида. Золото отделяют обработкой азотной кислотой, как указано на стр. 386. Осаждению сурьмы можно воспрепятствовать введением в раствор винной кислоты. Другие элементы целесообразно отделять до осаждения селена.
Ход определения. Смесь окислов, в которой содержится не более 0,25 г селена или теллура, растворяют в 100 мл холодной концентрированной соляной кислоты. При непрерывном перемешивании и следя за тем, чтобы температура не поднималась выше 30° С, в раствор вводят 50 мл холодной концентрированной соляной кислоты, насыщенной сернистым газом при обыкновенной температуре 3, и оставляют раствор стоять до полного выделения красного селена. Осадок отфильтровывают через взвешенный тигель Гуча, промывают сначала холодной концентрированной соляной кислотой, затем водой до исчезновения реакции на хлор, а под конец — спиртом и эфиром «. Сушат красный селен сначала 3—4 ч при 30—40° С для удаления эфира, а затем 2 ч при 120—130° С. Охлаждают в эксикаторе и взвешивают в виде Se.
Если требуется определить теллур, фильтрат и промывные воды (кислоту и воду) сохраняют.
Восстановление солянокислым гидроксиламином
Селен количественно осаждается и отделяется от- теллура восстановлением солянокислым гидроксиламином в солянокислой, виннокислой или лимоннокислой среде. Сернокислый гидроксиламин неприменим.
Ход определения. Для осаждения из солянокислого раствора 0,5 г смеси окислов или менее (содержание теллура не должно превышать 0,2 а) растворяют в 35—40 мл холодной соляной кислоты и разбавляют раствор до 100 мл. Наиболее приемлемой концентрацией соляной кислоты является 5 н. (около 42% по объему). К раствору прибавляют 10 мл 25 %-ного раствора солянокислого гидроксиламина й нагревают при 90° С в продолжение 4 ч. Выделившийся осадок элементарного селена переносят во взвешенный тигель Гуча, промывают водой, затем спиртом, сушат при 110° С и взвешивают. Если требуется определить теллур/ фильтрат сохраняют и поступают, как указано на стр. 392.
Для осаждения селена в виннокислой среде смесь окислов (содержащую не более 0,25 г ТеОг) растворяют в возможно меньшем количестве раствора едкого натра, прибавляют 25%-ный раствор винной кислоты
1 Согласно нашим опытам, превращение одной модификации селена в другую не отражается на ходе анализа, если в растворе содержится только один селен.
2 Если совместно с селеном осаждается теллур, то одновременно выпадает и палладий в виде теллурида.
3 Образование сернистого ангидрида в растворе путем прибавления сульфита нежелательно, так как при этом может выделиться сера. Применение других восстановителей, как, например, FeSO4, TiCl3, Н3РО3, SnCl2 или металлов, не дает хороших результатов вследствие большей или меньшей окклюзии их осадком.
1 Согласно J. S е a t h и F. Е. Beamish [Ind. Eng. Chem., Anal. Ed., 9, 373 (1937)], промывку осадка спиртом и эфиром можно опустить. Промытый водой селен (или теллур) высыхает в течение 45 мин, если сушить его в атмосфере СО2.
Методы определения
391
до растворения выделяющегося вначале осадка теллура, а затем еще 25 мл избытка. Раствор разбавляют до 100 мл, нагревают до 90° С, прибавляют 10 мл 25 %-ного раствора солянокислого гидроксиламина и в дальнейшем поступают так же, как при осаждении селена из солянокислого раствора.
Осаждение селена из лимоннокислого раствора проводят следующим образом. Смесь окислов растворяют в возможно меньшем количестве едкого натра, раствор разбавляют до 100 .мл, 5%-ным раствором лимонной кислоты, прибавляют 10 мл 25 %-ного раствора солянокислого гидроксиламина, нагревают до 90° С и заканчивают определение, как указано при осаждении селена из солянокислого раствора.
* В результате проверки 1 ряда методов определения селена были сделаны следующие выводы. При выпаривании солянокислых растворов, особенно досуха, потери селена неизбежны. Нельзя выпаривать досуха и соляно-азотнокислые растворы и азотнокислые растворы, но кипятить их можно в продолжение нескольких часов, не опасаясь потерь селена. Даже солянокислые растворы можно кипятить, применяя обратный холодильник или непрерывно добавляя перекись водорода. Присутствие натриевых солей, как это отмечено и авторами настоящей книги, не гарантирует от потерь. С другой стороны, выпаривание солянокислых или азотнокислых растворов с избыточным количеством серной кислоты до появления ее паров вполне допустимо. При очень малых количествах селена не следует, однако, слишком долго нагревать после появления паров серной кислоты.
Автор находит, что наиболее благоприятными условиями определения селена являются следующие. 20 г измельченной руды обрабатывают на холоду 125 мл азотной кислоты (пл. 1,4 г!см3, прибавляя ее небольшими порциями в продолжение 10 мин. Затем нагревают до кипения и по охлаждении вводят 60 мл серной кислоты (пл. 1,84 г!см3, причем происходит кристаллизация сернокислых солей. Выпаривают до появления паров серной кислоты, охлаждают, разбавляют 300 мл воды, к разогревшемуся при этом раствору прибавляют 20 мл соляной кислоты (пл. 1,19° С) и нагревают до растворения солей. Разбавив раствор до 400 мл, отфильтровывают нерастворившуюся породу, нагревают до 60° С, пропускают 10 мин SO2, прибавляют 3 мл 10 %-ного раствора гидразина и оставляют стоять до охлаждения. Затем отфильтровывают загрязненный другими элементами селен через небольшой плотный фильтр, который вместе с осадком переносят в коническую колбу и обрабатывают 10 .мл соляной и несколькими каплями азотной кислоты, нагревая при температуре водяной бани и взбалтывая до распадения фильтра. Разбавляют водой, фильтруют и обрабатывают раствор 2 мл раствора гидразина. По охлаждении до комнатной температуры отфильтровывают очищенный „селен через взвешенный фильтровальный тигель, промывают водой, сушат в течение получаса при 105° С и взвешивают. Доп. ред*
Объемные методы
Большинство объемных методов определения селена требует предварительного отделения теллура или внесения поправки на его содержание. Из объемных методов нужно упомянуть следующие: 1) иодометриче-
1 К. Bruckner, Z. anal. Chem., 94, 305 (1933),
392
Гл. XIX. Селен и теллур
ский, согласно которому селеновую кислоту восстанавливают соляной или бромистоводородной кислотой, освобождающийся галоген поглощают раствором иодида калия и выделяющийся при этом иод титруют арсенитом или тиосульфатом х; 2) восстановление селена иодистоводород-ной кислотой с последующим титрованием освободившегося иода тиосульфатом 1 2, 3) восстановление тиосульфатом, заключающееся в обработке солянокислого раствора селенистой кислоты титрованным раствором тиосульфата, избыток которого затем оттитровь^вают раствором иода 3; 4) окисление селенистой кислоты в сернокислом растворе титрованным раствором перманганата. Избыток перманганата затем определяют потенциометрическим титрованием сульфатом железа (II) или введением определенного количества щавелевой кислоты, избыток которой титруют перманганатом 4.
Теллур не влияет на определение селена потенциометрическим титрованием селенистой кислоты сульфатом титана (III). При этом не мешают также железо, медь и небольшие количества серной кислоты 5.
ТЕЛЛУР
Восстановление сернистой кислотой и гидразином
Теллур, подобно селену, обычно взвешивают в виде элемента. Восстановление, как правило, осуществляется в фильтрате, полученном после отделения селена. Восстановление одним сернистым ангидридом протекает слишком медленно и выделяющийся осадок настолько тонко раздроблен, что теллур легко окисляется, несмотря на промывку спиртом и эфиром.
1 W. Muthmann, J. Schafer, Вег., 26, 1008 (1893); J. Т. Norton Jr., ? Am. J. Sci., [4], 7, 287 (1899); 0» Pettersson, Z. anal. Chem., 12, 287 (1873);
F. A. G о о c h и сотрудники, Am. J. Sci., [3], 50, 254, 400, 402 (1895); [4], 1, 31 (1896). :
Об усовершенствованном приборе cm. G. C. Soth, J. E. Ricci, Ind. Eng. Chem., Anal. Ed., 12, 328 (1940). ;
2 Применение этого метода для определения селена в нержавеющих сталях марки ? 18—8 см. G. G. Marvin, W. С. Schu mb, Ind. Eng. Chem., Anal. Ed., 9, 109 v
(1936). A. E. P a v 1 i s h и R. W. S i 1 v e r t h о r n [J. Am. Ceram. Soc., 23, 116 J
(1940)] приводят метод определения селена в стекле, согласно которому аналивйруемую пробу сплавляют с карбонатом натрия, плав растворяют в разбавленной (1 : 1) серной а
кислоте, отгоняют бромид селена (IV) из серно-бромистоводороднокислого раствора <
и титруют иодид тиосульфатным методом. i
3 J. F. N orr is, Н. Fa у, Am. Chem. J., 18, 703(1896); 23, 119(1900); J. T. N or-l ton Jr., цит. выше W. C. Coleman, C. R. M c Croskey, Ind, Eng. Chem., Anal. Ed., 9, 431 (1937). A. L. С u r 1 и R. A. О s b о г n [J. Assoc. Offic. Agr. Chemists, 21, 228 (1938)] описывают метод определения селена в пищевых продуктах; этот; метод заключается в переводе селена в селенистую кислоту обработкой при нагревании HNO3— H2SO4—HgO и титровании растворами тиосульфата натрия и иода.
4 F. A. Gooch, С. Е. Clemons, Am. J. Sci., [3], 50, 51 (1895); W. T. Schrenk, B. L. В г о w n i n g, J. Am. Chem. Soc., 48, 2550 (1926); Z. L i t-t m a n, Chem. Ztg., 51, 323 (1927); L. Moser, там же, 720. Метод определения селенитов или селенистой кислоты в присутствии селенатов окислением селенит-ионов до селенат-ионов обработкой азотнокислого раствора избыточным количеством титрованного раствора бромата с последующим титрованием раствором арсенита в присутствии красителя прочного красного в качестве индикатора приводят D. F. Adams и L. I. Gilbertson [Ind. Eng. Chem., Anal. Ed., 14, 926 (1942)].
* H. H. Willard, F. Fenwick, J. Am. Chem. Soc., 45, 933 (1923)"
Методы, определения 393
Чтобы избежать этого, рекомендуют 1 для восстановления теллура вводить в раствор одновременно сернистый ангйдрид и солянокислый гидразин. При этом осаждение теллура происходит почти мгновенно, если концентрация его в растворе достаточно высока и концентрация соляной кислоты соответствует примерно 3 н. Этот метод применим к теллуритам и теллуратам.
Ход определения. Раствор, содержащий примерно 0,2 г теллура (VI) и (IV) в 50 мл 3 н. соляной кислоты (примерно 25 %-пая по объему) нагревают до кипения и прибавляют сначала 15 мл насыщенного раствора сернистого ангидрида, затем 10 мл 15%-ного раствора солянокислого гидразина и, наконец, еще 25 мл насыщенного раствора сернистого ангидрида. Продолжают кипятить, пока осадок не приобретет форму, в которой он легко отфильтровывается и промывается. Для этого требуется не более 5 мин. Дают отстояться, осадок отфильтровывают через взвешенный тигель Гуча и немедленно промывают горячей водой до удаления хлоридов. Затем во избежание окисления возможно быстрей отмывают воду спиртом и сушат 2 при 105° С.
При необходимости осадить теллур в солянокислом фильтрате после отделения селена концентрацию кислоты в растворе снижают упариванием на паровой или водяной бане при температуре ниже 100° С. Если селен осаждали гидроксиламином .в виннокислом или лимоннокислом растворе, фильтрат упаривают до 50 мл, затем обрабатывают 25 мл насыщенного раствора сернистого ангидрида, 10 мл концентрированной соляной кислоты, 15 мл 15 %-ного раствора солянокислого гидразина и в дальнейшем поступают так, как указано выше.
Объемные методы
Селен препятствует применению почти всех объемных методов определения теллура, за исключением приводимого ниже бихроматного метода. Из числа известных объемных методов можно указать следующие:
1) йодометрический, согласно которому теллур (IV) окисляют избыточным количеством титрованного раствора перманганата. Избыток перманганата определяют титрованием арсенитом после введения в раствор иодида. Другой йодометрический метод заключается в восстановлении теллура (VI) галогеноводородной кислотой, поглощении освобождающегося галогена раствором иодида и титровании выделяющегося иода арсенитом или тиосульфатом 3;
2) окисление четырехвалентного теллура избыточным количеством перманганата и дальше, как при определении селена 4;
1 V. Lenher, A. W. Hornberger, J. Am. Chem. Soc., 30, 387 (1908). О. E. С 1 a u d e r [Z. anal. Chem., 89, 270 (1932)] описывает другие способы восстановления теллура, приводящие к образованию кристаллических осадков, а также методы отделения Те от Sb, Bi, Pb, Си, Fe, Со и Ni.
2 См. сноску 4 на стр. 390.
3 J. F. Norris, Н. Fay, Am. Chem. J., 20, 278 (1898); F. A. Gooch, C. A. Peters, Am. J. Sci., [4], 8, 122 (1899); F. A. G о о c h, J. Howland, там же, [3], 48, 375 (1894).
4 В. В r a u n e r, J. Chem. Soc., 59, 238 (1891); F. A. G о о c h и сотрудники, Am. J. Sci., [3], 44, 301 (1892); [3], 50, 51 (1895); [4], 8, 122 (1899); W. T. S c h r e n k, B. L. Browning, цит. выше; Z. L i 11 m a n, цит. выше; L. Moser, цит. выше, 729.
394
Гл. XIX. Селен и теллур
« 3) восстановление избытком титрованного раствора хлорида олова (II), с последующим титрованием иодом х;
4) окисление избытком титрованного раствора бихромата и титрование бихроматом, после введения в раствор избыточного количества титрованного раствора сульфата железа (II), или же титрование тиосульфатом после добавления иодида 1 2.
* Колориметрический метод определения теллура и селена
Для колориметрического определения селена и теллура А. С. Шахов 3 применил реакцию, разработанную ранее В. К. Земмелем 4 и основанную на восстановлении этих элементов до элементарного состояния раствором хлорида олова (II) в кислой среде. Растворы гидрозоля селена окрашены в оранжевый, а теллура — в темнокоричневый цвет. В. К. Земмель для стабилизации коллоидных растворов селена и теллура применял гуммиарабик. А. С. Шахов нашел, что эти растворы достаточно устойчивы, поэтому нет необходимости вводить в них защитный коллоид. Им установлено, что коллоидные растворы селена подчиняются закону Ламберта — Бера1 в интервале концентраций от 0,05 до 0,5 мг, а теллура от 0,025 мг до 0,5 .на в 50 мл. Определение селена и теллура по методу А. С. Шахова сводится к следующему. К раствору, содержащему 0,05—0,5 мг селена или 0,025—0,5 мг теллура, прибавляют 20 мл разбавленной (1 : 1) соля-' ной кислоты и 1, мл 10%-ного раствора SnCl2 • 2Н2О в соляной кислоте (пл. 1,14 г!см3) (реакция восстановления идет на холоду). Интенсивность появляющейся при этом окраски измеряют в фотоколориметре с голубым светофильтром.
Погрешность определения в среднем составляет ~3%. Доп. перев*
1 В. В г a u п е г, I. Chem. Soc., 59, 58 (1891).
2 В. Brauner, цит. выше, 62; V. Lenher, Н. F. W a k е f i е 1 d, J. Am. Chem. Soc., 45, 1423 (1923); W.T. Schrenk, B. L. Browning, цит. выше, 48, 139 (1926). Последние используют метод Lenher’a для потенциометрического титрования теллура, которое применимо в присутствии железа (III) и небольших количеств селена и меди. Кроме того, см. Z. Li tt man, а также L. Moser, цит. выше. Обзор объемных методов приведен в статье L. Moser, R. М i k s с h, Monatsh, 44, 335 (1923). ' '
3 А. С. III а х о в, Зав. лаб., 11, 893 (1945).
4 В. К. Земмель, Зав. лаб., 5, 1433 (1936).
БЛАГОРОДНЫЕ МЕТАЛЛЫ
ЗОЛОТО, ОСМИЙ, РУТЕНИЙ, ПЛАТИНА, ПАЛЛАДИЙ, РОДИЙ И ИРИДИЙ
Глава XX
ПЛАТИНОВЫЕ МЕТАЛЛЫ
Шесть платиновых металлов — осмий, рутений, платина, палладий, родий и иридий — встречаются в природе главным образом в металлическом состоянии в виде многочисленных сплавов, содержащих обычно большинство (если не все) из этих шести металлов совместно с золотом, а также железом, медью и некоторыми другими неблагородными металлами’ например никелем и кобальтом. Эти сплавы обычно ассоциируются друг с другом и нередко с самородным золотом. Наиболее часто встречаются сплавы, в которых преобладает платина. В следующих по распространенности сплавах основными компонентами являются осмий и иридий, так называемые осмистый иридий и иридистый осмий. Наиболее редко встречается рутений, содержащийся главным образом в сплавах иридия и осмия. Осмистый иридий и иридистый осмий, как правило, находятся совместно с платиновыми сплавами, но иногда встречаются и самостоятельно. Встречаются также более или менее чистый самородный иридий, сплав его с платиной и относительно чистый палладий. Известен самородный сплав золота с палладием, называемый пор-п е з и т о м. Найдены также сплавы золота с родием и палладия с ртутью ( п о т а р и т).
Платиновые металлы встречаются в природе 1 также в виде соединений с мышьяком, серой, сурьмой и, возможно, с селеном. Считают, что в рудах Садбери, имеющих промышленное значение, платина содержится в виде арсенида PtAs2, известного под названием сперрилита, а палладий, сопровождающий платину в этих рудах почти в равных
1 Подробнее о платиновых минералах и условиях их залегания см. Platinum and Allied Metals, published by the Imperial Institute, London, 1936.
396
Гл. XX. Платиновые металлы
количествах, как предполагают, находится в виде селенида. Известно два природных сульфида платины — куперит PtS и браггит (Pt, Pd, Ni) S. Стибиопалладинит является природным соединением палладия и сурьмы. Лаурит представляет собой редко встречающийся сульфид рутения RuS2.
Основным источником получения платиновых металлов служат самородные сплавы, залегающие главным образом в виде мелких зерен с небольшим количеством более крупных частиц и случайными самородками. Эти сплавы находятся во вторичных отложениях, образовавшихся в результате разрушения основных вулканических пород. Такие месторождения широко распространены, но лишь немногие из них имеют промышленное значение. Наиболее богатые залежи платиновых металлов находятся на Урале и в Колумбии. Сравнительно недавно к числу стран, производящих платину, причислены Соединенные Штаты Америки благодаря россыпям, обнаруженным на Аляске. Крупные месторождения платины в виде металла и сульфидно-мышьяковистых минералов находятся в Южной Африке. Некоторые из первичных отлоЖений в Южной Африке достаточно богаты для их разработки с целью добычи платиновых металлов.
В последние годы Канада заняла одно из первых мест по производству платиновых металлов благодаря присутствию их в канадских сульфидных никелевых и медных рудах, из которых они извлекаются в процессе электролитического рафинирования. Организация производства платиновых металлов в Канаде сделала рутений и родий значительно более доступными, чем они были до сего времени. Относительно небольшие количества платиновых металлов извлекают из сульфидных медных руд и в других частях света.
Сведения о распространении золота в природе здесь не приводятся, поскольку они общеизвестны и их нетрудно найти. Не дается также описание пробирного анализа руд на золото, а приводятся лишь «мокрые» методы его отделения и определения.
ОБЩИЕ ЗАМЕЧАНИЯ
Обычные содержащие платину материалы
Из материалов, содержащих платиновые металлы, с которыми приходится сталкиваться аналитику, наибольшее значение имеют два минерала: зернистая платина и осмистый иридий. Природные продукты, как правило, подвергаются механическому обогащению, в результате которого получаются концентраты с небольшой частью посторонних минералов, таких, как кварц, ильменит, хромит и магнетит. Эти неметаллические компоненты обычно известны под собирательным названием «песка». Такие концентраты можно анализировать непосредственно. Первичные же руды, содержащие малые количества платиновых металлов, обычно предварительно концентрируют плавкой в тигле с глетом и т. п., как это делается при анализе продуктов, содержащих золото и серебро. ,
Из числа продуктов производства и рафинировки обычно анализируются более или менее очищенные металлы, главным образом платина и палладий, в форме губки или компактного металла. Помимо того, при
Общие замечания
397
ходится анализировать платиново-иридиевые сплавы и платино-родиевые сплавы, применяемые в электротехнике, ювелирном деле и для изготовления лабораторной посуды и инструментов для научных работ; золотые сплавы, применяемые в зубоврачебной технике, содержащие платину или палладий либо обй эти металла вместе, и, наконец, различные сплавы для ювелирных изделий, в которых преобладает платина (иногда палладий), но могут содержаться и другие благородные металлы. Анализируются также биметаллический лом, состоящий из платиновых сплавов, и предметы, покрытые родием или другими платиновыми металлами. К этому следует добавить различные побочные продукты, остатки производства и рафинировки, а также платину, извлекаемую из каталитических масс и остатков осмистого иридия, и синтетические сплавы, из которых изготовляются перья к автоматическим ручкам.
Поведение платины в ходе анализа
В аналитической практике часто применяют платиновую посуду, и поэтому очень важно учитывать поведение платины при групповых разделениях элементов. Содержанием примесей других платиновых металлов в посуде можно пренебречь либо полагать, что они ведут себя аналогично платине. Платина осаждается сероводородом из кислых растворов, причем допускается изменение концентрации кислоты в растворе в весьма широких пределах. В растворе сульфида аммония сульфид платины растворяется не полностью, и реакция эта усложняется присутствием аммиака, с которым платина, подобно кобальту, образует различно растворимые аммиакаты. Эти соединения обычно достаточно устойчивы, и свойства их растворов, кислых или щелочных, заметно отличаются от свойств растворов хлорида платины, или, точнее, хлороплатината. Так, например, аммиакаты платины реагируют с сероводородом настолько медленно, что из их растворов практически невозможно выделить платину количественно. Из растворов аммиакатов платина не осаждается также прй восстановлении водородом или органическими восстановителями, хотя слабое взаимодействие с этими реагентами имеет место. Отсюда следует, что если в процессе анализа проводилась обработка аммиаком, то при каждой последующей операции осаждения может происходить частичное выделение платины в осадок. Это происходит уже при самой обработке раствора аммиаком, когда основная масса платины, если введен большой избыток аммиака, спустя непродолжительное время переходит в раствор, а часть ее задерживается в осадке других металлов. Таким образом, если платина в растворе находится в виде аммиакатов, то ее необходимо перевести в другие соединения.
Одним из способов разложения аммиакатов является обработка серной кислотой с небольшим количеством азотной кислоты и выпаривание раствора до появления обильных паров серной кислоты. Нагревание при температуре кипения серной кислоты следует продолжать 10— 15 мин. При необходимости избыток серной кислоты можно удалить выпариванием. После такой обработки обычно необходимо прибавить немного царской водки для растворения образовавшейся металлической платины. Более надежно раствор аммиакатов (не добавляя серной кислоты) выпарить досуха, сухой остаток осторожно прокалить и затем, для переведения платины в раствор, обработать небольшим количеством
398
Гл. XX. Платиновые металлы
царской водки. В присутствии хлоридов щелочных металлов часть платины может, не восстанавливаясь до металла, непосредственно превратиться в хлороплатинат.
Из раствора хлоридов аммиак может частично осадить платину в виде относительно малорастворимого хлороплатината аммония. Однако при анализе любых материалов, за исключением платиновых, содержание платины в растворе редко превышает растворимость хлороплатината аммония.
Платина реагирует с избытком едкого натра с образованием растворимых платинатов. При этой реакции, однако, образуются промежуточные нерастворимые соединения платины, которые могут задержаться в осадке, если одновременно происходит осаждение других элементов.
Необходимо иметь ₽ виду, что при введении в раствор некоторых органических реагентов или других восстановителей платина может выделиться из раствора в виде металла. Значительно труднее, однако, заметить действие таких восстановителей, которые восстанавливают платину (IV) не до металла, а до двухвалентного состояния. Эта реакция не легко выявляется в разбавленных растворах. Но эта же реакция может явиться причиной ошибки в объемном анализе вследствие повышенного расхода восстановителя, если непосредственно по нему вычисляется содержание определяемого элемента в растворе, либо вследствие окисления при последующем титровании окислителем образовавшейся в процессе восстановления платины (II). Это' наблюдается, например, при титровании некоторых элементов окислителем после предварительного восстановления их сернистым ангидридом. >. В этом случае, кроме того, происходит вторичная реакция между платиной (II) и сернистым ангидридом, в результате которой образуются комплексные сульфиты. Эти соединения устойчивы и лишь медленно превращаются в хлориды после удаления избытка сернистого ангидрида из раствора. При оксидиметри-Ческом титровании таких растворов дополнительные ошибки возможны за счет окисления сернистого ангидрида, медленно выделяющегося из этих сульфитов, или за счет непосредственного окисления группы SO3 в комплексном соединении платины.
Все изложенное ясно указывает на склонность платины к образованию комплексных соединений в растворах. Многие из этих соединений настолько устойчивы, что очень слабо или вовсе не обнаруживают свойств, характерных для «платины», которые в действительности являются свойствами иона платины или хлороплатината [PtCl6 ]2-. Некоторые из комплексных соединений платины можно перевести в хлороплатинаты или другие соединения, но эти процессы крайне продолжительны, в отличие от большинства, используемых в аналитической химии реакций, которые протекают почти мгновенно. Поэтому в тех случаях, когда в процессе анализа для сплавления или проведения других операций применяется платиновая посуда и платина таким образом может быть введена в анализируемый раствор, целесообразно предварительно отделять ее осаждением сероводородом из солянокислого или сернокислого раствора.
В случае замены платиновой посуды соответствующими изделиями из золота или из сплавов золота с палладием или платиной необходимо учитывать поведение всех этих металлов, так как они могут быть введены в анализируемые растворы. Золото и палладий количественно осаждаются сероводородом из кислых растворов. Восстановителями золото
Общие замечания
399
осаждается из раствора значительно легче, чем платина. Оно частично выделяется даже при выпаривании его раствора досуха на паровой бане х. Аммиаком золото осаждается, образуя гремучее золото. При введении избыточного количества едкого натра в раствор золото не осаждается, но выделяющиеся при этом другие элементы частично его захватывают. Поведение палладия в ходе анализа во многом сходно с поведением платины. Палладий имеет меньшую склонность к образованию устойчивых комплексных соединений. В присутствии избытка аммиака образуется растворимый аммиакат палладия, но если происходит осаждение других элементов, то часть палладия удерживается этим осадком. Едкий натр осаждает гидроокись палладия. Палладий, подобно платине, реагирует с сернистой кислотой с образованием растворимых простых или двойных сульфитов. Он осаждается иодидом и диметилглиоксимом. Последний при нагревании осаждает также частично и платину. Золото осаждается диметилглиоксимом в виде металла даже из холодных растворов.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ И СПЛАВОВ
Отдельно взятые кислоты действуют только на палладий и осмий. Азотная кислота медленно растворяет палладий и действует на осмий только в том случае, если он тонко измельчен. В таком виде осмий растворяется также в концентрированной серной кислоте и в царской водке, но медленнее, чем в дымящей азотной кислоте. Палладий и золото легко растворяются в царской водке, а платина несколько труднее. Царская водка слабо действует на родий, иридий и рутений (а также на компактный осмий), но платиновые сплавы, содержащие небольшие количества этих металлов, растворяются полностью. Указанные металлы заметно замедляют растворение платины, а 30%-ный сплав иридия с платиной, например, практически нерастворим в царской водке.
Лучшим способом растворения сплавов, в которых преобладает содержание платины, является обработка царской водкой при нагревании, если пренебречь продолжительностью этой операции. Необходимо при этом иметь в виду, что большая часть осмия теряется вследствие летучести четырехокиси осмия, если прибор, в котором проводится растворение, не снабжен приемником для конденсации или поглощения этого соединения. Для обработки таких сплавов применяют смесь, состоящую из четырех объемов соляной кислоты, одного объема азотной кислоты и одного объема воды. Растворение платиновых сплавов в царской водке часто идет настолько медленно, что целесообразно предварительно увеличить поверхность образца расплющиванием или прокаткой. Для растворения губчатых золота, платины и палладия или черней этих металлов применяют разбавленную царскую водку. В процессе разложения платиновых минералов царской водкой происходит отделение сплавов, с преобладающим
1 При быстром выпаривании раствора золота в царской водке досуха (максимальная температура 120° С) или при добавлении к такому раствору серной кислоты и выпаривании до появления ее густых паров золото частично улетучивается. Так, например, в результате выпаривания раствора, содержавшего 0,1 г золота, в стеклянном дистилляционном приборе в первом случае в дистилляте было найдено 1,5 мг золота, а во втором случае 2,5 мг. Потерь золота не наблюдалось, когда раствор такого же количества золота в царской водке обрабатывался 15 мг 70%-ной хлорной кислоты, а затем выпаривался до появления обильных паров этой кислоты (максимальная температура 205° С).
400
Гл. XX. Платановые металлы
» I
содержанием платины, а также самородных палладия и золота от осмиево-иридистых сплавов, которые в царской водке не растворяются.
Такие материалы, как осмистый иридий, или другие, содержащие рутений, осмий, родий и иридий не в виде сплавов с большим избытком платины или палладия, лучше всего разлагаются сплавлением со щелочными окислителями. Если металл не измельчен тонко, он разлагается плавнями крайне медленно. Крупнозернистые материалы, например природный осмистый иридий, могут быть измельчены сплавлением с десятикратным количеством цинка. Охлажденный слиток, для удаления избыточного цинка, обрабатывают соляной кислотой. При сплавлении металлические зерна осмистОго иридия переходят в порошкообразную смесь сплавов цинка с платиновыми металлами.
Сплавление проводят следующим образом. Смесь анализируемого материала с цинком покрывают слоем хлорида цинка и сплавляют в кварцевом тигле при красном калении не менее 1 ч. Плав перемешивают графитовой палочкой, следя за тем, чтобы металл не прилипал к ней. Для сплавления применяют цинк, практически свободный от свинца, так как при обработке плава соляной кислотой большая часть введенного с цинком свинца остается в нерастворимом остатке. Перед сплавлением со щелочью тонко раздробленный остаток, содержащий благородные 'металлы, промывают и сушат, но не прокаливают \ В качестве плавня применяют перекись бария, перекись натрия либо едкий натр, к которому примешивают 25% перекиси натрия или нитрата натрия 1 2. Сплавление с перекисью натрия или со смесью едкогр натра и перекиси натрия проводят в железных, никелевых или, лучше, в серебряных чашках. Для сплавления же со смесью едкой щелочи и нитрата лучше пользоваться золотой посудой, так как эти реагенты меньше действуют на золото, чем на серебро, и, кроме того, при сплавлении в золотой посуде плав не имеет склонности всползать вверх по стенке чашки.
Для разложения пробы можно применять также обработку перекисью натрия в никелевом или серебряном тигле методом «взрыва» 3. Этот способ имеет то преимущество, что тигель мало корродирует, и поэтому в раствор попадают лишь небольшие количества постороннего металла, но при этом расходуется значительно больше реактива.
Для обычного сплавления на 1 часть анализируемого материала требуется не менее 4 частей плавня. Массу, получаемую в результате сплавления с едкой щелочью или перекисью натрия, по охлаждении выщелачивают водой. Соединения металлов, образующиеся при сплавлении, частично растворяются в воде, а частично остаются в нерастворенном остатке. Осмий и рутений значительно более склонны к образованию растворимых в воде соединений, чем иридий 4, но степень, в какой эти металлй образуют водорастворимые соединения, зависит от температуры сплавления и, возможно, от других условий. По данным более старой литературы, различ
1 Такого типа материалы при прокаливании иногда взрываются.
2 В более старой литературе имеются указания, что едкое кали и нитрат калия действуют сильнее, чем соответствующие соединения натрия. Однако применение их для сплавления имеет то неудобство, что при подкислении раствора образуются относительно мало растворимые соли калия.
3 W. F. Muehlbeijg, Ind. Eng. Chem., 17, 690 (1925). См. стр. 912.
* Сомнительно, чтобы иридий в этих условиях находился в виде истинного раствора, по крайней мере в устойчивой при нагревании форме.
Разложение минералов и сплавов
401
ная способность давать водорастворимые соединения обычно использовалась для отделения осмия и рутения от иридия. В настоящее время, когда имеются более усовершенствованные методы, нет надобности останавливаться на этом способе, который не дает количественного разделения указанных металлов. После полного разложения раствор кипятят для разрушения перекиси водорода (если для сплавления применялась перекись натрия), затем сильно подкисляют соляной кислотой и нагревают до полного превращения гидроокисей или солей оксикислот в хлоросоединения. В присутствии осмия кислота вводится после перенесения разложенного плава в дистилляционную колбу, отводная трубка которой соединена с приемником для улавливания четырехокиси осмия, что особенно необходимо, если при сплавлении вводился нитрат.
Применение для разложения пробы перекиси бария вместо едкого натра перекиси натрия имеет известные преимущества. В этом случае происходит скорее спекание, чем сплавление, что вызывает меньшую коррозию тигля, но требует более тщательного перемешивания анализируемой пробы с плавнем, чтобы обеспечить максимальный переход металлов в растворимые соединения. Кроме того, при обработке спекшейся массы водой соединения платиновых металлов остаются в нерастворимом осадке, тогда как избыток перекиси бария, перешедшей в гидроокись, растворяется и может быть отделен фильтрованием. Фильтрат, прежде чем его отбросить, следует проверить на содержание платиновых металлов (подкислением соляной кислотой, нагреванием «и осаждением сероводородом). В результате обработки нерастворимого в виде остатка соляной или бромистоводородной кислотой при нагревании платиновые металлы, находящиеся частично в виде бариевых солей оксикислот, превращаются в растворимые хлоро- или бромосоединения. После такой обработки всегда, остается небольшой нерастворимый остаток, состоящий из неразложенных платиновых металлов и сульфата бария.
Продажная перекись бария, как правило, содержит примесь сульфата и, кроме того, может быть впоследствии загрязнена соединениями серы, если для нагревания применяется газовое пламя. Сера может попасть и из электрических печей, которые ранее использовались для обработки продуктов, содержащих серу. Нерастворимый остаток прокаливают и снова спекают с перекисью бария. Сульфат бария, если требуется, можно удалить сначала прокаливанием остатка в фарфоровом тигле в атмосфере водорода и последующим выщелачиванием образовавшегося сульфида (или сульфита) бария разбавленной соляной кислотой. В случае применения в качестве «плавня» перекиси бария нерастворимый остаток, остающийся после выщелачивания водой спекшейся массы, обрабатывают так же, как раствор и остаток, получающийся в результате сплавления материала со щелочами. При этом следует учесть, что совместно с платиновыми металлами остаются значительные количества бария, которые могут оказать влияние на ход последующих операций. Барий можно отделить осаждением из подкисленного раствора точно рассчитанным количеством серной кислоты. Введения в раствор избытка серной кислоты следует избегать, так как она влияет на некоторые используемые в дальнейшем реакции.
Независимо от характера применяемого плавня однократным сплавлением обычно не удается полностью перевести платиновые металлы в растворимые соединения. Степень разложения анализируемого материала зависит от его природы и измельчения, а также от температуры
26 Заказ 522.
402
Гл. XX. Платиновые металлы.
и продолжительности сплавления. Обычно проба разлагается тем полнее, чем выше температура и чем дольше проводится нагревание. В случае применения для разложения перекиси бария тигель со смесью анализируемого материала и реагента можно оставлять в печи при 750—800° С в течение нескольких часов. При сплавлении же с едким натром или перекисью натрия расплав нельзя оставлять такое продолжительное время без наблюдения. В некоторых случаях при сплавлении со щелочами при температуре красного каления полный перевод металлов в растворимую форму по существу заканчивается в пределах одного часа. Имеются указания 1 на то, что получаемые после, купелирования родиево-иридиевые остатки полностью разлагаются в течение 10 мин сплавления их с перекисью натрия при темно красном калении. Характер плавня и условия обработки, требующиеся для разложения различного типа материалов, необходимо устанавливать опытным путем.
Оксисоединения платиновых металлов, образующиеся в результате сплавления с щелочными окислительными плавнями, не всегда достаточно легко растворяются в соляной кислоте, особенно если преобладает содержание иридия. Эти соединения значительно легче разлагаются бромистоводородной кислотой, применение которой в отдельных случаях может быть рекомендовано.
Материалы, богатые родием, иногда весьма устойчивы по отношению к щелочным плавням. В таких случаях неразложившийся остаток можно обработать методом, изложенным ниже. •
Платиновые металлы в виде губки можно перевести в раствор тщательным растиранием с хлоридом натрия и нагреванием смеси в атмосфере хлора до начала плавления. При этом образуются растворимые хлоросоли платиновых металлов с натрием. Этот метод в особенности пригоден для переведения в раствор богатых родием материалов. Он может быть с успе- , хом использован также при щелочном сплавлении для разложения роди-ево-иридиевых остатков. Следует вводить двукратное количество хлорида натрия по отношению к теоретически необходимому для образования двойных солей. Таким путем редко достигается полное разложение и метод мало пригоден для анализа необработанных продуктов, так как почти всегда происходят потери некоторых компонентов за счет улетучивания, если не приняты соответствующие меры для их улавливания.
Рутений и осмий растворимы в растворах гипохлоритов щелочных металлов. Основанный на этом свойстве метод рекомендован 2 3 для переведения в раствор металлических порошков, содержащих рутений. В растворе должна находиться свободная щелочь для предотвращения окисления металла до летучей четырехокиси. Относительно чистый рутений растворяется в гипохлорите довольно быстро, но извлечение рутения из смесей протекает медленно и не количественно, если нерастворимые металлы, как иридий, тесно с ним смешаны, что имеет место, например, при совместном выделении из раствора двух или нескольких металлов в виде губки или при прокаливании смеси солей.
I1 F. Е. В eamish, М. Scott, Ind. Eng. Chem., Anal. Ed., 9, 460 (1937).
2 J. L. Howe, F. N. Mercer, J. Am. Chem. Soc., 47 , 2926 (1925).
3 C. L. Gordon, J. Research NBS, 30, 107 (1943); E. W i c h e r s, W. G. S chi ech t, C. L. G о r d on, там же, 33, 363 (1944); E. W i c h e r s, W. G. Schlech t,C. L. Gor do»n, там же, 451; C. L. Gordon, W.G. Schlecht, E. Wichers, там же, 457.
Разложение минералов и сплавов
403
Следует упомянуть ряд работ, разрешивших наиболее трудную в аналитической химий платиновых металлов задачу 3. Было обнаружено, что трудно разлагающиеся платиновые продукты можно полностью перевести в раствор нагреванием их при высокой температуре в запаянной стеклянной трубке с соляной кислотой, содержащей небольшие количества подходящего окислителя. Скорость разложения материала в большой степени зависит от состава растворителя, а также от температуры, при которой проводится реакция. Удовлетворительные результаты получаются при применении смеси, состоящей из 20 объемов концентрированной соляной кислоты и одного объема дымящей азотной кислоты или эквивалентного количества хлората натрия, хлорной кислоты или хлора. Такая смесь при температуре около 300° С быстро растворяет осмистый иридий и даже металлический иридий. Образцы осмистого иридия средней крупности достаточно держать в печи при этой температуре в продолжение 24 ч. Для разложения более крупнозернистых или исключительно стойких материалов может потребоваться более продолжительное нагревание.
Большинство материалов удовлетворительно разлагается обыкновенной концентрированной соляной кислотой (около 36% НС1 по массе), но для переведения в раствор необычно крупных частиц и особо стойких продуктов следует применять более концентрированную кислоту. Концентрацию соляной кислоты можно повысить (до 50% НС1 по массе) дистилляцией обычной кислоты, если охлаждать холодильник очень холодной водой, а приемник держать погруженным в лед. Еще более концентрированную кислоту можно получить конденсацией сухого хлористого водорода на поверхности льда. Разложение пробы следует проводить таким количеством кислоты, которое обеспечивает наиболее благоприятное соотношение между кислотой и окислителем, а именно: 20 г хлористого водорода на 1 г хлора. Для растворения 1 г пробы требуется не менее 4,2 г хлористого водорода, что соответствует 10 мл обычной концентрированной соляной кислоты. При добавлении большого количества кислоты, до 20 мл, растворение ускоряется. Растворение иридия при 250—300° С протекает лучше всего при введении на каждый миллилитр емкости трубки 0,025 г хлора или же соответствующего количества другого окислителя, действие которого обусловлено выделением хлора.
Если реакция проводится при температуре выше 250° С, для окисления наиболее целесообразно пользоваться хлорной кислотой, содержащей 70% НС1О4 (по массе) х. При более низких температурах хлорная кислота реагирует с соляной кислотой слишком медленно, и в таких случаях в качестве окислителя применяют азотную кислоту или хромат калия, если следует избегать образования нитрозосоединений, например HgtRuC^NO], На каждый грамм пробы, состоящей преимущественно из иридия, осмия или платины, по стехиометрическому расчету требуется 0,22 мл хлорной кислоты (70% НСЮ4 по массе). Такому количеству хлорной кислоты эквивалентны 0,27 мл обычной азотной кислоты (70% HNOg по массе) или 0,19 мл дымящей азотной кислоты (91 % HNOg по массе). Этому соответствует также 0,37 г хлората натрия или 0,74 г хлора. Если в анализируемой пробе содержатся значительные количества родия, рутения и палладия, следует вводить большие количества окислителя из расчета на металл
1 Следует помнить, что если в анализируемом материале присутствуют органические вещества, то в результате взаимодействия их с хлорной кислотой может произойти взрыв.
21*
404
Гл. XX. Платиновые металлы
с наименьшим эквивалентным весом. Для окисления 1 г чистого родия требуется 1,05 г хлора.
Необходимо отметить, что в случае применения хлорной кислоты некоторые количества ее могут остаться неразложенными, если раствор нагревают при температуре значительно ниже 300° С в продолжение не более 24 ч. Наличие хлорной кислоты в растворе может оказаться нежелательным в дальнейшем ходе анализа. Так, например, в процессе упаривания раствора в ее присутствии могут иметь место потери осмия и рутения вследствие летучести их четырехокисей. При анализе иридиево-платиновых сплавов, растворение которых достигается при 100—150° С, применение хлорной кислоты не рекомендуется. В этом случае для окисления на каждый грамм металла вводят 27 мл обычной азотной кислоты или 0,37 г хлората натрия.
Температура, при которой проводится разложение, обычно не должна быть выше, чем это требуется для полного растворения материала в продолжение приемлемого для анализа промежутка времени, например 18— 24 ч. Температура 300° С требуется для растворения лишь особо устойчивых материалов, если величина их частиц не чрезмерно велика. Рассматривая содержимое трубок после некоторого времени нагревания, устанавливают, следует ли повысить температуру, чтобы достигнуть полного разложения пробы в течение требуемого времени. Для разложения менее устойчивых сплавов, состоящих в основном из платины, достаточно нагревания при 110° С в течение ночи. В этом случае предохранительного внешнего давления для стеклянной трубки не требуется.
Трубка должна быть небольшого размера для того, что применяемое для растворения материала количество кислоты заняло половину или две трети ее емкости. Для растворения 1,0—1,5 г пробы вполне пригодна толстостенная трубка (2,5 мм) из стекла пайрекс с внутренним диаметром 15 мм и длиной около 20 см со стержнем, внутренний диаметр которого равен 4 мм, а толщина стенок 2 мм. Для разложения больших навесок целесообразнее увеличивать длину трубки, чем ее диаметр, или же распределять анализируемый материал в нескольких трубках. Для растворения 100 мг пробы рекомендуют пользоваться толстостенной трубкой из стекла пайрекс длиной около 20 см с внутренним диаметром 4 мм, которую заполняют кислотой до половины ее объема. Для микроанализа следует пользоваться капиллярными трубками с диаметром капилляра 1 мм и толщиной стенок 1 мм из стекла пайрекс или кварца. Установлено, что при 300° С давление внутри трубки, заполненной до половины своего объема, равно 240— 270 атм. Толстостенную трубку с внутренним диаметром 4 мм и длиной 20 см можно без опасения нагревать до 300° С. Трубки большего размера помещают в стальную бомбу, в которой находится взвешенное количество твердой углекислоты, необходимое для уравновешивания давления.
Запаянную трубку с содержимым заключают в стальной кожух, снабженный колпачком и прокладкой. В этот кожух предварительно вводят достаточное для нейтрализации всей содержащейся в трубке кислоты количество карбоната кальция на тот случай, если трубка неожиданно разобьется. Затем прибавляют твердую углекислоту в количестве, необходимом для создания давления, которое требуется при той температуре, при которой проводится реакция. Создающееся при нагревании кислых смесей давление примерно соответствует давлению пара применяемой соляной кислоты, при условии, что соотношение между кислотой и окисли
Разложение минералов и сплавов
405
телем равно по крайней мере 10 : 1. Отсюда можно полагать, что при применении соляной кислоты, содержащей 23; 37 и 48% НС1 (по массе) в количестве немного меньшем, чем 0,5 мл на каждый миллилитр емкости трубки, при 300° С создается давление по крайней мере 136; 247 и 322 атм соответственно. Такое давление при 300° С обеспечивает твердая углекислота в количестве, соответствующем 0,14; 0,24 и 0,31 г СО2 в 1 мл свободного пространства, окружающего запаянную трубку. После введения СО2 быстро навинчивают колпачок и кожух погружают в воду для испытания на герметичность. Кожух несколько раз опрокидывают для равномерного размещения анализируемого материала в трубке, затем помещают в горизонтальном положении в печи и нагревают до нужной температуры. По окончании реакции кожух охлаждают до комнатной температуры и трубку вынимают.
При загрузке трубки сначала вводят анализируемый материал через открытый конец. При использовании соляной кислоты, особенно повышенной концентрации, трубку следует погрузить в лед. Окислитель вводят в последнюю очередь. Если применяют хлорат натрия, трубку следует охладить до такой степени, чтобы заморозить соляную кислоту и сохранить ее в таком состоянии до тех пор, пока трубка не будет запаяна. Для охлаждения трубки можно пользоваться твердой углекислотой или, лучше, массой, состоящей из твердой углекислоты и смеси одной части хлороформа и одной части четыреххлористого углерода.
Для запаивания сужают и оттягивают небольшую HacTb трубки, а затем оттянутый конец обрабатывают обычным путем. Можно также запла-вить отверстие узкой части трубки нагреванием на окислительном газовом пламени и затем нагреть трубку, чтобы под давлением газов изнутри трубки конец ее принял округленную форму. Таким путем получается более прочное запаивание, чем оттягиванием трубки, если в последнем случае ему не придают полусферическую форму. Во всех случаях необходимо следить за тем, чтобы не оставалось капиллярного отверстия, через которое в трубку может проникнуть газ, применяемый для уравновешивания давления. Этот газ может не выйти из трубки, если отверстие закупорится продуктами реакции, и, таким образом, в трубке создастся чрезмерно высокое давление, вследствие чего она может разорваться во время отрезания запаянного конца, когда наружное давление не будет уравновешивать давление внутри. Как бы трубка ни была запаяна, ее следует испытать на отсутствие в ней капиллярного отверстия, опрокинув тщательно вытертую трубку над кусочком лакмусовой бумаги.
По окончании разложения материала нижний конец трубки погружают й стакан с теплой водой. При этом пары, конденсируясь в верхней части трубки, вскоре смывают раствор с ее стенок. Прежде чем открыть трубку, целесообразно охладить ее охлаждающей смесью или твердой углекислотой для конденсации оставшегося хлора, а также для уменьшения давления азота, который находится в трубке, когда в качестве окислителя применяется азотная кислота. Предварительное охлаждение трубки служит также для предотвращения потерь летучих продуктов, которые могут образоваться в процессе разложения материала. По охлаждении отбивают конец трубки и опрокидывают ее в стакан, содержащий воду или какую-нибудь другую жидкость для разбавления концентрированного анализируемого раствора.
Необходимо следить за скоростью таяния массы в открытой трубке, содержащей избыток окислителя, особенно хлора, который был заморожен.
406
Гл. XX. Платиновые металлы
Если верхний запаянный конец опрокинутой трубки нагревается раньше нижнего, выделяющийся хлор может выбросить находящуюся под ним все еще замороженную смесь. Это можно предотвратить, если направить струю теплой дистиллированной воды из промывалки через отводную капиллярную U-образную трубку на нижнюю часть застывшей массы. При этом основание столба, этой массы в трубке расплавляется, хлор начинает выделяться с равномерной скоростью и исчезает причина, вызывавшая увеличение давления.
Если в результате разложения в трубке не появляются другие летучие продукты, помимо примененных для разложения материала, ее можно открыть следующим образом. Конец трубки нагревают на слабом пламени горелки, пока небольшая часть поверхности стекла не размягчится и за счет разности давления внутри трубки и внешнего не выдуется отверстие. При продолжительном нагревании отверстие может снова заплавиться. Это явление может повториться и самопроизвольно. Поэтому, пока конец трубки мягок, пламя сначала держат против стекла под заплавленной частью трубки, причем выдувается большой тонкий пузырь. В результате нагревания верхней части пузыря он лопается и оплавляется вокруг конца трубки, образуя раструб. При таком способе открывания трубки анализируемый материал не загрязняется кусочками отбиваемого стекла, что особенно важно в тех случаях, когда в нерастворимом остатке требуется определять кремний, титан и другие элементы.
Если в анализируемом материале содержатся осмий или рутений, то, чтобы предотвратить улетучивание их в виде четырехокисей, открытую трубку погружают в 6 н. соляную кислоту, насыщенную сернистым ангидридом и содержимому дают вытечь.
МЕТОДЫ РАЗДЕЛЕНИЯ
Разделение платиновых металлов и золота
В приводимых ниже методах анализа и разделения предполагается, если нет других указаний, что платиновые металлы и золото находятся в виде «хлоридов» или, точнее, в виде хлорокислот. Платина, например, в растворах образует хлороплатиновую кислоту H2PtCle и в реакциях ведет себя как часть комплексного аниона. При анализе металлов платиновой группы и золота исходные растворы чаще всего содержат именно эти соединения. Поэтому в основе методов разделения обычно лежат реакции, свойственные этим комплексным анионам или ионам, образующимся в результате разложения таких комплексов. В отдельных случаях при анализе используются также и другие соединения этих металлов. Так, например, при отделении рутения дистилляцией или при отделении родия от иридия восстановлением солями титана (III)' целесообразнее оперировать с растворами, в которых эти металлы находятся в виде сульфатов, а для успешного отделения многих неблагородных металлов от платиновой группы гидролитическим осаждением прибегают к предварительному переведению платиновых металлов в комплексные нитриты.
Методы, приведенные в этом разделе, разработаны в предположении, что неблагородные металлы в растворе отсутствуют.
Отделение золота от платиновых металлов. Золото можно отделить от платиновых металлов различными реагентами, выбор которых зависит
Методы разделения
407
от обстоятельств, главным образом от предполагаемого хода определения остающихся в растворе металлов. Сульфат железа (II) и сернистый ангидрид (или сульфиты) полностью осаждают золото, причем кислотность раствора может колебаться в широких пределах и не требуется полного удаления азотной кислоты из раствора. Однако применение сульфата железа (II) часто нежелательно, так как это связано с введением в раствор железа. При осаждении сернистым ангидридом раствор лишь немного загрязняется сульфатом, но в этом случае осадок золота увлекает небольшие количества платиновых металлов'и для получения точных результатов его необходимо переосадить. Часто применяемый способ выделения золота щавелевой кислотой требует полного удаления из раствора азотной кислоты и значительно более тщательного регулирования кислотности раствора, чем при осаждении сульфатом железа (II) или сернистый ангидридом. Для отделения золота от платины и палладия рекомендуют также осаждение гидрохиноном из холодного примерно 1,2 н. по концентрации НС1 раствора *. Отделение золота от малых количеств платины и палладия можно осуществить осаждением хлоридом тетраэтиламмония 1 2.
Установлено 3, что применение нитрита натрия в качестве реагента для отделения золота от платиновых металлов имеет ряд преимуществ. Так, однократного осаждения обычно достаточно даже при проведении очень точных анализов. Металлическое золото осаждается в виде легко отфильтровывающегося и промывающегося осадка, а платиновые металлы остаются в растворе в виде комплексных нитритов, которые можно перевести в хлоросоли нагреванием с соляной кислотой. Восстановление нитритом натрия начинается при относительно высокой кислотности, но для обеспечения количественного осаждения золота раствор необходимо довольно точно нейтрализовать до pH — 8. Для нейтрализации раствора почти до нужной степени может быть использован сам реагент.
При использовании любого восстановителя, за исключением гидрохинона, раствор необходимо кипятить в течение нескольких минут для достижения полноты осаждения золота и лучшей коагуляции осадка. Некоторыми реагентами, в частности щавелевой кислотой, золото выделяется в виде Очень тонкого осадка. Во всех случаях во избежание потерь металла при фильтровании следует пользоваться плотными фильтрами и, если нужно, вводить мацерированную бумагу.
Прежде чем приступить к анализу фильтрата, независимо от применявшегося для Осаждения золота реагента, избыток его необходимо разрушить, а платиновые соли перевести в хлоросоли. Золото можно также отделить от платиновых металлов экстрагированием эфиром 4 5 * или этил-ацетатом 8 из солянокислых растворов. Эти методы, однако, не имеют никаких преимуществ перед восстановлением золота до металла, за исключением некоторых особых случаев.
Систематический ход разделения платиновых металлов. Все платиновые металлы, кроме платины, образуют нерастворимые гидроокиси при
1 F. Е. Beamish, J. J. Russel, J. S е a t h, Ind. Eng. Chem., Anal. Ed., 9, 174 (1937).
2 J. L. Maynard, Ind. Eng. Chem., Anal. Ed., 8, 368 (1936).
3 R. Gilchrist, J. Research NBS, 20, 745 (1038).
4 F. Mylius, Z. anorg. Chem., 70, 203 (1911).
5 А. Нойес, В. Брэй. Качественный анализ редких элементов, ОНТИ
Гл. ред. хим. лит., 1936, стр. 103.
408
Гл. XX. Платиновые металлы
нейтрализации раствора их хлорокислот гидроокисями, карбонатами или бикарбонатами и кипячением раствора в течение нескольких минут Ч Пределы концентраций гидроксильных ионов в растворе, при которых достигается количественное осаждение этих элементов, несколько различны. Для осаждения платиновых металлов пригодны не все реагенты, способные создать соответствующую концентрацию гидроксильных ионов в растворе, так как некоторые из них, как, например, нитриты, фосфаты, аммиак, переводят хлоросоли этих металлов в другие соединения, а не в нерастворимые гидроокиси. В некоторых случаях гидролитическое осаждение можно с большим успехом применять к растворам, содержащим сульфатные комплексы, чем к растворам хлоросолей.
Хотя осмий и рутений отделяются от платины совместно с другими металлами платиновой группы, но иногда целесообразно применять дистилляцию, используя способность этих элементов образовать летучие четырехокиси. Этот простой способ удаления двух металлов группы упрощает разделение палладия, родия и иридия после отделения их от платины гидролитическим осаждением 1 2.
Реакции, применяемые для определения этих металлов после их разделения, приведены в разделе «Методы определения» (стр. 416). Раздел «Систематический ход разделения и определения платиновых металлов» (стр. 423) содержит подробное описание пригодных для рядовых анализов методов разделения и определения платиновых металлов, приспособленных для анализа корольков, в которых эти металлы сконцентрированы 3, 4.
Отделение осмия.. Для отделения осмия от остальных металлов платиновой группы во всех случаях применяется отгонка его в виде четырехокиси из раствора, содержащего азотную кислоту. Осмий полностью отгоняется, если щелочной плав анализируемого материала растворить в азотной кислоте, концентрацию свободной кислоты довести до 10% (по объему) и раствор кипятить 5 6 1 ч. В этих же условиях осмий удаляется, когда он находится в виде бромоосмата, но при содержании его в растворе в виде хлороосмата для полного его удаления может потребоваться более
1 При отсутствии в растворе окислителя иридий осаждается не сразу. Возможно, что радикал хлороиридата, подобно хлороплатинату, гидролизуется медленно. В осадке, выделяющемся в присутствии бромата, иридий находится в более высокой степени окисления, чем четырехвалентная.
2 Более подробное описание этих реакций см. R. Gilchrist, Е. Wichers, Trabajos IX Congr. intern, quim. pura у aplicada, 6, 32 (1934), Madrid.
3 См. сноску 2 на стр. 402.
4 F. E. Beamish, J. J. Russell, Ind. Eng. Chem., Anal. Ed., 8, 141 (1936). E. C. F о r b e s, F. E. В e a m i s h, там же, 9, 397 (1937). S. О. Thompson, F. E. Beamish, M. Scott, там же, 420; J. J. Russell, F. E. Beamish, J. S e a t h, там же, 475; J. Seath, F. E. Beamish, там же, 10, 535, 639 (1938); F.E. Beamish, J. Dale, там же, 697; J. Seath, F.E. Beamish, там же, 12, 169 (1940); W. J. Rogers, F. E. Beamish, D.S. Russell, там'же, 561; F. E. Beamish, Can. Mining J., 62, 146, 233 (1941); R. Thiers, W. Graydon, F. E. Beamish, Anal. Chem., 20, 831 (1948); W. F. Allen, F. E. Beamish, там же, 22, 451 (1950). M. A. Hill, F. E. Beamish, там же, 590.
Определение металлов платиновой группы в никелевых рудах и концентратах методами пробирного анализа см. F. Е. Lathe, Can. J. Research, 18, 333 (1940).
Микрохимическое титрование золота и платиновых металлов в связи с анализом корольков см. в статьях: W. В. Pollard, Analyst, 62, 597 (1937); 67, 184 (1942); Bull. Inst. Mining Met., 400, 401, 402, 406, 409, 410, 411, 417, 497 и 500, c 1938 до 1948 г.
6 Из растворов, содержащих едкий натр и спирт, таким способом осмий полностью не удаляется.
Методы разделения
409
продолжительное кипячение (до 8 ч). В этом случае, если возможно, отгонку лучше осуществлять из сернокислого раствора. Отгонка осмия из сернокислого раствора бромоосмата, в отличие от хлороосмата, идет очень медленно и большая часть осмия выделяется из раствора в виде черного осадка. Добавление к сернокислому раствору небольшого количества азотной кислоты ускоряет отгонку осмия. Однако при наличии в растворе рутения добавлять азотную кислоту не рекомендуется, так как в этих условиях медленно образуется четырехокись рутения. В кипящем сернокислом растворе, не содержащем азотной кислоты, четырехокись рутения не образуется и осмий полностью отделяется от рутения, так же как и от других платиновых металлов. Осмий количественно отделяется от рутения также и при дистилляции азотнокислого раствора (содержащего немного серной дислоты или-вовсе ее не содержащего), если концентрация азотной кислоты в растворе не превышает 40% по объему.
Отделение рутения,. Обычно принятый способ отделения рутения от других платиновых металлов, за исключением осмия, заключается в отгонке четырехокиси рутения, образующейся при насыщении щелочного раствора хлором.
При использовании этого способа присутствие в растворе иридия вызывает значительные затруднения. В процессе нагревания насыщенный хлором щелочной раствор становится сначала нейтральным, а затем постепенно слегка кислым, причем образующийся вначале гипохлорит натрия переходит в хлорат. В этих условиях иридий осаждается в виде гидроокиси, которая обладает свойством каталитически разлагать хлорат (и гипохлорит) на хлорид и свободный кислород. Не улетучившаяся в процессе отгонки часть четырехокиси рутения в таком растворе может восстановиться с образованием соединения рутения (IV). Поэтому раствор необходимо охладить, прибавить едкую щелочь, насытить хлором и продолжить отгонку. Для полного удаления рутения может потребоваться многократное повторение этой операции.
При окислении рутения не в щелочном, а в слабокислом растворе иридий осаждается значительно труднее (если он присутствует в концентрациях, обычно встречающихся в аналитической работе), и отгонку четырехокиси рутения можно выполнить без особых затруднений. Для этого достаточна 2 н. концентрация серной кислоты в растворе при условии, что платиновые металлы предварительно переведены в сульфаты выпариванием раствора с серной кислотой до температуры ее кипения. Платина, большая часть которой осаждается в виде металла при обработке серной кислотой, не препятствует последующей дистилляции рутения и в процессе окисления снова переходит в раствор. Весьма эффективным окислителем является бромноватая кислота, вводимая в виде бромата натрия. При окислении бромноватбй кислотой, которая относительно устойчива в разбавленной серной кислоте, дистилляция рутения заканчивается в течение двух часов.
Четырехокись рутения можно удалить также отгонкой из раствора, содержащего 10% азотной кислоты. Следовательно, эта операция может быть выполнена непосредственно простым добавлением бромата натрия после отгонки осмия. Основным недостатком этого способа является то, что совместно с рутением отгоняется много азотной кислоты, вследствие чего в поглотителе, содержащем насыщенный раствор сернистого ангидрида образуются значительные количества серной кислоты, наличие
410
Гл. XX. Платиновые металлы
которой, в свою очередь, нежелательно при последующем выделении рутения из раствора.
Дистилляция рутения из солянокислого или бромистоводороднокислого раствора является не только неподходящей, но и недопустимой, так как даже при небольшом содержании этих кислот в растворе бромат разлагается с выделением значительных количеств свободного брома. Кроме того, в их присутствии при не очень точном соблюдении температуры дистилляции на стенках перегонного аппарата образуется пленка двуокиси рутения. Эта пленка удаляется с трудом и ее образования следует избегать.
Отделение палладия, родия и иридия от платины. Удобным способом отделения палладия, родия и иридия от платцны является гидролитическое осаждение их в присутствии бромата. Гидроокиси палладия (II), родия (III) и иридия (III) хотя и осаждаются также количественно, но медленнее оседают и труднее отфильтровываются, чем осадки, образующиеся в присутствии бромата. Кроме того, бромат замедляет взаимодействие между хлороплатинатом и гидроксил-ионами, хотя специально для этой цели вводить в раствор бромат не требуется, так как, при обычной для аналитической практики концентрации платины, гидролиз хлороплатината идет настолько медленно, что не препятствует отделению указанных элементов. Во всяком случае, в начальной стадии гидролиза хлороплатината нерастворимых соединений не образуется.
С этой медленной реакцией хлороплатината не следует отождествлять реакцию других соединений платины, например сульфата. Так, в процессе выпаривания раствора с серной кислотой платина в основном выделяется в виде металла, но часть ее остается в растворе, переходя в сульфат, который при нейтрализации раствора образует осадок.
Для отделения палладия, родия и иридия от платины методом гидролитического разложения их хлоро-комплексов в присутствии бромата раствор рекомендуют нейтрализовать до pH = 8, главным образом для количественного выделения палладия и родия, что не препятствует также полному осаждению иридия. Если гидроокиси осаждаются в результате нейтрализации кислых растворов их можно полностью отмыть от щелочных солей, не переводя в коллоидную форму. Такие осадки очень легко отфильтровываются и содержат настолько незначительные количества платины, что при выполнении рядовых анализов вполне достаточно1 однократного осаждения. Переосаждение осадка может понадобиться лишь в тех случаях, когда требуется исключительно точное разделение.
Разделение палладия, родия и иридия. Палладий количественно отделяется от родия и иридия однократным осаждением диметилглиоксимом из кислого раствора.
Отделение родия от иридия достигается восстановлением титаном (III), лучше в кипящем растворе, содержащем оба металла в виде сульфатов. Металлический родий, который при этом образуется, обычно несколько загрязняется иридием, но его можно растворить в кипящей серной кислоте и переосадить. Если в фильтрате требуется определить иридий, введенный титан необходимо предварительно удалить, для чего можно воспользоваться осаждением купфероном. Осадком титана захватываются лишь незначительные количества иридия, которые извлекаются переосажде-нием этого осадка.
Различные методы разделения платиновых металлов. При анализе материалов, содержащих платиновые металлы, для отделения платины
Методы разделения
411
от других элементов платиновой группы (а также от золота и неблагородных металлов) наиболее часто йрименяется осаждение в виде хлороплатината аммония. Отделение это основано на том, что родий и палладий в наиболее характерном для них валентном состоянии не образует нерастворимых двойных солей с хлоридом аммония. Четырехвалентные осмий, рутений и иридий дают соли, изоморфные с солью платины и обладающие примерно такой же растворимостью, как хлороплатинат аммония.
Этот способ, разделения обычно применяют для анализа смесей, которые могут быть богаты иридием, но содержат лишь ничтожные количества осмия и рутения. В некоторых случаях предотвращают выделение иридия вместе с платиной, восстановив его предварительно до трехвалентного состояния, а иногда обе соли осаждают совместно, с целью отделения их от палладия и родия. Родий, который в солянокислом растворе всегда находится в трехвалентном состоянии, и палладий (II) не образуют нерастворимых двойных солей с хлоридом аммония, но они увлекаются солью платины, причем родий с исключительным постоянством. С другой стороны, достигнуть этой реакцией количественного осаждения платины фактически невозможно. Лишь продолжительная обработка большим избытком хлорида аммония приводит к почти количественному выделению хлороплатината аммония, но это способствует также соосаждению других металлов. Таким образом, количественно отделить платину в виде хлороплатината аммония от других металлов платиновой группы практически не представляется возможным, хотя результаты определения «платины» иногда бывают близки к истинным за счет взаимной компенсации ошибок. •
В тех случаях, когда описанной выше реакцией выделяют платину совместно с иридием, смесь их солей прокаливают до получения губки, которую затем обрабатывают царской водкой с целью отделения платины (переходящей в раствор) от иридия. Хотя чистая иридиевая губка не реагирует с царской водкой, но вследствие того, что большая часть иридия и платины находятся в виде смешанных кристаллов, растворимость которых в царской водке зависит от относительного содержания этих металлов в смеси, таким путем достигается лишь очень неточное разделение платины и иридия.
Палладий количественно осаждается из раствора его хлорида в виде иодида, если не вводить слишком большой избыток реагента Ч Другие платиновый металлы, за исключением родия 1 2, не осаждаются в этих условиях. Палладий можно осадить также в виде цианида введением в раствор цианида ртути (II). Однако этот метод, так же как и иодидный, редко предпочитают методу осаждения диметилглиоксимом. Описан способ отделения палладия от платины, основанный на осаждении этиленом 3. Опубликованные результаты, однако, не дают возможности судить о точности этого способа.
При анализе сплавов иридия с платиной для отделения иридия от родия и палладия, которые могут одержаться в таких сплавах, можно
1 F. Е. Beamish, J. D ale, Ind. Eng. Chem., Anal. Ed., 8, 697 (1936).
2 В. В. Лебединский [Известия Института платины Академии наук СССР, 5, 364 (1927)] описывает способ разделения родия и иридия, основанный на осаждении иодида родия. Однако при испытании этой реакции мы не достигли количественного осаждения родия.
3 S. С. Ogburn Jr., W.C. В г a s t о w, J. Am. Chem. Soc., 55, 1307 (1933).
412
Гл. XX. Платиновые металлы
с успехом применять сплавление анализируемого материала с десятикратным количеством свинца при 900—1000° С. Избыток свинца и сплавы свинца с платиной, родием и палладием растворяют последовательной обработкой азотной кислотой, а затем разбавленной царской водкой. Иридий не образует сплава со свинцом и не растворяется в царской водке, но он загрязняется рутением, железом и, возможно, осмием, если эти элементы присутствуют в сплаве. Подробный ход выполнения этого исключительно точного разделения приведен в разделе «Методы определения» (стр. 416). Способ этот применим также к анализу губок, состоящих из платины и иридия. Наличие цинка, который мог быть введен, например, для выделения платиновых металлов из раствора, приводит к растворению некоторого количества иридия. *
Отделение платиновых металлов и золота от других металлов
Осаждение в виде металлов. Платиновые металлы и золото вытесняются из кислых растворов неблагородными металлами, например цинком, магнием, алюминием и др. Получающиеся при этом осадки почти всегда содержат примененный для осаждения металл в количествах, колеблющихся от следов, в случае применения магния, до значительных, как в случае применения цинка. Осадки могут быть загрязнены также и другими осаждающимися металлами, содержавшимися в анализируемом растворе или же введенными с осадителем. Достигнуть цементацией количественного осаждения иридия практически невозможно. Кроме того, осадок металлов, содержащий иридий и родий, растворяется с большим трудом. Поэтому в настоящее время способом вытеснения платиновых металлов неблагородными металлами пользуются редко, отдавая предпочтение другим более эффективным аналитическим методам.
Благородные металлы осаждаются также молекулярным водородом, но эта реакция протекает медленно. Скорость осаждения можно повысить, проводя процесс под давлением, но для этого требуется специальная аппаратура.
Для осаждения благородных металлов, главным образом платины и палладия, часто применяют органические восстановители. Одним из наиболее приемлемых реагентов для этой цели является муравьиная кислота, которая выделяет металлы из растворов, кислотность которых понижают добавлением ацетата натрия или аммония х. Однако при этом всегда следует учитывать хорошо известные адсорбционные свойства платиновой черни. Установлено, например, что платиновой чернью, выделенной муравьиной кислотой, увлекаются значительные количества меди.
Осаждение в виде сульфидов. Осаждение сероводородом из кислых растворов может служить для отделения платиновых металлов и золота от большинства других элементов, исключая серебро, медь, кадмий, ртуть, индий, германий, олово, свинец, мышьяк,- сурьму, висмут, молибден, селен, теллур и рений.
Все платиновые металлы, за исключением иридия, довольно легко выделяются в виде сульфидов из растворов, приблизительно 0,5 н. по кон-
1 J. S е a t h и F. Е. Beamish [Ind. Eng. Chem., Anal. Ed., 9, 373 (1937)] для отделения золота от теллура и селена рекомендуют пользоваться гидрохиноном.
Методы, разделения
413
центрации соляной кислоты. Осаждение платиновых металлов, особенно платины и родия, должно быть проведено из нагретого почти до кипения раствора. Насыщения раствора сероводородом в течение 30 мин обычно вполне достаточно. В некоторых условиях рутений частично остается в растворе вследствие восстановления его до промежуточной валентности, что характеризуется синей окраской, но при продолжительной обработке раствора сероводородом рутений и в этой форме осаждается количественно.
Для полного осаждения сульфида иридия требуется значительно более высокая концентрация кислоты в растворе (около 3 н. по соляной кислоте) и более продолжительная обработка раствора сероводородом (примерно 2—3 ч при слабом кипении). Даже в этом случае осаждение может быть не количественным, если в растворе не присутствуют ионы, способствующие коагуляции коллоидного сульфида рутения. Для этой цели можно ввести на каждые 100 мл раствора по 0,5 л хлорида алюминия (шестиводную соль). Коагуляция осадка вызывается также и солями двухвалентных элементов, таких, как магний или кальций. Соосаждения этих металлов как будто можно не опасаться.
Сульфиды платиновых металлов легко растворяются в царской водке, но образующиеся комплексные соединения содержат иногда тесно связанные с металлом сульфатные группы, которые могут повлиять на последующие реакции. Это наблюдалось главным образом в отношении иридия. Раствор сульфида иридия в царской водке (или других окислителях, например перекиси водорода и азотной кислоте) в таких процессах, как осаждение хлоридом аммония или превращение в комплексные нитритные соли, ведет себя совершенно иначе, чем хлороиридат.
Применимость способа отделения платиновых металлов в виде сульфидов зависит от последующих аналитических операций.
Из перечисленных выше элементов, от которых платиновые металлы не могут быть отделены сероводородом, серебро (I) и ртуть (I) можно отделить осаждением в виде хлоридов. Медь, кадмий, индий, олово, свинец и висмут можно отделить гидролитическим осаждением описанным в следующем разделе. Отделение мышьяка, сурьмы и германия можно осуществить дистилляцией этих элементов с соляной кислотой, как описано в соответствующих главах. Молибден можно' удалить совместно с золотом экстракцией эфиром из солянокислого раствора. Селен (IV) и теллур (IV) можно отделить, также совместно с золотом, осаждением сернистым ангидридом. Этот реагент можно использовать и для отделения золота от молибдена, а извлечение азотной кислотой служит для отделения селена и теллура от золота.
Аналитическая химия рения недостаточно изучена, чтобы можно было судить о том, какие реакции применимы для отделения его от платиновых металлов. Результаты проведенных работ показывают, что все платиновые металлы, за исключением платины, можно отделить от рения осаждением в виде гидроокисей. Рений отгоняется из растворов в таких же условиях, как осмий и рутений, и его, несомненно, можно отделить таким путем от платины.
Осаждение в виде гидроокисей. Все металлы платиновой группы, за исключением золота и платины (IV), осаждаются в виде гидроокисей из почти нейтральных растворов. Этот способ применим для отделения платиновых металлов, кроме платины, от щелочных металлов и магния.
414
Гл. XX. Платиновые металлы
Было показано х, что палладий совместно с родием и рутением можно та-> ким же образом отделить от серебра в сернокислом растворе. Выделение в виде гидроокисей имеет то преимущество перед осаждением сероводородом, что образующиеся соединения легко превращаются в галогениды, которые требуются для последующих операций. Поскольку этот метод наиболее применим для отделения платины от палладия, родия и иридия, он более детально излагается в разделе «Систематический ход разделения и определения платиновых металлов» (стр. 423).
Отделение неблагородных металлов гидролитическим осаждением их из растворов, содержащих платиновые металлы в виде комплексных нитритов. При обработке раствора, содержащего хлоросоединения платиновых металлов, нитритом натрия эти соединения превращаются в растворимые комплексные нитриты. Превращение это происходит быстро в горячем растворе, кислотность которого не превышает pH = 1,5.
Комплексные нитриты платины, родия и иридия вполне устойчивы в растворах средней щелочности. Они не разлагаются в кипящих растворах, имеющих pH = 10 и даже несколько больше. Комплексный нитрит палладия устойчив в кипяших растворах, имеющих pH не выше 8, и начинает разлагаться1 2 с образованием гидроокиси при pH около 10. При более высокой концентрации щелочи нитрит палладия быстро разлагается, причем палладий практически полностью выделяется из раствора. Неблагородные металлы, исключая никель и кобальт, не склонны к образованию комплексных нитритов. Комплексный нитрит никеля полностью разлагается при pH = 8, а кобальта — при pH = 10. Таким образом, переведением платиновых металлов в комплексные нитриты и регулированием pH раствора возможно отделить их от тех элементов, которые осаждаются в виде гидроокисей или карбонатов из нейтральных или слабощелочных растворов 3.
Если горячий нитритный раствор нейтрализовать едким натром по тимолфталеину (pH = 10) (или, что практически равноценно, но удобнее, по тимоловому синему) до изменения желтой окраски в синюю, индий, медь, цинк, никель, кобальт, марганец, хром и железо количественно осаждаются и отделяются4 таким образом от платиновых металлов, за исключением палладия, комплексное соединение которого в этих условиях неустойчиво. Переосаждением осадок освобождается от следов платиновых металлов, кроме палладия; последний затем легко отделяется осаждением диметилглиоксимом. •
При нейтрализации раствора едким натром до появления синей окраски тимолового синего или ксиленолового синего (п-ксиленолсульфо-фталеин), желтая окраска которого переходит в синюю при рЙ = 8, кадмий и висмут полностью не осаждаются, но если вместо едкого натра применять бикарбонат натрия, оба элемента выделяются количественно. Свинец едким натром полностью не осаждается, независимо от щелочности раствора, а в присутствии карбоната выделяется количественно
1 F. Е. Beamish, J.J. Russell, Ind. Eng. Chem., Anal. Ed., 8, 141 (1936).
2 Возможность замедления или предотвращения разложения введением большого избытка нитрита не изучалась.
3 Более детальное описание гидролитического осаждения, а также его преимуществ см. R. Gilchrist,!. Research NBS, 30, 89 (1943), а также Chem. Rev., 32, 277 (1943).
1 R. Gilchrist, J. Research NBS, 20, 745 (1938).
Методы разделения
415
при нейтрализации раствора до перехода окраски ксиленолового синего.
Ртуть (II) не осаждается ни тогда, когда нейтрализацию проводят до появления синей окраски ксиленолового синего, ни тогда, когда в качестве ийдикатора применяют тимоловый синий, независимо от того, прибавляют ли в качестве осадителя бикарбонат натрия или едкий натр. Не осаждаются также мышьяк, ванадий, молибден и вольфрам в анионной форме, если каждый из этих элементов находится в растворе один. Не полностью осаждается сурьма, а также магний, кальций, стронций и барий.
Осаждение бериллия начинается примерно в точке перехода окраски бромкрезолового зеленого и заканчивается в пределах pH, соответствующих переходу окрасок хлорфенолового краеного и тимолового синего. При гидролитическом разложении нитрата уранила полное осаждение желтого соединения достигается в точке перехода ксиленолового синего. Присутствие карбонатов препятствует полному осаждению урана.
Наиболее низкое значение pH, при котором происходит количественное осаждение гидроокиси алюминия, соответствует точке перехода бромкрезолового пурпурного. При pH выше 7,5 (примерно в середине между точками перехода окрасок бромтимолового синего и крезолового красного) растворимость гидроокиси алюминия значительно возрастает. Четырехвалентный церий, а также торий полностью гидролитически осаждаются примерно при pH = 3. Осаждение трехвалентных элементов группы редкоземельных металлов начинается при pH около 6 и, в зависимости от основности индивидуального окисла, простирается до pH = 14 (щелочности, необходимой для осаждения лантана). Торий количественно осаждается при значении pH, > соответствующем переходу окрасок ксиленолового синего или тимолового синего. При этой же величине pH происходит, по-видимому, осаждение гафния, циркония и титана.
Галлий количественно осаждается в интервале pH от 4 до 6, но при pH, соответствующем переходу окраски ксилецолового синего и более высоком, он полностью переходит в раствор. Двуокись германия растворима в кипящей воде, и в интервале pH от 1,5 до 10 не наблюдается образования осадка.
Рений из раствора перрената не выделяется в пределах pH от 1,5 до 10. Он не осаждается даже после обработки раствора перрената соля-.ной кислотой, нитритом или сернистым ангидридом с последующей нейтрализацией до pH = 10 и выше.
Некоторые неблагородные металлы осаждаются в виде гидроокисей из растворов, кислотность которых достаточно высока, чтобы не происходило выделение платиновых металлов (исключая осмий), даже если они не находятся в виде нитритных комплексов. Можно достигнуть, например, удовлетворительного отделения олова1 от платиновых металлов нагреванием раствора хлоридов, pH которого равен 1,5.
После отделения неблагородных металлов способом, описанным в предыдущих разделах, 'для проведения последующих операций нитритные комплексы платиновых металлов обычно следует перевести в хлоросоединения. Это можно осуществить выпариванием раствора с соляной кислотой. При этом, однако, необходимо учитывать, что некоторые из
1 R. Gilchrist, J. Research NBS, 20, 745 (1938).
416
Гл. XX. Платиновые металлы
нитритных комплексов, особенно комплекс иридия, настолько устойчивы, что для полного их разложения может потребоваться продолжительное нагревание остатка с разбавленной (1 : 1) соляной кислотой.
Осаждение в виде двойных хлоридов (хлоросолей). Об осаждении хлороплатината аммония и хлороиридата аммония уже упоминалось в разделе «Методы разделения» (стр. 406). Подобные же относительно малорастворимые соли образуют и другие металлы этой группы, за исключением родия, который в соединениях с галогенами не бывает четырехвалентным. Соли палладия (IV) легко разлагаются на соответствующие соединения палладия (II) и свободный хлор. Поэтому осаждение палладия в виде хлоросоли следует проводить в присутствии сильного окислителя. Хлорид осмия (хлороосмиевая кислота) склонен к гидролизу и, следовательно, при осаждении хлороосмата концентрация соляной кислоты в растворе должна быть достаточно высокой (3 н.). Имеется указание, что для предотвращения частичного гидролитического разложения хлороруте-ната кислотность раствора должна быть 6 н. Гидролиз этого соединения, однако, ограничивается замещением одного атома хлора в радикале. Аммоний в выделяемых соединениях может быть заменен на калий, рубидий, цезий или таллий (I), а хлор — на бром.
Обычно эти соли не имеют резко выраженной склонности к захватыванию других тяжелых металлов, за исключением металлов платиновой группы. Но некоторые металлы, в особенности олово, могут загрязнять хлоросоли, так как в четырехвалентном состоянии они образуют аналогичные двойные хлориды. В случае высокой концентрации натриевых солей в растворе, из которого проводится осаждение, осадок до некоторой степени загрязняется натрием. Заметная растворимость хлоросолей ограничивает их использование в аналитической практике, тем более, что для отделения платиновых металлов в настоящее время имеются лучшие методы.
Концентрирование в металлическом свинце. Из руд и других твердых веществ малые количества платиновых металлов можно извлечь металлическим свинцом, подобно тому, как извлекают золото и серебро в пробирном анализе руд. Если требуется затем удалить свинец купеллпрова-нием, для удержания платиновых металлов можно прибавить серебро. Не все платиновые металлы растворяются в серебре, но они все удерживаются им, за исключением осмия, большая часть которого, если не весь, теряется в процессе купеллирования. Вопрос о влиянии платиновых, металлов на корольки серебра и золота освещен в литературе х. Описывается также действие этих металлов на свойства поверхности королька серебра 1 2.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Общие замечания
Для определения золота и элементов платиновой группы обычно применяются весовые методы, основанные на взвешивании в вине металлов. В последние годы появилось несколько объемных методов, но, как пра-
1 J. L. Byers, Trans. AIME, 102, 286 (1932); Bull. Mich. Coll. Min. Tech., 6, 1 (1933).
2 E. C. F о r b e s, F. E. Beamish, Ind. Eng. Chem., Anal. Ed., 9, 397 (1937).
Методы определения
417
вило, они не дают таких точных результатов, как весовые. Эти .методы могут быть применены лишь после отделения определяемого элемента от остальных металлов группы, и поэтому сомнительно, чтобы их можно было рекомендовать для практической работы даже на том основании, что они просты и удобны.
Методы, которые описаны для отделения золота, могут служить одновременно и для его определения. Выделенное золото следует только тщательно отмыть от раствора, из которого оно было осаждено, и затем прокалить при невысокой температуре. Для промывания осадка, во избежание его диспергирования, следует применять разбавленный раствор электролита, например разбавленную (1 : 99) соляную кислоту. Фильтровать нужно через плотный фильтр с мацерированной бумагой. Цвет прокаленного металла является показателем его чистоты. Даже небольшие количества платины или палладия заметно его обесцвечивают.
Платину можно осадить в виде металла различными восстановителями, но одним из лучших является муравьиная кислота. Осажденный металл собирают на фильтре, прокаливают и взвешивают. Для осаждения платины из раствора лучше пользоваться муравьиной кислотой, чем цементацией металлами, за исключением некоторых особых случаев. При необходимости провести осаждение металлом лучше пользоваться магнием, чем цинком или алюминием, которые, особенно цинк, загрязняют осадок, в связи с чем получаются повышенные результаты. В некоторых случаях платину предпочитают осаждать в виде сульфида, который затем прокаливают до металла и взвешивают. При восстановлении до металла платина, а также и другие платиновые металлы склонны прилипать к стенкам стакана. Это может происходить также и с сульфидом, но в последнем случае небольшое количество осадка, которое не удается стереть с поверхности стекла фильтровальной бумагой, можно растворить прибавлением в стакан небольшого количества царской водки и нагреванием. Таким же способом можно перевести в раствор металлические палладий и платину, но не родий и иридий. Сульфид платины следует вначале прокаливать при возможно более низкой температуре и свободном доступе воздуха, в противном случае небольшие количества серы могут задержаться в осадке.
Для определения платины обычно нецелесообразно непосредственно прокаливать хлороплатинат аммония, так как при прокаливании его не в восстановительной атмосфере могут происходить потери платины, возможно, вследствие летучести хлорида PtCl2. Платиновая губка имеет также склонность удерживать небольшие количества летучих компонентов. Кроме того, хлороплатинат аммония может быть загрязнен солями щелочных металлов, если они содержались в исходном растворе.
Платину можно осадить электролитически, но трудно обеспечить необходимые для этого условия.
В результате прокаливания на воздухе сульфидов и гидроокисей рутения, родия и иридия образуются безводные окислы, которые, однако, не могут, быть использованы в качестве весовой формы для определения этих элементов вследствие не вполне определенного их состава. Окись, образующаяся при прокаливании гидроокиси палладия на воздухе, также не имеет постоянного состава. Прокаливания сульфида палладия, если только количество его не очень мало, следует избегать, так как сера очень прочно удерживается осадком и полностью удалить ее не удается. Даже
27 Заказ 522.
418
Гл. XX. Платиновые металлы
при прокаливании самих металлов происходит частичное их окисление, поэтому окисленные осадки необходимо прокаливать в атмосфере водорода, чтобы восстановить их до металла и взвесить. Соединения осмия следует превращать в металл сразу в восстановительной атмосфере, так как при прокаливании на воздухе осмий окисляемся до летучей четырех-окиси.
Те количества водорода, которые адсорбируются губками рутения, родия и иридия, не влияют на результаты определения этих элементов. В момент прекращения тока водорода, когда восстановленный металл приходит в соприкосновение с воздухом, часто можно наблюдать кратковременную вспышку, являющуюся следствием каталитического окисления водорода. Развивающейся в процессе реакции теплоты достаточно для испарения образующейся при этом на металле воды. Однако в случае определения осмия каталитическое окисление приводит к заметной Потере металла в виде четырехокиси, и поэтому, прежде чем металл придет в соприкосновение с воздухом, водород следует вытеснить струей какого-нибудь инертного газа, например двуокиси углерода или азота. Палладий же поглощает значительные количества водорода, и поэтому результаты определения будут вообще неправильны, если не удалить водород, лучше всего, кратковременным прокаливанием металла в атмосфере инертного газа.
Окислы палладия мало устойчивы, и частично окисленный осадок, который получается при прокаливании соединений палладия на воздухе, может быть превращен в металл прокаливанием и охлаждением в атмосфере двуокиси углерода, что избавляет от необходимости восстановления осадка водородом.
Палладий 1 можно определить прокаливанием, любого из тех соединений, которые служат для его отделения, за исключением сульфида. Соединение палладия с диметилглиоксимом имеет определенный состав и может быть высушено и взвешено в таких же условиях, как и соответствующее соединение никелй. Взвешивание диметилглиоксимина палладия часто более удобно, чем его прокаливание, которое необходимо проводить постепенно и осторожно во избежание потерь металла.
При определении родия и иридия более удовлетворительные результаты получаются при осаждении их в виде сульфидов или гидроокисей, чем в виде металлов. Оба эти соединения можно непосредственно прокалить до окислов (непостоянного состава), которые затем восстанавливают' до металла кратковременным прокаливанием в токе водорода. Чтобы избежать возможного воспламенения, рекомендуется к гидроокиси перед прокаливанием добавлять небольшое количество концентрированного раствора хлорида аммония.
1 Для весового определения палладия (2—30 мг) J‘. R. Hayes и G. С. С h a n d-1 е е (Ind. Eng. Chem., Anal. Ed., 14, 491 (1942)] рекомендуют осаждать его p-фурфур-альдоксимом в виде соединения Pd(C4H3OCHNOH)2Cl2, что имеет некоторые преимущества перед осаждением диметилглиоксимом. В частности, это соединение имеет более низкое содержание Pd, легче отфильтровывается, а реагент, p-фурфуральдоксим, растворим в воде.
Малые количества палладия (0,5—50 мкг) Т.Н. Yoe и L. G. Overholser [J. Am. Chem., Soc., 61, 2058 (1939)] рекомендуют определять колориметрическим методом, основанным на образовании темно-красного окрашивания с п-нитрозодифенил-амином в нейтральном или слабокислом растворе. Этой реакции мешают золото,, серебро, окислители, цианиды и иодиды..
Методы, определения
419
Условия осаждения гидроокисей и сульфидов платиновых металлов приведены в разделе «Систематический ход разделения и определения платиновых металлов» (стр. 423).
Осмий1 и рутений 2 наиболее удобно определять выделением их в виде гидроокисей и последующим прокаливанием до металла. Эти соединения осаждаются при нейтрализации кислых растворов хлоридов, так же как родий, палладий и иридий, с той лишь разницей, что при этом не следует вводить бромат или другие окислители во избежание образования летучих четырехокисей вместо гидроокисей осмия (III и IV) и рутения (III) и (IV). Для осаждения осмия раствор следует нейтрализовать до pH = 4, а для осаждения рутения — до pH = 6.
Гидроокиси рутения и осмия удобнее всего осаждать из солянокислых растворов четырехокисей этих элементов, полученных в процессе разделения по способу, описанному в разделе «Методы разделения» (стр. 406). Они могут быть осаждены также нейтрализацией растворов, полученных после поглощения летучих четырехокисей этих элементов щелочью. Образующиеся в этих условиях осадки, однако, труднее обрабатываются и почти всегда загрязнены солями щелочных металлов. В тех случаях, когда используются щелочные растворы, вероятно, целесообразнее сначала подкислить их соляной кислотой в присутствии достаточного для восстановления нитратов количестве сернистого ангидрида, затем нагреть с соляной кислотой для удаления избытка сернистого ангидрида и разложения сульфитов и лишь после этого провести осаждение.
Рутений количественно не осаждается в виде гидроокиси, если в растворе находится нитрозохлорид рутения. Это весьма устойчивое комплексное соединение рутения легко образуется при обработке раствора азотной кислотой или нитритом натрия.
Осажденную гидроокись рутения можно собрать на бумажном фильтре и прокалить на воздухе. Гидроокись осмия необходимо сушить и прокаливать в атмосфере водорода, поэтому лучше собирать ее в тигле Мунро или каком-либо другом фильтрующем тигле. Оба осадка, пока они влажные, обрабатывают небольшим количеством хлорида аммония, чтобы воспрепятствовать их воспламенению во время прокаливания. Гидроокись осмия сушат, прокаливают и охлаждают в атмосфере водорода, который следует затем вытеснить двуокисью углерода, прежде чем открыть доступ воздуху.
Ход анализа
' Золото. Осаждение сернистым ангидридом. В анализируемом растворе, подготовленном для осаждения золота, могут находиться почти все элементы, кроме селена, теллура, свинца и щелочноземельных металлов. В солянокислый раствор, содержащий в 100 мл не более 5 мл соляной кислоты и не более 0,5—1 г золота, прибавляют на каждые 100 мл по 25 мл
1 Колориметрическое определение малых количеств осмия (4—37 мкг) с тиомочевиной см. Е. В. S а п d е 11, Ind. Eftg. Chem., Anal.^Ed., 16, 342 (1944).
2 Микрохимическое определение рутения осаждением его Д-аминонафталидом тиогликолевой кислоты (тионалидом) после отгонки из разбавленного сернокислого раствора, содержащего бромат натрия, и поглощения 3%-ным раствором перекиси водорода см. W.J. Kogers, F. Е. В е a m i s h, D. S. R u s s e 11, Ind. Eng. Chem., Anal. Ed., 12, 169 (1940).
27*
420
Гл. XX. Платиновые металлы
насыщенного раствора сернистого ангидрида и нагревают на водяной бане 1 ч. Прибавляют еще 5—10 мл раствора сернистого ангидрида и оставляют стоять до охлаждения. Если охлажденный раствор имеет сильный запах сернистого ангидрида, осаждение золота можно считать количественным. Если предполагают, что однократного осаждения недостаточно, то отстоявшийся раствор сливают через плотный фильтр с мацерированной бумагой, перенося на фильтр возможно меньшее количество осадка. Осадок тщательно промывают декантацией горячей разбавленной (1 : 99) соляной кислотой и тщательно исследущт фильтрат, чтобы удостовериться в том, что частички золота не прошли сквозь фильтр. Если в растворе •содержатся платиновые металлы, то для осаждения более целесообразно применять нитрит натрия. Если же в этом случае применяют сернистый ангидрид, то в присутствии платины и палладия для получения точных результатов осадок золота необходимо переосадить. Для второго осаждения можно пользоваться нитритом натрия или щавелевой кислотой.
Осаждение щавелевой кислотой. Если осадок, выделенный сернистым ангидридом, надо переосадить, фильтр с осадком переносят обратно в стакан и для растворения 1 г золота или мейее вводят 8 мл соляной кислоты, 2 мл азотной кислоты и 10 мл воды. По окончании растворения металла бумагу отфильтровывают и тщательно промывают горячей разбавленной (1 : 99) соляной кислотой. Фильтрат выпаривают досуха на водяной бане, добавляют 2—3 мл соляной кислоты и снова выпаривают досуха. Эту операцию повторяют еще дважды для полного удаления азотной кислоты. Остаток обрабатывают 3 мл соляной кислоты, 5 каплями серной кислоты и 75 мл воды из расчета на 1 г или менее присутствующего золота. Не обращая внимания на наличие небольшого количества металлического золота, которое может при этом выделиться, вводят на каждые 75 ли раствора по 25 мл насыщенного раствора щавелевой кислоты и кипятят не более 15 мин. Добавляют еще,5—10 мл раствора щавелевой кислоты и снова кипятят 1—2 мин. Если дальнейшего образования осадка не наблюдается, раствор нагревают на водяной бане не менее 4 ч, золото отфильтровывают и стакан тщательно вытирают фильтровальной бумагой, чтобы полностью извлечь осадок. Золото прокаливают в фарфоровом тигле при 900° С и взвешивают в'виде металла.
Осаждение нитритом натрия1'2. Следующая методика может быть применена для осаждения и определения золота в растворах, содержащих только одно золото или золото и платиновые металлы, а также золото, платиновые металлы и некоторые неблагородные металлы, как, например, медь, цинк и никель. Описанная здесь методика касается только определения золота, поэтому она упрощена и несколько отличается от методики, приводимой в цитируемой работе, в которой предусматривается также определение и других металлов.
Солянокислый раствор, содержащий не более 1 г золота в 100 мл, нейтрализуют едким натром до появления красно-оранжевой окраски тимолового синего (pH около 1,5). Нагревают раствор до кипения и прибавляют 10 мл 10%-ного раствора нитрита натрия. Кипятят 2—3 мин для коагуляции осажденного металла. Снова нейтрализуют до появления
1 Определение золота в сплавах, как, например, в «золоте», применяемом в зубоврачебном деле, см. R. Gilchrist,!. Research NBS, 20, 745 (1938).
2 Определение золота в ювелирных сплавах см. Н. Holzer, Е. Zaussin-g е г, Z. anal. Chem., ill, 321 (1938).
Методы определения
421
красно-оранжевой окраски тимолового синего, добавляют 20 мл раствора нитрита и продолжают нейтрализацию горячего раствора, добавляя по каплям при непрерывном перемешивании разбавленный раствор едкого натра до бледно-синей окраски ксиленолового синего или бледно-розовой окраски крезолового красного (pH около 8). Осторожно кипятят 5 мин, после чего осажденное золото отфильтровывают, тщательно промывают сначала горячей водой, затем горячей разбавленной (1 : 99) азотной кислотой и снова горячей водой. Фильтр с осадком прокаливают в фарфоровом тигле при 900° С и взвешивают в виде металлического золота.
Платина. Метод определения платины осаждением сероводородом и муравьиной кислотой описан в разделе «Систематический ход разделения и определения платиновых металлов» (стр. 418).
В некоторых случаях может оказаться целесообразным осадить платину магнием. Для этого к не слишком сильно кислому раствору (не содержащему других вытесняемых магнием металлов) добавляют кусочки магния до прекращения осаждения. При этом раствор должен оставаться достаточно кислым, чтобы не происходило гидролитического осаждения неблагородных металлов. Нагревают на водяной бане, пока осажденный металл не скоагулирует и раствор не станет прозрачным, затем фильтруют. Осадок промывают разбавленной (1 : 99) соляной кислотой и прокаливают в фарфоровом тигле.
Определение иридия в платиновых сплавах1. Анализируемый сплав сплавляют с десятикратным по массе количеством пробирного свинца в графитовом тигле в течение 1 ч примерно при 1000° С. Расплавленному металлу дают затвердеть в тигле, охлажденный слиток вынимают из тигля и слегка очищают от прилипшего к нему углерода. Слиток обрабатывают затем в накрытом стакане при нагревании на водяной бане разбавленной (1 : 4) азотной кислотой р количестве 5 мл на каждый грамм свинца. По растворении слитка раствор разбавляют равным объемом воды и затем сливают декантацией через плотный фильтр, в который вложен фильтр из менее плотной бумаги. Осадок в стаканеТпромывают горячей водой, сливая промывную жидкость через фильтр. Фильтры с небольшим количеством попавшего в них осадка опускают в стакан, содержащий главную часть осадка, и добавляют на каждый грамм анализируемого сплава сначала 15 мл воды, затем 5 мл соляной кислоты и, наконец, 0,8 мл азотной кислоты. Нагревают на водяной бане до тех пор, пока черная более легкая часть осадка не растворится и останется только плотный серый осадок металлического иридия (обычно около 1,5 ч). Разбавляют раствор равным объемом воды и фильтруют через такой же двойной фильтр, как в первый раз. Тщательно переносят плотный осадок на фильтр, ополаскивая струей воды из промывалки и вытирая кусочками беззольной бумаги стенки стакана. Осадок и фильтр хорошо промывают горячей разбавленной (1 : 99) соляной кислотой, следя за тем, чтобы осадок не прошел через фильтр. Фильтр с осадком высушивают и затем прокаливают в фарфоровом тигле. После озоления фильтра иридий сильно прокаливают сначала на воздухе, а затем кратковременно в токе водорода. Охлаждение тигля с осадком производят также в атмосфере водорода. Полученный осадок (металлический иридий) взвешивают. Для внесения поправки на содержание
1 R. Gilchrist, Bur. Sci. Papers, 19, 325 (1924); J. Am. Chem. Soc., 45, 2820 1923).
422 Гл. XX. Платиновые металлы
рутения, осмия и железа, которые обычно сопровождают иридий, осадок переводят в раствор и затем отделяют иридий от этих элементов соответствующими методами, приведенными в этой главе.
Другие платиновые металлы. Методы, рекомендуемые для определе-ления палладия осаждением диметилглиоксимом, а также для выделения иридия, родия, осмия, рутения в виде гидроокисей и родия в виде сульфида, описаны в разделе «Систематический ход разделения и определения платиновых металлов» (стр. 423).
В некоторых случаях может оказаться целесообразным определение иридия осаждением его сульфида с последующим прокаливанием осадка. Для этой цели на каждые 100 мл раствора прибавляют в качестве коагулянта по 0,5 г хлорида алюминия (А1С13-6Н2О). Раствор подкисляют соляной кислотой с таким расчетом, чтобы концентрация ее была Зн., и затем при слабом кипячении пропускают медленный ток сероводорода в течение 2,5—3 ч. Охлаждают, не прекращая тока сероводорода, и фильтруют. Осадок промывают разбавленной (1 : 99) соляной кислотой ц прокаливают в фарфоровом тигле. Окисленный осадок восстанавливают в атмосфере водорода и взвешивают в виде металлического иридия. Фильтрат проверяют на полноту осаждения.
Новые методы анализа
Объемные методы. Один из предложенных1 объемцых методов определения платины заключается в восстановлении платины (IV), находящейся в виде PtClg-, до двухвалентного состояния хлоридом меди (I). После окисления избытка меди (I) воздухом хлороплатинит титруют до хлороплатината перманганатом калия.
Платину (IV) рекомендуют 2 также восстанавливать до двухвалентного состояния определенным количеством железа (II), а избыток последнего титровать NH4VO3 в присутствии фенилантраниловой кислоты в качестве индикатора. Концентрацию ионов железа (III) в растворе понижают введением фосфата или фторида. Предложен3 метод, в котором родий (III) в виде сульфата окисляют до «пятивалентного» состояния висмутатом натрия и затем восстанавливают солью Мора.
Для определения палладия описан4 объемный метод, основанный на восстановлении хлоропалладата солью Мора и последующем титровании избытка восстановителя раствором К2Сг2О7, солью ванадия (V) или Ce(SO4)2, с применением в качестве индикаторов фенилантраниловой кислоты. Эти же авторы5 опубликовали метод, в котором органическое комплексное соединение палладия с p-фурфуральдоксимом окисляют ванадием (V), избыток которого титруют солью Мора.
Предложен также потенциометрический метод определения платины и иридия титрованием хлоридом меди (I)8. Для той же
1 А. А. Гринберг, Ц. Е. Гольбрайх, ЖОХ, 14, 808 (1944).
< 2. В. С. Сырокомский, Н. Н. Прошенкова, ЖАХ, 1, 83 (1946).
3 В. С. Сырокомский, Н. Н. Прошенкова, ЖАХ, 2, 247. (1947).
4 В. С. Сырокомский, С. М. Губельбанк, ЖАХ, 4, 146 (1949).'.
б-В. С. Сырокомский, М. С. Губельбанк, ЖАХ, 4, 203 (1949).
8 Д. И. Рябчиков, С. В. Нерсесова, Известия АН СССР, сектор платины, 18, 100 (1945).
Методы определения
423
цели разработан1 2’3 метод, являющийся развитием ранее известного способа 3.
Для определения иридия предложен также потенциометрический метод, основанный на реакции между хлороиридатом и гидрохиноном4.
Методы с применением органических реагентов. Интерес; проявляемый в последние годы к органическим реагентам, привел к разработке нескольких методов, в которых эти реагенты используются для анализа платиновых металлов.
Изучены5 реакции аллилтиомочевины, фенилтиомочевины, S-ди-о-толилтиомочевины, тиофенола и S-фенилтиогидантоиновой кислоты, тио-барбитуровой кислоты и S-дифенилтиомочевины с растворами платиновых металлов в разбавленной соляной кислоте и предложены методы определения платины с тиофенолом и родия с тиобарбитуровой кислотой.
Рекомендуется6 весовой метод определения родия, основанный на осаждении его из уксуснокислого раствора 2-меркаптобензоксазолом и прокаливании выделенного соединения до металла. Позднее7 эта реакция была использована для микроопределения родия. В этом случае комплексное соединение родия растворяют в ацетоне и заканчивают определение колориметрическим путем. Описан метод8, в котором в качестве осадителя благородных металлов применяется меркаптобензотиазол. - '
Диоксимы, в частности 1,2-циклогександйондиоксим9 и а-фурилдиоксим10, образующие, подобно диметилглиоксиму, внутрикомплексные соли с палладием (II), используются в методах определения палладия. Кроме того, для определения палладия рекомендуется применять 1,1О-фе-нантролин 11.
СИСТЕМАТИЧЕСКИЙ ХОД РАЗДЕЛЕНИЯ Н ОПРЕДЕЛЕНИЯ ПЛАТИНОВЫХ МЕТАЛЛОВ
Ниже описывается систематический ход разделения платиновых металлов в отсутствие золота и других металлов, а также методы определения каждого из шести элементов платиновой группы. Реакции, используемые в методах разделения, были описаны в разделе «Методы разделения» (стр. 406), а реакции, примененные для определения этих элементов, — в разделе «Методы определения» (стр. 416),
1 А. А. Гринберг, Е. А. Максимюк, Известия сектора платины и других благородных металлов, ИОНХ АН СССР, 20, 149 (1947).
2 А. А. Гринберг, _ Е. А. Максимюк, Б. В. Птицын, ДАН СССР, 51, 687 (1946).
3 Е. Muller, К. Н. Т a n z 1 е г, Z. anal. Chem., 89, 339 (1932).
4 Д.И. Рябчиков, Известия сектора платины и других благородных метал-
лов, ИОНХ АН СССР, 22, 35 (1948).
6 J. Е. Curr ah, W. А. Е. М с В г у d е, A. J. С г u i k s h а п к, F. Е. В е а-
m i s h, Ind. Eng. Chem., Anal. Ed., 18, 120 (1946).
8 R. L. Haines, D. E. Ryan, Can. J. Research, 27B, 72 (1949).
, ’ D. E. R yah, Anal. Chem., 22, 599 (1950).
8 W. B. Pollard, Bull. Inst. Mining. Met., 497, 9 (1948); 500, 44 (1948). Cm. также I. Ubaldini, Gazz. chim. ital., 78, 293 (1948); Ann. chim. appl., 38, 241 (1948).
9 R. С. V о t e г, С. V. В a n к s, H. D i e h 1, Anal. Chem., 20, 652 (1948),.
19 S. A. Reed, С. V. Banks, Proc. Iowa Acad. Sci, 55, 267 (1948).
11 D. E. R у a n, P. F a i n e r, Can. J. Research, 27B, 67 (1949).
424
Гл. XX. Платиновые металлы
Подробности этих методов, а также данные, показывающие точность получаемых результатов, можно найти в оригинальной работе1, из которой этот ход анализа взят.
Рекомендуемый ход анализа
Отделение и определение осмия. Аппарат для дистилляции. Аппарат для дистилляции, который показан на рис. 26, состоит из трех основных частей: дистилляционной колбы емкостью 700 мл, трех соединенных между собой поглотительных колб емкостью по 300 мл каждая и ряда соединительных трубок. Трубка, закрытая краном и помещенная между первой и второй поглотительными колбами, служит для введения в погло-
Рис. 26. Прибор для перегонки.
титель сернистой кислоты, если перегоняются слишком большие количества азотной кислоты, а также для ополаскивания (по окончании операции) трубки, соединяющей обе колбы. Аппарат лучше всего сделать из стекла пайрекс. Соединения должны быть тщательно пришлифованы. Весьма удобно, если они сделаны так, чтобы колбы были взаимозаменяемы.
Необходимо отметить, что эти соединения должны быть закрыты пленкой воды, а не смазаны жиром, так как жир может вызвать восстановление некоторого количества четырехокиси осмия до двуокиси, которую затем трудно будет определить. Смазывать жиром необходимо только кран в трубке, служащей для введения азотной кислоты в дистилляционную колбу. В процессе дистилляции эту трубку часто промывают струей воды, чтобы удалить четырехокись осмия, которая может в эту трубку проникнуть.
Четырехокись осмия полностью поглощается в первой поглотительной колбе, и в последующих двух колбах осмий обычно не обнаруживается.
Отделение осмия дистилляцией в виде четырехокиси. В первую поглотительную колбу наливают 150 мл свеженасыщенной сернистым ангидридом разбавленной (1 : 1) соляной кислоты. В последующие две колбы наливают по 50 мл того же реагента. В дистилляционную колбу вводят
1 R. Gilchrist, Е, Wickers, J. Am. Chem. Soc., 57, 2565 (1935).
Систематический ход разделения и определения
425
раствор, содержащий платиновые металлы, и проверяют правильность соединения отдельных частей аппарата. Раствор, если это необходимо, разбавляют в дистилляционной колбе водой до объема 100 мл и через вводную трубку прибавляют 40 мл разбавленной (1:1) азотной кислоты. Трубку и кран промывают 10 мл воды. Через аппарат пропускают слабый ток воздухд-и нагревают’раствор в дистилляционной колбе до кипения. Перегонку продолжают 1 ч, что достаточно для полного удаления осмия из растворов, в которых он находится в виде осмата или бромоосмата щелочного металла. Если же осмий в исходном растворе находится в виде хлороосмата, то для его дистилляции требуется 7—8 ч. В этом случае для ускорения процесса дистилляцию целесообразно проводить из растворов в концентрированной серной кислоте, или если отсутствует рутений, — из растворов в концентрированной серной кислоте, содержащих несколько миллилитров азотной кислоты.
Раствор, который остается в дистилляционной колбе, сохраняют для выделения рутения.
Выделение осмия осаждением его в виде гидроокиси. Растворы из поглотительных колб объединяют и выпаривают, насколько возможно, на водяной бане в чистом с неразъеденной поверхностью стакане. При гидролитическом осаждении платиновых металлов последнее условие весьма существенно, так как в противном случае на стенках стакана образуются пятна осадков, которые не всегда легко удалить. К сухому остатку приливают 10 мл соляной кислоты, нагревают 15 мин и затем снова выпаривают.
Эту операцию повторяют еще 3 раза для того, чтобы обеспечить полное разложение сульфитов осмия. По окончании выпаривания остаток растворяют в 150 мл воды. Раствор нагревают до кипения и добавляют профильтрованный 10%-ный раствор бикарбоната натрия до появления быстро коагулирующегосй осадка. В горячий раствор вводят несколько капель 0,04%-ного раствора бромфенолового синего, желтая окраска которого изменяется в синюю при pH = 4. Затем к раствору прибавляют по каплям бикарбонат натрия до появления слабой синеватой окраски индикатора. После этого раствор кипятят 5—6 мин, чтобы обеспечить полное осаждение гидрата двуокиси оСмия.
Определение осмия в виде металла. Раствор с осажденным гидратом двуокиси осмия фильтруют через платиновый тигель Мунро \ осторожно сливая сначала отстоявшуюся жидкость. Затем переносят в тигель осадок и вытирают стакан и стеклянную палочку хорошо смоченной резинкой, чтобы осадок к ней не прилипал. Необходимо иметь в виду, что в данном случае для удаления осадка со стенок стакана не следует применять фильтровальную бумагу, хотя при перенесении осадков других пяти платиновых металлов ею пользоваться можно. Тщательно промывают осадок горячим 1%-ным раствором хлорида аммония и затем покрывают его этой же кристаллической солью, для чего хлорид аммония смачивают несколькими каплями промывной жидкости и при помощи отсасывания насыщают им осадок.
1 При использовании тигля Гуча с асбестовым фильтром возникают затруднения при прокаливании двуокиси осмия до металла. Даже в том случае, когда тигель Гуча для предохранения от пламени горелки помещают в фарфоровый тигель большего размера на асбестовый кружок, не всегда удается достаточно легко погасить пламя водорода. Кроме того, восстановленный металл не вполне предохраняется от действия воздуха, который проникает через отверстие в дне тигля.
426 Гл. XX. Платиновые металлы
Для пропитывания осадка можно применять также насыщенный раствор хлорида аммония. Отсасывание продолжают до тех пор, пока дно тигля не покроется* твердым слоем хлорида аммония. Этот слой соли вытирают и надевают платиновый колпачок на дно тигля. Затем покрывают тигель крышкой Розе, лучше из кварца, поджигают струю водорода, выходящего из трубки Розе (также кварцевой), и регулируют ток водорода так, чтобы пламя было невелико. После этого трубку вводят в отверстие крышки. Если водород при этом погаснет, его вновь поджигают, подведя на мгновение пламя горелки под тигель. Когда водород загорается, пламя выходит из-под крышки по краю тигля. Горящйй водород выделяет достаточно тепла, чтобы, не сжигая, дегидратировать соединение осмия. Спустя 5 мин постепенно накаливают тигель пламенем горелки до полного улетучивания хлорида аммония. Осадок осмия сильно прокаливают 10 мин в атмосфере водорода, затем удаляют горелку и дают тиглю несколько охладиться. После этого гасят пламя водорода, прервав на мгновение его ток, и охлаждают тигель до комнатной температуры. Наконец, вытес-^ няют водород током двуокиси углерода, предохраняя осадок даже от мгновенного действия воздуха. Если водород не вытесняют каким-нибудь инертным газом, например двуокисью углерода, восстановленный металл при первом же соприкосновении с воздухом быстро подвергается его воздействию, вследствие чего происходят заметные потери осмия. Полученный металлический осмий взвешивают.
Отделение и определение рутения. Приготовление раствора. Раствор после удаления осмия дистилляцией выпаривают досуха на водяной бане, добавляют 5—10 мл соляной кислоты и снова выпаривают. Выпаривание с соляной кислотой повторяют до тех пор, пока не прекратится выделение окислов азота. Затем остаток растворяют в 20—30 мл воды, прибавляют 10 мл серной кислоты и осторожно выпаривают раствор до появления паров серной кислоты. Раствор с осадком платины, который может выделиться, переносят в дистилляционную колбу, применявшуюся для перегонки четырехокиси осмия, и разбавляют водой до 100 мл.
Отделение рутения дистилляцией в виде четырехокиси. Наливают 150 мл свеженасыщенной сернистым ангидридом разбавленной (1 : 1) соляной кислоты в первую поглотительную колбу и по 50 мл того же реагента в каждую из двух последующих колб. Через трубку вводят в дистилляционную колбу 100 мл профильтрованного 10%-ного раствора бромата натрия. Эту трубку время от времени промывают водой, чтобы извлечь четырехокись рутения, которая растворяется в остающейся в трубке воде. Для того чтобы сохранять избыток сернистого ангидрида в поглотителе, время от времени в него вводят воду, насыщенную сернистым ангидридом, через трубку, расположенную между первой и второй поглотительными колбами. Через аппарат пропускают слабый ток воздуха и нагревают раствор в дистилляционной колбе до кипения. Перегоняют 1,5 ч, добавляют 25 мл раствора бромата и продолжают дистилляцию еще 1 ч.
Выделение рутения в виде гидроокиси. Объединяют растворы из поглотительных колб и выпаривают на водяной бане. Влажный остаток обрабатывают 10 мл соляной кислоты и нагревают раствор в закрытом стакане на водяной бане 30 мин. Прибавляют 50 мл воды и нагревают до кипения для растворения образующихся при выпаривании малорастворимых соединений рутения. По окончании растворения фильтруют и промывают фильтр разбавленной (1 : 99) соляной кислотой. Этим фильтрованием
Систематический ход разделения и определения 427
удаляют небольшие количества кремнекислоты, которая может здесь присутствовать. Раствор, содержащий рутений, разбавляют до 200 мл, нагревают до кипения и затем добавляют профильтрованный 10%-ный раствор бикарбоната натрия до начала образования осадка. Вводят 3—4 капли 0,04%-ного раствора бромкрезолового пурпурного и заканчивают нейтрализацию избытка кислоты, добавляя по каплям растврр бикарбоната до изменения желтой окраски индикатора в синюю. Раствор кипятят 5 мин для коагуляции осадка.
Образующийся осадок представляет собой гидроокись рутения, возможно, трехвалёнтного. Обычно по внешнему виду осадок сходен с гидроокисями трехвалентных форм других платиновых металлов. Он менее компактен и не так быстро оседает, как осадки, образующиеся при гидролизе соединений рутения (IV), но осаждается количественно, и при последующей работе с этим осадком никаких затруднений не возникает.
Определение рутения в виде металла. Осадок гидроокиси рутения отфильтровывают и остатки его со стенок стакана и стеклянной палочки снимают небольшим кусочком беззольной фильтровальной бумаги. Фильтр и осадок тщательно промывают - горячим 1 %-ным раствором сульфата аммония, а под конец 3—4 раза холодным 2,5%-ным раствором той же соли. Затем фильтр помещают в фарфоровый тигель, сушат и медленно обугливают. Высушенный фильтр обычно обугливается полностью, если только юн начал дымиться. Эту операцию следует проводить осторожно, чтобы избежать потери рутения вследствие воспламенения фильтра. Сильно прокаливают осадок сначала на воздухе, а затем в атмосфере водорода так, как это описано при определении осмия. Металл охлаждают в атмосфере водорода, после чего выщелачивают горячей водой для удаления следов растворимых солей. Целесообразно выщелачивание провести сначала в тигле, а затем осадок перенести на фильтр. Полученную металлическую губку вместе с фильтром прокаливают сначала на воздухе, а затем в токе водорода, после чего металлический рутений охлаждают также в токе водорода и взвешивают.
Отделение и определение плнтины. Раствор, остающийся в дистилляционной колбе после удаления рутения, . содержит платину, палладий, родий и иридий, а также серную кислоту, сульфат или бисульфат натрия, бром и неразложенный бромат. Опыт показывает, что платина, частично выделяющаяся при приготовлении раствора для перегонки рутения, полностью растворяется в 1 процессе дистилляции. К концу перегонки иногда осаждаются следы иридия в виде двуокиси.
Приготовление раствора. Обработка раствора, остающегося после дистилляции. Содержимое дистилляционной колбы переносят в стакан емкостью 1 л и осторожно разлагают бромат соляной кислотой. При этой операции следует соблюдать соответствующие предосторожности во избежание механических потерь вследствие сильного выделения газа. По прекращении бурной реакции раствор выпаривают й удостоверяются в том, что оставшийся бромат разложился в процессе упаривания с соляной кислотой. На дистилляционной колбе вблизи уровня раствора иногда остаются пятна двуокиси иридия, поэтому целесообразно ополаскивать колбу 5—10 мл царской водки. Эту жидкость выпаривают с соляной кислотой для разложения нитрозосоединений и затем присоединяют к основному раствору. После этого раствор,
428.
Гл. XX. Платиновые металлы
насколько возможно, выпаривают на водяной бане и разбавляют водой до 200 мл.
Обработка раствора, не содержащего осмия и рутения. При отсутствии в анализируемом растворе осмия и рутения операции, выполняемые для их отделения, исключаются и анализируемый раствор выпаривают на водяной бане до получения влажного остатка. Если раствор Содержал азотную кислоту, обрабатывают 5 мл соляной кислоты и снова выпаривают. Эту операцию повторяют до полного разложения нитрозосоединений. Прибавляют 2 г хлорида натрия, 5 мл соляной кислоты и на этот раз выпаривают на водяной бане досуха. Остаток обрабатывают 2 мл соляной кислоты и разбавляют водой до 300 мл.
Отделение платины совместным осаждением палладия, родия и иридия в виде гидроокисей. Раствор, содержащий платину, палладий, родий и иридий, нагревают до кипения и прибавляют 20 мл профильтрованного 10%-ного раствора бромата натрия. Затем осторожно вводят профильтрованный 10%-ный раствор бикарбоната натрия1 до появления в темнозеленом растворе заметного неисчезающего осадка. Время от времени проверяют кислотность раствора, давая капле2 0,01 %-ного раствора бромкрезолового пурпурного стекать по вынутой из анализируемого раствора стеклянной палочке, на кончике которой остается капля анализируемой жидкости. Когда желтая окраска индикатора переходит в синюю, добавление бикарбоната прекращают. По окончании нейтрализации к раствору добавляют еще 10 мл раствора бромата и кипятят 5 мин. Слегка повышают pH раствора, осторожно добавляя по каплям раствор бикарбоната до появления бледно-розовой окраски при испытании капли анализируемого раствора с каплей 0,1 %-ного раствора крезолового красного или бледно-синей окраски — с раствором ксиленолового синего. После этого снова добавляют 10 мл раствора бромата и кипятят 15 мин.
Когда прекращают нагревание, осадок быстро оседает. Платина остается в растворе. Раствор фильтруют под вакуумом через толстостенный фарфоровый тигель с пористым дном. Если предстоит переосаждение осадка, весьма желательно избежать применения фильтровальной бумаги. Продукты, входящие в состав бумаги, реагируют с кислотами и, возможно, образуют небольшие количества трудно гидролизующихся органических соединений с платиновыми металлами. Двуокись иридия, переходящая в раствор, значительно труднее, чем двуокиси палладия и родия, имеет склонность окрашивать мацерированную бумагу, которую потом не всегда удается отмыть. Эти затруднения устраняются при использовании для фильтрования фарфорового тигля, удобного еще и тем, что для растворения осадка можно пользоваться концентрированной соляной кислотой, что
1 При гидролитическом осаждении палладия, родия и иридия для нейтрализации раствора вместо бикарбоната натрия можно пользоваться свежеприготовленным раствором едкого натра. В случае применения едкого натра, в процессе нейтрализации раствор следует непрерывно перемешивать, чтобы избежать местного пересыщения его щелочью.
2 Ввиду того что нейтрализуемый раствор сильно окрашен или содержит осадок, для определения конечной точки нейтрализации рекомендуется способ, в котором капля 0,01%-ного раствора индикатора реагирует с каплей раствора на кончике стеклянной палочки. Это не является обычным методом с применением «внешнего индикатора». Для выполнения этой операции палочку поднимают над уровнем раствора и испытывают приставшую к ней каплю жидкости, а затем палочку вновь опускают в стакан, избегая таким образом потерь раствора.
Систематический ход разделения и определения
429
значительно сокращает продолжительность подготовки раствора для последующей операции.
В фильтрующий тигель сначала сливают отстоявшуюся жидкость, а затем переносят осадок. Ополаскивают стакан и промывают осадок горячим 1 %-ным раствором хлорида натрия, нейтральным по крезоловому красному или ксиленоловому синему (pH около 8). Содержащие платину фильтрат и промывную жидкость сохраняют. Тигель с осадком, а также стеклянную палочку помещают в стакан, в котором проводилось осаждение. Небольшое количество осадка, в процессе фильтрования приставшее к краю стакана, лучше снять влажными кристаллами хлорида натрия, положенными на палец, чем бумагой или резинкой. Стакан накрывают часовым стеклом и вводят 10—20 мл соляной кислоты, наливая большую часть ее в тигель. Закрытый стакан ставят на водяную баню. Соединения родия и палладия растворяются быстро, а двуокись иридия — значительно медленнее. Осторожно поднимают тигель стеклянной палочкой, обмывают его водой и помещают в стакан емкостью 250 мл. Наливают в тигель 5 мл соляной кислоты, накрывают стакан часовым стеклом и ставят на водяную баню. Этой обработкой обычно выщелачивают небольшие количества хлоридов из пор дна тигля. Для полного извлечения операцию повторяют со свежей порцией кислоты. Соединяют. эту жидкость с основным раствором, добавляют 2 г хлорида натрия и выпаривают на водяной бане досуха. Остаток обрабатывают 2 мл соляной кислоты, разбавляют раствор до 300 мл и повторяют осаждение гидроокисей. Двукратного осаждения обычно бывает достаточно для полного отделения платины от палладия, родия и иридия. Снова растворяют осадок, как было указано выше, и сохраняют раствор для отделения палладия.
Выделение платины осаждением ее сероводородом. К каждому фильтрату, полученному после гидролитического осаждения двуокисей палладия, родия и иридия, прибавляют по 20 мл соляной кислоты. Растворы осторожно нагревают, пока не прекратится реакция, немного упаривают их, затем объединяют и выпаривают досуха. Для того чтобы бромат полностью разрушился, повторяют выпаривание с соляной кислотой. Прибавляют небольшое количество воды и полученный желтый раствор хлороплатината фильтруют. Фильтр промывают разбавленной (1 : 99) соляной кислотой. Фильтрат разбавляют водой до 400 мл и добавляют соляную кислоту с таким расчетом, чтобы в 100 мл раствора содержалось 5 мл кислоты.
Платину осаждают из горячего раствора сильной струей сероводорода. Чтобы обеспечить полноту осаждения, продолжают пропускать сероводород и тогда, когда раствор немного охладится.
Определение платины в виде металла. Осадок отфильтровывают и промывают разбавленной (1 : 99) соляной кислотой. Фильтр с осадком сушат и прокаливают в фарфоровом тигле. Полученный металл выщелачивают разбавленной соляной кислотой, переносят на фильтр, тщательно промывают горячей водой и снова сильно прокаливают на воздухе. После этого металлическую платину взвешивают. Полученный таким образом металл обычно содержит небольшие, но заметные количества серы, которая не удаляется прокаливанием ни на воздухе, ни в атмосфере водорода.
Для получения более точных результатов металлическую платину, образовавшуюся в результате прокаливания сульфида, растворяют в царской водке. Нитрозосоединения разлагают выпариванием с соляной
430
Гл. XX. Платиновые металлы
кислотой. Фильтруют раствор в чистый с неразъеденной внутренней поверхностью стакан и промывают фильтр разбавленной (1 : 99) соляной кислотой. Разбавляют раствор до 100 мл водой, нагревают до кипения и прибавляют раствор, содержащий 3 г ацетата натрия1 и 1 мл муравьиной кислоты на каждые 0,25 г платины, после чего слабо кипятят до тех пор, пока осадок металлической платины не скоагулирует, а жидкость не обесцветится. Металл отфильтровывают и промывают горячим 1%-ным раствором хлорида аммония. Фильтр с металлической губкой помещают в фарфоровый тигель и сильно прокаливают на воздухе. Для удаления растворимых солей прокаленный металл выщелачивают и промывают, как указано выше. После этого осадок снова прокаливают на воздухе. Металлическую платину, освобожденную таким- образом от серы, взвешивают.
Отделение и определение палладия. Осаждение палладия диметилглиоксимом. Раствор, полученный после растворения осадка гидроокисей палладия, родия и иридия, фильтруют, разбавляют приблизительно до 400 мл и осаждают палладий 1 % -ным раствором диметилглиоксиМа в 95 %-ном этиловом спирте. Для осаждения 1 г палладия требуется примерно 2,3 г диметилглиоксима. Чтобы обеспечить необходимый для полного выделения палладия избыток реагента, на каждые 100 мг палладия следует ввести 25 мл раствора диметилглиоксима. После добавления осадителя раствор цставляют при комнатной температуре в течение 1 ч и затем фильтруют. Полноту осаждения палладия проверяют, добавляя к фильтрату диметил глиоксим. Применяемый способ фильтрования зависит от того, в какой форме надлежит определить палладий. Осадок промывают сначала разбавленной (1 : 99) соляной кислотой, а затем водой, температура которой не должна быть выше 85° С. Однократного осаждения достаточно для количественного отделения палладия от родия и иридия. Фильтрат и промывные воды сохраняют для отделения родия.
Определение палладия в виде диметилглиоксимата палладия. Соединение палладия с диметилглиоксимом имеет определенный состав и достаточно устойчиво, благодаря чему его можно, высушив, непосредственно ' взвешивать. Когда палладий определяют таким путем, осадок диметилглиоксимата палладия отфильтровывают под вакуумом через стеклянный или фарфоровый фильтрующий тигель, промывают, как указано выше, и сушат при 110° С в течение 1 ч. Содержание палладия вычисляют, пользуясь теоретическим фактором 0,3167.
Определение палладия в виде металла. При определении палладия в виде металла, что иногда бывает удобнее, осадок отфильтровывают через беззольный фильтр. Вытирают стенки стакана и стеклянную палочку небольшим кусочком беззольной бумаги. Фильтр с осадком завертывают в другой фильтр, помещают в фарфоровый тигель, сушат и осторожно нагревают на воздухе лишь до такой степени, чтобы фильтр слабо дымился. Обугленный осадок сильно прокаливают сначала на воздухё, затем в токе
1 Установлено, что при осаждении платины муравьиной кислотой проще и целесообразнее нейтрализовать раствор едким натром по бромфеноловому синему (pH — 4). В этом случае металл осаждается полностью при нагревании на водяной бане и нет необходимости раствор кипятить. Осаждение может быть осуществлено в растворах, содержащих значительные количества солей натрия, что недопустимо при использова- -нии ацетата натрия. Кроме того, таким способом часто можно избежать предварительного осаждения сероводородом.
Систематический ход разделения и определения
431.
водорода и, наконец, 2 мин в атмосфере двуокиси углерода1. По охлаждении (также в токе двуокиси углерода) металлический палладий взвешивают.
Отделение и определение родия. Приготовление раствора. Раствор, содержащий родий и иридий в виде хлоридов и избыток диметилглиоксима, оставшийся после осаждения/палладия, упаривают до небольшого объема и переносят в коническую колбу емкостью 500 мл. Закрывают колбу воронкой с короткой трубкой, вводят 10 мл серной кислоты, 2—3 мл азотной кислоты и выпаривают до появления густых паров серной кислоты. Для полного разложения органических веществ время от времени добавляют небольшие количества азотной кислоты и под конец нагревают на голом пламени, непрерывно перемешивая раствор. По охлаждении разбавляют раствор 20 мл воды и снова выпаривают до появления паров серной кислотц. Целью такой обработки является разложение нитрозосоединений, кбторые могут*препятствовать осаждению родия хлоридом титана (III).
Осаждение родия хлоридом титана (III). Сернокислый раствор переносят в чистый с неразъеденными стенками стакан, разбавляют до 200 мл и нагревают до кипения. Вводят по каплям раствор хлорида титана (III) (обычно 20%-ный) до тех пор, пока отстоявшаяся жидкость не окрасится в слабо пурпурный цвет2’3.' Конечную точку значительно легче наблюдать, если раствор поместить над лампой мощностью 100 ватт и перемешивать его. Выделяющийся металлический родий быстро коагулирует, образуя губчатую массу. '
В присутствии больших количеств иридия конечную точку можно определить по прекращению образования осадка и появлению оранжевой окраски раствора. Раствор кипятят 2 мин и фильтруют. Стенки стакана и стеклянную палочку вытирают беззольной фильтровальной бумагой. Фильтр и осадок тщательно промывают холодной разбавленной (2,5 : 97,5) серной кислотой. Фильтрат и промывные воды, содержащие иридий, сохраняют.
Фильтр с осадком переносят в коническую колбу емкостью 500 мл, вводят 10 мл серной кислоты и осторожно его обугливают. Затем добавляют 5 мЛ азотной кислоты и нагревают раствор на горйчей плите. Полного растворения родия достигают, нагревая колбу на голом пламени при непрерывном перемешивании раствора. Следует добиваться разрушения всех органических веществ и разложения нитрозосоединений. Если остаются неразложившиеся черные частички, то раствор разбавляют, фильтруют и фильтр опускают обратно в колбу, содержащую фильтруемый раствор. Стенки стакана вытирают беззольной фильтровальной бумагой. Вводят в колбу 5 мл серной кислоты, обугливают бумагу и раз-' рушают органические вещества азотной кислотой, после чего раствор
1 Палладий обладает свойством поглощать значительные количества водорода, поэтому трудно получить постоянную массу, если металл впоследствии не прокалить в атмосфере инертного газа.
2 Н. Н о 1 z е г и Е. Z a u s s i n g e r (Z. anal. Chem., Ill, 321 (1938)] рекомендуют применять значительно более разбавленный раствор хлорида титана (III) и вводить метиленовую синюю в качестве индикатора.
3 Н. К. Пшеницын [Известия сектора платины и других благородных металлов ИОНХ АН СССР, 22, 16 (1948)] предлагает метод, основанный на восстановлении родия хлоридом хрома (II). Реакция проводится в 10 %-ном по содержанию соляной кислоты растворе без доступа воздуха.
432
Гл. XX. Платиновые металлы
нагревают до появления густых паров серной кислоты. При этой обработке металл переходит в раствор и остается лишь небольшой бесцветный осадок кремнекислоты.
Затем осаждают родий вторично, как описано выше. Фильтрат и промывные воды, содержащие небольшие количества иридия, сохраняют. Металлический родий снова растворяют, как было указано, сернокислый раствор разбавляют 20 мл воды и 10 мл соляной кислоты, а затем кипятят 15 мин. При этой обработке родий переводится в форму, необходимую для количественного осаждения его сероводородом. В процессе обработки желтая окраска раствора переходит в розовую. Раствор фильтруют и промывают фильтр разбавленной (1 : 99) соляной кислотой. Фильтрат разбавляют до 400—500 мл.
Выделение родия осаждением сероводородом. Родий осаждают в виде сульфида сильной струей сероводорода из нагретого до кипения раствора. Раствору дают немного охладиться, не прекращая тока' сероводорода.
Определение родия в виде металла. Осадок сульфида отфильтровывают и промывают сначала разбавленной (2,5 : 97,5) серной кислотой, а затем разбавленной (1 : 99) соляной кислотой. Фильтр с осадком переносят в фарфоровый тигель, высушивают и осторожно прокаливают на воздухе, после чего окисленный осадок прокаливают и охлаждают в атмосфере водорода. Полученный в результате этого металлический родий взвешивают.
Выделение и определение иридия. Иридий можно определить одним из следующих двух способов. Если раствор, содержащий родий и иридий, можно разделить на аликвотные части, определение иридия значительно упрощается и осаждения титана купфероном можно избежать. В одной части раствора родий и иридий осаждают гидролитически, как описано при отделении от платины (стр. 408). Осадок гидроокисей родия и иридия промывают горячим 1 %-ным раствором хлорида аммония, нейтральным по бромтимоловому синему (pH = 7). Фильтр с осадком высушивают в фарфоровом тигле и затем во избежание воспламенения пропитывают несколькими каплями насыщенного раствора хлорида аммония. Осторожно прокаливают до получения безводных окислов, которые последующим прокаливанием в токе водорода переводят в металл. По охлаждении в атмосфере водорода смесь металлов взвешивают.
Для вычисления содержания иридия вычитают находящееся в осадке количество родия, которое определяют в отдельной порции раствора, как описано выше. Если иридий таким образом определить не представляется возможным, следует установить его содержание в фильтрате после осаждения родия хлоридом титана (III).
Отделение титана осаждением купфероном. Объединенные филь-траты'после осаждения родия хлоридом титана (III) разбавляют до 800 мл и охлаждают во льду. Прибавляют охлажденный, профильтрованный, свежеприготовленный 6%-ный раствор купферона (аммонийная соль нитрозофенилгидроксиламина C5HeN(NO)ONH4) в небольшом избытке. Осадок титана отфильтровывают и промывают охлажденной разбавленной (2,5 : 97,5) серной кислотой, содержащей немного купферона. Фильтрат и промывные воды/содержащие иридий, сохраняют. Купферонат титана обычно бывает слегка загрязнен иридием, количества которого, однако, не превышают 1 мг при содержании иридия порядка 0,2 г в исходном растворе. Фильтр с осадком переносят обратно в стакан, в котором про
Систематический ход разделения и определения
433
водилось осаждение, прибавляют 20 мл азотной кислоты и нагревают до тех пор, пока большая часть осадка не разложится. Затем вводят 20 мл серной кислоты и выпаривают до появления паров серной кислоты. Оставшиеся органические вещества разрушают нагреванием с азотной кислотой. Полученный раствор разбавляют до 800 мл и повторяют осаждение титана. Оба фильтрата и промывные воды объединяют и выпаривают до тех пор, пока не останется приблизительно 10 мл серной кислоты. В полном разрушении органических веществ убеждаются способом, описанным ранее; раствор несколько разбавляют и фильтруют.
Выделение иридия в виде гидрата двуокиси. Раствор, содержащий иридий, разбавляют водой до 200 мл и нейтрализуют большую часть кислоты свежеприготовленным раствором едкого натра. Нагревают до кипения и заканчивают нейтрализацию едким натром или бикарбонатом натрия по бромкрезоловому пурпурному, как описано при отделении от платины. После этого вводят 20 мл профильтрованного 10%-ного раствора бромата натрия и кипятят 20—25 мин. Удостоверяются в том, что в растворе содержится достаточное количество бромата для окисления иридия до четырехвалентного состояния. Выделившийся осадок отфильтровывают и тщательно промывают горячим 1%-ным раствором хлорида аммония.
Определение иридия в виде металла. Помещают фильтр с осадком в фарфоровый тигель, немного подсушивают и затем смачивают несколькими каплями насыщенного раствора хлорида аммония. Осторожно прокаливают осадок сначала на воздухе, а затем в токе водорода. Полученный металл выщелачивают разбавленной соляной кислотой, переносят на фильтр, промывают горячей водой и снова прокаливают на воздухе и в токе водорода. Металлический иридий охлаждают в атмосфере водорода и взвешивают.
Описанные здесь методы анализа рассчитаны на получение наиболее точных результатов, но в тех случаях, когда можно ограничиться меньшей точностью определения, ход анализа может быть значительно сокращен. Так, например, в некоторых стадиях анализа можно исключить пере-осаждение осадков. В частности, можно ограничиться однократным гидролитическим осаждением палладия, родия и иридия при отделении их от платины. Определение платины можно закончить непосредственным прокаливанием сульфида, пренебрегая незначительными количествами серы, которые при этом задерживаются в осадке. Можно исключить и вторичное восстановление родия хлоридом титана (III), а также избежать осаждения титана купфероном, определив родий и иридий в аликвотных частях раствора.,Необходимо, однако, иметь в виду, что при этом могут иметь место ошибки, величина которых зависит от относительного содержания металлов, находящихся в анализируемой пробе.
28 8акаа >22.
ГРУППА СУЛЬФИДА АММОНИЯ
ЖЕЛЕЗО, НИКЕЛЬ, КОБАЛЬТ, ЦИНК, МАРГАНЕЦ, ВАНАДИЙ, УРАЙ, ТАЛЛИЙ, ИНДИЙ, ГАЛЛИЙ, АЛЮМИНИЙ, БЕРИЛЛИЙ, ХРОМ, ТОРИЙ, СКАНДИЙ, РЕДКОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ, ЦИРКОНИЙ, ТИТАН, НИОБИЙ И ТИНТАЛ
ЭЛЕМЕНТЫ, ОБРАЗУЮЩИЕ ПРИ ДЕЙСТВИИ (NH4)2S РАСТВОРИМЫЕ В КИСЛОТАХ СУЛЬФИДЫ
ЖЕЛЕЗО, НИКЕЛЬ, КОБАЛЬТ, ЦИНК, МАРГАНЕЦ, ВАНАДИЙ, УРАН, ТАЛЛИЙ, ИНДИЙ, ГАЛЛИЙ
Глава XXI
ЖЕЛЕЗО
Железо по своему содержанию в земной коре следует сейчас же за алюминием — самым распространенным металлом. Оно находится практически во всех горных породах, особенно в тех, которые содержат амфиболы, пироксены, слюды и оливин.' Железо является составной частью сотен различных минералов. Исключая пириты, марказиты и редко встречающиеся металлическое самородное железо и его сплавы, железо в природе находится в двухвалентном и трехвалентном состояниях. Это справедливо и в отношении магнитной окиси железа Fe3O4, которая образует при растворении в условиях, исключающих окисление, определенные количества соединений железа (II) и (III). Большое значение методов определения железа не требует доказательств.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород предварительная обработка анализируемой пробы приводит к получению железа в трехвалентном состоянии, в котором оно количественно осаждается аммиаком. Общее содержание железа может быть затем определено, как указано в разделе «Методы определения» (стр. 439), после растворения прокаленного и взвешенного осадка. Для некоторых целей удобнее определять железо из отдельной навески или же разделить фильтрат, полученный после отделения кремнекислоты, на две равные части: одну — для определения железа, другую — для осаждения аммиаком.
Если железо первоначально присутствует в двухвалентном и трехвалентном состояниях, то обычно в одной навеске пробы определяют общее содержание железа, а в другой — либо двухвалентное, либо трехвалент-
Разложение минералов, содержащих железо 435
ное железо. Можно также провести эти две операции и с одной навеской, определив объемным путем сначала двухвалентное или трехвалентное железо, а затем и общее его содержание \
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ЖЕЛЕЗО
Типы минералов, содержащих железо, настолько разнообразны, что из многочисленных возможных методов их разложения каждый находит применение. Некоторые минералы растворимы в воде. Многие окисленные минералы, нерастворимые в воде, разлагаются соляной кислотой, азотной кислотой или царской водкой, часто лишь после очень тонкого измельчения и продолжительного действия кислоты. Для разложения многих кислотоупорных минералов требуется сплавление с различными плавнями, указанными на стр. 919. В качестве плавней могут применяться как щелочно-окислительные смеси, так и пиросульфаты и даже кислые фториды. Выбор плавня зависит от природы анализируемого материала и намеченной цели. При анализе сульфидов и арсенидов щелочное сплавление часто предпочитают кислотной обработке, потому что при выщелачивании плава водой достигается количественное отделение серы, мышьяка, фосфора, ванадия и молибдена от многих основных металлов. Вот почему при определении серы в пиритсодержащих рудах кислотной обработке предпочитают метод щелочного сплавления.
Один вариант обработки щелочами применялся одним из нас с успехом при анализе некоторых растворимых в соляной кислоте железных минералов и руд, например щелочных минералов группы ярозита K[Fe(OH)2]3(SO4)2 — основных щелочно-железных сульфатов. Последние при нагревании с концентрированными растворами едких щелочей быстро разлагаются, образуя легко отделяемый фильтрованием остаток гидроокиси железа и раствор, содержащий все сульфат-ионы. Гиллебранд впервые применил этот метод к анализу смеси ярозита и мышьякового пироморфита. Последний содержит Р2О5, As2O6 и С1, переходящие в раствор вместе с SO|~ , что облегчает анализ. Вдумчивый аналитик часто будет применять для анализа специальные методы обработки, вроде изложенного выше.
Обычно, когда железо отделяют от других металлов аммиаком или иными осадителями, часто после предварительного отделения кремния и элементов сероводородной группы1 2 желательно иметь его в растворе в виде хлорида. Поскольку такие растворы получаются при анализе горных пород и силикатов, они описаны в гл. LIII (см. стр. 919 и 936),
1 В статье J.O. Percival [Ind. Eng. Chem., Anal. Ed., 13, 71 (1941)] описан метод раздельного определения двухвалентного и трехвалентного железа, присутствующих в разбавленном сернокислом растворе. Сначала титруют железо (II) перманганатом, затем раствор сильно встряхивают в течение 2 мин с порошком меди (осажденная медь, диаметр частиц от 10 до 35 .ик), фильтруют и титруют перманганатом общее количество железа.
2 Следует помнить, что платина несколько растворяется при действии солянокислых растворов, содержащих железо; поэтому такие растворы, если содержание железа в них велико, нельзя кипятить или выпаривать в платиновых чашках. При таком выпаривании происходит некоторое восстановление трехвалентного железа в двухвалентное. Восстановление не происходит при выпаривании в фарфоровой посуде, а при выпаривании сернокислых растворов сульфата железа (III) — нп в платиновой, ни в фарфоровой посуде.
28*
436
Гл. XXI. Железо
а получение таких растворов при анализе сульфидов и арсенидов — на стр. 927. Многие железосодержащие минералы, стойкие в отношении соляной кислоты, разлагаются при действии азотной кислоты, царской водки, хлора или брома и затем могут быть переведены в требуемое состояние выпариванием с соляной кислотой.
Азотнокислые, а иногда й сернокислые растворы также вполне пригодны, но соляная кислота гораздо менее мешает в следующих операциях и ее, как правило, нужно предпочесть при первоначальном отделении группы сероводорода. В последнем случае фильтрат от осаждения сульфидов содержит железо (II), которое можно сразу осаждать сульфидом аммония. Но если предстоит осаждение аммиаком или ацетатом, то фильтрат надо освободить от избытка сероводорода кипячением и затем для окисления железа обработать окислителем — хлором, бромом или перекисью водорода. Избыток окислителя, если он в дальнейшем может причинить затруднения, как, например, в присутствии марганца, затем удаляют (обычно кипячением). Азотная кислота является менее пригодным окислителем, чем перечисленные выше \
Материалы, применяемые для производства некоторых сортов стекла, особенно оптического, содержат очень незначительные количества железа. Поэтому при определении железа в песках, идущих на производство стекла, песок обычно обрабатывают фтористоводородной и серной кислотами для удаления кремния и затем нагревают до удаления всей фтористоводородной кислоты и большей части серной кислоты. Было отмечено 1 2, что при этой операции некоторые входящие в состав таких песков минералы не разлагаются, например магнетит, ильменит, турмалин и ставролит, и что получаемые этим методом результаты определения железа выражают только часть действительно находящегося в песке железа. Остаток после обработки песка фтористоводородной и серной кислотами и после выпаривания до появления густых паров серной кислоты следует собрать, сплавить с пиросульфатом калия, и если сплав будет все еще содержать черные частицы (ставролит), то их нужно сплавить с карбонатом натрия.
При анализе стандартного образца песка для производства стекла (№ 81, Бюро Стандартов США) оказалось, что лучше проводить эту операцию в обратном порядке, т. е. сначала сплавлять с карбонатом натрия,, затем с пиросульфатом. Некоторые минералы, например циркон, лишь слабо поддаются действию пиросульфата, но разлагаются при сплавлении с карбонатом натрия с образованием нерастворимых соединений, которые легко переходят в раствор после сплавления их с пиросульфатом. Применяемые в анализе реактивы должны быть, конечно, свободными от железа, а при пользовании платиновой посудой отсутствие в ней железа является важнейшим условием. Тщательно проведенные «холостые» опыты со всеми реактивами могут устранить ошибку, вызванную наличием в них железа, но не дают точной поправки на железо, попадающее в раствор из платиновой посуды.
Железо (III) можно быстро восстановить до двухвалентного состояния следующим образом. К анализируемому раствору приливают раствор
1 С. F. Bonilla [Ind. Eng. Chem., Anal. Ed., 4, 128 (1932)] исследовал скорости растворения железа в царской водке и в различных «видоизмененных царских .водках».
2 J. В. Ferguson, J. Ind. Eng. Chem., 10, 941 (1918).
Методы, отделения
437
аммиака до появления неисчезающего осадка, затем медленно прибавляют 1 н. раствор соляной кислоты, пока цвет анализируемого раствора не перейдет в чисто желтый (не оранжевый или красновато-коричневый). После этого разбавляют раствор до 200—300 мл, прибавляют 5—15 г тиосульфата натрия, осторожно перемешивают, нагревают до кипения и кипятят 5 мин.
МЕТОДЫ ОТДЕЛЕНИЯ
Главными, методами отделения железа от остальных элементов являются: 1) обработка сероводородом в кислом растворе (стр. 83), в результате которой металлы группы сероводорода, например висмут или мышьяк, осаждаются, а железо остается в растворе; 2) осаждение сульфидом аммония в растворе, содержащем тартрат аммония (стр. 115); при этом железо осаждается в виде сульфида железа, а алюминий, титан и другие элементы остаются в растворе; 3) осаждение едким натром (стр. 109), в результате которого железо переходит в осадок и отделяется от ванадия, вольфрама, молибдена, мышьяка, алюминия и фосфора; 4) сплавление с карбонатом натрия с последующим выщелачиванием плава водой (стр. 511), дающее практически тот же результат, что и предыдущий метод, с тем лишь различием, что алюминий в этом случае обычно отделяется не полностью, хром окисляется и переходит в раствор, а уран частью остается в остатке, частью переходит в раствор; 5) извлечение эфиром из разбавленного солянокислого раствора (стр. 161), которое применяется главным образом для удаления большей части железа, если оно присутствует в таких больших количествах, что создаются затруднения при определении других элементов.
Если от щелочных и щелочноземельных металлов аммиаком отделяется одно железо (III) или железо в сопровождении только титана и циркония и раствор не содержит фосфора в количестве, превышающим то, какое может быть связано железом, точная нейтрализация, необходимая для полного осаждения алюминия, значения не имеет. Не требуется также присутствия значительных количеств аммонийных солей, если только не приходится принимать во внимание наличия магния. Но если присутствует алюминий или требуется отделение железа от магния, цинка, марганца, никеля и кобальта, то нужно применить метод, описанный для отделения алюминия 1 * * * * * * В (стр. 565). Бумажную массу следует прибавлять при последнем из двух или нескольких переосаждений железа. Она полезна тем, что делает осадок Fe2O3 после прокаливания более тонко измельченным
1 G. Е. F. Lundell и Н. В. Knowles [J. Am. Chem. Soc., 45, 676 (1923)] тщательно исследовали точность этого метода при использовании его для отделения алюминия, железа и других аналогичных элементов от марганца, цинка, никеля,
кобальта и меди. Они пришли к следующим выводам: 1) умеренные количества железа и алюминия могут быть отделены от марганца и никеля аммиаком так же удовлетворительно, как и ацетатом натрия или карбонатом бария; 2) при указанных выше условиях
отделение железа и алюминия от кобальта, меди и цинка неполно; большой избыток
хлорида аммония улучшает это отделение; 3) избыток аммиака и хлорида аммония
дает лучшее отделение от меди и цинка, но при этих условиях осаждение алюминия
неполно и отделение от марганца, никеля и кобальта менее удовлетворительно; 4) фос-
фор и ванадий мало мешают отделению, если железо или алюминий преобладают.
В противном же случае они образуют нерастворимые соединения с марганцем и мешают не только при отделении аммиаком, но и при^отделении ацетатами или карбонатом бария.
438
Гл. XXI. Железо
и легче растворимым. Следует помнить, что, кроме железа, аммиаком осаждаются многие другие элементы (см. стр. 102) и что осадок может захватить с собой вольфрам, ванадий, уран, мышьяк и фосфор.
Вероятно, одним из лучших методов.отделения железа от других элементов при анализе горных пород и подобных им материалов является осаждение его сульфидом аммония в присутствии тартратом (стр. 115) после предварительного отделения сероводородной группы сероводородом в растворе, содержащем минеральную и винную кислоты х. Этим методом железо может быть отделено от алюминия, титана, циркония, ниобия, тантала, урана, ванадия и фосфора. Элементы, сопровождающие железо при этом разделении, — никель, кобальт, цинк и маранец (частично) — редко встречаются в горных породах и легко отделяются, например никель и марганец, осаждением железа аммиаком. Сульфид железа для дальнейшей обработки нужно растворить. Для этого возможно два метода:
1) При последующем определении железа весовым способом сульфид железа разлагают вместе с бумагой фильтра, как указано в п. 2, или помещают фильтр с осадком в стакан подходящего объема, покрывают часовым стеклом и осторожно растворяют осадок в разбавленной (1:1) соляной кислоте. Нагревают до исчезновения всех черных частичек, измельчают бумагу сильным перемешиванием стеклянной палочкой с резиновым наконечником, окисляют железо несколькими каплями концентрированной азотной кислоты или небольшим количеством бромной воды, разбавляют, если необходимо, водой и осаждают железо в виде гидроокиси. Так как алюминий здесь отсутствует, избытка аммиака опасаться не приходится.
2) Если железо в дальнейшем йредполагают определять объемным способом, фильтр с осадком помещают в коническую колбу, прибавляют 10 мл азотной кислоты, встряхивают колбу до размельчения бумаги и затем прибавляют 5 мл серной кислоты. Осторожно перемешивают содержимое колбы, нагревая ее на голом пламени, и выпаривают до появления паров серной кислоты. Если раствор не станет прозрачным и бесцветным, его охлаждают немного, прибавляют еще 5 мл азотной кислоты и снова выпаривают. Наконец, несколько охлаждают и для разрушения всех азотистых соединений очень осторожно прибавляют по каплям насыщенный раствор перманганата калия до заметного его избытка. Охлаждают, разбавляют раствор до 100 мл водой и определяют железо, как указано на стр. 440.
Методы осаждения железа йвиде основного ацетата (стр. 103), а также добавлением сукцината натрия или карбоната бария (стр. 106,108) очень хороши в тех случаях, когда надо отделить железо от цинка, марганца, никеля и кобальта, особенно если последние присутствуют в больших количествах.
1 При выполнении такого осаждения к первоначально кислому анализируемому раствору прибавляют винную кислоту и делают его аммиачным, чтобы убедиться в том, что винной кислоты было прибавлено достаточное количество и что мешающие вещества (например, магний вместе с фосфат-ионами) отсутствуют. Если раствор остается прозрачным, его снова подкисляют и обрабатывают сероводородом. При получении раствора аналитик должен выяснить, можно ли избежать выпадения осадка добавлением большего количества вйнной кислоты или, если осадок снова образуется, содержит ли этот осадок компоненты, способные помешать дальнейшим операциям.
Методы, определения 439
Описан метод 1 отделения железа от марганца, кобальта, никеля, меди, цинка, кадмия, магния, кальция и бария, основанный на гидролитическом осаждении формиата железа (III) путем гидролиза мочевины.
Осаждение купфероном, как указано 2 на стр. 143, редко может применяться для отделения одного железа, но этот метод очень удобен для отделения малых количеств железа вместе с титаном, цирконием и ванадием от больших количеств алюминия, хрома и марганца. Обычно осадок в таких случаях прокаливают, смесь окисей взвешивают и затем определяют железо и сопровождающие его элементы соответствующими методами после сплавления прокаленного остатка окисей с пиросульфатом.
Платина, перешедшая в раствор при сплавлении или выпаривании в платиновых сосудах, создает затруднения при применении многих методов определения железа. Ее можно полностью удалить осаждением сероводородом, лучше всего из разбавленных сернокислых растворов, не содержащих аммонийных солей. Вполне удовлетворительное отделение железа от платины может быть достигнуто осаждением его аммиаком или едким натром. Если выделившийся при этом осадок велик, его надо пере-осадить.
Фтористоводородная кислота мешает в тех растворах, где надо сохранить железо в двухвалентном состоянии, так как в ее присутствии происходит быстрое окисление железа кислородом воздуха 3. В этих случаях перед восстановлением железа фтористоводородную кислоту надо удалить выпариванием с серной кислотой или превратить в безвредную фгороборную кислоту HBF4 прибавлением борной кислоты либо перед восстановлением железа, либо после его восстановления, но до соприкосновения полученного раствора с воздухом (стр. 994).
Железо может быть отделено от алюминия, хрома, циркония и бериллия осторожным нагреванием смеси окисей этих металлов в токе смеси сухого хлористого водорода и хлора 4.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Весовой метод
Весовым способом общее содержание железа в минерале определяется взвешиванием в виде Fe2O3 независимо от того, в каком состоянии — двухвалентном или трехвалентном — железо первоначально находилось.
Наиболее полное отделение железа от веществ, которые его обычно сопровождают, достигается следующим способом. Удаляют сероводородную группу осаждением ее сероводородом в кислом растворе, затем осаждают железо сульфидом аммония в присутствии тартрата аммония, растворяют сульфид железа в кислоте, окисляют железо до трехвалентного ненова осаждают его, но на этот раз аммиаком. Прямое определение железа
1 Н. Н. Willard, J. L. Sheldon, Anal. Chem., 22, 1162 (1950)
2 См. также G. Е. F. L u n d е 11, J. Am. Chem. Soc., 43, 847 (1921).
3 R. Peters, Z. phys. Chem., 26, 193 (1898); W. F. H ill ebrand., U. S. Ceol. Survey Bull., 700, 199.
' F.S. Havens, A. F. W a y, Am. J. Sci., 8, 158, 217 (1899).
440
Гл. XXI. Железо
осаждением его аммиаком или купфероном редко бывает возможным вследствие присутствия других, осаждаемых этими реактивами элементов.
Получение конечного осадка гидроокиси железа было описано в предшествующих разделах; здесь остается только указать дальнейшую его обработку.
Фильтр с осадком помещают в платиновый или фарфоровый тигель, постепенно нагревают, пока фильтр не высохнет, обугливают бумагу фильтра, следя за тем, чтобы она не воспламенилась, и затем сжигают уголь в хороших для окисления условиях при возможно более низкой температуре, после чего закрывают тигель крышкой и прокаливают1 2 при 1000— 1100° С. Это прокаливание должно происходить в окислительной атмосфере, поэтому при пользовании горелкой закрытый тигель надо наклонить под некоторым углом и защитить от пламени асбестовым щитом.
Прокаленная окись железа обычно содержит кремнекислоту из реактивов и стеклянной или фарфоровой посуды. Если предварительные отделения не были проведены тщательно, например было прибавлено недостаточное количество винной кислоты для удержания алюминия в растворе, то могут присутствовать и другие «кислы. Кремнекислота легко может быть удалена обработкой прокаленного остатка фтористоводородной кислотой с последующим выпариванием и осторожным прокаливанием окиси железа перед взвешиванием. Потери железа вследствие улетучивания не наблюдается 2.
Объемные методы
Титрование растворами окислителей
Оксидиметрические методы определения железа могут применяться либо для определения железа, находящегося в анализируемой пробе в двухвалентном состоянии, либо для определения общего содержания железа после отделения его, как описано в разделе «Методы отделения» (стр. 437), от мешающих элементов и восстановления до двухвалентного состояния.
Сернокислые и солянокислые растворы солей железа (II) при комнатной температуре окисляются кислородом воздуха лишь очень слабо3. Так, например 4, после пропускания тока воздуха в течение трех часов через подкисленные растворы сульфата железа (II) или соли Мора последу-
1 При температурах выше 1100° С может образоваться магнитная окись железа Fe3O4 вследствие потери кислорода, а при температурах выше 1200° С некоторое количество окиси железа, находящейся в контакте с платиной, может восстановиться до металла и сплавиться с платиной [R. В. Sosm an, J. С. Hostetter, J. Am. Chem. Soc., 38, 807, 1188 (1916)]. Этим частично объясняется наблюдаемое в аналитической работе загрязнение платиновых тиглей железом.
2 Например, в опытах, проведенных J. I. Hoffman, два образца окиси железа (III) весили 0,1176 г и 0,1177 г после обработки HF -b H2SO4, выпаривания и прокаливания; 0,1176 е и 0,1179 г после растворения в HF, выпаривания и прокаливания и 0,1176 г и 0,1179 г после обработки H2SO4, выпаривания и прокаливания.
3 J. W. McBain, J. Phys. Chem., 5, 623 (1901).
4 С. Baskerville, R. Stevenson, J. Am. Chem. Soc., 33, 1104 (1911).
Методы определения
441
тощим титрованием 0,5 и. раствором перманганата не было обнаружено окисления железа; проба же роданидом аммония показала, что из 0,7 г присутствовавшего в растворе железа (II) было окислено приблизительно 0,1 мг. Скорость окисления увеличивается с повышением в растворе концентрации соли железа (II) и кислорода. Концентрация свободной кислоты имеет сравнительно малое значение, серная кислота несколько замедляет окисление, соляная кислота ускоряет его. Окисление железа ускоряется также некоторыми другими соединениями, например азотной кислотой и более заметно — фтористоводородной кислотой (стр. 994) и медью х.
Для восстановления железа чаще всего применяют следующие восстановители: хлорид олова (II), амальгамированный цинк, сернистый ангидрид и сероводород. Хлорид титана (III) обычно применяется для прямого титрования железа (III), о чем будет Подробно сказано ниже, в разделе «Титрование растворами восстановителей» (стр. 449). Восстановление 'амальгамированным цинком или сернистым ангидридом дает более точные результаты, чем применение других восстановителей, но только тогда, когда эти восстановители применяются для анализа растворов чистых соединений железа. Амальгамированный цинк восстанавливает такое большое число соединений, что в обычных случаях анализа надо предварительно провести их отделение или ввести в результаты определения соответствующие поправки. При использовании сернистого ангидрида, хлорида олова (II) или сероводорода восстановлению мешает меньшее число веществ.
Обычно применяемыми окислителями для титрования железа (II) являются перманганат калия и бихромат калия1 2. Титруемые растворы могут содержать или сульфат железа или хлорид железа. При титровании сернокислых растворов оба указанные окислителя дают очень хорошие результаты; при титровании солянокислых растворов хлоридов применение перманганата может привести к получению повышенных результатов, если не был предварительно прибавлен сульфат марганца для противодействия окислению соляной кислоты, сопровождающемуся выделением хлора. Бихромат калия может применяться без стабилизатора, и приготовление его раствора требует меньшего труда и времени. О приготовлении титрованных растворов перманганата' калия и бихромата калия см. стр. 214 и 218.
Восстановители
Хлорид олова (II). В обычных случаях анализа хлорид олова (II) является наиболее удобным восстановителем железа, потому что восстановление им протекает быстро и избыток SnCl2 легко удалить; определению мешает небольшое число веществ, и для большинства целей результаты получаются достаточно точными. Из мешающих веществ чаще всего встре
1 Е. Р о s n j a k, Am. Inst. Min. and Met. Eng. (1927).
2 * О применении ванадата аммония для определения железа (а также меди, марганца, хрома, молибдена, серы, фосфора, кальция, свинца, натрия и других щелочных металлов, платиновых металлов, перхлоратов, гидразина и некоторых органических веществ) см. В. С. Сырокомский, Ю. В. Клименко. Ванадатометрия, Металлургиздат, Свердловск — Москва, 1950. Прим, ред.*
442
Гл. XXI. Железо
чаются платина 1 и ванадий. Первая вносится в раствор вследствие проведения предварительных операций в платиновой посуде, второй является частым спутником железа.
Платина мешает титрованию как бихроматом, так и перманганатом, ванадий мешает при титровании КМпО4, но не мешает при титровании К2Сг2О-. Из других мешающих определению элементов следует отметить золото, молибден, мышьяк 2, сурьму и вольфрам 3. Все мешающие вещества лучше удалять перед прибавлением хлорида олова (II), потому что восстановление их не протекает количественно и нельзя вычислить поправку, даже если их количества известны. Уран хлоридом олова (II) не восстанавливается.
Ход определения. Восстановление проводится, как описано ниже, на стр. 447. Главные предосторожности, которые необходимо соблюдать, состоят в следующем. Нужно убедиться в отсутствии мешающих веществ, надо следить за тем, чтобы анализируемый солянокислый раствор имел малый объем, был горячим, чтобы количество прибавленного избытка раствора хлорида олова (II) не превышало 0,2 мл', между прибавлением хлорида ртути (II) и титрованием следует выждать 5—10 мин.
Амальгамированный цинк. Восстановление железа амальгамированным цинком можно проводить в холодных и горячих, сернокислых и солянокислых растворах и желательно в редукторе Джонса (стр. 135). Метод этот быстрый и при анализе растворов, содержащих только одно железо, дает более точные результаты, чем восстановление хлоридом олова (II). Главное препятствие для его применения во всех случаях анализа заключается в том, что многие другие элементы и соединения также вос-
1 Следующие данные [J. Knop, J. Am. Chem. Soc., 46, 268 (1924)] показывают влияние платины на определение железа титрованием бихроматом после восстановления хлоридом олова (II) и обработки хлоридом ртути (II). В каждом случае для титрования брали 20 мл приблизительно 0,1 н. раствора FeCl3:
Платина, мг....................... 0 0,1 0,1 0,3 0,5 1,0
Объем 0,1 н. раствора К2Сг2О7, израсходованного на титрование,
20,20 20,27 20,33 20,40 20,59 20,90
Разность, мл +0,07 +0,13 +0,20 +0,39 +0,70
В бюро Стандартов США в опытах, где железо восстанавливали хлоридом олова (II) и непосредственно титровали потенциометрически бихроматом [хлорида ртути (II) не прибавляли], были получены следующие результаты:
Прибавлено платины, мг ........ 0 1 3 5
Найдено Fe2O3, мг....................... 7,28 7,58; 8,28 7,86; 7,64 8,88; 8,00
9,66; 8,66
2 Мешающего влияния мышьяка можно избежать, если сначала провести восстановление металлическим синцом при кипячении раствора, профильтровать, а затем дополнительно восстановить железо несколькими каплями раствора хлорида олова (II) [Н. W. J ones, Chemist Analyst, 18, 11 (1929)].
3 Вольфрам мешает мало, если только он не присутствует в таких количествах, которые после восстановления препятствуют титрованию своей окраской. Влияние его меньше, чем можно было бы предполагать, считая, что восстановление его происходит от WO3 до W2O5.
Например, при анализе растворов, содержавших 0,0528 г железа, было получено 0,0530 г — когда было прибавлено 2,5 мг вольфрама, и 0,0534 г — при прибавлении 5 мг вольфрама. При добавлении 50 мг вольфрама раствор после восстановления получился настолько синим, что невозможно было титровать. Молибден медленно восстанавливается до соединения с валентностью его ниже пяти. Золото образует кассиев ПУРПУР, который мешает как восстановлению, так и последующему титрованию.
Методы, определения
443
станавливаются цинком. К ним относятся: титан, ниобий, ванадий, хром, молибден, уран, вольфрам, мышьяк, нитраты и некоторые азотистые соединения Ч
Нитраты и азотистые соединения могут быть введены в раствор в. процессе его анализа, например при недостаточном промывании осадка от аммиака, полученного из раствора, первоначально содержавшего азотную кислоту, или при разложении фильтровальной бумаги и осадка азотной и серной кислотами. Эти вещества перед восстановлением следует удалить. Что касается перечисленных выше мешающих элементов, то их надо либо отделить перед восстановлением, либо, определив их содержание в отдельной пробе, собрать прошедший через редуктор раствор под раствором сульфата железа (III) и вычислить затем поправку на присутствие этих восстанавливаемых цинком элементов.
Соединения титана, молибдена, ванадия и хрома, полученные при восстановлении железа цинком, настолько легко окисляются кислородом воздуха, что внесение поправок, основанных на предположении об определенной степени их восстановления, может привести к ошибочным результатам, если прошедший через редуктор раствор не собирать в приемнике под раствором сульфата железа (III) (стр. 137). Например, когда раствор, содержавший 0,2 г железа и 0,002 г ванадия, собирали после восстановления в пустой приемник, а затем титровали перманганатом и результаты были вычислены, исходя из предположения, что ванадий окислялся перманганатом от V2O2 до V2O5, то результаты получились пониженными на 1,5 мг. С другой стороны, поправка на содержание урана может быть введена только тогда, когда прошедший через редуктор раствор собирают в пустой приемник (стр. 530).
Мешающее влияние титана может быть устранено прибавлением окиси меди, сульфата меди или окиси висмута [эти соединения окисляют титан (III), но не окисляют железо (II)], фильтрованием и затем титрованием перманганатом калия * 2.
Мешающее влияния умеренных количеств титана (меньше 10 мг) можно также избежать, аэрируя раствор или прибавляя к нему проаэрированную воду и перемешивая 2—5 мин. При такой обработке титан (III) переходит в титан (IV), а железо (II) не окисляется 3. Если при этом в качестве катализатора применять хлорид ртути (II), то можно легко вновь окислить большие количества титана, не затрагивая восстановленного железа4. Если конечное титрование проводится перманганатом, необходимо прибавление раствора Рейнгарда (стр. 446).
Восстановленные цинком хром и титан можно также вновь окислить добавлением 3 мл 0,0001 М раствора сульфата меди и пропусканием воздуха через раствор в течение 10—15 мин. Затем можно определить железо титрованием перманганатом, как обычно 5.
Ход определения. Восстановление может быть проведено в горячем или холодном, солянокислом или сернокислом растворах, как описано в разделе о редукторе Джонса (стр. 135). Кислотность может колебаться в пределах от 3 до 15% соляной кислоты или от 1 до 5% серной кислоты (и то и другое — по объему).
' 1 Если при восстановлении железа в кислом растворе цинком в редукторе Джонса образуется окрашенное соединение, то это служит признаком присутствия примеси других элементов. Разбавленный раствор солей железа (II) бесцветен, в то время как восстановленные соединения молибдена, хрома и урана — зеленого цвета, вольфрама и ниобия — коричневого цвета, а титана и ванадия — соответственно фиолетового и бледно-лилового цветов.
2 F. A. Gooch, Н. D. Newton, Am. J. Sci, [4], 23, 365 (1906).
3 E. Truog, R. W. Pearson, Ind. Eng. Chem., Anal. Ed., 10, 631 (1938). См. также W. M. Thornton, R. R о s e m a n, J. Am. Chem. Soc., 57, 619 (1935).
4 W. M. M c N a b b, H. S k о 1 n i k, Ind. Eng. Chem., Anal. Ed., 14, 711 (1942).
5 F. S. Grimaldi, R.E. Stevens, M. K. Carron, Ind. Eng. Chem., Anal. Ed., 15, 387 (1943).
444
Гл. XXI. Железо
Кислые растворы солей железа (II) настолько медленно окисляются кислородом воздуха, что нет необходимости защищать от него раствор в приемнике.
Серебряный редуктор. Применение в редукторе серебра вместо цинка (стр. 139) имеет преимущество, если железо надо определять в присутствии хрома или титана. Присутствие ванадия также не отражается на результатах титрования, если в качестве индикатора применяют комплексное соединение о-фенантролина с железом (II) (см. стр. 454).
Сернистый ангидрид. Восстановлецие железа сернистым айгидридом может также давать более точные результаты по сравнению с получаемыми при использовании хлорида олова (II), но эта операция протекает гораздо медленнее, так как приходится принимать особые меры предосторожности, чтобы быть уверенным в полноте восстановления железа и в удалении избыка сернистого газа. Вещества, мешающие восстановлению, здесь не так многочисленны, как при восстановлении хлоридом олова (II). В их число входят платина х, некоторые металлы сероводородной группы (например, медь, мышьяк, сурьма) и ванадий.
Растворы сернистого ангидрида или сульфита щелочного металла следует применять только свежеприготовленные, потому что после некоторого промежутка времени эти растворы, кроме сернистой кислоты или сульфита, содержат также и другие окисляемые перманганатом калия вещества. Сернистый ангидрид удобнее получать из баллонов с сжиженным газом или, за отсутствием их, нагреванием колбы с раствором сернистого ангидрида или.сульфита, к которому была прибавлена серная кислота. Сернистый ангидрид из баллона предварительно следует пропустить через серную кислоту.
Восстановление сернистым ангидридом требует низкой кислотности раствора. Солянокислые растворы хлоридов восстанавливаются быстрее, чем сернокислые растворы сульфатов. Полученные после восстановления растворы, содержащие сернистый ангидрид, не должны оставляться более чем на 24 ч во избежание образования других окисляемых перманганатом калия веществ. Удаление сернистого ангидрида из раствора ускоряется кипячением с одновременным пропусканием быстрого тока свободной от кислорода двуокиси углерода. Если двуокись углерода получают из мрамора, то для удаления сероводорода ее следует предварительно пропустить через U-образную трубку, содержащую стеклянные бусы и раствор сульфата меди.
Ход определения. Восстановление лучше всего проводить в приборе, состоящем целиком из стекла, например в стеклянной промывалке с притертыми частями. Приготовляют солянокислый или сернокислый анализируемый раствор, содержащий железо, и удаляют элементы сероводородной группы, если они присутствовали. Кипятят для удаления сероводорода и прибавляют раствор перманганата калия до устойчивой розовой окраски. Затем приливают аммиак, пока образующийся осадок не будет при перемешивании растворяться лишь с трудом, но раствор при этом все же должен оставаться прозрачным. Раствор разбавляют до 50—100 мл водой, пропускают через него 2—3 мин сернистый ангидрид и затем, продолжая пропускание газа, постепенно нагревают до кипения. Когда раствор
1 Например, на титрование 3,7 мг и 3,3 мг платины в растворе было израсходо-вано^соответственно 0,47 мл и 0,38 мл 0,1 н. раствора перманганата калия.
Метода определения
445
обесцветится (15—ЗОлшн), прекращают ток сернистого ангидрида, пропускают через раствор промытую двуокись углерода и сильно кипятят до удаления сернистого ангидрида (15—30 мин), контролируя это пропусканием выходящего газа в течение 30 сек через разбавленный раствор серной кислоты, содержащий 2—З капли 0,1 н. раствора перманганата калия. Затем ток двуокиси углерода усиливают, раствор охлаждают, прибавляют еще кислоты и титруют, как указано ниже.
Сероводород. В настоящее время сероводород не применяется так часто для восстановления железа, как это было прежде, потому что реакция восстановления железа сероводородом сопровождается образованием довольно устойчивых соединений серы, которые титруются вместе с железом, отчего получаются повышенные результатых. К этому надо прибавить, что метод восстановления сероводородом кропотлив и присутствие ванадия мешает его применению.
Восстановление должно проводиться в небольших объемах растворов, содержащих немного кислоты, лучше всего 2—2,5% серной кислоты. Полное восстановление железа медленно протекает в холодных растворах и совсем не происходит в кипящих растворах, содержащих значительно более 1% кислоты.
Ход определения. Приготовляют 50—100 мл холодного анализируемого раствора, свободного от ванадия и содержащего 2—2,5% серной кислоты по объему. Пропускают через раствор ток промытого сероводорода в течение 30 мин и затем еще 15 мин, одновременно нагревая раствор до кипения. Если выделились сульфиды элементов сероводородной группы, их отфильтровывают через бумажный фильтр, промытый предварительно разбавленной серной кислотой, для удаления органических веществ 2 и затем промывают фильтр с осадком сероводородной водой, подкисленной серной кислотой. К фильтрату прибавляют 15 мл разбавленной (1 :1) серной кислоты, пропускают через раствор ток двуокиси углерода, свободной от кислорода, и кипятят 30—60 мин, пока объем раствора не уменьшится приблизительно до 50 мл. Затем ток двуокиси углерода усиливают, раствор охлаждают, разбавляют дистиллированной водой до 200 мл и титруют, как указано ниже 8.
Окислители
Перманганат калия. Определение в сернокислом растворе. Для получения наилучших результатов при титровании железа перманганатом калия анализируемый раствор должен содержать сульфат железа (П), свободную серную кислоту в концентрации 1 : 20 и не должен содержать других окисляемых перманганатов веществ. Такие растворы получаются после отделений, описанных в разделе «Методы отделения» (стр. 437), и восстановления цинком или сернистым ангидридом (стр. 442 и 444) или, что менее удовлетворительно, сероводородом (см. выше). Объем раствора перед титрованием должен соответствовать содержанию в нем железа. Титрование обычно проводят на холоду. В присутствии ванадия
1 G. Е. F. L u n d е 11, Н. В. Knowles,!. Am. Chem. Soc., 43, 1560 (1921). ! R. S. Me Bride, J. A. Scherrer, J. Am. Chem. Soc., 39, 928 (1917). * Применяя этот метод восстановления и титруя перманганатом, Lundell и Knowles получили 0,0046 г Fe2O3 вместо 0,0044 г; 0,0256 г вместо 0,0254 г; 0,1313 г вместо 0,1310 е я 0,1766 г вместо 0,1765 г.
446
Гл. XXI. Железо
титрование лучше проводить в горячем растворе, так как ванадий окисляется перманганатом калия очень медленно.
Окраска в конце титрования бывает различной, от обычной слабо-розовой, если железа мало, до темно-желтой, если присутствует большое количество железа и для уничтожения этой окраски не была прибавлена фосфорная кислота. Во всех случаях окраска в конце титрования не должна изменяться при перемешивании в течение 30 сек. В израсходованный на титрование объем перманганата калия вносят поправку на «холостой» опыт, проведенный с одними реактивами.
Определение в солянокислом растворе. Титрование железа перманганатом в солянокислом растворе обычно следует за восстановлением его хлоридом олова (II), как описано на стр. 441. Имеется большое число вариантов этого метода, из которых наиболее удовлетворительный известен под именем метода Циммермана — Рейнгардта х.
Ввиду того что этот метод широко применяется, мы даем подробное его описание.
Для определения требуются следующие специальные растворы:
Раствор Рейнгардта. Растворяют 200 г MnSO4.4H2O в 1000 мл воды и прибавляют охлажденную смесь 400 мл серной кислоты, 1200 мл воды и 400 мл Сиропообразной фосфорной кислоты.
Раствор хлорида олова (II). Растворяют 50 г SnCl2-2Н2О в 100 мл соляной кислоты и разбавляют водой до 1000 мл.
Раствор хлорида ртути (II). Приготовляют насыщенный раствор HgCl2 в воде.
Анализируемый раствор, содержащий железо в виде его хлорида в 25 мл разбавленной (1 : 1) соляной кислоты, нагревают и восстанавливают железо, приливая по каплям и при перемешивании раствор хлорида олова (II) до обесцвечивания жидкости 1 2, охлаждают и быстро прибавляют в один прием 10 мл раствора хлорида ртути (II). По истечении 5—10 мин переносят раствор в большую фарфоровую чашку, в которую предварительно наливают 25 мл раствора Рейнгардта и 400 .чл воды; ополаскивают колбу, где был раствор железа, умеренным количеством воды и титруют по каплям перманганатом, при постоянном перемешивании, до появления неисчезающей розовой окраски. В результат титрования необходимо ввести соответствующую поправку на «холостой» опыт, проведенный со всеми реактивами.
Результаты, получаемые этим методом, несколько повышены, но достаточно точны для обычных анализов, как это можно видеть из следующих данных:
Взято жс-ле- 0,2496 0,2496 0,1248 0,1248 0,0250 0,0250 0,0125 0,0125
за, г
Найдено же- 0,2499 0,2500 0,1249 0,1249 0,0251 0,0252 0,0125 0,0125
леза, г
Ошибка, г +0,0003 +0,0004 +0,0001 +0,0001 +0,0001 +0,0002 +0,0000 +0,0000
1 С. Zimmermann, Ann., 213, 305 (1882); С. Reinhardt, Chem. Ztg., 13, 323 (1889); G. C. J о n e s, J. H. J e f f e r y, Analyst, 34, 306 (1909).
2 Следует избегать избытка раствора хлорида олова (II), превышающего 0,2 мл. Большой избыток этого раствора приводит к образованию соединений, окрашенных в черный цвет, которые совершенно портят анализ, или же образуется большой избыток хлорида ртути (I), что ведет к получению повышенных результатов. Если хлорид олова (II) прибавлен в требуемом количестве, то после приливания раствора хлорида ртути (II) получается небольшой шелковистый осадок хлорида ртути (I).
Методы определения
447
Быстрое прибавление перманганата приводит к более повышенным результатам. Например, при титровании 0,2496 г железа результаты получались в среднем повышенными на 1 мг, если 0,1 н. раствор перманганата прибавляли со скоростью 20 мл в минуту; величина ошибки достигала +2 мг, когда все требуемое количество перманганата калия, без 1 мл, прибавляли сразу, после чего титрование продолжали обычным способом.
Бихромат калия. Титрование бихроматом калия может проводиться в солянокислом или сернокислом растворах. Если раствор содержит только одну серную кислоту, следует предпочесть титрование перманганатом, за исключением тех случаев, когда раствор содержат органические вещества, так как они легче окисляются перманганатом, чем бихроматом (см. «Анализ горных пород, Железо (II)», стр. 998). Обычно титрование бихроматом следует за восстановлением хлоридом олова (II), и его можно проводить как с внутренним, так и с внешним индикатором или же потенциометрически .
Определение с внутренним индикатором. В качестве внутреннего индикатора обычно применяют дифениламин х. Этот индикатор окрашивается в фиолетово-синий цвет при добавлении 1 капли 0,1 н. раствора бихромата калия к. солянокислому или сернокислому его раствору, содержащему железо (III). Желательно также присутствие фосфорной кислоты для уничтожения окраски соли окисного железа 1 2. Прибавление железа (II) обесцвечивает индикатор, и потому возможно обратное титрование. Наилучшие результаты 3 получаются, если объем раствора невелик (не более 100 мл). Как только появятся первые следы фиолетовой окраски, добавляют еще несколько капель раствора бихромата и устанавливают конечную точку титрования прибавлением раствора соли железа (II) и затем снова раствора бихромата калия 4 *.
Соли ванадия (V) окисляют дифениламин с образованием соединения синего цвета, в то время как соли ванадия (IV) оставляют дифениламин бесцветным 6. Следовательно, ванадий не мешает определению железа
1 J. Knop, J. Am. Chem. Soc., 46, 263 (1924). Раствор дифениламина приготовляют растворением 1 г дифениламина в 100 мл концентрированной серной кислоты. Трех капель раствора достаточно для титрования в объеме 100—250 мл. Раствор дифениламина пригоден для применения в течение по меньшей мере 8 мес. со времени его приготовления, даже если он стал коричневым. Согласно Н. М. State [Ind. Eng.
Chem. Anal. Ed., 8, 259 (1936)], индикатор может быть быстро приготовлен расплавлением дифениламина (т. пл. 52,9° С), добавлением к полученной прозрачной жидкости требуемого количества концентрированной серной кислоты и взбалтыванием до полного растворения (15—30 сек).
3 Кноп рекомендует проводить титрование в растворах, содержащих 1 мл фосфорной кислоты и 5 мл моляной кислоты или 2,5 мл серной кислоты в 100 мл. * Роль фосфорной кислоты при титровании в присутствии дифениламина не ограничивается уничтожением желтой окраски солей железа (III). Согласно очень подробному исследованию С. J. SchoellenbergerfJ. Am. Chem. Soc., 53, 98 (1931)], для получения точных результатов количество фосфорной (или плавиковой) кислоты в растворе должно по крайней мере в 2 раза превышать требуемое для исчезновения желтой окраски. Это объясняется тем, что окислительный потенциал раствора, содержащего ионы железа (II) и (III), настолько возрастает к концу титрования, когда сильно увеличивается отношение Fe3+/Fe2+, что синее окрашивание дифениламина может появиться слишком рано, т. е. еще до того, как все железо (II) окислилось. Прибавление фосфорной или плавиковой кислоты значительно уменьшает концентрацию ионов Fe3+ в растворе.*
3 L. A. Sarver, J. Am. Chem. Soc., 49, 1473 (1927).
4 Вместо дифениламина можно применять 0,2—0,5%-ный раствор дифениламин-
сульфоната калия. (* Этот индикатор дает более резкий переход окраски в конце титрования и меньшую поправку на окисление индикатора*).
6 N. Н. Furman, Ind. Eng. Chem., 17, 314 (1925).
448
Гл. XXI. Железо
в растворах, содержащих железо в двухвалентном состоянии, а ванадий в четырехвалентном1.
Определение с внешним индикатором. До введения дифениламина титрование бихроматом калия почти всегда проводилось с применением разбавленного раствора гексацианоферрата (III) калия K3[Fe(CN)6] в качестве внешнего индикатора. Этот индикатор реагирует с железом (II) с образованием окрашенного в синий цвет соединения и не реагирует с железом (III). Конечную точку титрования определяют, отбирая через определенные промежутки времени (в начале титрования редко, в конце его — часто) каплю анализируемого раствора и испытывая ее на пластинке, куда помещают каплю индикатора. Вблизи конечной точки титрования соединенные капли защищают от действия света тигельной крышкой, для появления окраски дают постоять по меньшей мере 3 мин. Целесообразно иметь две пробы для титрования; при титровании первой пробы определяют приблизительно требуемое количество титрованного раствора; при титровании второй пробы вливают сразу почти все необходимое его количество, а затем приступают к испытаниям на пластинке с индикатором.
Очевидно очень важно, чтобы применяемый гексацианоферрат (III) калия не содержал гексацианоферрата (II) калия, иначе синяя окраска получится и с двухвалентным и с трехвалентным железом. Для приготовления реактива кристаллы чистого гексацианоферрата (III) калия промывают несколько раз водой для удаления гексацианоферрата (II) калия, который мог образоваться вследствие восстановления первого случайно попавшей на него пылью, и затем растворяют с таким расчетом, чтобы получить 1%-ный раствор. Растворы гексацианоферрата (III) калия при стоянии медленно восстанавливаются, поэтому приготовляют очень малые количества этого раствора. На солнечном свету гексацианоферрат (III) калия и железо (III) реагируют друг с другом с образованием окрашенного в синий цвет соединения. При определении железа этим методом нельзя для его восстановления применять металлический цинк, потому что образующиеся в растворе соли цинка будут реагировать с гексацианоферратом (III) калия с выделением осадка, мешающего пробе. Должны отсутствовать также и другие элементы, образующие нерастворимые гексацианоферраты (III), как, например, медь, никель и марганец. Присутствие фтористоводородной кислоты в растворе приводит к замедлению образования синего окрашивания и к получению неправильных результатов. Для устранения ее мешающего влияния надо прибавить борную кислоту 2 * * s.
Ванадий (IV) в обычных условиях не дает окраски с гексацианоферратом (III) калия и, следовательно, не мешает определению, если восстановление железа было проведено сероводородом, сернистым ангидридом или хлоридом олова (II).
Определение методом потенциометрического титрования. Точность потенциометрического метода определения железа 2 равна точности метода
1 * Это утверждение не вполне справедливо. Как показали исследования
Ю. Ю. Лурье [Труды VI Менделеевского съезда, т. II, вып. 2, 1933], при большом
количестве ванадия (IV) в растворе наблюдается явление, обратное отмеченному Schoellenberger ддя ионов железа (III) (см. сноску 1, стр. 447). При большой величине отношения Vlv/Vv восстановительный потенциал ванадия (IV) настолько велик, что ванадий обесцвечивает синее окисленное соединение дифениламина; поэтому на титрование железа расходуется излишнее количество бихромата калия и результаты определения железа получаются повышенными. По этой причине при титровании железа в присутствии ванадия надо вводить в раствор фторид-ионы, связывающие ванадий (IV) в комплексные анионы. Прим, ред.*
8 О. L. В а г и е b е у, J. Am. Chem. Soc., 37, 1829 (1915).
s J. Н. Hildebrand,!. Am. Chem. Soc., 35, 871 (1913); J. B. Ferguson, J. Ind. Eng., Chem., 9, 941 (1917); J. B. Ferguson, J. С. Hostetter, J. Am. Ceram. Soc., 2, 608 (1919); J. С. H о s t e 11 e r, H. S. R о b e r t s, J. Am. Chem. Soc., 41, 1337 (1919); H. S. R о b e r t s, тамже, 1358;„ J. Am. Ceram. Soc., 10, 100 (1927); G. E. F. L u n d e 11, H. B. Knowles, там же, 11, 119 (1928). См. также -«Потенциометрическое титрование» (стр. 229).
Методы определения
449
Циммермана — Рейнгардта; потенциометрический метод особенно удобен для определения^малых количеств железа. Преимуществом потенциометрического титрования является то, что оно возможно в присутствии восстановителей, например хлорида олова (II) или хлорида ванадия.
Сульфат церия (IV). Для титрования сульфата железа (II) можно также применять сульфат церия (IV) (стр. 219). Определению не мешают умеренные количества хлоридов и соляной кислоты, некоторые спирты и такие органические кислоты, как уксусная, щавелевая, винная и лимонная.
* Другие окислители. Для титрования восстановленного до двухвалентного состояния железа можно также применять раствор метаванадата аммония NH4VO3 в приблизительно 6 н. серной кислоте х. Индикатором служит 0,1 %-ный раствор фенилантраниловой кислоты — переход окраски из бледно-голубой в вишнево-красную.
Предложен1 2 также .метод титрования солей железа (II) раствором хлорамина (натриевой соли n-толуолсульфонхлорамида) с индикатором 0,2%-ным раствором индигокармина в присутствии фторид-ионов, связывающих ионы железа (III). Доп. ред. *
Титрование растворами восстановителей
Титрование солями титана (III). Применение хлорида титана (Ш) для определения железа было предложено давно 3. Можно применять 4 5 также и раствор сульфата титана (III). Титрование этими растворами отличается от описанных выше титрований тем, что в этом случае железо титрующим раствором не окисляется, а восстанавливается. Конечную точку титрования находят, применяя внутренние индикаторы: метиленовую синюю, которая, восстанавливаясь при прибавлении избытка раствора соли титана (III), обесцвечивается, или же роданид аммония, окрашивающий раствор в красный цвет, пока в нем еще присутствуют ионы железа (III). В обычных случаях анализа следует предпочесть последний индикатор, потому, что метиленовая синяя не чувствительна при температурах ниже 35° С, если не ввести в раствор салициловую кислоту или ее натриевую соль 6, прибавление же органических веществ не дает возможности провести обратное титрование перманганатом.
Этим методом можно определять общее содержание железа или то железо, которое находится в пробе в трехвалентном состоянии. В первом случае перед титрованием надо окислить все железо до трехвалентного, лучше всего перманганатом в сернокислом растворе. Определению мешают:
1 В. С. Сыр ок омский, Ю. В. Клименко, Ванадатометрия, Метал-лургиздат, Свердловск — Москва, 1950; В. С. С ы р о к о м с к и й, Ю. В. Климе н к о, Зав. лаб., 7, 1093 (1938); И. П. А л и м а р и н, Б. И. Фрид, там же, 9, 1217 (1940); В. С. С ы р о к о м с к и й, К. Н. Жукова, там же, 11, 373 (1945).
2 Б. Н. А ф а и а с ь е в, А. В. У р а л ь с к а я, Зав. лаб., 15, 407 (1949).
3 Е. К п е с h t, Вег., 36, 166 (1903); Е. Knecht, Е. Hibbert, там же, 1549. См. также Р. S. В г a i 11 е г, Ind. Eng. Chem., 19, 846 (1927).
4 W. М. Thornton, J. E. Chapman, J. Am. Chem. Soc., 43, 91 (1921).
5 E. Knecht, E. Hibbert, New Reduction Methods in Volumetric Analysis, 1925, p. 64; H. Lauterbach, Ber. d. 7-ten Internat. Verg. d. Chem. Koll, 33 (1922).
29 Заказ 522.
450
Гл. XXI. Железо
азотная кислота, фтористоводородная кислота (но не в присутствии
борной кислоты), некоторые
Рис. 27. Прибор для хранения раствора сульфата титана (III) и титрования этим раствором:
1 — аппарат Киппа для получения водорода; 2 — бутыль; 3 — бюретка;
4 — трехходовой кран.
органические соединения, медь, сурьма, платина (и, вероятно, другие элементы сероводородной группы), вольфрам и ванадий (восстанавливающийся в условиях титрования до трехвалентного). Титрование рекомендуется проводить 1%-ным раствором хлорида титана (III), который можно получить или разбавлением продажного 20%-ного раствора (свободного от сероводорода и значительных количеств железа) или же восстановлением раствора соли четырехвалентного титана в редукторе Джонса. Приготовленный титрованный раствор, не должен подвергаться действию прямого солнечного света, и его следует постоянно защищать от кислорода воздуха, так как титан (III) легко окисляется К
Эти условия легко выполнить, Пользуясь прибором,1 2 изображенным на рис. 27.
Стеклянная бутыль 2 наполнена до шейки раствором соли титана (III). Трехходовой кран 4 поворачивается так, что раствор может входить в бюретку 3 из бутыли 2, а затем при новом повороте крана он выливается из бюретки. Водород, выходящий из аппарата Киппа 1 (или какого-нибудь др^ого прибора), полностью
вытесняет воздух из системы, после чего бюретку снова наполняют раствором, и тогда она пригодна для применения. Защищенный таким способом титрованный раствор хлорида титана (III)
1 Согласно A. S. R u s s е 1 1 [J. Chem. Soc., 129, 497 (1926)], соль титана (III) более устойчива в 4 н. растворе серной кислоты. Russell также утверждает, что сульфат ванадия (II) VSO4 является более сильным восстановителем, чем сульфат титана (III), находясь по своему восстанавливающему действию между металлическим кадмием и оловом в 2 н. растворе серной кислоты. 0,1 н. раствор соли ванадия (II) в 10 н, серной кислоте можно сохранять в течение 1 ч в бюретке без применения особых мер предосторожности для устранения окисления. См. также Р. С. Banerjee, J, Indian Chem., Soc., 19, 30 (1942). W. M. Thornton и J. F. Sadusk [Ind. Eng. Chem., Anal. Ed., 4, 240 (1932)] утверждают, что 0,1 н. раствор сульфата хрома (II), растворенного в серной кислоте, концентрация которой не превышает 0,18 н., если его правильно сохранять, не изменяется в течение двух месяцев. Такой раствор может применяться для потенциометрического определения железа в холодном разбавленном сернокислом растворе, содержащем небольшое количество роданида калия. О приготовлении растворов сульфата хрома (II) см. Н. W. Stone, С. Beeson, Ind. Eng. Chem., Anal. Ed., 8, 188 (1936) иЕ. Zintl, G. Rienacker, Z. anorg. allgem. Chem., 161, 374 (1927). О потенциометрическом титровании раствором хлорида хрома (II) см. R. F 1 a t t, F. Sommer, Helv. Chim. Acta, 27, 1518 (1944).
2 W. M. Thornton, J. E. Chapman, J. Am. Chem. Soc., 43, 91 (1921). Другие приборы, в которых такие растворы остаются без изменения в течение месяцев, описаны в статьях Е. Zintl, G. R i е n а с k е г (Z. anorg. allgem. Chem., 161, 374 (1917)] и G. F. Smith, C. A, G e t z [Ind. Eng. Chem. Anal. Ed., 6, 252 (1934)].
Методы определения
451
не подвергается значительным изменениям в течение 1—2 мес. 1 При выливании раствора из бюретки и соприкосновении его с воздухом происходит только незначительное его окисление. Ошибку можно достаточно хорошо уравновесить, если титр раствора устанавливать таким же способом, и ее можно совершенно устранить, если титрование проводить в койбе, закрытой пробкой с тремя отверстиями, в которые вставляют конец бюретки, вводную и отводную трубки, и пускают по этим трубкам ток двуокиси углерода.
Установку титра раствора можно проводить по чистой соли Мора. Навеску соли Мора растворяют в разбавленной (5 : 95) серной кислоте. Титруют раствором перманганата калия до обычной конечной точки титрования и сильно кипятят для удаления растворенного в жидкости кислорода и разложения незначительного избытка перманганата. Раствор затем охлаждают, приливают 10 мл 10%-ного раствора роданида аммония и титруют раствором соли титана (III) до полного обесцвечивания 2.
При определении железа титрование должно проводиться при комнатной температуре в растворах, имеющих объем от 100 до 500 мл и содержащих приблизительно 10% (по объему) серной или соляной кислоты, 10— 25 мл 10%-ного раствора роданида аммония и не содержащих мешающих определению веществ.
По окончании титрования роданид можно осадить нитратом серебра 3, прибавляя его в избытке, восстановленное железо оттитровать в присутствии роданида серебра перманганатом, как описано, на стр. 445, и внести поправку на «холостой» опыт, проведенный с одними реактивами после окисления титанового раствора трехокисью висмута и отфильтровывания избытка последней 4 5.
* Титрование другими восстановителями. Кроме солей титана (III), для титрования железа (III) были предложены следующие восстановители:
Соли хрома (II), являющиеся сильными восстановителями Б: окислительно-восстановительный потенциал системы Ст3+/Сг2+ имеет значение от —0,40 до —0,44 в, т. е. он близок к потенциалу металлического кадмия (—0,40 в) и ниже потенциалов металлических свинца, олова и никеля. В качестве индикатора лучше всего применять фенилантраниловую кислоту.
После титрования железа (III) солью хрома (II) раствор можно взболтать [при этом избыток хрома (II) окисляется кислородом воздуха до хрома (1П)1 и оттитровать все железо [как то, которое восстановилось до двухвалентного при титровании хромом (II), так и то, которое в самом начале было в двухвалентном состоянии ] титрованным раствором перманганата калия или сульфата церия (IV).
1 В статье H.W. Stone и С. Beeson (Ind. Eng. Chem., Anal. Ed., 8, 188 (1936)] описан прибор, в котором двуокись углерода подается из баллона и очищается пропусканием через подкисленный раствор сульфата хрома (II).
2 Об установке титра раствора хлорида титана (III) по гексацианоферрату (III) калия или гексацианоферрату (II) калия см. в статье G. F. Smith, С. A. Getz, Ind. Eng. Chem., Anal. Ed., 6, 252 (1934).
3 См. сноску 2 на стр. 411.
4 H. D. N ewt on, Am. J. Sci, [4], 25, 343 (1908).
5 В. С. Сыр окомс кий, К. H. Жукова, Зав. лаб.5 11, 373 (1945), 29*
452
Гл. XXI. Железо
Хлорид олова (II) в присутствии какотелина в качестве индикатора При добавлении капли раствора соли олова (II) в избытке какотелин окрашивается в фиолетовый цвет. Анализируемый раствор должен быть 2 н. по содержанию соляной кислоты и титровать его надо горячим.
Нитрат ртути (I) 1 2. В качестве индикатора применяют раствор роданида аммония. Мешают ионы хлора, образующие с ионами чртути недиссоциированное соединение.
Аскорбиновая кислота 3. К анализируемому раствору прибавляют точную навеску х. ч. аскорбиновой кислоты и избыток ее оттитровывают раствором иода в присутствии крахмала. Доп. ред. *
Колориметрические методы
Из колориметрических методов определения железа широкое распространение получил метод, основанный на образовании окрашенного в красный цвет роданида железа (III). Простой способ прибавления роданида калия или роданида аммония к кислому раствору соли железа (III) не является идеальным, так как интенсивность получаемой окраски ни в коей мере не пропорциональна концентрации железа 4 *, образующаяся окраска ослабевает при стоянии раствора8 и, кроме того, равновесие реакции образования окрашенного соединения зависит от концентрации ионов водорода в растворе и от присутствия веществ, реагирующих с ионами железа (III) с образованием более или менее устойчивых слабоокрашенных или бесцветных комплексных соединений.
Следует добавить, что многие вещества мешают применению этого метода своей собственной окраской., К мешающим веществам относятся: соли серебра, меди, висмута, платины, никеля, кобальта, титана, фториды, фосфаты, арсенаты, молибдаты и, в меньшей степени, сульфаты [* а также соли ртути (I), ртути (II); урана, рутения, осмия, мета- и пирофосфаты, оксалаты, большие количества солей кадмия, цинка, сурьмы (III) и марганца*]. Мешающее действие некоторых из них можно устранить, прибавляя к раствору роданид калия или аммония роданид ртути (II) или экстрагируя полученное окрашенное соединение эфиром или амиловым спиртом. При анализе солянокислых растворов хлорида железа (III) получаются лучшие результаты, чем при анализе сернокислых растворов 6 *, содержащих сульфат железа (III).
1 М. Л. К у ч м е и т, А. И. Г е и г р и н о в и ч, Зав. лаб., 11, 267 (1945).
2 С. А. Бабушкин, М. Л. Погребинская, Зав. лаб., 14, 1182 (1948).
8 В. В. Птицын, В. А. К о з л о в, ЖАХ, 4, 35 (1949).
4 См. Е. К a h a n е, Bull. Soc. chim., 41, [4] Т, 1403 (1927).
6 С. A. Peters, М. М. MacMasters и С. L. French [Ind. Eng. Chem., Anal., Ed., 11, 502 (1939)] утверждают, что ослабления окраски (которое приписывается медленному''восстановлению железа роданид-ионами) можно избежать при соблюдении следующих условий: анализируемый раствор должен быть 0,01—0,1 н. по содержанию соляной кислоты, 0,2—0,4 и. по содержанию роданида натрия и 0,001— 0,0025 и. по содержанию перекиси водорода. * См. также С. A. Peters, С. L. French, Ind. Eng. Chem., Anal. Ed., 13, 551 (1941) *.
6 * Согласно H. A. Daniel и H. J. Harper (J. Assoc. Offic. Agr. Che-
mists, 17, 286 (1934)], более удовлетворительные результаты улучаются в растворе,
1 %-ном по сбдержанию азотной кислоты и не содержащем соляной кислоты и хлоридов. Прим, перев.*
Методы определения
453
* В зависимости от концентрации роданид-ионов в растворе образуются различные комплексные ионы при очень малой концентрации роданид ионов преобладают ионы [Fe(CNS)]2+, при 0,1 М концентрации роданида получаются преимущественно ионы [Fe(CNS)2]+, при 0,2 М концентрации роданида и выше образуются отрицательно заряженные ионы [Fe(CNS)4]_, [Fe(CNS)5]2- и [Fe(CNS)e]3-. Все эти комплексы окрашены в красный цвет приблизительно одинакового оттенка (при увеличении концентрации роданид-ионов в растворе происходит сдвиг максимума поглощения света от 450 до 480 ммк). Доп. ред. *
Если содержание железа очень мало, то перед колориметрическим определением надо провести специальную операцию его концентрирования, а также удалить все мешающие вещества 1 2.
Проведение колориметрического определения железа с роданидом не в водной среде, а в 2-метоксиэтаноле (метилцеллосольве) приводит к лучшим результатам3, потому что: 1) этот органический растворитель имеет меньшую диэлектрическую проницаемость; 2) роданид аммония в нем хорошо растворяется, образуя бесцветный раствор; 3) получаемые в этой среде окраски подчиняются закону Бера и на 85% интенсивнее, чем окраски в водной среде; 4) при стоянии растворов окраски заметно не изменяются в течение нескольких недель.
Ход определения. Анализируемый раствор, занимающий объем приблизительно 2 мл, должен быть 2,5 н. по содержанию соляной кислоты и должен содержать в 1 мл примерно 0,02 мг железа в виде хлорида железа (III). Если имеются сомнения в том, что все железо в растворе трехвалентно, прибавляют 30—50 мг персульфата аммония или добавляют 2 капли 0,1 н. раствора перманганата калия и дают постоять до исчезновения окраски перманганата. Затем приливают реактив 4 в четырехкратном объеме по отношению к анализируемому раствору, содержащему железо. Полученную окраску сравнивают с окрасками стандартных растворов, подготовленных таким же способом.
Из других методов колориметрического определения железа следует отметить методы, основанные на:* 1) желтом окрашивании раствора хлорида железа (III) в соляной кислоте, имеющей постоянную точку кипения 5 6,
1 А. К. Бабко, ЖОХ, 16, 1549 (1946). См. также Н. S. Frank, R. L. О s-w а 11, J. Am. Chem. Soc., 69, 1321 (1947); T. C. J. О v е n s t о n, С. A. Parker^ Anal. Chim. Acta, 3, 277 (1949).
2 В статьях H. N. Stokes.и J. R. Cain [NBS Sci. Paper, 53; J. Am. Chem. Soc., 29, 409, 443 (1907)] описан точный метод определения очень малых количеств железа, основанный на предварительном его отделении и концентрировании, растворении в родановой кислоте l экстрагировании роданида железа (III) амиловым спиртом и эфиром.
8 Н. W. W i n s о г, Ind. Eng. Chem., Anal. Ed., 9, 453 (1937). J. T. Woods и M. G. Mellon [там же, 13, 551 (1941)] утверждают, что той же цели можно достичь, проводя определение в 60%-ном (по объему) ацетоне.
4 Для приготовления реактива 10 г роданида аммония помещают в колбу емкостью 250 мл, снабженную притертой пробкой, наполняют колбу 2-метоксиэтанолом и взбалтывают до растворения соли. Перед применением этого раствора дают ему
постоять 24 ч. За это время должно исчезнуть розовое окрашивание, которое могло появиться от следов железа в реактиве. Если таким способом окрашивание не уничтожается, растворитель надо предварительно перегнать при 125° С в приборе, в котором все стеклянные части притерты друг к другу. Приготовленный раствор реактива надо сохранять в темноте, так как он подвергается фотохимическому изменению, приводящему к образованию соединения, окрашенного в желтый цвет.
6 J. С. Hostetter, J. Am. Chem. Soc., 41, 1531 (1919).
454
Гл. XXI. Железо
2) интенсивном красном окрашивании комплексных ионов 1 2 3 4, образуемых о-фенантролином с железом (II); 3) аметистово-фиолетовом окрашивании комплекса, образуемого ионами железа (III) с салициловой кислотой в уксуснокислых растворах 2; 4) красновато-пурпурном окрашивании, получаемом в результате реакции меркаптоуксусной (* тиогликолевой *) кислоты с железом (II) в щелочных растворах 3 (pH — 10); 5) сине-зеленом окрашивании комплекса, образуемого 7-иод-8-оксихинолин-5-сульфо-кислотой («ферроном») о железом (III) в растворах, кислых по метиловому оранжевому4; 6) красно-оранжевом окрашивании, получаемом при обработке солей железа (III) салицилальдоксимом5 (при рН=7), и, наконец, 7) красновато-пурпурном окрашивании, получаемом при добавлении 2,2'-дипиридила6 к раствору, содержащему ионы железа (II) (меньше 1 мг). Опубликованы сравнительные данные для большинства из этих реактивов 7.
* Заслуживает вниманйя также колориметрический метод определения железа с сульфосалициловой кислотой 8. В аммиачных растворах этот реактив образует окрашенное в желтый цвет соединение как с ионами железа (III), так и с ионами железа (II). Таким способом определяется суммарное содержание обеих форм железа. В кислых растворах тот же реактив образует окрашенное в красный цвет соединение9 только с ионами железа (III). По разности между результатами обоих определений находят
1 Этот метод лучше всего подходит для определения очень малых количеств железа (0,001—0,05 мг). Железо (III), которое обычно получается при подготовке раствора к анализу, надо предварительно восстановить до двухвалентного состояния. Лучший восстановитель для этой цели — солянокислый гидроксиламин. См. L. G. S а-у w е 11, В. В. Cunningham, Ind. Eng. Chem., Anal. Ed., 9, 67 (1937); F. С. H u m-m e 1, H. H. Willard, там же, 10, 13 (1938); W. В. F or t une, M. G. Mellon, там же, 60. В последней статье дано спектрофотометрическое исследование этой реакции. Применение этого метода для определения железа в железных рудах см. в статье J. Р. М е h 1 i g, Н. R. Hulett, там же, 14, 869 (1942).
2 Применение этого реактива для спектрофотометрического определения железа см. J. Р. М е h 1 i g, Ind. Eng. Chem., Anal. Ed., 9, 162 (1937), * J. P. M e h 1 i g, Ind. Eng. Chem., Anal. Ed., 10, 136 (1938) *. 4
3 Этот метод служит для определения железа в присутствии таких анионов, как фторид-, фосфат- и тартрат-ионы [Н. W. S wank, М. G. Mellon, Ind. Eng. Chem. Anal. Ed., 10, 7 (1938)]. * См. также J. P. M e h 1 i g, M. J. Shepherd, Chemist Analyst, 35, 8 (1946).*
4' J. H. Y о e, J. Am. Chem. Soc., 54, 4139 (1932); N. A. Clark, D. H. S i e-1 i n g, Ind. Eng. Chem., Anal. Ed., 8, 256 (1936). Чувствительность, по данным авторов, 0,1 мг/л. Окраска устойчива на свету, но разрушается сильными кислотами и щелочами. Мешают соли, легко гидролизующиеся и образующие в растворе окрашенные ионы, если только они не присутствуют в очень малых концентрациях. * См. также
Н. W. S w a n g, М. G. М е 11 о п, там же, 9, 406 (1937).*
6 D. Е. Howe, М. G. Mellon, Ind. Eng. Chem., Anal. Ed., 12, 448 (1940). Этим методом можно определять железо в концентрациях от 0,05 до 10 мг!л\ мешающие вещества легко удаляются.
6 М. L. Moss, М. G. Mellon, Ind. Eng. Chem., Anal. Ed., 14,»862 (1942).
7 J.T. Woods, M. G. Mellon, цит. выше.
8 L. L о r b e r, Biochem. Z., 181, 391 (1927); С. H. P о з а н о в, Г. А. Марко в а, Е. А. Федотова, Зав. лаб., 4, 639 (1935); В. М. Пешкова, А. Д. Его-р о в, там же, 4, 885 (1935); А. Т h i е 1, О. Р е t е г, Z. anal. Chem., 103, 161 (1935); Е. Е. 3 у с е р, Зав. лаб., 8, 1182 (1939); Е. И. Никитина, там же, 9, 629 (1940); В. И. Кузнецов, там же, 12, 278 (1946); Е.И. Фогельсон, Н. В. Калмыкова, там же, 12, 973 (1946).
9 О составе образующихся комплексов см. В. И. Кузнецов, цит. выше; А. К. Бабко, А.Т. Пилипенко, Колориметрический анализ, Госхимиз-дат, 1951.
Методы определения
455
содержание железа (II). Определение железа этим методом можно проводить в присутствии больших количеств фосфат-ионов *. Получаемые окраски подчиняются закону Ламберта-Бера и очень устойчивы. В отличие от роданидного метода и метода с применением феррона, этот метод дает хорошие результаты при анализе гальванических ванн хромирования 1 2. Доп. ред. *
Трудности, связанные с точным определением малых количеств железа в песках, и точность различных методов анализа иллюстрируются данными 3 табл. 20.
ТАБЛИЦА 20
Определение железа в песке для производства стекла
Методы 1 Процентное содержание Ге20з, найденное
аналитиками стекольной промышленности Н. В. Knowles
первые анализы конечные анализы
Потенциометрический2 0,068 0,070 0,0714
0,073 0,072 0,0726
0,083 0,0726 —
0,095 0,073 —
0,097 — —
Весовой3 0,076 0,076 0,0720
0,100 — 0,0724
— — 0,0724
— — 0,0724
— — 0,0728
Колориметрический4 0,06 0,067 0,068
0,065 0,068 0,069
0,067 — 0,070
Восстановлением сероводородом6 0,066 0,077 0,0769
0,097 0,079 0,0785
- 0,077 0,080 —
Восстановлением сернистым ангид-
рядом6 — — 0,0745
— — 0,0749
1 Во всех методах песок был полностью разложен HF + H2SO4 и нерастворимый остаток сплавлен.
8 Восстановление хлоридом олова (II) и потенциометрическое титрование перманганатом.
3 Осаждение в растворе (NH4)2S + (NH4)2G4H4Oe, разложение FeS и осаждение аммиаком.
4 Осаждение аммиаком, растворение в соляной кислоте и обработка роданидом калия.
8 Восстановление сероводородом в сернокислом растворе и титрование перманганатом после удаления сероводорода двуокисью углерода.
в Подобно предыдущему, но восстановление проводилось сернистым ангидридом.
Данные табл. 20 получены авторами указанной в сноске статьи и аналитиками стекольной промышленности.
1 С. Н. Розанови др., цит. выше.
2 Н. Pfeiffer, Z. anal. Chem., 126, 81 (1943).
3 G. Е. F. L u n d e 11, H. В. Knowles,!. Am, Ceram. Soc., 11, 119 (1928),
Глава XXII
НИКЕЛЬ
Никель (обычно вместе с кобальтом) встречается в самородном виде . и в сплавах с железом — в метеоритах и в минералах аваруите Ni2Fe иджозефините. Никель часто обнаруживают и в изверженных породах, где он присутствует, вероятно, в качестве составной части оливина; его находят преимущественно в силикатах, сульфидах, арсенидах, антимонидах и в виде теллурида никеля, реже — в некоторых других минералах, например в фосфатах, где он часто сопровождается цинком, медью и хромом х. Присутствие никеля особенно характерно для магнезиальных изверженных пород, в которых он обычно связан с хромом. Очевидно, что тщательное испытание на никель желательно при анализе всех пород и минералов. Постоянная необходимость определения никеля в различных металлургических материалах хорошо известна и не нуждается в комментариях.
ОБЩИЕ ЗАМЕЧАНИЯ
В общем ходе анализа горных пород, если все осаждения производить двукратно, никель пройдет через весь анализ незамеченным и останется в последнем фильтрате. В осадке от аммиака никель будет отсутствовать, в осадке оксалата кальция он может находиться лишь в виде слоев, и наибольшее его количество, но все же очень ничтожное по сравнению с общим его содержанием в растворе, будет присутствовать вместе с магнием в фосфатном осадке 1 2. Если в анализируемой пробе присутствует малое количество никеля, его лучше всего выделить осаждением сульфидом аммония (стр. 90) после отделения железа, алюминия и подобных им элементов аммиаком. Если присутствуют большие количества никеля, то обычно осаждают железо, алюминий и другие элементы в виде основных ацетатов и никель затем выделяют в виде сульфида перед осаждением щелочноземельных металлов.
1 Более подробные сведения даны в статьях A. Grammont, Bull. Soc. chim. France, 35, 405, 1351 (1924).
2 H. F. Harwood и L. S. Theobald [Analyst, 58, 673 (1933)] отмечают, что в условиях обычного хода анализа горных пород большее или меньшее количество никеля будет в осадке от аммиака и что его совсем не будет в осадке фосфата магния и аммония, если при осаждении последнего в растворе находились в избытке оксалат-ионы.
Методы отделения
457
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ НИКЕЛЬ
Минералы, в состав которых входит никель, можно перевести в раствор обработкой кислотами с последующим сплавлением, если необходимо, нерастворимого остатка с карбонатом натрия или пиросульфатом калия. Обычный ход работы следующий. Переносят 0,5—1,0 г измельченного в порошок минерала в чашку, покрывают часовым стеклом и приливают 40 мл азотной кислоты. Дают постоять несколько минут, кипятят до удаления бурых паров и затем осторожно время от времени прибавляют по нескольку кристалликов хлората калия. Когда разложение закончится и выделение газа прекратится, снимают часовое стекло, обмывают его и выпаривают раствор досуха. К сухому остатку прибавляют соляной кислоты, снова выпаривают досуха и смачивают остаток той же кислотой. Разбавляют водой, фильтруют и промывают нерастворимый остаток. Фильтр с остатком сжигают и прокаливают, остаток сплавляют с карбонатом натрия или пиросульфатом калия, растворяют плав в кислоте и прибавляют полученный раствор к первоначальному фильтрату х.
МЕТОДЫ ОТДЕЛЕНИЯ
Наилучшим методом отделения никеля от остальных элементов является осаждение его диметилглиоксимом (CHS)2C2(NOH)2. Применение этого реактива было впервые предложено Л. А. Чугаевым 1 2. Для осаждения никеля обычно прибавляют раствор диметилглиоксима к почти кипящему слабокислому анализируемому раствору с последующим добавлением раствора аммиака в очень небольшом избытке. Если анализируемый раствор содержит элементы, осаждаемые аммиаком, например железо или алюминий, то перед прибавлением раствора аммиака в анализируемый раствор вводят винную или лимонную кислоту. В присутствии большого количества кобальта, для более полного осаждения и получения более чистого осадка, вместо аммиака к слабокислому анализируемому раствору прибавляют в избытке ацетат натрия. Вследствие этого свободная минеральная кислота замещается слабо диссоциированной уксусной кислотой, в которой этот осадок нерастворим 3. Этот же метод дает более чистые осадки и в присутствии значительных количеств цинка и марганца.
Осаждению никеля диметилглиоксимом мешают лишь очень немногие элементы, из которых важнейший — палладий, осаждающийся и в слабокислых, и в слабоаммиачных растворах. Платина и золото загрязняют осадок диметилглиоксимата никеля, равно как и железо (II), если
1 С. F. Bonilla [Ind. Eng. Chem., Anal. Ed., 4, 128 (1932) исследовал кинетику растворения никеля в царской водке и различных «видоизмененных царских водках». Из последних наилучшие результаты дала смесь 3 частей соляной кислоты и 40 частей азотной кислоты.
2 L. А. Т s с h u g а е f f, Z. anorg. Chem., 46, 144 (1905); Вег., 38, 2520 (1905).
3 Вполне удовлетворительное отделение никеля диметилглиоксимом от больших количеств кобальта может быть также достигнуто, если до прибавления диметилглиоксима окислить кобальт до трехвалентного состояния. Для этого к аммиачному анализируемуму раствору прибавляют перекись водорода или персульфат аммония и кипятят для разрушения избытка этих реактивов и удаления большей части аммиака. Еще один метод основан на прибавлении к аммиачному анализируемому раствору цианида калия в избытке, окислении перекисью водорода, добавлении твердого диаметил-глиоксима и обработке формальдегидом [F. F е i g 1, Н. J. К а р u 1 i t z a s, Z. anal. Chem., 82, 417 (1930)].
458
Гл. XXII. Никель
осаждение проводится в аммиачном растворе. Малые количества меди, мышьяка и молибдена, какие могут содержаться в обыкновенной углеродистой стали, не причиняют затруднений. Если медь присутствует в большом количестве, то она должна быть удалена, так же как и другие члены сероводородной группы х.
Осаждение а-бензилдиоксимом (С6Н5)2 С2 (NOH)a равноценно осаждению диметилглиоксимом как в отношении полноты выделения никеля, так, вероятно, и в отношении чистоты получаемого осадка. Можно также применять водный раствор а-фурилдиоксима (С4Н3О)2 С2 (NOH)2; утверждают, что он является более чувствительным реактивом на никель, чем другие оксимы 1 2.
Малые количества никеля можно отделить от больших количеств кобальта, вливая холодный слабокислый раствор, содержащий около 1 г кобальта, в 200 мл холодного раствора 5 г хлорида аммония, 5 г фосфата аммония и 25—30 мл концентрированного раствора аммиака. Затем все перемешивают, нагревают до кипения и, если выделится аморфный осадок, прибавляют еще аммиак. После этого кипятят 1—2 мин, оставляют на водяной бане в течение 15 мин, фильтруют через неплотный фильтр и промывают осадок холодной водой. Фильтрат слегка подкисляют уксусной кислотой и определяют в нем никель осаждением диметилглиоксимом, как обычно 3 4.
Другие методы отделения никеля не выдерживают сравнения с описанным выше. Это групповые отделения, которые в лучшем случае оставляют никель все еще связанным с кобальтом, от которого его высвобождают осаждением никеля диметилглиоксимом или осаждением кобальта нитритным (стр. 471) или нитрозо-p-нафтоловым методами (стр. 473). Групповые разделения идут обычно по следующему пути:
1) Отделение элементов группы сероводорода осаждением их этим газом в кислом растворе (стр. 83)1.
2) Отделение железа, алюминия, титана и других элементов осажде-, нием их путем установления определенной концентрации ионов водорода в растворе — аммиаком (стр. 102), ацетатом натрия (стр. 103), сукцинатом натрия (стр. 106), окисью цинка (стр. 108) или карбонатом бария (стр. 108).
3) Отделение никеля от щелочных металлов, магния и щелочноземельных металлов осаждением его бесцветным сульфидом аммония в присутствии -хлорида аммония (стр. 90) 5 6.
Отделение железа и других элементов осторожным добавлением аммиака вполне удовлетворительно, если эти элементы и никель присутствуют в умеренных количествах, например 0,1 г В2О3 и такое же коли
1 J. Ranedo [Anales Soc. espan, fis у quim., 32, 611 (1934); там же, 33, 951 (1935)] утверждает, что мешающего влияния меди можно избежать, добавляя гипо-сульфат натрия Na2S2O4 перед прибавлением диметилглиоксима.
2 В. A. S о u 1 е, J. Am. Chem. Soc., 47, 981 (1925).
3W. R. Schoeller, Analyst, 69, 8 (1944).
4 Однако олово (IV) при осаждении его сероводородом склонно увлекать с собой
никель.
6 Э. А. Остроумов [Зав. лаб., 7, 20 (1938)]; Э. А. Остроумов и Г. С. Масленникова [там же, 7, 267 (1938)] рекомендуют проводить осаждение сероводородом из горячего раствора, содержащего пиридин и солянокислую соль пиридина, потому что в этих условиях сульфиды никеля и кобальта выделяются в крупнокристаллическом виде и хорошо отделяются от марганца, кальция и магния. Пиридин не мешает последующим осаждениям кальция в виде оксалата и магния в виде фосфата.
Методы отделения
459
чество никеля х. Если же присутствуют большие количества, то лучше применять методы осаждения ацетатом натрия, сукцинатом натрия или карбонатом бария. Добавление цианида аммония в избытке удерживает никель в растворе, что может быть использовано для разделений. Ни одним из указанных выше методов никель от цинка и кобальта не отделяется.
Отделение цинка от никеля лучше всего проводить осаждением его сероводородом из сернокислых растворов, 0,01 н. по содержанию серной кислоты (стр. 482), или из муравьинокислых растворов (стр. 482). Это осаждение должно быть повторено, если количество того или другого элемента превышает 0,1 г.
Отделение никеля (и кобальта) от марганца лучше всего проводить электролитическим осаждением никеля из аммиачного раствора, содержащего сульфат аммония (стр. 463), если количество марганца не превышает 0,01 г. При наличии большего количества марганца его лучше осадить кипячением азотнокислого раствора с хлоратом калия (стр. 494). Отделение кобальта и никеля от марганца осаждением их сероводородом в горячем уксуснокислом растворе, содержащем ацетат аммония, не вполне удовлетворительно, поскольку это осаждение редко бывает полным, и при выполнении точных анализов необходимо извлечение остатков никеля и кобальта из фильтрата 1 2. Никель (но не кобальт, за исключением тех случаев, когда малые количества кобальта присутствуют вместе с большими количествами никеля) может быть количественно отделен от мышьяка (V) электролитическим осаждением из аммиачного раствора 3.
Интересный способ, заслуживающий большого внимания, представляет осаждение никеля в кипящем растворе едкой щелочью, свободной от аммиака и содержащей персульфат, перекись, гипобромит или гипохлорит какого-нибудь щелочного металла. Лучшее отделение получается, если анализируемый раствор почти полностью нейтрализовать раствором едкого натра и затем медленно влить его при перемешивании в горячий щелочной раствор персульфата калия или персульфата натрия (но не аммония), взятый в избытке. Если присутствует хром, то его лучше сначала окислить персульфатом в кислом растворе, перед нейтрализацией последнего. Для осаждения никеля в присутствии урана применяют смесь перекиси натрия и карбоната натрия (стр. 110). Если осадок велик или нужна особенно большая Точность, то требуется переосаждение. В результате получается плотный, легко отделяемый фильтрованием осадок гидро
1 G. Е. F. L u n d е 1 1, Н. В. Knowles, J. Am. Chem. Soc., 45, 676 (1923). Согласно H. Brearley [The Chemical Analysis of Steel-Works Materials, by F. Ibbotson, 1920, p. 3], возможно совершенное отделение 1 дециграмма никеля даже от 1 г железа (III), если холодный слабокислый раствор, содержащий железо и никель, обработать цианидом калия' в количестве, несколько превышающем требуемое для образования K2[Ni(CN)4], затем медленно и при постоянном перемешивании влить этот раствор в раствор аммиака, взятый в избытке, и профильтровать. Если потом в фильтрате предполагается определить никель электролизом или осаждением диметилгли-оксимом, то фильтрат надо подкислить и удалить синильную кислоту кипячением под хорошей тягой.
2 Согласно М. М. Haring, М. Leatherman [J. Am. Chem. Soc., 52, 5135 (1930)] иМ. M. Haring, В. В. Westfall [там же, 5141], оптимальные значения pH для осаждения сульфидов кобальта и никеля равны соответственно 3,93 и 4,4. Осаждение надо проводить в горячих растворах с добавлением буферной смеси уксусной кислоты и ацетата аммония, отвечающей этим значениям pH.
8 N. Н. Furman, J. Am. Chem. Soc., 42, 1793 (1920).
460
Гл. XXII. Никель
окиси никеля (III). В то же время достигается полное отделение никеля от таких элементов, как хром, ванадий, фосфор, уран и, вероятно, цинк х.
Из других методов следует отметить’ отделение больших количеств железа от малых количеств никеля извлечением железа эфиром из холодного разбавленного солянокислого раствора (стр. 161) и малых количеств железа, титана, циркония и ванадия от никеля осаждением их купфероном в растворе, содержащем 10% по объему серной кислоты (стр. 143).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Осаждение диметилглиоксимом
При осаждении никеля диметилглиоксимом выделяется алого цвета осадок, настолько объемистый, что оперировать с количествами никеля, превышающими 0,02—0,03 г, очень неудобно. Осадок нерастворим в разбавленных растворах аммиака, в растворах аммонийных солей, в избытке осаждающего реактива и в разбавленных растворах уксусной кислоты, содержащих ацетат натрия. Он растворим в свободных минеральных кислотах (даже в том количестве кислоты, которое освобождается при выделении осадка из нейтрального раствора), в растворах цианидов, в спиртовых растворах, содержащих более 50% спирта по объему, и в концентрированных аммиачных растворах солей кобальта. Большие количества аммиака, аммонийных солей, кобальта или меди замедляют осаждение.
Палладий и золото частично осаждаются диметилглиоксимом из слабоаммиачных растворов. Из слабокислых растворов палладий осаждается количественно, золото — частично. Как общее положение, можно принять, что перед осаждением никеля диметилглиоксимом лучше удалить из раствора элементы группы сероводорода, несмотря на то, что умеренные количества меди, мышьяка, молибдена и, вероятно, некоторых других членов этой группы не мешают осаждению. Надо помнить, что присутствующее в растворе железо перейдет после обработки сероводородом в двухвалентное состояние и его следует затем окислить, так как железо (II) в аммиачных растворах реагирует с диметилглиоксимом с образованием имеющего красный цвет соединения, что приводит к повышенным результатам, если железо присутствует в больших количествах. Кремний и вольфрам в количествах, не превышающих нескольких миллиграммов, осаждению не мешают. Если эти элементы находятся в больших количествах, то они должны быть удалены обычными способами.
Осаждение обычно проводится прибавлением осадителя к горячему слабокислому раствору соли никеля и затем добавлением аммиака в небольшом избытке. Таким способом получаются легче отделяемые фильтрованием осадки, чем при прямом осаждении из холодных или аммиачных растворов. Применяемый аммиак должен быть свободен от карбонатов; раствор должен содержать аммонийные соли, если присутствуют цинк, марганец, магний или щелочноземельные металлы, и винную ки-
1 Ни двухвалентный, ни трехвалентный никель не могут быть количественно отделены от алюминия осаждением едким натром. Однако отделение это улучшается, если одновременно присутствует железо. При обработке, например, таким способом раствора, содержавшего 200 мг железа, 40 мг алюминия и 40 мг никеля, в осадке от едкого натра оказалось менее 0,5 мг алюминия [ср. Н. А._ Bright, R. М. F о w-1 е г, J. Research NBS, 10, 330 (1933)].
Методы определения
461
слоту, если присутствует железо и подобные ему элементы. Диметилгли-оксим почти нерастворим в воде, и его прибавляют в виде 1 %-ного раствора либо в спирте, либо в концентрированном растворе аммиака (13 : 7). Требуется лишь небольшой избыток реактива по сравнению с теоретически рассчитанным количеством: две части диметилглиоксима на одну часть никеля. Больший избыток реактива не мешает при условии, что-вместе с ним не вводится более 50% по объему спирта.
Когда осаждение проводится в аммиачном растворе,, содержащем значительные количества кобальта, меди или цинка, то требуется добавочное количество реактива, так как эти элементы реагируют с ним, образуя различные растворимые соединения. В таких случаях для осаждения требуется и добавочное время, например для осаждения очень малых количеств никеля в солях кобальта необходимо около двух дней.
Удовлетворительные результаты получаются в присутствии значительных количеств кобальта, марганца или цинка и в отсутствие железа, алюминия и аналогичных им элементов при выполнении анализа следующим методом. Раствор делают слабокислым, нагревают до кипения, прибавляют осаждающий реактив, затем добавляют раствор ацетата натрия до образования осадка и сверх того избыток ацетата в 1—2 г. Осаждение может быть проведено в присутствии железа после восстановления efo до двухвалентного, причем оно . должно оставаться двухвалентным в продолжение всего осаждения. Лучше, однако, сначала выделить железо в виде основного ацетата (стр. 103) и затем отделить никель от кобальта нагреванием раствора до кипения, прибавлением диметилглиоксима и, если необходимо, добавочного количества ацетата.
Утверждают \ что можно полностью осадить никель из растворов, содержащих железо, никель и умеренные количества кобальта, и при этом получить чистый осадок никеля, если операции осаждения проводить в следующем порядке: обработка кислого анализируемого раствора сернистой кислотой или сульфитом натрия, добавление винной кислоты, нейтрализация большей части кислоты раствором аммиака, кипячение, добавление диметилглиоксима, осаждение никеля прибавлением ацетата аммония или раствора аммиака. Если кроме железа, кобальта и никеля в растворе присутствует также медь, то осаждение надо проводить в уксусно-ацетатном растворе. При отсутствии кобальта никель можно осаждать и из аммиачного раствора и в добавлении сульфита нет необходимости.
Ход определения в отсутствие значительных количеств кобальта, марганца или цинка. Приготовляют раствор, содержащий не более 0,03 г никеля и свободный от элементов сероводородной группы, за исключением тех, которые не загрязняют осадка. Убеждаются в том, что железо находится в трехвалентном состоянии и что нет избытка окислителей. Добавляют Кислоту в таком количестве, чтобы при последующем добавлении аммиака в растворе оказалось достаточное количество аммонийных солей, а затем приливают профильтрованный раствор винной кислоты, если присутствуют элементы, осаждаемые аммиаком. Разбавляют раствор до 200 мл, нагревают почти до кипения и прибавляют раствор аммиака до небольшого его избытка. Если появился осадок, подкисляют раствор, прибавляют еще винной кислоты и снова прибавляют аммиак. Подготовленный таким способом раствор должен быть совершенно прозрачным. Прибавляют соляную кислоту до очень слабокислой реакции, нагревают до 60—80° С и затем прибавляют по меньшей мере 5 мл 1 %-ного спиртового *или аммиачного раствора диметилглиоксима на каждую 0,01 г
1 G. В а 1 z, Z3 anorg. u. allg. Chem., 231s 15 (1937).
462
Гл. XXII. Никель
никеля. Перемешивают и сейчас же прибавляют разбавленный раствор аммиака, пока осаждение не закончится, и затем еще небольшой его избыток. Раствор аммиака надо прибавлять по каплям, вливая его не по стенкам стакана, а прямо в середину анализируемого раствора. Дают постоять на паровой бане 1 ч или дольше, если никеля мало или кобальт и медь преобладают. Охлаждают до комнатной температуры, снова дают постоять 1 ч (а если осадок очень мал, то оставляют на ночь) и фильтруют через тигель Гуча или Мунро. Можно также применять и стеклянные фильтрующие тигли и другие пористые тигли. Фильтрат испытывают на полноту осаждения, промывают осадок холодной водой х, высушивают при 110—120° С в течение 45 мин, охлаждают и взвешивают в виде NiC8H14O4N4, содержащего 20,314% никеля 1 2.
Если полученные осадки велики или требуется повышенная точность определения, высушивание лучше проводить при 150° С до постоянной массы, чтобы улетучилось то небольшое количество реактива, которое могло быть захвачено осадком. Осадок возгоняется приблизительно при 250° С, и следовательно, сжигание бумажного фильтра (если фильтровали через бумажный фильтр) с осадком до NiO должно проводиться с особыми предосторожностями. Прокаливать осадок (хотя это вообще не рекомендуется) нужно следующим образом. Влажный фильтр с осадком завертывают в другой фильтр, осторожно нагревают до обугливания бумаги, следя за тем, чтобы она не воспламенялась, потом постепенно повышают температуру до полного сгорания угля и, наконец, прокаливают до красного каления.
Ход определения в присутствии значительных количеств кобальта, марганца или цинка. В присутствии значительных количеств кобальта, марганца или цинка осаждение никеля лучше проводить в растворе, содержащем уксусную кислоту и ацетат натрия.
Приготовляют раствор, содержащий не более 0,1 г всех металлов в 100 мл. Раствор должен быть освобожден от элементов сероводородной группы, за исключением тех, которые не загрязняют осадка, и от элементов, осаждающихся в виде основных ацетатов. Если должны быть выполнены оба отделения, сероводородом и в виде основных ацетатов, то железо после осаждения сероводородом надо окислить до трехвалентного состояния. Если было проведено осаждение в виде основных ацетатов, то фильтрат нагревают до кипения, регулируют кислотность раствора так, чтобы он содержал не более 2 мл 50%-ной уксусной кислоты и приблизительно 5 г ацетата натрия, и затем прибавляют диметилглиоксим в таком количестве, чтобы его хватило на образование диметилглиоксиминов никеля и кобальта и оставался избыток. Если же основных ацетатов не выделяли, то выпаривают раствор до малого объема и разбавляют его
1 Осадок заметно растворим в горячем маточном растворе и в горячей промывной воде [Р. N u k a, Z. anal. Chem., 91, 29 (1932—1933); W. J. Boyer, Ind. Eng. Chems Anal. Ed., 10, 175 (1938)]. Осадки, выделенные из растворов, содержащих растворимые соли в высоких концентрациях, рекомендуется растворять в разбавленной соляной кислоте и снова осаждать диметилглиоксимом.
2 Диметилглиоксимат никеля не гигроскопичен. Точность результатов, получаемых при анализе чистых растворов никеля, иллюстрируется следующими данными: были анализированы растворы, содержавшие 0,0910; 0,0916 и 0,0921 г никеля. Получено соответственно: 0,09083; 0,09162 и 0,09207 г. При проверке выпаренных фильтратов на содержание в них никеля получились лишь очень бледные окраски.
Методы определения
463-
или нейтрализуют так, чтобы он содержал не более 1 мл свободной соляной или серной кислоты в 200 мл. Свободная азотная кислота нежелательна, так как она разлагает реактив. Раствор нагревают до кипения, прибавляют раствор диметилглиоксима, перемешивают и приливают концентрированный раствор ацетата натрия до появления осадка и сверх того около 2 г избытка. Раствор ацетата натрия надо вливать прямо в середину анализируемого раствора, а не по стенке стакана. Нагревают (на паровой бане) по меньшей мере полчаса, охлаждают, фильтруют, осадок промывают и высушивают, как указано выше (см. «Ход определения в отсутствие значительных количеств кобальта, марганца или цинка», стр. 461).
' Вполне удовлетворительно, хотя, возможно, и менее точно, определить никель в присутствии значительных количеств кобальта можно окислением кобальта перекисью водорода, персульфатом аммония или другим подходящим окислителем в аммиачном растворе, содержащем аммонийные соли. Затем раствор кипятят для разрушения избытка окислителя и удаления большей части аммиака и осаждают никель диметйлглиок-симом.
Вместо диметилглиоксима рекомендовали 1 а-бензилдиоксим, который является более чувствительным реактивом на никель, и осаждение им идет быстрее. Согласно автору этого метода, удовлетворительные результаты получаются в присутствии железа, марганца,, цинка, магния и хрома. Осаждение лучше всего проводить прибавлением 0,02%-ного раствора реактива в спирте к аммиачному раствору солей никеля. Спиртовой раствор реактива должен содержать небольшое количество аммиака. Осаждение из уксуснокислых растворов не так удовлетворительно вследствие склонности а-глиоксима переходить в р-форму, и, кроме того, во всех случаях надо следить за тем, чтобы довольно плохо растворимый осаждающий реактив не был прибавлен в слишком большом избытке и не выпал в осадок.
а-Бензилдиоксим ((дифенилглиоксим) применяют 2, кроме того, как индикатор в цианидном методе определения никеля. В качестве реактива на никель рекомендуется также 3 применение а-фурилдиоксима.
Электролитический метод
Никель не осаждается электролизом из сильнокислых растворов. Осаждение в слабокислых растворах неполно, и для количественного анализа оно интересно лишь тем, что подчеркивает необходимость электролитического осаждения меди в сильнокислом растворе во избежание загрязнения осадка меди никелем и потери никеля. Из аммиачных растворов никель и кобальт осажддются легко и количественно, если принять некоторые простые меры предосторожности. Электролиз рекомендуется проводить в сильноаммиачном растворе, содержащем сульфат никеля и в некотором избытке — сульфат аммония. Если присутствует кобальт, осаждение облегчается прибавлением ацетата натрия или, лучше,
1 F. W. At ack, Chem. Ztg., 37, 773 (1913); Analyst, 38, 316 (1913); Z. anal. Chem., 53, 620 (1914).
2 G. L. К e 11 e y, J. В. С о n a n t, J. Ind. Eng. Chem., 8, 803 (1916).
3 B. A. S о u 1 e, J. Am. Chem. Soc., 47, 981 (1925),
464
Гл. XXII. Никель
сульфита натрия, хотя последний несколько загрязнет осадок серой х. Электролиз можно проводить и в растворах, содержащих хлориды вместо сульфатов. Нитраты должны отсутствовать, хотя одному или двум экспериментаторам удалось получить осадки и в их присутствии. Соли калия не оказывают влияния, так как и малые количества марганца и хрома в присутствии бисульфита натрия х. Хотя малые количества осаждающихся гидроокисей таких элементов, как железо и др., не окклюдируются в значительной степени отложившимся на катоде никелем, лучше все же их предварительно удалять повторным осаждением аммиаком, если они присутствуют в малых количествах, и ацетатным методом, если количество их велико. Серебро, медь, мышьяк и цинк также осаждаются, следовательно сероводородную группу и цинк следует всегда предварительно удалять. Ванадий и вольфрам нежелательны: ванадий, видимо, не мешает осаждению одного никеля или одного кобальта, но мешает их совместному осаждению; вольфрам не мешает осаждению никеля, но препятствует осаждению кобальта или никеля и кобальта вместе. Соли железа (II), хроматы, тартраты и молибден очень мешают осаждению 1 2. Электролиз не должен быть слишком продолжительным вследствие некоторой тенденции анода растворяться с последующим осаждением платины на катоде. О полноте осаждения можно судить по пробе с тиокарбонатом калия. Осаждение никеля редко бывает полным, и при выполнении точных анализов требуется дополнительное выделение его из электролита. При однократном осаждении никеля из аммиачного раствора, содержащего сульфат аммония, получаемые осадки обычно весят на несколько десятых долей миллиграмма больше, чем это следовало бы, несмотря на неполноту осаждения никеля.
Проводя электролиз нескольких порций раствора соли никеля, в которых содержание никеля было равно 0,30027 г, при плотности тока в 0,2 а на Ли2, W. Jukkola (Бюро Стандартов США) получил массу выделенного никеля равным в среднем 0,30032 г. После осаждения никеля электролиты выпаривали до небольших объемов и в них определяли остаточный никель диметилглиоксимом. В среднем каждая проба дала таким образом еще 0,00041 г никеля, т. е. общее его содержание было получено равным 0,30073 г вместо 0,30027 г. Платиновые аноды показали потерю в массе, равную для каждого анода в среднем 0,23 .иг. Вторичное осаждение никеля обратным током при замене электролита свежим раствором не привело к заметному улучшению. *
Ход определения. Из анализируемого раствора удаляют элементы сероводородной группы, окисляют железо (II) до железа (III) и выделяют его и подобные ему элементы повторным осаждением аммиаком (стр. 102), если количестве этих элементов мало, и ацетатным методом (стр. 103) — при значительном их содержании. Если присутствует цинк, его удаляют осаждением сероводородом из 0,01 н. по содержанию серной кислоты раствора или из муравьинокислого раствора (стр. 482). Прибавляют достаточное количество серной кислоты, чтобы связать присутствующие основания, и затем еще столько, чтобы после нейтрализации в растворе образовалось около 20 г сульфата аммония. Выпаривают до появления
1 Следует отметить, что прибавление сульфита предупреждает загрязнение осадка марганцем, но приводит к загрязнению его серой. Например, осадок, полученный в результате электролиза раствора, в котором было 0,3 г никеля, 5 мг марганца и 2 г сульфита натрия, содержал 2,2 мг серы и не содержал марганца. При выделении же никеля из раствора, в котором было 0,3 г никеля и 2 мг марганца и не было добавлено сульфита, получился осадок, содержавший 0,74 мг марганца.
2 G. Е. F. L u n d е 11, J. I. Hoffman, J. Ind. Eng. Chem., 13, 541 (1921)
Методы, определения 465
--------------------------------------------------------------------------
паров серной кислоты, охлаждают, несколько разбавляют водой, нейтрализуют раствором аммиака и прибавляют его избыток в 35 мл х.
Разбавляют до 150—200 мл, прибавляют 2 г бисульфита натрия, если присутствует кобальт, и ведут электролиз 6—8 ч с сетчатыми электродами (см. «Медь», стр. 286) при плотности тока 1 2 0,2—0,3 а на 1 дм2. Не прерывая тока, опускают стакан с электролитом, обмывают электроды холодной водой, затем промывают катод спиртом, высушивают несколько минут при 100° С, охлаждают и взвешивают 3.
Электролит, в котором обычно остается от 0,1 до 1 мг никеля и кобальта (главным образом кобальта), должен быть испытан следующим образом. Если присутствует марганец, то сначала кипятят раствор с персульфатом аммония, прибавленным в избытке, фильтруют и промывают осадок двуокиси марганца. Фильтрат обрабатывают сероводородом, нагревают его на краю паровой бани 1 ч, фильтруют через маленький фильтр и промывают осадок холодной водой, содержащей немного хлорида аммония и бесцветного сульфида аммония. Осадок затем прокаливают; сумму окисей никеля и кобальта взвешивают. Этот осадок должен весить не более 1 — 2 мг, и, при таком малом его количестве, для перечисления на металл можно умножить его массу на 0,75 и результат прибавить к массе осадка на катоде (факторы для перечисления NiO, СоО и Со3О4 на металлы равны соответственно 0,786, 0,787 и 0,734). Если масса окисей превышает 2 мг, то следует считать, что электролиз был проведен неудовлетворительно, и поправку вводить по массе металла, полученного прокаливанием окисей в токе водорода (стр. 432).
Если присутствует кобальт и к электролиту прибавляли сульфит, то осадок металлов и окислы, полученные из электролита, растворяют в концентрированной азотной кислоте, разбавляют раствор, охлаждают, доводят объем его точно до 100 мл и берут две аликвотные части по 40 Лл. Одну часть выпаривают с соляной кислотой и определяют в ней содержа-, ние серы (стр. 803). Другую часть выпаривают с 1—2 мл серной кислоты, охлаждают, разбавляют водой и определяют никель, как описано выше (стр. 462 и 463). Массу кобальта находят вычитанием найденных масс никеля и серы из полученной раньше суммарной массы.
Для отделения малых количеств никеля от таких больших количеств кобальта, как 1 г, Т. R. Cunningham и Т. R. McNeill из Электрометаллургической компании предложили следующий метод. Осадок на катоде растворяют в 10 лл азотной кислоты, выпаривают раствор до сиропообразной консистенции и приливают 10 мл воды. Затем прибавляют раствор карбоната натрия в небольшом избытке, нейтрализуют последний 50%-ной уксусной кислотой и прибавляют избыток ее в 10 мл. Нагревают до кипения и прибавляют малыми порциями и при непрерывном и сильном перемешивании 10— 12 г твердого нитрита калия. Через 10 мин прибавляют 50 мл горячей воды и оставляют стоять раствор при 50—60° С, время от времени перемешивая. Прибавляют бумажной массы, фильтруют и промывают осадок холодным раствором, содержащим 10 г нитрита калия и 2 мл 50%-ной уксусной кислоты в 1 л. Фильтрат с промывными водами
1 R. М. Fowler утверждает,’ что прибавление 1 г мочевины на 100 мл электролита приводит к более полному выделению никеля.
> 2 Высокие плотности тока приводят к получению относительно больших поло-
жительных ошибок.
3 Полное удаление никеля с платинового катода иногда бывает затруднительным, вследствие того что никель имеет склонность становиться «пассивным», Если никель частично останется на платине, то последующее нагревание электрода приведет к образованию на нем трудно удалимых пятен. Мы достигали полного растворени я ни келя, погружая платиновые электроды в кипящую концентрированную азотную кислоту,
30 Заказ 522.
466
Гл. XXII. Никель
обрабатывают 25 лм азотной кислоты, нагревают до кипения, прибавляют малыми порциями 5 г хлората калия и кипятят 10 мин для разложения азотистой кислоты. Затем осаждают никель обычным методом диметилглиоксимом, фильтруют, растворяют осадок в 25 мл кипящей разбавленной (1 : 1) соляной кислоте и повторяют осаждение.
Цианидный метод
Цианидный метод не вполне пригоден для единичных точных определений никеля. Однако метод этот очень удобен для массовых определений никеля в металлургических продуктах, и при соблюдении необходимых условий он дает достаточно точные результаты. Метод заключается в прибавлении титрованного раствора цианида калия к аммиачному раствору соли никеля до образования комплексного цианида K2[Ni(CN)a]. Конец титрования определяют по исчезновению мути иодида серебра, прибавленного в качестве индикатора. В присутствии железа или хрома прибавляют лимонную кислоту. Главные мешающие элементы — медь кобальт и цинк — обычно находятся в таких малых количествах, что ими можно пренебречь. Если же эти элементы присутствуют в больших количествах или если требуется большая точность, то никель выделяют сначала осаждением диметилглиоксимом, как описано на стр. 462, осадок растворяют в горячей азотной кислоте, диметил-глиоксим разрушают путем кипячения с персульфатом или хлоратом и затем титруют обычным способом.
Некоторое количество раствора цианида расходуется, без сомнения, на растворение осадка иодида серебра, и поэтому количество раствора иодида, вводимого в анализируемый раствор, должно быть точно отмерено и введена соответствующая поправка. Этого затруднения, а также и недостаточной четкости конечной точки титрования можно избежать, применяя в качестве индикатора а-бензилдиоксим Е
Ход определения. Если присутствует один никель в кислом растворе, то прибавляют аммиак в избытке 1—5 мл, разбавляют до 250 мл, приливают 10 мл раствора иодида калия (8 г в 1 л) и 10 мл раствора нитрата серебра (0,5 г в 1 л).
Подготовленный таким способом раствор титруют раствором цианида калия (4,6 г в 1 л), сначала быстро, пока муть усиливается, а затем более медленно до ее исчезновения. Титр раствора цианида калия устанавливают титрованием известных количеств никеля, причем в израсходованный на титрование объем раствора цианида калия следует вводить поправку на количество раствора, потребляемое иодидом серебра. Для определения этой поправки к оттитрованному раствору соли никеля прибавляют по 10 мл растворов иодида калия и нитрата серебра, титруют раствором цианида калия, операцию эту повторяют и берут среднее из близко совпадающих отсчетов.
Титрование никеля в сталях, содержащих немного меди, кобальта или вольфрама (или вовсе не содержащих этих элементов), может быть проведено после растворения 1 г стали в смеси азотной и соляной кислот, добавления небольшого количества серной кислоты для получения более резкой конечной точки титрования, 12 г лимонной кислоты и, наконец, аммиака в небольшом избытке. Более точное определение можно провести, осаждая никель диметилглиоксимом, растворяя осадок, разрушая диме-тилглиоксим и затем поступая, как описано выше х.
1 G. L. К е 11 е у, J. В. С о n a n t, J. Ind. Eng. Chem., 8, 803 (1916).
Методы определения
467
* Объемный метод (титрование диметилглиоксимом)
Объемный метод определения никеля титрованием его раствором диметилглиоксима 1 с применением в качестве индикатора диметилглиок-симной бумаги совмещает, по-видимому, точность весового диметилглиок-симного метода с быстротой цианидного метода. Раствор подготовляют к анализу так же, как и для определения никеля весовым диметилглиок-симным методом. Медь предварительно удаляют осаждением ее электролизом, металлическим цинком или алюминием, сероводородом или тиосульфатом. Железо окисляют до трехвалентного состояния перекисью водорода или персульфатом аммония и затем связывают в комплексное соединение. Для последней цели можно применять винную кислоту, лимонную кислоту, соли этих кислот, фториды и пирофосфат натрия. При добавлении фторида натрия выпадает бесцветный осадок, который не мешает последующему титрованию.
После добавления аммиака до слабого его запаха титруют 0,1 М или 0,04 М раствором диметилглиоксима [11,6 г (или 4,6 г) диметилглиоксима растворяют в 100 мл 2 н. раствора едкого натра и разбавляют до 1 л водой], пока взятая стеклянной палочкой капля раствора не перестанет давать красное пятно на индикаторной бумаге. Для приготовления последней полоску фильтровальной бумаги пропитывают насыщенным спиртовым раствором диметилглиоксима и высушивают на воздухе. Капли анализируемого раствора наносят на непосредственно на индикаторную бумагу, а на положенную поверх нее полоску чистой фильтровальной бумаги 2. Тогда присутствующий в растворе осадок диметилглиокси-мина никеля остается на верхней бумаге, а раствор, если он содержит еще неосажденный никель, проникая в индикаторную бумагу, образует красное пятно. Доп. ред*
Другие методы
Из весовых методов определения никеля следует отметить еще два: 1) осаждение никеля в виде гидроокиси никеля (III) едким кали и бромной водой с последующим прокаливанием осадка до NiO и 2) осаждение никеля в виде сульфида и прокаливание осадка до окиси никеля. Первый метод не точен вследствие невозможности отмыть весь КОН из осадка и загрязнения осадка кремнекислотой, а при применении более или менее сложных операций, необходимых для очистки осадка, метод становится очень продолжительным и мало удобным. Осаждение никеля сероводородом в виде сульфида никеля в уксуснокислом растворе, содержащем ацетат натрия, обычно неполно, а осаждение никеля сульфидом аммония затруднительно вследствие склонности сульфида никеля переходить в этих условиях в раствор. Применению обоих методов мешает присутствие многих элементов. Металлический никель, получаемый прокаливанием окиси никеля в токе водорода при 1000° С, пирофорен 3.
1 В. Д. Б о г а т с к и й, Л. Я. Д зюбина, Р. К о с а я, Зав. лаб., 9, 472
(1940); А. К. Б а б к о, М. В. К о р о т у н, там же, 11, 896 (1945); Р. И. А г л а д з е,
Н. Т, Гофман, там же, 12, 243 (1946); М. И. Левина, И. В. Т а н а н а е в, там же, 12, 245 (1946); Е. И. Г р ен б е р г, М. Я. Г е н и с, там же, 14, 491 (1948).
2 А. К. Б а б к о, М. В. К о р о т у н, цит. выше.
3 М. М. Н а г i n g, В. В. W е s t f а 11, J. Am. Chem. Soc., 52, 5141 (1930
30*
468
Гл. XXII. Никель
Малые количества никеля удобно определять колориметрическими методами, основанными, например, на синем окрашивании 1 ионов Ni(NH3)l+ или на окраске растворимого комплексного соединения, образующегося при обработке ионов никеля (III) диметилглиоксимом в аммиачном растворе 2.
Предложен метод колориметрического определения никеля в сталях и магниевых сплавах добавлением а-фурилдиоксима и извлечением полученного соединения 1,2-дихлорбензолом 3.
1 G. Н. А у г е s, F. S m i t h, Ind. Eng. Chem., Anal. Ed., 11, 365 (1939). Растворы, содержащие от 100 до 1500 мг!л никеля в 1,5 М растворе аммиака, практически не изменяют своего цвета при хранении даже в течение 59 недель на рассеянном свету в склянках из стекла пайрекс, закрытых притертыми пробками [J. Р. М е fall g, R. Е. К i t s о п, там же, 15, 606 (1943)].
2 А. Р. R о 11 е t, С. г., 183, 212 (1926). Спектрофотометрическое исследование этого метода описано в статье А. М. М i t с h е 11, М. G. Mellon, Ind. Eng. Chem., Anal. Ed., 17, 380 (1945). Приложение этого метода для определения никеля в стали см. J. В. Culbertson, R. М. Fowler, Testing Stainless Steel, Steel, 122, № 21, 108 (1938). *K. В. Троицкая, Ученые записки Казанского государственного университета, 97, 67 (1937); В. М. Пешкова, Зав. лаб., 8, 921 (1939); О. Я. Яковлева, там же, 11, 471 (1945); А. М. Д ы м о в, О. А. Володина, там же, 12, 53? (1946); А. К. Бабко, ЖАХ, 3, 284 (1948); М. Д. Ч е х о в и ч, Д. П. Щ е р б-о в, Зав. лаб., 16, 405 (1950). См. также Е. Passamaneck, Ind. Eng., Chem., Anal. Ed., 17, 257 (1945); A. J. H a 11, R. S. Young, Analyst, 71, 479 (1946) (применение органических растворителей); P. S ii e, Ann. chim. anal, appl., 28, 26 (1946) (осаждение диметилглиоксимата никеля на сульфате бария).*
3 A. R. G a h 1 е г, А. М. М i t с h е 11, М. G. М е 11 о n, Anal. Chem., 23, 502 (1951).
Глава XXIII
КОБАЛЬТ
Кобальт менее распространен в природе, чем никель, с которым он главным образом связан. Эти элементы встречаются в перидотитах (в оли-' вине), в сульфидах, например в пирите и пирротине, в арсенидах, в роговой обманке и биотите. Кобальт находится в железных рудах и часто присутствует в марганцевых рудах, что очень важно для аналитика ввиду вредного влияния его при определении марганца висмутатным методом. При анализе горных пород и металлургических продуктов определение кобальта по сравнению с определением никеля требуется значительно реже.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород кобальт ведет себя подобно никелю, и если количество его не велико и все осадки в ходе анализа переосаждаются, то большая часть кобальта останется в последнем фильтрате. В отличие от никеля небольшое количество кобальта переходит в осадок от аммиака. В несколько большем количестве, чем никель, он также захватывается осадками оксалата кальция и фосфата магния, особенно последним. Малые количества кобальта, присутствующие в горных породах, обычно выделяются вместе с марганцем, никелем и цинком обработкой сульфидом аммония (стр. 89) перед осаждением кальция. При выполнении особо точных работ и в присутствии больших количеств кобальта отделение его следует проводить перед осаждением аммиаком железа, алюминия и подобных им элементов или же проводить осаждение этих элементов ацетатным методом.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ КОБАЛЬТ
Как общее правило, кобальтовые минералы разлагают предварительной обработкой соляной кислотой с последующим добавлением азотной кислоты и, если это необходимо, сплавлением нерастворимого остатка с пиросульфатом калия или карбонатом натрия. Руды, содержащие серебро, лучше обрабатывать азотной и серной кислотами. В исключительных случаях можно применить сплавление с перекисью натрия (стр. 918). Эта операция может применяться и в тех случаях, когда необходимо определить серу, наравне с кислотными методами разложения, приведенными при описании анализа пирита или стибнита (см. «Сера», стр. 794).
470
Гл. XXIII. Кобальт
МЕТОДЫ ОТДЕЛЕНИЯ
Групповое отделение кобальта от других элементов совпадает в основном с описанным для никеля. Здесь также для отделений применяют сероводород, сульфид аммония, ацетат натрия, сукцинат натрия, карбонат бария, окись цинка, едкое кали, бром, эфир и соляную кислоту, купферон и электролиз.
Кобальт, однако,, увлекается оловом (IV) и виде смешанного сульфида, даже если осаждение сероводородом проводить из концентрированного по содержанию соляной кислоты раствора. Олово (II) при осаждении сероводородом не захватывает кобальта. Специальные меры должны быть также приняты для предупреждения соосажденпя кобальта с сульфидом цинка (см. стр. 479).
Увлечения кобальта осадком сульфида олова (IV) можно избежать, проводя осаждение сероводородом при 60° С из 1 М по содержанию соляной кислоты раствора, содержащего акролеин в концентрации 0,5 мл на 100 лл раствора [J. F. Flagg, J. Am. Chem. Soc., 63, 3150 (1941)].
Важное исключение, однако, мы имеем в групповом осаждении аммиаком: это осаждение нельзя применять для отделения железа, алюминия и других элементов от кобальта, как это делается в отношении умеренных количеств никеля. Захват кобальта осадком от аммиака значителен, даже при наличии малых количеств кобальта и при двукратном осаждении железа и алюминия х.
При анализе сталей осаждение окисью цинка ведут следующим способом. Навеску растворяют в разбавленной соляной или серной кислоте, окисляют железо азотной кислотой и удаляют большую часть кислот выпариванием раствора почти досуха. Затем раствор разбавляют, переносят в мерную колбу емкостью 500 мл и разбавляют приблизительно до 300 лм. После этого вносят в колбу, порциями по 5 мл, свежеприготовленную суспензию тонко измельченной окиси цинка (ее приготовляют тщательным взбалтыванием 50 г тонко измельченного реактива с 300 мл воды). При каждом добавлении суспензии окиси цинка раствор в колбе сильно взбалтывают. Эту операцию заканчивают, когда осадок становится светло окрашенным или когда жидкость над осадком после длительного отстаивания принимает молочный цвет. Тогда раствор в колбе разбавляют до метки, тщательно перемешивают и отбирают аликвотную часть, фильтруя через сухой фильтр, но не промывая осадка. Для лучшего отделения, в особенности в присутствии кобальта или больших количеств никеля, рекомендуется вторичное осаждение. В этом случае разбавления до определенного объема не проводят, фильтруют весь раствор, осадок растворяют, осаждают его снова окисью цинка и оба фильтрата соединяют. Если анализируемая сталь содержит значительные количества хрома, его лучше отогнать из хлорнокислого раствора (стр. 591) перед обработкой раствора окисью цинка. Дальнейшие подробности см. в гл. «Хром» (стр. 589).
Для конечного отделения кобальта от никеля, цинка и марганца нет реакции, равноценной реакции отделения никеля диметилглиоксимом. Осаждение нитрозо-Р-нафтолом для отделения кобальта применяется редко. Наиболее часто пользуются способом, являющимся обратным кобальтинитритному методу определения калия и основанным на осаждении кобальта в уксуснокислом растворе нитритом калия. Этот метод требует предварительного отделения сероводородной группы и проводится, как описано ниже (стр. 471).
При анализе горных пород, минералов и руд можно удовлетворительно отделить^кобальт от железа, титана, циркония и гафния осажде-
1 Например, после двукратного осаждения гидроокиси алюминия аммиаком, как описано в гл. «Алюминий» (стр. 565), из раствора, содержавшего первоначально 0,1 г А12О3 и 0,05 г кобальта, в осадке гидроокиси алюминия все еще осталось 0,0012 г кобальта [G. Е. F. L u n d е 11, Н. В. К п о w 1 е s, J. Am. Chem. Soc., 45, 680 (1923)].
Методы определения
471
нием фосфат-ионами в уксуснокислом растворе х, в котором концентрация ионов водорода приводится к pH «=«3,5.
Кобальт может быть отделен от алюминия, хрома, ванадия, урана, вольфрама, молибдена, мышьяка, титана, цинка, марганца, кальция и магния осаждением в слабоаммиачном растворе 1 2 фенилтиогидантоиновой кислотой NH2—CS—N(C6H5)—СН2СООН. Недостатками этого метода являются частичное осаждение железа и никеля и очень неприятный запах применяемого реактива.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Электролитический метод
Электролитический метод является наиболее удовлетворительным методом определения любых, но не слишком малых количеств кобальта, несмотря на то, что метод этот не дает отделения кобальта от никеля и что содержание кобальта вычисляют по разности после анализа осадка на катоде и определения в нем никеля и других элементов, например меди, серебра и серы. Последняя неизменно присутствует в осадке на катоде в виде сульфида кобальта, если к электролиту был прибавлен сульфит натрия. Если электролит содержал только сульфаты, в осадке на катоде может быть лишь самое незначительное количество серы или ее вовсе не будет. Ход определения совпадает с описанным для никеля (стр. 464).
Предложен также3 следующий ход определения, при котором от 15 до 160 мг кобальта можно осадить в течение 30 мин. Применяют быстро вращающийся анод (800—1000 об!мин), сетчатый катод и электролит, содержащий 50 мл раствора аммиака, 5 г хлорида аммония и 0,3—0,4 г бисульфита натрия в объеме 80—100 мл. Плотность тока 4—7 а на 100 сл2. Бисульфит натрия надо прибавлять к щелочному раствору и вводить его в большем количестве при малом содержании кобальта (0,4 г при 15— 80 мг кобальта), чем при большом его содержании (0,3 г при 100—160 мг кобальта). Если электролит не обесцветится через 2 мин, добавляют еще 10—20 мг бисульфита.
Выделенный на катоде осадок может содержать немного серы (не более 0,3 мг) и немного платины (0,2 мг, если электролиз продолжался 30 мин, и 0,5 мг, если проведение электролиза было растянуто до 60 мин), перешедшей на катод в результате некоторого растворения Знода.
Калиево-нитритный метод
Очень хорошим методом отделения кобальта от никеля является осаждение его в виде нитрокобальтиата (III) калия 2K3[Co(NO2)e]-3H2O прибавлением раствора нитрита калия в уксусной кислоте. Свободные минеральные кислоты и окислители должны отсутствовать, а также и элементы сероводородной группы. Кроме того, раствор не должен содержать значительных количеств элементов, осаждаемых аммиаком. По этой причине осаждение обычно проводится после отделения железа и др. ацетатным методом и концентрирования полученного фильтрата. Осадок
1 V. N о г t h, R. С. Wells, Ind. Eng. Chem., Anal. Ed., 14, 859 (1942).
2 H. H. W i 11 а г d, D. Hall, J. Am. Chem. Soc., 44, 2219 (1922).
3 D. H. Brophy, Ind. Eng. Chem , Anal. Ed., 3, 363 (1931).
472
Гл. XXIII. Кобальт
нитрокобальтиата (III) калия хорошо растворяется в воде и в разбавленных минеральных кислотах, но нерастворим в спирте и в разбавленных растворах, содержащих немного уксусной кислоты и либо ацетат калия, либо нитрит калия. Осадок никогда не взвешивают как таковой и не титруют, как это делают в некоторых модификациях аналогичного метода определения калия. Этот метод наиболее пригоден для количественного отделения нескольких миллиграммов кобальта цт никеля, и в этом случае осадок нитрокобальтиата (III) калия обычно растворяют, вторично осаждают кобальт в виде сульфида и затем взвешивают его в виде сульфата. При определении больших количеств кобальта осадок растворяют в серной кислоте, раствор делают сильноаммиачным и подвергают его электролизу.
Осаждение нитрокобальтиата (III) калия. Кислый анализируемый раствор йе должен содержать аммонийных солей и элементов сероводородной группы (за исключением тех, которые не мешают определению). Желательно также отсутствие элементов, осаждающихся в уксуснокислом растворе, содержащем ацетат натрия (стр. 103). Если в предыдущих операциях были введены сколько-нибудь значительные количества посторонних солей, выделяют сначала кобальт осаждением его сероводородом в горячем растворе, содержащем уксусную кислоту и ацетат натрия, или лучше, если обстоятельства это позволяют, осаждают кобальт едким кали и бромом в горячем растворе. Осадок растворяют в соляной кислоте (в первом случае — с добавлением окислителя), выпаривают раствор досуха, увлажняют сухой остаток 4—5 каплями соляной кислоты и растворяют соли в возможно меньшем количестве воды.
Отдельно приготовляют раствор 2—3 г нитрита калия (соль должна содержать по крайней мере 85% KNO2) в возможно меньшем количестве разбавленной уксусной кислоты (10%-ной по объему) и прибавляют этот раствор к анализируемому раствору кобальта при сильном перемешивании. Оставляют раствор (объем которого не должен превышать 100 мл) на 24 ч, время от времени его перемешивая. Когда жидкость над осадком станет прозрачной, проверяют на полноту осаждения, прибавляя еще немного осадителя. Затем фильтруют через маленький фильтр, не стараясь полностью перенести осадок на фильтр, и промывают сосуд, в котором проводилось осаждение, фильтр и осадок 5%-ным раствором ацетата калия или нитрита калия, подкисленным 1 мл уксусной кислоты на 1 л. Если количество никеля было очень велико, осадок растворяют и осаждение повторяют.
Конечное определение в виде CoSO* (при определении малых количеств кобальта). После предварительного промывания осадка уксуснокислым раствором ацетата калия его переносят полностью на фильтр и промывают 95%-ным спиртом, пока из осадка и фильтра не будет отмыт осадитель. Фильтр с осадком потом прокаливают в фарфоровом тигле при возможно более низкой температуре, чтобы избежать плавления осадка. Остаток переносят из тигля в маленький стакан, обрабатывают его соляной кислотой и несколькими кристаллами хлората калия, выпаривают раствор досуха и растворяют остаток в 3—5 каплях соляной -кислоты. Раствор разбавляют холодной йодой, прибавляют около 1 г ацетата натрия, нагревают до кипения и осторожно кипятят 1 ч для того, чтобы осадить железо и алюминий.
Если появится осадок, его отфильтровывают через маленький фильтр и промывают горячим 2%-ным раствором хлорида аммония. Осадок
Методы определения
отбрасывают, фильтрат обрабатывают аммиаком до щелочной реакции, нагревают до кипения и прибавляют бесцветный сульфид аммония в небольшом избытке. Дают постоять на паровой бане, пока осадок не осядет, затем отфильтровывают его и промывают холодным 2 %-ным раствором хлорида аммония, содержащим немного бесцветного сульфида аммония, до удаления щелочных солей. Затем очень осторожно высушивают фильтр» с осадком во взвешенном платиновом тигле, усиливают нагревание так, чтобы бумага фильтра обуглилась, но не воспламенилась и, наконец, прокаливают до удаления углерода. Охладив тигель, прибавляют достаточное количество разбавленной (1 : 1) соляной кислоты для растворения окиси кобальта, осторожно нагревают до удаления избытка кислоты и охлаждают. Наконец, прибавляют серной кислоты в количестве, необходимом для превращения хлорида кобальта в сульфат, осторожно нагревают до удаления ее избытка и затем прокаливают несколько минут при , темно-красном калении (приблизительно 500° С). Если осадок весит более» 1 мг, увлажняют его 1—2 каплями воды и снова осторожно выпаривают и прокаливают для удаления последних следов свободной серной кислоты. Охлаждают в эксикаторе и взвешивают в виде CoSOi.
Конечное определение электролизом (при содержании кобальта больше нескольких миллиграммов'). Фильтр с осадком переносят в сосуд, где проводилось осаждение, прибавляют 20 мл азотной кислоты, взбалтывают, пока бумага не распадется на Ьолокна, и затем прибавляют 5 мл серной кислоты, выпаривают до появления ее паров, охлаждают и, если органические вещества не разрушились, прибавляют азотной кислоты и. повторяют выпаривание. Наконец охлаждают, обмывают внутренние стенки колбы водой и снова выпаривают до появления паров серной кислоты, т. е. до удаления всей азотной кислоты. Снова охлаждают, разбавляют и поступают дальше так, как указано при электролизе никеля (стр. 463). Если для более быстрого осаждения был прибавлен бисульфит натрия, то осадок будет содержать большее или меньшее количество серы в виде-сульфида кобальта \
а-Нитрозо^-нафтоловый метод
При а-нитрозо-р-нафтоловом методе определения кобальта, так же как и при калиево-нитритном методе, требуется предварительное удаление железа, меди и некоторых других элементов группы сероводорода.. Получаемый в этом методе осадок также нельзя взвешивать непосредственно, как и осадок нитрокобальтиата (III) калия. Отделение малых количеств кобальта от больших количеств никеля однократным осаждением а-нитрозо-р-нафтолом дает менее удовлетворительные результаты, чем нитритный метод. В отношении количества кобальта, для определения которого этот метод лучше всего применим (0,1 г), он занимает среднее положение между электролитическим и нитритным методами.
Образование осадка1 2 нитрозо-Р-нафтолята кобальта (III)
[С10Н6О (NO) ]3Со происходит после прибавления свежеприготовленного
1 Например, A. Isaacs (Бюро Стандартов США) нашел 0,5 мг серы в осадке кобальта, весившем 0,9692 г, и 0,7 мг — в другом осадке, весившем 0,2446 г.
2 М. A. I 1 i n s к у, G. von К п о г г е, Вег., 18, 699 (1885); G. von К п о г г е, Z. angew. Chem., 264 (1933).
474
Гл. XXIII. Кобальт
раствора реактива в 50%-ной уксусной кислоте 1 .к горячему разбавленному солянокислому раствору соли кобальта. Осадок исключительно устойчив по отношению к кислотам, щелочам, окислителям и восстановителям. Он растворяется в феноле, анилине, заметно растворим в концентрированном спирте, меньше — в разбавленном спирте и только очень слабо растворим в разбавленной соляной кислоте. Железо, медь, висмут, серебро, хром, цирконий, титан, ванадий, олово и азотная кислота мешают определению. Алюминий, бериллий, свинец, кадмий, фосфат-ионы, арсе-нат-ионы, аммонийные соли, купферон и умеренные количества никеля не мешают 2. Если содержание никеля превышает содержание кобальта или если никель находится в значительном количестве, то осадок следует прокалить, остаток растворить в соляной кислоте и снова осадить кобальт а-нитро’зо-Р-нафтолом. Осадок можно прокаливать и взвешивать в виде Со3О4 только если содержание кобальта не превышает нескольких миллиграммов, так как при прокаливании осадка получается окисел неопределенного состава. При определении большего количества кобальта следует восстановить полученный окисел до металла в струе водорода (стр. 432) или же заканчивать определение электролизом или превращением этого окисла в сульфат кобальта, как в нитритном методе 3.
Утверждают 3, что осадок можно высушить при 130° С и взвесить в виде дигидрата [C10H6O(NO)i3 Со-2НгО, если: 1) окислить кобальт до трехвалентного состояния перед прибавлением а-нитрозо-р-нафтола; 2) осаждать не более 25 мг кобальта; 3) проводить осаждение в уксуснокислом растворе. Окисление кобальта в минеральнокислом растворе, свободном от аммонийных солей, можно провести, обрабатывая холодный анализируемый раствор последовательно перекисью водорода (пергидролем), едким натром до щелочной реакции и уксусной кислотой до кислой реакции. Этот метод может применяться в присутствии алюминия, цинка и умеренных количеств никеля.
Ход определения. Приготовляют слабо солянокислый раствор сульфатов или хлоридов кобальта и никеля, содержащий не больше 0,1 г кобальта и 0,2 г никеля и не содержащий мешающих определению перечисленных выше веществ. Разбавив солянокислый раствор до 200 мл,
1 Применяют свежеприготовленный раствор 1 г а-нитрозо-р-нафтола в 15 мл ледяной уксусной кислоты. Качество применяемого реактива следует проверить, так как некоторые продажные препараты содержат какое-то органическое соединение, которое захватывается выделяющимся осадком и которое почти невозможно потом сжечь. Другие препараты имеют в своем составе железо, переходящее в уксуснокислый раствор реактива и затем осаждающееся в солянокислом растворе. Н. A. F а 1 е s [Inorganic Quantitative Analysis, 1925, примечание к § 230] утверждает, что некоторые препараты а-нитрозо-р-нафтола осаждают никель. Осаждающая способность применяемого реактива может быть определена следующим способом. Растворяют 1 г реактива в 15 мл ледяной уксусной кислоты и медленно при сильном перемешивании вливают в этот раствор 300 мл разбавленной (5 : 95) соляной кислоты, содержащей 0,25 г кобальта в виде хлорида или сульфата. Дают постоять, фильтруют, промывают осадок, прокаливают его и взвешивает, как описано в ходе определения. Теоретически 1 г реактива должен осадить 0,1 г кобальта.
2 В. S. Evans [Analyst, 62, 363 (1937)] утверждает, что а-нитрозо-р-нафтол, растворенный в сиропообразной фосфорной кислоте, не осаждает железа (III), но осаждает кобальт, медь и железо (II). Раствор реактива, который надо приготовлять не ранее чем за 2—3 дня до его применения, получают, всыпая 3,5 г а-нитрозо-р-нафтола в маленький стакан, прибавляя 50 мл сиропообразной фосфорной кислоты, помещая смесь на горячую плитку, перемешивая термометром, пока температура не достигнет 60° С, и охлаждая раствор. Подробности осаждения кобальта см. в оригинальной статье.
3 С. Mayr, F. Feigl, Z. anal. Chem., 90, 15 (1932).
Методы определения
475
прибавляют такое количество соляной кислоты, чтобы общее ее содержание равнялось 10 мл, и нагревают до 60° С. Затем медленно и при непрерывном перемешивании прибавляют осадитель в 1,5-кратном количестве по сравнению с требуемым для осаждения кобальта, но не меньше 10 мл. Сильно перемешивают еще 1 мин, дают постоять, пока раствор не охладится до комнатной температуры (около 1 ч), и фильтруют через фильтр средней плотности. Осадок на фильтре тщательно промывают по крайней мере 100 мл горячей разбавленной (1 : 19) соляной кислоты. Если проходящий через трубку воронки раствор прозрачен, то на осадок, который потом выпадает в фильтрате, не следует обращать внимания, так как он не содержит кобальта.
Фильтр с осадком переносят во взвешенный фарфоровый тигель высокой формы (емкость 30 мл) и прокаливают сначала осторожно, потом при 750—850° С до постоянной массы. Взвешивают в виде Со3О4, содержащего 73,4% кобальта.
Окись Со3О4 начинает разлагаться1 при температуре выше 900° С, и переход ее в СоО при 1000° С происходит почти количественно. Если СоО нагревать при 750° С, то состав окисла снова приближается к Со3О4. При выполнении анализов высшей точности окись кобальта можно прокаливать в атмосфере водорода в тигле Розе (стр. 56), затем охлаждать в атмосфере водорода до комнатной температуры и взвешивать в виде металла. Полученный таким способом металл не пирофорен.
Для растворения Со3О4 ее переносят в высокий тигель или в стакан, приливают концентрированную соляную кислоту, покрывают часовым стеклом для предупреждения разбрызгивания и нагревают на паровой бане. Прокаленная окись кобальта должна полностью раствориться (что показывает отсутствие в ней органических загрязнений), при добавлении аммиака к полученному раствору не должен выпадать осадок (отсутствие железа) и при подкислении этого раствора и пропускании через него сероводорода также не должен выделяться осадок (отсутствие меди). Прокаленную окись кобальта можно также перевести в раствор сплавлением с пиросульфатом калия в тигле, покрытом крышкой. Если в анализированной пробе было много никеля, то прокаленную Со3О4 следует растворить в соляной кислоте, перенести раствор в стакан емкостью 200 мл, разбавить или подкислить раствор так, чтобы в объеме 100 мл было 5 мл кислоты, и снова осадить кобальт а-нитрозо-[}-нафтолом.
При выполнении точных анализов, в которых определяется более 0,01 г кобальта, окись кобальта надо восстановить в токе водорода и взвесить кобальт в виде металла (стр. 432). Можно также превратить окись кобальта в сульфат кобальта и закончить определение, как описано на стр. 472 и 473.
При определении малых количеств кобальта (меньше 0,6 мг) осадок можно растворить в хлороформе и закончить определение фотометрическим методом 2.
Колориметрические методы
Для определения очень малых'гколичеств кобальта (микрограммов) предложен колориметрический метод’3, основанный на красном окрашивании, которое образует кобальт с нитрозо-В-солью (1,2-нитрозонафтол-3,6-дисульфонат натрия). Мешающее влияние меди, никеля и железа в значительной мере устраняется, если после образования кобальтового
1 J. I. Hoffman, J. Research NBS, 7, 885 (1931).
2 L. W а 1 d b а п е г, N. М. Ward, Ind. Eng. Chem., Anal. Ed., 14, 727 (1942).
3 H. S. Van Kloos t er, J. Am. Chem. Soc., 43, 746 (1921); *M. И. Троиц-к а я, T. В. 3 а г л о д и и а, Зав. лаб., 13, 145 (1947); Д. П. М а лю г а, ЖАХ, 1, 176 (1946); 2, 323 (1947); Д. П. Щ е р б о в, Зав. лаб., 15, 1399 (1949).*
476
Гл. XXIII. Кобальт
комплекса раствор кипятить с определенным количеством азотной кислоты. В таких условиях десятые доли миллиграмма кобальта можно определить в присутствии 10—12 мг меди х.
*Состав образующегося при этом соединения отвечает формуле Co[C10H4ONO(SO3Na)2]3. Реакция проводится в уксусно-ацетатном растворе (pH не ниже 5,5). Марганец и цинк не мешают определению. Измерение йветопоглощения полученного раствора рекомендуется проводить в фотоколориметре с применением зеленых светофильтров (максимум светопоглощения 520— 530 ммк), устраняющих мешающее влияние собственной окраски реактива. В присутствии железа следует увеличить количество прибавляемой нитрозо-В-соли. Доп. ред.*
Колориметрические определения кобальта можно также проводить роданидном способом 1 2. К 40 мл нейтрального раствора хлорида кобальта прибавляют 5 г роданида аммония и 50 мл ацетона. Разбавляют точно до 100 мл и сильно взбалтывают. Полученное синее окрашивание сравнивают с окраской стандартного раствора, содержащего не меньше половины и не больше двукратного количества кобальта по сравнению с тем, которое присутствует в анализируемом растворе, и обработанного таким же образом. Если присутствует никель, надо белую пластинку под пробирками колориметра покрыть блестящей желтой желатиновой бумагой. Удовлетворительные результаты в присутствии никеля получаются, если хлорид никеля и хлорид кобальта присутствуют каждый в концентрации 8 • 10-3 М на 1 л. При анализе более разбавленных растворов солей кобальта можно допустить присутствие относительно большого количества никеля.
*Для устранения мешающего влияния железа было рекомендовано добавление фторида аммония 3. Было показано 4 5 далее, что при введении в раствор пирофосфата натрия и соблюдении требуемых условий (отношении количества роданида в растворе к количеству пирофосфата, равном 5 : 1, и надлежащем pH раствора — исчезновении окраски фенолфталеина при добавлении роданида аммония) кобальт можно определять в присутствии очень большого количества никеля (метод был предложен для определения кобальта в металлическом никеле). Добавление пирофосфата натрия может быть использовано также и для связывания железа при анализе руд, содержащих железо, кобальт и никель в. Доп. ред.*
Другие методы
Из других методов определения кобальта можно указать на: 1) осаждение его в виде сульфида сероводородом из уксусно-ацетатного рас
1 A. J. Н а 11, R. S. Young, Anal. Chem., 22, 497 (1950).
2 Е. S. Т omul a, Z. anal. Chem., 83, 6 (1931). См. также V. North, R. Z. Wells, Ind. Eng. Chem., Anal. Ed., 14,859 (1942).* Ю. А. Чернихов (см. в книге В. Ф. Гиллебранд, Г. Э. Лендель, Практическое руководство по неорганическому анализу, ОНТИ, 1935, стр. 358); Ю. Ю. Л урь е, М. И. Троицкая, Mikrochemie, 22, 101 (1937); В. М. Звенигородская, Зав. лаб., 7, 1350 (1938); Ю.-Ю. Лурье, Э. М. Т а л ь, там же, 8, 383 (1939); Н. М. Р u t s с h ё, W. F. М о 1 о о 1 у, Anal. Chem., 19, 236 (1947); А. К. Бабко, О. Ф. Драко, Зав. лаб., 16, 1162 (1950).*
3 Ю. А. Чернихов, цит. выше.
4 Ю. Ю. Лурье, М. И. Троицкая, цит. выше.
5 В. М. Звенигородская, цит. выше, Ю. Ю. Лурье, Э. М. Таль, цит. выше.
Методы определения
477,
твора или сульфидом аммония с последующим прокаливанием осадка до Со3О4 и 2) метод титрования цианидом. Первый из этих методов связан с затруднениями в достижении полноты осаждения кобальта и в доведении получаемого прокаливанием окисла до определенного состава. Поэтому метод применим лишь для определения малых количеств кобальта и при его выполнении надо всегда проверять полноту осаждения. Цианидный объемный метод не так хорош для определения кобальта, как для определения никеля, и так как его применение требует большого числа модификаций для разных случаев анализа, мы его не будем здесь рассматривать Ч Метод титрования гексацианоферратом (III) калия 1 2 K.3[Fe(CN)e] чаще применяется в массовых анализах металлургических продуктов, поскольку он не требует предварительного отделения никеля и железа. Метод основан на окислении кобальта (II) до трехвалентного состояния вливанием анализируемого раствора в аммиачный цитратный раствор, содержащий титрованнный раствор K.3[Fe(CN)e] в некотором избытке. Этот избыток затем оттитровывают обратно потенциометрически нитратом кобальта. Марганец также титруется, поэтому его надо определить отдельно и внести соответствующую поправку в результат определения кобальта.
*Объемный потенциометрический метод является одним из лучших методов определения кобальта, получившим в настоящее время широкое распространение. К сказанному о нем выше следует добавить, что никель не реагирует с K3[Fe(CN)6] и не мешает титрованию кобальта, даже когда присутствует в большом количестве (например, при анализе металлического никеля). Не мешают также цинк, медь и мышьяк (V). Мешают железо (II), мышьяк (III) и значительные количества железа (III). Для устранения мешающего влияния последнего его связывают в комплекс винной кислотой'или ее солью. При титровании надо соблюдать следующие условия: температура раствора должна быть не выше 25° С; раствор должен содержать 20—25 мл концентрированного раствора аммиака и не менее 5 г хлорида аммония в 100 мл; следует добавлять 10 мл 30%-ного раствора лимонной кислоты или цитрата аммония; содержание кобальта не должно превышать 0,05 г в 150—180 мл; концентрация применяемого титрованного раствора гексацианоферрата (III) калия должна быть не ниже 0,05 М. Кроме указанного выше обратного титрования, применяется 3 и прямое титрование раствором K3[Fe(CN)e].
Был предложен также метод 4, в котором сначала титруют потенциометрически марганец в кислой среде раствором перманганата калия в присутствии фторид-ионов [образуется комплексный фторид марганца (III)], а затем определяют кобальт, как описано выше. Доп. ред.*
1 В статье В. S. Evans [Analyst, 62, 363 (1937)] описывается подробно метод титрования цианидом и приложение этого метода для определения кобальта в сталях после предварительного отделения от никеля и железа осаждением специальным образом приготовленным нитрозонафтолом и от меди — осаждением последней сероводородом.
2Р. Dickens, G. Maassen, Archiv Eisenhiittenw., 9, 487 (1936); G. J. S t e-e 1 e, J. J. Phelan, Gen. Elec. Rev., 42, 218 (1939); *B. M. Звенигородская, Зав. лаб., 11, 1010 (1945); В. M. Звенигородская, Р. Г. Готсди-и е р, там же, 12, 142 (1946); М. Н. Ч е п и к, там же, 15, 1470 (1949).*
3 М. Н. Че пи к, Зав. лаб., 15, 1470 (1949).
4 В. М. Звенигородская, Р. Г. Готсдинер, Зав. лаб., 12, 142(1946).
Глава XXIV
ЦИНК
Цинк не является редким элементом. Он довольно широко распространен в земной коре, хотя помимо своих руд он встречается лишь в очень малых количествах. Наиболее распространенной рудой цинка является сульфид цинка — сфалерит ZnS. В больших количествах встречаются также карбонат цинка — смитсонит ZnCOз и силикат цинка — каламин Zn2(OH)2SiO3. Цинк находят иногда в гранитах и основных горных породах. В металлургических продуктах, в частности в различных продуктах цветной металлургии, цинк встречается часто, и его содержание в них обычно приходится определять. Исключение составляют лишь те сплавы, в которых цинк является одной из главных составных частей, нацример латунь, при анализе которых определяют все остальные металлы, составляющие сплав, а содержание цинка вычисляют по разности.
ОБЩИЕ ЗАМЕЧАНИЯ
Цинк входит в состав некоторых сортов стекла. Поэтому, когда определяют очень малые его количества или требуются анализы высокой точности, особое внимание надо обращать на то, в какой стеклянной посуде проводится анализ. Кроме того, при анализе материалов, содержащих очень малые количества цинка, нельзя просеивать их через латунные сита.
В обычном ходе анализа горных пород, содержащих небольшие количества цинка, большая часть его пройдет через весь ход анализа и оста-' нется в последнем фильтрате, особенно если все осаждения проводятся двукратно. Незначительная часть цинка перейдет, однако, в осадок от аммиака и будет принята за алюминий, другая малая часть присоединится к фосфатному осадку и будет принята за магний. В осадке оксалата кальция может быть лишь самое ничтожное количество цинка, а скорее всего его там вовсе не будет. Когда цинк находится в небольшом количестве, его обычно определяют, осаждая сульфидом аммония (стр. 89) из фильтрата, полученного после двукратного осаждения аммиаком. Большие количества цинка лучше выделять сероводородом из слабокислых растворов, как описано в разделе «Методы определения» (стр. 481), перед осаждением железа и других элементов аммиаком.
Методы отделения
479
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ЦИНК
Разложение цинковых минералов не представляет затруднений и обычно проводится либо одной соляной кислотой, либо соляной кислотой с последующим добавлением азотной кислоты, либо, наконец, одной азотной кислотой. Нерастворимый в кислотах остаток, обычно не содержащий цинка, может быть затем разложен сплавлением с карбонатом натрия и растворением полученного плава в кислоте.
МЕТОДЫ ОТДЕЛЕНИЯ
Не существует методов отделения цинка, которые одной операцией удовлетворительно отделили бы его от большинства других элементов, как это имеет место в отношении марганца (метод с азотной кислотой и хлоратом калия) и никеля (диметилглиоксимный метод).
Наилучшим методом отделения цинка является осаждение его сероводородом из слабокислых растворов после предварительного выделения элементов сероводородной группы также сероводородом, но из более кислых растворов.
Имеется большое число научных работ, посвященных нахождению наилучших условий проведения этих разделений. Элементы сероводородной группы очень хорошо отделяются от цинка осаждением их сероводородом из растворов, 2 н. по содержанию свободной серной кислоты, если фильтрование после осаждения будет проведено немедленно, и 9 н.— если фильтрование будет отложено.
Осаждение из слабо солянокислых растворов менее удовлетворительно.
В опытах, проведенных Н. В. Knowles, достигалось полное отделение 0,1 г меди от 0,1 г цинка в следующих условиях: 1) сульфаты обоих металлов были растворены в серной кислоте, концентрация которой была в пределах от 2 до 18 н.; 2) температура растворов была от 25 до 100° С; 3) пропускание сероводорода через растворы продолжалось 60 мин', 4) фильтрование проводили немедленно после осаждения сульфида меди и осадки промывали насыщенными сероводородом горячими растворами серной кислоты той же концентрации, в какой проводили осаждение.
Во втором опыте через нагретый до 100° С раствор, содержавший по 0,05 г цинка, меди, висмута, мышьяка, сурьмы и олова, в 300 мл 9 н. серной кислоты был пропущен сероводород -в течение 60 мин и выделившийся осадок был сейчас же отфильтрован. В нем оказалось только 0,2 мг цинка. 4
В третьем опыте раствор, содержавший 0,1 г меди и 0,2 г цинка в 200 мл 9 н.. серной кислоты, был обработан сероводородом в течение 15 мин при 100° С; пропускание сероводорода продолжали еще 5 ч, в то время, когда раствор охлаждался, затем раствор оставили на ночь, после чего отфильтровали и промыли осадок, — цинка в нем обнаружено не было.
При осаждении меди и кадмия сероводородом даже из 0,4 н. солянокислого раствора (т. е. при двукратной концентрации соляной кислоты по сравнению с той, какая обычно применяется) цинк захватывается осадком. Поэтому выделенный осадок сульфидов всегда надо растворять и повторять осаждение еще раз или даже несколько раз, в зависимости от величины осадка (см. также стр. 296).
Для отделения цинка от железа, марганца, хрома, алюминия и никеля (но не от кобальта) было предложено 1 осаждение его сероводородом из горячего раствора сульфата цинка, приведенного к pH =1,65 добавлением буферного раствора сульфата и бисульфата. Хорошее отделение цинка от тех же элементов может быть также достигнуто 2 и однократным
1 С. Е. Р. J е f f г е у s, Е. Н. S w i f t, J. Am. Chem. Soc., 54, 3219 (1932).
2 J. R. С a 1 d w e 1 1, H. V. M о у e r, J. Am. Chem. Soc., 57, 2372, 2375 (1935); 59, 90 (1937).
480
Гл. XXIV. Цинк
осаждением из холодного раствора, если после обработки сероводородом к раствору прибавить 10 мл 0,02 %-ного раствора желатины, дать постоять 15 мин, отфильтровать осадок и затем промыть его холодной водой. Желатина в этом случае способствует коагуляций коллоидного сульфида цинка.
Для отделения от кобальта применяется тот же метод, но, кроме того, перед обработкой сероводородом и прибавлением желатины вводят в раствор 0,2 мл акролеина. Акролеин реагирует с сероводородом на поверхности частиц сульфида цинка и этим уменьшает «последующее осаждение» сульфида кобальта. Если введение в раствор сульфат-ионов недопустимо, то можно пользоваться буферным раствором хлоруксусной кислоты и ацетата натрия (pH =2,8).
Всеми указанными выше методами нельзя отделить цинк от индия, таллия и галлия А
Для отделения малых количеств цинка от никеля был предложен метод 1 2, основанный на относительной устойчивости комплексного цианида никеля по сравнению с комплексным цианидом цинка. Через раствор, содержащий эти цианиды, слегка подкисленный уксусной кислотой, пропускают сероводород, — осаждается сульфид цинка.
Цинк вместе с железом, никелем и кобальтом может быть отделен от щелочных металлов, магния и щелочноземельных металлов осаждением сульфидом аммония (стр. 89), и от всех этих элементов, а также и от алюминия, титана, циркония, ниобия и др. — осаждением сульфидом аммония в присутствии тартрат-ионов. Отделение от олова, сурьмы, мышьяка и др. может быть проведено осаждением цинка сульфидом натрия или сульфидом калия. Аммиак часто применяется для отделения цинка от железа, алюминия и некоторых других элёментов. К сожалению, при обычной обработке аммиаком 3 (стр. 102) цинк частично увлекается осадком, и отделение это можно применять только в тех случаях, когда осадок мал, кремнекислота отсутствует и осадок переосаждают не менее трех раз или же когда определяют только цинк и можно применять особый метод обработки 4. При осаждении железа и других элементов аммиаком
1 Н i s a j i Kato [Sci Rep. Tonoku Imp. Univ., Ser. 1, 26 (4) 714, 733 (1938)] утверждает,-что отделение цинка от никеля и кобальта осаждением его сероводородом лучше всего проводить: от никеля при pH = 2,4, от кобальта — при pH = 2,3. К раствору надо прибавить соответствующий буферный раствор монохлоруксусной кислоты и ее натриевой соли, обработать при комнатной температуре быстрым током сероводорода в чистой стеклянной посуде с гладкой внутренней поверхностью и дать постоять перед фильтрованием.
2 В. S. Evans, Analyst, 60, 464 (1935).
3 Например, G. Е. F. L u n d е 1 1 и Н. В. К nowles [J. Am. Chem. Soc., 45, 680 (1923)] проводили однократное и двукратное осаждение алюминия из смесей, содержавших 0,1 г А1 и 0,05 г Zn, и получали в осадках гидроокиси алюминия: в первом случае — 0,0216 г, во втором случае — 0,0108 г цинка. Если и аммиак и хлорид аммония прибавляли в избытке (вследствие чего осаждение алюминия становилось неполным), то захват цинка осадком гидроокиси алюминия после двукратного его осаждения уменьшался до 0,0005 г.
4 При определении цинка в рудах Е. G. R. А г d a g h и G. R. Bongard [Ind. Eng. Chem., 16, 297 (1924)] получали удовлетворительное отделение 0,2 г цинка от такого же количества железа или алюминия однократным осаждением последних следующим способом: солянокислый раствор хлоридов этих элементов выпаривают приблизительно до 2—3 мл, прибавляют 5 г хлорида аммония, перемешивают, приливают 10 мл концентрированного раствора аммиака и 25 мл воды, фильтруют и промывают осадок раствором, содержащим 5 г хлорида аммония и 5 мл аммиака в 100 мл.
Методы определения 481
или едкой щелочью из растворов, содержащих цинк и хром (III), выделяющийся осадок может захватить с собой цинк в виде нерастворимого хромита цинка. Хорошее отделение железа, титана, циркония, ванадия ' и олова от больших количеств цинка можно провести с помощью купферона (стр. 143). Следует упомянуть об отделении малых количеств цинка от больших количеств железа и подобных ему элементов, основанном на осаждении последних карбонатом бария (стр. 108), ацетатным (стр. 103) и сукцинатным (стр. 106) методами.
Отделения меди от цинка электролизом, олова — нагреванием с азотной кислотой, кадмия — осаждением сероводородом из относительно кислого раствора, никеля — диметилглиоксимом, таллия — осаждением его в виде иодида таллия и индия — осаждением карбонатом бария рассматриваются в главах, посвященных этим элементам.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наилучший метод точного определения цинка состоит в осаждении его в виде сульфида цинка из слабо сернокислых Или муравьинокислых растворов с последующим превращением сульфида цинка в его окись или сульфат. Осаждение цинка в виде фосфата с последующим прокаливанием и взвешиванием в виде пирофосфата также является хорошим методом, но этот метод не может так широко применяться, как первый х.
Объемные методы определения цинка менее точны, чем весовые, и могут применяться лишь для массовых определений цинка аналитиками, опытными в проведении именно этих определений. Иэ объемных методов следует упомянуть только два: гексацианоферратный, более удовлетворительный, и метод с сульфидом натрия, применяющийся главным образом в Европе 1 2. Электролитическое определение цинка не так точно, как предыдущие методы, и применяется главным образом для отделения цинка от некоторых других элементов. Этот метод нельзя, как правило, применять для количественного определения цинка без предварительных отделений. Из других методов следует отметить: 1) ртутно-родановый метод 3’4; 2) осаждение цинка карбонатом натрия с последующим прокаливанием до его окиси и 3) взвешивание цинка в виде его сульфида. Последний метод не заслуживает одобрения. Осаждение же карбонатом натрия пригодно только для грубой массовой работы.
Осаждение в виде сульфида цинка
Перед осаждением цинка в виде сульфида следует удалить все элементы сероводородной группы. Осаждение цинка можно проводить из слабо сернокислых 5 или муравьинокислых растворов. В обоих случаях желательно присутствие в растворе электролита, например сульфата
1 Фосфатный метод и метод осаждения цинка сероводородом из слабо сернокислого раствора приводят к неудовлетворительным результатам, если применяются непосредственно к фильтрату, полученному после отделения никеля диметилглиоксимом.
2 *В СССР этот метод не нашел применения. Прим, ред.*
8 G. Е. F. L u n d е 11, N. К. В е е, Trans Am. Inst. Metals, 8, 146 (1914); С. A., 8, 3404 (1914).
4 G. S. Jamieson, J. Am. Chem. Soc., 40, 1036 (1918).
s Осадки, получаемые из сернокислых растворов сульфата цинка, значительно компактнее и легче отделяются фильтрованием, чем осадки, полученные из солянокислых растворов.
31 Заказ 522.
482
Гл. XXIV. Цинк
аммония или роданида аммония, вследствие «высаливающего» действия таких солей. Наиболее подходящая концентрация водородных ионов для осаждения цинка лежит в интервале значений pH от 2 до 3. В более кислых растворах осаждение неполно, а в более щелочных растворах образуется слизистый осадок. Во всех случаях следует помнить о возможности присутствия в осадке сульфида цинка редких элементов — таллия, индия и галлия.
Осаждение из 0,01 и. сернокислого раствора. Осаждение сульфида цинка из 0,01 н. по содержанию свободной серной кислоты раствора никогда не бывает совершенно полным вследствие растворимости осадка. Кроме того, осадок будет загрязнен железом, никелем и кобальтом, если эти элементы присутствовали в растворе. Потеря цинка не превышает нескольких десятых миллиграмма и может быть почти полностью устранена регулированием кислотности в начале осаждения таким образом, чтобы при последующем выделении кислоты при образовании сульфида цинка кислотность не поднялась выше 0,01 н. Загрязнение железом и другими элементами не может быть полностью устранено регулированием кислотности, поэтому осадки необходимо переосаждать один или несколько раз, в зависимости от количества подлежащих отделению примесей.
Ход определения. Приготовляют прозрачный сернокислый раствор сульфата цинка, свободный от элементов сероводородной группы, сероводорода и элементов, образующих легко гидролизующиеся соли. Прибавляют несколько капель метилового оранжевого или метилового красного, затем раствор едкого натра или карбоната натрия до нейтральной реакции и разбавляют водой. Объем раствора после разбавления должен быть не менее 250 мл и раствор должен содержать не более 0,1 г цинка в 100 мл. Подкисляют раствор так, чтобы кислотность его стала по возможности близкой к 0,01 н. (приблизительно 0,25 мл серной кислоты на 1 л), охлаждают до комнатной температуры и пропускают быстрый ток сероводорода в течение 30—45 мин. Затем дают постоять около 30 мин, фильтруют и промывают осадок холодной водой. Осадок обрабатывают далее, как указано на стр. 483 и 484.
Осаждение из муравьинокислого раствора. Осаждение сульфида цинка из растворов, содержащих свободную муравьиную кислоту, цитрат аммония и формиат аммония в качестве буферов для поддержания требуемой концентрации ионов водорода во все время обработки сероводородом *, оказалось особенно пригодным для определения цинка в Присутствии больших количеств алюминия, например при анализе алюминиевых сплавов. Точность метода, по данным его авторов, колеблется в пределах от 0,5 до 1 части на 1000. Отделение цинка от марганца этим методом почти совершенное. Загрязнение сульфида цинка железом незначительное, но в тех случаях, когда содержание железа в растворе превышает одну десятую содержания цинка, следует прибавлять избыток в 20 мл 23,6 М муравьиной кислоты. Прибавление роданида аммония не улучшает отделения. Избыток муравьиной кислоты требуется также и в присутствии никеля и кобальта, но однократное осаждение, особенно в присутствии кобальта, не дает таких удовлетворительных результатов, как при отделении от железа.
‘ Н. A. Pales, G.M. Ware, J. Am. Chem. Soc., 41, 487 (1919).
Методы определения
483
Ход определения.. Кислый раствор, свободный от элементов сероводородной группы, выпаривают до объема 125 мл и прибавляют аммиак до появления неисчезающего при перемешивании осадка. Прибавляют 25 мл 20%-ного раствора лимонной кислоты и приливают раствор аммиака до нейтральной реакции по метиловому оранжевому. Затем добавляют 25 мл «муравьинокислой смеси» х, раствор переливают в коническую колбу и разбавляют до 200 мл. Если количество цинка в растворе превышает 0,2 г, объем должен быть пропорционально увеличен. Нагревают до 60° С и закрывают колбу пробкой с двумя отверстиями, в которые вставлены отводная трубка, оканчивающаяся у самой пробки, и вводная трубка, доходящая почти до уровня жидкости. Присоединяют источник сероводорода 1 2 и вытесняют воздух из колбы медленным током этого газа, нагревая одновременно колбу до 90—100° С. Когда сероводород начнет выходить из колбы, прекращают нагревание, закрывают выходное отверстие и дают раствору охладиться, насыщая его таким способом сероводородом под повышенным давлением. Колбу с раствором изредка взбалтывают и оставляют так на 20—40 мин. Затем раствор фильтруют и промывают осадок холодным 0,1 М раствором муравьиной кислоты (4 мл 23,6 М муравьиной кислоты разбавляют до 1 л), насыщенным сероводородом. Осадок обрабатывают, как указано ниже. Если некоторое количество осадка пристало, к .стенкам колбы, растворяют его в горячей разбавленной серной кислоте, нейтрализуют раствор аммиаком, слабо подкисляют муравьиной кислотой и насыщают сероводородом под давлением. Нагревают до кипения для коагуляции осадка и фильтруют через фильтр, содержащий промытый главный осадок. В дальнейшем промывании нет необходимости, и вся эта операция занимает не более 15 мин.
Осаждение в аммиачном растворе. Метод этот неудобен в тех случаях, когда определяются большие количества цинка, ввиду того что осадок получается слизистым. Он применяется главным образом для отделения малых количеств цинка от магния и щелочноземельных металлов при анализе пород и минералов. Ход определения описан на стр. 89.
Обработка осадка сульфида цинка. Чистый осадок сульфида цинка можно: 1) прокаливанием превратить в окись цинка и взвесить как таковую; 2) превратить в сульфат цинка и 3) прокалить и взвесить в виде сульфида. Последний способ наименее удобен, первые два одинаково хороши.
Превращение в окись цинка. В первой стадии прокаливания осадка сульфида цинка он почти полностью превращается в сульфат цинка, разложение которого начинается около 675° С и заканчивается приблизительно при 950° С. Окись цинка очень мало улетучивается при 1000° С, но ее не следует продолжительное время прокаливать при этой температуре и совсем нельзя прокаливать при более высоких температурах 3 4. Цинк
1 200 мл муравьиной кислоты (23,6 М~), 250 г сульфата аммония и 30 мл раствора аммиака (15 М) разбавляют до 1 л.
2 Можно рекомендовать установку, состоящую из прибора Киппа, соединенного последовательно с промывной склянкой и манометром, имеющим трубку такой длины, что когда уровень жидкости достигнет нижней части манометра, давление в колбе, где происходит осаждение, будет равно одной трети атмосферного.
3 Например, при прокаливании 0,3723 г окиси цинка в течение 2 ч при 1000° С в трубке, через которую пропускали медленный ток чистого воздуха, потеря была равна 0,2 мг. Когда же подняли температуру до 1100° С и прокаливали при этой температуре
4 ч, окись цинка потеряла в массе еще 1,3 мг.
31*
484
Гл. XXIV. Цинк
может также улетучиваться и теряться при более низкой температуре, если прокаливание усиливают, когда углерод еще не весь выгорел, или ведут прокаливание в восстановительной атмосфере. Прокаленная окись цинка гигроскопична, и поэтому ее следует охлаждать над сильно высушивающими веществами, например над серной кислотой или фосфорным ангидридом. Взвешивать надо в плотно закрытом тигле. При определении фильтр с осадком высушивают при низкой температуре в открытом фарфоровом или платиновом тигле, затем обугливают фильтр и дают частицам угля выгореть при возможно более низкой температуре, после чего постепенно повышают температуру приблизительно до 900° С. Покрывают тигель крышкой, прокаливают при 1000° С в течение 5—10 мин, приподнимают крышку для удаления окислов серы, снова прокаливают несколько минут, охлаждают над сильно высушивающим веществом и взвешивают в виде ZnO. Если применяется тигель Гуча, то он должен быть предварительно прокален при 1000° С и взвешен; тигель с сульфидом цинка следует прокаливать в муфельной печи или в радиаторе.
Превращение в сульфат цинка. Многие аналитики предпочитают превращать сульфид цинка в сульфат. Эта операция не более точна, чем операция, описанная выше (см. «Превращение в окись цинка»), и требует больше времени и работы, если соблюдать все необходимые меры предосторожности. Обычно влажный сульфид цинка помещают в стакан, покрытый часовым стеклом, растворяют в разбавленной соляной кислоте, раствор затем кипятят до удаления сероводорода, переносят во взвешенный платиновый тигель и выпаривают с серной кислотой, прибавленной в небольшом избытке х. Если сульфид цинка был превращен сперва в окись цинка, то обработку надо проводить разбавленной серной кислотой, причем очень осторожно, во избежание механических потерь. Некоторые авторы 2 предпочитают сначала прокаливать осадок при низкой температуре до образования сульфата, как описано выше (см. «Превращение в окись цинка»), затем прибавляют малыми порциями серную кислоту и нагревают до удаления ее избытка. Сульфат цинка, полученный при выпаривании с серной кислотой, подобно образующимся таким же образом сульфатам марганца и кадмия, упорно удерживает при прокаливании небольшое количество сёрной кислоты 3. Поэтому, во избежание получения повышенных результатов, прокаленный остаток сульфата цинка следует охладить, смочить несколькими каплями воды, выпарить и снова прокалить при температуре около 500° С. Для проверки можно после взвешивания прокалить сульфат цинка до окиси цинка. Ход превращения сульфата цинка в окись цинка и окончание этого процесса можно наблюдать по изменению окраски. Сульфат цинка имеет белый цвет, окись же цинка в горячем состоянии желтая. Изменение окраски начинается с краев и заканчивается в центре.
Взвешивание в виде сульфида'цинка. Цинк редко взвешивают в виде его сульфида, потому что этот метод хлопотлив и не более точен, чем опи-
~________I—
1 Е. С. Sullivan, W. С. Taylor, J. Ind. Eng. Chem., 1, 476 (1909).
2 H. A. F a 1 e s, G. M. Ware, J. Am. Chem. Soc., 41, 487 (1919).
3 A. Gu tbier, K. S t a i b, Z. anal.'Chem., 61, 100 (1922). Например, Knowles растворял чистый цинк в количествах, отвечающих 0,2509 и 0,2521 г сульфата цинка, и получил 0,2515 и 0,2538 г ZnSO4 — после выпаривания с серной кислотой и прокаливания в радиаторе, и 0,2508 и 0,2525 г — после смачивания прокаленного остатка водой и вторичного прокаливания. '
Методы определения
485
санные выше. Ход определения следующий. Осадок высушивают и переносят, насколько возможно полно, во взвешенный тигель Розе. Фильтр сжигают отдельно, прибавляют золу к осадку в тигле, всыпают в тигель чистую серу в количестве, примерно равном одной трети осадка, и хорошо перемешивают. Сверху покрывают еще слоем чистой серы, закрывают тигель крышкой и пускают ток водорода. По удалению воздуха медленно нагревают до улетучивания серы и слабого покраснения тигля. Охлаждают, усиливая ток водорода, переносят тигель в эксикатор и взвешивают остаток в виде ZnS.
Осаждение в виде фосфата цинка
Определение цинка взвешиванием его в виде пирофосфата является превосходным методом, но требует большого внимания при подготовке раствора к осаждению и при промывании осадка. Раствор не должен содержать значительных количеств солей щелочных металлов, если не имеется в виду двукратное осаждение. Перед осаждением раствор надо очень точно сделать слабокислым, так как фосфат цинка растворим и в щелочах, и в сильных кислотах. Для осаждения не нужно применять фосфат натрия или фосфат калия, так как эти реактивы дают осадки, загрязненные натрием или калием *. Следует применять 10%-ный раствор двузамещенного фосфата аммония, но его нельзя хранить продолжительное время и перед применением его надо привести к надлежащему составу, растворяя соль в воде, прибавляя фенолфталеин и затем раствор аммиака до появления розовой окраски. Для промывания осадка лучше всего Применять горячий 1 %-ный раствор двузамещенного фосфата аммония, приготовленный из того же тщательно нейтрализованного реактива. Потеря цинка в фильтрате и в промывных водах не должна превышать 0,1 или 0,2 мг.
Ход определения. К холодному кислому раствору соли цинка, свободному от элементов, образующих нерастворимые фосфаты, например от кадмия или висмута, и от элементов, соли которых в условиях определения легко гидролизуются, прибавляют несколько капель метилового оранжевого, 5 мл соляной кислоты (если в растворе не присутствуют уже в достаточном количестве аммонийные соли), затем аммиак в небольшом избытке и, наконец, разбавленную соляную кислоту до слабокислой реакции. Разбавляют до 150—200 мл водой, нагревают почти до кипения и медленно прибавляют из пипетки 10—15-кратное количество (по сравнению с теоретически требуемым) свежеприготовленного раствора (NH4)2HPO4. Раствор нагревают на водяной или паровой бане и перемешивают, пока осадок не станет кристаллическим и не будет быстро оседать на дно, после чего нагревание прекращают, дают постоять 2—4 ч и фильтруют через тигель Гуча или Мунро 1 2 (стр. 127). Осадок промывают горячим 1 %-ным раствором двузамещенного фосфата аммония (приготовленным разбавлением нейтрализованного раствора реактива) до удаления
1 Н. D. Dakin. Z. anal. Chem., 39, 273 (1900),
2 Dakin (цит. выше) утверждает, что некоторые сорта асбеста несколько растворимы в растворах (NH4)2HPO4. Для проверки, не произошло ли такое растворение, при проведении точных анализов надо взвешенный осадок растворить в разбавленной азотной кислоте, промыть асбест водой, прокалить тигель, как указано в ходе определения, и снова его взвесить.
486
Гл. XXIV. Цинк
из осадка хлоридов и затем несколько раз 50%-ным спиртом для удаления фосфатного раствора. После этого тигель с осадком осторожно высушивают в радиаторе, очень медленно нагревают до удаления воды и аммиака и затем прокаливают приблизительно при 900° С до постоянной массы. Взвешивают1 в виде Zn2P2O7. Осадок можно также высушить при 100—105° С и взвесить в виде ZnNH4PO4, но этот метод, хуже 2.
Осаждение в виде роданомеркуриата цинка
Осаждение цинка в виде белой комплексной соли Zn[Hg(CNS)4] успешно применяется для определения от 1 до 100 мг цинка в некоторых материалах 3.
Осаждение можно проводить из нейтральных или слабокислых рас-, творов, содержащих в 100 мл не более 1 мл соляной или азотной кислоты (последнюю следует предпочесть) и 0,5 мл серной кислоты. Осадок роданомеркуриата цинка слегка растворим в воде, спирте и эфире. Растворимость его в промывном растворе, содержащем очень малое количество осаждающего реактива, ничтожна, и то количество этого реактива, которое присоединяется к осадку с последней порцией промывного раствора, соответствует не более чем 0,02 мг цинка.
В присутствии некоторых элементов цвет выпадающего осадка сильно изменяется. Малые количества меди придают ему шоколадно-коричневый цвет (*от еще меньших количеств меди он окрашивается в темнофиолетовый цвет*), а большие — темнозеленую окраску; в присутствии кобальта осадок принимает синий цвет, тем более интенсивный, чем больше кобальта. Изменяется цвет осадка и в присутствии никеля, железа (III) й железа (II).
Осадок можно взвешивать непосредственно или растворять его в кислоте и определять содержание цинка косвенно, титруя в полученном растворе роданид-ионы йодатом калия или определяя их методом Фольгарда; прибавляя нитрат серебра в избытке и оттитровывая этот избыток раствором роданида.
Применению этого метода мешает сравнительно небольшое число веществ. Растворимые хлориды, нитраты и сульфаты натрия, калия, аммо-ния, магния, кальция, стронция, бария, алюминия, железа (III) (в умеренных количествах), свинца, мышьяка (III), мышьяка (V), сурьмы (III), сурьмы (V) и олова (IV) не оказывают заметного влияния. Соли железа (III) изменяют цвет осадка, но их присутствие в небольшом количестве мало отражается на получаемых результатах.
Мешают определению медь, которая полностью осаждается, железо (II), кобальт, никель, марганец, висмут и кадмий, осаждающиеся частично.
1 Точность метода иллюстрируется следующими результатами: при анализе двух навесок по 0,1001 г 99,997%-ного цинка Knowles получил 0,1004 и 0,1003 г цинка.
2 Z. Н. Pan и С. Н. С h i a n g [J. Chinese Chem. Soc., 3, 118 (1935)] утверждают, что разложение ZnNH4PO4 начинается при 350° С и полностью заканчивается при 520° С.
8 G. Е. F. L u n d е 11, N. К. Bee, Trans. Am. Inst. Metals, 8, 146 (1914); С. A., 8, 3404 (1914); * Ю. Ю. Лурье, Минеральное сырье, № 7 (1931); Зав. лаб. 3, 222 (1934); Ю. Ю. Лурье, В. Ф. Неклютина, там же, 4, 1174 (1935); 5, 587 (1936); Ю. К). Лурье, Л. Б. Гинзбург, там же, 7, 1345 (1938); Ю. Ю. Лурье, Н. А. Филиппова, там же, 8, 1047 (1939).*
Методы определения
487
Серебро и ртуть (II) осаждаются в виде роданидов и мешают весовому, но не объемному окончанию определения. Мешающего влияния ртути (II) при весовом окончании можно избежать, прибавляя в избытке роданид-ионы. Таким же способом можно уменьшить и мешающее влияние марганца, но не устранить его полностью.
Ход определения. Приготовляют разбавленный азотнокислый раствор, свободный от перечисленных выше мешающих веществ и содержащий не более 1 мл свободной азотной кислоты и не более 0,05 г цинка в виде его нитрата в 100 мл. При сильном перемешивании прибавляют по каплям раствор роданомеркуриата калия 1 до начала образования осадка и, не прекращая перемешивания, медленно вливают по 20 мл осаждающего реактива на каждые 100 мг цинка, присутствующего в растворе. Дают постоять, изредка перемешивая, в течение 30 мин. При желании, можно оставить раствор и на ночь.
Весовое окончание. Через предварительно взвешенный фильтрующий тигель сначала декантируют прозрачную жидкость и промывают осадок два раза декантацией промывным раствором 2, после этого его переносят в тигель, где умеренно промывают, и дают промывному раствору возможно полнее стечь. Затем высушивают 30 мин при 105—110° С, охлаждают и взвешивают. Фактор пересчета осадка на цинк равен 0,1312.
Объемное окончание. 1. Метод Фольгарда. Переносят осадок на бумажный фильтр или в фильтрующий тигель, промывают его и дают стечь промывному раствору. Затем фильтр с осадком переносят в колбу, обрабатывают 50 мл воды и ,10 мл азотной кислоты, сильно перемешивают и медленно прибавляют из бюретки титрованный раствор нитрата серебра, пока он не будет прибавлен в избытке. Тогда добавляют раствор железных квасцов и титруют, как указано при описании метода Фольгарда (стр. 238). При достижении точки эквивалентности получаемая окраска при перемешивании раствора должна сохраняться 30 сек 3.
Происходящие реакции могут быть выражены следующими уравнениями:
Zn[ Hg( CN S )4 ] + 2 Ag+ = Zn2+ -J- Hg( CN S )3 + 2 AgC N S
Избыток серебра связывается роданидом:
Ag+ + CNS" = AgCNS
1 Для приготовления осаждающего реактива растворяют 39 г роданида калия или 33 г роданида натрия в 200 мл воды и прибавляют при сильном перемешивании 27 г измельченного в порошок хлорида ртути (II), после чего постепенно разбавляют водой до 1000 мл. Если не получился прозрачный раствор, фильтруют. Роданид калия или роданид натрия нельзя заменить роданидом аммония, потому что роданомеркуриат аммония не дает полного осаждения цинка.
2 Приготовляется прибавлением 20 мл осаждающего реактива к 1000 мл воды.
3 Можно также применять и следующий, более быстрый и во многих случаях достаточно точный метод. Осадок обрабатывают 50 мл кипящей воды, 10 мл азотной кислоты и 5 мл раствора железных квасцов, после чего медленно титруют раствором нитрата серебра до тех пор, пока после продолжительного и сильного перемешивания ни осадок, ни раствор не будут иметь розового оттенка.
Происходящие при этом титровании реакции могут быть выражены следующими уравнениями:
3Zn[Hg(CNS)4] + 2Fe3+ = 2Fe(CNS)3 + 3Zn2++3Hg(CNS)2
Fe(CNS)3+ 3Ag+ = Fe3+ + 3AgCNS
488
Гл. XXIV. Цинк
Титрование йодатом калия х. Промытый осадок растворяют в разбавленной соляной кислоте и титруют титрованным раствором йодата калия.
*В первой части титрования идет реакция с выделением иода 5ZnHg(CNS)4+24IO~ + 4H++8H2O =
= 5Zn2+ + SHg1 2 + + 20SO2- +12 Ia + 20HCN
который окрашивает хлороформ, применяющийся в качестве индикатора. При дальнейшем добавлении йодата окрашивание исчезает, так как иод переходит в нерастворимый в хлороформе хлорид иода:
1212 + 6Ю;+36Н+4-30СГ = 301С1 + 18Н2О
Всего на 1 атом цинка расходуется 6 молекул йодата калия. Максимальная окраска хлороформа достигается тогда, когда прибавляют 4/5 всего требуемого количества йодата. Уловив начало ослабления окраски хлороформа, можно приблизительно определить конечную точку титрования и подходить к ней с требуемой осторожностью. Концентрация соляной кислоты в растворе должна быть не ниже 12 %-ной, иначе IC1 будет разлагаться с выделением иода. Доп. ред*
При определении фильтр, содержащий осадок роданомеркуриата цинка, сворачивают и переносят в склянку емкостью 250—300 мл, снабженную притертой пробкой. Затем осторожно приливают охлажденную смесь 35 мл соляной кислоты, 10 мл воды и 7 мл хлороформа. Сейчас же начинают титровать раствором йодата калия (1 мл которого соответствует 0,002 г цинка), прибавляя раствор вначале быстро и вращая склянку, чтобы её содержимое хорошо перемешивалось. Когда выделившийся вначале иод исчезнет из водного раствора, склянку закрывают пробкой и взбалтывают 30 сек. Затем титрование продолжают медленно, сильно взбалтывая закрытую пробкой склянку после каждого добавления раствора йодата, пока окраска иода не исчезнет из слоя хлороформа.
Если конец титрования не будет достигнут после прибавления 50 мл раствора йодата, то для поддержания требуемой кислотности следует добавить еще 10—15 мл соляной кислоты и продолжать титрование.
Титр раствора йодата калия лучше всего устанавливать, проводя раствор, содержащий известное количество цинка, через все стадии анализа 2.
*0писаны 3 методы определения цинка в виде роданомеркуриата цинка в присутствии мешающихся веществ и в различных объектах: рудах, продуктах флотации, алюминиевых сплавах и т. п. Железо в этих методах связывается лимонной или винной кислотой, фторидами, фосфорной кислотой и др. Медь или определяют отдельно и содержание ее вычитают из результата анализа, или отделяют в виде роданида.
1 G. S. Jamieson, J. Am. Chem. Soc., 40, 1036 (1918); G. S. Jamieson, Volumetric Iodate Methods, New. York, 1926.
2 В статье A. C. Titus и J. S. Olsen [Ind. Eng. Chem., Anal. Ed., 12, 133 (1940)] описан эмпирический метод массового определения цинка, в котором рода-ноМеркуриат цинка растворяют в определенном количестве титрованного раствора иодида калия, избыток которого затем оттитровывают раствором нитрата ртути (II).
3 Ю. Ю. Л у р ь е и сотрудники, см. сноску 3 на стр. 486.
Методы определения
489
В методе, предложенном для анализа цинковых руд и продуктов их флотации * 1, анализируемый раствор нейтрализуют щелочью до слабокислой реакции, нагревают до 70—80° С, приливают 6 мл насыщенного раствора сернистого ангидрида, нагревают до восстановления железа и осаждают медь 2%-ным раствором роданида калия. Осадок роданида меди (I) отфильтровывают и промывают 1 %-ным раствором сульфата калия. Фильтрат нагревают до удаления сернистой кислоты, окисляют двухвалентное железо, добавляя 5—10 капель пергидроля, кипятят несколько минут, выпаривают или разбавляют до объема 50 мл, прибавляют 5 мл фосфорной кислоты (пл. 1,69г/сл43)и 2 мл той же кислоты, предварительно нейтрализованной едким натром по фенолфталеину, и осаждают цинк указанным выше раствором роданомеркуриата калия. Дают постоять 1—1,5 ч, время от времени перемешивая раствор, и фильтруют.
Заканчивают определение весовым или объемным способами, как описано выше. Доп. ред*
Гексацианоферратныи метод
Гексацианоферратный метод заключается в образовании нерастворимого гексацианоферрата (II) калия и цинка K2Zn3[Fe(CN)6]2 при титровании солянокислого анализируемого раствора, содержащего цинк и хлорид аммония, титрованным раствором гексацианоферрата (II) калия. Конечную точку титрования можно определять потенциометрически или при помощи внутреннего или внешнего индикатора. Так как метод этот дает точные результаты только в опытных руках, то его нельзя рекомендовать ни в качестве арбитражного метода, ни даже для обычных анализов, если аналитик не освоился хорошо со всеми предосторожностями, которые следует соблюдать.
Титрования этим методом должны проводиться при одинаковых условиях: температуры (предпочтительно 70° С), кислотности (3 мл свободной соляной кислоты в общем объеме 200 мл), объема раствора (200 мл), количества хлорида аммония (полученного нейтрализацией 13 мл концентрированного раствора аммиака соляной кислотой) и скорости титрования (медленно, при постоянном перемешивании). Обычно применяющиеся индикаторы: 10%-ный раствор нитрата уранила в качестве внешнего индикатора и гексацианоферрат (III) калия с сульфатом железа (II) в качестве внутреннего индикатора 2. В последнем случае окраска раствора
х. Ю. Ю. Лурье, Н. А. Ф и л и п п о в а, Зав. лаб., 8, 1047 (1939).
2 F. С. Breyer [Intern. Congr. Appl. Chem., 25, 13 (1912)] дает следующие указания для приготовления этого раствора. Растворяют 44 г гексацианоферрата (II) калия в 1 л воды и дают постоять в темноте в закрытом сосуде 1 месяц или более. 1 мл этого раствора отвечает приблизительно 0,01 г цинка. Непосредственно перед применением этого раствора надо прибавить 0,3 г гексацианоферрата (III) калия на
1 л. Раствор постепенно изменяет свой титр вследствие окисления гексацианоферрата (II) растворенным кислородом воздуха. Согласно I. М. Kolthoff и Е. A. Pearson [Ind. Eng. Chem., Anal. Ed., 3, 381 (1931)], растворы гексацианоферрата (II) калия должны содержать 0,2% карбоната натрия и хранить их надо в темных склянках. В этих условиях растворы устойчивы в течение по крайней мере 1 мес. Изменение Температуры (вплоть до 50° С) и присутствие малых количеств (0,2%) гексацианоферрата (III) не оказывают влияния на устойчивость раствора, если он имеет щелочную
реакцию. Устойчивость раствора падает с увеличением кислотности, усилением дей-
ствия света и в присутствии гексацианоферрата (III) (в кислом или нейтральном растворе). 1
490
Гл. XXIV. Цинк
изменяется от бирюзово-синей до горохово-зеленой. Гексацианоферрат (III) калия реагирует сперва с сульфатом железа (II), образуя синий гексацианоферрат (III) железа (II), а затем избыток гексацианоферрата (II), в конце титрования реагирует с окрашенным в синий цвет соединением, образуя гексацианоферрат (II) железа (II), вследствие чего цвет изменяется сначала до зеленого, потом до желто-зеленого *.
Нитраты, окислители, большинство тяжелых металлов, кадмий, марганец и избыточные количества железа должны отсутствовать 1 2. Тяжелые металлы могут быть удалены кипячением с тиосульфатом натрия в разбавленном сернокислом растдоре или добавлением к 0,5 — 1 %-ному по содержанию свободной серной кислоты анализируемому раствору нескольких кусочков алюминия, кипячением в течение получаса, фильтрованием через фильтр, на дно которого положено несколько кусочков алюминия, и промыванием холодной водой. Ни одним из этих методов полное отделение кадмия не достигается, и его надо удалять либо электролизом из раствора, содержащего 5% серной кислоты по объему, при силе тока 1 а, на катоде, предварительно покрытом кадмием, либо осаждением сероводородом. Последнее производится насыщением сероводородом раствора, содержащего 10—12% по объему серной кислоты. Если осадок не появляется, прибавляют по каплям аммиак до образования осадка и затем снова пропускают сероводород в течение нескольких минут. Раствор затем нагревают до 70—90° С, продолжая пропускать сероводород, фильтруют, промывают осадок холодным 8—10%-ным по объему раствором серной кислоты и, наконец, водой.
Малое количество кадмия, остающееся после этого в растворе, компенсируется цинком, увлеченным осадком. Если осадок велик, его надо растворить, снова осадить и оба фильтрата объединить. Железо и марганец отделяют осаждением аммиаком с бромом, перекисью водорода или персульфатом или осаждают цинк в виде сульфида. Последний способ рассмотрен в разделе «Осаждение в виде сульфида» (стр. 481), а первый описан в гл. «Марганец» (стр. 496), и его можно применять только тогда, когда осадок мал, так как имеется большая опасность потерять цинк в осадке марганца, если не применять специальные методы обработки 3.
Приводим ход установки титра раствора гексацианоферрата (II) калия. (В тех же условиях проводится и титрование анализируемого раствора после удаления мешающих элементов.) Растворяют 0,25 г чистого
1 W. Н. Cone и L. С. С a d у [J. Am. Chem. Soc., 49, 356 (1927)] рекомендуют применение в качестве индикатора нескольких капель 1-%ного раствора дифенилбен-зидина. В начале титрования окраска синяя, затем она переходит в пурпурную и дальше — в зеленовато-желтую. Согласно I. М. Kolthoff [Chem. Weekblad, 24, 203 (1927)], I. М. Kolthoff и E. A. Pearson [Ind. Eng. Chem., Anal. Ed., 4, 147 (1931)], лучшие результаты получаются в сернокислых растворах, содержащих сульфат аммония и нагретых до 60° С. Конечную точку титрования можно также определять потенциометрически, как рекомендуют F. R. Bichowsky [Ind. Eng. Chem., 9f 668 (1917)] и G. G. ReissansfZ. anal. Chem., 69, 450 (1926)]. ^Лучшие результаты получаются при обратном титровании прибавленного в избытке гексацианоферрата (II) калия титрованным раствором сульфата цинка [см. И. М. Кольт-гоф и В. А. Стенгер, Объемный анализ, т. 2, Госхимизат, 1952, стр. 373].*
2 Согласно А. С. Аруиной [Зав. лаб., 8, 565 (1939)], мешающего влияния железа можно избежать, прибавляя пирофосфат натрия Na4P2O„ образующий с железом устойчивое комплексное соединение.
3 Е. G. R. А г d a g h, G. R. В о n g а г d, Ind. Eng. Chem., 16, 297 (1924).
Методы определения
491
цинка в 10 мл соляной кислоты, прибавляют 13 мл аммиака, затем соляную кислоту до нейтрализации и сверх того ее избыток в 3 мл, после чего разбавляют до 200 мл.
Если в качестве внешнего индикатора применяют нитрат уранила, то больше ничего прибавлять не требуется; если же предполагают применять указанный выше внутренний индикатор, то прибавляют 0,3 мг железа [0,3—0,4 мл раствора сульфата железа (II), содержащего 1 г железа в 1л]. Нагревают до начала кипения, отливают третью часть раствора в маленький стакан, титруют оставшийся раствор гексацианоферратом (II) калия до конечной точки и прибавляют избыток титрованного раствора в 20%. Перемешивают несколько минут и приливают оставшуюся часть анализируемого раствора, оставляя в резерве еще несколько миллилитров его. Продолжают титрование, снова переходя за конечную точку титрования, но не больше, чем на несколько десятых долей миллилитра. Сновав перемешивают, прибавляют последнюю порцию анализируемого раствора, ополаскивают стакан и продолжают титрование до конечной точки.
Другие методы
Метод титрования цинка раствором сульфида натрия менее удовлетворителен, чем предыдущий. Титр раствора сульфида натрия не постоянен, и его надо часто проверять х.
Электролитические методы определения цинка можно разделить на две группы: 1) методы, в которых применяются щелочные электролиты; 2) методы, требующие кислых электролитов. В первых методах осадки цинка получаются значительно легче, но результаты определения обычно повышены и менее точны, чем при осаждении цинка из кислых электролитов. В обоих случаях необходима специальная аппаратура и надо предварительно удалить большое число мешающих элементов. Эти методы мало применимы в практике анализа. Если же особые условия создают возможность их применения, читателю следует обратиться к специальной литературе 1 2.
1 Описание этого метода можно найти в книгах: F. Sutton, Volumetric Analysis, 1924 и Верль — Лунге, Химико-технические методы исследования, ОНТИ, Химтеорет, 1936—1940.:
2 F. S р i t z е г, Z. Elektrochem., 11; 391 (1905); Е. В. Spear, Е.Е. Wells, В. D у е г, J. Am. Chem. Soc., 32, 530 (1910); S т i t h, Electro Analysis, 6th ed., 1918; А. Классен, Электроанализ, Госхимиздат, 1934. Для осаждения из кислых цитратных растворов R. Winchester и L. F. Yntema [Ind. Eng. Chem., Anal. Ed., 9, 254 (1937)] получают следующим образом. Приготовляют раствор сульфата цинка, не содержащий других металлов, кроме щелочных, алюминия, хрома, магния и олова. Разбавляют до 175 мл, прибавляют 1,5 г лимонной кислоты и нейтрализуют 40 %-ным раствором едкого натра до нейтральной или очень слабокислой реакции, пользуясь индикатором метиловый красный -J- метиленовая синяя (pH = 4—5). Разбавляют до 200 мл и ведут элёктролиз при плотности тока 1 а!дм? с медным или покрытым медью катодом и вращающимся платиновым анодом. Электролиз продолжают 1,5—2 ч или пока при добавлении к 1 мл электролита 0,5 мл насыщенной сероводородом воды не получится только слабая опалесценция. Опуская вниз стакан с электролитом, быстро промывают катод, затем вынимают его из гнезда, погружают в ацетон, высушивают при 85° С и взвешивают. Определению мешают: нитрат-иовы, ди-метилглиоксим, мочевина, сурьма, мышьяк, висмут, кадмий, медь, свинец, ртуть, серебро, железо, кобальт, никель и марганец.
492
Гл. XXIV. Цинк
Определение малых количеств цинка
При определении малых количеств цинка особо важно, чтобы анализ проводился в посуде, изготовленной из стекла, не содержащего в своем составе этого элемента. Очень малые количества цинка лучше всего определять осаждением в виде сульфида цинка методом, описанным в разделе «Осаждение в виде сульфида» (стр. 481), после прибавления небольшого количества хлорида ртути (II), который, образуя осадок сульфида ртути, служит коллектором, захватывающим с собой сульфид цинка. Осадок сульфидов затем осторожно прокаливают — сульфид ртути при этом улетучивается, а сульфид цинка превращается в окись цинка. Последнюю можно взвесить непосредственно или растворить в соляной кислоте, обработать гексацианоферратом (II), как указано в разделе «Гексацианоферратный метод» (стр. 489), и полученную суспензию сравнить в нефелометре со стандартным раствором, содержащим известное количество цинка и обработанным таким же способом. Так же заканчивают определение 1 и после предварительного осаждения сульфида цинка из лимоннокислого раствора, содержащего нитрат кальция. В качестве в высшей степени специфичного флуоресцирующего качественного реактива на цинк, обнаруживающего 10 мкг цинка и более, был предложен 2 бензоин С6Н5—СНОН—СО—С5Нв. Флуоресценция появляется в щелочном растворе в присутствии гидроокиси магния и силикат-ионов. Единственными мешающими элементами являются бериллий, бор и сурьма. Окрашенные анионы должны отсутствовать, а также платиновые металлы, ртуть, серебро и золото, которые, если их не удалить, восстанавливаются до металлического состояния.
* Следует отметить также оксихинолиновый метод, в котором цинк осаждают оксихинолином из уксуснокислого раствора, содержащего ацетат натрия. Определение заканчивают или весовым способом 3, взвешивая осадок в виде Zn(C9HeON)2 после высушивания при 130—140° С, или объемным способом, добавляя в избытке бромат-бромидную смесь и определяя избыток йодометрическим методом, или, наконец, колориметрическим методом: с диазотированной сульфаниловой 4 или нафтионовой кислотой 5. Для определения очень малых количеств цинка наибольшее применение имеет дитизоновый метод 6. Тысячные и десятитысячные доли процента цинка в металлическом никеле высших марок рекомендуется7 сначала осаждать сульфатом акридина, образующим с цинком и роданид-ионами соединение Zn(CNS)2(C13H9N-HGNS)2, а затем заканчивать определение дитизоновым методом. Доп. ред. *
1 М. В odansky, J. Ind. Eng Che$n., 13, 696 (1921).
2 С. E. White, M. H. Neustadt, Ind. Eng. Chem., Anal. Ed., 15, 599 (1943). О применении дитизона см. Ф. Ф а й г л ь, Капельный анализ, ОНТИ, 1937; F. F е i g 1, Specific Selective Sensitive Reaction, New York, 1949.* E. Б. С e н д э л, Колориметрическое определение следов металлов, Госхимиздат, 1949.*
3 Р. Берг, Применениео-оксихинолина в аналитической химии, ОНТИ, 1937.
4 Л. М. К у л ь б е р г, Вопросы питания, 8, 75 (1935); Л. М. К у л ь б е р г, Ф. Юровская, Зав. лаб., 9, 295 (1940); С. К). Ф а й н б е р г, Т. В. 3 а г л о-д и н а, там же,- И, 1109 (1945); Е-. И. Фогельсон, Н. В. Калмыкова, там же, 13, 114 (1947); Ю. Ю. Л у р ь е, А. И. Рыбникова, Методы химического анализа сточных вод, Госстройиздат, 1953, стр. 117.
8 А. С. А р у и н а, Ю. А. Чернихов, Зав. лаб., 13, 33 (1947).
8 См. Н. J. W i с h m а п п, Ind. Eng. Chem., Anal. Ed., 11, 66 (1939); П. A. К o-л о д у б, Зав. лаб., 9, 514 (1940); J. C h о 1 a k, D. M. Hubhar d, R. E. Burkey, Ind. Eng. Chem., Anal. Ed., 15, 754 (1943); L. G. Bricker, S. Weinberg, K. L. Proctor, там же, 17, 661 (1945).
7 H. А. Ф и л л и п о в а, Ю. Ю. Лурье, Зав. лаб., 16, 912 (1950).
Глава XXV
МАРГАНЕЦ
Марганец находится в железно-магнезиальных минералах почти во всех горных породах, хотя в результате изменения этих пород он бывает иногда в более или менее окисленном состоянии, особенно на поверхности известняков и песчаников. Марганец чаще встречается в породах, богатых железом, чем в породах с высоким содержанием магния', и редко присутствует в количествах, превышающих 0,5%. Чаще всего он встречается в силикатах, окисях и карбонатах и реже — в сульфидах, фосфатах, вольфраматах и ниобатах. Наиболее распространенными марганцевыми минералами являются двуокись марганца — пиролюзит МпОг и п с и-л о м е л а н Мп2МпО4 • пН2О. Марганец широко применяется в промышленности, и методы его определения имеют первостепенное значение.
ОБЩИЕ ЗАМЕЧАНИЯ
Если в обычном ходе анализа горных пород не учесть присутствия марганца, то он в большей своей части будет найден вместе с кальцием и магнием. В осадке от аммиака, если осаждение проводится двукратно и в условиях, требуемых для полного выделения алюминия (стр. 565), могут остаться лишь следы марганца или его вовсе не будет. Значительно большее количество выпадает вместе с магнием и взвешивается в виде пирофосфата марганца Мп2Р2О7. Метод выделения марганца вместе с другими элементами аммиаком с добавлением окислителей (брома или персульфата) нежелателен, если присутствует большое количество марганца и анализ заканчивается весовым способом, так как прокаленная окись марганца имеет неопределенный состав. Как общее правило, марганец лучше всего удалять непосредственно после осаждения железа и алюминия аммиаком. Для этой цели малые'количества марганца лучше всего осаждать, сульфидом аммония, а большие — хлоратно-азотнокислым методом.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ МАРГАНЕЦ
Разложение марганцевых минералов не вызывает затруднений и может быть проведено обработкой одной кислотой или смесью кислот, либо сплавлением с карбонатом натрия или пиросульфатом калия. При кислотной обработке следует добиваться полного разложения минерала. Для этой цели нерастворившийся остаток надо отделить, промыть, прокалить,
494
Гл. XXV. Марганец
сплавить, плав растворить и раствор присоединить к первоначальному раствору.
Окисленные руды лучше всего обрабатывать соляной кислотой в покрытом стеклом стакане или чашке до прекращения выделения хлора. Затем, если требуется определение кремнекислоты, стекло снимают и раствор выпаривают досуха. Если же кремнекислоту определять не нужно, раствор кипятят до удаления хлора, переносят в платиновую чашку и обрабатывают фтористоводородной и серной кислотами.
Если в окисленной руде надо определить только один марганец, то лучше всего разложить руду нагреванием с разбавленной азотной кислотой (пл. 1,135 г/см3) и перекисью водорода, раствор профильтровать, не-растворившийся остаток промыть, прокалить, сплавить с пиросульфатом, обработать плав разбавленной азотной кислотой и полученный раствор присоединить к основному раствору.
МЕТОДЫ ОТДЕЛЕНИЯ
Марганец, подобно хрому, обычно определяют из отдельной навески методами, которым мешают только очень немногие элементы. Поэтому отделение марганца от других элементов чаще применяют для удаления его, чем для выделения в чистом виде.
Лучший метод отделения марганца от других элементов состоит в осаждении его в виде двуокиси марганца кипячением раствора с азотной кислотой и хлоратом калия х. Осаждение это не вполне количественное, и малые количества марганца (1—2 мг) могут из раствора не выпасть. Однако при осаждении больших количеств марганца в растворе остается1 2 не более 0,1—0,3 мг.
Определение нельзя заканчивать взвешиванием двуокиси марганца, так как она загрязнена кремнием, вольфрамом, ниобием и танталом, частично выделяющимися во время выпаривания азотной кислоты, а также железом, кобальтом, сурьмой и ванадием, если значительные количества этих элементов находились в первоначальном растворе. При объемном методе определения марганца присутствие всех этих примесей не имеет такого большого значения, как при весовом. Ход отделения следующий.
Раствор (желательно не содержащий других кислот, кроме азотной) выпаривают до сиропообразного состояния и затем прибавляют 50 мл
1 I. В. Н annay, J. Chem. Soc., 33, 269 (1878); S. A. For d,' Trans. AIME, 9, 397 (1880—1881). L. H. James [Ind. Eng. Chem., Anal. Ed., 3, 31 (1931)] утверждает, что окисление марганца можно провести в присутствии хлорной кислоты следующим образом. К анализируемому раствору, свободному от органических веществ, прибавляют 20 мл 60%-ной хлорной кислоты и кипятят до появления ее паров и начала выделения солей. Затем охлаждают, растворяют выделившиеся соли добавлением 30 мл азотной кислоты и нагревают на горячей плитке до начала кипения. Тогда добавляют несколько кристаллов хлората натрия, чтобы, предупредить чрезмерное вспенивание, и осаждают марганец, прибавляя хлорат натрия четырьмя порциями по 2 г. Всыпать каждую новую порцию хлората натрия надо только после того, когда вспенивание от предыдущей порции полностью прекратилось. На все это требуется около 20 мин, в течение которых раствор должен не перегреваться и быть все время насыщенным свободной хлорноватой кислотой. Затем стакан с раствором снимают с горячей плитки, пока вспенивание от последней прибавленной порции хлората натрия еще не прекратилось, фильтруют и промывают осадок, как обычно.
2 Возможно, что осаждение будет значительно менее полным, если присутствуют значительные количества некоторых других элементов, например молибдена.
Методы отделения
495
азотной кислоты. Покрывают часовым стеклом, кипятят несколько минут для удаления окислов азота и затем, поддерживая температуру, близкую к кипению, вводят в течение 10—20 мин очень малыми порциями 5 г хлората калия, приоткрывая часовое стекло и быстро всыпая реактив при помощи стеклянной или фарфоровой ложечки. После прибавления всего хлората калия продолжают кипятить, пока вспенивание почти не прекратится, но некоторое количество хлорноватой кислоты будет все же оставаться в растворе. Обмывают часовое стекло и стенки стакана небольшим количеством воды, прибавляют 40 мл холодной воды и быстро охлаждают. Фильтруют через плотный фильтр или асбест и промывают стакан и осадок на фильтре малыми порциями холодной бесцветной азотной кислоты или ледяной водой. Фильтрат и промывные воды испытывают на присутствие в них марганца висмутатным методом. Выделенная двуокись марганца может быть оттитрована, как указано ниже (стр. 502), или же определена весовым методом после предварительной очистки, выполняемой следующим образом.
Фильтр с осадком двуокиси марганца переносят в стакан/в’котором проводилось осаждение, и прибавляют 10—40 мл концентрированного раствора сернистой кислоты. Затем фильтруют, собирая фильтрат в стакан емкостью 200 мл, промывают остаток горячей водой и прибавляют к фильтрату 2—3 мл соляной кислоты. Нагревают до удаления сернистого ангидрида, прибавляют бромную воду до сильного окрашивания раствора и кипятят до удаления всего свободного брома. Затем, прибавив разбавленный (1 : 1) раствор аммиака до щелочной реакции по метиловому красному, кипятят 2 мин и сейчас же фильтруют, собирая фильтрат в стакан емкостью 600 мл.
Осадок промывают 2—3 раза горячей водой и фильтрат вместе с промывными водами сохраняют. Затем растворяют осадок в горячей разбавленной (1 : 3) соляной кислоте, собирая раствор в стакан, в котором проводилось осаждение. Промывают фильтр горячей водой, нагревают раствор до кипения, вновь осаждают по-прежнему аммиаком, фильтруют, собирая фильтрат в стакан с сохранением первым фильтратом, и умеренно промывают осадок. Соединенные фильтраты подкисляют уксусной кислотой, нагревают до кипения и пропускают 10—15 мИн сероводород. Дают постоять 15 мин на краю паровой бани, фильтруют и промывают осадок сероводородной водой, слабо подкисленной уксусной кислотой и содержащей небольшое количество хлорида аммония. Фильтрат, в котором находится весь марганец, теперь готов для определения в нем марганца методом, описанным на стр. 502.
Нами был приготовлен ряд растворов, содержавших каждый 0,0113 г марганца и 5 лг одного из перечисленных ниже элементов. Затем марганец выделяли в виде двуокиси, осадок очищали описанным методом и заканчивали определение марганца, взвешивая его в виде фосфата. Были получены следующие результаты:
Прибавленный элемент Введено марганца г Получено марганца г Прибавленный элемент Введено марганца г Получено марганца г
V 0,0113 0,0112 Ti 0,0113 0,0111
Ni 0,0113 0,0112 Zr 0,0113 0,0110
Со 0,0113 0,0113 Mo 0,0113 0,0109
W 0,0113 0,0113 Sn 0,0113 0,0112
Ст 0.011& 0,0111 Sb 0,0113 0,0111
496
Гл. XXV. Марганец
В другой серии опытов был взят ряд растворов, содержавших каждый 0,0108 г марганца и 5 мг одного из перечисленных ниже элементов, затем марганец осаждали в виде двуокиси, осадок растворяли (без очистки его) в титрованном растворе сульфата железа (II) и избыток последнего оттитровывали перманганатом. Получены следующие результаты:
Прибавленный Введено Получено Прибавленный элемент Введено Получено
элемент марганца г марганца г марганца 8 марганца 8
V 0,0108 0,0108 Ti 0,0108 0,0110
Ni 0,0108 0,0107 Zr 0,0108 0,0108
Со 0,0108 0,0108 Mo 0,0108 0,0105
W 0,0108 ' 0,0103 Sn 0,0108 0,0108
Сг' 0,0108 0,0110 Sb 0,0108 0,0105
Железо, алюминий и другие элементы можно количественно отделить от марганца осаждением их аммиаком из кипящего раствора при условии, что раствор, после осторожного добавления аммиака по индикатору, кипятят не более 2 мин и затем сразу фильтруют х. Этим методом, совпадающим с тем, который рекомендуется для определения алюминия (стр. 569). можно отделить двукратным осаждением 0,2—0,3 г железа или алюминия от марганца, находящегося в растворе в количестве, превышающем 1 г, при условии, что фосфор и ванадий присутствуют в таких малых количествах, какие полностью захватываются осадком железа или алюминия.
Отделение ацетатным методом (стр. 103) более хлопотливо и нисколько не лучше, чем отделение аммиаком, за исключением тех случаев, когда осадки от аммиака настолько велики, что с ними трудно работать, или когда невозможно применить индикатор из-за окраски раствора.
Марганец от кальция и магния в общем ходе анализа горных пород и минералов лучше всего отделять сульфидом аммония, как описано на стр. 96. '
Марганец может быть осажден бромом из слабокислых (обычно уксуснокислых) растворов, содержащих ацетат натрия (фильтрат от осаждения ацетатным методом), или из аммиачных растворов, но этот метод в присутствии других металлов хуже сульфидного метода: трудно достигнуть полного осаждения марганца и, кроме того, если присутствуют цинк и щелочноземельные металлы, осадок может содержать манганиты этих металлов 1 2. Присутствие аммонийных солей при недостатке свободного аммиака вредно, поскольку аммонийные соли окисляются бромом и увеличивают этим содержание в растворе свободной кислоты. Поэтому, как правило, перед осаждением марганца аммонийные соли следует разрушать или удалять. Если количество их мало, осаждение марганца можно проводить в их присутствии, но на это потребуется больше брома и надо следить за тем, чтобы раствор все время был щелочным.
Марганец можно отделить от большого числа элементов осаждением сероводородом. Так, осаждение сероводородом в растворах, содержащих минеральные кислоты (стр. 83), служит для отделения элементов сероводородной группы от марганца. Осаждение в уксуснокислом или муравьинокислом растворах (стр. 85) служит для отделения цинка и, наконец, как
1 G. Е. F. L u n d е 11, Н. В. Knowles,.!. Am. Chem. Soc., 45, 676 (1923).
2 Если присутствуют малые количества марганца и не предполагается проводить осаждение его сульфидом аммония, то лучше всего включить марганец в осадок от аммиака, прибавляя немного бромной воды или персульфата, иначе, оставшись в растворе, марганец загрязнит осадки кальция и магния.
Методы определения
497
уже было указано, осаждение марганца сульфидом аммония очень хорошо отделяет его от щелочноземельных металлов.
Хорошим методом отделения марганца от молибдена, ванадия и подобных им элементов является осаждение его едким натром (стр. 109). Если, кроме едкого натра, применяют еще и перекись натрия или раствор перед осаждением обрабатывают персульфатом калия или персульфатом натрия, то этим методом можно отделить марганец и от хрома. Той же цели можно достигнуть сплавлением с перекисью натрия, выщелачиванием плава водой, кипячением с небольшой прибавкой перекиси натрия, если образуется перманганат, и фильтрованием.
Марганец можно также отделить от хрома отгонкой последнего в виде хлористого хромила из кипящего хлорнокислого раствора, к которому время от времени добавляют хлорид натрия или соляную кислоту (стр. 590) При этом некоторое количество марганца также улетучивается, но оно так незначительно, что им можно пренебречь, если не проводят исключительно точного анализа Ч
Из других методов отделения ряда элементов от марганца следует отметить: осаждение купфероном (стр. 143), в результате которого железо, титан,' цирконий и ванадий могут быть количественно отделены от марганца; электролиз с ртутным катодом в разбавленном сернокислом растворе (стр. 165), при котором осаждаются железо, хром, никель и молибден, а марганец остается в растворе; извлечение железа и молибдена из солянокислых растворов из хлоридов эфиром (стр. 161) и осаждение железа, алюминия и хрома карбонатом бария.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Большинство методов определения марганца является объемными методами, основанными на окислении его до марганцевой кислоты с последующим восстановлением ее титрованным раствором восстановителя. Исключения составляют: метод Фольгарда, в котором марганец (II) титруют раствором перманганата до четырехвалентного состояния, и методы, в которых марганец сперва выделяют в виде двуокиси марганца и затем количественно восстанавливают сульфатом железа (II), перекисью водорода, щавелевой кислотой и др.
Весовые методы определения марганца применяются редко. Обычно марганец взвешивают или в виде пирофосфата, или в виде сульфата марганца. Определение малых количеств марганца в породах весовыми методами почти всегда приводит к получению повышенных результатов. Колориметрические методы определения марганца могут применяться только для определения малых его количеств, такие обычно и встречаются в большинстве пород и минералов.
Руды, содержащие пиролюзит, следует высушивать при 120° С и анализировать всю высушенную пробу, так как во время открывания сосуда для взятия навески руда поглощает заметное количество влаги из воздуха.
Объемные методы
Висмутатный метод. Висмутатный метод можно применять во всех случаях, когда требуется определять марганец; он является самым точным методом определения этого элемента. Прежде полагали, что висмутатным
1 J. I. Н о f f m a ns G, Е. F. L u n d е 11, J. Research NBS, 22, 465 (1939).
32 Заказ 522.
498 1 Гл. XXV. Марганец
методом можно определять не больше 50 мг марганца, но было доказано1, что при соблюдении соответствующих мер предосторожности можно определять и десятикратное количество (0,5 г) марганца при сохранении той же высокой точности определения. Таким образом висмутатный метод может применяться при анализе материалов с высоким содержанием марганца, как пиролюзит или ферромарганец.
Висмутатный метод заключается в окислении марганца (II) до марганцевой кислоты висмутатом натрия в азотнокислом растворе, фильтровании через асбест, промывании не вступившего в реакцию висмутата натрия разбавленной азотной кислотой, прибавлении отмеренного количества сульфата железа (II) (в избытке) и титровании избытка последнега перманганатом. Происходящие реакции протекают быстро, и все определение может быть закончено в несколько минут.
Установлено 2, что висмутатный метод может применяться и при анализе растворов, содержащих серную кислоту вместо азотной. Мы этот вариант метода широко не применяли, но он удобен в тех случаях, когда хром отсутствует, содержание марганца не превышает 20 мг в 100 мл и нет возможности охладить раствор до 10—15° С, как это необходимо в присутствии азотной кислоты. Согласно Park, требуется соблюдение следующих условий: кислотность в пределах 2—4 н. (5,5—11 мл H2SO4 в 100 мл), концентрация марганца предпочтительно не выше 0,1 мг в 1 мл, температура 15— 50° С, взбалтывание с висмутатом, взятым в избытке, в течение 1 мин и промывание не вступившего в реакцию висмутата 3%-ной H2SO4.
. Марганец полностью не окисляемся висмутатом в присутствии одной хлорной кислоты, но окисление протекает нормально в растворе, содержащем 15 мл 60 %-ной хлорной кислоты и 12 мл азотной кислоты в объеме 50 мл.
Условия применения висмутатного метода3 состоят в следующем.
Концентрация азотной кислоты в растворе не должна быть менее 11% (пл. 1,062 г/см3) и желательно, чтобы она была близка к 22% (пл. 1,135 г/см3). Концейтрация марганца в растворе имеет большое значение, если должно быть окислено значительное его количество. Раствор, содержащий 0,05 г марганца в 100 мл, обладает наибольшей устойчивостью, но количество марганца в растворе может быть доведено до 0,1 г на 100 мл без опасности разложения образующейся марганцевой кислоты, если фильтрование будет проведено без замедления. Требуется приблизительно 26 г 80%-ного висмутата натрия на каждый грамм марганца, т. е. на 60% более теоретически рассчитанного количества. При отсутствии хрома окисление может быть проведено при любой температуре ниже 25° С, но лучше при более низких температурах; обычно ^его проводят при 10—20° С. Марганец количественно окисляется при взбалтывании раствора с прибавленным висмутатом натрия в течение 1 мин. Этот период может быть без вреда увеличен даже до 30 мин, но в этом нет необходимости. Фильтровать следует через асбест, стеклянные пористые пластинки и т. п.; остаток непрореагировавшего висмутата обычно промывают холодной разбавленной азотной кислотой.
При выполнении массовых определений небольших количеств марганца применяют 0,1 н. — 0,03 н. растворы сульфата железа (II) и перманганата калия, в зависимости от концентрации определяемого марганца. Если определение марганца проводят изредка или когда присутствует
1 Т. R. Ctinningham, R. W. Col t m an, Ind. Eng. Chem., 16, 58 (1924).
2 В. Park, Ind. Eng. Chem., 18, 597 (1926).
3 W. Blum, J. Am. Chem. Soc., 34, 1395 (1912); G. E. F. Lundell, там же, 45, 2600 (1923); T. К. Cunningham, R. W. С о 11 m a n, цит. выше.
Методы определения
499
более 0,05 г марганца, лучше прибавлять точно отвешенные порции сухой соли Мора, при условии, что кристаллы ее очень мелки и одинаковой чистоты. Во всех случаях количество прибавленного сульфата железа (П) должно быть рассчитано так, чтобы на обратное титрование было израсходовано не больше 10 мл раствора перманганата. Титр перманганата обычно устанавливают по оксалату натрия, как описано на стр. 214. Для установки титра можно применять также оксалат ма’рганца1 или безводный сульфат марганца 2, если в лаборатории имеются эти соли в чистом виде.
Висмутатный метод, как он описан выше, является методом исключительно точным и свободным от мешающего влияния других веществ. Исключение составляют лишь азотистая и фтористоводородная кислоты, хлорид-ионы, церий, кобальт и хром, мешающие определению.
Азотистую кислоту, фтористоводородную кислоту и хлорид-ионы нетрудно удалить 3. Редко присутствующий церий окисляется висмутатом до четырехвалентного, восстанавливается затем до трехналентного сульфатом железа (II) и не окисляется вновь перманганатом 4. Следовательно, в присутствии церия получаются повышенные результаты определения марганца. Церий надо удалять или при титровании заменять сульфат железа (II) титрованным раствором арсенита, не восстанавливающего церий (IV). . • •
Висмутатный метод неприменим в присутствии кобальта, потому что этот Элемент окисляется висмутатом до' соединения неопределенного состава, и это соединение реагирует не только с сульфатом железа (II), но и с марганцевой кислотой5, как только при титровании начнут в растворе получаться ионы марганца (II). Марганец можно отделить от кобальта кипячением с азотной кислотой и хлоратом калия (стр. 494) или вполне удовлетворительно определить его в присутствии кобальта персульфат-ным методом.
Хром мешает определенью, потому что он медленно окисляется марганцевой кислотой даже на холоду. Можно получить достаточно точные для обычных целей результаты6, если присутствует не более 2 мг хрома в 100 Л1Л раствора и реакции проводят при 10—20 °C. При выполнении особо точных анализов хром должен быть отделен, например, повторным осаждением марганца перекисью натрия. При проведении массовых анализов можно получить достаточно удовлетворительные результаты непосредственным титрованием образовавшейся марганцевой кислоты
1 R. W. С о 11 m a n, Ind. Eng. Chem., 16, 606 (1924).
2 W. Blum, цит. выше.
3 A. Isaaks указывает, однако, что примесь до 2,5 мг хлора в виде NaCl не оказывает влияния при колориметрическом определении марганца в соли Мора при условии, что колориметрическое сравнение проводится немедленно после отфильтровывания избытка висмутата натрия. Окраска ослабевает очень медленно.
4 F. J. М е t z g е г, J. Am. Chem. Soc., 31, 523 (1909).
8 G. Е. F. L u n d е 11, цит. выше; A. A. Blair, The Chemical Analysis of Iron, 8th ed., Philadelphia, 1918, p. 208.
8 Например, при анализе четырех навесок по 1 г стали, содержавшей 0,635% марганца и не содержавшей хрома, было получено 0,637; 0,648; 0,652 и 0,659% мар-l ганца после прибавления к анализируемому раствору соответственно 2,5; 5; 7,5 и 10 мг хрома и окисления марганца при 20° С. Когда температура раствора во время окисления ( ' была снижена до 10° С, то при прибавлений 10 мг хрома было получено 0,644% мар-г ганца.
32*
500
Гл. XXV. Марганец
раствором, не реагирующим с хроматом, например раствором арсенита натрия.
Присутствие ванадия приводит к получению повышенных результатов, если не принять специальных мер предосторожности при определении конца титрования, так как ванадий восстанавливается сульфатом железа (II) и окисляется обратно разбавленным раствором перманганата на холоду, только очень медленно.
В присутствии сурьмы получаются несколько пониженные результаты. Например, было получено 0,0109 г марганца вместо 0,0111 г в присутствии 0,05 г сурьмы.
Серебро вызывает образование окислов азота; в его присутствии получаются, чуть-чуть повышенные результаты, например вместо 0,0111 г марганца было получено 0,0112 г в присутствии 0,01 г серебра и 0,0113 г— в присутствии 0,1 г серебра.
Фосфорной кислоты следует избегать во время окисления марганца, хотя она не оказывает большого влияния, если присутствует в количестве, не превышающем 1 г РгОв на 100 мл раствора.
Умеренные количества сульфата аммония, например 10 г, или вольфрама до 0,25 г на 100 мл определению не мешают. Молибден, уран, титан, свинец и платина влияния не оказывают.
Ход определения. Приготовляют азотнокислый раствор, содержащий приблизительно 22% по массе азотной кислоты, не более 0,05 г марганца в 100 мл и не содержащий упомянутых выше мешающих определению веществ. Раствор охлаждают, прибавляют по 0,26 г висмутата натрия на каждую 0,01 г присутствующего в растворе марганца, перемешивают 1 мин, разбавляют равным объемом холодной воды и фильтруют через асбест, который должен быть предварительно подвергнут специальной обработке (см. стр. 126). Остаток не вступившего в реакцию висмутата промывают холодной 3%-ной азотной кислотой до удаления марганцевой кислоты1 и прибавляют к фильтрату сульфат железа (II) в виде точной навески сухой соли или в виде титрованного ее раствора. Соль железа (II) прибавляют в таком количестве, чтобы ее хватило на восстановление всей марганцевой кислоты и остался избыток, требующий примерно 10 мл раствора перманганата на обратное титрование. Основательно перемешивают до обесцвечивания марганцевой кислоты и растворения всего сульфата железа (II), если последний был прибавлен в твердом виде. Затем сразу же титруют 0,1 н. раствором перманганата до обычной конечной точки титрования. Одновременно проводят «холостой» опыт через все стадии анализа, прибавляя то же количество сульфата железа (II), какое было прибавлено в анализируемый раствор, и титруют перманганатом. Разность между числом миллилитров раствора КМпО4, израсходованного на оба титрования, показывает количество раствора КМпО4, эквивалентное марганцевой кислоте, образовавшейся при окислении марганца висмутатом. Содержание марганца вычисляют по титру
1 Когда фильтруют раствор, содержащий большое количество марганцевой кислоты, остаток висмутата натрия упорно удерживает последнюю при его промывании. Для извлечения этой марганцевой кислоты надо отсосать жидкость из остатка на асбестовом фильтре, дать постоять 20—30 мин и снова обработать его промывным раствором. При анализе нескольких навесок по 0,3 г марганцевой руды, содержавшей 60% марганца, когда не проводилась указанная выше дополнительная обработка, остаток висмутата удерживал 0,03% марганца.
Методы, определения
501
раствора перманганата, выраженному в граммах содержащегося в нем марганца.
Персульфатный метод. Персульфатный метод определения марганца в настоящее время настолько усовершенствован, что им можно определять до 100 мг марганца с точностью, не уступающей точности висмутатного метода. При этом надо учитывать, что при использовании персульфатного метода сурьма, умеренные количества церия, кобальт и хром не оказывают мешающего влияния. Только хром несколько маскирует конечную точку титрования, если присутствует в количествах, превышающих 15 мг в 100 мл раствора.
Метод заключается в окислении марганца до марганцевой кислоты персульфатом аммония в разбавленном азотнокислом растворе, содержащем нитрат серебра и фосфорную кислоту (допустимо также присутствие серной кислоты), и в титровании полученной марганцевой кислоты раствором арсенита натрия. Уолтерс1, первый применивший этот метод для анализа стали, нашел, что окисление сухой солью протекает неудовлетворительно и что она должна быть немного увлажнена за несколько дней до ее употребления. В настоящее время персульфат аммония обычно прибавляют в виде его 25%-ного раствора. Эта соль иногда продается плохого качества, поэтому перед применением надо определить ее окислительную способность методом, описанным в разделе «Реактивы» (стр. 61).
Нитрат серебра прибавляют в качестве катализатора реакции окисления марганца2. Он также препятствует образованию двуокиси марганца, если концентрация марганца в растворе не превышает 5 мг в 100 мл. Допустимую концентрацию марганца можно, однако, довести до 50 мг в 100 мл добавлением фосфорной кислоты, хотя в практике анализа она обычно не больше 15 мг в 100 мл.
Оптимальные концентрации реактивов, требуемых для окисления 10 мг марганца в 100 мл раствора, равны: 0,05 г серебра (0,079 г нитрата серебра), 2,5 г персульфата аммония и 1 мл фосфорной кислоты (если присутствует железо, фосфорной кислоты надо прибавить 4 мл). Количества азотной и серной кислот могут варьировать без заметного влияния на результаты анализа, особенно если титр раствора арсенита устанавливается при тех же концентрациях этих кислот, какие имеются в анализируемом растворе. При других постоянных условиях время, требуемое для количественного окисления марганца до марганцевой кислоты, зависит от температуры. Если персульфат аммония прибавить к холодному анализируемому раствору, быстро довести его до кипения и прокипятить 1 мин, то окисление марганца происходит полностью.
Для титрования марганцевой кислоты применяют арсенит натрия, поэтому что он не реагирует с примесями, которые могут присутствовать в растворе, например с хромовой кислотой. Окисление арсенита марганцевой кислотой не происходит стехиометрически по уравнению реакции, и потому титр раствора арсенита надо устанавливать по материалу с известным содержанием марганца, проведя его через все стадии анализа. Титр раствора арсенита можно также устанавливать по титрованному
1 Н. Е. Walters, Proc. Eng. Soc. West. Penn., 17, № 7 (1901); Chem. News, 84, 239 (1901).
2 * В. И. Кузнецов и Л. M. Буданова [ЖАХ, 8, 55 (1953)] предложили взамен нитрата серебра применять соль кобальта. Прим, ред.*
502 Гл. XXV. Марганец
раствору перманганата, как указано ниже, или восстанавливая его, а затем вновь окисляя персульфатом аммония, как при анализе пробых. На конечную точку титрования некоторое влияние оказывают концентрации реактивов в растворе, но если они приблизительно постоянны, конечная точка вполне воспроизводима.
Приводим ход определения, предложенный1 2 для анализа чугунов! и сталей.
Ход определения. Навеску в 1,000 г чугуна или стали помещают в коническую колбу емкостью 500 мл и обрабатывают 30 мл «кислотной смеси» (100 мл концентрированной серной кислоты прибавляют к 525 мл воды, охлаждают, добавляют 125 мл 85%-ной фосфорной кислоты и 250 мл концентрированной азотной кислоты и перемешивают). Нагревают до полного растворения пробы и кипятят до удаления окислов азота 3. Затем приливают 100 мл горячей воды, 10 мл 0,8%-ного раствора нитрата серебра, и 10 мл 25%-ного раствора персульфата аммония, нагревают до кипения и кипятят 1 мин. Охладив до 25 °C или ниже, приливают 75 мл холодной воды и быстро титруют раствором арсенита до светло-желтой окраски, не изменяющейся при дальнейшем прибавлении арсенита. Конец титрования можно определять и потенциометрическим методом.
Приготовление раствора арсенита натрия и установку его титра ведут следующим способом. 6 г окиси мышьяка (III) помещают в стакан емкостью 600 мл, прибавляют 250 мл воды, 15 г едкого натра и перемешивают до растворения. Затем насыщают раствор двуокиси углерода и разбавляют водой до 5 л. Для установки титра в коническую колбу емкостью 500 мл наливают 30 мл указанной выше кислотной смеси, прибавляют 100 мл воды, 10 мл раствора персульфата аммония, 10 мл раствора нитрата серебра и кипятят 1 мин для удаления окислов азота. Охладив затем раствор до комнатной температуры, разбавляют его 75 мл воды, приливают точно 20 мл титрованного 0,03 н. раствора перманганата калия и титруют устанавливаемым раствором арсенита натрия. Концом титрования является появление желто-зеленого окрашивания, не изменяющегося при дальнейшем прибавлении арсенита. Здесь не появляется та светло-желтая окраска, какая получается при анализе пробы, содержащей железо. Разница в оттенках незначительная, и при некотором опыте она не вызывает затруднения.
Другие объемные методы. Из остальных объемных методов следует отметить: 1) метод потенциометрического титрования нитратом ртути (I) после окисления марганца висмутатом натрия или персульфатом аммония4; 2) выделение двуокиси марганца с последующим восстановлением ее в кислом растворе титрованным раствором сульфата железа (II), щавелевой кислоты или перекиси водорода, взятым в избытке, и титрованием
1 Е. В. Sandell, I. М. Kolthoff и J. J. Lingan е [Ind. Eng. Chem., Anal. Ed., 7, 256 (1935)] установили, что реакция титрования происходит полностью и в конце титрования получается бесцветный раствор, если марганцевую кислоту титровать раствором содержащим арсенит натрия и нитрит натрия в эквивалентных количествах. В этом варианте метода перед титрованием серебро надо осадить в виде хлорида серебра. Малые количества хрома, ванадия, никеля и молибдена не мешают. Согласно Н. D. Hillson [там же, 16, 560 (1944)], можно определять до 0,05 г марганца окислением его персульфатом в растворе, содержащем серную и фосфорную кислоты и нитрат серебра, и титрованием раствором арсенита, проходящим точно по уравнению реакции, если прибавить в качестве катализатора осмиевую кислоту. * Подробности арсенитнонитритного метода см. в статье В. А. Г е н е р о з о в, Зав. лаб., 6, 1431 (1937) и в руководстве А. М. Дымов, Технический анализ руд и минералов, 5-е изд., Металлургиздат, 1949.*
2 Н. А. В г i g h t, С. P. L а г г a b e e, J. Research NBS, 3, 573 (1929).
3 При анализе чугунов полученный раствор фильтруют через быстро фильтру-
ющий фильтр для удаления графита и промывают водой. Фильтрат разбавляют, если* надо, до 125 мл и продолжают работу, как при анализе сталей.
Методы определения , . 503
перманганатом избытка восстановителя* 1 и 3) окисление марганца (II) .до четырехвалентного состояния титрованием при температуре около 80 °C раствором перманганата в слабощелочной среде, получаемой после осаждения железа и других элементов окисью цинка2.
Первый метод вполне удовлетворителен и может применяться в присутствии хрома и ванадия. Восстановление двуокиси марганца титрованным раствором восстановителя, прибавленным в избытке, с последующим титрованием перманганатом не проходит точно по уравнению реакции. Поэтому приходится пользоваться эмпирическим титром раствора перманганата, установленным по известному количеству марганца, обработанному таким же способом. Эмпирическим титром приходится пользоваться и при применении метода Фольгарда3, кроме того, при титровании по Фольгарду трудно определить конечную точку.
Весовые методы
Взвешивание в виде пирофосфата марганца. Пирофосфатный метод был предложен4 еще в 1867 г. и затем несколько позже усовершенствован5 * 7. Вероятно, он является наилучшим методом весового определения марганца. Метод основан на осаждении фосфата марганца и аммония в слабоаммиачном растворе, содержащем избыток аммонийных солей, и требует предварительного отделения марганца от элементов, также осаждающихся в этих условиях. Его обычно применяют после предварительного отделения марганца в виде двуокиси марганца и очистки осадка от примесей®, как описано на стр. 494. При выполнении очень точных анализов вводят поправки, определяя колориметрически марганец в фильтратах и промывных водах, полученных после выделения сначала двуокиси Марганца, потом фосфата марганца. Эти поправки обычно не превышают 0,3 мг марганца в первом фильтрате и 0,2 мг — во втором фильтрате.
Осаждение фосфата может быть проведено из горячего раствора8’® или же однократным или двукратным осаждением из холодного раствора,
* G. L. К е 11 е у, М. G. S р е n s е г, С. В. 111 i n g s w о г t h, Т. G г а у, J. Ind. Eng. Chem., 10, 19 (1918).
1 F. Н. Williams, Trans, AIME, 10, 100 (1881—1882); G. v о n Knorre, Z. angew. Chem., 14, 1149 (1901). Первый автор выделял двуокись марганца обработкой -азотной кислотой и хлоратом калия, как описано на стр. 494, а второй — кипячением -с персульфатом аммония анализируемого раствора, содержащего марганец в виде сульфата и не больше 4% свободной серной кислоты.
2 J. Volhard, Ann. Chem., 198, 318 (1879).
3 Например, в проведенных (Бюро Стандартов США) опытах, в которых марганец определялся методами Форда-Вильямса,. Кнорре и Фольгарда [Bur. Standards, Circ. 26, Analyzed Iron and Manganese Ores, p. 17], было найдено, что эмпирические титры раствора при применении первых двух методов оказались равными соответственно 0,4959 и 0,5058 вместо теоретического 0,4918, а при применении третьего метода — 0,2984 вместо 0,2951.
4 W. Gibbs, Am. J. Sci„ [2], 44, 216 (1867).
5 F. A. Gooch, M. Austin, Am. J. Sci., [4], 6, 233 (1898); A. A. Blair, The Chemical Analysis of Iron, 8th ed., Philadelphia, 1918, p. 106.
• Например, кобальт частично осаждается вместе с марганцем; добавление цитрата (стр. 718) приводит к неполному осаждению марганца.
7 A. A. Blair, цит. выше.
8 W. Heintz [Ann. Phys., 74, 449 (1849)] указал на то, что при осаждении на холоду образуется фосфат марганца Мп3(РО<)2, который при кипячении или продол-.жительном стоянии превращается в MnNH4P04.
504
Гл. XX V. Марганец
как описано в гл. «Магний» (стр. 721)1. В последнем случае первое осаждение может длиться 12 ч или долее, а второе во избежание получения повышенных результатов должно всегда продолжаться не более 4 ч. Осаждение из горячего раствора проводят следующим способом.
' Ход определения. К кислому раствору, занимающему объем 100— 200 мл, содержащему марганец в концентрации не выше 0,001 г в 1 мл и не содержащему мешающих определению веществ, прибавляют 20 г хлорида аммония и 1—2 г двузамещенного фосфата аммония (NH4)2HPO4. Нагревают раствор до кипения, прибавляют соляную кислоту, если это необходимо для просветления раствора, и затем приливают по каплям и при непрерывном перемешивании разбавленный раствор аммиака. Как только появится осадок, добавление аммиака прекращают и перемешивают, пока осадок не станет кристаллическим. Затем прибавляют еще каплю раствора аммиака, перемешивают и так продолжают, пока осадок не перестанет выпадать и его шелковистый вид не будет оставаться без изменения. Всю операцию надо вести при кипячении. Наконец прибавляют избыток аммиака в 0,5 мл и охлаждают раствор, лучше всего в ледяной воде. Дают постоять 2—3 ч, фильтруют через беззольный бумажный фильтр и промывают осадок холодным слабоаммиачным 10%-ным раствором нитрата аммония до удаления хлоридов. Высушивают осторожным нагреванием и прокаливают сначала при возможно низкой температуре до выгорания углерода, а затем приблизительно при 1000 °C до постоянной массы Взвешивают2 в виде Мп2Р2О4.
Другие весовые методы. Кроме пирофосфатного метода, для определения марганца рекомендуются весовые методы, основанные на взвешивании его в виде Mn3O4, MnS или MnSO,. Лучший из этих методов — последний.
Первый метод совершенно неудовлетворителен, поскольку состав взвешиваемой окиси зависит от температуры прокаливания и от состава газов, окружающих осадок во время прокаливания3.
1 При осторожном осаждении марганца из раствора, полученного восстановлением 50 мл перманганата, эквивалентных 0,1381 г Mn2P,O7, Knowles получил 0,1378 г, применяя метод Blair, и 0,1385 г, применяя метод Gooch and Austin. Поправки на потерю марганца в фильтратах и промывных водах не определялись. При однократном 4-часовом осаждении из холодного раствора, как описано в гл. «Магний» (стр. 721), было получено 0,1383 г; при двукратном осаждении, из которых первое продолжалось 12 ч, а второе 4^ч, было получено 0,1382 г. При однократном осаждении фильтрат и промывные воды содержали 0,12 мг марганца, при двукратном осаждении — 0,15 мг. Однократное осаждение из горячего уксуснокислого раствора, содержащего ацетат аммония, с последующим охлаждением и стоянием в течение 4 ч дало 0,1351 г Мп2Р207 и 1,3 мг Мп в фильтрате и промывных водах. В результате однократных осаждений из растворов (NH.)2HPO4 -I- NH.H2PO4 методом, описанным в гл. «Цинк», получилось 0,1388; 0,1389 и 0,1390 г.
2 Осадок фосфата магния и аммония можно взвесить в виде MnNH4PO4-Н2О. Для этого его собирают в предварительно взвешенном стеклянном фильтрующем тигле, промывают в конце эфиром, дают постоять несколько минут в эксикаторе и взвешивают [R. С. Crippen, Chemist Analyst, 29 (3), 54 (1940)]. Если определяют лишь несколько миллиграммов марганца, как это обычно имеет место при анализе горных пород, L. A. Dean и Е. Truog [Ind. Eng. Chem., Anal. Ed., 7, 383 (1935)] рекомендуют осаждать его вместе с магнием в виде фосфата после осаждения группы «R2O3» при pH = 6,2 и кальция при pH = 4,4.
3 При тщательном регулировании условий прокаливания Р. N. Raikow иР. Tischkow [Chem. Ztg, 35, 1013 (1911)] не могли получить большей точности, нем 1 : 200.
Методы определения
505
Перед тем как взвешивать марганец в виде его сульфида, надо прокалить осадок до окиси марганца в тигле Розе (стр. 56), затем превратить его в сульфид марганца осторожным нагреванием под слоем чистой серы в токе сухого водорода и, наконец, прокалить при 900 °C и охладить, не прекращая пропускания водорода.
Для определения марганца взвешиванием в виде сульфата надо сначала отделить его от других элементов, образующих с серной кислотой нелетучие соединения, а затем прокалить1 при 450—500 °C до постоянной массы. Для превращения сульфида марганца в сульфат растворяют его в серной кислоте, взятой в небольшом избытке, переводят раствор во взвешенный платиновый тигель, выпаривают до удаления кислоты, охлаждают, увлажняют остаток водой, снова нагревают при 450—500 °C до постоянной массы и взвешивают в виде MnSO4. Если взвешенный остаток имеет коричневый оттенок, прибавляют несколько капель сернистой и серной кислот и прокаливание повторяют.
Колориметрические методы
Малые количества марганца можно точно и быстро определить, окисляя его до марганцевой кислоты перйодатом 2, персульфатом, висмутатом или двуокисью свинца и сравнивая окраску полученного раствора со стандартом. Лучшим следует считать первый метод, потому что в полноте окисления перйодатом можно быть всегда уверенным, не требуется фильтрования раствора после окисления и в присутствии небольшого избытка перйодата марганцевая кислота очень устойчива. Авторы этого метода не обнаружили никаких изменений в обработанном перйодатом растворе после стояния его в- течение трех месяцев в закрытой колбе.
Перйодат железа (III) нерастворим в концентрированной азотной кислоте, но растворяется в серной и фосфорной кислотах. В присутствии большого количества железа может применяться любая из последних двух кислот, но лучше добавлять фосфорную кислоту, так как она обесцвечивает растворы солей железа (III). Церий и хром также окисляются (в различной степени) перйодатом в кислом раствбре.
Ход определения. Раствор, содержащий марганец, освобождают от восстановителей кипячением с азотной кислотой с прибавлением небольшого количества персульфата аммония, если присутствуют соединения углерода, как, например, при анализе сталей. Если в растворе присутствуют хлориды, то его выпаривают с азотной и серной кислотами до появления паров Последней. Затем раствор обрабатывают таким образом, чтобы он содержал в 100 мл приблизительно 20 мл азотной кислоты, кроме того, 10—15 мл серной кислоты или 5—10 мл сиропообразной фосфорной кислоты (или же смесь обеих), прибавляют по 1 г К1О4 на каждую 0,1 г марганца (или эквивалентные количества NaIO4 или Na3H2IO6), кипятят 1 мин, поддерживают высокую температуру еще 5—10 мин, охлаждают, разбавляют до требуемого объема и сравнивают со стандартным раствором марганца, обработанным таким же способом. Перед сравнением окрасок раствор не должен содержать более 1 мг марганца в 50 мл, иначе окраска
1 W. В 1 u m. J. Am. Chem. Soc., 34, 1379 (1912).
2 Н. Н. Willard, L. Н. Great house, J. Am. Chem. Soc , 39 r 2366 (1917).
.'506 Гл. XXV. Марганец
будет слишком темной1. Светопоглощение полученного раствора можно измерять фотометрически при 525 ммк (или при 575 ммк, если присутствует хромат).
1 Если желают закончить определение объемным методом, надо удалить избыток перйодата. Согласно Н. Н. Willard и J. J. Thompson [Ind. Eng. Chem., Anal. Ed., 3, 399 (1931)J, это можно сделать, прибавляя в избытке нитрат ртути (II), фильтруя через большой асбестовый фильтр, собирая фильтрат в колбу, содержащую титрованный раствор сульфата железа (II), взятый в избытке, и 10 мл разбавленной (1 : 1) серной кислоты, промывая асбестовый фильтр 4 или 5 раз холодной водой и титруя избыток сульфата железа (II) раствором перманганата. Реакция лучше всего проходит ;В фосфорнокислом растворе; в этом случае хром в количествах, не превышающих 1 мг, не мешает. Кобальт, церий и хлорид-ионы должны отсутствовать.
Глава XXVI
ВАНАДИЙ
Установлено, что в земной коре ванадий широко распространен не только в рудах и углях, но и в глинах, известняках, песчаниках и изверженных породах1. В значительных количествах (до 0,08% в расчете на У2О3 и выше) он встречается в основных изверженных и метаморфических породах, но, очевидно, отсутствует или содержится лишь в виде следов в высококремнистых породах. Некоторые из образующих эти породы ферро-и алюмосиликаты содержат более 0,13% V2O3, как, например, ^биотит, выделенный Из пироксенового гнейса. Таким образом, ванадий обычно приходится определять в породах, содержащих менее 60% кремнекислоты. Время, затраченное на определение ванадия, в этих породах не всегда себя оправдывает, но необходимо учитывать, что в присутствии ванадия в результаты определения алюминия и железа (II) и (III) вносится ошибка, которая для железа может достигать значительной величины. Тот факт, что в породах ванадий обычно находится в сочетании с ферроалюмосили-катами, а также существование минерала роскоэлита, классифицируемого как ванадиевая слюда, указывает, что в природйых соединениях ванадий играет такую же роль, как алюминий и железо (III), и его нужно рассматривать как элемент, их замещающий. Поэтому результаты определения ванадия следует пересчитывать на V2O3, а не на V2O5.
Валентность ванадия в соединениях, входящих в состав пород вторичного происхождения, вроде глин, известняков, песчаников, углей и железных руд, пока еще точно не установлена. В. Гиллебранд одно время считал, что ванадий в этих дородах находится в пятивалентном состоянии, однако исследование некоторых содержащих ванадий песчаников 2 3 Западного Колорадо, в которых ванадий оказался трехвалентным, показало несостоятельность этого взгляда. >
ОБЩИЕ ЗАМЕЧАНИЯ
При анализе горных пород обычно принятым методом в процессе •обезвоживания кремнекислоты, выпариванием раствора с соляной кислотой ванадий восстанавливается от пятивалентного до четырехвалентного состояния. Содержащиеся в породах небольшие его количества практи-
1 W. F. Hillebrand, Am. J. Sci., [4], 6, 209 (1898); Chem. News, 78, 216
(1898); U.S. Geol. Survey Bull., 700, 184 (1924).
3 W. F. Hillebrand, F. L. Ransome, Am. J. Sci., [4], 10, 120 (1900),
508
Гл. XXVI. Ванадий.
«
чески полностью переходят в осадок от аммиака, если сумма полуторных окислов (R2O3) велика и в ней преобладает содержание Fe2O3. Если же преобладает содержание А12О3, ванадий переходит в осадок не полностью. • При прокаливании этого осадка ванадий окисляется до пятивалентного-и, следовательно, взвешивается в виде V2O5. Если перед определнением железа объемным методом ванадий не отделяют, то его влияние зависит от применяемого способа восстановления. Сероводород, сернистый ангидрид и хлорид олова (II) восстанавливают ванадий до четырехвалентного состояния, и в этом случае 1 мг V2O5 соответствует 0,878 мг Fe2O3. Цинком ванадий восстанавливается до двухвалентного, и в этом случае 1 мг V2O5 соответствует 2,63 мг Fe2O3. Отсюда очевидно, что если пренебречь наличием ванадия, то при восстановлении его до четырехвалентного как для железа, так и для алюминия получаются положительные ошибки. В случае же восстановления ванадия до двухвалентного состояния для железа получаются повышенные результаты, а для алюминия — пониженные. В обычных условиях ванадий (II) настолько неустойчив, что для точного учета его влияния на результаты определения железа раствор после восстановления следует сразу же вливать в раствор, содержащий избыточное количество сульфата железа (III). Ванадий оказывает влияние также на колориметрическое определение титана и на определение фосфора.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ВАНАДИЙ
Полный анализ ванадиевых минералов лучше всего проводить, пользуясь отдельными навесками пробы. Большинство ванадиевых минералов легко разлагается кислотами. Полное извлечение ванадия достигается при обработке материала сначала соляной, а затем азотной кислотой. Для полного разложения руды нерастворимый остаток сплавляют с карбонатом натрия, плав растворяют в соляной кислоте и присоединяют к основному раствору. В тех случаях, когда не требуется определять кремнекислоту, нерастворимый остаток можно обработать фтористоводородной кислотой, которую затем удаляют выпариванием с серной или какой-либо другой кислотой. Обычно принятую обработку фтористоводородной и серной кислотами (с последующим выпариванием и прокаливанием для удаления кремнекислоты) не следует применять к остаткам, содержащим ванадий, так как при красном калении могут образоваться летучие фториды или оксифториды ванадия.
Ванадиевые минералы также легко сплавляются с карбонатом натрия (или калия) и с перекисью натрия. При использовании карбоната целесообразно прибавить немного селитры. Если пользуются перекисью натрия, то нет необходимости нагревать до полного расплавления смеси,, так как реакция проходит полностью и в случае спекания.
Сплавление с пиросульфатом применяется редко, главным образом в тех сдучаях, когда требуется определять - малые количества ванадия в остатках, получаемых в процессе анализа.
Для определения валентности ванадия в минерале, например в роско- ’ элите, разложение можно осуществить нагреванием свободного от воздуха порошка минерала с разбавленной серной кислотой в запаянной трубке,. j из которой воздух вытеснен током двуокиси углеродах. i
1 W. F. Н i 11 е Ь г a n d, Am, J. Sci., 7, 451 (1899). ч
' 1
Методы отделения
509
Имеются указания, что при нагревании растворов, содержащих ванадий, при температуре выше 100 °C наблюдаются его потери. Мы, однако, не могли обнаружить потерю ванадия при выпаривании солянокислых растворов досуха и нагревании остатка при 175° С, а также в случае нагревания сернокислых растворов ванадия (V) и (IV) в течение 5 ч при той же температуре. Это можно объяснить неустойчивостью летучих хлоридов ванадия в присутствии влаги.
Небольшие количества ванадия соосаждаются с вольфрамовой кислотой (приблизительно 1 часть ванадия на 100 частей вольфрама) и должны быть потом определены в ней, если осадок велик.
Небольшие количества ванадия в вольфрамовой кислоте можно определить колориметрическим путем в виде желтой ванадиевовольфрамовой кислоты следующим спо-•собом1. Вольфрамовую кислоту растворяют в 15 мл 4%-ного раствора едкого натра, фильтруют, если нужно, разбавляют до 75—100 мл, подкисляют 5 мл фосфорной кислоты (пл. 1,37 г/см3) и через 1—2 ч Сравнивают окраску с окраской, полученной при добавлении стандартного раствора ванадиевовольфрамовой кислоты к такому же объему воды. Аммонийные соли должны отсутствовать. Стандартный раствор содержит 0,1 мг ванадия и 5 мг вольфрама в 1 мл. Для его приготовления соответствующие количества ванадата и вольфрамата натрия растворяют в воде, добавляют 25 мл фосфорной кислоты (пл. 1,37 г/см3) и разбавляют до 500 мл.
МЕТОДЫ ОТДЕЛЕНИЯ
Большинство методов отделения ванадия можно классифицировать в зависимости от того, служат ли они для переведения ванадия в осадок или в фильтрат. Так, например, ванадий обычно переходит в осадок вместе с другими элементами при осаждении аммиаком; он осаждается вместе с фосфоромолибдатом аммония,- при выпаривании с азотной кислотой, а также при осаждении нитратом ртути (I), ацетатом свинца и купфероном. В раствор ванадий переходит при сплавлении с перекисью натрия или карбонатом натрия с селитрой и последующем выщелачивании плава водой, при осаждении едким натром или сероводородом из кислого раствора. Кроме того, для отделения ванадия от других элементов используются электролиз с ртутным катодом, экстракция эфиром из разбавленного (1 : 1) солянокислого раствора (при которой отделяются железо и молибден) и отгонка ванадия в струе сухого газообразного хлористого водорода.
Один ванадий аммиаком не осаждается; его наличие в осадке от аммиака обусловлено образованием нерастворимых соединений ванадия с железом и алюминием.
Однако при осаждении аммиаком ванадий не всегда полностью переходит в осадок, и поэтому содержание его обычно определяют из отдельной навески. Осаждение ванадия аммиаком представляет интерес главным образом в связи с предосторожностями, которые следует соблюдать при анализе осадка от аммиака2.
Давно известно, что осадки фосфоромолибдата аммония увлекают ванадий (V), приобретая при этом оранжевую или коричнево-красную
1 Н. Н. W i 11 а г d, Р. Young, Ind. Eng. Chem., 20, 764 (1928).
2 Опыты H. В. Knowles показали, что 1 мг пяти- или четырехвалентного ванадия полностью захватывается 100 мг алюминия, тогда как 10 мг ванадия, неза-
висимо от его валентности, таким количеством алюминия не удерживаются. С железом ванадий соосаждается несколько лучше. Так, 100 мг железа полностью увлекают 10 мг ванадия (V) и почти полностью, если он находится в четырехвалентном состоянии.
510
Гл. XXVI. Ванадий
окраску. Если содержание фосфора в 5—10 раз превышает содержание ванадия, последний осаждается полностью1. Однако в том случае, если ванадий находится в четырехвалентном состоянии и осаждение производится при комнатной температуре, то соосаждение его с осадком фосфоро-молибдата не происходит 2. При соосаждении ванадия с фосфором достигается его отделение от тех же элементов, от которых отделяется в этих условиях фосфор (стр./789). Результаты нескольких опытов показывают, что отделение таким образом 5 мг ванадия (V) от 100 мг урана (VI) происходит вполне удовлетворительно. Для выполнения этого отделения раствор подготовляют таким же способом, как и для осаждения фосфоромо-либдата аммония. Прибавляют десятикратное по отношению к содержанию ванадия количество фосфора и несколько большее, чем обычно, количество нитрата аммония. Затем вводят аммиак почти до нейтральной реакции, нагревают раствор до кипения, прибавляют обычные количества раствора молибдата аммония и встряхивают несколько минут. Осадок отфильтровывают и промывают декантацией горячим (80° С) раствором кислого сульфата аммония (1000 мл воды, 15 мл аммиака и 25 мл серной кислоты).
При выпаривании азотнокислого раствора досуха и последующем выщелачивании остатка водой происходит отделение ванадия в виде пиро-ванадиевой кислоты H4V2O7 совместно с железом и алюминием от щелочных металлов и урана, переходящих при этом в раствор 3. Это разделение не количественное 4, так как небольшие количества ванадия, железа и алюминия переходят в раствор, а уран частично захватывается осадком, особенно если присутствует фосфор.
Для группового отделения таких небольших количеств ванадия, хрома, молибдена, вольфрама, фосфора и мышьяка, какие встречаются в породах, давно используется способ осаждения их нитратом ртути (I) из растворов, содержащих небольшие количества карбоната натрия 5. Метод этот йрименяется после разложения пробы сплавлением ее с карбонатом натрия и селитрой. Осторожно сплавляют 5 г измельченной породы с 20 г карбоната натрия и 3 г нитрата натрия. Выщелачивают плав водой, марганец восстанавливают спиртом и затем раствор фильтруют. В том слу- • чае, если проба полностью не разложилась или присутствуют большие количества ванадия, осадок прокаливают и сплавление повторяют, а фильтраты объединяют. В раствор вводят разбавленную (1:1) азотную кислоту почти до нейтральной реакции, предварительно устанавливая требуе- ! 4 мое для этого количество кислоты на таком же количестве реактивов, j какое было израсходовано для разложения пробы. При нейтрализации нельзя переходить за точку нейтральности, так как в кислом растворе j хром и ванадий восстанавливаются образующимся в процессе сплавления ; нитритом. Раствор выпаривают почти досуха, разбавляют 100 мл воды, на-гревают до перехода в раствор растворимых солей и фильтруют. Остаток ч кремнекислоты и гидроокиси алюминия обрабатывают фтористоводород- J -----*---
1 J. R. Cain, J. С. Hostetter, J. Ind. Eng. Chem., 4, 250 (1912); J, Am. . Chem. Soc., 43, 2552 (1921).
2 J. R. Cain, F. H. Tucker, J. Ind. Eng. Chem., 5, 647 (1913). .
3 C. Friedel, E. C u m e n g e, C. r., 128, 532 (1899). S
4 W. F. Hillebrand, F. L. Ransome, Am. J. Sci., [4], 10, 136 (1900).
6 5V. F. Hillebrand, J. Am. Chem. Soc., 20, 454 (1898); U. S. Geol. Survey ч
Bull. 700, 185. |
Методы отделения
511
ной и серной кислотами, выпаривают досуха и сплавляют с карбонатом натрия. Плав растворяют 'в 100 мл воды, раствор доводят почти до-нейтральной реакции азотной кислотой, кипятят несколько минут и фильтруют. Фильтрат присоединяют к основному раствору. После этого в холодный щелочной раствор вводят по каплям почти нейтральный раствор нитрата ртути (I) до прекращения образования осадка.
Если осадок продолжает выделяться, и после того как на дне сосуда его собралось большое количество (что указывает на слишком высокое содержание карбоната в растворе), вводят по каплям разбавленную (1 : 1) азотную кислоту до тех пор, пока''отстоявшийся раствор не перестанет мутнеть после добавления капли раствора нитрата ртути. (I). Нагревают до кипения, давдт осадку осесть на дно, после чего его отфильтровывают и промывают горячей водой, содержащей несколько капель раствора нитрата ртути (I). Осадок высушивают и снимают с фильтра, чтобы предотвратить потерю молибдена во время прокаливания, а также порчу тигля вследствие восстановления мышьяка. Осадок осторожно нагревают, в платиновом тигле под тягой до удаления ртути, затем прокаливают при температуре не выше 400—500 °C и сплавляют с Небольшим количеством карбоната натрия в окислительных условиях. Плав выщелачивают водой и испытывают раствор на ванадий, как указано в разделе «Методы определения» (стр. 513, учитывая при этом, что в растворе содержатся весь хром и молибден, а также некоторые количества фосфора, мышьяка и вольфрама.
Осаждение ванадия в виде ванадата свинца может служить лишь для erb предварительного отделения, обычно группового, так как осадок нельзя непосредственно взвешивать и, кроме того, совместно с ранадием осаждаются молибден, вольфрам и хром (VI). Этот метод Применяют после переведения ванадия в раствор в виде ванадата щелочного металла. Раствор слабо подкисляют азотной кислотой, вводят ацетат свинца в небольшом избытке, нагревают до кипения и перемешивают до коагуляции осадка. Затем осадок отфильтровывают, промывают сильно разбавленной уксусной кислотой, растворяют в азотной кислоте и отделяют свинец в виде сульфата после выпаривания с серной кислотой.
Осаждение купфероном (стр. 143), является хорошим способом отделения ванадия (V) от многих элементов, особенно урана (VI), хрома, мышьяка, фосфора и алюминия *. Осаждение можно проводить непосредственно после переведения породы в раствор, если количества ванадия, железа, циркония и титана невелики. В случае же высокого содержания последних трех элементов осаждение купфероном можно осуществить в объединенных и подкисленных растворах, полученных после выщелачивания карбонатно-нитратных плавов водой, как и при осаждении нитратом ртути (I). При этом хром следует восстановить до трехвалентного перекисью водорода, которую затем разрушают кипячением
Сплавление с перекисью натрия или карбонатом натрия и селитрой с последующим выщелачиванием плава водой служит для отделения железа, титана и циркония от ванадия, фосфора, мышьяка, молибдена, хрома, урана и большей части алюминия и кремния. Эти разделения таким
1 Согласно S. G. Clarke [Analyst, 52, 466, 527 (1927)], ванадий этим способом можно отделить и от вольфрама, если осаждение проводить в присутствии фтористоводородной кислоты. - -
512
Гл. XXVI. Ванадий
образом являются групповыми, и для дальнейшего отделения элементов друг от друга требуется применение других методов. Это же относится и к осаждению едким натром.
Осаждение урана в виде фосфата из щелочного раствора 1 2 является хорошим методом отделения урана от ванадия. Проводится это следующим образом.
К разбавленному сернокислому раствору прибавляют приблизительно по 0,5 г фосфата аммония на каждые 0,25 г USO8, нагревают до кипения и затем вводят аммиак до щелочной реакции. После кипячения в течение нескольких минут осадок отфильтровывают и промывают горячей водой, содержащей немного сульфата аммония.
Ванадий не осаждается сероводородом из кислых растворов, но имеет склонность соосаждаться с элементами сероводородной группы, если в растворе не содержится винная кислота..
Весьма удовлетворительным методом отделения ванадия от различных элементов является электролиз разбавленного сернокислого раствора с ртутным катодом 9 (стр. 165). При этом железо, хром, кобальт, никель, медь и молибден осаждаются на ртути и отделяются таким образом от ванадия, урана, алюминия и фосфора. Мышьяк частично улетучивается, а частично остается вместе с ванадием в растворе.
Экстракция железа и молибдена эфиром из холодного разбавленного солянокислого раствора (стр. 161) для отделения от ванадия применяется главным образом при анализе феррованадия и специальных сталей. Соединения ванадия (V) заметно растворимы в эфире. Растворимость соединений ванадия (IV) значительно ниже, ею можно пренебречь при определении нескольких миллиграммов ванадия 3.
В продуктах, богатых ванадием, для отделения его основной массы перед определением других элементов удобно пользоваться отгонкой в токе хлористого водорода 4 * *. В этом методе струю сухого газообразного хлористого водорода пропускают над сухой пробой, находящейся в лодочке, помещенной в стеклянной трубке, которую для лучшего удаления ванадия можно слегка нагревать. Летучий оксихлорид ванадия может быть поглощен водой и затем количественно определен. При прохождении хлористого водорода ванадий частично восстанавливается и перестает отгоняться. Поэтому содержимое лодочки целесообразно окислить выпариванием с азотной кислотой, после чего отгонку ванадия продолжить. Эту операцию повторяют до тех пор, пока не прекращается образование коричневого дистиллята. Молибден и мышьяк отгоняются совместно с ванадием. Железо также сопровождает ванадий, если слишком сильно нагревать трубку. Этот метод может сочетаться с операцией обработки исходной пробы азотной кислотой. В этом случае высушенный нерастворимый остаток и выпаренный досуха азотнокислый фильтрат лучше обрабатывать хлористым водородом порознь в.
1 A. N. F i в в, J. Am. Chem. Soc., 28, 1443 (1906).,
2 J. R. C a i в, J. Ind. Eng. Chem., 3, 476 (1911).
3 В опытах, проведенных H. A. Bright с 2,25 г железа и 24 мг ванадия, эфиром извлекалось 2 мг ванадия (V) и менее 0,03 мг ванадия (IV). При экстрагировании же раствора, содержавшего 0,67 г железа и 0,33 г ванадия (IV), в эфирном экстракте было найдено 1,6 мг ванадия.
4 Е. F. Smith, J. G. Hibbs, J. Am. Chem. Soc., 16, 578 (1894).
6 Об отгонке ванадия в токе СО2 4- СС14 см. Р. Jannasch, Z. prakt. Chem.,
97, 93, 141, 154 (1918).
Методы определения
513
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Для определения ванадия предложено большое число как весовых, так и объемных методов. Весовому определению ванадия мешает значительное число элементов, и поэтому часто предпочитают пользоваться объемными методами. Весовой формой ванадия обычно является его пяти-окись (V2O5), представляющая собой красновато-коричневое кристаллическое вещество, плавящееся при 658 °C и не возгоняющееся при этой температуре. В присутствии органических веществ V2O5 в процессе прокаливания может частично восстановиться до V2O4. Поэтому осадок целесообразно обработать несколькими каплями азотной кислоты и затем снова прокалить.
Восстановление сернистым ангидридом
Наиболее удовлетворительный метод определения больших и малых количеств ванадия заключается в восстановлении его сернистым ангидридом и титровании горячего раствора перманганатом после вытеснения избытка сернистого ангидрида двуокисью углерода. Восстановленное соединение ванадия вполне устойчиво в соляно- и сернокислых растворах х. Число элементов, мешающих определению, как, например, железо, мышьяк, сурьма, невелико и они обычно легко отделяются. Присутствие хрома нежелательно, так как в горячих растворах он частично окисляется перманганатом, вследствие чего приходится вводить поправку 2, а в холодных растворах окисление ванадия протекает медленно и конечная точка титрования недостаточно резка. Платину следует отделять, так как в ее присутствии получаются повышенные результаты за счет образования соединений платины (II) и, кроме того, она препятствует полному удалению сернистого ангидрида. Если для осаждения платины или других металлов применяют сероводород, его необходимо затем полностью удалить кипячением и для разрушения политионовых соединений раствор обработать перманганатом до появления- розовой окраски. Как указано в некоторых работах, сульфат натрия на определение не влияет.
Ход определения. Раствор соли ванадия в разбавленной (2 : 98) серной кислоте, свободный от элементов, способных окисляться перманганатом после восстановления их сернистым ангидридом, нагревают до кипения, вводят концентрированный раствор перманганата калия до появления розовой окраски и насыщают раствор 5—10 мин сернистым ангидридом 3. После этого пропускают сильный ток двуокиси углерода до полного удаления сернистого ангидрида, что определяют, пропуская струю газа через сильно разбавленный, подкисленный раствор перманганата. Раствор охлаждают до 60—80 °C и титруют раствором перманганата
1 Например, L. Е. Stout и G. С. Whita к е г [Ind. Eng. Chem., 20, 210 (1928)] установили, что сернокислый раствор сульфата ванадия не изменяется в течение 6 мес.
2 W. F. Hillebrand, U. S. Geol. Survey Bull., 700, 199.
3 Растворы сернистой кислоты или сульфита щелочного металла можно применять только свежеприготовленные, так как по прошествии некоторого времени в них образуются другие окисляющие вещества, помимо сульфита. Сернистый ангидрид удобнее всего получить из баллона со сжиженным газом. В случае отсутствия баллона сернистый ангидрид получают нагреванием раствора сернистой кислоты или сульфита, в который по каплям вводят серную кислоту.
33 заказ 522.
514
Гл. XXVI. Ванадий
калия, титр которого устанавливают по оксалату натрия. 1 мл 0,1 н. раствора перманганата калия соответствут 0,0051 г ванадия. В присутствии хрома лучше титровать холодный раствор до тех пор, пока появившаяся от капли перманганата окраска не перестанет исчезать после непрерывного перемешивания раствора в течение. 1 мин. Восстановление и титрование можно повторить 1 или 2 раза. После многократного повторения этих операций накапливающийся в растворе сульфат марганца делает конечную точку титрования не четкой. Если результаты титрования указывают на содержание очень малых количеств ванадия, то после удаления таких мешающих элементов, как молибден и титан, и после упаривания раствора до небольшого объема наличие ванадия должно быть проверено реакцией с перекисью водорода, как указано в разделе «Колориметрический метод» (стр. 516).
Метод с применением сульфата железа (II) 1
и персульфата аммония 1
В холодных кислых растворах, не содержащих нитрата серебра, персульфат аммония окисляет железо (II) и лишь очень медленно реагирует с ванадием (IV), перманганатом, марганцем (II) и хромом (III). На этом основан быстрый метод определения ванадия, который заключается в восстановлении ванадия (V) сульфатом железа (II), в окислении избытка последнего персульфатом и последующем титровании восстановленного ванадия, перманганатом. Определению мешает вольфрам. Хром, никель, кобальт, молибден, мышьяк и уран не оказывают влияния на результаты.
Персульфатный метод не так точен, как предыдущий, особенно при содержании малых количеств ванадия, но вполне его заменяет, когда необходимо быстро получить результаты, причем с вполне приемлемой точностью. Другими преимуществами персульфатного метода является то, что определению не мешает железо и что этот метод можно применять после определения хрома, как описано в гл. «Хром» (стр. 592).
Ход определения. Анализируемый раствор должен быть свободен от вольфрама и веществ, окисляющихся перманганатом после последовательной обработки сульфатом железа (II) и персульфатом аммония. Если присутствует железо, то для обесцвечивания раствора добавляют фосфорную кислоту в достаточном для этого количестве и затем раствор перманганата до появления неисчезающей розовой окраски. К раствору, содержащему 10% серной кислоты, при непрерывном перемешивании прибавляют приблизительно 0,1 н. раствор сульфата железа (II) (1 мл-0,1 н. раствора соответствует 5,1 мг V) 2 до тех пор, пока капля анализируемого раствора не окрасит раствор гексацианоферрата (III) калия в синий цвет, после чего прибавляют избыток этого раствора в 3—5 мл. Раствор перемешивают 1 мин, прибавляют 8 мл персульфата аммония (свежеприготовленный 15 %-ный раствор 95 %-ной соли) и снова энергично перемешивают 1 мин. Титруют 0,03 н. или более концентрированным раствором перманганата до появления отчетливой розовой окраски, не исчезающей после перемешивания раствора в течение 30 сек.
1 Н. L. Hamner, Met. Chem. Eng., 17, 206 (1917).
* Приготовляют растворением 39,2 а (NH4)2Fe(SO4)2 ’6^0 в 1л разбавленной’ (1 : 19) H2SO4.
Методы определения
515
Осаждение купфероном
Ванадий (V) количественно осаждается купфероном из холодных растворов в разбавленной (1 : 9) серной кислоте (стр. 143). Этот метод, однако, редко используется для определения ванадия, так как при этом требуется предварительное отделение железа, титана и циркония. Он применяется главным образом для группового отделения нескольких элементов, содержащихся в небольших количествах х.
Потенциометрическое определение
Потенциометрические методы определения ванадия дают очень хорошие результаты и особенно удобны для массовой работы. Титрование обычно основано на восстановлении пятивалентного ванадия до четырехвалентного отмеренным количеством раствора сульфата железа (II) 1 2. Другие восстанавливающиеся железом (II) вещества должны осутствовать или должно быть учтено влияние их на результаты определения. Из мешающих элементов чаще всего приходится сталкиваться с хромом, так как при окислении ванадия в начальной стадии диализа могут образоваться некоторые количества хромата. Имеется указание 3, что при использовании азотной кислоты такой концентрации, при которой не происходит окисление хрома, ванадий окисляется только на 99%. Полное окисление ванадия достигается при кипячении раствора с азотной кислотой и последующей обработке перманганатом, взятым в небольшом избытке 4. Образующийся при этом хромат разрушают кипячением уксуснокислого раствора с перборатом натрия.
Ванадий количественно окисляется до пятивалентного при действии бромата калия на растворы, содержащие аммонийные соли и соляную кислоту соответствующей концентрации 5 * * В. В этих условиях фольфрам, молибден и хром переходят в шестивалентное состояние, а марганец остается двухвалентным. На 200 мл разбавленного сернокислого раствора, содержащего 4 г железа, 10 мг ванадия и 40 мг хрома, требуется 6 мл соляной кислоты, 5 г сульфата аммония и 2 г бромата калия. Раствор нагревают до 25—60 СС в течение 15 мин, затем кипятят 10 мин для разрушения избытка бромата
1 Точность метода доказывается тем, что в аликвотных частях одного и того же
• раствора найдено V2O6: осаждением купфероном и осторожным прокаливанием осадка is при 800° С — 0,2564 г, а потенциометрическим методом — 0,2560 г. Прокаливание
купферонового осадка следует проводить в хороших окислительных условиях, ста-p. раясь, чтобы углерод полностью сгорел до начала плавления осадка, который затем I прокаливают при температуре несколько более высокой, чем температура плавления
окиси ванадия.
|, 2 Применяя, например, прибор, подобный предложенному Kelley (стр. 231),
Г; погружают каломельный и платиновый электроды в холодный разбавленный азотно-
го, сернокислый раствор, содержащий ванадий в пятивалентном состоянии, и титруют
В раствором сульфата железа (II), пока луч света, отраженный от зеркальца гальвано-
ft метра, после некоторого периода покоя не придет снова в движение. Затем прибавляют
ft титрованный раствор хромата калия эквивалентной концентрации до тех пор, пока
ft двигающийся обратно к своему прежнему положению луч света на некоторое время не
К: остановится. После этого снова прибавляют раствор сульфата железа (II) до заметного
смещения луча. Израсходованный объем раствора хромата вычитают из введенного
К объема раствора сульфата железа (II) и содержание ванадия вычисляют, основываясь
К- на том, что IFe соответствует IV. Хром и вольфрам определению не мешают.
ft 3 G. L. К е 11 е у, J. A. W i 1 е у, R. F. В о h n, W. С. W г i g h t, Ind. Eng. К Chem., 11, 632 (1919).
№ 4 H. H. Willard, F. Fenwick, J. Am. Chem. Soc., 45, 84 (1923).
К ‘ И. H. Willa r.d, P. Young, Ind. Eng. Chem., 20, 764 (1918).
К 33*
516
Гл. XXVI. Ванадий
и обрабатывают 25 мл серной кислоты (пл. 1,5 г/см3). Перед потенциометрическим титрованием сульфатом железа (II) раствор охлаждают до 5 °C. Вольфрамовую кислоту можно растворить в растворе едкого натра и присоединить к основному раствору, в который предварительно вводят раствор сульфата железа (III) в количестве, соответствующем 5 г железа, после чего окисляют ванадий.
Колориметрический метод
Колориметрический метод, основанный на окислении перекисью водорода в кислом растворе, даёт менее удовлетворительные результаты при определении ванадия, чем при определении титана, так как возникающую окраску (от красновато-желтой до коричневой, в зависимости от содержания ванадия) трудно сравнивать с эталоном. Эта реакция используется главным образом для качественного обнаружения ванадия, хотя ее можно с успехом применять и для количественного определения ванадия в растворах, содержащих 15—20% серной кислоты, не более 0,1 мг/мл ванадия и свободных от элементов, образующих в этих условиях окрашенные соединения. Ванадий при этом должен содержаться в растворе в пятивалентном состоянии, так как при низшей его валентности окраска развивается очень медленно. Интенсивность окраски увеличивается в присутствии сильных кислот и не разрушается фтористоводородной кислотой. Это дает возможность определить ванадий в присутствии титана, перекисное соединение которого обесцвечивается при введении в раствор фторида (стр. 655). Окраску, присущую солям железа (III), можно уничтожить добавлением фосфорной или фтористоводородной кислоты. При действии перекиси водорода молибден также образует окрашенные соединения, и поэтому он должен быть предварительно отделен.
Ход определения. К разбавленному (1 : 4) сернокислому раствору, содержащему ванадий (V) и свободному от молибдена, титана и других элементов, образующих с перекисью водорода окрашенные соединения, прибавляют 1—2 мл 30%-ной перекиси водорода и разбавленной (1 : 4) серной кислотой доводят раствор до определенного объема. Интенсивность полученной окраски сравнивают с окраской стандартного раствора ванадия методом, описанным в главе «Титан» (стр. 656) *.
*3начительно более чувствительным, чем пероксидный метод, является метод А. П. Виноградова 1 2, основанный на образовании желтой растворимой фосфоровольфрамованадиевой кислоты при добавлении фосфорной кйслоты и вольфрамата натрия к кислому раствору ванадата. При этой реакции молекулярные соотношения между фосфорной кислотой и вольфраматом натрия могут колебаться в пределах от 3 : 1 до 20 : 1. Концентрация вольфрамата натрия должна быть в пределах от 4 до 34 г!л.
1 Согласно J. Меуег и А. Р a w 1 е 11 a [Z. anal. Chem., 69, 15 (1926)], определение лучше всего проводить в растворе, содержащем 15—20% серной кислоты, при соотношении V : Н2О2, равном 1:1. При большем содержании перекиси водорода в растворе появляется желтая окраска вместо красно-коричневой. J. Lukas и A. J i 1 е к [там же, 76, 348 (1929)] утверждают, что ванадий модщо открыть в присутствии умеренных количеств вольфрамата или молибдата, если щелочной раствор подкислить борной и щавелевой кислотами (прибавляемыми в твердом виде), а затем добавить 30%-ную перекись водорода.
2 А. П. Виноградов, ДАН СССР, 1931 А, 249; см. также Е. В. S a n d е 11, Ind. Eng. Chem., Anal. Ed., 8, 336 (1936); E. R. Wri g ht, M. G. Mellon, там же, 9, 251 (1937).
Методы определения
517
В довольно широких пределах может колебаться и кислотность раствора1. При проведении реакции на холоду получается коричневатая окраска (чем выше кислотность раствора, тем сильнее коричневый оттенок), которая через несколько часов переходит в нормальную желтую. Кипячение ускоряет достижение равновесного состояния. Однако при малых концентрациях ванадия (меньше 10 мг!л), отсутствии железа и при кислотности раствора около 0,5 н. можно раствор не кипятить.
. Определению мешают: 1) катионы, образующие малорастворимые фосфоровольфраматы — калий и, особенно, аммоний; 2) элементы, образующие малорастворимые фосфаты — титан, цирконий, висмут, сурьма и олово; 3) молибден в концентрации, превышающей 500 мг/л (дает желтое окрашивание); 4) иодиды, роданиды и другие восстановители, восстанавливающие фосфоровольфрамовую кислоту, и 5) элементы, образующие окрашенные в растворе ионы — медь, кобальт, никель, хром и т. п.
Железо (III) в концентрации до 100 мг!л не мешает, если раствор после добавления реактивов прокипятить — тогда коричневатая окраска, вызванная присутствием железа, исчезает.
Определение выполняют следующим способом. Анализируемый раствор подкисляют серной, азотной или соляной кислотой до 0,5 н. концентрации кислоты, после чего добавляют 1,0 мл разбавленной (1 : 2) фосфорной кислоты и 0,5 мл 0,5 М раствора вольфрамата натрия на каждые 10 мл анализируемого раствора. Затем нагревают до кипения, кипятят несколько секунд, охлаждают, разбавляют до определенного объема и измеряют светопоглощение раствора в фотоколориметре при длине волны 400 ммк. При малом содержании ванадия и отсутствии железа кипячение раствора можно опустить. Заканчивать определение можно также и визуально методом колориметрического титрования.
Фосфоровольфраматный метод был применен к определению ванадия в сталях и чугунах 2.
Позднее был разработан метод 3, в котором фосфорно-ванадиево-воль-фрамовый комплекс восстанавливают хлоридом олова (II) с образованием соединения, окрашенного в красно-фиолетовый цвет; интенсивность получаемой окраски пропорциональна содержанию ванадия. Доп. ред*
*Метод А. П. Виноградова был применен также для анализа алюминия 4. Ванадий концентрируют и отделяют от алюминия и других мешающих колориметрическому определению элементов извлечением карбамата ванадия хлороформом. При содержании ванадия от 2 мкг и выше он извлекается количественно из кислых 0,1—0,2 н. растворов. Последующее удаление ванадия из экстракта осуществляется обработкой разбавленной (1:1) азотной кислотой.
Для микроколориметрического определения ванадия (V) в минералах, горных породах и рудах была применена реакция ванадия (V) с бензидином в концентрированной форсфорной кислоте, сопровождающаяся появлением желтого окрашивания, свойственного продуктам окисления
1 Г. А. Панченко, Р. И. Мардер, Труды центральной лаборатории завода «Большевик», 1940, стр. 203.
2 Б. А. Г е н е р о з о в, Н. К. Кускова, ЖАХ, 1, 325 (1946).
3 А. Л. Д а в ы д о в, 3. М. Вайсберг, Зав. лаб., 9, 715 (1940); А, Л. Д а-в ы д о в, 3. М. Вайсберг, Фотоэлектрические методы анализа черных, цветных металлов и руд, Изд. АН УССР, 1943.
4 Ю. А. Чернигов, Б. М. Добкина, Зав. лаб., 16, 402 (1950).
518
Гл. XXVI. Ванадий
бензидина х. Реакция эта позволяет обнаружить присутствие ванадия при его концентрации 0,0002 мг!мл. Аналогичную реакцию с бензидином дают хроматы, манганаты, персульфаты, йодаты, броматы, нитриты, церий (IV), кобальт (III), таллий (III) и золото (III). Перекись водорода мешает реакции вследствие образования надванадиевой кислоты.
Нитраты, сульфаты, вольфраматы не оказывают влияния, и только в присутствии больших количеств этих соединений выпадают белые кристаллические осадки.
Молибден (VI), уран (VI) и железо (III), а также хлораты, хлориды и фториды определению не мешают.
В присутствии восстановителей, например соли железа (II) или хлорида олова (II), желтое соединение обесцвечивается.
Влияние хроматов, манганатов и церия (IV) можно устранить путем восстановления их солью железа (II) и нитритом натрия, избыток которого затем связывают мочевиной. Органические растворители: бензол, хлороформ, диэтиловый эфир, четыреххлористый углерод и изоамиловый спирт не экстрагируют окрашенного продукта реакции ванадия с бензидином. Доп. перво. *
Другие методы
Большинство других методов определения ванадия основано на титровании его перманганатом после восстановления различными способами. Из этих методов можно указать: 1) метод, в котором ванадий восстанавливают до четырехвалентного выпариванием с соляной кислотой, лучше в присутствии железа (III) и серной кислоты. После этого к раствору прибавляют, если это нужно, серную кислоту, выпаривают до появления паров последней и титруют ванадий в сернокислом растворе 1 2>3; 2) метод, основанный на восстановлении ванадия в редукторе Джонса до двухвалентного состояния и вливания этого раствора в раствор, содержащий избыточное количество сульфата железа (III) (стр. 137) 4 *; 3) восстановление ванадия до четырехвалентного сероводородом, избыток которого удаляют кипячением, при непрерывном продувании через раствор тока двуокиси углерода 6 * * 9; 4) восстановление ванадия до четырехвалентного встряхиванием со ртутью солянокислого или сернокислого анализируемого раствора, содержащего достаточное количество хлорида натрия, чтобы связать образующуюся ртуть (I). Раствор затем фильтруют и титруют перманганатом 6; 5) восстановление перекисью водорода в горячем сернокислом
1 И. П. А л и м а р ин, ЖПХ, 17, 83 (1944).
2 Е. Campagne, С. г., 137, 570 (1903).
3 Нагревать не следует выше температуры кипения серной кислоты, и после появления ее паров выпаривание надо прекратить не позднее чем через 10 мин.
4 Соединение ванадия (II) в обычных условиях весьма легко окисляется, но
согласно A. S. Russell [J. Chem. Soc., 129, 497 (1926)], оно вполне устойчиво в холодных 10 н. растворах H2SO4.
6 G. Е. F. L u n d е 11 и Н. В. К n owl es [J. Am. Chem. Soc., 43, 1566
(1921)] установили, что при восстановлении образуются политионовые кислоты,
вследствие чего получаются немного повышенные результаты.
9 L. W. М с С а у, W. Т. Anderson, Jr., J. Am. Chem. Soc., 43, 2372 (1911); 44, 1018 (1922). При восстановлении ртутью мешают железо, сурьма и молибден, тогда как титан, мышьяк и уран не влияют на определение.
Методы определения
519
растворе 1. Интерес представляет также метод, основанный на восстановлении пятивалентного ванадия до четырехвалентного титрованным раствором соли Мора с применением гексацианоферрата (III) калия 2 или дифениламина 3 (стр. 447) в качестве внутреннего индикатора 4 5. Дифениламин является очень чувствительным индикатором для определения точки эквивалентности при реакции ванадиевой кислоты с сульфатом железа (II) в растворах, содержащих соляную или серную кислоту и [если присутствует железо (III)] фосфорную кислоту. Железо (III), мышьяк (V) и уран (VI) не влияют на определение. При титровании необходимо вводить 'небольшую поправку на то количество ванадиевой кислоты, которое расходуется на окисление индикатора.
Всем описанным выше методам мешают многие элементы, поэтому перед выполнением анализа необходимо знать, какие элементы присутствуют и в связи с этим, какие реакции могут иметь место в анализируемом растворе.
*Метод Фурмана исследовался многими авторами6. В частности, изучались свойства индикатора дифениламина и продуктов его окисления, значение фосфорной кислоты и фторидов при титровании и т. д. Установлены оптимальные условия титрования (кислотность среды, разбавление и т. д.) и разработаны многочисленные прописи применительно к анализу руд и специальных сталей различных марок. Из теоретических положений необходимо отметить лишь следующее. Роль фосфорной кислоты в растворе 6 не ограничивается обесцвечиванием присутствующего железа (III). Для получения точных результатов количество фосфорной кислоты должно по крайней мере вдвое превышать то, которое требуется для исчезновения желтой окраски. Это объясняется тем, что окислительный потенциал раствора, содержащего ионы железа (II) и (III), настолько возрастает к концу титрования вследствие резкого увеличения отношения концентраций [Fe3+]/[Fe2+], что синее окрашивание дифениламина может или появиться слишком рано (если титрование проводится бихроматом калия), т. е. до того, как все железо (II) окислилось, или исчезнуть слишком поздно (если титрование проводится солью Мора), т. е. после того, когда весь ванадий восстановится. В обоих случаях для ванадия получаются повышенные результаты. Фосфорная кислота, связывая ионы Fe3+ в комплекс, устраняет это явление.
В противоположность этому, большая величина отношения [VIT]/[Vv] в конце титрования обусловливает настолько большое значе
1 J. R. Cain, I. С. Hostetter, Ind. Eng. Chem., 4, 250 (1912).
2 С. M. Johnson, Chemical Analysis of Special Steels, 3d ed., 1920, p. 6.
3 N. H. Furman, Ind. Eng. Chem., 17, 314 (1925).
* *Титровайие ванадия может проводиться или непосредственно солью Мора или добавлением избытка соли Мора и окислением этого избытка титрованным раствором К2Сг2О7. Прим, ред.*
5 I. М. К о 11 h о f f, L. A. Sarver, J. Am. Chem. Soc., 49, 1473 (1927); Ind, Eng. Chem., Anal. Ed., 2, 125(1930); J. Am. Chem. Soc., 52, 4179 (1930); там же, 53, 2906 (1931); Z. S z e b e 11 e d у, Z. anal. Chem., 81, 97 (1930); C. Schoellen-b e r g e r, J. Am. Chem., Soc., 53, 98 (1931); H. W i 11 a r d, P. Young, Ind. Eng. Chem., 20, 764 (1928); там же, 20, 760 (1928); Ind. Eng. Chem., Anal. Ed. 4, 187 (1932); там же 5, 158 (1933); 6, 48 (1934); Ю. Ю. Лурье, Зав. лаб., 1, 34 (1932); Труды VI Менделеевского съезда, т. 2, вып. 2, 1933.
•С. Schoellenberger, цит. выше.
520
Гл. XXVI. Ванадий
ние соответствующего восстановительного потенциала, что при титровании солью Мора синее производное дифениламина восстанавливается слишком рано х, т. е. когда в растворе еще имеются ионы ванадия (V), а при титровании К,Сг2О7 — слишком поздно, т. е. когда весь избыток соли Мора уже окислился. В обоих случаях результаты для ванадия получаются пониженные. Влияние избытка ионов ванадия (IV) может быть устранено добавлением фторидов х.
На окисление индикатора расходуется около 0,05 мл 0,1 н. раствора К2Сг2О7. Ниже приводим ход анализа при определении ванадия в шлаках и специальных сталях.
Определение ванадия в шлаках 1 2
Навеску 1,5—2,0 г, в зависимости от содержания ванадия, разлагают фтористоводородной и серной кислотами в платиновом тигле и полученный раствор выпаривают досуха. Остаток сплавляют с пиросульфатом калия, плав растворяют в 2 н. серной кислоте и раствор разбавляют в мерной колбе водой до 250 мл. Отбирают 100 мл раствора и добавляют на холоду 0,1 н. раствор КМпО4 до появления неисчезающей в течение 1 мин розовой окраски. После этого вводят несколько капель разбавленной (1 : 1) соляной кислоты и кипятят 10—15 мин. По охлаждении добавляют к раствору 3 г NH4F или эквивалентное ему количество фтористоводородной кислоты, 3 капли раствора индикатора (1 г дифениламина в 100 мл концентрированной H2SO4) и титруют 0,1 н. раствором соли Мора до перехода ярко-синей окраски индикатора в зеленую. К израсходованному на титрование количеству миллилитров соли Мора делают поправку на окисление индикатора, прибавляя еще 0,05 мл. Титр раствора соли Мора устанавливают по К2Сг2О7. Навеску К2Сг2О7 растворяют в воде, добавляют серную кислоту и дифениламин и титруют раствором соли Мора. Пересчет производят на основании отношений 1Сг : 3Fe и IFe : IV.
Определение ванадия в специальных сталях и чугунах
Первый вариант2
Навеску стали 2—5 г, в зависимости от содержания ванадия, помещают в коническую колбу емкостью 500—600 мл, обрабатывают 30—40 мл разбавленной (1 : 1) серной кислоты и, прикрыв колбу часовым стеклом, нагревают до прекращения реакции. Затем добавляют по каплям азотную кислоту, стараясь не вводить большого ее избытка (обычно достаточно 2—3 мл кислоты). Кипятят до удаления окислов азота, охлаждают под краном и разбавляют до 200—250 мл. К холодному раствору прибавляют 0,1 н. раствор перманганата калия до появления неисчезающего розового окрашивания. Избыток перманганата разрушают добавлением нескольких капель HG1 и кипячением. По охлаждении добавляют 3 г фторида аммония и определяют ванадий, как в шлаках.
1 Ю. Ю. Лурье, Зав. лаб., 1, 34 (1932).
2 Ю. Ю. Л у р ь е, Зав. лаб., 1, 34(1932); Труды VI Менделеевского съезда, т. 2j вып. 2, 1933,
Методы определения 521
Второй вариант1
Обрабатывают 4—5 г стали в стакане емкостью 600 мл 30—40 мл воды и затем серной кислотой в количестве 1,5 мл на каждый грамм стали и 3 мл избытка. По разложении кипятят, выпаривают до малого объема, приливают 30—40 мл воды, нагревают до кипения и окисляют железе 3—3,5 мл азотной кислоты, избегая ее избытка. Удаляют кипячением окислы азота, охлаждают до комнатной температуры, приливают 25 мл фосфорной кислоты (пл. 1,37 г/сл«3), воды до 300 мл и 0,1 н. раствор перманганата до появления неисчезающего розового окрашивания. Прибавляют еще 3—4 капли перманганата, дают постоять 2 мин и разрушают его избыток, добавляя из бюретки 0,05 М раствор нитрита натрия до зеленого окрашивания и сверх того еще 5 мл, а затем 2 г мочевины. Перемешивают и оставляют на 5 мин, после чего вводят 15 г ацетата натрия, дают соли раствориться (если образуется осадок, его растворяют в нескольких каплях серной кислоты), добавляют 0,5 мл 0,1%-ного раствора индикатора и титруют ванадий 0,025 н. раствором сульфата железа (II). Доп. ред*
1 Н, Willard, Р. Young, Ind. Eng. Chem., Anal. Ed., 5, 154, 158 (1933)^
Глава XXVII
УРАН
Уран относится к числу редких элементов и в качестве основного компонента входит в состав лишь немногих минералов, из которых главными являются уранинит U3O8, карнотит K2(UO2)2(VO4)2 • ЗН2О ж самарскит (Fe, Са, UO2)3(Ce, Y)2(Nb, Та)6О21. Он находится почти исключительно в сильно кремнистых породах. Так, урансодержащие фосфаты — отенит Ga(UO2)2(PO4)2 • 8Н2О и торбе-н и т Cu(UO2)2 • (РО4)2 • 8Н2О встречаются в граните. То же относится к ураниниту, тогда как карнотит иногда обнаруживают в осадочных песчаниках. Следы урана присутствуют во многих минералах, особенно содержащих ниобий, тантал, торий, цирконий и некоторые редкоземельные элементы Ч
Уранинитом называется кристаллический урановый минерал, разновидности которого были названы бреггеритом, клевеитом и нивенитом еще до того, как было установлено их отношение к ураниниту. Уранинит встречается в некоторых пегматитовых жилах, насколько известно, главным образом в Норвегии и Северной Америке. Все разновидности уранинита характеризуются высоким содержанием урана в четырехвалентном и шестивалентном состояниях, содержанием редкоземельных элементов и главным образом тория и свинца. Подвергшийся наименьшему изменению минерал, найденный в Бранчвилле (Коннектикут), содержит 72% UO2 ж 13% UO3. В наиболее измененных разновидностях — клевеите и ниве-ните содержание UO2 значительно ниже, даже более низкое, чем содержание UO3. По-видимому, первоначальный минерал имел формулу UO2, подобно ThO2 в торианите.
Урановая смолка, часто неправильно называемая уранинитом, представляет собой аморфное соединение урана. Она находится в некоторых минеральных жилах совместно с сульфидными минералами и почти свободна от тория, редкоземельных элементов и гелия.
Оба эти минерала легко растворяются в азотной кислоте с окислением содержащейся в них UO2. При обработке горячей разбавленной серной кислотой они с большей или меньшей скоростью переходят в раствор с выделением азота и гелия, если эти газы присутствовали в мине-
1 М. A. Northrup {Ind. Eng. Chem., Anal. Ed., 17, 664 (1945)] приводит результаты изучения возможности использования флуоресценции фторидного королька для определения урана непосредственно в минералах, содержащих свыше 1% урана.
Общие замечания
523
рале. Ураниниты полностью разлагаются фтористоводородной кислотой, причем фторид урана (IV) выделяется в осадок совместно со фторидами свинца и редкоземельных металлов.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород поведение урана в значительной мере зависит от наличия двуокиси углерода и ванадия. В их отсутствие уран количественно осаждается аммиаком; если не ввести поправку на его содержание, точность анализа будет зависеть от метода, применяемого для определения железа. Наибольшая ошибка получается при определении железа, если последнее проводится титрованием перманганатом после восстановления цинком, который восстанавливает уран, частично даже ниже, чем до четырехвалентного состояния. Поскольку при титровании перманганатом эквивалент БегОз меньше эквивалента U3O8, то рассчитанное по разности содержание алюминия также будет не совсем точным (несколько повышенным). При восстановлении же железа сернистым ангидридом, сероводородом или хлоридом олова (II) ошибка получается только лишь в рассчитанном по разности содержании алюминия, так как уран этими реагентами не восстанавливается.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ УРАН
Большинство урановых руд разлагают обработкой азотной кислотой с добавлением в некоторых случаях соляной кислоты. При разложении руд, содержащих значительные количества кремнекислоты, если ее определение не требуется, лучше добавлять смесь соляной и фтористоводородной кислот. В тех же случаях, когда надо определять крёмнекис-лоту, нерастворимый в кислотах остаток сплавляют с карбонатом натрия.
Шведские руды разлагают обработкой концентрированной серной и фтористоводородной кислотами и нагреванием до появления паров серной кислоты. Если после вторичной обработки фтористоводородной кислотой проба полностью не растворяется, раствор разбавляют и фильтруют, а остаток прокаливают и сплавляют с пиросульфатом калия. Плав растворяют в основном растворе. Уранинит можно разложить обработкой азотной и серной кислотами Е
Титанониобаты и подобные им минералы разлагают сплавлением с пиросульфатом натрия.
О растворении карнотита см. гл. «Ванадий» (стр. 508), а методы обработки при анализе уранинита изложены в специальных статьях 2.
Для определения азота в урановых минералах Гиллебранд растворял пробу в разбавленной (1 : 6) серной кислоте при продолжительном кипячении (при 100° С) или смешивал минерал с карбонатом калия-натрия в фарфоровой лодочке (которую помещал в открытый с обоих концов цилиндр из платиновой фольги) и нагревал в трубке для сжигания, как при определении общего азота.
- 1 W. R. В е п п е t t, J. Am. Chem. Soc., 56, 277 (1934).
J W. F. Hillebrand, U. S. Geol. Survey Bull., 78 (1889—1890); Am. J. Sci., 46, 384 (1890).
524
Гл. XXVII. Уран
МЕТОДЫ ОТДЕЛЕНИЯ
Необходимо иметь в виду, что применению всех обычно принятых методов отделения урана мешают двуокись углерода и ванадий. Так, уран количественно осаждается аммиаком, свободным от карбоната аммония, но если аммиак поглотил некоторое количество двуокиси углерода из воздуха, уран осаждается только частично и совершенно не выделяется растворами карбоната аммония. Подобно этому, уран полностью' осаждается едким натром, свободным от карбоната натрия, но при условии, если отсутствует ванадий. В присутствии ванадия уран осаждается не количественно или вовсе не выделяется из раствора. Большинство операций отделения урана целесообразно повторять 2 или 3 раза.
Важнейшие методы отделения урана основаны на применении карбонатов щелочных металлов. Такие элементы, как железо и алюминий, отделяются от урана осаждением карбонатом аммония. Эта операция всегда проводится в горячих растворах, причем необходимо следить за тем, чтобы карбонат аммония полностью не разрушился и уран не выделился из раствора. При использовании карбоната натрия или карбоната калия для лучшего отделения от указанных элементов целесообразно вводить в раствор также и перекись натрия.
Уран можно отделить от элементов сероводородной группы осаждением последних сероводородом из кислого раствора, содержащего винную кислоту, а от элементов группы мышьяка — осаждением урана сульфидом аммония. В присутствии карбоната аммония уран сульфидом аммония не осаждается, что используется для отделения от него железа, алюминия, титана и других элементов. В виде сульфида уран не осаждается также из растворов, содержащих тартрат и сульфид аммония, и отделяется таким образом от железа, кобальта и других элементов.
Рекомендуется1 отделять уран от фосфатов и арсенатов следующим способом. Выделяют кремнекислоту выпариванием раствора с соляной кислотой, затем к слабокислому раствору прибавляют избыток гексацианоферрата (II) калия и насыщают хлоридом натрия. Загрязненный осадок гексацианоферратов промывают декантацией раствором хлорида натрия, затем переносят на фильтр и тщательно промывают тем же-раствором. Гексацианоферраты (II) разлагают обработкой на холоду разбавленным раствором едкого кали. Осадок гидроокисей промывают водой, содержащей немного аммиака и хлорида аммония, один раз декантацией, а затем на фильтре до тех пор, пока в подкисленной промывной жидкости не исчезнет реакция на гексацианоферрат-ионы. Гидроокиси, свободные от фосфора и мышьяка, растворяют в соляной кислоте, раствор, если нужно, упаривают, нейтрализуют аммиаком почти до нейтральной реакции и прибавляют небольшой избыток карбоната аммония. Дают постоять, затем осадок отфильтровывают и промывают водой, содержащей карбонат аммония. Фильтрат и промывные воды нагревают до разложения большей части карбоната аммония, затем подкисляют и полностью удаляют двуокись углерода кипячением. Кипящий раствор насыщают сероводородом, осадок отфильтровывают и в фильтрате осаждают уран обычными способами.
*Предложен метод осаждения урана сероводородом в присутствие гексаметилентетрамина 2. При этом выделяется кристаллический осадок непостоянного состава, который легко отделяется фильтрованием; этот осадок отличается незначительной адсорбционной способностью. Таким образом происходит хорошее отделение урана от щелочных и щелочно
1 R. Fresenius., Е. Huntz, Z. anal. Chem., 34, 437 (1895).
’ Э. А. Остроумов, P. И. Бомштейн, Зав. лаб., 8, 558 (1939).
Методы отделения
525
земельных металлов. Осаждение проводится в следующих условиях. Раствор соли уранила, почти нейтрализованный раствором аммиака, нагревают до 60° С и, прибавив 2 г гексаметилентетрамина, насыщают •сероводородом в течение 15 мин. Затем снова нагревают и, часто встряхивая колбу, продолжают пропускать струю сероводорода еще 15—20 мин. После отстаивания в течение 15 мин осадок отфильтровывают и промывают 3%-ным раствором нитрата аммония, который подщелачивают раствором аммиака. Доп. перев.*
Интересный способ отделения некоторых элементов от урана (VI) основан на применении в качестве осадителя купферона. Уран (IV) также осаждается купфероном, и его в свою очередь можно отделить таким способом от целого ряда других элементов. Так, например, ванадий, 7келезо, титан и цирконий легко отделяются от урана (VI) осаждением их купфероном из сернокислого раствора (стр. 143), а затем, восстановив в фильтрате уран до четырехвалентного, можно выделить его тем же реагентом. При этом уран отделяется от алюминия, хрома, марганца и фосфора.
Уран количественно отделяется от тантала, ниобия и титана при осаждении этих элементов таннином из полунасыщенного хлоридом аммония слабокислого раствора, содержащего оксалат х. Полученный осадок отфильтровывают и затем выделяют уран из фильтрата, прибавив к нему еще некоторое количество таннина и сделав его аммиачным.
От больших количеств железа уран можно отделить экстракцией солянокислого раствора эфиром 1 2 (стр. 161) и от железа и хрома электролизом с ртутным катодом в разбавленном сернокислом растворе (стр. 165).
Уран (IV) образует нерастворимый фторид и может быть таким образом отделен от урана (VI) (стр. 622) *. Это свойство было использовано для раздельного определения урана (IV) и урана (VI) в минералах 3. Авторы нашли, что двойной фторид урана (IV) и аммония значительно менее растворим, чем фторид урана (IV), и практически совсем не растворяется в присутствии избытка фторида аммония. Осаждение урана (IV) рекомендуется проводить в ацетатной среде избыточным количеством фторида аммония. Доп. перев*
Для разделения урана (IV) и урана (VI) можно применить также осаждение гипофосфорной кислотой Н4Р2О6 из раствора, содержащего свободную минеральную кислоту 4.
Методы отделения урана от ванадия описаны в гл. «Ванадий» (стр. 510), от редкоземельных элементов на стр. 622 и от ниобия и тантала на стр. 666.
Для отделения бериллия от урана предложено 5 использовать осаждение карбонатом аммония и солянокислым гидроксиламином, которое проводится следующим образом. К солянокислому раствору, содержащему бериллий и уран, прибавляют 5 г хлорида аммония, 5 г солянокислого гидроксиламина и затем концентрированный раствор карбоната •аммония до тех пор, пока образующийся вначале осадок полностью не растворится. Нагревают раствор до кипения и после появления тяжелого
1 W. R. Schoeller, Н. W. Webb, Analyst, 58, 143 (1933).
2 Е. F. Kern, I. Am. Chem., Soc., 23, 689 (1901).
3 В. Г. X л опин, Э. К. Г е р и н г, ЖОХ, 6, 1701 (1936).
4 F. Н е с h t, Н. К г a f f t-E b i n g, Z. Anal. Chem., 106, 321 (1936).
s P. H. Br int on, R. В. E 11 e s t a d, J. Am. Chem. Soc., 45, 395 (1923).;
526
Гл. XXVII. Уран
белого осадка основного карбоната бериллия продолжают кипятить-30—60 сек. Слишком продолжительного кипячения следует избегать. Осадок тотчас же отфильтровывают и тщательно промывают холодной водой. Свободный от урана осадок сохраняют и присоединяют к нему небольшое количество гидроокиси бериллия, которое дополнительно извлекают из фильтрата. Для этого фильтрат подкисляют соляной кислотой и кипятят до удаления двуокиси углерода. Затем вводят 1 г солянокислого гидрокси ламина, охлаждают и прибавляют раствор аммиака в небольшом избытке. Холодный раствор фильтруют и осадок гидроокиси бериллия промывают 2 %-ным раствором нитрата аммония, к которому добавлено несколько кристалликов солянокислого гидррксиламина и аммиак до щелочной реакции. Если осадок имеет желтый оттенок, его растворяют в соляной кислоте и переосаждают. Объединенные осадки бериллия прокаливают до ВеО.
Для осаждения урана фильтраты, если осаждение бериллия проводилось двукратно, объединяют, слегка подкисляют соляной кислотой и окисляют гидроксиламин обработкой 75—100 мл 3%-ной перекиси водорода при кипячении до прекращения выделения кислорода и уменьшения объема раствора до одной трети. Если при этом образуется осадок, его растворяют в соляной кислоте. Окисление гидроксиламина можно, осуществить также добавлением твердого бромата калия или бромата натрия до начала выделения свободного брома, который затем удаляют-кипячением. Уран осаждают аммиаком, как описано в разделе «Весовое определение после осаждения аммиаком».
Уран можно также отделить почти от всех посторонних элементов экстракцией высушенных нитратов диэтиловым эфиром или некоторыми другими органическими растворителями Ч
МЕТОДЫ ОПРЕДЕЛЕНИЯ 2
Весовой метод, согласно которому уран осаждают в виде ураната аммония (NH4)2U2O7 и взвешивают в виде закиси-окиси урана U3O8, а также метод, основанный на восстановлении урана цинком и титровании перманганатом, дают достаточно точные результаты, если соблюдать некоторые несложные предосторожности. При использовании этих методов для анализа сложных продуктов основная задача сводится к отделению мешающих элементов, как, например, ванадий и железо, которые и в том и в другом методе препятствуют определению урана.
Наиболее быстрый способ определения урана, так же как и тория, основан на природной радиоактивности этих элементов. К сожалению, способ этот применим только в том случае, если в анализируемом материале содержится или один уран или только торий. Кроме того, необходимо, чтобы определяемый элемент находился в равновесии с продуктами его радиоактивного распада, что имеет место не во всех рудах и мине---------- i
ЛЕ. Р е 1 i g о t, Ann. chim. phys., [3], 5, 5 (1842); W. F. H i 11 e b г a n d, U. S. Geol. Survey Bull., 78, 47 (1891); R. C. W e 11 s, J. Wash'., Acad. Sci., 20, 146 (1930); J, I. Hoffman., там же, 38, 233 (1948).
’Подробный обзор методов определения урана, разработанных во время второй мировой войны, приводит С. J. Rodden, Analytical Chemistry of the Manhattan Project, McGraw-Hill Book Co., 1950, p. 3—159. * Аналитическая химия урана и тория, Издатинлит, 1956.*
Методы определения
527
ралах. Это равновесие, например, не достигнуто в содержащих карнотит песчаниках Колорадо. Из этих соображений, а также и в связи с тем, что для выполнения радиоактивных измерений требуется дорогостоящее оборудование и квалифицированный персонал, радиометрические методы здесь не приводятся 1;
Весовое определение после осаждения аммиаком
Определение урана взвешиванием в виде U3O8 после осаждения аммиаком дает достаточно точные результаты, вопреки многим исследованиям, в которых указывается на ряд недостатков этого метода. Основной йеудобство метода заключается в том, что его не всегда можно применить непосредственно вследствие наличия других, осаждающихся аммиаком элементов 2. Предосторожности, которые следует соблюдать, сводятся главным образом к удалению органических веществ и карбонатов, препятствующих выделению урана аммиаком. Для получения U3O8 вполне достаточно прокаливания осадка в хороших окислительных условиях и нет необходимости в конечном прокаливании его в атмосфере кислорода или переведении в UO2 нагреванием в токе водорода, тем. более что полное восстановление до UO2 в тигле практически не выполнимо.
Проведенные опыты3 показали, что оеадки, выделенные из аликвотных частей стандартного раствора урана, весили 0,1138 и 0,1138 г после прокаливания на воздухе,. 0,1136 и 0,1136 г после прокаливания в кислороде и 0,1137 и 0,1136 г после того, как прокаленные на воздухе осадки были растворены в азотной кислоте, растворы выпарены досуха и остатки снова прокалены на воздухе. Результаты определения UOa во взвешенных осадках U3O8 показали, что содержание ее может отклоняться от теоретического в пределах 1 мг. Такое отклонение вызывает колебание в массе U3O8 менее чем на 0,1 мг, что при обычных взвешиваниях не принимается во внимание. После осторожного прокаливания в атмосфере водорода масса осадков составляла 0,1118 и 0,1115 г, что соответствует восстановлению до UO3 примерно на две третьих.
Ход определения. В разбавленный сернокислый раствор, содержащий не более 1 % сульфата уранила и свободный от двуокиси углерода, органических веществ и элементов, осаждающихся аммиаком или образующих в аммиачной среде соединения с ураном, вводят несколько капель раствора метилового красного и нагревают до кипения. Затем прибавляют разбавленный раствор аммиака, пока окраска индикатора не станет отчетливо желтой,' вводят мацерированную бумагу в количестве, соответствующем половине 9-сантиметрового фильтра, и перемешивают. Осадок отфильтровывают и промывают горячим 2 %-ным раствором нитрата аммония 4. Высушивают в платиновом тигле, прокаливают в хороших окислительных условиях, сначала при возможно более низкой температуре до сгорания углерода, а затем, поместив тигель в наклонном
1 См. С. J. R odden, Anal. Chem., 21, 331 (1949).
2 Часто повторяемые утверждения, что уран имеет склонность захватывать натрий, лишены основания. Так, например, Н. В. Knowles в растворах, содержащих в 100 мл по 5 г NaCl и 0,2389 г U3O8 (в виде нитрата), определил 0,2386; 0,2386 и 0,2384 г U3O8 соответственно однократным, двукратным и трехкратным осаждением аммиаком.
3 G. Е. F. L u n d е 11, Н. В. К п о w 1 е s, J. Am. Chem. Soc., 47, 2637 (1925).
4 Согласно Kin'ichi Someya [Sci. Rep. Toboku Imp. Univ., 15 [1], 411 (1926); Z. anorg. Chem., 152, 378 (1926)], можно получить кристаллический осадок и избежать введения мацерированной бумаги, если прибавить аммиак до оранжево-красной окраски индикатора, затем раствор прокипятить 10 мин и прибавить еще аммиак до отчетливо желтой окраски индикатора.
528
Гл. XXVII. Уран
положении, прокаливают на полном пламени горелки Теклю (1000° С). Под конец тигель закрывают и прокаливают еще 1 мин. Прокаленный «статок охлаждают в эксикаторе и взвешивают в виде UsOg.
Теоретически взвешенный остаток ПзО8 должен иметь состав UO2 2иОз. Для проверки полученных результатов целесообразно определить в этом остатке содержание UO2 и общего количества урана, особенно если в исходном растворе находились элементы, которые могли полностью не отделиться от урана и загрязнить таким образом осадок. Эти определения можно осуществить следующим способом. Взвешенный прокаленный остаток в платиновом тигле обрабатывают на холоду 10 мл фтористоводородной кислоты. Разбивают комочки остатка платиновой или бакелитовой палочкой и перемешивают 3—5 мин. Полное растворение не происходит вследствие образования нерастворимого фторида урана (IV). Раствор обрабатывают 10—20 мл разбавленной (1 :1) серной кислоты, перемешивают в течение нескольких секунд и затем переносят в стакан, содержащий 100 мл холодного насыщенного раствора борной кислоты. Перемешивают до растворения солей и титруют раствором перманганата. Вычитают количество перманганата, расходуемое на окрашивание раствора после достижения точки эквивалентности, и вычисляют содержание UO2, считая, что при титровании он окисляется до UOs.
Оттитрованный раствор переносят в платиновую чашку и выпаривают до появления паров серной кислоты. По охлаждении разбавляют водой с таким расчетом, чтобы концентрация серной кислоты в растворе составляла 5% по объему, и определяют общее количество урана объемным методом, описанным в разделе «Объемное определение восстановлением цинком и титрованием перманганатом» (стр. 529). Если при определении UO2 для нахождения поправки (количества перманганата, затраченного на окрашивание раствора) был введен сульфат железа (II), то эквивалентное ему количество перманганата следует вычесть из общего количества перманганата, израсходованного в данном случае на титрование всего урана. Кроме того, нужно ввести поправку на количество перманганата, которое затрачивается на титрование реактивов и на окрашивание анализируемого раствора в конечной точке титрования. Общее содержание урана вычисляют, исходя из того, что UO2 окисляется до UOs. Теоретически объем раствора перманганата, необходимый для окисления общего количества урана, должен в 3 раза превышать объем, израсходованный на титрование UO2. Принимая во внимание высокий эквивалентный вес урана, весьма важно, чтобы как при первом, так и при втором титровании были внесены все указанные поправки.
Способ установления величины поправок при определении общего содержания урана изложен в разделе «Объемное определение восстановлением цинком и титрованием перманганатом» (стр. 529). Расход перманганата на окрашивание раствора в точке эквивалентности при первом титровании не постоянен и, как правило, больше, чем при втором титровании. Величина этой поправки в обоих случаях может быть найдена одним и тем же методом, только при первом титровании объем вводимого для этого раствора сульфата железа (II) следует тщательно измерить, чтобы можно было ввести поправку на содержание- железа в растворе при определении общего количества урана.
Содержание UO2 в осадке U3O8 можно определить также титрованием перманганатом после разложения осадка обработкой разбавленной (1 : 6)
Методы определения
529
серной кислотой при 150—175° С в запаянной трубке, из которой воздух вытеснен двуокисью углерода *.
Вполне вероятно, что при испытании на чистоту взвешенного U3O8 приведенными выше способами результаты определения UOa соответствуют истинному содержанию ее в прокаленном остатке. Общее количество урана также определяется правильно, если находящиеся в рас-створе примеси в процессе восстановления урана не восстанавливаются, как, например, алюминий или цирконий. В случае же, когда примеси восстанавливаются вместе с ураном и затем окисляются перманганатом, как, например, примеси ванадия и титана, результаты определения общего содержания урана получаются повышенными и превышающими возможное содержание урана в осадке, если этот осадок свободен от других загрязнений, не окисляющихся перманганатом в условиях титрования урана.
По-видимому, во многих случаях соотношение между UIV и UVI в прокаленном остатке зависит от природы находящихся в нем примесей, так как мало вероятно, например, чтобы такие элементы, как алюминий и титан, не оказывали влияния на восстановление урана в процессе прокаливания.
* Э. А. Остроумов 1 2 3 рекомендует осаждать уран пиридином, что имеет известные преимущества перед осаждением аммиаком. Пиридин, являясь слабым основанием, не поглощает двуокись углерода, благодаря чему ибключается возможность неполноты выделения урана и соосажде-ния карбонатов щелочноземельных металлов. При осаждении пиридином происходит отделение урана от щелочноземельных и щелочных металлов, магния, кобальта и никеля. В этих условиях выделяются металлы, гидролитическое осаждение которых происходит при pH меньше 6, в частности цирконий, титан, железо, хром, алюминий.
Большие количества сульфата мешают осаждению урана.
При определении к растйору, содержащему 5—10 г нитрата аммония, при перемешивании прибавляют 20 %-ный раствор пиридина до нейтральной реакции по метиловому красному, после чего вводят избыток пиридина в 10 мл. Раствор нагревают, и если он при этом становится кислым по индикатору, прибавляют еще пиридин. Осадок отфильтровывают и промывают 3 %-ным раствором нитрата аммония, содержащим немного пиридина. Доп. перев.*
Объемное определение восстановлением цинком и титрованием перманганатом
Амальгамированным цинком в кислых растворах уран частично восстанавливается до трехвалентного, а в основном до четырехвалентного состояния, причем достигнуть определенного соотношения между образующимися количествами UIV и Uni не удается При действии воздуха,
1 W. F. Hillebrand, U. S. Geol. Survey Bull., 78, 47 (1891).
3 Э. А. О с т р о у м о в, Зав. лаб., 6, 16 (1937); Z. anal. Chem., 106, 244 (1936).
3 Согласно Kin'ichi Someya [Sci. Rep. TShoku Imp. Univ., 15 (1), 411 (1926); Z. anorg. Chem., 152, 378 (1926)], уран легко восстанавливается до трехвалентного цинковой амальгамой в 2—6 н. солянокислом растворе и точно до четырехвалентного висмутовой амальгамой в 6—10 н. НС1 или 10—12 н. растворе H2SO4. Алюминием в разбавленных сернокислых растворах уран также восстанавливается до четырехвалентного состояния [Н. V. Churchill, R. W. Bridges, A. L. Miller, Ind. Eng. Chem., Anal. Ed., 8, 348 (1936)].
34 Заказ 522.
530
Гл. XXVII. Уран
однако, уран весьма быстро полностью переходит в четырехвалентное состояние. Для этого достаточно, например, раствор, содержащий 0,25 г урана, после восстановления перемешивать на воздухе или пропускать через него струю воздуха в течение нескольких минут. Уран (IV) вполне устойчив в холодных сернокислых растворах, а в горячих растворах медленно окисляется х. На холоду окисления урана (IV) не наблюдается, даже если оставить раствор на воздухе на 4 ч, изредка его перемешивая, или пропускать через раствор струю воздуха в течение 25 мин. Таким образом, если после восстановления урана в сернокислом растворе амальгамированным цинком при комнатной температуре раствору дать постоять достаточное время на воздухе, уран можно определить оксидиметриче-ским методом с не меньшей точностью, чем ванадий, титан, железо и молибден.
Ход определения. К раствору, содержащему 5% (по объему) серной кислоты и не более 1 % урана и свободному от элементов, восстанавливающихся в редукторе Джонса а, прибавляют перманганат до появления, устойчивой розовой окраски. Раствор охлаждают до 20—25° С и пропускают через редуктор Джонса (стр. 135) со скоростью 50—100 мл в минуту. Затем через раствор продувают очищенный воздух в течение 5 мин или же сливают раствор в. фарфоровую чашку и перемешивают 5 мин. Титруют раствором перманганата, титр которого установлен по чистому оксалату натрия (стр. 214), и вычитают то количество перманганата, которое расходуется на титрование реактивов и на окрашивание желтого раствора в точке эквивалентности. 1 мл 0,1 н. раствора перманганата соответствует 0,01191 г урана.
Для определения количества перманганата, расходуемого на реактивы, пропускают раствор применяемых реактивов через редутор,, затем титруют его перманганатом и вычитают незначительный объем раствора перманганата, который затрачивается на окрашивание такого же объема раствора. Количество пермангайата, которое расходуется на окрашивание анализируемого раствора в точке эквивалентности, устанавливают следующим образом. В оттитрованный раствор вводят несколько капель 0,1 н. раствора сульфата железа (II), прибавляют 0,5—1 г персульфата аммония, перемешивают 1 мин и затем титруют раствором перманганата до появления розовой окраски такой же интенсивности, какая была при титровании урана. О титровании урана бихроматом см. ссылку в сноске 2' на стр. 526.
1 G. Е. F. L u n d е 11, Н. В. Knowles [J. Am. Chem. Soc., 47, 2637 (1925)]. Согласно G. Vort m a n n и F. Binder [Z. anal. Chem., 67, 269 (1925)], сернокислый раствор U(SO4)2 достаточно устойчив для применения его в качестве титрованного'-раствора восстановителя.
> Азотная кислота должна быть полностью удалена, в противном.случае в редукторе образуются восстановители; на которые затем расходуется перманганат. Согласно J. A. Holladay и Т. R. Cunningham [Trans. Am. Electrochem. Soc., 43, 329 (1923)], азотная кислота прочно удерживается ураном и повторным выпариванием с серной кислотой до появления ее паров полностью удалить азотную кислоту не удается. Они рекомендуют избегать введения азотной кислоты и переводить соединения урана в раствор обработкой одной серной кислотой или сплавлением с пиросульфатом. Азотную кислоту, вероятно, можно удалить осторожным введением раствора перманганата в горячил концентрированный сернокислый раствор, как это рекомендуется при определении молибдена' (стр. 362).
Методы определения
531
* Описаны методы,, основанные на титровании восстановленного урана раствором ванадата в присутствии фенилантраниловой кислоты в качестве индикатора 1 и на титровайии урана (VI) раствором сульфата титана (III) с применением в качестве индикаторов красителя новой синей Д (с), вольфрамата натрия или сафранина 2. Доп. перев.*
Весовое определение после осаждения купфероном
В разделе «Методы отделения» (стр. 524) было указано, что в солянокислых и сернокислых растворах купферон образует нерастворимое соединение с ураном (IV). Уран (VI) при этом не осаждается. Поэтому в некоторых случаях целесообразно определять уран следующим образом. Сначала проводят осаждение купфероном из раствора, содержащего уран в шестивалентной форме. Осадок отфильтровывают и в фильтрате, после разрушения купферона и восстановления цинком, как это описано в разделе «Объемное определение восстановлением цинком и титрованием перманганатом» (стр. 529), осаждают уран (IV) купфероном. Таким путем железо, ванадий, титан и цирконий отделяются от урана, а затем уран в свою очередь отделяется от алюминия и фосфора. Хром (II) также частично осаждается купфероном, но его влияние можно устранить, подвергнув раствор действию воздуха, как указано выше (стр. 529).
\
\ Другие методы
Из других методов определения урана следует указать на взвешивание в виде Na2U2O7, (UO2)2P2O5 и V2O5 • 2UO3. Осаждение урана едким натром служит лишь для его отделения, так как при определении урана взвешиванием в виде диураната натрия, как правило, получаются повышенные результаты 3. Определение урана в виде фосфата не имеет никаких преимуществ перед взвешиванием его в виде U3O8 и применяется лишь в рядовых анализах, чтобы избежать предварительного отделения ванадия 4. То же относится и к осаждению урана в виде аммоний-уранил-ванадата с последующим прокаливанием до V2O8 • 2UO3.
В расплавленном фториде натрия под действием ультрафиолетовых лучей уран дает интенсивную и хорошо воспроизводимую желтую флуоресценцию. Методы, основанные на этой флуоресценции, с успехом используются для определения следов урана в бедных рудах, песках, минералах и других материалах. Поскольку железо, медь, хром и марганец понижают интенсивность -флуоресценции, уран обычно предварительно отделяют от других компонентов пробы экстракцией нитрата уранила соответствующими органическими растворителями, которые затем удаляют выпариванием. Сухой остаток сплавляют с фторидом натрия и карбонатом калия в золотой чашке. Плав извлекают из чашки, подвер
1 В. С. Сырокомский, Ю. В. Климе н-к о, Зав. лаб., 9, 1077 (1940).
1 В. Г. X л о п и н, Л. Э. К а у ф м а н, Редкие элементы, вып. 6 (1934).
9 Согласно И. Т. S. В г i 11 о n [J. Chem. Soc., 127, 2110 (1925)], осадок, выделяемый едким натром, состдит в основном из гидроокиси, а не из диураната натрия.
4 Количественное разделение уранила и ванадия не достигается даже переосажде-нием фосфата. Так, после двукратного осаждения в трех аликвотных частях раствора, содержащего только нитрат уранила, найдено 0,3052; 0,3056 и 0,3058 г
а в других трех порциях раствора, в которые было введено по 0,2 г ванадия в виде сульфата, определено 0,3081; 0,3102 и 0,3147 г (UO2)2P2O7.
34*
532
Гл. XXVII. Уран
гают действию ультрафиолетового света и интенсивность флуоресценции измеряют электрофотометром х.
Урановые соли, активированные облучением их ультрафиолетовыми лучами с длиной волны ниже 3000 А, также обнаруживают интенсивную флуоресценцию в растворах.' На этом свойстве основан количественный метод определения урана 1 2.
Небольшие количества урана можно определить колориметрически, способом, подобным способу определения хрома (стр. 595) в растворах, содержащих едкий натр и перекись натрия, сравнением окраски раствора с окраской обработанного таким же способом стандартного раствора урана 3 или фотометрическим измерением светопоглощения раствора при 425 ммк 4.
*Пероксидный метод определения урана впервые был предложен И. Е. Стариком s; метод основан на реакции урана с перекисью в карбонатной среде ®. Рекомендуемый автором метод сводится к следующему. Из раствора, освобожденного от всех элементов, кроме щелочных и щелочноземельных металлов, осаждают уран аммиаком, не содержащим карбонатов. Осадок растворяют в небольшом количестве 5%-ной азотной кислоты, избыток которой удаляют выпариванием на водяной бане. Остаток растворяют в 1 н. растворе карбоната натрия или калия. Раствор переносят в мерную колбу емкостью 10—25 мл, прибавляют перекись натрия в количестве 0,2—0,5 г, в зависимости от содержания урана, и разбавляют до метки тем же раствором карбоната. Окраску раствора сравнивают в колориметре с окраской стандартного раствора урана, подготовленного таким же способом.
Наиболее удобной для колориметрирования концентрацией урана является 0,1 мг в 1 мл.
Этот метод не отличается большой чувствительностью (предел чувствительности метода 0,01 % урана), но применению его мешает относительно небольшое число элементов. Основными элементами, влияющими на определение урана, являются, помимо хрома, молибден (VI) и ванадий (V), которые также дают окраску с перекисью водорода в карбонатной среде, хотя значительно менее интенсивную, чем уран. Имеются указания 4 на то, что ванадий не мешает колориметрированию урана в растворе, содержащем едкий натр и перекись натрия. Значительное влияние оказывает марганец, что обусловлено заметной окклюзией урана двуокисью марганца и каталитическим разложением перекиси. Большие количества железа также каталитически разлагают перекись; кроме того, выделяющимся осадком захватывается некоторая часть урана. Для исключения мешающего влияния железа колориметрирование рекомендуется осуществлять в аммиачной среде в присутствии тартрата.
Определение урана фотометрическим методом в карбонатной среде рекомендуется проводить в следующих условиях. К 15 мл нейтрализован
1 М. A. Northrup, Ind. Eng. Chem., Anal. Ed., 17, 664 (1945); С. E. W h i t e, Anal. Chem., 21, 104 (1949).
2 C. W. S i 11, H. E. P e t e r s о n, Anal. Chem., 19, 646 (1947).
8 О мешающих определению элементах см. G. Е. F. L u n d е 11, J. I. Hof f-m a n, Outlines of Methods of Chemical, Analysis, 1938, p. 172.
4 См. сноску 2 на стр. 481.
6 См. в книге: «Анализ минерального сырья», под ред. Б. Т. Карпова, Ю. Н. Книпович и Ю. В. Морачевского, ОНТИ, Химтеорет, 1936.
8 A. Rosenheim, Н. Daehr, Z. anorg, Chem., 181, 177 (1929),
Методы определения
533
ного раствора, содержащего 0,2—0,8 JteU3O8, прибавляют 5 мл 10 %-ного раствора карбоната натрия, вводят 2 мл 3%-ной перекиси водорода и тщательно перемешивают. Разбавляют раствор до 25 мл и измеряют светопоглощение при 450 ммк.
Помимо пероксидного, описан еще ряд других колориметрических методов определения урана, из которых можно указать следующие.
Гексацианоферратный метод х. Для определения малых количеств урана в бедных рудах Ю. А. Чернихов и Е. И. Гульдина разработали колориметрический метод 1 2, основанный на реакции урана с гексацианоферратом (II). Отделение урана от железа и других металлов, дающих с гексацианоферратом (II) окрашенные растворы или нерастворимые соединения, осуществляется электролизом с ртутным катодом. Из раствора после электролиза [реакция на железо с K3Fe(GN)e должна быть отрицательной] осаждают уран свободным от карбонатов раствором аммиака в присутствии небольшого количества перекиси водорода. Отфильтрованный осадок промывают горячим 3 %-ным раствором сульфата аммония, содержащим несколько капель раствора аммиака, и затем растворяют в 10 мл горячей 2 %-ной (по объему) серной кислоты. Раствор разбавляют в мерной колбе до 100 мл водой, а в случае содержания ванадия уран переосаждают в виде фосфата. Для этого раствор нейтрализуют аммиаком до появления слабой мути, которую растворяют в нескольких каплях 1 н. раствора серной кислоты, разбавляют до 40 мл и прибавляют 5 мл 6 н. раствора уксусной кислоты и 15 мл 1/з М раствора NazHPCh.
Если раствор содержит очень мало алюминия, перед осаждением прибавляют 1—2 мл раствора алюминиевых квасцов (5 мг АЬОз в 1 мл). Нагревают до кипения, дают осадку отстояться 10 мин, отфильтровывают его и промывают 5—6 раз 1 н. раствором нитрата аммония. Осадок фосфатов растворяют в 10 мл горячей 2 %-ной (по объему) серной кислоты и затем в мерной колбе разбавляют раствор водой до 100 мл.
Для колориметрирования отбирают 10 мл раствора в градуированную пробирку и прибавляют 5 З1л10%-ного раствора K4[Fe(CN)e], содержащего 1% КагЭОз. Окраску раствора через 5—10 мин сравнивают со шкалой стандартных растворов, для приготовления которой в серию пробирок вводят строго отмеренные объемы типового раствора урана 3, содержащего в 1 мл 0,1 мг U3O8. Растворы в пробирках разбавляют до 10 мл 0,2 %-ной (по объему) серной кислотой и одновременно с анализируемым раствором вводят в них по 5 мл раствора гексацианоферрата (II).
Результаты изучения 4 реакции урана с гексацианоферратом (II) показали, что ее чувствительность в большой степени зависит от pH раствора. Установлено, что наибольшая интенсивность окраски достигается при pH от 4 до 6. Соответствующую концентрацию ионов водорода
1 J. Tschernichow, Е. Guldina, Z. anal. Chem., 96, 17 (1934); см. также в книге В. Гиллебранд, Г.Лендель. Практическое руководство по неорганическому анализу, ОНТИ, 1935, стр. 410.
2 A. Bruttini, Gazz. chim. ital., 23, 251 (1893); Z. anal. Chem., 44, 432 (1905).
8 Из соли урана (свободной от железа) приготовляют сначала раствор, содержащий в 1 мл 1 мг U3O8. Титр раствора устанавливают весовым путем, осаждением аммиаком (свободным от карбонатов) в'присутствии перекиси водорода. Отбирают 50 мл этого раствора и осаждают аммиаком, как при подготовке раствора для колориметрирования. Промытый осадок растворяют в 50 мл 2 %-ной (по объему) серной кислоты и полученный раствор разбавляют в мерной колбе водой до 500 мл. Этот раствор служит для приготовления шкалы стандартных растворов.
4 См. сноску 2 на стр. 526.
534
Гл. XXVII. Уран
в растворе рекомендуется устанавливать введением формиатного буферного раствора. Другие буферные растворы, как, например, ацетатный, тартратный и цитратный, понижают интенсивность окраски, возникающей при реакции урана с гексацианоферратом (II). Такое же действие оказывают и анионы, образующие комплексные соединения с ураном, например карбонаты и фториды. В присутствии формиатного буферного раствора устойчивость окраски повышается.
Колориметрирование в присутствии формиата натрия рекомендуется проводить следующим способом. Аликвотную часть раствора, содержащую 0,05—1,1 мг урана, выпаривают досуха, избегая кипения. По охлаждении вводят 0,5 мл разбавленной (1 : 6) серной кислоты и нагревают до прекращения выделения ее паров. Охлаждают и обрабатывают остаток дистиллированной водой (не более 30 мл). Прибавляют несколько капель серной кислоты, нагревают 10 мин и вводят 10 мл формиатного буферного раствора х.
При этом величина pH раствора должна стать равной 5,0 ± 0,2. Если кислотность раствора выше, прибавляют соответствующее количество разбавленного (1 : 6) раствора'аммиака. После этого раствор фильтруют в мерную колбу емкостью 100 мл, тщательно ополаскивают стакан и промывают фильтр. Объем полученного раствора не должен превышать 65 мл. Прибавляют точно 25 мл 10%-ного раствора гексацианоферрата (II) калия и разбавляют раствор до метки дистиллированной водой. Нулевой раствор приготовляют разбавлением 25 мл раствора гексацианоферрата (II) и 10 мл буферного раствора до 100 мл в мерной колбе. Измерение интенсивности окраски раствора проводят в фотоколориметре с синими светофильтрами, не позднее чем через 15 мин после приготовления раствора. Содержание урана вычисляют по калибровочной кривой, которую следует возобновлять с каждым вновь приготовленным раствором гексацианоферрата (II).
Метод с Z-аскорбиновой кислотой. Уран в ацетатном растворе дает красно-коричневую окраску с Z-аскорбиновой кислотой 1 2. Окраска эта устойчива, и на реакцию мало действуют такие обычно мешающие колориметрированию ионы, как Fe3+, Сг3+ и РЬ2+. Влияние меди устраняется введением пиридина. Наиболее благоприятным является pH = ~4,6, который устанавливают добавлением ацетатного буфера. В отсутствие ацетата окраска более интенсивна, но неустойчива. Вредное влияние оказывают сульфат-, фторид- и фрсфат-ионы. Метод применяется после экстракции нитрата урана эфиром. В полученный экстракт вводят 10 мл воды и удаляют эфир выпариванием. К водному слою прибавляют 8 мл буферного раствора (160 мл 1 н. раствора ацетата аммония, 10 мл 5 н. уксусной кислоты и 180 жл. воды), 2 мл 10%-ного раствора пиридина и 10 мл 12%-ного раствора Z-аскорбиновой кислоты. Раствор, pH которого доводят до 4,6 ± 0,1 аммиаком или соляной кислотой, разбавляюгт до 50 мл и измеряют светопоглощение при 410 ммк. Содержание урана вычисляют по калибровочной кривой. ,
Метод с диэтилдитиокарбаматом натрия. В нейтральных растворах с диэтилдитиокарбаматом натрия уран образует устойчивое, окрашенное
1 Для приготовления формиатного буферного раствора 68 г формиата натрия растворяют в 500 мл воды, прибавляют 2 мл муравьиной кислоты, перемешивают и доводят pH раствора до 5,0, прибавляя формиат натрия или муравьиную кислоту.
2 См. сноску 2 на стр. 526.
Методы определения
535
в желтый цвет комплексное соединение, которое используется для его колориметрического определения х. Соединение это нерастворимо в четыреххлористом углероде, но растворяется в этилацетате и в эфире, хотя еще лучшим растворителем является хлороформ х. Колебание величины pH в пределах 6—8 не влияет на оптическую-плотность раствора. Определению мешает большое число элементов, как например, железо, медь, никель, кобальт и другие, от которых уран перед его определением должен быть отделен.
Хлориды, нитраты и сульфаты не влияют.на определение, а фосфаты вызывают осаждение урана, который затем не экстрагируется диэтилдитиокарбаматом натрия и хлороформом.
Определение проводят следующим способом: раствор, свободный от мешающих ионов и содержащий 2 мг урана, 5 мл 20%-ного раствора ацетата аммония и 20 мл воды, переносят в делительную воронку. Вводят 6 мл 2 %-ного раствора диэтилдитиокарбамата натрия и после доведения pH до 6,8 раствор экстрагируют последовательно 4 порциями четыреххлористого углерода, составляющими в общей сложности 30 мл. После этого водную фазу экстрагируют 3 раза хлороформом, порциями по 10 мл, экстракты фильтруют в мерную, колбу емкостью 50 мл и разбавляют хлороформом до метки. Светопоглощение раствора измеряют при 380 ммк, применяя для сравнения раствор. реактивов, который проводят через те же операции.
Если мешающие элементы отделены экстракцией эфиром, то можно измерять светопоглощение уранового комплекса с диэтилдитиокарбаматом в водном растворе, величина pH которого равна 6,5—7,2. Чувствительность реакции при этом почти такая же, как и при применении хлороформа, и окраска устойчива по меньшей мере 2 часа. Доп. перед.*
1 См, сноску 2 на стр. 526.
Глава XXVIII
ТАЛЛИЙ
В качестве основного компонента таллий содержится в редко встречающихся минералах крукезите (Си, Tl, Ag)2Se, лорандите T1AsS2, врбаите TlAs2SbS6 и гутчинсоните — сульфиде таллия, серебра, меди, свинца и мышьяка. Он встречается также в незначительных концентрациях совместно с щелочными металлами, железом, цинком, свинцом, медью и марганцем в некоторых породах и минералах, как, например, в карналлите, алюните, пиритах, халькопиритах, цинковой обманке 1 и брауните. В процессе переработки таких минералов таллий концентрируется в некоторых продуктах и отходах производства, в которых содержание его может достигать поддающихся определению количеств, как, например, в илах и пыли сернокислотных заводов, в цинке, меди и теллуре, а иногда в товарных серной и соляной кислотах.
ОБЩИЕ ЗАМЕЧАНИЯ
Поведение таллия в обычном ходе анализа зависит от его валентности. Таллий (I), когда он содержится в небольших количествах, не осаждается аммиаком и остается незамеченным, если только фильтрат от осаждения аммиаком не обрабатывают сульфидом аммония перед осаждением кальция. При наличии больших количеств таллия он может выделиться в виде хлорида 2 Т1С1 в процессе обезвоживания кремнекислоты выпариванием с соляной кислотой, что замечается по внешнему виду осадка или по его плавлению во время прокаливания. При обычных условиях прокаливания некоторая часть таллия может задержаться в осадке в виде силиката, но основная его масса теряется вследствие улетучивания.
В тех случаях, когда при обработке анализируемой пробы образуются соли таллия (III), основная масса таллия переходит в осадок от аммиака. При сильном прокаливании этого осадка некоторая часть таллия, если не весь, переходит в одновалентное состояние. Если железо в осадке окислов определяется титрованием перманганатом после обработки такими восстановителями, как сероводород и сернистый ангидрид,
1 W. N. Н а г 11 е у, Н. Ramage, Chem. Soc., 71, 533 (1897).
3 Согласно М. R and ell м К. S. С h a n g [J. Am. Chem. Soc., 50, 1535-(1928)] в 1л воды при 25° С растворяется 3,863 г Т1С1.
Разложение минералов, содержащих таллий 537
которые восстанавливают таллий до одновалентного, он частично учитывается вместе с железом, а частично с алюминием.
Таллий может быть открыт в минералах спектроскопическим методом. Для этого тонко растертую в агатовой ступке пробу смачивают соляной кислотой и на платиновом колечке постепенно вводят во внешнюю часть пламени бунзеновской горелки. При этом появляется характерная ярко-зеленая линия, которая сохраняется в течение нескольких секунд. Если погасить яркую линию натрия, то можно открыть 0,0002% таллия в 0,03 г материала х.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ТАЛЛИЙ
Таллиевые минералы легко разлагаются следующим способом. 1 г тонко растертой пробы помещают в коническую колбу емкостью 250— 500 мл и обрабатывают 2 г сульфата калия и 10 мл серной кислоты при нагревании на голом пламени до полного разложения. По охлаждении прибавляют 100 мл воды, кипятят, снова охлаждают и фильтруют. В фильтрат вводят карбонат натрия до нейтральной реакции и затем 2 г избытка. Раствор разбавляют до 200 мл, прибавляют 3 г цианида натрия, свободного от сульфидов, нагревают до кипения и оставляют на 12 ч. После этого осадок отфильтровывают и промывают 1 %-ным раствором карбоната натрия. Фильтрат обрабатывают раствором сульфида аммония (бесцветным) до прекращения выделения черного осадка сульфидов.
По охлаждении осадок отфильтровывают и промывают разбавленным раствором сульфида аммония (бесцветным), следя за тем, чтобы фильтр все время оставался влажным. Сульфид таллия растворяют в разбавленной (1 : 4) серной кислоте, раствор кипятят до удаления сероводорода, затем'вводят несколько капель сернистой кислоты, нейтрализуют карбонатом натрия, фильтруют, если нужно, и осаждают таллий, как указано в разделе «Методы определения» (стр. 540).
Для определения таллия в пирите или цинковой обманке 25—100 г тонко растертой пробы обрабатывают соляной кислотой при кипячении, добавляя время-от времени азотную кислоту до полного разложения пробы. Затем прибавляют в избытке разбавленную (1:1) серную кислоту и нагревают до появления ее паров. По охлаждении разбавляют водой и фильтруют. В фильтрате восстанавливают таллий, медь, кадмий и другие аналогичные элементы до металла избыточным количеством металлического цинка. Раствор оставляют стоять в течение ночи. Если при этом кислота полностью израсходуется, снова подкисляют раствор до возобновления растворения цинка. Фильтруют через бумажный фильтр, содержащий несколько кусочков цинка, и быстро промывают осадок небольшим количеством горячей воды. Из осадка таллий извлекают горячей разбавленной серной? кислотой и полученный раствор обрабатывают карбонатом натрия и цианидом, как указано выше 1 2.
1 R. Berg, W. Roebling [Вег., 68, В. 403 (1935)] и R. В е г g, Е. S. Fahrenkamp [Z. anal. Chem., 109, 305 (1937)] указывают, что р-аминонафталид тио-гликолевой кислоты («тионалид») является специфическим реагентом для открытия и определения таллия в щелочных цианидных растворах, содержащих тартраты или не содержащих их. Этот метод особенно применим для макро- и микротоксикологиче-ских исследований.
2 При исследовании органических веществ осадок лучше разлагать мокрым путем, например обработкой серной и азотной кислотами, так как в процессе прокаливания даже при низкой температуре могут происходить потери таллия.
538
Гл. XXVIII. Таллий
МЕТОДЫ ОТДЕЛЕНИЯ
Известны соединения одновалентного и двухвалентного таллия. По химическим свойствам таллий (I) сходен со щелочными металлами, а таллий (III) — с тяжелыми металлами. В кислых .растворах таллий (III) легко восстанавливается сернистой кислотой или сероводородом до одновалентного состояния. Таллий (I) окисляется хлором, бромом1 и царской водкой, но не окисляется азотной кислотой. Из сильнокислых растворов таллий, если он один, не осаждается сероводородом, но выделяется совместно с другими металлами группы сероводорода, образуя соединения с такими элементами, как мышьяк, сурьма, олово и медь. Из растворов, содержащих разбавленную минеральную кислоту, таллий сероводородом осаждается не полностью, но выделяется количественно в виде T12S из уксуснокислых растворов или при осаждении сульфидом аммония. Так как T12S на воздухе легко окисляется, фильтрование следует проводить возможно быстрее, следя за тем, чтобы фильтр все время оставался влажным. Промывание осадка заканчивают разбавленным раствором сульфида аммония (бесцветным). х
Свинец легко отделяется от таллия повторным осаждением в виде •сульфата (стр. 262).
Лучшим методом отделения серебра от таллия, независимо от валентности последнего, является осаждение серебра в виде хлорида из разбавленного азотнокислого раствора. При наличии в растворе таллия (I) осадок хлорида серебра целесообразно прокипятить с царской водкой и раствор затем разбавить или же для осаждения применять соляную кислоту, насыщенную хлором.
Таллий можно отделить от кадмия, железа, алюминия, хрома, кобальта, никеля, цинка, марганца, щелочноземельных металлов, магния и щелочных металлов в виде иодида таллия (I) обработкой холодного •слабокислого раствора сульфатов этих элементов сернистым ангидридом и иодидом калия^ прибавленным в небольшом избытке. В этих условиях осаждаются также медь и серебро. Медь можно перевести в раствор обработкой осадка разбавленным аммиаком. Ртуть вначале образует нерастворимый красный иодид ртути (II), который при дальнейшем добавлении иодида калия вновь переходит в раствор.. В присутствии ртути иодид калия следует добавлять до тех пор, пока осадок иодида таллия не приобретет чисто желтый цвет.
Установлено 2, что марганец и свинец можно отделить от таллия (I) •осаждением их двузамещенным фосфатом аммония в аммиачном растворе, •содержащем сульфосалицилат аммония. Иэ - фильтрата таллий можно выделить в виде хромата и отделить таким образом от железа, алюминия и хрома, которые не осаждаются хроматом в щелочных растворах в при-
1 Р. Е. Browning, Ind. Eng. Chem., Anal. Ed., 4, 417 (1932).
2 L. M о s e г, А. В r u k 1, Monatsh., 47, 709 (.1926). В более поздней работе [там же, 52, 343 (1929)] L. Moser nW. Reif описывают метод отделения алюми-ния, хрома и железа (III) от таллия (I), который заключается в следующем. Кислый раствор сульфатов обрабатывают карбонатом натрия почти до нейтральной реакции. Раствор нагревают до 40 °C, добавляют 20 мл 7%-ного раствора нитрита аммония, 20 мл метилового спирта и осторожно кипятят 20 мин. Затем вводят еще некоторое количество тех же реактивов и продолжают кипятить, чтобы обеспечить полноту осаждения. Осадок отфильтровывают и промывают 2%-ным раствором нитрата аммония. •Фильтрат упаривают до 100—200 мл, вводят аммиак и осаждают таллий в виде хромата.
Методы отделения
539
-сутствии сульфосалицилата. Кроме того, хроматом таллий можно отделить: 1) от селена, кадмия, цинка, никеля и кобальта, если предварительно введено достаточное количество аммиака, чтобы удержать их в растворе; 2) от меди, ртути и серебра в присутствии цианида и 3) от мышьяка и сурьмы, если предварительно окислить их до пятивалентного состояния перекисью водорода в аммиачной среде. Висмут можно отделить от таллия (I) осаждением двузамещенным фосфатом аммония из нейтрального раствора их солей.
Галлий отделяют от таллия осаждением аммиаком из кипящего раствора после предварительного восстановления таллия сернистым газом в почти нейтральном растворе.
Известный интерес представляет метод отделения таллия от ряда -элементов, основанный на экстрагировании хлорида таллия (III) эфиром из раствора в 6 н. соляной кислоте 1 (стр. 161). Таллий количественно выделяется в виде металла при восстановлении металлическим цинком 2 или магнием в слабосолянокислом или сернокислом растворе и отде-.ляется таким образом от многих элементов. Образующаяся при этом металлическая губка легко окисляется воздухом или растворенным кислородом, и ее следует спрессовать в комок стеклянной палочкой. Большую часть раствора сливают и осадок быстро промывают декантацией свеже-' прокипяченной водой. Таллий можно отделить от ванадия, алюминия и циркония электролизом разбавленного сернокислого раствора, с применением катода из цинковой амальгамы 3.
*Следует отметить, что метод экстракции хлорида таллия (III) эфиром из солянокислых растворов мало специфичен. Наряду с таллием в эфирный слой переходит значительное число других элементов, в частности золото, железо (III), сурьма, олово, галлий, германий.
Значительно лучшее отделение таллия от посторонних элементов, :за исключением золота, достигается экстракцией бромида таллия (III) из раствора в 1 н. бромистоводородной кислоте 4. В этих условиях в эфир-ный слой совместно с таллием (и золотом) переходят лишь очень незначительные количества некоторых других-элементов, которые легко извлекаются из эфирного экстракта взбалтыванием с 1 н. бромистоводородной кислотой. Экстракция бромида таллия эфиром успешно использована 5 для отделения от посторонних металлов микрограммовых количеств таллия при определении его в пылях цинкового, свинцового и медного шроизводств.
При анализе продуктов переработки руд цветных металлов для •отделения таллия от посторонних элементов применен также ионообменный метод ®. Согласно этому методу, таллий извлекается катионитом {марки СБС) из слабокислого раствора (pH =3—5), содержащего винную или лимонную кислоту и пирофосфат натрия, тогда как элементы, образующие в этих условиях комплексные соединения (железо, медь,
1 А. А. No у-е s, W. С. В г а у, Е. В. Spear, J. Am. Chem. Soc., 30, 489, 515, 559 (1908).
2 H. В. Knowles, Anal. Chem., 21, 1539 (1949).
8 G. W. Morden, J. Am. Chem. Soc., 31, 1045 (1909).
4 I. Wada, R. Ishii, Bull. Inst. Phys. Chem. Research Tokyo, 13, 264 (1934).
6 С. Д. Гурьев, Обогащение, металлургия цветных металлов и методы анализа, «Сборник Гинцветмет, № 10, 371 (1955).
• Л. Б. Г и н з б у р г, Э. П. Шкробот, Зав. лаб., 21, 1289 (1955).
540
Гл. XXVIII. Таллий
* цинк, кадмий, свинец, сурьма), удерживаются в растворе. Из катионита таллий извлекается разбавленной (1 : 1) соляной кислотой. Доп. перев.*
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Практически все методы определения таллия требуют предварительного восстановления его до одновалентного состояния. Восстановление легко осуществляется обработкой кислого раствора сернистым ангидридом, избыток которого затем удаляют кипячением. Вероятно, наиболее-удовлетворительным является метод, основанный на взвешивании таллия (I) в виде хромата Т12СгО4. Взвешивание в виде окиси таллия ТЪОз также дает хорошие результаты, но эта операция сложнее, в особенности когда определяют малые количества таллия. Методы, основанные на осаждении в виде иодида таллия ТП, обычно дают пониженные результаты, вследствие заметной растворимости этой соли х.
Определение в виде хромата таллия
Метод определения таллия в виде хромата 1 2 сводится к следующему. Раствор, концентрация таллия (I) в котором составляет около 0,001 г в 1 мл, не содержащий большого избытка аммонийных солей и веществ, которые осаждаются аммиаком, восстанавливают хромат, а также реагируют с таллием или хроматом калия в аммиачной среде, нейтрализуют разбавленным (2 : 1) раствором аммиака и затем на каждые 100 мл раствора прибавляют по 3 мл избытка раствора аммиака. Нагревают до 70— 80° С и медленно при перемешивании вводят по 20 мл 10%-ного раствора хромата калия на каждые 100 мл раствора. Охлаждают и оставляют стоять не менее 12 ч. Осадок отфильтровывают через тигель Гуча и промывают сперва 1 %-ным раствором хромата калия, а затем умеренно 50 %-ным спиртом. Сушат при 120—130° С в течение 1 ч и взвешивают в виде THCrOa.
Определение в виде окиси таллия (III)
Соли таллия (I) при обработке едким кали и гёксацианоферра-том (III) калия окисляются, причем выделяется гидроокись таллия (III). Эта реакция была положена в основу метода определения таллия 3, который впоследствии был видоизменен 4 следующим образом. Анализируемый раствор должен быть свободен от восстановителей, соединений,, осаждающихся гексацианоферратом (III) или гексацианоферратом (II) калия из растворов, содержащих едкое кали, и от соединений, которые реагируют с таллием, как, например, иодид калия. Кислый раствор соли таллия (I) в объеме 50—100 мл нейтрализуют 5%-ным раствором едкого-кали и затем прибавляют еще 25 мл раствора едкого кали и 25 мл 8 %-ного
1 Обзор методов определения таллия приведен в статье: W. Strecker, Р. de la Р е n a, Z. anal. Chem., 67, 256 (1925—1926).
2 Р. Е. Browning, G. Р. Hutchins, Am. J. Sci., [4], 8, 460 (1899); F. M a c h., W. L e p p e r, Z. anal. Chem., 68, 36 (1926); L. M oser, А. В r u k 1, Monatsh., 47, 709 (1926).
8 P. E. В г о w n i n g, H. E. Palmer, Am. J. Sci., (4), 27, 380 (1909).
4 F. Mach, W. L e p p e r, Z. anal. Chem., 68, 41 (1926).
.Методы определения
541
раствора гексацианоферрата (III) калия. Оставляют стоять на 18 ч, после чего осадок отфильтровывают через тигель Гуча и промывают горячей водой. Сушат при 200° С в течение 1 ч (не дольше), желательно в атмосфере, свободной от двуокиси углерода. Взвешивают' в виде 1 TI2O3.
Определение в виде иодида таллия (I)
, Метод определения таллия 2 в виде иодида таллия (I) вследствие заметной растворимости осадка 3 4 * 6 менее надежен, чем приведенные ранее.
При осаждении таллия иодидом раствор должен быть свободен от хлоридов, от свинца и серебра, которые образуют нерастворимые иодиды, а также от элементов, соли которых гидролизуются в уксуснокислых растворах, как, например, титан. Этот метод сводится к следующему. 50—100 мл раствора, содержащего не более 0,5 г таллия, обрабатывают сернистым ангидридом, если присутствует таллий (III), и избыток SO2 удаляют кипячением. Раствор нейтрализуют, затем прибавляют 2 мл уксусной кислоты, нагревают до кипения и вводят раствор иодида калия в таком количестве, чтобы в 100 мл анализируемого раствора содержался 1 г избытка иодида калия. Оставляют стоять 12—18 ч, после чего осадок отфильтровывают через тигель Гуча и промывают сначала 1 %-ным раствором иодида калия в 1 %-ной уксусной кислоте до удаления растворимых солей, а затем, для удаления промывной жидкости, 80 %-ным ацетоном. Сушат при 120—130° С и взвешивают8 в виде T1L
Другие методы
Из весовых методов для определения таллия рекомендуются методы, в которых таллий взвешивается в виде сульфостанната 8 Tl4SnS4, в виде кислого сульфата T1HSO4 или среднего сульфата8 T12SO4, а также в виде хлорплатината таллия7 Tl2PtCle, который является наименее растворимым из хлороплатинатов.
* Рекомендуется также8 метод определения таллия, основанный на осаждении таллия (I) тетрародандиамминхромиатом аммония. При этом образуется комплексное соединение Tl[Cr(CNS)4 • (NH3)2] красно
1 Этим методом в растворах, содержащих 0,4206; 0,1120 и 0,0280 г таллия, Mach и L е р р е г (цит. выше) определили соответственно 0,4203; 0,1122 и 0,0278 г таллия. Если присутствуют малые количества таллия, образуется мелкозернистый осадок, который с трудом отделяется фильтрованием. Прокаленный осадок может поглощать двуокись углерода и при температурах выше 800 °C восстанавливаться до окиси таллия (I).
2 G. Wertner, Z. anal. Chem., 3, 1 (1864); Н. Baubigny, С. г., ИЗ, 544 (1891); J. Н. Long, Z. anal. Chem., 30, 342 (1891).
3 Согласно F. Kohlrausch [Z. phys. Chem., 64, 168 (1908)], в 1 л воды при 26 °C растворяется 0,0847 г иодида таллия (I). Это соединение менее растворимо в растворах, содержащих иодид калия, спирт или немного уксусной кислоты, и почти нера-створймо в растворах, содержащих аммиак или тиосульфат натрия.
4 Этим методом Mach и Lepper (цит. выше) в растворах, содержавших
0,4206; 0,1120 и 0,0280 г таллия, определили соответственно 0,4185; 0,1113 и 0,0277 г таллия.
6 L. F. Hawley, J. Phys. Chem., 10, 654 (1906); J. Am. Chem. Soc., 29, 1011 (1907).
6 P. E. Browning, Am. J, Sci., (4], 9, 137 (1900).
7G. Neumann, Ann., 244, 349 (1888); A. S. Cushman, Am. Chem. J., .24, 222 (1900).
8 И. Л. Б a r б а н л ы, П. M и p з о e в а, ДАН Аз. ССР, 9, № 7, 373 (1953).
542
Гл. XXVIII. Таллий.-
малинового цвета, весьма характерное для таллия. Чувствительность-этой реакции значительно превосходит чувствительность реакции осаждения таллия в виде иодида или хромата.
Авторы указывают, что при установленных ими оптимальных условиях можно определить 2 мг таллия с достаточной точностью. Доп. перев*
Из объемных методов определения таллия заслуживает внимания метод, основанный на окислении таллия (I) (в виде сульфата) до таллия (III) перманганатом калия *, броматом калия 1 2 или сульфатом церия 3 (IV).
Для определения от 0,005 до 0,2 мг таллия в присутствии 0,5—1 г серебра, меди, кадмия, сурьмы, хрома и железа предлагают 4 таллий (I) экстрагировать раствором дифенилтиокарбазона (дитизона) в хлороформе, затем окислять таллий до трехвалентного, восстанавливать в кислом; растворе иодидом калия в присутствии крахмала и глицерина и появляющуюся при этом иодокрахмальную окраску сравнивать с эталонами, обработанными таким же способом, как и анализируемый раствор. ,
*Таллий колориметрически можно определить также по интенсивности окраски молибденовой сини, образующейся при нагревании раствора соединения таллия с тионалидом, обработанного фосфорномолиб-деновольфрамовой кислотой в присутствии формамида. Для этой цели таллий осаждают тионалидом из щелочного тартратноцианидного раствора, причем он практически отделяется от всех посторонних металлов 5 * 7. Осадок переводят в раствор обработкой серной кислотой и этиловым спиртом.
Турбидиметрически или нефелометрически таллий определяют по реакции с фосфорномолибденовой кислотой ®. При добавлении 0,2 мл 5 %-ного раствора фосфорномолибденовой кислоты к 5 мл раствора, содержащего 10—50 ммк таллия и азотную кислоту в 0,1 н. концентрации, образуется золь фосфоромолибдата таллия желтого цвета. Светопоглощение можно измерять через 5 мин.
Определению не мешают свинец, кадмий, висмут и ртуть (II). Соли ртути (I) образуют незначительный осадок, который растворяется при добавлении достаточного количества азотной кислоты. Калий и аммоний должны отсутствовать, так как они образуют относительно малорастворимые молибдаты.
В растворах, свободных от элементов, образующих малорастворймые сульфиды, таллий можно определять нефелометрически в виде сульфида в нейтральной или аммиачной среде
Сульфид таллия имеет коричневую окраску. Присутствие в растворе цианида не мешает образованию этого осадка.
1 L. F. Н a w 1 е у, J. Am. Chem. Soc., 29, 300 (1907); см. также R. S. Beale, A. W. Hutchison, G. С. С h a n d 1 е е, Ind. Eng. Chem., Anal. Ed., 13, 240-(1941). ' •
2 H. Marshall, J. Soc. Chem., Ind., 19, 994 (1900); E. Z i n 11, G. Rien- 4 acker, Z. anorg. Chem., 153, 276 (1926). -
8 H. H. Willard, P. Young, J. Am. Chem. Soc., 52, 36 (1930).
4 L. A. Haddock, Analyst, 60, 394 (1935). i
6 R. Berg, W. Roebling, Angew. Chem., 48, 430, 597 (1935); R. В e r
E. S. Fahrenkamp, W. Roebling, Mikrochemie, Festschr, von Hans MoliscL 1936, p. 42-51. - ft
eF. Pavelka, H. Morth, Mikrochemie, 5, 30 (1932).
7 C. Stich, Pharm. Ztg., 74, 27 (1929). .
Методы определения
543
Описан 1 2 также спектрофотометрический метод одновременного определения таллия, висмута и свинца, основанный на обнаруженной ранее z способности хлорокомплексов этих элементов давать характерный максимум светопоглощения в ультрафиолетовой области спектра. В условиях определения таллий (I) показывает максимум светопоглощения при 245 ммк, висмут (III) при 327 ммк, а свинец (II) при 271 ммк. При наличии мешающих ионов таллий, висмут и свинец рекомендуется предварительно выделять в виде дитизонатов из раствора цианидов. Влияние-олова (II), которое при этом не отделяется, можно свести к минимуму окислением его до четырехвалентного состояния. Для определения малых количеств таллия, порядка микрограммов, успешно применяется иодо-метрическое 3 и броматометрическое 4 * титрование 0,001 н. растворами. Доп. персе.*
*Интересный колориметрический метод определения таллия, основанный на том, что ионы Т1СЦ образуют с метиловым фиолетовым комплексное соединение, был разработан С. Д. Гурьевым 6. Указанное комплексное соединение растворяется в толуоле, окрашивая его в синий цвет. Метод очень чувствителен: молярный коэффициент светопоглощения при X = 620 лак равен 64 300, при X = 570 лак равен 48 000. Доп. ред*
1 С. Merritt Jr., Н. М. Н er she ns on, L. В. R о g е г s, Anal. Chem.,. 25, № 4, 572 (1953).
2 M. A. D e sesa, L. B. Rogers, Anal. Chim. Acta, 6, 534 (1952),
3 C. W. Sill, H. E. P e t e r s о n, Anal. Chem., 21, 1268 (1949): K. L о u-na m a a, Z. anal. Chem., 147, 196 (1955).
4 G. R i e n a c k e r, G. К n a u e 1, Z. anal. Chem., 128, 459 (1946).
* С. Д. Гурьев, Сборник трудов Гинцветмета № 10, М., 1955, стр. 371.
Глава XXIX
ИНДИЙ
Индий, подобно галлию, не входит в состав минералов в качестве основного компонента. Следы его находятся в большинстве цинковых обманок и в оловянных рудах. Он встречается во многих пиритах, в галените, смитсоните, сидерите 1 и марганцевых, железных и вольфрамовых рудах. В незначительных количествах индий иногда содержится в товарном цинке и, совместно с галлием, в остатках после дистилляции цинка.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа индий переходит в осадок при осаждении аммиаком. В процессе прокаливания этого осадка некоторые количества индия могут улетучиваться. Остающаяся в прокаленном остатке часть индия, если на нее не вводят соответствующую поправку, учитывается совместно с алюминием. Индий можно открыть после концентрирования раствора по окрашиванию пламени в ярко-голубой цвет, это окрашивание вызывает хлорид индия. Более надежным способом открытия индия является спектроскопическое исследование искрового спектра анализируемого материала. Наличие в спектре двух ярких голубых линий (X = О о
= 4511,55 А и Х = 4101,95 А) указывает на присутствие индия. Наилучшим методом определения индия является фотографирование дугового спектра.
Для концентрирования и открытия индия пробу обрабатывают соляной кислотой и после этого раствор нагревают с цинком. Индий выделяется из раствора совместно с другими восстанавливающимися в этих условиях элементами, как, например, медью и свинцом. Осадок отфильтровывают и растворяют в азотной кислоте. Для отделения свинца к раствору прибавляют серную кислоту и выпаривают до появления ее паров. Разбавляют таким количеством воды, чтобы концентрация серной кислоты в растворе составляла 10% по объему, осадок отфильтровывают и несколько раз промывают серной кислотой той же концентрации. Фильтрат нагревают до кипения, прибавляют раствор аммиака в небольшом избытке, осадок отфильтровывают и промывают 2%>-ным раствором нитрата аммония. Затем растворяют осадок в возможно меньшем количестве соляной кислоты, прибавляют раствор аммиака почти до нейтральной реакции и вводят 5 г бисульфита натрия. Кипятят 15 мин, фильтруют, промы
1 W. N. Hartley, Н. R a mage, J. Chem. Soc., 71, 533 (1897).
Методы отделения
545
вают осадок холодной водой и затем растворяют в соляной кислоте. Раствор упаривают до небольшого объема и испытывают на платиновой проволоке в пламени горелки Бунзена.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ИНДИЙ
Методы разложения минералов, содержащих индйй, зависят от состава исследуемых минералов. Так, цинковые обманки разлагают обработкой соляной кислотой с последующим добавлением азотной кислоты (стр. 479); для разложения пиритов требуется обработка царской водкой; оловянные руды следует сплавлять, например, с перекисью натрия (стр. 333). При выпаривании с соляной кислотой потерь индия (вследствие улетучивания) можно не опасаться.
Для концентрирования индия используются такие реакции, как восстановление избыточным количеством цинка в кислом растворе или осаждение ацетатом натрия, карбонатом бария и бисульфитом натрия.
МЕТОДЫ ОТДЕЛЕНИЯ
Индий образует соединения, в которых он одно-, двух- или трехвалентен, но в водных растворах устойчивы только соли индия (III).
Из сильнокислых растворов индий сероводородом не выделяется, но при осаждении из слабокислых растворов,' например 0,5 н. серной кислоты, захватывается некоторыми элементами группы сероводорода. В отличие от таллия, полное отделение индия от металлов сероводородной группы может быть достигнуто осаждением последних сероводородом при концентрации кислотььв растворе, равной 3,6 и. или выше. Желтый сульфид индия — 1пг8з осаждается сероводородом из уксуснокислых или слабо сернокислых (приблизительно 0,02 н.) растворов, что используется для отделения индия от железа и алюминия. Белый осадок образуется при’ насыщении сероводородом щелочных растворов или при введении сульфида аммония в нейтральный раствор. Из холодных растворов при действии NH4HS индий осаждается не полностью, a (NH4)2S осаждает его почти количественно. Из кипящих растворов эти реагенты выделяют очень небольшую часть индия или даже совсем его не выделяют.
Индий можно количественно отделить от пинка, марганца, никеля и кобальта осаждением свежеосажденным карбонатом бария (стр. 108) или ацетатом аммония из уксуснокислого раствора (стр. 103). Хорошее отделение индия от умеренных количеств меди, цинка и никеля может быть достигнуто двукратным осаждением раствором аммиака в присутствии хлорида аммония следующим способом. Раствор, содержащий элементы в виде хлоридов, разбавдяют до 200 мл, прибавляют 5 г хлорида аммония и нагревают до 60—80° С. После этого вводят аммиак до растворения образующейся в начале гидроокиси меди. Большого избытка аммиака следует избегать. Нагревают несколько минут, затем фильтруют и промывают фильтр и осадок горячим 1 %-ным раствором хлорида аммония. Осадок растворяют в горячей разбавленной (1 : 10) соляной кислоте и тщательно промывают бумагу фильтра горячей разбавленной (1 : 100) кислотой. Раствор разбавляют до 200 мл и осаждение аммиаком повторяют х.
1 R. Gilchrist, J. Research NBS, 20, 745 (1938).
35 Заказ 522.
546
Гл. XXIX. Индий-
Имеется указание, что при осаждении цианатом калия1 индий хорошо-отделяется от цинка, никеля, хрома (VI), а в случае некоторого изменения условий осаждения и от кобальта. В самом простом случае 200—400 мл слабокислого раствора обрабатывают шестикратным, по отношению к содержанию цинка, количеством хлорида аммония, вводят несколько капель метилового оранжевого и затем цианат калия до пожелтения раствора. Тотчас нагревают до .кипения, фильтруют, осадок промывают горячей водой и, если присутствуют большие количества цинка или никеля, растворяют и переосаждают. Если осадок предполагают прокалить, то его следует предварительно полностью отмыть от хлоридов.
*Э. А. Остроумов и Б. Н. Иванов-Эмин2 для отделения от марганца, кобальта, никеля, цинка, кадмия, меди, щелочноземельных и щелочных металлов рекомендуют осаждать индий пиридином в присутствии аммонийных солей. Авторами установлено, что при осаждении индия пиридином из растворов, свободных от солей аммония, образуются смешанные осадки, состоящие из индатов марганца, кобальта, никеля, цинка,, кадмия и меди и гидроокиси индия, вследствие чего отделения от этих металлов не происходит. Доп. перев*
Индий легко восстанавливается до металла цинком в разбавленных сернокислых растворах и отделяется таким образом от таких элементов, как галлий, алюминий и цирконий, остающихся при этом в растворе. В присутствии меди, кадмия, олова и таллия, которые в этих условиях также восстанавливаются до металла, этот метод может служить лишь для предварительного отделения индия.
Электролитическое осаждение индия из раствора его сульфатов в разбавленной (1 : 4) серной кислоте является хорошим способом отделения его от/Железа, однако достигнуть полного выделения индия таким путем затруднительно 3.
Свинец отделяют от индия осаждением в виде сульфата свинца, как описано на стр. 262.
При кипячении почти нейтрального раствора соли индия с избыточным количеством бисульфита натрия образуется нерастворимый основной сульфит индия 1п2(30з)з-21п(ОН)з-5Н2О. Эта реакция не может служить для окончательного отделения индия, если присутствуют олово, титан и цирконий, которые при этом также выделяются из раствора.
Метод отделения индия от галлия, основанный на осаждении едкой щелочью от кипящего раствора, не вполне удовлетворителен, так как небольшая часть индия остается в растворе, а некоторые количества галлия захватываются осадком, даже при двукратном осаждении. Отделение рекомендуют 4 осуществлять следующим образом. Раствор, содержащий приблизительно по 100 мг каждого элемента в 200 мл, нейтрализуют едким натром, затем вводят 1,5 г избытка едкого натра и кипятят несколько минут. Осадок отфильтровывают, растворяют в соляной кислоте и переосаждают, прежде чем приступить к определению по методам, изло-
1 L. Moser, F. Siegmann, Monatsh., 55, 14, (1930).
2 Э. А. Остроумов, Б. Н. Иване в-Э мин, Зав лаб., 11, 1034 (1945).
3 L. М. Dennis, W. С. Geer, J. Am. Chem. Soc., 26, 437 (1904); F. C. Mat-h e r s, там же, 29, 485 (1907); U. В. Bray, H. D. Kirschman, там же, 49, 2739 (1927).
4 L. M. Dennis, J. A. Bridgman, J. Am. Chem Soc., 40, 1552 (1918).
Методы определения
547
женным в разделе «Методы определения». Те же авторы отмечают, что осаждение роданомеркуриатом калия не дает достаточно удовлетворительного отделения цинка от индия.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Индий обычно взвешивают в виде окиси индия 1и2О3, после прокаливания1 при 1100—1200° С. В горячем виде окись имеет красно-коричневую окраску, в холодном — светложелтую. Она легко -растворяется в горячих кислотах.
Прокаливание до окиси следует проводить в хороших окислительных условиях, так как индий легко восстанавливается до металла.
Конечное осаждение обычно проводят аммиаком в виде 1и(ОН)з из растворов, содержащих нитрат аммония. Введения в раствор хлорида аммония следует избегать, чтобы не образовался хлорид индия, который улетучивается при последующем прокаливании осадка 2'.
Из других методов определения индия можно указать на способ, основанный на титровании его гексацианоферратом (II) калия 3, и электролитическое микроосаждение малых количеств индия (2 мг) в слабоаммиач-ных растворах, содержащих оксалат аммония4.
Описан метод определения малых количеств индия дитизоном 5 и исследовано влияние меди, свинца, железа, никеля, кобальта, цинка, галлия, олова, алюминия, марганца, кадмия и таллия на получаемые результаты.
Осаждение аммиаком
При этом осаждении должны отсутствовать все осаждающиеся аммиаком элементы. Введения большого избытка аммиака следует избегать вследствие заметной растворимости в нем осадка гидроокиси индия. Винная кислота препятствует осаждению.
Ход определения.. Анализируемый раствор, предпочтительно азотнокислый, свободный от органических соединений и не содержащий избыточного количества аммонийных солей и элементов, осаждаемых аммиаком, а также образующих нерастворимые соединения с индием в аммиачной среде, как, например, фосфора, разбавляют так, чтобы в 200 мл содержалось не более 0,1 г индия. Затем вводят 1 мл азотной кислоты, если в растворе ее еще нет, нагревают до кипения и медленно прибавляют разбавленный (1 : 1) раствор аммиака в небольшом избытке. Кипятят 1 мин, охлаждают приблизительно до 60° С, прибавляют немного мацерированной бумаги и фильтруют. Осадок промывают горячим слабоаммиач-ным 1 %-ным раствором нитрата аммония и помещают во взвешенный
1 Согласно L. Moser, F. Siegmann [Monatsh., 55, 14 (1930)], In2O3 не улетучивается при температуре пламени паяльной горелки и после такого сильного прокаливания не гигроскопична.
2 A. Thiel, Н. К о е 1 s с h, Z. anorg. Chem., 66, 288 (1910).
3 U. В. В г а у и Н. D. К irschman [J. Am. Chem. Soc., 49, 2839 (1927)1 рекомендуют потенциометрическое титрование, а Н. В. Н о р е, М. R о s s и J. F. Skelly [Ind. Eng. Chem., Anal. Ed., 8, 51 (1936)] применяют дифенил-бензидин в качестве окислительно-восстановительного индикатора.
4 G. L. Royer, Ind. Eng. Chem., Anal. Ed., 12, 439 (1940).
6 I. M a y, J. I. Hoffman, J. Wash. Acad. Sci., 38, 329 (1948).
35*
548
Гл. XXIX. Индий
платиновый тигель. Осторожно высушивают, бумагу обугливают, избегая воспламенения, и сжигают углерод в хороших окислительных условиях при возможно более низкой температуре. Прокаливание лучше всего проводить в муфеле. Если тигель прокаливают на пламени горилки, то целесообразно поместить его в отверстие асбестовой пластинки, чтобы предохранить от восстанавливающего действия газообразных продуктов пламени горелки. После полного сгорания углерода температуру повышают до 1100—1200° С и прокаливают 15 мин. Затем тигель закрывают крышкой и нагревают еще несколько 'минут. Охлаждают в эксикаторе и взвешивают. Прокаливание повторяют до постоянной массы.
* Колориметрический метод
Помимо упомянутого выше дитизонового метода, колориметрически индий определяют также по интенсивности желтой окраски раствора его оксихинолята в хлороформе1. Определение сводится к следующему. Доводят pH анализируемого раствора (содержащего около 0,5 мг индия) приблизительно до 3,2—4,5 добавлением ледяной уксусной кислоты или ацетата натрия. По охлаждении до комнатной температуры экстрагируют 0,15 %-ным раствором оксихинолина в хлороформе 5 раз порциямипо 5 мл. Объединенные экстракты разбавляют до 50 мл хлороформом и измеряют светопоглощение раствора при 400 ммк.
Мешают определению алюминий, галлий, таллий (III), олово (II), висмут, медь, железо (III), ванадий (V), молибден (VI), никель и, в меньшей степени, кобальт, также дающие окрашенные хлорофофмные экстракты в этих условиях.
Имеется указание1, что малые количества индия, подобно галлию, можно определять по флуоресценции хлороформных экстрактов. Флуоресцентный метод разработан2 применительно к определению индия в продуктах переработки руд цветных металлов. Для отделения ийдия от мешающих определению металлов авторы применяют экстракцию бромида индия эфиром из 5 н. бромистоводородной кислоты3. Для этой цели рекомендуется4 также ионообменный метод. Индий извлекается катионитом (марки СБС) из растворов, содержащих сульфосалициловую кислоту. При этом, как^тверждают авторы, индий отделяется практически от всех элементов, влияющих на флуоресценцию хлороформного раствора оксихинолята индия, за исключением галлия. Доп. перев*
1Т. Moeller, Ind. Eng. Chem. Anal. Ed., 15, 270 (1943); E. Б. Сендэл, Колориметрическое определение следов металлов, Госхимиздат, 1949.
2 С. Д-. Г у р ь е в, Л. Б. Г и н з б у р г, А. П. Ш е б а р е н к о в а, Обогащение, металлургия цветных металлов и методы анализа. Сборник трудов Гинцвет-мета № 10, 1955, стр. 387.
3 I. Wada, R. Ishii, Bull. Inst. Phys. Chem. Research Tokyo, 34,787 (1937— 1938).
4 Л. Б. Г и н з б у p г, Э. П. Ill к p о б о т, Зав. лаб., 21, 1289 (1955).
Глава XXX
ГАЛЛИЙ
Минералы, в состав которых галлий входит в качестве основного компонента, неизвестны. Он, однако, широко распространен в природе и в незначительных концентрациях встречается совместно с алюминием, железом, марганцем, цинком, свинцом и индием. Следы его находятся во многих цинковых обманках, железных рудах1 и каолине. Спектроскопические следы галлия всегда присутствуют в бокситах и почти во всех продуктах, содержащих алюминий2. До 0,02% галлия найдено в товарном алюминии.
ОБЩИЕ ЗАМЕЧАНИЯ
По своим свойствам галлий относится к аналитической подгруппе алюминия. В обычном ходе анализа он попадает в осадок от аммиака и учитывается совместно с алюминием, если его не определяют и на его содержание не вводят соответствующую поправку. Предварительное качественное испытание обычно проводят, искровым спектральным методом после концентрирования галлия.
'Установлено 3, что 0,0046 мг галлия и Ю, 0013 мг индия можно открыть визуальным исследованием искрового спектра солянокислого раствора их хлоридов и что относительно большие количества одного из этих элементов не влияют на чувствительность определения другого. Наилучшим методом предварительного концентрирования и качественного открытия галлия является осаждение его гексацианоферратом (II) калия из солянокислого раствора, как изложено в разделе «Методы отделения» (стр. 551).
Такие малые количества галлия, как 0,02 мг!л и выше, можно открыть и определить колориметрическим методом, используя реакцию с хинализарином, с которым он образует лак, окрашенный в розовый до аметистового цвет 4. Наилучшие результаты получаются в следующих условиях. Анализируемый раствор должен содержать 0,02—0,2 мг галлия и 0,5 г!л фторида натрия; он должен быть 1 н. по содержанию ацетата аммония
1 Согласно W. N. Н artley и Н. Ramage [Trans. Chem. Soc., 71, 533 (1897)], галлий был найден в 35 из 91 образца железных руд. Присутствие галлия отмечено главным образом в магнетитах. В сидеритах галлий не обнаружен.
2 F. W. Clarke, U. S. Geol. Survey Bull., 770, 16 (1924).
2 L. M. Dennis, J. A. Bridgman, J. Am. Chem. Soc., 40, 1534 (1918).
4 H. H. Willard, H. C. Fogg, J. Am. Chem. Soc., 59, 40 (1937).
550
Гл. XXX. Галлий
и 0,5 н. по содержанию хлорида аммония и иметь pH равным 5,0. Известно, что этой реакции мешают и, следовательно, должны быть предварительно отделены свинец, медь, олово, сурьма, индий, платина, германий, ванадий, молибден и более 1 мг железа или 10 мг алюминия.
Описан метод1 открытия 0,1 мкг галлия в 10 мл раствора в присутствии железа (II) и алюминия, основанный на флуоресценции хлороформного раствора оксихинолята галлия, осажденного при pH = 2,6. Этой реакции мешают железо (III), медь (II), ванадаты, молибдаты, фтор, литий, бериллий, скандий, индий и цинк. В присутствии этих элементов требуется, специальная обработка.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ГАЛЛИЙ
Утверждение, что хлорид галлия улетучивается в процессе выпаривания солянокислых растворов его солей2, не соответствует действительности, так как при выпаривании таких растворов на водяной бане досуха потерь галлия не наблюдается 3. Галлий не улетучивается и в процессе дистилляции при 200—220° С, когда в его раствор в серной или хлорной кислоте по каплям вводят соляную или бромистоводородную кислоту 4. Поэтому галлий можно переводить в раствор обычно принятыми для разложения анализируемого материала методами. Соляную кислоту и летучие хлориды не следует оставлять в соединениях галлия, подлежащих прокаливанию 5.
Металлический галлий легко растворяется при нагревании его со смесью, состоящей из двух частей концентрированной серной кислоты и одной части хлорной кислоты (72 %-ной) 6.
МЕТОДЫ ОТДЕЛЕНИЯ
В соединениях галлий бывает двух- или трехвалентным. Соли галлия (II), являющиеся восстановителями, не устойчивы, и в обычном хрде анализа приходится иметь дело, главным образом, с соединениями галлия (III).
В растворах, содержащих свободную минеральную или органическую кислоту, галлий не образует осадка с сероводородом, если он присутствует один, то он полностью осаждается сероводородом совместно с цинком, серебром, медью, марганцем, железом, мышьяком из слабокислых, например уксуснокислых или ацетатных растворов. Эти элементы (большей частью мышьяк) иногда вводятся в раствор с целью выделения малых
1 Е. В. S a n d е 1 J, Ind. Eng. Chem., Anal. Ed., 13, 844 (1941). *См. также С. Д. Гурьев, Л. Б. Гинзбург, А. П. Шебаренкова, Сборник трудов Гинцветмета № 10, М., 1955, стр. 387.*
2 Freseniu s-M i t с h е 11, Introduction to Qualitative Chemical Analysis, John, Wilev and Sons, 1921, p. 269.
3 E. H. Swift, J. Am. Chem. Soc., 46, 2377 (1924). *
4 J. I. Hoffman, G. E. F. Lundell, J. Research NBS, 22, 467 (1939).
5 Быстрый метод концентрирования, галлия (и германия), находящегося в окиси цинка, основанный на растворении пробы в кислоте, подщелачивании раствора, медленном введении некоторого количества анализируемой окиси цинка и фильтровании, описан в статье С. J a m е s, Н. С. F о g g, J. Am. Chem. Soc., 51, 1459 (1929). F. S e b-ba и W. Pugh [J. Chem. Soc., 1371 (1937)] установили, что галлий полностью извлекается из германита в раствор при сплавлении тонко измельченной руды с едким натром в железном тигле, при выщелачивании плава водой и фильтровании.
• 6 L. S. Foster, J. Chem. Soc., 61, 3122 (1939).
Методы отделения
551
количеств галлия. В таком случае мышьяк можно удались дистилляцией, как описано в гл. «Мышьяк» (стр. 303), после растворения сульфидов в царской водке и выпаривания раствора с серной кислотой до появления ее паров. Отделение сероводородом лучше применять лишь в тех случаях, когда оно может быть выполнено в сильнокислых растворах1.
Подобно алюминию, галлий можно отделить от никеля, марганца, щелочноземельных металлов, магния и щелочных металлов осаждением аммиаком в присутствии хлорида аммония. Введения избытка аммиака следует избегать, так как осадок заметно растворим в нем.
*Б. Н. Иванов-Эмин и Э. А. Остроумов2 рекомендуют отделять галлий от марганца, кобальта, никеля, цинка, кадмия и меди осаждением пиридином из раствора, содержащего аммонийные соли. Небольшой избыток пиридина и большие количества аммонийных ролей не повышают растворимости гидроокиси галлия. В отсутствие аммонийных солей, так же как в случае присутствия индия, разделение не происходит вследствие выделения галлитов этих элементов. Этим методом можно также отделить галлий от щелочных и щелочноземельных металлов и от магния. Доп. перев.*
Для отделения галлия от цинка, никеля, кобальта, марганца, кадмия, бериллия и таллия, так же как и для получения осадка, который после прокаливания можно взвесить в виде Ga2O3, рекомендуется осаждение таннином из кипящего 1—2%-ного уксуснокислого раствора, содержащего 2% нитрата аммония3. В кипящий раствор при перемешивании вводя по каплям 10%-ный раствор таннина до полного осаждения. Обычно для этого достаточно 10-кратного избытка таннина. При содержании же незначительных количеств галлия следует прибавить не менее 5 мл осадителя. Осадок промывают горячей водой, содержащей немного нитрата -аммония и 1—2 капли уксусной кислоты. Как правило, осадок целесообразно растворить в соляной кислоте и осаждение таннином повторить после нейтрализации кислоты аммиаком и введения в раствор уксусной кислоты. Осадок прокаливают в кварцевой или фарфоровой посуде. Таннином выделяются очень объемистые осадки, поэтому трудно оперировать более чем с 0,1 г Ga2O3. В случае содержания больших количеств галлия -основную массу рекомендуется выделить двукратным осаждением в виде основного ацетата, а остальную часть выделить из объединенных фильтратов таннином.
Галлий совместно с алюминием, бериллием и другими элементами можно отделить от индия, железа, титана и тому подобных элементов осаждением едким натром. Это разделение не вполне количественное, так как индий осаждается не полностью, а галлий может частично захватиться осадком. Железо полностью отделяется от галлия 4 однократным осаждением едким натром, если конечная концентрация щелочи в растворе не ниже 0,3 н. Отделение индия от галлия рекомендуют проводить приблизительно при 0,2 н. концентрации едкого натра в растворе5.
1 Галлий, например, не захватывается сульфидами меди, мышьяка и сурьмы при осаждении их из 0,36 н. сернокислого раствора.
2 Б. И. И в а н о в-Э мин, Э. А. Остроумов, Зав. лаб., 12, 674 (1946),
3 L. Moser, А. В г u k 1, Monatsh., 50, 181 (1928).
4 Е. Н. S w i f t, J. Am. Chem. Soc., 46, 2377 (1924).
6 L. M. Dennis, J. А. В r i d g m a n n, J. Am. Chem, Soc., 40, 1534 (1918), см, также гл. «Индий» (стр. 547).
552
Гл. XXX. Галлий
Осаждение карбонатом бария (стр. 108) с успехом служит для отделения галлия от марганца, но этой реакцией нельзя пользоваться в присутствии цинка, никеля и кобальта.
Имеются указания, что отделение галлия от свинца, кадмия, кобальта, никеля, марганца, цинка, железа, таллия (в присутствии SO2), бериллия, урана и редкоземельных элементов может быть успешно осуществлено кипячением с гидроокисью меди т.
Реакция осаждения гексацианоферратом (II) , калия отличается тем, что ею можно пользоваться как для качественного открытия галлия, так и для отделения его от других элементов 1 2. Элементы,-образующие осадки с гексацианофер'ратом (II) калия, как, например, цинк, цирконий, индий, а также нитраты а другие окислители, при.эТом должны отсутствовать. Эта реакция служит для отделения галлия от алюминия, хрома, марганца, кадмия, ртути, свинца, висмута и таллия. В разбавленный (1 : 3) солянокислый раствор хлоридов вводят в небольшом избытке гексацианоферрат (II) калия. Нагревают при 60—70° С в продолжение 30 мин, а затем оставляют на холоду в течение нескольких часов или дней, если в растворе содержатся незначительные количества галлия. Осадок промывают холодной разбавленной (1 : 3) соляной кислотой. Разложение гексацианоферрата (II) галлия наиболее целесообразно проводить путем сплавления с нитратом аммония и последующей обработкой едким натром для отделения железа.
Установлено 3 * * * * 8, что количественное разделение^галлия и железа можно осуществить следующим образом. К солянокислому раствору хлоридов добавляют раствор аммиака до появления небольшого неисчезающего осадка, который растворяют введением 1 капли соляной кислоты. Затем прибавляют 2—3 капли избытка соляной кислоты и достаточное количество ацетата аммония, чтобы связать всю свободную минеральную кислоту. Вводят избыток реактива (1 г а-нитрозо-р-нафтола растворяют в 50 мл 50%-ного раствора уксусной кислоты и фильтруют) и оставляют стоять в течение нескольких часов. Осадок отфильтровывают и промывают сначала холодным 50%-ным раствором уксусной кислоты, а затем водой. Высушивают, очень осторожно прокаливают в фарфоровом тигле и взвешивают в виде Fe2O3. Фильтрат выпарийают досуха, прибавляют разбавленный раствор аммиака до щелочной реакции и затем кипятят до тех пор, пока смоченная водой красная лакмусовая бумага не перестанет синеть в выделяющихся парах. Охлаждают, фильтруют и отмывают осадок от хлоридов 1%-ным раствором нитрата аммония. Прокаливают и взвешивают в виде Ga2O3, учитывая при этом, что окись галлия гигроскопична.
Алюминий можно отделить от галлия осаждением газообразным хлористым водородом из солянокислого раствора, к которому прибавляют
1 W. Crookes, Select Methods in Chemical Analysis, Longmans, Green and Co., London and New York, 1894.
2 Например, P. E. Browning и L. E. Porter [Am. J. Sci., [4], 44, 222
(1917)] установили, что гексацианоферратом (II) калия можно открыть 0,1 мг галлия
в 10 мл разбавленного (1 : 3) солянокислого раствора, если раствор после введения
реагента оставить стоять 1 ч. О применении этой реакции для потенциометрического титрования галлия см. Н. D. Kirsch m.a n, J. В. R a m s e у [J. Am. Chem. Soc.,
50, 1632 (1928)] и Sunao Ato [Sci. Papers Inst. Phys. Chem. "Research Токуо, 10,
1 (1929)]. ' ’ .
8 J. P a p i s h, L. E. Hoag, J. Am. Chem. Soc., 50, 2118 (1928)«
Методы отделения
553
равный объем эфира1 (стр. 161). Очень существенно при этом повторной обработкой раствора удостовериться в полноте осаждения алюминия.
Осаждение купфероном из разбавленного (7 : 93) сернокислого раствора, как описано в разделе «Осаждение купфероном» (стр. 143), является. хорошим методом выделения таких малых количеств галлия, какие содержатся, например, в алюминии2, а также для отделенияугаллия от индия, который в этих условиях не осаждается (см. стр. 554). В присутствии больших количеств алюминия или индия осадок купферата галлия следует растворить (стр. 143) и переосадить.
Установлено3, что галлий можно отделить: 1) от титана, циркония и тория — осаждением купфероном в 1 н. растворе щавелевой кислоты, содержащем оксалат аммония; 2) от циркония — осаждением фениларсоновой кислоты (стр. 638) из-горячего 2 н. сернокислого раствора; 3) от тория — осаждением щавелевой кислотой из солянокислого раствора, свободного от сульфат-ионов, и 4) от ванадия, молибдена и вольфрама — осаждением основной массы галлия оксихинолином из горячего разбавленного (5 : 95) аммиачного раствора. Для выделения остающейся в фильтрате части галлия раствор слабо подкисляют уксусной кислотой, прибавляют 1 мл насыщенного раствора карбоната аммония, кипятят до нейтральной реакции по лакмусу, дают постоять 2—3 ч и затем фильтруют. Полученный осадок обычно бывает- загрязнен примесями, и его лучше растворить и переосадить, прежде чем присоединить к основному осадку. Объединенные осадки растворяют в 2 н. серной кислоте и из раствора осаждают галлий купфероном.
Осаждение купфероном из раствора в 2 н. серной кислоте служит также для выделения галлия из фениларсоновокислого фильтрата и из фильтратов, содержащих оксалаты, которые предварительно разрушают выпариванием с серной кислотой.
Галлий (III) можно отделить от многих элементов повторной экстракцией его хлорида из холодного раствора в 5—6 н. соляной кислоте эфиром, свободным от спирта. Эфир предварительно 2—3 раза встряхивают с соляной кислотой той же концентрации 4 (стр. 161). В этих условиях эфиром вместе с галлием извлекаются также следы индия 5.
Хцорид галлия можно отделить от нелетучих хлоридов или от хлоридов, которые улетучиваются при значительно более высоких температурах, нагреванием в токе хлора. Так, хлорид галлия (т. кип. 215—220° С) отделяют6 от хлорида индия (III) (медленно возгоняется при 600° С) и хлорида цинка (т. кип. 730° С) дистилляцией безводных хлоридов в токе сухого хлора при температуре около 230—255° С.
1 Р. Е. Browning, L. Е. Porter, цит. выше, см. также сноску 2 на. стр. 552.
2 J. A. Scherrer, J. Research NBS, 15, 585 (1935).
3 А. В г u k 1, Monatsh., 52, 253 (1929).
4 Е. Н. S w i f t [J. Am. Chem. Soc., 46, 2375 (1924)1 установил, что при экстрагировании 50 мл раствора, содержащего 0,1 г галлия в 4,9—5,0 н. соляной кислоте, 50 мл эфира могут извлечь 97 % галлия. После трехкратного экстрагирования в водном слое остается менее 0,1 мг галлия. J. A. Scherrer [J. Research NBS, 15, 588 (1935)] считает целесообразным после последнего экстрагирования (для отделения водного слоя) оставлять воронку при комнатной температуре в течение 1 часа. *См. *-также И. П. А л и м а р и н, В. Н. И в а н о в-Э мин, ЖПХ, 9‘ вып. 6 (1936).*
И. Wada, SunaoAto, Sci. Papers Inst. Phys. Chem. Research Tokyo, 1, 70 (1922). z
• G. E. F. Lun dell, Nai Kim В he, Trans. Am. Inst. Metals, 8, 146.(1914).
554
Гл. XXX. Галлий
Цинк осаждается из разбавленных сернокислых или азотнокислых растворов роданомеркуриатом калия1 и отделяется таким способом от галлия 2 3. Галлий можно затем выделить из фильтрата после удаления ртути осаждением сероводородом из раствора, содержащего 5—7% соляной кислоты по объему. Для проверки сульфида ртути на содержание галлия осадок высушивают и ртуть удаляют прокаливанием. Нелетучий остаток сплавляют с карбонатом натрия, после чего плав растворяют в кислоте.
’ Прозрачный реактив для осаждения цинка приготовляют растворением 27 г хлорида ртути (II) и 39 а роданида калия в 1000 мл воды.
Анализируемый сернокислый или азотнокислый раствор, содержащий в 100 мл не более 0,05 г цинка и 1 мл кислоты, охлаждают до комнатной температуры. Затем по каплям при непрерывном перемешивании вводят реактив до появления осадка, после чего на каждую 0,1 г цинка добавляют еще по 20 мл осадителя. Оставляют стоять не менее 2 ч. Фильтруют через асбест и промывают осадок водой, содержащей 20 мл реактива в 1000 мл. В присутствии больших количеств цинка осадок на асбесте обрабатывают сначала несколькими каплями концентрированного растрора едкого натра, а затем соляной кислоты, после чего промывают асбест горячей водой. Раствор нейтрализуют едким натром, подкисляют, как указано выше, и цинк переосаждают. Фильтраты, в которых содержится весь галлий, объединяют.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Галлий всегда взвешивают в виде окиси Ga2O3 после осаждения аммиаком, купфероном, сульфитом аммония и др. из растворов его солей, где он находится в трехвалентном состоянии. Так как хлорид галлия GaCh летуч, осаждение лучше проводить из растворов, свободных от хлорид-ионов. Окись галлия, полученная Прокаливанием при температуре ниже 1000° С, гигроскопична, и с нею следует обращаться должным образом. В окислительной атмосфере галлий можно прокаливать без потерь при 1300° С. Образующаяся при такой температуре окись не гигроскопична з. Окись галлия белого цвета. Если она окрашена, то это указывает на наличие в ней примесей. Так, например, примесь окиси индия окрашивает ее в желтоватый цвет.
Осаждение аммиаком
При осаждении гидроокиси галлия следует избегать введения избытка раствора аммиака и необходимо принять меры к тому, чтобы полностью перенести осадок на фильтр, учитывая его склонность приставать к стенкам стакана.
Ход определения. Сернокислый раствор, содержащий галлий (III), свободный от органических веществ, избыточного количества аммонийных солей и осаждающихся аммиаком посторонних элементов, разбавляют до 200 мл, вводят несколько капель метилового красного и нагревают до кипения. Затем медленно прибавляют разбавленный и свободный от
1 G. Е. F. Lunde 11, NaiKimBee, Trans. Am. Inst. Metas, 8, 146(1914).
2 L. M., Dennis,!. A. Bridgman,!. Am. Chem. Soc., 40, 1534 (1918).
3 G. E. F. L u n d e 1 1, I. I. Hoffman,!. Research NBS, 15, 409 (1935),
-Методы определения
555
карбоната раствор аммиака до начала образования осадка. Когда выделение гидроокиси галлия прекратится, добавляют еще немного раствора, аммиака и нагревают раствор до кипения. Так продолжают до тех пор, пока окраска индикатора не станет желтой1. После этого вводят немного мацерированной бумаги, кипятят еще 1—2 мин, фильтруют и возможно полнее переносят осадок на фильтр, стирая его со стенок стакана и смывая горячим нейтральным 2%-ным раствором нитрата аммония. Покрыв воронку стеклом, быстро растворяют оставшийся на стенках стакана осадок в возможно меньшем количестве горячей разбавленной серной кислоты, осаждают аммиаком, как указано выше, и фильтруют через тот же фильтр. Осадок тщательно промывают 2%-ным раствором нитрата аммония.
Влажный фильтр с осадком переносят в платиновый тигель, взвешенный вместе с плотно прилегающей крышкой, и осторожно нагревают (не покрывая крышкой), пока осадок не высохнет. Затем прокаливают сначала при возможно более низкой температуре до сгорания углерода, а затем в хороших окислительных условиях при температуре около 1000° С. Закрывают тигель крышкой и продолжают прокаливать ‘еще несколько минут. Охлаждают в эксикаторе над серной кислотой или фосфорным ангидридом и плотно закрытый тигель взвешивают. Затем тигель снова прокаливают, охлаждают, как прежде, и, предварительно положив нужные разновески на весы, быстро взвешивают. Прокаливание продолжают до достижения постоянной массы. Взвешенный осадок следует исследовать на содержание в нем посторонних окислов, а при проведении особо точных анализов необходимо ввести поправку на содержание примесей, например, кремнекислоты, введенных с реактивами или извлеченных из стекла посуды.
•Осаждение купфероном
Галлий количественно ’осаждается купфероном из разбавленных (7 : 93) сернокислых растворов, содержащих значительный избыток реагента. Фильтровать следует через некоторое время после введения в раствор купферона. Содержание в растворе до 1 г винной кислоты не влияет на осаждение галлия, благодаря чему эту операцию можно осуществить после отделения посторонних элементов сероводородом из кислого раствора или сульфидом аммония из аммиачного раствора, содержащего тартрат аммония (стр. 89).
Ход определения. Фильтрат после осаждения сульфидом аммония подкисляют разбавленной (1:1) серной кислотой и затем кипятят до удаления сероводорода. Фильтруют, если имеется осадок, и разбавляют раствор до 375 мл. Нейтрализуют аммиаком, перемешивают и осторожно вводят 30 мл серной кислоты. Охлаждают раствор в ледяной воде и вводят охлажденный 6%-ный водный раствор купферона в количестве, на 15— 20 мл превышающем необходимое для осаждения содержащегося в растворе галлия. Прибавляют немного беззольной мацерированной бумаги, хорошо перемешивают и оставляют стоять в ледяной воде 1 ч. После этого осадок отфильтровывают и промывают холодной разбавленной (7 : 93)
1 При малом содержании галлия, порядка нескольких десятых миллиграмма^ раствор оставляют на ночь на краю водяной бани.
556
Гл. XXX. Галлий
серной кислотой, содержащей 1,5 г купферона в 1л. Фильтр с осадком помещают в платиновый тигель и осторожно высушивают. Затем в хороших окислительных условиях тщательно сжигают углерод и под конец прокаливают при 1000—1300° С. Охлаждают в эксикаторе и взвешивают.
Указанной обработкой можно отделить элементы сероводородной группы, например, олово, и элементы группы сульфида аммония, например железо, но при этом не отделяются титан, цирконий и ванадий, которые также осаждаются купфероном из кислого раствора. Если предварительным испытанием не установлено отсутствие этих элементов в анализируемой пробе или их отделение не обеспечивается предварительными специальными операциями, то прокаленный осадок необходимо исследовать на содержание посторонних окислов и ввести соответствующую поправку.
Осаждение бисульфитом аммония
Установлено1, что гидроокись галлия количественно осаждается бисульфитом аммония, причем осадок не пристает к стенкам стакана, как это имеет место при осаждении аммиаком.
Ход определения. К 100—200 мл солянокислого или, лучше, сернокислого раствора, содержавшего не более 0,1 г галлия (III) и свободного-от посторонних, осаждающихся аммиаком или бисульфитом аммония элементов, прибавляют раствор аммиака до слабощелочной реакции по лакмусу и затем 5 мл раствора бисульфита аммония (приготовленного насыщением раствора аммиака сернистым ангидридом). После этого раствор точно подкисляют по лакмусу разбавленной соляной кислотой, разбат вляют до 200 мл, покрывают часовым стеклом и сильно кипятят 5 мин. Фильтруют и в дальнейшем поступают, как указано в разделе «Осаждение аммиаком» (стр. 554). Для проверки полноты осаждения в фильтрат вводят 1—2 капли метилового красного и затем раствор аммиака точно до пожелтения индикатора.
* Колориметрические методы
Хинализариновый метод. Галлий можно определить колориметрическим методом, основанным на его реакции с хинализарином а, в результате которой образуется лак, окрашенный в розовый до аметистового цвет. Эта реакция весьма чувствительна (можно открыть 0,02 мг/л галлия), но крайне не специфична, и при ее применении требуется предварительное отделение от галлия многих посторонних металлов, Наилучшие результаты получаются при pH раствора, равном 5, и содержании в растворе ацетата аммония (Гн.) и хлорида аммония (0,5 н.). В этих условиях влияние алюминия, бериллия, титана, циркония, тория, редкоземельных металлов олова (IV), таллия (III) и других элементов можно устранить введением фторида который, однако, не препятствует реакции хинализарина, с железом (III), оловом (II), сурьмой (III), медью, свинцом, индием, германием, ванадием (IV) и (V) и молибденом (VI). При pH = 5 магний, марганец, железо (II), ртуть (II), таллий (III), кадмий, вольфрам, уран (VI)
l1 L. М. Dennis,!. A. Bridgman,!. Am. Chem. Soc., 40, 1534 (1918).
’ H. H. Willard, H. С. F о g g, J. Am. Chem. Soc., 59,-40 (1937).
Методы, определения
557
и мышьяк (V) не дают окраски с хинализарином. Никель, кобальт и др. мешают окраской своих ионов. Серебро, ртуть (I), висмут, ниобий и пр. осаждают гидролизом их солей или в виде хлоридов, а те количества этих металлов, которые остаются в растворе, не влияют на реакцию гал-.лия с хинализарином. Цитраты, оксалаты и тартраты препятствуют реакции, а фосфаты понижают ее чувствительность. . ,
Колориметрическое определение галлия сводится к следующему. Аликвотную часть раствора, свободного от мешающих элементов и содержащего соответстйующие количества хлорида и ацетата натрия, и 0,001—0,005 мг галлия переносят в несслеровскую пробирку. В другие такие же пробирки вводят соответствующие количества типового раствора галлия и тех же солей, какие содержатся в анализируемом растворе. Растворы, находящиеся в пробирках, нейтрализуют до pH = 5 (с помощью хингидронного электрода), в каждый из-них вводят по 1 мл 0,01%-ного спиртового раствора хинализарина1 2 и затем разбавляют до определенного объема. Интенсивность окрасок сравнивают через 2 мин.
Оксихинолиновый метод (флуоресцентный) а. Оксихинолят галлия из водного раствора, имеющего pH = 2,6—3,0, экстрагируют хлороформом и содержание галлия в экстракте устанавливают по интенсивной желтоватой флуоресценции, возникающей в ультрафиолетовом свете. Индий также реагирует при pH = 3, но относительно слабо (флуоресценция 1 мг индия соответствует флуоресценции 0,002 мг галлия). Алюминий^ и железо (II) определению не мешают.
Аналогичную реакцию дает также индий, но флуоресценции оксихинолята этого элемента значительно слабее флуоресценции галлия. Так, .при pH = 3 флуоресценция оксихинолята галлия примерно в 500 раз превосходит флуоресценцию оксихинолята индия, а при pH = 2,6 — в 10 000 раз. Оксихинолятыжелеза (III), ванадия (V), меди (II), молибдена (VI) при указанном значении pH растворяются в хлороформе с образованием окрашенных растворов, поглощающих ультрафиолетовый и видимый свет, и мешают тем самым определению галлия. Влияние железа и ванадия можно устранить восстановлением их солянокислым гидроксиламином.
Ослабляет флуоресценцию галлия также и цинк. Фтор понижает чувствительность реакции, если одновременно не присутствуют эквивалентные количества алюминия. Цитраты замедляют реакцию, а небольшие количества фосфатов лишь слабо влияют на ее чувствительность.
При анализе силикатных пород автор рекомендует отделять*, галлий от мешающих определению элементов экстракцией хлорида галлия эфиром из солянокислого раствора после восстановления железа серебром.
Ход определения. При отсутствии железа поступают следующим образом. Отбирают 5 или 10 мл анализируемого раствора и нейтрализуют .до pH = 3, если присутствует менее 0,05 мг индия, и до pH = 2,6, когда содержание индия составляет 0,05—1 мг. Добавляют 0,25 мл 0,1%-ного
1 Через неделю раствор хинализарина не пригоден к употреблению. Наилучшие результаты получаются при применении раствора 1—4-дневной давности.
2 Е. В. S a n d е 11, Ind. Eng. Chem., Anal. Ed., 13, 944 (1941); Anal. Chem., 19, 63 (1947). *Метод применен к анализу пылей производства цветных металлов [Q. Д. Г у р ь е в, Л. Б. Гинзбург, А. П. Шебаренкова, Обогащение, .металлургия цветных металлов и методы анализа, Сборник трудов Гинцветмета № 10, 1955, стр. 387].*
558
Гл. XXX. Галлий
раствора оксихинолина в 0,04 н. растворе уксусной кислоты 1 и энергично взбалтывают с 1,0—2,0 мл хлороформа в плоскодонной пробирке (18 X 150 мм) с притертой пробкой. Подобным же способом обрабатывают серию стандартных растворов, имеющих такую же концентрацию ионов водорода и такой же объем, как анализируемый раствор, и содержащих по возможности одинаковые с ним количества нейтральных солей и т. п.’ Для сравнения флуоресценции хлороформных экстрактов пользуются ртутной ультрафиолетовой лампой с баллоном из пурпурного (черного) стекла. Флуоресценцию 0,1 мкг галлия в 1 мл хлороформа можно легко заметить.
В присутствии железа к слабокислому раствору [содержащему в 5 мл не более 30—40 мг железа (III)] добавляют 0,5 г солянокислого гидроксиламина, затем раствор ацетата натрия до появления бурой окраски и оставляют стоять 10 мин для полного восстановления железа (за несколько минут раствор должен стать практически бесцветным). Доводят pH раствора до 3,0 или до 2,6, в зависимости от содержания индия, и в дальнейшем поступают, как указано выше. В этом случае раствор не следует слишком продолжительное время взбалтывать с хлороформом, так как при этом железо может снова окислиться. На дневном' свету хлороформ должен иметь лишь слабо буроватую окраску, обусловленную извлеченными следами оксихинолята железа (III).
В присутствии цинка стандартные растворы должны содержать его примерно в таких же количествах, как и анализируемый раствор.
Вместо метода стандартных серий можно пользоваться методом колориметрического титрования. Доп. перев*
1 0,10 г чистого оксихинолина нагревают с 1 млЬ н. раствора уксусной кислоты? • до растворения и разбавляют водой до 100 мл.
ЭЛЕМЕНТЫ, ОБРАЗУЮЩИЕ ПРИ ДЕЙСТВИИ СУЛЬФИДА АММОНИЯ ГИДРООКИСИ ИЛИ ОСНОВНЫЕ СОЛИ
АЛЮМИНИЙ, БЕРИЛЛИЙ, ХРОМ, ТОРИЙ, СКАНДИЙ, РЕДКОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ, ЦИРКОНИЙ, ТИТАН, НИОБИЙ И ТАНТАЛ
Глава XXXI
АЛЮМИНИЙ
Алюминий — наиболее распространенный в земной коре металл. В качестве основного компонента он входит в состав почти всех имеющих практическое значение пород, за исключением перидотитов, песчаников и известняков. Но даже в этих породах соединения алюминия^ являются обычной примесью. В самородном состоянии алюминий не встречается. В природе он всегда находится в трехвалентном состоянии. Алюминий содержится главным образом в силикатах, таких, как полевые шпаты, слюды, глины, но встречается также и в виде окиси — корунда А12О3, гидроокиси — боксита А1а0(0Н)4, в виде фторида в криолите Na3ALFB и в различных фосфатах и сульфатах.
ОБЩИЕ ЗАМЕЧАНИЯ
Определение алюминия в чистых солях обычно не вызывает особых затруднений, но установление точного содержания его в таких ‘материалах, как горные породы, минералы и керамические или металлургические продукты, является одной из наиболее сложных задач аналитической химии. В обычном ходе анализа алюминий попадает в осадок от аммиака совместно со многими другими элементами, такими, как железо, титан, цирконий, ванадий, фосфор и кремний. Содержание такой смеси часто принимают за «процентное содержание В2О3», что, естественно, может ввести в заблуждение. Если состав осадка неизвестен, его следует считать как «процентное содержание смешанных окислов». Неправильно также, как это часто практикуется, определять в осадке от аммиака только железо, иногда и титан, а остальное считать за алюминий. В большинстве случаев содержание алюминия целесообразно устанавливать по разности, после определения всех остальных компонентов во взвешенном прокален
560
Гл. XXXI. Алюминий
ном остатке. Задачу можно упростить, определив содержание группы элементов, как например, Fe2O3, TiO2, ZrO2 и V2O5, осаждением купфероном (стр. 143).
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ АЛЮМИНИЙ
Разложение минералов, содержащих алюминий, сплавлением изложено в разделе «Разложение при помощи плавнёй» (стр. 915).
Боцситы почти полностью разлагаются обработкой смесью фтористоводородной, азотной и серной кислот. Применение такого способа обработки более целесообразно, чем сплавление, так как при этом избегают введения посторонних солей и удаляют кремнекислоту. Фтористоводородную кислоту затем следует полностью удалить выпариванием с серной кислотой, в противном случае алюминий количественно не выделится из раствора при осаждейии аммиаком. Нерастворимый в кислотах остаток необходимо сплавить и перевести в раствор.
Для разложения некоторых тугоплавких алюминиевых материалов, как, например, прокаленной окиси алюминия, рекомендуют следующий способх. Обрабатывают 1 г пробы 20 мл фтористоводородной кислоты, 20 мл воды.и 50 мл разбавленного (1 : 1) раствора аммиака. Выпаривают почти досуха, прекращая нагревание, когда осадок еще влажен и не закончилось выделение паров. Эту операцию, если требуется, повторяют 2—3 раза. После этого добавляют 5 мл серной кислоты, нагревают до прекращения выделения паров и прокаливают при темнокрасном калении. Вводят 5 мл серной кислоты и снова выпаривают до прекращения выделения ее паров. Обрабатывают 3 мл серной кислоты, нагревают до появления паров серной кислоты, охлаждают и растворяют соли в воде. Этот, способ разложения удобен тем, что не вводятся нелетучие соли и раствор можно использовать для определения щелочных металлов, например для определения натрия в виде тройного ацетата (стр. 748).
Некоторые обожженные огнеупорные материалы с высоким содержанием окиси алюминия при обработке кислотами не разлагаются. Для переведения в раствор таких материалов, а также бокситов, когда требуется определение кремнекислоты, следует пользоваться методом сплавления. В таких случаях в качестве плавня пригодны карбонат натрия, перекись натрия 1 2 или пиросульфаты щелочных металлов. Для сплавления некоторых огнеупорных материалов требуется 7—10-кратное количество карбоната натрия и нагревание на полном пламени горелки Мекера не менее 1ч.'
Обожженный боксит удовлетворительно разлагается, сплавлением со смесью равных частей карбоната натрия и плавленой буры3. Плав можно растворить в разбавленной соляной или серной кислоте, н зависимости от того, какую кислоту предпочитают для обезвоживания кремнекислоты. Если заметно, что материал разложился не полностью после спла
1Н. V. Churchi 11, R. W. В г i d g е s, A. L. Miller, Ind. Eng. Chem., Anal. Ed,, 8, 348 (1936).
2 Рекомендуется применение метода взрыва с сахарным углем, как описано в разделе «Разложение при помощи плавней» (стр. 919).
8 Бор необходимо удалить перед определением кремния и алюминия, в противном случае часть его увлечется кремнекислотой и затем улетучится' в виде фторида BF3, .а часть захватится осадком от аммиака независимо от его переосаждения. Бор удаляют, как изложено! в гл. «Кремний» (стр. 753).
Методы отделения
561
вления или обработки кислотами, нерастворимый остаток отфильтровывают, прокаливают и сплавляют с пиросульфатом. Плав растворяют в основном анализируемом растворе и затем приступают к анализу.
Окись алюминия (и окись бериллия) можно перевести в раствор нагреванием с одной соляной кислотой или с добавлением окислителя, например азотной кислоты или хлора, при 250—300° С в запаянной стеклянной трубке, (стр. 402). При этом в исследуемый раствор не вводятся посторонние вещества.
Алюминий легко растворяется в некоторых кислотах и щелочах. Он быстро растворяется в концентрированной или не сильно разбавленной соляной кислоте, а также во фтористоводородной и других галогеноводородных кислотах. Реакция алюминия с разбавленной (2 : 1) серной кислотой сначала идет быстро, но затем значительно замедляется, если не ввести в раствор небольшого количества соли ртути. Растворение алюминия в азотной кислоте идет медленно, но также может быть ускорено введением ртути. Нередко для растворения алюминия применяют смесь трех кислот: азотной, соляной и серной. Растворы едких щелочей легко растворяют алюминий1.
МЕТОДЫ ОТДЕЛЕНИЯ *
Для отделения алюминия, когда он присутствует вместе с небольшим числом других элементов, известны достаточно удовлетворительные методы, но для отделения его из сложных смесей, в которых он обычно встречается, простых методов неизвестно. Так, например, осаждение фенилгидр-азином (стр. 154), которое является хорошим способом отделения алюминия от железа (II), может служить лишь предварительной ступенью, если присутствуют такие элементы, как титан, цирконий, фосфор и ванадий, как это обычно бывает.
Алюминий можно отделить от щелочных металлов (включая литий), щелочноземельных металлов, магния и небольших количеств никеля и марганца осаждением аммиаком в присутствии хлорида аммония (стр. 102). Алюминий полностью выделяется и отделяется от марганца и никеля при медленной нейтрализации раствора от pH = 3 в начале и до pH = 6,6— 6,7 в конце осаждения. Отделение таким способом от кобальта, цинка и меди не дает удовлетворительных результатов 2 *. Аммиаком осаждается настолько большое число элементов, что этот способ может служить лишь для предварительного разделения.
При осаждении алюминия по методу, изложенному в разделе «Осаждение аммиаком» (стр. 102) получены следующие результаты:
ог.гтг.т.пигттатппттта Количества сопровождающего элемента, найденные
Алюминий Со Рэлемент4™ в А1(ОН)а, г
г «.гл после однократного после двукратного
в осаждения осаждения ,
0,1 1,0 Мп 0,0017 0,00002
0,1 0,05 Ni 0,0006 Не обнаружено
0,1 0,05 Со 0,0041 0,0012
0,1 0,05 Си Ъ,0211 0,0077
0,1 .0,05 Zn 0,0216 0,0108
1 Н< V. Churchill and associates, Chemical Analysis of Aluminium 3d ed.,
Aluminium Research Laboratories, New Kensington, Pa, 1950.
’ W. Blum, Bur. Standards Sci. Paper, 286; J. Am. Chem. Soc., 38, 1282 (1916); G. E. F. Lu nd ell, H. В. К n о w 1 e s, там же, 45, 676 (1923).
36 Заказ 52 2.
562
Гл. XXXI. Алюминий
Более плотные осадки гидроокиси алюминия образуются при осаждении тиосульфатом натрия х, иодидом и йодатом калия 2 и нитритом натрия или аммония3. Эти реагенты вводятся в слабокислый раствор. Можно предполагать, что выделяющиеся при этом осадки содержат те же элементы, что и при осаждении аммиаком. В первом случае раствор обрабатывают избыточным количеством тиосульфата натрия, кипятят до удаления сернистого ангидрида и затем вводят аммиак в незначительном избытке для осаждения находящихся еще в растворе небольших количеств алюминия. Осадок всегда содержит серу. Содержание в нем железа зависит от способа обработки.
Так, при применении одного тиосульфата натрия железо соосаждается лишь в весьма незначительной степени, тогда как при добавлении аммиака выделяются заметные количества железа.
При осаждении алюминия по второму способу для достижения нужной величины pH (примерно 7,5) в раствор вводят в равных объемах 25%-ный раствор иодида калия и насыщенный раствор йодата калия (—7%-ный) и спустя 5 мин раствор обесцвечивают 20%-ным раствором тиосульфата натрия. После этого добавляют еще небольшие количества иодида и йодата и, если выделяется иод, прибавляют тиосульфат натрия, а затем раствор нагревают на водяной бане 30 мин. Этот осадок менее склонен захватывать соли щелочных металлов, чем осадок от аммиака, и, по имеющимся данным, алюминий, таким способом, количественно отделяется от бора.
При использовании нитритного метода раствор разбавляют приблизительно до 250 мл, прибавляют избыточное количество (20 мл) 6%-ного раствора чистого нитрита аммония 4, покрывают стакан часовым стеклом, кипятят до прекращения выделения окислов азота (около 20 мин) и затем нагревают на водяной бане 15—30 мин. Если в исходном растворе содержалось более 1% аммонийных солей, необходимо ввести аммиак в небольшом избытке.
Алюминий может быть количественно осажден в виде основного сукцината (соль янтарной кислоты) при добавлении к слабокислому анализируемому раствору мочевины и янтарной кислоты и кипячении®. При этом происходит отделение алюминия от ряда элементов. Успешность разделения обусловлена сочетанием четырех факторов: плотности осадка, медленного и постепенногб повышения pH раствора (в результате гидролиза мочевины, который сопровождается выделением аммиака), однородности раствора и небольшой величины pH раствора в конце реакции. Однократным осаждением таким способом достигается удовлетворительное отделение 0,1 й алюминия от такого же количества никеля и кобальта, или от 1 г кальция, бария, магния, марганца и кадмия, или же нескольких
1 G. Chancel, С. г., 46, 987 (1858); W. Gibbs, Z. anal. Chem., 3, 391 1
(1864). ;
2 A. S t ос к, Вег., 33, 548 (1900).'
3 G. W у n с о о p, J. Am. Chem., Soc., 19, 434 (1897); E. S c h i r m, Chem. Ztg., i
33, 877 (1909). |
4 Нитрит аммония иногда содержит барий. Если в анализируемом растворе содер-жатся сульфаты, к реактиву следует прибавить сульфат аммония и перед употребле- /Я нием его профильтровать. , ч
6 Н. Н. W i 11 а г d, N. К. Tang, Ind. Eng. Chem., Anal. Ed., 9, 357 (1937); 3
см. также стр. 439. 3
Методы отделения
563
миллиграммов алюминия от 1 г кобальта и никеля. Для отделения от цинка, меди и железа требуется особая обработках.
Метод осаждения алюминия в виде основного сукцината заключается в следующем. К 50—100 мл разбавленного солянокислого раствора, содержащего приблизительно 0,1 г алюминия и находящегося в высоком стакане емкостью 600 мл, прибавляют раствор аммиака до появления слабой мути. Вводят по каплям разбавленную (1 : 1) соляную кислоту до растворения мути и затем 1 каплю избытка. Раствор разбавляют до 400 лл, вводят 5 г янтарной кислоты, растворенной в 100 мл воды, 10 г хлорида аммония и 4 г мочевины и кипятят до тех пор, пока раствор не начнет мутнеть (40—45 мин). Продолжительность предварительного кипячения можно сократить введением в раствор аммиака до появления слабой опалесценции. Кипячение после этого продолжают еще 2 ч. В этой стадии pH раствора должен иметь значение 4,2—4,6. Дают отстояться, вводят немного мацерированной бумаги и фильтруют. Стакан, бумагу и осадок промывают 10 раз нейтрализованным по метиловому красному 1 %-ным раствором янтарной кислоты. Стенки стакана тщательно вытирают кусочками бумаги, которые присоединяют к основному осадку. Для полного извлечения приставшего к стакану осадка его растворяют в разбавленной соляной кислоте и переосаждают аммиаком, как указано в разделе «Осаждение аммиаком» (стр. 565). Осадок отфильтровывают через другой фильтр и затем оба фильтра с осадками прокаливают в платиновом тигле при 1200°С до постоянной массы.
Несколько менее удовлетворительно происходит отделение никеля, кобальта, цинка и меди, если янтарную кислоту заменить 1 г сульфата аммония, так как при этом в конце реакции значение pH раствора достигает 6,5—7,5.
Описан способ осаждения алюминия бензоатом аммония в связи с анализом магниевых сплавов 1 2.
Отделение, основанное на осаждении алюминия карбонатом или сульфидом аммония, не дает достаточно удовлетворительных результатов вследствие растворимости осадка в избытку реагентов, который обычно приходится вводить. При использовании ацетатного метода (стр. 103) также следует опасаться неполного осаждения алюминия, в особенности когда его содержание преобладает.
Метод отделения, основанный на осаждении карбонатом бария (стр. 108), дает хорошие результаты, но его редко можно применять, если осаждается один алюминий.
Необходимо иметь в виду, что сульфат бария, а возможно и сульфат свинца увлекают с собой значительные количества алюминия3.
Железо, титан, цирконий, редкоземельные элементы и марганец можно отделить от алюминия осаждением едким натром (стр. 109). Этот метод не пригоден в присутствии магния и никеля, которые увлекают алюминий, а также в том случае, когда значительно преобладает содержание титана, который при этом полностью не осаждается. Кальций не мешает. Фосфор, ванадий, вольфрам, молибден и другие элементы сопровождают алюминий.
Разделению обработкой смесью едкого натра и карбоната натрия препятствуют магний и марганец, тогда как кальций при этом не мешает.
Элементы сероводородной группы отделяют от алюминия осаждением их сероводородом из кислого раствора (стр. 83). Осаждение сульфидом
1 См. также A. J. Boyle, D. F. Musser, Ind. Eng., Chem., Anal. Ed., 15, 621 (1943);
2 V. A. Stenger, W. R. Kramer, A. W. Beshge t о or, Ind. Eng. Chem., Anal. Ed., 14, 797 (1942).
3 Так, например, при анализе стекла, содержавшего 17,5% РЬО и 1,4% ВаО, после растворения 10 г пробы в H2SO4 + HF и выпаривания раствора до появления * паров H2SO4 было определено 0,14% вместо 0,18% А12О3, причем не учитывался алюминий, захваченный осадком сульфата свинца и бария.
36*
564
Гл. XXXI. Алюминий
аммония из аммиачного раствора, содержащего винную кислоту (стр. 89), с успехом служит для отделения железа.
Кремнекислота выделяется в более компактной и легче отделяемой фильтрованием форме из сернокислого раствора, содержащего значительные количества алюминия, если в слегка охлажденный после выпаривания до появления паров серной кислоты раствор быстро влить немного холодной воды, затем разбавить его теплой водой, нагреть и перед фильтрованием ввести немного бумажной массы.
Осаждение купфероном (стр. 143) дает четкое отделение ряда элементов от алюминия. Этот метод особенно успешно применяется в тех случаях, когда требуется отделить малые количества железа, титана, циркония, ванадия, олова, ниобия и тантала от больших количеств алюминия, как, например, при анализе бокситов или металлического алюминия. Алюминий можно выделить из фильтрата добавлением еще некоторого количества купферона и нейтрализацией раствора до слабокислой реак-. ’ ции (pH около 5). Его можно осадить также из нагретого до 70° С фильтрата оксихинолином после добавления аммиака до щелочной реакции. В дальнейшем поступают, как указано на стр. 572.
Железо, никель, кобальт, хром, цинк, галлий, медь, олово и некоторые другие элементы успешно отделяются от алюминия электролизом с ртутным катодом (стр. 165) разбавленного сернокислого растворах. Железо можно также отделить ют алюминия экстракцией эфиром холодного разбавленного солянокислого раствора (стр. 161). Очень хороший метод отделения алюминия от хрома основан на окислении хрома до хромата нагреванием с хлорной кислотой дб появления обильных паров, разбавлении охлажденного раствора и осаждении алюминия аммиаком.
Насколько известно, единственным способом, который дает возможность одной операцией достаточно удовлетворительно отделить алюминий от железа, титана, редкоземельных элементов и бериллия, является осаждение алюминия в виде шестиводного хлорида 1 2 А1С13-6Н2О. Для отделения этим методом концентрированный раствор хлорида алюминия в смеси соляной кислоты и эфира насыщают газообразным хлористым водородом. Растворимость хлорида алюминия составляет приблизительно 5 частей АКЛд-бНгО (что соответствует содержанию 1 части А12О3) в 125 000 частях смеси. Показано2, что алюминий таким путем можно отделить также от железа, цинка, меди, ртути и висмута. Четкое отделение достигается от фосфора и, по всей вероятности, от многих других элементов. Хлориды щелочных металлов нерастворимы в эфирносолянокислой смеси и должны отсутствовать.
Метод заключается наследующем. Солянокислый раствор, свободный от щелочных металлов, выпаривают до небольшого объема (но так, чтобы не началась еще кристаллизация солей), примерно до 5—10 мл. Раствор охлаждают, разбавляют соляной кислотой до 12—25 мл и насыщают газообразным хлористым водородом, все время при охлаждении, не допуская, чтобы температура раствора поднялась выше 15° С. Затем прибавляют эфир в количестве, равном объему раствора, и снова при охлаждении насыщают хлористым водородом.
1 Относительно применения этого метода к растворам, содержащим алюминий, медь, железо, никель, марганец и хром, можно найти указания в статье: D. Н. В г о- , р h у, Ind. Eng. Chem., 16, 963 (1924).
2 F. A. Gooch, F. S. H a v e n s, Am. J. Sci., [4], 2, 416 (1896).
Методы определения
565
Одновременно или в то время, когда раствор охлаждается, приготовляют промывную жидкость, состоящую из равных объемов соляной кислоты и эфира, насыщенных хлористым водородом при 15° С.
Выпавший осадок хлорида алюминия отфильтровывают через тигель Гуча или, лучше,Мунро, предварительно пропустив через него немного промывной жидкости для охлаждения. Если осадок предполагают непосредственно прокалить и взвесить, для фильтрования применяют тигель, взвешенный после того, как он был промыт промывной жидкостью и прокален.
Осадок промывают холодной промывной жидкостью до удаления посторонних солейх. Если осадок непосредственно не прокаливают, его растворяют в .разбавленной соляной кислоте и затем раствор обрабатывают в соответствии с тем, предполагают ли переосадить алюминий в виде хлорида или осадить аммиаком в виде гидроокиси. Если осадок хлорида алюминия хотят перевести в А12О3, его покрывают тонким слоем чистой (беззольной) окиси ртути для избежания механических потерь, слабо нагревают до наступления гидролиза, затем повышают температуру, чтобы удалить ртуть, и под конец прокаливают при 1000—1100° С. Взвешивают в виде А12О3.
Алюминий количественно осаждается оксихинолином и в виде окси-хинолята отделяется: от фосфатов, арсенатов, фторидов и боратов в аммиачном растворе 1 2; от ванадия, молибдена, ниобия, тантала и титана в аммиачном растворе, содержащем перекись водорода2; от урана в растворе, содержащем карбонат аммония2, и от бериллия в уксуснокислом растворе 3 (стр. 583).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Осаждение аммиаком
Основным методом определения алюминия является осаждение его аммиаком в виде гидроокиси и взвешивание в виде окиси А12О3. При осаждении аммиаком в большинстве случаев совместно с алюминием осаждаются и другие металлы, поэтому почти всегда приходится сначала взвешивать сумму всех окислов, а затем, определив содержание посторонних компонентов в осадке, вычислять содержание алюминия по разности. Таким образом, в результат определения алюминия включаются ошибки определения всех сопровождающих его элементов.
Осаждение гидроокиси алюминия начинается приблизительно при pH = 3 и заканчивается при pH = 6,5—7,5, что соответствует примерно изменению окраски метилового красного или розоловой кислоты. В более щелочных растворах начинается растворение осадка, которое становится весьма заметным4 при pH = 10.
1 При особо точных анализах фильтрат и промывные воды следует упарить до небольшого объема' и снова обработать соляной кислотой и эфиром.
2 G. Е. F. Lu nd ell, Н. В. К п о w 1 е s, J. Research NBS, 3, 91 (1929). .
3 I. M. Kolthoff, E. В. S a n d e 11, J. Am. Chem. Soc., 50, 1900 (1928); H. B. Knowles, J. Research NBS, 15, 87 (1935).
4 В действительности, при проведении рядовых анализов введение избыточного количества аммиака в раствор больше отражается на результатах определения кальция и магния, чем алюминия, так как потери алюминия в фильтрате компенсируются наличием в осадке осаждающихся совместно с алюминием примесей, определение которых обычно опускают, тогда как остающийся в фильтрате алюминий, соосаждаясь частью с кальцием и в основном с магнием, увеличивает положительную ошибку, которая обычно имеет место при определении этих элементов.
566
Гл. XXXI. Алюминий
Наличие хлорида аммония в растворе предотвращает осаждение таких элементов, как магний, ограничивает повышение щелочности раствора и способствует коагуляции осадка. Утверждение, что наличие сульфатов в растворе приводит к неполноте осаждения гидроокиси алюминия, лишено основания х. В присутствии больших количеств фосфата аммония алюминий выделяется не количественно и осаждение может вовсе не наступить, если содержание алюминия очень мало.
Для промывания осадка нельзя применять горячую воду, так как гидроокись алюминия легко переходит в коллоидную форму2. Для этой цели обычно пользуются 2%-ным раствором хлорида или нитрата аммония. Если фильтрат и промывные воды предполагают пооде подкисления выпаривать в платиновой посуде, лучше применять хлорид аммония. При использовании нейтральных солей нет необходимости промывную жидкость нейтрализовать по метиловому красному.
Присутствие хлорида аммония в осадке не вызывает улетучивания алюминия в процессе прокаливания. Осадки, содержащие значительные количества железа, целесообразно под конец промыть 2%-ным раствором нитрата аммония. Потери алюминия не наблюдаются также, когда гидроокись или окись алюмйния обрабатывают соляной кислотой, выпаривают досуха и затем осторожно прокаливают.
При определении А12О3 в таких материалах, как бокситы или огнеупоры с высоким содержанием алюминия, основными источниками ошибок являются:
1. Неполное разложение материала. Если имеется сомнение в полноте извлечения алюминия, нерастворившийся остаток после растворения плава следует снова сплавить, плав растворить и полученный раствор присоединить к основному раствору.
2. Отбрасывание нелетучего остатка после обработки кремнекислоты фтористоводородной и серной кислотами. Этот остаток необходимо сплавить, плав растворить и раствор присоединить к фильтрату после отделения кремнекислоты.
3. Необнаружение некоторых соединений, перешедших в осадок от аммиакй. Так, например, в этом осадке могут быть щелочноземельные металлы, осажденные в виде фосфатов, если содержание фосфора превышает, то его количество, которое может быть захвачено алюминием и железом. Сюда же относятся ошибки в количественном определении окислов, сопровождающих алюминий при осаждении аммиаком. Проводим перечень окислов, выделяемых аммиаком при анализе Образца боксита № 69 (Бюро Стандартов .США); этот перечень характеризует степень погрешности, которая может быть допущена при определении алюминйй, если пренебречь присутствием какого-нибудь из этих окислов в осадке от аммиака3: Fe2O3 5,66%, TiO2 3,07%, ZrO2 0,08%, Р2О3 0,11%, V2O3 0,03% и Сг2О3 0,04%.
---------- г
1 Так, например, при анализе аликвотных частей раствора А1С13 осаждением аммиаком обычным путем в присутствии NH4C1 найдено 0,0969 и 0,0968 г А12О3, а осаждением аммиаком после введения в растворы по 5 мл H2SO4, выпаривания растворов до появления паров серной кислоты и разбавления водой определено 0,0968 и 0,0970 г. А1ЯО„
а Так, W. Blum [Bur. Standards Sci. Paper, 286; J. Am. Chem. Soc., 38, 1282 (1916)] после промывки 75 мл горячей воды осадков, содержание алюминия в которых соответствовало 0,1 г А12О3, нашел в промывных водах 0,5—2 мг А12О3.
8 Об анализе осадка от аммиака см. стр. 118.
Методы, определения
567
4. Соосаждение гидроокисей, для растворения которых требуется значительный избыток аммиака, например гидроокисей цинка, меди и кобальта. Эти гидроокиси не могут быть отделены переосаждением осадка, так как для переведения их в раствор требуется прибавление такого большого количества аммиака, при котором гидроокись алюминия также частично растворяетсях.
5. Осаждение щелочноземельных металлов в виде карбонатов. Выделение карбонатов щелочноземельных металлов при pH раствора 6,5— 7,5 за счет поглощения двуокиси углерода из воздуха в процессе кипячения раствора мало вероятно, но оно имеет место, когда для осаждения пользуются раствором аммиака, содержащим карбонат аммония.
6. Неудаление солей щелочных металлов, увлекаемых осадком от аммиака. Так, например, после сплавления 0,5 г боксита с 7—10 г карбоната натрия и отделения кремнекислоты в прокаленное осадке смешанных окислов было найдено методом Лоуренса Смита (стр. 1006) до 0,7 мг солей щелочных металлов после двукратного и 0,1 мг после трехкратного осаждения аммиаком. Ошибка за счет содержания щелочных металлов в осадке, таким образом, может достигать 0,14% А12О3 (при навеске 0,5 г) после двукратного осаждения аммиаком и должна быть еще значительно больше, если осадок от аммиака не переосаждается.
7. Неполное растворение осажденной гидроокиси алюминия. Пол-ностькхудалить гидроокись алюминия с фильтра обработкой кислотой очень трудно, поэтому тщательно промытый фильтр следует сохранить и затем сжечь вместе с конечным осадком гидроокиси алюминия. Можно также перед отфильтровыванием осадка, после вторичного осаждения алюминия аммиаком, ввести фильтр в раствор и разболтать его, пока он не распадается на волокна.
8. Несоответствующее прокаливание осадка от аммиака. Было показано 1 2, что А12О3, прокаленная до постоянной массы на паяльной горелке, не теряет в массе после дальнейшего прокаливания при 1440—1460° С. Установлено также, что для достижения постоянной массы осадок достаточно прокалить в пламени паяльной горелки в течение 5—10 мин. Это, однако, справедливо для небольших осадков, выделяемых из чистых растворов. Большие же осадки сложного состава, которые получаются при анализе бокситов или огнеупорных материалов, необходимо прокаливать до постоянной массы при температуре не ниже 1200° С. Например, осадок, содержавший 0,35 г А12О3, прокаленный 1 ч при 800° С, после дальнейшего прокаливания в муфеле при 900° С в течение такого же времени потерял только 0,1 мг. После, прокаливания же в течение 1 ч при 1050° С потеря составляла 1,4 мг, а после прокаливания в пламени паяльной горелки осадок потерял в массе еще 1,5 мг. В обоих случаях была учтена потеря в массе платинового тигля. Ошибка определения, вследствие неполного удаления воды из окиси алюминия, составляла, таким образом, 0,58% абсолютных (при навеске 0,5 г), если ограничиться прокаливанием осадка при 900°С, и 0,30% в случае прокаливания при 1050° С.
9. Недостаточное предохранение осадка от поглощения влаги перед взвешиванием или во время взвешивания. Окись алюминия, прокаленная
1 G. Е. F. L u n d е 11, Н-. В. К п о w 1 е s, J. Pesearch NBS, 3, 91 (1929); W. Blum, Bur. Standards Sci. Paper, 286; J. Am. Chem. Soc., 38, 1282 (1916).
2 W. Blum, цит. выше.
568
Гл. XXXI. Алюминий
при недостаточно высокой температуре (900—1000° С), гигроскопична и в течение первых 5 мин поглощает большую часть из того количества влаги, какое она способна абсорбировать в течение 24 ч. Это имеет существенное значение, так как атмосфера в эксикаторе непосредственно после того, как его открывают, немногим отличается от комнатной. Скорость поглощения влаги, однако, весьма невелика, если во время охлаждения и взвешивания тигель закрыть хорошо пригнанной крышкой, а если к тому же в эксикаторе находится сильный поглотитель влаги и взвешивание проводится достаточно быстро, то ошибка настолько незначительна, что ею можно пренебречь. Так, хорошо закрытый тигель заметно не изменился в массе, находясь на чашке весов в течение 5 мин, но масса его увеличилась на 1 мг через 5 мин после того, как он был открыт. Окись алюминия, прокаленная при 1200° С, менее гигроскопична, чем прокаленная при 1000° С.
10. Невнесение поправки на содержание кремнекислоты, соосажден-ной с гидроокисями. При осаждении аммиаком кремнекислота всегда захватывается осадком и содержание ее в осадке зависит от полноты ее выделения в начальной стадии анализа и от количеств кремнекислоты, вводимых впоследствии с реактивами, главным образом с раствором аммиака. Полностью выделить кремнекислоту из раствора не удается даже двукратным обезвоживанием с соляной или серной кислотой. При тщательном проведении анализа содержание кремнекислоты в осадке (при навеске 0,5 г) может соответствовать 0,05—0,3% А12О3.
11. Неправильное определение поправки на загрязнения, внесенные с реактивами и извлеченные из стекла посуды. Небольшой осадок, который выделяется аммиаком из раствора одних лишь применяемых реактивов при проведении «холостого» опыта, как правило, менее полно захватывает SiO2 и Р2О5, содержащиеся в растворе, чем значительно больший осадок, получаемый при анализе пробы. Так, например, при, анализах 0,5 г навесок боксита средняя поправка на загрязнения в реактивах при проведении «холостых» опытов составила 0,25 мг. После прибавления к реактивам 0,25 г А12О3 в виде чистого хлорида алюминия величина поправки составила 1,8 мг, когда содержание кремнекислоты в хлориде алюминия не было принято во внимание, и 0,9 мг после учета содержащейся в нем кремнекислоты. Таким образом, когда для определения величины поправки через все стадии анализа проводили «холостой» опыт с одними только реактивами, были получены повышенные результаты, которые отличались от истинных на 0,13%, а если в реактивы был введен хлорид алюминия, но содержание кремнекислоты в нем не учитывалось, результаты оказались пониженными на 0,18%.
Ход определения. В анализируемый раствор, содержащий не менее 5 г хлорида аммония в объеме 200 мл или эквивалентное ему количество соляной кислоты, вводят несколько капель метилового красного (0,2%-ный спиртовый раствор) и нагревают до кипения. Затем осторожно по каплям прибавляют разбавленный раствор аммиака до отчетливого пожелтения индикатора1 * *. Раствор кипятят 1—2 мин и тотчас, же филь4-
1 В присутствии больших количеств железа трудно заметить переход окраски, так как гидроокись железа выделяется прежде', чем раствор становится шелочным по
метиловому красному. Конечную точку нейтрализации легко установить, если после
прибавления достаточного для осаждения железа количества аммиака раствор кратко-
временно прокипятить и дать ему отстояться. В отстоявшейся жидкости отчетливо
Методы определения
569
труют через бумажный фильтр. Осадок тщательно промывают горячим 2%-ным раствором хлорида или нитрата аммония. Ебли это требуется, в зависимости от характера или количества присутствующих посторонних элементов, осадок растворяют в разбавленной (1 : 3) соляной кислоте и переосаждают аммиаком. После вторичного осаждения, если желательно, можно ввести в раствор мацерированную бумагу.
Влажный осадок завертывают в фильтр, сушат, бумагу фильтра обугливают и осадок прокаливают* 1 под конец в течение 5—10 мин при 1200° С. Охлаждают в эксикаторе, взвешивают и прокаливание повторяют до постоянной массы. Если осадок не очень мал, прокаливание на пламени бунзеновской горелки не обеспечивает полного обезвоживания окиси алюминия. Когда осадок содержит большие количества железа и других окислов, восстанавливающихся обугленной бумагой или теряющих кислород при сильном прокаливании, окончательное прокаливание следует проводить так, чтобы был обеспечен свободный доступ воздуха в тигель. Для этого удобна электрическая печь, так как в ней нет восстановительной атмосферы.
Взвешенная окись алюминия почти всегда содержит кремнекислоту. Количество последней колеблется в зависимости от того, насколько полно она была выделена в начале анализа и в какой посуде проводилось осаждение — в фарфоровой, стеклянной или платиновой, — а также от чистоты реактивов. Если осадок невелик, его можно непосредственно обработать каплей разбавленной (1 : 1) серной кислоты и 2—5 мл фтористоводородной кислоты, выпарить досуха, затем прокалить, медленно повышая температуру до прежней, и взвесить. В том случае, когда .осадок весит больше 50 мг, вводят большее количество серной кислоты, медленно выпаривают и, постепенно повышая температуру2, прокаливают.
Кремнекислоту в некоторых случаях можно отделить также сплавлением с пиросульфатом и выпариванием с серной кислотой, как описано в III части (стр. 955).
Осаждение в виде фосфата алюминия
Определение алюминия взвешиванием его в виде фосфата не дает вполне удовлетворительных результатов, так как при точном соблюдении необходимых условий осаждения в осадке всегда содержатся избыточные количества Р2О5. Определить полноту промывания осадка по реакции на
видна окраска индикатора. Затем, если это требуется, прибавлять еще раствор аммиака, а при его избытке нейтрализуют разбавленной кислотой. В том случае, когда в качестве индикатора применяют бромтимоловый синий, аммиак прибавляют к горячему раствору до изменения окраски в сине-зеленую. Раствор затем кипятят, пока окраска не станет чисто зеленой, если присутствует один алюминий, или желтовато-зеленой при содержании значительных количеств железа. При применении бромкрезолового пурпурного конечной точкой считается появление пурпурной окраски индикатора (pH = 6,8).
1 Присутствие в осадке небольших количеств хлорида аммония, введенных с промывной жидкостью, не приводит к потере алюминия в процессе прокаливания. Не улетучивается также и железо, если содержание хлорида ниже 1% [D a u d t, J. Ind. Eng. Chem., 7, 847 (1915)].
2 Воспроизводимость метода характеризуется следующими результатами: после двукратного осаждения, введения мацерированной бумаги при вторичном осаждении и удаления кремнекислоты непосредственной обработкой HF + H2SO4 было получено 0,0970; 0,0969 и 0,0968 г А12О3.
570
Гл. XXXI. Алюминий
Р2О5 в промывной воде не представляется возможным вследствие гидролиза А1РО4, который происходит независимо от того, проводится промывание чистой водой* или водой, содержащей электролит, например нитрат аммония. Установлено1, что промывание осадка облегчается введением в него мацерированной бумаги и что удаление избытка Р2О5 можно определить по исчезновению реакции на хлориды, которые прибавляют в начальной стадии анализа. При выполнении этих требований метод дает очень точные результаты, если определяют несколько миллиграммов А12О3; большие количества алюминия можно определить с точностью до 1—2%.
Фосфат алюминия осаждают из разбавленного уксуснокислого раствора, содержащего ацетат натрия (pH = 5—5,4), не менее чем пятикратным количеством реагента, лучше (NH4)2HPO4. При осаждении из щелочного раствора или меньшим количеством осадителя получаются пониженные результаты. В условиях осаждения алюминия количественно выделяются также железо, титан, цирконий и другие элементы, а частично — марганец, цинк и кальций. В присутствии железа возникают затруднения, независимо от того, восстанавливают ли его тиосульфатом nepeft осаждением или выделяют совместно с алюминием и затем его содержание вычитают, определив его и пересчитав на FePO4. В первом случае обычно получаются пониженные результаты для алюминия вследствие наличия ' в растворе сернистой кислоты, которая несколько растворяет фосфат алюминия.
Этого можно избежать, применив другой восстановитель, например сероводород. Вычитание рассчитанного количества FePO4 приводит к повышенным результатам, так как промытый осадок фосфата железа, как правило, содержит избыток Р2О8. В связи с этим железо, а также и другие мешающие элементы, лучше отделять перед осаждением алюминия.
Влияние кальция можно устранить, поддерживая величину pH раствора ниже 6, введением большого избытка хлорида аммония, а прц больших количествах кальция осадок фосфата следует переосаждать. Соли щелочных металлов и хроматы, даже и в больших количествах, не влияют на осаждение. Ванадий частично выделяется вместе с фосфатом алюминия, но после двукратного осаждения практически полностью отделяется от алюминия, если содержание последнего не превышает 50 мг. Многие элементы, влияющие на осаждение алюминия фосфатом, можно отделить от алюминия обработкой едким натром, а если присутствует кальций, то с добавлением карбоната натрия. Наиболее целесообразно поступать следующим образом. Раствор нейтрализуют едким натром (свободным от алюминия) и вливают в раствор, содержащий такое количество едкого натра, чтобы после осаждения гидроокисей остался избыток щелочи в 5—10%. Затем раствор разбавляют до определенного объема и после фильтрования отбирают половину для определения алюминия. В присутствии магния и никеля этот метод не пригоден.
Осадок фосфата алюминия можно промывать горячей водой или,, лучше, горячим 5%-ным раствором нитрата аммония. Промывание разбавленными кислотами, например уксусной, или аммиачными растворами приводит к пониженным результатам. Прокаливание следует проводить осторожно, так как осадок склонен растрескиваться, причем в меньшей
1 G. Е. F. Lun de 11, Н. В. К п о w 1 е s, Ind. Eng. Chem., 14,1136 (1922).
Методы, определения
571
степени, если была введена мацерированная бумага. Прокаленный остаток гигроскопичен, и поэтому его необходимо взвешивать быстро и в плотно закрытом тигле.
Ход определения. Анализируемый раствор, желательно содержащий одну только соляную кислоту, разбавляют до 400 мл, нейтрализуют с таким расчетом, чтобы в растворе осталось приблизительно 10 мл свободной соляной кислоты, и бтрибавляют 1 г (NH4)2HPO4 или больше, если этого количества недостаточно, чтобы обеспечить десятикратный избыток реагента. Вводят мацерированную бумагу, полученную из одного 11-сан-тиметрового фильтра, или половину таблетки готовой бумажной массы, затем 2 капли метилового оранжевого, нейтрализуют раствором аммиака и прибавляют 0,5 мл соляной кислоты, после чего окраска раствора должна опять стать розовой. Раствор нагревают до кипения, прибавляют 30 мл 25%-ного раствора ацетата аммония и продолжают кипятить 5 мин. Осадок отфильтровывают и промывают горячим 5%-ным раствором нитрата аммония до тех пор, пока 5 мл промывной жидкости не дадут лишь едва заметную опалесценцию с подкисленным раствором нитрата серебра. Во время фильтрования и промывания нельзя давать осадку высыхать. Фильтр с осадком высушиващт, осторожно нагревая в открытом платиновом или фарфоровом тигле, и сжигают уголь при низкой температуре. Под конец, закрыв тигель крышкой, прокаливают примерно при 1000° С до постоянной массы. Взвешивают в виде А1РО4.
Точность метода характеризуется следующими данными1:
Взято А12О3, г ............... 0,0019 0,0094 0,0943 0,1886
Определено А12О8 (в виде А1РО4), г 0,0019 0,0096 0,0964 0,1930
Повреждения платинового тигля никогда не наблюдалось, если углерод фильтра полностью сжигался при низкой температуре в хороших окислительных условиях до окончательного прокаливания осадка при 900—1000 °C.
Осаждение оксихинолином 1
Алюминий количественно осаждается оксихинолином 2 при значении pH раствора 4,2—9,8. Осадок имеет состав Al(C9HeNO)3 и может быть непосредственно взвешен. Кроме того, его можно прокалить и взвесить получающуюся в результате этого А12О9 или растворит в разбавленной соляной кислоте и оттитровать.
Осадок отличается склонностью захватывать некоторые количества реагента, в связи с чем результаты получаются несколько повышенные, если оксихинолят алюминия непосредственно взвешивать или титровать.
Осаждение из кислого раствора. Осаждение оксихинолином из слабокислого раствора с успехой используется для отделения алюминия от щелочноземельных металлов, магния и бериллия. Применение этой реакции для определения ограничивается, однако,, тем, что многие элементы осаждаются оксихинолином из кислого раствора. Пользоваться этим методом можно лишь в исключительных случаях, как, например, при анализе полевых шпатов, в которых содержание железа, титана, циркония и фосфора настолько незначительно, что ими можно пренебречь, или при
1 G. Е. F. L u n d е 11, Н. В. Knowles, Ind. Eng. Chem., 14, 1136 (1922).
’H. Goto, J. Chem. Soc. Japan, 54, 725 (1933).
572
Гл. XXXI. Алюминий
определении алюминия в растворах, из которых мешающие элементы предварительно удалены А
Ход определения. В анализируемый кислый раствор, содержащий в 200 мл 10 мл соляной кислоты и не более 0,1 г алюминия и св’ободный от мешающих элементов, перечисленных на стр. 148, вводят 15 мл раствора ацетата аммония, приготовленного растворением 30 г соли в 75 мл воды, и 8—10 капель 0,04%-ного раствора бромкрезоловбго пурпурного. Прибавляют разбавленный (1 : 1) раствор аммиака до отчетливо пурпурной окраски индикатора и затем при перемешивании медленно вводят из бюретки 2,5%-ный раствор оксихинолина в уксусной кислоте1 2, в количестве на 15—25% большем, чем это требуется по расчету (1 А1 соответствует 3 C9H7NO). Раствор нагревают до кипения, время от времени перемешивая, и слабо кипятят 1 мин. Охлаждают до 60° С и фильтруют3 (при слабом отсасывании) через стеклянный тигель № 4 с плотным пористым дном (5—15° С). Тигель и осадок промывают горячей /70—75° С) водой, 1—2 порциями по 10 мл, а затем холодной водой до тех пор, пока промывная вода не станет бесцветной. Объем промывных вод не должен превышать 100 мл.
Заканчивают определение алюминия весовым или объемным способом, как изложено на стр. 574 [см. «Титрование смесью бромида и бромата калия», «Взвешивание в. виде Al(C9HeON)3» или «Взвешивание в виде А12О3»].
При нейтрализации раствора аммиаком сначала выделяется гидроокись алюминия, которая затем переходит в оксихинолят. Если фильтрат надлежит отбросить или содержание в нем винной кислоты не препятствует дальнейшему его использованию, образование осадка гидроокиси алюминия можно предотвратить прибавлением винной кислоты в пятикратном по отношению к содержанию алюминия количестве. Винную кислоту вводят в раствор перед добавлением ацетата и нейтрализацией аммиаком 4.
Осаждение из щелочного раствора. Оваждением оксихинолином из аммиачного раствора можно отделить алюминий от фосфора, мышьяка, фтора и бора, а в присутствии перекиси водорода — от тантала, ниобия-, титана, ванадия, хрома и молибдена. От урана алюминий отделяют осаждением оксихинолином из раствора, содержащего карбонат аммония. От элементов, образующих комплексные ионы с цианидом, как, например, железо, медь, кобальт и никель, алюминий отделяют осаждением из аммиачного раствора, содержащего цианид щелочного металла. В связи с тем что в щелочной среде оксихинолином осаждаются многие элементы,
1 Влияние небольших количеств (<Ч мг) железа на осаждение алюминия оксихинолином из слабокислого раствора (pH = 4—6) можно устранить восстановлением железа до двухвалентного состояния сернистой кислотой или солянокислым гидроксиламином и введением комплексообразователя, например, а> а'-дипиридила или 1,10-фенантролина, которые образуют комплексные соединения с железом (II), устойчи-. вые при этой кислотности [Е. W. Koenig, Ind. Eng. Chem., Anal. Ed., 11, 532 (1939)].
2 Для приготовления раствора 12,5 г оксихинолина обрабатывают 25 мл ледяной уксусной кислоты при слабом нагревании до растворения. Полученный раствор вливают в 450 мл нагретой до 60° С воды. Охлаждают, фильтруют, если требуется, и затем разбавляют до 500 мл.
3 Осадок можно также отфильтровывать через плотный бумажный фильтр (синяя лента).
4 Подробно см. Н. В. Knowles, J. Research NBS, 15, 87 (1935).
Методы определения
573
метод этот для определения алюминия имеет ограниченное применение. Он используется главным образом в таких случаях, как анализ фильтратов после осаждения едким натром или растворов после выщелачивания щелочных плавов водой.
Ход определения. Определение в присутствии фосфора, мышьяка, фтора и бора. К 100—200 мл кислого раствора, содержащего не более 0,1 г алюминия и свободного от ванадия, тантала, ниобия, титана, молибдена, хрома и других перечисленных на стр. 148 элементов, прибавляют избыточное количество 2,5%-ного раствора оксихинолина в разбавленной уксусной кислотех. Затем раствор нейтрализуют разбавленным (1 : 1) раствором аммиака до щелочной реакции, после чего прибавляют еще по 10 мл раствора аммиака на каждые 100 мл анализируемого раствора. Нагревают до 60—70° С и продолжают нагревать при этой температуре до тех пор, пока осадок не станет плотным и кристаллическим. После этого охлаждают, лучше в ледяной воде, фильтруют через плотный бумажный фильтр или через пористый стеклянный тигель и тщательно промывают осадок холодным разбавленным (1 : 40) раствором аммиака, содержащим 25 мл предварительно нейтрализованного аммиаком реактива в 1 л. Если осадок следует непосредственно взвесить или после растворения титровать, его под конец промывают горячей водой для удаления реагента1 2
Определение в присутствии тантала, ниобия, титана, ванадия, хрома и молибдена. Единственным дополнением, которое следует ввести в метод, изложенный выше (см. •«Определение в присутствии фосфора, мышьяка, фтора и бора»), является добавление 10—15 мл 3%-ной перекиси водорода перед введением в раствор оксихинолина2.
Определение в присутствии урана. При необходимости отделить уран от алюминия, вместо нейтрализации аммиаком, как указано выше (см. «Определение в присутствии фосфора, мышьяка, фтора и бора»), раствор нейтрализуют насыщенным раствором карбоната аммония, после чего прибавляют еще по 25 мл раствора карбоната на каждые 100 мл раствора и нагревают приблизительно до 50° С (избегая бурного выделения газов, вызываемого слишком быстрым нагреванием)2.
Определение в присутствии элементов, образующих устойчивые комплексные цианиды3 (ж е-лезо, никель, кобальт, медь, молибден, хром4). Анализируемый раствор, содержащий свободную соляную и хлорную
1 Для приготовления раствора 12,5 г оксихинолина обрабатывают 25 мл ледяной уксусной кислоты при слабом нагревании до растворения. Полученный раствор вливают в 450 мл нагретой до 60s С воды. Охлаждают, фильтруют, если требуется, и затем разбавляют до 500 мл.
2 Подробно см. G. Е. F. L u n d е 11, Н. В. Knowles,!. Research NBS, 3, 91 (1929).
8 Т. Н е с z к о, Chem. Ztg., 58, 1032 (1934).
4 Хром задерживает осаждение оксихинолята алюминия из раствора, содержа-
щего тартрат и цианид. Если присутствует больше 25 мг хрома, "прибавляют 20 мл хлорной кислоты (если ее нет в растворе), выпаривают до появления паров хлорной кислоты и затем кипятят 5 мин для переведения хрома в хромовую кислоту. Разбавляют до 100 мл и снова кипятят несколько минут. Вводят в небольшом избытке 25 % -ный раствор нитрата свинца. Охлаждают раствор до 15 °C, фильтруют и промывают осадок холодной разбавленной (1 : 99) хлопной кислотой.
574
Гл. XXXI. Алюминий
кислоты, разбавляют примерно до 250 мл и прибавляют 50 мл 20%-ного раствора винной кислоты. Если раствор не был обработан для удаления хрома, вводят 20 мг нитрата свинца, который способствует последующему выделению марганца в виде сульфида. Прибавляют раствор аммиака в небольшом избытке, 10 г цианида калия и значительное количество беззольной мацерированной бумаги. Через раствор пропускают сильную струю сероводорода в течение 20—25 мин. Фильтруют через 11-сантиметровый фильтр, содержащий немного мацерированной бумаги и промывают осадок 8—10 раз сероводородной водой, содержащей по 2% тартрата аммония, цианида натрия и хлорида аммония.
Фильтрат кипятят 4—5 мин и затем медленно при энергичном перемешивании вводят в него избыточное количество 2,5 %-ного раствора оксихинолина в разбавленной уксусной кислоте (стр. 572). Прибавляют 1—2 мл аммиака й перемешивают механической мешалкой 15 мин. Фильтруют при слабом отсасывании через плотный фильтр (или пористый стеклянный тигель № 4), если осадок предполагают взвесить в виде оксихино-лята. Осадок промывают сначала 8—10 раз холодным водным раствором, содержащим по 2% тартрата аммония, цианида натрия и хлорида аммо-ния, затем 5—10 мл горячей воды (70—75° С) и, наконец, холодной водой до тех пор, пока промывная вода не станет бесцветной.
Титрование смесью бромида, и бромата калия. Тигель с осадком помещают в стакан емкостью 600 мл, вводят 200 мл разбавленной (1 : 4) соляной кислоты и слабо нагревают до растворения осадка. Обычно тигель можно оставлять в стакане. При выполнении особо точных анализов тигель вынимают из стакана, ополаскивают и затем для извлечения раствора, оставшегося в порах стеклянной пластинки, промывают его при отсасывании теплой разбавленной (1 : 9) соляной кислотой, которую присоединяют потом к основному раствору. Раствор охлаждают до 20 ± 3° С.
Вводят титрованный раствор смеси бромида и бромата калия1 в таком , количестве, чтобы после реакции с оксихинолином остался избыток его в 2—3 мл, что определяется по мгновенному исчезновению красной окраски капли 2%-ного спиртового раствора метилового красного или по расчету, если известно приблизительное содержание алюминия. Перемешивают и оставляют стоять 30—60 сек, чтобы обеспечить полноту бромирования. Затем вводят 15 мл 20%-ного раствора иодида калия, тщательно перемешивают, чтобы реакция между бромом и иодидом прошла полностью, и титруют 0,1 н. раствором тиосульфата натрия до изменения окраски раствора в бледную коричнев о-желтую 2. После этого прибавляют раствор крахмала (стр. 220) и титруют до исчезновения синего окрашивания.
Из введенного объема раствора бромат-бромида калия вычитают то количество его, которое эквивалентно израсходованному на титрование количеству тиосульфата натрия. Если прежде чем был введен необходимый избыток раствора смеси бромида и бромата пришлось прибавить больше 2—3 капель раствора метилового красного, то определяют расходуемое
1 Концентрация раствора бромат-бромида должна быть такова, чтобы на титрование расходовались приемлемые его количества. Наиболее удобными являются 0,,1 н. раствор при содержании до 10 мг алюминия и более концентрированный раствор до 1 н. для титрования растворов, содержащих 50—100 мг алюминия.
2 Следует отличать эту окраску от окраски, обусловленной наличием в растворе бромированного оксихинолина.
' Методы, определения
575
на реакцию с ним количество раствора смеси бромида и бромата калия и также вычитают.
Для вычислейия содержания алюминия умножают полученную разность, соответствующую объему раствора бромат-бромида калия, израсходованного на бромирование оксихинолина, на титр этого раствора1. . .
Приготовление и установка титра растворов смеси бромида и бромата калия и тиосульфата натрия описаны в разделе «Осазйдение оксихинолином» (стр. 148). Для рядовых определений алюминия титры этих растворов целесообразно устанавливать по материалам, подобным анализируемому (стандартным образцам), содержание алюминия в которых известно и не очень отличается от количеств, какие обычно приходится определять в повседневной работе. При установке титра навеску стандартного образца проводят через все стадии анализа.
Взвешивание в виде Al(C9HeON)3. Осадок сушат 1,5 ч при 135—140° С и взвешивают в виде Al(C9HeON)3; это соединение содержит 5,87% алюминия.
Взвешивание в виде А12О3. Разложение оксихинолята и превращение алюминия в окись А12О3 вызывает известные затруднения вследствие летучести оксихинолята алюминия. При применении этого способа осадок собирают на бумажном фильтре, покрывают 5 г безводной щавелевой кислоты, завертывают, и влажный фильтр с осадком помещают во взвешенный тигель. Бумагу и осадок покрывают свернутым 11-сантиметровым фильтром и прибавляют еще 2 г щавелевой кислоты. Затем постепенно нагревают до начала обугливания бумаги, а потом прокаливают как обычно до А12О3 (стр. 569). Щавелевую кислоту необходимо исследовать на содержание нелетучего остатка для введения соответствующей поправки в массу А12О3.
Более длительным, но более надежным способом переведения оксихинолята алюминия в окись алюминия является метод, в котором все органические вещества разрушают нагреванием осадка или осадка вместе с бумагой фильтра, после добавления азотной и серной кислот (стр. 91). Раствор затем разбавляют, алюминий осаждают аммиаком, отфильтровывают осадок гидроокиси алюминия и прокаливают его обычным способом. При этом обязательно надо найти поправки на примеси, вводимые с реактивами илц извлеченные из стекла посуды.
Определение малых количеств алюминия (< 0,1 мг)
В связи с тем что специфические реакции на алюминий неизвестны, открытие и определение малых количеств его вызывает известные затруднения. В тех случаях, когда в результате предшествующих операций в растворе остается менее 0,1 мг алюминия, для его определения нередко используют реакцию с ауринтрикарбоновой кислотой2 в слабокислом растворе, в результате которой образуется ярко-красный лак. Концентрацию __________ \
1 О непосредственном титровании солянокислого раствора броматом в присутствии катализатора, например осмиевой кислоты или хлорида магния, см. в статье G. F. S m i t h, R. L. May, J. Am. Ceram. Soc., 22, 31 (1939).
2 Качество красителя является важным фактором при определении алюминия этим методом. Приготовление и колориметрические свойства красителя описаны в в статье: W. Н. S m i t h, E. E. S a g e r, I. J. S i e v e г s, Anal. Chem., 21, 1334 (1949).
576
Гл. XXXI. Алюминий
ионов водорода в растворе при этом обычно регулируют ацетатными буферами А
Ауриновый краситель не является специфическим реагентом на алюминий; многие катионы и анионы мешают этой реакции (главным образом железо, бериллий, кремний, медь, хром, метафосфаты и фториды2). Влияние посторонних ионов уменьшается при измерении интенсивности окраски алюминиевого лака в слабощелочных растворах (pH = 7,1—9). В этих условиях менее интенсивна также и окраска самого красителя, что имеет известное преимущество при определении очень малых количеств алюминия визуальным способом. Однако высокие фотометрические свойства лака, проявляющиеся в слабокислых растворах, содержащих защитный коллоид, часто имеют более существенное значение, чем увеличение селективности реакции в щелочных растворах.
В большинстве современных фотометрических методов светопоглоще-ние лака измеряют фотометрически в слабокислых растворах, в которых он образуется.
Интенсивность окраски и устойчивость алюминиевого лака зависят от многих факторов, в частности от pH и температуры раствора, в котором лак образуется, продолжительности времени развития окраски, концентрации алюминия и применения защитных коллоидов. Реакцию образования лака поэтому необходимо проводить в строго определенных условиях, чтобы обеспечить ее воспроизводимость. Весьма существенно также избегать введения загрязнений, поэтому следует пользоваться исключительно чистой аппаратурой и реактивами высокой чистоты3.
Характерными примерами применения ауринового метода могут служить определения алюминия в железе4, стали5 и природных водах6. В последнем случае лак получают при pH = 4,4. Для устранения влияния небольших количеств железа (до 100 мг) вводят тиогликолевую 7 Я кислоту, которая связывает его в комплекс ’ .
1 L. Р. Hammett и С. Т. S о 11 е г у [I. Am. Chem. Soc., 47, 142 (1925)] установили, что, если раствор сделать слабощелочным, лак не разрушается в течение довольно продолжительного времени, хотя в щелочных растворах он не образуется. См. также G. Е. F. L u n d е 11, Н. В. Knowles, Ind. Eng. Chem., 18, no 1, 60 (1926).
2 J. H. Yoe, W. L. H i 11, J. Am. Chem. Soc., 49, 2395 (1927).
3 A. L. 0.1 s e n, E. A. G e e, V. McLendon, Ind. Eng. Chem., Anal. Ed., 16, 169 (1944).
4 H. G. Short, Analyst, 75, 420 (1950). В этом методе железо и другие мешающие элементы отделяются эфиром и последующей экстракцией купфероновых комплексов хлороформом при pH = 0,5. Затем получают алюминиевый лак при pH = 5' и комнатной температуре; спустя 15 мин измеряют светопоглощение при 530 ммк.
6 С. Н. С г a f t, G. R. Makepeace, Ind. Eng. Chem., Anal. Ed., 17, 206 (1945). Этот метод предназначается для анализа сталей, содержащих 0,04—1,5% алюминия. Железо отделяют изопропиловым эфиром. Лак образуется в ацетатном буферном растворе (pH = 5,4), содержащем желатину. Раствор нагревают 10 мин при 90—100° С. Хром в количестве менее 2% на реакцию не влияет.
° А. С. В о 1 f е, F. R. В u s s е 11, N. Т. Wilkinson,!. Appl. Chem., 1, 170 (1951).
’ Е. . М. Che пег у, Analyst, 73, 501 (1948).
8 С. L. Luke, К. С. Braun, Anal. Chem., 24, 1120 (1952); там же, 24, 1122 (1952). Фотометрическое определение алюминия в сплавах цветных металлов (а именно, в сплавах на свинцовой и оловянной основе, марганцевых бронзах, цинковом литье и магниевых сплавах).
Метод» определения 577
Фотометрическое измерение светопоглощения лака в кислом растворе Для определения необходимы следующие реактивы:
Соляная рсл Ота (5 н.). 250 мл соляной кислоты (пл. 1,18 г/см*) разбавляют равным объемом дистиллированной воды и перегоняют в стеклянном аппарате. Определяют концентрацию кислоты ц затем доводят ее до 5 н.
Раствор крахмала. Смешивают 1 г растворимого крахмала с 5 аил холодной дистиллированной воды, прибавляют 95-мл кипящей дистиллированной воды и нагревают до кипения. Через каждые 5 дней приготовляют свежий раствор и перед употреблением его фильтруют.
Раствор тиогликолевой кислоты. 2 мл тиогликолевой кислоты (90%-ной) разбавляют до 100 мл дистиллированной водой. Приготовляют свежий раствор через каждые 5 дней.
Раствор ацетата аммония (3,5 М). Приготовляют из ледяной уксусной кислоты и перегнанного аммиака следующим способом. Определяют концентрацию кислоты титрованием 1 н. раствором едкого натра по фенолфталеину. К 10 мл раствора аммиака прибавляют измеренное избыточное количество 1 н. раствора НС1 (150 мл). Титруют Избыток НС1 1 н. раствором NaOH. Отмеряют объем ледяной уксусной кислоты, эквивалентный 3500 мл 1 н. раствора, разбавляют приблизительно 100 мл дистиллированной воды и затем при перемешивании вводят перегнанный аммиак в количестве, соответствующем 3500 мл 1 н. раствора. Охлаждают и разбавляют до 1 л.
Ауринтрикарбоксилат аммония (алюминон). Растворяют 1 г соли приблизительно в 400 мл дистиллированной воды, фильтруют и разбавляют до 500 мл. Раствор можно употреблять через 24 ч после его приготовления.
Ход определения. Отбирают соответствующий объем анализируемой воды, содержащей не более 20 мг алюминия, и помещают в мерную колбу емкостью 50 мл. Вводят последовательно 2 мл раствора 5 н. соляной кислоты, 2 мл разбавленной тиогликолевой кислоты, 3 мл отфильтрованного раствора крахмала и 5 мл раствора ацетата аммония. Разбавляют приблизительно до 45 мл, прибавляют 3 мл раствора алюминона, разбавляют до метки и перемешивают. Погружают открытую колбу точно на 4 мин в кипящую воду. Колбу следует поместить на подставку высотой приблизительно 10—15 мм, чтобы избежать местного перегрева. Вынимают колбу из бани и постепенно в течение 1 ч охлаждают до комнатной температуры, а затем помещают в термостат, где держат ее 30 мин при 20° С (±0,1 град). После этого, если требуется, объем раствора в колбе доводят до метки.. Измеряют светопоглощение в 4-сантиметровой кювете, применяя светофильтр с областью пропускания 525 ммк, и помещая другую кювету с дистиллированной водой.
Содержание алюминия определяют, пользуясь калибровочным графиком, построенным по стандартным растворам алюминия, обработанным так же, как анализируемый раствор.
Вычитают количество алюминия, соответствующее отсчету, полученному при измерении светопоглошения в «холостом» опыте, в котором одни реактивы проводят через все стадии анализа.
Измерение интенсивности окраски алюминиевого лака и щелочном растворе визуальным методом 1 2. Согласно этому методу, алюминиевый лак образуется в ацетатном буферном растворе, имеющем pH = 4—5, после чего раствор подщелачивают приблизительно до pH = 8 раствором
1 А. С. R о 1 f е, F. R. R u s s е 11, N. Т. Wilkinson, цит. выше. * См. также А. К. Бабко, ЖПХ, 12, 1560 (1939); А. К. Б а б к о, Т. Н. Назаренко, ЖАХ, 9, 96 (1954).*
2 J. A. S herrer, W. D. Mogerman, J. Research, NBS, 21, 105 (1938).
37 Заказ 522.
578
Гл. XXXI. Алюминий
аммиака или раствором аммиака, насыщенным карбонатом аммония. В этих условиях сам краситель имеет слабо-желтую окраску. В присутствии алюминия окраска раствора изменяется от слабо-розовой до темно-красной, если содержание его не превышает 0,1 ata в 100 мл раствора. При более высокой концентрации алюминия красный лак выделяется из раствора в виде осадка. Следует вводить точно отмеренные количества щелочи и кислоты. Элементы, осаждающиеся в щелочном растворе, должны быть предварительно отделены. Такой же лак образует бериллий. Железо должно быть полностью отделено.
Цирконий, гафний, скандий, торий, иттрий, лантан, церий, неодим и эрбий образуют розовые или красноватые лаки в аммиачных растворах, не содержащих карбоната аммония. Галлий в количествах менее 0,1 мг, иридий и таллий (менее 2 мг) не влияют на реакцию. Небольшие количества ванадия (V) «1 мг) не сказываются на определении, а большие количества дают желтое окрашивание. Кальций, стронций и барий в количестве 10 мг Не оказывают влияния, а такие же количества магния дают розовую окраску, не исчезающую в присутствии карбоната аммония. Азотная кислота, сернистый ангидрид, сероводород, фтористоводородная кислота и более 25 мг фосфорной кислоты обесцвечивают лак х.
Ход определения. К 40—75 мл прозрачного кислого раствора, содержащего 0,02—0,08 мг алюминия, не более 5 мл серной или соляной кислоты и свободного от мешающих элементов, прибавляют 5 мл соляной кислоты, если в нем ее нет, затем 5 мл ледяной уксусной кислоты и 5 мл 0,2%-ного водного; раствора ауринтрикарбоксилата аммония соответствующего качества. После этого, тщательно перемешивая, осторожно вводят аммиак 1 2 до тех пор, пока раствор не станет прозрачным, но сохранит еще кислую реакцию по лакмусу и темную окраску. Помещают маленький кусочек лакмусовой бумаги на внутренней поверхности стакана и при непрерывном перемешивании вводят по каплям аммиак до посинения лакмуса.
Затем прибавляют 5 мл ледяной уксусной кислоты, дают раствору постоять 10 мин и медленно, со скоростью 1 капли в 2—3 сек, вводят раствор аммиака точно до посинения лакмусовой бумаги. После этого прибавляют избыток раствора аммиака в 5 мл. По охлаждении раствора до комнатной температуры сравнивают его окраску с окраской растворов с известным содержанием алюминия, обработанных таким же способом.
Если во всех стадиях определения применять меньшие объемы раствора, например такие, из которых получается 10 мл конечного раствора, то этот метод, без сомнения, может быть применен к растворам, содержащим такие малые количества алюминия, как 0,001 мг.
Другие методы
Желтая окраска раствора оксихинолята алюминия в хлороформе используется 3 для определения Малых количеств алюминия в стали. После отделения железа и др. осаждением на ртутном катоде в буферном ацетат
1 A. R. М i d d 1 е t о n, J. Am. Chem. Soc., 48, 2125 (1926); R. B. Corey and H. W. R о g e r s, там же, 49, 216 (1927); J. H. Y о e, J. Am. Chem. Soc., 54, 1022 (1932).
2 В присутствии редкоземельных элементов, скандия и циркония во всех стадиях определения следует прибавлять раствор аммиака, насыщенный карбонатом аммония.
3S. Е Wiberley, L. G. Bassett, Anal. Chem., 21, 609 (1949); о влиянии посторонних ионов см. также Т. Moeller, Ind. Eng. Chem., Anal. Ed., 15, 346 (1943) и С- H. R. G e n t r y, L. G. S h e r r i n g t о n, Analyst, 71, 432 (1946).
Методы, определения
579
ном растворе, имеющем pH = 6 ± 1, получают оксихинолят алюминия, который экстрагируют хлороформом. Интенсивность окраски конечного раствора измеряют при 390 ммк.
* Ю. Ю. Лурье и 3. В. Николаева 1 показали возможность отделения оксихинолята железа от оксихинолята алюминия экстракцией хлороформом при pH = 1,8—1,9. После отделения железа оксихинолят алюминия извлекают хлороформом из ацетатного раствора и определяют колориметрическим способом. Метод применен авторами для определения алюминия в природных и промышленных сточных водах. Доп. перев. *
Краситель эриохромцианин R образует с алюминием в ацетатном буферном растворе 2 3, имеющем pH = 5—6, красно-фиолетовый лак. В присутствии более 2,5 мг!л железа (III) получаются повышенные результаты з. Эта реакция применена 4 для определения алюминия в сталях после отделения железа электролизом с ртутным катодом.
Кислотный хром сине-черный Р ([2-оксинафталин-(1-азо-1)-2'-окси-нафталин-4'-сульфокислота], Colop index, № 202) в ацетатном буферном растворе при pH = 4,8 образует с алюминием лак, который дает красную флуоресценцию 5 6. Эта реакция очень чувствительна (0,5 мкг в 50 мл) и применена для определения алюминия в сталях, бронзах и минералах ®. Железо (III) гасит эту флуоресценцию. Титан влияет слабо, ванадий несколько больше. Цирконий и бериллий вовсе не оказывают влияния 7.
* Заслуживает внимания также метод 8 определения алюминия, основанный на образовании окрашенного соединения алюминия с реагентом «стильбазо», являющимся диаммониевой солью стильбен-2,2'-дисуль-фокислоты-4,4'-бис-(азо-4)-1,2-диоксибензола. Определение алюминия этим методом выполнимо в присутствии железа, бериллия и некоторых других элементов.
Кроме того, следует указать на колориметрический метод9, в котором используется реакция алюминия с «арсеназо» (бензол-2-арсоновая кислота-< 1-азо-7 > -1,8-диоксинафталин-3,6-дисульфокислота, натри-
евая соль). Доп. перев. *
1 Ю. Ю. Л у р ь е, 3. В. Николаева, Зав. лаб., 19, 152 (1953).
3 R. Fresenius, G. lander, Handbuch der analytischen Chemie, T. Ill, Bd. Ill, Springer, Berlin, 1942, S. 310.
3 W. E. Thrum, Anal. Chem., 20, 1117 (1948).
4 W. Koch, Arch. Eisenhuttenw., 12, 69 (1938). О применении эриохромциа-нина R для определения алюминия в цинке и стали см. также L. С. I k е n b е г г у,
A. ffhomas, Anal. Chem., 23, 1806 (1951).
6 С. Е. White, С. S. Lowe, Ind. Eng. Chem., Anal. Ed., 9, 430 (1937); J. A. Radley, Analyst 68, 369 (1943).
* A. W e i s s 1 e г, С. E. White, Ind. Eng. Chem., Anal. Ed., 18, 530 (1946).
7 Определение алюминия фотометрическим измерением интенсивности флуоресценции, возникающей в результате взаимодействия алюминия с морином, см. в статье С. Е. W h i t е, С. S. Lowe, Ind. Eng. Chem., Anal. Ed., 12, 229 (1940).
8 В. И. К у з н e ц о в, Г. Г. Каранович, Д. А. Драпкина, Зав. лаб., 16, 787 (1950).
’В И. Кузнецов, Р. Б. Голубцова, Зав. лаб., 21, 1422 (1955); Зав. лаб., 22, 161 (1956).
37*
Глава XXXII
БЕРИЛЛИЙ
Бериллий является относительно редким элементом. Он встречается главным образом в минерале берилле — двойном алюмосиликате H2BeeAl4Si12O37 (Na2, Li2, Cs2, изоморфные c Be). Другими минералами, содержащими бериллий, являются эвклаз — также алюмосиликат Be(AlOH)SiO4, хризоберилл — алюминат бериллия ВеА12О4, ф е-н а к и т — силикат бериллия Be2SiO4, бериллонит — бериллиево-натриевый фосфат BeNaPO^ и гамбергит — борат бериллия Ве(ВО2)2-Н2О. Бериллий встречается также совместно с иттрием, церием и цирконием в минералах, гадолините Be2Fe(YO)2(SiO4)2, м и-ромонтите и циртолите.
Вашингтон 1 отмечает: «было высказано предположение, что в некоторых случаях (когда порода содержала не обнаруженный берилл) бериллий осаждался и взвешивался вместе с алюминием, вследствие чего в результатах анализов получилось такое высокое содержание А12О3, что его было трудно согласовать с минералогическим составом анализированной породы. В недавнее время было показано, что бериллий принимался за алюминий при анализе минерала везувианита, в котором присутствие бериллия раньше не предполагали. Надо, очевидно, исследовать в этом отношении и некоторые другие минералы и горные породы».
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа бериллий сопровождает алюминий, и если его не определяют и не учитывают при подсчете результатов, он будет принят за алюминий. Учитывая это, аналитики, выполняющие анализы горных пород, должны всегда производить специальные испытания на присутствие в них бериллия.
Когда в распоряжении имеется мало материала, качественное и количественное определение бериллия может быть включено в общий ход анализа, как описано на стр. 120. При наличии достаточного количества материала качественную пробу на бериллий лучше выполнять следующим способом. '
Измельченную руду сплавляют с карбонатом натрия, плав растворяют в соляной кислоте и раствор выпаривают досуха. Остаток смачивают
1 Н. S. Washington, The Chemical Analysis of Rocks, 4th ed., John Wiley and Sons., 1930, p. 22; см. также Am. Mineral., 16, 37 (1931).
Общие замечания
581
5 мл соляной кислоты, прибавляют горячую воду и кипятят до растворения солей. Сразу же отделяют кремнекислоту фильтрованием и промывают остаток холодной разбавленной соляной кислотой. Фильтрат разбавляют до 200 мл, насыщают сероводородом и нагревают некоторое время при 40—50° С. Фильтруют для отделения олова и большей части германия. Осадок не промывают. Фильтрат кипятят до удаления сероводорода, прибавляют небольшое количество азотной кислоты и продолжают кипятить для окисления железа и уменьшения объема до 25—50 мл. Нейтрализуют раствор концентрированным раствором едкого натра, нагревают до кипения и медленно при перемешивании вливают в концентрированный раствор едкого натра, взятый в таком количестве, чтобы после смешения растворов получился 10%-ный раствор щелочи. Фильтруют для удаления титана, циркония и других элементов.
При отсутствии фосфора раствор подкисляют соляной кислотой и затем осаждают небольшим избытком аммиака. Фильтруют для отделения от накопившихся в растворе солей, промывают осадок горячим 2 %-ным раствором нитрата аммония и затем растворяют в возможно меньшем количестве горячей разбавленной соляной кислоты. В присутствии фосфора щелочной фильтрат подкисляют азотной кислотой и затем осаждают молибденовой жидкостью, как описано в гл. «Фосфор» (стр. 784). Осадок фосфоромолибдата отфильтровывают, фильтрат нагревают до кипения и осаждают алюминий, бериллий и т. п. небольшим избытком аммиака. Фильтруют для отделения молибдена и избытка щелочных солей. Осадок растворяют в соляной кислоте, нереосаждают аммиаком, снова фильтруют и растворют осадок в возможно меньшем количестве горячей разбавленной соляной кислоты. Раствор почти нейтрализуют едким натром и медленно при перемешивании вливают в горячий раствор, содержащий такое количество бикарбоната натрия, чтобы после введения анализируемого раствора получился 10%-ный его раствор. Быстро нагревают до кипения и кипятят 30 сек. Тотчас же фильтруют для отделения алюминия. Фильтрат подкисляют соляной кислотой, затем кипятят до полного удаления двуокиси углерода и прибавляют небольшой избыток раствора аммиака. Появление белого осадка указывает на присутствие бериллия. Осадок, полученный после осаждения бикарбонатом, содержит весь алюминий, если операция проведена тщательно, и не захватывает бериллия, за исключением тех случаев, когда в растворе содержатся значительные количества алюминия наряду с малыми количествами бериллия. В осадке могут присутствовать германий и галлий, поэтому дальнейшее его исследование должно проводиться с учетом этих элементов.
Для открытия и количественного определения малых количеств бериллия рекомендовано 1 применение хинализарина (1,2,5,8-тетраоксиантра-хинон, ализарин-бордо). В чистых растворах качественное испытание проводится при комнатной температуре следующим способом. Раствор, предпочтительно не содержащий аммонийных солей, нейтрализуют свободным от магния раствором чистого едкого натра, 10 мл нейтрализован
1 Wissenschaftliche Veroffentlichungen aus dem Siemens-Konzern, T. 2, 99 (1926). Эта статья содержит полное изложение методов открытия и определения бериллия. См. также G. R i е n а с к е г, Z. anal. Chem., 88, 29 (1932).
Открытие и определение 0,0002—0,0025% бериллия в силикатных породах по флуоресцентной реакции с морином в растворе, содержащем едкий натр и пирофосфат натрия, приводит Е. В. Sandell [Ind. Eng. Chem., Anl. Ed., 12, 674, 762 (1940)].
582
Гл. XXXII. Бериллий
ного раствора обрабатывают 5 мл 2 н. раствора едкого натра и затем вводят 2—3 капли 0,05%-ного раствора хинализарина в 0,25 н. растворе едкого натра или 10—15 капель 0,01 %-ного раствора этого же реагента в абсолютном спирте. Таким же способом приготовляют раствор из одних реактивов, которые вводят в 10 мл воды.
Поместив растворы на белом фоне, сравнивают их окраски, глядя сверху через весь столб жидкости. В присутствии бериллия раствор имеет чисто синюю окраску, тогда как раствор, содержащий одни реактивы, окрашен в фиолетово-красный цвет. Таким способом можно открыть 0,015 мг бериллия в 1 мл. Алюминий не мешает, а фосфаты, тартраты, железо и магний влияют на реакцию, и в присутствии их следует применять видоизмененный метод. Определение малых количеств бериллия может быть выполнено с этим же реагентом методом колориметрического сравнения или колориметрического титрования.
Можно открыть куркумином1 2 0,05 мг бериллия в 1 л. Этот реагент поглощается выпадающей Ве(ОН)г, которая при этом окрашивается в оранжево-красный цвет. Испытание проводится следующим образом. К 10 мл слабокислого раствора прибавляют 1 каплю 0,1 %-ного спиртового раствора куркумина, затем 0,5 мл 4 н. раствора NH4C1 и 6—7 капель 4 н. раствора аммиака. Магний понижает чувствительность этой реакции, а железо и алюминий ей мешают. Железо можно отделить осаждением едким натром. Влияние алюминия, а также и железа можно устранить введением NaF и фильтрованием через 1 ч.* Этот метод служит также для количественного определения бериллия 112’3.*
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ БЕРИЛЛИЙ
Разложение бериллиевых минералов в основном осуществляется так же, как описано в гл. «Алюминий» (стр. 559). Измельченный минерал сплавляют с карбонатом натрия, плав растворяют в соляной кислоте, выделяют кремнекислоту выпариванием, отфильтровывают ее и фильтрат осаждают раствором аммиака или бикарбоната натрия. Берилл можно сплавить с перекисью натрия в никелевом тигле. Фосфатные минералы можно разложить обработкой царской водкой, после чего раствор несколько раз выпаривают с азотной кислотой досуха для разложения хлоридов4. Нерастворимый остаток отделяют фильтрованием, прокаливают, сплавляют с карбонатом натрия и плав выщелачивают водой. Водный экстракт содержит фосфат, а нерастворимый осадок может состоять из титана и циркония. В дальнейшем поступают в зависимости от того, какие элементы следует определить. Если, например, требуется определить только фосфор, осадок отбрасывают, а фильтрат присоединяют к основному раствору.
МЕТОДЫ ОТДЕЛЕНИЯ
Методы отделения бериллия от других элементов сходны с методами, применяемыми для отделения алюминия. Гидроокись бериллия осаждается аммиаком при несколько более высоком значении pH раствора, чем гидро
1 I. М. Kolthoff,!. Am. Chem. Soc., 50, 393 (1928).
2 E. Б. С e н д э л, Колориметрическое определение следов металлов, Госхимиз-дат, 1949, стр. 157.
3 Э. А. Остроумов, Б. Н. Иванов-Эмин, Зав. лаб., И, 386 (1945).
4 Раствор хлорида бериллия можно выпарить досуха и затем остаток прокалить при 1100° С до получения свободной от хлорида окиси без потерь бериллия [L. F г е genius, М. F г о m m е s, Z.'anal. Chem., 93, 275 (1933)].
Методы отделения
583
окись алюминия. Тартраты в определенных условиях препятствуют осаждению гидроокиси бериллия, что дает возможность осуществить разделение бериллия и железа осаждением сульфидом аммония в присутствии тартрата, как изложено на стр. 115.
* Э. А. Остроумов и Б. Н. Иванов-Эмин 1 предлагают осаждать гидроокись бериллия слабым органическим основанием а-пиколином в присутствии хлорида аммония. В рекомендуемых авторами условиях осаждения в растворе устанавливается pH = 7. При этом происходит количественное отделение бериллия от кальция, стронция, бария, магния и щелочных металлов, а также от марганца, кобальта, никеля, цинка, образующих растворимые комплексные соединения с а-пиколином. Доп. перев. *
Бериллий совместно с алюминием, галлием, германием, фосфором, ванадием и другими элементами можно отделить от железа, циркония, титана и подобных элементов осаждением последних избыточным количеством едкого натра, как описано на стр. 109.
Лучшими методами разделения бериллия и алюминия являются осаждение алюминия оксихинолином (стр. 148) или газообразным хлористым водородом из холодного раствора хлоридов в смеси равных объемов соляной кислоты и эфира, как указано в гл. «Алюминий» (стр. 564). При осаждении хлористым водородом осадок хлорида алюминия не захватывает бериллий, но всегда можно опасаться неполноты выделения алюминия. Поэтому фильтрат следует концентрировать выпариванием и операцию повторять до тех пор, пока не прекратится образование осадка. Нередко эфирно-хлористоводородным методом пользуются для осаждения основной массы алюминия 2, а остающуюся в растворе часть затем дополнительно выделяют оксихинолином в слабокислой среде (стр. 148). Из горячего фильтрата бериллий можно осадить аммиаком в присутствии избытка оксихинолина, необходимо только перед фильтрованием аммиачный раствор охладить.
Для отделения алюминия от бериллия осаждением оксихинолином рекомендуется следующий метод 3. К раствору, содержащему в 200 мл не более 0,1 г алюминия и 10 мл соляной кислоты, прибавляют 15 мл раствора 30 г ацетата аммония в 75 мл воды. Вводят 8—10 капель 0,04%-ного раствора бромкрезолового пурпурного и затем разбавленный (1 : 4) раствор аммиака до появления отчетливой пурпурной окраски индикатора. После этого при перемешивании медленно из бюретки прибавляют 2,5%-ный раствор оксихинолина в уксусной кислоте 4 в количестве, на 15—25% превышающем теоретически необходимое для образования оксихинолята алюминия (1А1 соответствует 3C9H7NO). Нагревают до кипения, временами перемешивая раствор, и продолжают кипятить 1 мин. Дают охладиться до 60° С и фильтруют (при слабом отсасывании) через стеклянный тигель
1 Э. А. Остроу мо в, Б. Н. И в а н о в - Э м и н, Зав. лаб., 11, 386 (1945)в
2 Н. V. Churchill, R. W. В г i d g е s, М. F. Lee, Ind. Eng. Chem., Anal. Ed., 2, 405 (1930); J. I. Hoff man, G. E. F. L u n d e 11, J. Research NBS, 18, 8 (1937).
3 H. B. Knowles,!. Research NBS, 15, 94 (1935).
4 Этот раствор приготовляют следующим образом. 12,5 г оксихинолина обрабатывают 25 мл ледяной уксусной кислоты и слабо нагревают до растворения. Полученный раствор вливают в 450 мл воды, нагретой до 60° С. Охлаждают, фильтруют, если требуется, и затем разбавляют до 500 мл.
584
Гл. XXXII. Бериллий
диаметром 35 мм с плотным пористым дном. Осадок промывают 100 мл холодной воды.
В точных работах й во всех случаях, когда присутствуют большие количества алюминия, осадок растворяют в разбавленной (1 : 4) соляной кислоте и переосаждают. Фильтраты и промывные воды объединяют для определения бериллия. Определение алюминия можно закончить весовым или объемным способом (стр. 574).
Бериллий можно осадить в фильтрате аммиаком, как описано в разделе «Методы определения» (стр. 586), с той лишь разницей, что раствор перед фильтрованием необходимо охладить, чтобы обеспечить количественное выделение бериллия в присутствии оксихинолина.
В описанных выше реакциях, естественно, принимают участие и другие элементы (см. стр. 102), и если их присутствие мешает, они должны быть предварительно отделены, или же их влияние необходимо учитывать при конечном определении бериллия и алюминия после разделения.
Для отделения малых количеств алюминия от бериллия (например, при соотношении 1А1 : 500Ве) предложен метод экстрагирования оксихинолята алюминия хлороформом А
Некоторые авторы 1 2 предпочитают разделять бериллий и алюминий сплавлением предварительно выделенных фосфатов с карбонатом натрия и выщелачиванием плава водой в продолжение 15 ч.
Менее удовлетворительно происходит разделение алюминия и бериллия при: 1) осаждении алюминия из кипящего 10%-ного раствора бикарбоната натрия 3, 2) осаждении алюминия карбонатом аммония и 3), выделении бериллия сильным разбавлением раствора едкого натра или едкого кали, содержащего оба эти элемента.
Установлено 4, что алюминий достаточно удовлетворительно отделяется от бериллия осаждением таннином следующим способом. Слабокислый (сернокислый) раствор сульфатов этих элементов, объем которого равен 500 мл, если содержание алюминия в нем ниже 0,1 г, и 600—800 мл в случае более высокого содержания алюминия,' нагревают до 80° С и затем при энергичном перемешивании в один прием вливают в избытке горячий (80° С) прозрачный раствор таннина (3 ? чистого таннина растворяют в 400 мл насыщенного при комнатной температуре раствора ацетата аммония). Кипятят 2 мин, дают отстояться и испытывают на полноту осаждения добавлением еще некоторого количества раствора таннина. По охлаждении фильтруют и промывают осадок горячим 5 %-ным раствором ацетата аммония.
Если количество алюминия не превышает 0,06 г, осадок отфильтровывают через бумажный фильтр и прокаливают до А12О3. Окончательное прокаливание проводят после 1—3-кратной обработки осадка азотной кислотой. При более высоком содержании алюминия осадок отфильтровывают через стеклянный тигель с пористым дном и после промывания растворяют его в разбавленной (1 : 3) азотной кислоте. Раствор собирают в высокий стакан, покрывают часовым стеклом, кипятят, обрабатывают
1 С. J. Rodden, Analytical Chemistry of the Manhattan Project, Me Graw-Hill Book Co., 1950, p. 388. См. также T. Moeller, Ind. Eng. Chem., Anal. Ed., 15 346 (1943).
2 R. E. S t e v e n s, M. К. С а г г о n, U. S. Geol. Survey Bull., 950, 91 (1946).
2 C. L. P a r s о n s, S. К. В a r n e s, J. Am. Chem. Soc., 28, 1589 (1906).
4 L. Moser, M. Niessner, Monatsh., 48, 113 (1927).
Метод» отделения
585
дымящей азотной кислотой для разрушения таннина, а затем осаждают раствором аммиака, как обычно, для получения менее объемистого осадка. Фильтрат, полученный после осаждения таннином, кипятят, обрабатывают азотцой кислотой для разрушения таннина, отгоняют всю уксусную кислоту и осаждают бериллий раствором аммиака, как изложено в разделе «Методы определения» (стр. 586).
После удаления таннина и уксусной кислоты бериллий можно осадить также следующим образом. К слабокислому раствору прибавляют 20— 30 г нитрата аммония, разбавляют до 300—400 мл и нагревают до кипения. Вводят 10%-ный раствор таннина в таком количестве, чтобы соотношение между таннином и окисью бериллия составляло 10 : 1, и затем прибавляют по каплям раствор аммиака до полного осаждения гидроокиси бериллия. Дают отстояться, осадок отфильтровывают и промывают горячей водой. Если присутствуют соли щелбчных металлов, осадок растворяют в небольшом количестве горячей разбавленной соляной кислоты, раствор разбавляют и осаждение повторяют. Осадок помещают в платиновый или кварцевый тигель, обрабатывают несколькими каплями азотной кислоты, выпаривают и прокаливают до окиси ВеО.
С некоторыми изменениями танниновый метод служит для отделения бериллия от железа, хрома, титана, циркония, вольфрама и ванадия1.
*Танниновый метод, предлагаемый для разделения бериллия и алюминия, непригоден в присутствии железа, так как выделяющийся в этих условиях осадок железа всегда захватывает некоторые количества бериллия. Для отделения железа от бериллия рекомендуют проводить осаждение при несколько более высокой концентрации кислоты в растворе (1,5 мл 80%-ной уксусной кислоты на 100 мл раствора), а для предотвращения восстановления железа в раствор вводить несколько капель перекиси водорода (3%-ной). В этих условиях, однако, не достигается количественное выделение алюминия.
Ю. А. Чернихов и Э. А. Остроумов 2 3 предложили условия осаждения таннином, при которых происходит отделение от бериллия одновременно алюминия и железа. Согласно указаниям авторов, осаждение нужно проводить следующим способом. Анализируемый раствор нейтрализуют карбонатом натрия или аммония до появления неисчезающей мути, которую растворяют добавлением нескольких капель разбавленной серной кислоты, нагревают до 80° С и тонкой струей при перемешивании вливают в 250—300 мл горячей воды, содержащей требуемые для осаждения количества ацетата аммония и таннина. Выделяющийся в этих условиях осадок железа и алюминия свободен от бериллия. Доп. перев. *
Отделение бериллия от урана изложено в гл. «Уран» (стр. 526).
Железо можно отделить от бериллия нагреванием окислов в токе сухого хлористого водорода, содержащего немного хлора 8. Температура в начале реакции не должна превышать 200° С, чтобы избежать механических потерь бериллия вследствие слишком быстрого выделения хлорида железа (HI), а под конец для полного удаления железа нет необходимости поднимать ее выше 300° С.
1 L. Moser, J. Singer, Monatsh., 48, 671 (1927).,
2Ю. А. Чернихов, Э. А. Остроумов, Редкие металлы, 1, № 5, 25 (1933).
3 F. S. Havens, A- F-. Way, Am. J. Sci., [4], 8, 158, 217 (1899).
586
Гл. XXXII. Бериллий
Отделение бериллия от железа, хрома, меди и никеля может быть произведено электролизом с ртутным катодом 1 (стр. 165).
Бериллий можно отделить от элементов цериевой группы осаждением последних сульфатом натрия (стр. 631), а от элементов иттриевой группы — осаждением щавелевой кислотой из 0,5 н. солянокислого раствора хлоридов (стр. 621).
Известен метод отделения бериллия (не проверенный, однако, нами на смесях), заключающийся в продолжительном сплавлении с карбонатом натрия при высокой температуре и выщелачивании плава водой. Как указывают, бериллий количественно остается в осадке совместно с железом, титаном ит. и., тогда как хром (в виде хромата), фосфор и большая часть кремния и алюминия переходят в раствор 2.
Бериллий осаждается не полностью или не ойаждается вовсе фенил-гидразином (стр. 154). Он не осаждается также купфероном, благодаря чему его можно отделить от железа, титана, циркония, ванадия и урана (IV).
* Следует указать также на метод отделения бериллия от алюминия и железа, согласно которому гидроокиси всех этих элементов превращают в муравьинокислые соли двух- или трехкратным выпариванием с муравьиной кислотой на водяной бане досуха3. Полученный остаток нагревают в вакууме при 180—200° С, причем бериллий возгоняется и оседает на стенках отводной трубки в виде красивых кристаллов Ве4О(СНО2)6, а железо и алюминий остаются в нелетучем остатке. Кристаллы Ве4О(СНО2)6 растворяют в концентрированной азотной кислоте, осаждают бериллий аммиаком и взвешивают в виде ВеО. Титан и кремнекислота мешают этому отделению, так как удерживают бериллий в нелетучем остатке.
И. В. Тананаев и Ш. Талипов 4 разработали метод отделения бериллия от алюминия, железа, кальция и магния, основанный на обработке раствора определенным количеством фторида натрия, в результате чего образующиеся малорастворимые Na3AlF6, Na3FeF6, CaF2 и MgF2 выделяются в осадок, а фторобериллат натрия Na2BeF4 остается в растворе. Доп. перев. *
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Осаждение аммиаком
•
Бериллий обычно осаждают аммиаком в виде гидроокиси Ве(ОН)2 и взвешивают в виде окиси ВеО. Осаждению, естественно, должно предшествовать отделение катионов и анионов, которые выделяются совместно с бериллием из аммиачного раствора, а также органических веществ, препятствующих осаждению гидроокиси бериллия. В присутствии нелетучих солей, таких, как хлорид натрия, гидроокись бериллия необходимо
1 R. Е. Meyers, J. Am. Chem. Soc., 26, 1124 (1904).
2 M. Wunder, P. W e n g e r, Z. anal. Chem., 51, 470 (1912). С другой стороны, A. Stock, P. PraetoriusnO. Priess [Ber., 58, 1577 (1925)] не добились успеха при применении этого метода. В одном опыте Н. В. Knowles после сплавления с 6 г Na2CO3 в течение 3 ч приблизительно при температуре 1100° С из взятой навески 0,1 г ВеО обнаружил 0,6 мг ВеО в водной вытяжке плава.
3 Р. Adami, Ann. chem. appl., 23, 428 (1933).
4 И. В. Тананаев, Ш. Талипов, Изв. АН СССР, серия хим., № 2, 547 (1938); Зав. лаб., 8, 23 (1939).
Методы отделения
587
дважды или трижды переосадить. Осадок имеет склонность приставать к стенкам стакана. После удаления большей части осадка прилипшую к стакану гидроокись бериллия растворяют в возможно меньшем количестве кислоты и затем снова осаждают аммиаком. Гидроокись бериллия заметно растворима в чистой воде, вследствие чего ее необходимо промывать водой, содержащей электролит (лучше всего нитрат аммония). Окись слегка гигроскопична, и поэтому ее следует охлаждать и взвешивать, как указано в гл. «Алюминий» (стр. 567).
Осаждение бериллия аммиаком можно выполнить в фильтрате после отделения алюминия оксихинолином из слабокислого раствора, как описано на стр. 148. В этом случае нет необходимости разрушать оксихинолин, если нейтрализованный раствор перед фильтрованием охлаждать. Этим операциям, естественно, должно предшествовать удаление веществ, мешающих разделению алюминия и бериллия, как, например, фосфор х, или осаждению бериллия, как, например, магний и тартраты.
Осажденный фосфат бериллия нельзя после прокаливания взвесить в виде пирофосфата, так как осадок не имеет строго определенного состава.
Ход определения. Почти нейтральный солянокислый или азотнокислый раствор, содержащий бериллий, свободный от посторонних веществ, осаждающихся аммиаком, и содержащий не менее 5 г хлорида аммония в 200 мл, нагревают до кипения и к нему медленно прибавляют разбавленный раствор аммиака в незначительном избытке (pH = 8—9). Кипятят 1—2 дак, вводят небольшое количество мацерированной бумаги и фильтруют. Возможно полнее переносят осадок на фильтр, тщательно оттирая его со стенок стакана и смывая горячим 2%-ным раствором нитрата аммония. Покрывают воронку часовым стеклом и немедленно растворяют приставший к стенкам стакана осадок в небольшом количестве горячей разбавленной азотной кислоты. Раствор нагревают до кипения и осаждают аммиаком, как прежде. Фильтруют через тот же фильтр, счищая осадок со стенок стакана, и тщательно промывают раствором нитрата аммония. Перемешивают фильтрат и промывные воды и оставляют стоять в течение нескольких часов, чтобы удостовериться в прекращении выделения осадка. Фильтр с осадком помещают в платиновый тигель, взвешенный вместе с крышкой, сушат, нагревают при невысокой температуре до выгорания углерода фильтра, закрывают тигель крышкой и прокаливают приблизительно при 1000° С. Охлаждают в эксикаторе над серной кислотой или пятиокисью фосфора и взвешивают. Снова прокаливают, охлаждают и, предварительно положив необходимые разновесы на чашку весов, быстро взвешивают. Эту операцию повторяют до достижения постоянной массы. Взвешенный осадок необходимо исследовать на содержание алюминия, фосфора, галлия и германия 1 2.
* Имеются указания на то, что бериллий можно осаждать в виде гидроокиси 3 или фосфата 4 в присутствии алюминия, железа, кальция,
1 При наличии фосфора в растворе бериллий частично осаждается в виде фосфата при pH = 4,2, и помимо того, фосфор загрязняет осадок бериллия, выделенный из фильтрата.
2 Согласно A. Craig, окись бериллия можно перевести в раствор сплавлением с фторидом натрия и небольшим количеством кремнекислоты, обработкой плава серной кислотой и последующим нагреванием до начала выделения ее паров.
3 R. Pribil, J. Kucharsky, Coll. Czech. Chem. Comm., 15, 132 (1950).
4 I. Hure, M. Kremer, F. Berquier, Anal. Chim. Acta, 7, 36 (1952).
588
Гл. XXXII. Бериллий
магния, свинца, хрома, меди, марганца и др. из растворов, содержащих комплексон III, с которым указанные элементы образуют растворимые прочные комплексные соединения. В. Г. Горюшина 1 использовала этот метод для весового определения бериллия в виде фосфата в медно-берилли-евых бронзах. Осаждение фосфата бериллия производится из ацетатного раствора в присутствии комплексона III после отделения основной массы меди электролизом или в присутствии всей меди после введения достаточного количества комплексона III для удержания ее в растворе. Доп. перев. *.
Другие методы
Описан2 флуоресцентный метод определения бериллия, заключающийся в следующем. -Анализируемый материал сплавляют со смесью карбоната натрия и буры. Плав растворяют в соляной кислоте, вводят цитрат натрия и хинизарин (1,4-диоксиантрахинон), а зате^ нейтрализуют раствор едким натром. Прибавляют еще хинизарина и интенсивность флуоресценции сравнивают со стандартами, приготовленными аналогичным способом 3.
* Содержание бериллия в нейтральных растворах можно определить объемным путем, титрованием едкой щелочью по фенолфталеину4. В. М. Звенигородская и А. А. Гайгерова 5 при объемном определении бериллия в нейтральных растворах фторобериллата натрия Na2BeF4 для связывания фторид-ионов, мешающих титрованию, рекомендуют вводить в раствор хлорид кальция. Ю. А. Чернихов и Е. И. Гульдина 6 показали, что при наличии в растворе фторидов бериллий можно определять алкалиметрически также и в растворах, содержащих свободную кислоту и фторосиликаты. В этом случае после введения хлорида кальция раствор нейтрализуют сначала едкой щелочью по метиловому оранжевому, а затем титруют бериллий, как обычно по фенолфталеину. Доп. перев. *
1 В. Г. Горюшина, Зав. лаб., 21, 148 (1955).
2 М. Н. F 1 е t с h е г, С. Е. W h i t е, М. S. S h е f t е 1, Ind. Eng. Chem., Anal. Ed., 18, 179 (1946).
3 Фотометрический метод определения бериллия в алюминии и его сплавах, основанный на реакции с n-нитробензолазоарсином, см. W. S t г о s s, G. Н. Osborn, J. Soc. Chem. Ind., 63, 249 (1944). * n-Нитробензолазоарсин впервые был предложен для открытия бериллия А. С. Комаровскими Н. С. Полуэктовым [ЖПХ, 7, 839 (1934)]. *
4 В. В 1 е у е г, А. М о о г m a n, Z. anal. Chem., 51, 360 (1912).
5 В. М. Звенигородская, А. А. Гайгерова, Редкие металлы, № 5 (1933).
6 Ю. А. Чернихов, Е. И. Гульдина, Зав. лаб., 4, 487 (1935).
Глава XXXIII
ХРОМ
Хром очень широко распространен в природе. Его местонахождение почти полностью ограничивается ферромагниевыми породами, главным образом с высоким содержанием магния и низким содержанием кремния и, следовательно, изобилующими оливинами, как перидотит и дунит. Он встречается в виде хромита FeCr2O4 и пикотита (Fe, Mg) (Сг, А1)2О4 (хромовой шпинели). Хром находится также в некоторых авгитах, биотитах и оливинах. В горных породах может содержаться до 0,5% Сг2О3. В природе известны также немногие хроматы и некоторые содержащие хром силикаты. Однако эти минералы встречаются сравнительно редко. Хром является обычным компонентом многих промышленных продуктов, главным образом чугунов и сталей, которые только в редких случаях не содержат хрома. Поэтому методы его определения имеют важное значение.
ОБЩИЕ ЗАМЕЧАНИЯ
При анализе горных пород хром попадает в осадок от аммиака. В его присутствии получаются повышенные результаты определения алюминия даже в том случае, когда на содержание Сг2О3 в осадке вводится соответствующая поправка. Это объясняется тем, что в процессе прокаливания Сг2О3 всегда несколько окисляется. Если содержание хрома не учитывается, величина погрешности при определении алюминия, зависит от метода определения железа-. Если железо определяют восстановлением раствора прокаленного остатка цинком и титрованием перманганатом, то хром (III) сначала восстанавливается до хрома (II), а затем при титровании вновь окисляется до хрома (III). Поскольку 1 мл 0,1 н. раствора перманганата соответствует приблизительно 0,008 г Fe2O3 и 0,0076 г Сг2О3, результаты определения железа при этом получаются повышенными, а результаты определения алюминия — несколько пониженными. В том случае, когда железо восстанавливают сероводородом или сернистой кислотой,.ошибка полностью отражается на результате определения алюминия, так как соли хрома (III) этими реагентами не восстанавливаются.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ХРОМ
Для определения хрома в минералах удобно сплавлять пробу с перекисью натрия в фарфоровом тигле 1 следующим способом. Измельченную в тончайший порошок пробу высушивают при 105° С и 0,5 г ее смешивают
1 В случае применения никелевого или железного тигля повышается содержание соединений железа (III) или никеля, которые с трудом распадаются при кипячении. Это может не иметь места, если применяют «метод вспышки» (стр. 919).
590
Гл. XXXIII. Хром.
с 5—6 г сухой желтой перекиси натрия в фарфоровом толстостенном тигле емкостью 30—50 мл. Смесь покрывают 1—2 г перекиси натрия, закрывают тигель крышкой и осторожно сплавляют, постепенно вводя тигель в электрический муфель, нагретый до 600—700° С. Когда содержимое тигля расплавится, медленно вращая тигель, захватывают жидкой массой приставшие к стенкам непрореагировавшие частицы.
Если вместо муфеля для сплавления пользуются горелкой, то тигель помещают на треугольник и медленно, держа горелку с небольшим пламенем в руке, нагревают до температуры плавления реагирующей смеси.
По окончании сплавления поддерживают температуру темно-красного каления еще 5 мин, время от времени перешивая расплав платиновой проволокой. По охлаждении тигель и крышку помещают в стакан емкостью 600 мл. Покрывают стакан часовым стеклом, вливают 200 мл воды и нагревают до растворения плава. Вынимают тигель и крышку из стакана, ополаскивают их, после чего кипятят раствор до полного удаления перекиси водорода. К раствору прибавляют разбавленную серную кислоту почти до. нейтральной реакции, фильтруют через бумажный фильтр, промывают остаток горячей водой и прокаливают. Из прокаленного остатка извлекают хром вторичным сплавлением с перекисью натрия. Фильтраты объединяют.
Для обеспечения полного переведения хрома в шестивалентное состояние перед титрованием подкисляют раствор серной кислотой и окисляют персульфатом, как указано в разделе «Титрование сульфатом железа (II) и перманганатом» (стр. 592). При фильтровании через бумагу в раствор вводятся растворимые органические вещества, которые, однако, разрушаются при последующем кипячении с персульфатом.
Сплавление с карбонатом натрия и нитратом калия (стр. 927) в платиновом тигле менее удобно, но им целесообразно пользоваться, если требуется определить кремнекислоту или другие элементы, которые могут быть введены при сплавлении в фарфоровых, никелевых или железных тиглях.
Хромит можно разложить обработкой 0,5 г тонко измельченной пробы 50 мл разбавленной (1 : 4) серной кислоты и 5 мл хлорной кислоты (пл. 1,54 г/сл13) при нагревании до начала выделения паров * 1 H2SO4
МЕТОДЫ ОТДЕЛЕНИЯ
Полное отделение хрома от других элементов требуется лишь в редких случаях, так как основные методы его определения применимы в присутствии многих посторонних элементов.
Основной метод отделения хрома, хотя это групповое отделение, основан на переведении хрома в растворимый хромат натрия или калия окислительным сплавлением со щелочами или окислением в щелочном
1 Т. R. Си nningha m, Т. R. McNeill, Ind. Eng. Chem., Anal. Ed.,
1, 70 (1929). О переведении в раствор хромита обработкой смесью 8 частей 95%-ной серной кислоты и 3 частей 85 %-ной фосфорной кислоты см. G. F. S m i t h, С. A. Getz, там же, 9, 518 (1937). Метод полного анализа хромита после сплавления материала с пиросульфатом калия, растворения плава в кислоте, фильтрования и разложения нерастворимого остатка сплавлением с карбонатом натрия изложен С. F. J. vander Walt, Analyst, 63, 176 (1938). Анализ хромита см. также F. J. Bryant, Р. I. И ar d wick, Analyst, 75, 12 (1959) *и Ю. Ю. Лурье, Зав. лаб., 1, 21 (1932).*
Методы отделения
591
растворе. Так, при осаждении перекисью натрия 1 (стр. НО), так же как при сплавлении с перекисью натрия или с карбонатом натрия и селитрой и выщелачивании плава водой (стр. 919), хром совместно с алюминием, мышьяком, молибденом, вольфрама, ванадием и др. переходит в. раствор и отделяется таким образом от железа титана, циркония, никеля, кобальта, меди и многих других элементов. Для отделения хрома от железа и алюминия часто применяется метод, который состоит в окислении хрома до шестивалентного состояния и последующем осаждении алюминия и железа аммиаком.
Хром окисляется также до шестивалентного при кипячении с хлорной кислотой. Полное окисление достигается с трудом 2. Окисленный хром можно отогнать в виде хлорида хромила СгО2С12 добавлением по каплям соляной кислоты 3 или осторожным введением кристаллов хлорида натрия в кипящий раствор 4. В обоих случаях происходит частичное восстановление хрома до трехвалентного состояния, и поэтому нужно время от времени прекращать обработку и кипятить раствор, чтобы вновь окислить хром.
Если наличие в растворе свинца не влияет на дальнейший ход анализа, хром после окисления его хлорной кислотой можно выделять из раствора в виде хромата свинца. Для этого окисленный раствор разбавляют с таким расчетом, чтобы он был приблизительно 1 М по содержанию хлорной кислоты, и вводят свинец в таком количестве, чтобы концентрация перхлората свинца была 0,02 М. Выделенный хромат свинца промывают 0,1 М раствором НС1О4. Осадок не захватывает ванадия 5.
Другой групповой метод разделения, применяемый при анализе горных пород, основан на осаждении хромата ртути (I), как описано в гл. «Ванадий» (стр. 510).
Осаждение сульфид-ионами из кислого раствора служит для отделения от хрома элементов сероводородной группы, а осаждение из щелочного раствора — для отделения хрома от группы мышьяка, магния и щелочноземельных металлов. В присутствии тартрата аммония хром из щелочного раствора сероводородом не выделяется и отделяется таким образом от железа, никеля, кобальта и цинка.
Хром почти количественно отделяется от больших количетств железа экстракцией последнего эфиром из солянокислого раствора хлоридов (стр. 161).
Известный интерес представляет метод отделения ванадия, железа, циркония, титана, ниобия и тантала от хрома., основанный на осаждении этих элементов купфероном (стр. 143).
Заслуживает внимания также метод, основанный на электролизе с ртутным катодом в разбавленном сернокислом растворе, который используется для отделения больших количеств хрома от ванадия, урана и ряда других элементов.
1 Осаждение хрома (III) едким натром см. «Осаждение едким натром» (стр. 109).
3 Г. Лендель, Дж. Гофман и Г. Брайт, Анализ черных металлов. Госхимтехиздат, 1934; G. F. Smith, Ind. Eng. Chem., Anal. Ed., 6, 229 (1934).
3 J. I. H о f f m a n, G. E. F. L u n d e 1 1, J. Research NBS, 22, 465 (1939).
4 F. W. Smith, Ind. Eng. Chem., Anal. Ed., 10, 360 (1938).
* Метод осаждения CrVI в виде хромата свинца нитратом свинца из разбавленного азотнокислого раствора и отделение его таким образом от железа, алюминия, кадмия, меди, марганца и цинка см. D. Tschawdarov, N. Tschawdarowa, Z. anal. Chem., Ц0, 348 (1937).
592
Гл. ХХХШ. Хром
Из раствора, содержащего значительные количества хрома, малые количества кремнекислоты лучше всего выделять после добавления избыточного количества серной кислоты (20—30 мл) выпариванием при возможно более низкой температуре до появления паров серной кислоты. К слегка охлажденному раствору затем прибавляют воду при энергичном перемешивании.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наиболее удовлетворительными методами определения хрома являются колориметрические и объемный. Объемный метод основан на окислении хрома до хромата, прибавлении избуточного количества сульфата железа (II) и титровании избытка последнего перманганатом. Колориметрический метод пригоден для определения малых количеств хрома, какие обычно содержатся в горных породах. При значительном содержании хрома, когда колориметрические методы неприменимы, пользуются объемным методом.
Титрование сульфатом железа (II) и пермангатом
По нашим данным, наиболее удовлетворительный метод определения хрома заключается в окислении хрома до хромата, введении в раствор отмеренного количества сульфата железа (II) и оттитровывании избытка последнего перманганатом или бихроматом калия. Окисление хрома можно осуществлять в процессе сплавления или соответствующей обработкой кислого раствора. Оба эти метода нашли практическое применение.
Сплавлением с карбонатом натрия и нитратом калия, (стр. 928) пользуются при определении малых количеств хрома; когда определяют большие количества хрома, лучше применять сплавление с перекисью натрия, как описано в разделе «Разложение минералов, содержащих хром» (стр. 589). В кислом растворе хром можно окислить двуокисью свинца, хлоратом калия, перманганатом калия и персульфатом калия или аммония в присутствии нитрата серебра *.
Из этих методов наилучшим является последний. Ниже приводится его подробное описание.
В персульфатном методе присутствие нитрата серебра имеет существенное значение для полного окисления хрома. Нитрат серебра вводят в горячий кислый раствор перед прибавлением персульфата так, чтобы его содержание было не меньше, чем содержание хрома 1 2. При чрезмерно высокой концентрации кислоты в растворе достигнуть полного окисления хрома не удается. Наиболее благоприятными условиями является содер
1 М. Р h i 1 i р s, Stahl u. Eisen, 27, 1164 (1907); H. E. Walter s, J. Am. Chem. Soc., 27, 1550 (1965); Met. Chem. Eng., 12, 310 (1914).
2 D. M. Y о s t [J. Am. Chem. Soc., 48, 152 (1926)] показал, что скорость реакции между надсерной кислотой и сульфатом хрома в присутствии соли серебра в качестве катализатора пропорциональна концентрации ионов надсерной кислоты и серебра, не зависит от концентрации ионов хрома и мало зависит от концентрации ионов водорода. Он также установил, что серебро в сильнокислом растворе может существовать и в трехвалентной форме в виде твердой окиси или в виде соли. Полученные им данные показывают, что каталитическое действие серебра в реакции окисления хрома основано на образовании соли серебра (III), являющейся промежуточным продуктом.
Методы определения
593
жание 5—6 мл серной и 1 мл азотной кислот в 100 мл раствора. В присутствии железа и вольфрама целесообразно вводить 3—5 мл сиропообразной фосфорной кислоты, так как она удерживает вольфрам в растворе и, кроме того, обесцвечивая желтую окраску железа (III), делает конечную точку титрования ' более отчетливой.
Для окисления применяют свежеприготовленный раствор персульфата 95%-ной чистоты (стр. 61). Во всех случаях анализируемый раствор следует кипятить 8—10 мин, чтобы обеспечить полное окисление хрома и разложение избытка персульфата. Марганец при этом окисляется до перманганата или двуокиси марганца, которые также необходимо разрушить до прибавления сульфата железа (II). Для этого, после разложения персульфата кипячением, на каждые 300 мл раствора прибавляют по 5 мл разбавленной (1:3) соляной кислоты и продолжают кипятить до восстановления окисленных соединений марганца, после чего кипятят еще 5 мин для удаления хлора. Если до прибавления соляной кислоты персульфат был полностью разрушен, хромовая кислота при этой обработке не восстанавливается.
, Это подтверждается результатами опытов W. С. Fed de. Во всех случаях бихромат находился в 300 мл разбавленной (1 : 9) серной кислоты и титрование проводилось потенциометрическим методом.
КаСг>Оу Взято Определено г г
1. Непосредственное титрование раствором FeSO4......... 0,1939 0,1940
2. То же................................................. 0,1939 0,1938
3. Прибавлено 20 мл разбавленной (1 : 3) НС1. Кипячение в течений ЗОлши...................................... 0,1939 0,1937
4. То же................................................. 0,1939 0,1938
5. Прибавлено 10 мл раствора AgNO3 (2,5 г/л) + 1 г
(NH4).2S2O8. Кипячение 10 мин. Обработка 5 мл разбавленной (1:3) НС1 и кипячение еще 5 мин..............". 0,2131 0,2131
6. Прибавлен MnSO4. Дальнейшая обработка, как в п. 5 0,2143 0,2142
7. Прибавлено 5 мл 0,3 н. раствора КМнО4, восстановлен
FeSO4. Дальнейшая обработка, как в п. 5 ............... 0,2143 0,2145
8. Введено 5 мл 0,3 н. раствора КМнО4, 10 мл раствора
AgNO3 (2,5 г/л), 2 г (NH4)2S2O8 и 5 мл разбавленной (1 :3) НС1. Кипячение 10 мин .......................... 0,2040 0,1943
9. Так же, как в п. 8, но перед добавлением НС1 раствор
кипятили 10 мин....................‘................... 0,2052 0,2053
Большие количества хрома наиболее целесообразно восстанавливать взвешенным количеством однородных кристаллов соли Мора
FeSO4-(NH4)2SO4-6H2O. В случае же, когда содержание хрома невелико и особенно когда приходится одновременно проводить большое число определений, удобнее пользоваться разбавленным сернокислым раствором этой соли. В присутствии значительных количеств хрома определение конца титрования несколько затрудняется вследствие зеленой окраски раствора, но соответствующая поправка для конечной точки определяется легко. Все реакции протекают стехиометрически; следовательно, можно пользоваться теоретическими титрами, установленными по оксалату натрия.
Ванадий и мышьяк не мешают титрованию, так как они хотя и восстанавливаются сульфатом железа (II), но затем снова окисляются эквивалентным количеством перманганата. Небольшие количества вольфрама изменяют окраску раствора в точке эквивалентности, но не влияют на 38 Заказ 522.
594
Гл: XXXIII. Хром.
результаты титрования. Если содержание вольфрама настолько велико, что конечная точка титрования становится неотчетливой, его лучше предварительно отделить, а хром, захваченный осадком вольфрама, определить отдельно. Никель, кобальт, молибден и уран не оказывают влияния на титрование.
Ход определения. Раствор, полученный после разложения пробы сплавлением, как указано в разделе «Разложение минералов, содержащих хром» (стр. 589), или другим способом, свободный от хлорид-ионов и содержащий приблизительно 15—18 мл серной кислоты и 3 мл азотной кислоты в общем объеме 300 мл, нагревают до кипения. Прибавляют 2,5%-ный раствор нитрата серебра в количестве, соответствующем 0,01 г соли на каждую 0,01 г находящегося в растворе хрома. Нагревают до кипения и приливают 20 мл свежеприготовленного 10%-ного раствора персульфата аммония. Кипятят 10 мин и затем, если образуется перманганат или окислы марганца, вводят 5мл5%-ного раствора хлорида натрия или 5 мл разбавленной (1 : 3) соляной кислоты, снова нагревают до кипения и после восстановления соединений марганца продолжают кипятить еще 5 мин. Если при этом марганец не восстанавливается, вводят еще некоторое количество хлорида натрия или соляной кислоты и снова кипятят.
По охлаждении раствора прибавляют отмеренное избыточное количество раствора соли Мора или взвешенное, количество этой соли и затем титруют раствором перманганата, титр которого установлен по оксалату натрия. По окончании титрования отмечают израсходованное количество перманганата, нагревают раствор до кипения для разрушения избытка перманганата, охлаждают до комнатной температуры и затем снова титруют перманганатом до появления окраски такой же интенсивности, как при первом титровании. Израсходованный в этом случае объем перманганата вычитают из объема раствора, израсходованного при первом титровании1. Полученную разность умножают на количество соли Мора, эквивалентное 1 мл раствора перманганата. Это произведение вычитают из введенного количества соли Мора и затем вычисляют содержание хрома, исходя из соотношения 3Fe : 1Сг.
Для установления соотношения между солью Мора и перманганатом приготовляют раствор из такого же количества соли, какое было введено в анализируемый раствор; приготовленный раствор должен иметь такой же объем и такую же кислотность, как анализируемый раствор в момент титрования. Приготовленный раствор титруют перманганатом. Из израсходованного объема раствора перманганата вычитают количество его, которое требуется для окрашивания раствора в точке эквивалентности.
. При выполнении массовых анализов после сплавления с перекисью натрия нет необходимости окислять хром персульфатом. В этом случае плав растворяют в 200 мл воды, прибавляют 1 г перекиси натрия и сильно кипятят до разложения перекиси (приблизительно 10 мин). Фильтруют через асбест и промывают осадок горячей водой, следя за тем, чтобы он все время был влажным. Фильтрат и промывные воды подкисляют серной кислотой и затем вводят сульфат железа (II). При зтом следует избегать введения в раствор органических веществ (что происходит, например, при фильтровании через бумагу), так какзто приводит к получению пони-
1 Оттированный раствор может использовать для определения ванадия, как описано в гл. «Ванадий» (стр. 514).
Методы определения
595
женных результатов для хрома. Эта ошибка обусловлена не восстановлением хромата, как можно было бы предположить, а восстановлением перманганата в процессе титрования.
Легкость, с которой можно осуществить потенциометрическое титрование хрома, применяя соответствующую аппаратуру \ характеризуется следующим ходом анализа. В охлажденный до 20° С разбавленный сернокислый раствор, содержащий хром (VI), погружают каломельный и платиновый электроды; пучок лучей, отраженных зеркальным гальванометром устанавливают вблизи левого края шкалы. Затем в раствор постепенно вводят титрованный раствор сульфата железа (II), пока пучок лучей не начнет непрерывно передвигаться вправо. После этого прибавляют эквивалентный титрованный раствор хромата калия до прекращения перемещения пучка лучей влево. Снова вводят раствор сульфата железа (II), пока опять не начнется перемещение лучей вправо. Вычитают израсходованный объем раствора хромата из введенного объема сульфата железа и вычисляют содержание хрома, исходя из соотношения ICr : 3Fe. Ванадий также восстанавливается железом (II) и учитывается совместно с хромом. Вольфрам не влияет на титрование.
Колориметрические методы
Хроматный метод. Определение хрома колориметрическим методом преимущественно проводят сравнением интенсивности окраски хромата с окраской стандартного раствора в щелочной среде. При анализе горных пород для этого обычно используют водную вытяжку плава анализируемого материала со смесью карбоната натрия и нитрата калия. Обычно встречающиеся в горных породах элементы определению не мешают. При сплавлении пробы в платиновом тигле не следует вводить в плавень слишком больших количеств селитры и температура сплавления не должна быть слишком высокой, чтобы не вызвать порчу тигля, так как раствор может окраситься в желтый цвет за счет переходящей в него платины.
При сплавлении с перекисью натрия и выщелачивании плава водой некоторое количество железа может остаться в растворе и окрасить его. Этого можно избежать, если перед фильтрованием нагревать раствор на водяной бане не менее одного часа. Щелочной раствор окрашивается также в случае фильтрования его через бумажный фильтр, что, однако, может быть устранено, если предварительно тщательно промывать применяемый фильтр раствором щелочи или, лучше, применением для фильтрования этого раствора асбеста.
Бораты не мешают определению.
Уран в условиях колориметрирования хрома также дает желтую окраску, и, следовательно, в его присутствии метод неприменим.
Наиболее удобно сравнивать окраски растворов, содержащих приблизительно 0,01—0,1 мг Сг2О3 в 1 мл.
Ход определения. Приготовляют стандартный раствор бихромата, содержащий 0,1 мг Сг2О," в 1 мл. Для этого 0,1934 г перекристаллизованного бихромата калия К2Сг2О7 растворяют в воде и разбавляют до 1 л. Перед применением этого раствора для сравнения с анализируемым его следует» сделать щелочным.
1 G. L. К е 11 е у и др., J. Ind. Eng, Chem., 9, 780 (1917).
38*
596
Гл. XXXIII. Хром
Для определения хрома в горных породах 1—5 г пробы сплавляют со смесью карбоната и селитры, как описано в гл. «Ванадий» (стр. 510). Плав растворяют в воде, и если в растворе находится перманганат, прибавляют несколько капель этилового спирта для разрушения его окраски, а затем фильтруют через асбест. Если окраска раствора чрезмерно бледна, повышают концентрацию хрома упариванием раствора или осаждением нитратом ртути (I) и последующей обработкой, как указано в гл. «Ванадий» (стр. 510). t
При анализе других объектов, например осадков от аммиака, содержащих малые количества хрома, окйсление можно осуществлять сплавлением с перекисью натрия в фарфоровом тигле.
Во всех случаях полученный раствор хромата переносят в мерную колбу такой емкости, чтобы интенсивность окраски раствора после разбавления до метки соответствовала 0,01—0,1 мг Сг2О3 в 1 мл.
Заканчивают определение титрованием раствора такого же количества реактивов стандартным раствором бихромата калия до одинаковой с анализируемым раствором окраски или же каким-нибудь другим обычно применяемым колориметрическим или фотометрическим способом измерения.
Метод с дифенилкарбазидом. Малые количества хрома (микрограммы) можно определять по реакции с дифенилкарбазидом *, который окисляется бихроматом в слабокислом растворе с образованием соединения, окрашенного в красно-фиолетовый цвет.
Ванадий, если он присутствует в значительных количествах, мешает определению. Его можно удалить экстракцией хлороформом после переведения в оксихинолят 1 2.
Уран не влияет на реакцию хрома с дифенилкарбазидом.
Другие методы
Хром количественно осаждается аммиаком3 по методу, приведенному в гл. «Алюминий» (стр. 565). Осадок следует под конец прокаливать в атмосфере водорода, иначе получаются повышенные результаты вследствие окисления хрома в процессе прокаливания. В связи с этим, а также и потому, что хром почти всегда сопровождают посторонние, осаждающиеся аммиаком элементы, как, например, железо, алюминий, фосфор и ванадий, этим методом для определения хрома пользуются лишь в редких случаях 4.
1 С. F. J. van derWalt, A. J. vanderMerwe, Analyst, 63, 809 (1938). *См. также А. К. Бабкой А. Т. Пилипенко, Колориметрический анализ, Госхимиздат, 1951.*
2 Е. В. S a n d е 11, Ind.Eng. Chem., Anal. Ed., 8, 336 (1936).
3 В опытах, проведенных Н. В. Knowles, количественное осаждение хрома было достигнуто при' кипячении и нейтрализации аммиаком до отчетливо желтой окраски метилового красного 350 мл раствора, содержащего 0,06 г Cr (II). При введении избытка раствора аммиака в количестве 1 мл в растворе оставалась 0,1 мг Сг, а в количестве 5 мл—0,2 мг Сг. Когда же к раствору было прибавлено 25 мл НС1, а затем, по нейтрализации кислоты, 5 мл избытка раствора аммиака, потеря хрома составила 0,4 мг. При промывании осадка 200 мл 2%-ного раствора NH4C1 в раствор перешла 0,1 мг Сг.
4 Заслуживает внимания утверждение, что сильно прокаленная Сг2О3 растворяется в горячих разбавленных растворах бромата калия с образованием хромата JR. Lyden, Z. anorg. U. allgem. Chem., 223, 28 (1935)].
Методы определения
597
Осаждение хрома в виде хромата серебра Ag2CrO4, хромата ртути Hg2CrO4 и хромата бария ВаСгО4 представляет интерес главным образом для группового разделения и качественного испытания на хром, а не для количественного его определения, так как многие другие элементы также образуют нерастворимые соединения с этими реагентами.
Точные результаты получаются при определении хрома методом, основанным на восстановлении хромата иодистоводородной кислотой и титровании выделяющегося при этом иода раствором тиосульфата натрия. Этот метод, однако, не получил такого широкого распространения, как метод, описанный в разделе «Титрование сульфатом железа (II) и перманганатом», так как железо, медь, мышьяк, ванадий и молибден, которые в состоянии высшей валентности выделяют иод в кислых растворах иодида калия, должны отсутствовать.
* Известен колориметрический метод определения хрома с комплексоном III (этилендиаминтетраацетатом натрия1). Метод специфичен, мешают только окрашенные катионы (своей окраской), но сравнительно мало чувствителен (оптимальные концентрации хрома 5—80 мг/л). Светопогло-щение получаемого красно-фиолетового раствора измеряют, применяя зеленые светофильтры (длина волны 550 ммк). Доп. ред*
1R. Pribil, J. Klubalova, Chem. Listy, 43, 265 (1949); Coll. Czech. Chem. Comm., 15, 42 (1950); P. П p ш и б и л, Комплексоны в химическом анализе, Издатинлит, 1955, стр. 88; В. Г. Г о р ю ш и н а, Е. Я. Г а й л и с, Зав. лаб., 21, 642 (1955).
Глава XXXIV
ТОРИИ
Торий, по всей вероятности, входит в состав силикатных горных пород чаще, чем это обычно предполагают, причем он встречается главным образом в породах с высоким содержанием натрия. Совместно с ураном, цирконием, танталом, ниобием и редкоземельными элементами торий находится в таких минералах, как уранинит, титано- и танталониобаты и монацитовый песок. Подобно урану, торий отличается сильной радиоактивностью. Типичным ториевым минералом является торит — силикат тория ThSiOa, встречающийся всегда в более или менее' метаморфизированном виде. Торианит — окись тория и урана — отличается более высоким содержанием гелия, чем все остальные известные минералы. Основным источником получения тория является монацитовый песок, в котором торий встречается в различных количествах совместно с фосфатами элементов цериевой группы. Использование соотношения между содержанием свинца, урана и тория для установления возраста пород и минералов повышает интерес и значение методов определения тория.
ОБЩИЕ ЗАМЕЧАНИЯ
Для определения тория его целесообразно сперва отделить в виде оксалата совместно с скандием и редкоземельными элементами от циркония, титана и других обычно встречающихся вместе с торием элементов. В дальнейшем торий отделяют от скандия и редкоземельных элементов осаждением значительным избытком йодата калия из сильно азотнокислого раствора, свободного от соляной кислоты *. Для осуществления этого разделения требуются два раствора йодата: концентрированный раствор, состоящий из 15 г йодата калия, 50 мл азотной кислоты и 100 мл воды (раствор 1) и разбавленный раствор, содержащий в 450 мл воды 4 г йодата и 50 мл азотной кислоты (раствор 2). Испытание на присутствие тория проводят следующим способом.
Азотнокислый раствор, содержащий редкоземельные элементы, концентрируют и кипятят с небольшим количеством сернистой кислоты для восстановления церия. После этого 5 мл раствора обрабатывают 10 мл раствора 1, тщательно перемешивают, прибавляют 20 мл раствора 2 и кипятят. Некоторые количества цериевых и иттриевых земель могут выделиться при обработке раствором 1, но они вновь растворяются в процессе
1 R. J. М е у е г, Z. anorg. Chem., 71, 65 (1911).
Разложение минералов, содержащих торий
599
кипячения с раствором 2. Появление неисчезающего осадка указывает наТсодержание тория. Чувствительность метода — 0,1 мг тория в 1 мл исходного раствора.
Цирконий также осаждается в этих условиях. Если имеется сомнение в полноте предварительного отделения циркония, осадок йодата отфильтровывают, промывают, затем смывают в маленький стакан и кипятят с 50 мл 10%-ного раствора щавелевой кислоты. При этом цирконий переходит в раствор, а торий образует нерастворимый оксалат.
Торий можно открыть спектральным методом и «методом эманации»
В обычном ходе анализа торий попадает в осадок от аммиака и принимается за алюминий, если содержание последнего вычисляют по разности. В тех случаях, когда осадок от аммиака растворяют и затем обрабатывают фтористоводородной или щавелевой кислотой (стр. 621) для отделения редкоземельных металлов, торий также переходит в осадок, и если его предварительно не отделить, захватывается впоследствии элементами цериевой или иттриевой группы, в зависимости от применяемого способа обработки.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ТОРИЙ
Монацит разлагают следующим способом * 2. Обрабатывают 50 г монацитового песка 75 мл концентрированной серной кислоты в фарфоровой чаЩке емкостью 500 мл и, часто перемешивая, нагревают около 4 ч так, чтобы слабо выделялись пары серной кислоты. Когда масса превратится в пасту, ее охлаждают и затем небольшими порциями вводят в 400 мл холодной воды, находящейся в большом стакане, погруженном в ледяную воду. Не следует допускать, чтобы температура раствора поднималась выше 20° С. Жидкость декантируют в мерную колбу емкостью 1000 мл. Остаток выщелачивают небольшими порциями холодной воды и полученный водный экстракт сохраняют отдельно. Нерастворимый остаток высушивают, обрабатывают 10 мл концентрированной серной кислоты, нагревают до появления обильных ее паров и нагревание продолжают еще 1,5 ч.
По охлаждении пасту постепенно вводят в охлажденный водный экстракт, дают отстояться и затем раствор декантируют в мерную колбу, в которой находится основной раствор. Остаток тщательно выщелачивают разбавленной (1 : 10) серной кислотой, сливая каждый раз раствор декантацией в мерную колбу. Когда раствор примет комнатную температуру, его разбавляют до метки водой, тщательно перемешивают и отфильтровывают для каждого определения по 200 мл.
Каждую аликвотную часть раствора (соответствующую 10 г песка) разбавляют до 1000 мл и затем по каплям из делительной воронки сливают в 150 мл холодного насыщенного раствора щавелевой кислоты, энергично перемешиваемого механической мешалкой. Оставляют на ночь. Осадок отфильтровывают и промывают водой сначала декантацией, а затем на фильтре до удаления кислоты. Фильтр с осадком высушивают в воздушной бане и сохраняют. Фильтрат и промывные воды нейтрализуют аммиаком, подкисляют соляной кислотой и затем вводят 15 мл избытка кислоты. Выделившемуся осадку дают остояться в течение ночи, после чего
i,H. Н. Helmick, J. Am. Chem. Soc., 43, 2003 (1921).
2 Bur. Mines Bull., 212, 53 (1923).
600
Гл. XXXIV. Торий
его отфильтровывают и промывают разбавленной (1 : 1000) соляной кислотой, содержащей 2% щавелевой кислоты. Фильтр с осадком прокаливают в платиновой чашке, прокаленный остаток смачивают водой и растворяют в небольшом количестве соляной кислоты при нагревании. Фильтруют, промывают фильтр небольшими порциями разбавленной (1 : 99) соляной кислоты, раствор нейтрализуют точно до кислой реакции по метиловому красному, доводят его объем до 50 мл и приливают 50 мл насыщенного на холоду раствора щавелевой кислоты. Нагревают несколько минут и оставляют стоять 4 ч. После этого фильтруют, промывают осадок разбавленным солянокислым раствором щавелевой кислоты и прокаливают в платиновом тигле.
Сохраненные ранее высушенные оксалаты отделяют от фильтра и сжигают бумагу в том же тигле. Затем переносят в тигель высушенные оксалаты и осторожно прокаливают до постоянной массы. Эта масса соответствует общему содержанию окислов редкоземельных элементов. Взвешенный осадок помещают в стакан емкостью 600 мл, смачивают небольшим количеством воды, растворяют в разбавленной соляной кислоте (1 : 1) и отделяют торий, как указано в разделе «Методы отделения» (см. ниже).
Монацитовый песок и другие редкоземельные минералы, содержащие небольшие количества тория, можно также разложить следующим способом. Тщательно смешивают 0,5 г тонко измельченной пробы с 0,5 г фторида натрия и 10 г пиросульфата калия в большом платиновом тигле. Закрывают тигель крышкой и помещают над очень маленьким пламенем горелки. Постепенно повышают температуру, пока не прекратится выделение газов. По окончании сплавления плав охлаждают и обрабатывают при нагревании разбавленной (1 : 10) соляной кислотой до разложения. Дают отстояться и затем фильтруют. Смывают остаток обратно в стакан, нагревают с 10 мл соляной кислоты, разбавляют до 100 мл и снова фильтруют. К объединенным фильтратам прибавляют раствор аммиака почти до нейтральной реакции, затем осаждают оксалаты редкоземельных элементов общепринятым методом, описанным в разделе «Методы определения» (стр. 607).
Торит и другие силикаты, богатые торием и бедные редкоземельными элементами, можно разложить нагреванием в течение 1—2 ч тонко измельченной пробы с концентрированной соляной кислотой при температуре, близкой к кипению. Раствор затем выпаривают досуха, отделяют кремнекислоту, как обычно, и остаток кремнекислоты сплавляют с пиросульфатом. Плав выщелачивают разбавленной (Г: 1) соляной кислотой, фильтруют и присоединяют раствор к ранее полученному фильтрату.
МЕТОДЫ ОТДЕЛЕНИЯ
Заслуживают внимания следующие методы отделения тория:
1) осаждение в виде фторида или оксалата, в результате чего торий отделяется от обычных элементов, включая цирконий и титан, но не отделяется от редкоземельных элементов;
2) осаждение перекисью водорода, йодатом, гексамином или тиосульфатом, при котором происходит отделение тория от редкоземельных элементов;
3) осаждение йодатом или избыточным количеством фторида аммония, служащее для отделения тория от скандия.
Методы отделения
601
Оксалат тория растворим в растворах оксалатов щелочных металлов и в сильных минеральных кислотах. Осаждение его лучше всего проводить введением щавелевой кислоты в солянокислый раствор, содержащий менее 4% соляной кислоты по объему. Метод разделения тория и редкоземельных элементов, основанный на растворимости оксалата тория в оксалате аммония, не дает удовлетворительных результатов, независимо от того, нагревают ли выделенные оксалаты с оксалатом аммония, или же осаждение оксалатов осуществляют введением в раствор избыточного количества оксалата аммония \
Фторид тория нерастворим в разбавленных минеральных кислотах. Осаждение в виде фторида чаще используется для отделения тория, совместно с редкоземельными металлами, от других элементов, попадающих в осадок от аммиака в процессе анализа, чем для отделения больших количеств тория от обычно встречающихся металлов. Это разделение лучше всего осуществлять в условиях, обеспечивающих отсутствие других кислот, кроме фтористоводородной, как, например, обработкой промытого осадка от аммиака фтористоводородной кислотой в платиновой чашке и упариванием раствора до небольшого объема (стр. 623). Более полное осаждение тория, особенно в присутствии минеральных кислот, достигается, если вводить фторид аммония.
При осаждении тория гексамином, тиосульфатом или перекисью водорода, или при конечном выделении его в виде оксалата торий следует предварительно отделить от фосфора. Сплавление с карбонатом натрия или осаждение едким натром в данном случае не дает удовлетворительных результатов вследствие растворимости тория в щелочных растворах, содержащих карбонат. Отделение тория от фосфора может быть выполнено йодатным методом или осажденйем щавелевой кислотой из холодного разбавленного солянокислого раствора. При анализе монацитового песка (наиболее частый случай) отделение проводят следующим способом.
Разбавляют до 1 л 200 мл сернокислого раствора, полученного, как указано в разделе «Разложение минералов, содержащих торий» (стр. 599), и очень медленно при непрерывном перемешивании вливают в 150 мл холодного насыщенного раствора щавелевой кислоты. Оставляют стоять не менее 15 ч, после чего фильтруют, промывают осадок разбавленной (1 : 99) соляной кислотой, высушивают и сохраняют. Фильтрат и промывные воды нейтрализуют аммиаком, вводят 10 мл соляной кислоты и оставляют на 15 ч. Выделившийся осадок отфильтровывают, промывают раствором, содержащим 40 мл соляной кислоты и 25 г щавелевой кислоты в 1 л, затем сушат, осторожно прокаливают в фарфоровой чашке, остаток смачивают небольшим количеством воды, растворяют его в 10 мл соляной кислоты и фильтруют. Фильтрат разбавляют до 500 мл, нагревают до кипения и медленно вливают в него 100 мл горячего 10%-ного раствора щавелевой кислоты. Оставляют стоять 4—5 ч при комнатной температуре, после чего фильтруют, промывают осадок 2,5%-ным раствором щавелевой кислоты в разбавленной (4 : 96) соляной кислоте, затем сушат и присоединяют к сохраненному ранее осадку. Объединенные осадки прокаливают до ойислов, после чего отделяют торий от редкоземельных элементов.
Простейший метод отделения тория от редкоземельных элементов заключается, по-видимому, в осаждении гексаметилентетрамином (гекс-
1 Е. В е n z, Z. angew. Chem., 15, 297 (1902).
602
Гл. XXXIV. Торий
амин или уротропин CeH12N4) следующим способом х. Приготовляют 100 мл разбавленного солянокислого раствора, содержащего эти элементы (pH = 2—4), нагревают до 30° С и прибавляют сначала 5 г хлорида аммония, а затем по каплям при перемешивании 10%-ный раствор гексамина в небольшом избытке. В этот момент pH раствора должен иметь значение примерно 5,8. При добавлении 1 капли осадителя к отстоявшемуся про- ( зрачному раствору не должно происходить осаждения тория. Раствор декантируют через фильтр, затем переносят на фильтр осадок и промывают горячим 2%-ным раствором нитрата аммония. Фильтрат сохраняют для определения редкоземельных элементов. Осадок на фильтре растворяют в горячей 2 н. соляной кислоте и затем тщательно промывают фильтр горячей водой. Раствор разбавляют до 100 мл, вводят несколько капель метилового красного, а затем при перемешивании разбавленный (1 : 1) раствор аммиака до пожелтения индикатора. После этого прибавляют разбавленную (1 : 1) соляную кислоту до появления розовой окраски индикатора и хлорид аммония в таком количестве, чтобы содержание его в растворе составляло 5%. Раствор нагревают'до 30° С и осаждают торий \ гексамином. Осадок промывают, как указано ранее. Фильтрат и промывные воды присоединяют к сохраненному ранее раствору. Промытый осадок можно прокалить 1 2 и взвесить в виде ThO2 или растворить в соляной кислоте и осадить торий в виде оксалата, как указано на стр. 607.
Для отделения тория от небольших количеств церия и лантана можно пользоваться также осаждением перекисью водорода 3. Этот метод не испытывался на растворах, содержащих другие редкоземельные элементы и скандий. Цирконий и титан осаждаются совместно с торием. Осаждение проводят следующим способом. Нейтральный раствор нитратов тория, церия и лантана4 обрабатывают 10 г нитрата аммония, разбавляют до 1 100 мл, нагревают до 60—80° С и затем для осаждения тория прибавляют 20 мл перекиси водорода (3%-ной). Осадок отфильтровывают и промы- j вают горячим 2%-ным раствором нитрата аммония. Если осадок окрашен в желтый цвет, что указывает на неполное отделение церия, его следует переосадить. Для этого осадок растворяют в азотной кисл<йе, раствор | выпаривают досуха и затем растворяют нитраты в 100 мл 10%-ного рас- я твора нитрата аммония5. - Я
1 А. М. I s m a i 1, Н. F. Harwood, Analyst, 62, 185 (1937). • fl
2 Таким путем авторы метода успешно отделяли торий от церия (Ш), лантана, j
неодима, празеодима и иттрия. Скандий сопровождает торий. При анализе монаци- ''' тового песка С. J. Rodden в конечном осадке на обнаружил редкоземельных J элементов спектрохимическим методом. а
3 G, W у г о н b о f f, А. V е г п е н i 1, С. г., 126, 340 (1898);’ Е. Benz, Z.
angew. Chem., 15, 297 (1902). , . J
4 Нитраты можно приготовить непосредственно из оксалатов следующим спо- ,1 собой. Смывают оксалаты с фильтра в фарфоровую чашку возможно меньшим коли- <4 чеством воды. Фильтр промывают сначала разбавленной азотной кислотой, а затем горя- J чей водой. Раствор выпаривают почти досуха. Прибавляют 5—10 мл азотной кислоты, Д затем 20 мл дымящей азотной кислоты и закрывают чашку часовым стеклом. Нагре- Д вают на паровой бане до прекращения выделения газов. Снимают часовое стекло, опц- д ласкивают его и стенки чашки, а затем выпаривают раствор досуха. Смачивают оста- я ток небольшим количеством воды и снова выпаривают досуха. Я
5 Этим методом Benz определил 0,1468 г ThO2 однократным осаждением из -я раствора, содержащего 0,1470 г ThO2, 0,13 е Се’О2 и 0,2 г Ьа2О3; в растворе, содержащем 0,0735 г ThO2 и те же количества церия и .лантана, двукратным осаждением он опре- j| делил 0,0735 г ThO2. Я
Методы отделения
603
Торий можно легко отделить от церия (III), «дидима», обычных металлов и фосфорной кислоты осаждением йодатом калия из сильно азотнокислого раствора 1>2. Титан и цирконий осаждаются вместе с торием. Метод сводится к следующему. Окислы или гидроокиси тория и редкоземельных элементов растворяют в 150 мл разбавленной (1 : 2) азотной кислоты. Раствор охлаждают в ледяной воде 3, прибавляют охлажденный раствор 15 г йодата калия в 30 'мл воды и 50 мл азотной кислоты и время от времени перемешивают в течение 30 мин. После этого дают отстояться и фильтруют, раздавливая комочки осадка сплющенной стеклянной палочкой. Осадок промывают 250 мл холодного раствора, содержащего 8 г йодата и 200 мл разбавленной (1 : 1) азотной кислоты в 1 л. Когда жидкость стечет, осадок смывают 100 мл промывной жидкости обратно в стакан, энергично перемешивают и фильтруют через тот же фильтр. Дают жидкости стечь и снова смывают осадок обратно в стакан, на этот раз горячей водой. Нагревают почти до кипения и затем растворяют осадок, добавляя медленно при перемешивании 30 мл азотной кислоты. Разбавляют до 60 мл и прибавляют раствор 4 г йодата калия в небольшом количестве воды и разбавленной азотной кислоты. По охлаждении фильтруют через тот же фильтр, промывают, как прежде, декантацией и затем переносят осадок на фильтр. Смывают осадок в стакан и промывают фильтр горячей разбавленной соляной кислотой, ^содержащей небольшое количество сернистой кислоты. Раствор кипятят и, если требуется, прибавляют еще соляную или сернистую кислоты до полного растворения осадка. Раствор разбавляют и затем осаждают торий аммиаком. Осадок отфильтровывают, промывают горячей водой до удаления йодата, после чего растворяют в соляной кислоте и осаждают щавелевой кислотой 4, как указано ниже в разделе «Методы определения» (стр. 607).
Йодатный метод, описанный в разделе «Общие замечания» (стр. 598) для качественного открытия тория, в измененном виде может служить также для отделения тория от скандия 5. В этом случае редкоземельные элементы выделяют щавелевой кисло/ой, осадок прокаливают, окислы растворяют в соляной кислоте и отделяют торий и скандий осаждением тиосульфатом (см. ниже). Полученный осадок растворяют в азотной кислоте, фильтруют и выпаривают фильтрат досуха. Остаток обрабатывают 5 мл воды, содержащей несколько капель азотной кислоты, и затем прибавляют 5—10 мл раствора йодата (раствор 1, стр. 598), в зависимости от содержания тория. Перемешивают, прибавляют 10—20 мл раствора 2 (стр. 598), нагревают 15 мин при 60—80° С и затем дают отстояться на холоду. Фильтруют и промывают осадок отдельными порциями раствора 2. Фильтрат сохраняют. Осадок смывают обратно в стакан, прибавляют
1 R. J. М е у е г, Z. anorg. Chem., 71, 65 (1911).
2 R. J. M e у e r, M. S р е t е г, Chem. Ztg., 34, 306 (1910),
3 100 мл сернокислого раствора монацитового песка, полученного, как указано в разделе «Разложение минералов, содержащих торий» (стр. 599), можно непосредственно обработать азотной кислотой.
4 Этим методом Meyer и Speter определили 0,4994, 0,4990 и 0,4988 г ThO2 в растворе, содержащем 0,4995 г ThO2, и 0,4999 г ThO2 в растворе, содержавшем 0,4995 г ThO2 и 10 г нитрата церия. Было установлено, что празеодим и неодим также не влияют на осаждение тория. В монацитовом песке, в котором «наиболее точным методом» было определено 5,53% ThO2, этим методом было найдено 5,54; 5,57; 5,58 и 5,57% ThO2.
8 R. J. Meyer, Z. anorg. Chem., 71, 67 (1911).
604
Гл. XXXIV. Торий
в значительном избытке раствор аммиака, кипятят для переведения йодата в гидроокись и к концу кипячения вводят мацерированную фильтровальную бумагу. Фильтруют, осадок тщательно промывают для удаления йодата, сушат и прокаливают до ThO2. Сохраненный фильтрат нагревают, прибавляют избыточное количество раствора аммиака, кипятят, фильтруют, промывают осадок, так же как осадок тория, прокаливают и взвешивают в виде Sc2O3.
Разделение тория и скандия осаждением тория в виде фторида из раствора, содержащего большой избыток фторида аммония, изложено в гл. «Скандий» (стр. 614).
Торий можно весьма успешно отделить от церия (III), лантана, празеодима и неодима осаждением пирофосфатом натрия из раствора приблизительно 0,3 н. по содержанию соляной или серной кислоты х, лучше из солянокислого раствора 1 2.
Отделение тория (и скандия) от редкоземельных элементов осаждением тиосульфатом натрия не дает вполне удовлетворительных результатов, так как торий при этом не осаждается количественно и разделение недостаточно четко. Этот метод менее пригоден для отделения тория от иттриевых земель, чем от цериевых3. Другим недостатком метода является выделение серы в процессе осаждения, что при содержании малых количеств тория не дает возможности установить, имело ли место образование осадка гидроокиси тория.
Осаждение следует повторять до тех пор, пока результаты испытания тиосульфатного фильтрата раствором аммиака не покажут, что отделение от редкоземельных элементов прошло успешно. Помимо этого, после осаждения тиосульфатом торий необходимо дополнительно извлекать из фильтратов.
Такие элементы, как алюминий, титан и цирконий, в условиях осаждения тория также выделяются из раствора и, следовательно, должны быть предварительно отделены.
Метод заключается в следующем 4. Смесь окислов или гидроокисей, полученных после предварительного выделения щавелевой кислотой, растворяют в соляной кислоте. Раствор выпаривают досуха, остаток смачивают 5—10 мл воды и снова выпаривают досуха. Затем прибавляют 150 мл воды, 2 капли соляной кислоты и нагревают до кипения. Если предварительное осаждение щавелевой кислотой было проведено надлежащим образом, раствор при этом остается прозрачным. Постепенно вводят раствор 2 г тиосульфата натрия в 10 мл воды. Закрывают стакан часовым стеклом и кипятят 15 мин. Фильтруют и промывают осадок горячей водой. Фильтрат (фильтрат А) сохраняют для дополнительного извлечения из него тория.
1 R. J. С arney, Е. D. Campbell,!. Am. Chem. Soc., 36, 1140 (1914).
3 Согласно M. К о s s [Chem. Ztg., 36, 686 (1912)] и F. W i г t h [Z. angew. Chem., 25, 1678 (1912)], торий можно успешно отделить от церия, празеодима и неодима введением по каплям, при перемешивании, холодного раствора NaHPO3 в кипящий анализируемый раствор, содержащий 15—20% H3SO4. Раствор оставляют стоять несколько часов и затем выделившийся осадок Th(PO3)2 отфильтровывают.
3 G. Р. D г о s s Ь а с h [Z. angew. Chem., 14, 655 (1901)] установил, что осадком увлекаются значительные количества иттрия даже после четырехкратного пере-осаждения.
4 Е. В е n z, Z. angew. Chem., 15, 297 (1902); R. Fresenius, E. Hintz, Z. Anal. Chem., 35, 625 (1896).
Методы отделения
605
Фильтр развертывают на стеклянной пластинке, смывают осадок струей горячей воды в фарфоровую чашку и тщательно промывают бумагу сначала горячей разбавленной соляной кислотой, а затем небольшим количеством горячей воды. К раствору прибавляют 20 мл соляной кислоты, кипятят 5—10 мин, фильтруют и промывают осадок серы и фильтр разбавленной соляной кислотой и водой. Раствор выпаривают досуха и повторяют осаждение тиосульфатом. Осадок после второго осаждения тиосульфатом (осадок Б) сохраняют.
Фильтрат кипятят, прибавляют раствор аммиака в небольшом избытке, фильтруют, промывают осадок небольшим количеством горячей воды и сохраняют для определения в нем тория. Величина этого осадка (осадок В) показывает, следует ли осадок Б растворить и затем снова переосадить тиосульфатом. Осадок после конечного осаждения тиосульфатом растворяют в соляной кислоте, как это было указано, и раствор (раствор Г) сохраняют для осаждения тория в виде оксалата, как изложено в разделе «Методы определения» (стр. 607). Объединяют фильтраты (А и остальные, еще не обработанные раствором аммиака), нагревают до кипения, прибавляют избыточное количество раствора аммиака, фильтруют и затем осадок совместно с осадком В и другими, полученными ранее осаждением аммиаком, растворяют в соляной кислоте. Раствор выпаривают досуха, затем проводят осаждение тиосульфатом, как ранее, и фильтруют. Если требуется определение редкоземельных элементов., фильтрат сохраняют. Промытый осадок после осаждения тиосульфатом растворяют в соляной кислоте и полученный раствор присоединяют к раствору Г, в котором содержится основная масса тория Ч
Установлено1 2, что торий количественно осаждается при обработке кипящего нейтрального раствора небольшим избытком горячего почти насыщенного раствора себациновой кислоты и отделяется таким образом от церия, лантана, празеодима, неодима, самария и гадолиния. Реагент вводят медленно, непрерывно перемешивая раствор. По осаждении осадок тотчас же отфильтровывают, промывают, горячей водой и прокаливают до окиси ThO2. Недостатком метода является трудность разложения себациновой кислоты в фильтрате.
Суспензия чистого карбоната свинца 3 количественно осаждает торий, цирконий, церий (IV) и железо (III), а уран, хром (III) и алюминий не полностью. Церий (III), лантан, неодим, празеодим, иттрий, самарий и иттриевая группа карбонатом свинца не выделяются. Осаждение осуществляют введением избыточного количества карбоната свинца в холодный анализируемый раствор, после чего раствор оставляют стоять на 24 ч, время от времени перемешивая.
Карбонат, свинца приготовляют следующим способом. Чистый перекристаллизованный Pb(NO3)a растворяют в пятикратном по массе количестве воды. Раствор нагревают до 70° С, сильно перемешивают и затем вводят возогнанный (NH4)2CO3 до образования небольшого осадка. Время от времени часть мутной жидкости фильтруют, фильтрат подкисляют HN03 и испытывают добавлением K.CNS. Если присутствует железо, продолжают перемешивание. По охлаждении фильтруют и прозрачный фильтрат тонкой струей вливают в прозрачный насыщенный раствор возогнанного карбоната аммония. Осадку дают отстояться, промывают декантацией дистиллированной ___________ /
1 По существу тем же методом Benz определил 0,0436 г ThO2 и 0,2077 г СеОг в смеси, содержавшей 0,0443 г ThO2 и 0,2086 г СеО2.
2 Т. О. S m i t h, С. James, J. Am. Chem. Soc., 34, 281 (1912).
3 W. B. Giles, Chem. News, 92, 1, 30 (1905).
606
Гл. XXXIV. Торий
водой, затем переносят на фильтр и промывают до тех пор, пока промывная вода не покажет отчетливо кислую реакцию по метиловому оранжевому после прибавления к 500 мл 2 капель 1 н. HNO3. Жидкости дают стечь и затем осадок нагревают при 40— 50 °C до образования пасты. Сохраняют в широкогорлой склянке с притертой пробкой.
Для отделения тория от редкоземельных элементов, находящихся в монацитовом песке, было предложено 1 осаждение фениларсоновой кислотой CeH5AsO(OH)2 из кипящего уксуснокислого раствора, содержащего ацетат аммония. Церий долж'ен быть в трехвалентном состоянии. Цирконий при этом также осаждается.
Обработка сероводородом, вначале в сильнокислом растворе, который. затем разбавляют (стр. 83), служит для отделения элементов сероводородной группы от тория. Осаждением раствором аммиака (свободным от карбоната аммония) пользуются для отделения тория от магния, щелочноземельных и щелочных металлов. Гидроокись тория нерастворима в избытке едкого натра или кали, свободных от карбонатов, что дает возможность отделять торий от таких элементов, как алюминий и бериллий. Торий не осаждается аммиаком или сульфидом аммония в присутствии винной кислоты. Это используется для разделения тория и железа в сульфидном растворе2. ,
* Установлено3 *, что гидроокись тория количественно выделяется пиридином в виде плотной модификации, обладающей незначительной адсорбционной способностью. ХДорид и нитрат аммония ускоряют коагуляцию и отстаивание осадка. Сульфаты мешают осаждению вследствие образования сульфатоториатов. Влияние небольших количеств сульфатов можно устранить введением избыточного количества хлорида аммония. При осаждении пиридином происходит отделение тория от марганца, никеля, кобальта, меди, цинка и кадмия, образующих растворимые комплексные соединения с осадителем. Благодаря тому что пиридин не образует прочных карбонатов и не поглощает двуокиси углерода из воздуха, метод удобен для отделения тория от щелочноземельных металлов. Небольшая адсорбционная способность осадка обусловливает хорошее отделение тория от щелочных металлов. Доп. перев*
Торий, даже на холоду, осаждается свежеприготовленной суспензией карбоната бария (стр. 108) и отделяется таким способом от никеля и марганца.
Из растворов, в которых находится свободная минеральная кислота, купфероном торий осаждается не полностью*, но выделяется количественно из растворов, содержащих уксусную кислоту и ацетат аммония.
Цирконий можно отделить от тория двукратным осаждением в виде фосфата из разбавленного (1 : 10) сернокислого раствора (см. стр. 641). Отделение вольфрама от тория можно осуществлять отгонкой при 700° С в токе кислорода и сухого хлористого водорода, а в случае анализа металлов 5 — после предварительного окисления вольфрама одним кислородом.
1 А. С. R i с е, Н. С. F о g g, С. James, J. Am. Chem. Soc., 48, 895 (1926).
3 W. M. Thornton, Jr., Am. J. Sci., [4], 42, 151 (1916).
3Э. А. Остроумов, С. Беручьян, Зав. лаб., 12, 806 (1946).
< W. M. Thornton, ци,т. выше; G. E. F. L u n d e 11, H. В. Knowles, J. Ind. Eng. Chem., 12, 349 (1920).
•D. H. Brophy, C. VanBrunt, Ind. Eng. Chem., 19, 107 (1927).
Методы, определения.
607
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Осаждение в виде оксалата тория
Практически во всех методах определения тория необходимо конечное осаждение его в виде оксалата для обеспечения полного удаления циркония ц титана, обычно сопровождающих торий при всех предваритель-ных операциях. Осаждению тория в виде оксалата должны предшествовать операции, изложенные в разделе «Методы отделения» (стр. 600), для отделения обычных металлов, щелочноземельных металлов, редкоземельных элементов и скандия. Осаждение аммиаком, как описано в гл. «Алюминий» (стр. 565), с последующим прокаливанием до окиси вполне приемлемо для анализа растворов, свободных от других осаждаемых аммиаком элементов. В этом случае осадок лучше промывать нитратом аммония, чем хлоридом аммония, вследствие летучести хлорида тория.
Ход определения. Солянокислый раствор, свободный от фосфатов и редкоземельных элементов, включая скандий, и содержащий не свыше 4% соляной кислоты по объему и не более 1 мг ThO2 в 1 мл, нагревают до кипения. Непрерывно перемешивая раствор, медленно вводят кипящий 10%-ный раствор щавелевой кислоты в количестве, необходимом для связывания тория и получения избытка в 20 мл на каждые 100 мл раствора. Оставляют на ночь при комнатной температуре. Фильтруют, промывают осадок раствором, содержащим 40 мл соляной кислоты и 25 г щавелевой кислоты в литре, прокаливают приблизительно при 1100° С и взцешивают в виде ТЬОг.
* Осаждение в виде йодата тория
Первый метод. Ю. А. Чернихов и Т. А. Успенская 1 разработали условия выделения йодата тория постоянного состава, который после высушивания можно непосредственно взвешивать или определять в нем содержание тория косвенным путем, йодометрическим титрованием. Это дает возможность использовать выгодный фактор пересчета на торий и, следовательно, повысить точность определения, что особенно существенно при определении малых количеств тория. Авторы нашли, что при осаждении тория (от 0,07 до 16 мг) в приводимых ниже условиях осадок имеет состав 4Th(IO3)4• КЮ3- 18Н2О, и. таким образом содержание тория в нем составляет 21,76%, а при йодометрическом титровании один атом тория соответствует 25,5 эквивалентам иода.
.В условиях осаждения тория малорастворимые йодаты образуют также серебро, свинец, железо (II) и (III), таллий, цирконий, титан, марганец и церий (IV). От железа, марганца, титана и циркония торий можно отделить осаждением щавелевой кислотой совместно с редкоземельными элементами, причем полученные оксалаты можно потом непосредственно растворить в азотной кислоте, так как щавелевая кислота не препятствует выделению йодата тория. Влияние церия устраняется восстановлением его перекисью водорода в кислой среде.
Ход определения. К раствору тория в разбавленной (1 : 2) азотной кислоте, объем которого при содержании 4—16 мг тория не превышает
1 Ю. А. Ч е р н и х о в, Т. А, Успенская, Зав. лаб., 9, 276 (1940).
608
Гл. XXXIV. Торий
20 мл, а при содержании менее 2 мг тория — 5—6 мл, добавляют несколько капель перекиси водорода для восстановления церия, а затем 10%-ный раствор йодата калия в разбавленной (1:2) азотной кислоте в количестве, равном объему анализируемого раствора. Спустя 30—40 мин осадок йодата тория отфильтровывают через стеклянный тигель с пористым дном (№ 3 или 4), а в случае определения малых количеств тория (менее 1 мг) — через маленький асбестовый фильтр, вложенный в обычную воронку. Эту воронку вставляют в колбу Бунзена с отрезанным дном, края которой пришлифованы к пластинке толстого стекла, на которую ставят стакан для собирания фильтрата.
Осадок промывают сначала 5%-ным раствором йодата калия в разбавленной (1 : 2) азотной кислоте, чтобы не изменился состав фильтрата, который можно использовать для определения церия. Затем осадок промывают раствором, содержащим 8 г йодата калия и 50 мл концентрированной, азотной кислоты в 1л, 3—4 раза небольшими порциями 95%-ного спирта и под конец 1—2 раза эфиром. Высушенный при 40—50° С в течение 10—15 мин осадок смывают в коническую колбу, снабженную притертой пробкой. Приставшие ко дну тигля частицы осадка растворяют подкисленным раствором иодида калия, который просасывают через тигель. Раствор вливают в колбу, содержащую главную массу осадка, и выделившийся иод оттитровывают 0,1 н. раствором тиосульфата натрия. При определении малых количеств тория (менее 1 мг) осадок, не промывая эфиром и не высушивая, растворяют на асбестовом фильтре непосредственно после промывания спиртом. В этом случае полученный раствор титруют 0,01 н. раствором тиосульфата.
При определении малых количеств тория в присутствии значительных количеств церия однократным осаждением не удается достигнуть количественного разделения. В этом случае требуется двух- и трехкратное осаждение. Переосаждение осадка йодата проводится следующим способом. Промытый спиртом осадок вместе с асбестовым фильтром переносят в небольшой стакан и растворяют в 4 мл разбавленной (1 : 1) азотной кислоты с несколькими каплями перекиси водорода при нагревании в течение 1—2 мин. К раствору добавляют 2 мл воды, охлаждают и приливают 6 мл реактива. Спустя 30—40 мин осадок отфильтровывают через асбестовый фильтр, затем, промывают и титруют, как указано выше.
Восстановление двойного йодата 4Th(IO3)4-KIO3- 18Н2О протекает по уравнению
1710” 4- 85Г 4-Т02Н+ = 5112+51Н2О
Второй метод. При последующем изучении 1 йодатного метода определения тория было найдено, что при промывании йодата тория спиртом не удается полностью удалить из него промывную жидкость, что обусловливает повышенное содержание в осадке йодата. Указанные авторы уста- -новили, что при осаждении тория (175 мг) в обычно принятых условиях выделяется нормальный иодат тория ТЬ(Ю3)4, который можно промывать ледяной водой, не опасаясь его растворения или гидролитического разложения. Определение тория в монацитовых песках рекомендуют проводить следующим способом.
1 Т. М о е 11 е г, N. D. Fritz, Anal. Chem., 20, № И, 1055 (1948).
Методы определения
609
Ход определения. Нацеску в 10—20 г измельченного до 100 меш (d — = 0,147 мм) монацитового концентрата вводят в серную кислоту, пред--варительно выпаренную до появления ее паров. Смесь кипятят до полного разложения монацита. Полученную пасту охлаждают и вводят в нее кусочки льда, по растворении которого суспензию фильтруют через бумажную массу, помещенную в воронку Бюхнера. Осадок промывают водой. Фильтрат и промывные воды разбавляют в мерной колбе до 500 мл.
Отбирают аликвотную часть раствора, соответствующую 1—2,5 г монацита, и разбавляют до 100 мл. Прибавляют 50 мл азотной кислоты (пл. 1,42 г/см3) и несколько капель 30%-ной перекиси водорода, избыток которой разрушают кипячением. В охлажденный анализируемый раствор вводят 15 г йодата калия, растворенные в смеси 50 мл азотной кислоты и 30 мл воды. Полученную суспензию оставляют стоять 30 мин, часто перемешивая. Белый осадок от йодата калия отфильтровывают через бумажный фильтр средней плотности и промывают 20 мл раствора, содержащего 2 г йодата калия и 25 мл азотной кислоты в 225 мл воды. После этого осадок смывают горячей водой обратно в стакан, в котором проводилось осаждение. Суспензию нагревают до кипения и для растворения осадка прибавляют при перемешивании 30 мл азотной кислоты. По охлаждении до комнатной температуры осаждают торий добавлением раствора 4 г йодата калия в 7 мл азотной кислоты и 20 мл воды. Осадок отфильтровывают через тот же фильтр и, после того как жидкость стечет, промывают не более чем 100 мл ледяной воды, добавляемой небольшими порциями.
Фильтр с осадком переносят в коническую колбу емкостью 500 мл, обрабатывают 100 мл 4 н. серной кислоты и прибавляют около 50 мл воды и 30—35 мл 10%-ного раствора иодида калия. Выделившийся иод тотчас же титруют 0,2 н. раствором тиосульфата натрия, применяя крахмал в качестве индикатора. Содержание тория вычисляют, исходя из того, что 4107 соответствуют 1ТЪ4+.
*Колориметрические методы
Колориметрические методы определения тория сравнительно немногочисленны и основаны главным образом на образовании его окрашенных соединений с органическими реагентами.
Метод с применением торона Ч Этот метод 1 2 основан на реакции взаимодействия тория с реагентом уороном в минеральнокислой среде, в результате которой образуется красно-малиновый осадок, а в случае разбавленных растворов — розовое окрашивание. По данным В. И. Кузнецова, в солянокислых или азотнокислых растворах окрашенные соединения с реагентом, помимо тория, образуют только цирконий, гафний и титан. Редкоземельные элементы не дают цветной реакции с тороном в минеральнокислой среде, но при значительной их концентрации в растворе
1 Реакция тория с тороном была открыта В. И. К у з н е ц о в ы м [ЖОХ, 14, 914 (1944)]. В СССР тороном сокращенно называется бензол-2-арсоновая кислота-(1-азо-1)-2-оксинафталин-3,6-дисульфокислота. В иностранной литературе этот реагент называют также торин, торонол, нафтарсон и APANS. Имеются указания, что разные партии реагента могут несколько отличаться по составу, что обусловливает изменение окраски анализируемого раствора. Кроме того, иногда растворы реагента не совсем устойчивы и окраска их с течением времени ослабевает.
2 Р. Thomason, М. Perry, W. Byerly, Anal.- Chem., 21, № 10, 1239 (1949); J. Clinch, Anal. Chim. Acta, 14, № 2, 162 (1056).
39 Заказ 522.
t610 Гл. XXXIV. Торий
может иметь место некоторое изменение окраски реагента. Железо (III} мешает окраской своих солей, но его можно восстановить солянокислым гидроксиламином или цинковой пылью. Железо (II) при содержании до 10 мг/мл не оказывает влияния на реакцию. Препятствуют реакции вещества, образующие с торием комплексные соединения, как, напримерг фториды, сульфаты или органические оксисоединения. Влияние фтора можно устранить, связав его в комплекс с алюминием.
При использовании этой реакции для количественного спектрофотометрического определения тория было установлено х, что церий (IV) ослабляет образующуюся окраску и должен быть восстановлен до церия (III) перед введением в раствор торона. Влиянием редкоземельных элементов можно пренебречь, если содержание их в растворе не превышает 5 мг, за исключением тех случаев, когда требуется особо точное определение. Щелочные металлы^ аммоний и кальций понижают интенсивность окраски комплексного соединения тория с тороном. Барий не влияет на реакцию при его содержании до 2 мг!мл. При более высокой его концентрации образуется оранжево-красный осадок. Железо, даже двухвалентное, заметно влияет на светопоглощение раствора и поэтому его присутствия следует избегать. Олово, независимо от валентности, и уран (IV} должны отсутствовать.. Влияние ионов UOa+ незначительно. В условиях колориметрического определения при концентрации урана (VI) порядка 50 мг в 100 мл его действие эквивалентно 0,2 мг тория. » -
Из анионов, помимо перечисленных выше, на светопоглощение комплексного соединения тория с тороном заметное влияние оказывают также фосфат-ионы. Сернистая кислота, находясь в растворе, в условиях колориметрического определения, дает красную окраску, и следовательног и сульфит- и тиосульфат-ионы также мешают определению тория. Их влияние можно устранить предварительной обработкой раствора нейтральным раствором перманганата калия до появления устойчивой розовой окраски. После этого прибавляют 0,5 г гидроксиламина и соответствующие количества кислоты и торона. Двуокись углерода и-карбонатьг ослабляют окраску и должны быть предварительно удалены кипячением кислого раствора. Органические кислоты, если они присутствуют в значительных количествах, должны быть разрушены.
Окраска чистых растворов комплексного соединения тория с тороном ’ при комнатной температуре устойчива в течение продолжительного времени. Оптимальное значение pH раствора лежит в пределах 0,5—1,5, что соответствует 0,5—4 мл концентрированной соляной кислоты на 100 мл раствора.
Ход определения. Анализируемую пробу или выделенные в процессе анализа оксалаты обрабатывают при нагрёвании концентрированной азотной кислотой и небольшим количеством 70%-ной хлорной кислоты. Последнюю возможно полнее удаляют выпариванием, так как согласно имеющимся указаниям концентрированный раствор хлорной кислоты влияет на окраску соединения тория с тороном. По растворении полученной массы отбирают аликвотную часть раствора, содержащую не более 1,3 мг тория, в мерную колбу емкостью 100 мл. Раствор нейтрализуют разбавленным (1 : 1) раствором аммиака по фенолфталеину, вводят 10 мл разбавленной (1 : 4) соляной кислоты, 5 мл 10%-ного раствора гидррксил-
1 См. сноску 2 на стр. 609.
Методы определения
611
амина и 15 мл 0,1%-ного водного раствора торона. Одновременно приготовляют раствор для сравнения из таких же количеств кислоты, раствора солянокислого гидроксиламина и торона. Оба раствора разбавляют до метки водой, держат в термостате при 25° С в течение 15 мин и затем измеряют светопоглощение при 545 лшк.
Метод применен для определения тория в монацитовых песках.
Метод с n-арсоновой кислотой. С и-диметиламиноазофениларсоновой кислотой («n-арсоновая кислота») торий образует малорастворимое интенсивно окрашенное соединение, которое растворяется в растворе аммиака или едкого натра х. Интенсивность окраски раствора можно измерять спектрофотометрически х. Реакции мешает цирконий, соединение которого с n-арсоновой кислотой еще менее растворимо, чем соединение тория. Имеются указания \ однако, что небольшие количества циркония можно отделить от тория осаждением этим же реагентом из сильнокислого раствора (pH = 1—0). Метод позволяет определять 0,5—500 мкг тория.
Ход определения. Раствор соли тория в 43 мл воды, содержащей 4 капли НС1, нейтрализуют аммиаком по метиловому красному. Затем по каплям прибавляют разбавленную (7 : 100) соляную кисдоту до покраснения индикатора и сверх того 1,2 мл избытка. Прибавляют 5 мл раствора 0,1 г n-диметиламиноазофениларсоновой кислоты в 50 мл разбавленного (1 : 1) спирта, содержащего 10 г ацетата аммония. Смесь нагревают на водяной бане до коагуляции осадка (10—20 мин). Осадок отфильтровывают через асбест, вложенный в тигель Г-уча, и 4 раза, порциями по 10 мл, промывают жидкостью, состоящей из 10 г ацетата аммония, 12 мл разбавленной (7 : 100) соляной кислоты и 488 мл воды.
Промытый осадок растворяют в 30 мл теплого 4%-ного раствора едкого натра. Тигель один раз ополаскивают водой. Раствор разбавляют водой до 250 мл и измеряют его светопоглощение при 460 ммк. Содержание тория вычисляют по калибровочной кривой.
Определение по реакции восстановления йодата тория *. В результате восстановления йодата тория фосфорноватистой кислотой в присутствии серной кислоты выделяется иод, который при наличии четыреххлористого углерода тотчас растворяется в нем, окрашивая его в фиолетовый цвет. Поскольку количества восстановленного йодата тория и выделяющегося иода эквивалентны, содержание тория можно определить косвенным путем, измерив интенсивность окраски раствора иода в четыреххлористом углероде.
Малые количества тория, менее 1 мг, можно также определить нефелометрически 1 по количеству выпадающего осадка йодата тория.
Комплексометрические методы
Известный интерес представляют объемные комплексометрические методы определения тория 2 3 *. Из них- можно указать метод 8, основанный на том, что торий титруют комплексоном III в присутствии ализаринового
1 Аналитическая химия урана и тория, Издатинлит, 1956.
2 J. J. F г i t z, J. J. Ford, Anal. Chem., 25, 1640 (1953); H. Flaschka, K. ter H a a r, J. В a z e n, Mikrochim. Acta, 345 (1953); V. Suk, M.M ala t, O. R у b a, Chem. Listy, 48, 533 (1954); H. Malmitadt, A. Johrbandt, Anal. Chem., 26, 442 (1954).
3 J. J. F r i t z, J. J. F о r d, Anal. Chem., 25, 1640 (1953).
39*
612
Гл. XXXIV. Торий
красного S при pH = 2,3—3,4, подобно тому как рекомендуется 1 определять комплексон титрованием нитратом тория.
Авторы отмечают, что титрованию препятствуют ионы, образующие устойчивые комплексные соединения с комплексоном или реагирующие синдикатором, а именно: титанил, цирконил, церий (III), ванадил, железо (III), никель, медь (II), олово (II) и (IV), свинец и висмут. Должны отсутствовать также анионы, образующие с торием осадки или комплексные соединения, как, например, фосфат-, фторид-, оксалат-, сульфат- и ман-делят-ионы.
Ход определения. Раствор, содержащий 6—50 мг тория, разбавляют до 25 мл, вводят 4 капли ализаринового красного (0,05%-ный водный раствор), нейтрализуют разбавленным раствором аммиака до появления отчетливой розовой окраски (pH = —2,5), а затем титруют из бюретки емкостью 10 мл 0,025 М раствором комплексона III до тех пор, пока окраска раствора не начнет быстро бледнеть. После этого доводят pH раствора до 3,0, пользуясь для контроля pH-метром, и продолжают титровать до исчезновения последних следов розового оттенка и появления чисто желтой окраски.
При анализе продуктов, богатых торием, отбирают 100 мл раствора, содержащего 0,12—0,24 г тория, вводят 6 капель индикатора и титруют, как указано выше, из бюретки- емкостью 50 мл.
Для отделения тория от посторонних элементов рекомендовано пользоваться экстрагированием окисью мезитила, выполняемым следующим способом 2. К раствору нитрата тория в 1,2 М азотной кислоте прибавляют на каждые 10 мл по 19 г безводного нитрата алюминия и затем экстрагируют окисью мезитила. Однократной экстракцией торий полностью переводится в слой органического растворителя и отделяется почти от всех металлов, мешающих титрованию комплексоном, за исключением циркония и ванадия, которые можно отделить перед экстракцией. Доп. перев. *
1 К. ter Н а а г, J. В a z е n, Anal. Chim. Acta, 9, 235 (1953).
2 Н. Levine, F. S. Grimaldi, Atomic Energy Commission, AECD-3186 (February 1950).
Глава XXXV
СКАНДИЙ
Скандий считается одним из наиболее распространенных элементов в земной коре, хотя в качестве основного преобладающего компонента он входит в состав лишь одного минерала — тортвейтита (Sc, Y)2Si2O7, силиката скандия и элементов иттриевой группы х. Обычно скандий содержится в таких незначительных концентрациях, что открыть его можно только спектральными методами. Скандий встречается во многих редкоземельных минералах, в некоторых циркониевых минералах, берилле, титанатах, ниобатах, титанониобатах, слюдах и главным образом в касситерите и вольфрамите 1 2.
ОБЩИЕ ЗАМЕЧАНИЯ
Для открытия скандия обычно исследуют дуговой спектр раствора, полученного после концентрирования окиси скандия (как описано в разделах «Разложение минералов, содержащих скандий» и «Методы отделения») и растворения концентрата в соляной кислоте,о на характерные для скандия линии 3572,72; 3613,98; 3630,90 и 3642,96 А.
В обычном ходе анализа горных пород скандий попадает в осадок от аммиака и принимается за алюминий, если содержание последнего вычисляют по разности. В том случае, когда выделенный аммиаком осадок растворяют во фтористоводородной кислоте и раствор выпаривают для отделения фторидов редкоземельных металлов (стр. 623), скандий также выделяется в осадок и в дальнейшем, в зависимости от способа обработки, сопровождает торий, иттриевые или цериевые металлы.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ СКАНДИЙ
Если в минерале содержатся значительные количества скандия, как, например, в тортвейтите, 1 г тонко измельченной пробы сплавляют с 5—6 г карбоната натрия и затем или выделяют редкоземельные металлы иэ осадка аммиака (стр. 623) или же, отделив кремнекислоту выпариванием
1 G. Eberhard, Sitzber. kgl. preuss. Akad. Wiss., 38, 851 (1908); Chem. News, 99, 32 (1909), 102, 211 (1910).
2 H. S. Lukens [J. Am. Chem. Soc., 35, 1470 (1913)] указывает на содержание скандия в вольфрамите из Колорадо. При спектрохимическом исследовании силикатных пород на скандий А. К v а 1 h е i m и L. W. Strock [Spektorchim. Acta, 1, 221 (1939)] нашли в пироксенитах от 0,0056 до 0,0178% Sc2O3.
614
Гл. XXXV. Скандий
с соляной кислотой, непосредственно осаждают скандий совместно с цирконием, титаном и торием раствором тиосульфата, как указано в разделе «Метод отделения».
Редкоземельные минералы разлагают, как описано в гл. «Редкоземельные металлы» (стр. 619). При последующем разделении редкоземельных элементов сульфатом натрия скандий присоединяется к иттриевой группе.
Для разложения вольфрамита 10—100 г тонко измельченного минерала сплавляют с двух- или трехкратным количеством карбоната натрия. Скандий отделяют от основной массы вольфрама выщелачиванием плава горячей водой и последующим фильтрованием г. Осадок растворяют в соляной кислоте, обычным путем отделяют кремнекислоту и из солянокислого фильтрата осаждают фторид скандия фторосиликатом натрия, как указано в разделе «Методы отделения» (см. ниже). Тонко измельченную руду можно также обработать царской водкой при нагревании и отделить вольфрам, как описано в гл. «Вольфрам» (стр. 765).
Некоторые минералы, содержащие титан, тантал и ниобий, можно разложить обработкой избыточным количеством фтористоводородной кислоты и выпариванием на водяной бане досуха. Перед такой обработкой пробу следует тонко измельчить и смочить водой. Остаток обрабатывают разбавленной фтористоводородной кислотой, раствор фильтруют и фторид скандия промывают разбавленной (1 : 99) фтористоводородной кислотой.
МЕТОДЫ ОТДЕЛЕНИЯ
По некоторым свойствам скандий проявляет сходство с иттрием и лантаном, а по другим — с торием и цирконием. Подобно лантану, он образует нерастворимую двойную соль при обработке насыщенным раствором сульфата калия и осаждается щавелевой и фтористоводородной кислотами. Аналогично торию: 1) оксалат скандия растворим в оксалате аммония; 2) карбонат скандия на холоду растворяется в избыточном количестве карбонатов щелочных металлов и 3) скандий образует основной тиосульфат при кипячении нейтрального раствора с тиосульфатом натрия. Фторид скандия, так же как фторид циркония, растворим в избыточном количестве фторидов щелочных металлов.
Наиболее характерный метод отделения скандия заключается в выделении его в виде фторида ScF3 постепенным введением небольших порций фторосиликата натрия Na2SiF6 (2 г на 100 мл раствора) в энергична перемешиваемый кипящий раствор анализируемого материала в разбавленной 1 2 (1 : 10) соляной кислоте. После добавления реагента раствор кипятят 30 мин (сохраняя первоначальный объем раствора добавлением -горячей воды) и затем дают осадку отстояться. Прозрачный раствор сливают декантацией, после чего осадок переносят на фильтр и промывают 1%-ным раствором фторосиликата натрия в разбавленной (1 : 99) соляной кислоте. Помимо скандия, в осадке содержатся кремний и некоторая часть находившихся в растворе редкоземельных металлов и тория. Перед дальнейшей обработкой, которая может потребоваться, осадок нагревают с серной кислотой для удаления фтора.
1 R. I. М е у е г, Chem. News, 99, 86 (1909); Z. anorg. Chem., 60, 134 (1908).
2 R. J. Meyer, Chem. News, 99, 97 (1909).
Методы отделения
615
Осаждение сероводородом из раствора, содержащего минеральную кислоту, служит для отделения от скандия элементов сероводородной группы \ а осаждение аммиаком — для отделения скандия от магния и щелочноземельных металлов.
Осаждением щавелевой или фтористоводородной кислотой скандий можно отделить от посторонних элементов (стр. >621), за исключением тория и редкоземельных металлов. Оксалат скандия считается наиболее растворимым из оксалатов редких земель. Осаждение его не должно проводиться в присутствии минеральных кислот или оксалата аммония. •Фторид же скандия является наименее растворимым соединением при условии отсутствия в растворе избытка фтористоводородной кислоты, приводящего к образованию растворимых комплексных соединений, и наличия 2—4 мл соляной кислоты на 100 мл раствора.
Отделение скандия (и тория) от редкоземельных металлов проводится осаждением тиосульфатов натрия, как описано в разделе «Торий» (стр. 604). От цериевой группы скандий можно отделить насыщением раствора сульфатом натрия, в результате чего выделяются двойные сульфаты металлов цериевой группы (стр. 631).
*Э. А. Остроумов 1 2 рекомендует отделять скандий от лантана, церия (III), празеодима, неодима, самария, гадолиния, гольмия, эрбия, иттербия и иттрия осаждением буферным раствором, состоящим из смеси пиридина и его азотнокислой соли, причем в растворе устанавливается pH = 5,4. Автор отмечает, что в этих условиях гидроокись скандия выделяется количественно и что двукратного осаждения достаточно для удовлетворительного отделения от перечисленных элементов. .Доп. перев.*
Изучение способов отделения скандия от тория имело целью не столько количественное разделение этих элементов, сколько очистку скандия. Из этих методов можно указать на осаждение скандия медленным добавлением раствора аммиака к кипящему раствору, содержащему оба элемента 3 и большое количество тартрата аммония, а также на выделение скандия осаждением из кипящего 20%-ного раствора карбоната натрия с последующим фильтрованием горячего раствора 4. Для количественного разделения скандия и тория может применяться метод 5, состоящий в медленном вливании, при энергичном перемешивании, почти нейтрального раствора хлоридов этих элементов в горячий раствор фторида аммония, количество которого должно быть не менее восьмикратного по отношению к содержанию скандия. После этого раствор кипятят 10 мин в эбонитовом или бакелитовом стакане. Осадок отфильтровывают через эбонитовую или бакелитовую воронку и промывают небольшим количеством горячей воды. Скандий определяют затем осаждением аммиаком или щавелевой кислотой после выпаривания фильтрата с серной кислотой до появления ее паров.
1 Согласно R. J. Meyer (цит. выше), сульфид свинца захватывает скандий, и поэтому осаждение сероводородом следует начинать в сильнокислом растворе, который затем разбавляют.
2Э. А. Остроумов, ЖАХ, 3, 153 (1948).
3R. J. Meyer, Н. Goldenberg, Cem. News, 106, 13, (1912).
4 R. J. M e у e r, H. Winter, Chem. News, 102, 176 (1910).
5 R. J. Meyer, A. Wass juchn ow,N. Drapier, E. Bodlander, 2. anorg, Chem., 86, 257 (1914).
616
Гл. XXXV. Скандий
Торий можно также отделить от скандия осаждением йодатом калия из сильно азотнокислого раствора, как описано в< гл. «Торий» (стр. 607).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Скандий обычно взвешивают в виде окиси Sc2O3 после прокаливания выделенного аммиаком осадка. Скандий осаждают аммиаком в таких же условиях, как и алюминий (стр. 565). Осадок промывают 2%-ным раствором нитрата аммония, сушат в тигле, осторожно сжигают до получения окиси, после чего прокаливают приблизительно при 1000° С.
*Для определения скандия в тортвейтите предложен 1 следующий метод. Сплавляют 1 г тонко измельченной руды с 5—6 г едкого натра в серебряном или никелевом тигле при темно-красном калении. Во время сплавления перемешивают жидкую массу серебряной или никелевой палочкой. По охлаждении плав выщелачивают горячей водой. Остаток отфильтровывают, промывают горячей водой и растворяют в концентрированной серной кислоте при нагревании. Полученный раствор вливают в большое количество холодной воды, находящейся в стакане, и ополаскивают сосуд, в котором проводилось растворение осадка. К раствору прибавляют избыточное количество раствора аммиака, выделившийся осадок отфильтровывают, промывают и растворяют в азотной кислоте. Полученный раствор выпаривают досуха, нитраты растворяют в холодной воде, осаждают скандий сульфатом калия и оставляют на ночь. Отфильтрованный осадок промывают насыщенным раствором сульфата калия и затем обрабатывают холодным 25%-ным раствором карбоната аммония. Скандий при этом переходит в раствор в виде двойного карбоната. Осадок отфильтровывают и промывают холодным раствором карбоната аммония. Фильтрат кипятят до полного удаления аммиака, причем скандий осаждается в виде двойного карбоната. Осадок отфильтровывают, промывают горячей водой, прокаливают и взвешивают в виде Sc2O3. Доп. ред*
1 G, U г b a i п, Ct г., 174, 1310 (1922).
Глава XXXVI
РЕДКОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ
Под названием редкоземельных металлов объединена большая группа, как правило, трехвалентных элементов, соединения которых по свойствам весьма сходны между собой. В эту группу входят элементы с атомными номерами от 58 до 71 включительно. Ясно выраженными свойствами редкоземельных металлов обладают также скандий (21), иттрий (39) и лантан (57), вследствие чего их также причисляют к этой группе металлов. Торий обычно также рассматривается как один из членов этой группы вследствие сходства некоторых из его химических свойств и Потому что в природе он обычно встречается вместе с трехвалентными редкоземельными металлами. Все перечисленные металлы образуют основные окислы, имеющие, за исключением окиси тория, формулу М2О3. Некоторые из этих металлов образуют также и окислы с более высокой степенью окисления, из которых наиболее известна СеО2. В этом соединении церий проявляет сходство с единственной окисью тория ThO2.
Редкоземельные металлы широко распространены в природе1. Известны многочисленные редкоземельных минералы, которые весьма рассеяны в земной коре, но обычно их находят тесно смешанными с другими минералами. Наиболее распространены силикаты редкоземельных металлов, многочисленны также и фосфаты, а кроме того, встречаются галогениды, карбонаты, окислы, титанаты, уранаты, ниобаты, танталаты и цирконаты.
Редкоземельные минералы встречаются главным образом в гранитовых или пегматитовых жилах, в элеолитовых сиенитах или в происшедших от них песках 2. Насчитывается около 150 редкоземельных минералов, из которых наиболее известны: гадолинит — силикат бериллия, железа и иттриевой группы Be3Fe( YO)2(SiO4)2; ксенотим — ортофосфат иттрия YPO4, содержащий эрбий и элементы цериевой группы; эвксенит — титанониобат, содержащий титан, уран, торий, иттриевую и цериевую группы; фергусонит Y(Nb, Та)О4(Ег, Се, U и др., изоморфные с Y) — танталониобат иттрия, содержащий торий, цирконий, уран и элементы иттриевой и цериевой групп; самарскит — танталониобат R^R^11 (Nb, Та)6О21 (где Rn — Fe, Са, UO2, a Rin — редкоземельные металлы), содержащий уран, торий, цирконий и иттриевую и
1 В. S. Hopkins, Chemistry of the Rarer Elements, D, C. Heath and Co., New York, 1923, p. 96.
2 F. W. С 1 a r k e, U. S. Geol. Survey Bull., 770, 15, 21.
618
Гл. XXX VI. Редкоземельные металлы,
цериевую группы; монацит — фосфат цериевых металлов (Се, La, Nd, Рг)РО4 с силикатом тория ThSiO4; церит Н(Са, Fe)(Ce, La, Pr, Nd)3Si30i2 • H2O — гидратированный силикат цериевых металлов с большим или меньшим содержанием иттриевых металлов, кальция и железа; алланит RlIR3n(OH)(SiO4)3 (где Rn — Са, Be, Fe, а RnI — Al, Fe, Ce, La, Nd, Pr) — гидратированный силикат, содержащий, помимо элементов цериевой и иттриевой групп, кальций, бериллий, железо и алюминий; иттроцерит — фторид иттрия, церия и кальция (Y, Er, Ce)Ca6F13 • Н2О. Помимо обычных элементов — кальция, железа и алюминия, которые почти всегда связаны с вышеперечисленными редкоземельными элементами, в этих минералах более или менее часто встречаются также: уран, бериллий, фтор, бор, свинец, сурьма, мышьяк, олово, вольфрам, гелий, азот и, вероятно, некоторые другие элементы, например гафний.
ОБЩИЕ ЗАМЕЧАНИЯ
Все редкоземельные металлы образуют оксалаты, нерастворимые в щавелевой кислоте и разбавленных минеральных кислотах, и фториды, нерастворимые в разбавленной фтористоводородной кислоте. На использовании свойств этих солей основаны методы группового отделения редкоземельных элементов от большинства других элементов. Редкоземельные элементы количественно осаждаются аммиаком, что дает возможность отделять их от щелочных и щелочноземельных металлов и магния. Их гидроокиси нерастворимы в едком натре и едком кали, свойство, которое также может быть использовано в анализе.
Торий в природе практически всегда связан с редкоземельными металлами, и его оксалаты и фториды по отношению к щавелевой и фтористоводородной кислоте ведут себя так же, как соответствующие соединения редкоземельных элементовх. Гидроокись тория, подобно гидроокисям редкоземельных элементов, нерастворима в растворах едких щелочей. Цирконий также встречается во многих редкоземельных минералах, и при определении редкоземельных металлов и тория во всех операциях следует учитывать его присутствие. Поэтому в процессе анализа цирконий удобно обрабатывать совместно с группой редкоземельных элементов, хотя он и не образует нерастворимого оксалата и фторида. В некоторых реакциях цирконий ведет себя подобно церию (IV) и торию.
В обычном ходе анализа горных пород редкоземельные элементы попадают в осадок от аммиака и принимаются за алюминий.
Более основные редкоземельные элементы осаждаются полностью при pH = 6,5— 7,5, при котором обычно проводится осаждение алюминия. При некоторых условиях лантан вовсе не выделяется. В присутствии редкоземельных элементов целесообразно провести осаждение аммиаком, как обычно, осадок отфильтровать, а затем из фильтрата выделить неосажденные редкоземельные металлы дополнительным введением -аммиака. Таким способом алюминий извлекается полнее и осадок менее загрязняется марганцем. Лантан, церий и иттрий лучше извлекаются из фильтрата в этом дополнительном осаждении, когда: 1) сперва разрушают аммонийные соли нагреванием с кислотой (стр. 161); 2) избыток кислоты удаляют выпариванием раствора почти досуха;
1 Для многих редкоземельных элементов оксалат аммония является таким же хорошим осадителем, как щавелевая кислота, но некоторые оксалаты растворимы в избытке оксалата аммония, и поэтому его применение более ограничено, чем применение щавелевой кислоты.
Разложение редкоземельных минералов
619
5) разбавленный раствор обрабатывают достаточным количеством аммиака, чтобы полностью исчезла окраска малахитовой зелени (pH >> 14); 4) перед фильтрованием раствор нагревают до 50—60° С и 5) осадок промывают разбавленным (5 : 95) раствором аммиака, содержащим 1% нитрата аммония.
Утверждают1, что даже таким путем количественное осаждение редкоземельных металлов достигается с трудом и что лучший метод заключается во введении таннина перед Ъбработкой кипящего раствора избытком аммиака. В этом случае аммонийные соли и даже тартраты не влияют на осаждение.
Для редкоземельных элементов характерных реакций, за исключением окраски некоторых окислов, неизвестно. Поэтому для их обнаружения приходится прибегать к испытанию со щавелевой или фтористоводородной кислотой. Некоторые из редкоземельных элементов' образуют окрашенные окислы и растворы, но случаи, когда эти элементы встречаются в достаточно значительных количествах и не связаны с другими окрашенными соединениями, сравнительно редки. Присутствие редкоземельных элементов часто можно установить исследованием спектра света, отраженного гидроокисью или каким-либо другим соединением, или же. исследованием света, проходящего через раствор этих соединений. Наличие в спектре полос, характерных для неодима и празеодима, указывает на присутствие цериевой группы, а в случаях, когда наблюдаются полосы, свойственные эрбию, всегда присутствует иттриевая группа. Необходимо отметить, что при прокаливании на воздухе церий, празеодим и тербий образуют высшие окислы, вследствие чего получаются повышенные результаты для суммы окислов, если в массу прокаленного осадка от аммиака вводят поправку на содержание трехвалентных окислов редкоземельных металлов.
РАЗЛОЖЕНИЕ РЕДКОЗЕМЕЛЬНЫХ МИНЕРАЛОВ
В Природных минералах редкоземельные элементы встречаются в настолько разнообразных сочетаниях, что нельзя указать общих правил для их определения, которые оказались бы приемлемыми во всех случаях, даже в пределах, одной подгруппы. В лучшем случае можно привести лишь в общих чертах некоторые наиболее часто применяемые методы. Здесь больше чем в какой-либо другой области неорганического анализа аналитику раскрываются широкие возможности для проявления своей способности комбинировать различные методы анализа.
Прежде чем приступить к количественному анализу минерала, необходимо провести возможно более тщательное качественное его исследование (предпочтительно спектрографическое), насколько позволяет это имеющееся количество материала для анализа. Знание качественного состава и хотя бы приблизительного количественного соотношения между компонентами имеет весьма существенное значение для правильного выбора методов разложения пробы и разделения находящихся в ней элементов. Однако очень часты случаи, когда для предварительного исчерпывающего исследования не имеется достаточного количества материала, что впоследствии может явиться причиной более или менее серьезных ошибок. Существенную помощью в определении качественного состава анализируемой пробы во многих случаях может оказать исследование отраженного от поверхности света посредством ручного спектроскопа.
1 W. R. S с h о е 11 е г, Е. F. Wa terhouse, Analyst, 60, 284 (1935).
620
Гл. XXXVI.. Редкоземельные металлы
Хотя и существуют редкоземельные, а равно и тантало-ниобиевые минералы относительно простого состава, но нередки случаи, когда в состав минералов одновременно входят обе эти группы элементов, что крайне затрудняет их анализ. Для разложения такого рода минералов редко применяют одну азотную кислоту.
Единственными минералами, для разложения которых специально рекомендуется обработка азотной кислотой, являются, пожалуй, уранинит, бреггерит, клевеит, нивенит. урановая смолка и торианит, так как они полностью в ней растворяются, ит кроме того, одновременно происходит окисление четырехвалентного урана до шестивалентного.
Некоторые минералы, главным образом силикаты редкоземельных металлов, более или менее легко разлагаются соляной кислотой. В тех случаях, когда разложение происходит полностью, полученный раствор следует обработать так же, как раствор обычного силиката (стр. 940).
Другие минералы легко разлагаются обработкой фтористоводородной кислотой (например, самарскит), и почти все эти минералы разлагаются при сплавлении с кислыми фторидами щелочных металлов. Такого рода обработки обычно используются для разложения ниобатов, танталатов. и титанатов, которые не содержат кремнекислоты.
Для разложения минералов, содержащих бор, фтор и кремний, естественно, следует применять сплавление со щелочами, чтобы иметь возможность определить эти элементы.
Большинство минералов разлагается серной кислотой. Для разложения некоторых минералов, как, например, монацита, ксенотима и церита, требуется продолжительная обработка серной кислотой при 200° С или выше. Для определения в них фосфора, так же как и для разложения минералов, содержащих бор или фтор, целесообразнее применять однократное или многократное сплавление со щелочами.
Все эти минералы, по-видимому, разлагаются при сплавлении с пиро-сул#>фатами щелочных металлов, но так же, как и обработка с серной кислотой, этот способ скорее используется для технических проб на торий, чем для полного анализа. Для сплавления лучше пользоваться пиросульфатом натрия, чем пиросульфатом калия, вследствие большей растворимости некоторых образующихся в результате сплавления двойных сульфатов натрия. При определении кремния в тех случаях, когда минерал не разлагается кислотами, когда присутствует фтор или требуется определить также содержание бора или фтора, обычно применяют сплавление с карбонатами или едкими щелочами. Сплавлением с карбонатом натрия пользуются также при проведении полного анализа фосфатов. Для определения фтора в минералах:, растворимых в горячей концентрированной серной кислоте, можно пользоваться методом отгонки. В техническом анализе для разложения материала иногда применяют сплавление с едким натром или перекисью натрия, но при выполнении полного анализа оба эти реагента менее пригодны, чем карбонат натрия, так как они обычно менее чисты и, кроме того, слишком сильно действуют на сосуды, в которых проводят сплавление.
МЕТОДЫ ОТДЕЛЕНИЯ
Выбор методов отделения редкоземельных металлов от других элементов зависит от состава анализируемого минерала и от того, какой реагент был применен для его разложения. Силикаты редкоземельных
Методы отделения
621
металлов, не содержащие титана, ниобия и тантала, как правило, растворимы в соляной или азотной кислоте или разлагаются при сплавлении с карбонатами щелочных металов. В этих случаях кремнекислоту отделяют обычным путем — выпариванием с кислотой и фильтрованием, причем эту операцию повторяют не менее двух раз (стр. 940). После этого обычно проводят двукратное осаждение аммиаком (стр. 949).
При содержании значительных количеств редкоземельных металлов и кальция может потребоваться трех- или четырехкратное осаждение аммиаком, чтобы обеспечить полное отделение кальция. Осадок от аммиака содержит полностью редкоземельные металлы, торий, цирконий, титан и некоторые другие осаждающиеся аммиаком элементы и должен быть свободен от марганца, никеля, щелочноземельных металлов, магния и щелочных металлов. Его растворяют в соляной кислоте, раствор выпаривают, сухой остаток растворяют в воде, прибавляя соляную кислоту в количестве не большем, чем требуется для получения прозрачного раствора. Из этого раствора редкоземельные элементы осаждают насыщенным раствором щавелевой кислоты1, вводимый избыток которой зависит главным образом от-содержания железа (III) и урана (VI).
Изучалась 2 растворимость оксалатов лантана, церия, празеодима, неодима, самария и гадолиния в соляной, азотной, серной и щавелевой кислотах. Растворимость оксалатов этих элементов в насыщенных щавелевой кислотой растворах, содержащих небольшие количества соляной кислоты, уменьшается в приведенном порядке. Авторы считают, что для осаждения лантана и церия из чистых растворов следует прибавлять щавелевую кислоту почти до насыщения раствора, тогда как для осаждения всех остальных редкоземельных элементов концентрация щавелевой кислоты может быть 0,5 н. Концентрация же соляной кислоты в растворе во всех случаях должна быть 0,5 н. Авторы рекомендуют горячий концентрированный раствор щавелевой кислоты вливать при перемешивании в кипящий раствор редкоземельных металлов в 0,5 н. соляной кислоте и затем перед фильтрованием оставлять раствор в течение ночи.
Железо (III) и уран (VI) растворяюще действуют на оксалаты редкоземельных металлов, особенно более основных. Это действие устраняется при наличии в растворе щавелевой кислоты в количестве большем, чем требуется для образования оксалата’железа 3, уранилщавелевой кислоты 4 и для полного осаждения редкоземельных элементов. •-
Оксалаты редкоземельных металлов можно перевести в раствор: 1) прокаливанием оксалатов и растворением полученных окислов в кислоте; 2) обработкой Н2О2, если присутствуют большие количества СеОа: 3) кипячением с дымящей азотной кислотой и 4) кипячением с NaOH или КОН и растворением промытых гидроокисей в кислоте.
Ниобаты и танталаты большей частью нерастворимы в соляной или азотной кислоте, но некоторые из них более или менее легко разлагаются фтористоводородной кислотой. В тех случаях, когда анализируемая проба полностью разлагается фтористоводородной кислотой, фториды редкоземельных металлов, тория и урана (IV) осаждаются практически количественно и могут быть отмыты почти от всех посторонних элементов разбавленной фтористоводородной кислотой. Это особенно удобно для отделения редкоземельных металлов и урана (IV) от ниобия, тантала, титана, циркония и урана (VI), фторид которого растворим. После превраш фторидов в сульфаты и растворения в соляной кислоте осаждением аммиа
1 L. A. S а г v е г, Р. Н. В г i n t о n, J. Am. Chem. Soc., 49, 943 (1927).
2 L. A. S а г v er, P. H. В г i n t о n, цит. выше.
’ M. Dittrich, Ber., 41, 4373 (1908).
4 О. H a u s e r, Z. anal. Chem., 47, 677 (1908).
622
Гл. XXXVI. Редкоземельные металлы
ком освобождают редкоземельные элементы от щелочноземельных металлов, которые также могут выделиться в виде фторидов. Промытые гидроокиси редкоземельных металлов можно непосредственно прокалить до окислов, если в исходном растворе не присутствует U , или же раство-рить в соляной кислоте [в азотной, при наличии UIV], раствор йыпарить и редкоземельные металлы осадить избыточным количеством щавелевой кислоты.
Окислы некоторых редкоземельных металлов затруднительно прокалить до постоянной массы их, так как часто масса, упав до минимума, после последующих прокаливаний постепенно снова возрастает1. Окись празеодима, полученная из оксалата1, имеет формулу PreOu, когда прокаливание проводят на воздухе, и РггО3 в случае прокаливания в токе водорода при 900—1000° С. Церий после прокаливания при 1200Q С всегда взвешивают в виде двуокиси СеОг, которая не легко восстанавливается до Се2О3 при прокаливании в атмосфере водорода. Тербий, при прокаливании его окиси на воздухе, дает ТЬ4О7, а после прокаливания в токе водорода восстанавливается до ТЬгО3.
В более поздней работе те же авторы2 отмечают, что они не могли получить высших окислов лантана, неодима, самария, гадолиния, эрбия и иттербия. Они нашли, однако, что состав окиси празеодима, полученной прокаливанием на воздухе, зависит от условий прокаливания и охлаждения. При определении суммы редкоземельных элементов церий следует отделить перед прокаливанием остальных окислов редкоземельных элементов в токе водорода.
Обработка фтористоводородной кислотой, в том или ином виде, рекомендуется тогда, когда наряду с малыми количествами редкоземельных элементов присутствуют значительные количества посторонних, осаждающихся аммиаком элементов, например при анализе силикатов (стр. 949). В таких случаях осадок от аммиака непосредственно обрабатывают фтористоводородной кислотой, раствор выпаривают почти досуха, фториды редкоземельных элементов отфильтровывают, промывают разбавленной фтористоводородной кислотой и затем обрабатывают, как указано выше. Такую обработку осадка от аммиака можно, без сомнения, применять и в случае содержания в нем значительных количеств редкоземельных элементов вместо упомянутого ранее осаждения щавелевой кислотой, при котором происходит менее полное выделение этих элементов. Протактиний не осаждается фтористоводородной кислотой, церий (IV) осаждается и фтористоводородной и щавелевой кислотами, причем в последнем случае образуется соль церия (III).
Небольшие количества редкоземельных элементов, находящиеся в смеси с ниобием и танталом, можно отделить следующим способом3. Сплавляют 0,5 г окислов с 6 а бисульфата калия в кварцевом тигле. Плав растворяют в 60 мл 10%-ного раствора винной кислоты, раствор разбавляют до 400 мл, прибавляют 50 'мл концентрированной соляной кислоты и кипятят 3 мин. Дают отстояться и затем фильтруют при слабом отсасывании через бумажный фильтр, содержащий немного мацерированной бумаги. Фильтр с осадком переносят обратно в стакан, заливают подкисленной водой, перемешивают, снова отфильтровывают и отбрасывают. Фильтраты объединяют, прибавляют аммиак до щелочной реакции, нагревают до кипения, после чего вводят 10 г ацетата аммония и свежепригото
1 Р. Н. В г i n t о п, Н. А. Р a g е 1, J. Am. Chem. Soc., 45, 1460 (1923).
2 Н. А. Р a g е I, Р. Н. В г i n t о n, J. Am. Chem. Soc., 51, 53 (1929).
3 W. R. Schoeller, E. F. Waterhouse, цит. выше; W. R. Schoeller, The Analytical Chemistry of Tantalum and Niobium. Chapman and Hall, London, 1937, p, 98.
Методы отделения
623
вленный раствор 0,7 г таннина. Дают отстояться, затем осадок отфильтровывают и промывают 5%-ны1и раствором нитрата аммония, содержащим небольшое количество таннина и аммиака. Прокаливают в кварцевом тигле, сплавляют с 2 г бисульфата натрия, после чего плав растворяют в 50 мл 10%-ного раствора винной кислоты. Раствор разбавляют до ЮОлм, нагревают до кипения, перемешивают и медленно вводят 10 мл насыщенного раствора щавелевой кислоты. Оставляют стоять в течение ночи, затем осадок отфильтровывают, промывают раствором, содержащим 1 % щавелевой и 1% винной кислот, и прокаливают до образования окислов.
Минералы, на которые названные выше кислоты не действуют, обрабатывают серной кислотой либо сплавляют с пиросульфатами щелочных металлов или с бифторидом калия. При использовании бифторида калия фториды редкоземельных металлов осаждаются менее полно, чем при осаждении фтористоводородной кислотой, вследствие некоторой растворимости их в присутствии фторидов щелочных металлов. Перешедшую в раствор часть редкоземельных металлов иногда бывает необходимо дополнительно извлечь после выпаривания фильтрата с серной кислотой для удаления фтора. Обработанные серной кислотой растворы нагревают до удаления большей части избыточной кислоты, образующуюся пасту охлаждают и растворяют, вводя небольшими порциями в холодную воду (лучше в ледяную, чтобы уменьшить, возможность образования нерастворимых сульфатов). Растворение происходит медленно и должно быть ускорено частым перемешиванием, фильтруют после перехода в раствор всех растворимых солей. Остаток может состоять из нерастворившегося минерала, кремнекцслоты и окиси титана. Если проводился анализ нечистого-минерала (монацитовый песок), в остатке могут содержаться также посторонние минералы. -Нерастворимый остаток собирают на фильтре, прокаливают и повторяют обработку серной кислотой и т. д. до тех пор, пока под. увеличительным стеклом не перестанут различать частички неразложив-шегося минерала.
,В случае разложения минерала (за исключением фосфатов) азотной, соляной или серной кислотами кремнекислоту переводят в нерастворимую форму так же, как при анализе силикатов (стр. 939), и отфильтровывают. Если для разложения пробы, содержащей свинец, применяют серную кислоту, то совместно с кремнекислотой выделяется сульфат свинца, который до прокаливания остатка следует удалить обработкой ацетатом аммония или горячей соляной кислотой. В противном случае может образоваться силикат свинца, что приведет к неполному его извлечениюх. При разложении материала, в котором находятся олово и сурьма, азотной кислотой осадок кремнекислоты загрязняется окислами этих элементов. Титан, ниобий и тантал при выпаривании с этими кислотами также переходят в нерастворимую форму. После прокаливания и взвешивания выделенной смеси кремний можно отогнать выпариванием с фтористоводородной и несколькими каплями серной кислоты, а затем нелетучий остаток прокалить и взвесить, определив таким образом содержание кремнекислоты.
1 Подобные затруднения возникают во многих случаях. Их часто можно избежать применением соответствующей кислоты для обезвоживания. Так, если присутствует сурьма, следует пользоваться серной кислотой, а не соляной, так как с соляной кислотой образуются нерастворимые основные хлориды сурьмы, масса которых в процессе-прокаливания неопределенно изменяется. При наличии свинца лучше применять, хлорную кислоту, чем соляную или серную.
'624
Гл. XXXVI. Редкоземельные металлы
Нелетучий остаток следует потом сплавить с циросульфатом щелочного металла или карбонатом (в зависимости от состава осадка) и плав соответствующим образом обработать для разделения и определения находящихся в нем компонентов.
Не слишком кислый фильтрат после отделения кремнекислоты’обрабатывают на х<утоду сероводородом. Полученный осадок чаще всего содержит свинец и олово. К нему присоединяют свинец, сурьму и олово, которые были извлечены из осадка кремнекислоты или остались в нелетучем остатке после удаления кремнекислоты обработкой фтористоводородной кислотой, и затем обрабатывают соответствующими способами.
Фильтрат после удаления сероводорода, окисления азотной кислотой и, если требуется упаривания, осаждают аммиаком (стр. 565). Осадок растворяют в соляной кислоте и переосаждают аммиаком. Эту операцию повторяют до полного удаления . щелочноземельных металлов, магния и щелочных металловх.
Дальнейшая обработка осадка от аммиака зависит от его состава. Если содержание алюминия и железа значительно превышает содержание редкоземельных элементов, осадок целесообразно обработать фтористоводородной кислотой, после чего поступают следующим образом. Раствор выпаривают на водяной бане почти досуха. Остаток смачивают 0,5 мл фтористоводородной кислоты, прибавляют 25 мл воды, 0,5 мл соляной кислоты и после непродолжительного нагревания фильтруют. Осадок промывают водой, содержащей 2 мл фтористоводородной кислоты и 2 мл соляной кислоты2 в 100 мл. Фториды смывают с фильтра в платиновую чашку, фильтр сжигают и золу присоединяют к осадку. Осадок смачивают серной кислотой, выпаривают и избыток кислоты удаляют нагреванием в радиаторе (см. рис. 5, стр, 48). Остаток сульфатов растворяют в холодной воде. Из раствора редкоземельные металлы осаждают в виде оксалатов, которые промывают 1 %-ным раствором щавелевой кислоты, прокаливают при 1200° С и взвешивают. По цвету окислов можно получить некоторое представление об их составе. Осадок, если возможно, растворяют в соляной кислоте (если нет, то в серной), после чего производят соответствующую обработку для отделения и определения тория и церия. Фильтрат, после отделения фторидов редкоземельных металлов, выпаривают с серной кислотой до полного удаления фтора. Остаток растворяют в соляной кислоте, и затем железо, алюминий и другие элементы осаждают аммиаком (стр. 565). Осадок прокаливают, доводя температуру в конце прокаливания до 1200° С, и взвешивают. В этом осадке определяют железо (стр. 122), цирконий (стр. 122) и бериллий (стр. 121). В осадке можно определить также и титан, если содержание его не устанавливают в отдельной навеске пробы. Фосфор определяют в отдельной навеске. Содержание всех этих элементов вычитают из массы суммы смешанных окислов, а полученную разность считают за окись алюминия. 1
1 В минералах, о которых идет речь, магний и щелочные металлы редко содержатся в таких количествах, чтобы требовалось более однократного или двукратного осаждения аммиаком. То же относится к марганцу и никелю, если осаждение проводится надлежащим образом.
а Можно применять и следующий метод. Гидроокиси растворить в соляной кислоте, раствор выпарить досуха, остаток растворить в возможно меньшем количестве •соляной кислоты, разбавить до 50 лед и затем обработать щавелевой кислотой. Осадку дают отстояться ночь, после чего его отфильтровывают.
Методы отделения
625
Описанная обработка неприменима к минералам, состоящим в основном из фосфатов х. Для разложения таких минералов требуется однократное или многократное сплавление с карбонатом натрия, за исключением тех случаев, когда их исследуют на содержание одного лишь компонента (обычно тория). В водной вытяжке плава содержатся фосфор 1 2, мышьяк, сурьма, олово и вольфрам, а также большая часть кремния, алюминия и урана. Остаток тщательно промывают разбавленным раствором карбоната натрия, а фильтрат выпаривают с азотной кислотой для переведения кремнекислоты в нерастворимое состояние (при этом частично выделяются также вольфрам и сурьма). После выпаривания и отделения кремнекислоты фильтрат насыщают сероводородом для удаления свинца, мышьяка и оставшейся в растворе части сурьмы. Удалив сероводород и упарив раствор, осаждают фосфор молибденовой жидкостью (стр. 781) (которую предварительно проверяют на содержание алюминия и других осаждающихся аммиаком элементов) и заканчивают его определение, как указано в гл. «Фосфор» (стр. 784). Из фильтрата, выпаренного для удаления избытка азотной кислоты, выделяют алюминий двукратным осаждением аммиаком (стр. 565). Осадок промывают 2%-ным раствором нитрата аммония, прокаливают и взвешивают.
Остаток, полученный после выщелачивания карбонатного плава водой, растворяют в соляной кислоте и в дальнейшем поступают, как при анализе силикатов, или в соответствии с тем, что указано для определения кремния, алюминия, бериллия, титана, циркония, редкоземельных металлов, щелочноземельных металлов и магния, если те или иные из этих элементов не определялись из отдельных навесок пробы.
Церий можно отделить от молибдена, иттрия, гольмия, эрбия и диспрозия осаждением йодатом калия после окисления его до четырехвалентного 3. Торий, цирконий и, возможно, частично скандий выделяются при этом совместно с церием. Метод заключается в следующем.
Раствор нитратов редкоземельных элементов, содержащий 25 мл азотной кислоты и не более 0,15 г СеО2, разбавляют до 75 мл, прибавляют 0,5 г бромата калия и перемешивают до его растворения. Затем медленно при непрерывном перемешивании вводят раствор йодата калия (10 г йодата и 33,3 мл азотной кислоты в 100 мл) в 10—15-кратном количестве по отношению к теоретически необходимому для связывания находящегося в растворе церия. Дают отстояться, фильтруют через плотный фильтр и один раз ополаскивают стакан промывной жидкостью (8 г йодата и 50 мл азотной кислоты в 1 л). Жидкости дают стечь и немедленно смывают осадок с фильтра обратно в стакан. Разминают комки, взбалтывают смесь, снова фильтруют и дают жидкости стечь. После этого опять смывают осадок в стакан, на этот раз применяя около 50 мл горячей воды, нагревают до кипения и, непрерывно перемешивая, по каплям вводят азотную кислоту до растворения осадка.
1 При анализе таких минералов, когда сернокислый раствор минерала, например монацита (стр. 599), вливают в десятикратное по объему количество 95%-ного спирта, редкоземельные металлы осаждаются в виде сульфатов. Это разделение не количественное, так как редкоземельные элементы полностью не осаждаются, а некоторое количество фосфат-ионов захватывается осадком.
2 Редкоземельные металлы полностью осаждаются также едким натром и могут быть таким образом отделены от фосфора и подобных элементов.
3 Р. Н. В г i n t о п, С. J a m е s, J. Am. Chem. Soc., 41, 1080 (1919),
40 Заказ 522.
626 Гл. XXXVI. Редкоземельные металлы
Для растворения йодата, сответствующего 0,1 г СеО2, требуется 20—25 мл азотной кислоты. Введения избытка кислоты следует избегать. Объем раствора не должен превышать 75 мл и в нем должна находиться приблизительно одна треть по объему азотной кислоты. К этому раствору прибавляют 0,25 г бромата калия и почти такое же количество йодата, как при первом осаждении. Дают отстояться и по охлаждении фильтруют через тот же фильтр. Ополаскивают один раз стакан и смывают в него обратно осадок. Взбалтывают, затем осадок отфильтровывают и 3 раза промывают небольшими порциями промывной жидкости. Переносят фильтр с осадком в стакан, прибавляют 5—8 г щавелевой кислоты и 50 мл воды. Закрывают стакан часовым стеклом, осторожно нагревают и затем кипятят до прекращения выделения паров иода и удаления сублимата со стекла и краев стакана. После этого ополаскивают стекло и внутреннюю поверхность стакана и оставляют стоять несколько часов. Фильтруют, промывают осадок холодной водой и прокаливают до СеО2 на паяльной горелке.
*Показана возможность выделения осадка йодата церия постоянного состава, содержание церия в котором можно определить непосредственным взвешиванием или косвенным путем, йодометрическим титрованием1. Церий можно определять в фильтрате после отделения йодата тория (см. стр. 607). Для этого находящийся в фильтрате церий окисляют небольшим количеством бромата. Выделяющийся при этом желтый осадок йодата церия, состав которого, по данным авторов, соответствует формуле 2Се(Ю3)4 • КЮ3 • 8Н2О, отфильтровывают, промывают и титруют так же, как двойной иодат тория.
Реакция восстановления йодата церия иодидом протекает по уравнениям:
9Ю; + 451“ + 54Н+ = 2712 + 27Н2О
2Се4+ + 21“ = 2Сез+ + 12
Из этих уравнений можно вывести, что один атом церия соответствует 28 эквивалентам иода. Доп. перев*
Довольно удовлетворительно происходит отделение церия по методу Мосандера 2.
Метод Мосандера состоит в следующем. Окислы редкоземельных Металлов растворяют в соляной кислоте с небольшим количеством перекиси водорода. Кипятят до разрушения перекиси и затем обрабатывают раствор едким натром, прибавляя его в небольшом избытке. Кипятят в течение нескольких минут, дают отстояться, после чего жидкость сливают и осадок промывают декантацией. Осадок растворяют в возможно меньшем количестве соляной кислоты, раствор разбавляют приблизительно до 200 мл, нейтрализуют едким кали и прибавляют избыток щелочи в 2 г. Насыщают хлором до тех пор, пока раствор не перестанет показывать щелочную реакцию. Кипятят 5 мин, разбавляют до 400 мл и оставляют стоять 12 ч. После этого фильтруют, осадок гидроокиси церия (IV) промывают горячей водой, растворяют в соляной кислоте и повторяют всю операцию 5—10 раз. Но все же, полного отделения церия от редкоземельных металлов таким путем достигнуть не удается.
1 Ю. А. Чернихов, Т. А. Успенская, Зав. лаб., 9, 276 (1940).
2 С. G. М о s a n d е г, Phil. Mag., 23, 241 (1843); Р. Е. Browning, Е. J. Roberts [Am. J. Sci., [4], 29, 45 (1910)], а также S. J. J ohns t one [J. Soc. Chem. Ind., 33, 55 (1914)] рекомендуют заменять хлор бромом.
Методы отделения
627
Отделение ^ория и скандия приведено в соответствующих главах, посвященных этим элементам. Следующий метод может дать удовлетворительное отделение церия от редкоземельных металлов.
В анализируемом растворе окисляют церий, как указано выше (см. мелкий шрифт на стр. 626—627). Затем охлаждают до 50° С, прибавляют 1 г персульфата аммония и суспензию окиси цинка (стр. 108) до прекращения растворения окиси цинка. Кипятят, фильтруют и промывают осадок 2%-ным раствором нитрата аммония. Фильтрат испытывают с перекисью водорода на полноту осаждения церия. Если при этом образуется осадок, его отфильтровывают, промывают раствором нитрата аммония и присоединяют к основному осадку церия. Фильтрат сохраняют для определения других редкоземельных металлов. Осадок переносят в стакан емкостью 400 мл, причем оставшиеся на фильтре частицы растворяют в горячей 2 н. серной кислоте, после чего, фильтр тщательно промывают горячей разбавленной (1 : 100) серной кислотой. Раствор разбавляют до 200 мл и прибавляют разбавленную (1 : 1) серную кислоту до полного растворения осадка. Окисление персульфатом и описанную выше обработку повторяют, а затем, испытав фильтрат добавлением перекиси водорода, присоединяют его к сохраненному ранее раствору. Если содержание церия значительно преобладает, может потребоваться трехкратная обработка.
Для определения остальных редкоземельных элементов объединенные фильтраты обрабатывают раствором аммиака до растворения образующейся вначале гидроокиси цинка. Осадок отфильтровывают, промывают разбавленным (1 : 20) раствором аммиака, содержащим 2% нитрата аммония, растворяют в горячей разбавленной (1 : 5) соляной кислоте и тщательно промывают фильтр горячей водой. Полученный раствор выпаривают, и осаждают редкоземельные металлы щавелевой кислотой (см. сноску 2, стр. 624). Для определения церия осадок,• выделенный окисью цинка, растворяют в горячей разбавленной (1 : 5) соляной кислоте и затем осаждают аммиаком и щавелевой кислотой, как только что было указано х.
*Описан способ 1 2 отделения церия от других редкоземельных металлов осаждением в виде основного бромата при регулировании концентрации ионов водорода в растворе буферным раствором, состоящим из пиридина и его хлористоводородной соли. Автор указывает, чт;о однократным осаждением достигается достаточно четкое разделение. Доп. перев*
Европий, а возможно и иттербий, можно качественно открыть, отделяя от сопутствующих редкоземельных элементов, таких, как туллий и лютеций, в амальгаме, которую он образует, когда раствор его ацетата в водном растворе трехзамещенного цитрата калия встряхивают с амальгамой калия или подвергают электролизу с ртутным катодом. В присутствии самария, который также образует амальгаму, следует соблюдать известные предосторожности3.
Во время второй мировой войны для разделения редкоземельных металлов, образующихся при расщеплении тяжелых элементов, были
1 О применении ароматических оснований в качестве осадителей для редкоземельных элементов см. А. М. Jefferson, J. Am. Chem. Soc., 24, 540 (1902).
2 Э. А. Остроумов, ЖАХ, 2, 111 (1947).
3 Н. N. Me Coy, J. Am. Chem. Soc., 63, 3432 (1941). См. также К. Marsh, J. Chem. Soc., 8 (1943) и T. Moeller, H. E. Kremers, Ind. Eng. Chem. Anal. Ed., 17, 798 (1945).
40*
628
Гл. XXXVI. Редкоземельные металлы.
разработаны методы, основанные на ионном обмене. Эти Методы во многих случаях дают возможность значительно быстрее осуществлять разделение, чем обычно принятая фракционная кристаллизация. Раствор, содержащий смесь редкоземельных элементов, пропускают через колонку, наполненную катионитом (например, амберлитом). В результате ионного обмена редкоземельные металлы переходят в катионит, откуда их затем извлекают раствором лимонной кислоты и цитрата аммония, имеющим определенный pH. Комплексные цитраты отдельны^ редкоземельных металлов извлекаются из катионита с различными скоростями, благодаря чему, последовательно отбирая соответствующие порции промывной жидкости, пропущенной через колонку, можно достигнуть довольно четкого разделения редкоземельных элементов. Для определения момента полного извлечения того или иного элемента цользуются методом радиоактивных изотопов или другими способами. Обычно, прежде чем один из элементов оказывается количественно извлеченным из колонки, в растворе появляется следующий за ним по скорости извлечения элемент. В этом отношении метод, основанный на ионном обмене, напоминает метод дистилляции. Однако благодаря той легкости, с которой весь цикл можно повторить, этот метод имеет огромные преимущества перед старым методом разделения кристаллизацией. Методы, в которых используется ионный обмен, можно применить и для многих других очень сложных случаев разделения х.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Методы количественного разделения и определения редкоземельных элементов, как правило, мало совершенны. Лишь немногие элементы этой группы можно количественно выделить из смеси окислов относительно удобными методами. Предложено несколько спектрографических методов анализа смеси окислов редкоземельных металлов. Из них особый интерес представляет рентгеноспектральный метод1 2. Некоторые редкоземельные элементы дают характерные линии и полосы поглощения в видимой части спектра, что обычно используется для их открытия в присутствии других редкоземельных элементов3. Различные авторы4 рекомендуют спектры поглощения использовать также для определения этих элементов в смеси
1 По этому вопросу см. серию статей в J. Am. Chem. Soc., 69 2769—2881 (1947), * а также О. Самуэльсон, Применение ионного обмена в аналитической химии, Издатинлит, 1955. Подробный обзор методов исследования продуктов деления ядер дан в докладе К. А. Крауса и Ф. Нельсона, см. «Химия ядерного горючего» (Доклады иностранных ученых на международной конференции по использованию атомной энергии, Госхимиздат, 1956). *
2 Литература и исследования по определению La, Nd и Cd в присутствии других редкоземельных элементов приведены в статье Kenjiro Kimura, Bull. Chem. Soc. Japan, 13, 10 (1938).
3 Cm. J. Ba hr, R. Bunsen, Am. Chem., 137, 20 (1866); W. Muthmann, L. Stiitzel, Ber., 32, 2653 (1899); G. and H. К г ii s s, Kolorimetrische und Quantitative Spectralanalyse, Leopold Voss, Hamburg und Leipizig; W. P r a n d 11, K. Schei-n e r, Z. anorg. u. allgem. Chem., 220, 107 (1934).
4 Например, Haas [Beitr. Kenntnis Pr u. Nd Dissert., Berlin, 1920, S. 47, 51, 54] описывает метод разбавления, по которому раствор разбавляют до исчезновения некоторых характерных линий поглощения. По приведенным им данным для растворов двойных нитратов с аммонием в момент исчезновения соответствующих линий концентрация неодима соответствовала 0,0059% Nd2O3, а празеодима 0,0091% Рг2О3. См. также L. F. Yntema, J. Am. Chem. Soc., 45, 907 (1923); Е. D е 1 a u п е у, С. г., 185, 354 (1927); J. N. F г i е n d, D. А. Н a l 1, Analyst, 65, 152 (1940).
Методы определения
629
окислов редкоземельных элементов. Многие авторы1 предлагают также определять приблизительное содержание редкоземельных элементов по спектру излучения.
Церий в присутствии тория, лантана, неодима, празеодима, иттрия, эрбия, самария, гадолиния, титана и циркония можно определять объемным путем после окисления его до четырехвалентного состояния четырех-окисью висмута2, висмутатом натрия3 или персульфатами щелочных металлов 4. Чувствительный способ испытания на церий основан на появлении желтой до красно-желтой окраски при обработке нейтрального раствора нитрата церия (IV) 30%-ной перекисью водорода и последующем прибавлении солянокислого хинина 5 *.
Для определения в смеси редкоземельных элементов европия, который легко восстанавливается до двухвалентного состояния, солянокислый раствор хлоридов пропускают через редуктор Джонса (стр. 135) в приемник, содержащий избыточное количество раствора сульфата железа (III). Затем приливают смесь Рейнгардта (стр. 446) и образовавшееся железо (II), количество которого эквивалентно содержанию в растворе европия, титруют перманганатом®. Ни один из других редкоземельных элементов в этих условиях не восстанавливается и не титруется.
Европий (III) не менее легко, чем цинком, восстанавливается до двухвалентного состояния магнием, алюминием и амальгамой натрия. Иттербий (III) с цинком заметно не реагирует, но восстанавливается амальгамой натрия. Самарий (III) не восстанавливается в водной среде 7. Имеются указания, что самарий можно восстановить до двухвалентного состояния амальгамой кальция в растворах неводных растворителей 8 и что иттербий можно количественно определить восстановлением на ртутном катоде в кислом растворе и последующей обработкой железо-аммонийными квасцами9.
1 См., например, С. N. М с С а г t h у, L. R. Scribner, М. Lawr.enz, В. S. Hopkins, Ind. Eng. Chem., Anal. Ed., 10, 184 (1938).
2 A. Waegner, A. Muller, Ber., 36, 282 (1903). Согласно этому методу, церий окисляют встряхиванием холодного разбавленного (1 : 1) азотнокислого раствора с значительным избытком четырехокиси висмута. После этого раствор оставляют на 30 мин и затем разбавляют до определенного объема. По отстаивании в течение 1 — 2 ч раствор сливают декантацией через сухой фильтр, аликвотную часть его разбавляют равным объемом воды и обрабатывают титрованным раствором перекиси водорода, избыток которой определяют титрованием перманганатом.
3 F. J. Metzger,!. Am. Chem. Soc., 31, 523 (1909); К. S о m e у a, Z. anorg. u. allgem. Chem., 168, 56 (1927); N. H. F u г m a n, J. Am. Chem. Soc., 50, 755 (1928). По этому методу раствор сульфатов редкоземельных элементов, содержащий 20% по объему серной кислоты и 2% сульфата аммония, кипятят с избыточным количеством висмутата натрия. По охлаждении раствор разбавляют 50 мл разбавленной (2 : 98) серной кислоты и фильтруют через асбест. Осадок промывают разбавленной (2 : 98) серной кислотой. Фильтрат обрабатывают титрованным раствором сульфата железа (II), избыток которого титруют перманганатом.
4 G. von Knorr е, Z. angew. Chem., 10, 717 (1897); Ber., 33, 1924 (1900). Окисление персульфатом лучше осуществлять в присутствии нитрата серебра [Н. Н. W i I 1 а г d, Р. Young,!. Am. Chem. Soc., 50, 1379 (1928)] следующим способом. Раствор, содержащий 0,1—0,3 г церия и 2,5—10 мл серной или 5 мл азотной кислоты в 200 мл, обрабатывают 1—5 г (в зависимости от кислотности) (NH4)2S2Og и 2—5 мл 0,25%-ного раствора нитрата серебра. Кипятят 10 мин, охлаждают до комнатной температуры и титруют, лучше потенциометрически, раствором сульфата железа (II).
5 J. L и с a s, A. J i 1 е k, Z. anal. Chem., 76, 350 (1929).
« Н. N. McCoy, !. Am. Chem. Soc., 58, 1577 (1936); 61, 2455 (1939).
7 H. N. McCoy, J. Am. Chem. Soc.,58,1577 (1936);61,2455 (1939); 63,3432(1941).
8 А. В г u к 1, Z. angew. Chem., 52, 151 (1939).
9 А. В r u к 1, Z. angew. Chem., 50, 25 (1937).
630
Гл. XXXVI. Редкоземельные металлы
Кристаллический сульфат европия (II) заметно не изменяется на воздухе в течение месяцев. Сульфат иттербия (II) окисляется на воздухе, а также в воде в течение нескольких часов, а сульфат самария (II) полностью окисляется водой с выделением водорода уже в течение нескольких минут1.
Метод спектрофотометрического определения празеодима, неодима и самария, относящихся к цериевой группе, и диспрозия, гольмия, эрбия, тулия и иттербия, элементов иттриевой группы, основан на измерении светопоглощения растворов их нитратов 2. В этом методе редкоземельные элементы сначала отделяют от посторонних элементов, а затем разделяют цериевую и иттриевые группы сульфатом натрия. Элементы цериевой группы определяют в осадке, а иттриевой группы в растворе после переведения смеси окислов в нитраты. Светопоглощение измеряют при 446 ммк для празеодима, 512 и 798 ммк для неодима, 402 ммк для самария, 910 ммк для диспрозия, 643 ммк для гольмия, 521 и 653 ммк для эрбия, 684 ммк для тулия, 950 и 937 ммк для иттербия. Из элементов, которые дают слабо выраженные полосы поглощения, церий и еврдпий можно определить другими методами, а содержание лантана, гадолиния, тербия и иттрия вычисляют по разности.
Описан способ раздельного экстрагирования хлороформом соединений неодима и эрбия с 5,7-дихлор-8-оксихинолином и последующего спектрофотометрического определения этих элементов в хлороформном растворе3.
Более или менее полного разделения всех элементов редкоземельной группы, без сомнения, можно достигнуть, но только путем обработки большого количества материала и обычно посредством сотен и даже тысяч фракционирований. Но каждый аналитик может легко осуществить еще одну степень разделения: приближенно разделить редкоземельные элементы на две большие подгруппы, для чего их раствор насыщают сульфатом калия или натрия. В результате этого элементы так называемой цериевой подгруппы, скандий и торий (а также и цирконий) выделяются в виде двойных сульфатов.
Торий более полно осаждается сульфатом калия, чем натрия, а скандий натриевой солью не осаждается. Поэтому, если присутствует торий, обычно в качестве осадителя пользуются сульфатом калия, чтобы полностью перевести его в нерастворимый осадок. Если в осадке, выделенном сульфатом калия, содержатся торий и скандий, их можно разделить обработкой раствором сульфата натрия, в котором соединения скандия растворимы.
Редкоземельные металлы обычно делят на три подгруппы, а именно: 1) цериевую, состоящую из лантана, церия, празеодима, неодима, прометия и самария; 2) тербиевую, в состав которой входят европий, гадолиний и тербий, и 3) иттриевую, состоящую из иттрия, диспрозия, гольмия, эрбия, тулия, иттербия и лютеция. Диспрозий, гольмий, эрбий и тулий иногда называют эрбиевой подгруппой.
--------:— I
1 Н. N. McCoy, цит. выше.
2 С. J. Rodden, J. Research NBS, 26, 227 (1941); 28, 265 (1942).
3 Т. Moeller, D. E. Jackson, Anal. Chem., 22, 1393 (1950). Спектрофотометрическое определение празеодима, неодима и самария см. также A. W. Wylie, J. Soc. Chem. Ind., 69, 143 (1950).
Методы определения
631
*При описываемом способе разделения элементы тербиевой подгруппы распределяются между цериевой и иттриевой подгруппами.
Показано1, что посредством фракционного осаждения сульфатом натрия можно разделить редкоземельные элементы на все три подгруппы. Авторы приведенной работы указывают, что, пользуясь разработанной ими схемой, можно относительно легко выделить концентраты с содержанием свыше 80% элементов иттриевой или тербиевой подгруппы. Из растворов, в которых концентрируются элементы иттриевой подгруппы, •была получена окись иттрия 90—94% -ной чистоты.
Рекомендовано также 2 проводить разделение редкоземельных металлов фракционным осаждением миндальной кислотой. Автор предполагает, что избирательное осаждение редкоземельных элементов миндальной кислотой является результатом различной скорости образования внутрикомплексных соединений, а также различной растворимости этих комплексов. Доп. перев.*
Осаждение проводят следующим способом. Если редкоземельные элементы находятся в виде нитратов или хлоридов, их растворяют в возможно меньшем количестве воды, прибавляют 200—300 мл насыщенного раствора сульфата калия и затем еще 5 г измельченной в порошок соли. Тщательно перемешивают и оставляют стоять 12 ч. Затем фильтруют и промывают двойные сульфаты насыщенным раствором сульфата калия. Осадок обрабатывают 5 %-ным раствором едкого натра при нагревании.
Образующиеся при этом гидроокиси отфильтровывают, промывают горячей водой и растворяют в разбавленной соляной кислоте. Раствор выпаривают досуха и осаждение повторяют.
Когда редкоземельные элементы находятся в виде сульфатов, их нагревают приблизительно при 450° С до прекращения выделения паров серной кислоты, разминают массу и растворяют в возможно меньшем количестве ледяной воды. Разбавляют равным объемом воды, затем при перемешивании вводят измельченный в порошок сульфат натрия до насыщения раствора и оставляют на 12 ч. Двойные сульфаты отфильтровывают, промывают насыщенным раствором сульфата натрия и обрабатывают едким натром при нагревании. Гидроокиси растворяют в соляной кислоте, раствор выпаривают досуха и осаждение сульфатом калия или натрия повторяют.
Если редкоземельные элементы следует осадить в виде оксалатов и затем взвесить в виде окислов, двойные сульфаты необходимо снова перевести в хлориды, так как при осаждении в присутствии больших количеств натриевых солей оксалаты загрязняются натрием.
Хотя таким путем и удается отделить группу элементов одного конца ряда от элементов другого конца, т. е. отделить так называемую цериевую подгруппу от иттриевой, но растворимость двойных калиевых сульфатов возрастает настолько равномерно от наименее растворимой соли лантана, что достигнуть четкого разделения невозможно. Промежуточные элементы всегда находятся в обеих смесях двойных сульфатов. Изменив условия фракционирования, можно получить другие группы редкоземельных металлов, но разделение никогда не бывает количественным. Когда стремятся получить две основные подгруппы элементов, осаждение двойных сульфатов следует проводить при комнатной температуре, так как температура влияет на результаты разделения.
В литературе имеются указания, что фракционирование этилсульфатов дает более четкое разделение редкоземельных элементов на две
1Т. Moeller, Н. Е. Kremers, Ind. Eng. Chem., Anal. Ed., 17, 44 (1945).
2 Boyd, Weaver, Anal. Chem., 26, 476'(1954).
632
Гл. XXX VI. Редкоземельные металлы
подгруппы1, однако этот способ не получил широкого распространения в аналитической практике2.
Такое частичное разделение редкоземельных металлов на две или более подгруппы в количественном анализе имеет целью выяснение природы компонентов каждой подгруппы, а также установление того, какая из этих подгрупп преобладает в исследуемом минерале. Например, в церите и монаците преобладает содержание цериевой подгруппы, а в гадолините — иттриевой подгруппы.
О полезности проведения таких разделений, а также о воспроизводимости получаемых результатов можно судить по нижеследующим данным, приведенным в статье Гиллебранда 3.
Три минерала — гадолинит, иттриалит и роуландит — были найдены в округе Ллано (Техас) тесно связанными друг с другом, что внушало мысль об их общем происхождении. Эта мысль еще более подкрепляется редким совпадением для гадолинита и иттриалита результатов определения не только относительных содержаний трехвалентных редкоземельных металлов, но и их абсолютных количеств:
СваОз ' % И др., % YSO3 и др., '
Гадолинит (Гент) . . 2,65 5,22 44,35
Гадолинит (Гент) . 2,66 5,01 44,45
Гадолинит (Икин) . 2,62 5,22 41,55
Иттриалит (Гиллебранд) . . . 3,07 5,18 43,45
Роуландит (Гиллебранд) . . . 5,06 9,34 47,70
Такое совпадение результатов, полученных тремя химиками, является, кроме того, убедительным подтверждением пригодности примененного ими метода разделения редкоземельных элементов. Как видно из таблицы, почти такое же соотношение между трехвалентными редкоземельными элементами (как в гадолините и иттри-алите) имеется и в роуландите.
Характерным признаком преобладания той или иной подгруппы служит также окраска оксалатов и прокаленных окислов и, кроме того, приближенный средний атомный вес, полученный для смеси окислов. Определение приближенного атомного веса является простой операцией по сравнению с точным его определением и сводится к сравнению веса смеси окисей с весом безводных сульфатов тех же элементов4 *.
При определении среднего атомного веса смеси редкоземельных элементов по методу, основанному на переведении оксалатов в окислы и титровании оксалатов перманганатом, титр раствора перманганата устанавливают по чистому оксалату тех редкоземельных металлов, которые являются основными компонентами смеси6. Хотя определение атомного веса большей частью служит для контроля точных разделий, но оно представляет интерес и в рядовых анализах, так как может дать представление
1 G. Urbain, Am. Chim. phys., [7], 19, 184 (1900). J. Kleinberg, W. A. Taebel и L. F. Audriet h [Ind. Eng. Chem., Anal. Ed., 11, 368 (1939)) установили, что сульфаминовую кислоту можно применять для разделения смеси окислов, свободной от церия, на нерастворимую лантановую (La, Рг, Nd и Sm) и растворимую иттриевую (Y, Tb, Dy, Но, Er, Tu, Yb и Lu) подгруппы.
2 * Обзор методов разделения редкоземельных металлов см. Д. И. Рябчиков и Е. А. Терентьева, Усп. хим., 16, № 4, 961 (1947); 24, № 3, 260 (1955). Прим, перев. *
3 W. F. Н i 11 е b г a n d, Am. J. Sci., [4], 13, 150 (1902).
4 См. R. J. M e у e r, O. Hauser, Die Analyse der seltenen Erden und Erd-sauren, 1912, S. 216.
8 G. L. В a r t h a u e r, R. G. R u s s e 1, D. W. Pearce, Ind. Eng. Chem.,
Anal. Ed., 15, 548 (1943).
Методы определения
633
об относительном содержании некоторых металлов или подгрупп, особенно иттриевой группы, элементы которой мало различаются по физическим и химическим свойствам.
Характерным различием элементов цериевой и иттриевой подгрупп является цвет их окислов.
Окислы трехвалентных лантана, церия, гадолиния, тербия, иттрия, диспрозия, иттербия и лютеция белого цвета. Окись тулия белого цвета с зеленоватым оттенком, гольмия — бледно-желтая, празеодима — зеленовато-желтая, европия — бледно-розовая, эрбия — розовая и неодима — лиловая. ТЬ4О7 (полученная прокаливанием оксалата) — темно-коричневого цвета, Рг60ц (прокаленная на воздухе) — коричневаточерного цвета, а СеОг (полученная прокаливанием на воздухе) в горячем состоянии имеет желтую окраску, а по охлаждении становится белой.
Достаточно незначительного содержания окиси празеодима в виде твердого раствора с СеО2 для окрашивания цериевых земель в коричневый цвет. Чем светлее окраска смеси окислов редкоземельных элементов обеих подгрупп, тем выше относительное содержание в них итттриевой подгруппы.
Смеси окислов элементов иттриевой подгруппы, свободные от окислов элементов цериевой подгруппы и бедные эрбиевой подгруппой, имеют белый до бледно-желтого цвет. С увеличением концентрации элементов эрбиевой подгруппы окраска становится более, интенсивно желтой и даже красноватой1.
* Колориметрические методы определения церия
Для колориметрического определения церия обычно используют его способность окисляться в водной среде до четырехвалентного состояния. Колориметрирование проводится либо непосредственно по желтой окраске ионов церия (IV), либо косвенным путем по окраске продуктов, образующихся в результате окисления церием (IV) некоторых органических соединений.
Персульфатный метод. Этот метод основан на измерении интенсивности желтой окраски сернокислого раствора, содержащего церий (IV), полученный в результате окисления персульфатом аммония в присутствии нитрата серебра. Другие редкоземельные элементы не оказывают влияния на определение. Марганец и хром, окисляющиеся в этих условиях соответственно до перманганата и хромата, должны отсутствовать. Мешают определению также хлориды, фториды и фосфаты.
Колориметрирование сводится к следующему. Аликвотную часть анализируемого раствора, содержащую 0,5—5,0 мг церия и 1 мл разбавленной (1 : 1) серной кислоты, разбавляют до 10 мл, прибавляют 0,5 мл 0,1 %-ного раствора нитрата серебра и 0,2 г персульфата аммония. Осторожно кипятят 5 мин, затем охлаждают, разбавляют до определенного объема и измеряют светопоглощение раствора, пользуясь фиолетовым светофильтром. Содержание церия вычисляют по калибровочной кривой.
Бруциновый метод. Метод основан на реакции окисления бруцина церием (IV), в результате чего образуются продукты окисления бруцина, окрашенные в оранжево-красный цвет 2. Интенсивность окраски зависит
1 См. R. J. М е у е г, О. Hauser, Die Analyse der seltenen Erden und Erd-sauren, 1912, S. 27, 191.
2 Ф. M. Ш e м я к и н, В. А. Волкова, ЖОХ, 9, 698 (1939).
634
Гл. XXXVI. Редкоземельные металлы
от продолжительности стояния раствора после введения в него реагентов. Интенсивность получаемых окрасок отвечает закону Бера. Ионы других металлов не влияют на определение. Посторонние окислители должны отсутствовать. Определение выполняют следующим способом. Окисляют церий в растворе, как указано выше (см. «Персульфатный метод»). Затем, если требуется, добавляют к раствору воду и продолжают кипятить еще 10 мин до полного разрушения персульфата. По охлаждении до комнатной температуры в раствор вводят 0,25 мл 0,1%-ного раствора бруцина в 1 н. серной кислоте и разбавляют до определенного объема (если возможно до 10 мл).
Светопоглощение раствора измеряют спустя 10 мин, пользуясь синим светофильтром.
Другие методы. Для определения церия предложено также 1 использовать желтую окраску комплексного соединения, образующегося при обработке разбавленного раствора соли церия карбонатом калия, растворением выделившегося белого осадка добавлением избытка карбоната калия и стоянии полученного раствора на воздухе.
Описаны также колориметрические методы определения церия, основанные на его реакции с пирогаллолом2 или галловой кислотой3 в аммиачной среде, в результате чего образуются окрашенные растворы или осадки 4. Доп. перев*
1 J. Р 1 a nk, Z. anal. Chem., 116, 312 (1939).
2 Ф. М. ПТ е м я к и н, Z. anorg. allgem. Chem., 217, 272 (1934).
3 Ф. М. Шемякин, Зав. лаб., 3, 1090 (1934).
* Ф. М. Шемякин, Т. В. В а ш е д я е н к о, ЖОХ, 5, 607 (1935); Ф. М. Ш е м я к и н, А. В. Веселова, М. И. В л а д и м и р о в а, Зав. лаб., 5, 231 (1936).
Глава XXXVII
ЦИРКОНИЙ (ГАФНИЙ)
Микроскопические исследования горных пород показывают, что цирконий является одним из наиболее постоянных их компонентов. Он присутствует обычно в виде минерала циркона, а в таких случаях о приблизительном содержании его в породе можно судить по микроскопическим данным, и химический анализ часто становится ненужным. Однако в некоторых породах он встречается в виде других минералов, которые микроскопическим путем распознать не удается. Содержание цикония в породах может доходить до нескольких процентов, но оно редко достигает 0,2% и обычно бывает ниже 0,1%. Применение циркония в производстве огнеупорных материалов, эмалей, в металлургии и др. повышает интерес к методам определения его в рудах.' Основные минералы циркония — циркон (ZrSiO4) ибадделеит (ZrO2), но цирконий является также более или менее важным компонентом многих других минералов. Во всех циркониевых минералах обычно находится также и гафний х, содержание которого иногда достигает 1%. Установлено, что в земной коре содержится 4-10"4% гафния.
Для количественного разделения циркония и гафния достаточно удовлетворительных методов неизвестно. Для этой цели предложен метод ионного обмена. При соответствующем подборе катионитов и раствора для элюирования эти методы могут дать хорошие результаты в аналитической практике, но они еще недостаточно детально разработаны, чтобы их здесь можно было излагать. Комплексные оксалаты, а также фториды циркония и гафния были хроматографически разделены на анионите еильноосновного типа 1 2. Для очистки циркония и разделения циркония и гафния предложены 3>4 также и некоторые другие способы, основанные на ионном обмене. Для разделения этих элементов рекомендуется, кроме того, использовать различное давление паров их тетрахлоридов, а также их фосфорилхлоридов Б.
В последующем изложении все положения, касающиеся циркония, относятся также и к гафнию.
1 D. С о s t е г, G. Н е v е s у, Nature, 111, 182 (1923); Naturwissenschaften, 11, 133 (1923); Chem. a. Ind., 42, 258 (1923).
2 К. А. К r a u s, G. E. Moore, J. Am. Chem. Soc., 71, 3263 (1949).
3 А. А у r e s, J. Am. Chem. Soc., 69, 2879 (1947).
* G. T. Seaborg, J. Am. Chem. Soc., 70, 4268 (1948).
6 * О раздельном колориметрическом определении циркония и гафния при их совместном присутствии с помощью 2,4-дисульфобензаурин-3,3'-дикарбоновой кислотой см. В. И. К у з н е ц о в, А. А. Немод р у к, Тезисы докладов на совещании по спектрофотометрическим и колориметрическим методам, Изд. АН СССР, 1955, стр. 46. Прим. ред. *
636
Гл. XXXVII. Цирконий [гафний]
ОБЩИЕ ЗАМЕЧАНИЯ
При анализе силикатов цирконий подобно титану, частично выделяется совместно с кремнекислотой вследствие гидролиза его солей, в большей степени когда присутствуют фосфат-ионы, с которыми он дает нерастворимое соединение. После отгонки кремнекислоты цирконий можно Извлечь из нерастворимого остатка и присоединить к той части циркония, которая осталась в растворе. При осаждении аммиаком цирконий переходит в осадок и, когда присутствие его не учитывается, принимается за алюминий. Если осадок представляет собой только гидроокись циркония, его можно прокалить и взвесить в виде ZrOa после внесения поправки на содержание в нем небольших количеств SiO2 (стр. 644). Если же осадок от аммиака имеет сложный состав, цирконий можно отделить, но определять его лучше в отдельной порции пробы, в которой можно определить также барий и редкоземельные металлы (стр. 968).
Предложен1 почти специфический капельный метод открытия малых количеств циркония, основанный на образовании окрашенного в коричневый цвет соединения циркония с п-диметиламиноазофениларсоновой кислотой в солянокислом растворе (условия несколько изменяются в присутствии.некоторых соединений). Как указывают авторы, эту реакцию, помимо циркония, дает только тантал *.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ЦИРКОНИЙ
Небольшие количества циркония, находящиеся в горных породах, легко переводят в раствор методом, применяемым для разложения природных силикатов.
Циркониевые руды, содержащие циркон и бадделеит, не так легко разлагаются этим методом, но его тем не менее приходится применять при полном анализе фосфатных и фторсодержащих минералов. В этих случаях сплавлять с карбонатом натрия приходится продолжительное время, и часто разложение бывает неполным, несмотря на очень тонкое измельчение анализируемого материала. Нерастворимый остаток после выщелачивания плава водой обычно следует сплавлять повторно. С пиросульфатом калия легко сплавляется бадделеит, но медленно реагирует циркон.
Для разложения некоторых материалов целесообразно применять сплавление с карбонатом натрия, плав тщательно выщелочить водой, затем нерастворимый остаток промыть, сплавить с пиросульфатом калия жрастворить плав в разбавленной серной кислоте. После первого сплавления и выщелачивания остается нерастворимое соединение циркония2, которое разлагается и легко переходит в раствор после вторичного сплавления.
Для сплавления наждака было рекомендовано 3 применять пиросульфат натрия, так как эта соль в данном случае оказывает значительно более эффективное действие, чем пиросульфат калия. По аналогии с этим можно предполагать, что и циркониевые руды также будут более успешно разлагаться при сплавлении с пиросульфатом натрия, чем с пиросульфатом
1 F. Feigi, Р. Krumholz, Е. Rajman n, Microchemie, 9, 395 (1931); Analyst, 56, 615 (1931).
, 2 Это соединение часто принимают за цирконат натрия. Но это, очевидно, окись циркония, так как Н. В. Knowles после тщательного сплавения 0,1143 г ZrO2 с карбонатом'натрия, выщелачивания плава водой и прокаливания хорошо промытого осадка нашел массу его, равную 0,1148 г.
3 J. L. S m i t h, С. г., 31, 48—50, 191—93 (1850); Sill. Am. J. Sci., 10, 354 (1850).
Разложение минералов, содержащих цирконий
637
калия. Для разложения циркониевых руд предложено также сплавление с перекисью натрия 1. С целью предохранения платинового тигля, прежде чем ввести в него смесь анализируемой пробы с перекисью натрия, дно его и стенки покрывают сначала слоем расплавленного в том же тигле небольшого количества карбоната натрия, а затем слоем расплавленной перекиси натрия. Установлено, что циркон разлагается сплавлением его с едким натром 2 при 600° С.
Имеются указания, что хорошие результаты дает сплавление с бифторидом калия или со смесью 1 части бифторида калия и 10 частей пиросульфата, с последующей обработкой 1—2 мл серной кислоты и осторожным нагреванием при 900—1000° С. В обоих случаях фтор необходимо затем полностью удалить. Цирконий при этом не теряется в виде фторида, если сплавление проводить при температуре не более высокой, чем это требуется для поддержания плава в жидком состоянии 3. Это может показаться удивительным, поскольку при выпаривании соединений циркония с фтористводородной кислотой происходит улетучивание циркония в виде его фторида. Однако в процессе сплавления с фторидами щелочных металлов, по-видимому, образуются менее летучие двойные соли. При применении этих плавней исключается, конечно, возможность определения кремния или щелочных металлов. Для определения последних применяют сплавление с фторидом аммония при возможно более низкой температуре, если в этих условиях минерал полностью разлагается.
Для. сплавления циркона и бадделеита в Бюро Стандартов США с успехом применяется бура 4. Этот способ не может, конечно, применяться в тех случаях, когда надо определять щелочные металлы, тогда буру следует заменить борной кислотой (см. «Разложение при помощи плавней», стр. 929), если она разлагает анализируемый минерал при достаточно низкой температуре, при которой щелочные металлы не улетучиваются.
Сплавление с бурой проводят следующим способом. Расплавляют 4 г буры в платиновом тигле, охлаждают и затем на расплавленную массу помещают 0,3 г тонко измельченной анализируемой руды, просеянной через сито в 100 меш (d = 0,147 мм}. Закрывают тигель крышкой и сплавляют на горелке Мекера, время от времени перемешивая плав в тигле короткой негнущейся платиновой палочкой или проволокой. По окончании разложения руды, когда плав становится прозрачным, на что обычно требуется не более получаса, крышку и палочку переносят в стакан, в котором предполагают растворять плав. Во время охлаждения вращают тигель, чтобы расплавленная масса распределилась по его стенкам. Охлажденный плав растворяют в 150 мл разбавленной (1 : 5) соляной кислоты, поместив тигель в стакан с кислотой в лежащем положении и подложив под него у его отверстия крышку от тигля. Это дает возможность растворителю свободно циркулировать. Нагревают на водяной бане до полного растворения плава. Эта операция очень продолжительна, поэтому лучше оставить плав растворяться в течение ночи. Борная кислота ни в какой мере не мешает последующему определению циркония фосфатным
1 М. Travers, Chim, et ind., 2, 385 (1919); J. A. Holladay, Electro Me tallurgical Co. (частное сообщение).
2 M. A x t, Ing. chim., 23, 142 (1939).
3 Например, H. B. Knowles после сплавления 0,2311 и 0,2312 г ZrO2 с KHF2 получил 0,2309 и 0,2313 г.
4 G. Е. F. L u n d е 11, Н. В. Knowles, J. Am.-Chem. Soc., 42, 1439 (1920).
638
Гл. XXXVII. Цирконий [гафний]
или купфероновым методом. Если же по ходу анализа требуется затем провести осаждение аммиаком, борную кислоту необходимо предварительно удалить выпариванием с метиловым спиртом и соляной кислотой (стр. 753), так как она частично.улекается осадком гидроокисей даже после двукратного переосаждения.
Следует иметь в виду, что при анализе материалов, в которых одновременно присутствуют фосфор и цирконий, всегда возникают затруднения, связанные с нерастворимостью фосфата циркония. Такие материалы рекомендуется сплавлять с карбонатом натрия, плав выщелачивать водой, а затем раствор и осадок анализировать раздельно. Необходимо также учитывать, что соли циркония легко гидролизуются и цирконий может выделиться из раствора, если кислотность последнего недостаточно высока.
МЕТОДЫ ОТДЕЛЕНИЯ
Наиболее удовлетворительным методом отделения циркония от других элементов является осаждение его в виде двузамещенного фосфата из раствора, содержащего 10% по объему серной или соляной кислоты, а также перекись водорода, если присутствуют титан, ниобий или тантал. По всей вероятности, лишь очень немногие элементы, помимо гафния, протактиния, ниобия и тантала, влияют на осаждение фосфата циркония, если осаждение проводится в сернокислом растворе. При отсутствии циркония ниобий и тантал выделяются из растворов, содержащих серную кислоту, фосфорную кислоту и перекись водорода, лишь после продолжительного стояния при комнатной температуре, но в присутствии циркония они частично выпадают в осадок. Метод осаждения циркония фосфатом и последующей обработки полученного осадка приводится ниже (стр. 642). Другие элементы, например железо, титан, торий и редкоземельные металлы, можно затем отделить от фосфорной кислоты осаждением едким натром и определйть обычно принятыми методами.
Для полного отделения от тория цирконий следует переосадить из разбавленного (1 : 10) сернокислого раствора. Для этого первый осадок вместе с фильтром обрабатывают HNO3 и H2SO4 (стр. 438), раствор выпаривают до появления паров серной кислоты и после разбавления водой снова осаждают цирконий необходимым количеством (NH4)2HPO4 при нагревании1 при 40—50° С.
Осаждение фениларсоновой кислотой CeH5AsO(OH)2 из кипящего раз-бавлеиного'(1 : 9) сернокислого раствора служит для отделения циркония (и гафния) от большинства посторонних элементов. В присутствии титана и ниобия, так же как и при осаждении фосфата циркония, следует вводить перекись водорода. .При наличии тория или фосфора требуется пере-осаждение осадка 2. Тантал влияет на осаждение циркония при любых условиях.
♦Осаждение циркония фениларсоновой кислотой изучалось И. П. Алимарицым и О. А. Медведевой 3. Авторы применили этот метод для определения циркония в минералах, горных породах и стали.
1 Таким способом в растворах, содержащих цирконий в количестве, соответствующем 0,1024 г ZrP2O7 и 0,0232 г ThO2, в виде Th(SO4)2, Н. В. Knowles определил 0,1021 и 0,1022 г ZrP2O7. После однократного осаждения из растворов, содержавших такие же количества циркония и тория, было получено 0,1057 и 0,1058 г ZrP2O7.
2 А, С. R i с е, Н. С. F о g g, С. lame s, J. Am. Chem. Soc., 48, 895 (1926).
3И. П. Алимарин, О. А. Медведева, Зав. лаб., И, 254 (1945).
Методы отделения
639
Осаждение фениларсоновой кислотой было использовано 1 для определения циркония в высоколегированных сплавах, содержащих титан, ниобий, бор, ванадий, алюминий, медь и большие количества хрома и вольфрама. Автором разработаны условия определения циркония при одновременном присутствии титана и вольфрама. Доп. перев.*
Для отделения циркония от титана, алюминия, хрома, кобальта, никеля, меди, урана, ванадия, тория и молибдена, а также от таких малых количеств кремнекислоты и вольфрама, какие могут остаться в растворе после обезвоживания выпариванием с кислотой, применяют осаждение ге-пропиларсоновой кислотой из горячего разбавленного (3 : 100) солянокислого раствора и последующее нагревание раствора в течение 30— 60 мин. Осадок промывают горячей водой 2. Если присутствуют большие количества железа, как в случае анализа стали, осадок и фильтр разлагают осторожным нагреванием с 10 мл соляной кислоты, раствор разбавляют до 100 мл водой и цирконий осаждают’снова. Осадок можно прокалить в фарфоровом тигле до окиси ZrO2. Олово частично осаждается, но его можно отделить обработкой прокаленного осадка иодидом аммония, как указано на стр. 342. Если в анализируемом растворе присутствует достаточное для осаждения циркония количество фосфора, выделившийся осадок отфильтровывают и для отделения циркония от фосфат-ионов сплавляют с карбонатом натрия. Плав выщелачивают водой, нерастворимый остаток отфильтровывают, прокаливают, затем сплавляют с пиросульфатом и растворяют плав в воде, содержащей несколько капель серной кислоты.
Наиболее удовлетворительное отделение цирконий от титана, ниобия и тантала достигается 3 сплавлением смеси окислов этих металлов с пиросульфатом калия, растворением плава в насыщенном растворе оксалата аммония осаждением и таннином из слабокислого кипящего раствора, полунасыщенного хлоридом аммония. Полученный осадок промывают раствором, содержащим 5% хлорида аммония и 1% оксалата амммония, высушивают й прокаливают в кварцевом тигле. В этих условиях тантал, ниобий и Фитан осаждаются, тогда как берилий, алюминий, железо, торий и уран остаются в фильтрате совместно с цирконием. Если присутствуют лишь небольшие количества циркония, целесообразно предварительно отделить большую часть ниобия и тантала, для чего смесь окислов сплавляют с карбонатом калия, плав выщелачивают водой, содержащей небольшое количество едкого кали, нерастворимый остаток отфильтровывают и промывают 2%-ным раствором карбоната калия. Остаток вместе с фильтром обрабатывают затем при нагревании разбавленной соляной кислотой, осаждают аммиаком и фильтруют. Успешность разделения зависит от тщательности выполнения всех указаний. Протактиний, если он присутствуют один, не переходит в водный раствор после сплавления с карбонатом калия 4. Его поведение в присутствии других элементов неизвестно.
1 Р. Б. Голубцова, Зав. лаб., 17, 9'19 (1951).
2 Н. Н. G е i s t, G. С. С h a n d 1 е е, Ind. Eng. Chem., Anal. Ed., 9, 169 (1937).
3 W. ft. Schoeller, A. R. P о w e 11, Analyst, 55, 605 (1930); 57, 550 (1932); W. R. S c h о e 11 e г, H. W. W e b b, Analyst, 58, 143 (1933).
4 A. V. Grosse, J. Ain. Chem. Soc., 52, 1742 (1930).
640
Гл. XXXVII. Цирконий [гафний]
Цирконий можнц. количественно отделить от алюминия, железа, хрома, неодима, иттрия, урана, бериллия, марганца и никеля осаждением таннином из солянокислого раствора хлоридов этих элементов. Большая часть циркония выделяется из 0,25 н. (0,5 н., если присутствуют торий и ванадий) по концентрации соляной кислоты раствора, содержащего хлорид аммония. Небольшое, остающееся в растворе количество циркония извлекают прибавлением щелочи (пока концентрация кислоты в растворе не станет 0,1 н.) и отфильтровыванием выделяющегося при этом осадка. Титан и олово осаждаются совместно с цирконием х.
Отделение циркония от фосфора выполняют сплавлением анализируемой пробы с карбонатом натрия или перекисью натрия и выщелачиванием плава водой, как указано в разделе «Осаждение в виде фосфата циркония» (стр. 642), или такой же обработкой прокаленного осадка от аммиака.
При обработке раствора сероводородом, аммиаком или сульфидом аммония в присутствии винной кислоты, фенилгидразином, а также при экстрагировании эфиром (стр. 161) и электролизе с ртутным катодом (стр. 165) цирконий в основном ведет себя так же, как титан (стр. 652), алюминий (стр. 561) и др.
Заслуживают внимания методы, основанные: 1) на осаждении купфероном, как изложено в разделе «Купфероновый метод» (стр. 643), при котором в сильнокислой среде цирконий быстро и количественно отделяется от алюминия, хрома и урана (VI), и 2) на применении фтористоводородной или щавелевой кислоты (стр. 621—622); таким способом редкоземельные элементы отделяют от циркония.
Описано фракционированное осаждение гафния и циркония триэтилфосфатом а, однако таким путем трудно достигнуть достаточно четкого разделения этих элементов.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Осаждение в виде фосфата циркония
В виде двузамещенного фосфата цирконий количественно осаждается избытком осадителя из растворов, содержащих 10% по объему серной кислоты, при нагревании в течение нескольких часов при 40—50° С. Метод определения циркония, основанный на этой реакции, можно было бы считать почти совершенным, если бы не склонность осадка фосфата циркония к гидролизу и потере Р2О5 в процессе промывания. После продолжительного промывания и последующего прокаливания осадок содержит меньше Р2О5, чем это соответствует формуле пирофосфата ZrP2O7. При определении малых количеств циркония, например 2—3 мг, ошибка несущественна, но при значительных количествах циркония 3 она может достигать 1—2% от общего содержания ZrO2. В таких случаях прокаленный остаток следует разложить сплавлением с карбонатом натрия, отделить
1 W. R. Schoeller, Analyst, 69, 259 (1944).
2 Н. Н. W i 11 а г d, Н. Freund, Ind. Eng. Chem., Anal. Ed.5 18, 195 (1946).
3 Характерны следующие результаты:
Полученная масса ZrP2O- (в пересчете
HaZrO2)>a............................... 0,00065 0,00106 0,00185 0,0212 0,0426
Взято ZrO8, г............................. 0,00064 0,00107 0,00193 0,0216 0,0431
Методы определения
641
фосфор выщелачиванием плава водой и затем, после сплавления нерастворимого остатка с пиросульфатом калия и растворения плава в разбавленной (5 : 95) серной кислоте, выделить цирконий двукратным осаждением аммиаком.
Для отделения циркония от фосфора промытый осадок рекомендуют1 также дважды кипятить с 5%-ным раствором едкого натра. Хотя этот метод весьма привлекателен, но мы не можем рекомендовать его для точных анализов, так как он не дает удовлетворительных результатов, если осадки фосфата циркония обрабатывают не сейчас же после их выделения. К тому же гидроокись циркония несколько растворима в щелочных растворах, и, кроме того, полученный в результате обработки осадок иногда лишь с трудом удается перевести в раствор 2.
В присутствии титана перед добавлением фосфата необходимо ввести избыток перекиси водорода, который нужно сохранять в растворе в процессе осаждения 3. Для полного отделения от тория требуется двукратное осаждение. Ниобий и тантал частично осаждаются фосфатом, и поэтому их следует предварительно удалить (стр. 638). Отделение циркония от церия происходит более успешно, если церий находится в трехвалентном состоянии. Гафний, а также протактиний осаждаются фосфатом полностью 4.
Ход определения 5. К сернокислому раствору сульфата циркония, объем которого зависит от содержания циркония (например, 25 ль^ при содержании 0,5 мг и 200 мл при содержании 0,1 г циркония), прибавляют 1—2 капли перекиси водорода. В присутствии титана вводят избыточное количество чистой перекиси водорода, сохраняя ее в растворе до окончания процесса осаждения. Установив концентрацию серной кислоты в растворе несколько выше 10% по объему, прибавляют свежеприготовленный 10%-ный раствор (NH4)2HPO4 в 10—100-кратном количестве по отношению к необходимому По соотношению Zr : 2Р, причём больший избыток осадителя требуется при содержании малых количеств циркбния. Доводят концентрацию серной кислоты в растворе до 10% по объему и затем нагревают * при 40—50° С. Если по истечении двух часов выделяется значительный осадок фосфата циркония, вводят мацерированную бумагу,
1 Н. Р а р е, Inaugural Dissertation, Friedrich-Wilhelms Universitat, Berlin, 1917, S. 37.
2 Мы не можем согласиться с утверждением Раре о том, что после выделения циркония едким натром невозможно освободить его от солей натрия переосаждением аммиаком.
3 При отсутствии перекиси водорода титан почти .-полностью выделяется из раствора. Так, например, после обработки 200 мл раствора Ti(SO4)2 в разбавленной (5 : 95) серной кислоте (содержащего 0,1248 г TiO2) 100-кратным (по отношению к необходимому для образования фосфата титана) количеством Н3РО4 и нагревания при 30—40° С в течение ночи в фильтрате было найдено лишь 0,0066 г TiO2.
4 А. V. Grosse, .1. Am. Chem. Soc., 52, 1742 (1930); Ind. Eng. Chem., 27, 422 (1935); Ber., 61, 233 (1938).
8 G. E. F. L u n d e 11, H. B. Knowles, J. Am. Chem. Soc., 41, 1801 (1919).
8 Согласно G. HevesynK. К i m u r a ]J. Am. Chem. Soc., 47, 2540 (1925)], растворимость фосфата циркония в 6 н. растворе НС1 при 20° G составляет 0,00012 моля на 1л, а фосфата гафния — 0,00009. В 10 н. растворе НС1 растворимость этих соединений составляет соответственно 0,00023 и 0,00012 .иоль на 1 л. Эти данные относятся к чистым растворам фосфатов. Они несомненно уменьшаются в присутствии избытка фосфорной кислоты. Авторы полагают, что выделяющиеся фосфаты имеют состав ZrO(H2PO4)2 и HfO(H2PO4)2 и что после прокаливания их получаются ZrO(POs)2 и HfO(PO3)2.
41 Заказ 522.
642
Гл. XXXVII. Цирконий [гафний]
дают отстояться и фильтруют. Если же осадок невелик, оставляют на ночь при комнатной температуре, а при содержании очень малых количеств циркония стакан покрывают часовым стеклом и оставляют стоять на 2—3 дня. В том случае, когда осадок необходимо исследовать, фильтруют через бумажный фильтр, в противном случае для фильтрования можно пользоваться тиглем Гуча, обработанным до взвешивания так же, как в процессе анализа. Осадок промывают холодным 5%-ным раствором нитрата аммония до удаления избытка фосфата. Для промывания небольших осадков требуется примерно 300 мл. раствора нитрата аммония. Точно установить необходимое количество промывной жидкости невозможно, так как промывные воды никогда не бывают свободными от фосфора вследствие гидролитического разложения осадка. Чтобы избежать растрескивания осадка во время прокаливания, влажный фильтр с осадком сушат в тигле, осторожно нагревают до обугливания бумаги и затем, прикрыв почти все отверстие тигля крышкой, очень осторожно прокаливают ди сгорания угля. Под конец прокаливают на паяльной горелке. Прокаленный остаток взвешивают в виде ZrP2O7.
При проверке этого метода было найдено1, что: 1) винная кислота не влияет на осаждение; 2) цирконий прекрасно отделяется от А1, Си, Cd, Bi, Ni, Со, Ми, Mg, щелочных и щелочноземельных металлов, W, V, Мо и U; 3) большие количества железа препятствуют осаждению, а олово всегда вызывает затруднения при осаждении и 4) когда в растворах, содержащих Н2О2, присутствует Ti, удовлетворительные результаты получаются лишь при определении малых количеств циркония.
При содержании значительных количеств циркония или когда требуется цысокая точность определения, поступают следующим образом. Прокаленный остаток сплавляют с карбонатом натрия и выщелачивают плав водой. Нерастворимый остаток отфильтровывают, промывают сначала 1%-ным раствором карбоната натрия, затем водой и прокаливают. Сплавляют с пиросульфатом, плав растворяют в разбавленной серной кислоте, раствор кипятят и затем осаждают аммиаком. Отфильтрованный осадок от аммиака промывают горячим 2%-ным раствором нитрата аммония, прокаливают и взвешивают в виде ZrO2- В связи с трудностью количественного отделения фосфат-ионов сплавлением с карбонатом и выщелачиванием плава водой, а также удаления солей щелочных металлов однократным осаждением аммиаком обе эти операции целесообразно повторить.
Цирконий (а также и гафний) осаждается арсенатом аммония из кипящего 2,5 н. солянокислого или 3,75 н. азотнокислого раствора, свободного от серной кислоты 2. Перекись водорода мешает осаждению из азотнокислого раствора. Осадок можно прокаливать до окиси, если в начале прокаливания имеется достаточное количество углерода (желательно добавлять обугленный сахар). Церий (IV), торий, вольфрам, тантал и ниобий препятствуют осаждению из солянокислых растворов, содержащих перекись водорода, поэтому в присутствии этих элементов требуется специальная обработка 3.
'A. Claassen, J. Visser, Rec. trav. chim., 61, 103 (1942); C., 1942, 1, 2304.
2 W. C. S c h u m b, E. J. Nolan, Ind. Eng. Chem., Anal. Ed., 9, 371 (1937).
3 См. также L. Moser, R. Lessing, Monatsh., 45, 323 (1924); V. Coppie-t e r s, Ing. chim., 22, 179, 233 (1938). M. A x t, Ing. Chim., 23, 142 (1939).
Методы определения
643
Купфероновый метод
Осаждение циркония купфероном 1 с последующим прокаливанием осадка до окиси дает точные результаты. Этот метод удобен тем, что в результате прокаливания получается остаток определенного ' состава, который можно взвешивать, и, кроме того, при этом происходит полное отделение циркония от алюминия, хрома, урана (VI), борной кислоты и малых количеств фосфата. Однако определению циркония купфероновым методом препятствует многие элементы, например титан, торий, церий (и, возможно, другие редкоземельные металлы), большинство элементов сероводородной группы, железо, ванадий, ниобий, тантал, вольфрам, кремнекислота и уран (IV).
Осаждение купфероном проводят из сернокислого раствора после отделения кремнекислоты, вольфрама и элементов сероводородной группы. Другие элементы можно предварительно отделять или определять впоследствии в прокаленном остатке, в зависимости от того, что удобнее. Так, например, железо, если оно предварительно не было выделено сульфидом аммония из аммиачного раствора, содержащего винную кислоту, осаждается совместно с цирконием, и содержание его необходимо определить и вычесть из массы прокаленного остатка 2 3. Подобно этому, необходимо также ввести поправку на содержание ванадия, если он не был отделен обработкой щелочами и фильтрованием, например при осаждении едким натром или выщелачивании водой нлава с перекисью или карбонатом натрия. Редкоземельные металлы можно отделить от циркония осаждением фтористоводородной или щавелевой кислотой, но обычно их осаждают, подобно титану, совместно с цирконием, а затем определяют их содержание и вычитают из массы осадка. Осадок купферата циркония Zr [CeH6N(NO)O]4 нельзя высушить и непосредственно взвесить, а необходимо прокалить до окиси. Прокаливать следует крайне осторожно, особенно в начальной стадии, так как при нагревании влажный осадок расплавляется, и в дальнейшем, в процессе сжигания высушенного осадка, происходит обильное выделение газообразных продуктов. Окись циркония не гигроскопична.
Ход определения. Сернокислый раствор сульфата циркония разбавляют до 400 мл доводят концентрацию серной кислоты в растворе до 10% по объему ®, охлаждают до 5—10° С и осаждают избыточным количеством холодного 6%-ного водного раствора купферона. Наличие избытка реагента определяют по появлению тонкого белого растворяющегося осадка вместо нерастворимого творожистого осадка купферата циркония. По цвету осадка легко судить о количестве находящегося в нем титана. Осадок купферата циркония чисто белого цвета, а купферата титана имеет желтую окраску. Коричневый оттенок указывает на наличие в осадке железа или ванадия.
1 О. Baudish, Chem. Ztg., 33, 1298 (1909); W. M. Thornton, E. M. Hayden, Am. J. Sci., [4], 38, 137 (1914) ;G. E. F. L u n d e 11, H. B. Knowles, J. Ind. Eng. Chem., 12, 344 (1920).
2 Согласно С. Л. Ц и н б ергу [Зав. лаб., 4, 735 (1935)], железо отделяется от циркония осаждением оксихинолином из буферированного уксуснокислого раствора, содержащего винную кислоту. Цирконий можно затем полностью осаждать купфероном в присутствии избытка оксихинолина из сильно подкисленного фильтрата.
3 Цирконий осаждается количественно из растворов, содержащих 1% винной кислоты и до 40% серной кислоты по объему.
41*
644
Гл. XXXVH. Цирконий [гафний]
Спустя 5 мин, после прибавления купферона вводят мацерированную бумагу и фильтруют, слабо отсасывая, через бумажный фильтр, помещенный в платиновый конус. Осадок тщательно промывают холодной разбавленной (1 : 10) соляной кислотой. Иногда в фильтрате или промывных водах появляется муть или опалесценция, что указывает на неполноту осаждения. Это можно объяснить лишь применением старого или несоответствующим способом приготовленного реагента. Если после повторного фильтрования через плотный фильтр фильтрат непрозрачен, его упаривают до небольшого объема и разрушают органические, вещества обработкой азотной кислотой. Почти всю азотную кислоту удаляют выпариванием, по Охлаждении разбавляют, фильтруют, если требуется, и затем снова осаждают купфероном.
Осадок вместе с фильтром осторожно сушат и затем прокаливают во навешенном платиновом тигле, под конец приблизительно при 1200° С. Охлаждают в эксикаторе и взвешивают в виде ZrO2.
Поправка на кремнекислоту обычно не нужна, но в точных анализах и в тех случаях, когда цирконий приходится дополнительно извлекать из фильтрата после осаждения купфероном, кремнекислоту необходимо удалить обработкой прокаленного остатка фтористоводородной и серной кислотами. При этом следует прибавить достаточное количество серной кислоты, чтобы воспрепятствовать улетучиванию фторидов циркония и тирана, а при удалении серной кислоты необходимо следить за тем, чтобы не было механических потерь х.
Осаждение в виде основного селенита циркония
Взамен купферонового метода предложено 1 2 осаждать цирконий в виде основного селенита с последующим прокаливанием осадка до ZrO2. Этот метод не так тщательно изучен, как купфероновый, но он имеет то преимущество, что при его применении не требуется предварительного отделения железа й титана. Метод основан на осаждении циркония селенистой кислотой цз горячего разбавленного солянокислого раствора, фильтровании, промывании осадка горячей разбавленной соляной кислотой и прокаливании до ZrOz. Концентрация соляной кислоты в растворе должна составлять от 5 до 7 %, но не выше (по объему)3. Другие кислоты должны отсутствовать. Этим методом цирконий отделяется от алюминия, некоторых редкоземельных элементов, небольших количеств железа, а также от титана, если присутствует перекись водорода. Установлено, что тррий и фосфор загрязняют осадок. Подобным же образом ведут себя, по всей вероятности, тантал и ниобий, а также и некоторые другйе элементы, на что следует обратить внимание при дальнейшем изучении этого метода.
1 См. гл. «Ниобий и тантал» (стр. 663).
2 М. М. S m i t h, C. James, J. Am. Chem. Soc., 42, 1764 (1920).
3 Приводимые ниже результаты, полученные при анализе аликвотных частей раствора хлорида циркония, характеризуют точность этого метода по сравнению с аммиачным и купфероновым:
Аммиачный метод.............
Осаждение купфероном . . . .
Осаждение селенистой кислотой
0,2107 0,2111 — —
0,2102 0,2106 — -
0,2106 0,2108 0,2109 0,2109
Методы, определения
645
Ход анализа. Применительно к анализу циркона метод заключается в следующем. Смешивают 1 г цирконовой руды с 12—15 г бифторида калия 1 в платиновой чашке и осторожно нагревают на небольшом пламени. Когда смесь расплавится, перемешивают платиновой палочкой и постепенно повышают температуру, пока расплав не станет твердым и белым. После этого нагревают на сильном пламени горелки, не поднимая температуру выше, чем это требуется для получения жидкого прозрачного расплава. Охлаждают, прибавляют 50 мл разбавленной (1 : 1) серной кислоты и осторожно нагревают до удаления воды, а затем до появления обильных паров серной кислоты.
Охлаждают, прибавляют/воды и нагревают до растворения солей 2, снова охлаждают, разбавляют точно до 250 мл и перемешивают. Отбирают точно Ю0 мл раствора, разбавляют до 250 мл, нагревают до кипения, осаждают аммиаком и фильтруют. Промытый осадок вместе с фильтром переносят обратно в стакан, в котором проводилось осаждение, и обрабатывают сначала 36 мл соляной кислоты, а затем 40 мл воды. Кипятят до растворения осадка, разбавляют до 700 щл, нагревают до кипения, прибавляют 30—50 мл 12,5%-ного раствора селенистой кислоты 3 и затем кипятят несколько минут. Дают отстояться, после чего осадок отфильтровывают и промывают горячей разбавленной (3 : 97) соляной кислотой, содержащей небольшое количество селенистой кислоты. Фильтр с осад? ком сушат в платиновом или фарфоровом тигле и прокаливают до постоянной массы. Взвешенная ZrO2 почти всегда содержит ТЮг (если при осаждении не вводили перекись водорода), кроме того, ThO2, Nb2O5, Та2О5 и, возможно, окислы других элементов, например SnO2, если они содержались в исходной руде.
Рекомендуется 4 * выделить сначала цирконйй в виде основного селенита, затем осадок растворить в соляной кислоте и переосадить цирконий в виде фосфата. Преимущество этого метода заключается в том, что не требуется промежуточного сплавления и соосажденный торий можно легко отделить в кислом растворе селенита перед осаждением фосфата циркония. Кроме того, в результате двух осаждений разными методами во всех случаях получается более чистый конечный продукт.
Описан 6 также видоизмененный метод, согласно которому цирконий осаждают и определяют в виде нормального селенита Zr(SeO3)2- Этот метод применен для определения циркония и гафния в смеси их окислов’. Осаждают основные селениты 20%-ным раствором селенистой кислоты и затем переводят их в нормальные селениты нагреванием на водяной бане.
1 Для приготовления KHF обрабатывают KF фтористоводородной кислотой, взятой в небольшом избытке, и выпаривают на небольшом пламени горелки до получения прозрачной расплавленной массы. По охлаждении плав измельчают.
2 Если присутствуют фосфаты, нерастворимый остаток отфильтровывают, промывают, прокаливают и сплавляют с карбонатом натрия. Плав выщелачивают водой, остаток отфильтровывают, промывают, растворяют в соляной кислоте и присоединяют к основному раствору.
3 Раствор селенистой кислоты приготовляют следующим способом. Обрабатывают селен азотной кислотой до прекращения выделения окислов азота и затем выпаривают досуха. Полученную двуокись селена очищают возгонкой, после чего ее растворяют в воде и раствор разбавляют до требуемого объема.
4 S. G. S i m р s о п, W. С. S с h u m b, Ind. Eng. Chem., Anal. Ed., 7, 36 (1935).
’ A. С 1 a a s s e n, Z. anal. Chem., 117, 252 (1939).
’ W. C. S c h u m b, F. K. Pittman, Ind. Eng. Chem., Anal. Ed., 14, 512 (1942); E. M. L arsen, W. C. Fernenius, L. L. Quill, Inorganic Sintheses, III, McGraw-Hill Book Co., 1950, p. 69.
-646
Гл. XXXVII. Цирконий [гадбний]
Взвешенную аликвотную часть высушенных селенитов прокаливают до окислов: 0/„.п 374,86 х-0,35702 у
%ню2=----------------
х
тде х — масса окислов циркония и гафния; у — масса селенитов циркония и гафния.
Другие методы
Метод осаждения аммиаком дает хдрошие результаты, но он применим лишь к растворам чистых солей циркония. В несколько меньшей степени это относится к осаждению фенилгидразином и к осаждению сернистой кислотой из нейтрального раствора хлорида х, а также к осаждению йодатом 1 2. Цирконий в количестве микрограммов можно определить ализариновом красным S, с которым он образует розовый лак в растворе 0,2 н. кислоты 3.
* Показано 4, что цирконий, подобно торию и церию, можно выделить в виде йодата постоянного состава, отвечающего формуле 2Zr(IO3)4-KIO3-8H2O. При содержании циркония от 1 до 10 мг определение его с достаточной точностью можно выполнить йодометрическим титрованием этого соединения. При меньших количествах циркония получаются пониженные примерно на 15% результаты. Определение производится так же, как это указано -для тория (см. стр. 607), причем объем анализируемого раствора при содержании 1—4 мг циркония не должен превышать 10 мл, а при больших количествах циркония может достигать 20 Л1Л. Количество вводимого для осаждения йодата калия должно быть не менее 15—20-кратного по отношению к теоретически необходимому для образования йодата циркония.
Рекомендуется также метод 5, основанный на осаждении циркония из солянокислого раствора в виде манделята (соли миндальной кислоты) циркония (GeH6CHOHCOO)4Zr. Определение может быть выполнено в присутствии титана, железа, ванадия, алюминия, хрома, тория, церия, олова, бария, кальция, меди, висмута, сурьмы и кадмия. Содержание свободной серной кислоты в растворе не должно превышать 5 %. Аммонийные соли препятствуют осаждению вследствие образования, как предполагает автор, растворимого комплексного манделята аммония и циркония. Определение сводится к следующему. Раствор, содержащий хлорид или сульфат цирконила в соляной кислоте, в количестве 0,050—0,3 г в пересчете на окись циркония, разбавляют приблизительно до 20 мл концентрированной соляной кислотой. К раствору йрибавляют 50 мл 16 %-ной миндальной кислоты и разбавляют до 100 мл. Температуру раствора медленно повышают до 85° С и нагревают при этой температуре 20 мин. Выделившийся осадок отфильтровывают, промывают горячим раствором, содержащим 2% соляной кислоты и 5% миндальной кислоты, и прокаливают до окиси циркония.
(1894).
1 С. Baskerville, J. Am. Chem. Soc., 16, 475 (1894); Chem. News, 70, 57
2 I. T. D a v i s, Am. Chem. J., 11, 27 (1889).
3 В. H о k, Photometric Determination of Zirkonium, M. S. thesis University of Minnesota, 1949; D. E. Green, Anal. Chem., 20, 370 (1948).
4 Ю. A. Чери и.х OB, T. А. Успенская, Зав. лаб., 10, 248 (1941;.
6 С. А. К u m i n s, Anal. Chem., 19, № 6, 376 (1947).
Методы определения
647
Позднее было найдено1, что из солянокислых растворов миндальной кислотой количественно осаждаются также и малые количества циркония (до 0,1 мг). Имеются указания 2, что относительно небольшие количества циркония (3—30 мг) полностью выделяются из сернокислых растворов [3 мл H2SO4 (пл. 1,4 г/см3) на 65 мл раствора] при продолжительном стоянии раствора (не менее 18 ч) после введения в него осадителя.
Установлено 3, что цирконий количественно осаждается также фталевой кислотой из раствора в 0,3 н. соляной кислоте. В этом случае однократным осаждением цирконий отделяют от большинства элементов, в частности от тория, железа, алюминия, бериллия, урана, марганца, никеля и редкоземельных металлов цериевой группы. В присутствии олова, титана, ванадия и хрома требуется двукратное осаждение. Основанный на этой реакции метод определения циркония заключается в следующем. К раствору хлорида циркония прибавляют 30 мл насыщенного раствора нитрата аммония, а затем вводят достаточное количество 2 н. соляной кислоты, чтобы при последующих операциях после разбавления раствора до 200 мл концентрация соляной кислоты в нем была 0,3 н. Разбавляют до 100 мл, нагревают до кипения и, непрерывно перемешивая, вводят 100 мл кипящего 4%-ного раствора фталевой кислоты. Осторожно кипятят 2 мин, затем нагревают на водяной бане 2 ч, после чего оставляют при комнатной температуре 1 ч. Осадок отфильтровывают и промывают сначала один раз горячим 0,1%-ным раствором фталевой кислоты в 0,3 н. соляной кислоте, а затем 0,1%-ным раствором фталевой кислоты в 2%-ном растворе нитрата аммония. Прокаливают и взвешивают в виде ZrO2.
Метод осаждения фталевой кислотой с успехом применен 4 * для определения, циркония в магниевых и алюминиевых сплавах.
Заслуживает внимания объемный метод определения циркония, основанный на титровании комплексоном III. Показано 6, что цирконий можно определять прямым титрованием комплексоном в кислом растворе (pH = = 1—2) в присутствии эриохромцианина в качестве индикатора или же обратным титрованием введенного избытка комплексона раствором хлорида циркония по эриохромцианину или ализаролцианону. На результаты титрования оказывают влияние Ag+, 'Fe3+, Мо3+, Sb3+, Sn2+, Sn4+, Ti*+, Ti4+, Th4+, WO2-, МоО2-. He мешают перхлорат-, хлорид-, нитрат-и ацетат-ионы. Авторы предполагают, что в значительной степени должны мешать фосфат-, молибдат-, оксалат- и фторид-ионы. Определение проводится следующим способом.
Прямое титрование. Раствор, концентрация циркония в котором должна быть 0,003—0,005 М, нейтрализуют разбавленным раствором аммиака до тех пор, пока pH не станет равным 1,4. Вводят 2 капли 0,4%-ного водного раствора эриохромцианина и титруют 0,05 М раствором комплексона III до изменения окраски индикатора.
В присутствии железа вводят 10 г амальгамированного цинка в 50 мл раствора, содержащего 0,15—0,25 миллимоля циркония ине более 1 миллимоля железа. При этом величина pH раствора должна быть ниже 1,0.
1 R. В. Hahn, Anal. Chem., 21, 1579 (1949).
«С. С. W a s h Ь г о о k, Analyst, 78, 256 (1953).
3 A. Purushottam, Bh. S. V. Raghava Rao, Analyst, 75, 684 (1950).
4 P, С. В о л о д a p с к а я, 3. С. Мухина, Зав. лаб., 19, 782 (1953).
6 J, S. F r i t z, M. O. Fulda, Anal. Chem., 26, 1206 (1954).
648
Гл. XXXVII. Цирконий [гафний}
Перемешивают смесь в течение 5 мин, нейтрализуют раствор разбавленным раствором аммиака до pH = 1,2—1,4, прибавляют 2 капли эрио-хромцианина и, пропуская через раствор струю азота, титруют комплексоном до исчезновения розовой окраски.,
Обратное тит.ройание. Поступают так же, как выше (см. «Прямое титрование»), только титруют быстро и вводят комплексон в небольшом избытке. Доводят pH раствора до 1,4 разбавленным раствором аммиака, нагревают почти до кипения и избыток комплексона оттитро-вывают 0,05 М раствором хлорида циркония, применяя в качестве индикатора эриохромцианин или ализаролцианон, до появления устойчивой розовой окраски.
Медь мешает при прямом титрованиии, но не оказывает влияния, если перед обратным титрованием раствор нагревают несколько минут.
Влияние олова (IV) устраняется введением избыточного количества комплексона в довольно кислый раствор (pH 0,5). Титрование затем проводят при комнатной температуре в присутствии эриохромцианина. Таким же путем устраняется влияние титана (IV). В этом случке следует ввести комплексон в количестве большем, чем это требуется для связывания в комплекс циркония и титана. Раствор затем нагревают до кипения и избыток комплексона оттитровывают раствором хлорида циркония. В присутствии олова или титана конечная точка титрования менее отчетлива.
При наличии в растворе алюминия следует титровать, как указано в разделе «Прямое титрование» (стр. 647).
При комплексометрическом определении циркония избыток комплексона может быть также оттитрован хлоридом железа (III) в присутствии сульфосалициловой кислоты в качестве индикатора, причем величина pH раствора должна быть в пределах от 2,2 до 6. Точку эквивалентности можно определять как спектрофотометрически 1 при 520—525 ммк, так и визуально 2.
В качестве индикатора при комплексометрическом определеййи циркония применен 3 также хромазуроЛ S. Титрование осуществляется в солянокислой среде. Авторы отмечают, что перед титрованием раствор необходимо кипятить для полного переведения ионов Zr4+ в ионы ZrO2+, в противном случае получаются повышенные результаты. Доп. перев*
X
*Колориметрические методы
Помимо упомянутого (стр. 646) колориметрического метода определения циркония с ализариновым красным S, известны и другие методы, в которых также используются реакции образования окрашенных соединений циркония с органическими реагентами.
Определение с хлораниловой кислотой4. В растворе, содержащем хлорную кислоту в 1—2 М концентрации, цирконий реагирует с хлораниловой кислотой, образуя соединение, имеющее яркую красно-фиолетовую окраску. При 2-10-6—5-10"8 М концентрации циркония концентрация хлораниловой кислоты в растворе должна быть не ниже 10-4 М. Светопоглощение окрашенного соединения циркония можно измерять
1 Р. Sweetser, С. Bricker, Anal. Chem., 26, 1, 195 (1954).
2 W. Milner, P. Phennah, Analyst, 79, 941, 475 (1954).
3 A. M i s i 1, M. T h e i s, Z. anal. Chem., 144, 427 (1955).
4 B. J. T h a m e r, A. F. V о i g t, J. Am. Chem. Soc., 73, 3197 (1951).
Методы определения
649
при 330—350 ммк. Оно достигает максймальной величины спустя 15 мин после смешения растворов циркония и хлораниловой кислоты.
В 2 М хлорной кислоте аналогично цирконию с хлораниловой кислотой реагируют гафний, уран (IV), торий и олово (IV). Титан в этих условиях дает слабо-розовое окрашивание, а железо (III) — черно-фиолетовое. Заметной реакции между хлораниловой кислотой и ионами железа (II), хрома (VI), алюминия, меди, кобальта, марганца, бария и калия при этой кислотности не наблюдается.
Определение с тороном [бензол-2-арсоновая кислота-(1-азо-1)-2-окси-нафталин-3,6-дисульфокислота] х. Цирконий образует окрашенное соединение с тороном в минеральнокислой среде. Установлено, что окраска комплексного соединения циркония с тороном стабилизируется спустя 2 ч после введения в раствор реагента, но этот процесс значительно ускоряется при нагревании раствора. Так, окраска достигает максимальной интенсивности, если раствор нагреть до 75° С и затем держать при этой температуре в продолжение 5 мин.
На реакцию циркония с тороном не влияют кобальт, алюминий, медь, свинец, кадмий и титан. Хлорная кислота (70%-ная) не мешает в количествах 0,1—0,5 мл на 10 мл раствора при условии, если измерение проводится не позднее чем через 1 ч после нагревания раствора. При содержании больших количеств хлорной кислоты соединение циркония с то- роном вскоре выделяется в осадок.
Гафний реагирует с тороном аналогично цирконию.
Ход определения; Раствор, содержащий 10—100 мг циркония и 1,5— 2% соляной кислоты, помещают в мерную колбу емкостью 10 мл. В присутствии железа (III), олова (IV) или хромата вводят 5 капель 20%-ного раствора солянокислого гидроксиламина. Затем прибавляют 7 капель концентрированной соляной кислоты и 1,0 мл 0,2%-ного водяного раствора торона. Раствор разбавляют до метки водой, нагревают до 75° С и выдерживают при 75—80° С в течение 5 мин. По охлаждении измеряют светопоглощение раствора при 555 ммк, применяя для сравнения раствор, содержащий в 10 мл дистиллированной воды 7 капель концентрированной соляной кислоты и 1,0 мл реагента. Доп. перев*
* Для колориметрического определения циркония предложен также реактив арсеназо 1 2 (см. «Алюминий», стр. 579), В условиях проведения реакции (pH = 1,5—1,8) мешает ей только титан. Гафний определяется вместе с цирконием. В указанной статье 2 приведены также очень важные соображения авторов по аналитический химии циркония. Доп. ред.*
1 A. D. Horton, Anal. Chem., 25, 1331 (1953).
2 В. И. Кузнецов, Л. М. Буданова, Т. В. Матросова, Зав, лаб., 22, 406 (1956).
Глава XXXVIII
ТИТАН
Титан, считавшийся ранее редким металлом только вследствие отсутствия для него характерной качественной реакции, как выяснилось в настоящее время, является одним из наиболее распространенных в природе элементов. По распространенности он занимает десятое место в ряду элементов, образующих исследованную часть земной коры. Титан присутствует почти во всех изверженйых, метаморфических и осадочных породах, имеющих более или менее силикатный характер. Он распространен главным образом в так называемых основных силикатных породах, но, по-видимому, встречается также и в наиболее кислых породах.
Из многочисленных титановых минералов главными являются р у-т и л ТЮг, октаэдрит, или а н а с т а з (TiO2 другой кристаллической модификации), ильменит FeTiO3, титанит GaTiSiO6 (иногда Fe, Мп) и перовскит (Ga, Fe)TiO3. Титан входит также в состав других минералов и в меньших количествах находится во многих пироксенах, роговых обманках, биотитах, гранатах и в других железомагниевых минералах, а также в некоторых магнетитах и гематитах. Благодаря устойчивости своих соединений титан склонен концентрироваться в остаточных продуктах разложения многих пород, чем объясняется его высокое содержание в глинах, в которых титан редко присутствует в таких малых количествах, которые можно назвать следами. В подавляющем большинстве случаев содержание его не превосходит 1%, но иногда превышает 5%. На содержание титана приходится исследовать, часто количественно, и многие другие вещества, например руды, огнеупорные материалы и продукты металлургического производства, в связи с чем методы отделения и определения титана имеют первостепенное значение.
ОБЩИЕ ЗАМЕЧАНИЯ
В процессе анализа горных пород или силикатных минералов титан частично (но ни в коем случае не полностью) захватывается осадком кремнекислоты, откуда он извлекается и впоследствии количественно попадает в осадок от аммиака. До того как появился достаточно удобный метод определения титана, на его содержание обычно не проводилось даже ка-. чественного испытания. В связи с этим допускались две погрешности, а именно: отсутствие титана в перечне находящихся в анализируемом
Разложение минералов, содержащих титан 651
материале элементов и, как это станет ясным из последующего пояснения, повышенные результаты для алюминия или железа или для обоих этих элементов (в зависимости от метода, применяемого для определения железа, и от присутствующих количеств титана). Так, если железо определяют титрованием перманганатом после восстановления сероводородом или сернистым ангидридом, которые титан не восстанавливают, получаются повышенные результаты определения алюминия. В случае же применения цинка, который восстанавливает Ti4+ до Ti3+, получаются повышенные результаты для Fe2O3 и почти точные для А12О3, если содержание титана невелико, так как результаты пересчета титра перманганата на TiO2 и Fe2O3 почти совпадают. При наличии больших количеств титана окисление его кислородом воздуха перед титрованием^становится заметным, вследствие чего обычно получаются повышенные результаты и для А12О3, и для Fe2O3.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ТИТАН
Вде титановые минералы представляют собой в'значительной степени окисленные соединения. Они, как правило, не полностью разлагаются кислотами, за исключением фтористоводородной, применение которой, естественно, исключает возможность определения кремния. Для разложения этих минералов обычно применяют фтористоводородную кислоту совместно с серной или азотной кислотами. Избыток фтористоводородной кислоты необходимо полностью удалить нагреванием раствора до появления паров серной кислоты. После выпаривания, чтобы обеспечить полное удаление фтора, охлажденный раствор разбавляют водой, растворяют все твердые частицы, особенно находящиеся на стенках чашки (добавив, если это требуется, еще некоторое количество серной кислоты), и снова выпаривают •до появления паров серной кислотых. Эту операцию не следует продолжать до полного или почти полного удаления серной кислоты, чтобы не затруднить последующее растворение солей. При всех операциях надо учитывать склонность солей титана к гидролизу и, если требуется удержать его в растворе, необходимо обеспечить достаточно высокую концентрацию кислоты.
Искусственно полученную окись титана можно перевести в раствор нагреванием с концентрированной серной кислотой и сульфатом аммония или осторожным сплавлением с бисульфатом аммония.
Титановые минералы обычно разлагают сплавлением с реагентами из числа указанных в разделе «Разложение при помощи плавней» (стр. 915). При выборе плавня необходимо учитывать состав минерала, а также преследуемую4 цель.
Хорошие результаты дает сплавление с едким натром или со смесью едкого натра с карбонатом или перекисью натрия. Но так как сплавление с этими реагентами следует проводить в железных или никелевых тиглях, использование их ограничивается областью технического анализа. Для определения, отдельных компонентов это не является препятствием, но при выполнении полного анализа следует предпочесть сплавление с карбонатом натрия.
1 Нитраты и сульфаты титана, вопреки прежним указаниям, не действуют на платиновую посуду при выпаривании с азотной или серной кислотощ
652
Гл. XXXVIII. Титан.
Сплавление с пиросульфатом также, как правило, дает хорошие результаты, но при анализе силикатных пород этот метод менее эффективен *. При выщелачивании пиросульфитного плава холодной водой или разбавленной серной кислотой титан вместе с основаниями переходит в раствор, а большая часть кремнекислоты остается нерастворенной совместно с сульфатами щелочноземельных металлов и свинца. Тантал и ниобий также частично или полностью переходят в осадок, увлекая некоторые количества титана и циркония. Часть кремнекислоты под действием расплавленного пиросульфата образует силикат щелочного металла и при выщелачивании плава переходит в раствор 1 2. Этим обстоятельством пренебрегают в некоторых руководствах и рекомендуют разлагать силикатные материалы и очищать кремнекислоту сплавлением с пиросульфатом.
Титановые минералы полностью разлагаются при сплавлении с би-фторидами щелочных металлов, но при этом исключается возможность определения кремния и щелочных металлов 3. Имеются указания на то, что хорошие результаты дает сплавление с четырехкратным количеством обезвоженной буры с последующим растворением плава в 5 н. серной кислоте 4.
МЕТОДЫ ОТДЕЛЕНИЯ
Обычно нет необходимости в отделении титана от всех сопутствующих ему элементов, и часто вообще никакие разделения не требуются. Это вытекает из того, что двумя основными методами определения титана являются колориметрический, основанный на образовании в растворе окрашенной перекиси титана, и объемный, основанный на предварительном восстановлении титана цинком. Совершенно очевидно, например, что колориметрический метод можно непосредственно применять для определения титана в растворах известняка, не содержащего ванадия, а объемный — для определения титана в рутиле после отделения элементов, восстанавливающихся цинком.
Элементы сероводородной группы отделяют от титана осаждением сероводородом в кислой среде (стр. 83), лучше в присутствии винной кислоты. Если вводят винную кислоту, то дальнейшее отделение титана от железа, кобальта, никеля, цинка и большей или меньшей части марганца можно провести в фильтрате. Для этого фильтрат подщелачивают аммиаком, дополнительно насыщают сероводородом, затем прибавляют бисульфит аммония и фильтруют. Эту операцию лучше выполнять после восстановления железа в кислом растворе, в присутствии достаточного количества винной кислоты, чтобы удержать титан в растворе. Железо можно
1 Согласно G. W. S е а г s и L. Qu i 11 [J. Am. Chem. Soc., 47, 922 (1925) }r титановые руды лучше разлагаются сплавлением с пиросульфатом натрия, чем с пиросульфатом калия. Эти же авторы указывают, что соотношение между количествами анализируемого материала и плавня должно быть не менее 1 : 12,5 в случае анализа рутила и 1 : 35 в случае анализа титанита, а температура сплавления должна быть не выше 700° С.
2 W. F. Hillebrand,!. Am. Chem. Soc., 24, 368 (1902). Извлечение кремнекислоты из раствора см. «Кремний» (стр. 956).
8 При сплавлении с бифторидами титан не улетучивается. Так, например, после осторожного сплавления 0,1821 г TiO2 с KHF2 было определено 0,1820 и 0,1822 г TiO2.
4 Р. Ronchesse, Ann. Soc. sci. Bruxelles, B54, 53 (1934).
Методы отделения
653
почти количественно отделить от титана экстрагированием эфиром из разбавленного солянокислого раствора (стр. 161).
Осаждение купфероном (стр. 143) обычно служит лишь для группового отделения, так как цирконий, редкозмельные металлы (по крайней мере частично), некоторые элементы сероводородной группы, железо, ванадий, вольфрам и уран (IV) выделяются совместно с титаном. Этот метод позволяет, однако, отделять титан от алюминия, хрома, марганца, , никеля и небольших количеств фосфора.
Осаждение титана кипячением сильноуксуснокислого раствора, после предварительного отделения железа, неприемлемо в присутствии циркония. Фосфор при этом также переходит в осадок. Вполне вероятно, что некоторые редкоземельные металлы и другие элементы также осаждаются в этих условиях. Это относится и к осаждению титана кипячением разбавленного солянокислого раствора, содержащего сернистую кислоту.
Цирконий и торий выделяются в осадок при кипячении раствора, содержащего салициловую кислоту и салицилат натрия \ Титан при этом остается в растворе, на чем основано количественное отделение этих элементов от титана, которое выполняют следующим способом.
Азотнокислый раствор возможно более полно нейтрализуют карбонатом натрия, нагревают до кипения и затем по каплям вводят раствор 10 г салицилата аммония в 50 мл воды. Раствор кипятят 30—60 мин, разбавляют кипящей водой до 200 мл, немедленно фильтруют и промывают осадок кипящим концентрированным раствором салицилата аммония, пока он не станет белым или лишь слабо-желтым.
Отделение титана от молибдена, ванадия и фосфора, препятствующих его колориметрическому определению, а также от алюминия и бериллия удобно проводить осаждением едким натром. Титан при этом частично остается в растворе, но в присутствии железа, что обычно имеет место, количественно переходит в осадок. Осаждение титана осуществляют следующим способом.
Кислый раствор почти нейтрализуют едким натром и затем при перемешивании вливают в 200 мл кипящего 5%-ного раствора едкого натра. Кипятят 1—3 мин, фильтруют и промывают осадок горячим разбавленным раствором едкого натра, содержащим сульфат натрия. В присутствии больших количеств молибдена или ванадия осаждение необходимо повторить. Осадок с трудом растворяется в разбавленной кислоте, поэтому его лучше вместе с фильтром обработать азотной и серной кислотами или же прокалить, а затем сплавить с пиросульфатом и растворить плав в разбавленной (1 : 9) серной кислоте. Таким путем происходит также отделение хрома, если перед осаждением едким натром его окисляют персульфатом щелочного металла в слабосернокислом растворе или если к щелочному раствору прибавляют перекись натрия или перекись водорода. В случае введения перекиси раствор необходимо кипятить не менее 10 мин, так как вначале титан удерживается в растворе в виде пертитаната. Вместо осаждения едким натром можно применить сплавление с карбонатом натрия и выщелачивание плава водой. Если сплавление и выщелачивание требуется повторить,, нерастворимый в воде остаток перед прокаливанием целесообразно обработать при кипячении разбавленной соляной кислотой, а затем прибавить раствор аммиака в небольшом избытке и
1 М. Dittrich, S. Freund, Z. anorg. Chem., 56, 344 (1908).
654
Гл. XXXVIII. Титан
профильтровать. Сплавлением с едким натром с последующим выщелачиванием плава водой можно пользоваться лишь в присутствии железа, в противном случае имеют место значительные потери титана1.
Небольшие количества ниобия и тантала можно отделить от титана сплавлением смеси окислов с пиросульфатом калия и обработкой плава при кипячении раствором таннина в разбавленной серной кислоте. Осадок ниобия и тантала , отфильтровывают.
При содержании ниобия и тантала в количестве сотых долей грамма плав растворяют в растворе винной кислоты, затем кипятят с разбавленной азотной кислотой и выделяющиеся при этом окислы ниобия и тантала отфильтровывают. Более четкое отделение титана от ниобия и тантала достигается при растворении пиросульфатного плава в горячем концентрированном растворе оксалата аммония с последующей обработкой раствора сначала салициловой кислотой, а затем хлоридом кальция 2.
* Следует упомянуть также методы, основанные на гидролитическом осаждении титана слабыми органическими основаниями, например уротропином 3 и пиридином 4.
При осаждении уротропином в растворе устанавливается pH 5—5,5. В этих условиях титан отделяется от никеля, кобальта и марганца. При введении в раствор аммонийных солей происходит также отделение титана от редкоземельных элементов, не осаждающихся уротропином в присутствии солей аммония. Метод имеет довольно ограниченное применение, так как не позволяет отделять титан от таких элементов, как железо (III), алюминий, медь, хром, уран, цирконий, торий и бериллий, которые выделяются из раствора при pH ниже 5. Имеется указание 5 об использовании уротропина при анализе легированных сталей для совместного отделения титана и ниобия от железа, предварительно восстановленного до двухвалентного состояния. Применение пиридина, создающего в растворе pH около 6, предложено Э. А. Остроумовым 6 для отделения железа, алюминия., титана и других элементов от марганца, кобальта, никеля, щелочных и щелочноземельных металлов. Доп. перев*
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наиболее важными методами определения титана являются колориметрический, основанный на реакции с перекисью водорода, и объемный, заключающийся в восстановлении титана цинком и последующем титровании перманганатом.
Колориметрический метод применим к растворам, содержащим не более 0,1 мг TiO2 в 1 мл, и, следовательно, наиболее подходит для анализа горных пород и глин, в которых, как правило, находятся малые количества Читана. Объемным методом обычно пользуются, если в анализируе
1 F, A. Gooch, Proc. Am. Acad. Arts. Sci. [New Ser.J, 12, 436 (1884—1885); W. F. H ill e bran d, U. S. Geol. Survey Bull., 700, 132. См. также Z. Auger, C. r., 177, 1302 (1923).
2 Подробно см. W. R. Schoeller, The Analytical Chemistry of Tantalum and Niobium, 1937, p. 109; W. R. Schoeller, C. Jahn, Analyst, 57, 72 (1932); 54, 320 (1929).
3 P. R a у Z. anal. Chem., 86, 13 (1936).
4Э. А. Остроумов, Зав. лаб., 6, 16 (1937).
ЛК. Traub, Ind. Eng. Chem. Anal. Ed., 18, 122 (1946).
6 Э. А. Остроумов, цит. выше.
Методы определения
655
мой пробе содержится свыше 5% титана. Весовые методы, основанные на осаждении едкими щелочами, сульфидами щелочных металлов, купфероном, кипячением уксуснокислых 1 или солянокислых растворов 2, недостаточно селективны, чтобы ими можно было успешно пользоваться для анализа сложных продуктов.
При приготовлении растворов для определения титана необходимо учитывать, что в случае недостаточно высокой кислотности может происходить гидролитическое осаждение титана, особенно из нагретых растворов. Кроме того, следует иметь в виду, что фосфат титана трудно растворим в кислоте и склонен выделяться из растворов, в которых одновременно присутствуют титан и фосфор. При растворении гидроокиси титана на фильтре необходимо соблюдать известные меры предосторожности, так как значительные количества незаметного для глаза осадка могут оставаться на бумаге несмотря на тщательное ее промывание.
Колориметрический метод
Общие замечания. Колориметрический метод определения дитана основан на сравнении интенсивности окраски, появляющейся при добавлении перекиси водорода к разбавленному сернокислому раствору анализируемой пробы, с интенсивностью окраски стандартного раствора сульфата титана, в который также введена перекись водорода. При анализе горных пород это определение обычно проводят после определения железа в сернокислом растворе, полученном после сплавления прокаленного и взвешенного осадка от аммиака с пиросульфатом калия и растворения плава в разбавленной серной кислоте (см. гл. LIII, стр. 955). Испытание на титан, естественно, можно провести и до этой операции. При применении колориметрического метода необходимо соблюдать следующие условия.
Концентрацию применяемой перекиси водорода (которая не обязательно должна быть выше 3%-ной) следует определять титрованием перманганатом после откупоривания каждой склянки и затем проверять через определенные промежутки времени, в противном случае могут происходить серьезные ошибки вследствие разложения реагента. Перекись водорода иногда содержит фтор, поэтому отсутствие его должно быть тщательно установлено (стр. 63).
Перекись водорода.следует прибавлять к раствору, содержащему не менее 5% по объему серной кислоты, в противном случае титан может полностью не окислиться. Интенсивность окраски возрастает с повышением температуры, Поэтому сравниваемые друг с другом растворы должны иметь одинаковую температуру, лучше всего 20—25° С. .
Определению препятствуют следующие элементы: 1) железо, никель, хром и другие элементы, растворы солей которых окрашены; 2) ванадий, молибден, ниобий 3, а в определенных условиях и хром, образующие
1 F. A. Gooch, Proc. Am< Acad. Arts. Sci. [New Ser.), 12, 436 (1884—1885); T. M. C h a t a r d, Am. Chem. J., ‘13, 106 (1891); Chem. News, 52, 55, 68 (1885); 63, 267 (1891); W. F. H illebr a nd, U. S. Geol. Survey Bull., 700, 162.
2 C. Baskerville,!. Am. Chem. Soc., 16, 427, 475 (1894).
3 Ниобий дает бледно-желтое окрашивание с перекисью водорода в растворе, содержащем менее 25% (по массе) серной кислоты. Интенсивность окраски возрастает с повышением кислотности раствора и достигает максимума в 100%-ной кислоте (d15= = 1,845). Интенсивность же окраски соединения титана постепенно возрастает с повышением кислотности раствора до 25% и резко понижается, но не полностью исчезает в 100%-ной кислоте.
€56
ГЛ. XXXVIII. Титан
окрашенные соединения с перекисью водорода; 3) фтор (даже незначительные количества), большие количества солей щелочных металлов и фосфаты, обесцвечивающие растворы надтитановой кислоты \
Влияние элементов, дающих окрашенные растворы при невысоком их содержании, можно устранить введением таких же количеств этих элементов в стандартный раствор перед добавлением перекиси водорода. При значительном содержании железа в анализируемый и стандартный растворы, после прибавления перекиси водорода, вводят одинаковые количества фосфорной кислоты. Большие количества других окрашенных ионов следует предварительно отделять.
Из числа мешающих элементрв второй группы ванадий и молибден легко отделяются при осаждении титана едким натром в присутствии небольшого количества железа.
Из элементов, обесцвечивающих окраску титана, наиболее опасным является фтор, но его можно легко удалить повторным нагреванием с серной кислотой. Действие солей щелочных металлов менее заметно при содержании в растворе 10% (по объему) серной кислоты, чем при 5%. Поэтому, если присутствуют небольшие количества щелочных металлов, следует повысить концентрацию кислоты в анализируемом и стандартном растворах и, кроме того, в стандартный раствор вводить такое же количество этих солей. От значительных количеств солей щелочных металлов титан отделяют осаждением аммиаком, после чего осадок гидроокиси титана растворяют в серной кислоте.
Влияние малых количеств фосфора можно избежать введением соответствующего количества фосфорной кислоты в стандартный раствор. В присутствии больших количеств фосфорной кислоты, наряду со значительным содержанием титана, может оказаться затруднительным получение прозрачного раствора вследствие образования нерастворимого фосфата титана. В таких случаях необходимо предварительно осадить титан едким натром.
Приготовление стандартного раствора. Вполне успешно применяются стандартные растворы, приготовленные из стандартного образца двуокиси титана № 154 (содержащего 98,7% TiO2) (Бюро Стандартов США). Эти растворы удобно приготовлять следующим способом.
Отдельные навески двуокиси титана растворяют в смеси концентрированной серной кислоты и сульфата аммония, разбавляют, фильтруют, промывают фильтр и после этого раствор разбавляют до определенного объема.
Стандартный раствор можно приготовить также из фторотитаната калия K2TiF6-2H2O, для чего некоторое количество этой соли несколько раз перекристаллизовывают в платиновой чашке из кипящего водного раствора. Кристаллы высушивают и сохраняют в склянке с притертой пробкой. Грубо отвешивают несколько большее количество соли, чем это соответствует 1 г TiO2, обрабатывают в платиновой чашке 100 мл разбавленной (1:1) серной кислоты и выпаривают до появления обильных паров серной кислоты. По охлаждении ополаскивают стенки чашки и снова выпаривают. Эту операцию повторяют, чтобы обеспечить полное
1 Согласно R. D. Hall [J. Am. Chem. Soc., 26, 1241 (1904)], окраски титана обесцвечиваются лимонной кислотой. Действие винной и щавелевой кислот менее заметно. Янтарная кислота также заметно не влияет на окраску.
Методы определения.
657
удаление фтористоводородной кислоты. По охлаждении раствор разбавляют до 1 л. Отбирают несколько аликвотных частей раствора по 50 мл, разбавляют каждую из них до 200 мл, нагревают до кипения и прибавляют раствор аммиака до слабощелочной реакции. Кипячение продолжают 1—3 мин, затем фильтруют и промывают осадок горячей водой до удаления солей щелочных металлов. Влажный фильтр с осадком прокаливают (под конец при 1100—1200° С) до постоянной массы. Вычисляют содержание титана в 1 мл раствора и результат записывают на склянке, в которой хранят раствор. Пробка склянки должна быть смазана вазелином. Необходимое количество раствора не отливают, а отбирают сухой пипеткой. В приготовленном таким способом растворе солей щелочных металлов содержится так мало, что они не могут оказать влияния на окраску раствора после прибавления к нему перекиси водорода.
Полученный при этом титр раствора несколько повышен, так как гидроокись титана увлекает небольшие количества калия. Для рядовых анализов это, однако, не имеет значения. Для более точных анализов осаждение аммиаком лучше повторить 2—3 раза или же осадить титан купфероном. При такой обработке получаются близко совпадающие результаты. Так, например, после трехкратного осаждения аммиаком, причем первые два осадка растворялись в разбавленной соляной кислоте, а тщательно промытые фильтры сжигались совместно с окончательно полученным осадком, в двух порциях раствора по 25 г было определено 0,002008 и 0,002004 г TiOa на 1 г раствора, а однократным осаждением купфероном в двух порциях по 25 г того же раствора было найдено 0,002008 и 0,002002 г ТЮ2 на 1 г раствора.
Описан1 метод, согласно которому чистую TiOa получают из TiCl4 и затем растворяют кипячением с концентрированной серной кислотой и сульфатом аммония.
В качестве исходного вещества рекомендуют 2 пользоваться K2TiO(C2O4),- 2Н2О. Для этой цели продажную соль перекристаллизовывают один раз из водного раствора и сушат при комнатной температуре несколько дней. Смешивают 4,5 г соли (соответствует приблизительно 1 г TiOa) с 8 г сульфата аммония и обрабатывают в колбе Кьельдаля 100 мл концентрированной серной кислоты. Постепенно нагревают до кипения и кипятят 10—15 мин. По охлаждении разбавляют приблизительно до 1 л и, если раствор непрозрачен, фильтруют. Испытывают порцию раствора с перманганатом, чтобы удостовериться в отсутствии щавелевой кислоты, которая обесцвечивает окраску надтитановой кислоты, и затем определяют содержание титана обычным способом.
Приготовление анализируемого раствора. Желательно, чтобы титан находился в виде сульфата в сернокислом растворе, свободном от влияющих на"колориметрирование элементов, перечисленных в разделе «Общие замечания» (стр. 651). Для колориметрического определения можно использовать сконцентрированный раствор, сохраненный после определения железа в осадке от аммиака титрованием перманганатом (стр. 958), при условии, если в него не вводились другие кислоты, кроме серной. Присутствие марганца, введенного при титровании железа, не влияет на колориметрическое определение титана. Непосредственное использование этого раствора нежелательно, когда в нем содержатся ванадий и значительные количества фосфора. Из этих соображений, а также для отделения солей щелочных. металлов, введенных при сплавлении осадка от аммиака (стр. 955), титан лучше сначала выделить из анализируемого раствора едким натром (стр. 110). Если в анализируемом растворе находятся только соли щелочных металлов, удовлетворительные результаты получаются, когда в стандартный раствор вводят такое же количество
1 W. W. Plechner, J. М. J ar mus, Ind. Eng. Chem., Anal. Ed., 6, 447 (1934).
2 W. M. Thornton Jr., R. Roseman, Am. J. Sci., 20, 14 (1930).
42 Заказ 522.
658
Гл. XXXVIII. Титан
этих солей и сравнивают окраски растворов, содержащих 10% серной кислоты (по объему). Но, несмотря на это, лучше титан осадить аммиаком, осадок отфильтровать, промыть и после этого растворить в разбавленной серной кислоте.
Для колориметрического определения титана можно использовать растворы, полученные непосредственно после разложения анализируемой пробы или растворения некоторых взвешенных прокаленных остатков, учитывая при этом наличие мешающих элементов, перечисленных выше (стр. 655). Так, например, при использовании раствора после разложения породы фтористоводородной и серной кислотами необходимо обеспечить полное удаление фтора, а применяя раствор-купфератов, следует иметь в виду возможное присутствие ванадия.
Сравнение окрасок. Сернокислый раствор сульфата титана упаривают до объема менее 100 мл и затем полностью окисляют перекисью водорода. Если появляющаяся при этом окраска менее интенсивна, чем это соответствует 0,01 г ТЮг, прибавляют серную кислоту с таким расчетом, чтобы общее содержание ее в растворе было равно 5 мл, и разбавляют раствор в мерной колбе до 100 мл. Если же окраска раствора более интенсивна, разбавляют до большего объема и вводят серную кислоту в таком количестве, чтобы концентрация ее в растворе составляла 5% (по объему). Сравнение интенсивности окрасок затем можно легко выполнить в фотометрах, спектрофотометрах или простых колориметрах 1 (стр. 50). Если предполагают присутствие ванадия, то по окончании колориметрирования вводят в раствор фтористоводородную кислоту.. Окраска титана при этом исчезает, тогДЬ как коричневато-красная окраска, обусловленная присутствием ванадия, остается 2.
*Окрашенное соединение титана с перекисью водорода образуется не только в сернокислой среде, но и в растворах, содержащих другие минеральные кислоты, что дает возможность проводить определение титана в присутствии той кислоты, какую наиболее удобно применять для приготовления анализируемого раствора. Так, например, описано качественное открытие титана по реакции с перекисью водорода в азотнокислой среде 3. Э. И. Нагерова и А. Д. Лебедева при анализе цемента колориметрируют титан в солянокислом растворе 4 5. Некоторые авторы Б, определяя титан в легированных сталях, проводят колориметрирование в растворах, содержащих хлорную кислоту. Доп. перев.*
Имеются указания 6, что метод определения малых количеств титана « 1 мг Ti на 100 мл), основанный на появлении интенсивной красной
1 G. Н. Ayr е s и Е. М. V ienneau [Ind. Eng. Chem., Anal. Ed., 12, 96 (1940)] установили, что желтая окраска, появляющаяся при взаимодействии титана с перекисью водорода, не изменяется в продолжение по меньшей мере 2 лет.
2 М. Schenk [Analyst, 61, 872 (1936)] установил, что реакция титана с салициловой кислотой в 86%-ной (по массе) серной кислоте, сопровождающаяся появлением интенсивной красной окраски, в 5 раз чувствительнее реакции титана с перекисью водорода. Азотная, азотистая и марганцевая кислоты мешают этой реакции.
3 А. Н. Н о й е с, В. Брэй, Качественный анализ редких элементов, ОНТИ, 1936.
4 Э. И. Нагерова, А. Д. Лебедева, Зав. лаб., 8, 10 (1939).
5 С. М. Г у т м а н, Э. М. Загорацкая, 3. А. В ы р о п а е в а, Зав. лаб., 9, 1, 101 (1940); A. Weissler, Ind. Eng. Chem., Anal. Ed., 17, 775 (1945).
• P. К 1 i n g e r, W. Koch, Tech. Mitt. Krupp Forschungsber., 14, 179 (1939); Arch., Eisenhuttenw., 13, 127 (1939).
Методы определения
659
окраски при взаимодействии титана (IV) с хромотроповой кислотой, дает вполне удовлетворительные результаты. Этим методом определение титана в смеси окислов ниобия и тантала осуществляют следующим способом.
Осадок сплавляют с пиросульфатом калия, плав растворяют в 2%-ном растворе щавелевой кислоты и полученный раствор разбавляют до 80 мл. Вводят 10 мл 6%-ного водного раствора хромотроповой кислоты, разбавляют до 100 мл и определяют светопоглощение в фотоколориметре, пользуясь соответствующим светофильтром (470 ммк) и применяя раствор одних реактивов в качестве нулевого раствора. Содержанием титана вычисляют по калибровочной кривой, построенной по растворам, содержащим определенные количества титана. Олово (II), железо (III), уранил и нитраты мешают определению. Цирконий, молибден, ниобий и тантал не оказывают влияния на реакцию. В соответствующих условиях реакцию с хромотроповой кислотой можно использовать для определения титана в простых и легированных сталях *.
*Хромотроповая кислота первоначально была предложена для качественного открытия титана Н. А. Тананаевым и Г. А. Панченкоа, а затем Г. А. Панченко и М. В. Раецкий 3 на основе этой реакции предложили колориметрический метод определения титана. Эти авторы, применив свой метод к определению титана в чугуне и стали, устраняют влияние железа предварительным восстановлением его амальгамой цинка. Для этой цели можно пользоваться также сульфитом натрия4. Основную массу железа (II) рекомендуется 4 предварительно отделять осаждением титана купфероном. Доп. перев.*
Объемный метод
В сернокислом или солянокислом растворе титан быстро восстанавливается до трехвалентного состояния цинком в редукторе Джонса. Трехвалентный титан, однако, очень легко окисляется на воздухе, в связи с чем раствор после восстановления непосредственно вливают в раствор сульфата железа (III). В результате этого титан немедленно окисляется до четырехвалентного с образованием эквивалентного количества более устойчивого сульфата железа (II) Б.
Все элементы и соединения, образующие растворимые продукты восстановления, способные окисляться перманганатом, должны быть отделены, если их влияние на результаты титрования нельзя точно учесть. К числу восстанавливающихся цинком соединений и элементов относятся некоторые органические вещества, азотная кислота, олово, мышьяк, сурьма, молибден, железо, хром, ванадий, вольфрам, уран и ниобий.
1 Р. Klinger, W. Koch, Tech. Mitt. Krupp Forschungsber., 14, 179(1939); Arch. Eisenhiittenw., 13, 127 (1939).
2 H. A. T а н а н a e в, Г. А. П а н ч e н к о, Z. anorg. u. allg. Chem., 150, 163 (1926).
3 Г. А. П а н ч e н к о, M. . В. Раецкий, ЖПХ, 8, 4, 718 (1935).
4 Б. А. Генерозов, Зав. лаб., 18, 161 (1952).
5 A. S. R u s s е 11 [J. Chem. Soc., 129, 497 (1926)] рекомендует восстанавливать титан в 4 н. сернокислом растворе, так как в этих условиях титан (III) окисляется воздухом лишь очень медленно. Метод открытия титана (IV) в присутствии титана (III), основанный на образований окрашенного соединения титана (IV) с хромотроповой кислотой, пирокатехином и другими полифенолами в слабокислой среде, см. В. И, К у з-н е ц о в, ЖОХ, 14, 902 (1944).
42*
660
Гл. XXXVIII. Титан
Органические вещества и азотную кислоту можно разрушить выпариванием раствора с серной кислотой, обычно двукратным. В промежутке между выпариваниями раствор охлаждают и обмывают стенкй сосуда водой. Для разрушения некоторых стойких органических веществ в кипящий концентрированный сернокислый раствор анализируемой пробы осторожно по каплям через носик покрытого часовым стеклом стакана вводят насыщенный раствор перманганата.
Олово, мышьяк, сурьму и молибден можно легко удалить осаждением сероводородом из холодного раствора, кислотность которого должна быть достаточно высокой, чтобы воспрепятствовать гидролитическому осаждению титана. В результате введения сероводорода в растворе образуются политионовые соединения *, которые, после удаления сероводорода кипячением, разрушают введением перманганата до появления устойчивой розовой окраски.
Железо можно отделить осаждением сульфидом аммония из аммиачного раствора, содержащего винную кислоту, которую затем следует разрушить (стр. 90).
Хром, ванадий, уран и вольфрам, после окисления до высших степеней валентности, отделяются при осаждении титана (повторном, если требуется) едким натром.
В отсутствие железа титан едким натром осаждается не количественно, в связи с чем эта операция должна предшествовать отделению железа.
Достаточно удовлетворительный метод для отделения ниобия от титана неизвестен, поэтому в присутствии ниобия объемный метод определения титана неприменим.
Ход определения. Сернокислый или солянокислый (лучше сернокислый) раствор солей титана, приготовленный с учетом того, что изложено выше, восстанавливают, как указано на стр. 135, и титруют 0,1 или 0,05 н. раствором перманганата. Вычисляют объем перманганата, который расходуется на титрование реактивов, обработанных так же, как анализируемый раствор.
В тех случаях, когда размеры столба амальгамированного цинка меньше рекомендуемых, следует установить, какие факторы должны быть изменены, чтобы достигнуть полного восстановления титана. Так, например, при более низком столбе цинка следует с меньшей скоростью пропускать раствор через редуктор или нагреть раствор до 40—60° С. О применении цинковой амальгамы для восстановления титана см. стр. 141.
*Необходимо указать, что для объемного определения титана в присутствии ниобия рекомендуется 1 2 вводить в раствор фторид натрия (2,5 г NaF), который препятствует восстановлению ниобия и не влияет на восстановление титана. . Определение титана заканчивают титрованием в токе углекислого газа растворов соли железа (III) в присутствии роданида в качестве индикатора 3.
В присутствии ниобия предложено 4 * также восстанавливать титан •металлическим железом, не восстанавливающим ниобий. Для этой цели 0,2 г TiO2 растворяют в 10 мл концентрированной серной кислоты, содержащих 4 г (NH4)2SO4. По охлаждении раствор разбавляют 100 мл
1 G., Е. F. Lundell, Н. В. Knowles, J. Am. Chem. Soc., 43, 1563 (1921).
2 Z, Winterstein, Z. anal. Chem., 119, 385 (1940).
,8 См. сноску 2 на стр. 662.
4 F. В i s c h о f f, Z. anal. Chem., 130, 195 (1950); Microchim. Acta, 36/37,251
(1951).
Методы, определения
661
разбавленной (1 : 5) серной кислоты. В коническую колбу емкостью 750 мл, из которой воздух предварительно вытеснен током двуокиси углерода, вводят 4—5 г чистого металлического железа (порошок или проволоку). После этого пропускают ток двуокиси углерода в течение 10 мин, сливают анализируемый раствор в колбу и прибавляют еще 20 мл разбавленной (1 : 1) серной кислоты. Оставляют стоять при комнатной температуре 5—10 мин, часто перемешивая раствор, затем нагревают, при непрерывном перемешивании жидкости, до тех пор, пока все железо не растворится и раствор не закипит. Кипятят 5 мцн и охлаждают в токе двуокйси углерода и в этой же атмосфере титруют 0,1 н. раствором сульфата железа (III) в присутствии 10 мл 5%-ного раствора роданида калия. Индикатор целесообразно вводить к концу титрования, когда начинает исчезать фиолетовая окраска титана (III). Доп. перев.*
Осаждение купфероном
Титан количественно осаждается купфероном из разбавленных сернокислых растворов 1 и отделяется таким образом от алюминия, хрома, урана (VI), фосфора, никеля и щелочноземельных металлов. Единственным недостатком этого метода является то, что многие другие элементы также осаждаются купфероном. Например, железо, цирконий, ниобий, тацтал и ванадий осаждаются количественно, а некоторые редкоземельные металлы, вольфрам и элементы сероводородной группы — частично. Купфероновый и объемный методы в основном отличаются друг от друга тем, что применению объемного метода не препятствует цирконий, редкоземельные металлы и тантал, а применению купферонового метода — уран (VI).
Ход, определения. Приготовляют солянокислый или сернокислый раствор, свободный от элементов сероводородной группы и содержащий все элементы в их обычной валентности, а затем поступают, как описано на стр. 143, включая прокаливание и взвешивание осадка.
Когда состав анализируемой пробы неизвестен, осадок необходимо , исследовать на содержание посторонних элементов, которые не были предварительно удалены.
Гидролитическое осаждение из уксуснокислых или солянокислых растворов
Метод отделения титана от алюминия осаждением из кипящего ацетатного раствора 2, после удаления железа и фосфора, впоследствии несколько видоизмененный 3, долгое время являлся незаменимым в анализе горных пород. В настоящее время этим методом пользуются редко, отчасти потому, что имеются более надежные колориметрический и объемный методы, а отчасти потому, что цирконий препятствует полному осаждению титана 4.
Применению метода осаждения титана, основанного на кипячении слабо солянокислого раствора 5, препятствуют: цирконий, который
1 Титан количественно осаждается купфероном из растворов, содержащих до 40% по объему серной кислоты и 1% винной кислоты.
2 F. A. Gooch, Proc. Am. Acad. Arts. Sci. [New Ser.], 12, 436 (1884—1885).
3 T. M. C hat ar d, Am. Chem, J., 13, 106 (1891); W. F. Hillebrand, U. S. Geol. Survey Bull., 700, 132.
♦ W. F: Hillebrand, U. S. Geol. Survey Bull., 700, 163.
1 C. Baskerville, J. Am. Chem. Soc., 16, 427, 475 (1894).
662
Гл. XXXVIII. Титан
осаждается полностью или частично, в зависимости от кислотности раствора, фосфор и, вероятно, еще некоторые другие элементы, поведение которых не изучалось.
Другие методы
Из других методов определения титана важнейшими являются объемные методы, основанные на восстановлении титана цинком и последующем титровании метиленовой синью1 или сульфатом железа (III) в присутствии роданида калия в качестве индикатора 2. Из этих двух методов следует предпочесть титрование метиленовой синью, так как при этом получается более отчетливая конечная точка титрования. В обоих случаях возможны ошибки, если не принять особых Мер предосторожности, чтобы воспрепятствовать окислению трехвалетного титана до и во время титрования.
Эти методы менее надежны, чем объемный метод, изложенный на стр. 659, но они обладают тем преимуществом, что ими можно пользоваться в присутствии железа. При использовании для титрования метиленовой сини солянокислый раствор хлорида титана восстанавливают цинком, предпочтительно в редукторе Джонса (стр. 135). Полученный после восстановления раствор защищают от действия воздуха, создавая атмосферу двуокиси углерода, и титруют раствором метиленовой сини до появления неисчезающей голубой окраски. Восстанавливать и титровать лучше горячие растворы. Присутствие азотной и серной кислот нежелательно, так как они - затрудняют определение конечной точки титрования. Мешают титрованию также молибден, ванадий, вольфрам, хром и олово, которые реагируют с метиленовой синью. Метод применим в присутствии кремния, железа, алюминия, сурьмы, мышьяка и фосфора.
Раствор метиленовой сини приготовляют растворением 3,9—7,8 г реагента в 1 л воды. 1 мл раствора соответствует 1—2 мг Ti. Титр этого раствора устанавливают по титрованному раствору хлорида титана, обработанному, как указано в приведенном выше методе 3.
*Для усовершенствования объемного метода определения титана известный интерес представляют исследования 4, показавшие, что сульфат аммония и уксусная кислота в сернокислое растворе связывают ион титана (III) в прочный комплекс, благодаря чему происходит значительный сдвиг потенциала системы в положительную сторону и раствор восстановленного титана становится более устойчивым по отношению к кислороду воздуха. Таким образом, применив указанные комплексо-образователи, можно избежать необходимости проводить титрование титана в атмосфере инертного газа, что делает определение более простым и вместе с тем более надежным. Доп. перев*
1 Е. Hibbert, J. Soc. Chem. Ind., 28, 189 1(1909); В. Neumann, R. К. Murphy, Z. angew. Chem., 26, T. 1, 613 (1913).
2 Knecht, Hibbert, Ber., 36, 1550 (1903); G. Gallo, Atti Acad. nazl. Lincei, 16, pt. 1, 325 (1907); E. Knecht, Z. angew. Chem., 26, T. 1, 734 (1913).
3 Методы анализа различных материален, содержащих титан, подробно описаны в монографии W. М. Thornton Jr., Titanium, American Chemical Society Monograph Series, Chemical Katalog Co., New York, 1927.
4B. С. Сырокомский, E. В. Силаев, В. Б. Авилов, Зав. лаб., 15, 896 (1949).
Глава XXXIX
НИОБИЙ И ТАНТАЛ
Ниобий и тантал являются редкими кислотообразующими элементами. В природе они обычно встречаются совместно, образуя соли с железом, марганцем, кальцием, ураном и редкоземельными металлами. Типичными представителями минералов, содержащих ниобий и тантал, являются: колумбит (Fe, Mn)(NbO3)2, танталит (Fe, Мп)(ТаО3)2 и самарскит R3T R2n(Nb, Та)вО21, где R11 — Fe, Са, UO2, a R111 — редкоземельные металлы. Все эти минералы распространены главным образом в пегматитовых жилах \ но находятся также и в некоторых гранитах. Танталит иногда встречается совместно с фольфрамитом и касситеритом.
ОБЩИЕ ЗАМЕЧАНИЯ
По отношению к пятиокисям ниобия и тантала некоторыми авторами применяется термин «земельные кислоты». Подобно тому, как торий обычно рассматривается совместно с группой редкоземельных металлов, так и титан иногда относят к группе «земельных кислот» на том основании, что эти три элемента, помимо того, что тесно связаны друг с другом в природе, обладают некоторыми общими химическими свойствами, играющими важную роль в аналитической химии. Характерной особенностью этих металлов является сильная склонность их солей к гидролизу, что дает возможность отделять их от многих других элементов. Природные титанаты, свободные от ниобия и тантала, представляют собой обычное явление; ниобаты и танталаты также встречаются без титана, но как будто неизвестен в природе ниобат, совершенно свободный от тантала, так же как и танталат, не содержащий ниобия. В немногих, редко встречающихся минералах фосфор (V), мышьяк и сурьма частично замещают ниобий и тантал. Вольфрам и олово в тантало-ниобиевых минералах встречаются часто, но всегда в малых количествах.
Из других кислотных компонентов этих минералов важное значение имеет кремнекислота. Часто встречается цирконий, функционируя то как кислота, то как основание в соответствии с его амфотерным характером. Гафний, по всей вероятности, сопровождает цирконий. Встречаются
770.
1 F. W. С 1 а г k е, The Data of Geochemistry, 5th ed,, U, S, Geol, Survey Bull.,
664 Гл. XXXIX. Ниобий и тантал
также бар, фтор, хлор и двуокись углерода. В одном и том же минерале могут находиться-несколько кислотообразующих элементов. Из металлов для одних минералов характерно содержание железа и марганца (как, например, для относительно простых — колумбита и танталита), а для других — кальция (перовскит, титанит). Редкоземельные элементы и торий входят в состав многих минералов в качестве основных или второстепенных компонентов. Нередко встречаются также уран и бериллий. В минералах, содержащих торий и уран, возможно присутствие азота и гелия. Был найден также и германий. Большинство обычных элементов находится в этих минералах в рассеянном состоянии.
Можно отметить, что мышьяк и сурьма не встречаются в титановых минералах, не содержащих ниобия или тантала, тогда как бор скорее, связан с кремнием [каппеленит Ba3Y9(BO3)5(SiO3)9, гомилит (Са, Fe)3(BO)2(SiO4)2, тритотцит] или с титаном (ворвикит), чем с ниобием или танталом. Фтор (возможно и хлор) в этих минералах (за исключением простых фторидов и фторокарбонатов) обычно рассматривается как составная часть кремниевого или титанового комплекса. Некоторые минералоги причисляют фтор и хлор к основным оксикомплексам. Бор встречается преимущественно в форме боратов, но в случае наличия фтора не исключена возможность существования и комплексных фтороборатов.
Из изложенного следует, что многие минералы этой группы имеют чрезвычайно сложный состав й точно установить соотношение между образующими их компонентами крайне затруднительно. Многие фор- • мулы, которые приписывают этого типа минералам, являются гипотетическими и основаны на предположениях, которые часто не представляется возможным проверить.
Прежде чем приступить к количественному анализу минералов, в данном случае, более чем когда-либо, важно провести сначала тщательное качественное исследование материала, если возможно спектрографическое. Полученные при этом сведения могут иметь существенное значение для выбора наиболее рациональных методов разложения пробы и разделения содержащихся в ней элементов. Нередко, однако, имеющегося в наличии количества материала недостаточно для исчерпывающих предварительных испытаний, что может явиться причиной возникновения серьезных затруднений в процессе анализа. Аналитическое исследование тантало-ниобиевых минералов сопряжено с большими трудностями, чем анализ любых других минералов. Простых методов для количественного разделения ниобия и тантала неизвестно. Не имеется тдкже и точного метода для отделения этих элементов от титана. Титан, если он содержится в относительно небольших количествах, можно определить в присутствии ниобия и тантала колориметрическим методом.
По вопросам, Связанным с анализом тантало-ниобиевых минералов, в литературе имеется ряд указаний, которые в отдельных случаях возможно и соответствуют истине, но ни в коей мере не могут быть обобщены. Некоторые из_ них, однако, считаются справедливыми для всех случаев и, условий, что несомненно является причиной многочисленных недоразумений и ошибок, которые имеют место и поныне. В связи с этим многие опубликованные результаты анализов и формулы минералов не имеют никакой ценности не только с количественной Стороны, но и нередко неверна также основанная на этих анализах качественная характеристика минералов. Ошибки усугубляются еще тем, что не принимается
Общие замечания
665
во внимание взаимное влияние элементов на реакции, искажающее природные свойства этих элементов в случае совместного нахождения их в растворе; это влияние подобно тому, которое давно уже .известно относительно циркония и титана.
Наиболее ярким примером такого влияния, быть может, служит действие воды на пиросульфатный плав окисей тантала, ниобия и титана или содержащего эти окислы минерала. Предполагалось, что при обработке такого плава холодной водой титан количественно переходит в раствор, тогда как тантал и ниобий полностью остаются в. нерастворимом остатке. Оказалось, однако, что последнее справедливо лишь в отсутствие титана, а между тем на этой реакции основан метод отделения титана от ниобия и тантала, которым широко пользовались на практике. В настоящее время установлено, что не только титан, в зависимости от его содержания, влияет на растворимость тантала и главным образом ниобия, но что тантал в свою очередь, особенно большие количества его, препятствует полному переходу титана в раствор.
Далее предполагалось, что из смеси окислов., полученных после сплавления пробы с пиросульфатом, сульфид аммония (желтый) полностью извлекает вольфрам и олово, не затрагивая при этом окислов тантала и ниобия. Считалось, кроме того, что при выщелачивании водой плава прокаленных окислов с карбонатом натрия и серой также извлекаются только вольфрам и олово, а тантал и ниобий остаются в нерастворимом остатке. Однако ни одно из этих утверждений не соответствует действительности. Многократным сплавлением, особенно при высокой температуре, и выщелачиванием безусловно можно достигнуть полного извлечения вольфрама и олова, но при этом в раствор, перейдут также и некоторые количества ниобия и тантала. Растворимость окислов ниобия и тантала можно уменьшить, понизив температуру сплавления, но это влечет за собой неполное извлечение в раствор 1 вольфрама и олова.
Принимая во внимание сомнительность многих методов, наше описание их будет менее дидактическим, чем методов, хорошо изученных. С большими подробностями, чем обычно, мы будет излагать некоторые методы, которые были рекомендованы без достаточно веского подтверждения их универсальности. Такая система изложения нам кажется целесообразной, чтобы привлечь внимание к большому числу вопросов, требующих дальнейшего исследования.
В обычном ходе анализа горных пород большая часть ниобия и тантала выделяется совместно с кремнекислотой и остается в нелетучем . остатке после отгонки кремния выпариванием с фтористоводородной и серной кислотами. После сплавления нелетучего остатка с карбонатом натрия или с пиросульфатем трудно получить прозрачный раствор плава вследствие гидролиза соединений ниобия и тантала. В конечном счете ниобий и тантал попадают в осадок от аммиака, и большую часть их, если не полностью, принимают за алюминий. Если осадок от’ аммиака подвергают обработке, имеющей целью выделение кремнекислоты, ниобий и тантал снова переходят в осадок. В тех случаях, когда для определения железа проводят восстановление цинком, присутствие ниобия обнаруживается по появлению темно-коричневой окраски восстановленного соединения ниобия. Если наличием ниобия пренебречь, то
1 W. В. Giles,’ Chem. News, 95, 1, 37 (1907); 99, 1, 25 (1909).
666
Гл. XXXIX. Ниобий и тантал
получаются повышенные результаты для железа. При анализе материалов, содержащих ниобий и тантал, серьезные затруднения возникают в связи с тем, что соли этих элементов легко подвергаются гидролизу. Удержанию ниобия и тантала в растворе способствуют винная кислота х, щавелевая кислота 2, перекись водорода 3, фтористоводородная кислота 4 * и маннит б.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ,
СОДЕРЖАЩИХ1НИОБИЙ И ТАНТАЛ|
Разложение фтористоводородной кислотой 6 и последующая обработка
Метод разложения фтористоводородной кислотой применяется почти исключительно при анализе ниобатов и танталатов редкоземельных металлов. Кремнекислота при этом определяется из отдельной навески. В тех случаях, когда этот способ применим, им всегда следует пользоваться, так как он позволяет очень быстро, легко и почти полностью отделять нерастворимые фториды редкоземельных и щелочноземельных металлов от растворимых фторидов ниобия, тантала и других элементов. При этом в раствор не вводятся соли щелочных металлов. Кроме того, таким путем не только обнаруживается присутствие урана (IV), но происходит и количественное отделение его от урана (VI). Фторид урана UF4 нерастворим в разбавленной фтористоводородной кислоте, и его присутствие в количествах, превышающих следы, легко определяется по зеленой окраскц, даже тогда, когда он находится в смеси со значительным количеством фторидов редкоземельных металлов. В тех случаях, когда содержание урана (IV) не требуется определять, пробу можно обрабатывать фтористоводородной и азотной кислотами, а под конец выпаривать с одной фтористоводородной кислотой.
Некоторые природные ниобаты и танталаты очень легко растворяются во фтористоводородной кислоте (например, самарскит), другие же слабо поддаются действию этой кислоты (колумбит и танталит). Имеется указание в, что труднорастворимые минералы можно полностью разложить, если измельчить их в тончайший порошок и затем последовательно обрабатывать отдельными порциями кислоты. Результаты наших опытов, а также и других исследователей не позволяют, однако, полностью подтвердить это указание.
Растворение и предварительная обработка. Разложение минерала лучше всего проводить следующим способом. Измельченную в тонкий порошок и высушенную при 150° С прйбу помещают в платиновую чашку или большой тигель, смачивают водой и затем обрабатывают 5—10 мл фтористоводородной кислоты. Если при этом проба сразу не растворяется,
‘A. R. Powell, J. Soc. Chem. Ind., 37, 285 Т (1918); W. R. S c h о e 11 e г, A, R. Powell, J. Chem. Soc., 1192, 1928 (1921).
2 H. P i e d, С. r., 179, 897 (1924).
3 L. Weiss, M. Laudecker, Z. anorg. Chem., 64, 65 (1909).
4 F. Pisani, J. prakt. Chem., 102, 448 (1866).
- 6 O. Hauser, Z. anorg. Chem., 60, 231 (1908).
•J. L. Smith, Am. Chem. J., 5, 44, 73 (1883); W. F. Hillebrand, Colo. Sci. Soc. Proc., 3, 38 (1888); R. C. Wells, J. Am. Chem. Soc., 50, 1017 (1928).
Разложение минералов, содержащих ниобий и тантал
667
выпаривают почти досуха, слабо нагревая и время от времени перемешивая массу. Затем прибавляют свежую порцию фтористоводородной кислоты и снова выпаривают. Эту операцию повторяют до полного разложения минерала, после 4его раствор разбавляют с таким расчетом, чтобы в объеме 100мл содержалось 5—10 мл фтористоводородной кислоты.
Осадок фторидов переносят на бумажный фильтр, помещенный в платиновую, эбонитовую или бакелитовую воронку, и промывают разбавленной (1 : 20) фтористоводородной кислотой. Осадок смывают обратно в чашку, фильтр, сжигают и золу присоединяют к основному осадку. В осадке в виде фторидов могут содержаться редкоземельные металлы, уран (IV), частично свинец и цирконий, щелочноземельные металлы и, возможно, небольшие количества железа. Если присутствуют щелочные металлы, что случается очень редко, осадок может содержать также двойные фториды их с другими металлами.
Последующая обработка. Обработка осадка фторидов. Полученный осадок фторидов переводят в сульфаты выпариванием с серной кислотой и нагреванием до полного удаления фтора. В присутствии UF4 для его окисления прибавляют азотную кислоту х. Сульфаты растворяют в холодной воде и затем, если остается остаток, вращают жидкость в чашке таким образом, чтобы он собрался в центре чашки. Остаток, по-видимому, представляет собой сульфат свинца. Менее вероятно наличие в нем сульфата щелочноземельного металла. Он может также полностью или частично состоять из окислов ниобия и тантала, если фториды были недостаточно тщательно промыты. Фильтруют через бумажный фильтр. Фильтрат сохраняют. Остаток обрабатывают горячим раствором ацетата аммония и к полученному раствору прибавляют сульфид аммония. Если при этом появляется осадок сульфида свинца, его отфильтровывают и растворяют в горячей разбавленной азотной кислоте. Раствор выпаривают с серной кислотой, после чего свинец взвешивают в виде сульфата (стр. 262). Фильтраты после отделения сульфата и сульфида свинца испытывают на содержание щелочноземельных металлов. Если после обработки ацетатом аммония остается нерастворимый остаток, его прокаливают и взвешивают, а затем исследуют на содержание ниобия, тантала и щелочноземельных металлов.
Из сохраненного ранее фильтрата осаждают редкоземельные металлы и уран аммиаком. Осадок отфильтровывают и промывают 2 %-ным раствором хлорида аммония. Фильтрат упаривают и определяют в нем кальций (стр. 706), учитывая при этом возможное загрязнение осадка примесями.
Осадок от аммиака растворяют в небольшом избытке соляной кислоты и осаждают редкоземельные металлы щавелевой кислотой (см. «Редкоземельные металлы», стр. 621), добавив небольшое количество оксалата аммония для понижения кислотности раствора (избытка оксалата аммония не следует вводить, в противном случае могут раствориться оксалаты
1 По нашим данным, промытые фториды полностью свободны от ниобия и тантала. J. L. Smith, анализируя самарскит, после растворения сульфатов в воде обнаружил небольшой белый остаток, который он счел за окислы ниобия и тантала. Один из нас при анализе минерала, подобного самарскиту, также обнаружил белый остаток, но это оказался сульфат свинца без следов ниобия и танталаvЭто указывает на то, что фторид свинца, несмотря на его достаточную растворимость в фтористоводородной кислоте, может выделяться в осадок совместно с другими фторидами.
668
Гл. XXXIX. Ниобий и тантал
некоторых редкоземельных элементов). Осадок промывают 1 %-ным раствором щавелевой кислоты. Если присутствует уран, оксалаты переоса-ждают после предварительного переведения их в хлориды. Осадок сохраняют. Фильтраты выпаривают досуха и нагревайием разрушают щавелевую кислоту. Затем, если уран отсутствует, растворяют остаток и извлекают оставшиеся редкоземельные элементы осаждением щавелевой кислотой в очень небольшом объеме. Фильтрат исследуют на содержание железа и циркония. В присутствии урана остаток обрабатывают фтористоводородной кислотой, выпаривают почти досуха, фильтруют и промывают остаток разбавленной фтористоводородной кислотой. Фильтрат сохраняют. Остаток фторидов обрабатывают серной кислотой, переводят в оксалаты и присоединяют к ранее полученному осадку оксалатов редкоземельных металлов. Дальнейшая обработка объединенных оксалатов описана в гл. «Редкоземельные металлы» (стр. 628).
Сохраненный фильтрат, содержащий уран, выпаривают, удаляют фтористоводородную кислоту обработкой серной кислотой, сульфаты растворяют в разбавленной азотной кислоте и осаждают уран аммиаком. Осадок прокаливают на воздухе до U3Og и затем исследуют на содержание железа и циркония. Окончательную массу осадка пересчитывают на UO2, так как таким путем в минерале определяется только содержание урана (IV).
Обработка фильтрата от фторидов. Фильтрат после отделения фторидов редкоземельных металлов, содержащий ниобий, тантал и другие элементы, выпаривают почти досуха. Затем вводят серную кислоту и продолжают выпаривать до появление ее паров. По охлаждении разбавляют концентрированным раствором винной кислоты и насыщают сероводородом. Если при этом выделяются сульфиды, фильтруют, промывают осадок сероводородной водой и обрабатывают, как обычно. Присутствовать могут свинец, медь, олово и другие металлы. Фильтрат, оставшийся после осаждения сероводородом, обрабатывают, как описано на стр. 671. < »
к Разложение сплавлением с едким кали, карбонатом калия или перекисью натрия
Сплавление с едким кали в серебряном, золотом или никелевом тигле или сплавление с карбонатом калия в золотом или платиново'М тигле с успехом применяют для разложения ниобатов и танталатов. При сплавлении с едким кали расплавляют (лучше в тигле, помещенном в отверстие асбестового щита) 3 г плавня и продолжают нагревать до прекращения образования пузырьков. Затем крышку снимают и осторожно всыпают 0,5 г тонко измельченной, высушенной анализируемой пробы. Быстрым вращательным движением тигля перемешивают его содержимое и затем нагревают 5—10 мин при температуре темно-красного каления, снова перемешивают массу вращением тигля и продолжают сплавлять при ярко-красном калении 20—30 мин. Снимают с тигля крышку и охлаждают, не теряя приставший к ней расплав. Охлаждают расплав, держа тигель в наклонном положении, и затем обрабатывают его соответствующим растворителем.
В тех случаях, когда пдав выщелачивают водой с целью повторить сплавление, целесообразно из нерастворимого промытого остатка пред
Разложение минералов, содержащих ниобий и тантал
669
варительно извлечь задержавшиеся в нем соли щелочных металлов. Для этого его обрабатывают в стакане небольшим избытком соляной кислоты при 100° С. Затем, раствор делают аммиачным, фильтруют и промывают осадок до удаления хлоридов. Помимо солей щелочных металлов, фильтрат может содержать также и другие компоненты, поэтому его следует выпарить и присоединить к водной вытяжке плава.
Для сплавления с карбонатом калия смешивают в платиновом тигле 0,1—0,'5 г тонко измельченной высушенной пробы с 3 г карбоната калия. Нагревают при 1000—1100° С до получения прозрачного расплава, после чего тигель вращают, чтобы расплавленная масса застыла на его стенках. Расплав, содержащий значительные количества тантала, может быть слегка мутным. Для сплавленря минералов, которые разлагаются при более низкой температуре, можно пользоваться золотыми тиглями. По охлаждении в тигель вводят 0,5 г твердого едкого кали и достаточное количество воды, чтобы покрыть ею плав. Осторожно нагревают до полного разложения сплавленной массы и затем переносят в стакан емкостью 250 мл, употребив для этого возможно меньшее количество воды.
Для определения таких элементов, как сёра или кремний, следует применять сплавление с перекисью натрия. Серу извлекают кипячением плава с водой. Водную вытяжку фильтруют, подкисляют, затем делают аммиачной и снова фильтруют. Кремнекислоту вместе с некоторыми количествами окислов ниобия и тантала можно выделить обезвоживанием с серной кислотой после сплавления с перекисью натрия в тигле из чистого железа или с едким кали, или с карбонатом калия.
Отделение кремнекислоты от ниобия и тантала описано в разделе «Методы отделения».
Разложение сплавлением с пиросульфатом и дальнейшая обработка
Большинство ниобатов и танталатов разлагается при обработке тонко измельченной пробы горячей концентрированной серной кислотой х. Титанаты более устойчивы, но все они более или менее легко разлагаются сплавлением с пиросульфатами щелочных металлов 1 2. Для сплавления лучше пользоваться натриевой солью вследствие образования более легко растворимых продуктов реакции. Это относится главным образом к минералам, содержавшим редкоземельные металлы и цирконий.
Один из исследователей3 считает, что отделение ниобия и тантала от железа и марганца, а также тантала от ниобия можно выполнить следующим'способом. Танталат сплавляют с девятикратным количеством пиросульфата натрия при 835—875° С. Плав обрабатывают водой при нагревании, фильтруют и затем промывают осадок ниобйя и тантала горячей 3 н. соляной кисдотой для удаления железа и марганца. После этого осадок обрабатывают разбавленной (1:1) серной кислотой и кипятят до тех пор, пока объем раствора не уменьшится до одной трети. По охлаждении фильтруют через асбест и промывают осадок тантала холодной 6 н. серной кислотой до прекращения появления осадка в промывных водах после прибавления к ним аммиака.
Наши данные показывают, что таким путем не удается получить достаточно удо-летворительных результатов, особенно в присутствии титана и циркония.
1 G. И. Hoffman. Am. J. Sci., [3], 24, 475 (1882).
3 L. F. Nilson, Ber., 13, 1430 (1880).
3 G. W. Sears, J. Am. Chem. Soc., 48, 343 (1926).
670
Гл. XXXIX. Ниобий"гГтантал
В случае применения для разложения пробы серной кислоты или пиросульфатов исключается возможность определения: кремния, если присутствуют значительные количества фтора; фторщ за исключением тех случаев, когда применяется метод отгонки; бора, двуокиси углерода, серы и фосфора. Большая часть этих ограничений достаточно понятна.
Фосфор нельзя определять 1 вследствие возможности улетучивания при этом некоторых количеств Р2О5.
Необходимо отметить, что хотя сплавлением с пиросульфатом разлагаются, по-видимому, все тантало-ниобиевые минералы, но для разложения ниобатов и танталатов редкоземельных металлов целесообразнее пользоваться фтористоводородной кислотой. Для. определения кремния, естественно, следует применить какой-либо другой способ разложения. Пиросульфатное сплавление обладает тем недостатком, что однократного сплавления не всегда бывает достаточно для полного разложения пробы. Кроме того, осадок окислов ниобия и тантала, как правило, загрязняется при этом некоторыми содержащимися в анализируемом материале посторонними элементами, отделение которых часто сопряжено с большими трудностями. С другой стороны, в зависимости от относительного содержания ниобия и тантала, часть их переходит в раствор, где последующее определение этих элементов крайне затруднено.
Пиросульфатное сплавление является общепринятым, поэтому нами приводится только этот метод, тем более, что после сплавления с пиросульфатом и после разложения материала обработкой серной кислотой обычно следуют одни и те же операции.
Сплавление. Тонко измельченную пробу 2 сплавляют в закрытом кварцевом тигле с 5—10 г пиросульфата натрия при темно-красном калении, время от времени перемешивая расплав негнущейся платиновой проволокой. Если требуется, изредка охлаждают и прибавляют несколько капель серной кислоты, а затем снова нагревают 3. Разложение обычно заканчивается в течение 1 ч, если анализируемый минерал не смешан с другими более устойчивыми веществами.
Обработка пиросулъфатного плава. 1. Наилучший метод обработки пиросульфитного плава минерала или полученных в ходе анализа неочищенных окислов заключается в следующем 4. Охлажденный плав выщелачивают раствором 10 г винной кислоты в 50 мл воды и затем фильтруют. Если в нерастворимом остатке остается неразложенный минерал, сплавление и выщелачивание повторяют. Конечный нерастворимый остаток может состоять из кремнекислоты, сульфата свинца, касситерита и др. Его сплавляют и анализируют обычно принятыми методами. Фильтрат, обработанный с таким расчетом, чтобы в нем содержался 1 % (по объему) серной кислоты и 5% винной кислоты, насыщают сероводородом. Выде
1 W. F. Hillebrand, G. Е. F. L u n d е 11, J. Am. Chem. Soc., 42, 2609 (1920).
2 Обычно 1—2 г, но значительно больше, если для разделения ниобия и тантала предполагают применить фторидный метод Мариньяка (см. стр. 683).
3 Вместо платинового тигля следует пользоваться кварцевым, чтобы избежать загрязнения платиной, в присутствии которой значительно усложняется определение сурьмы, олова и вольфрама. Это может вызвать необходимость сплавления отдельной навески пробы в платиновом тигле для определения кремнекислоты. Однако если сплавление вести при возможно более низкой температуре, кварцевый тигель заметно не разрушается.
1 W. R. Soho е 11 е г, A. R. Powell, J, Chem, Soc., 1192, 1928 (1921),
Разложение минералов, содержащих ниобий и тантал
671
ляющийся при этом осадок отфильтровывают, промывают насыщенным сероводородом 5%-ным раствором винной кислоты в 1 %-ной (по объему) серной кислоте и исследуют на содержание элементов сероводородной группых. Сероводородный фильтрат подщелачивают аммиаком, снова насыщают сероводородом и фильтруют для отделения сульфидов железа, никеля, кобальта и частично марганца. Из фильтрата после подкисления можно, по всей вероятности, осадить купфероном ниобий, тантал, титан, цирконий, ванадий и некоторые редкоземельные металлы (стр. 147), а затем, разрушив купферон и винную кислоту выпариванием с серной и азотной кислотами, можно обычным путем определить алюминий, оставшиеся редкоземельные элементы, уран, бериллий и др. (стр. 145).
2. Для разложения пиросульфитных плавов минералов, содержащих редкоземельные металлы, ниобий и тантал, целесообразно пользоваться щавелевой кислотой, с которой элементы группы редких земель образуют труднорастворимые оксалаты, а ниобий и тантал — растворимые комплексные соединения * 1 2. Щавелевая кислота не препятствует последующему осаждению едким натром. Согласно имеющимся указаниям, пиросульфитный плив ристворяют в щивелевой кислоте и после нигревиния в продолжение соответствующего промежутки времени отделяют оксилиты редкоземельных метиллов фильтровинием. Титин определяют в филь-трите колориметрическим методом по ренкции с перекисью водороди3. После этого риствор можно обрнботить серной кислотой, выпирить для ризрушения щивелевой кислоты, ризбивить риствором винной кислоты и продолжить анализ, как указано в п. 1.
3. Пиросульфитный плав рекомендуется обрабатывать еще следующим образом. Плав разлагают водой, прибавляют раствор аммиака в небольшом избытке и фильтруют. Осадок тщательно промывают и растворяют во фтористоводородной кислоте. Этот способ удобен только в особых случаях, как, например, при анализе ниобитов и танталитов редкоземельных металлов, которые недостаточно легко разлагаются непосредственной обработкой фтористоводородной кислотой. При применении этого метода последующая обработка проводится так же, как при непосредственном разложении минерала фтористоводородной кислотой (см. раздел «Разложение фтористоводородной кислотой и последующая обработка», стр. 666). Кремнекислоту при этом определяют из отдельной навески. Необходимо указать, что осадок от аммиака следует тщательно отмыть от щелочных металлов, в противном случае при обработке фтористоводородной кислотой совместно с фторидами осаждаются фторосиликаты, фторотитанаты и фтороциркониаты щелочного металла, в особенности когда для сплавления пользуются пиросульфатом калия, а не натрия.
Содержащийся в минерале уран (IV) не выделяется совместно с фторидами редкоземельных металлов, так как в процессе сплавления он окисляется. [Фторид урана (VI) легко растворим. ]
J
1 Такой обработкой Н. В. Knowles успешно отделил сурьму, олово, свинец и медь от смеси 0,1 г Nb2O8 и 0,1 г Та2О5.
а Н. Pied, С. г., 179, 897 (1924).
3 R. D. Hall [J. Am. Chem. Soc., 26, 1241 (1904)] установил, что щавелевая и винная кислоты не влияют на окраску ТЮ2—Н2О2, тогда как лимонная кислота понижает ее интенсивность.
672
Гл. XXXIX. Ниобий и тантал
Разложение хлористой серой и последующая обработка
Заслуживает внимания метод разложения ниобатов, танталатов и вольфраматов однохлористой серой или смесью так называемой двухлористой серы и хлора х. Показано, что рутил и, до-видимому, большинство, если не все, вбльфраматы, ниобаты и танталаты полностью разлагаются при нагревании в токе паров этих реагентов, причем титан, ниобий, тантал, вольфрам, олово, молибден, сурьма, мышьяк и частично железо возгоняются в виде хлоридов или оксихлоридов, а в нелетучем остатке, помимо пустой породы, остаются в виде хлоридов или в неизмененном состоянии другие металлы, кремнекислота и, возможно, бор. При хлорировании одной двухлористой серой в приемнике выделяются больцхие количества серы, что вызывает известные затруднения в аналитической работе, но этого можно избежать, если пользоваться смесью двухлористой серы и хлора. Если предположение, касающееся поведения бора, справедливо, то этот метод является наиболее приемлемым для разложения танталониобиевых минералов, содержащих бор.
. При применении двухлористой серы и хлора навеску высушенной пробы насыпают в фарфоровую лодочку, которую помещают в соответствующую трубку, постепенно нагреваемую, сначала в пределах 150— 240° С в течение 40—60 мин, а затем при 240—280° С в продолжение такого же периода времени, пропуская при этом струю сухого хлора и двухлористой серы. Последнюю получают осторожным нагреванием реагента в колбе при 40—50° С. Отходящие газы конденсируют в воде. Под конец лодочку и прилегающие к ней части трубки нагревают 1 2 5 мин при 280— 550° С, пропуская при этом струю одного только хлора, чтобы удалить летучие продукты, сконденсировавшиеся возле лодочки. При анализе ниобатов и танталатов рекомендуется 3 дистиллят и промывные воды выпарить с серной кислотой, а затем остаток нагреть и обработать, как изложено в разделе «Разложение фтористоводородной кислотой и последующая обработка» (стр. 666) или в разделе «Разложение сплавлением с пиросульфатом и дальнейшая обработка» (стр. 669). Приводится также метод хлорирования с одной однохлористой серой, без хлора 4, согласно которому в приемник наливают азотную кислоту и под конец операции пропускают струю хлористого водорода. Образующиеся в приемнике комки серы по возможности разминают сплющенной стеклянной палочкой, затем прибавляют раствор аммиака и пропускают сероводород. При этом сера, вольфрам, олово и др. переходят в раствор, а железо в виде сульфида осаждается вместе с ниобием и танталом.
Последующую обработку осадка проводят по одной из схем, изложенных в разделах «Разложение сплавлением с едким кали, карбонатом калия или перекисью натрия» (стр. 668) и «Разложение сплавлением с пиросульфатам и дальнейшая обработка» (сур. 669), возможно с некоторыми изменениями.
1 F. Bourion, Ann. chim. phys., [8], 21, 89 (1910); E. F. Smith, J. Am.: Chem. Soc., 20, 289 (1898).
2 При применении более высокой температуры имеется опасность улетучивания некоторых хлоридов, например хлоридов циркония и тория.
3R. J. Meyer, О. Hauser, Die Analyse der seltenen Erden und Erdsaue-ren, 1912, S. 276.
4 C. W. B.. Hicks, J. Am. Chem. Soc., 33, 1492 (19’11).
Методы отделения
673
Аналитическая обработка остающегося в лодочке нелетучего остатка хлоридов, кремнекислоты и т. и. не требует специальных пояснений. В этом остатке, помимо кремнекислоты, редкоземельных металлов, марганца, части железа (если температура не была достаточно высокой, чтобы оно полностью улетучилось), алюминия, кальция, магния и щелочных металлов, могут содержаться свинец и другие осаждающиеся сероводородом металлы.
*Метод хлорирования в применении к анализу тантало-ниобиевых материалов получил некоторое развитие и в работах последних лет. Так, отделение ниобия и тантала от олова и титана рекомендуется 1 проводить хлорированием четыреххлористым углеродом в запаянной трубке. По окончании хлорирования отгоняют в вакууме фосген, избыток четыреххлористого углерода и легколетучие хлориды олова (IV) и титана (IV). После этого возгоняют более трудно летучие хлориды ниобия и тантала и взвешивают, а затем их переводят в окислы, снова взвешивают и вычисляют содержание ниобия и тантала* раздельно косвенным путем. Для определения ниобия и тантала в колумбитах и ауксенитах предлагается 2 хлорирование в токе смеси газообразного хлористого водорода и четыреххлористого углерода. Доп. перев*
МЕТОДЫ ОТДЕЛЕНИЯ
Для обработки неочищенной смеси окислов ниобия и тантала предложено несколько методов, из которых тот или иной может быть приемлем для определенной цели или отдельных определений, но ни один из них не является универсальным. Некоторые методы описаны в разделах, следующих ниже.
Наиболее простой способ отделения циркония от ниобия и тантала основан на сплавлении смеси окислов с карбонатом калия и выщелачивании плава водой. Тщательно перемешивают 0,25—0,5 г смеси окислов с 5—20-кратным количеством карбоната калия, осторожно нагревают, а затем сплавляют при 1200° С до прекращения образования пузырьков. Охлажденный плав выщелачивают горячей водой, вводят мацерированную бумагу, фильтруют и промывают осадок сначала горячим 2 %-ным раствором карбоната калия, а под конец небольшим количеством горячей воды. Фильтрат сохраняют. Осадок обрабатывают, как указано в разделе «Разложение сплавлением с пиросульфатом и дальнейшая обработка» (стр. 669), после чего фильтр с осадком прокаливают и сплавление и выщелачивание повторяют один раз, если преобладает содержание ниобия, и дважды, если преобладает содержание тантала. Фильтраты объединяют для определения ниобия и тантала. Титан частично растворяется и распределяется между осадком и раствором. Таким способом цирконий отделяется от ниобия успешно, а от тантала не вполне количественно.
Таннин полностью осаждает тантал, ниобий и титан из слабокислого оксалатного, полунасыщенного хлоридом аммония раствора. Цирконий, гафний, торий, уран, бериллий и алюминий в этих условиях не осаждаются.
1 S с h a f е г, Pietruck, Z. anorg. Chem.-, 264, H. 1, 2 (1951).
“Treadwell, Guyer, Hauser, Bischofberger, Helv. Chim. Acta, 35, 2248 (1952).
43 Заказ 522.
674
Гл. XXXIX. Ниобий и тантал
Таким способом достигается количественное отделение всех осаждающихся в этих условиях элементов от неосаждающихся х.
Метод количественного отделения ниобия и тантала от циркония изложен ниже 1 2.
Отделение малых количеств ниобия и тантала в присутствии больших количеств циркония
Смесь окислов сплавляют с пиросульфатом калия в кварцевом тигле. Плав выщелачивают насыщенным раствором такого количества оксалата аммония, какое было взято ранее пиросульфата для сплавления смеси окислов. Кипятят, вводят 0,2 г таннина, растворенного в горячей воде, а затем по каплям при непрерывном перемешивании разбавленный (1:1) раствор аммиака. По нейтрализации почти всей кислоты образуется осадок, беловатый при содержании очень малых количеств ниобия и тантала, в противном случае — оранжевый или красный. Раствор аммиака продолжают прибавлять до тех пор, пока окраска осадка не станет светлее вследствие начала выделения соединения циркония белого цвета. Затем нагревают при 50—70° С, пока жидкость не станет прозрачной, и фильтруют. Осадок промывают 2 %-ным раствором хлорида аммония и прокаливают в тигле, в котором проводилось сплавление. Прокаленный остаток сплавляют и плав растворяют, как это описано выше, применяя лишь меньшие количества пиросульфата и оксалата. Прозрачный раствор, объем которого не должен превышать 50 мл, кипятят, перемешивают и прибавляют аммиак до появления заметной мути, которую растворяют в минимальном количестве разбавленной (1 : 1) соляной кислоты. В нагретый до кипения прозрачный раствор вводят 1 г хлорида аммония, а затем свежеприготовленный 1 %-ный раствор таннина (обычно менее 10 мл) до коагуляции осадка и обесцвечивания раствора. Нагревают при 50—70° С, осадок отфильтровывают, промывают, прокаливают и взвешивают в виде NbaO6 + Та2О5.
Если имело место полное осаждение ниобия и тантала, то обработка кипящего фильтрата небольшим добавочным количеством таннина (и, когда это требуется, каплей аммиака) не вызывает никаких изменений в растворе или же вызывает осаждение беловатого осадка. Чтобы удостовериться в полном отделении циркония, взвешенный прокаленный остаток можно сплавить и операцию осаждения повторить.
Поведение в этих случаях титана и тория не .изучено.
Отделение больших количеств ниобия и тантала в присутствии любых количеств циркония
Смесь окислов сплавляют с 6 г карбоната калия в платиновом тигле при 1000—1200° С. Расплаву дают застыть на стенках тигля, после чего вводят 1 г чистого едкого кали и воду в количестве, достаточном, чтобы покрыть плав. Закрытый крышкой тигель нагревают на горячей плите
1 Подробное изложение метода см. W. R. Schoeller, A. R. Powell. Analyst, 57, 550 (1932) и W. R. Schoeller, Н. W. Webb, там же, 58, 143 (1933).
* W. R, Schoeller, Е. F. Waterhouse, Analyst, 53, 517 (1928),
Методы отделения
675
1—2 часа до полного разложения сплавленной массы. Содержимое тигля переносят в небольшой стакан, перемешивают с мацерированной бумагой и фильтруют, следя за тем, чтобы в фильтрате не было мути. Остаток промывают 2 %-ным раствором .карбоната калия, прокаливают в тигле, в котором производилось сплавление, и операцию повторяют. Полученный в результате этого остаток прокаливают, смачивают разбавленной серной кислотой, высушивают и извлекают оставшиеся ниобий и тантал, как указано, выше. Содержащие карбонат калия фильтраты подкисляют соляной кислотой и кипятят. Затем прибавляют раствор аммиака в небольшом избытке, вводят немного мацерированной бумаги и снова кипятят. Выделяющийся при этом осадок ниобия и тантала отфильтровывают, промывают 2 %-ным раствором нитрата аммония и осторожно прокаливают. Прокаленный остаток нагревают с 10 мл соляной кислоты, разбавляют 100 мл воды, кипятят, обрабатывают аммиаком и т. д., как было указано ранее. Осадок прокаливают и взвешивают в виде Nb2O5 + Та2О6.
Хорошего отделения ниобия и тантала от титана, но не от циркония и тория можно достичь повторным осаждением салициловой кислотой х. Ниобий и тантал при этом, однако, никогда не выделяются полностью и небольшие количества титана захватываются осадком1 2. Разделение обычно выполняют следующим способом. Смесь окислов сплавляют с 5 г карбоната калия. Плав обрабатываю! 200—300 мл воды при 60° С и полученный раствор медленно при перемешивании вливают в кипящий раствор, содержащий 15 г салициловой кислоты. Нагревают при 100° С в течение 3—4 ч, затем фильтруют и промывают осадок разбавленным раствором салициловой кислоты. Всю эту обработку повторяют до тех пор, пока раствор после введения салициловой кислоты не перестанет окрашиваться в желтый цвет при кипячении. Титай можно выделить из фильтрата осаждением избыточным количеством раствора аммиака при кипячении. Осадок титана, так же как и полученный ранее осадок ниобия и тантала, загрязнен солями щелочных металлов.
Относительно полное отделение ниобия и тантала от титана достигается при кипячении виннокислых комплексов этих элементов с азотной кислотой 2. Сплавляют 0,1—0,2 г смеси окислов с 2 г пиросульфата калия в кварцевом тигле. По охлаждении плав растворяют в концентрированном растворе 3 г винной кислоты, разбавляют до 300 мл, прибавляют 30 мл азотной кислоты и кипятят 15—20 мин. Выделившийся загрязненный осадок водных окислов ниобия и тантала, после введения мацерированной бумаги, отфильтровывают, промывают 1 %-ным раствором азотной кислоты, прокаливают и затем операцию повторяют.. Фильтраты, содержащие большую часть титана и незначительные количества ниобия и тантала, объединяют. Небольшие количества титана, удержанные осадком окислов ниобия и тантала, можно определить колориметрическим методом или отделить салицилатным методом. Из объединенных фильтратов, после разрушения винной кислоты (стр. 91), выделяют ниобий и тантал кипячением с салицилатом аммония, взятым в количестве большем, чем это требуется для связывания серной кислоты. Титан осаждаЮт
1 I. Н. Muller, J. Am. Chem. Soc., 33, 1506 (1911).
2 W. R. S c h о e 11 e r, E. C. Deering, Analyst, 52, 625 (1927). В этой статье дан прекрасный обзор методов, предложенных для отделения титана от ниобия и тантала.
43* '
676
Гл. XXXIX. Ниобий и тантал
из салицилатного фильтрата избыточным количеством раствора аммиака *.
*Для отделения ниобия и тантала от титана наиболее часто применяется так называемый «пиросульфатно-танниновый» метод1 2 3, основанный на гидролитическом осаждении ниобия и тантала в присутствии таннина. При этом происходит отделение ниобия и тантала также от большинства других сопутствующих элементов, за исключением вольфрама, олова, сурьмы. Метод заключается в выщелачивании пиросульфатного плава анализируемого материала, или предварительно выделенных и прокаленных окислов, 1 %-ным раствором таннина в 1 н. растворе серной кислоты при нагревании. Согласно указанию автора метода, таким путем происходит более четкое отделение ниобия и тантала от титана, чем при гидролизе без таннина, благодаря погашению адсорбционной способности водных окислов ниобия и тантала таннином, имеющим противоположный им заряд. Кроме того, титан образует в кислом растворе с таннином растворимые комплексные соединения, что также способствует удержанию его в растворе.
Однако и в этих условиях не удается достигнуть количественного отделения ниобия и тантала от титана. Полнота разделения зависит в основном от соотношения между ниобием и титаном. Большие количества титана препятствуют полнбму выделению ниобия в осадок, и известная часть его обычно остается в растворе (0,5—1 мг). В то же время значительные количества титана удерживаются осадком окислов ниобия и тантала. Для полной очистки осадка от титана требуется повторение операции, иногда многократное, что связано с возможной дополнительной потерей ниобия и тантала. Ю. А. Чернихов и Т. А. Успенская 8 показали, что наличие сульфатов щелочных металлов также препятствует цолноте выделения ниобия из раствора, и заменили пиросульфатное сплавление обработкой смесью фтористоводородной и серной кислот.
В. Быкова 4 * считает, что более полное отделение ниобия от титана, причем с меньшими потерями ниобия в растворе, происходит в солянокислой среде и рекомендует выщелачивать пиросульфатный плав солянокислым раствором таннина вместо сернокислого.
И. П. Алимарин и Б. И. Фрид 6 предлагают заменить таннин пирогаллолом, основываясь на том, что титан с пирогаллолом образует в кислой среде более устойчивое и лучше растворимое соединение, чем с таннином. При этом происходит отделение ниобия и тантала также от железа, циркония и ряда других элементов. По наблюдениям авторов при непосредственном выщелачивании пиросульфатного плава окислов ниобия, тантала и титана 1 %-ным солянокислым раствором пирогаллола происхо
1 Анализируя этим методом смесь, состоявшую из 0,0307 г Ti02 и 0,0620 г Nb20s4 4- Та2О6, один из нас (Лендель) определил 0,0321 г TiO2 и 0,0599 г окислов ниобия и тантала, а в смеси 0,0325 г TiO2 и 0,0602 г Nb3O6 4- Та2О6 он нашел 0,0344 г Т1О2 и 0,0584 г Nb2O5 + Та2О5. Смесь окислов ниобия и тантала была приготовлена из равных частей N b2Os и Та2О6.
2 W. R. Schoeller, The Analytical Chemistry of Tantalum and Niobium, London, 1937.
3 См. обзорную статью: Ю. А. Чернихов, В. Г. Г о р ю ш и н а, Зав. лаб., 11, 875 (1945).
4 В. Быкова, Материалы к геохимии Хибинских тундр, Кольская база АН
СССР, II. 1936, стр. 39.
» И, П, Алимарин, Б. И. Фрид, Зав. лаб., 7, 1109 (1938).
Методы отделения
677
дят потери ниобия вследствие образования гетерогенного комплексного сульфатосоединения титана и ниобия. Это соединение, однако, можно разрушить подщелачиванием раствора, причем после последующего подкисления оно вновь не образуется. Образованию комплекса ниобия с титаном препятствует также присутствие железа.
Для отделения ниобия и тантала от титана авторы рекомендуют следующий метод. Пиросульфатный плав анализируемого материала выщелачивают 100 мл 1 %-ного водного раствора пирогаллола. При наличии больших количеств титана и железа раствор окрашивается в темный коричнево-красный цвет. Если анализируемый материал содержит много титана и мало железа, то перед сплавлением к навеске прибавляют чистую окись железа с таким расчетом, чтобы общее содержание ее немного превышало содержание титана. На дне стакана собирается осадок пирогаллатов ниобия и тантала. К раствору прибавляют аммиак до явного запаха и затем нагревают до кипения, причем осадок пирогаллатов ниобия и тантала растворяется. Горячий раствор нейтрализуют соляной кислотой по индикаторной бумаге конго, после чего вводят избыток кислоты с таким расчетом, чтобы концентрация ее была 1 н. Кислый раствор кипятят 30—40 мин, добавляют мацерированную бумагу и оставляют стоять несколько часов. Осадок пирогаллатов ниобия и тантала отфильтровывают через плотный фильтр, промывают раствором пирогаллола, подкисленным соляной кислотой, и прокаливают в фарфоровом тигле. Полученные окислы сплавляют с пиросульфатом калия и снова обрабатывают пирогаллолом. Эту операцию повторяют до тех пор, пока окраска фильтрата не станет достаточно бледной. На задержавшиеся в осадке небольшие количества титана вводят поправку, определив их колориметрически.
Авторы отмечают, что отделение ниобия и тантала от циркония выщелачиванием пиросульфатного плава окислов 5%-ным раствором пирогаллола в 1 н. растворе соляной кислоты происходит значительно более успешно, чем от титана.
Метод осаждения ниобия и тантала подкислением соляной кислотой щелочного пирогаллового раствора в присутствии циркония не дает удовлетворительных результатов, так как при добавлении к пирогалловому раствору избытка аммиака выделяется гидроокись циркония, которая потом трудно растворяется в 1 н. растворе соляной кислоты и загрязняет осадок пирогаллатов ниобия и тантала. Доп. перев.*
Алюминий можно количественно осадить и отделить от ниобия и тантала следующим способом. К разбавленному сернокислоту раствору этих элементов, содержащему перекись водорода, прибавляют избыточное количество оксихинолина и такое количество раствора аммиака, чтобы на каждые 100 мл анализируемого раствора было 10 мл его избытка. В дальнейшем поступают так, как указано на стр. 148.
Отделение вольфрама от ниобия и тантала обычно, связано с большими затруднениями. Выщелачивание смеси окислов, выделенных аммиаком, сульфидом аммония или гидролизом из кислого раствора, или выщелачивание водой плава с карбонатом натрия и серой, так же как и кипячение щелочного раствора вольфрамата, ниобата и танталата, не дают удовлетворительных результатов \ Более того, ниобий и тантал препят
1 Критику этих методов см. W. R. Schoeller, С. Jahn, Analyst,- 52, 504 (1927).
678
Гл. XXXIX. Ниобий и тантал
ствуют количественному осаждению вольфрама цинхонином (стр. 770). Для разделения этих элементов можно использовать три метода х, в зависимости от сопровождающих вольфрам элементов. Из них магнезиальный рекомендуется в тех случаях, когда требуется отделить вольфрам от титана, ниобия, тантала и циркония. Этот метод заключается в следующем. Смесь окислов (0,2—0,5 г) сплавляют с 4 г карбоната калия в платиновом тигле на сильном пламени в течение 10—15 мин. Сплавленную массу выщелачивают 200 мл горячей воды, следя зй тем, чтобы полностью разложились комочки плава. Для этого их разминают стеклянной палочкой, а растворГ слабо кипятят. Горячий раствор обрабатывают свежеприготовленным реактивом (1 г кристаллического сульфата магния, 2 г хлорида аммония, 25 мл воды и 4 капли раствора аммиака). Покрывают часовым стеклом и оставляют стоять на закрытой водяной бане 1 ч. Хлопьевидный осадок переносят на неплотный фильтр диаметром 11 см и промывают раствором хлорида аммония (насыщенный раствор NH4C1 разбавляют в 4 раза водой).
Определение ниобия и тантала. Осадок смывают обратно в стакан той же промывной жидкостью. Фильтр помещают в платиновый тигель, предназначенный для прокаливания получаемого в дальнейшем осадка от таннина. Суспензию осадка от MgSO4 (150 мл) подкисляют соляной кислотой и нагревают на водяной бане 1 ч. После этого прибавляют равный обърм насыщенного раствора хлорида аммония и раствор аммиака до слабокислой реакции по лакмусу. Раствор обрабатывают 2 г ацетата аммония, нагревают до кипения и вводят свежеприготовленный 5%-ный раствор таннина до полной коагуляции осадка (осадок 1). По отстаивании на водяной бане осадок отфильтровывают при слабом отсасывании, промывают раствором хлорида аммония и прокаливают в том платиновом тигле, в котором поместили ранее фильтр, оставшийся после смывания с него осадка от MgSO4. Если осадок 1 надо для очистки переосадить, его снова сплавляют с карбонатом калия и повторяют описанную выше операцию, включая осаждение сульфатом магния. Фильтрат, полученный после переосаждения, присоединяют к ранее полученному фильтрату. Осадок переосаждают таннином, как при выделении осадка 1, причем получается осадок 2, который прокаливают, выщелачивают, как обычно 1 2, и взвешивают, как смесь окислов ниобия и тантала.
Определение вольфрама. Фильтраты от осаждения сульфатом магния обрабатывают так, как это требуется для осаждения вольфрама таннином и цинхонином. Это осаждение 3, как оно первоначально было описано, проводится из растворов, имеющих высокую концентрацию хлорида натрия. После отделения осадка от сульфата магния (перед осаждением комплексного соединения вольфрама) в раствор ^вводят профильтрованный, насыщенный при нагревании раствор 20—30 г хлорида аммония.
Большие количества кремнекислоты в присутствии малых количеств ниобия и тантала можно определить выпариванием с фтористоводородной и серной кислотами* как обычно. При наличии больших количеств нио
1 A. R. Powell, W. R. Schoeller, С. Jahn, Analyst, 60, 512 (1935).
2 Для очистки осадок нагребают с 5 %-ной соляной кислотой. Раствор делают аммиачным и фильтруют. Осадок промывают 2%-ным раствором нитрата аммония и снова прокаливают [W. R. Schoeller, Analyst, 56, 304 (1931)].
8 W. R. Schoeller, A. R. Powell, Analyst, 53, 262 (1928).
Методы отделения
679
бия и тантала непосредственная отгонка кремния не дает удовлетворительных результатов. В таких случаях поступают следующим образом. Прокаленные взвешенные окислы растворяют в платиновой чашке большого размера во фтористоводородной и серной кислотах при нагревании, после чего полностью удаляют фтор выпариванием до появления паров серной кислоты. По охлаждении влажную сильнокислую массу переводят в раствор, прибавляя перекись водорода и, если нужно, разбавленную серную кислоту. Если при стоянии появляется небольшой белый осадок, по-видимому, сульфата свинца, его переносят на фильтр Мунро, промывают разбавленной серной кислотой, содержащей перекись водород^; осторожно прокаливают, взвешивают и затем исследуют на содержание примесей. Фильтрат насыщают сернистым ангидридом, кипятят до полного разложения перекиси водорода и прибавляют избыточное количество раствора аммиака. Осадок отфильтровывают, промывают 2%-нЫм раствором нитрата аммония, прокаливают и взвешивают. Разность между первоначальной массой прокаленного остатка и полученной его массой дает содержание кремнекислоты, причем с большей точностью, если отсутствует свинец. Это объясняется тем, что полученная разность включает в себя также и величину, соответствующую содержанию свинца, которую точно определить не представляется возможным. Так, в случае, когда в прокаленных исходных окцслах свинец находится в виде сульфата, правильная поправка должна соответствовать найденной массе сульфата свинца. Если же прокаливание окислов ниобия и тантала привело к удалению SO2 и образованию ниобата свинца, то, чтобы получить правильные результаты для кремния, следует вычесть эквивалентное содержанию сульфата свинца количество РЬО.
Отделение кремния от ниобия и тантала можно осуществить т&кже следующим способом х. Смесь окислов сплавляют с 3 г или большим количеством пиросульфата калия. По охлаждении плав обрабатывают 50 мл водного раствора 3 г щавелевой или винной кислоты при нагревании на водяной бане. Осадок отфильтровывают, промывают горячей водой, прокаливают и взвепгивают. Затем его обрабатывают фтористоводородной и серной кислотами, выпаривают, прокаливают и снова взвешивают.
Тантал совместно с ниобием, титаном, цирконием, гафнием и оловом можно количественно выделить из раствора, полученного после разложения горной породы, осаждение^ фениларсоновой кислотой следующим способом. К 300 мл раствора 2—3 н. по концентрации соляной кислоты, содержащего небольшие количества серной и 5 г винной кислоты, прибавляют 5—10 г фениларсоновой кислоты. Вводят немного мацерированной бумаги; нагревают на водяной бане 2 ч и оставляют при комнатной температуре на 48 ч. После этого осадок отфильтровывают и промывают холодной разбавленной (1 : 10) соляной кислотой или холодным 2%-ным раствором нитрата аммония. Для полного разложения горной породы может потребоваться: 1) нагревание с фтористоводородной и серной кислотами, последующее удаление фтора, прибавление раствора винной кислоты и фильтрование; 2) сплавление осадка с карбонатом натрия, растворение в соляной кислоте и фильтрование; 3) сплавление остатка с пиросульфатом калия и-растворение плава в растворе винной кислоты1 2.
1 См. сноску 1 на стр. 677.
2 К. Rankama, Bull, comm, geol, Finland, 133 (1944),
680
Гл. XXXIX. Ниоб ий и тантал
♦Возможность осаждения ниобия и тантала фениларсоновой кислотой в присутствии винной кислоты впервые показали И. П. Алимарин и Б. И. Фрид1. На основе этой реакции ими разработан количественный метод определения Ниобия и тантала и отделения их от ряда посторонних элементов (алюминия, железа, ванадия, урана, марганцу и др.). Авторы указывают, что при осаждении ниобия и тантала фениларсоновой кислотой происходит также частичное отделение титана. Удерживаемые осадком количества титана, однако, сильно колеблются, главным образом в зависимости от температуры раствора и скорости фильтрования. В тех случаях, когда содержание титана в анализируемом материале невелико (менее 1 %), после двукратного осаждения в осадке остаются лишь незначительные количества титана, на которые можно ввести поправку после определения их колориметрическим методом.
Определение ниобия и тантала в присутствии посторонних примесей авторы проводят следующим способом. Исходный материал сплавляют в фарфоровом тигле с пиросульфатом калия. Плав выщелачивают 150 мл 1 н. раствора соляной кислоты и, не фильтруя, осаждают остающиеся при этом в растворе ниобий и тантал избыточным количеством 3%-ного раствора фениларсоновой кислоты при нагревании на песчаной плитке в течение 1 ч. Вводят небольшое количество мацерированной бумаги и через несколько часов или на. следующий день фильтруют. Осадок промывают 4 %-ным раствором нитрата аммония, подкисленного несколькими каплями азотной кислоты, прокаливают и полученные окислы снова сплавляют с пиросульфатом калия. Плав в этом случае растворяют в 10%-ном растворе винной кислоты и после добавления соответствующего количества соляной кислоты осаждение ниобия и тантала фениларсоновой кислотой повторяют.
Авторами метода установлено, что изменение концентрации соляной кислоты в растворе в пределах 0,3—3 и. не влияет на осаждение и что осадок быстро коагулирует и легко отфильтровывается только в том случае, если введен избыток осадителя (около 0,75 г фениларсоновой кислоты на каждые 10 мг М2О5). Доп. перев.*
МЕТОДЫ ОПРЕДЕЛЕНИЯ
В осадке окислов ниобия и тантала после отделения от посторонних примесей, как правило, задерживаются-еще некоторые количества титана. Наиболее целесообразно1 в конечной стадии анализа сосредоточить внимание на количественном разделении, ниобия и тантала, не считаясь с наличием титана, а затем колориметрически определить его содержание в каждой взвешенной окиси.
Во всех обычно принятых методах определение тантала заканчивают взвешиванием пятиокиси Та2О5. Ниобий можно определить в виде пяти-окиси 2 Nb2O5 или Же объемным способом (титрованием перманганатом после восстановления цинком в сернокислом растворе) 3.
1 И. П. Алимарин, Б. И. Фрид, Зав. лаб., 7, 913 (1938).’’
2 Пятиокиси ниобия и тантала можно прокаливать при 1100—1200° С без заметных потерь. Оба окисла не гигроскопичны. Ниобий и тантал улетучиваются при прокаливании непосредственно после вцпаривания с Одной фтористоводородной кислотой.
3 Критический обзор методов определения см. W. R. Schoeller, The Analytical Chemistry of Tantalum and Niobium. Chapman and Hall, London, 1937, см. также W. R. Schoeller, Analyst, 61, 806 (1936).
Методы определения
681
Отделение тантала от ниобия осаждением таннином в щавелевокислом растворе
Наилучшим способом разделения тантала и ниобия является, по всей вероятности, метод, основанный на различном поведении оксалатотанталовой и оксалатониобиевой кислот в разбавленных кислых растворах таннина Ч Оксалатотанталовая кислота не разлагается только в присутствии определенного количества свободной щавелевой кислоты. Ниобиевое комплексное соединение значительно более устойчиво. Таннино-вый осадок тантала окрашен в серно-желтый цвет. Ниобиевый осадок имеет яркую киноварно-красную окраску, которая достаточно интенсивна, чтобы обнаружить присутствие ниобия в осадке тантала. Титан 1 2,. вольфрам 3 и сурьма *, но не цирконий 4 *, частично осаждаются и препятствуют разделению тантала и ниобия. Титан в количествах, не превышающих 2% по отношению к содержанию окиси тантала, не вызывает затруднений при проведении этой операции 6.
Несомненно и другие элементы также препятствуют применению таннинового метода для разделения тантала и ниобия. Поэтому, прежде чем приступить к этой операции, целесообразно отделить ниобий и тантал от всех сопутствующих элементов.
В общих чертах метод сводится к следующему ®. Смесь окислов сплавляют с кислым сульфатом калия в кварцевом тигле. Плав растворяют в горячем насыщенном растворе оксалата аммония, фильтруют и, если нужно, повторяют обработку нерастворимого остатка. К кипящему раствору прибавляют определенное количество свежеприготовленного 2%-ного водного раствора таннина, лучше из бюретки, а затем — насыщенный раствор хлорида аммония. При этом выделяется желтый осадок (Та) или же окрашенный в оранжевый до красного цвет осадок (Та + Nb), который отфильтровывают и промывают. Фильтрат обрабатывают разбавленным раствором аммиака и таннином, в результате чего выпадает осадок, окрашенный в более темный оранжевый или красный цвет. Чиста желтую фракцию прокаливают в фарфоровом тигле и взвешивают как Та2О5, а оранжевые или красные осадки объединяют, прокаливают и сплавляют с бисульфатом. Плав выщелачивают, как прежде, и затем снова осаждают таннином. В результате многократного повторения этого процесса получается фракция тантала, которая загрязнена адсорбированными солями, кремнекислотой, титаном и незначительными количествами ниобия. Из объединенных фильтратов, после отделения тантала, выделяют ниобиевую фракцию осаждением таннином и раствором аммиака. Обе фракции следует подвергнуть очистке, а затем ввести поправку на содержание в них титана.
*Изучение реакции осаждения ниобия и тантала таннином в щавелевокислой среде 7 показало, что таким способом осуществить количе
1 W. R. Schoeller, Analyst, 57, 750 (1932).
2 W. R. Schoeller, A. R. Powell, Analyst, 53, 265 (1928).
3 W. R. Schoeller, C. Jahn, Analyst, 59, 465 (1934).
4 A. R. Powell, W. R. Schoeller, Analyst, 50, 494 (1925).
6 W. R. Schoeller, Analyst, 57, 755 (1932).
e Подробности этого метода, а также соответствующие примеры можно найти в оригинальной работе или в монографии W. R. Schoeller, The Analytical Chemistrv of Tantalum and Niobium, Chapman and Hall, London, 1937.x
7 H. T. S. Britton, P. L. Robinson, Analyst, 58, 368 (1933); J. Chem,. Soc., 419 (1933).
682
Гл. XXXIX. Ниобий и тантал
ственное разделение этих элементов в один прием не представляется возможным вследствие близости pH гидролитического разложения их оксалатных комплексов. (В кипящем растворе, содержащем щавелевую кислоту, тантал осаждается таннином при pH =2, а выделение ниобия начинается при pH = 3.) Некоторое усовершенствование в метод введено Куннингемом \ который при анализе стали, содержащей малые по сравнению с ниобием количества тантала, избегает выделения промежуточных фракций. Для этого он вводит таннин в раствор, нейтрализованный по феноловому синему (pH = 4,6). При кипячении такого раствора в осадок выделяется тантал с небольшими количествами ниобия, на которые вводится поправка после определения их объемным способом.
Независимо от Куннингема, Ю. А. Чернихов и Е. И. Венделыптейн 1 2 для анализа колумбита и продуктов его переработки также предложили разделять ниобий и тантал таннином только на две фракции. Эти авторы процесс осаждения контролируют по цвету осадка, а ниобий, попавший в осадок тантала, определяют колориметрически.
На различной устойчивости оксалатных комплексов тантала и ниобия основан лакже йодатный метод 3 разделения этих элементов. В кислых растворах, содержащих оксалат, тантал дает с яодатом нерастворимое соединение и отделяется таким образом от ниобия. Определение тантала заканчивают йодометрическим титрованием полученного соединения. Точный состав этого соединения авторами не установлен, но на основании ^полученных результатов тйтрования ими выведен эмпирический фактор, согласно которому отношение Та : 1О3" в осадке^равно 1 : 2.
Титан должен отсутствовать. Доп. перев.*
Отделение ниобия от тантала обработкой окислов оксихлоридом селена
Ниобий можно отделить от тантала выщелачиванием смеси окислов •оксихлоридом селена следующим способом 4. Сильно прокаленную смесь окислов ниобия и тантала (0,2—0,3 г, предпочтительно свободных от титана) обрабатывают в колбе 50 мл смеси равных частей оксихлорида селена 5 и серной кислоты при кипячении на Песчаной бане в течение 30 мин. Кипячение не должно быть настолько бурным, чтобы выделялись облака пара. По охлаждений и отстаивании жидкость сливают декантацией, отсасывая ее через взвешенный тигель Гуча с асбестовой прокладкой. Осадок, оставшийся в колбе, кипятят с 20 мл реактива 20 мин, после чего раствор также сливают декантацией через тот же тигель. Эту операцию повторяют до тех пор, пока в фильтрате, влитом в 1000 мл воды, при кипячении не появится лишь незначительный осадок, обусловленный следами растворенной окиси тантала. Необходимо следить за тем, чтобы в процессе декантации на фильтр попадало возможно Меньшее количество осадка. Обычно бывает достаточно трех- или четырехкратной обработки. Под
1 Th. R. Cunningham, Ind. Eng. Chem., Anal. Ed., 10, 233 (1938).
2 См. обзорную статью Ю. А. Чернихов, В. Г. Г о р ю ш и н а, Зав. .лаб., И, 875 (1945).
8Ю. А. Чернихов, Т. А. Успенс гСн-я, ДАН СССР, 28, № 9, 802 .(1940).
4 н. В. Merrill, J. Am. Chem. Soc., 43, 2378 (1921).
' 6 V. L e n h e r, J. Am. Chem. Soc., 43, 29 (1921).
Методы определения
683
конец струей воды пдлностыо переносят осадок на фильтр и промывают водой. Нет необходимости промывать осадок особо тщательно, так как продукты разложения реагента летучи. Осадок прокаливают и взвеши- • вают в виде ТааО5. При этой обработке титан переходит совместно с ниобием в раствор. Его содержание следует определить в отдельной навеске колориметрическим способом и вычесть из суммы окислов ниобия и титана, вычисленной по разности.
По данным автора метода1, максимальное расхождение между результатами анализа искусственно приготовленной смеси Nb2O5 и Та2ОБ составляет 3% относительных, причем большей Частью оно много ниже. Значительно менее совпадающие результаты должны .несомненно получаться, при применении итого метода к Смесям, содержащим посторонние примеси.
Разделение ниобия и тантала по методу Мариньяка
Старейшим методом разделения ниобия и тантала является классический метод Мариньяйа 2, весьма длительный и позволяющий провести лишь приближенное разделение.
При тщательной работе и применении больших навесок 3 (15—30 г) можно достигнуть точности до 0,5%. Метод Мариньяка основан на различной растворимости фторотанталата калия KaTaF7 и оксифторониобата калия KaNbOFd в фтористоводородной кислоте.
Метод был видоизменен некоторыми исследователями, главным образом применительно к контролю производства. Один из вариантов метода заключается в следующем 4. Свежеосажденную промытую смесь окис-лбв, предпочтительно свободную от титана и циркония, растворяют в точно необходимом количестве фтористоводородной кислоты, избегая введения ее избытка. Кипятят и затем медленно при непрерывном перемешивании прибавляют кипящий раствор двукратного по отношению к массе окислов количества фторида калия. Раствор упаривают до 10 мл, обмывают стенки чашки несколькими каплями воды и медленно охлаждают до температуры не выше 15° С. Прозрачный раствор сливают декантацией через маленький бумажный фильтр, помещенный в резиновой или бакелитовой воронке, и кристаллы фтортанталата калия промывают 4 раза небольшими количествами воды. Фильтрат и промывные воды упаривают до 5 мл и медленно охлаждают. Раствор сливают декантацией через другой малейький фильтр и промывают осадок, как прежде. Если заметны хлопья соли ниобия, промывают до ее растворения. Выпаривают фильтрат и промывные воды на водяной бане досуха. По охлаждении вводят 1 каплю фтористоводородной кислоты, а затем из бюретки 1—5 мл воды и нагревают до полного растворения массы. Прибавляют 0,1 г фторида калия, растворенного в 1 мл в (Уды, и'отмечают объем раствора (раствор А). Охлаждают 30—60 мин при 15° С, фильтруют, собирая фильтрат в маленькую
1 Н. В. Merrill, цит. выше.
2 С. Marignac, Ann. chim. et phys., [41, 8, 60 (1866).
3 R. J. Meyer, O. Hauser, Die Analyse der seltenen Erden und Erdsaue-ren, 1912. v
’Mellor, A Treatise on Quantitative Inorganic Analysis, Charles Griffin and Co., London, 1913, p. 421.
684
Гл. XXXIX. Ниобий и тантал
платиновую чашку, и промывают осадок 3—4 раза несколькими каплями холодной воды. Записывают объем промывных вод (раствор Б).
Для определения ниобия выпаривают фильтрат с серной кислотой до полного удаления фтора и по охлаждении разбавляют водой. Из полученного раствора осаждают ниобий небольшим избытком раствора аммиака при кипячении. Осадок отфильтровывают, промывают горячим 2%-ным раствором нитрата аммония и прокаливают. Затем прибавляют твердый карбонат аммония, закрывают тигель, осторожно нагревают, после чего прокаливают при 1000—1200° С и взвешивают. Вводят поправку на оставшееся с ниобием количество тантала, для чего из массы осадка ниобия вычитают по 0,002 г на каждый миллилитр кислого раствора А и по 0,00091 г на каждый миллилитр раствора Б (промывные воды). Содержание тантала вычисляют по разности, если известна масса смеси окислов, или же определяют непосредственно, так же как ниобий, причем к массе осадка тантала прибавляют величину, равную содержанию Та2О5 в осадке ниобия. В каждой окиси определяют содержание титана колориметрическим способом.
Согласно другому варианту метода \ смесь окислов растворяют в точно необходимом для образования двойных фторидов количестве фтористоводородной кислоты. Прибавляют избыточное количество насыщенного раствора хлорида калия и по охлаждении разбавляют до такого объема, чтобы оксифторониобат калия остался в растворе (растворимость оксифто-рониобата калия 1 : 12). Осадок отфильтровывают, промывают холодным насыщенным раствором хлорида калия и затем растворяют в воде, содержащей небольшое количество фтористоводородной кислоты. К раствору прибавляют еще некоторое количество раствора хлорида калия, охлаждают и фильтруют. Осадок промывают, как прежде, и затем нагревают с серной кислотой для удаления фтора. Остаток растворяют в воде, подкисленной соляной кислотой, и осаждают тантал аммиаком. Фильтраты, после отделения фторотанталата калия, выпаривают с серной кислотой, нагревают до удаления фтора и определяют ниобий, как в предыдущем методе. Если в смеси окислов присутствует титан, его определяют колориметрическим способом в обоих осадках — ниобия и тантала.
Предложена еще следующая модификация метода1 2. Растворяют 0,5—1 г окислов, предпочтительно свободных от титана, в точно необходимом количестве фтористоводородной кислоты. Если- введен избыток кислоты, его удаляют выпариванием и затем осадок растворяют в воде. Прибавляют фтористоводородную кислоту из расчета, чтобы избыток ее составлял 4,3%, а затем фторид калия в количестве, необходимом для образования KaNbOF5, считая, что взятый для анализа осадок полностью состоит из Nb2O5. Выпаривают досуха и обрабатывают остаток 0,75%-ной фтористоводородной кислотой при кипячении. Горячий раствор фильтруют. Осадок фторотанталата калия сразу же промывают горячей водой (двумя порциями по 10 мл) и сохраняют. Фильтрат выпаривают досуха, после чего нагревают при 120° С. Сухой остаток растворяют в избытке горячей воды и затем к раствору прибавляют 0,1 н. раствор едкого кали до растворения выделяющегося сначала осадка. Выпаривают, растворяют остаток в 10 мл воды и снова выпаривают. Растворение остатка в воде и вы
1 Е. Meimberg, Р. Winzer, Z. angew. Chem., 26, 158 (1913).
2 О. ,R u f f, E. Schiller, Z. anorg. Chem., 72, 348 (1911).
Методы, определения
685
паривание раствора повторяют еще 2 раза, в результате чего соль K4Ta4O5F14 стабилизуется и может бы, ь отфильтрована. Ниобий, как указано авторами, полностью остается при этом в фильтрате и может быть определен изложенным выше методом. Таким же способом можно установить содержание тантала в объединенных осадках.
Тантал можно отделить от ниобия, при содержании 0,01—0,09 г Nb2O5 в 0,1 г смеси пятиокисей, осаждением фениларсоновой кислотой GeH5AsO(OH)2 из сернокислого раствора, содержащего перекись водорода1. Этим способом не достигается строго количественное разделение ниобия и тантала, но, как указывают авторы, метод вполне приемлем для рядовых анализов, когда погрешность в ±5% относительных не имеет значения. См. также гл. «Цирконий» (стр. 638).
Описано хроматографическое отделение ниобия (>99%) от тантала (в растворах, содержащих НС1 + HF) с применением анионита сильно основного типа 2.
*Предложено, кроме того, хроматографическое разделение оксалатных комплексов ниобия и тантала на активной окиси алюминия 3, а для разделения фторидов ниобия и тантала и бтделения их от других элементов в качестве сорбента применена целлюлоза. Разделение производят последующим извлечением метилэтилкетоном, содержащим фтористоводородную кислоту 4.
Особого внимания заслуживают работы по разделению ниобия и тантала и отделению их от посторонних элементов экстрагированием органическими растворителями непосредственно из растворов, без использования хроматографии5. Этот процесс изучался главным образом применительно к разделению ниобия и тантала, однако он может быть весьма интересен и для отделения ниобия и тантала от титана, особенно при применении колориметрического метода определения тантала с пирогаллолом (стр. 691). Этот метод приобрел большое практическое значение. В условиях, колориметрического определения тантала чувствительность реакции пирогаллола с титаном почти в 5 раз выше, чем с танталом. В связи с этим погрешность анализа в Значительной мере зависит от степени очистки окислов ниобия и тантала от титана, а между тем, как уже было указано, при применении обычно принятых методов эта операция, помимо ее продолжительности, связана с известными потерями ниобия и тантала.
Разделение тантала и титана методом экстракции специально не изучалось. Однако имеются указания, что в определенных условиях (при извлечении диазопропилкетоном из раствора, 6 М по содержанию серной кислоты и 0,4 М по содержанию фтористоводородной кислоты)
1 Н. Fuck е, J. Daublander, Tech. Mitt. Krupp, 14, 174 (1939).
2 К. A. Kraus, G. E. Moore, J. Am. Chem. Soc., 71, 3855 (1949).
’ Tikhomirof f, C. r. Acad. Sci., 236, 1263 (1953).
4 A. F. Williams, J. Chem. Soc., 1952, 3155; R. A. Mercer, A. F. Willi a m s, J. Chem. Soc., 1952, 3399; F. H. Burstall, A. F. Williams, Analyst, 77, 983 (1952); R. A. Mercer, R. A. Wells, Analyst, 79, № 939, 1339 (1954).
5 G. W. Le d d i с о 11 e, F. L. Moore, J. Am. Chem. Soc., 74, № 6, 1618 (1952); P. C. S t i v e n s о n, H. G. Hicks, Anal. Chem., 25, № 10, 1517 (1953); H. G. Hicks, R. S. Gilbert, Anal. Chem., 26, № 7, 1205 (1954); J. Y. Eilenburg, G. W. Leddicotte, F. L. Moore, Anal. Chem., 26, .V; 6, 1045 (1954).
686 Гл. XXXIX. Ниобий и тантал
тантал и ниобий переходят в органическую фазу, тогда как титан совместно со многими другими элементами остается в водном слое1. Доп. перев.*
Объемное определение ниобия 2'
Количественное определение ниобия, основанное на восстановлении его амальгамированным цинком до трехвалентного состояния и последующем титровании перманганатом калия, обычно не дает достаточно точных и воспроизводимых результатов. Изучение различных факторов, влияющих на определение ниобия, показало, что существенное значений имеют условия амальгамирования цинка, продолжительность восстановления в присутствии избытка цинка, кислотность и температура раствора, а также концентрация ниобия в растворе.
Установлено3, что для успешного восстановления многих металлов требуется, чтобы цинк был покрыт лишь легким слоем ртути. Удовлетворительное амальгамирование цинка достигается в следующих условиях: 10Q0 г измельченного до 20 меш (d = 0,833 мм) металлического цинка (с низким содержанием железа) обрабатывают в прочной широкогорлой склянке 500 мл 2 %-ного водного раствора хлорида ртути (II), непрерывно встряхивая 45—60 сек, после чего жидкость сливают. Амальгамированный цинк промывают 5 раз дистиллированной водой, затем обрабатывают 500 мл теплой разбавленной (1 : 99) серной кислоты, перемешивают, кислоту сливают и снова тщательно промывают дистиллированной водой. Приготовленную амальгаму сохраняют в воде, подкисленной несколькими каплями соляной кислоты. Срок действия такой амальгамы ограничен, так как по мере использования она теряет свои свойства.
Исследования- показали, что для количественного восстановления ниобия концентраций серной кислоты в растворе должна составлять 20% и не менее 15% по объему. При содержании в растворе 15% кислоты обычно получаются пониженные, но все еще удовлетворительные результаты. В этих условиях раствор после восстановления прозрачен и имеет «аметистовую» окраску. При более низкой концентрации кислоты раствор после восстановления приобретает темный (черноватый) цвет и результаты для ниобия получаются пониженными.
Присутствие небольшого количества янтарной кислоты, вводимой для предотвращения гидролиза, на восстановление ниобия влияния не оказывает.
Результаты опытов показали, что существенное значение для восстановления ниобия имеют продолжительность соприкосновения раствора с амальгамой и количество активно действующего цинка, которое в свок> очередь зависит от величины активной поверхности амальгамы и от степени амальгамирования цинка. Весь раствор должен быть пропущен через редуктор в течение не менее 25 мин.
Точно установить оптимальные концентрации ниобия в растворе при использовании различных редукторов чрезвычайно затруднительно, так как в редукторе раствор разбавляется и, вследствие неравномерного сме
1 Р. С. Stivenson, Н. G. Hicks, Anal. Chem., 25} № 10, 1517 (1953),
2 Н. -В. Knowles, G. Е. F. L u n d е 11, J. Research NBS, 42, 405 (1949).
. 2 H. W. Stone, D. N. Hume, Ind. Eng. Chem., Anal, Ed., 11, 598 (1939);
H. A. Liebhaf sky, J. Am. Chem. Soc., 59, 452 (1937),
Методы определения
687
шения, недостаточно однороден. Хотя в длинном редукторе (—80 см} ниобий полностью восстанавливается при содержаний до 1,7 мг NbaO5 в 1 мл, однако исследования показали, что нецелесообразно увеличивать концентрацию Nb2O5 более чем до 1,0 мг в 1 мл. При применении короткого редуктора (~45 cjt) концентрация Nb2Os в' растворе не должна превышать 0,7—0,8 мг в 1 мл.
Нагревание раствора не является необходимым условием для количественного восстановления ниобия, однако во всех случаях этот процесс предпочитают проводить в растворах, имеющих температуру 65 ± 5° С. При этой температуре облегчается диффузия цинка через амальгаму, благодаря чему при прохождении раствора через редуктор реакция протекает более энергично.
В нижеследующей методике изложены условия, которые следует соблюдать при восстановлении ниобия в растворах, содержащих до 300 мг Nb2Os в редукторе, высота столба амальгамированного цинка в котором равна — 80 см.
Растворы, содержащие не более 175 Мг Nb2O6, можно восстанавливать в редукторе с высотой столба цинка —45 см.
Рекомендуемый метод. . Навеску высушенной взвешенной пробы, содержащую не более 300 .мг Nb2O5, сплавляют, с 3—5 г пиросульфата калия в закрытом кварцевом тигле емкостью 25 мл. По охлаждении переносят закрытый тигель в стакан емкостью 250 мл, прибавляют 20 мл серной кислоты и, закрыв стакан, слабо нагревают до растворения плава. Охлаждают, прибавляют 1—2'мл перекиси водорода (30%-ной) и осторожно разбавляют 100 мл воды. Затем ополаскивают тигель и крышку и вынимают их из стакана. Раствор снова охлаждают, вводят 20 мл серной кислоты, разбавляют до 200 мл водой, прибавляют 2 г янтарной кислоты, которую растворяют, слабо нагревая рдствор при перемешивании.
Ниобий восстанавливают в редукторе с высотой столба цинка —ВО см следующим способом. Сперва очищают и согревают редуктор, пропустив через него горячую разбавленную (1 : 19) серную кислоту, а затем горя; чую воду (90° С). Промывные жидкости отбрасывают. В приемник вводят трехкратный избыток раствора соли железа (III) и присоединяют приемник к редуктору, в котором находится 100 мл разбавленной (1 : 4) серной кислоты, нагретой до 65 ± 5° С. Применяя отсасывание, сливают через редуктор сначала эту кислоту, затем последовательно нагретые До 65 ± 5° С анализируемый раствор и 150 мл разбавленной (1 : 4) серной кислоты, содержащей 1% янтарной кислоты, и; наконец, 200— 250 мл холодной воды. Растворы пропускают непрерывно и с такой скоростью (равномерной), чтобы прохождение их через редуктор продолжалось не менее 25 мин. Во время проведения операции раствор восстановленного ниобия охлаждают, погрузив приемник в ледяную воду. Приемник отделяют от редуктора, ополаскивают трубку и дают стечь жидкости обычным способом. В полученный раствор вводят 10 мл фосфорной кислоты (85%-ной) и титруют его 0,1 н. раствором перманганата калия (1 мл соответствует 0,0066455 г Nb2O5), применяя ортофенантролин в качестве индикатора. Вводят поправку на количество перманганата, расходуемое на титрование реактивов, обработанных таким же способом, как и анализируемый раствор. Чтобы обеспечить отсутствие способных окисляться примесей, каждую кислую промывную жидкость в горячем состоянии слабо окрашивают несколькими каплями раствора перманганата калия.
«88
Гл. XXXIX. Ниобий и тантал
Непосредственное колориметрическое определение ниобия
В слабо сернокислых растворах ниобий дает с перекисью водорода светло-желтую окраску \ интенсивность которой возрастает с повышением концентрации кислоты ш достигает максимума в 100%-ной кислоте (пл. 1,845 г!см1 2 3 при 15° С).
Тантал с перекисью водорода окраски не дает. Окраска титана с повышением кислотности раствора бледнеет и в 100%-ной кислоте интенсивность ее становится настолько незначительной, что, внося поправку на содержание титана, можно определять ниобий.
Ход определения. Смесь окислов ниобия и тантала, предпочтительно в количестве 0,1 г, полученную методом, который обеспечивает отделение основной массы титана, прокаливают, взвешивают и затем сплавляют с 5 г пиросульфата калия. Плав растворяют в 10 мл серной кислоты, прибавив такое же количество перекиси водорода (3%-ной). По растворении вводят еще 20 мл перекиси водорода и разбавляют раствор до определенного объема. Конечная концентрация кислоты в растворе должна быть 10—20% по объему. Светопоглощение полученного раствора или аликвотной его части определяют в фотометре с ртутной лампой и с соответствующим светофильтром (436 ммк). Содержание титана вычисляют на основе результатов сравнения анализируемого раствора со стандартными растворами или по калибровочной кривой.
После этого раствор выпаривают до появления паров серной кислоты, разбавляют 100%-ной серной кислотой до определенного объема, вводят 1 мл перекиси водорода (30 %-ной) и определяют светопоглощение раствора или аликвотной его части, применяя свет ртутной лампы и соответствующий светофильтр (436 ммк). Содержание ниобия вычисляют по калибровочной кривой, построенной на основе результатов измерения светопогло-щения растворов с определенным содержанием ниобия. Для получения истинного содержания ниобия вводят поправку на титан из расчета, что 0,422 мг титана соответствует 0,70 мг ниобия. Содержание тантала вычисляют по разности 2.
*В присутствии титана предложено3 колориметрически определять ниобий в смеси 60% H2SO4 и 4% Н3РО4. В этих условиях чувствительность реакции на ниобий несколько снижается, но зато практически полностью устраняется влияние титана. В этом случае 10 мг Ti соответствуют лишь 0,23 мг Nb.
Установлена2 также возможность определения ниобия по реакции с перекисью водорода в растворах, содержавших в 50 мл раствора 25 мл серной кислоты и 5 мл фосфорной кислоты (85%-ной), если измерение све-топоглощения проводить при 342 ммк. Авторы метода отмечают, что в этих условиях чувствительность метода повышается, но зато резко сказывается влияние титана.
Имеются указания4 на возможность одновременного определения ниобия и тантала по реакции с перекисью водорода в концентрированной
1 Р. Klinger, W. Koch, Tech. Mitt. Krupp Forschungsber., 14, 179 (1939).
2 Спектрофотометрическое определение ниобия в ультрафиолетовой области спектра см. G. Т е 1 е р, D. F. Boltz, Anal. Chem., 24, 163 (1952).
8G. Thanheiser, Arch. f. Eisenhiittenw., 13, 41 (1940).
4 F. С. P a 11 i 1 a, N. Adler, C. F. H i s k e y, Anal. Chem., 25, № 6, 926 (1953).
Методы определения
689
серной кислоте (на 45 мл раствора пиросульфатного плава пятиокисей в свежевыпаренной серной кислоте 5 мл 30%-ной Н2О2). Измерение светопоглощения ниобиевого комплекса рекомендуется проводить при 365 ммк, а танталового комплекса — при 285 ммк. При этом следует учитывать взаимное влияние ниобия и тантала на результаты и вводить соответствующие поправки.
Наиболее чувствительным методом определения ниобия, который получил широкое практическое применение^ является роданидный, основанный на реакции образования окрашенного в желтый цвет комплексного соединения ниобия с родановодородной кислотой. Это соединение экстрагируется кислородсодержащими органическими растворителями (спиртами, эфирами, альдегидами и кетонами). В среде органического растворителя чувствительность реакции резко повышается.
На возможность колориметрического определения ниобия по его реакции с роданидом в солянокислых растворах, содержащих хлорид олова (II) и винную кислоту, впервые указали Л. Н. Моньякова и П. Ф. Федоров1. По их наблюдениям образующееся в этих условиях соединение экстрагируется эфиром, и содержание ниобия можно определить по интенсивности желтой окраски эфирного слоя. Механизм этой реакции и влияние на нее различных факторов, подробно изученные И. П. Алима-риным и Р. Л. Подвальной2, рассмотрены ниже; Титан также дает окрашенный в желтый цвет роданидный комплекс, но чувствительность реакции на титан во много раз меньше, чем на ниобий, и при соотношении Nb : Ti = 1 : 30 еще возможно достаточно точное определение ниобия при условии, если концентрация TiOa в анализируемом растворе не превышает 0,3 мг в 10 мл. Тантал в условиях определения ниобия дает с ро-данид-ионами бесцветный комплекс. Определению ниобия мешают молибден, фольфрам, уран, ванадий, железо, хром, кобальт, медь, золото и платина, образующие в этих условиях окрашенные соединения с роданидом. При экстрагировании эфиром устраняется влияние хрома, урана, железа и меди, которые остаются в водном слое. Совместно с ниобием эфиром извлекаются окрашенные роданиды молибдена, вольфрама, титана, кобальта и платины. Соединения золота, селена и теллура восстанавливаются до элементарного состояния и покрывают стенки сосуда, что мешает наблюдению окраски ниобиевого комплекса.
Препятствуют образованию желтого комплекса ниобия С2О1 , F , SO1 , РОГ и AsOl , причем наибольшее действие оказывают окса-лат-ионы. Нитрат-ионы реагируют с родановодородной кислотой с образованием кроваво-красного окрашивания. Не оказывают влияния на реакцию сг, в г, г, сю;, ЭОГи ВОГ-
Винная и лимонная кислоты замедляют реакцию образования окрашенного комплекса ниобия.
На образование роданидного комплекса ниобия значительное влияние оказывают концентрации соляной кислоты и роданида в растворе. Интенсивность окраски нарастает в течение 30—40 мин после введения реагентов и затем остается постоянной в продолжение нескольких часов,
1 Л. Н. Моньякова, П. Ф. Федоров, Бюллетень отдела изобретений Госплана при СНК СССР, 41 (1942).
2 И, П. Алимарин, Р. Л. Подвальная, ЖАХ, 1, 30 (1945)j
44 Заказ 522.
690
Гл. XXXIX. Ниобий и тантал
Изменение температуры в пределах 10—20° С на интенсивность окраски влияния не оказывает.
На основе результатов этих исследований разработан количественный метод определения ниобия, который в общих чертах сводится к следующему. 5—50 .из окислов ниобия и тантала, после отделения от посторонних примесей, сплавляют с 0,5—2 г пиросульфата калия. Плав растворяют в 10—20 мл 15 %-ного раствора винной кислоты. Раствор разбавляют в мерной колбе до 25—100 мл. К 2 мл анализируемого раствора, содержащим 0,018—0,2 мг Nb2O5, прибавляют 5 мл 20%-ного раствора хлорида олова (II) и 5 мл соляной кислоты (пл. 1,12 г/см3). После введения каждого реактива жидкость перемешивают. В конечном растворе концентрация роданида должна находиться в пределах 7—15%, а содержание соляной кислоты (пл. 1,12 г/см3) —30—50%. К раствору прибавляют 10 мл эфира и энергично встряхивают. Через 30—40 мин интенсивность желтой окраски эфирного слоя измеряют фотометрически, применяя светофильтр с областью пропускания 400—450 ммк.
Содержание ниобия можно определить также визуально сравнением окраски эфирного слоя по методу стандартных серий, причем в качестве стандартов можно пользоваться растворами соли ниобия, обработанными таким же способом, как и анализируемый раствор, или же раствором хромата калия, 30,8 мг которого соответствуют 1 мг NbaO6.
При использовании роданидного метода определения ниобия были сделаны некоторые дополнительные наблюдения, которые могут иметь известное практическое значение. Так, например, имеются указания \ что извлечение эфиром следует проводить не позднее чем через 5 мин после смешения анализируемого раствора с реагентами, в противном случае интенсивность окраски в водной среде постепенно нарастает и получаются невоспроизводимые результаты. Некоторые авторы1,2 считают, что в раствор лучше сначала вводить хлорид олова (II), затем соляную кислоту и в последнюю очередь роданид, так как при таком порядке введения реагентов быстрее нарастает окраска и получаются более устойчивые результаты. При содержании значительных количеств тантала рекомендуют1,2,3 повысить концентрацию винной кислоты в колориметрируемом растворе, чтобы воспрепятствовать гидролитическому осаждению тантала, при котором в осадок увлекаются некоторые количества ниобия.
Разработаны также условия определения ниобия в виде роданида без экстрагирования, в водно-ацетоновой среде 3. По этому способу осадок пятиокисей ниобия и тантала сплавляют с 2,5 г бисульфата калия. Плав растворяют в 200 мл 1,2 М винной кислоты и разбавляют до 500 мл. В мерную колбу емкостью 50 мл вводят 10 мл концентрированной соляной кислоты, 1 мл 2 М раствора хлорида олова (II), 5 мл воды и 10 мл ацетона, перемешивают и охлаждают 15 мин при 20° С. Затем вводят 10 мл 3 М раствора роданида калия и 10 мл анализируемого раствора. Снова охлаждают 5 мин, после чего разбавляют до метки и измеряют светопоглощение раствора при 385 ммк точно через 15 мин после введения в раствор роданида калия. В случае повышения концентрации соляной кислоты, роданида и ацетона в растворе может происходить выделение солей.
1 А. В. N. Law-Zech a, S. S. Lord Jr., D. N. Н u m е, Anal. Chem., 24, № 7 (1952). /
2 A. E. O. Marzys, Analyst, 79, № 939 (1954).
3 H. Freund, A. E. Lewi 11, Anal. Chem., 23, 1813 (1951).
Методы, определения
691
Авторы отмечают, что на результаты определения ниобия несколько влияет тантал, несмотря на то что он дает с роданидом бесцветный комплекс, и рекомендуют на его содержание вводить поправку. Результаты дальнейшего изучения метода1 показали, что в водно-ацетоновой среде более резко сказывается влияние титана, так как чувствительность реакции на него в этих условиях значительно выше, чем в случае экстрагирования эфиром, тогда как чувствительность определения ниобия, наоборот, заметно понижается в водно-ацетоновой среде. Доп. перев*
Фотометрическое определение тантала
Фотометрический метод определения тантала основан на образовании окрашенного в желтый цвет комплексного соединения с пирогаллолом в слабокислых растворах, содержащих оксалат аммония 2. Титан и молибден также образуют окрашенные комплексы с пирогаллолом. Кроме того, при применении этого метода следует учитывать окраску оксалата железа (Ш).
*Применение пирогаллола для колориметрического определения тантала в кислой среде и ниобия в щелочной среде впервые было предложено М. С. Платоновым, Н. Ф. КривоПглыковым и А. А. Маракаевым3. Основанный на реакции с пирогаллолом колориметрический метод определения тантала получил большое практическое значение. Определение выполняют следующим способом2. Прокаленные окислы ниобия и тантала в количестве 0,02 г сплавляют в фарфоровом тигле с 6 г бисульфата калия. Плав растворяют в 70 мл 4%-ного раствора оксалата аммония при нагревании. Полученный раствор разбавляют в мерной колбе до 100 мл водой (pH раствора должен находиться в пределах 1—2). К 10 мл раствора прибавляют 1,2 г пирогаллола и измеряют светопоглощение раствора при длине волны 436 ммк. Нулевым раствором служит анализируемый раствор, в который не введен пирогаллол. Содержание тантала вычисляют по калибровочной кривой.
В другой порции раствора определяют титан и на его содержание вводят поправку.
В этих условиях 1 мг TiO2 соответствует 4,75 мг Та2О5.
Вместо того чтобы вносить поправку на титан, можно, определив его содержание, вводить соответствующие количества в нулевой раствор, которым в данном случае должен служить раствор всех применяемых реагентов, включая пирогаллол.
В аликвотной части раствора, взятой для колориметрирования, допускается содержание до 20 мкг TiO2, но не более 10% по отношению к танталу. Судя по приведенным в работе результатам исследования, концентрацию пирогаллола в колориметрируемом растворе можно снизить до 2,5—3% без особого ущерба для интенсивности окраски.
Необходимо иметь в виду, что, помимо упомянутых титана и молибдена, окраску с пирогаллолом дают также железо, ванадий, уран и
-1 А. Е, О. Marz у s, Analyst, 79, № 939 (1954).
2G. Thanheiser, Mitt. Kaiser Wilhelm Inst. Eisenforsch., Dusseldorf, 22, 255 (1940).
3M. С. Платонов, H. Ф. Кривошлыков, А. А. Маракаев, ЖОХ, 6, 1815 (1936); 9, 539 (1939); M. С. Платонов, Н. Ф. Кривошлыков, Труды Всесоюзной конференции по аналитической химии, т. II, 1943, стр. 359, 44*
692
Гл. XXXIX. Ниобий и тантал
вольфрам. Кроме того, имеются указания \ что тантал и ниобий оказывают взаимное влияние на результаты колориметрического определения. Согласно приведенным данным, при содержании в анализируемом растворе (имеющем pH = 1—2) 4% оксалата аммония, 2% бисульфата калия, 1,6% пирогаллола и измерении светопоглощения при 400 ммк 1 мг Nb2O5 соответствует 0,0072 мг Та2О5. В условиях колориметрирования ниобия, а именно, при содержании в 50 мл раствора 5 мл 10%-ного раствора бисульфата калия, 20 мл 4%-ного раствора оксалата аммония, 20 мл 2%-ного раствора пирогаллола в 20%-ном растворе сульфата натрия и измерении светопоглощения при 410 ммк 1 мг Та,ОБ соответствует 0,03 мг Nb2OB.
Определение тантала по реакции с пирогаллолом рекомендуется 1 2 также проводить в 4 н. соляной кислоте. Согласно указаниям автора, в этих условиях снижается влияние титана и ниобия, а влияние железа устраняется введением в раствор олова (И). Доп. перев. ’
ПРОТАКТИНИЙ
Протактиний является крайне редким элементом. Его содержание в земной коре, поскольку это установлено в настоящее время, составляет 7-10-11%, т. е. он один из наименее распространенных в природе элементов. Те небольшие количества протактиния, которые были получены, выделены из осадков фосфата циркония, получающихся в процессе извлечения радия из урановой смолки.
По своему отношению к реагентам протактиний мало напоминает ниобий и тантал 3. По аналитическим свойствам он проявляет значительное сходство с цирконием. Так, например, он: 1) переходит в раствор при выщелачивании разбавленной серной кислотой пиросульфатного плава его окиси; 2) остается в нерастворимом осадке при выщелачивании водой плава его окиси с карбонатом калия; 3) полностью осаждается: а) при добавлении избытка перекиси водорода к горячему (40—60° С) раствору его в 2 %-ной серной кислоте; б) аммиаком; в) фосфорной кислотой из разбавленного сернокислого раствора, содержащего перекись водорода; 4) не образует осадка с фтористоводородной кислотой4.
Пятиокись протактиния — тяжелое белое порошковидное вещество, имеющее высокую температуру плавления, нерастворима в концентрированных серной, азотной и соляной кислотах. Этот окисел имеет отчетливые (хотя и слабые) основные свойства и не имеет кислотных свойств.
1 Е. С. Hunt, R. A. Wells, Analyst, 79, № 939, 345 (1954).
2 J. I. D i n n i n, Anal. Chem., 25, 1803 (1953).
3 A. V. Grosse, J. Am. Chem. Soc., 52, 1742 (1930); M. Bachelet, G. Bouissieres, Bull. Soc. chim., 11, 169 (1944).
4 Радиоактивный метод определения протактиния в земных силикатных породах и в метеоритах после осаждения его совместно с фосфатом циркония и очистки прокаленного осадка фосфатов приводят W. С. S с h u m b, R. ~D. Е vans и J. L. Hastings, J. Am. Chem. Soc., 61, 3451 (1939).
ЩЕЛОЧНОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ
И МАГНИЙ
КАЛЬЦИЙ, СТРОНЦИЙ, БАРИЙ, МАГНИЙ (РАДИЙ)
Гл а в a XL
ЩЕЛОЧНОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ — КАЛЬЦИЙ, СТРОНЦИЙ, БАРИЙ
Кальций является одним из наиболее распространенных в природе металлов, но, так же как и стронций и барий, он отсутствует в земной коре только в связанном состоянии. Кальций — один из основных компонентов многих породообразующих минералов и встречается в виде карбоната — известняка СаСО3, сульфата — гипса CaSO4 2Н2О, фторида — плавикового шпата CaF2, фторо- и хлорофосфата — апатита Ca5F(PO4)3 (Cl изоморфен с F) и во многих других минералах. Барий и стронций встречаются главным образом в полевошпатовых породах, причем барий почти всегда находится в большем количестве, чем стронций х. Барий находили в немногих характерных породах в количестве до 1%, считая на окись бария, но в большей части силикатных пород США содержание его значительно ниже 0,2%. Его находят также в виде сульфата — барита BaS04 и карбоната — витерита ВаСО3. Стронций обнаруживали в количестве до 0,3—0,4%, но обычно содержание его лишь порядка следов. Важнейшими минералами, содержащими стронций, являются сульфат стронция — целестин SrS04 и карбонат стронция — стронцианит SrCO3.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород определение щелочноземельных металлов не вызывает затруднений, если раствор содержит только
1 Есть некоторые указания на то, что барий чаще и в большем количестве находится в породах с высоким содержанием калия. Барий встречается в полевых шпатах, особенно в ортоклазе, в виде молекул цельзиана BaAlaSi2O8, в редком гиалофане K2BaAl4Si8O24, в некоторых цеолитах, а также' в немногих биотитах и мусковитах. В настоящее время мы не в состоянии сделать определенных выводов о характере пород, содержащих стронций, и поэтому аналитические исследования в этой области представляют интерес.
694
Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
хлориды и соляную кислоту. Другие кислоты или их соли могут создать осложнения при определении. Например, в присутствии серной кислоты и сульфатов барий (а иногда также стронций и кальций) будет в осадке вместе с кремнекислотой; если раствор содержит фториды, то кальций перейдет в осадок от аммиака вместе с железом и алюминием; если присутствуют карбонаты или фосфорная кислота в количестве, превышающем то, какое может быть связано железом и алюминием, тогда в осадке от аммиака будут все три металла — кальций, стронций и барий.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ЩЕЛОЧНОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ
Разложение минералов, содержащих щелочноземельные металлы, не представляет затруднений и проводится либо непосредственно кислотами, обычно соляной кислотой, либо сплавлением с карбонатом натрия с последующим растворением плава в кислоте, либо, наконец, предварительным растворением в кислоте, например, известняка с 'Последующим сплавлением нерастворимого остатка.
МЕТОДЫ ОТДЕЛЕНИЯ
Щелочноземельные металлы легко отделяются от мешающих их определению элементов обычными методами: осаждением сероводородом, аммиаком (не следует допускать большого его избытка) и сульфидом аммония. Следует, конечно, избегать введения сульфат-ионов, если присутствует барий или значительные количества стронция. Надо также остерегаться фосфат- и арсенаттионов, выделяющих щелочноземельные металлы из аммиачных растворов в том случае, если железо, алюминий и т. п. присутствуют в недостаточном количестве.
Отделение кальция от магния
Если из этих двух элементов преобладает кальций, то он обычно отделяется от магния двукратным осаждением оксалатом аммония, как описано на стр. 705. Можно провести удовлетворительное разделение этим методом и тогда, когда содержание магния в анализируемом растворе равно или не намного превышает содержание кальция, но тогда может потребоваться трехкратное осаждение кальция х. В таких случаях тот же результат может быть получен при меньшем числе осаждений, но с большим трудом, если, например, применять метод Ричардса 1 2, описанный на стр. 707.
Кальций можно количественно отделить от магния.осаждением молибдатом аммония из кипящего слабокислого или слабощелочного раствора 3.
1 Например, в результате трех анализов доломита, содержавшего 21,54% MgO, было получено окиси кальция после двукратного осаждения 30,63; 30,62 и 30,68%. После растворения осадков и третьего осаждения содержание окиси кальция понизилось до 30,48; 30,45 и 30,49%. После растворения этих осадков и осаждения в четвертый раз содержание кальция осталось тем же.
2 Например, при анализе доломита, о котором было сказано в предыдущей сноске, однократные осаждения этим методом дали 30,84; 30,79 и 30,78% окиси кальция; двукратные осаждения — 30,42 и 30,40%, а трехкратное осаждение — 30,44%.
3 R. С. Wiley, Ind. Eng. Chem., Anal. Ed., 3, 127 (1931).
Методы отделения
695
Осадок молибдата кальция можно отфильтровать через фильтрующий тигель, промыть горячей водой, высушить при 130° С и взвесить в виде СаМоО4.
Если требуется исключительно точное определение очень малых количеств кальция, особенно в присутствии большого количества магния, как, например, при анализе магнезита, то прямое осаждение кальция оксалатом не дает удовлетворительных результатов. Все применявшиеся раньше методы были основаны на осаждении кальция сперва в виде сульфата в спиртовом растворе, а затем в виде оксалата. Осаждение в виде сульфата предполагает, однако, отсутствие щелочных металлов, сульфаты которых большей частью трудно растворимы в спирте. Следовательно, эти методы не пригодны для анализа таких минералов, которые приходится разлагать сплавлением с карбонатом натрия, а к ним в первую очередь относится большинство, природных силикатов. Пользование некоторыми удовлетворительными методами1,2 требует применения сульфата лития, по другим 3 методам предполагается предварительное знание количества присутствующего кальция. Значительно лучший метод основан на предложенном В. Ф. Гиллебрандом способе извлечения из прокаленного остатка пирофосфата магния того малого количества кальция, которое осталось в фильтрате после осаждения кальция в виде оксалата и перешло затем в осадок при осаждении магния фосфатом. При применении этого метода обычное осаждение оксалатом опускают, кальций осаждают в виде фосфата вместе с магнием и определяют следующим образом.
Осаждают магний вместе с кальцием в виде фосфата по методу, изложенному в гл. «Магний» (стр. 719) 4, переносят взвешенный остаток после прокаливания в маленький стакан и растворяют в малом количестве серной кислоты, избегая избытка ее, большего чем 0,5 мл. Если прокаленный остаток растворяется с трудом, кипятят его с азотной кислотой и выпаривают до выделения обильных паров серной кислоты. На каждые 0,3 г первоначально присутствовавшего пирофосфата прибавляют по 100 мл 75%-ного (по объему) спирта и дают постоять раствору несколько часов или, лучше, в течение ночи, если количество кальция очень мало в. Фильтруют, промывают остаток сульфата кальция 75%-ным спиртом и высушивают. Затем растворяют остаток в очень небольшом количестве разбавленной соляной кислоты, осаждают кальций в возможно малом объеме раствора в виде оксалата и прокаливают до окиси кальция е. Если магний не подлежит определению, то осажденные фосфаты обоих металлов
1 С. St о] berg, Z. angew. Chem., 17, 741 (1904).
2 О. Kallauner, I. Preller, Chem. Ztg., 36, 449, 462 (1912).
3 E. M u r m a n n, Z. anal. Chem., 49, 688 (1910).
4 Барий также сопровождает магний, но осаждение бария не происходит вполне количественно, даже когда магний преобладает. Литий совершенно не захватывается осадком магния, если осаждение проводится двукратно и на холоду.
« В 100 мл смеси растворяется приблизительно 0,06 г окиси магния в виде MgSO4 и менее 0,1 мг окиси кальция в виде CaSO4. Для осаждения сульфата кальция можно также применять смесь, содержащую 90 частей этилового спирта, 10 частей метилового спирта и 1 часть серной кислоты; в 100 мл этой смеси растворяется MgSO4—0,46 г в пересчете на окись магния.
6 Исследование, проведенное Н. В. Knowles, показало, что кальций этим методом полностью извлекается из смесей, содержащих 0,0005 г СаО и 0,1 г MgO; 0,001 г СаО и 0,05 г MgO и, наконец, 0,05 г СаО и 0,05 г MgO.
696
Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
можно сразу растворить без прокаливания и взвешивания прокаленного остатка
Окись кальция можно экстрагировать 30%-ным раствором сахара 1 2 3 и таким образом количественно отделить ее от окиси магния, обрабатывая таким способом прокаленную смесь обеих окисей.
Отделения кальция, стронция и бария от других элементов
Для отделения трех щелочноземельных металлов друг от друга лучше, чтобы они*находились в виде хлоридов или безводных нитратов. Предварительно эти металлы надо отделить от других элементов. Некоторые из последних отделяются вначале, а отделение от других проводится совместным осаждением всей щелочноземельной группы.
Групповое отделение в виде оксалатов. Групповое отделение обычно проводят осаждением (не вполне количественным) щелочноземельных металлов в виде их оксалатов, а затем прокаливанием превращают оксалаты в окиси, как это описано для кальция (стр. 707).
Дальнейшая обработка описана ниже в разделе «Отделение кальция, стронция и бария друг от друга» (стр. 697).
' Надо хорошо понять различное поведение щелочноземельных элементов в отношении оксалата аммония. Ни один из получаемых оксалатов не является полностью нерастворимым в условиях осаждения. Оксалат стронция осаждается почти так же полно, как оксалат кальция, но оксалат бария осаждается очень неполно а. Если барий присутствует в количестве до 3—4 мг, то он никогда не будет найден в осадке оксалатов кальция и стронция после двукратного осаждения, а очень часто в этом осадке не обнаруживают бария, даже если он находился в больших количествах. Барий следует извлекать из соединенных фильтратов (после отделения оксалатов бывает два фильтрата, так как осаждение йроводят двукратно). Если, однако, количество бария настолько велико, что только в третьем осадке его совсем не будет, то уместно провести и третье осаждение. Трехкратное осаждение следует проводить также и в присутствии большого количества магния.
Во всех случаях фильтраты надо выпарить, удалить присутствующие аммонийные соли осторожным прокаливанием в платиновой или фарфоровой чашке или обработкой концентрированной азотной кислотой в фарфоровой чашке (имея над жидкостью широкую»воронку во избежание потери от разбрызгивания), прилить к остатку воду в количестве, едва достаточном для полного его растворения, и прибавить небольшое количество серной кислоты и спирт в объеме, равном объему раствора. Это обеспечит полное осаждение бария и стронция, но не всего того не
1 Об отделении кальция в виде сульфата из спиртового раствора перед определением магния в фосфатных породах см. J. I. Hoffman, J. Research NBS; 9, 487 (1932). ,
2 А. С. S h е a d, В. J. Heinrich, Ind. Eng. Chem., Anal. Ed., 2, 388 (1930); см. также A. C. Shead, R. K. Valla, там же, 4, 246 (1932).
3 W. F. Hillebrand, J. Am. Chem. Soc., 16, 83 (1894); C. A. Peters, Am. J. Sci., [4], 12, 216 (1901). Согласно второму автору, стронций почти полностью осаждается из раствора, пятую часть объема которого составляет 85%-ный спирт, а барий — из раствора, содержащего этот спирт в количестве, равном одной трети его объема.
Методы отделения
697
большого количества кальция, которое не выпало в осадок в виде оксалата. Оставшееся количество кальция извлекают из прокаленного остатка пирофосфата магния. Маленький осадок сульфатов следует отфильтровать, прокалить, превратить в нитраты и прибавить к главному раствору, полученному прокаливанием оксалатов до окисей и растворением их в азотной кислоте.
Прямое выделение бария осаждением его с фосфатом магния, как это применяется для кальция и стронция, недопустимо вследствие некоторой растворимости фосфата бария.
Групповое отделение в виде сульфатов. При некоторых условиях вся группа может быть отделена в виде сульфатов, как это описано для кальция (стр. 711). Этим методом получают большую полноту осаждения, чем описанным выше оксалатным методом, но он имеет недостатки в других отношениях. Сульфаты надо прокалить, сплавить с карбонатом натрия, выщелочить плав водой и растворить в соляной или азотной кислоте, избыток которой надо удалить выпариванием.
Дальнейшая обработка описана ниже.
Отделение кальция, стронция и бария друг от друга
Действительно хороших методов отделения кальция, стронция и бария друг от друга не существует. Все методы несовершенны и могут дать правильные результаты только вследствие взаимно компенсиру-' ющихся ошибок.
Разделение проводится одним из следующих двух способов ц зависимости от того, имеются ли указанные металлы в виде их нитратов или хлоридов.
1) Из смеси сухих нитратов извлекают нитрат кальция, пользуясь его растворимостью в смеси абсолютного спирта и абсолютного эфира или в концентрированной азотной кислоте. Из водного раствора оставшихся нитратов барий выделяют и взвешивают в виде хромата, а стронций определяют в фильтрате.
2) Из смеси хлоридов осаждают барий в виде хромата; из фильтрата осаждают кальций и стронций в виде карбонатов, которые превращают в нитраты и разделяют спирто-эфирной смесью, как описано ниже.
Отделения кальция от стронция и бария
Экстрагирование спирто-эфирной смесью. Растворимость нитрата стронция (высушенного при 130° С) в смеси равных объемов абсолютного спирта и абсолютного эфира при комнатной температуре равна 0,0023 г в 250 мл, тогда как при этих же условиях в 1 мл Указанного растворителя растворяется 0,37 г (92,5 г в 250 мл) нитрата кальция х. Определения, проведенные этим методом, показали, что для стронция получаются несколько повышенные результаты, а для кальция соответственно пониженные.
Мы приводим сначала метод обработки осадков, содержащих стронций и барий в малых количествах сравнительно с кальцием (что имеет
1 R. Fresenius, Z. anal. Chem., 32, 189 (1893).
698
Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
место в большинстве анализов горных пород), а затем — метод обработки осадков, содержащих большие количества бария и стронция.
Обработка осадка в присутствии малого количества стронция или бария или их обоих. Первая стадия метода заключается в превращении в нитраты той смеси карбонатов, которая получилась из смеси сульфатов (стр. 697) или взвешенных окисей, полученных после прокаливания оксалатов (стр. 696). Карбонаты прокаливают до окисей. Переносят окиси, полученные в том или другом случае, в маленькую колбу (колба емкостью 20—25 мл, как правило, вполне достаточна), добавляя, если нужно, 1—2 капли азотной кислоты, приливают достаточное количество той же кислоты для растворения смеси и выпаривают досуха, остерегаясь разбрызгивания. Спокойному выпариванию помогает продувание воздуха над раствором. Конечная температура должна быть 150—160° С, хотя по Фрезениусу 1 допустима температура и 180° С.
По охлаждении приливают к сухому остатку 10-кратное по массе количество абсолютного спирта, закупоривают колбу, осторожно перемешивают и оставляют на 1—2 ч. Затем прибавляют равный объем абсолютного эфира, закрывают колбу, снова осторожно перемешивают и оставляют на ночь (14—16 ч). Нерастворившиеся нитраты бария и стронция переносят на фильтр из тонкого асбеста или мелкораздробленной платины и осторожно отсасывают. Если применять бумажный фильтр, то размеры его должны соответствовать величине осадка и скорости промывания. Промывной жидкостью служит смесь равных объемов абсолютного спирта и абсолютного эфира. Если нерастворившийся в спирте и эфире остаток окажется значительным, то после промывания и высушивания его растворяют на фильтре в горячей воде, раствор выпаривают, высушивают остаток в колбе, как в первый раз, и повторяют обработку спиртов и эфиром. В соединенных фильтратах и промывных водах после выпаривания при низкой температуре определяют, если желательно, кальций одним из методов, приведенных в разделе «Кальций» (стр. 701 сл.)
Нерастворившиеся нитраты высушивают очень осторожным нагреванием и пропускают через фильтр горячую воду в количестве, достаточном для их растворения. Фильтр сжигают и взвешивают золу или тигель Гуча, если применялся последний. Масса остатка редко превышает (почти никогда) 0,5 мг, и обычно остаток не содержит щелочноземельных металлов. Следовательно, масса его должна быть вычтена из суммарной массы окисей, если только нельзя доказать, что эти примеси попали в остаток в результате выщелачивания стекла колбы, в которой выпаривался раствор нитратов. О дальнейшей обработке полученного раствора 2 см. стр. 700.
Обработка осадка в присутствии большого количества стронция или бария или их обоих. Нитраты высушивают в чашке при 130° С, измельчают их и экстрагируют пятью порциями по 5 мл указанной выше спирто-эфирной смеси. Перемешивают раствор после каждого добавления этой смеси и декантируют в маленькую колбу после оседания остатка. Колбу
1 Нитрат марганца разлагается ниже этой температуры с образованием черных окислов. Его следует поэтому отделять перед отделением стронция.
2 Knowles при однократном выщелачивании смесью эфира и спирта получил 0,0094 г SrO из смеси, содержавшей 0,0080 г SrO и 0,1 г СаО, и 0,0661 г SrO — из смеси 0,0641 г SrO и 0,05 г СаО.
Методы отделения
699
закрывают пробкой и отставляют ее. Остаток растворяют в воде, выпаривают раствор досуха и снова высушивают при 130° С. Затем остаток измельчают, переносят его в колбу и смывают в колбу, насколько возможно, нитраты, приставшие к чашке, тремя порциями растворителя по 5 мл каждая. Чашку сохраняют. Закрывают колбу пробкой, взбалтывают временами и оставляют на 24 ч. Затем фильтруют через маленький фильтр и промывают остаток декантацией 12 раз порциями растворителя по 5 мл. Если надо определить кальций, фильтрат обрабатывают, как указано выше.
Находящиеся в чашке, на фильтре и в маленькой колбе нитраты растворяют в воде, прибавляют спирт, осаждают серной кислотой и взвешивают в виде сульфатов.
Экстрагирование азотной кислотой. Был предложен 1 метод разделения, основанный на том, что нитрат кальция растворим в концентрированной азотной кислоте, тогда как нитраты стронция и бария практически в ней нерастворимы. Автор этого метода предпочитает применять азотную кислоту пл. 1,46 г/см3, но указывает, что кислота пл. 1,42 г!см3 также применима. В 100 мл первой кислоты растворяется приблизительно 0,7 г нитрата кальция, а в таком же объеме второй кислоты растворяется 7 г нитрата кальция.
К сухим нитратам щелочноземельных металлов приливают в избытке концентрированную азотную кислоту и некоторое время хорошо перемешивают. Кристаллы быстро оседают. Сливают прозрачную жидкость через тигель Гуча с платиновым фильтрующим сдоем или, если такого нет в лаборатории, через двойной бумажный фильтр, смоченный концентрированной азотной кислотой. Промывают малыми порциями этой же кислоты 2.
Осаждение азотной кислотой. Нитрат стронция можно полностью осадить из водного раствора в виде плотных кристаллов 3 и таким способом отделить стронций от кальция, магния, бериллия и 25 других элементов. Для этого надо к анализируемому раствору прибавлять по каплям и при сильном перемешивании 10%-ную азотную кислоту, пока не получится раствор, содержащий 79—81 % этой кислоты. Отделение от кальция проводят следующим способом.
Высушенные нитраты стронция и кальция растворяют в 10,0 мл воды, прибавляют из.бюретки по каплям, при механическом перемешивании, 26,0 мл 100%-ной азотной кислоты и дают постоять 30 мин. Затем фильтруют через взвешенный предварительно тигель Гуча, переносят осадок на фильтр с помощью 80 %-ной азотной кислоты и промывают его на фильтре 10 раз порциями примерно в 1 мл 80%-ной азотной кислоты.
1 S. G. Rawson, J. Soc. Chem. Ind., 16, 113 (1897).
2 В опытах Knowles, проведенных этим методом, нитраты были высушены при 130° С, экстрагированы 5—7,5 мл азотной кислоты (пл. 1,42 г/см3) в колбе с притертой стеклянной пробкой, затем раствор был перемешан и оставлен на ночь. Остаток был измельчен стеклянной палочкой, раствор был профильтрован через тигель с платиновым фильтрующим слоем и остаток промыт 20—25 мл концентрированной HNO3 после чего он был растворен в горячей воде, превращен в сульфат и взвешен. При анализе смесей, содержавших 0,0144 г SrO и 0,1 г СаО, было получено 0,0150 г и 0,0149 г SrO; при анализе смесей, содержавших 0,0055 г ВаО и 0,1 г СаО, было получено 0,0055 г и 0,0057 г ВаО.
3Н. Н. Willard, Е. W. Goodspeed. Ind. Eng. Chem., Anal. Ed., 8, 414 (1936).
700
Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
После высушивания в течение 2 ч при 130—140° С и охлаждения взвешивают в виде Sr(NO3)2.
Осаждение можно проводить при любой температуре в пределах от 20 до 70° С. Если присутствует менее 5 мг стронция, раствор надо перемешивать механической мешалкой в течение 45 мин. Если присутствует большое количество кальция, надо увеличить общий объем раствора (например, при осаждении 500 мг кальция надо прилить 30 мл воды и 78 мл азотной кислоты). Кроме того, когда содержание кальция превышает 50 мг, надо провести переосаждение: осадок растворяют в горячей воде, содержащей немного азотной кислоты, и вновь его осаждают таким же способом.
Барий и свинец мешают разделению,. так как барий полностью осаждается'при концентрации азотной кислоты, равной 76%, а свинец при концентрации ее, равной 84%. Умеренные количества соляной и хлорной кислот не мешают.
Отделение бария и стронция друг от друга
Отделение бария,в виде хромата. Нитраты, полученные, как указано на стр. 697, или хлориды обрабатывают для отделения и определения бария хроматным методом в том его единственном варианте \ которым можно .почти количественно отделить барий от кальция и стронция.
Для определения нужны следующие растворы:
Раствор бихромата аммония, свободного от серной кислоты; в 1 л раствора содержится 100 г соли.
Раствор ацетата аммония (раствор а): в 1 л раствора содержится 300 г нейтрализованной аммиаком соли.
Раствор ацетата аммония (раствор б): 20 мл раствора а разбавляют до 1 л.
Лучше, чтобы реакция ацетатных растворов была несколько щелочной, чем кислой. Ход разделения тех количеств, какие применялись авторами метода (по 0,135 г окиси каждого из щелочноземельных металлов, взятой в виде хлорида), состоит в следующем.
К нейтральному или слабокислому анализируемому раствору прибавляют в избытке раствор ацетата аммония (10 мл раствора а). Нагревают раствор до кипения и при перемешивании прибавляют 5 мл раствора бихромата аммония. После отстаивания и охлаждения декантируют прозрачную жидкость через фильтр и- промывают осадок раствором б, пока жидкость, стекающая с фильтра, не перестанет быть заметно окрашенной (около 100 мл промывной жидкости). Осадок смывают струей воды в стакан, где проводилось осаждение, помещают стакан под воронку, растворяют осадок, оставшийся на фильтре, горячей разбавленной азотной кислотой и промывают фильтр. Прибавляют еще кислоты, если необходимо для полного растворения осадка, и затем медленно при постоянном перемешивании приливают разбавленный раствор аммиака, пока образующийся осадок не перестанет при перемешивании переходить в раствор. Приливают 10 мл раствора а, нагревают жидкость, вращая ее в колбе, до кипения, дают медленно охладиться, фильтруют и промывают осадок декантацией раствором б. Фильтр с осадком высушивают, сжигают отдельно фильтр, присоединяют осадок, прокаливают и взвеши
1 A. Skrabal, L. Neustadt 1, Z. anal. Chem., 44, 742 (1905). J
Методы определения
701
вают желтый хромат бария. Если на прокаленном остатке хромата бария будут заметны зеленые пятна окиси хрома вследствие восстановления хромата, то продолжают прокаливание до полного окисления.
Для определения малых количеств бария, содержащихся в породах и некоторых минералах, достаточно, вероятно, однократного осаждения бихроматом, но если следует отделять значительные количества бария, однократного осаждения недостаточно. Авторы метода, проводя анализ смеси, содержавшей по 0,135 г окисей каждого из трех щелочноземельных металлов, получили при определении бария ошибки в ±0,2—0,5 мг.
Барий можно осадить и отделить от кальция и магния1, растворяя не больше 0,5 г смеси хлоридов этих элементов в возможно меньшем количестве горячей воды, прибавляя медленно и при перемешивании 25—50 .мл НС1, затем охлаждая и добавляя 5—10 мл эфира. Найдено 2, что этот метод пригоден также для отделения бария от стронция, если берут смесь 33%-ной соляной кислоты и эфира в отношении 4 : 1 (50— 75 мл этой смеси на 0,5 г хлоридов, содержащих не больше 0,3 г SrCla). Выделенный хлорид бария после фильтрования промывают кислотно-эфирной смесью, применяемой при отделении от кальция и магния.
Определение стронция в фильтрате от хромата бария. К соединенным фильтратам от хромата бария прибавляют немного азотной кислоты, выпаривают до малого объема и осаждают стронций в этом растворе аммиаком и карбонатом аммония. Затем осадок отфильтровывают, немного промывают его горячей водой, растворяют в необходимом количестве соляной кислоты и осаждают стронций серной кислотой, как описано на стр. 712. Таким способом стронций освобождается совершенно от загрязняющего его хромата.
/I
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Определение одного из щелочноземельных элементов в отсутствие других элементов этой группы — дело сравнительно простое, но совсем иное положение, когда в анализируемом растворе присутствуют два или все три щелочноземельных элемента, потому ^то выбор метода их разделения должен зависеть от их относительных количеств. Метод, пригодный для одного соотношения их в смеси, может оказаться непригодным для другого. Поэтому, прежде чем избрать метод количественного определения, надо заранее знать о присутствии или отсутствии того или другого из этих элементов и иметь хотя бы приблизительное представление об их относительных количествах 3.
КАЛЬЦИЙ
Общие замечания
Кальций почти всегда осаждают в виде оксалата СаС2О4 • Н2О после предварительной обработки, имеющей целью отделение его от всех других катионов, кроме ионов щелочных металлов, магния и остальных щелочноземельных металлов. Осаждение кальция в виде оксалата никогда
1 F. W. Mar, Am. J. Sci., (3), 43, 521 (1892).
2 F. A. Gooch, М. A. S о d е г т a n, Am. J. Sci., (4), 46, 538 (1918).
3 При очень маловероятном случае, когда в анализируемом веществе присутствуют весовые количества радия, он будет в каждом осадке, выделенном из раствора, содержащего сульфат-ионы. Описание электроскопического метода определения радия в карнотите и урановой смоляной руде см. в статье L. D. Roberts, Ind. Eng. Chem., Anal. Ed., 8, 5 (1936).
702
Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
не бывает вполне количественным, и первый осадок редко бывает чистым. Его обычно загрязняют щелочные металлы, магний, барий и стронций. Из других возможных загрязняющих веществ отметим здесь платину, кремнекислоту, марганец, алюминий и некоторые редкоземельные элементы х. Этих загрязнений следует ожидать, когда проводят обычные предварительные отделения. Если же предварительных отделений не проводили, то список возможных загрязняющих веществ можно значительно увеличить, так как оксалат аммония осаждает большое число элементов.
Из числа возможных загрязняющих веществ платина может оказаться только в том случае, если те или иные предварительные операции проводились в платиновой посуде. Кремнекислота может оказаться в осадке, если в ходе, анализа нагреванию в фарфоровой или стеклянной посуде подвергались аммиачные растворы. Марганец частично осаждается с кальцием, особенно если осаждение последнего проводится из аммиачного раствора. Многократное осаждение кальция в виде оксалата не освобождает осадок полностью от марганца, следовательно, марганец надо удалять перед осаждением кальция. Алюминий не должен присутствовать в осадке оксалата кальция, если предварительные операции были проведены надлежащим образом. Но если осаждение гидроокиси алюминия Проводили не совсем правильно, например прибавляли аммиак в избытке или осаждали алюминий в присутствии органических веществ или фторидов, то он может остаться в небольшом количестве в растворе и присоединиться к осадку оксалата кальция. Редкоземельные металлы не будут в осадке вместе с оксалатом кальция, если перед осаждением последнего было проведено осаждение аммиаком. Если же осаждение проводили ацетатным методом, в осадке оксалата кальция могут оказаться некоторые из редкоземельных металлов. В этих случаях загрязненный оксалат кальция прокаливают, выделяют алюминий и редкоземельные металлы, как описано ниже, и снова осаждают кальций в виде оксалата.
Из обычно загрязняющих осадок веществ щелочные металлы удаляют двукратным осаждением оксалата кальция. Таким же способом отделяют малые количества бария, часто встречающиеся в силикатных породах1 2. Если присутствует большое количество бария, то часть его будет в осадке оксалата, и его надо впоследствии удалить вместе со стронцием. Оксалат стронция почти так же нерастворим, как оксалат кальция, и в условиях, при которых осаждается кальций, его осаждение происходит полностью 3. Поэтому стронций взвешивают вместе с кальцием, затем его отделяют и массу его вычитают.
Наиболее обычной примесью, загрязняющей оксалат кальция, является магний. Оксалат магния окклюдируется оксалатом кальция, и степень окклюзии прямо пропорциональна концентрации недиссоции-рованного оксалата магния в растворе при осаждении. Оксалат магния также медленно адсорбируется на поверхности оксалата кальция, и адсорбированное его количество пропорционально времени, протекающему
1 Железо осаждается оксалатом в аммиачном растворе полностью, но при промывании осадка холодным раствором оксалата аммония большее или меньшее количество жейеза переходит в раствор. Алюминий, если он присутствует один, не осаждается оксалатом в аммиачном растворе.
2 W. F. Hillebrand, J. Am. Chem. Soc., 16, 83 (1894).
3 См. сноску 3, стр. 696.
Методы, определения
703
между осаждением и последующим фильтрованием. Концентрация недис-социированного оксалата магния увеличивается от прибавления любого из составляющих его ионов в избытке, например от прибавления большого избытка оксалата аммония во время осаждения. Концентрацию недиссо-циированного оксалата магния можно уменьшить добавлением ионов водорода, увеличением концентрации аммонийных солей (образующих с магнием комплексные ионы) или разбавлением раствора. К сожалению, все эти меры приводят также к замедлению осаждения кальция.
Хотя переосаждением можно удалить из осадка магний, это отражается на полноте выделения кальция. Поэтому при выполнении рядовых анализов не следует пытаться удалять магний полностью. Количество магния, переходящее в осадок оксалата кальция, примерно равно количеству кальция, которое остается в растворе и потом выделяется вместе 'с магнием в виде фосфата. Если проводят точный анализ, надо стремиться к полному отделению магния и определять затем то количество кальция, которое останется в растворе после его осаждения и потом окажется в прокаленном остатке пирофосфата магния. Так следует поступать потому, что выделение малых количеств кальция из фосфата магния значительно ^тегче, чем извлечение малых количеств магния из окиси кальция.
Величина потери кальция вследствие растворимости оксалата кальция зависит от условий осаждения и промывания осадка.
Количество чистого или загрязненного осадка оксалата кальция, переходящего в раствор при промывании той или иной промывной жидкостью, иллюстрируется следующими данными. Четыре порции по 0,1 г окиси кальция, введенные в раствор в виде соответствующего количества чистого хлорида кальция, были осаждены оксалатом аммония рекомендованным выше методом. Осадки оксалата кальция, отфильтрованные через четыре фильтра, промывались четырьмя различными промывными жидкостями: водой, 0,01%-ным раствором (NH4)2C2O4, 0,1%-ным раствором (NH4)2C2O4 и 1%-ным раствором (NH4)2C2O4. Промывные жидкости применялись холодными, и на каждое промывание было израсходовано по 1 л промывной жидкости. Затем то же было повторено с новыми порциями хлорида кальция, но промывные растворы применялись горячими. После этого были взяты четыре порции, каждая в составе 0,1 г СаО, 0,005 г MgO, 5 г NaCl и 5 г NH4C1. В каждой из них был выделен кальций однократным осаждением и четыре осадка загрязненного оксалата кальция промывались теми же промывными жидкостями в холодном виде. Наконец, новые четыре порции полученного таким же образом загрязненного оксалата кальция промывались горячими жидкостями. Все полученные 16 промывных вод выпаривались досуха в платиновой посуде, аммонийные соли удалялись прокаливанием, маленькие остатки растворялись в очень небольших количествах соляной кислоты и в полученных растворах определяли кальций обычным способом, взвешивая его в виде СаО. Полученные результаты представлены в табл. 21.
ТАБЛИЦА 21
Растворимость осадка оксалата кальция при промывании
Температура промывного раствора °C Осадок СаС2О4 Количество СаО в 1 л промывной жидкости, мг
воды 0,01 %-ного раствора (NH4)2C2O4 0,1 %-ного раствора (NH4)2C2O4 1 %-ного раствора (NH^gCgO^
25 Чистый 2,6 0,4 0,1 0,0
25 Загрязненный 2,7 0,5 0,1 0,1
100 Чистый 4,9 0,9 0,5 1,1
100 Загрязненный 4,3 1,7 1,2 1,2
Очевидно, что промывать надо холодным 0,1%-ным раствором оксалата аммония.
4
704 Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
Если осадок промывают холодным раствором оксалата аммония, то общая потеря не превышает 0,1 мг при каждом осаждении оксалата. Теряющийся таким образом кальций переходит в осадок фосфата магния и аммония и определяется, как описано в разделе «Отделение кальция от магния» (стр. 694).
Например, Н. В. Knowles получил только 0,2; 0,3 и 0,2 мг СаО во взвешенных прокаленных остатках М§2РгО7, полученных при анализе трех порций по 1 г доломита (30,5% СаО и 21,5% MgO), несмотря на то что в каждом анализе кальций четырехкратно осаждали в виде оксалата, чтобы быть уверенными в полном освобождении его от магния. Осаждение проводили каждый раз в объеме, приблизительно равном 250 мл, и осадок промывали 25—50 мл 1%-ного раствора оксалата аммония, причем общий объем соединенных фильтратов и промывных вод превышал 1000 мл. Практически вся потеря вследствие растворимости оксалата кальция, по-видимому, произошла при первом осаждении, потому что маеС& СаО, полученные после четырех осаждений, были точно такими же, как и веса, полученные после трех осаждений. Большее количество кальция, оставшегося в растворе после первого осаждения, без сомнения было вызвано более значительной концентрацией магния и аммонийных солей в растворе.
Кроме растворимости оксалата кальция, имеются еще и другие причины, могущие привести к потере кальция. Когда перед осаждением кальция проводят отделение других элементов аммиаком, то может произойти потеря кальция: 1) если осадок от аммиака был велик и проводилось только однократное осаждение; 2) если аммиак содержал карбонат аммония или если аммиачный раствор подвергался продолжительное время действию воздуха, содержащего двуокись углерода; 3) если анализируемый раствор содержал большое количество фосфатов и 4) если присутствовали фториды.
Потерь по первым двум причинам можно легко избежать. Выпадение кальция в виде фосфата во времй осаждения аммиаком зависит от соотношения между содержанием фосфат-ионов и содержанием ионов железа (III) и алюминия в растворе. Если железо находится в значительном избытке, До кальций не выпадает; если преобладают кальций и фосфат-ионы, что имеет место при анализе фосфатных пород, то практически весь кальций переходит в осадок. Опыты показали, что при двукратном осаждении железа или алюминия добавлением аммиака в небольшом избытке к раствору, содержащему 0,05 г окиси кальция, 0,005 г пятиокиси фосфора и десятикратное количество окисиЪкелеза|или окиси алюминия по отношению к Са2+, выделяющийся осадок не содержит кальция. Эти опыты, воспроизводящие обычные условия анализа горных пород, показывают, что в таких условиях потеря кальция при осаждении «полуторных окислов» аммиаком не произойдет. Если фосфор преобладает и желают определить только один кальций, то аналитик имеет для этого несколько способов, из которых, однако, ни один не свободен от возражений.
Например: 1) можно прибавить железо или алюминий в достаточном избытке; 2) можно осадить кальций серной кислотой и спиртом из раствора, слабо подкисленного соляной или азотной кислотой, как описано на стр. 711, или осадить в виде оксалата из раствора, содержащего оксалат аммония и свободную щавелевую кислоту (стр. 708); 3) можно осадить фосфат-ионы в виде фосфоромолибдата и затем определить кальций в фильтрате. Конечно, эта операция требует применения осадителя, не содержащего кальция. Молибден не мешает осаждению оксалата кальция в фильтрате ни из аммиачного, пи из щавелевокислого раствора.
Методы определения ' 705
Если анализируемая проба содержит фтор, то при повторном выпаривании с соляной или азотной кислотами (для отделения кремнекислоты) он полностью не удаляется и тогда кальций частично переходит в осадок от аммиака в виде фторида кальция. В таких случаях предварительное выпаривание следует проводить с серной или хлорной кислотой или с концентрированной азотной кислотой, но с прибавлением тонкого порошка чистого кремнезема х.
Отделение в виде оксалата
Осаждение в щелочном растворе. Описанный ниже метод отделения кальция от магния и щелочных металлов применим всегда, за исключением тех случаев, когда магния значительно больше, чем кальция, или кальций присутствует в очень малых количествах. Анализ большинства горных пород й силикатных минералов может быть проведен способом, описанным в данном разделе. Как уже было указано выше, для точного определения необходимо по крайней мере двукратное осаждение кальция. Оптимальное количество хлорида аммония в растворе неопределенно, потому что большой излишек его уменьшает соосаждение магния и бария, но, с другой стороны, замедляет осаждение кальция и особенно стронция. Если анализ проводится обычным способом, то нет необходимости удалять перед осаждением аммонийные соли. Если же в результате проведения каких-либо дополнительных операций в растворе скопилось большое количество аммонийных солей, то их надо удалить, как указано на стр. 161, или же выпариванием досуха подкисленного раствора в фарфоровой или платиновой посуде и дальнейшим осторожным прокаливанием так, чтобы поступающее тепло равномерно распределялось по внешней поверхности чашки и не вызывало слишком сильного выделения дыма. После этого смачивают остаток хлоридов или нитратов 2—3 мл соответствующей кислоты, растворяют соли добавлением небольшого количества воды М, если надо, фильтруют.
При улетучивании хлорида аммония имеется меньшая опасность потери, чем при улетучивании нитрата аммония. Последний плавится и выделяет пузырьки, которые лопаются, и жидкость разбрызгивается. Кроме того, разлагающийся нитрат аммония разъедает в некоторой степени платину, следовательно нельзя удалять нитрат аммония прокаливанием в платиновой посуде. Это, без сомнения, в еще большей мере относится к смеси нитрата и хлорида.
Первое осаждение. Приготовляют раствор, свободный от кремнекислоты, серы, элементов сероводородной группы и элементов, осаждающихся аммиаком или сульфидом аммония.
Оксалат кальция, осажденный в растворе, содержащем свободную-серу (например, в подкисленном фильтрате после осаждения сульфидом аммония, обработанном оксалатом аммония и затем постепенно нейтрализованном), нельзя прокаливать и взвешивать, вследствие того что сера при прокаливании окисляется и связывается с окисью кальция. Ошибк^.не устраняется растворением влажного осадка (т. е. до его прокаливания) и вторичным его осаждением, потому что тонкодисперсная сера проходит в фильтрат и снова осаждается. Это затруднение можно преодолеть прокаливанием осадка, растворением его и вторичным осаждением.
1 A. A. N о у е s, Technol. Quart,, 16, 101 (1903).
45 Заказ 522.
706 Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
Можно также удалить серу из первоначального раствора подкислением, кипячением, достаточно продолжительным для удаления сероводорода, но не для коагуляции серы, и затем окислением бромом. Если сера выделилась, раствор, конечно, надо профильтровать.х
Раствор разбавляют до 100—400 мл и делают его слабоаммиачным х. Ни в каком случае раствор не должен содержать перед осаждением больше' 1 мг СаО в 1 мл. Нагревают до кипения и медленно при перемешивании прибавляют горячий 4%-ный раствор оксалата аммония в количестве, достаточном для осаждения всего кальция и для обеспечения избытка — 1 г на 100 мл раствора.
Если в растворе присутствуют значительные количества магния, то для лучшего-Отделения и получения более крупнозернистого осадка осаждают кальций из кислого раствора следующим образом. Подкисляют раствор соляной кислотой, прибавляют 1 мл ее избытка на каждые 100 мл раствора и затем добавляют щавелевую кислоту или оксалат аммония в количестве, достаточном для того, чтобы связать весь кальций. Рекомендуется избегать большого избытка реактива. Нагревают раствор до кипения,, непрерывно перемешивают и прибавляют из пипетки по каплям разбавленный (1 : 3) раствор аммиака до начала образования осадка. С этого момента аммиак прибавляют еще медленнее и между каждым добавлением аммиака дают осадку образовываться возможно дольше. Так продолжают, пока окраски метилового красного не перейдет в желтую, и тогда прибавляют еще 2—3 г оксалата аммония. Дают раствору постоять при медленном охлаждении 4 ч, фильтруют и промывают осадок, как сказано в основном тексте.
Еще лучшее отделение можно получить, осаждая следующим образом1 2. Раствор-разбавляют настолько, чтобы концентрация магния была приблизительно равна 0,02 н., прибавляют 10 мл соляной кислоты, несколько капель метилового оранжевого и потом-раствор аммиака до нейтральной реакции. Затем прибавляютдцавелевую кислоту в количестве, достаточном для реакции с кальцием, вместе с трехкратным эквивалентным-ей количеством соляной кислоты. Нагревают раствор до кипения и затем медленно и малыми порциями приливают разбавленный (1 : 5) раствор аммиака к кипящему анализируемому раствору в течение 30 мин. К концу этого времени раствор должен стать точно нейтральным. Прибавляют 5 г оксалата аммония, дают раствору охлаждаться 4 ч, фильтруют и промывают осадок, как сказано в основном тексте.
Кипятят 1—2 мин, нагревают на паровой бане 30 мин, дают охладиться и чере!з 2 ч фильтруют. Фильтр с осадком промывают пятью порциями по 10 мСл холодного нейтрального 0,1 %-ного раствора оксалата аммония. Если надо определить магний или оставшийся в растворе кальций, сохраняют фильтрат и промывные воды.
Нет необходимости в прокаливании осадка перед вторым осаждением, если только аналитик не имеет основания полагать, что при осаждении присутствовали железо или редкоземельные металлы. Эти элементы не должны присутствовать, если перед осаждением оксалатом было проведено осаждение их аммиаком. Но они могут присутствовать, если осаждение было проведено ацетатным методом или если осаждение-аммиаком было проведено в неблагоприятных условиях, например в присутствии растворимых органических веществ. Если осадок оксалата был прокален, ставят открытый тигель прямо на дно стакана, содержащего небольшое количество воды., накрывают-стакан часовым стеклом и дают постоять некоторое время, пока известь не будет частично погашена. Затем смачивают осадок водой, растворяют в 50 мл разбавленной
1 В обычных анализах такой раствор получается при соединении фильтратов после двукратного выделения железа, алюминия и т. п. аммиаком, как описано на стр. 949. Если присутствуют марганец или цинк, то таким раствором будет фильтрат после отделения указанных элементов сульфидом аммония (стр. 961). Если содержание кальция в растворе очень мало, а аммонийных солей очейь много, то рекомендуется, удалять последние перед осаждением кальция.
2 Т, Ws Richards, Proc. Am. Acad. Arts and Sci., 36, 392 (1900—1901)..
Методы определения
707
(1:4) соляной кислоты, прибавляют несколько капель метилового красного, нагревают до кипения и приливают разбавленный раствор аммиака, пока раствор не окрасится в желтый цвет (стр. 567). Кипятят 1—2 мин и немедленно фильтруют. Фильтр с осадком промывают горячим 2 % -ным раствором хлорида аммония и сохраняют фильтрат и промывные воды для второго осаждения кальция. Если определение кальция является частью полного анализа, сжигают фильтр с осадком от аммиака, прокаливают, взвешивают и вводят соответствующие поправки в полученные раньше результаты определения FeaO3, А12О3 и т. п.
Второе осаждение. Промытый осадок оксалата кальция растворяют в'50 мл разбавленной (1 : 4) соляной кислоты, разбавляют раствор, прибавляют в избытке щавелевую кислоту, как указано выше (см. «Первое осаждение»), затем нагревают до кипения, непрерывно перемешивают и медленно прибавляют разбавленный раствор аммиака до щелочной реакции. Дают постоять, фильтруют и промывают осадок, как при первом осаждении Ч Фильтрат и промывные воды соединяют с фильтратом и промывными водами, полученными после первого осаждения.
Прокаливание осадка. Завертывают влажный осадок в бумагу фильтра, помещают его в платиновый тигель, взвешенный вместе с плотно прилегающей крышкой, и нагревают так, чтобы бумага обуглилась без воспламенения. По окончании обугливания усиливают пламя и, когда уголь в^сь выгорит, ставят тигель на треугольник, покрывают крышкой и прокаливают его 5 мин при 1200° С. Во время прокаливания рекомендуется защищать осадок от загрязнения его окислами серы из пламени горелки или от летучих веществ, выделяемых, муфелями, находящимися в общем употреблении. Снимают на момент крышку, чтобы дать выход оставшейся в тигле двуокиси углерода, помещают закрытый тигель в эксикатор, содержащий серную кислоту или пятиокись фосфора (но не хлорид кальция), и взвешивают, как только тигель охладится. Масса окиси кальция может заметно возрасти, если тигель долго будет в эксикаторе. Первое взвешивание является только предварительным. За ним следует короткое прокаливание и второе взвешивание, при котором заранее кладут разновески на чашку весов и быстро устанавливают рейтер. Если прокаленная окись кальция находится в тигле, хорошо закрытом крышкой, то масса ее не возрастает в течение 1 мин при обычных атмосферных условиях 1 2.
Присутствие марганца в прокаленной окиси кальция обычно узнается по окраске — от желтоватой до коричневой. Изредка окраска бывает зеленой от присутствия манганата кальция. Взвешенную окись кальция сохраняют для определения в ней стронция или бария, как описано на стр. 696. Об извлечении кальция, оставшегося в фильтрате после осаждения его одсалата, см. «Отделение кальция от магния» (стр. 694).
Некоторые замечания о прокаливании осадка. Часто., высказываемое утверждение, что необходимо продолжительное прокаливание при 1200° С для получения постоянной массы, неверно. Если пламя паяльной горелки
1 При выполнении точных анализов веществ, содержащих значительные количества магния, всю операцию надо повторить до получения постоянной массы окиси кальция. См. «Отделение кальция от магния» (стр. 694).
2 Можно также перенести осадок во взвешенный тигель Гуча или любой другой фильтрующий тигель, прокалить при 475—525° С и взвесить в виде карбоната кальция СаСО3 [Н. Н. Willard, A. W. Boldyreff, J. Am. Chem. Soc., 52, 1888 (1930)].
45*
708 Гл. XL. Щелочноземельные металлы — кальций,. стронций, барий
наклонено таким образом, что газы не могут проникнуть в тигель, то 5 мин, как правило, бывает вполне достаточно для прокаливания обычно определяемого количества окиси кальция.
Нагрев горелками Теклю, Мекера или видоизмененной Тирриллем бунзеновской горелкой является вполне достаточным, несмотря на то что пламя этих горелок окружает тигель. 50-минутное нагревание на горелке Тирриля 3 г осажденного карбоната кальция в платиновом тигле достаточно для превращения всей массы почти полностью в окись кальция настолько, что при последующем применении паяльной лампы дальнейшая потеря в массе составляет только 1 мг.
Эти замечания противоречат некоторым выводам .0. Брунка 1, сделанным им в результате сравнительного изучения методов определения кальция в виде окиси, карбоната, сульфата и фторида. Все его результаты, полученные методом, заканчивавшимся прокаливанием окиси кальция, оказались более высокими, чем результаты, полученные другими методами, а также непостоянными, и поэтому он заключил, что другие методы в среднем дают лучшие результаты.
В Геологической исследовательской лаборатории США при применении метода прокаливания до окиси кальция никогда не получались такие высокие или колеблющиеся результаты, какие получил Брунк. Результаты параллельных определений этим методом всегда совпадали, подобно тому как у Брунка совпадали результаты, полученные другими методами.
Брунк считал, что одним из источников ошибки при прокаливании до окиси кальция является потеря в массе платинового тигйя вследствие сильного его нагревания, необходимого для достижения постоянной массы прокаленного остатка. Возможно, что автор нагревал окись кальция значительно дольше, чем это действительно необходимо, или же он ошибался в причине потери платины. Подобно окиси бария, но в меньшей степени, окись кальция действует на платину, находясь в соприкосновении с ней при сильном прокаливании. Это легко можно продемонстрировать, растворяя в соляной кислоте окись кальция, которая прокаливалась на паяльной лампе в платиновом тигле в течение получаса, осаждая платину и взвешивая ее. Если Брунк считал, что разница между массой очищенного тигля, определенной до и после прокаливания в нем окиси кальция, представляет количество улетучившейся платины, то легко видеть, что его представление о причине потери ошибочно. Верно, что масса платиновых тиглей уменьшается при прокаливании их на пламени горелки с дутьем, особенно если платина содержит иридий, но при пятиминутном прокаливании, достаточном для достижения постоянной массы, заметная потеря может произойти только в редких случаях.
Осаждение в слабокислом (pH — 3,5—4,5) растворе. Было предложено большое число методов для осаждения кальция в виде оксалата из растворов, содержащих свободные органические кислоты, такие, как щавелевая, уксусная, лимонная и салициловая. Эти методы дают возможность прямо отделять кальций от железа, алюминия, титана, марганца (частично), магния, молибдена и фосфора и, хотя их следует рассматривать как быстрые методы для рядовых массовых анализов, они могут дать превосходные результаты. Были описаны методы осажде
1 О. В г u п с k, Z. anal. Chem., 45, 77 (1906).
Методы, определения
709
ния в присутствии свободной уксусной кислоты 1 и в присутствии свободной лимонной кислоты 2.
Мы применяли только метод осаждения кальция в растворе, содержащем свободную щавелевую кислоту и оксалат аммония. Этим методом анализируют фосфатные руды в Бюро Стандартов США. Тщательное его исследование, проведенное с растворами, содержащими только кальций, и с растворами, содержащими вместе с кальцием железо, алюминий, титан, цирконий, магний и большие количества фосфора, показало, что метод дает превосходные результаты и потеря вследствие растворимости оксалата кальция не превышает потери по той же причине, происходящей при применении обычного метода. Барий, если он присутствует в растворе в умеренных количествах, не переходит в конечный осадок оксалата кальция. Стронций, однако, распределяется таким образом, что часть его осаждается, а часть остается в фильтрате.
При проведении определения разбавляют раствор до 150 мл, обрабатывают 10 мл соляной кислоты и прибавляют несколько капель метилового красного или метилового оранжевого. Нагревают до 50° С, нейтрализуют аммиаком и прибавляют избыток его в 1 мл. Затем приливают 10%-ный раствор щавелевой кислоты до кислой реакции и сверх того еще 12 мл избытка, после чего кипятят 1—2 мин, сильно перемешивая. Прибавляют приблизительно 50 мл насыщенного раствора оксалата аммония (примерно 4%-ного), разбавляют до 250 мл, кипятят 1—2 мин и оставляют на паровой бане 1 ч. Дают охладиться до комнатной температуры, фильтруют и промывают осадок холодным щавелевокислым раствором оксалата аммония [2 г (NH4)2C2O4 • Н2О и 1 г Н2С2О4 • 2Н2О в 1 л]. При выполнении рядовых анализов промываю! осадок 5—10 малыми порциями холодной воды и определяют кальций объемным методом (стр. 710). В более тщательных анализах прокаливают оксалат кальция до окиси кальция, гасят последнюю водой и растворяют в 40 мл разбавленной (1 : 4) соляной кислоты. Затем разбавляют до 200 мл, прибавляют 0,005 г железа в виде FeCl3 и отделяют фосфор и марганец. Для этого раствор делают слабощелочным, прибавляя аммиак, затем добавляют 10 мл бромной воды и нагревают 15 мин при температуре немного ниже точки- кипения. Потом прибавляют еще 5 мл бромной воды, нагревают снова 15 мин и фильтруют. Осадок промывают горячим аммиачным раствором хлорида аммония (10 мл NH4OH и 10 г NH4C1 в 1 л) и отбрасывают его. Фильтрат подкисляют соляной кислотой, кипятят до удаления брома и вновь осаждают кальций в виде оксалата обычным способом (стр. 705).
Для объемного определения кальция в присутствии кремния, железа, алюминия, магния и фосфора, находящегося в растворе в количестве, отвечающем содержанию кальция или несколько большем, а также в присутствии малых количеств титана и
1 R. К. Me a d е, Chem. Eng., 1, 21 (1895). Кальций можно осаждать из уксуснокислого раствора, содержащего молибдат-ионы, например из фильтрата после осаждения фосфора в виде фосфоромолибдата аммония [R. С. Wiley, A. Y е d i n a k, Ind. Eng. Chem., Anal. Ed., 10, 322 (1938)]. Этим методом также успешно отделяют кальций от хромат-ионов.
2 М. Passon, Z. angew. Chen., 11, 776 (1898); 12, 48 (1899); 14, 285 (1901); Mellor, A Treatise on Quantitative Inorganic Analysis, 1913, p. 522.
710
Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
марганца, рекомендуется1 осаждение при рНл#4, проводимое следующим образом. Приготовляют 100 л«л раствора, содержащего 10 мл свободной соляной кислоты и около 0,07 г кальция в расчете на СаО. Нагревают до 90е С, приливают горячий (90° С) раствор 5 г (NH4)2C2O4 -Н2О в 100 мл и прибавляют несколько капель 0,1%-ного раствора метилового оранжевого. Осаждение начинают при температуре около 80° С, добавляя по каплям разбавленный (1 : 1) раствор аммиака в течение 5—10 мин при непрерывном и тщательном перемешивании раствора, пока pH его не станет равным 4,0 ± 0,3. Полученную розовато-желтую окраску сравнивают с окраской раствора-свидетеля (0,1 М раствор кислого фталата калия, занимающий такой же объем и содержащий такое же количество индикатора). Затем дают раствору охладиться в течение 20—30 да, фильтруют, умеренно промывают осадок небольшими порциями (всего не более 10 мл) холодной воды и титруют перманганатом (см. ниже)2.
Другие методы определения кальция после осаждения его в виде оксалата. Титрование перманганатом. Для перманганатометрического определения кальция оксалат его обрабатывают серной кислотой и освобожденную щавелевую кислоту титруют титрованным раствором перманганата калия. Очевидно, что если осадок оксалата кальция содержит некоторое количество оксалатов стронция, бария, магния или аммония, то они будут приняты за кальций.
При анализе материалов, содержащих только кальций, этот метод может дать точные результаты. Он имеет большое значение и широко применяется в качестве метода массового анализа таких продуктов, как цемент или алебастр. При, работе этим методом оксалат кальция лучше отфильтровывать через асбест, пористое стекло и т. п., для того чтобы избежать действия перманганата на бумагу 3. Если применяют бумажный фильтр, то осадок промывают (но не чрезмерно) холодной водой до удаления оксалата аммония, вынимают фильтр из воронки и расстилают на внутренней стенке стакана, на дне которого налито 100 мл разбавленной (1 : 10) серной кислоты. Затем смывают осадок в стакан небольшим количеством горячей воды, нагревают раствор до полного разложения оксалата и титруют при' 25—30° С раствором пермаганата до розового окрашивания (стр. 217). Тогда сбрасывают фильтр в стакан, обмывают стенки последнего и быстро заканчивают титрование.
Титр перманганата устанавливают по химически чистому оксалату натрия или по исландскому шпату известной чистоты, который растворяют в соляной кислоте и проводят через все стадии анализа.
Весовые методы. Большее или меньшее применение имеют также методы, в которых оксалат кальция превращают в карбонат, сульфат или фторид и взвешивают полученное соединение. Эти методы, подобно уже описанному объемному методу, могут применяться только в тех случаях, когда оксалат кальция свободен от стронция и бария или количества последних точно известны. Первый и второй методы являются старыми методами, которые, однако, полностью выдержали испытание временем и предпочитаются некоторыми аналитиками и в настоящее время.
О. Брунк 4 приводит самые благоприятные данные о всех этих трех методах, и, принимая во внимание прежнюю оценку их заслуженными аналитиками, нельзя не признать, что в умелых руках они дают превос
1 J. J. Lingane, Ind. Eng. Chem., Anal. Ed., 17, 39 (1945).
2 См. также H. D. Chapman, Soil. Sci., 26, 479 (1928).
3 R. S. McBride, J. A. Scherrer, J. Am. Chem. Soc?, 39, 928 (1917).
4.0. В r u n c k, Z. anal. Chem., 45, 77 (1906).
Методы определения
711
ходные результаты, особенно если количества определяемого кальция невелики. Тем не менее эти методы, главным образом первый и второй, требуют значительно большей осторожности и внимательности, чем метод прокаливания до окиси кальция, и поэтому они не могут широко рекомендоваться.
Вообще полное превращение одного твердого вещества в другое с помощью реактива, в котором первое вещество совсем или почти совсем нерастворимо, проводится не всегда легко, особенно если количество вещества велико. Это положение подтверждается опытами превращения хлорида свинца и мало растворимых фторидов в сульфаты с помощью серной кислоты. Как только при действии серной кислоты образуется новое нерастворимое соединение, некоторое количество первоначального •соединения не превращается даже после двух или более выпариваний со свежими порцйями кислоты.
Взвешивание в виде карбоната. Сначала сжигают отдельно фильтр, потом переносят в тигель осадок оксалата кальция и осторожно его прокаливают до превращения в карбонат кальция (* см. сноску 2, стр. 707 *). Прибавляют несколько капель раствора карбоната аммония, осторожно выпаривают жидкость, затем прокаливают остаток до начала красного каления и взвешивают. Обработку карбонатом аммония и т. д. повторяют до получения постоянной массы.
Взвешивание в виде сульфата. Осторожно гасят окись кальция, полученную после прокаливания оксалата кальция,’ и затем прибавляют серную кислоту в небольшом избытке. Концентрируют раствор выпариванием и удаляют избыток кислоты нагреванием в радиаторе (см. рис. 5, стр. 49). Охлаждают, прибавляют немного воды, снова выпаривают досуха и прокаливают безводный сульфат кальция до темно-красного каления. Прокаливание ведут недолго, затем охлаждают и взвешивают. Обработку кислотой повторяют. Слишком сильное прокаливание вызывает потерю SO3. ,
Взвешивание в виде фторида. Прокаленную окись кальция обрабатывают, как указано выше (см. «Взвешивание в виде сульфата»), применяя вместо серной кислоты фтористоводородную кислоту. Брунк считает этот способ самым простым из всех, но полагает, что в опытных фуках наилучшим является сульфатный метод.
Первоначальное осаждение в виде сульфата. Иногда обстоятельства позволяют Или требуют прямого осаждения кальция в виде сульфата в отсутствие или в присутствии других элементов, сульфаты которых нерастворимы. Раствор может быть нейтральным или слабо подкисленным соляной или азотной кислотой. Прибавляют разбавленную (1:1) > серную кислоту в десятикратном избытке и затем 4 объема спирта, перемешивают, оставляют на 12 ч, фильтруют, промывают осадок 75%-ным •спиртом, высушивают, озбляют отдельно (если они легко отделяются друг от друга) фильтр и осадок в платиновом тигле, прокаливают при темно-красном калении, охлаждают и взвешивают в виде CaSO4. При озолении фильтра может произойти восстановление незначительной части сульфата кальция до сульфида, но при последующем прокаливании происходит обратное окисление сульфида кальция до сульфата.
Если прокаленный остаток содержит сульфаты других металлов, например стронция, бария, свинца, его надо вновь растворить и выделить эти металлы одним из известных методов. Если присутствует свинец,
712
Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
то его надо отделять вначале, до прокаливания осадка, чтобы избежать восстановления свинца во время прокаливания х.
* Хорошим быстрым методом определения кальция в присутствии бария и магния является титрование его раствором двузамещенного этилендиаминтетраацетата натрия (называемого иначе комплексоном III, трилоном Б, версенатом натрия) с применением пурпурата аммония (мурексида) в качестве индикатора 1 2. Этим методом можно определять кальций и в присутствии стронция, если связать последний добавлением сульфата калия 3 4 5. Описан метод определения кальция,, в котором его осаждают в виде CaK2[Ni(NO2)6] и затем колориметрически определяют или никель *, или нитрит-ионы 6. Доп. ред.*
СТРОНЦИЙ
Осаждение и взвешивание в виде сульфата
Если стронций встречается в условиях, когда можно определить его в виде сульфата, то лучше всего осадить и взвесить стронций в этой форме. Такие условия получаются в обычном ходе анализа только после того, когда стронций отделен от бария и кальция, вместе с которыми его выделили перед этим в другой форме, например в виде оксалатов.
Раствор соли стронция (предпочтительно солянокислый) должен быть по возможности нейтральным. Прибавляют разбавленную (1 : 1) серную кислоту в десятикратном избытке и затем спирт в объеме, равном объему раствора. Перемешивают, оставляют на 12 ч, фильтруют и промывают сначала 50 %-ным спиртом, содержащим немного серной кислоты, а затем 95%-ным спиртом до удаления кислоты. Дальше продолжают, как описано для кальция (стр. 711), и взвешивают в виде SrSO4. Приведенные там же замечания (методы осаждения кальция) применимы и к осаждению стронция.
Осаждение в виде оксалата и взвешивание в виде окиси
Обычно стронций связан с кальцием и часто с барием, например при анализе минералов. В таких случаях сперва осаждают стронций вместе с кальцием в виде оксалата и прокаливают до окиси (стр. 707). Здесь можно отметить, что стронций по своему отношению к оксалату аммония значительно ближе к кальцию, чем к барию. Оксалат стронция немного
1 Е. R. Caley и Р. J. Elving [Ind. Eng. Chem., Anal. Ed., 10, 264 (1938)] предпочитают осаждать сульфат кальция прибавлением метанола к раствору солей кальция и магния в разбавленной серной кислоте (20 мл Н2О -|- 0,25 мл H2SO4 + 4- 180 мл СН3ОН). Этот метод дает удовлетворительные результаты при определении кальция в магнезитах, но не в известняках или в присутствии стронция и других элементов, образующих нерастворимые сульфаты. При отношении Mg : Са = 200 : 1 на осаждение требуется 24 ч, при еще большем относительном количестве магния осаждение неполно.
2Р. Пршибил, Комплексоны в химическом анализе, Издатинлит, 1955.
3 К. Cheng, Т. Kurtz, A. R. Bray, Anal. Chem., 24, 1640 (1952).
4 Б. С. Цивина, Зав. лаб., 15, 142 (1949).
5 Ю. Ю. Лурье, 3. В. Николаева, Зав. лаб., 16, 1058 (1950). В этой статье приводится также метод определения калия, основанный на выделении осадка,
такого же состава.
Методы определения
713
более растворим, чем оксалат кальция, и осадок оксалатов не будет полностью содержать всего стронция, если не будут приняты специальные меры для полного его выделения, например если не будет прибавлен спирт \ Однако добавление спирта, вполне .допустимое в присутствии одного стронция, вносит осложнения, если присутствуют другие элементы, например барий и магний. Если определение магния следует за определением стронция, то небольшое количество стронция, оставшееся в растворе после осаждения оксалатом аммония, будет найдено в прокаленном остатке пирофосфата магния. Обработка этого остатка, служащая для извлечения из него кальция (см. «Отделение кальция от магния», стр. 694), пригодна также и для извлечения стронция.
БАРИЙ
Осаждение и взвешивание в виде сульфата
Если позволяют обстоятельства, барий следует, подобно стронцию, осаждать и взвешивать в виде сульфата. Свинец, стронций и кальций мешают определению бария вследствие малой растворимости их сульфатов. Хлор, алюминий и железо, если они присутствуют в больших количествах, загрязняют осадок. Применяемые в таких случаях методы анализа приведены на стр. 694.
Сульфат бария обычно рассматривается как исключительно нерастворимое вещество — 1 часть его растворяется приблизительно в 400 000 частях холодной воды. Однако эта величина составляет 2,5 мг в 1 л. В горячей воде, в разбавленной соляной или азотной кислоте растворимость его больше; в растворах, содержащих избыток одного из составляющих BaSO4 ионов, растворимость меньше. Следовательно, осаждение должно проводиться в растворах, содержащих не более 1 % минеральных кислот, и промывание осадка даже холодной водой не должно быть продолжительным. Кроме того, хотя растворимость сульфата бария в разбавленной соляной кислоте [рекомендуемой часто для промывания сульфата бария, осажденного в присутствии большого количества железа (III)] и невелика, но промывной раствор насыщается сульфатом бария почти мгновенно, в противоположность промывным растворам, применяющимся для промывания других осадков.
К кипящему раствору, слабо подкисленному соляной кислотой (раствор должен иметь объем не больше 200 жл и не должен содержать более 1 мл соляной кислоты в 100 мл), прибавляют в избытке горячую разбавленную серную кислоту (так, чтобы в 100 мл раствора было не более 5 мл концентрированной H2SO4), нагревают на паровой бане, пока осадок хорошо не осядет, фильтруют, промывают осадок горячей водой, содержащей несколько капель серной кислоты, и затем небольшим количеством воды до удаления кислоты. Завертывают осадок в, бумагу фильтра, помещают его во взвешенный платиновый тигель, высушивают, обугливают и прокаливают, как указано для кальция (стр. 711). Здесь допускается более высокая температура, чем при прокаливании сульфата кальция, но все же паяльную горелку применять нельзя. Взвешивают в виде BaSCU, прибавляют каплю серной кислоты, медленно выпаривают досуха,.
1 С. A. Peters, Am. J. Sci., [4], 12, 216 (190-1).
'714 Гл. XL. Щелочноземельные металлы — кальций, стронций, барий
прокаливают и снова взвешивают. Если масса осадка заметно изменилась, обработку кислотой повторяют.
Обработка серной кислотой имеет целью превращение в сульфат бария того небольшого количества хлорида бария, который окклюдируется осадком BaSOa, а также и сульфида бария, который мог образоваться при обугливании фильтра. Окисление сульфида бария, вероятно, происходит и при одном прокаливании, без добавления серной кислоты.
Если хотят проверить чистоту прокаленного сульфата бария, растворяют его полностью в тигле в 5 мл горячей концентрированной серной кислоты, охлаждают, вливают полученный сернокислый раствор в 50 мл воды, разбавляют до 100 мл водой и нагревают 1 ч на паровой бане. Затем охлаждают, фильтруют, промывают осадок небольшим количеством горячей воды, озоляют по-прежнему и взвешивают. Если масса оказалась меньшей, чем она была раньше, то эта разность вызвана присутствием других металлов (например, кальция) в первоначальном растворе, откуда они перешли в осажденный сульфат бария. При желании, эти металлы могут быть извлечены из фильтрата и определены х.
*Барий можно количественно выделить в виде сульфата в присутствии всех других катионов, кроме стронция и кальция, если перед осаждением ввести в раствор комплексон III (этилендиаминтетраацетат натрия) 1 2. Осадок сульфата бария, можно очистить растворением в аммиачном растворе комплексона и вторичным осаждением.
Ход определения. К анализируемому раствору прибавляют комплексон III в количестве, достаточном для связывания всех катионов, доводят pH раствора до 4,5—5 прибавлением ацетатного буферного раствора (130 мл концентрированной уксусной кислоты смешивают с 200 мл концентрированного раствора аммиака и 200 мл воды) и при нагревании осаждают кипящим 10 %-ным раствором сульфата аммония, прибавляя его в достаточном избытке. Через 6 ч фильтруют, промывают осадок слабокислым 1 %-ным раствором комплексона, сжигают и прокаливают, как обычно. Если имеются сомнения в чистоте выделенного осадка, его на фильтре растворяют в горячем 3—5%-ном аммиачном растворе комплексона III, тщательно промывают фильтр горячей водой, осаждают вторично сульфат бария подкислением раствора соляной кислотой по метиловому красному, сжигают и прокаливают осадок. Доп. ред* ,
Осаждение и взвешивание в виде хромата
Применение хроматного метода определения бария практически ограничивается теми случаями, когда отделяют барий от кальция или стронция или от обоих (стр. 700).
1 Например, в опытах, проведенных Н. В. Knowles, прокаленная смесь BaS04 и CaSO4, весившая вначале 0,1641 г, после первой очистки весила 0,1525 г, после второй очистки — 0,1511 г и после третьей — 0,1511 г. Указанная смесь сульфатов была получена следующим образом. К $0 мл раствора чистого хлорида бария было прибавлено 0,00976 г СаО в виде СаС12, раствор был нагрет до кипения и было прибавлено 10 мл разбавленной (1 : 1) серной кислоты. После охлаждения раствора к нему прилили равный объем спирта, профильтровали и осадок промыли смесью равных объемов спирта и воды. Когда взяли две порции по 50 мл того же раствора хлорида бария и обработали ях так же, но без прибавления СаС1.2, то получили 0,1511 г и 0,1509 г.ВаЗО4,
а В, Р г i Ь i 1, D. Mari с о v a, Chem, listy, 46, 542 (1952)4
Тлава XLI
МАГНИЙ
Магний — один из наиболее распространенных в природе металлов. В изверженных породах он представлен амфиболами, пироксенами, слюдами и оливином. Обычными магнезиальными силикатами являются тальк H2Mg3(SiO3)4, хлорит H8(Mg, Fe)6Al2Si3O18 и серпентин H4(Mg, Fe)3Si2O8. Огромное распространение имеет также доломит CaMg(CO3)2 (Fe, Мп изоморфны с Mg) — карбонат магния и кальция. Соли магния находятся в морской воде и во многих минеральных источниках. В самородном виде магний не обнаружен х.
ОБЩИЕ ЗАМЕЧАНИЯ
Магний не создает затруднений в обычном ходе анализа горных пород за исключением тех случаев, когда присутствуют фосфаты и арсенаты в избытке по сравнению с тем их количеством, какое может быть свя&ано железом и алюминием при осаждении аммиаком. В таких случаях вместе с железом и алюминием осаждается и фосфат или арсенат магния; -если магний не будет определен в этом осадке, то результат его определения получится пониженным, а результат определения алюминия — соответственно повышенным.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ МАГНИЙ
Разложение минералов, содержащих магний, не представляет особых затруднений и проводится, как описано в гл. «Щелочноземельные металлы» (стр. 693).
МЕТОДЫ ОТДЕЛЕНИЯ
Методы отделения магния перед его определением большей частью просты. Обработка сероводородом в кислом растворе, затем последовательно аммиаком, сульфидом аммония и оксалатом аммония отделяет большинство элементов от магния и оставляет последний только вместе с аммонийными солями и щелочными металлами, удаление которых для определения магния необязательно. Барий, если он не был первоначально -связан с радикалом серной кислоты, также остается в растворе после
1 F. W. Clarke, U. S. Geol. Survey Bull., 770, 17 (1924).
716
Гл. XLI. Магний
всех этих отделений. Его можно удалить серной кислотой — методом, описанным в гл. XL (стр. 713).
Отделение магния от кальция не представляет затруднений, когда магний присутствует в значительном количестве. Отделение проводят так, как описано в гл. XL (стр. 694). Но если очень малые количества магния сопровождаются большими количествами кальция, то для отделения надо применять один из специальных методов. Из них наиболее удовлетворительным является следующий метод \ предложенный для отделения малых количеств магния от сульфатов щелочных металлов, хотя при применении этого' метода одновременное определение кальция в том же растворе становится невозможным. Применительно к анализу продажных солей кальция отделение проводится следующим образом. 10 г соли кальция переводят в раствор и разбавляют его до 100 мл. Если для растворения соли была применена кислота, то избыток ее после удаления кипячением СО2, SO2 и т. п. нейтрализуют едким натром. Прибавляют окись кальция (полученную прокаливанием 0,3—0,4 г карбоната кальция), нагревают до кипения и фильтруют, но не промывают осадка. Осадок растворяют в соляной кисйоте, удаляют кальций двукратным осаждением, как описано на стр. 705 и сл., и определяют магний в соединенных фильтратах, как описано в разделе «Определение в виде пирофосфата магния» (стр. 719). Об отделении малых количеств кальция от больших количеств магния см. стр. 694.
Может случиться, что при анализе силикатных минералов, разлагаемых кислотной обработкой, вследствие недостаточного количества пробы для анализа необходимо будет определять щелочные металлы, а также и другие основания, кремнекислоту и окись титана, все в одной и той же навеске пробы. Тогда после ряда отделений, исключающих применение реактивов, содержащих щелочные металлы, надо отделить магний от щелочных металлов. Для этой цели применяются следующие методы: осаждение магния окисью ртути, осаждение магния карбонатом аммония, осаждение щелочных металлов амиловым спиртом или спирто-эфирной смесью и осаждение магния оксихинолином.
В первом методе магний осаждают из растворов хлоридов влажной окисью ртути (II), свободной от щелочных металлов. Для осаждения удаляют аммонийные соли прокаливанием, растворяют остаток в небольшом количестве воды и прибавляют по каплям суспензию окиси ртути, перемешивая после каждого добавления реактива. (Суспензию окиси ртути приготовляют, как описано в разделе «Осаждение суспензиями карбонатов или окисей», стр. 108). Прибавление реактива ведут до тех пор, пока желтая окись ртути не перестанет переходить в раствор. Выпаривают досуха, прибавляют несколько капель воды, снова выпаривают и повторяют это несколько раз. Затем осторожно нагревают под сильной тягой до улетучивания ядовитого хлорида ртути (II). Удаление избытка окиси ртути прокаливанием на данной ступени анализа недопустимо в том случае, если надо определять щелочные металлы, так как последние могут улетучиться. Остаток обрабатывают горячей водой, фильтруют и промывают водой. При наличии более 1 % окиси магния растворяют остаток в соляной кислоте и повторяют операцию. Фильтр с остатком
1 Н. Neubauer, Z. anal. Chem., 43, 14 (1904); J. C. Hostetter, J. Ind. Chem., 6, 392 (1914); Chem. News, 110, 155 (1914).
Методы определения
717
высушивают во взвешенном платиновом тигле, озоляют его под тягой и взвешивают. Так как применение горячей воды для промывания остатка ведет к небольшому растворению окиси магния, то лучше это промывание проводить насыщенным раствором гидроокиси кальция. Небольшое количество кальция, остающееся в остатке окиси магния, должно быть затем отделено, как описано в гл. «Щелочноземельные металлы» (стр. 693), а тот кальций, который перейдет в фильтрат вместе с щелочными металлами, удаляют, как описано в гл. «Щелочные металлы» (стр. 729).
Метод осаждения карбонатом аммония 1 состоит в следующем. Приготовляют реактив, насыщая карбонатом аммония смесь, состоящую из 18 мл аммиака, 75 мл воды и 95 мл 95%-ного спирта. Анализируемый раствор, содержащий не более 0,4 г хлоридов магния и щелочных металлов, выпаривают до объема около 50 мл, прибавляют равный объем 95%-ного спирта и затем 50 мл приготовленного реактива. Перемешивают 5 мин и оставляют смесь на 20 мин. Если количество присутствующих солей щелочных металлов не велико (не больше 0,1 г), собирают осадок на асбестовой прокладке в тигле Гуча и промывают осаждающим реактивом. Если содержание щелочных солей значительно, сливают жидкость с осадка через асбестовый фильтр, осадок растворяют и вновь осаждают по-прежнему. Если хотят определить и магний и щелочные металлы, то осадок осторожно прокаливают и взвешивают в виде окиси магния. Тот факт, что литий частично выпадает вместе с магнием, не делает этот метод непригодным для анализа силикатных пород, так как литий не присутствует в заметных количествах почти ни в одной породе.
Амилово-спиртовый и спирто-эфирный методы отличаются от методов, описанных выше, тем, что в них вместо магния осаждаются щелочные металлы. При отсутствии лития амилово-спиртовый метод 2 может считаться вполне удовлетворительным. Он аналогичен методу Гуча для отделения лития от натрия и калия и требует тех же поправок на растворимость. Описание последнего метода см. в гл. «Щелочные металлы» (стр. 729). Спирто-эфирный метод 3 является видоизменением метода отделения лития от щелочных металлов, также описанного в гл. «Щелочные металлы» (стр. 738). Если присутствует литий, он будет сопровождать магний.
Единственное отклонение метода отделения магния от метода отделения лития заключается в применении для первой обработки 25 мл спирта и 25 мл эфира вместо 20 мл спирта и 60 мл эфира, как это требуется для отделения лития. Если первый осадок склонен желатинироваться, то прибавляют еще несколько миллилитров спирта, вращая при этом стакан. Фильтрат следует выпарить и осаждение повторить.
Магний можно отделить от щелочных металлов и от бария, стронция и кальция осаждением его оксихинолином 4. В присутствии кальция или
1 Е. A. Wulfing, Вег., 32, 2214 (1899); F. A. Gooch, Е. A. Eddy, Am. J. Sci., [4], 25, 444 (1908); Chem. News, 97, 280 (1908).
2 R. B. Riggs, Am. J. Sci., [3], 44, 103 (1892).
3 S. Palkin, J. Am. Chem. Soc., 38, 2326 (1916); 42, 1618 (1920).
4 F. L. Hahn, Chem. Ztg., 50, 754 (1926); F. L. Hahn, К. V i e w e g, Z. anal. Chem., 71, 122 (1927); R. Berg, там же, 70, 341 (1927); 71, 23, 171, 321, 369 (1927). Согласно L. Moser и К. Schutt [Monatsh., 51, 23 (1929)], наиболее удовлетворительное отделение магния от лития достигается осаждением его оксихинолином.
718 ' Гл. XLI. Магний
больших количеств других металлов требуется двукратное осаждение.. Осаждение магния оксихинолином может быть проведено вслед за осаждением кальция в виде оксалата; таким способом можно удалять магний; из раствора без введения в него нелетучих солей. Магний (а также медь,, кадмий и цинк) можно отделить также и от алюминия, если осаждение оксихинолином проводить из раствора, содержащего тартрат натрия и умеренное количество едкого натра. Применение оксихинолина для; отделения магния от кальция и алюминия не представляет каких-либо преимуществ по сравнению с обычными методами, за исключением разве случаев, когда надо отделить малые количества магния от больших количеств этих элементов. Описание метода см. в разделе «Осаждение оксихинолином» (стр. 725).
Магний.можно отделить от алюминия, железа, цинка, олова и многих, других элементов прямым осаждением его двузамещенным фосфатом аммония из аммиачного раствора, содержащего цитраты. Для этого поступают следующим образом. К кислому анализируемому/ раствору, занимающему объем приблизительно 100 мл и перенесенному в коническую^ колбу емкостью 300 мл, прибавляют 2 г лимонной кислоты и 15 мл-25%-ного раствора двузамещенного фосфата аммония. Приливают аммиак,, пока раствор не станет щелочным по лакмусу, и сверх того 10 мл избытка.. Затем всыпают 5—10 стеклянных-шариков, плотно закрывают колбу пробкой и помещают ее в аппарат для встряхивания на 1 ч, после чего оставляют в холодном месте на 4 ч или, лучше, на ночь. Отфильтровывают' осадок через плотный фильтр, содержащий немного бумажной массы, и тщательно промывают разбавленным (1 : 19) раствором аммиака, содержащим 50 г двузамещенного фосфата аммония в 1 л. Затем пропускают через фильтр 25 мл горячей разбавленной (1 : 19) соляной кислоты, собирая раствор в ту колбу, в которой проводилось осаждение. Переносят полученный раствор в стакан емкостью 150 мл и промывают колбу и фильтр разбавленной соляной кислотой, собирая промывные воды в тот-же стакан. К полученному раствору, занимающему объем 50—75 мл и отделенному от стеклянных шариков, прибавляют 0,5 мл 25%-ного раствора двузамещенного фосфата аммония, охлаждают и при перемешивании приливают аммиак, пока раствор не станет щелочным по лакмусу.. Перемешивают еще несколько минут, приливают 3—4 мл концентрированного раствора аммиака и дают постоять 4 ч или оставляют на ночь. Затем, переносят осадок на маленький фильтр, промывают его разбавленным (1 : 19) раствором аммиака и превращают прокаливанием в пирофосфат-магния, как обычно.
Если в анализируемом растворе присутствуют кальций, стронций или барий в значительных количествах, их надо, удалить перед осаждением магния. Если же содержание..этих элементов в анализируемом растворе незначительно, их можно осаждать вместе с магнием и затем определять в прокаленном осадке, как описано в разделе «Определение в виде пирофосфата магния» (см. ниже)
При анализе металлического алюминия и алюминиевых сплавов отделение магния можно проводить следующим методом. Навеску металла погружают в 200—500 мл воды и прибавляют 30%-ный раствор едкого
1 Дальнейшие подробности см. в статье J.. I- Hoffman, G. Е. F, Lun-dell, J. Research NBS, 20, 607 (1938).
Методы определения
719- .
натра порциями по 25 мл до растворения алюминия. После каждого прибавления едкого натра выжидают прекращения бурной реакции. Затем нагревают на паровой бане 1 ч, охлаждают и фильтруют. Осадок промывают 1 %-ным раствором, едкого натра Ч
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Определение в виде пирофосфата магния
Общие замечания. Магний почти всегда осаждают в виде фосфата-магния и аммония MgNH4PO4 • 6Н2О и взвешивают после прокаливания в виде пирофосфата магния Mg2P2O7. Так как многие другие элементы образуют нерастворимые фосфаты, то осаждение магния, как правило, должно проводиться после обычных отделений сероводородом, аммиаком, сульфидов аммония и оксалатом аммония. Надо иметь в виду, что при выполнении этих осаждений большее или меньшее количество магния может быть потеряно (см. стр. 694), особенно в тех случаях, когда присутствуют большие количества мышьяка или фосфора, или если осаждение аммиаком и оксалатом аммония проводится только однократно. Магний можно осаждать в виде фосфата в присутствии хромат-ионов.
Имеется еще много неясного относительно надлежащих условий определения магния в виде фосфата, несмотря на громадное число работ, посвященных этому вопросу 1 2.
Почти все экспериментаторы ставили своей целью получение осадка нормального состава при однократном осаждении. Это может быть достигнуто, если осаждение проводится из раствора, содержащего только один магний и в заранее известном количестве, но вряд ли можно надеяться на выделение такого осадка из растворов, полученных после ряда осаждений в ходе анализа. Такие растворы всегда содержат посторонние соли: хлориды щелочных металлов, хлорид аммония и оксалат аммония. Соли натрия, и особенно соли калия, загрязняют осадок магния, замещая группу аммония в MgNH4PO4 щелочным металлом 3. Осадок может быть также загрязнен фосфатами магния состава Mg3(PO4)2 или Mg(NH4)4(PO4)2. Примесь первого фосфата ведет к получению пониженных результатов, примесь второго — к получению повышенных результатов, если только образующийся при обычном прокаливании этого фосфата Mg(PO3)2 не прокаливать достаточно долго при более высокой температуре, чтобы он превратился в пирофосфат магния. Все неопределенности 1 в составе осадка устраняются при применении двукратного осаждения, так как после растворения первого осадка можно создать почти идеальные условия для второго осаждения. Повторное осаждение не
1 J. Research NBS, 18, 1 (1937).
2 Нужно отметить следующие статьи: Н. Neubauer (см. сноску 1, стр. 716); F. A. Gooch, М. Austin, Ань J. Sci., [4], 7, 187 (1899); К. Bube, Z. anal. Chem., 49, 587 (1910); К. К. J а г v i n е п, там же, 44, 335 (1905); D. В а 1 а-r е w, Z. anorg. Chem., 101—104 (1918—1919); Z. К ar a oglanow, Р. Dimi-trow, Z. anal. Chem., 57, 353 (1918).
3 При тщательном проведении однократных осаждений магния из растворов, содержавших 0,0741 г MgO в виде MgCl2 и 5 г' NaCl в 400 .ил,было найдено в среднем 0,0766 г MgO, когда образование осадка происходило при 20° С, и 0,0787 г MgO, когда осаждение проводилось из горячего раствора, а затем следовало отстаивание при 20° С.
720
Гл. XLI. Магний
вызывает потери магния 1 и почти не увеличивает времени, расходуемого аналитиком на определение.
Аммонийные соли, образовавшиеся в растворе в результате предварительных разделений, перед первым осаждением магния удалять необязательно, если количество магния в растворе не слишком мало и нет необычно больших количеств оксалата аммония 2. По нашему опыту, двузамещенный фосфат аммония (NH4)2HPQ4 является наилучшим осаждающим реактивом. Вероятно, также пригодна и фосфорная кислота, «ели она в достаточной степени чиста. Кроме этих веществ, в качестве осаждающих реактивов применяют NaNH4HPO4 и двузамещенный фосфат натрия Na2HPO4. Эти реактивы вполне удовлетворительны, если проводится двукратное осаждение, и для второго осаждения прибавляют лишь очень незначительное количество реактива. Если проводится однократное осаждение, получаются значительно повышенные результаты, особенно если присутствует большое количество магния или если осадок отстаивался в течение короткого промежутка времени, например 4 ч. При более продолжительном стоянии получаются лучшие результаты; очевидно, фосфат магния и натрия более растворим и он постепенно превращается в фосфат магния и аммония.
Прежде полагали необходимым проводить осаждение магния в присутствии очень большого избытка аммиака, но это оказалось ошибочным. Осадок практически нерастворим в растворе, содержащем 5—10% концентрированного раствора аммиака (по объему). Осадки, которым дают постоять перед фильтрованием 12 ч, немного менее растворимы, чем те, которые отстаиваются только 2—3 ч.
Если нет необходимости исследовать осадок после прокаливания на присутствие в нем примесей, то вместо бумажного фильтра' лучше применять тигель Мунро (стр. 127). Применение тигля Гуча не рекомендуется вследствие того, что некоторые разновидности асбеста немного растворяются под действием щелочных растворов, содержащих фосфаты. Прокаливание фильтра с осадком должно проводиться медленно, при возможно более низкой температуре, предпочтительно в муфеле, до удаления углерода 3, иначе пирофосфат магния частично восстановится и фосфор улетучится 4. Кроме того; уголь станет частично «огнеупорным» и может .не сгореть.
Из боязни испортить тигель вследствие восстановления некоторой части фосфата при озолении бумажного фильтра в тигле многие химики высушивают фильтр с осадком, отделяют насколько возможно осадок от фильтра, сжигают фильтр с приставшими к нему частицами осадка и затем прибавляют главную массу осадка. При таком способе всегда имеется опасность потери малого количества тонкого сухого порошка осадка, поэтому мы предпочитаем брать на себя очень небольшой риск
1 Например, в опытах, проведенных Н. В. Knowles, анализ аликвотных порций раствора чистого хлорида магния дал в средней 0,0729 г MgO после однократного осаждения, 0,0733 г'после двукратного осаждения и 0,0734 г после трехкратного осаждения, См. также гл. «Фосфор» (стр. 794).
2 О методе удаления аммонийных солей см. стр. 161.
3 Если время позволяет, можно поместить тигель с влажным фильтром, содержащим осадок, в холодный муфель и постепенно, в течение 3 ч, повышать температуру до 750° С, после чего провести окончательное прокаливание при 1100—1150° С.
4 К. D. Jacob, D. S. Reynolds, J. Assoc. Offic., Agr. Chemists, 11, 128 (1928).
Методы, определения
721
испортить тигель. Насколько он мал, видно из того, что, проводя многие тысячи анализов, мы только в двух случаях наблюдали повреждение тигля при совместном сжигании в нем осадка с фильтром; это произошло, вероятно, из-за присутствия в осадке неподозреваемых посторонних примесей. Если осадок имеет надлежащий состав, нет необходимости прокаливать его при температуре выше 1 1050—1100° С. Но в обычных случаях, особенно если образовался большой осадок (0,5 г), в результате Прокаливания при указанной температуре в осадке может остаться немного Mg(PO3)2; последний мог образоваться при прокаливании небольшого количества Mg(NH4)4(PO4)2, который мог получиться при осаждении магния. Поэтому желательно, особенно при проведении только однократного осаждения, прокаливать под конец в течение 5—15 мин, в зависимости от величины осадка, при 1150—1200° С для превращения Mg(PO3)2 в Mg2P2O7. При этой температуре осадок нельзя прокаливать до постоянной массы, потому что Mg2P2O7 тогда медленно теряет Р2О5 (см. стр. 786). Малые количества посторонних фосфатов, например фосфата кальция, значительно понижают температуру плавления пирофосфата магния и могут привести к сплавлению массы даже при 1000° С.
Применяемое Фрезениусом и другими исследователями смачивание прокаленного пирофосфата магния 1—2 каплями азотной кислоты, выпаривание и вторичное прокаливание с целью получения совершенно белого пирофосфата магния редко приносит какую-либу пользу, но иногда ведет к потерям. Растворение пирофосфата магния в азотной кислоте с последующим выпариванием и прокаливанием определенно приводит к потерям 2.
Меньше возражений вызывает рекомендованный некоторыми авторами метод, в котором растворяют в азотной .кислоте не пирофосфат, а первоначальный промытый осадок фосфата магния и аммония. Полученный раствор выпаривают во взвешенном платиновом тигле и прокаливают. Если выпаривание проводится осторожно, то заметной потери фосфора не наблюдается, а появление розовой окраски, когда осадок становится почти сухим, является чувствительной реакцией на загрязнение осадка марганцем.
Имеется еще предложение (направленное к той же цели) проводить конечное промывание осадка слегка аммиачным 10 %-ным раствором нитрата аммония. При не вполне выясненных условиях прокаливания В. Ф. Гиллебранд часто наблюдал, что продукт прокаливания не растворялся в соляной или азотной кислоте даже после продолжительного кипячения и что белые кусочки какого-то неизвестного фосфата долго не растворялись при обработке кислотой.
Осаждение и фильтрование. Первое осаждение. Обычно имеют дело с аммиачным раствором, из которого были удалены марганец, кальций и большинство других элементов. Раствор этот содержит достаточное
1 В действительности превращение осадка в Mg2P2O7 происходит даже при 250° С, но прокаливание при более высокой температуре необходимо для удаления других соединений.
* D. Campbell, Phil. Mag., [4], 24, 380 (1862); Е. Luck, Z. anal. Chem., 13, 255 (1874); Z. Karaoglanow, P.- D i mi trow, там же, 57, 353 (1918). Согласно W. M. McNabb [J. Am. Chem. Soc., 49, 893 (1927)], никакой потери не происходит, если азотнокислый раствор прокаленного пирофосфата магния сначала нейтрализовать аммиаком, потом выпарить и прокалить сухой остаток.
46 Зака» 522.
722
Гл. XLI. Магний
количество хлорида аммония, помимо оксалата аммония, и часто еще большое количество хлорида натрия Е
Этот аммиачный раствор слегка подкисляют соляной кислотой и прибавляют двузамещенный фосфат аммония в виде кристаллов чистой соли пли в виде свежеприготовленного 10%-ного раствора. Реактив прибавляют в таком количестве, чтобы обеспечить по крайней мере десятикратный его избыток, а лучше всего, чтобы избыток был равен 1 г (NH4)2HPO4 на каждые 100 мл раствора 1 2. Хорошо перемешивают раствор и медленно прибавляют аммиак до щелочной реакции. Прибавлять аммиак надо особенно медленно, когда начнет выделяться осадок. Затем прибавляют еще по 10 мл аммиака на каждые 100 мл раствора и оставляют раствор. Если содержание магния мало, то осаждение может быть ускорено сильным перемешиванием и охлаждением раствора до 0—10° С. Если пользуются стеклянной палочкой, то не следует скфести ею стенки сосуда, потому что быстро образующийся кристаллический осадок пристает к этим местам.
Дают раствору постоять в течение ночи, фильтруют через бумажный фильтр, не пытаясь перенести весь осадок на фильтр, и промывают сосуд, где проводилось осаждение, осадок и фильтр 5—6 раз холодным разбавленным (5 : 95) раствором аммиака.
Второе осаждение. Осадок на фильтре растворяют в горячей разбавленной (1 : 4) соляной кислоте, собирая раствор в сосуд, где проходило осаждение и где осталась часть осадка. Стеклянную палочку, которая служила для перемешивания, также обмывают кислотой 3. Если осадок магния был небольшим, переносят раствор в маленький стакан. Разбавив полученный солянокислый раствор до 50—150 мл, в зависимости от количества присутствующего магния, прибавляют 0,1—0,3 г двузамещенного фосфата аммония и затем медленно по каплям при непрерывном перемешивании прибавляют аммиак, пока он не будет в очень небольшом избытке и кристаллический осадок MgNH4PO4 хорошо не сформируется. Тогда прибавляют еще по 5 мл аммиака на каждые 100 мл раствора. Дают постоять 4 ч (можно оставить на ночь), фильтруют через новый фильтр и промывают осадок холодным разбавленным (5 : 95) раствором аммиака, очищая одновременно сосуд и палочку от приставшего к ним осадка.
Прокаливание осадка. Высушивают осадок, завернув его во влажный фильтр, в платиновом тигле, медленно обугливают бумагу фильтра, не давая ей воспламениться, и сжигают уголь, постепенно усиливая пламя, которое, однако, не должно накаливать тигель сильнее, чем до слабокрасного каления. Наконец, прокаливают 30 мин при 1000—1100° С,
1 Если количество аммонийных солей в растворе, особенно оксалата аммония, очень велико или присутствует очень небольшое количество магния, то аммонийные соли перед первым осаждением следует разрушить выпариванием и осторожным прокаливанием или мокрой обработкой, описанной на стр. 160.
2 Это особенно необходимо, если раствор содержит большие количества хлорида аммония, если количество магния мало или если объем раствора большой. Растворы фосфата аммония разъедают стекло. Надо применять или свежеприготовленные растворы или растворы, подкисленные перед их хранением соляной кислотой.
3 Если в ходе анализа силиката платина не была удалена и если раствор был обработан сульфидом аммония, но не был подкислен осаждением кальция, то на данной ступени анализа может выделиться некоторое количество сульфида платины, который должен быть удален фильтрованием.
Методы определения
723
охлаждают в эксикаторе, взвешивают в виде Mg2P2O7 и повторяют прока ливание до постоянной массы.
Удаление и определение веществ, обычно загрязняющих пирофосфат магния. Опыт показывает, что многие аналитики при определении магния обычно получают повышенные результаты. Одна из причин ошибок заключается в несовершенном отделении других элементов, сопровождающих магний. После обычных отделений следует ожидать осаждения вместе с магнием бария и малых количеств стронция и кальция, если они первоначально присутствовали, а если осаящение сульфидом аммония было опущено, то и большей части марганца. Осаждение кальция и марганца протекает полностью, осаждение стронция и бария практически также полно, если магний присутствует в значительном количестве и концентрация аммонийных солей в растворе не слишком высока. Если раствор не нагревают и не выпаривают, то литий не осаждается. Даже при соблюдении всех требуемых при предварительных осаждениях условий малые количества некоторых элементов оста!отся в растворе и взвешиваются в виде фосфатов вместе с магнием.
Даже при самой тщательной работе аналитик должен провести проверочные пробы на присутствие загрязняющих веществ в прокаленном осадке Mg2P2O7, хотя бы только для того, чтобы убедиться в реальности этого источника ошибок и в ограниченности наших методов анализа.
Барий. В обычных анализах барий при отсутствии сульфат-ионов сопровождает магний и в конце определения осаждается вместе с ним и взвешивается в виде Ва2Р2О7 или в виде Ва3(РО4)2. Если пирофосфат магния исследуется на присутствие в нем кальция, как указано ниже, то барий отделяется вместе с кальцием в виде сульфата. Вследствие неопределенности состава фосфата бария и некоторого сомнения в полноте его осаждения барий лучше удалять серной кислотой перед конечным осаждением магния.
Кальций. Осаждение кальция в виде оксалата никогда не проходит полностью, и поэтому если кальций первоначально присутствовал в растворе, то очень небольшое количество его будет найдено в пирофосфате магния в виде смеси Са8(РО4)2 и Са2Р2О7. Количество последнего относительно невелико, и серьезной ошибки не будет, если при расчетах принять, что весь кальций в прокаленном осадке Mg2P2O7 присутствует в виде 1 Са3(РО4)2. Кальций и барий могут быть определены и содержание их вычтено из массы нечистого Mg2P2O7, как описано в гл. «Щелочноземельные металлы» (стр. 693).
При анализе горных пород масса полученной окиси кальция должна быть в среднем меньше 0,5 мг; ее прибавляют к уже найденной массе окиси кальция (осажденной в виде оксалата) и вычитают после пересчета на Са3(РО4)2 из массы пирофосфата магния. Таким способом получают правильный результат определения магния. Спиртовый фильтрат, остав-
1 Например, в опытах Н. В. Knowles были получены следующие результаты:
СаО взято г MgO взято г Смесь фосфатов г СаО, пересчитанная на Са3(РО4), г Mg2PsO7 по разности г MgO вычислено г СаО, полученная из смеси фосфатов г
0,0014 0,0492 0,1380 0,0026 0,1354 0,0490 0,0015
0,0287 0,0158 0,0958 0,0526 0,0432 0,0156 —
0,0042 0,0492 0,1442 0,0077 0,1365 0,0494 0,0045
0,0070 0,0492 0,1489 0,0129 0,1360 0,0492 0,0072
46*
724
Гл. XLI. Магний
шийся после выделения кальция, можно выпарить, сухой остаток прокались для разрушения органических веществ и испытать на присутствие марганца, как описано ниже (см. «Марганец»).
Менее надежный путь отделения кальция, находящегося в прокаленном остатке пирофосфата магния, но вместе с тем допускающий открытие малых количеств железа и алюминия, которые могли раньше при осаждении' аммиаком не выпасть в осадок, заключается в следующем.
К прокаленному пирофосфату магния, растворенному в соляной кислоте, взятой лишь в небольшом избытке, прибавляют аммиак до щелочной реакции и затем по каплям уксусную кислоту до осветления раствора, который должен иметь к этому времени объем в 10—30 мл и не быть горячим. Иногда случается, что небольшой хлопьевидный осадок не растворяется. Его отфильтровывают, прокаливают, и если проверка показывает, что этот осадок не содержит магния (что часто случается), вычитают его массу из первоначально полученной массы Mg2P2O7 (см. ниже). Этот осадок большей частью или полностью состоит из фосфатов алюминия, железа и марганца и часто при прокаливании имеет красноватую окраску \ Если был прибавлен избыток уксусной кислоты, осторожно нейтрализуют большую ее часть аммиаком, затем прибавляют 1—2 капли раствора оксалата аммония и, если необходимо, оставляют стакан на 12 ч. Почти всегда появляется маленький осадок, который можно считать чистым оксалатом кальция, если он мелкозернист и не прилипает к стеклу, В противном случае осадок содержит оксалат магния или даже состоит из него в значительной своей части. В этом случае осадок отфильтровывают, прокаливают, вновь растворяют и осаждают.
Нужна большая тщательность, чтобы удовлетворительно выполнить эту операцию.
Марганец. Марганец осаждается фосфатом аммония, подобно магнию, и взвешивается в виде пирофосфата марганца Мп2Р2О7. Если предварительные отделения были проведены тщательно, то отделение друг от друга железа, алюминия и марганца, которые могут содержаться в хлопьевидном осадке (см. стр. 721), не имеет смысла.
Железо и алюминий в этом осадке обычно имеют своим источником те реактивы, которые вводились в раствор уже после осаждения этих элементов аммиаком; во всяком случае количество их в этом маленьком осадке ничтожно по сравнению с ранее определенным их общим содержанием в пробе. Но количество марганца в этом осадке является значительной частью общего содержания марганца в пробе, если не проводили предварительного выделения марганца специальным методом.
Для определения марганца растворяют указанный маленький осадок в азотной или серной кислоте и сохраняют раствор. В фильтрате после отделения этого осадка содержится магний и может также содержаться марганец. Фильтрат надо выпарить досуха, прокалить и выпарить еще 2—3 раза с азотной кислотой или 1 раз с серной кислотой для окончательного удаления следов хлора. Этот раствор соединяют с малым раствором, сохраненным прежде. Затем определяют марганец колориметрическим методом, как описано в гл. «Марганец» (стр. 505), и прибавляют
1 Одно железо не осаждается (NH4)2HPO4 из аммиачных растворов, содержащих оксалат аммония. Алюминий осаждается количественно, но частично потом переходит в раствор при промывании осадка 5%-ным раствором аммиака.
Методы определения
725
это количество к ранее найденному его содержанию, если только общее содержание марганца не определяли в отдельной навеске пробы. Во всяком случае это количество марганца, если оно значительно, должно быть пересчитано на пирофосфат марганца и вычтено из общей массы пирофосфата магния.
В табл. 35 (стр. 960) приведено распределение марганца между осадками окиси алюминия, окиси кальция и пирофосфата магния в том случае, когда осаждение алюминия проводилось аммиаком и марганец затем не был удален.
Осаждение оксихинолином
Очень хорошим методом определения магния является метод, в котором его осаждают из щелочного "раствора оксихинолином и или растворяют осадок в разбавленной соляной кислоте и титруют раствором бромата, или взвешивают осадок в какой-либо из следующих весовых форм: 1) в виде Mg(C9H6NO)2 • 2Н2О после высушивания при 105° С; 2) в виде Mg(C9H6NO)2 после высушивания при 150—160° С; 3) в виде MgO после прокаливания при 1000° С. Этот метод, особенно в его объемном варианте, быстрее, чем метод определения магния в виде пирофосфата.
Применяемым реактивом осаждается большое число элементов (см. стр. 148), поэтому определение магния должно проводиться после их удаления общими методами. Определение магния оксихинолином, реактивом, улетучивающимся при прокаливании, допускает последующее определение щелочных металлов в том же растворе, если щелочные металлы не вводились в раствор дополнительно в ходе анализа.
Какой бы вариант метода ни применялся, мешающие элементы: — медь, железо, алюминий, титан, марганец, цинк и кальций должны быть удалены. При объемном окончании определения можно кальций не удалять, но превращать его в оксалат кальция и, не фильтруя, проводить осаждение оксихинолята магния. Описанный ниже ход определения магния разработан для анализа цементов, не содержащих в заметных количествах элементов, которые не выпадают в осадок от аммиака: меди, цинка и марганца Ч Определение магния заканчивается объемным способом.
Ход определения. Навеску в 0,500 г пробы помещают в стакан емкостью 400 мл, смешивают с 10 мл воды и затем приливают 10 Л1л соляной кислоты. Осторожно нагревают и раздавливают расплющенным концом стеклянной палочки крупные частйцы, пока разложение пробы не закончится. Затем разбавляют жидкость до 150 мл горячей водой, прибавляют 3 капли 0,02%-ного спиртового раствора метилового красного и приливают раствор аммиака, пока раствор не станет желтым. Тогда вводят в него мацерированную фильтровальную бумагу и нагревают до кипения. Кипятят 1—2 мин, прекращают нагревание и дают постоять, пока осадок не осядет на дно. Фильтруют без замедления и тщательно промывают осадок горячим 2 %-ным раствором хлорида аммония.
К фильтрату приливают 5 мл соляной кислоты и затем 25 мл 4 %-ноГо раствора оксалата аммония (NH4)2C2O4 • Н2О. Нагревают до кипения, перемешивают и медленно прибавляют раствор аммиака, пока раствор
1 J. С. Redmond, J. Research NBS, 10, 823 (1933).
726
Гл. XLI. Магний
не станет щелочным. Продолжают кипячение 1—2 мин и дают постоять 10—15 мин, пока раствор не охладится приблизительно до 70° С.
Приливают 20 мл 1 прозрачного 1,25%-ного раствора оксихинолина (25 г реактива растворяют в 60 мл ледяной уксусной кислоты и разбавляют водой до 2 л) и прибавляют по 4 мл раствора аммиака на каждые 100 мл раствора. Перемешивают механической мешалкой 15 мин и дают постоять, пока осадок не осядет на дно. Фильтруют и промывают осадок горячим разбавленным (1 : 40) раствором аммиака.
Затем растворяют осадок в 200 мл горячей разбавленной (1 : 4) соляной кислоте, охлаждают приблизительно до 25° С и приливают из пипетки 25 мл титрованного (0,2 н.) бромид-броматного раствора (КВгО3 + КВг). Перемешивают и дают постоять около 30 сек для завершения бромирования. Затем приливают 10 мл 25%-ного раствора иодида калия, хорошо перемешивают и титруют 0,1 н. раствором тиосульфата натрия, пока окраска от выделившегося иода не станет бледно-желтой. Тогда прибавляют 2 мл раствора крахмала и титруют до исчезновения синего окрашивания. Для расчета содержания MgO вычитают объем израсходованного на титрование раствора тиосульфата из того объема раствора тиосульфата, который эквивалентен 25 мл бромид-броматного раствора, и разность умножают на титр раствора тиосульфата, выраженный в MgO.
Для приготовления 0,2 н. бромид-броматного раствора растворяют 20 г бромида калия и 5,57 г бромата калия в 200 мл воды и разбавляют до 1 л. Для приготовления 0,1 н. раствора тиосульфата натрия растворяют 25 г Na2S2O3-5H2O в 200 мл воды и разбавляют до 1 л (см. стр. 225).
Определение объема раствора тиосульфата, эквивалентного данному объему раствора бромата, см. в разделе «Осаждение оксихинолином» (стр. 148).
Установку титра раствора тиосульфата лучше всего проводить по стандартному образцу материала, подобного тому, который анализируется. Берут такую навеску стандартного образца, чтобы содержание в нем магния было близко к определяемому, и проводят его через все стадии анализа. Титр раствора тиосульфата затем находят, разделив количество MgO во взятой навеске стандартного образца на разность между объемом раствора тиосульфата, эквивалентным прибавленному бромид-броматному раствору, и объемом раствора тиосульфата, израсходованного на обратное титрование. Такой способ установки титра требует меньшего числа вычислений и, кроме того, он приводит к получению более точных результатов, если раствор тиосульфата приготовлен правильно и титр его проверяется каждую неделю.
Теоретически титр 0,1 н. растворов того и другого реактива равен 0,0005038 г!мл в расчете на MgO и 0,0003038 г!мл в расчете на Mg. Титр 0,2 н. растворов соответственно в 2 раза больше.
1 Этого количества достаточно для анализа навески в 0,5 г цемента, содержащего не более 6% окиси магния. При анализе цементов, содержащих большие количества магния, пропорционально увеличивают количество прибавляемого реактива. Оксихинолин несколько летуч при температурах выше 60° С. При выполнении осаждения надо следить за тем, чтобы не произошла потеря реактива прежде, чем образовался осадок. После выделения осадка растворы можно кипятить 1—2 мин, что способствует коагуляции осадка и удалению из него захваченного им реактива. Когда осадок затем растворяют в кислоте перед фильтрованием, температура раствора не должна быть выше, чем это необходимо для растворения осадка.
Методы, определения
727
Если выделенный осадок содержит только оксихинолят магния, его можно собрать во взвешенном стеклянном фильтрующем тигле и или высушить при 105° С и взвесить в виде Mg(C9H6NO)2 • 2Н2О или высушить при 150—160° С и взвесить в виде Mg(C9H6NO)2. Последнее следует предпочесть, если оксихинолин был прибавлен в значительном избытке. Малые осадки можно отфильтровывать через бумажный фильтр, осторожно сжигать и прокаливать до MgO.
Объемный метод
Для объемного определения магния были предложены различные методы. Наиболее часто применяемый из них 1-2 (если не считать описанного выше объемного варианта оксихинолинового метода) основан на реакции
MgNH4PO4 + H2SO4 = MgSO4+ NH4H2PO4
В этом методе сначала обычным способом осаждают фосфат магния и аммония, осадок отфильтровывают, промывают, удаляют из осадка аммиачную промывную жидкость, растворяют его в титрованном растворе серной кислоты, взятом в избытке, и титруют этот избыток обратно титрованным раствором едкого натра в присутствии метилового оранжевого до ясно-желтой окраски индикатора. Если растворы серной кислоты и едкого натра эквивалентны, то разность между израсходованными на титрование объемами этих растворов покажет количество серной кислоты, пошедшее на превращение трехзамещенного фосфата MgNH4PO4 в однозамещенные фосфаты NH4H2PO4 и Mg(H2PO4)2. Избыток осаждающего реактива (NH4)2HPO4 и промывной аммиачной жидкости должен быть удален, потому что на них расходуется серная кислота: на превращение (NH4)2HPO4 в однозамещенную соль NH4H2PO4 и на нейтрализацию NH4OH.
Рекомендуется 2 поступать следующим образом. Промытый осадок фосфата магния и аммония располагают на фильтре так, чтобы он занимал менее 2/з его объема, затем вынимают фильтр, развертывают его, отжимают как можно больше жидкости сухим фильтром и, наконец, высушивают в течение 45 мин при комнатной температуре или в течение 15 мин при нагревании не выше 60° С. Осадок можно высушить совершенно или прекратить высушивание, когда он высохнет на расстоянии 12 мм от края (тогда можно считать, что свободный аммиак удален). Можно фильтровать и через тигель Гуча; в этом случае высушивание облегчают, промывая осадок несколько раз спиртом. После высушивания помещают фильтр с осадком в подходящего размера стакан, обрабатывают отмеренным количеством 0,1 н. раствора серной кислоты, прибавляя его в избытке, и хорошо перемешивают до распадения бумаги фильтра и растворения осадка. Прибавляют 2 капли 0,1 %-ного раствора метилового оранжевого и затем еще кислоты, если раствор не станет розовым. Разбавляют до 100 мл водой и титруют обратно 0,1 н. раствором едкого натра до ясно-желтой окраски3. 1 мл 0,1 н. раствора серной кислоты соответствует 0,00202 г MgO.
1 F. S t о 1 b a, Jahresber., 985 (1876).
2 J. О. Handy, J. Am. Chem. Soc., 22, 31 (1900).
3P. J. Elving и E. R. Caley [Ind. iEng. Chem., Anal. Ed., 9, 558 (1937)] указывают на то, что магний можно количественно осадить в виде оксалата магния в 85%-ном растворе уксусной кислоты. Выделенный осадок можно прокалить до окиси магния или оттитровать перманганатом, а в фильтрате можно определить щелочные металлы. Недостатками метода являются малоудобная среда, в которой проводится осаждение, и большой объем осадка.
728 ' Гл. XLI. Магний
* Магний можно определять комплексометрически, титруя его раствором комплексона III (трилона Б)1 (см. стр. 157). В качестве индикатора применяют эриохром черный Т (хромоген черный специальный ЕТ-00), кислотный хром синий К или кислотный темно-синий 2. Одновременно с магнием титруется и кальций, что удобно, когда определяют жесткость природных вод. Если предварительно осадить кальций оксалатом, то, не фильтруя, можно титровать комплексоном III один магний 3.
Из колориметрических методов определения малых количеств магния заслуживает внимания метод с применением титановой желтой4. Доп. ред. *
1 См. Р. П р ш и б и л, Комплексоны в химическом анализе, Издатинлит, 1955.
2Т. Б. Стюнкель, Д. А. Савиновский, Е. М. Якимец, Известия, ВТИ, 21, 22 (1952).
8 * По данным некоторых авторов, если осадок не отфильтровывают, то результаты определения магния получаются несколько повышенными. Прим. ред. *
♦ С. С. Ш р а й б м а н, Зав. лаб., 13, 930 (1947); В. И. Адамович, Зав, лаб., 13, 935 (1947).
ЩЕЛОЧНЫЕ МЕТАЛЛЫ
ЛИТИЙ, НАТРИЙ, КАЛИЙ, РУБИДИЙ, ЦЕЗИЙ
Глава XLII
ЩЕЛОЧНЫЕ МЕТАЛЛЫ
Литий является одним из наиболее распространенных элементов; однако он никогда не содержится в породах в количествах, превышающих спектроскопические следы. Литий находится в щелочных полевых шпатах, мусковите, берилле и в литиевых минералах — лепидолите LiK[Al(OH, F)2 ] Al(SiOs)3, сподумене LiAl(SiO3)2, петалите LiAl(Si2O5)2, амблигоните Li(AlF)PO4, трифилине Li(Fe,Mn)PO4 илитиофилите Li(Mn, Fe)PO4. Есть основание предполагать, что литий- чаще залегает в породах с высоким содержанием натрия Ч Современными усовершенствованными спектральными методами обнаружены значительные количества лития в материалах, в которых прежде не предполагалось заметного содержания этого элемента. Например, стандартный образец огнеупорной глины из Clearfield Country, Ра., № 97 (Бюро Стандартов США) содержит 0,22% Li2O, что почти вдвое превышает содержание в нем окиси натрия. В связи с этим при определении щелочных металлов нельзя пренебрегать испытанием на литий.
Натрий является наиболее распространенным из щелочных металлов. Содержание его в литосфере 1 2 составляет 2,75%. В изверженных породах он входит в состав полевых шпатов (NaAlSigOg), минералов нефелиновой группы [NaAlSiO4 (иногда К и Са)1 и
1Н. S. Washington, Manual of the Chemical Analysis of Rocks, 4th ed., John Wiley and Sons, 1930, p. 31. О геохимии лития см. L. W. S t г 0 c k, Nachr. Ges. Wiss. Gottingen, Math.-physik. Klasse, vol. IV, 1936? S. 171. 0. Procke и R. Uzel [Mikrochim. Acta, 3, 105 (1938)] приводят специфическую реакцию на литий, основанную на низкой растворимости соединения, которое литий образует с железом (III) и иодной кислотой. Этой реакцией можно открыть несколько десятых микрограмма лития в капле раствора, насыщенного солями других щелочных металлов.
2 F. W. Clarke. U.S. Geol. Survey Bull., 770 (1924).
730
Гл. XLII. Щелочные металлы
некоторых пироксенов. Он широко распространен также в виде каменной соли и содержится почти во всех природных водах, главным образом в морской воде.
Калий также относится к распространенным в природе элементам группы щелочных металлов. Его содержание в литосфере составляет 2,58%. Он встречается во многих горных породах, главным образом в качестве составной части полевых' шпатов KAlSisO8, слюд H2KAl3(SiO4)3 и др. и лейцита KAl(SiO3)2. Почти все наземные воды содержат калий.
Рубидий находится в лепидолите и в ничтожных количествах во многих минералах, например в полевых шпатах, берилле, слюдах, лейците, сподумене и карналлите А
Цезий — наиболее редкий из щелочных металлов. Он содержится в поллуците — силикате алюминия и цезия H2Cs4Al4(SiO3)9 и, подобно рубидию, в лепидолите и некоторых других минералах.
ОБЩИЕ ЗАМЕЧАНИЯ
При изложении анализа горных пород, в разделе о щелочных металлах (стр. 1004 сл.), описан способ получения хлоридов щелочных металлов, свободных от всех прочих элементов, за исключением быть может незначительных количеств магния. Ни один из известных методов, по-видимому, не обеспечивает полного отделения магния. Небольшие количества магния, которые обычно переходят в фильтрат совместно с щелочными металлами и взвешиваются в виде хлорида, можно затем определить и вычесть массу их из общей массы хлоридов 1 2.
Когда для отделения кальция применяется осаждение одним карбонатом аммония, щелочные металлы содержат также несколько десятых миллиграмма хлорида кальция. В этом случае кальций следует дополнительно отделить осаящением небольшим количеством щавелевой кислоты из аммиачного раствора. Фильтрат затем выпаривают досуха и избыток оксалата аммония разрушают прокаливанием. Остаток смачивают каплей соляной кислоты, которую удаляют выпариванием, и после этого снова определяют массу хлоридов.
При обычной обработке по методу Лоуренса Смита (стр. 1006) литий не полностью попадает в остаток хлоридов щелочных металлов. Часть его остается в нерастворимом остатке после выщелачивания спекшейся массы водой, а некоторые количества увлекаются осадками карбоната кальция и оксалата кальция.
Содержание лития в горных породах обычно не достигает определимых количеств; определение его проводят, когда это возможно, хлоро-
1 W. R. Schoeller, A. R. Powell, The Analysis oi Minerals and Ores of the Rarer Elements, 2d ed., Charles Griffin and Co., London, 1940, p. 41.
2 Прекрасным методом отделения магния от щелочных металлов является, осаждение оксихинолином, как это описано на стр. 148; при наличии большого количества щелочных металлов рекомендуется повторное осаждение [R. Berg, Z. anal. Chem., 71, 32 (1928); F. L. Hahn, К. V i e w e g, там же, 126]. Этот метод служит также для отделения многих других элементов, например железа, алюминия, титана и циркония. Согласно L. Moser и К. Schutt [Monatsh., 51, 23 (1929)], наиболее удовлетворительное отделение магния от лития достигается осаждением оксихинолином.
Методы определения
731
платинатным методом, как указано в разделе «Хлороплатинатный метод» см. ниже.
В присутствии лития определение можно провести и методом с «-бутиловым спиртом и этилацетатом (стр. 734) или одним из методов, изложенных в разделе «Определение лития» (стр. 737). Хлороплатинатный метод применяется для определения последовательными операциями калия (рубидия и цезия), натрия и лития. Методы, изложенные в разделе «Определение лития», предназначаются в первую очередь для выделения лития и, если требуется, последующего определения сопровождающих его других щелочных металлов. Рубидий и цезий редко встречаются в горных породах Ч Если они содержатся в анализируемой породе, то попадают в осадок, содержаший калий, выделенный по одному из указанных выше методов. Их определяют методом, изложенным в разделе «Определение рубидия и цезия» (стр. 740). Наконец, в разделе «Определение одного калия» (стр. 744) приведены наиболее распространенные методы определения этого элемента.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Хлороплатинатный метод
В этом методе калий (вместе с рубидием и цезием) осаждают платинохлористоводородной кислотой из раствора хлоридов щелочных металлов. Для установления содержания натрия из массы смеси хлоридов вычитают количество хлорида калия, эквивалентное массе осадка хлороплатината. Метод вполне надежен, когда присутствуют только калий и натрий. Достоверность результатов зависит от точности определения калия и чистоты смеси хлоридов. В присутствии рубидия и цезия метод не пригоден. Если эти элементы не содержатся в анализируемом продукте, а присутствует литий, то, вычитая из массы суммы хлоридов содержание Хлорида калия, получают общую массу хлоридов натрия и лития, для раздельного определения которых необходимо провести разделение 1 2.
Хлороплатинатный метод, очень старый и хорошо изученный, и в настоящее время еще является наиболее широко распространенным в аналитической практике методом. Его недостатком являетсй возможность некоторого отклонения состава осадка от формулы K2PtCle. Отчасти из этих соображений, а также вследствие высокой стоимости реагента хлороплатинатный метод начинает иногда заменяться перхлоратным. Экономия от этого, однако, не очень велика, так как если сохранять осадки и фильтраты и время от времени их перерабатывать, можно избежать заметных потерь платины.
Определение калия. Приводимые ниже операции должны осуществляться в атмосфере, свободной от аммиака. К раствору хлоридов,
1 Цезий содержится в виде алюмосиликата в редком минерале поллуците и часто встречается в лепидолите. Рубидий также находится в лепидолите и нередко содержится наряду с калием в железных рудах [W. N. Hartley, Н. Ramage, Trans. Chem. Soc., 71, 533 (1897)].
2 Отделение калия от лития однократным осаждением в виде хлороплатината не всегда бывает количественным. В. Brauner [Coll. Czech. Chem. Comm., 2, 442 (1930)] нашел 23% лития в осадке хлороплатината калия, полученного однократным осаждением из раствора, содержавшего 0,1195 г NaCl, 0,0175 г КС1 и 0,0017 г LiCl.
732
Гл. XLII. Щелочные металлы
находящемуся в маленькой фарфоровой чашке1, прибавляют раствор Пла-тинохлористоводородной кислоты (см. «Реактивы», стр. 67), содержащий 10% платины, в количестве, достаточном для реакции с калием и натрием 2. Необходимое количество реактива легко вычислить, зная концентрацию платины в растворе и массу смеси хлоридов, которую при расчете условно принимают за хлорид натрия.
Полученный раствор разбавляют настолько, чтобы осадок, который может образоваться при введении реактива, полностью растворился при нагревании на водяной бане. Таким способом предупреждают увлечение маточного раствора внезапно выделяющейся массой кристаллов. Выпаривают до цолучения сиропообразной жидкости, затвердевающей при охлаждении. Не следует выпаривать досуха, так как это приводит к обезвоживанию соли натрия и делает ее менее растворимой в спирте.
Растворимость хлороплатината калия в 80%-ном спирте настолько незначительна 3, что нет необходимости вводить поправку, если объем раствора и промывных вод составляет 50—75 мл (в зависимости от величины осадка).
Растворимость K2PtCl6 в 80%-ном (по объему) С2Н5ОН при 20° С составляет приблизительно 10 мг на 100 мл, что соответствует около 4 1 мг К2О на 50 мл. Количество K2PtCl6, переходящее в раствор в процессе анализа, значительно меньше этой величины благодаря присутствию в растворе платинохлористоводородной кислоты и хлороплатината натрия. Кроме того, спирт, применяемый для промывания, при кратковременном соприкосновении с солью.не насыщается ею. Потеря калия, обусловленная растворимостью K2PtCl6, компенсируется, если калий содержится в реактивах, что обнаруживается при проведении «холостого» опыта.
Необходимо отметить, что хлороплатинат калия более растворим в метиловом спирте, чем в этиловом, а иногда рекомендуемое применение абсолютного этилового спирта может привести к разложению хлороплатината натрия и осаждению хлорида натрия 5. При осаждении натрия в виде тройного ацетата, а также в хлороплатинатном методе определения калия, 95%-ный этиловый спирт можно заменить спиртом, денатурированным 10% диэтилового эфира или ацетона 6.
Остаток смачивают 80%-ным (по объему) спиртом, фильтруют декантацией через маленький бумажный фильтр и промывают также декантацией еще некоторым количеством спирта, раздавливая при этом кристаллы маленьким пестиком или расширенным закругленным концом короткой стеклянной палочки. Фильтрат и промывные воды сохраняют для определения натрия. Осадок должен иметь золотисто-желтый цвет. Оранжевокрасная окраска указывает на неполное отделение натриевой соли. Нет необходимости переносить осадок на фильтр. Чашку и бумагу сушат в несколько мгновений для удаления оставшегося спирта. Затем осадок на фильтре растворяют в горячей воде, собирая раствор во взвешенный тигель или маленькую платиновую чашку. Выпаривают досуха и присоединяют соль, находящуюся в фарфоровой чашке. Если соль комковата, ее растворяют в воде и раствор снова выпаривают досуха. Нагревают 1 ч в воздушной бане при 130° С (очень малые количества
1 Лучше, чем в платиновой, так как избегается возможность образования нерастворимого соединения платины (II). См. также F. В о 1 m, Z. anal. Chem., 38, 349 (1899).
2 G. F. S m i t h и A. C. S h e a d [J. Am. Chem. Soc., 53, 947 (1931)] утверждают, что выделяющийся осадок имеет состав, более близкий к теоретическому! и что его можно нагревать при температурах до 260° С, если в качестве осадителя пользоваться хлороплатинатом лития вместо платинохлористоводородной кислоты.
8Н. Fresenius, Р. Н. Brinton, Z. anal. Chem., 50, 21 (1911).
4Е. Н. Archibald и сотрудники, J. Am. Chem. Soc., 30, 755 (1908).
5 J. М orozewicz, Anzeig. Akad. Krakau, 796 (1906).
• P. A. Webster, R. M. Crane, Ind. Eng. Chem., Anal. Ed., 15, 36 (1943).
Методы, определения 733
мелкозернистого осадка можно сушить при 100° С), вначале в закрытом тигле, так как осадок легко растрескивается. Высушенный осадок по охлаждении взвешивают в виде K2PtCle.
Сначала устанавливают, какому количеству КС1 эквивалентна масса осадка хлороплатината калия, с тем чтобы вычесть его из массы смеси хлоридов для вычисления содержания натрия, а затем пересчитывают на К2О или К.
При определении таких количеств калия, какие обычно содержатся в горных породах и минералах, для пересчета K2PtCle на КС1 вполне можно пользоваться теоретическим фактором 0,3067, основанным на общепринятом атомном весе платины. Применение эмпирического фактора 0,3056, предложенного Фрезениусом, Тредвеллом и др., при анализе описанным выше методом не рекомендуется.
Указывают \ что калий, рубидий и цезий можно количественно отделить от больших количеств натрия, железа, алюминия, марганца и других солей непосредственным осаждением платинохлористоводородной кислотой из раствбра в 60—70%-ном (по объему) спирте, при условии, что все содержащиеся в растворе элементы предварительно превращены в нитраты. Если щелочные металлы получены в виде хлоридов, непосредственное количественное осаждение их также можно осуществить добавлением платинохлористоводородной кислоты и 2—3 капель диэтилового эфира после растворения осадка щелочных металлов в 60—70%-ном спирте. Авторы утверждают, что осажденные таким образом хлороплатинаты чище, чем полученные выпариванием с платинохлористоводородной кислотой почти досуха и последующим выщелачиванием спиртом.
Определение натрия (лития). Известно лишь очень немного минералов и, пожалуй, ни одной горной породы, в состав которых из щелочных металлов входил бы один только натрий. Содержание натрия обычно вычисляют по разности после определения суммы всех щелочных металлов и остальных элементов всей группы. В тех случаях, когда желательно проверить полученные вычислением результаты содержания натрия, его можно определить в фильтрате после отделения хлороплатината калия. Для этого из фильтрата сначала удаляют платину, что можно осуществить несколькими способами, из которых метод Бунзена наиболее прост и надежен.
Спиртовый раствор помещают в колбу (обычно емкостью 100 мл), выпаривают до удаления большей части спирта, если требуется, прибавляют воды и закрывают колбу пробкой, снабженной вводной и отводной трубками. Последнюю трубку можно закрывать. Заполняют колбу водородом, пропуская его через вводную трубку. Закрывают выходное Отверстие трубки, не давая воздуху проникнуть в колбу, ставят колбу на горячую плитку или погружают в стакан с горячей водой и продолжают пропускать слабый ток водорода до тех пор, пока платина полностью не осядет и жидкость не станет бесцветной, как вода. Это можно установить, Осторожно взбалтывая раствор в колбе, что вызывает оседание суспендированного металла.
Вытесняют водород из колбы током индифферентного газа, фильтруют и промывают осадок водой. Фильтрат выпаривают до небольшого объема в фарфоровой или платиновой чашке, переносят во взвешенный
* W. J. O’L е а г у, J. Р а р i s h, Ind. Eng. Chem., Anal. Ed., 6, 107 (1934)
734
Гл. XLII. Щелочные металлы
платиновый тигель с хорошо прилегающей крышкой и выпаривают, по возможности, досуха. Выпариванию может способствовать смачивание сгущенной массы концентрированной соляной кислотой, которая вызывает осаждение мелкозернистого осадка. После смачивания выпаривать следует осторожно.
Когда осадок становится на вид сухим, помещают закрытый тигель на треугольник из платиновой проволоки, расположенной над очень слабым пламенем бунзеновской горелки. Высоту подбирают так, чтобы пламя, будучи в совершенно спокойном состоянии, не касалось дна и боков тигля и нагревало выступающий край крышки. Если при приближении уха к тиглю слышится потрескивание, немедленно оставляют на мгновение горелку. По прекращении потрескивания и выделения паров хлорида аммония (если эта соль присутствовала) можно усилить нагревание; под конец нагревают так, что соль начинает плавиться. Осадок взвешивают в виде NaCl и затем испытывают спектроскопически на содержание лития, если последний не отделялся.
Наконец, необходимо ввести еще поправку на небольшие количества хлорида магния, которые в процессе анализа некоторых продуктов могут не отделиться и сопровождают натрий. Для этой цели магний можно определить осаждением в виде фосфата магния и аммония, как описано в гл. «Магний» (стр. 719). Осаждение можно осуществить непосредственно в освобожденном от платины фильтрате после отделения хлороплатината калия при условии, если не будет проводиться проверочное определение натрия. Количество хлорида магния, вычисленное на основании полученной массы пирофосфата, вычитают из массы смеси всех хлоридов щелочных металлов.
Если желательно взвешивать натрий в виде сульфата, который можно нагревать до значительно более высокой температуры, чем хлорид, натриевую соль летучей кислоты выпаривают с серной кислотой до удаления избытка последней. Большую часть серной кислоты можно удалить нагреванием на голом пламени или в радиаторе (см. рис. 5, стр. 49), но небольшое ее количество удерживается настолько прочно, что для полного удаления необходимо прибавить карбонат аммония. Эту соль вводят в тигель маленькими порциями, не прекращая нагревания. Взвесив остаток, повторяют обработку карбонатом аммония до постоянной массы. Взвешивают в виде Na2SO4, который несколько гигроскопичен.
Натрий можно также определить непосредственно, как описано в следующем разделе.
Определение натрия с «-бутиловым спиртом и этилацетатом
Согласно этому методу \ хлориды щелочных металлов превращают в перхлораты, а затем натрий и литий отделяют от калия (рубидия и цезия) выщелачиванием смесью «-бутилового спирта и этилацетата. Полученную вытяжку выпаривают до удаления этилацетата и осаждают натрий в виде хлорида прибавлением «-бутилового спирта, насыщенного газообразным хлористым водородом. Раствор, содержащий литий, выпа
1 Н. Н. Willard, G. F. Smith, J. Am. Chem. Soc., 44, 2816 (1922); G. F. Smith, там же, 47, 762 (1925); G. F. S m i.t h, J. F. Ros s, там же, 47, 774, 1020 (1925).
Методы определения
735
ривают с серной кислотой и определяют литий взвешиванием в виде сульфата. Растворимость перхлоратов щелочных, щелочноземельных металлов, магния и аммония приведена в табл. 22.
ТАБЛИЦА 22
Растворимость перхлоратов щелочных, щелочноземельных металлов, магния и аммония1 (г/100 мл раствора при 25° С)
Соль Растворитель
вода метиловый спирт ЭТИЛОВЫЙ спирт н-бутиловый спирт этилацетат
NaClO4 113,88 35,833 11,134 1,495 8,425
LiC104 47,42 89,44 79,41 49,25 63,40
NH4C1O4 .... 21,91 5,268 1,488 0,0137 0,0286
КС1О4 2,0394 0,0830 0,0094 0,0036 0,0013
RbC104 1,328 0,0472 0,0071 0,0016 0,0014
CsC104 1,961 0,0734 0,0086 0,0048 Нерастворим
Ва(С1О4), .... 128,99 119,85 78,543 41,716 80,812
Са(С1О4)2 .... 112,34 113,68 89,551 68,419 57,377
Sr(C104)2 .... 157,51 113,95 100,01 71,205 76,67
Mg(C104)2 .... 73,453 37,749 18,398 44,638 54,173
1 Н. Н. Willard, G. F. Smith, J. Am. Chem. Soc., 45,
293 (1923).
Ход определения. Смесь хлоридов растворяют в воде и переносят в стакан из стекла пайрекс 1 емкостью 150 мл. Раствор обрабатывают хлорной кислотой, взятой в двух-трехкратном количестве по отношению к эквивалентному (в любом случае не менее 1 мл 60—70%-ной кислоты). Выпаривают досуха на горячей плитке (не выше 350° С) и удаляют кислоту, сконденсировавшуюся на стенках стакана, нагреванием голым пламенем. По охлаждении растворяют соли в 3—5 мл воды и снова выпаривают на плитке досуха 2. Охладив, прибавляют 10—20 мл смеси равных объемов w-бутилового спирта и этилацетата (оба безводные)3, нагревают при температуре, близкой к кипению, 2—3 мин и затем охлаждают до комнатной температуры.
Отстоявшуюся жидкость сливают декантацией в предварительно прокаленный и взвешенный тигель Гуча или, лучше, в тигель Мунро 4 и трижды промывают осадок декантацией спирто-ацетатной смесью, порциями по 5 мл. Фильтрат и промывные воды сохраняют 5. Остаток в тигле
1 Имеются указания на то, что горячая хлорная кислота растворяюще действует на некоторые сорта лабораторного стекла и фарфор. Стекло пайрекс ею не разъедается (G. F. Smith, J. F. Ros s, цит. выше).
2 Согласно G. F. Smith и J. F. Ross (цит. выше), перхлораты, образующиеся в растворе хлорной кислоты, после выпаривания при 350° С удерживают еще эту кислоту. Их следует растворить в воде и раствор снова выпарить.
3 и-Бутиловый спирт с температурой кипения в пределах от 116,0° С до 117,7° С при 760 мм рт. ст. должен быть приготовлен фракционированной дистилляцией химически чистого реактива [плотность d*° равна 0,8065] и высушен, лучше всего над металлическим кальцием в колбе с обратным холодильником. Безводный этилацетат должен быть свободен от этилового спирта и иметь концентрацию от 99,7 до 100%.
4 Фильтрующий прибор желательно приспособить так, чтобы фильтрат собирался непосредственно в маленький стакан.
5 В отсутствие натрия литий полностью отделяется от калия однократной экстракцией.
736
Гл. XLII. Щелочные металлы
растворяют в минимальном количестве воды, собирая раствор в стакан, в котором находится основная масса остатка, и снова выпаривают досуха. По охлаждении вводят 10 мл растворителя, нагревают и охлаждают, как прежде. Фильтруют через тот же тигель, который, естественно, должен быть высушен. Остаток взмучивают в спирто-ацетатной смеси и тонкой струей переносят в тигель и затем из промывалки промывают 10—15 раз той же смесью, порциями по 0,1—1 мл. Объединяют фильтраты и промывные воды, общий объем которых должен составлять приблизительно 55 мл, и сохраняют. Стакан, в котором проводилось отделение, высушивают и счищают в тигель приставшие к стенкам частички перхлората. Тигель с остатком сушат сначала в сушильном шкафу при 110° С, а затем нагревают 15 мин в муфеле при 350° С. Образуется КС1О4, который взвешивают после охлаждения
Полное обезвоживание КС1О4, перекристаллизованного из воды, происходит при 350° С. Содержание влаги в соли после высушивания при 150Q С может достигать 0,3%. Растворимость перхлората калия в смеси равных частей и-бутилового спирта и этилацетата при 25° С равна приблизительно 1 мг в 100 мл, что соответствует 0,6 мг КС1 или 0,34 мг окиси кадия. Если общий объем раствора не превышает 60 мл, то нет необходимости вводить поправку, так как потери возмещаются небольшими количествами перхлоратов натрия и лития, которые удерживаются солью калия.
Растворимость перхлоратов рубидия и цезия приблизительно вдвое меньше растворимости перхлората калия.
Объединенные фильтраты и промывные воды выпаривают на горячей плитке до удаления этилацетата, что происходит после сгущения раствора по крайней мере до 40% его первоначального объема. Продолжают выпаривание, пока объем раствора не уменьшится до 20 мл. Нагревают до 80—90° С и по каплям при перемешивании прибавляют 2 мл бутилового спирта, содержащего 20% хлористого водорода 1 2, а затем вводят еще 6 мл этого раствора. Охлаждают до комнатной температуры, переносят осадок в тигель Гуча или Мунро и промывают 8—10 раз порциями по 1—2 мл 6—7%-ного раствора [сГ‘ = 0,8425 — 0,8485] хлористого водорода в «-бутиловом спирДе (получают прибавлением 100 мл бутилового спирта к 40 мл 20%-ного раствора хлористого водорода). Фильтрат и промывные воды сохраняют для определения лития. Тигель с осадком сушат несколько минут при 110° С, а затем прокаливают 5 мин при 600° С в муфеле или до темно-красного каления на голом пламени.
Для определения натрия весовым путем тигель с загрязненным хлоридом натрия охлаждают в эксикаторе и взвешивают. Затем осадок в тигле обрабатывают тонкой струей воды до растворения хлорида натрия, тщательно промывают тигель, собирая раствор и промывные воды в стакан емкостью 150 мл. Тигель с нерастворимым остатком сушат 1 ч при 110° С, затем охлаждают и снова взвешивают. Потеря в массе соответствует содержанию хлорида натрия 3. Полученный таким образом результат можно проверить по методу Мора, как описано для титрования раство
1 Согласно G. F. Smith и J. F. Ross, цит. выше.
2 Приготовляют пропусканием тока сухого хлористого водорода в бутиловый спирт; 200 мл 20%-ного раствора (пл. 0,905 г! см3} можно приготовить в течение 2—3 ч.
3 Растворимость хлорида натрия в растворе хлористого водорода в и-бутиловом спирте при комнатной температуре приблизительно равна 1,4 мг в 100 мл, что соответствует 0,75 мг окиси натрия. Поэтому общий объем фильтрата и промывных вод не должен превышать 50 мл.
Методы определения
737
римых хлоридов (стр. 814), или же, не взвешивая осадка, можно непосредственно определить этим методом содержание натрия.
Фильтрат и промывные воды, сохраненные для определения лития, разбавляют водой на одну треть их объема, причем образуются два слоя. Накрывают стакан часовым стеклом, надев предварительно на стенки стакана согнутые под углом стеклянные палочки, и выпаривают на водяной бане, стараясь избежать при этом конденсации жидкости в верхней части стакана \ Под конец прибавляют 5—10 мл воды, чтобы полнее удалить органические вещества прежде чем начнут выделяться пары хлорной кислоты.
Когда появляется слабое коричневое окрашивание, удаляют стеклянные палочки, помещают стекло непосредственно на стакан и нагревают на проволочной сетке до появления паров хлорной кислоты. Если на стенках стакана остается коричневый осадок, его удаляют нагреванием стенок пламенем. Если прибавлено недостаточное количество хлорной кислоты для окисления органических веществ, прибавляют еще несколько капель. По разрушении коричневой окраски прибавляют 0,5 мл серной кислоты, удаляют часовое стекло и вьпщривают на горячей плитке или на сетке над маленьким пламенем досуха. Остаток охлаждают, прибавляют 5—10 мл воды, ополаскивают стекло и стенки стакана и затем переносят сульфат лития в платиновый тигель, прокаленный и взвешенный вместе с крышкой. Осторожно выпаривают досуха, накрывают тигель крышкрй и нагревают, лучше всего на кольцевой горелке, до удаления следов кислоты, а затем в муфеле при 600° С в течение 5—10 мин. Если под конец прокаливание проводится на голом пламени, может происходить восстановление некоторого количества сульфата до сульфида. По охлаждении остаток взвешивают в виде Li2SO4.
Имеются указания 1 2, что удовлетворительное определение лития (и других щелочных металлов) достигается при: 1) получение смеси хлоридов по комбинированному способу Лоуренса Смита — Берцелиуса; 2) превращении хлоридов в перхлораты; 3) осаждении калия, рубидия, цезия и натрия обработкой «-бутиловым спиртом, хлорной кислотой и хлористым водородом и 4) превращении лития, находящегося в фильтрате, в сульфат.
Определение лития
Методы, обычно применяемые для отделения лития от других щелочных металлов, основаны на большей растворимости хлорида лития по сравнению с хлоридами остальных металлов этой группы. Эти методы можно разделить на две группы, в зависимости от того, лежит ли в основе их извлечение хлорида лития или осаждение хлоридов других щелочных металлов.
В методе с «-бутиловым спиртом й этилацетатом, описанном выше (стр. 734), соединены оба эти принципа, причем сперва натрий и литий извлекают из смеси хлоридов щелочных металлов, ^ а затем натрий
1 Это можно осуществить, погрузив стакан в медный сосуд с закраинами вверху, установленный на кольцах водяной бани так, что ни на стенках стакана, ни на часовом стекле не происходит конденсации.
2 S. Kallmann, Ind. Eng. Chem.T Anal. Ed., 16, 712 (1944).
47 Заказ 522.
738 Гл- XLII. щелочные металлы
осаждают из спирто-ацетатной вытяжкй. Эфирно-спиртовый метод \ изложенный в разделе «Определение, со смесью спирта и эфира», является методом осаждения, тогда как ацетоновый метод (стр. 739) — методом экстракции. Амило-спиртовый 1 2 и пиридиновый 3 4 методы мало удобны и недостаточно надежны, чтобы их рекомендовать, поэтому описание их здесь опускается.
Количественное определение следов лития лучше всего осуществлять спектроскопическим методом А
Определение со смесью спирта и эфира. Первоначально эфирно-спиртовый метод Раммельберга отделения лития от натрия и калия являлся методом выщелачивания и в связи с этим обладал известными недостатками. Позднее® этот метод был превращен в метод осаждения, что значительно его усовершенствовало.
Сущность метода заключается в следующем. Хлориды растворяют в минимальном количестве воды (на 0,5 г соли .достаточно 1,5 мл воды) в высоком стакане, прибавляют 1 каплю концентрированной соляной кислоты и затем постепенно вводят 20 мл абсолютного спирта, вращая стакан и капая жидкость в центр стакана, а не на его стенки. Хлориды натрия и калия выпадают в виде однородного зернистого 'осадка. Вращая стакан, прибавляют 60 мл эфира (пл. 0,716—0,717 г/см3) и оставляют стоять 5 мин или пока осадок не осядет и жидкость не станет почти прозрачной. Стакан вращают время от времени. Фильтруют через тигель с пористым дном под стеклянным колоколом, собирая фильтрат в коническую колбу. Ополаскивают стакан и промывают осадок, а затем воронку, смесью 1 части спирта и 4—5 частей эфира. Снимают вещество, приставшее к стенкам стакана, стеклянной палочкой с резиновым наконечником. Тигель отставляют. Фильтрат выпаривают на паровой бане, пропуская ток воздуха. Остаток обрабатывают 10 мл абсолютного спирта и нагревают, если это необходимо, для полного растворения. Оставшуюся на стенках колбы пленку снимают палочкой с резиновым наконечником. Вращая колбу, прибавляют 50 мл эфира, а затем 1 каплю концентрированной соляной кислоты.-Перемешивают и оставляют стоять в течение 30 мин, продолжая изредка вращать колбу. Когда небольшой осадок осядет, фильтруют через тигель, в котором находится основная масса хлоридов натрия и калия, и промывают смесью спирта и эфира. Предварительно высушенный осадок осторожно прокаливают и взвешивают в виде хлоридов натрия и калия (а также рубидия и цезия, если они присут-
1 S. Palkin, J. Am. Chem., Soc., 38, 2326 (1916); C. R a m mels b erg, Pogg..Ann., 66, 79 (1845).
2 Gooch, Proc. Am. Acad. Arts. Sci., 22, 177 (1886); Am. Chem. J., 9, 33 (1887); Chem. News, 55, 18 (1887).
3L. Kahlenberg, F. C. Krauskopf, J. Am. Chem. Soc., 30, 1104 (1908).
4 См. определение лития no W. W. Skinner, W. D. Collins, U. S. Dept. Agr. Bur. Chem. Bull., 153. Турбидиметрический метод открытия и определения малых количеств лития, основанный на образовании нерастворимого стеарата лития в амилово-спиртовом экстракте, см. Е. R. С а 1 е у, J. Am. Chem. Soc., 52, 2754 (1930). L. В. Rogers и Е. R. Caley [Ind. Eng. Chem., Anal. Ed., 15, 209 (1943)] рекомендуют эмпирический метод, в котором литий осаждается в виде комплексного перйодата в щелочном растворе. Хлориды, нитраты, сульфаты и перхлораты не влияют на определение. Элементы, не относящиеся к группе щелочных металлов, а также небольшие количества аммонийных солей должны быть удалены. См. также F. R. Bacon, D. A. Starks, Ind. Eng. Chem., Anal. Ed., 17, 230 (1945).
Методы определения
739
ствуют), а затем проводят разделение хлороплатинатный методом (стр. 731). - ' . ,
Фильтрат осторожно выпаривают на паровой бане, остаток растворяют в небольшом количестве воды и прибавляют серную кислоту в небольшом избытке. Переносят в маленькую взвешенную платиновую чашку, жидкость выпаривают, удаляют избыток кислоты, прокаливают, как указано в разделе «Определение натрия с «-бутиловым спиртом и зтилаце-татом» (стр. 734), и взвешивают в виде Li2SO4. Обработку повторяют до постоянной массы.1
Определение с применением ацетона. Установлено 2, что хлорид лития легко растворим в ацетоне и может быть таким образом отделен от практически нерастворимых в ацетоне хлоридов натрия, калия, бария и стронция. Хлориды рубидия, цезия и магния несколько растворимы в ацетоне, а растворимость хлорида кальция настолько значительна, что он должен быть предварительно удален.
Метод сводится к следующему. Осторожно выпаривают в небольшом стакане раствор хлоридов щелочных металлов и затем нагревают до> удаления аммонийных солей. Растирают крисаллы в мелкий порошок закругленной палочкой или маленьким пестиком. Вводят около 25 мл сухого ацетона, 1 каплю концентрированной соляной кислоты и тщательно перемешивают. Дают отстояться в течение нескольких секунд, а затем декантируют прозрачную жидкость через маленький фильтр во взвешенную платйновую чашку. Промывают остаток в стакане и на фильтре ацетоном 2—3 раза порциями по 5 мл. Остаток растворяют в небольшом количестве воды и снова выпаривают. Затем растирают остаток в порошок и экстрагируют 25 мл ацетона и одной каплей кислоты, как ранее. Если масса смеси хлоридов превышает 0,5 г, то эту операцию повторяют в третий раз.
Объединенные фильтраты выпаривают на плитке или песчаной бане при невысокой температуре (< 50° С); полученный остаток осторожно нагревают для удаления органических веществ. По охлаждении смачивают хлориды 1—2 мл концентрированной серной кислоты. Необходимо следить за полнотой смачивания. Затем кислоту удаляют осторожным нагреванием; прокаливают, как указано выше (стр. 734), и взвешивают в виде Li2SO4.
При определении лития в огнеупорной глине (0,22% Li2O) были получены 3 прекрасные результаты, когда 1—3 г материала разлагали нагреванием с соляной кислотой в запаянной трубке (стр. 403), поело чего проводили следующие операции:
1) разбавление и фильтрование для удаления кремнекислоты;
2) упаривание до небольшого объема и обработку раствора, охлажденного льдом, газообразным хлористым водородом и эфиром с последующим фильтрованием для удаления основной массы алюминия;
3) двукратное осаждение аммиаком для отделения оставшегося в растворе алюминия, совместно' с другими осаждающимися при этом элементами;
1 Нет основания рекомендовать взвешивание лития в виде фторида или фосфата. Хлорид лития гигроскопичен.
2 М. Н. Brown, J. Н. Reedy, Ind. Eng. Chem., Anal. Ed., 2, 304 (1930).
3 J. I. Hoffman, неопубликованная работа (1940).
47*
740
Гл. XLII. Щелочные металлы
4) обработку оксалатом аммония, а затем оксихинолином для отделения кальция и магния (стр. 725);
5) выпаривание, прокаливание до хлорида и двукратное экстрагирование ацетоном для отделения лития;
6) обработку серной кислотой и прокаливание до Li2SO4.
Необходимо упомянуть также следующие методы экстракции: 1) хлорид лития экстрагируют чистым безводным диоксаном О = (СН2)4=О из прокаленной смеси хлоридов щелочных металлов. Экстрагирование ведут в аппарате, в котором соприкосновение осадка с влагой исключается, так как конденсирующийся диоксан непрерывно падает в подвешенный фильтрующий тигель, содержащий смесь хлоридов1; 2) хлорид лития экстрагируют из смеси хлоридов 2-этилгексанолом. Авторы утверждают, что никакой поправки на растворимость не требуется и что пары горячего раствора менее неприятны, чем пары изоамилового спирта2.
Определение рубидия и цезия
Минералы, содержащие одновременно определимые количества рубидия и цезия, почти неизвестны, что является весьма благоприятным фактом, так как неизвестны специфические реагенты, посредством которых можно было бы осуществить разделение этих металлов или отделение их от калия. Различная растворимость некоторых солей рубидия и цезия используется для их очистки, но основанные на этом аналитические методы ни в коей мере нельзя считать количественными. Рубидий и цезий образуют нерастворимые хлороплатинаты и перхлораты; каждый из этих элементов, когда он присутствует один или совместно с литием или натрием, можно определять методами, описанными для калия. Когда же они встречаются совместно или в сочетании с калием, необходимы дальнейшие отделения.
Первый метод. Следующий метод количественного разделения и определения калия, рубидия и цезия основан на последовательном применении 9-фосфорномолибденовой, кремневольфрамовой и платинохлористоводородной кислот. Осаждение 9-фосфорномолибденовой кислотой используется для отделения рубидия и цезия от калия, а кремневольфрамовой кислотой — для отделения цезия от рубидия, тогда как платинохлористоводородная кислота служит для конечного осаждения всех трех элементов 3. Этот метод не дает такого четкого разделения, как может быть желательно, но, как указывается, он более надежен, чем предложенные ранее.
Ход определения.. Навеску анализируемого материала, содержащую не более 0,08 г RbCl и 1,0 a КС1, растворяют в 100 мл разбавленной (1 : 3) азотной кислоты, раствор нагревают почти до кипения и обрабатывают 9-фосфорномолибденовой кислотой 4 до полного осаждения. Дают от
1 A. S i n k a, Z. anal. Chem., 80, 430 (1940). *
2 Е. R. С а 1 е у, Н. D. А х i 1 г о d, Ind. Eng. Chem., Anal. Ed., 14, 243 (1942).
3 W. J. O’L e a r y, J. P a p i s h, Ind. Erig. Chem., Anal. Ed., 6, 107 (1934).
4 Приготовляют следующим образом. Осторожно нагревают продажную, содержащую 12 МоО3 кислоту при непрерывном перемешивании, пока температура не достигнет 300—350° С и оранжевая окраска сухой кислоты не перейдет в зеленую. Продолжают нагревание, чтобы не осталось оранжевых частичек, затем охлаждают и выщелачивают водой. Фильтруют, еслй имеется муть, и к полученному зеленому раствору прибавляют небольшое количество бромной воды. Медленно выпаривают для получения коротких, толстых желтых призм лютеокислоты.
Методы определения
741
стояться, затем фильтруют и промывают осадок 1%-ным раствором нитрата натрия.
В фильтрате и промывных водах содержится весь калий, который можно определить осуждением в виде хлороплатината (стр. 731).
Осадок фосфоромолибдатов рубидия и цезия растворяют в возможно меньшем количестве 1 н. раствора NaOH. Раствор насыщают сероводородом, нагревают до кипения и добавляют разбавленную азотную кислоту до слабокислой реакции. Кипятят для коагуляции осадка, фильтруют, промывают осадок подкисленной сероводородной водой и отбрасывают. Если есть основание предполагать, что в растворе еще осталось немного молибдена, кипятят с небольшим количеством бромной воды, охлаждают, снова обрабатывают сероводородом, кипятят, фильтруют, промывают осадок и присоединяют фильтрат к первоначально полученному фильтрату.
Упаривают фильтрат (или объединенные фильтраты) приблизительно до 20 мл, вводят 60 мл 95%-ного спирта, обрабатывают платинохлористоводородной кислотой, прибавляемой в небольшом избытке, а затем приливают'2—3 мл этилового спирта. Осадку дают отстояться, после чего фильтруют через тигель Мунро (стр, 128) и промывают 80%-ным спиртом. Под тигель подставляют другой приемник и обрабатывают осадок 5 мл воды и 2—3 каплями гидразингидрата. Как только прекратится бурное выделение газа, включают вакуум, отсасывают и смывают хлориды щелочных металлов в приемник. Осторожно прибавляют царскую водку и кипятят для разрушения избытка гидразина.
Вводят соляную кислоту и выпаривают до небольшого объема. Эту операцию повторяют 2—3 раза, чтобы обеспечить полное удаление азотной кислоты. После этого разбавляют 50—75 мл 6 н. соляной кислоты. По охлаждении вводят 0,5—1,0 г кремневольфрамовой кислоты, растворенной в нескольких миллилитрах воды, 'и оставляют стоять в течение 12 ч. Фильтруют через тигель Мунро, промывают осадок 6 н. соляной кислотой и сохраняют его для определения цезия. К фильтрату, в котором должен находиться весь рубидий, прибавляют немного азотной кислоты и 5 мл платинохлористоводородной кислоты, а затем упаривают приблизительно до 10 мл. Вводят*трехкратный объем спирта, затем избыточное количество платинохлористоводородной кислоты и, наконец, 2—3 мл диэтилового зфира. Выделившийся осадок отфильтровывают, промывают 80%-ным спиртом, осторожно сушат при 120—130° С и взвешивают в виде Rb2[PtCle].
Сохраненный ранее осадок растворяют в возможно меньшем количестве едкого натра, полученный раствор слабо подкисляют азотной кислотой и разбавляют до 200 мл. Охлаждают и вводят 10%-ный раствор нитрата ртути (I) до тех поф, пока не закончится осаждение и осадок кремневольфрамата ртути (I) не станет хлопьевидным и не начнет быстро оседать. Фильтруют, промывают осадок 1 %-ным раствором нитрата ртути (I) и отбрасывают. Находящийся в фильтрате избыток ртути (I) окисляют кипячением с царской водкой. Затем раствор упаривают до 10 мл и обрабатывают трехкратным (по объему) количеством спирта, избыточным количеством платинохлористоводородной кислоты и эфиром, как при определении рубидия. Фильтруют, промывают осадок 80%-ным спиртом, осторожно сушат при 120—130° С и взвешивают в виде Cs2[PtCl6].
742 Гл. XLII. Щелочные металлы
Второй метод х. После безуспешных попыток применить существующие методы для определения редких щелочных металлов в горных породах и минералах был разработан следующий метод а. Исходным продуктом является смесь хлоридов, полученная в результате предшествующей обработки по методу Лоуренса Смита (стр. 1006). Методы разделения основаны главным образом на применении платинохлористоводородной кислоты, абсолютного спирта, эфира и сульфата аммония. Каждый щелочной металл взвешивают раздельно, и таким образом начальная масса смеси хлоридов служит только для контроля.
Разделение групп натрия, и калия. Это разделение осуществляется обработкой платинохлористоводородной кислотой, как описано на стр. 731. После взвешивания смеси хлороплатинатов калия, рубидия и цезия их превращают в хлориды. Для этого осадок растворяют в воде и осаждают платину горячей разбавленной муравьиной кислотой. Осадок удаляют фильтрованием. Этот раствор (1) сохраняют для обработки, как указано ниже (см. «Определение натрия» и «Отделение рубидия от цезия и калия»). Раствор, содержащий хлороплатинаты натрия и лития, также обрабатывают муравьиной кислотой и фильтруют для удаления платины. Полученный раствор (2) сохраняют для определения лития (см. ниже).
Определение лития. Отделение лития от натрия осуществляется по видоизмененному эфирно-спиртовому методу (стр. 738) следующим образом. Сохраненный раствор (2) хлоридов натрия и лития выпаривают досуха, лучше в маленькой, емкостью 30 мл конической колбе, снабженной стеклянной пробкой. Остаток растворяют в 0,4 мл воды, слегка нагревая, если требуется. По охлаждении прибавляют 0,01 мл концентрированной соляной кислоты, 5 мл абсолютного спирта и, вращая колбу, вводят 15 мл эфира. Оставляют стоять 15 мин и затем фильтруют через взвешенный стеклянный тигель. Осадок хлорида натрия тщательно промывают смесью, состоящей из 1 части спирта и 4 или 5 частей эфира. Тигель с осадком сохраняют (осадок 3).
Вторичной обработки фильтрата, содержащего литий, для дополнительного извлечения из него натрия обычно не требуется. Фильтрат выпаривают с серной кислотой, взятой в небольшом избытке, во взвешенной чашке и полученный сульфат Лития прокаливают до постоянной массы.
Определение натрия. Для непосредственного определения натрия • взвешивают сохраненный тигель с хлоридом натрия (осадок 3), полученным, как описано выше.
Отделение рубидия и цезия от калия. Сохраненный раствор (1), содержащий хлориды калия, рубидия и цезия, выпаривают досуха в конической колбе емкостью 30 мл, снабженной стеклянной пробкой, и затем нагревают при 106° С. По охлаждении прибавляют 0,4 мл воды, нагревают, снова охлаждают и затем насыщают газообразным хлористым водородом. Вводят 10 мл абсолютного спирта, также насыщенного газообразным хлористым водородом. Фильтруют под вакуумом через асбест, вложенный в маленькую воронку, или через стеклянный тигель. Промывают 2 мл смеси абсолютного спирта и эфира. Полученный осадок сохраняют (осадок 4) для извлечения оставшейся в нем части рубидия (см. «Оп-
1 Этим методом определяются также натрий и литий.
2 R. С. Wells, R. Е. Stevens, Ind. Eng. Chem., Anal. Ed., 6, 439 (1934).
Методы, определения
743
ределение рубидия», стр. 744). Фильтрат выпаривают досуха и затем осторожно прокаливают, под конец — только мгновение при 500° С. Охлаждают и взвешивают в виде хлоридов рубидия и цезия (осадок 5). Кроме того, в осадке содержится по 0,6 мг хлорида калия на каждую проведенную экстракцию спиртом с соляной кислотой при 25° С. Предполагается, естественно, что содержание хлорида калия в анализируемом продукте достаточно велико, чтобы обеспечить такую поправку.
Когда масса хлоридов не превышает 0,6 мг х, то рубидий и цезий или отсутствуют, или находятся лишь в спектральных следах. В случае незначительного содержания цезий почти полностью переходит в первый экстракт. При содержании же заметных количеств цезия необходима вто-.ричная экстракция. При этом значительная часть рубидия все еще может остаться в осадке калия (осадок 4).
Отмечают1 2, что при наличии в анализируемой пробе больших количеств рубидия и цезия, как, например, в поллуците, содержащем 30% окиси цезия, может потребоваться до шести экстракций. В таких случаях можно воспользоваться одним из следующих трех сцособов.
1) Взять небольшую аликвотную часть, если содержание калия в материале достаточно велико, чтобы было обеспечено извлечение по 0,6 мг КС1 в каждой экстракции.
2) При низком содержании калия заменить указанную выше экстракционную смесь 10 мл другой смеси, приготовленной из 1 части концентрированной соляной кислоты и 2 частей спирта; такая смесь растворяет 3,1 мг КС1 вместо 0,6 мг при 25° С.
3) Если содержание калия очень мало, совсем опустить экстракцию хлорида цезия из смеси хлоридов (раствор 1) и обработать ее, как «хлорид цезия».
Указывают также, что для отделения основной массы наименее растворимого соединения цезия смешанные хлороплатинаты можно подвергнуть нескольким фракционным кристаллизациям с последующим отделением цезия от рубидия и калия в фильтратах, содержащих их хлороплатинаты, по предложенному авторами3 методу.
Определение цезия. К сухим хлоридам щелочных металлов (осадок 5), полученным, как указано выше, прибавляют 0,1 мл 5%-ного раствора сульфата аммония и перемешивают до растворения хлоридов. Энергично перемешивая, вводят 5 мл спиртового раствора сульфата аммония4 5, первый миллилитр приблизительно по 1 капле в секунду, а затем с большей скоростью. Оставляют стоять на 30 мин, временами перемешивая. Фильтруют при слабом отсасывании через слой асбеста, собирая фильтрат, содержащий цезий, в маленький взвешенный платиновый тигель или чашку. Тщательно промывают осадок, стакан и фильтр тремя порциями по 0,5. мл промывной жидкости ®. Сохраняют осадок (осадок 6) для определения рубидия (см. ниже).
В тигель, содержащий фильтрат и промывные воды, вводят небольшое количество сульфата аммония, выпаривают на водяной бане до начала кристаллизации солей, а затем прибавляют несколько капель спирта. Продолжают выпаривание до удаления всей жидкости, прибавляют несколько капель абсолютного спирта и снова выпаривают. Закрывают
1 Количество хлорида калия, переходящее в раствор при однократной экстракции.
2 J. С. Hillyer, Ind. Eng. Chem., Anal. Ed., 9, 236 (1937).
3 R. C. Wells, R. E. Stevens, Ind. Eng. Chem., Anal. Ed., 9, 236 (1937).
4 Для приготовления растворяют 1 г сульфата аммония в 20 мл воды, перемешивают и медленно добавляют 100 мл 95%-ного спирта. Фильтруют и вводят в фильтрат несколько кристаллов сульфата аммония, чтобы раствор был насыщен солью.
5 Промывную жидкость приготовляют так же, как спиртовый раствор сульфата аммония, только в водный раствор соли, перед добавлением спирта, вводят 0,16 г хлорида аммония.
Цк Гл. XLII. Щелочные металлы
тигель стеклом и слабо нагревают в радиаторе до тех пор, пока хлорид аммония не сублимируется на стекле. Снимают стекло и осторожно, чтобы избежать потерь вследствие разбрызгивания, повышают температуру до начала расправления массы и возгонки сульфата аммония. По удалении сульфата аммония сразу же, вращая тигель, накаляют его докрасна. Охлаждают в эксикаторе, закрыв тигель стеклом, чтобы избежать растрескивания осадка. Взвешивают в виде Cs2SOi.
Повторяют прокаливание до получения постоянной массы. Содержание таких малых количеств цезия, как 0,1 мг, может быть подтверждено спектроскопически, для чего сульфат растворяют в 2—3 каплях воды, погружают в раствор платиновую спираль и накаливают в пламени горелки Бунзена.
Определение рубидия. Если масса цезия, пересчитанная на хлорид, плюс 0,6 мг хлорида калия на каждую проведенную экстракцию спиртом с соляной кислотой, равна массе хлоридов (осадок 5), полученных, как указано в разделе «Отделение рубидия от цезия и калия» (стр. 742), то рубидий отсутствует. Если же масса хлоридов (осадок 5) больше этой величины, то разность соответствует содержанию RbCl (масса 7) в хлоридах.
Вычисленная таким образом масса хлорида рубидия может быть проверена следующим образом. Растворяют сохраненный осадок (6), полученный, как указано в разделе «Определение цезия» (см. выше), раствор выпаривают досуха в другом взвешенном тигле, прокаливают и взвешивают для получения массы сульфатов рубидия и калия. Количество КС1, которое, как предполагается, должно находиться в экстрактах, пересчитывают на K2SO4 и вычитают из веса смеси сульфатов, а полученную разность пересчитывают на RbCl (масса 8).
Количество рубидия, полученное в результате любого из этих вычислений, не соответствует общему его содержанию, так как часть рубидия остается в сохраненном осадке 4. Для извлечения рубидия повторяют экстракцию этого осадка спиртом и соляной кислотой до тех пор, пока потеря в массе смешанных хлоридов после Каждой экстракции превышает 0,6 мг. Экстракты объединяют и выпаривают досуха. Остаток растворяют в небольшом количестве воды, прибавляют платинохлористоводородную кислоту в незначительном избытке и выпаривают почти досуха. Вводят 5 мл 15%-ного спирта, тщательно перемешивают и фильтруют. Полученный осадок (осадок 9) промывают 95%-ным спиртом, сушат при 130° С, взвешивают1 в виде Rb2[PtCle] и пересчитывают на RbCl.
Содержание RbCl соответствует сумме весов масс 9 и 7 или 8.
Определение калия. В случае умеренного содержания калия для вычисления его массы вычитают из первоначальной массы смеси хлороплатинатов массу хлороплатинатов цезия и рубидия. Если же процентное содержание калия мало, лучше взвесить его в виде хлорида или сульфата после отделения рубидия и цезия.
Определение одного калия
1 Восстановление хлороплатината калия. Вместо взвешивания хлороплатината калия давно уже применяется другой метод, который заклю
1 Некоторые количества цезия, не извлекшиеся при первоначальной экстракции, естественно, при этом взвешиваются в виде Cs2[PtCl6].
Методы определения
чается в восстановлении осадка и взвешивании платины. Таким способом можно определять калий в присутствии сульфатов, хлоридов, фосфатов, нитратов, боратов, растворенной кремнекислоты, солей натрия, щелочноземельных металлов, магния, железа и алюминия. Вероятно, единственными элементами, мешающими определению, помимо аммония, являются рубидий и цезий, а также некоторые органические вещества. Этот метод особенно удобен для анализа солей калия, рассолов и смешанных удобрений, когда интересуются только содержанием в них калия. Для выделения платины использовались различные восстановители, из которых наилучшим и наиболее простым, по-видимому, является металлический магний (в виде ленты).
Метод \ применяемой для исследования минеральных рассолов и буровых вод в U. S. Geological Survey, заключается в следующем.
Осаждают калий в виде хлороплатината обычным способом с‘ той лишь разницей, что перед добавлением платинового реактива вводят 5 мл соляной кислоты. Реактив прибавляют лишь в незначительном избытке против необходимого для образования хлороплатината калия, так как присутствие натрия и других солей не влияет на осаждение. Осадок измельчают, тщательно промывают 80—90%-ным (по объему) спиртом и затем растворяют в горячей воде. К раствору прибавляют 1 мл соляной кислоты и приблизительно по 0,5 г магниевой ленты (предварительна промытой водой) на каждые 0,2 г калия. Размешивают раствор и удерживают ленту на дне стакана стеклянной палочкой. Когда растворится почти весь магний, прибавляют несколько миллилитров разбавленной соляной кислоты и дают осадку платины осесть на дно. По окончании восстановления раствор становится прозрачным и бесцветным. Чтобы удостовериться в полноте восстановления платины, прибавляют еще немного магния и наблюдают, не потемнеет ли раствор. Прибавляют соляной кислоты, кипятят для растворения основных солей и фильтруют. Осадок платины промывают водой до отрицательной реакции на хлорид-ионы в промывной воде, прокаливают и взвешивают.
Содержание калия вычисляют на основе соотношения IPt : 2К.
Перхлоратный метод с бутиловым спиртом. Первоначально предложенный перхлоратный метод был впоследствии несколько видоизменен 1 2. Для выделения перхлората калия из раствора применен «-бутиловый спирт; при этом удается избежать недостатков, которыми обладают почти все методы экстракции. Поправку на растворимость перхлората кайия (0,38 мг К2О на 100 мл раствора) нужно вводить только в случае* отсутствия натрия или если содержание его крайне незначительно. Судя по опытам автора, хорошие результаты получаются при однократном осаждении, без введения поправки на растворимость перхлората калия, что обусловлено взаимной компенсацией положительных и отрицательных ошибок. При применении метода к смесям, состоящим из 0,14 г хлорида калия и 0,16 г хлорида натрия, результаты для.первого получились пониженные на 0,2 мг, а для последнего — повышенные на 0,6 мг.
Ход определения. Смесь хлоридов (свободную от* сульфатов) выпаривают с избыточным количеством хлорной кислоты досуха (при температуре не выше 350° С). Если количество хлоридов велико, эту операцию
1 W. В. Hicks, J. Ind. Eng. Chem., 5, 650 (1913); R. C. W e 1 1 s,. R. K. Bailey, J. G. Fairchild, там же, 16, 935 (1924).
2 G. F. S m i t h,. J. Am. Chem., Soc., 45, 2072 (1923).
746
Гл. XLII. Щелочные металлы
повторяют. В соответствии с количеством -калия прибавляют 2—3 мл воды и, осторожно покачивая сосуд над голым пламенем, растворяют соли. К горячему раствору перхлората, в зависимости от того, сколько было введено воды, 2 или 3 мл, медленно, энергично перемешивая, прибавляют 65 или 100 мл кипящего н-бутилового спирта х, содержащего 0,5—1% хлорной кислоты (70 %-ной). После прибавления реактива накрывают сосуд часовым стеклом, осторожно кипятят полминуты и охлаждают. Осадок переносят в тигель Мунро или Гуча 1 2 раствором осадителя и промывают тем же раствором 8—10 раз порциями по 1—2 мл. Фильтрат и промывные воды должны иметь общий объем около 125—150 мл. Сушат осадок при 350° С и взвешивают 3 в виде КС1О4.
Натрий можно определить в фильтрате, но лучше содержание его вычислить по разности.
Перхлоратный метод с этиловым спиртом. Приводимый ниже метод менее приемлем, чем метод, изложенный в разделе «Перхлоратный метод с бутиловым спиртом» (см. выше), так как он основан на процессе экстракции, а не осаждения и, кроме того, для его осуществления требуется абсолютный спирт. Анализируемый раствор должен быть свободен от сульфатов, и в нем может находиться не более 1 г смеси хлоридов. Желательно отсутствие аммонийных солей, но следы их не мешают определению. Для промывания применяются два раствора. Один из них представляет собой абсолютный спирт, содержащий 0,2% хлорной кислоты. Другой раствор приготовляют незадолго перед употреблением взбалтыванием первого раствора с кристалликами перхлората калия в течение 5—10 мин при комнатной температуре. Осадку дают осесть, после чего сливают прозрачную жидкость.
Ход определения. К концентрированному раствору хлоридов прибавляют 1—2 мл 60%-ной хлорной кислоты (свободной от серной кислоты) и выпаривают досуха на горячей плитке или на голом пламени. Вводят 10 лм. воды, снова добавляют 1—2 мл хлорной кислоты и выпаривают досуха при температуре не выше 350° С. В результате этой операции должно произойти полное превращение солей в перхлораты. Остаток обрабатывают 20 мл абсолютного спирта и оставляют стоять в течение нескольких минут. Прозрачный раствор фильтруют через тигель Гуча или, лучше, через тигель Мунро и промывают тигель с остатком 1 —2 раза первой промывной жидкостью. Отсосав досуха, растворяют остаток в воде, прибавляют 1—2 мл хлорной кислоты, выпаривают досуха и повторяют выщелачивание абсолютным спиртом. Фильтруют, переносят остаток во взвешенный тигель Гуча или Мунро, пользуясь второй промывной жидкостью; для переноса употребляют не более 100 мл, и отсасывают досуха. Сушат сначала при 120—130° С, затем прокаливают в течение нескольких минут при 350° С и взвешивают 4 в виде КСЮ4.
1 н-Бутиловый спирт ((d25 = 0,8065) с темп. кип. в пределах 112—118° С следует перегнать, причем первые и последние 5% отгона отбросить.
2 Диаметр отверстий тигля Гуча должен быть не больше 0,5 мм. В тигле должен находиться толстый слой асбеста, закрытый фарфоровым кружком с отверстиями. Асбест следует перед употреблением обработать смесью бутилового спирта и хлорной кислоты.
3 Определение и отделение калия в виде перйодата см. Н. Н. Willard, A. J. Boyle, Ind. Eng. Chem., Anal. Ed., 13, 137 (1941).
4 О термическом разложении перхлоратов лития, натрия, калия, кальция, магния, железа и алюминия см. G. G. Marvin, L. В. W о о 1 a v е г, Ind. Eng. Chem., Anal. Ed., 17, 474 (1945)s
Методы определения
747
Метод Линде-Глэддинга Ч Этот метод находит применение главным образом в случае присутствия сульфатов, нитратов, фосфатов и магния. Он отличается тем, что осадок хлороплатинатов сначала промывают 80 %-ным спиртом, затем специальной промывной жидкостью, содержащей хлорид аммония, и, наконец, снова 80%-ным спиртом. Для приготовления промывной жидкости растворяют 100 г хлорида аммония в 500 мл воды. К раствору прибавляют 5—10 г измельченного в порошок хлороплатината калия, оставляют стоять 6—8 ч, время от времени взбалтывая, а затем дают смеси отстояться в продолжение ночи и фильтруют.
Натриево-кобальтинитритный метод. Метод вполне приемлем для определения калия в некоторых материалах при условии, что детали операций разработаны для этого рода материалов 1 2. Так, например: 1) при исследовании растительных продуктов был разработан метод 3, согласно которому калий осаждают кобальтинитритом натрия не из уксуснокислого, а из слабо азотнокислого раствора, а затем определение заканчивают взвешиванием K2Na[Co(NO2)e ] • Н2О или титрованием перманганатом и оксалатом; 2) рекомендован полупрямой метод 4, который сводится к осаждению калия в виде кобальтинитрита после разложения при-родных силикатов или силикатных продуктов обработкой фтористоводородной и хлорной кислотами и к последующему переведению кобальтинитрита в перхлорат; 3) для определения малых количеств калия предложен фотометрический метод s *, основанный на образовании зеленого комплексного соединения, которое кобальт, находящийся в осадке, образует с солянокислым холином и гексацианоферратом (П) калия; 4) описан 8 колориметрический метод определения 0,002—0,40 мг калия в питьевой воде, основанный на осаждении кобальтинитритом серебра и последу-щем колориметрическом определении содержания нитрита в осадке. По данным авторов метода, кобальтинитрит серебра является наиболее чувствительным реагентом на калий 7. '
Опубликован подробный обзор по применению кобальтинитритного метода 8.
Отделение малых количеств калия от больших количеств натрия
В некоторых случаях, когда малые количества калия сопровождаются большими количествами натрия, как, например, в минеральных водах и соляных рассолах, бывает необходимо перед определением калия сконцентрировать его в растворе, который содержал бы лишь немного
1 Linde-Gladding, Chem. News, 53, 202, 296 (1886).
2 Состав- осадка кобальтинитрита калия и натрия колеблется в зависимости от условий осаждения, главным образом от температуры. Для получения наилучших результатов рекомендуется проводить «холостой» опыт, анализируя аналогичный материал с известным содержанием калия в нем.
3 L. V. Wilcox, Ind. End. Chem., Anal. Ed.,.9, 136 (1937).
4 E. W. Koenig, J. Am. Ceram. Soc., 22, 164 (1939).
5 A. Eden, Analyst, 68, 167 (1943).
’ R. J. Robinson, G. L. Putnam. Ind. Eng. Chem., Anal. Ed., 8, 211 (1936).
7 См. также L. L. Burgess, О. К a m m, J. Am. Chem. Soc., 34, 652 (1912);-O. Lutz, Z. anal. Chem., 59, 145 (1920); A. M. Ismail, H. F. Harwood. Analyst, 62, 443 (1937).
8 P. J. Van Rysselberge, Ind. Eng. Chem., Anal. Ed., 3. 3 (1931).
748
Гл. XLII. Щелочные металлы.
натрия. Кальций, магний и др. в виде хлоридов или сульфатов могут присутствовать.
В таких случаях, концентрируют, насколько возможно, водный раствор в конической колбе, но, не давая выделиться солям, помещают колбу в ледяную воду и насыщают хлористым водородом. Конец трубки, подводящей газ, должен быть расширен и не слишком глубоко погружен в жидкость. На каждые 100 мл раствора прибавляют по 2 мл воды, перемешивают и, когда соль осядет, сливают жидкость через воронку с обыкновенным фильтром (вложенным в платиновый конус) или, лучше, через тигель Гуча с прокладкой из мелко раздробленной платины; тигель должен быть достаточно большим, чтобы вместить всю соль. Переносят соль на фильтр, пользуясь ледяной водой, насыщенной хлористым водородом и содержащей хлорид натрия в той же концентрации, что и анализируемый раствор. Несколько раз промывают соль и отсасывают досуха.
В фильтрате содержится весь калий, некоторые количества хлорида натрия и соли других металлов, а также серная и борная кислоты, если в анализируемом материале находились сульфаты и бораты. Раствор выпаривают в платиновой чашке досуха, удаляют серную кислоту прокаливанием, обрабатывают остаток небольшим количеством соляной кислоты и после этого продолжают определение, как описано в разделах «Восстановление хлороплатината калия» (стр. 744) или «Метод Линде-Глэддинга» (стр. 747).
Определение одного натрия
Согласно этому методу, натрий осаждают из слабокислого раствора в виде тройного ацетата NaZn(UO2)3(CH3COO)9 • 6Н2О, который можно непосредственно взвесить или растворить, и, определив содержание урана объемным способом, пересчитать на натрий. Этот метод неприменим для анализов высокой точностй, за исключением особых случаев. Он, однако, удобен для .рядовых определений натрия, так как реакция вполне специфична, и обычно требуется лишь очень мало предварительных отделений или же они вообще не нужны.
Осадок тройного ацетата довольно объемистый, что ограничивает количества натрия, которые можно осаждать. Он также значительно растворим в воде. Растворимость осадка в растворе, содержащем избыток осадителя, ниже, чем в воде, но все же достаточно заметна, поэтому объем, в котором проводится осаждение, должен быть минимальным, и при перенесении и промывании осадка необходимо соблюдать особую осторожность.
Из мешающих определению веществ следует отметить фрсфаты, молибдаты, арсенаты, сульфаты (в присутствии калия), оксалаты, тартраты, стронций, литий 1 и калий в количествах, превышающих 50 мг на 1 мл раствора.
1Е. R. Caley и W. О. Baker [Ind. Eng. Chem., Anal. Ed., 11, 604 (1949)] утверждают, что при применении для осаждения лития того же уранил-ацетатного реактива, какой применяется для осаждения натрия, выпадает тройной ацетат аналогичного состава. Если же при действии этого реактива осаждается калий из концентрированных растворов его солей, то выпадает осадок состава KUO2(C2H3O2)3.
В статье Е. В. Caley и L. В. Rogers [там же, 15, 32 (1943)] приведен метод определения натрия в присутствии лития, в котором в качестве реагента применяется спиртовый раствор ацетата меди и ацетата урана.
Методы определения
749
Фосфаты мешают определять натрий в слабокислых растворах, получаемых при добавлении реактива к водному раствору смеси хлоридов. Умеренные количества фос-фат-ионов, порядка 2,5 мг, по-видимому, не влияют при осаждении из растворов разбавленной (0,2 н.) хлорной кислоты К Для отделения фосфат-ионов при анализе таких материалов, как зола от молока, поступают следующим образом 1 2. 10—15 Мл слабокислого (солянокислого) раствора, содержащего 2—8 мг натрия, обрабатывают избыточным количеством измельченного в порошок карбоната цинка и оставляют стоять 6 ч или на ночь. Фильтруют и промывают осадок и фильтр 5—6 раз холодной водой. Фильтрат и промывные воды упаривают до 1—5 мл; если образуется осадок, прибавляют каплю уксусной кислоты. Следует ввести поправку на «холостой» опыт, который проводят через все стадии анализа.
Для отделения фосфатов и арсенатов применяют ряд других реагентов, как, например, гидроокись кальция, ацетат свинца, хлорид бария, или же применяют ацетат цинка для отделения фосфатов и магнезиальную смесь для отделения арсенатов.
Влияние молибдена, который образует осадок с реагентом, можно устранить введением лимонной или винной кислоты в количестве не большем, чем это требуется для образования устойчивого комплексного иона 3.
Умеренные количества аммонийных солей, кальций, барий и магний не мешают определению. Если осаждение проводят в кислой среде, то не мешают также и такие элементы, как железо й алюминий, соли которых легко гидролизуются.
Для осаждения натрия рекомендуется 4 * 6 следующая методика.
Ход определения. К 1 мл нейтрального или слабокислого раствора, содержащего не более 8 мг натрия в виде хлорида и свободного от перечисленных выше мешающих веществ, прибавляют 10 мл раствора цинк-уранил-ацетата в. Тщательно перемешивают и оставляют стоять не менее 30 мин. Механическое перемешивание ускоряет осаждение, особенно в случае малых количеств натрия. Фильтруют при отсасывании через' взвешенный фарфоровый или стеклянный тигель средней пористости. Переносят осадок в тигель и удаляют большую часть раствора промыванием реактивом; промывание ведут порциями по 2 мл. Под конец тщательно промывают осадок и стенки тигля промывной жидкостью ®, 5— 10 раз порциями по 2 мл и затем 1 раз зфиром. Просасывают через тигель воздух до улетучивания эфира. Вытирают наружные стенки тигля слегка влажной тканью, помещают тигель с осадком в эксикатор на 10—15лшн, после чего взвешивают. После введения поправки на содержание натрия
1 Е. W. Koenig, J. Am. Ceram. Soc., 22, 24 (1939).
2 О. R. Overton, О. F. Garrett, Ind. Eng. Chem., Anal. Ed., 9, 72 (1937).
s С. H. Hale, там же, 15, 516 (1943).
4 H. H. Barber, I. M. Kolthoff, Am. Chem. Soc., 50, 1625 (1928);
51, 3233 (1929).
6 Для приготовления реактива растворяют 100 г UO2(C2H3O2)2 • 2Н2О и 27,8 г Zn(C2H3O2)2 • 2Н3О в 27 мл ледяной уксусной кислоты и 900 мл воды. Нагревают раствор на паровой баье или горячей плитке до растворения солей и затем охлаждают. Если при этом не образуется небольшой осадок соли натрия, которым загрязнены реактивы, прибавляют несколько миллиграммов хлорида натрия. Дают отстояться 24 ч, а затем фильтруют. Желательно, чтобы реактив применялся приблизительно при той же температуре, при которой проводилось его фильтрование. Повышение температуры на несколько градусов не имеет значения, но понижение температуры недопустимо, так как. это приводит к получению повышенных результатов.
• Для приготовления промывной жидкости прибавляют 1 мл уксусной кислоты (30%-ной) к 100 мл 95%-ного этилового спирта, встряхивают с небольшим количеством тройной соли при 25° С и фильтруют. Так же, Как осадителем, этим раствором лучше пользоваться при той температуре, при которой проводилось насыщение его тройной солью. При анализе некоторых продуктов нет необходимости применять реактив для перенесения осадка в фильтрующий тигель и предварительного промывания его, а при всех операциях можно пользоваться промывной жидкостью (см. Koenig, цит. выше).
750
Гл. XLII. Щелочные металлы
в реактивах, которые проводят через все стадии анализа, умножают на фактор: для Na2O — 0,02015, для Na — 0,01495.
Из объемных методов, которые рекомендуются для конечного определения, можно упомянуть титрование перманганатом после растворения осадка в кислоте и восстановления урана до четырехвалентного состояния 1 и растворение осадка в воде с последующим титрованием урана едким натром2. Описан 3 спектрофотометрический метод, основанный на образовании окрашенного в оранжевый, до красного, цвет соединения после обработки осадка карбонатом аммонии и перекисью водорода.
Разработан ряд вариантов цинк-уранил-ацетатного метода, при применении которых можно осаждать большие количества натрия 4; в этих вариантах увеличены объемы анализируемого раствора и осадителя 5, или температура раствора повышена до 40° С, или же повышена также кислотность раствора до 4 н. по серной кислоте 6 или 2 н. по хлорной кислоте 7 (1 : 5). Однако сколько-нибудь' значительное отклонение от обычно принятой методики может быть рекомендовано лишь в том случае, если оно обосновано результатами опытов, проведенных на ряде образцов того же типа, как и анализируемый 8.
Повышение температуры до 40° С допустимо при условии9, если: 1) реактив насыщают тройным ацетатом при той температуре, при которой он применяется; 2) период осаждения увеличивают до 30 мшТ, 3) в интервале между осаждением и фильтрованием температура остается неизменной; 4) температура во время этих операций контролируется с точностью до нескольких градусов.
Были исследованы тройные соли, содержащие другие двухвалентные металлы, как, например, медь, кадмий, ртуть, марганец, никель, кобальт, железо (II) и магний, но из них только соль магния нашла практическое применение.
Авторы 10, предложившие метод определения натрия в виде магниевой соли NaMg(UO2)3CH3COO • 6Н2О, нашли, что таким способом можно осаждать до 50 мг натрия. Эту соль можно высушить и непосредственно взвесить или же растворить, оттитровать магний в полученном растворе двузамещенным фосфатом натрия и пересчитать полученный результат
1 Н. V. Churchill, R. W. Bridges, A. L. Miller, Ind. Eng. Chem., Anal. Ed., 8, 348 (1936).
2 J. T. Dobbins, R. M. Byrd, J. Am. Chem. Soc., 53, 3288 (1931).
3 E. Arnold, A. R. Pray, Ind. Eng. Chem., Anal. Ed., 15, 294 (1943).
О методе, основанном на светопоглощении водным раствором тройного ацетата (с применением синих светофильтров), см. D. R. McCormic, W. Е. Carlson, Chemist Analyst, 31, 15 (1942).
4Е. W. Koenig (цит. выше) поднял верхний предел концентрации до 12 мг Na2O при конечном объеме раствора 25 мл.
5 Например, 5 мл раствора и по крайней мере 20 мл реактива (Е. W. Koenig, цит. выше).
s Н. V. Churchill, R. W. Bridges, A. L. Miller, цит. выше.
7 Koenig (цит. выше) указывает, что осадки, полученные в хлорнокислых растворах, содержащих алюминий, должны быть перенесены на фильтр и промыты реактивом для удаления перхлората алюминия, который переходит в осадок в случае непосредственного применения спиртовой промывной жидкости.
8 Применение уранил-цинк-ацетатного метода для прямого быстрого определения натрия в известково-натриевом стекле см. F. W. Glaze, J. Am. Ceram. Soc., 14, 450 (1931); применение того же реактива для прямогб определения натрия в цементах см. F. W. Glaze, Rock Products, 44, № 7, 42 (1941) и ASTM Specification С 114—47. Chemical Analysis of Portland Cement, p. 31.
9 Согласно Koenig, цит. выше.
10 E. R. С a 1 e у, C. W. F о u 1 k, J. Am. Chem. Soc., 51, 1664 (1929).
Методы определения
751
на натрий 1 или же восстановить уран в растворе, оттитровать его перманганатом и также пересчитать результат титрования на натрий 2.
Для осаждения натрия необходимы следующие реактивы: раствор магний-уранил-ацетата и свежеприготовленная промывная жидкость, которая представляет собой 95%-ный спирт, насыщенный при 20° С натрий-магний-уранил-ацетатом.
Раствор магний-уранил-ацетата получают из растворов I и II, которые приготовляют и смешивают следующим образом:
Раствор I Раствор II
UO2(CH3COO)2 • 2Н2О........ 90 г Mg(CH3COO)2 • 4Н2О........ 600 г
Ледяная уксусная кислота . . 60 г Ледяная уксусная кислота 60 г
Дистиллированная вода ... до 1000 мл Дистиллированная вода . 1 до 1000 мл
Каждый раствор в отдельности нагревают приблизительно до 70° С до полного-растворения соли, а затем оба раствора при этой же температуре смешивают. Дают охладиться до 20 + 1° С, держа при этой температуре не менее 1 ч, затем раствор фильтруют через сухой фильтр в сухую склянку. Раствор устойчив, если предохранять его-от действия прямого солнечного света и если не давать ему охлаждаться ниже 20° С.
Метод сводится к следующему. Объем нейтрального раствора, содержащего не более 25 мг натрия 3, лучше в виде хлорида, доводят до 5 мл или менее, если при этом не происходит выделение солей. Быстро прибавляют 100 мл реактива, если содержание натрия меньше 10 мг, а при наличии больших количеств — по 10 мл на каждый миллиграмм натрия. Растворы перемешивают, частично погружают в водяную баню, имеющую-температуру 20 ± 1° С, и сильно перемешивают от 30 до 60 мин, причем более продолжительное перемешивание желательно при содержании свыше 0,2 мг натрия. Тотчас же фильтруют через взвешенный тигель Гуча или стеклянный фильтрующий тигель средней пористости, применяя слабое отсасывание и держа раствор в легком движении, чтобы воспрепятствовать прилипанию осадка к стакану. Когда весь раствор перенесен, полностью отсасывают жидкость, очищают стакан и промывают осадок в тигле 4—5 последовательными порциями по 5 мл 95%-ного-спирта, свеженасыщенного тройным ацетатом при 20° С. Спиртовая промывная жидкость может вызвать осаждение в фильтрате солей (* на это не следует обращать внимания *). Тщательно отсасывают жидкость, сушат тигель с осадком в сушильном шкафу при 105—110° С в течение 30 мин, охлаждают и взвешивают. Осадок содержит 1,53% натрия4.
1 Е. R. Caley, J. Am. Chem. Soc., 52, 1349 (1930).
2 A. Nau, Bull. soc. phar. Bordeaux, 65, 67 (1927); E. Ka han e, Bull, soc. chim. [4], 47, 382 (1930). Потенциометрическое титрование перманганатом калия или сульфатом церия (IV) см. N.- Н. Furman, Е. R. Caley, I. С. Schoonover, J. Am. Chem. Soc., 54, 1344 (1932).
3 В случае содержания больших количеств натрия (свыше 50 мг) следует соблюдать особые предосторожности, а при малых количествах (<М мг) исходный объем раствора необходимо уменьшить до 1—2 мл.
4 Можно указать на следующие возможные случаи применения этого метода: 1) непосредственное определение натрия в карбонате кальция, предназначенного для использования в методе J. L. Smith [Е. R. Cale у, Ind. Eng. Chem., Anal. Ed., 1,191 (1929)]; 2) непосредственное определение натрия в товарном алюминии [Е. R. С а-1 е у, там же, 4, 340 (1932)]; отделение натрия от алюминия и хрома см. также Е. R. Caley, D. V. S i с k m a n, J. Am. Chem. Soc., 52, 4250 (1930) и 3) определение 0,05—2,0 мг натрия осаждением спиртовым раствором магний-уранил-ацетата, с последующим центрифугированием и измерением объема осадка в капиллярной трубке [Е. R. Caley, С. Т. Brown, Н. Р. Price, Ind. Eng. Chem., Anal. Ea., 6, 202 (1934)]. E. C. Elliott [там же, 12, 416 (1940)] установил, что метод может быть с успехом применен в присутствии бериллия, церия, лантана, неодима, таллия, тория, ванадия и циркония.
КИСЛОТООБРАЗУЮЩИЕ ЭЛЕМЕНТЫ
КРЕМНИЙ, ВОЛЬФРАМ, ФОСФОР, СЕРА, ХЛОР, БРОМ, ИОД, ФТОР, БОР, УГЛЕРОД, ВОДОРОД И АЗОТ
Глава XLIII
КРЕМНИЙ
Кремний составляет около 27,6% литосферы и идет вслед за кислородом, который является наиболее распространенным в природе элементом. Кремний находится в к в а р ц е SiO2, тридимите SiO2, о п а-л е Si02-reH20, во всех силикатах и является основным элементом вс^х важных горных пород, за исключением карбонатных пород.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа большая часть кремния отделяется в начальной стадии анализа обезвоживанием кремнекислоты в кислом растворе. Вся, или почти вся, оставшаяся часть кремнекислоты переходит в осадок от аммиака. Если предварительное отделение кремнекислоты проведено небрежно, значительная ее часть, особенно когда осадок от аммиака мал, 'может остаться в аммиачном фильтрате и загрязнить осадки оксалатов и фосфатов *. Кремнекислота, попавшая в осадок от аммиака, если она не обнаружена и не отделена, учитывается как алюминий.
РАЗЛОЖЕНИЕ ПОРОД И МИНЕРАЛОВ,
СОДЕРЖАЩИХ КРЕМНИЙ '
Разложение пород и минералов, содержащих кремний, обычно проводится, как указано в разделе «Разложение при помощи плавней» (стр. 915). В некоторых случаях разложение может быть осуществлено обработкой кислотами, в частности фтористоводородной кислотой, если опре-
1 В присутствии цинка перед осаждением аммиаком должны быть приняты особые меры для полного удаления кремнекислоты, в противном случае цинк переходит в осадок в виде силиката.
Методы отделения
753
деление кремнекислоты не требуется, и хлорной кислотой, когда присутствуют вещества, образующие нерастворимые соединения с соляной, серной или азотной кислотами.
МЕТОДЫ ОТДЕЛЕНИЯ
Количественное отделение кремния от других элементов редко бывает необходимо, так как содержание его обычно определяется по потере в массе, полученной после обработки взвешенной нечистой кремнекислоты фтористоводородной-и серной кислотами и прокаливания нелетучего остатка. Примеси, таким образом, не имеют значения, если они нелетучи, не изменяются в массе в результате обработки смесью кислот и не присутствуют в таких количествах, чтобы вызвать затруднения при выпаривании или последующем прокаливании нелетучего остатка.
Осадком кремнекислоты всегда увлекается бор, и при последующей обработке фтористоводородной и серной кислотами он улетучивается вместе с кремнием, вследствие чего результаты определения кремнекислоты получаются повышенными г. Поэтому бор необходимо предварительно отделять, что наиболее удобно осуществлять перед обезвоживанием^ кремнекислоты. Лучше всего удалять бор отгонкой в виде триметоксильного производного В(ОСН3)3 из раствора; не содержащего воду или содержащего ее лишь в небольших количествах. Применительно к плаву анализируемой пробы с борным ангидридом ход отделения состоит в следующем.
Помещают плав в большую фарфоровую или платиновую-чашку; накрывают часовым стеклом и обрабатывают метиловым спиртом, насыщенным хлористым водородом (реактив готовят насыщением- холодного метилового спирта сухим хлористым водородом в течение 1 —2 ч). По прекращении вскипания снимают часовое стекло и нагревают раствор почти до кипения на водяной бане в хорошем вытяжном шкафу .'Таким же образом обрабатывают тигель с остатками плава и присоединяют его содержимое к раствору в чашке. Прибавляют еще реактив, если требуется для полного разложения плава, и кипятят до тех пор, пока не останется небольшой объем раствора, а затем выпаривают на водяной бане досуха. Для удаления последних следов бора повторяют выпаривание на водяной бане при 80—85° С еще 2—3 раза с небольшими порциями реактива, обмывая каждый раз стенки чашки.
Определению кремнекислоты обычно принятым методом мешает фтор, с которым кремний образует летучий тетрафторид кремния в растворах, содержащих сильные кислоты. Потеря всегда бывает меньше, чем это соответствует количеству присутствующего фтора, и в рядовых анализах, если содержание фтора меньше 0,3 %, ею можно пренебречь.
Тетрафторид кремния неустойчив в присутствии влаги, и некоторые количества фтора улетучиваются, вероятно, в виде фтористоводородной кислоты, но большая
1 То же относится и к мышьяку, если выпаривание проводится с серной кислотой в присутствии кремнекислоты и мышьяка (III). Величина ошибки определения кремнекислоты, вызываемая наличием бора, характеризуется результатами 80,22 и 80,24% SiO2 (по сравнению с наиболее вероятным содержанием 80,13%), полученными при анализе стекла, содержавшего 12,67% В2О3, причем присутствие бора игнорировалось. Определение проводилось двукратным обезвоживанием с соляной кислотой и извлечением кремнекислоты из фильтрата обычным способом.
48 Заказ 522.
754 Гл. XLIII. Кремний
часть его задерживается, образуя фторосиликаты. Последние в процессе прокаливания совместно с кремнекислотой, по крайней мере, частично разлагаются и после обработки смесью кислот содержащиеся в них металлы (преимущественно натрий) будут взвешены в виде сульфатов, если они не улетучатся при температуре, при которой проводится прокаливание.
При анализе плавикового пшата можно1 в значительной мере избегать потерь кремния, если тонко измельченную пробу поместить в стакан из стекла пайрекс, прибавить 20%-ную хлорную кислоту, насыщенную борной кислотой при 50Q С, и нагревать до появления паров хлорной кислоты.
Установлено 2, что при определении кремния во фторсодержащих материалах, как, например, в криолите, хорошие результаты получаются при следующих условиях. Смешивают 1 г руды с 3 г плавленой буры и 14 г бисульфата калия (все измельченное в порошок) в платиновом тигле и, закрыв крышкой, нагревают сначала осторожно, а затем сильно, до окончания реакции. Охлажденный плав растворяют в разбавленной соляной кислоте, раствор выпаривают, а остаток сушат при 110° С, измельчают и снова нагревают до удаления соляной кислоты. Остаток под конец смачивают соляной кислотой, обрабатывают горячей водой, отфильтровывают и промывают, как обычно.
Меньшие количества кремния теряются, когда платиновая чашка закрыта платиновой крышкой и в процессе выпаривания пространство внутри чашки насыщено водяными парами. Еще менее значительны потери кремния, если плав растворить в концентрированном растворе борной кислоты и затем обработать, как указано выше, но при этом требуется отделение кремнекислоты от увлеченного ею бора.
При содержании больших количеств кремния наряду с малыми количествами фтора, как, например, в случае стекол, содержащих фтор, влияние фтора может, быть сведено к минимуму сплавлением пробы с борным ангидридом в течение 30 мин при 1200° С или выше и выделением кремнекислоты обычным способом, выпариванием с соляной кислотой. При содержании же больших количеств фтора и малых количеств кремния этот метод дает пониженные результаты. В этом случае необходимо пользоваться методом Берцелиуса, изложенным на стр. 1011. Сплавление со смесью карбоната натрия и буры или сплавление с одним карбонатом натрия и выщелачивание плава - насыщенным раствором борной кислоты, перед обработкой соляной кислотой, дает почти такие же хорошие результаты, как сплавление с борным ангидридом, в процессе которого ббль-шая часть фтора улетучивается в виде фторида бора BF3. В обоих случаях фтор, остающийся в растворе, при обезвоживании кремнекислоты связывается в Виде менее вредной фтороборной кислоты HBF<. Бор, перешедший в остаток кремнекислоты, должен быть удален обработкой метиловым спиртом перед прокаливанием этого остатка.
Вольфрам обычно сопровождает кремний и его, как правило, не отделяют, если требуется определение только одной кремнекислоты. Смесь кремнекислоты и окиси вольфрама прокаливают и обрабатывают серной и фтористоводородной кислотами, как обычно, только прокаливание неле-. тучего остатка следует проводить при температуре не выше 850° С вследствие некоторой летучести окиси вольфрама (стр. 766). Вольфрам не теряется при обработке смесью кислот и даже при обработке одной фтористоводородной кислотой. Некоторые количества вольфрама улетучиваются в процессе прокаливания смеси окислов йри температуре, необходимой для полного обезвоживания кремнекислоты.
1 W. Т. Schrenk, W. Н. О d е, Ind. Eng, Chem., Anal. Ed., 1, 200 (1929).
2 H. Spiel haczek, Z. anal, Chem., 119, 4 (1940).
Методы определения
755
Когда требуется определение обоих элементов, аналитику предоставляется выбор между получением повышенных результатов для кремния и пониженных результатов для вольфрама. Часто предпочитают первое, и это вызывает меньше затруднений, так как при совместном присутствии вольфрама и кремния содержание кремнекислоты обычно относительно невелико. Окись вольфрама можно растворить и отделить от кремния, как описано в гл. «Вольфрам» (стр. 766), или же нагреванием смеси окислов в токе хлористого водорода. Если для этой цели применяют платйновую лодочку, то газ должен быть сухим и свободным от воздуха и хлора *. В этих условиях некоторое количество вольфрамового ангидрида может восстановиться до окисла низшей валентности, который не образует летучего хлорида. Восстановления можно избежать, применив кварцевую лодочку и хлористый водород, смешанный с воздухом или кислородом. Когда пользуются платиновой лодочкой, восстановленную окись вольфрама следует вновь окислить и отогнать, для чего, поступают следующим образом. Охлаждают прибор, вытесняют хлористый водород воздухом, прокаливают, охлаждают, удаляют воздух током хлористого водорода и снова прокаливают.
Наилучшим методом отделения малых количеств кремния от больших количеств железа, как, например, в чистом железе, является следующий. Отделяют возможно большее количество кремнекислоты обычным обезвоживанием серной кислотой, последующим разбавлением и фильтрованием. Для извлечения остающейся при этом в растворе части кремнекислоты фильтрат выпаривают, остаток прокаливают с целью превращения сульфата железа (III) в окись, которую затем удаляют отгонкой, прокаливая при 400—700° С в трубке в токе сухого хлористого водорода. Нелетучий остаток обрабатывают для определения кремнекислоты 2.
От олова кремний лучше отделять обезвоживанием кремнекислоты соляной кислотой, а не серной или хлорной, с которыми при разбавлении олово может образовать нерастворимые соединения, или азотной, которая, безусловно, вызовет соосаждение олова.
Успешное разделение кремния и свинца достигается обезвоживанием серной кислотой и выщелачиванием сульфата свинца ацетатом или хлоридом аммония з. Если обработка проводится ацетатом аммония и присутствуют значительные количества свинца, целесообразно осадок смыть в чашку, нагреть с раствором ацетата и затем снова перенести на фильтр.
1 D. Н. Brophy, С. V а п В г u n t, Ind. Eng. Chem., 19, 107 (1927).; C. Friedheim, W. H. Henderson, A. Pinage 1, Z. anorg. Chem., 45, 398 (1905); F. P 5 r i 11 о n, Bull. soc. ind. Minerale (1884).
a R. M. Fowler, Ind. Eng. Chem., Anal. Ed., 4, 382 (1932).
8 Например, для анализа растворяли навеску 5 г бронзы (88% Си, 8% Sn, 1,5% РЬ, 2% Zn, 0,2% Sb плюс небольшое взвешенное количество чугуна с определенным содержанием кремния) в смеси НС1 и HNOS, обрабатывали раствор 15 мл H2S04 и выпаривали до появления' обильных паров. Затем анализ вели двумя методами: 1) добавляли 200 мл разбавленной (5 : 95) серной кислоты, фильтровали и промывали осадок последовательно разбавленной (2 : 98) НС1, горячей водой, горячим раствором CH8COONH4 (смешивали равные объемы Н2О и NH4OH и слабо подкисляли СН8СООН) и снова горячей водой, либо 2) разбавляли 150 мл воды, содержащей 25 г NH4C1, » и фильтровали. Были получены следующие результаты:
Метод.........'................. . 1
Прибавлено кремни^, %............... 0
Найдено кремния, % ................. 0
. 48*
1112 2
0,005 0,010 0,048 0,048 0,048
0,005 0,009 0,047 0,048 0,043
756
Гл. XLIII. Кремний
Ниобий и тантал всегда сопровождают кремнекислоту и иногда они переходят в осадок почти количественно *. Оба эти элемента остаются в виде пятиокисей в остатке после обработки фтористоводородной и серной кислотами. Если эти элементы первоначально присутствовали в значительных количествах, остается большой нелетучий остаток, что является некоторым указанием на возможное присутствие в- анализируемом материале тантала и ниобия. Определение кремния в присутствии ниобия и тантала см. гл. «Ниобий и тантал» (стр. 679). Титан и цирконий не сопровождают кремнекислоту в заметной степени, если при обработке раствора не создаются условия, благоприятные для гидролиза их солей, и в растворе не содержатся значительные количества фосфатов. При выпаривании со смесью фтористоводородной и серной кислот потерь титана и циркония не наблюдается 1 2.
В присутствии фосфатов удаление серной кислоты должно проводиться при возможно более низкой температуре во избежание потерь фосфорной кислоты 3.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Кремний обычно взвешивают в виде окиси SiOa после выделения ее обезвоживанием в кислом растворе (см. часть III, стр. 940 сл.). Полученная таким образом окись кремния загрязнена примесями, и истинное ее содержание определяется по потере в массе после обработки взвешенного остатка серной и фтористоводородной кислотами, прокаливания и взвешивания нелетучего остатка 4.
Однократным выпариванием с какой бы то ни было кислотой редко удается полностью выделить кремнекислоту из раствора 5.
Например, в 1-граммовых навесках известково-натриевого стекла, содержавшего 74,1% SiOa, однократным выпариванием с соляной кислотой на паровой бане и последующим высушиванием остатка при 110Q С в течение 1 ч было определено 73,4% SiOa. После однократного выпаривания с серной кислотой и с хлорной кислотой было обнаружено соответственно 72,4% и 73,3% SiO2.
Обычно некоторые количества кремния остаются в растворе даже после двукратного выпаривания с промежуточным фильтрованием.
1 Так, например, после сплавления окисей этих металлов с Na2CO3, обезвоживания кремнекислоты однократным выпариванием с соляной кислотой, нагревания остатка при 110° С, обработки его НС1 и горячей водой, фильтрования и промывания разбавленной (1 : 99) НС1 Н. В. Knowles обнаружил в остатке кремнекислоты 3,6 мг Та2О5 и 3,1 мг Nb2O5 при содержании в исходной окиси 3,9 мг Та2О5 и 4,3 мг Nb2O5.
а Лишь незначительные количества циркония теряются, если окись циркония обрабатывается одной фтористоводородной кислотой при осторожном прокаливании в хорошо закрытом тигле.
3 W. F. Hillebrand, G. Е. F. L u n d е 11, J. Am. Chem. Soc., 42, 2608 (1920).
4 При тщательном изучении нелетучих остатков, оставшихся после определения кремния в 16 финских горных породах, К. R a n k а ш а [С. г. soc. geol. Finlande, 14, 1 (1939)] спектрографически обнаружил Al, Ba, Be, Ca, Ce, Co, Cr, Fe, Ga, Ge, K, La, Mg, Mn, Mo, Na, Ni, Pb(Pt), Sc, Sn, Sr, Ti, W, V, Y, Zn и Zr, всего 28 элементов.
s R. W. Bunsen, Ann., 61, 265 (1847); E. Ludwig, Z. anal. Chem., 9, 321 (1870); С. M e i n e k e, Repert. Anal. Chem., 7, 215, 757 (1887); J. P. Gilbert, Technol. Quart., 3, 61 (1890); W. F. Hillebrand, J. Am. Chem. Soc., 24, 362 (1902); A. Cameron. Chem. News, 69, 171 (1894); M. Wunder, A. Su-1 e i m a n n, Ann. chim. anal, appl., 19, 45 (1914); V. Lenher, andE. T r u о g, J. Am. Chem. Soc., 38, 1050 (1916); R. M. Fowler, Ind. Eng. Chem., Anal. Ed., 4, 382 (1932). (
Методы определения
757
При этом остаток, полученный после первого обезвоживания, обрабатывают разбавленным раствором той же кислоты, какая применялась для обезвоживания, раствор фильтруют, остаток загрязненной кремнекислоты промывают и фильтрат с промывными водами снова выпаривают.
При анализе 0,5-граммовых навесок ферросилиция и обожженных огнеупорных материалов в растворе в среднем оставалось около 1 мг кремнекислоты после выпаривания с соляной кислотой и несколько больше этого количества после выпаривания с серной кислотой (см. также табл. 23, стр. 758).
Если выпаривание проводилось надлежащим образом, то после третьего выпаривания остаток кремнекислоты уже не образуется, так как при этом уже устанавливается равновесие растворимости. Оставшаяся в растворе кремнекислота переходит в осадок от аммиака в количестве, зависящем от величины этого осадка и от содержания кремнекислоты в растворе. Если масса прокаленного осадка от аммиака превышает 0,1 г и перед осаждением аммиаком проводилось двукратное выпаривание для выделения кремнекислоты, то в растворе после осаждения аммиаком остается не более 0,1—0,2 мг SiO2.
Небольшие количества кремнекислоты, остающиеся в растворе, могут быть удовлетворительно определены колориметрически сравнением окраски кремнемолибденовой кислоты, образующейся при добавлении к раствору молибдата аммония [2 мл 10%-ного раствора (КН4)гМоО4 и 2 капли HaSO4 на 50 мл раствора, содержащего не более 2,5 мг SiOa], с окраской искусственного стандартного раствора пикриновой кислоты1.
Описано применение растворов хромата калия, буферированных бурой, в качестве стандартов при определении малых количеств растворенной кремнекислоты по реакции образования желтого гетерополисоединения кремнемоЬгибденовой кислоты2 * *.
Для определения 0,01—1,0 мг SiO2 в 100 мл раствора рекомендуется 8 вводить (МН4)аМоО4 в таком количестве, чтобы молекулярная концентрация его в 12 раз превосходила концентрацию SiC>2, доводить pH раствора до 1,6—2,0 и спустя 10 мин окраску раствора сравнивать со стандартами буферированного раствора хромата или же измерять интенсивность окраски в фотоколориметре. Фосфаты и, несомненно, другие ионы, образующие комплексные соединения с молибдатом, мешают определению.
Для разрушения окраски фосфорномолибденовой кислоты при фотометрическом определении малых количеств кремния (§0 мг!л) в очищенных водах применяют* щавелевую кислоту. Рекомендован5 * фотометрический метод определения до 1 % кремнекислоты в алюминийсодержащих материалах, основанный на реакции образования молибденовой сини.
Если осадок от аммиака невелик и особенно если для выделения кремнекислоты проводили однократное выпаривание или пиросульфитный плав прокаленного осадка от аммиака растворяли в воде, а не выпаривали с серной кислотой (стр. 954), то потери кремнекислоты могут достигать нескольких миллиграммов.
Кремнекислота, перешедшая в осадок от аммиака, принимается за алюминий, если содержание последнего определяется по разности и кремнекислота из этого осадка не удаляется. Когда кремнекислота из осадка от аммиака извлекается, но не вводится поправка на содержание примесей
1 F. Dienert, F. Wandenbulcke, С. г., 176, 1478 (1923); Bull. Soc. chim., 33, 1131 (1923); Е. J. King, С. C. Lucas, J. Am. Chem. Soc., 50, 2^95 (1928).
2 H. W. Swank, M. G. Mellon, Ind. Eng. Chem., Anal. Ed., 6, 348 (1934).
R. J. Robinson, H. J. Spoor, там же, 8, 455 (1936); R. J. Robinson,
H. J. Spoor,, M. F. Adams, там же, 17, 542 (1945).
8 H. W. Knudson, C. J u d а у, V. W. M e 1 о c h e, Ind. Eng. Chem.»
Anal. Ed., 12, 270 (1940),
* M. C. Schwartz, Ind. Eng. Chem., Anal. Ed., 14, 893 (1942).
5 J. A. В r a b s о n, I. W. H a r w e y, G. E. Maxwell, O. A. Scha-
effer, Ind. Eng. Chem., Anal. Ed., 16, 705 (1944).
758
Гл. XLIII. Кремний
кремния и алюминия в реактивах, результаты определения этих элементов остаются ошибочными. Метод определения- алюминия и кремния в реактивах изложен ниже. В табл. 23 приведены типичные результаты, полученные при извлечении кремнекислоты из осадка от аммиака, выделенного после предварительного отделения кремнекислоты двукратным выпариванием с промежуточным фильтрованием х.
ТАБЛИЦА 23
Результаты извлечения кремнекислоты из осадка от аммиака, полученного после двукратного выделения кремнекислоты выпариванием с кислотой и фильтрованием
✓ Анализируемая проба Содержание SiO, в пробе % Приблизительная величина взятой навески г Примененная кислота Извлечено SiO* из осадка от аммиака, мг
Доломит ... 0,31 10 НС1 1—1,21
Полевой шпат 66,66 1 НС1 5,6
То же 66,66 0,5 НС1 0,9-1,1
Известково-натриевое стекло . . 74,1 1 НС1 0,3—0,82
То же .' 74,1 0,5 H2SO4 0,63
Свинцово-бариевое стекло . . . 65,36 0,5 НС1 0,3—0,4*
То же •. Обожженный огнеупорный мате- 65,36 0,5 НС1О4 0,4-1,Iе
риал 54,7 0,5 H.SO4 1
То же ' 32,4 0,5 H.SO4 0,5-0,6
» ............. 20,7 0,5 H,.SO4 0,7—0,8
Боксит 6,32 1 HaSO4 0,5
Очищенный кремний 96,8е 0,5 - HC1 0,6-1
То же 96,8е 0,5 H,SO4 1—1,5
* Осадок, выделенный аммиаком из подкисленного аммиачного фильтрата, к которому было предварительно добавлено 15 мг Fe,Os в виде FeCl., содержал не больше SiO2, чем введенные реактивы. Таким образом двукратным обезвоживанием и выделением совместно с осадком от аммиака кремнекислота была извлечена из раствора полностью.
* Количество S1O., выделенное при вторичном обезвоживании, колебалось, в пределах 3,3—7,6 мг.
3 При втором обезвоживании было выделено 9,4 мг SiOt.
* При втором обезвоживании было выделено S1O. порядка 3—6 мг.
• При втором обезвоживании было выделено SiO. порядка 0,6—0,9 мг.
• В пересчете на Si.
Обезвоживание кремнекислоты обычно осуществляется в солянокислых растворах, так как хлориды отличаются большей растворимостью, чем сульфаты или перхлораты, однако выбор кислоты должен определяться составом анализируемого материала 2. Помимо соляной кислоты,
1 Определение кремнекислоты во взвешенном осадке от аммиака непосредственной обработкой фтористоводородной и серной кислотами см. гл. «Алюминий» (стр. 569). Согласно L. R. Dawson и R. V. Andes [Ind. Eng. Chem., Anal. Ed., 12, 138 (1940)], рядовые определения R2O3 и SiO2 в цементах весьма успешно могут быть выполнены объединением осадков R2O3 и SiO2, прокаливанием, взвешиванием, обработкой после этого H2SO4 -f- HF, выпариванием, прокаливанием и повторным взвешиванием.
а Например, при анализе стекла, содержащего, помимо обычных окисей кремния, железа, алюминия, щелочных и щелочноземельных металлов, также и 5% окислов сурьмы, 2% свинца и 13% бора, применили тщательное двукратное выпаривание с промежуточным фильтрованием. В результате анализа найдено: в случае обезвоживания кремнекислоты азотной кислотой — 67,2% SiO2; в случае обезвоживания соляной кислотой — 68,2% и при обезвоживании серной кислотой — 68,8%. Во всех случаях определение заканчивалось обработкой осадка кремйекислоты смесью HF +
+ H2SO4. Наиболее близка» к истинному оказался результат, полученный после выпаривания с серной кислотой.
Методы определения
759
для этой цели пользуются также серной и хлорной кислотами. В некоторых случаях хорошие результаты получаются при применении азотной кислоты, как, например, при анализе известково-натриевого стекла, но она не всегда применима вследствие трудной растворимости многих нитратов в разбавленной кислоте. Во всех случаях требуется двукратное выпаривание и извлечение кремния из осадка от аммиака. Выделение кремнекислоты совместно с окисью титана нагреванием с муравьиной кислотой 1 не дает хороших результатов (Лендель).
При применении соляной кислоты для обезвоживания кремнекислоты выпаривание необходимо продолжать до тех пор, пока остаток не станет совершенно сухим. Это можно успешно осуществить на паровой бане, если образующиеся корки разбивать стеклянной палочкой 2. Более надежное обезвоживание, особенно в Присутствии магния, или марганца, достигается при нагревании остатка в муфеле или в шкафу, но при этом температура не должна быть выше 110° С и нагревание не следует продолжать более 1 ч, в противном случае кремнекислота снова соединится с окисью магния и перейдет в раствор.
В случае применения серной кислоты для обезвоживания кремне-кисЛЬты серную кислоту следует вводить в достаточном количестве, чтобы предотвратить образованно комков или пасты, так как это может вызвать "разбрызгивание, а также затруднить последующее растворение солей. Нагревание следует проводить до выделения паров серной кислоты. В присутствии таких элементов, как никель и хром, образующих малорастворимые сульфаты, нагревание при высокой температуре не должно быть слишком продолжительным. При наличии значительных количеств алюминия лучшие результаты получаются, когда к еще совершенно горячей кислоте прибавляют равный объем воды; после этого раствор нагревают до кипения и разбавляют кипящей водой.
Хлорная кислота для обезвоживания кремнекислоты должна вводиться в таком количестве,, чтобы в момент появления ее паров масса была жидкой, а не тестообразной или сухой. После появления паров хлорной кислоты следует осторожнб кипятить еще около 15 мин, чтобы обеспечить полное обезвоживание кремнекислоты (см. часть III, стр. 940).
Обезвоженная кремнекислота заметно растворима в кислых растворах 3 и даже в растворах, содержащих хлорид натрия или другие соли 4. Кислотность раствора, в котором нагревается обезвоженная кремнекислота, поэтому не должна быть выше той, которая требуется для растворения сопровождающих солей и для предотвращения гидролиза. Концентрация соляной или серной кислоты в растворе не должна превышать 5—10% по объему. Обезвоживание следует вести возможно менее продол-
1 A. L е с 1 е г с, С. г., 137, 50 (1903).
2 Согласно S. S h i n k a i [J. Soc. Chem. Ind. Japan, 29, 303 (1926)], NH4C1 превращает гидрозоль кремнекислоты в гидрогель, поэтому он рекомендует прибавлять 3—4 г NH4C1 после выпаривания солянокислого раствора карбонатного плава до малого объема, а затем выпаривать раствор досуха, часто перемешивая.
8 М. । Wunder,' A. Suleiman n, Ann. chim. anal, appl., 19, 45 (1914); V. Lenher, E. T r u о g, J. Am. Chem. Soc., 38, 1050 (1916); F. G. Hawley, Eng. Mining J., 103, 541 (1917). Последний нашел, что концентрированная соляная кислота действует менее активно, чем. 18-г25%-ная, и что количество растворяющейся кремнекислот'ы пропорционально количеству введенной кислоты.
4 V.i Lenher, Е. Т г и о g, J. Am. Chem. Soc., 38, 1050 (1916); см. также F. G. Hawley, цит. выше; H. А. В. Motherwell, Eng. Mining J., 103, 1155 (1917). i
760
Гл. XL 111. Кремний
. жительное время и никогда не долее 15 мин. Когда проводится двукратное выпаривание с соляной кислотой, первый остаток промывают горячей разбавленной (5 : 95) соляной кислотой, а второй остаток — холодной разбавленной (1 : 99) кислотой. При применении горячей кислоты соли, загрязняющие первый остаток, удаляются лучшие и это более чем компенсирует растворяющее ее действие на кремнекислоту. С другой стороны, холодной разбавленной кислоты вполне достаточно для предотвращения , гидролиза и для извлечения солей из меньшего остатка кремнекислоты, 1 полученного после второго выпаривания.
При применении фарфоровых или кварцевых чашек с них почти невозможно смыть приставшую к стенкам кремнекислоту. Поэтому, когда имеется возможность, следует пользоваться платиновыми или золотыми чашками. В случае применения фарфоровых или кварцевых чашек щелочной плав (после сплавления с карбонатами) лучше растворять в разбавленной кислоте, чем в воде, так как при этом на стенках чащки образуется меньшее количество плотной корки. Во всех случаях при выпаривании эти чашки разъедаются и введенная таким образом в раствор кремнекислота может оказаться в большем количестве, чем та, которую не удается удалить со стенок чашки соскребыванием х.
Необходимо проявлять особую осторожность при сжигании фильтра с осадком кремнекислоты и при обработке прокаленной кремнекислоты серной и фтористоводородной кислотами, чтобы избежать механических потерь. Лучше всего сначала медленно нагреть влажный фильтр с остатком кремнекислоты, выделенной после второго выпаривания, до обугливания бумаги, избегая ее воспламенения, а затем присоединить кремнекислоту, полученную при первом выпаривании, и также осторожно обуглить фильтр. После этого частично закрывают тигель, осторожно сжигают уголь и, наконец, плотно закрыв тигель, прокаливают, остерегаясь токов воздуха, особенно если прокаливание проводится на паяльной лампе или горелке Мекера 1 2. При прокаливании на паяльной горелке тигель целесообразно опустить на две трети высоты в отверстие, проделанное в асбестовом щите. Обработка прокаленного остатка водой или разбавленной кислотой может вызвать его распыление, поэтому жидкость лучше вводить из пипетки, которую наполняют и, слегка приподняв крышку тигля, осторожно выливают жидкость из пипетки так, чтобы она стекала по стенке тигля.
Часто утверждают, что для полного удаления воды, из кремнекислоты достаточно прокаливания при температуре около 1000° С. Это справедливо относительно умеренных количеств кремнекислоты, выделенной из растворов, свободных от солей. В обычном анализе, особенно при содержании больших количеств кремнекислоты, надежнее прокаливать 3 до постоянной
1 Например, в результате тщательно проведенного анализа известково-натрие-вого стекла, содержавшего 74,1% SiO2, было определено 74,1% при выпаривании в кварцевой чашке и 74,2% при выпаривании в фарфоровой чашке, несмотря на то, что приставшую к стенкам чашек кремнекислоту не удалось полностью снять.
2 При прокаливании влажного фильтра, содержащего кремнекислоту в закрытом тигле при такой низкой температуре, как 950° С, может образоваться карбид кремния [К. A. Krieger, Н. S. Lukens, Ind. Eng. Chem., Anal. Ed., 8, 118 (1936)].
3 W. F. H i 11 e hr an d, J. Am. Chem. Soc., 34, 372 (1902). Нам ни разу не удалось получить подтверждения наблюдениям Е. Jordis и Е. Н. Kanter [Z. anorg. Chem., 35, 16 (1903)] и Th. Bauer [Thonind Ztg., 37, 89 (1913)], согласно которым в обезвоженной соляной кислотой кремнекислоте хлор находится в виде химического соединения с кремнием.
Методы определения
761
массы при 1200° С. Такую температуру дает паяльная горелка или горелка типа мекеровской. Чистая обезвоженная кремнекислота не гигроскопична, но кремнекислоту, которую обычно получают, лучше помещать в плотно закрытый крышкой тигель-и охлаждать под хорошим поглотителем влаги.
Следует иметь в виду, что некоторые платиновые тигли, не только новые, но и бывшие долгое время в употреблении, систематически и весьма заметно теряют в массе при продолжительном прокаливании при 1200° С. Для тигля, обладающего таким дефектом, необходимо время от времени определять величину этой потери и вводить соответствующую поправку или же при вычислениях учитывать массу тигля после, а не до прокаливания осадка1. *
Как уже было указано, содержание кремния в прокаленном остатке обычно вычисляют по потере в массе после обработки этого остатка фтористоводородной кислотой2. Как правило, приходится вводить также и серную кислоту, чтобы воспрепятствовать улетучиванию, помимо SiF4, и некоторых других соединений, как, например, фторидов циркония и титана. Температура, при которой проводится прокаливание нелетучего остатка, должна быть достаточно высокой, чтобы образовались те же соединения, которые находились в загрязненной кремнекислоте, но обычно она может не превышать 1000° С.
В табл. 24 приведены температуры, при которых начинают разлагаться некоторые из обычно встречающихся сульфатов при прокаливании в слабом токе воздуха 3.
ТАБЛИЦА 24
Температуры разложения некоторых сульфатов '
Сульфат Температура, при которой Сульфат Температура, при которой
начинается разложение °C происходит энергичное разложение, °C начинается разложение °C происходит энергичное разложение, °C
FeSO4 . . . 167 480 NiSO4 . . . 702 764
Fe2(SO4)3 . . 530 — CoSO4 . . 720 770
Bi2(SO4)3 . . 570 639 CdSO4 . . 827 846
A12(SO4)3 . . 590 639 MgSO4 . . 890 972
PbSO4 . . . 637 705 Ag2SO4 . . 717 925
CuSOj . . . 653 670 CaSO4 . . 1200 —
MnSO4 . . . 699 1 790 BaSO4 . . . 15102 —
ZnSO4 . . . 702 720
1 Согласно W. Blum[J. Am. Chem. Soc., 34, 1389 (1912)], MnS04 начинает разлагаться при температуре 550—600° С.
2 В присутствии таких примесей, как S1O2, Fe2O3 температура разложения понижается [W. Mostowltsch, Metallurgy, 6, 450 (1909)]. Это безусловно справедливо и относительно других сульфатов.
Обработка одной фтористоводородной кислотой допустима, когда примеси при этом не улетучиваются и не образуют фторидов, которые не раз
1 Относительно этого см. R. W. Hall, J. Am. Chem. Soc., 22, 494 (1900) и G. A. Hulett, H. W. Berger, там же, 26, 1512 (1904). Потери, по-видимому, вызываются улетучиванием из сплава иридия.
2 Фтористоводородная кислота должна быть свободной от нелетучих примесей, в противном случае надо вводить поправку на содержание в ней этих примесей и учитывать их влияние при последующих операциях.
3 II. О. Н of man, W. Wan jukow, Bull. AIME, 889 (1912); Met. Chem. Eng., 10. 172, 695 (1912).
762
Гл. XLIII. Кремний
лагаются в процессе прокаливания. Это имеет место в случае окислов железа (III), алюминия или вольфрама. Бор и мышьяк (III) улетучиваются вместе с кремнекислотой, идюэтому они должны быть отделены перед обработкой остатка фтористоводородной кислотой *.
Большое значение имеет загрязнение кремнекислоты солями щелочных металлов, что легко происходит, когда перед выделением кремнекислоты пробу сплавляют с карбонатами щелочных металлов, а фильтр с остатком кремнекислоты затем недостаточно тщательно промывают. Наличие солей щелочных металлов в этом остатке искажает результаты определения кремния, так как масса их в большей или меньшей степениизме-няется при обработке фтористоводородной и серной кислотами и прокаливании нелетучего остатка. Так, в процессе прокаливания кремнекислоты образуется силикат щелочного металла, который разлагается при последующей обработке фтористоводородной и серной кислотами и щелочной металл взвешивается в виде сульфата, а не окиси, что является причиной получения пониженных результатов определения кремния. Если щелочной металл первоначально находился в остатке в виде хлорида и во время прокаливания кремнекислоты он не улетучился и не разложился, то после обработки серной и фтористоводородной кислотами он взвешивается в виде сульфата, что, несомненно, также приводит к получению пониженных результатов для кремнекислоты. И, наконец, когда прокаливание нелетучего остатка после обработки серной и фтористоводородной кислотами проводится при достаточно высокой температуре, чтобы сульфат щелочного металла полностью улетучился, то для кремнекислоты получаются повышенные • результаты.
Сульфаты, Как, например, сульфаты свинца и щелочноземельных металлов, также являются нежелательными примесями, так как при прокаливании кремнекислоты они образуют силикаты, вследствие чего получаются пониженные результаты для кремния (пропорционально количеству образовавшегося силиката) 1 2. Из числа других вредных примесей можно указать на хлорокиси висмута и сурьмы, оловянную и сурьмяную кислоты, которые неопределенно изменяют свою массу в процессе обработки остатка кремнекислоты фтористоводородной и серной кислотами и прокаливания нелетучего остатка. В присутствии таких элементов кремнекислоту следует обезвоживать кислотой, образующей с ними растворимые соли.
Неправильные результаты для кремнекислоты, как правило, получаются также и в случае содержания в прокаленном остатке SiO2 неразло-женной пробы. Это происходит даже и в том случае, когда крёмнекислота, находящаяся в неразложенной пробе, улетучивается при обработке серной и фтористоводородной кислотами, так как масса сопровождающих кремнекислоту веществ редко остается после этой обработки без изменения. Так, например, в результате обработки микроклина KAlSi3O8 фтористо
1 Согласно Т. . Н е с z к о [Z. anal. Chem., 77, 327 (1929)], избыток кислот можно выпарить значительно быстрее и с возможно меньшим разбрызгиванием нелетучего остатка, если в тигель вводить фильтровальную бумагу для поглощения жидкости.
2 Когда выделенная кремнекислота загрязнена сульфатом бария, заметного взаимодействия между этими двумя соединениями не наблюдается при условии, если температура прокаливания не превышает 1000° С. При 1200° Q SiO2 замещает SO3 в сульфате бария, вследствие чего получаются пониженные результаты для кремния, когда определение его проводится обычной обработкой загрязненной кремнекислоты фтористоводородной и серной кислотами.
Методы определения
763
водородной и серной кислотами и последующего прокаливания образуются не К2О и А12О3, a K2SO4 и А12О3, а после такой же обработки талька H2Mg3 (SiO3)4 получается MgSO4 вместо MgO.
Поправка на загрязнения, вносимые с реактивами или за счет разъедания посуды, может оказаться совершенно неправильной, если величина ее устанавливается обычным способом, проведением одних реактивов через все стадии анализа. Это вызвано тем, что осадки, получающиеся в ходе анализа, значительно больше по своей величине, чем осадки, выделяющиеся в «холостом» опыте. В первом случае эти осадки захватывают практически всю кремнекислоту, введенную с реактивами и перешедшую в раствор вследствие разъедания стеклянной посуды или по какой-либо другой причине, тогда как в осадках, выделяющихся при проведении «холостого» опыта, содержится лишь незначительная часть этой кремнекислоты.
Истинную величину поправки можно получить, если в раствор реактивов, который служит для установления этой поправки, ввести приблизительно такие же количества железа и алюминия, какие содержатся в анализируемой пробе *.
1 Так, например, при анализе 0,5 г боксита средняя поправка на содержание кремнекислоты в реактивах составляла менее 0,1 мг SiO2, когда «холостой» опыт проводился с одними реактивами, и 0,9 л«г, когда в реактивы было введено 0,25 г окиси алюминия в виде хлорида алюминия. Таким образом, если «холостой» опыт проводить с одними реактивами, то результаты определения кремнекислоты получаются повышенными на 0,18%.
* '
Глава XLIV
ВОЛЬФРАМ
Вольфрам распространен почти исключительно в сильно кремнекислых породах. Он встречается главным образом в виде вольфраматов железа, марганца, кальция, меди и свинца, образуя минералы ферберит FeWCU, гюбнерит MnWCh, вольфрамит (Fe, Mn)W04, шеелит CaW04, купрошеелит (Са, Cu)WOi, штольцит PbW04. Он встречается также в виде сульфида — тунгстенита WS2 — и в небольших количествах во многих ниобатах и танталатах. Вольфрамовые руды немногочисленны. Вольфрамит часто находится в оловянных рудах. Вольфрам входит в состав различных продуктов металлургического производства, и методы его определения имеют весьма важное значение.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород вольфрам осаждается совместно с кремнекислотой, и его присутствие обнаруживается по желтой окраске выделившейся вольфрамовой кислоты. Это осаждение не количественное и мбжет вовсе не произойти, если содержание вольфрама в пробе незначительно. Большая часть вольфрама, выделяющегося с кремнекислотой, теряется, если прокаливание остатка проводится, как обычно, при высокой температуре, так как вольфрамовая кислота начинает возгоняться при температуре около 800° С. Значительная часть вольфрама (если не полностью), оставшегося в растворе после выделения кремнекислоты, попадает в осадок от аммиака даже после многократного переосаждения в виде соединений его с железом, алюминием и главным образом со щелочноземельными металлами. Осажденный таким образом вольфрам при прокаливании осадка от аммиака, по-видимому, не улетучивается и принимается за алюминий, если для* определения железа проводится восстановление сероводородом или сернистым ангидридом. В случае же восстановления железа цинком или хлоридом олова (II) ошибка распределяется между железом и алюминием (если не обращается внимания на посинение раствора, вызванное присутствием восстановленного вольфрама).
Совершенно очевидно, что полное отделение вольфрама в начальной стадии анализа даже более желательно, чем отделение кремнекислоты.
Методы, отделения t 765
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ ВОЛЬФРАМ
Большинство вольфрамовых минералов удовлетворительно разлагается последовательной обработкой соляной и азотной кислотами, как изложено в разделе «Определение обработкой кислотами с добавлением цинхонина» (стр. 770), при условии измельчения анализируемого минерала не менее чем до 200 меш \d — 0,074 мм). При такой обработке шеелит-и гюбнерит разлагаются легко, ферберит несколько труднее й наиболее трудно разлагается вольфрамит.
При определении вольфрама следует по возможности избегать сплавления анализируемого материала с карбонатом натрия, перекисью натрия или с пиросульфатами щелочных металлов, ^гак как соли щелочных металлов препятствуют количественному выделению вольфрама кислотами и замедляют его осаждение цинхонином. Это, однако, не является препятствием для применения такого сплавления, если оно проводится в качестве предварительной операции перед определением других содержащихся в минерале элементов, например железа (содержание которого определяют в остатке после выщелачивания карбонатного плава водой).
Сплавлрние с карбонатом натрия проводится следующим образом. Тщательно смешивают 0,5 г тонко измельченной руды с 4 г карбоната натрия в платиновом тигле и нагревают на очень сильном пламени 30— 60 мин. Плав выщелачивают водой, нерастворимый остаток отфильтровывают и промывают сначала разбавленным раствором карбоната натрия, а под конец водой. Промытый остаток слабо прокаливают, снова сплавляют с карбонатом натрия и выщелачивают водой. В объединенных водных вытяжках содержатся вольфрам, молибден, ванадий, мышьяк, фосфор, хром, кремний и частично или полностью алюминий, сурьма, олово, ниобий и тантал. В осадке находятся железо, цирконий, марганец, кальций и т. п.
Разложение пиросульфатом осуществляют сплавлением 1 г тонко измельченной руды е 5 г пиросульфата в закрытом платиновом или кварцевом тигле при возможно более низкой температуре. При выщелачивании плава разбавленной (5 : 95) серной кислотой достигается тот же результат, что и при разложении пробы кислотами. В этом случае происходит лишь менее полное отделение вольфрама. Когда желают вольфрам удержать в растворе, плав растворяют в горячем 5%-ном растворе винной кислоты. В любом случае для более полного разложения пробы нерастворимый остаток отфильтровывают, промывают, прокаливают при низкой температуре, снова сплавляют и полученный плав выщелачивают.
Для разложения вольфрамитов, содержащих олово, целесообразно тоцко измельченную пробу сплавлять при температуре ярко-красного каления с безводным сульфитом натрия в фарфоровом тигле *. Если плав выщелачивают кипящей водой, раствор разбавляют до 700 мл, затем подкисляют и вводят избыток кислоты, не превосходящий 20 мл 1 н. кислоты, то, по утверждению автора, выделяющийся при этом осадок олова совершенно свободен от вольфрама.
МЕТОДЫ ОТДЕЛЕНИЯ
Основной метод отделения вольфрама основан на выделении его в виде вольфрамовой кислоты нагреванием с кислотами, причем для более полного осаждения обычно прибавляют цинхонин (стр. 770). Осадок воль-
1 М. Т ravers, С, г., 165, 408 (1917).
766 Гл. XLIV. Вольфрам,
' фрамовой кислоты, как правило, загрязнен примесями, которые следует отделять, как указано ниже.
При определении малых количеств вольфрама наиболее целесообразно кремнекислоту отделять обычным путем, обезвоживанием с разбавленной соляной кислотой \ затем из фильтрата осаждать вольфрам цинхонином при нагревании (стр^ 772) и полученный осадок присоединять к нелетучему остатку после обработки кремнекислоты фтористоводородной и серной кислотами. Объединенные осадки сплавляют затем с возможно меньшим количеством карбоната натрия, плав растворяют в разбавленной соляной кислоте, полученный раствор обрабатывают цинхонином и затем нагревают не менее 10 ч. »
Вольфрам, находящийся в виде вольфрамата в щелочном или аммиачном растворе, содержащем значительные количества хлоридов щелочных металлов, можно количественно осадить следующим спрсобом г. Раствор разбавляют до 100—200 мл, нагревают до 50° С, прибавляют свежеприготовленный раствор 0,5 г таннина, затем подкисляют разбавленной соляной кислотой по лакмусу и вводят немного мацерированной бумаги. Энергично перемешав раствор, прибавляют по каплям 5 мл 5%-ного раствора цинхонина в разбавленной (1 : 3) соляной кислоте. Оставляют стоять на холоду в течение нескольких часов,7лучше на ночь, после чего раствор сливают декантацией через бум.ажный фильтр средней плотности (белая лента). Смывают осадок, попавший на фильтр, обратно в стакан и тщательно промывают фильтр 100 мл холодного 5 %-ного раствора хлорида аммония, содержащего немного цинхонина. Осадок перемешивают, снова переносят на фильтр и промывают. Жидкости дают стечь, осадок переносят во взвешенный фарфоровый тигель, осторожно нагревают до обугливания бумаги, а затем прокаливащт до WO3, как описано на стр. 774.
Окись вольфрама и кремнекислоту можно количественно разделить, отгоняя последнюю обработкой фтористоводородной кислотой или смесью фтористоводородной и серной кислот. После этого прокаливание следует проводить при температуре ниже 850° С .вследствие летучести WO3. Эта температура слишком низка для обезвоживания окиси кремния, поэтому в случае присутствия зйачительных количеств последней или когда требуется получить очень точные результаты, определение кремния следует проводить из'отдельной навески 3. В этом случае перед взвешиванием загрязненную кремнекислоту прокаливают, как обычно, при 1200° С, а затем после обработки фтористоводородной и серной кислотами, нелетучий остаток прокаливают при 750—850° С. Вольфрам можно количественно
1 Ло нашим данным, введение фосфорной кислоты для удержания вольфрама в растворе при дегидратации кремнекислоты не дает достаточно удовлетворительных результатов.
2 W. R. Schoeller, С. Jahn, Analyst, 52, 504 (1927). См. также D. A. Lambie, *там же, 64, 481 (1939). О замене цинхонина антипирином см. там же, 70, 124 (1945). * О замене цинхонина другими, более доступными реагентами см. также Р. Б. Голубцова, ЖАХ, 2, 118 (1948)*.
3 Согласно J. С i о с h i n a [Z. anal. Chem., 72, 429 (1928)], вольфрам можно легко отделить от кремния и олова следующим образом. Свежеосажденные кислоты этих элементов промывают водой до удаления минеральной кислоты и затем, закрыв трубку воронки, наполняют фильтр горячим 25%-ным раствором вольфрамата натрия. Через несколько минут трубку открывают, дают жидкости стечь и промывают фильтр последовательно горячей водой, соляной кислотой и затем снова горячей водой. По нашим данным, при этой обработке в раствор переходит также и некоторое количество кремнекислоты.
Методы, отделения
767
отогнать и отделить таким образом от кремния, прокаливая смесь окислов этих элементов при 700° С в токе чистого газообразного хлористого водорода \ При этом в болыцей или меньшей степени отгоняются также тантал, титан, цирконий и алюминий. Отгонка этих элементов имеет место также при прокаливании в смеси хлористого водорода и кислорода, в хлоре, а также в смеси хлороформа с кислородом.
Из кислых растворов, содержащих только вольфрам, сероводород выделяет лишь небольшой осадок, тогда как совместно с сульфидом молибдена и другими сульфидами осаждаются значительные количества вольфрама. Это соосаждение можно предупредить, прибавляя винную или щавелевую кислоты. Сульфиды щелочных металлов дают темно-коричневые растворы тиовольфрамата, если отсутствуют такие элементы, как марганец, совместно с которыми вольфрам частично осаждается. Препятствует ли винная кислота соосаждению вольфрама в этих условиях, как в случае осаждения сероводородом из кислых растворов, нам 'не известно. Подкисление раствора тиовольфрамата приводит к неполному выделению коричневого хлопьевидного сульфида WS3 при условии отсутствия тартратов и оксалатов. В присутствии же этих реагентов осаждение не происходит.
Из разбавленных кислых растворов вольфрам осаждается а-бензоин-оксимом, как описано в гл. «Молибден» (стр. 364), Чем пользуются для выделения вольфрама из раствора и для отделения его от многих элементов. В этих условиях вольфрам осаждается количественно, если преобладает содержание молибдена, но когда вольфрам находится один, он нередко частично остается в растворе.
Для отделения таких элементов, как молибден, сурьма, мышьяк и олово, выделенную загрязненную вольфрамовую кислоту или окись вольфрама растворяют в растворе едкой щелочи, прибавляют 2—5 г винной кислоты, щелочной раствор насыщают сероводородом, после чего вводят кислоту в небольшом избытке или же, наоборот, раствор сначала подкисляют, а затем осаждают сероводородом элементы мышьяковой группы. От олова и сурьмы вольфрам можно отделить также сплавлением окислов всех этих металлов с 12—15-кратным количеством по массе цианида калия в фарфоровом тигле и выщелачиванием плава водой. Выделяющиеся при этом в виде металлов олово и сурьму отфильтровывают. Фильтрат подкисляют, кипятят под хорошей тягой и затем осаждают вольфрам цинхонином.
Разделение вольфрама и молибдена можно осуществить также обработкой влажных свежеосажденных вольфрамовой и молибденовой кислот серной кислотой, как описано в гл. «Молибден» (стр. 358), или нагреванием прокаленных окислов этих металлов (или солей соответствующих кислот и щелочных металлов) в токе сухого хлористого водорода при 250— 270° С. В этих условиях молибден отгоняется 1 2 в виде МоОз-2НС1. Температуру следует тщательно контролировать и поддерживать в указанных пределах, так как в случае ее повышения до красного каления отгоняется
1F. Perillon, Bull. cos. ind. Minerale, 1 (1884); C. Friedheim, W. H. Henderson, A. P i n a g e 1, Z. anorg. Chem., 45, 396 (1905); D. H. В r o-phy, C. Van Brunt, Ind. Eng. Chem., 19, 107 (1927).
2 E. Pechard, C. r., 114, 173 (1892); P. J. К о s k e y, Chemist Analyst, 29, 53 (1940).
768
Гл. XLIV. Вольфрам
также и вольфрам Ч В растворе вольфрамата натрия и молибдата натрия до 0,25 г вольфрама можно отделить 1 2 от таких же количеств молибдена в виде W2O5 следующим образом. Раствор разбавляют до 60—300 мл, в зависимости от содержания вольфрама, нагревают до кипения, вводят по 20 мл раствора хлорида олова (II) (50 г SnCl-2H2O в 200 мл концентрированной НС1) на каждые 0,15 г WO3 и кипятят 5 мин. Окрашенную в оранжевый цвет отстоявшуюся жидкость сливают декантацией через фильтр, а синий осадок W2O6 промывают декантацией отдельными порциями горячей разбавленной (1 : 20) соляной кислоты до удаления молибдена и затем переносят на фильтр. Осадок можно высушить и прокалить до WO3.
Мышьяк и фосфор отделяют от вольфрама осаждением магнезиальной смесью из аммиачного раствора, содержащего тартрат аммония, как указано в гл. «Фосфор» (стр. 780). Вследствие растворимости арсената магния и аммония отфильтрованный осадок целесообразно не промывать, а для более полного удаления вольфрама растворять в кислоте и повторять осаждение. Вольфрам можно освободить от малых количеств мышьяка ’ добавлением достаточного для восстановления мышьяка количества сернистой или бромистоводородной кислоты, прибавлением затем избыточного количества соляной кислоты и кипячением раствора до получения небольшого объема.
Установлено 3, что вольфрам можно отделить от титана сплавлением смеси окислов с карбонатом натрия и выщелачиванием плава 10%-ным раствором едкого натра. Нерастворимый остаток отфильтровывают и промывают полунасыщенным раствором хлорида натрия. Фильтрат следует проверить на содержание титана. Вольфрам осаждают из раствора таннином и цинхонином. Цирконий отделяют от вольфрама сплавлением с карбонатами йщлочных металлов и выщелачиванием плава водой.
Для отделения вольфрама от титана, циркония, ниобия и тантала смесь окислов сплавляют с карбонатом калия, водный раствор плава обрабатывают слабоаммиачным раствором сульфата магния и хлорида аммония. Осадок, содержащий все эти элементы, за исключением вольфрама, отфильтровывают, а вольфрам определяют в фильтрате таннин-цинхонино-вым методом. Смесь пятиокисей ниобия и тантала можно выделить осаждением таннином после растворения магнезиального осадка в кислоте; осадок отфильтровывают и затем прокаливают. Остаток после прокаливания, как правило, еще содержит небольшие количества вольфрама, для отделения которых его снова сплавляют с карбонатом и повторяют весь процесс. Уран частично попадает в осадок от сульфата магния, а часть его, перешедшая в раствор, остается в фильтрате после осаждения вольфрама таннином и цинхонинном 4.
1 F. Р ё г i 11 о n, Bull. soc. ind. Minerale (1884). Для отделения очень малых количеств тория от значительных количеств вольфрама D. Н. Brophy и С. Van Brunt (цит. выше) рекомендуют отгонять вольфрам при 700° С в токе смеси равных частей сухого хлористого водорода и кислорода. Об отгонке В атмосфере СС14 -ф- СО2 см. Р. lannasch и сотрудники, Z. prakt. Chem., 97, 93, 141, 154 (1918).
2 Е. Е. Marbaker, J. Am. Chem. Soc., 37, 86 (1915).
3 A. R. Powell, W. R. Schoeller, C. Jahn, Analyst, 60, 506 (1935), Детально метод описан в этой статье и в монографии W. R. Schoeller, The Analytical Chemistry of Tantalum and Niobium, Chapman and Hall, London, 1937, p. 91.
J1 Подробности см. A. R. Powell, W. R. Schoeller, C. Jahn, цит, выше.
Метода определения
769
Вольфрам можно отделить от небольших количеств олова, ниобия и тантала обработкой свежеосажденной вольфрамовой кислоты аммиаком, взятым в небольшом избытке, при нагревании. Отфильтрованный нерастворимый остаток следует тщательно проверить на содержание вольфрама, так как некоторые элементы, главным образом железо \ удерживают значительные его количества. Этим методом можно отделить также мышьяк, ванадий и фосфор, если они содержатся в таких небольших количествах, которые могут быть захвачены осадком от аммиака. Об отделении вольфрама от больших количеств ниобия и тантала см. стр. 677.
Сплавление загрязненной прокаленной окиси вольфрама с карбонатом натрия и последующее выщелачивание плава водой не дает удовлетворительных результатов в присутствии олова, ниобия и тантала, так как эти элементы распределяются между нерастворимым остатком и водной вытяжкой. Хром при этой операции окисляется и переходит в раствор совместно с вольфрамом. Небольшие количества железа, титана и т. п. можно отделить от вольфрама осаждением едким натром.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наиболее удовлетворительный метод определения вольфрама в различных материалах заключается во взвешивании его в виде WO3, после выделения вольфрамовой кислоты обработкой соляной и азотной кислотами при нагревании и добавления под конец цинхонина. Осаждение вольфрама в виде вольфрамата ртути Hg2WO4 и последующее прокаливание до окиси 1 2 является количественным методом, но при этом мешает такое большое число веществ, в частности ванадаты, хроматы, молибдаты, что им целесообразнее пользоваться для группового разделения, что это описано в гл. «Ванадий» (стр. 510).
Алкалиметрический метод, основанный на растворении вольфрамовой кислоты в избытке титрованного раствора едкой щелочи, который обратно титруют кислотой, дает менее точные результаты, чем весовой метод, даже при условии, что вольфрамовая кислота полностью очищена от примесей, а это редко выполнимо.
По нашим данным, методы, основанные на восстановлении вольфрама и последующем титровании перманганатом 3, мало надежны, так как трудно, если это вообще возможно, восстановить вольфрам до определенной валентности и полностью отделить его от таких элементов, как молибден и железо, которые также восстанавливаются 4 в условиях восстановления вольфрама.
1 Незначительные количества вольфрама можно выделить осаждением аммиаком после добавления избыточного количества соли железа (III).
2 J. J. Berzelius, Schweigger’s J., 16, 476 (1816); W. Gibbs, Am.
Chem. J. 1, 219 (1879); H. W. H u t chin, Analyst, 36, 398 (1911).
3 0. F r i e h, v. d. P f о r t e n, Ber., 16, 508 (1883); см. также E, Knecht,
E. Hibbert. Proc. Chem. Soc., 25, 227 (1909). M. L. Holt, A. G. Gray
[Ind. Eng. Chem., Anal. Ed., 12, 144 (1940)] установили, что вольфрам можно восстановить до трехвалентного состояния жидкой свинцовой амальгамой, затем окислить сульфатом железа (III), а образующееся при этом железо (III) титровать раствором бихромата калия, применяя дифениламин в качестве индикатора. Другие легко восстанавливающиеся элементы, как ванадий и молибден, естественно, должны отсутствовать.
4 Приготовление K3W2C19 восстановлением оловом в концентрированном солянокислом растворе и его применение в качестве восстановителя см. R. U z е 1, R. Р г i-b i 1, Coll. Czech. Chem. Comm., 10, 7, 330 (1938),
. 49 Заказ 522.
770
Гл. XLIV. Вольфрам
*.Ю. А Чернихов и В. Г. Горюшина 1 рекомендуют редуктометриче-ское определение вольфрама, при котором титрование проводится солью хрома (II). Авторы отмечают, что в солянокислой среде хром (II) восстанавливает вольфрам (VI) до вольфрама (V), и при потенциометрическом контроле процесса удается получить отчетливый скачок потенциала в точке эквивалентности. Доп. перев.*
Определение обработкой кислотами с добавлением цинхонина
Определение вольфрама .в чистых продуктах является относительно несложной задачей и может быть выполнено обработкой одними кислотами. В случае же анализа сложных материалов выделение вольфрамовой кислоты одними кислотами недостаточно надежно и в конце операции следует прибавлять цинхонин, который образует нерастворимое соединение с вольфрамом 2. Осаждению вольфрама обработкой кислотами при нагревании меШают натрий, калий, аммонийные соли, фосфор 3, молибден, мышьяк, фтор и органические вещества, как, например, винная кислота. Эти элементы замедляют также в известной мере осаждение цинхонином.
Осажденная вольфрамовая кислота обычно содержит ряд посторонних примесей. Из наиболее часто встречающихся примесей кремнекислота, олово, сурьма, ниобий, тантал • выделяются из раствора при обработке, которая применяется для осаждения вольфрамовой кислоты. Фосфор, молибден и ванадий соосаждаются, образуя нерастворимые соединения с вольфрамовой кислотой. И, наконец, такие элементы, как железо и хром, удерживаются вольфрамовой кислотой при однократном осаждении. Многие из этих примесей удаляются способами, рассмотренными ниже.
Влияние некоторых веществ приведено в табл. 25. Приведенные данные получены при обработке навесок ферберита по 1 г, к которым добавлялись указанные в таблице примеси. Определение вольфрама проводилось обработкой кислотами с добавлением цинхонина и прокаливанием выделенной загрязненной вольфрамовой кислоты.
Довольно трудной задачей является надлежащее прокаливание вольфрамовой кислоты, так как для ее полного обезвоживания требуется температура 750° С, а при 800° С начинается возгонка вольфрамового ангидрида. Скорость улетучивания вольфрамового ангидрида при температурах ниже 900° С, однако, невелика, и поэтому область 750—850° С оказывается вполне безопасной.
Чистый прокаленный WOs не гигроскопичен.
Ход определения. Пробу руды или минерала растирают в агатовой ступке не менее чем до 200 мещ (d — 0,074 мм). Навеску в 1 г помещают в стакан емкостью 400 мл, прибавляют 5 мл воды и слегка вращают стакан, чтобы равномерно распределить порошок по его дну. Затем прибавляют 100 мл соляной кислоты, покрывают стакан часовым стеклом и на-
1Ю. А. Чернихов, В. Г. Горюшина, Зав. лаб., И, 137 (1945); 12, 397 (1946). См. также R. Platt F. Sommer, Helv. Chim. Acta, 27, 1518 (1944).
2 Так, например, при анализе шеелита было найдено 58,4% WO3 при обработке только кислотой и 59,6% при добавлении цинхонина, а в ферберите в тех же условиях было определено соответственно 62,1% и 66,0% WO3.
3 См. S. G. Simpson, W. С. S chu m b, М. A. S i е m i n s k i, Ind. Eng, Chem., Anal. Ed., 10, 243 (1938).
Методы, определения
771
Влияние некоторых веществ на определение вольфрама
ТАБЛИЦА 25
Введенные примеси Содержание wo> % Получено загрязненной WO, % Примечания
1% р 10% р 10% Р 3% Р 5% As 25% NH4C1 25% NaCl 25% КС1 5% Mo 5% Mo 25% V 5% Bi 25% Cu 50% Pb 8,7% Mn 66,0 66,0 66.0 66,0 66,0 66,0 66,0 66,0 66,0 66,0 66,0 66,0 66,0 66,0 73,1 66,2 1 66,5 f 58,0 65,8 ' 65,9 65,8 65,6 65,7 66,8 65,7 65,7 66,0 65,8 65,7 73,0 WO3 содержит железо и фосфор При первом осаждении цинхонин не вводился Фосфор отделялся магнезиальной смесью после первого осаждения При первом осаждении цинхонин не вводился. Осадок содержал молибден
гревают 1 ч при температуре не выше 60° С, время от времени перемешивая, чтобы предотвратить образование комков на дне стакана. После этого поднимают часовое стекло, помещают его на согнутые под углом стеклянные палочки, повышают температуру и кипятят раствор, пока объем его не уменьшится до 50 мл. Перемешивают осевшую на дне стакана массу так, чтобы все собравшиеся в комки частицы разошлись, и затем прибавляют 40 мл соляной и 15 мл азотной кислоты.
Стакан покрывают часовым стеклом, которое снимают, когда исчезает опасность разбрызгивания, и продолжают кипятить, пока объем раствора не уменьшится приблизительно до 50 мл. Вводят 5 мл азотной кислоты, разбивают слежавшуюся массу и кипятят, пока объем раствора не станет равным 10—15 мл. Разбавляют 150 мл горячей воды, тщательно перемешивают и слабо кипятят раствор 30 мин. После этого вводят 5 мл раствора цинхонина 1 и нагревают на горячей плитке или водяной бане при температуре, близкой к кипению, не менее 30 мин.
Отстоявшуюся прозрачную жидкость сливают декантацией через фильтр диаметром 9 см, содержащий немного беззольной бумажной массы, и промывают осадок 3—4 раза декантацией горячей цинхониновой промывной жидкостью 2. Затем насколько возможно полнее переносят осадок
1 Приготовляют растворением 125 г цинхонина в смеси 500 мл соляной кислоты и 500 мл воды. *После введения в практику колориметрического метода определения вольфрама было неоднократно доказано, что при анализе природных вольфраматов кислотным разложением добавление органических осадителей излишне, так как после выделения вольфрамовой кислоты гидролизом в растворе обнаруживаются лишь ничтожные количества вольфрама, которые не имеют практического значения [Ю. А. Чернихов, В. Г. Горюшина, Зав. лаб., 12, 517 (1946)]. *
2 Приготовляют разбавлением 25 мл раствора цинхонина и 30 мл соляной кислоты до 1 л.
49*
П2
Гл. XLIV. Вольфрам
на фильтр, после чего тщательно ополаскивают стакан и промывают осадок на фильтре. К фильтрату и промывным водам (раствор А) прибавляют раствор цинхонина, хорошо перемешивают и оставляют стоять, чтобы удостовериться в полноте осаждения вольфрама. Промытый осадок струей воды переносят обратно в стакан, употребив для этого не более 25 мл воды. Вводят 6 мл раствора аммиака (достаточное количество, чтобы был небольшой избыток), покрывают стакан часовым стеклом и слабо нагревают несколько минут. Ополаскивают стенки стакана горячим разбавленным (1 : 9) раствором аммиака, содержащим 10 г хлорида аммония в 1 л. Раствор тщательно перемешивают и фильтруют через тот же фильтр, собирая фильтрат в стакан емкостью 400 мл. Ополаскивают стакан, в котором находился раствор, и промывают нерастворившийся остаток на фильтре указанной выше горячей аммиачной промывной жидкостью. Остаток (остаток Б) сохраняют.
Раствор упаривают или слабо кипятят до удаления большей части аммиака, затем прибавляют 20 лм соляной и 10 мл азотной кислоты и продолжают кипятить, пока объем раствора не уменьшится до 10—15 мл. После этого разбавляют до 150 мл горячей водой, прибавляют 10 лл раствора цинхонина, тщательно перемешивают и нагревают при 80—90° С не менее 30 мин. По охлаждении вводят мацерированную бумагу, полностью переносят осадок на фильтр диаметром 9 см и тщательно промывают горячей цинхониновой промывной жидкостью. Этот осадок основной массы вольфрама прокаливают совместно с дополнительными количествами вольфрама, выделенными, как будет указано далее.
Фильтрат и промывные воды (раствор В) тщательно перемешивают и затем оставляют стоять, чтобы удостовериться в полноте осаждения вольфрама.
Если анализируемая проба разложена полностью, небольшие количества вольфрама, задержавшиеся в сохраненном остатке (Б), обычно связаны с железом или алюминием и могут быть переведены в раствор обработкой остатка горячей разбавленной (1 : 9) соляной кислотой. Нераство-ряющийся при этом остаток отфильтровывают и промывают сначала небольшим количеством горячего 0,5%-ного раствора хлорида аммония, а затем аммиачной промывной жидкостью. Из раствора вольфрам осаждают обработкой кислотой и цинхонином (дополнительный осадок 1) так же, как из прокипяченной аммиачной вытяжки, с которой этот раствор не следует объединять.
Остающийся после обработки разбавленной кислотой и аммиаком остаток (остаток Г) обычно свободен от вольфрама. В нем могут находиться кремнекислота или неразложенные силикаты, касситерит и минералы, содержащие ниобий и тантал Этот остаток можно исследовать, как описано в гл. «Ниобий и тантал» (стр. 678), или следующим образом. Остаток (Г) вместе с фильтром, на котором он' находится, прокаливают при невысокой температуре в фарфоровом тигле (вследствие возможного содержания в нем олова), затем переносят в платиновый тигель и отгоняют кремний выпариванием с фтористоводородной и серной кислотами. Нелетучий остаток сплавляют с возможно меньшим количеством карбоната натрия и по охлаждении выщелачивают плав водой. Нерастворимый остаток от
1 Если раствор не содержит других элементов, осаждаемых цинхонином, помимо жжобия и тантала, то осаждение последних происходит очень медленно.
Методы, определения
773
фильтровывают. Фильтрат подкисляют соляной кислотой, кипятят до удаления двуокиси углерода и испытывают на содержание вольфрама, для чего прибавляют 5 мл раствора цинхонина, нагревают на краю водяной бани 30 мин, а затем оставляют при комнатной температуре в продолжение нескольких часов, лучше на ночь. Если в этом растворе, в двух сохраненных ранее фильтратах и промывных водах (растворы А и В), появляются осадки, их отфильтровывают через один и тот же фильтр и промывают цинхониновой промывной жидкостью. После этого выщелачивают аммиаком, осаждают вольфрам из раствора (дополнительный осадок 2) и промывают, как при получении предыдущего осадка.
Фильтры, содержащие основной и дополнительные осадки (1 и 2), помещают в большой взвешенный платиновый тигель и нагревают при невысокой температуре до полного сгорания углерода. По охлаждении смачивают остаток небольшим количеством фтористоводородной кислоты, выпаривают досуха для удаления кремния и затем прокаливают при 750— 850° С. Охлаждают в эксикаторе и взвешивают загрязненную WOs. Прокаливание повторяют до постоянной массы.
Полученную таким образом вольфрамовую кислоту нужно исследовать на содержание в ней таких примесей, как молибден, серебро, железо и фосфор.
Молибден увлекается из раствора вольфрамовой кислотой независимо от того, проводится осаждение обработкой кислотами или цинхонином. Когда цинхонин не вводят, осадок в меньшей степени загрязняется молибденом, но в этом случае вольфрам осаждается не количественно. Так, например, при анализе 1 г ферберита, содержавшего 65,8% WO3 и 5% МоО3, во взвешенном остатке было найдено 0,8% МоО3, когда при осаждении вольфрамовой кислоты добавлялся цинхонин, и 0,36% МоО3, когда цинхонин не вводился. В первом случае вольфрам выделился из раствора количественно, тогда как без добавления цинхонина в растворе осталось 0,4% WO3. При анализе 1 г ферровольфрама, содержавшего 75,2% W и 0,24% Мо, взвешенная окись вольфрама содержала 0,002 г МоО3, что составляет приблизительно половину всего молибдена. Вольфрамовую кислоту нельзя освободить от молибдена прокаливанием при температуре, при которой не происходила бы одновременно возгонка и вольфрама.
Молибден замедляет осаждение вольфрама цинхонином и в тех случаях, когда присутствуют значительные количества молибдена, как, например, в молибденововольфрамовых инструментальных сталях, чтобы достигнуть количественного выделения вольфрама, раствор следует фильтровать спустя 30—40 ч после начала осаждения.
Для этого исследования вольфрамовую кислоту сплавляют с возможно меньшим количеством карбоната натрия, плав выщелачивают водой, нерастворимый остаток отфильтровывают и промывают сначала горячим 1 %-ным раствором карбоната натрия, а затем горячей водой. Фильтрат сохраняют. Фильтр с остатком прокаливают и повторяют операцию. Фильтраты объединяют и отставляют. Тщательно промытый остаток прокаливают, охлаждают, взвешивают и его массу вычитают из ранее найденной массы вольфрамового ангидрида. В редких случаях, когда присутствует серебро, первый нерастворимый карбонатный остаток обрабатывают аммиаком при нагревацди, остающийся при этом остаток отфильтровывают, промывают, а затем прокаливают и снова сплавляют с карбонатом натрия. Аммиачный фильтрат обрабатывают сульфидом аммония, и если при этом образуется осадор, его прокаливают совместно с осадком, который выделяют из объединенных водных вытяжек карбонатных плавов.
Эти водные вытяжки подкисляют, кипятят до удаления двуокиси углерода и затем прибавляют избыточное количество аммиака. При этом раствор обычно остается прозрачным, но если выделяется осадок, его отфильт
774
Гл. XLIV. Вольфрам
ровывают, промывают, прокаливают и массу его вычитают из массы вольфрамового ангидрида. В прозрачный аммиачный раствор вводят 3—5 г винной кислоты, насыщают сероводородом, прибавляют серную кислоту в таком количестве, чтобы остался избыток в 1 % по объему, нагревают 1 ч при 40—60° С и затем фильтруют. Осадок промывают сероводородной водой, содержащей 5 г винной кислоты и 5 мл серной кислоты в 1 л. Фильтрат сохраняют, если предполагают провести определение фосфора и ванадия. Выделенные сульфиды могут содержать еще некоторые количества вольфрама, поэтому осадок растворяют в горячей разбавленной азотной кислоте, содержащей немного брома, избыток которого удаляют кипячением, прибавляют 1—2 а винной кислоты, делают раствор аммиачным, снова обрабатывают сероводородом и в дальнейшем поступают, как прежде. Осадок очень осторожно прокаливают при температуре не выше 600° С, по охлаждении взвешивают и полученную величину вычитают из массы вольфрамового ангидрида *.
* Колориметрический метод
Реакция вольфрама с роданидом и хлоридом олова (II), предложенная 1 2 для определения налета вольфрама на стенках колб электрических ламп, впервые была применена к анализу вольфрамовых руд Ю. А. Черни-ховым и Л. Аппельбаумом 3. Этот метод особенно ценен тем, что при его применении практически не требуется предварительного отделения вольфрама от сопутствующих ему элементов. Предполагалось, что определению вольфрама мешает молибден, однако многолетний опыт применения роданидного метода показывает, что благодаря малой устойчивости ро-дано-молибденового комплекса в сильнокислой среде даже относительно большие количества молибдена практически не оказывают влияния на колориметрическое определение вольфрама 4. Это подтверждается также данными ®, согласно которым в условиях колорйметрического определения вольфрама интенсивность окраски 1 мг молибдена соответствует интенсивности окраски 0,01 мг вольфрама.
Одним из элементов, мешающих определению вольфрама, является мышьяк. Его влияние объясняется тем, что в условиях колориметрирования возможно частичное' восстановление родинид-ионов до сероводорода, в связи с чем выделяется осадок сульфида мышьяка, который адсорбирует некоторые количества вольфрама. Мышьяк можно удалить предварительным обжигом анализируемого материала, сначала восстанови
1 Применение анти-i ,5-ди(п-метоксифенил)-1-гидроксиламино-3-оксиминопен-тена-4 для осаждения вольфрамат-ионов из 0,2 н. солянокислого раствора описано в статье J. Н. Yoe, A. L. Jones, Ind. Eng. Chem., Anal. Ed., 16, 45 (1944).
Колориметрический метод определения умеренных количеств вольфрама в сила- вах, основанный на появлении окраски при действии гидрохинона на сильно сернокислый раствор, содержащий вольфрам, см. G. Bogatski [Z. Anal. Chem., 114, 170 (1938)]. Титан и ниобий дают аналогичные реакции [см. С. W. Johnson, Iron Age, 157, № 14, 66 (1944)].
2 F. F e i g 1, P. Kru.mholz, Z. angew. Chem., 45, 674 (1932).
3 См. в книге В. Ф. Гиллебранд и Г. Э. Лендель, Практическое руководство по неорганическому анализу, ОНТИ, 1935, стр. 598. См. также Б е р л ь— Л у н г е. Химико-технические методы исследования, т. II, ч. 2, вып. 2, ГОНТИ, 1938, стр. 534.
4 Ю. А. Чернихов, В. Г. Горюшина, Зав. лаб., 12, 517 (1946).
6 F. S. Grimaldi, V. North, Ind. Eng. Chem., Anal. Ed., 15, 652 (1943).
Методы определения
775
тельным, а затем окислительным, или же методом \ основанным на восстановлении мышьяка гидразином в солянокислом растворе и отгонке его в виде AsGls.
Разработанный Черниховым и Аппельбаумом метод заключается в следующем. Навеску 0,3—0,5 а руды, в зависимости от содержания вольфрама, сплавляют с 2 г едкого натра в никелевом тигле. Плав выщелачивают водой, переносят в стакан, прибавляют несколько капель перекиси водорода для осаждения марганца и нагревают до полного разложения перекиси. Раствор вместе с осадком переносят в мерную колбу емкостью 100 мл, разбавляют до метки водой и дают отстояться. В 100 мл раствора должно содержаться не более 0,003 г вольфрама, в противном случае разбавляют до большего объема, увеличив соответственно и количество едкого натра с таким расчетом, чтобы концентрация его была 0,5 н. (около 2%).
Для колориметрирования отбирают 20 мл остоявшегося прозрачного раствора (жидкость должна быть бесцветной) в мерную колбу емкостью 50 мл, прибавляют 2,5 мл раствора роданида калия (25 г соли в 100 мл раствора) и разбавляют до метки раствором хлорида олова (II) (10 г SnCl2-•2НгО растворяют в 100 мл соляной кислоты пл. 1,19 г/сж3). В другую мерную колбу емкостью 50 мл вводят 2 мл стандартного раствора вольфрамата натрия, содержащего 1 мг вольфрама в 1 мл, разбавляют до 20 мл 2 %-ным раствором едкого натра, прибавляют 2,5 мл раствора роданида калия и разбавляют до метки раствором хлорида олова (II). Интенсивность окрасок анализируемого и стандартного растворов не должна различаться более чем в 2—3 раза, в противном случае в раствор для сравнения вводят соответственно другие количества раствора вольфрамата натрия. Спустя 45—60 мин окраски сравнивают в колориметре Дюбоска.
Сравнение окрасок можно провести также методом стандартных серий. Для этого анализируемый раствор и шкалу стандартов одновременно приготовляют в цилиндрах Эггерца, сохранив в растворах указанные выше концентрации роданида, хлорида олрва (II) и соляной кислоты.
Описано также фотоколориметрическое определение вольфрама этим методом. Измерение светопоглощения родано-вольфрамового комплекса проводится при 420 ммк. Для вычисления концентрации вольфрама пользуются калибровочной кривой 1 2 3.
Чувствительность метода можно значительно повысить экстрагированием окрашенного родано-вольфрамового соединения органическими растворителями, например эфиром, изоа^иловым спиртом, амилацетатом 3, которое проводится следующим образом.
Отбирают аликвотную часть раствора, содержащую не более 0,1 мг вольфрама, нейтрализуют, вводят 2 мл 10 %-ного раствора едкого натра и разбавляют приблизительно до 15 мл. Приготовляют стандартные растворы, содержащие такие же количества тех же реактивов, что и анализируемый раствор. Вводят в анализируемый раствор и одновременно в стандартные растворы по 1 мл 10%-ного раствора роданида калия и по 10 мл 5%-ного раствора хлорида олова (II) в концентрированной соляной кислоте. Разбавляют все растворы приблизительно до 30 мл и оставляют стоять при 25° С в продолжение 1 ч. По охлаждении до 20° С или ниже
1 П. Ф. Фалеев, Зав. лаб., 5, 1174 (1939).
2 К. И. Попов, М. Н. Дорохова, Зав. лаб., 9,1315 (1940); А. С. Шахов, Зав. лаб., 10, 470 (1941).
3 Е. В. S a n d е 11, Ind. Eng. Chem., Arial. Ed., 18, 163 (1946).
776
Гл. XLIV. Вольфрам
экстрагируют каждый раствор 5 мл эфира, встряхивая 5 сек. Сравнивают окраски эфирных экстрактов.
Реакция образования окрашенного родано-вольфрамого комплекса значительно ускоряется, если вместо хлорида олова (II) в качестве восстановителя применяют хлорид титана1 (III) или цинковую пыль2.
Рекомендуется 3 при применении в качестве восстановителя хлорида олова (II) для ускорения образования окрашенного роданидного комплекса вольфрама и стабилизации окраски вводить спирт и кратковременно нагревать раствор.
Необходимо отметить, что роданидный метод определения вольфрама приобрел большое значение и по существу является единственным, который широко применяется на практике для определения малых количеств вольфрама. Помимо приведенных, имеется еще целый ряд работ, посвященных этому методу 4. Доп. перев.*
1Ф. А. Ферьянчич, Зав. лаб., 3, 301 (1934); Z. Anal. Chem., 97, 332 (1934); Зав лаб., 6, 289 (1937).
2 О. А. С о н г и н а, П. А. Карпова, Зав. лаб., 10, 92 (1942).
3 Н. С. Полуэктов, Зав. лаб., 10, 92 (1941).
4 См., например, Ф. А. Ферьянчич, Д. Н. Иорданский, Зав. лаб., 7,866 (1938); Т. А. Вознесенский, Зав. лаб., 9, 25 (1940); G. S. Smith,. Ind. Chem., 21, 250 (1945); Я. Д. Ф р'и д м а н, О. А. С о н г и н а, Труды химического института Киргизского филиала АН СССР, вып. 1 (1946); О. А. С о н г и н а, М. Т. Козловский, Зав. лаб., 13, 677 (1947); Ю. А. Чернихов, Р. С. Т р а м м, Зав. лаб., 15, 15 (1949); Н. F г е u n d, М. L. W и g h t, R. К. В г о-о k s h i е г, Anal. Chem., 23, 781 (1951) и др.
Глава XLV
ФОСФОР
Фосфор больше всего распространен в более основных изверженных и метаморфических породах и практически всегда в них содержится, хотя бы в следах. Он чаще встречается в породах, богатых известью и железом, чем в магнезиальных породах. За одним или двумя редкими исключениями, фосфор в природных неорганических соединениях находится в виде фосфатов, число которых очень велико. Он распространен главным образом в виде минерала апатита Ga6 (F, G1, ОН)(РО1)3, но находится также в минералах ксенотиме (Y, Er, Ge)POi имонаците (Се, La, Nd, Pr)POi + Th3(POi)4. В метеоритах встречается фосфид железа х. Фосфор является также важной составной частью живых организмев. Он присутствует в многочисленных рудах и продуктах металлургического производства. Методы точного определения фосфора имеют исключительно' важное значение.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа горных пород кремнекислота, выделенная обезвоживанием кислотами, может быть загрязнена фосфатами. Это происходит в том случае, если анализируемая проба наряду с фосфором содержит титан, цирконий, торий или олово. Часть осажденного таким образом фосфора теряется и принимается за кремнекислоту, если отгонка кремния с фтористоводородной кислотой проводится в присутствии больших количеств серной кислоты и при высокой температуре 1 2. Оставшуюся часть фосфора можно снова перевести в осадок, для чего нелетучий остаток, после обработки кремнекислоты фтористоводородной и серной кислотами, сплавляют с карбонатом натрия и плав обрабатывают кислотой.
Целесообразнее, однако, отделить фосфор выщелачиванием плава водой, а нерастворимый остаток растворить в кислоте и раствор присоединить к фильтрату от кремнекислоты. В водной вытяжке карбонатного плава, помимо фосфора, могут находиться также и другие элементы, как, например, олово, вольфрам, ниобий, и поэтому ее следует тщательно исследовать. При обработке фильтрата от кремнекислоты аммиаком находящийся в растворе фосфор осаждается совместно с железом, алюминием и титаном. Когда содержание фосфора преобладает, он может вызвать также
1 F. W. Clarke, U.S. Geol. Survey Bull., 770, 18 (1924).
2 W. F. Hillebrand, G. E. F. Lundell, J. Am. Chem., Soc., 42., 2609 (1920).
778
Гл. XLV. Фосфор
осаждение и щелочноземельных металлов, в исключительных случаях даже количественное. Если влияние фосфора не учитывается, то результаты определения алюминия могут получиться ошибочными. Если же при осаждении аммиаком происходит соосаждение щелочноземельных металлов, то результаты определения последних в фильтрате после осаждения аммиаком будут также неправильными.
Потери фосфора вызываются главным образом небрежной или неправильной обработкой получаемых в ходе анализа нерастворимых соединений, которые часто содержат фосфор, как, например, кремнекислота, метаоловянная кислота, образующаяся при обработке азотной кислотой, и вольфрамовая кислота, выделяющаяся при кипячении с кислотами. Очень часто небольшие количества фосфатов остаются на фильтрах, которые кажутся на вид чистыми, и отбрасываются. Когда фосфоромолибдат аммония осаждают из растворов, содержащих титан, цирконий или олово, то легко потерять 0,1—1 мг фосфора в виде не заметного для глаза фосфата, остающегося на фильтре после выщелачивания осадка фосфоромолибдата аммиаком. Это же может происходить при растворении первого осадка MgNHiPOi в кислоте.
РАЗЛОЖЕНИЕ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ФОСФОР
При применении любого метода для определения фосфора в минералах, рудах или продуктах металлургического производства требуется, чтобы он находился в виде ортофосфата. Мета- или пирофосфаты должны быть превращены в ортофосфаты сплавлением со щелочами или нагреванием с сильной кислотой. За исключением некоторых фосфидов, находящихся в метеоритах, и одного сомнительного пирофосфата, все известные соединения, в виде которых фосфор встречается в минералах, являются ортофосфатами.
При разложении металлургических продуктов обычно принятыми методами также получаются ортофосфаты. Аналитику, таким образом, не приходится иметь дело с другими кислотами фосфора, за исключением тех случаев, когда пиро- или метафосфаты получаются в результате производимых им самим аналитических операций, как, например, при прокаливании одно- или двузамещенных ортофосфатов, или продолжительном сдла-влении их с пиросульфатами щелочных металлов при высокой температуре.
Примеров, когда анализируемый материал растворим в воде, очень немного, и они практически ограничиваются химическими реактивами. Применение соляной или серной кислоты в качестве растворителя нежелательно, за исключением тех случаев, когда непосредственно в полученном растворе проводится осаждение магнезиальной смесью или когда введение этих кислот необходимо для растворения анализируемой пробы, но при этом следует прибавлять возможно меньшее количество кислоты. Для переведения в раствор металлургических продуктов необходимо применять сильную окислительную обработку, в противном случае фосфор может улетучиться в виде фосфина РНз. В таких случаях лучше всего пользоваться одной азотной кислотой. Если приходится добавлять фтористоводородную кислоту, то ее затем удаляют (когда ее немного) или связывают борной кислотой.
Методы отделения
779
Минералы, нерастворимые в кислотах, или остатки, получаемые в процессе анализа, обычно сплавляют с карбонатом натрия или перекисью натрия (иногда после предварительного добавления кремнекислоты). Плав выщелачивают водой, нерастворимый остаток отфильтровывают и промывают 1 %-ным раствором карбоната натрия. При содержании больших количеств фосфора эту операцию обычно повторяют.
Соединения, содержащие фосфор, не должны подвергаться продолжительному сплавлению с пиросульфатом в открытых тиглях и при излишне высокой температуре. Не следует их также нагревать с серной кислотой при высокой температуре или до полного удаления серной кислоты *. При выпаривании с серной кислотой при низкой температуре до момента появления паров серной кислоты потерь фосфора не наблюдается, так же как и при выпаривании досуха растворов, содержащих азотную, соляную или фтористоводородную кислоту. При кратковременном сплавлении с пиросульфатом в плотно закрытом тигле, который нагревается лишь до такой степени, какая необходима для надлежащего сплавления, потери фосфора настолько незначительны, что Ими можно пренебречь. В процессе такого сплавления, однако, могут образоваться мета- или пирофосфаты, которые затем должны быть превращены в ортофосфаты кипячением с разбавленной (1 : 9) серной кислотой по меньшей мере в продолжение 1 2 ч. -
Как правило, смесь соединений фосфора с органическими веществами нельзя прокаливать в платиновой посуде, если не обеспечены хорошие окислительные условия.
МЕТОДЫ ОТДЕЛЕНИЯ
Основной метод отделения фосфора от других элементов заключается в осаждении его в виде фосфоромолибдата аммония из азотнокислого раствора, как описано в разделе «Осаждение фосфора в виде фосфоромолибдата аммония и обработка полученного осадка» (стр. 782). Метод имеет почти универсальное применение и пригоден также для удаления фосфора из раствора, когда его присутствие мешает.
Очень малые количества фосфора в присутствии больших количеств меди, никеля и хрома (VI) лучше выделять аммиаком, взятым в небольшом избытке, после введения в раствор 0,1—0,2 г алюминия или железа в виде какой-либо подходящей соли. Осадок отфильтровывают, растворяют в разбавленной азотной кислоте и затем осаждают фосфор из раствора молибдатом 2. Малые количества фосфора можно выделить также из раствора вместе с цирконием следующим образом. К солянокислому раствору ортофосфата прибавляют избыточное количество хлорида Циркония, выпаривают досуха, растворяют остаток в 30 мл соляной кислоты и 10 мл бромистоводородной кислоты, а затем кипятят для удаления мышьяка. После этого выпаривают досуха, прокаливают, обрабатывают остаток разбавленной (1:1) соляной кислотой, выпаривают до объема 20 мл и разбавляют 500 мл горячей воды. Дают постоять при 50° С и затем
1 W. F. Hillebrand, G. Е. F. L u n d е 11, цит. выше.
2 Метод извлечения фосфора нагреванием с оловом и азотной кислотой, сплавлением прокаленного осадка оловянной кислоты, содержащего фосфор, с цианидом калия и выщелачиванием плава водой не имеет в настоящее время практического значения, так как фосфор таким образом редко выделяется полностью, даже если первоначальное соотношение между оловом и фосфором равно 20 : 1.
780
Гл. XLV. Фосфор
фильтруют. Остаток прокаливают и извлекают из него фосфор сплавлением с карбонатом натрия и выщелачиванием плава водой.
Этот же метод может служить для извлечения части или всего фосфора из растворов, содержащих большие его количества. В первом случае раствор, в котором находится 10—20 мл соляной или серной кислоты, нагревают приблизительно до 50° С, затем медленно вводят раствор хлорида циркония до образования осадка. Дают отстояться и фильтруют. Для полного выделения фосфора к фильтрату добавляют небольшой избыток раствора хлорида циркония, нагревают до кипения и затем медленно при перемешивании вводят небольшой избыток аммиака. Прибавляют бумажную массу и фильтруют. В фильтрате определяют щелочноземельные металлы, магний и щелочные металлы.
Фосфор легко отделяется от железа, никеля, кобальта, титана, циркония и хрома (III) осаждением этих элементов (если требуется — двукратным) едким натром (стр. 108). Тот же результат достигается сплавлением с карбонатами щелочных металлов или с перекисью натрия, выщелачиванием плава водой и фильтрованием. При применении этих методов фосфор переходит в раствор совместно с ванадием и мышьяком, для отделения от которых требуется дальнейшая обработка, как будет описано ниже.
Отделение железа, кобальта, цинка и т. п. от фосфора осаждением сульфидом аммония (стр. 89) имеет ограниченное применение, так как при этом должны отсутствовать щелочноземельные металлы и магний. Цирконий, титан и торий не осаждаются, однако, из растворов фосфатов, содержащих сульфид аммония и виннокислый аммоний.
Элементы сероводородной группы, за исключением, быть может, молибдена, отделяются осаждением сероводородом из кислого раствора, как указано на стр. 83. Разделение молибдена и фосфора лучше всего осуществлять подкислением раствора, содержащего эти элементы и сульфид аммония, или повторным осаждением фосфора магнезиальной смесью, как описано в разделе «Осаждение фосфора в виде фосфоромолибдата аммония и обработка полученного осадка» (стр. 782).
Мышьяк можно также отделить от фосфора восстановлением до трехвалентного состояния и последующим кипячением либо с соляной, либо с фтористоводородной кислотой. Этот метод служит также и для удаления германия (IV). Вполне удовлетворительное отделение фосфора от вольфрама, ванадия (V) и молибдена достигается осаждением его магнезиальной смесью из ледяного аммиачного раствора, содержащего винную кислоту.
В этом случае прибавляют винной кислоты не более, чем это требуется до того, чтобы удержать основания в растворе, затем нагревают до 50q С, прибавляют в небольшом избытке аммиак и 10—25-кратный избыток магнезиальной смеси. Вводят стеклянные бусы и по охлаждении, предварительно сильно взболтав, прибавляют по 10 мл раствора аммиака на каждые 100 мл раствора. Снова охлаждают, сильно взбалтывают и оставляют стоять на холоду не менее 12 ч.
Полностью отделить фосфор таким путем несколько затруднительно, что подтверждается результатами определения фосфора в смеси Na2WO4 и (NH4)SHPO4, содержавшей 0,2 г W и приблизительно 0,04 г Р, в которой двукратным осаждением было получено 0,1589 г и 0,1587 г MgzPjO, вместо 0,1579 г.
Из числа других методов отделения следует упомянуть осаждение железа, меди, никеля, хрома и т. п. электролизом с ртутным катодом (стр. 165), осаждение купфероном в сернокислом или солянокислом растворе (стр. 143) и извлечение эфиром (стр. 161).
Методы определения
781
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Фосфор почти всегда определяют из отдельной навески и практически для всех методов определения требуется предварительное выделение его в виде фосфоромолибдата аммония, идеальный состав которого соответствует формуле 1 (NH4)3PO4-12MoO3-2HNO3-H2O. В обычных условиях определения фосфора, однако, трудно получить осадок такого состава. Для последующей обработки осадка фосфоромолибдата аммония предложены многочисленные методы, которые можно разделить на две группы. К одной из них можно отнести методы, для которых требуется получение чистого осадка, но любого состава, а к другой — методы, для которых требуются чистые осадки определенного состава. Известно лишь немного методов первого типа, и только один из них, заключающийся в конечном взвешивании фосфора в виде пирофосфата магния 2, получил широкое распространение.
Получение чистого осадка фосфоромолибдата постоянного состава требуется для методов, основанных на непосредственном взвешивании высушенного 3 или прокаленного 4 * * осадка, на титровании перманганатом предварительно восстановленного молибдена 8 и на алкалиметрическом титровании осадка фосфоромолибдата ®. Все эти методы в опытных руках дают хорошие результаты, но только последний из них получил всеобщее применение 7.
Если ввести в анализируемый раствор лимонную кислоту и прибавить магнезиальную смесь в достаточном избытке, то фосфор можно непосредственно осадить в виде фосфата магния и аммония в присутствии кальция, железа, алюминия, титана, циркония, ванадия и олова 8.
1F. Hundeshagen, Z. anal. Chem., 28, 141 (1889).
2 Этот метод первоначально был предложен F. L. Sonnenschein [J. prakt. Chem., 53, 339 (1851)] и затем видоизменялся многими авторами.
3С. R a m m elsberg, Вег., 10, 1776 (1877); R, Finkener, там же, 11,1640(1878); G. ,Р. Baxter, Am. Chem., J., 28, 298 (1902). Согласно последнему, наилучшими условиями осаждения являются следующие. Приготовляют раствор, содержащий около 0,1 г Р2О6 в 50 мл, и, если присутствует много HNO3, большую часть ее удаляют выпариванием. Выливают этот раствор при энергичном перемешивании в раствор молибдата, взятый в количестве, не менее чем на 50 мл превышающем теоретически необходимое. Оба раствора должны иметь комнатную температуру. Оставляют в течение 16 ч. Фильтруют через тигель Гуча, осадок промывают 10%-ным раствором NH4NO3, сушат при 300° С не менее 2 ч и взвешивают в виде (NH4)3PO4 НгМоОз- Осадок всегда содержит несколько большие количества МоО3, чем это соответствует формуле.
4 С. Mei л eke, Chem. Ztg., 20, 108 (1896); R. W о у, там же, 21, 441 (1897). Прокаливание проводят при 300—400° С, причем образуется соединение Р2О5 -24МоО3.
8 F. А. Е m m е г t о n, Trans. AIME, 15, 93 (1887); A. A. Blair, The Chemical Analysis of Iron, 8 th ed., J. B. Lippincott Co., 1918, p. 87.
®H. Pemberton, Chem., News, 46, 4 (1882); J. Am. Chem. Soc., 16, 278 (1894); ASTM Methods of Chemical Analysis of Metals, 1950, p. 88.
7 Рядовые определения фосфора можно осуществлять измерением объема осадка после его отстаивания или центрифугирования в градуированной трубке [V. Egger t z, J. prakt. Chem., [1], 79, 496 (1860); G. W. Goetz, H. Wedding, Stahl u. Eisen, 7, 118 (1887); W. E. В a u 1 i e u, Ind. Eng. Chem., 17, 908 (1925)].
8 G. E. F. Lu n'd ell, J. I. Hoffman, Ind. Eng. Chem., 15, 44, 171 (1923); J. I. Hoffman, G. E. F. L u n d e 11, J. Research NBS, 20, 607 (1938). *Лучшие результаты достигаются, если вместо лимонной кислоты вводить комплексон III (этилендиаминтетраацетат натрия) и тирон (пирокатехин-3,5-дисульфокислоту). *
782 Гл. XLV. Фосфор
Осаждение фосфора в виде фосфоромолибдата аммония и обработка полученного осадка
Осаждение в виде фосфоромолибдата аммония. Для осаждения фосфоромолибдата аммония требуется избыток осадителя. В обычных случаях в 100 мл раствора должно содержаться на 1,3 г МоОз больше, чем это теоретически требуется для образования фосфоромолибдата (56 мг МоОз на 1 мг Р). Больший избыток молибдата не мешает осаждению, но при этом образуется осадок, несколько более богатый МоО з. В некоторых условиях для полного осаждения фосфора может потребоваться вдвое большая концентрация реагента, как, например, в присутствии больших количеств титана, циркония, хлорида или сульфата. Для быстрого и полного осаждения необходимо, чтобы в растворе содержалось 5—10% нитрата аммония. Концентрация азотной кислоты может колебаться в пределах от 5 до 10% (по объему). Повышение кислотности приводит к неполноте осаждения, если одновременно не увеличить концентрацию нитрата аммония и молибдата в растворе. Умеренные количества хлорной кислоты (1 : 20) или перхлората аммония (10%) не мешают осаждению.
Нагревание раствора (40—50° С) и энергичное встряхивание ускоряет осаждение. Осадки, образующиеся при температуре выше 50° С, содержат избыточные количества МоО з и сильнее загрязняются такими веществами, как арсенаты, ванадаты и кремнекислота. В чистых растворах осаждение может закончиться в течение 10 мин. Более продолжительное время требуется, если осаждение проводится в холодных растворах или если присутствуют элементы, замедляющие осаждение, например ванадий (IV). При выполнении анализов, требующих высокой точности, раствору после введения осадителя следует дать постоять 12 ч. Более продолжительное стояние не оказывает влияния на состав осадка.
Из веществ, замедляющих осаждение или загрязняющих осадок, следует отметить серную, соляную и фтористоводородную кислоты, аммонийные соли этих кислот, мышьяк, ванадий, селен, теллур, вольфрам, кремний, титан, цирконий и некоторые органические соединения. При выполнении особо точных анализов присутствия этих трех минеральных кислот следует избегать. При выполнении рядовых анализов умеренные концентрации соляной кислоты (1 : 9) или серной кислоты (1 : 20) можно допустить, если прибавить большие количества нитрата аммония и осадителя и продлить время осаждения. Допустимо также и присутствие фтористоводородной кислоты (1 : 20), если она превращена во фтороборную кислоту HBF4 введением избыточного количества борной кислоты. Если время позволяет, рекомендуется сначала удалить большую часть фтористоводородной кислоты однократным или двукратным выпариванием с азотной кислотой.
Присутствие мышьяка нежелательно не только вследствие его замедляющего действия на осаждение фосфоромолибдата аммония, но и потому, что он реагирует с молибдатом, образуя нерастворимый арсеномолибдат. При 75—100° С мышьяк может выделиться количественно; при 20—25° С осаждается только небольшая его часть. Большие количества мышьяка лучше отделить в начальной стадии анализа восстановлением и дистилляцией с соляной кислотой (стр. 304) или осаждением сероводородом (стр. 305). Если присутствуют малые количества мышьяка и осаждение проводится при комнатной температуре, то примесью его можно пренебречь во всех случаях, за исключением особо точных анализов. При последующем
Методы определения
783
переосаждении фосфора в виде фосфата магния и аммония находившийся в осадке фосфоромолибдата мышьяк выделяется в виде арсената магния и аммония. В таких случаях обычно растворяют последний осадок в соляной кислоте и переосаждают магнезиальной смесью, отделив предварительно мышьяк осаждением сероводородом или (что менее надежно при содержании больших количеств мышьяка) восстановлением сернистой или бромистоводородной кислотой и кипячением с разбавленной (1 : 1) соляной кислотой.
Ванадий (V) также замедляет осаждение фосфора и загрязняет осадок. Он осаждается полностью, если фосфор присутствует в значительном из-, бытке. Ванадий (IV) также несколько замедляет осаждение, но не захватывается осадком фосфоромолибдата аммония, когда осаждение проводится 1 при 10—20° С.
Если содержание фосфора велико и при этом равно или превышает содержание ванадия (V), то полное осаждение его заканчивается в течение 2 ч. Для осаждения малых количеств фосфора требуется больше времени. Когда содержание ванадия (V) превышает содержание фосфора, осаждение значительно замедляется и никогда не бывает количественным спустя 2 ч после введения реагента, а при малых количествах фосфора не заканчивается и в продолжение 24 ч, иногда фосфор и совсем не осаждается. Замедляющее действие ванадця (V) можно до некоторой степени ослабить введением большого количества молибдата.
Соединения селена (IV) и (VI) и теллура (IV) и (VI) мешают определению, так как они замедляют осаждение фосфора молибдатом и загрязняют образующийся осадок. При их присутствии для полного осаждения фосфора раствор необходимо оставлять стоять на менее 4 ч и определение следует заканчивать осаждением в виде фосфата магния и аммония в присутствии лимонной кислоты.
Вольфрам захватывается осадком фосфоромолибдата аммония и должен быть отделен перед осаждением, за исключением, быть может, тех случаев, когда содержание его невелико и определение проводят молибдатно-магнезиальным методом. Отделение вольфрама осуществляют нагреванием с азотной и соляной кислотами и последующим фильтрованием. Фильтрат следует затем выпарить с азотной кислотой для удаления большей части соляной кислоты, а вольфрамовую кислоту необходимо исследовать на содержание в ней фосфора, лучше всего растворением в аммиаке, содержащем цитрат аммония, и осаждением магнезиальной смесью из ледяного раствора, как описано на стр. 788.
Кремний также замедляет осаждение фосфора и выделяется в виде силикомолибдата аммония. Большие количества кремнекислоты могут забить фильтр, поэтому ее лучше предварительно отделить обезвоживанием или выпариванием с фтористоводородной и азотной кислотами.
Осаждение фосфора молибдатом в присутствии титана и циркония обычно затруднено вследствие образования их малорастворимых фосфатов. Когда эти элементы присутствуют в значительных количествах, большая часть фосфора может выделиться в осадок при проведении предварительных операций и таким образом потеряется. Это происходит, если не прибегают к сплавлению нерастворимых остатков с карбонатом натрия и выщелачиванию плава водой. В присутствии циркония и титана следует прибавлять большее количество молибдата, чем обычно, и осадок должен отстаиваться дольше, не менее 12 ч. Титан и цирконий при этом частично
1 J. R. Cain, F. Н. Tucker, J. Ind. Eng. Chem., 5, 647 (1913).
784
Гл. XLV. Фосфор
выпадают в осадок, и их присутствие обнаруживается при растворении •осадка фосфоромолибдата в аммиаке. Когда содержание этих элементов не слишком велико, их можно перевести в раствор добавлением к аммиаку небольшого количества цитрата аммония. Если при этом осадок все же полностью не растворяется, его надо прокалить, сплавить с карбонатом натрия, плав выщелочить водой и водную вытяжку присоединить к аммиачному раствору. В присутствии умеренных количеств титана (0,025 г) фосфоромолибдат более полно осаждается из разбавленного (2:1) азотнокислого раствора, к которому добавляют раствор молибдата в количестве, равном половине объема анализируемого раствора.
Органические вещества обычно разрушают кипячением с перманганатом калия, избыток которого восстанавливают перед введением молибдата.
Осаждение в отсутствие ванадия. 100—200 мл раствора, содержащего 5—10% азотной кислоты (по объему), 5—15% нитрата аммония и не намного больше 0,05 г фосфора (полностью в виде ортофосфата), нагревают в коническойколбе до 40—50° С. Прибавляют 15 — 25-кратный избыток раствора молибдата (см. «Реактивы» стр. 66), закрывают колбу пробкой и энергично встряхивают 5—10 мин. Если содержание фосфора невелико или присутствуют вещества, замедляющие осаждение, вводят больший избыток реагента.
При выполнении точных анализов, когда фосфор впоследствии пере-осаждают в виде фосфата магния и аммония, раствор после введения молибдата оставляют на ночь. В рядовых анализах осадок фосфоромолибдата отфильтровывают через плотный фильтр спустя 10—30 мин после осаждения. Если определение заканчивают алкалиметрическим титрованием, можно фильтровать через асбест или бумажную массу; при этом применяют отсасывание для ускорения фильтрования и более тщательного промывания.
Состав применяемой промывной жидкости зависит от4 дальнейшей обработки осадка фосфоромолибдата. Если за этой операцией следует пере-осаждение фосфора в виде фосфата магния и аммония, осадок умеренно промывают 5%-ным раствором нитрата аммония. Если же определение заканчивают алкалиметрическим титрованием, осадок промывают 1 %-ным раствором нитрата калия до удаления свободной кислоты. Осадок фосфоромолибдата аммония несколько растворим в этой промывной жидкости, но не настолько, чтобы это заметно отразилось на результатах.
Осаждение в присутствии ванадия. Приготовляют раствор так же, как при определении в отсутствие ванадия (см. выше), охлаждают до 10—20° С, вводят достаточное количество сульфата железа (II), чтобы восстановить ванадий, а затем добавляют несколько капель сернистой кислоты. Если раствор не содержит железа, целесообразно прибавить около 1 г железа в виде нитрата железа (III), свободного от фосфора, для предотвращения восстановления молибдена. Вводят 20—30-кратный избыток холодного раствора молибдата и встряхивают раствор 5 мин. Оставляют стоять в продолжение ночи или, если заканчивают алкалиметрическим титрованием, не менее 30 мин. Фильтруют и в дальнейшем поступают, как указано в разделе «Осаждение в отсутствие ванадия» (см. выше).
Обработка осадка фосфоромолибдата аммония. Из известных методов обработки осадка фосфоромолибдата аммония наиболее распространены два метода. По первому из них фосфор осаждают в виде MgNH4PO4
Методы определения
785
после растворения осадка фосфоромолибдата аммония в аммиаке. Второй метод, так называемый алкалиметрический, заключается в обработке осадка фосфоромолибдата аммония определенным количеством титрованного раствора едкого натра, избыток которого обратно оттитровывают кислотой по фенолфталеину \
Весовое определение осаждением в виде фосфата магния и аммония и прокаливанием до пирофосфата. Осаждению фосфора в виде фосфата магния и аммония мешает такое большое число соединений, что ему должно обычно предшествовать отделение фосфора молибдатом аммония. Получение осадка MgNH4PQ4 • 6Н2О идеального состава не вызывает затруднений, и потому этот метод рекомендуется при выполнении точных анализов, особенно для определения больших количеств фосфора. В редких случаях, когда фосфор можно непосредственно осадить магнезиальной смесью, определение проводится в тех же условиях, как и после растворения осадка фосфоромолибдата аммония. Практически невозможно получить точные результаты однократным осаждением фосфата магния и аммония из холодного йли горячего раствора, даже когда он содержит только фосфат щелочного металла в приблизительно известных количествах. Когда содержание фосфора в растворе неизвестно или присутствуют посторонние примеси, однократное осаждение может дать правильные результаты только вследствие взаимной компенсации ошибок. Поэтому, как правило, надо проводить двукратное осаждение.
Потеря фосфора при повторном осаждении настолько ничтожна, что ею можно пренебречь1 2. ,
При анализе шести аликвотных частей стандартного раствора чистого двузамещенного фосфата аммония (без предварительного осаждения в виде фосфоромолибдата) было получено 0,2367 г и 0,2368 г Mg2P2O7 после однократного осаждения, 0,2370 г и 0,2368 г после двукратного осаждения и 0,2366 г и 0,2368 г после трехкратного осаждения. Как показали результаты тщательного определения фосфора в 10 фильтратах и промывных водах от 12 осаждений, при каждом осаждении в среднем теряется менее 0,03 мг P2O5. Совпадение результатов, полученных однократным и двукратным осаждением, является исключением и объясняется тем, что сначала фосфор определялся ' в растворе двукратным осаждением, причем было вычислено количество магнезиальной смеси, необходимое для получения избытка в 2 мл, а однократное осаждение затем проводилось в растворах, содержащих этот избыток осадителя, а также и то количество соляной кислоты, которое было израсходовано для растворения первого осадка при других определениях.
При первом осаждении магнезиальную смесь лучше прибавлять к кислому раствору, а затем медленно вводить аммиак до щелочной реакции. Прибавление магнезиальной смеси к щелочному раствору менее желательно.’ Вторичное осаждение необходимо проводить медленным прибавлением аммиака к кислому раствору первого осадка, к которому предварительно добавляют очень небольшое количество магнезиальной смеси.
1 При анализе образца фосфатной породы, содержавшей 31,33% Р2О6, алкалиметрическим методом (А), молибдатно-магнезиальным методом при однократном осаждении магнезиальной смесью (Б) и тем же методом, но при двукратном осаждении в виде MgNH4PO4 (В) опытными аналитиками были получены следующие результаты (содержание Р2Ое, %):
Метод А.........31,76
Метод Б.........31,41
Метод В.........31,29
30,68 31,42 31,75
31,44 31,56 31,61
31,30 31,35 31,34
2 G. Е, F. L u n d е 11, J. I. Hoffman, J. Assoc. Offic. Agr. Chemists, 8, 188 (1924). , '
50 Заказ 522.
786
Гл. XLV. Фосфор
Для первого осаждения достаточно кристаллизации осадка в течение 3—4 ч, если растворы относительно чисты и содержат большие количества ортофосфата. Двенадцатичасовой период осаждения желателен при выполнении точных анализов и необходим в тех случаях, когда концентрация фосфора очень мала или в растворе содержатся большие количества посторонних солей. Особые способы осаждения, как, например, осаждение из горячих растворов, не являются необходимыми и не способствуют получению лучших результатов. Загрязнения осадка оловом, ванадием, железом, титаном и цирконием можно полностью избежать, если проводить осаждение в присутствии цитрата аммония х, как описано на стр. 788.
Селеновая кислота не влияет на определение фосфора двукратным осаждением в виде фосфора магния и аммония. Селенистая кислота несколько мешает, а мешающее влияние теллуристой и теллуровой кислот очень велико. Однако ни одно из этих соединений не мешает, если перед первым осаждением фосфата в раствор ввести 1 г лимонной кислоты.
После двукратного осаждения фосфора, содержание которого было эквивалентно 0,2423 г Mg2₽2O7, получено 0,2424 г Mg2₽2O7, когда в раствор была введена 0,1 г НгйеОа и 0,2436 г в присутствии 0,1 г H2SeO3. Теллуриты и теллураты образуют с магнезиальной смесью белый осадок. В присутствии 0,1 г H2SeO3, НгЗеО4, НгТеО3 или НгТеО4 после двукратного осаждения, когда перед первым осаждением в раствор вводился 1 г лимонной кислоты, было получено соответственно 0,2420 а, 0,2424 а, 0,2424 а и 0,2421 г Mg2?2O7. 4
Определение в отсутствие мышьяка и значительных количеств олова или железа. Осадок фосфоромолибдата, полученный, как описано выше (стр. 781), растворяют в разбавленном растворе аммиака (1 : 2) и затем промывают фильтр последовательно горячей водой, разбавленной соляной кислотой и под конец снова раствором аммиака. Аммиачный раствор обрабатывают 0,5 г лимонной кислоты и, если он непрозрачен, фильтруют. Если в фильтрат проходит муть, вводят в него немного бумажной массы, кипятят и снова фильтруют. Если раствор был мутным, а в точных анализах даже в случае отсутствия заметной мути, хорошо промытый фильтр сжигают, золу сплавляют со щепоткой карбоната натрия, плав выщелачивают водой, фильтруют и подкисленную водную вытяжку присоединяют к основному раствору.
Слегка подкисляют раствор (лучше, если объем его не превышает 100 мл), охлаждают, прибавляют 10 мл магнезиальной смеси (см. «Реактивы», стр. 66) и сверх того по 0,1 мл на каждый миллиграмм предполагаемого содержания РгО5. Больший избыток осадителя не мешает.'Затем, непрерывно перемешивая, вводят разбавленный (1:1) раствор аммиака. Когда появляется белый кристаллический осадок фосфата, перестают прибавлять аммиак и продолжают перемешивать до прекращения образования осадка. После этого рводят еще несколько капель раствора аммиака, перемешивают и так продолжают до тех пор, пока раствор не станет щелочным. Затем добавляют еще по 10 мл разбавленного (1 : 1) раствора аммиака на каждые 100 мл раствора и оставляют на 4 ч или лучше на ночь.
Когда предполагают провести только однократное осаждение, раствор фильтруют, переносят осадок на фильтр, промывают разбавленным (1 : 20) раствором аммиака до удаления хлоридов и прокаливают, как бу-
1 G. Е. F. L u n d е 11, J. I. Hoffman, Ind. Eng. Chem., 15, 44, 171 <-1923).
Метода определения
787
дет описано ниже. Если же требуется переосаждение осадка, фильтруют, не перенося осадок на фильтр, и несколько раз промывают сосуд, осадок и фильтр разбавленным раствором аммиака. Попавший на фильтр осадок растворяют в 25 мл разбавленной (1 : 1) соляной кислоты, собирая раствор в стакан, в котором находится основная масса осадка. Фильтр тщательно промывают разбавленной (1 : 20) соляной кислотой *. Раствор разбавляют до 50—100 мл, вводят 1—2 мл магнезиальной смеси и медленно, при перемешивании, как ранее, прибавляют раствор аммиака, вводя под конец 10 мл его избытка. Раствор оставляют стоять не менее 2 ч, лучше на ночь.
Фильтруют, переносят осадок на фильтр и промывают разбавленным раствором аммиака до удаления хлоридов. Помещают фильтр с осадком во взвешенный платиновый тигель, сушат и затем осторожно, лучше в муфеле, обугливают бумагу, не допуская ее воспламенения. Сжигают уголь при возможно более низкой температуре, причем полностью, в противном случае, при последующем повышении температуры до 900° С, фосфат медленно восстанавливается и фосфор улетучивается 1 2. Затем прокаливают при 1050—1100° С до постоянной массы. Взвешивают в виде Mg2P2O7.
Осадки, имеющие надлежащий состав, достаточно прокаливать при 1000° С до постоянной массы. Если прокаливание проводить при температуре, превышающей 1100° G, осадок постепенно теряет в массе, вероятно, вследствие медленного разложения Mg2P2O на Mg3(PO4)2 и Р2О5.
Это иллюстрируется следующими данными, полученными при прокаливании приблизительно 0,25 г MgaPsO, [приготовленного двукратным осаждением чистого (NHJ2HPO4, как описано выше] в платиновом тигле. При прокаливании в течение 1 ч на паяльной горелке потери в массе не наблюдалось. После обычного прокаливания при 1000° С масса тигля оказалась равной 27,3394 г. В результате дальнейшего прокаливания масса изменялась следующим образом; при 1000° С в течение получаса масса стала равной 27,3392 г, спустя еще полчаса — 27,3391 г, еще спустя полчаса — 27,3392 г, в течение одного часа при 1100° С — 27,3392 г, полчаса при сильном дутье — 27,3386 г, еще один час в тех же условиях — 27,3382 г и еще спустя полчаса— 27,3380 г.
В другой серии опытов такой же осадок под конец прокаливался на паяльной горелке 4 раза по полчаса и 3 раза по одному часу, что дало среднюю потерю в массе 0,4 .иг в час. При этом потери в массе после четвертого и пятого часа прокаливания оказались одинаковыми. Скорость улетучивания была несколько выше при прокаливании осадка сразу на паяльной горелке, что можно объяснить наличием небольшого количества Mg(PO3)2, образовавшегося из соответствующего количества Mg(NH4)4(PO4)2, который присутствовал в исходном осадке.
Превращение метафосфата в пирофосфат, желательное при определении магния (стр. 721), несомненно, увеличивает ошибку, если определяют фосфор. В опытах, в которых Mg2P2O7 был получен предварительным осаждением фосфора в виде фосфоромолибдата и двукратным осаждением в виде MgNH4PO4, бсадки, весившие после прокаливания при 1000’ С до постоянной массы 0,2835 а, 0,2824 а и 0,2826 а, в результате прокаливания в течение 1 часа при 1100 °C потеряли в массе соответственно 0,4 мг, 0,3 мг и 0,1 мг. Вполне возможно, что эти потери происходили за счет улетучивания молибдена, который удерживался осадком, несмотря на двукратное осаждение магнезиальной
1 Обычно нет необходимости сохранять этот фильтр. Из предосторожности при выполнении особо ответственных анализов (и всегда, когда анализируемый материал содержит значительные количества циркония или титана) фильтр сжигают в платиновом тигле, сплавляют золу с 0,2 г или менее карбоната натрия, плав выщелачивают водой, фильтруют и присоединяют подкисленный фильтрат к основному раствору.
2 К. D. Jacob, D. S. Reynolds, J. Assoc. Offic. Agr. Chemists, Ц, 128 (1928).
50*
788
Гл. XLV. Фосфор
смесью. На это указывает также и тот факт, что средняя потеря в массе этих осадков при прокаливании на паяльной горелке составляла 0,7 мг в час в течение второго и третьего часа Ч
Обработка прокаленного остатка пирофосфата магния 1—2 каплями азотной кислоты и вторичное его прокаливание не ухудшают результатов, но редко приносят пользу. После растворения пирофосфата магния в азотной кислоте и последующего выпаривания и прокаливания получаются; пониженные результаты, так как невозможно предотвратить улетучивание фосфора и даже механические потери вследствие разбрызгивания остатка. Имеются указания 1 2 на то, что эта операция проходит успешно, если азотнокислый раствор перед выпариванием и прокаливанием нейтрализовать аммиаком. Хорошие результаты получаются также, если кислый раствор пирофосфата обработать 2—3 мл магнезиальной смеси, затем аммиаком и осадок отфильтровать и промыть, как обычно. Можно также с самого начала растворить осадок фосфата магния и аммония в азотной кислоте, раствор выпарить и затем прокалить сухой остаток.
Определение в присутствии мышьяка. Поступают так же, как указано выше (см. «Определение в отсутствие мышьяка и значительных количеств олова дли железа», стр. 786), вплоть до растворения первого осадка фосфата магния и аммония в разбавленной соляной кислоте. Если известно, что содержание мышьяка невелико, прибавляют 0,5—1 г бромида аммония, осторожно кипятят раствор, пока он почти полностью не выпарится, и обрабатывают остаток 25 мл разбавленной (1:1) соляной кислоты. Когда присутствуют большие количества мышьяка, обработку солянокислого раствора бромидом аммония и кипячением необходимо повторить 2—3 раза или лучше удалить мышьяк осаждением из солянокислого раствора сероводородом и фильтрованием. Фильтрат затем кипятят для удаления сероводорода и уменьшения объема до 50— 100 мл.
После удаления мышьяка любым из этих способов продолжают анализ, как указано в разделе «Определение в отсутствие мышьяка и значительных количеств олова или железа».
Определение в присутствии умеренных количеств железа, ал юминия, ванадия, цинка, олова, титана или циркония. К 100 мл раствора, содержащего фосфор в виде ортофосфорной кислоты, прибавляют 3—5 г лимонной кислоты и 25—50-кратный избыток магнезиальной смеси 3. Осаждают, как указано выше (см. раздел «Определение в отсутствие мышьяка и значительных количеств олова или железа», стр. 786), и оставляют раствор при комнатной температуре 12—24 ч. Осадок отфильтровывают, промывают и растворяют, как описано в том же разделе, а затем извлекают фосфат, оставшийся на фильтре. К раствору прибавляют 0,2—0,5 г лимонной кислоты, 3—4 мл магнезиальной смеси и продолжают анализ, как указано там же.
1 Изучение осаждения и прокаливания фосфата магния и аммония см. в статье J. I. Hoffman, G. Е. F. L u n d е 11, J. Research NBS, 5, 279 (1930).
2 W; М. McNabb, J. Am. Chem. Soc., 49, 893 (1927).
3 При введении большего количества лимонной кислоты необходимо прибавить больше магнезиальной смеси, особенно если присутствует ванадий.
Методы определения
789
Только в этом случае раствор перед фильтрованием оставляют стоять1 не менее 6 ч.
Объемное определение растворением осадка фосфоромолибдата аммония в титрованном растворе щелочи. Алкалиметрический метод является наиболее важным и широкораспространенным методом быстрого определения фосфора в рядовых анализах. Метод основан на допущении, что состав промытого осадка фосфоромолибдата соответствует формуле (NH4)3 РО4- 12МоО3 и что в результате титрования этого соединения едким натром по фенолфталеину образуются нормальные соли молибденовой кислоты R2MoO4 и двузамещенные соли фосфорной кислоты R2HPO4. Реакция может быть выражена уравнением
(NH4)3P04-12Mo03-|-23NaOH = llNa2Mo04-|-(NH4)2Mo04-|-NaNH4HP04+llH20
Это уравнение вполне справедливо для фосфоромолибдата, осажденного при температуре от 20 до 45° С, независимо от того, фильтровался ли раствор через 15 мин после добавления осадителя или спустя 15 ч. Осаждение ускоряется, если начинать его приблизительно при 40 °C. Ни при каких обстоятельствах нельзя нагревать раствор после прибавления молибдата.
Когда условия осаждения и промывания осадка не соблюдаются достаточно точно, отношение едкого гщтра к фосфору обычно бывает больше 23 : 1. Причины могут быть следующие:
1) осаждение при температуре выше 45° С, в результате чего образуется осадок с повышенным содержанием молибденовой кислоты;
2) недостаточно тщательное промывание осадка, вследствие чего в нем остается свободная кислота;
3) кипячение фосфоромолибдата с избыточным количеством едкого натра, при котором аммиак улетучивается и отношение NaOH : Р становится равным 26 : 1;
4) чрезмерно продолжительное стояние или взбалтывание щелочного раствора фосфоромолибдата (в особенности, когда осадок велик), ведущее к медленному улетучиванию аммиака;
5) присутствие карбонат-ионов в растворе, которые реагируют с едким натром.
Все указанное выше относится к чистым растворам. Помимо этих ошибок, есть еще другие ошибки, возникающие при наличии в растворе веществ, задерживающих осаждение фосфоромолибдата аммония за тот короткий промежуток времени, который обычно допускается при применении алкалиметрического титрования, или в присутствии веществ, загрязняющих осадок (как, например, мышьяк, селен, теллур), на которые затем расходуется щелочь.
1 Точность метода иллюстрируется следующими данными [G. Е. F. Lu п-d е 11, J. I. Hoffman, Ind. Eng. Chem., 15, 44, 171 (1923)]:
Ожидаемое количество Mg2P2O7 0,1048 г:
Введено загрязнений, г Нет Нет 0,005Fe 0,005А1 0,005Ti 0,005Ti 0,005Zr
Найдено Mg2P2O7, г . . 0,1048 0,1048 0,1044 0,1049 0,1044 0,1048 0,1043
Ожидаемое количество Mg2P2O7 0,0105 г:
Введено загрязнений, г ...... 0,005V 0,020Sn 0,250Zn
Найдено Mg2P2O7, г.................. 0,0107 0,0104 0,0107
790
Гл. XLV. Фосфор
Например, при алкалиметрическом определении фосфора в стали, содержавшей 0,096% фосфора, получено 0,167%, когда прибавляли 0,1 г H2SeO4; 0,109%, когда прибавляли 0,1 з Н2ТеО3 и 0,116%, когда прибавляли 0,1 г Н2ТеО4. Селенистая кислота H2SeO3 образовала объемистый осадок с одним молибдатом. Во всех приведенных четырех случаях было определено 0,0973%; 0,0971%; 0,0971% и 0,0970% фосфора, когда после осаждения фосфоромолибдата раствор оставлялся на 4 ч и затем осадок дважды переосаждался в виде фосфата магния и аммония.
Фенолфталеин в данном случае не является идеальным индикатором вследствие его высокой чувствительности к угольной кислоте и недостаточной чувствительности к аммиаку, однако лучший индикатор неизвестен. Примеси карбонат-ионов можно избежать правильным приготовлением раствора едкой щелочи.
Выделения некоторого количества аммиака в процессе титрования предотвратить нельзя, и это приводит к повышенным результатам определения фосфора. Влияние аммиака, однако, компенсируется нормальной отрицательной ошибкой, имеющей место, когда результаты титрования вычисляют на основе соотношения 23 : 1.
Авторы предпочитают устанавливать титр раствора едкого натра (свободного от карбоната) по‘бензойной кислоте или по бифталату калия (стр. 208), а "затем перечислять на фосфор, основываясь на соотношении 23 : 1. В этом случае при осаждении фосфора должны строго соблюдаться предписанные условия. Когда это невыполнимо, проще всего определять фосфор любым другим вариантом алкалиметрического метода, но применять тот же вариант при установке титра раствора едкого натра по стандартному образцу с определенным содержанием фосфора, имеющему одинаковый с анализируемым материалдм состав.
Определение в отсутствие ванадия. Осаждают фосфоромолибдат, как указано на стр. 784, но при температуре 30—45° С. Раствор можно нагреть непосредственно перед добавлением молибдата аммония или, проще, прибавлением определенного количества аммиака к раствору, содержащему определенный избыток азотной кислоты. После взбалтывания раствора в течение 5—15 мин и отстаивания в продолжение 10—15 мин фильтруют через плотный фильтр диаметром 11 см, стараясь перенести возможней меньшее количество осадка на фильтр.
При желании можно фильтровать через асбест или бумажную массу, применяя отсасывание. Колбу и осадок промывают 5 раз 1 %-ным раствором нитрата калия, порциями по 20 мл \ а затем промывают фильтр 5 раз такими же порциями той же промывной жидкости. Фильтр каждый раз тщательно промывают от краев к центру, давая каждой порции жидкости полностью стечь. Промывная жидкость и другие растворы должны быть свободны от угольной кислоты. Фильтр с осадком переносят обратно в колбу и прибавляют 0,1 н. раствор едкого натра в количестве, достаточном для растворения осадка, и чтобы остался избыток приблизительно ! в 2 мл. Разбавляют 25 мл воды, закрывают колбу пробкой и встряхивают до растворения осадка. Раствор разбавляют водой приблизительно до 150 мл, вводят 6 капель 1 %-ного раствора фенолфталеина и титруют кислотой, титр которой точно эквивалентен титру щелочи, до исчезновения окраски индикатора. Заканчивают титрование прибавлением титрован-
1 Если есть основание опасаться гидролиза, как, например, при анализе фосфористой бронзы, содержащей олово, осадок сперва промывают небольшими порциями разбавленной азотной кислоты, израсходовав в общей сложности не более 50 мл.
Методы, определения
791
-ного раствора щелочи, пока снова не появится розовая окраска. Вычитают израсходованный объем кислоты из общего объема введенной щелочи и полученную разность умножают на титр раствора щелочи, пересчитанный на фосфор.
Определение в присутствии ванадия. Осаждают, как указано на стр. 784. Оставляют раствор стоять не менее 30 мин, фильтруют и в дальнейшем поступают, как указано выше.
Непосредственное осаждение фосфора в виде фосфата магния и аммония
.Условия непосредственного осаждения фосфора в виде- фосфата магния и аммония изложены в приведенном ниже ходе определения фосфора в фосфатной породе, не содержащей заметных количеств нерастворимых в кислоте соединений фосфора Ч
Небольшие количества кальция не мешают осаждению фосфат-ионов магнезиальной смесью. Так, например, когда к анализируемому раствору или к магнезиальной смеси была добавлена 0,01 г СаО, осадок фосфата магния и аммония после однократного осаждения не был загрязнен кальцием, а введение даже 0,1 г СаО в анализируемый раствор не вызвало никаких затруднений при двукратном осаждении 1 2.
Ход определения. Помещают 0,5 г -высушенной при 105° С пробы в коническую колбу емкостью 300 мл и обрабатывают 15 мл соляной и 3 мл азотной кислоты. Покрывают колбу небольшим часовым стеклом и слабо кипятят 30 мин. Ополоснув стекло, удаляют его и, не фильтруя, вводят 30 г цитрата аммония, 10 мл соляной кислоты и 100 мл концентрированной магнезиальной смеси 3. Нейтрализуют раствор аммиаком по лакмусу и затем вводят избыток раствора аммиака в 3 мл. Разбавляют раствор водой до 225—250 мл, вводят несколько стеклянных бус, плотно закрывают колбу пробкой, встряхивают в аппарате для взбалтывания 30 мин и затем оставляют на ночь. Фильтруют через плотный фильтр и один раз промывают колбу и бумагу небольшим количеством разбавленного (1 : 19) раствора аммиака. Фильтрат отбрасывают.
Растворяют фосфат магния и аммония, оставшийся в колбе, в 50 мл горячей разбавленной (1 : 4) соляной кислоты и сливают раствор через фильтр, на который перенесен фосфат; в результате этой операции осадок растворяется и отделяется нерастворимый остаток, который может остаться после обработки кислотой. Промывают колбу и фильтр той же кислотой и затем прибавляют к раствору, объем которого должен быть около 100 мл, 0,3 г лимонной кислоты и 1 мл магнезиальной смеси. Перемешивая, медленно вводят аммиак до щелочной реакции по лакмусу, после чего прибавляют еще 5 мл избытка. В течение получаса время от времени перемешивают, а затем оставляют на 4 ч или на ночь. Осадок переносят на плотный фильтр диаметром 9 см, промывают холодным разбавленным (1 : 19)
1 J. I. Hoffman, G. Е. F. L u n d е 11, J, Research NBS, 19, 59 (1937).
2 G. E. F. L ив d e 11, J. I. Hoffman, J. Assoc. Offic. Agr. Chemists, 8, 188 (1924).
3 Концентрированную магнезиальную смесь приготовляют следующим образом. Растворяют 400 г MgCl2 -6Н2О и 300 г хлорида аммония в 1500 мл горячей воды. По •окончании растворения к раствору добавляют раствор аммиака до щелочной реакции жо лакмусу и оставляют стоять 1 ч. Затем фильтруют и подкисляют фильтрат соляной кжслотои по лакмусу. При правильном приготовлении объем раствора почти точно равен 2 л.
792
Гл. XLV. Фосфор
раствором аммиака и помещают во взвешенный платиновый или фарфоровый тигель. Обугливают бумагу без воспламенения, сжигают у£оль при температуре ниже 900® С‘и под конец прокаливают, лучше в муфеле, при 1050—1100° С до постоянной массы.
Продолжительность стояния при первом осаждении может быть снижена до 3 ч, а при втором — до 2 ч, если раствор перед встряхиванием или перемешиванием охлаждают и затем оставляют стоять в ледяной воде или в холодильнике при 5—10° С. При определении малых количеств фосфора (менее 10 мг Р2О8) необходимо оставлять стоять более продолжительное время, и при этом лучше раствор охлаждать и дополнительна встряхивать 2 или 3 раза \
Колориметрические методы
Малые количества фосфора можно определять колориметрически следующими способами: 1) превращением в синий комплекс, который фосфор> образует с молибденом, восстановленным хлоридом олова (II), и сравнением интенсивности окраски со стандартом визуально или в фотоколориметре а; 2) по реакции образования желтого комплекса с ванадием (V) и молибденом (VI) и измерением светопоглощения раствора при 450 ммк в спектрофотометре 3. Последний метод применим также к продуктам, содержащим значительные количества фосфора, как, например, фосфатные породы и основной фосфат кальция 4.
1 Методы анализа смесей орто-, пиро-, мета- и полифосфатов приводят L. F. Au d-rieth, R. N. Bell [Inorganic Syntheses, Vol. Ill, McGraw-Hill Book Co., 1950, p. 91]; R. N. Bell [Anal. Chem., 19, 97 (1947)1 и L. T. Jones [там же, 14, 536 (1942)].
2 Спектрофотометрическое изучение реакции образования синего комплекса приводят J. Т. Woods, М. G. Mellon [Ind. Eng. Chem., Anal. Ed., 13, 760 (1941)] и R. E. К i t s о n, M. G. Mellon [там же, 16, 466 (1944)]. C. W. Eddy и F. Deeds [там же, 9, 12 (1937)] применяли этот метод для определения таких малых количеств фосфора, как 0,001 мг. S. R. Dickman и R. Н. Bray [там же, 12, 665 (1940)] применили метод к солянокислым вытяжкам из почв и растений. J. L. Hague и Н. A. Bright [J. Research NBS, 26, 405 (1941)] при определении фосфора в чугуне и стали измеряют светопоглощение окрашенного раствора в фотоколориметре с желтым светофильтром. Влияние фторид-ионов на образование синего комплекса устраняется введением борной кислоты [L. Т. Kurt z, Ind. Eng. Chem., Anal. Ed., 14, 855 (1942)].
3 Метод анализа железных руд, в котором железо обесцвечивается, а кремнекислота отделяется обработкой хлорной кислотой перед образованием окрашенного фос-форованадомолибдата, рекомендуют Н. Н. Willard и Е. J. Center [Ind. Eng. Chem., Anal. Ed., 13, 81 (1941)]. Спектрофотометрическое изучение метода, преимущественно в применении к сталям, приводят R. Е. К i t s о п и М. G. Mellon (там же, 16, 379 (1944)].
4 A. Gee, V. R. Deitz, Anal. Chem. (1953).
Глава XLVI
СЕРА
Сера встречается в самородном состоянии и в виде многочисленных сульфидов и сульфатов. Важными породообразующими минералами, содержащими серу, являются только гаюин 3NaAlSiO4(Ca, Na2)SO4 и нозеан Na6Al3SO4(SiO4)3, которые находятся главным образом в более основных породах, преимущественно в породах, богатых натрием. Сульфидная сера широко распространена в форме минералов пирита FeS2 ипирротина (Fe, Ni)S. Реже встречаются халькопирит CuFeS2 и другие сульфиды. Сера находится в- минерале лазурите 3NaAlSiO4-Na2S3, также преимущественно в основных породах.
Очень распространенной ошибкой является пересчет результатов определения серы в силикатах, карбонатных породах, глинах и т. п. на SOs вместо S. Иногда сера может встречаться в обоих состояниях, но чаще она находится в виде сульфида.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа сера не создает затруднений, если только она не связана с такими элементами, как барий, свинец или стронций, (которые образуют нерастворимые сульфаты) илине присутствует в больших количествах совместно с кальцием. В первом случае, особенно при наличии бария, сера выпадает в осадок в виде сульфата бария вместе с кремнекислотой. Присутствие сульфата бария в остатке кремнекислоты узнается по характеру этого осадка и по размерам и внешнему виду нелетучего остатка после обработки кремнекислоты фтористоводородной и серной кислотами. Если обработка HF + H2SO4 опускается, то естественно,результаты определения кремнекислоты будут повышенными. Если же эта обработка проводится, получаются пониженные результаты для кремнекислоты, так как при том интенсивном прокаливании, которое требуется для обезвоживания кремнекислоты перед первым взвешиванием, образуется силикат бария. В результате обработки остатка фтористоводородной и серной кислотами перед вторым взвешиванием силикат бария снова переходит в сульфат.
Количество сульфата, которЬе выпадает вместе с кремнекислотой, зависит от растворимости этого сульфата. Осаждение никогда не бывает количественным, и сульфат, особенно сульфат кальция, обычно выделяется и при подщелачивании раствора аммиаком вследствие меньшей
794 Гл. XLVI. Сера
растворимости CaSO4 в щелочной среде. В этих случаях результаты определения алюминия, которые вычисляются по разности, получаются, повышенными, а результаты определения щелочноземельных металлов соответственно пониженными. Еще менее правильные результаты получаются, когда нелетучий остаток после удаления кремнекислоты взвешивают вместе с осадком от аммиака.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ СЕРУ
При подготовке пробы для анализа необходимо принять соответствующие меры для предотвращения окисления сульфидных минералов. Тонкое измельчение пирита приводит к потере небольших количеств серы в виде двуокиси серы и к заметному образованию сульфата \ И то, и другое является причиной получения пониженных результатов определения серы, а окисление до сульфата ведет, кроме того, к неправильным выводам относительно состояния, в котором находилась сера в исходном материале. Большей частью достаточно измельчить сульфид до 60—80 меш (d =0,18— 0,25 мм), причем пробу надо не растирать, а разбивать (стр. 891).
Необходимо также принять меры к тому, чтобы анализируемая проба в процессе разложения не загрязнилась серой из атмосферы. Применяемые реактивы должны быть свободными от серы или содержать ее лишь в таких незначительных количествах, чтобы можно было ввести соответствующую поправку на основании результатов «холостого» опыта. Загрязнение серой может быть вызвано нахождением растворов в течение некоторого времени в атмосфере, содержащей сероводород или двуокись серы, но чаще оно происходит вследствие применения для прокаливания и сплавления пламени газа, содержащего серу. Этого загрязнения можно большей частью достаточно успешно избежать защитой тигля от непосредственного соприкосновения с продуктами горения асбестовым щитом (стр. 48). При проведении анализов высокой точности для нагревания следует пользоваться электрическими приборами, например муфельной печью, или же спиртовой горелкой.
Некоторые сульфиды можно полностью окислить обработкой дымящей азотной кислотой при 125° С в запаянной трубке 1 2. Разложение пирита и последующее приготовление раствора для определения серы лучше всего проводить следующим образом \
Помещают 1,3736 г пирита, измельченного до 80 меш (d = 0,175 мм) и высушенного при 100° С, в сухой стакан из химически стойкого «текла емкостью 300 мл, прибавляют 10 мл смеси 2 частей (по объему) жидкого брома и 3 частей четыреххлористого углерода (и тот и другой должны
1 Е. Т. Allen, J. Johnston, Ind. Eng. Chem., 2, 196 (1910); W. S. A 1-1 e n, H. B. Bishop, 8th, Intern. Congr. Appl. Chem., 1—2 [I—II], 48, 1912. Последние авторы показали, что при измельчении пирита не более чем до 100 меш (d = 0,147 мм) большой ошибки не получается. Не наблюдается также окисления, если измельченную таким образом пробу нагревать при 100° С в течение даже 16 ч. Предварительное высушивание влажных образцов должно проводиться при 50—75° С.
2Е. Т. Allen и J. Johnston (цит. выше) вводили 4 мл дымящей азотной кислоты для окисления 0,5 г пирита. Для окисления соединений серы, дающих устойчивые сульфоны (*вещества типа R 'SOj •!<'*). V. С. R о g е г s и G. Dougherty [J. Am. Chem. Soc., 50, 1232 (1928)] рекомендуют применять дымящую азотную кислоту с небольшим количеством NaCl или КС1. Большинство органических веществ, содержащих серу, можно разложить методом взрыва с перекисью натрия и сахаром (стр. 919).
Разложение минералов, содержащих серу
795
быть свободными от серы) и накрывают часовым стеклом. Дают постоять 15 мин при комнатной температуре, время от времени перемешивая, прибавляют 15 мл азотной кислоты и оставляют смесь стоять еще 15 мин при комнатной температуре, изредка взбалтывая. После этого помещают стакан на край паровой бани и дают стоять до прекращения реакции и удаления большей части брома. Затем, подняв часовое стекло и поместив его на согнутые под углом стеклянные палочки, надетые на край стакана, выпаривают раствор непосредственно на паровой бане досуха. Прибавляют 10 мл соляной кислоты, тщательно перемешивают и, снова поместив часовое стекло на стеклянные палочки, выпаривают досуха.
Остаток кремнекислоты обезвоживают нагреванием при 100° С в воздушной бане, лучше в продолжение ночи. Смачивают высушенную массу 4 мл соляной кислоты, спустя 5 мин прибавляют горячую воду и тщательно ополаскивают стекло, стеклянные палочки и стенки стакана горячей водой. Снова надевают на край стакана стеклянные палочки, кладут на них часовое стекло и осторожно кипятят 5 мин. Через 5 мин, когда раствор немного охладится, прибавляют 0,2—0,3 г алюминия в виде порошка и осторожно взбалтывают до обесцвечивания раствора. По' охлаждении удаляют и ополаскивают часовое стекло и палочки холодной водой, а затем фильтруют в высокий стакан емкостью 2500 мл. Промывают стакан, фильтр и осадок 9 раз горячей водой. К фильтрату прибавляют 6 мл соляной кислоты, разбавляют его холодной водой до 1600 мл, тщательно перемешивают и осаждают, как указано в разделе «Определение в раство-, рах, содержащих большие количества сульфатов в присутствии железа и подобных ему элементов» (стр. 803).
Следующее видоизменение описанного метода было с успехом применено для растворения стибнита1. Навеску образца, равную факторному весу, помещают в чашку, накрывают часовым стеклом и вводят через носик чашки 10 мл 10%-ного раствора брома в четыреххлористом углероде. Затем медленно, часто взбалтывая, прибавляют 5 мл брома и оставляют стоять 30—60 мин, время от времени взбалтывая. Частично погружают чашку в стакан с холодной водой, прибавляют 15 мл концентрированной азотной кислоты и оставляют стоять еще 15—30 мин, временами взбалтывая. После этого вводят 15 Л4Л соляной кислоты, дают постоять при комнатной температуре 15— 30 мин, затем медленно нагревают для удаления четыреххлористого углерода и выпаривают до сиропообразной консистенции, но не досуха. Прибавляют еще 10 мл соляной кислоты и снова выпаривают до сиропообразной консистенции, после чего опять вводят 20 мл соляной кислоты. Нагревают до растворения всех растворимых веществ и переносят раствор в коническую колбу емкостью 500 мл. Объем раствора не должен превышать 100 мл. Прибавляют 5 г железной стружки, дают постоять 1—2 ч, фильтруют, тщательно промывают остаток и в дальнейшем поступают, как указано на стр. 803.
В следующем методе пирит окисляют и полностью отделяют железо без применения нелетучих реагентов 2. Приблизительно 0,5 г измельченной до 80 меш (d 0,175 мм) пробы помещают в стакан емкостью 250 мл и обрабатывают 12 мл смеси 3 частей азотной и 1 части соляной кислот, содержащей 4—5 капель брома. Накрывают стакан плотно прилегающим к нему часовым стеклом и оставляют при комнатной температуре на 30 мин. После этого осторожно нагревают на паровой бане до заметного ослабления реакции, снимают часовое стекло и выпаривают жидкость досуха. Затем прибавляют 5 мл соляной кислоты и, снова накрыв часовым стеклом, нагревают до прекращения разбрызгивания, после чего снимают стекло, ополаскивают его и стенки стакана и снова выпаривают досуха. Остаток
1 J. A. Scherrer (Бюро Стандартов"США).
2 А. М. Smoot, Eng. Mining J., 94, 412 (1912).
796
Гл. XLVI. Сера
обрабатывают 25 мл горячей воды при перемешивании. При отсутствии бария или значительных количеств свинца, кальция и стронция нерастворимый остаток не содержит больше серы и его можно не отфильтровывать. Переносят раствор в сосуд для электролиза и осаждают железо на ртутном катоде, как описано в разделе «Методы отделения».
Материалы, содержащие малые количества серы, лучше сплавлять, с хорошо перемешанной смесью карбоната натрия и селитры (12 : 1) в платиновом тигле, помещенном в асбестовом щите (стр. 48). После основательного сплавления (под конец в течение нескольких минут на паяльной горелке) плав выщелачивают водой, содержащей 1—2 капли спирта, для восстановления манганатов, фильтруют и определяют серу в фильтрате, как указано в разделе «Определение в растворах, содержащих умеренные количества сульфатов в присутствии солей щелочных металлов или аммонийных солей» (стр. 802), предварительно подкислив холодный раствор небольшим избытком соляной кислоты. Обычно нет необходимости повторять сплавление и выщелачивание или удалять кремнекислоту, хотя последнее может быть осуществлено выпариванием раствора досуха на паровой бане, смачиванием остатка небольшим количеством соляной кислоты и фильтрованием.
Раствор сульфатов в хлорной кислоте можно выпарить досуха без потерь серной кислоты при условии, что в растворе находится относительно большое количество элемента, образующего малорастворимую соль, например кальция. Выпаривание следует осуществлять при возможно более низкой температуре \
Заслуживает внимания метод разложения сульфатов и извлечения серы’ из некоторых продуктов, особенно содержащих вольфрам или молибден. Этот метод заключается в следующем 1 2. Тонко измельченную пробу нагревают при 1000° С в трубке, через которую проходит водород, предварительно пропускаемый через соляную кислоту. Этим методом сульфаты, например сульфат бария, могут быть полностью разложены с образованием хлоридов (хлорид бария) и выделением всей серы в виде сероводорода.
МЕТОДЫ ОТДЕЛЕНИЯ
Железо и другие элементы, которые осаждаются аммиаком в виде гидроокисей, а не основных сульфатов, можно отделить от сульфат-ионов двукратным осаждением, лучше из солянокислого раствора, избыточным количеством аммиака, как описано в гл. «Молибден» (стр. 359) 3. Образующийся при этом хлорид аммония можно удалить подкислением объединенных фильтратов соляной кислотой, выпариванием до небольшого объема, нагреванием с азотной кислотой (стр. 161) и, наконец, выпариванием с соляной кислотой на паровой бане для удаления азотной кислоты. Разложение нитрата аммония повторным выпариванием с соляной кисло
1 Например, Н. В. Knowles не наблюдал потерь серы, когда этим методом он анализировал известково-натриевое стекло, содержавшее 0,41% SO3, тогда как в растворе одного Na2SO4, содержание серы в котором соответствовало 0,1005 г BaSO4, после выпаривания с 10 мл 60%-ной НС1О4 было обнаружено только 0,0912 г BaSO4.
2 С. М. Johnson, Chemical Analysis of Special Steels, 3d ed., John Wiley and Sons, 1920, p. 122.
3 Пример такого отделения см. E. Hintz, H. Weber, Z. anal. Chemi, 45, 31 (1916).
Методы определения ' 1Q7
той (стр. 161) или сульфата аммония выпариванием с царской водкой достигается не так легко, но достаточно удовлетворительно для преследуемой в данном случае цели.
Для отделения железа, меди, хрома, молибдена и никеля перед осаждением сульфата бария рекомендуется 1 применять электролиз с ртутным катодом в слабокислой среде (стр. 165). При анализе пирита, в результате первоначальной обработки пробы «обратной царской водкой» (обратное соотношение между соляной и азотной кислотами по сравнению с обычно принятым) и бромом и выпаривания на паровой бане, как указано в разделе «Разложение минералов, содержащих серу» (стр. 794), остается достаточное для электролиза количество серной кислоты. Можно прибавить небольшое количество соляной или хлорной кислоты. Электролиз проводится при силе тока 0,8—1 а в течение 5—6 ч или при меньшей силе тока — в продолжение ночи. По окончании осаждения ополаскивают •стенки сосуда и возможно полнее сливают электролит декантацией в стакан емкостью 300 мл. Обмывают стенки сосуда для электролиза и ртуть небольшими порциями воды, которую сливают декантацией в основной раствор, и затем освобождают этот раствор от суспендированных веществ фильтрованием и промыванием остатка на фильтре.
При невозможности удалить железо значительно лучшие результаты получаются, если перед осаждением сульфата бария восстановить железо до двухвалентного состояния. Для восстановления можно применять алюминий, поступая, как описано в разделе «Разложение минералов, содержащих серу» (стр. 794), цинк или чистое железо. Этот способ применяется также для отделения некоторых других восстанавливающихся элементов, например свинца или сурьмы.
Наиболее простым методом отделения сульфат-ионов от магния и щелочноземельных металлов является сплавление с карбонатом натрия, выщелачивание плава водой, фильтрование и промывание нерастворимого остатка горячим 1 %-ным раствором карбоната натрия. Для полного извлечения сульфата эту операцию необходимо повторить один или несколько раз. Фильтраты и промывные воды объединяют.
Для отделения элементов сероводородной группы, а также для восстановления хлорида железа (III) рекомендуется 2, обработка сероводородом в слабосолянокислом растворе, однако этот метод нам кажется малонадежным вследствие возможного окисления сульфид-ионов.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Литература об определении серы в виде сульфата бария очень обширна и трудности, связанные с получением точных результатов, хорошо известны. Большинство исследований посвящено определению больших количеств серы, как, например, в имеющем промышленное значение минерала — пирите. В таких случаях хорошие результаты получаются только тогда, когда осаждение проводится в точно определенных условиях и имеющие место значительные положительные и отрицательные ошибки взаимно компенсируются.
При определении сульфат-ионов осаждением хлоридом бария из раствора, содержащего только серную кислоту, получается отрицательная
1 А. М. Smoot, цит. выше.
2 G. von Knorr е, Chem. Ind., 28, 9 (1905).
798
Гл. XLVI. Серег
ошибка вследствие растворимости сульфата бария в растворе и в промывной жидкости и положительная ошибка, связанная с соосаждением хлорида бария. Ошибки эти могут быть равны по величине (1—2 мг), когда осаждение проводится медленным прибавлением хлорида бария в умеренном избытке к горячему разбавленному раствору серной кислоты. Раствору затем дают отстаиваться в течение нескольких часов \ В анализах высокой точности следует вводить поправки на обе эти ошибки. Более правильно вовсе не вводить поправок, чем вводить поправку только на растворимость сульфата бария, как это часто делается.
Когда сульфат-ионы осаждаются из раствора, содержащего сульфаты щелочных металлов, возникают еще две ошибки, причем обе отрицательные, связанные с соосаждением среднего, или кислого сульфата щелочного металла. В обоих случаях пониженные результаты вызываются замещением ' бария во взвешиваемом осадке на элемент с меньшим атомным весом, а при осаждении кислого сульфата, кроме того, происходят потери серной кислоты в процессе прокаливания осадка. Когда присутствуют аммонийные соли, величина этих ошибок возрастает, так как при этом улетучивается и основание, и кислота. Потери серной кислоты вследствие улетучивания, очевидно, частично возмещаются тем хлоридом бария, который захватывается осадком сульфата бария. Однако в прокаленном осадке может содержаться лишь очень мало хлорида бария, если этим осадком были захвачены значительные количества кислого сульфата или сульфата аммония. В этом случае потеря за счёт растворимости сульфата бария не компенсируется наличием хлорида бария в прокаленном осадке, и таким образом при определении сульфат-ионов в растворах, содержащих сульфаты или хлориды, обычно получаются пониженные результаты.
Для установления 1 2 степени погрешности, вызываемой растворимостью сульфата бария и соосаждением сульфата натрия и кислого сульфата, взвешенное количество раствора сульфата натрия, достаточное для получения приблизительно 2 г BaSO4, обрабатывали соответствующими реактивами. Раствор разбавляли до 350 мл, нагревали до кипения и осаждали сульфат избыточным количеством в 1 —1,5 мл 10%-ного раствора хлорида бария, который вливали очень тонкой струей по стенкам стакана. Раствор затем отстаивали 18 ч. Осадок отфильтровывали через бумажный фильтр и промывали водой до тех пор, пока содержание хлоридов в 25 мл промывной воды не снижалось до 0,005 мг, затем сушили в платиновом тигле и осторожно прокаливали до постоянной массы.
При осаждении из растворов, содержавших в 350 мл 0,2; 1,5; 10; 20 и 100 мл соляной кислоты, потери вследствие растворимости сульфата бария в фильтрате и 350 мл промывной воды составляли соответственно 0,9; 1,50; 3,3; 3,9; 12,0 и 36,0 мг BaSO4. Средняя потеря, выраженная в миллиграммах BaSO4, обусловленная соосаждением Na2S04, составляла 8,1 мг, когда в растворе содержалось 0,2 мл соляной кислоты, и 11,4 мг, когда в растворе, кроме того, находилось 10 г NaCl. Средняя потеря, выраженная в миллиграммах BaSO4, вследствие улетучивания серной кислоты из соосажденного кислого сульфата натрия, равнялась 0,5 мг, когда в растворе сульфата натрия содержалось 0,2 мл соляной кислоты, и 8,8 мг при содержании 0,2 мл соляной кислоты и 10 г NaCl.
1 Так, например, Т. W. Richards и Н. G. Parker [Proc. Am. Acad. Arts Sci., 31, 67 (1895—1896)], осадив сульфат бария из 10,2107 г и 10,2189 г разбавленного раствора серной кислоты, получили 0,7804 г и 0,7821 г BaSO4, причем поправка на растворимость сульфата бария составляла 0,0011 г и 0,0017 г BaSO4, а на сооса-жденный хлорид бария соответствовала 0,0005 г и 0,0017 г BaSO4. Конечный правильный результат, таким образом, соответствует 3,213% и 3,215% H2SO4 в растворе. Точное среднее содержание H2SO4 в растворе, найденное тщательным алкалиметрическим титрованием составляет 3,214%.
2 Е. Т. Allen, J. Johnston, J. Am. Chem. Soc., 32, 588 (1910).
Методы определения 799!
* Все описанные авторами затруднения, связанные с тем, что осадок сульфата бария оказывается загрязненным хлоридом бария или сульфатами щелочных металлов, железа и т. п., исчезают с появлением возможности переосаждения сульфата бария \ поскольку был найден реактив, в котором этот осадок растворяется — щелочной раствор этилендиаминтетраацетата натрия (комплексона III, трилона Б)..Осадок сульфата бария растворяют на фильтре в горячем 3—5%-ном аммиачном растворе указанного реактива, тщательно промывают фильтр горячей водой, прибавляют 1—2 капли раствора хлорида бария и осаждают сульфат бария подкислением соляной кислотой по метиловому красному. Доп. ред*.
Растворимость сульфата бария возрастает с повышением кислотности раствора и с увеличением объема промывной жидкости. Она несколько выше в присутствии хлоридов. Степень соосаждения хлорида бария возрастает с повышением концентрации кислоты и сульфата в растворе и с увеличением скорости осаждения. Она значительнее, когда раствор сульфата вливают в- раствор хлорида бария, чем при обратном сливании растворов.
Количества сульфатов щелочных металлов и аммония, захватывающиеся осадком, увеличиваются с повышением концентрации хлоридов щелочных металлов в растворе и уменьшаются с повышением скорости осаждения и продолжительности настаивания раствора с осадком. Захват осадком кислых сульфатов возрастает с повышением кислотности и концентрации хлоридов щелочных металлов и аммония в растворе и уменьшается с увеличением скорости осаждения и продолжительности стояния раствора с осадком перед фильтрованием. Соли калия сильнее загрязняют осадок, чем соли натрия.
При содержании в растворе других веществ, помимо щелочных металлов, возникают еще большие затруднения. Некоторые из этих веществ, в частности нитраты, заметно окклюдируются осадком. В присутствии таких соединений, как сульфат хрома, сомнительна возможность достижения количественного осаждения.
В присутствии хрома (III) лучшие результаты получаются, если раствор после осаждения оставлять стоять в течение 12 ч или больше.
Для определения сульфатов в присутствии хроматов приготовляют раствор, содержащий приблизительно-1 г хромата и 2 мл соляной кислоты в 100 мл. Прибавляют 20 мл 1%-ного раствора хлорида бария и некоторое время нагревают. Фильтруют, осадок умеренно промывают, прокаливают и сплавляют с небольшим количеством карбоната натрия. Плав выщелачивают водой, фильтруют, фильтрат подкисляют соляной кислотой и восстанавливают хром спиртом. Регулируют кислотность и осаждают хлоридом бария, как обычно. Достаточно удовлетворительные результаты можно получить быстрее; для этого ведут осаждение хлоридом бария из уксусно-солянокислого раствора после предварительного восстановления всего хромата до трехвалентного состояния1 2. ‘
Сульфат железа (Ill) соосаждается с сульфатом бария, и при последующем прокаливании, с одной стороны, имеют место потери серной кислоты, а с другой — осадок загрязняется окисью железа. Такие соединения, как сульфат циркония, при низкой кислотности, которая требуется для осаждения сульфата бария, гидролизуются, и осадок значительно загрязняется окисью ццркония. В связи с этим необходимо стараться предварительно
1 R. Pribil, D. М а г ic о v a, Chem. Listy, 44, 97 (1950).
2 Подробнее см. Н. Н. Willard, R. Schneidewind, Trans. Am. Electrochem. Soc., 56, 333 (1929).
800 Гл. XL VI. Сера
тельио отделять тяжелые металлы и получать растворы, содержащие одну только серную кислоту или сульфат натрия и свободные от других примесей, кроме хлорида натрия. Осадок сульфата бария обычно собирают на бумажном фильтре, так как при этом удобно и легко исследовать взвешенный осадок. Можно фильтровать также через тигель Гуча или Мунро, если последующие операции это позволяют.
Промывание осадка сульфата бария не должно быть излишне продолжительным вследствие заметной его растворимости (2,4 мг в 1 л воды при 20° С). В точных анализах промывание следует прекращать, когда 25 мл промывных вод дают лишь слабую опалесценцию с нитратом серебра.
При прокаливании осадка сульфата бария необходимо следить за тем, чтобы фильтр сперва обуглился без воспламенения, а затем уголь медленно сгорал при возможно более низкой температуре. При быстром прокаливании может происходить распыление осадка. Если прокаливание фильтра с осадком проводится в окислительной атмосфере и углерод сгорает при температуре ниже 600° С, то восстановления сульфата можно не опасаться \ Окончательное прокаливание должно происходить при температуре не выше 350° С, если осадок получен из чистого раствора серной кислоты и нужно ввести поправку на окклюдированный хлорид бария, который при более высоких температурах частично разлагается. Когда •садок выделен из раствора сульфата натрия и необходимо ввести поправку на свободную кислоту и соосажденный сульфат натрия, предварительное нагревание должно проводиться приблизительно при 105° С, а окончательное прокаливание при 1000° С (см. мелкий шрифт на стр. 801), чтобы окклюдированный кислый сульфат полностью перешел в средний сульфат. Если не предполагают вводить поправку, то лучше прокаливать приблизительно при 900° С, хотя осадок заметно не диссоциирует при температурах до 1400° С, когда он чист и прокаливание проводится в атмосфере чистого сухого воздуха 1 2. При наличии примесей, например кремнекислоты или окиси железа, разложение сульфата с выделением серного ангидрида начинается примерно при 1000° С. Обрабатывать прокаленный осадок серной кислотой нет надобности, кроме того, это, естественно, лишает возможности вводить поправку на окклюдированный хлорид бария.
Ниже приводятся три метода анализа. Первый из них предназначен для определения сульфат-ионов в растворах, содержащих только серную кислоту, и для точных анализов растворов, не содержащих других веществ, кроме сульфатов и хлоридов щелочных металлов. При точных анализах серной кислотьт и во всех случаях, когда в растворах находятся соли щелочных металлов, необходимо вводить поправки, согласно приведенному ниже методу. Когда не предполагается вводить поправки, анализ растворов, содержащих умеренные количества сульфат-ионов в присутствии солей щелочных металлов или аммонийных солей, лучше проводить методом описанным на стр. 802, так как происходящие при этом потери возмещаются окклюзией хлорида бария. Метод, приведенный на стр. 803, предназначается в первую очередь для определения больших количеств сульфат-ионов в растворах, содержащих значительные количества железа (как, например, при анализе пирита) и умеренные количества других элементов, например алюминия, цинка или кальция.
1 W. Mostowitsch, Metallurgie, 6, 45 (1909); Chem. Zentr., 80, П, 1038 (1909); С. А., 5, 841 (1911).
' 2 W. Mostowitsch, цит. выше.
Методы определения
801
Определение в растворах, содержащих только серную кислоту
Раствор, содержащий не более 0,5 г серной кислоты, разбавляют с таким расчетом, чтобы концентрация кислоты составляла приблизительно 0,002 г в 1 мл. Нагревают до кипения и, непрерывно перемешивая, прибавляют по каплям из бюретки или из воронки для осаждения (имеющей трубку с оттянутым капиллярным концом) 10%-ный раствор хлорида бария в таком количестве, чтобы ввести 1—2 мл избытка. После осаждения раствор оставляют стоять в течение 18 ч, затем осадок отфильтровывают через бумажный фильтр и промывают водой до тех пор, пока 25 мл промывных вод не дадут лишь едва заметную опалесценцию с нитратом серебра. Влажный фильтр с осадком помещают во взвешенный платиновый тигель, высушивают, затем обугливают бумагу, без воспламенения, и осторожно сжигают уголь при хорошем доступе воздуха. Под конец прокаливают при 375° С до постоянной массы.
Предложен1 метод, в котором осадок становится хлопьевидным и может быть отфильтрован в течение 30 мин после осаждения. Для этого к раствору прибавляют 1 — 2 мл раствора агар-агара, содержащего 1 мг в 1 мл. Этот раствор приготовляют суспендированием 0,5 г агар-агара в 50 мл холодной воды, прибавлением 450 мл почти кипящей воды и последующим нагреванием до кипения при непрерывном перемешивании. Раствор агар-агара следует вводить по каплям, быстро перемешивая в течение 10 сек после добавления каждой капли.
Как уже отмечалось, результаты получаются ошибочными, если увлеченное осадком количество хлорида бария не компенсирует потери сульфата бария в фильтрате и промывных водах. В точных анализах для, устранения этой ошибки необходимо определять значения обеих поправок, как изложено на стр. 802 (см. мелкий шрифт).
В растворах, содержащих только сульфаты или сульфаты и хлориды щелочных металлов, сульфат-ионы можно осадить таким же образом после добавления избытка соляной кислоты в количестве 0,05 мл на каждые 100 мл раствора. Как было указано, в этих условиях получаются пониженные результаты и при выполнении точных анализов необходимо вводить все четыре поправки (см. мелкий шрифт).
При добавлении к отдельным порциям по 100 мл титрованного раствора серной кислоты по 10 г различных солей были получены следующие результаты:
Введенная соль............ — NaCl FeCl3 NH4C1 NH4NO3
Масса BaSO4 ........... . 0,5992 0,5883 0,5896 0,5849 0,6113
Несколько лучшие, но также неточные результаты были получены после обработки непрокаленного осадка BaSO4 соляной кислотой, разбавления или нейтрализа-, ции раствора до требуемой кислотности и прибавления еще небольшого количества ВаСЬ:
Введенная соль...................NaCl FeCl3 NH4C1 NH4NO3
Масса BaSO4, г................... 0,5896 0,5950 0,5872 0,6014
Получение повышенных результатов в присутствии NH4NO3 вызвано окклюзией осадком нитрата бария.
Поправка на растворимость сульфата бария. Выпаривают фильтрат и промывные воды в платиновой чашке досуха. Остаток обрабатывают небольшим количеством воды или, если требуется, сильно разбавленной соляной кислотой;
1 Е.; J. Bogan, Н. V, Moyer, Ind. Eng. Chem., Anal. Ed., 14, 849 (1942), 51 Заказ 522.
802
Гл. XLVI. Сера
той; фильтруют через маленький фильтр, выделившийся осадок промывают до удаления хлоридов и присоединяют к основному осадку сульфата бария.
Поправка на содержание в осадке BaSO4 хлорида бария. Эту поправку определяют одной из следующих операций:
1) осадок сплавляют с карбонатом натрия, плав выщелачивают водой, водную вытяжку подкисляют азотной кислотой и осаждают нитратом серебра. Массу ВаС1г вычисляют, исходя из полученной массы хлорида серебра;
2) обрабатывают осадок горячей серной кислотой, поглощая выделяющийся при этом хлористый водород раствором нитрата серебра, и в дальнейшем поступают1, как в п. 1.
Поправка на содержание в осадке сульфата бария сульфатов щелочных металлов. Осадок растворяют примерно в 15 мл серной кислоты в платиновой чашке. По охлаждении медленно при сильном перемешивании вливают раствор в 350 мл воды и для облегчения фильтрования нагревают. Фильтруют, не промывая осадка, и выпаривают прозрачный фильтрат до удаления всей серной кислоты. Остаток растворяют в воде, фильтруют через маленький фильтр во взвешенный платиновый тигель, выпаривают раствор досуха, осторожно нагревают до красного каления и взвешивают. Таким образом определяется примерно 90% окклюдированного сульфата щелочного металла. Оставшийся сульфат можно определить повторением операции или количество его можно эмпирически принять равным 10%.
Поправка на содержание свободной кислоты. Определение этой поправки является наиболее сложным. Оно осуществляется обработкой не прокаленного осадка. Рекомендуется 2 поступать следующим образом. Осадок высушивают при 105° С, переносят его во взвешенную платиновую лодочку и помещают в платиновую трубку для сжигания. Соединяют один конец трубки с источником сухого чистого воздуха, а другой конец — С двумя соединенными последовательно либихов-скимй кали-аппаратами, содержащими 1%-ный раствор пергидроля (30%-ной перекиси водорода). Пропускают слабую струю воздуха и постепенно нагревают трубку приблизительно до 1000° С. Охлаждают и взвешивают лодочку. Оба поглотительных раствора объединяют, выпаривают для удаления перекиси водорода, а затем осаждают хлоридом бария и вычисляют массу серной кислоты.
Нахождение величин всех поправок. Для того чтобы найти величины всех поправок с одним осадком BaSO4, проводят сначала описанную выше операцию определения потери серной кислоты вследствие улетучивания, взвешивают загрязненный сульфат бария, затем определяют хлорид-ионы отгонкой соляной кислоты из горячего сернокислого раствора неочищенного сульфата бария и, наконец, определяют окклюдированный сульфат щелочного металла в сернокислом растворе.
Определение в растворах, содержащих умеренные количества сульфатов в присутствии солей щелочных металлов или аммонийных солей 3
Водный или солянокислый раствор, содержащий 0,025 г серы в виде сульфат-ионов на 100 мл и предпочтительно свободный от других солей, кроме солей натрия, калия, или аммония, нейтрализуют по метиловому оранжевому, затем добавляют по 1 мл соляной кислоты на каждые 100 мл раствора и нагревают до кипения.
В другой стакан наливают 10%-ный раствор хлорида бария в количестве на 7—8 мл большем, чем это требуется для связывания всей серной кислоты, разбавляют до 100 мл, нагревают до кипения и быстро при сильном перемешивании вливают в этот раствор раствор сульфата. Дают постоять на краю паровой бани 30 мин и сливают прозрачный раствор декантацией через фильтр. Осадок промывают 3 раза декантацией горячей водой, переносят на фильтр и продолжают промывать до тех пор, пока при испытании промывных вод на хлориды не образуется лишь крайне
1 G. A. Hulett, L. Н. D u s с h a k, Z. anorg. Chem., 40, 196 (1904).
2 Е. Т. Allen, J. Johnston, J. Am. Chem. Soc., 32, .588 (1910).
3 Это в основном метод Е. Hi n t z и Н. Weber [Z. anal. Chem., 45, 31 (I960)].
Методы определения
803
слабая опалесценция. Помещадот влажный фильтр во взвешенный тигель, осторожно нагревают, пока он не высохнет, затем обугливают фильтр без воспламенения, осторожно сжигают углерод в сильно окислительных условиях и, наконец, прокаливают при 900° С до постоянной массы. Осадок можно собрать в тигле Гуча или Мунро и прокалить в муфеле j
При рядовых определениях серы в таких материалах этим или аналогичным методом можно получить более точные результаты, затратив немного труда на составление графика поправок. Для составления такого графика проводят ряд определений серы в растворах, содержащих известные количества сульфат-ионов и все те примеси, которые находятся в растворах анализируемого материала. Осаждение проводится методом, выбранным для анализа. Найденные массы сульфата бария наносят на график рядом с вычисленными на основании взятых для осаждения количеств сульфат-иона, и таким образом по графику можно прочесть исправленные результаты х.
Определение в растворах, содержащих большие количества сульфатов в присутствии железа и подобных ему элементов
Ход определения. Прозрачный раствор объемом около 1600 мл, содержащий 10 мл соляной кислоты, охлаждают до 20—25° С, накрывают часовым стеклом и затем прибавляют из прибора для осаждения (рис. 28), не перемешивая, 125 мл 5%-ного раствора хлорида бария со скоростью
Рис. 28. Прибор для осаждения.
Емкость 130 мл', выходное отверстие сделано такого диаметра, чтобы из воронки выливалось 5 мл жидкости в минуту.
Рис. 29. Прибор для фильтрования больших количеств жидкости.
5 мл в 1 мин. По окончании прибавления раствора осторожно перемешивают и дают осадку отстаиваться, лучше в течение ночи. Фильтруют через взвешенный тигель Гуча емкостью 35 мл или фильтрующий стеклянный тигель, отсасывая посредством прибора, изображенного на рис. 29. Прежде чем установить сифон и пробку, тигель наполняют водой почти до краев, затем выкачивают воздух из склянки и продолжают фильтровать до тех пор, пока большая часть жидкости не будет удалена сифонированием. Удаляют и промывают сифон и пробку, переносят осадок в тигель, соскребая
1 См. С. Е. Waters, Bur. Standards Tech. Paper, 177, 25. 51*
804
Гл. XLVI. Сера
и смывая его возможно меньшим количеством воды, промывают осадок 6 раз холодной водой, осторожно высушивают и прокаливают, лучше всего в печи при 900° С в течение 30 мин. Охлаждают в эксикаторе и взвешивают.
Объемные методы
Из объемных методов определения серы следует упомянуть бензидиновый метод, метод с хроматом бария и метод отгонки. Первый метод может быть применен для определения больших количеств серы, как, например, в пирите, второй — для определения средних количеств серы, например в угле, и третий — для определения малых количеств серы, как, например, в стали.
Все эти методы применяются главным образом в массовых анализах.
Наилучшие варианты бензидинового метода1 сводятся к осаждению сульфат-ионов солянокислым бензидином в нейтральной или слабокислой среде (pH = 4). Отфильтрованный осадок сернокислого бензидина промывают 5 раз водой, порциями по 3 мл, и титруют при 50° С раствором едкого натра по фенолфталеину2. Метод применим в присутствии железа (II), меди, кобальта, цинка, никеля, марганца и алюминия. Железо (III) мешает определению.
Согласно методу определения с хроматом бария 3, сульфат осаждают в очень слабо солянокислом растворе избыточным количеством солянокислого раствора хромата бария. После прибавления к раствору аммиака сульфат бария и избыток хромата бария отфильтровывают, а перешедший в раствор хромат, количество которого эквивалентно содержанию сульфата, определяют титрованием тиосульфатом (после прибавления иодида калия и подкисления фильтрата). Применению метода мешают значительные количества кальция, нитратов и хлорида аммония.
Так называемый метод отгонки 4 применяется для определения серы, находящейся в виде сульфида и количественно переходящей в сероводород при обработке анализируемого материала кислотой. Выделяющийся сероводород обычно поглощают аммиачным раствором хлорида кадмия или сульфата цинка и после прибавления кислоты титруют раствором иода или смеси иодида и йодата.
1 W. М filler, Вег., 35, 1587 (1902); F. R a s с h i g, Z. angew. Chem., 16, €17, 818 (1903); C. Friedheim, O. Nydegger, Z. angew. Chem., 20, 9 (1907); Chem. Ztg., 52, 318 (1938); G. v. К n о г r e, Chem. Ind., 28, 2 (1905); Chem. Ztg., 34, 405 (1910).
2 Согласно W. B. Meldrum и I. G. Newlin [Ind. Eng. Chem., Anal. Ed., 1, 231 (1929)], растворимость сернокислого бензидина на 1000 г раствора при 25° С составляет 0,098 г в воде; 0,542 г в 0,239 н. растворе НС1; 0,942 г в 0,53 н. растворе НС1 и 1,253 г в 1,009 н. растворе НС1. В 3,5 н. растворе НС1 растворимость солянокислого бензидина ниже, чем сернокислого бензидина.
3 С. W. Hinman, Am. J. Sci., [3], 14, 478 (1877); J. D. Pennock, D. A. Morton, J. Am. Chem. Soc., 25, 1265 (1903); M. H о 11 i g e r, Z. anal. Chem., 49, 84 (1910).* A. S. К о m а г о w s k y, Ch. Ztg. 39, 498 (1907); А. С. К o-маровский, А. Коган. Журнал научно-исследовательских кафедр в Одессе, 1, 11 (1924).*
4 Методы отгонки наиболее применимы для определения малых количеств серы ® сталях и подробно описаны в руководствах, посвященных анализу черных металлов [см. Лендель, Гофман и Брайт, Анализ черных металлов, Госхимтех-издат, 1934,* а также А. М. Дымов, Технический анализ руд и металлов, Метал-лургиздат, 1949*]. Метод, по которому для предотвращения окисления сероводорода высшими окислами в процессе растворения таких материалов, как цемент, применяется хлорид олова II), см. Н. A. Bright, J. Research NBS, 18, 137 (1937).
Методы определения
805
Когда сероводород поглощают раствором кадмия, очень важно, чтобы этот раствор не подвергался действию прямого солнечного света, в противном случае получаются пониженные результаты. Действие солнечного' света на сульфид цинка мало заметно.
Так, например, в образцах стали, содержавших 0,092% серы, при поглощении сероводорода на прямом солнечном свету аммиачным раствором, хлорида кадмия было найдено 0,046%, аммиачным раствором ацетата кадмия — 0,063%, а аммиачным раствором сульфата цинка — 0,090% серы. Дальнейшие исследования показали, что погрешность определения вызвана действием прямого солнечного света на сульфид кадмия, а не на раствор поглотителя.
Турбодиметрический и колориметрический методы
Небольшие количества растворимых сульфатов лучше всего определять сравнением в нефелометре (в одинаковых условиях) мути, образующейся при обработке хлоридом бария анализируемого и стандартного растворов х. Очень малые количества серы (0,001 мг), находящейся в виде легко разложимых сульфидов, удобно определять колориметрически, сравнением пятен, которые образуют анализируемый и стандартный растворы на бумаге, пропитанной ацетатом свинца 1 2.
Описан также фотометрический метод определения серы в металлах 3.
1 Cf. R. В. Rudy, J. Research NBS, 16, 555 (1936).
2 W. A. D r u s h e 1, C.M. Elston, Am. J. Sci., [4], 42, 155 (1916);
С. E. L a c h e 1 e, Ind. Eng, Chem., Anal. Ed., 6, 200 (1934).
3 C. L. Luke, Anal. Chem., 11, 1369 (1949). Металл растворяют в HC1 -f-
+ HNO3, Сульфат восстанавливают до сульфида иодистоводородной кислотой, отгоняют в виде H.,S и светопоглощение золя сульфида свинца в дистилляте измеряют при 370 ммк.
Глава XLVII
ХЛОР, БРОМ И ИОД
Хлор наиболее распространен в породах, богатых натрием, и особенно в породах с низким содержанием кремнекислоты, в состав которых входит нефелин. Иногда хлор встречается и в породах, не содержащих нефелина, а в редких случаях и в кварцевых рудах х. Хлор является важной составной частью некоторых породообразующих минералов, таких, как содалит (нозелит) Na4'(AlGl) Ah (SiO4)3 и скаполиты п (Ga4AleSieO2) + т (Na4Al3SieO24Cl) (re : т от 3:1 до 1: 2). Хлор обычно находится в виде хлорида натрйя в морской воде и в каменной соли и встречается также в ряде минералов, как, например, в карналлите KMgCl3-6H2O, кераргирите AgGl и в некоторых апатитах Са5 (Cl, F, ОН) (РО4)3. Концентрация хлора в горных породах редко превосходит 0,2—0,3%, но в содалитовых породах -содержание его может превышать 1%.
Бром в свободном состоянии в природе не встречается. Он обычно находится в соединении с щелочными и щелочноземельными металлами и поэтому содержится в морской воде, во многих минеральных источниках и в солевых отложениях. Второстепенное значение имеет нахождение брома в углях, в природной селитре и в минералах бромаргирите AgBr иэмболите Ag (Cl, Br).
Иод является наименее распространенйым элементом группы галогенов. Он находится в морской воде, в некоторых минеральных источниках и в некоторых редких минералах, особенно в виде иодидов серебра, меди и свинца. Лаутарит — иодат кальция — встречается в залежах чилийской селитры1 2.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа наличие галогенов редко вызывает затруднения. Исключение составляют анализы продуктов, содержащих такие элементы, как свинец и серебро, которые образуют нерастворимые хлориды, или ртуть и сурьма, образующие с галогенами летучие соединения. В таких случаях требуются специальные способы обработки, которые изложены в следующем разделе.
1 Н. S. Washington, Chemical Analysis of Rocks, 3d ed., John Wiley and Sons,- 1919, p. 20.
2 Об определении и нахождении иода в фосфатных породах см. W. L. Hill. К. D. Jacob, J. Assoc. Offic. Agr. Chemists, 16, 128 (1933).
Разложение соединений галогенов
807
РАЗЛОЖЕНИЕ СОЕДИНЕНИЙ ГАЛОГЕНОВ
Разложение пород и минералов, содержащих хлор, см. часть III (стр. 1017).
Нерастворимые в воде хлориды следует разлагать нагреванием с соответствующими растворителями или сплавлением. Хлорид свинца обрабатывают бикарбонатом натрия или калия при нагревании. Хлорид серебра сплавляют с карбонатом натрия или калия и плав выщелачивают водой, а хлорид ртути (I) обрабатывают едким натром или едким кали. Соединения, в которых хлор находится частично или полностью в неонизированном состоянии, следует разлагать осторожным прокаливанием, (например, перхлораты) или нагреванием с раствором карбоната натрия [например, хлорид ртути (II)].
При анализе стекла хлор был успешно определен одним из нас (Лен-дель) следующим образом. Стекло обрабатывали раствором сульфата серебра во фтористоводородной и хлорной кислотах при постепенном нагревании и выпаривании до появления паров хлорной кислоты. По охлаждении раствор разбавляли, осадок хлорида серебра отфильтровывали и очищали, для чего его растворяли в аммиаке и снова осаждали подкислением раствора азотной кислотой. Возможно, что такую обработку можно применять и для анализа некоторых минералов.
Методы разложения минералов, содержащих бром и иод, в общем аналогичны методам, описанным для минералов, содержащих хлор.
Для определения галогенов в органических соединениях описан метод, основанный на окислении анализируемого вещества дымящей серной кислотой и дистилляции галогена в приборе, состоящем из притертых друг к другу стеклянных частей х.
Описан1 2 также упрощенный метод, основанный на прокаливании анализируемой пробы с известью; по этому методу пробу взвешивают в желатиновой капсюле и прокаливают в трубке из стекла пайрекс, наполненной гашеной известью, а затем после растворения остатка в азотной кислоте определяют галогены титрованием методом Фольгарда в варианте Колдуэлла—Мойера (стр. 813).
При анализе галогенопроизводных дифенила и • дифенилбензола можно 3 содержащиеся в них галогены количественно перевести в Рало-гениды натрия методом взрыва с перекисью натрия и сахарным углем (стр. 919).
Из многих алифатических, ароматических, алициклических и гетероциклических соединений галогены можно удалить обработкой сплавом никель — алюминий в растворе едкой щелочи 4.
Описан 5 также метод, проверенный на 36 различных органических соединениях, содержащих галогены; в этом методе анализируемое вещество разлагают испарением в струе горючего газа, который горит в форсунке.
1 J. J. Thompson, U. О. Oakdale, J. Am. Chem. Soc. 52, 1195 (1930).
2 R. H. Kimball, L. E. Tufts, Ind. Eng. Chem., Anal. Ed., 10, 530 (1938).
3 F. E. Beamish, Ind. Eng. Chem., Anal. Ed., 6, 352 (1934).
4 E. Schwenk, D. Papa, H. Ginsberg, Ind. Eng. Chem., Anal, Ed., 15, 576 (1943).
5 P. K. Winter, Ind. Eng. Chem., Anal. Ed., 15, 571 (1943).
808
Гл. XLVII. Хлор, бром и иод
МЕТОДЫ ОТДЕЛЕНИЯ
Основной метод отделения хлорид-ионов основан на осаждении их в виде хлорида серебра. Применению этого метода мешают: 1) иодиды, бромиды, цианиды и роданиды, также осаждающиеся нитратом серебра; 2) хлориды олова и сурьмы, гидролизующиеся в нейтральных или слабокислых растворах; 3) хлорид платины (IV), который увлекается хлоридом серебра; 4) хлориды хрома и ртути, частично осаждающиеся нитратом серебра.
Олово можно отделить от хлорид-ионов разбавлением раствора до 200—300 мл, нейтрализацией раствора до щелочной ре^Хции по метиловому оранжевому, кипячением в течение 1—2 мин с 2—3 г нитрата аммония, фильтрованием и промыванием осадка 1 %-ным раствором нитрата аммония. Растворы хлорида платины (IV) следует выпарить с карбонатом натрия, взятым в небольшом избытке; остаток нагреть до начала плавления и плав выщелочить водой.
Хром и алюминий лучше всего отделять повторным осаждением аммиаком.
Сурьму и ртуть наиболее целесообразно отделять осаждением сероводородом.
Если хлорид-ионы надо определить после выделения элементов сероводородной группы, то в связи со склонностью некоторых сульфидов захватывать хлор осаждение сероводородом следует проводить в разбавленном растворе, который насыщают сероводородом и затем некоторое время нагревают. После фильтрования основную массу сероводорода можно удалить насыщением раствора углекислым газом, а оставшуюся часть удобно разрушить окислением перекисью водорода или перкарбонатом калия К2С2Ов, добавив предварительно аммиак до щелочной реакции. Раствор затем кипятят для разрушения окислителя, точно подкисляют азотной кислотой и обрабатывают, как описано в разделе «Хлор» (стр. 811).
Иодиды обычно отделяют от хлоридов окислением соответствующим реагентом в разбавленном сернокислом растворе и отгонкой выделяющегося иода. В качестве окислителя чаще всего применяют 1 азотистую кислоту или сульфат железа (III). Для осуществления разделения растворяют не более 0,5 г смеси галогенидов в конической колбе емкостью 1000 мл в 400 мл воды, прибавляют 10 мл разбавленной (1 : 1) серной кислоты и затем для окисления иодистоводородной кислоты прибавляют 2 г чистого нитрита натрия или насыщают раствор азотистой кислотой, которую получают из нитрита в отдельной колбе. Вставляют в колбу воронку с короткой трубкой и кипятят раствор до тех пор, пока он не обесцветится и лакмусовая бумажка не перестанет окрашиваться в серо-синий цвет при обработке ее парами этого раствора в течение 2 мин. Если для окисления иодистоводородной кислоты пользуются сульфатом железа (III) вместо нитрита, в раствор вводят 3 мл азотной кислоты и 2 г сульфата железа (III) [в виде железных квасцов или сульфата железа (III), полученного окислением сульфата железа (II) в концентрированном растворе кипячением с 0,3 мл азотной кислоты]. Иод удаляют, как описано выше, и затем, прибавив 1 мл азотной кислоты, проверяют полноту удаления иода, продолжая отгонку еще некоторое время.
1 F. A. Gooch, F. W. М а г, Am. J. Sci., [3], 39, 293 (1890),
Методы отделения
809'
Бром и (хлор) обычно отделяют от иода методом Гуча 1 следующим образом. Нейтральный раствор, содержащий не более 0,25 г каждого галоида в виде нелетучей соли и предпочтительно свободный от посторонних солей, разбавляют до 700 мл в конической колбе емкостью 1000 мл, прибавляют 2—3 мл разбавленной (1 : 1) серной кислоты и затем 2—3 г нитрита натрия, свободного от галогенов. Вставляют в колбу воронку с короткой трубкой и кипятят раствор до удаления иода. Это продолжается не долее 45 мин. Объем раствора при этом не должен уменьшиться более чем до 500 мл. В случае надобности выделяющийся иод можно количественно улавливать 2, для чего операцию проводят в закрытой колбе, снабженной стеклянной пробкой с вводной трубкой, доходящей до дна колбы, и с отводной трубкой, которую соединяют с охлаждаемыми колбами или пробирками содержащими равные объемы 5%-ного раствора едкого натра и 3%-ной перекиси водорода (оба раствора должны быть свободны от галогенов). Во время кипячения раствора пропускают через реакционную колбу слабую струю пара. По окончании отгонки иода объединяют щелочные растворы, прибавляют еще 50 мл раствора перекиси водорода для окисления нитрита и затем кипячением разрушают избыток перекиси. Раствор охлаждают, подкисляют разбавленной серной кислотой и обрабатывают сернистой .кислотой для превращения свободного иода в иодид.
Осаждение хлоридом палладия (II) в слабо подкисленном растворе смеси галогенидов щелочных металлов, как описано на стр. 816, с успехом служит для отделения иода от хлора и брома. Другой приемлемый метод отделения иода от двух других галогенидов основан на осаждении нитратом серебра и окислении смеси галогенидов серебра серной и хромовой кислотами 3, как описано на стр. 817.
Было показано 4, что бром, но не иод, реагирует с муравьиной кислотой или ее солями согласно уравнениям:
НСООН + Вг2 = СО2 + 2НВг
HCOONa + Br2 = CO2+HBr + NaBr
Эта реакция может служить для определения брома и иода при их совместном присутствии и иодида в присутствии бромида. В первом случае две аликвотные части раствора титруют тиосульфатом — одну после обработки иодидом калия (Вг2 + 12), а вторую после обработки формиатом натрия (только 12). Во втором случае прибавляют раствор брома в бромиде калия для выделения свободного иода, который титруют тиосульфатом после разрушения брома формиатом натрия.
Более сложной задачей является количественное отделение бромидов от хлоридов. Для этой цели рекомендуется 5 окисление бромистоводородной кислоты теллуровой кислотой, выполняемое следующим образом.
1 F. A. Gooch, J. R. Ensign, Am. J. Sci., [3], 40, 145 (1890).
2P. Jannasch, K. Aschoff, Z. anorg. Chem., 1, 144, 245 (1892),
3 Метод отделения и определения иода в природных водах см. Н. W. В г и Ь а-k е г, Н. S. V a n Bl аге о m, N. Н. Walker, J, Am. Chem. Soc., 48, 1502 (1926); Р. А. М е е г Ь и г g, Z. phys. Chem., 130, 105 (1927). Определение следов иода в органических веществах см. J. F. McClendon, J. Am. Chem. Soc., 50, 1093 (1928).
4 L. S р i t z е г, Ind. Eng. Chem., Anal. Ed., 8, 465 (1936).
5 F. A. G о о c h, H. I. С о 1 e, Am. J. Sci., [4], 37, 257 (1914).
.810
Гл. XLVII. Хлор, бром и иод
Помещают 0,5 г смеси галогенидов и 1 а теллуровой кислоты 1 в небольшую дистилляционную колбу с пришлифованной пробкой, в которую вставлена маленькая делительная воронка. Прибавляют 40 мл воды и 10 мл разбавленной (1 : 1) серной кислоты. Пропускают через колбу слабый ток двуокиси углерода и кипятят раствор, пока объем его не уменьшится до 17—18 мл.
Отделение брома от хлора можно осуществить также окислением следующим образом 2. Переносят 50 мл раствора, содержащих не более чем по 0,25 г хлорида и бромида, в колбу, описанную при отделении иода (стр. 808). Если используется раствор, оставшийся после удаления иода, как там изложено, прибавляют едкий натр точно до щелочной реакции и упаривают приблизительно до 50 мл. По охлаждении нейтрализуют разбавленной (1 : 2) чистой уксусной кислотой, прибавляют 65 мл избытка кислоты, а затем 1—1,5 г перманганата калия. Соединяют колбу с поглотительными пробирками, заполненными и охлаждаемыми, как при отгонке иода. Затем приблизительно 1 ч пропускают в колбу слабую струю пара .до удаления брома, щелочные растворы объединяют и в дальнейшем поступают, как при отделении иода.
Малые количества хлора лучше всего отделить от больших количеств брома 3 окислением бииодатом калия КН (1О3)2 в разбавленном азотнокислом растворе.
Для этого помещают смесь бромидов и хлоридов в колбу Кьельдаля емкостью 400—500 мл с шейкой длиной 20—25 см. Прибавляют рассчитанные количества 0,2 н. раствора йодата и 2 н. раствора азотной кислоты 4, после чего разбавляют до 200—250 мл. Устанавливают колбу в отверстие диаметром в 12 см, проделанном в асбестовом картоне, и наклоняют ее под углом 30° С к вертикали. Нагревают бунзеновской горелкой до слабого кипения и продолжают кипятить 5 мин, после чего оставляют на ночь. Снова нагревают и кипятят так, чтобы в продолжение 30—40 мин объем раствора уменьшился приблизительно до 90 мл. Прибавляют 100 мл воды и продолжают кипятить, пока не отгонится еще 50 мл жидкости, а затем пропускают выделяющиеся пары через узкую согнутую трубку, вставленную в чистую резиновую пробку, в пробирку, содержащую 10 мл 2%-ного раствора иодида калия, слабо подкисленного 0,1 н. соляной кислотой. Если жидкость в пробирке не пожелтеет в течение 40 сек., что указывает
1 Н. I. С о 1 е [Am. J. Sci., [4], 38, 265 (1914)] расширил применение теллуровой кислоты и использует ее для удаления из смеси трех галогенов — сначала иода, а затем брома. R. F. Newt они Е. R. N ewton [Ind. Eng. Chem., Anal. Ed., 6, 213 (1934)] описывают метод определения бромидов в присутствии большого избытка хлоридов и умеренных количеств иодидов, согласно которому бром удаляют хлорной водой, введенной в небольшом избытке, поглощают раствором сульфита натрия и титруют потенциометрически раствором бромата калия после окисления избытка сульфита воздухом.
2 Р. Jannasch, К. Aschoff, Z. anorg. Chem., 5, 8 (1894).
3 L. W. Andrews, J. Am. Chem. Soc., 29, 275 (1907); S. В u g a r s z k y, Z. anorg. Chem., 10, 387 (1895).
4 Для определения хлорида в неочищенном бромиде калия пользуются следующими навесками и количествами реактивов:
Содержание КС1, %....................
Навеска образца, г .................
Объем 0,2 н. раствора KJO3, мл . . .
Объем 2 н. азотной кислоты, мл . . .
5-10 1,5-5 0,25-1,5
0,6 1,8 3,6
36 96 186
20 26 35
Методы определения
811
на полное удаление брома, не прерывая кипячения, прибавляют к раствору в колбе 1—1,5 мл 25%-ного раствора фосфористой кислоты. Продолжают кипятить до удаления иода (около 5 мин), что определяется по цвету раствора, а затем еще 5 мин. Необходимо следить затем, чтобы объем раствора не становился меньше 90 мл. По охлаждении прибавляют к раствору 0,05 или 0,02 н. раствор нитрата серебра в небольшом избытке, затем кусочек фильтровальной бумаги и энергично встряхивают. Осадок отфильтровывают, умеренно промывают разбавленной (1 : 100) азотной кислотой и титруют избыток серебра методом Фольгарда, как описано в гл. «Серебро» (стр. 238).
Небольшие количества бромидов в присутствии больших количеств хлоридов можно определить окислением хромовой кислотой в холодном разбавленном (1 : 3) сернокислом растворе. При этом через раствор пропускают ток воздуха и выделяющийся бром улавливают отмеренным объемом титрованного раствора арсенита, содержащего бикарбонат х.
Описан 1 2 объемный метод определения малых количеств бромидов (0,003%) в присутствии больших количеств хлоридов, как, например, в хлориде натрия. Метод заключается в превращении бромидов в броматы гипохлоритом, разрушении избытка гипохлорита формиатом натрия, восстановлении броматов иодидом и титровании выделяющегося иода тиосульфатом.
Цианиды и хлориды обычно не отделяют друг от друга, так как сумму их можно определить методом Фольгарда, изложенным на стр. 813, а затем в отдельной навеске установить содержание цианида титрованием раствором нитрата серебра.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
ХЛОР
Важнейшим методом определения хлорид-ионов является весовой метод, по которому их взвешивают в виде хлорида серебра. Во всех случаях, за исключением особо точных анализов, можно пользоваться менее точным, но вполне удовлетворительным объемным методом, который заключается в осаждении хлорид-ионов нитратом серебра, избыток которого оттитровывается роданидом. Титрование нитратом серебра в нейтральном растворе в присутствии хромата калия в качестве индикатора удобно для массовых определений, но этот метод имеет ограниченное применение и является наименее точным из всех трех методов.
Взвешивание в виде хлорида серебра
Вещества, мешающие определению хлора в виде хлорида серебра, и способы устранения влияния этих веществ приведены в разделе «Методы отделения» (стр. 808).
В дополнение необходимо отметить, что ошибка определения может быть обусловлена растворимостью хлорида серебра в растворах, из которых он осаждается, а также загрязнением осадка посторонними соединениями, находящимися в растворе.
Растворимость хлорида серебра можно уменьшить прибавлением надлежащего избыточного количества нитрата серебра к разбавленному
1 В. S. Evans, Analyst, 56, 590 (1931),
2 I. М. К о 1 t h о f f, Н. Y u t z у, Ind. Eng. Chem., Anal. Ed., 9, 75 (1937).
812
Гл. XLVII. Хлор, бром и иод
раствору хлорида или соляной кислоты. Раствор должен быть свободным от мешающих определению веществ. Содержание азотной кислоты в нем должно быть крайне незначительным, лучше, если она отсутствует. Поправку на растворимость можно вычислить по данным растворимости хлорида серебра (стр. 237) или на основании нефелометрического определения серебра. Что касается загрязнения осадка, то следует избегать введения чрезмерно больших количеств нитрата серебра и нитратов щелочных и щелочноземельных металлов, так как они, с одной стороны, увлекаются хлоридом серебра, а с другой — увеличивают его растворимость т.
Когда в процессе осаждения хлорид серебра подвергается действию света, выделяется хлор, значительная часть которого снова переходит в хлорид-ионы. При наличии в растворе избытка нитрата серебра (как, например, при установке титра соляной кислоты) эти ионы вызывают дальнейшее осаждение хлорида серебра, что. приводит к получению повышенных результатов. Если же в анализируемом растворе нитрат серебра отсутствует (как в случае установки титра раствора нитрата серебра), хлорид-ионы остаются в растворе, вследствие чего получаются пониженные результаты.
При одинаковом по времени действии света на осадок положительная ошибка всегда больше отрицательной. При установке титров 0,1 н. растворов ошибка составляет приблизительно 1 часть на 1000 при осаждении серебра и 2 части на 1000 в случае осаждения хлоридов, если операция выполняется с нормальной скоростью при ярком свете (но не на прямом или отраженном солнечном свету). На сухой хлорид серебра свет оказывает незначительное действие. Ошибка обычно отрицательна и увеличивается, когда высушенный осадок прежде чем он попадает на свет, увлажняется или если осадок подвергается действию света прежде чем он вполне высох 1 2.
Ход определения. В последующих операциях осадок хлорида серебра следует по возможности предохранять от действия света.
К раствору, свободному от указанных выше мешающих веществ и содержащему в 200 мл приблизительно 0,1 г хлора (лучше в виде соляной кислоты или хлорида натрия), постепенно при перемешивании прибавляют 5%-ный раствор нитрата серебра (содержащий 1 мл азотной кислоты на 1 л) до коагуляции и прекращения образования осадка. Введение большого избытка раствора нитрата серебра следует избегать 3. В присутствии легко гидролизующихся солей прибавляют 1—2 мл азотной кислоты. Раствор нагревают приблизительно до 60° С, дают осадку отстояться и затем прибавляют еще несколько капель нитрата серебра. Если появляется осадок, повторяют операцию. По окончании осаждения ставят колбу в темное место на несколько часов, лучше на ночь.
Прозрачный раствор сливают декантацией через взвешенный тигель Гуча или Мунро (стр. 127) и промывают осадок декантацией раствором,
1 A. S. Cushman [Am. Chem. J., 26, 508 (1901)] и A. J. Berry [Proc. Cambridge Phil. Soc., 22, 367 (1923—1925)], вопреки литературным данным, нашли, что нитрат таллия (III) не оказывает растворяющего действия на хлорид серебра.
2 Дальнейшие подробности см. G. Е. F. L u n d е 11, J. I. Н о f f m a n, J. Research NBS, 4, 109 (1930). 1
3 При отсутствии посторонних веществ наиболее приемлемой концентрацией нитрата серебра в растворе после осаждения AgCl и в первой промывной жидкости является 0,05 г AgNO3 в 1 л [G. Р. В а х t е г, F. А. Н i 11 о n Jr. J. Am. Chem. Soc., 45 ,698 (1923)]. В таком растворе растворимость AgCl составляет 0,05 мг в 1 л при 20° С,
Методы определения
813
содержащим 0,05 г нитрата серебра в 1 л. Под конец переносят осадок в тигель, промывают очень небольшим количеством разбавленной (1 : 99) азотной кислоты для удаления нитрата серебра, а затем 2 раза водой, чтобы отмыть азотную кислоту. Сушат и прокаливают, как указано в гл. «Серебро» (стр. 238). Взвешивают в виде 1 AgCl.
Осаждение нитратом серебра и титрование роданидом
Метод определения хлоридов, основанный на осаждении избыточным количеством титрованного раствора нитрата серебра и последующем титровании этого избытка по методу Фольгарда, изложенному в гл. «Серебро» (стр. 238), большей частью вполне надежен и менее продолжителен, чем весовой. Этот метод имеет более широкое применение, чем метод Мора, описанный на стр. 814, так как определение им может быть выполнено в азотнокислых растворах.
Следует вводить лишь небольшой избыток нитрата серебра, чтобы уменьшить погрешность за счет окклюзии. Хлорид серебра надо отфильтровывать перед титрованием, так как он реагирует с роданистоводородной кислотой, вследствие чего получаются пониженные результаты. Так, в растворе хлорида натрия, в котором весовым методом было найдено 2,324 г хлора на 1 л, объемным способом было определено 2,276 г в присутствии хлорида серебра и 2,328 г, когда хлорид серебра был отфильтрован перед титрованием 2. Позже было установлено, что можно не удалять хлорид серебра фильтрованием, если перед осаждением нитратом серебра в раствор вводить по 1 мл нитробензола на каждые0,05 г хлорида. 3 Нитробензол покрывает осадок хлорида серебра не смешивающимся с водой слоем. Это настолько снижает скорость растворения хлорида серебра, что он не мешает титрованию роданидом. Кроме того, нитробензол уменьшает адсорбцию нитрата серебра осадком хлорида серебра и препятствует его потемнению на свету.
Йодиды, бромиды, цианиды и роданиды осаждаются нитратом серебра и мешают определению; ртуть (II) и палладий должны быть удалены, так как они образуют нерастворимые соединения с роданидом. Азотистая кислота и окислы азота разлагают роданистоводородную кислоту и вызывают преждевременное появление красной окраски раствора. Хлор в хлориде таллия Т1С13 нельзя определять методом Фольгарда, так как таллий (III) в азотнокислом растворе окисляет роданистоводородную кислоту, что вызывает повышенный расход роданида и, следовательно, пониженные результаты 4. Определение хлора в растворе Т1С1з можно проводить методом Мора при-условии предварительного добавления ацетата аммония 5.
1 В анализах высокой точности в массу хлорида серебра следует ввести поправку на часть его, оставшуюся в растворе или перешедшую в раствор при промывании осадка. Помимо того, незначительное количество влаги, остающееся в высушенном хлориде серебра, должно быть удалено сплавлением. Об этих поправках см. Т. W. R i с h а г d s,
R. С. Wells, Carnegie Inst. Wash. Pub., 1905, p. 25 или G. P. Baxter,
F. A. Hilton Jr., J. Am. Chem. Soc., 45, 694 (1923).
2 G. D r e c h s e 1, Z. anal. Chem., 16, 351 (1877); M. A. R о s a n о f f,
A. E. Hill, J. Am. Chem. Soc., 29, 269 (1907).
3 J. R. Caldwell, H.V, Moyer, Ind. Eng. Chem., Anal. Ed., 7, 38 (1935).
4 A. S. C u s h m a n, Am. Chem. J., 26, 508 (1901).
6 A. J. В e r r y, Proc. Cambridge Phil. Soc., 22, 367 (1923—1925).
814
Гл. XLVII. Хлор, бром и иод
Ход определения. Относительно применяемых растворов см. гл. «Серебро» (стр. 238).
Титр раствора нитрата серебра лучше (хотя при выполнении рядовых анализов это не обязательно) устанавливать по навескам чистого сухого хлорида натрия.
К 100—200 мл анализируемого раствора, содержащего разбавленную (1 : 99) азотную кислоту и приблизительно0,1 г хлора (лучше в виде соляной кислоты или хлорида щелочного металла) и свободного от перечисленных выше мешающих определению веществ, прибавляют титрованный 0,1 н. раствор нитрата серебра в таком количестве, чтобы осталось 1—3 мл избытка. Коагулируют осадок хлорида серебра, сильно взбалтывая в закрытой колбе, защищенной от света. Фильтруют через бумажный фильтр или слой асбеста или тонко измельченной платины. Промывают колбу, осадок и фильтр разбавленной (1 : 99) азотной кислотой, не содержащей азотистой кислоты. Титруют, как описано в гл. «Серебро» (стр. 239). Из прибавленного объема раствора нитрата серебра вычитают количество, эквивалентное израсходованному на титрование объему раствора роданида, разность и умножают на титр нитрата серебра, пересчитанный на хлор Ч
Метод Мора
Метод Мора 1 2 основан на значительно большей растворимости хромата серебра по сравнению с хлоридом серебра в нейтральных или слабокислых растворах, вследствие чего при прибавлении нитрата серебра к раствору, содержащему хромат- и хлорид-ионы, неисчезающий красный осадок хромата серебра появляется только после полного осаждения хлорида.
Минеральные кислоты, растворяющие хромат серебра, должны отсутствовать, в противном случае их следует нейтрализовать карбонатом кальция или же, нейтрализовав большую часть кислоты аммиаком, прибавить в избытке ацетат аммония 3.
Из веществ, мешающих определению, следует отметить: 1) иодиды, бромиды, роданиды, фосфаты, карбонаты и сульфиды, также образующие осадки с нитратом серебра; 2) цианиды и тиосульфаты, растворяющие осадок хлорида серебра; 3) органические или другие соединения, восстанавливающие нитрат серебра в нейтральном растворе. Желательно, чтобы
1 L. A. R е Ьег и W. М. М с N a b b [Ind. Eng. Chem., Anal. Ed., 9, 529 (1937)] указывают, что избыток нитрата серебра можно определить также титрованием титрованным раствором иодида калия в присутствии сульфата церия (IV) и аммония и крахмала после удаления осадка хлорида или бромида серебра фильтрованием. J. F. Ма-t е j с z у k [Chemist Analyst, 28, 54 (1939)] утверждает, что иодид серебра также должен быть отделен перед титрованием избытка нитрата серебра.
2 С. F. М о h г, Ann. 97, 335 (1856). Метод изучался A. J. В е г г у и J. Е. D г 1-v е г [Analyst, 64, 730 (1939)].
3 О применении флуоресцеина в качестве адсорбционного индикатора (розовая окраска на осадке галогенида серебра) вместо хромата при титровании галогенидов в нейтральных или слабокислых растворах см. I. М. Kolthoff, W. М. Lauer, С. J. Sunde, J. Am. Chem. Soc., 51, 3273 (1929). Титрование солянокислых органических оснований в спиртовых растворах описано в статье К. В a m b а с h,T. Н. R i-d е г, Ind. Eng. Chem., Anal. Ed., 7, 165 (1935).
W. T. Burg [Chemist Analyst, 28, 52 (1939)] рекомендует титровать 75 мл анализируемого раствора, содержащего 50 мл метилового спирта, 1 мл 0,5 н. раствора азотной кислоты и 1 каплю 1%-ного эозина, до окрашивания осадка в фиолетово-красный цвет.
Методы определения.
815
свинец, висмут, барий и железо также были удалены из раствора, так как они образуют нерастворимые хроматы, вследствие чего необходимо прибавлять большее количество индикатора.
Ход определения. Требуются титрованные, приблизительно 0,1 н. растворы нитрата серебра и хлорида натрия и 1 %-ный раствор хромата калия, свободного от хлоридов. Титрованные растворы приготовляют так, чтобы они были эквивалентными. Титр раствора нитрата серебра должен быть установлен по отдельным навескам сухого чистого хлорида натрия, который растворяют и титруют этим же методом.
Приготовляют 100 мл нейтрального или слабо уксуснокислого раствора, содержащего ацетат аммония. Раствор должен быть свободен от мешающих перечисленных выше веществ и должен содержать приблизительно 0,1 г хлорид-ионов. К раствору, переведенному в фарфоровую 'чашку, прибавляют 1 мл 1 %-ного раствора хромата калия (свободного от хлоридов) и затем медленно, хорошо перемешивая, прибавляют титрованный нейтральный раствор нитрата серебра. Под конец нитрат серебра вводят очень медленно, до появления неисче^ающей красной окраски. После этого, по каплям при сильном перемешивании прибавляют эквивалентный раствор хлорида натрия до исчезновения этой окраски % Вычитают объем израсходованного раствора хлорида натрия из объема раствора нитрата серебра, пошедшего на титрование, и разность умножают на титр раствора.
БРОМ
Бромид серебра менее растворим в воде, чем хлорид. В 1 л воды при 21° С растворяется 0,107 мг бромида серебра 1 2 и 1,54 мг хлорида серебра 3. Для определения брома применяются практически те же методы, что и для определения хлора. Защита от действия света для бромида серебра имеет еще большее значение, так как он светочувствительнее хлорида серебра. Титрование бромидов в смеси галогенидов см. стр. 817.
Осаждение в виде бромида серебра
Поступают так же, как описано для хлора (стр. 811), за исключением того, что раствор должен быть более разбавленным и что необходимо обра-щать особое внимание на полное удаление азотной кислоты из осадка 4.
1 После отсчета рекомендуется прибавить несколько капель избытка раствора хлорида натрия и затем пользоваться этим раствором для сравнения в последующих титрованиях («свидетель»). Согласно W. В. Meldrum и). С. Forbes [ J. Chem. Edus.,. 5, 205 (1928)], конечную точку титрования легко можно заметить, применяя следующий метод. Растворяют 0,4 г анализируемого хлорида в 75 мл Н2О, прибавляют метиловый оранжевый, разбавленную HNO3 до кислой реакции и затем 1 %-ный раствор NaHCO3 до перехода окраски индикатора в желтую. Вводят 5 капель 1 М раствора К2СгО4, титруют AgNO3 до появления окраски, кипятят 5 мин, охлаждают в ледяной воде и заканчивают титрование.
2 F. Kohlrausch, Z. phys. Chem., 50, 356 (1904).
3 G. S. W h i t Ь у, Z. anorg. Chem., 67, 108 (1910).
4 Ход работы при выполнении особо точных определений описан в статье G. Р. Baxter, Proc. Am. Acad., 42, 201 (1906—1907).
.816
Гл. XLVII. Хлор, бром и иод
Осаждение нитратом серебра и титрование роданидом
Поступают так же, как описано для £лора (стр. 813). При определении брома фильтрование перед титрованием менее необходимо, чем при определении хлора, и эту операцию во всех случаях можно опустить, кроме особо точной работы.
ИОД
Взвешивание в виде иодида серебра
Иодид серебра является наименее растворимым галогенидом серебра. В 1 л воды при 21° С растворяется1 всего лишь 0,0035 мг Agl. Осадок иодида серебра имеет тенденцию увлекать нитрат серебра и другие находящиеся в растворе соединения. Удалять эти примеси из иодида серебра значительно труднее, чем из хлорида. Осаждение поэтому должно проводиться медленным прибавлением при перемешивании сильно разбавленного, примерно 0,05 н. раствора нитрата серебра к такому же разбавленному аммиачному раствору иодида. По окончании осаждения вводят азотную кислоту в таком количестве, чтобы концентрация свободной кислоты составляла 1 % по объему. Осадок можно промывать азотной кислотой той же концентрации. Под конец осадок надо промывать водой до удаления азотной кислоты, которая во время высушивания разлагала бы иодид серебра. Для промывания следует, однако, употреблять небольшое количество воды, так как под ее действием осадок может перейти в коллоидный раствор. Остальные детали определения см. раздел «Хлор» (стр. 811) 2 3 *.
Осаждение в виде иодида палладия (II)8
Иодид палладия Pdb нерастворим в воде и в разбавленной (1 : 99) соляной кислоте, но он слегка растворяется в растворах, содержащих значительные количества галогенидов. Должны отсутствовать вещества, восстанавливающие палладий до металла, например спирт, а такще соединения, осаждающие палладий, например сульфиды и цианиды.
Палладий (IV) образует нерастворимые соединения с хлорид-ионами в присутствии солей калия и аммония. Мало вероятно, однако, чтобы соли палладия (IV) находились в реактиве или образовались в анализируемом растворе в условиях определения иодидов. Иодид палладия можно взвешивать непосредственно или прокаливать в токе водорода до образования металлического палладия. Следует предпочесть взвешивание металлического палладия, так как иодид палладия в процессе нагревания при 100° С медленно теряет иод.
Ход определения. К раствору галогенидов щелочных металлов, содержащему 1 % по объему соляной кислоты и свободному от восстановителей,
1 F. KohlrauschZ. phys. Chem., 50, 355 (1Ц04).
2 Описание методов, применяемых при анализах высокой точности, можно найти у G. Р. Baxter, Proc. Am. Acad., 40, 419 (1904); 41t 73 (1905); J. Am. Chem. Soc., 32, 1591 (1910). Иодид серебра постепенно теряет иод,- если прокаливание проводится при температуре выше его температуры плавления [G. Ps Baxter, А. С. Titus, J. Am. Chem. Soc., 62, 1862 (1940)].
3 J. L. Lassaigne, J. Chiem. Med., 1, 57 (1835); R. Strebinger,
I. Pollak, Microchemie 3, 38 (1925).
Методы определения
817
таких, как спирт, и желательно от солей других металлов, прибавляют раствор хлорида палладия (II) до прекращения образования осадка. Раствор оставляют стоять 24—48 ч при 20—30° С и затем фильтруют через взвешенный тигель Гуча. Осадок промывают сначала теплой водой, а под конец очень небольшим количеством спирта. Сушат при 90—95° С в течение 1 ч и взвешивают в виде Pdl2 или лучше постепенно прокаливают при 1000° С в атмосфере водорода и затем взвешивают металлический палладий. Ток водорода прекращают, как только удаляют пламя. По охлаждении смачивают металл спиртом и поджигают. Когда спирт выгорает, охлаждают в эксикаторе и взвешивают.
Окисление галогенидов серебра серной и хромовой кислотами
Наиболее удобным способом отделения и определения иода и определения хлора и брома в смеси этих трех элементов является видоизмененный Бекком 1 метод Бобиньи 2, который заключается в следующем.
Разделяют анализируемый раствор на две равные части. Каждую часть обрабатывают нитратом серебра, фильтруют и промывают, как указано в гл. «Серебро» (стр. 237).
Один из осадков сушат при 180° С и взвешивают; таким образом находят суммарную массу галогенидов серебра 3. Другой промытый осадок, содержащий не более 0,4 г смеси галогенидов серебра, обрабатывают раствором 2 г бихромата калия в 30 мл серной кислоты и нагревают 30 мин. при 95° для полного удаления хлора и брома и превращения иодида серебра в иодат. К концу операции продувают через раствор воздух. После этого разбавляют до .300—400 мл, фильтруют и восстанавливают в фильтрате иодат, прибавив концентрированный раствор сульфита натрия в таком количестве, чтобы спустя 10 мин еще чувствовался слабый запах сернистого ангидрида. Большой избыток SO2 может вызвать восстановление иодида серебра.
Осадок иодида серебра отфильтровывают, промывают, высушивают и взвешивают. В фильтрате и промывных водах осаждают серебро в виде иодида, фильтруют, осадок промывают и взвешивают. На основании массы суммы AgCl + AgBr -}- Agl, массы Agl, образовавшегося из йодата, и массы Agl, полученного из фильтрата в результате осаждения серебра, которое первоначально было связано с хлором и бромом, легко вычислить массу всех трех галогенов.
Метод Фольгарда
Иодиды можно определять по методу Фольгарда так же, как и хлориды (см. «Хлор», стр. 813), с той лишь разницей, что: 1) железо-аммонийные квасцы не следует прибавлять до тех пор, пока иодид серебра полностью не осядет; 2) раствор серебра надо прибавлять к разбавленному анализируемому раствору очень медленно и при сильном перемешивании; 3) нет необходимости фильтровать раствор перед титрованием роданидом.
1 J. В ekk, Chem. Ztg., 39, 405 (1915).
2 Н. В a u Ы g п у, С. г., 127, 1219 (1898), 128, 51 (1899).
8 Эти же взвешенные высушенные галогениды серебра можно использовать для дальнейшей обработки вместо влажного осадка этих солей, полученных из другой порции раствора. В Этом случае нагревание с хромовой кислотой должно продолжаться 2 ч.
52 Заказ 522.
818
Гл. XLVII. Хлор, бром и иод
Другие методы
Йодиды можно определять в разбавленном сернокислом растворе смеси галогенидов потенциометрическим титрованием раствором перманганата Ч
Иодид, бромид или сумму иодида, бромида и хлорида можно определить 1 2 следующим способом. В анализируемый раствор вводят цианид, затем тщательно регулируют кислотность раствора, в соответствии с определяемым галогеном, окисляют титрованным раствором\бромата калия до IGN, BrGN и C1CN, а затем, прибавив иодид калия, титруют раствором тиосульфата натрия. Описаны также 3 методы определения иодида и бромида, основанные на окислении их (в нейтральном растворе в случае иодида и в кислом растворе в случае бромида) титрованным раствором перйодата, прибавляемым в избытке, и титровании этого избытка арсенитом.
1 R. В е h г е n d, Z. phys. Chem., И, 466 (1893); W. S, Н en dr i х son, J. Am. Chem. Soc., 43, 14 (1921); I. M. К olthof f, E. J. A. H. Very zl, Rec. trav. chim., 42, 1055 (1923); H. H. W i 11 a r d, F. F e n w i c k, I. Am. Chem. Soc., 45, 623 (1923); F. L. H a h n, G. W e i 1 e r, Z. anal. Chem., 69, 417 (1926).
2 R. Berg, Z. anal. Chem., 69, 1, 369 (1926); см. также N. McCulloch, Chem. News, 60, 259 (1889); С. M e i nek e, Z. anorg. Chem., 2, 157 (1892); T. E w a n, J. Soc. Chem. Ind. 25, 1130 (1906); R. L a n g, Z. anorg. Chem., 144,75 (1925); G. A 1-sterberg. Biochem. Z. 166, 1 (1925); I. M. К о 11 h о f f, Chem. Zentr., 97 (1), 1858 (1926).
8 H. H. Willard, L. H. G r e a t h о u s e, J. Am. Chem. Soc., 60, 2869 1938).
Глава XLVIII
ФТОР
В последнем издании известного руководства по анализу горных пород Вашингтона отмечено \ «Фтор, являющийся компонентом апатитов, биотитов и т. п., не должен, как будто, иметь особой приверженности к магме, хотя в общем он чаще встречается в кремнекислых, чем в железных (железомагнезиальных) породах. Фтор большей частью находится в виде флюорита СаЕг (иногда G1 изоморфен с F) и в некоторых других более редких минералах, встречающихся в породах, содержащих нефелин, как, например, фойаиты и тингуаиты. Фтор является основным компонентом флюорита и большинства апатитов Са8 (F, G1, ОН)(РО«)з. Являясь одним из основных компонентов апатитов, фтор встречается почти повсеместно 1 2. В малых количествах фтор встречается в биотитах и других слюдах, в некоторых роговых обманках и авгите, а также в турмалине, топазе, хондродите и т. д.».
В горных породах фтор обычно находится в количествах, меньших 0,1 %, но так как при анализе пород взвешиваемый фторид кальция обычно очищают от загрязнений, указанное содержание фтора несомненно ниже истинного его содержания вследствие неточности метода отделения.
Некоторые авторы 3 полагают, что фтор необходим для существования живой клетки и утверждают, что он находится вместе с фосфором во всех животных и растительных тканях.
Определение фтора в природных водах, растительных продуктах, рудах, например плавиковом шпате, криолите и фосфатных породах, а также в керамических продуктах, как опаловые стекла и эмали, приобретает все возрастающее значение.
ОБЩИЕ ЗАМЕЧАНИЯ
В обычном ходе анализа присутствие фтора вызывает значительные затруднения. При анализе пород, богатых фтором, для получения точных результатов кремнекислоту следует выделять после отделения фтора,
1 Н, S. Washington, The Chemical Analysis of Rocks, 4th ed.,- John Wiley and Sons, 1930, p. 22.
2 См. также К. D. J а с о h, D. S. Reynolds, J. Assoc. Offic. Agr. Chemists, 11, 237 (1928).
3 A, G a u t i e г, P. Clansman П,С. r., 162, 105 (1916).
52*
820
Гл. XLVIII. Фтор
например .методом Берцелиуса (стр. 1011). На практике, однако, не часто приходится прибегать к этому продолжительному методу, так как содержание фтора обычно невелико, и если при выпаривании раствора, полученного после обработки карбонатного плава соляной кислотой, фтор даже полностью улетучится в виде тетрафторида кремния, то потеря кремния составляет лишь три четверти от массы фтора. Обычно, однако, потеря кремния меньше этой величины, потому что тетрафторид кремния неустойчив в присутствии влаги. Некоторая часть фтора поэтому отгоняется в виде фтористоводородной кислоты, а большая его часть связывается, образуя фторосиликаты. Эти соли при прокаливании с кремнекислотой частично разлагаются, а после обработки, прокаленного остатка фтористоводородной и серной кислотами металл, входивший в состав фторосиликата (обычно — натрий), взвешивается в виде сульфата, если он не улетучивается в процессе прокаливания.
Фтор, остающийся в фильтрате отделения после кремнекислоты, препятствует количественному осаждению алюминия Ч После разложения породы фтористоводородной и серной кислотами и выпаривания до появления паров серной кислоты иногда наблюдается неполнота осаждения алюминия аммиаком из сернокислых растворов. Это явление объясняется, несомненно, недостаточно полным удалением фтористоводородной кислоты 1 2. При выпаривании фторида алюминия с фтористоводородной кислотой и последующем осторожном прокаливании потери алюминия не происходит.
Помимо затруднений, которые связаны с определением алюминия, при осаждении аммиаком в присутствии фтора в большей или меньшей степени выделяется кальций в виде CaFn. Это несколько возмещает потерю алюминия, но результат определения кальция будет ошибочным и не только потому, что кальций частично попадает в осадок от аммиака, но также и потому, что при последующем осаждении кальция щавелевой кислотой, если в растворе остался фтор, часть кальция может выделиться в виде фторида.
В таких минералах, как апатит или флюорит, содержащих значительные количества фтора, открыть его нетрудно. Это вполне может быть сделано методом травления стекла, методом висячей капли или пробой на пламя.
Однако все эти методы недостаточно надежны в применении к материалам, содержащим лишь небольшие количества фтора, как, например, к горным прродам. И хотя в некоторых породах фтор можно легко открыть при содержании его порядка 0,1%, в породах другого типа даже значительно большие количества фтора можно не обнаружить 3.
Проба травлением общеизвестна. Она основана на разъедании стекла фтористоводородной кислотой, выделяющейся при нагревании испытуе
1 F. Р. Veitch, J. Am. Chem. Soc., 22, 246 (1900); F. W. H inrichsen, Ber., 40, 1497 (1907); Z. anorg. Chem., 58, 83 (1908); H. C a v a i g n a с, C. r., 158, 948 (1914).
2 E. S el c h, Z. anal. Chem., 54, 395 (1915).
8 A. G a u t i e г и P. С 1 a u s m a n [C. r., 154, 1670 (1912); 158, 1389, 1631 (1914); 162, 105 (1916)] опубликовали ряд работ об определении фтора в природных водах, минералах и других материалах. Авторы приводят разработанный ими, по-ви-Димому, исключительно чувствительный метод определения очень малых количеств фтора. Выводы авторов метода приводятся в последней ссылке. Ограниченность места не позволяет нам дать описание этого метода и требуемой аппаратуры.
Разложение соединений фтора
821
мого материала с серной кислотой, или при сплавлении его с кислым плавнем, например^ метафосфатом натрия. В присутствии кремния и бора чувствительность реакции понижается вследствие образования тетрафторида кремния и фторида бора, которые не разъедают стекло. Когда содержание фтора ниже содержания кремния или бора, реакция может дать отрицательный результат. В таких случаях фтор надо сперва отделить методом Берцелиуса (стр. 1011) и конечный промытый продукт подвергнуть этому испытанию х.
Метод висячей капли основан на помутнении капли воды, которое вызывается тетрафторидом кремния, образующимся при действии серной кислоты на сухую смесь фторида и измельченной кремнекислоты. Помутнение вызывается выделением кремнекислоты согласно уравнению
3SiF4+ЗН2О = H2S1O3 + 2H2SiFe
В применении к силикатным материалам, содержащим очень малые количества фтора, этот метод испытания более надежен, чем метод травления. В присутствии относительно больших количеств бора, так же как и при испытании методом травления, может получиться отрицательный результат, так как BF3 может не вызывать помутнения капли воды.
Испытание обычно проводится следующим образом. Смешивают приблизительно 0,5 г тонко измельченного высушенного испытуемого материала с 0,1 г сухой осажденной кремнекислоты. Смесь помещают в маленькую пробирку, закрытую пробкой, с одним отверстием, в которое вставлена маленькая стеклянная трубка, запаянная с одного конца и открытая с другого. В пробке сбоку сделана небольшая выемка. В открытом конце трубки помещают каплю воды, а затем трубку устанавливают так, чтобы этот конец ее находился на 2—3 мм ниже пробки. К смеси испытуемого материала с Кремнекислотой прибавляют 1—2 капли серной кислоты и, неплотно закрыв пробирку пробкой, осторожно нагревают на водяной бане 30 минут. Если в пробе присутствуют заметные количества фтора, у отверстия трубки образуется кольцо студенистой кремнекислоты.
Проба на пламя получила свое название благодаря зеленому окрашиванию пламени, которое вызывается фторидом бора, образующимся при внесении в пламя бунзеновской горелки перла, состоящего из кислого плавня, фторида и борсиликата.
Эту пробу лучше всего проводить следующим образом1 2. К 2 частям (по массе) KHSO4 прибавляют 1 часть боросиликата, например аксинита HCa3Al2B(SiO4)4, или борсодержащего стекла. Растирают в тонкий порошок и тщательно перемешивают. Смешивают 1 часть испытуемого вещества с 3 частями приготовленного таким образом плавня, захватывают петлей из платиновой проволоки немного смеси, сплавляют и по-лусплавленный перл вносят в наружную часть нижнего края пламени бунзеновской горелки. Можно также приготовить полусплавленный перл из одного плавня, а затем горячим перлом прикоснуться к испытуемому порошку и внести в пламя. Мгновенное зеленое окрашивание пламени, вызванное выделяющимся фторидом бора, указывает на присутствие фтора. Барий также окрашивает пламя в зеленый цвет.
РАЗЛОЖЕНИЕ СОЕДИНЕНИЙ ФТОРА
Некоторые силикаты и фториды выделяют весь содержащийся в них фтор в виде SiFi, когда их смешивают с тонко измельченной кремнекислотой и обрабатывают серной кислотой, как описано в разделе «Метод отгонки» (стр. 824).
1 A. G. Woodman и Н. Р. Т а 1 Ь о t [J. Am. Chem. Soc., 28, 1437 (1906)] дают подробное описание метода травления, чувствительность которого в отсутствие кремнекислоты равна 1 : 5 000 000.
2 Согласно F. В. Wilson, Chemist Analyst, 24, 23 (1918).
822
Гл. XLVIII. Фтор
Если силикат не разлагается серной кислотой, содержащийся в нем фтор можно перевести в растворимый фторид натрия сплавлением с карбонатом натрия при возможно более низкой температуре х, Для облегчения разложения тугоплавких фторидов их следует предварительно смешать с окисью кремния. Плав выщелачивают водой и силикат натрия, карбонат, фосфат и т. п. отделяют от фтора методом Берцелиуса (стр. 1011). Когда присутствуют значительные количества фтора или когда после выщелачивания плава водой остается большой остаток, операцию сплавления с карбонатом натрия и кремнекислотой необходимо повторить еще один или два раза, выщелачивая каждый раз плав водой.
В процессе выпаривания или кипячения нейтральных или аммиачных растворов фтор, если и теряется, то лишь в самом незначительном количестве х. (
Возможность практического использования непосредственного сплавления карбонатных пород с карбонатом натрия и калия и кремнекислотой, вследствие значительного преобладания в этих породах извести, несколько сомнительна. Наилучшие результаты, вероятно, дает следующая обработка. Измельченную в порошок пробу растворяют в разбавленной уксусной кислоте, насколько возможно, без кипячения, фильтруют и промывают остаток горячей водой. К фильтрату прибавляют раствор карбоната натрия в небольшом избытке, кипятят и затем обрабатывают осадок карбоната кальция и, возможно, фторида кальция методом Берцелиуса (стр. 1011). Большая часть фтора, который мог перейти в раствор при обработке уксусной кислотой, таким образом извлекается. Нерастворимый остаток сплавляют со смесью карбонатов натрия и калия и в дальнейшем поступают, как указано в методике.
Плавиковый шпат разлагается сплавлением с едким кали в золотом тигле или с карбонатом калия и селитрой в платиновом тигле и последующей обработкой плава водой или холодной разбавленной азотной кислотой. Полученные этими способами плавы растворяются значительно легче, чем плавы, получаемые в результате сплавления с соответствующими солями натрия. Когда не требуется определение фтора, плавиковый шпат можно, разлагать нагреванием тонкого порошка пробы с серной или хлорной кислотой 1 2 или же обработкой при нагревании разбавленной (1 : 1) соляной кислотой, содержащей борную кислоту.
Для разложения органических соединений, содержащих фтор (и бор), рекомендуется3 отвешенную в желатиновой капсюле пробу сжигать в бомбе Парра с окислительнойлсмесью, состоящей из перекиси натрия, хлората калия и сахара.
1 Согласно F.'G/Hawl ёу [Ind. Eng. Chem., 18, 573 (1926)], в присутствии сульфидов, например пирита, может произойти потеря фтора, что частично устраняется прибавлением к карбонату натрия селитры или перекиси натрия. Прокаливание после прибавления извести, как описано в разделе «Метод отгонки» (стр. 824), вероятно, совсем устраняет это затруднение.
2 W. Н. A d о 1 р h, The Quantitative Methods for Flurine, Thesis University of Pennsylvania, 1915, p. 12, Z. anal. Chem., 55, 395 (1916).
1 G.E. F. Lundell, J. I. Hoff man, J.. Research. NBS, 4, 671 (1929).
1 D. J. Pflaum, H. H. Wenzke, Ind. Eng. Chem., Anal. Ed., 4, 392 (1932).
Методы отделения
823
МЕТОДЫ ОТДЕЛЕНИЯ
В количественном анализе фтор обычно отделяют осаждением в виде фторида кальция CaFz или отгонкой в виде тетрафторида кремния SiF« или кремнефтористоводородной кислоты H2SiFe. Первый метод нельзя считать совершенным вследствие растворимости фторида кальция и необходимости отделения ряда соединений, например фосфатов, хроматов и сульфатов, которые также образуют нерастворимые соли с кальцием. Способы отделения этих компонентов подробно описаны при изложении метода Берцелиуса (стр. 1011). Отделение фтора отгонкой основано на обработке анализируемого материала серной или хлорной кислотой. Непосредственная обработка этими кислотами фторидных минералов, встречающихся в породах, редко бывает вполне успешной, так как большинство из них не полностью разлагается или вовсе не разлагается концентрированной серной или хлорной кислотой.
Полное отделение фтора не достигается даже в том случае, когда он находится в виде разлагаемых фторидов, если для образования летучих соединений применяется аморфная кремнекислота или силикаты х. Наилучшие результаты получаются при использовании тонко измельченного кварца, однако работы последнего времени показали, что при достаточно продолжительной отгонке полное отделение фтора достигается даже в присутствии студенистой кремнекислоты 1 2.
Фтор можно количественно отделить от ниобия и тантала следующим способом 3. Осаждают аммиаком, дают отстояться, сливают декантацией прозрачную жидкость и сохраняют ее. Осадок растворяют в небольшом количестве горячей разбавленной азотной кислоты, снова осаждают аммиаком, декантируют и еще один раз повторяют эту операцию. В объединенных фильтратах определяют фтор, а тантал и ниобий определяют в осадке, полученном при последнем осаждении.
Основной метод удаления фтора из растворов, в которых присутствие его нежелательно, состоит в отгонке его в виде фтористого водорода или, если присутствует кремний, в виде тетрафторида кремния или кремнефтористоводородной кислоты. Полное разложение фторидов иногда достигается с трудом.
Например, после обработки 1 г сульфата кальция фтористоводородной и серной кислотами, как при определении кремнекислоты4, не удалось полностью удалить фтор выпариванием раствора досуха и прокаливанием; не помогает даже четырехкратное прокаливание — дважды после последовательного прибавления воды и серной кислоты, и дважды — после прибавления одной серной кислоты, порциями по 5 мл.
Г. А. Брайт нашел, что фторид легче разлагается, если остаток растолочь и растереть с серной кислотой. Таким образом, ему удалось полностью удалить фтор двукратной обработкой серной кислотой порциями по 2 мл. Этим способом был полностью удален фтор из,1 г плавикового шпата.
Для' количественного удаления фтора обработкой более летучими кислотами, например азотной кислотой, рекомендуется5 вводить в раствор
1 К. D a n i е 1, Z. anorg. Chem., 38, 257 (1904).
2 Н. Н. W i 11 а г d, О. В. W i n t е г, Ind. Eng. Chem., Anal. Ed., 5, 7 (1933).
8 О. R u f f, E. S c h i 11 e r. Z. anorg. Chem., 72, 329^(1911).
4 R. W e 11 s, J. Am. Chem. Soc., 44, 2188 (1922).
6 A. A. Noyes, Technol. Quart., 16, 101 (1903); А. Нойес, В. Брэй, Качественный анализ редких элементов, ОНТИ, 1936.
824 Гл. XLVIII. Фтор
тонко измельченную кремнекислоту, которая усиливает действие азотной кислоты. Для этого, вероятно, может служить и борная кислота. Полное удаление фтора, без введения серной кислоты, можно также осуществить выпариванием с хлорной кислотой (см. «Реактивы», стр. 64). При применении этой кислоты лучше выпаривать досуха и в большинстве случаев целесообразно повторять эту операцию. Образующиеся перхлораты можно перевести в хлориды или основные хлориды осторожным нагреванием до прекращения выделения газа.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наиболее распространенными методами определения фтора в горных породах и минералах являются, вероятно, методы Берцелиуса 1 и Стей-гера 2. В первом из этих методов фтор осаждают и взвешивают в виде фторида кальция CaF2. В методе Стейгера содержание фтора определяется по обесцвечиванию им раствора соли титана, обработанного перекисью водорода. Метод Берцелиуса, обычно применяемый для определения значительных количеств фтора, всегда дает пониженные результаты. При содержании в 100 мл раствора менее 1 мг фтора этим методом его можно совсем не обнаружить. Если содержание фтора меньше 10 мг, следует предпочесть метод Стейгера. Предварительное разложение пробы, например горной породы, и последующая обработка обоими методами описаны в части III (стр. 1018).
В настоящее время некоторые аналитики считают целесообразным отделять фтор от мешающих элементов дистилляцией из сернокислого или хлорнокислого раствора. Последующий ход анализа для конечного определения фтора в дистилляте зависит от его содержания. Для определения 0,01—0,1 г фтора удобен метод осаждения в виде PbCIF, а при содержании фтора от следов его до 50 мг обычно применяют титрование нитратом тория. Последний метод особенно пригоден для определения очень малых количеств фтора. Все эти методы изложены в нижеследующих разделах.
Метод отгонки в виде H,SiF6 и титрования нитратом тория
В этом методе 3 водный раствор растворимого фторида или фторосиликата обрабатывают цирконий-ализариновой смесью, разбавляют равным объемом спирта и титруют раствором нитрата тория до появления розовой окраски цирконий-ализаринового лака. Вводят поправку на количество нитрата тория, расходуемого в «холостом» опыте на титрование реактивов. '
Если присутствуют вещества, мешающие определению, фтор предварительно отгоняют в виде кремнефтористоводородной кислоты. Отгонку проводят из колбы, в которую помещают кусочки стекла и серную или хлорную кислоту. Конечную температуру при отгонке поддерживают близкой к 135° С; для этого добавляют воду из капельной воронки. Нерастворимые соединения сплавляют с карбонатом натрия, и прежде чем перенести в дистилляционную колбу, плав обрабатывают небольшим ко
1 J. I. В е г z е 1 i u s, Schweigg J., 16, 426 (1816); Н. R о s е, Ann., 72, 343 (1849).
2 G. Steiger, J. Am. Chem. Soc., 30, 219 (1908); H. E. Merwin, Am. J. Sci.; [4] 28, 119 (1909).
3 См. сноску 2 на стр. 823.
Методы определения
825
личеством горячей воды. Если проба содержит большое количество кремнекислоты, ее надо перед отгонкой отделить обработкой перхлоратом цинка и карбонатом цинка г.
Обычно при небольшом объеме жидкости в дистилляционной колбе и высоких температурах (особенно в случае применения НС1О4 и Н3РО4) достигается более высокая чувствительность определения фтора в дистилляте. В случае присутствия значительных количеств алюминия требуется предварительная дистилляция с НгЗО4 при 162 ± 2° С, нейтрализация раствора едкой щелочью, концентрирование раствора до небольшого объема и вторичная дистилляция с H2SO4 при 137 ± 5° С. Применяемая для отгонки хлорная кислота должна быть свободной от летучих кислот.
Некоторые вещества мешают отгонке, другие — титрованию фтора. Из первых большие количества солей алюминия препятствуют полному улетучиванию фтора, а студенистая кремнекислота замедляет его отгонку.
Титрованию мешают ионы, образующие осадки или недиссоцииро-ванные соли со фтором или торием. К ним относятся сульфат-, фосфат-, арсенат-, хлорид-, нитрат-, перхлорат-, гипохлорит-ионы и в меньшей степени ионы щелочных металлов, борат- и сульфид-ионы. Арсенит-, хлорат- и силикат-ионы не оказывают влияния на титрование.
Предложены многочисленные видоизменения метода титрования фтора нитратом тория. В частности, было рекомендовано 3 применять в качестве ийдикатора один только ализаринсульфонат натрия, титрование проводить в водном, а не в спиртовом растворе и в раствор перед отгонкой вводить перхлорат серебра для связывания соляной кислоты.
Другие авторы 4 отмечают, что конечная точка титрования более отчетлива при 4-10 %-ной концентрации индикатора в 50 мл раствора, буферированного до pH = 3,5, как при применении монохлоруксусной кислоты.
Мешающее влияние фосфат-ионов можно устранить 4 предварительной отгонкой с серной кислотой при 150° С и последующей вторичной отгонкой дистиллята с хлорной кислотой при 135° С. Другие авторы 5 проводят отгонку, вводя перегретый пар в сернокислый раствор, находящийся в дистилляционной колбе, снабженной обратным холодильником и нагреваемой при 145° С в медном сосуде, в котором находится кипящий тетра-хлорэтан.
Хлорную кислоту применять нельзя, вследствие опасности, которая возникает, если колба лопнет.
Приводим вариант метода, разработанный для анализа фосфатных пород 3.
1 Исследование процесса отгонки фтора из сернокислого, хлорнокислого или фосфорнокислого раствора см. D. D a h 1 е, Н. J. Wichmann, J. Assoc. Offic. Agr. Chemists, 19, 313, 320 (1936); 20, 297 (1937).
2 W. D. Armstrong, J. Am. Chem. Soc., 55, 1741 (1933); Ind. Eng. Chem., Anal. Ed., 8, 384 (1936); R. J. RowleynH. V. Churchill [там же, 9, 551 (1937)] установили, что в водных растворах можно обработать 50 мг фтора, тогда как в спиртовых только 20 мг.
3 W. М. Н о s k i n s, С. A. F е г г i s, Ind. Eng. Chem., Anal. Ed., 8, 6 (1936).
4 H. V. Churchill, R. W. Bridges, R. J. Rowley, Ind. Eng. Chem., Anal. Ed., 9, 222 (1937).
1 2 3 4 5 W. K. Gilkey, H. L. Rohs, H. V. Hansen, Ind. Eng. Chem., Anal. Ed., 8, 150 (1936).
5 D. S. R e у n о 1 d s, W. L. H i 11, Ind. Eng. Chem., Anal. Ed., 11, 21 (1939).
826
Гл. XLVIII. Фтор
Ход определения. Помещают 0,5 г пробы и 15 мл разбавленной (2 : 1) хлорной кислоты в дистилляционную колбу для перегонки с паром и перегоняют при 125—150° С до получения 150 мл дистиллята.
При анализе шлаков, фосфатных пород и обожженных фосфатов стенки дистилляционной колбы постепенно покрываются слоем осадка кремнекислоты, который прочно удерживает фтор1. Если дать этому осадку накапливаться, могут возникнуть серьезные ошибки, когда одну и ту же колбу применяют для анализа продуктов, резко различающихся по содержанию фтора. Этот слой кремнекислоты можно удалить горячим концентрированным раствором едкой щелочи.
Установлено2, что выпаривание подщелоченного дистиллята для уменьшения его объема перед титрованием должно проводиться в платиновой посуде, так как в случае проведения этой операции в стеклянной или фарфоровой посуде получаются пониженные результаты.
^^Если в анализируемой пробе содержится пирит, смачивают навеску в дистилляционной колбе 2—3 мл насыщенного раствора перманганата калия и в соответствии с этим повышают концентрацию вводимой хлорной кислоты. Прибавляют к дистилляту несколько капель фенолфталеина и 1 М раствор едкого натра до щелочной реакции, а затем разбавляют точно до 250 мл. Отбирают 50 мл раствора, вводят 5 капель раствора индикатора (0,1%-ный водный раствор ализаринсульфоната натрия), а затем 0,1 М соляную кислоту до исчезновения розовой окраски. После этого в раствор прибавляют 2,5 мл монохлоруксусной кислоты (0,4 М), буферный раствор едкого натра (0,2 М) и титруют 0,04 н. раствором нитрата тория.
Титр раствора нитрата тория лучше устанавливать по специально очищенному фториду натрия, содержащему определенное количество фтора, или по стандартному образцу, имеющему одинаковый состав с анализируемым материалом и содержащему примерно такие же количества фтора. Навеску такого стандартного образца проводят через все стадии анализа 3.
Осаждение в виде фторохлорида свинца
Метод определения фтора в виде фторохлорида свинца проверялся многими исследователями 4. Он основан на осаждении соединения PbCIF, которое может быть непосредственно взвешено или оттитровано методом Фольгарда (стр. 813). Содержание фтора в этом соединении можно вычислить по найденному количеству хлора. Привлекательной чертой весового метода является низкий фактор пересчета для фтора. Объемный метод имеет то преимущество, что он может применяться в присутствии других осаждающихся веществ, при условии, что они не содержат хлора. Оба метода дают хорошие результаты при применении их к растворам чистых фторидов.
1 D. S. Reynolds, J. Assoc. Offic. Agr. Chemists, 18, 108 (1935).
2 F. J. McClure, Ind. Eng. Chem., Anal. Ed., 11, 171 (1939).
3 Чистый фторид натрия можно приготовить обработкой чистого карбоната или бикарбоната натрия фтористоводородной кислотой, удалением избытка последней выпариванием и прокаливанием остатка приблизительно при 600° С в атмосфере сухого азота или воздуха. Об очистке фтористоводородной кислоты см. J. I. Hoffman, G.E. Lunde 11, J. Research NBS, 3, сноска на стр. 582 (1929). *См. также Ю. В. Карякин, И. И. Ангелов, Чистые химические реактивы, Госхимиздат, 1955.*
4 G. Starck, Z. anorg. Chem., 70, 173 (1911); F. G. Hawley, Ind. Eng. Chem., 18, 573 (1926). См. также W. H. Adolph, цит. выше L. D. Hammond, Ind. Eng. Chem., 16, 928 (1924); J. I. H о f f m a n, G. E. F. L u n d e 11, J. Research NBS, 3, 581 (1929).
Методы определения
827
Осаждение проводят из растворов, имеющих pH в пределах от 3,6 до 5,6 в зависимости от условий. В присутствии даже таких малых количеств алюминия, как 0,5 мг, получаются пониженные результаты, так же как и при наличии значительных количеств бора О 0,05 г), аммонийных О0,5 г), натриевых и калиевых (> 10 г) солей. Железо также мешает определению, но на практике оно всегда отделяется при предварительной обработке. Когда осадок предполагается взвешивать, осаждение следует проводить в растворе, свободном от таких соединений, как карбонаты или фосфаты, которые также осаждаются хлоридом свинца или гидролизуются при той концентрации ионов водорода, которая требуется для осаждения PbCIF. При окончании определения объемным методом малые количества кремнекислоты не оказывают влияния, а значительные количества ее, более 0,05 г, забивают поры фильтра. Малые количества фосфатов или сульфатов (10 мг или менее) не мешают определению. В присутствии фосфатов фильтрат от PbCIF может быть мутным, но на это не следует обращать внимания.
Осадок заметно растворим в воде. В 1 л воды при 25° С растворяется 0,325 г PbCIF. Он менее растворим в ледяной воде и не растворяется в насыщенном растворе PbCIF. Когда осадок предполагается взвешивать, его собирают в тигле Гуча или Мунро и сушат при 120—150° С
Поправки на растворимость могут не соответствовать величинам, приводимым в таблицах растворимости. Опыты Дж. И. Гоффмана показали, что при промывании 0,5 г PbCIF описанным ниже методом растворяется не более 0,002 г (что соответствует 0,15 мг фтора). Растворимость, определенная на основании этих опытов, равна 0,320 г PbCIF в 1 л чистой воды при 21Q С, что хорошо согласуется с величиной, найденной Старком. Но когда 0,5 г PbCIF промывали на плотном (синяя лента) фильтре диаметром 9 см водой, порциями по 10 мл, причем при каждом промывании осадок сильно взмучивался, в 1 л промывных вод было найдено только 0,260 г PbCIF. Когда тот же осадок был промыт струей воды, направленной вдоль края фильтра и без взмучивания, перешедшее в раствор количество PbCIF снизилось до 0,220 г на 1 л.
Ход определения. Приготовляют нейтральный или щелочной раствор, содержащий 0,01—0,1 г фтора и свободный от перечисленных выше мешающих определению веществ. Приготовление такого раствора при анализе горных пород и минералов описано в разделе «Фтор» (стр. 1018). Описываемые далее операции можно при желании проводить в стеклянной посуде.
В раствор вводят 2 капли раствора индикатора бромфенолового синего \ 3 мл 10%-ного раствора хлорида натрия и разбавляют до 250 мл. Прибавляют разбавленную азотную кислоту до появления желтой окраски, а затем разбавленный раствор едкого натра — до перехода желтой окраски, а затем разбавленный раствор едкого натра — до перехода желтой окраски в синюю. После этого прибавляют 2 мл разбавленной (1 : 1) соляной кислоты, затем 5 г нитрата свинца Pb (NO3)2 и нагревают на паровой бане. Осторожно перемешивают до растворения нитрата свинца, после чего немедленно прибавляют 5 г ацетата натрия СН3СОО-ЗН2О. Сильно размешивают и дают постоять на паровой бане 30 мин, изредка помешивая. Оставляют на ночь при комнатной температуре. Фильтруют через плотный фильтр (синяя лента) и промывают осадок в стакане и на фильтре
1 Приготовляется растиранием 0,4 г сухого измельченного в порошок индикатора с 6 мл 0,1 н. раствора NaOH и последующим разбавлением до 100 мл.
828
Гл. XLVIII. Фтор
один раз холодной водой, затем 4—5 раз насыщенным раствором фторохлорида свинца 1 и, наконец, снова один раз холодной водой.
Фильтр с осадком переносят в стакан, в котором проводилось осаждение, превращают фильтр в бумажную массу, прибавляют 100 мл разбавленной (5 : 95) азотной кислоты и нагревают на паровой бане до растворения осадка (вполне достаточно 5 мин). Затем прибавляют титрованный 0,2 н. раствор нитрата серебра, нагревают на паровой бане 30 мин и охлаждают до комнатной температуры, защитив колбу от действия света. Осадок отфильтровывают и промывают холодной водой. В фильтрате титруют нитрат серебра раствором роданида калия, как описано в гл. «Серебро» (стр. 238). Из первоначально введенного в раствор количества серебра вычитают количество серебра, найденное в фильтрате. Разность соответствует количеству серебра, которое соединилось с хлором, находившимся в осадке PbCIF. По этой разности вычисляют содержание фтора. 1 мл 0,2 н. раствора нитрата серебра эквивалентен 0,00380 г фтора 2.
Колориметрические методы
Разработан целый ряд колориметрических методов определения малых количеств фтора в таких материалах, как природные воды, морская вода, хлебные злаки, продукты питания. Из них можно указать на методы, основанные на свойстве фторид-ибна обесцвечивать: 1) желтовато-красный цирконий-хинализариновый лак 3; 2) красновато-фиолетовый цирконий-ализариновый лак 4; 3) розовый лак оксихлорида циркония с пурпурином 5; 4) красную окраску роданида железа6 (III); 5) окрашенный в зеленый цвет «феррон» [комплексное соединение железа (III) с 7-иодо-8-окси-хинолин-5-сульфоновой кислотой] 7 и 6) желтую окраску надтитановой кислоты 8.
Описаны9 спектральные методы, визуальные и фотографические, фотоколориметрический метод, основанный на применении ацетилацетон-
1 Для приготовления промывной жидкости, насыщенной PbCIF, растворяют 10 г нитрата свинца в 200 мл воды и вливают этот раствор в 100 мл раствора, содержащего 1,0 г фторида натрия и 2 мл соляной кислоты. Тщательно перемешивают, дают отстояться, сливают жидкость декантацией и промывают осадок 4—5 раз декантацией водой, порциями по 200 мл. К осадку прибавляют 1 л воды, дают постоять в течение 1 ч, время от времени перемешивая, и фильтруют. Новые порции промывной жидкости можно получить, снова прилив к осадку чистую воду и перемешав.
2 J. I. Hoffman и G. Е. F. Lundell, [The Analysis of Phosphate Rock,-J. Research NBS 20, 607 (1938)] приводят применение этого метода к дистилляту, полученному по способу WillardnWinter (стр. 755).
3 О. М. Smith, Н. A. Dutcher, Ind. Eng. Chem., Anal. Ed., 6, 61 (1934) (природные воды).
4 T. G. T h о m p s о n, H. J. T а у 1 о r, Ind. Eng. Chem., Anal. Ed., 5, 87 (1933) (морская вода); J. M. Sanchis, Ind. Eng Chem., Anal. Ed., 6, 134 (1934) (природные воды) О. В. W i n t e r, J. Assoc. Offic. Agr. Chemists, 19, 362 (1936) (люцерна).
5 I. M. К о 11 h о f f, M. E. S t a n s b y, Ind. Eng. Chem., Anal. Ed., 6, 118 (1934). Авторы описывают простой метод дистилляции и утверждают, что чувствительность этого метода составляет 0,005 мг F в 1 мл.
6 J. G. F a i г с h i 1 d, J. Wash. Acad. Sci., 20,141 (1930) (породы); H. V. Churchill, Ind. Eng. Chem., 23, 996 (1931); M. D. Foster, Ind. Eng. Chem., Anal. Ed., 5, 234 (1933). Обзор различных методов см. Н. V. S m i t h, там же, 7, 23 (1935).
’ J. J. Fahey, Ind. Eng. Chem., Anal. Ed., 11, 362 (1939) (породы, минералы и природные воды).
8 Н. J. W i с h m a n n, D. D a h 1 е, J. Assoc. Offic. Agr. Chemists, 16, 612, 619 (1933) (фрукты и овощи); D. D a h 1 е, там же, 20, 505 (1937).
8 A. W. Р е t г е у, Ind. Eng. Chem., Anal. Ed., 6, 343 (1934).
Методы, определения
829
железного (III) реактива 1 и метод колориметрического титрования, в котором стандартный раствор, обработанный так же, как анализируемый раствор, содержащий торий-ализариновый лак, титруют фторидом натрия до одинаковой с анализируемым раствором окраски 2.
Другие методы
Из числа других методов определения фтора можно упомянуть объемный метод, основанный на титровании фторид-ионов хлоридом железа (III) 3, и весовой метод, в котором фтор осаждается в виде фторида тория 4 TI1F4. Первый метод, применимый к растворимым фторидам, основан на образовании «железного криолита» Na3FeFe. Титрование проводится в нейтральном по фенолфталеину растворе, содержащем роданид аммония в качестве индикатора, хлорид натрия и спирт для понижения растворимости осадка и эфир, который делает точку эквивалентности более отчетливой. Хлорид натрия можно заменить хлоридом калия. Можно применять потенциометрический метод титрования 5. Бор мешает определению.
Осаждение фтора в виде фторида тория не дает таких хороших результатов, как предыдущий метод. Осадок всегда загрязнен возможно такими соединениями, как Na2ThFe. Он заметно растворим в избытке осадителя и, кроме того, его необходимо прокаливать до ТЬОа. В растворе может остаться до 0,1 г фтора в 1 л6. В органических соединениях фтор можно количественно определять по потере в массе стекла колбы пайрекс, вызванной разъеданием стекла образующейся фтористоводородной кислотой. Анализируемая проба полностью разлагается нагреванием с раствором небольшого количества нитрата калия в серной кислоте 7.
1 L. V. W i 1 с о х, Ind. Eng. Chem., Anal. Ed., 6, 167 (1934).
2 D. D a hl e, R. U. В onn аг. H. J. Wi ch m ann, J. Assoc. Offic. Agr. Chemists, 21, 459, 468 (1938).
s P. G u у 0 t, C. r., 71, 274 (1870); A. G г e e f f, Ber., 46, 2511 (1913); I. Belin с c i, Ann. chim. appl., 1, 441 (1914); L. D. Hammond, Ind. Eng. Chem., 16, 938 (1924).
4 E. D e 1 a d г i e r, Chem. Weekblad, 1, 324 (1904); M. F. P i s a n i, C. r., 162,
791 (1916); F. A. G 00c h, Ц. К oh ay ashi, Am. J. Sci., 45, 370 (1918).
6 A. F. К 0 h 1, диссертация, 446, Technische Hochschule, Zurich, 1926; W. D. T r e-a,d w e 11» A. F. К 6 h 1, Helw. Chim. Acta, 9, 470 (1926).
6 Согласно L. D о m a n g e [C. r., 213, 31 (1914)], осаждение фтора в виде фторида висмута BiF3 имеет известные премущества, благодаря высокому молекулярному весу этого соединения, устойчивости его в водных растворах, меньшей растворимости, чем растворимость CaF2, и легкости отделения его фильтрованием и промывания.
’ D. Н, Brauns, J, Research NBS, 27, 105 (1941),
Глава XLIX
БОР
Бор, являющийся составной частью минерала турмалина (Н, Li, Na, К)9 Al3 (ВОН)2 Si4O18 + (БегОз, FeO, MgO, МпО) обычно находится в сильно кремнекислых породах. Он постоянно встречается также во многих метаморфических сланцах. Бор является также основной составной частью некоторых других силикатов, из которых важнейшим является датолит Са (ВОН) SiO<.
Соединения бора в промышленности получают из боратов, к которым относятся бура (тинкал) (Na2B4O7-ЮН2О), улексит (NaCaB5O9-6Н2О) и колеманит [НСа (ВО2)3-2Н2)О1, а также из природной ортоборной кислоты сассолина (Н3ВО3), которая находится в водах некоторых вулканических источников. В больших количествах бор добывается в некоторых щелочных озерах и лагунах, особенно в Калифорнии и в Тибете х.
Бор, без сомнения, иногда встречается в горных породах в количествах, превосходящих содержание некоторых из обычно определяемых компонентов. Однако недостаточная точность методов, которые применимы к анализу минеральных веществ, не растворимых непосредственно в кислотах (кроме фтористоводородной), приводит к тому, что этими методами обычно пользуются лишь тогда, когда заранее известно о наличии в анализируемом материале значительных количеств бора.
ОБЩИЕ ЗАМЕЧАНИЯ
Открытие малых количеств бора в породах является сложной задачей. Было найдено 1 2, что чувствительность непосредственной пробы на пламя равна 0,2%. Для выполнения испытания смешивают измельченный в порошок минерал с бисульфатом калия и фторидом кальция, а затем вносят эту смесь на чистой платиновой проволочке в несветящееся пламя бунзе-новской горелки. Присутствие бора узнается по зеленому окрашиванию пламени, быстро исчезающему при содержании небольших количеств бора, если в испытуемой смеси не присутствуют другие элементы, также окрашивающие пламя в зеленый цвет.
Показано 3, что испытание в спиртовом пламени становится более чувствительным, если применять его к борному эфиру, полученному в ре
1 F. W. Clarke, U.S. Geol. Survey Bull., 770, 14 (1924).
2 E. T. Wherry, W. H. C ha p i n, J. Am. Chem. Soc., 30, 1684 (1908).
• С. M a n n i c h, H. P r i e s s, Chem. Ztg., 32, 314 (1908); V. L e n h e r. J. S. C. Wells, J. Am. Chem. Soc., 21, 417 (1899).
Общие замечания
.831
зультате перегонки, как описано в разделе «Методы определения» (стр. 834). Барий мешает непосредственному испытанию со спиртом и кислотой Ч
Проба с куркумовой бумагой дает возможность 1 2 открыть бор в минералах, содержащих 0,08% В2О3. Метод заключается в следующем 3. Раствор солей бора, находящийся в плоской чашке (не из борсодержащего стекла), подкисляют соляной кислотой, вводят в него кусок куркумовой бумаги и испаряют досуха в эксикаторе. В отсутствие мешающих элементов, как, например, титана, и при не слишком высоком содержании соляной кислоты в растворе, бор окрашивает куркумовую бумагу в розоватокрасный цвет.
Непосредственно к раствору минерала метод неприменим. Для испытания пользуются дистиллятом, полученным, как описано в разделе «Методы определения» (стр. 834).
Метод испытания с куркумовой бумагой является наиболее чувствительным,. если соблюдать необходимые меры предосторожности, которые изложены в оригинальных статьях 3. Авторы применяли этот метод для открытия борного ангидрида в количестве от 0,1 до 0,0005 мг. Для испытания должны применяться специально приготовленные реактивы. Чувствительность метода подтверждена также данными, полученными другими авторами 4.
Метод заключается в сравнении длин окрашенцой части полосок куркумовой бумаги, погруженных в испытуемый и стандартный растворы, которые имеют одинаковый объем и находятся в градуированных цилиндрах. Раствор поднимается по бумажным полоскам капиллярными силами и испаряется на обнаженных концах этих полосок. По истечении нескольких часов эти полоски оказываются окрашенными, причем длина окрашенной поверхности зависит от концентрации бора в растворе. Когда имеется достаточное число стандартных растворов, испытание дает весьма точные количественные результаты. При обработке аммиаком окраска синеет.
Имеется указание 5, что: 1) реакция с куркумином (окрашивание в красный цвет) более чувствительна, чем с куркумовой бумагой; 2) окраска эта устойчива в течение не менее 10 ч; 3) окрашенное соединение растворяется без разложения в спирте и эфире и 4) при обработке щелочью получайся интенсивно синяя окраска.
Молоко и другие пищевые продукты обычно сначала озоляют, а затем испытывают следующим образом. Золу обрабатывают несколькими каплями: 1) разбавленной соляной кислоты; 2) насыщенного раствора щавелевой кислоты и 3) спиртового раствора куркумина. Затем выпаривают досуха на водяной бане и растворяют остаток в небольшом количестве спирта. Если содержание борной кислоты очень незначительно, анализируемую пробу перед выпариванием и озолением надо подщелочить раствором гидроокиси бария. Каустическая сода или поташ, а также большие . количества натриевых или калиевых солей в данном случае неприемлемы.
1 F. Krauss, Chem. Ztg., 51, 38 (1927).
2 W. Н. L о w, Chem. Ztg., 28, 807 (1906).
3 G. В e r t r a n d, H. A g u 1 h о n, C. r., 157, 1433 (1913); Bull. soc. chim. [4], 7, 90, 125 (1910); 13, 396 (1913).
* E. T. All en, E. G. Z i es, J. Am. Ceram. Soc., 1, 739 (1918).
* С. E. C a s s a 1, H. G e r r a n s, Chem, News, 87, 27 (1903).
832
Гл. XLIX. Бор
В обычном ходе анализа присутствие бора вызывает ряд затруднений. Основное из них заключается в том, что бор сопровождает кремнекислоту и улетучивается вместе с ней при обработке фтористоводородной кислотой, вследствие чего получаются повышенные результаты для кремния. Железо и алюминий не удается отделить от бора даже трехкратным осаждением их аммиаком. Бор, захваченный осадком от аммиака, при взвешивании принимается за алюминий. Насколько нам известно, умеренные количества бора, например 5%-ный раствор борной кислоты, не вызывают затруднений при определении кальция и магния.
РАЗЛОЖЕНИЕ МИНЕРАЛОВ, СОДЕРЖАЩИХ БОР
В процессе приготовления растворов для определения бора надо соблюдать особые меры предосторожности; в противном случае может произойти потеря бора в таких операциях, как выпаривание, высушивание, прокаливание, а при некоторых условиях и в процессе сплавления с карбонатами щелочных металлов. Возможные потери бора в процессе выпаривания растворов, в которых находится свободная борная кислота, иллюстрируются опытами *, в которых в результате выпаривания досуха на водяной бане двух растворов, содержащих 0,603 г и 0,0095 г В2О3 потери составляли соответственно 0,0522 г и 0,0071 г В2О3. Было отмечено также, что значительные количества бора теряются при нагревании твердой борной кислоты до образования борного ангидрида.
Летучесть буры при нагревании ее до не слишком высоких температур еще не установлена. С одной стороны, когда буру прокаливали * 2 при 800° С в электрической печи, никакой потери в массе не было отмечено. Но, с другой стороны, утверждают 3, что бура теряет некоторое количество окиси натрия, когда ее плавят в течение нескольких часов в электрическом муфеле при 700 —800° С. Имеются указания 4 на то, что бура возгоняется при нагревании на паяльной горелке. Было также найдено5, что бор улетучивается в процессе сплавления с карбонатом натрия стекол, содержащих более 10% борного ангидрида. Для устранения этого авторы последнего исследования рекомендуют пользоваться небольшими навесками и прибавлять перед сплавлением чистую кремнекислоту.
При применении 6 метода Чэпина (стр. 835) к анализу материала, свободного от фтора, и проведении всех операций в посуде из борсодержащего стекла, заметного загрязнения раствора бором не было обнаружено. Несмотря на это, когда надо определять очень малые количества бора или когда требуется исключительная точность, все операции лучше проводить в платиновой или кварцевой посуде. Реактивы всегда должны проверяться, так как содержание бора в них может быть довольно значительным. Так, в реактивах, израсходованных только на одно определение, было найдено 6 0,6 мг В2О3.
> Р. Tchijewski, Arch. Phys. Nat., [3], 12, 120 (1884).
2 I. M. К о I t h о f f, J. Am. Chem. Soc., 48, 1449 (1926).
3 H. V. A. Briscoe, P. L. Robinson, Nature, 118, 374 (1926); см. также H. V. A. Briscoe, P. L. Robinson, G. E. Stephenson, J. Chem. Soc., 127, 150 (1925).
4 S. W a 1 d b о 11, J. Am. Chem. Soc., 16, 410 (1894).
5 V. D i m bl e b y, W. E. S. Turner, J. Soc. Glass, Technol., 7, 76 (1923).
6 E. T. A 11 e n, E. G. Z i e s, цит. выше.
Методы отделения
833
Если минерал не растворяется в соляной кислоте, его можно, сплавить с карбонатом натрия или со смесью карбоната натрия и карбоната калия, а затем растворить в соляной кислоте при 15—30° С. В этом случае рекомендуется отвешивать плавень с точностью до 1 мг, чтобы можно было заранее установить количество кислоты, требуемое для растворения плава Ч
Для предварительного разложения анализируемой пробы рекомендуется 1 2 расплавить сначала 2 з безводного Na2HPO4 и 1 г НРО3 в платиновом тигле, а затем, прибавив 0,5 г тонко измельченной анализируемой пробы, осторожно нагревать до полного сплавления смеси. Если требуется, закрытый тигель можно нагревать несколько минут на паяльной лампе. По окончании сплавления плотно закрытый еще горячий тигель не полностью погружают в ледяную воду и отделившиеся кусочки плава помещают в дистилляционную колбу (см. раздел «Методы определения», стр. 836).
Частицы плава, приставшие к тиглю, удаляют нагреванием с сиропообразной фосфорной кислотой и переносят в дистилляционную колбу, куда вводят еще 10 мл фосфорной кислоты и несколько кусочков битого фарфора. Колбу соединяют с холодильником и нагревают до удаления воды и получения гомогенного раствора, который обрабатывают метиловым спиртом. Образовавшийся борный эфир отгоняют.
Разложение органических соединений, содержащих бор (и фтор), см. в гл. «Фтор» (стр. 822).
МЕТОДЫ ОТДЕЛЕНИЯ
Важнейший метод отделения бора от других элементов основан на отгонке борнометилового эфира В (ОСН3)3 из кислых растворов. Отгонка этого соединения проводится кипячением подкисленных растворов боратов щелочных или щелочноземельных металлов, в которые введен метиловый спирт. Метод служит как для извлечения бора перед его определением, так и для удаления из растворов, где присутствие его нежелательно. Метод, применяемый в первом случае, подробно изложен в разделе «Титрование едким натром» (стр. 834). Основным требованием при этом является полное извлечение бора, возможно менее загрязненного кислотой, для поглощения которой можно пользоваться различными реагентами, например хлоридом кальция или сиропообразной фосфорной кислотой. Если же целью обработки является только удаление бора из раствора, применение обезвоживающих реагентов недопустимо. В этом случае раствор выпаривают досуха, остаток обрабатывают 25 мл абсолютного метилового спирта или метилового спирта, насыщенного хлористым водородом, и затем, прикрыв раствор часовым стеклом, снова осторожно выпаривают досуха. Для полного удаления бора необходима 2—4-кратная обработка. Из разбавленных водных растворов бор полностью не удаляется.
1 Описание методов растворения некоторых металлов, содержащих бор, как, например, сталей, можно найти у N. Tschischewski [Ind. Eng. Chem., 18, 607 (1926)], который рекомендует обрабатывать сталь разбавленной серной кислотой в колбе с обратным холодильником, окислять 30%-ной Н2О2, после чего, нейтрализовав большую часть кислоты, подвергать раствор электролизу с ртутным катодом (стр. 165) и титровать едким натром, сначала по метиловому оранжевому, а затем по фенолфталеину, как указано в разделе «Методы определения» (стр. 834).
2 Р. J a n n a s с h, F. N о 11, J. prakt. Chem., 99, 1 (1919).
53 Заказ 522.
834
Гл. XLIX. Бор
Бор можно также отогнать из растворов, где присутствие его нежелательно, выпариванием с фтористоводородной кислотой 1 или со смесью серной и фтористоводородной кислот до появления паров серной кислоты 2.
Алюминий 3 4, и возможно также железо, титан, цирконий и т. п., можно количественно отделить от бора осаждением оксихинолином из аммиачного раствора (стр. 148).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Большинство методов определения бора основано на отгонке его в виде борнометилового эфира. В объемном методе, описанном в разделе «Титро-ванйе едким натром», отгоняемый эфир поглощается и омыляется раствором щелочи, а затем, после удаления спирта, борная кислота титруется раствором едкого натра. В весовом методе, описанном на стр. 841, эфир омыляют и борную кислоту связывают взвешенным количеством окиси кальция, которую затем снова взвешивают.
Объемный метод следует предпочесть.
Титрование едким натром
Водный раствор борной кислоты нельзя непосредственно титровать раствором едкого натра, так как неизвестен такой индикатор, окраска которого достаточно резко изменялась бы в точке эквивалентности, т. е. при pH, равном приблизительно И. Если, однако, к раствору прибавить какой-либо многоатомный спирт, например глицерин или маннит, то образуется соединение с более сильными кислотными свойствами, что дает возможность успешно осуществить титрование по фенолфталеину или а-нафтолфталеину. В присутствии маннита происходит более резкое изменение окраски индикатора, чем в присутствии глицерина А Маннит, кроме того, удобен еще тем, что при его применении не требуется соблюдения особых предосторожностей и существенно не изменяется объем анализируемого раствора. В рядовой работе можно пользоваться более дешевым глицерином.
В начальной стадии анализа обычно получается не борная кислота, а борат, поэтому основная задача сводится к количественному переведению бора в виде борной кислоты в раствор, не содержащий других свободных кислот. Это осуществляется титрованием соляной кислотой по такому индикатору, окраска которого изменяется при концентрации ионов водорода несколько более высокой, чем свойственная борной кислоте (pH = 6,5—7). Окраска индикатора не должна мешать определению точки эквивалентности при конечном титровании. Титрование до оранжево-красной окраски метилового оранжевого (pH —- 4) вполне отвечает этим требованиям.
Так как первое титрование проводится в среде метилового спирта, в которой переход метилового оранжевого недостаточно отчетлив, то в при-
1 Например, после обработки 0,2 г В2О3 20 мл HF в платиновом тигле, выпаривания и осторожного прокаливания нелетучий остаток не был обнаружен.
2 Отделение и определение малых количеств бора в вольфраме см. D. Н. В г о-р h у, J. Am. Chem. Soc., 47, 1856 (1925).
3 G. Е. F. L u n d е 1 1, Н. В. К п о w 1 е s, J. Research NBS, 3, 91 (1929).
4 J. A. M.VanLi empt,Z. anorg. u. allgem. Chem., Ill, 151 (1920); M. G. M e 1-1 о n, V. N. Morris, Ind. Eng. Chem., 16, 123 (1924).
Методы определения
835
водимом ниже методе в качестве индикатора применяется паранитрофенол (pH в конечной точке равен 7). Паранитрофенол чувствителен к борной кислоте 1 при высоких ее концентрациях, что приводит в таких случаях к пониженным результатам, если содержание В2О3 рассчитывается по теоретическому титру раствора едкого натра, установленного обычно принятыми методами. Это затруднение, а также и влияние следов карбоната, находящихся в растворе едкого натра, устраняется, если титр раствора едкого натра устанавливается по отдельным навескам чистого сухого борного ангидрида, равным приблизительно содержанию В2О3 в анализируемом веществе и обработанным согласно приводимому ниже методу.
Независимо от применяемого индикатора, некоторое количество едкого натра расходуется на переход от первой конечной точки титрования ко второй точке и тогда, когда раствор не содержит борной кислоты.
Было, например, найдено 2, что от точки перехода паранитрофенола до точки перехода фенолфталеина расходуется 0,08 мл 0,1 н. раствора едкой щелочи, что отвечает0,3 мг В2О3. Этот расход щелочи включается в поправку на «холостой» опыт, результаты которого всегда необходимо учитывать.
Приводим метод в его варианте, предложенном Чэпиным. Последовательные этапы развития этого метода приведены в сноске 3.
Для определения необходимы следующие реактивы:
Пара нитрофено л. 1 г паранитрофенола растворяют в 75 мл нейтрального этилового спирта и разбавляют водой до 100 мл.
Фенолфталеин. 1 г фенолфталеина растворяют в 100 мл этилового спирта и разбавляют водой до 200 мл.
Соляная кислота 0,1 н. раствор. Для приготовления реактива воду следует прокипятить до удаления двуокиси углерода.
Соляная кислота разбавленная (1:1). Капельная воронка прибора должна быть наполнена этой кислотой на случай необходимости применения этой кислоты при точном определении малых количеств бора.
Едкий натр 0,5 н. и 0,1 н. растворы. Эти растворы не должны соприкасаться с борсодержащим стеклом и лучше, если они свободны от карбоната (стр. 207). Титр их устанавливают по борному ангидриду, который приготовляют следующим образом. Расплавляют в платиновой чашке чистую борную кислоту. Разбивают еще горячий плав на куски и быстро переносят их в бюкс. Растворяют 1,741 г В 2О3 в 250 мл горячей свежепрокипяченной воды и по охлаждении разбавляют раствор до 500 мл. Каждый миллилитр этого раствора после обработки в соответствии с приводимым методом, должен отвечать приблизительно 1 мл 0,1 н. раствора едкого натра.
Маннит или глицерин. Лучше применять маннит.
1 W. Strecker, E.Kannappel, Z. anal. Chem., 61, 378 (1922).
2 См. сноску 4 на стр. 831.
3 В 1893 г. R. Т. Т h о m р s о n, [J. Soc. Chem. Ind., 12, 432 (1893)] ввел объемный метод, в котором борная кислота освобождается нейтрализацией раствора соляной кислотой по метиловому оранжевому и затем после прибавления глицерина титруется едким натром по фенолфталеину. G. W. S а т g е n t [J. Am. Chem. Soc., 21, 859 (1899)] применил этот метод после отгонки бора в виде эфира и получил удовлетворительные результаты при анализе турмалина. М. F. S с h a a k [J. Soc. Спет., Ind., 23, 699 (1904)] видоизменил метод тем, что ввел пропускание через раствор паров метилового спирта во время отгонки. В дальнейшем W. Н. L ow [J. Am. Chem. Soc., 28, 807 (1906)] еще улучшил этот метод тем, что добавлял безводный хлорид кальция в раствор бората • для предотвращения гидролитического действия воды на метиловый эфир и удалял последние следы двуокиси углерода под пониженным давлением. Наконец, W. Н. Ch а-р i n [J. Am. Chem. Soc., 30, 1691 (1908)] расширил область применения метода Low, применив его к анализу минералов, и показал, что борный эфир можно отгонять непосредственно из подкисленного раствора плава минерала с карбонатами. Видоизменение метода Chapin применительно к органическим продуктам, содержащим менее 0,0005% бора, см. L. V. Wilcox, Ind. Eng. Chem., Anal. Ed., 2, 358 (1930).
53*
836
Гл. XLIX. Бор
Метиловый спирт. Метиловый спирт высокой концентрации после кипячения в колбе с обратным холодильником в течение нескольких часов в контакте с окисью кальция должен быть перегнан над окисью кальция. Чем полнее обезвожен спирт, тем лучше. Если в спирте остаются летучие органические кислоты, то результаты определения бора получаются повышенными.
Хлорид кальция. Применяется гранулированная, безводная, свободная от бора соль.
Прибор для отгонки бора. Основные части прибора показаны на рис. 30. Колба 1 емкостью 500 мл для метилового спирта снабжена U-образной трубкой, содержащей немного ртути и служащей пред-охранитель'ным клапаном, и капиллярной трубкой «для кипения» диаметром 3 мм, запаянной внизу на высоте около 1 см от нижнего конца.
Рис. 30. Прибор для отгонки бора:
1 — колба для метилового спирта; 2 — колба для разложения минерала; 3 — стеклянный холодильник; 4 — приемная колба.
Колба 2 емкостью 250 мл помещена в водяной бане так, чтобы она не касалась дна бани; отходящая от этой колбы трубка должна иметь в высоту не меньше 12 см и довольно широкое отверстие. Конец трубки, находящийся внутри колбы, должен быть обрезан под углом, чтобы конденсирующаяся в ней жидкость падала обратно в колбу, а не подымалась вверх по трубке.
Длина стеклянного холодильника 40 см; диаметр его внешней трубки 3 см. Емкость приемной колбы 250 мл; две-три такие колбы с одинаковыми горлышками должны всегда находиться под рукой, чтобы можно было заменить один приемник другим, не прерывая перегонки. Присоединенная к приемнику небольшая U-образная трубка с водой служит для улавливания несконденсировавшегося метилбората.
Второй стеклянный холодильник (на рисунке не показан), подобный холодильнику 5, соединен с одной стороны с колбой, точно такой, как колба 2, но имеющей только одно отверстие в пробке и полностью погруженной в большую баню; в этой колбе должна быть также трубка «для
Методы определения
837
кипения»; другой конец этого холодильника соединен с приемником подобным приемнику 4, но без U-образного предохранителя. Этот прибор применяется для отгонки спирта после предварительного титрования.
Для фильтрования прибор соединяют с насосом. Между насосом и пробкой'колбы ставят маленькую предохранительную склянку для предотвращения перебрасывания воды из насоса в колбу.
Ход определения. Минерал измельчают в тонкий порошок и берут навеску с таким расчетом, чтобы содержание бора в ней не превышало 0,1г. Навеска должна быть не более 0,5 г, а когда содержание бора в анализируемом материале не превышает 10%, то не менее этого количества. Плавень, применяемый для предварительного разложения материала, необходимо взвесить с точностью до 1—2 jns, чтобы можно было вычислить количество кислоты, требуемое для разложения плава, и предотвратить таким образом введение чрезмерного ее избытка.
Если минерал растворяется в соляной кислоте, то навеску его в 1 г переносят в колбу 2, не давая ему прилипать к шейке колбы, обрабатывают не более чем 5 мл разбавленной (1 : 1) соляной кислоты и нагревают на водяной бане до полного растворения.
Если минерал не растворяется в соляной кислоте, его смешивают с точно шестикратным по массе количеством карбоната натрия или таким же количеством эквимолекулярной смеси карбонатов натрия и калия и сплавляют обычным способом. Не удаляя плав из тигля, разлагают его, постепенно добавляя вычисленное количество разбавленной (1 : 1) соляной кислоты. Эту операцию проводят, поместив тигель в чашку и по возможности держа его все время закрытым крышкой. К концу операции может потребоваться нагревание, однако необходимо иметь в виду, что применять кипячение нельзя, так как борная кислота может улетучиться с паром. Раствор переносят в колбу 2 и ополаскивают тигель очень небольшим количеством воды. Через бумажную воронку, чтобы не запачкать шейку колбы, всыпают чистый безводный хлорид кальция в количестве 1 г на каждый миллилитр раствора. Легким взбалтыванием смешивают хлорид кальция с водой, подымают баню настолько, чтобы колба погрузилась в воду, но не касалась дна бани, и затем начинают перегонку спирта из колбы 7, следя за тем, чтобы открытый конец капиллярной трубки «для кипения» был свободен от спирта, а U-образная трубка, соединенная с приемником, была заполнена водой.
Колбу 2, в которой происходит разложение, начинают нагревать лишь после того, как в ней сконденсируется 25 мл спирта.
Водяную баню нагревают на небольшом пламени, поддерживая в колбе температуру, достаточную для предупреждения дальнейшей конденсации спирта. Дистилляция не должна происходить слишком быстро во избежание потери борнометилового эфира.
По получении приблизительно 100 мл дистиллята заменяют приемник 4 другим таким же и продолжают перегонку. К первому дистилляту прибавляют содержимое U-образной трубки и проводят предварительное титрование следующим образом. Вводят в раствор каплю паранитрофенола, а затем титрованный раствор едкого натра до нейтрализации свободной минеральной кислоты. После этого прибавляют 0,2 мл раствора фенолфталеина и продолжают титрование до начала появления окраски индикатора. Конечная точка титрования не очень резко выражена, но количество щелочи, затраченное на титрование от точки перехода паранитрофенола до
838
Гл. ХЫХ. Бор
точки перехода фенолфталеина, соответствует приблизительно содержанию борной кислоты в дистилляте. Когда в минерале содержится более 3—4% В2О3, для титрования применяют 0,5 н. раствор едкого натра, чтобы вводить в дистиллят возможно меньшее количество воды и тем самым облегчить его последующее обезвоживание.
По окончании титрования к дистилляту прибавляют двукратное количество щелочи по отношению к израсходованному на титрование от точки перехода паранитрофенола до точки перехода фенолфталеина, затем переносят дистиллят во второй перегонный прибор и отгоняют спирт. Прибавление указанного количества едкого натра предотвращает потерю бора вследствие превращения нестойкого NaBO2 в устойчивый Na3BO3.
Одновременно с этим в первом аппарате отгоняют еще 100 мл жидкости. Если прибавленный метиловый спирт не содержит воды и введено достаточное количество хлорида кальция, то во втором дистилляте будет находиться оставшаяся часть бора *. Поэтому прежде чем удалить приемник, прекращают дистилляцию. Второй дистиллят обрабатывают так же, как первый, и затем оба дистиллята объединяют.
Если на титрование второго дистиллята расходуется менее 1 мл 0,1 и. раствора едкой щелочи, то перегонку можно считать законченной.
Когда раствор, из которого отгоняется спирт, перестает кипеть на водяной бане, его переводят в чашку, ополаскивают колбу 1—2 раза водой и затем нагревают на голом пламени, непрерывно двигая чашку, пока не испарится оставшийся спирт. Объем оставшейся жидкости должен быть небольшим — около 25 мл, если титрование проводилось 0,1 н. щелочью, и много меньше, если концентрация щелочи была 0,5 н. Если объем раствора меньше 25 мл, его соответственно разбавляют.
Раствор переносят обратно в колбу и, непрерывно перемешивая, вводят по каплям из капельной воронки разбавленную (1 : 1) соляную кислоту до исчезновения окраски обоих индикаторов, следя за тем, чтобы не была прибавлена лишняя капля кислоты. Затем помещают в колбу капиллярную трубку и нагревают на водяной бане 1—2 мин. Соединяют колбу с насосом и кипятят под пониженным давлением, пока жидкость не станет почти холодной и не будут появляться лишь отдельные пузырьки. Удалив таким образом двуокись углерода, прекращают действие насоса, охлаждают раствор в струе холодной воды (если требуется) и приступают к конечному титрованию.
Сперва нейтрализуют избыток соляной кислоты, осторожно прибавляя 0,5 н. раствор едкого натра до появления желтой окраски паранитрофенола. Затем доводят до кислой реакции 0,1 н. раствором соляной кислоты, после чего снова нейтрализуют 0,1 н. раствором едкого натра до появления слабо-желтой окраски индикатора, что соответствует строго нейтральной реакции. Одна капля 0,1 н. раствора кислоты должна пол-
1 В точных анализах, особенно в случае содержания больших количеств бора и кремнекислоты, остаток в колбе следует отфильтровать, умеренно промыть водой, прокалить, сплавить, провести через все стадии анализа и в полученный результат ввести поправку на «холостой» опыт с одними реактивами. Количества бора, извлеченные таким образом при анализе 1 г стандартного образца № 92 (Бюро Стандартов США), содержавшего приблизительно 75% SiO2 и 0,7% В2О3, соответствуют 0,05— 0,09% В2О3. При анализе 0,5 г навесок стандартного образца К» 128 (69,51 % SiO2 и 1,52% В2О3) и стандартного образца № 93 (80,60% SiO2 и 12,76% В2О3) найденные в остатке количества бора отвечали соответственно 0,04—0,07% В2О3 и 0,06—0,08% В2О3,
Методы определения
839
ностью уничтожить эту окраску. Прибавляют 1 г маннита \ отмечают уровень титрующего раствора в бюретке и продолжают титровать до появления окраски фенолфталеина. Вводят еще 1 г маннита и, если при этом окраска исчезает, продолжают титровать щелочью до ее появления. Из израсходованного на титрование количества щелочи вычитают то ее количество, которое расходуется на титрование одних реактивов («холостой» опыт). Полученную разность умножают на титр щелочи, выраженный в граммах В2О3, что дает содержание борного ангидрида в растворе. 1 мл точно 0,1 н. раствора соответствует 0,00348 г В2О3.
Следует обратить внимание на изменения окрасок индикаторов в процессе титрования. При прибавлении маннита (или глицерина) к раствору окраска паранитрофенола сразу исчезает. Это объясняется тем, что образующееся соединение реагента с борной кислотой является более сильной кислотой, чем борная кислота. В процессе титрования окраска снова появляется, постепенно усиливается и становится весьма интенсивной к моменту перехода окраски фенолфталеина. Появлению окраски фенолфталеина предшествует появление бледной коричневатойокраски, переходящей от одной капли 0,1 н. раствора щелочи в интенсивную коричнево-красную, которая и характеризует действительную конечную точку титрования 1 2.
По сравнений) с временем, которое затрачивается на определение бора весовым методом, определение объемным методом не является чрезмерно продолжительным. После установки прибора и приготовления реактивов легко можно последовательно выполнить четыре определения в течение 8 ч 3.
Метод Чэпина был очень тщательно проверен 4 применительно к определению бора в стеклах. Он оказался наиболее точным из всех методов, хотя при его применении приходится вводить небольшую (не более 1 мг), но всегда одинаковую поправку на «холостой» опыт. Поправка эта, по-видимому, вызвана частично содержанием бора в реактивах, а частично ошибкой титрования. Неизбежность введения поправки делает метод недостаточно надежным для определения очень малых количеств бора, которые могут содержаться в горных породах. Получаемые результаты, однако, настолько устойчивы, что если на титрование анализируемого раствора расходуется больше щелочи, чем на титрование одних реактивов, то это служит явным доказательством наличия бора в породе.
Найдено 4 также, что если в дистиллируемом растворе находится не свыше 0,5 мл свободной соляной кислоты, присутствие 1% ацетона
1 Если вместо маннита применяют глицерин, то в этой'стадии вводят 40 мл глицерина. Продажный глицерин содержит жирные кислоты, поэтому его надо предварительно обработать следующим образом. К 500 мл глицерина прибавляют 50 мл воды, 5 мл раствора фенолфталеина и затем 0,5 н. раствор едкого натра до появления розовой окраски индикатора. Если раствор при стоянии обесцвечивается, то перед употреблением его снова обрабатывают 0,1 н. раствором щелочи.
2 W. StreckernE.Kannappel [Z. anal. Chem., 61, 378 (1922)], указывают, что при применении а-нафтолфталеина вместо фенолфталеина конечная точка выражена более резко. В этом случае концом титрования считается появление зеленой окраски, что происходит вследствие смешения голубой окраски а-нафтолфталеина и желтой окраски метилового оранжевого или паранитрофенола.
3 В искусственных смесях, содержавших 0,035 г В2О3 и различные другие вещества, Chapin этим методом определил 0,0348 г, 0,0351 г и 0,0352 г В2О3. Воспроизводимость метода характеризуют также данные опытов AllennZies [J. Am. Ceram. Soc., 1, 739 (1918)], которые нашли 26,02%, 26,06% и 26,00% В2О3 в стекле с относительно высоким содержанием бора и 0,64%, 0,67% и 0,69% В2О3 в стекле с относительно низким его содержанием.
4 См. сноску 4 на стр. 831.
840
Гл. XLIX. Бор
в метиловом спирте не мешает определению, так же как и наличие до 10 мг мышьяковистого ангидрида. Большие количества мышьяковистой кислоты лучше окислять перекисью водорода, после того как к раствору прибавлен едкий натр до заметно щелочной реакции. Образующаяся при этом мышьяковая кислота определению не мешает. Было найдено также, что относительно большие количества фторида (0,2 г NaF) влияют на точность определения, однако не настолько заметно, чтобы сделать метод непригодным для рядовых анализов.
Автор метода нашел, что меньшие количества фторида (0,1 г KF) не вызывают затруднений.
Титрование едким натром после экстракции
Следующий метод основан на применении закона Генри (так называемого «закона распределения») и заключается в титровании борной кислоты, экстрагированной эфиром из водного раствора, содержащего, помимо борной кислоты, соляную кислоту и спирт !. Этот метод удобен для рядовых определений бора (в пределах 0,3—16% В2О3) в стекле. Кремний, кальций, барий, магний, алюминий, натрий, литий, железо, цинк, свинец и мышьяк в количествах, обычно встречающихся в стекле, не мешают определению. В присутствии фтора получаются пониженные результаты.
Метод, применяемый для определения борного ангидрида в стекле, сводится к следующему.
Ход определения. Тонко растирают в ступке 0,5000 г анализируемого стекла с 1 г карбоната натрия и переносят смесь в платиновый тигель. Сплавляют при возможно более низкой температуре и не дольше, чем это требуется для полного разложения пробы. По охлаждении ополаскивают крышку, собирая промывную воду в тигель, и обмывают внутренние стенки тигля горячей водой. Разлагают плав нагреванием на паровой бане при перемешивании платиновой проволокой. Концентрируют раствор приблизительно до 5 мл. По охлаждении осторожно нейтрализуют большую часть щелочи серной кислотой (приблизительно 5,8 н.), избегая разбрызгивания. Прибавляют 2 капли паранитрофенола 1 2 и заканчивают нейтрализацию, вводя кислоту по каплям, после чего прибавляют 1 мл серной кислоты (приблизительно 11,6 н.).
Содержимое тигля переносят в градуированный цилиндр емкостью 100 лл, снабженный притертой пробкой, разбавляют до 25 лл, а затем вводят 25 мл абсолютного этилового спирта и 50 мл эфира.
Эфир необходимо проверить следующим образом. Примерно 50 мл эфира смешивают с 5 мл 0,5 и. раствора едкого натра и 5 мл воды, а затем обрабатывают так же, как обрабатывают анализируемый раствор, когда из него удаляют эфир и этиловый спирт. Разбавляют до 35 мл и нейтрализуют 0,5 н. соляной кислотой по паранитрофенолу. Если окраска при нейтрализации не появляется, то это указывает на пригодность эфира; в противном случае порцию эфира встряхивают с асбестом, пропитанным щелочным раствором перманганата (5 лм насыщенного раствора КМпО4 смешивают с 15 мл 33%-ного раствора NaOH), затем медленно сифоиируют очень тонкой струей через столб такого же пропитанного KMnO4+ NaOH асбеста в склянку, также содержащую этот же асбест. Сохраняют в холодном, темном месте.
1 F. W. G 1 a z е, A. N. Finn, J. Research NBS, 16, 421 (1936); 27, 33 (1941).
2 Растворяют 1 г паранитрофенола в 75 мл 95%-ного этилового спрта и разбавляют до 100 мл дистиллированной водой. Согласно Р. A. Webs t ет и А. К. Lyle [J. Am. Ceram. Soc., 23, 235 (1940)], в этой и в последующих операциях паранитрофенол можно заменить одной каплей бромкрезолового пурпурного. При этом нейтрализовать следует до появления синей окраски.
Методы определения
841
Цилиндр периодически встряхивают в течение 20 мин и отмечают конечную температуру жидкости. Дают слоям разделиться, после чего отмечают их объемы. Отбирают пипеткой 50 мл эфирного слоя в коническую колбу емкостью 250 мл, вводят 2 капли паранитрофенола и титруют до перехода окраски индикатора 0,5 н. раствором NaOH, который не содержит карбоната и бора, и сохраняется в сосуде из свободного от бора материала. Отмечают объем раствора едкого натра в бюретке, прибавляют 1 мл раствора фенолфталеина 1 и продолжают титровать до появления окраски индикатора. Возможно быстрее удаляют эфир и этиловый спирт 2 кипячением, сначала на водяной или паровой бане, а под конец и на голом пламени, чтобы удалить последние следы этих веществ.
Раствор разбавляют затем водой до 35—45 мл, охлаждают и точно подкисляют 0,5 н. соляной кислотой. Нагревают на паровой бане 1 мин и затем кипятят при пониженном давлении для удаления двуокиси углерода. По охлаждении титруют 0,1 н. раствором гидроокиси бария по паранитрофенолу. Отмечают объем титрующего раствора в бюретке, прибавляют маннит и титруют борную кислоту.
Один миллилитр 0,1 н. раствора гидроокиси бария соответствует 0,00348 г В2О3. Вычитают количество щелочи, которое расходуется на титрование в «холостом» опыте, проводимом со стеклом, не содержащим бора.
Процентное содержание В2О3 (ж) в 0,5-граммовой навеске вычисляют по следующей формуле:
Х = 4 * * *(B2O3)50Et2O [vEt2o +
где (В2О3) ЕЛ 0 — количество ВаО3 в граммах, найденное в 50 мл эфирного слоя, взятых для анализа;
VEt 0 и V — соответственно объемы эфирного и водного слоев.
Значение к находят из равенства 3:
к = 0,417 — 0,00232 t: где t — температура.
Взвешивание в виде бората кальция
Ниже описан метод, усовершенствованный Гучем и Джонсом. Последовательные этапы развития этого метода приведены в сноске 4.
1 Растворяют 1 г фенолфталеина в 100 мл этилового спирта (95%-ного) и разбавляют до 200 мл дистиллированной водой.
2 В случае продолжительного нагревания на водяной или паровой бане раствор становится кислым и приобретает окраску, которая маскирует переход паранитрофонола при конечном титровании.
3 к — отношение концентраций борной кислоты в эфирном и водном слоях (к = = ^Et2o/^H2o)' изменяется в зависимости от температуры и количества плавня. Величина эта практически не зависит от присутствующего количества В2О3.
4 Первые количественные методы, основанные на омылении борнометилового эфира и взвешивании выделенной борной кислоты, поглощенной раствором щелочи, были одновременно предложены F. A. G о о с h [Am. Chem. J., 9, 23 (1887)] и T. R fl-sen Ы a d t [Z. anal. Chem., 26, 21 (1887)]. Для связывания бора первый пользовался
окисью кальция, а второй — окисью магния. Позднее F. A. G о осп и L. С. J ones
[Am. J. Sci., [4] 7, 34 (1899)] усовершенствавали метод. Весьма рациональное видо-
изменение метода, заключающееся в поглощении эфира аммиаком перед обработкой окисью кальция, см. S. L. Р е n f i е 1 d, Е. S. S р е г г у, Am. J. Sci., [3], 34, 222 (1887).
См. также статьи Н. Moissan, С. г., 116, 1087 (1893); Bull. Soc. chim. [3], 11, 955 (1894), который частично видоизменил дистилляционный аппарат Гуча.
842
Гл. XLIX. Бор
Прибор, показанный на рис. 31, состоит из реторты, парафиновой бани, холодильника и приемника. Реторта сделана из пипетки емкостью 200 мл с внутренним диаметром не менее 7 мм. Один конец трубки реторты согнут под прямым углом, а другой вытянут наподобие гусиной шеи. К первому концу припаяна стеклянная воронка с краном. Другой, дугообразно изогнутый конец трубки проходит через резиновую пробку в верх
нюю часть холодильника, соединенного в свою очередь с небольшой ко-
нической колбой посредством маленькой трубки с воронкообразным рас-
Рис. 31.(Прибор Гуча для отгонки бора.
ширением вверху. В резиновой пробке, в которую вставлена эта трубка, сделана выемка для свободной циркуляции воздуха.
В приемную колбу вводят окись кальция для улавливания борного ангидрида. Прежде чем приступить к определению, около 1 г чистой окиси кальция прокаливают в тигле, предназначенном для конечного выпаривания дистиллята, тщательно взвешивают и переносят в коническую колбу, служащую приемником, где гасят небольшим количеством воды. Тигель с приставшими к стенкам частицами окиси кальция помещают в эксикатор и сохраняют.
Для определения бора в минерале последний сплавляют с карбонатом, как описано в разделе «Титрование едким натром после экстракции» (стр. 840), плав выщелачивают водой и водную вытяжку выпаривают до небольшого объема. Слабо подкисляют раствор соляной кислотой, затем точно подщелачивают его едким натром и,
наконец, подкисляют уксусной кислотой по лакмусу. Полученный раствор вводят через воронку в реторту прибора, заполняя реторту
не более чем наполовину. Закрывают кран воронки и пускают воду
в холодильник. Нагревают парафиновую баню до 130—140° С и осторожно опускают в нее реторту так, чтобы вначале в парафин погрузилась только нижняя ее часть. При этом следят за тем, чтобы не было слишком бурного перехода жидкости в трубку, изогнутую дугой. Нако
нец, полностью погружают реторту в парафин и перегоняют жидкость.
По окончании дистилляции поднимают реторту, охлаждают и смачивают сухой остаток 10 мл метилового спирта (свободного от ацетона и предпочтительно абсолютного), затем снова осторожно погружают реторту в баню и отгоняют спирт. Эту обработку повторяют три раза. После этого смачивают остаток 2—3 мл воды и прибавляют 1—2 капли уксусной кис-
лоты до исчезновения щелочной реакции на лакмус, которая вызвана гидролитическим разложением ацетата. После трехкратной обработки метиловым спиртом поднимают реторту, для того чтобы парафин не раздавил ее при затвердевании, удаляют коническую колбу, закрывают ее пробкой, энергично взбалтывают и оставляют на 1—2 ч.
Возможно полнее переносят жидкость в платиновую чашку емкостью 200 мл и дают спирту медленно испариться при температуре ниже его точки
Методы, определения
843
кипения; это делают, чтобы избежать потери раствора от разбрызгивания. По удалении спирта осторожно нагревают чашку на голом пламени и, наконец, слабо прокаливают для разрушения всегда образующегося ацетата кальция. Охлаждают и переносят остаток небольшим количеством воды в тигель, в котором взвешивалась окись кальция. Растворяют в азотной кислоте приставшие к конической колбе и платиновой чашке частицы вещества и полученный раствор переносят в тот же тигель. Выпаривают содержимое тигля на водяной бане досуха и, накрыв крышкой, прокаливают сперва осторожно, а затем более сильно до постоянной массы. Увеличение в массе соответствует содержанию 1 В2О3.
Другие методы
Видоизмененный 2 метод Уэрри 3 вполне пригоден для массового контроля, но не приемлем для точных анализов, так как борная кислота при его применении частично задерживается в остатках. Применительно к анализу стекла ход определения этим методом состоит в следующем.
Сплавляют 0,5 г стекла с 3 г карбоната натрия. По расплавлении массы продолжают нагревать еще 1—2 мин. Разлагают плав 20—30 мл горячей воды, остаток отфильтровывают и промывают. Фильтрат и промывные воды переносят в круглодонную колбу емкостью 250 мл, прибавляют около 7 мл соляной кислоты, нагревают почти до кипения и вводят сухой осажденный карбонат кальция в небольшом избытке. Соединяют колбу с обратным холодильником, сильно кипятят 10 мин, фильтруют при отсасывании через маленькую воронку Бюхнера и промывают осадок несколько раз горячей водой. Объем фильтрата и промывных вод должен быть менее 100 мл. Переводят фильтрат обратно в колбу, прибавляют щепотку карбоната кальция и нагревают до кипения. После этого соединяют с фильтрующим насосом через предохранительную склянку, удаляют пламя и продолжают отсасывать почти до прекращения кипения. Охлаждают до комнатной температуры и, если остаток имеет красную окраску, фильтруют. Вводят в фильтрат 4—5 капель фенолфталеина и медленно прибавляют 0,1 н. раствор едкого натра до появления розового окрашивания. Затем вводят 1 г маннита, взбалтывают и продолжают титровать до появления розовой окраски. Прибавляют еще 1 г маннита и, если требуется, еще щелочи до появления устойчивой розовой окраски.
Борную кислоту можно так же быстро и точно определять потенциометрическим титрованием, предварительно отделив вещества, мешающие титрованию щелочью 4. Описаны также интересные методы, в которых подкисленные, свободные от двуокиси углерода растворы бората титруют до определенного pH, а затем, обработав маннитом, титруют до того же значения pH. Титрование можно осуществлять потенциометрически 5
1 Точность, на которую можно рассчитывать при применении этого метода, иллюстрируется результатами, полученными Gooch и Adams:
Взято В2О3, г....... 0,2065 0,2067 0,2077 0,1791
Получено В2О3, г . . . 0,2062 0,2070 0,2075 0,1795
1 Е. С. S u 11 i v a n, W. С. Т а у 1 о г, J. Ind. Eng. Chem., 6, 897 (1914).
3 Е. Т. Wherry, W. Н. Chapin, J. Am. Chem. Soc., 30, 1687 (1908),
4 J. H. Hildebrand, J. Am. Chem. Soc., 35, 861 (1913).
5 L. V. Wilco x, Ind. Eng. Chem., Anal. Ed., 4, 38 (1932); 12, 341 (1940).
844
Гл. X.LIX. Бор
и колориметрически *. В первом случае титруют до величины pH порядка 7,3, а во втором случае в качестве индикатора применяют феноловый красный и титруют до pH около 7,6.
Такие методы основаны на значительной разнице в силе самой борной кислоты и кислоты, образуемой борной кислотой с маннитом. Борная кислота является настолько слабой кислотой, что соли ее почти полностью гидролизуются при значении pH = 7,3 тогда как соли маннитоборной кислоты вполне устойчивы. Ошибка, вводимая за счет частичной нейтрализации борной кислоты при pH = 7,3 (около 12%), может быть устранена установкой титра едкой щелочи по таким же количествам борной кислоты, проведенным через все стадии анализа. Необходимо, естественно, чтобы раствор не был заметно буферирован и чтобы в нем не содержались основания или другие кислоты, реагирующие с маннитом. Этим условиям отвечает большинство природных вод и вытяжек из почв. Поведение фторидов и значительных количеств кремнекислоты или фосфатов не изучалось.
Для открытия и колориметрического определения малых количеств бора в почвах и растениях был предложен метод 1 2, основанный на реакции бора с хинализарином в растворе, содержащем 93% (по массе) серной кислоты, в результате которой розовая окраска красителя переходит в синюю. В соответствующих условиях можно открыть такие малые количества бора, как 0,0001 мг, Фториды, германий, нитраты, гексацианоферраты (III) и другие окислители мешают реакции 3.
В анализе растительных продуктов , содержащих малые количества бора, при дистилляции с метиловым спиртом для обезвоживания применена ортофосфорная кислота, а бор в дистилляте определяют по окраске с куркумовым экстрактом 4. В применяемых реактивах допустимо содержание лишь ничтожных количеств бора, а части аппаратуры, которые соприкасаются с растворами или с дистиллятом, должна быть изготовлены из кварца или из свободного от бора стекла. Определение выполняется следующим образом.
а) Переносят 10 мл разбавленного дистиллята в маленькую (7,5 см) фарфоровую чашку и прибавляют 5 мл 0,1 н. суспензии гидроокиси кальция 5. Отбираемая аликвотная часть дистиллята может быть уменьшена или увеличена, в зависимости от содержания бора. Жидкость в чашке должна иметь щелочную реакцию, в противном случае прибавляют достаточное для этого количество суспензии гидроокиси кальция. Смесь выпа
1 F. J. F о о t е, Ind. Eng. Chem., Anal. Ed., 4, 39 (1932).
2 К. С. В e r g e r, E. T r u о g, Ind. Eng. Chem., Anal. Ed., 11, 540 (1939). *Cm. также G. A. Rudolph, L. C. Flickinger, Steel, 112, 114 (1943); S. W e i n-b e r g, K. L. Proctor, О. M i 1 n e r, Ind. Eng. Chem., Anal. Ed., 17, 419 (1945); Б. А. Г e н e p о з о в, Зав. лаб., 12, 25 (1946); 3. С. Мухина, А. Ф. Алешин, там же, 11, 23 (1945). *
3 Ализарин S дает окраску, которую легче сравнивать с окраской стандартного раствора, чем окраску от хинализарина [D. Dickinson, Analyst, 68, 106 (1943)].
4 К. L. R о b i n s о n, Analyst, 64, 324 (1939); см. также J. A. N a f t e 1, Ind. Eng. Chem., Anal. Ed., 11, 407 (1939). Дистилляционно-объемный метод для определения бора в стали и чугуне описан J. L. Н a g и е и Н. A. Bright, [ J. Research NBS, 21, 125 (1938)].
5 Окись кальция (высокой чистоты), не содержащую бора, измельчают в агатовой ступке и 2,8 г ее вводят в 1 л дистиллированной воды. Окись кальция удобно приготовлять из металлического кальция, свободного от бора или с незначительным его содержанием. Суспензию сохраняют в сосуде, изготовленном из материала, свободного от бора.
Методы, определения.
845
ривают досухана водяной бане, затем охлаждают до комнатнойтемпаратуры и прибавляют 1 мл свежеприготовленного раствора щавелевой кислоты \ Вращая чашку, смачивают весь остаток раствором щавелевой кислоты. Затем вводят 2 мл куркумового экстракта 1 2 и, вращая чашку, тщательно перемешивают ее содержимое. Помещают чашку в водяную баню или термостат и нагревают при 55 ± 3° С. Когда остаток станет сухим, продолжают высушивать еще 30 мин, но не дольше, так как продолжительность нагревания имеет существенное значение. По охлаждении прибавляют из промывалки приблизительно 5 мл этилового спирта (95%-ного). Перемешивают массу палочкой с резиновым наконечником, затем переносят в пробирку емкостью 15 мл, ополаскивают чашку 95%-ным этиловым спиртом и жидкость сливают в ту же пробирку, после чего центрифугируют в течение примерно 10 мин при 1700 об/мин. Прозрачную жидкость сливают в мерную колбу емкостью 25 мл, разбавляют до метки этиловым спиртом (95%-ным) и перемешивают.
б) Светопоглощение анализируемого раствора удобно измерять в фотоколориметре с зеленым (525 ммк) светофильтром или в спектрофотометре при 540 ммк. В качестве нулевого раствора помещают раствор, полученный в «холостом» опыте, в котором 5 мл суспензии Са (ОН)2 проводят через все стадии анализа, описанные в п. а. Измерив светопоглощение анализируемого раствора, находят содержание бора по калибровочной кривой, построенной на основании результатов измерений светопоглощения различных количеств стандартного раствора 3 борной кислоты (0,5—2 мкг), проведенных через все стадии анализа, указанные в п. а.
В найденное содержание бора вводят поправку, соответствующую све-топоглощению раствора реактивов, проведенного через все стадии анализа.
1 Раствор щавелевой кислоты приготовляют, смешивая 20 мл НС1 (пл. 1,18 г/см3) и 80 мл 20%-ного раствора щавелевой кислоты. Если щавелевая кислота кристаллизуется, нагревают раствор до растворения кристаллов. Ежедневно перед употреблением приготовляют свежий раствор.
2 Для приготовления куркумового экстракта помещают 1 г порошка куркумы в склянку из стекла, не содержащего бора, прибавляют 10 мл 95%-ного этилового спирта и перемешивают 2—3 ч кварцевой или стеклянной (но из стекла, не содержащего бора) палочкой, после чего фильтруют. Фильтрат сохраняют в закрытом пробкой сосуде из стекла, также не содержащего бора.
3 В 1 мл стандартного раствора борной кислоты содержится 2 мкг бора. Помещают 5,72 мг борной кислоты в мерную колбу емкостью 500 мл, разбавляют до метки предварительно прокипяченной водой и перемешивают. Раствор сохраняют в сосуде из стекла, свободного от бора.
Глава L
УГЛЕРОД И ВОДОРОД
Углерод в минералах находится в кристаллической форме в виде графита и алмаза, в аморфном состоянии — в каменном угле; в виде углеводородов — в природных газах, нефти и асфальте. Двуокись углерода является обычной составной частью атмосферного воздуха. Угольная кислота и карбонаты присутствуют в большинстве природных вод; огромные горные массивы состоят из карбонатов кальция, магния и железа. Углерод содержится в немногих силикатах, из которых только канкринит H6Na6Ca (NaCO3)2Als(SiO4)g имеет петрографическое значение.
Водород встречается в природе почти повсюду. В свободном состоянии водород содержится в очень малых количествах в атмосфере, в некоторых гранитах и других породах В
В большей части минералов, а следовательно, практически во всех горных породах, водород находится либо в виде неконституционного водорода, как, например, в гигроскопической влаге, либо в виде конституционного водорода, в радикале гидроксила, кристаллизационной воде и т. п. (см. «Вода», часть III, стр. 896). Все органические вещества содержат водород, и в связи с этим он является существенной составной частью таких веществ, как природный газ, нефть, асфальт и уголь.
ОБЩИЕ ЗАМЕЧАНИЯ
Следующие замечания относятся главным образом к определению углерода и водорода в горных породах. Относительно определения этих элементов в специальных случаях, как, например,в сталях, следует обратиться к соответствующим руководствам 1 2.
Карбонаты и другие вещества, содержащие углерод, в обычном ходе анализа не вызывают затруднений, но в некоторых случаях, как, например, при осаждении железа аммиаком из раствора известняка, двуокись углерода должна быть предварительно удалена. Иногда, если присутст
1 A. Gautier, С. г., 131, 647 1276 (1900); 132. 58 (1901); Ann. chim. phys., [7], 22, 5 (1901); F. W. С 1 а г k е, U. S. Geol. Survey Bull., 770, 17, 48 (1924).
2 Например, Г. M ей ер, Анализ и определение строения органических веществ, ОНТИ, 1937; Houben, Weyl, Methoden der organischen Chemie, G. Thieme Verlag Band II, Stuttgart, 1953; L. G a t t e r m a n n, W. B. S c h о b e r, Practical^Methods of Organic Chemistry, MacMillan Co., New York; Г. Лендель, Дж. Г о'ф м а н, Г. Брайт. Анализ черных металлов. Госхимтехиздат, 1934; ASTM Methods of Chemical Analysis of Metals, 1950; ASTM Spec. D271—48, Loboratory Sampling and Analysis of Coal and Coke, ASTM Standards, pt. 5, 1949, p. 583. *A. M. Дымов, Технический анализ руд и металлов, Металлургиздат, 1949.*
Разложение веществ, содержащих углерод
847
вуют углеродсодержащие вещества, необходимо проводить некоторые предварительные операции, например приходится прокаливать пробу перед сплавлением ее с карбонатом натрия или применять окислительный плавень при сухой обработке пробы, или же проводить выпаривание с азотной и серной кислотами или окисление перманганатом в случае мокрой обработки пробы. Углеродсодержащие вещества вызывают затруднения также и при анализах некоторых материалов, содержащих элементы в их низших степенях валентности, например при определении железа (II) в карбонатных породах (стр. 986).
Свободный водород встречается в породах в таких малых количествах, что он не может оказать заметного влияния на результаты определения воды. В процессе подготовки пробы к анализу существенное значение имеет поведение водорода, входящего в состав минерала в виде кристаллизационной воды или связанного в форме гидроксила (см. часть III, стр. 896). Водород, входящий в состав органических веществ, участвует в том мешающем влиянии, которое эти последние оказывают на определение элементов, находящихся в низших степенях валентности, в частности железа (II).
РАЗЛОЖЕНИЕ ВЕЩЕСТВ, СОДЕРЖАЩИХ УГЛЕРОД
Углерод, присутствующий в пробе в виде карбонатов, обычно определяют разложением пробы разбавленной соляной кислотой, как сказано в разделе «Определение углерода, присутствующего в виде карбонатов» (стр. 848), а общее содержание углерода определяют прямым сжиганием (см. раздел «Определение общего содержания углерода и водорода прямым сжиганием» (стр. 850).
В особых случаях может оказаться целесообразным применять другие методы разложения, как, например, растворять сталь в растворе хлоридов меди и калия для предварительного отделения углерода или при определении углерода в алюминии проводить «мокрое сжигание» обработкой серной и хромовой кислотами, как описано в разделе «Определение общего содержания углерода ,,мокрым сжиганием11 (стр. 856). Для определения в органических веществах таких компонентов, как галогены, сера, фосфор и азот, анализируемую пробу можно окислить дымящей азотной кислотой при высоких температурах и давлениях в запаянной стеклянной трубке
При анализе неорганических веществ водород, находящийся в них в виде неконституционной воды или входящий в состав летучих органических веществ, обычно удаляют при температуре между 100 и 110° С с тем, чтобы определить содержание «гигроскопической влаги» или чтобы, приступая к анализу, иметь продукт определенного состава 1 2. Тот водо
1 L. С а г i u s, J. Н о и Ь е n. Die Methoden der organiscbc Chemie, Bd. 1, Georg Thieme, Leipzig, 1925, S. 59—63. Об изменениях, введенных в метод с целью уменьшения опасности взрыва, см. С. L. G о г d о n, J. Research NBS, 30, 107 (1943); C.L. G о r-d on, L. A. Greenberg, Ind. Eng. Chem. Anal. Ed., 16, 308 (1944).
2 При анализе материалов, содержащих конституционный водород в виде гидроксила, достижение постоянного состава при 110° С может оказаться затруднительным. Например, при стандартизации образца боксита пришлось материал сушить в течение 2 ч при 140° С. чтобы получить соответствующую пробу для анализа, а пиролюзит надо было сушить при 120° С.
848
Гл. L. Углерод и водород
род, который остается в виде конституционной воды или нелетучих органических веществ, обычно удаляют нагреванием при более высокой температуре и включают таким образом в величину «потери при прокаливании».
МЕТОДЫ ОТДЕЛЕНИЯ
Отделение органических веществ обычно не является необходимым и к нему прибегают лишь в особых случаях, например, при анализе сланцев, как описано в части III (стр. 1016).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Определение углерода, присутствующего в виде карбонатов
' Углерод, находящийся в пробе в виде карбонатов, лучше всего определять обработкой пробы кислотой и поглощением выделяющейся при этом двуокиси углерода натронным асбестом.
Рис. 32. Схема установки для поглощения двуокиси углерода:
1 — патронный асбест; 2 — Mg (СЮ,)2 .ЧН2О; 3 — пемза, пропитанная раствором сульфата меди; 4 — концентрированная серная кислота.
Для растворения пробы лучше всего применять соляную кислоту, чем другие кислоты, несмотря на ее летучесть, так как при этом образуются растворимые хлориды. В особых случаях может оказаться целесообразным применение разбавленной хлорной кислоты. Выделяющуюся двуокись углерода надо освобождать от других газообразных веществ, поглощаемых натронным асбестом. Ее следует количественно переводить в поглотитель током воздуха, свободного от двуокиси углерода, причем влажность непоглощенного газа при прохождении его через прибор не должна изменяться. В большинстве случаев эти требования достаточно хорошо обеспечивает установка, изображенная на рис. 32.
Все трубки этой установки компактно располагают вдоль края доски 15 X 22 см, поддерживаемой на треножнике на высоте 12 см от поверхности
Методы определения
849
стола. Установка состоит из небольшого холодильника для конденсации
большей части воды и соляной кислоты, за которым следуют: промывалка 4,
содержащая концентрированную серную кислоту для обезвоживания газа;
трубка 3 с безводным сульфатом меди для поглощения соляной кислоты,
хлора и сероводорода; трубка 2, содержащая Mg(C104)2-3H20 для окончательного высушивания газа; склянка 1 для поглощения СО,, содержащая натронный асбест и Mg(C104)2-ЗН2О и, наконец, вторая взвешенная склянка для поглощения СО2, которая одновременно является предохра-
нительной, поглощающей двуокись углерода, не поглощенную в первой склянке, и контрольной, указывающей на утрату поглотительной способности натронного асбеста. Необходимо отметить, что конец делительной воронки должен быть загнут кверху для предотвращения утечки двуокись углерода через трубку воронки. Как показано на рисунке, в установке сделаны ртутные затворы. При применении таких затворов для протягивания газа через установку лучше пользоваться давлением, чем отсасыванием. Предлагаемые поглотители могут быть заменены другими. Так, например, вместо сульфата меди можно пользоваться арсенитом серебра (см. «Реактивы», стр. 70), а вместо трехводного перхлората магния — безводным перхлоратом магния.
Для жидких поглотителей рекомендуется прибор, изображенный на рис. 33. Из такого прибора можно легко удалять истощенные поглотители и заменять их свежими К
Ход определения.. Навеску пробы от 1 до 5 г помещают в колбу прибора (см. рис. 32) и заливают водой. Закрывают колбу пробкой, в которую вставлены делительная воронка и холодильник. Последний соединяют с деталями 4, 3 и 2 и пропускают через систему воздух, освобожденный от СО2, до полного удаления двуокиси углерода из прибора. Закрывают кран делительной воронки и
Рис. 33. Трубка для поглощения окислов серы.
включают в установку
предварительно взвешенные поглотительные сосуды. Наполняют делительную воронку до половины разбавленной (1 : 1) соляной кислотой, присоединяют трубку, через которую проходит воздух, и удостоверяются в свободном прохождении газа через установку.
Открывают кран делительной воронки и вводят в колбу кислоту, медленно — при содержании больших количеств двуокиси углерода, в противном случае — быстро. Когда выделение пузырьков газа ослабевает.
а в случае содержания малых количеств двуокиси углерода тотчас после добавления кислоты, пускают воду в холодильник и слабо нагревают колбу так, чтобы жидкость кипела непрерывно, но спокойно. По окончании вы-
деления двуокиси углерода прекращают нагревание и усиливают ток
1 Когда этот прибор применяется для поглощения окислов серы при определении углерода в сталях прямым сжиганием в токе кислорода, его левую часть неплотно заполняют асбестом, а в правую часть наливают серную кислоту, насыщенную хромовым ангидридом.
54 Заказ 522.
850
Гл. L. Углерод и водород
воздуха для полного удаления этого газа из раствора. Отделяют взвешенные сосуды для поглощения СО2, закрывают вводную и отводную трубки и помещают эти сосуды в шкафчик весов. По охлаждении открывают на мгновение краны поглотительных сосудов и взвешивают каждый из них, поставив на другую чашку весов такой же сосуд в качестве противовеса.
Определение общего содержания углерода и водорода прямым сжиганием
При определении общего содержания углерода и водорода прямым сжиганием необходимо принять меры для того, чтобы сжигание было полным, чтобы двуокись углерода не связывалась и не удерживалась золой, чтобы не образовалась окись углерода, чтобы вся двуокись углерода и вода поглотились абсорбентами, чтобы были удалены галогены, окислы серы и азота, а также другие соединения, помимо СО2, которые могли бы быть поглощены применяемыми поглотителями, чтобы влажность воздуха при входе и выходе из поглотительной системы была одинакова и, наконец, чтобы воздух, вводимый в систему, был свободен от углеродсодержащих веществ, двуокись углерода, водорода и воды.
Определение в горных породах и минералах. Сжигание углерода и водорода в горных породах и минералах облегчается, а сера и галогены связываются, если анализируемую пробу сплавить с гранулированным хроматом свинца.
Ход определения. Навеску пробы от 1 до 5 г тщательно перемешивают со смесью 10 частей прокаленного и измельченного в порошок хромата свинца и" 1 части хромата калия. Смесь переносят в фарфоровую лодочку и помещают в трубку для сжигания, часть которой неплотно заполняют гранулированным хроматом свинца, как указано на стр. 855. Отводной конец этой трубки соединяют с трубкой, содержащей Mg(C104)-ЗН2О для высушивания газа. Пропускают через систему очищенный воздух до полного удаления двуокиси углерода и затем присоединяют взвешенный поглотитель двуокиси углерода и второй предохранительный поглотитель, как указано в разделе «Определение углерода, присутствующего в виде карбонатов» (стр. 848).
Пропуская слабый ток воздуха, нагревают хромат свинца в конце трубки до 300—400° С, а затем постепенно нагревают хромат, находящийся вблизи лодочки, до более высокой температуры, но так, чтобы он не расплавился. Наконец, сильно нагревают ту часть трубки,в которой находится лодочка. Когда можно предполагать, что сожжение закончено, прекращают нагревание и пропускают ток воздуха до полного удаления двуокиси углерода из системы. Закрывают поглотительные приборы, снимают их и взвешивают, как указано на стр. 849.
В случае присутствия в пробе азотсодержащих соединений, помещают в отводном конце трубки восстановленную медную спираль, как указано на стр. 855. При этом вначале нагревают, не пропуская тока воздуха, до тех пор, пока сжигание не будет практически закончено, спирали дают несколько охладиться, а затем пропускают ток воздуха и сжигание заканчивают, как указано выше Ч
1 О трудностях, связанных с определением углерода в горных породах и минералах, а также описание мокрого метода сжигания, основанного на применении фосфорной и хромовой кислот, см. В. Е. Dixon [Analyst, 59, 739 (1934)] и С. J. S с h о е 1-1 enberger [J. Ind. Eng. Chem., 8, 1126 (1916)].
Методы определения
851
Определение в органических соединениях. Методы определения общего углерода (и водорода) в органических соединениях прямым сжиганием видоизменяются в зависимости от состава анализируемого материала и его летучести. В случае анализа веществ, содержащих галогены, серу или азот, в трубке для сжигания должны находиться специальные реагенты. Летучие вещества следует взвешивать и сжигать, соблюдая особые предосторожности.
В простейшем случае, когда анализируемое вещество представляет собой нелетучее соединение, содержащее углерод, водород и кислород, ход определения состоит в следующем.
Предварительные операции. Для печи 1 длиной 75 см требуется тугоплавкая трубка для сжигания длиной 90—95 см, внутренним диаметром 10 —15 мм и толщиной стенок 2 2 мм. Приготовляют три плотных свертка из медной сетки, свободно входящих в трубку для сжигания. Два свертка должны иметь длину 1—2 см, а третий—10 —15 см. Свертки должны быть снабжены ушками, позволяющими легко вынимать их из трубки. Вдвигают один из коротких медных свертков на расстояние 10 см от одного из концов трубки (называемого в дальнейшем отводным концом), затем помещают крупную окись меди, приготовленную из медной проволоки или сетки, в таком количестве, чтобы образовался слой в 45 см. Окись меди удерживают на месте вторым коротким медным свертком3. Вводят трубку в печь так, чтобы отводной конец ее находился вне печи на расстоянии 10 см, и закрывают трубку пробкой, сквозь которую проходит стеклянная трубка с таким же внешним диаметром, как у бокового отростка поглотительной трубки, служащей для поглощения воды. Внутри трубки для сжигания стеклянная трубка не должна выходить из пробки, а с внешней стороны она должна выступать 4 на 5 см. Входное отверстие трубки для сжигания закрывают пробкой, скозь которую проходит трубка с одноходовым стеклянным краном. Пробки защищают асбестовыми пластинками с круглыми отверстиями в центре, надеваемыми на трубку у концов печи. Для предохранения от перегрева во время сжигания пробки охлаждают полосками мокрой ткани, которые обертывают вокруг концов трубки. Чтобы ткань не высыхала, концы ее погружают в воду, находящуюся непосредственно под трубкой.
Когда будет налажена трубка для ожигания, подготовляют трубки для поглощения влаги и двуокиси углерода, для очистки кислорода,
1 Очень удобна электрическая печь, состоящая из трех соединяющихся друг с другом частей. Такая конструкция дает возможность нагревать любую часть трубки, что облегчает успешное сжигание.
2 О портативной микро- и полумикроустановке для определения углерода и водорода, в которой 2,5—70 мг образца можно сжечь в продолжение 40—55 мин, см. S. Natelson, S. S. Brodie, Е. В. Conner, Ind. End. Chem., Anal. Ed.. 10, 276, 609 (1938). *См. также M. О. Коршун, H. Э. Гельман, Новые методы элементарного микроанализа, Госхимиздат, 1949; Дж. Н и д е р л ь, В. Н и дер ль, Микрометоды количественного органического анализа, Госхимиздат, 1949.*
3 Окись меди должна быть свободна от хлоридов меди, щелочных и щелочноземельных металлов.
4 Целесообразно оттянуть отводной конец трубки, чтобы можно было непосредственно присоединить к нему поглотительную трубку, если только аналитик достаточно опытен, чтобы не повредить при этом трубку и не сделать стенка ее у оттянутого конца слишком тонкими. О применении металлических трубок для сжигания — медных для определения углерода и водорода и никелевых для определения азота — см. S. A v ег у, D. Н а у m a n, Ind. Eng. Chem., Anal. Ed., 2, 336 (1930).
54*
852
Гл. L. Углерод и водород
а также поглотительные приборы. Для поглощения влаги можно пользоваться трехводным перхлоратом магния Mg(Cl4)2-3H2O, фосфорным ангидридом или серной кислотой (стр. 71). Эти вещества можно поместить в любую легкую стеклянную трубку, в которой обеспечивается хороший контакт между высушивающим веществом и газами. Трубка должна иметь приспособление, позволяющее легко изолировать ее содержимое от воздуха при удалении трубки из поглотительной установки. Для экономии поглотителя целесообразно, чтобы трубка имела сбоку, вблизи печи, соответствующее шарообразное расширение, в котором могла бы конденсироваться большая часть воды.
Лучшим поглотителем для двуокиси углерода является натронный асбест (стр. 69), который можно применять вместе с Mg(C104)2-3H20 или с форсфорным ангидридом в приборе, подобном тому, который изображен на рис. 32 (стр. 848) или в специальных приборах, например в видоизмененном приборе Флеминга. Прибор для очистки кислорода должен содержать те же поглотители, какие поставлены в поглотительной цепи, и они должны быть расположены в том же порядке.
С целью максимальной экономии реактивов можно перед основными поглотителями помещать менее энергичные и более дешевые реактивы, например концентрированный раствор едкого кали (стр. 68) перед натронным асбестом и хлорид кальция перед Mg(C104)2-ЗН3О. Если для сжигания применяется кислород или если применяемый воздух может содержать углеводороды (когда воздух подается, например, из компрессора), прежде чем газ ввести в очистительные трубки, его надо пропустить через трубку, содержащую окись меди, нагретую приблизительно до 700° С, в небольшой печи.
Присоединение предохранительной трубки после взвешенных поглотительных приборов не обязательно, кроме тех случаев, когда определение должно быть исключительно точным или когда сжигание протекает медленно и длится продолжительное время. Для предохранения системы применяют U-образные трубки со стеклянными кранами, содержащие Mg(C104)3-ЗН2О в той части, через которую поступает газ, и натронный асбест в другой ее части.
Перед началом определения в трубку для сжигания вводят длинный сверток медной сетки до того места, где находится окись меди, соединяют вводный конец трубки с очистителями, пропускают слабый ток воздуха и постепенно нагревают до темно-красного каления. Удаляют воду, собравшуюся в отводном конце трубки, касаясь трубки небольшим синим пламенем. Прекращают нагревание трубки, за исключением той ее части, которая прилегает к отводному концу, где находится окись меди, причем передняя часть трубки должна оставаться холодной. В то время когда трубка для сжигания нагревается и охлаждается, взвешивают приборы для поглощения воды и двуокиси углерода. Эти приборы взвешивают, естественно, после наполнения их газом, пропускаемым в процессе сжигания, и охлаждения до температуры весовой комнаты. В качестве тары пользуются такими же приборами, которые помещают на другую чашку весов.
При анализе твердых и нелетучих материалов берут навеску 0,15 — 0,3 г в фарфоровой, кварцевой или платиновой лодочке, предварительно сильно прокаленной, охлажденной в эксикаторе и взвешенной. Когда имеется основание предполагать, что анализируемый материал сжигается с трудом, смешивают его с измельченной до 100—200 меш (d = 0,15 —
Методы определения
853
0,07 мм) окисью меди, предварительно прокаленной и охлажденной в эксикаторе
Сжигание. По охлаждении вводного конца трубки вынимают из нее длинный медный сверток с помощью толстой проволоки с изогнутым в виде крючка концом, приняв меры к тому, чтобы не загрязнить при этом сверток. Вдвигают лодочку с анализируемой пробой настолько далеко, насколько позволяет слой окиси меди, и затем помещают медный сверток на расстоянии 1 см от лодочки (рис. 34). Тотчас же соединяют трубку с очистительной системой, с взвешенными поглотительными приборами и с предохранительной трубкой.
Все соединения должны быть сделаны встык с помощью туго охватывающих толстостенных резиновых трубочек без шва 1 2.
Через установку пропускают слабый ток воздуха (1—2 пузырька в секунду) и усиливают нагрев отводного конца трубки так, чтобы темпе-
10-15см* Медная сетка
Медная сетка
медная сетка
— 6-5 см Опись меди
Рис. 34. Трубка для сжигания соединений, содержащих углерод, водород и кислород.
ратура окиси меди поднялась до 600° С, а та часть трубки, в которой находится лодочка, осталась совершенно холодной. Нагревание затем постепенно распространяют по направлению к лодочке и одновременно начинают нагревать часть трубки у вводного конца.
Эта стадия анализа является наиболее трудной. Пока окись меди в отводном конце не нагревается до красного каления, не должно происходить ни улетучивания, ни сгорания.
Сжигание все время должно происходить не слишком быстро. Часть трубки, в которой находится лодочка, постепенно нагревают до 600— 800° С и, если сжигание протекает слишком медленно и неполно, пускают струю кислорода.
По окончании сжигания прекращают нагревание и воду, сконденсировавшуюся в отводном конце трубки для сжигания или в соединительной трубке, перегоняют в поглотительный прибор, касаясь трубок небольшим синим пламенем. Продолжают пропускать воздух или кислород до тех пор, пока вся двуокись углерода или вода не будут поглощены в поглотительных приборах, которые к концу сжигания должны быть наполнены тем же газом, что и в начале этой операции. Снимают и закрывают приборы с поглотителями, охлаждают их в шкафчике весов и взвешивают, пользуясь первоначально применявшимися противовесами. Перед
1 J. МеуегиН. Tischbierek [Z. anal. Chem., 80, 241 (1930)] установили что смешение с пятиокисью ванадия способствует сжиганию трудно сгорающих углеродсодержащих веществ. Для этой же цели служат металлическая медь и свежеприготовленная РЬ3О4 (свободная от С или СО2).
2 Об аналитических аппаратах для сжигания, служащих для макроопределения углерода и водорода в углеводородах или в соединениях, содержащих углерод, водород и кислород, а также о воспроизводимости и точности, достигаемых при сжигании бензойной кислоты высокой чистоты, см. D. D. W a g m a n, F. D. R о s s i n i [J. Research NBS, 32, 95 (1944)] и W. II. S m i t h, Ch. P. S а у 1 о r, H. J. W i n g [J. Research NBS, 10, 479 (1933)].
854
Гл. L. Углерод и водород
взвешиванием приборов их на мгновение открывают для уравновешивания давления.
Содержание водорода и углерода вычисляют по привесу соответствующих поглотительных приборов.
Сжигание летучих или возгоняющихся веществ. Сжигание летучих или возгоняющихся веществ следует проводить с большими предосторожностями.
Относительно труднолетучие вещества можно взвешивать в лодочке. Часть трубки, в которую помещают эту лодочку, необходимо нагревать в этих случаях с еще большей осторожностью, чем при анализе нелетучих веществ.
Умеренно летучие вещества можно взвешивать в очень маленькой склянке с притертой стеклянной пробкой, которую затем помещают в наклонном положении в лодочку и открывают непосредственно перед введением лодочки в трубку.
Легколетучие жидкости взвешивают в маленьких стеклянных ампулах с тонким оттянутым капилляром. Эти ампулы очищают, высушивают, взвешивают и затем наполняют анализируемой жидкостью. Для наполнения ампул их нагревают, погружают капилляр в жидкость и держат в таком положении до охлаждения пузырька. Это повторяют до тех пор, пока в ампулу не наберется достаточное количество жидкости. Затем капилляр освобождают от жидкости, запаивают и ампулу снова взвешивают. Когда все для сжигания подготовлено, окись меди в отводном конце трубки накалена до красного каления, а вводный конец трубки остается холодным, ставят ампулу наклонно в лодочку, обламывают капилляр, быстро вводят лодочку в трубку, немедленно вслед за ней кладут медную сетку и закрывают трубку пробкой.
При анализе возгоняющихся веществ необходимо принять соответствующие меры, чтобы воспрепятствовать возгонке части вещества вблизи пробки и у вводного конца трубки. Это достигается тем, что лодочку помещают возможно дальше от этого конца, по возможности сильнее нагревают часть медной сетки, прилегающую к пробке, прежде чем нагреть лодочку, и пускают несколько более сильную струю воздуха, чем обычно х.
Сжигание веществ, содержащих галогены и серу. При сжигании веществ, содержащих галогены или серу, образуются соединения, которые полностью не удерживаются окисью меди при красном калении. Замена окиси меди гранулированным прокаленным хроматом свинца приводит к удовлетворительным результатам при условии, что прилегающая к лодочке часть хромата не нагревается до плавления, а часть хромата, наиболее близкая к отводному концу, нагревается не выше 300—400° С. В таких случаях анализируемое вещество смешивают с измельченным в тонкий порошок прокаленным хроматом свинца в большой лодочке или в самой трубке.
1 Н. D. W i 1 d е Jr., и Н. L. L о с h t е [J. Am. Chem. Soc., 47, 440 (1925)] приводят быстрый метод сжигания органических соединений в калориметрической бомбе, содержащей кислород под давлением и известное количество титрованного раствора гидроокиси бария. О поглощении летучих, взрывчатых и легко карбонизующихся органических жидкостей инфузорной землей перед сжиганием см. М. G. S е v a g, Ind. Eng. Chem., Anal. Ed., 1, 16 (1929).
Методы определения
855
Неплавящуюся смесь хромата свинца и окиси свинца можно приготовить следующим образом !. Измельченный в тонкий порошок хромат свинца замешивают с водой в кашицу, а затем тщательно смешивают с красной окисью свинца, взятой в количестве одной четверти от массы хромата свинца. Переносят смесь на фильтр и слабо отсасывают до получения компактной массы. Влажную массу порциями величиной с горошину переносят лопаточкой в фарфоровый тигель и нагревают до красного каления. По охлаждении разбивают в фарфоровой ступке на кусочки величиной с пшеничное зерно. Отсеивают мелкоизмельченный материал, который для смешения с образцом измельчают еще тоньше. Тонкоизмельченный и грубозернистый материал нагревают до 150° С, а затем охлаждают в эксикаторе. В обычных условиях одна порция хромата может быть использована для трех или четырех сжиганий 1 2.
Сжигание веществ, содержащих азот. При сжигании азотсодержащих веществ образуются окислы азота, которые необходимо восстановить перед выходом из печи; в противном случае результаты определения углерода получаются повышенными. Восстановление легко осуществляется с помощью сетки из восстановленной меди, помещаемой у отводного конца трубки, причем загрузку трубки и способ сжигания образца следует видоизменить. Чтобы освободить место для медной сетки, слой окиси меди укорачивают с 45 до 35 см. При этом у отводного конца трубки остается свободное пространство длиной в 20 см, куда помещают компактный, плотный рулон медной сетки длиной 10—15 см; медь восстанавливают следующим образом. Наливают 1 мл метилового спирта в пробирку, достаточно широкую, чтобы поместить сверток медной сетки, и свободно закрепляют пробирку в отвесном положении. Медную сетку нагревают до красного каления, держа ее тигельными щипцами над большим пламенем паяльной лампы, и затем возможно быстрее опускают в пробирку. По окончании восстановления закрывают пробирку маленьким стаканчиком. Остывший сверток медной сетки вынимают из пробирки и осторожно высушивают, проводя его через небольшое синее пламя горелки.
Для предохранения свертка медной сетки от окисления в начальной стадии сжигания нельзя пропускать воздух, поэтому необходимо тщательно смешать анализируемое вещество с измельченной в достаточно тонкий порошок прокаленной окисью меди. К концу операции часть трубки, в которой находится восстановленная медная сетка, можно охладить и сжигание закончить в токе воздуха или кислорода.
Смешивание анализируемого вещества с окисью меди может быть выполнено в большой медной лодочке или непосредственно в трубке, в пустом пространстве перед окисью меди. Лодочку наполняют следующим образом. Достаточное количество окиси меди, просеянной через сито в 200 меш (d = 0,074 мм), нагревают в фарфоровом тигле и охлаждают в эксикаторе. Ставят на лист гладкой черной бумаги маленькую агатовую ступку, всыпают в нее немного окиси меди, затем навеску образца, покрывают другой порцией окиси меди и тщательно перемешивают пестиком, не раздавливая смесь. Необходимо следить за тем, чтобы не было потерь от распыления. Содержимое ступки переносят в лодочку и счищают ступку, пестик и бумагу некоторым количеством окиси меди. Если навеску вводят непосредственно в трубку (без лодочки), то ее смешивают с окисью меди в закрытой пробкой склянке, имеющей диаметр, соразмерный с диаметром трубки для сжигания. Осторожно высыпают смесь в трубку и очищают склянку окисью меди.
1 R. d е R о ode, Am. Chem. J., 12, 226 (1890).
2 См также Е. R е г у 1 и др., Rer., 61в, 83 (1928).
856
Гл. L. Углерод и водород
Определение общего содержания углерода «мокрым сжиганием»
Окисление органических веществ нагреванием с серной и хромовой кислотами 1 2 в настоящее время используется реже, чем это было в то время, когда сжатый кислород был мало доступен. Этот метод, однако, и теперь иногда находит применение, несмотря на то, что трудно достигнуть полного разложения анализируемого вещества без выделения серного ангидрида.
Процесс проходит более успешно, когда для удержания серного ангидрида применяют электрический осадитель 3. В этом осадителе газы проходят через трубки, в которых с помощью индукционной катушки поддерживается высокое напряжение между медной сеткой, находящейся с внешней стороны трубок, и медной проволокой внутри них. Так как в процессе сжигания может образоваться некоторое количество окиси углерода, целесообразно, кроме того, пропускать газы через трубку для сжигания, нагретую до 800—900° С и наполненную окисью меди. При применении электрического осадителя для соединения трубок следует пользоваться ртутными затворами вместо резины, так как в электрическом осадителе образуется озон. Двуокись углерода из колбы удаляют током воздуха, если только в колбе не образуется водород, что бывает при непосредственном растворении некоторых сплавов. В этих случаях вместо воздуха пользуются азотом. Основной частью аппарата служит колба типа Корлейса 3 с длинной шейкой и притертой стеклянной пробкой, в которую впаян внутренний водяной холодильник. Перечисленные в предыдущих разделах очистители и поглотители применимы и в данном случае.
Ход определения. В колбу вводят 60 мл 50%-ного раствора хромового ангидрида СгО3, 200 мл разбавленной (1 : 1) серной кислоты и соединяют колбу с очистительными трубками и печью. Нагревают печь до 800— 900° С, включают электрический осадитель, пускают воду в холодильник и кипятят раствор 30 мин, пропуская слабый ток воздуха. Включают в систему поглотитель двуокиси углерода и продолжают кипятит!, еще 30 мин, не прекращая тока воздуха. После этого вынимают поглотительный прибор и по охлаждении взвешивают. Снова включают поглотитель в установку и продолжают кипячение, пропуская ток воздуха 2 ч, после чего поглотительный прибор снова взвешивают. Увеличение массы прибора при проведении «холостого» опыта не должно превышать 0,5 мг.
Раствор в колбе охлаждают, включают в установку поглотительную трубку, помещают в колбу навеску образца, пускают воду в холодильник и пропускают через установку слабый ток воздуха. Постепенно нагревают и под конец кипятят раствор не менее 1 ч после растворения анализируемой пробы. Удаляют поглотительный прибор, снова взвешивают его и из привеса вычитают массу, соответствующую массе, полученной в «холостом» опыте 4.
1 R. Е. Rogers, W. В. Rogers, Sill. Am. J., [2], 6, 110 (1848); С. В г и в-п е г, Pogg. Ann., 95, 379 (1855).
2 F. Oschwald, Beitr. Bestimmung Kohlenstoffes Aluminium, Eidgenosi-schen Technischen Hochschule, Zurich, 1923, p. 31.
8 E. С о r 1 e i s, Stahl u. Eisen, 14, 581 (1894).
4 Определение общего углерода в почвах по видоизмененному методу, в котором применяется специальная аппаратура; в этом методе содержание углерода определяется титрованием после поглощения ее избыточным количеством титрованного раствора щелочи, см. J.E. Adams, Ind. Eng. Chem., Anal. Ed., 6, 277 (1934)*
Методы определения
857
Другие методы определения углерода
Вместо поглощения двуокиси углерода сухим взвешенным поглотителем, как описано выше (стр. 848, 850 и 856), некоторые аналитики предпочитают пользоваться для поглощения газа раствором гидроокиси бария, а затем: 1) взвешивать промытый и прокаленный карбонат бария \ 2) промытый карбонат бария растворять в титрованном растворе кислоты и избыток кислоты оттитровывать раствором едкой щелочи по метиловому оранжевому 1 2 или 3) определять количество непрореагировавшей гидроокиси бария титрованием кислотой по фенолфталеину3 или измерением изменения электрического сопротивления раствора, которое зависит от его концентрации 4 *. При применении всех этих методов необходимо тщательно соблюдать требуемые меры предосторожности, чтобы устранить возможность загрязнения двуокиси углерода из атмосферы. Во втором методе нет необходимости очищать двуокись углерода от таких веществ, как серный ангидрид или хлористый водород.
Некоторые авторы 6 предпочитают превращать карбонат бария в сульфат после поглощения двуокиси углерода холодным аммиачным раствором хлорида бария [50 г ВаС12 • 2Н2О и 250 мл разбавленного NH40H (пл. 0,88 г /см3) в 1 л раствора]. В результате этой реакции образуется карбамат бария Ba(NH2CO2)2, который растворим в умеренных количествах. Частичное превращение в нерастворимый карбонат бария имеет место в концентрированных растворах, а полное превращение происходит при кипячении раствора в течение 1 мин. Основным преимуществом применения такого раствора является то, что его требуется меньшее количество, так как хлорид бария более растворим^ чем гидроокись бария, и отсутствие необходимости соблюдать особые предосторожности при фильтровании и промывании карбоната бария. Этот раствор перед его применением кипятят 1 мин для осаждения (в виде карбоната бария) того СО2, который он мог поглотить ранее. Затем его фильтруют горячим через асбест в поглотительную склянку, воздух в которой свободен от двуокиси углерода, и, прежде чем приступить к анализу, охлаждают. По окончании поглощения, продолжая пропускать струю воздуха, свободного от двуокиси углерода, быстро нагревают раствор и кипятят его 1 мин. Охлажденный раствор фильтруют и промывают трубку, колбу и осадок сначала холодной, а затем горячей водой. Небольшие количества двуокиси углерода, поглощенные из воздуха во время фильтрования и промывания, остаются в растворе. Полученный осадок, вместе с осадком, приставшим к трубке и колбе, растворяют в соляной кислоте и осаждают барий в растворе серной кислотой (стр. 713), как обычно.
Газометрический метод определения СО2, применяемый в газовом анализе 6, редко может быть использован в анализе негазообразных неорганических веществ, так как получаемый объем двуокиси углерода очень мал по сравнению с объемом кислорода, воздуха или азота, примененным для ее улавливания.
К определению окиси углерода приходится прибегать редко; ее определяют главным образом в тех случаях, когда надо исследовать, вреден
1 Fresenius — Cohn, Quantitative Chemical Analysis, vol. 1, John Wiley and Sons, 1904, p. 482; Chemists U.S. Steel Corp., Sampling and Analysis of СагЬом and Alloy Steels, Reinhold Publishing Corp., New York, 1938, p. 54.
2 J. R. C a i n, J. Ind. Eng. Chem., 6, 465 (1914); J. R. Cain, L. C. Maiwell, там же, 10, 520 (1918).
3 W. Brady, J. Ind. Eng. Chem., 6, 843 (1914); Methods of the U. S. Steel Corp., цит. выше, стр. 56. J. Linder [Z. anal. Chem., 72, 135 (1927)] установил, что этот метод дает несколько повышенные результаты.
4 E.L. Bennet, J. Н. Harley, R. М. Fowler, Anal. Chem., 22, 445 (1940); К. Gardner, W. J. Rowland, H. Thomas, Analyst, 75, 173 (1950).
* J. McFarlane, A. W. Gregory, Chem. News, 94, 133 (1906).
• Л. M. Д e н н и с, M. H и к о л ь с, Газовый анализ, Госхимтехиздат, 1934.
858
Гл. L. Углерод и водород
ли воздух для дыхания людей и животных. При применении в качестве поглотителя силикагеля, пропитанного раствором палладия и молибдена, можно открыть 1 часть окиси углерода в 500 000 000 частях воздуха. Этим методом можно определить физиологически значительные количества окиси углерода в течение примерно 1 мин, а 0,001% СО по объему открывают быстрее, чем за 1 мин. При поглощении окиси углерода окраска силикагеля резко изменяется от ярко-желтой до ярко-зеленой и, наконец, до синевато-зеленой. В лабораторных или полевых условиях силикагель удобно применять в маленьких стеклянных трубках. Колориметрическое определение проводится сравнением с окрасками силикагеля, находящегося в таких же трубках и подвергавшегося воздействию определенных количеств окиси углерода. Еще удобнее пользоваться таблицей стандартных окрасок *.
Определение водорода в горных породах и минералах
К методам определения водорода в горных породах и минералах относятся: 1) методы определения неконституционного водорода; 2) методы определения конституционного водорода; 3) методы определения водорода в органических соединениях и 4) методы определения общего содержания водорода. Первые два метода описаны в разделе «Вода» (стр. 903 и стр. 908), третий метод изложен в разделе «Двуокись углерода; углерод» (стр. 1014), четвертый — в разделе «Определение общего содержания углерода и водорода прямым сжиганием» (стр. 850).
1 Подробности см. М. Shepherd, Ind. Eng. Chem., Anal. Ed., 19, 77 (1947). *Другие методы определения окиси углерода в воздухе см. М. В. А л е к с е-е в а, Б. Е. Андронов, С. С. Гурвиц, А. С. Житкова, Определение вредных веществ в воздухе производственных помещений, Госхимиздат, 1954.*
Глава LI
АЗОТ
Азот является преобладающим элементом атмосферы, в которой он находится в свободном состоянии. Он содержится также в органических веществах, например в угле. Нитраты содержатся в почве и в некоторых районах с сухим климатом встречаются в огромных количествах. В горных породах азот в несвязанном состоянии содержится в ничтожных количествах, но в виде аммиака и аммонийных солей он в некоторых породах находится часто и легко в них обнаруживается.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Классическим методом определения азота является метод, впервые предложенный Дюма К В этом методе анализируемое вещество смешивают с окисью меди и сжигают в трубке, содержащей окись меди и металлическую медь. Выделившийся азот измеряют в нитрометре после поглощения остальных газов раствором едкой щелочи. Этот метод применим для анализа всех азотсодержащих соединений.
Для анализа большинства материалов пригоден более простой метод Кьельдаля или многочисленные его видоизменения 1 2. Определение можно проводить быстро и просто микроаналитическими вариантами обоих методов. Если анализируемый материал достаточно однороден по составу, чтобы можно было пользоваться маленькими навесками, эти методы следует предпочесть в тех случаях, когда имеется ограниченное количество материала, а также и в рядовой работе 3. Очень малые количества азота, находящиеся в пробе в виде аммиака или нитратов, лучше всего определять колориметрическим методом.
Метод Дюма
Основные трудности при применении этого метода связаны с необходимостью поддерживать атмосферу чистой двуокиси углерода в продолжение всей операции и с количественным улавливанием и измерением азота.
1 J. В. Dumas, Ann. chim. phys., 2, 198 (1831).
1 J. Kjeldahl, Z. anal. Chem., 22, 366 (1886); J. W. Cunning, там же, 28, 188, (1889); H. W i 1 f а г t h, Chem. Zentr., 56, 17, 113 (1885).
3 Подробное изложение таких методов см. F. Р г е g 1, Н. Roth, Quantitative Organic Microanalysis, translated by E. B. Daw. P. Blakiston’s Son and Co., Philadelphia, 1937; A. Steyermark, Quantitative Organic Microanalysis, Blakiston Co., Philadelphia, 1951.
860
Гл. LI. Азот-
Прежде атмосфера двуокиси углерода обеспечивалась применением закрытой трубки, наполненной с одного конца магнезитом, нагреванием которого в начальной и конечной стадиях анализа получался необходимый газ. Позднее стали применять трубку, открытую с обоих концов, а двуокись углерода получали извне смешением насыщенного раствора карбоната натрия или калия с разбавленной (1 : 1) серной кислотой в соответствующем сосуде Еще более удобно приспособление, в котором удаление воздуха облегчается попеременным откачиванием и наполнением трубки двуокисью углерода, получаемой вначале из любого чистого источника, находящегося вне прибора, а затем из бикарбоната натрия, помещенного внутри прибора, как изображено на рис. 35.
Рис. 35. Передняя часть прибора для определения азота методом Дюма:
1 — бикарбонат натрия; 2 — отвод к вакууму; 3 — отвод к источнику двуокиси углерода; 4 — отвод к сосуду с ртутью.
Для удаления воздуха из прибора у вводного и отводного концов трубки для сжигания устанавливают краны. Затем вводный конец присоединяют к трубке, которая соединена: 1) с трубкой, ведущей к источнику двуокиси углерода (баллону со сжатым газом или аппарату Киппа); 2) с согнутой достаточно длинной, погруженной в ртуть трубкой, служащей манометром; 3) с трубкой, йедущей к насосу для разрежения, и 4) с большой трубкой, наполненной бикарбонатом (свободным от азота). При работе с этим приспособлением отводной конец трубки для сжигания закрывают, выкачивают из системы воздух, вытесняют оставшийся воздух током двуокиси углерода, получаемой из баллона или в генераторе, затем снова отсасывают и повторяют эту операцию несколько раз. В последний раз очистку и наполнение трубки проводят двуокисью углерода, получаемой нагреванием трубки с бикарбонатом натрия.
Когда система заполнена двуокисью углерода и присоединена к наполненному нитрометру, нагревание бикарбоната прекращают и крап вводного конца трубки закрывают. Затем обычным способом проводят сжигание, пока не прекратится появление пузырьков газа в течение 2 мин. После этого снова начинают нагревать бикарбонат, пока пузырьки двуокиси углерода не начнут энергично пробиваться через ртуть в манометр. Слегка приоткрывают крап у вводного конца трубки и пускают слабый ток двуокиси углерода до полного вытеснения азота из системы. Предложено множество схем для использования сухого льда в качестве источника двуокиси углерода при определении азота методом Дюма1 2.
Установка трубки для сжигания и обработка пробы проводятся так же, как при определении углерода и водорода (стр. 852). Навеска анализируемой пробы должна быть взята с таким расчетом, чтобы содержание в ней азота не превышало количество, которое может быть измерено
1 Подробное описание такого метода приведено Р. Н. В г i n t о n, F. М. S li е г t z, W. G. С г о с k е t t, Р. Р. М е г k е 1, J. Ind. Eng. Chem., 13, 636 (1921).
2 См. W. S. I d e, Ind. Eng. Chem., Anal. Ed., 7, 442 (1935); W. H. H a m ill, J. A. A 1 i c i n о, там же, 9, 290 (1937); F. S hea, С. E. Watts, там же, 11, 333 (1939),
Методы определения
861
в нитрометре. Пробу надо тщательно перемешать с достаточным для полного ее окисления количеством окиси меди, так как в процессе сжигания ни воздух, ни кислород пропускать через систему нельзя Выводной конец трубки для сжигания присоединяют непосредственно к нитрометру типа Шиффа с ртутным затвором, помещенным по середине между вводной трубкой и отростком для уравнительного сосуда, наполненного 50 %-ным раствором едкого кали (рис. 36). Нитрометр не наполняют раствором до тех пор, пока воздух практически полностью не удален, и к сжиганию приступают лишь после удаления из всей системы газов, не поглощающихся раствором едкого кали. Сжигание проводят обычным способом, причем азот полностью переводят в нитрометр слабым током двуокиси углерода. Применяемая для этого двуокись углерода должна быть свободна от газов, не поглощаемых раствором едкой щелочи.
В заключение собранный азот пере- _ тр
г Рис. 36. Конечная часть приводят в нитрометр, помещенный в водяную бора для определения а3ота муфту, тщательно отсчитывают объем и методом Дюма,
отмечают температуру и барометрическое
давление. Массу азота вычисляют, пользуясь таблицами, в которых приводится масса 1 мл влажного азота при различных температурах и давлениях, или по следующей формуле
х = 0,04493
И (В-Ж) (273 + 0 “
где х — содержание азота, % ;
V — объем полученного азота, мл;
В — барометрическое давление, мм рт. ст.;
W — давление водяного пара, мм рт. ст. (см. табл. 26); t — температура, °C;
а — навеска образца, г1 2 3.
Метод Кьельдаля
В наиболее простом виде метод Кьельдаля сводится к разложению пробы и связыванию азота в виде сульфата аммония кипячением с серной кислотой. Свободные галогены или кислородные соединения галогенов вызывают потерю азота в процессе нагревания по Кьельдалю, поэтому
1 Сжигание относительно мало устойчивых жидких соединений см. F. Shea,
С. Е. Watts, Ind. Eng. Chem., Anal. Ed., 11, 333 (1939), а сжигание трудно сжигаемых веществ см. J. R. S р i е s, Т. Н. Н а г г i s, там же, 9, 304 (1937).
3 Точность метода иллюстрируется результатами, полученными Brinton и его учениками (цит. выше), которые при анализе специально очищенных пикриновой кислоты и динитробензола определили 18,45 и 16,70% азота при теоретическом содержании в этих соединениях 18,35 и 16,64% азота.
862
Гл. Ы. Азот.
Давление водяного пара при температурах от 15 до 35° С
рт. ст.) 1
ТАБЛИЦА 2 С
°C 0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9
15 12,788 12,870 12,953 13,037 1.3,121 13,205 13,290 13,375 13,461 13,547
16 13,634 13,721 13,809 13,898 13,987 14,076 14,166 14,256 14,347 14,438
17 14,530 14,622 14,715 14,809 14,903 14,997 15,092 15,188 15,284 15,380
18 15,477 15,575 15,673 15,772 15,871 15,971 16,071 16,171 16,272 16,374
19 16,477 16,581 16,685 16,789 16,894 16,999 17,105 17,212 17,319 17,427
20 17,535 17,644 17,753 17,863 17,974 18,085 18,197 18,309 18,422 18,536
21 18,650 18,765 18,880 18,996 19,113 19,231 19,349 19,468 19,587 19,707
22 19,827 19,948 20,070 20,193 20,316 20,440 20,565 20,690 20,815 20,941
23 21,068 21,196 21,324 21,453 21,583 21,714 21,845 21,977 22,110 22,243
24 22,377 22,512 22,648 22,785 22,922 23,060 23,198 23,337 23,476 23,616
25 23,756 23,897 24,039 24,182 24,326 24,471 24,617 24,764 24,912 25,060
26 25,209 25,359 25,509 25,660 25,812 25,964 26,117 26,271 26,426 26,582
27 26,739 26,897 27,055 27,214 27,374 27,535 27,696 27,858 28,021 28,185
28 28,349 28,514 28,680 28,847 29,015 29,184 29,354 29,525 29,697 29,870
29 30,043 30,217 30,392 30,568 30,745 30,923 31,102 31,281 31,461 31,642
30 31,824 32,007 32,191 32,376 32,561 32,747 32,934 33,122 33,312 33,503
31 33,695 33,888 34,082 34,276 34,471 34,667 34,864 35,062 35,261 35,462
32 35,663 35,865 36,068 36,272 36,477 36,683 36,891 37,099 37,308 37,518
33 37,729 37,942 38,155 38,369 38,584 38,801 39,018 39,237 39,457 39,677
34 39,898 40,121 40,344 40,569 40,796 41,023 41,251 41,480 41,710 41,941
35 42,175 42,409 42,644 42,880 43,117 43,355 43,595 43,836 44,078 44,320
1 International Critical Tables, III, 212 (1928).
их присутствие недопустимо. Полученный раствор кипятят с едким натром и поглощают выделяющийся аммиак титрованным раствором серной кислоты, избыток которой оттитровывают обратно едкой щелочью. В настоящее время неизвестен вариант метода, который был бы применим для анализа любого азотсодержащего соединения. Для облегчения разложения пробы серной кислотой обычно добавляют окись ртути и сульфат калия, но в некоторых случаях добавления этих реагентов не требуется. Взамен окиси ртути, сульфата калия и даже серной кислоты были предложены многие другие вещества.
В большинстве случаев сульфат калия может быть заменен эквивалентным количеством сульфата натрия !.
Продолжительность разложения может быть значительно сокращена1 2 добавлением 18 г Na2SO4 и 0,7 г Hg и сильным прокаливанием. Найдено 3, однако, что такая замена не дает удовлетворительных результатов при разложении ппридин-цинк-хло-рида никотиновой кислоты и оксихинолина. Неудовлетворительной оказалась также замена сульфата калия такими соединениями, как Na2HPO4, Na4P2O7, CuSO4, NiSO4, KA](SO4), ZnCl2, MnCl2, MnO2, H2WO4, H2MnO4, H2TiO3 и HVO3. Рекомендуется также применение других окислителей, например CuSO4, КМпО4 и Н2О2. Действие сульфата меди достигает цели при продолжительном нагревании. Перманганат следует добавлять спустя 2 мин после прекращения нагревания, в противном случае получаются пониженные результаты 4. Разложение облегчается и потерь азота пе паблю-
1 R. В. D е е m е г, J. Assoc. Offic. Agr. Chemists, 3, 303 (1920); T. D. J a г г e 1 1, там же, 3, 304 (1920); W. L. La tshaw, J. Ind. Eng. Chem., 8, 586 (1916).
2 О. M. S h e 1 d, J. Assoc. Offic. Agr. Chemists, 10, 507 (1927).
3 I. K. Phelps, H. W. D a u d t, J. Assoc. Offic. Agr. Chemists, 3, 306 (1920); 4, 72 (1920).
4 W. F r e a r, W. Thomas, H. D. Edmiston, J. Assoc. Offic. Agr. Chemists, 3, 220 (1919).
Методы определения
863
дается 1 при обработке 30%-ной Н2О2 и H2SO4. К обугленному, охлажденному раствору органического вещества рекомендуется2 добавлять в умеренном избытке сухой, свободный от аммонийных солей K2S2O8.
Быстрое разложение достигается 3 при обработке 25 мл H2SO4, 1 г CuS04 и 2 мл НС1О4. Замена части H2SO4 фосфорной кислотой не имеет заметных преимуществ.
Разложение органических веществ происходит быстрее, если применять селен или хлорокись селена в качестве катализатора 4 5. Для этой цели рекомендуется раствор, содержащий 10 г сульфата натрия или калия, 25 мл серной кислоты и 0,1—0,2 г селена. При этом перед дистилляцией нет необходимости вводить сульфид. Продолжительность разложения сокращается 6 7 при применении по 0,25 г селена и сульфата железа (II). Введение больших количеств селена приводит к потере азота. Добавление окиси ртути (II) пе дает никаких преимуществ и приводит к полному окислению селена до селеновой кислоты в концентрированном сернокислом растворе 6.
Для поглощения аммиака некоторые исследователи рекомендуют ~ пользоваться 50 мл 4%-ного раствора борной кислоты вместо титрованного раствора серной кислоты из тех соображений, что раствор борной кислоты нет необходимости точно отмерять и при этом не требуется титрованного раствора едкой щелочи. При использовании подходящего индикатора, например бромфенолового синего (лучше при искусственном освещении), и учете данных тщательно проведенного «холостого» опыта получаются вполне удовлетворительные результаты. Вместо отгонки аммиака рекомендуется 2 обрабатывать слабо подщелоченный анализируемый раствор избыточным количеством титрованного раствора гипо-бромита, а затем, подкислив раствор и добавив иодид, титровать тиосульфатом.
Ход определения. Определение в отсутствие нитратов, нитритов, азосоединений, гидразина, цианидов и др. Помещают 0,35—3,5 г, в зависимости от содержания азота, измельченного до частиц размером 35 меш (d = 0,42 мм) или тоньше материала в колбу Кьельдаля емкостью 500—600 мл. Прибавляют 10 г измельченного в порошок сульфата калия, 0,5—0,7 г окиси ртути [приготовленной мокрым способом (стр. 109), но не из нитрата ртути], или 0,6—0,65 г металлической ртути и 20—25 мл серной кислоты. Тщательно перемешивают жидкость и помещают колбу в отверстие в асбестовом картоне, наклонив ее под углом около 60° С. Отверстие в картоне должно быть такого размера, чтобы при нагревании пламя не охватывало колбу выше уровня жидкости. Нагревают при температуре ниже точки кипения кислоты до прекращения образования пены.
Сильное вспенивание может быть предотвращено введением маленького кусочка парафина. Постепенно усиливают нагревание, пока жидкость не начнет энергично кипеть, и продолжают кипятить 15—30 мин
1 F. С. К о с h, Т. L. М с М е е k i n, J. Am. Chem. Soc., 46, 2066 (1924).
2 Н. Н. W i 11 а г d, W. Е. С а к е, J. Am. Chem. Soc., 42, 2646 (1920).
3 B.Mears, R. E. Hussey, Ind. Eng. Chem., 13, 1054 (1921).
* M. F. Lauro, Ind. Eng. Chem., 3, 401 (1931).
5 R. B. Bradstreet, Ind. End. Chem., 10, 696 (1938).
6 A. Sreenivasan, V. Sadasivan, Ind. Eng. Chem., 11, 314 (1939).
7 L. W. Winkler, Z. angew. Chem., 26, 231 (1913); F.M.Scales,A. P. Harrison, J. Ind. Eng. Chem., 12, 350 (1920); H. D. Spears, J. Assoc. Offic. Agr. Chemists 5, 105 (1921); K. S. Markley, R. M. Hann, там же, 8, 455 (1925); J. L. H a g u e, R. A. P a u 1 s о n, H. А. В r i g h t, J. Research NBS, 43, 201 (1949); A. Eisner и E. C. Wagner [Ind. Eng. Chem. Anal. Ed., 6, 473 (1934)] установили, что 4%-ный раствор борной кислоты не изменяется при хранении в сосуде из стекла пирекс.
864
Гл. LI. Азот
после обесцвечивания раствора или после того момента, когда можно полагать, что разложение закончилось (через 1—1,5 ч). Ослабив нагревание так, чтобы раствор продолжал слабо кипеть, продолжают нагревать еще 1—3 ч. Изредка прибавляют серную кислоту, чтобы объем ее в процессе нагревания не уменьшился более чем до 20 мл.
По охлаждении осторожно разбавляют раствор 200 мл воды. Если требуется устранить толчки, вводят несколько кусочков пемзы, гранулированного цинка или битого фарфора. Затем, взбалтывая, осторожно прибавляют 25 мл раствора, содержащего 80 г тиосульфата натрия или 40 г сульфида калия или натрия в 1 л. Это делается для связывания ртути и в случае ее отсутствия может быть исключено. Прибавляют достаточное количество 35%-ного раствора едкого натра, чтобы раствор сделался сильнощелочным (обычно достаточно 50. мл). Едкую щелочь приливают по стенкам сосуда, чтобы она не сразу смешалась с кислым раствором Ч
Ставят колбу в вертикальное положение, присоединяют насадку Кьельдаля или другое подходящее приспособление для предохранения от загрязнения брызгами едкого натра (каплеуловитель) и соединяют с вертикальным холодильником, желательно покрытым оловом. В коническую колбу наливают отмеренное количество титрованной серной кислоты, достаточное, чтобы связать выделяющийся аммиак, и устанавливают колбу так, чтобы конец холодильника был едва погружен в кислоту. Осторожно взбалтывая, перемешивают содержимое колбы Кьельдаля, затем нагревают до кипения и полностью перегоняют аммиак в титрованный раствор серной кислоты. Достаточно перегнать 150—200 мл жидкости. Опускают коническую колбу, удаляют пламя и ополаскивают холодильник водой. Прибавляют к дистилляту метиловый красный или кошениль и титруют раствором едкой щелочи, эквивалентным титрованному раствору кислоты. Объем щелочи, израсходованной на титрование, вычитают из объема взятого титрованного раствора кислоты и в полученную разность вводят поправку на «холостой» опыт, в котором 1 г органического вещества, не содержащего азота, например сахара, проводится через все стадии определения. 1 мл 0,5 н. раствора кислоты соответствует 0,007 г азота 1 2.
Этим методом были получены 2 превосходные результаты при анализе таких соединений, как монометиламин, триметиламин, солянокислый глюкозамин, иодид тетраметил аммония, солянокислый холин, изатин, атропин, кокаин, никотин-цинк-хлорид, никотиновая кислота, солянокислый р-эйкаин, оксихинолин, цинхонидин, стрихнин, бруцин, папаверин, наркотин, морфин, гидрастинин, кофеин, лофин, амарин, солянокислый гистидин, 2-метил-4-хиназолон, 2-метил-3-фенил-4-хиназолон. Неудовлетворительные результаты были получены при анализе азосоединений и соединений гидразина.
1 Удобный прибор для приливания раствора едкого натра и для продувания воздуха через раствор при перегонке приводят I. К. Р h е 1 р s и Н. W. D a u d t [J. Assoc, Offic. Agr. Chemists, 3, 312 (1920). Применение несколько видоизмененных приборов для продувания воздуха описано также W. В. М el dru m, R. М е 1 a m р у, W. D. Myers, Ind. Eng. Chem., Anal. Ed., 6, 63 (1934). F. T. Adriano [Philippine Agr., 17, 509 (1929)], J. Green [Ind. Eng. Chem., Anal. Ed., 3, 160 (1931)] и J. M. Fife [там же, 8, 316 (1936)] предпочитают проводить дистилляцию паром вместо нагревания газом или электричеством. Описание видоизмененной ловушки Кьельдаля см. G. Н. W. L u с a s, Ind. Eng. Chem., Anal. Ed., 1, 140 (1929).
2 I. K. Phelps, H. W. D a и d t, цит. выше.
Методы определения
865
Исследовано 1 поведение некоторых органических веществ при различных методах обработки.
Опубликован 2 обзор методов, в которых продолжительность нагревания по Кьель-далю уменьшена до 30 мин применением серной кислоты, содержащей ртуть, двуза-мещенный фосфат калия и сульфат железа (III);
Определение в присутствии нитратов. Предыдущий метод непригоден, если азот находился в виде нитратов. В этом случае можно пользоваться специальным вариантом этого метода, изложенным ниже (см. «Метод с сульфосалициловой кислотой»), но более быстро и точно определение может быть выполнено методом Деварда (стр. 866). Метод с применением нитрометра 3, в котором нитрат разлагают хлоридом железа (II) в солянокислом растворе и затем измеряют объем образующейся окиси азота, дает пониженные результаты, если он не базируется на результатах, полученных для чистых солей. Этот метод рекомендуется только для массовых контрольных анализов.
1. Метод с сулъфосалициловой кислотой 4. Растворяют 10 г нитрата в воде и разбавляют точно до 500 мл. Отбирают 25 мл раствора, переносят в колбу Кьельдаля емкостью 650 мл и выпаривают досуха при 90— 100° С. Прибавляют 25 мл сульфосалициловой кислоты (40 г салициловой кислоты растворяют в 1000 мл серной кислоты пл. 1,84г/с«и3и ополаскивают горло колбы. Часто взбалтывая, нагревают до полного растворения пробы и начала реакции, причем температуру не следует поднимать выше, чем может терпеть рука при соприкосновении с колбой. Оставляют не менее, чем на 1 ч, временами взбалтывая. Прибавляют 5 г тиосульфата натрия, нагревают при невысокой температуре до прекращения вспенивания (около 5 мин), затем прибавляют 10 г сульфата натрия или калия и 1 г окиси ртути, и в дальнейшем поступают, как при анализе в отсутствие нитратов (стр. 863).
2. Метод Деварда. Метод Деварда 5 не является модификацией метода Кьельдаля, но приводится здесь потому, что он применяется для определения азота в нитратах. Метод основан на восстановлении нитратов алюминием и цинком в щелочной среде. Обычно применяют сплав, содержащий 50 весовых частей Си, 45 весовых частей А1 и 5 весовых частей Zn. Медь придает сплаву хрупкость и способность легко измельчаться в порошок; в присутствии меди устраняются толчки при конечной перегонке. Метод неприменим в присутствии азотсодержащих органических веществ 6. Находящиеся в анализируемом материале аммонийные соли могут быть отогнаны из щелочного раствора перед добавлением сплава.
1 Р. Fleury, Н. Levaltieri, J. pharm. Chim., 30, 265 (1924); Bull. Soc. Chim., 37, 330 (1925); C. A., 18, 1625 (1924); 19, 2002 (1925).
2 F. M. S t u b b 1 e f i e 1 d, E. E. de Turk, Ind. Eng. Chem., Anal. Ed., 12, 396 (1940).
3 J. J. S c h 1 о s i n g, C. r., 37, 858 (1853); Jahresber., 654 (1854); F. T i e m a n n, Ber., 6, 1041 (1873); P. W a g n e r, Chem. Ztg., 7, 1710 (1883); 8, 475 (1884); Верль — Л у н г e, Химико-технические методы исследования, ОНТИ, Хпмтеорет, 1936.
4 О. Forster, Chem. Ztg., 13, 229 (1889); 14, 1674 (1890); Н. С. Moore,
J. Ind. Eng. Chem., 12, 669 (1920); J. Assoc. Offic. Agr. Chemists, 8, 411 (1924—1925).
6 A. D e varda, Chem. Ztg., 16, 1952 (1892); Z. anal. Chem., 33, 113 (1894); J. Assoc. Offic. Agr. Chemists 8, 410 (1924—1925).
6 При определении общего азота в растворах, содержащих органический и нитратный азот, В. S. Da visson и J. Т. Parsons [Ind. Eng. Chem., И, 306 (1919)] получили очень хорошие результаты, сначала восстанавливая нитраты сплавом Деварда в разбавленном щелочном растворе при поглощении аммиака серной кислотой, а затем разлагая органическое вещество, как указано на стр, 863.
55 Заказ 522.
866
Гл. Ы. Азот
Растворяют навеску в 10 г анализируемой пробы в воде и разбавляют раствор точно до 1000 мл. Переносят 50 мл раствора в коническую колбу емкостью 600 мл и перемешивают с 60 мл воды, 5 мл спирта и 40 мл раствора едкого кали (пл. 1,3г/сл43). Вводят 2—2,5 г сплава Деварда, измельченного в тонкий порошок, и немедленно соединяют колбу с трубкой и холодильником, конец которого погружен в определенное количество титрованного раствора кислоты. Слабо нагревают 30 мин, а затем перегоняют, вначале очень осторожно.
Вводят поправку на «холостой» опыт, который следует проводить с каждой новой партией сплава Деварда.
По Деварда, вся операция может быть выполнена в продолжении 1ч. Точность метода доказывается результатами, полученными Деварда при анализе химически чистых нитратов калия и натрия, в которых он определил 13,88% и 16,46% при теоретическом содержании 13,86 и 16,46% N. Точность метода по сравнению с сульфосали-цилатным характеризуется следующими средними результатами, полученными 20 химиками х:
Метод Деварда Метод сульфосалицилатный
ртуть связана Na,S ртуть связана Na2S гО>
15,55 15,55 15,57
12,49 12,45 12,48
15,13 15,08 15,17
Технический NaNO3, %N . .
Технический KNO3, %N . . .
Чистый KNO3, %N............
Оптимальными условиями восстановления 1 г нитрата являются добавление 3 г сплава и 2 г едкого натра приблизительно на 250 мл воды 1 2 3 4 . Приводится 3 видоизмененный метод, в котором едкая щелочь заменена окисью магния; аммиак и нитратный азот этим методом можно определять в одном и том же растворе.
Азот, находящийся в пробе в виде нитрата, нитрита, сульфонитрата или нитроклетчатки, можно определять обработкой тончайшим алюминиевым порошком (матовосерым, а не блестящим, который применяется для «серебряной» окраски), медленным добавлением раствора едкого натра (пл. 1,320 г/с.те3) и поглощением аммиака титрованной серной кислотой4.
Определение в присутствии азосоединений5. Растворяют 0,2—0,4 г анализируемого вещества в 20 мл спирта, прибавляют 5 мл раствора 40 г хлорида олова (II) в 100 мл соляной кислоты и нагревают в колбе с обратным холодильником при температуре начала кипения до обесцвечивания раствора, а затем еще 7 мин (в общей сложности не менее 15 мин). По охлаждении прибавляют равный объем воды и 30 мл серной кислоты, после чего нагревают до удаления воды и прекращения вспенивания. Вводят 0,7 г окиси ртути (II) и 10 г сульфата калия и в дальнейшем поступают, как описано на стр. 863. Если улетучивается такая значительная часть серной кислоты, что сульфат олова (II) начинает давать толчки, прибавляют еще 5—10 мл серной кислоты.
Определение в присутствии соединений гидразина. Соединения гидразина и семикарбазида не разлагаются описанными выше методами.
Имеется указание 5, что гидразин, фенилгпдразин, фенилметилгид-разин и т. и. соединения разлагаются при обработке спиртовых раство-
1 J. Assoc. Offic. Agr. Chemists, 8, 415 (1924—1925).
2 M. В. D о n a 1 d, Analyst, 61, 250 (1936).
3 J. Davidson, A. Krasnitz, Ind. Eng. Chem., Anal. Ed., 6, 315 (1934).
4 A. Seyewetz, Bull. soc. Chim., [4], 45, 463 (1929).
6 I. K. Phelps, H. W. D and t, J. Assoc. Offic. Agr. Chemists, 3, 306 (1920). Согласно данным этих авторов, метод был успешно применен для анализа азобензола, гидразобензола, толуол-азо-паратолуидина, метилового красного, дпэтплового красного, дипропилового красного, бензол-азо-р-нафтиламина, понсо 4R и конго квасного.
Методы определения
867
ров этих соединений формальдегидом, цинковой пылью и концентрированной соляной кислотой при кипячении (не менее 30 мин) с обратным холодильником и добавлением спустя 15 мин небольшого количества хлорида олова (II) для ускорения реакции между цинком и кислотой. Раствор после этого разбавляют равным объемом воды и обрабатывают предыдущим методом. Цинковая пыль должна быть очень тонкоизмельченпой и по меньшей мере 90%-ной чистоты.
Осаждение в виде азотнокислого нитрона
Нитрон [продажное наименование 1,4-дифенил-3,5-(эндоанил)-ди-гидро-1,2,4-триазола] 1 является органическим основанием, которое образует малорастворимые соединения с некоторыми кислотами. Наиболее важными из них в порядке растворимости (начиная с наиболее растворимого) являются бромид, нитрит, хлорат, хромат, роданид, иодид, нитрат, перхлорат и пикрат. Имеется указание, что щавелевая и лимонная кислоты также образуют более или менее мало растворимые соли с нитроном.
Нитрон находит применение главным образом для определения азотной, хлорной и пикриновой кислот, органических нитратов, азотнокислых эфиров, нитросоединений и окислов азота 2.
Из соединений, не мешающих определению, следует упомянуть декстрин, желатину, сахарозу, йодаты, алюминий, сульфаты магния и аммония, фосфат калия и хлорид магния. Серная,соляная, фосфорная, борная, муравьиная, уксусная, бензойная и винная кислоты образуют с нитроном растворимые соли.
Мешают кислоты, образующие нерастворимые соли, гексацианоферраты (II) и (III), пептон и большие количества хлоридов.
Ход определения,. Раствор, содержащий не более 0,1 г нитрат-ионов и свободный от перечисленных выше мешающих веществ, разбавляют до 80—100 мл, прибавляют 0,5—0,75 мл разбавленной (2 : 3) серной кислоты или 1 мл ледяной уксусной кислоты и нагревают до кипения. В один прием вводят 10—12 мл раствора нитрона 3, перемешивают и оставляют стоять 30—45 мин, а затем охлаждают 1,5 ч в ледяной воде. Фильтруют через взвешенный тигель при умеренном отсасывании, используя фильтрат для перенесения осадка в тигель и ополаскивания стакана.
1 Формула соединения:
пли C2oH4gN4
Азотнокислый нитрон имеет формулу C20H1(iN4 • HNO3.
2 Применение нитрона для определения рения см. стр. 343. Подробный обзор методов применения интрона в анализе неорганических и органических продуктов см. W. С. Соре, J. В а г a b, J. Am. Chem. Soc., 39, 504 (1917).
3 Приготовляют следующим образом. Растворяют 1 г нитрона в 10 мл 5%-ной уксусной кислоты, фильтруют через фильтрующий тигель, переносят в склянку из оранжевого стекла и сохраняют в холодном месте.
55*
868
Гл. LI. Азот
Под конец промывают осадок 5 раз ледяной водой, порциями по 2 мл. Чтобы удостовериться в полноте осаждения, нагревают фильтрат до кипения, прибавляют еще немного осадителя и охлаждают, как прежде. Тигель с осадком сушат при 105° С в течение 1 ч и по охлаждении в эксикаторе взвешивают. Эмпирический фактор пересчета на HNOa равен 0,1679.
Результаты определения нитрат-ионов обычно получаются несколько пониженными Ч
Другие методы
Для определения общего содержания азота в цианамиде кальция рекомендуется ? смачивать 1 г пробы в колбе Кьельдаля водой, смешивать с 1 г порошка меди и осторожно обрабатывать на холоду 30 мл холодной 50%-ной серной кислоты. После этого раствор нагревают 1 ч при невысокой температуре, затем 30—60 мин при более высокой температуре, и, наконец, отгоняют, как обычно 3.
Имеются указания 4, что при определении азота в таких соединениях, как солянокислый фенилгидразин, антипирин, бруцин, „м-динитробензол, цианамид кальция и нитраты, хорошие результаты получаются при применении следующего метода. Помещают 0,15 г пробы в сухую колбу Кьельдаля и растворяют в 5 мл воды. Прибавляют хорошо перемешанную смесь 7 г железных опилок или порошка железа на 1 г окиси меди, тщательно смешивают взбалтыванием в течение 1 мин так, чтобы все железо осело на дно колбы. После этого вводят 20 мл хорошо охлажденной разбавленной (1 : 1) серной кислоты и снова тщательно взбалтывают. Если реакция протекает бурно, то колбу охлаждают. Оставляют стоять 1 ч, прибавляют 30 мл серной кислоты, осторожно нагревают 15 мин, а затем, как обычно, в продолжение 1—3 ч в зависимости от скорости разложения соединения. Обесцвечивание раствора не является признаком полноты разложения. На присутствие остатка серы, непрореагировавшего железа и сульфатов меди и железа не следует обращать внимания.
Имеется указание 5, что азот алифатических нитритов, циансодержащих замещенных эфиров, амидов и имидов может быть количественно связан в виде хлорида аммония нагреванием с избыточным количеством соляной кислоты в запаянной трубке в течение 2—3 ч при температуре 200° С. Азот гликоля количественно связывается при этой температуре только по истечении 3—4 ч. После разложения раствор выпаривают досуха па водяной бане и остаток нагревают в сушильном шкафу 5 мин при 110—112° С для полного удаления соляной кислоты 6.
Содержание азота вычисляют на основе результатов титрования хлорида нейтральным титрованным раствором нитрата серебра по методу Мора (стр. 814).
1 Результаты исследования этого метода приведены в статье J. Е. Heck, Н. Hunt, М. G. Mellon. Analyst, 59, 18 (1934).
2 К. Scherrer, Chem. Ztg.', 49, 237, 243 (1925).
3 Определение мочевины и нитратного азота в присутствии цианамида см. Е. G. Fox, W. J. G е 1 d а г d [Ind. Eng. Chem., 15, 743 (1923)] и К. D. Jacob [там же 1175].
4 К. Kiirscher, Z. anal. Chem., 68, 209 (1926).
5 W. A. Drushel, М. М. Brand ege е , Am. J. Sci., [4], 39, 398 (1915).
6 W. A. Drushel и M. M. Brandegee нашли, что при нагревании 1,2377 г NH4C1 в течение 10 мин при ПО—112° С потеря в массе составляла всего лишь 0,0002 г, а после получасового нагревания при 105—108° С — только 0,0005 г.
Методы определения
869
Азот многих органических соединений может быть превращен в аммиак нагреванием в токе водорода и пропусканием смеси газов через асбест, покрытый никелем 4.
Определение очень малых количеств азота, находящегося в виде аммиака или нитрата
Для определения очень малых количеств аммиака или нитратов, как, например, в питьевой воде, применяют колориметрические методы 1 2: метод Несслера 3 4 5 для определения аммиака и фенолдисульфановый метод 4 для определения нитратов.
Определение очень малых количеств свободного аммиака. Сначала приготовляют реактив Несслера следующим образом. Растворяют около 13 г иодида калия в 25 мл воды и прибавляют при непрерывном перемешивании холодный насыщенный раствор хлорида ртути (II) до прекращения растворения образующегося осадка. Необходимо следить за тем, чтобы в раствор не попали кристаллы хлорида ртути (II). Остающийся нерастворенным осадок должен быть крайне незначительным. Раствор фильтруют и прибавляют раствор едкого кали.
Для приготовления раствора едкого кали растворяют 55 г едкого кали в 150 мл воды. Раствор оставляют стоять в закрытой пробкой колбе, пока он не станет прозрачным, после чего его сливают декантацией.
После добавления едкого кали разбавляют раствор до 250 мл, размешивают и прибавляют по каплям, непрерывно перемешивая, насыщенный раствор хлорида ртути (II) до появления едва заметного желтоватого иеисчезающего осадка. Дают отстояться в закрытой склянке и затем сливают прозрачный раствор декантацией. Сохраняют раствор в закрытой склянке в темном месте. Приготовленный таким образом реактив должен иметь желтоватый цвет. По прибавлении 0,5 мл реактива к 10 мл дистиллированной воды, содержащей 0,5 мл стандартного раствора хлорида аммония (0,00001 г азота в виде NH3 в 1 мл), в течение 1 мин должна появиться буроватая окраска, а после прибавления 0,5 мл реактива к 10 мл воды, содержащей 8 мл раствора хлорида аммония, не должно происходить помутнения 5.
1 Н. ter Meulen, Rec. trav. chim., 43, 643 (1924); 44, 271 (1925).
2 Подробное описание этих методов можно найти в Standard Methods for the Examination of Water and Sewage 9th ed., American Puhlic Health Association, Boston, 1946 или Official and Tentative Methods of Analysis of Association of Official Agricultural Chemists, 7th ed., Washington, D. C. 1950.
3 W. A. M i 1 1 e r, Z. anal. Chem., 4, 459 (1865); J. N e s s 1 e г, там же, 7, 415 (1868); E. F r a n k 1 a n d, H. E. Armstrong, J. Chem. Soc., [2], 6, 77 (1868); H. Trommsdorff, там же, 8, 356 (1869).
4 H. S p r e n g e 1, Pogg. Ann., 121, 188 (1864); A. Grandval, H. Lajoux, C. r., 101, 62 (1885); E. M. C h a m о t, D. S. P r a t t, J. Am. Chem. Soc., 31, 922 (1909); 32, 630 (1910); E. M. C h a m о t, D. S. P r a t t, H. W. Redfield, там же, 33, 366, 381 (1911).
6 A. P. V a n s e 1 о w [Ind. Eng. Chem., Anal. Ed., 12, 517 (1940)] рекомендует приготовлять реактив следующим образом. Растворяют 45,5 г иодида ртути (II) и 34,9 г иодида калия в возможно меньшем количестве воды, прибавляют 112 г едкого кали [140 .мл почти насыщенного раствора (d45 = 1,538)], разбавляют до 1 л и перед употреблением оставляют стоять в продолжение 1 недели. При проведении анализа
5 мл этого раствора прибавляют к 100 мл анализируемого раствора и перед сравнением со стандартами раствор оставляют стоять в течение 30 мин.
870
Гл. LI. Азот
Ход определения,. Наливают 50 мл воды в тщательно очищенную реторту, прибавляют 2 мл насыщенного раствора карбоната натрия, соединяют реторту с вертикальным холодильником, отгоняют 30 мл жидкости и затем собирают 10 мл дистиллята в цилиндр Несслера. По охлаждении до комнатной температуры прибавляют к раствору в цилиндре 0,5 мл реактива Несслера и оставляют стоять 5 мин. Если при этом окраска не появляется, то это указывает на отсутствие в приборе аммиака. Если же раствор желтеет, прибавляют в реторту еще воды и повторяют отгонку до полного удаления аммиака.
После этого вводят в реторту 100 мл анализируемого раствора и перегоняют со скоростью 10 мл в 2 мин, собирая по 10 мл дистиллята в цилиндры Несслера такой же емкости. После отгонки 5 порций жидкости по 10 мл собирают шестую порцию, добавляют к ней реактив Несслера и ставят в горячую воду (30—40° С). Если желтая окраска не появляется, прекращают перегонку, а в противном случае собирают в общей сложности 8 фракций. По охлаждении дистиллятов до комнатной температуры прибавляют к каждому из них по 0,5 мл реактива Несслера, в первую очередь к последней порции. Если в третьем дистилляте появляется интенсивная окраска, то надо полагать, что в первом и втором дистиллятах содержатся слишком большие количества аммиака для непосредственного определения. В этом случае разбавляют второй дистиллят точно до 50 мл, перемешивают и испытывают 10 мл этого раствора. Если окраска слишком интенсивна, разбавляют 10 мл уже разбавленного раствора до 50 мл. Подобным же образом обрабатывают первый дистиллят.
Определяют содержание аммиака в каждой фракции дистиллята отдельно следующим образом. В цилиндры Несслера емкостью 10 мл вводят 0,5, 1, 2, 3, 4 и 5 мл раствора хлорида аммония, содержащего 0,00001 г азота в 1 мл. Разбавляют до метки водой, свободной от аммиака, прибавляют в каждый цилиндр по 0,5 мл реактива Несслера, тщательно перемешивают и спустя 5 мин сравнивают с этой серией стандартов окраску анализируемых растворов. Все растворы должны иметь одинаковую температуру. Интенсивность окраски растворов, содержащих равные количества аммиака, должна быть, естественно, одинаковой.
Определение очень малых количеств нитратов. Фенолдисульфоновый реактив не должен содержать моносульфоновой кислоты. Его приготовляют следующим образом. Растворяют 25 г чистого белого фенола в 150 мл чистой концентрированной серной кислоты, прибавляют 75 мл олеума (13% SOs) тщательно перемешивают и нагревают 2 ч при температуре около 100° С. 2 мл этого реактива достаточно для открытия 50 мг/л нитрат-ионов.
Ход определения. Переносят в фарфоровую чашку 100 мл анализируемого раствора, а в случае содержания больших количеств нитратов такой объем, в котором содержится от 0,01 до 0,05 мг нитратного азота. Прибавляют 0,04 н. или 0,02 н. серную кислоту в количестве, достаточном для почти полной нейтрализации щелочи, а затем добавляют титрованный раствор сульфата серебра (свободного от нитратов и содержащего 4,3969 г Ag2SO4 в 1 л) с таким расчетом, чтобы в растворе осталось около 0,5 мг хлора (содержание хлора предварительно определяют в отдельной порции раствора).
Операцию отделения хлоридов можно опустить, если содержание их в анализируемом растворе ниже 30 мг/л. Нагревают раствор до кипения,
Методы, определения
871
прибавляют немного пасты гидроокиси алюминия \ фильтруют и промывают осадок небольшими количествами воды. Фильтрат выпаривают досуха, прибавляют 2 мл фенолдисульфонового реактива и растирают стеклянной палочкой. Если получается компактный или стекловидный остаток, нагревают несколько минут на водяной бане, разбавляют дистиллированной водой и медленно прибавляют концентрированный 10—12 н. раствор едкого кали до получения окраски максимальной интенсивности. Переносят раствор в цилиндр для колориметрирования, фильтруя, если это требуется, и сравнивают окраску раствора с окраской стандартных растворов нитрата калия.
Стандарты можно приготовить следующим образом. Растворяют 0,72 г чистого перекристаллизованного KNO3 в 1 л воды и выпаривают 10 мл этого раствора досуха на водяной бане. Быстро и тщательно смачивают остаток 2 мл фенолдисульфоновой кислоты и разбавляют до 1 л. 1 мл такого раствора соответствует 0,001 мг нитратного азота. Стандарты для сравнения приготовляют, прибавляя по 2 мл раствора едкого кали к различным количествам указанного раствора KNO3 (например, 0, 0.,05, 1,5, 2,4, 6, 8, 10, 15, 20 и 40 мл) и разбавлением до 50 мл в цилиндрах Несслера. Такие стандартные растворы сохраняются без изменения в продолжение нескольких недель.
Нитриты мешают определению, если присутствуют в количестве, превышающем 1 мг!л нитритного азота. Их следует превратить в нитраты кратковременным нагреванием анализируемого раствора и добавлением несколько раз по несколько капель перекиси водорода (свободной от нитратов). На образовавшийся при этом нитрат необходимо ввести поправку.
1 Приготовляют осаждением А1(ОН)3 из раствора сульфата алюминия едким натром, который вводят в незначительном избытке. Осадок гидроокиси алюминия отфильтровывают, промывают, растворяют в НС1 и затем повторяют осаждение и промывание осадка.
Часть третья
АНАЛИЗ
СИЛИКАТНЫХ ПОРОД
Глава LII
ВВЕДЕНИЕ В АНАЛИЗ СИЛИКАТНЫХ ПОРОД
ЗНАЧЕНИЕ ПОЛНЫХ АНАЛИЗОВ
Изучение состава различных минералов, многие из которых образуют горные породы и являются основными ингредиентами земной коры, было излюбленной темой крупнейших химиков первой половины XIX столетия: современные минералогия и геология очень многим обязаны усердию и точности Берцелиуса, Велера и других исследователей. Если принять во внимание их ограниченные возможности в отношении лабораторного оборудования и чистоты реактивов, то достигнутые ими прекрасные результаты кажутся почти чудесными. Впоследствии, в тесной связи с анализом минералов, появился и анализ более или менее сложных смесей их — горных пород. В течение ряда десятилетий многие химики трудились над исследованием этих веществ, выполняя ежегодно сотни более или менее полных и точных анализов, чтобы помочь геологии и петрографии в деле изучения горных пород. По мере роста и необыкновенного развития органической химии неорганическая химия стала постепенно отходить как бы на задний план. Во многих даже лучших европейских учебных заведениях курс минерального анализа, хотя и удержался как часть программы обучения, но стал своего рода введением ко все расширяющемуся изучению соединений углерода. Число этих соединений быстро увеличивалось и, таким образом, открывались доступные и удобные объекты для оригинальных исследований. Это казалось более привлекательным для молодых химиков, чем разработанная, порою кажущаяся исчерпанной, область неорганических соединений. На одного студента, посвящающего себя исследовательской работе в области неорганической химии, приходилось, может быть, пять-десять, работавших над сооружением огромного здания современной химии углерода. Обучение студента минеральному анализу ограничивалось указанием простых методов разделения наиболее обычных составных частей, встречающихся в значительных количествах, причем мало обращалось внимания на возможность присутствия некоторых элементов в виде «следов» и еще меньше делалось попыток установить, действительно ли охватывает принятый список все элементы, содержащиеся в данном минерале или породе.
Введение в петрографию улучшенных методов исследования, особенно применение тонких шлифов и тяжелых жидкостей, оказало существенную помощь немногочисленным химикам, занятым исследованием
876
Гл. LII. Введение в анализ силикатных пород
горных пород: с одной стороны, оказалось возможным довольно уверенно идентифицировать отдельные минералы, входящие в состав горной породы, а с другой — явилась возможность химического исследования отдельных ингредиентов, более или менее полно отделенных от других составных частей. Во многих случаях применение микроскопа устранило необходимость в скучных и утомительных качественных испытаниях, а применение тяжелых жидкостей, позволившее проводить обогащение п выделение определенных составных частей, облегчило открытие таких элементов, существование которых долго оставалось неизвестным.
Между тем по мере развития химии были постепенно открыты новые реактивы и разработаны новые методы качественного распознавания и количественного разделения и определения. Была доказана несостоятельность существовавшего мнения о достаточности нескольких хорошо установившихся методов анализа. Некоторые из этих методов пришлось отбросить совершенно, другими пользуются до сих пор после внесения изменений. В этом освещении стало, таким образом, возможным объяснение многих до тех пор непонятных колебаний состава некоторых видов и типов горных пород, обнаруженных в более ранних анализах: в значительном числе случаев оказалось, что отсутствие определений одного или нескольких элементов затемнило понимание минералогического состава породы, что и выяснилось при более тщательном исследовании (стр. 877—878). Следствием этого явилось чувство недоверия ко многим из более старых работ, возникшее среди тех химиков и петрографов, которые более всего были в состоянии судить о вероятном качестве этих работ. Это обстоятельство, а также недостаточная полнота почти всех более ранних анализов (к сожалению, и некоторых современных), как показывает сильно разросшийся перечень элементов, относительно которых теперь известно, что они входят в нормальный состав горных пород, делают старые материалы все менее и менее способными удовлетворять растущие требования петрографии г.
И все же эти требования за немногими исключениями были не так строги, как им следовало бы быть. Часто анализ доверяли студенту, не имевшему другого опыта, кроме приобретенного анализом двух-трех искусственных солей и немногих сравнительно простых минералов, причем руководил им химик, опыт которого в анализе горных пород мало превосходил опыт самого студента. Другими словами, одну из наиболее трудных задач практического анализа давали новичку, а результаты принимали благосклонно и широко публиковали без критического рассмотрения.
Даже для химиков, хорошо знакомых с анализом горных пород, он является сложной задачей и часто большим для них испытанием. Хотя долгая практика дает возможность аналитику проводить некоторые части работы почти механически, но все же возникают недоуменные вопросы, требующие для их разрешения развитой способности аналитического мышления и рассуждения; здесь еще можно найти большой простор для
1 Н. S. Washington (U. S. Geol. Survey «Professional Papers», № 99 и № 28) проделал весьма важную работу, пересмотрев громадное число данных, накопленных в тысячах анализов, опубликованных с 1869 г. После систематического критического исследования он дал каждому анализу определенную оценку по пятибалльной системе. Многие из этих оценок могут быть неверны, но в большинстве случаев они, несомненно, служат хорошим мерилом для оценки отдельных анализов.
Значение полных анализов
877
серьезно!! работы над некоторыми количественными определениями, обычно считающимися весьма простыми. Если результаты должны представлять известную ценность для научного использования их, они должны быть получены работником, способным справиться с трудностями анализа, в котором подчас приходится выделять и определять с наибольшим приближением к точности от 15 до 25 составных частей; само собой разумеется, что начинающий аналитик не может надеяться достигнуть этого. Добросовестный химик должен иметь живой интерес к своему делу. Он должен работать, имея в виду двоякую цель: облегчение труда последующих работников, предоставление им возможности использовать его работу с меньшим числом дополнительных исследований и повышение своей собственной репутации, чтобы заслужить благоприятную оценку работе, когда последняя выдержит испытание временем.
Как мало понимались раньше основные положения анализа таких сложных веществ и необходимость точного выделения и определения их составных частей, даже когда эти части сравнительно немногочисленны, видно из следующих фактов. Из специалистов химиков-аналитиков были созданы комиссии для исследования методов, применяемых в различных областях технической химии, в том числе методов анализа цинковых руд, шлаков, получаемых при выплавке медных руд, глинистых известняков и цементов. Оказалось, что получить сходящиеся результаты их анализов было невозможно, и это даже тогда, когда анализы проводились наиболее опытными в каждой области химиками. В результате последующих исследований, предпринятых этими комиссиями, и выработанных ими инструкций было достигнуто некоторое улучшение в промышленном анализе силикатов; в будущем можно ожидать дальнейшего улучшения. Значительно большего удалось добиться в отношении анализов, проводимых в научных учреждениях с целью научного исследования, однако до настоящего времени постановка обучения аналитической химии в наших учебных заведениях далеко не удовлетворительна и требуется более внимательное к ней отношение.
Петрографы, со своей стороны, должны добиваться того, чтобы нужные им анализы проводились насколько возможно полно, а не довольствовались бы, как это часто случается, определениями кремнекислоты, окиси алюминия, окислов железа, кальция, магния, щелочных металлов и воды. Такие сокращенные анализы, правда, имеют иногда свои основания, так как их, несомненно, можно использовать для некоторых целей. Однако при таких неполных анализах не только может остаться незамеченным многое, что представляло бы большую ценность для исследователя, но, что еще важнее, могут быть сделаны совершенно неверные заключения. Нам пришлось наблюдать достаточное число примеров таких неверных заключений, и мы имеем веские основания настаивать на большей полноте анализов горных пород и минералов, проводимых с чисто научной целью х.
Большое значение изложенных выше положений подтверждается данными табл. 27, где приведены результаты двух анализов одной горной породы. Один из этих анализов был проведен давно, другой — в настоящее время. Хотя между двумя анализами был большой промежуток
1 Подробнее по этому вопросу с точки зрения петрографии см. Н. S. W a s h i n g-t о n, Manual of the chemical analysis of rocks, 1919, p. 7—17.
878
Гл. LII. Введение в анализ силикатных пород
Анализы образцов, взятых из одного и того же массива горной породы в разное время
ТАБЛИЦА 27
Составные части Более ранний анализ % Анализ, проведенный в настоящее время 1 % Составные части Более ранний анализ % Анализ, проведенный в настоящее время 1 %
SiO2 .... 54,42 53,70 Н2О (ниже 110° С) 0,80
TiO2 — 1,92 Н2О (выше 110° С) 2,76 3 2,61
А120з .... 13,37 11,16 СО2 1,82 —
Сг2О3 .... — 0,04 Р2О5 — 1,75
Fe2O3 .... 0,61 2 3,10 so3 — 0,06
FeO 3,52 2 1,21 F — 0,44
MnO —. 0,04 Cl — 0,03
СаО 4,38 3,46
SrO — 0,19
BaO — 0,62 99,58 100,40
MgO 6,37 6,44 Уменьшение со-
K2o 10,73 11,16 держания О за
Na2O .... 1,60 1,67 счет F . . . . — 0,19
Li2O следы следы
100,21
1 Еще более новый анализ другого образца породы показал, что, по-видимому, приведенный здесь более поздний анализ сам является отчасти неполным и неточным: неполным—потому, что материал содержит еще 0,2% или больше Zr02; неточным—отчасти из-за ошибки в содержании А12Оэ, происшедшей вследствие того, что ZrO2 была принята за А12О3, и еще потому, что в присутствии циркония титан осаждается не полностью по примененному методу Гуча. Эта последняя ошибка влияет на результат определения TiO2 и А12Оз (стр. 967).
2 Тот факт, что повторные определения окислов железа в этой и исходных породах из той же местности всегда показывали значительное преобладание окиси железа (III) по сравнению с окисью железа (II), дает основания предполагать, что результаты, полученные для обоих окислов в первом анализе, были случайными.
3 Из опубликованных результатов анализа неясно, вся ли это вода, или, что более вероятно, только та, которая остается после высушивания при 100° С.
времени и проводились эти анализы разными лицами, все же можно быть уверенным, что пробы были взяты из одного и того же массива горной породы, в котором относительное содержание различных составляющих эту горную породу минералов хотя и могло колебаться в некоторых пределах, но не настолько, чтобы это могло объяснить расхождения между двумя анализами.
Другой пример подобного же рода приведен в табл. 28. Здесь опять некоторые различия могут быть объяснены и естественными изменениями соотношений минералов, составляющих породу, но вряд ли можно сомневаться в том, что TiO2, BaO, SrO, P2OS и SO3 содержались в обоих образцах приблизительно в одинаковых количествах. В более раннем анализе определения некоторых составных частей, которые считались не имеющими значения, были намеренно пропущены, или эти составные части определены только качественно, и в результате были получены цифры, которые могут только помешать полному выяснению минералогической природы данной горной породы.
Кларк показал, что содержание окислов титана и фосфора в горных породах земной коры при выводе среднего из сотен анализов в сумме достигает 0,8%. Когда определение этих окислов пропускается, всю ошибку следует относить за счет окиси алюминия. Если по количеству
Значение полных анализов
879
Анализы образцов, взятых из одного и того же массива горной породы в разное время
ТАБЛИЦА 28
Составные части Более ранний анализ % Анализ, проведенный в настоящее время % Составные части Более ранний анализ % Анализ, проведенный в настоящее время о/ /О
SiO2 44,31 44,65 Na2O 4,45 5,67
TiO2 не опр. 0,95 Ы2О .— следы
А12О3 .... 17,20 13,87 Н2О(ниже 110° С) 0,77 0,95
Fe2O3 .... 4,64 6,06 Н2О (выше 110 °C) — 2,10
FeO 3,73 2,94 Н2О (при прока-
MnO 0,10 0,17 лпвании) . . 3,30
CaO 10,40 9,57 со2 — 0,11
SrO — 0,37 1 Р2О5 — 1,50
BaO — 0,76 С1 — следы
MgO 6,57 5,15 SO3 — 0,61
K2O 3,54 4,49
99,11 99,92
1 Не совсем свободна от окиси кальция.
окиси алюминия вычисляют содержание полевого шпата, то легко увидеть, что ошибка в содержании последнего может доходить до нескольких процентов по отношению к горной породе.
Чтобы еще более резко подчеркнуть значение полноты анализа, можно сослаться на факты, обнаруженные сотнями анализов, выполненных в лаборатории Геологического института США (U. S. Geological Survey). Было показано самым убедительным образом, что барий и стронций почти всегда присутствуют в изверженных породах США и во многих продуктах их изменения. Количество окисла каждого из этих металлов в породах обычно ниже 0,1%, но более высокое содержание отнюдь не является редкостью. Далее, содержание бария почти всегда превосходит содержание стронция; но еще более важно то, что в изверженных породах области Скалистых Гор, насколько было исследовано, в среднем содержание обоих металлов гораздо выше, чем в породах из более восточных и более западных частей США. Следующие примеры служат для иллюстрации некоторых типов изверженных пород Скалистых Гор. Из семи пород, полученных из Колорадо, шесть содержали от 0,13 до 0,18% ВаО, а седьмая содержала 0,43%. Содержание SrO колебалось от 0,07 до 0,13% для шести пород и доходило до 0,28% для породы с наивысшим содержанием ВаО. В тринадцати геологически родственных горных породах из Монтана, включающих как основные, так и кислые и промежуточные виды породы, колебания в содержании ВаО были от 0,19 до 0,37 %, в среднем 0,30%. Три другие породы из той же серии содержали 0,1% ВаО и меньше, а семнадцатая имела 0,76% ВаО. Содержание SrO колебалось от 0,37'16 в семнадцатой породе до 0,06% в среднем в шестнадцати других. Некоторые особые породы из Уйоминга содержали от 0,62 до 1,25% ВаО и от 0,02 до 0,33% SrO. Несомненно, эта концентрация некоторых химических элементов в известных географических зонах имеет значение,
880
Гл. LII. Введение в анализ силикатных пород
объяснить которое предстоит будущим геологам, если современные не смогут это сделать.
Ванадий также является элементом, присутствие которого в изверженных породах пытались обнаружить лишь немногие химики, хотя давно известно, что он встречается в магнетитах и других железных рудах. А. А. Хейс в 1875 г. отметил его присутствие во многих породах и рудах. Как указывает «Химический словарь» Торпе, «по-видимому ванадий вместе с титаном рассеян во всех первичных гранитных породах (Дьёлафэ) и был найден Девилем в боксите, рутиле и многих других минералах, а Бечи и другими — в золе растений и глинистых известняках, сланцах и песках». Далее, было отмечено присутствие ванадия в виде пятиокиси в колическах, достигающих 0,1 %, во многих французских и австралийских глинах \ от 0,02 до 0,0396 в некоторых базальтах, 0,2496 в угле неизвестного происхождения и 0,45% в угле из Перу. Еще более поздние исследования (проведенные в лаборатории Геологического института США) примерно 100 горных пород, главным образом изверженных, покрывающих почти всю территорию США, показывают не только широкое распространение ванадия, но и то, что он преобладает в породах, бедных кремниевой кислотой, и отсутствует или почти отсутствует в породах, богатых ею. В некоторых более основных породах ванадий встречается в таких количествах, что можно серьезно повлиять на результаты определения окислов железа, если его не определили отдельно и не приняли в расчет (см. стр. 988); это обстоятельство очень важно, потому что петрографы придают большое значение точности определения окислов железа.
Те же исследования пролили некоторый свет на распространение молибдена. Этот элемент, по-видимому, сосредоточен в более богатых кремниевой кислотой породах и встречается в количествах значительно меньших, чем ванадий.
Наконец, если бы не стало обычаем в последние годы исследовать горные породы на содержание в них серы, даже в тех случаях, когда на глаз сульфидов не было видно, могло бы остаться незамеченным почти неизменное присутствие серы в форме сульфидов и связанное с этим и долгое время остававшееся необъясненным расхождение результатов определений железа (II) методом Мптчерлиха и методом с фтористоводородной кислотой (см. стр. 989).
Настойчиво подчеркивая необходимость более точной работы, хотя бы и за счет ее количества, мы далеки от мысли требовать, чтобы на каждый анализ затрачивалось слишком большое количество времени, не соответствующее объекту исследования. Однако мы настаиваем, чтобы все составные части, присутствие которых в количествах, достаточных для определения в обычно берущихся для анализа навесках 1 2, является вероятным, были определены — хотя бы качественно — в обычном ходе количественного анализа, и их присутствие или отсутствие было отмечено в таблице результатов анализа. Если известно, что какая-либо составная часть присутствует в количестве, немногим превышающем следы, то
1 J. С. Н.М i n g а у е [Recordsgeol. Survey,’New South Wales 7 (3), 213 (1903)] подтвердил его широкое распространение в австралийских горных породах, углях и т. д.
2 Навеска должна быть величиной в 1 г — для определения SiO3, А12О3 и т. д., 2 г — для определения других компонентов.
Составные части силикатных горных пород
881
одного этого факта уже может быть достаточно, потому что часто более важно знать, присутствует ли данный элемент или нет, чем быть в состоянии сказать, что его содержится точно 0,02 или 0,06%. При сведении результатов анализов в таблицы следует всегда сделать специальное указание на то, что умышленно или случайно не было обращено внимания на элементы, присутствие которых вероятно. Пренебрегая этим, аналитик может подвергнуть себя нежелательной критике, когда в будущем его работа будет пересмотрена и его пропуски будут открыты последующими анализами.
Насколько это возможно, химическому анализу должно предшествовать тщательное микроскопическое исследование шлифа горной породы. Оно может оказать химику величайшую помощь, указывая на присутствие необычных компонентов или необычно больших количеств постоянно присутствующих компонентов, и тем самым дает возможность внести необходимые изменения в применяемый ход анализа1. Как для геолога, так и для химика лучше сделать небольшое количество тщательных исследований, чем много таких, ценность результатов которых может оказаться сомнительной.
СОСТАВНЫЕ ЧАСТИ СИЛИКАТНЫХ ГОРНЫХ ПОРОД
Почти все рудные месторождения США обязаны своим происхождением распаду материнских изверженных пород или продуктов их превращения путем того или иного процесса концентрации. Некоторые группы концентратов, вероятно, выделились из магмы в жидком состоянии до ее затвердевания. Отсюда естественно и неизбежно следует, что достаточно тщательное исследование этих горных пород обнаружило бы в них содержание почти всех известных элементов, необязательно всех в одной данной породе, но гораздо больше, чем было найдено кем-либо до настоящего времени 2 3. На пути к экспериментальному доказательству этого стояли до настоящего времени механические и аналитические трудности, обусловленные ничтожным содержанием многих элементов. В исключительных случаях, может быть, следует подвергать исследуемый материал исчерпывающему анализу из больших навесок, например при изучении происхождения рудных месторождений. Обычно, однако, требования петрографии и геологии удовлетворяются знанием количественных соотношений тех составных частей, которые могут быть определены в небольшом количестве материала, например от 0,5 до 2 г или иногда до 5 г. В настоящем руководстве рассмотрение анализа силикатов ограничено теми методами, которые могут потребоваться при анализе изверженных, метаморфических и осадочных пород сложного минералогического состава, в которых большинство, а возможно и все составные части
1 См. также Н. S. Washington, Manual of the Chemical analysis of rocks, 6—7 (1919). Приведенные таблицы и сопровождающие их замечания, а также некоторые
объяснения, предшествующие таблицам, по большей части взяты пз литературы [см. J. Am. Chem. Soc., 16, 90—93 (1894); Chem. News, 69, 1163 (1894)]. О распределении и количестве ванадия и молибдена в горных породах США см. Am. J. Sci., [4], 6, 209 (1898); Chem. News, 78, 216 (1898); Bull. U. S. Geol. Survey, j\° 167, 49 (1900).
3 Исследования F. Sandberger показали, до какой степени это верно в отношении большого количества элементов, участвующих в заполнении рудоносных жил, а L. Dieulafait в своих тщательных качественных исследованиях показал, как широко распространены медь, цинк, барий, стронций и т. п. в первичных горных породах.
56 Заказ 322.
882
Гл. LII. Введение в анализ силикатных пород
из приводимого ниже списка могут^ содержаться в весомых или легко определимых количествах:
SiO2, TiO2, ZrO2, А12О3, Fe2O3, Сг2О3, V2O3, FeO, MnO, NiO, CoO, MgO, CaO, SrO, BaO, ZnO, ;CuO, K2O, Na2O, Li2O, H2O, P2O5, S (cm. chock}1 2)
SO3, С (см. сноску 2), CO2, F, Cl, N.
К этому списку можно прибавить еще группу так называемых редкоземельных элементов, олово, платину, тантал, ниобий, бор, бериллий и гелий. Некоторые из них встречаются иногда в определимых количествах, но их легко не обнаружить в ходе анализа по причине отсутствия точных методов их идентификации. Торий, церий и другие редкоземельные элементы, вероятно, встречаются в силикатных горных породах гораздо чаще, чем это обычно полагают. Их присутствие и количество могут быть легко и точно установлены методами, указанными в своем месте, так что нет более причин пропускать их определение, как это было до настоящего времени, особенно если микроскопическим анализом или каким-либо иным образом доказано присутствие минералов, которые могут содержать эти элементы.
Как это установлено спектрографическими исследованиями 3 некоторых минералов и систематическим изучением горных пород на присут-
ТАБЛИЦА 29 Элементы, присутствующие в малых количествах в некоторых горных породах, % Тире в этой таблице означает, что содержание данного элемента ниже чувствительности примененного спектрографического метода; отточия означают, что анализ на данный элемент не проводился
Элемент Горная порода
базальтовый андезит 1 диабаз 8 гранит * риолит 4 нефелиновый сиенит *
Сг 0,23 0,013 0,003
Ni 0,015 0,0081 — — —
Со 0,0017 0,0025 0,0003 — —
Sc 0,0019 0,0055 0,0002 — 0,0005
V 0,013 0,022 0,0026 — 0,0062
Си 0,0023 0,0089 0,0015 0,0003 —
Мп 0,099 0,13 0,014 0,054 0,13
Sr 0,044 0,014 0,012 0,0034 0,019
Ba 0,074 0,038 0,13 0,013 0,0086
Pb — — 0,0022 0,0005 0,0016
Zr . 0,013 0,0096 0,027 0,028 0,069
Ga 0,0013 0,0012 0,0018 0,0024 0,0022
Y — 0,0025 0,011 0,010
La — 0,017 0,0094 0,033
Mo — 0,0007 — 0,0012
Be — 0,0002 0,0004 0,0003
В — 0,001 — — —
Nb — — — 0,0045 0,017
1 Лава из вулкана Paricutin, Мексика.
- Centerville, Virginia.
J Westerley, Rhode Island.
4 Pulaski County, Arkansas.
‘ Jemez Mountain, New Mexico.
1 Обычно в виде пирита, иногда в виде лазурита, нередко в виде пирротита.
2 В виде графита или угля.
3 W.N. Hartley, Н. Ramage, Trans. Chem. Soc., 79, 61 (1901),
Суммирование результатов анализа и пределы допустимых ошибок
883
ствие в них рассеянных элементов, к приведенному списку можно прибавить еще ряд элементов. Такое изучение проводится Геологическим управлением США. В табл. 29 показаны некоторые из полученных там результатов.
ПРЕДВАРИТЕЛЬНЫЙ КАЧЕСТВЕННЫЙ АНАЛИЗ
Проведение полного качественного анализа горной породы перед ее количественным анализом в большинстве случаев является напрасной тратой времени. Можно иногда проделать специальную качественную пробу на присутствие того или иного компонента, но в общем, если принять, что большинство возможных компонентов имеется в анализируемом материале, и соответственно этому провести количественный анализ, то в результате получится значительная экономия времени.
Это утверждение не должно быть истолковано так, что качественный анализ вообще не имеет значения. При исследовании руд, отдельных минералов, природных вод и т. п. предварительный качественный анализ часто необходим; им пользуются также для проверки чистоты осадков и фильтратов, получаемых в ходе количественного анализа.
Изучающий качественный анализ найдет в специальной литературе 1 обширный запас сведений не только о выполнении аналитических разделений, но также, что особенно важно, о полноте этих разделений и о границах возможности качественного открытия того или иного элемента. Содержание указанных статей обширнее, чем можно было бы думать, судя по их названиям, потому что в них рассматриваются и такие элементы, как бериллий, уран, ванадий, титан, цирконий и торий.
СУММИРОВАНИЕ РЕЗУЛЬТАТОВ АНАЛИЗА
И ПРЕДЕЛЫ ДОПУСТИМЫХ ОШИБОК
Как хорошо известно, полный анализ силикатной породы, который дает в сумме менее 100%, вообще является менее удовлетворительным, чем анализ, показывающий в сумме несколько больше, чем 100%. Это справедливо по многим причинам. Почти все реактивы, даже тщательно очищенные, еще содержат в себе или извлекают из применяемых сосудов загрязнения, которые иногда частично взвешиваются вместе с определяемыми составными частями породы. Пыль, попадающая в раствор во время хода анализа, может иметь заметную массу, промывание осадков может быть неполным, а если применяются большие фильтры для собирания на них небольших осадков, то они часто могут оказаться плохо промытыми.
При применении чистейших из доступных реактивов, полного набора платиновой посуды и хорошего оборудования и при работе в достаточно чистой лаборатории опытный химик имеет мало оправданий, если сумма полученных им результатов анализа не достигнет величины, лежащей
1 A. A. N о у е s, W. С. В г а у, Е. В. S р е а г, J. Am. Chem. Soc., 28, 137 (1907); 30, 481 (1908); Chem. News, 95, 89 (1907); 98, 6 (1908). См. также A. H о й е с, В. Б р э й, Качественный анализ редких элементов, ОНТИ, Гл. ред. хим. лит., 1936. * Ряд указаний по качественному анализу минералов и горных пород можно найти в книге В. А. Назаренко, Н. С. Полуэктов, Полумикрохимический анализ минералов и руд. Госхимиздат, 1950, *
56*
884
Гл. LII. Введение в анализ силикатных пород
в пределах от 99,75 до 100,50%. В табл. 30 приведены результаты анализов и их суммы, полученные аналитиками, работавшими вместе с одним из нас (Лендель). Если в нескольких из ряда сходных между собой анализов получаются суммы ниже 100 %, то это является убедительным свидетельством того, что что-то в анализе было упущено. Если получится сумма свыше 100,5%, то следует повторить отдельные части анализа, чтобы найти место, где была сделана ошибка, потому что неверно было бы считать, что ошибка в сторону увеличения равномерно распространяется на все определявшиеся составные части породы. Вполне вероятно— в действительности более чем вероятно, — что ошибка была в одном каком-либо определении, и быть может в таком, которое можно для критического суждения о горной породе с точки зрения петрографии.
ТАБЛИЦА 30
Суммы результатов анализа, которые можно ожидать при тщательной работе (в %)
Составные части Доломит X Полевой шпат Бок-СИТ Огнеупорные изделия Стекла
X + + + + известко-во-натри-евое х свинцово-бариевое X
SiO„ . 0,311 66,68 66,63 6,32 54,68 32,49 20,78 74,09 65,36
А12б5 . 0,069 18,01 18,04 55,00 37,63 59,30 69,86 0,32 0,181
Fe2O3 . 0,086 0,031 0,031 5,63 2,35 0,86 0,73 0,064 0,048
MgO . . 21,54 0,02 0,006 0,015 0,59 0,43 0,52 3,23 0,029
СаО . . 30,48 0,052 0,085 0,047 0,25 0,27 0,32 4,65 0,21
Na2O 0,08 2,37 2,38 0,18 0,36 0,89 0,54 16,63 5,73
К2О . . 0,03 12,58 12,58 0,05 1,43 2,10 2,90 0,04 8,38
TiO2 . 0,004 0,002 0,002 3,04 2,23 2,97 3,34 0,02 0,011
ZrO, . — — — 0,068 0,074 0,095 0,12 0,003 0,003
Р2о5 . 0,002 0,009 0,014 0,116 0,07 0,45 0,61 — 0,22
so3 . . 0,069 — — 0,063 — — — 0,41 0,031
MnO . 0,009 SrO 0,01 S 0,013 0,001 ВаО 0,033 0,001 ВаО 0,035 0,55 Сг2О3 0,031 СиО 0,03 V2O5 0,02 v205 0,029 V2o 0,038 0,003 As.,O3 0,031 As2O5 0,068 0,089 РЬО 17,50 ВаО 1,41
Другие компоненты Потеря при прокалива- со, 47,25 С 0,08 Н 0,008 — — v205 0,025 — — — Cl 0,047 As2O3 0,031 As»O5 0,39 Cl 0,051
НИИ . . . . 47,52 0,21 0,23 28,81 0,26 0,23 0,27 0,30 0,30
Сумма . . . . 100,04 99,99 100,03 99,99 99,91 100,10 99,97 99,92 99,98
х Аналитик Н. В. Knowles; + аналитик J. I. Hoffman.
Вашингтон расширяет допустимые пределы до 99,50—100,75%. Если анализ проведен не в платиновой посуде и не были поставлены тщательные «холостые» опыты, то, конечно, получение суммы результатов анализа выше 100,50% может быть допустимо, но приемлемость результатов, имеющих в сумме меньше 99,75%, весьма сомнительна, потому что, как уже было указано, всегда существует тенденция к получению
Суммирование результатов анализа и пределы допустимых ошибок
885
ТАБЛИЦА 31
Воспроизводимость, возможная при тщательных анализах силикатных пород (помещенные в скобках результаты получены другими методами)
1 Составные части Доломит % Полевой шпат % Боксит % Огнеупорные изделия, % Стекло, %
№ образца извест-ково-нат-риевое свинцово-бариевое
1 2 3
SiOo 0,311 66,53; 66,76; 66,66 6,34 54,56 32,40 20,74 74,07 65,35; 65,36;
0,311 66,65; 66,55; 66,65 6,33 54,68 32,58 20,82 74,17 65,35
0,310 66,65; 66,63; 66,62 6,30 54,80 74,03 (65,38; 65,38;
74,10 65,36)
А1,03 0,069 18,05; 18,07; 17,93 54,95 37,62 59,40 69,80 0,325 0,180
0,068 18,16; 18,11; 18,14 55,01 37,76 59,30 70,00 0,313 0,182
0,070 (18,01; 17,99; 17,96) (59,94) (37,56) (59,32) (69,78) 0,321
(18,00; 17,99) (55,09) — (59,18) (69,84) 0,313 г'4
Fe2O3 0,086 0,032; 0,030; 0,030; 5,57 2,33 0,82 0,76 0,064 0,048; 0,058
0,086 0,028 5,63 2,39 0,89 0,70 0,062 0,048', 0,040
0,085 (0,032; 0,031) 5,65 2,31 0,066
5,65 2,36 0,064
MgO 21,54 0,024; 0,019 0,015 0,59 0,41 0,51 3,22 0,029
21,56 (0,005; 0,007) 0,014 0,58 0,44 0,52 3,22 0,028
21,53 3,23 0,028
3,24 0,0296
3,25
3,22
3,23
3,23
СаО 30,47 0,051; 0,050! 0,053 0,044 0,25 0,26 0,32 4,71 0,211
30,47 0,053; 0,051 0,050 0,25 0,27 0,32 4,67 0,210
30,01 (0,088! 0,082) 4,61 0,210
4,63 0,208
Na,О 0,08 2,35; 2,39; 2,41 0,19 0,36 0,91 0,52 16,64 5,72
0,06 2,35 0,17 0,35 0,87 0,56 16,62 5,76
0,10 5,70
К2О 0,04 12,56; 12,58; 12,60 0,04 1,40 2,10 2,87 0,04 8,38
0,03 12,58; 12,58 0,05 1,47 2,10 2,92 0,04 8,39
0,03 0,04 8,38
0,03
TiO, 0,004 0,002; 0,002; 0,003 3,06 2,19 2,93 3,37 0,03 0,0136
0,004 0,003 3,03 2,20 2,95 3,32 0,02 0,0102
3,04 2,26 3,01 3,32 0,02 0,0102
2,25 2,99 3,35 0,02 0,0085
ZrO2 0,069 0,075 0,099 0,12 0,0027
0,065 0,071 0,090 0,12 0,0027
0,069 0,076 0,093 0,13 0,0027
0,072 0,097 0,11 0,0018
886
Гл. LII. Введение в анализ силикатных пород
Продолжение табл. 31
Составные части Доломит % Полевой шпат % Боксит % Огнеупорные изделия, % Стекло, %
№ образца извест-ково-нат-риевое свинцово-бариевое
1 2 3
Р2О6 0,002 0,002 0,009; (0,015 0,009; 0,010 0,013 0,016; 0,014) 0,114 0,120 0,115 0,07 0,06 0,07 0,44 0,47 0,44 0,64 0,61 0,59 0,22 0,22
so. 0,069 0,069 0,070 0,064 0,062 0,404 0,402 0,404 0,413 0,419 0,416 0,422 0,033 0,029
МпО 0,0086 0,0088 0,001; 0,001; 0,001 0,001 0,545 0,559 0,547 0,003 0,003 0,091 0,086
С02 47,24; 47,25 47,20; 47,27 47,28; 47,28 ВаО 0,035 0,038 0,037 Сг2О3 0,031 0,030 С1 0,047 0,047 С1 0,051 0,051
S 0,013 0,013 v203 0,025 0,025 V2O 0,02 0,02 v2o5 0,027 0,030 V2O6 0,038 0,038 ВаО 1,408 1,409 1,412 1,411
SrO 0,01 0,01 РЬО 17,48 17,48 17,50 17,52
С 0,081 0,083 0,078 0,078 As, О3 0,030 0,032 0,029 ASoOg 0,031 0,031
Потеря при прокаливании Н 0,0084 0,0080 0,0080 0,0087 0,0078 47,55 47,52 47,49 0,22; 0,19; 0,21; 0,20 0,25; 0,25 28,78 28,82 28,83 28,84 28,78 0,25 0,27 0,22 0,24 0,27 0,26 As,O-, 0,068 0,068 0,070 0,27 0,28 0,33 0,32 AS9O5 0,39 0.39 0,30 0,30 0,29
Представление результатов анализа
887
более высоких результатов, и сумма меньшая 99,75% при работе не в платиновой посуде указывает на довольно грубую ошибку аналитика.
Для допустимых колебаний результатов параллельных определений одного какого-нибудь компонента нельзя установить строгих границ. Вашингтон 1 предлагал следующие допустимые пределы отклонений, с которыми в основном согласуются и экспериментальные данные Диттриха 2: для SiO2 и других компонентов, содержание которых в породе не меньше 30 %, — от 0,2 до 0,3 %; для А12О3 и других компонентов, содержание которых лежит в пределах от 10 до 30%, — от 0,1 до 0,2?6; для компонентов, содержание которых равно 1—10%, расхождение между параллельными определениями не должно превышать 0,05—0,1%. Указанные допустимые колебания даны в процентах от массы всей исследуемой породы, а не от содержания данного компонента. Табл. 31 показывает, каких совпадений между результатами параллельных определений можно ожидать при очень тщательной работе. Были опубликованы 3 результаты анализов гранита и диабазовой горной породы, которые проводились 34 химиками, работавшими в 25 лабораториях различных стран мира. Воспроизводимость определений 15 компонентов оказалась очень мало удовлетворительной. Это говорит о том, как много еще надо сделать для дальнейшего развития аналитической химии горных пород и искусства аналитиков.
Несмотря на то что иногда желательно, а для начинающих аналитиков и необходимо, проводить два параллельных определения, совпадение результатов этих определений нельзя считать доказательством их правильности, если только определения не были выполнены различными методами.
Особое внимание надо обратить на то как важно проверять все получаемые в ходе анализа окончательные осадки и фильтраты, чтобы получить уверенность, во-первых, в том, что выделенньге соединения действительно те, за которые их принимают, а во-вторых, в том, что определяемая составная часть породы выделена полностью. Никаким другим способом аналитик не может приобрести необходимого доверия к себе самому и к своим методам анализа.
ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ АНАЛИЗА
В течение многих лет в лаборатории Геологического управления США было принято располагать компоненты горных пород в таблице результатов анализа в том порядке, в каком их определяли, начиная с SiO2 как главной составной части породы, и группируя вместе все химически родственные окислы, как показано, например, в табл. 27 и 28 (стр. 878).
Со строго научной точки зрения классификация, при которой основные окислы записывают отдельно от кислотных, была бы более рациональной, по пока мы еще не знаем, с какими радикалами кремниевой кислоты и в каких количествах мы имеем дело, и сколько имеется свободной кремнекислоты, бесполезно применять такую систему расположения, хотя она и оказалась ценной при выражении результатов анализа природных вод.
1 Washington, Manual of the Chemical Analysis of Rocks, 1919, p. 127.
2 Dittrich, Nenes Jahrb., 2. 69 (1903).
3 U. S. Geol. Survey Bull., 980, 7 (1951); см. также W. G. Sclileclit, Anal. Chem., 23, 1568 (1951).
888
Гл, LII. Введение в анализ силикатных пород
В настоящее время петрографы требуют (имея для этого достаточные основания) такого расположения результатов анализа, «которое давало бы наглядно необходимую химическую характеристику породы, как процентный, так и молекулярный ее состав — выпукло и сжато — так, чтобы общий химический характер породы и отношения между различными ее составными частями могли быть видны сразу» Ч
В соответствии с этим требованием в настоящее время принято расположение, предложенное Пирсоном и позднее настойчиво защищавшееся Вашингтоном, а именно:
SiO2, А12О3, Fe2O3, FeO, MgO, СаО, Na2O, К2О, Н2О (выше 105—110° С), Н2О (ниже 102—110° С), СО2, TiO2, ZrO2, Р2О5, SO3, Cl, F, S (FeS2), Cr2O3, V2O3, NiO, CoO, CuO, MnO, SrO, BaO, Li2O, C, NH3.
При таком расположении в самом начале списка помещаются девять компонентов горной породы, которые в огромном большинстве случаев определяют ее характер. Таким образом значительно облегчается сравнение анализов различных пород, особенно если, как это рекомендует Вашингтон, непосредственно за величинами, выражающими процентные содержания различных окислов, поместить рассчитанные их молекулярные отношения. Порядок расположения остальных компонентов определяется следующими соображениями: содержание СО2 помещается сейчас же за содержанием НгО, потому что оба эти окисла показывают метаморфические изменения, которые претерпела порода. Содержания TiOa и ZrOa, естественно, следуют за содержанием СО2 по химическим соображениям, a SO3 и С1, являющиеся постоянными компонентами содалитовой группы, удобно помещать рядом.
О применении термина «следы» следует сказать, что им обозначается такое количество компонента, которое не может быть количественно определено во взятой для анализа навеске пробы. В анализах, претендующих на полноту и точность, следовало бы термином «следы» обозначать количества меньшие 0,01 %.
ПРОДОЛЖИТЕЛЬНОСТЬ ВЫПОЛНЕНИЯ АНАЛИЗА
Часто ставится вопрос: «сколько времени требуется для проведения полного анализа породы?». Это зависит, конечно, от минералогической сложности анализируемой породы и от того, как работает выполняющий анализ аналитик. Если в лаборатории имеется препаратор, который проводит измельчение, и если не требуется определения плотности, то после долгой практики можно научиться так экономить каждую минуту рабочего дня, чтобы при обилии платиновой посуды и возможности непрерывного пользования, днем и ночью, воздушными и водяными или паровыми банями и при условии отсутствия случайных задержек, — через каждые три дня после выполнения первого анализа заканчивать по одному анализу из серии образцов горных пород сходного характера, содержащих каждый от 18 до 20 определяемых количественно компонентов. В число последних не входят фтор, углерод, азот, металлы сероводородной группы и кобальт.
Такой ход работы предполагает, однако, редкую независимость от случайных задержек, с которыми обычно сталкивается каждый химик.
1 Н. S. Washington, Am. J. Sci., [4], 10, 61 (1900).
Глава LIII
МЕТОДЫ АНАЛИЗА СИЛИКАТНЫХ ПОРОД
ПРЕДВАРИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
Принятый далее порядок описания различных химических разделений не учитывает химического сродства составных частей горной породы; в каждую группу здесь объединены компоненты горной породы, которые удобно определять из одной навески пробы. В главной навеске определяют SiO2, TiO2, MnO, NiO, СаО, SrO, MgO, общее содержание железа и суммарную массу смеси А12О3, TiO2, Р2О5, ZrO2, всего железа в виде Fe2O3 и почти всего (если не всего) ванадия в виде V2O5, а также и редкоземельных металлов, если они присутствовали в пробе. В отдельной навеске определяют FeO, а также и все железо и ВаО, если желают таким образом проверить результаты определения этих компонентов в других навесках. Для определения щелочных металлов нужна еще одна навеска. В отдельной навеске удобно определять ZrO2, ВаО, редкоземельные металлы и общее содержание серы. Для определения V2O3 и Сг2О3 берут еще одну, обычно значительно большую навеску. СО2, С, Н2О, F и С1 лучше определять из отдельных навесок, хотя здесь возможны различные комбинации, например, можно определять в одной навеске СО2 и Н2О, С и Н2О, F и С1.
' В действительности при надлежащем выборе и комбинировании методов можно иногда провести анализ, даже имея только 4 г анализируемого материала, без пропуска какого-либо важного определения, по в этом случае на анализ будет затрачено времени больше, чем если брать для исследования большее количество пробы.
В качестве иллюстрации преимущества, достигаемого вдумчивым комбинированием методов, может служить определение серы, бария, циркония и редкоземельных металлов из одной навески. Многие химики, определяя количественно серу, не определяют из той же навески остальных перечисленных выше компонентов, хотя констатирование их присутствия или отсутствия, если оно проводится, как указано ниже (стр. 967), требует лишь немногим большей работы, чем определение содержания одной серы.
За небольшими исключениями, почти все компоненты силикатных пород, перечисленные на стр. 882, в случае их присутствия могут быть определены в навесках измельченной породы, не превышающих 1 г каждая.
890
Гл. LIII. Методы анализа силикатных, пород
Такую навеску удобно брать для главной порции, в которой определяют кремнекислоту, окись алюминия и др., щелочноземельные металлы и магний; но более 1 г брать не следует, потому что если взять большую навеску, то осадок гидроокиси алюминия и др. будет слишком объемистым. Навеску не следует и слишком уменьшать, если требуется точное определение марганца, никеля и стронция. Для определения щелочных металлов очень удобна навеска в 0,5 г. В общем можно принять за правило не брать для анализа более 2 г пробы, если ее будут сплавлять с карбонатами щелочных металлов, как это требуется при определении серы, фтора и хлора. Для определения СОг навеска может быть увеличена до 5 г или даже более, если содержание этого компонента очень мало. При этом на определение расходуется не больше времени, чем при навеске в 1 г, а результаты получаются значительно более точными. Для определения ванадия также обычно нужна навеска, превышающая 2 г.
Чтобы получить правильные результаты, необходимые для точной классификации горной породы по принятой системе х, нужно особое внимание обратить на тщательность проведения всего анализа. Это означает не только выполнение всех обычных операций, применяемых в анализе, но и проведение J двукратных осаждений (где они необходимы) и исследование всех фильтратов и осадков, чтобы получить уверенность, с одной стороны, в полноте извлечения всех определяемых компонентов, с другой стороны — в освобождении их от всего того, чего они содержать не должны. Эти предосторожности особенно необходимы для химиков с небольшим опытом. Всегда следует помнить, что анализ в лучшем случае может дать только приближенные числа, которые тем более будут приемлемыми, чем больше труда было затрачено на их получение 1 2.
ПРИГОТОВЛЕНИЕ ПРОБЫ ДЛЯ АНАЛИЗА
Величина средней пробы, поступающей в лабораторию
Количество анализируемого материала, которое необходимо взять для приготовления аналитической пробы, может быть различным. В большинстве случаев в лабораторию поступают средние пробы горных пород, состоящие лишь из небольшого числа кусков относительно малого размера. Но если исследуется порода с резко выраженной порфировой структурой, то необходимо отбирать большую пробу, отвечающую величине находящихся в породе кристаллов, и эта большая проба поступает в лабораторию. Ее целиком измельчают и из нее квартованием берут окончательную аналитическую пробу. Если этого не сделать, то анализ даст результаты, ни в коей мере не отражающие среднего состава этой горной породы. Геолог должен следить за тем, чтобы была приготовлена действительно средняя проба, иначе работа химика, как бы она ни была тщательна, может потерять свой смысл.
1 Cross, Iddings, Pirsson, Washington, Quantitative Classification of Igneous Rocks, University of Chicago Press (1903).
2 О применении микроаналитических методов в анализе силикатных пород см. F. Hecht, Mikrochim. Acta, 2, 120, 188 (1937), *а также И. П. А л и м а р и н, Зав. лаб., 9, 365, 545, 983 (1940); И. П. А л и м а р и н, Б. И. Фрид, Зав. лаб., 9, 1217 (1940); 10, 252 (1941); И. П. А л и м а р и н, ЖПХ, № 1—2, 83 (1944); И. П. А л п-м арин, А. Я.Шескольская, Зав. лаб., И, 141, 259 (1945); ЖАХ, 3, 166 (1946); И. П. А л н м а р п н, Б. И. Фр п д, Зав. лаб., 18, 1300 (1952). *
Приготовление пробы для анализа
891
Измельчение
Рис. 37. Ступка Эллиса для измельчения крупнозернистых материалов: 1 — подставка; 2 — пестик; 3 — цилиндр.
должен точно соответ-
При выполнении точных анализов нельзя применять механические стальные дробилки и ступки ввиду опасности загрязнения пробы частицами металла и невозможности очистки шероховатых поверхностей этих приборов, какими они становятся после кратковременной работы. Извлечение при помощи магнита введенных при измельчении частиц стали совершенно недопустимо, потому что сами породы почти без исключения содержат магнитные минералы.
Методы, принятые в Геологическом управлении США. Крупные куски породы удобнее разбивать до кусков небольшой величины, пользуясь толстой стальной плиткой с особо закаленной поверхностью и закаленной колотушкой. Только нужно, чтобы закалка ее была сделана особенно тщательно.
Небольшое количество породы может быть хорошо измельчено в ступке, предложенной Эллисом (механиком геофизической лаборатории института Карнеджи); ступка эта совершенно устраняет потерю измельчаемого материала вследствие отлетания обломков. Как проводится работа в этой ступке, видно из рис. 37. Подставка, цилиндр и пестик сделаны из лучшей закаленной инструментальной стали, поверхность которой проце-ментирована насколько возможно. Размеры подставки 12,5 X 12,5 X 6 см; в середине ее имеется выемка глубиной 0,6 см. Вышина пестика 20 см, диаметр у основания 3,5 см. Вышина цилиндра 12,5 см; внешний диаметр 5 см, внутренний диаметр 4,4 см. Цилиндр ствовать углублению в подставке.
Цилиндр плотно вделан в углубление в подставке, как в обыкновенной «алмазной» ступке, но диаметр пестика меньше, чем внутренний
диаметр цилиндра.
Дробление проводится пестиком“без помощи молотка и может быть, если нужно, доведено до такой степени измельчения (см. стр. 892), что после просеивания через тонкое сито, сделанное лучше всего из шелковой ткани, получается порошок, пригодный для анализа без дальнейшего измельчения в агатовой ступке. При исследовании очень многих пород и минералов порошок, прошедший через сито, в 900 отверстий на 1 см2 (75 меш), может быть непосредственно применен для анализа. Порошок в значительной своей части состоит из частиц более мелких, чем отверстия сита, если даже интервалы между последовательными измельчениями и просеиваниями будут очень короткими.
Ни в коем случае не следует применять металлические сита, если нужно определять те металлы, из которых сделаны проволоки сита.
При измельчении пробы этим методом больше всего нужно избегать малейшего вращательного и трущего движения пестика. Даже кварц можно при отвесно направленных ударах измельчить настолько, что дальнейшее его измельчение можно проводить в агатовой ступке. Окраска
892
Гл. LIII. Методы анализа силикатных пород
его при этом нисколько не изменяется. Но если при измельчении допустить малейшее трение, то после измельчения порошок в ступке становится значительно темнее. Даже самая твердая сталь несколько истирается и менее твердыми минералами, чем кварц. В этой ступке проба должна быть доведена до такой степени измельчения, чтобы при дальнейшем истирании в агатовой ступке от движения пестика не происходило отскакивания и потери отдельных частиц породы, так как последние могут иметь иной состав, чем остающиеся в ступке, если только анализируемая порода не является стекловидной или очень мелкозернистой массой.
Если дробление проведено с достаточной тщательностью, то загрязнение пробы сталью настолько ничтожно, что оно не может вызвать заметной ошибки. Сталь в большинстве случаев истирается не в виде осколков металла, но частицы ее покрывают зерна измельченного минерала тонкой пленкой. Присутствие такой пленки можно легко обнаружить, если растолченную пробу растереть под водой в очень тонкий порошок. Пленочки металла тогда отделяются от зерен, расплющиваются все тоньше и оказываются в конце концов в виде пены на поверхности воды, откуда их при некоторой осторожности можно извлечь при помощи магнита. Такое поведение металлических листочков можно было бы, пожалуй, использовать для исследования породы на присутствие в ней металлического железа. В этом случае, естественно, нельзя было бы применять стальную ступку для измельчения пробы.
Если раздробленный материал не требуется дальше растирать, то подбирают подходящее сито, чтобы прошедший через него порошок полностью или почти полностью разлагался при 20-минутном кипячении с разбавленной плавиковой кислотой (см. ниже «Растирание» и стр. 987). Чтобы получить возможно меньшее количество «муки», т. е. слишком тонко измельченного порошка, отсеивание проводят через короткие промежутки времени. С другой стороны, если присутствуют пластинчатые минералы, как, например, слюды, то некоторое количество этой тонкой «муки» облегчает получение однородной пробы. Если сначала удалить муку от такого содержащего слюду порошка, то сколько ни перемешивать потом оставшийся крупнозернистый порошок, нельзя получить пробы, где бы слюда была равномерно распределена, а не легла бы слоями. Окончательно полученную пробу тщательно перемешивают на глянцевой бумаге или осторожным встряхиванием в чистой банке.
Метод Вашингтона. Породу сначала разбивают методом, описанным выше (см. «Методы, принятые в Геологическом управлении США», стр. 891), на кусочки, которые вносят в «алмазную» стальную ступку с полусферическим дном и с соответствующим этому дну концом пестика (ступка Платтнера). Кусочки породы разбивают в ступке, один за другим, несколькими ударами полуфунтового молотка (деревянной колотушкой по Диттриху). Затем высыпают содержимое ступки в цилиндрический стеклянный сосуд вышиной около 3,5 см, внутренним диаметром 7,5 см и с толщиной стенок 2 мм. Когда вся проба (или взятая квартованием ее часть, если сама проба велика) будет таким образом измельчена, натягивают на открытый конец стеклянного сосуда кусок лучшей шелковой ткани для просеивания, имеющей около 25 отверстий на один линейный сантиметр, и крепко прижимают латунным кольцом высотой около 1 см. Затем этот сосуд перевертывают и осторожно отсеивают порошок на лист глянцевой бумаги. Порошок, оставшийся на сите, снова измельчают в ступке и опять просеивают; эти операции продолжают до тех пор, пока почти все не пройдет через сито. Оставшуюся в конце очень маленькую порцию растирают в агатовой ступке. Не следует, конечно, ничего
Приготовление пробы для анализа
893
отбрасывать, так как иначе состав анализируемого вещества будет определен неверно.
Вашингтон находит, что таким путем устраняется опасность загрязнения металлическим железом (см. мелкий шрифт на стр. 892) и что ошибка, происходящая от попадания в пробу волокон ткани, ничтожна. Последнее нельзя, однако, сказать о методе, применяемом часто в Европе; немецкое «просеивание», по крайней мере, как оно проводилось в лаборатории Бунзена, существенно отличается от того осторожного просеивания, которое предлагает Вашингтон.
Растирание
В обычных случаях особенно тонкого измельчения пробы для анализа вовсе не требуется. Так, тонко измельчают пробу только в тех случаях, когда предстоит определение щелочных металлов, или же когда из пробы нужно удалить растворимые в кислотах компоненты. Тогда, взяв небольшую часть пробы, измельченной одним из способов, описанных в разделе «Измельчение» (стр. 891), можно ее окончательно растереть перед самым взвешиванием.
Тонкое растирание пробы в большинстве случаев не только не нужно, но и вредно, так как при таком истирании могут удалиться содержащиеся в породе газы. Как было показано *, для правильного определения железа (II) надо брать для анализа возможно более крупнозернистый порошок, лишь бы его можно было полностью разложить принятым методом. В. Ф. Гиллебранд давно уже показал 1 2, что то же самое имеет место и при определении воды, которая не выделяется при 100—110° С. Было найдено 3, что чем тоньше измельчать пробу, содержащую железо (II), тем меньшие по величине получаются результаты определения этого компонента. Во всех опытах с горными породами и выделенными из этих пород железосодержащими минералами было замечено, что при истирании пробы происходит окисление железа (II). Наибольшее отклонение доходило до 40% от всего содержания железа (II) (3,13% его было найдено в самом тонком порошке и 5,13% — в самом крупнозернистом). Это совершенно неожиданное обстоятельство было затем нами подтверждено (стр. 987). Очевидно, что применявшийся до сих пор способ определения железа (II) в особо тонком порошке безусловно надо отбросить.
Хотя приготовление двух проб — одной крупнозернистой для определения железа (II), другой более тонкой для определения других компонентов — иногда неизбежно, все же против этого можно возражать по многим причинам: 1) Чем тоньше порошок, тем больше он содержит гигроскопической влаги; содержание ее в мелких зернах может быть в 5— 20 раз больше, чем в крупных. Чем тоньше измельчен порошок породы,
1 R. Mauzelius, Sveriges geol. Undersokn., Arsbok 1, № 3 (1907).
3 О влиянии тонкого измельчения на содержание воды и железа (II) в минералах и горных породах см. в статьях: W. F. Hillebrand, J. Am. Chem. Soc., 30, 1120 (1908); Chem. News, 98, 205 (1908). J. W. M a 11 e t [Phil. Trans., 171, 1017 (1880)], по-видимому, первый обратил внимание на то, что при измельчении может испариться вода из содержащих ее минералов (квасцы). Он приписал это влиянию нагревания, развиваемого при измельчении, — мнение, с которым мы согласны. М. С. Lea [Phil. Mag., 34, 36 (1892); 37, 31 (1894)] считает, однако, что действующим фактором являются здесь механические силы, особенно срезывающие силы.
3 R. Mauzelius, цит. выше.
894
Гл. LIII. Методы анализа силикатных пород
тем больше он также содержит воды, которая не выделяется при 100° С. Если приготовлены две аналитические пробы (крупнозернистая и тонко измельченная), то в обеих пробах нужно определить содержание воды, выделяющейся как ниже, так и выше 100° С, и при определении других компонентов в тонко измельченной пробе ввести соответствующие поправки. 2) Состав нерастертой пробы может также отличаться от состава топко измельченной пробы тем, что последняя загрязняется кремнекисло-топ пз ступки. Смотря по твердости анализируемых минералов и продолжительности растирания, это загрязнение может составлять от 0,5 до 1% от массы пробы (стр. 895).
Чтобы избежать необходимости приготовлять две пробы, были проведены опыты для нахождения условий, при которых можно было бы добиться тонкого измельчения пробы и в то же время избежать окисления. Число проведенных опытов пока еще недостаточно, чтобы можно было дать положительное заключение и формулировать точные указания. Однако уже теперь можно утверждать, что при растирании в неокисляющей среде, например в абсолютном спирте, получаются вполне удовлетворительные результаты. Вода менее пригодна для этого, хотя растворимость кислорода в ней и незначительна *. Спирт, по-видимому, не оказывает восстанавливающего действия на железо (III) в измельчаемых в нем минералах.
Эти опыты показали также, что окисление железа зависит не только от увеличения поверхности, соприкасающейся с воздухом; ему, вероятно, способствует местное разогревание, происходящее от трения и разламывания кусочков под пестиком. Вероятно, растирание в каком-нибудь инертном газе дало бы более удовлетворительные результаты. При применении механического растирателя это можно было бы устроить, но если растирать надо руками, то такое требование невыполнимо. Во всяком случае такой способ был бы очень неудобен и труден. Если применяют спирт, то его нужно приливать понемногу за один прием, лишь бы смочить порошок до очень жидкой массы. Так как спирт быстро улетучивается, то приходится его иногда прибавлять еще раза два. Когда растирание закончено, спирт оставляют свободно испаряться под часовым стеклом, помещенным на несколько сантиметров выше ступки и пестика, чтобы устранить попадание пыли и в то же время допустить возможность свободной циркуляции воздуха. Когда спирт совершенно испарится, полученный тонкий порошок осторожно снимают со ступки гибким лезвием ножа и ссыпают в банку.
Раньше в лаборатории Геологического управления США все пробы горных пород после их раздробления доводились до порошкообразного состояния растиранием от руки с затратой большого количества времени и труда. Теперь, за исключением некоторых особых случаев, эта работа производится механическими растирателями, приводимыми в движение маленьким электромотором. Из предыдущего, однако, ясно, что при пользовании такими истирателями следует быть очень осторожным.
1 W. F. Н i 11 е b г a n d, J. Am. Chem. Soc., 30, 1120 (1908); Chem. News, 98, 205 (1908).
Приготовление пробы для анализа
895
Истирание ступки и пестика
Об истирании дробильных аппаратов уже было сказано в разделе «Измельчение» (стр. 891).
Ступка и пестик истираются значительно сильнее при применении механических истирателей, чем при ручном растирании, несмотря на то, что движение в механических истирателях является в сущности подражанием ручной работе. В результате такого истирания в анализируемые пробы, без сомнения, попадает значительное количество кремнекислоты. Однако по сравнению с тем ее количеством, которое всегда находится в изверженных горных породах, это едва ли может иметь значение. Горные породы, не содержащие или почти не содержащие кремнекислоты, например железные руды, обычно мягче, чем изверженные породы, и, следовательно, слабее действуют на приборы для растирания.
С целью получить представление о количестве кремнекислоты, переходящей в исследуемую породу от истирания измельчающих приборов, Е. Аллен растирал 200 г кварцевого песка небольшими порциями до тех пор, пока вся проба не прошла через сито с 150 отверстиями на линейный дюйм (60 отверстий на 1 см). Для этого потребовалось 19,5 ч. Ступка (массой в 635 г) потеряла в массе0,189 г, а пестик (весивший 268 г) потерял 0,102 г. Общая потеря составила 0,291 г, или 0,145%, от массы взятого песка. Полученный порошок в этом опыте был менее тонким, чем это требуется для анализа горных пород, но, с другой стороны, истиранию подвергался один только кварц, т. е. минерал более твердый, чем большая часть компонентов обычных горных пород.
Было экспериментально исследовано 1 влияние истирания на ступки пз агата, стекла, чугуна и закаленной стали и оказалось, что для измельчения веществ большой твердости, например стекла, закаленная сталь гораздо лучше, чем агат, и что даже ступки и пестики из обыкновенного зеленого бутылочного стекла истираются меньше, чем агат. При растирании 10 г стекла в тонкий порошок агатовая ступка и пестик, весившие 416 г, потеряли в массе 0,052 г (или 5 мг на каждый грамм стекла), тогда как ступка и пестик из закаленной стали при том же весе потеряли только 0,005 г, несмотря на то, что ими до того уже пользовались некоторое время.
Хотя эти наблюдения и говорят в пользу применения стали, тем не менее, как было указано выше, применение металла в анализе горных пород совершенно недопустимо. Также недопустимо применение стеклянных ступок и пестиков вследствие сложного состава стекла. Если нельзя избежать загрязнения, то лучше, чтобы загрязнение это было одним каким-либо веществом, отражающимся на определении только одного элемента пли окисла из состава породы.
Масса пробы
После измельчения проба должна весить не менее 10 г, а лучше 20 г, на тот случай, если окажется необходимым повторить некоторые определения, требующие больших навесок вещества, например определение СО2. Анализ горных пород имеет преимущество перед анализом минера-
1 W. Н е m р е 1, Z, angew, Chem,, 14, 843 (1901).
896
Гл. LIII. Методы анализа силикатных пород
лов в том отношении, что материала для анализа здесь почти всегда достаточно, так что можно взять любое число навесок. Другое дело при анализе минералов, где аналитик часто принужден определять ряд, а иногда и все составные части пробы в одной, часто очень маленькой навеске. Это влечет за собой потерю времени и требует применения более сложных методов разделения, чем те, которыми обычно пользуются при анализе горных пород.
ВОДА. ОБЩИЕ ЗАМЕЧАНИЯ
Роль водорода в минералах 1
Так как целью этого раздела является прежде всего инструктирование аналитиков, то мы нашли целесообразным сократить рассмотрение роли водорода в минералах, ограничиваясь теми данными, которые могут помочь при толковании результатов анализа. Более полное освещение этого вопроса можно найти в литературе 2. Нужно заметить, однако, что изменения во взглядах о строении кристаллов, твердых растворов и т. п., происшедшие со времени опубликования этих работ, делают выводы более ранних работ сомнительными.
Формы, в которых водород может содержаться в минералах. Приведенная ниже схема представляет классификацию форм, в которых водород (главным образом водород, выделяющийся в виде воды) может присутствовать в минералах. При пользовании этой схемой нужно принять во внимание, что ее подразделения не являются строго разграниченными и что между ними существуют известные переходы; что минерал может содержать водород в нескольких формах и что водород, содержащийся в минерале в одной какой-либо форме, может перейти в другую при измельчении или нагревании.
Классификация форм нахождения водорода в минералах, агрегатах минералов и порошках минералов
А. Водород как одна из главных составных частей минерала, принимающий участие в расположении атомовв молекулярной или кристаллической структуре
I. Содержащийся в определенных стехиометрических отношениях'.
а) В виде водорода, называемого кислотным, по аналогии с его поведением в растворах.
1 Этот раздел был опубликован в Bulletin 700 of the U. S. Geological Survey on the Analysis of Silicate and Carbonate Rocks и был подготовлен E. T. Wherry, H. E. M er w i n и F. Q. Adams на основании материала по данному вопросу, изложенного в более раннем Bulletin 422.
2 Н. Ее Chatelier, Z. phys. Chem., 1, 396 (1887); С. г., 104, 1443—1517 (1887); Bull. Soc. franc, min., 10, 204 (1887); A. Hamberg, Geol. Foren, Forth. Stockholm, 12, 591 (1890); G. T a m m a n n, Z. phys. Chem., 10, 255 (1892); 27, 323 (1898); Z. anorg. Chem., 15, 319 (1897); J. M. Van В e m m e 1 e n, Z. anorg. Chem., 13, 233 (1897) и след, тома; G. Tschermak, Z. phys. Chem., 53, 349 (1905); Z. anorg. Chem., 63, 169 (1909); U. P a n i c h i, Pubbl. Inst, studi pract. etc., Sez di sci. fis. e nat., 1 (1908). Обширное извлечение — в Neues Jahrb., стр. 2 (1910), 2 реферата; F. Zambonini, Mem. Accad. sci. fis. mat., Napoli, 14, [2], № 1, 127 (1908); E. Lowenstein, Z. anorg. Chem., 63, 69 (1909); S. J. Thugutt, Centralbl. Mineralogie, 677 (1909); A. Beutell, K. Blaschke, Centralbl. Mineralogie, 4, 195 (1915); I. L a n g m u i r, J. Am. Chem. Soc., 40, 1361 (1918).
Вода. Общие замечания
897
б) В виде гидроксила — водород, называемый также основным, по аналогии с растворами.
в) В виде воды, именно кристаллизационной воды.
II. Не содержащийся в стехиометрических отношениях в минерале как в целом, но присутствующий в виде одной из вышеуказанных форм в одном или нескольких компонентах смешанных кристаллов, включая сюда изоморфные смеси.
Б. Водород, содержащийся в минералах, но не характерный для их состава (в виде воды или других соединений)
I. В жидком растворе, особенно в расплывающихся порошках, а также в аморфных телах, включая переохлажденные жидкости.
II. Водород, удерживаемый поверхностными силами'.
я) Адсорбированный поверхностями в виде пленок молекулярной толщины:
1. На стенках пустот внутри зерен агрегатов.
2. На поверхности зерен.
б) Удерживаемый капиллярными силами в жидкой форме (вода):
1. В коллоидах с сетчатым строением.
2. В неопределенных пустотах внутри зерен агрегатов.
3. В промежутках между отдельными зернами.
III. Водород включений'.
а) В виде жидких капель в пустотах.
б) Включенный в виде зернышек минералов, принадлежащих одному из перечисленных выше классов.
Следующие примечания имеют целью пояснить классификацию
А-I. Исследования при помощи рентгеновских и инфракрасных лучей показывают, что такие группы, как ОН и НгО, можно идентифицировать в кристаллах, так что разделение водорода, составляющего существенную часть минерала и содержащегося в определенных стехиометрических отношениях, на три класса — а, б и в — следует считать справедливым1.
А-П. В этот класс должны войти такие минералы, как топаз, понимаемый как изоморфная смесь двух крайних членов — одного водного, а другого безводного: A12(OH)2SiO4 и Al2F2SiO4. Так как соотношение между этими двумя частями может широко варьировать, содержание водорода может также изменяться; но даже если оно не будет стехиометрическим по отношению к топазу как целому, оно всегда останется таким в отношении его гидратного компонента, и потому водород может считаться существенной составной частью минерала. Однако нет необходимости, чтобы компоненты минерала были изоморфны в строгом смысле слова, т. е. чтобы их формулы были построены аналогично. Например, минерал тургит считают смесью двух компонентов (образующих смешанные кристаллы 2): гематита Fe2O3 и гетита FeO(OH) в различных отношениях.
Б-I. Сюда относится жидкость, окружающая расплывающиеся кристаллы. Часть воды опала может быть растворенной в его веществе, но, вероятно, главная масса ее относится к классу Б-П-б, 1, к которому принадлежит вода, входящая в состав большинства водных «аморфных минералов». Здесь надо также упомянуть такие вещества, как жидкие углеводороды и вулканические газы, хотя, строго говоря, они не являются настоящими минералами.
В-П-а. Адсорбция воды поверхностями в виде очень тонких (молекулярных) пленок считается общим свойством твердых тел, и если такая поверхность велика по отношению к количеству вещества, то количество адсорбированной воды приобретает значение для анализа. Возрастание количества воды, обнаруживаемое у многих минералов после тонкого их измельчения, соответствует случаю Б-П-а, 2. Известно немного примеров, где вода, относящаяся к разделу Б-П-а, 1, присутствует в заметном
1 W. W. Coblenz, Publ. Carnegie Inst. Washington, № 65 (1906); Bull. Bur. Standards, 2, 457 (1906); C. Schaefer und M. Schubert, Ann. Phys., 50, 339 (1916); A. Johnsen, Phys. Z., 5, 712 (1914).
2 E. Posnjak, H. E. Merwin, Am. J. Sci., [4], 47, 325—337 (1919.)
57 Заказ 5 22.
898
Гл. L1II. Методы анализа силикатных пород
количестве. Сюда относится волокнистая форма гетита, известная иод общим названием лимонита1.
Б-И-б, 1. Обращаясь к этому классу, необходимо отметить, что исследования в области коллоидов, к которым относятся также многие «аморфные минералы», показали, что они часто имеют неразличимую в микроскоп сетчатую структуру; вода или другая жидкая фаза задерживается в промежутках этой сетки. Возможно, что некоторые кристаллические вещества, в частности некоторые цеолитные минералы, могут также сдержать воду в таком состоянии, но это еще не доказано с достаточной убедительностью. Классы 2 и 3 этого подразделения имеют особое значение в связи с существованием тонковолокнистых минералов, один из которых был уже упомянут (гетит), и которые часто содержат значительные количества капиллярной воды.
Б-Ш. Жидкие включения воды или углеводородов и твердые включения водных минералов могут быть причиной значительного содержания водорода.
Влияние характера водорода на выбор метода анализа. Поведение водорода при измельчении минерала. Тонкое измельчение минерала может повести как к увеличению, так и к уменьшению общего содержания водорода (см. раздел «Изменение содержания воды при измельчении», стр. 900). Повышение содержания водорода есть результат раздробления осколков минерала и связанного с ним увеличения поверхности, на которой может адсорбироваться пли каким-либо иным способом удерживаться вода. Наоборот, понижение содержания большей частью является следствием разложения от местного разогревания — при раздавливании или просто при изменении величины зерен.
Вообще говоря (однако бывают и исключения), температура разложения минералов, содержащих кристаллизационную воду, ниже, чем минералов, принадлежащих к другим классам (в которых вода содержится в иной форме), и если при измельчении минерала происходит заметная потеря водорода, это следует объяснить вероятным присутствием водорода в виде кристаллизационной воды (класс A-I-в). Однако минералы, принадлежащие к другим классам, иногда имеют низкую температуру разложения, и потому не всегда с уверенностью можно руководствоваться этим признаком. Уменьшение содержания водорода, конечно, может быть также вызвано испарением воды или другой содержащей водород жидкости, заключенной первоначально в минерале в виде капелек и вскрытой затем при его измельчении (класс Б-Ш-а). Из высказанных соображений вытекает, что образец минерала, взятый для определения воды, должен быть измельчен насколько возможно меньше, однако настолько, чтобы вода могла выделиться в сравнительно короткий промежуток времени и при не слишком высокой температуре. Влияние измельчения на содержание воды может иметь значение по крайней мере в качестве одного из доводов при окончательном заключении о роли водорода в веществе.
Поведение водорода во время высушивания при постоянной температуре. Если минерал, содержащий стехиометрический водород, выделяющийся в виде воды, находится в атмосфере, в которой парциальное давление водяных паров непрерывно уменьшается, то этот минерал теряет воду. Водород класса А-П может, однако, вести себя различно; этот класс требует дальнейших исследований.
Поведение водорода, присутствующего в минерале не в стехиометрических отношениях, может быть различно, в зависимости от класса,
1 Е. Р о s п j a k, Н. Е. М е г w i n, Am. J. Sci [4], 47, 311 (1919).
Вода. Общие замечания
899
к которому он принадлежит. Если это водород включений (класс Б-Ш, а и б), потерь ожидать нельзя. Если он удерживается действием капиллярных сил (класс Б-П-б), ход потери будет зависеть от величины и формы капиллярных отверстий. Вода, адсорбированная поверхностью крупинок минерала, вероятно, теряется постепенно, по мере уменьшения давления водяного пара, но обезвоживание не бывает полным ни при каком определенном давлении. Можно ожидать, что водород, растворенный в «аморфных» минералах (класс Б-I), будет вести себя подобным же образом.
Процесс высыхания, изложенный здесь, часто является обратимым: вода, выделившаяся при уменьшении давления водяного пара, поглощается снова, когда это давление возрастает. Такая обратная гидратация часто может пойти значительно быстрее, чем первоначальная потеря воды, и этим определяется непрерывность или, наоборот, скачкообразное течение процесса. При тщательном изучении роли водорода в минерале нужно получить данные как по обезвоживанию, так и обратно по гидратации и представить их графически, потому что изучение таких кривых часто дает возможность лучше всего выяснить отношения, существующие в минерале.
Поведение водорода (удаляемого в виде воды) при нагревании1. При удалении водорода в виде воды нагреванием можно различить три случая.
1) Нагревание до приблизительно постоянной массы при каждой из возрастающего ряда температур, в атмосфере с постоянным или регулярно изменяющимся и обычно небольшим давлением водяных паров.
Минералы, содержащие водород в определенном стехиометрическом отношении, теряют его, по-видимому, скачками, т. е. при более или менее определенных температурах. Температуры, при которых наступает обезвоживание в сухом воздухе, зависят главным образом от элемента или элементов, с которыми связан водород, гидроксил или вода. Но, с другой стороны, в классе А-1-а температура обезвоживания «кислотного» водорода довольно высока, в классе A-I-6 («основной» водород) она несколько ниже, а в классе A-I-в кристаллизационная вода удаляется при совсем низких температурах, причем прежде всего удаляется вода из ряда гидратов. В этом случае вода из двух или более состояний гидратации может выделиться при одной и той же температуре, если в окружающей атмосфере не имеется тщательно регулируемого довольно высокого содержания водяных паров.
Удаление воды из смешанных кристаллов, вероятно, происходит постепенно, по мере повышения температуры, но экспериментальных данных для этого случая еще нет.
Водород, содержащийся в минералах в неопределенных отношениях, по большей части также удаляется постепенно, но здесь бывают и исключения. Например, вода включений может удаляться скачками, по мере того как будут вскрываться пустоты, содержащие ее (растрескивание минерала); даже капиллярная вода может удаляться скачками, если нагревание изменяет размеры пор.
2) Нагревание при непрерывно возрастающей температуре; давление водяного пара определяется в основном состоянием дегидратируемого вещества.
1 При термическом обезвоживании может выделиться также некоторое количество других соединений водорода, например НС1 из MgCl2-6H2O или А1С12-6Ы2О.
57*
900
Гл. LIII. Методы анализа силикатных пород
Характер потери воды, как он определяется частым взвешиванием 1 или методом Ле Шателье, может быть и непрерывным и прерывистым, в зависимости от уже указанных условий. Однако в этом случае температура, при которой происходят скачки, значительно сильнее подвержена влиянию различных факторов, как, например, скорость нагревания, степень измельчения минерала, метод перемешивания его во время нагревания; эти скачки, однако, более заметны, особенно в случае ряда гидратов.
3) Исследование кривых давления водяного пара в случае обратимых изменений в содержании воды.
Исследования такого рода дают ясное представление о состоянии гидратации вещества, но этот метод не так распространен, как аналитический метод.
Влияние обезвоживания на физические свойства минерала. Кристаллографические, оптические и другие свойства минерала, вообще говоря, при обезвоживании изменяются коренным образом, когда из минерала удаляется водород, принимающий участие в строении самой молекулы. Наоборот, свойства эти изменяются лишь немного, если водород содержался в минерале в случайных отношениях к его составу. Но здесь, как это бывает и в других случаях, существуют значительные исключения из этого правила. В частности, некоторые цеолитовые минералы, в которых вода входит, вероятно, частично в состав молекулы, могут обнаруживать только небольшие изменения в свойствах, когда вода из них частью или даже полностью удалена. В минералах класса А-П изменения в свойствах так же, несмотря на потерю воды, могут быть незначительными. С другой стороны, если минералы содержат много воды, не входящей в молекулу, могут произойти заметные изменения свойств при ее удалении 2.
Заключение. Из всего изложенного ясно, что хотя определение состояния водорода в минералах является далеко не простой задачей, все же большое число сведений о нем может быть получено путем применения соответствующих аналитических методов. Не следует делать каких-либо заключений на основании одного наблюдения, но когда несколько методов исследования дают согласные результаты, становится вполне возможным для большинства минералов сделать вывод о состоянии, в котором находится содержащийся в них водород.
Изменение содержания воды при измельчении
Прежде чем рассматривать методы определения воды, необходимо еще раз вернуться к влиянию тонкого измельчения минерала на его способность поглощать влагу из воздуха и удерживать при температуре выше 100° С весьма значительные количества ее, иногда даже более 1% (стр. 894). В табл. 32 содержатся весьма показательные данные по этому вопросу. Более подробные сведения см. в уже цитированных статьях 3.
С другой стороны, при продолжительном измельчении из минералов можно удалить кристаллизационную воду. Так, содержание воды в одном
1 W. F. Hillebr a’n d, Н. Е. М'е г w i n, F. Е. Wright, Proc. Am. Philos Soc., 53, 45 (1914); Z. Kryst., 54, 221 (1915).
2 E. S. Lars en, E. T. Wh er r y, J. Washington Acad. Sci., 7, 178—208 (1917).
3 См. сноску 2 на стр. 893.
Вода. Общие замечания
901
ТАБЛИЦА 32
Содержание воды в порошках минералов с различной величиной зерен, измельченных на воздухе
№ пп. Материал Измельчение Содержание воды, %
ниже 100е С выше 100° С
la ] 16 } Диабазовый норпт или габбро 1 Менее 60 отв. на 1 см < Растирался 30 мин 0,03 0,10 0,66 0,66
1в J 1 » 120 мин 0,74 1,00
2а 1 Измененный базальтиче- J Менее 40 отв. на 1 см 0,22 3,13
2б / ский грюнштейн ( Растирался 120 мин 1,70 4,19
За 1 Андезит ( Менее 60 отв. на 1 см 1,01 3,40
36 / [ Растирался 30 мин 1,18 3,54
4а 1 Кварц I Менее 60 отв. на 1 см нет 0,06
46 J [ Растирался 120 мин 0,35 0,45
5а 1 56 / Морская пенка f Грубое [ Растиралась 600 лши 5,76 6,33 6,79 9,81
6а 1 Неглазурованный берлин- ( Грубое нет нет
66 J ский фарфор 1 Растирался (?) мин 0,62 0,18
ТАБЛИЦА 33
Определение влаги в 1 г измельченного в порошок и выставленного на воздух материала
[< обозначает: тоньше, чем; >обозначает: крупнее, чем]
Материал Величина зерен (число отверстий на 1 дюйм) Влага г
Ортоклаз (естественное стекло) AbjAns (искусственное стекло) AbjAn5 (искусственный кристалл) АЬ^щ (искусственное стекло) АЬдАщ (искусственный кристалл) Ab (естественный кристалл) . . < 150 Отобранные, крупные < 100 > 120 < 100 > 120 < 100 > 120 ( Крупные [ <150 0,0006 0,0001 0,0010 0,0007 0,0010 0,0006 0,0069
Ортоклаз (естественный кристалл) » (тот же образец) . . . <120 >150 ( < 150 • ( Еще тоньше 0,0011 0,0031 0,0059
Ортоклаз (искусственное стекло) » (тот же образец) . . . Все> 100 > 150 0,0065 0,0022
образце гипса упало с 20 до 5% х. Из этого следует, что кристаллизационную воду надо определять в нерастертом минерале, если это возможно.
Следующая выдержка из работы о полевых шпатах 1 2 имеет прямое отношение к разбираемому вопросу:
1 См. сноску 2 на стр. 893.
2 Day, Allen; Publ. Carnegie Inst. Washington, No. 31, p. 36; Am. J. Sci.f 4th ser., 19, 93 (1905). Cp. R. Mauzelius [цит. выше] и W. F. Hillebrand [цит. выше, см. стр. 894].
902
Гл. LIII. Методы анализа силикатных пород
«Еще есть один источник ошибок при точных определениях плотности порошке^ минералов, несмотря на принятые предосторожности. Соприкосновение вещества с воздухом в период измельчения дает возможность атмосферной влаге осаждаться на крупинках минерала и тем самым изменять их массу. Количество влаги заметно изменяется в зависимости от величины крупинок минерала, как показывает табл. 33, и (вероятно) от степени насыщения атмосферы водяными парами и от времени соприкосновения с ней.
В двух последних группах в табл. 33 можно заметить, что в одинаковых пробах того же самого минерала содержание влаги зависит от величины зерен порошка.
Мы подтверждаем далее наблюдение Бунзена1, что эта поглощенная влага не может быть снова выделена при температуре несколько выше100° С, но что для этого необходимо от 600 до 800° С, т. е. темно-красное каление.
Некоторые образцы после определения влаги были оставлены на несколько недель в закрытых пробирках, после чего влага в них была снова определена — и были получены прежние числа. В связи с этим следует указать, что определенные здесь количества абсорбированной воды — того же самого порядка, как и количества, обыкновенно указываемые в анализах полевых шпатов, и что в этих анализах ошибка, могущая произойти от абсорбированной воды, тем больше, чем тоньше была измельчена проба для анализа. Возможно, что часть, а может быть и вся влага, обычно находимая в анализах, абсорбирована и указание на ее присутствие является ошибочным».
Применение для анализа воздушно-сухой пробы
Старинная привычка высушивать растертую в порошок пробу до взятия навески не имеет серьезных оснований. Ее цель — обеспечить однообразные гигроскопические условия для удобного сравнения результатов анализов, потому что порошки горных пород содержат различное количество влаги, которое зависит прежде всего от степени измельчения породы. Однако можно быть вполне уверенным, что вещество, прежде чем оно будет взвешено, вновь поглотит некоторое количество влаги, которое в большинстве случаев будет невелико, но иногда может быть весьма значительным. Ясно также, что при каждом открывании стаканчика в него проникает содержащий влагу воздух, который остается вместе с сухим порошком. Поэтому легко может случиться, что после многочисленных открываний стаканчика, особенно если его не закрывать тотчас же, высушенная проба будет такой же влажной, какой она была до высушивания. Аналогичной точки зрения придерживается и Диттрих. Должно быть ясным, конечно, что здесь речь идет не о веществах, расплывающихся при поглощении влаги.
Лучше брать навеску воздушно-сухой пробы и отдельно определять ее влажность. Если отвесить необходимые для анализа порции одну за другой или по крайней мере в тот же день, то будет устранена ошибка, которая может возникнуть вследствие различного содержания влаги в воздухе при разной погоде и которой можно совершенно пренебречь для большей части отдельных определений. Только в главной порции, где будут определяться кремнекислота и большая часть оснований, она может иметь значение. Особенно можно пренебречь этой ошибкой, имея дело с более крупным порошком, например с проходящим через сито с 30 отверстиями на линейный сантиметр и остающимся на сите с 60 отверстиями. У таких порошков влажность бывает часто ниже 0,1%, в то время как у порошков, которые подвергались растиранию в течение 1—2 ч, она часто превышает 1%.
1 Wiedemann’s Annalen, 24, 327 (1885).
Вода. Методы определения
903
Включение гигроскопической влаги в таблицы результатов анализа
Возникает вопрос: «Если так называемая гигроскопическая влага не всегда действительно является таковой, а часто включает также и связанную воду, то зачем ее определять и помещать в таблицу результатов анализа? Почему делается различие там, где в действительности его нет?» Необходимость включения в таблицу результатов анализа потери при 100—110° С обосновывается следующими соображениями.
Желание петрографов, чтобы все анализы были отнесены к безводному веществу, может показаться справедливым, так как в этом случае анализы становятся вполне сравнимыми между собой. Тогда гигроскопическая влага должна быть вычеркнута из списка перечисляемых компонентов. Это было бы правильно, если бы можно было быть уверенным в том, что потеря при 100—110° С действительно представляет собой только механически удерживаемую воду. Но так как сюда часто входит и связанная вода и так как установление в каждом отдельном случае, присутствует ли она или нет, не всегда возможно и сопряжено с потерей времени, то, по-видимому, все же необходимо указывать содержание гигроскопической влаги в результатах анализа. Если этого не делать, то могут возникнуть большие ошибки, как это совершенно очевидно из следующего, правда, редкого случая. Некоторые горные породы из Уиоминга в измельченном состоянии потеряли от 1 до 2% влаги при 110° С. Что здесь в действительности не было гигроскопической влаги в сколько-нибудь заметном количестве, следует совершенно определенно из того, что потеря в массе неиз-мельченного материала оказалась такой же; многие химики и петрографы по привычке удалили бы всю эту воду до начала анализа, и таким образом тогда был бы упущен очень важный характеризующий породу признак. В этом случае аналитик дал бы неточный анализ, который мог бы привести к неверным выводам, даже к путанице. (См. также примеры, приведенные на стр. 905.)
Отсюда следует, что определение потери при 100 или 110° С и включение ее величины в таблицы результатов анализа желательно, так как это дает литологу указание на характер минералов, входящих в состав горной породы, и таким образом может подтвердить микроскопическое исследование или побудить к дальнейшему исследованию в этом направлении. Заметная потеря воды при 100° С грубо измельченными порошками (за исключением некоторых вулканических стекол) может быть истолкована как положительный признак присутствия цеолитов или других минералов, характеризующихся большим содержанием слабо связанной воды. Возражение, что содержание гигроскопической влаги изменяется в зависимости от степени измельчения пробы, от состояния атмосферы, от продолжительности взвешивания, и что поэтому ее не следует включать в результаты анализа, не имеет достаточных оснований, так как в крупнозернистых порошках эти колебания обычно совсем не велики.
ВОДА. МЕТОДЫ ОПРЕДЕЛЕНИЯ
При работе с грубо измельченными порошками, необходимыми для получения истинной величины содержания водорода в минералах п горных породах, тот или иной из описанных в этой главе методов может оказаться в известной мере неудовлетворительным, особенно при анализе
904
Гл. LIII. Методы, анализа силикатных пород
веществ, теряющих водород при очень высоких температурах или с трудом разлагаемых плавнями. К сожалению, в настоящее время еще нельзя дать указаний, подходящих ко всем таким случаям. Аналитик должен научиться правильно выбирать метод анализа. Во всех случаях необходимо проводить параллельные определения, пока приобретенный опыт не позволит делать иногда исключения из этого правила. Сравнительную оценку методов прокаливания можно найти у М. Диттриха *.
Косвенные методы
Общие замечания. Методы, применяемые для косвенного определения воды, различаются в зависимости от того, проводится ли определение общего содержания воды или определение ее по фракциям.
В первом случае обычно просто прокаливают навеску, во втором — навеску подвергают действию или водоотнимающих веществ или различных температур. Эти методы в том виде, как они применяются, хотя и просты, но ведут часто к большим ошибкам, на которые в соответствующих местах будет указано. Если нужна большая точность, то эти методы применять нельзя, за исключением тех случаев, когда известно,что ошибки незначительны, или когда в метод введены изменения, устраняющие ошибки. Еще одно осложнение, иногда серьезное, вносится тем обстоятельством, что минералы в виде грубо измельченного порошка значительно медленнее отдают воду, чем если бы они были взяты в виде тонкого порошка.
Методы, основанные на применении водоотнимающих веществ. Обычно для этой цели в минеральном анализе применяют хлорид кальция, серную кислоту и фосфорный ангидрид. Кроме этих веществ, заслуживают внимания безводный и трехводный перхлораты магния (см. «Реактивы», стр. 71). Чаще всего пользуются концентрированной серной кислотой, а если имеют в виду детальное исследование процесса постепенной отдачи воды минералами, то берут серную кислоту различной известной концентрации. Было показано 1 2, что для полного обезвоживающего действия серной кислоты в эксикатор нужно наливать кислоту максимальной концентрации. Кислота, долго стоявшая в эксикаторах, которые были в постоянном употреблении, далеко не так активна, как свежая концентрированная серная кислота. Кроме того, серная кислота начинает темнеть от попадания в нее органических веществ из пыли или из смазки с крышки эксикатора. И то и другое является причиной появления заметных количеств сернистого ангидрида.
Для полного удаления воды из минералов, теряющих над концентрированной серной кислотой значительное ее количество, может потребоваться много дней и даже недель, Если взвешивания повторять изо дня в день, то кажущийся предел отдачи воды может быть достигнут еще задолго до того, как вся вода, действительно способная выделиться в этих условиях, будет поглощена серной кислотой. Каждый раз, когда тигель после взвешивания снова ставят в эксикатор, он оказывается не в сухой, а в более или менее влажной атмосфере, и содержимое тигля, даже если последний покрыт крышкой, иногда поглощает часть этой влаги и удержи
1 М. D 1 t t г i с h, Z. anorg. Chem., 78, 191 (1912).
2 W.F. H ill ebr and, Am. Chem., J., 14, 6 (1892).
Вода. Методы определения
905
вает ее так упорно, что в течение ближайших 24 ч серная кислота оказывается не в состоянии освободить его от этой влаги. Для этого требуется большее время. Опубликованный много лет тому назад опыт с тиролитом (табл. 34) отчасти иллюстрирует сказанное.
ТАБЛИЦА 3 4
Опыт с высушиванием 1 г тиролита над концентрированной серной кислотой
Промежутки времени часы Потеря массы г Промежутки времени часы Потеря массы г
18 0,0231 24 0,0002
26 0,0083 24 0,0003
23 0,0029 48 0,0006
24 0,0012 24 0,0002
23 0,0008
24 0,0001
25 0,0003 283 0,0380
Казалось бы, имелось достаточно оснований считать опыт законченным после 138 ч, так как в течение последних 24 ч потеря мсссы соста вила только 0,1 мг. Тем не менее еще в течение 6 дальнейших дней происходила потеря в массе примерно по 0,3 мг в день. Вероятно, она продолжалась бы и дальше, если бы опыт не был прекращен А Проба была в виде тонкого порошка; будь она более крупнозернистой, скорость обезвоживания была бы значительно меньше. Продолжительность высушивания может быть конечно значительно сокращена применением вакуума.
Косвенный метод определения влаги можно сделать более точным, нагревая минерал или горную породу во взвешенной трубке, через которую пропускают ток воздуха или какого-нибудь другого газа (сухого или с определенным содержанием влаги, в зависимости от цели анализа) и затем определяя потерю массы трубки с ее содержимым. Если газ высушивается над фосфорным ангидридом, то рационально сначала пропускать газ через хлорид кальция или серную кислоту; в противном случае активность фосфорного ангидрида, превращающегося с поверхности в стекловидную метафосфорную кислоту, быстро падает.
Методы, основанные на применении нагревания. Фракционные определения. Для высушивания с целью определения гигроскопической влаги1 2 в различные времена разными химиками применялись температуры от 100 до 110° С. При анализе большей части горных пород совершенно безразлично, какая из этих температур выбрана, так как при более продолжительном действии низшей температуры получается тот же результат, как и при более кратковременном действии высшей температуры.
В лаборатории Геологического управления США в настоящее время принято пользоваться толуоловой баней, дающей температуру приблизительно 105° С. Если потеря воды при этой температуре оказывается необычно большой, то пробу снова нагревают приблизительно до 125° С,
1 См. yJ. W. Mallet [Phill. Trans., 171, 1006 (1880)] пример медленной потери воды квасцами на воздухе в опыте, продолжавшемся 6 мес.
2 Исчерпывающее обсуждение методов определения влаги, особенно в углях, см. в Report of the International Commitee on Analysis of the VIII Intern. Congress of Appl. Chemistry, 1912, p. 77—131.
906
Гл. LIII. Методы анализа силикатных пород
чтобы установить, увеличивается ли потеря в массе с повышением температуры, так как знание это важно для объяснения результатов определения. Если нужно получить очень точные результаты, то никогда не следует проводить определение в тиглях или чашках, которые перед взвешиванием должны охлаждаться в эксикаторе. Одного примера будет достаточно. 1 г смеси минералов, содержавшей около 17% воды, из которых около 3% выделилось при 100° С, а 8—9% при 280° С, нагревали несколько часов при 280° С, затем оставили в эксикаторе над серной кислотой и тотчас по охлаждении взвесили. Проба прибавила в массе 1,5%, хотя эксикатор был плотно закрыт и тигель покрыт крышкой.
Один образец тиролита потерял 10,34% воды при 280° С, а в другой раз 14,33%. В последнем случае высушивание и нагревание при постоянном повышении температуры до 280° С продолжалось всего 528 ч. Взвешивание проводилось обычно ежедневно. В первом же случае продолжительность опыта была значительно меньше и промежуток между отдельными взвешиваниями составлял только несколько часов.
Одно очень важное исследование 1 ясно показало, каковы бывают ошибки при определении такой легко выделяющейся воды. Было найдено, что некоторые цеолиты, будучи сильно обезвожены (но не нагреты до разрушения их молекулярной структуры), могли затем поглощать вместо воды различные сухие газы, в атмосферу которых их помещали, например двуокись углерода, аммиак, сероуглерод и др., даже воздух в больших количествах, а также некоторые жидкости. На основании этого наблюдения можно полагать, что упомянутое выше большое прибавление в массе (в 1,5%) у частично обезвоженного минерала происходило за счет воздуха, который был поглощен из эксикатора, а не от поглощения влаги, как полагали прежде. Отсюда следует, что опасно принимать потерю в массе за меру количества выделенной воды, а также, что во многих случаях этот способ определения воды совершенно непригоден. Какой метод необходимо применить в каждом отдельном случае, должно быть полностью предоставлено выбору работающего; в этом выборе он часто будет руководствоваться минеральным составом породы на основании внешнего вида ее или исследования под микроскопом.
Известен способ определения истинной величины потери воды в минералах, подобных цеолитам, без собирания выделенной воды. Из обезвоженной и взвешенной пробы, при соблюдении необходимых предосторожностей, удаляют весь воздух, который она могла поглотить во время высушивания и охлаждения, измеряют объем этого воздуха, вычисляют соответствующую ему массу, который прибавляют к кажущейся потере в массе, и получают таким образом истинное содержание воды.
Кроме вышеуказанных возражений, против применения непрямого способа определения воды говорит еще одно важное обстоятельство. Обычно пользуются воздушной баней, между тем это никогда не приводит к верным результатам, потому что высушивание проходит в атмосфере, которая сама по себе вовсе не суха, и вещество поэтому ни в коем случае не может выделить всей своей влаги. Например, в двух пробах было найдено непрямым способом определения воды 0,12 и 0,75%, а прямой метод дал соответственно 0,17 и 1,00%. Подобные наблюдения делались много раз и не подлежат сомнению. Они относятся как к горным породам, так и к мине-
2 G. Friedel, Bull. Soc. mineral., 19, 14-94 (1896); С. г., 122, 1006 (1896).
Вода. Методы определения
907
ралам. Поэтому, если определение содержания влаги имеет большое значение, то надо или совершенно устранить применение непрямого способа или пользоваться им так, чтобы обеспечить постоянную смену сухого воздуха.
Чтобы достичь этого, порошок породы нагревают во взвешенной трубке, через которую пропускают ток сухого воздуха, охлаждают трубку и повторным взвешиванием определяют потерю воды. Степень высушивания пробы зависит от того, какой реактив применялся для высушивания проходящего воздуха. Серная кислота высушивает сильнее, чем хлорид кальция, а фосфорный ангидрид является наиболее действенным из всех высушивающих реагентов. Если применяют последний, то рекомендуется сначала подсушить воздух каким-либо из первых двух реагентов, так как обезвоживающая способность фосфорного ангидрида убывает очень быстро, когда он покрывается с поверхности стекловидной метафосфорной кислотой. Если высушивание ведут при комнатной температуре, то нет необходимости в воздушной бане; при применении более высоких температур трубку вставляют в соответствующий термостат.
Определение общего содержания воды по «потере от прокаливания». В некоторых случаях простое определение потери при прокаливании достаточно точно показывает общее содержание воды. Однако в огромном большинстве случаев имеется так много источников возможных ошибок, что на этот старинный способ редко когда можно положиться.
Например, следующие результаты были получены этим методом при анализе ферберита FeWO4, который был измельчен, просеян и нагрет, как указано ниже:
Фракция 48—65 меш (d = 0,3—0.2 лип)
Время прокаливания, ч............ 3 3* 2 1 2 * 1 1* 1*
Температура, °C....................110 НО 150 200 200 600 600 600
Потеря, %........................ 0,005 0,015 0,050 0,090 0,020 2,410 — —
Привес, % ..........................— — — — — — 0,060 0,040
Фракция меньше 100 меш-(d< 0,5 лии)
Время прокаливания, ч............ 3 3* 2 1 2 * 1 1* 1*
Температура, °C....................НО 110 150 200 200 600 600 600
Потеря, %......................... 0,130 0,050 0,010 0,040 0,010 1,193 — —
Привес, %...........................— — >— — — — 0,070 0,090
* Добавочно.
Только когда горная порода не содержит фтора, хлора, серы, углерода, СО2 и нелетучих окисляющихся (FeO) или восстанавливающихся (МпОг) компонентов, потеря при прокаливании может считаться истинной мерой содержания воды. Однако редко бывает, чтобы проба удовлетворяла этим условиям, особенно в отношении отсутствия железа (II). Если проба содержит из указанных выше веществ только СО2, то можно, установив точно содержание последней, правильно определить содержание воды прокаливанием пробы при 1100—1200° С; при этом должно соблюдаться условие, что СОа происходит от карбонатов щелочноземельных металлов, а не от карбонатов железа и марганца. Другим источником ошибок является то, что водород из некоторых минералов не может быть удален полностью даже при прокаливании на паяльной горелке, а между тем высокая температура может вызвать улетучивание других компонентов горной породы, которые при более низкой температуре не улетучиваются, например щелочных металлов. В частности, это происходит при анализе
908
Гл. LIII. Методы анализа силикатных пород
глинистых известняков и других пород, в состав которых входят силикаты, содержащие щелочные металлы, и карбонаты щелочноземельных металлов.
Давно установилось мнение, что в присутствии железа (II) можно довольно точно определить содержание воды, если к потере при прокаливании прибавить столько кислорода, сколько необходимо для окисления всего присутствующего железа (II). Это неверно, так как в полноте окисления железа (II) нельзя быть уверенным, особенно при анализе легкоплавких пород. Кроме того, при температуре паяльной горелки происходит и обратный процесс — частичное восстановление высших окислов Ч Если содержание железа велико, то проба после прокаливания иногда становится магнитной, вероятно от присутствия магнитной окиси железа, которая даже при сильном прокаливании полностью не окисляется, особенно если она находится в виде крупных зерен. Имеются указания на то, что окисление магнитной окиси железа не достигается и выпариванием с азотной кислотой и вторичным прокаливанием.
В этом случае может быть рекомендован следующий метод 1 2 определения воды. В общую потерю при прокаливании вносится поправка на двуокись углерода, если она присутствует, и на железо (II), путем определения последнего в пробе как до, так и после ее прокаливания. Вычтя количество гигроскопической влаги, найденное в отдельном определении, получают содержание более прочно удерживаемой породой воды. Разумеется, этот метод требует большой точности в определении влаги и железа (II).
Прямые методы
Метод Пенфильда. Анализ минералов, легко отдающих воду. Если кроме воды никаких других летучих веществ в породе нет, то можно рекомендовать изящный и простой метод, введенный Брашем и усовершенствованный Пенфильдом 3. Метод не оставляет желать лучшего в отношении точности при условии, что выделение воды происходит при сравнительно низких температурах. Этот метод состоит в простом нагревании растертой в порошок пробы в узкой трубке из стойкого стекла, расширенной на запаянном конце и имеющей, кроме того, еще одно или два расширения в середине для предохранения от обратного стекания конденсирующейся воды и растрескивания горячего стекла. От потери воды в воздух защищает вытянутая в капилляр вторая стеклянная трубочка, присоединенная с помощью резиновой трубки к первой. Трубку укрепляют в горизонтальном положении и нагревают расширение у запаянного конца до нужной температуры на бунзеновской или паяльной горелке. Часть трубки, служащую холодильником, обматывают мокрой фильтровальной бумагой или холстом, чтобы обеспечить полную конденсацию воды. По окончании выделения влаги отнимают нагретый конец, охлаждают трубку и взвешивают ее, предварительно очистив снаружи. Затем удаляют воду продува
1 Н. W а г t h [Chem. News, 84, 305 (1901)] также упоминает об этом и обращает внимание на влияние окиси алюминия, которая не только обесцвечивает окись железа (III), когда обе окиси прокаливаются вместе, но и уменьшает восстановление последней до магнитной окиси железа.
2 Р. G. W. В а у 1 у, Australasian Association for the Advancement of Science, Adelaide meeting, 1907.
3 S. L. P e n f i e 1 d, Am. J. Sci., [3], 48, 31 (1894); Z, anorg. Chem., 7, 22 (1894).
Вода. Методы определения
909
7
Рис. 38. Трубки Пенфильда для определения воды в минералах:
1,2, 3 — различные формы таких трубок; 4 — ершик, с помощью которого вводят в трубку анализируемую пробу; 5 — вытянутая в капилляр трубочка, служащая пробкой.
нием воздуха и снова взвешивают. Для определения незначительных количеств воды, содержащихся в большинстве горных пород, расширение в средней части трубки едва ли необходимо.
На рис. 38 изображены различные формы трубок, применяемых в методе Пенфильда. Перед употреблением «эти трубки нужно тщательно высушить изнутри, даже если они кажутся сухими, что лучше всего достигается пропусканием тока воздуха через нагретую трубку при помощи тонкой стеклянной трубочки, доходящей до дна».
В работе Пенфильда 1 подробно излагается, как в этом простом приборе можно точно определять содержание воды даже в тех случаях, когда анализируемая порода содержит карбонаты.
Вполне свободными от других летучих компонентов, кроме воды, являются лишь сравнительно немногие породы; поэтому при выполнении очень точных анализов применение этого метода в описанной выше простой установке возможно только в ограниченном числе случаев. Однако метод можно применять с добавлением окисей кальция, свинца или висмута для удержания фтора, серы, хлора и т. п. Этим методом находят, конечно, общее содержание воды.
Анализ минералов, с трудом отдающих воду. В присутствии минералов, которые даже при температуре паяльной горелки не выделяют воду полностью (например, талька, топаза, хондролита, ставролита), Пенфильд рекомендует применять простую печь из огнеупорного кирпича, нагреваемую древесным углем, и вводить в трубку вещество, задерживающее фтор, если это нужно.
Нагревающаяся часть трубки должна быть защищена цилиндром из платиновой жести, плотно окружающим ее конец. Среднюю часть трубки обертывают влажным полотном или мокрой фильтровальной бумагой и предохраняют от жара печи асбестовым картоном. Конец трубки обкладывают со всех сторон древесным углем и направляют на него горизонтальное пламя горелки. Таким образом можно достичь наивысшей температуры, но платиновая жесть при этом долго не выдерживает и разрушается.
М. Диттрих 2 считает описанный выше метод непригодным, как дающий непостоянные результаты при сравнении его с другими методами, но, по-видимому, он не применял поглотителей для фтора и серы.
Независимо от способа применения описанного прибора, метод дает общее содержание воды, из которого, если нужно, может быть вычтена гигроскопическая вода, определенная отдельно высушиванием при 105° С.
Прямой метод с поглотительными трубками. Общие замечания. Вообще следует отдать предпочтение методам, основанным на собирании и взвешивании воды, выделившейся из горной породы или минерала, в
1 S. L. Р е п t i е 1 d, цит. выше.
2 См. сноску 1 на стр. 902.
910
Гл. LIII. Методы анализа силикатных пород
соответствующих трубках. Эти методы особенно пригодны для определения всей воды или воды, выделяющейся при температуре выше 105° С. Менее пригодны эти методы для тех фракционных определений, когда выделение воды происходит медленно при разных температурах, потому что при этом поглотительные трубки могут получить некоторое приращение массы независимо от поглощения воды из минерала и тем ввести заметную ошибку в определение. В таких случаях необходимо проводить параллельно «холостой» опыт и вносить в результаты соответствующие поправки.
Поглощение не рекомендуется применять в тех случаях прокаливания без плавней, когда минерал или порода, кроме воды и двуокиси углерода, содержат еще другие летучие примеси. Во всяком случае тогда нужно применять вещества, задерживающие эти примеси, прежде чем они попадут в трубки, поглощающие воду. Но даже и при соблюдении этих условий при высоких температурах возможны ошибки. Поэтому при анализе минералов, отдающих воду только при очень высоких температурах, применение плавней обязательно.
Слишком часто оставляют без внимания следующее обстоятельство: почти не следят за тем, чтобы поглотитель, служащий для высушивания проходящего воздуха (который надо всегда нагнетать, а не отсасывать), был бы тем самым, что и в поглотительной трубке, в которой собирается вода пз пробы, причем должно быть то же самое высушивающее вещество с той же поглотительной способностью. Сказанное особенно важно по отношению к хлориду кальция. Поэтому следует часто возобновлять его, наполняя трубки для поглощения влаги и для предварительного высушивания воздуха хлоридом кальция из одного и того же запаса. Через свеже-наполненные хлоркальциевые трубки целесообразно пропустить ток двуокиси углерода, избыток которого перед употреблением трубок следует вытеснить током воздуха. Далее необходимо, чтобы трубки соединялись всегда встык (стекло к стеклу), с возможно меньшим обнажением резины внутри соединения. При длинном резиновом соединении возможна очень значительная ошибка в сторону увеличения. Важно также, чтобы направление тока воздуха через поглотительные трубки было при поглощении всегда одно и то же.
Нужно следить, чтобы разновременные взвешивания проводились при приблизительно одинаковых условиях температуры и влажности воздуха. Может получиться значительная разница от того, взвешивается ли сосуд в тот же день или на следующее утро. Проистекающей отсюда ошибки можно избежать до некоторой степени посредством тарирования поглотительной трубки другой трубкой примерно того же объема и веса.
Наконец, нельзя пренебрегать и электризацией трубки, которая появляется, если вытирать сосуд перед взвешиванием. При вытирании трубки чистым льняным полотенцем, по нашим наблюдениям, отклонение может достигнуть 0,01—0,02 г при применении весов, стоящих на стеклянной доске (всегда прибавка в массе). Заряд можно удалить многократным прикосновением руки, но он медленно исчезает и сам по себе. Сильная электризация становится заметной по явной неправильности колебаний стрелки. Хотя это явление чаще происходит в холодное время, но наблюдается оно не только зимой г.
1 Об электризации см. также Е. Borneman n, Chem. Ztg., 125, 220 (1908).
Вода. Методы, определения
911
Наполненные поглотительные трубки в общем не должны весить больше 30 г каждая. Серная кислота в качестве поглотителя не имеет заметных преимуществ перед хлоридом кальция, если прибор для высушивания проходящего воздуха содержит хлорид кальция, имеющий такую же поглотительную способность, как и хлорид кальция, находящийся во взвешенной поглотительной трубке.
Нагревание пробы. Нагревание пробы лучше всего проводить в электрических печах или муфелях, снабженных терморегуляторами и указателями температуры.
Рис. 39. Видоизмененная форма платинового тигля Гуча для определения воды. Четверть натуральной величины. Масса около 75 г:
1 — цилиндрическая часть тигля; 2 — тубус; 3 — желобок.
Предложен прибор!, в котором проба нагревается до 110° С в трубке, погруженной в толуоловую баню, а также описана 1 2 газовая печь для температур до 300° С. Для прокаливания при температурах 1000—1050°С рекомендуется3 трубка из плавленого кварца, длиной 45 см, диаметром 22 мм и со стенками толщиной 0,5 мм, оттянутая у выводящего конца и снабженная пробкой и отводящей трубкой из того же материала. Пробка и широкий конец трубки должны быть хорошо отшлифованными и натертыми графитом, чтобы обеспечить герметичность. С той же целью на пробке и на конце трубки имеются кварцевые крючочки, на которые можно надеть пружинки из проволоки. Навеску пробы смешивают с 5—6 г сухого карбоната натрия и помещают в платиново-иридиевую (10% иридия) лодочку длиной 12 см, снабженную крышкой из того же сплава. Для защиты трубки от разбрызгивания содержимого лодочки применяется такой же платиново-иридиевый цилиндрический щит. Плавление проводится или накаливанием в электрической печи пли прокаливанием на ряде горелок, усиливаемом к концу операции паяльной лампой. Для одновременного определения воды п двуокиси углерода при 1400° С и без плавней те же авторы рекомендуют применение платпново-иридпевой трубки меньших размеров, снабженной серебряной спиралью на выводящем конце.
Одним из лучших, по нашему мнению, приборов, предназначенных для работы с применением плавней, является платиновый тигель
Гуча 4 специальной формы. На рис. 39 тигель Гуча показан в форме, несколько отличающейся от оригинальной, предложенной автором, тем, что трубки для соединения этого тигля с высушивающим прибором и поглотительным сосудом сделаны из платины, а не из свинцового стекла. Если длина приводящей и отводящей трубок такая, как показано на рис. 39, то нет никакой опасности, что концы их нагреются и резиновые соединения перегорят или размягчатся.
Для удобства пользования прибором нужно иметь еще обыкновенный железный штатив с двумя небольшими передвигающимися кольцами и передвижную кольцевую горелку с вводами для газа и воздуха. На верхнем кольце штатива (см. рис. 41, стр. 913) находится треугольник из толстой платиновой проволоки, образующий в центре кольца надежную подставку для верхней части самого тигля. Как кольца, так и горелка могут быть закреплены на любой высоте.
1 G. S t е i g е г, U. S. Geol. Survey Bull., 700, 81.
2 Т. М. С h a t а г d, Am. Chem. J., 13, ПО (1891).
3 M. D i t t г i c h, W. E i t e 1, Z. anorg. Chem., 75, 373 (1912).
4 F. A. G о о c h, Am. Chem. J., 2, 247 (1880); Chem. News, 42, 326 (1880).
912
Гл. LIII. Методы анализа силикатных пород
Порошок анализируемой горной породы перемешивают в цилиндрической части тигля (7, рис. 39) не больше чем с 3—4 г совершенно обезвоженного карбоната натрия 1 или, если предполагают одновременно определять углерод, — с несколько большим количеством хромата свинца. Вставляют тигель в треугольник 2 (рис. 40), находящийся на верхнем кольце 3, закрывают крышкой с тубусом (рис. 39), которую соединяют с одной стороны с приспособлением для высушивания воздуха, состоящим из колонок с хлоридом кальция (перед которыми, если определяют также и ССЬ, помещают трубку с едким кали), с с другой стороны — с кали-аппаратом 1
Рис. 40. Прибор для определения воды по Гучу во время высушивания:
1 — водяная или толуоловая баня; 2 — треугольник; 3 — кольпо, подкладываемое под треугольник; 4 — кольцо для установки бани; о — кольцевая горелка с дутьем; 6 — асбестовая ширма.
(рис. 41), содержащим серную кислоту. Затем находящийся на краю тигля желобок 3 (рис. 39), в который входит край крышки, засыпают порошком вольфрамата натрия 2, не содержащего мышьяка (иначе край тигля скоро разрушился бы). Поднимают с помощью нижнего кольца 4 (рис. 40 и 41) металлический сосуд с холодной водой так, чтобы тигель был погружен достаточно глубоко, и направляют пламя обыкновенной паяльной го-
1 Для разложения бикарбоната, который почти всегда присутствует в карбонате натрия в качестве примеси, нагревают последний на голом огне или в воздушной бане почти до температуры плавления и ставят в эксикатор. После такой обработки карбонат натрия становится не очень гигроскопичным. Пенфильд нашел, что 2,5 г такого карбоната натрия, выставленные на часовом стекле на воздух, увеличились в массе в течение 15 мин только на 0,0002 г. Карбонат калия и смесь карбонатов калия и натрия слишком гигроскопичны и потому применяться не могут.
2 По данным Е. W, Morley, вольфрамат натрия можно вполне заменить карбонатом натрия. Если это так, то следует предпочесть последний в тех случаях, когда содержимое тигля применяют затем для других определений.
Вода. Методы определения
913
Рис. 41. Прибор для определения воды по Гучу во время сплавления:
1 — кали-аппарат с серной кислотой, служащий счетчиком пузырьков и одновременно защищающий поглотительную трубку от влаги воздуха; 2 — поглотительная трубка; 3 — защитная асбестовая пластинка; 4 — кольцо штатива; 5 — кольцевая горелка с дутьем; 6 — асбестовая ширма.
редки на край тигля, чтобы вольфрамат натрия расплавился; как только это произойдет, удаляют пламя. По застывании вольфрамата натрия образуется герметический затвор для тигля. В том, что он надежен, можно убедиться во время остывания тигля по неподвижности столба серной кислоты в соединенном с тиглем кали-аппарате, обусловленной сжатием воздуха в платиновом приборе.
После большего или меньшего высушивания в токе воздуха при 105° С в продолжение 2 ч, при помощи воздушной или толуоловой бани, как показано на рис. 40, включают поглотительную трубку 2 (рис. 41) между платиновым тиглем и кали-аппаратом с серной кислотой и, пропуская через весь прибор медленный ток воздуха, сплавляют содержимое тигля постепенным нагреванием передвижной кольцевой горелкой 5 с дутьем (рис. 40 и 41). Небольшая асбестовая пластинка 3 (рис. 41) снабженная соответствующим вырезом, защищает плавленый вольфрамат натрия от пламени. Хорошо начинать сплавление с верхнего слоя смеси в тигле и постепенно опускать горелку, пока она не достигнет дна. Таким образом избегают опасности закупоривания отводящей трубки плавом, увлекаемым в нее выделяющейся двуокисью углерода.
Момент, когда сплавление кончается, узнается в случае
с карбонатом натрия по прекращению выделения пузырьков газа в кали-аппарате, присоединенном к поглотительной трубке. Тогда прекращают нагревание и охлаждают прибор в токе воздуха.
При высоких температурах прибор слегка проницаем для газообразных продуктов сгорания светильного газа, что является недостатком. Это можно устранить тем, что описанный выше тигель Гуча вставляют в обыкновенный платиновый тигель, наполненный смесью карбонатов натрия и калия. Такой защитный тигель, однако, скоро становится непригодным для применения в других целях, так как он портится от попеременного расширения и сжатия карбоната, если плав карбоната не выливать из этого тигля после каждого сплавления прежде, чем он затвердеет.
Если, однако, определение проводить быстро и высокую температуру применять только в течение нескольких минут, то, как установлено, ошибка от проникновения водяных паров в тигель Гуча становится незначительной. Для того чтобы при такой быстрой работе быть уверенным в получаемых результатах, целесообразно в среднюю часть U-образной поглотительной трубки помещать очень тонко измельченный хлорид кальция или 58 Заказ 522.
914
Гл. LIII. Методы анализа силикатных пород
трехводный перхлорат магния (стр. 73) с целью устранить широкие воздушные каналы.
При применении как этого прибора, так и других, основанных на подобном же принципе, ток воздуха должен всегда прогоняться через прибор давлением, а не выкачиванием. Продолжительность определения можно значительно сократить, направляя струю теплого воздуха на отводную трубку вблизи ее соединения с поглотительной трубкой.
В этом приборе следовало бы определять только воду, выделяющуюся выше 100—110° С. Надежного определения гигроскопической влаги в нем сделать нельзя, потому что для герметизации прибора приходится прогревать вольфрамат натрия на прямом пламени, а в это время значительное количество водяного пара может проникнуть в прибор и частично поглотиться сухим карбонатом натрия.
Для определения воды в водных сульфатах, которые могут при прокаливании потерять часть серной кислоты, был предложен метод который может найти применение и для анализа других водных солей. Для нагревания минерала служит трубка из тугоплавкого стекла. Навеску смешивают с шестикратным по массе количеством паравольфрамата натрия 1 2 и помещают в фарфоровую или платиновую лодочку, которую вдвигают в трубку, предварительно хорошо высушенную продуванием сухого воздуха. В течение 15 мин прокаливают осторожно, при пропускании сухого воздуха со скоростью трех пузырьков в секунду. Затем температуру постепенно повышают, пока смесь не образует прозрачный сплав. При работе этим методом, конечно, должны быть соблюдены все обычные предосторожности для высушивания воздуха и поглощения воды, выделяющейся из минерала.
Этот метод был проверен на сульфатах меди, алюминия, никеля и на квасцах, калиево-алюминиевых и калиево-хромовых. Метод дает хорошие результаты, и радикал серной кислоты количественно удерживается паравольфраматом.
Описанные выше методы Гуча и Диттриха—Эйтеля позволяют в одной и той же порции материала определить одновременно с водой и другие компоненты, если это необходимо. При применении хромата свинца вместо карбоната натрия можно одновременно с водой быстро определить и графит или углерод органических веществ. Если, кроме графита или органических веществ, присутствуют карбонаты, то получают общее содержание углерода.
Если приобрести навык в работе с прибором Гуча и снабдить его приспособлением для высушивания и подвешивания, то с учетом указанных ограничений его можно признать, вероятно, самым удобным прибором для определения воды в горных породах. Высокая стоимость прибора в сравнении со стеклянными или кварцевыми трубками с течением времени окупается продолжительным сроком его работы.
1 S. В. Kuzirian, Am. J. Sci., [4], 36, 401 (1913); Chem. News, 109, 173 (1914); Z. anorg. Chem., 85, 127 (1914).
2 Чтобы очистить продажный паравольфрамат натрия, обычно содержащий соду, его плавят на паяльной горелке в большой платиновой чашке, прибавляя постепенно чистую трехокись вольфрама, пока не прекратится выделение пузырьков СО2. * Паравольфраматы — комплексные соединения, содержащие по сравнению с обыкновенными вольфраматами избыток WO3. Состав их отвечает формулам 5Na2O х 12WO3 х X п Н2О или 3Na^O • 7WO3 • п Н2О. Строение их, если принять вторую формулу, лучше всего можно изобразить в виде [W(WO4)e]Nae.*
Вода. Метода определения
915
На рис. 42 показан прибор, рекомендованный1 для определения воды
в горных породах, содержащих из летучих компонентов только воду и двуокиси углерода. Пробирка 1 из кварцевого стекла вставляется в резиновую пробку 2 так, чтобы она была слегка наклонена по сравнению со строго горизонтальным положением. Стаканчик 3 должен плотно наде-
ваться на пробку. В хлоркальциевую трубку 6 помещают от 4 до 5 г хлорида кальция.
Прибор собирают, как показано на рисунке, и нагревают дно пробирки 1 при той температуре, при которой проводят определение, и затем охлаждают. Для определения поправки на «холостой» опыт поступают следующим образом. Снимают стаканчик для взвешивания, быстро закрывают его пробкой или крышкой, взвешивают и снова помещают в прибор. Затем постепенно повышают температуру в течение 30 мин до требуемого ее значения и поддерживают эту температуру 90 мин. После этого охлаждают, снова снимают стаканчик для взвешивания, закрывают его пробкой и взвешивают. Само определение проводят, помещая в платиновую лодочку 2 г пробы, перенося
фильда:
1 — пробирка; 2 — резиновая пробка;
3 — стаканчик для взвешивания; 4 — хлорид кальция; 5 — капиллярная трубка; в — хлоркальциевая трубка; 7 — платиновая лодочка для навески пробы;
8 — асбестовая ширма.
последнюю в пробирку 1 и продолжая анализ, как при проведении «холостого» опыта. Стаканчик для взвешивания надо всегда ставить
на весы за 15 мин до взвешивания. На это время пробирку 1 закрывают таким же, но пустым стаканчиком. При анализе пород, выделяющих СО2, стаканчик для взвешивания 4 раза быстро поднимают вверх и опускают и лишь потом закрывают его пробкой. Подобным же образом поступают и при проведении «холостого» опыта 2.
МЕТОДЫ РАЗЛОЖЕНИЯ ГОРНЫХ ПОРОД
В анализе силикатных горных пород кремнекислоту, титан и большую часть основных элементов чаще всего определяют в одной и той же навеске пробы после сплавления ее с карбонатом натрия. Для определения других компонентов могут потребоваться другие методы разложения. Эти методы, а также и те вещества, которые применяются взамен карбоната натрия при сплавлении, будут описаны далее в разделе «Обычные плавни» (стр. 917).
Разложение при помощи плавней
Общие замечания. В этом разделе рассматриваются те плавни, которые находят применение в анализе минералов при исследовании различных их типов и классов; применение их может иметь своей целью: простое
1 С. О. Harvey, Bull. Geol. Survey Great Brit., 1, 8 (1939).
2 Микроаналитические методы определения воды в горных породах рассматриваются в статье F. Н е с h t, Mikrochim. Acta, 1, 127 (1938). *См. также И. П. Алима р и н, Зав. лаб., 9, 983 (1940).*
58*
916
Гл. LIII. Методы анализа силикатных пород
разложение анализируемого вещества, окисление одного или нескольких его компонентов или и то и другое вместе. Плавни и полуплавни, которые находят только какое-либо одно применение, например смесь карбоната кальция и хлорида аммония, применяемая при определении щелочных металлов в силикатах, — будут рассмотрены в соответствующих местах в связи с их специфическим применением.
За исключением борной кислоты, обычные плавни являются соединениями щелочных металлов: это или щелочные плавни — гидроокиси, перекиси, карбонаты и бораты или кислотные плавни — бисульфаты, пиросульфаты и бифториды. Сульфаты и фториды иногда применяются совместно с целью вызвать образование плавиковой кислоты и таким образом повысить разлагающее действие плавня. При температуре плавления перекиси щелочных металлов являются одновременно и сильными окислителями и разлагающими реактивами, слишком сильными для применения в платиновой посуде, если они не разбавлены другими веществами.
Сплавление с карбонатами сопровождается также окислением многих элементов, входящих в состав минералов, главным образом потому, что при сплавлении происходит более тесное соприкосновение с воздухом, чем при простом прокаливании, без применения плавня. Чтобы ускорить это окислительное действие, к карбонатам иногда прибавляют нитраты, хлораты или перекиси щелочных металлов, но обычно с течением времени окисление доходит до конца и без прибавления окислителей. Примером этому может служить минерал хромит, в котором хром (III) окисляется до хрома (VI), независимо от того, будет или не будет прибавлен окислитель. Действие сульфатов часто также оказывается окисляющим, но оно значительно слабее, чем действие гидроокисей или карбонатов. Сульфаты сравнительно легко окисляют железо (II) до железа (III), но не окисляют ни марганца (II), ни хрома (III).
Кроме обычных плавней, иногда применяют окись свинца, карбонат свинца и основной нитрат висмута, особенно при разложении силикатов.
Одним из преимуществ, которые имеют эти реактивы (а также и борная кислота) по сравнению с карбонатами щелочных металлов, является легкость удаления после выполнения ими своего назначения. Они допускают, таким образом, более совершенное выделение различных компонентов пробы, не вызывая осложнений, связанных с присутствием нескольких граммов посторонних нелетучих солей, которые особенно мешают при выделении кремния, алюминия, железа, кальция и магния. Другим преимуществом этих плавней является то, что при сплавлении с ними можно в одной и той же навеске анализируемой пробы определить, кроме кремнекислоты и обычно определяемых оснований, также и щелочные металлы. Если количество имеющейся для анализа пробы ограничено, как это часто бывает при анализе минералов, это является очень важным преимуществом, могущим превысить все отрицательные стороны этих плавней. Однако при анализе горных пород, где материала для анализа обычно бывает достаточно, применение таких плавней редко оправдывается. Еще одним преимуществом этих плавней является легкость получения их в чистом виде, не содержащими загрязнений по сравнению с карбонатами щелочных металлов.
Из трех названных соединений карбонат свинца, осажденный карбонатом аммония, и основной нитрат висмута, образующие при прокаливании окислы этих металлов, которые и служат плавнями, нужно предпо
Методы разложения горных пород
917
честь окиси свинца, потому что первые легче могут быть получены в чистом виде. В отсутствие восстанавливающих веществ и при условии защиты от продуктов сгорания светильного газа сплавление с этими плавнями можно безопасно проводить в платиновых тиглях даже небольших размеров, под хорошей тягой.
Однако применение некоторых из этих вообще мало применяемых плавней все же вызывает возражения. Сплавление с ними требует иногда очень большой затраты времени на предварительное растирание пробы в тончайший порошок; при этом возможно введение в пробу значительного количества загрязнений из применяемых для растирания приборов (см. раздел «Растирание», стр. 893), а также может произойти окисление части железа (II) (см. раздел «Железо», стр. 987). Сами плавни иногда нуждаются в тонком измельчении от руки (борный ангидрид).
С этими возражениями можно считаться гораздо меньше при анализе минералов, чем при анализе горных пород, так как задача, разрешаемая в первом случае — большей частью установление формулы — оправдывает большую затрату времени и необходимость проведения работы особенно тщательно.
Следует учитывать, что некоторые из этих плавней не могут быть применены во всех случаях, потому что существуют минералы, которые не разлагаются ими полностью в обычных условиях.
Описывать здесь простые способы сплавления с этими необычными плавнями, по-видимому, нет необходимости. Интересующихся их применением мы можем направить к оригинальным работам х.
Обычные плавни. Карбонат натрия. Чаще всего в качестве плавня применяют безводный средний карбонат натрия. За исключением некоторых особых случаев, например когда определяют хлор и фтор, нет никакого преимущества в применении более легкоплавкого KNaCO3 или смеси нормальных карбонатов калия и натрия, потому что температуры, требуемые для сплавления большей части минералов, много выше той, при которой плавится смесь карбонатов, и кроме того, соли калия, как известно, легче увлекаются осадками, чем соли натрия.
То обстоятельство, что так называемый химически чистый безводный карбонат натрия почти всегда содержит немного бикарбоната и потому при прокаливании выделяет воду, нисколько не уменьшает его пригодности; некоторые аналитики 1 2 даже советуют применять для сплавления бикарбонат натрия, который легче получить в чистом виде. При сплавлении с бикарбонатом натрия получается меньше брызг на крышке тигля, чем при сплавлении с нормальной солью. Учитывая, однако, что бикарбоната приходится брать в количестве 12—15 а на 1 г анализируемой породы, а карбоната требуется только 4—8 г, преимущество большей чистоты бикарбоната значительно снижается.
Сплавление с карбонатом натрия следует проводить только в сосудах из платины или ее сплавов.
Карбонат калия. Гигроскопичность карбоната калия делает его для сплавления минералов менее пригодным, чем карбонат натрия. Однако карбонат калия изредка применяют, главным образом потому,
1 G. В о n g, Bull. Soc. chim. France, 29, 50 (1878); W. H e m p e 1, Z. anal. Chem.. 20, 496 (1881); P. J a n n a s c h, J. L о с k e, V. anorg. Chem., 6, 321 (1894); P. J anna s c h, там же, 12, 219 (1896).
2 С. H о 1 t h о f f. Z. anal. Chem., 23, 499 (1884).
918
Гл. LIII. Методы анализа силикатных пород
что некоторые элементы дают после сплавления с ним более растворимые продукты, чем после сплавления с карбонатом натрия. Карбонат калия применяют чаще всего в некоторых методах анализа танталатов и ниобатов, а в смеси с карбонатом натрия — в тех случаях, когда надо иметь сплав с более низкой температурой плавления, чем у обоих карбонатов в отдельности. Перед применением карбоната калия его, конечно, надо обезвоживать.
Гидроокиси щелочных металлов. Гидроокиси натрия и калия являются энергичными плавнями, но редко применяются в полном анализе горных пород, потому что даже лучшие продажные препараты едких щелочей не так чисты, как карбонат и бикарбонат натрия; кроме того, для сплавления с едкими щелочами приходится применять серебряные и золотые тигли вместо платиновых; тигли из этих металлов не допускают применения таких высоких температур, при которых проводят сплавления в платиновой посуде; хотя золотые и серебряные тигли разрушаются меньше, чем платиновые, все же значительное количество металла переходит в плав и должно быть затем удалено.
Гидроокиси щелочных металлов, особенно гидроокись калия, при их плавлении более или менее обильно выделяют воду, что сопровождается вспениванием и разбрызгиванием; поэтому не следует смешивать анализируемую породу с плавнем до тех пор, пока расплав одного плавня не станет совершенно спокойным. Тогда, если известно, что реакция не будет бурной, порошок породы можно вносить в жидкий плавень небольшими порциями, в противном случае жидкости надо дать затвердеть, всыпать на застывший плавень измельченный порошок пробы и затем снова осторожно начать нагревание Б
Перекись натрия. Перекись натрия энергично разлагает минералы, все же ее не так часто применяют в полном анализе силикатов, как в техническом анализе, при определении отдельных элементов; в последнем случае ее применяют с целью перевести определяемый элемент в требуемую, более высокую степень окисления и одновременно отделить его от других элементов, которые могли бы помешать его определению.
Так, например, отделяют хром от железа с последующим его определением, во многих сульфидах определяют серу и др. Когда для этой цели применяют перекись натрия, ее полное освобождение от посторонних веществ не имеет значения, лишь бы она не содержала элемента, подлежащего определению. Сплавления с перекисью натрия, так же как и сплавления с едкими щелочами, нельзя проводить в платиновой посуде (в обычных условиях). Для этого требуются железные 1 2 или никелевые тигли. Однако тигли эти сильно разрушаются при обычном способе сплавления и потому быстро выходят из строя. Большинство элементов, входящих в состав металлов, из которых сделаны тигли, переходит в раствор при сплавлении, и этим ограничивается возможность последующего анализа.
1 Согласно С. J. vanNieuwenburg и Н. Н. Dingemans [Chem. Weekblad, 25, 266 (1928)], разложение некоторых промышленных силикатов может быть с успехом проведено сплавлением с КОН и NaOH приблизительно при 400° С в никелевом тигле; тигель очень мало разрушается, если он защищен от прямого соприкосновения с пламенем. См. также Е. W. К о е n i g, Ind. Eng. Chem., Anal. Ed., 11, 532 (1939).
2 Очень хороши железные тигли № 20 (толщиной 0,8 мм), штампованные из листов литого железа. Эти тигли содержат приблизительно 99,9% железа и только следы кремния, хрома и меди.
Методы разложения горных пород
919
В некоторых случаях можно взвесить тигель до и после сплавления и внести поправку, зная химический состав тигля. Можно применять и платиновые тигли, если внутренняя поверхность их покрыта толстым слоем плавленого карбоната натрия, затем тонким слоем плавленой перекиси натрия, а сплавление проводится при возможно более низкой температуре п не слишком продолжительное время.
При сплавлении с перекисью натрия навеску следует хорошо перемешать с 5—15 г сухой перекиси натрия, покрыть слоем перекиси натрия в 1—2 г, покрыть тигель крышкой и нагревать в электрическом муфеле при 600—700° С или на голом пламени. В первом случае постепенно вдвигают тигель в муфель и, когда содержимое тигля расплавится, осторожно поворачивают тигель, чтобы захватить жидкой массой еще неразложив-шиеся частицы, после чего нагревают еще 5 мин. Во втором случае сначала нагревают тигель с содержимым на горячей плитке 5—10 мин, чтобы удалить воду из перекиси натрия, а затем осторожно плавят содержимое тигля, держа тигель щипцами и медленно двигая им вокруг наружной части пламени, пока содержимое тигля, расплавившись, не опустится спокойно вниз. При этом надо остерегаться поднимать температуру слишком быстро, чтобы не произошло разбрызгивания. Когда содержимое тигля станет спокойно плавиться, медленно поворачивают тигель, чтобы жидкая масса захватила неразложившиеся еще частицы на дне и стенках тигля, и продолжают нагревание приблизительно при 600° С около 5 мин, после чего нагревают 1 мин при температуре около 700° С. Охлаждают сплав и затем вынимают его в виде плотной лепешки, постукивая дном тигля, пока он еще теплый, по железной плитке.
Был рекомендован 1 простой способ сплавления с перекисью натрия, при котором никелевые тигли очень мало разрушаются. Анализируемую пробу смешивают с сахарным углем и перекисью натрия и сплавляют в тигле, погруженном в воду во время сплавления и последующего охлаждения. После охлаждения сплав легко вынимается из тигля; в крайнем случае достаточно легкого удара, чтобы освободить его полностью. После сплавления обычно остается маленький несплавившийся остаток. Последний, однако, быстро оседает после растворения плава в кислоте и может быть легко перенесен на фильтр и снова сплавлен таким же образом с перекисью натрия или с карбонатом натрия в платиновом тигле. Хотя опасность сильного взрыва незначительна, все же во время сплавления смеси следует надевать очки.
При работе этим способом 0,5—1 г тонко растертой пробы и 0,5—0,7 г сахарного угля (готовится прокаливанием кристаллического сахара в закрытом фарфоровом тигле) вносят в никелевый тигель высотой 52 мм и диаметром 44 мм, имеющий вместимость около 60 мл. Всыпают в тигель 15 г свежей, сухой, желтой перекиси натрия, хорошо перемешивают, поворачивая тигель левой рукой вокруг шпателя, который держат погруженным в смесь под углом, и, наконец, счищают кисточкой в тигель приставшие к шпателю частицы. Смесь плотно утрамбовывают, покрывают тигель крышкой, имеющей в центре небольшое отверстие диаметром 5—6 мм, и погружают тигель на две трети его высоты в воду. Если проводят сразу много сплавлений, то тигли лучше всего помещать на металлический лист
1 W. F. М u е h 1 b е г g. Ind. Eng. Chem., 17, 690 (1925).
920
Гл. L1II. Методы анализа силикатных пород
с рядом отверстий для тиглей. Немедленно берут тигельными щипцами кусок хлопчатобумажного шнура длиной около 10 см, зажигают его конец и вводят в тигель через отверстие в крышке. Когда сплав охладится, резко ударяют тигель об стол, если плав не отстал сам от стенок и дна тигля, и переводят содержимое тигля в сухой стакан емкостью 600 мл, который покрывают часовым стеклом. Наполнив тигель водой, выливают затем полученный раствор в стакан, отодвинув закрывающее стакан часовое стекло в сторону, и тотчас же снова закрывают стакан.
Когда бурная вначале реакция пойдет спокойнее, смывают оставшиеся частицы плава с тигля и крышки в стакан и разбавляют раствор до 300 мл. Приливают в избытке соляную кислоту при сильном перемешивании раствора. При приближении к точке нейтрализации соляную кислоту добавляют очень медленно, чтобы избежать слишком сильного вспенивания. Если остается нерастворимый остаток, то дают ему осесть, фильтруют, остаток умеренно промывают, высушивают и прокаливают в платиновом тигле. Затем сплавляют остаток с возможно меньшим количеством карбоната натрия, охлаждают, растворяют плав в разбавленной кислоте и приливают полученный раствор к главному раствору.
При исследовании этого метода были сделаны следующие выводы х. 1) В большинстве случаев анализа рекомендуется применение 15 г смеси, состоящей из 150 г перекиси натрия и 10,0 г растертого до 100 меш (d = 0,147 лл) сахарного угля; 2) с трудом сплавляющиеся минералы, например корунд, следует растереть до порошка, проходящего через спто в 100 меш (d = 0,147 .м); 3) несплавившийся остаток при обработке навески в 0,5—1 г не должен весить больше 2 мг,— это не подвергшиеся разложению частички, отброшенные на крышку тигля в результате разбрызгивания плава; 4) температура сплавления, по-впдимому, превышает 1450 °C; 5) после многократного применения никелевого тигля никакой потери в его массе нельзя было обнаружить.
Бура. Бура является очень энергичным, эффективно разлагающим горные породы плавнем. Применяется она главным образом при анализе некоторых трудносплавляемых минералов, например хромита и циркона. К сожалению, при ее применении почти всегда необходимо полностью удалять бор после сплавления и лишь затем приступать к обычным аналитическим определениям (см. стр. 929, а также «Кремний», стр. 752). Если сплавления с бурой приходится делать часто, то лучше всего приготовить запас плавленой соли. Если бура применяется изредка, то, поместив в платиновый тигель необходимое количество кристаллической соли, сначала обезвоживают ее и плавят, затем охлаждают и на охлажденный и затвердевший плав высыпают навеску анализируемой пробы. Так как в расплавленном состоянии бура обладает очень вязкой консистенцией, рекомендуется помещать в тигель во время сплавления короткую толстую платиновую проволоку, которой время от времени перемешивают массу.
Борный ангидрид. Подобно буре, борная кислота является также сильнодействующим неокисляющим плавнем, который обладает теми же достоинствами, какие имеются у буры, но применение борной кислоты создает то же затруднение: после сплавления бор надо удалить и лишь потом можно приступить к анализу. Практически борную кислоту применяют в анализе силикатных горных пород и кислотоупор-
1 G. G. М а г v i п, W. С. S с h u m b, J. Am. Chem. Soc., 52, 574 (1930).
Метода разложения горных пород
921
ных минералов только с целью определения в них щелочных металлов
Яннаш и Вебер 1 2 заметили, что при сплавлении с борной кислотой происходит, по-видимому, полное выделение фтора без потери кремнекислоты, что придает этому плавню особый интерес. Их наблюдение было подтверждено последующими работами 3, в которых описано успешное применение борной кислоты при анализе криолита и для удаления фтора из искусственных фторофосфатов и фторованадатов, чтобы облегчить определение в них фосфора и ванадия. Однако исследование Гофмана и Ленделя 4 не подтвердило результатов предыдущих работ.
Если в одной и той же пробе одновременно с кремнекислотой и пр. должны быть определены и щелочные металлы, то необходимо применять для сплавления борную кислоту, совершенно этих металлов не содержащую; ее можно приготовить двойной или тройной перекристаллизацией продажного препарата. Очищенную таким образом кристаллическую борную кислоту обезвоживают плавлением в большом платиновом тигле. Тигель затем быстро охлаждают, чтобы полученный борный ангидрид распался на куски, которые легко могут быть растерты в порошок. Куски эти сохраняют в плотно закрывающемся сосуде и измельчают каждый раз только необходимое для определения количество, потому что безводный борный ангидрид гигроскопичен. При условии очень тонкого измельчения почти все силикаты легко разлагаются сплавлением с этим плавнем. Авторы рекомендуют измельчать пробу массой от полуграмма до грамма в продолжение 0,5—1 ч.
Бисульфаты и пиросульфаты. Для сульфатного сплавления применяют бисульфаты и пиросульфаты калия и натрия. Раньше рекомендовали применение бисульфатов, но так как бисульфаты становятся активными только тогда, когда они при плавлении превращаются в пиросульфаты, а пиросульфаты теперь имеются в продаже, то лучше применять последние, а не бисульфаты. Другим доводом против широкого применения бисульфатов является то, что при сплавлении с ними вначале надо строго следить, чтобы не произошло выбрасывания массы из тигля, потому что выделение большого количества воды сопровождается вспениванием и вещество поднимается к крышке тигля.
Сплавление с пиросульфатами протекает значительно спокойнее. Если пиросульфатов нет под рукой, а есть бисульфаты, то последние легко могут быть превращены в пиросульфаты предварительным плавлением их в платиновой чашке; расплавленное состояние поддерживают, пока не прекратится вспенивание и не начнут выделяться белые пары. Таким способом за одну операцию можно приготовить значительный запас пиросульфата. Если плав разлить по другим чашкам, он застынет тонким слоем или кусочками, которые можно разбить и хранить в банке для дальнейшего
1 Р. Jannasch, О. Heidenreich, Z. anorg. Chem., 12, 208 (1896). Этот метод разложения горных пород с целью определения содержания в них щелочных металлов был первоначально предложен Н. Davy, Phil. Trans., 231 (1805).
У. Jannasch и Heidenreich оригинален способ освобождения от введенной борной кислоты.
3 Р. Jannasch, Weber, Вег., 32, 1670 (1899).
3 A. Travers, Bull. Soc. chim. France, 31, 293 (1922); 33, 297 (1923).
♦ J. I. Hoffman, G. E. F. L u n d e 1 1, J. Research NBS, 3, 581 (1929).
922
Гл. LIII. Методы анализа силикатных пород
употребления. Если нельзя достать хороший препарат бисульфата, то нужно только смешать и сплавить вместе эквивалентные количества серной кислоты и нормального сульфата, пока не испарится вода и не закончится превращение в пиросульфат. Рекомендуется 1 2 применение натриевой соли вместо калиевой.
Пиросульфат натрия плавится быстрее, чем пиросульфат калия, и образует более растворимые двойные соли с алюминием и с некоторыми другими металлами. Разложение минералов пиросульфатом натрия идет не так быстро вследствие большой склонности натриевой соли создавать корку на поверхности плава; указанная особенность пиросульфата натрия является его небольшим недостатком.
Для сплавления минералов с пиросульфатами применяют платиновые тигли Нагревание проводится лишь до степени, достаточной для поддержания плава в жидком состоянии, пока не растворится весь порошок минерала. Этот момент не всегда отчетлив, потому что жидкость часто теряет прозрачность, даже если она не покрыта коркой. В этом случае тигель подносят к свету и дают ему охладиться. В некоторый определенный момент охлаждения жидкость становится снова прозрачной, и тогда ясно видно дно тигля и неразложившийся остаток, если он имеется. Такие явления чаще наблюдаются при применении соли калия, чем при применении соли натрия. Необходимо обращать больше внимания на отношение между величиной навески исследуемого вещества и взятым количеством пиросульфата, а также и на температуру сплавления, чем это обычно делают 3.
При очень длительном сплавлении иногда бывает необходимо возвратить плавню его первоначальную активность. Для этого плав охлаждают, прибавляют 1 каплю или более концентрированной серной кислоты, осторожно нагревают и затем снова продолжают сплавление. Такое прибавление кислоты может быть также целесообразным в конце обычного сплавления, если разложение плавня дошло до образования нейтрального сульфата; дело в том, что при сплавлении может произойти осаждение солей одного или нескольких компонентов минерала, которые затем с трудом растворяются в последующих операциях. Это особенно относится к титану и цирконию, не говоря уже о тантале и ниобии.
При сплавлении с пиросульфатами платиновый тигель всегда разъедается в большей или меньшей степени, тем больше, чем дольше продолжается операция сплавления. Необходимо помнить, что перешедшую в раствор платину 4 нужно удалить в одной из последующих стадий анализа.
Бифториды. Бифториды, обычно калиевая соль KHF2, применяются для разложения некоторых с трудом сплавляемых минералов, например танталатов, ниобатов и циркона. Эти плавни применяются чаще для определения одного какого-нибудь компонента, чем для полного анализа. В анализе силикатов плавление с бифторидами не может, конечно, применяться в тех случаях, когда хотят определить кремний,
1 J. L. S m i t h, Am. J. Sci., 40, 248 (1865).
2 Для некоторых работ их можно заменить кварцевыми.
3 G. W. S ears, L. Q u i 1 1, J. Am. Chem. Soc., 47, 922 (1925); G. W. S e a r s, там же, 48, 343 (1926); 51, 122 (1922). См. «Титан» (стр. 650) и «Ниобий и тантал» (стр. 663).
4 В среднем около 3 мг при обычном сплавлении.
Методы, разложения горных пород
923
но и при анализе материалов, содержащих кремний, этот метод может быть очень полезным для определения других компонентов, в особенности потому, что при сплавлении происходит удаление кремния.
Сплавление с бифторидами можно проводить только в платиновой посуде, причем платина во время сплавления не переходит в раствор. Температура, требуемая для сплавления, очень низка, и операция сплавления заканчивается в значительно более короткий срок, чем при применении других плавней. Особых указаний для проведения сплавления не требуется, но нужно помнить, что фториды многих металлов более или менее летучи и что поэтому температура и продолжительность сплавления должны быть доведены до возможного минимума, особенно когда анализируют минералы, содержащие тантал, ниобий, титан и цирконий.
Выбор плавня. Выбор плавня определяется составом породы и целью анализа. Если имеют в виду определение только одного элемента, то может быть пригоден плавень, который не годился бы в тех случаях, когда необходимо определять несколько компонентов в одной навеске минерала или породы.
Для разложения большей части из обширного списка силикатов и других окисленных минералов, нерастворимых в кислотах, наилучшим плавнем является карбонат натрия. Это справедливо также и в отношении многих растворимых в кислотах минералов, например фторофосфатов кальция и фосфатов вообще. При анализе многих из таких веществ сплавлением с карбонатами не только получаются все анионы в водной вытяжке, в которой они могут быть осаждены или иным образом определены, но одновременно достигают и отделения катионов от мешающих анионов, так что становится возможным определение ряда элементов в той части плава, которая не растворяется в воде.
Невозможно, да и бесполезно указывать все случаи, когда может оказаться необходимым или желательным применение сложных плавней, описываемых на стр. 928, или отдельных плавней, достаточно лишь нескольких замечаний; дополнительные указания можно найти в главах, посвященных отдельным элементам и группам минералов. Плавни, описываемые на стр. 928, применяются главным образом при анализе сульфидов и арсенитов; перекись натрия и хлорат натрия, если их применять в чистом виде, без разбавления, слишком энергично действуют на такие вещества. Целью применения этих плавней является окисление серы до сульфата, а мышьяка и сурьмы до арсената и антимоната. Большую часть упомянутых выше минералов можно, правда, окислить и мокрым путем, но сплавление со щелочами имеет, как выше указано, то преимущество, что при обработке плава водой происходит отделение анионов от многих элементов, которые могли бы помешать впоследствии их определению. Кроме того, этот метод делает возможным непосредственное определение некоторых элементов без предварительного их осаждения, например мышьяка без выделения его сероводородом.
Карбонат калия применяется редко. Им пользуются иногда только потому, что некоторые из продуктов сплавления с ним более растворимы, чем соответствующие соединения натрия.
Тетраборат натрия и борный ангидрид применяются только при анализе окисленных веществ, нелетучих при требуемой для сплавления температуре. Как правило, прежде чем приступить к анализу, из раствора полученного плава следует удалить бор. Борный ангидрид имеет то
924
Гл. Ы1Г Методы, анализа силикатных пород
преимущество перед тетраборатом натрия, что при его применении не вводится нелетучее твердое вещество.
Хлорат калия никогда не применяется один из-за возможности взрыва.
Перекись натрия может применяться одна, но при работе с неокислен-ными веществами необходима большая осторожность.
Пиросульфаты применяются только при анализе окисленных веществ или когда нужно в самом начале удалить фтор из веществ, не содержащих кремния. Несмотря на сильное разлагающее действие этих плавней даже на некоторые силикаты (циркон), существует много других силикатов, которые при сплавлении с рассматриваемыми плавнями разлагаются медленно и не полностью, например полевые шпаты. Чаще всего пиросульфаты применяются для первоначального разложения танталатов и ниоба-тов, но в последующем ходе анализа ими часто пользуются для переведения в раствор прокаленных смешанных окислов с целью определения их компонентов.
Бифторпды применяют очень редко, только для разложения некоторых тугоплавких веществ, свободных от кремнекислоты или в которых кремнекислоту предполагают определять после разложения другими способами. Улетучивание некоторых фторидов металлов во время сплавления препятствует более широкому применению бифторидов.
Ход работы при сплавлении с карбонатом натрия. Сплавление окисленных минералов в отсутствие заметных количеств хлора, фтора и сульфидной серы. Предполагается, что небольшое количество серы, которое может присутствовать в пробе, будет определено в отдельной навеске. 4—6 г карбоната натрия тщательно смешивают с 1 г порошка горной породы или минерала в платиновом тигле емкостью 20—30 мл 1. Не следует применять значительно большие количества плавня, рекомендуемые некоторыми авторами, за исключением, быть может, некоторых особых случаев, потому что это приводит к большему загрязнению и необходимости дольше промывать осадки.
В действительности при анализе многих материалов лучше применять меньшие количества плавня, чем в обычных сплавлениях. Рекомендуется2 следующий ход разложения алюмосиликатов. В платиновом тигле в течение по крайней мере 5 мин, пользуясь платиновой палочкой, тщательно смешивают 0,5 г растертой в порошок анализируемой пробы с 0,6 г также измельченного в порошок безводного карбоната натрия. Затем покрывают тигель крышкой и нагревают при 875° С в течение 2 ч. Охлаждают плав, приливают 1—2 мл воды, дают постоять около 10 мин и перемешивают или раздавливают все кусочки, если нужно, чтобы измельчить их возможно полнее. Затем содержимое тигля переносят в стакан или чашку, применяя для обмывания тигля не более 50 .тел воды. Нагревают до кипения, непрерывно перемешивая, и быстро приливают 20 .м концентрированной соляной кислоты, раздавливают снова возможно лучше все оставшиеся кусочки, выпаривают досуха и продолжают дальнейшее определение крем-некпслоты, как обычно3.
Тигель покрывают крышкой и помещают над маленьким пламенем, которое затем постепенно увеличивают до максимума (приблизительно
1 При анализе легкоразлагаемых материалов, например кальциево-натриевого стекла, сплавление можно проводить в платиновой чашке типа Раупе, в которой затем можно растворить плав и выпарить раствор для отделения кремнекислоты, не перенося его в какой-либо другой сосуд.
2 A. N. Finn, J. F. К 1 е к о t к a, J. Research NBS, 4, 813 (1930).
3 См. также J. I. Hoff man, J. Research NBS, 25, 379 (1940).
Методы, разложения горных пород
925
1000° С), и держат его так, пока расплавленная масса не станет спокойной; бурной реакции не должно быть. Содержимое тигля в случае большого содержания кремнекислоты будет тогда представлять собой вязкую жидкость, иногда почти прозрачную, но чаще более или менее мутную, которая при переносе тигля на пламя паяльной горелки (приблизительно 1200° С) не дает вскипания или выделяет лишь небольшое число пузырьков газа. Плавы такого характера легко разлагаются водой. При анализе веществ, содержащих меньшие количества кремнекислоты, сплавление происходит менее отчетливо, но это вовсе не означает неполного разложения. Вообще желательно, а иногда и необходимо нагревать эти менее плавкие смеси приблизительно до 1200° С; при Этом часто бывает отчетливо заметно дальнейшее выделение двуокиси углерода; крышку тигля следует приоткрывать осторожно, чтобы избежать потерь от вскипания. Это вскипание вызывается не только дальнейшим действием карбоната натрия на анализируемые минералы, но чаще разложением карбонатов щелочноземельных металлов, которые реагируют затем с другими компонентами плавня и минерала или породы с образованием сложных силикатов и, возможно, алюминатов, если присутствуют кремний или алюминий.
Большое заблуждение считать, как это многие делают, что щелочноземельные металлы, магний, железо и марганец после сплавления при 1200° С присутствуют в виде карбонатов. Они редко находятся в этом состоянии, даже если сплавление произведено при 900—1000° С.
Некоторые с большим трудом сплавляемые минералы (например, хромит или циркон) могут потребовать нагревания при 1200° С в течение 1—2 ч и даже дольше.
Как общее правило, пламя паяльной горелки должно быть направлено не вертикально на дно тигля, а под углом сбоку; оно не должно охватывать всего тигля. Этой предосторожностью раньше часто пренебрегали; ее, однако, надо всегда иметь в виду, когда в породе присутствуют восстанавливающиеся вещества. Если пренебречь этой предосторожностью, то внутри тигля не создастся необходимая окислительная атмосфера и наступит восстановление, которое может иметь серьезные последствия. Особенно часто это происходит в тех случаях, когда проба (например, силикатная горная порода) содержит пирит или другие сульфиды в количествах, превышающих следы. Тогда после очцстки и прокаливания тигля может появиться потемнение на его внутренней поверхности, происходящее от того, что восстановленное железо, сплавившееся с платиной, вновь окисляется. В исключительных случаях это железо может весить даже несколько миллиграммов. Его можно удалить, прокаливая тигель, обрабатывая потом соляной кислотой или (при высокой температуре) бисульфатом, снова прокаливая и повторяя эту обработку несколько раз.
Чтобы избежать указанного затруднения, а также применения селитры при сплавлении пород, содержащих пирит или углеродистые вещества, следует сначала осторожно обжечь навеску в тигле, который будет служить для сплавления. При этом тигель несколько раз повертывают, чтобы измельченная проба возможно полнее соприкасалась с воздухом. При анализе пород с очень большим содержанием пирита обжиг пробы целесообразно вести в фарфоровом тигле. В этом случае после обжига переносят пробу в платиновый тигель и, если после очистки кисточкой из верблюжьей шерсти в фарфоровом тигле останутся еще
926
Гл. LIII. Методы анализа силикатных пород
приставшие к его стенкам частицы, «смывают» их небольшим количеством сухого карбоната натрия *. Если в плаве предполагают определять сульфаты, то прокаливание следует проводить в муфеле или каким-либо способом удалять продукты сгорания светильного газа, содержащие серу 1 2.
Иногда цвет охлажденного плава указывает на отсутствие марганца, хотя в действительности он присутствует в количестве, обычно дающем яркое окрашивание плава. Два плава порошка одной и той же горной породы, полученные при одинаковых, по-видимому, условиях, могут в одном случае показать малое содержание марганца или отсутствие его, а в другом — значительное. (Поэтому отсутствие сине-зеленого окрашивания охлажденного плава не может служить указанием на отсутствие марганца, что, однако, бывает только при малом его содержании.) Это различие мы можем объяснить только тем, что в одном тигле была нейтральная или восстановительная атмосфера, а в другом — окислительная. Появления зеленой окраски можно ожидать, естественно, не раньше, чем совершенно окислятся все другие окисляющиеся компоненты породы, например сульфиды, железо (II) и органические вещества. Это, однако, происходит очень быстро, если воздух имеет доступ к поверхности плава.
Плав может оказаться желтым от присутствия хромата натрия, если в анализируемой пробе был хром, а содержание марганца оказалось недостаточным, чтобы маскировать окраску хромата. Водная вытяжка плава будет зеленой, если присутствует марганец, и желтой, если в растворе находится один хром без марганца. Если присутствуют оба, то желтую окраску хрома можно выявить, нагрев раствор с несколькими каплями спирта и, если нужно, отфильтровав выпавший осадок соединении марганца 3.
Если плаву осторожно придать вращательное движение, пока он охлаждается, и затем как следует охладить, то его можно обычно отделить от тигля, постукивая перевернутым тиглем по дну чашки, в которой предполагают растворять плав. В исключительных случаях, когда это не удается, отделению плава от стенок тигля помогают осторожным сдавливанием последнего, недостаточным, однако, для того, чтобы деформиро
1 Dnp arc [Bull. Soc. franc, mineral., 42, 156 (1919)] приписывает сплавление платины с железом восстанавливающему действию газов пламени, которые проникают через платину, и предлагает для защиты от них сплавление в муфеле. При этом, однако, появляется другой источник ошибок — сильное разъедание тигля по краям расплавленной щелочью и происходящее вследствие этого введение платины в анализируемый раствор. Исследования, проведенные Е. Wichers (Бюро Стандартов США), показали, что такое разъедание происходит при сплавлении в муфеле, но не на пламени паяльной горелки. Объясняется это, вероятно, тем, что при нагревании на голом Пламени всползающий по стенкам плавень остается в виде карбоната, а в муфеле он превращается в едкую щелочь. Этим, быть может, объясняются и противоречивые утверждения о действии расплавленного карбоната натрия на платину.
2 Не следует думать, что применение муфельной печи для сплавления дает гарантию отсутствия загрязнения серой. В нашей практике наблюдались случаи загрязнения щелочных плавов соединениями серы из атмосферы внутри муфеля, если последним раньше пользовались для прокаливания сульфидов или других летучих соединений серы.
3 Выщелачивание содовых плавов водой с последующим фильтрованием и промыванием остатка водой далеко не всегда приводит к четкому отделению кремния, алюминия и олова в виде их растворимых соединений от свинца, образующего в этих условиях нерастворимое соединение. На распределение этих элементов между остатком и фильтратом часто влияют другие сопутствующие им элементы.
Методы разложения горных пород
927
вать тигель. Другой простой способ освобождения плава состоит в том, что его медленно охлаждают, затем быстро нагревают тигель до 300— 400° С и тотчас же погружают его на две трети в холодную воду. Еще один прием состоит в том, что плаву дают затвердеть, как в предыдущем способе, наполняют охлажденный тигель до его половины водой и затем через минуту осторожно нагревают (но не до кипения раствора) по краю плава маленьким пламенем. Когда плав по краям отпадет от стенок, осторожно нагревают дно тигля, пока весь плав не отделится от тигля. Если потеря времени не имеет значения, можно поместить в плав согнутый конец толстой платиновой проволоки, пока плав еще не затвердел, затем после остывания снова нагреть, чтобы расплавился тонкий слой плава у стенок и дна тигля и вынуть плав или большую его часть, потянув за проволоку щипцами Ч
Сплавление окисленных минералов, содержащих значительные количества хлора или фтора при отсутствии заметных количеств сульфидной серы. При анализе хлорсодержащих минералов, допускающих сплавление их с карбонатами (галогениды серебра нельзя, например, сплавлять с карбонатами), или минералов, содержащих фтор в количествах, достаточных для его определения или для того, чтобы он помешал определению кремния и алюминия (см. стр. 940—946), в качестве плавня применяют эквимолекулярную смесь карбонатов калия и натрия. Пламя паяльной горелки не следует применять, если достаточна более низкая температура. Чтобы уменьшить опасность потери хлоридов и фторидов щелочных металлов вследствие их улетучивания, целесообразно применять тигель Смита (см. рис. 45, стр. 1007).
Следует отметить, что некоторые фториды (плавиковый шпат CaF2) и некоторые фосфаты [апатит или Са3(РО4)2] разлагаются не настолько полно, чтобы их анионы целиком перешли в фильтрат при выщелачивании плава водой. Для извлечения этих анионов надо перед сплавлением прибавить к измельченной пробе, кроме плавня, еще немного кремнекислоты. Эти случаи будут рассмотрены в разделах об определении фтора и фосфора.
Сплавление сульфидов и арсенидов. Если сплавлению с карбонатом натрия подлежат минералы, содержащие сульфидную серу или мышьяк, то при сплавлении добавляют немного нитрата, хлората или перекиси щелочного металла, свободных от серы и мышьяка; прибавление этих веществ имеет целью окисление отдельных компонентов пробы, а не облегчение сплавления. При этом следует соблюдать осторожность, чтобы отношение количества прибавленного окислителя к общему количеству плавня не оказалось слишком большим, что могло бы привести к повреждению тигля (если применяется платиновый тигель) или вызвало бы слишком бурную и быструю реакцию, связанную с потерей вещества вследствие выбрасывания и улетучивания. Последний источник ошибок особенно опасен при сплавлении арсенидов, когда теплота происходящей экзотермической реакции может вызвать улетучивание части вещества прежде, чем оно начнет окисляться, что можно обычно сразу узнать по характерному
1 Для облегчения растворения содовых плавов Р. S. Y о u n g [Ind. Eng. Chem., Anal. Ed., 16, 590 (1944)] рекомендует выливать расплавленную массу в углубления, сделанные в специальной плитке из монель-мета л ла. Образующиеся плоские лепешки твердого плава легко растворяются.
928
Гл. LIII. Методы анализа силикатных пород
запаху. Возникновения слишком сильной реакции можно избежать, нагревая вначале очень осторожно и, если нужно, по временам совсем прекращая нагревание тигля. Следует ограничиваться таким количеством окислителя, которое было бы лишь на немного больше необходимого для полного окисления всех окисляющихся компонентов пробы.
Сплавление со с месью карбоната натрия с нитратом калия. 1 вес. часть анализируемого минерала1 (обычно 0,5—1 г в зависимости от содержания серы) тщательно перемешивают в тигле с 10 вес. частями смеси карбоната натрия и нитрата калия п покрывают слоем плавня. Нитрат калия лучше нитрата натрия главным образом потому, что он не гигроскопичен. Отношение массы нитрата к массе карбоната обычно рекомендуется равным 1:2, но некоторые авторы советуют 1 : 4. Можно брать еще меньше нитрата калия, если анализируемый минерал содержит малоокисляющихся веществ. Чем ниже содержание нитрата в смеси, тем меньше портится тигель, что очень важно, если он платиновый. Если определяют только серу или мышьяк, то можно применять никелевые тигли.
Нужно принимать особые меры предосторожности против попадания продуктов сгорания светильного газа внутрь тигля, применяя свободное от серы горючее (например, спирт) или устанавливая приспособление, показанное на рис. 6 (стр. 49). Сначала нагревают осторожно, затем более сильно, пока содержимое тигля не расплавится, и еще 15—20 мин после плавления. Если плав или его раствор нужно нагревать с соляной кислотой, то этого нельзя делать в платиновой посуде.
Сплавление со смесью карбоната натрия с перекисью натрия. Перекись натрия в смеси с карбонатом натрия применяется в анализе минералов только для определения серы и хрома. Для определения других компонентов она применяется редко, потому что с ней в плав вводится больше загрязнений, чем с нитратом калия. С другой стороны, она действует даже более активно, чем смесь карбоната с нитратом, и для сплавления требуется меньше времени. Здесь также необходимы указанные выше меры предосторожности против попадания продуктов сгорания светильного газа внутрь тигля.
Сплавляют 1 вес. часть минерала приблизительно с 10 вес. частями перекиси натрия и 8 вес. частями карбоната натрия в никелевом тигле с защитным приспособлением, указанным выше (стр. 924), в течение 10 мин и затем нагревают 20 мин на полном пламени горелки. Можно пользоваться платиновым тиглем, если сначала покрыть всю внутреннюю поверхность тигля расплавленным карбонатом натрия и дать ему остыть; этим способом тигель будет предохранен от разъедания, при условии, что сплавление будет проводиться при возможно более низкой температуре и не слишком долго.
Сплавление со смесью карбоната натрия с хлоратом калия. Сплавляют 1 вес. часть анализируемого минерала с 5 вес. частями карбоната натрия и 1 вес. частью хлората калия. Сплавление ведут сначала осторожно, затем более сильно, пока не перестанет выделяться кислород.
1 Не следует очень тонко растирать пробу из-за опасности потерь вследствие окисления. Например, пирит при его растирании выделяет немного двуокиси серы (стр. 794).
Методы разложения гор пых пород
929
При применении этой смеси можно пользоваться платиновыми тиглями, что позволяет определять большее число компонентов, чем при работе в никелевых или железных тиглях.
Ход работы при сплавлении с борным ангидридом. Сплавление легкоразлагаемых силикатов. Смешивают 1 вес. часть тонко измельченного минерала с 3—8 вес. частями плавня, в зависимости от природы анализируемого силиката, в платиновом тигле емкостью 40 — 65 мл. Для выделения воды сначала нагревают на маленьком пламени 5 —10 мин. Затем величину пламени постепенно увеличивают до полного пламени горелки, температура при этом повышается. Насколько возможно, препятствуют вскипанию и поднятию плава, перемешивая его платиновой палочкой, которая по длине своей не должна выступать за край тигля и которую не вынимают во время сплавления. Продержав массу некоторое время в расплавленном состоянии в закрытом тигле, нагревают на паяльной горелке 20—30 мин или более, если это нужно, до полного исчезновения несплавившихся частиц.
Сплавление тру д нораз лаг аемых силикатов. Для сплавления таких минералов, как андалузит, кианит, топаз, которые не вполне разлагаются при применении обычной паяльной горелки, применяют газо-кислородное пламя х. В паяльную горелку с отверстиями в 2,5 мм дают много газа (авторы метода берут газ из пяти или шести обычных газовых кранов) и устанавливают широкое и несветящееся пламя. К минералу, который был сначала прокален, как указано выше (стр. 924), но с значительно большим количеством плавня — 30 вес. частей на 1 вес. часть минерала, прибавляют еще несколько граммов борного ангидрида и нагревают тигель газо-кислородным пламенем 10 — 15 мин пока плав не станет прозрачным, как стекло.
Обработка после сплавления. Начиная с этого момента, дальнейшая обработка в обоих случаях одинакова 1 2 и проводится следующим способом.
Горячий тигель (закрытый, чтобы избежать потери от разбрызгивания частичек плава) охлаждают в воде и содержимое его переносят в болыпу фарфоровую или платиновую чашку. Покрыв чашку часовым стеклом, обрабатывают плав насыщенным раствором хлористого водорода в мети^ ловом спирте 3.
Сняв часовое стекло, нагревают жидкость до кипения на асбестовой сетке небольшим пламенем (высотой 2,5 см) при постоянном перемешивании или представляют массу самой себе, нагревая чашку на еще меньшем пламени или на водяной бане, нагретой почти до кипения. Приставший к тиглю остаток плава обрабатывают подобным же образом и присоединяют его к главной массе в чашке. Если прибавлять время от времени немного раствора хлористого водорода в метиловом спирте (хлористого метила), то через 10—15 мин все растворяется. Затем кипячением доводят жидкость до небольшого объема и выпаривают досуха на водяной бане. Остаток обрабатывают еще 3 или 4 раза на водяной бане при 80 — 85° С раствором того же эфира для удаления последних следов бора в виде
1 *В паяльную горелку вместо воздуха подается кислород. Прим, ред*
2 См. сноску 2 на стр. 921.
3 Приготовляется пропусканием сухого хлористого водорода в охлаждаемый метиловый спирт в течение 1—2 ч.
59 Заказ 5 22.
930
Гл. LIII. Методы, анализа силикатных пород
борнометилового эфира. При этом каждый раз надо стараться смыть хлористым метилом борную кислоту, приставшую при выпаривании к стенкам чашки. Последующую обработку силиката см. ниже (стр. 940).
Недостатки метода. 1) Борнометиловый эфир, отгоняемый в воздух в значительных количествах, тотчас разлагается при соприкосновении с влагой, и борная кислота осаждается на всех окружающих предметах. Поэтому для таких выпариваний необходимо устроить особый вытяжной шкаф, иначе борная кислота всегда может попасть в другие сосуды и вызвать невероятную путаницу в анализах. 2) Второй недостаток связан с применением газо-кислородного пламени в тех случаях, когда после сплавления предполагают определять щелочные металлы. Что этот метод допускает определение последних, является одним из основных его преимуществ, однако нельзя сомневаться в том, что при высокой температуре газо-кислородного пламени соли щелочных металлов могут частично улетучиться. Борат натрия может медленно, но пол-ностьТо улетучиться даже на обыкновенном пламени паяльной горелки; поэтому есть основания опасаться, что при более высокой температуре могут произойти значительные его потери. 3) Неполное удаление борного ангидрида может привести к ошибкам в последующем анализе.
Когда не имеют в виду определения щелочных металлов, безусловно лучше применять не борный ангидрид, а плавленую буру, потому что первый действует менее энергично.
Разложение кислотами
Ход анализа, в котором алюминий и подобные ему элементы отделяются обычными методами после предварительного разложения породы плавиковой и серной кислотами, а кремнекислота определяется в отдельной навеске, — давно уже был оставлен одним из нас (В. Ф. Гиллебранд) после продолжительного его применения. Привлекательный в принципе этот метод приводит, однако, иногда к затруднениям, вызываемым неполным удалением фтора и также (в меньшей степени) присутствием в полученном растворе сульфатов вместо хлоридов. Удаление фтора можно сделать более полным, если раствор, выпаренный до выделения паров серной кислоты, охладить, обмыть края чашки небольшим количеством воды и снова выпарить до выделения паров серной кислоты, прибавив в небольшом избытке чистую измельченную кремнекислоту \ лучше всего искусственно приготовленную. В этом случае полученный раствор надо профильтровать, остаток кремнекислоты прокалить, обработать серной и плавиковой кислотами, раствор выпарить до полного удаления плавиковой кислоты и полученный остаток присоединить к главному раствору.
В некоторых случаях, особенно когда присутствуют щелочноземельные металлы, разложение лучше проводить плавиковой и хлорной кислотами, потому что тогда получаются растворимые перхлораты, которые не затрудняют дальнейшего анализа. Однако при применении хлорной кислоты несколько труднее удаляется фтористый водород. Если обстоятельства позволяют, то нагревание следует продолжать до получения сухого остатка. В последней стадии выпаривания иногда необходимо
1 A. A. N о у е s, Technol. Quart., 16 (2), 101 (1903).
Кремний
931
прибавление кремнекислоты, так же как и при выпаривании с серной кислотой.
Яннаш 1 помещает тонко растертый порошок породы в платиновую пробирку емкостью 26 мл, обливает его небольшим количеством несколько разбавленной (4 : 1) соляной кислоты, закрывает открытый конец пробирки не герметически прилегающим к ней колпачком и вставляет эту пробирку в большую, наполненную такой же соляной кислотой пробирку из калийного стекла. Последнюю он запаивает и вставляет в поставленную наклонно цельнотянутую стальную трубку (трубку Маннесмана), содержащую эфир или бензин для уравнения давления, и нагревает до любой желаемой температуры в пределах до 400° С.
Главным недостатком этого метода является, по-видимому, неполное разложение пробы, которое, без сомнения, происходит от наклонного по необходимости положения трубки. По этой причине порошок пробы собирается у нижнего конца платиновой пробирки и не так совершенно разлагается, как если бы он был распределен равномерно по ее длине. Кроме того, кислота сильно действует на платину, если воздух в обеих пробирках (стеклянной и платиновой) не будет заменен на двуокись углерода. Но даже если это сделано, все же несколько миллиграммов платины переходит в раствор.
Несмотря на все сказанное выше, описанный метод может оказаться очень полезным, если в лаборатории имеются необходимые платиновая пробирка и стальная трубка, особенно в тех случаях, когда надо экономить анализируемую пробу.
Многие силикаты, керамические материалы, огнеупорные окислы и с трудом разлагающиеся породы, содержащие платину, могут быть разложены без загрязнения получаемого раствора щелочными солями. Для этого такие вещества нагревают с кислотами при высоких температурах в запаянных стеклянных трубках, в которых внутреннее давление компенсируется внешним давлением двуокиси углерода, находящейся в стальном контейнере, в который эту трубку помещают 2.
Каолин, на который разбавленная соляная кислота действует очень медленно, может быть быстро разложен, если его предварительно прокалить примерно при 700° С в течение 15 мин и потом обработать кислотой. Вся кремнекислота остается при этом практически нерастворимой 3.
КРЕМНИЙ
Специальные случаи
Прежде чем дать общий обзор анализа силикатов, мы рассмотрим здесь два особых, более простых случая.
Анализ кварца и минералов с большим содержанием кремнекислоты
Иногда может появиться необходимость определения элементов, присутствующих в кварце, халцедоне и других минералов, богатых кремнекислотой. В таких случаях, когда содержание этих примесей в анализи-
1 Р. Jannasch, Вег., 24, 273 (1891); Z. anorg. Chem., 6, 72 (1894).
2 Е. Wichers, W. G. S c h 1 e c h t, C. L. Gordon, J. Research NBS, 33, 363, 451, 453 (1944); см. также C. L. G о r d о n, там же, 30, 107 (1943).
3 J. I. Hoffman, R. T. Leslie, H. J. C a u 1, L. J. Clark, J. D. Hoffman,!. Research NBS, 37, 409 (1946).
59*
932
Гл. LIII. Методы анализа силикатных пород
руемом минерале незначительно, удобнее определять сначала кремний косвенным методом, отгонкой с плавиковой и серной кислотами, вместо значительно более длительного способа, который обычно применяется. Отгонке в этом случае должно предшествовать определение потери при прокаливании; тем самым вводится Основное ограничение в применении - этого метода — состав нелетучих веществ не должен изменяться, когда остаток после отгонки кремния снова прокаливают при той же температуре, какая применялась в первом прокаливании. Таким образом, малые количества сульфидов, если они присутствуют, будут, по всей вероятности, превращены в окислы при первом прокаливании и потому не вызовут ошибки. Но если проба содержит элементы, образующие нераз-лагаемые прокаливанием сульфаты (щелочные и щелочноземельные металлы), то это приводит к неверным результатам.
После определения потери при прокаливании в платиновом тигле смачивают порошок пробы водой, обливают его концентрированной плавиковой кислотой, совершенно свободной от нелетучих примесей, и медленно выпаривают на паровой бане или в радиаторе. Обработку повторяют, добавляя новую порцию плавиковой кислоты, пока анализируемый минерал не разложится полностью. Для достижения этого может потребоваться многократная обработка, потому что плавиковая кислота значительно медленнее реагирует с кристаллическими формами кремнекислоты, чем с большинством силикатов, и особенно с осажденной кремне-кислотой. Конец операции можно узнать по окончательному исчезновению песчинок, что обнаруживается при осторожном трении платиновой или бакелитовой палочкой. После окончательного выпаривания досуха прибавляют каплю концентрированной серной кислоты, выпаривают ее и прокаливают остаток до полного разложения сульфатов железа и алюминия. Последнее важно, когда требуется определение самой кремнекислоты, а не только примесей в ней. Второе взвешивание показывает массу улетучившейся SiO2 и массу оставшихся примесей в виде окислов, если сульфаты полностью разложились. Состав примесей и количество их можно затем определить обычными методами. Если эти компоненты входили первоначально в состав силикатов, то связанная с ними кремнекислота улетучится вместе с свободной кремнекислотой.
Определение так называемой растворимой кремнекислоты
Очень часто при обработке минералов кислотами кремнекислота выделяется в зернистой или студенистой форме, и иногда бывает нужно удалить эту кремнекислоту для отделения от неразложившейся части пробы и определить ее, т. е. провести таким способом различие между растворимой и нерастворимой кремнекислотой в породе. Обычно для этой цели применяют кипящий раствор карбоната натрия, хотя некоторые авторы рекомендуют применение едких щелочей. Было убедительно доказано 1) что кварц не является настолько нерастворимым в растворах едких щелочей, как это предполагали раньше; напротив, при достаточной степени измельчения его можно полностью перевести в раствор; 2) что обработкой едкими щелочами и карбонатами невозможно точно отделить кварц от опаловой кремнекислоты; 3) что нагревание на наро-
1 G. Lunge. С. М. М i 1 1 Ь е г g. Z. angew. Chem.. 393, 425 (1897).
Кремний
933
вой бане в течение 15 мин с 5%-ным раствором карбоната натрия является единственным хорошим способом отделения непрокаленной осажденной кремнекислоты от кварца, и то только в том случае, если самые мелкие частицы были предварительно удалены отмучиванием.
Авторы говорят:
«Если, однако, содержание такой «муки» не превышает получаемого при обычных операциях измельчения в порошок и просеивания через шелковое сито с тончайшими отверстиями, то ошибка, происходящая от вышеуказанной обработки, так мала, что ею можно пренебречь; она достигает 0,1%, самое большее 0,2% от всего количества кремнекислоты. На эту величину содержание кварца будет найдено повышенным, а содержание аморфной кремнекислоты соответственно пониженным».
Те же авторы утверждают, что растворяющее действие едких щелочей на кварц становится очень заметным только тогда, когда анализируемую пробу доводят до такой совершенно неощутимой тонины, какая практически никогда не достигается при ее приготовлении для анализа. Поэтому следует рекомендовать применение разбавленного раствора едкого натра, когда подлежит определению кремнекислота, отделенная обработкой кислотами от одной из нескольких составных частей породы. Даже когда раствор едкой щелочи очень сильно разбавлен, растворение происходит почти тотчас же и, как только оно закончится (что узнается по изменению внешнего вида осадка), раствор разбавляют холодной водой и немедленно фильтруют. Затруднений при фильтровании часто можно избежать слабым подкислением, которое имеет еще то дополнительное преимущество, что тотчас же задерживает дальнейшее действие щелочи. Если разбавление достаточно, то выделения кремнекислоты при этом не происходит. Для промывания остатка применяется также очень разбавленная кислота. Когда обработка проводилась карбонатом натрия, то промывали разбавленным раствором последнего с добавлением спирта; получались прозрачные фильтраты.
Определение кварца в присутствии силикатов
Определение с фтороборной кислотой. Метод 1 основан на действии фтороборной кислоты, которая растворяет многие силикаты, но разрушает кварц так слабо, что на ее действие можно ввести соответствующую поправку. В течение 2—8 дней достигается количественное разложение волластонита, биотита, ортоклаза, альбита, мусковита, пироксена, андалузита, кордиерита, талька, амфибола и цоизита. Полностью не разлагаются: форстерит, гранат, дюмортьерит, силлиманит, берилл и циркон.
В благоприятных условиях точность определения равна примерно 1 % от содержания кварца.
1 W. R. Line, Р. W. Aradine, Ind. Eng. Chem., Anal. Ed., 9, 60 (1937). Авторы утверждают, что применять фтороборную кислоту лучше, чем кремнефтористоводородную, которая была рекомендована для той же цели A. Knopf [U. S. Pub. Health Rep., 48, 183 (1933)], потому что она растворяет большее число силикатов, растворение происходит быстрее и поправка равна 0,34% на каждые 24 ч обработки при 50° С, в то время как при применении кремнефтористоводородной кислоты поправка равна 0,7% па 24ч обработки при комнатной температуре. См. также Н. .Tung, Naturwissenschaften, 30, (1942) и F. Н. Goldman, Ind. Eng. Chem., Anal. Ed., 13, 789 (1941).
934
Гл. LIII. Методы анализа силикатных пород
Ход анализа. Разложение силиката. Измельчают пробу так, чтобы она проходила через сито в 100 меш (d = 0,147 ля), переносят 0,15—0,20 г в платиновый тигель и прибавляют 5 мл фтороборной кислоты \ 1 мл фосфорной кислоты (пл. 1,39 г/слг3) и 2 мл 2 М раствора хлорида железа (III). Нагревают при 50° С в течение 48 ч, добавляя еще хлорид железа (Ш), если желтая окраска раствора ослабевает. Затем фильтруют, перенося остаток на беззольный фильтр, и промывают его 4 раза 1 н. раствором соляной кислоты и 5 раз горячей водой. Если остаток не слишком ничтожен по величине, переносят его вместе с фильтром в тигель и сжигают фильтр, нагревая тигель только до темно-красного каления. Затем остаток снова подвергают той же обработке фтороборной кислотой еще 48 ч. Снова фильтруют, промывают и определяют массу остатка. Такую 48-часовую обработку повторяют до тех пор, пока потеря в массе не будет равна только 1—2 мг. Это указывает на полное растворение силиката. Некоторые кремнистые материалы могут полностью раствориться быстрее, чем за 48 ч, тогда обработку фтороборной кислотой надо прекратить. В других случаях даже 12-дневная обработка не приводит к полному разложению.
Определение свободной кремнекислоты. Остаток после обработки фтороборной кислотой обрабатывают 2—Змл 48%-ной плавиковой кислоты, выпаривают досуха, прокаливают, взвешивают и повторяют эту обработку до достижения постоянной массы остатка. Потеря в массе отвечает содержанию свободной кремнекислоты (кварца) в остатке, если все силикаты были разложены предыдущей обработкой.
В найденный результат надо ввести поправку на растворение свободной кремнекислоты при обработке фтороборной кислотой. Эта поправка равна 0,34% от массы кварца за 24 ч обработки 1 2 * * * * * 8. Результат определения кварца получится, конечно, повышенным, если в пробе после обработки фтороборной кислотой останутся неразложенные силикаты.
Сплавление с пиросульфатом. Было отмечено 3, что петрографические методы определения кварца в огнеупорных глинах могут привести только к результатам средней точности, а химические методы, основанные на обработке кремнефтористоводородной или фтороборной кислотами, слишком медленны и ненадежны. Указанные авторы 3 рекомендуют метод, в котором анализируемая глина сплавляется с пиросульфатом. Кварц остается без изменения, а образующие глину минералы разлагаются с выделением кремнекислоты, которая потом растворяется в горячем растворе едкого
1 Приготовляется следующим образом. В платиновую чашку емкостью 125 мл вливают 75 мл чистой 48%-ной плавиковой кислоты, охлаждают в ледяной ванне и прибавляют маленькими порциями 32 г перекристаллизованной борной кислоты. Каждую следующую порцию всыпают только тогда, когда предыдущая растворилась. Для растворения последних порций борной кислоты смесь нагревают на водяной бане. Затем концентрируют раствор примерно до 50 мл, охлаждают до 0-—5° С и фильтруют. Полученная кислота должна иметь плотность около 1,45 г!см? и проба ее на содержание фторидов должна дать отрицательный результат. Хранить ее надо в восковых или резиновых сосудах, но фильтровать ее можно в стеклянной посуде.
2 Е. Kaplan и W. Т. Fales [Ind. Eng. Chem., Anal. Ed., 10, 388 (1938)] pe-
y .j. a
комендуют вычислять эту поправку по формуле сложных процентов (* т. е. х —-
где х — действительное содержание кварца; а — содержание кварца, найденное от-
гонкой с фтористоводородной кислотой; п — число суток, в течение которых проба
подвергалась обработке фтороборной кислотой*).
8 L. J.Trostel, D. J. Wynne, J. Am. Ceram Soc., 23, 18 (1940).
Кремний
935
натра. Преимуществами этого метода являются: 1) применение простых доступных реактивов; 2) то, что определение можно сделать в течение 8 ч или еще быстрее; 3) что сумма ошибок не превышает 0,15%.
Точность этого метода основана на взаимной компенсации отрицательной ошибки, происходящей от растворения кварца, и положительной ошибки, происходящей от загрязнения кварца неразложившимися минералами. Сумма обеих ошибок будет в границах от — 15%, когда глина содержит около 50% кварца, до +0,13%. когда содержание кварца менее 10%. Если содержание кварца в глине равно 25%, сумма обеих ошибок практически равна нулю.
Ход анализа. В тигель из прозрачного кварца помещают навеску в 0,5 г анализируемой глины, предварительно измельченной до 150 меш (d = 0,104 мм) и высушенной. Прибавляют 10—15 г пиросульфата калия и осторожно сплавляют, нагревая сначала очень медленно для предупреждения потери SO3, а потом при 900—1000° С. Нагревание не следует доводить до момента, когда на поверхности плава или на стенках тигля начинают выкристаллизовываться соли. Плав затем охлаждают, переносят в стакан емкостью 400 мл и растворяют в 150—200 мл горячей воды. Затем прибавляют 12 г гранулированного едкого натра, вводя одновременно по 1—2 гранулы. Выделившаяся при сплавлении кремнекислота растворяется. Стакан оставляют на горячей плитке 30 мин, поддерживая температуру жидкости 85—90° С.
Затем быстро фильтруют через неплотный фильтр и тщательно переносят весь остаток кварца на фильтр при помощи горячей воды. Фильтр с остатком промывают 10 раз горячей водой, затем 5—10 раз горячей разбавленной (1 : 1) соляной кислотой для растворения железа и т. п. и снова 5 раз горячей водой. После этого переносят фильтр с остатком в предварительно взвешенный платиновый тигель, прокаливают, охлаждают в эксикаторе и снова взвешивают. Таким образом находят содержание свободного кварца.
Иногда бывает, что кварц остается загрязненным другими компонентами породы, не разложившимися при сплавлении или не растворившимися при последующей обработке. Поэтому остаток кварца надо исследовать под микроскопом, а затем обработать 2 каплями разбавленной (1 : 1) серной кислоты и 10 мл фтористоводородной кислоты, выпарить досуха, прокалить и взвесить. Если масса полученного остатка превышает 0,001 г, результат определения нельзя признать правильным и определение необходимо повторить.
Другие методы. Из других методов определения свободной кремнекислоты (кварца) следует отметить: 1) измененный петрографический иммерсионный метод *; 2) метод подсчета зерен, применяемый для быстрого определения кварца в полевых шпатах, идущих на изготовление эмалей 1 2; 3) рентгеноскопический диффракционный порошковой метод определения кварца в производственных пылях 3.
* Назовем также быстрый способ определения кварца в присутствии силикатов 4, основанный на нагревании анализируемой смеси с ортофос-
1 Н. L. Ross, F. W. S е h 1, Ind. Eng. Chem., Anal. Ed., 7, 30 (1935).
2 G. H. M с 1 n t у г e, M. В о z s i n, Ind. Eng. Chem., Anal. Ed., 12, 326 (1940).
3 J. W. Ballard, H. I. О s h г у, H. H. Schrenk, U.S. Bur. Mines Rep. Invest., 3520 (1940).
4 N. A. T a 1 v i t i, Analyt. Chem., 23, № 4 (1951).
936
Гл. LIII. Метода анализа силикатных пород
форной кислотой при 248—250° С. При этой температуре образуется пиро-фосфорная кислота, действующая как селективный растворитель — силикаты при этом процессе растворяются, а кварц практически не затрагивается. Позже 1 в качестве растворителя была применена пирофосфор-ная кислота и определение стали выполнять в кварцевых тиглях вместо платиновых. Доп. ред.*
Анализ силикатов
После переведения в раствор силикатных минералов или различных искусственных силикатов прежде всего определяют кремнекислоту, а затем в той же навеске пробы определяют ряд других ее компонентов; число последних различно в зависимости от природы и сложности анализируемого вещества и часто очень велико. К ним относятся, во-первых, элементы, осаждаемые сероводородом, затем осаждаемые аммиаком или сульфидом аммония, потом кальций и стронций и, наконец, магний. Общий ход анализа, который обычно применяют, в схематической форме приведен в литературе 2.
Та или другая из указанных там операций может в отдельных случаях оказаться ненужной, а иногда могут потребоваться п изменения в общем ходе анализа. Нужно подчеркнуть, что не все элементы той пли иной группы надо определять в выделенном групповом осадке. Некоторые из них лучше определять в отдельных навесках пробы, но, как правило, их надо выделять вместе со всей группой и взвешивать, как часть сложного осадка. Это особенно относится к группе, обычно осаждаемой аммиаком, которая иногда бывает очень сложной.
В качестве иллюстрации можно привести пример осадка от аммиака в анализе горных пород. Вместе с алюминием, железом, титаном и небольшим количеством кремния, которые являются почти постоянными компонентами этого осадка, он содержит также фосфор, хром, цирконий и т. п., которые могли присутствовать в породе, затем большее или меньшее количество ванадия и марганец. После нахождения массы этого сложного осадка обычно определяют в нем или в отдельных навесках пробы все перечисленные элементы, кроме алюминия. Так поступают из-за отсутствия точного метода определения алюминия в такой смеси.
Большая часть горных пород может быть исследована хорошо разработанными аналитическими методами, так как главные компоненты во всех горных породах одни и те же. Хотя горные породы в общем сложнее по составу, чем отдельные виды минералов, число компонентов горных пород, встречающихся в них в количествах, определимых в навеске 1—5 г вещества, редко превосходит 25, а в большей части горных пород их не больше 3 15. Состав силикатных минералов значительно более разнообразен в отношении их главных компонентов и требует соответственно большего разнообразия методов анализа. Однако многие методы, применяемые при анализе горных пород, в основном применимы также и к от
1 В. В. Добровольская, Тезисы докладов на Всесоюзном совещашш но промышленно-санитарной химии, Медгиз, 1954.
2 G. Е. F. L u n d е 1 1, J. I. Hoffman, Outlines of Methods of Chemical Analysis, 1938, p. 30—78.
3 Это утверждение ни в какой мере не противоречит положению, что в изверженных горных породах может присутствовать любой элемент.
Кремний
937
дельным минералам. При исследовании осадка от аммиака встречаются, как правило, очень большие возможности выбора разных методов анализа и их вариантов.
Так как анализ сложной силикатной породы включает в себя большое число операций и разделений, мы приводим здесь только схему такого анализа. В этом схематическом описании мы стремились прежде всего указать на те предосторожности и поправки, которые необходимы при выполнении работы, претендующей на точность. Ход работы при определении кремния мы излагаем здесь более подробно. Точное определение кремнекислоты не может быть проведено сразу, одной операцией, как это раньше предполагалось. Приходится вводить поправки на примеси, присутствующие в первоначально определенной нечистой кремнекислоте, и кроме того, извлекать кремнекислоту из других осадков, получаемых в ходе анализа. Методы определения других компонентов, кроме кремнекислоты, не будут излагаться подробно в этом описании — будут даны только ссылки на соответствующие главы, где эти методы изложены. Особые случаи анализа, когда кремний является второстепенным компонентом или случайной примесью в анализируемом минерале, рассмотрены в других главах, например в гл. «Ниобии и тантал» (стр. 663).
Разложение силикатов
Обработка кислотами. Кремний подлежит определению. Когда анализируемый силикатный минерал может быть разложен кислотами, то обычно для разложения навески, в которой определяют кремнекислоту, пользуются соляной кислотой. Ее предпочитают азотной кислоте потому, что при выпаривании солянокислого раствора для переведения кремнекислоты в нерастворимое состояние не так часто образуются малорастворимые соли, как это бывает при выпаривании азотнокислого раствора. Серная кислота также редко применяется, потому что она образует очень малорастворимые сульфаты свинца и щелочноземельных металлов, загрязняющие выделяемую кремнекислоту. Для разложения силикатов свинца обычно применяют азотную или хлорную кислоту; иногда эти кислоты применяют и в других случаях. Для разложения некоторых титано-силикатов может быть лучше вместо соляной кислоты применять хлорную или серную кислоты, потому что при достаточном избытке этих кислот титан можно удержать в растворе в то время, когда кремнекислота переводится в нерастворимое состояние.
В отношении концентрации кислоты, применяемой для разложения, нельзя дать определенных указаний. Если разбавленная кислота приводит к получению желаемых результатов, то ее следует предпочесть концентрированной, потому что тогда легче избежать преждевременного выделения кремнекислоты, которая, обволакивая остаток анализируемой пробы, препятствует полному ее разложению, а также потому, что некоторые хлориды (ВаС12, РЬС12) нерастворимы в концентрированной кислоте.
Не может быть также предписана определенная температура разложения. Некоторые силикаты разлагаются без нагревания; другие требуют нагревания, часто в течение продолжительного времени. Некоторые силикаты содержат двуокись углерода; в этом случае сосуд, в котором проводится разложение пробы, нужно прикрывать часовым стеклом,
938
Гл. LIII. Методы анализа силикатных пород
пока не выделится вся СО2. Прикрывать часовым стеклом следует также и в тех случаях, когда раствор надо кипятить, особенно когда применяется концентрированная соляная кислота.
г Конец разложения, если он не заметен на глаз, может быть определен по исчезновению всех песчинок, что обнаруживается осторожным надавливанием стеклянной палочкой.
Для быстрого определения кремнекислоты в искусственных силикатах, разлагаемых соляной кислотой, например в цементах, был рекомендован метод \ в котором пробу смешивают с хлоридом аммония, обрабатывают соляной кислотой и нагревают на паровой бане 30 мин. Затем фильтруют и кремнистый остаток промывают горячей водой.
Здесь уместно предостеречь от применения хода работы, на первый взгляд подходящего для анализа чистых гомогенных минералов, которые при атмосферном давлении лишь с трудом разлагаются соляной или серной кислотами. Этот ход анализа состоит в том, что навеску минерала подвергают продолжительному действию кислоты, фильтруют и анализируют фильтрат, учитывая и ту кремнекислоту, которая выделилась в свободном и нерастворимом состоянии при действии кислоты. Нерастворимый остаток, отделенный от этой освобожденной кремнекислоты, взвешивают, чтобы определить по разности массу перешедшей в раствор части навески. Этот метод основан на предположении, что процентные соотношения компонентов в разложенной части анализируемого минерала те же, что и оставшейся неразложенной части, и что, следовательно, анализ разложенной части дает состав всей анализируемой пробы. Неоднократно указывалось, что такое допущение не соответствует действительности 1 2.
Для разложения трудноразлагаемых минералов, требующих длительной обработки, следует применять платиновые или фарфоровые сосуды вместо стеклянных, потому что из стекла, и даже из фарфора, всегда переходят в раствор кремний и другие составные части этих материалов. Но платину нельзя применять при разложении веществ, выделяющих под действием соляной кислоты хлор, а также минералов, содержащих железо (III), если их приходится нагревать с соляной кислотой в течение долгого’ времени. Никогда не следует скрести палочкой, особенно платиновой, дно и стенки чашки, потому что чашка может от этого пострадать и, кроме того, таким способом можно соскоблить измеримое количество платины, которая будет затем принята в ходе анализа за какой нибудь компонент породы.
Когда минерал разложится, выпаривают раствор досуха и обрабатывают остаток, как указано на стр. 940.
Кремний не подлежит определению. Прежде чем метод разложения силикатов для определения в них щелочных металлов, предложенный Лоуренсом Смитом, получил всеобщее распространение, было принято поступать следующим образом. Анализируемую породу или минерал, устойчивые к действию соляной, азотной и серной кислот, обрабатывали плавиковой кислотой с добавлением такого количества серной кислоты, какое необходимо для превращения в сульфаты всех присутствующих
1 Е. Е. Maczkowske, J. Research NBS, 16, 549 (1936).
2 См. особенно L. D u р а г с, Bull. Soc. franj. mineral., 42, 138 (1919). Он сделал интересное наблюдение, что при анализе силикатов, быстро разлагаемых кислотами, часто остается остаток, очень устойчивый к действию кислот. Значение этого наблюдения в связи с изложенным выше методом анализа очевидно.
Кремний
939
в пробе оснований А Этот метод позволял определять в одной навеске не только щелочные металлы, но и другие основания, хотя он, конечно, исключал возможность определения кремния. Метод этот находит иногда применение и в настоящее время (с заменой серной кислоты хлорной кислотой), но имеет тот недостаток,' что при его применении невозможно полностью осадить алюминий аммиаком 1 2, пока не удалены последние следы фтора. Полное удаление фтора 3 может быть достигнуто растворением солей (остающихся после первого выпаривания) в разбавленной cepHoii кислоте и повторным выпариванием до сильного выделения паров серной кислоты; при работе с хлорной кислотой выпаривают досуха.
Сплавление с различными плавнями. Для разложения силикатных горных пород, неразлагаемых полностью соляной кислотой, и практи-, чески для разложения всех нерастворимых силикатов применяют метод сплавления. Сплавление необходимо также и для разложения растворимых силикатов, содержащих фтор, поскольку речь идет об определении в них кремния. Для этой цели почти универсальное применение имеет безводный карбонат натрия, хотя были предложены и другие реактивы, например смесь окиси свинца с борным ангидридом, карбонат свинца, бура и борный ангидрид (см. «Разложение при помощи плавней», стр. 915).
Сплавление с карбонатом натрия. Хотя для очень большой точности анализа и требовалось бы, чтобы все силикаты, содержащие фтор, обрабатывались способом, описанным ниже (см. «Анализ в присутствии значительного количества фтора»), все же, когда фтор содержится в очень малых количествах, как это имеет место в большинстве горных пород, не стоит црибегать к этому кропотливому методу. Так можно поступать потому, что фтор вызывает потерю SiO2 в количестве меньшем, чем три четверти его массы, и то только в том случае, если весь он улетучится в виде тетрафторида кремния, когда раствор выпаривают с соляной кислотой. На практике, однако, потеря бывает меньше, так как тетрафторид кремния разлагается водой. Часть фтора при этом, может быть, улетучивается в виде фтористого водорода, а большая часть его задерживается в виде фторосиликатов. Последние, при прокаливании их с остатком кремнекислоты, подвергаются по крайней мере частичному разложению, а при обработке прокаленной кремнекислоты фтористоводородной и серной кислотами связанный в этих фторосиликатах металл (обычно натрий) будет взвешен в виде сульфата, если не улетучится при прокаливании.
Во всех случаях получаемая ошибка не имеет большого значения, потому что она приходится на компонент, который присутствует в пробе в наибольшем количестве.
Ход анализа при отсутствии значительного количества фтора рассмотрен в разделе «Разложение при помощи плавней» (стр. 924).
Анализ в присутствии значительного количества фтора. При анализе пород или минералов, содержащих
1 Если перед выпариванием фтористоводородной кислоты подвергнуть навеску пробы обработке фтористоводородной кислотой на холоду в течение ночи, — это способствует ее разложению.
2 См. также F. Р. V е i t с h, J. Am. Chem. Soc., 22, 246 (1900); F. W. H i n г i c h-s e n, Ber., 40, 1407 (1907); Z. anorg. Chem., 58, 83 (1908); H. Cavaignac, C. r., 158, 948 (1914). См. также стр. 751.
3 E. S e 1 c h, Z. anal. Chem., 54, 395 (1915).
940
Гл. LIII. Методы анализа силикатных пород
фтор в количествах, достаточных для его определения или для того, чтобы вызвать большую потерю кремния при определении последнего или неполное осаждение алюминия, — поступают, как описано в разделе «Фтор» (стр. 1018).
Кремний может быть также достаточно точно определен 1 в силикатных горных породах, содержащих фтор, сплавлением с едким кали, выщелачиванием плава водой, осаждением фторидом калия и хлоридом калия, прибавляемым в избытке к подкисленному раствору, и титрованием осажденного фторосиликата калия титрованным раствором едкого кали. Метод этот не пригоден в присутствии титана и циркония и требует изменений, когда применяется к силикатам, содержащим алюминий.
Сплавление с борным ангидридом. См. стр. 920 и 929.
Кремнекислота, выделяющаяся в присутствии борной кислоты, всегда содержит бор, отчего получаются повышенные результаты вследствие улетучивания бора в виде BF3 (если бор предварительно полностью не удалить). Бор может быть удален обработкой плава метиловым спиртом и соляной кислотой, как описано на стр. 930, или (менее удовлетворительно) обработкой той же смесью влажной загрязненной кремнекислоты. Величина ошибки, происходящей от присутствия бора, иллюстрируется следующими результатами, полученными М. О. Lamar. Анализ стандартного образца стекла (Бюро Стандартов США) показал содержание в нем 74,63 и 74,52% кремнекислоты вместо истинного ее содержания 74,1%, когда 0,5 г пробы были сплавлены с 3 г карбоната натрия и 1 г борного ангидрида и кремнекислота была затем определена растворением плава в соляной кислоте и обычным двукратным выпариванием с промежуточным фильтрованием. Подобным же образом два анализа стандартного образца боксита (Бюро Стандартов США), который содержал 6,30% кремнекислоты, показали 6,49 и 6,44% когда проба была сплавлена с карбонатом натрия и борным ангидридом, растворена в серной кислоте и выпарена дважды до выделения паров серной кислоты с промежуточным фильтрованием.
Выделение и определение кремнекислоты
Обезвоживание. Описанные ниже методы применяются при анализе силикатных минералов, свободных от фтора или пород, не содержащих более 0,3% фтора.
Применение соляной кислоты. Содержимое тигля переносят в высокий стакан, содержащий небольшое количество воды. Зеленая окраска раствора плава указывает на присутствие марганца, а желтая — на присутствие хрома. Покрывают стакан часовым стеклом и через носик стакана, не снимая стекла, постепенно прибавляют разбавленную (1 : 1) соляную кислоту, вводя ее в избытке. Если появляется быстро исчезающая розовая окраска, это указывает на присутствие марганца, а интенсивность окраски показывает приблизительно, как велико его содержание. Ставят стакан на паровую баню и ускоряют распадение кусочков, надавливая оплавленным концом стеклянной палочки. Когда все растворится, за исключением хлопьевидной кремнекислоты, содержимое стакана переводят в большую чашку и выпаривают на бане досуха. Можно обойтись без стакана и провести всю операцию сразу в чашке. Обычно применяют платиновую чашку, но если минерал содержит марганец, хром или ванадий в количествах больших, чем те, какие обычно встречаются в породах, то нужно брать фарфоровую чашку, потому что при сплавлении эти элементы окисляются соответственно до манганатов, хроматов и ванадатов, выделяющих хлор из соляной кислоты. Фарфоровую чашку следует
1 Travers, С. г., 173, 714 (1921).
Кремний
941
брать также и в случае анализа веществ с большим содержанием железа, потому что хлорид железа (III) в соприкосновении с платиной медленно восстанавливается с переходом платины в раствор в виде хлорида платины.
Начиная с этой стадии, общий ход анализа одинаков, независимо от того, был ли силикат разложен сплавлением с карбонатом натрия, растворением в соляной кислоте или сплавлением с борным ангидридом с последующим удалением бора. При втором или третьем методах выделение кремнекислоты будет полнее, чем при первом методе, вследствие отсутствия большого количества хлорида натрия.
Чашка, содержащая растворенный содовый плав или разложенный соляной кислотой минерал, должна обладать такой емкостью, чтобы обеспечить достаточную поверхность распределения остатку, который остается после выпаривания. Когда разложение пробы проводится сплавлением, удобна чашка емкостью 0,5 л; в других случаях можно пользоваться чашками меньших размеров. Температура паровой бани достаточна для выпаривания досуха. Если анализ начался утром, первое фильтрование может быть окончено в полдень. Продолжительное выдерживание на бане после выпаривания досуха ничего не дает, так как несколько более полное выделение растворимой кремнекислоты связано при этом с очень большой потерей времени. Ничего не достигают и растиранием в порошок остатка, полученного после сплавления, тем более, что при этом чашку легко поцарапать. Количество кремнекислоты, остающееся в растворе, обычно колеблется в пределах от 1 до 3% от массы кремнекислоты, и после 20-часового высушивания оно немногим меньше, чем после высушивания в течение одной десятой этого времени.
Покрывают чашку часовым стеклом, осторожно прибавляют к сухой и охлажденной массе 10 мл соляной кислоты и затем через 1—2 мин 100 мл горячей воды. Чашку с остатком от растворения плава следует покрывать часовым стеклом, потому что быстрое прибавление кислоты может повлечь за собой вылетание пылеобразных частиц. Оставляют стоять на бане 5—10 мин, перемешивая раствор время от времени, и фильтруют сначала декантацией, оставляя почти всю кремнекислоту на дне чашки. Если частицы кремнекислоты очень крупны, их измельчают пестиком. Если присутствует большое количество железа, то в этой стадии анализа уместно прибавить еще разбавленной (1 : 1) соляной кислоты и нагреть. Затем переносят кремнекислоту на фильтр. Если на стенках чашки останется пленка кремнекислоты, приставшая настолько прочно, что ее нельзя снять пером, то ее нужно удалять, так как при втором выпаривании она исчезнет. Если же второго выпаривания не предполагают делать, то обрабатывают эту пленку аммиаком, нагревают, выпаривают раствор, прибавляют к остатку небольшое количество соляной кислоты и снова выпаривают, чтобы выделить таким образом это очень небольшое количество оставшейся кремнекислоты. Главную часть кремнекислоты промывают горячей разбавленной (5 : 95) соляной кислотой, пока цвет бумаги фильтра не покажет удаление большей части железа. В конце промывания соляную кислоту лучше заменить горячей водой и перед добавлением новой порции отсасывать возможно полнее каждую порцию промывной жидкости. Фильтр с остатком сохраняют.
Фильтрат и промывные воды выпаривают досуха в той же чашке. Если паровая баня работает ночью, выпаривание обычно заканчивается
942
Гл. LIII. Методы анализа силикатных пород
на следующее утро. Когда весь раствор выпарится, переносят чашку на воздушную баню и нагревают 1 ч при 110° С. Возможно, что более высокая температура в большинстве случаев не окажет вредного влияния, кроме увеличения количества нерастворимых примесей в кремнекислоте, особенно в случае анализа минералов, богатых железом и титаном. Покрывают чашку часовым стеклом, смачивают остаток 5 мл соляной кислоты, прибавляют 50 мл горячей воды, нагревают на бане 5—10 мин и тотчас же фильтруют через новый фильтр меньших размеров, промывают остаток на фильтре холодной разбавленной (1 : 99) соляной кислотой и под конец горячей водой.
Фильтр с остатком, а также фильтрат и промывные воды сохраняют. Во втором фильтрате обычно не должно быть больше 2—3 мг кремнекислоты. Третье выпаривание является напрасной тратой времени, если второе было проведено как следует, потому что после второго выпаривания обычно достигается равновесие. Кремнекислота, еще остающаяся в растворе, может быть извлечена позднее, если проводится осаждение аммиаком и при этом получается достаточно большой осадок. Второе фильтрование с сопровождающими его операциями занимает меньше времени, чем первое.
Вторая порция кремнекислоты, особенно при анализе горных пород, окрашена сильнее, чем первая, и если анализируемый минерал или порода содержат даже умеренные количества титана, то второй остаток кремнекислоты всегда будет сильно загрязнен.
При анализе минералов, подобных цеолитам, или таких искусственных продуктов, как портландцемент, непосредственно растворимых в соляной кислоте, первое обезвоживание может быть значительно ускорено. Для этого покрытую часовым стеклом чашку, после первоначального высушивания на водяной бане, переносят на песочную баню или на треугольник, лежащий на горячей плитке. Температуру в чашке можно поднять до 200° С, а по мнению некоторых авторов она должна быть не ниже 200° С. После выдерживания при этой температуре в течение часа в фильтрат перейдет, вероятно, меньше кремнекислоты (в отсутствие магния), чем после продолжительного высушивания при температуре паровой бани, но это приводит обычно к большему загрязнению остатка кремнекислоты. Так как при анализе пород и минералов с высоким содержанием титана и железа это загрязнение очень велико (стр. 945), аналитик должен быть осторожным в выборе температуры высушивания.
Как уже было указано, полученный остаток кремнекислоты не содержит всю кремнекислоту, находящуюся в минерале или породе, и не всегда он бывает чистым. Оставшуюся небольшую, но измеримую часть кремнекислоты находят и определяют в осадке, получаемом при осаждении аммиаком, как указано на стр. 952. Если металлов, осаждаемых аммиаком, в растворе нет, то для выделения оставшейся кремнекислоты лучше всего прибавить 0,1 г железа или алюминия в виде их хлоридов, осадить аммиаком и определить кремний в осадке, как описано на стр. 952.
Обезвоживание хлорной кислотой. Для обезвоживания кремнекислоты рекомендуют также применять хлорную кислоту х. Остаток кремнекислоты в этом случае получается чище, чем при применении соляной кис-
1 Н. Н, Willard, W. Е. С a k е, J. Am. Chem. Soc., 42, 2208 (1920),
Кремний
943
лоты, вся операция требует меньшего времени и образующиеся соли легко растворимы. Можно непосредственно применять продажную 60— 70%-ную хлорную кислоту, если обрабатываемая проба не содержит органических веществ и в этой кислоте растворима. В противном случае хлорную кислоту прибавляют после растворения анализируемой пробы в другой кислоте или после разложения ее сплавлением с карбонатом натрия. Результаты опытов, проведенных с силикатами, цементами, чугунами, сталями и другими металлами, показывают, что по этому методу получаются данные, совпадающие с полученными обычным методом х. В литературе нет указаний о влиянии присутствия хлорной кислоты на последующий ход анализа (стр. 947). Поэтому в настоящем, руководстве ход анализа изложен в предположении, что обезвоживание кремнекислоты проведено с соляной кислотой.
При навеске в 1 г обезвоживание с хлорной кислотой проводится следующим способом. Растворимые минералы обрабатывают смесью 10 мл хлорной кислоты и 20 мл воды или же для разложения применяют соляную или азотную кислоты, а потом прибавляют требуемое количество хлорной кислоты. Если содержание кремнекислоты очень велико, то берут больше хлорной кислоты. Неразлагаемые кислотами минералы сплавляют с карбонатами натрия, обрабатывают плав водой, пока он не разложится, и растворяют его в достаточном количестве хлорной кислоты. Выпаривают до сильного выделения паров хлорной кислоты и затем осторожно кипятят 10—15 мин. Чтобы избежать чрезмерной потери кислоты, кипячение проводят в стакане или глубокой чашке. Содержимому сосуда не дают затвердевать, иначе выделение кремнекислоты не будет полным. Если большое количество перхлоратов выпадает из раствора, то это значит, что либо температура нагревания не была правильно отрегулирована, либо прибавили мало хлорной кислоты. Однако в присутствии большого количества алюминия из горячего раствора всегда выделяется немного нерастворимого перхлората. При охлаждении весь раствор обычно затвердевает; к нему приливают 4—5 объемов воды, нагревают до кипения, фильтруют, промывают кремнекислоту разбавленной (1 : 99) соляной кислотой и под конец водой. Прокаливают и взвешивают, как описано ниже (см. «Прокаливание кремнекислоты»).
Поправка на присутствие примесей обычно бывает незначительной. Если при сплавлении применяли карбонат калия или соли калия были введены в раствор в ходе анализа каким-нибудь другим образом, то может выделиться перхлорат калия. Поэтому следует избегать применения солей этого металла. Перед прокаливанием остатка кремнекислоты его (а также и фильтровальную бумагу) надо хорошо отмыть от перхлоратов.
Прокаливание кремнекислоты. Оба фильтра, содержащих кремнекислоту, помещают в платиновый тигель, смачивают несколькими каплями
1 При анализе свинцово-бариевого стекла (навески по 0,5 г), содержавшего 17,5% РЬО и 1,41% ВаО, Н. В. Knowles получил 65,38, 65,38 и 65,36% SiO2, применяя метод с хлорной кислотой, и 65,35, 65,35 и 65,36% SiO2 — методом с соляной кислотой. В обоих случаях проводилось двукратное выделение кремнекислоты и остаток ее определяется в осадке от аммиака. Количество кремнекислоты, полученной при втором обезвоживании, достигало 3—6 мг при применении соляной кислоты и 0,6—0,9 мг при применении хлорной кислоты, а количество кремнекислоты, выделенной из осадка от аммиака, составляло 0,3—0,6 мг при применении НС1 и 0,4—1,1 мг при применении HCIO4.
944
Гл. LIII. Методы анализа силикатных пород
разбавленной (1 : 4) серной кислоты и осторожно нагревают так, чтобы кремнекислота медленно высохла и бумага фильтров обуглилась без воспламенения. Частично прикрывают тигель крышкой, медленно увеличивают пламя, пока не сгорит весь уголь, потом совсем закрывают тигель и прокаливают приблизительно при 1200° С в течение 10—30 мин х. Охлаждают над хорошо высушивающим веществом, взвешивают закрытый тигель и повторяют прокаливание, пока масса тигля не станет постоянной. Крышка тигля должна его плотно закрывать, чтобы не было потери кремнекислоты вследствие распыления. Если применяют паяльную горелку, то тигель должен быть опущен на две трети его глубины в отверстие асбестового картона.
Поправка на примеси в кремнекислоте. В найденную массу кремнекислоты надо обязательно внести поправку на примеси, которые всегда в ней содержатся. Поправку находят обработкой полученной кремнекислоты фтористоводородной и серной кислотами (обе должны быть свободными от нелетучих примесей), выпариванием, прокаливанием в течение 1—2 мин и вторичным взвешиванием. Прокаливания на паяльном пламени здесь не требуется, так как остаток в тигле очень незначительный. Не следует, однако, на этом основании пренебрегать вторым взвешиванием, даже если остаток кажется совсем ничтожным. При анализе горных пород он никогда не бывает невесомым, и, кроме того, этим вторичным взвешиванием исправляется ошибка, происходящая от потери в массе самого тигля при прокаливании кремнекислоты. Перед приливанием фтористоводородной кислоты надо смочить содержимое тигля водой, хотя при обработке HF прокаленной кремнекислоты реакция идет не так бурно, как она проходит с кремнекислотой, которая не была прокалена на паяльной горелке. Однократной обработки несколькими миллилитрами фтористоводородной кислоты достаточно, чтобы удалить всю кремнекислоту.
В присутствии кварца дело обстоит иначе, так как кварц разлагается плавиковой кислотой значительно труднее, чем осажденная кремнекислота. При анализе пород, содержащих 60—80% кремнекислоты, достаточно прибавления 1—2 капель разбавленной (1 : 1) серной кислоты, но при анализе менее кремнеземистых пород количество прибавленной серной кислоты должно быть увеличено не только для того, чтобы ее было достаточно для превращения всех оснований в сульфаты, но и для того, чтобы предупредить возможность потери титана (если он присутствует) в результате его улетучивания в виде фторида. При анализе горных пород содержание титана в этом остатке обычно тем больше, чем сильнее выражен основной характер этих пород.
Если присутствуют только окиси железа и алюминия, то достаточно одной фтористоводородной кислоты. В присутствии окислов циркония и титана лучше прибавлять обе кислоты — фтористоводородную и серную, хотя потеря от улетучивания
1 Нужно особо подчеркнуть обязанность каждого аналитика определить те температуры, которые дают применяемые им горелки и муфели. Расходжденпе многих результатов и утверждений по различным вопросам аналитической химии может быть приписано разнице в температуре прокаливания, применявшейся различными авторами.
Хорошее пламя паяльной горелки должно легко плавить несколько сотых грамма порошка ортоклаза, находящегося на дне маленького платинового тигля; температура при этом равна около 1200° С.
Кремний
945
с одной HF в хорошо закрытых тиглях не так велика, как обычно думают. Это показывают следующие данные, полученные Д. И. Гофманом:
Взвешивание после прокаливания, обработки HF4-HtSO< Взвешивание после обработки одной HF и вторичного Взвешивание после обработки HF + HNOi и вто-
и вторичного прокаливания прокаливания ричного прокаливания
г г г
0,0159 TiO2 0,0155 1 ...
0,0158 TiO2 0,0591 ZrO, 0,0591 2 0,01551
0,0591 ZrO‘ . . . 0,0591 2
0,1176 Fe2O3 0,1176 s —
0,117 Fe2O3 0,1179 s —
0,1056 A13O3 0,1056 3 —
При выполнении рядовых анализов получаемый в дальнейшем ходе анализа осадок окисей алюминия и др. обычно прокаливают в тигле, содержащем остаток от кремнекислоты. В более точных анализах этот остаток следует сплавить с щепоткой карбоната натрия, плав растворить и полученный раствор прибавить к фильтрату от кремнекислоты.
При анализе кремнетитанатов, по-видимому, следовало бы предпочесть иной метод, потому что большое количество титана может вместе с кремнекислотой перейти в нерастворимый остаток. В таких случаях лучше смыть, насколько возможно, всю нечистую кремнекислоту с фильтров в чашку, сжечь фильтры, прокалить при возможно более низкой температуре, перевести золу фильтров в ту же чашку, выпарить воду, прибавить концентрированную серную кислоту, нагреть до выделения ее паров, чтобы перевести в раствор титан, разбавить несколько водой, подержать недолго на паровой бане, профильтровать, остаток промыть водой, подкисленной серной кислотой, и затем прокалить и взвесить выделенную таким способом кремнекислоту. Возможно, что полученная кремнекислота еще не будет совершенно свободной от титана, но количество последнего будет так мало, что не помешает обычной обработке фтористоводородной и серной кислотами. Фильтрат должен быть затем обработан в основном так же, как и главный фильтрат, но отдельно от него, хотя соответствующие осадки, выделяемые в ходе анализа, соединяют и обрабатывают вместе.
Точность определения кремнекислоты. При соблюдении приведенных выше указаний — применения платиновой посуды, повторных выпариваний и фильтрований, надлежащего прокаливания и введения поправки на посторонние примеси — определение кремнекислоты, которое раньше часто проводилось с большими ошибками, превращается в одно из самых точных определений. Надо только всегда извлекать, как будет описано далее (стр. 955), то небольшое количество кремнекислоты, которое неизбежно проходит в фильтраты. При тщательной работе отклонения в 0,1% результатов параллельных анализов горных пород составляют исключение 4.
Состав осадка после удаления кремнекислоты с HF. Остаток, выделенный из горных пород. Качественный состав остатка, получаемого после удаления кремнекислоты, изменяется у различных горных пород в меньшей степени, чем количественный его состав. Остаток всегда содер
1 Белая пленка на стенках тигля и крышке и немного снаружи.
2 Очень тонкий слой на крышке тигля.
3 Нет признаков улетучивания.
4 См. сноску 1 на стр. 943 и табл. 31 на стр. 885.
60 Заказ 522.
946
Гл. LIII. Методы анализа силикатных пород
жит окислы алюминия, железа, титана и фосфора, если порода содержала эти элементы. Если содержание последних трех, особенно последних двух элементов в пробе невелико, то остаток должен быть незначительным по массе, часто меньше 1 мг. Обычно он весит значительно больше и может достигать 2 и даже 3% при анализе основных пород, богатых титаном и фосфором. Он может содержать до одной трети всего присутствовавшего в породе количества титана. Понятно, что остаток может содержать также цирконий и торий, если они присутствовали в породе, особенно при большом содержании в ней фосфора. В этом остатке будут находиться также ниобий и тантал.
Можно было бы предположить, что в остатке будет содержаться большая часть бария при анализе тех пород, в которых этот элемент присутствует вместе с сульфидами или сульфатами. Однако это не так, потому что сульфат бария заметно растворим в горячей соляной кислоте, и в большинстве горных пород барий встречается лишь в очень незначительных количествах. Если часть BaSOi присутствует в остатке, то его выделение и определение в этой стадии анализа не является необходимым, так как его гораздо лучше определять позже вместе с кремнекислотой, сопровождающей осадок окисей алюминия и т. п. (стр. 954). Если в самом начале разложение породы было полным, кальций очень редко входит в состав этого остатка. Когда анализ проводится надлежащим образом, остаток после растворения его затем количественно осаждается аммиаком в присутствии аммонийной соли. Этот факт, а также специальные исследования, произведенные одним из нас (В. Ф. Гиллебранд), опровергают утверждение некоторых, что остаток может содержать кальций, магний и щелочные металлы. Магний находили случайно, но количество его не превышало 0,3 мг MgO. Утверждение, что присутствие хлорида натрия является одной из причин наблюдаемых иногда небольших потерь в массе при прокаливании кремнекислоты, опровергается нашими исследованиями, а также наблюдениями других авторов \ Противоположные результаты опытов, проведенных некоторыми исследователями 1 2, следует, вероятно, приписать неполному разложению породы при сплавлении ее с карбонатами щелочных металлов.
Остаток, выделенный из минералов. Можно сказать вполне определенно, что примеси в кремнекислоте, выделенной при анализе минералов, сильно отличаются от примесей в кремнекислоте, выделенной из силикатных горных пород. Это различие находится, конечно, в зависимости от состава анализируемых минералов и иногда оно очень незначительно или совсем отсутствует, но в большинстве случаев оно велико, особенно при анализе минералов, содержащих титан, цирконий, олово, вольфрам, сурьму, ниобий и тантал. При анализе таких минералов нужно применять особые методы выделения кремнекислоты перед окончательным ее определением; например, в случае присутствия сурьмы обезвоживание кремнекислоты надо проводить с серной кислотой, а не с соляной.
ХОД АНАЛИЗА ПОСЛЕ ВЫДЕЛЕНИЯ КРЕМНЕКИСЛОТЫ
В предыдущем разделе описано определение в силикатах всего содержащегося в них кремния (кроме небольшого его остатка) применительно
1 Lenher, Truog, J. Am. Chem. Soc., 38, 1050 (1916).
2 W. R. Bloor, J. Am. Chem. Soc., 29, 1603 (1907); Aurousseau, J, Wash. Acad. Sci., 13, 330 (1923).
Ход анализа после выделения кремнекислоты
947
к анализу горных пород. В настоящем разделе описание анализа продолжается, но дается оно только в общих чертах, без подробностей. Многое из приведенного здесь относится не только к анализу силикатов; эти же положения могут найти применение при анализе многих других минералов, если, отсутствуют мешающие элементы. Однако раздел этот написан как непосредственное продолжение раздела «Кремний» в применении к горным породам, потому что анализ последних составляет значительную часть работы многих аналитиков, занимающихся минеральным анализом.
Когда кремнекислота с наивозможной полнотой выделена, перед аналитиком встает вопрос, нужно ли проводить обработку сероводородом? В анализе горных пород эта обработка обычно не нужна и является напрасной тратой времени, потому что единственным элементом сероводородной группы, который может здесь выделиться (при анализе навески в 1 г) является платина, попавшая в раствор из тиглей и чашек, применявшихся в ходе анализа; платину эту можно оставить в растворе, так как она не создает в дальнейшем никаких затруднений. При анализе минералов или искусственных силикатов неизвестного состава не следует пренебрегать обработкой сероводородом, потому что она проста и может привести к открытию и отделению элементов, которые иначе были бы пропущены и могли бы вызвать ошибки, если бы остались в растворе.
Прежде чем продолжать анализ после выделения кремнекислоты или осаждения группы сероводорода, если последнее было проведено, нужно позаботиться о том, чтобы все железо находилось в состоянии трехвалентного. Если применяли обработку сероводородом, в растворе всегда будет железо (II). В незначительных количествах железо (II) может присутствовать и тогда, когда обработка сероводородом не проводилась, но выпаривание с соляной кислотой для выделения кремнекислоты проводилось в платиновой чашке. Для окисления железа (II) прибавляют в избытке бром и кипятят раствор для удаления этого избытка или же кипятят для удаления сероводорода (если он применялся), прибавляют азотную кислоту и снова кипятят раствор.
Дальше анализ можно вести по одному из нескольких возможных путей, в зависимости от поставленной цели. В обычных случаях осаждают железо, алюминий и пр. осторожной нейтрализацией раствора аммиаком. Если pH раствора при этом не превышает 7 и раствор фильтруют сейчас же после нейтрализации, то опасность одновременного осаждения некоторого количества марганца очень мала. В настоящее время нет такой необходимости в применении ацетатного метода в случае присутствия значительных количеств марганца, как это было раньше, когда обращали меньше внимания на условия осаждения аммиаком (стр. 561). В старых методах анализа отделение марганца было далеко не совершенным и получаемая ошибка распределялась между алюминием, кальцием и магнием, когда все количество марганца не превосходило нескольких десятых долей процента. Получаемые при работе старыми методами ошибки иллюстрируются данными, приведенными на стр. 959 (см. мелкий шрифт). В присутствии больших количеств марганца обычно рекомендовали ацетатный метод.
При том и другом методе марганец остается в растворе вместе с никелем, цинком, кальцием и магнием. В анализе горных пород следующей ступенью поэтому является отделение марганца, цинка и никеля осажде-60*
948
Гл. LIII. Методы, анализа силикатных пород
нием сульфидом аммония, после чего осаждают кальций в виде оксалата и затем магний в виде фосфата. При таком ходе анализа стронций сопровождает кальций, а барий выделяется вместе с магнием.
Иногда бывает желательным получение всего марганца в осадке от аммиака; этого достигают, проводя осаждение аммиаком с добавлением брома или персульфата аммония. Никель и цинк, если они присутствовали в анализируемом веществе, остаются тогда в растворе.
Препятствием для применения большего числа методов на этой ступени анализа является неполное выделение кремнекислоты вначале. Если пренебречь кремнекислотой, еще остающейся в растворе, то можно применять разнообразные варианты хода анализа. Например, фильтрат после осаждения сероводородом можно обработать винной кислотой, затем подщелочить аммиаком и выделить таким способом железо, цинк, никель, кобальт и большее или меньшее количество марганца в виде их сульфидов. Фильтрат можно затем подкислить и обработать купфероном для выделения ниобия, тантала, циркония, титана и ванадия. Можно, при желании, в фильтрате от кремнекислоты или фильтрате от осаждения сероводородом окислить железо и затем разделить его на’ две части: в одной провести осаждение аммиаком, как обычно, во второй — осаждение купфероном. В первом случае будут выделены окислы АПОз, Р2О6, ЕегОз, ТЮг, ZtO2h V2O8, а во втором — только последние четыре окисла. Таким образом можно более точно определить по разности содержание алюминия.
При отсутствии магния и никеля можно применить еще один метод, при котором в фильтрате после отделения кремнекислоты или после осаждения сероводородом окисляют железо, ванадий и т. п. и затем прибавляют в небольшом избытке едкий натр. Таким способом железо, титан, цирконий и т. п. отделяют от алюминия, ванадия и фосфора (см. «Осаждение едким натром», стр. 109).
Металлы, осаждаемые сероводородом
В большинстве горных пород значительные количества металлов, осаждаемых сероводородом, содержатся редко, поэтому необходимость их удаления также встречается редко. Другое дело — анализ многих минералов, когда часто необходимо выделение этих металлов методом, описанным в разделе «Осаждение посредством сульфид-ионов» (стр. 83).
Определению или даже открытию в горных породах ничтожных количеств металлов сероводородной группы пока еще мало уделяют внимания. По этому вопросу см. раздел «Составные части, встречающиеся в породах в виде следов» (стр. 1033).
Если предполагают определять медь или медь и цинк, то пробу не следует просеивать через медные или латунные сита.
Фильтрат от кремнекислоты почти всегда содержит заметные количества платины. Она переходит в раствор только в ничтожных количествах или даже совсем не переходит при сплавлении с карбонатами щелочных металлов, за исключением тех случаев, когда сплавление проводится в муфеле, а не на пламени горелки (см. стр. 925) или когда в качестве окислителя добавляют слишком большие количества селитрых.
1 Само собой разумеется, что при применении селитры последующее выпаривание надо проводить в фарфоровой чашке.
Ход анализа после выделения кремнекислоты
949
В больших количествах (косвенным путем) платина попадает в раствор от действия соляной кислоты на манганаты, хроматы и ванадаты натрия, образующиеся при сплавлении; наконец, если присутствует большое количество железа, то немало платины переходит в раствор вследствие восстановления FeCls до FeCL платиной чашки, в которой проводили выпаривание. Второй и третий источники загрязнения исключают, применяя для выпаривания фарфоровые чашки; так следует всегда поступать, когда присутствуют другие элементы группы сероводорода и когда их необходимо определить в той же навеске пробы.
Присутствие одной платины, однако, не создает осложнений в анализе горных пород, так как ее можно удалить и в более поздней стадии анализа, не прибегая к выделению сероводородом. В дальнейшем изложении предполагается, что платина отсутствует или, если присутствует, то ее не надо удалять на этой ступени анализа. Если присутствуют другие металлы, осаждаемые сероводородом, подразумевается, что они удалены вместе с платиной.
Нужно помнить, что если присутствует большое количество титана, то часть его может быть увлечена осадком сульфидов.
Совместное осаждение железа, алюминия, титана, циркония, хрома, редкоземельных металлов, фосфора и ванадия вместе с марганцем и без него
Выделяющийся в следующей стадии анализа осадок имеет очень сложный состав, и в связи с этим получение его и последующая обработка связаны с большими затруднениями. Осадок должен или может содержать алюминий, железо, титан, фосфор и остаточную кремнекислоту, а также хром, ванадий, цирконий и редкоземельные элементы. В качестве возможных компонентов следует указать также бериллий, галлий и индий, хотя в результате анализа горных пород присутствие этих последних почти никогда не отмечается.
Впрочем, нет ни необходимости, ни действительной возможности определить все перечисленные элементы в одном и том же осадке. Для определения по крайней мере некоторых из них приходится брать отдельные навески, но суммарная масса этих элементов должна быть определена в данной навеске.
Если в осаждении сероводородом не было необходимости, то обрабатывают непосредственно фильтрат от кремнекислоты (в противном случае предварительно кипятят раствор для удаления сероводорода), окисляют железо бромной водой 1 и кипячением удаляют избыток брома.
Дальнейший ход анализа зависит от того, нужно ли на этой ступени анализа осадить марганец или нет.
Осаждение без марганца. Осаждение аммиаком. Двукратного осаждения аммиаком из кипящего раствора достаточно, чтобы количественно отделить железо (III), алюминий 2, титан, фосфор, ванадий, хром (III),
1 Если присутствует большое количество железа и выпаривание для выделения кремнекислоты проводилось с соляной кислотой в платиновой чашке, то окисление железа нужно даже в том случае, когда не было обработки сероводородом, потому что небольшая часть железа могла быть восстановлена во время выпаривания.
2 Следует помнить, что в присутствии фтора алюминий осаждается аммиаком далеко не полностью.
950
Гл. LIII. Методы анализа силикатных пород
редкоземельные элементы, цирконий, а также бериллий, галлий, индий (в случае их присутствия) от марганца, никеля, щелочноземельных металлов и магния. Точное разделение достижимо лишь при тех соотношениях, в которых все эти элементы обычно встречаются в горных породах, и при условии, что аммонийные соли, чаще всего хлорид аммония, содержатся в растворе в достаточном количестве. Это последнее условие особенно важно для отделения от магния. Прежде аналитики мало обращали внимания на это условие, что и было, по-видимому, причиной частого получения ими повышенных результатов при определении алюминия, особенно когда они удовлетворялись однократным осаждением. Необходимое количество хлорида аммония лучше получать в самом растворе из чистых аммиака и соляной кислоты, чем прибавлять твердую соль, которая редко бывает достаточно чистой.
Аналитик должен быть всегда уверен в том, что анализируемый раствор содержит аммонийные соли в количестве, достаточном для удержания в растворе в присутствии аммиака наибольшего возможного в данной породе количества магния. При очень высоком содержании магния обычно требуется третье осаждение.
Для полного осаждения алюминия и точного отделения марганца нужно тщательно избегать прибавления избытка аммиака (см. «Алюминий», стр. 565 и «Осаждение аммиаком», стр. 102). Для соблюдения требуемых условий осаждения поступают следующим образом. К раствору, содержащему в 200 мл по крайней мере 5 г хлорида аммония или эквивалентное количество свободной соляной кислоты и не содержащему окисляющих веществ (например, азотной кислоты), которые могли бы разрушить индикатор, прибавляют несколько капель метилового красного и нагревают раствор почти до кипения. Осторожно прибавляют по каплям разбавленный (1 : 1) раствор аммиака, пока окраска раствора не станет явственно желтой. Кипятят раствор 1—2 прибавляют еще
аммиак, если окраска раствора изменилась в оранжевую или красную, и тотчас же фильтруют. О полном осаждении редкоземельных металлов см. стр. 618. Осадок тщательно промывают горячим 2%-ным раствором хлорида аммония. Фильтрат и промывные воды сохраняют. Осадок растворяют в горячей разбавленной соляной кислоте, промывают фильтр горячей водой и сохраняют его. Затем снова осаждают алюминий и др., фильтруют и промывают осадок, как раньше. Последнее осаждение лучше делать с прибавлением мацерированной фильтровальной бумаги (стр. 124), и раствору нужно дать хорошо стечь с осадка.
Аммиачные фильтраты и промывные воды соединяют для обработки, описанной на стр. 952, и прокаливают осадок, как описано на стр. 953.
Осаждение ацетатным методом. В присутствии умеренных количеств марганца, никеля и железа аммиачный метод осаждения в том виде, как он только что был описан, значительно лучше, чем ацетатный метод (см. «Осаждение путем регулирования концентрации ионов водорода», стр. 100), прежде пользовавшийся большой популярностью. Тем не менее последний метод может быть полезен, когда в анализируемом растворе присутствует большое количество железа вместе с цинком или кобальтом или когда присутствуют большие количества марганца или никеля, особенно для первого осаждения (второе можно производить аммиаком).
Главное возражение против ацетатного метода, кроме его малой пригодности для осаждения алюминия в отсутствие большого количества
Ход анализа после выделения кремнекислоты
951
железа, заключается в том, что последние следы марганца часто бывает невозможно удалить даже многократным повторением осаждения. Осадок, получаемый ацетатным методом, содержит в общем те же элементы, какие осаждаются аммиаком. Наиболее существенное различие состоит, может быть, в том, что некоторые из редкоземельных элементов не осаждаются этим методом. По этой причине, если при подщелачивании аммиаком соединенных фильтратов неожиданно появляется большой осадок, его надо тщательно исследовать.
При одном анализе пьемонтита (из Мериленда) свыше 2% редких земель, в том числе церий и другие неидентифицированные элементы, остались в растворе и были количественно таким образом отделены от железа, алюминия и т. п. С другой стороны, указывают *, что ванадий (V) ацетатным методом осаждается полностью, что является ценным преимуществом метода.
После осаждения растворяют осадок в соляной кислоте и переосаждают аммиаком, как описано ниже. Фильтраты обрабатывают отдельно, как описано на стр. 952.
Осаждение вместе с марганцем. Осаждение аммиаком и персульфатом аммония. Осаждение этими реактивами имеет своей целью совместное выделение марганца с осадком гидроокисей железа, алюминия и др., что упрощает последующие операции. Способ этот, однако, не пригоден при анализе пород, содержащих значительные количества бария или стронция, по причине частичного осаждения одного из этих элементов или обоих в виде сульфатов. Кальций во всех случаях можно полностью удержать в фильтратах двукратным осаждением 1 2.
Если количество стронция не настолько велико, чтобы он перешел в осадок, то против этого метода не возникает существенных возражений, потому что барий может быть снова выделен, как описано на стр. 957.
При работе этим методом следует учитывать указанные ограничения. Ход анализа следующий:
К фильтрату от кремнекислоты, находящемуся в платиновой чашке, приливают достаточное количество соляной кислоты, чтобы предупредить осаждение магния, когда раствор будет подщелочен аммиаком (стр. 949), затем индикатор (метиловый красный) и, наконец, аммиак, пока индикатор не окрасится в желтый цвет. Прибавив около 1 г персульфата аммония. свободного от мешающих примесей (см. ниже петит), нагревают до кипения. Когда жидкость начнет кипеть, осторожно прибавляют разбавленный аммиак до слабого избытка его по запаху и продолжают кипячение несколько минут 3. Дают осадку осесть, быстро фильтруют, промывают осадок 3 или 4 раза 2%-ным раствором хлорида аммония и отсасывают возможно полнее. Затем осадок растворяют в небольшом количестве горячей соляной кислоты, содержащей несколько капель сернистой кислоты, прибавляют мацерированную фильтровальную бумагу и снова осаждают, фильтруют и промывают осадок, как раньше. Соединенные фильтраты обрабатывают, как описано на стр. 952, а осадок прокаливают, как указано ниже (см. стр. 953). Марганец взвешивают в виде Мп3О4.
1 Р о р е, Trans. АШЕ, 29, 372 (1899).
2 A. W. Epperson, Ind. Eng. Chem., 17, 744 (1925).
3 Нужно прибавлять больше аммиака, чтобы нейтрализовать кислоту, освобождающуюся при разложении персульфата. Метиловый красный не может быть применен в качестве индикатора, так как он слишком быстро разрушается.
952
Гл. LIII. Методы анализа силикатных пород
Весь хром, какой был в породе, будет окислен персульфатом и перейдет в фильтрат, но его следует снова выделить вместе со следами алюминия, как подробно описано на стр. 953, и затем прибавить к главному осадку. Колориметрическое определение его в этом фильтрате не рекомендуется.
Продажный персульфат аммония всегда следует испытать на содержание в нем (NH^jSaOg, а также на присутствие алюминия и других примесей (см. «Реактивы», стр. 61).
Реактив очищают следующим способом1. К горячему насыщенному раствору реактива (не выше 95 °C) приливают раствор аммиака до запаха (через некоторое время прибавляют еще аммиака, потому что непрерывное разложение персульфата приводит к освобождению серной кислоты). Когда осадок алюминия и пр. осядёт, фильтруют насколько возможно быстро (через асбест в фарфоровой воронке, потому что концентрированный раствор персульфата заметно разрушает бумагу и стекло). По охлаждении фильтрата приблизительно до 30 °C вливают его в двойное по объему количество спирта и охлаждают стакан снаружи льдом. Через полчаса фильтруют, промывают спиртом, затем эфиром и высушивают на воздухе. Спирт должен быть тщательно удален.
Продажный реактив, очищенный таким образом, не содержал мешающих примесей, хотя не был свободен от щелочных металлов и, кроме того, содержал большое количество сульфата аммония. Вследствие разложения реактива при его очистке описанным методом и происходящей отсюда потери лучше применять для анализа отвешенное количество продажного, относительно чистого реактива и вводить поправку на примеси по результату «холостого» опыта.
Не установлено, действует ли сильно разбавленный раствор персульфата, какой образуется в ходе анализа, заметным образом на стекло, но, по-видимому, не действует, насколько это показывает наш ограниченный опыт.
Осаждение аммиаком и сульфидом аммония. Хотя старый метод осаждения аммиаком и сульфидом аммония и может применяться в специальных случаях, но вообще в силикатном анализе в настоящее время избегают им пользоваться, и поэтому он здесь не будет рассматриваться.
Этот метод часто оказывается полезным при анализе минералов, хотя осаждение им алюминия, как правило, не бывает полным.
Извлечение из фильтратов неосажденных железа и алюминия. Извлечение после осаждения аммиаком. Алюминий, железо и т. д. обычно осаждаются аммиаком полностью, если применяется метод, описанный на стр. 949. В случае сомнения и, особенно, если хотят разрушить аммонийные соли перед осаждением марганца или кальция, переводят соединенные аммиачные фильтраты в стакан, подкисляют соляной кислотой и выпаривают до небольшого объема. Покрывают стакан часовым стеклом и нагревают с азотной кислотой, как указано в разделе «Удаление аммонийных солей» (стр. 161). Когда последние будут разрушены и выделение газа прекратится, удаляют и обмывают часовое стекло, разбавляют водой не больше чем нужно, чтобы удержать соли в растворе, и нагревают до кипения. Слабо подщелачивают аммиаком, как указано на стр. 950, кипятят 2 мин и, если появился осадок, тотчас же фильтруют через маленький фильтр. Промывают осадок горячим 2%-ным раствором хлорида аммония, прокаливают, взвешивают и прибавляют массу этого осадка к массе главного осадка.
Фильтрат и промывные воды сохраняют для обработки, как описано на стр. 959 и 964.
Извлечение после осаждения ацетатным методом. Первый фильтрат выпаривают досуха или почти досуха. Приливают горячей воды в коли
1 J annasch, Praktische Leitfaden der Gewichtsanalyse, (2), 179 (1904).
Ход анализа после выделения кремнекислоты
953
честве не большем, чем нужно для удержания всех солей в растворе, и снова ставят чашку на баню на короткое время. Собирают осадок (который обычно бывает значительно большим, чем полученный по описанному выше способу) на маленьком фильтре, а фильтрат собирают в колбу емкостью 150—200 мл. Одновременно с выпариванием первого ацетатного фильтрата выпаривают и второй аммиачный фильтрат, который затем обрабатывают аммиаком, как описано выше. Этот раствор служит в качестве первой промывной жидкости для промывания другой чашки и осадка на фильтре. Наконец, обмывают обе чашки и фильтр 2%-ным раствором хлорида аммония. Аммиачный фильтрат содержит достаточно нитрата аммония, чтобы предупредить осаждение магния при сливании фильтратов.
Если на поверхности чашки осела двуокись марганца, удаляют ее, добавляя соляную кислоту и 1—2 капли сернистой кислоты, пропускают смесь через фильтр и собирают фильтрат в маленький стаканчик. Снова осаждают аммиаком, собирают осадок на тот же самый фильтр, а фильтрат — в колбу, содержащую первый фильтрат. Если присутствует большое количество марганца, может потребоваться переосаждение аммиаком этого маленького осадка. В тех случаях, когда первоначальное количество марганца мало, он переходит полностью в фильтрат. Конечный осадок и фильтрат или соединенные фильтраты обрабатывают, как указано выше.
Извлечение после осаждения аммиаком и персульфатом аммония. Соединенные фильтраты подкисляют, восстанавливают хроматы, если они присутствуют, прибавлением 1—2 капель перекиси водорода и выпаривают, как описано в разделе «Извлечение после осаждения аммиаком» (стр. 952), для разрушения аммонийных солей. Разбавляют раствор до 100 мл, осаждают добавлением аммиака в небольшом избытке и ставят на паровую баню, пока выпавший малый осадок не соберется в хлопья. Осадок переносят на фильтр диаметром 7 см и сохраняют фильтрат. Затем растворяют осадок в соляной кислоте, снова осаждают аммиаком и присоединяют его к главной порции, полученной, как описано на стр. 951. Аммиачный фильтрат прибавляют к ранее сохраненному фильтрату и обрабатывают, как описано на стр. 959 и 964.
Прокаливание соединенных осадков гидроокисей алюминия, железа и других элементов. Соединенные осадки гидроокисей алюминия и т. и., полученные, как указано выше (см. стр. 949, 951 и 952), прокаливают без предварительного высушивания их в платиновом тигле, взвешенном с плотно прилегающей крышкой. Осадок, полученный с добавлением мацерированной бумаги, как это предложил Диттрих, дает при прокаливании остаток в виде порошка, что способствует легкому окислению восстановившегося железа и вместе с тем значительно ускоряет последующее растворение.
Имеются указания 1 на то, что применение бумажной массы при осаждении гидроокисей железа и алюминия допустимо только при проведении последнего из двух (или большего числа) переосаждений.
Вместо применения мацерированной бумаги можно 2 смачивать промытый и высушенный осадок, отделенный от фильтра, спиртовым раство
1 О. Н а с k 1, Chem. Ztg., 43, 70 (1919). См. также Е. Wilk e-D б г f u г t, Е. L о с h е г, Z. anal. Chem., 64, 436 (1924).
2 М. F. Connor, Congr. Geol. Intern. 12е Sess. Canada, 885 (1913).
954
Гл. LIII. Методы анализа силикатных пород
ром нитрата аммония, снова его высушивать и прокаливать; фильтр следует тогда прокаливать отдельно.
Окись алюминия в количествах, обычно присутствующих в породах и многих минералах, не может быть полностью обезвожена на полном пламени обычной горелки Бунзена (см. «Алюминий», стр. 566). Ее надо прокаливать на пламени паяльной горелки (1200° С) до получения постоянной массы. Если присутствует большое количество железа, то нужно обеспечить доступ воздуха в тигель во время прокаливания. Тигель с содержимым следует охлаждать над хорошим высушивающим веществом и взвешивать закрытым плотно прилегающей крышкой.
Для таких продолжительных прокаливаний очень удобна электрическая печь вследствие отсутствия в ней восстановительной атмосферы, при условии, что температура в ней не настолько высока (около 1200° С), чтобы могло произойти восстановление окиси железа (III) до магнитной окиси железа или образование сплава платины с железом, сопровождающееся освобождением кислорода в местах соприкосновения окиси железа со стенками тигля (см. «Железо», стр. 438).
Сам тигель мог измениться в массе с того времени, как в нем прокаливали кремнекислоту. Поэтому правильную массу тигля находят после обработки его содержимого, как описано в следующем разделе.
Определение остаточной кремнекислоты (и бария) в соединенных осадках. Вследствие крайней трудности прямого определения окиси алюминия в смеси окислов при рассматриваемых условиях ее обычно определяют по разности. Для этого вычитают из массы осадка, полученного при осаждении аммиаком, ацетатом натрия или аммиаком и персульфатом аммония, сумму масс всех других окислов, содержащихся в этом осадке. Из последних в данной навеске пробы определяют только окись железа (представляющую все железо анализируемой породы или минерала), двуокись титана, незначительное количество кремнекислоты, которое осталось в растворе после выделения ее методом, подробно описанным на стр. 940, и в редких случаях незначительное количество бария. Окиси фосфора, ванадия, хрома, редкоземельных металлов, циркония, титана (при желании), а иногда и марганца, лучше определять из отдельных навесок пробы, иногда больших, чем те, которые обычно берут для определения главных компонентов (см. главы о соответствующих элементах). То же можно сказать и о бериллии, уране, галлии и индии, если эти элементы присутствуют в исследуемом веществе. Вследствие их редкости и малых количеств, в которых они могут встретиться, на них обычно не обращают внимания, хотя, без сомнения, эти элементы могут содержаться в некоторых породах, в особенности в сильно кремнеземистых, типа гранита.
При таком способе определения окиси алюминия все ошибки, сделанные в отдельных определениях других компонентов смеси окислов, относятся к окиси алюминия, но эти ошибки часто взаимно уравновешиваются. Во всяком случае, вероятная ошибка едва ли будет выше, чем получаемая при прямом определении окиси алюминия, принимая во внимание сложность методов удовлетворительного отделения ее от других компонентов смеси (операция, которая, кроме того, сильно удлинила бы время, требуемое для выполнения каждого анализа).
Ход анализа после выделения кремнекислоты,
955
Фосфор не может быть определен в остатке прокаленных окислов после сплавления его с пиросульфатом (см. ниже), потому что при такой обработке часть фосфора, по-видимому, улетучивается \
Остаточная кремнекислота. Первой ступенью анализа прокаленных окислов является переведение их в подходящий для дальнейшей работы раствор. Прежде некоторые авторы рекомендовали растворение окислов в соляной кислоте, но это наименее эффективный способ разложения окислов. Полное растворение достигается следующим методом.
Взвешенный остаток переносят насколько возможно полно из тигля, где проводилось прокаливание, в другой тигель емкостью 25—30 мл, если первый тигель был меньших размеров или если желательно установить потерю в массе тигля 1 2. Небольшой приставший к стенкам тигля остаток переводят в раствор сплавлением с малым количеством пиросульфата калия или натрия. Такое сплавление требует нескольких минут и не должно продолжаться дольше, чем необходимо, иначе будет невозможно после очистки и прокаливания определить правильную массу тигля, которую он имел после прокаливания окиси алюминия и пр. 3. Горячую жидкость выливают на главную массу осадка, находящуюся в другом тигле, прибавляют еще пиросульфат (всего до 7 г) и возобновляют сплавление, продолжая его в закрытом тигле при температуре не выше, чем требуется для поддержания плава в жидком состоянии, пока все не растворится.
Общая масса пиросульфата должна быть приблизительно известна, чтобы позже внести поправку на влияние этой соли при колориметрическом определении титана (см. «Титан», стр. 655), за исключением тех случаев, когда предполагают провести осаждение аммиаком, чтобы удалить щелочные металлы; тогда указанная предосторожность становится излишней.
Применение натриевой соли вместо калиевой дает превосходные результаты 4 5. Соль натрия действует быстрее, чем соль калия, и образует более растворимую соль с алюминием, но имеет тот небольшой недостаток, что за ходом разложения прокаленной массы не так легко проследить, потому что соль натрия может легко образовать корку на поверхности плава.
Расплавленный пиросульфат несколько растворяет платину тигля. Чтобы избежать этого, предложено 6 сплавлять прокаленные окислы приблизительно с 1 г кислого фторида калия. Масса плавится на очень слабом пламени, потом затвердевает. Раствор плава затем надо выпаривать с серной кислотой, чтобы удалить большую часть фтора и определить железо титрованием.
Применение фторида недопустимо, однако, при выполнении очень точных анализов силикатов, потому что в этом случае нельзя определить то небольшое количество кремнекислоты, которое присутствует в прокаленном остатке окислов. Кроме того, перед сплавлением с бифторидом калия прокаленный остаток окислов надо тонко измельчить, а следовательно, взять для сплавления только аликвотную его часть. Удаление фтора выпариванием с серной кислотой должно быть проведено очень тщательно, если затем в растворе предполагают определять титан.
При использовании пиросульфата калия конец растворения всегда легко определить (даже когда жидкость имеет темно-красную окраску
1 W. F. Н i 1 1 е b г а n d, G. Е. F. L u и d el 1, J. Am. Chem. Soc., 42, 2607 (1920).
2 Этот тигель может быть золотым, так как золото меньше разрушается при последующей обработке, чем платина. Если применяют золотой тигель, то замечания в тексте, касающиеся выделения растворенной платины, приложимы и к растворенному золоту.
3 Долгое прокаливание на пламени паяльной горелки или в печи может привести к значительному уменьшению массы тигля, откуда вытекает необходимость проверки массы пустого тигля на этой стадии анализа.
4 J. L. S m i t h, Am. J. Sci., 40, 248 (1865).
5 E. D e u s s e n, Z. anorg. Chem., 44, 423 (1925).
956
Гл. LIII. Методы анализа силикатных пород
и непрозрачна), перенеся тигель к свету и дав ему охладиться; во время охлаждения жидкость на одно мгновение становится прозрачной, и, таким образом, можно разглядеть дно тигля. При применении пиросульфата натрия это явление выражено не так резко.
Когда весь осадок растворится, выливают расплавленную массу в большую сухую платиновую чашку, остатки плава в обоих тиглях растворяют в разбавленной (1 : 9) серной кислоте и сливают полученный раствор и промывные воды в ту же чашку. Прибавляют столько разбавленной серной кислоты, чтобы получился объем 100 мл, и нагревают чашку, пока все не растворится. Выпаривают раствор, насколько это возможно, на паровой бане, затем постепенно повышают температуру, пока не начнется обильное выделение паров серной кислоты. Во время этого нагревания масса иногда темнеет от восстановления и выделения растворенной платины из тигля. Серную кислоту надо прибавлять в таком количестве, чтобы после охлаждения масса была тестообразной, а не твердой, потому что тогда она легко растворяется при нагревании в воде, которую теперь следует прибавить; соль натрия растворяется значительно легче, чем соль калия.
Чашку ставят на баню и, как только на дне ее станет видна скоагу-лировавшая кремнекислота, собирают последнюю на маленьком фильтре, хорошо промывают горячей водой, сжигают в платиновом тигле и взвешивают. Так как полученная таким образом кремнекислота редко бывает чистой, вводят поправку, прибавляя несколько капель плавиковой кислоты и каплю серной кислоты, выпаривают, прокаливают и снова взвешивают тигель. Небольшой остаток, который может оказаться в тигле, переводят в раствор сплавлением с пиросульфатом и присоединяют его к главной порции для дальнейшей обработки, которую проводят, как описано на стр. 957; если присутствует сульфат бария, его предварительно отфильтровывают (см. ниже «Барий»).
Количество выделяемой таким образом кремнекислоты (см. табл. 23, стр. 758) обычно находится в пределах от 0,5 до 5 мг, если главная масса кремнекислоты была выделена двукратно, как описано выше. Массу этого последнего остатка кремнекислоты прибавляют к массе основной ее части. Выделение остатка кремнекислоты из прокаленного осадка окислов может быть проведено только описанным выше способом Ч Прежде считали, что нерастворившийся остаток после обработки плава водой или разбавленной кислотой представляет собой всю присутствовавшую в плаве кремнекислоту. Но это лишь небольшая часть всей кислоты, захваченной осадком гидроокисей алюминия и др., так как большая ее часть образует во время сплавления силикаты щелочных металлов и остается затем в растворе, если ее не выделить и не перевести в нерастворимое состояние указанной выше обработкой.
Следует отметить, что небольшое количество кремнекислоты может остаться в растворе и после выделения ее с осадком от аммиака. Величина этого количества зависит от того, сколько кремнекислоты избежало выделения вначале, при выпаривании с соляной кислотой, и от размера осадка от аммиака. При однократном выделении кремнекислоты в начале анализа масса кремнекислоты, оставшейся невыделенной, может достигать нескольких миллиграммов. Отсюда ясно, почему так настоятельно
1 Доказательство этого см. в J. Am. Chem. Soc., 24, 368 (1902).
Ход анализа после выделения кремнекислоты
957
требуется возможно более полное выделение кремнекислоты в самом начале анализа.
В случае, если не требуется выделения остаточной кремнекислоты, можно применять метод Траутмана 1 для удаления ее из окиси алюминия, особенно когда масса прокаленных окислов незначительна. Этот метод состоит в обработке прокаленных окислов в тигле фтористоводородной и серной кислотами, выпаривании, вторичном прокаливании и взвешивании. При применении этого метода не следует спешить с удалением серной кислоты, потому что можно опасаться неполного превращения фторидов в сульфаты и вследствие этого потери от улетучивания безводного фторида. Имеются, однако, указания 2 на то, что при применении этого метода можно не опасаться заметной потери алюминия вследствие улетучивания его в виде фторида; это подтверждается и нашим опытом.
Барий. Если осадок прокаленных окислов алюминия и др. содержит барий 3, он будет обнаружен как загрязнение кремнекислоты, выделенной после сплавления с пиросульфатом. Если взвешенный остаток после удаления этой кремнекислоты плавиковой кислотой сплавить с небольшим количеством пиросульфата и охлажденный плав растворить в разбавленной серной кислоте, то весь барий останется в виде сульфата. Иногда вместе с ним оказываются частицы металлической платины. В этом случае их можно достаточно удовлетворительно разделить методом отмучивания.
Определение железа в осадке смеси окислов. Объемное определение в отсутствие ванадия. Охлажденный фильтрат, полученный, как описано на стр. 953, обрабатывают методами, приведенными в гл. «Железо» (стр. 444), если присутствует титан, или методом, описанным на стр. 442, в отсутствие титана. Затем титруют полученное железо (II) перманганатом (стр. 445) и определяют титан способом, описанным в гл. «Титан» (стр. 655).
Можно также, если титан отсутствует или его определение в этом растворе не требуется, оттитровать железо в трехвалентном состоянии солью титана (III) (см. «Железо», стр. 449).
Объемное определение в присутствии ванадия. Описанные дальше способы основаны на предположении, что ванадий присутствует только в очень малых количествах, как это бывает в силикатных породах, глинах и кремнистых известняках. В таких случаях в найденном общем содержании железа будет небольшая ошибка, независимо от того, какой был применен восстановитель (см. «Железо», стр. 441). Принимая во внимание вышеуказанное, рекомендуется в присутствии небольшого количества ванадия пользоваться только методом восстановления сернистым ангидридом (см. «Железо», стр. 444) даже при отсутствии титана. Когда количество ванадия известно, можно внести поправку, предполагая, что весь ванадий содержится в исследуемом осадке, что, однако, нуждается в доказательстве. Ряд авторов подтверждает выпадение ванадия в осадок вместе с алюминием и железом при осаждении аммиаком или ацетатом аммония, но имеются указания 4 и на то, что при повторном осаждении аммиаком,
1 Trautman n, Z. angew. Chem., 26, 702 (1913).
2 См. сноску 3 на стр. 939.
3 При анализе горных пород в нашей практике не обнаруживали содержания бария за исключением случаев, когда применялся персульфат аммония (см. стр. 952).
4 Carnot, С. г., 104, 1803 (1887).
958
Гл. LIII. Методы анализа силикатных пород
карбонатом аммония или кислым сульфидом аммония ванадий отделяется от железа.
Было проведено тщательное исследование 1, в какой мере ванадий соосаждается с различными металлами, и были получены многочисленные цифровые данные, показывающие, что приблизительно 90% ванадия переходит в осадок (когда осаждение происходит в условиях, обычно применяемых при анализе шлаков), остальное остается в фильтрате. Из оставшегося небольшая часть осаждается с кальцием, а большая — с магнием. Таким образом, для всех практических целей можно принять, что те небольшие количества ванадия, которые встречаются в горных породах, полностью переходят в осадок гидроокисей алюминия и железа.
Если количество ванадия неизвестно и нужна большая точность, осторожность требует, чтобы или определение общего количества железа было проведено в отдельной навеске после удаления ванадия или чтобы после осаждения аммиаком в сернокислом растворе железа, алюминия, титана, ванадия и т. д. было отделено железо от ванадия прокаливанием и сплавлением прокаленных окислов с карбонатом натрия, выщелачиванием плава водой, фильтрованием, переведением нерастворимого остатка в сернокислый раствор и титрованием, как в случае отсутствия ванадия. Но если при этом не соблюдать определенных предосторожностей, то здесь может быть сделана ошибка большая, чем та, которой старались избежать.
В противоположность общему мнению, водная вытяжка после сплавления с содой содержит небольшую, но все же заметную часть железа. Это железо выделяется вместе с алюминием (и кремнекислотой, если таковая присутствует) при обычном методе нейтрализации щелочного раствора и может быть обнаружено, если осадок, полученный таким образом, обработать едкой щелочью или снова сплавить с карбонатом натрия и затем плав выщелочить водой; тогда железо останется полностью или частично нерастворенным. Необходимо собрать это железо и присоединить его к главной порции перед титрованием.
Метод определения железа и ванадия при совместном их присутствии 2, в котором ванадий определяют по разности, мало пригоден для анализа горных пород вследствие очень малого содержания в них ванадия.
Весовое определение после выделения железа в виде сульфида. Ввиду ошибок, получаемых при объемном определении железа, при выполнении очень точных анализов рекомендуется определять его весовым способом, осаждая аммиаком после предварительного выделения в виде сульфида иэ раствора, содержащего тартрат аммония (см. «Осаждение сульфидом аммония», стр. 90), роль которого — удержать в растворе большую часть других элементов.
Истинное содержание железа (III) в горных породах и минералах. Чтобы найти количественное содержание железа (III) в горных породах и минералах, содержащих также железо (II), принято вычитать из общего содержания железа, перечисленного на БегОз, количество окиси железа (III), эквивалентное содержанию железа (II), определенному отдельно.
Если бы содержание железа (III) можно было определить так же точно, как определяется содержание железа (II), последнее можно было бы находить вычитанием уже определенной окиси железа (III) из общего
1 Ridsdale, J. Soc. Chem. Ind., 7, 73 (1868).
2 Edgar, Am. J, Sci., 26, 9 (1908).
Марганец, никель, кобальт, медь и цинк
959
содержания железа, перечисленного на ГегОз, и пересчетом разности на FeO. При точной работе результаты и в том и в другом случае должны получаться удовлетворительными, если нет мешающих элементов. Последние, однако, часто присутствуют, особенно в горных породах и глинах. В горных породах обычно содержатся небольшие количества сульфидов, почти всегда пирит.
Результат определения железа (III), найденный по разности, только в том случае не будет ошибочным, если известно количество пиритного железа, перечисленное на окись, и если оно вычтено из общего количества железа (III). (Предполагается, что содержание пиритного железа представляется в отдельной графе таблицы результатов анализа). Это количество может быть определено по содержанию серы, если она присутствует только в виде пиритной, — факт, который обычно может быть установлен с достоверностью. Но если присутствующие сульфиды растворимы в серной или плавиковой кислоте, то сероводород, выделяющийся при растворении пробы во время определения железа (И) или железа (III), вероятно, восстановит небольшое количество соли железа (III) и таким образом увеличит количество железа (II) и уменьшит количество железа (III). Влияние таких сульфидов не может быть определено. Мешают также незначительные количества ванадия, присутствующие в большей части горных пород и глин. Поэтому очевидно, что результат определения окиси железа (III) может быть только приблизительным.
Определение других составных частей в осадке окислов (алюминия и т. и.). Здесь нет необходимости рассматривать методы, которые можно применить для определения других возможных компонентов осадка, перечисленных на стр. 949. Достаточно сослаться на главы о различных элементах и на параграф о разделении элементов в осадке от аммиака (стр. 112).
МАРГАНЕЦ, НИКЕЛЬ, КОБАЛЬТ, МЕДЬ И ЦИНК
Затруднения, встречающиеся при точном определении марганца весовым способом
Весовое определение небольших количеств марганца, по-видимому, представляет больше затруднений для среднего химика, чем определение почти всех других элементов, встречающихся часто в анализе минералов. Большая часть трудностей вызывается неполным предварительным отделением элементов, которые потом осаждаются вместе с марганцем. Поэтому ошибка обычно бывает в сторону увеличения и часто превосходит в несколько раз действительное содержание марганца. Отсюда ясно, насколько важно наиболее полное выделение компонентов, рассмотренных в предыдущих разделах.
Неопытному аналитику осадок может показаться состоящим исключительно из МпзО«, так как очень малые количества марганца, осажденного в виде гидроокисей высшей степени его окисления, могут маскировать значительные количества окиси алюминия и других неокрашенных окислов.
На этом основании Вашингтон считает, что при анализе пород малоопытным аналитикам лучше не проводить определения марганца, а распределять получаемую ошибку между алюминием, кальцием и магнием. Относительный порядок этого распределения был определен Г. Стейгером (лаборатория Геологического управления
960
Гл. LIII. Методы анализа силикатных пород
США) при анализе ряда известняков, часть которых отличалась высоким содержанием кремнекислоты. Результат (табл. 35) получился весьма поучительным. Повсюду применялось двукратное осаждение, и железо с алюминием осаждались добавлением аммиака в небольшом избытке. Если прибавление аммиака прекращали, когда значение pH достигало 7, и раствор фильтровали через 1—3 мин, то осадок от аммиака содержал очень мало марганца или совсем его не содержал.
ТАБЛИЦА 3 5
Распределение марганца при весовом определении
м пробы Содержание, % Колориметрически определенное содержание марганца в расчете на МпО, %
-A-ljOj, FetOi СаО MgO всего в осадках
AljOj, FetO3 (по разности) СаО MgjPtOj
957 2,03 10,60 6,30 0,193 0,085 0,011 0,097
973 9,35 11,84 2,81 0,311 0,036 0,023 0,252
974 4,80 50,51 1,04 0,700 0,301 0,087 0,312
975 12,71 11,98 4,30 0,442 0,088 0,016 0,338
1126 0,58 30,54 20,41 0,281 0,030 0,030 0,221
1128 0,98 29,69 19,07 0,245 0,019 0,055 0,171
ИЗО 3,49 3,99 0,92 0,016 0,016 Нет Нет
1131 1,00 28,04 19,11 0,574 0,032 0,101 0.441
Учитывая исследования Стейгера в настоящее время в Геологическом управлении США применяют такой метод осаждения алюминия и т. и., при котором марганец не осаждается. Метод этот состоит в том, что общее содержание марганца определяют в отдельной навеске, а в массу пирофосфата магния вносят поправку на содержащийся в нем марганец, который определяют колориметрическим способом. Этот метод допустим, потому что, как указывает табл. 35, большая часть марганца остается в растворе, пока не осадят вместе с магнием в виде фосфата, а ошибки, приходящиеся на окись алюминия и окись кальция, очень малы. Загрязнение прокаленного оксалата кальция даже малым количеством марганца обнаруживается обычно по коричневому окрашиванию, которое придают ему окислы марганца, а иногда по зеленому цвету манганата кальция.
Если, однако, тщательно следовать описанному ниже ходу работы, то можно не опасаться загрязнения осадка марганца алюминием, а также и магнием, при условии, что было Добавлено достаточное количество аммонийных солей. Нужно только принять во внимание возможность присутствия небольшого количества редкоземельных металлов, которые остаются в растворе после осаждения ацетатным методом (стр. 950), потому что тогда они выделяются на этой ступени анализа.
Осаждение всей группы и разделение ее составных частей
Метод осаждения сульфидом аммония. Преимущества и недостатки метода. Обычные методы отделения марганца от щелочноземельных металлов и магния добавлением брома или сульфида аммония несовершенны, отчасти вследствие того, что марганец осаждается не полностью, отчасти по причине соосаждения небольших количеств других металлов. Первая ошибка, хотя она по абсолютной величине и незначительна, вероятно имеет большее значение в анализе горных пород, чем вторая ошибка. В отношении полноты осаждения бром не имеет преимуществ перед сульфидом аммония, преиму
Марганец, никель, кобальт, медь и цинк
961
ществом же последнего реактива является то, что при одной обработке вместе с марганцем от щелочных и щелочноземельных металлов отделяются также никель, кобальт, медь и цинк, если они присутствуют. Не следует опасаться при этом потери части никеля или меди, так как в этих условиях они осаждаются полностью. Это преимущество и является главным основанием, по которому метод осаждения сульфидом аммония заслуживает предпочтения. Небольшое количество марганца, которое остается в растворе после осаждения сульфидом аммония, взвешивается потом вместе с магнием в виде пирофосфата и может быть легко определено колориметрическим методом, описанным на стр. 724, и учтено.
Метод осаждения марганца из щелочного раствора перекисью водорода 1 казался простым и точным и, кроме того, допускающим одновременное отделение от цинка, но затем была доказана 2 его непригодность.
Осаждение сульфидом аммония и отделение марганца и цинка от никеля, кобальта и меди. К находящемуся в колбе раствору, содержащему марганец, щелочноземельные металлы и т. п. (стр. 952), прибавляют 2—3 мл раствора аммиака и насыщают сероводородом. Марганец, никель, кобальт, медь, цинк и немного платины (из платиновой чашки) осаждаются. Затем прибавляют еще такое же количество аммиака, наполняют колбу до горла водой, закрывают пробкой и оставляют стоять по крайней мере 12 ч (лучшее 24 ч или еще дольше). Фильтруют через небольшой фильтр, промывают осадок водой, содержащей хлорид аммония и сульфид аммония, и выщелачивают осадок разбавленной соляной кислотой, содержащей сероводород [1 объем соляной кислоты (пл. 1,11 г/см3 * *) на 5 объемов сероводородной воды]. Марганец и цинк, если они присутствуют, переходят в раствор.
О дальнейшей обработке фильтрата от осаждения сульфидом аммония см. стр. 963.
Марганец и цинк
Солянокислый фильтрат выпаривают досуха и разрушают аммонийные соли выпариванием с несколькими каплями раствора карбоната натрия. Избыток карбоната разлагают и выпавшую двуокись марганца переводят в раствор добавлением соляной кислоты и одной капли сернистой кислоты. После удаления выпариванием соляной кислоты осаждают марганец карбонатом натрия в кипящем растворе. Если присутствует цинк, его можно отделить от марганца после взвешивания осадка. Для тех малых количеств марганца, с которыми обычно имеют дело, способ осаждения карбонатом натрия следует предпочесть методам осаждения бромом и фосфатом натрия как более быстрый и одинаково точный. При определении малых количеств марганца, которые обычно присутствуют в горных породах, ошибкой, вызванной адсорбцией щелочных металлов выпадающим осадком, можно пренебречь.
Марганец взвешивают в виде МпзСП и пересчитывают на МпО или, если количество его относительно велико, взвешивают в виде сульфата марганца 8. Но с гораздо большей уверенностью и точностью марганец
1 Р. J а n n a s с h, Е. V. С 1 о е d t, Z. anorg. Chem.. 10, 405 (1895).
2 C. Fri edheim, E. Briihl, Z. anal. Chem., 38, 681 (1899).
* J. V о 1 h a r d, Ann. Chem., 198, 329 (1879); F. A. G о о c h, M.Austin, Am.
J. Sci., [4], 5, 209 (1898); Z. anorg. Chem., 17, 264 (1898).
61 Зака» 622.
962
Гл. LIII. Методы анализа силикатных пород
можно определить колориметрическим методом, как описано ниже, если только количество его не выходит за пределы, обычные для горных пород. В последнем случае можно в отсутствие цинка применять для колориметрического определения солянокислый раствор сульфата марганца, удалив предварительно соляную кислоту выпариванием с азотной или серной кислотами.
Никель, кобальт и медь
Фильтр, на котором находится осадок сульфидов никеля, кобальта и меди, сжигают в фарфоровом тигле, растворяют золу в нескольких каплях царской водки и раствор выпаривают с соляной кислотой. Медь и платину осаждают при нагревании сероводородом, а из подщелоченного аммиаком фильтрата осаждают также сероводородом никель и кобальт. После пропускания сероводорода слабо подкисляют этот раствор уксусной кислотой и оставляют на некоторое время. Затем осадок сульфидов никеля и кобальта отфильтровывают, фильтр с осадком сжигают и взвешивают в виде NiO -f- СоО. Количество этих окислов всегда очень мало, редко больше 0,3—0,4 мг. Затем проводят пробу на кобальт с перлом буры. Присутствие или отсутствие никеля и кобальта легко узнается также по окрашиванию последних капель солянокислого раствора, когда разрушают царскую водку и затем выпаривают раствор. При этом рекомендуется удалять аммиаком небольшие количества возможного здесь железа, снова подкислять раствор и обрабатывать его сероводородом, как указано выше х.
Следы меди, выделенные в этой стадии анализа, нельзя с уверенностью приписать присутствию меди в анализируемой породе, если в ходе анализа выпаривание проводили, как это обычно делают, на медных воздушных или водяных банях или применяли воду, которая была перегнана из медного куба, хотя бы и луженого. Если присутствие меди в анализируемой породе установлено несомненно, то по указанной выше причине, а также и потому, что выделенный в этой стадии осадок сульфида меди всегда бывает загрязнен платиной, лучше определять медь в отдельной навеске анализируемой пробы (см. стр. 948). Особенно осторожным следует в этом отношении быть, когда при подготовке пробы к анализу применяют медные или латунные сита.
Колориметрическое определение марганца
Как было показано выше (стр. 959), весовое определение марганца нередко приводит к большим ошибкам, даже если его проводят очень тщательно. Это происходит от различных причин: от неполноты отделения алюминия и железа, неполного осаждения марганца сульфидом аммония и загрязнения осадка другими веществами. Так как определяемые количества марганца обычно очень малы, то относительная величина этих ошибок может быть весьма значительной и нельзя быть уверенным, что они будут компенсировать друг друга. Поэтому лучше определять общее содержание марганца в отдельной навеске, а в главной навеске определять
1 Об определении малых количеств никеля и кобальта в горных породах см. О. Н а с k 1, Chems Ztg., 46, 385 (1922).
Кальций, стронций [барий]
963
только ту его часть, которая взвешивается вместе с пирофосфатом магния (только для введения поправки при определении магния). Определение марганца в отдельной навеске пробы особенно легко выполнимо при анализе карбонатных горных пород, которые легко можно перевести в раствор в течение нескольких минут без сплавления и отделения кремнекислоты. При анализе силикатов это требует большого труда, но само колориметрическое определение дает такие же точные результаты, какие получаются при анализе карбонатных пород.
Предварительная обработка проводится следующим образом. 1,0— 0,5 г измельченной пробы разлагают в маленькой платиновой чашке или во вместительном тигле (помещенном в радиаторе, показанном на рис. 5, стр. 49) смесью фтористоводородной и серной кислот. Когда все растворится, удаляют плавиковую кислоту повторным выпариванием с небольшими количествами серной кислоты. Затем приливают свободную от хлора серную или азотную кислоту и воду и нагревают до перехода возможно большей части остатка в раствор. Нерастворившийся остаток обычно состоит из сульфата бария и сульфата кальция. Его отфильтровывают через маленький фильтр, собирая фильтрат в небольшой стакан или колбу, и определяют в растворе марганец, как описано в гл. «Марганец» (стр. 507).
КАЛЬЦИЙ, СТРОНЦИЙ (БАРИЙ)
Прежде чем приступить к дальнейшему анализу, надо внимательно прочитать разделы «Общие замечания», «Методы отделения» и «Методы определения» в гл. «Щелочноземельные металлы» (стр. 693).
Вся платина, перешедшая в раствор из платиновой чашки при выделении кремнекислоты, находится теперь в фильтрате от сульфида марганца, полученного, как описано на стр. 961, кроме небольшого ее количества, которое могло осесть вместе с марганцем. Удаление платины здесь совершенно не нужно; обычно не требуется также удаления хлорида аммония, так как раствор не должен содержать слишком больших его количеств, особенно если первое осаждение железа и алюминия было сделано ацетатным методом. Поэтому можно немедленно осадить кальций п стронций оксалатом аммония, не разрушая также и сульфида аммония. После осаждения оксалатом аммония раствор фильтруют и осадок промывают 0,1 %-ным раствором этого реактива, как описано в гл. «Щелочноземельные металлы» (стр. 705). Фильтрат сохраняют и затем соединяют его с фильтратом от второго осаждения. Осадок, полученный при первом осаждении, часто бывает темного цвета от выделившегося сульфида платины. Содержащаяся в нем платина может быть отделена фильтрованием после растворения осадка оксалатов или после прокаливания этого осадка и растворения полученных окисей кальция и стронция в разбавленной соляной кислоте. Во всех случаях осадок оксалатов следует переосадить, промыть и взвесить, как описано в гл. «Щелочноземельные металлы» (стр. 707). Фильтрат от второго осаждения соединяют с ранее полученным фильтратом и определяют магний, как описано ниже.
Полученный в результате всех этих операций прокаленный и взвешенный осадок переводят в колбу емкостью 20 мл, растворяют в разбавленной азотной кислоте и выпаривают раствор досуха. Потом прокаливают сухой остаток при 130—160 °C и отделяют стронций от кальция извлечением последнего смесью спирта и эфира, как описано в гл. «Щелочноземельные металлы» (стр. 697).
61*
.9 64
Гл. LIII. Методы анализа силикатных пород
Описанный выше ход анализа применим почти ко всем силикатным породам. Однако когда необходимо исключительно точное определение очень малого количества кальция в присутствии большого количества магния, надо применять метод, в котором кальций сначала осаждают в виде сульфата, а затем уже в виде оксалата (см. «Щелочноземельные металлы», стр. 697).
МАГНИЙ
Прежде чем приступить к осаждению магния, нужно внимательно прочитать разделы «Методы отделения» и «Методы определения» гл. «Магний» (стр. 715 и 719).
Предварительное осаждение магния может быть сделано непосредственно в соединенных фильтратах от определения кальция, если этот раствор не содержит слишком большого количества аммонийных солей, особенно оксалата аммония. Объем раствора не должен превышать 600 мл, в противном случае раствор следует подкислить и упарить кипячением. Если в растворе присутствует чрезмерно большое количество аммонийных солей, особенно когда содержание в нем магния мало, надо подкислить раствор, выпарить его и разрушить аммонийные соли прокаливанием или обработкой азотной кислотой, как описано в разделе «Удаление аммонийных солей» (стр. 161).
На каждые 100 мл анализируемого раствора следует прибавить двузамещенный фосфат аммония (NH4)2HPO4 в количестве, приблизительно равном 2 г. К концу осаждения раствор должен содержать примерно 10% по объему аммиака, и перед фильтрованием его нужно оставить стоять 12 ч. При фильтровании следует сначала декантировать прозрачную жидкость через быстро фильтрующий фильтр, затем перенести на фильтр осадок и промыть стакан и осадок 3 или 4 раза небольшими порциями разбавленного (5 : 95) раствора аммиака. Фильтрат с промывными водами перемешивают и оставляют стоять, чтобы посмотреть, не выпадет ли еще осадок.
В анализах, в которых при выделении кремнекислоты применяли платиновую чашку, а для выделения марганца — сульфид аммония, первый осадок фосфата магния и аммония может быть загрязнен сульфидом платины. Это, однако, не имеет значения, так как последний остается на фильтре, когда фосфат растворяют в соляной кислоте перед вторым осаждением. Сначала растворяют осадок, приставший ко дну и стенкам сосуда, где происходило осаждение, в горячей разбавленной (1 : 9) соляной кислоте, затем пропускают этот раствор через фильтр и собирают фильтрат в стакан емкостью 200 мл. Когда осадок на фильтре полностью растворится, стакан, фильтр и воронку тщательно промывают водой. Второе осаждение и прокаливание фосфата проводят, как описано в гл. «Магний» (стр. 722), причем не надо забывать прибавить к раствору немного осадителя и ввести указанные там поправки.
ТИТАН
Общие замечания
В последние годы начали усиленно заниматься аналитической химией титана не столько в применении к анализу горных пород, сколько в связи с анализом титановых руд и металлического титана, а также полу
Т итан
965
чаемых из него сплавов. До известной степени то же может быть сказано и в отношении циркония. Так как цирконий является очень распространенным, хотя и второстепенным компонентом горных пород и в значительной части своих химических свойств подобен титану, то нельзя описывать какой-либо метод осаждения и выделения титана, не указывая постоянно на поведение при этом циркония, особенно если речь идет об определении титана весовыми методами.
Когда титан и цирконий не сопровождаются другими элементами, осаждаемыми аммиаком, лучше всего осадить их этим реактивом. С другой стороны, при благоприятных условиях колориметрический метод определения титана (стр. 655) дает результаты, не уступающие результатам, получаемым лучшими из весовых методов, и при этом в значительно более короткое время, особенно при определении тех малых количеств титана, какие обычно встречаются в горных породах, глинах и почвах (менее 1% и лишь изредка до 2—3% и более). Ошибка при применении этого метода не должна превышать 2% в широких пределах концентраций х.
Наиболее благоприятными условиями для колориметрического определения, о которых сказано выше, являются: отсутствие больших количеств железа и фосфора, солей щелочных металлов, ванадия и даже следов фтора. Применение колориметрического метода, когда в растворе присутствует одна или несколько из указанных выше примесей в количествах, способных помешать определению, настолько затруднено, что в таких случаях лучше или совсем отказаться от этого метода или применять его только после отделения титана от мешающих элементов одним из способов, описанных на стр. 959 (см. «Определение других составных частей в осадке окислов»). В некоторых случаях, например при отсутствии циркония, можно удовлетвориться взвешиванием осадка титана, выделенного одним из таких способов, но вообще следует заканчивать определение колориметрическим методом или титрованием. Когда аналитик ближе узнает недостатки и преимущества всех этих методов, он научится сам разбирать, какому из этих методов следует отдать предпочтение в каждом отдельном случае.
Из весовых методов определения титана купфероновый метод, более простой, чем метод Гуча, в настоящее время, по-видимому, в значительной мере вытеснил последний. Присутствие циркония в пробе в количествах, превышающих следы, также ограничивает возможность применения метода Гуча. Старинный метод осаждения титана длительным кипячением почти нейтрального сернокислого раствора можно совсем не рассматривать.
Большим преимуществом объемных методов является то, что они применимы в присутствии алюминия и циркония, и потому эти методы широко используются в анализе руд и технических продуктов.
В общем, можно посоветовать для окончательного определения титана после отделения мешающих элементов применять либо колориметрический (стр. 655), либо объемный (стр. 659) методы. Выбор метода зависит также от цели, которую имеют в виду. Если требуется определение процентного содержания только одного титана, то наиболее удобным может оказаться метод, который вообще нельзя ввести в общую схему полного анализа силикатов.
1 R. С. W е 11 s, J. Am. Chem. Soc., 33, 501 (1911); Z. anorg. Chem., 70, 399 (1911).
966
Гл. LIII. Методы анализа силикатных пород
Приведенные выше замечания сделаны в предположении, что надо определить только общее содержание титана, безотносительно к степени его окисления. В громадном большинстве случаев в минералах, содержащих титан, последний, несомненно, находится в четырехвалентном состоянии, отвечающем окислу TiO2. Но есть некоторое основание предполагать, что титан может присутствовать и в трехвалентном состоянии, отвечающем окислу ТлгОз. Определение содержания титана различных степеней валентности представляет в настоящее время почти непреодолимые трудности, главным образом из-за повсеместного присутствия железа. Якоб1 приводит анализы двух флогопитов и одного биотита, показывающие, что в этих минералах присутствуют примерно одинаковые количества четырехвалентного и трехвалентного титана.
Колориметрическое определение с перекисью водорода
Подробные указания см. в гл. «Титан» (стр. 655).
Если предполагают применить колориметрический метод, то можно предварительно получить приблизительную оценку содержания титана, обрабатывая сернокислый раствор пиросульфатного плава осадка от аммиака перекисью водорода, прежде чем выпаривать этот раствор для определения содержания кремнекислоты в этом плаве (стр. 955). Титан удобно определять в растворе, который служил для определения титрованием общего количества железа (стр. 957).
Непосредственное применение этого раствора требует, однако, при точной работе специальных мер для устранения мешающего влияния содержащихся в нем железа, сульфатов щелочных металлов и иногда ванадия. Вредное влияние железа может быть устранено прибавлением раствора сульфата железа (III) к стандартному раствору, еще до прибавления перекиси водорода, пока не сравняются окраски обоих растворов. Влияния солей щелочных металлов можно избежать, если предварительно осадить титан и пр. аммиаком, умеренно промыть осадок и растворить его в 5 %-ной серной кислоте. Мешающего влияния ванадия можно избежать, осаждая титан, железо и др. едким натром вместо аммиака (стр. 655) или сплавляя прокаленный осадок от аммиака с содой, выщелачивая ванадий водой и переводя остаток в сернокислый раствор.
Ошибки, происходящие от присутствия сульфатов щелочных металлов, можно также избежать, если определять титан в другой навеске горной породы, а не в той, которая указана в предыдущем разделе. Это определение может быть соединено с определением бария разложением порошка породы серной и фтористоводородной кислотами; последнюю удаляют повторным выпариванием с серной кислотой, остаток растворяют в разбавленной серной кислоте, отфильтровывают сульфат бария и т. д. и в фильтрате определяют титан колориметрически.
Нужно принять во внимание, что выпаривание с одной фтористоводородной кислотой, сопровождаемое прокаливанием, влечет за собой потерю титана вследствие улетучивания и что такой потери не происходит, если присутствует избыток серной кислоты.
В случае анализа кислых пород растворение в разбавленной серной кислоте происходит хорошо. При анализе основных пород его можно ускорить, проводя обработку в маленьком стакане и при осторожном кипячении. Полученный таким образом оста
1 J. J а к о Ь, Chemigche Analyse der Gesteine und silikatischen Mineralien, 1952, S. 73, Basel.
Титан
967
ток может содержать, помимо сульфата бария, немного гипса, циркон, андалузит, топаз и, возможно, следы титана в какой-либо форме. Поэтому остаток надо сплавить с карбонатом натрия, выщелочить плав водой, затем сплавить остаток с пиросульфатом калия, растворить в разбавленной серной кислоте, раствор профильтровать и фильтрат присоединить к главному раствору. После этого нерастворимый остаток состоит преимущественно из сульфата бария. Исследование его см. стр. 968.
Удалять фтор надо очень тщательно, иначе результат определения титана будет пониженным, как было указано выше (стр. 655); не всегда легко достигнуть такого полного удаления фтора, хотя время, требуемое для этого, по-видимому, ни в какой мере не зависит от природы фторидов, которые приходится разлагать. Долго еще после того, как весь фтор кажется удаленным, под образующейся коркой на поверхности раствора скопляется газообразный фтористый водород, который легко обнаружить по запаху, если разбить эту корку Е
Влияние железа и щелочных металлов исключается, если определение титана проводить в осадке TiOa и ZrO2, полученном одним из методов, указанных на стр. 959 (см. «Определение других составных частей в осадке окислов»).
Остаток от сплавления навески породы с карбонатами щелочных металлов, полученный, как указано на стр. 969, можно использовать для определения титана до и после отделения циркония (стр. 971).
При определении титана необходимо ввести поправку на железо, содержащееся в породе, независимо от того, был ли цирконий удален сначала или нет. Если он был удален сначала, то следует также ввести поправку на сульфат щелочного металла, образующийся от прибавления фосфата натрия, применяемого для осаждения циркония, а также, может быть, и на фосфорную кислоту, содержащуюся в породе и прибавленную. Однако в большинстве случаев количество фосфата натрия, необходимое для двух осаждений циркония, не вызывает заметной ошибки в результате, получаемом для титана. Не надо еще забывать соединять оба фильтрата от фосфата циркония, чтобы определить весь присутствующий титан.
Выбор того или иного хода анализа в каждом отдельном случае должен проводиться опытным аналитиком в зависимости от условий и удобства работы. Естественно, что следует предпочесть тот вариант метода, при котором не требуется внесения поправок, но для получения точных результатов было бы неразумно рисковать введением других ошибок (значения которых неизвестны и не всегда одинаковы), если есть возможность внести точную поправку, хотя это и требует некоторой дополнительной работы.
При анализе горных пород, если поправок даже совсем не вводить, то ошибка в найденном значении содержания титана будет так мала, что ею можно в большинстве случаев пренебречь, особенно если известно, что ошибка эта всегда одного знака. Разумеется, каждая ошибка в определении окиси титана вызывает равную ей, но противоположную по знаку ошибку в результате определения окиси алюминия.
Весовые методы
Когда титан присутствует в количествах, превышающих 4—5%, или если по какой-либо другой причине желательно применение весового метода, можно воспользоваться либо купфероновым методом (стр. 143),
1 На стр. 821 описан метод ускоренного удаления фтора.
968
Гл. ЫН. Методы анализа силикатных пород
либо методом Гуча (стр. 661). Первый метод требует меньшего труда; в этом методе цирконий количественно осаждается вместе с титаном. Во втором методе цирконий препятствует полному осаждению титана, особенно когда содержание последнего невелико1.
Метод Гуча применим ко всякому раствору горной породы, свободному от кремнекислоты, железа, циркония и от элементов, соли которых легко гидролизуются, например от ниобия и тантала.
Купфероновый метод можно применять к любому раствору горной породы, не содержащему кремния, элементов группы сероводорода и больших количеств фосфора. Обычно этот метод служит для отделения титана вместе с цирконием, железом, ванадием и пр. (стр. 145) от алюминия, хрома, а также фосфора, за исключением тех случаев, когда последний присутствует в значительных количествах и сопровождается цирконием, торием или титаном. Тогда сначала сплавляют пробу с карбонатом натрия, выщелачивают плав водой, остаток переводят в сернокислый раствор (иногда применяя для этого сплавление с пиросульфатом) и в этом растворе проводят осаждение купфероном. Тем же способом удаляют и ванадий. Металлы сероводородной группы могут быть удалены из сернокислого раствора обработкой сероводородом (стр. 83), после чего удаляют железо прибавлением винной кислоты и сульфида аммония (стр. 90). Эти методы отделения служат для удаления всех мешающих веществ, кроме циркония. Фильтрат после отделения сульфида железа подкисляют, осаждают титан и цирконий купфероном, осадок прокаливают и взвешивают сумму окислов обоих металлов. Содержание титана находят затем по разности после сплавления смеси окислов с пиросульфатом, растворения плава в серной кислоте и определения циркония в виде пирофосфата (стр. 640).
Объемные методы
Из объемных методов определения титана следует предпочесть метод, в котором проводят восстановление в редукторе Джонса (стр. 135), собирают прошедший через редуктор раствор под раствором сульфата железа (III), взятым в избытке, и титруют перманганатом (стр. 659). В этом методе мешает присутствие железа, ванадия, хрома и азотной кислоты, которые также восстанавливаются в редукторе Джонса. Поэтому его следует применять после отделения этих элементов от титана или вводить поправки на их присутствие в получаемый результат.
БАРИЙ
(ЦИРКОНИЙ, РЕДКОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ, ОБЩЕЕ СОДЕРЖАНИЕ СЕРЫ, ХРОМ)
Много времени и труда можно сберечь, если все эти пять компонентов породы определять в одной навеске пробы величиной около 2 г. Если, однако, нужно определить также и ванадий, то тогда определение ванадия и хрома лучше проводить в отдельной навеске (стр. 982). Выше было сказано (см. «Щелочноземельные металлы», стр. 695), что только
1 Например, при анализе некоторых горных пород присутствие 0,2% ZrO, вызвало неосаждение 0,3% TiO2; остальное количество титана выделилось не в виде хлопьевидного осадка, как это бывает в отсутствие циркония, и его было очень трудно отфильтровать.
Барий [цирконий, редкоземельные металлы, общее содержание серы, хром] 969
в совершенно исключительных случаях, после дву- или трехкратного осаждения кальция и стронция в виде оксалатов, барий может оказаться вместе с ними. Обычно он переходит количественно в фильтрат вместе с магнием, откуда и может быть выделен осаждением серной кислотой после удаления аммонийных солей. Прибавив при этом немного спирта, можно одновременно выделить следы стронция, если анализируемая порода была им богата. Но нельзя быть уверенным, что отделенный таким образом от магния барий представляет все количество этого элемента, содержавшееся в породе. Найденные таким путем величины почти всегда оказывались ниже истинных, вероятно потому, что в ходе анализа создаются благоприятные условия для небольших потерь бария.
Общий метод
Разложение пробы проводят сплавлением 2 г порошка анализируемой породы со свободным от серы карбонатом натрия и с таким количеством селитры, которое не может повредить тиглю (0,5—1 г). Нагревают сначала на горелке Бунзена, а затем косо направленным пламенем паяльной горелки, поместив тигель в отверстие пластинки из платиновой жести или асбестового картона для защиты от содержащих серу газов пламени (см. рис. 6, стр. 49). Если в этой навеске серу не определяют, то применение селитры и защитной пластинки отпадает. После тщательной обработки плава водой, к которой для восстановления марганцевистой кислоты прибавляют несколько капель метилового или этилового спирта, раствор фильтруют и остаток промывают сильно разбавленным раствором карбоната натрия, не содержащего бикарбоната. Последнее необходимо, чтобы избежать получения мутных промывных вод. Желтая окраска фильтрата указывает на присутствие хрома.
После восстановления манганата спиртом фильтрат содержит кремнекислоту, алюминий и все другие кислотные компоненты, за исключением титана, а также очень небольшие количества железа, но их отделение от остальных компонентов не всегда бывает количественным, равно как и отделение кремнекислоты и алюминия. Какая часть общего содержания этих последних переходит в водную вытяжку, сказать нельзя, так как это в значительной мере зависит от относительного их содержания в исходной пробе и от количества и характера присутствующих в пробе оснований. Например, при анализе щелочного полевого шпата значительные количества кремнекислоты и алюминия переходят в раствор, а при анализе чистого каолина в раствор переходит лишь очень небольшая их часть. При анализе большинства пород как остаток, так и фильтрат содержат значительные количества и алюминия и кремнекислоты.
Дальнейшая обработка фильтрата описана в гл. «Сера» (стр. 1028) и «Хром» (стр. 978).
Не снимая фильтр с воронки, смывают остаток в небольшой стакан и нагревают его с несколько большим количеством разбавленной серной кислоты, чем зто необходимо для полного растворения всех растворимых частей (если определяется только барий, можно взять более концентрированную кислоту и в большем количестве). Если хотят определить цирконий, то нужно измерить количество кислоты, прибавленной в этой и последующей стадиях анализа, чтобы иметь возможность получить в .дальнейшем точную ее концентрацию, указанную ниже (стр. 972). Для раство
970 Гл. LIII. Методы анализа силикатных пород
рения коричневой гидроокиси марганца прибавляют несколько капель сернистой кислоты. Остается больший или меньший нерастворимый остаток. Надо остерегаться нагревать с кислотой слишком долго, чтобы избежать желатинирования растворенной кремнекислоты. Жидкость фильтруют через прежний фильтр и собирают фильтрат в коническую колбу емкостью 100—150 мл. Фильтр с остатком промывают и затем сжигают, золу обрабатывают фтористоводородной и серной кислотами, выпаривают раствор досуха и остаток нагревают с небольшим количеством горячей разбавленной серной кислоты. Остается нерастворенным весь барий, а также немного стронция, а иногда и довольно большое количество кальция. Остаток собирают на маленьком фильтре, а фильтрат прибавляют к первому фильтрату, который теперь будет содержать весь цирконий и редкоземельные металлы (о его дальнейшей обработке см. далее стр. 971 и 974).
Полученный последний остаток прокаливают, сплавляют с карбонатом натрия, выщелачивают плав водой, а нерастворимый в воде остаток растворяют на фильтре в нескольких каплях соляной кислоты. Из этого раствора осаждают барий серной кислотой. Достаточно однократного растворения сульфата бария в концентрированной серной кислоте и пере-осаждения разбавлением водой, чтобы удалить следы кальция, которые могут его загрязнять, если порода была богата кальцием. Даже стронций при такой обработке редко задерживается осадком в количествах, имеющих значение. Если бы, однако, это случилось, что бывает, когда порода содержит SrO и ВаО в количествах около 0,2% первого и 0,4% второго, то единственным удовлетворительным способом анализа является превращение сульфатов в хлориды с последующим применением к смеси хлоридов метода разделения хроматом аммония (см. «Щелочноземельные металлы», стр. 700).
Сульфаты бария и стронция можно перевести в форму, удобную для спектроскопического их исследования, восстанавливая осадок или его часть в платиновой петле непродолжительным нагреванием в светящемся пламени горелки Бунзена, смачивая затем соляной кислотой и подвергая обычному испытанию. Этой пробой не следует пренебрегать.
Метод определения бария в силикатных горных породах и отделения его от кальция и стронция, описанный выше, дает наиболее удовлетворительные результаты при наименьшей затрате времени. Наша долголетняя практика показала1, что если даже не предпринимать отделения следов стронция, загрязняющих осадок бария и обратно, ошибка обычно не имеет большого значения. Это обусловлено тем, что относительная ошибка даже в 25% от содержания вещества, составляющего только 0,1—0,2% породы, менее важна в сравнении с тем, что присутствие данного компонента вообще констатируется, хотя бы с приблизительной точностью.
При определении малых количеств бария, обычно находящихся в горных породах, сомнительно, можно ли применить следущий метод2 отделения бария от кальция и магния. При этом методе пользуются растворяющим действием концентрированной соляной кислоты (содержащей 10% эфира) на хлориды кальция и магния, хотя при определении больших количеств бария этот метод, по-видимому, дает хорошие результаты
1 Подробности см. в статьях: W. F. Hillebrand, J. Am. Chem. Soc., 16, <83 (1894); Chem. News, 69, 147 (1894).
2 Mar, Am. J. Sci., 3d ser., 43, 521 (1892)..
Цирконий
971
и осуществление его не представляет затруднений. К тому же указанный метод не приводит к полному отделению примеси стронция, и поэтому он не имеет никаких преимуществ перед методом, описанным выше.
Если серу и цирконий определять не нужно, то простейший способ состоит в следующем. Пробу разлагают фтористоводородной и серной кислотами (стр. 966) и полученный остаток сульфата бария очищают, как описано выше.
ЦИРКОНИЙ
Цирконий является одним из наиболее постоянных компонентов горных пород. Обычно он содержится в породах в виде минерала циркона; в этом случае его содержание может быть приблизительно определено под микроскопом и химическое его определение становится почти излишним; но иногда цирконий встречается в виде других минералов, и тогда его присутствие не может быть установлено микроскопическим исследованием. В совершенно исключительных случаях, цирконий может содержаться в породе в количестве нескольких процентов, изредка его содержание достигает 0,2%, но обычно оно значительно ниже 0,1%.
Метод Гиллебранда
Предварительные замечания. Во всех случаях, когда необходимо сделать пробу на цирконий, можно рекомендовать для его открытия и определения следующий способ, основанный на использовании метода которым при тщательной работе можно открыть малейшие следы циркония, например 0,02% в 1 г пробы. Реактивом является растворимый ортофосфат.
Предварительная обработка измельченной горной породы полностью описана в разделе «Барий» (стр. 968), где указано, как проводится отделение циркония от бария и получение его в малом объеме раствора, содержащего небольшое количество свободной серной кислоты.
Прежде утверждали, что осаждение циркония следует проводить в растворе, содержащем очень малое количество серной кислоты, по более 1 %. Однако было показано 1 2, что полное осаждение происходит и в присутствии 3% кислоты, а позже было обнаружено 3, что цирконий осаждается полностью даже при содержании серной кислоты, равном 20% (по массе). Такая высокая кислотность имеет то преимущество, что устраняет загрязнение осадка титаном, железом и хромом. Полное отделение циркония от алюминия достигается в растворе, содержащем 10% серной кислоты3.
В Бюро Стандартов США было найдено, что состав осадка зависит от количества прибавленного осадителя и от способа промывания осадка. Если осадителя прибавлено не менее чем в 10-кратном количестве по сравнению с теоретически требуемым, то состав осадка таков, что после промывания раствором нитрата аммония он дает при прокаливании 7гРгО7, содержащий 46,5% 7г0г. Если же осадитель прибавлен в меньшем количестве, состав осадка колеблется, хотя полное осаждение цирко
1 G. Н. В a i 1 е у, J. Chem. Soc., 59, 149, 481 (1886).
2 G. S t е i g е г, J. Washington, Acad. Sci., 8, 637 (1918).
8 P. Nicolardot, A. Reglade, C. r., 168, 348 (1919).
972
Гл. LIII. Методы анализа силикатных пород
ния происходит уже при двойном по сравнению с теоретически требуемым количеством осадителя. Было установлено также, что при промывании осадка чистой водой происходит извлечение фосфора из осадка. Указанные выше авторы осаждали цирконий добавлением 1 г фосфата аммония, который они предпочитают фосфату натрия, хотя последний также полностью осаждает цирконий.
Ход анализа. К раствору, находящемуся в маленькой колбе или стакане и содержащему 10% (по объему) серной кислоты, прибавляют перекись водорода для окисления титана и затем по крайней мере 10-крат-ное по сравнению с теоретически требуемым количество оротофосфата в водном растворе. Предпочтительнее применять аммонийную соль. Обычно прибавляют 1 г осадителя. Раствор затем нагревают при 40—50 °C в течение 2 ч. Если спустя некоторое время окраска раствора побледнеет, то прибавляют еще перекиси водорода.
В таких условиях выпадает фосфат циркония и собирается в виде хлопьевидного осадка. Если можно принять, что он свободен от титана и железа, то его отфильтровывают, тщательно промывают водой, содержащей 5% нитрата аммония, прокаливают и взвешивают в виде ZrP2O7 (46,5% ZrO2). При определении таких малых количеств циркония, какие обычно содержатся в горных породах, фосфат циркония можно прокаливать без особых предосторожностей, но когда количество циркония велико, например при анализе циркониевых руд, во время прокаливания происходит заметное растрескивание осадка, что заставляет вести прокаливание первое время в плотно закрытом тигле. Даже и тогда возможны небольшие потери. (Обработка фильтрата описана ниже, см. «Редкоземельные металлы», стр. 974).
Если имеется сомнение в чистоте осадка, то, отфильтровав его, прокаливают, сплавляют осаток с карбонатом натрия и выщелачивают плав водой. Не растворившийся в воде остаток собирают на фильтре, промывают, прокаливают и сплавляют с пиросульфатом калия или пиросульфатом натрия. Плав обрабатывают разбавленной (1 : 9) серной кислотой, прибавляют немного перекиси водорода и повторяют осаждение фосфатом. Объем раствора перед осаждением берут в зависимости от количества циркония. В большинстве случаев при анализе пород достаточно 20 мл. После такой повторной обработки титан почти всегда отсутствует и вскоре выделяется бесцветный хлопьевидный осадок циркония, который отфильтровывают, прокаливают и взвешивают в виде пирофосфата.
В случае сомнения в нормальном составе прокаленного остатка, особенно когда последнего много, или если желательно качественно идентифицировать цирконий,применяют следующий способ. Прокаленный остаток ZrP2O7 сплавляют с карбонатом натрия, выщелачивают плав водой, нерастворимый остаток прокаливают, сплавляют с пиросульфатом, снова осаждают, но не фосфатом, а аммиаком, прокаливают и взвешивают в виде ZrOa. Для доказательства присутствия циркония в прокаленном остатке переводят его в раствор, осаждают цирконий аммиаком, растворяют осадок в соляной кислоте, выпаривают до объема в 1—2 капли и испытывают куркумовой бумагой или проводят микрохимическую реакцию на цирконий. При определении очень малых количеств циркония проба с куркумовой бумагой может не дать никакой окраски, хотя окраска легко появляется от 1 мг ZrCh, а при очень тщательном проведении пробы — даже от 0,3 мг. Кроме гафния, ниобия, тория и тантала, никакие
Цирконий
973
другие элементы не могут содержаться в виде примесей в полученном таким образом осадке.
Описанный выше метод приложим для выделения циркония, если он имеется, из смеси его с титаном, полученной методом осаждения купфероном (стр. 967).
В первоначальном методе1 осаждение проводилось не прибавлением фосфата, а одной только перекисью водорода; осадок представлял собой гидрат перекиси циркония 2 Zr2O6 или ZrO3. Наши попытки (а также и других авторов) получить осадок в кислых растворах сульфата циркония при помощи одной только перекиси водорода потерпели неудачу; осаждение происходит лишь из очень концентрированных растворов и то только с 30 %-ной перекисью водорода. Если бы можно было выделить цирконий свободным от фосфат-ионов, это было бы, конечно, большим улучшением описанного метода.
Другие методы
Имеются указания3 4 * 6 на то, что полное отделение титана от железа и циркония может быть достигнуто кипячением нейтрализованного раствора сульфатов этих металлов с уксусной кислотой, взятой в большом избытке (50%). Способ этот рекомендуют время от времени, но не приводят никаких данных, доказывающих его пригодность. Единственное разделение, проведенное авторами этого метода, было далеко не совершенным.
Полное отделение циркония от алюминия, но не от железа было достигнуто 1 осаждением его в виде оксииодата из нейтрального кипящего раствора хлоридов. Однако способ этот не был дальше изучен.
Был предложен8 метод отделения циркония от железа и алюминия, подобный методу отделения титана от этих элементов, предложенному тем же автором. Метод основан на способности циркония осаждаться из нейтрализованного раствора хлоридов при двухминутном кипячении в присутствии сернистой кислоты. По-видимому, это — очень хороший метод. Так как титан постоянно присутствует вместе с цирконием и также полностью осаждается, то в дальнейшем эти два элемента следует отделить друг от друга добавлением перекиси водорода и растворимого фосфата. Еще не выяснено, пригоден ли этот способ для определения таких малых количеств циркония, какие обычно встречаются в анализе горных пород, но для определения больших количеств этот метод был успешно использован® в измененном виде.
В той же работе описан метод прямого отделения циркония от титана осаждением салицилатом аммония.
Купфероновый метод, описанный в разделе «Титан» (стр. 967), можно применять и для осаждения циркония, одного или вместе с титаном.
1 G. Н. В a i 1 е у, J. Chem. Soc., 49, 149, 481 (1886).
2 G. Н. Bailey, Chem. News, 60, 6 (1889).
3 G. S itei t, В. Franz, J. prakt. Chem. 108, 65 (1869).
4 I. T. Davis, Am. Chem. J., 11, 27 (1889).
°C. Baskerville, J. Am. Chem. Soc., 16, 475 (1894); Chem. News, 70, 57 (1894),
6 M. Dittrich, S. Freund, Z. anorg. Chem., 56, 344 (1907).
974
Гл. LIII. Методы анализа силикатных пород
РЕДКОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ
При необходимости исследовать породу на присутствие в ней редкоземельных металлов рекомендуется следующий ниже ход анализа, которым без особых затруднений можно получить удовлетворительные результаты1.
Соединения железа (III), алюминия, хрома и урана препятствуют осаждению церия и, вероятно, других редкоземельных металлов в виде их оксалатов, если не прибавить очень большого избытка осадителя. Но если прибавить такой избыток, то осаждение будет полным. Соли указанных металлов растворяют оксалат церия, но избыток щавелевой кислоты или оксалата аммония осаждает его снова. В приводимом здесь методе анализа это мешающее влияние железа (III), алюминия, хрома и урана не может проявиться.
Первый метод
Фильтрат от фосфата циркония (стр. 971) или, если цирконий не определялся, полученный ранее раствор (стр. 969) обрабатывают избытком едкого кали для осаждения гидроокисей редкоземельных металлов, железа и титана; кремнекислота и алюминий при этом остаются в растворе. После отстаивания прозрачную жидкость сливают через фильтр, а осадок промывают 1—2 раза декантацией, переносят его на фильтр и промывают немного на фильтре. Осадок смывают с фильтра в маленькую платиновую чашку, обрабатывают плавиковой кислотой и выпаривают жидкость почти досуха. Затем приливают немного воды с несколькими каплями фтористоводородной кислоты, собирают оставшиеся нерастворенными фториды редкоземельных металлов (нечистые) на маленьком фильтре, вставленном в дырчатый платиновый или резиновый конус, и промывают водой, слабо подкисленной фтористоводородной кислотой. После этого смывают осадок в маленькую платиновую чашку, фильтр сжигают, золу переносят в ту же чашку и выпаривают с серной кислотой досуха. Образовавшиеся сульфаты растворяют в разбавленной соляной кислоте, осаждают гидроокиси редкоземельных металлов аммиаком, прибавляя его в небольшом избытке (см. стр. 618), снова растворяют в соляной кислоте, раствор выпаривают досуха и нагревают остаток с несколькими каплями раствора щавелевой кислоты. Все, что могло присутствовать в качестве примесей, при этом легко растворяется, в то время как оксалаты редкоземельных металлов остаются нерастворенными. Если есть сомнение в полноте их отделения от щелочноземельных металлов, то прокаленный осадок оксалатов можно растворить в соляной или азотной кислоте, снова осадить аммиаком, растворить в какой-нибудь кислоте, выпарить раствор досуха, снова осадить оксалаты, прокалить осадок и, наконец, взвесить образовавшиеся окислы.
Количества редкоземельных металлов, содержащиеся обычно в горных породах, настолько невелики, что дальнейшее их разделение на отдельные группы невозможно. Однако прпсутствие церия легко узнается при нагревании прокаленных окислов с концентрированной серной кислотой по появлению интенсивного желтого окрашивания.
iM. Dittrich, Вег., 41, 4373 (1908).
фосфор
975
Второй метод
Измельченную породу полностью разлагают многократным выпариванием с фтористоводородной кислотой. Остаток фторосиликатов и фторидов всех редкоземельных металлов, кроме циркония, собирают на платиновом конусе, промывают водой, слабо подкисленной плавиковой кислотой, и снова смывают в чашку или в тигель. Затем выпаривают для удаления всего фтора с достаточным количеством серной кислоты, сжигают отдельно фильтр и присоединяют полученную золу. Осторожным нагреванием удаляют избыток серной кислоты и растворяют сульфаты в разбавленной соляной кислоте. Потом осаждают аммиаком редкоземельные металлы, может быть вместе с небольшим количеством алюминия, осадок промывают и снова растворяют в соляной кислоте. Раствор выпаривают досуха, остаток растворяют в воде с добавлением одной капли соляной кислоты, прибавляют, чтобы нейтрализовать последнюю, немного ацетата аммония и затем осаждают редкоземельные металлы щавелевой кислотой (не оксалатом аммония, потому что тогда не осадится торий). Этим способом можно определить 0,03% редкоземельных металлов в 2 г анализируемой пробы.
ФОСФОР
Предварительные замечания
Иногда весь фосфор можно извлечь из породы простым нагреванием ее с азотной кислотой. Однако часто, если даже не в большинстве случаев, это не удается и приходится прибегать к одному из описанных ниже более длительных методов. Каким бы методом ни пользовались, нужно работать очень тщательно, если хотят получить точные результаты.
Только при необходимости крайней экономии анализируемого материала фосфор определяют в смеси окислов, полученной, как описано на стр. 949; обычно же его рекомендуется определять в отдельной навеске породы. Кроме того, фосфор никогда не следует определять в растворе, полученном в результате растворения плава анализируемой породы (или окислов) с бисульфатом или пиросульфатом или после выпаривания с серной кислотой, потому что при этих операциях происходит потеря Р2О5 (см. стр. 955) отчасти вследствие улетучивания в виде метафосфорной кислоты, отчасти вследствие превращения в метафосфорную кислоту без улетучивания. В последнем случае соответствующей обработкой не удается вполне превратить метафосфорную кислоту в ортофосфорную, между тем как фосфор осаждается магнезиальной смесью только из последней.
Метод, применяемый при большом количестве материала для анализа
Предварительная обработка. Первый метод. В платиновой чашке или вместительном тигле разлагают около 1 г порошка породы смесью разбавленных азотной и фтористоводородной кислот. Как только исчезнут все песчаные крупинки, выпаривают досуха и затем снова выпаривают 2 или 3 раза с одной азотной кислотой, чтобы разложить большую
976
Гл. LIII. Методы, анализа силикатных пород
часть фторидов и фторосиликатов Е Остаток обрабатывают на бане разбавленной азотной кислотой и, если нужно, отфильтровывают нерастворимый остаток. Из предосторожности последний всегда следует прокалить, сплавить с небольшим количеством карбоната натрия, выщелочить плав водой, подкислить раствор азотной кислотой и присоединить к главному раствору. Дальнейшая обработка осадка описана ниже.
Второй метод. Сплавляют породу с карбонатом натрия, выделяют кремнекислоту однократным выпариванием с азотной кислотой, фильтруют, прокаливают, обрабатывают кремнекислоту фтористоводородной и азотной кислотами, выпаривают до удаления фтористоводородной кислоты и повторяют выпаривание с одной азотной кислотой 2 или 3 раза. Небольшой остаток переводят в раствор кипячением с азотной кислотой и присоединяют полученный раствор к главному раствору.
Дальнейшая обработка. К раствору, полученному после предварительной обработки пробы, прибавляют 5 г нитрата аммония, 1—2 г борной кислоты и затем раствор молибдата аммония, как описано в гл. «Фосфор» (стр. 781). Оставляют стоять при температуре не выше 40 °C в течение 12 ч или пока не будет уверенности в полноте осаждения.
Фильтруют через бумажный фильтр и промывают осадок водой, содержащей 5% нитрата аммония и 1% азотной кислоты, после чего смывают его струей воды в маленький стакан, который затем ставят под воронку с фильтром. Промывают фильтр разбавленным раствором аммиака и 2 или 3 раза чистой водой. Если пропущенного через фильтр аммиака недостаточно, чтобы растворить весь желтый осадок в стакане, прибавляют еще аммиака, и если прозрачный раствор все еще не получается, пропускают жидкость через тот же фильтр, чтобы собрать нерастворимое в аммиаке вещество.
Муть, часто наблюдаемая при растворении в аммиаке осажденного и промытого фосфоромолибдата, происходит от присутствия какого-то соединения фосфора. Если прибавления небольшого кристалла лимонной пли винной кислоты недостаточно для растворения мути, то ее нужно отфильтровать и сплавить с карбонатом натрия, выщелачить плав водой и фильтрат обработать, как описано выше, чтобы выделить содержащийся в нем фосфор.
Аммиачный раствор затем обрабатывают магнезиальной смесью и продолжают дальше, кончая взвешиванием фосфора в виде Mg2P2O7, как описано в гл. «Фосфор» (стр. 785). Повторного осаждения фосфата магния не требуется, если осадок невелик, как это обычно бывает при анализе горных пород.
Если присутствует ванадий, то он не только осаждается молибденовой жидкостью вместе с фосфором, но и замедляет осаждение фосфора и может даже сделать последнее неполным. Кроме того, смешанный осадок относится иначе к промывным растворам, чем чистый фосфоромолиб-дат. Присутствие ванадия обнаруживается по оранжевому цвету фосфоромолибдата. Однако ванадия в горных породах содержится обычно так
1 Метод первоначально описан Вашингтоном (см. Washington, Manual of the Chemical Analysis of Rocks). Позднее этот метод был изменен Вашингтоном и Виллебрандом (независимо друг от друга) в том отношении, что отделение осадка фторосиликатов и фторидов фильтрованием перед дальнейшим выпариванием было выпущено.
Фосфор
977
мало, что его влиянием можно пренебречь. Если ванадий имеется в большом количестве, то осаждение должно быть сделано на холоду после восстановления ванадия, как описано в гл. «Фосфор» (стр. 785).
Метод, применяемый при малом количестве материала для анализа
Следующий метод позволяет в одной и той же навеске определить не только фосфор, но и барий, железо, ванадий, хром и титан (последние два либо колориметрическим, либо весовым способами). Метод этот отчасти взят из работы Чатарда1.
Удаляют сначала кремнекислоту обработкой фтористоводородной и серной кислотами, а затем отгоняют избыток фтора 2 (стр. 939). Остаток, насколько возможно, переводят в раствор при помощи горячей разбавленной (1 : 10) серной кислоты, фильтруют, прокаливают остаток, сплавляют его с карбонатом натрия, растворяют плав в соляной кислоте, восстанавливают хроматы, если цвет раствора указывает на их присутствие, и после осаждения и отделения бария присоединяют этот раствор к гавному раствору. Затем проводят осаждение аммиаком, чтобы избавиться от солей магния, которые обычно присутствуют; благодаря этому последующее сплавление с карбонатом натрия проходит более гладко.
Осажденные А12О3, Р2О5, V2O5, Cr2O3, Fe2O3, ZrO2 и TiO2 растворяют в горячей соляной кислоте и раствор фильтруют, собирая фильтрат в большой платиновый тигель; фильтр сжигают изолу присоединяют к раствору; раствор выпаривают до пастообразного состояния, растворяют соли добавлением небольшого количества воды и всыпают малыми порциями сухой карбонат натрия. Тщательно перемешивают, чтобы предупредить образование комков при последующем сплавленпп, которое продолжается полчаса. Прибавление нитрата натрия излишне.
Полученный плав обрабатывают водой при кипячении, фильтруют и промывают нерастворимый в воде остаток 1 %-ным раствором карбоната натрия. В остатке можно определить железо, титан и цирконий ранее описанными методами3. В фильтрате можно определить колориметрически хром, если он присутствует в количестве достаточном, чтобы придать раствору заметную окраску (см. ниже, стр. 978). После этого, или тотчас же, если не проводили колориметрического определения хрома, прибавляют достаточное количество нитрата аммония, чтобы весь карбонат мог вступить с ним в реакцию, и нагревают на водяной бане, пока не будет удалена большая часть карбоната аммония. При этом осаждается весь или почти весь алюминий вместе с фосфором и частью присутствующего ванадия. Осадок промывают разбавленным раствором нитрата аммония, пока желтый цвет хромата не исчезнет совершенно из промывных вод, после чего растворяют осадок в азотной кислоте и осаждают фосфор раствором молибдата аммония. Фильтрат от алюминия и фосфора, содержащий хром в виде хромата и большее или меньшее количество ванадия, может быть обработан, как описано ниже.
1 Т. М. С h a t а г d, Am. Chem. J., 13, 106 (1891); U.S. Geol. Survey Bull., 78, 87 1891); Chem. News, 63, 267 (1891).
2 Удаление фтора надо проводить без усиленного выпаривания серной кислоты, чтобы избежать потери Р2О5 (см. стр. 955).
3 См. стр. 957 об извлечении того малого количества железа, которое может перейти в щелочной фильтрат.
62 Заказ 522.
978
Гл. LIII. Методы анализа силикатных пород
ХРОМ
При полном или почти полном отсутствии ванадия, как это бывает в тех богатых магнием горных породах (перидотитах) которые содержат большие количества хрома, приведенный ниже метод выделения и весового определения хрома дает хорошие и согласующиеся результаты; но в присутствии ванадия (а лучше всего предполагать его присутствие) нужно отдавать предпочтение колориметрическому методу.
Весовые методы
Метод Гиллебранда. С полученным, как указано выше (см. «Фосфор») раствором хромата, свободного от всех других элементов, кроме небольшого количества алюминия, поступают следующим образом.
Раствор, если нужно, концентрируют и прибавляют свежеприготовленный раствор сульфида аммония или пропускают в раствор сероводород. Хром восстанавливается и появляется в виде осадка гидроокиси хрома вместе с гидроокисью алюминия. Этот осадок обрабатывают следующим образом1. Растворяют в азотной кислоте, выпаривают раствор почти досуха, нагревают с концентрированной азотной кислотой и хлоратом калия и, наконец, выпаривают досуха, чтобы удалить кислоту. Этим очень быстро достигают полного окисления хрома. По разбавлении холодной водой прибавляют в небольшом избытке бикарбонат натрия п через 2 или 3 ч отфильтровывают выделившуюся гидроокись алюминия. Из фильтрата затем осаждают хром свежеприготовленным раствором сульфида аммония, осадок снова растворяют и переосаждают, чтобы освободить его от щелочных металлов, и взвешивают. Полученный результат немного выше истинного вследствие частичного окисления окиси хрома во время прокаливания (стр. 596), но это не имеет значения, потому что количества хрома, о которых идет речь, очень невелики.
Метод Якоба. Якоб 2 рекомендует следующий метод осаждения алюминия [а также железа (III), титана, циркония и марганца] и отделения его от хрома. К солянокислому раствору, содержащему амомипий в количестве, эквивалентном 0,1—0,2 г АПОз, и занимающему объем от 20 до 100 мл в конической колбе емкостью 100—300 мл, прибавляют по каплям свежеприготовленный раствор едкого натра (примерно 5%-ный) до тех пор, пока образовавшаяся гидроокись алюминия не растворится снова; избытка щелочи избегают. Затем раствор кипятят, прибавляя воду, насыщенную бромом. Сначала бромную воду добавляют по каплям, чтобы не слишком понизить температуру раствора и не получить вследствие этого слизистого осадка. К концу операции бромную воду можно приливать быстрее, пока раствор не станет красным. Избыток брома затем удаляют кипячением, жидкость фильтруют и осадок промывают горячей водой. Осадок, приставший к стенкам колбы, растворяют в капле азотной кислоты и снова осаждают аммиаком. Некоторое количество хрома осаждается вместе с алюминием, но его можно удалить после декантирования жидкости кипячением в ченпе нескольких минут с водой, содер
1 Н. Baubigny, Bull. Soc. chim. (New Ser.), 42, 291 (1884); Chem. News, 50 18 (1885).
2 W. J a k о b, Z. anal. Chem., 52, 651 (1913).
Хром
979
жащей нитрат аммония и аммиак. Возможно, что небольшое количество алюминия может при такой обработке вновь перейти в раствор. Соединенные осадки прокаливают и взвешивают.
Хром может быть определен в фильтрате описанным выше методом (стр. 978).
Магний и цинк, если они присутствуют, частично осаждаются вместе с алюминием. Якоб утверждает, что небольшие количества этих элементов могут быть удалены обработкой нитратом аммония и аммиаком. В присутствии сульфатов или боратов этот метод не дает удовлетворительных результатов.
Колориметрический метод
При применении этого очень точного и наиболее быстрого метода определения хрома в горных породах и тех рудах, где количество его не превышает нескольких процентов \ нужно сплавить породу с карбонатом натрия, выщелочить плав водой (например, методом описанным на стр. 977) и окраску полученного раствора сравнить с окраской стандартного раствора.
Приготовление стандартного раствора. Стандартный раствор приготовляют растворением 0,1935 г или 0,3870 г чистого бихромата калия в 1л воды, которую немного подщелачивают карбонатом натрия; 1 мл этого раствора соответствует 0,1 или 0,2 мг окиси хрома (Сг2О3). Более концентрированными растворами пользоваться не рекомендуется.
Приготовление исследуемого раствора. Плав, полученный сплавлением пробы с карбонатом натрия, выщелачивают водой и до фильтрования приливают несколько капель этилового или метилового спирта для разрушения окраски манганата натрия. Если желтая окраска фильтрата очень слаба, ее можно усилить концентрированием раствора и тогда можно точно определить около 2 мг окиси хрома в 1 г пробы. Если содержание хрома еще меньше, следует взять навеску пробу в 3—5 г и концентрировать хром путем осаждения его нитратом ртути (I), как описано в разделе «Ванадий» (стр. 982). Иначе из-за наличия больших количеств карбоната может оказаться трудным или совсем невозможным получение такого малого объема фильтрата, чтобы желтая окраска его была ясно видна.
Если при сплавлении применяли селитру и при этом тигель несколько подвергся разъеданию, то желтая окраска фильтрата может происходить от присутствия в растворе платины; ввиду этого следует регулировать как количество прибавляемой селитры, так и температуру пламени паяльной горелки, чтобы избежать разъедания тигля плавом. Скоро исчезающая желтоватая окраска фильтрата, которая может происходить от этой или иных причин 1 2, не должна быть приписана хрому.
При применении растворов, полученных, как указано выше (см. «Метод Гиллебранда» или «Метод Якоба», стр. 978), нет необходимости в добавлении спирта, так как марганец в них отсутствует.
1 W. F. Hillebrand, J. Am. Chem. Soc., 20, 454 (1898); Chem. News, 78, 227, 239 (1898); U. S. Geol. Survey Bull., 167, 37 (1900). Этот метод был впервые применен L. de К о n i n g h [Nederland Tijdschr. Pharm. Chem. Tox. (1889)] для определения хрома в пищевых продуктах.
О применении дифенилкарбазидного метода для определения хрома в горных породах см. сноски 1 и 2 на стр. 596.
2 См. R.S. Me Bride, J. A. Scherrer, J. Am. Chem. Soc., 39, 928 (1917).
62*
980
Гл. LIII. Методы анализа силикатных пород
Сравнение окрасок. Готовый раствор переносят в мерную колбу и доливают до метки водой. Колбу выбирают такой емкости, чтобы анализируемый раствор в ней был слабее окрашен, чем стандартный раствор. Затем отмеренное количество последнего разбавляют водой, приливаемой из бюретки, пока окраски обеих жидкостей не сравняются.
При определении очень малых количеств хрома могут оказаться более удобными пробирки Несслера, которые применяют при определении аммиака.
Описанный способ, как и вообще колориметрические методы, дает при определении малых количеств хрома более точные результаты, чем при определении больших количеств, но и в последнем случае можно достичь удовлетворительных результатов, если проделать большое число сравнений с одним раствором х.
Сравнительные данные
В табл. 36 сопоставлено небольшое число результатов определения хрома, полученных Гиллебрандом колориметрическим и весовым методами.
Определение (%) содержания хрома (Сг2О3)
ТАБЛИЦА 36
Весовым методом Колориметрическим методом Весовым методом Колориметрическим методом
Следы 0,018 Следы 0,013
0,05 0,051 Нет 0,0080
0,14 0,12 Нет 0,0067
0,08 0,083
Колориметрические определения сделаны спустя несколько месяцев, а иногда даже и через несколько лет после весовых.
Результаты оказались несколько неожиданными, так как едва ли можно было ожидать, что продолжительные и трудные количественные отделения моглц быть сделаны настолько удачно. Следует отметить, что весовые определения проводились из навесок в 1 а или, самое большее — в 2 г вещества. Это объясняет, почему в двух случаях весового определения хром не был обнаружен.
ВАНАДИЙ (ХРОМ) И МОЛИБДЕН
Распространение ванадия и молибдена
Широкое распространение ванадия в земной коре было установлено (см. «Ванадий», стр. 507) не только в рудах и углях, но и в глинах, известняках, песчаниках и изверженных породах 1 2. Одним из нас (Гиллебранд) было показано, что ванадий встречается в заметных количествах в извер
1 D. W. Horn [Am. Chem. J., 35, 253 (1906); 36, 196 (1906)] исследовал чувствительность колориметрических методов определения хрома.
2 W. F. Hillebrand, Am. J. Sci., [4], 6, 209 (1898); Chem. News, 78, 216 (1898); U. S. Geol. Survey Bull., 167, 49 (1900).
Ванадий [хром] и молибден
981
женных п метаморфических породах более основного характера (до 0,08% и более V2O3), но что он, по-видимому, отсутствует или почти отсутствует в породах с высоким содержанием кремнекислоты.
Некоторые железо-алюминиево-силикатные минералы, образующие первые породы, обнаруживают еще более высокое содержание ванадия; например, в биотите, выделенном из пироксенового гнейса, было найдено 0,13% V2O3. С другой стороны, распространение молибдена в количествах, доступных их открытию, ограничено, по-видимому, более кремнекислыми породами и, за исключением, быть может, очень редких случаев, в них не удается количественно определить молибден при навеске в 5 г исходного материала. Отсюда следует, что количественное определение ванадия надо обычно проводить только в породах, содержащих менее 60% кремнекислоты. Даже и при соблюдении этого условия поиски ванадия не всегда оправдывают затраченное на них время, но нужно помнить, что если его определение опустить, то в результаты определения железа (III) и (II) будут введены ошибки, которые в некоторых случаях могут достичь заметной величины (см. стр. 957 и стр. 999).
Валентность ванадия в породах
Указанная выше и в других местах связь ванадия с железисто-алюми-ниевыми силикатами наравне с существованием минерала роскоелита K2H8(Mg, Fe)(Al, У)4(8Юз)12, который классифицируют как ванадиевую слюду, приводит к предположению, что ванадий встречается в породах в той же степени окисления, как железо (III) и алюминий, и способен замещать каждый из этих элементов или оба вместе. Поэтому результаты определения ванадия следовало бы представлять в виде V2O3, а не V2O5.
Остается открытым вопрос, в какой степени окисления ванадий содержится в породах вторичного происхождения, как, например, в глинах, известняках, песчаниках, углях и железных рудах. Некоторое время Гиллебранд полагал, что ванадий находится в этих породах в пятивалентном состоянии. Однако исследование замечательных песчаников западного Колорадо \ содержащих ванадий, в которых он, несомненно, присутствует в виде трехвалептного (V2O3), поколебало это мнение.
Следует отметить, что уже Чуднович1 2, учитывая чрезвычайную трудность выделения ванадия из железных руд сплавлением с карбонатами, а также легую восстанавливаемость ванадиевой кислоты солями железа (II) в условиях, при которых, вероятно, образовались в природе бурые железняки, — считал, что ванадий в таких рудах должен присутствовать в низшей степени окисления (V2O3). Противоположное мнение Линдемана 3 в отношении некоторых железных руд, основанное на том, что при сплавлении их с карбонатом натрия без прибавления селитры ванадий извлекается в виде V2O5, не убедительно, так как это присходило бы, вероятно, также и в том случае, если бы ванадий находился первоначально в руде в виде V2O3.
1 W. F. Hillebrand. F. L. Ransome, Am. J. Sci., [4], 10, 120 (1900); U. S. Geol. Survey Bull., 262, 9 (1905).
2 C. Czudn owicz. Pogg. Ann., 120, 20 (1863).
3 O. Lindemann, диссертация, Иепа, 1878, цит. no Z. anal. Chem., 18, 99 (1879).
982
Гл. LIII. Методы анализа силикатных пород
Метод Гиллебранда
Описание метода. Предлагаемый метод не содержит ничего совершенно нового, кроме того, что .в этом методе при одновременном присутствии хрома и ванадия нет необходимости отделять их друг от друга, а можно определять их в одном и том же растворе, первый — колориметрическим методом, описанным выше (стр. 979), а второй — объемным методом *.
Сплавляют на пламени паяльной горелки 5 г анализируемой породы с 20 г карбоната натрия и 3 г нитрата натрия. После выщелачивания плава водой и восстановления марганца спиртом, по-видимому, нет необходимости снова сплавлять остаток, если первое сплавление было проведено тщательно, хотя было показано 1 2, что при анализе некоторых магнетитов и других руд, содержащих большие количества ванадия, это может быть необходимо. Водную вытяжку нейтрализуют почти до конца азотной кислотой и выпаривают почти досуха. Количество кислоты, которое необходимо прибавить для нейтрализации, удобно устанавливать проведением «холостого» опыта с точно отвешенными 20 г карбоната натрия. При подкислении раствора нужно остерегаться переходить точку нейтрализации вследствие восстанавливающего действия азотистой кислоты, выделяющейся в кислой среде из нитрата, образующегося при сплавлении. Хром и иногда ванадий увлекаются в небольших количествах осевшими кремнекислотой и гидроокисью 'алюминия.
Применение нитрата аммония вместо азотной кислоты для превращения карбоната натрия в нитрат, по-видимому, не улучшает дела. Поэтому во всех случаях, когда, кроме ванадия, надо определить и хром, остаток кремнекислоты и гидроокиси алюминия следут обработать фтористоводородной и серной кислотами, выпарить досуха, остаток сплавить с небольшим количеством карбоната натрия, обработать плав водой, водную вытяжку снова почти нейтрализовать азотной кислотой, прокипятить в течение нескольких минут, профильтровать и полученный фильтрат присоединить к главному фильтрату.
Затем к этому холодному щелочному раствору прибавляют достаточное количество нитрата ртути (I), чтобы образовался значительный по величине осадок карбоната ртути (I), содержащий ванадий, молибден, вольфрам, фосфор и мышьяк, находившиеся в анализируемой породе. Карбонат ртути (I) служит также для связывания кислоты, которая могла бы образоваться вследствие разложения нитрата ртути (I). Таким образом, проводя осаждение в слабощелочном растворе вместо нейтрального, избегают прибавления осажденной окиси ртути для регулирования кислотности. Образование чрезмерно большого осадка указывает на то, что реакция раствора была слишком щелочной, и тогда можно уменьшить щелочность раствора, осторожно прибавляя азотную кислоту, пока прибавленная капля раствора нитрата ртути (I) не перестанет вызывать образования мути.
После нагревания и фильтрования осадок прокаливают в платиновом тигле при температуре не выше 500 °C, предварительно высушив его и
1 W. F. Н i 11 е b г a n d, J. Am. Chem. Soc., 20, 461 (1898); Chem. News, 78, 295 (1898); U. S. Geol. Survey Bull., 167, 44 (1900).
2 E doClassen, Am. Chem. J., 8, 437 (1886).
Ванадий [хром) и молибден
983
сняв с фильтра, чтобы предупредить возможную потерю молибдена и повреждение тигля вследствие восстановления мышьяка. Прокаленный остаток сплавляют с небольшим количеством карбоната натрия, выщелачивают плав водой и, если раствор окрашен в желтый цвет, фильтруют, собирая фильтрат в мерную колбу емкостью 25 мл или более. В этом растворе можно затем точно определить хром в течение нескольких минут сравнением со стандартным щелочным раствором бихромата калия (стр. 979). После этого или— в отсутствие хрома —тотчас же после фильтрования прибавляют к раствору в небольшом избытке серную кислоту и осаждают сероводородом 1 молибден и мышьяк вместе со следами платины.
Если цвет осадка указывает на отсутствие мышьяка, фильтр с содержимым осторожно сжигают во взвешенном фарфоровом тигле при температуре не выше 500 °C, взвешивают и проводят реакцию на молибден с серной кислотой; остаток в тигле смачивают одной каплей концентрированной серной кислоты и нагревают, пока серная кислота почти полностью не выпарится. Появление при охлаждении красивого синего окрашивания указывает на присутствие молибдена.
В присутствии мышьяка молибден можно определить колориметрическим методом после растворения смеси окислов, как указано в гл. «Молибден» (стр. 368). Мышьяк следует определить в отдельной навеске породы методом отгонки из солянокислого раствора, как описано в гл. «Мышьяк» (стр. 303).
Фильтрат объемом 25—100 мл кипятят для удаления сероводорода и титруют при 70—80 °C очень разбавленным раствором перманганата, 1 мл которого соответствует приблизительно 1 мг V2O5; нужное разбавление можно вычислить из титра перманганата, выраженного по железу, считая, что одна молекула V2O5 соответствует одной молекуле ЕегОз. Рекомендуется для верности проводить два-три контрольных определения, для чего в оттитрованном растворе снова восстанавливают ванадий током сернистого ангидрида, удаляют избыток последнего кипячением 2 и повторяют титрование. Последующие титрования обычно дают немного более низкие результаты, чем первое титрование, и их следует считать более правильными.
Высокие результаты первого титрования объясняются образованием политионовых соединений при восстановлении ванадия сероводородом 3, а также извлечением окисляемых веществ из фильтровальной бумаги. Первого избежать трудно; второе легко может быть устранено почти пол
1 Из сернокислого раствора выделение платины и молибдена сероводородом идет быстрее и лучше, чем из солянокислого.
2 Водные растворы сернистого ангидрида и сульфитов щелочных металлов можно применять только свежеприготовленными, так как по прошествии некоторого времени эти растворы содержат другие окисляющиеся вещества, помимо сернистой кислоты или сульфита. Сернистый ангидрид лучше всегда получать перед его применением, нагревая раствор сернистой кислоты или обрабатывая раствор сульфита серной кислотой. Удаление последних следов сернистого ангидрида можно ускорить, если к концу этой операции одновременно с кипячением раствора пропускать через него быстрый ток двуокиси ангидрида. При продолжительном пропускании углекислого газа небольшое количество примешанного к нему воздуха может вызвать незначительное окисление ванадия [О. М a n a s s е, Ann. Chem. und Pharm., 240, 23 (1887); Z. anal. Chem., 32, 225 (1893)].
3 G. 11. F. Lundell, H. B. Knowles,!. Am. Chem. Soc., 43, 1560 (1921).
984
Гл. LIII. Методы анализа силикатных пород
ностью предварительным пропусканием через фильтр 25 мл воды и полностью устранено предварительным промыванием фильтра большим количеством воды при применением тиглей Гуча с асбестовым пли платиновым фильтрующим слоем (стр. 127).
Качественные реакции для подтверждения присутствия ванадия. Если израсходованное на титрование количество раствора перманганата так мало, что возникает сомнение в действительном присутствии ванадия, то следует проделать качественную реакцию, чтобы удостовериться в присутствии последнего.
Лучше всего эту пробу проводить следующим образом. Раствор выпаривают и нагревают для удаления избытка серной кислоты. Остаток растворяют в 2—3 мл воды и нескольких каплях разбавленной азотной кислоты и затем прибавляют 1—2 капли перекиси водорода. Характерное коричневатое окрашивание указывает на присутствие ванадия. Если большая часть свободной серной кислоты не была удалена, то окрашивание иногда появляется не сразу или оказывается слабее; отсюда вытекает необходимость придерживаться описанного выше хода работы. Далее необходимо, чтобы азотная кислота присутствовала в значительном избытке; в нейтральном пли только слабокислом растворе окрашивание получается слабое.
Можно также проводить и следующую пробу на ванадий, хотя она несколько менее надежна, чем предыдущая. Раствор концентрируют приблизительно до 10 мл, прибавляют аммиак в избытке и насыщают сероводородом. Интенсивное вишнево-красное окрашивание ванадия в растворе сульфида аммония значительно отчетливее, чем окрашивание, вызываемое в кислых растворах перекисью водорода. Однако при действии аммиака часть ванадия или он весь осаждается с хромом или алюминием (которые могли присутствовать в растворе) или с введенным при титровании марганцем, и сульфид аммония не может полностью извлечь ванадий из этого осадка. Обычно, однако, некоторое окрашивание происходит, и при подкислении профильтрованного раствора осаждается бурый сульфид ванадия; его можно отфильтровать, прокалить и, если нужно, исследовать дальше.
Применение метода в присутствии сравнительно большого количества хрома. В описанной выше простейшей форме метод применим при одном условии: содержание хрома не должно превышать определенной умеренной величины. Это ограничение вытекает из того, что при титровании в горячем растворе, как это рекомендуется для определения ванадия (стр. 513), в присутствии хрома отчетливое изменение окраски раствора наступает лишь при прибавлении слишком большого количества перманганата. В холодном растворе, содержащем сульфат хрома, требуется значительно меньше перманганата для получения своеобразной темной окраски без следов зеленоватого оттенка; эта окраска ясно указывает на наличие избытка перманганата. При нагревании, особенно при температуре кипения, хром сам настолько быстро окисляется, что надо прибавить значительный избыток перманганата, чтобы получить отчетливое изменение окраски.
Несмотря на все это, можно довольно точно определить 1—2 мг-пятпокиси ванадия в присутствии 30 мг окиси хрома. Для этого только необходимо ввести простую поправку, величину которой определяют, вливая перманганат в раствор сульфата хрома, занимающий такой же-
Ванадий [zpo.ii] и молибден
985-
объем, какой имеет анализируемый раствор, и содержащий приблизительно такое же количество хрома. Этот раствор надо нагреть до той же температуры, до которой нагревали анализируемый раствор при титровании.
В табл. 37 и 38 приведены результаты большого числа опытов. Из табл. 38 видно, какая степень точности может быть достигнута в присутствии большого количества хрома при введении указанной выше поправки, а также видна величина этой поправки.
Л ТАБЛИЦА 37-
Определение ванадия в присутствии хрома
Окись хрома мг Пятиокись ванадия мг Пятиокись ванадия, найдено Л12 Ошибка мг
1 9,37 9,22 —0,15-.
1 0,94 1,04 +0,10
0,98 +0,04
1,5 5,25 5,49 +0,24
5,43 -'-0,19
2 5,62 5,50 —0,12
5,50 -0,12
3 4,68 4,78 +0,10
4,78 +0,10
4,83 +0,15
3 5,62 5,58 —0,04
5,58 -0,04
3,5 18,74 18,89 +0,15
18,97 +0,23.
6 5,60 6,10 +0,50
6 4,68 4,78 +0,10
6 5,62 5,58 -0,04
10 5,62 5,58 -0,04
10 23,52 23,81 +0,29
23,71 +0,19
10 46,85 46,98 +0,13
47,20 +0,35
25 23,52 23,65 +0,13
87,5 23,52 23,75 +0,23
23,71 +0,10
Несмотря на то, что величина поправки в большинстве опытов, результаты которых приведены в табл. 38, составляет сравнительно большую долю израсходованного и на титрование раствора перманганата, полученные результаты следует признать удовлетворительными, если принять во внимание малые количества присутствующего в растворе ванадия. Эти результаты показывают, что в умелых руках этот способ при некотором навыке дает падежные результаты.
Пользуясь методом, в котором осадок, содержащий метаванадат свинца, хромат свинца, арсенат свинца и т. п., нагревают с концентрирован-
986
Гл. LII1. Методы анализа силикатных пород
ТАБЛИЦА 38
Поправки при определении хрома
(Применение при больших количествах хрома поправки, полученной прибавлением перманганата калия к раствору сульфата хрома, соответствующему анализируемому раствору по объему и содержанию в нем хрома)
Окись хрома ,мг Пятиокись ванадия мг Найдено пятиокиси ванадия Ошибка мг Объем раствора мл
без поправки Л13 с поправкой Л13
20 0,94 1,59 0,99 +0,05 50-100
20 1,87 2,69 2,09 +0,22 50-100
2,39 1,79 -0,08
2,59 1,99 +0,12
.20 18,74 19,4 18,73 -0,01 50-100
19,3 18,63 -0,11
19,3 18,63 -0,11
30 1,87 2,99 2,14 +0,27 Около 100
2,79 1,94 +0,07
2,79 1,94 +0,07
2,69 1,84 —0,03
2,69 1,84 -0,03
30 1,87 2,69 1,79 -0,08 200
2,89 2,09 +0,22
2,89 2,09 +0,22
2,79 1,99 +0,12
62 46,85 48,90 47,60 +0,75 200
ним раствором карбоната калия, по-видимому, можно хорошо отделить мышьяк, фосфор, хром, вольфрам и молибден от ванадия.
Согласно литературным данным, при такой обработке метаванадат свинца остается совершенно нетронутым, а другие соли свинца полностью разлагаются. Однако применимость этого метода для отделения малых количеств ванадия, встречающихся обычно в породах и рудах, еще не изучена. В этом случае требуется получение удовлетворительных результатов при возможно меньшей затрате времени, и если хром присутствует в незначительном количестве, то эту задачу можно считать решенной.
Однако хром почти никогда не бывает преобладающей составной частью в глинах, железных рудах и других породах, в которых до сих пор находили ванадий х.
ЖЕЛЕЗО
Железо (II)
Правильное определение содержания железа (II) в минералах обычно сопряжено с большими затруднениями, которые начинаются с самого приготовления пробы к анализу, и их следует обсудить подробнее. Измельчение 1 2 на воздухе порошков минералов, содержащих железо (II), умень
1 В статье Е. В. Sandell [Ind. Eng. Chem., Anal. Ed., 8, 336 (1936)] описан колориметрический метод определения таких малых количеств, как 0,001 % ванадия и хрома и 0,0001 % молибдена, после обычного сплавления навески в 1 г силикатной породы с карбонатом натрия.
2 В. Mauzelius, Sveriges Geol. Undersokning, Arsbok 1, № 3 (1907); W. F. Hillebrand, J. Am. Chem. Soc., 30, 1120 (1908).
Железо
987
шает в них количество железа (II) и соответственно увеличивает содержание железа (III). Количество железа (II) может, таким образом, уменьшиться в течение 2 ч измельчения на 20—30% и даже больше. Были сделаны попытки предотвратить это химическое действие воздуха растиранием пробы под водой, спиртом или четыреххлористым углеродом. Они отчасти были успешны, однако не в такой мере, чтобы можно было предложить для общего применения измельчение пробы под одной из этих жидкостей.
Если не полностью, то все же в большей части можно избежать окисления, растирая порошок пробы в атмосфере индифферентного газа, например азота (но не двуокиси углерода, которая в этих условиях, по-видимому, образует карбонаты), однако в обычных условиях работы это практически невыполнимо. Кроме того, при этом не устраняется поглощение измельченным порошком влаги (гигроскопической и прочно связывающейся) при последующем выставлении его на воздух, что является результатом увеличения поверхности порошка. Из того факта, что окисление идет менее энергично при измельчении породы под одной из названных выше жидкостей (безразлично, оставляют ли потом порошок па воздухе или нет), можно вывести, что главной причиной окисления является не увеличение поверхностп, но, вероятно, местное разогревание, происходящее при раздавливании зерен пестиком. Это зависит, по-видимому, также от твердости входящих в состав породы железосодержащих минералов и сопутствующих им и, быть может еще больше зависит от химической природы самих железосодержащих минералов. В цитированной выше статье 1 приведены характерные данные.
Опыты, проведенные главным образом с силикатами, показали следующее: 1) хотя сравнительно быстрое измельчение на воздухе (15— 30 мин) и продолжительное измельчение под спиртом и не всегда приводит к заметному окислению порошка породы (а иногда, по-видимому, не происходит никакого окисления), однако эти нельзя считать правилом. Поэтому нельзя рекомендовать проводить измельчение в той или другой из этих сред во всех случаях; 2) спирт, несмотря на его большую растворяющую способность в отношении кислорода, по-видимому, несколько лучше защищает железо (II) от окисления, чем вода. Он имеет еще и то преимущество, что может быть быстро удален из вещества после измельчения; 3) из примененных органических веществ спирт оказался более действенным средством, чем четыреххлористый углерод; 4) довольно большие расхождения результатов параллельных анализов получаются в случае присутствия в породе трудно разлагаемых железосодержащих минералов (гранат и др.), если последние не измельчены до очень тонкого порошка; 5) совпадение результатов параллельных определений, проведенных как методом Пратта, так и методом Кука, получается превосходное при работе с тонкими порошками пробы, а при анализе крупных порошков — только в тех случаях, когда они легко поддаются разложению фтористоводородной кислотой; 6) так как измельченная в порошок порода, прошедшая через сито с 30 или даже 60 отверстиями на 1 линейный сантиметр, часто содержит меньше 0,1% влаги, то если в этой породе нет веществ, чувствительных к влаге, можно заключить, что при ее измельчении имело место такое же малое окисление железа (II), как и поглощение влаги.
1 W. F. Hillebrand, цит. выше.
988
Гл. LIII. Методы анализа силикатных пород
Этот вывод справедлив при условии, что найденное небольшое количество влагп происходит действительно вследствие увеличения поверхности во время измельчения, а не содержалось в породе раньше. Поэтому если при такой степени измельчения порошок породы легко разлагается фтористоводородной кислотой, можно быть уверенным, что определение в нем железа (II), даст результат, очень близкий к действительному его содержанию, при условии, что определение будет проведено с необходимыми предосторожностями и что никаких других источников ошибок не будет; 7) у различных минералов после многочасового измельчения при одинаковых условиях можно наблюдать различные степени окисления — от нескольких процентов до 45% всей FeO; ине всегда тот минерал, который a priori рассматривается как легко окисляющийся, действительно показывает наиболее сильное окисление. Мягкий или вязкий минерал, измельченный вместе с твердым, подвергается большему окислению, чем если он измельчается один.
Из сказанного выше должно быть ясным, что результаты почти всех анализов минералов и горных пород, проведенных до того, как выяснилось влияние измельчения, заключают в себе более или менее грубые ошибки не только в отношении окислов железа, но и в отношении влаги, причем ошибки эти тем больше, чем дольше проводилось измельчение образца. Очепь многие результаты анализов указывают на присутствие железа (III) в таких минералах, которые по всем данным не должны были бы его содержать, а также на присутствие небольших количеств воды, для которой нет места ни в какой вероятной формуле этого минерала.
В настоящее время этим несоответствиям можно дать простое и во многих случаях вероятное объяснение. Большая часть анализов прежнего времени, особенно анализов железосодержащих минералов, должна быть переделана. Однако прежде чем такие анализы смогут провести, надо позаботиться о том, чтобы в дальнейшем не повторять старых ошибок.
Ниже описаны методы, которые в свете современного знания кажутся для этого наиболее подходящими. Но нужно еще проделать большую работу, состоящую как в проведении многих обычных анализов, так и исследовательскую, прежде чем можно будет установить правила, пригодные для всех возможных случаев. Чтобы избежать недоразумений, следует здесь прибавить, что ошибки прежних анализов не должны быть непременно такого же порядка, как показанные в работе Гиллебранда последние явились следствием двухчасового или многочасового измельчения, тогда как лишь очень немногие аналитики применяли раньше постоянно такое длительное растирание пробы.
Приготовление пробы для анализа. Исходным материалом для определения железа (II) служит порошок минерала или горной породы, полученный измельчением его без растирания (см. стр. 891). Если предварительное испытание показало, что после осторожного кипячения его в течение 20 мин с фтористоводородной кислотой совсем ничего не остается или остается лишь незначительный по величине остаток, то такую пробу можно непосредственно применить для определения в ней железа (II). Если же остаток после обработки фтористоводородной кислотой
См. сноску 2 на стр. 986.
Железо
989
значителен, то отвешенное количество пробы (0,5—1 г) растирают под абсолютным спиртом в большой агатовой ступке столько времени, сколько нужно, чтобы при обработке фтористоводородной кислотой не осталось никакого остатка или последний был бы ничтожно мал. Затем пробу оставляют на воздухе, чтобы спирт свободно испарился. Когда исчезнут последние следы спирта, переносят порошок пробы, смывая его тонкой струей воды, в платиновый тигель емкостью от 20 до 100 мл. Частицы, приставшие к ступке и пестику, смывают также в этот тигель.
Дальнейшая обработка приведена при описании отдельных методов определения (см. ниже, стр. 991), которые основаны на разложении фтористоводородной кислотой. Метод Митчерлиха (стр. 991) почти всегда требует очень тонкого измельчения, поэтому, а также и по некоторым другим причинам, он редко применяется. Те варианты метода с плавиковой кислотой, которые требуют более тонкого измельчения пробы, применяются реже, чем варианты этого метода, допускающие анализ относительно крупных порошков пробы. Там, где непременно требуется очень тонкое измельчение, по-видимому, еще нет в настоящее время никакой возможности ввести поправку на окисление, происходящее во время измельчения. В таких случаях лучше всего измельчить под спиртом небольшую навеску пробы и применить ее для анализа, вместо того чтобы сначала измельчать большое количество пробы, а затем брать от нее невеску. В последнем случае пришлось бы снова отдельно определять свободную и связанную воду, чтобы можно было исправить полученный результат определения железа (II) в соответствии со значительным количеством воды, поглощенным во время измельчения (стр. 900).
Сравнение метода разложения в запаянной трубке с методом разложения фтористоводородной кислотой (методом Кука). В анализе горных пород ничто не доставляет столько забот химику, и особенно минералогу и петрографу, как определение железа (II).
Метод Митчерлиха, заключающийся в разложении пробы серной кислотой в запаянной трубке под давлением, в течение долгого времени был единственным применявшимся при исследовании минералов. Теоретически он превосходен, так как при его применении легко достигается полное устранение кислорода. Однако недостатком этого метода, прежде всего обнаруженным, является невозможность обеспечить полноту разложения железосодержащих минералов, и часто даже невозможность удостовериться, разложился ли минерал или нет.
Прибавление фтористоводородной кислоты к серной кислоте, находящейся в трубке, чтобы обеспечить полное разложение, является в большей части случаев вспомогательным средством весьма сомнительной ценности, так как действие фтористоводородной кислоты на стекло может быть настолько сильным, что в результате в раствор попадает значительное количество железа из стекла, и кроме того, фтористоводородная кислота может оказаться израсходованной на разложение стекла раньше, чем она разрушит более стойкий анализируемый минерал. Тем не менее, если разложение может быть достигнуто действием одной серной кислоты, этим методом получаются хорошие и согласующиеся друг с другом результаты. При анализе горных пород они, однако, бывают обычно более высокими, чем получаемые одним из вариантов метода с фтористоводородной кислотой.
990
Гл. LIII. Методы анализа силикатных пород
Разница не очень велика, если анализируются породы, содержащие 1—2% железа (II), но она возрастает с повышением содержания последнего так сильно, что там, где метод разложения в запаянной трубке показывает содержание 12% железа (II), другие методы могут дать результаты, не превышающие 10%. Опыты с растворимыми солями железа известного состава, например с сульфатом железа (II) или с солью Мора, не внесли ясности в этот вопрос, так как с этими солями все методы дают одинаково точные результаты.
Ключ к решению вопроса был найден 1 в 1882 г., но эта работа осталась неизвестной химикам. Вторично он был обнаружен в 1901 г. 2 при исследовании действия солей железа (III) на пирит и другие сульфиды. Несколько ранее 3 было найдено, что при этом происходит окисление пирита и восстановление железа (III), но тогда еще не было замечено, что это взаимодействие идет очень легко и что пирит окисляется полностью: не только содержащееся в нем железо, но и большая часть серы. Сам по себе чистый пирит при кипячении с разбавленной серной или фтористоводородной кислотой или смесью обеих разлагается крайне медленно, но как только прибавляют какую-нибудь соль железа (III), картина резко меняется.
Окончательное решение проблемы было достигнуто на основе сделанного Гиллебрандом наблюдения, что почти все породы и многие минералы содержат пирит или пирротин, или оба эти минерала вместе, иногда в значительных количествах, иногда в следах. В двухграммовой навеске породы можно почти всегда обнаружить присутствие серы.
Опыты показали (стр. 996), что при определении железа (II) обработкой фтористоводородной кислотой под обычным атмосферным давлением и при температуре кипения можно пренебречь влиянием небольших количеств сернистых соединений, обычно присутствующих в изверженных породах. С возрастанием содержания сульфидов влияние их становится все более и более заметным, потому что действию содержащегося в породе железа (III) подвергается большая поверхность этих сульфидов. С другой стороны, в условиях применения метода разложения в запаянной трубке, при температуре 150—200° С и даже выше, при высоком давлении, более продолжительном времени действия и невозможности удаления образующегося сероводорода сера сульфидов окисляется почти полностью до серной кислоты. Это окисление происходит за счет железа (III) (присутствующего в анализируемом минерале или горной породе) с образованием эквивалентного количества железа (II) [в добавление к тому железу (II), которое образуется из самого сульфида].
Посмотрим, какое влияние на результат анализа оказывают эти следы сульфидов, когда они полностью окисляются. Для полного превращения одного атома серы (32) в серный ангидрид требуется три атома кислорода или три молекулы ЕегОз (480), которые при восстановлении образуют шесть молекул FeO (432). Другими словами, 0,01 % серы может увеличить содержание FeO на 0,135%, а 0,1% серы — вызвать ошибку в 1,35%. Еще хуже обстоит дело, если сера выделяется из растворимых сульфидов
1 L. L. de Koninck, Ann. Soc. Geol. Belg.; 10, 101 (1882—1883).
2 H. N. S t о k e s, U.S. Geol. Survey Bull., 186 (1901); Am. J. Sci., [4], 12, 414 (1901).
3 J. H. L. Vogt, Z. prakt. Geol., 250 (1899).
Железо
991
в виде сероводорода, так как тогда для указанных выше количеств серы ошибки в определении FeO составят соответственно 0,18 и 1,8%.
Ошибка, вызываемая присутствием сульфидов, возрастает с увеличением содержания как самих сульфидов, так и железа (III). Богатые железом породы содержат обычно и железа (III) больше, чем бедные породы; они также содержат и сравнительно много пирита и пирротина. Поэтому в полном соответствии с приведенным выше объяснением находится наблюдение, что расхождение результатов, получаемых двумя указанными методами, возрастает с увеличением содержания железа в породех.
Углистые вещества в условиях метода Митчерлиха также, конечно, восстанавливают серную кислоту и приводят к получению неверных результатов при определении железа (II). Таким образом, метод Митчерлиха как общий метод, применимый для анализа всех пород и минералов, не заслуживает полного доверия, но он все еще остается наилучшим методом анализа пород, совершенно свободных от сульфидов и полностью разлагающихся. Поэтому в следующих далее разделах приводятся условия, при которых этот метод дает наилучшие результаты.
Описание методов. Метод разложения в запаянной трубке. Наполнение, запаивание и нагревание трубки. Метод разложения в запаянной трубке Митчерлиха, в его оригинальном и обычном виде, требует применения смеси из 3 вес. частей серной кислоты и 1 части воды, что соответствует приблизительно отношению объемов 3:2, иногда применяют и более концентрированную кислоту. В некоторых случаях, а быть может даже и в большинстве случаев, можно достигнуть гораздо лучшего разложения силикатов, если взять обратное отношение количества кислоты к количеству воды, во всяком случае применить значительно более разбавленную кислоту. Этим избегают выделения солей, трудно растворимых в концентрированной серной кислоте, а разлагающее действие кислоты на минерал при этом, по-видимому, не уменьшается.
Очень тонко измельченную пробу всыпают в трубку из жароустойчивого стекла, не содержащего железа (II), и вытягивают открытый конец трубки так, чтобы остался канал для вливания кислоты. Затем вливают очень немного воды и осторожно нагревают до кипения для освобождения порошка анализируемой породы от воздуха. После этого вливают разбавленную серную кислоту, которую сначала приготовляют несколько более разбавленной, чем это нужно, а затем кипячением доводят до требуемой концентрации; таким образом освобождают ее от воздуха. Кислоту вливают в таком количестве, чтобы она заполнила трубку на три четверти ее длины. После этого из аппарата Киппа или баллона пропускают очищенную от сероводорода двуокись углерода в свободное пространство над серной кислотой. Для этого применяют узкую трубку, сделанную из того же стекла, что и трубка для разложения. Предварительно, однако, выпускают некоторое время двуокись углерода в воздух. Вытеснение воздуха из трубки происходит быстро. Потом узкую трубку, через которую подавался газ, приплавляют к трубке для разложения. Ток газа при этом не прерывают до последнего момента, когда дальнейшее его пропускание может раздуть стекло. Ток газа легко прервать в нужный момент, если при
1 Подробности опытов см. в статьях W. F. Hillebrand, Н. N. S t о k е s,. Je Am. Chem. Soc., 22, 625 (1900): Z. anorg. Chem.,25, 326 (1900).
992
Гл. LIII. Методы анализа силикатных пород
сплавлении держать рукой подводящую газ стеклянную трубку в месте соединения ее с подающей газ резиновой трубкой, которую и зажимают, когда нужно, указательным и большим пальцами. Никогда не было случая, чтобы в результате такого спаивания обеих трубок основная трубка лопнула при последующей работе х.
Трубку помещают в горизонтальном положении в печь и нагревают .до желаемой температуры, например до 200 °C, пока разложение не закончится или не дойдет до максимальной степени. Это можно определить, вынимая время от времени трубку из печи и исследуя ее содержимое под лупой с небольшим увеличением. Чтобы достигнуть более полного разложения очень стойких минералов, нужно поднять температуру значительно выше допустимой при описанных условиях; этого можно достичь, помещая трубку в плотно закрывающуюся гильзу из прочной стали, содержащую немного бензина или эфира для выравнивания давления снаружи п внутри стекла.
Титрование железа. Наносят напильником царапину на верхнем конце трубки значительно выше уровня жидкости, отламывают этот конец, смывают содержимое трубки в стакан или чашку, ополаскивая также и отломанный конец трубки, и титруют холодный раствор перманганатом, как описано в гл. «Железо» (стр. 445). Результаты параллельных определений совпадают почти точно.
При отсутствии в растворе других способных восстанавливаться соединений можно затем определить и все железо объемным методом — восстановительным титрованием хлоридом титана (III), как описано в гл. «Железо» (стр. 449). Это может служить контролем результата, полученного при анализе главной навески пробы.
Метод Кука с фтористоводородной кислотой. Б методе, предложенном Куком 1 2 3 * * * *, порошок минерала разлагают фтористоводородной кислотой в присутствии серной кислоты без доступа воздуха при атмосферном давлении и титруют в полученном растворе железо (II) перманганатом. Обычно при проведении этого метода допускаются две ошибки противоположных знаков, но не равной величины; о существовании второй и большей из этих ошибок долгое время не подозревали 8.
Железо (II) можно титровать в присутствии серной кислоты и 5— 7 мл 40 %-ной плавиковой кислоты в объеме раствора 200—400 мл почти так же точно, как в присутствии одной серной кислоты, при условии, что раствор, содержащий железо, разбавлен водой и что титрование проводится непосредственно после прибавления плавиковой кислоты и по
1 Раньше воздух обычно удаляли перед запаиванием трубки введением в нее нескольких кристалликов карбоната натрия, предполагая, что двуокиси углерода, образующейся при взаимодействии карбоната с кислотой, достаточно, чтобы вытеснить весь воздух. Что вытеснение воздуха при этом было далеко не полным и что ошибка достигала значительной величины, доказано Гиллебрандом [W. F. Hillebrand, U. S. Geol. Survey Bull., 78, 50 (1891)]; Chem. News, 64, 232 (1891)].
2 J. P. Cooke, Am. J. Sci., [2], 44, 347 (1867).
3 Неясно, имеют ли какое-либо значение для данного метода некоторые наблюдения Е. Deussen [Monatsh., 28, 163 (1907)]. Он утверждал, что окись железа (III),
растворенная в фтористоводородной кислоте, может частично восстанавливаться
с образованием железа (II) и перекиси водорода:
2Fe2O8+8HF = Fe3Fg+FeO + ЗН2О + H2OJ
Если это верно, то можно предположить, что оба эти вещества будут нейтрализо-
вать друг друга в отношении потребления перманганата.
Железо
993
возможности быстро. Это условие не может быть выполнено в ходе анализа, так как в обычно применяемом приборе кислота должна быть в соприкосновении с минералом продолжительное время, и избежать при этом небольшого окисления, по-видимому, почти невозможно. Неизвестно, можно ли после титрования железа (II) в растворе, содержащем плавиковую кислоту, оттитровать затем все железо хлоридом титана (III), как это делается при применении метода разложения в запаянной трубке К
Недостатки метода Кука. Способность марганца (II) окисляться перманганатом в присутствии фтористоводородной кислоты. Фторид марганца (III), в противоположность сульфату марганца (III), в разбавленном растворе почти совершенно не диссоциирован, и то, что ион марганца (III) тотчас же после своего образования переходит в недиссоциированное состояние или в комплексный ион 2, объясняет, почему в присутствии HF марганец (II) легко окисляется перманганатом. По этой причине в присутствии большого количества HF (больше 7 мл 40%-ной HF) нельзя получить четкого конца титрования. Получается кратковременное розовое окрашивание всей жидкости, но оно сразу исчезает, притом тем быстрее, чем больше содержание в растворе плавиковой кислоты или соли марганца (II).
К растворам, содержащим уже в самом начале сульфат марганца (II) в присутствии фтористоводородной кислоты, можно прибавлять перманганат целыми миллилитрами, получая только временную окраску. В то же время раствор принимает все более и более интенсивный красно-бурый цвет, характерный для солей марганца (III). Обесцвечивание перманганата, вызываемое этой причиной, является, следовательно, гораздо более выраженным при анализе пород с большим содержанием железа (II), чем при анализе пород с малым его содержанием. В первом случае из перманганата при его действии на соли железа (II) образуется соответственно большее количество соли марганца (II); кроме того, оно возрастает с увеличением содержания в растворе плавиковой кислоты. Если анализируемая проба содержит небольшое количество железа (II), например 0,02 г, и прибавлено 5—7 мл плавиковой кислоты, окраска, получающаяся от одной избыточной капли перманганата, некоторое время остается, однако она становится все менее устойчивой по мере увеличения содержания железа (II), а следовательно, и образующейся соли марганца (II).
Гиллебрандом были поставлены многочисленные опыты с целью определить ошибку, которая может быть вызвана окислением марганца (II) при определении тех количеств железа, которые обычно встречаются в породах. К определенным количествам сульфата железа (II) и серной кислоты прибавляли различные количества плавиковой кислоты и затем по возможности быстро проводили титрование. При добавлении не более 7 мл 40 %-ной фтористоводородной кислоты была отмечена очень небольшая тенденция к получению повышенных результатов; в большинстве случаев полученные результаты были совершенно тождественны с полученными в присутствии одной серной кислоты (без плавиковой кислоты). Когда количество плавиковой кислоты было увеличено до 10 мл, можно было отчетливо обнаружить повышение получаемых результатов, которое при 15 мл плавиковой кислоты становилось еще больше, достигая в сред-
1 См. стр. 992.
2 Е. М u 11 е г, Р. Корре. Z. anorg. Chem., 68, 160 (1910).
Г"
994
Гл. LIII. Методы анализа силикатных пород
нем 0,2 мл в том случае, когда на титрование шло 20 мл раствора перманганата, титр которого, выраженный по Fe, равен 0,0032 г!мл FeO.
Поэтому для получения возможно более точных результатов нужно после разложения минерала удалять избыток фтористоводородной кислоты пли же устранять ее влияние.
С последней целью было исследовано большое число предохраняющих веществ 1 и было установлено, что лучше всего действует борная кислота. При прибавлении ее (в твердом виде или в растворе) фтор становится неактивным вследствие образования фтороборной кислоты HBF4, анионы которой практически почти не диссоциируют с образованием фторид-ионов.
Способность железа (II) легко окисляться кислородом воздуха в присутствии фтористоводородной кислоты или фторидов. Сравнение двух серий опытов, проведенных в приборах Кука, одна — с сульфатом железа (II) и серной кислотой, другая — с сульфатом железа (И) и смесью серной и фтористоводородной кислот, показало, что во второй серии опытов получились без исключения пониженные результаты. Это может быть объяснено следующим образом 2. Фторид железа (III), как и фторид марган-ца(Ш), в растворе почти совершенно не диссоциирован, в то время как сульфат железа (III) значительно диссоциирует с образованием ионов Fe3+, которые противодействуют окислению ионов Fe2+ свободным кислородом. Поэтому окисление железа (II) в сернокислом растворе на воздухе идет медленно, а в присутствии фтористоводородной кислоты — очень быстро. В то время как раствор сульфата железа (II) в разбавленной серной кислоте, будучи выставлен на воздух в открытой чашке, в течение часа почти не изменяется в отношении содержания в нем железа (II), такой же раствор, но содержащий кроме серной кислоты еще и фтористоводородную, уже за четверть часа претерпевает значительное окисление. Изменение его титра может быть отмечено уже через несколько минут 3 *.
В упомянутых выше опытах, проведенных в приборе Кука, когда на титрование шло 20 мл раствора перманганата, отрицательная ошибка не превосходила 0,1—0,2 мл, что отвечает 0,5—1% от содержания Fe11.
. Но ошибка возрастает, если при определении не обращают серьезного внимания на применение, насколько возможно, совершенно свободной от воздуха двуокиси углерода и на исключение возможности проникновения воздуха в прибор каким-либо другим образом. Ошибка, кроме того, возрастает в известных пределах с увеличением концентрации титруемого раствора.
В целях возможного уменьшения ошибки этоот метод был изменен следующим образом. Двуокись углерода было предложено пропускать только вначале, а затем ток газа прекращать, как только вода в водяной бане начинала сильно кипеть, после чего тигель с горячей бани прямо переносить в сосуд для титрования. Результаты улучшились, хотя и незначительно.
Так как при применении приборов Кука, Барнеби или Тредвелла аналпзируемый минерал долгое время остается в соприкосновении с кис
1 О. L. Barnebey, J. Am. Chem. Soc., 37, 1481 (1915).
2 R. Peters, Z. phys. Chem., 26, 193 (1898).
3 Окисление сульфата железа (II) также сильно ускоряется в присутствии суль-
фата меди [Е. Р о s n j a k, Am. Inst. Min. and Met. Eng. (1927)].
Железо
995
лотами, а только что упомянутые опыты дали благоприятные результаты, возникла мысль об изменении в том же направлении более простого метода Пратта К Метод этот не требует никаких приборов, кроме вместительного платинового тигля, снабженного удобной для введения двуокиси углерода крышкой с отверстием в ней. Пратт кипятит в тигле порошок анализируемого минерала со смесью серной и фтористоводородной кислот, причем разложение сильно ускоряется перемешиванием и более высокой температурой и обычно заканчивается в течение 5 или 10 мин. Предлагаемое изменение состоит в том, что двуокись углерода вводится только вначале, а затем пары кипящего раствора препятствуют проникновению воздуха в тигель. Пратт сам пробовал применить этот видоизмененный метод, но получил довольно низкие результаты, быть может потому, что он применял тигель меньших размеров, чем это нужно.
Результаты, приведенные в табл. 39, получены при работе с сульфидом железа (II) при применении тигля емкостью 100 мл', они показывают, какие точные данные можно получить этим методом.
, ТАБЛИЦА 39
Определение железа (II) измененным методом Пратта
(титр перманганата: 0,0032 г FeO в 1 мл)
Продолжительность разложения -мин Израсходовано на титрование раствора перманганата Л1Л Должно было израсходоваться перманганата Л1Л Продолжительность разложения мин Израсходовано на титрование раствора перманганата мл Должно было израсходоваться перманганата мл
10 4,9 4,8 10 19,4 19,2
10 4,8 4,8 10 19,3 19,2
10 5,0 4,8 15 19,25 19,2
10 4,9 4,8 15 19,1 19,2
10 9,6 9,6 15 19,3 19,2
10 9,6 9,6 20 19,2 19,2
10 9,6 9,6 20 19,3 19,2
10 19,2 19,2
Результаты не оставляют желать лучшего: они показывают либо правильные, либо слегка повышенные значения вместо пониженных, чаще всего получаемых при применении метода в его первоначальном виде. Этот метод в течение двух лет сравнивали с методом Кука и получили хорошие результаты. Его надо предпочесть не только потому, что он прост и быстро выполним, но также и потому, что он допускает анализ менее тонкого порошка пробы, чем методы Кука, Барнеби и Тредвелла. Так как теперь известно, однако, что определение железа (II) требует измельчения пробы до возможно более грубого порошка, следует проводить разложение более продолжительное время, чем это делал Пратт, подвергая анализу тонкие порошки пробы.
Разложение трудноразлагаемых минералов ускоряется путем добавления 0,5 г не очень тонко измельченного кварца (но не осажденной кремнекислоты) 1 2. Действие
1 J. Н. Pratt, Am. J. Sci., [3], 48, 149 (1894).
2 M. Dittrich, Вег., 44, 990 (1911).
63*
996
Гл. LIII. Методы анализа силикатных пород
кварца состоит в разделении им зерен минерала и в предупреждении спекания их на дне тигля, что приводит к более эффективному действию на них кислоты.
Влияние сульфидов, ванадия и углистых веществ на определение железа (II) с фтористоводородной кислотой. Темная окраска нерастворимых фторидов и фторосиликатов может происходить от присутствия пирита, графита или углистых веществ. Первый из этих компонентов мало влияет на получаемые результаты определения железа (II), второй, вероятно, совсем не влияет. Эти примеси можно отличить от углистых веществ по их отношению к азотной кислоте. Присутствие углистых веществ делает определение железа (II) невозможным. Все перечисленные примеси в минералах, в отличие от горных пород, встречаются редко.
Сульфиды. В малых количествах, в которых пирит обычно встречается в изверженных породах, он, вероятно, не оказывает серьезного влияния на определение железа (II) ни по одному из вариантов метода с фтористоводородной кислотой. Этот сульфид при отсутствии кислорода очень стоек по отношению ко всякому воздействию, как показывает тот факт, что если пирит присутствует в сколько-нибудь заметных количествах, его можно обнаружить в остатке после титрования. Во всяком случае нельзя на основании одного предположения, что пирит может разложиться, вводить какую-либо поправку в полученные результаты, которые надо всецело отнести за счет присутствовавших в пробе кислородных соединений железа (II).
Но если в пробе присутствуют растворимые в серной кислоте сульфиды, то надо учитывать две возможные ошибки, происходящие: 1) от восстановления железа (III) образующимся сероводородом; 2) от железа (II), входящего в состав самих сульфидов, особенно если присутствует пирротин.
Первой ошибкой, быть может, можно пренебречь, так как большая часть сероводорода, вероятно, улетучивается, не успев восстановить железа.
Вторую ошибку можно приблизительно учесть, если известно, что пирротин является единственным присутствующим растворимым сульфидом и его содержание было определено по количеству сероводорода, выделяющемуся при кипячении породы с соляной кислотой в токе двуокиси углерода газа. В этом случае можно ввести поправку в результат титрования всего железа (II) (см. также стр. 1031).
Чтобы получить количественные данные о влиянии присутствующего пирита на определение железа (II) с фтористоводородной кислотой, были поставлены следующие опыты. Часть хорошо образованного кристалла пирита была тонко измельчена и прокипячена с разбавленной серной кислотой. Последняя извлекла из пирита значительное количество железа (II), вероятно, вследствие растворения примешанного к пириту или совместно с ним выкристаллизовавшегося пирротина, так как после вторичного кипячения со свежей порцией серной кислоты в полученном растворе железа (II) не было обнаружено. После этого промытый водой, а затем спиртом и эфиром порошок был высушен и снова измельчен. 0,25 г его было обработано методом Кука в большом платиновом тигле серной и фтористоводородной кислотами, и полученный раствор был быстро профильтрован через бумажный фильтр, помещенный в платиновом конусе. На титрование этого раствора потребовалось только 2 капли перманганата, титр которого был эквивалентен 0,0032 г!мл FeO.
Железо
997
Так как было найдено \ что окисляющее действие солей железа (III) на пирит и другие сульфиды значительно больше, чем до сих пор думали (см. стр. 990—991), то для определения вероятной ошибки, которая может произойти от этого возаимодействия в существующих при анализе силикатов условиях, были проделаны следующие опыты. По 1 г роговообман-кового сланца, не содержащего серы и содержащего (в среднем из многих определений) 10,09% FeO и 4,00% ГегОз, смешали в большом платиновом тигле емкостью 100 мл с 0,02; 0,025 и 0,10 г вышеописанного (очищенного от пирротина) порошка пирита и обработали фтористоводородной и серной кислотами методом Кука в течение 1 ч на кипящей водяной бане. По охлаждении содержимое тигля вливали в платиновую чашку с водой и быстро титровали почти до конца реакции. Затем для удаления пирита, который мог бы из-за его восстанавливающего действия на перманганат маскировать конец реакции, раствор фильтровали, как указано выше, и прозрачный фильтрат дотитровывали до конца. Было получено: 10,02; 10,16 и 10,70% FeO.
Самая меньшая из этих прибавок пирита все же в несколько раз превосходит количество его, которое лишь в редких случаях может встретиться в изверженных горных породах. Из этих опытов ясно вытекает, что в практике анализа влиянием пирита на определение железа (II) методом Кука можно пренебречь. В то же время следует иметь в виду, что с возрастанием содержания железа (III) увеличивается количество разложенного пирита и интенсивность этой реакции зависит от степени измельчения порошка пирита, который, с другой стороны, сам подвергается окислению при измельчении.
Ванадий. Если находящийся в породе ванадий присутствует в трехвалентном состоянии, то он вызывает ошибку в результате титрования железа (II), пропорциональную его содержанию в породе. Если это содержание известно, можно легко рассчитать и ввести соответствующую поправку.
Углистые вещества. Как уже было сказано выше (стр. 996), присутствие в пробе веществ органического происхождения, кроме углерода в форме графита, делает результат определения железа (II) совершенно ненадежным.
Ход анализа по различным вариантам метода с фтористоводородной кислотой. Метод Пратта (видоизмененный).В платиновый тигель емкостью 80—100 мл помешают 0,5—1 г возможно более грубо измельченной пробы 1 2 3, смачивают 1—2 мл освобожденной от воздуха воды и прибавляют 10 мл разбавленной (1 : 3) серной кислоты. Если порода содержит карбонаты, то происходит вспенивание; поэтому кислоту следует приливать осторожно и тигель держать покрытым крышкой, пока не кончится реакция. Не следует опасаться, что железо (II), в случае перехода его в раствор, может на этой стадии анализа окислиться. Затем наполняют тигель по крайней мере до половины свободной от воздуха горячей водой, покрывают крышкой и вставляют в треугольник, помещенный над малень
1 Н. N. S t о k е s, U. S. Geol. Survey Bull., 186 (1901); Am. J. Sci., [4], 12,
414 (1901).
3 Степень измельчения зависит от состава анализируемой породы. Большинство гранитных пород требует только умеренного измельчения; породы же, содержащие большие количества железистых и трудноразлагаемых минералов, как, например, турмалина, обычно нуждаются в очень тонком измельчении.
998
Гл. LIII. Методы анализа силикатных пород
ким, защищенным от воздушных токов, пламенем. После этого быстро удаляют воздух из тигля током двуокиси углерода, которую подводят под приподнятую с одной стороны крышку.
Раствор в тигле быстро нагревают до кипения, и как только пары воды начнут выделяться из-под крышки тигля, слегка отодвигают крышку тигля и вливают в один прием 7 мл фтористоводородной кислоты из маленького платинового тигля. На это требуется не более 1 сек1.
Как только раствор снова закипит, удаляют трубку, подводящую двуокись углерода, плотно закрывают тигель крышкой и осторожно кипятят 12—15 мин. После этого быстро переносят тигель вместе с его содержимым и крышку в сосуд для титрования (он может быть стеклянным), содержащий 300 мл освобожденного от воздуха раствора борной кислоты 2, некоторое количество твердой борной кислоты, 10 мл разбавленной (1 : 3) серной кислоты и почти все количество (за исключением 1—2 мл) титрованного раствора перманганата, какое требуется Гдля окисления железа (II). Заканчивают титрование обычным способом.
Бихромат калия значительно труднее реагирует с органическими веществами, чем перманганат; поэтому и предложено 3 применять этот реактив с дифениламином в качестве индикатора. Для определения железа (II) в силикатах автор поступает следующим образом. К обычному платиновому тиглю подбирают плотно прилегающую крышку из прозрачного бакелита, снабженную бакелитовой трубкой для введения двуокиси углерода и маленькой бакелитовой воронкой для вливания кислоты и удаления паров. В тигель помещают 0,5 г грубо измельченной пробы, смачивают водой и обрабатывают 10 мл 12 н. соляной кислоты или 18 н. серной кислоты. Опускают в тигель маленькое колечко из платиновой проволоки, чтобы предупредить толчки, плотно закрывают крышкой и пропускают двуокись углерода по крайней мере 10 мин, пока смесь не дойдет постепенно до кипения. Ток двуокиси углерода прекращают, как только начнет выделяться пар. Тогда тотчас же прибавляют через воронку 7 мл 48%-нои фтористоводородной кислоты и осторожно кипятят 10—15 мин. Когда разложение закончится, что можно видеть через прозрачную крышку, снова пропускают ток двуокиси углерода и охлаждают до комнатной температуры. Прибавляют через воронку отмеренное количество титрованного раствора бихромата калия (в избытке), снимают крышку, обмывают ее над тиглем и выливают раствор на твердую борную кислоту, находящуюся в фарфоровом тигле, покрытом изнутри церезином. Титруют тотчас же титрованным раствором сульфата железа (II), применяя дифениламин в качестве индикатора4.
Даже и без добавления борной кислоты, при определении малого количества железа, окраска сохраняется некоторое время, однако с повышением содержания железа, а также фтористоводородной кислоты скорость исчезновения окраски возрастает. При проведении второго контрольного определения рекомендуется, прежде чем вливать в сосуд для титрования анализируемый раствор, налить в него раствор перманганата в количестве несколько меньшем, чем его потребуется для титрования.
Неопытному работнику можно посоветовать проделать сначала опыт с раствором сульфата железа (II), титр которого был установлен без прибавления фтористоводородной кислоты. Важно не кипятить раствор слишком долго, потому что тогда могут выделиться малорастворимые соли
1 Фтористоводородная кислота часто содержит частички того материала, из которого сделан содержащий ее сосуд (например, парафина). Их следует избегать. Не следует также применять фтористоводородную кислоту, загрязненную растворимыми органическими веществами.
2 См. сноску 1 на стр. 994.
3 L. A. Sarver, J. Am. Chem. Soc., 49, 1472 (1927).
4 См. также С. J. Schollenberger, J. Am. Chem. Soc., 53, 88 (1931) и стр. 447.
Железо
999
и, кроме того, с возрастанием температуры, происходящим вследствие увеличения концентрации серной кислоты, становится заметным и окисляющее действие последней.
Если после титрования обнаружится нера.зложившийся остаток,
то ему дают полностью осесть, декантируют раствор и промывают остаток один раз декантацией водой. Затем смывают остаток струей воды из про
мывалки в маленькую агатовую ступку, снова дают ему осесть, сливают
большую часть воды и растирают остаток под оставшейся водой в течение нескольких минут. Растертый остаток смывают в большой тигель и повторяют обработку в течение более короткого времени с меньшим количеством
серной и фтористоводородной кислот, чем в первый раз. Последующее затем титрование можно провести в самом тигле, наполнив его предварительно почти до краев холодной, освобожденной от воздуха водой. Обычно таким образом достигают полного разложения. Если же разложение прошло не полностью, то всю операцию повторяют еще раз. Не нужно опасаться, что в течение короткого времени, которое требуется для измельчения остатка под водой, произойдет заметное окисление.
Данные Праттом указания1, как проводить обработку нерастворимого остатка, полученного при первом разложении трудно разлагаемых минералов, могут быть применены только к анализу однородных веществ, но не горных пород, так как у последних^ отношение содержания железа (II) к содер-
Рпс. 43. Прибор Кука для определения железа (II).
жанию железа (III) в неразложенной части, несомненно, отличается от
величины этого отношения в разложенной части.
Метод Кука2. Изображенный на рис. 43 прибор состоит из маленькой, с одним только отверстием водяной бани, которая накрыта стеклянной воронкой со срезанной у начала конуса трубкой. Воронка входит краем в желобок, сделанный в крышке бани. Из склянки с тубусом в этот желобок все время капает вода, которая образует водяной герметический затвор, а стекая затем внутри бани, поддерживает в нем постоянный уровень. Для более успешной изоляции от воздуха рекомендуется применять свежепрокипяченную воду. Через узкую металлическую трубку пропускают внутрь водяной бани (но над поверхностью воды)
двуокись углерода, которая, выходя через выемки на внутреннем крае отверстия крышки, наполняет воронку и тигель3.
Обрабатывают 0,5—1 г порошка анализируемой породы точно так, как при определении методом Пратта (стр. 997), пока не прекратится вскипание, вызванное возможным присутствием карбонатов. Затем сни
1 J. Н. Pratt, Am. J. Sci., [3], 48, 150 (1894).
2 J. P. Cooke, Am. J. Sci., [2], 44, 347 (1867).
3 Двуокись углерода, получаемая из новой порции мрамора, даже белого на вид, нужно тщательно испытывать на присутствие в нем сероводорода. Его нужно пропускать через большую U-образную трубку со стеклянными бусами и раствором сульфата меди.
1000
Гл. LIII. Методы анализа силикатных пород
мают крышку тигля, обмывают ее, если это нужно, и помещают открытый тигель в отверстие водяной бани; ставят воронку на место, пускают ток двуокиси углерода и ток воды из резервуара, после чего зажигают горелку под наполненной заранее баней. Как только можно будет с уверенностью считать, что воздух из воронки и тигля вытеснен, вливают в тигель через платиновую, эбонитовую или бакелитовую воронку отмеренное количество (5—7 мл) концентрированной фтористоводородной кислоты и вводят в него через трубку защитной воронки платиновую палочку для перемешивания. Если для вливания кислоты не имеетбя специальной воронки, то можно поднять на мгновение защитную воронку, однако лишь тогда, когда тигель совершенно наполнится двуокисью углерода.
Когда из конца воронки пойдет сильная струя пара, прерывают пропускание двуокиси углерода и оставляют прибор на 1 ч, время от времени перемешивая содержимое тигля. Когда зернистые частицы больше не будут ощущаться (или если из опыта известно, что за протекшее время разложение должно закончиться), снова пропускают ток двуокиси углерода, гасят пламя под баней и увеличивают приток воды до максимума пропускной способности отводной трубки. Через 15 мин тигель и его содержимое должны охладиться. Удаляют платиновую палочку, помещают ее в сосуд для титрования и титруют, как описано выше (стр. 998).
Пока аналитик не приобретет достаточного опыта, он всегда должен проводить два параллельных определения. Надо заметить, что содержимое тигля увеличивается в объеме за время разложения вследствие конденсации водяных паров.
Вместо того чтобы охлаждать содержимое тигля в струе двуокиси углерода, можно его тотчас же перенести в чашку для титрования и прибавлять перманганат, как указано выше. При тщательной работе, как показывают упомянутые опыты, это изменение метода дает, по-видимому, * несколько лучшие результаты, чем получаемые после охлаждения в токе двуокиси углерода.
Аналитик, незнакомый с методом в том или другом его варианте, должен непременно испытать его сначала на растворе сульфата железа (II), титр которого был установлен без прибавления HF. Если есть возможность, то следует проводить два параллельных определения.
Метод Б а р не би. Барнеби 1 описал упрощенную форму прибора Кука. В фарфоровую чашку диаметром 20 см помещают несколько стеклянных бус и фарфоровую пластинку из эксикатора, имеющую диаметр 12,5 см и стоящую на ножках. Поверх пластинки ставят стеклянную пли фарфоровую подставку для поддержки платиновой чашки, в которой проводится разложение. Над пластинкой помещают стеклянную воронку диаметром 15 см, как в приборе Кука. Нижний край воронки опускают в чашку ниже уровня той жидкости, которая будет налита в чашку; таким образом создается гидравлический затвор. При определении железа (II) Барнеби пользуется в качестве такой жидкости разбавленным (1:2) раствором фосфорной кислоты. Этот раствор служит не только для создания гидравлического затвора, но и для вытеснения парами воды воздуха из воронки и платиновой чашки. Баню нагревают на голом пламени или плитке. По мере надобности вместо выкипевшей воды добавляют еще
1 О. L. Barnebey, J. Am. Chem. Soc., 38, 374 (1916).
Железо
1001
свободную от воздуха воду, которую приливают по платиновой палочке. В остальном можно сослаться на описание прибора Кука.
Конечно, отсутствие приспособления для пропускания двуокиси углерода делает необходимым перевод раствора навески в сосуд для титрования в еще горячем виде. Кроме того, и в этом методе необходимость тонкого измельчения пробы является довольно серьезным затруднением, как и при применении прибора Кука. Последний имеет, однако, преимущество, состоящее в том, что пропускаемая двуокись углерода удаляет из прибора воздух быстрее, чем может это сделать пар, играющий ту же-роль в приборе Барнеби.
Метод Тредвелла. Тредвелл1 применяет парафиновую баню, в которую вставляется свинцовый ящик со свинцовой же крышкой, снабженной двумя отверстиями: одно сбоку для введения двуокиси углерода, другое в центре для удаления пара и помещения палочки для перемешивания. В ящике под центральным отверстием стоит на подставке-маленькая чашечка, содержащая навеску пробы.
Хотя главной целью этого метода было осуществить удаление избытка фтористоводородной кислоты повышением температуры до 120° С к концу разложения пробы и тем исключить ошибку при титровании, все же, как показал наш ограниченный опыт, при этом полное удаление HF не достигается, а большая продолжительность определения (около 2 ч) приводит к получению пониженных результатов по сравнению с теми, какие дают ранее описанные методы, особенно метод Пратта.
Метод Соула. Соул2 рекомендует растворять навеску в колбе из стекла пайрекс или, лучше, из кварца, потому что в этом случае легко следить за ходом реакции и раствор не приходится переливать. При этом нужно, конечно, вводить поправку на каждый грамм растворившегося стекла. Для определения этой поправки взвешивают колбу, обрабатывают в ней данным методом навеску чистой соли Мора, титруют и снова взвешивают опорожненную колбу. Разность между объемом раствора перманганата, израсходованного в этих условиях, и объемом того же раствора, требуемым для непосредственного титрования такой же навески соли Мора, делят на массу растворившегося стекла.
Ход определения следующий. Очищают колбу, взвешивают ее, беря другую такую же колбу в качестве противовеса, наполняют колбу двуокисью углерода и вводят навеску для анализа. Затем прибавляют 15 мл воды, 10 мл разбавленной (1:3) серной кислоты и от 5 до 8 мл фтористоводородной кислоты (40%-ной). Ставят колбу на горячую плитку и осторожно кипятят, пропуская слабый ток двуокиси углерода. Когда при перемешивании раствора не будет заметно неразложившихся частиц, снимают колбу с плиты и тотчас же прибавляют 100 мл свежеприготовленного холодного раствора, содержащего 6 г борной кислоты и 5 мл разбавленной (1 : 1) серной кислоты. Охладив раствор приблизительно до 15° С, титруют 0,05 н. раствором перманганата. Затем колбу моют, высушивают, снова взвешивают и вводят поправку в величину израсходованного на титрование объема раствора перманганата соответственно количеству растворившегося стекла.
1 F. Р. Treadwell, Kurzes Lehrbuch analytischen Chem., [6], 2, 425 (1913).
2 В. A. S о u 1 e, J. Am. Chem. Soc., 50. 1691 (1928).
1002
Гл. LIII. Методы анализа силикатных пород
В более поздней статье 1 Соул указывает на то, что железо (II) в маг-
нетитах и материалах с высоким содержанием кремнекислоты можно быстро и точно определить потенциометрическим титрованием 0,05 н. раствором сульфата церия (IV) (стр. 219). В этом случае навеску пробы (0,3—0,4 г) разлагают, обрабатывая ее смесью 10 мл соляной кислоты и 3 мл фтористоводородной кислоты в колбе стекла пайрекс, наполненной двуокисью углерода, и нагревая при 40—50° С в течение 3—5 мин. Затем к раствору приливают 100 мл воды, содержащей 5 г борной кислоты и 5 мл разбавленной (1 : 6) серной кислоты, и титруют указанным выше раствором сульфата церия (IV) с применением биметаллического (Pt—Ag) электрода. Мышьяк, главный восстановитель, переходящий в раствор
из стекла, определению не мешает.
*М етод Шеленбергера. В 1931 г. был предложен 2 метод определения железа (II) в горных породах; по этому методу титрование проводится бихроматом калия в присутствии дифениламина в качестве внутреннего индикатора. В этом случае присутствие фтористоводородной кислоты в растворе не только не мешает получению резкой и правильной конечной точки титрования, как это имеет место при титровании перманганатом, но, наоборот, способствует ей (см. гл. «Железо», стр. 446 и сноску 1 на стр. 447). После титрования железа (II) можно, прибавив борную
Рис. 44. Платпповый тигель с крышкой из бакелита и промывалкой.
кислоту, оттитровать все железо раствором хлорида титана (III).
Разложение пробы удобно проводить в платиновом тигле емкостью 50 мл с крышкой из бакелита и промывалкой для вводимой двуокиси углерода. Тигель и промывалка показаны на рис. 44.
Крышка должна быть отполирована и должна плотно прилегать к краю тигля, почти не пропуская газа. Сквозь крышку проходит трубка из бакелита для введения двуокиси углерода. Другое отверстие в крышке закрывают пробкой. Это отверстие служит для вливания реактивов, перемешивания и наблюдения за титрованием. Промывалка служит также для наблюдения за скоростью пропускания двуокиси углерода.
Поместив навеску пробы в тигель, плотно закрывают его крышкой, вставляют пробку и помещают тигель в отверстие, проделанное в листе асбестового картона, укрепленного над микрогорелкой. Присоединяют промывалку посредством резиновой трубки, как показано на рис. 44,
пропускают двуокись углерода для удаления воздуха и продолжают пропускание углекислого газа во всех дальнейших операциях. Вынув пробку, прибавляют 6 мл свежепрокипяченной и охлажденной воды, после этого осторожно вливают 6 мл разбавленной (1 : 1) серной кислоты, снова вставляют пробку и нагревают до кипения, продолжая пропускать ток двуокиси углерода. Затем опять вынимают пробку, прибавляют 6 мл фтористоводородной кислоты из небольшого тигля, в котором можно отмерить ее с достаточной точностью, и вставляют пробку обратно. Осторожно
1 В. A. S о u 1 е, J. Am. Chem. Soc,, 51, 2117 (1929).
2 С. J, Schoellenberger, J- Am. Cbem. Soc., 53, 96 (1931).
Железо
1003
нагревают до слабого кипения и время от времени перемешивают содержимое тигля, слегка покачивая асбестовый картон.
По окончании растворения прекращают нагрев и дают тиглю остыть. Когда тигель остынет настолько, что до него можно будет дотронуться рукой, вынимают его из картона и ставят в чашку с водой для более быстрого охлаждения. Когда он охладится до комнатной температуры, вынимают пробку и прибавляют столько воды, чтобы разбавить раствор приблизительно до 25 мл. Затем приливают 2 к^пли раствора дифениламина [0,5 г дифениламина растворяют в 50 мл серной кислоты (пл. 1,84 г/c.w3)] и титруют 0,05 н. раствором бихромата калия, медленно и при перемешивании платиновой палочкой, до появления непсчезающей синей окраски. Вводят поправку на объем раствора бихромата, израсходованного на титрование в «холостом» опыте, и полученную разность умножают на титр раствора бихромата, пересчитанный на FeO (1 мл 0,05 н. раствора бихромата соответствует 0,003592 г FeO).
После титрования железа (II) можно в той же пробе определить и общее содержание железа, для чего добавляют 4 г борной кислоты, растертой в пасту с небольшим количеством воды, перемешивают до растворения, прибавляют 5 мл 10%-ного раствора роданида калия и титруют титрованным раствором хлорида титана (III) (стр. 449), пока не исчезнет красная окраска, а оставшаяся желтая окраска не будет больше ослабевать при прибавлении одной капли титрующего раствора. Если емкость тигля недостаточна для проведения в нем этого титрования, его можно поместить в стакан, перевернуть и продолжать титрование, не вынимая тигля.
В присутствии органических веществ результаты определения железа (II) получаются несколько лучшие, чем при титровании перманганатом, но все же слишком высокие. Доп. ред*
Известен метод 1 определения железа (II) в силикатных материалах, растворимых в холодной соляной кислоте (или смеси соляной и фтористоводородной кислот), содержащей однохлористый иод (IC1); в этом методе выделившийся иод титруют йодатом калия.
Для определения железа (II) в силикатных минералах, нерастворимых в кислотах, был предложен 2 3 в качестве плавня метафтороборат натрия NaBCh • NaBOF2. В новом варианте этого метода 3 плавление проводится в кварцевой трубке при 950° С в атмосфере двуокиси углерода и определение заканчивается или титрованием перманганатом в присутствии борной кислоты или указанным выше методом с применением однохлористого иода (IC1).
Ненадежность определения, железа (II). Из предыдущего ясно, что, несмотря на крайнюю тщательность в работе, точное определение железа (II) в горных породах сопряжено с чрезвычайными затруднениями и его результаты ненадежны. Только при отсутствии разлагаемых сульфидов и углистых веществ, точном знании количества находящегося в породе ванадия и его валентности и анализе относительно грубоизмельченного порошка породы можно считать результаты определения безупречным.
1 М. Н. Неу, Mineralog. Mag., 26, 116 (1941).
2 Н. Р. R о w 1 е d g е, J. Royal Soc. W. Australia, 20, 165 (1934).
3 А. В. Г p о в с, Анализ силикатов, Издатинлит, 1953.
1004
Гл. LIII. Методы анализа силикатных пород
Железо (III)
Прямое определение в присутствии железа (II). Возможность непосредственного применения в анализе минералов метода титрования хлоридом титана (III) (см. «Железо», стр. 449) для прямого определения в минералах железа (III) в присутствии соединений железа (II) еще не изучена. Если этот метод пригоден для анализа минералов, легко растворимых в серной или соляной кислоте, то он может оказаться непригодным для анализа йЬрод и минералов, которые приходится разлагать фтористоводородной кислотой в отсутствие воздуха; условия такой обработки резко отличаются от тех, в которых работали авторы этого метода.
Во всяком случае при растворении минерала и титровании железа необходимо тщательно следить за тем, чтобы были приняты все меры предосторожности, приведенные при описании методов определения железа (II), и в полученном результате могут быть ошибки того же порядка, но только противоположного знака.
Истинная величина содержания железа (III) в породах и минералах. Если в веществе, подлежащем анализу, железо содержится и в виде двухвалентного и в виде трехвалентного, и кроме общего количества железа (выраженного в расчете на ЕегОз) была определена только одна из этих форм [обычно железо (II)], то. остается только из общего содержания железа вычесть количество ЕегОз, эквивалентное найденному содержанию железа (II), чтобы получить содержание ЕегОз, присутствовавшей в породе. Это наиболее благоприятный случай.
При анализе минералов и горных пород, содержащих сульфиды железа или других элементов, происходят две ошибки. Одна из них зависит от содержания железа в сульфидах, другая — от неопределимого восстанавливающего действия на железо (III) того сероводорода, который выделяется в свободном состоянии при определении железа (II) (стр. 996). К этим двум источникам ошибок нужно прибавить третий, вызванный присутствием ванадия (стр. 997). Поэтому во всех таких случаях окончательные результаты определения как железа (III), так и железа (II) не совсем надежны.
ЩЕЛОЧНЫЕ МЕТАЛЛЫ
Общие замечания
Очень редко бывает, чтобы один из металлов щелочной группы встретился в породе, не сопровождаемый большим или меньшим количеством других металлов той же группы. Чаще всего приходится иметь дело только с натрием и калием, которые в минералах иногда сопровождаются заметными количествами лития. В горных породах литий широко распространен, но обычно в виде следов, открываемых только при помощи спектроскопа. Рубидий и цезий находятся только в очень немногих редких минералах, которые, концентрируясь местами в глубоких слоях земной коры, без сомнения, обусловливают содержание этих элементов в некоторых природных минеральных водах.
Первая стадия определения щелочных металлов в минералах состоит в том, чтобы собрать эти металлы и взвесить их в виде хлоридов. Литий и калий (рубидий и цезий) определяют затем отдельно. Содержание натрия находят по разности.
Щелочные металлы
1005
Чаще всего применяемые методы определения рассматриваемой группы требуют, чтобы сначала были удалены все другие металлы, а также неметаллы, кроме хлора и иногда сульфат-ионов. В исключительном случае, когда присутствует только один из щелочных металлов, можно взвесить его в виде сульфата. Однако не всегда, вернее очень редко, бывает известно заранее, что минерал не содержит других металлов этой группы, а потому лучше иметь раствор, в котором можно провести все требуемые разделения, а таковым является раствор хлоридов.
Растворимые сульфаты переводят в .хлориды обработкой хлоридом бария; другие соли обычно переводят в хлориды повторным выпариванием с соляной кислотой, которую заменяют метиловым спиртом, насыщенным хлористым водородом, когда хотят превратить в хлориды растворимые бораты х. Бромиды и иодиды лучше всего превращать в хлориды выпариванием с соляной кислотой, содержащей перекись водорода (но не в платиновой посуде) 1 2.
Существуют только два хорошо известных и применимых во всех случаях метода получения щелочных металлов в виде их хлоридов, когда анализируемый минерал нерастворим в соляной кислоте. Один из них — старый — был предложен Берцелиусом 3, другой — более новый — дан Лоуренсом Смитом 4. Оба метода находят применение при анализе силикатов. Первый начинается с разложения пробы плавиковой и серной кислотами и удаления кремния и избытка фтора, затем следует удаление всех металлов, кроме щелочных, и превращение оставшихся сульфатов в хлориды. В методе Лоуренса Смита минерал разлагают нагреванием порошка его со смесью хлорида аммония и карбоната кальция, причем происходит полное разложение, и щелочные металлы превращаются в хлориды, которые могут быть извлечены водой. В обычном случае ничего другого при этом не извлекается, кроме большого количества кальция и сульфат-ионов, если они присутствовали. И тот и другие легко удалимы, так что в конце концов остается только раствор хлоридов щелочных металлов. В анализе необычных соединений, например боросиликатов или сложных стекол, могут перейти в раствор и другие вещества, например бораты, и потребуется специальная обработка для их удаления.
В других, реже используемых для разложения методах применяются окиси бора, свинца и висмута. Первый из этих методов и последующий способ обработки описаны выше (стр. 929), другие не нуждаются в подробном описании. Они связаны с отделением больших количеств бора, свинца или висмута из полученных растворов, но они очень удобны в тех случаях, когда вследствие недостаточного количества материала для анализа требуется, чтобы другие металлы были определены в той же навеске, в кото
1 Величина ошибки при определении щелочных металлов, происходящая от присутствия бора, иллюстрируется следующими данными. Когда анализ стекла, содержавшего 4,15% Na2O и 12,7% В2О3, провели методом Лоуренса Смита, не обращая внимания на присутствие бора, то вместо указанного содержания Na2O получили 5,01 и 4,73%. (Масса остатка «хлоридов» вместо действительной величины 0,0404 г была равна соответственно 0,0486 и 0,0458 г. После обработки этих «хлоридов» СН3ОП -|- IIC1 масса остатка хлоридов оказалась равной 0,0403 и 0,0401 г).
2 См. G. Е. F. L u n d е 11, J. I. Н о f f m a n, Outlines of Methods of Chemical Analysis, 1938, p. 76.
3 J. J. Berzelius, Pogg. Ann., 1, 169 (1824).
4 J. L. S with. Am. J. Sci., [2], 50, 269 (1871); Am. Chemist, 1 (1871); Ann. Chem. und Pharm., 159, 82 (1871).
1006
Гл. LIII. Методы анализа силикатных пород
рой определяют щелочные металлы, или когда данный минерал может быть разложен только одним из этих окислов и не разлагается другими. Какой из этих методов следует выбрать в каждом отдельном случае, определяется больше личными вкусами или же выбор проводится на основании опыта. Здесь нельзя дать определенных правил х.
Метод Берцелиуса долго был единственным практическим методом определения щелочных металлов, но в настоящее время он в значительной мере вытеснен методом Смита. Метод Смита имеет несколько существенных преимуществ, наибольшим из которых является то, что магний при работе этим методом не сопровождает щелочные металлы в водном растворе, вследствие чего устраняются затруднения и ошибки, связанные с отделением этого металла и других металлов, кроме кальция. Большая часть бора также остается нерастворенной в виде бората кальция. Кроме того, метод Смита не требует осаждения большого количества сульфата бария, при котором можно всегда опасаться окклюзии солей щелочных металлов. Наконец, операции после смешения навески с указанными реактивами в методе Смита проще, чем в методе Берцелиуса. Поэтому метод Смита предпочитают теперь все, кто пользовался обоими методами. Его рекомендуют даже для анализа силикатов, растворимых в соляной кислоте, особенно тех, которые содержат магний.
Метод Лоуренса Смита
Реактивы. Навеску пробы разлагают прокаливанием ее с равным по весу количеством хлорида аммония и восьмикратным количеством осажденного карбоната кальция. Применяемый для этого определения хлорид аммония лучше всего приготовлять медленной возгонкой продажной соли, растворением сублимата в воде в платиновой чашке, фильтрованием, если нужно, и выпариванием на паровой бане (не следует нагревать газовыми горелками). Когда начнется выделение кристаллов, раствор перемешивают, чтобы они получились мелкими и легко растирающимися. Сливают горячий маточный раствор, переносят кристаллы на пористый фильтр и высушивают на воздухе. Сохраняют препарат в маленькой банке с хорошо притертой пробкой.
Карбонат кальция лучше всего приготовлять из чистого кальцита растворением его в соляной кислоте, осаждением чистым карбонатом аммония и аммиаком и тщательным промыванием горячей водой. После высушивания его следует хранить в хорошо закрытой банке, отдельно от хлорида аммония. Полученный таким способом карбонат кальция никогда не бывает совершенно свободным от хлоридов щелочных металлов. Поэтому их надо раз навсегда определить, проведя с приготовленным препаратом «холостой» опыт, чтобы можно было вносить соответствующую поправку в результаты анализа. 8 г карбоната кальция содержат обычно от 0.0012 до 0,0016 г хлоридов щелочных металлов, главным образом хлорида натрия, если во время всего определения применялась платиновая посуда. Это количество может быть снижено наполовину очень длительным промыванием осажденного карбоната.
1 Для определения калия в почвах кобальтинитритным методом J. Е. Giese-k i n g и Н. J. Snider [Ind. Eng. Chem., Anal. Ed., 9, 232 (1927)] рекомендуют предварительное сплавление почвы со смесью 2 частей карбоната натрия и 1 части карбоната лития при температуре 500—600° С.
Щелочные металлы
1007
Рис. 45. Тигель Лоуренса Смита для определения щелочных металлов.
Необходимость введения указанной поправки может быть принята на первый взгляд за недостаток метода, однако она вводится без затруднений и, если «холостой» опыт был проведен тщательно, дает точные результаты. Возможно, что не все щелочные металлы, найденные в «холостом» опыте, имеют своим источником карбонат кальция: в ходе анализа применяют большое количество горячей воды, а горячая вода извлекает весовые количества солей щелочных металлов даже из лучшего стекла.
Имеются указания1 на то, что лучшее разложение пробы достигается, если смесь карбоната кальция и хлорида аммония приготовляется в шаровой мельнице. В фарфоровую шаровую мельницу с шариками из кремня всыпают 1 часть возо-гнанного хлорида аммония и 8 частей осажденного карбоната кальция и растирают 5 ч. Эффективность действия приготовленной таким способом смеси сохраняется 12 месяцев, и в течение такого же срока не изменяется результат проводимого с этой смесью «холостого» опыта 2.
Аппаратура. Для разложения порошка минерала или горной породы лучше всего применять специально для этой цели предназначенный платиновый тигель удлиненной формы, снабженный закрывающим его колпачком (рис. 45). Такой тигель допускает применение более высокой температуры, чем это возможно с тиглями другой формы. Таким образом достигается более полное разложение порошка породы; при этом не требуется очень тонкого растирания последнего, как это было бы необходимо при применении обычных тиглей. Наиболее подходящие размеры тигля, служащего для обработки навески в 0,5 г минерала. следующие: высота 8 cjt, ширина верхнего края 1,8 СЛ1, ширина у дна 1,5 см. Для обработки 1 г минерала применяются более широкие тигли: при той же высоте ширина у верхнего края 2,5 см,
ширина у дна 2,2 см. Масса таких тиглей соответственно равна 25 г и 40 г. В настоящее время можно приобрести также и никелевые тигли такой же формы; они дают вполне удовлетворительные результаты, если колпачок тигля плотно его закрывает 3.
Прокаливание можно проводить и в закрытом тигле обыкновенной формы емкостью 20—30 мл, как это практиковалось Гиллебрандом, когда
1 М. О. La та г, W. М. Hazel, W. J. O’L е а г у, Ind. Eng. Chem., Anal. Ed., 7, 429 (1935).
2 S. R. S c h о 1 e s, J. E. Wessels [Chemist Analyst, 25, 38 (1936); W. P u-k a 11 [Sprechsaal, 66, 23 (1933)] и D. J a p h e [Sklarske Rozhl, 14, 123 (1937)] рекомендуют применение смеси CaO и CaCl2. E. M a k i n e n [Z. anorg. Chem., 74, 74 (1912)] рекомендует взамен смеси хлорида аммония и карбоната кальция применять хлорид кальция (плавленый, измельченный и сохраняемый в банке с хорошо притертой пробкой). При применении этого реагента происходит полное плавление, что, по данным автора, приводит к более совершенному разложению пробы. Для анализа минералов, содержащих сульфаты, R. Е. Stevens [Ind. Eng. Chem., Anal. Ed., 12, 413 (1940)] рекомендует применение смеси ВаС12 и СаСО3.
3 A. W. Epperson, R. В. Rudy, Ind. Eng. Chem., 17, 35 (1925).
1008
Гл. LIII. Методы, анализа силикатных пород
-он впервые начал применять метод Смита, а также и самим Смитом. Чтобы при употреблении обыкновенного тигля применить высокую температуру прокаливания без потери щелочных металлов, нужно вставить тигель в отверстие, проделанное в чистом асбестовом картоне, так, чтобы приблизительно одна четверть тигля опустилась ниже картона х. В качестве дальнейшей предосторожности крышку тигля можно заменить маленькой платиновой чашкой, плотно прилегающей к тиглю и наполненной водой.
Обработка порошка минерала. Растирают 0,5 г измельченной пробы (см. «Приготовление пробы для анализа», стр. 890) в большой гладкой агатовой ступке, пока при растирании между пальцами не перестанет ощущаться зернистость порошка 2, прибавляют 0,5 г хлорида аммония и основательно перемешивают дальнейшим растиранием. Затем прибавляют в несколько приемов почти все 4 г карбоната кальция и перемешивают все возможно тщательнее в агатовой ступке. Посыпают дно тигля небольшим количеством карбоната кальция, чтобы предотвратить прилипание массы к тиглю при прокаливании, и переносят смесь в тигель при помощи гибкого, не слишком широкого шпателя. Остатком карбоната кальция обмывают ступку и пестик, перенося все это также в тигель. Все эти операции надо делать над листом'глянцевой бумаги, желательно черной. Если проба богата железом, относительное количество карбоната кальция должно быть увеличено, чтобы предотвратить образование слишком сильно спекшейся массы или даже лепешки плава, которая трудно поддается выщелачиванию. Для получения хорошо выщелачиваемой спекшейся массы было*рекомендовано 3 прибавлять 1 г кварца и растирать его вместе с пробой. После этого массу выщелачивают водой, остаток переносят в агатовую ступку, измельчают и переносят в тот же раствор для дальнейшего выщелачивания.
Закрывают длинный тигель колпачком, осторожно постукивают по нему, чтобы порошок осел на его дно, и помещают тигель в наклонном положении в глиняный цилиндр, снабженный отверстием, отвечающим форме тигля (см. рис. 45), или же в отверстие, проделанное в листе толстого асбестового картона, поставленного в слегка наклонном положении. Нагревают часть тигля, находящуюся внутри цилиндра или выступающую книзу от картона, плоским пламенем (в виде рыбьего хвоста), находящимся значительно ниже тигля и защищенным от сквозного ветра, в течение приблизительно 15 мин или до тех пор, пока не перестанет ощущаться запах аммиака. Нагревание не должно быть настолько сильным, чтобы вызвать разложение и улетучивание хлорида аммония. Затем пламя в виде рыбьего хвоста удаляют и заменяют пламенем горелки Мекера, поднимают температуру до 1000—1100° С в течение 20—30 мин и прокаливают при этой температуре еще 90 мин.
1 Th. D б г i n g, Z. anal. Chem., 49, 158 (1910); Е. Wilke-Dorfurt, там же. 51, 755 (1912).
2 М. О. Lamar (цит. выше) утверждает, что это «перестанет ощущаться при растирании между пальцами» не может служить хорошим критерием и указывает на то, что 1) сравнимые результаты разложения можно получить только тогда, когда все Пробы измельчаются до одинаковых по размеру частиц; 2) пробы с высоким содержанием алюминия и низким содержанием кремния (например, плавленая окись алюминия) должны быть измельчены так, чтобы олп полностью проходили через сито в 200 меш (d = 0,074 мм).
3 W. Р. Е с h d a h 1, Chemist Analyist, 28, 47 (1939).
Щелочные металлы
1009
Спекшаяся или полусплавленная масса обычно легко отстает от тигля или же ее можно извлечь легким надавливанием палочкой. Когда масса остынет, переносят ее в фарфоровую чашку диаметром 12,5 сл{, наливают в тигель горячую воду и нагревают, пока все оставшееся в тигле вещество можно будет смыть в чашку. Массу в чашке обливают сначала 1—2 мл воды в течение 10—15 мин, затем приливают еще 50 мл воды и нагревают на паровой бане до полного ее распадения. Если масса из тигля не извлекается, ее выщелачивают в самом тигле. Когда масса распадется и раствор насытится гидроокисью кальция, фильтруют, декантируя через фильтр диаметром 9 см, и собирают фильтрат во вместительную чашку, желательно платиновую. Остаток, насколько возможно, оставляют в первой чашке. Прибавляют к нему еще 50 мл воды, растирают кусочки осторожным надавливанием пестиком, дают осесть и, когда раствор снова насытится гидроокисью кальция, опять декантируют. Выщелачивание повторяют еще 3 раза или более, в зависимости от содержания в пробе щелочных металлов. Если магния в пробе совсем нет или его мало, то промывают остаток на фильтре и в чашке 50—100 мл горячей воды; если магния много, промывание проводят насыщенным раствором гидроокиси кальция.
Остаток после выщелачивания должен полностью раствориться в соляной кислоте, не оставляя даже следов неразложенного минерала, хотя иногда несколько черных частиц железной руды или других руд, если они присутствовали в анализируемой пробе, растворяются медленно. При тщательной работе редко случается, чтобы остаток содержал еще щелочные металлы, вопреки утверждению некоторых химиков *, считающих, что при выполнении анализа этим методом теряется от 2 до 3 мг этих элементов. Неудовлетворительные результаты, полученные ими, могли иметь своей причиной недостаточно высокую температуру при спекании или слишком грубое измельчение порошка пробы; нам же редко приходилось проводить повторную обработку 1 2. В сомнительных случаях остаток следует высушить, смешать еще раз с хлоридом аммония, но без прибавления карбоната кальция, и повторпть обработку. В этом случае водную вытяжку из спекшейся массы следует обрабатывать отдельно от того момента, когда будет удален весь кальций.
Удаление кальция, и сулъфат-ионов. Раствор, полученный в результате обработки, описанной в разделе «Обработка порошка минерала» (стр. 1008), содержит большое количество кальция, большее или меньшее количество сульфатов и боратов, если они были в пробе, и иногда магний при высоком его содержании в анализируемом веществе. В последнем
1 J. W. Mellor, Quantitative Inorganic Analysis, p. 225, M. Dittrich, Neues Jahrb., 2, 69 (1903); M. F. Connor, Congr. Geol. Intern. 12 Sess. Canada^ 885 (1913).
2 Например, J. I. Hoffman, исследуя полевой шпат, содержавший 12,58% К2О и 2,37% Na2O и огнеупорные материалы, содержавшие от 1,43 до 2,90% К2О и от 0,36 до 0,89% Na2O, получил совершенно одинаковые результаты как при однократной, так и при двукратной обработке. Н. В. Knowles подобные же результаты получил при исследовании известково-натриевого стекла, содержавшего 16,63% Na2O и 0,04% К2О. Однако при анализе диорита он получил 3,55% К2О при однократном сплавлении и еще 0,08%, или всего 3,63%, при двукратном сплавлении. Для Na2O однократная обработка дала 4,16%, а двукратная еще 0,05%, всего 4,21%. Следует отметить, что спекшаяся масса, получающаяся при анализе огнеупорных материалов с высоким содержанием алюминия, с трудом разлагается водой.
64 Заказ 522.
1010
Гл. LIII. Методы анализа силикатных пород
случае, чтобы быть уверенным в удалении магния, выпаривают раствор приблизительно до 100 мл, фильтруют, промывают остаток насыщенным раствором гидроокиси кальция, обрабатывают фильтрат, как будет описано ниже, и присоединяют остаток к первоначальному остатку, если этот последний будет проверяться на возможное содержание в нем щелочных металлов.
Кальций удаляют осаждением карбонатом аммония и аммиаком при температуре кипения в большой чашке (если возможно, платиновой), которую ладо держать покрытой часовым стеклом ввиду разбрызгивания, вызываемого разложением карбопата аммония в горячем растворе. Фильтруют, промывают осадок горячей водой, снова растворяют его в соляной кислоте, взятой в небольшом избытке, и повторяют осаждение и фильтрование. Соединенные фильтраты выпаривают досуха, осторожно удаляют аммонийные соли при температуре, близкой к темно-красному калению, растворяют остаток в 25 мл воды и осаждают последние следы кальция прибавлением нескольких кристаллов оксалата аммония и 1—2 капель раствора аммиака. Оставляют стоять на краю паровой бани по крайней мере на 30 мин, затем фильтруют через маленький фильтр и промывают осадок 0,1 %-ным раствором оксалата аммония, собирая фильтрат в платиновый тигель (его взвешивать не надо) или в маленькую чашку. Выпаривают жидкость досуха и прокаливают остаток при температуре, близкой к темно-красному калению. Затем смачивают остаток каплей соляной кислоты, чтобы перевести в хлориды те карбонаты, которые могли образоваться от действия оксалата аммония при прокаливании, снова выпаривают, покрывают чашку часовым стеклом, нагревают при 110° С в течение 30 мин и затем осторожно (не снимая часового стекла) продолжают нагревание, медленно водя вокруг чашки голым пламенем до начала плавления солей.
Охлаждают в эксикаторе и взвешивают х. Затем растворяют соли в очень малом количестве воды, переносят нерастворимый остаток (несколько десятых долей миллиграмма) на маленький фильтр и промывают, собирая фильтрат в фарфоровую 1 2 чашку диаметром 7,5 см. Фильтр с остатком сжигают в том же тигле или чашке, в котором прокаливали и взвешивали хлориды, снова прокаливают и взвешивают и таким образом по разности находят массу всех хлоридов щелочных металлов. При этом может быть взвешен и хлорид магния, попавший в фильтрат, несмотря па все операции его отделения. Если в этом отношении возникнут сомнения,
1 Очень трудно удалить весь хлорид аммония из большого количества хлоридов щелочных металлов и избежать при этом улетучивания последних. В таких случаях лучше растворить взвешенные хлориды в воде, снова выпарить раствор, прокалить остаток и взвесить. Прокаливание не следует проводить при температуре выше 500° С. Хлорид калия медленно улетучивается даже из не совсем сплавленной соли. Хлорид лития очень гигроскопичен, хлорид натрия — слегка гигроскопичен. Если присутствует литий, смесь хлоридов надо во время взвешивания защищать от возможности проникновения влаги. Хлориды калия, рубидия и цезия не гигроскопичны, если они чисты и хорошо высушены. Следы примесей, например хлорида магния, могут заметно увеличить их гигроскопичность. Отмечено [G. F. Smith, F. М. Stubblefield, Е. В. М i d d 1 е t о n, Ind. Eng. Chem., Anal. Ed., 6, 314 (1934)], что смесь хлоридов удерживает некоторое количество воды даже при 550° С; нагревать надо до плавления соли, но не допускать улетучивания.
2 Фарфоровая чашка лучше, чем платиновая, потому что в некоторых, пока не выясненных, условиях может образоваться нерастворимое соединение платины (II). См. также F. В о 1 m, Z. anal. Chem., 38, 349 (1899).
Щелочные металлы
1011
следует проделать соответствующие испытания и ввести поправку (см. стр. 733). Сказанное относится и к проведению «холостого» опыта.
Если анализируемая проба содержит серу, то последняя будет находиться, полностью или частично, вместе с хлоридами щелочных металлов в виде сульфат-ионов. Поэтому если сера присутствует в заметном количестве, ее нужно удалить прибавлением соответствующего количества 10%-ного раствора хлорида бария (одной капли его часто более чем достаточно) перед вторым осаждением кальция. Избыток прибавленного бария затем удаляют карбонатом аммония, а остаток кальция — оксалатом аммония, как указано выше. Если серу не удалить таким образом, то масса хлоридов будет несколько выше истинной, и в дальнейшем ходе анализа осадок хлороплатината калия почти наверняка будет загрязнен сульфатом натрия. Слабая реакция на сульфат-ионы обычно получается всегда, когда выпаривание проводят на водяной бане, обогреваемой газом.
Метод Берцелиуса
Общие замечания.. Для обработки пробы методом Берцелиуса — смесью фтористоводородной и серной кислот, пробу, как правило, можно не измельчать так тонко, как этого требуется для обработки методом Смита. Метод Берцелиуса, т. е. разложение фтористоводородной кислотой (серная кислота здесь применяется только для превращения образующихся фторидов и фторосиликатов в сульфаты), применим для анализа большей части силикатов.
К числу сравнительно немногих силикатов, разлагаемых этим способом не полностью, можно отнести андалузит, топаз и некоторые разновидности турмалина. Шпинель, графит и пирит, которые находятся в некоторых горных породах, также трудно разлагаются, но они не являются силикатами и не содержат щелочных металлов, поэтому их присутствием можно пренебречь, если они будут обнаружены. Для анализа первых трех упомянутых выше минералов Яннаш рекомендует сильно прокалить порошок минерала в платиновом тигле, разложить его сплавлением с фторидом аммония, удалить избыток последнего прокаливанием и превратить фториды в сульфаты нагреванием с серной кислотой *.
Для анализа минералов, полностью разлагаемых фтористоводородной кислотой, применяют разложение и первую часть последующей обработки, описанные ниже.
Ход анализа составлен в предположении, что присутствуют элементы, требующие применения всех перечисленных ниже операций, как, например, при анализе силикатных горных пород. При отсутствии некоторых элементов ход анализа может быть сокращен, но это условие встречается редко.
Разложение. Около 1 г измельченной пробы обрабатывают в платиновом тигле или маленькой чашке 5 мл разбавленной (1:5) серной кислоты, перемешивают шпателем или толстой платиновой проволокой, прили
1 А. С. S h е a d и G. F. Smith [J. Am. Chem. Soc., 53, 483 (1931)] рекомендуют для разложения указанных устойчивых силикатов сплавление их с фторидом аммония. Этот же метод предлагается для определения (по разности в массе до и после обработки) кремнекислоты в песках, идущих для производства стекла.
64*
1012
Гл. LIII. Методы анализа силикатных пород
вают 5 мл чистой концентрированной фтористоводородной кислоты 1 и выпаривают, пока не начнется испарение серной кислоты. Если на этой стадии анализа еще ощущаются при перемешивании платиновым шпателем крупные зернышки породы, то повторяют выпаривание с большим количеством фтористоводородной кислоты, пока минерал полностью не разложится. Затем удаляют большую часть серной кислоты нагреванием в радиаторе (стр. 49). Удаление фтора должно быть проведено тщательно, для чего может оказаться необходимым прибавление еще небольшого количества серной кислоты и вторичное удаление большей ее части. Было предложено 2 растворять остаток сульфатов в разбавленной кислоте до начала второго выпаривания; автор утверждает, что таким образом можно достигнуть полного удаления фтора двукратным выпариванием.
Следует помнить, что долго после того, как фтор уже нельзя обнаружить по запаху, образование корки на поверхности медленно испаряющегося раствора иногда вызывает накопление под ней такого количества фтористого водорода, что присутствие его становится ясно заметным по своеобразному острому запаху при пробивании этой корки.
Начиная с этой стадии, обработку можно вести различными путями. 1) Прежде аналитики не удаляли серную кислоту настолько, чтобы могли образоваться окислы или основные сульфаты, но оставляли остаток влажным, чтобы при нагревании с водой образовался прозрачный раствор 3. Некоторые прибавляли при этом немного соляной кислоты, чтобы облегчить растворение. 2) Чтобы сократить время, требуемое для последовательного удаления всех элементов, кроме щелочных металлов, Лоу 4 предложил нагревать остаток сульфатов почти до прекращения выделения паров серной кислоты, а Кришнайя 5 — пока выделение последних не прекратится совсем. При этом окислы и соли железа, алюминия, титана и фосфора (из обычно встречающихся элементов) становятся нерастворимыми в воде.
Дальнейшая обработка. Обычный способ. Получив прозрачный раствор остатка после выпаривания, разные аналитики ведут дальнейшую обработку по-разному. Чаще всего, вероятно, применяется метод Бунзена, который в описании Диттриха, когда определяют только щелочные металлы, в основных чертах заключается в следующем.
Осаждают сульфат-ионы раствором хлорида бария, прибавляемым в небольшом избытке, выпаривают жидкость, не фильтруя, досуха, обрабатывают остаток небольшим количеством горячей воды и прибавляют свободную от щелочных металлов гидроокись бария до появления щелочной реакции. Снова выпаривают досуха, не фильтруя, чтобы сделать гидро
1 В США концентрированную чистую фтористоводородную кислоту можно получать в церезиновых или полиэтиленовых сосудах. При отсутствии такой чистой кислоты можно перегнать продажную кислоту из платиновой реторты или провести с нею «холостой» опыт для определения содержания в ней щелочных металлов. Небольшое количество органических веществ в нёй здесь не мешает, как это имеет место при определении железа (II).
2 См. сноску 3 на стр. 939.
3 Если нет бария и других щелочноземельных металлов, сульфаты которых остаются в виде белого остатка. Если такой остаток получится, его отфильтровывают, промывают холодной водой и отбрасывают. Или если остаток этот должен послужить для определения бария, его либо взвешивают в виде BaSO4, если он чист, либо очищают, если он загрязнен.
4 А. Н. Low, Chem. News, 67, 185 (1893).
3 Н. V. Krishnayya, Chem. News, 107, 100 (1913).
Щелочные металлы
1013
окись магния менее растворимой, перемешивают остаток с горячей водой, фильтруют и промывают остаток разбавленным раствором гидроокиси бария, не содержащим щелочных металлов.
Удаление магния происходит неполностью вероятно потому, что осажденная гидроокись магния частично растворяется в промывной жидкости. Возможно, что гидроокись кальция в виде известкового молока была бы лучшим осадителем, чем гидроокись бария. Рекомендуется Применить следующий способ. К нейтральному водному раствору хлоридов или сульфатов, находящемуся в мерной колбе емкостью 125 мл, прибавляют каплю фенолфталеина и затем столько известкового молока (свободного от щелочных Металлов), чтобы получился раствор, насыщенный гидроокисью кальция. Наполняют колбу до метки водой и тщательно перемешивают. Раствор должен быть интенсивно красным, что указывает на насыщение. Через полчаса фильтруют через сухой фильтр в сухой сосуд. На то, что известкового молока было прибавлено достаточное количество, указывает появление пленки карбоната кальция на поверхности жидкости в фильтрате и в воронке. К аликвотной части раствора, взятой без учета объема, занимаемого осадком, например к 100 мл, прибавляют щавелевую кислоту, нагревают и затем медленно приливают аммиак до небольшого его избытка. Оставляют стоять 1—2 ч и фильтруют. При определении большого количества щелочных металлов осадок следует растворить, снова осадить и фильтраты соединить вместе.
Фильтрат нагревают и осаждают большую часть избытка бария аммиаком и карбонатом аммония 1 2. Нагревают полчаса, фильтруют, осадок промывают, выпаривают фильтрат досуха в платиновой чашке и удаляют последние следы аммонийных солей осторожным прокаливанием. Остаток обрабатывают горячей водой, снова осаждают аммиаком и карбонатом аммония, фильтруют и т. д., повторяя все эти операции, пока не перестанет появляться осадок карбоната бария. Затем окончательно полученный фильтрат выпаривают досуха, смачивают остаток каплей соляной кислоты, выпаривают, очень осторожно прокаливают и взвешивают хлориды щелочных металлов, которых еще могут содержать немного магния в виде основного хлорида. Этот последний образуется из хлорида магния при его разложении во время предшествующего прокаливания. Если при растворении хлоридов в небольшом количестве воды будет обнаружен остаток, его отфильтровывают, прокаливают, взвешивают и вычитают найденную массу из веса смеси хлоридов. Оставшийся еще в растворе хлорид магния можно определить позже и массу его вычесть.
Варианты метода. Рекомендуют 3 после нагревания сульфатов до прекращения выделения паров серной кислоты разбивать плотный остаток солей, кипятить их с небольшим количеством воды несколько минут и затем, не фильтруя, продолжать анализ обычным методом. Опубликованные результаты превосходно согласуются с результатами, полученными при применении обычной обработки. Лоу 4 рекомендует нагревать сульфаты почти до исчезновения паров серной кислоты, смачивать остаток небольшим количеством концентрированного аммиака и кипятить. Осадок от аммиака в этих условиях получается компактным и хо
1 Н. Neubauer, Z. anal. Chem., 43, 14 (1904).
2 Чтобы определить эффективность этого метода, когда присутствует заметное количество лития, Н. В. Knowles провел три определения с растворами, содержавшими 0,0035 г Li2O, 0,01 г СаО; 0,005 г MgO и 0,2 г А12О3. Он нашел 0,0035 г, 0,0036 г и 0,0037 г Ы2О и установил спектроскопически, что все выделенные в ходе анализа осадки были свободны от лития. Он нашел также, что литий не захватывается ни алюминием, когда его осаждают аммиаком пз раствора хлоридов или сульфатов, ни магнием, когда последний осаждают в виде фосфата магния и алюминия в холодном растворе.
3 Н. V. Krishnayya, цит. выше.
4 А. Н. Low, цит. выше.
1014
Гл. LIII. Методы, анализа силикатных пород
рошо отфильтровывается. Когда плотный остаток в чашке вполне размягчится и распадется, его отфильтровывают, промывают небольшим количеством горячей воды, подкисляют фильтрат соляной кислотой и продолжают анализ по обычному методу, минуя, однако, обработку гидроокисью бария. Это упрощение можно допустить для скорых и не очень точных анализов, но его очень трудно оправдать, если анализируемое вещество содержит очень большое количество магния или если нужны очень точные результаты, несмотря на хорошее совпадение результатов двух параллельных определений, приведенных в работе Лоу х.
Для определения калия в силикатах был предложен следующий метод 1 2. Пробу разлагают фтористоводородной и хлорной кислотами, раствор выпаривают до появления паров хлорной кислоты, проводят отгонку, как описано на стр. 824, для полного удаления фтора, выпаривают до удаления воды и большей части хлорной кислоты, после чего в безводном растворе этилацетата осаждают калий в виде его перхлората. То незначительное количество фтора, которое может остаться после двух выпариваний с хлорной кислотой, не мешает определению натрия методом осаждения в виде тройного ацетата. В этом случае отгонку можно опустить и проводить осаждение прямо в водном растворе того остатка, который остается после вторичного обезвоживания с хлорной кислотой. (Последнее надо остановить, когда остаток еще влажный.)
Для определения щелочных металлов в полевых шпатах было рекомендовано 3 сначала проводить разложение одной лишь фтористоводородной кислотой, затем выпаривать досуха, растворять остаток в воде и осаждать окисью кальция алюминий, железо, магний, фтор и оставшийся кремний. После фильтрования и промывания осадка горячей водой большую часть кальция удаляют из фильтрата осаждением карбонатом аммония и фильтрованием. Для удаления оставшейся части кальция проводят осаждение оксалатом аммония и фильтруют. Фильтрат снова подкисляют соляной кислотой, выпаривают досуха и сухой остаток осторожно прокаливают для удаления аммонийных солей, как обычно. В этом методе, как и в методе Смита, массу смеси хлоридов лучше находить, обрабатывая остаток небольшим количеством воды, фильтруя раствор во взвешенную платиновую чашечку, промывая остаток горячей водой, выпаривая досуха, прокаливая до начала плавления солей, охлаждения и снова взвешивая.
Специальные методы отделения магния. Для отделения магния разработаны некоторые специальные методы, которые описаны в гл. «Магний» (стр. 715).
ДВУОКИСЬ УГЛЕРОДА; УГЛЕРОД
Качественное испытание
При предварительном качественном испытании на двуокись углерода надо помнить о том, что кальцит выделяет двуокись углерода уже при обработке холодной кислотой, в то время как доломит и сидерит в этих условиях двуокиси углерода не выделяют. Поэтому нельзя пренебрегать
1 А. Н. Low, цит. выше.
2 Н. Н. Willard, L. М. Liggett, Н. Diehl, Ind. Eng. Chem., Anal. Ed., 14, 234 (1942).
3 F. W. Koenig, Ind. Eng. Chem., Anal. Ed., 7, 314 (1935).
Двуокись углерода", углерод
1015
нагреванием, так как иначе легко можно не уловить нескольких десятых процента двуокиси углерода. Кроме того, порошок породы, перед обработкой его соляной кислотой, надо предварительно взболтать с небольшим количеством горячей воды, чтобы совершенно удалить приставшие пузырьки воздуха, иначе воздух ошибочно можно принять за СОг.
Чтобы не пропустить даже следов СО 2, опыт лучше всего проводить в пробирке: кипятят порошок сначала с водой, охлаждают и приливают разбавленную соляную кислоту. Если сразу произойдет вспенивание, то присутствие кальцита можно считать доказанным. Если выделение пузырьков начинается только при нагревании, то кальцита нет, а присутствуют другие карбонаты. Для того чтобы сделать пробу совершенно отчетливой, наблюдают в лупу, не поднимаются ли вдоль верхней стенки косо поставленной пробирки крошечные пузырьки. Конечно, не следует принимать за двуокись углерода выделяющийся сероводород.
Количественное определение
Для количественного определения двуокиси углерода применяют собранный для постоянного пользования прибор, различные видоизменения которого описаны многими авторами. Одна из форм его приведена в гл. «Углерод и водород» (стр. 848).
В обычном случае помещают точную навеску от 1 до 5 г порошка породы в колбу и заливают горячей водой. Колбу закрывают пробкой со вставленными в нее делительной воронкой и холодильником и присоединяют последний к поглотительной цепи, не включая пока взвешенного поглотителя. Пропускают через систему воздух, свободный от двуокиси углерода, до полного удаления СОг из всей системы, закрывают кран делительной воронки п вводят в цепь взвешенный поглотитель. Наполняют делительную воронку до ее половины разбавленной (1 : 1) соляной кислотой, снова вставляют пробку с трубкой, приводящей воздух, и наблюдают, чтобы воздух свободно проходил через всю систему. Открывают кран делительной воронки и наливают в колбу кислоту медленно, если СО2 много, и быстро — если ее мало. Когда выделение пузырьков уменьшится в первом случае, и тотчас же по прибавлении кислоты — во втором случае, зажигают горелку и пускают воду через холодильник. Нагревание регулируют так, чтобы жидкость кипела спокойно и непрерывно, и уменьшают ток воздуха, но не прекращают его совсем. Когда можно будет считать, что вся двуокись углерода выделилась, т. е. через 5—10 мин кипячения, прекращают нагревание, увеличивают ток воздуха и вытесняют всю СО2 во взвешенный поглотитель. Затем объединяют поглотитель из цепи, останавливают ток воздуха и помещают поглотитель в шкафчик весов. Когда он охладится, открывают его на мгновение и взвешивают, тарируя другим таким же поглотителем.
Об одновременном определении карбонатов и углерода, .содержащегося в углистых веществах, см. ниже.
Как уже было указано в разделе «Вода» (стр. 914), в случае необходимости, например при недостатке материала для анализа, определение двуокиси углерода можно соединить с определением воды сплавлением породы с хроматом свинца (при отсутствии свободного углерода и органических веществ). При проведении таких определений поглотительные трубки для СОг помещают вслед за хлоркальциевой трубкой.
1016
Гл. LIII. Методы, анализа силикатных пород
Было отмечено \ что некоторые минералы, содержащие двуокись углерода, например скаполиты, разлагаются соляной кислотой с трудом. Когда такие минералы содержатся в породе, прибавляют к соляной кислоте 30 мл смеси из 1 части концентрированной плавиковой кислоты и 4 частей воды. Через хлорид кальция, перед его применением, следует пропустить хлористый водород, чтобы нейтрализовать щелочь, которая в нем может быть, и предотвратить поглощение ею двуокиси углерода.
Определение углерода
Для определения углерода графита или углистых веществ в породах и рудах обычно применяют сплавление с хроматом свинца; можно получить хорошие результаты и методом кипячения пробы со смесью хромовой и серной кислот.
Если одновременно присутствуют карбонаты, то они могут быть определены, как описано выше (стр. 1015); затем в другой навеске пробы определяют все содержание углерода (присутствовавшего в обеих формах) в виде СО 2 и рассчитывают содержание графита по разности. Можно также 1 2 разложить карбонаты (как описано в разделе «Количественное определение», стр. 1015), применяя ортофосфорную кислоту вместо соляной кислоты, после чего прибавить хромовую кислоту, окислить углерод и взвесить выделившуюся СО2.
Для определения углерода и водорода горючих веществ, содержащихся в сланцах, глинах и известняках, был предложен метод 3, который, по-видимому, дает вполне удовлетворительные результаты. В этом методе не принимаются в расчет летучие и растворимые компоненты горючих веществ, которые могут быть потеряны во время предварительной обработки породы.
Ход анализа следующий: навеску от 0,2 до 1 а тонко растертой породы помещают в платиновую чашку емкостью 100 мл, приливают 15 мл соляной кислоты и нагревают, не доводя до кипения, на горячей плитке в течение 10 мин. Затем приливают 20 льг фтористоводородной кислоты и нагревают при температуре начала кипения, пока не разложатся неорганические вещества пробы. На это требуется обычно от получаса до двух часов. Объем жидкости должен быть больше 15 мл, для этого время от времени прибавляют смесь соляной и фтористоводородной кислот в отношении 3 : 4. Когда разложение закончится, выпаривают раствор до объема 15 мл, прибавляют 25 мл соляной кислоты и нагревают до кипения. К кипящему раствору прибавляют равный объем горячей воды, снова нагревают до кипения и фильтруют горячим через фильтр из прокаленного асбеста или губчатой платины. Остаток промывают горячей водой до исчезновения реакции на хлор. Затем переносят фильтр с остатком в фарфоровую или платиновую лодочку, высушивают 2 ч при 105° С и быстро переносят в трубку для сожжения (см. гл. «Углерод и водород», стр. 853) так, чтобы вещество не могло снова поглотить влагу. Содержание углерода и водорода определяют обычным способом.
1 L. Н. Borgstrom, Z. anal. Chem., 53, 685 (1914).
2 G. T. Morgan, J. Chem., Soc. 85, 1001 (1904).
3 A. C. F i e 1 d n e r, W. A. Selv ig, G. В. T а у 1 о r, Bur. Mines Tech. Paper., 212, 1919.
Хлор
1017
ХЛОР
Состояние хлора в горных породах
Хлор может содержаться в породах в растворимой в воде форме, в составе минералов, разлагаемых азотной кислотой, и в минералах, которые этой кислотой не разлагаются. Растворимая в воде форма несомненно является поваренной солью, отложившейся в породе или представляющей первичное включение в одном или нескольких минералах, входящих в состав породы.
В растворимой в азотной кислоте форме хлор находится в минералах содалитовой группы и иногда в апатите; в нерастворимой в азотной кислоте форме хлор присутствует главным образом в скаполитах. Если качественной пробой обнаруживается в водной вытяжке определимое количество хлора, то нужно растворимый в воде хлор определить отдельно и вычесть из найденного в другой навеске пробы общего количества хлора. Иногда можно пробу после выщелачивания водой применить для определения в ней остального хлора.
Определение растворимого в воде хлора
Достаточную по величине навеску измельченной в порошок пробы, иногда несколько граммов, выщелачивают водой. Фильтрат часто оказывается мутным; этого во многих случаях можно избежать, если применить двойной фильтр или прибавить к промывной жидкости немного какой-нибудь соли, не содержащей хлора, например нитрата натрия. Если, несмотря на эти меры, фильтрат все же остается мутным, можно, не обращая внимания на муть, осадить хлор в виде хлорида серебра обычным методом после подкисления азотной кислотой. Затем раствор оставляют на ночь (чтобы фильтрат получился прозрачным) и фильтруют. Осадок загрязненного первоначальной мутью хлорида серебра собирают на маленьком фильтре, промывают водой, слабо подкисленной азотной кислотой, высушивают и озоляют в маленьком фарфоровом тигле так, чтобы фильтр не воспламенился. Большая часть хлорида серебра при этом восстанавливается до металла. После полного выгорания угля прибавляют к золе каплю азотной кислоты, выпаривают, затем прибавляют каплю соляной кислоты, осторожно нагревают тигель (не до плавления хлорида серебра) и, наконец, взвешивают. После этого хлорид серебра растворяют в нескольких каплях горячего раствора аммиака и отделяют фильтрованием примеси силикатов. Последние прокаливают и взвешивают, чтобы определить таким образом истинную массу хлорида серебра.
Этот же ход анализа можно применять и тогда, когда первоначальная водная вытяжка совершенно прозрачна, только тогда все заканчивается взвешиванием хлорида серебра.
Определение хлора, переходящего в раствор при обработке кислотами
Обработка азотной кислотой. Если азотная кислота разлагает все хлорсодержащие минералы или желательно определить отдельно хлор, переходящий в раствор при обработке кислотами, и хлор, остающийся в нерастворимом остатке, то кипятят измельченную в порошок породу несколько минут с разбавленной, свободной от хлора азотной кислотой.
-1018
Гл. LIII. Методы, анализа силикатных пород
Азотная кислота должна быть сильно разбавленной, чтобы не произошла потеря свободного хлора. По той же причине, а также, чтобы избежать желатинирования кремнекислоты, освобождающейся из растворимых в кислотах силикатов, следует кипятить по возможности короткое время. Разбавленная (1 : 40) азотная кислота легко разлагает апатит и, вероятно, также минералы содалитовой группы. Но необходимо ли действительно такое сильное разбавление, пока еще не доказано.
Фильтрат не надо выпаривать досуха для удаления растворенной кремнекислоты, его можно сразу же осадить нитратом серебра. Выделенный осадок обрабатывают, как указано в разделе «Определение растворимого в воде хлора» (стр. 1017), или же, если количество его достаточно велико, можно собрать его в тигле Гуча.
Обработка азотной и фтористоводородной кислотами. Во многих случаях, чтобы определить весь содержащийся в породе хлор, достаточно разложить навеску пробы (которую обычно для этой цели надо очень тонко измельчить) азотной и плавиковой кислотами (свободными от хлора) на холоду при перемешивании. После фильтрования через бумажный фильтр, вставленный в резиновую воронку или в поместительный платиновый конус, осаждают хлориды нитратом серебра. Присутствие азотной кислоты необходимо, потому что без нее железо (II) в присутствии фторид-ионов восстанавливает нитрат серебра с выделением металлического серебра. Осадок хлорида серебра рекомендуется растворять на фильтре в аммиаке и снова осаждать его добавлением азотной кислоты и одной капли нитрата серебра.
Определение хлора сплавлением со щелочами
Вследствие указанных выше затруднений для большей уверенности в определении всего хлора, содержащегося в анализируемой породе, лучше порошок ее сплавить со свободным от хлора карбонатом калия-натрия или даже с одним карбонатом натрия. Сначала нагревают полным пламенем обыкновенной горелки, затем в течение 1—2 мин пламенем паяльной горелки. Полученный плав выщелачивают горячей водой. Раствор затем охлаждают, прибавляют метиловый оранжевый и слабо подкисляют азотной кислотой. Если раствор совершенно прозрачен, то проводят осаждение хлоридов. Если раствор непрозрачен, то оставляют его на ночь, прибавляют аммиак в небольшом избытке, кипятят, фильтруют и промывают осадок горячей водой. Фильтрат охлаждают, подкисляют азотной кислотой и осаждают хлориды Ч
ФТОР (КРЕМНЕКИСЛОТА В ПРИСУТСТВИИ ФТОРА)
Общие замечания
Не существует, к сожалению, прямой качественной пробы, которая с уверенностью указывала бы на присутствие или отсутствие фтора в породах. Известные качественные пробы: 1) прокаливание при помощи паяльной трубки смеси испытуемого порошка с метафосфатом натрия, помещен-
1 Согласно Е. W i 1 k e-D б г f и г t [Z. angew. Chem., 40, 1478 (1927)], иод, содержащийся в породах, можно отогнать в виде 12 или HI обработкой измельченной породы серной кислотой в приборе, состоящем только из стеклянных частей, притертых друг к другу, и нагреванием в течение 3 ч при температуре 220—230° С при пропускании через жидкость тока воздуха.
Фтор
1019
ной на согнутом кусочке платиновой жести, вставленном в один конец узкой стеклянной трубочки, или 2) прокаливание в закрытой с одного конца трубке и суждение о присутствии или отсутствии фтора по разъеданию стекла в том месте, где при нагревании конденсируется вода (после высушивания трубки), — не всегда дают надежные результаты. В некоторых горных породах этими способами легко можно открыть малые количества фтора (до 0,1 %), в других породах гораздо большие количества фтора часто остаются неоткрытыми.
Так же обстоит дело и с методами прямого количественного определения фтора. Метод Берцелиуса почти всегда дает пониженные результаты и может даже показать отсутствие фтора при малом его содержании. Для определения малых количеств фтора (меньше 10 мг) химики, работавшие в этом направлении, предпочитают поэтому непрямые методы определения, один из которых описан далее (см. «Непрямое определение малых количеств фтора» (стр. 1025). Поскольку предварительная обработка породы и дальнейшее ее исследование до известного момента одинаковы при применении обоих методов и так как одним методом можно воспользоваться для контроля другого, мы описываем дальше подробно оба метода определения.
Большинство фторсодержащих минералов, встречающихся в горных породах, разлагается серной или хлорной кислотой только отчасти, а многие минералы совсем не разлагаются. Поэтому ни один из методов, основанных на отгонке фтора после разложения этими кислотами, не может быть использован непосредственно для определения фтора в породе без предварительного ее сплавления. При анализе разлагаемых кислотами фторидов или после сплавления пробы с карбонатом натрия фтор можно определить отгонкой, но если присутствует аморфная кремнекислота, ее надо предварительно удалить (стр. 824) или же собрать большее количество дистиллята, чем это необходимо, когда кремнекислота присутствует в виде порошка кварца (стр. 822). Обычно применяют метод Берцелиуса, по которому пробу сплавляют со смесью карбонатов натрия и калия и экстрагируют фтор из плава водой. Этот способ дает возможность одновременно определять и кремнекислоту. Следует, однако, отметить, что при анализе таких материалов, которые одновременно со фтором содержат большие количества кальция и фосфат-ионов г, например фосфатных пород, таким способом полностью экстрагировать фтор не удается. При анализе этих пород, по-видимому, единственным способом отделения фтора является его отгонка.
Прямое определение фтора
Метод Берцелиуса. Ход определения. Сплавляют 2 г измельченной в порошок породы с четырех- или пятикратным количеством свободного от фтора карбоната калия-натрия, по возможности избегая нагревания на паяльной горелке (см. стр. 927). При анализе минералов с высоким содержанием фтора и низким — кремния для полного разложения фторидов может оказаться необходимым прибавление перед сплавлением чистой кремнекислоты.
1 D. S. Re yn olds, К. D. I а с о Ь, Ind. Eng. Chem., Anal. Ed., 3, 366 (1931) и J. I. Hoffman, G. E. F. L u n d e 11, J. Research NBS, 20, 607 (1938).
1020
Гл. LIII. Методы анализа силикатных пород
Однако при обычном анализе горных пород такого прибавления не требуется. Плав выщелачивают 100 мл воды, фильтруют и промывают остаток горячей водой. Если требуется очень большая точность или количество фтора велико, сохраняют нерастворимый остаток и прокаливают его вместе с осадками, полученными далее от карбоната аммония и окиси цинка. К водной вытяжке плава, содержащей фтор и обычно значительную часть кремнекислоты (а также и другие соединения), прибавляют 2—5 г карбоната аммония, дают постоять некоторое время при умеренной температуре (40° С) и, охладив, прибавляют еще немного карбоната аммония х.
После 12-часового стояния осадок отфильтровывают и промывают водой, содержащей карбонат аммония. Фильтрат освобождают от избытка последнего выпариванием почти досуха и немного разбавленный раствор доводят по предложению Тредвелла почти до нейтрального состояния следующим образом. К раствору прибавляют несколько капель фенолфталеина, затем азотной кислоты (не соляной, см. ниже), пока не исчезнет розовая окраска. Кипятят раствор, охлаждают и снова прибавляют кислоту до исчезновения окраски. Это делают до тех пор, пока не потребуется добавление только 1—1,5 мл 2 н. раствора кислоты, чтобы окраска исчезла. Наконец, прибавляют 1—2 мл аммиачного раствора карбоната цинка и кипятят жидкость до полного удаления аммиака. Осадок содержит остаток кремнекислоты и некоторое количество фосфора, не осевшее вместе с алюминием при осаждении карбонатом аммония. Фильтруют и промывают осадок 2%-ным раствором нитрата калия.
Аммиачный раствор карбоната цинка приготовляют растворением окиси цинка в растворе, содержащем аммиак и карбонат аммония. При анализе пород сильноосновного характера содержание кремнекислоты в растворе щелочного плава может быть так мало, что прибавление карбоната аммония можно опустить и сразу после нейтрализации раствора описанным выше способом прибавить раствор окиси цинка. Чем меньший избыток окиси цинка при этом вводится, тем лучше. Вместо аммиачного раствора окиси цинка рекомендуют 1 2 раствор Шаффготша 3 (* 230 г карбоната аммония растворяют в 180 мл раствора аммиака пл. 0,92 г! см3 и разбавляют до 1 л *), в 1 л которого на холоду растворены 20 г свежеосажденной HgO. На каждые 0,2 г растворенной SiOa надо брать 100 мл этого раствора. Затем раствор выпаривают досуха, остаток растворяют в воде, фильтруют и промывают; фильтрат снова нейтрализуют соляной кислотой (при анализе горных пород — азотной кислотой) и снова выпаривают после прибавления реактива, чтобы удалить последние остатки кремнекислоты. Там, где можно на этой стадии анализа определить кремнекислоту, данный реактив имеет перед окисью цинка то преимущество, что простым прокаливанием осадка (под хорошей тягой!) непосредственно находят содержание кремнекислоты. В анализе горных пород его применение, очевидно, возможно только в тех случаях, когда алюминий предварительно удален осаждением карбонатом аммония.
Указанное выше применение азотной кислоты для нейтрализации вместо соляной кислоты основано на том, что необходимо еще удалить фосфор, который присутствует почти всегда, и хром, а это можно сделать только в азотнокислом растворе следующим образом. К еще щелочной жидкости приливают в избытке нитрат серебра; выпадают фосфат, хромат и хлорид серебра (последний, если присутствовали хлорид-ионы), а также
1 Для выделения кремнекислоты и алюминия нельзя рекомендовать применение нитрата или хлорида аммония вместо карбоната аммония из-за потери фтора при последующем выпаривании (Н. Rose).
2 F. S eemann, Z. anal. Chem., 44, 343 (1905).
3 Schaffgotsch, U. S. Geol. Survey Bull., 700, p. 213.
Фтор
1021
гидроокись или карбонат серебра. В результате реакции между кислыми фосфатами и нитратом серебра могла бы выделиться свободная кислота, но она будет нейтрализована карбонатом серебра или его гидроокисью, а сохранение нейтральной реакции необходимо для полноты осаждения хромата и фосфата серебра.
По осаждении слегка нагревают раствор, фильтруют и промывают осадок небольшим количеством воды. Для удаления избытка серебра из фильтрата добавляют хлорид натрия, фильтруют и промывают осадок небольшим количеством воды. К фильтрату приливают 2 мл 2 н. раствора карбоната натриях, выпаривают, чтобы уменьшить объем раствора до 100 мл, и, если нужно, фильтруют. Прибавляют к горячему раствору по каплям 15 мл 20%-ного раствора хлорида кальция СаСП-бНзО (достаточно для осаждения 0,1 г фтора), нагревают почти до кипения в течение нескольких минут и охлаждают раствор. В растворе теперь не должно быть никаких аммонийных солей, иначе фторид кальция может осесть не полностью. Фильтруют через беззольный фильтр, умеренно промывают осадок водой и высушивают фильтр с осадком при 110° С.
Затем с возможной полнотой переносят осадок в платиновую чашку 1 2. Приливают 10 мл воды и потом 10%-ный раствор уксусной кислоты, пока не закончится выделение газа 3 4. Выпаривают на горячей плитке, пока не исчезнет запах уксусной кислоты, и затем продолжают нагревание 30 мин, чтобы перевести фторид кальция в зернистую форму. Обрабатывают остаток 10 мл воды и перемешивают, чтобы растворить ацетат кальция. Затем декантируют раствор через беззольный фильтр, промывают остаток 10 мл воды и переносят его на фильтр с помощью очень небольших порций воды. Продолжают промывание малыми порциями воды до тех пор, пока промывные воды не перестанут давать муль с раствором фторида натрия. Фильтр с остатком переносят в платиновый тигель, высушивают, прокаливают при низкой температуре, пока не выгорит уголь, и затем при 800— 900° С до постоянной массы *.
Так как содержание фторидов в породах обычно очень невелико, то достаточно однократной обработки, описанной выше. При анализе минералов, содержащих большое количество фтора, необходимо повторить обработку уксусной кислотой 2 и даже 3 раза, и в промежутках каждый
1 Совместное осаждение карбоната кальция и фторида кальция было первоначально предложено Н. Rose [Ann., 72, 343 (1849)]. Чтобы получить осадок менее растворимый и легче отделяемый фильтрованием, G. Starck и Е. Thorin [Z. anal. Chem., 51, 14 (1912)] предпочитают прибавлять определенное количество оксалата патрия и затем в избытке — хлорид кальция. В массу полученного осадка они затем вносят поправку на количество оксалата кальция, эквивалентное количеству введенного оксалата натрия. По нашему опыту этот метод оказался гораздо труднее выполнимым, чем это можно было ожидать по литературным данным.
2 Если осаждение вели в платиновой чашке, то нет необходимости очищать ее от приставших частиц осадка; содержимое фильтра смывают обратно в чашку, жидкость в ней выпаривают и золу фильтра всыпают туда же.
3 В 10 мл 10%-ного раствора уксусной кислоты можно растворить приблизительно 1,5 мг чистого свежеосажденного фторида кальция, но количество это значительно меньше в присутствии ацетата кальция, образующегося при обработке смеси фторида и карбоната кальция уксусной кислотой. Приблизительно 1,6 мг фторида кальция растворяется в 100 мл воды.
4 Прокаленный фторид кальция следует всегда обработать фтористоводородной кислотой и снова взвесить.
1022
Гл. LIII. Методы анализа силикатных пород
раз отфильтровывать и прокаливать остаток. Было найдено х, что если сразу прибавить уксусную кислоту в большом избытке, то получаются более низкие результаты, чем если обрабатывать несколько раз меньшими порциями уксусной кислоты. При определении больших количеств фтора, когда в результате ряда обработок достигают того, что между двумя последовательными обработками потеря в массе не превышает 0,5 мг, следует считать правильной предпоследнюю массу 1 2.
Исследование фторида кальция. Полученный этим методом хорошо промытый и слабо прокаленный фторид кальция следует потом превратить в сульфат кальция для того, чтобы проверить его чистоту и чтобы одновременно качественно (по характерному запаху выделяющегося газа) установить, что это действительно был фторид кальция. Если присутствие фтора будет таким образом доказано, но масса сульфата кальция окажется не соответствующей массе фторида кальция, то нужно растворить сульфат кальция в горячей азотной кислоте и испытать на фосфор молибдатом аммония. Если фосфат не будет обнаружен, то загрязнением могла быть кремнекислота или силикат кальция, но которое вещество из них, — решить трудно.
В первом случае действительное количество фторида кальция можно точно вычислить на основании массы полученного сульфата кальция, во втором случае этого сделать нельзя. При анализе пород, богатых серой, может случиться, что вместе с фторидом кальция в осадок будет увлечен также и сульфат кальция, однако последний должен быть полностью удален основательным промыванием осадка. Если это не удалось и если имеется уверенность, что кроме сульфата кальция других загрязняющих веществ нет, то можно после превращения фторида кальция в сульфат вычислить содержание фтора (х) по следующему уравнению:
CaS04—CaF2 b— а а — а
2F ~~Г~ ПЛИ
где а — масса нечистого фторида кальция, b — масса сульфата кальция, полученного из этого фторида кальция.
Полное соответствие между массой фторида кальция и массой полученного из него сульфата наблюдается в исключительных случаях, и при определении малых количеств фтора, обычно встречаемых в породах, эта ошибка по отношению к содержанию фтора может быть значительной.
Точность метода. Из изложенного следует, что если не принимать чрезвычайных мер предосторожности для избежания всякого загрязнения фторида кальция, то ошибка при определении малых количеств, о которых идет речь, может стать очень большой. Если осадок загрязнен фосфатом кальция, не следует предпринимать для проверки выпаривание с серной кислотой, потому что при этом выпаривании и последующем прокаливании может улетучиться метафосфорная кислота.
Другой источник ошибок заключается в растворимости самого фторида кальция в воде и уксусной кислоте. Было исследовано 3 растворяющее на него действие как названных, так и других реактивов. Оказалось, что из фильтрата можно выделить еще некоторое количество фтора по
1 S. L. Р е n f i е 1 d, J. С. М i п о г, Am. J. Sci., [3], 47, 389 (1894).
1 F. P. Tread wall, A. A. Koch, Z. anal. Chem., 43, 469 (1904).
3 F. S e e m a n n, Z. anal. Chem., 44, 343 (1905).
Фтор
JO23
вторным осаждением фторида кальция в присутствии карбоната натрия. При выделении 5 мг или меньше фторида кальция ошибка, происходящая в результате его растворимости, составляет 0,0015 г СаРг на каждые 100 мл жидкости (включая и промывную воду). Из этого количества только 0,0009 г СаРг в 100 мл раствора могут быть затем обнаружены качественно. Пересчитывая это последнее количество СаРг на фтор, мы можем заключить, что при навеске в 1 г и содержании фтора 0,04—0,05% (т. е. 0,0004—0,0005 г) он может совершенно ускользнуть от определения.
Не всем известно, что источником ошибки может также явиться сжигание фторида кальция при соприкосновении с бумагой. При этом небольшое количество фтора улетучивается и замещается кислородом. Реакция эта происходит, несомненно, под влиянием водяного пара, а не кислорода воздуха.
Было проведено подробное исследование 1 всех известных способов определения фтора на одном и том же образце плавикового шпата. Оказалось, что метод Берцелиуса из-за больших и не всегда одинаковых потерь (причину которых не удалось удовлетворительно объяснить) во многом уступает методам, основанным на отгонке фторида кремния. Методом Берцелиуса не удалось извлечь более 87—89% присутствовавшего в пробе фтора. Однако проведенные Гиллебрандом исследования (подтвержденные и другими химиками) не подтвердили этих неблагоприятных выводов. Мы постоянно извлекали от 95 до 98% присутствовавшего фтора. Однако для этого нужно было: 1) остаток, остающийся после выщелачивания плава с карбонатом натрия, сплавить вторично вместе с кремнекислотой, осажденной карбонатом аммония и окисью цинка, 2) обработать фильтрат от осаждённых карбоната и фторида кальция новыми порциями карбопата натрия и хлорида кальция и 3) повторно выпарить уксуснокислый раствор осажденного карбоната кальция.
Одновременное определение кремнекислоты при применении метода Берцелиуса. Несколько осадков, полученных при осаждении карбонатом аммония и карбонатом цинка вместе с нерастворимым остатком от первоначального сплавления с карбонатом калия-натрия, обрабатывают следующим образом. Прежде чем перевести все осадки в раствор и продолжать анализ, как описано в разделе «Кремний» (стр. 940), удаляют цинк, перешедший в осадок, от окиси цинка. Для этого осадок, содержащий цинк, обрабатывают азотной кислотой, выпаривают совершенно досуха, растворяют остаток в азотной кислоте, фильтруют, промывают кремнекислоту и соединяют ее с кремнекислотой, выделенной из других осадков после растворения их в соляной кислоте и дальнейшей обработки раствора обычным методом, применяемым для выделения кремнекислоты (стр. 940).
Метод имеет тот недостаток, что небольшая часть фтора удерживается осадком, вследствие чего происходит небольшая потеря кремнекислоты, когда осадок растворяют в кислоте и раствор выпаривают для обезвоживания кремнекислоты. Это особенно справедливо в отношении таких материалов, как фосфатные породы, содержащие большие количества кальция и фосфат-ионов (см. стр. 1019). В этих случаях может получиться большая ошибка.
Если, как это обычно бывает, проводят полный анализ минерала или породы, то необходимо выделить и определить молибдатным методом (см. «Фосфор», стр. 781) тот фосфор, который может быть в растворе нитрата цинка, а также фосфор и хром, которые могут содержаться в фильтра-
1 Seemann, Z. anal. Chem., 44, 343 (1905).
1024
Гл. LIII. Методы анализа силикатных пород
те от силиката цинка. В большинстве случаев хром лучше определять колориметрическим методом (см. «Хром», стр. 595), а затем в том же самом растворе, подкислив его HNO3, можно осадить фосфор молибдатом аммония и определить его. Соединенные массы хрома, пересчитанного на Сг2О3, и фосфора, полученного из двух фракций, пересчитанного на Р2О5, должны быть присоединены к массе сложного осадка от аммиака, полученного в обычном ходе анализа осадков, о которых говорилось выше.
Метод Гофмана и Ленделя х. В следующем методе определения кремния и фтора во фторидных минералах кремнекислоту выделяют видоизмененным методом Берцелиуса, более скорым и таким же точным, а фтор затем осаждают в виде хлорофторида свинца и определяют объемным методом, являющимся вариантом метода, первоначально предложенного Штарком 1 2. Осадок растворяют в азотной кислоте и титруют, вместо того, чтобы его взвешивать, потому что тогда примеси в осадке не оказывают влияния, если они не содержат хлорида, присутствие которого мало вероятно.
Ход определения. Навеску 3 в 0,5000 г высушенной пробы сплавляют с 5 г карбоната натрия, выщелачивают охлажденный плав горячей водой и, после полного распадения плава, фильтруют. Полученный фильтрат (фильтрат А) сохраняют. Нерастворимый остаток смывают струей воды обратно в чашку, прибавляют 50 мл 2%-ного раствора карбоната натрия, кипятят несколько минут, фильтруют и тщательно промывают остаток (остаток Б) горячей водой 4. Остаток Б сохраняют для определения кремнекислоты, а фильтрат соединяют с ранее полученным фильтратом А. К соединенным фильтратам, которые должны иметь объем приблизительно 300 мл, прибавляют раствор 1 г окиси цинка в 20 мл разбавленной (1 : 9) азотной кислоты. Раствор кипятят 1 мин, фильтруют и тщательно промывают осадок (осадок В) горячей водой. Осадок В сохраняют для определения кремнекислоты. К фильтрату прибавляют несколько капель метилового красного, почти нейтрализуют его азотной кислотой и выпаривают до объема 200 мл, следя за тем, чтобы раствор оставался щелочным в течение всего процесса выпаривания. Немного охлаждают и прибавляют по каплям разбавленную (1 : 1) азотную кислоту до появления слаборозовой окраски. Затем прибавляют 1,0 а окиси цинка, растворенной в аммиаке при добавлении небольшого количества карбоната аммония, и кипятят в покрытой чашке (лучше всего платиновой) до исчезновения запаха аммиака. Обычно при этом раствор упаривается приблизительно до 50 мл. После удаления аммиака прибавляют около 50 мл горячей воды, перемешивают, дают осадку (осадок Г) осесть в течение нескольких минут и фильтруют (фильтрат Д). Осадок Г промывают холодной водой. Фильтрат Д сохраняют.
Кремнекислота. Струей разбавленной (1 : 20) соляной кислоты смывают с фильтров остаток Б и осадки В и Г в сосуд, в котором проводили последнее осаждение. Сжигают все фильтры и золу их переносят туда же. Приливают 25 мл соляной кислоты и определяют кремнекислоту
1 J. I. Hoffman, G. Е. F. L u n d е 11, Research NBS, 3, 581 (1929).
2 G. S t а г с k, Z. anorg. Chem., 70, 173 (1911).
3 Величина навески должна быть подобрана таким образом, чтобы количество фтора в растворе не выходило из пределов от 0,01 до 0,1 г.
4 Такая обработка нерастворимого остатка способствует полному извлечению фтора. Ее можно не проводить, если предполагают прокаливать этот остаток вместе с первым осадком (от нитрата цинка) и снова сплавлять с карбонатом натрия.
Фтор
1025
двукратным выпариванием с фильтрованием в промежутке, как это описано в разделе «Кремний» (стр. 940). При втором выпаривании и обезвоживании следует прибавить 10 мл серной кислоты и выпарить до появления паров H2SO4, потому что трудно высушить хлорид цинка при отсутствии заметных количеств кремнекислоты. Небольшое количество кремнекислоты (обычно менее 1 мг) остается в растворе и может быть выделено следующим образом. К фильтрату, полученному после второго выпаривания, прибавляют около 0,04 г алюминия в виде хлорида алюминия и 10 г хлорида аммония. Нагревают до кипения, осаждают аммиаком и фильтруют. Осадок растворяют в 50 мл разбавленной (1 : 10) серной кислоты, выпаривают до появления паров H2SO4, разбавляют водой, фильтруют и прибавляют выделенный небольшой остаток кремнекислоты к полученному ранее.
Фтор. Фильтрат Д обрабатывают, как описано в гл. «Фтор» (стр. 826).
“"Определение фтора этим методом можно значительно упростить1, если вместо обычного сплавления с карбонатом натрия проводить спекание со смесью окиси цинка и карбоната натрия, взятых в отношении 3:1. Спекание можно проводить в фарфоровых тиглях, и в фильтрате не требуется выделения кремнекислоты, так как она вся или почти вся остается в остатке после выщелачивания водой. При определении больших количеств фтора (свыше 5%) спекание надо повторить, иногда даже два раза. Доп. ред*
Непрямое определение малых количеств фтора
Метод Стейгера. Предложенный Стейгером 2 непрямой метод определения фтора очень удобен, хотя он и не пригоден для определения больших количеств фтора.
Метод основан на хорошо известном факте, что в присутствии фтора желтая окраска растворов титана, содержащих перекись водорода, обесцвечивается. К анализируемому раствору, содержащему фторид-ионы, прибавляют определенные количества перекиси водорода и раствора соли титана, после чего разбавляют до определенного объема. Этот раствор затем сравнивают в колориметре с другим раствором, содержащим в том же объеме такие же количества титана и перекиси водорода. По обесцвечиванию первого раствора (по сравнению со вторым) рассчитывают содержание в нем фторид-ионов.
Хотя получаемые результаты не так точны, как результаты определения многих других элементов, но принимая во внимание трудности определения фтора, а также и то, как много времени и труда расходуется при определении малых количеств фтора другими методами, этот косвенный метод следует признать заслуживающим широкого распространения.
Следы фтора (порядка нескольких сотых долей процента) не только легко можно открыть этим методом, но и приблизительно определить их количественно. При определении нескольких десятых долей процента фтора метод, по-видимому, дает более надежные результаты, и если содержание фтора не превышает 2%, полученные результаты в отношении точности не уступают результатам, получаемым стандартными методами.
1 Ю. А. Ч е р н и х о в, см. в книге В. Ф. Гиллебранд, Г. Э. Лендель, Практическое руководство по неорганическому анализу, ОНТИ, 1935, стр. 855.
2 G. S t е i g е г, J. Am. Chem. Soc., 30, 219 (1908).
6 > Заказ 522.
1026
Гл. LIII. Методы, анализа силикатных пород
Однако там, где надо определять значительные количества фтора, было бы неправильным ожидать от колориметрического метода, который требует лишь нескольких миллиграммов определяемого вещества, чтобы он был близок по точности к весовым методам, в которых перерабатывают гораздо большие количества анализируемого вещества и имеют дело со значительным содержанием фтора.
В присутствии большого количества солей натрия колориметрический метод показывает несколько пониженное содержание фтора х, однако не настолько, чтобы это серьезно повлияло на получаемые результаты. Кремнекислота в количестве до 0,1 г влияет мало, а предварительная обработка уменьшает ее содержание в растворе самое большее до 0,02— 0,03 г. Алюминий, который даже в малых количествах оказывает заметное мешающее влияние, легко удаляется предварительной обработкой. Фосфорная кислота, подобно фтору, также обесцвечивает окраску раствора, но определению мешает только присутствие ее в количествах больших, чем те, которые встречаются в анализе горных пород.
Метод Стейгера, видоизмененный Мервином, Общие замечания. В цитированной выше работе Мервин изучил не только влияние солей щелочных металлов на окраску растворов, содержащих титан и перекись водорода, но также и влияние кислотности раствора и его температуры. Соли щелочных металлов вызывают ослабление интенсивности окраски, тогда как прибавление кислоты в значительной степени противодействует этому ослаблению. Было также замечено, что стандартный раствор титана, содержащий мало кислоты, нельзя сравнивать с побледневшим анализируемым раствором. Так, нельзя было сравнить окраски, возникающие от 3 мг фтора в присутствии 2% серной кислоты, но в присутствии 10% кислоты сравнение возможно даже для 5 мг фтора.
Мервин дает формулы для вычисления количества фтора в присутствии известных. количеств кислоты и сульфатов щелочных металлов. Он несколько изменил прежние методы получения фтора (в виде его растворимого соединения) для его определения и теперь применяют следующий способ.
Ход определения. Сплавляют 2 г измельченной в порошок породы с 8 г смеси карбонатов калия и натрия. Полученный плав обрабатывают горячей водой.
По растворении, не фильтруя, прибавляют 3—4 г карбоната аммония в порошке. Смесь подогревают несколько минут, а затем помещают на водяную баню, где нагревают до разрушения карбоната аммония и значительного упаривания раствора. Таким способом осаждают кремнекислоту, которая могла бы вызвать помутнение раствора при его колориметрировании, а также гидроокись алюминия и гидроокись железа, мешающие определению. Разрушение карбоната аммония необходимо, так как присутствие сульфата аммония в окончательном растворе вызывает побледнение окраски. После фильтрования к фильтрату, объем которого не должен превышать 75 мл, прибавляют 3—4 мл перекиси водорода и затем очень осторожно приливают 10 мл стандартного раствора титана 2 (содержащего 0,01 г TiOs). После этого нейтрализуют присутствующие в растворе карбонаты.
1 Н. Е. Merwin, Am. J. Sci., [4], 28, 119 (1909).
2 Перекись водорода предотвращает осаждение титана карбонат-ионами, присутствующими в анализируемом растворе.
Фтор
1027
Для нейтрализации карбонатов требуется около 3,5 мл концентрированной серной кислоты, включая и ту кислоту, которая уже содержится в растворе. Как только реакция раствора станет нейтральной, он примет светло-оранжевую окраску. Нейтральность раствора проверяют прибавлением небольшого количества карбоната натрия, от чего окраска исчезает; затем снова прибавляют 1—2 капли кислоты, чтобы восстановить окраску. Дальнейшая обработка зависит от предполагаемого содержания фтора и от типа применяемого колориметра.
Отношение окрасок
1 — 3 мл концентрированной серной кислоты; 2 — 6 мл концентрированной серной кислоты; 3 —12 мл концентрированной серной кислоты.
Количество кислоты, которое нужно прибавить к нейтральному раствору, зависит от количества фтора. Поэтому сначала прибавляют 3 мл концентрированной серной кислоты, доводят объем раствора до 100 мл при 22° С и сравнивают окраску раствора при этой же температуре с окраской 100 мл другого раствора, содержащего 0,01 г TiOa, 4 мл Н2О2 и около 3 мл концентрированной серной кислоты. После этого содержание фтора можно определить по кривым, изображенным на рис 46.
Если указанное в этой диаграмме отношение будет больше 0,40, то количество фтора в пробе можно найти точно (пользуясь диаграммой), если же это отношение меньше 0,40, т. е. если фтора больше, то нужно прибавить еще кислоты к анализируемому раствору, проделать новое сравнение окиси и новый отсчет по диаграмме Ч Диаграмма построена в предположении, что количество щелочных металлов соответствует 8 г смеси карбонатов калия и натрия (взятым в отношении 1 : 1) или 7 г карбоната натрия, взятым для первоначального сплавления навески. По оси абсцисс диаграммы отложены средние величины отношения
1 Для некоторых других систем колориметров необходима поправка на объем анализуемого раствора, который в эти колориметры вводится.
65*
1028
Гл. LIII. Методы анализа силикатных пород
окрасок, полученных при 8 г карбонатов. Применяемые карбонаты надо испытывать на содержание в них фтора тем же методом, каким анализировали профильтрованный раствор породы. Найденные отношения отличаются от помещенных в диаграмме, вероятно, не больше чем на 0,03. Эта разница соответствует 0,5 мг фтора, что уже выходит за пределы точности метода при количествах фтора, меньших 0,006 г. При содержании 0,025 г фтора этот предел равен приблизительно 0,002 г.
При пользовании диаграммой необходимо учесть следующее:
_ толщина слоя стандартного раствора
Отношение окрасок -------------------1-----£--------
толщина слоя анализируемого раствора
Каждый раствор обработан перекисью водорода, доведен до 100 п содержит 0,01 г TiOa. Анализируемый раствор содержит указанное у конца кривой число миллилитров концентрированной серной кислоты и показанное слева количество фтора: кроме того, в его состав входят сульфаты, эквивалентные 4 г карбоната натрия, и 4 г карбоната калия. Стандартный раствор содержит неопределенное количество серной кислоты (от 3 до 5 мл). Эта диаграмма построена для растворов, имеющих температуру 22° С, но она существенно пе изменяется при всех температурах в интервале от 19 до 25° С при условии, что анализируемый и стандартный растворы приведены к совершенно одинаковой температуре. Если температура анализируемого раствора на 5° С превышает температуру стандартного раствора, то кривые смещаются влево на 0,02, если присутствуют 12 мл серной кислоты, и на 0,01 в присутствии 3 мл кислоты. При изменении количества воды на 10% окраски растворов изменяются незначительно.
Из сказанного выше совершенно ясно, что аналитик должен принимать во внимание содержание солей щелочных металлов и кислоты, а также проводить все отсчеты при одной и той же температуре. Каждый, кому часто приходится проводить такое непрямое определение фтора, должен составить собственную диаграмму поправок, соответствующую применяемым им условиям работы.
Метод Стейгера — Мервина был в дальнейшем еще изменен и усовершенствован Для измерения интенсивности окраски применили фотометр и получили более точные данные об оптимальных значениях pH для получения наиболее интенсивной окраски. Также подробнее было выяснено влияние алюминия, фосфора и сульфатов щелочных металлов.
Методом Мервина было проведено 2 большое число анализов почв на содержание в них фтора. Найденные количества доходили до 0,05%. Метод этот оказался «недостаточно точным, чтобы с уверенностью определять фтор, когда содержание его меньше 0,01%», но даже если это и так, то, по сравнению с весовым методом, он представляет большие преимущества при определении таких малых количеств.
СЕРА
Определение различных форм, в которых сера находится в горных породах
Прежде чем приступить к определению серы, следует установить, в виде каких соединений она содержится в анализируемой породе. Для этого служит как микроскопическое исследование породы, когда оно возможно, так и качественный ее анализ. Большое значение микроскопи-
1 Н. J. W i с h m a n n, D. D a h 1 е, J. Assoc. Offic. Agr. Chemists, 16, <612 (1933); 20, 505 (1937).
2 L. A. S t e i n k 6 n i g, J. Ind. Eng. Chem., 11, 463 (1919).
Сера
1029
ческого исследования нужно особо подчеркнуть, так как оно дает аналитику наиболее ценные указания.
Если при кипячении породы с разбавленной соляной кислотой выделяется сероводород, то это является определенным указанием на присутствие растворимого в кислотах сульфида, обычно пирротина, но иногда и лазурита. Если можно при помощи магнита извлечь частицы, дающие реакцию на серу, то это доказывает, что причиной выделения сероводорода, по крайней мере частичной, является присутствие пирротина (магнитного колчедана). Реакция на серную кислоту в профильтрованном растворе указывает на присутствие в породе растворимого в кислоте сульфата, большей частью в виде силикатных сульфатов: нозеана и гаюина. Если хорошо промытый остаток после обработки разбавленной соляной кислотой обработать царской водкой или соляной кислотой с бромом и в результате этой обработки в растворе снова образуются сульфат-ионы, то это указывает на вероятное содержание в пробе пирита. Если в полученном при такой обработке растворе можно обнаружить присутствие мышьяка, то возможно, что в породе был арсенопирит, хотя в изверженных породах он встречается крайне редко.
Присутствие серы в породе в форме простых сульфатов, даже при значительном содержании в ней пирита, чрезвычайно редко (за исключением встречающихся иногда растворимых в воде следов). Барий в первичных породах, и даже в породах умеренно разложенных, содержится, по-видимому, всегда в виде силиката, а не в виде сульфата бария.
В анализе минералов следует по возможности избегать сплавления со щелочами, однако не надо упускать из виду, что при прямой обработке минерала соляной кислотой, одной или с добавлением окислителя, в раствор переходит большое количество солей, из которых некоторые могут создать непреодолимое препятствие для получения приемлемых результатов анализа.
Определение серы в горных породах
Определение общего содержания серы. Прежде чем приступить к количественному определению серы в породах, надо прочитать гл. «Сера» (стр. 726).
В горных породах сера редко присутствует в больших количествах, поэтому вряд ли нужно принимать специальные меры предосторожности при осаждении сульфата бария, или применять его очистку, тем более, что осаждение сульфата бария обычно проводится в отсутствие железа.
Хотя сера сульфидов иногда может быть точно определена мокрым путем — извлечением царской водкой или другими сильными окислителями, тем не менее это удается не во всех случаях. Поэтому гораздо лучше сплавлять пробу со свободным от серы карбонатом натрия и небольшим количеством селитры, нагревая на бунзеновской горелке, а потом короткое время на паяльной горелке. В течение всего нагревания следует пользоваться установкой, показанной на рис. 6 (стр. 49), чтобы защитить плав от попадания в него серы из газов пламени. Определение может быть очень удобно соединено с определением бария, титана, циркония и редкоземельных металлов (стр. 968).
После полного разложения плава водой, к которой добавляют 1—2 капли спирта для восстановления и осаждения всего марганца, перешед
1030
Гл. LIII. Методы анализа силикатных пород
шего в раствор в виде манганата, фильтруют и промывают остаток разбавленным раствором карбоната натрия. К фильтрату (занимающему объем от 100 до 250 мл), после подкисления его на холоду соляной кислотой, прибавляют (при кипячении или на паровой бане) в избытке хлорид бария для осаждения сульфата бария. Выпаривать досуха с кислотой для выделения кремнекислоты не нужно, потому что при таком объеме раствора кремнекислота (даже в самых ничтожных количествах) почти никогда не увлекается сульфатом бария Е Это очень удобно, так как при выпаривании раствора на водяной бане, обогреваемой газом (для выделения кремнекислоты), можно вследствие содержания серы в светильном газе ввести в определение ошибку, которая во многих случаях может оказаться равной по величине определяемому количеству серы. При применении паровой бани это затруднение исключается. Если есть подозрение, что сульфат бария загрязнен следами кремнекислоты, то последнюю можно удалить до взвешивания сульфата бария прибавлением капли фтористоводородной и серной кислот и прокаливанием.
С целью значительного уменьшения ошибки, вызываемой присутствием хлорида натрия, образующегося в растворе в результате сплавления с карбонатом натрия, было предложено 2 выпаривание раствора до небольшого объема (25 мл), насыщение его газообразным хлористым водородом или прибавление концентрированной соляной кислоты (5 объемов кислоты на 1 объем раствора, сконцентрированного до 10 мл), фильтрование через тигель Гуча, промывание осадка хлорида натрия концентрированной соляной кислотой, выпаривание фильтрата, вторичное фильтрование, если при выпаривании выпал осадок, и, наконец, осаждение хлоридом бария.
Ошибок, зависящих от растворяющего действия соляной кислоты на сульфат бария и от присутствия аммонийных и других солей в растворе, обычно можно избежать, вводя поправку следующим образом. Фильтрат от сульфата бария выпаривают, разрушают и удаляют аммонийные соли, если они присутствовали, остаток обрабатывают водой, слегка подкисленной соляной кислотой, отфильтровывают маленький остаток сульфата бария, затем очищают его и взвешивают.
Определение отдельных соединений серы. Если сера содержится в породе в нескольких формах, то обычно желательно узнать относительные их количества; поэтому приходится определять отдельные ее формы.
Если присутствуют следы растворимых в воде сульфатов, то они при желании могут быть экстрагированы кипящей водой.
Для того чтобы сульфатную серу всех растворимых сульфатов количественно перевести в раствор, достаточно простого кипячения с соляной
1 С. W. S t о d d а г t [J. Am. Chem. Soc., 24, 852 (1902)] получал при определении серы в углях методом Эшка часто повышенное содержание серы, если кремнекислота не была удалена обычным способом. Это, однако, противоречит всему нашему опыту по анализу силикатов. Результаты параллельных определений (проведенных с выделением кремнекислоты и без ее выделения) у нас обычно точно совпадали, а если этого иногда и не было, то более низкие результаты получались иногда при отделении кремнекислоты, иногда же когда кремнекислота не была отделена. Объяснение этому расхождению можно найти в том, что Стоддарт свои растворы подкислял горячими или при этом применял слишком концентрированную соляную кислоту, вследствие чего выпадало немного кремнекислоты.
J W. A. Turner, Am. J. Sci., [4], 38, 41 (1914); Z. anorg. Chem., 8, 412 (1914); Chem. News, 111, 100 (1915).
Бор
1031
кислотой. Если одновременно присутствует пирит или другие окисляющиеся сульфиды, кипячение следует проводить в атмосфере двуокиси углерода и по возможности быстрее, чтобы уменьшить ошибку, происходящую от окисления сульфидной серы до серной кислоты под действием переходящего в раствор железа (III).
Если присутствуют одновременно растворимые и нерастворимые сульфаты и сульфиды, то, выщелачивая породу соляной кислотой, можно определить 1 растворимые сульфиды путем поглощения выделяющегося сероводорода, в растворимые сульфаты — в кислотной вытяжке, осаждая их в виде BaSO4. В остатке можно определить сульфидную серу нерастворимых сульфидов после соответствующей окислительной обработки, а в остатке от этой обработки определить еще и серу растворимых сульфатов после сплавления этого остатка с карбонатом натрия. Серу нерастворимых сульфатов можно, однако, вычислить, вычитая содержание всех ранее найденных видов серы из общего содержания серы, определенного в отдельной навеске пробы.
Ошибкой, которая может произойти при указанном выше определении серы в растворимых сульфидах до окисления освобождающегося сероводорода солями железа (III), по-видимому, можно пренебречь. Большая часть сероводорода выделится из раствора прежде, чем может начаться такая реакция, и, вероятно, прежде, чем соли железа (III) перейдут в раствор в сколько-нибудь заметном количестве, но, конечно, небольшое количество элементарной серы, выделившейся из пирротина, не будет определено. Однако содержание пирротина можно с достаточной точностью рассчитать, принимая для его состава формулу Fe;S8.
Очевидно, что описанный выше ход анализа заключает в себе большое число источников возможных ошибок, особенно при анализе сложных смесей; как бы тщательно ни были сделаны отдельные определения, все же если присутствует много сульфидов, то конечный результат определения двухвалентного и трехвалентного железа, выведенный с поправкой, рассчитанной по найденному содержанию сульфидов, будет только приблизительным (см. стр. 996).
БОР
Общие замечания
Бор, без сомнения, встречается иногда в горных породах в больших количествах, чем некоторые из других обычно определяемых элементов. Однако недостаточная точность методов, которые вообще применимы к веществам, растворимым только в фтористоводородной кислоте, является причиной того, что до настоящего времени никто не пытался их применить к горным породам, разве только, если заранее были указания на значительное содержание бора.
Для определения бора (если его много) породу обычно сплавляют с карбонатом натрия и затем применяют метод Розенбладта — Гуча в каком-либо из его многочисленных вариантов.
1 Если присутствует пирротин, то небольшая часть содержащейся в нем серы (одна восьмая, если принять, что пирротин имеет формулу Fe7S8) выделится в виде элементарной серы, а не в виде сероводорода,
1032
Гл. LIII. Методы, анализа силикатных пород
В этом методе бор отгоняют из кислого раствора дистилляцией с метиловым спиртом в виде метилового эфира борной кислоты, последний собирают и определяют бор либо весовым, либо объемным методом. При анализе простых боратов, особенно растворимых, этот метод дает удовлетворительные результаты, если были соблюдены все необходимые меры предосторожности; удовлетворительные результаты также при анализе трудно разлагаемых боросиликатов были получены Чэпиным 1.
До работ Чэпина было принято щелочной плав минерала выщелачивать водой и в водной вытяжке определять бор. Причину многочисленных п больших расхождений результатов, полученных различными авторами, часто даже результатов параллельных определений, проведенных одним аналитиком, следует, вероятно, искать в том, что сплавление и выщелачивание плава проводилось ими только один раз, тогда как многими авторами 2 было доказано, что и то и другое надо повторять. Чэпин, по-видимому, достиг значительного улучшения метода; он показал, что нет необходимости удалять алюминий и железо, а сплав породы с карбонатами можно непосредственно растворять в кислоте; метод при этом не теряет в своей точности. Присутствие хлора, и даже фтора, по-видимому, не мешает определению. Описание метода см. в гл. «Бор» (стр. 834).
Качественное испытание
Открытие в породах небольших количеств бора является далеко не легкой задачей. Было установлено 3, что предел чувствительности реакции на пламя составляет 0,2%. Открытие бора этим методом проводится смешением порошка анализируемого минерала с бисульфатом калия и фторидом кальция и введением этой смеси на чистой платиновой проволочке в бесцветное пламя бунзеновской горелки. Зеленое окрашивание пламени (в виде отдельных исчезающих вспышек, если содержание бора близко к пределу чувствительности метода) указывает на присутствие бора, если нет других элементов, сообщающих пламени такую же зеленую окраску.
Дистиллят, полученный, как описано в разделе «Общие замечания» (стр. 830), можно исследовать на присутствие в нем бора по окраске пламени горящего спирта, зависящей от присутствия борного эфира. Рядом авторов 4 было показано, что эта проба значительно чувствительнее предыдущей.
При помощи куркумовой бумаги по Лоу 5 бор был открыт 6 в минерале, где было только 0,08% ВгОз. Ход работы этим методом следующий. В анализируемый раствор, подкисленный соляной кислотой и находящийся в плоской чашке (только не из боросиликатного стекла), помещают полоску куркумовой бумажки. Раствор затем выпаривают досуха
1 W. Н. С h а р i n, J. Am. Chem. Soc., 40. 1691 (1908).
2 S. L. P e n f i e 1 d, H. W. Foote, Am. J. Sci., [4], 7, 97 (1899); G. W. S a r-g e n t, J. Am. Chem. Soc., 21, 858 (1899); W. E. Ford, Am. J. Sci., [4], 14, 195 (1903).
3 E. T. Wherry, W. H. C h a p i n, J. Am. Chem. Soc., 30, 1684 (1908).
4 C. Mannich, H. Pries s, Chem. Ztg., 32, 314 (1908); V. Lenhe r, J. S. C. Wells, J. Am. Chem. Soc., 21, 417 (1899).
5 W. W. Low, J. Am. Chem. Soc., 28, 807 (1906).
6 E. T. W h e r r y, W. H. C h a p i n, цит. выше.
Компоненты,, встречающиеся в породах в виде следов
1033
и помещают чашку в эксикатор. В присутствии бора бумага принимает розовато-бурую окраску, если нет мешающих элементов, например титана, и если не было взято слишком большого количества соляной кислоты. Этим способом нельзя открывать бор непосредственно в растворе анализируемого минерала; следует пользоваться дистиллятом, полученным, как описано в разделе «Общие замечания» (стр. 1031). На основе этой качественной пробы был разработан точный количественный метод определения малых количеств бора (см. стр. 843).
Куркумовая проба может быть наиболее чувствительным из всех качественных методов открытия бора, если только проводить ее со всеми требуемыми предосторожностями. Подробности даны в оригинальной статье х. Авторы ее открывали и количественно определяли этим способом борную кислоту при содержании ее от 0,1 до 0,0005 мг. Для такого чувствительного открытия бора нужны специально для этой цели приготовленные реактивы.
В основном метод состоит 2 в сравнении длины окрашенных частей маленьких полосок куркумовой бумаги, опущенных концами с испытуемый раствор и в несколько стандартных растворов, содержащих борную кислоту в различных концентрациях. Все растворы должны быть разбавлены до одинакового объема. Растворы поднимаются по полоскам бумаги вследствие капиллярности и, высыхая на выступающих концах этих полосок, через несколько часов окрашивают полоски на различную длину в зависимости от содержания борной кислоты в разных растворах. При достаточном числе стандартных растворов можно получить вполне точные количественные результаты. Аммиак изменяет окраску куркумы в синий цвет.
КОМПОНЕНТЫ, ВСТРЕЧАЮЩИЕСЯ В ПОРОДАХ В ВИДЕ СЛЕДОВ
Иногда может случиться, как это было в исследованиях Зандбергера, изучавшего происхождение рудоносных жил, что приходится анализировать горные породы на содержание в них следов золота, серебра и других элементов, которые обычно в породах не определяют. В таких случаях нужно брать для анализа большие навески (50 г и больше). Это требует применения больших количеств реактивов, на чистоту которых в таких случаях надо обращать особое внимание. В статье Зандбергера s мало освещена аналитическая часть его исследования, и вообще литература по этому вопросу очень невелика 4.
1 G. Bertrand, Н. A g u 1 h о n, С. г., 157, 1433 (1913); Bull. Soc. chim., 7. 90 (1910); 13, 396 (1913).
2 Е. Т. А 11 е n, Е. G. Z i е s, J. Am. Ceram. Soc., 1, 739 (1918).
3 F. Sandberger, Jahrb. Bergakad., Leoben. und Pribram, 363 (1887). См. также статью H. von Foulton, помещенную в том же сборнике.
1 S. F. Е m m о n, U. S. Geol. Survey Mon., 12, Приложение В, 592—596 (1886) (золото, серебро, свинец, цинк и др.); J. S. Curtis, там же, 7, 120 (1884) (золото, серебро); J. D. Robertson, Rep. Missouri Geol. Survey, 7, 740 (свинец, цинк, медь в силикатных и карбонатных породах); J. R. D о n, Trans. AIME, 27, 564 (1897) (золото); L. Wagoner, там же, 31, 808 (1901) (золото); О. Н а с k 1, Chem. Ztg., 45, 1169 (1921); 46, 385 (1922). В последней работе даны точные указания, как проводить открытие п определение следов мышьяка, никеля и кобальта в силикатных горных породах; автор определяет мышьяк после отгонки его в токе двуокиси*углерода, насыщенной бромом.
1034
Гл. LIII. Методы анализа силикатных пород
Из сообщения Зандбергера, по-видимому, следует, что анализированные им породы последовательно обрабатывали водой, уксусной кислотой, кипящей разбавленной соляной кислотой в течение 2 дней и, наконец, фтористоводородной кислотой. Каждую вытяжку и окончательный остаток фторидов (а также и пирита) отдельно исследовали на тяжелые металлы. Неожиданным оказался факт, наблюдавшийся им во всех случаях, что пирит, даже при такой обработке, оставался совершенно нетронутым. Это говорит в пользу точности метода определения железа (II) в силикатах обработкой их смесью фтористоводородной и серной кислот, если нет других сульфидов помимо пирита (см. стр. 996).
Из новых работ отметим работу Сендэла и Перлиха 1 по определению никеля и кобальта в силикатных породах. Определение никеля -основано на осаждении его диметилглиоксимом из аммиачно-тартратного раствора анализируемой породы, экстрагировании полученного соединения хлороформом, взбалтывании хлороформного слоя с соляной кислотой для переведения никеля в водную фазу и конечном его определении колориметрическим методом с диметилглиоксимом (см. стр. 468, сноска 2) при концентрации его, не превышающей 6 мкг в 1 мл. Этим методом можно обнаружить 0,0001% никеля в 0,5 г пробы; медь, кобальт, марганец, хром и ванадий в количествах, в каких эти элементы встречаются в большинстве изверженных горных пород, определению никеля нс мешают.
Описан 2 прямой метод определения больших количеств никеля (порядка 0,01%), в котором навеску в 2 г тонко измельченной породы разлагают плавиковой п серной кислотами (сплавляя остаток, если это необходимо) и двукратно осаждают никель дц-метилглиоксимом из аммиачно-цитратного раствора. Если содержание нпкеля (в расчете на NiO) меньше 0,02% , то вместо диметилглиоксима применяют а-фурплдиокспм.
Определение кобальта основано на экстрагировании его раствором дитизона в четыреххлористом углероде из аммиачно-цитратного раствора пробы и на выпаривании экстракта досуха, прокаливании остатка, растворении его в царской водке, восстановлении хлоридом олова (II) и колориметрическом определении роданидно-ацетоновым методом (стр. 476). Этим способом можно обнаружить 0,0001% кобальта в 1 г пробы.
Сендэл3 описывает также колориметрические методы определения меди, цинка, свинца и кадмия в силикатных породах, основанные на предварительном извлечении этих элементов обработкой раствором дитизона в четыреххлористом углероде. Этими методами можно определить 0,002% меди, 0,0025% цинка, 0,0005% свинца и 0,00005% кадмия.
ГАЗЫ И ПАРЫ, ВЫДЕЛЯЮЩИЕСЯ ПРИ НАГРЕВАНИИ
То, что породы и минералы при нагревании могут выделять большие количества газов и паров, является давно известным фактом. Многочисленные анализы показали, что летучие вещества обычно состоят преимущественно из водорода и двуокиси углерода и, кроме того, содержат небольшие количества окиси углерода, метана, азота, сероводорода и т. и. Общий объем выделяемых газов может во много раз превосходить объем твердого вещества.
1 Е. В. S a n d е 11, R. W. Р е г 1 i с h, Ind. Eng. Chem., Anal. Ed., 11, 309 (1939).
2 H. F. Harwood, L. T. T he о Ь a 1 d, Analyst, 58, 673 (1933).
3 E. B. S a n d e 11, Ind. Eng. Chem., Anal. Ed., 9, 464 (1937); 11, 364 (1939). •См. E. B. S a 11 d e 11, Colorimetric Determination of Trances, of Metals, 2d ed., New York (1950).
Газы и пары, выделяющиеся при нагревании
1035
До известной степени, а иногда и в значительной мере (например, в случае выделения двуокиси углерода) источником этих газов и паров являются жидкие включения, образовавшиеся одновременно с образованием содержащих их минералов. Кроме того, газы и пары могут содержаться в горных породах в растворенном состоянии, особенно в таких породах, которые не полностью закристаллизовались.
Однако рядом исследований1 было несомненно установлено, что часто газы образуются в результате тех химических изменений, которые происходят во время нагревания породы, проводимого с целью их выделения. Так, водород может образоваться вследствие восстановления воды минералами, способными окисляться при высокой температуре, как, например, силикатами, содержащими железо (II); окись углерода может получиться подобным же образом из угольного ангидрида или вследствие восстановления СО а водородом, образовавшимся в результате первой реакции; метан может получиться при взаимодействии воды с карбидами металлов и т. д.2
Свободный кислород не упоминается среди выделяющихся газов, чего, судя по его природе, и нельзя было ожидать.
Азот, который в свободном состоянии в земной коре найден лишь в сравнительно малом количестве, в виде аммиака или аммонийных солей встречается в некоторых группах пород в сравнительно больших количествах и легко может быть открыт, что уже давно известно. Так было найдено 3, что смоляной камень при нагревании выделяет аммиачную воду. Находим аммиак и в горных породах 4. Аммиак выделяется при обработке некоторых минералов из древних изверженных пород едкими щелочами 5. Присутствие сульфата аммония было установлено 6 в одном образце барита из Миссури, что потом подтверждено Гплле-брандом.
При анализе ряда руд, кровельных сланцев и изверженных пород в лаборатории Геологического управления США в трех различных случаях было найдено, что при прокаливании выделяется хлорид или сульфат аммония или даже свободный аммиак. Появление этих соединений не ограничивалось отдельными или немногими образцами, напротив, оно наблюдалось во всех образцах указанной серии, а также не зависело от того, исследовалась ли неизмельченная или растертая в порошок порода. Так как условия взятия этих проб точно не известны, то нельзя быть вполне уверенным, что аммиак не мог получиться от более позднего их загрязнения какими-либо органическими веществами (особенно в отношении кровельных сланцев); однако нужно полагать, что и более безупречный способ отбора материала не изменил бы общего результата.
1 М. W. Travers, Proc. Roy. Soc., 64, 130 (1899); A. Gautier, C. r., 131, 647 (1900); 132, 58, 189 (1901); 136, 16 (1903); Ann. chim. phys., {7], 22, 97 (1901); Ann. mines, [10], 9, 316 (1906); К. H ii t n e r, Z. anorg. Chem., 43, 8 (1905); R. T. Cha into e r 1 i n, The gases in rocks, Carnegie Institution of Washington (1908). Последняя работа представляет очень обстоятельный труд, содержащий большое число экспериментальных данных.
2 Методы собирания этих газовых смесей и их анализа описаны в цитированной выше статье Gautier и в статье С. М о u г е u, С. г., 142, 44 (1906).
3 Н. Rose, Quantitative Analyse, р. 673.
4 A. Delesse. Ann. mines, 18, 151 (1860).
3 H. Erdmann, Ber., 29, 1710 (1896).
°C. Luedeking, H. A. Wheeler, Am. J. Sci., [3], 42, 495 (1891).
1036
Гл. LIII. Методы анализа силикатных пород
Количество аммиака иногда легко можно было определить колориметрическим методом, и оно в некоторых сланцах достигало 0,04%.
Большинство проб не содержало органических веществ, но они несомненно некогда присутствовали в породах. В этом случае аммиак, по крайней мере, частично выделялся в свободном состоянии, так как сгущавшаяся в верхней части трубки вода имела сильнощелочную реакцию. Если в породе присутствовали сульфиды, фториды или хлориды, то происходила возгонка сульфата, фторида или хлорида аммония. Тот факт, что в одном образце лавы с вулкана Этна был обнаружен нитрид железа1, вызывает предположение, что аммиак может быть обязан своим происхождением действию воды на нитриды металлов.
ОСОБЫЕ ИССЛЕДОВАНИЯ
Иногда нужно установить состав той части породы, которая растворяется в специальной смеси реактивов или в каком-нибудь одном реактиве точно определенной концентрации. Для таких случаев нельзя дать точных указаний. Метод обработки будет изменяться в зависимости от характера компонентов породы и от цели анализа, и только в отдельных случаях разделение может быть точным. Многое зависит от степени измельчения порошка породы и продолжительности действия растворителя.
Открытие нефелина в присутствии оливина
Для подтверждения результатов микроскопического исследования Пирсон2 предложил способ, которым можно открыть нефелин в присутствии оливина, например в нефелиновых базальтах. Этот способ основан на очень легкой растворимости нефелина (по сравнению с оливином) при кипячении в течение только 1 мин с сильно разбавленной (1 : 40) азотной кислотой.
Если после фильтрования фильтрат застывает при выпаривании в студень, то это указывает на присутствие нефелина. Но если оливина очень много, то эта проба не может считаться окончательной, так как некоторые, если не все, оливины значительно больше растворимы в азотной кислоте указанной концентрации, чем это мог предполагать Пирсон на основании своих исследований. Поэтому, если при выпаривании фильтрата обнаруживается большое количество железа, то можно считать, что желатинизация частью или полностью произошла от оливина, и в этом случае следовало бы определить отношение между кремнекислотой и суммой железа и магния. Далее, нужно иметь в виду, что все другие очень легко растворимые силикаты (если они присутствуют в породе) могут при такой обработке разложиться в большей или меньшей степени и что апатит при этом растворяется полностью или большей частью. Может быть, при применении еще более разбавленной азотной или какой-либо другой кислоты удалось бы уменьшить разложение оливина без одновременного ослабления действия кислоты на нефелин и т. и. В соединении с количественным анализом получаемого раствора этот способ
1 О. Silvestri, Gazz. chim. ital., 5, 301 (1875).
2 L. V. Pirsson, Am. J. Sci., [4], 2, 142 (1896).
Особые исследования
1037
может найти более широкое применение, чем в том специальном случае, для которого он был разработан. Вопрос безусловно заслуживает дальнейшего изучения 1-2.
Определение «растворимой» кремнекислоты х>2
В практике анализа осадочных горных пород иногда приходится определять так называемую растворимую (аморфную) кремнекислоту. Л1етод ее определения основан на сравнительно легкой растворимости как искусственно приготовленного геля кремневой кислоты, так и кремнекислоты минералов группы опала в растворах едких щелочей и карбонатов щелочных металлов.
Ход определения. Навеску породы в 1—5 г, измельченную несколько более крупно, чем обычно, помещают в стакан из йенского стекла или в платиновую чашку емкостью 200—250 мл. Навеску смачивают несколькими каплями воды, приливают 50 мл 5%-ного раствора соды и ставят на полчаса на кипящую водяную баню. Стакан прикрывают часовым стеклом. Через полчаса, в течение которых содержимое стакана через каждые 10 мин перемешивают, стакан снимают с водяной бани и раствор сейчас же фильтруют 3 через плотный фильтр, стараясь не взмучивать осадка. Рекомендуется фильтровать в высокий стакан, в который предварительно наливают 5%-ную соляную кислоту. В этом случае одновременно с фильтрованием происходит подкисление содового раствора соляной кислотой, которое протекает довольно спокойно, и почти устраняется соприкосновение со стеклом горячего щелочного раствора. Стакан, в который фильтруют, прикрывают часовым стеклом. К оставшемуся в стакане осадку приливают 20 мл горячей воды, перемешивают и,, дав осадку отстояться, сливают через тот же фильтр. Фильтрат переносят в фарфоровую чашку и выпаривают для выделения кремнекислоты. Операцию определения SiO2 проводят, как при обычном силикатном анализе. Под воронку с фильтром подставляют стакан, из которого производилось фильтрование, фильтр осторожно протыкают стеклянной палочкой и частицы осадка с фильтра тщательно смывают 50 мл 5%-ного раствора соды обратно в стакан, в котором находится главная масса осадка.
Операцию выщелачивания породы повторяют снова с соблюдением тех же условий. Фильтры от всех проводящихся фильтрований сохраняют; в дальнейшем их обрабатывают в отдельном стакане горячей водой, которую используют для промывания последнего осадка.
При фильтровании в большинстве случаев первая порция фильтрата бывает мутной. Поэтому рекомендуется поступать следующим образом. Конец трубки воронки закрывают пальцем, наполняют воронку жидкостью и дают ей стечь в тот же стакан, в котором находится фильтруемая жидкость. После того как стекающая с носика воронки жидкость станет прозрачной, ее собирают в другой чистый стакан. Осадок на фильтре промывают 2 раза. Во избежание взмучивания и прохождения частичек осадка через фильтр промывание проводят не из промывалки, а приливая
1 Об определении так называемой «растворимой» кремнекислоты см. также раздел «Кремний» (стр. 932).
2 Подробно см. А. И. Пономарев, Методы химического анализа минералов и горных пород, т. I, изд. АН СССР, 1951.
3 Воронка должна быть быстро фильтрующей.
1038
Гл. LIII. Методы анализа силикатных пород
промывную жидкость по палочке. Промывание может вестись горячим 1 %-ным раствором азотнокислого аммония, а также, как рекомендует Лунге, горячим раствором соды, к которому для получения прозрачных фильтратов прибавлено немного спирта.
Трудно сказать, сколько таких вытяжек необходимо сделать из навески, чтобы быть уверенным в полноте извлечения растворимой кремнекислоты. Для разных пород вопрос решается по-разному. Так, например, для трепелов, чтобы полностью извлечь растворимую кремнекислоту, обычно бывает достаточно 2—3 таких вытяжек, для опок — около 3—5. Для того чтобы определить окончание обработки, поступают следующим ооразом. Очередной фильтрат содовой вытяжки, которым предполагают закончить операцию, выпаривают досуха, остаток высушивают на водяной бане и выделившуюся кремнекислоту отфильтровывают. Осадок прокаливают и взвешивают. Если количество кремнекислоты окажется незначительным, дальнейшую вытяжку не проводят. Убедившись таким путем в полноте извлечения из породы растворимой кремнекислоты, соединяют накопившиеся фильтраты (вытяжки) и выпаривают на водяной бане для выделения кремнекислоты. Доп. ред*
Часть четвертая
АНАЛИЗ
КАРБОНАТНЫХ ПОРОД
\
Глава LIV
ВВЕДЕНИЕ В АНАЛИЗ
КАРБОНАТНЫХ ПОРОД
КАЧЕСТВЕННОЕ СРАВНЕНИЕ КАРБОНАТНЫХ ПОРОД С СИЛИКАТНЫМИ
Анализ совершенно чистого карбоната любого металла крайне прост. Встречающиеся в природе карбонаты никогда не бывают совершенно чистыми, но иногда они достигают большой чистоты, например когда находятся в виде хорошо образованных кристаллических минералов, как кальцит, магнезит, сидерит или церрусит. Но даже и в таких случаях обычно оказывается, что кристаллы содержат небольшие включения посторонних веществ или представляют собой изоморфную смесь нескольких карбонатов, и тогда разделение может потребовать значительной затраты времени и труда. Большие отложения карбонатов в земной коре никогда не имеют такого простого состава. Даже ослепительно белый мрамор содержит помимо карбоната кальция другие компоненты, и степень его чистоты может изменяться от почти однородного карбоната кальция до того несколько неопределенного состояния, когда породы называются уже не известняками или доломитами, а глинистыми сланцами, известковыми песчаниками и т. и.
Вещества, не представляющие собой карбонатов и могущие содержаться в этих породах, являются главным образом первоначальными образованиями, попавшими в породы во время отложения последних. Они могут состоять из остатков прежних геологических отложений в виде песка или глины, которые в течение дальнейшего времени претерпели очень незначительные изменения или вовсе не изменились, и из углеродистых веществ, образовавшихся из морских флоры и фауны, существовавших во время отложения карбонатной породы. Но кроме указанных веществ, в породе могут находиться еще и вторичные минералы, которые произошли вследствие химических превращений, происходящих внутри самой породы, самостоятельно или под действием динамических сил, например продолжительных колебаний земной коры или интрузии горячих изверженных пород. Изменения, производимые такими силами, могут быть чрезвычайно глубокими, и в результате этих изменений могут образоваться разнообразные минералы, совершенно отличающиеся от первоначальной породы.
Одновременно с такими изменениями могло произойти проникновение в породу или примешивание к ней новых веществ, но если этого и
66 Заказ 522.
1042
Гл. LIV. Введение в анализ карбонатных пород
не было, то во всяком случае химические компоненты продуктов разрушения первоначальных примесей становились при этом частью . карбонатной породы. Эти компоненты имеют много общего с компонентами силикатных пород земной коры, из чего следует, что карбонатные породы в качественном отношении существенно не отличаются от силикатных пород, за исключением того, что углистые вещества являются в первых более обычными примесями. Полный анализ карбонатных пород требует поэтому большинства разделений, описанных в части III этой книги. В некотором отношении ход анализа будет, однако, отличаться, потому что здесь в большинстве случаев преобладают вещества, легко растворимые в минеральных кислотах. Это обстоятельство или позволяет совершенно исключить сплавление пробы с карбонатами, или допускает применение в таких сплавлениях гораздо меньшего количества плавня, что упрощает последующую обработку. Анализ упрощается также и тем, что элементы, сильнее всего затрудняющие анализ силикатов, здесь обычно содержатся лишь в незначительных количествах. Некоторые из них, как, например, кремний, алюминий и щелочные металлы, отсутствуют только в самых чистых карбонатных породах, другие, более редкие компоненты, например цирконий, барий и стронций, часто совсем не присутствуют в определимых количествах.
Дальнейшее изложение относится главным образом к тем мощным образованиям карбонатов, называемых известняками, магнезиальными или доломитовыми известняками и доломитами, из которых многие имеют большое и все возрастающее экономическое значение. Даже у геологов нет определенных правил для строгого их разграничения, а из соотношения окислов следует, что всякое разграничение будет произвольным, так как известняки и доломиты образуют совершенно непрерывный ряд.
Строго говоря, доломитом следовало бы называть только эквиатом-ную двойную соль кальция и магния CaMg(CO3)2, и так считают многие геологи. С другой стороны, многие придерживаются противоположной крайности, утверждая, что все известняки, содержащие только 1—2% карбоната магния, следует считать доломитами. Однако большинство геологов х, по-видимому, предпочитает все известняки, содержащие от 5 до 18% карбоната магния, называть магнезиальными известняками, те, у которых содержание карбоната магния ниже 5%, — называть известняками и, наконец, те, у которых содержание карбоната магния превышает 18%, — доломитами.
Ряд геологов, принимая это последнее разделение, предлагает, однако, породы с содержанием карбоната магния в 20% и выше, приближаясь к формуле CaMg(CO3)2, называть доломитовыми известняками, оставляя название доломиты только за породами, точно отвечающими этой формуле. Пфафф 1 2 называет известняки, содержащие от И до 46% MgCOs, доломитами, потому что такие встречаются чаще всего, тогда как известняки с 7—11% MgCOs, по его словам, очень редки, если они вообще когда-либо встречаются.
Детальный анализ карбонатных пород требуется редко. Большинство анализов проводится в целях технического использования этих пород и в этих случаях в них определяют только наиболее важные компоненты.
1 См. Н. R i е s, Bull. N. Y. State Mus., 44, 644 (1901).
2 F. W. Pfaff, Neues Jahrb. Beilage, Bd. 23, 529 (1907).
Минеральный состав карбонатных пород
1043
Тогда прямое определение двуокиси углерода совсем опускают и либо вычисляют ее содержание по общему содержанию кальция и магния, либо принимают за содержание двуокиси углерода потерю в массе при прокаливании. Нерастворимый в кислоте остаток часто принимают за кремнекислоту. При определении железа последние пересчитывают на Fe2O3; на воду и углистые вещества совсем не обращают внимания, так же как и на титан, фосфор и более редкие компоненты, а серу почти всегда представляют в результатах анализа в виде SO3.
Такой ход анализа приводит к большим ошибкам: ряд элементов и их содержание в таблице результатов совсем отсутствует, а кроме того, найденные значения содержания отдельных компонентов оказываются неправильными. Ошибки эти, хотя иногда и не имеют существенного значения, если анализ проводится для технических целей, однако значительно понижают ценность анализа с точки зрения геолога, так как последний стремится по результатам анализа вывести возможно более точные данные о возрасте или о происхождении ценных в экономическом отношении рудных месторождений, которые могут представлять собой или остатки известняка, разложившегося под атмосферным воздействием, или отложения руд свинца, цинка, ванадия и т. п., образовавшихся в недрах земной коры при участии циркулирующих вод.
МИНЕРАЛЬНЫЙ СОСТАВ КАРБОНАТНЫХ ПОРОД
Главные минералы, участвующие в образовании карбонатных пород, — это, конечно, прежде всего карбонат кальция, образующий собственно известняки, и затем двойной карбонат кальция и магния (минерал доломит), количество которого в известняках относительно мало, больше в магнезитовых известняках и всего больше в природных доломитах. В качестве примесей эти минералы почти всегда содержат другие карбонаты, в особенности карбонаты железа и марганца, образующие изоморфные смеси с тем или другим из названных минералов. Примеси эти обычно присутствуют в значительно меньших количествах, чем главные компоненты, но иногда их содержание может достигать нескольких процентов. Если же они преобладают, то порода уже будет не известняком, а железной или марганцевой рудой.
Пренебрежение этими второстепенными карбонатными компонентами пород бывает часто причиной ошибок при подведении итогов результатов анализа, в которых нередко в виде карбонатов приводят только кальций и магний, присутствующие одновременно даже значительные количества железа представляют в виде Fe2O3, а марганца — в виде какого-нибудь из его высших окислов, хотя почти с полной уверенностью можно полагать, что эти элементы в преобладающем большинстве случаев присутствуют в виде карбонатов железа (II) и марганца (II). Так ли это или нет, часто-может раскрыть точное определение содержания СО 2 в породе, ибо если железо и марганец действительно присутствуют в виде карбонатов, то СО2 будет обнаружено больше, чем это отвечало бы найденным количествам СаО и MgO. Но, с другой стороны, если избытка СО2 не окажется, то отсюда никак нельзя с уверенностью вывести, что карбоната железа и карбоната марганца нет совсем, так как очень часто небольшая часть MgO может присутствовать в породе в составе силикатов, а в некоторых породах небольшое количество СаО может находиться в виде гипса. Последнее мало вероятно в породах средней твердости, применяемых 66*
1044
Гл. LIV. Введение в анализ карбонатных пород
в производстве цемента. В таких породах сера, представляемая в результатах анализа в виде SOs, в действительности большей частью находится в виде пирита или в составе органических веществ. Из сказанного легко понять, что точное соотношение между карбонатами кальция и магния установить невозможно, разве только при отсутствии марганца и железа. В некоторых исключительных случаях это можно сделать, обрабатывая породу разбавленной соляной кислотой и анализируя отдельно полученный раствор и нерастворимый остаток.
Самым распространенным в известняках сульфидом является пирит, хотя присутствие его часто бывает трудно установить вследствие исключительно высокой его дисперсности или одновременного присутствия в породе органических веществ. В метаморфированных известняках, особенно таких, которые были изменены интрузией изверженных пород, нередко встречаются и другие сульфиды, например сфалерит, галенит. Иногда они образуют значительные скопления или же в результате окисления превращаются в руды еще более ценные. Сильно метаморфированные кремнеземистые известняки редко, однако, являются объектом химического анализа, особенно для промышленных целей.
Силикатные компоненты карбонатных пород могут быть самого разнообразного характера. Одни их названия могли бы заполнить целую страницу. Достаточно отметить здесь, что в метаморфированных породах могут присутствовать гранат, везувианит, волластонит, тремолит, диопсид, скаполиты, турмалин, аппатит и двуокись кремния в виде кремня. Известняки, содержащие кремень, встречаются чрезвычайно часто, кремнекислота вошла в их состав, несомненно, во время их метаморфического превращения. Главнейшим источником находимой в неметаморфированных известняках кремнекислоты являются, как сказано выше, песок и глина. Последняя всегда содержит в себе воду, и на этот факт в рядовых анализах обычно не обращают внимания. Иногда в таблице результата анализа вода совсем не приводится, хотя одновременно отмечено наличие большого количества глины.
Песок состоит обычно главным образом из кварца, но может частично содержать и другие минералы, часто находимые в песках, например слюду, гранат и циркон. Последние можно часто обнаружить даже в наименее кремнеземистых и глинистых известняках, растворяя большое количество пробы в соляной или уксусной кислоте и подвергая остаток микроскопическому исследованию после сжигания углистых веществ, если последние присутствовали. Непрокаленный остаток содержит, конечно, весь пирит и другие сульфиды, не поддающиеся действию разбавленных кислот.
Остаток этот в случае анализа некремнеземистых пород может состоять в значительной мере даже из окиси железа, в большинстве случаев, вероятно, в форме одного из гидратов окиси, но часто в форме гематита. Иногда присутствие такого компонента в породе может быть обнаружено по красноватому ее оттенку при условии, что углистых веществ не так много, чтобы они могли маскировать этот оттенок. Присутствие окиси железа (III) не всегда несовместимо с одновременным наличием таких углистых веществ.
Пурпурно-красное окрашивание породы может зависеть, как в некоторых сланцах, от одновременного присутствия окиси или гидроокиси железа (III) и углистых веществ. Одни углистые вещества придают породе часто встречающуюся серую или черную окраску. Зеленоватая окраска
Распознавание различных карбонатов
1045
осадочного известняка может происходить от присутствия некоторых минералов из группы слюд, например хлоритов.
В виде каких соединений присутствует в карбонатных породах фосфор, когда содержание его незначительно, с уверенностью определить нельзя. В виде следов он может иногда находиться в нерастворимом остатке по-видимому, в форме тяжелых фосфатов, например монацита или ксенотима, в других случаях он связан с железом и алюминием. В породах, очень богатых фосфатами, фосфор в преобладающем большинстве случаев присутствует в виде фосфорита или апатита, который может даже стать главным компонентом породы.
Известно, что ванадий является постоянным, хотя и второстепенным компонентом как известняков, так и песчаников и изверженных пород. Анализ навесок известняка величиной в 100 г из мощного его месторождения, находящегося в северо-западной Мексике, показал, что эта порода, помимо ванадия, содержит также молибден и никель. В виде каких соединений эти элементы находятся в известняках, точно не известно, хотя вполне вероятно, что ванадий был первоначальным компонентом алюмосиликатов глины и что никель, цинк и свинец присутствуют преимущественно в виде карбонатов или сульфидов.
Углистые вещества часто считаются распространенным компонентом второстепенного значения. Они отсутствуют в белых сортах мрамора и в некоторых окрашенных декоративных камнях, но в состав других известняков входят в значительных количествах.
Некоторые сорта битуминозных известняков отличаются тем, что при дроблении или растирании распространяют неприятный запах. Последний вызывается в большинстве случаев сероводородом, который или присутствовал в известняке со времен его образования или получился при разложении органических веществ. Иногда присутствие сероводорода несомненно; в одном из издававших неприятный запах известняков из Канады сероводород был определен в жидких включениях в количестве 0,02%.
Все сказанное выше никоем образом не исчерпывает вопроса о минеральном составе известняков (это наименование применено здесь и в других местах как родовое и включает магнезиальные их разновидности). Однако эти замечания могут служить достаточным руководством для химиков, которые предполагают заниматься анализом этих пород. Незнакомство с второстепенными их компонентами бывает причиной многочисленных ошибок в анализе.
РАСПОЗНАВАНИЕ РАЗЛИЧНЫХ КАРБОНАТОВ
ПО ИХ ОТНОШЕНИЮ К РАЗНЫМ РЕАКТИВАМ
Для аналитика, работающего в хорошо оборудованной лаборатории, методы качественного различия отдельных разновидностей карбонатных пород (помимо обычных методов качественного анализа) имеют малое значение. Все же некоторые методы, позволяющие быстро различать породы, могут оказаться очень полезными, особенно в полевых условиях, когда трудно или невозможно брать с собой большие количества жидких реактивов или громоздкие приборы.
Различная растворимость карбонатов в винной и лимонной кислотах и в бисульфате калия. Этот вопрос, временами затрагивавшийся прежними
1046
Гл. LIV. Введение в анализ карбонатных пород
исследователями, был впервые изучен Болтономх, и много времени спустя многие из его выводов были подтверждены последующими исследователями 1 2. Твердые органические кислоты, как лимонная и винная, вследствие различного их действия на разные карбонатные минералы оказались очень хорошими реактивами для отличия последних. О действии бисульфата калия как реактива для различения карбонатов имеются как ранние данные 3, так и полученные более поздними исследователями 2. Здесь достаточно сослаться на указанные оригинальные работы, особенно на статьи Болтона, где приведены данные о растворимости самых разнообразных минералов как в указанных выше реактивах, так и в других (неорганических) растворителях.
Различие между кальцитом и арагонитом. Для качественного отличия кальцита от арагонита применение одних только химических методов исследования не решает вопроса4 s, «в особенности там, где эти методы чаще всего применяются, а именно для характеристики тонко измельченного материала, который может содержать вещества, мешающие испытанию», поэтому описание этих методов здесь опускается. Оптические методы в опытных руках заслуживают большего доверия.
Различие между кальцитом и доломитом. Надо помнить, что магнезиальные и доломитовые известняки являются, как правило, смесями двойного карбоната — доломита с кальцитом, а не смесями кальцита с магнезитом.
Кальцит легко растворим в разбавленной соляной или уксусной кислотах, доломит и магнезит в них почти нерастворимы. Поэтому под действием этих кислот кальцит сильно вскипает, даже если обрабатывать неизмельченные его куски, в то время как доломит дает лишь очень слабую реакцию или не показывает никакой реакции. На этом основан простой метод различения указанных двух минералов. Доломитовые известняки, соответственно содержанию в них кальцита, также разлагаются, хотя и слабо, так что в порошкообразной смеси нетрудно осуществить приблизительное отделение кальцита от доломита и магнезита, но только не от водных карбонатов магния. Последние, впрочем, никогда не являются существенной составной частью мощных известковых образований.
Отношение этих карбонатов к высокой температуре также иногда может служить отличительным признаком, потому что доломит так легко выделяет свою двуокись углерода, что порошок с силой выбрасывается из тигля, если его нагревают без соблюдения мер предосторожности, в то время как кальцит можно сильно прокаливать сразу, не опасаясь потерь твердого вещества.
Было найдено 8 также, что порошок кальцита принимает фиолетовую окраску при обработке его раствором хлорида алюминия и гематоксилина (отвар кампешевого дерева) и что, напротив, доломит остается неизмененным. Для приготовления указанного реактива растворяют 4 весовые части
1 Н. С. Bolton, Ann. New York Acad. Sci., 1, 1 (1877—1880); 2, 1; Proc. Am. Assoc. Advancement Sci., 31, 271 (1883); Chem. News, 36 , 37 , 38, 43, 47 (1877—1883); Brit. Assoc. Advancement Sci. Rep., 506 (1880); Mineralog. Mag., 4, 181 (1880—1881); Ber. dent. chem. Ges., 13, 726 (1880).
2 J. W. Richards, N. S. Powell, J. Am. Chem. Soc., 22, 117 (1900).
3 E. J a n n e t t a z, C. r., 77, 838 (1873); 78, 852 (1874).
4 J. Johnston, H. E. Merwin, E. D. Williamson, Am. J. Sci., [4], 41, 473 (1916). Эта статья содержит сводку литературы по данному вопросу.
s J. Lemberg, Z. deut. geol. Ges., 40, 357 (1888).
Распознавание различных карбонатов
1047
сухого хлорида алюминия в 60 частях воды, прибавляют 6 весовых частей кампешевого дерева и кипятят, подливая воду (вместо выкипающей), в течение 25 мин. Темно-фиолетовый раствор затем фильтруют. Грубо измельченный исландский шпат и каррарский мрамор принимали фиолетовую окраску после погружения в этот раствор на 5—10 мин и омывания от приставшей жидкости. В измельченном доломитовом известняке можно было с помощью этого реактива легко отличить зерна кальцита от зерен доломита. Не рекомендуется затягивать обработку слишком долго, потому что по истечении 20 мин на доломитовых участках могут выступить бледно-голубые пятна. Реакция основана на том, что на кальците осаждается гидроокись алюминия, образующая с красящим веществом (гематоксилином) окрашенный лак.
Было установлено далее х, что известняк (кальцит) в 10 %-ном растворе хлорида железа (III) выделяет СО2 при комнатной температуре, а доломит — только при нагревании. В качестве реактива можно также применять раствор сульфата меди, из которого кальцит осаждает основной карбонат меди, а доломит не осаждает.
Возможно, что эти последние методы к1 2 могут вызвать такие же возражения, как и методы отличия кальцита от арагонита.
1 F. Hinden, Verhandl. naturforsch. Ges. Basel. 15, 1 (1905).
2 .1. Lemberg, Z. deut. geol. G-es., 40, 357 (1888).
Глава LV
ТОЧНЫЕ МЕТОДЫ АНАЛИЗА КАРБОНАТНЫХ ПОРОД
КРЕМНЕКИСЛОТА, ОТДЕЛЕНИЕ ЕЕ ОТ ОКИСИ АЛЮМИНИЯ И ДРУГИХ ОКИСЕЙ
Методы разложения породы
Лишь очень немногие известняки бывают настолько чистыми, что при обработке их соляной кислотой не оставляют никакого остатка. Обычно получаемый остаток может состоять из кварца, глины и других силикатов, углистых веществ, пирита и т. п., и способ разложения породы в каждом отдельном случае зависит от количества нерастворимых веществ, а также от того, предполагают ли анализировать их отдельно от растворившейся части породы или нет.
Неорганический остаток анализируют отдельно. Смачивают 1 г измельченной в порошок породы 1 водой и в покрытом часовым стеклом стакане обрабатывают соляной кислотой или (если опасаются, что она разложит заметное количество силикатов) разбавленной уксусной кислотой, пока совершенно не прекратится выделение газа. Если выделение газа идет так слабо, что это указывает на доломитовый характер анализируемой породы, то необходимо умеренное подогревание. Раствор фильтруют затем через фильтр (диаметр 7 см) и промывают остаток водой или горячей разбавленной соляной кислотой, если присутствует гипс, а силикаты этой кислотой не разлагаются. Фильтр вместе с его содержимым сжигают мокрым в платиновом тигле и прокаливают (применение паяльной горелки необходимо только в том случае, когда нерастворимый остаток значителен по величине). После взвешивания остаток сплавляют с карбонатом натрия и обрабатывают дальше, как силикатную породу (стр. 939 и дальше).
Если остаток содержит гидратные минералы, то первоначальное содержание в нем воды лучше всего определить в отдельной навеске пробы, а не (как это иногда делается) высушиванием фильтра с нерастворимым
1 Для определения компонентов, содержание которых превышает 5%, берут отдельные навески, высушивают при 105—110° С в стаканчике для взвешивания, взвешивают, высыпают насколько возможно полнее в сосуд, где будет проводиться растворение, и снова взвешивают стаканчик.
Кремнекислота, отделение ее от окиси алюминия
1049
остатком, взвешиванием его (на вторую чашку весов при этом помещают пустой фильтр) и прокаливанием. Найденную массу воды надо прибавить к массе прокаленного остатка, чтобы получить таким образом истинный вес нерастворимой части пробы.
Если прокаленный остаток состоит только из кварца или количество его незначительно, его можно тотчас же обработать каплей серной кислоты и несколькими каплями плавиковой кислоты. Кислоты удаляют потом в радиаторе (см. рис. 5, стр. 48). Если после этого остается еще видимый маленький остаток, то хорошо повторить обработку кислотами и выпаривание, так как следует подчеркнуть, что кварц устойчивее по отношению к действию фтористоводородной кислоты, чем многие силикаты, и что иногда к цели приводит только многократное выпаривание (или предварительное измельчение кварца в чрезвычайно тонкий порошок). Если после пятиминутного прокаливания на полном пламени горелки вес больше не изменяется, то потеря в весе показывает содержание кремнекислоты. Если после этого остается еще небольшой остаток, то его нужно сплавить с небольшим количеством карбоната натрия, растворить плав в соляной кислоте и полученный раствор соединить с первым солянокислым фильтратом, который затем обрабатывают для определения железа, алюминия и пр. лучше всего, как описано на стр. 1052.
Если первоначальный остаток мал и нужно точно определить его состав, то для анализа следует обработать кислотой несколько граммов породы. В дальнейшем ходе анализа следует взять весь фильтрат от нерастворимого остатка для определения перешедших в раствор железа, алюминия и марганца (стр. 1052), а затем после отделения этих элементов отобрать аликвотную часть фильтрата для определения кальция и магния.
Если, как это иногда бывает, отфильтрованный от нерастворимого остатка раствор заметно окрашен органическими веществами, то последние должны быть совершенно разрушены до осаждения железа и алюминия, потому что иначе один из этих металлов или оба они могут осесть не полностью. Обычными окислителями не всегда удается полностью удалить эти мешающие вещества, поэтому раствор надо выпарить и прокалить остаток до обугливания. При этом, во избежание потери железа вследствие улетучивания хлорида железа (III), нужно выпарить раствор раза два досуха с азотной кислотой и затем только осторожно нагреть остаток до нужной температуры на голом пламени. Если это почему-либо кажется нежелательным, то лучше для проведения анализа обработать новую навеску пробы по одному из способов, приведенных ниже.
Неорганический остаток не анализируют отдельно. Чаще всего отдельный анализ неорганического остатка не делают. Как было выше сказано, остаток может состоять из глины или других силикатов, углистых веществ, пирита и т. п. с кварцем или без кварца. Несгораемая часть нерастворимого остатка может быть переведена в раствор двумя способами.
Растворение в кислоте после сильного прокаливания. Этот метод оказывается самым лучшим в тех случаях, когда отношение содержания нерастворимой части к содержанию растворимой части не меньше, чем в тех глинистых известняках, которые без дальнейшей обработки могут быть обожжены для получения портланд-цемента, т. е. когда содержание кремнекислоты не на много превышает 15%, а содержание окислов железа, алюминия и титана вместе составляет не более 6%. Допустимые пределы здесь еще точно не установлены, а также неизвестно, каково значение этого
1050
Гл. LV. Точные методы анализа карбонатных пород
отношения в доломитах и доломитовых известняках. Вопрос требует дальнейшего исследования.
Оказывается, известняки, в которых содержание кремнекислоты, окиси алюминия и т. п. веществ не превышает только что указанного, прокаливанием в течение 10—15 мин на хорошей паяльной горелке, дающей пламя с температурой в 1100—1200° С, можно превратить в форму, совершенно растворимую в соляной кислоте, при условии, что порода была предварительно измельчена в очень тонкий порошок.
Прокаливают 1 г измельченной породы в закрытом тигле при 1100— 1200° С. При анализе известняков можно, не боясь потерь, применять паяльную горелку сразу или после короткого прокаливания при 700—900°С. Но известняки, богатые магнием, следует прокаливать с величайшей осторожностью, если вообще можно применять к ним этот способ, так как их температура разложения лежит гораздо ниже температуры разложения обыкновенных известняков. Часто порода начинает с силой выбрасываться из тигля еще задолго до достижения температуры красного каления.
Сильное прокаливание прекращают обычно через 10—15 мин. Остаток после прокаливания имеет вид спекшейся или твердой массы, которая большей частью легко отделяется от тигля.
Во время прокаливания удаляется вся СОа, вся вода и все органические вещества; пирит окисляется, а его сера удерживается полностью в виде сульфата кальция. При продолжительном прокаливании вся трехокись серы постепенно выделилась бы из сульфата кальция, а потом улетучились бы щелочные металлы, которые можно полностью удалить прокаливанием в течение часа или даже более короткого времени на сильной паяльной горелке. При наклонно стоящем пламени паяльной горелки соли щелочных металлов частью осаждаются на нижней поверхности крышки тигля и образуют там растворимый налет, обладающий сильнощелочной реакцией, который иногда весит несколько миллиграммов. При указанной выше продолжительности прокаливания потери щелочных металлов, по-видимому, не происходит.
Остаток после прокаливания переносят в стакан или чашку и смачивают водой. Приставшие к тиглю частицы растворяют в разбавленной (1 : 1) соляной кислоте, и полученный раствор присоединяют к главной массе. Осторожно нагревая и раздавливая крупные куски плоским концом стеклянной палочки, в течение нескольких минут переводят в раствор большую часть спекшейся массы. Некоторое количество кремнекислоты может остаться нерастворенным в виде хлопьев; это, однако, не имеет значения. Когда исчезнут все зернистые частицы, раствор, если он находится в стакане, переносят в чашку и выпаривают досуха. Если растворение велось с самого начала в чашке, то раствора в ней должно быть лишь несколько миллилитров и для выпаривания требуется непродолжительное время. Для выпаривания вполне достаточно чашки значительно меньших размеров, чем те, которые применяются в анализе силикатов, потому что в этом случае раствора мало и разложение проводилось без прибавления нелетучих солей.
Растворение в кислоте после сплавления с карбонатом натрия. Когда силикаты присутствуют в таком количестве, что сильным прокаливанием нельзя получить массы, полностью растворимой в кислоте, необходимо измельченную породу смешать с карбонатом натрия и разложить силикаты сплавлением на паяльной горелке. Способ этот при желании может быть применен ко всем известнякам, содержащим силикаты. Требуемое количество плавня значительно меньше того, какое применяется в анализе
Кремнекислота, отделение ее от окиси алюминия
1051
силикатов. Достаточно 0,25 или 0,5 г на 1 г известняка, потому что образующаяся при прокаливании известь сама является сильным плавнем, и нет никакой необходимости в получении жидкого плава. Достаточно только спекания. Указанное количество плавня достаточно также для известняков, богатых MgO, и для цементных пород, которые содержат только 50% СаО.
Отделение кремнекислоты
Приступая к отделению кремнекислоты, следует тщательно просмотреть замечания, приведенные на стр. 940.
Так как здесь отсутствуют большие количества солей щелочных металлов, а также кремнекислоты, процесс выпаривания солянокислого раствора, полученного, как указано выше (стр. 1048 и 1049), занимает меньше времени по сравнению со временем, необходимым для анализа силикатов. Когда раствор выпарится (или почти выпарится) досуха на паровой бане, чашку можно поставить на воздушную баню или (покрыв ее) на платиновый треугольник, лежащий на плитке, и нагревать 1 ч при температуре не выше 110° С. Более высокой температуры или более продолжительного нагревания следует избегать, так как иначе кремнекислота и окись магния могут снова соединиться, в результате чего при последующей обработке кислотой кремнекислота снова перейдет в раствор.
Имеются указания 1 на то, что при нагревании до 200° С кремнекислота полностью выделяется после однократной обработки, но мы не могли подтвердить этого, хотя количество кремнекислоты, переходящей в раствор после прибавления кислоты, очень мало, и редко превышает 2 или 3 мг. Поэтому при точной работе, если кремнекислота находится в количестве 2—4% и выше, ее следует отфильтровать после переведения в нерастворимое состояние, и раствор выпарить снова. Для этого смачивают сухой остаток 10 мл соляной кислоты, затем прибавляют 100 мл горячей воды, чашку покрывают часовым стеклом и ставят на баню на 10 мин. Затем переносят кремнекислоту на фильтр подходящей величины, тщательно промывают ее разбавленной (1 : 99) соляной кислотой и потом дважды водой. Фильтрат выпаривают снова досуха, остаток обрабатывают так же, как и раньше, но половинным количеством соляной кислоты и воды и в течение нескольких минут. Раствор затем фильтруют еще раз через второй, меньший по размерам фильтр, фильтр и осадок промывают сначала холодной разбавленной (1 : 99) соляной кислотой, а затем горячей водой. Оба фильтра с их содержимым медленно высушивают, озоляют и прокаливают в платиновом тигле, под конец в течение 10 мин при 1200° С. Чтобы исследовать прокаленный остаток на чистоту, прибавляют 6 мл фтористоводородной кислоты и 1—2 капли разбавленной (1 : 1) серной кислоты, выпаривают и продолжают работу, как описано на стр. 943. Остаток, который получается после прокаливания, всегда значительно меньше, чем находимый при анализе силикатных пород. После удаления серной кислоты почти всегда достаточно прокалить его 1—2 мин на полном пламени горелки (1000° С). По качественному составу он сходен с остатком, получаемым при анализе силикатов. Он никогда не содержит кальция и магния п состоит главным образом из окиси алюминия и небольшого количества
1 В. Blount, J. Soc. Chem. Ind., 21, 1217 (1902).
1052
Гл. LV. Точные методы анализа карбонатных пород
окислов железа, титана и фосфора. Его следует сплавить с небольшим количеством карбоната натрия, плав растворить в соляной кислоте и полученный раствор присоединить к фильтрату от кремнекислоты.
АЛЮМИНИЙ, ОБЩЕЕ СОДЕРЖАНИЕ ЖЕЛЕЗА, ТИТАН (КРЕМНЕКИСЛОТА, МАРГАНЕЦ); ОСАЖДЕНИЕ ВМЕСТЕ С ФОСФОРОМ 1
Методы дальнейшей обработки значительно отличаются друг от друга в зависимости от общего содержания железа, алюминия и других элементов и от того, предполагают ли осаждать марганец вместе с указанными элементами или же после их отделения. Опытные аналитики часто расходятся в выборе метода, и мы не намерены предписывать здесь определенный ход анализа, которому всякий должен безусловно следовать; в дальнейшем описаны также не все методы разделения, могущие дать хорошие результаты.
Есть такие методы, которые может быть и заслуживают доверия, но еще не настолько изучены, чтобы их можно было здесь привести. Может случиться, что метод, который при одном сочетании элементов оказывается хорошим, становится негодным, если к этим элементам будет прибавлен хотя бы еще один элемент 2.
Осаждение алюминия, железа и аналогичных элементов
Совместное осаждение алюминия, железа, марганца, титана и фосфора сульфидом аммония. Если содержание в породе этих элементов в общей сложности не превышает 2—3%, то, сконцентрировав фильтрат от кремнекислоты (стр. 1051), переносят его в колбу емкстью 150 мл и прибавляют столько соляной кислоты, чтобы потом, когда соляная кислота будет нейтрализована аммиаком, образовалось достаточно хлорида аммония для предупреждения осаждения магния. Последнее очень важно. Затем прибавляют раствор аммиака, свободный от карбоната аммония, до сильно щелочной реакции, насыщают раствор сероводородом и прибавляют еще столько аммиака, сколько было прибавлено раньше сверх количества, необходимого для нейтрализации соляной кислоты. Наполняют колбу прокипяченной водой до горла, закрывают ее пробкой и оставляют на 12—24 ч. Фильтруют через небольшой фильтр (диаметр 7 см) и промывают осадок водой, содержащей немного сульфида аммония и хлорида аммония. Осаждение того или иного компонента осадка может оказаться неполным, однако количества, которые могут быть еще найдены в фильтрате, обычно бывают очень малы. (О дальнейшей обработке фильтрата см. ниже, стр. 1054.)
После промывания хлоридом аммония фильтр вместе с его содержимым осторожно озоляют и прокаливают. При этом дают свободный доступ воздуху. После прокаливания в течение самое большее 5 мин косо направленным пламенем паяльной горелки осадок взвешивают и получают сумму А12О3 + Fe2O3 + Mn304 + TiO2 + Р2ОВ + другие окислы этой
1 См. также стр. 949.
2 Например, осаждение едким натром и карбонатом натрия, прибавляемым в избытке, дает удовлетворительные результаты при отделении железа от алюминия в присутствии кальция, но непригодно в присутствии магния.
Алюминий, общее содержание железа, титан
1053
группы, например ZrC>2, если они присутствовали. (Об их разделении см. стр. 1054.)
Метод этот неприменим к анализу фосфатных известняков, содержащих слишком мало алюминия, чтобы полностью связать весь присутствующий фосфор, так как в этом случае в осадок перейдут также фосфаты кальция и магния. В таких случаях можно воспользоваться методами, описанными ниже, если алюминия и железа вместе достаточно, чтобы осадить фосфор; но в большинстве случаев приходится добавлять отвешенное количество чистой соли железа (III), затем осаждать аммиаком или ацетатом натрия и из результата, полученного для железа, вычитать прибавленное его количество.
Осаждение алюминия, железа, титана и фосфора аммиаком. Фильтрат от кремнекислоты после прибавления нескольких капель бромной воды или 2—3 капель концентрированной азотной кислоты кипятят в стакане из устойчивого стекла, пока не удалятся последние следы брома или хлора. Затем, если нужно, прибавляют столько соляной кислоты, чтобы после нейтрализации аммиаком образовалось достаточное количество хлорида аммония для предотвращения осаждения магния. Добавляют несколько капель метилового красного и нагревают раствор (объем 100—200 мл) до начала кипения. После этого нейтрализуют аммиаком (под конец разбавленным), пока окраска раствора не станет желтой. Наконец кипятят раствор 1—2 мин, дают отстояться, фильтруют, немедленно промывают осадок 2—3 раза 2%-ным раствором хлорида аммония и отсасывают досуха (см. стр. 950). Затем осадок растворяют в горячей соляной кислоте умеренной концентрации. Полученный раствор кипятят для удалениия следов хлора, который мог образоваться при растворении выпавшей в осадок двуокиси марганца, и повторяют описанное выше осаждение'2—3 раза, в зависимости от количества содержащегося в породе марганца. Если гидроокиси осаждаемых металлов выделились в большом количестве, то перед последним осаждением прибавляют мацерированную бумагу. Окончательный осадок промывают раствором хлорида аммония (об обработке соединенных аммиачных фильтратов см. ниже).
Влажный осадок прокаливают в том тигле, который содержит остаток после удаления кремнекислоты фтористоводородной кислотой (если только этот остаток не был сплавлен и растворен). Этот остаток, прибавленный к нему главный осадок от аммиака и, наконец, следы гидроокисей, которые выделяются позже (см. ниже, раздел «Обработка фильтратов от гидроокисей алюминия и других элементов»), содержат все железо в виде Fe2O3, всю А12О3, TiO2, Р2О5 и может быть другие окислы, например ZrO2 (разделение прокаленных окислов см. ниже, раздел «Растворение и разделение окислов»).
Осаждение алюминия, железа, титана и фосфора ацетатным методом. Этот метод разделения реже применяется в анализе известняков, чем в анализе силикатов, потому что в известняках элементы, осаждаемые ацетатным методом, присутствуют в относительно малых количествах и легче могут быть отделены аммиаком от значительных количеств марганца. О применении ацетатного метода см. стр. 950. Нужно при этом иметь в виду что в известняках мы имеем дело с меньшими количествами железа и марганца, чем в силикатных породах.
1054
Гл. LV. Точные методы анализа карбонатных пород
Обработка фильтратов от гидроокисей алюминия и других элементов
Соединенные фильтраты, полученные, как указано на стр. 1052 (или на стр. 1053), и содержащие, кроме всего кальция и магния, также марганец и обычно следы алюминия и даже железа, после подкисления соляной кислотой выпаривают в платиновой чашке до небольшого объема и обрабатывают аммиаком до слабощелочной реакции по метиловому красному. Если образуется осадок, его отфильтровывают, слегка промывают, растворяют снова в соляной кислоте (с прибавлением 1—2 капель сернистой кислоты, если окраска осадка показывает, что в нем есть марганец) и снова осаждают его аммиаком при нагревании до кипения в очень маленьком стаканчике. Если нужно, переосаждение повторяют, но это требуется редко. Окись алюминия и, возможно, также ничтожные следы железа, полученные таким путем, присоединяют к ранее найденным (стр. 1052). Дальнейшая обработка фильтрата описана далее (стр. 1055).
Растворение и разделение окислов
Обработка не зависит от того, какой из способов, описанных выше, был применен при осаждении железа, алюминия и пр. В зависимости от количества прокаленных окислов, их сплавляют в закрытом тигле на маленьком пламени с одним или несколькими граммами пиросульфата калия или лучше пиросульфата натрия х. При применении пиросульфата вместо бисульфата значительно сокращается время сплавления, особенно если окислы находятся в тонкоизмельченном состоянии, как это бывает после осаждения в присутствии мацерированной бумаги.
После полного разложения охлажденный плав растворяют в 50 мл горячей разбавленной (5 : 95) серной кислоты, переводят раствор в платиновую чашку и выпаривают, насколько возможно, на водяной бане. Затем приливают 10 мл разбавленной (1 : 1) серной кислоты и продолжают выпаривание на воздушной или песчаной бане, или на голом огне, но так осторожно, чтобы совершенно избежать разбрызгивания; выпаривание продолжают до обильного выделения паров серной кислоты. После охлаждения масса должна быть пастообразной, но не твердой и сухой. Ее растворяют при нагревании в 100 мл воды, и раствор оставляют на 10—15 мин на водяной бане. Обычно при этом на дне чашки появляются хлопья кремнекислоты. Фильтруют через фильтр диаметром 7 см, даже если не заметно осадка кремнекислоты, и промывают фильтр горячей водой, пока стекающая вода не будет свободна от солей.
Кремнекислота (барий). Фильтр с кремнекислотой озоляют, прокаливают и взвешивают. Прокаленный остаток обрабатывают одной каплей серной кислоты и несколькими каплями фтористоводородной кислоты, прокаливают и взвешивают тигель снова. Потеря в массе представляет кремнекислоту, которая осталась в растворе после всех предыдущих отделений. Ее массу прибавляют к массе, найденной ранее.
Если остаток после удаления кремнекислоты имеет красноватую окраску, то его сплавляют с небольшим количеством пиросульфата. Раствор полученного плава, если он прозрачен, присоединяют к фильтрату,
1 О приготовлении его см. стр. 61.
Марганец
1055
который содержит главную массу железа. Если же раствор мутный, то это может зависеть от присутствия следов сульфата бария, которые, конечно, должны быть отфильтрованы, прокалены, взвешены и масса их должна быть вычтена из общей массы окиси алюминия и других окислов.
Железо. В этом растворе железо определяют титрованием перманганатом калия после восстановления, как это описано на стр. 957 (см. «Объемное определение в отсутствие ванадия»). Количество присутствующего ванадия, насколько известно, всегда так мало, что нет необходимости применять способ, описанный в разделе «Объемное определение в присутствии ванадия», стр. 957.
Титан. После определения железа титрованием раствор обрабатывают так, как описано на стр. 960, и определяют титан колориметрическим методом (стр. 966).
Алюминий. Содержание алюминия вычисляют по разности между массой всех окислов и массой окислов железа и титана, найденных, как указано выше, а также фосфорного ангидрида, определенного в отдельной навеске пробы (см. ниже, стр. 1057) и Мп304, если последнюю намеренно осаждали вместе с гидроокисями железа и алюминия методом, описанным на стр. 1052 (Обсуждение преимуществ и недостатков прямых и косвенных методов определения алюминия см. на стр. 953 и 960).
МАРГАНЕЦ
Определение в фильтратах после осаждения
Фильтраты, содержащие кальций, магний и марганец, собирают в колбу емкостью 150 мл, прибавляют 2 мл раствора аммиака и насыщают сероводородом. Затем прибавляют еще 2 мл раствора аммиака. Колбу наполняют свежепрокипяченной водой, закрывают пробкой и оставляют стоять от 12 до 24 ч. Осадок (он может быть окрашен в темный цвет сульфидом платины или сульфидами железа и других тяжелых металлов х) собирают на фильтре (диаметром 7 см) и промывают разбавленным раствором хлорида аммония, содержащим немного сульфида аммония 1 2. (О дальнейшей обработке фильтрата см. ниже, стр. 1057).
Если марганец не собираются определять в отдельной навеске, то очищают колбу от приставших к ней частиц сульфидов разбавленной на половину соляной кислотой, смешанной с пятикратным объемом сероводородной воды. Этот раствор пропускают затем через фидьтр, чтобы растворить сульфид марганца. Если марганец хотят далее определить весовым способом, то фильтрат обрабатывают, как описано на стр. 961.
Принимая во внимание большие ошибки, которые могут быть в результатах, полученных этим методом, если содержание марганца очень мало
1 Железа здесь не должно быть больше ничтожных следов. Но если органические вещества известняка растворимы в кислотах и был применен метод прямого растворения, то легко может случиться, что некоторая часть железа (а также и алюминия) ускользнет от осаждения аммиаком. Тогда эти металлы выпадут вместе с марганцем. Поэтому в случае присутствия органических веществ навеску породы перед ее растворением обязательно надо прокаливать.
2 Промывную жидкость лучше всего приготовлять следующим образом. К небольшому количеству соляной кислоты в маленьком стаканчике прибавляют аммиак в значительном избытке, затем приливают сероводородную воду или несколько минут пропускают сероводород и, если нужнод разбавляют водой.
1056
Гл. LV. Точные методы анализа карбонатных пород
(см. стр. 959), гораздо надежнее солянокислый раствор выпарить с азотной или серной кислотами до удаления последних следов хлора и затем применить колориметрический метод определения марганца (стр. 962).
Если марганец определяют в отдельной навеске пробы, то осадок сульфида марганца отбрасывают, но тогда не стоит и выделять его, если известно, что он присутствует лишь в количестве нескольких сотых процента.
Отдельное определение
Малые количества марганца лучше всего определять колориметрическим методом. Определение более точно, если для его выполнения берут отдельную навеску пробы. Измельченную в порошок породу, если она без остатка разлагается кислотами, растворяют в разбавленной, свободной от хлора азотной кислоте, раствор фильтруют, если нужно, и обрабатывают полностью или аликвотную его часть, как описано на стр. 962. Если анилизируемая порода не вполне разлагается кислотой или раствор получается окрашенным органическими веществами, то лучше всего порошок породы прокалить с карбонатом натрия (взятым в количестве, равном половине его массы и не содержащем марганца) в косо направленном пламени паяльной горелки. Охлажденный плав следует обработать при нагреваний азотной кислотой, пока остаток, который может оказаться, не станет совершенно бесцветным, профильтровать, собирая фильтрат в колбу подходящей емкости, и определить в нем марганец.
МЕДЬ, НИКЕЛЬ, КОБАЛЬТ, СВИНЕЦ, РЕДКОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ, ХРОМ, ВАНАДИЙ И МОЛИБДЕН
Обычно эти элементы бесполезно искать в той небольшой навеске, в которой проводят основные определения. Для этого нужно взять 50— 500 г измельченной породы. Эту навеску растворяют в соляной кислоте и отфильтровывают нерастворимый остаток. Если хотят провести анализ этого остатка, то лучше его анализировать отдельно, сплавляя с карбонатом натрия, выщелачивая плав водой и т. д. Хром, ванадий и молибден будут тогда в водной вытяжке, а другие из перечисленных в заголовке металлов — в остатке. Первые можно определить, как описано на стр. 980, последние — обычными методами.
Солянокислый раствор, содержащий некоторые или все перечисленные выше металлы, лучше всего подвергнуть обработке, описанной на стр. 1052, а проводимое здесь кратное описание может служить руководящей схемой анализа.
Осадок от сульфида аммония растворяют, осаждают сероводородом медь, и, возможно, некоторое количество молибдена, обрабатывают фильтрат окислителем, выпаривают его досуха и кипятят остаток с раствором щавелевой кислоты. Выпадают редкоземельные металлы, возможно загрязненные кремнекислотой; их промывают разбавленным раствором щавелевой кислоты, прокаливают и взвешивают. Осадок редкоземельных металлов может быть далее исследован более подробно методами, описанными в гл. «Редкоземельные металлы» (стр. 661). Фильтраты освобождают от щавелевой кислоты выпариванием и осторожным прокаливанием, остаток растворяют и раствор исследуют на присутствие в нем никеля,
Фосфор
1057
кобальта, цинка и хрома общепринятыми методами. Надо помнить, что фильтрат от первоначального осаждения сульфидом аммония может содержать большую часть ванадия и молибдена и может быть никель, которые следует выделить в осадок подкислением уксусной кислотой.
Ванадий обычно можно открыть и определить с достаточной точностью в 5-граммовой навеске известняка сплавлением с карбонатом натрия и дальнейшей обработкой, описанной на стр. 982.
КАЛЬЦИЙ, СТРОНЦИЙ, БАРИИ, МАГНИЙ [МАРГАНЕЦ] i
Фильтрат, полученный, как описано на стр. 1054 или на стр. 1055, обрабатывают в основном так же, как и в анализе силикатов (стр. 963).
При анализе доломитовых известняков и доломитов обычно невозможно внести поправку на те малые количества кальция, которые содержатся в пирофосфате магния, как это описано во втором методе, приведенном в гл. «Магний» (стр. 722). В таких анализах следует применять метод, описанный на стр. 694 (см. гл. «Щелочноземельные металлы»).
Точное определение бария в известняке — задача более трудная, чем определение его в силикатной породе. Быть может лучше всего выделять барий (если он присутствует) из фильтратов от оксалата кальция. Фильтраты соединяют, выпаривают, освобождают от аммонийных солей (стр. 161), остаток растворяют в возможно меньшем количестве соляной кислоты, прибавляют, не фильтруя, несколько капель серной кислоты и оставляют на несколько часов. Осадок отфильтровывают, промывают, прокаливают и для удаления кремнекислоты выпаривают с одной каплей серной кислоты и несколькими каплями плавиковой кислоты. Полученный остаток растворяют в небольшом количестве горячей концентрированной серной кислоты, вливают этот раствор в холодную воду, взятую в объеме нескольких миллилитров, и тщательно обмывают тигель холодной водой, протирая его стенки палочкой с резиновым наконечником. Если теперь появляется осадок, то это сульфат бария; его отфильтровывают, промывают и после прокаливания взвешивают.
К фильтрату после осаждения бария приливают достаточное количество соляной кислоты, чтобы после прибавления аммиака не выпал магний. Прибавляют аммиак в небольшом избытке и кипятят несколько минут в платиновой чашке. Очень часто при этом появляется незначительный осадок гидроокиси алюминия. Его отфильтровывают, растворяют, пере-осаждают и полученный фильтрат присоединяют к первому фильтрату. Затем осаждают магний вместе с марганцем, который мог остаться в растворе (см. стр. 964).
ФОСФОР
Для определения фосфора породу растворяют в разбавленной азотной кислоте, раствор фильтруют, остаток сплавляют с небольшим количеством карбоната натрия и прибавляют азотнокислый раствор полученного плава к главному раствору.
1 В гл. «Щелочноземельные металлы» (стр. 694) были приведены лучшие методы отделения небольших количеств кальция от больших количеств магния, как это требуется при анализе важного для промышленности минерала — магнезита и продуктов его обжига.
67 заказ 522.
1058
Гл. LV. Точные методы анализа карбонатных пород
Если проводится анализ довольно глинистый породы, можно сплавить порошок породы на сильной паяльной горелке с половинным по массе количеством карбоната натрия. Плав растворяют в азотной кислоте и раствор выпаривают досуха. Таким образом, главную часть кремнекислоты переводят в нерастворимое состояние. Сухой остаток обрабатывают азотной кислотой, фильтруют, остаток промывают, прокаливают, обрабатывают плавиковой и азотной кислотами и выпаривают досуха. Для удаления фтора выпаривают еще 1—2 раза с азотной кислотой, растворяют остаток в азотной кислоте и прибавляют полученный раствор к главному раствору.
К полученному первым или вторым способом раствору прибавляют раствор молибдата аммония и обрабатывают выделившийся осадок одним из известных методов (см. «Фосфор», стр. 784).
При анализе известняков, бедных фосфором, часто бывает нужно сконцентрировать фосфор, выделяя его из большой навески пробы. Рекомендуется 1 следующий способ.
К азотнокислому раствору прибавляют аммиак до начала помутнения. [Если железа и алюминия в породе нет, то надо предварительно прибавить несколько капель раствора хлорида железа (III)]. После этого прибавляют 0,5 г чистого осажденного карбоната кальция и кипятят 5 мин. Затем фильтруют, растворяют осадок (который содержит весь фосфор, извлеченный из раствора) в азотной кислоте и осаждают фосфор в виде фосфоромолибдата аммония.
ЖЕЛЕЗО [II]
Присутствие в известняках углистых веществ часто делает точное или даже приблизительное определение железа (II) невозможным. Несмотря на это, даже в присутствии углистых веществ могут быть иногда получены приемлемые результаты, если количество этих веществ невелико и при растворении породы в кислоте не получается окрашенный раствор. Но даже и тогда можно определить только переходящее в раствор железо, которое присутствовало в породе преимущественно (если не полностью) в виде карбоната. Иногда известняки содержат двуокись марганца, которая также мешает определению.
Определение в отсутствие углистых веществ
Присутствует железо (II), переходящее в раствор при обработке серной кислотой. Кипятят 1—2 г измельченной в порошок породы в колбе емкостью 200—250 мл с небольшим количеством воды, пока не будет удален весь воздух. Продолжая кипячение, приливают понемногу разбавленную серную кислоту, пока не кончится выделение СО2, а затем еще некоторый избыток кислоты. Осаждается сульфат кальция, а железо переходит в раствор. Затем пламя удаляют и колбу закрывают пробкой, в которую вставлена небольшая капельная воронка. После охлаждения наливают в воронку холодную воду, осторожно открывают кран воронки и впускают воду в колбу, подливая ее в то же время в воронку, пока объем раствора в колбе не достигнет 100—150 мл.
'F. Hinden, Z. anal. Chem., 54, 214 (1915).
Железо [77]
1059
Такая тщательная изоляция от воздуха в большинстве случаев может быть и не нужна, так как в присутствии серной кислоты железо (II) окисляется кислородом воздуха чрезвычайно медленно. Обычно бывает совершенно достаточным, открыв кран после охлаждения, выровнять давление снаружи и внутри, после чего надо удалить пробку и влить холодную воду. Затем ставят колбу под бюретку с разбавленным раствором перманганата и немедленно титруют железо.
Карбонаты, растворимые без нагревания, можно обрабатывать кислотой на холоду, в атмосфере двуокиси углерода, благодаря чему уменьшается опасность разложения силикатов, если таковые присутствуют. Если предпочитают титровать бихроматом калия, то в отсутствие двуокиси марганца можно для разложения пробы брать соляную кислоту. В этом случае, конечно, не происходит образования нерастворимой соли кальция, что упрощает последующее определение железа (II) в нерастворимом остатке породы. Можно считать, что большая часть определенного таким способом железа (II), если не все, содержалось в породе в виде карбоната.
Железо (II) находится в нерастворимом остатке. Оттитрованный раствор фильтруют через уплотненный фильтр средней величины и промывают нерастворимый остаток водой. Затем смывают остаток с фильтра в поместительный платиновый тигель, выпаривают большую часть воды и определяют в остатке железо (II), как описано на стр. 996.
Все железо (II), переходящее в раствор при обработке серной кислотой и находящееся в нерастворимом остатке, пересчитывают на Fe2O3 и вычитают из общего количества железа, также пересчитанного на Fe2O3. Таким образом получают содержание Fe2O3 в породе. Точно так же можно вычислить содержание Fe2O3 отдельно в растворимой и нерастворимой в кислоте частях породы, если они исследовались отдельно.
Общее количество железа (II). Если не требуется отдельных определений, то измельченную породу смачивают водой в вместительном платиновом тигле и осторожно обрабатывают разбавленной серной кислотой, пока не прекратится выделение С02. Затем, прибавив 5 мл фтористоводородной кислоты, помещают тигель в один из приборов, описанных на стр. 999 и далее, и проводят разложение силикатов в атмосфере двуокиси углерода п титрование, как там указано. ]
Определение в присутствии нерастворимых углистых веществ
Разложение проводят в колбе, обрабатывая пробу разбавленной серной кислотой в атмосфере двуокиси углерода. Если анализируют известняки и разложение проводят при энергичном взбалтывании, то можно обойтись без нагревания, но при разложении доломитов нагревание необходимо. Затем раствор быстро фильтруют (если много железа, то в атмосфере С02), промывают фильтр с остатком несколько раз и тотчас же титруют раствором перманганата. Если раствор окрашен органическими веществами, то результат может получиться неправильным.
В отсутствие двуокиси марганца можно, как сказано выше, применить соляную кислоту для растворения пробы и бихромат калия для титрования. В обоих случаях важно, чтобы кислота действовала не дольше, чем это необходимо, и чтобы фильтрование проводилось быстро. Определения железа (II) в нерастворимом остатке обычно не стоит предпринимать ввиду присутствия в нем органических веществ.
67*
1060
Гл. LV- Точные методы анализа карбонатных пород
ЩЕЛОЧНЫЕ МЕТАЛЛЫ
Щелочные металлы в карбонатных породах являются, без сомнения, компонентами силикатных примесей. Так как они обычно присутствуют в небольших количествах, то следует брать для обработки не менее 1 г породы и следовать методу Смита почти во всех его деталях (стр. 1006). Так как уже с самого начала в породе присутствует большое количество карбоната кальция, прибавление сверх того еще осажденного карбоната кальция может показаться излишним. Что это, однако, не так, следует из опытов, проведенных с прибавлением и без прибавления карбоната кальция. В первом случае было найдено немного больше щелочных металлов, чем во втором; по всей вероятности это объясняется тем, что искусственно приготовленный тонкоизмельченный карбонат кальция лучше реагирует с хлоридом аммония, чем природный кристаллический карбонат кальция. Вполне достаточно, однако, брать половинное количество карбоната кальция по сравнению с тем, какое применяется в анализе силикатов.
ДВУОКИСЬ УГЛЕРОДА1, УГЛЕРОД (ВОДА)
Определение углерода и водорода углистых веществ
Природа углистых веществ известняков, насколько нам известно, еще не была предметом подробных исследований. Кажется, есть все-таки основание считать, что эти вещества стоят ближе к углям, чем к неопределенным веществам гумуса. Хотя иногда при растворении породы в разбавленной кислоте запах выделяющегося газа указывает на присутствие органических соединений, тем не менее улетучивающиеся при этом вещества составляют лишь незначительную долю общего количества органических веществ. Если порошок породы кипятят с кислотой, то иногда раствор получается окрашенным; однако если для растворения брать разбавленную кислоту и фильтровать тотчас же, как только прекратится действие кислоты на карбонаты, то в раствор, вероятно, или совсем не перейдет, или перейдет лишь очень малое количество органических веществ. Поэтому для определения углерода может быть применен следующий метод 2.
От 0,2 до 1 г анализируемой пробы обрабатывают 30 мл разбавленной (1:2) соляной кислоты и нагревают до полного разложения карбонатов. Разбавляют затем раствор водой, фильтруют через прокаленный асбест и промывают остаток водой, содержащей немного соляной кислоты, пока щелочноземельные металлы не будут полностью удалены. Переносят асбест с нерастворимым остатком в платиновую чашку емкостью 100 мл, обрабатывают 15 мл соляной кислоты и нагревают при температуре, близкой к кипению, на плитке в течение 10 мин. Затем прибавляют 20 мл фтористоводородной кислоты и нагревают при температуре начинающегося. кипения, пока не разложатся неорганические вещества. Для этого требуется обычно от 30 мин до 2 ч и объем жидкости нужно все время
1 Определение двуокиси углерода проводится точно по указаниям, данным на стр. 1014.
2 А. С. F i е 1 d п е г, W. A. S е 1 v i g, G. В. Т а у 1 о г, Bur. Mines Tech. Paper, 212 (1919).
Хлор
1061
держать близким к 15 мл, прибавляя время от времени смесь соляной и фтористоводородной кислот в отношении 3 : 4.
Когда разложение закончится, выпаривают раствор до объема 15 мл, прибавляют 25 мл соляной кислоты и нагревают до кипения. К кипящему раствору прибавляют равный объем горячей воды, снова нагревают до кипения и фильтруют горячим через фильтр из прокаленного асбеста или губчатой платины. Остаток промывают горячей водой до исчезновения реакции на хлор, переносят фильтр с остатком в фарфоровую или платиновую лодочку, высушивают 2 ч при 105° С и переносят непосредственно в трубку для сжигания так, чтобы вещество не могло снова поглотить влагу.
Содержание углерода и водорода определяют, как описано в главе «Углерод и водород» (стр. 850), соблюдая необходимые предосторожности в отношении серы, если в известняке присутствовали сульфиды.
Одновременное определение воды и общего содержания углерода
В присутствии сульфидов или растворимых органических веществ для одновременного определения воды и углерода карбонатов и органических веществ может быть применен следующий метод. Прокаливают 1 г анализируемой породы в токе воздуха в трубке из очень тугоплавкого стекла или в специальном тигле Гуча (см. рис. 39, стр. 911) и собирают выделившиеся воду и двуокись углерода. Сера сульфидов будет связана в виде сульфата кальция. Для успешного применения этого метода необходимо, чтобы температура была достаточно высока для выделения двуокиси углерода из карбонатов, но, с другой стороны, не настолько высока, чтобы разложился сульфат кальция. Найденная вода является суммой воды, содержавшейся в минералах породы, и воды, образовавшейся из водорода органических веществ при их сжигании.
После отдельного определения двуокиси углерода карбонатов (см. стр. 1015) можно вычислением определить количество углерода органических веществ.
Можно также 1 в одной и той же навеске определить последовательно углерод в обеих его формах, если сначала определить двуокись углерода, как описано на стр. 1015, только с ортофосфорной кислотой вместо соляной кислоты, а затем, взвесив поглотительные трубки и включив их снова, следует прибавить хромовую кислоту и снова кипятить для окисления органических веществ и выделения их углерода в виде СО2.
ХЛОР
Несколько граммов анализируемой породы растворяют в свободной от хлора разбавленной азотной кислоте на холоду или же при возможно более слабом нагревании, раствор фильтруют, прибавляют нитрат серебра, отфильтровывают осадок и обрабатывают его, как обычно (см. стр. 1017-1018).
Если предполагают присутствие в породе хлорсодержащих силикатов, то сплавляют породу в платиновом тигле с равным по массе количе
1 G. Т. Morgan, J. Chem. Soc., 85, 1001 (1904).
1062
Гл. LV. Точные методы анализа карбонатных пород
ством свободного от хлора карбоната натрия. Нагревают обыкновенной горелкой или паяльной горелкой средней силы, избегая при этом слишком длительного прокаливания. Полученный плав извлекают горячей водой, фильтруют и фильтрат подкисляют холодной разбавленной азотной кислотой (можно сразу обработать плав азотной кислотой и потом профильтровать раствор). Дальнейшая обработка указана выше.
ФТОР
Точное определение фтора в карбонатных породах, как и определение его в силикатных породах, затруднительно. Из всех методов определения фтора, по-видимому, наилучшим является метод отгонки (стр. 824) с последующим титрованием фтора в отгоне раствором нитрата тория (стр. 825) или колориметрическим его определением в том же отгоне одним из вариантов метода Стейгера — Мервина. Если анализируемая порода не полностью растворяется в кислоте, как это требуется при применении указанного метода, то надо или исследовать отдельно остаток после обработки кислотой на присутствие в нем фтора, или же всю навеску пробы сплавлять с карбонатом натрия и потом проводить отгонку1.
СЕРА
Установление формы соединений серы.
Определение сульфатной серы
В огромном большинстве карбонатных пород сера находится в виде сульфидов, обычно в форме пирита. В каком соединении находится сера, можно легко обнаружить. Почернение бумажки, смоченной ацетатом свинца и поднесенной к открытому концу пробирки, в которой пробу породы кипятят с разбавленной соляной кислотой, указывает на присутствие растворимых в кислоте сульфидов. Их редко находят в количествах, превышающих следы. Если фильтрат от нерастворимого в кислоте остатка после прибавления к нему аммиака до почти полной нейтрализации и затем хлорида бария выделяет осадок, то присутствуют сульфаты. Если определенное, как описано в разделе «Описание общего содержания серы» (см. ниже), общее количество серы больше количества сульфатной серы и растворимых сульфидов вместе взятых, то разность можно уверенно пересчитать на пирит.
В присутствии больших количеств сульфидной и сульфатной серы извлечение последней следовало бы делать в атмосфере двуокиси углерода, но обычно эта предосторожность оказывается излишней. Подробности определения серы см. на стр. 1029.
Определение общего содержания серы
Превращение сульфидов в сульфаты. Прокаливание без добавления плавня. При анализе пород, содержащих не слишком большое количество сульфидов, рекомендуется следующий метод, дающий хорошие результаты даже в присутствии 1% органических веществ; границы его применимости, однако, еще с достаточной точностью не установлены.
1 J. I. Hoffman, G. Е. F. L u n d е 11, J. Research NBS, 20, 610 (1938).
Вода
1063
Платиновый тигель, содержащий 1—2 г измельченной в порошок породы и помещенный в отверстие асбестового щита (рис. 6, стр. 49), прокаливают 15 мин на обыкновенной горелке или 10 мин в косо направленном пламени паяльной горелки умеренной силы. Как показывает наш опыт, вовсе не нужно постепенно поднимать температуру, начиная со слабого нагревания. Вся сульфидная сера окисляется в сульфатную и связывается известью в сульфат кальция, причем не происходит никаких потерь от улетучивания, все органические вещества сгорают, а силикаты, если они не содержатся в избытке, становятся растворимыми в кислоте (см. стр. 1049).
Прокаливание с добавлением карбоната натрия. Смешивают 1—2 г измельченной в порошок породы в платиновом тигле с половинным по массе количеством свободного от серы карбоната натрия и прокаливают (в отверстии асбестового щита), как описано в разделе «Прокаливание без добавления плавня». Даже в присутствии довольно большого количества органических веществ прибавление селитры едва ли необходимо.
Так как количество плавня так мало, что оно вызывает лишь спекание нагреваемой смеси, то воздух, проникающий в пористую массу после удаления большей части двуокиси углерода, производит быстрое и полное окисление.
Несмотря на это, при анализе очень нечистых известняков может оказаться необходимым увеличение количества плавня, а также прибавление небольшого количества селитры.
Дальнейшая обработка после прокаливания. Прокаленная масса, полученная, как описано выше, обычно легко отстает от стенок тигля. Ее переносят в маленький стакан и обливают водой. Тигель обмывают разбавленной соляной кислотой над стаканом. Затем прибавляют еще кислоты, пока на холоду или при слабом нагревании все не разложится. Фильтруют, если нужно, разбавляют раствор до 150—200 .мл 1—2%-ной (по объему) соляной кислотой, нагревают до кипения пли просто помещают на паровую баню, прибавляют хлорид бария и осажденный сульфат бария отфильтровывают, промывают и взвешивают, как это описано в гл. «Сера» (стр. 801).
Можно совершенно не проводить выпаривания раствора досуха для выделения растворенной кремнекислоты. После разложения кислотой раствору не следует давать желатинизироваться, и нужен лишь небольшой навык, чтобы всегда избегать этого. Если желатинизирование все же произошло, то самое лучшее — начать сначала; но в случае, если раствор содержит мало свободной кислоты, можно выделившуюся студенистую кремнекислоту растворить прибавлением свободной от серы едкой щелочи и снова подкислить на холоду соляной кислотой.
ВОДА
Гигроскопическая вода
Один или несколько граммов воздушно-сухого порошка породы нагревают при 100—105° С в стеклянной трубке в струе высушенного хлоридом кальция воздуха. Выделяющуюся воду улавливают в хлоркальциевой трубке. Можно также воздух сушить серной кислотой или трехводным перхлоратом магния и воду поглощать таким же высушивающим
1064 Гл. LV. Точные методы анализа карбонатных пород
веществом, какое было применено для осушения воздуха. При таком прямом определении воды получают, несомненно, более высокие и более точные результаты, чем если высушивают породу в тигле при указанной выше температуре и определяют содержание гигроскопической воды по потере в массе.
Связанная вода
Связанную воду обычно определяют следующим образом. Воздушносухую пробу нагревают в трубке для сжигания, и выделяющийся водяной пар улавливают с указанными выше предосторожностями в трубке с хлоридом кальция.
Можно также смешать порошок пробы с сухим карбонатом натрия и прокалить в приборе Гуча (стр. 911). Чтобы получить содержание связанной воды, надо из найденного общего содержания воды вычесть определенное раньше количество гигроскопической влаги. Нужно при этом помнить, что результат может получиться больше истинного на то количество воды, которое при сжигании образуется из водорода органических веществ, содержащихся в известняке.
Глава LVI
СОКРАЩЕННЫЙ АНАЛИЗ
КАРБОНАТНЫХ ПОРОД
Для многих целей, особенно технических, нет необходимости в подробном анализе, проводимом с крайней тщательностью и точностью. В настоящее время число анализов, выполняемых без строго научной точности, значительно превосходит число анализов, требующих применения методов, описанию которых посвящены многие главы части II этой книги. Считаясь с требованиями тех, кто нуждается в анализах для технических целей, мы ниже даем описание сокращенного анализа, в котором определяются основные компоненты карбонатных пород. Отдельные операции описываются без пояснений, так как обоснования того или иного хода работы можно найти в соответствующих главах части II.
РАЗЛОЖЕНИЕ И РАСТВОРЕНИЕ
Если анализируемая проба может быть переведена прокаливанием при 1000—1200° С в состояние, в котором она совершенно растворяется в соляной кислоте, то навеску от 0,5 до 1 г тонкоизмельченного порошка пробы сильно прокаливают в закрытом платиновом тигле при 1200° С в течение 15 мин или еще дольше, если горелка не дает указанной температуры.
При анализе известняков, сильно загрязненных силикатами, навеску в 0,5—1 г смешивают с половинным от ее массы количеством чистого карбоната натрия и прокаливают на горелке до образования хорошо спекшейся массы.
Продукт прокаливания, полученный тем или иным способом, переносят в выпарительную чашку, лучше всего платиновую, и смачивают несколькими миллилитрами воды. Тигель обмывают разбавленной соляной кислотой, перенося раствор в ту же чашку, которую, если при прокаливании применяли карбонат натрия, надо держать покрытой часовым стеклом, и прибавляют еще соляной кислоты. Процесс растворения можно ускорить растиранием спекшейся массы сплющенным концом стеклянной палочки. Затем раствор выпаривают на водяной бане досуха.
КРЕМНЕКИСЛОТА
Остаток обрабатывают 5—10 мл концентрированной соляной кислоты и разбавляют 50 мл горячей воды. Чашку покрывают часовым стеклом и нагревают 10 мин на водяной бане, после чего фильтруют раствор через
1066
Гл. LVI. Сокращенный анализ карбонатных пород
фильтр диаметром 7 см. Промывают кремнекислоту горячей разбавленной (5 : 95) соляной кислотой, затем небольшим количеством горячей воды. Фильтрат выпаривают досуха на паровой бане и затем нагревают 1 ч при 105—110° С. Остаток обрабатывают кислотой и водой, но в меньших количествах, и нагревают на водяной бане, как раньше. Собирают вторую порцию кремнекислоты, имеющую обычно темную окраску, на другом маленьком фильтре и промывают сначала холодной разбавленной (1 : 99) соляной кислотой, а затем горячей водой. Фильтрат (фильтрат А) сохраняют.
Оба фильтра с их содержимым медленно нагревают в платиновом тигле и прокаливают сначала на обыкновенной горелке, затем 5—15 мин (в зависимости от количества кремнекислоты) при 1200° С. Прокаливание повторяют до достижения постоянной массы. Прокаленную кремнекислоту смачивают водой, прибавляют 1 каплю разбавленной (1 : 1) серной кислоты и от 5 до 10 мл чистой фтористоводородной кислоты. Затем ставят тигель в радиатор (см. рис. 5, стр. 49) и жидкость выпаривают. Остаток нагревают 1 мин при 1000—1100° С и по охлаждении взвешивают.
Поправки (на нечистоту выделенной кремнекислоты) лучше не находить, если не имеют в виду выделить и определить также и ту кремнекислоту, которая всегда выделяется вместе с гидроокисями железа и алюминия, потому что при малом количестве кремнекислоты в известняках нелетучий остаток более или менее компенсируется кремнекислотой, остающейся в растворе после второго выпаривания. Однако не следует пренебрегать этой поправкой на нечистоту SiO2, если имеют в виду ввести поправку и на растворимость кремнекислоты. Если первая поправка введена и остаток весит более 1—2 мг, надежнее сплавить его с небольшим количеством карбоната натрия и прибавить раствор полученного плава к фильтрату от кремнекислоты, чем взвешивать этот остаток вместе с осадком от аммиака.
Результаты определения кремнекислоты, получаемой после однократного выпаривания, всегда бывают пониженными, если обработка серной и фтористоводородной кислотами проведена, даже и в том случае, когда определено содержание кремнекислоты в осадке от аммиака.
Например, после однократного выпаривания кремнекислоты в пробе стекла, содержавшего 74,1% SiO2, было найдено 73,7%, если никаких поправок не вводили, 73,5%, если проводилась обработка HF + H2SO4, и 73,7%, если в последний результат вводилась поправка на SiO2, выпавшую с осадком от аммиака. Такой же ход анализа, примененный к глинистому известняку, содержавшему 18,08% SiO2, дал соответственно 17,80, 17,71 и 17,90% SiO2. Очевидно, что мы очень мало или ничего не выигрываем, если вводим поправки после однократного выпаривания, и что не вся кремнекислота, оставшаяся в растворе после определения ее однократным выпариванием, захватывается осадком от аммиака.
АЛЮМИНИЙ, ЖЕЛЕЗО И ДРУГИЕ ЭЛЕМЕНТЫ
К фильтрату А (см. «Кремнекислота», стр. 1005), объем которого при анализе очень чистых известняков не должен превышать 100 мл, прибавляют несколько капель метилового красного, нагревают до кипения и медленно приливают разбавленный (1 : 1) раствор аммиака, пока окраска индикатора не перейдет в желтую. Кипятят 1—3 мин, тотчас же фильтруют, обмывают стакан и фильтр 1—2 раза горячим 2%-ным раствором хлорида аммония и сохраняют фильтрат (фильтрат Б). Если осадок
Марганец
1067
мал и хорошо промыт, повторного осаждения не нужно. Если он велик, ставят стакан, .в котором было проведено осаждение, под воронку, растворяют осадок на фильтре горячей разбавленной (1 : 1) соляной кислотой, тщательно промывают фильтр горячей водой и отставляют его в сторону. Раствор разбавляют приблизительно до 75 мл, прибавляют метиловый красный, осаждают аммиаком, как ранее, опускают в раствор сохраненный фильтр, хорошо размешивают и фильтруют через новый фильтр. Осадок полностью переносят на фильтр и промывают горячим раствором хлорида аммония.
Полученный фильтрат соединяют с фильтратом Б. Третье осаждение требуется редко.
Фильтр с осадком помещают в платиновый тигель, медленно озоляют фильтр, постепенно усиливая нагревание, и прокаливают при хорошем доступе воздуха приблизительно при 1200° С в течение 5—10 мин. Масса прокаленного остатка является суммарной массой окислов железа, алюминия, титана, фосфора и т. п., но не марганца.
Если взвешенный прокаленный остаток мал, как это часто бывает, поправка на содержание в нем кремнекислоты может быть найдена следующим образом. Остаток обрабатывают каплей разбавленной (1 : 1) серной кислоты, затем 5—10 мл фтористоводородной кислоты и выпаривают досуха. Нагревают сначала осторожно, пока сульфаты не разложатся, затем 3—5 мин приблизительно при 1000° С и взвешивают.
Железо и титан во взвешенной смеси окислов могут быть определены после сплавления ее с пиросульфатом, как описано на стр. 957, но обычно их можно определить быстрее в отдельных навесках пробы, так же как и фосфор. Содержание окиси алюминия находят по разности — вычитанием массы всех определенных окислов из массы их суммы. Если вычитают содержание одной лишь окиси железа, то содержание окиси алюминия может получиться совершенно неверным. При проведении только однократного осаждения аммиаком масса суммы окислов получается несколько повышенной, и соответственно повышенным получают и содержание алюминия.
Например, при однократном осаждении аммиаком после однократного выпаривания для выделения кремнекислоты при анализе известково-натриевого стекла, содержавшего 0,42% окислов «R2O3», содержание их было найдено равным 0,58% , когда был взвешен один осадок, 0,78% , когда он был взвешен вместе с нелетучим остатком от кремнекислоты, и 0,69%, когда в последний результат была введена поправка на содержание кремнекислоты. При такой же обработке, примененной к глинистому известняку, содержавшему 7,86% «R2O3», найдено 8,09%, если нелетучий остаток не учитывали, и 7,91%, если в этот результат вводили поправку на кремнекислоту.
МАРГАНЕЦ
Марганец, если он присутствует в известняке и не удален предварительно, выпадает частично вместе с кальцием, а в большей части вместе с магнием. Он может быть осажден и удален обработкой фильтрата Б (см. стр. 1066) сульфидом аммония или же выделен с осадком от аммиака следующим образом: разбавляют фильтрат А (см. стр. 1066) до 150—200 мл, прибавляют 10—15 мл соляной кислоты, если она не была введена раньше, нейтрализуют почти полностью аммиаком, прибавляют 1 г твердого персульфата аммония и продолжают прибавлять аммиак до появления щелочной реакции на лакмус. Нагревают раствор до кипения, кипятят 2 мин
1068
Гл. LVI. Сокращенный анализ карбонатных пород
и прибавляют аммиак, пока снова не будет достигнута щелочная реакция на лакмус Ч Тотчас же фильтруют, умеренно промывают осадок горячим 2%-ным раствором хлорида аммония и сохраняют фильтрат (фильтрат В). Осадок растворяют в горячей разбавленной соляной кислоте, раствор охлаждают, снова осаждают, как в первый раз, фильтруют, промывают осадок и полученный фильтрат соединяют с сохраненным ранее фильтратом В.
Само определение марганца лучше всего проводить, как описано в главе «Марганец» (стр. 497 или стр. 505), в отдельной навеске пробы, и полученный результат, пересчитанный на Мп3О4, вычесть из массы смеси окислов, если при осаждении их пользовались персульфатом аммония.
КАЛЬЦИЙ
Сохраненный фильтрат В концентрируют, если нужно, до объема 200—250 мл, прибавляя время от времени по нескольку капель аммиака. Если появится маленький осадок, его собирают на небольшом фильтре, промывают горячим 2%-ным раствором хлорида аммония, растворяют в горячей разбавленной соляной кислоте, снова осаждают, фильтруют и прибавляют полученный осадок к главному осадку гидроокисей железа и алюминия. Соединенные фильтраты кипятят и медленно приливают 40 мл (для навески в 1 г; при навеске в 0,5 г приливают 20 мл) горячего 4%-ного раствора оксалата аммония; продолжают кипятить, пока осадок оксалата кальция не перейдет в зернистую форму, и оставляют охлаждаться 1 ч. Фильтруют, и осадок промывают несколько раз 0,1 %-ным раствором оксалата аммония. Полученный фильтрат (фильтрат Г) сохраняют. Подставив стакан под воронку с осадком, растворяют последний на фильтре в 25—50 мл горячей разбавленной (1 : 9) соляной кислоты и промывают фильтр горячей водой. Полученный раствор разбавляют до 100 мл, прибавляют 5 мл 4%-ного раствора оксалата аммония и затем при перемешивании приливают аммиак до небольшого его избытка.
Через 1 ч фильтруют, умеренно промывают стакан, фильтр и осадок сначала разбавленным раствором оксалата аммония, а затем горячей водой, приливая последнюю в количестве не большем, чем это надо для отмывания избытка оксалата (всего приблизительно 50 мл, маленькими порциями). Дают жидкости стечь, соединяют ее с фильтратом Г и сохраняют для определения магния. Осадок смывают, насколько возможно, обратно в стакан и раскладывают фильтр на внутренней стенке стакана в верхней его части. Прибавляют серной кислоты и титруют перманганатом, как описано в главе «Щелочноземельные металлы» (стр. 710). При желании можно осадок оксалата кальция прокалить и полученную СаО взвесить, как описано на стр. 707.
Результаты, получаемые при однократном осаждении оксалата кальция, обычно повышены как при весовом, так и при объемном окончании определения. Весовой вариант дает несколько более высокие результаты,
1 Лакмус не является идеальным индикатором при осаждении алюминия, но к нему приходится прибегать, потому что другие индикаторы (с более подходящим интервалом перехода окраски), например метиловый красный, разрушаются в горячем растворе, содержащем окислитель. Количество алюминия, остающееся в растворе при применении этого индикатора, незначительно, и в обычных анализах им можно пренебречь.
Магний
1069
чем объемный Е Осадок оксалата кальция содержит также и весь стронций, бывший в анализируемой пробе, часть марганца, если он не был удален предварительно, и не содержит бария. Если стронций нужно определить, то осадок оксалата прокаливают, взвешивают, превращают в нитраты и выделяют стронций смесью спирта и эфира, как описано в главе «Щелочноземельные металлы» (стр. 697).
МАГНИЙ
К фильтрату Г, сохраненному от определения кальция, прибавляют 2 г двузамещенного фосфата аммония (NH4)2HPO4 и сверх того еще по 1 г на каждые 100 мл раствора. Перемешивают, пока все не растворится, прибавляют аммиак в избытке, равном 5% (по объему), снова тщательно перемешивают и оставляют по крайней мере на 4 ч (лучше на ночь, если осадок мал или если объем раствора превышает 400 мл). Фильтруют, умеренно промывают стакан, фильтр и осадок разбавленным (5 : 95) раствором аммиака и отбрасывают фильтрат. Подставив стакан под воронку с осадком, растворяют осадок на фильтре в 25—50 мл горячей разбавленной (1 : 9) соляной кислоты. К полученному раствору прибавляют 0,1 г (NH4)2HPO4, разбавляют водой до 75 мл, охлаждают и затем прибавляют по каплям, при постоянном перемешивании, аммиак до щелочной реакции раствора. После этого приливают еще 5 мл раствора аммиака и оставляют стоять 4 ч. Декантируют раствор через новый фильтр, промывают осадок 1 раз декантацией холодным 5%-ным раствором аммиака, количественно переносят осадок на фильтр и продолжают промывание до полного удаления хлоридов.
Влажный фильтр с осадком свертывают, высушивают во взвешенном платиновом тигле, медленно озоляют фильтр, не давая ему гореть, сжигают уголь, постепенно увеличивая пламя, и, наконец, прокаливают 15 — 30 мин при 1000—1050° С. Охлаждают в эксикаторе и взвешивают в виде Mg2P2O7.
Взвешенный осадок содержит СаО, оставшуюся в растворе после осаждения оксалатом аммония, весь барий, содержавшийся в пробе, и ^практически весь марганец, присутствовавший в фильтрате после осаждения кальция.
Результаты, получаемые при однократном осаждении магния, обычно бывают повышенными как при осаждении из горячего, так и при осаждении из холодного раствора 1 2.
При серийном выполнении большого числа определений получаемые осадки можно отфильтровать, как описано в главе «Магний» (стр. 727).
1 Например, при анализе известково-натриевого стекла после однократного осаждения оксалатом аммония, следовавшего за однократным выпариванием для выделения кремнекислоты и однократным осаждением аммиаком, содержание СаО было найдено равным 5,11% при весовом и 4,81% — при объемном окончании определения, вместо истинного содержания 4,65%. При анализе известняка после такой же обработки весовое определение дало 38,7% СаО вместо действительного содержания 37,7%.
2 Например, при анализе известково-натриевого стекла, в котором выделение SiO2, R2O и СаО проводили однократным осаждением, при однократном же осаждении магния было найдено 3,31% MgO вместо истинного содержания 3,23%. При анализе известняка таким же методом было найдено 5,46% вместо 5,08%.
1070
Гл. LVI. Сокращенный анализ карбонатных пород
ОПРЕДЕЛЕНИЕ ДРУГИХ КОМПОНЕНТОВ
Щелочные металлы определяют, как указано на стр. 1060; определение двуокиси углерода — см. стр. 1060; определение серы — стр. 1062; определение воды — стр. 1063.
ПОТЕРЯ В МАССЕ ПРИ ПРОКАЛИВАНИИ
Определение потери в массе при прокаливании не имеет большого значения, хотя оно и позволяет сделать более или менее правильное заключение о содержании карбонатов в породе. Однако поскольку в лабораториях технического анализа обычно проводят это определение, ему следует здесь уделить место.
При анализе карбонатных пород, так же как и в силикатных, потеря в массе при прокаливании составляет алгебраическую сумму увеличений и потерь, соответствующих отдельным химическим реакциям; величина общей потери сильно зависит от температуры прокаливания. При прокаливании в умеренно сильном пламени паяльной горелки (1100° С) в закрытом тигле выделяются полностью двуокись углерода, вода и сгорают все углистые вещества. Сульфиды окисляются до сульфатов, и вся сера остается в виде сульфата кальции. На сильном пламени паяльной горелки при 1200—1300° С сульфат кальция постепенно разлагается, иногда настолько, что вся сера может улетучиться. При этой температуре начинают улетучиваться также и щелочные металлы в виде окисей (калий относительно сильнее, чем натрий) и могут частично осесть, образуя налет на внутренней стороне крышки тигля. При долгом прокаливании они могут улетучиться полностью. Этим потерям противопоставляется небольшой привес от кислорода, связываемого железом пирита и железом (II) и марганцем (II) карбонатов этих элементов. Надлежащей регулировкой температуры нетрудно предотвратить улетучивание серы и щелочных металлов, и за этим нужно внимательно следить при выполнении определения.
От 0,5 до 1 г измельченной в порошок породы вносят в платиновый тигель емкостью 20—25 мл, тигель вставляют на три пятых его высоты в отверстие асбестовой или платиновой пластинки (см. рис. 6. стр. 49) и 15—20 мин нагревают дно закрытого тигля косо направленным пламенем сильной паяльной горелки. Если вторично прокалить тигель в течение 5 мин, то дальнейшая потеря в массе обычно не наблюдается. Параллельные определения не должны расходиться более чем на 0,2%, а после некоторой практики расхождение становится меньше 0,1%. Вместо паяльной горелки можно пользоваться муфелем. Важно подчеркнуть, что аналитик должен знать температуру, которую дает его паяльная горелка или муфель. Источник высокой температуры подходит для желаемой цели, если после прокаливания вся сера оказывается связанной в виде сульфата.
Международные атомные веса
1071
МЕЖДУНАРОДНЫЕ АТОМНЫЕ ВЕСА
Название Символ Атомный номер Атомный вес Название Символ Атомный номер Атомный вес
Азот N 7 14,0067 Неон Ne 10 20,183
Актиний .... Ас 89 [227] Нептуний .... Np 93 [237]
Алюминии . . . А1 13 26,9815 Никель Ni 28 58,71
Америций .... Ат 95 [243] Ниобий Nb 41 92,906
Аргон Аг 18 39,948 Нобелий .... No 102 [256]
Астат At 85 [210] Олово Sn 50 118,69
Барий Ва 56 137,34 Осмий Os 76 190,2
Бериллий .... Be 4 9,0122 Палладий .... Pd 46 106,4
Беркелий .... Bk 97 [247] Платина .... Pt 78 195,09
Бор В 5 10,811 Плутоний .... Pu 94 [242]
Бром Вт 35 79,904 Полоний .... Po 84 [210]
Ванадий .... V 23 50,942 Празеодим . . . Pr 59 140,907
Висмут Bi 83 208,980 Прометий .... Pm 61 [145]
Водород Н 1 1,00797 Протактиний . . Pa 91 [231]
Вольфрам .... W 74 183,85 Радий Ra 88 [226]
Гадолиний . . . Gd 64 157,25 Радон Rn 86 [222]
Галлий Ga 31 69,72 Рений Re 75 186,2
Гафний Hf 72 178,49 Родий Rh 45 102,905
Гелий Не 2 4,0026 Ртуть Hg 80 200,59
Германий .... Ge 32 72,59 Рубидий .... Rb 37 85,47
Гольмий .... Но 67 164,930 Рутений Ru 44 101,07
Диспрозий . . . dY 66 162,50 Самарпй .... Sm 62 150,35
Европий .... Eu 63 151,96 Свинец Pb 82 207,19
Железо Fe 26 55,847 Селен Se 34 78,96
Золото Au 79 196,967 Сера S 16 32,064
Индий In 49 114,82 Серебро Ag 47 107,868
Иод I 53 126,9044 Скандий Sc 21 44,956
Иридий Ir 77 192,2 Стронции .... Sr 38 87,62
Иттербий .... Yb 70 173,04 Сурьма Sb 51 121,75
Иттрий Y 39 88,905 Таллий T1 81 204,37
Кадмий Cd 48 112,40 Тантал Ta 73 180,948
Калий К 19 39,102 Теллур Те 52 127,60
Калифорний . . . Cf 98 [249] Тербий Tb 65 158,924
Кальций .... Ca 20 40,08 Технеций .... Tc 43 [99]
Кислород .... 0 8 15,9994 Титан Ti 22 47,90
Кобальт Co 27 58,9332 Торий Th 90 232,038
Кремний .... Si 14 28,086 Тулий Tu 69 168,934
Криптон .... Kr 36 83,80 Углерод .... C 6 12,01115
Ксенон Xe 54 131,30 Урай u 92 238,03
Кюрий Cm 96 [247] Фермий Fm 100 [253]
Лантан La 57 138,91 Фосфор P 15 30,9738
Лауренсий . . . Lr 103 [257] Франций .... Fr 87 [223]
Литий Li 3 6,939 Фтор F 9 18,9984
Лютеций Lu 71 174,97 Хлор Cl 17 35,453
Магний Mg 12 24,312 Хром Cr 24 51,996
Марганец .... Mn 25 54,9380 Цезий Cs 55 132,905
Медь Cu 29 63,546 Церий Ce 58 140,12
Менделевии . . . Md 101 [256] Цинк Zn 30 65,37
Молибден .... Mo 42 95,94 Цирконий .... Zr 40 91,22
Мышьяк .... As 33 74,9216 Эйнштейний . . Es 99 [254]
Натрий Na И 22,9898 Эрбий Er 68 167,26
Неодим Nd 60 144,24
в ск°бках обозначают массовое число изотопа с наиболее продолжительным периодом 11UJ1 у раСиаДа»
ИМЕННОЙ УКАЗАТЕЛЬ
Авилов В. Б. 662
Агладзе Р. И. 467
Адамович В. И. 728
Айдиньян Н. X. 254
Алексеев Р. И. 240, 255
Алексеева М. В. 858
Алексеевская О. А. 348, 350
Алешин А. Ф. 844
Алимарин И. П. 73, 348, 350, 352, 360, 449, 518, 553, 638, 676, 680, 689, 890, 915
Аллен Е. 895
Альтерзон Г. С. 185
Ангелов И. И. 826
Андронов Б. Е. 858
Анфимова Е. А. 77 Аппельбаум Л. 774, 775 Аруина А. С. 490, 492 Архангельская 3. В. 158 Афанасьев Б. Н. 449
Бабко А. К. 174, 368, 453, 454,; 467, 468, 476, 577, 596
Бабушкин С. А. 452
Багбанлы И. Л. 541 Барнеби 995, 1000 Беззаботнова А. П. 56 Бекк 817
Берг Р. 148, 492
Берль 491, 774, 865
Беручьян С. 111, 606
Берцелиус 737, 754, 820, 821, 822, 823, 824, 1005, 1006, 1011, 1019, 1023, 1024
Бечи 880
Блюм 308
Боберкова О. Т. 361
Бобиньи 817
Богатский В. Д. 467 Болтон 1046
Большакова Г. А. 171, 343
Бомштейн Р. И. 88, 111, 524
Брайт Г. 591, 804, 823, 846
Браш 908
Бриттон X. Т. 102
Брунк О. 105, 708, 710
Брэй В. 65, 85, 386, 388, 407 658, 823, 883
Буданова Л. М. 501, 649 Бунзен 733, 902, 1012 Бусев А. И. 269, 279, 280 Быкова В, 676 Бюлов К. 246
Вайсберг 3. М. 517 Васильев А. 328, 329, 330 Вашедченко Т. В. 634 Вашингтон 580, 884, 887, 888, 892, 893, 959, 976
Вебер 921
Вельтман Р. П. 185 Венделыптейн Е. И. 682 Веселова А. В. 634 Веселовский С. Ф. 43 Вильм Т. 246
Виноградов А. П. 345, 516, 517
Владимирова М. И. 634 Вознесенский Т. А. 776 Волкова В. А. 633 Володарская Р. С. 647 Володина О. А. 468 Выропаева 3. А. 658
Гайгерова А. А. 588 Гайлис Е. Я. 597 Гей-Люссак 237, 238 Гейровский Я. 173 Гельман Н. Э. 851 Генгринович А. И. 452 Генерозов Б. А. 502, 517, 659, 844
Генис М. Я. 467
Гент 632
Геринг Э. К. 525
Герлях В. В. 177
Гиллебранд В. Ф. 476, 507,
533, 632, 695, 721, 774,
893, 930, 946, 971, 976,
978, 979, 980, 988, 990,
992, 993, 1007, 1023, 1025, 1035
Гильтнер В. 231
Гинзбург Л. Б. 170, 273, 278, 295, 316, 355, 369, 486, 539, 548, 550, 557
Голл В. Т. 221, 291
Голубцова Р. Б. 359, 579, 639, 766
Гольбрайх Ц. Е. 422
Горюшина В. Г. 371, 588, 597, 676, 682, 770, 771, 774
Готсдинер Р. Г. 477
Гофман Д. И. 591, 804, 827, 846, 921, 945, 1024
Гофман Н. Т. 467
Гренберг Е. И. 467
Гринберг А. А. 422, 423
Гровс А. В. 1003
Губельбаник С. М. 422
Гульдина Е. И. 533, 588
Гурвиц С. С. 858
Гурьев С. Д. 355, 539, 543, 548, 550, 557
Гутман С. М. 658
Гутцайт 313
Гуч 717, 841, 878, 914, 965, 968, 1031
Давыдов А. Л. 368, 517
Данкворт П. В. 176
Даценко О. В. 289
Девард 865, 866
Девиль 880
Деннис Л. М. 857
Джилес 101
Джонс 841
Дзюбина Л. Я. 467
И менной указатель
1073
Диттрих 887, 902, 904, 909, 914, 953, 1012
Добкина Б. М. 293, 316, 375, 517
Добровольская В. В. 936
Дорохова М. Н. 775
Драко О. Ф. 476
Драпкина Д. А. 579
Дымов А. М. 468, 502, 804, 846
Дюма 859, 860
Егорова А. Д. 454
Житкова А. С. 858
Жукова К. Н. 449, 451
Заглодина Т. В. 475, 492
Загорацкая Э. М. 658
Зайчикова Л. Б. 267, 369
Зандбергер 1033, 1034
Звенигородская В. М. 476, 477, 588
Земмель В. К. 394
Золотухина А. П. 331
Зотов Г. 56
Зусер Е. Е. 454
Иванов-Эмин Б. Н. 111, 351, 352, 546, 551, 553, 583
Икин 632
Илькович 172
Иорданский Д. Н. 776
Йоу Д. Г. 50, 175
Калмыкова Н. В. 316, 454, 492
Каранович Г. Г. 579
Каргин В. А. 231, 328
Кариус 244
Карпов Б. Т. 532
Карпова И. А. 776
Карякин Ю. В. 198, 826
Кауфман Л. Э. 531
Кирк И. 186
Кирсанов А. В. 213
Кларк 198, 878
Классен А. 264, 285, 491
Клименко Ю. В. 441, 449, 531
Книпович Ю. Н. 532
Кноп 447
Коган А. 804
Козлов В. А. 452
Козловский М. Т. 776
Кокорин А. И. 331
Колодуб П. А. 328, 492
Колосов В. И. 170, 273
Кольтгоф И. М. 35, 173, 198, 200, 202, 204, 206, 225, 227, 230, 231, 490
Комаровский А. С. 588, 804 68 Заказ 522.
Константинова-Шлезингер М. А. 176
Копченова Е. В. 186
Коренман И. М. 187, 301
Корец И. И. 279
Коротун М. В. 467
Коршун М. О. 851
Косая Р. 467
Краус К. А. 628
Крешков А. П. 56
Кривошлыков Н. Ф. 691 Кришнайя 1012
Кузнецов В. И. 143, 330. 331, 454, 501, 579, 609, 635, 649, 659
Кузятина Н. С. 255
Кук 987, 989, 992, 993, 995, 996, 997, 999
Кульберг Л. М. 143, 185, 241, 492
Куннипгем 682
Кускова Н. К. 517
Кучмент М. Л. 452
Кьельдаль 859, 862, 865
Лабе 198
Лайтинен Г. А. 200, 231
Лебедева А. Д. 658
Лебединский В. В. 411
Левина М. И. 467
Леднева А. М. 241
Лендель Г. Э. 138, 476, 533, 591, 676, 759, 774, 804, 846, 884, 921, 1024, 1025
Ле Шателье 900
Лингейм Дж. Дж. 173
Линде-Глэддинг 747
Линдеман 900
Лоу 1012, 1013, 1014, 1032
Лохвицкая А. П. 368
Лунге 49, 491, 774, 865, 1038
Лурье Ю. Ю. 170, 253, 259, 261, 273, 278, 291, 295, 298, 314, 315, 330, 369, 448, 476, 486, 488, 489, 492, 519, 520, 579, 590, 712
Ляликов Ю. С. 231
Максимюк Е. А. 423
Мальцев В. Ф. 368
Малюга Д. И. 475
Маракаев А. А. 691
Мардер Р. И. 517
Мариньяк 670, 683
Маркова Г. А. 454
Масленникова Г. С. 86, 458
Матросова Т. В. 649
Медведева О. А. 638
Мейер Г. 846
Мервин 1026, 1027, 1028, 1061
Мирзоева И. 541
Митташ А. 106
Митчел А. Д. 153
Митчерлих 880, 989, 991
Мицеловский Э. С. 184
Моньякова Л. Н. 689
Морачевский Ю. В. 532
Мосандер 626
Мухина 3. С. 647, 844
Нагерова Э. И. 658
Назаренко В. А. 344, 883
Назаренко Т. Н. 577
Неклютина В. Ф. 486
Нельсон Ф. 628
Немодрук А. А. 635
Нерсесова С. В. 422
Неустроева М. В. 265
Нпдерль В. 851
Нидерль Дж. 851
Никитина Е. И. 294, 301, 454
Николаева 3. В. 579, 712
Никольс М. 857
Нойес А. 65, 85, 386, 388, 407, 658, 823, 883
Ноульз Г. Б. 29, 297
Оленин С. С. 56
Остроумов 3. А. 86, 88, 111, 271, 273, 280, 458, 524, 529, 546, 551, 582, 583, 585, 606, 615, 627, 654
Панченко Г. А. 517, 659
Пенфильд 908, 909
Перлих 1034
Петров А. Д. 265
Пешкова В. М. 454, 468
Пилипенко А. Т. 174, 454, 596
Пирсон 1036
Платонов М. С. 691
Погребинская М. Л. 452
Подвальная Р. Л. 689
Полежаев Н. Г. 256
Полуэктов Н. С. 351, 353, 380, 588, 776, 883
Полянский В. Н. 360
Пономарев А. И. 1037
Попов К. И. 775
Попов М. А. 289
Пратт 987, 995, 997, 999,: 1001
Прехт 67
Прошенкова Н. Н. 422
Пршибил Р. 157, 255, 597, 712, 728
Птицын Б. В. 423
Птицын В. В. 452
Пфафф 1042
Пшеницын Н. К. 431
Рабинович А. И. 231
Раецкий М. В. 659
1074
Именной указатель
Райхипштейн Ц. Г. 293
Ричардс 51, 694
Родден 36
Розанов С. Н. 454, 455
Розенбладт 1031
Романов Д. В. 184
Рощина Р. Н. 343
Рудо Н. 32
Русанов А. К. 177
Рыбникова А. И. 261, 315, 492
Рябчиков Д. И. 422, 423,632
Сабинина Л. Е. 331
Савиновский Д. А. 158, 728
Самуэльсон О. 184, 628
Сауков А. А. 254
Сендэл Е. Б. 35, 157, 175, 227, 240, 255, 280, 301, 492, 548, 582, 1034
Серебряный С. Б. 241
Силаев Е. В. 662
Синякова С. И. 159
Смит Л. 25, 46, 82, 130, 180, 567, 730, 737, 742, 938, 1005, 1006, 1008, 1011, 1014, 1060
Сойферман И. А. 344
Сонгина О. А. 776
Соул 1001, 1002
Старик И. Е. 532
Стейгер 824, 959, 960, 1025, 1026, 1028, 1062
Стенгер В. А. 198, 202, 204, 490
Степин В. В. 213
Стюнкель Т. Б. 158, 728
Суворовская Н. А. 168
Супрунович И. Б. 255
Сырокомский В. С. 213, 422, 441, 449, 451, 531, 662
Талипов Ш. 586
Таль Э. М. 476
Тананаев И. В. 467, 586
Тананаев Н. А. 185, 368, 659
Танатар С. 63
Тараторин Г. А. 259, 289
Терентьева Е. А. 632
Тиновская Е. С. 149
Трамм Р. С. 776
Траутман 957
Тредвелл Ф. П. 221, 733, 995, 1001, 1020
Троицкая К. В. 468
Троицкая М. И. 170, 298, 475, 476
Уолтерс 501
Уорд А. М. 153
Уральская А. В. 449
Усатенко Ю. Н. 289
Успенская Т. А. 320, 607, 626, 646, 676, 682
Уэрри 843
Фаворский Л. 56
Файгль Ф. 185, 246, 294, 492
Файнберг С. Ю. 261, 267, 289, 294, 299, 316, 329, 341, 492
Фалеев П. Ф. 775
Федоров П. Ф. 689
Федотова Е. А. 454
Ферьянчич Ф. А. 776
Филиппова Н. А. 330, 486, 489, 492
Фишер А. 56
Фогельсон Е. И. 316, 454, 492
Фрайберг С. М. 267
Фрезениус 721, 733
Фрид Б. И. 449, 676, 680, 890
Фридман Я. Д. 776
Функ В. 105
Фурман Н. 230, 231, 519
Хан Г. А. 77
Хейс А. А. 880
Хлопин В. Г. 525, 531
Хлопин Н. Я. 231
Худынцев А. 329
Цивина Б. С. 316, 712
Цинберг С. Л. 643
Чатард 977
Чепик М. Н. 477
Черкасов В. П. 213
Черкашина Т. В. 371
Чернихов Ю. А. 171, 293, 320, 328, 343, 375, 476, 492, 517, 533, 585, 588, 607, 626, 646, 676, 682, 770, 771, 774, 775, 776, 1025
Чеховпч М. Д. 468
Чугаев Л. А. 457
Чуднович 981
Чэпин 832, 835, 839, 1032
Шаврин А. М. 56
Шалфеев В. М. 56
Шапиро М. Я. 329
Шахов А. С. 394, 775
Шварцбуд Л. Е. 344
Шведов В. П. 261
Шебаренкова А. П. 355, 548, 550, 557
Шейбе Г. 177
Шеленбергер 1002
Шемякин Ф. М. 184, 359, 633, 634
Шескольская А. Я. 890
Шик И. Р. 294
Шкробот Э. П. 539, 548
Шлейхер А. 56
Шрайбман С. С. 728
Штарк 1024
Шуб М. Е. 329, 330
Щербов Д. П. 468, 475
Эйтель 914
Эшка 1030
Юровская Ф. 492
Якимец Е. М. 158, 728
Якоб В. 978, 979
Якоб Я. 966
Яковлева О. Я. 468
Яннаш 48, 921, 931, 1011
Abbott J. L. 182
Abrahams Н. J. 306
Adami Р. 586
Adams 843
Adams D. F. 392
Adams F. Q. 896
Adams J. E. 856
Adams J. R. 231
Adams M. F. 757
Adams R. 244
Adamson D. С. M. 318
Adler N. 688
Adolph W. H. 822, 826
Adriano F. T. 864
Agulhon H. 831, 1033
Ahrens L. H. 178
Aitkenhead W. C. 346
Alber С. M. 56
Alexander H. 267
Alicino J. A. 860
Allen 839, 901
Allen E. T. 154, 259, 303, 794, 798, 802, 831, 832, 1033
Allen H. O. 293
Allen W. F. 408
Allen W. H. R. 339
Allen W. S. 312, 313, 320,
794
Allison E. R. 348
Alsberg C. L. 221
Alsterberg G. 818
Amin A. 158
Anderegg F. O. 237
Anderson A. C. 206
Anderson W. T. 518
Andes R. V. 758
Andrew G. 255
Andrews L. W. 239, 810
Именной указатель
1075
Aradine Р. W. 933
Archibald Е. Н. 250, 732
Ardagh Е. G. R. 480, 490
Armstrong Н. Е. 869
Armstrong W. D. 825
Arnold Ed. 185
Arnold E. 750
Aschoff K. 809, 810
Ashley S. E. Q. 164, 212
Atack F. W. 463
Ato S. 162, 552, 553
Audrieth L. F. 211, 632, 792
Auger V. 84, 143, 654
Aughey H. 180
Aurousseau 946
Austin M. 300, 503, 504, 719, 961
Avery S. 851
Axelrod J. 164
Axilrod H. D. 740
Axt M. 637, 642
Ayres A. 635
Ayres G. H. 468, 658
Babler B. J. 378
Bachelet M. 692
Bacon F. R. 738
Baeckstrom S. A. 230
Bahr J. 628
Bailey G. H. 971, 973
Bailey R. K. 745
Baker I. 344
Baker W. O. 748
Balarew D. 719
Ballard A. E. 255
Ballard J. W. 935
Balz G. 461
Bambach K. 814
Banerjee P. C. 450
Bangerter F. 157
Banks С. V. 423
Barab J. 867
Barber H. H. 749
Barbier P. 117
Barnebey O. L. 224, 448, 994, 1000
Barnes J. W. 312
Barnes R. B. 176
Barnes S. K. 584
Barthauer G. L. 632
Barton C. J. 293
Barueby O. 56
Baskerville C. 117, 440, 646, 655, 661, 973
Bassett L. G. 340, 578
Bates R. G. 194
Baubigny H. 541, 817, 978
Baudisch O. 143, 285, 643
Bauer Th. 760
Baulieu W. E. 781
Bawden A. T. 231
Baxter G. P. 76, 237, 781, 812, 813, 815, 816 68*
Baylis J. R. 231
Bayly P. G. W. 908
Bazen J. 158, 611, 612
Beale R. S. 542
Beamish F. E. 390, 402,
407, 408, 411, 412, 414,
416, 419, 423, 807
Bean L. 180
Beck O. 334
Bee N. 481, 486, 553, 554
Beebe A. C. 267
Beeson C. 450, 451
Behrend R. 818
Beilstein F. 300
Bekk J. 817
Bell R. K. 278, 286
Bell R. N. 792
Bellucci I. 829
Bendigo В. B. 278
Bennet E. L. 857
Bennett W. R. 523
Benoit J. R. 35
Benz E. 601, 602, 604, 605 Berg O. 372
Berg R. 148, 149, 280, 297,
537, 542, 717, 730, 818
Berger H. W. 761
Berger К. C. 844
Berk L. H. van. 211
Berman H. 186
Bernewitz M. von 77
Berquier F. 587
Berry A. J. 812, 813, 814
Bertrand G. 831, 1033
Beryl E. 855
Berzelius J. J. 769, 824,
1005
Beshgetoor A. W. 244, 563
Beutell A. 896
Bichowsky F. R. 490
Biedermann W. 157
Biltz W. 373
Binder F. 530
Birks L. S. 182
Birnbaum N. 139, 371
Bischof G. 293
Bischofberger G. 673
Bischoff F. 660
Bishop H. B. 320, 794
Black O. F. 312
Blair A. A. 360, 499, 503, 504, 781
Blalock T. L. 190
Blank H. R. 349
Blaschke K. 896
Blasdale W. C. 276
Bleesen M. 56
Bleyer B. 588
Blodgett G. 303
Bloom A. 240
Bloor W. R. 946
Blount B. 1051
Blum W. 214, 498, 499, 505, 561, 566, 567л 761
Blumenthal H. 323
Bodansky M. 492
Bodlander E. 615
Boehm G. 57
Bogan E. J. 801
Bogatski G. 774
Bogue R. H. 180
Bohm 358
Bohn R. T. 515
Boldyreff A. W. 251, 707
Bolm F. 732, 1010
Bolton H. C. 1046
Boltz D. F. 688
Bong G. 917
Bongard G. R. 480, 490
Bonilla C. F. 436, 457
Bonnar R. U. 829
Bonner W. D. 202, 210, 212
Booth H. 57, 73, 76,253, 254
Borchardt L. F. 167
Borck H. 118
Borgstrom L. H. 1016
Bornemann E. 910
Bornemann K. 118
Bottger W. 240
Bouissieres 692
Bourion 672
Bower J. H. 75
Boyd T. 148, 631
Boyer W. J. 462
Boyle A. J. 563, 746
Bozsin M. 935
Brabson J. A. 757
Bradstreet R. B. 863
Bradt W. E. 108, 384
Brady W. 857
Braile N. 260
Brailler P. S. 449
Brandegee M. M. 868
Brandt W. W. 213
Brastow W. C. 411
Braun К. C. 576
Brauner 393, 394, 731
Brauns D. H. 829
Bray A. R. 712
Bray R. H. 792
Bray U. B. 546, 547
Bray W. C. 65, 85, 162, 216, 539, 883
Brearley H. 459
Brewer F. M. 1345
Breyer F. C. 489
Bricker С. E. 158, 167, 648
Bricker L. C. 492
Bridges R. W. 529, 560, 583, 750, 825
Bridgman J. A. ; 551, 554, 556 546, 549,
Bright H. A. 110, 166, 215,
216, 278, 293, 460, 502,
512, 792, 804, 844, 863
Brinton P. H. 36, 367, 525,
&|621, 622, 625, 862 732, 860,
1076
Именной указатель
Briscoe Н. V. А. 832
Britton Н. Т. S. 100, 531, 681
Brode W. R. 177
Brodie S. S. 851
Brooks E. J. 182
Brookshier R. K. 776
Brophy D. H. 166, 471, 564, 606, 755, 767, 768, 834
Brown С. T. 751
Brown D. J< 261
Brown E. R. 293
Brown M. H. 739
Browning B. L. 392, 393, 394
Browning К. C. 254
Browning P. E. 538, 540, 541, 551, 553, 626
Brubaker H. W. 809
Briickner K. 384, 391
Briihl E. 961
Bruhns G. 289 '
Brukl A. 154, 386, 538, 540, 551, 553, 629
Brunck O. 105, 279, 708, 710
Bruner L. 84
Brunner C. 856
Bruttini A. 533
Bryant F. J. 590
Bube K. 719
Buchheit H. A. 317, 339
Buckley F. 172
Bugarszky S. 810
Biilmann E. 231
Biilow K. 246
Bull I. C. 267
Bunsen R. W. 628, 756
Burford M. G. 250, 342
Burg W. T. 814
Burgess G. K. 45
Burgess L. L. 747
Burkey R. E. 492
Burris; R. 388
Burstaill F. H. 685
Busker D. 177
Butler С. M. J. 211
Butler L. I. 293
Byerly W. 609
Byers H. G. 382, 385
Byers J. L. 416
Byrd R. M. 750
•Cade' G. N. 64, 297
Cady L. C. 490
Cain J. R. 108, 166, 453, 510, 512, 519, 783, 857
Cake W. E. 863, 942
Caldwell C. W. 165
Caldwell G. C. 126
Caldwell J. R. 288, 479, 813
Caley E. R. 53, 250, 342, 712, 727, 738, 740, 748, 750, 751
Calhape O. 56
Calhaue D. F. 56
Callan T. 293
Cameron A. 756
Campagne E. 518
Campbell D. 721
Campbell E. D. 154, 604
Campbell M. 158
Carey F. P. 303
Carey W. M. 218
Carhart H. S. 64
Carius L. 847
Carlson W. A. 167
Carlson W. E. 750
Carney R. J. 604
Carnot A. 957
Carpenter R. O’b 178
Carr F. H. 200
Carton M. K. 443, 584
Case О. P. 315
Caseneuve M. P. 255
Cassal С. E. 831
Cassil С. C. 312
Castanares A. P. 271
Caul H. J. 931
Cavaignac H. 820, 939
Celsi S. 170
Center E. J. 792
Challis H. J. 385
Chamberlin R. T. 1035
Chamot E. M. 187, 869
Chancel G. 562
Chandlee G. C. 156, 272, 280, 334, 418, 542, 639
Chang K. S. 536
Chapin R. M. 221, 222, 223,
224, 227, 228
Chapin W. H. 830, 835, 839, 843, 1032
Chapman H. D. 710
Chapman J. E. 449, 450
Chapman R. P. 218
Charpentier P. 238
Chatard T. M. 366, 655, 661,
911, 977
Cheftel H. 261
Chenery E. M. 576
Cheng K. 712
Chiang С. H. 486
Chirnside R. 166
Chism R. E. 243
Cholak J. 255, 301, 492
Churchill J. R. 178
Churchill H. V. 529, 560, 561, 583, 750, 825, 828
Ciochina J. 766
Claassen A. 642, 645
Clabaugh W. S. 39
Claessens J. 146, 336
Clark A. B. 198
Clark A. R. 282
Clark L. J. 931
Clark N. A. 454
Clark P. E. 124
Clark R. E. D. 344
Clark W. M. 198, 201, 208, 230
Clarke B. L. 168, 184, 185, 260
Clarke F. W. 89, 98, 235, 296, 302, 321, 345, 549, 617, 663, 715, 729, 777, 830, 846
Clarke S. G. 144, 329, 338, 511
Classen A. 118, 167, 328, 343
Classen E. 982
Clauder О. E. 393
Clausmann P. 819, 820
Clegg D. 184
Clemons C. F. 392
Clifford P. A. 267
Clinch J. 609
Clippinger D. R. 231
Cloedt E. V. 961
Cluley H. J. 348, 351, 354
Coase S. A. 345
Coblentz W. W. 897
Cohen A. 200
Cohn A. I. 244, 857
Cole H. 809, 810
Coleman W. C. 392
Collin E. M. 168
Collins W. D. 738
Coltman R. W. 498, 499
Conant J. B. 463, 466
Cone W. H. 490
Conner E. B. 851
Connor M. F. 953, 1009
Conrady A. E. 38
Conroy J. T. 46
Convey J. 180
Cooke J. P. 992, 999
Cope W. C. 867
Coppieters 642
Corey R. B. 578
Corleis E. 856
Coster D. 635
Cox D. C. 231
Cox H. 368
Craft С. H. 576
Craig A. 587
Craig D. N. 204, 263
Craig K. A. 334
Crane R. M. 732
Cremer E. 56
Crippen R. 504
Croad G. F. 205
Crockett W. G. 860
Crookes W. 552
Cross W. 890
Crossley H. E. 312, 314
Crowell W. R. 289
Cruikshank A. J. 423
Culberston J. B. 173, 468
Cumenge E. 510
Cunning J. W. 859
Именной указатель
1077
Cunnigham В. В. 454
Cunnigham Т. R. 138, 146,
465, 498, 530, 590, 682
Curl A. L. 392
Currah J. Е. 423
Curtis J. S. 1033
Cushman A. S. 297, 541, 812, 813
Czudnowicz С. 981
Daehr Н. 532
Dahls D. 825, 828, 829,
1028
Dakin H. D. 485
Dale J. 408, 411
Daniel H. A. 452
Daniel K. 823
Darbinian M. B. 154
Das-Gupta P. N. 154
Daublander J. 685
Daudt 569
Daudt H. W. 862, 863, 866
Dauncey L. H. 166
Davey W. P. 183
Davidson J. 866
Davies G. R. 347
Davis L. T. 646, 973
Davisson B. S. 865
Davy H. 921
Dawson E. C. 260
Dawson L. R. 758
Dawson P. R. 315
Day A. L. 901
Dean L. A. 504
Deeds F. 792
Deemer R. B. 315, 862
Deering E. C. 675
Degering E. F. 148
Deitz V. R. 792 4
Deladrier E. 829
Delano P. H. 264
Delauney E. 628
Delesse A. 1035
Demorest 70
Demorest D. J. 283, 285,
292 ,
Dennis L. M. 346, 347, 349,
546, 549, 551, 554, 556
Dennstedt M. 70
Deribfcre M. 176
Dernies J. 145, 271
Desesa M. A. 543
Deussen E. 955, 992
Devarda A. 865
Diamond J. J. 180
Dibbits H. C. 72
Deckens P. 477
Dickinson D. 844
Dickman S. R. 792
Diehl H. 165, 298, 423,
1014
Di6nert F. 757
Dietert H. W. 178
Dieulafait L. 881
Dimbleby V. 832
Dimitrow P. 719, 721
Dingemans H. H. 918
Dinnin J. I. 692
Dittrich M. 117, 124, 621, 653, 887, 904, 911, 974, 995, 1009
Divers E. 389
Divine R. E. 152
Dixon В. E. 850
Dobbins J. T. 750
DoleJVL 230, 231
Dolezal J. 159
Domange L. 829
Don J. R. 1033
Donald M. B. 866
Donan J. 187
Donath E. 303, 320, 334
Doring Th. 1008
Dougherty G. 794
Dover M. V. 76
Drabkin D. L. 293
Drapier N. 615
Drechsel G. 813
Driver J. E. 814
Drossbach G. P. 604
Drushel W. A. 805, 868
Dubois E. 178
Dudley H. C. 385
Duke F. R. 219
Dulin R. S. 285
Dumas J, B. 859
Dunbar-Poole A. G. 327
Duparc L. 926, 938
Durham E. J. 300
Durst D. 293
Duschak L. H. 802
Dutcher H. A. 828
Dyer B. 491
Easly H. F. 167
Eberhard G. 613
Echdahl W. P. 1008
Eden A. 747
Eddy C. W. 792
Eddy E. A. 717
Edgar G. 958
Edmiston H. D. 862
Edmonds S. M. 139
Edwards J. W. 176
Eegriwe F. 331
Eggertz V. 781
Ehret W. F. 36
Ehrlich J. 304, 305
Eisner A. 349, 863
Eitel W. 911
Eilenburg J. Y. 685
Ellestad R. B. 525
Elliott E. C. 751
Elliott J. A. 253
Elston С. M. 805
Elving P. J. 712, 727
Emich F. 187
Emmerton F. A. 781
Emmon S. F. 1033
Engemann H. 272
Ensign J. R. 809
Ephaim E. 294
Epperson A. W, 46, 951, 1007
Erdmann H. 285, 1035
Eschka A. 243
Esteve E. 242
Eubank W. R. 180
Euler H. 370
Evans B. S. 184, 272, 294,
296, 308, 316, 32% 338,
340, 341, 474, 477, 811 480,
Evans R. D. 692 Evenson R. F. 289 Evers E. W. 68, 70
Evers N. 293
Ewan T. 818
Fahey J. J. 244, 828 ;
Fahrenkamp E. S. 537, 542
Fainer P. 423
Fairbanks C. 370
Fairchild J. G. 745, 828
Fales H. A. 474, 482, 484
Fales W. T. 934
Fanchon M. L. 330
Fankuchen I. 183
Fassel V. A. 179
Fay H. 392, 393
Fedde W. C. 593
Feigl F. 84, 143, 176, 185, 245, 246, 260, 280, 294, 457, 474, 492, 636, 774
Feisel J. P. 240
Feit W. 372
Feldman C. 179
Fenwick F. 230, 231, 360, 371, 383, 392, 515’, 818
Ferguson J. B. 436, 448
Ferguson W. C. 205
Fernenius W. C. 645
Ferris C. A. 825
Fethenheuer B. 56
Field J. 221
Fieldner A. C. 1016, 1060
Fife J. G. 168
Fife J. M. 864
Finkener R. 781
Finn A. N. 39, 512, 840, 924
Fischer H. 240, 301, 334
Flagg J. F. 143, 470
Flaschka H. 157, 158, 611
Flatt R. 450
Fleming W. R. 68
Fletcher M,H. 588
Fleury P. 865
Flickinger L. C. 844
Flora С. P. 300
Fogg H. C. 346, 549, 550,-556, 606, 638
1078
Именной указатель
Foote F. J. 351, 844 Gard E. L. 78 Green D. E. 646
Foote H. W. 288, 289, 1032 Forbes E. C. 408, 416 Forbes G. S. 237 Gardner K. 857 Garrett O. F. 749 Gatterman L. 846 Green H. S. 52 Green J. 864 Greenberg L. A. 847
Forbes J. C. 815 Ford J. J. 158, 611 Ford S. A. 494 Ford W. E. 1032 Forster 0. 865 Gautier A. 819, 820, 846, 1035 Gebhardt H. T. 295 Gee A. 792 Gee E. A. 576 Greenough M. L. 165 Gregory A. W. 857 Grilling E. P. 221 Griffith F. S. 280 Grimaldi F. S. 443, 612, 774
Fortune W. B. 174, 454 Foster L. S. 550 Foster M. D. 828 Geer W. C. 546 Geilmann W. 374, 376 Geist H. H. 639 Grimm C. 190 Grosscup 352 Grosse A. V. 639, 641, 692
Foulk C. W. 204, 210, 224, 231 750 Foulton H. von 1033 Geldard W. J. 868 Gentry С. H. R. 578 Germuth F. G. 145 Guess H. A. 265 Guichard M. 223 Guillaume Ch. Ed. 188
Fowler R. M. 110, 173, 215, Gerrans H. 831 Guiterman K. S. 84
460, 465, 468, 755, 756, Gerritsma K. W. 255 Guldina E. 533
857 Fox E. G. 868 Frank G. 219 Frank H. S. 453 Getz C. A. 219, 450, 451, 590 Gewecke J. 56 Gibbs R. S. 344 Gutbier A. 484 Guyer 673 Guyot P. 829 Guzman J. 56, 170
Franke K. W. 388 Frankland E. 869 Franz B. 973 Frear W. 862 Frederick W. G. 331 Fredga A. 384 Freedman L. D. 331 Gibbs W. 503, 562, 769 Gibson F. H. 231 Gibson J. A. 327 Gieseking J. E. 1006 Gilbert J. P. 756 Gilbert R. S. 685 Gilbertson L. I. 392 Gyory S. 328 Haar K. ter 158, 611, 612 Haas 628 Haber F. 231 Hackl O. 953, 962, 1033 Haddock L. A. 293, 542
French C. L. 452 Gilchrist R. 100, 261, 283, Haen F. de 285
Fresenius C. R. 244, 550, 335, 343, 407, 408, 414, Hague J. 293, 792, 844, 863
857 415, 420, 421, 424, 545 Hahn F. L. 148, 316, 717,
Fresenius H. 732 Fresenius L. 582 Giles W. B. 101, 108, 605, 665 730, 818 Hahn R. B. 647
Fresenius R. 524, 579, 604, 697 Gilkey W. K. 825 Gilman H. 344 Haines R. I. 423 Hakomori S. 141
Freund H. 640, 690, 776 Ginsberg H. 807 Hale С. H. 749
Freund S. 117, 653, 973 Frevel L. K. 183 Frick C. 272 Friedel C. 510 Friedel G. 906 Gjems O. 158 Gladstone M. 164 Glaze F. W. 750, 840 Glazunov A. 185 Goetz G. W. 781 Hall A. J. 468, 476 Hall D. 359, 471 Hall D. A. 628 Hall N. F. 230 Hall R. D. 656, 671
Friedheim C. 370, 755, 767, 804, 961 Goldenberg H. 615 Goldman F. H. 933 Hall R. W. 761 Haller H. C. 320
Frieh O. 769 Gooch F. A. 118, 125, 240, Hallett R. L. 337
Friend J. N. 628 285, 306, 370, 388, 392. Hamberg A. 896
Fritz H. 185 393, 443, 503, 504, 564^ Hamill W. H. 860
Fritz J. J. 158, 611 654, 655, 661, 701, 717, Hammett L. P. 139, 218,
Fritz J. S. 647 Fritz N. D. 608 Froehde A. 334 719, 738, 808, 809, 829, 841, 843, 911, 961 Goodspeed E. W. 699 576 Hammond L. D. 826, 829 Hammond W. A. 76
Frommes M. 582 Fryling C. F. 139 Fucke H. 685 Fudge J. F. 199 Fulda M. O. 647 Funk W. 105 Gordon C. L. 402, 847, 931 Gordon L. 102 Gorman J. G. 184 Goto H. 571 Graham R. P. 166 Grammont A. 456 Hamner H. L. 514 Hampe W. 285 Hana wait J. D. 183 Handy J. O. 727 Hann R.M. 863 Hannay J. B. 494
Furman N. H. 93, 142, 146, Grandval A. 869 Hansen H. V. 825
147, 167, 198, 219, 229, Grant J. 176 Hanson V. F. 255
231, 260, 284, 320, 336, Grassl G. 84 Hardwick P. J. 590
383, 447, 459, 519, 629, 751 Gray A. G. 769 Gray T. 503 Graydon W. 408 Hardy L. V. 78 Hardy V. R. 205 Haring M. M. 459, 467
Gahler A. R. 468 Gallo G. 662 Greathouse L. H. 505, 818 Greeff A. 829 Harkins W. D. 313 Harley J. H. 857
Именной указатель
1079
Harley J. Н. А. 127
Harper Н. J. 452
Harris М. 267
Harris Т. Н. 861
Harris W. 157
Harrison А. Р. 863
Harrison G. R. 177
Hartley W. N. 536, 544, 549, 731, 882
Hartong В. D. 231
Hartshorne N. H. 187
Harvey С. E. 179
Harvey C. 0. 915
Harwey I. W. 757
Harwood H. F. 456, 602, 747, 1034
Hasler M. F. 178
Hassler F. 70
Hastings J. L. 692
Hauser 0. 621, 632, 633, 666, 672, 683
Hauser R. 673
Havens F. S. 118, 439, 564, 585
Hawley F. G. 759, 822, 826
Hawley L. F. 541, 542
Hayden E. M. 643
Hayes J. R. 280, 418
Hayman D. 851
Haywood F. W. 175
Hazel W. M. 1007
Headden W. P. 313
Heath F. H. 285
Heberling J. B. 164
Hecht F. 187, 525, 890, 915
Hecht 0. 236
Heck J. E. 868
Heczko T. 573, 762
Hehman P. L. 369
Heidenreich 0. 921
Heinrich B. J. 696
Heintz W. 503
Helmick H. H. 599
Hempel W. 267, 895, 917
Henci V. 159
Henderson J. A. 293
Henderson W. D. 64
Henderson W. H. 755, 767
Hendrixson W. S. 64, 203, 210, 217, 818
Henz F. 322, 326, 328, 343
Herlich K. 183
Hermance H. W. 185
Hershenson H. M. 543
Hess W. H. 154
Hevesy G. 635, 641
Hey M. H. 1003
Heyl P. R. 102, 109, 160
Heyrovsky J. 173
Hibbert E. 449, 662, 769
Hibbs J. G. 512
Hickman К. C. D. 199
Hicks C. W. B. 672
Hicks H. G. 685, 686
Hicks W. B. 745
Higgs D. G. 184, 294, 338, 340
Hildebrand J. H. 167, 230,
231, 448, 843
Hill A. E. 813
Hill M. A. 408
Hill W. L. 576, 806, 825
Hillebrand W. F. 77, 132,
370, 439, 507, 508, 510,
513, 523, 526, 529, 632,
652, 654, 655, 661, 666,
670, 696, 702, 756, 760,
777, 779, 893, 894, 900,
901, 904, 955, 970, 979,
980, 981, 982, 986, 987,
991, 992
Hillis T. E. 289
Hillson H. D. 502
Hillyer J. C. 743
Hilton F. A., jr. 237, 812,
813
Hinden F. 1047, 1058
Hinmann C. W. 804
Hinrichsen F. W. 820, 939
Hintz E. 284, 604, 796, 802
Hippie J. A. 184
Hiskey C. F. 139, 173, 374,
688
Hoag L. E. 552
Hoffman G. C. 669
Hoffman H. 0. 334
Hoffman J. D. 931
Hoffman J. I. 54, 66, 83,
98, 109, 165, 166, 204,
208, 232, 311, 368, 374,
378, 379, 383, 440, 464,
475, 497, 526, 532, 547,
550, 554, 583, 591, 696,
718, 739, 781, 785, 786,
788, 789, 791, 812, 822,
826, 828, 884, 921, 924,
931, 936, 1005, 1009,
1019, 1024, 1062
Hofman H. 0. 761
Hoitsema C. 238
Hok B. 646
Holladay J. A. 138, 146, 362, 530, 637
Hollander M. 351
Holliger M. 804
Hollingsworth M. 210
Holloway G. T. 243
Holmes S. 143
Holness H. 348
Holt M. L. 769
Holthoff C. 917
Holzer H. 420, 431
Hommel W. 358
Hoover C. R. 205
Hope H. B. 142, 291, 547
Hopkins B. S. 617, 629
Hopkinson F. J. 231
Horn D. W. 980
Horn M. J. 388
Hornberger A. W. 393
Hornichova E. 158
Horton A. D. 649
Horton P. G. 224
Hoskins W. M. 825
Hostetter J. C. 193, 230, 440, 448, 453, 510, 519, 716
Houben J. 846, 847
Hough G. J. 221
Howe D. E. 454
Howe J. L. 402
Howland J. 393
Hubbard D. M. 255, 280, 301. 492
Hugg H. 240
Hughes W. S. 231
Hulett G. A. 202, 210, 212, 761, 802
Hullett H. R. 454
Hume D. 135, 686, 690
Hummell F. C. 454
Hundeshagen F. 781
Hunt E. C. 692
Hunt H. 868
Huntz E. 524
Hurd L. C. 282, 369, 374, 378
Hure I. 587
Hurley F. H. 207
Hussey R. E. 863
Hutchin H. W. 769
Hutchins G. P. 540
Hutchison A. W. 542
Hussey R. E. 863
Hutner K. 1035
Huttig G. F. 128
Huttner C. 162
Hutton R. S. 388
Hybbinette A. G. 353
Hymas F. C. 164
Iddings 890
Ide W. S. 860
Ikenberry L. C. 579
Illinsky M. 473
Ilkovic D. 172
Illingsworth С. B. 503
Irving H. 255
Isaacs A. 58, 473, 499
Ishii R. 539, 548
Ishimaru S. 202
Ismail A. M. 602, 747
Jackson D. E. 630
Jacob K. D. 720, 787, 806, 819, 868, 1019
Jahn C. 654, 677, 681, 766, 768
Jakob J. 966
Jakob W. 978
James C. 346, 550, 605, 606, 625, 638, 644
1080
Именной указатель
James Е. J. 184
James L. Н. 369, 494
Jamieson G. S. 227, 291, 328, 481, 488
Jander G. 579
Jannasch P. 359, 389, 512, 768, 809, 810, 833, 917, 921, 931, 952, 961
Jannettaz E. 1046
Japhe D. 1007
Jarmus J. M. 657
Jarrell T. D. 862
Jarvinen К. K. 719
Jawein L. 300
Jefferson A. 627
Jeffery J. H. 446
Jeffreys С. E. P. 479
Jensen M. A. 230
Jilek A. 516, 629
Jirkovsky R. 185 ’
Johnsen A. 897
Johnson С. M. 66, 519, 774, 796
Johnson E. B. 347, 349
Johnson F. M. 74, 75
Johnston J. 794, 798, 802, 1046
Johnstone S. J. 626
Johrbandt A. 611
Jones A. L. 774
Jones C. 135
Jones G. C. 446
Jones H. W. 442
Jones L. C. 841
Jones L. T. 792
Jones P. T. 230
Jordis E. 760
Juday C. 757
Jukkola W. 424
Jung H. 933
Kahane E. 452, 751
Kahlenberg L. 738
Kallauner O. 695
Kallmann S. 737
Kaminska H. 206
Kamm O. 145, 747
Kankanian A. G. 154
Kannappel E. 835, 839
Kano N. 141, 221
Kanter E. H. 760
Kao С. H. 389
Kaplan E. 934
Kapron M. 369
Kapulitzas H. J. 457
Karaoglanov Z. 266
Karaoglanow Z. 719, 721
Kaspin B. L. 125
Kassner E. E. 225
Kassner J. L. 225, 261, 263
Kato, Hisaji 480
Kato T. 214
Kaufman H. S. 183
Keefe W. H. 271
Keller E. 389
Kelley G. L. 68, 69, 70, 231, 463, 466, 503, 515, 595
Kellog A. M. 208
Kellog H. B. 208
Kemp J. W. 178
Kenny W. R. 199
Kern E. F. 161, 162, 525
Kestranek V. 231
Kharasch M. S. 164
Kieft L. 272
Kikuchi S. 141
Kilpatrick M. 222, 225
Kilpatrick M. L. 63, 225
Kilpatrick M. K. 225
Kimball R. H. 807
Kimura, Kenjiro 628, 641
King E. J. 757
King V. L. 285'
King W. B. 344
King W. J. 369
Kinnunen J. 158
Kirschman H. D. 546, 547® 552
Kitson R, E. 353, 468, 792
Kieldahl J. 859
Kleinberg J. 632
Kiekotka J. F. 924
Klemensiewwicz Z. 231
Kling A. 146, 336
Klinger P. 658, 659, 688
Klubalova J. 158, 597
Kanpper J. S. 334
Knauel G. 543
Knecht E. 449, 662, 769
Knop J. 442, 447
Knopf A. 933
Knorre G. von. 473, 503, 629, 797, 804
Knowles D. C. 386
Knowles H. B. 100, 137,
138, 139, 143, 147, 148,
248, 281, 282, 333, 361,
365, 374, 377, 378, 437,
445, 448, 455, 459, 470,
479, 480, 484, 486, 496,
504, 509, 518, 527, 530,
539, 561, 565, 567, 570,
571, 572, 573, 576, 583,
586, 596, 606, 636, 637,
638, 641, 643, 660, 671,
686, 695, 698, 699, 704,
714, 720, 723, 756, 796,
834, 884, 943, 983, 1009, 1013
Knox J. 270
Knudson H. W. 757
Kobayashi M. 829
Kober P. A. 50
Koch A. A. 1022
Koch F. C. 863
Koch W. 579, 658, 659, 688
Koch W. W. 211
Koelsch H. 547
Koenig E. W. 572, 747, 749, 750, 918, 1014
Kohl A. F. 829
Kohlrausch F. 250, 541, 815, 816
Kollock L. G. 240
Kolthoff I. M 84, 107, 148,
173, 198, 200, 202, 204,
205, 206, 211, 216, 217,
225, 231, 249, 280, 290,
489, 490, 502, 519, 565,
582, 749, 811, 814, 818,
828, 832
Komarowsky A. S. 804
Kondaiah K. 323
Koninck L. L. de 990
Koningh L. de 979
Корре P. 993
Koppel I. 358
Kopper F. 70
Koppius O. G. 180
Koskey P. J. 360, 767
К oss M. 604
Koten I. A. 244, 245
Kott A. E. 219
Koudela Z. 158
Kraft-Ebing H. 525
Kramer W. R. 563
Krarup J. 231
Krasnitz A. 866
Kraus K. A. 635, 685
Krauskopf F. C. 368, 738
Krauss F. 831
Kremer M. 587
Kremers H. E. 627, 631
Krieger K. A. 760
Krishnayya H. V. 1012, 1013
Krivohlavy J. 185
Krumholz P. 636, 774
Kriiss G. 628
Kriiss H. 628
Krutwig J. 240
Kucharsky J. 158, 587
Kuhnt O. 66
Kumins C. A. 646
Kunin R. 183, 184
Kunos F. 305
Kunz A. H. 226
Kiirscher K. 868
Kurtz L. T. 142, 792
Kurtz T. 712
Kiister F. W. 240
Kuzirian S. B. 914
Kvalheim A. 613
Lachele С. E. 315, 805
Laitinen H. A. 216, 217
Laioux H. 869
Lakin H. W. 185
Lamar M. O. 940, 1007, 1008
Lambie D. A. 276, 342, 359, 766
Именной указатель
1081
Lamphere R. W. 165
Lamphrey H. 155
Lang J. 84
Lang R. 216, 818
Lange G. 374
Langmuir A. C. 161, 162
Langmuir I. 986
Larrabee С. P. 502
Larsen E. M. 645
Larsen E. S. 186, 900
Lassaigne J. L. 816
Lassieur A. 146, 336
Lathe F. E. 408
Latshaw W. L. 862
Laubengayer A. W. 349
Laudecker M. 666
Lauer W. M. 814
Laug E. P. 130
Lauro M. F. 863
Lauterbach H. 449
Lawrenz M. 629
Law-Zecha A. B. N. 690
Lea M. C. 893
Leatherman M. 459
Le Chatelier H. 896
Leclerc A. 759
Leddicotte G. W. 685
Ledebur A. 161
Lee M. F. 583
Lemberg J. 1046, 1047
Lemkin W. 222, 225
Lenher V. 382, 386, 388, 389, 393. 394, 682, 756, 759, 830. 946, 1032
Leopold! G. 240, 301
Lepper W. 540, 541
Leslie R. T. 931
Lessing R. 642
Letonoff T. V. 265
Levaltieri H. 865
Levine H. 612
Levol A. 306
Levy L. H. 291
Lewitt A. E. 690
Liebhafsky H. A. 182, 686
Liggett L. M. 1014
Lighty P. E. 162
Linde-Gladding 747
Lindemann O. 981
Linder J. 857
Line W. R. 933
Lingane J. J. 165, 173, 216, 217, 249. 502, 710
Linstead R. P. 199
List F. 154
Littman Z. 392, 393, 394
Lizius J. L. 200
Locher E. 953
Lochte H. L. 854
Locke J. 917
Long J. H. 541
Loofbourow J. R. 177
Lorber L. 454
Lord 70
Lord R. C. 177
Lord S.S. 690
Loughrey J. H. 267
Lounamaa K. 543
Low A. H. 244, 265, 267, 285, 288, 293, 334, 337, 1012, 1013, 1014
Low W. H. 831, 835, 1032
Low W. W. 1032
Lowe C. S. 177, 579
Lowe J. 49
Lowenstein E. 896
Lubs H. A. 198, 201
Lucas С. C. 757
Lucas G. H. 864
Lucas J. 516, 629
Luck E. 721
Luckow C. 240, 264
Ludwig E. 756
Luedeking C. 1035
Luff G. 84
Luke C. L. 158, 168, 576, 805
Lukens H. S. 55, 613, 760
Lumsden J. 339, 340
Lundegardh H. G. 178
Lundell G. E. F. 27, 54, 66,
78, 83, 98, 100, 137, 138,
139, 143, 148, 165, 166,
232, 281, 337, 361, 368,
374, 377, 378, 379, 383,
437, 439, 445, 448, 455,
459, 464, 470, 480, 481,
486, 496, 497, 498, 499,
518, 527, 530, 532, 550,
553, 554, 561, 565, 567,
570, 571, 573, 576, 583,
591, 606, 637, 641, 643,
660, 670, 686, 718, 756,
777, 779, 781, 785, 786,
788, 789, 791, 812, 822,
826, 828, 834, 921, 936,
955, 983, 1005, 1019,
1024, 1062
Lunge G. 206, 932
Luther R. 198
Lutz O. 307, 747
Lyden R. 596
Lydersen D. 158
Lyle A. K. 840
Lyons R. E. 108, 384
Maassen G. 477
Mach F. 540, 541
Maclnnes D. A. 230, 231
MacMasters M. M. 452
Maczkowske E. E. 938
Mahr C. 254, 255, 261, 278, 279, 295, 301
Makepeace G. R. 576
Makinen E. 1007
Makowka O. 285
Malat M. 158, 611
Mallet J. W. 893, 905
Malmitadt H. 611
Malouf F. E. 378
Manasse O. 983
Manchot W. 84
Manly J. J. 36
Mannich C. 830, Ю32
Mar F. W. 701, 808, 970
Marbaker E. E. 768
Mardem J. W. 76
Maren T. H. 331
Maricovd D. 714, 799
Mariel C. S. 145
Marignac C. 683
Markley K. S. 863
Marsh K. 627
Marshall H. 542
Martin F. 144
Marvin G. G. 388, 392, 746, 920
Marzys A. E. O. 690, 691
Mascazzini A. 328
Mason G. W. 187.
Mason W. B. 147
Matejczyk J. F. 814
Mathers F. C. 546
Matyska В. B. 158
Mauzelius R. 893, 901, 986
Maxwell G. E. 757
Maxwell J. A. 166.
Maxwell L. C. 857
Maxymowicz W. 109, 128, 269, 386
May I. 547
May R. L. 151, 152, 575
May W. C. 265
Maynard J. L. 407
Mayr C. 474
McBain J. W. 440
McBride R. S. 445, 710, 979
McBryde W. A. E. 423
McCarthy C. N. 629
McCay L. W. 46, 89, 93, 99, 260, 284, 307, 310, 367, 518
McChesney E. W. 330
McClendon J. F. 809
McClure F. J. 826
McCormic D. R. 750
McCoy H. N. 627, 629, 630
McCroskey C. R. 392
McCrumb F. R. 199
McCulloch N. 818
McDonnell С. C. 228
McDuffie B. 167
McFarlange J. 857
McIntyre G. H. 935
McIntyre L. H. 57, 73, 76
McLendon V. 576
McLennan I. C. 150
McMeekin T. L. 863
McNabb W. M. 240, 443, 721, 788, 814
McNeill T. R. 465, 590
Meade R. K. 709
1082
Именной указатель
Mears В. 55, 863
Medway H. E. 240, 300
Meene G. H. P. v. d. 290
Meerburg P. A. 809
Meggers W. F. 177
Mehlig J. P. 293, 454, 468
Meimberg E. 684
Meineke C. 756, 781, 818
Melampy R. 864
Melaven A. D. 166
Meldrum W. B. 804, 815, 864
Mellon M. G. 174, 175, 188, 353, 453, 454, 468, 516, 757, 792, 834, 868
Mellor J. W. 683, 709, 1009
Meloche V. W. 139, 180, 374, 757
Mercer F. N. 402
Mercer R. A. 685
Mericanto B. 158
Merkel P. P. 860
Merrill H. B. 682, 683
Merritt C. 543
Merwe A. J. v. d. 596
Merwin H. E. 824, 896, 897, 898, 900, 1026, 1046
Metzger F. J. 499, 629
Metzl A. 85, 322, 326
Metzler D. E. 217
M eulen H. ter 869
Meyer J. 323, 516, 853
Meyer R. J. 598, 603, 614, 615, 632, 633, 672, 683
Meyers R. E. 586
Michov M. 266
Middelton E. B. 1010
Middleton A. R. 346, 578
Middleton G. 164
Miksch R. 394
Milbauer J. 293
Millberg С. M. 932
Miller A. L. 529, 560, 750
Miller С. C. 150
Miller E. H. 300
Miller M. 344
Miller W. A. 869
Millner T. 305
Milner O. 844
Milner О. I. 293
Milner W. 648
Milobedzki T. 206
Mingaye J. С. H. 880
Minor J. C. 1022
Minozzi A. 242
Misil A. 648
Mitchell A. M. 468
Mitchell R. L. 148, 179
Mittasch A. 106
Moeller T. 548, 578, 584, 608, 627, 630, 631
Moerk F. X. 198
Mogerman W. D. 286, 343, 577
Mohr C. F. 814
Moissan H. 841
Molooly W. F. 476
Moody S. E. 109
Moore F. L. 685
Moore G. E. 635, 685
Moore H. C. 865
Moorman A. 588
Moran R. F. 27, 142
Morden G. W. 539
Morey G. W. 203, 210
Morgan G. 347
Morgan G. T. 1016, 1061
Morley E. W. 72, 76, 912
Morozewicz J. 732
Morris V. N. 834
Morrison G. 360
Morse H. N. 190
Morth H. 542
Morton D. A. 804
Morton D. S. 349
Mosander C. G. 626
Moser L ,. 109, 128, 152,
153, 154, 269, 279, 304,
305, 392, 393, 394, 538,
540, 546, 547, 551, 584,
585, 642, 717, 730,
Moss J. A. 261
Moss M. L. 454
Mostovitz B. 107
Mostowitsch W. 761, 800
Motherwell H. A. B. 759
Moulton J. D. 162
Moureu C. 1035
Moyer H. V. 148, 479, 801, 813
Muehlberg W. F. 400, 919
Muir 279
Muller A. 629
Miiller E. 993
Miiller E. 423
Muller H. 265
Muller J. H. 99, 306, 348, 349, 675
Muller M. 389
Muller О. H. 167
Muller R. H. 174
Muller W. 804
Mullin H. R. 179
Munro L. A. 75
Munroe С. E. 127
Murmann E. 695
Murphy R. K. 662
Murray C. W. 267, 312
Murray W. M. 142, 164
Musser D. F. 563
Muthmann W. 392, 628
Myers R. J. 217
Myers W. D. 864
Mylius F. 162, 407
Nachtrieb N. H. 177
Naftel J. A. 844
Nakazono T. 141
Natelson S. 851
Nau A. 751
Neher F. 98
Neller J. R. 312
Nessler J. 869
Nette M. 128
Neubauer H. 127, 716, 719,
1013
Neumann B. 662
Neumann G. 541
Neu may er K. 154
Neustadt M. H. 492
Neustadtl L. 700
Newell I. L. 271
Newlin I. G. 804
Newton E. R. 810
Newton H. D. 443, 451
Newton R. C. 383
Newton R. F. 810
Nichols M. L. 153
Nichols M. S. 221
Nicholson D. G. 65
Nickoils L. C. 277
Nicolardot P. 971
Niessner M. 153, 584
Nieuwenburg C. J. van 918
Nilson L. F. 669
Noddack W. 372
Nolan E. J. 642
Noll F. 833
Norris J. F. 392, 393
North V. 471, 476, 774
Northrup M. A. 522, Norton J. T. 392 Norton R. H. 68, 70 532
Noyes A. A. 65, 85. 162,
539, 705, 823, 883, 930
Nuka P. 462
Nydegger O. 804
Oakdale U. O. 807
Ode W. H. 754
Odinot L. 84
Ogburn S. C. 411
Okac A. 171
Okell F. L. 339. 340
Oldfield J. H. 180
O’Leary W. J. 733, 740,
1007
Olsen A. L. 576
Olsen J. S. 488
Ordelt H. 280
Orlik W. 289
Osborn G. H. 588
Osborn R. A. 392
Oschwald F. 856
Oshry H. I. 935
Oswalt R. L. 453
Ovenston T. C. J. 453
Overholser L. G. 278, 418
Overton O. R. 749
Page R. W. 300
Pagel H. A. 622
Именной указатель
1083
Palilla F. С. 688
Palitzsch S. 201
Palkin S. 717, 738
Palmer H. E. 540
Palmer R. M. 312, 313
Pan Z. H. 486
Panajotow G. 84
Panichi U. 896
Panouse-Digeaud L. 261
Papa D. 807
Pape H. 641
Papish J. 345, 346, 552, 733, 740
Pappenhagen J. A. 204
Parish J. A. 552
Park B. 289, 498
Parker C, A. 453
Parker H. C. 231
Parker H. G. 798
Parker R. C. 165
Parle W. 276
Parr S. W. 285
Parrodi G. 328
Parsons C. L. 584
Parsons J. T. 865
Pass A. 297
Passamaneck E. 468
Passon M. 709
Pattison M. M. 279
Paulson R. A. 863
Pavelka F. 542
Pavlish A. E. 392
Pawletta A. 516
Pearce D, W. 632
Pearson E. A. 489, 490
Pearson R. W. 443
Pochard E. 767
Pekola J. S. 147
Peligot E. 526
Pelouze J. 267
Pemberton H. 781
Pena P. de la 540
Penfield S. L. 841, 908, 909, 1022, 1032
Pennock J. D. 804
Percival J. O. 435
Perillon F. 755, 767, 768
Perlich R. W. 1034
Perry M. 609
Peter O. 454
Peters C. A. 393, 452, 696, 713
Peters R, 439, 994
Peterson H. E. 532, 543
Petrey A. W. 828
Pettersson O. 392
Pfaff F. W. 1042
Pfeiffer H. 455
Pflaum D. J. 822
Pforten v. d. 769
Phelan J. J. 477
Phelps I. K. 862, 864, 866
Phelps M. A. 306
Phennah P. J. 648
Philips M. 592
Pied H. 147, 666, 671
Pierce A. W. 388
Pierre W. H. 199
Pierson G. G. 143
Pietruck 673
Pinagel A. 755, 767
Pine P. R. 55
Pinkus A. 144, 145, 146, 271 336
Pirsson L. V. 890, 1036
Pisani F. 666
Pisani M. F. 829
Pittman F. K. 645
Plank J. 634
Platow A. M. 221, 225
Platt R. 770
Plechner W. W. 657
Ploetz A. O. 142
Pollak I. 816
Pollard W. B. 408, 423
Pollitt A. A, 368
Pope F. J. 951
Popoff S. 222, 226
Porter L. E. 552, 553
Posniak E. 441, 897, 898, 994
Pouzergues J. 280
Powell A. R. 337, 360, 639, 666, 670, 674, 678, 681, 730, 768
Powell N. S. 1046
Praetorius P. 586
Prandtl W. 628
Pratt D. S. 869
Pratt J. H. 995, 999
Pray A. R. 750
Precht H. 67
Pregl F. 187, 859
Preller I. 695
Pribil R. 158, 159, 587, 597, 714, 769, 799
Price H. P. 751
Priess H. 830, 1032
Priess O. 586
Procke O. 729
Proctor K. L. 492, 844
Pugh W. 550
Pukall W. 1007
Pulman O. S. 370
Purushottam A. 647
Putman G. L. 747
Putscbe 476
Quill L. L. 645, 652, 922
Rabinowitz J. 173
Raghava Rao Bh. S. V. 647
Radley J. A. 176, 579
Raikow P. N. 504
Raj P. 654
Rajmann E. 636
Ramage H. 536, 544, 549, 731, 882
Rammelsberg C. 738, 781
Ramsey J. B. 552
Randall L. D. 138, 361
Randell M. 536
Rane M. B. 323
Ranedo J. 458
Rankama K. 679, 756
Ransome F. L. 507, 510, 981
Raschig F. 804
Rath G. von 94
Ratnam M. K. 323
Rawson S. G. 699
Reber L. A. 814
Redfield H. W. 869
Redmond J. C. 725
Reed S. A. 423
Reedy J. H. 65, 739
Rees A. 260
Reglade A. 971
Reif W. 294, 538
Reiff О. M. 63
Reinhardt C. 446
Reinhold J. G. 265
Reis M. A. 343
Reissans G. G. 490
Reith J. F. 255
Remington W. J. 148
Reynolds D. S. 720, 787, 819, 825, 826, 1019
Reynolds F. 369
Rice A. C. 606, 638
Rice F. O. 63, 222, 225
Ricci J. E. 392
Richards J. W. 1046
Richards T. W. 35, 50, 127, 205, 237, 250, 706, 798, 813
Richter F. P. 213
Rider T. H. 814
Ridsdale С. H. 958
Rieman W. R. 183, 351
Rienacker G. 450, 542, 543, 581
Ries H. 1042
Riggs R. B. 717
Rinn H. W. 183
Risdon E. J. 255
Rittenberg S. R. 289
Rivot L. E. 285
Roark R. C. 228
Roberts E. J. 626
Roberts H. S. 193, 230, 231, 448
Roberts L. D. 701
Robertson J. D. 1033
Robinson H. 240
Robinson K. L. 844
Robinson P. L. 681, 832
Robinson R. J. 747, 757
Robinson W. O. 385
Robitschek J. 148
Rodden C. J. 148, 315, 526, 527, 584, 602, 630
Roebling W. 537, 542
1084
Именной указатель
Rogers Н. W. 578
Rogers L. В. 543, 738, 748
Rogers R. E. 856
Rogers V. C. 794
Rogers W. B. 856
Rogers W. J. 408, 419
Rohs H. L. 825
Rolfe A. C. 576, 577
Rollet A. P. 468
Ronchese P. 652
Roode R. de 855
Rosanoff M. A. 813
Rose H. 279, 824, 1020,
1021, 1035 Roseman R. 443, 657 Rosenbaum С. K. 75 IQ
Rosenbladt T. 841 Rosenheim A. 532 Rosenquist I. T. 258, 260
Rosin J. 58
Ross H. L. 935
Ross J. F. 734, 735, 736
Ross M. 291, 547
Rossini F. D. 853
Roth H. 187, 859
Roth W. A. 230
Rothe J. W. 161
Rowland W. J. 857
Rowledge H. P. 1003
Rowley R. J. 825
Royer G. L. 547
Rudolph G. A. 844
Rudy R. B. 46, 805, 1007
Rue S. O. 227
Ruegenberg M. J. 358
Ruff O. 684, 823
Rupp E. 244, 248
Rusgel D. S. 408, 419
Russel J. J. 407, 408, 414
Russel R. G. 632
Russell A. S. 450, 518, 659
Russell F. R. 576, 577
Russell W. W. 127
Ryan D. E. 423
Ryba O. 611
Sadasivan V. 863
Sadler B. 313
Sadusk J. F. 450
Sager E. E. 575
Sale P. D. 45
Sanchis J. M. 828
Sand H. J. S. 165, 167, 168
Sandberger F. 881, 1033
Sandell E. B. 148, 175, 347, 353, 354, 419, 502, 516, 550, 557, 565, 581, 596, 775, 986, 1034
Sandilands J. 244
Sanger C. R. 312, 327
Sargent G. W. 835, 1032
Sarver L. A. 447, 519, 621, 998
Satterlee H. S. 303
Sawyer R. A. 178
Saylor Ch. P. 853
Saywell L. G. 454
Sazerac R. 280
Scales F. M. 863
Schaak M. F. 835
Schaefer C. 897
Schaeffer O. A. 757
Schafer 392, 673 Schaffgotsch 1020 Scheiner K. 628 Schenk M. 658
Scherrer J. A. 49, 78, 96, 286, 318, 337, 445, 553, 577, 710, 795, 979
Scherrer K. 868
Schiller E. 684, 823
Schindler C. 267
Schirm E. 562
Schirmeister H. 118
Schlecht W. G. 402, 887, 931
Schleicher A. 56, 164, 168
Schloesser W. 190
Schlosing J. J. 865
Schneeberger A. 84
Schneider P, 158
Schneider W. 354
Schneidewind R. 799
Schober W. B. 846
Schoder W. P. 265, 267, 288, 293, 334, 337
Schoellenberger С. J. 447, 448, 519, 850, 998, 1002
Schoeller W. R. 151, 152, 276, 342, 359, 360, 389,
458, 525, 619, 622, 639,
640, 654, 666, 670, 674,
675, 677, 678, 680, 681,
730, 766, 768
Schoep A. 359
Scholes S. R. 1007
Schoonover I. C. 58, 751
Schreiber N. E. 253, 254
Schrenk H. H. 935
Schrenk W. T. 264, 392, 393, 394, 754
Schricker J. A. 315
Schubert L. 184
Schubert M. 897
Schuhmann S. 255
Schulek F. 328
Schumb W. C. 388, 392, 642, 645, 692, 770, 920
Schutt K. 717, 730
Schwartz M. C. 757
Schwarzenbach G. 157
Schwenk E. 807
Scott M. 402, 408
Scott R. O. 148, 179
Scribner B. F. 177, 179
Scribner L. R. 629
Seaborg G. T. 635
Sears G. W. 652, 669, 922
Seath J. 390, 407, 408, 412
Sebba F. 550
Sebelien J. 206
Sedivec V. 158, 159, 255, 293
Sedlar V. 158
Seemann F, 1020, 1022, 1023
Sehl F. W. 935
Sekerka J. 159
Selch E. 820, 939
Selvig W. A. 1016, 1060
Sevag M. G. 854
Seyewetz A. 866
Sharwood W. J. 177
Shaw J. A. 210
Shea F. 860, 861
Shead A. C. 696, 732, 1011
Sheftel M. S. 588
Sheld О. M. 862
Sheldon J. L. 439
Shempf J. M. 153
Shepherd M. 184, 255, 858
Shepherd M. J. 454
Sherrington L. G. 578
Shertz F. M. 860
Shimose M. 389
Shinkai S. 759
Short H. G. 576
Short M. N. 186
Shreve R. N. 148
Sickman D. V. 751
Siegmann F. 546, 547
Sieling D. H. 454
Sieminski M. A. 770 .
Sievers I. J. 575
Sill C. W. 532, 543
Silver S. H. 289
Silverthorn R. W. 392
Silvestri O. 1036
Simon V. 159
Simpson С. T. 156
Simpson S. G. 645, 770
Singer J. 152, 154, 585
Sinka A. 740
Skelly J. F. 547
Skinner W. W. 738
Skolnik H. 443
Skowronski S. 285
Skrabal A. 700
Skraup Z. H. 148
Smith D. P. 388
Smith E. F. 102, 109, 160, 240, 285, 300, 358, 512, 672
Smith F. 468
Smith F. W. 591
Smith G. B. L. 386
Smith G. F. 64, 72, 73, 76, 78, 142, 151, 152, 165, 205, 211, 213, 219, 220, 328, 450, 451, 575, 590, 591, 732, 734, 735, 736, 745, 1010, 1011
Именной указатель
1085
Smith G. М. 155, 376
Smith G. S. 776
Smith H. V. 828
Smith J. D. M. 132
Smith J. G. 127
Smith J. L. 159, 160, 161, 636, 666, 667, 751, 922, 955, 1005
Smith M. M. 644
Smith О. M. 828
Smith T. 0. 605
Smith W. H. 575, 853
Smither F. W. 39
Smoot A. M. 362, 795, 797
Snell С. T. 175
Snell F. D. 175
Snelling W. 0. 127
Snider H. J. 1006
Soderbaum H. G. 285
Soderman M. A. 701
Someya Kin’ichi 141, 527, 529, 629
Sommer F. 450, 770
Sommer H. H. 295
Sommer L. 171
Sonnenschein F. L. 781
Sorensen S. P. L. 206
Soroos H. 76
Sosman R. B. 440
Soth G. C. 392
Sottery С. T. 576
Soule B. A. 208, 458, 463, 1001, 1002
Spacu G. 295
Spear E. B. 162, 491, 539, 883
Spears H. D. 863
Speller F. N. 161, 162
Spencer M. G. 503
Sperry E. S. 841
Speter M. 603
Spielhaczek H. 754
Spies J. R. 861
Spiher A. T. 289
Spitzer F. 491
Spitzer L. 809
Spoor H. J. 757
Sprengel H. 869
Springemann W. 200
Springer V. F. 139
Spuhr W. P. 365
Sreenivasan A. 863
Staehler A. 127
Staib K. 484
Stanek V. 293
Stanford 318
Stansby M. E. 828
Starck G. 826, 1021, 1024
Starks D. A. 738
Starkweather H. W. 76
Stas J. S. 237
State H. M. 447
Steele G. J. 477
Steiger G. 824, 911, 971, 1025
Steinbeck Dr. 285
Steinkonig L. A. 1028
Steinkuhler W. 359
Steinwehr H. 240
Stelling E. 336
Stenger V. 107, 563
Stephenson G. E. 832
Sterba J. 358
Sterling C. 365
Sterner J. 178
Stetser J. B. 68, 70
Stevens R. E. 185, 443, 584, 742, 743, 1007
Stevenson R. 440
Steyermark A. 859
Stich C. 542
Stillman T. B. 270
Stivenson P. C. 685, 686
Stock A. 254, 562, 568
Stock C. R. 176
Stoddart C. W. 1030
Stokes H. N. 221, 453, 990, 991, 997
Stolba F. 727
Stolberg C. 695
Stone G. C. 270
Stone H. W. 135, 450, 451, 686
Stone K. G. 219
Stoppel A. F. 367
Stout L. E. 513
Strafford N. 158, 344
Strain H. H. 184
Strebinger R. 816
Strecker W. 540, 835, 839
Streit G. 973
Strock L. W. 55,382,613, 729
Stross W. 588
Struthers J. D. 260
Stuart A. 187
Stubblefield F. M. 865,
1010
Stumpf L. F. 340
Stiitzel L. 628
Sue P. 468
Suk V. 611
Suleimann A. 756, 759
Sullivan E. C. 484, 843
Sullivan J. D. 186
Sullivan V. R. 219
Sunde ,C. J. 814
Sutton F. 491
Svestka L. 159
Swanger W. H. 285, 290
Swank H. W. 454, 757
Swartz С. E. 368
Sweet T. 157
Sweetser P. 158, 648
Swift E. H. 162, 164, 217, 479, 550, 551, 553
Swinne R. 307
Szebelledy Z. 519
Таске I. 372
Taebel W. A. 632
Talbot H. P. 821
Talviti N. A. 935
Tammann G. 896
Tang N. K. 562
Tanzler K. 423
Taylor G. B. 1016, 1060
Taylor H. J. 828
Taylor J. K. 165, 172
Taylor W. C. 484, 843
Tschakirian A. 351
Tchijewski P. 832
Teitelbaum M. 280
Telep G. 688
Thamer B. J. 648
Thanheiser G. 688, 691
Theis M. 648
Theobald L. S. 456
Theobald L. T. 1034
Thiel A. 200, 454, 547
Thiers R. 408
Thomas A. 579
Thomas H. 857
Thomas W. 862
Thomason P. 609
Thompson J. J. 251, 506,
807
Thompson R.!T. 835
Thompson S. O. 408
Thompson T. G. 828
Thorin E. 1021
Thornton C. D. W. 255
Thornton W. M. 127, 643
Thornton W. M. Jr. 443,
449, 450, 606, 657, 662
Thrum W. E. 579
Thugutt S. J. 896
Teitelbaum M. 280
Tiemann F. 865
Tietze W. 289
Tikhomiroff 685
Tischbierek H. 853
Tischkow P. 504
Titus A. C. 212, 488, 816
Todoroff T. 168
Tokos J. V. 278
Tomicek F. 377
Tomicek O. 244, 377
Tomula E. S. 476
Tongeren W. van 334, 339
Tooley F. V. 139
Topf G. 225
Torrance S. 168
Tougarinoff B. 334
Townsend F. E. 297
Traub K. 654
Trautmann W. 957
Travers 940
Travers A. 921
Travers M. 637, 765
Travers M. W. 1035
Treadwell F. P. 1001, 1022
1086
Именной указатель
Treadwell W. D. 84, 135, 673, 829
Tribalat S. 375
Trommsdorff H. 869
Trostel L. J. 934
Truog E. 443, 504, 756, 759, 844, 946
Tschawdarov D. 591
Tschawdarowa N. 591
Tschermak G. 896
Tschernichow J. A. 320, 533
Tschischewski N. 833
Tschugaeff L. A. 457
Tucker F. H. 510, 783
Tufts L. E. 807
Turk E. E. de 865
Turner W. A. 1030
Turner W. E. S. 832
Ubaldini I, 423
Uhl A. 231
Ullgren C. 167
Uorlichek J. 158
Urbain G. 616, 632
Urk H. W. van 200
Uslar H. von 240
Uspenskaya T. A. 320
Uzel R. 729, 769
Valla R. K. 696
Van Bemmelen J. M. 896
Van Blarcom H. S. 809
Van Brunt C. 606, 755, 767, 768
Vance J. E. 288, 289
Van der Meulen P. A. 231
Van Dik J. C. 84
Van Klooster H. S. 475
Van Liempt J. A. M. 834
Van Name R. G. 285
Van Rysselberge P. J. 747
Vanselow A. P. 869
Van Straten F. W. 36
Vasa к V. 158, 159, 255, 293
Veitch F. P. 820, 939
Verneuil A. 602
Vernon W. H. J. 38
Veryzl E. J. A. H. 818
Vienneau E, M. 658
Vieweg K. 148, 717, 730
Vinal G. W. 263
Vincent H. B. 178
Visser J. 642
Vogt J. H. L. 990
Voigt A. 376, 648
Volhard J. 109, 160, 238, 249, 503. 961
Vorhes F. A. 267
Vortmann G. 85, 236, 322, 326, 530
Vostrebal J. 358
Voter R. C. 423
Wada I. 141, 162, 539,548, 553
Waddell J. 265
Waegner A. 285, 629
Wagman D. D. 853
Wagner E. C. 863
Wagner P. 865
Wagoner L. 1033
Wakefield H. F. 394
Waldbauer L. 475
Waldbott S. 832
Walden G. H. 139, 218, 371
Walker N. H. 809
Walker P. H. 39
Walt C. F. J. van der 590,
596
Waltenberg R. G. 45
Walters H. E. 501, 592
Walton J. H. 75, 76
Wandenbulcke F. 757
Wanjukow W. 761
Ward A. M. 297
Ward N. M. 475
Ware G. M. 482, 484
Warren H. N. 334
Warren R. D. 76
Warth H. 908
Washbrook С. C. 647
Washburn E. W. 193, 222, 224 228
Washington H. S. 345, 580, 729, 806, 819, 876, 877, 881, 887, 888, 890, 976
Wassjuchnow A. 615
Waterhouse E. F. 276, 619, 622, 674
Waters С. E. 44, 803
Watts С. E. 860, 861
Way A. F. 439, 585
Weaver E. R. 209, 631
Webb H. W. 525, 639, 674
Weber H. 796, 802, 921
Weber H. С. P. 67
Webster P. A. 732, 840
Wedding H. 781
Wegscheider R. 285
Weibke F. 374
Weiler G. 818
Weinberg S. 492, 844
Weinig A. J. 265, 267, 288, 293, 334, 337
Weinstein B. 35
Weiser H. B. 263, 300, 367
Weiss L. 666
Weissler A. 177, 579, 658
Welcher F. J. 143
Weller A. 328
Wells E. E. 491
Wells H. L. 36, 291
Wells J. S. C. 830, 1032
Wells P. V. 50, 175
Wells R. A. 685, 692
Wells R. C. 50, 237, 388, 471, 476, 526, 666, 742, 743, 745, 813, 823, 965
Wenger P. 586
Wenzke H. H. 822
Wernimont G. 27, 231
Werther G. 541
Wessels J. E. 1007
West P. W. 278
Westfall В. B. 459, 467
Weyl 846
Wheaton F. 56
Wheeler H. A. 1035
Wherry E. T. 830, 843, 896, 900, 1032
Whitaker G. C. 513
Whitby G. S. 237, 240, 815
White С. E. 176, 177, 492, 532, 579, 588
White M. G. 378
Whitehead T. H. 282
Whitman J. L. 222, 226
Wiberley S. E. 578
Wicher D. 290
Wichers E. 39, 58, 108, 167, 285, 402, 408, 424, 926, 931
Wichmann H. J. 267, 492, 825, 828, 829, 1028
Wilcox C. S. 142
Wilcox L. V. 747, 829, 835, 843
Wilcoxon F. 231
Wilde H. D., Jr. 854
Wiley J. A. 231, 515
Wiley R. C. 267, 301, 694, 709
Wilfarth H. 859
Wilhelm H. A. 179
Wilke — Dorfurt E. 953, 1008, 1018
Wilkie J. M 267
Wilkinson N T. 576, 577
Willard H. H. 64, 73, 155,
218, 219, 230, 231, 251,
261, 263, 360, 371, 376,
383, 392, 439, 454, 471,
505, 506, 509, 515, 519,
521, 542, 549, 556, 562,
629, 640, 699, 707, 734,
746, 792, 799, 818, 823,
828, 863, 942, 1014
Williams A. F. 685
Williams F. H. 503
Williams J. B. 261
Williams К. T. 382, 385
Williams W. E. 165
Williamson E. D. 1046
Wilson F. B. 821
Winchester R. 491
Wing H. J. 853
Winkler L. W. 267, 863
Winsor H. W. 453
Winter H. 615
Winter K. 154
Winter О. B. 823, 828
Winter P. K. 807
Winterstein Z. 660
Именной указатель
1087
Winzer Р. 684
Wirth F. 604
Withrow J. R. 76
Wood A. A. R. 175
Woodman A. G. 821
Woods J. T. 453, 454, 792
Woodson T. T. 255
Woolaver L. B. 746
Wooten L. A. 168, 260
Woy R. 781
Wright A. H. 231
Wright E. R. 516
Wright F. E. 900
Wright T. A. 77
Wright W. C. 515
Wught M. L. 776
Wulfing E. A. 717
Wunder M. 586, 756, 759
Wurm O. 280, 297
Wylie A. W. 630
Wyncoop G. 562
Wynne D. J. 934
Wyrouboff G. 602
Yagoda H. 185
Yedinak A. 709
Yntema L. F. 491, 628
Yochida I. 225
Yoe J. H. 278, 283, 418,
454, 576, 578, 774
Yost D. M. 592
Youden^W. J. 27
Young I. G. 173
Young P. 218, 509, 515, 519, 521, 542, 629
Young R. S. 468, 476, 927
Yutzy H. 811
Zachariason R. H. 180
Zambonini F. 896
Zaussinger E. 420, 431
Zawadski J. 84
Zies E. G. 259, 303, 831, 832, 839, 1033
Zimmermann C. 446
Zintl E. 450, 542
Zinzadze Ch. 315
Zwik K. G. 253, 254
предметный указатель
Абсорбционный рентгеновский анализ 181, 182
Адсорбционные индикаторы 202
Азорезорцинсульфаниловой кислоты натриевая соль (Тропеолин 0) 196, 202
Азот 859 сл.
определение 859 сл.
в виде аммиака 869
— — нитратов 865, 866, 869
весовое, нитроном 867
в присутствии азосоединений 866
— — нитратов 865
— — соединений гидразина 866
в урановых минералах 523
в цианамиде кальция 868 колориметрическое 869 сл. методом Деварда 865 — Дюма 859
— Кьельдаля 859, 862
осаждением в виде азотнокислого нитрона 867
Азотная кислота 60
установка титра 202
Акридинсульфат 492
Ализарин-бордо см. Хинализарин
Ализариновый красный S 646, 648, 844
Алкалиметрия 193
Аллилтиомочевина 423
Алунд 128
Алюминиевые сплавы, анализ 718
Алюминий 559 сл.
металлический, анализ 718, 751
— растворение 561
окись
адсорбент 71, 74, 76
разложение 561
определение 565 сл.
алюминоном 577
арсеназо 579
в бронзах 579
в виде окисла 565 сл.
— — фосфата 569
весовыми методами 565 сл.
в карбонатных породах 1052, 1066
в минералах 579
Алюминий
определение
в осадке от аммиака 121, 952
в силикатных породах 952, 953
в сплавах 576
в сталях 579
в стекле 563
колориметрическое 577, 579
малых количеств 575
морином 177, 579
о-оксихинолином, весовое 575
— колориметрическое 578
— объемное 575
стильбазо 579
флуоресцентное 176, 579
фотометрическое 577
эриохромцианином R 579
осаждение
аммиаком 561, 565 сл.
бензоатом аммония 107, 563
в виде хлорида 117, 552, 584
иодид-иодатной смесью 562
карбонатом бария 563
купфероном 147, 564
нитритом 562
о-оксихинолином 148, 149, 571 сл., 583
слабыми органическими основаниями 111
таннином 152, 153, 584
тиосульфатом натрия 562
фенилгидразином 155, 561
фторидами 586
янтарной кислотой 562
отделение 154, 458, 496, 517, 524, 538,
552, 561 сл., 583 сл., 952, 977, 1053
поведение в ходе анализа 559
разложение Al-содержащих минералов 559 сл.
экстрагирование оксихинолята 584
Алюминон (Ауринтрикарбоновая кислота) 575 сл.
Амальгама(ы)
висмутовая 141
жидкие 141
Предметный указатель
1089
Амальгама (ы)
свинцовая 141, 769
цинковая 135, 136, 141, 442, 529, 686 Р-Аминонафталид тиогликолевой кислоты (Тионалид) 419, 537, 542
Аммиак концентрация продажного 59 определение реактивом Несслера 869 осаждение гидроокисей 102 титрование растворов 197 хранение растворов 59
Аммоний
бензоат 107, 563 гексанитроцерат (IV) 219 метаванадат 449 молибдат 66, 267 персульфат 61, 951 полисульфид 93
роданид, приготовление раствора 453 салицилат 118
сульфид как осадитель 87, 90 сл., 115, 249
сульфит, приготовление 90 тетрародандиаминхромиат (соль Рей-неке) 254, 285, 294, 541
Аммонийные соли, удаление 161 Анализ весовой 25 воспроизводимость результатов 27, 885
выбор метода 80 капельный 184 карбонатных пород 1041 сл. качественный 25 количественный 25 микрохимический 185 объемный 25, 188 сл.
отбор проб 77 полярографический 177 сл.
приготовление проб 79 сл., 82, 820 сл. рентгеновский 181 сл.
силикатных пород 875 сл. смесей 26
спектрохимический 177 сл. точность результатов 27 сл., 883 сл. флуоресцентный 176 фотометрическими методами 173 сл. хроматографический 183 чистых веществ 26
электролитическими методами 164 сл.
Аити-1,5-ди-(га-метоксифенил)-1-гидр-окси лам ино-3-окс им инопен тен-4 774
Антипирин 766
Аппараты для перегонки воды 53
Арагонит, идентификация 1046
Арсеназо 579
Арсениды, разложение 303, 927
Арсенопирит, растворение 303 га-Арсоновая кислота (га-Диметиламино-азофениларсиновая кислота) 611, 636
Асбест 125
натронный 69, 848
69 Заказ 522.
Аскарит 70, 848
Z-Аскорбиновая кислота 452, 534
Ауринтрикарбоновая кислота (Алюминон) 575 сл.
Ацидиметрия 193
Бакелитовые изделия 56
Баллоны газовые 68
Бани
воздушные (радиаторы) 47
паровые 52
Барий 693 сл., 713, 968, 1057 карбонат как осадитель 108, 545, 552, 563
нахождение в горных породах 878 окись, адсорбент 71, 73, 76
определение
в виде сульфата 713, 797—805, 969, 1031, 1062
весовое 713
в карбонатных породах 1057 в силикатных породах 701, 968, 969 осаждение
фосфата 723
хромата 700, 714
отделение
от кальция и магния 701
от кальция и стронция 791
от стронция 700
перхлорат, адсорбент 72, 76
поведение в обычном ходе анализа 693
Бензидин солянокислый 157, 517, 804 а-Бензилдиоксим (Дифенилглиоксим) 458, 463
Бензоин 177, 492
Бензойная кислота, для установки титра оснований 196, 209
а-Бензоиноксим 157, 285, 294, 361, 364, 767
Бензол-2-арсоновая кислота-(1-азо-1)-2-оксинафталин-3,6-д исульфокис-лота (Торон) 609, 649
Берилл, разложение 582
Бериллиевые минералы, разложение 580, 582
Бериллий 580 сл.
возгонка формиата 586 обнаружение
в силикатных породах 581
морином 581
окись, разложение 587 определение 586 сл.
в виде фосфата 587 весовыми методами 586 в осадке от аммиака 121 га-нитробензоларсином 588 объемное 588 флуоресцентное 581, 588 хинализарином 581 хинизарином 588
осаждение
аммиаком 582, 584, 586 гидролитическим методом 415 карбонатом аммония 525
1090
Предметный указатель
Бериллий
осаждение
карбонатом бария 108
а-пиколином 583
таннином 152, 153, 584, 585 отделение 525, 582 сл.
поведение в общем ходе анализа 580 разложение бериллиевых минералов 580, 582
Бокситы, разложение 559, 560, 566 Бор 830 сл., 1031 сл.
обнаружение
в минералах 830, 1031
в пищевых продуктах 831
куркумой 831, 845, 1032
пробой на пламя 830, 1032 определение 834 сл.
в горных породах 1031 сл.
весовое 841
взвешиванием в виде бората кальция 841
в растениях 844
в стекле 839, 843
колориметрическое 843
методом Уэрри 843
объемными методами 834, 843
титрованием до определенного pH 843
— едким натром 834, 840
флуоресцентное 177
хинализарином 844
отгонка борноэтилового эфира 833, 1032
отделение 833
поведение в обычном ходе анализа 830 разложение борсодержащих минералов 832, 1031
Борная кислота, титрование 196, 843 Борный ангидрид 75, 920
Бром 807 сл.
определение 811 сл.
весовое 811
в присутствии иода 809
— — хлора 809, 810
в хлориде натрия 810
объемными методами 809, 810, 816, 817
потенциометрическим титрованием 809
титрованием роданидом 816 осаждение в виде бромида серебра 808, 815
отделение
от иода 809
от хлора 809, 810
разложение Вг-содержащих минералов 807
Броматы как осадители 109 Бромид-броматная смесь 109, 151 Бромистоводородная кислота 60 Бромкрезоловый пурпурный (Дибром-о-крезолсульфофталеин) 195, 197, 199, 200
Бромтимоловый синий 200
Бромфеноловый синий (Тетрабромфенол-сульфофталеин) 195, 196, 197, 199, 200
Бруцин 301, 633
Бумага
ацетатносвинцовая 315 куркумовая 831, 972, 1032 пропитанная хлоридом ртути, приготовление 313
фильтровальная 123
Бумажная масса 124
Бунзена горелка 133
Бура
для установки титра кислот 207 плавень 920, 923
«-Бутиловый спирт, обезвоживание 734
Буферные растворы 201
Ванадиевые минералы, разложение 508
Ванадий 507 сл.
восстановление пятивалентного 508, 513, 518
нахождение в изверженных породах 879
обнаружение 984
окисление до пятивалентного 515 определение 513 сл.
весовое 513
в карбонатных породах 1056
в вольфрамовой кислоте 509
в осадке от аммиака 121, 509
восстановлением сернистым ангидридом и титрованием перманганатом 513, 518, 983
в присутствии хрома 984
в силикатных породах 980
в сталях и чугунах 520
в шлаках 520
колориметрическое 509, 516
микроколориметрическое с бензидином 517
объемное 513 сл., 982
окислением перекисью водорода 516 иерсульфатным методом 514 по Гиллебранду 982
по Фурману 519
потенциометрическим титрованием 515
фосфоровольфраматным методом 516 осаждение'
аммиаком 509
ацетатом свинца 509
купфероном 115, 144, 511, 515
нитратом ртути (I) 510, 511
сульфидом аммония 90
отгонка в токе НС1 509, 512
отделение 509 сл.
из горных пород 510
сплавлением с Na2O2 или Na2COs и NaNO3 509, 510, 511 электролизом на ртутном катоде 509, 511
экстрагированием эфиром 509, 512 поведение в общем ходе анализа 505
Предметный указатель
1091
Ванадий разложение ванадиевых минералов 508 сл.
Вата из кремнекислоты 127
Версен 157
Версенат см. Комплексон III
Весовой анализ 25
Весы аналитические 31 сл.
Взвешивание 36 сл., 79
Викор 41, 42
Висмут 268 сл.
выделение внутренним электролизом 273
определение 274 сл.
в виде окисла 274, 275
— — сульфида 279
— — фосфата 276
— — хлорокиси 274
весовыми методами 274 сл., 280
в меди 277
в свинце 278
дитизоном 280
иодидом калия 277 йодометрическое 280 колориметрическое 273, 277, 278 о-оксихинолином и иодидом калия 280
пирогаллолом 280 роданохромиатом калия 279 тиомочевиной 273, 278 фосфорномолибденовольфрамовой кислотой 280
осаждение
в виде хлорокиси (или бромокиси) 109, 269
галловой кислотой 272 купфероном 271, 280 о-оксихинолином и иодидом калия 280
сероводородом 84, 88, 271
хинальдином 280
отделение 94, 258, 269 сл., 282, 538
от кадмия 271, 272
от меди 269, 270
от ртути 269, 271
от свинца 269, 271—273 поведение в общем ходе анализа 268 разложение Bi-содержащих минералов 268
Внутренний электролиз 165, 167 сл. без диафрагмы 170, 273, 285, 299 с применением защитной пленки на аноде 171, 343
Вода
дистиллированная 53, 58 нахождение в минералах 896 масса различных объемов 189 определение 903 сл.
в карбонатах 1061
в минералах 903 сл.
водоотнимающими веществами 904 косвенными методами 904 сл.
по Гучу 911 сл.
по Диттриху — Эйтелю 911, 914
69*
Вода
определение
по Пенфильду 908, 915
по потере при прокаливании 907 прямыми методами 908 сл.
фракционное 905
удаление
из минералов 898 сл.
при измельчении минералов 900 сл.
при сушке минералов 898 сл.
Водород 846 сл.
определение
в горных породах 847, 858, 896 сл.
в неорганических соединениях 847, 896 сл.
в органических соединениях 848, 851
мпкро- и полумикрометодами 851 общего содержания прямым сжиганием 850
удаление
при измельчении минералов 898
при сушке минералов 898
форма нахождения в минералах 896 сл.
Водородный электрод 230
Вольфрам 764 сл.
восстановление 769, 776
определение 769 сл.
алкалиметрическим титрованием 769
весовое 766, 770 сл.
гидрохиноном 774
в виде вольфрамата ртути 769 колориметрическое 774 объемными методами 770 роданидным методом 774, 776 таннином и цинхонином 678, 766, 768
титрованием перманганатом 769
— солью хрома (II) 770
цинхонином 765, 769, 770
осаждение
аммиаком 769
анти-1,5-ди-(га-метоксифенил)-1-гидр-оксиламино-3-оксиминопен-теном-4 774
антипирином 766 а-бензоиноксимом 767 кислотами 765
цинхонином 678, 765, 766, 769
отгонка 766
отделение 336, 358, 765 сл.
от кремнекислоты 766
от молибдена 767
от ниобия и тантала 677, 768
от фосфора и мышьяка 768
поведение в общем ходе анализа 764
разложение вольфрамовых минералов 764, 770
Вольфрамат, разложение 614, 771
Вольфрамовые^ минералы, разложение
1092
Предметный указатель
Вольфрамовый электрод 230
Воронки 123
Восстановление 96, 101, 133 сл., 139 сл., амальгамами 135 сл.
Высушивающие вещества 57, 71 сл., 904 поглотительная способность 76
Газовое оборудование 52, 133
Галлий 549 сл.
извлечение из германита 550 концентрирование 549, 550 металлический, растворение 550 обнаружение
гексацианоферратом (II) калия 548, 552
о-оксихинолином 550, 557 окись как коллектор урана 179 определение 554 сл.
весовыми методами 555
колориметрическим методом 549, 556
о-окспхинолином 557 флуоресцентное 550, 557 хинализарином 549, 556 осаждение
аммиаком 551, 554
бисульфитом аммония 556
гексацианоферратом (II) калия 552 гидролитическим методом 415 гидроокисью меди 552 едким натром 551 карбонатом бария 551 купфероном 146, 553, 555 а-нитрозо-р-нафтол ом 552 пиридином 111, 551 сероводородом 551 таннином 153, 154, 551 фениларсоновой кислотой 553 отгонка хлорида 553 отделение 247, 539, 550 поведение в обычном ходе анализа 549 разложение Ga-содержащих материалов 550
экстракция хлорида эфиром 553
Галловая кислота 272, 634
Галогены анализ смеси 817 определение в органических соединениях 807, 822
поведение в обычном ходе анализа 806
Гафний 635 сл.
определение колориметрическое 635 осаждение
в виде арсената 642
— — фосфата 638, 640
тетраэтилфосфатом 642
отделение от циркония 635
Гексаметилентетрамин (Уротропин) 88, 524, 601, 602, 654
Германиевые минералы, разложение 346 сл.
Германий 345 сл.
извлечение хлорида эфиром 348 определение 348 сл.
Германий
определение
в виде окиси 348
— — ортогерманата магния 349 весовыми методами 348—350 в окиси цинка 346 йодометрическое 351 колориметрическое 352 молибдатом аммония 352 объемными методами 351 о-оксихинолином 350
титрованием маннитогерманиевой кислоты 351
фенилфлуороном 352, 354, 355
электролитическим восстановлением до GeH4 345
осаждение
сероводородом 282, 346, 347, 348 таннином 348
отгонка хлорида 95, 346, 347 отделение 95, 346, 347 сл.
поведение в общем ходе анализа 345 разложение германиевых минералов 346 сл.
спектрографическое открытие 345
Гидразинсульфат 96, 293
Гидролитическое осаждение 100, 117
бериллия 415
железа (III) 439
иридия 406, 433
платиновых металлов 406, 408, 410, 414 сл., 433
титана 653, 661
Гидроокиси, pH осаждения 101
Гидрохинон 407, 412, 774
«Гиперол» (твердая перекись водорода) 63
Гольмий, спектрофотометрическое определение 630
Горелки
Бунзена 133
газовые 52, 132, 133
Мекера 52, 56, 133
паяльные 133
Тирриля 133, 708
Гуча тигли 127, 128, 131, 132, 911
Двуокись углерода
определение
в карбонатах 848, 857, 1060
в силикатных породах 1014 газометрическое 857
поглощение 64—71, 848, 857, 1015, 1060
Деварда сплав 866
Дистиллированная вода 53, 58
Дибром-о-крезолсульфофталеин (Бромкрезоловый пурпурный) 195, 197, 199
Диметиламиноазобензол (Метиловый желтый) 195, 196, 197, 201
Диметил аминоазобензол-о-карбоновая кислота (Метиловый красный) 195, 196, 199, 201
П редметный указатель
1093
Д имети л аминоазоб ензопсу л ьфон ат на-
трия (Метиловый оранжевый) 195, 196, 197, 199, 201
п- Диметил аминоазофениларсиновая кислота (n-Арсоновая кислота) 611, 636
Диметилглиоксим 68, 410, 430, 457, 459, 467, 468
Диметилдиаминотолуолфеназин (Нейтральный красный) 195, 196, 202 1-Диметил-3,4-димеркаптобензол (Дитиол) 344
Ди-Р-нафтилтиокарбазон 255, 301
1,4-Диоксиантрахинон (Хинализарин) 588
2,2'-Дипиридил 454
Дипиридил-сульфат железа (II) 213
Диспрозий, спектрофотометрическое определение 630
2,4-Дисульфобензаурин-3,3'-дикарбоно-вая кислота 635
Дитизон (Дифенилтиокарбазон) 155, 158, 240, 255, 267, 280, 301, 492
Дитиол (1 -Метил-3,4-димеркаптобензол) 344
S-Ди-о-толилтиомочевина 423
Дифениламин 200
Дифениламиносульфоновая кислота (Ва-или Na-соль) 213,
Дпфенилбензидин 213
Дпфенилбепзидинсульфоновая кислота (Na-соль) 213
Дифенилглиоксим (а-Бензил диоксим)
458, 463
Дифенилкарбазпд 255, 596
Дифенилкарбазон 255
Дпфенплтиокарбазон (Дитизон) 155, 158, 240, 255, 267, 280, 301, 492
Диффракционный рентгеновский порошковый анализ 181, 182
5,7-Дихлор-8-оксихинолин 630
Р,Р'-Дихлорэтпловый эфир 164
Диэтилдитиокарбамат натрия 159, 293, 375, 534
Доломит, анализ 1046
Дюробакс, состав 43
Европий
обнаружение 627
объемное определение 629
Железо 434 сл., 957 сл., 987 сл., 1004 см. также Железо двухвалентное, Железо трехвалентное
отделение 258, 437 сл.,458, 496 524, 585
поведение в обычном ходе анализа 434 разложение Fe-содержащих минералов 435
Железо двухвалентное 987 сл., 1058 определение 440 сл.
влияние ванадия 996, 997
— сульфидов 996
— углистых веществ 997
Железо двухвалентное определение
в запаянной трубке 987, 989
в карбонатных породах 1058
в силикатных породах 949, 986
2,2'-дипиридилом 454
объемными методами 440 сл., 957, 992
по Барнеби 994, 1000
по Куку 987, 992, 993, 994, 999
по Митчерлиху 989, 991
по Пратту 994, 995, 997
по Соулу 1001, 1002
по Тредвеллу 994, 1001
по Шелленбергу 1002
потенциометрическим титрованием 442, 448, 455, 1002
сульфосалициловой кислотой 454 тиогликолевой кислотой 454
титрованием метаванадатом аммония 449
— бихроматом 441, 442, 447, 1002 — йодатом 1003
— перманганатом 441, 446, 957, 992
— сульфатом церия 140, 449, 1002 — хлорамином 449 о-фенантролином 454
Железосодержащие минералы, разложение 435 сл.
Железо трехвалентное 437 сл., 441 сл., 994
восстановление 436, 441 сл.
амальгамой цинка 442
серебром в редукторе 139, 444 сернистой кислотой 444, 455, 958 сероводородом 445, 455 хлоридом олова 441, 452, 455
истинное содержание в минералах 958 1004
определение 441 сл.
аскорбиновой кислотой 452 весовое 28, 438, 439, 455, 958 в карбонатных породах 1055, 1066 в осадке от аммиака 122 в песке 436, 455
в присутствии железа (II) 1004
в силикатных породах 952, 953, 957, 958, 1004
7-иодо-8-оксихинолин-5-сульфо-кис-лотой 454
колориметрическими методами 452 сл., 455
объемными методами 440 сл., 449
по Циммерману — Рейнгардту 446, 449
роданидом 452
салицилальдоксимом 454
салициловой кислотой 454 сульфосалициловой кислотой 454 титрованием нитратом ртути (I) 452 — хлоридом олова (II) 452 — хлоридом титана (III) 449, 957 — хлоридом хрома (II) 451
1094
Предметный указатель
Железо трехвалентное осаждение аммиаком 437, 496, 952 бензоатом аммония 107 в виде основного ацетата 103, 438 гидролитическое, мочевиной 439 купфероном 145, 439 слабыми органическими основаниями 111
сукцинатом натрия 106, 112, 438 сульфидом аммония 438, 958 фенилгидразином 114
отгонка с соляной кислотой и хлором 118
экстрагирование эфиром 161, 453, 460, 512, 525
Земельные кислоты 663
Золото восстановление нитритом натрия 407 как заменитель платины 56 определение
в виде металла 417, 419, 420
хлоридом тетрафениларсония 155 осаждение
гидрохиноном 407, 412 нитритом натрия 407, 420 сернистым ангидридом 407, 419 сульфатом железа (II) 407 хлоридом тетраэтиламмония 407 щавелевой кислотой 420 отделение 386, 406 сл.
от платиновых металлов 407 растворение металлического 399
Йенское стекло, состав 43
Измельчение образцов горных пород методом геологического управления США 891 по Вашингтону 892
Индигокармин 198
Индий 544 сл.
восстановление цинком до металла 546
концентрирование 544, 545
определение 547 сл.
весовое 547
дитизоном 547
колориметрическими методами 547 о-оксихилолином 547
титрованием гексацианоферратом (II) калия 547
флуоресцентное 548 осаждение
аммиаком 545, 547
ацетатом аммония 545
едкими щелочами 546
карбонатом бария 545
пиридином 111, 545 сероводородом 545 цианатом калия 546 отделение 545
поведение в обычном ходе анализа 544 разложение In-содержащих минералов 544
Индий
экстракция бромида эфиром 548
электролитическое осаждение 546
Индикаторы 193 сл.
адсорбционные 202
влияние «солевого эффекта» 200
— спирта на константы диссоциации 200
выбор для титрования кислот и оснований 197
интервал перехода окраски 195
кривые диссоциации 198
набор Кларка и Лабса 198
нейтральные 194
окислительно-восстановительные 202, 212, 213
приготовление растворов 201 смешанные 196
универсальные 200
чувствительные к кислотам и основаниям 194, 197
Иод 806 сл.
для установки титров 223, 224 определение 816 сл.
весовое 816, 817
в природных водах 809
в присутствии брома 809 объемное 817, 818
по Бобиньи 817
по Фольгарду 817
потенциометрическим титрованием 818
осаждение
в виде иодида палладия 816
— — иодида серебра 808, 809, 817 отгонка 808
отделение от хлора и брома 808, 809 очистка 223
разложение иодсодержащих минералов 807
установка титра раствора 222 сл. Иодид-иодатная смесь 109, 222 Иодистоводородная кислота 60 Иодометрия 220
7-Иод-8-оксихинолпн-5-сульфокпслота (Феррон) 454, 828
Ионообменное разделение 539, 628, 635 Иридий
определение
в виде металла 433
весовое 418, 432, 433
в платиновых сплавах 421
потенциометрическим титрованием 422
осаждение
в виде гидроокиси 418, 428
— — сульфида 413, 418
гидролитическим методом 408, 430 отделение
от платины 410
от родия и палладия 410, 430 Иттербий
восстановление 629
обнаружение 627
П редметный указатель
1095
Иттербий
спектрофотометрическое определение 630
Иттриевая группа 630
Кадмиевые минералы, разложение 296 Кадмий 296 сл.
карбонат, осадитель 108
определение 299 сл.
бруцином 301
в виде молибдата 301
— — фосфата 300
взвешиванием сульфата 299
внутренним электролизом 298, 301 динафтилтиокарбазоном 301 дитизоном 156, 301 колориметрическое 301 комплексометрическое 157 электролитическое 300
осаждение
fJ-нафтохинолином 297, 301
сероводородом 83, 300
отделение 296 сл.
от висмута 271
от меди 282, 284
от свинца 298
от цинка 297, 298
поведение в общем ходе анализа 297 разложение кадмиевых минералов 296 электролитическое отделение 298
Кали едкое, осадитель 118
— — поглотитель СО2 68
Калибровка объемно-аналитических приборов 189, 191, 192
— разновеса 34
Калий 729 сл.
бииодат 211, 227
бифталат 208
бихромат, установка титра 218, 227
бромат 109, 151, 227
гексацианоферрат (II), установка титра 489
гексацианоферрат (III) 227, 477
иодат 227, 252
определение
в виде перхлората 734
весовое 731, 736, 744, 745, 746, 747
восстановлением хлороплатината и весовым определением платины 744
в питьевой воде 747
в почвах 1006
колориметрическое 747
натриево-кобальтинитритным методом 747, 1006
объемными методами 747
перхлоратным методом с бутиловым спиртом 745
— — с этиловым спиртом 746
по методу Линде-Глэддинга 747 фотометрией пламени 180 хлороплатинатным методом 731
отделение
от лития 732, 734, 739
Калий
отделение
от натрия 733, 734, 747
от рубидия и цезия 740, 743
перйанганат
установка титра 214 сл.
— — по железной проволоке 217
— — по мышьяковистому ангидриду 216
— — по оксалату натрия 214
— — по соли Мора 217
— — по стандартным образцам (нормалям) 217
— — по щавелевой кислоте 217 персульфат 61
пиросульфат 115, 469, 916, 921 сл.
роданид, приготовление раствора 487 роданомеркуриат 554
сульфид 87, 93, 246
хлороплатинат, растворимость 731
цианид, установка титра 466
Кальций 693 сл.
карбонат, осадитель 108, 261
окись
адсорбент 71, 75, 76
экстрагирование раствором сахара 696
оксалат
прокаливание 711
растворимость 703
определение 701 сл.
в виде карбоната 710, 711
— — сульфата 710, 711
— — фторида 710, 711
в доломите 694
весовое 696, 701 сл., 710 сл.
в карбонатных породах 1057, 1068
в магнезите 694, 712
в силикатных породах 963 колориметрическое 712 комплексометрическое 157, 712 объемное 709
титрованием перманганатом 710
осаждение
молибдатом аммония 694
оксалата 695, 696, 701 сл., 705 сл.
сульфата 695, 696, 697, 711
фосфата 723
отделение 361, 694 сл.
в виде нитрата 696
— — оксалата 705 сл.
от магния 694, 706 сл., 724
от стронция и бария 697, 698
экстрагированием азотной кислотой 699
— спирто-эфирной смесью 697
поведение в обычном ходе анализа 693 хлорид 71, 76, 904
Кальцит, идентификация 1046
Капельный анализ 184
Карбонатные породы 1041 сл.
минеральный состав 1043 определение
воды 1064
1096
П редметный указатель
Карбонатные породы
определение
потери при прокаливании 1070
разложение и растворение 1045, 1065 сл.
сокращенный анализ 1065 сл.
сравнение с силикатными породами 1041
точный анализ 1048 сл.
«Кароксайт» поглотитель СО2 70
Качественный анализ 25
полярографический 171
силикатных пород 883 сл.
Кварц, определение в силикатах 931 сл., 935
Кварцевое стекло, состав 43
Кислотный хром сине-черный Р 579
Кислоты
концентрация имеющихся в продаже 60
установка титра 196, 202 сл.
методами весового анализа 203, 204 объемными методами 204
по тетраборату натрия 206
Кобальт 469 сл.
определение 471 сл.
весовое в виде Со3О4 474
в карбонатных породах 1056
в силикатных породах 962, 1034
в сталях 470
гексацианоферратом (III) калия 477
калиево-нитритным методом 471
колориметрическими методами 475, 747
комплексометрическим титрованием 159
а-нитрозо-0-нафтолом 473
нитрозо-К-солью 475
объемное 477
потенциометрическим титрованием 477
роданидным способом 476
холином солянокислым 747
электролитическое 471, 473
осаждение
нитритом калия 470, 471
сероводородом 84, 86, 470
фосфатами 471
феиилтиогидантоиновой кислотой 471
отделение 458, 459, 469, 470^сл.
поведение в общем ходе анализа 469
Кобальтовые минералы разложение 469
Колбы мерные 190, 192
Количественный анализ 25
полярографический 171
Коллекторы 260
Колокол
для титрования без доступа двуокиси углерода 209
для фильтрования в вакууме 126
Колориметрия 173
Колориметры 50
Комплексометрическое титрование 157 649, 728
Комплексные анионы сульфо-анионы 88 фторо-анионы 89, 99 цианиды 89
Комплексон II 157
Комплексон III 157, 274, 611, 647, 712 714, 728
Комплексообразователп 82, 158 Кондуктометрическое титрование 229 Концентрирование следов элементов 179 Крахмал 220
Крезоловый красный 199, 200
Крезолфталеин (Ортокрезолфталеин) 199
Кремневольфрамовая кислота 740 Кремнекислота обезвоживание 756, 758 сл., 940 серной и фтористоводородной кислотами 323, 756, 758, 761 соляной кислотой 758, 937, 940 фтористоводородной кислотой 436 679 хлорной кислотой 323, 759, 942 определение в искусственных силикатах 936, 941, 954 в карбонате натрия 60 в карбонатных породах 1051, 1054, 1065
в осадке от аммиака 120, 756, 936
в присутствии фтора 1018
в свинцово-бариевом стекле 943 в силикатных породах 932, 934, 935, 954 сл.
«растворимой» 932, 1037 прокаливание 943 удаление обработкой HF 932, 945 Кремний 752 сл.
определение 756 сл.
в виде SiO2 756 сл.
в железе 755
во фторсодержащих материалах 754, 939
в силикатных породах 931 сл., 954 сл.
колориметрическое 757 молибдатом аммония 752 отгонка 753, 932 отделение 120, 323, 437, 679, 753 сл. поведение в общем ходе анализа 752 разложение кремнийсодержащих пород 753
Кристаллический фиолетовый 330
Ксиленолцианол красный 199
Купфераты, экстрагирование 147
Купферон 115, 143 сл., 261, 271, 280, 285, 336, 343, 432, 439, 511, 515, 525, 531, 553 сл., 640, 644, 661, 965, 973
повышение устойчивости раствора 144 Куркума 582, 831, 845, 972, 1032
Лакмус 202
Предметный укааатель
1097
Лантановая группа 632 Литий 729 сл. ц нахождение в породах 729 обнаружение железом (III) и иодной кислотой 729
определение
в виде стеарата 738
— — сульфата 737, 742
весовое 737 сл.
в огнеупорной глине 739 спектроскопическим методом 738 турбидиметрическое 738
отделение
от других щелочных металлов 737 от калия 731, 738, 739
от натрия 738, 739
экстрагирование 739, 740
Литр 188
Лодочка для взвешивания 49
Лупы 51
Лютео-кислота (9-Фосфорномолибдено-вая кислота) 344, 740
Магнезиальная смесь 66, 98, 306, 791 Магний 715 сл., 964, 1069
окись 109
определение 719 сл.
ацидиметрическим титрованием 727 в виде оксалата 727
— — пирофосфата 719 гсл. весовое 719 сл., 725, 727 в карбонатных породах 1057, 1069 в силикатных породах 726, 964 йодометрическое 726 колориметрическое 728 комплексометрическое 157, 728 объемными методами 726, 727 о-оксихинолином 726 титановой желтой 728
осаждение
карбонатом аммония 717
окисью ртути (II) 716
оксалата 727
о-оксихинолином 148 сл., 717, 718, 725, 730
фосфата 718 сл.
отделение 713 сл., 730
от алюминия 718
от кальция 694, 705, 724
от лития 717
от щелочных металлов 717, 718, 730
перхлорат 71, 73, 76, 848, 849, 904 поведение в обычном ходе анализа 715
разложение Mg-содержащих минералов 715
Магний-уранил-ацетат, приготовление раствора 751
Мазда, состав 43
Маннит 834, 835
замена глицерином при определении бора 839
Марганец 493 сл.
Марганец
окисление
висмутатом натрия 497, 505 двуокисью свинца 505 перйодатом 505 персульфатом 501, 505
определение 497 сл.
в виде пирофосфата 503, 724
— — сульфата 504, 505
— — сульфида 504
весовыми методами 497, 503, 959 висмутатным методом 497
в карбонатных породах 1055, 1067 восстановление двуокиси 503 в силикатах 949, 959, 961
в чугуне 502
колориметрическое 159, 505, 962 объемными методами 497 сл., 505 персульфатный методом 501 потенциометрическим титрованием
502
по Фольгарду 497, 503 осаждение
аммиаком в присутствии окислителей 493, 949
в виде двуокиси 494 сероводородом 496 сульфидом аммония 493, 496, 960 хлоратно-азотнокислым методом
493, 494
отделение 494 сл.
от железа и алюминия 496
от хрома 497
от цинка 496
поведение в общем ходе анализа 493 разложение марганцевых минералов 493
Марганцевые минералы, разложение 493 сл.
Масс-спектрометрия 184
Медные руды, разложение 282 Медь 281 сл.
определение 285 сл.
аммиачным методом 293 а-бензоиноксимом 294 весовыми методами 285—290 в карбонатных породах 1056 внутренним электролизом 170, 285 в силикатных породах 962 гидразином сернокислым 293 диэтилдитиокарбаматным методом
293
йодометрическое 285, 287 колориметрическое 159, 293, 295 объемными методами 291 пиридинродановым методом 295 роданидным методом 285, 290 салицилальдоксимом 285, 294 солью Рейнеке 285, 294 сульфидным методом 285, 292 электролитическим методом 286 осаждение
бензоиноксимом 285
в виде сульфида 87, 88, 282, 285
1098
Предметный указатель
Медь
осаждение
купфероном 285
а-нитрозо-0-нафтолом 284
электролитическое 95, 284
отделение 87, 95, 269, 271, 272, 282 сл.,
479, 481, 962
поведение в обычном ходе анализа 281
разложение медных руд 282
Мекера горелка 52, 132, 133
2-Меркаптобензоксазол 423
Меркаптоуксусная кислота (Тиоглико-левая кислота) 454
Метиленовая синяя 213, 662
Метиловый желтый (Диметиламиноазобензол) 194, 195, 196, 201
Метиловый красный (Диметиламиноазо-бензол-о-карбоновая кислота) 194, 195, 197, 198, 199
Метиловый оранжевый (Диметиламино-азобензолсульфонат натрия) 194, 195, 196, 198, 201
Метиловый фиолетовый 330, 543
Механический растиратель пород 894
«Микобайт», поглотитель 70
Микроскопия 185
Микрохимия 185
Миллилитр 188
Миндальная кислота 631, 647
Молибден 356 сл.
нахождение в горных породах 880.
определение 361 сл.
а-бензоиноксимом 361, 364
в виде молибдата свинца 361, 366
----МоО8 361, 367
----MoS2 361
колориметрическое 368
нитратом ртути (I) 370
потенциометрическим титрованием сульфатом титана (III) 371
титрованием ацетатом свинца 371 — перманганатом 28, 361 — солью хрома (II) 371 роданидным методом 369 цериметрическое 140
осаждение р-нафтохинолином 359 сероводородом 357 сл., 367
отгонка 767
отделение 100, 357
от вольфрама 767
от группы мышьяка 100, 359
от кальция 359
от рения 374, 375, 379
от сурьмы 321, 359, 360
от хрома 360
поведение в общем ходе анализа 356 разложение молибденовых руд 356 сл., 361
экстракция эфиром из солянокислого раствора 360
Молибденовые руды, разложение 356 сл., 360
Монацит, разложение 599, 601
Морин 177, 579, 581
Мунро тигель 127
Муравьиная кислота, титрование 196 Мышьяк 302 сл.
металлический, растворение 303 определение 309 сл.
весовое в виде сульфидов As2S3 и
As2S5 309, 310
гипофосфитным методом 316
колориметрическое 315
отгонкой в виде AsH3 312
титрованием иодом 311
— по Фольгарду 310
— броматом калия 316
осаждение
магнезиальной смесью 306, 321
сероводородом 306
отгонка 303, 304, 308, 312
отделение 87, 96, 98, 282, 303 сл., 321, 360, 788
поведение в обычном ходе анализа 302 разложение мышьяковых минералов
303
Мышьяковистая кислота, титрование 196
Мышьяковистый ангидрид 229 применение для установки титров 216, 218, 224
раствор в едком натре 228
Мышьяковые минералы, разложение 303
Насосы водоструйные 53
Натр едкий 68
осадитель 109, 116, 122
поглотитель СО2 68, 69, 70
приготовление растворов 207
установка титра 207
Натрий
арсенит, установка титра 227 сл.
ацетат 104, 112, 117
висмутат 61, 62, 498
гипосульфит 117
гипофосфит 316
карбонат 60, 116, 204, 205
оксалат 214, 223
определение
в виде натрий-магний-уранил-ацетата 750 в
— — натрий-цинк-уранил-ацета-та 748
— — перхлората 735
— — сульфата 734
— — хлорида 734, 738, 742 весовое 733, 736, 739, 742, 748 сл. в карбонате кальция 751
в стекле 750
в товарном алюминии 751
в цементах 750
объемное 751
спектрофотометрическое 750
фотометрией пламени 180
паравольфрамат, очистка 914 перекись 333, 918, 924, 928 пиросульфат 652, 916, 921 сл.
П редметный указатель
1099
Натрий
отделение
от калия, рубидия и цезия 733, 734, 742
от лития 738
сукцинат 107, 112, 458
сульфид 87, 93 тетраборат см. Бура тиосульфат 225 сл., 266 янтарнокислый 107, 112, 438
Натронная известь 69
Натронный асбест 69, 848
Нафтарсон см. Торон
Нафтионовая кислота 492 0-Нафтохинолин 297, 301, 359 Нейтральный красный (Диметилдиами-нотолуолфеназин) 194, 195, 202
Неодим, спектрофотометрическое определение 630
Неокупферон 143
Несслера реактив 869
Нефелин, идентификация 1036 Нефелометрия 173, 175 Нефелометры 50 Никель 456 сл.
определение 460 сл.
весовое а-бензилдиоксимом 463
в виде окиси 467
весовыми методами 460, 463, 467 в карбонатных породах 1056 в силикатных породах 962, 1034 диметилглиоксимом 460 сл., 467 колориметрическое 159, 467 объемными методами 466, 467 титрованием диметилглиоксимом
467
цианидным методом 466 электролитическое 463
осаждение
а-бензилдиоксимом 458 в виде сульфида 86, 87, 458 диметилглиоксимом 457, 460 едкой щелочью в присутствии окис-
лителя 459
фосфатом аммония 458 а-фурилдиоксимом 458, 463
отделение 457 сл.
поведение в обычном ходе анализа 456
разложение Ni-содержащих минералов 457
электролитическое осаждение 459, 463 Никельсодержащие минералы, разложение 456
Н иобаты
разложение
сплавлением 669, 674
фтористоводородной кислотой 665
хлористой серой 672
растворимость 621
Ниобий 663 сл.
восстановление 686 определение 680 сл.
весовое 678, 680
Ниобий
определение
в минералах 663 сл.
колориметрическое 688 сл.
объемное 680, 686
роданидным методом 689
спектрофотометрическое в ультрафиолетовой области 688
с перекисью водорода 655, 688
титрованием перманганатом 686 сл.
осаждение
купфероном 147
пирогаллолом 676
салициловой кислотой 675
таннином 654, 673, 674 сл., 681
фениларсоновой кислотой 680, 685
отделение 525, 673 сл.
от вольфрама 677, 769
от тантала 681 сл.
от титана 681
от циркония 673
хроматографическое 685
экстрагированием 685
Нитрамин (2,4,6-Тринитрофенилметил-
нитрамин) 196, 202
Нитраты
определение малых количеств 865, 868, 869 сл.
превращение в сульфаты и карбонаты п-Нитробензоларсин 588
п-Нитрозодифениламин 418
а-Нитрозо-0-нафтол 157, 284, 473, 552
проверка качества 474
Нитрозо-Б-соль (1,2-Нитрозонафтол-3,6-
дисульфонат натрия) 476
Нитрозофенилгидроксиламина аммонийная соль см. Купферон
Нитрон [1,4-Дифенил-(3,5-эндоанил)-ди-гидро-1,2,4-триазол] 157, 376, 867
Нитрофениларсоновая кислота 334 Нитро-о-фенантролинсульфат железа
(II) 213
Нитроферроин 213, 220
Обезвоживание минералов 896
Объем, измерение 188
Объемный анализ 25, 188 сл.
применяемая посуда 190
Окисления-восстановления методы
212 сл.
Окислительно-восстановительные индикаторы 212, 213
потенциал перехода окраски 213
Окислительно-восстановительный по-
тенциал, сдвиг 159
n-Оксифениларсоновая кислота 156 о-Оксихинолин 148 сл., 280, 350, 492, 548, 500, 553, 557, 565, 574 сл., 583, 717, 718, 725, 730
приготовление раствора 149, 557, 575
Оксихиноляты, титрование 150
Оливин, идентификация 1036
1100
Предметный указатель
Олово 332 сл.
восстановление
алюминием и цинком 338, 340, 341
никелем 337, 340
свинцом 337, 338, 340
фосфорноватистой кислотой 341
обнаружение 334
определение 337
в виде двуокиси 341
весовое 341, 343
внутренним электролизом 343
в органических соединениях 344
гидролизом солей 343
дитиолом 344
йодометрическое 338
колориметрическое 344
силикомолибденовой кислотой 344 осаждение
аммиаком 334, 342
купфероном 147, 336, 343
сероводородом 98, 282, 335, 342
фениларсоновой кислотой 334
отгонка в виде хлорида 96, 333, 336, 344
отделение 96, 98, 283, 332, 334 сл.
поведение в общем ходе анализа 332 разложение оловянных руд 332 сл. хлорид (II), восстановитель 441, 452, 455
Оловянные руды, разложение 332 сл.
Органические соединения
обнаружение
ртути 254
селена 384
определение
водорода 848, 851
галогенов 807, 822
олова 344
углерода 850
сжигание 850 сл.
Ортокрезолфталеин (Крезолфталеин) 199
Осадки
высушивание 131
прокаливание 131
промывание 128
Осаждение
аммиаком 102
ацетатов по Брунку — Функу 105
— по Митташу 106
ацетатом 103, 117, 950
бензоатом аммония 107
броматами 109
гидролитическое см. Гидролитическое осаждение
группы меди и мышьяка 84
дитизоном см. Дитизон
едким кали и перекисью водорода 118
•едким натром 109, 116, 122
иодатами 109
карбонатом бария 108, 545, 552
купфероном см. Купферон
окисью цинка 108, 458, 470, 503
л-оксифениларсоновой кислотой 156
Осаждение
о-оксихинолином см. о-Оксихинолин путем изменения pH раствора 101 сл. салициловой кислотой и салицилатом аммония 118
сероводородом 83 сл.
слабыми органическими основаниями
111, 627, 654
сукцинатом натрия 106, 112, 438
сульфидами щелочных металлов 87 сл. сульфидом аммония 90 сл., 115, 952, 960 сл.
суспензиями карбонатов и окисей 108 таннином см. Таннин
фениларсоновой кислотой 156, 334, 553, 626, 638, 680, 685
фенилгидразином 114, 154
хлоридом тетрафениларсония 154, 375, 376
электролитическое см. Электролитическое осаждение
Осмий
определение 424 сл.
в виде металла 419, 425
дистилляцией 424
колориметрическое, тиомочевиной 419
осаждением гидроокиси 425 отгонка 399, 406, 424
отделение от металлов платиновой группы 409, 425
Основания, установка титра 197, 207 сл.
по бензойной кислоте 209
по бифталату калия 208
по титрованному раствору кислоты 210
Отгонка
бора 833 сл., 1032
ванадия 509, 512
вольфрама 766
галлия 550
германия 95, 345, 346
железа 117
иода 808
кремния 753, 933
молибдена 767
мышьяка 303, 304, 309, 312
олова 96, 332, 337, 344
осмия 399 , 406 , 424
рения 374
ртути 242
рутения 406, 408, 409, 426
селена 384, 388
сурьмы 96, 99, 317, 327
теллура 384, 388
фтора 820, 821, 823 сл., 1019
хлористого хромила 497, 591
циркония 635
Пайрекс, состав 42, 43
Палладий
определение 430 сл.
в виде металла 430
диметилглиоксимом 430
П редметный указатель
1101
Палладий
определение
колориметрическое 418 71-нитрозодифениламином 418 объемное 422
1,10-фенантролином 423 а-фурилдиоксимом 423 Р-фурфуральдоксимом 418, 422
1,2-цикл огександиондиоксимом 423 осаждение
диметилглиоксимом 410, 430
иодидом 411
цианидом 411
отделение
от золота 407
от платины 410
от родия и иридия 410
от серебра 236
растворение в кислотах 399
Пемза 70
Перекись водорода 63, 118
твердая 63
Перйодаты, определение 155
Перренаты, определение 155
Перхлоратоцераты 219
Перхлораты, определение 155
а-Пиколин 111, 583
Пипетки
измерительные 191
обыкновенные 190
Пиридин 87, 111, 273, 529, 546, 551, 654
соли 87, 111, 112, 273, 615
Пирит
анализ 537, 794, 795
разложение 537, 794
Пирогаллол 280, 634
Пирослизевая кислота 208
Плавиковая кислота см. Фтористоводородная кислота
Плавни 915 сл.
выбор 923
кислотные 916
обычные 917
применение для разложения силикатных пород 915 сл.
щелочные 916
Пластинки
для тиглей 49
фильтрующие, стеклянные 128
Платина 393
восстановление до металла 417
губчатая 127
извлечение из остатков 67
комплексные соединения 398
определение
в виде металла 417, 429
объемными методами 422
потенциометрическим титрованием 422
тиофенолом 423
хлоридом тетрафениларсония 155
осаждение
аммиаком 397
в виде хлороплатината аммония 410
Платина
осаждение
муравьиной кислотой 417
сероводородом 397, 412, 429
электролитическое 417
отделение 410, 427 сл., 733
поведение в ходе анализа 397 разложение сплавов 399 сл. растворение 399
Платиновые изделия 32, 44, 53, 55, 127, 132
растворение 435
Платиновые металлы 395 сл.
взаимодействие с органическими реактивами 423
концентрирование в металлическом свинце 416
методы определения 416 сл.
осаждение
в виде двойных хлоридов (хлорсо-лей) 416
в виде металлов 412, 416, 425, 427, 429, 432, 433
гидроокисей 407, 413, 426, 427
сероводородом 397, 412, 429
отделение от других металлов 410 сл.
поведение в общем ходе анализа 396 разделение 406 сл., 423 сл.
различными методами 410
систематическое 407, 423
разложение минералов и сплавов 399 сл.
Платинохлористоводородная кислота 731 сл.
Поглотители
влаги 57, 71 сл., 904
двуокиси углерода 68 сл.
Показатель ионов водорода 194
Полярограммы 172
Полярограф 172
Полярографический анализ 171 сл.
Полярографическое титрование 229
Посуда
бакелитовая 56
золотая и серебряная 53, 55
кварцевая 54
платиновая 44, 53, 127, 132
полиэтиленовая 59, 63
стеклянная 39, 128
устойчивость 39 сл.
фарфоровая 39, 44, 128
церезиновая 59, 63
Потенциал полуволны 172
Потенциометр 51
Потенциометрическое титрование 210 сл.
Празеодим, спектрофотометрическое определение 630
Приборы
для внутреннего электролиза 169, 171
— восстановления жидкими амальгамами 141
— восстановления мышьяка 314
— определения азота методом Дюма 860
1102
Предметный указатель
Приборы для определения воды в минералах по Пенфильду 915
— — воды по Гучу 911 сл.
— — железа (II) по Куку 999
— осаждения BaS04 803
— отгонки
-----бора 836, 842
-----AsClg 304
— перегонки воды 53
— потенциометрического титрования по Гиллебранду 229
— разделения As, Sb, Sn перегонкой 96
— титрования без доступа СО3 209
— фильтрования в вакууме 126
— — концентрированного NaOH 208
— экстрагирования, Роте 163
— электролиза, Мелавена 166
Пробы для анализа воздушно-сухие 902 измельчение 891 сл. отбор 77 сл. 890 растирание 893 сл.
Прокаливание осадков 131
Промывалки 53
Промывание осадков 128 полнота 130
Промывные растворы 129
л-Пропиларсиновая кислота 157
Протактиний 692
Радиаторы для испарения жидкостей 47
Радий, электроскопическое определение 701
Разделение в группе меди 94 мышьяка 95 группы меди и мышьяка 87, 93 платиновых металлов 406 сл., 423 редкоземельных элементов 621 сл., 628
с применением комплексообразовате-лей 88, 99
щелочных металлов 731 сл.
электролизом 88 элементов в осадке от аммиака 112, 118 сл. от ацетата и сукцината натрия 112 сл.
от сероводорода 92 сл.
Разновес аналитический 32 сл.
проверка 35
Растирание проб для анализа 893 сл.
Реактивы 58 сл.
газообразные 68
качество 58
растворы 66 сл.
Редкоземельные минералы, разложение 600, 619 сл.
Редкоземельные элементы 617 сл., см. также Торий, Церий иттриевая группа 630
Редкоземельные элементы лантановая группа 632 обнаружение 618, 619 окраска окислов 632 оксалаты, растворимость 621 определение 628 сл.
весовое 631
в осадке от аммиака 123 рентгеноспектральное 628 спектрофотометрическое 630 осаждение
аммиаком 618, 624, 627 едким натром 625, 626 лантановой группы сульфамиповой кислотой 632
миндальной кислотой 631 органическими основаниями 627 сульфатом натрия 631 фтористоводородной кислотой 618, 622
щавелевой кислотой 618, 621, 631 отделение 620 сл.
групповое 618
ионообменным методом 628 лантановой группы от иттриевой 632
от ниобия и тантала 622
от циркония 644
фракционированным осаждением 631
цериевой группы от иттриевой 631 церия 625 сл.
поведение в обычном ходе анализа 618
разложение природных минералов 619 сл.
тербиевая группа 630 цериевая группа 630 эрбиевая подгруппа 630
Редуктор Джонса 135, 443, 450, 518, 530, 968 серебряный 139, 443
Рейнгардта раствор 446
Рейнеке соль (Тетрароданодиаминохро-миат аммония) 254, 285, 294, 541
Рений 372 сл. возгонка 374 обнаружение 372 определение 375 сл.
в виде «перрената нитрона» 376 — — металла 375 весовыми методами 375 каталитическим методом 380 колориметрическое 378 объемными методами 377 роданидным методом 378 титрованием перманганатом 378 — щелочью 377 хлоридом тетрафениларсония 375, 376
электролитическое 375, 380 осаждение
аммиаком 372
Предметный указатель
1103
Рений
осаждение
диэтилдитиокарбаматом 375
оксалатом аммония 373
сероводородом 373
фосфатом аммония 372
отгонка хлоридов 374
отделение 373 сл., 379
поведение в общем ходе анализа 372 разложение Re-содержащих минералов 373
электролитическое осаждение 374, 380
Рентгеновский анализ 181 сл.
Рентгено-флуоресценция 181, 182
Родамин В 331
Родий
определение
в виде металла 410, 418, 431 а-меркаптобензоксазолом 423 тиобарбитуровой кислотой 423
осаждение
гидролитическим методом 408
сероводородом 431
хлоридом титана 410, 431
отделение
от иридия 410, 411, 431
от платины 410
Ртуть 242 сл.
обнаружение 254
в органических жидкостях 254
окись 109
определение 248 сл.
в виде металла 251
— — перйодата 251
— — сульфида 249
— — хлорида 250
в воздухе 256
весовыми методами 249, 251
дитизоном 156, 255
колориметрическое 255 малых количеств 253 сл. объемное 248, 251
титрованием роданидом 248
осаждение сероводородом 84, 245
отгонка из минералов 242
отделение 84, 88, 93, 245 сл., 271, 282, 386
очистка 166
поведение в обычном ходе анализа 242
разложение соединений ртути 242 сл., 253
сульфид, разложение 243
Рубидий 729 сл.
определение
в виде хлорида по разности 742
— — хлороплатината 740, 744
осаждение 9-фосфорномолибденовой кислотой 740
отделение
от калия 740, 742
от натрия 733, 735
от цезия 740
Рутений
определение 426
в виде металла 427
микрохимическое, тионалидом 419 осаждение гидроокиси 419, 428 отгонка 406, 408, 409, 426 отделение 409, 426
Салицилальдоксим 285, 294, 454
Салициловая кислота 118, 454, 653, 658, 675
Самарий восстановление 629 спектрофотометрическое определение 630
Сверхпайрекс, состав 43
Свинец 100, 257 сл.
карбонат 108, 605
металлический, анализ 168, 272, 279 определение 262 сл.
в виде молибдата 263
— — сульфата 258, 262, 269 весовыми методами 262 сл.
в силикатных породах 258
гексацианоферратным способом 267 дитизоном 267 колориметрическое 265, 267 объемными методами 265, 267 титрованием молибдатом аммония 267
хроматным методом 261, 265 электролитическое 262, 264 осаждение
едким натром в присутствии окислителя 261
купфероном 261
молибдатом 263 хроматом 261
отделение 258 сл.
от железа 261
от индия 544, 546
от кальция, бария и стронция 258, 259, 266
от меди, кадмия и ртути 94
от серебра и висмута 260
от сурьмы 323
поведение в обычном ходе анализа 257 разложение сульфидных руд 258 электролитическое выделение 260, 264
Свинцово-бариевое стекло, анализ 943
Свинцово-натриевый сплав 76
Себациновая кислота 605
Селен 382 сл.
восстановление сернистым ангидридом 385, 389
выделение в элементарном состоянии 385
определение 389 сл.
броматометрическим титрованием 392
взвешиванием элементарпого 389 восстановлением солянокислым гидроксиламином 389, 390
в пищевых продуктах 392
1104
Предметный указатель
Селен
определение
в рудах 389
в сере 388
в стекле 392
колориметрическое 388, 394 объемными методами 391 сл. потенциометрическое 392
осаждение сероводородом 386 отгонка хлоридов 383, 388 отделение 98, 100, 283, 385 сл.
от теллура 387, 388, 389
поведение в общем ходе анализа 382 разложение Se-содержащих минералов 383 сл.
содержание в органических соединениях 385
Селенистая кислота, приготовление 645
Сера( 793 сл., 1029
нахождение в горных породах 882, 1028, 1062
определение 797 сл.
бензидином 804
в виде сульфата барияJ797 сл., 966, 1030, 1062
весовое 797 сл., 1030, 1062
в минералах 794, 1029
в пирите 794, 795
в присутствии железа 803
----солей щелочных металлов или аммония 802
в растворах, содержащих только серную кислоту 801
в сталях 804
в угле 1028
колориметрическое 805
объемными методами 804
отгонкой в виде H2S 804 сульфидной серы 1031, 1062 турбидиметрическое 805 Ц хроматом бария 804
отделение 796
поведение в общем ходе анализа 793
разложение серусодержащих минералов 794
Серебро 236 сл.
восстановитель 139, 444
выделение из минералов 236
иодид 808, 816
обнаружение формазилкарбоксило-вой кислотой 241
определение 237 сл.
весовое в виде AgCl 237, 811
внутренним электролизом 169
в товарном свинце 168
дитизоном 240
колориметрическое 240
нефелометрическое 240
по Мору 240
титрованием роданидом калия или аммония 238
о-толидином 240
электролитическое 240
осаждение сероводородом 84, 236
Серебро
отделение 93, 236, 246, 258, 282, 538
поведение в общем ходе анализа 235
электролитическое отделение 236
Серебряный редуктор 139, 444
Серная кислота 60
Сернистая кислота, титрование 196
Сернистый ангидрид 100
Сжигание соединений
в калориметрических бомбах 854
летучих или возгоняющихся 854
мокрое 856
органических 853
содержащих азот 855
содержащих галогены 854
Силикагель 76, 858
Силикатные породы 875 сл.
анализ 875 сл., 889 сл., 936 сл.
измельчение 891
определение кварца 933 сл., 935
ошибки анализов 883
предварительный качественный анализ 883, 946
разложение
в запаянной трубке 987, 989
кислотами 930, 937, 989, 992
при помощи плавней 915 сл., 939
растирание 893
составные части 881, 949 сл.
ход анализа после выделения кремнекислоты 946 сл.
Силикомолибденовая кислота 344
Синильная кислота, титрование 196
Скандий 613 сл.
обнаружение в силикатных породах 613
определение весовое 616
осаждение
аммиаком 615
карбонатом натрия 614
пиридином и его азотнокислой солью 615
слабыми органическими основаниями 111, 615
сульфатом калия 614, 631
тиосульфатом натрия 614, 615
фтористоводородной кислотой 614, 615
щавелевой кислотой 614, 615
отделение 614 сл.
поведение в обычном ходе анализа 613
разложение Sc-содержащих минералов 613
«Солевой эффект» 200
Соляная кислота
концентрация 60
растворы с постоянной температурой кипения 210, 211
установка титра 202
Спектрографы 178
Спектроскоп 51
Спектрофотометр 173, 174
Спектрофотометрия 173
П редметный^указатель
1105
Спектрохимический анализ 177
Сплав
Вуда 143
Деварда 866
свинцово-натриевый 76
Сплавление с бифторидами 922 борным ангидридом 920, 923, 928 бурой 920, 923
карбонатами калия и натрия 116, 917, 923, 924 сл., 928
карбонатом натрия и серой 333, 346
перекисью натрия 333, 918, 923, 924, 928
пиросульфатом 114, 119, 469, 916,
921 сл., 934
цианидом 384
щелочами 55, 333, 357, 361, 916, 917
Стекло химическое 39 сл., 188 американское 42 состав 42, 43
Стеклянный электрод 230
Стильбазо 579
Стронций 693 сл., 712 сл.
нахождение в изверженных породах 879
определение весовое 712 в карбонатных породах 1057 в силикатных породах 963
осаждение
азотной кислотой 699
оксалата 696, 712
сульфата 697, 712
отделение
от бария 700
от кальция и бария 697
Ступки 50
агатовые 895
истирание и износ 895
стальные 895
Эллиса для крупнозернистых материалов 891
Сульфаминовая кислота 211, 632
Сульфаты, превращение
в карбонаты 159, 160
в тартраты и оксалаты 160
в хлориды 159
Сульфиды
разложение 794, 927 растворимость 84
Сульфосалициловая кислота 454, 865
Сурьма 317 сл.
восстановление свинцом 323, 360 выделение на медной фольге
324
металлическая, растворение 318 определение 324 сл.
в виде сульфида 325, 326
----четырехокиси 324, 326
весовыми методами 326
иодидным методом 330 колориметрическое 330
70 Заказ 522.
Сурьма
определение
отгонкой в виде сурьмянистого водорода 327
пиридин-иодидным методом 329 по образованию молибденовой сини 331
родаминовым методом 331
с кристаллическим фиолетовым или метиловым фиолетовым 330
титрованием броматометрическим методом 328
— иодом 328
— перманганатом 324, 325
— тиосульфатом натрия 328 электролитическое 328
осаждение
аммиаком 329
сероводородом 83, 321 сл., 325 отгонка 289, 298, 96, 99
отделение 83, 89, 96, 99, 321 сл., 359
поведение в общем ходе анализа 317 разложение Sb-содержащих минералов 317 сл.
сульфид, растворение 318
хлорид (III), отгонка 317
Сурьмяная кислота, растворение 320
Сурьмяные руды, разложение 317 сл.
Таллий 536 Тел.
восстановление цинком до металла 539 ионообменный метод отделения 539 обнаружение
в пирите 537
в цинковой обманке 537
по реакции с тионалидом 537, 542 спектральным методом_ 537
окисление 538, 543
определение 540 сл.
броматометрическое 542, 543
в виде иодида 541
---окиси таллия (III) 540
--- хромата 540
весовыми методами 540 сл.
йодометрическое 543
колориметрическими методами 542 сл.
объемными методами 542
по окраске молибденовой сини 542
с метиловым фиолетовым 543 спектрофотометрическое 543 тетрародандиамминхромиатом аммония 541
турбидиметрическое 542
осаждение
в виде иодида 438, 541
--- хромата 438, 540
тионалидом 542
отделение 247, 538 сл.
поведение в ходе анализа 536
экстрагирование трехвалентного эфиром 539
электролитическое отделение 539
1106
Предметный указатель
Таннин 151, 347, 525, 551, 585, 640, 654,
673 сл., 678, 681, 767
Тантал 663 сл.
обнаружение п-диметиламиноазофе-ниларсиновой кислотой 636
определение 680 сл.
весовое 147, 677, 680
в минералах 663 сл.
йодометрическое 682
колориметрическое 684, 691 объемное 682
пирогаллолом 685, 691
осаждение
йодатом 682
пирогаллолом 676
салициловой кислотой 675
таннином 654, 673 сл., 681
фениларсоновой кислотой 679, 685
отделение 525, 673 сл.
от ниобия 680—683, 685
от титана 653, 675
от циркония 673 сл.
— — хроматографическое 685
экстрагированием органическими растворителями 685
Танталаты
разложение сплавлением с плавнями 669 фтористоводородной кислотой 666 хлористой серой 672
растворимость 621
Тантало-ниобиевые минералы, разложение 619, 664, 669
Теллур 382 сл.
восстановление сернистой кислотой и гидразином 392
определение 392 сл.
в рудах 389
йодометрическое 393
колориметрическое 394
объемными методами 393
потенциометрическое 394
титрованием бихроматом 394
элементарного 393
осаждение
сероводородом 386
сульфидом аммония 387
отгонка хлоридов 383, 388
отделение 98, 100, 283, 385 сл.
от селена 387, 388
поведение в общем ходе анализа 382 разложение Те-содержащих минералов 383 сл.
Температура, стандартная 188
Тербиевая группа 630
Тетрабромфенолсульфофталеин (Бромфеноловый синий) 195, 196, 197, 199
Тетраметилдиаминодифенилметан 265
1,2,5,8-Тетраоксиантрахинон (Хинализарин) 549, 556, 581
Тетрафениларсония хлорид 155, 375,
377
Тетраэтиламмония хлорид 407
Тетраэтилфосфат 640
Тигли
Гуча 127, 131, 132
для определения воды в минералах 911
железные 57
золотые 55
Мунро 131
никелевые 57
платиновые 45, 46, 53, 131
Розе 56
Смита для определения щелочных металлов 1007
фарфоровые 132
фильтрующие 125, 126, 131
Тимоловый синий (Тимолсульфофта-
леин) 195, 196, 199
Тимолфталеин 195, 196, 202
Тиобарбитуровая кислота 423
Тиогликолевая кислота (Меркаптоук-сусная кислота) 454
Тиомочевина 261, 273, 278, 419
Тионалид (Р-Аминонафталид тиоглико-
левой кислоты) 419, 537, 542
Тиофенол 423
Тирриля горелка 133, 708
Титан 650 сл.
восстановление 143, 659, 662, 967
двуокись, получение чистой 657
обнаружение 658
определение 654 сл.
весовыми методами 145, 156, 661,
965, 966, 967
в карбонатных породах 1055
в легированных сталях 658
в осадке от аммиака 122
в силикатных породах 965
в цементе 658
колориметрическими методами 654, 655 сл., 966, 967, 1055
малых количеств 658
методом Гуча 661, 968
объемными методами 654, 655,
659 сл., 662
n-оксифениларсоновой кислотой 156 с перекисью водорода 653, 654, 966 титрованием метиленовой синью 662 — перманганатом 654, 968
— сульфатом железа (III) 662
хромотроповой кислотой 659
осаждение
аммиаком 965
гидролитическое 655, 661
едким натром 653
купфероном 144, 432, 661, 967
пиридином 111, 654
уротропином 654
отделение 432, 652 сл.
от железа и циркония 973
от урана 525
поведение в общем ходе анализа 650
разложение титановых минералов 651 сл.
хлорид (III), установка титра 449
Титановая желтая 728
Предметный указатель
1107
Титановые минералы, разложение 651 сл.
Титанониобаты, разложение 523
Титрование 202 сл.
в отсутствие двуокиси углерода 209 комплексометрическое 157, 645, 728 полярографическое 229 потенциометрическое 229 сл.
о-Толидин 241
Торий 598 сл., 618
нитрат, приготовление титрованного раствора 826
обнаружение
йодатом калия 603
методом эманации 599
спектрографическое 599
определение 607 сл.
весовыми методами 607 сл.
п-диметиламиноазофениларсиновой кислотой 611
колориметрическими методами 609 сл.
комплексометрическим титрованием 611
тороном 609
осаждение
ацетатом 117
в виде оксалата 598, 600, 607 гексаметилентетрамином 600 йодатом калия 598, 600, 603, 607 карбонатом свинца или бария
605
перекисью водорода 600, 602
пиридином 111, 606
салициловой кислотой 118 себациновой кислотой 605 тиосульфатом натрия 600, 604 фенилгидразином 154
фторидом аммония 600
отделение 598, 600 сл.
от редкоземельных элементов 604, 606
от скандия 598, 604
от церия и лантана и др. 603 сл. поведение в обычном ходе анализа
598
разложение Th-содержащих минералов 599
Торит, разложение 598
Торон (Бензол-2-арсоновая кислота-(1-азо-1)-2-оксинафталин-3,6-ди-сульфокислота) 609, 649
Тортвейтит, разложение 613, 616
Трилон Б см. Комплексон III
2,4,6-Тринитрофенилметилнитрамин (Нитрамин) 196, 202
2,3,7-Триокси-9-фенил-6-флуорон (Фе-нилфлуорон) 352, 354, 355
Тропеолин 0 (Натриевая соль азорезорцинсульфаниловой кислоты) 196, 202
Тулий, спектрофотометрическое определение 630
Турбидиметрия 173, 175
Турмалин 81
70*
f Углерод 846 сл.
нахождение в минералах 846
определение 848 сл.
в горных породах и минералах 846, 847, 850
в летучих веществах 854
в органических соединениях 851
в почвах 856
в присутствии азота 851
---- галогенов 854
----серы 854
в силикатных породах 1016
в сланцах 1016
микро- и полумикрометодами 851
«мокрым сжиганием» 856
прямым сжиганием 850
поведение в обычном ходе анализа 846
Углерода двуокись см. Двуокись углерода
Углерода окись, определение в воздухе 857
Угольная кислота, титрование 197
Уксусная кислота 60
Уран 522 сл.
восстановление до трехвалентного 526
определение 526 сл.
1-аскарбиновой кислотой 534
в виде U3O8 527
весовое 115, 146, 525, 526 сл., 531
в осадке от аммиака 119
гексацианоферратным методом 533 диэтилдитиокарбаматом натрия 534 колориметрическими методами
532 сл.
объемное 529
пероксидным методом 532
по природной радиоактивности 526
титрованием ванадатом 531
— перманганатом 527, 529
— сульфатом титана (III) 531
флуоресцентными методами 522, 531, 532
осаждение
аммиаком 524, 527
в виде аммоний-уранил-ванадата 531
— — фосфата 531
---- фторида 525
едким натром 524
купфероном 115, 146, 525, 531
пиридином 111, 529
сероводородом в присутствии гексаметилентетрамина 524
отделение 359, 511, 524 сл.
урана (IV) от урана (VI) 525
поведение в общем ходе анализа 523
Урановые руды
определение азота 523
разложение 523
Уротропин (Гексаметилентетрамин) 88,
524, 601, 602, 654
Установка титра
арсенита натрия 227 сл.
бихромата калия 218, 227
1108
Предметный указатель
Установка титра
едкого натра 207
кислот 202 сл.
нитрата тория 826
оснований см. Основания, установка титра
перманганата калия см. Калий перманганат, установка титра
раствора иода 222
серной кислоты 203
соляной кислоты 202, 204
тиосульфата натрия 225, 266
фтористоводородной кислоты 196
хлорида титана (III) 449
хлорной кислоты 211
цианида калия 466
Фактор пересчета 30
Фарфор 44, 128
н-Фенаптраниловая кислота 213
о-Фенантролин 213, 423, 454
о-Фенантролин-сульфат железа (II) 202 Фениларсоновая кислота 156, 334, 553, 639, 679, 685
Фенилгидразин 114, 154
Фенилгидразинсульфит 155
S-Фенилтиогидантоиновая кислота 423, 471
Фенилтиомочевина 423
Фенилфлуорон (2,3,7-Триокси-9-
фенил-6-флуорон) 352, 354, 355
Фенолдисульфоновый реактив 869
Феноловый красный (Фенолсульфофталеин) 195, 196, 199
Фенолфталеин 195, 196, 202
Феррон (7-Иод-8-оксихинолин-5-суль-фодовая кислота) 454, 828
Фильтровальная бумага 123
Фильтрование 123
Фильтрофотометр 174, 180
Фильтрующие материалы 123 сл.
Флуоресцентный анализ 176
Фолина и Дениса реактив (Фосфорномо-либденовольфрамовая кислота) 280
Формазилкарбоновая кислота 241
Фосфатная порода, анализ 785
Фосфор 777 сл.
определение 781 сл.
алкалиметрическим титрованием 789
в виде пирофосфата магния 781, 787
весовое 781, 785
в карбонатных породах 1057
в осадке от аммиака 121
в присутствии ванадия 784, 791
— — железа, алюминия, ванадия, цинка, олова, титана или циркония 788
— — мышьяка 788
в силикатных породах 977 сл.
в стали 790, 792
колориметрическими методами 792
Фосфор
определение объемное 789
по окраске восстановленного фосфоромолибдата аммония 792, 978
— — фосфорованадатомолибдата 795
осаждение
фосфата магния и аммония 781, 784, 788, 791, 977
фосфоромолибдата аммония 779, 780,. 781 сл.
хлоридом циркония 779 отделение 779 сл.
поведение в обычном ходе анализа горных пород 777 сл.
разложение фосфорсодержащих соединений 778
Фосфорная кислота, концентрация 60 титрование 196
9-Фосфорномолибденовая кислота (Лю-тео-кислота) 344
приготовление 740
Фосфорномолибденовольфрамовая кислота (Реактив Фолина и Дениса) 281
Фосфорномолибденовый реактив 331 Фосфорный ангидрид 71, 72, 904 Фотоколориметры 174
Фотометрия пламени 180 сл.
Фотометрические методы 173 сл.
Фотометры 50
Фталевая кислота 647
титрование 196
Фтор 819 сл., 1018 сл.
нахождение в горных породах и минералах 819, 1018
обнаружение
методом висячей капли 821 — травления 820, 1019
пробой на пламя 821
определение 824 сл.
в виде фторида тория 829 весовыми методами 823, 825, 826, 829, 1021
в карбонатных породах 1062
в почвах 1028
в природных водах 820, 828
в силикатных породах 1018 сл.
в фосфатных породах 825
в шлаках 826
колориметрическими методами 828 объемными методами 824, 825, 826, 829, 1024
по Берцелиусу 824, 1019, 1023 по Гофману и Ленделю 1024 по обесцвечиванию надтитановой кислоты 824, 828, 1026, 1028
— — цветных лаков 828
по Стейгеру 824, 1025, 1029
по Стейгеру—Мервину 1026, 1028 спектральным методом 828 титрованием нитратом тория 824
Предметный указатель
1109
Фтор
осаждение
в виде фторида висмута 829
— — фторида кальция 823, 1021
— — фторхлорида свинца 824, 826, 1024
отгонка 820, 821 сл., 1019
отделение 823 сл.
поведение в обычном ходе анализа 819
разложение F-содержащих минералов 821 сл., 1019
удаление из силикатных пород 939 Фтористоводородная кислота (Плавиковая кислота) 63 концентрация продажной 60 применение для разложения горных пород 752, 820, 830, 989 сл., 993, 999
титрование 196
хранение 56, 63
Фторборная кислота 933
Фториды, разложение 821
а-фурилдиоксим 423, 458, 463, 468 Р-Фурфуральдоксим 418, 422
Хинализарин (1,2,5,8-Тетраоксиантрахи-нон) 549, 556, 581, 844
Хинальдин 280
Хингидронный электрод 230
Хинизарин (1,4-Диоксиантрахинон) 588
Хинин солянокислый 629
Хлор 807 сл.
определение 811 сл.
в бромиде натрия 810
весовое 811 сл.
в карбонатных породах 1061
в силикатных породах 1017
в стекле 807
методом Мора 813
объемное 813
осаждением AgCl и титрованием роданидом 813
осаждение
в виде хлорида серебра 808, 811 сл.
хлоридом палладия 809
отделение 808 сл.
от брома 809, 810
от иода 808
содержание в горных породах 807 Хлорамин (Натриевая соль паратолуол-сульфонхлорамида) 449 Хлораниловая кислота 648 Хлориды
превращение в окислы 160 в нитраты, карбонаты 160
разложение нерастворимых в воде 806
Хлорная кислота 60, 64, 211
действие на стекло 735
Хлороплатиновая кислота 67
Холин солянокислый 864
Хром 589 сл.
окисление до шестивалентного 589 сл.
Хром
определение 592 сл.
весовыми методами 597, 978
в минералах 590 сл.
в осадке от аммиака 120
в силикатных породах 978
дифенилкарбазидом 596
колориметрическими методами 592, 595, 597, 979
комплексоном III 158, 597
объемными методами 592 сл., 597
по Гиллебранду 978
титрованием сульфатом железа (II)
и перманганатом 592
— тиосульфатом натрия 597
хроматным методом 595, 597 осаждение .
аммиаком 596
в виде хромата бария 597
— — хромата ртути (I) 591, 597
— — хромата свинца 591
— — хромата серебра 597
едким натром 109, 590, 978
пиридином 111
сульфидом аммония 978
отгонка хлористого хромила 497, 591
отделение 497, 538 сл., 590, 978
поведение в общем ходе анализа 589 разложение хромсодержащих мине-
ралов 589
Хроматографический анализ 183
Хроматография на бумаге 184
Хромит, разложение 590
Хромовая кислота, титрование 196, 218
Хромотроповая кислота 659
Цезий 729 сл.
определение
в виде сульфата 743
— — хлороплатината 740
осаждение кремневольфрамовой
кислотой 740, 741
отделение
от калия 740, 742
от натрия 733, 734
от рубидия 740
Цементация 412
Центрифуга 51
Цериевая группа 630
Церий см. также Редкоземельные элементы
обнаружение 630
окисление до четырехвалептного 629
окись 219
определение
бруциновым методом 633
в виде йодата церия 626
весовое 626
галловой кислотой 634
йодометрическое 626
колориметрическими методами 633 объемными методами 626, 629 персульфатным методом 633
1110
Предметный указатель
Церий
определение
пирогаллолом 634
титрованием перманганатом 629
четырехвалентного потенциометрическим титрованием 629
осаждение
в виде основного ацетата 103
— — основного бромата 627
йодатом калия 626
отделение от редкоземельных элементов 627 сл.
сульфат (IV) 219
Цериметрия 219
в растворах хлорной кислоты 220 Цианамид кальция, определение азота 868
1,2-Циклогександиондиоксим 423
Цинк 478 сл.
амальгамирование 136, 142, 686
окись 108, 458, 470, 503, 627
определение 481 сл.
бензоином 492
в виде роданомеркуриата 486
— — сульфида 481, 490
— — фосфата 485
весовые 481 сл.
в силикатных породах 961
гексацианоферратным методом 489 дитизоном 156, 492 колориметрическое 491
малых количеств 492
нефелометрическое 492
объемными методами 481, 487, 489, 491
оксихинолином 492
титрованием сульфидом натрия 491
электролитическое 491
осаждение
сероводородом 479, 481
сульфатом акридина 492
отделение 479 сл.
от галлия 554
от кадмия 297
от марганца 496
от меди 479, 481
от никеля 457
от олова 481
поведение в общем ходе анализа 479
разложение цинковых минералов 479
хлорид 76
Цинковые минералы, разложение 479
Цинковые обманки, анализ 537, 544
Цинковые руды 480, 489
Цинк-уранил-ацетат, приготовление раствора 749
Цинхонин 678, 766, 768, 770
Циркониевые руды, разложение 636 сл.
Цирконий 635 сл.
обнаружение
ализариновым красным S 646
в виде манделята 646
— — селенита 644
— — фосфата 640
Цирконий
обнаружение
в высоколегированных сталях 639
весовыми методами 636, 640 сл., 971
в силикатных породах 971
• п-диметиламиноазофениларсиновой кислотой 636
определение 640 сл.
2,4- дисульфобензаурин-3,3 '-дикарбоновой кислотой 635
колориметрическими методами 648
комплексометрическим титрованием 648
купфероном 644
по Гиллебранду 971
тороном 649
хлораниловой кислотой 648
осаждение
аммиаком 646
в виде арсената 642
— — ортофосфата 889
— — основного ацетата 105
— — фосфата 638, 640, 645
карбонатом свинца 101
купфероном 115, 143, 640, 643, 973
миндальной кислотой 646
n-оксифениларсоновой кислотой 156
перекисью водорода 973
салициловой кислотой 653
селенистой кислотой 644
слабыми органическими основаниями 111
таннином 639
тетраэтилфосфатом 640
фениларсоновой кислотой 638
фенилгидразином 154, 646
фталевой кислотой 647
отгонка тетрахлорида 635
отделение 638 сл.
ионообменным способом 635
от гафния 635
от железа и алюминия 973
от тантала и ниобия 673 сл.
от титана 652, 973
от тория 607
поведение
в обычном ходе анализа 618
при анализе силикатов 636
разложение циркониевых руд 636 сл.
хроматографическое отделение от гафния 635
Чашки платиновые 54
Шведские руды, разложение 523
Щавелевая кислота, титрование 196
Щелочноземельные металлы 693 сл.
определение 701 сл.
в карбонатных породах 1057
в силикатных породах 963
отделение 694 сл., 747
в виде оксалатов 696
Предметный указатель
1111
Щелочноземельные металлы
в силикатных породах
в виде сульфатов 697
Щелочные металлы 729 сл.
определение 731 сл.
в карбонатных породах 1060
в силикатных породах 1004 сл.
по Берцелиусу 1005, 1011 сл.
хлоридов по Лоуренсу Смиту, 730, 737, 1005, 1006
осаждение
амиловым спиртом 717
спирто-эфирнои смесью 717, 738
хлорной кислотой и хлористым водородом 737
отделение от магния 717, 718, 730. превращение хлоридов в перхлораты 737
разделение хлороплатинатным методом 731 сл., 747
растворимость перхлоратов 735
Щипцы тигельные 47
ЭДТА см. Этилендиаминтетрауксусная кислота 157
Экстрагирование купфератов 147
Экстракторы 57
Роте 163
Фридриха 164
Экстракция эфиром
бромидов индия 548
хлоридов ванадия 509, 512
— галлия 553
— германия 348
— железа 161, 453, 460, 512, 525
— молибдена 360
— таллия 539
Электроды 55, 230, 254
Электролиз
внутренний 165, 167 сл., 273, 285, 298, 301, 343
из растворов смеси азотной и фтористоводородной кислот 93
с контролируемым потенциалом 165
с ртутным катодом 166, 512
Электролитические определения кадмия 300 кобальта 471, 473 меди 285, 286 олова 343 рения 375, 380 ртути 254 свинца 262, 264 серебра 240 сурьмы 328
Электролитическое осаждение 93, 164 сл.
ванадия 510, 511
индия 545
меди 95, 284
платины 417
свинца 260, 264
серебра 240
Электроприборы лабораторные 52
Эмиссионный спектральный анализ рентгеновский 181
Эрбиевая подгруппа 630
Эрбий, спектрофотометрическое определение 630
Эриохромцианин R 579
Этилендиаминтетрауксусная кислота 157 — — двунатриевая соль 157, 611, 647, 712, 728
04.05.05
Отсканировал <TXEMON
E-maiC: Demon_L@pocfita.ru
Редактор Л. Н. Одерберг
Техн, редактор В. В. Коган
Подписано к печати 11/VIII 1966 г. Уч.-изд. л. 89,0. Формат 70 X 1001/Ц. Усл. печ. л. 89,66. Бумага JSB 2.
Тираж 10 000 экз. с. 1112. Цена 6 р. 58 к. Заказ 522.
ЕЗ И 20- 1966 г. пор. N 8.
Ленинградская типография Ki 14 «Красный Печатник» Главполиграфпрома Комитета по печати при Совете Министров СССР.
Московский проспект, 91.