/


































































































































































Текст
Маргарита Николаевна Храмкина
ПРАКТИКУМ ПО ОРГАНИЧЕСКОМУ СИНТЕЗУ
Редактор издательства В И Поз и на
Технический редактор Ф Т Черкасская
Корректор Б Н Тамаркина
ИБ № 668
Поди в печ с матриц 21 02.77. Формат бума! и Ы)Х90’/1г Бумага
тип N 3 Уел печ л 20,0 Уч-изд л 4,41 Тираж 11 500 экз Зак 543
Изд К 13 5 Цеиа 85 коп
Издате 1Ь(_тво «Химия» Ленинградское отделение 191186,
Ленинград Д 186, Невский пр , 28
Ордена Трудового Красного Знамени Ленинградская типография У» 2
имени Евгении Соколовой Союзполиграфпоома при Государственном
комитете Совстт Министров СССР по делам издательств полиграфии
и книжной торговли 198052, Ленин! рад Л 52, Измайловский проспект 29
М. Н. ХРАМКИНА
ПРАКТИКУМ
ПО ОРГАНИЧЕСКОМУ
СИНТЕЗУ
Издание четвертое, исправленное
Под редакцией до кт. хим. наук
проф. X. В. Вальяна
ЛЬпущено Министерством высшего и среднего
специального образования СССР в качестве
учебного пособия для химических техникумов
ИЗДАТЕЛЬСТВО «ХИМИЯ»
Ленинградское отделение
1977
547
Х89
УДК 547 (076.5)
Храмкина М. Н.
Х89 Практикум по органическому синтезу. Изд.
4-е, испр. Л., «Химия», 1977.
320 стр., 97 рис.
В книге подробно описаны синтезы органических соедине-
ний, аппаратура, применяемая в лаборатории органического син-
теза, обшие приемы и методы лабораторной работы. Приводятся
основные сведения по технике безопасности.
Книга является учебным пособием по органическому син-
тезу для учащихся химических техникумов. Она может быть
полезна также работникам заводских лабораторий.
20504—197
Х 050(01)-77
Б 3-88-10-76
547
© Издательство «Химия», 1974.
Оглавление
Предисловие ....................................................... 8
ЧАСТЬ I
МЕТОДЫ РАБОТЫ ПРИ ПРОВЕДЕНИИ ОРГАНИЧЕСКОГО СИНТЕЗА
Глава I. Организация работы и техника безопасиости................. 9
1. Общие правила работы в лаборатории органического синтеза ... 9
2. Меры предосторожности и первая помощь при несчастных случаях 10
Работа с ядовитыми и едкими веществами.................... 10
Работа с легковоспламеняющимися и взрывоопасными веществами 12
Правила обращения со стеклом...............................12
Первая помощь при ожогах, отравлениях и других несчастных слу-
чаях ....... ............................................. 13
Тушение местных загораний и горящей одежды..........-... 13
3. Основная лабораторная химическая посуда...................14
4. Сборка приборов...........................................22
5. Мытье и сушка химической посуды...........................23
6. Пользование литературой и правила составления отчета .... 25
Глава II. Основные операции при работе в химической лаборатории . . . 28
1. Нагревание.................................................28
2. Охлаждение.................................................31
3. Измерение и регулирование температуры.....................31
4. Измельчение и перемешивание................................32
5. Растворение и свойства некоторых органических раствори гелей . . 35
Этиловый спирт.............._......................................36
Метиловый спирт........................................... ..... 39
Диатиловый эфир .... .... 39
Петролейный эфир . . . .........41
Ацетон........................... . ...........41
6. Сушка и основные осушители ... 42
Осушивание газов...................... . ..............42
Высушивание органических жидкостей ................43
Высушивание твердых веществ .... ... . . 44
Основные осушители........................ . . .... 45
Г. Фильтрование . . . ............................................. 46 -
Фильтрование при обычном давлении . . ..................47
Фильтрование под вакуумом..........................................49
3
Глава III. Методы очистки органических веществ........................51
1. Кристаллизация . ........................................... 51
Выбор растворителя............................................51
Проведение перекристаллизации................................ 53 .
Отделение кристаллов ........................................ 54
2. Возгонка (сублимация)....................................... 55
3. Экстракция...................................................56
4. Перегонка................................................ . 60
Простая перегонка при атмосферном давлении...................60
Перегонка с водяным паром.................................... 62
Перегонка при пониженном давлении.............................66
Фракционная (дробная) перегонка...............................70
Ректификация..................................................72
5. Хроматография................................................74
Адсорбционная хроматография.................................. 74
Распределительная хроматография...............................77
Хроматография на бумаге • . ................................. 78
. Ионообменная хроматография . ....................................80
Глава IV. Определение важнейших констант органических соединений ... 80
1. Температура плавления.........................................81
2. Температура кипения..........................................83
3. Относительная плотность......................................85
4. Показатель преломления.......................................88
5. Молекулярный вес............................................ 89
Глава V. Работа со сжатыми и сжиженными газами....................90
1. Газовые баллоны и обращение с ними...........................90
2. Дозирование газов............................................93
3. Очистка и введение газов в прибор............................94
4. Правила техники безопасности при работе с газовыми баллонами 95
Глава VI. Количественный элементный анализ органических веществ . . 96
1. Определение углерода и водорода полумикрометодом.............96
Сборка установки ............................................ 97
Выполнение анализа...........................................100
2. Определение азота полумикрометодом (по Дюма).............. . 102
Сборка установки ........................................... 102
Выполнение анализа ... 104
3. Определение углерода и водорода микрометодом................105
Сборка установки . ..........................................106
Выполнение анализа ..........................................110
ЧАСТЬ II
СИНТЕЗЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
Глава VII. Реакции галогенирования.......................... ; 113
1. Замещение гидроксильной группы спирюв галогеном...........113
2. Замещение гидроксильной группы кислот галогеном...........115
3. Присоединение галогена по кратной связи.....................115
4. Прямое замещение водорода галогеном........................116
5. Примеры синтезов............................................117
Бромистый этил.............................................. 117
а-Бромнафталин и бромистый этил...............................119
Йодистый этил................................................122
Бромистый бутил ........................................... 124
4
' Хлористый ацетил ..........................................126
Хлористый бензоил . 128
1,2- Дибромэтан............................................ 129
Бромбензол . . ........................................... 132
а-Бромнафталин .............................................135
и-Броманизол . ............................................ 137
Глава VIII. Реакции алкилирования..................................* 138
1. Алкилйрование ароматических углеводородов спиртами в присут-
ствии серной кислоты...........................................139 »
2. Получение простых эфиров...................................139
3. Примеры синтезов...........................................141
втор-Бутилбензол............................................141
Дибутиловый эфир............................................142
Изоамиловый эфир.......................................... 143
Дифениловый эфир........................................... 145
Фенетол.....................................................146
Этиловый эфир Р-нафтола (неролин новый, бромелин)...........147
Анизол......................................................148
Глава IX. Реакции ацилирования ....... i ... ;........150
1. Ацилирование спиртов и аминов карбоновыми кислотами .... 150
2. Ацилирование спиртов, фенолов и аминов хлорангидридами кислот 153
3. Ацилирование спиртов, фенолов и аминов ангидридами кислот . . 154
4. Примеры синтезов..........................................155
Уксусноэтиловый эфир.......................................155
Уксусноизоамиловый эфир....................................157
Этиловый эфир хлоруксусной кислоты.........................159
Диэтиловый эфир щавелевой кислоты.............................• 160
Этиловый эфир бензойной кислоты............................163
Бензанилид . . 164
Аспирин (ацетилсалициловая кислота) ............................ 165
Р-Нафтилацетат.............................................167
Ацетанилид . ....................................................168
Глава X. Реакции Фриделя — Крафтса .г................................170
1. Алкилирование ароматических соединений.....................171
2. Ацилирование ароматических соединений......................172
3. Примеры синтезов......................................... 174
Изопропилбензол ........................................... 174
Дифенилметан .............................................. 176
Ацетофенон .................................................177
Бензофенон..................................................180
Глава XI. Реакции окисления ............................................182
1. Окисление по двойной связи....................................183
2. Окисление первичных и вторичных спиртов до альдегидов или ке-
тонов ........................................................ 183
3. Окисление альдегидов и кетонов до кислот............184
4. Окисление метильных и метиленовых групп..............185
5. Получение хинонов окислением..................................185
6. Примеры синтезов . .........................................185
Ацетальдегид......................................... .. ... 185
Пропионовый альдегид.......................................190
Изовалериановый альдегид . 191
Бензофенон..................................................194
Изомасляная кислота.........................'..............195
Валериановая кислота . .................................... 196
$
Бензойная кислота .......................................... 198
Бензохинон . ................................................200
Антрахинон...................................................201
Глава XII. Реакции нитрования........................................203
1. Нитрование углеводородов жирного ряда.................. 204
2. Нитрование углеводородов ароматического ряда................204
3. Примеры синтезов . ........................................ 206
Нитрометан...........................:.......................206
Нитробензол . ...............................................208
о- и п-Нитротолуол...........................................210
о- и и-Ннтрофенол............................................212
а-Нитронафталин..............................................215
Глава XIII. Реакции аминирования.....................................217
1. Получение "аминов жирного ряда ...... . 217
2. Получение аминов ароматического ряда . ... 218
3. Примеры синтезов.......................................... 220 '
Метиламин ... ... .......... ... 220'
Анилин .................................................... 222
о- и п-Толуидин............................................ 226
а-Нафтпламин............................................. . . 229
Глава XIV. Реакции сульфирования.................................231
1. Сульфирование ароматических соединений......................232
2. Примеры синтезов . ..........................................234
Р-Нафталинсульфокислота (натриевая соль).....................234
Бензолсульфокислота (натриевая соль)....................... 237
и-Толуолсульфокислота........................................239
Сульфаниловая кислота ...................................... 241
Глава XV. Реакции диазотирования и азосочетаиия......................244
1. Реакции солей диазония, сопровождающиеся выделением азота . . 246
2. Реакции солей диазония, идущие без выделения азота..........248
3. Примеры синтезов..................................... ..... 249
Фенол............................................... ..... 249
Иодбензол .................................................. 252
Гелиантин ............................................... . . 254
Р-Нафтолоранж ...............................................257
Глава XVI. Реакции Гриньяра 258
1. Получение углеводородов.....................................260
Количественное определение активного водорода по Чугаеву — Це-
ревитннову........................ ... . . 261
2. Получение карбоновых кислот................................ 263
3. Получение спиртов...........................................264
4. Примеры синтезов............................................265
Фенилуксусиаи кислота ...................................... 265
Трифенилкарбинол ........................................... 268
Дифеннлкарбинол (бензгидрол).................................270
Глава XVII. Реакция Канниццаро.......................................273
Синтез бензойной кислоты и бензилового спирта .............. 273
Глава XVIII. Реакция Клайзена........................................276
Примеры синтезов.............................................278
Ацетоуксусный эфир......................................... 278
Бензоидацетоц.................................... . . . . 28Q
6
Глава XIX. Реакции полимеризации и поликонденсации..................282
1. Полимеризация........................ . . ............282
2. Поликонденсация.............................................285
3. Примеры синтезов........................................... 289
Паральдегид . 289
Полистирол...................................................290
Полиметнлметакрилат..........................................292
Сополимер стирола с метилметакрилатом ...................... 294
Метилметакрилат (из полиметнлметакрилата)....................295
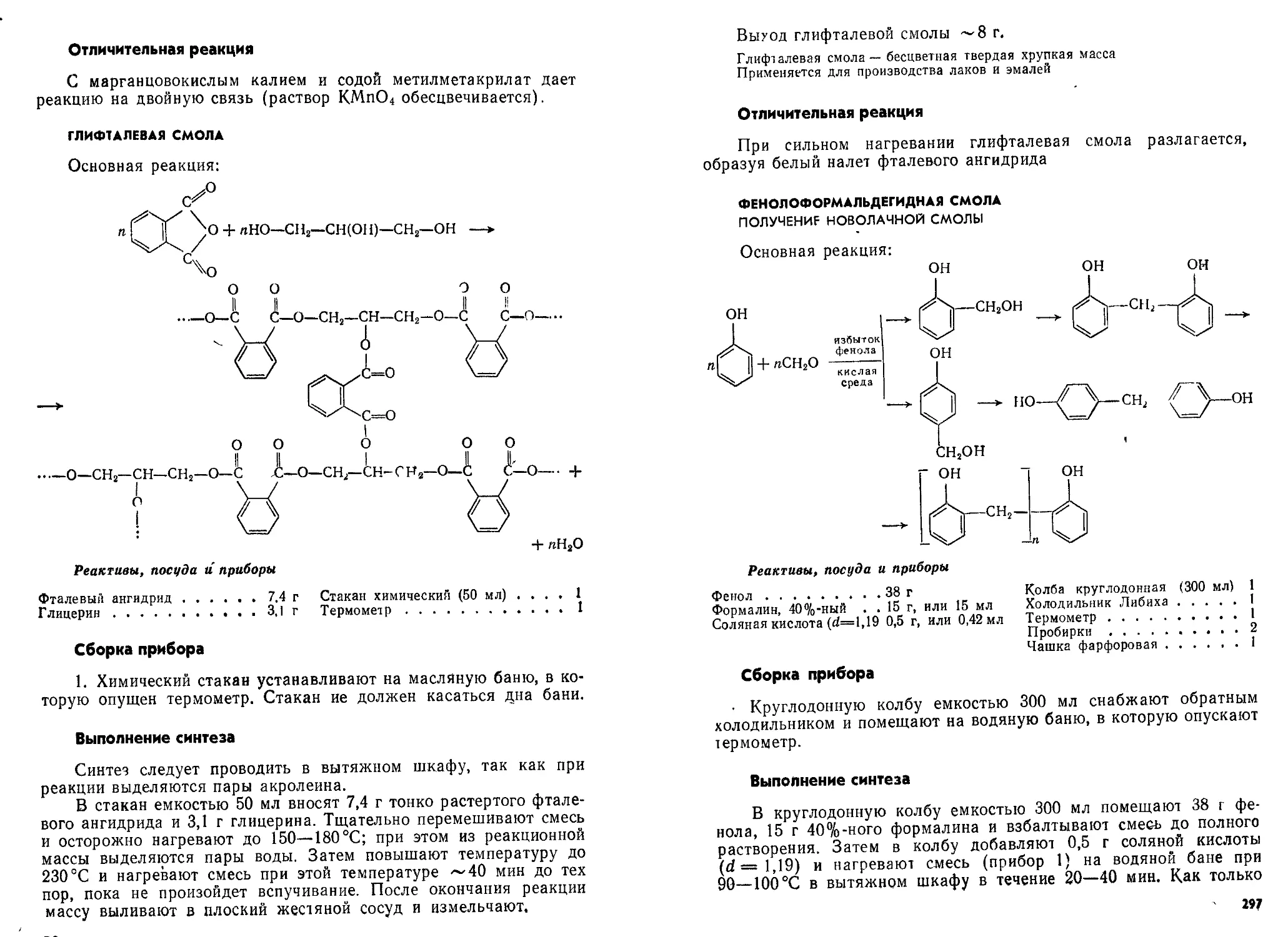
Глифталеваи смола............................................296
Феноло-формальдегндная смола.................................297
Глава XX. Идентификация ; 300
1. Цредварительные испытания.................................300
2. Качественные реакции ... 303
3. Получение производных.................................. 305
Рекомендуемая литература..........................................309
Приложения........................................................310
I. Осушители для органических соединений....................310
2. Давление пара воды при различных температурах...........310
3. Давление сжиженных газов в баллонах.....................311
4. Цвет баллонов со сжатыми газами . ......................311
5. Плотность растворов серной кислоты (20 °C)..............311
6. Плотность растворов соляной кислоты (20 °C)............ 313
7. Плотность растворов азотной кислоты (20 °C)............ 313
8. Плотность растворов едкого натра (20 °C) 315
9. Плотность растворов едкого кали (20 °C)................ 315
10. Физические свойства спиртов и их производных............ 317
11. Физические свойства фенолов и их производных.............317
12. Физические свойства альдегидов и их производных......... 318
13. Физические свойства кетонов и их производных.............318
14. Физические свойства карбоновых кислот и их производных . . 318
15. Физические свойства первичных н вторичных аминов и Их произ-
водных .....................................................319
16. Физические свойства алкилгалогенндов и их производных .... 319
Предисловие
Учебной программой практических занятий по органи-
ческому синтезу предусматривается изучение учащимися тех-
никумов основных методов синтеза органических веществ,
применяемых в заводской и лабораторной практике, приоб-
ретение ими навыков для работы в лабораториях предприя-
тий и научно-исследовательских учреждениях.
При выполнении работ по органическому синтезу уча-
щиеся техникумов должны практически ознакомиться с ос-
новными химическими реакциями, приобрести определенные
экспериментальные навыки.
В книге дано краткое (изложение современных теорети-
ческих представлений органической химии в той мере, в ка-
кой это необходимо для сознательного проведения синтеза.
Для более подробного изучения теоретических вопросов, в
частности касающихся механизмов реакций, следует обра-
титься к учебникам по органической химии.
В учебном пособии приводится большое количество син-
тезов, причем наибольшее' внимание уделяется лабораторным
работам, имеющим практическую и методическую ценность.
В случае необходимости можно заменить одну лабораторную
работу данной темы другой, сходной по характеру, примени-
тельно к будущей специальности учащихся. Кроме того, боль-
шое число синтезов позволит преподавателю выбрать зачетные
синтезы для выяснения качества усвоения материала.
При составлении учебного пособия были использованы
некоторые руководства по органическому синтезу.
В третье издание учебного пособия включены новые ма-
териалы по механизму отдельных реакций и введена глава
по идентификации органических соединений.
8
МЕТОДЫ РАБОТЫ
ПРИ ПРОВЕДЕНИИ
ОРГАНИЧЕСКОГО СИНТЕЗА
ЧАСТЬ
Глава I
ОРГАНИЗАЦИЯ РАБОТЫ
И ТЕХНИКА БЕЗОПАСНОСТИ
Приступая к работе по органическому синтезу, в первую оче-
редь необходимо твердо усвоить правила работы в лаборатории
органического синтеза, правила техники безопасности, знать меры
предупреждения и предотвращения несчастных случаев, помнить,
что беспорядочность, поспешность, неряшливость могут привести
к порче работы и даже к несчастным случаям.
1. ОБЩИЕ ПРАВИЛА РАБОТЫ
В ЛАБОРАТОРИИ ОРГАНИЧЕСКОГО СИНТЕЗА
1. Во время работы" в химической лаборатории соблюдайте
тишину, порядок, чистоту, рационально стройте свою работу, ве-
дите ее точно, аккуратно, быстро, но без спешки.
2. Не приступайте к работе без разрешения преподавателя
или лаборанта.
3. Запрещается работать в лаборатории одному.
4. Каждый учащийся должен работать на закрепленном за
ним месте или на специально отведенном для данной работы.
5. Содержите рабочий стол в чистоте и не загромождайте
его.
6. Экономьте газ, воду, электричество, реактивы.
7. Во время работы надевайте халат; имейте в лаборатории
мыло, полотенце.
8. Аккуратно и осторожно обращайтесь с химической посу-
дой, реактивами и приборами. Во избежание несчастных случаев
из-за возможного выброса реакционной смеси не заглядывайте в
пробирку или колбу сверху.
9. Не работайте с грязной посудой, не оставляйте ее не-
мытой.
10. Не пускайте ни одного прибора без предварительной про-
верки. Не оставляйте действующий прибор без присмотра.
Ц, Не взносите из лаборатории приборы, посуду и реактивы.
12. Работу с ядовитыми веществами проводите в вытяжном
шкафу.
13. Соблюдайте меры предосторожности при работе с взрыво-
опасными и легковоспламеняющимися веществами.
14. Не выливайте в раковины остатки кислот, щелочей, огне-
опасных жидкостей и т. д. Сливайте эти вещества в специальные
склянки, помещенные в вытяжном шкафу. Не бросайте в раковину
бумагу, песок и другие твердые вещества.
15. Растворы, содержащие концентрированные кислоты и ще-
лочи, перед тем, как выливать в канализационную систему, ней-
трализуйте. Остро пахнущие и ядовитые вещества должны быть
обезврежены химической обработкой или сожжены в специально
отведенном месте вне пределов лаборатории, желательно на воз-
духе.
16. Не оставляйте никаких веществ в посуде без этикеток.
17. Работайте с кислотами и щелочами только на столах со
специальным покрытием.
18. Не путайте пробки от склянок, содержащих разные реак-
тивы, во избежание загрязнения последних.
19. При взвешивании сухих реактивов не высыпайте их прямо
на чашку весов.
> 20. Запрещается курить и принимать пищу в лаборатории.
21. При возникновении пожара немедленно выключите газ во
всей лаборатории, уберите из помещения все горючие вещества,
засыпьте песком или закройте одеялом очаг пожара и сообщите
дежурному пожарной охраны о случившемся. Усвойте и соблю-
дайте правила противопожарной безопасности.
22. Уходя из лаборатории, проверьте, выключены ли газ, вода
И электричество.
2. МЕРЫ ПРЕДОСТОРОЖНОСТИ И ПЕРВАЯ ПОМОЩЬ
ПРИ НЕСЧАСТНЫХ СЛУЧАЯХ
При работе в лаборатории органического синтеза необходимо
строго соблюдать все меры предосторожности; малейшая неосмот-
рительность может привести к тяжелым последствиям.
РАБОТА С ЯДОВИТЫМИ И ЕДКИМИ ВЕЩЕСТВАМИ
Большинство химических соединений более иди менее ток-
сичны. Поэтому каждый приступающий к синтезу должен хорошо
знать токсические свойства тех веществ, с которыми он работает,
и, если возникает необходимость, уметь оказать помощь постра^
давшему.
При работе с ядовитыми и едкими веществами следует со-
блюдать следующие правила:
1. Иметь в помещении противогаз, защитные лицевые щитки,
предохранительные очки,
2. Работу с особо опасными веществами (бром, концентриро-
ванные кислоты и др.) проводить под наблюдением преподавателя
или лаборанта.
3. Все работы с ядовитыми и горючими газами и парами сле-
дует проводить в вытяжном шкафу.
Во время работы в вытяжном шкафу:
а) не закрывать плотно дверцы и оставить их на указанном
преподавателем уровне;
б) не влезать в вытяжной шкаф с головой;
в) не разбирать прибор, пока из него под тягой не будет вы-
теснен водой или воздухом ядовитый газ или пары;
г) работая с хлором, бромом и другими ядовитыми веще-
ствами, иметь наготове противогаз.
4. Измельчение, едких щелочей, натронной извести, иода,
хромпика, солей анилина проводить в вытяжном шкафу и наде-
вать защитные лицевые щитки или очки.
5. При обращении с концентрированными кислотами, олеумом
и аммиаком следует соблюдать следующие меры предосторож-
ности:
а) разливать указанные жидкости только через воронку и
под тягой;
б) при разбавлении концентрированной серной- кислоты вли-
вать кислоту порциями в воду и слегка перемешивать;
в) не разбавлять олеум водой;
г) при растворении концентрированной серной кислоты в
воде, при изготовлении хромовой смеси, при смешивании концен-
трированных серной и азотной кислот можно пользоваться только
тонкостенной химической посудой.
6. Не вливать горячих жидкостей в толстостенную посуду и
приборы.
7. Работать с бромом следует в вытяжном шкафу, при этом
необходимо:
а) остерегаться вдыхать его пары, беречь глаза, руки;
б) при наливании брома тщательно снимать каплю с края
горлышка склянки о край сосуда;
в) при переливании больших количеств брома надевать рези-
новые перчатки.
8. При работе с металлическим натрием необходимо соблю-
дать особую осторожность:
а) для защиты лица и головы следует применять очки и
экраны из органического стекла;
б) хранить натрий нужно под слоем керосина в банке, закры-
той корковой пробкой;
в) резать натрий следует только на сухой бумаге;
г) не бросать обрезки натрия в раковину и не оставлять их
на столе открытыми, а сразу же собирать в банку с керосином;
д) брать натрий только пинцетом или щипцами;
е) не сушить натрием галогенпроизводные жирного ряда, если
они не находятся в растворе;
11
Ж) не допускать соприкосновения натрия с водой и четырех-
хлористым углеродом;
з) небольшие остатки непрореагировавшего натрия уничто-
жают добавкой небольшими порциями бутилового или амилового
спирта;
и) не проводить реакций с металлическим натрием на кипя-
щей водяной бане, а применять в этих случаях песочную или мас-
ляную баню.
РАБОТА С ЛЕГКОВОСПЛАМЕНЯЮЩИМИСЯ
И ВЗРЫВООПАСНЫМИ ВЕЩЕСТВАМИ
В препаративной органической химии часто приходится поль-
зоваться огнеопасными растворителями, такими как эфиры,
спирты, бензол, ацетон и т. п. Работать с этими веществами нужно
особенно осторожно, так как их пары могут легко воспламениться.
Необходимо помнить и выполнять следующее.
1. Не держать эти растворители вблизи огня, в теплом месте
или вблизи нагревательных приборов.
2. Не нагревать их на открытом огне, на сетке, вблизи огня
или в открытых сосудах, а только на водяной бане с обратным
водяным холодильником.
3. Не хранить в тонкостенной посуде с плотно закрытой проб-
кой.
4. Не выливать в раковину.
5. Не держать на рабочем месте больших их количеств.
6. Перегонять только на водяной бане с водяным холодильни-
ком на специальном лабораторном столе. При работе с эфиром
нагревание водяной бани должно быть произведено вдали от ме-
ста перегонки.
7. Не перегонять долго хранившийся эфир без предваритель-
ной проверки на присутствие перекисей.
8. Если в лаборатории по какой-либо причине оказалось про-
лито значительное количество легковоспламеняющейся жидкости,
то необходимо погасить все горелки и электронагревательные при-
боры, открыть окна и собирать пролитую жидкость тряпкой или
полотенцем.
ПРАВИЛА ОБРАЩЕНИЯ СО СТЕКЛОМ
1. При разламывании надрезанных напильником стеклянных
трубок или палочек стараться как бы разорвать трубку, чтобы не
порезать руки краями стекла.
2. Вставляя стеклянную трубку, холодильник, капельную во-
ронку, термометр в пробку, нужно держать их рукой как можно
ближе к вставляемому концу и не вдавливать в пробку, а слегка
ввинчивать. В случае применения резиновых пробок следует сма-
зать их глицерином, а затем избыток глицерина снять.
<2
3. Не нагревать толстостенную посуду.
4. Осторожно работать при стеклодувных работах, беречь
глаза.
ПЕРВАЯ ПОМОЩЬ ПРИ ОЖОГАХ, ОТРАВЛЕНИЯХ
И ДРУГИХ НЕСЧАСТНЫХ СЛУЧАЯХ
1. При легких термических ожогах кожу следует обмыть
спиртом, а затем смазать глицерином или вазелином. При более
сильных ожогах обожженное место после обмывания концентриро-
ванным раствором перманганата калия и спиртом необходимо
смазать мазью от ожогов (например, сульфидиновой эмульсией).
2. При ожогах бромом нужно тщательно обмыть пораженное
место бензолом или спиртом, а затем 10% раствором тиосульфата
натрия. После вдыхания паров брома следует понюхать разбав-
ленный раствор аммиака и выйти на свежий воздух.
3. При ожогах жидким фенолом необходимо растирать побе-
левший участок кожи глицерином до тех пор, пока не восстано-
вится нормальный цвет кожи, затем промыть пораженный участок
водой и наложить компресс из ваты или марли, смоченной глице-
рином.
4. При ожогах крепкими кислотами требуется немедленно
обмыть обожженное место большим количеством воды, а затем
3% раствором соды или нашатырного спирта.
5. При ожогах крепкими щелочами кожу надо промыть водой,
а затем нейтрализовать 1 % раствором борной кислоты. Аммиак
почти не действует на кожу, однако при попадании в глаза может
вызвать сильное повреждение и даже слепоту.
6. При случайном попадании реактивов внутрь рекомендуется
выпить побольше воды. Наряду с этим необходимо: а) при отрав-
лении кислотами выпить стакан 2%-ного раствора двууглекислой
соды, б) при отравлении щелочами выпить стакан 2%-ной уксус-
ной или лимонной кислоты.
7. При отравлении необходимо вывести пострадавшего на
свежий воздух, сделать искусственное дыхание и вызвать врача. •
8. При неосторожном изгибании трубок, вставлении трубки
или термометра в отверстие колбы, возможны порезы и ранения.
При порезах в первую очередь нужно удалить из раны осколки,
края раны дезинфицировать 3% спиртовым раствором иода, а
затем наложить стерильную повязку. При сильных кровотечениях
следует наложить выше раны жгут и вызвать врача или направить
пострадавшего в амбулаторию (поликлинику).
ТУШЕНИЕ МЕСТНЫХ ЗАГОРАНИЙ
И ГОРЯЩЕЙ ОДЕЖДЫ
1. В случае воспламенения горючей жидкости следует пога-
сить все горелки, прикрыть пламя асбестовым полотенцем или
засыпать его песком, или воспользоваться огнетушителем с угле-
кислым газом.
13
2. Растворимые в воде огнеопасные вещества, такие как
спирт, ацетон и другие, можно тушить водой.
3. Если горит нерастворимое в воде вещество (например,
эфир, бензол, бензин, скипидар), то воду применять для тушения
пожара нельзя, так как он не только не будет ликвидирован, но
даже может усилиться. В этом случае пламя следует тушить пес-
ком или использовать огнетушитель.
4. В случае воспламенения одежды не следует бежать; надо
набросить на пострадавшего халат, пиджак, брезент, шерстяное
или войлочное одеяло, которое должно всегда лежать на видно'м
и доступном месте.
3. ОСНОВНАЯ ЛАБОРАТОРНАЯ ХИМИЧЕСКАЯ ПОСУДА
Ответственным моментом в подготовке к синтезу является
подбор соответствующей посуды и сборка прибора, которая долж-
на проводиться с особым вниманием, тщательностью и осторож-
ностью, в противном случае может произойти потеря части ве-
щества, а также пожар или взрыв. Перед тем как приступить к
сборке прибора, необходимо
П Н П подготовиться к выполнению
а 5 6 где
Рнс. 1. Колбы:
а—узкогорлая круглодонная; б—широкогор-
лая круглодонная; в —со шлифом; г—шнроко-
горлая плоскодонная; д —узкогорлая плоско-
донная; е—коническая.
данной работы, т. е. овладеть
теоретическим материалом, от-
носящимся к данному синтезу,
хорошо усвоить описание ра-
бЬты, ясно представлять ход
предстоящих операций и твер-
до знать правила техники без-
опасности.
Колбы и стаканы — это ос-
новная лабораторная посуда;
наиболее употребительным материалом для их изготовления слу-
жит жаростойкое стекло. Чаще всего приходится проводить син-
тезы в колбах, которые бывают разнообразной емкости и формы.
В тех случаях, когда реакция идет при нагревании реакционной
смеси до кипения, следует пользоваться круглодонными колбами,
так как они устойчивы к толчкам, возникающим при кипении
жидкости. Круглодонные колбы бывают широкогорлые, узкогорлые,
со шлифами и без них (рис. 1). Круглодонные колбы применяют
для проведения в них синтезов, для перегонки при атмосферном
давлении и с водяным паром, а также в качестве приемников при
вакуум-перегонке.
Специальные круглодонные колбы, например перегонные кол-
бы Аншютца и Вюрца (рис. 2), колбы Клайзена, Фаворского,
двух- или трехгорлые колбы (рис. 3) и другие, применяют для
перегонки жидкостей. Колбы Аншютца употребляют при перегонке
быстро затвердевающих веществ, колбы Клайзена — при пере-
гонке в вакууме. Двух- или трехгорлые колбы очень удобны для
одновременного проведения нескольких операций, например, при
14
нагревании с обратным холодильником требуется равномерное пе-
ремешивание реакционной смеси и медленное введение компонен-
тов через капельную воронку. Если же в лаборатории отсутствуют
такие колбы, то употребляют круглодонные колбы с насадками-
форштоссами (рис. 4).
Рис. 2. Перегонные колбы:
о—колба Аншютца: б, в—колбы Вюрца.
в 9 t
Рнс. 3. Специальные круглодонные
колбы:
а—колба Клайзена; б—колба Фаворского;
в—двугорлая колба; г—трехгорлая колба.
Плоскодонные колбы, широкогорлые и узкогорлые (см.
рис. 1,а, д), не используют для работы под вакуумом, так как они
не‘выдерживают наружного давления и разрываются. Не приме-
няют эти колбы также для работы при высоких температурах.
Их следует употреблять для приготовления реактивов, проведения
реакций, проходящих при температурах не выше 100°C, когда не
Рис. 5. Колба Бунзена (а) и
стаканы химические (б, в).
Рнс. 4. Форштоссы:
а, б—двурогие; в—трехрогнй.
нужно изолировать процесс от влияния влаги и воздуха, а также
в качестве приемников при перегонке с водяным паром или при
атмосферном давлении.
Конические колбы (см. рис. 1,е) применяют для кристалли-
зации, приготовления реактивов, проведения простых операций,
когда нет необходимости защищать процесс от доступа влаги и
воздуха, в качестве приемников и для некоторых других целей.
Колбы Бунзена (рис. 5, а) служат для отсасывания под вакуу-
мом и реже в качестве приемников при перегонке ц, вакууме. Они
бывают различной емкости и формы, чаще всего используют
колбы конической формы, так как они наиболее устойчивые.
Колбы Бунзена изготавливают из толстого ртекла, иначе они
1J
могут быть раздавлены атмосферным давлением. Перед работой
новые колбы Бунзена проверяют, выдерживая их под вакуумом в
течение 15 мин. При испытаниях и работе с колбами Бунзена ре-
комендуется завернуть их в полотенце или поместить в металли-
ческую сетку.
Стаканы (рис. 5,6, в) применяют для проведения реакций при
температурах ниже 100 °C. Основным же назначением стаканов
является использование их в качестве вспомогательных сосудов.
Стаканы нельзя употреблять для работы с низкокипящими и огне-
опасными растворителями, так как эти жидкости летучи.
Холодильники (рис. 6) служат для охлаждения и конденсации
паров при проведении реакций с органическими соединениями.
Рнс. 6. Холодильники:
а—воздушный; б—Либиха; в —ша-
риковый, г—змеевиковый.
Рис. 7. Применение воздушного
холодильника:
а—как обратного; б—как нисходящего.
Холодильники называют обратными, когда конденсирующиеся в
них пары возвращаются в реакционную смесь (рис. 7,а). Нисхо-
дящими, или прямыми, их называют в том случае, если при пере-
гонке конденсат из холодильника поступает в приемник (рис. 7,6).
Охлаждающими агентами для холодильников являются воздух
(воздушный холодильник) и вода (водяной холодильник).
Самым простым холодильником является воздушный, который
может применяться как в качестве обратного, так и нисходящего,
причем для жидкостей с температурой кипения 150 °C и выше;
водяные холодильники для таких жидкостей не употребляются,
так как вследствие резкого перепада температур трубка холо-
дильника может лопнуть *.
Широко распространенным холодильником, применяемым в
лаборатории, является холодильник Либиха, который используется
в качестве нисходящего и обратного. Холодильник Либиха состоит
из наружной рубашки, которая припаяна к внутренней трубке
или соединяется с ней при помощи отрезков резиновой трубки.
* Для жидкостей с температур.ой кипения 140—-160 °C можно пользоваться
и водяными холодильниками, но без циркуляции воды,
16
Наружная рубашка имеет два отростка. На эти отростки наде-
вают резиновые трубки, одну из которых присоединяют к водо-
проводному крану, а другую отводят в раковину. При этом надо
помнить следующее правило: если холодильник Либиха исполь-
зуется как нисходящий, то воду подают по принципу противо-
тока, т. е. вода должна двигаться навстречу парам охлаждаемой
жидкости (рис. 8,а). Если же холодильник употребляется как
обратный, то принцип противотока не соблюдается и воду подают
в нижний отросток, чтобы полностью заполнить водой холодиль-
ник и эффективно сконденсировать пары реакционной жидкости
(рис. 8, б, в).
Рис. 8. Применение холодильника Либиха, как нисходя-
щего (а) и как обратного (б, в):
/—внутренняя трубка; 2—соединительные резиновые трубки;
3 — наружная рубашка; 4 — отростки.
В лабораториях часто применяют холодильники и других ти-
пов— шариковый и змеевиковый. Шариковый холодильник
(см. рис. 6, в) чаще всего употребляется как обратный, так как
шаровидные расширения внутренней трубки значительно улуч-
шают его охлаждающее действие. Змеевиковый холодильник
(см. рис. 6, г) всегда применяется только как нисходящий для
низкокипящих веществ. Он никогда не используется как обратный,
так как конденсат, плохо стекающий по сгибам, может быть вы-
брошен из холодильника.
Холодильники могут нормально работать только при постоян-
ном напоре воды, поэтому всегда необходимо следить за тем,
чтобы в холодильнике не прерывался ток охлаждающей воды,
в противном случае может произойти авария.
Капельные воронки (рис. 9) служат для приливания жидкости
к реакционной смеси. Перед работой с капельной воронкой шлиф
стеклянного крана нужно слегка смазать вазелином; это дает воз-
можность легко открывать кран, что очень важно, так как если
кран открывается туго, то можно, открывая, сломать его или
17
повредить весь прибор. После смазывания крана следует прове-
рить, не пропускает ли воронка воду (при закрытом кране).
Дефлегматоры разнообразных конструкций (рис. 10) приме-
няют часто при фракционной перегонке (см. стр. 71). Дефлег-
Рис. 10. Дефлегматоры.
Рис. 9. Капельные
воронки.
Маторы вставляют нижним концом в пробку, укрепленную в
горле колбы, в верхнее отверстие помещают термометр, а отвод-
ную трубку дефлегматора соединяют с холодильником (рис. 11).
Рис. 11. Прибор для фракционной Рис. 12. Хлоркальцневые труб-
' перегонки: кн: 4
/—реакционная колба; 2—дефлегматор; а—простая; б, е—V-образные.
5—термометр; 4—холодильник; 5—алонж;
6—приемник.
Алонж применяют для отвода дистиллята во время перегонки.
Он крепится к холодильнику при помощи пробки.
Хлоркальциевые трубки (рис. 12) используют, если необходимо
реагирующие вещества защищать от действия влаги воздуха.
Хлоркальциевая трубка содержит слой хлористого кальция, в виде
зерен; 1 —1,5 см до верха трубки оставляют свободными, а сверху
и снизу кладут небольшой слой ваты.
1»
Фарфоровая посуда более прочна, чем стеклянная, не боится
сильного нагревания. Изделия из фарфора имеют и недостатки:
они тяжелы, непрозрачны и значительно дороже стеклянных. Из
фарфора делают стаканы (рис. 13,а), выпарные чашки (рис. 13,6),
Рис. 13. Фарфоровая посуда:
а—-стакан; б — выпарная чашка; в — ступка с пестиком;
г —тигель; д—воронка Бюхнера.
ступки, пестики к ним (рис. 13,в), тигли (рис. 13,а), воронки
Бюхнера (рис. 13, д), ложки-шпатели, лодочки для прокаливания,
трубки и др.
Рис. 14. Пробкомялка (а), нож для точки сверл (б) и
набор св^рл (в).
Пробки (корковые и резиновые) применяют для соединения
частей стеклянных приборов и для плотного закупоривания сосу-
дов. Обычные корковые пробки хорошего качества обеспечивают
достаточную герметичность сосуда или прибора, однако надо по-
мнить, что бывшие в употреблении корковые пробки уже загряз-
нены и никогда не удается хорошо отмыть их из-за пористости.
Пробку подбирают таким образом, чтобы ее диаметр был немного
больше диаметра закрываемого отверстия, и входить в отверстие
она должна с небольшим усилием. Чтобы придать корковой
пробке эластичность и несколько уменьшить в диаметре, ее
19
следует обмять специальной пробкомялкой (рис. 14,«). Когда не
удается подобрать пробку, приходится ее подгонять, пользуясь но-
жом или напильником, а иногда тем и другим.
Отверстия в пробках просверливают при помощи ручных
(рис. 14, в) или механических сверл, которые должны быть всегда
хорошо наточены специальным ножом (рис. 14,6). Выбранное
сверло должно быть немного меньшего диаметра, чем диаметр
трубки, которую необходимо вставить в пробку. Перед сверлением
резиновой пробки ее следует слабо нагреть, на водяной бане в
2—3% растворе щелочи, затем обмыть водой и вытереть, а сверло
Рис. 16. Соединение реакционной колбы
с приборами:
а—путем сверления нескольких отверстий
в пробке; б —посредством форштосса.
Рис. 15. Машина для
сверления пробок.
смазать вазелином или обмыть глицерином для уменьшения
трения.
Сверлить пробку нужно не спеша, и обязательно начинать с
ее узкого основания, так как эта часть будет находиться в сосуде
и должна быть ровной. При сверлении в левую руку берут пробку,
а в правую — сверло, причем придают ему такое положение,
чтобы ось его совпала с осью пробки, а полученное отверстие в
пробке должно быть параллельно стенкам горла сосуда, что зна-
чительно облегчит правильную сборку сосуда. Сверлить начинают,
нажимая на пробку и медленно вращая сверло, однако сильно
надавливать на сверло не нужно, так как при этом у корковых
пробок получаются неровные отверстия с рваными краями, а у
резиновых — сильно уменьшенные в диаметре отверстия.
Резиновые пробки удобнее сверлить при помощи специальной
машинки для сверления пробок (рис. 15), пользуясь которой
можно получить ровно просверленное отверстие, что не всегда
удается при ручном сверлении. Сделав отверстие в пробке, сверло
вынимают и находящиеся внутри него остатки пробки выталки-
вают стержнем, который имеется в каждом наборе сверл. Если
при сверлении корковой.пробки получилось слишком узкое отвер-
20
стие, то его можно расширить круглым напильником. Часто при
работе необходимо соединять колбу с несколькими приборами
(холодильником, капельной воронкой, термометром и т. п.), тогда
применяют широкогорлую колбу и в пробке сверлят несколько
отверстий или употребляют форштоссы (рис. 16).
Для придания пробкам большей устойчивости к действию
разъедающих паров или газов (щелочей, кислот, окислов азота,
брома, хлора и т. п.) их надо специально обработать. Корковые
пробки нагревают 15—20 мин при 50°C в растворе, состоящем из
3 ч. желатины, 5 ч. глицерина и 100 ч. воды. Затем пробки высу-
шивают и пропитывают расплавленной смесью из 12 ч. вазелина
и 42 ч. парафина, при этом все время переворачивают их стеклян-
ной палочкой. После такой обработки корковые пробки вынимают
и высушивают.
Если резиновые пробки долго находились в работе, то они
затвердевают, растрескиваются и делаются непригодными к даль-
нейшему употреблению. Поэтому старые резиновые пробки кла-
дут в парафин, нагретый до 100°C, и выдерживают там не более
1 мин. Для обновления старых корковых пробок их обливают го-
рячей водой, затем помещают в смесь из 15 ч. воды и 1 ч. сали-
циловой кислоты, промывают водой и сушат на воздухе.
Необходимо иметь в виду, что корковые и резиновые пробки
очень неустойчивы к действию высоких температур и многих хи-
мических реактивов. Так, резиновые пробки плохо выдерживают
температуры выше 140°С, а под действием бензола, толуола, эфи-
ра, ацетона, галогенпроизводных и других веществ легко набу-
хают и разрушаются, загрязняя при этом продукты реакции. Кор-
ковые пробки неустойчивы к сильным кислотам и щелочам и в
силу своей пористости непригодны для работы в вакууме.
Стеклянные трубки разных диаметров применяют для соеди-
нения отдельных частей прибора. Чтобы разрезать узкую стеклян-
ную трубку, делают надрез на ее поверхности напильником или
ножом для резки стекла, затем, взяв трубку в обе руки, прижи-
мают большие пальцы к противоположной стороне ее против над-
реза и быстрым движением ломают трубку, при этом одновре-
менно слегка растягивая ее. Широкие и толстые трубки не ло-
мают, для их разрезания делают поперечный надрез ножом и к
месту надреза прикладывают нагретую докрасна стеклянную па-
лочку или проволоку, трубка лопается, образуя ровное сечение.
Обрезанные концы трубок должны быть обязательно оплавлены.
Обычно это делают в пламени газовой горелки. Благодаря оплав-
лению устраняется возможность ранения рук, трубка легко и без
повреждения надевается на пробку и не царапает стеклянную
посуду.
При обработке стеклянных трубок в пламени горелки трубки
должны быть совершенно сухими и чистыми. И нагревание, и
охлаждение следует производить постепенно, так как при резком
изменении температуры стекло легко трескается Трубку при по-
стоянном вращении оплавляют сначала в -коптящем, а затем в
21
сильном некоптящем пламени горелки. По окончании нагревания
раскаленную трубку немного охлаждают в коптящем пламени го-
релки и дают остыть, положив ее на асбест.
Для сгибания стеклянных трубок необходимо иметь специаль-
ную насадку для горелки типа «ласточкин хвост» (рис. 17), даю-
щую плоское пламя. Трубку, один конец которой закрыт, напри-
мер кусочком асбеста, равномерно и сильно нагревают на участке
длиной 5—8 см, держа за холодные концы и медленно вращая ее
в пламени горелки. Как только трубка размягчится настолько,
что начнет гнуться сама, ее вынимают из пламени и очень плавно
а 6 8
Рис. 17. Сгибание стеклянной трубки:
а—нагревание на горелке с насадкой; б—пра-
вильно согнутая трубка; в —неправильно со-
гнутая трубка.
сгибают, поднимая концы квер-
ху и вдувая слегка воздух в
трубку. Затем согнутое место
следует нагреть в коптящем
пламени горелки и дать мед-
ленно остыть.
Капилляры очень часто ис-
пользуются в лабораторной
практике. Они бывают двух на-
значений. Одни служат для оп-
ределения температуры плав-
ления органических веществ, а
другими пользуются при нагревании и перегонках жидкостей, что-
бы избежать перегрева и создать условия равномерного нагрева.
. Чтобы получить первые, стеклянную трубку нагревают при
постоянном вращении в плоском пламени. Когда трубка размяг-
чится и начнет в нагретом месте сужаться, ее вынимают из пла-
мени и, не переставая вращать, не очень быстро растягивают.
Как только обработанная трубка остынет, ее разрезают на куски
длиной ~4—5 см, заплавляют тонкий конец и используют капил-
ляры для определения температуры плавления.
Капилляры, используемые по второму назначению, изготов-
ляются аналогично, с той лишь разницей, что размягченную труб-
ку растягивают быстро. Длина капилляров зависит.от величины
нагреваемой колбы. Один конец капилляра также заплавляется.
Капилляры для перегонок под вакуумом изготовляют из спе-
циадьных капиллярных трубок. Их оставляют открытыми с обоих
концов.
4. СБОРКА ПРИБОРОВ
- Прежде чем налить в колбу жидкость или наполнить ее ка-
ким-либо другим веществом, нужно собрать прибор и, только
убедившись в правильности сборки, приступать к выполнению син-
теза. Соединять отдельные части прибора надо осторожно во из-
бежание их поломки. Подгонку пробок и других соединений сле-
дует производить до закрепления прибора в штативе.
В настоящее время часто используют аппаратуру на шлифах
(рис. 18). При работе под вакуумом шлифы следует смазывать
22
жиром, вазелином или специальной вакуумной смазкой, но не
слишком жирно, иначе смазка может попасть в реакционную
смесь или в полученный продукт.
До вставления пробки в колбу необходимо соединить отвер-
стия пробки с соответствующими приборами (капельной ворон-
кой, холодильником и т. п.), причем в случае применения рези-
новой пробки следует предварительно легко смочить ее отверстия
глицерином. Затем пробку соединяют с сосудом, при этом посуду
нельзя ставить на стол или держать за дно. Следует держать со-
суд за горло как можно ближе к тому месту, куда вставляется
пробка, иначе можно поранить руки.
Вынимать и надевать пробку нуж- л
но не слишком энергично, осторож- Jy
но вращая ее.
При сборке прибора приходится /(j/
надевать на стеклянные части рези- . /[(/
новые трубки. Для уменьшения тре- f, /(J/
ния нужно слегка смочить трубку \ \ /г(/
водой или глицерином. Не рекомен- \) Z k
дуется смазывать резиновую трубку Ur
маслом или вазелином, так как по- Wj
следние впитываются резиной, кото- |
рая разбухает и становится менее \Х/
эластичной. При надевании резино-
вой трубки на стеклянную следует Рис- 18‘ Прибор на шлифах,
взять резиновую, трубку у самого
конца и надвигать на трубку не прямо, а несколько сбоку или
снизу. Собирая прибор, надо не только наблюдать за плотностью
н правильностью соединения отдельных его частей, но и строго
следить за тем, чтобы прибор всегда имел сообщение с атмосфе-
рой, во избежание повышения в нем давления в результате нагре-
вания или выделения газов.
После того как собраны основные части прибора, его укреп-
ляют в штативах. Чтобы избежать поломок в собранном приборе,
необходимо всегда обращать внимание на наличие прокладок на
зажимах и захватах лапок. Крепить колбы в зажимах следует
не за середцну горла, а около пробки. Аппаратуру больших раз-
меров нельзя закреплять слишком жестко. Мешалки, дефлегма-
торы необходимо закреплять в строго вертикальном положении.
После окончания сборки прибора следует тщательно осмот-
реть аппаратуру и убедиться в правильности сборки.
5. МЫТЬЕ И СУШКА ХИМИЧЕСКОЙ ПОСУДЫ
Химическая посуда должна быть чистой, так как грязь может
резко изменить ход синтеза. Необходимо твердо усвоить: грязную
посуду следует мыть сразу же после окончания работы. При этом
следует руководствоваться правилами, изложенными на стр. 9—12.
Стеклянная посуда считается чистой, если на стенках ее не
23
образуется отдельных капель и вода оставляет равномерную тон-
кую пленку. Удалять загрязнения со стенок сосудов можно раз-
личными методами: механическими, физическими, химическими
и т. п.
Если химическая посуда не загрязнена смолами, жирами и
другими не растворяющимися в воде веществами, то ее можно
мыть теплой водой, применяя щетки и ерши. Для удаления жиро-
вых загрязнений лучше мыть посуду струей водяного пара, но этот
способ очень длителен, поэтому применяется довольно редко. Для
удаления из посуды продуктов перегонки нефти (парафин, керо-
син, воск, масло) и других нерастворимых в воде органических
веществ часто пользуются органическими растворителями: диэти-
ловым эфиром, ацетоном, спиртом, бензином, скипидаром и дру-
гими. Большинство органических растворителей — огнеопасные
жидкости, поэтому работать с ними нужно осторожно, вдали от
огня. Загрязненные органические растворители следует собирать,
а затем очищать их перегонкой (см. стр. 60).
Для мытья посуды можно также применять мыло, 10% рас-
твор тринатрийфосфата и современные синтетические моющие
средства; ни в коем случае нельзя пользоваться для очистки по-
суды песком, так как он царапает стекло, которое при нагревании
может лопнуть.
Для очистки посуды химическими методами чаще всего при-
меняют хромовую смесь, перманганат калия, смесь соляной кис-
лоты и перекиси водорода, серную кислоту, растворы щелочей.
Хромовая смесь является сильным окислителем и используется
для мытья посуды, загрязненной смолистыми и другими нераство-
римыми в воде веществами, однако ее не употребляют для удале-
ния продуктов перегонки нефти, а также солей бария, так как
последние образуют трудноудаляемый осадок сернокислого бария.
При работе с хромовой смесью следует соблюдать осторожность,
так как она действует на кожу и одежду. Для приготовления
хромовой смеси берут концентрированную серную кислоту и до-
бавляют 5 вес.% в расчете на кислоту тонкоизмельченного дву-
хромовокислого калия, который растворяют осторожным нагрева-
нием этой смеси в фарфоровой чашке или фарфоровом стакане.
После мытья хромовой смесью посуду ополаскивают водой, а за-
тем наливают до */з объема сосуда подогретую на горячей водя-
ной бане до 45—50°C хромовую смесь и смачивают ею стенки
сосуда. Слив* всю смесь обратно в тот же сосуд, в котором она
хранится, промывают посуду теплой водой. Признаком непригод-
ности хромовой смеси для мытья служит изменение ее цвета от
темно-оранжевого до темно-зеленого.
Очень удобным окислителем, который часто применяется для
очистки посуды, является подогретый до 50—60°C 5% раствор
перманганата калия. Образовавшийся после мытья посуды налет
на стенках легко удаляется ополаскиванием посуды 5% раство-
ром кислого сернистокислого-натрия NaHSO3, растворами серно-
кислого закисного железа FeSO^ а также щавелевой кислотой,
24
Хорошим средством для мытья посуды является сМесь, состоя-
щая из равных объемов соляной или уксусной кислоты и 5—6%-
ного раствора перекиси водорода. Смесь нагревают до 30—40°C,
обмывают ею стенки посуды, затем выливают обратно в тот же
сосуд, в котором она хранится, а посуду моют водой.
При длительном употреблении холодильников на внутренней
поверхности водяной рубашки образуется красноватый налет окис-
лов железа, которые попадают с водой из водопроводных труб.
Для очистки рубашку холодильника ополаскивают 10—16%-ной
соляной кислотой. После растворения окислов железа кислоту вы-
ливают, а через холодиль-
ник пропускают воду в тече-
ние 5—10 мин.
Для очистки посуды от
загрязнений веществами
можно применять концен-
трированные серную кисло-
ту или щелочи, при этом не-
обходимо соблюдать все
меры предосторожности,
указанные на стр. 10—12.
Очень многие органиче-
ские реакции проходят в от-
сутствие следов влаги, по-
этому после тщательной
очистки и мытья посуду необходимо хорошо высушить. Обычно вы-
мытую посуду высушивают в специальной сушилке (рис. 19).
Если же последняя отсутствует, то надевают посуду на колышки
и оставляют до высыхания. Для ускорения сушки часто через
сосуд при помощи груши и стеклянной палочки продувают воз-
дух, а также высушивают посуду в сушильном шкафу при 80—
100 °C.
6. ПОЛЬЗОВАНИЕ ЛИТЕРАТУРОЙ
И ПРАВИЛА СОСТАВЛЕНИЯ ОТЧЕТА
Весьма важной и существенной частью работы является до-
машняя подготовка по учебникам, руководствам, справочной, а
иногда и оригинальной литературе. Ни в крем случае не следует
превращать лабораторию в читальный зал и тратить лаборатор-
ные часы на первоначальное ознакомление с материалом, однако
пользоваться литературой в лаборатории можно при возникнове-
нии того или иного вопроса в ходе работы. При этом следует
принять за правило просматривать литературу как можно более
полно и тщательно. Домашнюю работу надо начинать с подго-
товки теоретических вопросов по данному синтезу по учебникам и
руководствам, затем ознакомиться с исходными веществами, ко-
торые используются в данном синтезе, т. е. отыскать в справочной
и учебной литературе физические константы и данные о свойствах
25
йтих соединений, особое внимание обратить на токсичность приме-
няемых реагентов. После этого приступить к проработке данного
синтеза по руководству.
В процессе подготовки и выполнения синтеза необходимо со-
ставить отчет по каждой работе. Каждый отчет следует начинать
с даты, номера работы и названия синтеза. Отчет должен содер-
жать уравнение основной реакции, по которой проводится расчет,
и уравнения побочных реакций, если они протекают. Затем необ-
ходимо ддть краткую характеристику исходным веществам, про-
извести их перерасчет, если предлагается исходить из иных
количеств, чем указано в руководстве, и составить план работы,
где записываются все последовательные операции, подлежащие
выполнению при том или ином синтезе.
В описании экспериментальной части работы надо подробно
остановиться на аппаратуре, условиях проведения реакции и на
особенностях ее протекания. Здесь же отмечается все, на что было
обращено внимание во время работы (изменение окраски, появле-
ние характерного запаха и т. д.). При этом нужно усвоить, что
план работы и описание экспериментальной части ни в коем слу-
чае не должны быть пересказом методики синтеза, данного в ру-
ководстве.
По окончании работы следует записать количество чистого
продукта, произвести расчет процентного выхода данного веще-
ства от теоретически возможного, а затем дать ему характери-
стику. Отчет заканчивается выводами, вытекающими из проде-
ланной работы. В конце дается список использованной литера-
туры.
Ниже приводится образец записи в рабочем журнале.
Образец записи в рабочем журнале
Дата...................Работа №
Получение бромистого этила
1. Основные реакции:
KBr + H2SO4 —> KHSO4 + HBr
C2H5OH + HBr C2H6Br + H2O
СНз—CH2—ОН + НО— SO2-OH ч=± СН3—СН2—О—so2—он + Н2О
СН3—СН2—О—SO„—ОН + НВг —> СН3—СН2Вг + H2SO4
2. Побочные реакции-
2НВг + H2SO4 —> Br2 + 2H2O+SO2
СНз—СН2—О—SO2-OH + НО—СН2—СН3 —>
—► H2SO4 -f- СН3—СН2—О—СН2—СНЭ
26
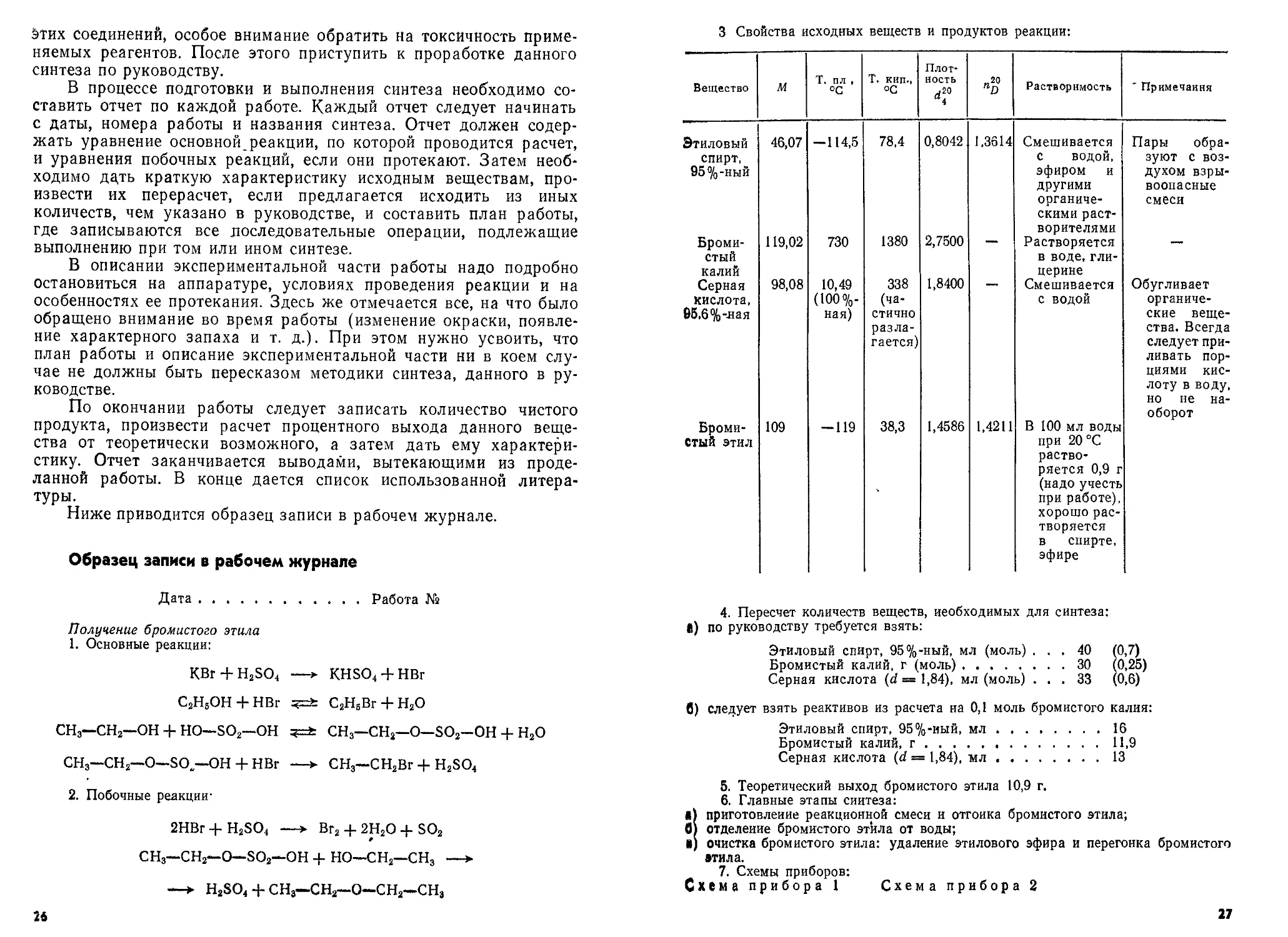
3 Свойства исходных веществ и продуктов реакции:
Вещество м Т. пл , °C Т. кип., °C Плот- ность j20 di «20 Растворимость 'Примечания
Этиловый спирт, 95%-ный 46,07 -114,5 78,4 0,8042 1,3614 Смешивается с водой, эфиром и другими органиче- скими раст- ворителями Пары обра- зуют с воз- духом взры- воопасные смеси
Броми- стый калий 119,02 730 1380 2,7500 —“ Растворяется в воде, гли- церине —•
Серная кислота, 95,6%-яая 98,08 10,49 (100%- ная) 338 (ча- стично разла- гается) 1,8400 Смешивается с водой Обугливает органиче- ские веще- ства. Всегда следует при- ливать пор- циями кис- лоту в воду, но не на- оборот
Броми- стый этил 109 -119 38,3 1,4586 1,4211 В 100 мл воды при 20 °C раство- ряется 0,9 г (надо учесть при работе), хорошо рас- творяется в спирте, эфире
4. Пересчет количеств веществ, необходимых для синтеза:
а) по руководству требуется взять:
Этиловый спирт, 95%-ный, мл (моль) ... 40 (0,7)
Бромистый калий, г (моль)............ 30 (0,25)
Серная кислота (d = 1,84), мл (моль) ... 33 (0,6)
в) следует взять реактивов из расчета на 0,1 моль бромистого калия:
Этиловый спирт, 95%-иый, мл..............16
Бромистый калий, г.......................11,9
Серная кислота (d = 1,84), мл............13
5. Теоретический выход бромистого этила 10,9 г.
6. Главные этапы синтеза:
а) приготовление реакционной смеси и отгоика бромистого этила;
в) отделение бромистого этила от воды;
В) очистка бромистого этила: удаление этилового эфира и перегонка бромистого
этила.
7. Схемы приборов:
Схема прибора 1 Схема прибора 2
27
8 Описание хода синтеза (описание операций, наблюдения, объяснения),
9. Выход чистого бромистого этила:
а) 8,7 г; б) 80% от теоретического количества.
Дата окончания работы.......................«
Отзыв и подпись преподавателя ......................
Глава II
ОСНОВНЫЕ ОПЕРАЦИИ
ПРИ РАБОТЕ
В ХИМИЧЕСКОЙ ЛАБОРАТОРИИ
Распространенными операциями в лаборатории органического
синтеза являются нагревание и охлаждение, так как многие реак-
ции в органической химии зависят от строгого соблюдения тем-
пературного режима.
1. НАГРЕВАНИЕ
Большинство реакций органической химии идут при комнат-
ной температуре весьма медленно. Чтобы увеличить скорость та-
ких реакций, повышают температуру, считая, что обычно при по-
„ вышении температуры на 10 °C скорость реакции возрастает при-
мерно в 2—3 раза. Повышение скорости химических реакций при
нагревании связано с увеличением числа столкновений реагирую-
щих молекул в единицу времени и с увеличением числа активных
молекул, т. е. таких молекул, которые по сравнению с другими
обладают повышенным запасом энергии.
В химической лаборатории нагревание можно проводить элек-
тронагревательными приборами, газовыми горелками или водяным
паром. Из электронагревательных приборов наибольшее распро-
странение получили плитки, термостаты, бани, сушильные шкафы,
печи, колбонагреватели. Наряду с ними в последнее время все
чаще для обогревания перегонных и реакционных колб применяют
лампы накаливания, излучающие инфракрасные лучи. Электро-
колбонагреватели (закрытые) обычно применяют в тех случаях,
когда требуется нагреть легколетучие органические вещества.
Применение же водяного пара для нагревания целесообразно
лишь в том случае, когда лаборатория имеет возможность поль-
зоваться паром от какого-либо парового хозяйства. При прове-
дении реакций непосредственное нагревание реакционного сосуда
электричеством или газовым пламенем не рекомендуется ввиду ма-
лой устойчивости стекла к резким изменениям температуры и не-
равномерности такого нагревания. Вследствие местного перегрева
28
возможно частичное разложение органических соединений. Кроме
того, правилами техники безопасности запрещается нагревание на
открытом пламени горючих жидкостей, так как в случае трещин
в сосуде они способны воспламеняться и даже взрываться. Откры-
тым пламенем нагревают фарфоровую, шамотную, кварцевую и
другую посуду, большей частью при прокаливании, а также фарфо-'
ровые глазурованные чашки для выпаривания водных растворов
или посуду из жаростойкого стекла.
При нагревании химической посуды выше 100°С часто поль-
зуются асбестовыми сетками или куоком тонкого листового асбеста,
при этом достигается большая равномерность обогрева, чем при
нагревании на голом огне. Для поддержания заданной наружной
температуры обогрева применяют разного рода бани, из них наи-
более употребительными являются водяные, глицериновые, мас-
ляные, парафиновые, воздушные,
песочные, из смеси H2SO4 и
K2SO4 (в соотношении 3:2), из
легкоплавких металлов, сплавов
и других материалов. При этом I k J
следует усвоить, что бани необхо-
димо применять при всех реак-
циях, которые Проводятся При Рис- 20. Водяные бани,
строго определенной температуре.
Обязательно нужно пользоваться банями при перегонке в вакууме
и при работе с легковоспламеняющимися жидкостями.
Для нагревания до температуры, не превышающей 100°C,
применяют водяные бани (рис. 20). Водяные бани покрыты сверху
кольцами; это дает возможность подбирать диаметр отверстия
бани соответственно размерам нагреваемого сосуда, который по-
гружают в воду так, чтобы он не касался дна бани.
Для нагревания до 220°C применяют масляные бани. Для
этого миску или кастрюлю до половины наполняют минеральными
маслами, получаемыми из нефти, и нагреваемый сосуд помещают
в баню таким образом, чтобы уровень вещества в сосуде был на
одном уровне с маслом. Максимальная температура, достигаемая
с помощью таких бань, зависит от сорта применяемого масла. При
сильном нагревании масла могут частично разлагаться и «ды-
мить», поэтому работа с ними проводится в вытяжном шкафу.
Особенно надо следить за тем, чтобы в такие бани не попа-
дала вода, так как в противном случае масло при нагревании на-
чинает пениться, выливаться наружу, вызывая пожары. Поэтому
обратные холодильники всегда должны иметь около нижнего кон-
ца манжетку из фильтровальной бумаги. После работы сразу же
следует осторожно обтирать тряпкой, бумагой поверхность колбы,
удаляя еще горячее масло. При длительном нагревании до высо-
кой температуры масло в бане может вспыхнуть. Вспыхнувшее
масло нельзя тушить ни водой, ни песком. Следует накрыть баню
листом асбеста или добавить в сосуд порцию холодного масла.
Во время работы опасность воспламенения масла можно уменьшить,
29
прикрывая баню двумя половинками асбестового картона, выре«
занного по размеру бани в виде кольца с отверстием для нагре-
ваемого сосуда.
Иногда вместо масляных бань применяют глицериновые и па-
рафиновые. На глицериновых банях обогрев ведут до температуры
не выше 200°C, а на парафиновых — не выше 220°C. Нагревание
глицериновой бани следует вести на асбестовой сетке, а не на го-
лом огне, так как при
церина с образованием
Рис. 21. Воронка Бабо:
1—жестяной конус; 2 —жестя*
мая пластинка, частично закры-
вающая нижнее отверстие во-
ронки; 3 — полоски асбеста;
4—отверстие для выхода горя-
чих газов.
перегревании возможно разложение гли-
слезоточивого акролеина. А в остальном
все сказанное о масляных банях относит-
ся и к глицериновым и парафиновым.
Простейшей воздушной баней являет-
ся воронка Бабо (рис. 21), нагреваемая
газовой горелкой. Она состоит из жестя-
ного конуса, на внутренней стенке кото-
рого укреплено несколько асбестовых по-
лосок, чтобы нагреваемый сосуд не со-
прикасался с горячей жестью. В нижней
части конуса укреплена жестяная пла-
стинка, предохраняющая дно колбы от
непосредственного соприкосновения с
пламенем. Воздушные бани позволяют
проводить нагревание практически до
любой температуры, однако такое нагревание оказывается менее
равномерным.
При нагревании веществ до 325 °C можно применять бани из
смеси H2SO4 и K2SO4, до 400°C — песочные бани, а до 600°C. и
выше — бани из легкоплавких металлов и сплавов. При нагрева-
нии необходимо строго соблюдать все меры предосторожности.
Работая с веществами, которые при нагревании могут разбрызги-
ваться, нужно надевать защитные очки.
При работе с огнеопасными жидкостями (эфир, ацетон, бен-
зол, спирт и другие) следует сначала вдали от прибора нагреть
водяную баню, потом погасить горелку, а затем уже постепенно
погрузить нагреваемый сосуд с огнеопасной жидкостью в баню.
Сосуд с жидкостью следует погружать в баню таким образом,
чтобы уровень этой жидкости в нем был на одном уровне с водой
в бане. Кроме того, необходимо помнить, что водяные бани нельзя
использовать при работе с металлическими натрием и калием.
При нагревании жидкостей выше температуры кипения может
произойти перегрев и даже взрыв. Этого 'можно избежать, приме-
няя «кипелки», т. е. кусочки обожженного неглазурованного фар-
фора, мелкие кусочки кирпича или длинные стеклянные капил-
ляры, запаянные с одного конца. Открытыми концами капилляры
погружаются в жидкость, а другими они должны выступать над
жидкостью и входить в горло колбы. «Кипелки» или капилляры
при нагревании выделяют небольшое количество воздуха мелкими
пузырьками и обеспечивают перемешивание и равномерное кипе-
ние. При этом следует помнить, что их нужно вносить только в
30
холодную жидкость. Ни в коем случае нельзя бросать «кипелкй»
в уже нагретую до кипения жидкость, так как внезапное парооб-
разование может вызвать разбрызгивание жидкости из колбы.
Если перегонка прервалась, то прежде чем возобновить ее, сле-
дует погасить гЬрелку и добавить в охладившуюся колбу свежие
«кипелки», так как при охлаждении жидкость заполнила все их
поры и они потеряли свою действенность.
2. ОХЛАЖДЕНИЕ
При проведении экзотермических реакций в результате выде-
ления большого количества тепла может произойти перегрев реак-
ционной смеси, что приводит к снижению выхода продукта. В та-,
ких случаях необходимо охлаждение этой смеси.
Самым дешевым и удобным средством охлаждения является
Водопроводная вода, температура которой колеблется в зависи-
мости от времени года от 4 до 20 °C. Обычно реакционный сосуд
охлаждают под краном проточной водой или периодически погру-'
жая его в холодную воду. Если же реакционную колбу необхо-
димо охладить в приборе, то ее помещают в большую воронку со
шлангом и поливают проточной водой. При охлаждении паров для
их конденсации применяют различные типы холодильников (см.
рис. 6), в .рубашках которых циркулирует холодная вода.
Для охлаждения до 0°С пользуются льдом, который размель-
чают до размеров грецкого ореха, а до температуры ниже 0°С —
охлаждающими смесями. Для получения температуры примерно
ОТ —5 до —20 °C применяют смесь льда с поваренной солью, ко-
торую готовят из 3 ч. тонкоизмельченного льда и 1 ч. технической
поваренной соли. Более низкие температуры (до —50°C) можно
получить, применяя смесь из 5 ч. кристаллического хлористого
кальция и 4 ч. мелкоизмельченного льда. Температуру до —70 °C
можно получить, пользуясь твердой двуокисью углерода (сухим
льдом). При смешении твердой двуокиси углерода с абсолютным
Втиловым спиртом можно получить температуру до —72 °C, с эфи-
ром до —77 °C, с ацетоном до —78 °C.
Измельчение сухого льда желательно проводить в металли-
ческой ступке, при этом следует надевать защитные очки. Добав-
ление сухого льда к спирту,. ацетону, эфиру нужно проводить
осторожно, так как происходит сильное вспенивание. Если охлаж-
дающее действие указанных выше смесей недостаточно, то для
Охлаждения используют жидкий воздух и жидкий азот.
3. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ ТЕМПЕРАТУРЫ
Для -измерения температуры реакции в пределах от —35 до
•4-350 °C обычно применяют ртутные термометры. Температуру от
350 до 600 °C можно измерить при помощи ртутных термометров,
наполненных азотом. Для контроля за температурой от —35 до
—60 °C употребляют термометры, наполненные подкрашенным
31 .
Фолуолом или спиртом. Высокие температуры измеряют термопа-
рами. Термометр обычно вводят в реакционную смесь или опу-
скают в баню. Пользуясь масляными, глицериновыми и парафи-
новыми банями, всегда следует помещать в них термометр, так
как они, в отличие от кипящей водяной~~бани, не обладают по-
стоянной температурой. Некоторого регулирования температуры
можно добиться путем ограничения подвода тепла к бане, т. е.
путем изменения величины газового пламени или включением
электронагревательного прибора через сопротивление. Для регу-
лирования напряжения можно применять лабораторный авто-
трансформатор (ЛАТР), однофазный регулятор напряжения
(РНО) и специальные регулирующие приспособления.
4. ИЗМЕЛЬЧЕНИЕ И ПЕРЕМЕШИВАНИЕ
Твердые материалы можно измельчать вручную, а также при
помощи различных дробилок, мельниц, истирателей и пр. Для
ручного измельчения применяют различные ступки: стальные, чу-
гунные, бронзовые, фарфоровые, агатовые и т. п. Больше всего в
лабораториях органического синтеза распространены фарфоровые
Рис. 22. Мешалки:
а, б, в —стеклянные, г — лопастная металлическая, д—центробежная
металлическая; е—металлическая мешалка Хершберга.
ступки (см. рис. 13,в). Вещество, подлежащее измельчению, на-
сыпают на */з объема ступки и осторожно пестиком разбивают
крупные куски до размеров горошины, а затем растирают их. При
измельчении сильно пылящих и вредных веществ работу следует
проводить в вытяжном шкафу.
Перемешивание является ответственной операцией, не только
ускоряющей реакцию, но и обусловливающей возможность ее про-
ведения. Очень важно хорошо перемешивать реакционную смесь
в том случае, когда одно из реагирующих веществ нерастворимо,
а также когда один из реагентов прибавляют к реакционной смеси
постепенно. Благодаря размешиванию добиваются быстрого и
равномерного распределения вещества по всему объему раствора,
что позволяет избежать местных перегревов и повышения кон-
центрации. При работе с малыми количествами, а также в тех
32
случаях, когда реакция идет быстро и проводится в открытых со-
судах, часто оказывается достаточным перемешивание от руки
или встряхивание реакционного сосуда. При работе с большими
крличествами и при реакциях, протекающих в течение длитель-
ного периода времени, пользуются
мешалками различного типа (рис.
22).
Эффективность перемешивания
во многом зависит от конструкции
мешалок. Очень часто пользуются
Мешалками, изготовленными из
толстых стеклянных палочек (рис.
22, а, б, в); они очень удобны, так
как перед опытом им можно при-
дать' любую форму в зависимости
Рис. 23. Двигатели для мешалок:
а—электродвига гель; б—водяная тур-
бинка.
от величины реакционного сосуда,
ширины горла и других требований, предъявляемых в данных
условиях. Для перемешивания больших количеств применяют ме-
таллические мешалки (рис. 22, г, д), а для размешивания тяжелых
Рне. 24. Крепление ме-
шалки к электродвига-
осадков или вязких жидкостей — мешалки
Хершберга (рис. 22, е).
Обычно мешалки приводятся в движе-
ние электродвигателями (рис. 23,а), кото-
рые можно крепить в штативе, а также на
специальных деревянных стойках. Скорость
вращения мотора следует регулировать при
помощи реостата или регулировочного
трансформатора (РНО). Перед включением
мешалки ее следует прокрутить рукой, что-
бы убедиться, что при движении она не
касается стенки сосуда или термометра и
что ее не «заедает». Многие электродвига-
тели имеют муфту крепления мешалки. Если
же у двигателя нет муфты, то, чтобы ме-
шалка не проскальзывала, ее соединяют с
валом при помощи двух отрезков вакуум-
ного шланга и стеклянной палочки (рис.
24), при этом следят за тем, чтобы вал
Телю при помощи двух
отрезков вакуумного
шланга:
/—отрезки вакуумного шлан-
га; 2—стеклянная палочка.
электродвигателя и мешалки составлял
одну прямую.
При работе с легковоспламеняющимися
веществами (например, сероуглерод, эфир)
можно применять электродвигатели с длин-
ным гибким шлангом, но целесообразнее использовать водяные
турбинки (рис. 23,6). Чтобы водяная турбинка работала, ее
прочно крепят на штативе, затем один из отростков ее посред-
ством шланга соединяю?- с водопроводным краном, а на другой
надевают водоотводящую трубку, которую опускают в раковину.
Открывая водопроводный кран, приводят в движение турбинку и
2 М Н. Храмкина
33
регулируют вращение ротора турбинки силой струи воды
(рис. 25).
При гидрировании, работе в высоком вакууме и в некоторых
других случаях применяют магнитные мешалки,
Для равномерной и бесшумной работы мешалки необходимо
хорошо фиксировать положение ее оси. Для этого обычные ме-
шалки монтируют следующим образом: стержень мешалки поме-
щают в стеклянную трубку, выполняющую роль подшипника, ко-
торую смазывают вазелином или глицерином. Эту трубку встав-
ляют в резиновую или корковую пробку; последнюю зажимают в
Рис. 25. Работа водяной
турбинки.
Рис. 26. Мешалка с ртут-
ным затвором.
лапку штатива. На верхний конец мешалки при помощи короткой
резиновой трубки надевают деревянный шкив с канавкой, который
посредством ремня соединяют с валом турбинки или электродви-
гателя (см. рис. 25).
В тех случаях, когда необходимо изолировать реакционную
смесь от действия влаги или воздуха, применяют затворы. Самое
простое и обычное уплотнение мешалки заключается в соедине-
нии стержня мешалки с подшипником при помощи небольшого
куска резиновой трубки. В таких случаях для уменьшения трения
резиновую трубку внутри смазывают 'вазелином, а подшипник
соединяют с реакционным сосудом посредством пробки. Практи-
чески полная герметичность достигается применением ртутного
затвора (рис. 26).
Для защиты от вредных паров ртути надо поверх ртути на-
лить слой глицерина. В студенческих лабораториях вместо ртут-
ных затворов лучше применять глицериновые.
34
5. РАСТВОРЕНИЕ И СВОЙСТВА
НЕКОТОРЫХ ОРГАНИЧЕСКИХ РАСТВОРИТЕЛЕЙ
Растворение — это процесс образования раствора (прозрачная
гомогенная жидкость) в результате смешения какого-либо веще-
ства (газ, жидкость, твердое тело) с другой жидкостью, называе-
мой растворителем. Основным условием этого процесса является
отсутствие химического взаимодействия между растворяемым ве-
ществом и растворителем, следовательно, после удаления раство-
рителя, исходное вещество должно остаться неизменным.
Процесс растворения твердых и жидких веществ всегда связан
с затратой энергии, т. е. с поглощением тепла; это объясняется
тем, что молекулы вещества распределяются в большем объеме.
При растворении газов происходит обратное явление — выделение
тепла; оно связано с уменьшением объема, в котором распреде-
ляются молекулы газа. Однако при растворении твердых и жид-
ких веществ часто наблюдается выделение тепла, которое указы-
вает, что теплота гидратации данного твердого вещества больше,
чем теплота растворения соответствующего гидрата.
Скорость растворения твердого вещества зависит от размеров
его частиц. Чем мельче отдельные частицы вещества, тем больше
поверхность контакта между жидкой и твердой фазами и, следо-
вательно, тем быстрее происходит растворение. Однако бывают
случаи, когда тонкоизмельченное вещество при высыпании в воду
не смачивается и плавает на поверхности воды; в таких случаях
порошок вначале обливают небольшим количеством чистого спир-
та, а затем уже высыпают его в воду. Разумеется, что применять
спирт можно лишь в том случае, если он не оказывает химиче-
ского действия на вещество или его раствор.
Часто растворимость твердого вещества можно повысить, если
раствор нагревать. Но при этом нужно учесть, что некоторые соли
не подчиняются этому правилу—растворимость их или пони-
жается с повышением температуры, или повышается незначи-
тельно.
Почти все газообразные вещества способны в той или иной
мере растворяться в воде или органических растворителях. Так,
например, аммиак и хлористый водород очень хорошо погло-
щаются водой, а кислород и водород обладают меньшей раство-
римостью в воде. Растворимость газов в жидкостях обычно умень-
шается с повышением температуры или понижением давления.
Поэтому для удаления растворенного газа жидкость обычно на-
гревают или вакуумируют.
Процесс растворения в химической лаборатории используют
для многих целей: для получения гомогенной (однородной) среды,
т. е. для создания более благоприятных условий, обеспечивающих
наибольшую поверхность соприкосновения реагирующих веществ;
для отделения веществ друг от друга и очистки продуктов реак-
ции путем перекристаллизации; для изменения скорости или на-
правления реакции; для проведения реакции в отсутствие воды и пр.
2* 3S
Различают водные и неводные растворы. К водным относятся
растворы большинства солей, щелочей, кислот; к неводным при-
надлежат растворы в органических растворителях, например в
спиртах, эфирах, ацетоне и др.
Органические растворители широко применяются для раство-
рения органических жидких и твердых веществ, например масел,
жиров, смол и т. д. Кроме того, они используются для растворе-
ния неорганических веществ и в практике аналитической химии
для неводного титрования. Обычно в лабораторной практике
употребляются чистые органические растворители, которые полу-
чают путем очистки дешевых технических продуктов.
Очень многие органические растворители огнеопасны и ядо-
виты. Поэтому при работе с ними всегда следует соблюдать все
меры предосторожности.
ЭТИЛОВЫЙ СПИРТ
Этиловый 95,6%-ный спирт-ректификат имеет температуру
кипения 78,3 °C, = 0,7936 и является постоянно кипящей
азеотропной смесью, содержащей 4,4% воды. Для многих целей
необходим этиловый спирт 100 или 99,9%-ный, так называемый
«абсолютный», который впервые был получен русским академи-
ком Ловицем. Вода, содержащаяся в этиловом спирте-ректифи-
кате, не может быть удалена простой перегонкой. В лаборатории
обезвоживание спирта можно производить при нагревании с легко-
гидратирующимися веществами, такими как окись кальция или
безводная сернокислая медь(II). Получение обезвоженного 99,5%-
ного этилового спирта с помощью окиси кальция и безводной сер-
нокислой меди проводится в колбе с обратным'холодильником,
снабженным хлоркальциевой трубкой для защиты спирта от попа-
дания влаги из воздуха. Товарную окись кальция в виде кусков
размером с лесной орех перед употреблением прокаливают в элек-
трической печи в течение 1—2 ч. Тотчас по охлаждении переносят
окись кальция в хорошо закрывающуюся банку.
Круглодонную колбу емкостью 1 л заполняют 500 мл 95,6%-
ного этилового спирта и 125 г свежепрокаленной окиси кальция.
Смесь кипятят на водяной бане в течение 6 ч, а затем оставляют
на ночь; за это время куски окиси кальция большей частью пре-
вращаются в порошкообразную гидроокись кальция. После охла-
ждения спирт отгоняют на приборе для перегонки высушенных
низкокипящих растворителей, при этом приемником служит склян-
ка, в которой будет храниться абсолютный спирт (рис. 27). Пе-
регонку спирта сначала ведут на слабокипящей водяной бане,
к которой добавляют немного поваренной соли, а затем — на ки-
пящей, причем первые 15 мл дистиллята отбрасывают. Перегонка
идет довольно медленно и значительное количество спирта удер-
живается твердым остатком. Такая обработка позволяет довести
крепость спирта до 99,5%, что достаточно для большинства работ.
36
При применении для обезвоживания спирта сернокислой
меди(П) поступают следующим образом. Кристаллогидрат
CuSO4-5H2O нагревают при перемешивании в никелевой или фар-
форовой чашке до 220 °C до тех пор, пока он не превратится в
белый порошок. Полученную обезвоженную сернокислую медь
после охлаждения прибавляют к спирту в количестве 200—250 г
на 1 л спирта. Смесь кипятят с обратным холодильником и хлор-
кальциевой трубкой в течение
6 ч, оставляют стоять на ночь,
а на следующий день отгоняют
(см. рис. 27). Такую обработку
повторяют еще раз. Для обез-
воживания спирта непригодны
хлористый кальций и серная
кислота, так как хлористый
кальций образует со спиртом
кристаллогидрат, а серная кис-
лота — эфиры.
Для получения безводного
абсолютного этилового спирта
можно применять магний, на-
трий; в настоящее время ши-
роко применяется абсолютиро-
вание спирта методом азео-
тропной смеси с бензолом. Рассмотрим эти случаи.
Действие магния:
Рис. 27. Прибор для перегонки высу-
шенных низкокипящих растворителей.
2С2Н5ОН + Mg —> (C2H5O)2Mg + Н2
(C2H6O)2Mg + 2Н2О —> Mg(OH)2 + 2С2Н5ОН
В круглодонную колбу емкостью 2 л, снабженную обратным
холодильником с хлоркальциевой трубкой, помещают 5 г стружки
магния, 0,5 г иода и приливают 70—75 мл обезвоженного этило-
вого спирта. Смесь нагревают до тех пор, пока окраска иода не
исчезнет. После этого вливают в колбу 900 мл обезвоженного эти-
лового спирта и кипятят смесь в течение 30 мин. Затем колбу
охлаждают, заменяют обратный холодильник нисходящим, при-
соединяют к нему приемник при помощи алонжа с отводом, сое-
диненным с хлоркальциевой трубкой для предохранения от влаги
воздуха, и перегоняют. При соблюдении всех мер предосторожно-
сти против влаги воздуха получают 99,95 %-ный спирт.
Действие натрия. Натрий легко реагирует с водой, содержа-
щейся в спирте, и частично со спиртом. Образовавшийся едкий
натр подвергается алкоголизу, в котором устанавливается равно-
весие между едким натром и спиртом, с одной стороны, и про-
дуктами их взаимодействия — этилатом натрия и водой — с дру-
гой:
2Na + 2H2O —> 2NaOH + Н2
2NaOH + 2С2Н5ОН 2C2H5ONa + 2Н2О
37
При обработке диэтиловым эфиром фталевой кислоты можно
образовавшийся едкий натр заставить необратимо реагировать
с избытком высококипящего сложного эфира.
При последующей перегонке из колбы с небольшим дефлегма-
тором этиловый спирт легко отгоняется от сложного эфира:
С6Н4(СООС2Н5)2 + 2NaOH —► C6H4(COONa)2 + 2С2Н5ОН
Круглодонную коЯбу емкостью 2 л снабжают обратным холо-
дильником, с которым при помощи изогнутой трубки соединяют
нисходящий холодильник с присоединенным к нему, как описано
выше, приемником. В колбу помещают 1 л обезвоженного спирта
и вносят 7 г чистого сухого натрия. После того как натрий про-
реагирует, приливают 30 г диэтилфталата и равномерно кипятят
реакционную смесь в течение 2 ч Затем выпускают воду из муф-
ты обратного холодильника и перегоняют спирт на водяной бане,
используя в качестве насадки неохлаждаемый водой обратный
холодильник.
Абсолютирование спирта методом азеотропной смеси с бензо-
лом. Азеотропные, или нераздельно кипящие, растворы перего-
няются без изменения состава и температуры кипения, т. е. обыч-
ной перегонкой их нельзя разделить на составные части. Д. П. Ко-
новалов в 1884 г. показал, что азеотропные смеси существуют
в таких системах, у которых зависимость давления пара раствора
от его состава при постоянной температуре или зависимость тем-
пературы кипения от состава при постоянном давлении выра-
жаются кривыми, имеющими точки минимума или максимума.
В соответствии с этим различают азеотропные смеси: а) с мини-
мумом температуры кипения (с максимумом давления пара) и б) с
максимумом температуры кипения (с минимумом давления пара).
Многокомпонентные смеси, как и бинарные, часто имеют азео-
тропные точки, характеризующиеся преимущественно максиму-
мом давления и минимумом температуры кипения. Минимум
температуры кипения у тройных смесей наблюдается в тех слу-
чаях, когда все три или, по крайней мере, две бинарные смеси,
содержащие те же компоненты, имеют минимумы температуры
кипения. При этом у тройных смесей наблюдается более глубо-
кий минимум температуры кипения, чем у бинарных смесей, со-
ставленных из этих же компонентов. Так, например, из компонен-
тов тройной смеси — спирта, бензола и воды — могут быть обра-
зованы три бинарные смеси: 1) спирт — вода; 2) бензол — вода;
3) спирт — бензол, Минимумы температур кипения этих смесей
равны соответственно 78,2; 69; 68,3°C, а тройная смесь кипит при
еще более низкой температуре 64,9°C.
Этим свойством тройной азеотропной смеси можно иногда
воспользоваться для разделения бинарных смесей, имеющих азео-
тропные точки. Так, для абсолютирования С2Н5ОН, т. е. для от-
гонки чистого этилового спирта из бинарной смеси этиловый
спирт — вода, достаточно прибавить к этой смеси такое количе-
ство бензола, которое требуется для образования тройной смеси.
38
Отгонка абсолютного этилового спирта возможна по той причине,
что после прибавления бензола образовалась тройная смесь, ко-
торая имеет более глубокий минимум температуры кипения и со-
держит в азеотропной точке больше воды, чем азеотропная смесь
этиловый спирт — вода. Прибавление третьего компонента для
разделения бинарных азеотропных смесей часто используется при
азеотропной ректификации.
Содержание воды в полученном абсолютном спирте устанав-
ливают путем определения его плотности и нахождения соответ-
ствующего ему состава по данным таблицы, которая имеется в
любом химическом справочнике. Абсолютный 99,95—100%-ный
этиловый спирт очень гигроскопичен, поэтому его нужно хранить
в склянке с хорошо притертой пробкой
Этиловый спирт — наркотик При длительном воздействии вы-
зывает тяжелые заболевания сердечно-сосудистой системы, пище-
варительного аппарата и т. д.
МЕТИЛОВЫЙ СПИРТ
Синтетический метиловый спирт с температурой кипения
64,7 °C и d\5 = 0,7961 пригоден для большинства работ Обычно он
содержит 1—2% воды, следы ацетона (до 0,1%) и формальдегида.
Удобный способ удаления ацетона заключается в следующем:
смесь 500 мл метилового спирта, 25 мл фурфурола и 60 мл 10%
водного раствора едкого натра нагревают с обратным холодиль-
ником 6—12 ч; при этом образуется смола, содержащая дифурфу-
ральацетон и другие высококипящие вещества:
Затем метиловый спирт отгоняют с дефлегматором (см. рис. 11)
или на колонке, отбрасывая первые 15—20 мл, в которых содер-
жатся следы формальдегида При работе с метиловым спиртом не-
обходимо соблюдать все меры предосторожности, так как он очень
легко воспламеняется и образует с воздухом взрывоопасные смеси.
Кроме того, следует помнить, что метиловый спирт очень токсичен:
длительное вдыхание его паров вызывает повреждение зрения,
перебои сердца, потерю сознания, а прием внутрь даже небольших
количеств приводит к слепоте и смерти.
ДИЭТИЛОВЫЙ ЭФИР
Товарный диэтиловый эфир с температурой кипения
34,6 °C и d\5 = 0,7192 содержит небольшое количество воды и эти-
лового спирта При длительном хранении на свету и при доступе
39
воздуха эфир медленно окисляется с образованием альдегидов,
кислот и нестойких, взрывоопасных перекисных соединений:
СН3—СН—О—ОН СН,—СН2—О—О—СН2—СНз
ОН
Гидроперекись оксиэтила Перекись этила
сн3—сн—о—о—сн—он3 сн3—сн; I
I 1 чо
ОН он
Перекись диоксидиэтила Перекись этилидена
Следует знать, что все простые эфиры (особенно диизопропи-
ловый) образуют перекиси. Часто в лабораториях происходят
сильные взрывы, вызванные перегонкой неочищенного эфира. По-
этому прежде чем начать работу с эфиром, следует обязательно
проверить его на наличие перекисных соединений.
Присутствие перекисей можно обнаружить, встряхивая не-
сколько миллилитров эфира с равным количеством 2°/о-ного рас-
твора йодистого калия (или йодистого натрия), подкисленного не-
сколькими каплями соляной или серной кислоты. Появление через
некоторое время бурого окрашивания эфирного слоя указывает на
присутствие перекисей.
Другой способ обнаружения перекисей заключается в следую-
щем: растворяют 1 мл двухромовокислого натрия в 1 мл воды,
добавляют каплю разбавленной серной кислоты, пробу эфира и
взбалтывают. Синее окрашивание эфирного слоя указывает на на-
личие перекисных соединений.
Удаление перекисей из эфира можно производить несколькими
способами. Самый простой способ — встряхивание эфира с по-
рошкообразным едким кали (на 1 л эфира 70 г едкого кали). Дру-
гой способ — промывание эфира раствором сернокислой соли за-
киси железа. Для промывания 1 л эфира достаточно 10—20 мл
раствора, состоящего из 60. г кристаллической сернокислой соли
закиси железа, 6 мл концентрированной серной кислоты и ПО мл
воды. Перекиси можно также удалять, встряхивая 1 л эфира с
20—25 мл насыщенного на холоду раствора сульфита натрия
в 50 мл воды. После отстаивания эфир отделяют и промывают
0,5% раствором перманганата калия для превращения образовав-
шегося ацетальдегида в уксусную кислоту. Выделенный эфир за-
тем промывают 5% раствором едкого натра, водой и сушат в те-
чение суток, вводя 150—200 г гранулированного хлористого каль-
ция на 1 л эфира. Потом хлористый кальций отфильтровывают
через складчатый фильтр, а эфир собирают в склянку темного
цвета С корковой пробкой, следя при этом за тем, чтобы все
горелки поблизости были потушены.
40
Для получения абсолютного эфира в склянку с высушенным
эфиром добавляют тонко нарезанный металлический натрий — на
1 л эфира 5 г натрия. Склянку плотно закрывают корковой проб-
кой со вставленной в нее хлоркальциевой трубкой. После 24 ч
стояния вносят еще 2,5 г натрия и через 12 ч эфир переливают
в склянку из темного стекла с хорошей корковой пробкой. Для
предохранения эфира от образования перекисей к нему добавляют
ничтожное количество дифениламина или фосфорный ангидрид
или помещают несколько кусков медной проволоки.
Диэтиловый эфир является чрезвычайно легко воспламеняю-
щимся растворителем, поэтому необходимо следить за тем, чтобы
на расстоянии не менее 3 м от места работы с эфиром не было
огня. Наливать эфир нужно при помощи небольшой воронки с ши-
рокой отводной трубкой; перегонять эфир необходимо на водяной
бане в приборе для перегонки низкокипящих безводных раствори-
телей (см. рис. 27).
Диэтиловый эфир является наркотиком и действует раздра-
жающе на дыхательные пути.
ПЕТРОЛЕЙНЫЙ ЭФИР
Петролейный эфир — это фракция нефти, кипящая при 40—
70 °C. Он содержит ненасыщенные и ароматические углеводороды;
очищают его встряхиванием в течение 20 мин с двумя или тремя
порциями концентрированной серной кислоты (на 1 л петролей-
ного эфира 100 мл кислоты). После отделения нижнего кислотного
слоя эфир промывают в делительной воронке несколькими пор-
циями концентрированного раствора перманганата калия в
10%-ной серной кислоте до неисчезающего фиолетового окраши-
вания водного слоя. Потом петролейный эфир промывают водой
10% раствором едкого натра, снова водой, сушат безводным
хлористым кальцием, как диэтиловый эфир (см. стр. 40), и пере-
гоняют (см. рис. 27) на водяной бане с закрытым электрообогре-
вом. Хранят полученный эфир над металлическим натрием.
Петролейный эфир образует с воздухом взрывоопасные смеси
и имеет низкую температуру вспышки, следовательно, необходимо
соблюдать особую осторожность при работе с ним. Пары петро-
лейного эфира могут вызвать отравления.
АЦЕТОН
Чистый ацетон с температурой кипения 56,2 °C и <4° = 0,7908
можно выделить из технического ацетона, который содержит воду,
метиловый спирт и уксусную кислоту. Для очистки и получения
абсолютного ацетона можно применить несколько способов. Один
из них заключается в нагревании ацетона с порошкообразным
перманганатом калия. Технический ацетон (1 л) помещают в
колбу, снабженную обратным холодильником, вносят туда пор-
циями 6—7 г растертого в порошок перманганата калия и кипятят
41
на водяной бане в течение 5—6 ч до неисчезающего окрашивания
жидкости. Затем заменяют обратный холодильник нисходящим и
отгоняют ацетон, который уже освобожден от метилового спирта,
но еще содержит воду. Для высушивания закрывают обратный
холодильник хлоркальциевой трубкой, вносят в колбу безводный
хлористый кальций (120 г на 1 л ацетона) и кипятят на водяной
бане в течение 4—5 ч. После охлаждения быстро переливают аце-
тон на свежий хлористый кальций и снова кипятят его 4—5 ч.
После этого заменяют обратный холодильник нисходящим
(рис. 27) и отгоняют ацетон в охлаждаемый льдом приемник из
коричневого стекла.
Другой способ очистки ацетона заключается в получении со-
единения ацетона с иодистым натрием (NaI-ЗСНзСОСНз), кото-
рое при нагревании разлагается с выделением ацетона. В колбу,
снабженную обратным холодильником, помещают 100 г чистого
тонко растертого йодистого натрия, вносят 400 мл ацетона и ки-
пятят на водяной бане до полного растворения осадка. Затем
охлаждают раствор до —8 °C смесью льда и соли, выделившиеся
кристаллы отфильтровывают на воронке Бюхнера, переносят
в перегонную колбу, соединенную с нисходящим холодильником,
и при слабом нагревании перегоняют ацетон в приемник, охлаж-
даемый льдом.
При работе с ацетоном должны приниматься меры предосто-
рожности, так как он очень легко воспламеняется и образует с
воздухом взрывоопасные смеси.
Ацетон обладает токсическим действием, пары его раздражают
верхние дыхательные пути.
6. СУШКА И ОСНОВНЫЕ ОСУШИТЕЛИ
Сушкой называется процесс освобождения вещества в любом
агрегатном состоянии от любой примеси жидкости. Чаще всего под
сушкой понимают освобождение от влаги или органических рас-
творителей.
Многие реакции в органической химии проводятся при отсут-
ствии влаги, в таких случаях следует высушивать исходные веще-
ства, применять абсолютированные растворители и предохранять
реакционную среду от попадания влаги из воздуха. Осушитель
должен быстро действовать, не растворяться в органических
жидкостях, не взаимодействовать с высушиваемым веществом.
ОСУШИВАНИЕ ГАЗОВ
Большинство газов, получаемых в лаборатории, а также мно-
гие сжатые газы из баллонов могут быть осушены концентриро-
ванной серной кислотой или твердыми осушителями, такими как
хлористый кальций, натронная известь, фосфорный ангидрид и др.
42
Серной кислотой можно- осушить воздух и следующие наибо-
лее часто применяемые газы: кислород, водород, азот, двуокись
и окись углерода, хлор, хлористый водород, сернистый газ. Для
Рис. 28. Промывные склянки:
а —склянка Дрекселя; б—склянка Тищенко; в—склянка Алифанова.
осушивания газ
осушивания газ пропускают через промывные склянки Дрекселя,
Тищенко или Алифанова, в которые налита на х/3
ная серная кислота (рис. 28). Обычно промыв-
ная склянка соединена с источником газа и при-
бором посредством двух пустых предохранитель-
ных склянок, роль которых выполняют склянки
Дрекселя или Тищенко (рис. 29). Осушивание
концентрирован-
Рис. 29. Схема прибора для ввода газа в реакцион- Рис. 30. Осуши-
ный сосуд: тельная колонка.
/— баллон с газом; 2, 4—-предохранительные склянки, 3 —про-
мывная склянка;5—сосуд для проведения реакции.
газов твердыми осушителями проводят в осушительных колонках
(рис. 30), а для защиты газа от влаги воздуха прибор закрывают
хлоркальциевой трубкой (см. рис. 12).
ВЫСУШИВАНИЕ ОРГАНИЧЕСКИХ ЖИДКОСТЕЙ
Органические жидкости сушат обычно твердыми неорганиче-
скими осушителями, при этом следует брать небольшое количество
последних, чтобы избежать потерь от адсорбции вещества осуши-
телем. Сначала встряхивают органическую жидкость с неболь-
шим количеством осушающего вещества (до 3% от массы раство-
ра), через некоторое время выделяется небольшой слой водного
43
раствора осушителя, если были взяты для сушки вещества, обра-
зующие с водой гидраты (хлористый кальций, сернокислый натрий,
едкий натр, сернокислый магний). Жидкость сливают, снова вносят
свежую порцию осушителя и так повторяют до тех пор, пока осу-
шитель не перестанет поглощать воду, например, хлористый каль-
ций не будет расплываться, фосфорный ангидрид не станет сли-
паться и т. п. После такой обработки органическую жидкость по-
мещают в колбу, которую закрывают пробкой с хлоркальциевой
трубкой и оставляют стоять на ночь с новой порцией осушителя.
Перед перегонкой высушенную органическую жидкость отфильтро-
вывают или чаще всего декантируют.
ВЫСУШИВАНИЕ ТВЕРДЫХ ВЕЩЕСТВ
Легколетучие примеси могут быть удалены из негигроскопич-
ных твердых веществ высушиванием на фильтровальной бумаге,
Рис. 31. Эксикаторы:
а—обыкновенный; б, в —вакуум-эксикаторы.
термически устойчивые вещества могут быть высушены в сушиль-
ных шкафах. Часто для сушки твердых веществ применяют
Рис. 32. Схема соединения вакуум-эксикатора с водо-
струйным насосом:
1 — вакуум-эксикатор; 2—манометр; 3 — предохранительная
склянка.
обыкновенные и вакуум-эксикаторы (рис. 31). Последние имеют
отверстие, в которое на резиновой пробке вставляют трубку с кра-
ном. Это дает возможность соединить эксикатор с водоструйным
44
насосом, между которыми помещают манометр и предохранитель-
ную склянку (рис. 32).
Иногда под вакуумом эксикаторы взрываются, поэтому перед
включением насоса их необходимо обернуть полотенцем. При от-
крывании вакуум-эксикатора, чтобы избежать распыления вы-
сушенного вещества воздухом, следует очень осторожно и мед-
ленно поворачивать кран. Только после того как давление будет
уравнено, можно открывать притертую крышку вакуум-эксика-
тора.
Осушающий агент подбирают в зависимости от химических
свойств высушиваемого вещества. Чаще всего в качестве осуши-
телей для эксикаторов применяют хлористый кальций, натронную
известь, едкий натр, едкое кали, фосфорный ангидрид, концентри-
рованную серную кислоту. При этом нужно помнить, что серную
кислоту нельзя применять для высушивания в вакууме, ее исполь-
зуют только в обыкновенных эксикаторах для поглощения влаги,
остатков спирта, эфира, ацетона, анилина, пиридина. Для адсорб-
ции углеводородов, особенно гексана, лигроина, бензола и его го-
мологов, в качестве заполнителя для эксикатора применяют пара-
фин; для удаления веществ кислого характера применяют едкий
натр или едкое кали. Вода и спирты хорошо поглощаются фосфор-
ным ангидридом, натронной известью.
ОСНОВНЫЕ ОСУШИТЕЛИ
Безводный хлористый натрий — дешевый широко применяе-
мый осушитель, обладает высокой осушительной способностью.
Однако высушивает он медленно и непригоден для сушки спиртов,
фенолов, аминов, аминокислот, амидов, нитрилов кислот, сложных
эфиров, некоторых кетонов и альдегидов, так как образует с ними
соединения. Кроме того, хлористый кальций содержит в качестве
примеси известь, следовательно, его нельзя употреблять Для сушки
веществ кислотного характера. Применяется он для предваритель-
ной сушки предельных, этиленовых углеводородов, ацетона, про-
стых эфиров и других соединений от воды.
Безводный сернокислый магний — один из лучших осушающих
нейтральных агентов, обладающий большой скоростью поглощения
воды и хорошей поглотительной способностью; применяется для
высушивания наибольшего числа соединений.
Безводный сернокислый натрий — дешевый нейтральный осу-
шитель, который применяется для предварительного удаления
больших количеств воды, однако действует он медленно и не свя-
зывает всю воду. Его нельзя использовать для сушки бензола,
толуола, хлороформа.
Едкий натр и едкое кали — хорошие и быстрые осушители, но
они находят лишь весьма ограниченное применение, исключительно
для аминов и простых эфиров.
45
Гигроскопическая вата, предварительно высушенная в сушиль-
ном шкафу при 100 °C, является отличным осушающим средством
и применяется в хлоркальциевых трубках.
В Приложении 1 приведены осушители, применяемые для суш-
ки различных классов органических соединений.
7. ФИЛЬТРОВАНИЕ
зуются фильтрованием.
Рис. 33. Сливание жидкости
по стеклянной палочке.
ся более полно отмыть
В лабораторной практике для механического разделения
твердых и жидких компонентов какой-либо смеси обычно поль-
Однако в простейшем случае можно ис-
пользовать сливание жидкости с отстояв-
шегося осадка, т. е. декантацию. Реко-
мендуется использовать оба приема, а
именно: сначала отделить жидкость и
промыть несколько раз осадок деканта-
цией, а затем уже применить фильтро-
вание.
Промывание с применением декан-
тации заключается в том, что осадок за-
ливают водой или специально приготов-
ленной промывной жидкостью, взбалты-
вают при помощи стеклянной палочки
и дают отстояться. Затем жидкость осто-
рожно, во избежание разбрызгивания,
сливают с осадка при помощи стеклян-
ной палочки на фильтр в воронке, при
этом осадок должен оставаться в сосуде
(рис. 33). Промывку осадка повторяют
несколько раз. Путем декантации удает-
осадок от маточного раствора; при филь-
тровании же сделать это удается не всегда, в силу того, что оса-
док легко слеживается. Промывание нужно проводить возможно
малым количеством жидкости, так как абсолютно нерастворимых
веществ нет и каждый раз при промывании свежей порцией
жидкости часть осадка, хотя и незначительная, переходит в рас-
твор. При промыващш осадка наливать жидкость на фильтр
следует в таком количестве, чтобы она полностью покрывала
осадок и не доходила до краев фильтра на 3—5 мм; кроме того,
выливать новую порцию жидкости на фильтр нужно после того,
как предыдущая будет полностью отфильтрована.
Фильтрование характеризуется скоростью и полнотой отделе-
ния осадка от жидкости. На эффективность фильтрования влияют
следующие факторы:
1) вязкость (чем выше вязкость раствора, тем труднее фильт-
ровать) ; ,
2) температура (чем выше температура раствора, тем легче
фильтровать);
46
3)давление (чем выше давление, тем быстрее фильтруется
жидкость);
4) величина частиц твердого вещества (чем больше размер
частиц вещества по сравнению с размером пор фильтра, тем легче
идет фильтрование).
Из фильтрующих средств в лаборатории чаще всего приме-
-няют фильтровальную бумагу, ткани, пористое стекло, асбест
и т. п. Необходимо учесть, что для фильтров нельзя применять
материалы, каким-либо образом взаимодействующие с фильтруе-
мой жидкостью.
ФИЛЬТРОВАНИЕ ПРИ ОБЫЧНОМ ДАВЛЕНИИ
Этот способ фильтрования является наиболее простым и часто
применяемым. Фильтрованием при обычном давлении называется
процесс, в котором жидкость проходит через фильтрующий мате-
риал только под давлением столба филь-
труемой жидкости.
В обычную стеклянную воронку вкла-
дывают простые или складчатые фильт-
ры из фильтровальной бумаги
Для изготовления простого
(рис. 35) квадратный кусок
вальной бумаги складывают
свободный угол полученного
(рис. 34).
фильтра
фильтро-
вчетверо,
квадрата
а
Рис. 34. Стеклянные ворон-
ки с фильтрами:
а—простой фильтр; б — склад-
чатый.
при пользовании склад-
6
обрезают ножницами по пунктирной ли-
нии. Отделив один слой бумаги, расправ-
ляют готовый фильтр, который прини-
мает вид конуса.
Фильтрование значительно ускоряется
чатым фильтром, так как фильтрующая поверхность его больше,
чем у простого фильтра. Од-
нако складчатый фильтр ис-
пользуют лишь в том случае,
когда остающийся на фильтре
осадок не нужен, и если его
немного.
Для приготовления склад-
чатого фильтра (рис. 36) сна-
Рис. 35. Приготовление простого чала, как и в предыдущем
фильтра. случае, складывают фильтро-
вальную бумагу вчетверо и
срезают угол, чтобы получился круг. Круглый фильтр складывают
вдвое, а затем вчетверо; сгиб 2, 1 накладывают на 2, 4, a 2, 3 — на
2, 4. Образуются новые сгибы 2, 5 и 2, 6. Сгибы 2, 3 и 2, 1 по оче-
реди накладывают соответственно на 2, 5 и 2, 6, сгибают и полу-
чают новые сгибы 2, 7 и 2, 8. Таким же образом получают сгибы 2,
10 и 2, 9. Затем складывают гармошкой весь фильтр по линии сги-
ба и после развертывания получают складчатый фильтр.
47
Фильтр следует подбирать таким образом, чтобы величина его
соизмерялась с количеством осадка, при этом край фильтра в
. 4 воронке должен быть всегда ниже
Рис. 36. Приготовление складчатого
фильтра.
края воронки на 3—5 мм. Фильтр
должен плотно прилегать к стен-
кам воронки, причем при вкла-
дывании необходимо следить,
чтобы не прорвалась его верхуш-
ка. Перед фильтрованием фильтр
нужно смочить в воронке чистым
растворителем. Уровень фильтру-
емой жидкости в воронке должен
быть ниже края бумаги.
Условием быстрого фильтро-
вания является наличие жидкости
в трубке воронки. Для этого при смачивании наливают в воронку
растворитель выше края фильтра, а затем немного приподнимают
фильтр и быстро опускают, при этом образуется столб жидкости
в трубке. В тех случаях, когда жидкости имеют большую вязкость,
Рис. 38. Воронки для фильтрования с охла-
ждением.
Рис. 39. Воронки
с пористым стек-
лянным фильтром.
а также при перекристаллизации (см. стр. 53) фильтрование про-
водят при нагревании. Обычно для этой цели применяют воронки
48
для горячего фильтрования (рис. 37). Для фильтрования веществ
с низкой температурой плавления (например, уксусная кислота,
бензол) применяют специальные воронки с охлаждением (рис. 38).
В присутствии сильных щелочей и кислот, ангидридов, окислите-
лей и других веществ, разрушающих фильтровальную бумагу,
осадки фильтруют через пористые стеклянные фильтры (рис. 39).
ФИЛЬТРОВАНИЕ ПОД ВАКУУМОМ
Сущность фильтрования под вакуумом заключается в том, что
в приемнике создают уменьшенное давление, вследствие чего
жидкость фильтруется под давлением атмосферного воздуха. Это
ускоряет процесс фильтрования.
Прибор для отсасывания со-
стоит из фарфоровой воронки
Бюхнера, колбы Бунзена, предох-
ранительной склянки и водоструй-
ного насоса (рис. 40).
Размер воронки Бюхнера дол-
жен соответствовать количеству
отфильтровываемого вещества —
кристаллы должны полностью по-
крывать поверхность фильтра,од-
нако слишком толстый их слой
Рис. 40. Прибор для отсасывания:
/—•воронка Вюхнера; 2—колба Бунзена;
3 — предохранительная склянка.
затрудняет отсасывание и промывание. Между колбой Бунзена и
вакуум-насосом помещают предохранительную склянку, так как
при падении давления в водопроводной сети вода из насоса при
отсутствии предохранительной склянки попадает в колбу Бунзена.
Предохранительную склянку соединяют с водоструйным насо-
сом при помощи толстостенной резиновой трубки, стенки которой
не сжимаются при наличии в трубке разрежения.
Чаще всего в химической лаборатории применяются водоструй-
ные вакуум-насосы, которые работают по принципу увлечения
частиц газа струей жидкости (рис. 41). Они бывают стеклянные и
металлические. Их прикрепляют к водопроводному крану при по-
мощи насадки (рис. 42).
На верхний конец насоса надевают толстостенную резиновую
трубку или прорезиненный шланг длиной 10 см, который закреп-
ляют мягкой железной проволокой, чтобы не просачивалась вода.
Другой конец трубки или шланга соединяют с насадкой крана и
также стягивают проволокой. Затем проверяют насос. Для этого
открывают водопроводный кран, а отверстие бокового отростка
насоса закрывают пальцем. Если палец присасывается, значит на-
сос для работы годен. На боковой отросток водоструйного насоса
надевают толстостенную резиновую трубку, которую соединяют с
предохранительной склянкой.
Чисто вымытую воронку Бюхнера вставляют в колбу на рези-
новой пробке (корковые пробки применять не рекомендуется из-за
их пористости). На сетчатую перегородку воронки помещают
49
кружок фильтровальной бумаги, диаметр которого на 1 мм меньше
внутреннего диаметра воронки. Чтобы вырезать такой кружок, бе-
рут вдвое сложенный лист фильтровальной бумаги, накладывают
сверху на воронку и слегка нажимают ладонью. На бумаге полу-
чается отпечаток круга верхнего диаметра; затем ножницами под-
гоняют кружок до нужного размера. Уложив бумажный фильтр
в воронку, смачивают его растворителем и включают насос с тем,
чтобы присосать фильтр ко дну воронки. В случае хорошо поло-
женного фильтра слышится спокойный шумящий звук, если же
фильтр положен неплотно и имеется подсос воздуха, — свистящий
а, б —стеклянные; в—металлический.
Рис. 42. Насадка к
водопроводному кра-
ну для крепления
водоструйного ва-
куум-насоса.
звук. После проверки фильтра, не выключая насос, в воронку на-
ливают до половины ее высоты фильтруемую смесь.
При фильтровании необходимо следить, чтобы на поверхности
осадка не образовывалось трещин, так как это ведет к неравно-
мерному, неполному отсасыванию и к загрязнению осадка в ре-
зультате испарений растворителя. Кроме того, нужно следить,
чтобы в колбе не собиралось слишком много фильтрата, иначе он
будет втягиваться в насос. При фильтровании огнеопасных жид-
костей необходимо соблюдать соответствующие меры предосто-
рожности (см. гл. II). Чтобы удалить остатки маточного рас-
твора, кристаллы промывают на фильтре небольшими порциями
растворителя. Для этого осадок на фильтре пропитывают раство-
рителем, а затем включают насос.
Промытые кристаллы на фильтре отжимают плоской частью
стеклянной пробки до тех пор, пока не перестанет капать маточ-
ный раствор. Затем вынимают воронку вместе с пробкой из колбы
й вытряхивают фильтр вместе с осадком на фильтровальную бу-
50
магу. Очистив бумажный кружок и стенки воронки лопаточкой от
приставших кристаллов, отжимают полученный продукт в филь-
тровальной бумаге и высушивают на воздухе или' другими спосо-
бами (см. стр. 44).
Глава III
МЕТОДЫ ОЧИСТКИ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ
1. КРИСТАЛЛИЗАЦИЯ
Кристаллизацией называется процесс образования и роста
кристаллов из расплава, раствора или газовой фазы.
Важнейшая задача органического синтеза — получение чистых
веществ. Обычно органические соединения, получаемые в резуль-
тате той или иной реакции, представляют собой «сырые» продукты,
загрязненные примесями. Большинство твердых органических ве-
ществ способно кристаллизоваться и выделяться в более или менее
чистом виде при охлаждении реакционной смеси или после упари-
вания раствора. Полученный продукт требует дальнейшей очистки,
поэтому подвергается перекристаллизации.
Перекристаллизацию проводят так. Растворяют неочищенное
вещество в горячем растворителе, отфильтровывают горячий рас-
твор от взвешенных нерастворимых частиц и охлаждают раствор;
в результате из него обычно выкристаллизовывается более чистое
вещество. Перекристаллизация, однако, дает большие потери ве-
щества в маточном растворе, поэтому рекомендуется предвари-
тельно подвергнуть продукт очистке каким-либо другим способом,
например обычной перегонкой, перегонкой с водяным паром, отде-
лением от примесей при помощи растворителей или поверхностно-
активных веществ.
Из маточных растворов и промывных жидкостей можно извлечь
добавочное количество вещества, если отогнать из них часть рас-
творителя, охладить раствор и произвести кристаллизацию. Полу-
ченные при этом кристаллы обычно бывают менее чистыми, чем
первая порция, их следует перекристаллизовать еще раз.
Если перекристаллизация вещества велась из воды, то маточ-
ный раствор можно упаривать до половины в фарфоровой чашке,
а затем охлаждать для кристаллизации.
ВЫБОР РАСТВОРИТЕЛЯ
Для процесса перекристаллизации большое значение имеет
правильный выбор растворителя. При выборе растворителя необ-
ходимо учитывать химические свойства очищаемого вещества.
51
Вещество должно плохо растворяться в выбранном растворителе на
холоду и хорошо при нагревании; примеси должны либо обла-
дать более хорошей растворимостью в холодном растворителе,
либо плохой — в горячем. Кроме того, растворитель не должен
реагировать с кристаллизуемым веществом, должен способствовать
образованию устойчивых кристаллов и легко удаляться с поверх-
ности кристаллов при отмывании и высушивании. При выборе
растворителя часто можно руководствоваться правилом «подобное
растворяется в подобном». Так, например, фенолы, карбоновые
кислоты, низшие спирты и другие вещества, содержащие гидро-
ксильную группу, легко растворяются в воде; высшие сложные
эфиры — в низших, высшие спирты —в низших спиртах и т. д.
Однако это правило верно только для веществ простого строения,
для сложных соединений оно не всегда соблюдается. Окончатель-
ный выбор растворителя можно произвести лишь опытным путем
в пробирках с малыми количествами вещества, подвергаемого
очистке. При этом отмечают растворимость вещества при нагре-
вании и на холоду, кристаллическую форму выделяющегося осад-
ка, его чистоту и т. п.
Кончиком шпателя берут пробу вещества, помещают ее в про-
бирку и прибавляют несколько капель растворителя. Если веще-
ство растворяется уже на холоду, то данный растворитель непри-
годен для перекристаллизации. Если вещество на холоду раство-
ряется плохо или совсем не растворяется, то пробирку осторожно
нагревают до кипения. В необходимом случае добавляют еще
несколько капель растворителя, чтобы добиться полного раство-
рения. Раствор фильтруют горячим. Если после охлаждения из
раствора выпадут кристаллы, температура плавления которых
совпадает с известной температурой плавления чистого вещества
(часто это удается достигнуть только при многократной кристалли-
зации), то растворитель пригоден для проведения кристаллизации.
Признаком пригодности растворителя для перекристаллизации
вещества, температура плавления которого неизвестна, является
постоянство температуры плавления при повторных кристалли-
зациях. Если вещество не кристаллизуется при охлаждении горя-
чего насыщенного раствора, либо, наоборот, нерастворимо или
мало растворимо даже при нагревании, то данный растворитель
непригоден.
В качестве растворителей при кристаллизации применяют
воду, этиловый и метиловый спирты, бензол, толуол, ацетон, эти-
ловый эфир, ледяную уксусную кислоту, хлороформ, уксусно-
этиловый эфир и др. Легколетучие растворители, как, например,
эфир, сероуглерод, неудобны в употреблении, так как они слиш-
ком легко улетучиваются с поверхности раствора или кристаллов.
Применяя легковоспламеняющиеся растворители (эфир, спирт,
ацетон и др.), растворение следует вести очень осторожно в колбе
с обратным водяным холодильником. Находящиеся поблизости
горелки и электронагревательные приборы с открытым обогревом
должны быть выключены.
52
В тех случаях, когда не удается подобрать индивидуальный
растворитель для перекристаллизации, применяют смеси, состоя-
щие из двух, а иногда и трех растворителей. Для этого вещество
растворяют в том растворителе, в котором оно растворяется очень
легко, и к полученному раствору по каплям добавляют горячий
второй растворитель, плохо растворяющий данное вещество, до
тех пор, пока не образуется устойчивая муть. Эту смесь нагре-
вают до получения прозрачного раствора и оставляют для кри-
сталлизации. Растворители, применяемые совместно, должны сме-
шиваться друг с другом во всех отношениях. В большинстве слу-
чаев применяют следующие смеси: спирт — вода; спирт—бензол;
спирт — ледяная уксусная кислота; ацетон — вода; эфир — аце-
тон— бензол; хлороформ — петролейный эфир и др.
Рекомендуется провести перекристаллизацию загрязненного
ацетанилида (т. пл. 115°C).
ПРОВЕДЕНИЕ ПЕРЕКРИСТАЛЛИЗАЦИИ
Чтобы перекристаллизовать какое-либо вещество, его поме-
щают в колбу, снабженную обратным холодильником (см.
рис. 8,6), если же растворителем служит вода, то обратный холо-
дильник не нужен. Во избежание перегрева жидкости и толчков
при кипячении раствора, в колбу перед нагреванием вносят «ки-
пелки» (см. стр. 30). После этого в колбу вливают растворитель
в несколько меньшем количестве, чем это необходимо для полного
растворения вещества, и нагревают смесь до кипения, так как
обычно кривая растворимости вблизи точки кипения растворителя
резко поднимается вверх. Нагревание ведут на водяной бане, за
исключением тех случаев, когда работают с высококипящими
растворителями. Затем через обратный холодильник осторожно
добавляют такое количество растворителя, чтобы при кипячении
все вещество полностью растворилось. Прежде чем возобновить
кипячение, после внесения холодного растворителя следует «ки-
пелки» заменить новыми, так как они прекращают свое дейст-
вие, если раствор охлаждается ниже температуры кипения (см.
стр. 31).
Иногда твердое неочищенное вещество содержит окрашенные
примеси смолистых продуктов; такие загрязнения трудно отде-
ляются перекристаллизацией. В подобных случаях для обесцвечи-
вания раствора добавляют активированный уголь. Чтобы избе-
жать адсорбции основного продукта, не следует брать слишком
много угля. Обычно его добавляют 1—2% от количества очищае-
мого вещества. Перед внесением активированного угля нужно
несколько охладить раствор, так как уголь выделяет много воз-
духа, а это может привести к сильному вспениванию и выбросу
жидкости. Затем смесь кипятят 5—10 мин с обратным холодиль-
ником и фильтруют еще горячий раствор. Если полученный горя-
чий насыщенный раствор содержит какие-либо механические при-
меси или муть, его отфильтровывают как можно быстрее через
53
складчатый фильтр, смоченный растворителем на воронке для го-
рячего фильтрования.
Приемником при фильтровании может служить фарфоровая
чашка, коническая колба или стакан.
Если же полученный раствор совершенно прозрачен и не со-
держит механических примесей, то фильтрование излишне, так как
оно связано с потерей некоторого количества кристаллизуемого
вещества.
ОТДЕЛЕНИЕ КРИСТАЛЛОВ
После того как все механические примеси будут удалены
фильтрованием, горячий прозрачный раствор либо оставляют
стоять в сосуде, накрытом часовым стеклом для медленного охла-
ждения, либо быстро охлаждают раствор, поместив колбу в хо-
лодную воду или снег. При быстром охлаждении получаются мел-
кие кристаллы, а при медленном — крупные.
Обычно стараются получить кристаллы среднего размера, так
как крупные кристаллы содержат включения маточного раствора,
а мелкие образуют густую кашицу, между отдельными кристал-
лами которой также прочно удерживается маточный раствор.
Некоторые вещества очень трудно кристаллизуются даже при
охлаждении раствора. Это явление чаще всего связано с очень
медленным ростом кристаллов или очень медленным образова-
нием центров кристаллизации. В первом случае кристаллизацию
ведут долго. Если же в растворе медленно образуются центры
кристаллизации, то их можно создать искусственно, внося «за-
травку» в виде нескольких кристаллов того же самого чистого ве-
щества. Кристаллизацию можно ускорить, если потереть стеклян-
ной палочкой стенки сосуда с раствором.
Часто при кристаллизации вещества с низкой температурой
плавления выделяются из растворов в виде масла. Для кристал-
лизации такого вещества раствор разбавляют небольшим количе-
ством чистого растворителя, нагревают до растворения масла и
очень медленно охлаждают. Ускорить кристаллизацию можно, если
потереть стенки сосуда стеклянной палочкой или внести «за-
травку».
Кристаллизация считается законченной, когда дальнейшего вы-
падения кристаллов не наблюдается. Образовавшиеся кристаллы
отделяют от маточного раствора фильтрованием с отсасыванием
на воронке Бюхнера (см. рис. 40) под уменьшенным давлением.
После отсасывания вакуум отключают, заливают кристаллы не-
большим количеством растворителя, перемешивают их стеклянной
палочкой, снова включают вакуум и отсасывают раствор. Обычно
бывает достаточно двукратной промывки кристаллов холодным
растворителем. После отсасывания и промывки осадок отжимают
на воронке широкой стеклянной пробкой, а затем переносят на
фильтровальную бумагу и сушат в зависимости от свойства веще-
ства или на воздухе, прикрыв кристаллы сверху другим листом
фильтровальной бумаги, или одним из ранее описанных методов.
54
Вещества гигроскопические сушат в эксикаторах, а вещества,
устойчивые к действию воздуха и температуры, — в сушильных
шкафах, температура которых должна быть на 20—50 °C ниже
температуры плавления данного вещества.
Чистоту полученного при кристаллизации вещества определяют
по температуре его плавления после высушивания. Если вещество
плавится при более низкой температуре, чем указано в справоч-
нике, то повторяют кристаллизацию до тех пор, пока не получат
вещество с указанной температурой плавления. Следует заметить,
что с каждой новой перекристаллизацией количество вещества
будет уменьшаться, так как потери при кристаллизации неиз-
бежны, как бы тщательно она ни велась.
2. ВОЗГОНКА (СУБЛИМАЦИЯ)
Возгонка — это процесс испарения твердого вещества с по-
следующей конденсацией пара в твердое состояние, минуя жид-
кую фазу. Этим способом пользуются для очистки твердых ве-
ществ, имеющих достаточно большое давление пара при сравни-
тельно невысокой температуре. Возгонка применима особенно в
тех случаях, когда очистка твердого ор-
ганического вещества от смолистых при-
месей путем кристаллизации не дости-
гает цели. Чаще всего возгонка пред-
ставляет собой относительно медленный
процесс. Скорость возгонки прямо про-
порциональна давлению пара вещества
при данной температуре и обратно про-
порциональна внешнему давлению в
приборе. Чем меньше разность ме-
жду внешним давлением и давлением
пара вещества, тем больше скорость воз-
гонки. Кроме того, скорость возгонки
прямо пропорциональна величине поверх-
ности испаряемого вещества и по-
этому препарат надо очень тонко из-
fl 5
Рис. 43. Приборы для воз-
гонки:
а—при атмосферном давлении;
б—в вакууме.
мельчать.
Вещества, имеющие относительно высокое давление пара, при
нагревании могут приобрести давление пара, равное атмосфер-
ному, при температуре, лежащей ниже температуры плавления.
Температура плавления при нагревании этих веществ не дости-
гается, они непосредственно переходят в парообразное состояние,
т. е. возгоняются.
Температура, при которой давление пара над твердым веще-
ством равно внешнему давлению, называется температурой воз-
гонки.
Возгонка проводится как при атмосферном давлении, так и
в вакууме. Для возгонки при атмосферном давлении вещество
помещают на небольшую фарфоровую чашку, покрывают ее куском
55
фильтровальной бумаги с отверстием (чтобы возгон не падал
обратно на возгоняемое вещество) и затем накрывают фарфоро-
вую чашку опрокинутой стеклянной воронкой (рис. 43, а). Отвод-
ную трубку воронки закрывают тампоном из стеклянной ваты.
Чашку с веществом нагревают на маленьком пламени горелки
или на песочной бане. Для охлаждения на внешнюю поверхность
воронки помещают кусочек влажной ваты или ткани. Нагревать
чашку с веществом нужно осторожно; небольшое перегревание
может способствовать быстрому термическому разложению возго-
няемого вещества. Описанным выше путем рекомендуется возго-
нять бензойную кислоту или нафталин.
При возгонке в вакууме (рис. 43, б), снижая давление, можно
добиться того, чтобы и у тех твердых веществ, которые при обыч-
ном давлении плавятся, температура возгонки была ниже темпе-
ратуры плавления.
Рекомендуется очищать возгонкой в вакууме бензойную кис-
лоту, /щавелевую кислоту (кристаллогидрат), ализарин.
Навеску бензойной или щавелевой кислоты (1 г), или взятую на кончике
шпателя пробу ализарина помещают в прибор для возгонки в вакууме. Собирают
прибор, хорошо смазав шлиф. Включают водяное охлаждение После создания
вакуума с помощью масляного насоса прибор медленно нагревают на парафино-
вой бане. Температуру бани необходимо контролировать; температуру начала
возгонки следует записать в журнале. По окончании возгонки прибору дают
охладиться и затем осторожно открывают. Если шлиф не удается открыть, нужно
обратиться к стеклодуву.
3. ЭКСТРАКЦИЯ
Для очистки веществ от примесей и для разделения смеси
веществ часто применяют метод экстрагирования (извлечения).
В зависимости от того, в каком виде находится выделяемое ве-
щество, его можно экстрагировать как из смеси твердых веществ,
так и из растворов.
Очень важной операцией в лаборатории органической химии
является экстракция веществ из растворов (большей частью из
водных), которая заключается во встряхивании раствора в опре-
деленном растворителе (обычно воде) с другим растворителем, не
смешивающимся с первым.
Для экстракции органических соединений из водных растворов
часто применяют диэтиловый и петролейный эфиры, бензин, бен-
зол, хлороформ и др. При этом выбирают такой растворитель,
в котором лучше всего растворяется экстрагируемое вещество или
который легче удалить из вытяжки. Практически вещества, плохо
растворимые в воде, следует извлекать из водного раствора петро-
лейным эфиром или бензином, а вещества, растворимые в воде,—
диэтиловым эфиром, обладающим низкой температурой кипения
и большой способностью растворять органические соединения Во
время работы с огнеопасными растворителями следует погасить
все близко расположенные горелки.
56
Экстрагирование веществ из водных растворов и разделение
несмешивающихся жидкостей осуществляют с помощью делитель-
ных воронок разнообразной формы (рис. 44). Перед началом ра-
боты верхнюю пришлифованную пробку и нижний кран делитель-
ной воронки смазывают вазелином. Однако избытка смазки сле-
дует избегать, так как при соприкосновении с эфиром она может
перейти в раствор. Затем наливают в делительную воронку рас-
твор и добавляют туда растворитель (от ’А до ’/з объема рас-
твора); при этом следят, чтобы количество
не превышало 2/3 ее объема. Делительную
пробкой и, одной рукой придерживая проб-
ку, а другой — кран, осторожно встряхи-
вают ее, плавно переворачивая вверх и
вниз в течение 5—15 мин. Ни в коем случае
нельзя энергично взбалтывать содержимое
воронки, так как при этом образуются
стойкие эмульсии, которые трудно разру-
шаются.
Обычно при встряхивании повышается
давление внутри воронки (часть раствора
превращается в пар). Для выравнивания
его после кратковременного осторожного
встряхивания, держа воронку в переверну-
том положении, открывают кран. Собрав-
шиеся пары эфира выходят через открытый
кран, и давление уравнивается с атмосфер-
ным. Затем кран закрывают и вновь мед-
ленно взбалтывают содержимое воронки,
после этого снова открывают кран. Так по-
вторяют несколько раз. По окончании
жидкости в воронке
воронку закрывают
Рис. 44. Делительные во-
ронки:
а—цилиндрическая; б—ко-
ническая.
встряхивания делительную воронку укрепляют на штативе и дают
жидкости полностью расслоиться на два слоя, причем оба слоя
(эфирный и водный) должны быть прозрачны. Затем открывают
пробку и осторожно поворачивают кран, давая медленно стекать
нижнему слою в сосуд, при этом следят, чтобы вместе с нижним
слоем не слить часть верхнего. Верхний же слой всегда выливают
через верхнее отверстие делительной воронки в приемник.
Обычно органические продукты реакции переходят в верхний
эфирный слой, а неорганические соли, кислоты, основания — в вод-
ный слой. Однако в сомнительных случаях необходимо уточнить,
какой из двух слоев водный; для этого отбирают несколько капель
одного из слоев и добавляют их в пробирку с водой; если капли
не растворились, значит это эфирный слой.
Для боле? полного извлечения экстрагируемого вещества вод-
ный слой снова переносят в делительную воронку и извлекают ве-
щество новой порцией эфира, поступая точно так же, как и в пер-
вый раз. Вещества, плохо растворимые в воде, экстрагируют двумя
порциями растворителя, для хорошо растворимых в воде со-
единений экстракцию надо проводить раза четыре. Следует
57
учитывать, что при проведении экстрагирования целесообразнее
извлекать вещество несколько раз небольшими порциями раствори-
теля, чем сразу обрабатывать раствор большим его количеством
Оптимальные условия экстрагирования определяют исходя из
закона распределения Нернста, согласно которому отношение кон-
центраций вещества, которое растворено в двух несмешивающихся
и находящихся в равновесии растворителях (например, в воде и
эфире), при данной температуре является величиной постоянной:
Рис. 45. Прибор для непрерыв-
ной экстракции растворов.
1 — круглодониая колба: 2—пробир-
ка; 5 —пароотводная трубка; 4—во-
ронка; 5 —обратный холодильник,
А—растворитель, В —экстрагируе-
мый раствор.
где К — коэффициент распределения;
СА и Св — концентрации вещества, растворенного в обоих раство-
рителях.
Экстракция вещества легко осуществима, если вещество рас-
творяется в экстрагирующем растворителе значительно легче, чем
в другом растворителе, и коэффициент
распределения, следовательно, значи-
тельно отличается от 1. Для вещества
с коэффициентом распределения К <
< 100 недостаточно однократной экс-
тракции. В этом случае прибегают к
многократной повторной экстракции
свежим растворителем.
Соединенные эфирные вытяжки
обычно очищают от посторонних ве-
ществ, чаще всего кислот или основа-
ний, увлеченных с растворителем при
экстракции. Для этого их промывают,
т. е. встряхивают в делительной ворон-
ке один-два раза с разбавленным вод-
ным раствором щелочи (сода или би-
карбонат натрия) или кислоты и в за-
ключение один-два раза промывают
водой. При этом всегда следует по-
мнить, что при промывании экстракта
содой может выделяться двуокись
углерода и давление в делительной во-
ронке может повыситься, поэтому вы-
деляющийся газ надо осторожно вы-
пускать через кран при его верхнем
положении.
Затем экстракт высушивают подходящим осушителем (см.
стр. 43). После сушки вытяжку декантируют или фильтруют через
складчатый фильтр, удаляют эфир, отгоняя его на водяной бане
(см. стр. 40), а остаток очищают путем кристаллизации, возгонки
или перегонки.
Часто при экстрагировании из жидкостей, особенно из водных
растворов, образуются трудно разделяющиеся эмульсии. Смеси,
58
Рис. 46. Аппарат1 Сокслета:
/-круглодониая колба; 2-гильза;
3—труба для стока экстракта;
4—экстрактор;
5—пароотводная трубка; 5—обрат-
ный холодильник.
прибор. пригодный для ра-
склонные к образованию эмульсий, в делительной воронке сильно
не встряхивают, а только слегка взбалтывают. Эмульсии возни-
кают по разным причинам. Одна из причин — наличие ничтожного
количества легкого осадка, собирающегося на границе раздела
слоев. Другой причиной может быть большое поверхностное натя-
жение в месте раздела двух жидкостей и, кроме того, малое раз-
личие в их плотностях.
Для разрушения эмульсии пользуются различными приемами
в зависимости от причин ее возникновения. Образующуюся эмуль-
сию можно разрушать добавлением
нескольких капель этилового спирта,
уменьшающего поверхностное натяже-
ние, путем фильтрования с отсасыва-
нием (см. рис. 40), путем длительного
отстаивания, а также насыщением
раствора поваренной солью для уве-
личения плотности водного слоя.
Во многих случаях при экстрагиро-
вании вещества из водного раствора
рекомендуется предварительно насы-
тить этот раствор какой-либо неорга-
нической солью, например поваренной
солью или сернокислым аммонием.
При этом растворимость большинства
органических соединений в воде пони-
жается и в то же время уменьшаются
потери растворителя, так как раство-
римость последнего в воде также сни-
жается. В тех случаях, когда экстра-
гируемое вещество лучше растворяет-
ся в воде, чем в органических раство-
рителях, и извлечение в делительной
воронке не дает хороших результатов,
применяют непрерывную экстракцию
растворов.
На рис. 45 изображен простейший
боты с небольшими количествами раствора. Растворитель, нали-
тый в круглодонную колбу, нагревают на водяной бане до кипе-
ния. Пары его через пароотводную трубку попадают в обратный
холодильник, где конденсируются. Конденсат стекает в воронку,
вставленную в широкую пробирку, содержащую экстрагируемую
жидкость. Так как плотность жидкости выше, чем плотность рас-
творителя, то последний, вытекая из воронки, проходит сквозь
слой экстрагируемого раствора, извлекает растворенное в нем ве-
щество, поднимается на поверхность раствора и стекает обратно
в колбу.
В лаборатории довольно часто приходится проводить экстраги-
рование твердых веществ. Для этой цели используют так назы-
ваемые аппараты Сокслета (лучше всего на шлифах, рис, 46).
59
Аппарат состоит из круглодонной колбы, экстрактора и обратного
холодильника. К колбе с растворителем присоединяют экстрактор,
в который вводят экстрагируемое вещество, плотно завернутое в'
фильтровальную бумагу или помещенное в специальную гильзу.
Растворитель в колбе нагревают на водяной бане до кипения.
Пары его через пароотводную трубку поступают в экстрактор, а
затем в холодильник, где конденсируются. Конденсат стекает в
гильзу с экстрагируемым веществом, извлекает требуемое соеди-
нение и через трубку для отвода экстракта переливается обратно
в колбу. При этом происходит постепенное накапливание извле-
ченного вещества в колбе, причем количество жидкости практи-
чески не изменяется. Это позволяет ограниченным объемом рас-
творителя извлечь неограниченное количество экстрагируемого
вещества, так как оно все время обрабатывается чистым раство-
рителем. По окончании экстракции закрывают воду, снимают
холодильник, отнимают экстрактор, дают стечь из него остаткам
жидкости в колбу, затем отгоняют растворитель.
4. ПЕРЕГОНКА
Одним из важнейших методов очистки и выделения органи-
ческих веществ является перегонка, представляющая собой про-
цесс разделения жидкой смеси на составные части путем нагре-
вания жидкости до кипения и конденсации ее паров в виде ди-
стиллята в холодильнике.
Над всеми жидкостями в результате их испарения устанавли-
вается равновесие между жидкостью и паром, а следовательно, и
определенное давление пара. Величина этого давления зависит от
природы жидкости и температуры. С повышением температуры
давление пара над жидкостью сильно возрастает.
Температуру, при которой давление пара становится равным
внешнему давлению, называют температурой кипения. Любая
жидкость, не разлагающаяся при нагревании до температуры, при
которой давление пара становится равным 760 мм рт. ст., имеет
свою характерную температуру кипения при обычном давлении.
Поскольку температура кипения сильно зависит от давления,
всегда надо указывать давление, при котором эта температура
кипения наблюдалась. Если давление не указано, то подразуме-
вается атмосферное давление.
Содержащиеся в веществе примеси могут по-разному влиять
на температуру кипения, поэтому она менее пригодна для иден-
тификации жидкостей и характеристики их частоты, чем темпера-
тура плавления для твердых веществ.
ПРОСТАЯ ПЕРЕГОНКА ПРИ АТМОСФЕРНОМ ДАВЛЕНИИ
В тех случаях, когда перегоняемое вещество достаточно
устойчиво к нагреванию и практически не разлагается при темпе-
ратуре кипения, для очистки пользуются простой перегонкой при
60
Рис. 47. Прибор для ки-
пячения смеси при одно-
временном введении
жидкости и перемеши-
вания.
атмосферном давлении. Обычно этот способ перегонки целесо-
образно применять для жидкостей с температурой кипения от 40
до 150 °C, так как выше 150 °C многие вещества заметно разла-
гаются, а жидкости с температурой кипения ниже 40 °C перего-
няются со значительными потерями. Часто при перегонке темпе-
ратура кипящей жидкости, вследствие перегрева, несколько выше,
чем температура пара. Перегревы, возникающие при отсутствии
центров кипения в перегоняемой жидкости, приводят к сильным
толчкам, в результате которых вещество вместе с примесями и за-
грязнениями может быть переброшено в приемник. Существуют
различные способы предотвращения или
ослабления толчков при кипении. Чаще все-
го в колбу с жидкостью, подвергаемой пе-
регонке, вносят так называемые «кипелки»
(см. стр. 30).
Часто перед тем как выделить вещество
путем перегонки, приходится в течение мно-
гих часов нагревать при температуре кипе-
ния смесь реагирующих веществ, чтобы
между ними прошла реакция. В таких слу-
чаях применяют кипячение жидкости в при-
боре с обратным холодильником. Если же
приходится вести реакцию с одновремен-
ным введением в колбу жидкости и переме-
шиванием, то используют трехгорлую колбу
или применяют двурогие или трехрогие
форштоссы (рис. 47).
В качестве перегонного сосуда обычно употребляют круглодон-
ные колбы, чаще всего колбы Вюрца. Для перегонки низкокипя-
щих жидкостей берут колбу Вюрца с высоко припаянной отводной
трубкой, для высококипящих— с низко припаянной. Температура
кипения обычно контролируется термометром, ртутный шарик ко-
торого должен полностью омываться парами кипящего вещества,
т. е. верхний край шарика следует устанавливать примерно на
0,5 см ниже отверстия отводной трубки колбы. Иногда нет необ-
ходимости строго контролировать температуру паров при перегон-
ке; в этих случаях горло перегонной колбы Вюрца закрывают
пробкой без термометра или применяют круглодонную колбу, ко-
торую соединяют с холодильником посредством изогнутой стек-
лянной трубки.
Объем перегонной колбы выбирают в зависимости от количе-
ства перегоняемой жидкости и от температуры ее кипения. Жид-
кость должна занимать не более 2/з объема колбы, в противном
случае возможен переброс жидкости во время кипения в прием-
ник. Колба не должна быть слишком большой, особенно при пере-
гонке высококипящих жидкостей, так как в ней остается большое
количество перегоняемого вещества.
Колбу укрепляют в штативе, зажимая ее лапкой выше отвод-
ной трубки. Во избежание загрязнения вещества дистиллят должен
61
по возможности меньше соприкасаться с пробками, поэтому
отводную трубку перегонной колбы соединяют с холодильником
так, чтобы конец ее выступал из пробки в холодильник не менее
чем на 4—5 см и доходил до той части холодильника, которая
охлаждается водой. Размер холодильника выбирают в зависимо-
сти от скорости перегонки и температуры кипения отгоняемой
жидкости. Более подробно о холодильниках см. стр. 16.
Пары веществ, легко кристаллизующихся при комнатной тем-
пературе, не должны охлаждаться в холодильнике до темпера-
туры затвердевания. Для этого холодильник можно периодически
прогревать струей теплой воды.
Жидкости, кипящие в пределах 200—300°C, перегоняют без
холодильника, функцию которого в этом случае может выполнять
отводная трубка колбы Вюрца.
Чтобы по возможности снизить потери вещества за счет испа-
рения дистиллята д приемнике, последний присоединяют к фор-
штоссу холодильника при помощи алонжа. В качестве приемника
обычно употребляют конические или круглодонные колбы. Для бо-
лее полной конденсации паров низкокипящих жидкостей приемник
помещают в сосуд с охлаждаемой смесью.
Когда весь прибор собран, его тщательно проверяют и только
тогда начинают нагревание. В зависимости от температуры кипе-
ния нагревание ведут газовой горелкой через асбестовую сетку,
на закрытом электронагревательном приборе или на бане (стр. 29).
Скорость перегонки обычно выбирают такую, чтобы стекало не бо-
лее 1—2 капель дистиллята в секунду. Во многих работах часто
приходится сначала отогнать легкокипящий растворитель. Эту опе-
рацию следует проводить на водяной бане, так как большинство
растворителей огнеопасно (см. гл. I), а также для того, чтобы
вещество не подвергать лишний раз термическому воздействию,
если в этом нет необходимости. После отгонки растворителя бро-
сают в охлажденную колбу несколько «кипелок» и производят пе-
регонку вещества обычным образом.
ПЕРЕГОНКА С ВОДЯНЫМ ПАРОМ
Перегонка с водяным паром — один из распространенных ме-
тодов разделения и очистки органических веществ. Этот метод
широко используется не только в лаборатории, но и в промыш-
ленности. Перегонка с паром применяется к веществам, которые
практически не смешиваются ^с водой и химически не взаимодей-
ствуют с ней.
Перегонку с паром ведут в следующих случаях.
1. Для выделения из смесей и очистки многих веществ, кото-
рые кипят при очень высокой температуре или вообще не перего-
няются без разложения.
2. Для очистки веществ, загрязненных большим количеством
смолистых примесей.
62
3. Для разделения смесей веществ, из которых только одно
вещество отгоняется с водяным паром (например, природные
смолы и масла могут быть разделены этим методом на летучие
с паром и нелетучие фракции).
4. Для выделения нелетучих с паром твердых веществ из их
растворов в растворителях с высокой температурой кипения (та-
ких, как нитробензол с температурой кипения 210 °C, который пе-
регоняется с водяным паром при 99°C).
5. Для выделения мало растворимых в воде веществ, имею-
щих при температуре около 100 °C заметное давление пара.
С повышением температуры давление паров воды и давление
паров не смешивающегося с нею вещества возрастают практически
независимо одно от другого.
Кипение начинается, когда сумма парциальных давлений насы-
щенного пара будет равна внешнему давлению. Следовательно,
согласно закому Дальтона, суммарное давление пара Р является
суммой парциальных давлений паров перегоняемого! вещества А
и воды В: Р = Ра + Рв. Таким образом, эта смесь перегоняется
при более низкой температуре, чем чистая вода. В дистилляте бу-
дут одновременно пары воды и перегоняемого вещества.
Относительное количество вещества, отгоняемого с водяным
паром, можно найти по следующему уравнению:
Qa . Л Ла
Qb Рв-Мв
где Qa—количество вещества в дистилляте;
Qb — количество воды в дистилляте-
Л1А— молекулярный вес вещества;
Мв — молекулярный вес воды;
Ра — давление паров вещества при температуре перегонки;
Рв— давление паров воды при температуре перегонки.
Давление паров воды Рв при температуре перегонки в каж-
дом случае может быть найдено из табл. 2 Приложения. Давле-
ние паров вещества РА, следовательно, равно 760 — Рв.
Отсюда можно определить весовое количество воды, необхо-
димое для перегонки 1 г вещества:
УВ Ма(760 -Рв)
Так, например, смесь нитробензола (т. кип. 210 °C) и воды пе-
регоняют при 99 °C.
Используя данные табл. 2, находим
733-18
—-----= 4
- 123*27
т. е. для отгона 1 г нитробензола требуется 4 г воды.
Перегонку с водяным паром проводят в приборе (рис. 48), со-
стоящем из парообразователя, перегонной колбы, холодильника и
приемника.
63
Парообразователь (паровик)—это металлический сосуд (его
можно заменить обычной круглодонной колбой емкостью 1,5—
2' л), который имеет предохранительную и водомерную трубки.
Предохранительная трубка доходит почти до дна парообразователя
и предохраняет систему от резкого повышения давления, вызван-
ного сильным нагреванием. Обычно при повышении давления
вода поднимается по этой трубке, иногда происходит даже выброс
воды наружу. В таких случаях нужно убавить огонь.
Парообразователь соединен с перегонной колбой при помощи
резиновой трубки. В качестве перегонной колбы можно применять
колбу Вюрца (с высокоприпаянной трубкой) или обычную кругло-
донную колбу. Трубка, по которой пар вводится в колбу, должна
Рис. 48. Прибор для перегонки с водяным паром:
/ — парообразователь; 2—перегонная колба; 3—холодильник; 4—при-
емник.
доходить почти до самого дна колбы. Необходимо не допускать
переброса перегоняемой жидкости в приемник. Для этого колба
должна быть с длинным горлом и располагать ее надо несколько
наклонно, чтобы брызги не попадали в пароотводную трубку, ко-
торая лишь немного выступает из пробки. Другим концом трубку
соединяют с холодильником. Колбу наполняют жидкостью не бо-
лее чем на !/з ее объема. Между парообразователем и колбой по-
мещают стеклянный тройник. На его боковой отросток надевают
резиновую трубку с винтовым зажимом. Этот тройник позволяет
разбирать прибор после того, как остынут колба и парообразова-
тель и, кроме того, выполняет роль водоотделителя. Дело в том,
что пар из парообразователя идет влажный и перегонная колба
может быстро заполниться водой. В боковом отростке тройника
происходит накапливание воды, которая время от времени спу-
скается.
Перегонку с водяным паром ведут следующим образом: паро-
образователь заполняют водой приблизительно на 2/3 его объема
и нагревают до температуры кипения. Одновременно нагревают
перегонную колбу. Все это время резиновая трубка, надетая на
тройник, открыта.
Предварительный нагрев перегонной колбы необходим потому,
что иначе пары воды, поступающие в колбу, будут охлаждаться
и конденсироваться, увеличивая объем жидкости. Если же смесь
предварительно нагреть, объем жидкости почти не изменяется.
64
Когда вода в парообразователе' закипит, закрывают резино-
вую трубку и начинают перегонку. Образующиеся пары конден-
сируются в холодильнике и поступают в приемник в виде
эмульсии.
Если вещество осаждается в виде кристаллов в холодильнике,
то на короткое время прекращают подачу охлаждающей воды, и
пары вещества, идущие из колбы, расплавляют кристаллы. Нужно
стараться не допустить, чтобы несконденсировавшийся пар увлек
с собой перегоняемое вещество. Впускание холодной воды в хо-
лодильник нужно производить осторожно.
Рис. 49. Пароперегреватель
с термометром.
Рис. 50. Прибор для пе-
регонки малых коли-
честв веществ с водя-
ным паром:
t—круглодонная колба; 2 —
пробирка с двумя трубками;
3—холодильник.
Перегонку ведут до тех пор, пока из холодильника не начнет
вытекать чистая вода. После окончания перегонки сначала от-
крывают зажим, а затем гасят горелки, иначе при охлаждении
парообразователя в нем создается вакуум и жидкость из перегон-
ной колбы может быть втянута в паровик.
Погон разделяют в делительной воронке, затем остаток веще-
ства экстрагируют из воды каким-либо растворителем (см. стр. 58).
Для увеличения выхода отгоняемого вещества, когда плот-
ность данного вещества близка к плотности воды, рекомендуется
прибавлять к воде хлористый натрий. Добавление его снижает
растворимость органического вещества в воде, а также способ-
ствует расслаиванию, так как увеличивается плотность водного
раствора.
Для перегонки веществ, давление паров которых при 100 °C
столь незначительно, что они очень плохо перегоняются с водя-
ным паром, можно использовать перегретый водяной пар, кото-
рый получается в так называемых пароперегревателях (рис. 49).
В качестве пароперегревателя используют свернутую в" спираль
медную трубку, которую в'ключают между парообразователем и
колбой, для перегонки и нагревают горелкой до необходимой
3 М. Н. Храмкина И
1емпературы. Колбу для перегонки помещают на масляную баню,
нагретую до температуры примерно на 10 °C выше температуры
подаваемого перегретого пара. Малые количества вещества можно
перегонять с водяным паром в приборе, изображенном на рис. 50.
ПЕРЕГОНКА ПРИ ПОНИЖЕННОМ ДАВЛЕНИИ
При уменьшении давления температура кипения жидкости
уменьшается. Этим свойством пользуются очень часто как в ла-
боратории, так и на производстве. Некоторые вещества при нагре-
вании их до температуры кипения при нормальном давлении раз-
лагаются. Поэтому для очистки таких веществ применяют пере-
гонку при пониженном давлении (в вакууме). Перегонку проводят
Рис. 51. Прибор для перегонки в вакууме:
/ — колба Клайзена; 2—термометры; 5 —капилляр; 4— холодильник;
5—приемник; ‘6—манометр; 7—тройник; 8—предохранительная склянка.
обычно при остаточном давлении от 50 до 1 мм рт. ст. В некоторых
случаях применяют давление менее 1 мм рт. ст., например при вы-
соковакуумной и молекулярной перегонке.
Понижение температуры кипения с уменьшением давления
проявляется значительно сильнее в области низких давлений. При
уменьшении атмосферного давления на 10 мм рт. ст. температура
кипения жидкости снижается менее чем на 1 °C. При умень-
шении внешнего давления вдвое температура кипения пони-
жается примерно на 15 °C. Так, вещество при обычном давлении
(760 мм рт. ст.) кипит при 180 °C, при 380 мм рт. ст. — около
165 °C, а при 190 мм рт. ст.— около 150 °C.
Прибор для перегонки в вакууме (рис. 51) состоит из колбы
Клайзена, снабженной капилляром и термометром, холодильника,
приемника, манометра, тройника, предохранительной склянки и
водоструйного или масляного насоса. Лучше применять приборы
на шлифах, предохраняющих дистиллят от загрязнения. Если в
лаборатории нет аппаратуры на шлифах, то следует употреблять
резиновые пробки и толстостенные резиновые трубки. Резиновые
пробки должны входить в отверстия приборов на глубину от !/з до
V2 высоты пробки. Очень полезно слегка смазать эти пробки гли-
церином, но лишь настолько, чтобы поверхность пробки не имела
жирного блеска. Недостатком резиновых пробок является их спо-
собность легко поглощать пары многих органических веществ, в
результате чего они разбухают и разрушаются. Иногда можно
66
Рис: 52.
мометра
в колбе
помощи
Крепление тер-
и капилляра
Клайзена при
отрезков рези-
новой трубки.
применять и корковые пробки, пропитанные раствором жидкого
стекла или парафина (см. гл. I).
Резиновые трубки, предназначенные для перегонки в вакууме,
перед употреблением следует очистить кипячением в разбавленном
растворе едкого натра или уксусной кислоты в течение 10 мин,
затем эти трубки тщательно промывают струей воды и сушат, про-
дувая или просасывая через них воздух. Обработанные резиновые
трубки рекомендуется хранить вдали от растворителей и масел.
Перед тем как надеть толстостенную резиновую трубку на оплав-
ленную стеклянную трубку, нужно смазать
поверхность последней тонким слоем глице-
рина.
Колбу Клайзена погружают в баню,
обеспечивающую равномерное нагревание.
Температура бани должна быть примерно
на 20—30 °C выше температуры кипения
вещества. Колбу погружают в баню таким
образом, чтобы уровень перегоняемой
жидкости был немного ниже уровня жидко-
сти в бане. Форма колбы Клайзена такова,
что возможность переброса перегоняю-
щейся жидкости в дистиллят при вспени-
вании или разбрызгивании минимальна.
Применение двугорлой колбы Клайзена
дает возможность укреплять в горлах кол-
бы при помощи резиновых пробок термо-
метр и капиллярную трубку. Если нужно
избежать соприкосновения паров вещества с
колбы Клайзена, у которых горла на концах оттянуты; в таких
колбах термометр и капиллярную трубку укрепляют при помощи
коротких отрезков резиновых трубок, которые надевают на горла
(рис. 52).
Для поддержания равномерного кипения в колбу вставляют,
стеклянную трубку, которая на конце оттянута в тонкий капилляр,
доходящий почти до дна колбы (1—2 мм от дна). Через эту ка-
пиллярную трубку просасывают все время мелкие пузырьки воз-
духа (если вещество взаимодействует с кислородом, то пропускают
аргон, азот или двуокись углерода). Количество воздуха, поступаю-
щего через капилляр в колбу, можно регулировать при помощи
винтового зажима? надетого на толстостенную резиновую трубку,
насаженную на капилляр. Кроме того, рекомендуется вставить
внутрь резиновой трубки нитку или тонкую проволоку, чтобы по-
перечное сечение трубки не могло полностью закрыться.
Капилляр изготавливают из стеклянной трубки диаметром 4—
8 мм. Для этого трубку сначала вытягивают в пламени горелки в
довольно тонкий капилляр, который вторично нагревают, оттяги-
вая на небольшом пламени в тонкую нить. Наличие просвета про-
веряют, опуская капилляр в пробирку с эфиром и продувая через
него воздух; пузырьки должны выходить медленно, по одному, так
3* 67
как большие количества проходящего через капилляр вбЗдуХа сни-
жают вакуум.
К отводной трубке колбы Клайзена при перегонке летучих ве-
ществ, имеющих температуру кипения ниже 150 °C, необходимо
присоединять короткий водяной холодильник, так как неполная
конденсация паров дистиллята приводит к потерям перегоняемого
вещества. Жидкости, кипящие в пределах 150—200 °C, не требуют
большого охлаждения, в этом случае достаточно охлаждать при-
емник струей воды (рис. 53).
В качестве приемников, когда вещество перегоняется без раз-
деления на фракции, пользуются колбами Вюрца или толстостен-
ными колбами для отсасывания. Для собирания нескольких фрак-
ций дистиллята применяют алонж с отводами, или так называемый
Рис. 53. Прибор для перегонки Рис. 54. «Паук»,
высококипящих веществ в вакууме.
«паук» (рис. 54). К отводам на резиновых пробках присоединяют
пробирки или круглодонные колбы, предназначенные для сбора
отдельных фракций. У верхнего конца алонжа имеется отводная
трубка, через которую производится отсасывание воздуха. Чтобы
собрать фракции при вакуум-перегонке, поворачивают алонж во-
круг оси, при этом пробка должна быть слегка смочена глицери-
ном. Для создания вакуума в лаборатории чаще всего применяют
водоструйные насосы или специальные вакуум-насосы, создающие
высокое разрежение.
Водоструйный насос понижает давление приблизительно до
10 мм рт. ст. в зависимости от температуры воды. Для измерения
разрежения при перегонке в вакууме служит ртутный манометр
(рис. 55), который включают в прибор между приемником и предо-
хранительной склянкой водоструйного насоса. Предохранительная
склянка присоединяется к прибору, чтобы избежать переброса
воды из насоса в манометр или прибор (например, при внезапном
падении давления воды в водопроводной сети). Между манометром
и предохранительной склянкой ставят стеклянный тройник с на-
детыми на резиновые трубки винтовыми зажимдми. Тройник по-
зволяет соединять прибор с насосом или атмосферой.
И
Приступая к перегонке под вакуумом; необходимо соблюдать
все меры предосторожности и надевать защитные очки или лице-
вые щитки из пластмассы.
Успешность вакуум-перегонки в значительной степени зависит
от правилньости сборки прибора, при этом особое внимание обра-
щается на герметичность аппаратуры. Чтобы проверить, хорошо
ли «держит вакуум» прибор, плотно закрывают зажимом резино-
вую трубку, присоединенную к трубке капилляра, включают насос
и наблюдают, насколько быстро создается необходимое разреже-
ние. Если поворотом зажима отключают прибор
ртути в манометре должен оставаться без из-
менения. Если разрежение не достигается,
проверяют все места соединений, более плотно
вставляют пробки, более глубоко надевают
резиновые трубки и т. д.
После того как прибор проверен на герме-
тичность, колбу Клайзена наполняют через
горло, предназначенное для термометра, пере-
гоняемой жидкостью до !/2 объема. Затем
включают насос и, после того как в приборе
установится нужное давление, начинают по-
степенно нагревать колбу на водяной или мас-
ляной бане или бане со сплавом Вуда. Тем-
пература бани, измеряемая термометром,
должна превышать температуру кипения ве-
щества на 20—30 °C. При помощи зажима ре-
от насоса, уровень
Рис. 55. Ртутный ма-
нометр.
гулируют впуск воздуха через капилляр так,
чтобы обеспечить равномерное кипение. Когда жидкость начнет
кипеть, регулируют нагревание таким образом, чтобы в приемник
капало не более двух капель в секунду.
В случае обильной конденсации паров жидкости на стенках
колбы Клайзена и слишком медленной перегонки колбу теплоизо-
лируют слоем асбеста. Если во время вакуум-перегонки возникла
необходимость очень сильно нагреть перегонную колбу., то повы-
шение температуры до нужного предела следует проводить посте-
пенно, так как резкий подъем температуры приведет к быстрому
парообразованию, а образующаяся при этом смесь паров перего-
няемого вещества с воздухом окажется взрывоопасной.
После окончания перегонки прежде всего прекращают нагре-
вание, удаляют баню, чтобы колба Клайзена могла быстро остыть,
поворотом зажима отключают прибор и манометр от вакуум-на-
соса, выключают последний и, медленно открывая винтовой за-
жим, который зажимает резиновую трубку на капиллярной трубке
колбы Клайзена, дают воздуху проникнуть внутрь прибора для
выравнивания давления. Если же при сборке прибора был исполь-
зован очень тонкий капилляр и винтового зажима для регулирова-
ния впуска воздуха не было, то воздух в прибор впускают, осто-
рожно открывая зажим у тройника.
69
Силу просасывания воздуха контролируют по манометру, при-
чем ртуть должна медленно переходить из открытого колена в за-
крытое. Если впустить сразу много воздуха, ртуть может пробить
запаянное колено и манометр выйдет из строя; кроме того, воз-
можно разбрызгивание остатков жидкости в колбе Клайзена и
загрязнение ими дистиллята в приемнике. После того как давле-
ние в приборе уравняется с атмосферным, можно приступить к
разборке аппаратуры. Прежде всего отсоединяют приемник, затем
колбу Клайзена. Из колбы сначала осторожно вынимают термо-
метр, потом капилляр.
ФРАКЦИОННАЯ (ДРОБНАЯ) ПЕРЕГОНКА
При простой перегонке по мере испарения смеси (если смесь
состоит из двух компонентов) содержание низкокипящего компо-
нента в отходящих парах непрерывно уменьшается, максимальное
содержание низкокипящего компонента в них —в начальный мо-
мент перегонки. При этом можно получать несколько фракций
различного состава, раздельно собирая их. Способ перегонки с
разделением смеси на несколько фракций, в различной степени
обогащенных низкокипящим компонентом, называется фракцион-
ной перегонкой.
В основе теории фракционной перегонки лежат два закона фа-
зового равновесия в системе жидкость — пар, разработанные в
1881—1884 гг. Д. П. Коноваловым.
Законы Коновалова в современной формулировке читаются
так:
1) в двойной жидкой системе пар относительно богаче тем
компонентом, прибавление которого повышает общее давление
пара.
2) максимуму или минимуму давления пара двойной жидкой
системы соответствует жидкая смесь, насыщенный пар которой
обладает одинаковым с ней составом.
Из законов Коновалова вытекают два следствия:
I) в двойной жидкой системе пар относительно богаче тем
компонентом, прибавление которого понижает температуру ки-
пения;
2) максимуму или минимуму температуры кипения двойной
жидкой системы соответствуют жидкость и пар с одинаковым ка-
чественным и количественным составом.
Смеси полностью смешивающихся жидкостей разделяются на
два класса. Смеси первого класса могут быть разделены на чи-
стые компоненты, они не образуют нераздельно кипящих смесей.
Смеси второго класса образуют нераздельно кипящие смеси, кото-
рые, несмотря на различные температуры кипения чистых веществ,
не могут быть разделены перегонкой на чистые компоненты.
‘Смеси первого класса имеют давление пара, по величине яв-
ляющееся промежуточным между давлениями паров обоих чистых
компонентов (рассмотрим для простоты перегонку бинарных сме-
70
сей). Низкокипящая жидкость имеет высокое давление пара, кото-
рое становится равным атмосферному при относительно низкой
.температуре, в то время как высококипящая жидкость имеет низ-
кое давление пара, которое становится равным атмосферному
лишь при относительно высокой температуре.
Смеси первого класса выделяют пары, в которых содержание
легколетучего компонента всегда больше, чем в исходной смеси.
Поэтому жидкость в колбе при перегонке все более и более обо-
гащается высококипящим компонентом.
Для увеличения эффективности разделения смеси и уменьше-
ния числа перегонок следует пользоваться дефлегматорами (см.
рис. 10). Сущность действия дефлегматоров состоит в том, что
вследствие охлаждения наружным воздухом часть паров перего-
няемой смеси конденсируется. Сконденсировавшиеся пары, назы-
ваемые флегмой, стекают обратно в колбу, обогащаясь высококи-
пящим компонентом, а пары, которые летят дальше, обогащены
низкокипящим компонентом. Устройство дефлегматоров обеспечи-
вает хороший контакт между стекающей вниз жидкостью (флег-
мой) и поднимающимся вверх паром. В образовавшейся противо-
точной системе происходит непрерывный тепловой и материальный
обмен, в результате пар обогащается низкокипящим компонентом,
а стекающая вниз жидкость — высококипящим.
Отношение количества конденсата, возвращающегося в дефлег-
матор в виде орошения, к общему количеству получаемого за тот
же промежуток времени отгона называется флегмовым числом. По
этой величине можно определить эффективность всего процесса,
так как она характеризует точность разделения смеси на компо-
ненты и продолжительность разгонки. Например, если флегмовое
число большое, разделение смеси будет тем более полным, чем
меньше скорость перегонки.
Рассмотрим разделение смеси бензола и толуола путем фракционной (дроб-
ной) перегонки. Эта смесь удобна тем, что ее компоненты имеют различные тем-
пературы кипения.
Для опыта выдается определенное весовое количество смеси толуола (т. кип.
110,6 °C; d — 0,86) и бензола (т. кип. 80,2 °C; d = 0,88). Приступая к разгонке,
нужно записать в рабочий журнал веса колбы и смеси или общий объем (в мл)'.
Для фракционной перегонки собирают прибор, состоящий из круглодонной
колбы, дефлегматора, термометра, холодильника, алонжа и приемника (см.
рис. 11). Круглодонную колбу берут такой емкости, чтобы перегоняемая жидкость
занимала не более 2/з объема колбы. В колбу, помещенную иа песочную баню
и закрепленную в штативе, при помощи пробки вставляют дефлегматор, причем
его подбирают с учетом свойств жидкостей, образующих смесь. Так, для веществ,
кипящих ниже 150 °C, берут более длинный дефлегматор, для веществ, кипящих
выше 150 °C, — более короткий. В данном случае (бензол + толуол) нужен длин-
ный дефлегматор. Дефлегматор также прикрепляют к штативу, затем его соеди-
няют с холодильником и уже после этого вставляют в дефлегматор пробку
с термометром. Напоминаем, что для веществ, кипящих ниже 150 °C, употребляют
водяной холодильник Либиха; для веществ, кипящих выше 150 °C, — воздушный.
В данном случае следует пользоваться водяным холодильником. Термометр
укрепляют таким образом, чтобы участок шкалы между 75 и 120 °C не был за-
крыт пробкой, и верхний край шарика ртути был примерно на 0,5 см ниже
отверстия отводной трубки дефлегматора. Шарик ртути не должен прикасаться
к стеклу. Затем к холодильнику крепят алонж и подставляют приемник. Готовят
три приемника (плоскодонные колбы), предварительно взвешенные вместе с Проб-
ками иа технических весах с точностью до 0,1 г и пронумерованные (можно
также мензуркой измерить объем фракций и расчет вести в миллилитрах).
Первая разгонка. В колбу помещают всю смесь, «кипелки» и начинают
осторожно нагревать. Нагревание колбы во время перегонки регулируют таким
образом, чтобы дистиллят поступал в приемник со скоростью 30—40 капель в
минуту.
Последовательно меняя приемники, собирают три фракции в определенных
температурных интервалах, которые определяют следующим образом. Находят
разность температур кипения чистых веществ, составляющих данную смесь, и
делят эту разность на три, т. е. на число намеченных к отбору фракций. Напри-
мер, температура кипения чистого бензола 80,2 °C, чистого толуола 110,6 °C.
Разность температур кипения этих веществ составляет 30 °C. Разделив эту раз-
ность на три, получают интервалы по 10 °C, в которых отбирают каждую из
фракпий.
Следовательно, при разгонке смеси из бензола и толуола надо брать фрак-
ции в следующих температурных интервалах: / — 80—90 °C; II— 90—100 °C;
III—100—110,6 °C. Фракция I содержит главным образом бензол, фракция
II — смесь бензола и толуола, III — в основном толуол. Приемники при отборе
фракций меняют, как только термометр покажет верхнюю границу температурного
интервала фракции. Перегонку прекращают, когда в колбе остается 1—2 мл
перегоняемой жидкости, так как нельзя перегонять вещество досуха. Окончив
разгонку, взвешивают или измеряют объем полученных фракций и остатка из
перегонной колбы. Вычисляют общую массу (или объем) продуктов перегонки,
затем рассчитывают (в %) массу (или объем) каждой фракции к общей массе
(объему) продуктов отгона. Все эти данные заносят в рабочий журнал.
Вторая разгонка. В чистую и сухую перегонную колбу помещают фракцию I
и перегоняют ее в прежних температурных интервалах, т. е. 80—90 °C. В первый
приемник собирают отгон до температуры 90 °C, выключают горелку, дают колбе
слегка остыть и приливают через дефлегматор фракцию II к остатку в перегонной
колбе. Опять нагревают колбу, при этом в первый приемник собирают все то,
что перегоняется до 90 °C, а во второй приемник — то, что перегоняются в пре-
делах 90—100 °C. Затем выключают горелку и по охлаждении колбы приливают
фракцию III к остатку в перегонной колбе. Возобновляют перегонку, собирая в
первый приемник отгон до 90 °C; во второй — отгон до 100 °C; в третий — до
110,6 °C. Окончив вторую разгонку, взвешивают (измеряют объем) полученные
фракции и остаток от перегонки, вычисляют общую массу (объем) продуктов
второй разгонки и процентное содержание полученных фракций от общей массы
(объема) продуктов второй разгонки. Эти данные также заносят в рабочий
журнал.
Подобным образом можно провести и последующие разгонки, однако при
большом количестве перегонок получаются большие потери вещества.
По окончании последней разгонки следует указать выход фракции I (бен-
зольной) в процентах к количеству бензола, находившемуся в смеси, и выход
фракции II (толуольной) в процентах к количеству толуола, находившемуся
в данной смеси.
РЕКТИФИКАЦИЯ
Как простая, так и фракционная перегонка не дают возмож-
ности провести полное разделение компонентов смеси и получить
их в чистом виде. Оба компонента являются летучими и потому
оба переходят в пары, хотя и в различной степени. Для достиже-
ния наиболее полного разделения компонентов применяют ректи-
фикацию.
Ректификацией называется многократная дистилляция, осуще-
ствляемая при противотоке пара и жидкости. При соприкоснове-
нии поднимающихся паров со стекающей жидкостью происходит
Рис. 56. Ректификацион-
ная колонка:
/ — насадка; 2— изоляция;
3—’термометр; 4—холодиль-
ник.
частичная конденсация паров и частичное испарение жидкости.
При этом из паров конденсируется преимущественно высококипя-
щий компонент, а из жидкости испаряется преимущественно низ-
кокипящий. Таким образом, стекающая жидкость обогащается
высококипящим компонентом, а поднимающиеся пары — низкоки-
пящим, в результате чего выходящие из аппарата пары представ-
ляют собой почти чистый низкокипящий компонент. Эти пары кон-
денсируются. Часть конденсата возвращается на орошение обратно
в колонку в виде флегмы, другая часть от-
водится в качестве дистиллята.
Для ректификации применяют лабора-
торные ректификационные колонки раз-
личных конструкций (например, рис. 56),
которые присоединяют к перегонной колбе.
С помощью ректификационных колонок
удается разделять жидкости, температу-
ры кипения которых отличаются лишь
на 2 °C.
Эффективность колонки зависит от ее
высоты, характера насадки, количества
флегмы и качества теплоизоляции. Часто
в лаборатории употребляют упрощенные
ректификационные колонки, наполненные
обрезками стеклянных трубок, стеклянными
бусами и т. д.
Однако не всегда вещества, кипящие
при различных температурах, могут быть
разделены при помощи перегонки. Эти ве-
щества образуют так называемые нераз-
дельно кипящие, или азеотропные, смеси.
Таковы, например, смеси: этиловый спирт —
бензол (32:68); этиловый спирт — хлоро-
форм (7:93); этиловый спирт — четырех-
хлористый углерод (16:84); этиловый
спирт — вода (95:5); хлороформ — вода
(97 : 3) и т. д. Существуют двойные и трой-
ные смеси с определенным соотношением компонентов, у которых
состав насыщенного пара и жидкости одинаков.
Такие смеси кипят при температуре более низкой или более
высокой, чем температура кипения каждого компонента смеси
в отдельности. Азеотропные смеси встречаются очень часто, при-
чем большинство из них имеет температуру кипения более низкую,
чем температура кипения каждого компонента смеси в отдель-
ности. Так, например, смесь, состоящая из 7 ч. этилового спирта
и 93 ч. бензола, кипит при 60 °C, несмотря на то, что спирт кипит
при 78 °C, а бензол при 80 °C.
Явление азеотропии обусловлено сложным взаимодействием
молекул в жидкости, осн.ованным на силах сцепления, ассоциации
и сольватации. Азеотропия находит разнообразное применение. Ею
73
пользуются для выделения и очистки различных веществ, для уда-
ления воды из реакционной смеси, для абсолютирования раствори-
телей и т. д.
Для выделения одного из компонентов азеотропной смеси
можно пользоваться вымораживанием, а также химической реак-
цией, в которой участвует один из компонентов смеси.
5. ХРОМАТОГРАФИЯ
В начале этого века русский ученый М. С. Цвет открыл способ
адсорбционного хроматографического анализа веществ и положил
начало одному из эффективных методов разделения и очистки ве-
ществ — хроматографии.
Этот метод основан на том, что при пропускании исследуемого
раствора или газообразных продуктов через какой-либо адсорбент,
вещества поглощаются им с различной силой. В зависимости от
взаимодействия между адсорбентом и находящимся в растворе
веществом различают три вида хроматографии: адсорбционную,
распределительную и ионообменную. Обычно приходится иметь
дело со смешанными процессами при преобладании одного из них.
Так, при адсорбционной хроматографии имеют значение процессы
ионного обмена, а при распределительной хроматографии — про-
цессы адсорбции и ионного обмена.
Хроматография в настоящее время широко применяется пре-
жде всего для разделения малых количеств веществ, близких по
своему составу и свойствам; для очистки соединений, которые об-
ладают высокой температурой кипения или же термически неустой-
чивы, а также для разделения некоторых природных веществ, на-
пример природных красителей, аминокислот и т. п.
АДСОРБЦИОННАЯ ХРОМАТОГРАФИЯ
Принцип адсорбционной хроматографии заключается в том,
что при пропускании исследуемого раствора или газообразных
продуктов через высокую и сравнительно узкую колонку, напол-
ненную мелко раздробленным или порошкообразным адсорбентом,
вещества поглощаются в соответствии с их склонностью к адсорб-
ции в определенной последовательности.
Образуется так называемая «хроматограмма», которую «про-
являют», пропуская через колонку некоторое количество чистого
растворителя, обычно того же, в котором растворено исследуемое
вещество.
Наиболее часто применяемые для проявления адсорбционных
хроматограмм растворители можно расположить в следующем
порядке: петролейный эфир, бензин, сероуглерод, четыреххлори-
стый углерод, бензол, хлороформ, эфир, этилацетат, ацетон, про-
пиловый спирт, этиловый спирт, метиловый спирт, вода, пиридин.
При промывании колонки растворителем происходит распределе-
ние растворенных веществ по колонке, при этом образуются цо-
74
лосы или зоны, перемещающиеся вниз по пути движения прояв-
ляющей жидкости со скоростью, которая зависит от способности
данного вещества адсорбироваться находящимся в колонке ад-
сорбентом. Обычно это свойство у разных веществ различно, по-
этому зоны более или менее четко отделяются друг от друга.
При разделении окрашенных или окрашивающихся при ад-
сорбции веществ можно наблюдать возникновение окрашенных зон
непосредственно на бесцветных или слабоокрашенных адсорбентах.
Если же визуально трудно определить положение зон на столбике
адсорбента, то применяют ультрафиолетовые лучи, так как многие
вещества флуоресцируют в ультрафиолетовом свете. Следова-
тельно, рекомендуется иметь колонки из кварцевого стекла, про-
пускающего эти лучи.
Если же имеют дело с бесцветными и нефлуоресцирующими
веществами, то применяют флуоресцирующие адсорбенты. Для
этого к обычному адсорбенту добавляют какое-либо вещество,
флуоресцирующее в ультрафиолетовом свете, например сернистый
цинк, какой-либо флуоресцирующий органический краситель.
Для выделения веществ из проявленной хроматограммы при-
меняют различные приемы. В тех случаях, когда работают с окра-
шенными веществами или когда можно легко различить границы
Отдельных зон по флуоресценции в ультрафиолетовом свете, осто-
рожно выталкивают адсорбент с поглощенными веществами из ко-
лонки и, разрезав его на зоны, экстрагируют вещества из каждой
зоны отдельно.
Другой способ обработки хроматограммы заключается в том,
что адсорбированные вещества элюируют, т. е. вымывают раство-
рителями из колонки и собирают в виде отдельных фракций рас-
творов (элюатов). Затем в этих фракциях обнаруживают или
количественно определяют разделенные вещества соответствую-
щими химическими или физическими методами. При снятии такой
«жидкой хроматограммы» более сильно адсорбируемые вещества
появляются в элюате позже, чем менее сильно адсорбируемые.
При так называемом вытеснительном проявлении для элюиро-
вания применяют растворитель, который вытесняет из колонки
подлежащие разделению вещества в порядке уменьшающейся
прочности их связей с адсорбентом. Кроме этих описанных мето-
дов выделения веществ из хроматограмм, существуют самые раз-
нообразные способы, частично использующие каждый из этих
приемов.
' В лабораторных условиях для хроматографирования обычно
пользуются простым прибором, изображенным на рис. 57. В каче-
стве хроматографической колонки используют стеклянную трубку,
суженную в нижней части. Иногда для поддержания заданной
температуры при элюировании применяют колонку, устроенную по
типу обычного холодильника Либиха, т. е. омывающуюся водой
с определенной температурой. Размеры трубки устанавливают в
зависимости от условий опыта и от предъявляемых требований. Во
всяком случае, трубка не должна быть слишком широкой, так как
75
Рис. 57. Прибор для ад-
сорбционной хромато-
графии:
/—адсорбент; 2—пробка нз
ваты; 3 — растворитель.
при этом уменьшается четкость разделения зон при проявлении
хроматограммы. В нижнюю часть трубки вставляют неплотный
тампон из обычной или стеклянной ваты для удержания адсорбен-
та, а в верхнюю часть трубки при помощи резиновой пробки кре-
пят капельную воронку.
Наиболее распространенными адсорбентами являются окись
алюминия, используемая для разделения нейтральных и основных
веществ, и активированный уголь, применяемый для адсорбции ве-
ществ из водных или спиртовых растворов.
Реже применяются силикагель, окись маг-
ния, гидроокись кальция, углекислые и сер-
нокислые соли щелочноземельных и ще-
лочных металлов, а также глюкоза, лак-
тоза и др.
Для успешного разделения смеси боль-
шое значение имеет наполнение хромато-
графической колонки достаточно однород-
ным адсорбентом, при этом чем меньше
размер частиц, тем лучше происходит раз-
граничение зон при проявлении хромато-
граммы, но одновременно с этим увеличи-
вается и сопротивление колонки. Чтобы из-
бежать этого недостатка, иногда рекомен-
дуется примешивать к измельченному ад-
сорбенту некоторые сорта инфузорной зем-
ли, адсорбционная способность которой
очень незначительна. На результаты хрома-
тографии отрицательно влияет наличие пу-
зырьков воздуха и трещин в адсорбенте, а
также неравномерность засыпки последнего.
Наполнение колонки может произво-
диться двумя способами: сухим и влажным.
Сухой способ наполнения. Колонку промывают горячей хромо-
вой смесью, затем дистиллированной водой и высушивают. Тща-
тельно просеянный измельченный адсорбент вносят в колонку в
сухом виде равномерными маленькими порциями и утрамбовы-
вают плотно входящим пестиком. Заполнение колонки ведут та-
ким образом, чтобы */з трубки была незаполнена.
По окончании наполнения колонки адсорбент увлажняют рас-
творителем, снова прессуют пестиком, затем выравнивают его по-
верхность и помещают сверху адсорбента кусочек ваты, предо-
храняющий верхний слой столбика адсорбента от размыва раство-
рителем. Поверх ваты наливают слой растворителя в 5—7 см
высотой, в противном случае образуются трещины в адсор-
бенте.
Влажный способ наполнения. Этот метод заключается во введе-
нии в колонку суспензии адсорбента в соответственно подобран-
ном растворителе. Адсорбент и растворитель хорошо перемеши-
вают механической мешалкой и через делительную воронку при
7*
постоянном помешивании вносят образовавшуюся суспензию в ко-
лонку. Затем на расстоянии 1 см от поверхности адсорбента по-
мещают рыхлый кусок ваты, поверх которого наливают слой рас-
творителя в 5—7 см высотой.
' В подготовленную колонку осторожно из капельной воронки
вводят раствор, состоящий из смеси веществ в подходящем рас-
творителе. Соотношение количества данной смеси к адсорбенту
должно составлять примерно 1 : 100. Как только раствор впи-
тается, вливают в воронку чистый растворитель и начинают прояв-
ление хроматограммы. В современных лабораториях элюат
собирают в виде большого числа маленьких порций по 0,5—10 мл.
Этот процесс осуществляется автоматически с помощью специаль-
ных коллекторов. Затем растворитель из элюата удаляют пере-
гонкой или выпариванием, выделяя чистое вещество. Если выде-
ленный таким путем компонент смеси является жидкостью, то ее
перегоняют и определяют температуру кипения, показатель луче-
преломления, плотность. Если выделено твердое вещество, то опре-
деляют температуру плавления и при необходимости проводят
перекристаллизацию.
Разделение о-, м- и n-нитроаинлинов. Хроматографическую колонку (см.
рис. 57) заполняют 50 г гидроокиси кальция по сухому способу наполнения.
Раствор смеси о-, м- и n-иитроаиилинов (по 0,15 г каждого) в 50—60 мл петро-
лейиого эфира пропускают через адсорбент со скоростью 1—2 капли в секунду.
Хроматограмму проявляют 200 мл петролейиОго эфира, при этом появляются три
полосы: верхняя — светло-желтая (п-нитроаиилин); средняя — желтая (л-нитро-
аиилии); нижняя — желто-коричиевая (о-нитроанилии).
Для выделения иитроаиилииов проводят элюирование полос смесью из 4 мл
Метилового спирта и 200 мл бензола, прибавляя ее по каплям из капельной во-
ронки По мере вымывания полос собирают фракцию элюата в разные приемники.
Затем от каждой фракции отгоняют растворитель, определяют выход и темпера-
туру плавления каждого из выделенных веществ и, если нужно, перекристалли-
зовывают их.
РАСПРЕДЕЛИТЕЛЬНАЯ ХРОМАТОГРАФИЯ
Распределительная хроматография заключается в фракцион-
ном разделении смеси, компоненты которой имеют различные ко-
эффициенты распределения между двумя несмешивающимися
растворителями.
В качестве носителя обычно применяют силикагель, реже крах-
мал, клетчатку, так как они обладают небольшой адсорбцией ко
многим веществам.
Одним из растворителей пропитывают твердый носитель, но
так, чтобы он казался на вид все еще сухим. Этот растворитель
называется неподвижным. Исследуемую смесь растворяют (или
суспендируют) во втором растворителе, называемом подвижным,
и пропускают равномерно раствор (или жидкую кашицу) через
колонку (рис. 58). После того как раствор впитается, колонку про-
мывают чистым подвижным растворителем до тех пор, пока ком-
поненты смеси веществ не образуют в колонке зоны или пока ве-
щества не окажутся в растворителе, капающем из нижнего конпа
7/
колонки (это обнаруживают по специфическим реакциям). Затем
компоненты последовательно элюируют и по мере вымывания зон
собирают фракции элюата в разные приемники. Растворитель из
фракций удаляют перегонкой или выпариванием, выделяя чистые
вещества.
В отличие от адсорбционной хроматографии распределитель-
ная хроматография особенно пригодна для разделения близких
членов гомологических рядов, отличающихся главным образом
растворимостью.
ХРОМАТОГРАФИЯ НА БУМАГЕ
Частный случай распределительной хроматографии — хрома-
тография на бумаге, используемая главным образом для каче-
ственного анализа смесей органических веществ. Этот вид хрома-
Рис. 58. Прибор для рас-
пределительной хромато-
графии:
t — растворитель; 2—пробка
пз ваты; 5—силикагель: 4 —
пробирки для собирания
элюата.
тографии выполняется весьма просто и к
тому же с очень малыми количествами ве-
ществ.
В качестве носителя для неподвижного
растворителя применяют чистую целлюлозу
в виде специальной фильтровальной бу-
маги. Она должна обладать высокой чисто-
той и равномерной плотностью. Неподвиж-
ным растворителем в бумажной хромато-
графии в большинстве случаев является
вода, всегда присутствующая в фильтро-
вальной бумаге. Однако неподвижной фа-
зой на бумаге может быть и другой рас-
творитель. В качестве подвижных раство-
рителей чаще всего используют бутиловый
и амиловый спирты, фенол, крезолы, изо-
масляную кислоту или их смеси, предвари-
тельно насыщенные водой.
Техника бумажной хроматографии со-
стоит в следующем: каплю раствора иссле-
дуемой смеси наносят на бумагу и высуши-
вают. Затем бумагу помещают в закрытый
сосуд, в котором она непрерывно смачи-
вается растворителем. Этот растворитель
равномерно движется по бумаге в одном
направлении, при этом вещества, входящие
в состав исследуемой смеси, перемещаются по бумаге в том же
направлении с различной скоростью, образуя отдельные зоны или
пятна. Для получения хороших результатов при хроматогра-
фировании на бумаге необходимо соблюдать следующие ус-
ловия:
1. Применять очень чистые растворители.
2. Наносить пятна размером 0,5—1 см при больших дозах и
0,2 см при малых.
78
3. Обеспечить газонепроницаемость хроматографической каме-
ры и насыщение ее парами обеих фаз,
4. Поддерживать во время опыта постоянную температуру,
чтобы не происходило расслоения фаз жидкости.
5. Соблюдать чистоту, чтобы избежать загрязнения влажных
хроматограмм посторонними примесями при их перемещении.
Идентификация каждого компонента смеси по окрашенному
пятну производится на основании расчета величины коэффициента
распределения Rf. Коэффициент распределения характеризуется
отношением расстояния х
(мм), пройденного веществом
от линии нанесения вещества
(линия старта) до центра пят-
на, к расстоянию у (мм),прой-
денному растворителем от той
же линии старта до фронта
растворителя. Значение Rf вы-
числяют по формуле:
= 7
Коэффициент распределе-
ния для каждого соединения
является характерной величи-
ной. Для большого числа ве-
ществ значения Rf приведены
в специальных таблицах, по-
этому легко можно определить,
Рис. 59. Проведение восхо,гя1цей хро-
матографии иа бумаге
1— бумага до хроматографии; 2 — бумага пос те
хроматографии; 5 —прибор для проведения
восходящей хроматографии;
А—линия старта; 5 —граница фронта раство-
рителя; В— растворитель.
какому веществу соответствует
найденная величина. Значения Rf существенно зависят от темпе-
ратуры, растворителя, качества бумаги и других факторов. По-
этому при идентификации вещества следует для сравнения парал-
лельно хроматографировать пробу чистого препарата этого же ве-
щества— «свидетеля». По‘этому способу на расстоянии 3—4 см
ту и другую сторону от капли смеси на той же линии старта на-
носят капли «свидетеля». После хроматографирования и выявле-
ния пятен сопоставляют положение пятен компонентов исследуе-
мой смеси с положением пятен «свидетеля».
Известны разные способы осуществления бумажной хромато-
графии, характеризующиеся той или иной техникой выполнения:
восходящая хроматография, нисходящая хроматография и круго-
вая хроматография.
Простейший случай хроматографии на бумаге — восходящая
хроматография с водой в качестве неподвижного растворителя.
Она заключается в следующем. Вырезают полоску фильтроваль-
ной бумаги, соответствующую размерам применяемого для хрома-
тографии цилиндра (обычно полоску вырезают шириной 15—30 мм
и длиной 300—-500 мм). На расстоянии 3 см от нижнего края по-
лосы бумаги проводят карандашом линию старта. На этой линии
ца расстоянии 2—2,5 см друг от друга и от краев полоски
79
помечают кружками диаметром 3 мм точки старта. Исследуемый
раствор вещества в воде или легколетучем растворителе должен
иметь примерно 1%-ную концентрацию по отношению к каждому
компоненту смеси. При помощи специальной пипетки или капил-
ляра наносят в каждую точку старта капли исследуемого ве-
щества.
Затем наливают подвижный растворитель на дно цилиндра
(высота слоя ~ 2 см), подвешивают высушенную полоску таким
образом, чтобы она не касалась ни стенок, ни жидкости (рис. 59),
и оставляют на ночь для насыщения атмосферы цилиндра парами
данного растворителя.
На следующий день нижний край полоски погружают на 0,5 см
в подвижный растворитель, который в течение 12—18 ч под дей-
ствием капиллярных сил поднимается вверх приблизительно на
20—25 см. Затем вынимают полоску, отмечают карандашом верх-
нюю границу фронта растворителя и высушивают хроматограмму.
Если пятна не окрашены и не флуоресцируют в ультрафиолетовом
свете, их проявляют, опрыскивая из пульверизатора реактивами-
индикаторами, дающими окрашивание с соответствующими компо-
нентами.
ИОНООБМЕННАЯ ХРОМАТОГРАФИЯ
Ионообменная хроматография основана на обмене между
ионами, находящимися в растворе, и ионами некоторых погло-
тителей, называемых ионитами. Примерами таких веществ могут
служить сульфированный уголь, полиметакриловая кислота, про-
дукты конденсации различных замещенных фенолов и амино-
фенолов с формальдегидом и др. Многие иониты применяются как
в водной среде, так и в органических растворителях. Перед упо-
треблением их измельчают и подвергают очистке, для этого после-
довательно обрабатывают ионит кислотой и щелочью. Полученный
чистый и сухой ионит суспендируют в воде и наполняют им хро-
матографическую колонку, как указано выше (стр. 76). Через эту
колонку пропускают раствор исследуемой смеси со скоростью
1 мл/мин. Затем проявляют хроматограмму путем элюирования.
Ионообменная хроматография имеет большое значение для
исследования родственных друг другу соединений и применяется
в основном в аналитической химии.
Глава IV
ОПРЕДЕЛЕНИЕ ВАЖНЕЙШИХ КОНСТАНТ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Для идентификации органических веществ и доказательства
чистоты того или иного вещества используют методы определения
физических констант органических соединений. Чаще всего для
80
выяснения степени чистоты кристаллического вещества достаточно
определить температуру его плавления, а для жидкости — плот-
ность, температуру кипения и показатель лучепреломления.
1. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
Температурой плавления считается та температура, при кото-
рой замечается первое появление жидкой фазы. Разность между
температурой, при которой появляется жидкая фаза, и темпера-
турой полного расплавления вещества
для чистых соединений не должна превы-
шать 0,5 °C. Незначительные загрязнения
вещества иногда сильно понижают тем-
пературу его плавления и плавление про-
исходит в более широком интервале тем-
ператур. Такое явление используют
для установления идентичности двух ве-
ществ с одинаковой температурой плав-
ления.
Для этого тщательно смешивают
равные количества двух веществ. Если
температура плавления этой «смешанной
пробы» остается неизменной, то делают
заключение об идентичности обоих ве-
ществ. Понижение же температуры плав-
ления пробы служит признаком неиден-
тичности этих веществ. Оценка идентич-
ности исследуемого вещества по темпе-
ратуре плавления смешанной пробы яв-
ляется настолько общепринятой, что этот
прием часто считают вполне достаточным
для вынесения окончательного решения.
Многие органические вещества пла-
вятся с разложением, которое обнаружи-
вается обычно по окрашиванию расплава
или выделению газов. Разложение яв-
ляется химическим процессом и темпе-
ратура, при которой оно происходит, зависит, в первую очередь,
от продолжительности и скорости нагревания, а также от плот-
ности набивки вещества в капилляре, диаметра капилляра и т. п.
Нередко в качестве характеристики веществ, которые плавятся
с разложением, в справочной литературе приводится величина
температуры плавления с дополнением «разл.».
Между температурой плавления вещества и его строением су-
ществует зависимость. Так, симметрично построенные молекулы
плавятся при более высокой температуре, чем их изомеры.
Температуру плавления органического кристаллического веще-
ства можно определить в капилляре. Испытуемое вещество в
а 6
Рис. 60. Прибор для опре-
деления температуры пла-
вления:
1—термометр с капилляром,
помещенный в пробирку; б,—
крепление на термометре ка-
пилляра с веществом.
81
количестве ~ 0,001 г помещают в маленький капилляр, запаянный
с одного конца. Капилляр вытягивают из хорошо вымытой и вы-
сушенной тонкостенной стеклянной трубки диаметром 10—12 мм.
Исследуемое вещество тщательно растирают в ступке или на ча-
совом стекле. Открытым концом капилляра набирают в него не-
много вещества и бросают его запаянным концом вниз в стеклян-
ную трубку длиной 80—90 см, поставленную вертикально на лабо-
раторный стол. Эту операцию наполнения повторяют несколько
раз до получения
в
Воздух
Рис. 61. Прибор Тиля
для определения тем-
пературы плавления.
капилляре хорошо уплотненного столбика ве-
щества высотой ~ 2 мм. Наполненный та-
ким образом капилляр закрепляют на тер-
мометре так, чтобы проба вещества находи-
лась на уровне шарика термометра. Для
прикрепления капилляра к термометру
обычно применяют резиновое кольцо.
Температуру плавления возгоняющихся
веществ определяют в капиллярах, запаян-
ных с обоих концов.
Наиболее простой прибор для опреде-
ления температуры плавления изображен
£ща рис. 60. Он состоит из круглодонной
колбы, имеющей боковые отверстия для
испарения обогревающей жидкости. Капил-
ляр вместе с термометром помещают в про-
бирку, вставленную в эту колбу. Шейка
колбы должна быть длинной, чтобы пред-
отвратить
уменьшить
мометра
ртути,
описанным
разбрызгивание жидкости и
погрешность в показании тер-
на выступающий столбик
способом гораздо лучше прбво-
По сравнению с
дить определение температуры плавления в приборе Тиля (рис. 61),
в’котором осуществляется более равномерный перенос тепла.
В качестве теплопередающей среды жидкости используют
воду, если известно, что температура плавления исследуемого ве-
щества ниже 100°C, или концентрированную серную кислоту, ко-
торая дает возможность определять температуру плавления до
250°C. При длительном пользовании серная кислота темнеет; для
обесцвечивания вносят в нее кристаллик селитры. Определений
температуры плавления в приборах с серной кислотой требует
осторожности, так как в случае поломки прибора горячая серная
кислота представляет опасность. Поэтому при работе с серной кис-
лотой следует надевать защитные очки. Кроме воды и серной
кислоты употребляют парафиновое масло (температура разложе-
ния 220°C). Однако парафиновое масло пр сравнению с серной
кислотой менее теплопроводно, вследствие чего может быть не-
равномерное нагревание прибора. Лучше всего пользоваться сили-
коновым маслом. При работе с веществом, плавящимся выше
300°C, нельзя применять масло, так как оно довольно быстро
82
темнеет, в этом случае пользуются смесью 54,5% азотнокислого
калия и 45,5% азотнокислого натрия. Такая смесь плавится при
218 °C и допускает нагревание до 600 °C.
После того как прибор собран, начи- л
нают медленно нагревать его на асбестовой
сетке маленьким пламенем горелки и вни-
мательно следят за повышением темпера-
туры и состоянием столбика вещества в
капилляре. Наблюдая за веществом в капил-
ляре, отмечают все его изменения — пере-
мену окраски, разложение, слипание, спека-
ние, намокание и т. п. Когда исследуемое
вещество начинает заметно сжиматься и
мокнуть, горелку удаляют. Началом плав-
ления считают появление первой капли в
капилляре, а окончанием — исчезновение
последних кристалликов вещества. Для
определения температуры плавления ве-
ществ, плавящихся выше 300 °C, желатель-
но употреблять металлический блок (рис.
62), изготовленный из латуни или меди.
Нижняя часть блока нагревается горелкой.
В цилиндрический канал помещается тер-
Рис. 62. Металлический
блок для определения
температуры плавления:
1 — канал для термометра;
2—термометр; 3 — капилляр;
4—смотровые окна.
мометр, а в пазы — два капилляра с веществом. Наблюдение за
плавлением в капиллярах ведут через смотровые окошки, закры-
тые небольшими стеклами.
2. ТЕМПЕРАТУРА КИПЕНИЯ
Температура кипения — это температура, при которой давле-
ние пара жидкости становится равным внешнему давлению. Тем-
пература кипения в значительной степени зависит от молекуляр-
ного веса вещества, его строения, состава, межмолекулярного
взаимодействия и давления.
Самый простой прибор для определения температуры кипе-
ния— это обычный прибор для перегонки, состоящий из кругло-
донной колбы, термометра, холодильника, алонжа и приемника.
В круглодонную колбу наливают до *Д объема колбы жидкость,
температуру кипения которой нужно определить. Шарик термо-
метра находится на небольшом расстоянии от поверхности жид-
кости. Если же определяют температуру кипения раствора, то
шарик термометра опускают в жидкость. Чтобы избежать сильного
перегрева жидкости, для нагревания при опредёлении темпера-
туры кипения применяют соответствующие бани. Разность темпе-
ратур начала и конца кипения для чистых веществ не должна
превышать 0,5 °C. Кипение жидкости в широком интервале гово-
рит о смеси жидкостей.
При определении температуры кипения следует помнить о по-
правке на отклонение от нормального давления. Для многих
83
веществ в справочной литературе есть таблицы поправок темпера-
тур кипения при разных давлениях для приведения их к
760 мм рт. ст. Если же таблиц нет, то можно вычислить поправку
на отклонение от атмосферного давления исходя из того, что тем-
пература кипения многих веществ при 760 мм рт. ст. изменяется
приблизительно на 3/8о с изменением давления на 1 мм рт. ст.
Если давление А, при котором идет определение температуры
кипения, меньше 760 мм рт. ст., то поправку щ на отклонение
можно вычислить с достаточной степенью точности по уравнению:
л, =-^-(760-А)
Если давление А выше, то поправку п2 получают из урав-
нения:
«2 = 4-(А-760)
При работе с малыми количествами вещества используют ме-
тод Сиволобова. По этому методу 0,5 мл исследуемой жидкости
Рис. 63. Способ крепле-
ния трубки с веществом
к термометру при опре-
делении температуры
кипения методом Сиво-
лобова.
наливают в стеклянную трубку диаметром ~6 мм, в которую
погружают незапаянным концом вниз капилляр для определения
температуры плавления. Трубку прикрепляют к термометру, как
показано на рис. 63. Термометр помещают в прибор для опреде-
ления температуры плавления. При медленном нагревании сна-
чала наблюдается слабое, а затем и бурное выделение пузырьков
пара из капилляра. Температурой кипения считают показания тер-
мометра в тот момент, когда начинается бурное образование пу-
зырьков. Ошибки этого метода могут составлять ±5 °C,
84
Точное определение температуры кипения возможно с помощью
эбулиоскопов (рис. 64). Принцип их действия основан на том, что
жидкость нагревают с обратным холодильником до кипения и из-
меряют температуру; при соответствующей конструкции исклю-
чаются тепловые потери и перегрев паров. Однако эти приборы
требуют обычно больших количеств вещества (несколько милли-
литров).
3. ОТНОСИТЕЛЬНАЯ ПЛОТНОСТЬ
Относительная плотность — одна из важнейших физико-хими-
ческих характеристик вещества, и ее определение представляет со-
бой одну из наиболее обычных операций в лабораторной прак-
тике. Плотность имеет большое значение .для распознавания
многих жидких изомеров, характеристики смесей, вычисления мо-
лекулярной рефракции и т. п.
Плотностью вещества называют отношение массы этого веще-
ства к его объему:
где т—масса вещества;
V — объем вещества.
Обычно в лабораторной практике пользуются относительной
плотностью, представляющей собой отношение плотности данного
вещества к плотности другого вещества при определенных усло-
виях. Относительную плотность d вещества обычно определяют
по отношению к плотности дистиллированной воды:
где р — плотность вещества;
рв — плотность дистиллированной воды при 4 °C.
Относительная плотность выражается отвлеченным числом.
Так как все тела изменяют свой объем в зависимости от темпе-
ратуры, то ясно, что и величина плотности будет колебаться в ре-
зультате изменения температуры, при которой ее определяют. По-
этому всегда приводят температуру, при которой делали определе-
ние, и температуру воды, объем которой принят за единицу.
Обычно относительную плотность определяют при 20 или 15°C и
обозначают при помощи соответствующих индексов, например d't';
приведенное обозначение указывает, что относительная плотность
определена при 15 °C и за единицу для сравнения взята плотность
воды при 4 °C.
Относительную плотность можно определить при помощи
ареометров, пикнометров, гидростатических весов и т. п. В про-
стейших случаях для быстрого определения относительной плотно-
сти жидкости применяют так называемые ареометры (рис. 65).
Определение плотности при помощи ареометров основано на том,
что если тело, погруженное в жидкость, не тонет, а плавает, то
85
глубина его погружения зависит от плотности данной жидкости.
Ареометр представляет собой стеклянную трубку, расширяющуюся
внизу и имеющую на конце стеклянный шарик, заполненный дро-
бью или специальной массой, чтобы во время измерения ареометр
находился в вертикальном положении. В верхней узкой части
ареометра помещена шкала с делениями. Чем меньше отно-
сительная плотность жидкости, тем глубже погружается в нее
а 6
Рис. 65. Ареомет-
ры:
а—без термометра,
б —с термометром.
а
Рис. 66. Положе-
ние ареометра при
измерении плот-
ности.
ареометр, поэтому на его шкале вверху нанесено наименьшее зна-
чение относительной плотности, внизу наибольшее. Иногда арео-
метры снабжены термометрами (рис. 65, б), что позволяет одно-
временно измерять температуру, при которой проводится опре-
деление.
Для определения относительной плотности при помощи арео-
метра жидкость наливают в стеклянный цилиндр емкостью не ме-
нее 0,5 л и осторожно погружают в него ареометр. Ареометр дол-
жен находиться в центре цилиндра, ни в коем случае не касаться
стенок или дна цилиндра (рис. 66). Деление, против которого
установился верхний мениск жидкости, характеризует величину
плотности.
Для определения относительной плотности жидкостей с точно-
стью до четвертого знака пользуются пикнометрами (рис. 67).
Сначала на аналитических весах с точностью до 0,0001 г опреде-
ляют массу пустого пикнометра, потом с водой, а затем с иссле-
86
дуемой жидкостью и находят массы равных объемов исследуемой
жидкости. Взяв отношение этих масс, получают значение относи-
тельной плотности d-.
где р — масса пустого пикнометра;
Pi — масса пикнометра с исследуемой жидкостью;
р2— масса пикнометра с водой.
Измерения следует производить дважды, причем при одной и
той же температуре.
Рис. 67. Пикнометры,
Для определения относительной плотности жидкости и твер-
дого тела с успехом можно применять гидростатические весы. В ка-
честве таких весов могут служить обыкновенные аналитические
весы. Чтобы определить относительную плотность жидкости, на
левую чашку весов помещают цилиндрический сосуд с исследуе-
мой жидкостью, а к крючку, на котором висит дужка с чашкой,
подвешивают цилиндрическое стеклянное грузило.
Сначала взвешивают грузило на воздухе, затем определяют
вес его в дистиллированной воде, а затем в исследуемой жидкости
и по формуле находят относительную плотность данной жидкости:
Ро ~ Рв
где ро — вес грузила в воздухе;
рв — вес грузила в дистиллированной воде;
р — вес грузила в исследуемой жидкости.
Для определения относительной плотности твердого тела по-
следнее прикрепляют вместо грузила и взвешивают его на воздухе
и в дистиллированной воде. Если тело на воздухе уравновешивают
грузом р, а при погружении этого тела в воду — грузом рв, то
относительная плотность тела будет равна:
т. е. весу тела, деленному на вес вытесняемой им воды.
97
4. ПОКАЗАТЕЛЬ ПРЕЛОМЛЕНИЯ
Показатель преломления широко используется для идентифи-
кации жидких веществ и проверки их чистоты, при анализе двой-
ных жидких растворов, для которых известна зависимость показа-
теля преломления от состава раствора, при определении концен-
трации сахара в водных растворах и во многих других случаях.
Показатель преломления п — величина постоянная для дан-
ного вещества, равная отношению синусов угла падения света на
Рис. 68. Прелом-
ление луча света
на границе двух
прозрачных сред.
Рис. 69. Ход лучей в призмах
рефрактометра Аббе.
I — осветительная призма, 2—изме-
рительная призма; 3— исследуемая
жидкость: 4— поле в зрительной
трубе при измерении в проходящем
свете, 5—-то же в отраженном
свете, 6—-ход лучей при измерении
в проходящем свете, 7—ход лучей
при измерении в отраженном свете.
поверхность раздела двух сред и угла преломления света (рис. 68):
Обычно первоначальной средой является воздух, т. е. падение
света на преломляющую среду идет через воздух.
Показатель преломления сильно зависит от температуры и
сильно изменяется в зависимости от длины волны света. Так, у ор-
ганических жидкостей с ростом температуры на 1 °C он падает
на 4>10~4. Поэтому измерение показателя преломления проводят в
монохроматическом свете при постоянной температуре, при этом
индексом указывают принятое буквенное обозначение спектраль-
ной линии, в свете которой проводилось измерение, или длину вол-
ны, а показателем — температуру, например п® (обычно показа-
тель преломления п дается для спектральной D-линии желтого
натриевого пламени).
Простым и распространенным прибором для измерения пока-
зателя преломления является рефрактометр Аббе. Измерительная
призма снабжена осветительной призмой, зазор между которыми
заполнен 1—2 каплями исследуемой жидкости (рис. 69). Рефрак-
тометр Аббе снабжен компенсатором, позволяющим проводить
99
измерения при освещении дневным или электрическим светом. Точ-
ность измерения рефрактометром Аббе составляет 1 • 10~3. Чтобы
добиться такой точности, во время измерения следует поддержи-
вать постоянную температуру, что достигается с помощью термо-
стата.
5. МОЛЕКУЛЯРНЫЙ ВЕС
Молекулярный вес — одна из самых существенных характери-
стик вещества. Он лежит в основе определения таких величин, как
молярные концентрации, объем, теплоемкость, электропроводность,
теплота реакции и т. д. Молекулярный вес используется для уста-
новления строения вещества, применяется во всех расчетах по хи-
мическим формулам и уравнениям.
Существует много различных методов, дающих возможность
определять молекулярный вес веществ органических соединений
любого класса. Наибольшее распространение получили криоскопи-
ческий, эбулиоскопический методы и метод Раста.
Для определения молекулярного веса криоскопическим мето-
дом берут отвешенное количество чистого растворителя и изме-
ряют его температуру замерзания. Затем вновь в том же сосуде
расплавляют растворитель, вносят в него навеску определяемого
вещества и измеряют температуру начала кристаллизации. По
эбулиоскопичеекому методу повышение температуры кипения
растворителя, обусловленное введением определенного количества
испытуемого вещества, измеряют термометром Бекмана, а затем
по формулам рассчитывают молекулярный вес. (Подробное описа-
ние криоскопии и эбулиоскопии дается в руководствах по физико-
химическому практикуму.)
В настоящем пособии более подробно остановимся на методе
Раста, позволяющем определять молекулярный вес органических
соединений в приборе для определения температуры плавления.
Этот метод применим для веществ, растворимых в расплавленной/
камфоре и устойчивых до 190 °C. Метод состоит в определении по-
нижения температуры плавления камфоры при растворении в ней
определенного количества исследуемого вещества. Для определе-
ния молекулярного веса требуется всего 10—60 мг вещества; точ-
ность определения 5—10%.
Рассмотрим в качестве примера определение молекулярного
веса, нафталина по понижению температуры плавления камфоры.
В небольшую чистую сухую пробирку помещают 60—70 мг нафта- ч
лина и около 0,7—0,8 г камфоры. Осторожно, на маленьком пламе-
ни, нагревают пробирку для сплавления этих веществ. Затем уда-
ляют из пламени пробирку, перемешивают полученный расплав
проволочкой, сплющенной на конце. После того как расплав за-
твердевает, извлекают большую его часть той же проволочкой на
часовое стекло. Массу растирают и набивают ею капилляр диамет-
ром ~2 мм (см. стр. 82). В другой такой же капилляр набирают
пробу чистой камфоры. Оба капилляра крепят при помощи
ао
маленького резинового кольца к термометру с делениями 0,1 или
0,2 °C. Затем начинают медленно нагревать прибор, внимательно
наблюдая за состоянием проб через лупу, и в момент плавления
последних кристаллов смеси камфоры с нафталином записывают
температуру, принимая ее за температуру плавления смеси. Про-
должая нагревание, определяют температуру плавления чистой
камфоры в другом капилляре. Молекулярный вес исследуемого ве-
щества (в данном случае нафталина) рассчитывают по формуле:
40 • lOOOmi
М =---П----7Т
т2 (б —t2)
где т{— навеска исследуемого вещества;
т2— навеска камфоры;
/1—температура плавления чистой камфоры;
/2—температура плавления смеси;
40 — криоскопическая константа.
Для контроля рекомендуется повторить определение с другой
парой капилляров.
Глава V
РАБОТА СО СЖАТЫМИ
И СЖИЖЕННЫМИ ГАЗАМИ
1. ГАЗОВЫЕ БАЛЛОНЫ И ОБРАЩЕНИЕ С НИМИ
Многие газы поступают в лабораторию в стальных баллонах
в сжатом или сниженном состоянии. В сжиженном состоянии в
баллоне может находиться только такой газ, критическая темпе-
ратура которого выше обычной комнатной температуры (двуокись
углерода, хлор, сернистый газ, аммиак и др.). В этом случае дав-
ление газа остается постоянным, пока в баллоне еще есть жидкая
фаза. Наоборот, газы, имеющие низкую критическую температуру
(кислород, водород, азот, воздух и другие), не обращаются в жид-
кость при обыкновенных условиях и накачиваются в баллоны в
сжатом состоянии под давлением в 150—200 кгс/см2; по мере рас-
ходования газа давление в баллоне постепенно падает. Величины
давления, под которым находятся в баллонах некоторые сжижен-
ные газы, указаны в Приложении 3.
По конструкции баллоны бывают двух типов: для сжатых и
для сжиженных газов; последние внутри имеют сифонную трубку.
Наиболее распространены баллоны емкостью 20 и 40 л. Все бал-
лоны для сжатых и сжиженных газов периодически испытываются
на заводах-изготовителях; так, например, баллоны для сероводо-
90
рода проверяются один раз в год; для хлора фосгена — один раз в
два года; остальные — раз в 3 года.
Баллоны для сжатых газов представляют собой стальные ци-
линдры (рис. 70), имеющие днище, горловину, башмак. В горло-
вину баллона ввернут вентиль для впуска и выпуска газа. Кроме
того, на горловину насажено кольцо с резьбой, на которое навер-
тывается предохранительный колпак, защищающий вентиль от по-
вреждений. Колпак пломбируется, а на сферической части баллона
Рис. 70. Устрой-
ство простого га-
зового баллона:
/—корпус: 2—кольцо;
3—башмак; 4— гор-
ловина; 5— вентиль;
в—Колпак.
Рис. 71. Разрез Рис. 72. Подстав- Рис. 73. Разрез
баллона с сифо- ка для газового ацетиленового
ном: баллона: баллона:
/—корпус; 2—сифон- 1 — баллон; 2—под- / — подставка; 2—кор-
ная трубка; 3—вен- ставка. пус баллона; 3— гор-
тиль; 4—колпак. ловииа; 4—вентиль.
высекается марка завода-изготовителя, тип и номер баллона, вес
его, дата изготовления, срок следующего испытания, рабочее Р и
пробное П давления, емкость баллона И клеймо ОТК завода.
Баллоны для хлора или фосгена (рис. 71) состоят из корпуса,
навинчивающегося колпака и вентиля. Внутри такого баллона от
горла и почти до дна проходит сифонная трубка, через которую
жидкий хлор или жидкий фосген поступают в вентиль, где испа-
ряются. Такое устройство затрудняет регулировку тока хлора или
фосгена и замедляет их испарение, таю как при быстром поступ-
лении их вентиль охлаждается и покрывается инеем. Чтобы избе-
жать этого, следует устанавливать баллоны данного типа в лабо-
ратории вентилем вниз, для чего служат специальные подставки
(рис. 72). При таком положении по сифонной трубке в вентиль по-
ступает газ, и ток его можно точно регулировать. В противном слу-
чае при открывании вентиля из баллона будет выходить жидкость,
а не газ.
Для хранения ацетилена применяют специальные баллоны
(рис, 73), Чистый ацетилен уже при давлении 2 кгс/см2 может
91
легко взорваться даже от сотрясения. Растворенный же ацетилен
дая^е под давлением 25 кгс/см2 безопасен. Поэтому баллоны для
ацетилена наполняют какой-либо пористой массой (активирован-
ным углем, диатомитом, хризотил-асбестом и др.), которую про-
Рис. 74. Редукционный вен-
тиль:
/—регулировочный винт; 2—от-
росток, 3— накидная гайка; 4 —
манометр, указывающий, под
каким давлением газ находится
в баллоне; 5 —манометр, пока-
зывающий рабочее давление.
питывают ацетоном, и в этом ацетоне растворяют ацетилен под
давлением до 16 кгс/см2. Если даже где-нибудь внутри -баллона и
произойдет взрыв, то пористая масса не даст ему распространиться
на весь газ и тем предохранит баллон от разрыва.
Во избежание ошибок при пользовании газами в баллонах по-
следние имеют опознавательную окраску (см. Приложение,
табл. 4). Кроме того, баллоны, содержа-
щие водород, метан, этан, этилен и дру-
гие горючие газы, отличаются тем, что
боковой штуцер' вентиля имеет «левую»
резьбу; этим устраняется возможность
опасной ошибки как при наполнении
баллонов, так и при работе со сжатыми
газами.
Для регулирования тока газа, вы-
пускаемого из баллона, следует пользо-
ваться редукционными вентилями (рис.
74), которые позволяют иметь на выходе
постоянное давление. Они бывают раз-
личных конструкций и отличаются друг
от друга пропускной способностью, вели-
чиной рабочего давления и принципом
действия. Лабораторные редукционные
вентили имеют небольшую пропускную
способность — до 1 м3/ч и снабжены ре-
при помощи которого можно устанавли-
вать точный расход газа. Более совершенные вентили имеют два
манометра, один из которых показывает давление газа в баллоне,
а другой — давление струи выходящего газа. Обычно редукцион-
ные вентили окрашивают в тот цвет, в который окрашен газовый
баллон.
Приступая к отбору газа из баллона, в первую очередь нужно
хорошо укрепить баллон, снять пломбу, а затем ключом отвернуть
и снять колпак, осмотреть вентиль и боковой штуцер баллона; при
этом на вентиле не должно быть следов жира или масла, а резьба
бокового штуцера не должна иметь повреждений. Если вентиль
исправен, то производят его продувку, открывая вентиль быстрым
поворотом маховика, и тотчас закрывают; при продувке горючего
газа все источники огня в лаборатории устраняются и включается
гулировочным винтом 1,
тяга.
Редукционный вентиль привинчивается к боковому штуцеру
баллона при помощи накидной гайки 3, причем, в зависимости от
того, для какого газа предназначен баллон, эта гайка имеет пра-
вую или левую резьбу. Затем поворотом налево вывинчивают регу-
лировочный винт, запирая таким образом редукционный вентиль,
92
а кран завинчивают до отказа. На отросток 2 надевают резино-
вый шланг для отвода газа. После этого открывают вентиль бал-
лона поворотом маховика против часовой стрелки. Если при этом
слышится шипение выходящего газа, вывинчивают еще винт 1.
Если газ проходит у гайки 3, то ее подвинчивают.
Когда выход газа прекратится, манометр 4 покажет давление
газа в баллоне. Затем осторожно ввинчивают винт 1, пока мано-
метр 5 не покажет нужное рабочее давление или не начнется вы-
пуск газа с требуемой скоростью. После этого медленно вывинчи-
вается кран и из отростка 2 начинает выходить газ. Необходимо
помнить, что выпускать весь газ из баллона нельзя, обычно отбор
газа прекращают, когда остаточное давление в баллоне составляет
1—1,5 кгс/см2. После окончания работы вентиль плотно закрывают,
поворачивая маховик по часовой стрелке, затем закрывают выход
из редукционного вентиля, ослабляя регулировочный винт, т. е.
поворачивая его налево.
2. ДОЗИРОВАНИЕ ГАЗОВ
При работе с газами в лабораторий скорость тока газа кон-
тролируется пропусканием газа через счетчик пузырьков, т. е. не-
большой сосуд, где газ пробулькивает через жидкость. В качестве
жидкости применяют концентрированную серную кислоту, а при
Рис. 75. Схема водяных
газовых часов:
1 — корпус; 2 —полая ось;
3— выпуклые лопасти.
Рис. 76. Реометры:
1 — капилляр; 2—жидкост-
ный дифференциальный ма-
нометр.
работе с аммиаком —50%-ный раствор едкого кали. Если тре-
буется одновременно измерить скорость тока газа и высушить его,
применяют в качестве счетчика пузырьков промывную склянку
Тищенко, которую присоединяют к редукционному вентилю при
помощи резинового шланга.
Количество газов измеряют по их объему или массе. Объемы
газов либо отмеривают прямо в калиброванных сосудах — газомет-
рах, либо измеряют газовыми часами. Чаще всего пользуются
так называемыми жидкостными газовыми часами с водяным на-
полнением (рис. 75), в которых газовый поток вращает барабан,
связанный с отсчетным устройством. Эти часы состоят из
93
Рис 77. Ротаметр:
1—вертикальная
трубка; 2 — волчок.
цилиндрического корпуса, в котором вращается полая ось с выпук-
лыми лопастями, соединенная со стрелкой на циферблате. Газ по-
ступает в часы через полую ось и попадает под одну из лопастей,
при этом начинает вращаться ось. Заполнив пространство под одной
лопастью, газ начинает поднимать следующую лопасть и т.д. Одна-
ко газовые часы не применяют для работы с агрессивными газами
или с газами, хорошо растворимыми в воде, так как газовые часы
изготовлены из металла и наполнены водой.
При пропускании через реакционный сосуд
равномерного тока газа дозирование осуществ-
ляют путем измерения скорости движения газо-
вого потока в единицу времени. Чаще всего для
этой цели применя'ют реометры и ротаметры.
В реометрах (рис. 76) на пути газового потока
при прохождении его через сужение (капилляр)
возникает разность давлений с обеих сторон уз-
кого отверстия. Перепад давлений, пропорцио-
нальный количеству протекающего газа, изме-
ряют при помощи параллельно подключенного
U-образного манометра со шкалой, прокалибро-
ванной для непосредственного отсчета скорости
газа.
Ротаметры (рис. 77) состоят из градуирован-
ной стеклянной трубки, сужающейся книзу.
В трубке находится волчок, изготовленный из
эбонитц или другого легкого, и химически устойчивого материала.
Газ пропускают через ротаметр снизу вверх, при этом волчок под-
нимается по трубке силой давления газа на такую высоту, кото-
рая соответствует скорости потока газа.
3. ОЧИСТКА И ВВЕДЕНИЕ ГАЗОВ В ПРИБОР
Перед тем как ввести газ в реакционный сосуд, требуется его
очистить, пропуская через промывную склянку, наполненную той
или иной жидкостью.
Электролитический водород в баллонах достаточно чист и мо-
жет применяться для гидрирования без предварительной очистки.
Водород, полученный из водяного газа, может содержать различ-
ные примеси: предельные и непредельные углеводороды, кислород,
азот, окись и двуокись углерода, мышьяковистый водород, серово-
дород и другие. Для очистки такой водород пропускают через 50%
раствор едкого кали, затем через две промывные склянки с рас-
твором марганцовокислого калия (для окисления сероводорода и
мышьяковистого водорода), одну склянку с щелочным раствором
гидросульфита натрия, через трубку с медной сеткой или с плати-
нированным асбестом, нагретую до 350—400 °C (для удаления
кислорода) и, наконец, через склянку Тищенко (для сухого веще-
ства) или U-образную трубку с хлористым кальцием,
94
Азот, находящийся в баллонах, обычно содержит около 1%’
кислорода, от которого освобождаются пропусканием газа через
щелочной раствор пирогаллола (15 г пирогаллола в 100 мл 50%
раствора едкого кали) или через щелочной раствор гидросульфита
натрия.
Двуокись углерода содержит примеси кислорода, окиси угле-
рода и иногда следы сернистого ангидрида и сероводорода. Удале-
ние этих примесей (кроме кислорода) достигается пропусканием
газа через трубку с окисью меди, нагретую до темно-красного ка-
ления, потом через раствор двууглекислого натрия и, наконец, че-
рез нейтральный раствор марганцовокислого калия.
Очищенные газы после промывной склянки нельзя сразу же
вводить в прибор, так как может возникнуть опасность засасыва-
ния реакционной жидкости в газопроводящую трубку. Поэтому
перед прибором, в который вводится газ, следует установить пу-
стую предохранительную склянку; также надо устанавливать пре-
дохранительный сосуд и после газового баллона (см. рис. 29).
Кроме того, между промывными склянками с щелочами и кисло-
тами всегда необходимо помещать пустую склянку. Всю аппара-
туру для проведения реакции в токе газа следует тщательно про-
верить перед использованием. Прибор, присоединяемый к баллону
со сжатым газом, должен всегда иметь сообщение с атмосферой,
так как в наглухо закрытом приборе давление быстро возрастет
и прибор взорвется.
4. ПРАВИЛА ТЕХНИКИ БЕЗОПАСНОСТИ
ПРИ РАБОТЕ С ГАЗОВЫМИ БАЛЛОНАМИ
Большинство применяемых сжатых газов в смеси с воздухом,
а особенно с кислородом, легко взрываются. К этим газам отно-
сятся водород, ацетилен, метан, нефтяные газы и др. Кислород
также относится к числу огнеопасных газов, так как энергично
поддерживает горение. Кроме перечисленных газов, которые мо-
гут вызвать взрыв и пожар, есть газы, которые могут привести
‘к отравлениям (например, хлор и фосген). Чтобы избежать не-
счастных случаев при пользовании газовыми баллонами, необхо-
димо соблюдать все меры предосторожности и руководствоваться
следующими правилами.
1. Устанавливать баллоны в специальных стойках и как мож-
но дальше от источников тепла.
2. Оберегать баллоны от резких толчков, ударов, падения.
3. Не смазывать редукционные вентили для кислорода жи-
рами и органическими смазывающими веществами и не гермети-
зировать их при помощи прокладок из органических материалов,
так как органические вещества при соприкосновении с кислоро-
дом, находящимся под давлением, воспламеняются со взрывом.
4. Вентиль баллона открывать постепенно; если он откры-
вается туго, то нельзя пользоваться для открывания его молотком,
так как это представляет большую опасность.
95
5. Приступая к работе, убедитесь по окраске баллона и над-
писи на нем, что в баллоне именно тот газ, с которым предстоит
работать.
6. Запрещается самостоятельно исправлять вентиль.
7. Нельзя пользоваться неисправным баллоном.
8. Периодически отправляйте баллоны на проверку.
Глава VI
КОЛИЧЕСТВЕННЫЙ ЭЛЕМЕНТНЫЙ АНАЛИЗ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ
Определение количественного содержания отдельных элемен-
тов в органических веществах называется элементным анализом,
который может проводиться макро-, полумикро- и микрометодами.
При макроанализе берут для сжигания навеску в 0,15—2 г, при
полумикроанализе — в 20—30 мг, а при микроанализе — в 2—5 мг.
В настоящее время широко распространен полумикрометод, кото-
рый позволяет работать с малыми количествами вещества и про-
водить довольно быстро анализ. Определение основных элемен-
тов — углерода, водорода, азота, кислорода — чаще всего произ-
водят сжиганием навески вещества в трубке из тугоплавкого стек-
ла или кварца, причем определение углерода и водорода произ-
водят одновременно.
1. ОПРЕДЕЛЕНИЕ УГЛЕРОДА И ВОДОРОДА
ПОЛУМИКРОМЕТОДОМ
Сущность метода состоит в том, что навеску исследуемого ор-
ганического вещества сжигают в кварцевой трубке в токе воздуха
и кислорода. Газообразные продукты разложения проходят над
катализатором (окись меди или хромовокислый свинец), находя-
щимся в трубке, в результате чего углерод окисляется до двуокиси
углерода, а водород — до воды. Воду, выделяющуюся при сожже-
нии, поглощают в трубке с хлористым кальцием или перхлоратом
магния Mg(C104)2, жадно соединяющимися с водой; двуокись
углерода поглощают в трубке с натронной известью. Взвешивая
трубки до и после опыта, устанавливают количество образовав-
шейся воды и двуокиси углерода. Из этих данных можно вычис-
лить процентное содержание углерода и водорода во взятом для
исследования веществе.
96
СБОРКА УСТАНОВКИ
Для проведения определения углерода и водорода пользуются
установкой, которая изображена на рис. 78. Все приборы этой
установки по выполняемым функциям можно разделить на три
группы. К первой группе относятся приборы, подающие кислород
и воздух и оснащенные приспособлениями для регулирования дав-
ления, ко второй — трубка для сожжения и нагревательные при-
боры и к третьей — поглотительные аппараты для поглощения
воды и двуокиси углерода и склянка Мариотта, при помощи кото-
рой регулируют давление в системе. Кислород и воздух, при-
меняемые при сожжении органического вещества, должны быть
Рис. 78. Схема установки для определения углерода и водорода полу-
микрометодом:
/'•—регуляторы давления, 2 —хлоркальциевые трубки; 5—трехходовой кран;
4—счетчик пузырьков; 5—-U-образная осушительная трубка; 6—трубка для
сожжения; 7 —поглотительные приборы; 5—хлоркальциевая трубка, 9-скляика
Мариотта.
собраны в газометры. Подробное описание отдельных частей при-
бора приведено ниже.
Наполнение газометров кислородом и воздухом. Обычно поль-
зуются стеклянными газометрами, состоящими из сосуда для со-
бирания газа и воронки с жидкостью, служащей для вытеснения
газа через трубку верхнего тубуса сосуда. Перед наполнением
газометра кислородом или воздухом все краны и шлиф воронки
смазывают вазелином, нижний тубус закрывают пробкой, и в га-
зометр, вынув воронку, наливают воду до полного вытеснения
воздуха. Затем вставляют воронку с открытым краном и удаляют
из нее остатки воздуха, нагнетая воду вдуванием воздуха через
отводную трубку. После удаления воздуха из трубки в воронку
подливают воду и последний пузырек воздуха удаляют осторож-
ным приподниманием воронки.
После того как газометр наполнен водой, закрывают все краны,
открывают нижний тубус и вводят в него трубку, по которой идет
кислород из баллона. Газ вытесняет воду, которая вытекает
из этого же тубуса. Как только вода перестанет вытекать из
газометра, трубку немедленно вынимают и закрывают тубус
пробкой.
4 М Н Храмкина 97
При наполнении газометра воздухом открывают кран верхнего
тубуса и выпускают воду через нижний тубус.
Регулирование давления. Чтобы исключить влияние колебаний
давления газов в газометрах и, следовательно, поддержать по-
стоянной скорость газа во время сожжения, применяют регуля-
торы давления для воздуха и кислорода. Регулятор давления со-
стоит из колоколообразного сосуда с двумя трубками, погружен-
ного в цилиндр, до половины наполненный водой с небольшим
количеством едког9 кали.
Регуляторы давления соединяют с газометрами резиновыми
трубками. Ввиду того, что резина может выделять летучие орга-
нические соединения, трубки подвергают искусственному старе-
нию, для чего их в течение часа нагревают в сушильном шкафу
до 100—110°С при одновременном просасывании через них воз-
духа.
На соединительных резиновых трубках имеются винтовые за-
жимы, при помощи которых регулируют ток газа из газометра
таким образом, чтобы из регулятора давления выделялся один
пузырек газа не чаще чем через каждые 10—15 сек. Отводные
трубки регуляторов давления соединены с трехходовым краном,
позволяющим подавать в установку кислород или воздух при по-
мощи резиновых трубок, подвергнутых также искусственному ста-
рению. >
Для поддержания постоянной концентрации раствора едкого
кали в счетчике пузырьков, который включен за трехходовым кра-
ном, между регуляторами давления и трехходовым краном поме-
щают хлоркальциевые трубки, наполненные крупнозернистым хло-
ристым кальцием.
Подготовка к работе счетчика пузырьков и осушительной
U-образной трубки. Счетчик пузырьков служит для контроля ско-
рости тока газов; к нему припаяна U-образная осушительная
трубка с двумя пришлифованными пробками, наполненная натрон-
ным асбестом (асбест, пропитанный натронной известью) и хлори-
стым кальцием. Трубка служит для высушивания газов.
U-образную трубку со счетчиком пузырьков промывают сна-
чала хромовой смесью, затем водой и спиртом, сушат и наполняют
следующим образом. Вынимают пришлифованную пробку U-об-
разной трубки и в отводную трубку, припаянную к счетчику
пузырьков, временно вводят через шлиф ватный тампон при по-
мощи изогнутой стальной проволочки. Затем в самую нижнюю
часть трубки помещают кусочек ваты, а на него при постукивании
насыпают слой хлористого кальция (обычно не слишком прока-
ленного) высотой 0,5 см, который прикрывают маленьким ватным
тампоном, а затем натронный асбест почти до отводной трубки.
Прикрыв слой натронного асбеста ватой, заменяют ватный тампон
в отводной трубке очень рыхлой ватной -прокладкой. Затем поверх
асбеста вплоть до шлифа трубку заполняют более крупными кусоч-
ками хлористого кальция и снова закрывают кусочком ваты.
Тщательно вытирают шлиф и вставляют стеклянную пробку, сма-
98
занную вазелином, следя за тем, чтобы шлиф был прозрачным.
Счетчик пузырьков наполняют через открытую отводную трубку
при помощи оттянутой стеклянной трубочки 50%-ным непеня-
щимся раствором едкого кали так, чтобы кончик вводной трубки
был едва погружен в щелочь, после этого отводную трубку прочи-
щают внутри комочком ваты и тщательно вытирают снаружи. Для
получения непенящегося раствора едкого кали раствор встряхи-
вают с тонкоизмельченным едким баритом (1 г на 200 г щелочи),
после чего фильтруют через сухой фильтр.
Другое колено осушительной трубки наполняют следующим
образом. Сначала вводят просушенный рыхлый ватный тампон в
отводную трубку, закрывают ее резиновым колпачком и напол-
няют колено трубки при постукивании до уровня шлифа кусоч-
ками предварительно высушенного в сушильном шкафу при 180—
200 °C хлористого кальция. Затем прикрывают слой хлористого
кальция кусочком просушенной ваты, вытирают шлиф, смазывают
его вазелином и закрывают. Счетчик пузырьков соединяют с трех-
ходовым краном отрезком резиновой трубки, подвергнутой искус-
ственному старению.
Осушительную U-образную трубку соединяют с трубкой для
сожжения 6 при помощи толстостенного капилляра, который при-
соединяют к отводной трубке осушительного аппарата посред-
ством кусочка вакуумной резиновой трубки, пропитанной расплав-
ленным вазелином в вакууме.
Подготовка к работе трубки для сожжения. Перед употребле-
нием трубку для сожжения моют хромовой смесью, дистиллиро-
ванной водой и высушивают. Через широкую часть трубки в су-
женный конец ее вводят серебряную проволоку толщиной 1 мм,
один конец которой должен выступать наружу, а другой, сверну-
тый в спираль, находиться в трубке. Серебряная спираль препят-
ствует конденсации воды в трубке вследствие хорошей теплопро-
водности. Затем вводят слой серебряной ваты толщиной 2 мм и
пробку из свежепрокаленной асбестовой ваты толщиной примерно
5 мм, которая снижает скорость проходящего через трубку газо-
вого потока. Асбест уплотняют при помощи медной проволоки.
За асбестовой пробкой помещают слой мелкой окиси меди,
при этом трубку держат вертикально, уплотняют слой окиси меди,
похлопывая по стенкам ладонью. Слой окиси меди закрывают
асбестовой пробкой, которую вносят тремя порциями, причем
каждую порцию слегка утрамбовывают стеклянной палочкой. Эта
пробка необходима для предотвращения проскока через наполнен-
ную трубку паров несожженного вещества.
За пробкой помещают слой серебряной ваты длиной 2 см, за-
тем рыхлую асбестовую пробку и при вращении трубки насыпают
слой в 14 см окиси меди с нанесенным на нее хроматом свинца и
закрепляют этот слой также рыхлой асбестовой пробкой: После
этого трубку тщательно прочищают большим ватным тампоном.
Наполнение поглотительных приборов. Для поглощения воды
и углекислого газа, образующихся при сожжении, употребляют
4*
99
поглотительные трубки с пришлифованными полыми пробками,
при этом одна труока наполняется хлористым кальцием, другая —
натронным асбестом. Пробку хлоркальциевой трубки превращают
в «водяной мешок», для чего в дне пробки делают маленькое от-
верстие, к которому припаивают капиллярную трубку. Назначе-
ние «водяного мешка» — улавливать сконденсировавшиеся капли
воды.
При наполнении чистой и сухой трубки хлористым кальцием
сначала пробку слегка смазывают вазелином, оставляя верхнюю
ее часть (около 2 мм) несмазанной. Закрывают трубку пришли-
фованной пробкой с капилляром, помещают на последнюю малень-
кий ватный тампон, насыпают слой крупнозернистого хлористого
кальция высотой 1 см и покрывают его маленьким кусочком ваты.
Затем наполняют трубку почти до шлифа на противоположном ее
конце кусочками прокаленного хлористого кальция величиной с
просяное зерно. Слой закрепляют высушенным кусочком ваты и
закрывают трубку смазанной вазелином пришлифованной пробкой.
При наполнении чистой и сухой трубки натронным асбестом
сначала вставляют смазанную вазелином пришлифованную проб-
ку, в полость которой помещен кусочек ваты. Маленький ватный
тампон также кладут на пробку, а затем наполняют трубку на
2/з натронным асбестом, накрывают маленьким кусочком ваты, по-
верх которого насыпают слой в 0,5 см хлористого кальция (обычно
не очень хорошо высушенного) и закрывают этот слой ватным
тампоном. После этого набивают трубку почти до шлифа кусоч-
ками высушенного при 180—200 °C хлористого кальция величиной
с просяное зерно. Слой закрывают тампоном высушенной ваты и
закрывают трубку смазанной вазелином пришлифованной пробкой.
Поглотительные трубки соединяют друг с другом отрезком ва-
куумной резиновой трубки длиной 2 см, а с трубкой для сожже-
ния— такой же трубкой длиной 1,5 см; при этом резиновые соеди-
нительные трубки должны-быть предварительно обработаны ва-
зелином в вакууме, чтобы уменьшить диффузию газов через их
стенки.
Для поддержания в поглотительных приборах определенного
и легко изменяемого пониженного давления включают склянку
Мариотта. Между склянкой Мариотта и поглотительными прибо-
рами имеется маленькая хлоркальциевая трубка.
ВЫПОЛНЕНИЕ АНАЛИЗА
Для точного определения привеса необходимо поглотительные
приборы взвешивать через одинаковые промежутки времени. Сна-
чала каждую из поглотительных трубок в отдельности тщательно
вытирают влажной фланелью, а затем замшей, помещают их на
проволочную подставку'весов и точно отмечают время, когда труб-
ка поставлена к весам.
При взвешивании сначала определяют нулевую точку весов.
Затем открывают на короткое время кран, выравнивают давление
100
с атмосферным и переносят трубку при помощи специальных щип-
цов на левую чашку весов. Через 10 мин после вытирания трубки
определяют ее приблизительную массу, а через 15 мин — точную.
Вещество взвешивают в фарфоровой лодочке, гигроскопиче-
ское вещество —в специальном сосуде с колпачком (рис. 79).
Электрическую цепь целесообразно включить в начале взвешива-
ния и пропускать через трубку для сожжения воздух с той же
скоростью, что при проведении анализа.
После взвешивания поглотительных приборов на отводную
трубку поглотительного прибора с хлористым кальцием надевают
резиновую соединительную трубку длиной 1,5 см. Другую отвод-
ную трубку присоединяют к отводной трубке поглотительного
аппарата с натронным ас- *4^
бестом. Концы отростков
соединяют встык. Про-
веряют по часам число ""
пузырьков воздуха, про- рис. 79, Сосуд для взвешивания гигроскопи-
ходящих за 10 сек, и, если ческих веществ,
нужно, изменяют положе-
ние регулятора давления. Затем соединяют поглотительный аппа-
рат с хлористым кальцием с трубкой для сожжения, а трубку
с натронным асбестом присоединяют к хлоркальциевой трубке
склянки Мариотта. Потом вынимают резиновую пробку из трубки
для сожжения, вносят лодочку с веществом, открывают трех-
ходовой кран и впускают воздух. После этого открывают краны
поглотительных приборов и кран склянки Мариотта и присту-
пают к сожжению. Во время проведения анализа необходимо сле-
дить за наполнением газометра, счетчиком пузырьков и за ве-
ществом.
После окончания пропускания воздуха через трубку закры-
вают кран склянки Мариотта и краны поглотительных приборов и
отсоединяют последние от трубки для сожжения. Трубку закры-
вают резиновым колпачком и оставляют охлаждаться, после чего
она готова к следующему анализу. Поглотительные приборы взве-
шивают, как указано ранее. По разнице в массе поглотительных
приборов до и после анализа находят количество воды и углекис-
лого газа, образовавшихся при сожжении вещества.
Содержание углерода и водорода (в %) в исследуемом веще-
стве находят по формулам:
Ь 12,011-100
с ~ а ’ 44,022
с 2,016-100
Н ~ а ’ 18,016
где а — навеска вещества;
b — найденное количество СО2;
с — найденное количество Н2О.
Предельная ошибка определения: для углерода ±0,3%, для
водорода от 4-0,2 до —0,1 %.
tot
2. ОПРЕДЕЛЕНИЕ АЗОТА
ПОЛУМИКРОМЕТОДОМ (ПО ДЮМА)
Метод определения азота основан на сжигании навески веще-
ства посредством раскаленной окиси меди в 'трубке, наполненной
двуокисью углерода. Выделяющийся свободный азот улавливается
над раствором едкого кали и определяется по объему.
СБОРКА УСТАНОВКИ
Установка для полумикроопределения азота по Дюма состоит
из аппарата Киппа для получения двуокиси углерода, не содержа-
щего воздуха, трубки для сожжения и полумикроазотометра
(рис. 80).
Рис. 80. Схема установки для определения азота полумикроме-
тодом (по Дюма):
1 — полумикроазотометр; 2—регулировочный кран; 3 — трубка для сож-
жения; 4—Z-образиая стеклянная трубка; 5 —аппарат Киппа.
Подготовка аппарата Киппа для получения двуокиси угле-
рода. Для определения азота нужна двуокись углерода, совер-
шенно свободная от примеси воздуха’. Поэтому нельзя пользовать-
ся газом из баллонов, а лучше получать его в лаборатории.
В фарфоровой чашке обливают разбавленной (1:1) соляной
кислотой (d = 1,18) маленькие кусочки мрамора. После оконча-
ния бурной реакции сливают собравшуюся сверху грязь, промы-
вают водой кусочки мрамора и загружают их до половины в сред-
ний шар аппарата Киппа. В тубус среднего шара вставляют
резиновую пробку со стеклянным краном. К трубке крана, находя-
щейся внутри аппарата, посредством короткой резиновой трубки
присоединяют стеклянную изогнутую вверх трубку для удаления
в первую очередь более легких газообразных примесей. Затем в
аппарат наливают столько разбавленной соляной кислоты, чтобы
1Щ
последняя заполнила весь нижний шар и половину верхнего.
Бросают в трубку воронки два маленьких кусочка мрамора, при
этом бурно выделяющаяся двуокись углерода вытесняет воздух,
растворенный в кислоте.
Обычно вновь заряженный аппарат Киппа дает чистую дву-
окись углерода только через 2 или 3 дня стоянки, когда воздух,
поглощенный поверхностью стекла и резиновыми трубками, пере-
ходит в атмосферу двуокиси углерода. Аппарат Киппа соединяют
с трубкой для сожжения при помощи изогнутой Z-образной стек-
лянной трубки. Один ее конец, оттянутый в толстостенный капил-
ляр, соединяют с трубкой для сожжения, другой конец, расширен-
ный снизу и заполненный асбестовой ватой для улавливания тума-
нообразной соляной кислоты, присоединяют к крану аппарата
Киппа.
Подготовка трубки для сожжения. Для наполнения чистой и
сухой трубки окисью меди сначала в оттянутую ее часть поме-
щают немного серебряной ваты, затем 2—3 мм асбестовой ваты
для создания пробки. После этого насыпают слой длиной 12 см
крупной окиси меди, постукивая ладонью по стенкам трубки, по-
том слой в 6 см мелкой окиси меди и наконец слой в 10 см круп-
ной окиси меди. Все это закрепляют асбестовой пробкой.
В наполненную трубку через ее широкий конец направляют
ток водорода, пропущенный через склянку с подкисленным рас-
твором перманганата калия для вытеснения воздуха. После этого
восстанавливают окись меди в слое длиной 6 см, нагревая слой
горелкой. Охлаждают трубку в медленном токе водорода. Затем
трубку для сожжения прокаливают на всем ее протяжении в элек-
трической печи в токе двуокиси углерода и оставляют охлаждать-
ся под давлением двуокиси углерода из аппарата Киппа.
Наполнение и присоединение полумикроазотометра. Полу-
микроазотометр предназначен для собирания азота, образовав-
щегося во время сожжения навески вещества, имеет цену деления
0,02 мл, емкость 8—10 мл, что соответствует навеске исследуемого1
вещества в 20—30 мг. Трубка, по которой газ поступает в полу-
микроазотометр, имеет стеклянный кран 2 для регулирования вы-
хода газа. Полумикроазотометр соединяют с трубкой для сожже-
ния при помощи капиллярной трубки, согнутой под тупым углом.
Для соединения служит слегка смазанный глицерином кусочек
вакуумной резиновой трубки.
Перед наполнением полумикроазотометр моют хромовой
смесью и дистиллированной водой. Резиновую трубку, соединяю-
щую уравнительную грушу с полумикроазотометром, прикрепляют
проволокой. Сначала через грушу в полумикроазотометр прили-
вают столько чистой ртути, чтобы ее уровень был на 1—2 мм
выше отверстия вводной трубки. Для наполнения полумикроазото-
метра применяют 50% непенящийся раствор едкого кали.
Уравнительную грушу закрывают резиновой пробкой с корот-
кой вытянутой в капилляр стеклянной трубкой.
<03
ВЫПОЛНЕНИЕ АНАЛИЗА
В маленькую пробирку с пришлифованной пробкой вносят не-
большое количество окиси меди, насыпают 20—30 мг вещества и
взвешивают. Чтобы внести твердое исследуемое вещество в трубку
для сожжения, в нее вставляют загрузочную воронку, которую
можно изготовить из широкой пробирки, и насыпают сначала слой
крупной (7 см), а затем слой мелкой (0,5 см) окиси меди, при этом
постукивают по стенкам вертикально поставленной трубки для
уплотнения окиси меди. К навеске вещества, находящейся в про-
бирке для взвешивания, прибавляют слой мелкой окиси меди вы-
сотой 2 см, закрывают пробирку корковой пробкой, встряхивают и
затем пересыпают ее содержимое через загрузочную воронку в
трубку для сожжения.
Трубку помещают в электрическую печь таким образом, чтобы
слой окиси меди выступал из печи на 2 см со стороны оттянутой
части трубки. Для защиты этой части трубки от нагревания стен-
ку печи заслоняют маленьким асбестовым экраном. На другой
конец трубки надевают муфту из проволочной сетки длиной 5 см
и для защиты резиновой пробки от нагревания помещают асбе-
стовый экран. Затем включают электрическую печь и соединяют
трубку для сожжения с аппаратом Киппа. Открыв кран последнего
пропускают через трубку в течение нескольких минут двуокись
углерода, после чего соединяют трубку для сожжения с полу-
микроазотометром, в котором щелочь должна быть переведена в
уравнительную грушу, установленную в возможно более низком
положении.
Двуокись углерода пропускают еще в течение 2 мин, затем за-
крывают регулировочный кран полумикроазотометра, наполняют
последний щелочью и оставляют уравнительную грушу в низком
положении. После этого осторожно открывают регулировочный
кран и устанавливают ток двуокиси углерода так, чтобы в полу-
микроазотометр проходили 1—2 пузырька в секунду. Как только
через полумикроазотометр начнут проходить микропузырьки, за-
крывают кран у аппарата Киппа и полностью открывают регули-
ровочный кран. Одновременно надвигают проволочную муфту на
слой окиси меди, находящейся в широком конце трубки, и нагре-
вают горелкой.
После того как электрическая печь раскалится, на что тре-
буется 10—20 мин, и прекратится выделение газов, вызванное
нагреванием трубки горелкой, закрывают регулировочный кран,
поднимают уравнительную грушу выше верхнего крана полумик-
роазотометра и поворачивают этот кран в ту и другую сторону,
выпуская при этом собравшийся в полумикроазотометре газ.
Закрыв верхний кран полумикроазотометра, опускают уравни-
тельную грушу вниз, открывают регулировочный кран и подви-
гают на несколько миллиметров ближе к печи проволочную муфту,
причем горелка должна находиться на противоположном конце
муфты. Таким образом продвигают муфту и горелку и в дальней-
104
шем следят за тем, чтобы в полумикроазотометр поступало не бо-
лее 2 пузырьков газа в 3 сек.
Как только горелка будет придвинута к электрической печи,
иа что требуется 10—25 мин, закрывают регулировочный кран,
полностью открывают кран аппарата Киппа и регулировочным
краном снова устанавливают скорость прохождения газа 2 пузырь-
ка в 3 сек. Затем еще раз сильно в течение 10 мин прокаливают
горелкой через проволочную, муфту сменяемый слой окиси меди.
После этого выключают горелку, а спустя 5 мин — электрическую
печь. Как только через полумикроазотометр начнут проходить
микропузырьки, закрывают регулировочный кран, разъединяют
трубку для сожжения и оставляют ее охлаждаться под давлением
двуокиси углерода.
Полумикроазотометр оставляют в прохладном помещении для
охлаждения, при этом следует установить уравнительную грушу
на одном уровне с жидкостью в полумикроазотометре. Через
10 мин отмечают объем азота, при этом мениск уравнительной
груши держат на одном уровне с мениском в измерительной труб-
ке. Отсчет производят по нижнему краю мениска, затем отмечают
температуру вблизи азотометра и показание барометра.
Содержание азота в исследуемом веществе An (в %) вычис-
ляют по формуле:
1,2517-273 (Р-Рв)
%N= (273 +1) 760а 0,1
где 1,251 — масса 1 мл чистого азота при обычных условиях, мг;
V — найденный объем азота, мл;
Р— атмосферное давление, мм рт. ст.;
Рв—давление паров воды при данной температуре,
мм рт. ст.;
t — температура, °C;
а — навеска вещества, г.
3. ОПРЕДЕЛЕНИЕ УГЛЕРОДА
И ВОДОРОДА МИКРОМЕТОДОМ
Большое распространение получил количественный элемент-
ный микроанализ органических веществ. Основоположником этого
метода является австрийский профессор Фриц Прегль. Хотя со
времен Прегля появилось очень много различных работ, в мето-
дики определения углерода и водорода в органических веществах
не введено принципиальных изменений.
В последнее время в СССР широкое распространение получил
принципиально отличный от классического, но столь же надежный
скоростной метод определения, предложенный М. О. Коршун и
В. А. Климовой.
Сущность метода состоит в том, что навеска исследуемого
органического вещества сжигается в полузамкнутой зоне в
быстром токе кислорода в ненаполненной трубке в условиях,
10$
обеспечивающих длительный и полный контакт вещества, его па-
ров и продуктов разложения с горячим кислородам. Углерод при
этом количественно окисляется до двуокиси углерода, а водород —
до воды. Продукты окисления других элементов, если они содер-
жатся в веществе, улавливаются соответствующими соединениями
и не мешают определению.
Вода поглощается безводным перхлоратом магния (ангидро-
ном), а двуокись углерода — щелочным поглотителем (аскаритом).
Поглощенные вещества определяют по привесу поглотительных
аппаратов.
СБОРКА УСТАНОВКИ
Для определения углерода и водорода микрометодом поль-
зуются установкой, изображенной на рис. 81.
Рис. 81. Прибор для определения углерода и водорода микрометодом:
/—газометр; 2—осушительная колонка; 3—кран с нарезкой; 4—катализаторная трубка;
5—сосуд для охлаждения змеевика катализаторной трубки; 6, 7—U-обпазные трубки;
8t 9t 10, 11—электрические печи. 12—узкая часть трубки для сожжения; 13 — аппарат для
поглощения воды; 14 — аппарат для поглощения дзуокиси углерода; 15—заключительная
трубка; /5—скляика Мариотта; /7 —цилиндр.
Аппаратура состоит из трех основных частей: системы подачи
и очистки кислорода, трубки для сожжения, системы для погло-
щения продуктов горения (рис. 82).
Многие из частей аппарата для определения углерода и водо-
рода выпускает завод «Гослаборприбор» (г. Клин). Подробное
описание отдельных частей установки приведена ниже.
Система подачи и чистки кислорода
Кислород можно подавать из газометра или непосредственно
из баллона. Для регулировки скорости газа баллон должен быть
снабжен прецизионным вентилем и маностатом (рис. 83). Газо-
метр может быть любым. Наиболее удобен газометр, состоящий
.106
из двух 10-литровых бутылей, расположенных одна под другой и
соединенных толстостенной стеклянной трубкой.
Рис. 82. Аппаратура для определения углерода и во-
дорода микрометодом:
1— U-образная трубка; 2, 3, 4, 5, 6, 7—сосуды для навесок;
5, Р, 10—трубка для сожжения; 11 — кран с нарезкой; 12—погло-
тительный аппарат; 13 — катализаторная трубка.
—К прибору
От баллона.
Обычно кислород в баллонах загрязнен и увлажнен, поэтому
его необходимо сначала осушить, пропуская через одну или две
колонки с осушителями (силикагель, хлористый кальций, аскарит).
Лучше всего колонку наполнить сили-
кагелем, пропитанным раствором бро-
мида или хлорида кобальта и высу-
шенным при 210—240 °C. Силикагель
марки КСМ или ШСМ измельчают,
просеивают через сито и отбирают
фракцию 0,5—1,0 мм. Отобранный си-
ликагель нагревают 2—3 ч с концен-
трированной соляной кислотой па во-
дяной бане, хорошо промывают водой
и сушат при 100 °C. После этого нагре-
вают при 200 °C в течение 2—3 ч.
Сухой силикагель (10 ч.) смеши-
вают с 20 мл 7,5%-ного спиртового
или водного раствора гексагидрата
хлорида кобальта и снова сушат 1—
2 ч при 180—200 °C. Полученный готовый силикагель окрашен в
ярко-синий цвет. При поглощении воды он меняет окраску от си-
ней через фиолетовую до розовой (в зависимости от числа молей
присоединенной воды):
Рис. 83 Маностат.
Число молей Н2О 0
Цвет...........Синий
1 1,5 2 4 6
Сине- Темный, Розово- Красный Розовый
фиоле- сиие-фио- фиоле-
товый летовый товый
Такое изменение цвета позволяет определить, когда требуется
сменить силикагель. Увлажненный препарат регенерируют сушкой
и нагреванием при 200 °C до получения синей окраски.
107
Можно также использовать продажный препарат — силика-
гель-индикатор. Его измельчают, отбирают фракцию 0,5—1,0 мм
и просушивают при 200 °C.
После осушки кислород очищают в катализаторной трубке,
при этом примесь органических веществ окисляется до двуокиси
углерода и воды, которая улавливается аскаритом и ангидроном
в U-образных трубках. Для соединения катализаторной трубки
с осушительной колонкой используют трубку из вакуумной резины,
в середину которой вставляют кран с нарезкой для регулирования
скорости тока кислорода. Нарезки на кране в виде двух запятых
(рис. 84) делаются бритвой.
Катализаторная трубка (см. рис. 82) состоит из двух частей.
Нижняя часть служит холодильником, а верхнюю наполняют
окисью меди и затем заплавляют. Напол-
нение трубки окисью меди проводят сле-
дующим образом: закрывают боковой от-
вод трубки маленькими неплотными тампо-
L I нами прокаленного асбеста и насыпают
I | окись меди не до верха, оставив 1 см. Про-
тирают ненаполненную часть трубки зам-
Рис. 84. Кран с нарезкой, шей, чтобы убрать следы окиси меди, так
как при заплавлении они могут разрушить
кварцевое стекло. Затем вынимают асбестовый тампон из внутрен-
ней трубки и заплавляют верхний конец катализаторной трубки
на горелке с кислородным дутьем. После этого соединяют верхнюю
часть с холодильником замазкой Кренига. Для приготовления за-
мазки Кренига сплавляют в тигле смесь из 1 ч пчелиного воска
и 4 ч. канифоли. Эта замазка при нагревании размягчается. Ею
заплавляют те шлифы, которые не поворачивают в процессе
работы.
Катализаторная трубка обычно служит долго, если слой окиси
меди не слишком плотный, а температура нагревающей печи не
превышает 800°C*.
Верхняя часть катализаторной трубки, наполненная окисью
меди, нагревается вертикально-горизонтальной электропечью.
С помощью трубки из вакуумной резины катализаторная трубка
встык присоединяется к U-образной.
U-образную трубку наполняют наполовину аскаритом и на-
половину ангидроном, располагая аскарит между двумя слоями
ангидрона. Между слоями и по концам трубки делают прокладки
из прокаленного асбеста или стеклянной ваты. Еще лучше поста-
вить две U-образные трубки, при этом трубки заполняют на две
трети аскаритом и на одну треть ангидроном и располагают их
так, чтобы аскарит был посередине. Для того чтобы кислород по-
* При слишком плотном наполнении трубка при нагревании 'лопнет; при
слишком сильном нагревании начинает быстро разрушаться кварц.
108
ступал в трубку для сожжения с той же влажностью, с какой он
выходит из установки, необходимо, чтобы ангидрон был в -конце
всей системы очистки кислорода.
Трубки для сожжения
Трубки для сожжения бывают различными в зависимости от
применяемого метода; чаще всего применяются трубка для сожже-
ния со вспышкой и трубка для сожжения с пиролизом.
Трубка для сожжения со вспышкой состоит из двух частей,
соединенных между собой шлифом или резиновой трубкой. Основ-
ная часть трубки длиной около 500 мм представляет собой широ-
кую пробирку, в запаянный конец которой вставлена более узкая
трубка. Один конец ее отогнут внутрь к стенке пробирки, другой
присоединяется к системе для очистки кислорода. Заплавленный
конец пробирки служит зоной разложения. Вторая часть трубки
для сожжения — прямая, заканчивающаяся оттянутым концом, к
которому присоединяют поглотительные аппараты. На эту часть
трубки надвинуты две печи: печь на 950 °C — дополнительная зона
окисления, печь на 400—600 °C — зона нагревания серебра, кото-
рая улавливает галоген и серу, если эти элементы имеются в ис-
следуемом органическом веществе. Зона окисления находится в
том месте пробирки, где в нее поступает кислород. Эта зона на-
гревается пламенем горелки или специальной разъемной печью.
Трубка для сожжения с пиролизом прямая, имеет с одной сто-
роны оттянутый конец, у другого конца — боковой отвод, внешний
диаметр которого равен диаметру отводов U-образных трубок.
Через этот боковой отвод подается кислород. На трубку для сож-
жения на расстоянии 320 мм от оттянутого конца надвигают печь
на 400—600 °C, нагревающую серебро, затем ставят печь на
950 °C — зона окисления. Зона разложения находится в 80 мм от
бокового отвода, ее можно нагревать пламенем горелки или разъ-
емной печью.
Наполнение поглотительных приборов
Поглотительная система состоит из нескольких поглотитель-
ных аппаратов, заключительной трубки, склянки Мариотта и мер-
ного цилиндра.
Аппарат для поглощения воды наполняют следующим обра-
зом: сначала помещают слой прокаленного асбеста или стеклян-
ной ваты (3—4 мм), затем насыпают ангидрон, слегка постукивая
по аппарату, и наконец снова слой прокаленного асбеста или стек-
лянной ваты. Пробку и шлиф аппарата слегка нагревают на сла-
бом пламени горелки. Затем прикасаются нагретой пробкой к ку-
сочку замазки Кренига диаметром 2—3 мм, вставляют пробку в
аппарат и поворачивают ее несколько раз, так чтобы весь шлиф
стал прозрачным. Если шлиф окажется плохо смазанным, надо его
слегка подогреть, снять смазку ватой и повторить все сначала.
109
Аппарат для поглощения двуокиси углерода наполняют сле-
дующим образом: сначала помещают тампон из асбеста, затем
насыпают слой ангидрона (2—2,5 см), снова асбестовый тампон,
аскарит и опять асбестовый тампон. Смазывают шлиф и закры-
вают аппарат как указано выше.
Подготовка аппаратуры к работе
Для соединения U-образной трубки с катализаторной и труб-
кой для сожжения берут трубку из вакуумной резины длиной 4 см
с внутренним диаметром 2 мм. Для соединения между собой по-
глотительных приборов с трубкой сожжения и заключительной
трубкой применяют трубку из вакуумной резины с внутренним
диаметром 1 мм и длиной 2 см. Внутреннюю поверхность резино-
вых трубок протирают и смазывают глицерином.
Заданную температуру в печах поддерживают с помощью рео-
стата. Газометр собирают и проверяют его герметичность: напол-
няют газометр кислородом, отмечают уровень воды и проверяют
изменение объема через 2—3 ч — объем не должен измениться.
К газометру присоединяют осушительную колонку, затем кран
с нарезкой и катализаторную трубку. Последнюю ставят верти-
кально. Катализаторную трубку соединяют с U-образной трубкой,
которую встык присоединяют к трубке для сожжения. Надвигают
на сжигательную трубку печь и присоединяют к ней встык погло-
тительные аппараты. Сначала присоединяют аппарат с ангидро-
ном, затем аппарат с аскаритом, присоединяют последнюю трубку
и склянку Мариотта, наполненную водой, опускают носик склянки
в цилиндр, открывают краны у газометра, кран с нарезкой и кран
склянки Мариотта. Затем убеждаются, что кислород проходит че-
рез систему, закрывают кран газометра и проверяют герметич-
ность системы. При закрытом кране газометра вода из носика
склянки Мариотта очень быстро перестает вытекать и может лишь
слегка капать.
После того как прибор собран, устанавливают скорость тока
кислорода 25—30 мл/мин.
ВЫПОЛНЕНИЕ АНАЛИЗА
Чтобы взять навески необходимо иметь лодочки (см. рис. 82),
капилляры, кварцевые пробирки. Для сожжения веществ, об-
разующих золу, берут большие кварцевые пробирки. Навеску ле-
тучих жидких соединений берут в капилляры, которые не запаи-
вают. Гигроскопичные или неустойчивые твердые соединения берут
в пробирки с притертыми пробками. Для сожжения веществ, не
образующих золы, используют маленькие пробирки.
Величина навески обычно 5—7 мг. Навески твердых веществ
берут при помощи микрошпателя, который можно сделать, рас-
плющив молотком кончик алюминиевой или медной проволоки
110
диаметром 2—3 мм. Навески жидкости берут при помощи капил-
ляра, вязкие жидкости — при помощи стеклянных пипеток.
Навеску в лодочке, маленькой пробирке и капилляре поме-
щают в заплавленный конец сжигательной трубки — в зону разло-
жения. (Перед тем как внести пробирку в трубку, пробку выни-
мают.) Капилляр лучше помещать в лодочку, причем открытый
конец его должен быть направлен к запаянному концу трубки.
После внесения навески начинают сожжение. Подставляют
печь под ту часть трубки, где в нее поступает кислород. Вторую
печь подставляют почти под навеску, После того как трубка в
месте поступления кислорода нагрелась, печь медленно передви-
гают к навеске до тех пор, пока вещество не вспыхнет. После окон-
чания горения печь ставят так, чтобы она закрыла всю расширен-
ную часть трубки. Сожжение длится 2—3 мин, затем печь мед-
ленно, в течение 5—7 мин, передвигают по всей длине трубки для
полного вытеснения продуктов сожжения. Снимают печь, закры-
вают кран склянки Мариотта. Аппараты снимают, йротирают и
взвешивают на полумикровесах (например, марки ВМ-20).
Время от времени необходимо проводить контрольные взвеши-
вания.
По результатам анализа контрольного вещества устанавли-
вают поправку, которую вводят при расчетах.
Расчет. Поправку п (в мг) вычисляют по формулам:
Ag , Bg
«со2 — а~“лГ’ пн2о — ь~м-
где а — привес СО2, мг;
А — масса СО2, рассчитанная по реакции при окислении
грамм-молекулы контрольного вещества, г;
g— навеска, мг;
М — молекулярный вес контрольного вещества;
Ь — привес Н2О, мг;
В — масса Н2О, рассчитанная по реакции при окислении
грамм-молекулы контрольного соединения, г.
Содержание углерода и водорода (в %) вычисляют по фор-
мулам;
*с = Ксо2 • 100 -- £СО?~ Хн = *н2О * 100
Mr 12,01 Мн
где ^ = -^- = ^ = 0,2729 W =
kuUj ' 4124-
Пример. Контрольное вещество — сахароза С12Н22О11, М = 342,31. Навеска
(5,325 мг; получено; СОа 9,800 мг, Н2О 3,750 мг.
111
Теоретически из 6,325 мг сахарозы должно получиться (в мг):
- тяг •Ы25 - 9'758
Н2О = 183°26зу--- • 6.325 = 3,662
Расчет поправки (в мг):
пс02 = 9,800 — 9,758 = 0,042
nHjO = 3,750 — 3,662 = 0,088
Навеска 5,280 мг; получено: СОа 8,180 мг, Н2О 3,150 мг.
Содержание углерода и водорода (в %) с учетом поправки:
Хс = 0,2729 • 100 8’189.~Т9--Р1? = 42,05
G 5,280
V14OU
СИНТЕЗЫ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ
ЧАСТЬ
II
Глава VII
РЕАКЦИИ ГАЛОГЕНИРОВАНИЯ
Галогенсодержащие органические соединения нашли широкое
применение как растворители, антисептики, наркотические сред-
ства, инсектициды, стимуляторы роста растений, хладагенты, ле-
карственные препараты, мономеры в производстве полимеров и
исходные вещества в производстве различных органических ве-
ществ. Галогенпроизводные углеводородов широко используются
в различных синтезах вследствие способности их атома галогена
замещаться на различные группы (аминогруппа, карбоксильная
и гидроксильная группы и т. д.).
Наибольшее значение имеют следующие способы введения
атома галогена в молекулу органического вещества:
1) замещение галогеном гидроксильной группы в спиртах и
кислотах, а также атома кислорода в альдегидах и кетонах;
2) присоединение галогена и галогенводорода по кратной
связи;
3) прямое замещение водорода галогеном.
1. ЗАМЕЩЕНИЕ ГИДРОКСИЛЬНОЙ ГРУППЫ СПИРТОВ
ГАЛОГЕНОМ
Для замещения гидроксильной группы в спирте галогеном
применяют такие реагенты, которые содержат атомы или атомные
группы, способные связать гидроксильную группу. Чаще всего
такое замещение может быть достигнуто при действии галоген-
водородных кислот (HCl, НВг, HI), галогенных соединений фос-
фора (Р13, РВг3 и РС15), а также хлористого или бромистого тис-
нила (SOCI2, SOBrs). Легче всего спирты реагируют с иодистово-
дородной кислотой, которая обычно применяется для получения
иодпроизводных высших спиртов. Труднее всего идет реакция с
хлористоводородной кислотой, при этом в случае первичных спир-
тов необходимо прибавлять водоотнимающее вещество, например
безводный хлористый цинк. Из спиртов наиболее интенсивно всту-
113
пают в реакцию третичные (достаточно смешать спирт с концен-
трированным водным раствором галогенводородной кислоты), наи-
более трудно — первичные. Действие на спирты галогенводородных
кислот можно представить следующим уравнением:
ROH + HX ч=± RX + H2O
где X —галоген (Cl, Br, I).
Приведенная реакция обратима, так как вода разлагает полу-
чающееся галогенпроизводное на исходные вещества. Чтобы реак-
ция прошла практически до конца, необходимо удалять воду или
галогеналкил из реакционной смеси. Поэтому для получения гало-
генпроизводного удобнее применять не галогенводородную кисло-
ту, а ее соль и концентрированную серную кислоту (в момент
образования галогенводородная кислота действует активнее). Этим
способом можно получить, например, бромистые метил, этил, бутил
и др.
Механизм реакции образования галогенпроизводных из спир-
тов и галогенводородов можно представить следующим образом:
R—СН2—О—Н + Н—Вг —► R—СН2—6—Н + Вг-
I
Н
н
R—СНз + Вг" —► R—СН2Вг
Существует также способ, который дает возможность синте-
зировать два галогенпроизводных одновременно, например при
действии брома на легко бромирующийся углеводород в присут-
ствии первичного спирта. Таким путем можно получить а-бром-
нафталин и бромистый этил, а-бромнафталин и бромистый метил,
а-бромнафталин и бромистый пропил и др.
Замещение галогеном гидроксильной группы в спирте может
быть достигнуто также при действии тригалогенида и пентагало-
генида фосфора. Из тригалогенидов фосфора обычно применяют
РВгз и PI3. При этом они реагируют со спиртами в момент своего
образования из фосфора и галогена:
2Р + ЗХ2 —► 2РХ3
3ROH + PX3 —> 3RX + Р(ОН)3
гдеХ—I, Вг.
Необходимо отметить, что ибдпроизводные получают почти
исключительно указанным методом. Применение треххлористого
фосфора ограничено. Его используют только при хлорировании
третичных спиртов и фенолов, так как с первичными спиртами он
дает эфиры фосфористой кислоты, а со вторичными — ненасыщен-
ные углеводороды:
3RCH2OH+PC13 —> P(OCH2R)3 + ЗНС1
3R—CH(OH)~CHj—R' + PCI3 T-* 3R—CH=CH—R' + 3HC1 + H3PO3
114
Из пентагалогепидов фосфора применяют PClg и РВГ5, кото-
рые реагируют со спиртами следующим образом:
ROH + PC16 —> RC1 + РОС13 + НС1
ROH + PBrs —> RBr + РОВг3 + НВг
Реакция взаимодействия пятихлористого фосфора со спиртами
(первичными, вторичными и третичными)—наиболее универсаль-
ный способ получения хлорпроизводных из спиртов. Образующаяся
хлорокись фосфора (РОС13) при нагревании также реагирует со
спиртами:
3ROH + РОС13 —> 3RC1 + H3PO4
С этой целью применяют и хлористый тионил SOC12:
roh + soci2 —> rci + so2 + hci
2. ЗАМЕЩЕНИЕ ГИДРОКСИЛЬНОЙ ГРУППЫ КИСЛОТ
ГАЛОГЕНОМ
При замещении гидроксильной группы в кислотах галогеном
образуются галогенангидриды кислот. Для их получения приме-
няют галогениды фосфора и хлористый или бромистый тионил
(SOC12, SOBr2).
Широкое применение из галогенангидридов кислот находят
хлорангидриды. Это объясняется большой подвижностью атома
хлора. Для получения хлорангидридов кислот в основном приме-
няют треххлористый фосфор, хлористый тионил (SOCI2) и пяти-
хлористый фосфор:
RCOOH4-PCI5 —► RCOC1 + РОС13 + НС1
Выбор реагента зависит от многих факторов: температуры
кипения хлорангидрида, легкости, с которой реагирует кислота,
•но чаще всего имеет значение способ отделения хлорангидрида от
побочных продуктов. С этой точки зрения удобен хлористый тио-
нил, дающий только газообразные легкорастворимые в воде про-
дукты SO2 и НС1:
R—СООН + SOC12 —> R— СОС1 + SO2 + НС!
Треххлористый фосфор используют для получения хлорангид-
ридов низших кислот, пятихлористый фосфор —для получения
хлорангидридов высших кислот. Бромангидриды кислот получают
аналогично хлорангидридам, т. е. с применением бромистых соеди-
нений фосфора. Иодангидриды кислот получают действием смеси
иода и фосфора на кислоты.
3. ПРИСОЕДИНЕНИЕ ГАЛОГЕНА ПО КРАТНОЙ СВЯЗИ
Непредельные углеводороды способны присоединять галогены
к двойной и тройной связи. В лабораторной практике чаще всего
применяют бромирование этиленовых углеводородов, при этом
115
получают дибромпроизводные с атомами брома у соседних атомов
углерода:
СН2=СН2 + Вг2 —> СН2Вг—СН2Вг
Реакция присоединения брома к непредельным углеводородам
широко применяется в органическом анализе для открытая и коли-
чественного определения этиленовых и ацетиленовых связей. Коли-
чество брома в граммах, присоединяющегося к 100 г органиче-
ского вещества, получило название бромного числа. Скорость
реакции присоединения галогена к непредельным углеводородам
зависит от строения углеводорода, катализатора, температуры и
других факторов.
По кратной связи присоединяются НС1, НВт, HI с образова-
нием моногалогеналкила, например:
СН3СН=СН2 + НВг —> СН3СНВг
сн,
4. ПРЯМОЕ ЗАМЕЩЕНИЕ ВОДОРОДА ГАЛОГЕНОМ
При реакциях прямого замещения атомов водорода галогеном
часто получается смесь различных продуктов. При этом происхо-
дит последовательное замещение нескольких атомов водорода с
образованием смеси моно-, ди- и тригалогенпроизводных и смеси
изомеров. Полученные смеси трудно разделить, так как темпера-
туры кипения компонентов близки. Поэтому реакция прямого за-
мещения удобна лишь в тех редких случаях, когда в молекуле
исходного вещества один атом водорода способен замещаться га-
логеном значительно легче, чем другие, т. е. имеется атом водо-
рода в a-положении электроноакцепторной группы. Отсюда сле-
дует, что легко галогенируются карбоновые кислоты, альдегиды,
кетоны и простые эфиры жирного ряда.
Реакция прямого замещения водорода галогеном широко
используется также при синтезах галогенпроизводных ароматиче-
ского ряда. Прямое замещение водорода галогеном во многом
зависит от температуры, света и катализатора (железо, соли алю-
миния, фосфор, иод и т. д.). При подборе соответствующего ката-
лизатора, температуры и на свету нужный продукт будет полу-
чаться в преобладающем количестве. В зависимости от этих усло-
вий замещение водорода галогеном может идти в боковой цепи
или в бензольном ядре. При нагревании и действии света без ка-
тализатора замещение идет в боковой цепи (например, получение
хлористого бензила из толуола). В присутствии катализатора за-
мещение идет в ароматическом ядре (например, получение бром-
бензола в присутствии железа). На процесс замещения водорода
галогеном в ароматическом ядре влияет также наличие в ядре
гидроксила или аминогруппы, которые увеличивают подвижность
атомов водорода в о- и «-положении. Например, при действии
бромной воды на анилин три атома водорода бензольного кольца
116
легко замещаются атомами брома, в результате чего образуется
симметричный триброманилин:
Легко получается а-бромнафталин при проведении реакции
замещения в органическом растворителе (уксусная кислота или
четыреххлористый углерод):
Вг
Галоген, связанный непосредственно с бензольным или нафта-
линовым ядром, более прочно связан с ним, чем галоген, стоящий
в боковой цепи.
5. ПРИМЕРЫ СИНТЕЗОВ
БРОМИСТЫЙ ЭТИЛ
Формула:
С2Н5Вг
Основные реакции (механизм реакции см. стр. 114):
KBr + H2SO4 —> КН&О4 + НВг
С2Н5ОН + НВг <=* С2Н6Вг + Н2О
СН3СН2ОН 4- НО—SO2—ОН СН3СН2—О—SO2—ОН + Н2О
Этилсерная кислота
(этилгидросульфат)
СН3СН2—О—SO2—ОН + НВг —► СН3СН2Вг + H2SO4
Побочные реакции:
2HBr + H2SO4 —> Br2 + 2Н2О + SO2
СН3СН2—О—SO2—ОН + НО—СН2СН3 —> H2SO4 + сн3—сн2—о—СН2СН3
Реактивы, посуда и приборы
Этиловый спирт, 95-%ный . . 28 мл
(0,5 моль)
Бромистый калий.............30 г
(0,25 моль)
Сериая кислота (d = 1,84) . . 33 мл
(0,6 моль)
Колбы круглодонные (100 и 300 мл) 2
Колба коническая (200 мл) . . . . 1
Воронка делительная .......... 1
Воронка капельная ............ 1
Холодильник Либиха............1
Дефлегматор...................1
Термометр.....................1
Алонж........................ 1
117
Сборка приборов
1. В песочную баню помещают круглодонную колбу емкостью
300 мл, соединяют ее с дефлегматором и
к которому присоединяют алонж. Конец
1,5 см в смесь воды со льдом, налитую в
длинным холодильником,
алонжа опускают на 1 —
колбу-приемник, которую
погружают в ледяную ба-
ню (рис. 85).
Так как бромистый
этил летуч, то для умень-
шения потерь погон соби-
рают в приемник с водой
и льдом (несколько ку-
сочков).
2. В водяную баню
погружают круглодонную
колбу на 100 мл, в кото-
рую вносят «кипелки»
(см. стр. 30). Колбу снаб-
жают дефлегматором с
термометром и водяным
холодильником с алон-
Рис. 85. Прибор для получения бромистого
этила:
/ — песочная баня; 2—круглодоииая колба; 3 — дефлег-
матор; 4 —холодильник; 5 —алоиж; 6—колба-прием-
иик; 7—ледяная баня.
жем, который опускают в приемник, охлаждаемый снаружи льдом.
Так как бромистый этил кипит при низкой температуре (38—
39°C), то пользуются возможно более длинным холодильником и
пропускают через него сильную струю холодной воды.
Выполнение синтеза
В реакционной колбе емкостью 300 мл смешивают 28 мл эти-
лового спирта с 20 мл холодной воды, добавленной для уменьше-
ния образования побочного продукта (диэтилового эфира), со-
кращения потерь бромистоводородной кислоты, которая обладает
большой летучестью, и для частичного разбавления серной кис-
лоты, чтобы избежать окислительно-восстановительной реакции
образования брома. Для более полного использования образую-
щейся бромистоводородной кислоты спирт берут в избытке.
При постоянном перемешивании реакционной смеси и охлаж-
дении колбы проточной водой к смеси осторожно добавляют 33 мл
серной кислоты, а затем 30 г тонкорастрртого бромистого калия.
Собирают прибор 1 и нагревают реакционную смесь на песочной
бане большим пламенем. Вскоре начинает отгоняться бромистый
этил, который собирается под слоем воды со льдом. Если реак-
ционная смесь в колбе пенится, то на короткое время уменьшают
нагревание.
Реакционную смесь нагревают до тех пор, пока из холодиль-
ника не перестанет стекать не растворяющийся в воде бромистый
этил. Перегонку ведут с такой скоростью, чтобы в приемник не-
прерывно поступали маслянистые капли бромистого этила, а жид-
кость из приемника не поднималась бы в холодильник.
118
Иногда при перегонке налитая в приемник вода втягивается
обратно в алонж. В этом случае приемник опускают пониже, что-
бы освободить конец алонжа. Когда вода стечет в приемник,
алонж устанавливают в первоначальное положение.
После окончания реакции переливают содержимое приемника
в делительную воронку, отделяют тяжелый (нижний) слой броми-
стого этила в коническую колбу емкостью 200 мл, помещают в со-
суд с мелко раскрошенным льдом и при легком встряхивании по
каплям приливают из капельной воронки концентрированную сер-
ную кислоту до тех пор, пока жидкость не расслоится.
Промывку бромистого этила серной кислотой проводят для
удаления диэтилового эфира, который получается при реакции как
побочный продукт. При этом серная кислота действует и как водо-
отнимающее вещество, высушивая бромистый этил. Так как про-
исходит выделение тепла (что может вызвать потери легколету-
чего бромистого этила), то колбу все время держат в охладитель-
ной ванне (мелко раскрошенный лед).
Содержимое колбы переносят в делительную воронку и отде-
ляют нижний сернокислотный слой. Высушенный серной кислотой
неочищенный бромистый этил перегоняют из круглодонной колбы
емкостью 100 мл (прибор 2).
Неочищенный бромистый этил окрашен в желтый цвет. Это
свидетельствует о наличии в нем примеси растворенного брома.
Выход бромистого этила 22 г.
Бромистый этил (бромэтан) — бесцветная жидкость с запахом эфира; в 100 г
воды при 20°C растворяется 0,914 г (это надо учесть и не брать в приемник
много воды); хорошо растворяется в спирте, эфире, обладает свойствами, типич-
ными для галогенпроизводных предельных углеводородов. На свету желтеет
вследствие выделения свободного брома. Мол. вес 108,98; т. пл. —119 °C;
т. кип. 38,3 °C; dff = 1,4580; = 1,4211.
Применяется как наркотик, хладоагент, вместе с СНзС1 и C2H5CI для полу-
чения этилбензола, этилмалонового эфира, для ингаляции, а также в синтезе
Гриньяра.
При повышенных концентрациях бромистый этил вызывает поражения нерв-
ной системы. Хранят его в запаянных ампулах или в толстостенных склянках
с притертой пробкой.
Отличительная реакция
При внесении в пламя горелки медной проволоки, смоченной
бромистым этилом, появляется ярко-зеленая окраска пламени
вследствие образования летучего соединения меди с бромом. Эта
окраска пламени характерна для галогенпроизводных (проба
Бейльштейна).
п-БРОМНАФТАЛИН И БРОМИСТЫЙ ЭТИЛ
Формулы:
Вг
а-С10Н7Вг или
С2Н5Вг
419
Основные реакции:
Вг
+ НВг
С2Н6ОН + НВг 4=* С2Н6Вг + Н2О
Реактивы, посуда и приборы
Нафталин.....................16 г
(0,12 моль)
Этиловый спирт, 95%-ный ... 8 мл
(0,14 моль)
Бром.........................8 мл
(0,16 моль)
Колба круглодонная широкогорлая
(250 лл)......................1
Колба круглодонная (50 мл) . . . I
Колба коническая (100 мл) . . . . 1
Форштосс трехрогяй............1
Дефлегматор...................I
Воронка капельная ................ I
Воронка делительная ............. I
Холодильник Либиха .............. I
Алонж.........................I
Прибор для перегонки с перегре-
тым водяным паром.............1
Прибор для перегонки в вакууме 1
Сборка приборов
1. В водяную баню помещают круглодонную широкогорлую
колбу емкостью 250 мл и закрывают ее пробкой с трехрогим фор-
штоссом. К реакционной колбе заранее подбирают пробку с труб-
кой для перегонки а-бром-
цафталина с перегретым во-
дяным паром. В прямое гор-
ло форштосса вставляют
мешалку, а в два других —
капельную воронку и не-
большой дефлегматор, со-
единенный с нисходящим хо-
лодильником.- К последнему
присоединяют алонж, конец
которого на 1—1,5 см погру-
жают в воду со льдом, на-
Рис. 86. Прибор для получения а-бром-
нафталина и бромистого этнла:
/—водяная баня; 2—круглодонная широкогорлая
колба; 3—трехрогнй форштосс; 4—капельная»
воронка; 5—мешалка; 6— дефлегматор; 7—холо-
дильник; 3 —алонж; Р—колба-приемник.
3. Прибор для перегонки с
(см. рис. 48, 49).
4. Прибор
(см. рис. 51).
ходящуюся в конической
колбе (рис. 86).
2. Прибор 2 на стр. 118
(в данном случае приме-
няется круглодонная колба
50 мл).
перегретым водяным паром
для перегонки в вакууме из колбы Клайзена
120
Выполнение синтеза
В круглодонную широкогорлую колбу емкостью 250 мл, уста-
новленную в соответствии с описанием прибора 1, загружают 16 г
тонко растертого нафталина, вливают 8 мл этилового спирта, пе-
ремешивают содержимое колбы и осторожно нагревают на водя-
ной бане до 40—50°C. При непрерывном помешивании реакцион-
ной смеси прибавляют из капельной воронки 8 мл брома (см.
стр. 11) с такой скоростью, чтобы температура смеси не превы-
шала 50 °C.
При введении в реакционную смесь метилового или пропилового спирта
можно получить соответственно а-бромнафталин и бромистый метил, а-бромиаф-
талнн и бромистый пропил. При введении брома в реакционную смесь необхо-
димо поддерживать температуру кипения соответствующего бромистого алкила
После того как весь бром будет введен в колбу, нагревают
водяную баню до 60 °C. Чтобы содержимое колбы не пенилось,
необходимо все время регулировать нагревание. Реакционную
смесь нагревают до тех пор, пока в приемник не перестанут сте-
кать маслянистые капли бромистого этила, опускающиеся на дно.
После окончания реакции отделяют при помощи делительной
воронки бромистый этил от воды, спуская его в коническую колбу.
Для высушивания и освобождения бромистого этила от примеси
спирта колбу помещают в сосуд со льдом и при встряхивании
прибавляют из капельной воронки по каплям концентрированную
серную кислоту (см. стр. 11). Как только жидкость расслоится,
переливают ее в делительную воронку. Слой серной кислоты,
который собирается внизу, отделяют от бромистого этила, а по-
следний (см. стр. 119) переносят в перегонную колбу емкостью
50 мл и перегоняют на водяной бане (прибор 2) при 35—40°C.
Выход бромистого этила ~5 г.
В круглодонной колбе на 250 мл остаток представляет собой
смесь а-бромнафталина, непрореагировавшего нафталина и не-
большого количества бромистоводородной кислоты. При перегонке
с перегретым до 130°C водяным паром удаляются нафталин, бро-
мистоводородная кислота и небольшое количество а-бромнафта-
лина.
Чтобы избежать потери а-бромнафталина, его отделяют от
воды, вносят обратно в реакционную колбу и снова перегоняют
с водяным паром. Как только в холодильнике перестанут появ-
ляться кристаллы нафталина, колбу охлаждают, отделяют в дели-
тельной воронке а-бромнафталин (нижний слой) от воды и сушат
его хлористым -кальцием.
Высушенный а-бромнафталин перегоняют в вакууме из колбы
Клайзена при 132—135°С и остаточном давлении 12 мм рт. ст.
(1-я фракция) и при 145—148 °C и остаточном давлении 20 мм рт. ст.
(2-я фракция).
Перегнанный при атмосферном давлении а-бромнафталин
темнеет при хранении, так как частично разлагается.
Выход а-бромнафталина ~13 г.
121
а-Бромнафталин — желтоватая жидкость с характерным запахом; нераство-
рим в воде; смешивается со спиртом, эфиром, бензином Мол вес 207,07; т. кип.
281,2 °C, т. пл 6,1 °C; d|5= 1,488Э, Пр == 1-6600-
Применяется в качестве теплоносителя в сушильных шкафах, определения
показателя преломления минералов, а также для синтеза а-нафтойной кислоты
и других целей.
Отличительная реакция
а-Бромнафталин дает реакцию Бейльштейна на галогены (см.
стр. 119).
ЙОДИСТЫЙ этил
Формула:
С2Н61
Основные реакции:
2Р + 312 —> 2Р13
ЗС2Н6ОН + Р13 —> ЗС2Н61 + Р(ОН)3
Побочная реакция:
ЗС2Н6ОН + Р13 —> (С2Н5О)3Р + 3HI
Йодистый этил нельзя получать действием йодистого калия и
серной кислоты на спирт, так как образующаяся иодистоводород-
ная кислота восстанавливает серную кислоту до сернистого газа:
H2SO4 + 2HI —> I2 + SO2 + 2Н2О
Поэтому для получения йодистого этила применяют трехиоди-
стый фосфор, образующийся при взаимодействии иода с фосфо-
ром. Иодистоводородная кислота в отличие от бромистоводородной
часто восстанавливает серную кислоту до свободной серы.
Реактивы, посуда и приборы
Этиловый спирт, абсолютный 17 мл
(0,3 моль)
Иод........................32 г
(0,25 г-атом)
Красный фосфор.............3,1 г
(0,1 г-атом)
Кислый серннстокислый натрий
Колба круглодонная (100 мл) . . . 1
Колба Вюрца (50 мл)............1
Колба коническая (50 мл) . . •. . 1
Форштосс двурогий..............1
Воронка делительная .......... 1
Термометр......................1
Алонж..........................1
Приемник..................... 1
Сборка приборов
1. В круглодонную колбу емкостью 100 мл, помещенную на
водяную баню, вставляют двурогий форштосс, вертикальный от-
росток которого закрывают резиновой пробкой, а боковой отросток
соединяют с водяным обратным холодильником.
2. Реакционную колбу на 100 мл посредством изогнутой стек-
лянной трубки соединяют с нисходящим холодильником, к концу
которого присоединяют алонж, опущенный почти до дна прием-
ника, охлаждаемого снаружи холодной водой.
122
3. В колбу Вюрца на 50 мл помещают «кипелки» (см. стр. 30),
вставляют термометр и присоединяют нисходящий холодильник
с алонжем, который погружают в приемник.
Выполнение синтеза
Работу следует проводить в вытяжном шкафу!
В реакционную колбу прибора 1, собранного в вытяжном шка-
фу, вносят 3,1 г красного фосфора, просушенного в эксикаторе
над серной кислотой, и приливают 17 мл абсолютного этилового
спирта. (Для более полного использования дорогостоящего иода
фосфор и спирт берут в избытке по сравнению с теоретическими
количествами.)
Отвешивают 32 г тонко растертого в ступке иода. Поместив
реакционную колбу в -ледяную воду и включив обратный холо-
дильник, в течение 10—15 мин вводят в колбу небольшими пор-
циями иод через вертикальное горло двурогого форштосса. После
каждого добавления плотно закрывают пробку и хорошо встряхи-
вают колбу.
Затем реакционной смеси дают постоять 1 ч, изредка встряхи-
вая колбу. Смесь нагревают 2 ч на горячей водяной бане. Реак-
ционную колбу охлаждают, соединяют с нисходящим холодильни-
ком (прибор 2) и отгоняют иодистый этил, нагревая колбу на
кипящей водяной бане. Так как в конце перегонки иодистый этил
отгоняется очень медленно, то водяную баню оставляют, выти-
рают колбу полотенцем и отгоняют остаток йодистого этила,
непрерывно обводя колбу слабым коптящим пламенем горелки.
Полученный погон содержит некоторое количество спирта и
окрашен в коричневый цвет из-за примеси иода. Дистиллят пере-
носят в делительную воронку, отделяют верхний водный слой, за-
тем иодистый этил промывают для удаления этилового спирта
2—3 раза водой; для удаления иода к последней порции воды
прибавляют ~1 мл раствора кислого сернистокислого натрия и
наконец для нейтрализации окончательного обесцвечивания встря-
хивают иодистый этил с разбавленным раствором щелочи:
ЗЫаОН
I2 + NaHSO3 4- Н2О —► NaHSO4+2HI -------► Na2SO4 + 2NaI + ЗН2О
Нижний слой сливают в коническую колбу емкостью 50 мл,
сушат гранулированным хлористым кальцием и перегоняют из
колбы Вюрца (прибор 3), собирая фракцию, кипящую при 72—
72,3 °C. Если хлористый кальций плавает на поверхности йоди-
стого этила, то алкилгалогенид переливают через воронку со стек-
лянной ватой.
Выход йодистого этила ~30 г.
Иодистый этил (иодэтан)—тяжелая бесцветная жидкость; мало растворим
в воде. ^4° — 1,9371; Пд = 1,5168; т. кип. 72,3 °C. Применяется для получения фе-
нетола, этилбензола и этилмалоновой кислоты. Хранят его в хорошо закупорен-
ной склянке темного цвета.
113
БРОМИСТЫЙ БУТИЛ
Формула:
С4Н9Вг
ПОЛУЧЕНИЕ БРОМИСТОГО БУТИЛА
С ПРИМЕНЕНИЕМ БРОМИСТОГО КАЛИЯ
Основные реакции:
KBr + H2SO4 —> HBr + KHSO4
С4Н9ОН + НВг <=> С4Н9Вг + Н2О
Побочные реакции:
H2SO4
СН3(СН2)2СН2ОН -----> СН3СН2СН=СН2 4- Н2О
2НВг 4- H2SO4 —> Вг2 4-SO2 4-2Н2О
СН3(СН2)2СН2ОН 4- HOSO2OH —> CH3(CH2)2CH2OSO2OH 4- Н2О
CH3(CH2)2CH2OSO2OH + СН3(СН2)2СН2ОН —>
—> СН3(СН2)2СН2ОСН2(СН2)2СН3 4- H2SO4
Реактивы, посуда и приборы
Бутиловый спирт.............28 мл
(0,32 моль)
Бромистый калий............ 30 г
(0,25 моль)
Едкий натр. 10%-ный .... 20 мл
Хлористый кальций...........5 г
Серная кислота (d = 1,84) . . 23 мл
(0,43 моль)
Колба круглодонная (300 мл) . . . 1
Колба Вюрца (50 мл).............1
Холодильник Либиха..............1
Воронка коническая..............1
Воронка делительная ........... 1
Сетка асбестовая................1
Приемники ..................... 2
Кислый сернистокислый натрий
1. В круглодонную колбу емкостью 300 мл помещают несколь-
ко «кипелок» и соединяют ее с обратным водяным холодильником.
Последний неплотно закрепляют в штативе (это необходимо для
встряхивания колбы при перемешивании).
2. Прибор 2 на стр. 118, (в данном случае используется круг-
лодонная колба емкостью 300 мл).
3. Прибор 3 на стр. 123.
Выполнение синтеза
В круглодонную колбу прибора 1 вливают 30 мл воды, 28 мл
бутилового спирта и вносят 30 г бромистого калия. Колбу сна-
ружи охлаждают проточной водой и медленно добавляют "23 мл
концентрированной серной кислоты, хорошо перемешивая смесь
встряхиванием колбы. Содержимое колбы нагревают до кипения
небольшим пламенем на асбестовой сетке в течение 2 ч. Затем
собирают прибор 2 и быстро отгоняют бромистый бутил, который
содержит примеси воды, бутилового эфира, бутилена, бутилового
спирта, брома.
124
Дистиллят переносят в делительную воронку, промывают 20 мл
воды с небольшим количеством кислого сернистокислого натрия
для устранения окраски, вызванной присутствием следов брома,
сливают нижний слой бромистого бутила в чистую колбу. Дели-
тельную воронку моют, сушат и опять переносят в нее бромистый
бутил, к которому добавляют (при наружном охлаждении) рав-
ный объем концентрированной серной кислоты для удаления бути-
лового эфира. Происходит образование двух слоев: верхний —
бромистый бутил, нижний — сернокислотный слой. Разделяют слои
и промывают бромистый бутил 20 мл 10% раствора едкого натра
(для удаления следов серной кислоты). Мутный бромистый бутил
сушат 5 г хлористого кальция, слегка нагревая на водяной бане.
Как только содержимое колбы станет прозрачным, сливают
высушенный бромистый бутил в колбу Вюрца емкостью 50 мл и
перегоняют, собирая фракцию, кипящую в пределах 99—103°С.
Выход бромистого бутила ~23 г.
ПОЛУЧЕНИЕ БРОМИСТОГО БУТИЛА
С ПРИМЕНЕНИЕМ БРОМА И КРАСНОГО ФОСФОРА
Основные реакции:
2Р + ЗВг2 —> 2РВг3
ЗС4Н9ОН + РВг3 —> ЗС4Н9Вг + Р(ОН)3
Реактивы, посуда и приборы
Бутиловый спирт .... 19 г или 23,5 мл
(0,25 моль)
Красный фосфор .... 3,9 г
(~0,12 г-атом)
Бром....................31,4 г или 10 мл
(~0,2 моль)
Кислый сернокислый натрий
Хлористый кальций
Колбы круглодонные (100 н
250 мл).................2
Колба коническая .........1
Форштосс двурогий.........1
Холодильник Либиха . . . . 1
Воронка коническая . . . . 1
Сборка приборов
Воронка капельная.........1
Воронка делительная .... 1
Прибор для перегонки с во-
дяным паром.............1
Дефлегматор...............1
Приемники ............... 2
1. В охладительную баню помещают круглодонную колбу ем-
костью 250 мл, в которую вносят «кипелки». К колбе заранее
подбирают пробку с двумя трубками. Это необходимо для после-
дующей перегонки с водяным паром.
Колбу снабжают двурогим форштоссом с обратным холодиль-
ником и капельной воронкой. Лапку штатива, удерживающую
холодильник, закрепляют неплотно, чтобы .лучше встряхивать
колбу.
2. Прибор для перегонки с водяным паром (см. рис. 48). Во
избежание потерь бромистого бутила необходимо пользоваться
длинным, хорошо охлаждаемым холодильником.
125
3. В круглодонную колбу на 100 мл вносят «кипелки», соеди-
няют ее с дефлегматором, который снабжают термометром и
нисходящим холодильником с алонжем, опущенным в приемник.
)
/
Выполнение синтеза ч
Работу необходимо проводить в вытяжном шкафу!
В круглодонную колбу прибора 1 помещают 3,9 г красного
фосфора, высушенного в эксикаторе над серной кислотой, и
23,5 мл бутилового спирта. Колбу охлаждают и из капельной во-
ронки медленно (в течение 1 ч) при частом встряхивании добав-
ляют 10 мл брома (см. гл. I). После введения всего количества
брома реакционную массу 3 ч нагревают на водяной бане до
исчезновения паров брома, содержимое колбы при этом должно
энергично кипеть. После окончания реакции бромистый бутил от-
гоняют с водяным паром из той же реакционной колбы. Дистил-
лят переносят в делительную воронку, встряхивают его с 10%-ным
кислым сернистокислым натрием для удаления следов брома:
Вг2 4-NaHSO3 4-Н2О —> NaHSO44-2HBr
В чистую колбу сливают нижний слой бромистого бутила, су-
шат гранулированным хлористым кальцием до тех пор, пока со-
держимое колбы не станет прозрачным. Через воронку со стеклян-
ной ватой сливают высушенный бромистый бутил в круглодонную
колбу емкостью 100 мл и перегоняют (прибор 3), собирая фрак-
цию, кипящую в пределах 99—103 °C.
Выход бромистого бутила -~28 г.
Бромистый бутил — бесцветная жидкость; хорошо растворим в спирте, эфи-
ре. Мол. вес 137,03; т. кип. 101,6 °C; т. пл. — 112,4 °C; ^°= 1,2829; 1,4398.
Применяется как алкилирующее средство и прн синтезе нитрилов.
Отличительная реакция
Бромистый бутил дает реакцию Бейльштейна на галогены (см.
стр. 119).
ХЛОРИСТЫЙ АЦЕТИЛ
Формула:
СН3—С—С1
О
Основная реакция:
ЗСН3—С—ОН 4-РС13 —> ЗСН3—С—С1 4-Н3РО3
А А
Побочная реакция:
ЗСН3СООН 4- РС13 (СН3СОО)3Р 4-ЗНС1
116
Реактивы, посуда и приборы
Уксусная кислота, ледяная 23 г илн 22 мл
(0,38 моль)
Треххлористый фосфор . . 18 г или 11 мл
(0,13 моль)
Хлористый кальций
Колба круглодониая (200 мл) 1
Колба Вюрца (200 мл) . . . I
Холодильник Либиха ' , . . . 1
Воронка капельная ....... 1
Колба коническая (100 мл) 1
Дефлегматор...............1
Хлоркальциевые трубки ... 2
Термометр.................1
Алонж с боковым отростком 1
Сборка приборов
1. Колбу Вюрца емкостью 200 мл, помещенную на водяную
баню, соединяют с капельной воронкой и нисходящим холодиль-
ником. Внутреннюю трубку холодильника при помощи алонжа
с хлоркальциевой трубкой соединяют с конической колбой (рис. 87).
Рис. 87. Прибор для получения хлористого аце-
тила:
/—водяная баня! 2 — колба Вюрца; 3—капельная во-
ронка; 4—хлоркальццезая трубка; 5—холодильник; £ —
алонж с хлоркальциевой трубкой; 7—приемник.
Хлористый ацетил легко разлагается влагой воздуха, поэтому
его не следует собирать в открытом приемнике.
2. Круглодонную колбу емкостью 200 мл, помещенную на во-
дяную баню, соединяют с дефлегматором, термометром и нисхо-
дящим водяным холодильником с алонжем. Последний имеет
боковой отросток, на который надевают хлоркальциевую трубку.
Алонж соединяют с приемником через пробку.
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
В колбу прибора 1 вносят 22 мл ледяной уксусной кислоты
(осторожно! ледяная уксусная кислота легко воспламеняется, об-
разует с воздухом взрывоопасные смеси; пары ее действуют на
слизистые оболочки и кожу; при попадании кислоты на кожу сле-
дует пораженное место промыть большим количеством воды).
Колбу помещают в баню с холодной водой. Через капельную
127
воронку постепенно, приливают 11 мл треххлористого фосфора,
время от времени встряхивая колбу. После внесения всего коли-
чества треххлористого фосфора реакционную смесь нагревают в те-
чение 30 мин на водяной бане при 40—50 °C. Нагревание продол-
жают до тех пор, пока не прекратится сильное выделение хлори-
стого водорода и в жидкости не образуется два слоя. Хлористый
ацетил образует верхний слой, а фосфористая кислота — нижний.
Водяную баню нагревают до кипения и отгоняют хлористый ацетил.
Для полной очистки дистиллят еще раз перегоняют (при-
бор 2). Собирают хлористый ацетил при 50—53°C.
Выход хлористого ацетила ~ 18 г.
Хлористый ацетил (хлорангидрид уксусной кислоты) — бесцветная, дымя-
щая на воздухе жидкость с резким запахом, нерастворим в воде; хорошо раство-
рим в органических растворителях. Мол. вес 78,54; т. кип. 51,8 °C; т. пл— 112 °C;
^2°= 1,1051; 4° = 1,3897.
Применяется для ацетилирования, в производстве красителей, лекарственных
веществ и для других целей.
Отличительные реакции
1. Хлористый ацетил дает реакцию Бейльштейна на галогены
(см.стр. 119).
2. Дымит на воздухе, подвергаясь гидролизу под действием
влаги воздуха.
3. При взбалтывании с водой происходит гидролиз хлористого
ацетила; полученный раствор дает кислую реакцию по конго крас-
ному.
ХЛОРИСТЫЙ БЕНЗОИЛ
Формула:
С6н5—С—С1
Основная реакция:
С6Н5—С—ОН + РС15 —> СвН5—С— С1 + РОС13 + НС1
А А
Реактивы, посуда и приборы
Бензойная кислота..........12,2 г Колба круглодонная (100 мл) . . . 1
(0,1 моль) Колба Вюрца (100 мл)............1
Пятнхлористый фосфор ... 21 г Ко-лба коническая...............1
(0,1 моль) Холодильник воздушный..........1
Холодильник Либиха.............1
Сборка приборов
1. Круглодонную колбу емкостью 100 мл при помощи резино-
вой пробки соединяют с обратным воздушным холодильником.
2. Колбу Вюрца емкостью 100 мл соединяют с термометром
и нисходящим водяным холодильником.
128
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
В круглодонную колбу вносят 12,2 г бензойной кислоты и 21 г
пятихлористого фосфора (прибор 1). Затем встряхивают колбу
до тех пор, пока смесь не станет жидкой.
Собирают прибор 2, сначала отгоняют хлорокись фосфора при
НО °C, а затем, сменив водяной холодильник на воздушный, при
199°C перегоняют хлористый бензоил.
Выход хлористого бензоила ~ 10 г.
Хлористый бензоил — бесцветная, слегка дымящая на воздухе жидкость с
резким запахом; хорошо растворим в эфире, хлороформе и бензоле. Мол. вес
140,57; т. кип. 197,9 °C; т. пл. — 0,6 °C; d*° = 1,219; n°D = 1,5535.
С помощью хлористого бензоила открывают спирты, амины, фенолы; его
используют для синтетических целей. Хранят в хорошо закупоренных склянках
с притертыми пробками или в ампулах. Пары хлористого бензоила раздражают
слизистые оболочки, вызывая слезотечение.
Отличительная реакция
При растирании 2 г углекислого аммония в фарфоровой чаш-
ке с 1 г хлористого бензоила образуется осадок бензамида:
С6Н6-С-С1 + (NH4)2CO3
—> С6Н5—С—NH2 + NH4C1 + Н2СО3
1,2-ДИБРОМЭТАН
Формула:
СН2Вг—СН2Вг
Основные реакции:
СН3—СН2—ОН 4-НО—SO2—ОН «=> СН3—СН2—О—SO2—ОН 4-Н2О
160-170 °C
сн3—сн2—о—so2—он -------> сн2=сн2 + H2SO4
СН2=СН2 + Вг2 —> СН2Вг—СН2Вг
«
Побочные реакции:
СН2=СН2 + Вг2 + Н2О —> СН2—СН24-НВг
J)H Вг
CH3-CH2-O-SO2-OH + но-сн2—сн3 —»
—> сн3—сн2—о—сн2—сн3 + H2SO4
5 М. Н. Храмкина
129
Реактивы, посуда и приборы
Этиловый спирт, 95%-ный 45 мл
(0,7 моль)
Бром..................31,4 г, или 10 мл
(0,2 моль)
Серная кислота (d = 1,84) 90 мл
(1,7 моль)
Сернокислый алюминий,
безводный.............10 г
Едкий натр
Хлористый кальций
Песок крупнозернистый, сухой
Колба круглодонная (500 мл) I
Колба Вюрца (100 мл) . , . . 1
Колба коническая ......... 2
Холодильник Либиха.........1
Воронка капельная..........1
Термометр..................1
Склянка Дрекселя...........3
Склянка Тищенко............1
Стакан химический..........3
Тройннк ...................1
Алонж......................1
Сборка приборов .
1. Круглодонную колбу емкостью 500 мл помещают на песоч-
ную баню. Отверстие колбы плотно закрывают пробкой, в которую
вставляют капельную воронку с длинной трубкой, термометр и
согнутую под прямым углом газоотводную трубку. Последнюю
Рис. 88. Прибор для получения 1,2-дибромэтана'.
/—песочная баня; 2—колба круглодонная; 3—термометр; 4— капельная воронка;
5—склянка Дрекселя; 6—стакан химический; 7 —тройннк; в—склянка Тищенко; ?—ко-
ническая колба.
соединяют с промывной склянкой Дрекселя, содержащей концен-
трированную серную кислоту (для очистки этилена от паров
спирта и эфира). Эту склянку помещают в стакан с холодной
водой. Чтобы избежать потери этилена, промывную склянку охла-
ждают снаружи, так как при повышенной температуре этилен
взаимодействует с серной кислотой, образуя этилсерную кислоту.
Промывную склянку посредством тройника соединяют со
склянкой Тищенко, где находится 4 н. раствор едкого натра, ис-
пользуемого для очистки этилена от сернистого газа. Тройник при
помощи согнутой трубки соедйняйт с верхним отверстием капель-
ной воронки для выравнивания давления в приборе.
Давление нужно выравнивать, так как иначе этилен не будет
проходить через серную кислоту, а пойдет через капельную ворон-
ку, поскольку вес столба жидкости в воронке меньше, чем сум-
марный вес столба жидкости в промывных склянках.
Предохранительную склянку Тищенко соединяют с двумя по-
глотительными склянками Дрекселя, заполненными бромом й по-
130
мещенные в стаканы с холодной водой, которую в течение реакции
меняют (охлаждение склянок необходимо, так как реакция соеди-
нения брома с этиленом идет с выделением тепла). Отводную
трубку последней склянки Дрекселя опускают в коническую колбу
с 2 н. раствором едкого натра (для поглощения паров брома, уле-
тучивающихся из склянок с бромом), не погружая трубку в жид-
кость. Для проверки полноты поглощения этилена бромом время
от времени трубку опускайт на 0,5 см в раствор едкого натра. При
правильном ведении реакции пузырьки этилена только изредка
будут выходить из трубки.
Перед началом работы нужно обратить внимание на правиль-
ность сборки прибора (рис. 88) и проверить газонепроницаемость
поглотительных склянок. Газонепроницаемость поглотительных
склянок проверяют при помощи промывной склянки с серной кис-
лотой, которую соединяют с поглотительными склянками и газо-
метром, наполненным азотом или воздухом. Пропуская через по-
глотительную систему азот, следят за прохождением пузырьков
газа.
2. Колбу Вюрца емкостью 100 мл соединяют с термометром и
нисходящим холодильником с алонжем, который опускают в ко-
ническую колбу на 50 мл.
Выполнение синтеза
В круглодонную колбу прибора 1 вливают 15 мл этилового
спирта и осторожно при перемешивании и наружном охлаждении
добавляют 45 мл концентрированной серной кислоты. Затем вно-
сят 20 г сухого крупнозернистого песка, отсеянного от мелких
частиц для ускорения отщепления воды и 10 г безводного серно-
кислого алюминия, действующего каталитически. В капельную во-
ронку помещают смесь 30 мл этилового спирта и 45 мл концен-
трированной серной кислоты. При этом нужно следить, чтобы
трубка капельной воронки была целиком заполнена смесью спирта
и серной кислоты. Перед тем как вставить воронку в колбу, от-
крывают кран капельной воронки, погружают отводную трубку
последней в приготовленную смесь спирта и серной кислоты и
осторожно засасывают ее до тех пор, пока не заполнится вся от-
водная трубка. Быстро закрывают кран, вставляют пробку в кол-
бу и вливают остаток смеси в капельную воронку.
Колбу так нагревают на песочной бане, чтобы температура не
превышала 170°С, и постепенно приливают смесь спирта и серной
кислоты из капельной воронки. Скорость прибавления смеси нуж-
но регулировать таким образом, чтобы этилец выделялся быстро
(но не сплошной струей) и в колбе не происходило сильного вспе-
нивания нагреваемой массы. Выделяющийся этилен проходит
через промывную склянку Дрекселя с концентрированной серной
кислотой, склянку Тищенко с 4 н. раствором едкого натра и две
поглотительные склянки Дрекселя, содержащие по 5 мл брома,
поверх которого наливают воду слоем в 1 см (для уменьшения
потерь брома за счет испарения).
5*
131
Через 2 ч после начала реакции, когда исчезает окраска
брома в обеих поглотительных склянках, сразу же разъединяют
склянки, иначе при охлаждении содержимое их может переме-
шаться.
Сырой дибромэтап переносят в делительную воронку, промы-
вают водой, затем разбавленным раствором едкого натра до пол-
ного обесцвечивания и еще несколько раз водой. Продукт сушат
над хлористым кальцием и перегоняют (прибор 2) при 130—
132 °C.
Выход 1,2-дибромэтана ~30 г.
1,2-Дибромэтап— бесцветная жидкость; хорошо растворим в органических
растворителях Мол. вес 187,89; т. кип. 131,5 °C; т. пл. 9,97 °C; d\a = 2,1806; =
= 1,5380 1,2 Дибромэган ядовит, вызывает поражение кожи.
Применяется для получения этилендиамина, в качестве добавок к мотор-
ному топливу и для синтеза лекарственных препаратов, например пиперазина.
БРОМБЕНЗОП
Формула:
С6НбВг
Основная реакция:
Fe
С|Н6 + Вг2 --> С6Н5Вг + НВг
Побочная реакция;
С6Н5Вг + Вг2
Бромирование бензола идет в присутствии катализатора (же-
леза), при этом возможны два механизма:
а) с предварительной ионизацией брома
2Fe + ЗВг2 —> 2FeBr3
FeBr3 + Br2 —> Br+ + FeBr?
СГ-комплекс
<з-нс>ммекс
+ HBr + FeBr3
132
б) без предварительной ионизации
+ ГеВгз
<3*- комплекс
+ НВг + Тевг3
Реактивы, посуда и приборы
Бензол................19,4 г, или 22 мл
(0,25 моль)
Бром..................31,4 г, или 10 мл
(0,2 моль)
Железные опилки ... 0,5 г
Хлористый кальций . . 12 г
Колба круглодонная (200 мл) 1
Колба Вюрца (50 мл) .... 1
Форштосс двурогий........1
Холодильник Либиха.......1
Холодильник воздушный . . . 1
Воронка капельная........1
Воронка коническая ...... 1
Воронка делительная ..... 1
Прибор для перегонки с водя-
ным паром................1
Чашка фарфоровая.........1
Стакан химический........1
Алонж....................1
Термометр................1
Приемники................2
ПОЛУЧЕНИЕ БРОМБЕНЗОЛА
С ПРИМЕНЕНИЕМ ПЕРЕГОНКИ С ВОДЯНЫМ ПАРОМ
Сборка приборов
1. Круглодонную колбу емкостью 200 мл при помощи двуро-
гого форштосса соединяют с капельной воронкой и обратным во-
дяным холодильником.
Выход реакции во многом зависит от правильной сборки и
герметичности прибора. Бром разъедает резиновые и корковые
пробки, поэтому наиболее чистый продукт реакции получается при
работе в приборе на шлифах. В случае применения корковых про-
бок их следует пропарафинить, резиновые — смазать слегка вазе-
лином (см. гл. I).
В верхний конец холодильника вставляют стеклянную изогну-
тую трубку, которую соединяют при помощи резиновой трубки
с опрокинутой стеклянной воронкой, установленной в стакане над
поверхностью холодной воды для поглощения образующегося при
реакции бромистого водорода. Воронка должна находиться на
расстоянии 1—2 см от поверхности воды. Установку (рис, 89) сле-
дует поместить в вытяжном, шкафу!
133
2. Прибор для перегонки с водяным паром (см. рис. 48).
3. В колбу Вюрца на 50 мл вставляют термометр и соеди-
няют колбу с воздушным холодильником, на конец которого наде-
вают алонж.
Выполнение синтеза
В реакционную колбу емкостью 200 мл вносят 0,5 г железных
опилок, приливают 22 мл бензола, предварительно высушенного
над хлористым кальцием. Затем из капельной воронки при встря-
- 1
Рис. 89. Прибор для получения бромбеизола:
/—круглодонная колба: 2—-двурогий форштосс;
3 —капельная воронка; 4—холодильник; 5—воронка;
б—стакан с водой.
хивании медленно при-
бавляют бром (см. гл. I).
Если бром, бензол и по-
суда хорошо высушены,
то реакция начинается
сразу, в случае задержки
реакции можно смесь
слегка нагреть на водя-
ной бане. После того как
начнется выделение бро-
мистого водорода, прили-
вают оставшееся количе-
ство брома с такой ско-
ростью, чтобы реакция
протекала равномерно и
интенсивно, но не становилась бурной, так как быстрое введение
брома и повышение температуры способствуют образованию ди-
бромбензола.
После введения всего количества брома для завершения реак-
ции нагревают содержимое колбы в течение 20—30 мин на во-
дяной бане, постепенно повышая температуру с 15—25 до 60—
70°C, пока над жидкостью не исчезнут бурые пары брома. Затем
в колбу наливают воду, встряхивают полученную смесь и осто-
рожно сливают раствор из колбы в делительную воронку, наблю-
дая, чтобы железные опилки не попали в воронку. Снова промы-
вают жидкость водой и перегоняют с водяным паром (прибор 2).
Сначала отгоняется бромбензол; с появлением в холодильнике
кристаллов n-дибромбензола меняют приемник и собирают и-ди-
бромбензол до тех пор, пока не будет конденсироваться только
вода. Отогнанный бромбензол отделяют в делительной воронке от
воды, сушат над хлористым кальцием в течение 1 ч и перегоняют
из колбы Вюрца емкостью 50 мл, применяя воздушный холодиль-
ник (прибор 3).
Для сушки бромбеизола вносят бромбензол и хлористый кальций в колбу,
снабженную обратным воздушным холодильником. Нагревают смесь на водяной
бане до 60—70 °C (часто встряхивая) до тех пор, пока жидкость в колбе не
станет прозрачной.
Получают фракцию, кипящую в пределах 140—170 °C, а оста-
ток выливают из колбы Вюрца в фарфоровую чашку и после
134
застывания присоединяют к п-дибромбензолу, полученному пере-
гонкой с водяным паром.
Осадок n-дибромбензола высушивают на фильтровальной бу-
маге (для отделения жидкого о-дибромбензола) и перекристалли-
зовывают из небольшого количества спирта (см. стр. 53).
Фракцию, кипящую в пределах 140—170 °C, перегоняют вто-
рично (прибор 3), собирая чистый бромбензол при 152—158°C.
Выход бромбеизола ~ 14 г.
ПОЛУЧЕНИЕ БРОМБЕИЗОЛА ПРОСТОЙ ПЕРЕГОНКОЙ
Сборка приборов
1. Прибор для получения бромбеизола (см. рис. 90).
2. Прибор 3 на стр. 134.
Выполнение синтеза
Первую часть синтеза (процесс бромирования) проводят, как
в предыдущем случае. Полученный продукт переносят в делитель-
ную воронку, промывают водой, переливают в сухую колбу, сушат
прокаленным хлористым кальцием, после чего перегоняют (при-
бор 2), собирая фракцию, кипящую в пределах 140—170°С. Со-
бранную фракцию перегоняют вторично при 154—160°С. С остат-
ком, находящимся в колбе Вюрца, поступают так же, как указано
на стр. 134—135.
Выход бромбеизола ~13 г, а n-дибромбензола ~1 г.
Бромбензол — тяжелая прозрачная жидкость с запахом, напоминающим за-
пах бензола; слабо растворим в воде, хорошо — в спирте, эфире, хлороформе,
бензоле Мол. вес 157,02, т кип 156,2 °C; т. пл. — 30,6 °C, d™ = 1,495; Пд° = 1,5602
Применяется при реакциях Гриньяра и Фиттига, в качестве теплоносителя
для сушильных шкафов и для других целей.
п-Днбромбензол — бесцветные кристаллы с т. пл 89 °C; хорошо растворим
в спирте, эфире, плохо — в воде. Используется как промежуточный продукт в
синтезах.
а-ВРОМНАФТАЛИН
Формула:
a-C!0H7Br или
Основная реакция:
Вг
Вг
+ НВг
135
Побочная реакция:
1,4-Дибром-
нафталин
Реактивы, посуда и приборы
Нафталин..........25,6 г
(0,2 моль)
Бром..............31,4 г, или 10 мл
(0,2 моль)
Хлористый кальций
Стакан химический (200 мл) . . . 1
Прибор для перегонки с перегре-
тым водяным паром..............1
Прибор для перегонки в вакууме 1
Воронка капельная .............. 1
Воронка делительная ............ 1
Термометр........................1
Сборка приборов
1. Стакан емкостью 200 мл помещают на водяную баню. На
штативе крепят термометр и капельную воронку, трубка которой
доходит почти до дна стакана.
2. Прибор для перегонки с перегретым водяным паром (см.
рис. 48, 49).
3. Прибор для перегонки в вакууме (см. рис. 51).
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
В стакан на 200 мл помещают 25,6 г тонко растертого нафта-
лина, 40 мл воды и при помешивании, нагревая смесь на водяной
бане до 40—50°C, добавляют по каплям 10 мл брома из капель-
ной воронки с такой скоростью, чтобы температура реакционной
смеси не превышала 50 °C. После прибавления всего количества
брома перемешивание продолжают до исчезновения желтой окрас-
ки реакционной массы. Затем содержимое стакана переносят в
делительную воронку и отделяют нижний масляный слой от вод-
ного. Переносят масло в колбу на 250 мл и перегоняют с пере-
гретым до 130 °C водяным паром (прибор 2) для удаления от
а-бромнафталина неизмененного нафталина и других примесей,
при этом должно отогнаться около 70—80 мл жидкости. Пере-
гонку с паром продолжают до исчезновения в холодильнике кри-
сталлов нафталина. Оставшееся в реакционной колбе масло сушат
хлористым кальцием и перегоняют в вакууме из колбы Клай-
зена (прибор 3), собирая фракцию, кипящую при '145—148 °C
(20 мм рт. ст. или при 132—135°C 12 мм рт. ст.).
Выход а-бромнафталина ~20 г (см. стр. 122),
136
Синтез re-бром нафталина можно провести в трехгорлой колбе с термомет-
ром, капельной воронкой и обратным холодильником, который соединен с изо-
гнутой стеклянной трубкой, опущенной в стакан с водой таким образом, чтобы
она немного не доходила до поверхности воды. В этом случае улавливается
бромистый водород.
п-БРОМАНИЗОЛ
Формула:
ВгС6Н4ОСН3;
Основная реакция:
Побочная реакция:
О-СН3
+ Вг2
Реактивы, посуда и приборы
Анизол.........14,5 г, или 14,6 мл
(~0,13 моль)
Бром...........22 г, или 7 мл
(~0,14 моль)
Этиловый эфир..............70 мл
Хлористый кальций, безводный
Колба Вюрца (150 мл)............1
Колба круглодонная (200 мл) . . . 1
Колба коническая................1
Форштосс двурогий...............1
Холодильник Либиха..............1
Воронка капельная ............ 1
Воронка делительная .......... 1
Термометр ..................... 1
Холодильник воздушный...........1
Сборка приборов
1. Прибор для получения бромбензола (см. рис. 89).
2. Колба Вюрца с термометром и нисходящим водяным холо-
дильником.
437
Выполнение синтеза
В реакционную колбу емкостью 200 мл помещают 14,6 мл
анизола, слегка нагревают ее и через капельную воронку медлен-
но вводят 7 мл брома (см. гл. I). После прибавления всего коли-
чества брома смесь нагревают в течение 15 мин, затем охлаждают
и вносят 70 мл эфира. После этого эфирный раствор промывают
в делительной воронке водой, сушат безводным хлористым
кальцием. Эфир отгоняют на водяной бане (прибор 2), а затем,
сменив водяной холодильник на воздушный, собирают п-бромани-
зол при 213—223 °C.
Выход n-броманизола ~15 г. n-Броманизол получается с при-
месью о-броманизола, очистка от которого затруднена, так как
оба изомера имеют близкие температуры кипения.
п-Броманизол — тяжелая жидкость с приятным запахом. Т. кип. 213—
223 °C, т. пл 11,5 °C, zip = 1,5605,
Глава VIII
РЕАКЦИИ АЛКИЛИРОВАНИЯ
Алкилирование представляет собой процесс введения алкиль-
ных радикалов (метил, этил, пропил, бутил, амил и т. д.) в орга-
нические соединения.
Реакции алкилирования широко применяются в промышленно-
сти. На их основе в настоящее время осуществляется синтез ком-
понентов высокооктанового моторного топлива, исходных продук-
тов для производства синтетических каучуков и смол, моющих
средств, пластических масс, лаков, растворителей, гомологов бен-
зола, простых и сложных эфиров, жирноароматических кетонов,
аминов, а также соединений аммония и сульфония.
Реакции алкилирования можно разделить на несколько групп.
К одной из них можно отнести реакции введения алкила в угле-
родную цепь, например, получение втор-бутилбензола, изооктана;
к другой группе реакций можно отнести введение алкилов вместо
водорода в гидроксильную группу, приводящее к образованию
простых эфиров, и введение алкилов в амины с образованием
алкиламинов, третичных жирноароматических аминов.
К реакциям алкилирования относится и реакция Фриделя —
Крафтса для ароматических углеводородов, которая будет рас-
смотрена ниже (см. гл. X).
Алкилирование можно проводить различными агентами, из
них наибольшее распространение получили олефины, спирты, гало-
генпроизводные, диалкилсульфаты
139
1. АЛКИЛИРОВАНИЕ АРОМАТИЧЕСКИХ
УГЛЕВОДОРОДОВ СПИРТАМИ
В ПРИСУТСТВИИ серной кислоты
Ароматические углеводороды можно алкилировать спиртами
в присутствии серной кислоты (а также А1С13, HF, Р2О5, Н3РО4).
При этой реакции хорошие результаты получаются с высшими
алифатическими спиртами, главным образом с третичными. Протон
серной кислоты (Н+) с молекулой спирта дает карбкатион
СН3—СН2—СН2—CHJ, который изомеризуется в стабильный ион
+
СН3—СН2—СН—СН3. Последний и является алкилирующим аген-
том.
Алкилирование зависит от природы заместителей, находящих-
ся в ядре. Присутствие галогенов, нитро- и сульфогрупп затруд-
няет введение алкильного радикала. Присутствие же алкильной
группы в ядре способствует дальнейшему алкилированию, причем
главным образом получаются пара-замещенные; например, из то-
луола и изобутилового спирта образуется 1-метил-4-трет-бутил-
бензол:
СН3
+ СН3—СН—СН2ОН
+ Н2О
I ,
сн3—с-сн3
сн3
При реакции алкилирования ароматических углеводородов
спиртами в присутствии серной кислоты последнюю нагревают до
70—80 °C и при перемешивании, медленно, в течение 3—5 ч, при-
ливают смесь ароматического углеводорода и спирта.
2. ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ
Простые эфиры получают дегидратацией спиртов, действием
галогеналкилов на алкоголяты и феноляты, диалкилсульфатов на
феноляты и другими способами.
Образование простых эфиров дегидратацией спиртов проис-
ходит только в присутствии водородных ионов, которые выпол-
няют роль катализаторов. В зависимости от свойств спирта обра-
зующегося эфира нужно применять тот или иной кислый реагент.
Чаще всего для этой цели применяют серную кислоту, но упо-
требляют также хлористый водород, сульфокислоты, фосфорную
кислоту и др.
13J-
Например, образование дибутилового эфира можно предста-
вить схемой:
н+
СН3—-СН2—СН2—-СН2—О—Н -----> СН3—сн2—сн2—сн2 —о—и
i I
; Н
—> СН3—СН2—СН2—СН2
н-о—с4н9
СН3—СН2—СН2—СН2—6—С4Н9 —>
I
н
—> СНз—СП,-СН2—СН2— О—СН2—СН2—СН2—СН3 + н+
При получении простых эфиров в качестве побочных продук-
тов образуются олефины:
(И;
СН3—СН2—(!н—СН2 —> н+ + СНз—СН2—сн=сн2
Одновременно протекают реакции полимеризации, обуглива-
ния и восстановления серной кислоты до SO2.
Реакция взаимодействия галогеналкилов с алкоголятами или
фенолятами имеет большое значение, так как, пользуясь ею, мож-
но превратить различные органические соединения со спиртовыми
и фенольными гидроксильными группами в простые и смешанные
эфиры, а также в жирноароматические эфиры. Из галогеналкилов
лучше всего применять йодистые (иодиды > бромиды>хлориды).
Обычно реакцию выполняют в колбе с обратным холодильником
и конец реакции определяют по исчезновению щелочной реакции
на влажную лакмусовую бумагу.
Жирноароматические эфиры получают из фенолятов (этилат
натрия 4- фенол) и галогеналкилов, при этом реакция протекает
так же легко, как с алкоголятами. Реакция замещения водорода
гидроксильной группы фенола на алкильную группу может быть
легко достигнута действием диалкилсульфатов (диметил- или ди-
этилсульфатов) на фенол в присутствии щелочи.
Обычно фенол растворяют в необходимом количестве водного
раствора едкого натра и при перемешивании или встряхивании
раствора добавляют с небольшим избытком диалкилсульфат.
В большинстве случаев продукт реакции выделяется в виде
осадка или масла. Реакция протекает легко на холоду или при
слегка повышенной температуре, но при этом в реакцию вступает
только одна алкильная группа диалкилсульфата.
Так как эфиры серной кислоты, и особенно диметилсульфат,
очень ядовиты, то все операции с ними следует проводить под
тягой, соблюдая большую осторожность.
<40
3. ПРИМЕРЫ СИНТЕЗОВ
втор-БУТИЛБЕНЗОЛ
Формула:
Основная реакция:
СН—СН2~СНз
СН,
+ Н?О
Побочная реакция:
+ НО—SO2—ОН
Реактивы, посуда и приборы
Бензол..................29 г, или 33 мл
(0,37 моль)
Бутиловый спирт .... Иг, или 14 мл
(0,15 моль)
Серная кислота, 85%-ная 75 мл
Колба трехгорлая, круглодон-
ная (500 мл)................1
Холодильник Либиха ........ 1
Мешалка.....................1
Воронка капельная ......... 1
Воронка делительная ..... 1
Прибор для фракционной пере-
гонки .....................1
Приемники ................ 2
Сборка приборов
1. Трехгорлую круглодонную колбу емкостью 500 мл, поме-
щенную на водяную баню, снабжают обратным холодильником,
капельной воронкой и мешалкой.
2. Прибор для фракционной разгонки (см. рис. 11).
Выполнение синтеза
В трехгорлую круглодонную колбу емкостью 500 мл поме-
щают 75 мл 85%-ной серной кислоты и из капельной воронки
в течение 1 ч при энергичном перемешивании и нагревании на
водяной бане при 70—80 °C по каплям вносят смесь, состоящую
из 33 мл бензола и 14 мл бутилового спирта.
1. Необходимо поддерживать строго определенную концентра-
цию кислоты, так как это сильно влияет на выход продукта.
2. Опыт ведется при интенсивном перемешивании, чтобы реак-
ционная смесь не разделялась на два несмешивающихся слоя сер-
ной кислоты и бензола.
141
9 Необходимо строго следить за температурным режимом,
так как при 55 °C реакция идет очень медленно, а при повышении
ее до 95 °C продукты начинают сульфироваться.
После внесения смеси размешивание продолжают еще в тече-
ние 5 ч при той же температуре.
Работать с бензолом следует осторожно, так как длительное
воздействие паров бензола вызывает головные боли, утомляемость,
сонливость.
После окончания реакции переливают содержимое реакцион-
ной колбы в делительную воронку, отделяют верхний углеводо-
родный слой, два раза промывают его водой, высушивают над
хлористым кальцием, а затем переносят в круглодонную колбу и
проводят фракционную разгонку, отбирая две фракции: до 172 °C
и 172—172,5 °C. Последняя фракция представляет собой чистый
зтор-бутилбензол.
Выход втор-бутилбензола 9 г.
вгор-Бутилбензол — жидкость с т. кип. 170—174 °C; df = 0,8605 ч- 0,8639;
/$= 1,4885 -J- 1,4902.
ДИБУТИЛОВЫЙ ЭФИР
Формула: СН3(СН2)2—СН2—О-СН2— (СН2)2—СН3.
Основная реакция (механизм реакции см. стр. 140):
H2SO4
2СН3—СН2—СН2—СН2ОН --->
—> сн3—сн2—сн2—сн2—о-сн2-сн2—сн2-сн3 + Н2О
Побочная реакция:
H2SO4
СН3—СН2-СН2-СН2-ОН -----> СН3-СН2-СН=СН2 + Н2О
Реактивы, посуда и приборы
Бутиловый спирт .... 50 г, или 62 мл Колбы круглодонные (100 и
(0,67 моль) 200 мл)....................2
Серная кислота (d = 1,-84) 7 мл Форштосс двурогий..........1
(0,13 моль) Дефлегматор................1
Едкий натр, 3 н. раствор 200 мл Термометр..................1
Хлористый кальций Холодильник Либиха.........1
Металлический натрий Воронка капельная..........1
Воронка делительная.......1
Алонж......................1
Приемники ................ 2
Цилиндр мерный .......... 1
Сборка приборов
1. Круглодонную колбу емкостью 200 мл, снабженную «ки-
пелками», соединяют с двурогим форштоссом. В одно колено по-
следнего вставляют капельную воронку, в другое — дефлегматор
142
(с термометром), соединенный с нисходящим холодильником при
помощи алонжа, конец которого опускают в приемник.
2. Прибор 3 на стр. 126.
Выполнение синтеза
В круглодонную колбу прибора 1 вносят 62 мл бутилового
спирта и при перемешивании приливают 7 мл концентрированной
серной кислоты. Смесь спирта и кислоты осторожно нагревают
через асбестовую сетку, следя за тем, чтобы температура отходя-
щих паров не превышала 100—101 °C (при нарушении темпера-
турного режима могут образоваться бутилен и продукты его поли-
меризации, обугливания и восстановления серной кислоты до SO2).
Происходит медленная отгонка дистиллята. Время от времени
дистиллят отделяют от воды и переносят в капельную воронку, из
которой вводят его по каплям обратно в реакционную колбу. Воду
сливают в мерный цилиндр. После 3—4 ч, когда отгонится 10 мл
воды, отделяют дистиллят от воды, вносят его обратно в колбу,
кипятят еще 15—20 мин, а затем прекращают нагревание
Содержимое колбы охлаждают, при перемешивании и охла-
ждении вносят 30 мл 3 н. раствора едкого натра и переносят в
делительную воронку. Промывание раствором щелочи ведут до
тех пор, пока промывные воды не будут показывать щелочную
реакцию. Затем эфирный слой промывают 30 мл воды и 30 мл
насыщенного раствора хлористого кальция. Тщательно отделив
эфирный слой в сухую склянку, сушат его хлористым кальцием.
Высушенный эфирный слой отфильтровывают и перегоняют из
круглодонной колбы емкостью ПО мл с дефлегматором, собирая
погон до 135 °C. Затем остаток в колбе охлаждают, вносят туда
кусочек металлического натрия (защитные очки!) и перегоняют
дибутиловый эфир, собирая его в пределах 140—145 °C (не пере-
гонять досуха ввиду опасности взрыва!).
Выход дибутилового эфира ~25 г.
Дибутиловый эфир — бесцветная жидкость; мол. вес 130,22, т. кип.
141,97 °C; т. пл. —95,37 °C, d™ = 0,7688; п^= 1,3992.
Применяется как растворитель, используется в синтезе Гриньяра. При
длительном хранении дибутилового эфира образуются взрывчатые перекиси,
в связи с чем перед употреблением берут пробу на их содержание.
ИЗОАМИЛОВЫЙ ЭФИР
Формула: (СН3)2СН—СН2—СН2—О—СН2—СН2—СН(СН3)2.
Основная реакция (механизм реакции см. стр. 140):
h2so4
2(СН3)2СН-СН2-СН2ОН --->
—> (СН3)2СН—СН2—СН2—О—СН2—СН2--СН(СНз)2 + Н2О
Побочная реакция:
H2SO4
(СН3)2СН—СН2—СН2ОН ---► (СН3)2СН—СН=СН2 + Н2О
143
Реактивы, посуда и приборы
Изоамиловый спирт . . . 50 г, 62 мл
(0,57 моль)
Сериал кислота (<7 = 1,84) 2 мл
(0,04 моль)
Углекислый калий
Колба круглодонпая (200 мл) . . . 1
Насадка для отделения воды . . . 1
Холодильник Либиха...............1
Прибор для перегонки с водяным
паром.........................1
Воронка делительная ........... 1
Колба Вюрца (100 мл)...........1
Холодильник воздушны.!...........1
Термометр........................1
Приемники ..................... 2
Сборка приборов
1. Круглодонную колбу емкостью 200 мл при помощи корко-
вых пробок соединяют с насадкой для отделения воды и обратным
холодильником (рис. 90).
2. Прибор для перегонки с водяным паром (см. рис. 48,
стр. 64).
3. Перегонную колбу Вюрца на 100 мл, снабженную термо-
метром, соединяют с нисходящим воздушным хо-
лодильником, конец которого опускают в прием-
. ник.
Рис. 90. Прибор
с насадкой для от-
деления воды.
Выполнение синтеза
В круглодонную колбу емкостью 200 мл по-
мещают 62 мл свежеперегнанного изоамилового
спирта и 2 мл концентрированной серной кис-
лоты. Образовавшуюся смесь слабо кипятят в
течение нескольких часов до тех пор, пока в
водоотделителе не соберется 4 мл воды. Содер-
жимое колбы охлаждают до 100 °C и перегоняют
с водяным паром. Перегонку ведут до тех пор,
пока в дистиллят не перестанут переходить мас-
лянистые капли. Дистиллят переносят в дели-
тельную воронку, отделяют эфир от водного
слоя в сухую склянку и сушат его неболь-
шим количеством прокаленного углекислого ка-
лия.
Высушенный эфир помещают в колбу Вюрца па 100 мл, со-
единенную с воздушным холодильником. Начинают медленно на-
гревать колбу. При 21 °C отгоняется немного амилена, при 128°С
начинает отгоняться не вошедший в реакцию изоамиловый спирт,
а в пределах 165—172 °C отгоняется изоамиловый эфир.
Изоамиловый эфир получается с примесями. Для получения
чистого продукта его кипятят с 1 г амида натрия, отгоняют, встря-
хивают в делительной воронке с разбавленной серной кислотой,
сушат хлористым кальцием и перегоняют над металлическим на-
трием.
144
Выход пзоамилового эфира ~25 г.
Изоампловый эфир - - жидкое |ь с т. кип. I /2 <7'г’= - 0,7р07.
ДИФЕНИЛОВЫЙ ЭФИР
Формула:
СГ)Н&-О-Со11ь
Основные реакции:
2С0Н,ОН + К2СО3 +=± 2С6Н5—ок + н2о + со2
Си
С6Н6— ОК + С6Н6Вг > С6н6-О-Сьн5 + КВг
Реактивы, посуда и приборы
Фейол 17 г
(0,18 моль)
Бромбензол . . . 15,7 г, пли 10,5 мл
(0,1 моль)
Углекислый калий 15 г
Медь в порошке 0,2 г
Едкий натр, 10%- пып растпор
Сборка приборов
Колба круглодоппая (250 мл) . . . 1
Холодильник воздушный.........1
Прибор дли перегонки с водяным
паром.........................1
Колба Бунзена.................1
Воронка Бюх юра ................1
Предохранительная сктяпка . . . 1
Термометр.....................1
Приемники ........................ 2
1. Круглодонную колбу емкостью 250 мл, снабженную длин-
ным обратным воздушным холодильником, помещают на масля-
ную баню, в которую погружен термометр.
2. Прибор для перегонки с водяным паром (см. рис. 48).
3. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
В круглодонную колбу емкостью 250 мл помещают 17 г фе-
нола (осторожно! фенол явл ется ядом и вызывает сильные ожоги
кожи; работать с фенолом надо в перчатках) и нагревают на
асбестовой сетке до 42—45 °C. Когда фенол расплавится, нагрева-
ние прекращают, в колбу вносят 15 г прокаленного и тонкоизмель-
ченного углекислого калия (можно применять КОП, с NaOH реак-
ция протекает плохо), 10,5 мл сухого свежеперегнапного бром-
беизола и 0,2 г порошка меди. Нагревают смесь на масляной бане
при 210°C в течение 2 ч. Затем колбу охлаждают и для вымыва-
ния фенола из сырого дифениловою эфира добавляют к реакцион-
ной смеси 10% раствор едкого натра до щелочной реакции.
Собирают прибор 2 и начинают перегонять смесь с водяным
паром. Сначала перегоняется не вступивший в реакцию бром-
бензол. Как только в холодильнике начнут появляться кристаллы
дифенилового эфира, меняют приемник и собирают фракцию. По-
лученный дифениловый эфир отсасывают на воронке Бюхнера и
сушат на фильтровальной бумаге.
145
Выход дифенилового эфира ~ 14 г.
Дифениловый эфир, загрязненный фенолом, желтеет на свету
и плохо кристаллизуется. Для очистки дифениловый эфир может
быть перекристаллизован из 50%-ного этилового спирта.
Дифениловый эфир (феиоксибензол, дифенилоксид) — бесцветные кристал-
лы с запахом герани; нерастворим в воде, хорошо растворим в эфире, бензоле.
Мол. вес. 170,2; т. кип. 259,3°С, т пл. 26—27°С; d’° = 1,066; «д = 1,5762.
Применяется/в качестве теплоносителя и для отдушки мыла.
ФЕНЕТОЛ
Формула:
С6Н5—О-С2Н5
Основные реакции:
2C2H5OH + 2Na —> 2C2H5ONa + Н2
С6Н5ОН + C2H5ONa <=* C6H5ONa + С2Н5ОН
C6H5ONa + C2H5I —> С6Н5—О-С2Н5 + Nal
Реактивы, посуда и приборы
Фенол..............9,4 г Колба круглодонная (100 мл) 1
(0,1 моль) Колба Вюрца (100 мл) . . . . 1
Натрий металлический 2,3 г Холодильник Либиха........1
(0,1 г-атом) Воронка делительная . . . . 1
Йодистый этнл .... 20 г, или 15,4 мл Колба коническая.........1
(0,13 моль) Холодильник воздушный . . . 1
Этиловый спирт абсо- Алонж....................1
лютный.............30 мл Термометр.................1
Едкий натр, 10%-ный раствор Приемники.................2
Хлористый кальций
Диэтиловый эфир
Сборка приборов
1. Круглодонную колбу емкостью 100 мл соединяют с обрат-
ным холодильником и помещают колбу на водяную баню.
2. Круглодонную колбу на 100 мл при помощи пробки с,изо-
гнутой стеклянной трубкой соединяют с водяным нисходящим хо-
лодильником, а последний с алонжем, помещенным в приемник.
3. Прибор 3 на стр. 144.
Выполнение Синтеза
В круглодонную колбу прибора 1 помещают 30 мл абсолют-
ного этилового спирта (реакцию ведут в спиртовом растворе, так
как этиловый спирт хорошо растворяет фенолят натрия и иодистый
этил, обеспечивая тем самым однородность среды и легкое проте-
кание реакции) и постепенно прибавляют 2,3 г металлического
натрия. Затем вносят в колбу 9,4 г фенола, растворенного в абсо-
лютном спирте, и 20 г йодистого этила. Смесь нагревают на во-
дяной бане с воздушным холодильником не до кипения, так как
иодистый этил летуч, а до тех пор, пока спиртовый раствор не
146
перестанет показывать щелочную реакцию на лакмус. После этого
собирают прибор 2 и отгоняют этиловый спирт, а к остатку при-
бавляют воду для растворения выделившегося при реакции йоди-
стого натрия. Содержимое колбы переносят в делительную во-
ронку и извлекают фенетол диэтиловым эфиром.
Верхний слой эфирного раствора отделяют и для удаления
непрореагировавшего фенола взбалтывают эфирный слой в дели-
тельной воронке с раствором ёдкого натра, снова отделяют эфир-
ную вытяжку, переносят ее в коническую колбу и сушат хлори-
стым кальцием. Отгоняют эфир при 34—36°C из колбы с водяным,
холодильником, а затем перегоняют фенетол при 167—172 °C (при-
бор 3).
Выход фенетола ~9 г.
Фенетол (фенилэтиловый эфир) — бесцветная жидкость с ароматным запа-
хом; нерастворим в воде; смешивается со спиртом и эфиром. Мол. вес 122,16;
т. кип 172 °C при 760 мм рт. ст; т. пл. —29,5 °C; d?a = 0,9651; Пд = 1,5076.
Применяется в смеси с этиленгликолем при омылении трудно расщепляю-
щихся гидроокисью калия эфиров и как растворитель для кристаллизации орга-
нических соединений На организм фенетол действует Наркотически.
Отличительная реакция
При нагревании фенетола до кипения с 48%-ной бромисто-
водородной кислотой происходит его разложение на фенол и уле-
тучивающийся бромистый этил.
ЭТИЛОВЫЙ ЭФИР р-НАФТОЛА
(НЕРОЛИН НОВЫЙ, БРОМЕЛИЯ]
Формула:
О—сн2—СН3
Основная реакция:
СН3—СН2ОН +
h2so4
----->.
—СНз
Oj
Реактивы, посуда и приборы
₽-Нафтол....................25 г
(0,17 моль)
Этиловый спирт, абсолютный 38 мл
(0,6 моль)
Серная кислота (d = 1,84) . . 5,5 мл
(0,09 моль)
Едкий натр, 5%-цый раствор 90 мл
Колба круглодонная (100 мл) ... 1
Колба коническая, широкогорлая
(250 мл)......................1
Холодильник Либиха..............1 >
Мешалка........................1
Воронка Бюхнера ...............1
Колба Бунзена..................1
Предохранительная склянка ... 1
447
Сборка приборов
1. Круглодоппую колбу емкостью 100 мл соединяют с обрат-
ным холодильником п помещают ее на глицериновую или воздуш-
ную баню.
1. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодоппую колбу емкостью 100 мл помещают 25 г
р-пафтола, вливают 38 мл абсолютною этилового спирта (см.
стр. 37) и осторожно встряхивают колбу до растворения большей
части р-нафтола, затем добавляют 5,5 мл концентрированной сер-
ной кислоты, (наблюдается сильное разогревание смеси). Колбу
помещают на водяную баню и нагревают при кипении в течение
3—4 ч. После окончания нагревания теплый раствор выливают в
коническую колбу емкостью 250 мл, где содержится 90 мл 5%-ного
раствора едкого натра, нагретого до 50°С; при этом неролин осе-
дает в виде темного масла, которое мешалкой перемешивают с
раствором щелочи до тех пор, пока неролин полностью не затвер-
деет. Чтобы избегать моментального затвердевания сырого перо-
липа в момент влияния реакционной смеси в раствор щелочи,
нужно последний хорошо нагреть, причем нагревание конической
колбы следует вести осторожно на водяной бане.
Осадок грязно-песочного цвета отфильтровывают на воронке
Бюхнера и снова обрабатывают, как и в первый раз, таким же
количеством раствора едкого натра. Неролин отсасывают, промы-
вают водой до исчезновения щелочной реакции на лакмус и сушат
при температуре не выше 50 °C.
Высушенный продукт перегоняют с перегретым до 150 °C во-
дяным паром. В случае необходимости продукт можно очистить
перекристаллизацией из спирта или перегонкой в вакууме (т. кип.
140 °C при 12 мм рт, ст.).
Выход этилового эфира р-нафтола ~24 г.
Этиловый эфир 0-нафтола — белые пластинки с т. кип 282 °C; т пл 37,5 °C.
Растворим в большинстве органических раствори гелей Применяется для от-
душки мыла.
АНИЗОЛ
Формула:
148
Реактивы, посуда и приборы
Фенол...........12 г Колба круглодонная (150 мл) . . • 1
(0,12 моль) Холодильник Либиха.............1
Диметилсульфат 8 г, или 5,8 мл Воронка делительная............1
(0,05 моль) Колба круглодонная (100 мл) . . . 1
Бензол..........13 мл Дефлегматор....................1
Едкий натр .... 5,5 г Термометр......................1
Хлористый кальций 1,5 г Алонж..........................1
Приемники ..................... 2
Сборка приборов
1. Прибор 1 на стр. 148 (в данном приборе колба круглодон-
ная емкостью 150 мл).
2. Прибор 3 на стр. 126.
Выполнение синтеза
В круглодонной колбе прибора 1 готовят раствор 5,5 г едкого
натра в 40 мл воды и в нем растворяют 12 г фенола. После
охлаждения содержимого колбы до 15 °C к полученному раствору
прибавляют 2 мл диметилсульфата (Диметилсульфат — яд! Ра-
боту необходимо проводить в вытяжном шкафу). Колбу закры-
вают корковой пробкой, снабженной термометром и имеющей
боковую прорезь, и встряхивают в течение 20 мин, охлаждая ее
время от времени (температура смеси не должна превышать 40°C).
После встряхивания в течение 20 мин вносят еще 2 мл диме-
тилсульфата и повторяют проделанную процедуру в течение еще
30 мин. Затем вносят остаток диметилсулъфата и нагревают реак-
ционную смесь 1 ч на кипящей водяной бане в колбе с обратным
воздушным холодильником для окончания реакции и гидролиза
непрореагировавшего диметилсульфата. Затем отделяют в дели-
тельной воронке анизол, из нижнего водного слоя экстрагируют
бензолом частично оставшийся анизол (см. стр. 56).
Бензольную вытяжку соединяют (^отделенным ранее анизолом
и сушат хлористым кальцием. После этого отгоняют бензол на
кипящей водяной или песочной бане (прибор 2) при 80—85°C и
перегоняют анизол, собирая его при 154—156 °C.
Выход анизола ~7,5 г.
Анизол (метоксибепзол, фенилметиловый эфир) — бесцветная жидкость с
ароматическим запахом; нерастворим в воде, легко растворяется в спирте, эфи-
ре, бензоле; легко перегоняется с водяным паром. Мол. вес 108,13; т. кип.
155 °C; т. пл. -37,4 °C; d.’0 = 0,9940; я™ = 1,5170.
Применяется как растворитель в парфюмерной промышленности и для
кристаллизации.
Отличительные реакции
1. При нагревании с 5%-ным раствором селенистой кислоты
в концентрированной серной кислоте анизол дает темно-зеленое
окрашивание.
2. С ванилином и соляной кислотой дает вишнево-красное
окрашивание.
149
Глава IX
РЕАКЦИИ АЦИЛИРОВАНИЯ
Реакцией ацилирования называется процесс замещения атома
водорода некоторых функциональных групп — гидроксильной, ами-
ногруппы и других — на остаток карбоновой кислоты — ацил
R—С—. В результате ацилирования спиртов получаются сложные
О
эфиры, при ацилировании аминов — одно- и двузамещенные
амиды.
Кроме спиртов и аминов, легко ацилируются и другие соеди-
нения с подвижными атомами водорода, например меркаптаны,
фенолы. К реакциям ацилирования особого типа можно отнести
и получение ароматических кетонов взаимодействием ароматиче-
ских углеводородов с ангидридами или хлорангидридами кислот
в присутствии хлористого алюминия (реакция Фриделя—Крафтса).
В качестве ацилирующих агентов применяют карбоновые кис-
лоты или чаще более реакционноспособные эфиры, ангидриды или
хлорангидриды кислот. Смешанные ангидриды кремневой и карбо-
новой кислот, например (CH3COO)4Si, являются также эффектив-
ными ацилирующими средствами.
1. АЦИЛИРОВАНИЕ СПИРТОВ
И АМИНОВ КАРБОНОВЫМИ КИСЛОТАМИ
При ацилировании спиртов карбоновыми кислотами обра-
зуются сложные эфиры и вода:
R—+ HOR' ?=± R—С/ + Н2О
Ч)Н XOR'
Эта реакция называется реакцией прямой этерификации.
Кинетика реакции этерификации подробно исследована
Н.А. Меншуткиным и его учениками в ряде работ (1877—1891 гг.).
Реакция этерификации — пример обратимой реакции.
Опыт показывает, что если в реакцию вводить эквимолекуляр-
ные количества исходных веществ, то*в-начале реакции, согласно
закону действующих масс, скорость прямой реакции будет боль-
шая, а скорость обратной реакции незначительная. По мере обра-
зования сложного эфира и воды скорость обратной реакции бы-
стро возрастает и наконец наступает момент динамического рав-
новесия, при котором количество образующегося эфира и воды
будут равны количеству спирта и кислоты, получающихся при гид-
150
ролизе эфира в единицу времени. В силу этого только около 2/з
кислоты и спирта реагируют, образуя сложный эфир и воду.
Чтобы сместить равновесие в сторону образования продукта,
т. е. повысить выход эфира, следует брать для реакции избыток
кислоты или спирта, или удалять из сферы реакции образующий-
ся эфир или воду. При получении низкокипящих сложных эфиров
(например, уксусноэтилового, т. кип. 77 °C) отгоняют из реакцион-
ной колбы эфир, а при получении высококипящих сложных эфи-
ров (уксуснризоамилового, т. кип. 142°C) — воду.
Часто, особенно в случае нестойких соединений, лучше уда-
лять воду азеотропной отгонкой (азеотропной этерификацией), по-
скольку при этом можно довольствоваться меньшими количест-
вами катализатора и не применять избытка кислоты или спирта.
Выбор растворителя, с которым отгоняется вода, производят с
учетом температуры кипения самого низкокипящего компонента
реакционной смеси. Обычно для удаления воды в виде азеотроп-
ной смеси применяют хлороформ, четыреххлористый углерод, бен-
зол, толуол.
Взаимодействие между кислотами и спиртами при комнатной
температуре протекает медленно. Это объясняется малой актив-
ностью карбонильной группы в кислотах. Карбонильная группа,
связанная с гидроксильной в кислотах, становится менее реакци-
онноспособной по сравнению с той же группой в альдегидах и
кетонах. Это уменьшение реакционноспособности карбонильной
группы связано с влиянием гидроксильной группы, которая приво-
дит к уменьшению положительного заряда карбонильного угле-
рода. Скорость этерификации карбоновой кислоты тем выше, чем
больше положительный заряд карбонильного углерода, т. е. чем
сильнее кислота. Так, например, муравьиная и щавелевая кислоты
(сильные кислоты) быстро реагируют со спиртами.
Для увеличения скорости реакции повышают температуру и
применяют катализаторы. Такими катализаторами являются ионы
водорода (протоны) которые получаются при диссоциации силь-
ных минеральных кислот, чаще всего концентрированной серной
кислоты и сухого хлористого водорода. В присутствии минераль-
ных кислот скорость образования эфира возрастает в той же сте-
пени, что и скорость его гидролиза. Поэтому в присутствии ката-
лизатора устанавливается точно такое же равновесие, как и в его
отсутствие. Таким образом, катализаторы увеличивают скорость
реакции, но не сдвигают равновесие. Это положение относится ко
всем обратимым реакциям, протекающим в присутствии катали-
заторов.
Концентрированная серная кислота очень часто применяется
при реакциях этерификации, так как, являясь сильной кислотой,
она служит источником ионов водорода. Уже небольшая добавка
ее действует каталитически, а введение серной кислоты в больших
количествах в реакционную среду связывает образующуюся воду,
что положительно влияет на выход эфира. Однако в сильнокислой
среде возрастает склонность к образованию простых эфиров и
151
олефинов из спиртов. Чаще всего серная кислота применяется в
количестве 5—10% от взятого в реакцию спирта.
Роль кислоты (катализатора) заключается в присоединении
протона к карбонильному кислороду, что приводит к увеличению
положительного заряда па атоме углерода карбоксильной группы
и способствует более активному присоединению к нему нуклео-
фильного реагента — спирта:
, он R'
Н+ + R'—ОН | |
R—С—ОН <=* R—С—ОН R—С—О—Н
II I I +
О он он
I
Полученный комплекс I способен обратимо распадаться с от-
щеплением воды и образованием карбкатиона сложного эфира:
ОН R'
I I
R—С—О—II
I +
Н2О + R—С—О—R'
I
ОН
II
Карбкатион II при диссоциации образует сложный эфир и про-
тон (т. е. регенерируется катализатор):
R-C—О—R'
ОН
R—С—О—R' + Н+
Если серная кислота может вызвать побочную реакцию, вме-
сто нее можно применять ароматические сульфокислоты или луч-
ше всего — сухой хлористый водород, который пропускают через
реагирующий спирт.
Реакция ацилирования спиртов карбоновыми кислотами дает
хорошие результаты только с первичными спиртами и низкомоле-
кулярными кислотами. Вторичные спирты дают около 40% выхода
эфира, а третичные — только 3%. Таким образом, получение слож-
ных эфиров при непосредственном действии карбоновых кислот на
спирты практически ограничивается применением первичных и
вторичных спиртов.
152
Ацилирование аминов карбоновыми кислотами идет по сле-
дующей схеме:
R_NH2 + R'-C^ R-NH-C-R'+ Н2О
\он
Реакция протекает скорее, если кислота имеет низкий моле-
кулярный вес.
2. АЦИЛИРОВАНИЕ СПИРТОВ, ФЕНОЛОВ
И АМИНОВ ХЛОРАНГИДРИДАМИ КИСЛОТ
При ацилировании спиртов и фенолов хлорангидридами кис-
лот получают сложные эфиры по следующей схеме:
R'OH + RCOCl «=> RCOOR'+HCl
В эту реакцию легко вступают с образованием сложных эфи-
ров лишь первичные и вторичные спирты, третичные же при дей-
ствии хлорангидридов кислот часто образуют хлорзамещепные
углеводороды:
R3COH + R'COCl —> RjCCl + R'COOH
Хлорангидриды карбоновых кислот обычно легко реагируют
со спиртами и фенолами. Если реакция протекает слишком бурно,
необходимо охлаждать смесь или применять растворители (бен-
зол, толуол и др.). Остаток не вступившего в реакцию хлор-
ангидрида удаляют, обрабатывая смесь раствором углекислого
натрия. Полученный эфир очищают перегонкой или кристалли-
зацией.
Очень часто хорошие результать! получают, применяя в каче-
стве растворителя пиридин (иногда хинолин). Этот способ особен-
но удобен для ацилирования соединений, содержащих несколько
гидроксильных групп (многоатомных спиртов, сахаров, фенолов
и т. п.). Обычно спирт или фенол растворяют в пиридине, охла-
ждают раствор и прибавляют к нему хлорангидрид. Через не-
сколько часов стояния при комнатной температуре реакционную
смесь, окрашенную в темно-красный цвет, выливают в разбавлен-
ную серную кислоту, к которой предварительно добавили немного
льда. При этом ацильные производные выделяются в виде масла
или в твердом состоянии. Продукт реакции извлекают диэтиловым
эфиром или хлороформом и промывают полученную вытяжку раз-
бавленной кислотой для полного удаления пиридина. Если же
применяют хлорангидрид плохо растворимой кислоты, то избыток
ее удаляют промыванием разбавленным раствором углекислого
натрия.
Однако в случае применения пиридина часто образуются по-
бочные продукты, которые могут загрязнить основной продукт
реакции.
153
Ацилирование хлорангидридами, стойкими к воде, проводят в
разбавленных растворах NaOH или КОН. Обычно к раствору или
взвеси спирта или фенола в 10% щелочи приливают постепенно
хлорангидрид кислоты 5 случае разогревания смесь охлаждают
водой со льдом.
Хлорангидрид берут в избытке, который в щелочной среде
медленно превращаете*! в водорастворимую натриевую соль соот-
ветствующей кислоты. В процессе реакции эфир выделяется в
виде масла илц осадка.
Ацилирование спиртов и фенолов хлорангиДридами кислот на-
ходит широкое применение в препаративной органической химии,
особенно для идентификации спиртов и фенолов.
При ацилировании аминов хлорангидридами кислот образует-
ся ацильное производное и солянокислая соль амина:
RCOC1 + 2R'NH2 —> RCONHR'+ R'NH2 • НС1
Отделение солянокислой соли амина от его ацильного произ-
водного основано на их различной растворимости. Обычно реак-
цию ведут в том растворителе, в котором соль амина нераство-
рима. Кроме того, если ацильное производное амина нерастворимо
в воде, то солянокислую соль легко удалить промыванием реак-
ционной смеси водой. Обычно этот метод ацилирования аминов
хлорангидридами кислот применяют для получения бензоильных
и арилсульфонильных производных ароматических аминов.
3. АЦИЛИРОВАНИЕ СПИРТОВ, ФЕНОЛОВ И АМИНОВ
АНГИДРИДАМИ КИСЛОТ
Ангидриды кислот реагируют со спиртами и фенолами, обра-
зуя сложные эфиры по схеме: •»
ROH + (R'CO)2O —► ROCOR' + R'COOH
Однако это взаимодействие идет медленнее, чем ацилирова-
ние спиртов и фенолов хлорангидридами.
Ацилирование проводят, действуя ангидридом на спирт или
фенол непосредственно или в среде растворителя (например, при
получении аспирина). Реакцию можно ускорить, добавляя неболь-
шое количество концентрированной серной кислоты, хлористого
цинка, ацетата натрия или пиридина. Длительность реакции и
температура зависят от природы ацилируемого соединения.
Циклические ангидриды двухосновных кислот ведут себя при
этой реакции аналогично.
Ацилирование аминов ангидридами кислот протекает очень
быстро и с хорошими выходами. Эту реакцию можно осуществить
путем нагревания амина с уксусным ангидридом в течение не-
скольких минут. Продукт реакции выливают в воду и отсасывают
кристаллическое ацильное производное. Кроме того, реакцию мож-
но проводить также в среде бензола При этом амин растворяют
в бензоле и постепенно приливают ангидрид. Реакция экзотермич-
154
на, поэтому растворитель закипает {внимание! возможен выброс).
Если ацильное производное плохо растворимо в бензоле, то после
охлаждения оно выпадает в осадок.
4. ПРИМЕРЫ СИНТЕЗОВ
УКСУСНОЭТИЛОВЫЙ ЭФИР
Формула:
сн2—с—о—сн2—сн3
Основная реакция (механизм реакции см. стр. 152):
h2so4 _
СН3—СН2ОН + СН3—(Г СН3—С—О-СН2—СН3 + н2о
- Х)Н
Побочные реакции:
нагревание
2СН3—сн2он ----- сн3-сн2-о-сн2-сн3 + Н2О
г12о04
Этот метод получения уксусноэтилового эфира основан на свя-
зывании воды водоотнимающим средством (серной кислотой), од-
нако можно применять и азеотропную отгонку воды, используя в
качестве растворителя хлороформ или четыреххлористый углерод.
Уксусноэтиловый эфир легко воспламеняется и образует с воз-
духом взрывоопасные смеси.
Реактивы, посуда и приборы
Этиловый спирт, 95%-ный . . 22,5 мл
(0,37 моль)
Уксусная кислота, ледяная 20 мл\
(0,3 моль) \
Серная кислота (d = 1,84) . . 2,5 мл
Хлористый кальций..........8 г
Углекислый натрий, 2%-ный раствор
Сернокислый натрий, безводный
Колба Вюрца (100 мл)...........1
Воронка капельная..............1
Холодильник Либиха ......... , 1
Термометр......................1
Алонж..........................1
Воронка делительная .......... 1
Приемники......................2
Сборка приборов
1. В горло перегонной колбы Вюрца емкостью 100 мл встав-
ляют капельную воронку, а отводной отросток колбы соединяют
с нисходящим холодильником, снабженным алонжем, опущенным
в приемник. Колбу помещают на масляную баню, туда же встав-
ляют термометр.
2. В колбу Вюрца емкостью 100 мл, помещенную на водяную
баню, вставляют термометр и соединяют колбу с нисходящим
холодильником. Последний снабжают алонжем, конец которого
опускают в приемник.
155
Выполнение синтеза
В перегонную колбу Вюрца на 100 мл помещают 2,5 мл эти-
лового спирта, 2,5 мл концентрированной серной кислоты и нагре-
вают па масляной бане до 140°C. Нагревание можно вести на
песочной бане, при этом в колбу нужно поместить термометр, опу-
стив шарик его в жидкость; нагревать следует до 110—120 °C.
По достижении температуры 140 °C начинают постепенно при-
ливать из капельной воронки смесь 20 мл этилового спирта и
20 мл ледяной уксусной кислоты (см. стр. 127) с такой скоростью,
с какой отгоняется образующийся уксусноэтиловый эфир; при
этом необходимо постоянно поддерживать заданную температуру
140 °C, так как при более высокой температуре образуется диэти-
ловый эфир.
Вместе с уксуспоэтиловым эфиром отгоняется некоторое количество этило-
вого спирта, полому спирт берут в избытке и выход продукта рассчитывают
на взятое количество уксусной кислоты.
После окончания реакции, которая длится около 2 ч, дистил-
лят, содержащий кроме уксусноэтилового эфира не вошедшие в
реакцию уксусную кислоту и спирт, для удаления уксусной кис-
лоты встряхивают в делительной воронке с 2% раствором угле-
кислого натрия до тех пор, пока верхний слой не покажет ней-
тральную или слабощелочную реакцию на лакмус. (Раствор угле-
кислого натрия следует прибавлять постепенно, так как жидкость
сильно вспенивается выделяющейся двуокисью углерода.) Затем
отделяют нижний водный слой, а верхний эфирный слой встряхи-
вают с насыщенным раствором хлористого кальция (8 г хлористого
кальция в 8 мл воды) для удаления примеси этилового спирта.
Снова отделяют верхний эфирный слой и сушат его безводным
сернокислым натрием. После высушивания продукт перегоняют на
водяной бане (прибор 2).
Хлористый кальций дает с первичными спиртами кристаллическое соедине-
ние СаСЬ-гСзНбОН, которое нерастворимо в уксусноэтиловом эфире.
При 71—75 °C отгоняется смесь спирта с уксусноэтиловый
эфиром, а при 75—78 °C — сравнительно чистый уксусноэтиловый
эфир.
Выход уксусноэтилового эфира ~20 г. Вследствие потерь
эфира из-за его летучести и хорошей растворимости в воде прак-
тический выход обычно не превышает 70% от теоретического.
Уксусноэтиловый эфир (этилацетат) — бесцветная жидкость с фруктовым
запахом; растворим в воде; смешивается со спиртом, хлороформом, бензолом
и другими органическими растворителями. Мол. вес 88,10; т. кип. 77,2 °C; т. пл.
—83,6 °C, = 0,9008; 4?= 1,3724.
~ Применяется как растворитель нитроклетчатки, целлулоида, жиров, воска,
смол, а также в аналитической химии и тля получения ацетоуксусного эфира и
ацетилацетона. На организм Действует как наркотик, раздражает слизистые
оболочки.
156
УКСУСНОИЗОАМИЛОВЫЙ ЭФИР
Формула:
о
СН —с—О-СП —СП2—СП—СНз
I
СНз
Основная реакция:
СН3 О
I II H2SOt
СНз—СН—СН2—СН2ОН + СН3—С—ОН
О сн3
II I
СНз—с—о—СН2—СН2—СП—СНз + Н2о
Обращаться с уксуспоизоамиловым эфиром следует осторож-
но, так какой легко воспламеняется (температура вспышки36°C)!
ПОЛУЧЕНИЕ УКСУСНОИЗОАМИЛОВОГО ЭФИРА
ПУТЕМ УДАЛЕНИЯ ВОДЫ
АЗЕОТРОПНОЙ ОТГОНКОЙ С БЕНЗОЛОМ
Реактивы, посуда' и приборы
Изоамиловый спирт ... 26 мл. 22 г
(0,25 моль)
Уксусная кислота, 80%-пая 20 мл
(0,28 моль)
Бензол......................25 мл
Серная кислота (cl = 1,84) . . 0,5 мл
Кислый углекислый натрий,
насыщенный раствор
Колбы круглодоппые (100 мл) . . 2
Насадка («ловушка») для отделе-
ния воды......................1
Холодильник Лпби.ха............1
Воронка делительная .......... 1
Колба Вюрца (100 мл)...........1
Приемники ..................... 4
Термометр . ....................1
Сборка приборов
1. Круглодопную колбу емкостью 100 мл соединяют через на-
садку-отделитель («ловушку») для отделения воды с обратным
холодильником (см. рис. 90). Колбу помещают на песочную баню.
2. Колбу Вюрца емкостью 100 мл, помещенную на песочную
баню, снабжают термометром и нисходящим водяным холодиль-
ником' с алонжем, конец которого опускают в приемник.
Выполнение синтеза
В круглодонную колбу (прибор 1) помещают 26 мл изоамило-
рого спирта, 20 мл уксусной кислоты, 25 мл бензола и 0,5 мл кон-
центрированной серной кислоты. (Бензол применяется как раство-
ритель, с которым отгоняется вода в виде азеотропной смеси.)
Колбу нагревают на песочной бане (прибор 1) до силь-
ного кипения. Конденсат, состоящий из образовавшегося эфира,
157
непрореагировавшей кислоты, бензола и выделившейся во время
реакции воды, попадает из обратного холодильника в «насадку»,
где расслаивается на бензольный (верхний) и водный (нижний)
слои. Водный слой время от времени сливают в мерный цилиндр
и измеряют (всего за реакцию выделяется около 8—9 мл), а бен-
зольный раствор по мере накопления в «насадке» стекает обратно
в реакционную колбу. (Реакция считается законченной, когда
слой воды в насадке перестанет изменться в объеме.)
После окончания реакции, которая длится 3—4 ч, бензольный
раствор, собравшийся в «насадке», сливают обратно в колбу, со-
бирают прибор 2 и отгоняют бензол от реакционной смеси при
80—81 °C. Остаток переносят из колбы в делительную воронку,
промывают 50—60 мл воды и нижний водяной слой отделяют.
Оставшийся в делительной воронке эфирный слой встряхивают
последовательно с 50 мл воды, 20—25 мл разбавленного раствора
кислого углекислого натрия до нейтральной реакции и вновь с
25 мл воды. Отделив от воды эфир, высушивают его безводным
сернокислым натрием или хлористым кальцием. Если водяной слой
недостаточно хорошо отделяется от эфирного, следует добавить
5 г поваренной соли и тщательно перемешать. Этот процесс вы-
саливания снижает растворимость сложного эфира в воде. Затем
в приборе 2 отгоняют две фракции: 1) до НО °C, состоящую глав-
ным образом из остатка бензола; 2) 136—142 °C. Если температур-
ный интервал второй фракции будет больше, то эту фракцию под-
вергают вторичной перегонке, собирая уксусноизоамиловый эфир
при 138—142 °C.
Выход уксусноизоамилового эфира 25—27 г.
Рассмотренным выше методом можно получить уксуснобути-
ловый эфир, температура кипения которого 124—125 °C.
ПОЛУЧЕНИЕ УКСУСНОИЗОАМИЛОВОГО ЭФИРА
ПУТЕМ ОТГОНКИ ВОДЫ БЕЗ БЕНЗОЛА
Реактивы, посуда и приборы
Изоамиловый спирт ... 22 г, или 26 мл
(0,25 моль)
Уксусная кислота, ледя-
ная ....................18 г, или 17 мл
(0,3 моль)
Сериал кислота (d = 1,84) 0,5 мл
Кислый углекислый натрий, насыщенный
раствор
Колбы круглодонные (100 мл) 2
Насадка («ловушка») для от-
деления воды.............1
Холодильник Либиха ....... 1
Воронка делительная........1
Дефлегматор................1
Приемники..................2
Сборка приборов
1. Прибор 1 на стр. 157.
2, Прибор 2 на стр, 157.
158
Выполнение синтеза
В круглодонную колбу прибора 1 помещают 26 мл изоамило-
вого спирта, 17 мл ледяной уксусной кислоты и 0,5 мл серной кис-
лоты. Колбу нагревают на песочной бане до тех пор, пока «& ло-
вушке» не образуется два слоя: нижний (водный) и верхний (об-
разовавшийся эфир и непрореагировавшие уксусная кислота и
спирт), который стекает обратно в реакционную колбу. Конец
реакции определяют по количеству выделившейся воды (4,5 мл)
или ведут реакцию до тех пор, пока «в ловушке» не перестанет
увеличиваться,слой воды, отделяют в делительной воронке отводы
эфир, высушивают его безводным сернокислым натрием или хло-
ристым кальцием, а затем работу проводят, как указано на
стр. 157.
Выход уксусноизоамилового эфира ~27 г.
Уксусноизоамиловый эфир (изоамилацетат) — бесцветная жидкость с фрук-
товым запахом, в 100 г воды растворяется 0,25 г, смешивается со спиртом,
эфиром, хлороформом Мол. вес 130,18, т кип 142 °C; т пл —78,5 °C;
d™ = 0,8719; п% = 1,4053.
Применяется как растворитель нитроцеллюлозы, а также в пищевой Про-
мышленности под названием грушевой эссенции Кроме того, используется как
растворитель в аналитической химии Наркотически действует иа организм, раз-
дражает слизистые оболочки глаз, носа и дыхательных путей.
Отличительная реакция
В пробирку помещают 2—3 мл уксусноизоамилового эфира,
3 мл 1%-ного раствора хлорного железа в 10 н. соляной кислоте.
После сильного встряхивания пробирки происходит расслаивание.
Верхний слой эфира окрашен в яркий буро-желтый цвет вслед-
ствие перехода в него почти всей соли железа. Нижний слой окра-
шен лишь в бледно-желтоватый цвет.
ЭТИЛОВЫЙ ЭФИР ХЛОРУКСУСНОЙ кислоты
Формула:
о
II
СН2С1—С—О—С2Н5
Основная реакция:
О
н2зо4 И
СН2С1—СООН + С2Н5ОН СН2С1—С—О—С2Н5 + Н2О
Работать с этиловым эфиром хлоруксусной кислоты следует
осторожно, так как он раздражает слизистые оболочки'.
159
Реактивы, посуда и приборы
Хлоруксусиая кислота . . 20. г
(0,2 моль)
Этиловый спирт, абсолют-
ный ..................12 г, пли 15 мл
(0,26 моль)
Серная кислота (d = 1,84) 1 мл
Кислый углекислый на-
трий, насыщенный рас-
твор .................. 5 мл
Хлористый кальций ... 2 г
Колба круглодонпая (100 мл) 1
Холодильник Либиха..........1
Трубка хлоркальциевая . . . 1
Воронка делительная . . . . 1
Прибор для фракционной пе-
регонки ..................1
Стакан химический ......... 1
Приемники . ................2
Сборка приборов
1. Круглодонную колбу емкостью 100 мл соединяют с обрат-
ным водяным холодильником, к которому крепится хлоркальцие-
вая трубка, необходимая для защиты реакционной среды от до-
ступа влаги из воздуха.
2. Прибор для фракционной перегонки (см. рис. 11).
Выполнение синтеза
В круглодонную колбу емкостью 100 мл помещают 20 г хлор-
уксусной кислоты (осторожно] хлоруксусная кислота вызывает
ожоги кожи!), 15 мл абсолютного этилового спирта (см. гл.. II),
1 мл концентрированной серной кислоты и нагревают смесь на
песочной бане в течение 5 ч.
После окончания реакции смесь охлаждают, а затем выли-
вают в стакан со 100 мл ледяной воды. Из стакана смесь перено-
сят в делительную воронку, отделяют нижний водный слой, а эфир-
ный слой нейтрализуют 5 мл насыщенного раствора кислого
углекислого натрия и промывают водой до нейтральной реакции.
После этого сырой этиловый эфир хлоруксусной кислоты сушат в
течение 10—12 ч над безводным хлористым кальцием, затем со-
бирают прибор 2 и отгоняют фракцию, кипящую при 142—143 °C.
Выход этилового эфира хлоруксусной кислоты ~19 г.
Этиловый эфир хлоруксусной кислоты можно также получить
азеотропной этерификацией.
Этиловый эфир хлоруксусной кислоты — бесцветная подвиж-
ная жидкость с острым фруктовым запахом; т. кип. 144 °C;
«20= 1,4227. Применяется для получения эфира фенилметилгли-
цидной кислоты.
ДИЭТИЛОВЫЙ ЭФИР ЩАВЕЛЕВОЙ КИСЛОТЫ
Формула:
СООС2Н5
СООС2ТГ9
160
Основная реакция:
соон на COOC2HS
I + 2С2Н6ОН =₽=* 1 +2Н2О
СООН СООС2Н5
Реактивы, посуда и приборы
Щавелевая кислота, безводная 30 г Колба круглодонная короткогорлая
(0,4 моль) (150 мл)................................................I
Этиловый спирт, абсолютный 45 мл Форштосс двурогий..............1
(0,75 моль) Холодильник Либиха.............1
Углекислый натрий, кристал- Трубка хлоркальциевая...........1
лический............100 г Трубка газопроводная............1
Газообразный хлористый водород Прибор для получения хлористого
Хлористый кальций водорода....................1
Промывная склянка..............1
Предохранительная склянка ... 1
Воронка делительная .......... 1
Колба Вюрца (100 мл)...........1
Термометр......................1
Холодильник воздушный..........1
Приемники .................... 2
При получении диэтилового эфира щавелевой кислоты путем
нагревания щавелевой кислоты со спиртом и серной кислоты по-
лучаются пониженные выходы, так как щавелевая кислота при на'
гревании с серной кислотой разлагается, об-
разуя двуокись и окись углерода и муравьи-
ную кислоту. Поэтому лучше применять в
качестве катализатора и поглотителя воды
хлористый водород.
Сборка приборов
1. Круглодонную короткогорлую колбу
емкостью 150 мл, помещенную на охлади-
тельную баню, снабжают двурогим форш-
тоссом, в прямое колено которого встав-
ляют изогнутую под прямым углом стеклян-
ную газопроводную трубу, холодящую по-
чти до дна колбы. К боковому колену форш-
тосса присоединяют обратный холодильник,
верхний конец которого закрывают хлор-
кальциевой трубкой. Газопроводную труб-
ку при помощи резиновых трубок соединяют
Рис. 91. Прибор для
получения хлористого
водорода.
последовательно с пустой предохранительной склянкой н с про-
мывной склянкой, содержащей концентрированную серную кис-
лоту, а затем с прибором для получения’хлористого водорода.
Хлористый водород можно получить в аппарате Киппа при
действии концентрированной серной кислоты на крупные куски
сплавленного нашатыря. Кроме того, удобно получать хлористый
водород, приливая из капельной воронки по каплям концентриро-
ванную серную кислоту к концентрированной соляной кислоте, в
б М. Н. Храмкина
161
приборе изображенном на рис. 91. Хлористый водород высуши-
вают, пропуская через промывную склянку с концентрированной
серной кислотой. За промывной склянкой надо обязательно ста-
вить пустую предохранительную для устранения опасности пере-
броса насыщаемой жидкости из приемника в промывную склянку
с серной кислотой.
2. Колбу Вюрца на 100 мл снабжают термометром и соеди-
няют с нисходящим воздушным холодильником, конец которого
опускают в приемник.
Выполнение синтеза
В круглодонную короткогорлую колбу прибора 1 помещают
30 г растертой в порошок безводной щавелевой кислоты (осторож-
но! щавелевая кислота действует раздражающе на кожу и слизи-
стые оболочки дыхательных путей!), приливают 45 мл абсолют-
ного этилового спирта (см. гл. II) и пропускают сильный ток су-
хого хлористого водорода. Реакционная смесь при этом сильно
разогревается и после 10-минутного пропускания НС1 ее охлаж-
дают до 0 °C.
Безводную щавелевую кислоту получают из товарной кристаллической
[(СООН)г-2Н2О]; последнюю растирают в порошок, помещают в фарфоровую
чашку и высушивают в течение нескольких часов в сушильном шкафу при
95 °C.
Хлористый водород продолжают пропускать при 0°С до тех
пор, пока он не перестанет поглощаться и не начнет выходить че-
рез хлоркальциевую трубку, а также пока не растворится вся
щавелевая кислота. После этого постепенно, при перемешивании
выливают содержимое колбы на смесь 225 г толченого льда и
100 г измельченного кристаллического углекислого натрия. Выде-
лившийся эфир отделяют при помощи делительной воронки, про-
мывают небольшим количеством воды, сушат хлористым кальцием
и перегоняют из колбы Вюрца при 186 °C.
Иногда образуется эмульсия, которая долго не расслаивается
и затрудняет отделение слоя диэтилового эфира щавелевой кис-
лоты. Тогда переливают смесь в делительную воронку и 2—3 раза
экстрагируют диэтиловым эфиром (по 30 мл). Соединенные эфир-
ные вытяжки промывают водой и сушат хлористым кальцием. За-
тем, отогнав диэтиловый эфир на водяной бане, перегоняют диэти-
ловый эфир щавелевой кислоты.
Выход диэтилового эфира щавелевой кислоты ~ 24 г.
Диэтиловый эфир щавелевой кислоты (диэтилоксалат) — бесцветная
жидкость, обладающая слабым приятным запахом; мало растворим в воде;
смешивается со спиртом, атиловым эфиром. Мол. вес 146,1; т. кип. 185,4 °C;
т. пл. — 40,6°С; df= 1,0785;1,4101.
Применяется как растворитель эфиров целлюлозы и в органическом син-
тезе, в частности может быть использован при получении щавелевоуксусного
эфира. Диэтилоксалат действует раздражающе на кожу и слизистые оболочки
дыхательных путей.
162
Отличительная реакция
При кипячении слабощелочного раствора диэтилового эфира
щавелевой кислоты образуется щавелевокислый натрий, который
обнаруживается по образованию осадка с солями кальция (после
подкисления раствора уксусной кислотой).
ЭТИЛОВЫЙ ЭФИР БЕНЗОЙНОЙ кислоты
Формула:
С6Н5СООС2Н5,
Основная реакция:
С6Н6СООН + с2н5он
H2SO4
CSH5COOC2H5 + Н2О
Реактивы, посуда и приборы
Бензойная кислота.........12,2 г Колба круглодонная (150 мл) . . v 1
(0,1 моль) Холодильник Либиха..............I
Этиловый спирт, 95%-ный . . 35 мл Воронка делительнар................1
(0,6 моль) Колба Вюрца (50 мл).............1
Серная кислота (d — 1,84) . . 4 мл Термометр..........................1
Углекислый натрий Алонж..............................1
Диэтиловый эфир Холодильник воздушный...........1
Хлористый кальций Стакан химический..................2
Приемники .................... 1
Сборка приборов
1. Круглодонную колбу емкостью 150 мл соединяют с обрат-
ным водяным холодильником и помещают на водяную баню.
2, Круглодонную колбу емкостью 150 мл, помещенную на во-
дяную баню, снабжают термометром и нисходящим водяным холо-
дильником с алонжем, конец которого опускают в приемник.
3. В горло колбы Вюрца емкостью 50 мл вставляют термо-
метр, а отросток соединяют с нисходящим водяным холодильником
с алонжем, опущенным в приемник, охлаждаемый льдом. Колбу
помещают на водяную баню.
4. Колбу Вюрца емкостью 50 мл снабжают термометром и ко-
ротким воздушным нисходящим холодильником с алонжем, кото-
рый опускают в приемник.
Выполнение синтеза
В круглодонную колбу емкостью 150 мл помещают 12,2 г бен-
зойной кислоты (осторожно! бензойная кислота раздражает кожу),
35 мл 95%-ного этилового спирта и прибавляют 4 мл концентриро-
ванной серной кислоты. Смесь нагревают в течение 3—4 ч на водя-
ной бане (прибор 1). После окончания реакции отгоняют избыток
этилового спирта (прибор 2),
6*
163
Так как этиловый эфир бензойной кислоты, имеющий высокую темпера-
туру кипения, трудно удалить из сферы реакции и применяется ие абсолютный
этиловый спирт, то для смещения равновесия вправо приходится брать значи-
тельный избыток этилового спирта.
Остаток охлаждают и для растворения оставшегося спирта и
бензойной кислоты выливают в стакан с 60 мл холодной воды. До-
бавляют порциями углекислый натрий до слабощелочной реакции.
Бензойная кислота извлекается диэтиловым эфиром, а натриевая соль бен-
зойной кислоты, которая образуется при внесении углекислого натрия, ие из-
влекается. Поэтому нужно убедиться в щелочности среды при внесении угле-
кислого натрия.
Выделившийся в виде масла этиловый эфир бензойной кис-
лоты извлекают диэтиловым эфиром, отделяют при помощи дели-
тельной воронки эфирный слой и сушат его несколько часов хло-
ристым кальцием. После отгонки эфира (прибор 3) остаток этило-
вого эфира бензойной кислоты перегоняют (прибор 4), собирая
фракцию, кипящую при 210—212 °C.
Выход этилового эфира бензойной кислоты ~ 10 г.
Этиловый эфир бензойной кислоты (этилбензоат) — бесцветная жидкость с
фруктовым запахом; нерастворим в воде; смешивается со спиртом, эфиром, хло-
роформом. Мол. вес 150,17; т. кип: 212,4°C; т. пл. —34,7°С; dj5 = 1,0511; Пд —
= 1,5029.
БЕНЗАНИЛИД
Формула:
C6H6NHCOCeH5;
Основная реакция:
CeH5NH2 + С6Н6СООН «=* C6H6NHCOCeH5 + Н2О
Реактивы, посуда и приборы
Бензойная кислота . .
Анилин...............
Соляная кислота, 1 ш
раствор .............
Едкий натр, 1 н. рас-
твор ................
16,7 г
(0,13 моль)
18,6 г, или 18 мл
(0,2 моль)
250 мл
250 мл
Колба круглодоииая (150 мл) 1
Холодильник Либиха ....... 1
Колба Бунзена..............1
Воронка Бюхнера............1
Предохранительная склянка 1
Чашка фарфоровая...........1
Стакан химический ........ 1
Ступка ................... 1
Приемники..................2
Сборка приборов
1. Круглодонную колбу емкостью 150 мл соединяют с нисходя-
щим водяным холодильником с алонжем, конец которого опускают
в приемник. Колбу помещают на масляную баню, туда же
опускают термометр.
2. Прибор для отсасывания (см. рис. 40).
Ш
Выполнение синтезе
В круглодонную колбу прибора 1 помещают 13 мл анилина
(осторожно! анилин ядовит), 16,7 г бензойной кислоты и нагре-
вают смесь при 180—190 °C до тех пор, пока не перестанут от-
гоняться анилин и вода. Затем доводят температуру до 225 °C и
поддерживают ее на этом уровне до прекращения перегонки. Уда-
ляют масляную баню, дают колбе немного охладиться, добавляют
еще 5 мл анилина и повторяют нагревание, как описано выше.
После прекращения отгонки анилина и воды содержимое кол-
бы выливают в фарфоровую чашку. Остывший и затвердевший
продукт растирают в ступке, переносят в стакан, где размешивают
со 125 мл 1 н. раствора соляной кислоты. С осадка сливают
жидкость и снова повторяют эту операцию, чтобы удалить из
осадка не вступивший в реакцию анилин. Затем таким же образом
промывают продукт водой, два раза 1 н. раствором едкого натра
для удаления непрореагировавшей бензойной кислоты и, наконец,
ещещесколько раз водой.
После промывки осадок отсасывают на воронке Бюхнера и
,сушат сначала на воздухе, а потом в сушильном шкафу при 1и0юС.
Выход сырого бензанилида ~ 16 г.
Сырой бензанилид можно очистить кристаллизацией из этило-
вого спирта с добавкой активированного угля.
Бензанилид — бесцветное кристаллическое вещество, растворим в спирте,
эфире, бензоле; нерастворим в воде. Мол. вес 197,23, т кип 117—119*С при
10 мм рт. ст.; т. пл. 163 °C; = 1,3200.
Применяется как пластификатор в лакокрасочной промышленности.
Отличительная реакция
Бензанилид окрашивает раствор хинона в красный цвет, а
сернокислый раствор хромовокислого калия — в фиолетовый.
▲СПИРИН (АЦЕТИЛСАЛИЦИЛОВАЯ КИСЛОТА)
Формула:
-s^COOH
^^ОСОСНз
Основная реакция:
асоон сн3-со. ^^.соон
+ Х° ~[ Г + СНзСООН
ОН СН3-СО/ ^-^ОСОСНз
16»
ПОЛУЧЕНИЕ АСПИРИНА НЕПОСРЕДСТВЕННЫМ ДЕЙСТВИЕМ
УКСУСНОГО АНГИДРИДА НА САЛИЦИЛОВУЮ КИСЛОТУ
Реактивы, посуда и приборы
Салициловая кислота 12,5 г
(0,09 моль)
Уксусный ангидрид . . 10,2 г, или 10 мл
(0,1 моль)
Серная кйслота (d= 1,84) 0,5 мл
Толуол
Колба круглодонная (100 мл) 1
Холодильник воздушный . . . 1
Трубка хлоркальциевая . . . 1
Стакан химический (300 мл) 1
Воронка Бюхнера.............1
Колба Бунзена...............1
Предохранительная склянка 1
Термометр...................1
Сборка приборов
1. Круглодонную колбу емкостью 100 мл соединяют с обрат-
ным воздушным холодильником, снабженным хлоркальциевой
трубкой, и помещают на водяную баню.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную колбу прибора 1 помещают 12,5 г салицило-
вой кислоты, 10 мл уксусного ангидрида (осторожно! уксусный
ангидрид огнеопасен, раздражает кожу) и 0,5 мл концентрирован-
ной серной кислоты. Смесь нагревают (прибор 1) на водяной бане
при 60 °C в течение I ч. После этого доводят температуру до 90—
95 °C и выдерживают реакционную смесь при этой температуре
20 мин. Затем при помешивании дают жидкости остыть. После
охлаждения жидкость выливают в 20 мл воды, перемешивают, от-
сасывают на воронке Бюхнера выделившийся аспирин и промы-
вают его небольшим количеством холодного толуола.
Выход аспирина ~16 г.
'Аспирин для очистки можно перекристаллизовать из разбав-
ленной (1:1) уксусной кислоты, бензола, хлороформа или этило-
вого спирта.
ПОЛУЧЕНИЕ АСПИРИНА ДЕЙСТВИЕМ УКСУСНОГО АНГИДРИДА
НА САЛИЦИЛОВУЮ КИСЛОТУ В СРЕДЕ РАСТВОРИТЕЛЯ
Реактивы, посуда и приборы
Салициловая кислота 12,5 г
(0,09 моль)
Уксусный ангидрид 10 г, или 9 мл
(0,1 моль)
Беизол, безводный . . 30 мл
Колба круглодонная (100 мл) . . . 1
Холодильник водяной .............. 1
Трубка хлоркальцневая...........1
Воронка для горячего фильтрова-
ния ...........-..............1
Воронка Бюхнера . 1
Чашка фарфоровая..............1
Колба Бунзена.................1
Предохранительная склянка . . . 1
Термометр.....................1
- 166
Сборка приборов
1. Круглодонную колбу емкостью 100 мл соединяют с обрат-
ным водяным холодильником (без циркуляции или со слабой цир-
куляцией воды) с хлоркальциевой трубкой и помещают на водя-
ную баню,
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную колбу прибора 1 помещают 15 мл сухого бен-
зола (бензол следует высушить хлористым кальцием), 12,5 г сухой
салициловой кислоты, 9 мл свежеперегнанного уксусного ангидрида
(уксусный ангидрид перегоняют над безводным ацетатом натрия)
и нагревают смесь в течение 4 ч на кипящей водяной бане (при-
бор 1), время от времени встряхивая колбу. Затем берут пробу
реакционной жидкости на наличие в ней салициловой кислоты, ко-
торая с водным раствором хлорного железа дает характерное
фиолетовое окрашивание.
Если салициловая кислота не обнаружена, то содержимое
колбы фильтруют через складчатый фильтр на воронке для горя-
чего фильтрования (см. стр. 48) и оставляют фильтрат стоять в
фарфоровой чашке на сутки.
На следующий день чашку помещают на 0,5 ч в охладитель-
ную баню (температура от —5 до —10°C), после чего выпавший
осадок аспирина отсасывают на воронке Бюхнера, промывают су-
хим, охлажденным до 6°C бензолом до исчезновения запаха ук-
сусной кислоты и сушат. Можно использовать фильтрат для из-
влечения оставшегося в растворе аспирина.
Сначала от фильтрата при нагревании на водяной бане отго-
няют с холодильником бензол, а затем в вакууме (20 мм рт. ст.) —
уксусную кислоту и уксусный ангидрид.
Выход аспирина ~13 г.
Аспирин (ацетилсалициловая кислота) — бесцветные кристаллы в виде игл
или пластинок слабокислого вкуса; плохо растворим в воде, хорошо — в вод-
ных растворах щелочей. Мол. вес 180,16; т. пл. 136,5 °C.
Широкое применение находит в медицине как жаропонижающее и боле-
утоляющее средство.
Отличительная реакция
При нагревании со щелочами аспирин распадается на салици-
ловую и уксусную кислоты.
Р-НАФТИЛАЦЕТАТ
Формула:
167
Основные реакции:
он
ONa СНз—СО
•ОСОСНз
+ CHsCOONa
СН3—СО
Реактивы, посуда и приборы
Р-Нафтол
10 г
(0,07 моль)
Уксусный ангидрид 11,4 г, или 10,5 йл
(0,11 моль)
Колба круглодонная (500 мл) 1
Холодильник воздушный . . . 1
Воронка Бюхнера.............I
Едкий натр.........5 г
Лед
Колба Бунзена..............1
Предохранительная скляика 1
. 1
Сборка приборов
1. Круглодонную колбу емкостью 500 мл соединяют с обрат-
ным воздушным холодильником.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонной колбе емкостью 500 мл растворяют 10 г чи-
стого р-нафтола в 50 мл 10%-ного раствора едкого натра.
P-Нафтол можно очистить путем перекристаллизации из воды с разбавлен-
ным этиловым спиртом или из четыреххлористого углерода.
К раствору прибавляют 125 г измельченного льда и 10,5 мл
уксусного ангидрида. Колбу встряхивают в течение 15—20 мин
(прибор 1). Выпавший р-нафтилацетат отсасывают на воронке
Бюхнера, промывают водой и высушивают на воздухе. Выход
р-нафтилацетата ~13 г.
Для очистки р-нафтилацетат перекристаллизовывают из раз-
бавленного этилового спирта.
}
р-Нафтилацетат — бесцветное кристаллическое вещество с т. пл. 71 °C. Хо-
рошо растворяется в эфире и хлороформе.
Применяется в органическом синтезе.
АЦЕТАНИЛИД
Формула:
•NH—с—СН8
C|H5NHCOCHs;
1М
Основные реакции:
4 CeH5NH2 + НС! —> [CeHsNH3l+Cr
/
Солянокислый
анилин
СН3—СО.
[CeHeNH3]+Cr + ;о —> CeH6NHCOCH3 + СНзСООН + НС!
СНз—со/
CH3COONa + НС! —► СН3СООН + NaCl
Реактивы, посуда и приборы
Анилин.....................9 мл, 9,3 г
(0,1 моль)
Уксусный ангидрид..........12,5 мл41,«,г
(0,13 моль)
Соляная кислота (d = 1,19) . . 8,5 мл
(0,1 моль)
Уксуснокислый натрий, кри-
сталлический ........ 15 г
Стакан химический (500 мл) 1
Мешалка ...................1
Воронка Бюхнера........ 1
Колба Бунзена............1
Предохранительная склянка 1
Термометр................I
Сборка приборов
1. Химический стакан на 500 мл, снабженный мешалкой, по-
мещают на водяную баню.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В стакан с 250 мл воды приливают 8,5 мл концентрированной
соляной кислоты и при перемешивании мешалкой прибавляют
9 мл свежеперегнанного анилина.
Бесцветный раствор нагревают на водяной бане до 50 °C, при-
ливают 12,5 мл уксусного ангидрида и перемешивают (осторожно!
возможен выброс).
Если раствор окрашен, то прибавляют 2 г активированного
угля, перемешивают несколько минут и фильтруют.
Затем вносят раствор 15 г уксуснокислого натрия в .50 мл
воды, хорошо перемешивают и охлаждают льдом. Выпавшие кри-
сталлы ацетанилида отсасывают на воронке Бюхнера, промывают
ледяной водой и сушат.
Ацетанилид можно перекристаллизовать из воды.
Выход ацетанилида ~10 г.
Ацетанилид (фениламид уксусной кислоты) — белое кристаллическое ве-
щество без запаха, плохо растворяется в холодной воде, хорошо — в эфире,
спирте, хлороформе, ацетоне, анилине.
Применяется В качестве жаропонижающего и болеутоляющего средства в
медицине, полупродукта при синтезе красителей и стабилизатора перекиси во-
дорода.
Отличительная реакция
Нагревают 0,2 г ацетанилида и 1 мл соляной кислоты в тече-
ние 2 мин, затем добавляют кристаллик фенола, 5 мл воды и 2 мл
раствора хлорной извести — смесь окрашивается в грязно-фиолето-
вый цвет. При добавлении к этой смеси избытка аммиака окраска
становится синей (индофенольная реакция).
Глава X
РЕАКЦИИ ФРИДЕЛЯ —КРАФТСА
В 1877 г. Ш. Фридель и Д. Крафтс показали, что бензол и
его гомологи в присутствии безводного хлористого алюминия спо-
собны к алкилированию под действием галогеналкилов, при этом
происходит отщепление хлористого водорода и замещение атома
водорода ароматического ядра на соответствующий радикал.
Позднее было установлено, что указанная реакция возможна для
самых различных ароматических соединений — бензола, его гомо-
логов и производных, нафталина и его производных, ряда,гетеро-
циклических соединений и т. д. >
В настоящее время под реакцией Фриделя — Крафтса пони-
мают алкилирование или ацилирование ароматических соедине-
ний галогеналкилами или галогенангидридами карбоновых кислот
в присутствии специальных катализаторов, например безводного
хлористого алюминия:
А1С13
C6H6 + RC1 --> C6H5R+HC1
А1С1з
CeH6 + RCOC1 ---> C6H5COR + НС1
Первая реакция применяется в основном для получения алки-
лированных ароматических углеводородов; вторая — является луч-
шим способом получения ароматических кетонов и имеет большое
значение.
Чаще всего реакцию Фриделя — Крафтса проводят в присут-
ствии хлористого алюминия, но иногда применяют и другие ката-
лизаторы (FeCl3, BF3, ZnCl2, H2SO4, Н3РО4), активность которых
зависит от природы алкилирующего агента.
Обычно хлористый алюминий употребляется в виде готового
твердого продукта (порошка), при этом он должен быть чистым
и сухим. Желательно перед употреблением очистить его методом
возгонки. Хлористый алюминий можно получать и в реакционном
сосуде во время реакции металлического алюминия с газообраз-
ным хлористым водородом (катализатор Радзивановского),
1. АЛКИЛИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Некоторые ароматические соединения в присутствии хлори-
стого алюминия могут легко алкилироваться галогеналкилами,
олефинами и спиртами. Скорость реакции зависит от природы
галогена и алкилируемого агента.
В лаборатории алкилирование по Фриделю—Крафтсу имеет
ограниченное значение. Так, при алкилировании бензола полу-
чается смесь моно-, ди- и полизамещенных углеводородов. Это
объясняется тем, что скорость реакции алкилирования самого бен-
зола меньше, чем скорость реакции алкилирования образующегося
на первой стадии алкилбензола. Под влиянием хлористого алюми-
ния может происходить не только замещение алкильным радика-
лом водорода ароматического ядра, но и дегидрирование, гидриро-
вание, изомеризация и полимеризация продуктов. Течение реакции
алкилирования бензола частично можно регулировать путем под-
бора соответствующих количеств реагентов. Если хотят получить
моноалкилзамещенный продукт бензола, то берут большой избы-
ток последнего, а для получения полиалкилзамещенных производ-
ных бензола применяют избыток алкилирующего агента. Однако
нельзя полностью избежать образования продуктов разной сте-
пени замещения, что снижает выход и представляет определенные
трудности при выделении основного продукта.
Во время взаимодействия ароматического соединения с пер-
вичными галогеналкилами, имеющими неразветвленную цепь угле-
родных атомов, могут получаться производные с разветвленной
боковой цепью. Например, из бензола и хлористого н-пропила
получается изопропилбензол, из бромистого н-бутила и бензола —
етор-бутилбензол.
Алкилирование в присутствии хлористого алюминия, на при-
мере образования изопропилбензола можно представить следую-
щим образом.
В безводном хлористом алюминии атом алюминия имеет лишь
шесть электронов на внешней орбитали. Он дополняет эту груп-
пировку до октета за счет оттягивания электронов хлора хлори-
стого пропила:
AlCIs б+ б-
СН3СН2СН2С1 ---> СН3СН2СН2—С1- А1С13 —>
—> СН3СН2СН2--- А1С17
+
Карбкатион СН3СН2СН2 изомеризуется в более стабильный
ион:
СН3СН2СН2 ч=* СН3СНСН3
171
Последний и является электрофильным алкилирующим
агентом:
А1С1,
CHjCHCHj • AlClf
Л-комплекс
Л1СГ4
er-комплекс
Перед синтезом по методу Фриделя — Крафтса все реагенты
должны быть тщательно высушены, так как хлористый алюминий
и другие катализаторы этой реакции очень легко подвергаются
гидролизу. Аппаратура для проведения реакции также должна
быть высушена и защищена от доступа влаги.
2. АЦИЛИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИИ
Ацилирование ароматических углеводородов по Фриделю —
Крафтсу — важнейший метод синтеза кетонов различного строе-
ния. Это объясняется тем, что введение в молекулу ароматиче-
ского соединения карбонильной группы затрудняет дальнейшее
ацилирование, вследствие чего образуются однородные продукты
реакции с хорошими выходами. В качестве ацилирующих агентов
применяют хлорангидриды жирных и ароматических кислот, ан-
гидриды кислот, фосген и карбоновые кислоты.
Выбор катализатора определяется реакционной способностью
ароматического соединения. Чаще всего применяют хлористый
алюминий и только для очень реакционноспособных веществ (на-
пример, тиофена) используют хлористый цинк, серную кислоту
и др.
В отличие от реакции алкилирования по Фриделю — Крафтсу,
для которой достаточно присутствия небольшого количества хло-
ристого алюминия, реакция ацилирования протекает с удовлетво-
рительным результатом только в том случае, если количество
хлористого алюминия будет значительно больше, чем при полу-
чении алкилбензолов. Причиной этого является образование комп-
лексного соединения хлористого алюминия с кислородсодержа-
щими реагентами, в частности с хлорангидридами, ангидридами
кислот, кетонами и кислотами.
m
Например, образование дифенилкетона можно представить
схемой:
СвН5—С—Cl + А1С1, —>
гс6н5—ci+Aici;
II
I о]
©'-комплекс
А1СЦ
Полученный комплекс разлагают ледяной водой в присутствии
соляной кислоты:
—С=О • А1С1»
С6Н5
Н2О
НС1
С6Н5—С=О + А1С13
С6Н5
бензофенон
Бензофенон, растворенный в органическом растворителе, всплы-
вает наверх, а хлористый алюминий остается в водном растворе.
Хлористый алюминий, связанный с кетоном, не участвует в ре-
акции, поэтому нужно применять его в таком количестве, чтобы
после образования комплекса оставался еще некоторый избыток,
необходимый в качестве катализатора для реакции ацилирования
по Фриделю—Крафтсу. При ацилировании ангидридами кислот
необходимо еще большее количество хлористого алюминия, так
как образующиеся в результате реакции кетон и уксусная кислота
связывают по одному эквиваленту катализатора. Поэтому при
синтезе кетонов следует применять более I моль хлористого алю-
миния на 1 моль хлорангидрида кислоты и более 2 моль хлори-
стого алюминия на 1 моль ангидрида кислоты.
Особый интерес представляет взаимодействие с ангидридами
двухосновных карбоновых кислот. При этом получаются кетокис-
лоты, которые дальше могут быть превращены в дикетоны; напри-
мер, из фталевого ангидрида’и бензола образуется о-бензоилбен-
зойная кислота (о-карбоксибензофенон), которая дальше может
быть конденсирована в антрахинон:
1Я
Ацилирование по Фриделю — Крафтсу можно применять к
одно- и многоядерным ароматическим углеводородам, моноалкил-
и моногалогенпроизводным бензола, эфирам фенолов, реакцион-
носпособным гетероциклам (например, тиофену, фурану). Нитро-
соединения и фенолы не вступают в эту реакцию.
Выбор растворителя в синтезе Фриделя — Крафтса довольно
ограничен, так как очень многие органические соединения реаги-
руют с хлористым алюминием. В качестве растворителя преиму-
щественно применяют избыток ароматического углеводорода, а
также сероуглерод, нитробензол и петролейный эфир.
Способ проведения реакции ацилирования в основном не отли-
чается от способа, применяемого при реакции алкилирования.
Реакция взаимодействия ароматических соединений с ацилирую-
щими агентами также экзотермична, и поэтому хлорангидрид или
ангидрид кислоты следует вводить в смесь по каплям. Закончив
добавление ацилирующего агента, реакционную смесь нагревают
на водяной бане от 1 до 3 ч, затем выливают на лед и обрабаты-
вают соляной кислотой. Выделившийся кетон извлекают раствори-
телем, отделяют водный слой, сушат и перегоняют.
3. ПРИМЕРЫ СИНТЕЗОВ
ИЗОПРОПИЛБЕНЗОЛ
Формула:
сн.
-СН.
^сн
Основная реакция (механизм реакции см. стр. 171):
СН;
I] + СН3СН2СН2С1
А1С13
СН
СНз + НС1
Реактивы, посуда и приборы
Бензол .
100 г, или 114 мл
(1,28 моль)
Хлористый пропил . . 10 г или 11 мл
(О; 12 моль)
Хлористый алюминий,
безводный . . . .
Едкий натр
Сернокислый магний
Лед
Соляная кислота
Колба Вюрца (200 мл) . . . . 1
Колба круглодонная (300 мл) 1
Форштосс двурогий...........1
Холодильник ............... 1
Трубка хлоркальцневая . . . . 1
Воронка капельная .
Воронка делительная
Колба коническая .
Термометр..........
Стакан химический .
<74
Сборка приборов
1. Круглодонную колбу на 300 мл помещают на водяную
баню, соединяют при помощи двурогого форштосса с капельной
воронкой и обратным водяным холодильником, снабженным хлор-
кальциевой трубкой, к наружному концу которой присоединяют
изогнутую стеклянную трубку, опущенную в колбу с водой для
поглощения хлористого водорода; конец трубки должен нахо-
диться на расстоянии примерно 1 см от поверхности воды, иначе
вода может быть втянута внутрь прибора. В водяную баню по-
мещают термометр.
2. Колба Вюрца на 200 мл с нисходящим водяным холо-
дильником.
Выполнение синтеза
В круглодонную колбу прибора 1 помещают 80 мл свобод-
ного от тиофена сухого бензола и 2 г безводного тщательно из-
мельченного хлористого алюминия (осторожно! хлористый алю-
миний раздражающе действует на кожу, сухой препарат реагирует
с водой со взрывом!).
1. Неочищенный бензол содержит около 0,15% тиофена. Для очистки бен-
зола от тиофена смешивают 1 л бензола с 80 мл концентрированной серной
кислоты и энергично перемешивают в течение 30 мин Образовавшийся темно-
окрашенный слой серной кислоты отделяют и повторяют перемешивание с сер-
ной кислотой до тех пор, пока последняя почти не перестанет окрашиваться.
Бензол осторожно перегоняют
Для высушивания бензола его перегоняют и отбрасывают первые 10% по-
гона. Вся влага отгоняется в виде азеотропной смеси с бензолом.
2. Все операции с безводным хлористым алюминием следует проводить по
возможности быстро вследствие его большой гигроскопичности
К содержимому колбы прибавляют по каплям раствор 11 мл
хлористого пропила в 34 мл бензола, причем температуру реак-
ционной смеси поддерживают около 80 °C (при температуре выше
указанной и применении больших количеств хлористого алюминия
образуется л-диизопропилбензол).
После прибавления всего количества хлористого пропила на-
гревание продолжают, пока выделение хлористого водорода не
прекратится.
Конец реакции определяют: а) по исчезновению покраснения
синей лакмусовой бумажки; б) по отсутствию дыма при поднесе-
нии пробирки с аммиаком к концу трубки или при пропускании
выделившегося хлористого водорода через предварительно взве-
шенную колбу с водой. Реакцию прекращают, когда поглотится
рассчитанное количество хлористого водорода.
Затем реакционную смесь выливают на лед, соляной кислотой
растворяют гидроокись алюминия, отделяют в делительной во-
ронке верхний слой изопропилбензола в бензольном растворе,
промывают его разбавленным раствором едкого натра и водой до
нейтральной реакции, после чего сушат сернокислым магнием или
хлористым кальцием и фракционируют (прибор 2), отбирая фрак-
цию изопропилбензола в интервале 151—153°C.
Выход изопропилбензола -—11 г.
Изопропилбензол (кумол) — бесцветная жидкость, нерастворим в воде;
смешивается со спиртом, эфиром, ацетоном, хлороформом, бензолом. Мол. вес
120,19; т. кип. 152,4 °C; т. пл. —96,03 °C; d,° = 0,8618; «д = 1,49ГЗ. Легко вос-
пламеняется (температура вспышки 38 °C) и образует взрывоопасные паро-воз-
душные смеси.
Применяется в качестве растворителя лаков и красок, добавки к авиацион-
ным бензинам, а также в производстве фенола и ацетона.
ДИФЕНИЛМЕТАН
Формула:
Основная реакция:
А1С1а
Реактивы, посуда и приборы
Бензол................43 г, или 50 мл Колба круглодонная, двугор-
(0,56 моль) лая (200 мл)................1
Хлористый бензил . . . 12,7 г, или 12 мл Холодильник Либиха........1
(0,1 моль) Трубка хлоркальциевая . . . . 1
Хлористый алюминий Воронка капельная...........1
безводный............5 г Вороика делительная........1
Соляная кислота (d₽=l,!9) Колба коническая............1
Едкий натр - Колба Вюрца (100 мл) . , . . 1
Лед Термометр...................1
Алоиж.......................1
Приемники ................. 2
Сборка приборов
1. Круглодонную двугорлую колбу емкостью 200 мл снаб-
жают капельной воронкой и обратным водяным холодильником
с хлоркальциевой трубкой. Последнюю соединяют с изогнутой
стеклянной трубкой, конец которой опускают в колбу с водой
(примерно на 1 см над поверхностью воды). Прибор помещают
в вытяжной шкаф!
2. Колбу Вюрца, помещенную на водяную баню, снабжают
термометром и нисходящим холодильником с алонжем, опущен-
ным в приемник.
Выполнение синтеза
В круглодонную двугорлую колбу прибора 1 наливают 50 мл
сухого бензола и 5 г безводного, тщательно измельченного хло-
ристого алюминия (см. стр. 175). Затем прибавляют по каплям
12 мл хлористого бензила (работать с хлористым бензилом сле-
дует в вытяжном шкафу, так как пары его обладают слезоточи-
вым действием и раздражают дыхательные пути!).
После прекращения выделения хлористого водорода в колбу
вносят 40 г толченого льда и немного соляной кислоты. Затем ре-
акционную смесь переносят в делительную воронку и отделяют
верхний бензольный слой, содержащий дифенилметан; его промы-
вают водой, раствором едкого натра и снова водой до нейтраль-
ной реакции. Отогнав от бензольного раствора бензол (прибор 2),
снимают холодильник, отводную трубку колбы Вюрца вставляют
в приемник, погруженный в чашку с холодной водой, и отгоняют
дифенилметан на воронке Бабо (стр. 30). Если дифенилметан
окрашен или не застывает при охлаждении, его следует перегнать
еще раз с 2—3 кусочками металлического натрия.
Выход дифенилметана ~ 10 г.
Дифенилметан — бесцветные кристаллы, с запахом апельсиновой кожуры;
нерастворим в воде, хорошо растворим в спирте, эфире, хлороформе, бензоле.
Мол. вес 168,5; т. кип. 261—262 °C; т. пл. 26—27 °C; d“= 1,0060; /$= 1,5788.
Применяется как растворитель TiaKOB и красок и для отдушки мыла.
АЦЕТОФЕНОН
Формула: Основная реакция: снэ—СОч А1С1 [ Ц+ 7° СНз—СО7 Побочная реакция: С6Н5СОСЩ ф CSH8COCH5 - ^СОСН3 ^s^CQCHs * Г Т +сн3соон —> с6н6—с=снсос6н6 + Н2О сна
17»
Реактивы, посуда и приборы
Бензол, безводный ... 35 г. или 40 мл
(0,45 моль)
Уксусный ангидрид . . 10,2 г, или 10 мл
(0,1 моль)
(0,24 моль)
Хлористый алюминий,
безводный.............32 г
Диэтиловый эфир ... 35 мл
Едкий натр, 10%-ный 25 мл
Соляная кислота, 10%-ная
Хлористый кальций, безводный
Колба круглодонная, трехгор-
лая (300 мл)..............I
Мешалка механическая .... 1
Холодильник Либиха ........ 1
Воронка капельная ......... 1
Воронка коническая ........ 1
Воронка делительная ....... 1
Трубки хлоркальциевые ... 2
Стаканы химические.........2
Колба Вюрца (100 мл) . . . . 1
Термометры..................2
Алонж.......................1
Холодильник воздушный . . . 1
Приемник .................. 1
Сборка приборов
1. Круглодонную трехгорлую колбу емкостью 300 мл (или
круглодонную колбу с двурогим форштоссом) помещают на водя-
ную баню и снабжают мешалкой, йапель-
Рис. 92. Прибор для полу-
ной воронкой и обратным водяным холо-
дильником. Холодильник и капельную
воронку закрывают хлоркальциевыми
трубками. К наружному концу хлоркаль-
циевой трубки холодильника присоеди-
няют изогнутую стеклянную трубку, со-
единенную с опрокинутой стеклянной
воронкой. Последнюю опускают в стакан
с водой так, чтобы края ее находились
на расстоянии примерно 1 см от поверх-
ности воды. В водяную баню помещают
термометр (рис. 92).
Образующаяся в результате реакции уксус-
ная кислота реагирует с хлористым алюми-
нием:
СН3СООН +А1С13 —> СН3СООА1С12 + НС1
Выделяющийся хлористый водород необхо-
димо поглощать водой.
чения ацетофенона:
/—водяная баня; 2—круглодон-
мая трехгорлая колба; 3 — тер-
мометр; 4—капельная воронка;
5—хлоркальциевая трубка; 6 —
мешалка; 7—холодильник; 8 —
воронка; 9—стакан.
2. Колбу Вюрца на 250 мл, поме-
щенную на водяную баню, снабжают тер-
мометром и нисходящим водяным холо-
дильником с алонжем, конец которого
опускают в приемник.
3. Колбу Вюрца на 100 мл соединяют с термометром и нисхо-
дящим воздушным холодильником.
В связи с гигроскопичностью хлористого алюминия вся аппаратура должна
быть тщательно высушена.
178
Выполнение синтеза
Следует избегать вдыхания паров ацетофенона, так как они
обладают снотворным действием.
В круглодонную трехгорлую колбу прибора 1 помещают 40 мл
сухого фензола (см. стр. 175) и 32 г тонкоизмельченного свеже-
возогнанного хлористого алюминия. Включают мешалку и по кап-
лям через капельную воронку в течение 30 мин приливают 10 мл
чистого уксусного ангидрида (см. стр. 178), охлаждая колбу хо-
лодной водой. Для завершения реакции колбу нагревают на водя-
ной бане при 80—85 °C в течение 45 мин. При более продолжитель-
ном нагревании выход ацетофенона уменьшается, так как обра-
зуется высококипящий продукт — дипион
CSH6—C=CHCOCSH5
СН3
Охлажденную реакционную смесь выливают в стакан, содер-
жащий 80 г воды со льдом; в случае выделения осадка основной
соли алюминия (при гидролизе комплексного соединения хлори-
стого алюминия с ацетофеноном) добавляют разбавленную соля-
ную кислоту до растворения этого осадка.
Смесь переносят в делительную воронку, обрабатывают 20 мл
диэтилового эфира и отделяют эфирно-бензольный слой, а водный
извлекают еще раз 15 мл эфира. Объединенные эфирные вытяжки
промывают водой, затем 25 мл раствора едкого натра, снова во-
дой и сушат безводным хлористым кальцием.
После отгонки растворителей (эфира и бензола) на водяной
бане (прибор 2) перегоняют ацетофенон (прибор 3), собирая
фракцию, кипящую при 199—203 °C. Ацетофенон можно отгонять
также в вакууме (12 мм рт. ст.) при 88 °C.
Выход ацетофенона ~8г.
Ацетофенон (метилфенилкетон) — бесцветная, иногда желтоватая масляни-
стая жидкость или крупные легко плавящиеся кристаллы с запахом черемухи;
мало растворим в воде, хорошо растворим в спирте, эфире, хлороформе, бен-
золе. Мол. вес 120,14; т. кип. 202,3 °C при 760 мм рт. ст. или 88 °C при
12 мм рт ст.; т пл. 20,5 °C; == 1,0281; re|j = 1,5342.
Применяется в парфюмерии.
Отличительные реакции
1. К 2 мл 0,5% раствора нитропруссида натрия прибавляют
2 капли ацетофенона и затем 2 капли 10% раствора едкого натра;
появляется красное окрашивание раствора. При прибавлении ук-
сусной кислоты окраска переходит в синюю.
2. Ацетофенон хорошо растворяется в концентрированной сер-
ной кислоте, при этом появляется оранжево-желтая окраска. По-
сле прибавления воды ацетофенон снова выделяется.
179
БЕНЗОФЕНОН
Формула:
О
tОсновная реакция (механизм реакции см. стр. 173):
Реактивы, посуда и приборы
О
СУ"О+на
Бензол, безводный ... 48 г, или 55 мл Колба круглодонная (500 мл) I
(0,62 моль) Форштосс двурогий ..........1
Хлористый бензоил . . 14 г, или 11,5 мл Холодильник Либиха........1
(0,11 моль) Воронка капельная...........1
Хлористый алюминий, Воронка коническая........1
безводный..........15 г Трубки хлоркальциевыё ... 2
Соляная кислота (d = l,19) Стакан химический.........1
Диэтиловый эфир Термометр.................1
Едкий натр, 5%-ный . . 25 мл Прибор для перегонки с води-
Хлористый кальций, гранулированный ным паром............ . 1
Воронка делительная ....... 1
Колба Вюрца (100 мл) . . . . 1
Алонж.......................1
Термометр...................1
Колба Вюрца (50 мл) с низко-
припаянной отводной трубкой 1
Сборка приборов
1. Круглодонную колбу емкостью 50 мл, помещенную на водя-
ную баню, соединяют при помощи двурогого форштосса с обрат-
ным холодильником и капельной воронкой, которые закрывают
хлоркальциевыми трубками. К хлоркальциевой трубке холодиль-
ника присоединяют изогнутую под углом стеклянную трубку, ко-
торую соединяют с опрокинутой стеклянной воронкой, опущенной
в стакан с водой таким образом, чтобы края ее находились на
расстоянии 1 см от поверхности воды. В водяную баню помещают
термометр.
2. Прибор для перегонки с водяным паром (см. рис. 48).
3. Колбу Вюрца емкостью 100 мл, помещенную на водяную
баню, соединяют с термометром и нисходящим холодильником
с алонжем, опущенным в приемник.
180
Выполнение синтеза
В круглодонную колбу прибора 1 вносят 15 г безводного,
тщательно измельченного хлористого алюминия (см. стр. 175) и
55 мл сухого бензола (см. стр. 175). Затем при взбалтывании
приливают из капельной воронки 11,5 мл хлористого бензоила с
такой скоростью, чтобы реакция протекала не слишком бурно,.
Далее колбу нагревают на водяной бане при 50 °C до полного
прекращения выделения хлористого водорода (обычно 2—3 ч).
После охлаждения к содержимому колбы, окрашенному в темно-
коричневый цвет, осторожно, при перемешивании, добавляют
150 мл воды, несколько кусочков льда для разложения комплекса
бензофенона с хлористым алюминием и небольшое количество кон-
центрированной соляной кислоты, чтобы перевести основную соль
алюминия в раствор. После этого отгоняют бензол с водяным па-
ром (прибор 2) в течение 20 мин.
Оставшуюся в колбе смесь охлаждают и смешивают с эфиром
в делительной воронке. Эфирную вытяжку промывают 25 мл вод-
ного раствора едкого натра, сушат хлористым кальцием (хлори-
стый кальций можно заменить безводным сульфатом магния или
карбонатом калия) и отгоняют эфир (прибор 3). Остаток перего-
няют из колбы Вюрца с низкоприпаянной отводной трубкой без
холодильника при обычном давлении, собирая фракцию, кипящую
при 297 °C; если нужно получить более чистый продукт, остаток
перегоняют в вакууме, собирая фракцию, кипящую при 170—
175°С (15 мм рт. ст.). ,
Выход бензофенона ~ 13 г.
Бензофенон (дифенилкетон) — белое блестящее кристаллическое вещество
со своеобразным запахом, нерастворим в воде, растворим в хлороформе Мол.
вес 182,21 Известен в стабильной н лабильной формах Стабильная форма:
т кип. 305 °C, т пл 48 °C, 1,0976; Пд = 1,5975. Лабильная форма: т. кип.
305 °C; т. пл 26 °C; d = 1,108; п^= 1,6060.
При перегонке обычно получают лабильную форму бензофенона, которая
в момент перегонки представляет собой жидкость и в отсутствие затравки очень
медленно затвердевает в бесцветные кристаллы При трении о стенки или вне-
сении кристаллов стабильной формы жидкость быстро затвердевает, превра-
щаясь в стабильную форму
Бензофенон применяется в парфюмерной промышленности, в производстве
красителей и дли других целей
Отличительные реакции
1. В концентрированной серной кислоте растворяется с жел-
той окраской. После прибавления воды бензофенон выделяется
снова.
2. При осторожном сплавлении с металлическим натрием по-
является темно-синяя окраска.
Глава XI
РЕАКЦИИ ОКИСЛЕНИЯ
Окисление — это процесс отдачи электронов. Соединения окис-
ляются тем легче, чем выше их склонность к отдаче электронов.
Окислители представляют собой вещества, имеющие сродство к
электронам.
Окисление органических соединений чаще всего осуществляется
при помощи следующих окислителей: кислорода воздуха, перман-
ганата калия, хромовой смеси (получаемой растворением бихро-
мата калия или натрия в серной кислоте), хромового ангидрида,
азотной кислоты, озона, двуокиси свинца, окиси серебра, трет-бу-
тилата алюминия и др. Действие окислителя на органическое со-
единение зависит от характера окисляемого вещества и от хими-
ческой природы самого окислителя. Так, например, при окислении
анилина хромовой смесью образуется бензохинон; бертолетовой
солью или хромпиком в присутствии катализатора (соли ванадия,
меди или железа) — анилиновый черный (краситель сложного
строения); перманганатом калия в нейтральной среде—азобензол,
в щелочной — нитробензол; хлорноватой кислотой — п-аминофе-
нол:
Азобензол
Нитробензол
я-Амннофенол
Бензохинон
Окисляемость органического соединения связана с наличием
атомов водорода, так как окисление идет с отдачей атомов водо-
рода или с присоединением атомов кислорода. Например, толуол
легко окисляется
С6Н5-СН3 —> C6HS-CH2OH —+ С6Н5-С\ —> сен5-с(
чт хон
R
I
а третичные спирты R—С—ОН окисляются в жестких условиях
i
с разрывом углеродной цепи. Частный случай реакции окисле-
ния — дегидрирование, т. е. отщепление, водорода. В промышлен-
ности важнейшим методом получения альдегидов и кетонов яв-
182
ляется каталитическое дегидрирование первичных и вторичных
спиртов:
R—СН2ОН «=> R-C=O + H2
I
н
R—СН—ОН R—С=О + Н3
R' R'
В качестве катализаторов используют металлическую медь,
серебро, хромокись меди и окись цинка. Дегидрирование — про-
цесс эндотермический, поэтому катализатор постоянно нагревают.
1. ОКИСЛЕНИЕ ПО ДВОЙНОЙ СВЯЗИ
Двойная связь очень чувствительна к хромовой смеси и пер-
манганату калия. Под воздействием окислителя сначала происхо-
дит присоединение двух гидроксильных групп; образовавшийся
гликоль окисляется дальше с расщеплением углеродной связи, при
этом получаются кетоны или кислоты:
R\
;c=ch-r"
RZ
R'
R'
V—СН
ОН ОН
R\
)с=О + R"—COOH
RZ
Эта реакция используется для получения гликолей, кетонов и
кислот из непредельных углеводородов, а также для установления
строения непредельных соединений. Окисление по двойной связи
можно производить действием озона с целью получения альдеги-
дов и кетонов, если их нельзя получить другими методами, а так-
же чтобы определить положение двойных связей и структуру не-
насыщенных соединений. Озон присоединяется по' двойной связи,
образуя очень неустойчивые, взрывчатые озониды, которые под
'действием воды легко разлагаются до альдегидов или кетонов:
R\
V=CH—R"
RZ
н2о
R'\
—* н2о2 + )с=о + R"-qf
RZ ХН
2. ОКИСЛЕНИЕ ПЕРВИЧНЫХ И ВТОРИЧНЫХ СПИРТОВ
ДО АЛЬДЕГИДОВ ИЛИ КЕТОНОВ
Для окисления первичных алифатических спиртов до альдеги-
дов в лаборатории обычно применяют хромовую смесь, дву-
окись марганца с серной кислотой и азотную кислоту. Однако
183
образующийся альдегид может подвергаться дальнейшему окисле-
нию в соответствующую кислоту, и, таким образом, выход продук-
тов реакции уменьшается. Поэтому полученный альдегид необхо-
димо как можно быстрее удалять из сферы реакции. Это дости-
гается, во-первых, медленным приливанием окислителя и, во-вторых,
пропусканием через реакционную смесь тока двуокиси углерода,
который увлекает с собой легколетучий альдегид.
Для получения ненасыщенных и ароматических альдегидов
применяют при окислении трет-бутилхромат в петролейном эфире,
бензоле и двуокись марганца в ацетоне, разбавленной серной кис-
лоте или четыреххлористом углероде. Окисление вторичных спир-
тов до кетонов идет легче, чем окисление первичных спиртов, и
дает лучшие выходы. Это объясняется тем, что реакционная спо-
собность вторичных спиртов выше, чем первичных, и получаю-
щийся кетон более устойчив к окислителям, чем альдегид. Обра-
зующиеся кетоны удаляют из реакционной смеси органическими
растворителями и таким образом защищают от дальнейшего окис-
ления.
3. ОКИСЛЕНИЕ АЛЬДЕГИДОВ И КЕТОНОВ ДО КИСЛОТ
Все окислители, которые окисляют первичные спирты до аль-
дегидов, могут быть использованы и для получения карбоновых
кислот с тем же числом углеродных атомов из альдегидов или
спиртов. Однако окисление одноатомных первичных спиртов в со-
ответствующие кислоты, минуя .стадию образования альдегидов,
лучше всего проводить с помощью перманганата калия в ще-
лочной среде, которую создает образующееся во время реакции
едкое кали
2КМпО4 + Н2О —> 2МпО2 + 2КОН + 30
При окислении первичных спиртов хромовой смесью и перман-
r-анатом калия в кислой среде в качестве побочных продуктов по-
лучаются сложные эфиры.
В некоторых случаях хорошие выходы наблюдаются при при-
менении азотной кислоты, которая используется главным образом
для получения карбоновых кислот, выделяющихся из реакционной
смеси в виде трудно растворимых в воде солей.
При окислении вторичных спиртов и кетонов нельзя получить
кислоту с тем же числом углеродных атомов, так как кетоны бо-
лее устойчивы к окислению, чем альдегиды, ш окисляются только
с разрывом углеродной цепи, для чего требуется более сильное и
длительное воздействие окислителей. Алифатические кетоны и
вторичные спирты окисляются хромовой смесью или азотной кис-
лотой, при этом образуется смесь жирных карбоновых кислот.
Большое практическое значение имеет окисление алицикличе-
ских спиртов и кетонов до двухосновных карбоновых кислот. Так,
адипиновую кислоту получают окислением циклогексанола,
184
4. ОКИСЛЕНИЕ МЕТИЛЬНЫХ И МЕТИЛЕНОВЫХ ГРУПП
Неразветвленные насыщенные углеводороды — наиболее труд-
но окисляемые органические соединения. Окисление начинается
лишь в жестких условиях, например под действием горячей хромо-
вой смеси. При этом получаются кетоны, кислоты и другие про-
дукты окисления. Поэтому окисление насыщенных углеводородов
не является препаративным методом получения определенных со-
единений.
Метильная группа окисляется легче, если она связана с двой-
ной связью или ароматическим ядром. Метильные группы, находя-
щиеся при бензольном ядре, окисляются хромовой смесью, азот-
ной кислотой, перманганатом калия до карбоксильных. Напри-
мер, при окислении n-нитротолуола получают п-нитробензойную
кислоту:
OjN——СН3 + К2Сг2О7 + 4H2SO4 —-*
—> O2N—fy- СООН + Cr2(SO4)3 + K2SO4 + 5H2O
Метиленовые и метильные группы, соседние с карбонильной
Труппой или с двойной и тройной связью, могут быть окислены
двуокисью селена SeOj.
5. ПОЛУЧЕНИЕ ХИНОНОВ ОКИСЛЕНИЕМ
Некоторые ароматические соединения могут быть окислены
до о- и n-хинонов различными окислителями. Например, гидро-
хинон окисляется в n-бензохинон под действием двухромовокис-
лого натрия или бромноватокислого натрия в присутствии серной
кислоты.
Образование хинонов из ароматических соединений облег-
чается в тех случаях, когда в получающихся соединениях двойные
связи в а, ^-положении к карбонильной группе соединены с ал-
кильными или ароматическими группами. Например, 2-метилна-
фталин легко окисляется в 2-метилнафтохинон-1,4 благодаря на-
личию в последнем метильной и ароматической групп в а, р-поло-
жениях к карбонильной группе.
6. ПРИМЕРЫ СИНТЕЗОВ
АЦЕТАЛЬДЕГИД
Формула:
СН,-С\
<81
Основные реакции:
ЗСНз—СН2ОН + K2Cr2O7 + 4H2SO4 —>
—> ЗСН3-С^ + K2SO4 + Cr2(SO4)3 + 7Н2О
\н
ЗСНз—С^ + 3NH3 —> СНз—CH-NH—СН-СНз
\Н I ।
NH—СН—NH + ЗН2О
СНз
СНз—сн—NH—сн—сн3 + ЗН2О + 3H2SO4 —> зсн3—с; +3(NH4)HSO4
I I \н
NH—СН—NH
Побочные реакции:
СН3—СН2ОН + H2SO.
СНз—СН2—О—SO3H + Н2О
СНз
О
// H2SO
ЗСНз—С—Н -----’
СН
Н3С—Сх /СН-СНз
о
сн3- с; + о
О
СН3-СН2ОН + СНз—с
\он
H2SO4 II
СНз—С—О—СН2—СНз + Н2О
Ацетальдегид действует раздражающе на слизистые оболочки.
Образует с воздухом взрывоопасные смеси!
Реактивы, посуда и приборы
Этиловый спирт, 95%-ный . . 25 мл
(0,4 моль)
Двухромовокислый калий . . 35 г
(0,12 моль)
Серная кислота.............35 мл
Этиловый эфир..............60 мл
Углекислый газ
Аммиак из баллона
Хлористый кальций
Хлористый натрий
Лед
Колбы круглодонные широкогор-
лые (250 и 500 мл)............2
Холодильник Либиха .............. 1
Холодильник змеевиковый . . . . 1
Воронки капельные.............2
Баллон с углекислым газом или
аппарат Киппа...............1
U-образная трубка ............. 1
Промывная склянка.............1
Колба Бунзена.................1
Воронка Бюхнера................1
Термометр (от —10 до +100 °C) 1
Колба коническая (300 мл) . . . . 1
Стаканы химические.............5
Перегонная колба..............1
186
Сборка приборов
1. Круглодонную широкогорлую колбу емкостью 500 мл за-
крывают резиновой пробкой с тремя отверстиями. В одно из от-
верстий при помощи изогнутой насадки вставляют обратный холо-
дильник, в другое — капельную воронку с длинной оттянутой труб-
кой, а в третье—изогнутую стеклянную трубку, доходящую почти
до дна колбы. Посредством этой трубки прибор соединяют с бал-
лоном с углекислым газом или с аппаратом Киппа.
со2
Рис. 93. Прибор для получения ацетальдегида:
1 — круглодонная колба, 2—тройннк; 3—-капельная воронка;
4—зажим; 5—алонж; 6—холодильник Лнбиха; 7 — термо-
метр; 8— хлоркальциезая трубка; Р—змеевиковый холодиль-
ник; 10—склянка Дрекселя; 11— ледяная баня.
Для преодоления избыточного давления верхний тубус аппа-
рата Киппа закрывают пробкой, через которую проходит трубка,
наполненная разбавленной соляной кислотой.
Для выравнивания давления верхнее отверстие капельной во-
ронки соединяют с трубкой для ввода газа. Верхний конец холо-
дильника соединяют с U-образной трубкой, наполненной прока-
ленным хлористым кальцием. Последнюю соединяют через зме-
евиковый холодильник с промывной склянкой, содержащей 60 мл
эфира.
Если в приборе развивается повышенное давление, которое препятствует
приливанию раствора хромовой смеси, то приливание ведут под давлением угле-
кислого газа. Для этого к трубке, подводящей углекислый газ, крепят тройннк,
свободный конец которого при помощи резиновой трубки с винтовым зажимом
соединяют с тубусом капельной воронки. Открывая больше или меньше винто-
вой зажим, регулируют скорость подачи раствора хромовой смеси.
Промывную склянку погружают в свежеприготовленную смесь
соли и льда (температура смеси должна быть не больше—10°C).
В верхний конец холодильника помещают термометр, прикреплен-
ный при помощи нитки к пробке. Шарик термометра должен на-
ходиться посредине внутренней трубки холодильника (рис. 93).
187
2. Прибор для отсасывания (см. рис. 40).
3. Круглодонную широкогорлую колбу емкостью 250 мл, поме-
щенную на водяную баню, плотно закрывают 'резиновой пробкой
с четырьмя отверстиями. В этих отверстиях крепят капельную во-
ронку, доходящую почти до дна колбы, стеклянную трубку для
ввода углекислого газа, широкую трубку, которая через U-образ-
ную хлоркальциевую трубку соединяет колбу с вертикально по-
ставленным змеевиковым холодильником, и термометр. Холодиль-
ник при помощи стеклянной трубки присоединяют к приемнику,
охлаждаемому смесью льда и соли (приемник необходимо охлаж-
дать, так как ацетальдегид кипит при 20,8°C).
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу.
В круглодонную широкогорлую колбу (прибор 1) вливают 25 мл
этилового спирта и */з смеси, состоящей из 35 мл концентрирован-
ной серной кислоты и 70 мл воды. Смесь нагревают до кипения на
водяной бане. К оставшемуся количеству разбавленной серной кис-
лоты добавляют еще 35 мл воды и растворяют в ней 35 г двухро-
мовокислого калия (осторожно! двухромовокислый калий ядовит!)
и теплый раствор вливают в капельную воронку, следя за тем,
чтобы вся трубка воронки была заполнена жидкостью. После
этого начинают постепенно приливать хромовую смесь к кипящему
спирту, причем скорость подачи раствора регулируют таким обра-
зом, чтобы реакционная смесь непрерывно кипела, а термометр
в холодильнике показывал 25—30 °C. Так как реакция экзотерми-
ческая, то смесь продолжает кипеть без внешнего подогрева.
Одновременно с введением хромовой смеси пропускают через
жидкость''струю углекислого газа с такой скоростью, чтобы можно
было считать проходящие пузырьки газа.
Во избежание дальнейшего окисления ацетальдегида через реакционную
смесь пропускают слабый ток углекислого газа, который и уносит образующийся
ацетальдегид вместе с некоторым количеством паров спирта, воды, ацеталя и
этилсерной кислоты
Большая часть ацетальдегида успевает сконденсироваться в
холодильнике, а прошедшие через холодильник пары ацетальде-
гида поглощаются эфиром, охлаждаемым смесью льда и соли.
Через 20 мин приливание хромовой смеси заканчивается. После
этого еще в течение 10 мин пропускают углекислый газ, который
полностью вытесняет ацетальдегид из колбы. Затем отделяют про-
мывную склянку от прибора и только после этого прекращают по-
дачу углекислого газа.
Ацетальдегид нельзя отделить от эфира путем перегонки, по-
этому его переводят в кристаллический альдегидаммиак. Для этого
содержимое промывной склянки переливают в химический ста-
кан, который помещают в охладительную смесь, и через широкую
стеклянную трубку, доходящую до дна колбы, подают сухой ам-
миак до тех пор, пока раствор не станет сильно пахнуть,
188
Если в лаборатории нет баллона с аммиаком, то его получают кипячением
концентрированного раствора аммиака в круглодонной колбе, снабженной об-
ратным холодильником (горелку необходимо загородить предохранительным
экраном) Для высушивания пропускают выделяющийся аммиак через колонку,
наполненную едким кали и натронной известью в стакан 6 с эфирным раство-
ром ацетальдегида (рис 94).
Рис. 94. Прибор для получе-
ния аммиака:
/—•круглодонная колба; 2 — шари-
ковый холодильник; З-поглотитель-
ная колонка с CaCls; 4—зоронка;
5—ледяная баня; d—стакан с эфир-
ным раствором ацетальдегида.
Чтобы реакционную смесь можно было перемешивать трубкой
для ввода аммиака и чтобы уменьшить испарение эфира, колбу
необходимо закрыть куском картона
с отверстием.
Для полного связывания альде-
гида необходимо около 6 л аммиака.
Эфирный раствор ацетальдегида, насы-
щенный аммиаком, оставляют стоять
в охладительной смеси в течение 1 ч.
Смесь отсасывают на воронке Бюх-
нера, остаток промывают небольшим
количеством эфира и сушат сначала
на фильтровальной бумаге, а затем в
эксикаторе над серной кислотой. Чи-
стый альдегидаммиак можно хранить
длительное время в хорошо закупорен-
ной склянке, загрязненный препарат
темнеет и разлагается через несколько
дней.
Выход кристаллического альдегид-
аммиака ~ 9 г.
Для получения свободного аце-
тальдегида собирают прибор 3. В круг-
лодонную колбу емкостью 250 мл по-
мещают полученный альдегидаммиак
и вливают 10 мл воды. В капельную воронку вносят охлажденную
смесь, состоящую из 7 мл концентрированной серной кислоты и
20 мл воды, и по каплям в течение 30 мин приливают ее к рас-
твору альдегидаммиака.
Выделяющийся ацетальдегид перегоняют, нагревая колбу на
водяной бане. Чтобы предохранить ацетальдегид от окисления,
прибор перед перегонкой заполняют углекислым газом (ввиду вы-
сокого давления паров ацетальдегида во время перегонки угле-
кислый газ не пропускают, иначе будут большие потери ацеталь-
дегида) .
К концу перегонки через прибор снова пропускают (медленно)
углекислый газ в течение короткого промежутка времени. Соби-
рают ацетальдегид в приемник, охлаждаемый смесью льда и соли.
Ацетальдегид (уксусный альдегид) — бесцветная жидкость с резким удуш-
ливым запахом, хорошо смешивается с водой, спиртом, эфиром, хлороформом.
Мол вес 44,05; т. кип 20,8 °C; т пл —123,5 °C, dj8 = 0,783; = 1,3316.
Применяется в производстве уксусной кислоты, бутадиена, ацетальдоля,
альдегидных синтетических смол, в лабораторной практике н для других целей.
189
Отличительные реакции
1. К фуксинсернистой кислоте прибавляют 1% водного рас-
твора ацетальдегида — появляется сине-фиолетовое окрашивание.
2. К 2 мл водного раствора ацетальдегида прибавляют 2 мл
0,5% водного раствора нитропруссида натрия, затем две капли
10% раствора щелочи — появляется темно-красное окрашивание.
ПРОПИОНОВЫЙ АЛЬДЕГИД
Формула:
сн3—сн2—с^°
Ж
Основная реакция:
ЗСН3—СН2—СН2—ОН + К2Сг2О7 + 4H2SO4 —>
—> ЗСН3-СН2-С^ + K2SO4 + Cr2(SO4)3 + 7Н2О
Побочная реакция:
сн3—сн2—с; +0 —> СН3-СН2-С^
'Н х>н
Реактивы, посуда и приборы
Пропиловый спирт...........31 мл Колба круглодонная..........1
(0,4 моль) Мешалка.......................1
Двухромовокислый калий ... 19 г Воронка капельная...........1
(0,06 моль) Форштосс трехрогий...........1
Серная кислота (г/= 1,84) . . 36 мл Холодильник шариковый.......1
Сернокислый натрий, безводный Холодильник Либиха............1
Алонж............................1
Колба Вюрца (100 мл)..........1
Термометр ................... 1
Приемники ................... 2
.Сборка приборов
1. В круглодонную колбу емкостью 500 мл вставляют трехро-
гий форштосс, снабженный мешалкой, капельной воронкой и ша-
риковым обратным холодильником, через который пропускают
нагретую до 60 °C воду. Шариковый холодильник закрепляют под
углом в 45°.
К верхнему концу шарикового холодильника присоединяют
нисходящий холодильник, через который циркулирует холодная
вода. Конец второго холодильника снабжают алонжем, который
погружают в приемник, охлаждаемый ледяной водой.
Колбу помещают на водяную баню (рис. 95).
2. Перегонную колбу Вюрца емкостью 100 мл, помещенную на
водяную баню, снабжают термометром и нисходящим водяным хо-
лодильником с алонжем, который погружают в охлаждаемый при-
емник.
190
Выполнение синтеза
В круглодонную колбу прибора 1 помещают 31 мл пропило-
вого спирта и нагревают его до кипения. Затем через капельную
воронку медленно, в течение 15—20 мин вносят хромовую смесь
из 19 г двухромовокислого калия (см. стр. 188), 36 мл концентри-
рованной серной кислоты и 300 мл воды. Во время приливания
содержимое колбы перемешивают и поддерживают сильное кипе-
ние. После внесения всего количества хромовой смеси содержимое
2
Рис. 95. Прибор для получения пропио-
нового альдегида:
/—водяная баня; 2—круглодонная колба;
3—мешалка; 4 —-трехрогий форштосс; 5—ка-
пельная воронка; 6—шариковый холодильник;
7—холодильник Либиха; 3—алонж; 9—прием-
ник; /0—ледяная баня-
колбы кипятят 15—20 мин, чтобы отогнать следы пропионового
альдегида. Затем альдегид сушат безводным сернокислым нат-
рием. Высушенный пропионовый альдегид отгоняют при 50—55 °C
на водяной бане (прибор 2).
Выход пропионового альдегида ~ 10 г.
Пропионовый альдегид — бесцветная жидкость с характерным удушливым
запахом; растворяется в воде; смешивается со спиртом, эфиром. Мол. вес 58,08;
т. пл —81 °C; т. кип 48,8 °C; cZ*0 = 0,8066; п% = 1,3635.
Применяется как полупродукт в органическом синтезе.
Отличительная реакция
К одной капле пропионового альдегида прибавляют 10 мл
1 % раствора нитропруссида натрия, 0,5 г пиперазина — появляется
синее окрашивание.
ИЗОВАЛЕРИАНОВЫЙ АЛЬДЕГИД
Формула:
СНзх .0
)СН-СН2-(Г
сн3/ \н
191
Основная реакция!
СНзХ
3 ;СН-СН2—СН2-ОН + К2Сг2О7 + 4H2SO4 —►
СНЭ7
СНзЧ .0
—> 3 ;СН-СН2-С( + K2SO4 + Cr2(SO4)3 + 7Н3О
СН37 хн
Побочные реакции:
СНзХ Л СНзХ
)сн—сн2—+ о —> ;сн-сн2-
СНз7 СНз7 ХОН
СНз\ /СНз
;сн-сн2-С\ + но-сн2-сн2-сн(ч —>
СНз7 Х0Н 7СНз
СНзк /° /СНз
'СН— сн2—С-0—сн2—сн2—сн( + Н2о
СН87 ХСНз
Реактивы, посуда и приборы
Изоамиловый спирт..........13,2 г
(0,15 моль)
Двухромовокислый калий ... 16,5 г
(0,055 моль)
Серная кислота (rf == 1,84) . . 16 мл
Углекислый натрий
Кислый сернистокнслый натрий
Эфир '
Хлористый кальций
Колбы Вюрца (100 и 500 мл) ... 2
Воронка капельная ............ 1
Воронка делительная .......... 1
Холодильник Либиха.............1
Воронка Бюхнера................1
Колба Буизена..................1
Термометр . . . . -............1
Алонж..........................1
Приемники .................... 2
Сборка приборов
1. Перегонную колбу Вюрца емкостью 500 мл, помещенную
на родяную баню, закрывают пробкой с двумя отверстиями. В одно
из них вставляют капельную воронку, в другое — термометр. Боко-
вой отросток колбы соединяют с нисходящим холодильником с
алонжем, конец которого погружают в приемник.
2. Прибор для отсасывания (см. рис. 40).
3. Перегонную колбу на 100 мл снабжают термометром и нис-
ходящим холодильником с алонжем, который опускают в приемник.
Выполнение синтеза
В перегонную колбу прибора 1 помещают смесь, состоящую
из 16,5 г двухромовокислого калия (см. стр. 168), 160 мл воды и
16 мл концентрированной серной кислоты. Смесь нагревают на
192
водяной бане до 90 6С и осторожно, по каплям приливают из ка-
пельной воронки изоамиловый спирт, время от времени встряхивая
колбу. Так как окисление спирта — экзотермическая реакция, то
приливать спирт необходимо с такой скоростью, чтобы не происхо-
дило бурного вскипания и выбрасывания содержимого колбы. По
окончании приливания спирта колбу нагревают еще 15 мин на ки-
пящей водяной бане', при этом начинает отгоняться изовалериа-
новый альдегид.
Ввиду низкой растворимости изовалериаиового альдегида в воде скорость
его дальнейшего окисления в изовалернаиовую кислоту снижается. Поэтому нет
.необходимости пропускать углекислый газ через реакционную смесь.
Убирают баню, отбирают колбу снаружи и через асбестовую
сетку нагревают смесь до кипения на горелке, при этом отгоняется
изовалериановый альдегид с примесями.
Вследствие того, что изовалериановый альдегид имеет высо-
кую температуру кипения (92,5°C), его удаление из реакционной
смеси затруднено. Поэтому в продуктах реакции наряду с альде-
гидом и небольшим количеством непрореагировавшего спирта при-
сутствуют также изовалериановая кислота и ее сложный эфир с
изоамиловым спиртом.
Для удаления следов кислоты разделившийся на два слоя ди-
стиллят обрабатывают углекислым натрием до щелочной реакции
по лакмусу. Верхний слой, состоящий из изовалериаиового альде-
гида и непрореагировавшего спирта, отделяют при помощи дели-
тельной воронки. Затем взбалтывают изовалериановый альдегид
с равным объемом свежеполученного насыщенного раствора кис-
лого сернистокислого натрия. Выпавшие кристаллы производного
изовалериаиового альдегида отсасывают на воронке Бюхнера, про-
мывают небольшим количеством эфира, отжимают между листами
фйльтровальной бумаги и сушат в эксикаторе над хлористым
кальцием.
, Выход производного изовалериаиового альдегида ~ 15 г.
Для выделения изовалериаиового альдегида его производное
обрабатывают раствором углекислого натрия. Альдегид отделяют
при помощи делительной воронки, суша! хлористым кальцием и
перегоняют (прибор 3).
Изовалериановый альдегид — бесцветная жидкость с неприятным запахом;
плохо растворимое воде, хорошо — в спирте, эфире. Мол. вес 86,14; т. кип.
92,5 °C; т. пл. —51 °C, 0,802; /$=* 1,3902.
Отличительные реакции
1. Изовалериановый альдегид с фуксинсернистой кислотой
дает фиолетовое окрашивание.
2. К 5 мл 1—2% раствора 2,4-динитрофенилгидразина в
95%-ном спирте прибавляют несколько капель изовалериаиового
альдегида — появляются оранжево-красные иглы динитрофенил-
гидразона (т. пл. 123°C).
7 М Н Храмкина
193
БЕНЗОФЕНОН
Формула:
C6H6-C-CeHs
Основная реакция:
О
II
ЗС6Н5-СН-С6Н6 + К2Сг2О7 + 4H2SO, —>
он
—> 3CeH5-C-CeH5 + K2so4 + Cr2(SO4)3 + 7Н2О
Реактивы, посуда и приборы
Бензгидрол...................10 г
(0,06 моль)
Двухромовокислый калий ... 12 г
(0,04 моль)
Серная кислота (d = l,84) ... 6 мл
(0,1 моль)
Беизол.................... 75 мл
Углекислый калий, безводный
Колба круглодонпая трехгорлая
(200 мл)......................i
Колба Вюрца (100 мл)...........1
Меш алка........................1
Холодильник Либиха..............1
Термометр ..................... 1
Воронка делительная .......... 1
Алонж..........................1
Колба коническая...............1
Стакан химический ............ 1
Сборка приборов
1. Круглодонную трехгорлую колбу на 200 мл, снабженную
мешалкой и термометром, помещают на водяную баню.
2. Колбу Вюрца на 100 мл, помещенную на водяную баню, со-
единяют с термометром и нисходящим холодильником с алонжем,
конец которого опущен в приемник (коническую колбу).
3. К отводной трубке перегонной колбы Вюрца емкостью
100 мл подставляют маленький стакан.
Выполнение синтеза
В круглодонной трехгорлой колбе прибора 1 приготовляют
хромовую смесь последовательным внесением в колбу 12 г дву-
хромовокислого калия (см. стр. 188), 60 мл воды и 6 мл концен-
трированной серной кислоты. Смесь нагревают до 40 °C на водяной
бане и вносят через свободное отверстие порциями, при перемеши-
вании 10 г бензгидрола. После того как весь бензгидрол будет вве-
ден в реакционную смесь, содержимое колбы перемешивают 1 ч
при 40—50 °C (при этом синтезе следует строго соблюдать темпе-
ратурный режим).
Собирают прибор 2 и отгоняют бензол при 80—90 °C. Остав-
шийся в колбе Вюрца бензофенон перегоняют без холодильника,
подставив стакан к концу отводной трубки колбы.
Выход бензофенона ~ 9 г.
Свойства бензофенона см. стр. 181.
194
Отличительная реакция
Бензофенон растворяется в концентрированной серной кис-
лоте, давая желтое окрашивание.
ИЗОМАСЛЯНАЯ КИСЛОТА
Формула:
СНз\
;сн—
сн3/ /он
Основные реакции:
СН3\
3 Vh—СН2—ОН + 4КМпО4 —>
СН3/
СНзЧ ^0
—> 3 /СН—+ 4МпО2 + КОН + 4Н2О
сн3/ /ок
СНз\ .0 СНз\ хО
2 ;сн—С/ +H2SO4 —>2 /СН—С^ +K2so4
СН3/ Х0К СН3/ хон
Реактивы, посуда и приборы
Изобутиловый спирт . . 14,8 г, 18 мл Колба круглодонная (1000 мл) . . 3
(0,2 моль) Колба Вюрца (100 мл)............1
Марганцовокислый калий 42 г Колба Бунзена...................1
(0,27 моль) Воронка Бюхнера................1
Углекислый натрий ... 12 г Воронка делительная.............1
Серная кислота, 50%-ная Холодильник Либиха..............1
Эфир диэтиловый ... 50 мл Холодильник воздушный...........1
Сернокислый магний, безводный Алонж...........................1
Термометр......................1
Стаканы химические.............1
Приемники....................• . 2
Сборка приборов
1. Круглодонную колбу емкостью 1000 мл помещают в баню
с водой и льдом.
2. Прибор для отсасывания (см. рис. 40).
3. Перегонную колбу Вюрца емкостью 100 мл помещают на
водяную бащо, соединяют ее с термометром и водяным нисходя-
щим холодильником с алонжем, который погружают в приемник.
4. Перегонную колбу Вюрца, снабженную термометром, соеди-
няют с воздушным холодильником и нагревают колбу небольшим
пламенем горелки.
Выполнение синтеза
В круглодонную колбу прибора 1 вносят 18 мл изобутилового
спирта, 45 мл воды и 12 г углекислого натрия (получение изо-
масляной кислоты ведут в щелочной среде, что уменьшает
7» 195
образование побочных продуктов). Образовавшуюся смесь охлаж-
дают и при непрерывном помешивании, постепенно, небольшими
порциями *вносят в нее предварительно охлажденный раствор, со-
стоящий из 42 г марганцовокислого калия и 800 мл'воды. Колбу
при этом охлаждают, поддерживая температуру реакционной
смеси в пределах 12—15 °C. По окончании приливания марганцо-
вокислого калия реакционную смесь охлаждают до 2 °C и выдер-
живают 10—12 ч при комнатной температуре.
Конец реакции окисления определяют по обесцвечиванию
раствора марганцовокислого калия. Если обесцвечивания не про-
изошло, нужно продолжать перемешивание до тех пор, пока не
исчезнет малиновое окрашивание. Если смесь остается все же
окрашенной, то прибавляют немного этилового спирта или щаве-
левой кислоты.
Выделившийся осадок двуокиси марганца отсасывают на во-
ронке Бюхнера, а фильтрат упаривают на водяной бане до 40 мл. ,
Упаренный раствор охлаждают, переливают в делительную во-
ронку, прибавляют 50%-ную серную кислоту до кислой реакции по
конго красному и вносят 20 мл эфира. Содержимое делительной
воронки встряхивают, дают расслоиться и отделяют водный слой,
который дважды обрабатывают эфиром (по 10 мл). Соединенные
эфирные вытяжки сушат безводным сернокислым магнием.
Безводный сернокислый магний является хорошим нейтральным осушите-
лем для многих органических соединений Его можно заменить более дешевым
осушителем — безводным сернокислым натрием, однако он высушивает медленно
и ие до конца.
После высушивания собирают прибор 3 и отгоняют на водяной
бане эфир, остаток перегоняют (прибор 4), собирая фракцию, ки-
пящую при 150—158 °C.
Выход изомасляной кислоты ~ 14 г.
Изомасляная кислота — бесцветная жидкость с запахом прогорклого масла;
растворяется в воде (20 г в 100 мл воды прн 20 °C); смешивается со спиртом,"
эфиром и хлороформом. Мол. вес 88,10; т. кип. 154,7 °C; т. пл. —46,1 °C;
d™ = 0,9527; п£°= 1,3930.
Отличительная реакция
К раствору кальциевой соли изомасляной кислоты, насыщен-
ному на холоду, прибавляют равный объем ацетона. Через 1 ч
выпадают игольчатые кристаллы.
ВАЛЕРИАНОВАЯ КИСЛОТА*
Формула:
' СН3(СН2)зСООН
* Изовалериановую кислоту получают аналогичным образом из изоамилового
спирта (продолжение примечания см, на стр. 197).
Основные реакции:
ЗСН8(СН2)зСН2ОН + 4КМпО4 —>
—> ЗСНз(СН2)зС^ + 4МпО2 + КОН + 4Н2О
ХОК
2СН3(СН2)зСХ + H2SO4 —> 2СН3(СН2)з<\ + K2SO4
ХОК ОН
Реактивы, посуда и приборы
Амиловый спирт.........12 мл 10 г
(0,11 моль)
Марганцовокислый калий 24 г
(0,14 моль)
Едкий иатр.............4,3 г
Сернокислый натрий, без-
водный ..............4 г
Серная кислота, 50%-ная 18 мл
Четыреххлористый угле-
род .............. ... 12 г
Лед
Колба круглодонная шнрокогорлая
(500 мл)......................1
Колба Вюрца (100 мл)...........1
Форштосс трехрогий.............1
Мешалка........................1
Воронка капельная ............ 1
Колба Бунзена..................1
Воронка Бюхнера................1
Холодильник Либиха ........... 1
Холодильник воздушный..........1
Алонж..........................1
Термометр .................... 1
Приемники......................3
Сборка приборов
1. Круглодонную широкогорлую колбу на 500 мл закрывают
пробкой с трехрогим форштоссом, который снабжают мешалкой
и капельной воронкой.
2. Прибор для отсасывания (см. рис. 40).
3. В горло перегонной колбы на 100 мл вставляют термометр,
а боковой отросток соединяют с нисходящим холодильником Ли-
биха с алонжем, конец которого опущен в приемник. Колбу по-
мещают на водяную баню.
4. Перегонную колбу емкостью 100 мл соединяют с термомет-
ром и воздушным нисходящим холодильником.
СНзх
3 CH—СН2—СН2ОН + 4КМпО4 >
СНз/
сн3\ ^0
--> 3 ГН-СН2—+ 4МпО2 + КОН + 4Н2О
CHj/ 4>к
СНзх Ю СНзх уэ
2 CH—СН2— с' +H2SO4 > 2 'СН-СНг-С^ + K2SO4
СНз/ хж СН3/ Ч)Н
Применяемые при этом реактивы: изоамиловый спирт (22 мл); марганцово-
кислый калий (42 г); едкий иатр; сернокислый натрий; 50%-ная серная кислота;
четыреххлористый углерод, лед.
Выход изовалериановой кислоты ~ 16 г.
<97
Выполнение синтеза
В круглодонную широкогорлую колбу прибора 1 помещают
24 г марганцовокислого калия, 10 мл воды и 4,3 г едкого натра.
Содержимое колбы хорошо перемешивают и медленно, в течение
1 ч приливают из капельной воронки 12 мл амилового спирта, од-
новременно прибавляя в реакционную смесь 160 г измельченного
льда; при этом температура реакционной смеси должна поддержи-
ваться в пределах 18—24 °C. По окончании приливания амилового
спирта смесь перемешивают еще 30 мин и выдерживают 10—12 ч
при комнатной температуре.
Выделившийся осадок двуокиси марганца отсасывают на во-
ронке Бюхнера. Фильтрат упаривают на водяной бане до объема
30 мл, затем охлаждают и для выделения валериановой кислоты
из ее калиевой соли вносят 18 мл 50 %-ной серной кислоты до кис-
лой реакции по конго красному.
Жидкость переносят в делительную воронку, отделяют слой
выделившейся валериановой кислоты. Из водного слоя извлекают
три раза четыреххлористым углеродом остатки кислоты. (Осто-
рожной четыреххлористый углерод нельзя сушить металлическим
натрием ввиду опасности взрыва; четыреххлористый углерод раз-
дражает слизистые оболочки, вызывает головные боли.)
Соединенные вытяжки присоединяют к основной порции вале-
риановой кислоты и сушат над 4 г безводного сернокислого нат-
рия. Затем при 80 °C отгоняют четыреххлористый углерод (при-
бор 3), и остаток перегоняют (прибор-4), собирая валериановую
кислоту при 184—187 °C.
Выход валериановой кислоты ~ 8 г.
Валериановая кислота — бесцветная жидкость с характерным неприятным
запахом; плохо растворима в воде; хорошо растворяется в спирте, эфире, че-
тыреххлористом углероде. Мол. вес 102,14; т. пл. —35 °C; т. кип. 186,4 °C;
d = 0,939; = 1,4086.
Применяется в пищевой промышленности, в парфюмерии и для синтетиче-
ских целей.
БЕНЗОЙНАЯ КИСЛОТА
Формула:
соон
Основные реакции:
+ 2КМпО4
+ 2МпО2 + КОН 4- Н2О
КОН + НС1 —> KCl+HjO
Окисление толуола идет в щелочной среде, которую создает
образующееся во время реакции едкое кали.
Реактивы, посуда и приборы
Толуол.................2 г, или 2,5 мл Колба круглодонная (150 мл) 1
(0,02 моль) Холодильник Либиха........1
Марганцовокислый калий 3,2 г Мешалка....................1
(0,02 моль) Форштосс двурогий.........1
Соляная кислота Термометр..................1
Алоцж.....................1
Воронка Бюхнера ..........1
Колба Бунзена.............1
Приемник ................ 1
Сборка приборов
1. Круглодонную колбу емкостью 150 мл, помещенную на пе-
сочную или водяную баню, снабжают двурогим форштоссом с об-
ратным холодильником и мешалкой.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную колбу прибора 1 помещают 2,5 мл толуола
(осторожно'. вдыхание паров толуола действует на нервную систе-
му!), 3,2 г тонкоизмельченного марганцовокислого калия и 75 мл
воды. Содержимое колбы нагревают на кипящей водяной бане
(или песочной бане) в течение 3 ч при постоянном перемешивании
до обесцвечивания раствора. Если раствор после нагревания
остается окрашенным в розовый цвет, то добавляют несколько ка-
пель спирта или немного щавелевой кислоты (эти добавки оконча-
тельно восстанавливают марганцовокислый калий, и раствор обес-
цвечивается) .
По окончании реакции и охлаждении отфильтровывают выпав-
шую двуокись марганца на маленькой воронке Бюхнера и два раза
промывают небольшим количеством горячей воды. Соединенный
фильтрат выпаривают на водяной бане (или в чашке на песочной
бане*) до объема 15—20 мл. Снова отфильтровывают выпавшую
двуокись марганца и промывают один раз 5 мл горячей воды.
Фильтрат — раствор бензойнокислого калия действием разбавлен-
ной (1:1) соляной кислоты (ее добавляют до явно кислой реакции
по конго красному) переводят в свободную бензойную кислоту.
Выпавшую бензойную кислоту отсасывают на маленькой во-
ронке Бюхнера, промывают небольшим количеством холодной
воды и сушат/
Выход бензойной кислоты ~ 2 г.
Бензойная кислота — бесцветное кристаллическое вещество, трудно раство-
рима в холодной воде (0,2 г в 100 мл воды при 18 °C и 2,2 г при 75 °C),
* При работе с большими, чем в приведенном примере, количествами исход-
ных продуктов следует по окончании реакции отогнать непрореагировавший то-
луол из той же колбы, заменив обратный холодильник нисходящим.
199
хорошо растворима в хлороформе, ацетоне, четыреххлористом углероде, бензоле,
метиловом спирте Мол вес 122,12, т. пл. 122,3 °C, т. кип. 250 °C
Бензойная кислота и ее производные применяются в пищевой промышлен-
ности, производстве красителей, фармацевтических препаратов, в парфюмерии
и дли других целей.
Отличительная реакция
К 1 мл водного раствора бензойной кислоты прибавляют по
капле 0,3% раствор перекиси водорода и 1—3% раствор хлорного
железа. При погружении пробирки в кипящую воду быстро появ-
ляется красно-фиолетовое окрашивание (при окислении образуется
салициловая кислота, которая с хлорным железом дает окраску).
БЕНЗОХИНОН
Формула:
п-Бензохинон — яд, раздражает слизистые оболочки, на коже
оставляет коричневые пятна.
Реактивы, посуда и приборы
Гидрохинон...................10 г
(0,09 моль)
Бромноватокислый калий ... 5,5 г
(0,03 моль)
Серная кислота, 5 н. раствор 5 мл
Колба круглодонная (200 мл) . . . 1
Термометр.......................I
Колба Бунзена...................1
Воронка Бюхнера.................1
Сборка приборов
1. Круглодонную колбу емкостью 200 мл, помещенную на во-
дяную баню, снабжают термометром, который крепится в лапке
штатива.
2. Прибор для отсасывания (см. рис. 40).
200
Выполнение синтеза
В круглодонную колбу прибора 1 вносят 10 г гидрохинона и
100 мл воды, нагретой до 50 °C. После растворения гидрохинона
раствор охлаждают до 20 °C и медленно, небольшими порциями
прибавляют 5 мл серной кислоты, выполняющей роль катали-
затора.
В случае применения ие совсем чистого гидрохииоиа сразу же после вне-
сения серной кислоты образуется черный клейкий осадок. Для освобождения
от этого осадка жидкость фильтруют черед складчатый фильтр еще до внесения
окислителя, в данном случае бромноватокислого калия
В реакционную смесь осторожно вносят 5,5 г бромноватокис-
лого калия, нагревая колбу на водяной бане до 60 °C. Сразу же
начинается реакция с образованием зеленовато-черного осадка —
хингидрона.
При окислении гидрохинона ь п-бензохинои образуется промежуточное со-
единение молекулы гидрохинона с молекулой л-бензохинона — так называемый
хингидрон, углубление окраски которого связано с наличием в молекуле одно-
временно хиноидного и бензольного ядра.
Подогревание прекращают, так как температура самопроиз-
вольно повышается до 75 °C. Реакция окисления считается закон-
ченной, как только черный цвет реакционной массы изменится до
ярко-желтого цвета бензохинона. Реакционную смесь нагревают
при 80 °C до полного растворения бензохинона, затем охлаждают
до 0 °C. Выпавший n-бензохинон отфильтровывают на воронке
Бюхнера, промывают небольшим количеством ледяной воды и
сушат.
Выход n-бензохинона (т. пл. 116 °C) ~8г.
п-Бензохинон — золотисто-желтое кристаллическое вещество с характерным
запахом, мало растворим в воде на холоду, хорошо растворим в горячей воде,
спирте, эфире, бензоле. Мол. вес 108,09, т. пл 115,7 °C; dj°= 1,318. Хранят
n-бензохиион в банках темного стекла с притертыми пробками
Применяется для получении гидрохинона, используется как окислитель и
как дубящий агент.
Отличительная реакция
В две пробирки наливают по 3 мл лигроина, затем в одной
растворяют немного n-бензохинона, в другой — такое же количе-
ство фенола. Смешивают оба раствора и охлаждают — выпадают
красные иглы фенохинона.
АНТРАХИНРН
Формула:
201
Основная реакция:
ОСХ>сго>
о
СС)О+Нг0+Сг2°з
Окисление антрацена в антрахинон протекает очень легко при
воздействии различных окислителей (хромового ангидрида в уксус-
ной кислоте, перманганата калия и др.). Даже азотная кислота
действует на антрацен как окислитель, а не как нитрующий агент.
Реактивы, посуда и приборы
Антрацен...................2,5 г
.(0)014 моль)
Уксусная кислота, ледяная 150 мл
(2,5 моль)
Хромовый ангидрид..........10 г
(0,1 моль)
Едкий натр
Колба круглодонная (300 мл) . . . 1
Холодильник.....................1
Воронка капельная ............. 1
Форштосс двурогий...............1
Стакан химический...............1
Воронка Бюхнера.................1
Колба Бунзена...................1
Сборка приборов
1. Круглодонную колбу на 300 мл снабжают двурогим фор-
штоссом с обратным холодильником и капельной воронкой.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную колбу прибора 1 к раствору 2,5 г тонкоиз-
мельченного антрацена в ПО мл ледяной уксусной кислоты (осто-
рожно! антрацен вызывает раздражение слизистых оболочек), в
течение 1 ч при кипячении прибавляют из капельной воронки рас-
твор 10 г хромового ангидрида в 10 мл воды и 40 мл ледяной ук-
сусной кислоты. После прибавления всего количества хромового
ангидрида (окрашивание реакционной смеси в зеленый цвет ука-
зывает на окончание реакции) смесь переносят в стакан, охлаж-
дают и разбавляют ее 300 мл воды. Выдерживают 1 ч, затем от-
сасывают выпавшие кристаллы антрахинона, промывают их водой,
разбавленным раствором щелочи для нейтрализации уксусной
кислоты, затем снова водой и сушат.
Выход антрахинона ~2 г.
Если для окисления был взят чистый антрацен, то антрахинон
очищают возгонкой; если же технический антрацен, то антрахинон
нагревают до 100 °C в течение 10 мин с пятикратным (по массе)
количеством 10 %-ной серной кислоты, а затем смесь оставляют
на 12 ч во влажном месте; после выливания смеси в воду выпав-
шие кристаллы чистого антрахинона отфильтровывают, промы-
вают раствором углекислого натрия, водой и сушат,
202
Антрахинон — светло-желтое кристаллическое вещество; плохо растворим в
воде, спирте, эфире, хорошо растворим в концентрированной серной кислоте,
горячем растворе ацетата натрия, анилине, нитробензоле и горячем толуоле.
Мол. вес 208,20, т. пл 285—286 °C; т. кип 378—381 °C' d = 1,438.
Применяется для производства кубовых красителей, лаков и для других
целей.
Отличительная реакция
В пробирку помещают несколько кристалликов антрахинона
и 2—3 мл разбавленного раствора щелочи, нагревают смесь до
кипения, затем вносят немного цинковой пыли, нагревают еще
1—2 мин и охлаждают. По окончании выделения пузырьков водо-
рода появляется ярко-красное окрашивание, при встряхивании
раствор обесцвечивается.
Глава XII
РЕАКЦИИ НИТРОВАНИЯ
Нитрованием называется процесс введения нитрогруппы в мо-
лекулу органического соединения. Реакция нитрования ароматиче-
ских соединений проходит в общем легко и является одной из
важнейших реакций органической химии.
Нитросоединения ароматического ряда служат полупродук-
тами в производстве красителей и лекарственных веществ, приме-
няются также в парфюмерной промышленности. Многие нитросо-
единения ароматического ряда используются как взрывчатые ве-
щества, например тринитротолуол, тринитробензол, пикриновая
кислота.
Нитросоединения жирного ряда применяются как раствори-
тели синтетических смол, смазочных масел, красителей, нитроцел-
люлозы, как промежуточные продукты в многочисленных синтезах,
например карбоновых кислот, гидрокбиламина, оксимов, нитро-
спиртов, нитроолефинов и т. д.
Введение нитрогруппы в молекулу органического соединения
осуществляется с помощью различных нитрующих агентов.
Основными нитрующими агентами являются:
1) азотная кислота различной концентрации;
2) нитрующая смесь (смесь концентрированных азотной и
серной кислот);
3) нитраты щелочных металлов в присутствии серной кислоты;
4) окислы азота;
5) нитраты металлов в присутствии уксусного ангидрида и
уксусной кислоты;
203
6) смесь азотной и серной кислот с ледяной уксусной кисло-
той или уксусным ангидридом;
7) ацетилнитрат и бензоилнитрат;
8) эфиры азотной кислоты.
1. НИТРОВАНИЕ УГЛЕВОДОРОДОВ ЖИРНОГО РЯДА
Углеводороды жирного ряда при обычной температуре не реа-
гируют с концентрированной азотной кислотой; при нагревании
концентрированная азотная кислота окисляет их — в этом случае
происходит разрыв углеродной цепи с образованием карбоновых
кислот.
Для нитрования углеводородов жирного ряда применяют раз-
бавленную азотную кислоту, которая не обладает нитрующим
действием, а служит лишь источником двуокиси азота, являю-
щейся подлинным нитрующим агентом. Нитрование идет по сле-
дующей схеме:
HNOs (разб.)
" по-Ъч? R-no2+h2o
Эта реакция называется реакцией Коновалова, так как была
Открыта и детально изучена в 90-х гг. прошлого столетия М. И. Ко-
новаловым.
Для препаративных целей обычно используют действие ни-
трита серебра на галогенпроизводные жирного ряда. При этом
образуется незначительное количество побочного продукта (слож-
ного эфира азотистой кислоты), который легко отделяется.
2. НИТРОВАНИЕ УГЛЕВОДОРОДОВ
АРОМАТИЧЕСКОГО РЯДА
Для ароматических соединений характерна способность при
взаимодействии с азотной кислотой превращаться в нитросоеди-
нения. Нитрование ароматического соединения можно представить
следующей схемой:
* Ar—Н + HNO3 —> Ar—NO2 + Н2О
Как видно из приведенной схемы реакции, процесс нитрования
ароматических соединений азотной кислотой идет с выделением
воды, т. е. происходит снижение концентрации азотной кислоты.
Разбавленная азотная кислота является сильным окислителем, что
приводит к образованию побочных продуктов. Чтобы избежать
этого, для нитрования обычно применяют нитрующую смесь
(смесь концентрированных азотной и серной кислот).
Серная кислота способствует образованию иона нитрония
(NO*), который является истинным нитрующим агентом при ни-
тровании ароматического ядра:
HNO3+2HjSO4 ч=* NOJ +Н3О+ + 2HSQ;
нитроний- гидроксоний-
катиои нон
204
При исследовании спектра раствора HNO3 в сильных кислотах
было установлено существование иона нитрония в большой кон-
центрации. Факт, что ион нитрония действительно участвует в ре-
акции нитрования нитросмесью, доказан измерением скорости
реакции.
С разбавлением нитрующей смеси водой уменьшается концен-
трация ионов нитрония из-за диссоциации серной кислоты с обра-
зованием ионов гидроксония и бисульфат-ионов:
H2SO4 + H2O H3O+ + HSO7
Нитроний-катион взаимодействует с ароматическим углеводо-
родом, в результате образуется нитросоединение:
4- HSO4
St-комплекс
G-комплекс
Установлено, что лучшие выходы продуктов нитрования полу-
чаются при использовании 90%-ной серной кислоты. Для оценки
места вступления нитрогруппы в ароматическое ядро используют
приближенные правила замещения:
1) заместители I рода (—ОН, —NH2, —NHR, —N = N—,
—СН2С1, —CH2R, —СНз, F, Cl, Вг, —I) направляют нитрогруппу
преимущественно в орто- и пара-положение и облегчают (кроме
галогенов) ее введение в ядро;
2) заместители II рода (—SO3H, —NO2, —СООН, —COOR,
+ +
—CN, —СС13, —NH3, —NH2R) направляют нитрогруппу преиму-
щественно в мета-положение и затрудняют ее введение в ядро.
Так, вторая нитрогруппа при введении в ароматическое ядро
становится в мета-положение к уже имеющейся. При введении
второй нитрогруппы в ароматическое ядро требуются более жест-
кие условия — повышение температуры и увеличение концентра-
ций кислот.
Фенол, толуол нитруются значительно легче бензола, так как
содержат заместители I рода (—ОН, —СН3), которые облегчают
вступление нитрогруппы в ядро. Так, нитрование толуола идет
при 20—30°C, а нитрование фенола можно осуществить разбав-
ленной азотной кислотой в интервале 0—20 °C. Характер замести-
телей, имеющихся в ядре бензола, влияет не только на условия
205
нитрования и место вступления второго заместителя, ио и на ско-
рость реакции. Заместители —СН3, —ОСН3, —OC2IU, —ОН (груп-
пы расположены в порядке возрастания их активною воздей-
ствия) ускоряют процесс нитрования, а заместители —СООН,
—SO3H, —NO2 замедляют его. Например, толуол нитруется в
24 раза быстрее (благодаря наличию заместителя —СН3), а нит-
робензол в 105 раз медленнее (из-за наличия заместителя —NO2),
чем бензол.
3. ПРИМЕРЫ СИНТЕЗОВ
НИТРОМЕТАН
Формула:
Основные реакции:
СНз—no2
2С1СН2СООН + Na2CO3 —> 2ClCH2COONa + СО2 + Н2О
ClCH2COONa + NaNO2 —> NO2CH2COONa + NaCl
NO2CH2COONa + H2O —> CH3NO2 + NaHCO3
Побочная реакция:
ClCH2COONa + NaHCO3 —> HOCH2COONa + NaCl + CO2
Нитрометан — сильный яд!
Реактивы, посуда и приборы
Монохлоруксусная кислота . . 18,9 г
(0,2 моль)
Азотистокислый натрий .... 14 г
(0,2 моль)
Углекислый натрий..........11 г
Хлористый кальций
Колба перегонная (200 мл) . . . . 1
Колба перегонная (50 мл).........1
Холодильник .................. 1
Колба коническая...............1
Ворон'ка делительная...........1
Термометр......................1
Алонж..........................1
Приемник.......................1
Сборка приборов
Реакционную колбу на 200 мл соединяют с термометром, ша-
рик которого должен находиться в жидкости, и с хорошо дей-
ствующим нисходящим холодильником, снабженным алонжем.
Приемником служит коническая колба, охлаждаемая водой.
Выполнение синтеза
В реакционную колбу емкостью 200 мл помещают 18,9 г моно
хлоруксусной кислоты {осторожно! монохлоруксусная кислота вы-
зывает ожоги кожи!), 50 мл холодной воды и постепенно, тща-
тельно перемешивая смесь стеклянной лопаточкой, нейтрализуют
206
11 г углекислого натрия до щелочной реакции (проба с фенол-
фталеином). При нейтрализации температура не должна превы-
шать 20°C. Затем в реакционную колбу приливают раствор 14 г
азотистокислого натрия в 40 мл воды, собирают прибор и мед-
ленно нагревают колбу слабым пламенем на сетке.
При 80—85 °C, как только начинают выделяться пузырьки
углекислого газа, горелку удаляют. При этой температуре реакция
продолжается без внешнего подогрева, и образующийся нитроме-
тан отгоняется с водой в виде тяжелых маслянистых капель.
Нитрометан начинает отгоняться при температуре около 90 °C.
При этом на 5 мл нитрометана выделяется 15 мл воды.
По мере отгонки содержимое колбы буреет и температура по-
вышается до 100 °C. Когда реакция в основном закончится, реак-
ционную смесь осторожно нагревают в течение 10 мин до 110 °C.
Отгонку прекращают, когда в приемник начнет переходить вода
без масла. Полученный дистиллят переносят в делительную во-
ронку и отделяют нижний слой нитрометана от верхнего водного
слоя.
Нитрометан сушат хлористым кальцием и перегоняют из ма-
ленькой перегонной колбы при 98—102 °C.
Выход нитрометана ~ 4 г.
/
Чистый нитрометан — бесцветная жидкость со своеобразным запахом, на-
поминающим запах горького миндаля. Мол. вес 61,042; т. кип. 102 °C при
760 мм рт. ст.; т. пл. -29,0 °C; df = 1,13816; = 1,38188.
Применяется как растворитель, например для эфироцеллюлозных лаков, ви-
ниловых смол; как горючее для реактивных двигателей; в качестве добавки
к дизельному топливу.
Отличительная реакция
Несколько капель нитрометана смешивают с 1 мл воды и 2—
3 каплями концентрированного раствора щелочи. Образовавшуюся
жидкость-охлаждают холодной водой. Затем вносят 1 мл раствора
азотистонатриевой соли и по каплям разбавленную серную кис-
лоту до тех пор, пока раствор, окрасившийся в красный цвет, не
примет желтоватый оттенок. Потом снова добавляют щелочь до
появления ярко-красного окрашивания. Эти реакции связаны с
таутомерией нитросоединений жирного ряда:
щелочь
СН3—\О2 CH2^NO2H
Ациформа
В щелочной среде образуется соль CH;j=NOoNa, которая мо-
жет реагировать с HNO2(NaNO2 + H2SO4):
CH2=NO2Na + HONO —> Н2О + ON-CH=NO2Na
С азотистой кислотой реагируют первичное и вторичное нитро-
роединения, третичное — не реагирует.
307
НИТРОБЕНЗОЛ
Формула:
Основная реакция (см. стр. 205):
+ HNO3
H2SO4
----->.
Реактивы, посуда и приборы
Бензол..................18 мл, или 15,6 г
(0,2 моль)
Азотная кислота (d— 1,4) 28 г, 20 мл
(0,28 моль)
Серная кислота (d=l,84) 46 г, 25 мл
(0,45 моль)
Углекислый натрий
Хлористый кальций
Колба круглодонная (250 мл) 1
Колба коническая (50 мл) . . 1
Колба Вюрца (100 мл) . . . . 1
Мешалка....................1
Форштосс трехрогий.........1
Воронка делительная........1
Холодильник воздушный . . . 1
Термометр ................ 1
Приемники...................2
Сборка приборов
1. Круглодонную колбу емкостью 250 мл снабжают трехрогим
форштоссом, одно горло которого закрывают пробкой с термомет-
ром, доходящим почти до дна колбы, во второе вставляют обрат-
ный воздушный холодильник (препятствует улетучиванию бензо-
ла) и в третье — механическую мешалку.
2. Собирают прибор для перегонки нитробензола, состоящий
из колбы Вюрца на 100 мл, термометра и обращенного вниз воз-
душного холодильника (нитробензол лучше перегонять в вакууме).
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу \ В реак-
ционную колбу прибора 1 вносят 20 мл концентрированной азот-
ной кислоты, к которой осторожно, при наружном охлаждении хо-
лодной водой и перемешивании, приливают 25 мл концентрирован-
ной серной кислоты (защитные очки!).
К охлажденной до 15 °C смеси кислот при перемешивании при-
бавляют через верхнее отверстие холодильника бензол порциями
по 2—3 мл. Нужно следить за тем, чтобы температура смеси не
поднималась выше 50 °C, так как может образоваться большое ко-
личество динитробензола. Температуру регулируют скоростью при-
ливания бензола, а в необходимых случаях колбу время от вре-
мени охлаждают, Погружая в баню с холодной водой и прекращая
приливание бензола. Часто при внесении новой порции бензола
появляется бурая окраска (окиси азота). Скорость внесения бен-
зола должна быть такой, чтобы не выделялись бурые пары окис-
лов азота.
Реакционная смесь является гетерогенной системой, поэтому
процесс нитрования практически происходит только на границе
двух фаз — кислотной и бензольной. Для успешного проведения
реакции необходимо тщательное перемешивание, при этом проис-
ходит образование эмульсии, которая обеспечивает тесное сопри-
косновение реагентов.
После добавления всего количества бензола колбу погружают
в водяную баню, нагретую не более чем до 60 °C, и ведут реакцию
при этой температуре 30—45 мин, продолжая перемешивание. За-
тем реакционную смесь охлаждают и переливают в делительную
воронку. Дают жидкости расслоиться и тщательно отделяют ниж-
ний кислотный слой от верхнего слоя — нитробензола (смесь кис-
лот нельзя непосредственно выливать в канализацию, предвари-
тельно ее необходимо нейтрализовать известью). Нитробензол про-
мывают водой, встряхивая не слишком сильно в делительной
воронке, так чтобы не образовалась стойкая эмульсия Если эмуль-
сия все же образовалась, то вносят каплю спирта, изменяя таким
образом поверхностное натяжение.
После,отстаивания нижний слой нитробензола спускают через
кран, а водный слой сливают через верхнее отверстие воронки.
Затем нитробензол снова вносят в делительную воронку и для
очистки от следов кислот промывают разбавленным раствором
углекислого натрия до прекращения выделения двуокиси углерода
(верхнее отверстие делительной воронки не закрывать!). После
промывки спускают нижний слой нитробензола в сухую кониче-
скую колбу емкостью 50 мл и сушат над безводным гранулирован-
ным хлористым кальцием.
Если на дне колбы образуется слой водного раствора хлори-
стого кальция, то нитробензол осторожно сливают в сухую колбу,
снова прибавляют к нему гранулированный хлористый кальций и
высушивают по описанному выше способу.
Для ускорения процесса сушки колбу закрывают пробкой со
вставленной в нее стеклянной трубкой (в качестве обратного воз-
душного холодильника) и нагревают на водяной бане в течение
30 мин до тех пор, пока жидкость не станет совсем прозрачной.
Время от времени коническую колбу встряхивают.
Высушенный нитробензол переливают в колбу Вюрца на
100 мл Собирают прибор 2. Колбу Вюрца нагревают на воздуш-
ной бане или на асбестовой сетке. Сначала отгоняют небольшое
количество непрореагировавшего бензола, затем при 207—211 °C
собирают нитробензол.
Выход нитробензола ~ 21 г.
Нитробензол и другие нитросоединения нельзя перегонять до-
суха, так как ди- и полинитросоединения (в данном случае динит-
робензол) могут самовоспламеняться и даже взрываться. Пере-
гонку нужно прекращать, когда в колбе останется еще немного
жидкости или температура кипения достигнет 214 °C.
Нитробензол (мирбановос масло)—слегка желтоватая маслянистая жид-
кость с запахом горького миндаля. Мол вес 123,11; т. кип. 210,9 °C; d%g = 1,2055;
п“ = 1,5525. Растворим в спирте, бензоле, эфире; в воде почти не растворим.
Нитробензол является ядом центральной нервной системы, нарушает обмен ве-
ществ, вызывая заболевания печени Длительное вдыхание паров нитробензола
вызывает головную боль, а попадание его на кожу — ожоги. Пораженное нитро-
бензолом место кожи нужно потереть спиртом, а затем обмыть водой с мылом.
Применяется для получения анилина, бензидина, л-динитробензола, л-ди-
нитрохлорбензола, в производстве красителей, в качестве растворителя и для
других целей.
Отличительная реакция
Качественно нитробензол открывают восстановлением до ани-
лина и окислением последнего разбавленным раствором хлорной
извести или гипохлорита натрия. Вследствие окисления образовав-
шегося анилина возникает интенсивная фиолетовая окраска.
о- И «-НИТРОТОЛУОЛ
Толуол нитруется легче бензола, причем согласно правилу за-
мещения образуется главным образом смесь о- и п-нитротолуолов.
Реактивы, посуда и приборы
Толуол.................23 г, или 27 мл
(0,24 моль)
Азотная кислота (d = 1,4) 18 мл, 25 г
(0,25 моль)
Серная кислота (d = 1,84) 21,5 мл
(0,39 моль)
Хлористый кальций, гранулированный
Колбы круглодонные (100 и
200 мл)...................2
Воронка капельная ............ 1
Воронка делительная .......... 1
Колба коническая............1
Термометр на 360 °C.........1
Колба Вюрца (100 мл) . . . . 1
Холодильник воздушный . . . 1
Стакан химический (100 мл) . . 1
Прибор для отсасывания . . . 1
Приемники...................1
Холодильник Либиха..........3
Сборка приборов
1. Круглодонную колбу емкостью 200 мл, снабженную термо-
метром на 360 °C, доходящим почти до дна колбы, и капельной
воронкой, помещают в водяную баню.
2. Собирают прибор для перегонки непрореагировавшего то-
луола. Колбу Вюрца на 100 мл соединяют с нисходящим водяным
холодильником и термометром.
3. Прибор для отсасывания (см. рис. 40).
4. Прибор для перегонки о- и n-нитротолуолов. Колбу Вюрца
на 100 мл соединяют с воздушным нисходящим холодильником и
термометром на 360 °C.
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу\ Нитрую-
щую смесь готовят в круглодонной колбе на 100 мл. В колбу вно-
сят 18 мл азотной кислоты и постепенно, небольшими порциями,
при встряхивании приливают 21,5 мл концентрированной серной
кислоты (защитные очки!).
В круглодонную колбу прибора 1 помещают 27 мл чистого то-
луола, к которому порциями по 1—2 мл через верх воздушного
холодильника при перемешивании приливают нитрующую смесь.
Приливание смеси ведут с такой скоростью, чтобы температура
реакционной смеси не превышала 60 °C. Если же температура под-
нимется выше 60 °C, то колбу охлаждают холодной водой, чтобы
избежать образования побочных продуктов — полинитропроиз-
водных.
После добавления всего количества нитрующей смеси колбу
нагревают на водяной бане при 60 °C в течение 30 мин. Затем ре-
акционную смесь охлаждают и переносят в делительную'воронку.
Дают жидкости расслоиться и отделяют нижний слой, состоящий
из серной и азотной кислот. Верхний слой промывают водой. После
отстаивания нижний маслянистый слой сливают в сухую кониче-
скую колбу и сушат гранулированным хлористым кальцием до тех
пор, пока жидкость не станет прозрачной.
Собирают прибор 2 и перегоняют жидкость, нагревая ее на
сетке до 150 °C; при этом отгоняется непрореагировавший толуол
211
(в результате нитрования при низкой температуре часть толуола
не используется в процессе). Остаток после перегонки переносят
в стакан на 100 мл и выдерживают 10—12 ч в охладительной
смеси (лед + соль); при этом выделяются кристаллы /г-нитрото-
луола, которые отсасывают на воронке Бюхнера и перегоняют
(прибор 4) в интервале 232—238 °C. (Нитротолуол нельзя перего-
нять досуха, иначе может произойти взрыв!)
Масляный слой перегоняют (прибор тот же) в пределах 216—
222 °C.
Тщательное разделение о- и n-нитротолуолов лучше всего до-
стигается фракционной перегонкой в вакууме.
Выход о- и и-нитротолуолов 30—31 г.
Наряду с о- и n-нитротолуолами образуется около 4% .м-ни-
тротолуола.
о-Нитротолуол — светло-желтая жидкость; хорошо растворим в спирте,
эфире Мол. вес 137,14, т кип 222,3 °C; п™ = 1,5472.
/г-Нитротолуол — бесцветные кристаллы, хорошо растворим в спирте и
эфире, т кип 238 °C, т пл 57 °C
Применяются в производстве красителей, промежуточных продуктов и в
аналитической практике Нитротолуолы ядовиты, окисляют гемоглобин в мета-
гемоглобин.
Отличительная реакция
В пробирку вносят 2 мл нитротолуола и 5 мл петролейного
эфира. Затем при энергичном взбалтывании всыпают 0,5 г порош-
кообразного едкого кали. Появляется желто-коричневое окраши-
вание.
о- И п-НИТРОФЕНОЛ Формулы: Основная реакция: ОН 6 н он yN0’ no2 он ^^/NO2 НМОз((/=1,П) no2
212
Побочная реакция:
он он
no2
он
no2
Фенол нитруется очень легко даже на холоду разбавленной
азотной кислотой (d=l,ll), образуя о- и n-нитрофенол. Вслед-
ствие того что в разбавленной азотной кислоте отсутствует ион ни-
трония, нитрование фенолов идет через предварительное образо-
вание нитрозофенола с последующим окислением его в нитрофе-
нол азотной кислотой.
Реактивы, посуда и приборы
Фенол......................28,2 г
(0,3 моль)
Азотная кислота (d = 1,11) . . 185 мл
(0,6 моль)
Этиловый спирт
Активированный уголь ,
Соляная кислота, 2%-ная
Колба круглодонная (300 мл) . . . '1
Колба коническая (150 мл) . . . . 1
Термометр на 100 °C............1
Прибор для перегонки с водяным
паром..........................1
Прибор для отсасывания.........1
Прибор для перекристаллизации . . 1
Сборка приборов
1. Прибор для перегонки с водяным паром (см. рис. 48).
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В коническую колбу емкостью 150 мл помещают 28,2 г фе-
нола, 5 мл воды и нагревают содержимое колбы до плавления.
В круглодонную колбу емкостью 300 мл вносят 185 мл азотной
кислоты, к которой постепенно, при постоянном встряхивании
и перемешивании, прибавляют расплавленный фенол. При этом
нужно следить за тем, чтобы температура реакционной смеси
не превышала 20 °C, иначе могут образоваться ди- и тринитрофе-
нолы.
После введения всего количества фенола темноокрашенную
смесь выдерживают 2—3 ч в ледяной воде, часто и сильно встря-
хивая колбу. Затем выливают смесь в двойной объем воды и вы-
держивают до тех пор, пока маслообразный частично осмелив-
шийся продукт реакции не отделится от водяного слоя и не осядет
на дно колбы. Водный слой осторожно сливают, а остаток^два-три
раза промывают водой для удаления следов азотной кислоты.
Применение в качестве нитрующего агента разбавленной азот-
ной кислоты приводит к образованию смолоподобных побочных
продуктов окисления и полимеризации, снижающих выход
213
продукта реакции. Поэтому лучше нитровать фенол смесью ни-
трата натрия или калия с копцетрировапной серной кислотой
в водном растворе.
После промывки маслянистый слой переносят в колбу на
500 мл для перегонки с водяным паром. При этом отгоняется
только о-нитрофенол в виде желтого быстро кристаллизирующе-
гося масла, а и-нитрофенол остается в колбе, так как он не пере-
гоняется с водяным паром. Перегонка считается законченной, если
проба (несколько миллилитров дистиллята) при охлаждении не
выделяет кристаллов.
Приемник необходимо охлаждать ледяной водой. Если о-нит-
рофенол начнет кристаллизоваться в холодильнике и забьет его
внутреннюю трубку,4 то на некоторое время прекращают подачу
воды в холодильник. Горячий конденсат расплавит застывший
продукт, который стечет в приемник. После этого нужно медленно
включить воду в холодильник, иначе последний может лопнуть.
Выпавшие кристаллы о-нитрофенола отсасывают на воронке
Бюхнера, отжимают между листами фильтровальной бумаги и
высушивают на воздухе (о-нитрофенол нельзя сушить в сушиль-
ном шкафу, так как он обладает большой летучестью).
Если о-нитрофенол получается недостаточно чистым, что можно определить
по темному окрашиванию и несоответствующей температуре плавления, то его
следует повторно перегонять с водяным паром или перекристаллизовать из раз-
бавленного спирта.
Перекристаллизацию из разбавленного спирта ведут следующим образом.
Растворяют о-нитрофенол в небольшом количестве кипящего спирта в колбе
с обратным воздушным холодильником Затем осторожно, при перемешивании,
по каплям добавляют горячей воды до тех пор, пока не появится слабое по-
мутнение. Снова вносят несколько капель горячего спирта до исчезновения мути
и быстро охлаждают раствор. Выпавшие чистые кристаллы-о-нитрофенола от-
фильтровывают на воронке Бюхнера и сушат между листами фильтровальной
бумаги.
n-Нитрофенол, оставшийся в колбе после отгонки о-нитрофе-
нола с водяным паром, охлаждают, отделяют от воды, переносят в
колбу с 350 мл 2 н. раствора едкого натра, добавляют тонко рас-
терый активированный уголь, кипятят и фильтруют. Фильтрат
упаривают на водяной бане до тех пор, пока капля раствора не
будет застывать при охлаждении, добавляют к нему 30 мл
30%-ного раствора едкого натра и охлаждают. Выделившийся
и-нитрофенолят натрия отсасывают, промывают на воронке не-
сколько раз большими количествами 10%-ного раствора едкого
натра.
Полученный препарат переносят в стакан и разлагают
10 %-ной соляной кислотой при нагревании. Выделившийся масло-
образный n-нитрофенол застывает при охлаждении. Его отделяют
и перекристаллизовывают из горячей 2%-ной соляной кислоты.
Образуются бесцветные иглы.
Выход о-нитрофенола ~9 г, n-нитрофенола ~4г.
о-Нитрофенол — желтое кристаллическое вещество с острым ароматиче-
ским запахом; мало растворим в холодной воде; хорошо растворим в горячей
2(4
воде, спирте, эфире Мол. вес 139,11; т пл 45 °C; т кип 217°С; d$°= 1,2712
Соли о-нитрофенола имеют оранжево-красную окраску
«-Нитрофенол — бесцветные кристаллы, без запаха; хорошо растворим в
горячей воде, спирте и эфире. Т пл. 114 °C; т. кип 279 °C (с разложением)
d2/1 — 1,480. Соли ^«-нитрофенола имеют желтую окраску. Нитрофенолы приме-
няются в производстве сернистых красителей, взрывчатых веществ, инсектици-
дов и гербицидов. «-Нитрофенол применяется также в качестве индикатора при
колориметрическом определении pH.
Отличительная реакция
С хлорным железом n-нитрофенол дает, в отличие от о-нитро-
фенола, красно-фиолетовое окрашивание.
а-НИТРОНАФТАЛИН
Формула:
Основная реакция:
+ Н2о
Углеводороды ряда нафталина нитруются легче, чем углеводо-
роды ряда бензола. При нитровании нафталина нитрогруппа
вступает преимущественно в а-положение (p-нитрофенол обра-
зуется в очень небольших количествах).
Реактивы, посуда и приборы
Нафталин....................16,6 г Стаканы химические (200 и
(0,13 моль) 500 мл).......................2
Азотная кислота (rf = 1,4) . . 25 г, 18 мл Термометр....................1
(0,25 моль) Прибор для отсасывания , . . 1
Серная кислота (d = 1,84) . . 14,3 мд
Спирт
215
Сборка приборов
1. Стакан на 200 мл, помещенный в водяную баню с холод-
ной водой, снабжают термометром, доходящим почти до дна
стакана.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В стакан емкостью 200 мл помещают 18 мл азотной кислоты
и при охлаждении и перемешивании осторожно добавляют 14,3 мл
серной кислоты. Затем к смеси кислот небольшими, порциями, при
перемешивании вносят 16,6 г тонко растертого нафталина. Добав-
ление последнего ведут с такой скоростью, чтобы температура
реакционной массы не/превышала 50 °C, в случае необходимости
стакан охлаждают проточной водой.
1. Так как нафталин нитруется при температуре, лежащей ниже темпера-
туры его плавления, перед реакцией необходимо тщательно растереть его; в
противном случае выход продукта окажется пониженным
2. При несоблюдении температурного режима наряду с а-нитронафталином
могут получиться в значительном количестве 1,5- и 1,8-динитронафталииы.
После добавления всего количества нафталина реакционную
массу-выдерживают в течение 1 ч при 60 °C, постоянно перемеши-
вая содержимое стакана. Затем реакционную массу выливают в
стакан на 500 мл, содержащий 200 мл холодной воды; при этом
а-нитронафталин застывает в виде лепешки, плавающей на по-
верхности раствора. Водно-кислотный слой сливают, а сырой а-ни-
тронафталин кипятят несколько раз по 15 мин в стакане с 100 мл
воды. После каждого кипячения воду сливают. Продукт кипятят
до тех пор, пока жидкость не перестанет показывать кислую реак-
цию. Расплавленный а-нитронафталин при энергичном перемеши-
вании выливают тонкой струей в стакан с 200 мл холодной воды,
в которой он застывает в виде красновато-желтых шариков.
Осадок отфильтровывают на воронке Бюхнера, отжимают ме-
жду листами фильтровальной бумаги и сушат на воздухе. Для по-
лучения чистого продукта его перекристаллизовывают из разбав-
ленного спирта. Перекристаллизацию а-нитронафталина из спирта
осуществляют так же, как перекристаллизацию о-нитрофенола
(см. стр.'214). '
Выход а-нитронафталина ~20 г.
а-Нитройафталин — кристаллы в виде желтых игл, нерастворим в воде,
хорошо растворяется в спирте, эфире, сероуглероде Мол вес. 173,17; т. пл.
61,5 °C, т кип 304 °C
Применяется в производстве красителей, для получения нафтиламина.
Отличительная реакция
а-Нитронафталин с диметиланилином дает кровавооранжевое
окрашивание.
216
Глава XIII
РЕАКЦИИ АМИНИРОВАНИЯ
Амины можно рассматривать как продукты замещения орга-
ническими радикалами одного, двух или трех атомов водорода в
аммиаке. Соответственно различают амины первичные R-—NH2,
R\ R\
вторичные ^NH и третичные R'—N. По характеру радикалов,
R'/ R"/
связанных с атомом азота, амины бывают жирного, алицикличе-
ского, ароматического, гетероциклического и смешанного рядов.
Амины обладают основными свойствами, т. е. способны давать
с кислотами соли Даже плохо растворимые в воде амины, основ-
ность которых нельзя определить при помощи индикатора, обра-
зуют соли с минеральными кислотами Поэтому их можно отли-
чить от нейтральных азотсодержащих веществ, например амидов,
нитрилов, при помощи простой пробы с кислотой. Амины пред-
ставляют собой практически очень важный класс соединений, так
как являются полупродуктами в производстве красителей, многих
лекарственных веществ (сульфаниламидные препараты, п-амино-
салициловая кислота — ПАСК), высокомолекулярных соединений
(поливинилкарбазол) и др.
1. ПОЛУЧЕНИЕ АМИНОВ ЖИРНОГО РЯДА
Один из основных методов синтеза аминов жирного ряда —
реакция Гофмана — заключается во взаимодействий галогенпро-
изводных с аммиаком, приводящем одновременно к образованию
смеси аминов и четвертичной аммониевой соли. Например, иоди-
стый метил дает смесь солей моно-, ди- и триметиламина и тетра-
метиламмония:
CH3L+NH3 —> (СН3-NH3JI
сн31 + [сн3—nh3]i + nh3 —>
ГСН34
;nh2
-CH3Z
I 4- NHJ
CH3I +
'CH3X
>н4
,CH3Z
I + NH3
-СНзХ -
СНз-NH
-СН3/ -
I + NHJ
-СНзХ - СНз -
сн31 + СНз—NH I + NH3 —> СНз-N- СН3 I + NHJ
-СНз/ - СНз -
21?
Современный промышленный метод получения первичных,
вторичных и третичных аминов жирного ряда заключается во
взаимодействии спиртов с аммиаком над окисью алюминия. Пер-
вичные амины могут быть получены также при обработке амидов
кислот бромом и щелочью.
Иногда первичные амины получают также восстановлением
нитрозосоединений, нитрилов, оксимов, азидов, гидразонов.
Для распознавания и разделения аминов жирного ряда при-
меняют азотистую кислоту. При действии последней на первичные
амины жирного ряда выделяется азот и образуется соответствую-
щий спирт. Вторичные жирные амины с азотистой кислотой дают
нитрозамины (R2NNO), третичные амины при обычной темпера-
туре не изменяются.
2. ПОЛУЧЕНИЕ АМИНОВ АРОМАТИЧЕСКОГО РЯДА
Многие ароматические амины получают восстановлением со-
ответствующих нитросоединений атомарным водородом (в момент
выделения):
[Н]
Аг—NO2 ---> Ar—NH2 + 2H2O
Впервые реакция восстановления нитробензола в анилин была
изучена Н. Н. Зининым в 1841 г.; в качестве восстановителя он
использовал сернистый аммоний:
C6H5-NO2 + 3(NH4)2S —-> C6H5-NH2 + 6NH3 + 3S + 2H2O
Это открытие имело исключительное значение в развитии ор-
ганической химии, особенно в области производства красителей,
лекарств и фотореактивов.
В настоящее время в качестве восстановителей используют
железо и соляную иди серную кислоту, олово и соляную кислоту,
сероводород и сернистые щелочи, кислые сернистокислые соли,
каталитически возбужденный водород. Кроме того, восстановле-
ние можно осуществить электролитическим методом.
В зависимости от характера среды в результате восстановле-
ния можно получить различные продукты. Ароматические амины
получаются обычно как конечные продукты в кислой среде. Боль-
шое промышленное значение приобрел способ восстановления ни-
тробензола водородом в момент выделения в кислой среде, напри-
мер железом (оловом, цинком) и соляной кислотой:
С6Н5—NO2 + 3Fe + 6НС1 —> CeH5-NH2 + 3FeCl2 + 2H2O
Практически для проведения реакции берут значительно
меньше кислоты, чем требуется по уравнению. Это объясняется
тем, что наряду с основной реакцией происходит гидролиз хлори-
стого железа с образованием соляной кислоты и гидроокиси же-
218
леза (II) Ге(ОН)г легко восстанавливает нитробензол в апидин,
окисляясь при этом в Fe(OH)3.
Fe+2HC1 —> FeCl2 + 2Н
FeCl2 + 2Н2О Fe (ОН)2 + 2НС1
C6H5NO2 + 6Fe (ОН)2 + 4Н2О —> C6HBNH2 + 6Fe (ОН)3
В реакционной среде может образоваться и смешанный оки-
сел железа Fe3O4.
Таким образом, роль кислоты сводится к инициированию ре-
акции. Суммарно реакцию можно записать так:
НС1
C6H5NO2 + 2Fe +4Н2О -----> C6H5NH2 + 2Fe(OH)3 или
HCI
4C6H5NO2 + 9Fe + 4Н2О ----> C6H5NH2 + 3Fe3O4
Анилин образует соль с соляной кислотой, которую по оконча-
нии восстановления разлагают прибавлением углекислого натрия
или щелочи:
[С6Н5—NH3]+CF + NaOH —> СВН5—NH2 + NaCl + Н2О
В качестве промежуточных продуктов образуются нитрозобен-
зол и фенилгидроксиламин.•
При проведении реакции в кислой среде промежуточные продукты не
удается выделить, в нейтральной среде, как, например, при действии цинка в
присутствии хлористого аммония получают фенилгидроксиламин, который при
нагревании изомеризуется в /г-аминофенол
В щелочной среде процесс идет более сложно Промежуточные продукты —
нитробензол и фенилгидроксиламин — взаимодействуют с образованием азо-
ксибензола (азоксисоединение), затем азосоединения (азобензол), гидразосоеди-
нения (гидразобензол) и наконец амина:
+2Н +2Н
С6НВ—N=N—СВНВ > С6НВ—N=N—-С6Н5 -------►
~ Н2°
О
Азоксибензол Азобензол
+ 2Н
—> C6HB-NH-NH-C6H5 ---------> 2СВН5—NH2
Гидразобензол Анилин
Восстановление оловом не применяется в промышленных усло-
виях из-за высокой стоимости олова, однако в лабораторной
практике этот метод имеет большое распространение, так как
олово обладает сильным восстанавливающим действием. Суммар-
ное уравнение процесса восстановления нитробензола оловом в
кислой среде:
2C6H5NO2 + 3Sn + 121IC1 —> 2CeHBNH2 + 3SnCl4 + 4Н2О
Ароматические амины чаще всего получают из галогенпр'оиз-
водных ароматического ряда и аммиака:
давление,
катализатор
СВН5С1 + 2NH3 --------C6H5NH2 + NH4C1
Вследствие малой подвижности атома галогена, связанного с
ароматическим ядром, реакцию приходится вести в автоклавах
при высоких давлениях и температурах в присутствии катализа-
торов (меди и ее соединений).
Хороший способ получения чистых вторичных жирных аминов
состоит в нитрозировании диалкиланилинов с последующим гидро-
лизом продукта реакции: •
CeH5NR2 + HNO2 —> n-ONCeH4NR2 —> n-ON'CeH4OH + RaNH
Основные свойства ароматических аминов выражены значи-
тельно слабее, чем у аминов жирного ряда. Это объясняется влия-
нием фенильной группы. Почти все ароматические амины нестой-
ки, если они не абсолютно чисты. Они окрашиваются в серый,
фиолетовый или черный цвет, по-видимому, в результате окис-
ления.
Очистку ароматических аминов можно провести следующим
образом. Растворяют амин при 50 °C в разбавленной соляной кис-
лоте. Добавляют 10% (от массы амина) хлористого олова и про-
пускают сероводород до полного осаждения олова. Для осажде-
ния хлопьевидного осадка сернистого олова в смесь добавляют
небольшое количество хлористого натрия. Сернистые соединения
отфильтровывают, фильтрат кипятят для удаления сероводорода
и подщелачивают щелочью. Если амин труднорастворим, то его
фильтруют; если легкорастворим или жидкий, то его извлекают
эфиром.
Очищенные таким образом ароматические амины могут хра-
ниться длительное время без изменения.
3. ПРИМЕРЫ СИНТЕЗОВ
МЕТИЛАМИН
Формула:
CH3-NH2
Основные реакции:
CH3-CO-NH2 + Br2 + 4КОН —> CHs-NH2 + К2СО3 + 2КВг + 2Н2О
CHS—NH2 + HC1 —>
СН3—NH2. НС1 или
Солянокислый
метиламин
[CH3NH3]+Cr
Хлористой
метиламмоний
Реактивы, посуда и приборы
Ацетамид.....................15 г
(0,25 моль)
Бром.........................13 мл
(0,26 моль)
Едкое кали...................65 г
Соляная кислота (</=1,19) . . 40 мл
Спирт абсолютный
Колба круглодонная, двугорлая
(500 мл).......................1
Колба Вюрца (150 мл)............1
Воронка капельная ............. 1
Холодильник воздушный...........1
Термометр.......................I
Прибор для перегонки с водяным
паром..........................1
Колба Бунзена.................. 1
Воронка Бюхнера.................1
Чашка фарфоровая................1
220
Сборка приборов
1. Круглодонную двугорлую колбу емкостью 500 мл погру-
жают в водяную баню, снабжают капельной воронкой и обратным
воздушным холодильником. В лапке штатива крепят термометр,
конец которого опускают в водяную баню.
2. Прибор для перегонки с водяным паром (см. рис. 48).
К холодильнику присоединяют алонж, конец которого погружен на
1 см в находящуюся в приемнике соляную кислоту.
3. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
В круглодонной колбе прибора 1 смешивают 15 г ацетамида
и 13 мл брома (см. стр. И). Встряхивая смесь и хорошо охлаж-
дая ее в воде, приливают понемногу раствор 25 г едкого кали в
175 мл воды до тех пор, пока красно-бурая окраска раствора не
превратится в светло-желтую, т. е. до исчезновения окраски брома.
Собирают прибор 1. Полученный раствор при помощи капель-
ной воронки медленно приливают к раствору 40 г едкого кали в
75 мл воды, который находится в колбе на 500 мл. Затем реак-
ционную смесь нагревают на водяной бане при 70—75 °C, не до-
пуская перегрева. Нагревают смесь до тех пор, пока она не обес-
цветится (15—30 мин).
Образовавшийся метиламин перегоняют с водяным паром, со-
бирая его в приемник с 30 мл концентрированной соляной кислоты
и 20 мл воды в виде солянокислой соли. Перегонку прекращают,
как только конденсат в холодильнике перестанет давать щелоч-
ную реакцию. Содержимое приемника переносят в фарфоровую
чашку и выпаривают досуха на водяной бане. Для полного высу-
шивания чашку с сухим остатком оставляют на 10—12 ч в ва-
куум-эксикаторе или в сушильном шкафу при 100°С.
Для отделения хлористого аммония от солянокислой соли ме-
тиламина сухой остаток тонко растирают и кипятят- его с абсо-
лютным спиртом (см. стр. 37); при этом хлористый аммоний не
растворяется. Отфильтровав осадок хлористого аммония, прозрач-
ный фильтрат, содержащий солянокислый метиламин, упаривают
на водяной бане до небольшого объема. Затем раствор быстро
охлаждают и выпавшие мягкие кристаллы солянокислого метил-
амина отсасывают на воронке Бюхнера, промывают небольшим
количеством спирта и сушат в эксикаторе.
Выход солянокислой соли метиламина 7—10 г.
Свободный метиламин можно выделить при действии щелочи
на солянокислый метиламин. В перегонную колбу Вюрца на
150 мл помещают 3 г солянокислой соли метиламина и закрывают
пробкой со вставленной в нее капельной воронкой. Отводную
трубку колбы при помощи резиновой трубки соединяют с изогну-
той под углом стеклянной трубкой. Из капельной воронки
221
прибавляют 35 мл 50%-кого раствора едкого кали, смесь осто-
рожно подогревают. Происходит выделение свободного метил-
амина в виде газа с характерным резким запахом:
CH3-NH2 • НС! + КОН —> СНз-NHj + КС! + Н2О
Солянокислая соль метиламина— белый кристаллический порошок; легко
растворим в воде и спирте; нерастворим в эфире, ацетоне и хлороформе. Мол.
вес 67,52; т. пл 227—228 °C (возгонка); т. кип 225—230 °C при 15 мм рт. ст.
Метиламин — газ с резким тошнотворным запахом, легко горит, легко сме-
шивается со спиртом, эфиров, бензолом. Выпускается в промышленности в виде
хлоргидрата (солянокислой соли) или водных растворов. Мол. вес 31,06; т. пл.
—93,6°С; т. кип —6,5°С; </“”=0,696. Метиламин ядовит, в организме окис-
ляется с образованием муравьиной кислоты, кроме того угнетает функции ды-
хания.
Применяется в производстве фармацевтических препаратов, красителей ан-
трахинонового ряда, фотореактивов, поверхностно-активных веществ и для дру-
гих целей
Для идентификации первичных аминов жирного ряда проводят следующие
реакции:
СН3—NH2 • НС1 + NaNOj —> СН3—ОН + NaCl + N2 + Н2О
0,5 г солянокислого метиламина растворяют в 2 мл воды и прибавляют
2—3 мл 10%-ного раствора азотистокислого натрия, при этом выделяются пу-
зырьки азота.
Отличительная реакция
Первичные амины дают фиолетовое окрашивание при добав-
лении нитропруссида натрия и ацетона.
АНИЛИН
Формула:
ВОССТАНОВЛЕНИЕ НИТРОБЕНЗОЛА ЖЕЛЕЗОМ
В ПРИСУТСТВИИ КИСЛОТЫ ’
Основные реакции:
С6Н5—NOj + 3Fe + 7НС1 —► С6Н5—NH2 • НС1 + 3FeCl2 + 2H2O
C6H5—NH2 • НС1 + NaOH —► C6H5—NH2 + NaCl + H2O
* Для уменьшения продолжительности работы синтез ведется в присутствий
большого количества соляной кислоты.
Реактивы, посуда и приборы
Нитробензол.............18 г, или 15 мл
(0,15 моль)
Соляная кислота (d = 1,19) 80 мл
(1 моль)
Железо (опилки) .... 30 г
(0,55 г-агом)
Едкий натр..............45 г
Диэтиловый эфир .... 120 мл
Едкое кали..............5 г
Хлористый натрий
Колба круглодопная (500 мл) 1
Колба переюпная (250 мл) 1
Воронка делительная (500 мл) 1
Холодильник воздушный . . . 1
Холодильник Либиха ....... 1
Термометр..................1
Алонж.......................1
Прибор для перегонки с водя-
ным паром.................1
Сборка приборов
1. Круглодонную колбу емкостью 500 мл, снабженную воз-
душным холодильником, помещают в водяную баню.
2. Прибор для перегонки с водяным паром (см. рис. 48).
3. Перегонную колбу емкостью 250 мл, снабженную термо-
метром, соединяют с воздушным холодильником, на конец кото-
рого надет алонж (для перегонки эфира применяют водяной хо-
лодильник).
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
В колбу прибора 1 вносят 15 мл нитробензола, 30 г железных
опилок и вливают порциями по 1—2 мл 80 мл соляной кислоты.
Перед восстановлением рекомендуется железо кипятить с кислотой для
Повышения его активности, при этом имеющаяся на поверхности железа тонкая
окисная пленка растворяется.
После каждого прибавления кислоты содержимое колбы хо-
рошо перемешивают. Если реакция идет слишком бурно, колбу
охлаждают холодной водой. После введения половины соляной
кислоты остальное ее количество вносят крупными порциями, по
10—20 мл. Затем колбу нагревают на водяной бане в течение
30 мин.
Реакция считается законченной, когда исчезнет характерный
запах нитробензола и часть железа превратится в красно-корич-
невую закись-окись железа. Далее к горячей реакционной смеси
осторожно добавляют 30 мл воды и небольшими порциями прили-
вают раствор 45 г едкого натра в 60 мл воды до щелочной ре-
акции.
Выделившийся анилин отгоняют из горячей жидкости с водя-
ным паром. В приемнике собирается водная эмульсия анилина, ко-
торая расслаивается. Когда из холодильника пойдет мутноватая
жидкость, приемник меняют. Перегонку ведут до тех пор, пока
дистиллят не станет совершенно прозрачным. В делительной во-
ронке отделяют нижний слой анилина, собравшегося внизу пер-
вого приемника, а верхний слой смешивают с содержимым второго
приемника. Из объединенного дистиллята анилин высаливают
223
хорошо измельченным хлористым натрием, добавляя на каждые
100 мл дистиллята 20 г хлористого натрия.
Анилин плохо растворим в воде (в 100 г воды растворяется 3 г анилина).
Для более полного выделения его из водного раствора дистиллят насыщают
хлористым натрием, в концентрированном растворе которого анилин нерас-
творим.
После растворения хлористого натрия отделяют маслянистый
слой анилина в делительной воронке и добавляют его к первой
порции, водный слой обрабатывают эфиром (см. стр. 57). Эфир-
ную вытяжку добавляют к ранее отделенному анилину, сушат не-
сколькими кусочками твердого едкого кали (для этой цели можно
также применять окись кальция, окись бария и натронную из-
весть; хлористый кальций не употребляется для высушивания ами-
нов, так как образует с ними соединения типа кристаллогидратов)
и отгоняют эфир на водяной бане с водяным холодильником. За-
тем перегоняют анилин с воздушным холодильником (для обесцве-
чивания анилин можно перегнать над небольшим количеством
цинковой пыли) на сетке, собирая фракцию, кипящую при 184 °C.
Выход анилина ~ 11 г.
ВОССТАНОВЛЕНИЕ НИТРОБЕНЗОЛА ЖЕЛЕЗОМ
В ПРИСУТСТВИИ МАЛОГО КОЛИЧЕСТВА СОЛЯНОЙ КИСЛОТЫ
Реактивы, посуда и приборы
Нитробензол..........31 г, или 25,7 мл Колба круглодонная (1000 мл) I
(0,25 моль) Колба перегонная (500 мл) . . 1
Железо (опилки) .... 60 г Воронка капельная ....... I
(1,1 г-атом) Форштосс двурогий..........1
Соляная кислота (d=l,19) 10 мл Холодильник Либиха.........I
(0,13 моль) Воронка делительная........1
Углекислый натрий . . . 7—10 г Термометр...................1
Диэтиловый эфир ... 50 мл Алонж.......................1
Хлористый натрий Прибор для перегонки с водя-
Едкое кали ным паром.................1
Сборка приборов
1. Круглодонную колбу емкостью 1000 мл, помещенную на
песочную баню, соединяют с двурогим форштоссом, который снаб-
жают капельной воронкой и обратным водяным холодильником.
Чтобы можно было вести перемешивание, холодильник соединяют
с форштоссом через резиновую трубку.
2. Прибор для перегонки с водяным паром (см. рис. 48).
3. Прибор 3 на стр. 223.
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
В круглодонную колбу прибора 1 помещают 60 г мелких же-
лезных опилок, 75 мл воды и 26 мл нитробензола. Затем прили-
вают через верх холодильника 10 мл соляной кислоты. При нагре-
224
вании в течение 4—4,5 ч на песочной бане нитробензол восстанав-
ливают в анилин После окончания восстановления в реакционную
смесь вносят 7—10 г углекислого натрия (или соответствующее
количество щелочи) и отгоняют анилин с водяным паром. Дальше
работу ведут, как указано на стр. 223—224.
Выход анилина ~18 г.
ВОССТАНОВЛЕНИЕ НИТРОБЕНЗОЛА ОЛОВОМ
В ПРИСУТСТВИИ СОЛЯНОЙ кислоты
Основные реакции:
2CbH5NO2 + 3Sn + 14НС1 —> 2C6H5NH2 • НС1 + 3SnCl4 + 4H2O
C6H5NH2 • HC1 + NaOH —> C6H5NH2 + NaCl + H2O
Реактивы, посуда и приборы
Нитробензол...........18,5 г, илн 15 мл Колба круглодонная (500 мл) 1
(0,15 моль) Колба перегонная (250 мл) . . I
Олово гранулированное 36 г Воронка делительная (500 мл) 1
(0,3 г-атом) Холодильник воздушный . . . 1
Соляная кислота (</=1, 19) 80 мл Холодильник Либиха..........1
(1 моль) Прибор для перегонки с водя-
Едкий натр............45 г ным паром...................1
Диэтиловый эфир ... 120 мл Алонж.......................1
Едкое кали............5 г
Хлористый натрий
Сборка приборов
1. Круглодонную колбу емкостью 500 мл помещают на водя-
ную баню и снабжают воздушным холодильником.
2. Прибор для перегонки с водяным паром (см. рис. 48).
3. Перегонную колбу на 250 мл соединяют с термометром и
воздушным холодильником (в случае перегонки эфира — холо-
дильник водяной), на конец которого надет алонж.
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
В круглодонную колбу прибора 1 вносят 36 г гранулирован-
ного олова и 15 мл нитробензола.
Если в лаборатории нет готового гранулированного олова, то его можно
приготовить следующим образом расплавляют куски олова в железной ложке
с деревянной ручкой в пламени горелки и выливают расплавленную массу по
каплям в ведро с водой с высоты 1,5 м ,
К, смеси постепенно и осторожно приливают 10 мл концентри-
рованной соляной кислоты. Содержимое колбы хорошо взбалты-
вают, через некоторое время смесь разогревается и начинает ки-
петь за счет тепла реакции. Колбу охлаждают холодной водой, но
так, чтобы реакция не прекратилась совсем Затем постепенно, при
постоянном взбалтывании, вносят небольшими порциями 70 мл
8 М Н Храмкина
Оставшейся соляной кислоты; при этом должно поддерживаться
энергичное кипение смеси
После добавления всего количества кислоты колбу нагревают
на водяной бане еще в течение 1 ч, поддерживая слабое кипение.
К еще теплому раствору осторожно приливают небольшими пор-
циями 45 г едкого натра в 90 мл воды до сильнощелочной реак-
ции для нейтрализации соляной кислоты и разложения солянокис-
лого анилина. Затем колбу включают в систему для перегонки с
водяным паром. Анилин перегоняется вместе с водой и собирается
на дне приемника светло-желтоватыми маслянистыми каплями.
Дальнейшую обработку проводят так, как описано на стр. 223—
224.
Выход анилина ~12 г.
Анилин (аминобензол) — бесцветная или бледно-желтая жидкость со свое-
образным запахом, темнеет при действии света и воздуха, в 100 г воды рас-
творяется 3,6 г, хорошо смешивается со спиртом, эфиром, бензолом Мал. вес
93,13, т. кип 184,4 °C; т пл.—6,15 °C, df = 1,0217, г$= 1,5863.
Анилин является сильным ядом крови Отравления возможны как в резуль-
тате вдыхания паров, так и при попадании жидкого анилина на кожу
Применяется в производстве промежуточных продуктов и красителей, в тек-
стильной промышленности (при черноанилиновом крашении), в фармацевтиче-
ской промышленности (ацетанилид, сульфамиды, новарсенол), в производстве
искусственных смол, для получения проявителей для фотографии, ускорителей
вулканизации каучука (меркаптобензтиазол) и в лабораторной практике
Отличительные реакции
1. В пробирке растворяют 4 капли анилина в 10 мл воды, вно-
сят несколько капель соляной кислоты и растворяют несколько
кристалликов двухромовокислого калия и сернокислой меди. При
медленном нагревании возникает темно-зеленая окраска и выпа-
дают черные хлопья.
2. В пробирку помещают 2 капли анилина, 20 мл воды и при-
бавляют 2—3 капли раствора хлорной извести. Появляется фиоле-
товое окрашивание (в отличие от о-толуидина, дающего в этих
условиях желтовато-бурую окраску).
3. Каплю раствора, содержащего солянокислую соль анилина,
наносят на кусочек газетной бумаги или бумажной салфетки
(в них содержится много лигнина)—появляется желто-оранже-
вое пятно. Фильтровальная бумага или ватман, которые не содер-
жат лигнина, окрашиваться не будут.
о- И п-ТОЛУИДИН
Формулы:
СН;
NH2
226
Основные реакции (см. стр. 218 — 219).
о- и n-CH3CeH4NO2 + 3Fe + 7НС1 —->
—> о- и n-CH3C6H4NH2 • НС1 + 3FeCl2 + 2Н2О
о- и n-CH3CaH4NH2 • НС1 + Na2CO3 —>
—> о- и n-CH3CaH4NH2 + 2NaCl + Н2СО3
Реактивы, посуда и приборы
Смесь о- и «-нитротолуола . . 18 г
(0,13 моль)
Железо (опилки)............20 г
(0,36 г-атом)
Соляная кислота (d = 1,19) . . 2 мл
(0,02 моль)
Углекислый натрий, безвод-
ный ......................3 г
Едкое кали.................10 г
Бензол.....................130 мл
Цинковая пыль..............0,5 г
Едкий натр
Колба круглодонная, трехгорла я
(500 мл)......................1
Колба перегонная (50 мл) .... 1
Холодильник воздушный...........1
Холодильник Либиха ............ 1
Термометр ..................... 1
Мешалка.........................1
Воронка делительная ........... 1
Воронка капельная ............. 1
Прибор для перегонки с водяным
паром.........................1
Алонж...........................I
Приемники ......................2
Сборка приборов
1. Круглодонную трехгорлую колбу емкостью 500 мл снаб-
жают термометром, воздушным холодильником, мешалкой и по-
мещают на водяную баню.
2. Прибор для перегонки с водяным паром (см. рис. 48).
3. Перегонную колбу емкостью 50 мл соединяют с капельной
воронкой, водяным нисходящим холодильником с алонжем и по-
гружают в водяную баню.
4. Перегонную колбу на 50 мл снабженную термометром, со-
единяют с нисходящим воздушным холодильником с алонжем, опу-
щенным в приемник.
Выполнение синтеза
Работу необходимо^ проводить в вытяжндм шкафу!
В круглодонную трехгорлую колбу прибора 1 помещают 20 г
мелких железных опилок, 150 мл воды и 2 мл концентрированной
соляной кислоты и нагревают колбу на водяной бане до 70 °C. За-
тем через отверстие, в которое вставлен воздушный холодильник,
вливают 1 мл смеси о- и n-нитротолуола и включают механиче-
скую мешалку. Температура смеси сразу же повышается-за счет
тепла реакции. Остальное количество о- и «-нитротолуола вливают
постепенно по 1—2 мл через верх воздушного холодильника при
энергичном помешйвании и с такой скоростью, чтобы температура
реакционной смеси не превышала 95 °C. Реакция считается закон-
ченной, как только проба смеси при добавлении к ней разбавлен-
ной соляной кислоты перестанет давать характерного для нитро-
соединения запаха горького миндаля.
8*
По окончании реакции в смесь осторожно вносят 3 г углекис-
лого натрия для нейтрализации и выделения толуидинов из их со-
лей. Собирают прибор для перегонки с водяным паром и перего-
няют реакционную смесь до тех пор, пока проба дистиллята с
концентрированным раствором едкого натра не перестанет мутнеть.
К дистилляту добавляют хлористый натрий (20 г соли на каждые
100 мл дистиллята) и встряхивают до тех пор, пока почти весь
хлористый натрий не растворится. (Хлористый натрий необходим
для более полного выделения толуидинов из водного раствора.)
Дистиллят сливают с оставшегося хлористого натрия в дели-
тельную воронку и несколько раз извлекают бензолом толуидины.
Объединенные бензольные вытяжки сушат над едким кали (см.
стр. 45) и собирают прибор 3. Переносят высушенные вытяжки
в капельную воронку, установленную в колбе Вюрца, и перего-
няют бензол на кипящей водяной (или песочной) бане, подливая
постепенно в колбу раствор из капельной воронки. После того как
весь бензол будет отогнан, заменяют капельную воронку термо-
метром (прибор 4), вносят в колбу 0,5 г цинковой пыли для обес-
цвечивания толуидинов и отгоняют толуидины на открытом огне
при 194—195 °C.
Выход смеси толуидинов ~11 г.
о-Толуидин — бесцветная жидкость; темнеет под действием света и воз-
духа; смешивается со спиртом, эфиром и другими органическими растворите-
лями. При 25 °C в 100 г воды растворяется 1,5 г. Мол. вес 107,15; т. кнп.
200,4 °C; т. пл. -16,4 °C; df = 1,0028; п™ = 1,5728.
п-Толуидин— бесцветные блестящие пластины; хорошо растворим в спирте,
эфире, сероуглероде. При 20 °C в 100 г воды растворяется 0,74 г. Т. кип.
200,5 °C; т. пл. 43,7 °C; = 0,9538; Пд = 1,5532. Толуидины — ядовитые веще-
ства.
Применяются толуидины в производстве красителей, в качестве добавки к
моторному топливу, в лабораторной практике и для других целен.
Отличительные реакции
о-Толуидин. 1. Растворяют 2 капли о-толуидина в 2 мл
50%-ной серной кислоты, подогревают до 50°C и прибавляют
3 капли насыщенного раствора бихромата калия. Смесь взбалты-
вают и через 1 мин выливают в 500 мл воды. Раствор перемеши-
вают до тех пор, пока он не перестанет изменять своего цвета,
затем подщелачивают аммиаком. При прибавлении бихромата воз-
никает красноватое окрашивание, переходящее в темно-синее.
После разбавления водой и перемешивания раствор приобретает
оранжево-желтый цвет. После подщелачивания аммиаком раствор
становится голубым.
2. При взбалтывании эфирного раствора о-толуидина с раз-
бавленным раствором хлорной извести водный слой окрашивается
в коричневый цвет.
п-Толуидин. 1. Проба с бихроматом (см. о-Толуидин, п. 1).
После добавления бихромата возникает красно-коричневое окра-
228
шивание, при разбавлении водой и перемешивании раствор жел-
теет, а после подщелачивания аммиаком происходит незначитель-
ное потемнение раствора.
2. В отличие от о-толуидина не дает окраски с разбавленным
раствором хлорной извести.
а-НАФТИЛАМИН
Формула:
Основные реакции:
Реактивы, посуда и приборы
а-Нитронафталин..............13,8 г
(0,08 моль)
Железо (опилки)..............20 г
(0,36 г-атом)
Соляная кислота (d = 1,19) . . 2 мл
Этиловый спирт...............50 мл
Углекислый натрий
Стакан толстостенный (500 мл) . . 1
Мешалка.........................1
Воронка капельная (100 мл) . . . . 1
Термометр ..................... 1
Воронка Бюхнера.................1
Колба Бунзена ;.................1
Прибор для перегонки в вакууме 1
Сборка приборов
1. Толстостенный стакан емкостью 500 мл, погруженный в
водяную баню, снабжают термометром, мешалкой и капельной
воронкой.
2. Прибор для отсасывания (см. рис. 40).
3. Прибор для перегонки в вакууме (см. рис. 51).
Выполнение синтеза
I
В толстостенный стакан емкостью 500 мл помещают 20 г же-
лезных опилок и наливают раствор 2 мл концентрированной со-
ляной кислоты в 75 мл воды, предварительно нагретый до 50°C.
Готовят раствор 13,8 г а-нитронафталина в 50 мл этилового спир-
та и помещают в капельную воронку, из которой в течение 1 ч
229
небольшими порциями вносят его в толстостенный стакан, энер-
гично перемешивая реакционную смесь. При этом температура
смеси не должна превышать 75 °C. Реакция считается законченной,
когда проба полностью растворится в разбавленной соляной кис-
лоте.
1. Для восстановления а-нитронафталина железные опнтки должны быть
мелкими, чистыми и обезжиренными. Для обезжиривания их нужно промыть
эфиром и высушить на воздухе.
2 а-Нитронафталин нерастворим в воде, поэтому реакцию проводят в вод-
но-спиртовом растворе.
По окончании реакции смесь нагревают 15 мин, затем подще-
лачивают углекислым натрием (до слабощелочной реакции). Для
выделения а-нафтиламина раствор разбавляют водой.
а-Нафтиламин нерастворим в воде, но частично растворим в
спирте, поэтому его можно выделить путем разбавления спирта,
находящегося в реакционной смеси, водой.
Выделившийся а-нафтиламин отсасывают на воронке Бюхнера
вместе с оставшимися железными опилками. Осадок сушат на воз-
духе, а затем перегоняют в вакууме.
Выход а-нафтиламина ~ 10 г.
ПОЛУЧЕНИЕ СОЛЯНОКИСЛОЙ СОЛИ а-НАФТИЛАМИНА
Формула:
Основная реакция:
Реактивы, посуда и приборы
а-Нитронафталин..............10,5 г
(0,06 моль)
Цинк гранулированный .... 16 г
(0,25 моль)
+ 3ZnCl2 + 2Н2О
Соляная кислота (</= 1.19) . . 45 мл
(0,55 моль)
Спирт этиловый..............50 мл
Колба круглодонная (200 мл) . . . 1
Воронка Бюхнера.................1
Колба Бунзена................. . 1
Термометр.......................1
Сборка прибора
Прибор для отсасывания, состоящий из воронки Бюхнера и
колбы Бунзена (см. рис. 40).
2Э0
Выполнение синтеза
В круглодонную колбу емкостью 200 мл вносят 50 мл этило-
вого спирта и 10,5 г а-нитронафталина. К образовавшейся смеси
добавляют 45 мл концентрированной соляной кислоты и слегка
подогревают колбу на водяной бане. В полученный раствор по-
степенно, небольшими порциями вносят 16 г гранулированного
цинка. Температура реакционной смеси не должна превышать 75 °C.
По окончании реакции горячий раствор фильтруют через
скадчатый фильтр на воронке для горячего фильтрования
(стр 48). Фильтрат охлаждают и отсасывают выпавшие кристал-
лы солянокислой соли а-нафтиламина, которые промывают не-
большим количеством спирта и сушат на воздухе.
Выход солянокислой соли а-нафтиламина ~8г.
а-Нафтиламин—бесцветное кристаллическое вещество с неприятным запа-
хом, на воздухе темнеет вследствие окисления, мало растворим в воде; хорошо
растворим в спирте, кислотах, эфире. Мол. вес 143,18; т. кип. 301 °C; т. пл. 50 °C;
d = 1,120.
Солянокислая соль а-нафтиламина — бесцветное вещество с кристаллами в
форме игл. Плохо растворима в холодной воде, растворима в спирте и эфире.
а-Нафтиламин широко применяется в синтезе красителей, а также’ для синтети-
ческих целей в лабораторной практике.
Отличительная реакция
1. При внесении в подкисленный уксусной кислотой водный
раствор а-нафтиламина сульфаниловой кислоты и капли раствора
азотистокислого натрия возникает розовое окрашивание.
2. При нагревании подкисленного соляной кислотой а-нафтил-
амина с хлорным железом выделяются сине-зеленые хлопья.
Глава XIV
РЕАКЦИИ СУЛЬФИРОВАНИЯ
Сульфированием называется процесс введения сульфогруп-
пы — SO2OH в органическое соединение. Продуктами реакции яв-
ляются сульфоновые кислоты (сульфокислоты). Сульфирование
широко, используется в лабораторной практике и промышленности.
Сульфокислоты и их производные находят разнообразное при-
менение: они являются полупродуктами в производстве фенолов '
и нафтолов; многие производные сульфокислот используются в
производстве азокрасителей, моющих средств, лекарственных пре-
паратов, поверхностно-активных веществ, применяются также как
алкилирующие агенты и дезинфицирующие средства.
Для введения сульфогруппы применяются различные сульфи-
рующие агенты: серная кислота различных концентраций, олеум,
231
серный ангидрид, сернистый ангидрид и кислород, сернистая
кислота в виде солей щелочных металлов, сернистый ангидрид и
хлор, хлорсульфоновая кислота и др.
Парафиновые углеводороды устойчивы к действию обычных
сульфирующих агентов. Высшие парафины (от гексана) сульфи-
руются 15°/о-ным олеумом при температуре кипения:
С6Н(, + HO-SO2-OH —> CeHl3-SO3H + H2O
Большое промышленное значение имеет сульфирование пара-
финов и циклопарафинов смесью сернистого газа и хлора (сульфо-
хлорирование), а также смесью сернистого газа и кислорода
(сульфоокисление).
1. СУЛЬФИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИИ
Одно из характерных отличий ароматических углеводородов
от парафиновых — легкость, с которой ароматические соединения
сульфируются серной кислотой.
Наиболее легко сульфируются полициклические ароматические
углеводороды (антрацен, фенантрен и др.), труднее — нафталин,
еще труднее — бензол.
Сульфирование серной кислотой — обратимый процесс:
Ar—Н + HOSO3H ч=* Ar—SO3H + Н2О
Вода, выделяющаяся при реакции, понижает концентрацию
серной кислоты, которая при этом теряет свои сульфирующие
свойства, и вызывает обратную реакцию — гидролиз образующейся
сульфокислоты. Поэтому при сульфировании применяют большой
избыток серной кислоты (от двух- до пятикратного) или же ис-
пользуют олеум с таким содержанием серного ангидрида, который
достаточен для связывания выделяющейся воды.
При исследовании кинетики реакции сульфирования было
установлено, что сульфирующим агентом является ион сульфония
SO3, образующийся по реакции:
2H2SO4 SO3 + Н3О++ HSO7
Легкость сульфирования производных ароматических углево-
дородов зависит также от характера их заместителей. Заместители
I рода, расположенные в порядке убывания их влияния,
ОН > OR > NH2 > NHCOR > R (где R — алкил) облегчают вве-
дение сульфогруппы; заместители II рода NO2 > SO3H > СО >
> СООН, а также галогены затрудняют введение сульфогруппы.
Для получения полисульфокислот ароматического ряда суль-
фируемое соединение обрабатывают либо сразу необходимым ко-
личеством серной кислоты или олеума, либо проводят сульфирова-
ние ступенчато, выбирая для каждой «ступени» оптимальную
температуру и подходящую концентрацию; при этом требуются
уже более жесткие условия, чем для получения моносульфокислот.
232
Важным фактором, влияющим на течение реакции сульфиро-
вания, является температура. Повышение температуры не только
ускоряет процесс, но и способствует образованию различных по-
бочных продуктов (полисульфокислот, сульфонов, продуктов реак-
ции окисления и реакции конденсации). Однако не только поэтому
при каждом процессе сульфирования должна поддерживаться
строго определенная оптимальная температура. Часто температур-
ный режим обусловливает место вхождения сульфогруппы в аро-
матическое ядро. При сульфировании соединений с заместителями
I рода повышение температуры способствует увеличению выхода
пара-изомера. Так, например, при О °C из толуола образуются о-
и n-толуолсульфокислота примерно в равных количествах, а при
100°C получается 79% пара-изомера и лишь 13% орто-изомера.
Сульфирование фенола при комнатной температуре приводит к
образованию о-фенолсульфокислоты, а при 100 °C — к параизомеру.
Направление реакции сульфирования нафталина также в ос-
новном зависит от температуры. При сульфировании 100%-ной
серной кислотой при 80—90 °C получается главным образом а-на-
фталинсульфокислота; наряду с ней образуется p-изомер в соотно-
шении 96:4. В результате сульфирования нафталина при 160 °C
получается преимущественно р-нафталинсульфокислота, но наряду
с ней образуются также а-изомер и продукты дальнейшего суль-
фирования дисульфокислоты. Объясняется это тем, что при суль-
фировании нафталина протекают две обратимые реакции с обра-
зованием а- и р-изомеров:
C10H8 + H2SO4 Cl0H7SO2OH + Н2О
При низких температурах скорость образования а-нафталин-
сульфокислоты в несколько раз выше, чем p-изомера, поэтому
главным образом и получается а-нафталинсульфокислота с при-
месью p-изомера. При повышении температуры а-нафталинсуль-
фокислота гидролизуется в 16 раз быстрее, чем p-изомер, следова-
тельно, количество а-нафталинсульфокислоты уменьшается, а
р-изомера — все более увеличивается. Таким образом, при высокой
температуре равновесие реакции сульфирования нафталина сдви-
нуто для а-изомера влево, а для p-изомера, как более устойчивого
к гидролизу, — вправо.
В некоторых случаях место вступления сульфогруппы зависит
от катализатора. Так, при сульфировании антрахинона без ката-
лизатора образуется главным образом р-антрахинонсульфокпс-
лота, а в присутствии незначительного количества солей ртути
получается а-антрахинонсульфокислота.
Сульфокислоты являются трудно характеризуемыми вещества-
ми, так как большинство из них не имеет определенных темпера-
тур плавления и кипения. Кроме того, при выделении свободных
сульфокислот трудно освободиться от неорганических примесей
В связи с этим сульфокислоты выделяются обычно в виде солей,
которые могут быть очищены кристаллизацией, и многие из них
имеют характерные температуры плавления.
233
2. ПРИМЕРЫ СИНТЕЗОВ
0-НАФТАЛИНСУЛЬФОКИСЛОТА (НАТРИЕВАЯ СОЛЬ)
Формула:
Основные реакции:
+ НС1
Побочные реакции:
Ди-Р-нафтнлсульфон
Реактивы, посуда и приборы
Нафталин...................12,8 г
(0,1 моль)
Серная кислота (d = 1,84) . . 12 мл
(0,21 моль)
Хлористый натрий...........38 г
Двууглекислый натрий .... 8 г
Колба круглодониая (100 мл) . . . 1
Стаканы химические ............ 2
Воронка Бюхнера.................1
Колба Бунзена...................1
Термометр.......................1
234
Сборка приборов
1. В круглодонную колбу емкостью 100 мл вставляют термо-
метр так, чтобы его шарик был погружен в реакционную массу, и
помещают колбу на песочную баню. Горло колбы закрывают проб-
кой с боковой прорезью.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную колбу прибора 1 помещают 12 мл концентри-
рованной серной кислоты и постепенно, при перемешивании, пор-
циями вносят 12,8 г тщательно измельченного нафталина. После
прибавления всей кислоты нагревают реакционную массу в тече-
ние 4 ч на песочной бане при 160—170°C (термометр в колбе),
наблюдая, чтобы температура не поднималась выше, иначе могут
иметь место побочные процессы — образование полисульфокислот,
сульфонов, реакция окисления.
По окончании реакции смесь немного охлаждают и постепенно,
при перемешивании переливают ее в стакан, содержащий 200 мл
холодной воды. При этом выделяется осадок, состоящий из не-
прореагировавшего нафталина и небольшого количества побочного
продукта — ди-р-нафталинсульфона, который отсасывают на во-
ронке Бюхнера или отделяют декантацией (побочный процесс
можно исключить, если применять большой избыток серной кис-
лоты) .
Избыток исходной серной кислоты нейтрализуют постепенным
добавлением 8 г углекислого натрия. Для получения натриевой
соли р-нафталинсульфокислоты сульфомассу постепенно, при по-
мешивании деревянной лопаточкой вливают в насыщенный на хо-
лоду раствор хлористого натрия, который находится в стакане,
охлаждаемом льдом (насыщенный раствор хлористого натрия го-
товят растворением 38 г NaCl в 125 мл горячей воды).
Безводная р-нафталинсульфокислота сильно гигроскопична. Соединяясь с
водой, она разбухает и кристаллизуется с тремя молекулами воды, поэтому ее
выделяют в виде натриевой соли — устойчивого соединения, которое может
использоваться для дальнейшей переработки.
р-Нафталинсульфокислота является сильной кислотой и легко вытесняет
соляную кислоту из раствора хлористого натрия:
4- NaCl
Сад +на
। Для полного выделения натриевой соли р-шафталинсульфокис-
лотную массу оставляют на 30 мин во льду.
Выпавшую натриевую соль р-нафталинсульфокислоты с при-
месями натриевой соли а-нафталинсульфокислоты отфильтровы-
вают на воронке Бюхнера и промывают небольшим количеством
воды. Соль высушивают в сушильном шкафу при 100 °C.
235
Для очистки натриевой соли р-нафталинсульфокислоты от хло-
ристого натрия нужно провести перекристаллизацию из абсолют-
ного этилового спирта.
Выход сырой натриевой соли р-нафталинсульфокислоты 15 г.
По описанному методу получают натриевую соль р-нафталин-
сульфокислоты с небольшими примесями натриевой соли «-изо-
мера. Отделение примеси а-изомера основано на различной рас-
творимости в воде кальциевых солей обеих кислот. Соль р-нафта-
линсульфокислоты менее растворима, а следовательно, раньше вы-
падает в осадок.
Натриевая соль р-нафталинсульфокислоты получается в виде мелких стек-
ловидных пластинок, в смеси с а-изомером кристаллизуется в виде очень мел-
ких блестящих пластинок. Смесь обоих изомеров хорошо растворяется в воде.
Присутствие примеси а-изомера в сырой соли обнаруживается следующим
образом. Нагревают сырую роль р-нафталннсульфокислоты с бромной водой.
При действии брома натриевая соль а-нафталинсульфокислоты превращается
в 1,5-дибромнафталин, выделяющийся в виде белой мути:
+ 2Вг2 4- Н2О —>
+ NaHSO4 + 2НВг
Натриевая соль р-нафталинсульфокислоты на бром не замечается, а обра-
зующаяся 4-бромнафталин-2-сульфокислота растворима в воде:
Безводная Р-нафталинсульфокислота имеет т. пл. 91 °C; одноводный гид-
рат — т пл 124 °C; кристаллогидрат — т. пл. 83 °C.
Р-Нафталинсульфокислота служит для получения Р-нафтола, Н-кислоты и
является промежуточным продуктом для синтеза красителей. Кроме того, Р-на-
фталннсульфокислота образует различные функциональные производные (эфиры,
хлорангидриды, амиды), которые используются для идентификации.
Отличительная реакция
Для идентификации р-нафталинсульфокислоты используют ее
натриевую соль с n-толуидином. Для этого 1 г полученной натрие-
вой соли растворяют в небольшом количестве кипящей воды и
вносят 0,5 мл n-толуидина. После того как раствор станет про-
зрачным, добавляют 2 мл концентрированной соляной кислоты.
Пробирку с раствором охлаждают во льду и, потирая палочкой
236
о стенки пробирки, вызывают кристаллизацию образовавшейся
соли n-толуидина и р-нафталинсульфокислоты с т. пл. 217—218 °C
СН3
NH2 + HC1 —> СН
Полученные кристаллы отсасывают на маленькой воронке, про-
мывают водой и перекристаллизовывают из горячей воды или раз-
бавлфшого этилового спирта.
БЕНЗОЛСУЛЬФОКИСЛОТА (НАТРИЕВАЯ СОЛЬ)
Формула:
Основные реакции:
+ Н2О
4-NaCl
Побочные реакции:
л-Дисульфобеизол (льбеизолдисульфоквслота)
Реактивы, посуда и приборы
Бензол.......................5 мл
(0,05 моль)
Серная кислота, моногидрат . . 10 мл
Хлористый натрий.............10 г
Колба (50 мл)...................1
Стакан химический (100 мл) . . . 1
Колба Бунзена...................1
Воронка Бюхнера.................1
237
Выполнение синтеза
Работу необходимо проводить-в вытяжном шкафу\
В стакане приготовляют насыщенный раствор чистого хлори-
стого натрия (10 г NaCl на 35—40 мл воды) и охлаждают его
ледяной водой.
В колбу наливают 10 мл серной кислоты (моногидрат) и при-
бавляют осторожно маленькими порциями 5 мл бензола, встря-
хивая колбу после каждого приливания бензола. Новую порцию
добавляют после растворения предыдущей добавки.'Если реакция
идет слишком быстро, то нужно колбу охлаждать снаружи ледя-
ной водой. После растворения всего внесенного бензола и охлаж-
дения раствора его приливают малыми порциями к насыщенному
холодному раствору хлористого натрия.
Через некоторое время, особенно при легком протирании сте-
нок стеклянной палочкой и при охлаждении, выпадает натриевая
соль бензолсульфокислоты в виде блестящих листочков, которую
отсасывают на воронке Бюхнера, промывают небольшим количе-
ством насыщенного раствора хлористого натрия. Сушат натриевую
соль бензолсульфокислоты сначала на воздухе, а затем в сушиль-
ном шкафу при ПО °C.
Выход натриевой соли бензолсульфокислоты 8—9 г.
Натриевая соль бензолсульфокислоты содержит примесь хло-
ристого натрия. Ее можно очистить перекристаллизацией из
спирта.
Бензолсульфокислота — бесцветные кристаллические пластинки, мол. вес
168,18. Безводная кислота имеет т. пл. 171—172 °C; одноводный гидрат — т. пл
45—60 °C; кристаллогидрат — т. пл. 43—44 °C. Бензолсульфокислота хорошо рас-
творима в воде и спирте, плохо — в бензоле.
Применяется для производства фенола, бензонитрила и в качестве катали-
затора в некоторых реакциях конденсации и полимеризации.
Отличительная реакция
Смешивают небольшое количество бензолсульфокислоты и тио-
нилхлорида (SOCh), образующийся бензолсульфохлорид выпари-
вают досуха в микротигле. При обработке бензолсульфохлорида
4 каплями спиртового раствора гидроксиламина образуется бен-
золсульфогидроксамовая кислота ]C6H5SO(OH) = NOH], которую
подщелачивают 5% раствором углекислого натрия. Сода разла-
гает бензолсульфогидроксамовую кислоту на бензолсульфиновую
кислоту и остаток нитроксила
CsH5SO(OH)=NOH —> C6H5SO2H + /NOH
затем вводят две капли ацетальдегида и получают ацетогидрокса-
мовую кислоту:
^О х .NOH
СН3—ОН —> СНз—
\н Z Ч)Н
23В
После охлаждения раствор нейтрализуют соляной кислотой и
прибавляют две капли разбавленного водного раствора хлорида
железа — появляется красное окрашивание, т. е. гидроксамовые
кислоты при взаимодействии с хлоридом железа FeCl3 дают ин-
тенсивно окрашенные комплексы железа.
/г-ТОЛУОЛСУЛЬФОКИСЛОТА
Формула:
Основная реакция:
Побочные реакции:
+ Н2О
+ HO-SO3H
Толуол сульфируется очень легко, так как метильная группа
является заместителем I рода, облегчает введение сульфогруппы,
причем преимущественно в орто- и пара-положение.
239
ПОЛУЧЕНИЕ НАТРИЕВОЙ СОЛИ ТОЛУОЛСУЛЬФОКИСЛОТЫ
n-CH3C6H4SO3H + NaCl ?=* n-CH3C6H4SO3Na + НС1
Реактивы, посуда и приборы
Толуол......................16 мл
(0,15 моль)
Серная кислота (d = 1,84) . . 9,5 мл
(0,18 моль)
Углекислый натрий..........8 г
Хлористый натрий............20 г
Колба круглодонная длинногорлая
(100 мл)......................I
Холодильник обратный .......... 1
Стакан химический ............. 1
Воронка Бюхнера.................1
Колба Бунзена...................1
Сборка приборов
1. В длинногорлую круглодонную колбу на 100 мл, снабжен-
ную обратным холодильником, помещают несколько «кипелок».
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную колбу прибора 1 помещают 16 мл толуола
и осторожно вносят 9,5 мл концентрированной серной кислоты.
Реакционную жидкость в течение 1 ч слабо кипятят на асбесто-
вой сетке, время от времени встряхивая колбу (при перемешива-
нии слоев).
При правильном нагревании толуол кипит равномерно. Чрезмерное нагре-
вание выводит толуол из зоны реакции и способствует образованию дисульфо-
производных. Непрерывное встряхивание ускоряет реакцию и способствует
хорошему выходу.
Реакция считается законченной, когда слой толуола почти ис-
чезает и из холодильника изредка стекают капли конденсата. За-
тем теплую реакционную смесь выливают в стакан с 70 мл воды
(если смесь начнет кристаллизоваться, ее необходимо подогреть)
и колбу ополаскивают из промывалки. Кислый раствор осторожно
нейтрализуют 8 г углекислого натрия, прибавляя его небольшими
порциями, добавляют в раствор 20 г хлористого натрия и нагре-
вают смесь до кипения (если хлористый натрий не растворяется,
добавляют еще немного воды). Раствор охлаждают водой со
льдом.
Выпавшие кристаллы натриевой соли толуолсульфокислоты
отсасывают на воронке Бюхнера и отжимают между листами филь-
тровальной бумаги.
Выход натриевой соли толуолсульфокислоты с примесью хло-
ристого натрия ~ 8 г.
ПОЛУЧЕНИЕ СВОБОДНОЙ п-ТОЛУОЛСУЛЬФОКИСЛОТЫ
Для получения кристаллической n-толуолсульфокислоты на-
гревают избыток толуола с концентрированной серной кислотой
(d=l,84), причем вода, выделяющаяся во время реакции, уда-
ляется из реакционной смеси, поэтому реакция почти доходит до
конца и количество n-толуолсульфокислоты возрастает до 85%.
240
Сборка приборов
1. Круглодонная колба, снабженная обратным холодильником
и насадкой для отделения воды.
2. Прибор для отсасывания с воронкой со стеклянным филь-
тром.
Выполнение синтеза
В круглодонную колбу прибора 1 вносят 45 мл толуола, 8 мл
концентрированной серной кислоты и кипятят смесь в течение 5 ч
так, чтобы пары толуола доходили до холодильника. Вода, выде-
ляющаяся при реакции, конденсируется в обратном холодильнике
и стекает в насадку. После 5 ч кипячения собирается около 3,5 мл
воды, причем из-за влажности применяемых реагентов количество
выделившейся воды бывает несколько больше рассчитанного.
По окончании нагревания дают реакционной массе охладиться
и добавляют к ней 2,5 мл воды. Толуолсульфокислота при этом
выкристаллизовывается в виде гидрата. Сливают непрореагиро-
вавший толуол, отсасывают кристаллы на воронке со стеклянным
фильтром, хорошо отжимая их стеклянной пробкой; при этом по-
бочно образовавшаяся о-толуолсульфокислота остается в филь-
трате.
Для очистки' гидрат n-толуолсульфокислоты растворяют в не-
большом количестве горячей воДы (20 мл) и прибавляют к рас-
твору тройной объем концентрированной соляной кислоты. Рас-
твор охлаждают в ледяной воде, выпавшие кристаллы отсасывают
на воронке со стеклянным фильтром и промывают их небольшим
количеством холодной концентрированной соляной кислоты. Та-
кую очистку повторяют дважды. Затем гидрат п-толуолсульфокис-
латы сушат в эксикаторе над твердым едким натром до тех пор,
пока реакция на соляную кислоту не будет отрицательной (НС1
с AgNO3 образует муть AgCl).
Выход n-толуолсульфокислоты ~8 г.
/г-Толуолсульфокислота кристаллизуется с одной молекулой воды
(n-CHsCeHtSOsH-HaO) Кристаллы имеют вид бесцветных призм с т. пл. 104—
105 °C; при 150 °C подвергаются гидролизу.
Применяется при получении n-крезолов и при титровании аминов.
Для идентификации n-толуолсульфокислоты получают ее соль с п-толуидн-
ном, имеющую т. пл 197 °C (см. получение соответствующей соли 0-нафталин-
сульфокислоты, стр. 236).
СУЛЬФАНИЛОВАЯ КИСЛОТА
Формула
NH2
SO3h
241
Основные реакции:
NH2.H2SO4 nh2
SO3H
Побочная реакция:
Для получения сульфаниловой кислоты и других моносульфо-
кислот первичных ароматических аминов обычно применяется ме-
тод, основанный на том, что при нагревании в течение нескольких
часов при 180—190 °C кислые сернокислые соли многих первичных
аминов переходят в сульфокислоты, преимущественно в пара-по-
ложение. Анилин сульфируется легко, при этом в качестве побоч-
ного продукта образуется ортаниловая кислота (о-аминобензол-
сульфокислота), которую с хорошим выходом можно получить при
сульфировании анилина хлорсульфоновой кислотой в органических
растворителях.
Реактивы, посуда и приборы
Анилин свежеперегнанный 9,3 г, или 9 мл
(0,1 моль)
Сериая кислота (d=l,84) 16,3 мл
(0,3 моль)
Едкий натр...............4 г
Соляная кислота, 2 н. раствор
Активированный уголь
Ступка фарфоровая ........ 1
Чашка фарфоровая...........1
Колба Бунзеиа..............1
Воронка Бюхнера............1
Термометр..................1
Выполнение синтеза
Работу необходимо проводить в вытяжном шкафу!
Вариант 1. В фарфоровую ступку помещают 5,5 мл кон-
центрированной серной кислоты и постепенно, небольшими пор-
циями приливают 9,3 г свежеперегнанного анилина. Образуется
24?
кислая сернокислая соль анилина, которую тщательно растирают
пестиком и переносят в фарфоровую чашку. Последнюю накрывают
куском асбестового картона и через отверстие в картоне опускают
в соль анилина термометр. При нагревании на воздушной бане
в течение 3—4 ч при 175—180°С образуется сухая твердая сульф-
аниловая кислота. Реакция считается законченной, когда разбав-
ленная раствором щелочи проба сульфомассы не будет выделять
капелек анилина.
По окончании реакции горячую сульфаниловую кислоту из-
мельчают в ступке и для очистки растворяют в растворе 4 г ед-
кого натра в 40 мл воды. Затем раствор кипятят 5 мин с активи-
рованным углем для обесцвечивания; фильтруют и выделяют
сульфаниловую кислоту, подкисляя соляной кислотой до кислой
реакции (по конго красному). Раствор охлаждают холодной водой
и через 20 мин отсасывают выпавшие кристаллы сульфаниловой
кислоты через воронку Бюхнера (см. рис. 40). Затем кристаллоги-
драт сульфаниловой кислоты (n-H2NCeH4SO3H-2H2O) перекри-
сталлизовывают из воды и сушат.
Выход сульфаниловой кислоты ~ 11 г.
Вар.иант 2. В круглодонную колбу на 100 мл помещают
9,3 г анилина и постепенно при постоянном встряхивании добав-
ляют 16,3 мл концентрированндй серной кислоты. Смесь нагревают
в вытяжном шкафу на масляной бане при 180—190 °C до тех пор,
пока разбавленная водой проба при прибавлении раствора едкого
натра не перестанет выделять анилин (3—4 ч).
Сернокислый анилин растворим в щелочи; если реакция не закончилась,
то из сернокислого анилина при этом выделяется свободный анилин, который
из-за плохой растворимости вызывает помутнение
Охлажденную реакционную смесь выливают при помешивании
в стакан с небольшим количеством холодной воды, отфильтровы-
вают выпавшие кристаллы сульфаниловой кислоты и промывают
их небольшим количеством воды. Перекристаллизовывают кислоту
из горячей воды (в случае надобности с применением активиро-
ванного угля).
Выход сульфаниловой кислоты ~ 10 г.
Сульфаниловая кислота (п-аминобензолсульфокнслота)—бесцветное кри-
сталлическое вещество. Мол. вес 173,18, температура разложения 280—300 °C
Из водного раствора выкристаллизовывается двуводный гидрат при 0—21 °C с
мол. вес. 209, моногидрат — при 21—40 °C, при более высокой температуре —
безводная сульфаниловая кислота.
Применяется в промышленности для производства азокрасителей, в лабо-
ратории — для определения нитрилов, обнаружения осмия, рутения и др Амиды
сулфаннловой кислоты применяются в производстве лекарственных веществ.
Отличительная реакция
При обработке бромной водой в горячем водном растворе
сульфаниловая кислота превращается в триброманилин, который
выпадает в виде мелкокристаллического осадка.
243
Глава XV
РЕАКЦИИ ДИАЗОТИРОВАНИЯ
И АЗОСОЧЕТАНИЯ
Реакция диазотирования, открытая в 1858 г. Гриссом, заклю-
чается в образовании солей диазония при взаимодействии первич-
ных ароматических аминов с азотистой кислотой в кислой среде:
нх +
Ar—NH2 + НО—NO —> [Ar—N]X~ + 2Н2О
III
N
где Аг—ароматический радикал,
X — анион минеральной кислоты.
Эта реакция очень характерна для первичных ароматических
аминов, имеет большое промышленное значение и широко приме-
няется в лабораторной практике. Лабораторное значение этой ре-
акции заключается в том, что с ее помощью можно осуществить
множество синтезов и отличить первичные амины жирного ряда
от первичных ароматических аминов. В промышленности реакция
диазотирования применяется для синтеза промежуточных продук-
тов, красителей и фармацевтических препаратов.
Для реакции диазотирования применяется не свободная азо-
тистая кислота, которая очень неустойчива, а кислота, выделяю-
щаяся при взаимодействии азотистокислого натрия или калия с
минеральной кислотой (чаще всего соляной или серной). При этом
образуется соль диазония, диссоциирующая на катион диазония
и анион кислоты:
ArNH2 + 2НХ + NaNO2 —► [Ar-NJX-+ NaX + 2Н2О
Скорость реакции диазотирования зависит от строения амина
и природы аниона участвующей в реакции кислоты.
Амины, содержащие в ароматическом ядре заместители П-го
рода (—NO2, —SO3H, —СООН и т. п.), диазотируются быстрее
аминов, содержащих заместители I рода (—СН3, —ОН, —ОСН3
и т. п.). Заместители II рода понижают основность аминогруппы.
Соли таких аминов легко гидролизуются водой (например, нитро-
анилины). Поэтому обычные условия диазотирования в водной
среде здесь неприменимы. В этом случае реакцию диазотирования
проводят в среде концентрированной серной кислоты твердым азо-
тистокислым натрием.
Диазотирование двух аминогрупп в одном бензольном ядре
требует создания специальных условий, а диазотирование трех
244
аминогрупп провести не удается — образуются только продукты
осмоления.
Для получения солей диазония чаще всего применяют амино-
производные бензола, нафталина, антрахинона, аминобензол- и
аминонафталинсульфокислот.
Для диазотирования 1 моль ароматического амина теорети-
чески необходим 1 моль азотистокислого натрия и 2 моль одно-
основной минеральной кислоты, причем 1 моль кислоты идет для
образования соли ароматического амина и 1 моль — для образо-
вания азотистой кислоты*:
ArNH2 + HX —► [ArNH3]X~
[ArNH3]X‘ + NaNO2 + HX —► [Ar-NJX* + 2H2O + NaX
Практически же применяют значительно большее количество
кислоты. Для аминов типа анилина применяют 2,5—3 моль мине-
ральной кислоты на 1 моль амина и 1 моль азотистокислого нат-
рия, более слабоосновные амины требуют более высокой концен-
трации кислот.
Избыток минеральной кислоты необходим для предотвраще-
ния сочетания образовавшейся соли диазония с еще не прореаги-
ровавшим свободным амином. Так, в слабокислой среде легко про-
текает побочная реакция между солью диазония и первичным ами-
ном с образованием диазоаминосоединений:
[ArNJX' + ArNH2 —► ArNH—N=N—Ar + HX
При действии же избытка минеральной кислоты диазоамино-
соединения распадаются на соль диазония и соль амина:
ArNH—N=N—Ar + 2НХ —► [ArNJX’ + [ArNH3]X‘
Реакцию диазотирования проводят в толстостенном стакане
при постоянном перемешивании реакционной смеси, чтобы избе-
жать местных перегревов.
Соли диазония очень неустойчивы и легко разлагаются водой
при повышении температуры, поэтому реакцию диазотирования
следует проводить при низких температурах (0—5°C), для чего
стакан снаружи охлаждают льдом и в случае необходимости к ре-
акционной массе добавляют кусочки льда.
Азотистая кислота взаимодействует с амином постепенно, так
как эта реакция не является ионной. Поэтому азотистокислый
* Установлено, что диазотированию подвергается свободный амин, а не его
соль.
245
натрий следует приливать медленно (по каплям) во избежание
накопления свободной азотистой кислоты.
Диазотирование нужно вести так, чтобы над поверхностью
раствора не появлялись бурые пары окислов азота, так как распад
азотистой кислоты с образованием окислов азота влечет за собой
сохранение в реакционной смеси избытка соли амина, которая
может связать диазосоединение.
Контролируют реакцию диазотирования обычно по присут-
ствию свободной азотистой и других минеральных кислот. Для
этого капли реакционного раствора наносят на иодкрахмальную
бумагу * и бумагу конго. Бумага конго в присутствии сильных ми-
неральных кислот синеет, иодкрахмальная бумага синеет под дей-
ствием свободного иода, выделяющегося в результате взаимодей-
ствия йодистого калия с азотистой кислотой.
Если реакция диазотирования идет нормально, то иодкрах-
мальная бумага окрашивается в серо-синий цвет, а бумага конго—
в голубой. При избытке азотистой кислоты на иодкрахмальной
бумаге образуется темно-бурое пятно. Если в конце реакции азо-
тистой кислоты недостаточно, то добавляют азотистокислый нат-
рий до появления устойчивой, но не сильной окраски на иодкрах-
мальной бумаге.
Если во время диазотирования выделился окращенный осадок
диазоаминосоединения, его можно разложить добавлением мине-
ральной кислоты.
Растворы солей диазония нельзя оставлять на длительное вре-
мя и нельзя держать на солнечном свету.
Соли диазония обычно не удается выделить из водных раство-
ров. В твердом состоянии их можно выделить лишь при проведе-
нии реакции в спиртовом растворе, однако сухие соли диазония
очень опасны —они взрываются при нагревании или ударе. По-
этому реакции с солями диазония всегда проводят в их водных
растворах сразу же после образования.
Соли диазония обладают большой реакционной способностью.
Для них характерны два типа реакций: 1) сопровождающиеся вы-
делением азота, и 2) идущие без выделения азота, в том числе и
реакции восстановления, в результате которых образуются арома-
тические гидразины.
1. РЕАКЦИИ СОЛЕЙ ДИАЗОНИЯ,
СОПРОВОЖДАЮЩИЕСЯ ВЫДЕЛЕНИЕМ АЗОТА
Большинство ароматических диазосоединений — неустойчивые,
легко разлагающиеся вещества. Если реакцию разложения вести
в определенных условиях, то диазогруппу можно заменить мно-
гими другими группами (ОН, CN, С1, Вг, I и т. д.).
* Иодкрахмальная бумага — это фильтровальная бумага, пропитанная рас-
твором йодистого калия и крахмальным клейстером и высушенная.
24«
При нагревании солей диазония в водных кислых растворах
происходит реакция замещения диазогруппы гидроксильной груп-
пой, сопровождающаяся бурным выделением азота и образова-
нием фенолов:
[Ar—N]X" + Н—ОН —► АгОН + N2 + НХ
Замещение диазогруппы на иод осуществляется при нагрева-
нии раствора соли диазония с раствором йодистого цалия:
[Ar-N]Cl’ + KI [Ar-N]I* + КС1
III III
N N
[Ar—N]I" —► Ari + N2
N
В качестве побочного продукта реакции получается фенол:
[Ar—NJCF + Н—ОН —► Аг—ОН + N2 + НС1
Присутствие избытка йодистого калия препятствует побочной
реакции. Замещение диазогруппы на бром или хлор аналогичным
образом не удается, так как реакции протекают с невысокими вы-
ходами. Выходы можно увеличить, если в качестве катализатора
применять однохлористую медь или медный порошок (реакция
Зандмейера):
' + Си2СЬ
[Ar-N]Cr ----> ArCl + N2
Для замены диазогруппы нитрильной группой недостаточно
действия солей синильной кислоты; реакция идет с хорошими вы-
ходами только в присутствии цианистой меди или медного по-
рошка:
+ Cu2(CN)2, KCN
[Ar-N]Cl' -----------> ArCN + N2 + КС1
Для этой реакции применяют цианиды натрия или калия, не-
обходимые для образования растворимой комплексной соли
Na3[Cu(CN)4].
247
2. РЕАКЦИИ СОЛЕЙ ДИАЗОНИЯ,
ИДУЩИЕ БЕЗ ВЫДЕЛЕНИЯ АЗОТА
Наибольшее значение из этого типа реакций имеют реакции
азосочетания, т. е. реакции взаимодействия солей диазония с аро-
матическими аминами или фенолами, приводящие к образованию
соединений Аг—N = N—Аг по общей схеме:
[Аг—N]X' + HArY —► Ar—N=N—ArY + НХ
где X — анион кислоты;
Y—NH2; —NHAr, —NHA1K; -N(A1K)2; —ОН; —OA1K (по-
добные группы в красителях называются ауксохромами).
Продукты реакции азосочетания — азосоединения весьма проч-
ны и обычно ярко окрашены. Окраска азосоединений обусловлена
наличием в молекуле хромофорной группы, в данном случае азо-
группировки —N — N—, соединенной с двумя ароматическими яд-
рами. Однако не всякое окрашенное вещество является красите-
лем. Последний помимо окраски должен обладать способностью
легко закрепляться на различных материалах. Эту способность
придают красителю так называемые ауксохромные группы.
Реакция азосочетания широко используется в технике для по-
лучения азокрасителей.
Для каждой реакции азосочетания имеется определенное опти-
мальное значение pH. В сильнокислой среде реакция, как правило,
не идет ни с ароматическими аминами, ни с фенолами, так как
очень понижены концентрации свободного амина (из-за образова-
ния соли диазония) и иона фенолята (диссоциация фенола силь-
но подавлена). В сильнощелочной среде из соли диазония выде-
ляется свободное основание — гидроокись диазония, которая затем
перегруппировывается в диазогидрат:
[Ar—NJC1" —> [Ar—N]OH~ —► Ar—N=N—ОН
Диазогидрат является слабой кислотой и в щелочной среде об-
разует не способную к азосочетанию соль Аг—N = N—ONa.
Таким образом, для азосочетания в случае применения аминов
наиболее благоприятной является слабокислая, а в случае приме-
нения фенолов — слабощелочная среда.
Оттенок азокрасителей заметно изменяется в зависимости от
счепени кислотности или Щелочности среды. Так, например, гели-
антин в щелочной среде имеет желтый, в нейтральной — оранже-
вый, а в кислой — красно-розовый цвет; цвет конго красного в
щелочной среде — красный, а в кислой — синий.
248
Амины, образующие соли диазония, обычно называются диазо-
компонентами красителя, а амины и фенолы, вступающие в азо-
сочетание, — азокомпонентами
Если в азокомпоненте пара-положение по отношению к амино-
или оксигруппе свободно, то азосочетание происходит в этом по-
ложении; если же в азокомпоненте уже имеются заместители в
пара-положении, то азосочетание происходит в орто-положении
к амино- или оксигруппе. Степень легкости азосочетания опреде-
ляется природой реагирующих веществ легче оно протекает с
фенолами, труднее — с аминами Активность диазокомпонентов
увеличивают галогены, карбоксильная, карбонильная, нитро- и
сульфогруппы. Эти же заместители понижают активность азоком-
понентов. Поэтому, подбирая условия реакции, необходимо учиты-
вать реакционную способность диазо- и азокомпонентов.
3. ПРИМЕРЫ СИНТЕЗОВ
ФЕНОЛ
249
Реактивы, посуда и приборы
Анилин..................9,3 г, или 9 мл
(0,1 моль)
Серная кислота (d — 1,84) 10 мл
(0,18 моль)
Азотистокислый натрий . . 7,25 г
(0,11 моль)
Хлористый натрий .... 7,5 г
Эфир.....................60 мл
Сернокислый натрий ... 7 г
Лед
Стакан химический толсто-
стенный (300 мл) ... 1
Колба круглодониая (500 мл) 1
Термометр ................. 2
Колба Вюрца (150 мл) .... 2
Воронка капельная ......... 1
Вороика делительная ....... 1
Прибор для перегонки с водя-
ным паром..................I
Холодильник Либиха..........1
Холодильник воздушный ... I
Приемники . . .'............2
Сборка приборов
1. В толстостенный стакан емкостью 300 мл, помещенный в
баню с накрошенным льдом, опускают капельную воронку, конец
которой должен быть погружен в жидкость на 1—2 см (для пре-
дотвращения потерь азотистой кислоты вследствие ее разложения
на поверхности жидкости с выделением окислов азота).
2. Круглодонную колбу емкостью 500 мл соединяют с обрат-
ным воздушным холодильником и помещают на водяную бйню, в
которую опущен термометр.
3. Прибор для перегонки с водяным паром (см. рис. 48).
4. Колбу Вюрца емкостью 150 мл, помещенную, на водяную
баню, соединяют с термометром и нисходящим водяным' холодиль-
ником с алонжем, конец которого опускают в приемник.
5. Колбу Вюрца емкостью 150 мл соединяют с термометром и
нисходящим воздушным холодильником, к другому концу которого
подставляют приемник.
Выполнение синтеза
В толстостенный стакан на 300 мл наливают 50 мл воды и при
перемешивании прибавляют 10 мл концентрированной серной кис-
лоты.
В среде серной кислоты скорость реакции диазотирования меньше, чем в
среде соляной кислоты, однако в последнем случае происходит образование
хлорпроизводных в качестве побочных продуктов.
К горячему раствору медленно при перемешивании приливают
9 мл свежеперегнанного анилина, который должен полностью рас-
твориться. Если раствор непрозрачен, его следует нагреть до пол-
ного растворения анилина.
Полученный раствор охлаждают до комнатной температуры,
а затем постепенно прибавляют 75 г тонкоизмельченного льда, до-
водя температуру раствора до 0°С. Во время добавления льда
нужно энергично перемешивать содержимое стакана для того, что-
бы частично выделяющийся сернокислый анилин был мелкокри-
сталлическим, так как только в таком виде он растворяется при
диазотировании.
250
К охлажденной смеси из капельной воронки медленно прили-
вают при сильном перемешивании охлажденный до 0—5 °C рас-
твор 7,25 г азотистокислого натрия в 30 мл воды. Во время диазо-
тирования температура реакционной смеси не должна превышать
8 °C. В случае необходимости к смеси добавляют кусочки льда.
Прибавив большую часть раствора азотистокислого натрия, за-
крывают кран капельной воронки, перемешивают смесь еще 5 мин,
затем берут пробу на присутствие свободной азотистой кислоты.
Если при нанесении капли жидкости на иодкрахмальную бумагу
не образуется темного пятна, то можно прибавить новую порцию
раствора азотистокислого натрия, так как азотистая кислота уже
полностью вступила в реакцию. Если на бумаге сразу же обра-
зуется темное пятно, т. е. азотистая кислота еще неполностью ис-
пользована для диазотирования, нужно подождать несколько ми-
нут, а затем снова взять пробу на содержание свободной азотистой
кислоты. Одновременно нужно следить, чтобы раствор имел кис-
лую реакцию (проба на бумагу конго), и в случае необходимости
прибавить несколько капель концентрированной серной кислоты.
Реакцию диазотирования можно считать законченной, если
через несколько минут после прибавления последней порции рас-
твора азотистокислого натрия в реакционной смеси обнаружи-
вается свободная азотистая кислота. Вследствие уменьшения кон-
центрации реагирующих веществ перед окончанием диазотирова-
ния реакция замедляется, поэтому перед взятием пробы следует
подождать несколько минут. Наряду с присутствием свободной
азотистой кислоты признаком окончания реакции является полный
переход сернокислого анилина в раствор.
Полученный прозрачный раствор соли диазония переносят в
круглодонную колбу емкостью 500 мл и оставляют на 15—20 мин
при комнатной температуре. Находящийся в водном растворе сер-
нокислый фенилдиазоний при этом постепенно разлагается с выде-
лением азота и образованием фенола. Затем для ускорения разло-
жения раствор нагревают на водяной бане (прибор 2) при 40—
50 °C, время от времени взбалтывая содержимое колбы до тех пор,
пока не прекратится выделение пузырьков азота (15—20 мин).
После этого образовавшийся фенол отгоняют с водяным паром
(прибор 3) до тех пор, пока проба дистиллята будет давать лишь
слабое помутнение с бромной водой вследствие образования три-
бромфенола. К дистилляту добавляют 7,5 г хлористого натрия и
встряхивают до почти полного растворения его, затем в делитель-
ной воронке из дистиллята извлекают фенол эфиром (2 раза по
30 мл). Объединенные эфирные вытяжки собирают в колбе, сушат
над безводным сернокислым натрием или магнием (можно над
хлористым кальцием) и отгоняют эфир на водяной бане с водяным
холодильником (прибор 4)' Фенол 'перегоняют, нагревая колбу
(прибор 5) на асбестовой сетке, и собирают фракцию, кипящую
при 179—183 °C. Фенол можно очистить перегонкой в вакууме.
После охлаждения фенол должен закристаллизоваться.
Выход фенола ~6г.
251
Фенол — бесцветное кристаллическое вещество, легко растворим в спирте,
эфире, бензоле. Мол. вес 94,11; т. кип. 182,2 °C; т. пл. 40,9 °C; djs= 1,0545;
Пд5 = 1,5402. Фенол образует кристаллогидрат СвЬЬОНО.бНаО с т. пл. ~17°С.
Фенол является ядом для нервной системы, вызывает поражения кожи.
В случае попадания его на кожу пораженное место следует промыть извест-
ковой водой.
Отличительная реакция
1. Прибавляют к водному раствору фенола несколько капель
разбавленного раствора FeCl3 — появляется фиолетовое окраши-
вание вследствие образования сильнодиссоциированного комплекс-
ного фенолята трехвалентного железа:
6С6Н5ОН + FeCI3 —> 6Н++ ЗСГ + [Fe(OCsH5)6]3-
При подкислении окрашенного раствора окраска исчезает.
2. К разбавленному водному раствору фенола прибавляют по
несколько капель разбавленного раствора аммиака и хлорной из-
вести. Затем смесь нагревают 1 мин — появляется синяя окраска,
постепенно усиливающаяся за счет образования красящего веще-
ства — индофенола:
НО—СУ—N=( )=О
При прибавлении к смеси соляной кислоты синяя окраска
переходит в ярко-красную.
3. Раствор фенола в воде подкисляют разбавленной соляной
кислотой и добавляют постепенно по каплям бромную воду — вы-
падает белый осадок трибромфенола:
ОН
Вг
Аналогичным способом можно диазотировать толуидины,
ксилидины, аминофенолы, л-хлор- и ж-нитроанилины.
ИОДБЕНЗОЛ
Формула;
2»
Основные реакции:
Побочные реакции:
СГ+Н-ОН —>
+ N2 + НС1
+ NaOH
Реактивы, посуда и приборы
Анилин.................9,3 г, или 9,2 мл Стакан химический толсто-
(0,1 моль) стенный (300 мл).............1
Соляная кислота (d=l, 19) 25 мл Воронка капельная........1
(0,35 моль) Колба круглодонная (500 мл) 1
Азотистокислый натрий 8 г Холодильник воздушный . . . 1
(0,12 моль) Прибор для перегонки с водя-
Иодистый калий .... 20 г ным паром..................1
(0,12 моль) Воронка делительная......1
Едкий натр Колба коническая.........1
Хлористый кальций, безводный Колба Вюрца (50 мл)......1
Лед Термометр................1
Приемники ................. 2
Сборка приборов
1. В толстостенный стакан емкостью 300 мл, помещенный в
баню со льдом, опускают термометр и капельную воронку, конец
которой должен быть погружен в жидкость на 1—2 см.
2. Круглодонную колбу емкостью 500 мл соединяют с обрат-
ным воздушным холодильником и помещают на водяную баню.
3. Прибор для перегонки с водяным паром (см. рис. 48).
4. Колбу Вюрца емкостью 50 мл соединяют с термометром и
нисходящим воздушным холодильником, к другому концу которого
подставляют приемник.
253
Выполнение синтеза
В толстостенном стакане прибора 1 смешивают 25 мл концен-
трированной соляной кислоты и 25 мл воды и к раствору прибав-
ляют 9,3 г свежеперегнанного анилина. Затем к раствору, охлаж-
денному льдом до 1—2 °C, при непрерывном перемешивании при-
ливают постепенно из капельной воронки охлажденный раствор
8 г азотистокислого натрия в 20 мл воды (подробнее см. стр. 250).
Диазотирование ведут при постоянном охлаждении; в случае
повышения температуры к реакционной смеси можно добавить не-
сколько кусочков льда.
Окончание реакции'юпределяют с помощью иодкрахмальной
бумаги. Конец реакции диазотирования следует строго контролиро-
вать, так как избыток азотистой кислоты может привести к умень-
шению выхода иодбензола.
Полученный раствор соли диазония смешивают в круглодон-
ной колбе емкостью 500 мл с раствором 20 г йодистого калия в
15 мл воды (избыток йодистого калия препятствует образованию
побочного продукта — фенола) и оставляют смесь на несколько
часов охлаждать. Затем колбу нагревают на слабо кипящей водя-
/ ной бане (прибор 2) до прекращения выделения азота.
После этого раствор подщелачивают концентрированным рас-
твором едкого натра до резко щелочной реакции, чтобы связать
образовавшийся в качестве побочного продукта фенол в виде
фенолята натрия, и отгоняют иодбензол с водяным паром до тех
пор, пока из холодильника не перестанут стекать маслянистые
капли. Фенолят натрия, в отличие от фенола,.с водяным паром не
перегоняется. Дистиллят переносят в делительную воронку и от-
деляют нижний слой иодбензола от воды в коническую колбу.
Сырой иодбензол должен быть светло-желтым; если же он буро-
го цвета (от наличия свободного иода), то его встряхивают в де-
лительной воронке с небольшим количеством раствора сульфита
или бисульфита натрия до осветления и снова отделяют нижний
слой. Иодбензол сушат над безводным хлористым кальцием, пере-
гоняют, нагревая кОлбу (прибор 4) на асбестовой, сетке, и соби-
рают фракцию, кипящую при 185—190 °C. Можно перегонять иод-
бензол в вакууме.
Выход иодбензола ~16 г.
Иодбензол — жидкость со своеобразным запахом; нерастворим в воде, хо-
рошо растворяется в спирте. Мол: вес 204,01; т. кип. 188,45 °C; т. пл —31^35 °C;
d‘5= 1,8382, п£ = 1,6213.
Применяется для синтезов этилбензола и бензойной кислоты.
гелиантин
Формула:
NaO3S—Ч—N=N—
\=/ \==/ \СНз
254
Основные реакции:
Реактивы, посуда и приборы
Сульфаниловая кислота . . 5 г
(0,025 моль)
Азотистокислый натрий ..2г
Стаканы химические (125 и
500 мл)....................2
Колба Бунзена...............1
Воронка Бюхнера.............1
Предохранительная склянка 1
(0,03 моль)
Едкий натр, 2 н. раствор . . 12,5 мл
Соляная кислота, 2 н. рас-
твор .....................12,5 «л
Соляная кислота 1 н. рас-
твор .....................25 мл
Диметнланилнн.............3 г, или 3 мл
(0,025 моль)
Выполнение синтеза
В стакане емкостью 125 мл растворяют при легком нагрева-
нии 5 г кристаллической сульфаниловой кислоты в 12,5 мл 2 н.
раствора едкого натра. Поскольку сульфаниловая кислота плохо
растворима в воде, ее переводят в растворимую натриевую соль
NaO3S——NH2. После растворения всей кислоты жидкость
должна иметь щелочную реакцию (по лакмусу). Полученный рас-
твор охлаждают водой, добавляют 2 г азотистокислого натрия в
25 мл воды и перемешивают до полного его растворения. Далее
раствор охлаждают льдом и приливают его при помешивании в
стакан емкостью 500 мл, содержащий 12,5 мл 2 н. раствора соля-
ной кислоты. Через несколько минут выделяется белый порошко-
образный осадок соли диазония в виде биполярного иона
25S
Полученный продукт не отделяют, а используют в виде взвеси.
Он более устойчив, чем другие соли диазония, и может храниться
несколько часов.
В маленьком стаканчике растворяют 3 мл свежеперегнанного
диметиланилина в 25 мл 1 н. раствора соляной кислоты. Необхо-
димо следить, чтобы весь диметиланилин растворился, о чем сви-
детельствует отсутствие маслянистого слоя над раствором. По-
лученный раствор приливают к взвеси диазотированной сульфани-
ловой кислоты, находящейся в стакане емкостью 500 мл, и хорошо
перемешивают. Через 5—10 мин образуется густая паста красной
устойчивой в кислоте модификации красителя, для превращения
которого в натриевую соль к пасте добавляют 2 н. раствор едкого
натра до резко щелочной реакции, хорошо перемешивают и нагре-
вают до кипения; при этом большая часть красителя переходит в
раствор.
В щелочной среде гелиантии имеет желтый, в нейтральной — оранжевый,
а в кислой — красный цвет Он изменяет цвет при pH = 3,2—4,4 вследствие
превращения одной хромофорной группы хиноидной группиров-
ки} в другую (—N = N—азогруппу). Итак, красный гелиантин имеет хиноид-
ную группировку
сн3-|
Ох^Пз
xchJ
сг
при прибавлении к нему щелочи образуется желтый гелиантин с азогруппой
Смесь охлаждают в ледяной воде, выделившиеся оранжевые
кристаллы натриевой соли красителя отфильтровывают на воронке
Бюхнера *.
Продукт может быть сразу перекристаллизован из небольшого
количества воды.
Гелиантин (метилоранж; метиловый оранжевый; натриевая соль 4-диме-
тиламиноазобензол-4'-сульфокислоты; оранж Ш) — кристаллы в виде листочков
или порошок. Мол. вес 327,34.
Гелиантин является одним из общеупотребительных кислотно-щелочных
цветных индикаторов.
* Для ополаскивания стакана лучше пользоваться насыщенным раствором
хтористого натрия.
Отмывать руки следует теплым, слабо подкисленным серной кислотой рас-
твором марганцовокислого калия, пока они не окрасятся двуокисью марганца,
а затем нужно удалить окраску теплым разбавленным раствором кислого сернн-
стокислого натрия.
256
Отличительная реакция
При прибавлении к не слишком разбавленному раствору кра-
сителя соляной кислоты раствор становится розовым и выпадает
красно-коричневый кристаллический осадок.
Р-НАФТОЛОРАНЖ
Формула:
Основные реакции:
O?S
NaO3S
NH3 + NaOH —► NaO3S
NH2 + H2O
NH2 + NaNOs-4- 2HC1 —►
N=N + 2H2O + 2NaCl
Реактивы, посуда и приборы
Сульфаниловая кислота ... 5 г
(0,025 моль)
Азотистокислый натрий ... 2 г
(0,03 моль)
р-Нафтол....................3,6 г
(0,025 моль)
Едкий натр, 2 н. раствор . . 12,5 мл
Соляная кислота, 2 н. рас-
твор .....................12,5 мл
Едкий натр..................2 г
Хлористый натрий............25 г
Стаканы химические ............ 2
Колба Бунзена...................1
Воронка Бюхнера.................1
Предохранительная склянка . . . 1
9 М. Н. Храмкина
157
Выполнение синтеза
Диазотирование сульфаниловой кислоты проводят так же, как
при получении гелиантина (см. стр. 255). В стакане растворяют
2 г едкого натра в 40 мл воды, в полученный раствор вносят 3,6 г
р-нафтола и приливают при перемешивании взвесь диазотирован-
ной сульфаниловой кислоты. Перемешивание продолжают в тече-
ние 30 мин. Затем для уменьшения растворимости красителя при-
бавляют 25 г хлористого натрия и оставляют стоять стакан на
льду 1 ч, время от времени перемешивая смесь. Выпавший краси-
тель отсасывают (см. рис. 40), промывают небольшим количеством
холодной воды, плотно отжимают на воронке, сним-ают с фильтра
и сушат на воздухе.
Выход p-нафтолоранжа ~8 г.
p-Нафтолоранж (кислотный оранжевый, оранж II)—кристаллическое ве-
щество ярко-оранжевого цвета, хорошо растворимое в воде; мол. вес. 350,34.
Благодаря яркости оттенка и довольно хорошей прочности этот краситель
имеет практическое применение.
Глава XVI
РЕАКЦИИ ГРИНЬЯРА
Реакции Гриньяра — это реакции синтеза ряда органических
соединений с помощью смешанных магнийгалогенорганических
соединений.
Начало применению магния в органическом синтезе было по-
ложено в 1899 г. Барбье для синтеза третичных спиртов, ранее
впервые полученных А. М. Бутлеровым через цинкорганические
соединения. В 1900 г. ученик Барбье В. Гриньяр усовершенствовал
эту реакцию и нашел, что магний в среде сухого эфира реагирует
со многими галогеналкилами и галогенарилами, образуя смешан-
ные магнийгалогенорганические соединения (реактивы Гриньяра),
которые обычно называются магнийорганическими соединениями:
абсолютный эфир
R—X + Mg --------------> R-Mg—X
где R— алкил, арил и др.;
X — галоген.
Эти соединения отличаются большой химической активностью
и способны вступать в разнообразные реакции.
Основное условие проведения реакции Гриньяра — чистота
прибора и тщательная очистка реактивов. Особенно необходимо
удалить вещества, содержащие подвижный (активный) атом во-
дорода (прежде всего воду, кислоты, спирты). Для этого галоген-
258
производное промывают несколько раз водой, сушат над хлори-
стым кальцием и перегоняют непосредственно перед введением его
в реакцию.
Магнийорганические соединения разлагаются водой с образо-
ванием соответствующих углеводородов:
RMgX + н2О —RH + Mg/
Х)Н
Аналогично действуют спирты и карбоновые кислоты.
Наиболее часто употребляемым растворителем для реакции
Гриньяра является абсолютный диэтиловый эфир. Его следует
хранить в склянках из темного стекла над натрием, поверхность
которого должна быть блестящей. Кроме диэтилового эфира мож-
но применять также дипропиловый, диизопропиловый, дибутило-
вый, диамиловый и диизоамиловый эфиры в том случае,' если
требуется нагревание реакционной смеси до более высокой тем-
пературы, чем температура кипения диэтилового эфира. Эфиры
являются не только растворителями для магнийорганических со-
единений, но, присоединяясь к последним, они образуют комплекс-
ные соединения, которые могут быть выделены в кристаллическом
виде. Эти соединения, называемые эфиратами, хорошо раство-
ряются в этиловом эфире и тем самым освобождают поверхность
магния, продолжающего реагировать.
Магнийорганические соединения находятся в эфирном раство-
ре в состоянии подвижного равновесия и рассматриваются как
смесь смешанных RMgX и полных RsMg магнийорганических со-
единений:
2RMgX R2Mg + MgX2
В зависимости от условий реакции равновесие сдвигается то
в одну, то в другую сторону. Оно зависит от величины радикала:
чем больше молекулярный вес алкила магнийорганического со-
единения, тем больше равновесие сдвинуто в сторону образования
диалкилмагния R2Mg.
Скорость взаимодействия алкилгалогенов в реакции Гриньяра
падает от иодидов к хлоридам. Однако хлориды и бромиды дают
лучшие выходы, чем иодиды. Это объясняется протеканием побоч-
ной реакции, причем с наибольшей скоростью в случае иодидов:
RMgl + Rl —> MgI2 + R2
Исключение составляет иодистый метил СН31, широко исполь-
зуемый для реакции Гриньяра.
При работе с легко реагирующими бромистыми и иодистыми
алкилами магний применяют в виде стружки, для более трудно
реагирующих веществ применяют мелкие опилки или даже поро-
шок.
Во многих случаях, особенно при введении в реакцию галоген-
арилов, магний необходимо активировать. Обычно в качестве
активирующего вещества применяют иод. К рассчитанному
9*
259
количеству магния в абсолютном эфире прибавляют немного эфир-
ного раствора галогенпроизводного и, если реакция не начинается
даже при подогревании колбы, вносят кристаллик иода, при этом
раствор мутнеет и эфир закипает от выделяющегося при’реакции
тепла. Затем вводят по каплям эфирный раствор галогенпроизвод-
ного с такой скоростью, чтобы эфир продолжал равномерно
кипеть.
Активировать магний в процессе реакции можно также и дру-
гим способом, а именно при помощи реакционноспособных гало-
геналкилов, например бромистого этила. Это необходимо в тех
случаях, когда малоактивные галогенпроизводные образуют с
магнием плохо растворимые в эфире магнийорганические соеди-
нения, покрывающие поверхность магния и препятствующие даль-
нейшему вступлению магния в реакцию с исходным галогенпроиз-
водным. При введении бромистого этила, который реагирует с
магнием, образуя этилмагнийбромид (C2H5MgBr), происходит
очистка поверхности магния от малорастворимого магнийоргани-
ческого соединения. При проведении этой реакции магний берется
в избытке по отношению к исходному галогенпроизводному.
Магнийорганические соединения в силу своей способности
вступать в реакцию с различными органическими и неорганиче-
скими соединениями имеют очень большое препаративное значение
в органической химии, особенно для получения спиртов, кислот,
альдегидов, кетонов, углеводородов.
1. ПОЛУЧЕНИЕ УГЛЕВОДОРОДОВ
Магнийорганические соединения легко реагируют с вещества-
ми, содержащими подвижный атом водорода, такими как вода,
спирты, фенолы, оксимы, амины (в том числе и гетероциклические
соединения — пиррол и индол), кислоты, амиды, меркаптаны, аце-
тилен и моноалкилацетилены, образуя углеводороды. Например:
RMgX + Н2О —> RH+MgXOH
2RMgX + NH4C1 —> 2RH + MgXCl + MgXNH2
Эта реакция пригодна также для получения таких магнийорга-
нических соединений, которые обычным путем, т. е. действием
магния на соответствующие галогенпроизводные, получаются с
трудом или совсем не получаются. Так, например, взаимодей-
ствием ацетилена с бромистым или иодистым этилмагнием полу-
чают магнийбром- или магнийиодпроизводное ацетилена:
СН=СН + 2C2HsMgBr —> BrMgfeCMgBr + 2С2Нв
В 1902 г. Л. А. Чугаев предложил применять иодистый метил-
магний для количественного определения гидроксильных и других
групп, содержащих подвижный атом водорода:
ROH + CH3MgI —> ROMgl + СН4
260
Ф. В. Церевитинов, основываясь на этих реакциях, разработал
метод (прибор) количественного определения подвижного водорода
в органических соединениях по объему выделяющегося метана.
В настоящее время этот метод определения активного (под-
вижного) атома водорода называется методом Чугаева — Цереви-
тинова. Позднее этот метод был несколько видоизменен А. П. Те-
рентьевым.
Таким образом, реакции между магнийорганическими соеди-
нениями и соединениями с подвижным атомом водорода имеют
важное значение. Они используются для качественного и количе-
ственного определений функциональных групп, содержащих под-
вижный атом водорода, для получения углеводородов из галоген-
производных и магнийорганических соединений ряда ацетилена,
флуорена, пиррола, индола и т. п.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ АКТИВНОГО ВОДОРОДА
ПО ЧУГАЕВУ — ЦЕРЕВИТИНОВУ
Метод основан на взаимодействии магнийиодметила с иссле-
дуемым веществом:
RH + CH3MgI —> RMgl + СН4
При этом выделяется эквимолекулярное количество метана,
объем которого измеряют бюреткой. Обычно реакцию проводят в
среде амилового эфира, анизола, ксилола, пиридина. Если реак-
ция протекает легко при 20 °C, то можно применять диэтиловый
эфир.
Все реактивы, используемые для анализа, должны быть тща-
тельно обезвожены, так как магнийиодметил разлагается в при-
сутствии влаги.
Реактивы, посуда и приборы
Анизол.............19,9 г, или 20 мл Колба круглодонная (100 мл) 1
(0,18 моль) Холодильник Либиха..........1
Магниевые стружки ..2г (0,08 г-атбм) Трубка хлсркальциевая . . . . 1
Приемник .................. 1
Йодистый метил .... 6,9 г, или 3 мл
(0,04 моль)
Иод
Получение реактива Гриньяра (магнийиодметила). В кругло-
донную колбу емкостью 100 мл помещают 20 мл анизола, 2 г маг-
ниевых стружек, высушенных в сушильном шкафу в течение
10 мин, 3 мл йодистого метила и несколько кристалликов иода.
Если при комнатной температуре реакция не начинается, смесь
слегка /подогревают. После окончания реакции смесь нагревают
в течение 1 ч на кипящей водяной бане, присоединив к колбе об-
ратный холодильник с хлоркальциевой трубкой. Затем, заменив
обратный холодильник нисходящим, отгоняют не вошедший в
реакцию иодистый метил. Раствор магнийиодметила сливают с
261
оставшегося после реакции магния и хранят в склянке, хорошо за-
крытой корковой пробкой, залитой парафином (срок хранения 3—-
4 недели).
Выполнение синтеза
Рис. "96. Прибор для коли-
чественного определения
активного водорода по Чу-
гаеву — Церевитинову:
/ — реакционный сосуд; 2—ре-
зервуар для магнийиодметила;
5—водяная баня; 4—газовая
бюретка; 5—-отводная " трубка;
водяная рубашка газовой
бюретки; 7—кран; в—уравни-
тельный баллои; 9,10—воронки.
Прибор для определения активного водорода (рис. 96) состоит
из реакционного сосуда 1 с резервуаром 2 для магнийиодметила.
Сосуд 1 соединен при помощи резиновой трубки с газовой бюрет-
кой 4, в которой собирают выделяющийся метан и измеряют его
объем. Реакционный сосуд 1 и газовая
бюретка 4 помещены в водяные бани 3 и
6 с одинаковой температурой.
Перед началом анализа через реак-
ционный сосуд в течение 15 мин пропу-
скают сухой воздух или сухой азот, так
как для получения точных результатов
прибор должен быть абсолютно сухим.
Укрепив реакционный сосуд 1 на
штативе в вертикальном положении, вво-
дят в него 100—200 мг исследуемого ве-
щества через воронку 9. Затем наливают
в сосуд через ту же воронку 3,5 мл ани-
зола, стараясь смыть с воронки в сосуд
остатки вещества. Когда вещество рас-
творится, осторожно вносят в резервуар
2 при помощи изогнутой воронки 10 5 мл
приготовленного реактива Гриньяра. Хо-
рошо закрывают реакционный сосуд ре-
зиновой пробкой с отводной трубкой 5
и соединяют эту трубку с газовой бюрет-
кой 4.
После этого для создания постоян-
ной температуры реакционный сосуд опу-
скают в водяную баню 3. Когда темпера-
тура воды в ней и водяной бане 6, в ко-
торую помещена бюретка 4, уравняется
(приблизительно через 10 мин), быстро
вынимают кран 7 и тотчас же вставляют
его обратно. Это необходимо для созда-
ния в реакционном сосуде атмосферного давления. Затем кран / по-
ворачивают так, чтобы соединить бюретку с атмосферой, и подни-
мают наполненный ртутью уравнительный баллон 8, который вы-
тесняет из бюретки весь воздух.
Поворачивают кран 7 на 90°, опускают уравнительный баллон
и укрепляют его в штативе. Убирают водяную баню и наклоняют
реакционный сосуд так, чтобы весь магнийиодметил перетек из
резервуара 2 в сосуд 1, где находится исследуемое вещество. Про-
исходит выделение метана, под давлением которого понижается
уровень ргути в бюретке. Как только падение уровня ртути в бю-
ретке замедлится, для завершения реакции сосуд 1 нагревают на
водяной бане до 90 °C. После охлаждения его помещают на водя-
ную баню комнатной температуры. Через 10 мин уровни в бюретке
и баллоне 8 выравнивают, измеряют объем собранного газа, тем-
пературу и барометрическое давление. Объем газа приводят к нор-
мальным условиям.
Содержание активного водорода х (в вес. %) рассчитывают
по формуле:
0,000719-У- 1,008
-------------100
где 0,000719 — масса 1 мл метана при 0°С и 760 мм рт. ст.;
У —объем метана, приведенный к 0°С и 760 мм рт. ст.;
1,008 — атомный вес водорода;
16 — молекулярный вес метана;
а — навеска исследуемого вещества, г.
Допустимая ошибка — до 10%.
2. ПОЛУЧЕНИЕ КАРБОНОВЫХ КИСЛОТ
Карбоновые кислоты образуются при выливании реактива
Гриньяра на сухой лед или при пропускании двуокиси углерода
через эфирный раствор магнийорганического соединения:
/> /Ом*х н.о /О
Двуокись углерода необходимо тщательно высушить и пода-
вать в реакционную среду очень медленно, в противном случае
происходит побочная реакция, которая приводит к образованию
кетонов и третичных спиртов:
n OMgX R
I RMgX | RMgX
(Г + RMgX —> С=О -------► R—С=О ------►
I
R
R R
I н2о |
—> R—С—OMgX -------► R—С—ОН
R R
Кроме того, в первой стадии реакции образуется углеводород
__R.
RX + RMgX —> R—R + MgX2
При разложении реакционной смеси водой образуется углево-
дород RH:
RMgX + Н2О —> RH + MgXOH
Выход продукта можно увеличить, если вести реакцию при •
сильном охлаждении. Для этого следует вводить в эфирный
263
раствор магнийорганического соединения кусочки твердой двуокиси
углерода. Для отделения образовавшейся карбоновой кислоты от
побочных веществ (кетона, третичного спирта и углеводородов)
весь продукт реакции обрабатывают водной щелочью до щелочной
реакции Кислота в виде соли переходит в водный раствор В дели-
тельной воронке отделяют его от нерастворимых примесей, добав-
ляют минеральную кислоту до явно кислой реакции, извлекают
органическую кислоту эфиром и дальше обрабатывают обычным
образом.
3. ПОЛУЧЕНИЕ СПИРТОВ
Спирты образуются при взаимодействии магнийорганических
соединений с альдегидами, кетонами и сложными эфирами, в кото-
рых имеется карбонильная группа—С=О со смещенным к атому
П
кислорода электронным облаком —С=О. Следовательно, у атома
углерода возникает некоторый положительный заряд, а у атома
кислорода — частичный отрицательный заряд В магнийгалогенал-
киле RMgBr радикал R ведет себя как нуклеофильная частица,
которая легко присоединяется к углероду карбонильной группы:
R' R'
RMgBr + О=С—R" —► BrMg—О—С—R"
R
Полученный магнийбромалкоголят гидролизуется водой с об-
разованием спирта:
* *
R"—C-O-MgBr + Н—ОН —> R"-C—ОН + MgBr(OH)
I I Основная соль
R R магния
Если в реакцию с магнийорганическими соединениями всту-
пают кетоны, то получащтся третичные спирты:
СН3 СН3
| | н-он
C2H5MgBr + O=C—СН3 —> BrMg—О—С—СН3 ------------►
Магнийбром Ацетон I
ЭТИЛ С2ГТ5
Магнийбромапкоголят
третичного амилового
спирта
СН3
I
—> НО—С—СН3 + MgBrOH
С2н5
Тре гичиый ами
ловый спирт
264
Если в качестве исходного карбонильного соединения в реак-
цию вводят формальдегид, то продуктами реакции являются пер-
вичные спирты
н—он
C2HsMgBr + О=СН2 —> BrMg—О—СН2—C2HS --------->
Магнийброма лкоголят
пропилового спирта
—> НО—СН2—C2HS + MgBrQH
Пропиловый
спирт
Если же в реакцию вводят другие альдегиды, то образуются
вторичные спирты-
н—он
C2HsMgBr + О=СН—СН3 —> Br—Mg—О—СН—С2Н5 ------------>
СН3
Магнийбромалкоголят
вторичного бутилового спирта
—> С2Н5—СН—СН3 + MgBrOH
он
Вторичный бутиловый спирт
4. ПРИМЕРЫ СИНТЕЗОВ ФЕНИЛУКСУСНАЯ КИСЛОТА Формула: —СН2СООН Основные реакции: /) \ абсолютный эфир / \—CH2C1 + Mg > У—CH2MgCl + C^Q —> О —СН2—С—О—MgCl + НС1 —> —CH2MgCl О II -СН2—С—О—MgCl О II —СН2-С—ОН + MgCl2
265
Побочные реакции*:
Реактивы, посуда и приборы
Хлористый бензил . . 12,7 г, или 11,5 мл Колба круглодонная (250 мл) 1
(0,1 моль) Воронка капельная........1
Магний металлический Воронка делительная.....1
(стружка, опилки) 2,4 г (0,1 г-атом) Форштосс двурогий.......1
Холодильник Либиха..........1
Абсолютный диэтило- Склянки Тищенко.........2
вый эфир..........70 мл Трубки хлофкальциевые ... 2/
Соляная кислота Колба Бунзена .............. 1
(d = 1,19)........10 г, или 8,4 мл Воронка Бюхнера.........1
Диэтиловый эфир . . 40 мл Предохранительная склянка 1
Иод кристаллический 0,1—0,2 г Аппарат Киппа...........1
Углекислый газ
Сборка приборов
1. В водяную баню помещают круглодонную колбу (250 мл),
плотно закрывают ее корковой пробкой (резиновые пробки на-
бухают от эфира) с двурогим форштоссом. В прямом горле форш-
тосса укрепляют капельную воронку, а в боковом — обратный
* Приведенные ниже реакции возможны, но ие обязательны,
Ж
холодильник. Как холодильник, так и капельная воронка должны
быть закрыты хлоркальциевыми трубками. Весь прибор должен
быть абсолютно сухим Не следует пробки смазывать глицерином.
• 2 Капельную воронку заменяют заранее подогнанной газопри-
водной трубкой, один конец которой доходит почти до дна колбы,
а другой через склянки Тищенко соединяют с аппаратом Киппа
(или с газовым баллоном).
3 Прибор для отсасывания (см рис. 40).
Выполнение синтеза
В круглодонную колбу емкостью 250 мл помещают несколько
кристалликов иода, 2,4 г магниевых опилок и через капельную во-
ронку по каплям приливают раствор 12,7 г чистого хлористого бен-
зила в 70 мл абсолютного эфира
1 При введении в реакцию трудно реагирующих галогеналкилов магний
приходится активировать иодом Следует остерегаться попадания иода на кожу
2 Магний надо сушить сначала 30 мин при 60—80 °C, затем в вакуум-
эксикаторе и хранить в герметически закупоренных стеклянных банках
3 Хлористый бензил раздражает слизистые оболочки, в больших концен-
трациях обладает общим токсическим действием
После введения */з раствора хлористого бензила начинается
самопроизвольная экзотермическая реакция, которая сопровож-
дается кипением эфира, помутнением раствора и исчезновением
окраски иода Задержка начала самопроизвольной реакции сви-
детельствует о недостаточной чистоте реактива. В этом случае
следует слегка подогреть баню, с началом реакции нагревание
нужно прекратить
Дальнейшее введение эфирного раствора хлористого бензила
следует производить с такой скоростью, чтобы эфир слегка кипел.
При этом конденсат эфира должен стекать из обратного холодиль-
ника не непрерывной струей, а быстрыми каплями. Необходимо
остерегаться введения больших порций раствора, так как это мо-
жет вызвать слишком бурную реакцию, в результате которой
образуется большое количество побочных продуктов и, следова-
тельно, уменьшается выход фенилуксусной кислоты
После введения всего количества эфирного раствора хлори-
стого бензила кипение постепенно прекращается Содержимое кол-
бы нагревают на водяной бане еще 0,5 ч. При этом неизменным
может остаться только незначительное количество магния, равное
взятому избытку. Большое количество непрореагировавшего маг-
ния в остатке свидетельствует о слишком бурной реакции или же
о недостаточной очистке и сушке реактивов В этих случаях само-
произвольная реакций обычно начинается с опозданием
Затем колбу снаружи сильно охлаждают смесью льда и соли
(или водой с измельченным льдом). Через эфирный раствор маг-
нийорганического соединения (обычно мутный, серый или корич-
невый) пропускают в течение 2—3 ч углекислый газ из аппарата
Киппа или из баллона, тщательно осушенный пропусканием через
267
две склянки Тищенко с концентрированной серной кислотой. Угле-
кислый газ нужно подавать очень медленно, иначе возможна по-
бочная реакция.
1 Насыщение углекислым газом следует проводить при температуре
—10—0 °C, так как ею взаимодействие с магнийорганическим соединением экзо-
термично и может привести к испарению большого количества эфира.
2 Выход фенилуксусной кислоты можно повысить, вводя в реакцию твер-
дую двуокись углерода, при этом реактив Гриньяра сливают на сухой лед
3 За счет тепла, выделяющегося при реакции, эфир начинает кипеть, окон-
чание его кипения служит признаком конца реакции
Колбу с раствором вновь охлаждают, вводя в нее 15—20 г
льда. После окончания реакции газоприводную трубку заменяют
капельной воронкой. Продукт реакции из капельной воронки гидро-
лизуют подкисленной водой— 10 г НС1 (d — 1,19) в 20 мл воды —
до кислой реакции по конго красному. Затем добавляют немного
эфира, если он сильно испарился во время реакции. Происходит
разделение реакционной смеси на два прозрачных слоя, которые
отделяют друг от друга в делительной воронке. Водный раствор
дважды экстрагируют эфиром (по 20 мл). Соединенные эфирные
растворы фенилуксусной кислоты обрабатывают разбавленным
раствором едкого натра до щелочной реакции, освобождая этим
кислоту от побочных продуктов.
При добавлении раствора едкого натра фенилуксусная кис-
лота переходит в соль, растворимую в воде, нейтральные же про-
дукты, являющиеся примесями, остаются в эфирном растворе.
Нижний щелочной раствор отделяют от эфира, подкисляют
10% раствором НС1 и охлаждают. Перед подкислением щелочной
раствор полезно несколько упарить для освобождения от раство-
ренного в нем эфира. Выпавшую фенилуксусную кислоту отсасы-
вают на воронке Бюхнера и, если продукт недостаточно чист, пере-
кристаллизовывают из горячей воды. _
Выход фенилуксусной кислоты ~8 г. | "о
Фенилуксусная кислота — светло-желтый или светло-зеленый кристалличе-
ский порошок Мол вес 136,15; т пл 72—77 °C, т кип 265,5 °C Фенилуксусная
кислота трудно растворяется в холодной воде, лучше — в горячей, хорошо рас-
творима в спирте и эфире
Применяется в органическом синтезе
Отличительная реакция
При нагревании с концентрированной серной кислотой фенил-
уксусная кислота разлагается с выделением углекислого газа.
ТРИФЁНИЛКАРБИНОЛ
Формула;
ОН
СвН5-С-С6Нб
I
СвНа
168
Основные реакции:
абсолютный эфир
С6Н6Вг + Mg----------------> C6H5MgBr
С6Н5—С—C6HS + C6H5MgBr —>
OMgBr
C6H6-C-C6HS
I
c6H5
OMgBr
I
C6H5-C-C6H5 + HC1
I
c6H5
OH
C6H5-C-CeH6+MgClBr
I
c6H5
Побочная реакция:
CeH5MgBr + CeH5Br
Реактивы, посуда и приборы
—> СеН5—CsHs + MgBr2
Дифенил
Бромбензол............20 г, или
(0,12
Магний металлический
(стружки) ............3 г (0,12 г
Абсолютный диэтиловый
эфир..................80 мл
Бензофенон............18,2 Г (0,1
Диэтиловый эфир .... 40 мл
Иод
Соляная кислота
Кислый углекислый натрий
13 мл Колба круглодонная трехгор-
моль) лая (300 мл)....................1
Мешалка..................1
-атом) Холодильник Либиха........1
Воронка капельная ........... 1
Воронка делительная ......... 1
Трубка хлоркальциевая . . . . 1
моль) Колба Вюрца (300 мл) . . . . 1
Термометр................1
Алонж....................1
Приемник .................... 1
Прибор для перегонки с водя-
ным паром.................1
Колба Бунзена............1
Воронка Бюхнера...........1
Предохранительная склянка 1
Сборка приборов
1. Трехгорлую круглодонную колбу емкостью 300 мл, помещен-
ную на водяную баню, снабжают мешалкой, капельной воронкой
и обратным водяным холодильником с хлоркальциевой трубкой.
2. Колбу Вюрца емкостью 300 мл помещают на водяную баню,
снабжают термометром и нисходящим водяным холодильником с
алонжем, конец которого погружают в приемник.
3- . Прибор для перегонки с водяным паром (см. рис. 48).
4. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную трехгорлую колбу емкостью 300 мл помещают
3 г стружек магния и несколько кристалликов иода. Туда же вли-
вают 15 мл абсолютного эфира и ’/з раствора (13 мл) бромбензола
в 40 мл абсолютного эфира Через несколько минут начинается са-
мопроизвольная реакция и эфир закипает (см. стр. 267). Задержка
269
начала реакции свидетельствует о недостаточной чистоте реакти-
вов. После начала самопроизвольной реакции в колбу через ка-
пельную воронку постепенно приливают остаток эфирного рас-
твора бромбеизола, таким образом регулируя его подачу, чтобы
эфир кипел интенсивно, но не слишком бурно. После введения
всего эфирного раствора бромбеизола реакционную смесь нагре-
вают на водяной бане, поддерживая легкое кипение эфира в тече-
ние 30 мин.
Реакция считается законченной, когда остается лищь не-
сколько маленьких кусочков магния (см. стр. 267) и раствор ста-
нет мутным или коричневым.
Затем колбу с полученным магнийбромфенилом снаружи
охлаждают холодной водой со льдом и медленно приливают охла-
жденный раствор 18,2 г бензофенона, предварительно растворен-
ного в 25 мл эфира, после чего колбу нагревают на водяной бане
30 мин. Затем колбу снова охлаждают водой со льдом и при по-
стоянном помешивании, не разбирая прибора, гидролизуют про-
дукт реакции охлажденной водой, после чего приливают мини-
мальное количество наполовину разбавленной концентрированной
соляной кислоты, необходимое для полного растворения осадка
основной соли магния.
Получившиеся водный и эфирный слои разделяют в делитель-
ной воронке и из водного слоя делают одну эфирную вытяжку
20 мл обычного эфира. Эфирную вытяжку промывают раствором
кислого углекислого натрия для удаления желтой окраски, затем
отгоняют эфир на водяной бане (прибор 2), после чего подвергают
смесь перегонке с водяным паром для удаления непрореагировав-
шего бромбеизола и побочно образовавшегося дифенила.
Оставшийся в колбе в виде кристаллической массы трифенил-
карбинол отсасывают на воронке Бюхнера и сушат.
Выход трифениЛкарбинола ~ 13 г.
Трифеиилкарбииол — бесцветный кристаллический продукт с т. пл. 162 °C,
который легко перекристаллизовывается из спирта, бензола или лигроина.
Отличительная реакция
При внесении кристаллика трифенилкарбинола в пробирку
с 2 мл концентрированной серной кислоты появляется желтое
окрашивание:
(С3Н5)3СОН + H2SO4 =?=> [(C6H5)3C]+HSO7 + Н2О
ДИФЕНИЛКАРБИНОЛ (БЕНЗГИДРОЛ)
Формула:
С6Н6-СН—свн»
он
270
Основные реакции:
абсолютный эфир
с3н6Вг + Mg ---------------------------:----> C3HsMgBr
ceH5MgBr + с6н5-с;
C3H5v
yZH—OMgBr
С6Н5/
C6HS4
'CH-OMgBr + HC1 —>
C6HS/
C6H6-CH-C6HS + MgClBr
OH
Побочная реакция:
C3HsMgBr + C3HsBr
—> C3HS-C3HS +MgBr2
Дифенил
Реактивы, посуда и приборы
Бромбензол............23,5 г, или 16 мл
(0,14 моль)
Магний металлический
(стружки).............3,6 г (0,14 моль)
Бензальдегид..........11,9 г, или 11 мл
(0,11 моль)
Абсолютный диэтило-
вый эфир..............100 мл
Соляная кислота
(d = 1,19).............12 мл
Диэтиловый эфир ... 40 мл
Кислый сернокислый
натрий, 40%-ный рас- ~
твор.....................10 мл
Колба круглодонная, трехгор-
лая (500 мл)..............1
Мешалка ...................1
Воронка капельная..........1
Воронка делительная ... 1
Холодильник Либиха.........1
Трубка хлоркальциевая . . 1
Колба Вюрца (300 мл) . . . . 1
Термометр..................1
Алонж.................... . 1
Приемник ................. 1
Кислый углекислый натрий
Иод
Сборка приборов
1. Круглодонную трехгорлую колбу емкостью 500 мл, по-
мещенную на водяную баню, снабжают мешалкой, капельной во-
ронкой и обратным водяным холодильником с хлоркальциевой
трубкой.
2. Колбу Вюрца емкостью 300 мл помещают на водяную баню
и снабжают термометром и нисходящим водяным холодильником
с алонжем, опущенным в приемник.
Выполнение синтеза
В круглодонную трехгорлую колбу емкостью 50 мл прибора 1
помещают 3,6 г магниевых стружек, приливают 20 мл абсолютного
диэтилового эфира и прибавляют кристаллик иода. Затем из ка-
пельной ворбнкй приливают 7з раствора, содержащего 16 мл
бромбеизола в 50 мл абсолютного эфира, и ждут или легким на-
греванием вызывают самопроизвольную реакцию (см. стр. 267).
Затем из капельной воронки приливают оставшийся эфирный
271
раствор бромбензола, чтобы эфир умеренно кипел. После прибав-
ления всего раствора бромбензола колбу нагревают на водяной
бане в течение 0,5—1 ч. К концу реакции в колбе остаются только
незначительные остатки магния.
К полученному охлажденному раствору магнийбромфенила
постепенно, из капельной воронки, при перемешивании и наруж-
ном охлаждении приливают раствор 11 мл свежеперегнанного
бензальдегида в 30 мл абсолютного диетилового эфира. Не следует
допускать избытка бензальдегида, в противном случае идут по-
бочные реакции с образованием кетона, а также первичного и вто-
ричного спиртов.
После прибавления всего бензальдегида колбу нагревают на
водяной бане в течение 30 мин, чтобы довести реакцию до конца.
Снова охлаждают колбу снаружи водой со льдом, прибавляют к
реакционной смеси 40 г льда и полученное соединение разлагают
раствором 12 мл соляной кислоты в 12 мл воды.
При использовании более концентрированной соляной кислоты может про-
изойти дегидратация дифенилкарбинола с образованием дибензгидрилового
эфира:
CeHj СвН5
2C6HSCH(OH)C6HS —> сн—о—сн + н2о
<Lhs ieHs
Получившиеся два слоя, верхний — эфирный и нижний — вод-
ный, разделяют в делительной воронке, из водного слоя делают
две эфирные вытяжки — по 20 мл эфира каждая.
Объединенные эфирные вытяжки промывают 10 мл 40%-ного
раствора кислого сернистокислого натрия для удаления непро-
реагировавшего бензальдегида, затем раствором кислого углекис-
лого натрия для удаления следов сернистой кислоты и сушат без-
водным хлористым кальцием, после чего эфир отгоняют на водя-
ной бане (прибор 2):
п 0Н
C6HS—С; +NaOSO2H —> CeH6-C-SO3Na
\н I
н
Небольшой остаток эфирного раствора переносят в фарфоро-
вую чашку; после испарения эфира образуется твердая масса
дифенилкарбинола.
Если дифенилкарбинол из-за примесей не затвердевает,
остается в виде густого масла, то его следует поместить в колбу и
отогнать примеси с водяным паром. Оставшийся в перегонной
колбе дифенилкарбинол после охлаждения выделяется в твердом
виде. Его отфильтровывают и перекристаллизовывают из лигроина
или смеси бензина с петролейным эфиром (1:1 по объему).
Выход дифенилкарбинола ~15 г.
Дифенилкарбинол (бензгидрол) — бесцветное кристаллическое вещество;
т. пл 68 °C.
272
Глава XVII
РЕАКЦИЯ КАННИЦЦАРО
В 1853 г. Канниццаро установил, что ароматические альде-
гиды под действием концентрированного раствора щелочи претер-
певают окислительно-восстановительное превращение (диспропор-
ционирование). Примером этой реакции может служить превраще-
ние бензальдегида в бензиловый спирт и бензойную кислоту:
2С6Н5С< +НСГ —> СвН5СН2ОН + С5Н5СОО
\н
В реакцию Канниццаро вступают не только ароматические
альдегиды, но и муравьиный альдегид и те альдегиды, у которых
карбонильная группа связана с третичным атомом углерода, на-
пример:
(СНз)3С-С:
Большинство альдегидов жирного ряда не вступает в эту ре-
акцию, так как в слабощелочной среде они конденсируются по типу
альдольного уплотнения.
Широкое применение находит так называемая «перекрестная»
реакция Канниццаро, в которой ароматический и гетероцикличе-
ский альдегиды восстанавливаются до соответствующего спирта
под действием муравьиного альдегида, окисляющегося при этом
в муравьиную кислоту:
Ч—+Н-С^ + NaOH —> —СН2ОН + Н—
\==/ \н \н \=/ XONa
Эта «перекрестная» реакция Канниццаро используется для
восстановления большого числа различных альдегидов. Так, на-
пример, этой реакцией можно получить фурфуриловый спирт из
фурфурола. /
СИНТЕЗ БЕНЗОЙНОЙ КИСЛОТЫ И БЕНЗИЛОВОГО СПИРТА
Формулы:
СООН
•СНгОН
273
Основные реакции}
Реактивы, посуда и приборы
Бензальдегид .............. 20 г
(~ 0,2 моль)
Едкое кали.................18 г
Этиловый эфир .............45 мл
Кислый сернистокислый на-
трий, 40%-ный раствор ... 10 мл
Углекислый натрий
Сернокислый натрий, безводный
Соляная кислота
Реактивная склянка или плоско-
донная колба (100 мл)..........1
Холодильник воздушный.........1
Воронка делительная .......... 1
Колбы Вюрца (100 мл)...........2
Холодильник Либиха.............1
Термометр......................1
Алоиж..........................1
Приемники .................... 2
Колба Бунзена..................1
Воронка Бюхнера................1
Предохранительная склянка . . . 1
Сборка приборов
1. Колбу Вюрца емкостью 100 мл, помещенную на водяную
баню, соединяют с термометром и нисходящим водяным холодиль-
ником с алонжем, который опускают в приемник.
2. Колбу Вюрца емкостью 100 мл снабжают термометром и
нисходящим воздушным холодильником, который опускают в при-
ёмник.
3. Прибор для отсасывания (рис. 40).
Выполнение синтезе
В плоскодонную колбу или реактивную склянку емкостью
100 мл помещают 20 г свежеперегнанного бензальдегида и при
охлаждении вносят холодный раствор 18 г едкого кали в 12 мл
воды. Закрывают колбу пробкой с боковой прорезью и содержи-
мое колбы энергично встряхивают до тех пор, пока не образуется
стойкая эмульсия. После этого смесь оставляют на 10—12 ч.
Затем к образовавшейся смеси бензилового спирта и кристалли-
ческой массы бензойнокислого калия прибавляют небольшое коли-
чество воды, необходимое для растворения выделявшихся кристал-
лов, и переливают раствор в делительную воронку. Следует
избегать большого количества воды, тдк как бензиловый спирт
частично растворяется в воде (в 100 мл воды растворяется 4 г при
17°C). Из этого раствора извлекают бензиловый спирт диэтило-
вым эфиром, делая 2—3 эфирные вытяжки, по 15 мл каждая.
Эфирные вытяжки соединяют и встряхивают, добавляя дважды по
5 мл 40 %-кого раствора кислого сернистокислого натрия.
274
Эфирные вытяжки обрабатывают концентрированным раствором бисуль-
фита натрия для удаления иепрореагировавшего бензойного альдегида, который
образует бисульфитное производное, не растворяющееся в эфире:
Затем промывают эфирный слой раствором углекислого нат-
рия для освобождения от сернистой кислоты, Которая может быть
в растворе бисульфита натрия. Перелив органический слой из де-
лительной воронки в колбу, сушат его безводным сернокислым
натрием или безводным углекислым калием. Высушенный эфир-
ный раствор переливают в колбу Вюрца емкостью 100 мл, тща-
тельно отгоняют эфир на водяной бане (прибор 1), а затем пере-
гоняют бензиловый спирт (прибор 2), нагревая колбу горелкой
через асбестовую сетку или небольшим коптящим пламенем. Со-
бирают фракцию, кипящую в пределах 204—206°С.
Выход бензилового спирта ~8г.
Бензиловый спирт (фенилкарбинол) — бесцветная жидкость со слабым
приятным запахом; плохо растворим в воде, хорошо растворяется в спирте,
хлороформе, этиловом эфире. Мол. вес 108,13, т. кип. 205,8 °C при 750 мм рт. ст.
или 93 °C при 10 мм рт. ст.; т. пл. —15,3 °C; d2° = 1,0455; — 1,5396.
Применяется как растворитель лаков, а также в парфюмерной промыш-
ленности.
Воднощелочной раствор, слитый из делительной воронки после
экстрагирования эфиром, подкисляют соляной кислотой. Выпав-
шую бензойную кислоту отсасывают на воронке Бюхнера и сушат.
Для получения более чистой бензойной кислоты следует перекри-
сталлизовать ее из кипящей воды.
Выход бензойной кислоты ~9 г.
, Бензойная кислота — бесцветные блестящие листочки, плохо растворяется
в холодной воде, лучше — в горячей (в 100 мл воды растворяется 0,2 г при
17°C и 2,2 г При 75°C), хорошо растворяется в хлороформе, ацетоне, четырех-
хлористом углероде, бензоле, метиловом спирте Мол. вес 122,05; т. кип. 250 °C; ,
т. пл. 122,3 °C; Д'5 = 1,2659.
Бензойная кислота и ее производные широко применяются в синтезе кра-
сителей, фармацевтических препаратов, в пищевой промышленности.
Отличительные реакции бензилового спирта
1. При окислении бензилового спирта хромовой смесью или
марганцовокислым калием образуется бензойная кислота.
2. К 2 мл разбавленной азотной кислоты прибавляют 1 каплю
бензилового спирта и погружают пробирку в кипящую воду — об-
разуется желтоватая эмульсия и появляется сильный запах бенз-
альдегида,
275
Отличительная реакция бензойной кислоты
К 1 мл раствора бензойной кислоты прибавляют по 1 капле
0,3% раствора перекиси водорода и раствора хлорного железа.
Пробирку с этой смесью погружают в кипящую воду. Вследствие
образования салициловой кислоты появляется красно-фиолетовая
окраска.
Глава XVIII
РЕАКЦИЯ КЛАЙЗЕНА
Реакция Клайзена относится к типу альдольных реакций и за-
ключается в конденсации сложных эфиров с соединениями, содер-
жащими активную метиленовую группу (сложные эфиры, кетоны,
нитрилы), в присутствии основных катализаторов (металлический
натрий, амид натрия и некоторые магнийорганические соедине-
ния). Так, например, при конденсации сложных эфиров образуют-
ся р-кетоэфиры:
NaOC2H5
R—СН2—COOR' + R—СН2—COOR' ; R—СН2—С—СН—COOR' + R'OH
При конденсации сложных эфиров с кетонами образуются
р-дикетоны:
NaOC2Hs
Rj—COOR' + R-CH2— C—R2 .....Rj-C-CH-C—R2 + R'OH
Во всех случаях получения p-кетоэфиров и р-дикетонов необ-
ходимо применять чистые безводные исходные вещества и тща-
тельно высушенную аппаратуру, защищенную от доступа влаги.
Классическим примером сложноэфирной конденсации являет-
ся синтез ацетоуксусного эфира из уксусноэтилового эфира в при-
сутствии металлического натрия или этилата натрия.
В настоящее время установлено, что конденсирующим сред-
ством является не металлический натрий, а алкоголят натрия (для
получения ацетоуксусного эфира — этилат натрия). Металличе-
ский натрий, вводимый в реакцию, взаимодействует с этиловым
спиртом, следы которого всегда имеются а уксусноэтиловом эфире.
Выделяющийся во время реакции этиловый спирт постепенно рас-
творяет натрий и, таким образом, этилат натрия все время обра-
зуется по мере течения реакции:
2C2H5OH + 2Na —► 2C2HsONa + Н2
276
Однако наличие больших количеств этилового спирта в уксуС*
ноэтиловом эфире вредно, так как сложноэфирная конденсация
обратима: ацетоуксусный эфир под действием этилового спирта и
этилата натрия распадается на две молекулы исходного сложного
эфира.
Под влиянием этилата натрия образуется натрийзамещенный
уксусноэтиловый эфир:
О
СН3—С—ОС2Н5 + C2H6ONa —>
О
..nCll
СН2—С—ОС2Н5
Na+ + С2Н6ОН
Полученное соединение реагирует с другой молекулой уксусно-
этилового эфира, образуя комплекс I:
О
.. n С II
СН2-С-ОС2Н6
о
II
Na++ СН3—С—OC2HS —»
Комплекс I отщепляет молекулу С2Н5ОН, образуя натрий-
замещенный эфир [CH3COCHCOOC2H5]~Na+. При действии на него
уксусной кислоты образуется свободная енольная форма ацето-
уксусного эфира, переходящая в кетонную до положения равно-
весия:
[СН3—СО—СН—COOC2H5]"Na+ + СН3СООН —►
—> СН3—С=СН—COOC2HS + CH3COONa
I
ОН
СН3—С=СН—COOC2HS СН3—СО—СН2—COOC2HS
он
Аналогичным образом при конденсации сложных эфиров с ке-
тонами получаются 0-дикетрны.
Примером этого типа конденсации является синтез бензоил-
ацетона из ацетофенона при действии металлического натрия.
Растворителем при сложноэфирной конденсации обычно слу-
жит избыток сложного эфира, однако в некоторых случаях при-
меняют диэтиловый эфир, бензол или толуол.
При синтезе ацетоуксусного эфира избыток уксусноэтилового
эфира не только служит растворителем, но и препятствует обра-
зованию побочных продуктов а-дикетона и ацилоина, которые мо-
гут получаться в присутствии больших количеств натрия.
Сложноэфирная конденсация — частный случай реакции кон-
денсации, которая очень распространена и разнообразна. Целый
ряд органических соединений,' как в синтетической химии, так и
в природе, является результатом конденсации.
хп
ПРИМЕРЫ СИНТЕЗОВ
АЦЕТОУКСУСНЫЙ ЭФИР
Формула:
О О
II II
СН3—С—СН2—С—O-C2HS
Основная реакция (см. стр. 276):
О 0 0
II NaOCjHg II 1|
2СН3—С—О—с2н6 ------->- сн3-с-сн2-с-о-с2н5 + С2Нб0Н
Побочные реакции:
0 0 ОН О
СНз—<5—СН2—С—OC2HS СН3—С=СН—(5—ОС2Н.
о
О II
II с—сн3
/С—OC2HS I
НС/ сн2 _______
II + I
Н3С—Сч /С=0
ХОН с2н5ох
о
II о
с II
нсх хсн—с—СНз
—> [ + 2С2Н5ОН
Н3С—Сх. /С=о
о
Дегидрацетовая
кислота
Реактивы, посуда и приборы
Уксусноэтиловый эфир .... 56 мл, 50 г Колба круглодонная (250 мл) 1
(0,57 моль) Форштосс двурогий.........1
Металлический натрий .... 5 г Холодильник Либиха........1
(0,22 г-атом) Трубка хлоркальциевая . . . 1
Уксусная кислота, 50%-иая . . 30 мл Термометр . ..............1
Хлористый натрий Воронка делительная..............1
Кислый углекислый натрий Колба Вюрца (100 мл).............1
Хлористый кальций, безводный Алонж............................1
Приемники.......................2
Прибор для перегонки в вакууме 1
Сборка приборов
1. В круглодонную колбу емкостью 250 мл вставляют двуро-
гий форштосс, соединенный с наклонно поставленным обратным
холодильником, верхний конец которого закрыт хлоркальциевой
трубкой; прямое горло форштосса закрывают пробкой. Колбу по-
278
мещают на песочную баню. Прибор и исходные продукты должны
быть тщательно высушены.
2. Колбу Вюрца емкостью 100 мл снабжают термометром и
нисходящим холодильником с алонжем, конец которого опускают
в приемник.
3. Прибор для перегонки в вакууме (см. рис. 51).
Выполнение синтеза
В сухую круглодонную колбу (250 мл) (прибор 1) помещают
56 мл уксусноэтилового эфира и через второе отверстие форштосса,
приоткрывая пробку, вносят 5 г мелко нарезанного металличе-
ского натрия. Постепенно начинается слабое выделение водорода
и спустя некоторое время жидкость умеренно закипает.
1. Уксусноэтиловый эфир не должен содержать влаги, но может содержать
2—3% спирта. Если содержание спирта в эфире мало, то реакция идет медлен-
но, если же оно очень велико, то уменьшается выход ацетоуксусного эфира.
Уксусноэтиловый эфир сушат над хлористым кальцием и перегоняют. Работать
с ним следует осторожно, так как уксусноэтиловый эфир огнеопасен и образует
с -воздухом, взрывоопасные смеси.
2. При работе с металлическим натрием необходимо соблюдать все меры
предосторожности. Резать его следует в защитных очках. Не вводить в реак-
цию больших количеств натрия, иначе могут образоваться побочные продукты
(а-дикетон и ацилоин).
После того как течение реакции замедлится, колбу нагревают
на песочной баие в течение 2—3 ч, поддерживая слабое кипение
смеси. Если в колбе осталось немного кусочков натрия, то следует
перевести их в этилат, добавив 2—3 мл этилового спирта. Обра-
зовавшийся окрашенный прозрачный раствор при перемешивании
подкисляют 30 мл 50 %-ной уксусной кислоты до кислой реакции
по лакмусу.
Если в уксусноэтиловом эфире было много влаги, то получается желатино-
образная маеса (едкий натр).
Затем для расслаивания добавляют насыщенный раствор хло-
ристого натрия в количестве, равном объему реакционной смеси.
Если при этом выпадает осадок, его растворяют небольшим коли-
чеством воды при перемешивании. После этого в делительной во-
ронке отделяют верхний слой, состоящий из ацетоуксусного и
уксусноэтилового эфиров, и встряхивают этот слой с небольшим
количеством насыщенного на холоду раствора бикарбоната нат-
рия. Отделенный органический раствор сушат над хлористым
кальцием, а затем отгоняют избыток уксусноэтилового эфира, вы-
кипающего до 95 °C, осторожно нагревая колбу на песочной бане
(прибор 2).
Остаток в колбе перегоняют в вакууме, собирая фракцию с
т. кип. 86—90°C при 30 мм рт. ст. или 69—73 °C при 12 мм рт. ст.
В колбе после отгонки ацетоуксусного эфира остается кри-
сталлизующаяся при охлаждении дегидрацетовая кислота.
Выход ацетоуксусного эфира ~11г,
279
Ацетоуксусный эфир — бесцветная жидкость с освежающим запахом; плохо
растворим в воде, смешивается с органическими растворителями. Мол. вес.
130,14; т. кип. 181 °C (с разложением) при 760 мм рт. ст. или 69—73 °C при
12 мм рт. ст., т. пл. -45 °C; == 1,023; /$=1,4198.
Применяется в производстве красителей, лекарственных веществ и для син-
тетических целей.
Отличительная реакция
Ацетоуксусный эфир с водным раствором хлорного железа
дает красно-фиолетовую окраску.
Ацетоуксусный эфир состоит из'фЗ’Уо кетонной и 7% енольной формы. При- -
сутствие енольной формы может бытЬ-~обнаружено характерной цветной реак-
цией с хлорным железом. Несколько капель ацетоуксусного эфира растворяют в
воде и прибавляют 1—2 капли раствора хлорного железа. Появляется красно-
фиолетовое окрашивание. Если прибавлять бромную воду, то окраска исчезает,
так как бром реагирует с енольной формой эфира. Однако через некоторое вре-
мя окрашивание вновь появляется. Это объясняется тем, что свободная еноль-
ная форма постепенно образуется из кетонной формы эфира до установления
положения равновесия.
БЕНЗОИЛАЦЕТОН
Формула:
с6н3—с—сн2—с—сн3
II II
о о
Основные реакции:
2С2Н6ОН + 2Na 2C2H5ONa + Н2
’ 0 0
C2H3ONa
хсн3 хсн3 Механизм реакции: C2H6ONa /> - СвН6—(Г + ОС2Н5 —> хсн3 с6н5—с—сн2 + С2Н£ 0 ОС2Н5 —свн6-с—сн2-с—сн3 —> А А CeH5—С—СН2—С—СН3 <=2 II II 0 0 Кетонная форма Na+ - ОС2Н5 0 II - свн5—с—сн2 + С2Н5ОН 6+ 0—с—сн3 —> *06- CjHs—с—сн2—с—сн3 + 6с2н5 II II 0 о : CeH6—С—СН=С—СН3 II 1 0 он Енольная форма.
Ж
Реактивы, посуда и приборы
Уксусноэтиловый эфир 18 г, или 20,0 мл
(0,2 моль)
Ацетофенон...............Юг, или 9,7 мл
(0,08 моль)
Натрий металлический 2 г
(0,09 г-атом)
Абсолютный эфир ... 60 мл
Уксусная кислота
Колба круглодонная (250 мМ) 1
Холодильник Либиха..........1
Трубка хлоркальциевая . . . . 1
Воронка Бюхнера.............1
Колба Бунзена...............1
Предохранительная склянка 1
Стакан химический ......... 1
Эксикатор...................1
Сборка приборов
1. Круглодонную колбу емкостью 250 мл, помещенную на во-
дяную баню, соединяют с обратным холодильником, верхний конец
которого закрывают хлоркальциевой трубкой.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную колбу емкостью 250 мл приливают 10 г аце-
тофенона, 18 г уксусноэтилового эфира, 60 мл абсолютного эфира
(см. гл. II) и прибавляют нарезанный тонко металлический нат-
рий.
Ацетофенон — снотворное средство. При работе необходимо
избегать вдыхания его паров.
Уксусноэтиловый эфир обладает наркотическим действием и
раздражает слизистые оболочки.
Через некоторое время эфир закипает. После того как кипение
эфира уменьшается, колбу нагревают на водяной бане в течение
45 мин (прибор 1).
После окончания реакции охлаждают реакционную смесь и
отсасывают на воронке Бюхнера выделившийся натрийбензоил-
ацетон, который промывают сухим эфиром и отжимают между
листами фильтровальной бумаги. Затем натрийбензоилацетон
растворяют в 100 мл воды, фильтруют и, подкисляя раствор уксус-
ной кислотой при одновременном охлаждении льдом, осаждают
бензоилацетон. Выделившиеся кристаллы отсасывают, промывают
водой и сушат в эксикаторе.
Выход бензоилацетона ~8г.
Бензоилацетон (1-фенил-1,3-бутандион) — кристаллы с сильным запахом,
хорошо растворимы в спирте и эфире. Мол. вес 162,19; т. кип. 261—262 °C при
760 мм рт. ст. или 134—136 °C при 16 мм рт. ст.; т. пл. 61 °C; d’°'5 = 1,044.
Бензоилацетон применяется в парфюмерной промышленности.
Отличительная реакция
С водным раствором хлорного железа бензоилацетон дает
красное окрашивание.
181
Глава XIX
РЕАКЦИИ ПОЛИМЕРИЗАЦИИ
И ПОЛИКОНДЕНСАЦИИ
Синтетические высокомолекулярные соединения получают в
результате двух типов реакций: полимеризации и поликонденса-
ции. Эти реакции имеют большое техническое значение, с их по-
мощью получают синтетические смолы, волокна, каучук, широко
применяемые в народном хозяйстве.
1. ПОЛИМЕРИЗАЦИЯ
Полимеризацией называется процесс соединения молекул мо-
номера, приводящий к образованию макромолекул:
пМ —► Мп
где М — молекула мономера;
Мп — макромолекула, состоящая из п мономерных звеньев;
п — степень полимеризации.
Молекулы мономера, вошедшие в состав макромолекул, ста-
новятся ее мономерными звеньями. Элементарный состав макро-
молекул (без учета концевых групп) не отличается от состава мо-
номера.
Известно большое число соединений с кратными связями, ко-
торые склонны к полимеризации, протекающей путем насыщения
валентностей исходных мономерных молекул в результате разрыва
в них кратной связи
;с=с
с=с—с=с
—C=N
или раскрытия цикла в трехчленных циклических окисях.
Полимеризация, в которую вступают молекулы одного веще-
ства, называется гомополимеризацией, например полимеризация
стирола:
282
Если в реакцию полимеризации вступают различные мономе-
ры, она носит название сополимеризации, например сополимери-
зация стирола с метилметакрилатом:
сн3 СНз
сн2=сн + сн2=с —► —сн2—сн—сн2—<i—сн2—сн—
Обязательными стадиями полимеризации являются иницииро-
вание и рост макромолекул. При инициировании в системе возни-
кают активные частицы (А), способные начать реакцию роста с
последовательным присоединением молекул мономера к активной
частице:
А + М —> AM
AM + М —► АМ2 ..,
AMra-i + М —> АМП
Механизм полимеризации определяется химической природой
частиц типа АМП, являющихся промежуточными продуктами по-
лимеризации.
Различают два основных вида полимеризации: цепную и сту-
пенчатую.
Цепная полимеризация. В этом случае начальные продукты
полимеризации не представляют собой устойчивых молекул и не
могут быть изолированы. Продуктами реакции являются вещества
с большим молекулярным весом:
СН=СН2 + СН=СН2 —>----СН—СН2—СН—СН2---
II II
R Ri R Ri
Ступенчатая полимеризация. Продукты реакции могут быть
изолированы на различных стадиях процесса:
R\
r—сн=сн2 + ;с=сн2
н/
R—СН2—СН2—С=СН2
R
В зависимости от механизма реакции полимеризация может
быть радикальной и ионной.
Радикальная полимеризация вызывается (инициируется) веще-
ствами, способными в условиях реакции распадаться на свобод-
ные радикалы (перекиси, персульфаты, диазо- и азосоединения и
другие), а также действием тепла и света.
Радикалы инициаторов входят в состав молекулы полимера,
образуя его конечную группу Обрыв цепи происходит при
столкновении молекулы растущего полимера либо с молекулой
283
специально добавляемого регулятора роста цепи, либо с молекулой
мономера. По этому механизму идет цепная полимеризация:
СвН6СО—О
I —► 2СвН5СОО • —> 2СО2 + 2С6Н6 •
С6Н6СО—О
Перекись бензоила Свободный
(инициатор) фенильный
радикал
свнБ • + сн2=сн —> сБнБ—сн2—сн
I I
R R
с6н6—сн2-сн + сн2=сн —> С6Н6—сн2—сн—сн2—сн
Ионная полимеризация происходит благодаря образованию из
молекулы мономера реакционноспособных ионов в присутствии
катализаторов (кислоты, безводные хлориды алюминия, бора и
г. п.). При ионной полимеризации катализатор регенерируется и
не входит в состав полимера.
Ионная полимеризация может проходить как по цепному, так
и по ступенчатому механизму.
Существует несколько методов осуществления полимеризации:
в блоке, в растворе, в эмульсии. Разновидностью полимеризации
в эмульсии является суспензионная полимеризация.
Полимеризация в блоке — это полимеризация мономера в от-
сутствие растворителя. Если реакцию ведут до полного превраще-
ния мономера, то получают монолит (блок), имеющий форму
сосуда, в который был залит исходный мономер. В данном случае
можно использовать как инициаторы радикальной, так и катали-
заторы ионной полимеризации, растворимые в мономере. Труд-
ность этого способа связана с обеспечением быстрого отвода теп-
ла, выделяющегося при реакции.
Полимеризация в растворе происходит либо в жидкости, сме-
шивающейся с мономером и с образующимся полимером (лаковый
способ), либо в среде, растворяющей только мономер. В послед-
нем случае образующийся полимер выпадает из раствора и может
быть отделен фильтрованием. При этом способе применяют ради-
кальные инициаторы и катализаторы ионной полимеризации, рас-
творимые в реакционной среде. Преимущество этого способа —
легкость отвода тепла; недостаток — трудность отделения от рас-
творителя и необходимость гранулирования полимера.
Полимеризация в эмульсии — наиболее распространенный про-
мышленный способ получения полимеров, при котором полимери-
зацию проводят в жидкой среде (чаще всего в воде), не раство-
ряющей ни мономер, ни полимер. Для стабилизации эмульсии
используют мыла (олеаты, пальмиаты, натриевые соли ароматиче-
ских и высокомолекулярных жирных сульфокислот и др.), а также
поливиниловый спирт, карбоксиметилцеллюлозу и некоторые дру-
284
гие вещества. При этом способе полимеризации обычно применяют
водорастворимые низкотемпературные инициаторы. Наряду с ини-
циаторами в систему вводят регуляторы — буферные вещества (би-
карбонаты, фосфаты, ацетаты щелочных металлов) для поддержа-
ния постоянного pH среды. При эмульсионной полимеризации по-
лимер образуется в виде латекса с размером частиц ~1СН см.
Преимущество этого способа — легкость отвода тепла и получение
продукта высокого молекулярного веса; недостаток — необходи-
мость отмывания полимера от эмульгатора.
Суспензионная полимеризация идет в присутствии эмульгатора
и инициатора. Полимер образуется в виде мелких гранул. Недо-
статок этого способа полимеризации — необходимость отмывания
полимера от стабилизатора.
Очень часто при нагревании происходит процесс, обратный по-
лимеризации, который называется деполимеризацией. Некоторые
циклические димеры, кольца которых образованы лишь углерод-
ными атомами, деполимеризуются очень легко. Однако подавляю-
щее большинство других димеров и полимеров деполимеризуются
лишь при очень жестких условиях.
2. ПОЛИКОНДЕНСАЦИЯ
Поликонденсацией называется процесс образования высокомо-
лекулярных соединений, сопровождающийся одновременным выде-
лением какого-либо низкомолекулярного продукта (вода, галоген-
водород, аммиак, спирт и т. д.):
па—А—а + nb—В—b —► а—(—А—В—)я—b + (2Я — 1) ab
где а и b — функциональные группы (ОН), СООН, NH2, и др).
В отличие от продуктов полимеризации состав элементарного
звена полимера, полученного в результате реакции поликонденса-
ции, не соответствует элементарному составу исходного мономера.
Проведение реакции поликонденсации зависит от химического
строения исходных веществ и получаемых продуктов, от их физи-
ческих свойств, природы побочных продуктов и константы скоро-
сти реакции. Процесс поликонденсации возможен лишь в том слу-
чае, когда исходные вещества имеют не менее двух функциональ-
ных групп, способных участвовать в реакции.
Если в исходном мономере содержатся две функциональные
группы, то в результате реакции получается термопластичный и
плавкий линейный полимер, растворимый в органических раство-
рителях. Если хотя бы в одном из исходных реагентов имеются
три или более функциональные группы, то образуется неплавкий
полимер пространственного строения, нерастворимый в органиче-
ских растворителях. Поэтому поликонденсацию, при которой могут
образоваться пространственные полимеры, следует вести таким;
образом, чтобы не допускать затвердевания продукта в самом
реакционном сосуде. Для получения твердой смолы нужно
285
перелить жидкую смолу в формы, из которых легко извлечь за-
твердевший полимер.
При поликонденсации одноверменно с ростом цепи полимера
протекает обратный процесс — деструкция, которую можно осла-
бить, тщательно удаляя низкомолекулярные соединения из сферы
реакции в соответствии с законом действующих масс.
Скорость реакции поликонденсации можно регулировать, из-
меняя температуру реакционной среды. Так, для замедления реак-
ции нужно охладить реакционный сосуд, Ь котором происходит
процесс.
Ниже приведены несколько типов реакции поликонденсации.
Поликонденсация многоосновных кислот с многоатомными
спиртами. При поликонденсации многоосновных карбоновых
кислот с многоатомными спиртами получаются высокомолекуляр-
ные сложные полиэфирные смолы. Наиболее известный полимер
этой группы — глифталевая смола, которую получают поликонден-
сацией фталевой кислоты (иди ее ангидрида) с глицерином.
В зависимости ют соотношения компонентов и условий реак-
ций можно получать различные по свойствам полиэфиры. Так, при
150 °C фталевый ангидрид в результате взаимодействия с глице-
рином образует вначале линейный полиэфир
• —СОСвН4СООСН2СНОНСН2ООСС6Н4СО—•
представляющий собой мягкую плавкую смолу, растворимую в
спирте, ацетоне и хлороформе. При этой температуре только пер-
вичные спиртовые группы глицерина взаимодействуют с фтале-
вым ангидридом, вторичные же остаются свободными. При даль-
нёйшем нагревании (до 220 °C) и при избытке фталевого ан-
гидрида эти линейные полиэфиры постепенно связываются
(сшиваются) между собой. Сшивание идет за счет вторичных
спиртовых групп глицерина и карбоксильных групп уже образо-
вавшихся линейных полиэфиров. При этом отщепляется вода и
образуется трехмерная жесткая пространственная структура гли-
фталевой смолы.
...—OOC—С6Н4СООСН2СН—СН2—ООС—С6Н4—СООСН2—СН—СН2—ооссвн4—соо—
оос Аос
А0Н4 А0Н4
ооА ооА
Ан 1
Grig :
coo—Ан
А0Щ СНз—ООСС0Н4-СОО—...
СОО--
Пространственный полимер не растворяется в ацетоне, не пла-
вится, но хорошо прессуется. Он обладает большой твердостью,
механической прочностью и стойкостью к действию воды.
286
Поликонденсация фенолов с альдегидами. Реакция между фе-
нолами и альдегидами протекает при нагревании в присутствии
катализаторов (кислот или щелочей). Так, например, при получе-
нии фенолоформальдегидных смол в начальной стадии поликон-
денсации фенола с формальдегидом в присутствии кислот обра-
зуются изомерные диоксидифенилметаны
2,4'«Дноксндифенилметан
2,2'«Д иокс ид ифе и н лме таи
4,4'-Диоксидифеннлметан
в присутствии же щелочей образуются о- и п-оксибензиловые
спирты:
При дальнейшем нагревании эти промежуточные продукты
реакции превращаются в смолы.
В щелочной среде при избытке формальдегида первоначаль-
ный продукт поликонденсации представляет собой плавкое и рас-
творимое в органических растворителях вещество — резольная
смола, резол:
Резольная смола
Резольная смола при нагревании переходит через промежуточ-
ный продукт резитол, который не плавится, не растворяется, но
287
набухает, в резит — неплавкий, нерастворимый и не набухающий
конечный продукт поликонденсации:
Резит
В стадии резита фенолоформальдегидные смолы имеют про-
странственное строение и используются в технике для получения
прессованных изделий.
При поликонденсации фенола с формальдегидом в присут-
ствии кислого катализатора и при избытке фенола образуется
легкоплавкая и растворимая в органических растворителях ново-
лачная смола, новолак:
Новолачная смола
Кислоты, однако, применяются в качестве катализаторов
только в тех случаях, когда нет опасности преждевременного от-
верждения смолы, так как они сильно ускоряют реакцию поликон-
денсации.
Поликонденсация диаминов' и хлорангидридов двухосновных
кислот. В результате этой реакции образуются полиамиды, кото-
рые при соответствующей обработке дают прочные и эластичные
нити. Так, при поликонденсации гексаметилендиамина с хлор-
ангидридом адипиновой кислоты образуется полиамид — най-
лон:
• • -CO(CH2)4CONH(CH2)6NHCO(CH2)4CONH(CH2)6NH—•
288
3. ПРИМЕРЫ СИНТЕЗОВ
ПАРАЛЬДЕГИД
Формула:
Основная реакция:
О
II
ЗСН3С—н
H2SO4
------>.
СНз
сн
(д \)
I I
НС сн—сн,
Нзс/ \сн3
Реактивы, посуда и приборы
Ацетальдегид........50 г, или 64,0 мл Колба круглодонная (500 мл) 1
(1,13 моль) Воронка делительная.......1
Хлористый кальций ..Юг Холодильник Либиха.........1
Серная кислота Колба Вюрца (50 мл)........1
(d = 1,84)........0,3 мл или 0,5 г Дефлегматор.................1
Лед Термометр..................1
Алонж.....................1
Приемник ................ 1
Сборка приборов
1. Круглодонную колбу емкостью 500 мл помещают на водя-
ную баню, снабжают обратным холодильником.
2. Колбу Вюрца емкостью 50 мл снабжают дефлегматором,
соединенным с термометром и нисходящим холодильником с алон-
жем, конец которого опущен в приемник.
Выполнение синтеза
В круглодонную колбу емкостью 500 мл помещают 64 мл све-
жеперегнанного ацетальдегида. Колбу охлаждают водой со льдом
и через холодильник, интенсивно охлаждаемый сильной струей
воды, сверху приливают по каплям 0,3 мл серной кислоты. Смесь
в колбе закипает, так как реакция полимеризации экзотермична.
После окончания реакции содержимое колбы переливают в дели-
тельную воронку, несколько раз промывают водой и сушат над
хлористым кальцием. Затем в приборе 2 отгоняют паральдегид при
122—124 °C.
Выход паральдегида ~37 г.
Паральдегид — жидкость с характерным запахом, плохо растворимая в
воде и хорошо растворимая в спирте, эфире.
10 М. Н. Храмкина
289
Отличительная реакция
При нагревании на водяной бане с небольшим количеством
серной кислоты паральдегид деполимеризуется до ацетальдегида.
ПОЛИСТИРОЛ
Формула:
—СН2—СН—СН2—СН-----
А6Н6 свн6
Основная реакция:
пС6Н5СН=СН2 —►-----СН2—СН—СН2—СН—СН2—СН----
<Lh5 С6Н5 С6Н5
ПОЛУЧЕНИЕ ПОЛИСТИРОЛА
СУСПЕНЗИОННОЙ ПОЛИМЕРИЗАЦИЕЙ СТИРОЛА
В ПРИСУТСТВИИ ИНИЦИАТОРА —ЙЕРЕКИСИ БЕНЗОИЛА
И ЭМУЛЬГАТОРА —ПОЛИВИНИЛОВОГО СПИРТА
Реактивы, посуда и приборы
Стирол...........10 г, или 11 мл Реактор с боковым тубусом 1
(~0,1 моль) Мешалка...................1
Перекись бензоила,, ... 0,2 г Холодильник обратный . . . . 1
Поливиниловый спирт . . 0,234 г Термометр..................1
Воронка Бюхнера...........1
Колба Бунзена.............1
Предохранительная склянка 1
Сборка приборов
1. Реактор снабжают механической мешалкой и обратным хо-
лодильником и помещают на водяную баню (рис. 97), в которую
Рис. 97. Прибор для
суспензионной полимери-
зации:
/ — реакционный сосуд; 2~
мешалка; 3—холодильник;
4—водяная баня.
погружают термометр.
2. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В реактор заливают раствор 0,234 г по-
ливинилового спирта в 60 мл воды и смесь,
состоящую из 10 г свежеперегнанного стиро-
ла и 0,2 г перекиси бензоила.
Стирол может полимеризоваться даже при ком-
натной температуре, поэтому при хранении и транс-
портировке к нему добавляют ингибиторы (обычно
гидрохинон), которые отмывают перед полимериза-
цией.
Осторожно! Перекись бензоила взрыво-
опасна!
Реактор помещают на водяную баню, включают мешалку и
при 80 °C ведут полимеризацию 4—5 ч, строго следя за темпера-
турой и работой мешалки. Как только гранулы полимера станут
290
твердыми, полимеризацию заканчивают. Затем гранулы отфиль-
тровывают, промывают водой и сушат на фильтровальной бумаге.
ПОЛУЧЕНИЕ ПОЛИСТИРОЛА
ПОЛИМЕРИЗАЦИЕЙ СТИРОЛА В РАСТВОРИТЕЛЕ
В ПРИСУТСТВИИ ИНИЦИАТОРА —ПЕРЕКИСИ БЕНЗОИЛА
Реактивы, посуда и приборы
Стирол ...............
Перекись бензоила . . .
Бензол................
Петролейный эфир
20 г, или 2,2 мл
(——0,2 моль)
0,4 г
20 г, или 22,5 мл
Колба круглодонная (150 мл)
Холодильник Либиха.........
Чашка фарфоровая............1
Термометр...................1
Сборка приборов
Круглодонную колбу емкостью 150 мл, снабженную обратным
холодильником, помещают на водяную баню, в которую опускают
термометр.
Выполнение синтеза
В круглодонную колбу емкостью 150 мл помещают 20 г све-
жеперегнанного стирола и 0,4 г перекиси бензоила, затем туда же
добавляют 20 г бензола. Колбу нагревают в течение 4 ч (при-
бор 1) при 90—95 °C. После окончания полимеризации содержи-
мое колбы охлаждают, выливают в фарфоровую чашку и добав-
ляют петролейный эфир. Растворитель сливают, а выпавший оса-
док полистирола помещают в нагретый до 80 °C ваккум-сушильный
шкаф при 500—600 мм рт. ст.
ПОЛУЧЕНИЕ ПОЛИСТИРОЛА ПОЛИМЕРИЗАЦИЕЙ В БЛОКЕ
Реактивы, посуда и приборы
Стирол ...............5 г, или 5,5 мл Пробирка (150X75 мм) . . , , 1
(—'0,05 моль)
Перекись бензоила . . . 0,05 г
В широкую пробирку помещают 5 г стирола, прибавляют
0,05 г перекиси бензоила и осторожно нагревают пробирку на во-
дяной бане в течение 1—1,5 ч. Образуется стекловидный’полимер.
Осторожно! Перекись бензоила взрывоопасна!
Полистирол легко воспламеняется и горит ярким коптящим пламенем. Он
растворяется в диоксане, бензоле, четыреххлористом углероде; не растворяется
в спирте, лигроине; набухает в ацетоне. Молекулярный вес полистирола —
30 000—100 000.
Полистирол применяют в качестве электроизоляционного материала, анти-
коррозионного покрытия химической аппаратуры и аккумуляторов, для изго-
товления предметов домашнего обихода, тары для фармацевтических препара-
тов. Полистирольные лаки применяют как антикоррозионные и электроизоля-
ционные покрытия.
10*
291
Отличительная реакция
При нагревании до 300 °C полистирол деполимеризуется.
ПОЛИМЕТИЛМЕТАКРИЛАТ
Формула:
СН3 СНз
---СН2—С—СН2—С----
Ь)ОСН3 СООСНз
Основная реакция:
СНз СН3
пСН2=С—СООСН3 —>------СН2—<L—СН2— С
СНз СООСНз (к)ОСН3
ПОЛУЧЕНИЕ ПОЛИМЕТИЛМЕТАКРИЛАТА
ПОЛИМЕРИЗАЦИЕЙ В ЭМУЛЬСИИ
В ПРИСУТСТВИИ ИНИЦИАТОРА ПЕРСУЛЬФАТА АММОНИЯ
Реактивы, посуда и приборы
Метилметакрилат ... 10 г, или 10,58 мл Колба круглодонная, трехгор-
. (~0,1 моль) лая (250 мл)...........................................1
Надсернокислый аммо- Холодильник Либиха..........1
' ний...................1г Воронка капельная...........1
Мешалка механическая . . . . 1
Термометр.............'. . . 1
Парообразователь............1
Колба Бунзена...............1
Воронка Бюхнера.............1
Предохранительная склянка 1
Сборка приборов
1. Трехгорлую круглодонную колбу емкостью 250 мл, снаб-
женную обратным холодильником, механической мешалкой и ка-
пельной воронкой, помещают на водяную баню, в которую опу-
скают термометр.
2. Трехгорлую круглодонную колбу (250 мл), снабженную
механической мешалкой и обратным холодильником, соединяют
при помощи газоприводной трубки, доходящей до дна колбы, с
парообразователем.
3. Прибор для отсасывания (см. рис. 40).
Выполнение синтеза
В круглодонную трехгорлую колбу емкостью 250 мл при-
бора 1 наливают 100 мл дистиллированной воды, в которой рас-
творяют 1 г надсернокислого аммония (NH^SsOs. Колбу нагре-
вают на водяной бане до 80 °C При этой температуре и энергии;
292
ном размешивании приливают по каплям из капельной воронки
10 г свежеперегнанного метилметакрилата. Через 4—6 ч нагрева-
ние прекращают и в эмульсию пропускают пар из парообразова-
теля (прибор 2) до коагуляции полимера.
Если полимер не выпадает в осадок, то добавляют небольшое
количество 10%-ного раствора хлористого'натрия или соляной
кислоты. Осадок полимера отфильтровывают на воронке Бюхнера,
промывают и высушивают на воздухе.
Если для коагуляции полимера применялся хлористый натрий,
то осадок полимера промывают до отрицательной реакции на ион
хлора.
ПОЛУЧЕНИЕ ПОЛИМЕТИЛМЕТАКРИЛАТА
СУСПЕНЗИОННОЙ ПОЛИМЕРИЗАЦИЕЙ В ПРИСУТСТВИИ
ИНИЦИАТОРА —ПЕРЕКИСИ БЕНЗОИЛА —
И ЭМУЛЬГАТОРОВ — БУФЕРНЫХ ВЕЩЕСТВ
Реактивы, посуда и приборы
Метилметакрилат ... 50 г, или 52,9 мл
(~0,5 моль)
Динатрийфосфат .... 0,85 г
Моноиатрийфосфат . . 0,05 г
Перекись бензоила . . 1 г .
Полиметакрилат натрия 1 г
Колба круглодонная трехгор-
лая (500 мл).............I
Холодильник ................ J
Мешалка механическая . . . !
Термометр..................i
Фильтр стеклянный, пористый 1
Сборка прибора
1. Круглодонную трехгорлую колбу емкостью 500 мл, снаб-
женную обратным холодильником, термометром и механической
мешалкой, помещают на водяную баню.
Выполнение синтеза
В трехгорлую круглодонную колбу емкостью 500 мл вливают
раствор 1 г полиметакрилата натрия в 150 мл воды, добавляют
буферный раствор из 0,85 г динатрийфосфата и 0,05 г натрий-
фосфата в 5 мл воды, а затем раствор 1 г перекиси бензоила в
50 г свежеперегнанного метилметакрилата. Реакцию ведут в тече-
ние 45 мин при 80—82 °C и постоянном перемешивании.
Скоростью вращения мешалки регулируют размер капель (со-
ответственно, размер гранул). После окончания реакции получен-
ные гранулы отфильтровывают на стеклянном пористом фильтре,
промывают водой и высушивают на воздухе.
Полиметилметакрилат (плексиглас, органическое стекло) растворяется в
ацетоне, бензоле, этилацетате, диоксане, уксусной кислоте и не растворяется
в бензине, эфире, этиловом спирте, четыреххлористом углероде и воде. Он имеет
молекулярный вес порядка 100 000—200 000.
Полиметилметакрилат перерабатывается в изделия литьем под давлением
и экструзией.
293
Отличительная реакция
При нагревании до 400 °C полиметилметакрилат начинает раз-
мягчаться, а при 250—300 °C почти нацело деполимеризуется.
СОПОЛИМЕР СТИРОЛА С МЕТИЛМЕТАКРИЛАТОМ
Формула сополимера:
СНз
---СН2—СН—СН2—i—СН2—СН---
Основная реакция:
СНз
СНз
nCH2=CH + mCH2=i
с6н5
соосн3
—> —СН2—СН—СН2—С—СН2—СН—••
С6Н5 СООСНз i6H5
Свн5
Реактивы, посуда и приборы
Метилметакрилат ... 10 г, или 10,58 мл Колба круглодонная, трехгор-
Стирол
(~0,1 моль)
Надсернокислый аммо-
50 г, или 55 мл
(~ 0,5 моль)
ний ..............
Поливиниловый спирт
Хлористый натрий,
1,45 г
2 г
10%'Ный раствор . .
100 г
лая (500 мл)...............
Холодильник Либиха ........
Воронка капельная .........
Мешалка механическая . . . .
Термометр..................
Воронка Бюхнера ...........
Колба Бунзена .............
Предохранительная склянка
Сборка приборов
1. Колбу круглодонную трехгорлую емкостью 500 мл, снаб-
жают обратным: холодильником, капельной воронкой, механиче-
ской мешалкой и помещают на водяную баню, в которую опускают
термометр.
2. Прибор для отсасывания (рис. 40).
Выполнение синтеза
В круглодонную трехгорлую колбу емкостью 500 мл поме-
щают 2 г поливинилового спирта и 240 мл достиллированной воды.
Поливиниловый спирт вводится в реакцию в качестве эмульгатора.
В полученный раствор вводят 1,45 г надсернокислого аммония
(инициатор), нагревают содержимое колбы до 80 °C и прибавляет
по каплям из капельной воронки смесь 50 г стирола и 10 г метил-
метакрилата Реакция сополимеризации длится 5 ч. После этого
полученную эмульсию разрушают при той же температуре, добав-
294
ляя 10% раствор хлористого натрия до полного осаждения сополи-
мера Эмульсию можно разрушить также обработкой ее водяным
паром.
После отстаивания сополимер отфильтровывают на воронке
Бюхнера, промывают водой и высушивают.
МЕТИЛМЕТАКРИЛАТ (ИЗ ПОЛИМЕТИЛМЕТАКРИЛАТА) *
Формула:
СН2=С—СООСН3
Ан3
Нагревание
-------->• пСН2=С—СООСНз
I
СНз
Основная реакция:
СНз СНз
.—СН2—d—СН2—С—••
I I
СООСНз СООСНз
Реактивы, посуда и приборы
Полиметилметакрилат .... 12,5
Сборка прибора
1. Колбу Вюрца емкостью
нисходящим холодильником с
г Колба Вюрца (50 мл)...........1
Холодильник Либиха.............1
Алонж..........................1
Приемник . '...................1
Термометр .................... 1
50 мл соединяют с термометром и
алонжем, конец которого опускают
в приемник, охлаждаемый водой со льдом.
Выполнение синтеза
В колбу Вюрца емкостью 50 мл помещают 12,5 г измельчен-
ного полиметилметакрилата и нагревают колбу (прибор 1) на не-
большом светящемся пламени горелки или на воронке Бабо. Выше
300°C полиметилметакрилат размягчается и деполимеризуется в
метилметакрилат, который и отгоняется в приемник. Перегонку
ведут до тех пор, пока в колбе не останется небольшое количество
черного остатка Полученный дистиллят еще раз перегоняют при
100—102 °C
Чтобы сохранить метилметакрилат в течение некоторого вре-
мени, к нему прибавляют 0,1 г гидрохинона, который является ин-
гибитором полимеризации.
Выход метилметакрилата ~ 10 г.
Метилметакрилат — бесцветная жидкость с приятным эфирным запахом,
хорошо растворяется в воде
* Получение мономера из обрезков или отходов полиметилметакрилата пу-
тем деполимеризации.
29S
Отличительная реакция
С марганцовокислым калием и содой метилметакрилат дает
реакцию на двойную связь (раствор КМпО4 обесцвечивается).
ГЛИФТАЛЕВАЯ СМОЛА
Основная реакция:
Фталевый ангидрид...........7,4 г
Глицерин....................3,1 г
Стакан химический (50 мл) .... 1
Термометр.......................I
Сборка прибора
1. Химический стакан устанавливают на масляную баню, в ко-
торую опущен термометр. Стакан ие должен касаться дна бани.
Выполнение синтеза
Синтез следует проводить в вытяжном шкафу, так как при
реакции выделяются пары акролеина.
В стакан емкостью 50 мл вносят 7,4 г тонко растертого фтале-
вого ангидрида и 3,1 г глицерина. Тщательно перемешивают смесь
и осторожно нагревают до 150—-180°С; при этом из реакционной
массы выделяются пары воды. Затем повышают температуру до
230°C и нагревают смесь при этой температуре ~40 мин до тех
пор, пока не произойдет вспучивание. После окончания реакции
массу выливают в плоский жестяной сосуд и измельчают.
Выход глифталевой смолы ~8г.
Глифталевая смола — бесцветная твердая хрупкая масса
Применяется для производства лаков и эмалей
Отличительная реакция
При сильном нагревании глифталевая смола разлагается,
образуя белый налет фталевого ангидрида
ФЕНОЛОФОРМАЛЬДЕГИДНАЯ СМОЛА
ПОЛУЧЕНИЕ НОВОЛАЧНОЙ СМОЛЫ
Основная реакция:
ОН
избыток
фенола
кислая
среда
ОН он он
Реактивы, посуда и приборы
Фенол..................38 г Колба круглодонная (300 мл) 1
Формалин, 40 %-ный . . 15 г, или 15 мл Холодильник Либиха......I
Соляная кислота (4=1,19 0,5 г, или 0,42 мл Термометр...............1
Пробирки................. 2
Чашка фарфоровая...........1
Сборка прибора
• Круглодонную колбу емкостью 300 мл снабжают обратным
холодильником и помещают на водяную баню, в которую опускают
термометр.
Выполнение синтеза
В круглодонную колбу емкостью 300 мл помещают 38 г фе-
нола, 15 г 40%-ного формалина и взбалтывают смесь до полного
растворения. Затем в колбу добавляют 0,5 г соляной кислоты
(d — 1,19) и нагревают смесь (прибор 1) на водяной бане при
90—100 °C в вытяжном щкафу в течение 20—40 мин. Как только
29?
начнется бурная реакция, нагревание прекращают, при этом реак-
ция продолжается самопроизвольно. После бурного периода реак-
ции снова продолжают нагревание до тех пор, пока смесь не раз-
делится на два слоя: верхний — водный и нижний — густой светло-
коричневый. Содержимое колбы сливают в фарфоровую чашку,
дают отстояться и сливают верхний водный слой. Оставшуюся в
чашке жидкую смолу промывают теплой водой до нейтральной
реакции по метиловому оранжевому и высушивают, постепенно
нагревая до 200 °C.
Новолачная смола применяется для получения пресс-порошков, идущих на
изготовление изделий технического и бытового назначения
298
Резит
Реактивы, посуда и приборы
Фенол.................8,4 г
Формалин, 40%-ный . . 9,4 г, или 9 мл
Аммиак, 25%-ный ... 0,6 г, или 0,6 мл
Колба круглодонная (100 мл) 1
Холодильник Либиха.........1
Чашка фарфоровая...........1
Термометр ................ 1
Сборка прибора
1. Круглодонную колбу емкостью 100 мл соединяют с обрат-
ным холодильником и помещают на водяную баню, в которую опу-
щен термометр.
Выполнение синтеза
В круглодонную колбу вносят 8,4 г фенола, 9,4 г 40%-ного
формалина, 0,6 г 25%-ного раствора аммиака и нагревают смесь
на водяной бане до 90°C (прибор I). Через 30—40 мин реакцион-
ная смесь мутнеет и разделяется на два слоя: верхний — водный
и нижний — смоляной. Эту смесь нагревают при 90 °C еще 1 ч, за-
тем переливают в фарфоровую чашку, охлаждают и сливают
верхний водный слой. Смоляной слой промывают водой до ней-
тральной реакции на лакмус и нагревают на водяной бане в те-
чение 30 мин при 60°C. Полученный в виде клейкой прозрачной
массы резол выдерживают еще 4—6 ч на воздушной бане при
75 °C. Получается твердая плотная смола — резит.
Выход резита ~ 9 г,
Резольные смолы применяют для изготовления текстолита, пресс-порошков,
и антикоррозионных лаков.
Отличительная реакция
Резит нагревают в пробирке до 300°С, он не плавится,-а раз-
лагается. Выделяющийся формальдегид определяют пробкой с
фуксинсернистой кислотой.
299
Глава XX
ИДЕНТИФИКАЦИЯ
Современные физико-химические методы Исследования — опти-
ческие, спектральные (УФ, ИК, масс-спектроскопия, ЯМР и др.),
рентгеноструктурные — позволяют в короткий срок установить
строение органических соединений.
Наряду с ними находят применение также и химические ме-
тоды определения структуры органических веществ При идентифи-
кации любого вещества сначала следует провести предварительные
испытания, чтобы определить, к какому классу относится исследуе-
мое вещество Предварительные выводы подтверждают соответ-
ствующими качественными реакциями для данного класса.
Для окончательной идентификации исследуемого вещества
следует получить твердое кристаллическое производное с постоян-
ной температурой плавления и определить ее. Рекомендуется по
возможности получить производное с наиболее высокой температу-
рой плавления.
В Приложениях 10—16 приведены температуры плавления
ряда производных некоторых соединений важнейших классов.
Для .проведения работ по идентификации необходимо иметь
набор посуды для работы с малыми количествами вещества, т. е
небольшую воронку Бюхнера, склянку (или пробирку) для отса-
сывания, колбу с обратным холодильником, прибор для перегонки.
Многие реакции можно проводить в пробирках. Для экстрак-
ции лучше пользоваться пробиркой и пипеткой. Перед тем как
отфильтровать кристаллический осадок на воронке Бюхнера,
можно часть осадка отжать на фильтровальной бумаге, промыть
несколькими каплями растворителя и определить температуру
плавления.
Рекомендуется проводить идентификацию веществ, принадле-
жащих к следующим классам: спирты и фенолы, альдегиды и ке-
тоны, карбоновые кислоты и их производные, амины и нитросоеди-
нения, углеводороды и галогенпроизводные.
1. ПРЕДВАРИТЕЛЬНЫЕ ИСПЫТАНИЯ
Делают некоторые выводы на основании запаха и цвета ве-
щества. Затем определяют физические константы. Температуру
плавления определяют в приборе Тиле, температуру кипения — в
простой перегонной аппаратуре. Для небольших количеств лучше
пользоваться микрометодом, который состоит в следующем. 5—
10 капель исследуемого вещества помещают в пробирку. В жид-
кость погружают капилляр, который перепаян в 4—5 мм от ниж-
него открытого конца. Пробирку прикрепляют резиновым колеч-
300
ком к термометру и нагревают в бане (масло, глицерин). Вначале
из капилляра выходят отдельные пузырьки В тот момент, когда
начинает идти непрерывная цепочка пузырьков, нагрев прекра-
щают и дают бане’охладиться.
Температура, при которой выделяется последний пузырек и
жидкость втягивается в капилляр, и есть искомая температура
кипения
После определения температуры кипения проводят дальней-
шие предварительные испытания.
Проба на -сожжение. Несколько кристаллов или капель веще-
ства помещают на металлическую ложечку и медленно сжигают
в пламени горелки Наблюдения во время сжигания позволяют
сделать предварительное заключение, в какому классу принадле-
жит вещество.
Ненасыщенные соединения (в том числе ароматические) го-
рят с большим выделением сажи (коптящим пламенем). Алифати-
ческие углеводороды сгорают светящимся пламенем и с неболь-
шим образованием сажи Спирты, т. е. кислородсодержащие ве-
щества, горят слабо светящимся пламенем. Несгораемый остаток
на шпателе дает основание предположить, что присутствуют ме-
таллы Вещества, содержащие серу, можно узнать по запаху
двуокиси серы.
Проба Бейльштейна. Прокаленную медную проволоку погру-
жают в исследуемое вещество и затем вносят в бесцветное пламя
горелки. Зеленое окрашивание пламени свидетельствует о присут-
ствии галогена в органическом веществе.
Реакция с серной кислотой. В пробирке сильно встряхивают
2 мл холодной концентрированной серной кислоты с 1 мл иссле-
дуемого вещества Не растворяются насыщенные и ароматические
углеводороды, а также их нитро- и галогенпроизводные. Ненасы-
щенные углеводороды, спирты, простые и сложные эфиры раство-
ряются обычно без окрашивания Алифатические карбонильные
соединения растворяются с потемнением.
Перманганатная проба (по Вагнеру). В 2 мл воды, спирта
или ацетона растворяют 0,1—0,2 г исследуемого вещества и по
каплям, при энергичном встряхивании добавляют водный
1—3%-ный раствор марганцовокислого калия (перманганата ка-
лия) Если присутствуют ненасыщенные углеводороды, альдегиды,
муравьиная кислота, многоатомные фенолы, аминофенолы, то окра-
ска перманганата, как правило, исчезает и образуется осадок
двуокиси марганца. Пробу следует считать положительной, если
быстро обесцветилось не менее 10 капель раствора перманганата.
Реакция с бромом. К раствору 0,1—0,2 г исследуемого веще-
ства в 2 мл четыреххлористого углерода прибавляют по каплям
при хорошем охлаждении 4%-ный раствор брома в четыреххло-
ристом углероде. Коричневая окраска брома исчезает в присут-
ствии ненасыщенных соединений в результате реакции присоеди-
нения Если обесцвечивание сопровождается выделением броми-
стого водорода (пузырьки), это служит признаком того, что идет
301
реакция замещения. В данных условиях к замещению водорода
на бром способны фенолы, ароматические амины.
Определение растворимости. В пробирку помещают 0,1—0,2 г
исследуемого вещества и постепенно прибавляют 3 мл раствори-
теля, встряхивая энергично после каждого прибавления. В необхо-
димых случаях подогревают, чтобы добиться растворения.
Поскольку не исключена возможность гидролитического рас-
щепления исследуемого вещества, особенно если нагревают с кис-
лотами или щелочами, после растворения вещество надо по воз-
можности выделить снова и идентифицировать его по температуре
плавления или кипения.
Испытывают растворимость в воде, диэтиловом эфире, 5%-ной
щелочи, 5 %-ном растворе кислого углекислого натрия и 5 %-ной
соляной кислоте. По растворимости в воде и диэтиловом эфире
органические вещества можно разделить на четыре группы:
Группа I (в диэтиловом эфире растворимы, в воде нераст-
воримы или труднорастворимы): углеводороды, галогенпроизвод-
ные углеводородов, высшие спирты, большинство карбонильных
соединений, аминов, карбоновых кислот и их производных, фенолы,
простые и сложные эфиры.
Группа II (в воде растворимы, в диэтиловом эфире нерас-
творимы или труднорастворимы): ацетон, метилэтилкетон, му-
равьиный, уксусный и пропионовый альдегид, муравьиная, уксусная
и пропионовая кислота, многоатомные спирты, амиды кислот,
соли, оксикислоты, ди- и трикарбоновые кислоты.
Группа III (растворимы и в воде, и в диэтиловом эфире):
карбонильные соединения с небольшим молекулярным весом, али-
фатические оксисоединения, алифатические нитрилы, карбоновые
кислоты, оксикислоты, кетокислоты, многоатомные фенолы, амины.
Группа IV (труднорастворимы как в воде, так и в диэтило-
вом, эфире): высокомолекулярные соединения, высшие амиды кис-
лот, антрахиноны.
Такие соединения, как ангидриды и галогенангидриды кислот,
разлагающиеся гидролитически при более или менее продолжи-
тельном действии воды, можно найти в виде продуктов гидролиза
в одной из четырех групп.
В разбавленной соляной кислоте растворяются все вещества,
растворимые в воде (группы II и III), и кроме того вещества
основного характера, т. е. первичные, вторичные и третичные али-
фатические амины, первичные ароматические амины, жирноаро-
матические амины, содержащие не более одного ароматического
остатка. Некоторые кислородсодержащие соединения, например
диэтиловый эфир, бутиловый спирт, низкомолекулярные сложные
эфиры, растворяются в 5 %-ной соляной кислоте.
В разбавленном растворе кислого углекислого натрия раство-
римы все сильнокислые вещества, например карбоновые кислоты.
В разбавленной щелочи растворяются фенолы, первичные и вто-
ричные нитросоединения, арилсульфонильные производные первич-
ных аминов.
302
2. КАЧЕСТВЕННЫЕ РЕАКЦИИ
Ангидриды и' галогенангидриды кислот. Три капли исследуе-
мого вещества вводят в реакцию с пятью каплями анилина. Если
исследуемое вещество твердое, то берут 0,1 г его, растворяют в
1 мл горячего бензола и к прозрачному раствору прибавляют
анилин.
Ангидриды и галогенангидриды кислот реагируют с анилином
с выделением тепла. Продуктом реакции являются твердые веще-
ства — анилиды. Если кристаллы не образуются, кристаллизацию
вызывают потиранием палочкой.
Кислоты. Четыре капли вещества (или 0,1 г) растворяют в
2 мл воды или водного спирта. Раствор дает кислую реакцию на
лакмус; эта реакция сохраняется и после добавления капли
10%-ного раствора соды. Так же ведут себя ангидриды, галоген-
ангидриды кислот и некоторые сложные эфиры.
Амины. Растворимые в воде амины узнают по щелочной реак-
ции их водного раствора (4 капли вещества в 2 мл воды). Щелоч-
ная реакция на лакмус сохраняется и после добавления одной
капли ледяной уксусной кислоты.
Нерастворимые в воде амины, имеющие углеродную цепь из
6—10 атомов, можно узнать по их растворимости в 5%-ной соля-
ной кислоте (4 капли или 0,1 г вещества в 2 мл соляной кислоты).
Нерастворимые в воде амины, имеющие в цепи более 10 атомов
углерода, встряхивают с 5%-ной соляной кислотой (0,1 г веще-
ства, 2 мл соляной кислоты) и фильтруют. К прозрачному филь-
трату добавляют равный объем 10%-ного раствора соды. Образо-
вание осадка или сильное помутнение указывает на присутствие
амина.
Установив наличие амина, необходимо выяснить, является ли
он первичным, вторичным или третичным. Только первичные и
вторичные амины способны к реакциям ацилирования, что можно
использовать для их идентификации. Чтобы отличить третичный
амин от первичного или вторичного, можно использовать реакцию
с хлористым ацетилом.
Если к 0,5 мл первичного или вторичного амина прибавлять
пипеткой по каплям хлористый ацетил, то наблюдается энергич-
ная реакция. Действие хлористого ацетила на третичный амин со-
провождается лишь едва заметным нагреванием.
Твердые амины перед проведением реакции растворяют в бен-
золе.
Фенолы. Кацлю или 0,02 г исследуемого вещества растворяют
в 1 мл этилового спирта, прибавляют 1 мл воды и затем несколь-
ко капель 5%-ного раствора хлорного железа. Присутствие фенола
узнают по появлению зеленого, фиолетового или красного окра-
шивания.
Поскольку само хлорное железо имеет желтый цвет, появле-
ние желтой или коричневой окраски следует считать отрица-
тельным результатом пробы. В случае тимола, гидрохинона,
03
4-оксидифенила, о-нитрофенола и пикриновой кислоты проба дает
отрицательный результат. Некоторые амины и уксуснокислый
натрий дают красноватое окрашивание
Спирты. В сухую пробирку помещают 0,5 мл исследуемого
вещества и измеряют температуру Перемешивая термометром,
добавляют две капли хлористого ацетила Если происходит энер-
гичная реакция (разогревание), можно сделать вывод о наличии
первичного или вторичного спирта. Если реакции не наблюдается,
то добавляют еще пять капель хлористого ацетила, и наблюдают
за температурой Повышение температуры более чем на 10°C ука-
зывает на присутствие третичного спирта.
Если исследуемое вещество твердое, то растворяют 0,5 г его
в 1 мл теплого бензола, охлаждают до 20 °C, измеряют темпера-
туру и при помешивании прибавляют 7 капель хлористого аце-
тила. Повышение температуры более чем на 5 °C следует считать
показателем присутствия спирта.
Вода, фенолы и амины также дают эту реакцию.
Альдегиды. К 1 мл фуксинсернистой кислоты прибавляют кап-
лю исследуемого вещества Альдегиды немедленно дают интенсив-
ное фиолетовое окрашивание
Имея дело с веществами, нерастворимыми в воде, берут две
капли (или 0,05 г твердого вещества) растворяют в 1 мл спирта
и прибавляют к раствору 1 мл фуксинсернистой кислоты. Резуль-
тат считают отрицательным, ес'ли фиолетовое окрашивание не по-
является в течение 15 с; при более длительной выдержке окраску
может вызвать и кислород воздуха
Сложные эфиры. В пробирке смешивают 1 мл 1 н. раствора
едкого кали в этиловом спирте и 3 капли водного 40%-ного рас-
твора хлоргидрата гидроксиламина. Сливают прозрачный раствор
с образовавшегося осадка и к раствору прибавляют 2 капли (или
0,05 г) исследуемого вещества Нагревают минуту до кипения, за-
тем охлаждают, прибавляют каплю 5%-ного раствора хлорного же-
леза и 10 капель 2 н. соляной кислоты для растворения осадка,
который может образовываться в некоторых случаях. Положи-
тельной реакцией на сложный эфир считается появление красной
или пурпурной окраски.
Кетоны. Можно получить семикарбазон или 2,4-динитрофе-
нилгидразон (см стр 306)
Нитрилы и амиды. При нагревании с водным раствором ед-
кого кали происходит гидролиз, сопровождающийся выделением
аммиака или амина
Растворяют 5 гранул едкого кали в 5 каплях воды, прибав-
ляют 2 мл диэтиленгликоля и 8 капель (или 0,2 г) исследуемого
вещества. Осторожно нагревают до кипения, проверяя реакцию
паров влажной лакмусовой бумажкой; если образуется аммиак
или алифатический амин, то красный лакмус синеет. Капли ще-
лочного раствора, попадая на индикаторную бумагу, могут вве-
сти в заблуждение.
304
Аналогичный результат получается и при нагревании-со ще-
лочью солей алифатических аминов
Нитросоединения. При восстановлении в нейтральном раст-
воре образуются гидроксиламины, способные выделять металличе-
ское серебро из аммиачного раствора азотнокислого серебра
В 10 мл 50%-ного спирта растворяют 0,3 г исследуемого ве-
щества, прибавляют 0,5 г хлористого аммония и 0,5 г цинковой
пыли Смесь встряхивают и нагревают до кипения в течение 2 мин.
После охлаждения фильтруют и прибавляют аммиачный раствор
нитрата серебра. Выделение металлического серебра (серый хло-
пьевидный осадок) указывает на присутствие нитро- или нитро-
зогруппы.
Галогенпроизводные. Если исследуемое вещество нераство-
римо в воде, концентрированной соляной и концентрированной сер-
ной кислотах, а при предварительных испытаниях доказано при-
сутствие в веществе галогена, можно сделать вывод, что вещество
является ароматическим или насыщенным алифатическим галоген-
производным
Углеводороды. Исследуемое вещество представляет собой
обычно углеводород в том случае, если ею нельзя отнести на
основании проведенных испытаний ни к одному из перечисленных
выше классов
Насыщенные алифатические углеводороды нерастворимы в
воде, а также в концентрированной серной кислоте при нагре-
вании Ненасыщенные (алкены) углеводороды растворяются в
концентрированной серной кислоте и присоединяют бром.
Для определения исследуемое вещество растворяют в четы-
реххлористом углероде К раствору прибавляют бром, также рас-
творенный в четыреххлористом углероде Если происходит немед-
ленное обесцвечивание, то весьма вероятно присутствие двойной
связи. При медленном обесцвечивании при появлении тумана бро-
мистого водорода алкенов в пробе нет.
Ароматические углеводороды не растворяются в концентриро-
ванной серной кислоте на холоду, но могут растворяться при на-
гревании Кроме того, ароматические углеводороды подвергаются
нитрованию.
3. ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ
ПРОИЗВОДНЫЕ СПИРТОВ
Эфиры бензойной, п-нитробензойной или 3,5-динит роб ензой-
ной кислот
R—ОН + С1—ОС у"/—n°2 —* R—о—ОС—NO; + НС1
К 0,5 г (или двадцати каплям) спирта или фенола в пробирке
добавляют 20 капель бензоилхлорида или 0,5 г «-нитро- либо
3,5 динитробензоилхлорида Раствор при встряхивании на-
гревают 3—5 мин в кипящей водяной бане, затем охлаждают и
305
встряхивают с 10 мл 5%-ного рас+вора соды. Твердый осадок от-
фильтровывают, промывают водой и перекристаллизовывают из
разбавленного этилового спирта. Если спирт чувствителен к дей-
ствию кислот (третичные спирты), то его растворяют в 1 мл пири-
дина и после этого прибавляют хлорангидрид.
ПРОИЗВОДНЫЕ ФЕНОЛОВ
Эфиры бензойной, п-нитробензойной и 3,5-динитробензойной
кислот получают, как это описано выше для спиртов.
Бромпроизводные
Вг
Небольшое количество фенола растворяют в воде или ацетоне,
при встряхивании прибавляют по каплям раствор брома (2 мл
брома, 5 г бромистого калия в 50 мл воды) до тех пор, пока не
прекратится обесцвечивание. Полученный осадок отсасывают и
хорошо промывают водой. Перекристаллизовывают из 96%-ного
или 50%-ного этилового спирта.
Феноксиуксусная кислота
NaOH ^/О-СН2-СООН
Г [J +С1—СН2—СООН -----> Гн +НС1
К раствору 1 г хлоруксусной кислоты и 0,6 г фенола в 2 мл
воды прибавляют 1,5—2 г едкого натра, растворенного в 3 мл
воды. При необходимости добавляют еще воды до полного раство-
рения. Нагревают 30—60 мин при 80—100°C. После этого охлаж-
дают, нейтрализуют разбавленной соляной кислотой по конго
красному или метиловому оранжевому и экстрагируют феноксиук-
сусную кислоту эфиром. Эфирный слой промывают водой и затем
встряхивают с разбавленным раствором соды. Феноксиуксусная
кислота в виде натриевой соли переходит в раствор соды, из кото-
рого ее выделяют подкислением соляной кислотой. Феноксиуксус-
ную кислоту отсасывают и перекристаллизовывают из воды.
ПРОИЗВОДНЫЕ АЛЬДЕГИДОВ И КЕТОНОВ
п-Нитрофенилгидразоны или 2,4-динитрофенилгидразоны
R\ лл
А=о + h2n-nh—/ \—nq2 —>
R/Z \==/
я-нитрофенилгидразин
R\ / /ЛА
—> V=N—NH—" у—NO2 + H2O
R'/ \==/
Л-низрофенилгидразоц
306
В круглодонной колбе с обратным холодильником нагревают
до кипения смесь 0,7 г n-нитрофенил- или 2,4-динитрофенилгидра-
зина с 1 г (или 1 мл) карбонильного соединения и 25 мл спирта.
Через холодильник добавляют катализатор — 1 мл концентриро-
ванной соляной кислоты.
Производное должно выпасть после 15-минутного кипячения
и последующего охлаждения раствора. Перекристаллизовывают из
спирта.
Семикарбазоны
R\ Rx
С=О + H2N—NH—СО—NH2 —> /C=N—NH— CO—NH2 + H2O
R'Z R'Z
В пробирке растворяют 0,5 г хлоргидрата семикарбазида и
1 г ацетата натрия в 3 мл воды. К раствору добавляют 20 капель
(или 0,5 г)карбонильного соединения и такое количество спирта,
чтобы получить гомогенный раствор. После этого нагревают
10 мин в кипящей водяной бане, затем охлаждают, образовав-
шийся осадок отфильтровывают и перекристаллизовывают из раз-
бавленного спирта.
ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ
Анилиды
R—СООН + РС15 —> R—СО—Cl + POClj + НС1
R—СОС1 + H2N-R' —► R-CO—NH—R'+ НС1
Смесь 0,2—0,3 г безводной кислоты с небольшим избытком пяти-
хлористого фосфора нагревают несколько минут в горячей водяной
бане. Нагревание ведут в вытяжном шкафу. Охлажденную реак-
ционную смесь растворяют в нескольких миллилитрах петролей-
ного эфира. Для удаления хлорокиси фосфора встряхивают
с небольшим количеством (несколько капель) воды. Полученный
раствор хлорангидрида кислоты немедленно используют для даль-
нейшей реакции.
Для получения анилида к раствору хлорангидрида в петро-
лейном эфире при встряхивании прибавляют примерно 20 капель
анилина. По окончании реакции встряхивают с несколькими мил-
лилитрами разбавленного раствора едкого натра. Полученный про-
дукт реакции отфильтровывают, промывают один раз небольшим
количеством разбавленной соляной кислоты и разбавленным рас-
твором кислого углекислого натрия. Перекристаллизовывают из
небольшого количества спирта.
Ангидриды и хлорангидриды кислот
(R—CO)2O + H2N——* R—СО—NH—+ RCOOH
R—СОС1 + H2N—/ > —> R-CO-NH—+ HC1
307
К исследуемому веществу (20 капель или около 0,5 г) осто-
рожно прибавляют 20 капель анилина и 5 мл 10%-ного раствора
едкого натра Перемешивают, нагревают минуту в кипящей водя-
ной бане и охлаждают Анилид хорошо выкристаллизовывается,
если нет избытка анилина Если кристаллизации не происходит,
то отделяют получившееся масло и промывают его разбавленной
соляной кислотой. Полученные кристаллы перекристаллизовы-
ваются из этилового спирта или его смеси с водой
ПРОИЗВОДНЫЕ ПЕРВИЧНЫХ И ВТОРИЧНЫХ АМИНОВ
Ацетамиды (из аминов, содержащих более пяти атомов угле-
рода)
R-NH2 + О(ОС—СН3)2 —► R—NH—СО—СНз + НООС—СН3
В пробирке смешивают 20 капель (или 0,5 г) амина с 20 кап-
лями уксусного ангидрида и нагревают несколько минут на водя-
ной бане при 80—90°C После охлаждения выливают смесь в не-
большое количество воды Потиранием стеклянной палочкой вы-
зывают кристаллизацию продукта реакции Кристаллы промывают
водой и перекристаллизовывают из спирта, водно-спиртовой смеси
или циклогексана
ПРОИЗВОДНЫЕ НИТРОСОЕДИНЕНИЙ
Цветная реакция с хлорным железом и щелочью (для первич-
ных и вторичных алифатических нитросоединений):
R-CH2—NQ2 + NaOH —>
R—CH—NO2
3NaCl + [R—CH=NO2]JFe,+
х/°”|
r—CH=N
I
eCl3
Na+ + H2O
Нитросоединение действием небольшого количества концентри-
рованного раствора едкого натра переводят в натриевую соль
ацц-формы, раствор которой в воде дает при добавлении 3%-ного
раствора хлорного железа кроваво-красное окрашивание
ПРОИЗВОДНЫЕ ГАЛОГЕНАЛКИЛОВ
Анилиды
c0h5nco
RX + Mg —> RMgX ---------->
XMg—N—CQR
H2o
--------->
—MgfOHJX
Магниевую стружку (0,3—0,5 г) активируют нагреванием С
кристаллом иода Прибавляют 5—10 мл абсолютного этилового
эфира и 1 мл галогенпроизводного. После начала реакции нагре-
зоя
йают 20 мин на водяной бане, так чтобы эфир слабо кипел *.
После того как растворится почти весь магний, раствор филь-
труют, и к фильтрату добавляют 0,5 мл фенилизоцианата в не-
большом количестве эфира Через некоторое время охладившийся
раствор выливают в несколько миллилитров холодной воды, в ко-
торой предварительно добавляют около 1 мл концентрированной
соляной кислоты Эфирный слой отделяют, сушат сернокислым
натрием, после чего отгоняют эфир. Остаток кристаллизуют из
этилового спирта.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Воскресенский? И Техника лабораторных работ Изд 10 е М «Хи-
мия», 1973 720 с
Потапов В М ТатаринчикС Н Органическая химия М «Химия»,
1972 510 с
Препаративная органическая химия Изд 2 е М «Химия», 1964 907 с
* Реакцию надо проводить в колбе с обратным холодильником и хлоркаль-
циевои трубкой
ПРИЛОЖЕНИЯ
Приложение 1
Осушители для органических соединений
Органические соединения
Углеводороды .......................
Галогенпроизводные .................
Спирты.........'....................
Эфиры ..............................
Альдегиды...........................
Кетоны..............................
Органические кислоты................
Амины...............................
Нитросоединения.....................
Фенолы..............................
Осушители
СаС12, CaSO4, Р2О5, Na
СаС12, Na2SO4, MgSO4 Р2О5
MgSO4, CaSO4, К2СО3, CaO
СаС12, CaSO4, Na
CaCl2, MgSO4, Na2SO4
MgSO4, Na2SO4, K2CO3
MgSO4, Na2SO4, CaSO4
KOH, NaOH, K2CO3, CaO
CaCl2, Na2SO4
Na2SO4
Приложение 2
Давление пара воды лри различных температурах
Темпера- тура, °C Давление, мм рт. ст. Темпера- тура, °C Давление, мм рт. ст. Темпера- тура, °C Давление, мм рт ст. Темпера- тура, °C Давление» мм рт ст.
60 149,3 70 233,7 80 355,1 90 525,8
61 156,4 71 243,9 81 369,7 91 546,0
62 163,8 72 254,6 82 384,9 92 567,0
63 171,4 73 265,7 83 400,6 93 588,6
64 179,3 74 277,2 84 416,8 94 610,9
65 187,5 75 289,1 85 433,3 95 633,9
66 196,1 76 301,4 86 450,9 96 657,6
67 205,0 77 314,1 87 468,7 97 682,1
68 214,2 78 327,3 88 487,1 98 707,3
69 223,7 ' 79 341,0 89 506,1 99 733,2
310
Приложение 3
Давление сжиженных газов в баллонах
Газ Температура Кипения. °C Критическая температура, °C Избыточное давление при 20 °C, кгс/см2
Углекислый газ .... -78,2 ’ +31,1 55,25
Аммиак -33,4 + 132,4 7,46
Сероводород -60 + 100,4 16,7
Хлор -33,6 + 144,0 5,62
Приложение 4
Цвет баллонов со сжатыми газами
Газ Цвет баллона s Цвет надписи
Азот Черный Желтый
Аммиак Темно-желтый Черный
Ацетилен Белый Красный
Водород Темно-зеленый Красный
Кислород Светло-голубой Черный
Сернистый газ Черный Белый
Углекислый газ Черный Желтый
Приложение 5
Плотность растворов серной кислоты (20 °C)
d % моль/л г/л d % моль/л г/л
1 000 0,261 0,0266 2,608 1,095 14,04 1,567 153,7
005 0,986 0,1010 9,906 100 14,73 1,652 162,0
010 1,731 0,1783 17,49 105 15,41 1,735 170,2
015 2,485 0,2595 25,45 НО 16,08 1,820 178,5
020 3,242 0,3372 33,07 115 16,76 1,905 186,8
025 4,000 0,4180 41,99 120 17,43 1,990 195,2
030 4,746 0,4983 48,87 125 18,09 2,075 203,5
035 5,493 0,5796 56,85 130 18,76 2,161 211,9
040 6,237 0,6613 64,86 135 19,42 2,247 220,4
045 6,956 0,7411 72,69 170 23,95 2,857 280,2
050 7,704 0,8250 80,92 175 24,58 2,945 288,8
055 3,415 0,9054 88,80 180 25,21 3,033 297,5
060 9,129 0,9856 96,67 185 25,84 3,122 306,2
065 9,843 1,066 104,6 190 26,47 3,211 314,9
070 10,56 1,152 113,0 195 27,10 3,302 323,9
075 11,26 1,235 121,1 200 27,72 3,391 332,6
080 11,96 1,317 129,2 205 28,33 3,481 341,4
085 12,66 1,401 137,4 210 28,95 3,572 350,3
090 13,36 1,484 145,6 215 29,57 3,663 359,3
>11
Продолжение
d % моль/л Г/Л d % моль/л г/л
1,220 30,18 3,754 368,2 1,490 59,24 9,000 882,7
225 30,79 3,846 377,2 495 59,70 9,100 892,5
230 31,40 3,938 386,2 500 60,17 9,202 902,5
235 32-.01 4,031 395,4 575 60,62 9,303 912,4
240 32,61 4,123 404,4 510 61,08 9,404 922,3
245 33,22 4,216 413,5 515 61,54 9,506 932,3
250 33,82 4,310 422,7 520 62,00 9,608 942,4
255 34,42 4,404 431,9 525 62,45 9,711 952,5
260 35,01 4,498 441,2 530 62,91 9,813 962,5
265 35,60 4,592 450,4 535 63,36 9,916 972,6
270 36,19 4,686 459,6 540 63,81 10,02 982,8
275 36,78 4,781 468,9 545 64,26 10,12 992,6
280 37,36 4,876 478,2 550 64,71 10,23 1003
285 37,95 4,972 487,6 555 65,15 10,33 1013
290 38,53 5,068 497,1 560 65,59 10,43 1023
295 39,10 5,163 506,4 565 66,03 10,54 1034
300 39,68 5,259 515,8 570 66,47 10,64 1044
305 40,25 5,356 525,3 575 66,91 10,74 1053
310 40,82 5 452 534,7 580 67,35 10,85 1064
315 41,39 5,549 544,2 585 67,79 10,96 1075
320 41,95 5,646 553,8 590 68,23 11,06 1085
325 42,51 5,743 563,3 595 68,66 11,16 1095
330 43,07 5,840 572,8 600 69,09 11,27 1105
335 49,62 5,938 582,4 605 69,53 11,38 1116
340 44,17 6,035 591,9 610 69,96 11,48 1126
345 44,72 6,132 601,4 615 70,39 11,59 Ц36
350 45,26 6,229 610,9 620 70,82 11,70 1148
355 45,80 6,327 620,6 625 71,25 11,80 1157
360 46,33 6,424 630,1 630 71,76 11,91 1168
365 46,86 6,522 639,7 635 72,09 12,02 1179
370 47,39 6,620 649,3 640 72,52 12,13 1290
375 47,92 6,718 658,9 645 72,95 12,24 1200
380 48,45 6,817 668,6 650 73,37 12,34 1210
385 48,97 6,915 678,2 655 73,80 12,45 1221
390 49,48 7,012 687,7 660 74,22 12,56 1232
395 49,99 7,110 697,3 665 74,64 12,67 1243
400 50,50 7,208 707,0 670 75,07 12,78 1253
405 51,01 7,307 716,7 675 75,49 12,89 1264
410 51,52 7,406 726,4 680 75,92 13,00 1275
415 52,02 7,505 736,1 685 76,34 13,12 1287
420 52,51 7,603 745,7 690 76,77 13,23 1298
425 53,01 7,702 755,4 695 77,20 13,34 1308
430 53,50 7,801 765,1 700 77,63 13,46 1320
435 54,00 7,901 774,9 705 78,06 13,57 1331
440 54,49 8,000 784,6 710 78,49 13,69 1343
445 54,97 8,099 794,3 715 78,93 13,80 1354
450 55,45 8,198 804,1 720 79,37 13,92 1365
455 55,93 8,297 813,8 725 79,81 14,04 1377
460 56,41 8,397 823,6 730 80,25 14,16 1389
465 56,89 8,497 833,4 735 80,70 14,28 1401
470 57,36 8,598 843,3 740 81,16 14,40 1412
475 57,84 8,699 853,2 745 81,62 14,52 1424
480 58,31 8,799 863,0 750 82,09 14,65 1437
485 58,78 * '8,899 872,8 755 82,57 14,78 1450
Продолжение
d % МОЛЬ/Л Г/Л d % МОЛЬ/Л Г/Л
1,760 83,06 14,90 1461 1,822 91,56 17,01 1668
765 83,57 14,98 1469 823 91,78 17,06 1673
770 84,08 15,17 1488 824 92,00 17,11 1678
775 84,61 15,31 1502 825 92,25 17J7 1684
780 85,16 15,46 1516 826 92,51 17,22 1689
785 85,74 15,61 1531 827 92,77 17,28, 1695
790 86,35 15,76 1546 828 93,03 17,34 1701
795 86,99 15,92 1561 829 93,33 17'40 1707
800 87,69 16,09 1578 830 93,64 17^7 1713
8о5 88,43 16,27 1596 831 93,94 17,54 1720
810 89,23 16,47 1615 832 94,32 17,62 1728
815 90,12 16,68 1636 833 94,72 17,70 1736
820 91,11 16,91 1659 834 95,12 17'79 1745
821 91,33 16,96 1663 835 95,72 17'91 1757
Приложение 6
Плотность растворов соляной кислоты (20 °C)
d % МОЛЬ/Л г/л d % МОЛЬ/Л г/л
1,000 0,360 0,0987 3,599 1,095 19,41 5,829 2125
005 1,360 0,3745 13,65 100 20,39 в; 150 224'2
010 2,364 0,6547 23,87 105 21,36 6'472 236 0
015 3,374 0,939 34,24 НО 22,33 6,796 247 8
020 4,388 1,227 44,74 115 23,29 7; 122 2597
025 5,408 1,520 55,42 120 24,25 7,449 2716
030 6,433 1,817 66,25 125 25,22 7,782 283 7
035 7,464 2,118 77,22 130 26,20 8; 118 3960
040 8,490 2,424 88,27 135 27,18 5',459 308,4
045 9,510 2,725 99,35 140 28,18 8,809 321 2
050 10,52 3,029 110,4 145 29,17 9,159 3333
055 11,52 3,333 121,5 150 30,14 9,505 346 6
060 12,51 3,638 132,6 155 31,14 9,863 359 6
065 13,50 3,944 143,8 160 32,14 10,225 372,8
070 14,495 4,253 155,1 165 33,16 10,59а 38б'3
075 15,485 4,565 166,4 170 34,18 10,97 3993
080 16,47 4,878 177,8 180 36,23 11,73 427 7
085 17,45 5,192 189,3 190 38,32 12,50 455,8
090 18,43 5,5096 200,9 198 40,00 13,14 479Д
Приложение 7
Плотность растворов азотной кислоты (20 °C)
d % моль/л г/л d % моль/л г/л
1,000 0,3296 0,0523 3,295 015 3,073 0,4950 31,19
005 1,255 0,2001 12,61 020 3,982 0,6445 40,61
010 2,164 0,3468 21,85 025 4,883 0,7943 5005
31?
313
Продолжений
d % МОЛЬ/Л г/л d % моль/л г/л
1,030 5,784 0,9454 59,57 1,305 49,21 10,19 642,1
035 6,661 1,094 68,93 310 50,00 10,39 654,7
040 7,530 1,243 78,32 315 50,85 10,61 668,5
045 8,398 1,393 87,77 320 51,71 10,83 682,4
050 9,259 1,543 97,22 325 52,56 11,05 696,3
055 10,12 1,694 106,7 330 53,41 11,27 710,1
060 10,97 1,845 116,3 335 54,27 11,49 724,0
065 11,81 1,997 125,8 340 55,13 11,72 738,5
070 12,65 2,148 135,3 345 56,04 11,96 753,6
075 13,48 2,301 145,0 350 56,95 12,20 768,7
080 14,31 2,453 154,6 355 57,87 12,44 783,8
085 15,13 2,605 164,1 360 58,78 12,68 799,0
090 15,95 2,759 173,8 365 59,69 12,93 814,7
095 16,76 2,913 183,5 370 60,67 13,19 831,1
100 17,58 3,068 193,3 375 61,69 13,46 848,1
105 18,39 3,224 203,1 380 62,78 13,73 865,1
ПО 19,19 3,341 213,0 385 63,72 14,01 882 8
115 20,00 3,559 223,0 390 64,74 14,29 900,4
120 20,79 3,666 232,9 395 65,84 14,57 918,1
125 21,59 3,854 242,8 400 66,97 14,88 937,6
130 22,38 4,012 252,8 405 68,10 15,18 956,5
135 23,16 4,171 262,8 410 69,23 15,49 976,0
140 23,94 4,330 272,8 415 70,34 15,81 996,2
145 24,71 4,489 282,9 420 71,63 16,14 1017
150 25,48 4,649 292,0 425 72,86 16,47 1038
155 26,24 4,810 303,1 430 74,09 16,81 1059
160 27,00 4,970 313,2 435 75,35 17,16 1081
165 27,76 5,132 323,4 440 76,71 17,53 1105
170 28,51 5,293 333,5 445 78,07 17,90 1128
175 29,25 5,455 343,7 450 79,43 18,28 1152
180 30,00 5,618 354,0 455 80,88 18,68 1177
185 30,74 5,780 364,2 460 82,39 19,09 1203
190 31,47 5,943 374,5 465 83,91 19,51 1229
195 32,21 6,110 385,0 470 85,50 19,95 1257
200 32,04 6.273 395,3 475 87,29 20,43 1287
205 33,68 6,440 405,8 480 89,07 20,92 1318
210 34,41 6,607 416,3 485 91,13 21,48 1353
215 35,16 6,778 427,1 490 93,49 22,11 1393
220 35,93 6,956 438,3 495 95,46 22,65 1427
225 36,70 7,135 449,6 500 96,73 23,02 1450
230 37,48 7,315 460,9 501 96,98 23,10 1456
235 38,25 7,497 472,4 502 97,23 23,18 1461
240 39,02 7,679 483,8 503 97,49 23,25 1465
250 40,58 8,049 507,2 504 97,74 23,33 1470
255 41,36 8,237 519,0 505 97,99 23,40 1474
260 42,14 8,426 530,9 506 98,25 23,48 1479
265 42,92 8,616 542,9 507 98,50 23,56 1485
270 43,70 8,808 555,0 508 98,76 23,63 1488
275 44,48 9,001 567,2 509 99,01 23,71 1494
280 45,27 9,195 579,4 510 99,26 23,79 1499
285 46,06 ,9,394 591,9 511 99,52 23,86 1503
290 46,85 9,590 604,3 512 99,77 23,94 1508
295 47,63 9,789 616,8 513 100,00 24,01 1513
300 48,42 9,990 629,5
Приложение 8
Плотность растворов едкого натра (20 °C)
d % моль/л г/л d % моль/л г/л
1,000 0,159 0,0398 1,592 1,270 24,64 7,824 313,0
005 0,602 0,151 6,040 275 25,10 8,000 320,0
010 1,04 0,264 10,56 280 25,56 8,178 327,1
015 1,49 0,378 15,12 285 26,02 8,387 334,3
020 1,94 0,494 19,76 290 26,48 8,539 341,6
025 2,39 0,611 24,44 295 26,94 8,722 348,9
030 2,84 0,731 29,24 300 27,41 8,906 356,2
035 3,29 0,851 34,04 305 27,87 9,092 363,7
040 3,74 0,971 38,84 310 28,33 9,278 371,2
045 4,20 1,097 43,88 315 28,80 9,466 378,6
050 4,65 1,222 48,88 320 29,26 9,656 386,2
055 5,11 1,347 53,88 325 29,73 9,847 393,9
060 5,56 1,474 58,96 330 30,20 10,04 401,6
065 6,02 1,602 64,08 335 30,67 10,23 409,2
070 6,47 1,731 69,24 340 31,14 10,43 417,2
075 6,93 1,862 74,48 345 31,62 10,63 425,2
080 7,38 1,992 79,68 350 32,10 11,83 433,2
085 7,83 2,123 84,92 355 32,58 11,03 441,2
090 8,28 2,257 90 28 360 33,06 11,24 449,6
095 8,74 2,391 95,64 365 33,54 11,45 458,0
100 9,19 2,527 101,1 370 34,03 11,65 466,7
105 9,64 2,664 106,6 375 34,52 11,86 474,4
ПО 10,10 2,802 112,1 380 35,01 12,08 483,2
115 10,55 2,942 117,7 385 35,50 12,29 491,6
120 11,01 3,082 123,3 390 36,00 12,51 500,4
125 11,46 3,224 129,0 395 36,49 12,73 509,2
[30 11,92 3,367 134,7 400 36,99 12,95 518,0
135 12,37 3,510 140,4 405 37,49 13,17 526,8
140 12,83 3,655 146,2 410 37,99 13,39 535,6
145 13,28 3,801 152,0 415 38,49 13,61 544,4
150 13,73 3,947 157,9 420 38,99 13,84 553,6
155 14,18 4,095 163,8 425 39,40 14,07 562,8
160 14,64 4,244 169,8 430 40,00 14,39 572,0
165 15,09 4,395 175,8 435 40,51 14,53 581,2
170 15,54 4,545 181,8 440 41,03 14,77 590,8
175 15,99 4,697 187,9 445 41.55 15,01 600,4
180 16,44 4,850 194,0 450 42,07 15,25 610,0
185 16,89 5,004 200,2 455 42,59 15,49 619,6
190 17,34 5,160 206,4 460 43,12 15,74 629,6
195 17,80 5,317 212,7 465 43,64 15,98 639,2
200 18,25 5,476 219,0 470 44,17 14,23 649,2
205 18,71 5,636 225,4 475 44,69 16,48 659,2
210 19,16 5,796 231,8 480 45,22 16,73 669,2
215 19,62 5,958 238,3 485 45,75 16,98 679,2
220 20,07 6,122 244,9 490 46,27 17,23 689,2
225 20,53 6,286 251,4 495 46,80 17,49 699,6
230 20,98 6,451 258,0 500 47,33 17,75 710,0
205 21,44 6,619 264,8 505 48,85 18,00 720,0
220 21,90 6,788 271,5 510 48,38 18,26 730,4
235 22,36 6,958 278,3 515 48,90 18,52 740,8
250 22,82 7,129 285,2 520 49,44 18,78 751,2
255 23,27 7,302 292,1 525 49,97 19,05 762,0
260 23,73 7,475 299,0 530 50,50 19,31 772,4
265 24,19 7,650 306,0
314
315
Приложение 9
Плотность растворов едкого кали (20 °C)
d % моль/л г/л 1 d % моль/л г/л
1,000 0,197 0,035 1,964 1,270 28,29 6,40 359,1
005 0,743 0,133 7,463 275 28,77 6,54 367,0
010 1,295 0,233 13,07 280 29,25 6,67 374,3
015 1,84 0,333 18,68 285 29,73 6,81 382,1
020 2,38 0,433 24,30 290 30,21 6,95 390,0
025 2,93 0,536 30,07 295 30,68 7,08 397,3
030 3,48 0 639 35,85 300 31,15 7,22 405,1
035 4,03 0,744 41,75 305 31,62 7,36 413,0
040 4,58 0,848 47,58 310 32,09 7,49 420,3
045 5,12 0,954 53,53 315 32,56 7,63 428,1
050 5,66 1,06 59,48 320 33,03 7,77 ' 436,0
055 , 6,20 1,17 65,65 325 33,50 7,91 443,8
060 . 6,74 1,27 71,26 330 33,97 8,05 451,7
065 7,28 1,38 77,43 335 34,43 8,19 459,5
070 7,82 1,49 83,60 340 34,90 8,33 467,7
075 8,36 1,60 89,78 345 35,36 8,48 475,8
080 8,89 1,71 95,95 350 35,82 8,62 483,7
085 9,96 1,82 102,1 355 36,28 8,76 491,4
090 9,43 1,94 108,9 360 36,73' 8,90 499,4
095 10,49 2,05 115,0 365 37,19 9,05 507,8
100 11,03 2,16 121 2 370 37,65 9,19 515,7
105 11,56 2,28 127,9 375 38,10 9,34 524,1
ПО 12,08 2,39 134,1 380 38,56 9,48 531,9
115 12,61 2,51 140,8 385 39,01 9,63 540,3
120 13,14 2,62 147,0 390 39,49 9,78 > 548,8
125 13,66 2,74 153,7 395 39,92 9,93 557,2
130 14,19 2,86 160,5 400 40,37 10,07 565.0
135 14,70 2,97 166,6 405 40,82 10,22 573,4
140 15,22 3,09 173,4 410 41,26 10,37 581,9
145 15,74 3,21 180,1 . 415 41,71 10,52 590,3
150 16,26 3,33 186,6 420 42,15 10,67 598,7
155 16,78 3,45 193,6 425 42,60 10,82 607,1
160 17,29 3,58 200,9 430 43,04 10,97 615,5
165 17,81 3,70 207,6 435 43,48 11,12 623,9
- 170 18,32 3,82 214,3 440 43,92 11,28 632,9
175 18,84 3,94 221,1 445 44,36 11,42 640,8
180 19,35 4,07 228,4 450 44,79 11,58 649,7
185 19,86 4,19 235,1 455 45,23 11,73 658,2
190 20,37 4,32 242,4 460 45,66 11,88 666,6
195 20,88 4,45 249,7 465 46,09 12,04 675,6
200 21,38 4,57 256,4 470 46,53 12,19 684,0
205 21,88 4,70 263,7 475 46,96 12,35 693,0
210 22,38 4,83 271,0 480 47,39 12,50 701,4
215 22,88 4,95 277,7 485 47,82 12,66 710,4
220 23,38 5,08 285,0 490 48,25 12,82 719,3
225 23,87 5,21 292,3 495 48,67 12,97 727,7
230 24,37 5,34 299,6 500 49,10 13,13 736,7
235 23,86 5,47 306,9 505 49,53 13,29 745,7
240 25,36 5,60 314,2 510 49,95 13,45 754,7
245 25,85 5,74 322,1 515 50,38 13,60 763,1
250 26,34 5,87 329,4 520 '50,80 13,76 772,1
255 26,83 6,00 336,7 525 51,22 13,92 781,1
260 23,82 6,13 344,0 530 51,74 14,08 790,0
265 27,80 6,27 351,8 535 52,05 14,24 799,0
316
П риложение 10
Физические свойства спиртов и их производных
Спирт Т, °C Г. пл., °C
п-нитро беизоат 3.5 динитро беизоат
Метиловый 65 96 (180 мм) * 108
Этиловый 78 57 93
Изопропиловый 82 110 123
трет-Бутиловый 83 116 143
Пропиловый 9.7 35 74
Аллиловый 97 29 50
втор-Бутиловый 99 26 76
трет-Амиловый 102 85 117
Изобутиловый 108 ' 69 88
Пентанол-3 116 17 98
Бутиловый 118 36 (70 мм) 64
втор-Амиловый - 120 17 62
Амиловый 138 11 46
Гексиловый 157 5 61
Циклогексанол 161 50 113
Этиленгликоль 198 141 169
Бензиловый 205 86 113
Бутандиол-1,4 230 175 —
Г лицерин ... 1 290 188 —
* Здесь и далее в скобках указано дазление в миллиметрах ртутного столба.
Приложение 11
Физические свойства фенолов и их производных
Т пл,, °C
Соединение т. пл., °C Т. кип., °C бензоат 1 ° । со О S S X О О о. а. о ю и X арилокси- уксусные кислоты о *- О. «5 Е- О § i 7 Ф о,и-ДИ нитро- бензоат
Этилсалицилат 1 234 87 — — 108 —
о-Бромфенол . . 5 195 86 Три- : 95 143 — —•
о-Хлорфенол . . 7 176 — Ди-: 76 115 143
.и-Крезол .... 12 203 54 Три-: 84 102 90 —
о-КрезоЛ .... 31 192 — Ди-: 56 52 94 138
.и-Бромфенол . . 32 236 86 — 108 — —
Гваякол 32 205 —— Три-: 116 — 93 141
.и-Хлорфенол . . 33 214 — — — 99 156
я-К.резол .... 36 202 — Тетра-: 108 — 98 187
Фенол 42 183 68 Три-: 95 99 127 146
я-Хлорфенол . . 43 (37 мм) 217 93 Ди-; 90 156 171 186
о-Нитрофеьол . . 45 216 59 Ди-: 117 — 141 155
я-Бромфеиол . . 64 236 102 Ди-: 95 157 180 191
а-Нафтол .... 94 280 56 Ди-: 105 192 143 217
л-Нитрофенол . . 97 194 (70 мм) 95 Ди-: 91 — 174 159
Пирокатехин . . 105 246 Ди-: 81 Тетра- : 192 — 169 152
Резорцин .... НО 280 Ди-: 117 Ди-: 116 195 182 201
я-Нитрофенол . . 114 —. 142 118(142 мм) — 159 188
-Нафтол .... 123 286 107 84 155 169 210
Пирогаллол . . . 133 301 Три-: 90 Ди-: 158 — 230 205
Гидрохинон . . . 169 286 Ди-: 199 Ди-: 186 — 258 317
Флороглюцин . . 219 Три-: 173 Три-: 151 — 283 162
>17
П риложение 12
Физические свойства альдегидов и их производных
Альдегид Т. кип., °C Г пл , °C
я-иитрофе- иилгидразон 2,4-дииитрО- феннлгидра- зон семикарба ЗОИ
Муравьиный —21 182 167 169
Уксусный 21 129 168(147 мм) 162
Пропионовый 49 124 155 89 (154 мм)
Изомасляный 64 132 182 125
Масляный 74 92 122 106
Валериановый 103 — 98 (107 мм) —-
Капроновый 128 —» 104 106
ФурФУР0Л 161 54 214 (230 мм) 202
Янтарный 170 —— 280 —
Бензальдегид 179 192 237 222
Салициловый 196 223 248 231
Приложение 13
Физические свойства кетонов и их производных
Кетон Т. кип , °C Г. пл. °C
п-нитрофе- нилгидразон 2,4 дииитро- фенилгидра- зои семикарба- зон
Ацетон 56 150 128 189
Метилэтилкетон 80 129 117 146
Метилизопропилкетон .... 94 109 120 114
Диэтилкетон • 102 144 156 139
Диизопропилкетон 124 — 96 160
Приложение 14
Физические свойства карбоновых кислот и их производных
Кислота Т. пл , ОС Т. кип , °C Т. пл. анилида, °C Т. кип., °C
хлор- ангидрида ангидрида
Муравьиная .... 8 100 50 — —
Уксусная 17 118 113 52 138
Акриловая 13 140 105 76 —
Пропионовая .... 140 105 80 168
Изомасляная .... 155 105 92 82
Масляная 163 96 100 98
Валериановая . . 186 63 — —
Дихлоруксусная . . 194 119 107 215
Капроновая .... 205 95 — •—
Олеиновая 14 223 (10 мм) 41 —
Лимонная 100 53 (199 мм) —
Щавелевая 101 — 90(149 мм) 64
Адипиновая .... 153 — 161 148 (9 мм)
Салициловая .... 158 — 175 — —
Фталевая 200. — 226 190 250
318
Приложение 15
Физические свойства первичных и вторичных аминов
и их производных — ацетамидов
Амин Т. пл , °C Т кип., °C Т пл. ацет- амида, °C Амии Т. пл., °C Т. кип., °C Т. пл. ацет- амида, °C
Метиламин . . . — -6 28 о-Толуидин . . . 200 111
Диметиламин . . 7 — .и-Толуидин . . — 203 65
Этиламин .... — 17 — .и-Броманилнп 18 251 87
Диэтиламин . . . — 56 — о-Броманилин 32 229 99
Бутиламин . . . 77 — я-Толуидин . . . 45 200 147
Изоамиламин . . 96 — а-Нафтиламин 50 300 159
Амиламин .... 104 — Дифениламин. . 54 302 101
Бензиламин . . . — 184 65 п-Броманилин 66 245 168
Анилин — 184 114 о-Нитроанилин 71 93
П риложение 16
Физические свойства галогеналкилов и их производных — анилидов
Галогена тки л Т. пл ани- лида, °C Галогеналкил Т. пл. ани- лида, °C
Метил- 114 трег-Бутил- 128
Этил- 104 ИзоОутил- 109
Изопропил 103 Бутил- 63
Пропил- 92 108
Аллил- 114 Амил- 96